Embed Size (px)
Citation preview

F. Hahn. Die Bestimmung von Jodid neben Bromid und Chlorid 75
Die Bestimmung von Jodid neben Bromid und Chloridl) Von FRIEDRICH L. HAHN
Mit 2 Figuren im Text
Vor kurzem haben S. W. GORBATSCHEFF und I. A. KASSATKINA eine Arbeit veroffentlicht2), die sich mit der genannten Frage be- schiiftigt. Sie haben unter anderem auch die Titration von Jodid mit Permanganat unter potentiometrischer Erkennung des Endpunktes untersucht. Dabei ist es ihnen anscheinend entgangen, da13 ich schon vor einigen Jahren gemeinsam mit G. WEILER dieses Verfahren durch Zugabe von Tetrachlorkohlenstoff zu der titrierten Losung ganz wesentlich verbessern k ~ n n t e . ~ ) Der Potentialsprung am Ausgleich (Aquivalenzpunkt) wird wesentlich steiler, ist also vie1 leichter zu fassen, als ohne diesenzusatz, bzw. es durfen groBereMengen an Bromid oder Chlorid anwesend sein, ohne daB der dem Jodid entsprechende Umschlag verdeckt wird. Wir haben damals stets den Punkt zu fassen gesucht, an dem aller Jodwasserstoff zu freiem Jod oxydiert, also 3 Val KMnO, auf 1 Mol Jodid verbraucht ist. GORBATSCHEFF und KASSATKINA, die ohne Zugabe von Tetrachlorkohlenstoff gearbeitet haben, berichten, daB noch ein zweiter, wesentlich deutlicherer Poten- tialsprung beim Verbrauch von 2 Val KMnO, auf 1 Mol Jodid auf- trete, dem Ubergang des freien Jods in Chlorjod entsprechend. Sie empfehlen, so weit zu titrieren, und stellen uberdies fest, daB dieser Punkt auch ohne Potentiometer gefaBt werden kann. Fugt man namlich der titrierten Losung etwas Chloroform oder Tetrachlor- kohlenstoff zu, so nimmt dieser bis zum Verbrauch von 1 Val Per- manganat auf 1 Mol Jodid das freiwerdende Jod auf, ffrbt sich also standig dunkler; von da an wird das Jod weiter oxydiert, so daB sich die Farbe des Tetrachlorkohlenstoffs aufhellt, und beim Verbrauch von 2 Val KMnO, auf 1 Mol Jodid ist der Tetrachlorkohlenstoff
l) VII. Mitteilung zur Auswertbarkeit potentiometrisoher Titrationen -
2, S. W. GOFLBATSCHEFF u. I. A. KASSATKINA, Z. anorg. u. allg. Chem. 191
3, F. HAHN u. G. WEILER, Fr. 69 (1927), 417.
VI. Mitteilung: Z. phys. Cbem. A. 151 (1930). 80.
(1930), 104.

76 Zeitschrift fur anorganische und allgemeine Chemie. Band 195. 1931
wieder vollig entfarbt. Bei der starken Flirbekraft kleinster Jodmengen sollte dieser Umschlag tatsiichlich den Endpunk t vorzuglich anzeigen, wenn die Umsetzung zwischen Chlor (KMnO, in saurer Losung bei Gegenwart eines Riesenuberschusses an Chlorid) und Jod zu Chlorjod gegenuber der Reaktion zwischen Chlor und Bromid zu Rrom und Chlorid so stark begunstigt ist, dai3 die Entfarbmg tatslichlich stets am richtigen Punkt eintritt. Leider geben nun GORBATSCHEIF und KASSATKINA keinerlei Versuchszahlen, aus denen man sich ein Bild uber die Genauigkeit und Obereinstimmung der verschiedenen Ver- fahren machen konnte; aus zwei Griinden schien es deshalb wichtig, die Frage erneut zu uberprufen: Erstens kommt wegen der bekannten physiologischen ,, Jodfrage" der Ermittelung kleinster Jodidmengen gerade im Gemisch mit anderen Haloiden eine grol3e praktische Be- deutung zu, zweitens hat seit unsrer ersten Arbeit die Auswertbarkeit potentiometrischer Titrierungen sehr erhebliche Fortschritte gemacht, und wenn etwa die zwei Wendepunkte der Volumen-Potentialkurve tatsachlich dem Ausgleich der Reaktionenl)
und
entsprechen sollten, d a m muBte sich allerlei theoretisch Wichtiges aus den Kurven herauslesen lassen.
Fig. 1 zeigt zunlichst den schon fruher untersuchten EinfluS des Tetrachlorkohlenstoffzusatzes auf den Potentialgang; man sieht, daS bei gleicher Empfindlichkeit des benutzten Potentiometers unter An-
J' + JC1 fr J, + C1'
J, + Br,[+ nCl'] fr 2 JCl + 2Br'[+ (n - 2)CYJ
1) Die Formulierung soll folgendes ausdriicken: Wird bei Gegenwart reich- licher Mengen an Bromid und Chlorid Jodwasserstoff durch Permanganat oxydiert, so wird, auch m h d e m 1 Val Permanganat auf 1 Mol Jodid zugegeben ist, die Konzentration an Permanganat in der Losung sich nicht merklich von Null ent- fernen, sondern es wird jetzt ein weiteres Oxydationsprodukt in meBbaren Kon- zentrationen auftreten, nach Annahme von GARBATSCHEFF u. KASSAT~INA ist dies JCl. An dieser Stelle iindert sich also die Konzentration an Jodid (abnehmend) und die an Chlorjod (zunehmend); das heiBt, wir komen die Konzentrations- Lnderungen am ersten Wendepunkt durch die Fiktion beschreiben, daB eine LGsung von Jodid mit Chlorjodid titriert werde. Entsprechend begimt (immer nach den Amhmen von GARBATSCHEW u. KASSATIUNA) am zweiten Ausgleich, an dem das freie Jod unter Bildung von Chlorjod aufgebraucht ist, als nachstes Oxydationsprodukt Brom aufzutreten; wir konnen also hier Jod 81s Stoff axwehen, dessen Konzentration am Ausgleich stark absinkt, Brom, dessen Konzentration am Ausgleich stark ansteigt, als Reagens. Chlorionen gehen zwar auch in die Reaktionsgleichung ein, sind aber in solehem Uberschul3 vorhanden, daB ihre Konzentration trotzdem als konstant gelten kann.
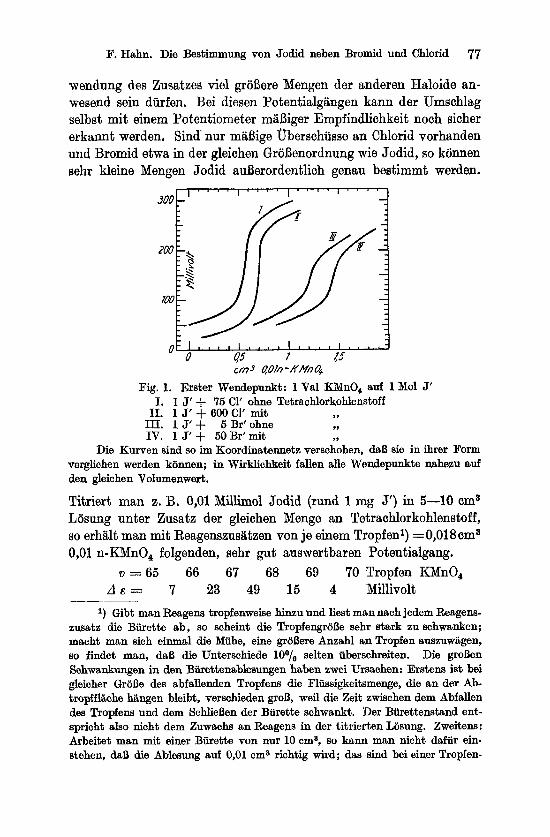
F. Hahn. Die Bestimmung von Jodid neben Bromid und Chlorid 77
wendung des Zusatzes vie1 groBere Mengen der anderen Haloide an- wesend sein diirfen. Bei diesen Potentialgangen kann der Umschlag selbst mit einem Potentiometer m5Biger Empfindlichkeit noch sicher erkannt werden. Sind nur miiBige uberschusse an Chlorid vorhanden und Bromid etwa in der gleichen GroBenordnung wie Jodid, so konnen sehr kleine Mengen Jodid auBerordentlich genau bestimmt werden.
cm3 qOln-KMn4
I. 1 J' f 75 C1' ohne Tetrachlorkohlenstoff Fig. 1. Erater Wendepunkt: 1 Val KMnO, auf 1 Mol J'
11. 1 J' + 600 C1' mit ,, 111. 1 J'+ 5 Br'ohne ,7
IV. 1 J' + 60 Br'mit ,, Die Kurven sind so im Koordinatennetz verschoben, daB sie in ihrer Form
verglichen werden klinnen; in Wirklichkeit fallen alle Wendepunkte naheeu auf den gleichen Volumenwert.
Titriert man z. B. 0,Ol Millimol Jodid (rund 1 mg J') in 5-10 ema Losung unter Zusatz der gleichen Menge an Tetrachlorkohlenstoff, so erhalt man mit Reagenszusiitzen von je einem Tropfen') = 0,018 ema 0,Ol n-KMnO, folgenden, sehr gut auswertbaren Potentialgang.
d E = 7 23 49 15 4 Millivolt v = 6 5 66 67 68 69 70 Tropfen KMnO,
1) Gibt man Reagens tropfenweise hinzu und liest man nach jedem Reagens. zuaatz die Burette ab, so scheint die 'I'ropfengroBe sehr stark zu schwanken; macht man sich einmal die Muhe, eine gr6Bere Anzahl an Tropfen auseuwiigen, so findet man, daB die Unterschiede 10% selten uberschreiten. Die grolen Schwankungen in den Burettenablesungen haben zwei Ursachen: Erstens ist bei gleicher GroBe des abfallenden Tropfens die Flussigkeitsmenge, die an der Ab- tropfflbhe hLngen bleibt, verschieden SOB, weil die Zeit zwischen dem Abfallen des Tropfens und dem SchlieBen der Biirette schwankt. Der Biirettenstand ent- spricht also nicht dem Zuwachs an Reagens in der titrierten LSsung. Zweitens: Arbeitet man mit einer Biirette von nur 10 cma, so kann man nicht dafiir ein- stehen, daB die Ablesung auf 0,Ol cms richtig wird; das sind bei einer Tropfen-

78 Zeitschrift fur anorganische und allgemeine Chemie. Band 196. 1931
Wendepunkt v, =67,3 Tropfen=1,23 em3. Mittelwert aus 13 Be- stimmungen: 1,244 em3; Grenzwerte: 1,22 und 1,27 em3.
Die Schwankungen liegen also durchaus innerhalb der Fehler der Volumenmessung ; durch Verwendung von MiGro- oder Wageburetten mussen sie sich noch erheblich verkleinern lassen. Auch ohnedies be- tragen sie nur etwa 40/0 bei der Bestimmung von 1 mg Jod.
Bei steigenden Uberschussen an Br' und C1' mussen die Reagens- zusiitze vergroBert werden, bei Anwesenheit von 2 Millimol Br' und 25 Millimol Cl', das ist J':Br': C1' = 1 :200:2500, auf etwa das Zehn- fache.
Beis piel: v =40 50 60 70 80 90 Tropfen KMnO,
A E = 10 17 47 11 4 Millivolt v, = 63,l Tropfen = 1,20 em3
Man kann hier also das Reagens schon aus einer feinen Burette in Anteilen von etwa 0,2 em3 zugeben oder tropfenweise mit 0,l n- Losung titrieren; die Schwankungen im End punkt betragen dann etwa einen Tropfen. Der Verbrauch ist im Durchschnitt etwas geringer als in reinen Jodidlosungen: 1 em3 J' = 1,14 em3 KMnO,.
Sehr interessant sind die Erscheinungen, die man beim Weiter- titrieren uber den ersten Ausgleich hinaus erhalt. 1st beliebig vie1 Chlorid aber kein Bromid zugegen, so bleibt die Farbe des Jods im Tetraohlorkohlenstoff bestehen und die waBrige Schicht nimmt immer starker die Permanganatfarbe an ; sttirkeres Ansauern mit Schwefel- saure Bndert daran nichts. Es wurden bis ZU 2,5 Val KMnO, auf 1 Mol J' zugegeben; beide Schichten waren stark violett. Auf Zugabe von wenig Bromid trat rasch Entfarbung ein.
1st von vornherein Bromid und Chlorid zugegen und titriert man uber den ersten Umschlag hinaus, so nimmt die Farbtiefe des Tetra- chlorkohlenstoffs allmiihlich ab ; die vollige Entfarbung kann auf 1-2 Tropfen 0,Ol n-Losung genau festgelegt werden. Um etwa den doppelten Betrag schwanken bei wiederholten Versuchen die Ergeb- groBe von 0,03 om3 iiber 30% Fehlermoglichkeit. Man findet vielfach Messungen im Sohrifttum, bei denen offensichtlich tropfenweise titriert, aber der Reagens- zusatz mit 0,03--0,05 om3 in Rechnung gestellt wurde. Werden mit derart ,,ge- messenen" Reagenszusatzen ,,DifferenZenquotienten" gebildet, so kann das zu groben Tauschungen fiihren. Genauer zlnd zugleich wesentlich einfacher ist es, entweder von Anfang an oder nach geniigender Annaherung an den Umschlag die Tropfen zu zahlen und den Volumenwert des einzelnen Tropfens dadurch zu ermitteln, daf3 man vor Beginn und nach Beendigung des ZLhlens den Biiretten- stand ahliest und durchdividiert.

F. Hahn. Die Bestimmung von Jodid neben Bromid und Chlorid 79
nisse. Im Mittel von 13 Bestimmungen wurden fur 1 em3 J’ 2,5 em3 KMnO, verbraucht, also genau doppelt soviel wie bis zum ersten potentiometrischen Umschlag in reinen Jodidlosungen.
Die Hoffnung aber, dafi der Zusatz von Tetrachlorkohlenstoff auch den zweiten potentiometrischen Umschlag verbessern und daher eine sehr genaue Bestimmung kleinster Jodidmengen neben groBten aberschiissen der anderen Haloide ermoglichen wurde, hat sich nicht verwirklicht. Erstens erfolgt dieser zweite Umschlag wesentlich zu spat und iiberdies schwanken die Werte aufierordentlich. Verbraucht wurden 2,6-3,0 em3 KMnO, auf 1 em3 J’. Die Schwankungen sind vielleicht in der folgenden, sehr storenden Erscheinung begrundet : Jeder Reagenszusatz erzeugt in der Umschlagsgegend zuniichst einmal eine starke Potentialanderung; sie geht aber wieder zuruck und zwar rasch um einen betrgchtlichen Teil, dann langsamer noch weiter. Die schlieBlich verbleibenden Potentialschritte sind nicht groB, schwanken stark in ihren Verhiiltnissen zueinander und ergeben Kurven von wechselnder Form, manchmal stark unsymmetrisch, die sich schwer auswerten lassen. Vermutlich hangt trotz peinlichster Gleichhaltung der Versuchsbedingungen (die im Ernstfalle, beim Arbeiten mit un- bekannten Stoffmengen, nicht einmal zu erreichen ware) die Gleich- gewichtseinstellung in diesem heterogenen System von allerlei Zu- fiilligkeiten ab. Versucht man, die sofort nach jedem Reagenzzusatz eintretenden grofien Potentialiinderungen zu kompensieren, also den Potentialgang moglichst ohne Wartezeit aufzunehmen, so kommt man dem richtigen Endpunkt niiher, aber dies Verfahren wiire naturlich ganz unzuverliissig.
Hier verdirbt also der Zusatz von Tetrachlorkohlenstoff die po- tentiometrische Endpunktserkennung, denn ohne diesen Zusatz laBt sich der zweite Ausgleich tatsachlich erfassen. Die gemessenen Kurven sind symmetrisch und lassen sich gut auswerten. Fig. 2, Kurve I und I1 zeigt zwei derartige Messungen. Die gemessenen Punkte sind als Kreuze eingetragen; die stark ausgezogenen Kurven sind aber nicht etwa mit dem Kurvenlineal moglichst gut durch die MaBpunkte hindurchgezogen (dann liel3e sich ein noch vie1 besseres Zusammenfallen erreichen), sondern in der weiter unten angegebenen Weise theoretisch berechnet.
Vergleicht man die Kurven mit I1 und IV aus Fig. 1, die ungefahr den gleichen Verhaltnissen, aber am ersten Umschlag entsprechen, so findet man, da13 der Potentialgang steiler ist als dort, d. h. der groBte Potentialschritt ist, a n s ein en N a c h b a r n geme s s en , grofier, und er wiire deshalb leichter zu erkennen, wenn nicht die gesamte
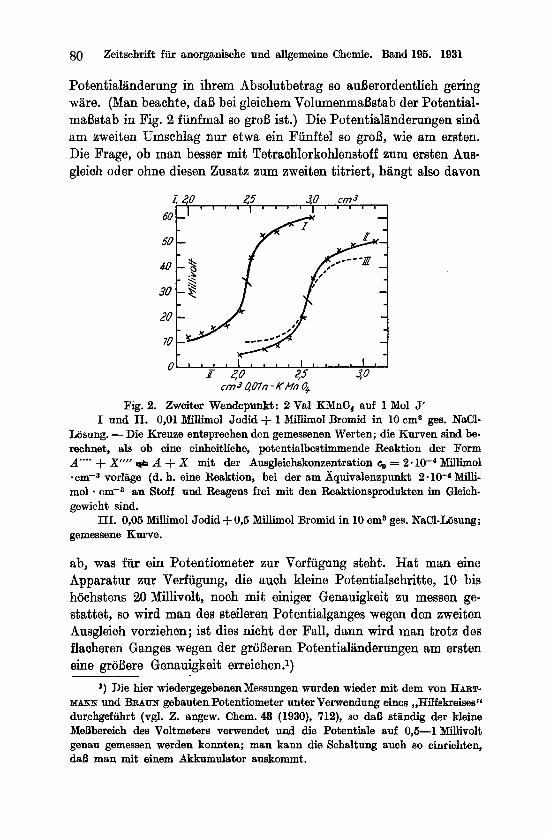
80 Zeitschrift fur anorganische und allgemeine Chemie. Band 195. 1931
60 I " " '
30 <xJ: 20 - - i$ I #fl* - - -'- -lz -
I .' _----- 70 - - -

F. Hahn. Die Bestimmung yon Jotlid neben Bromid uncl Chlorid 81
Der steile Potentialgang und die ungewohnlich kleine Potentiai- anderung am zweiten Ausgleich hat nun auch noch ein merkIiches theoretisches Interesse. Man sieht zunachst, daB die Kurven symme- trisch sind; dreht man sie im Wendepunkt um 1800, so gehen sie wieder in sich selbst uber. Das spriclit fur eine Reaktion, bei der Stoff und Reagens sich im Molarverhtiltnis 1 :1 nmsetzen. Der Poten- tialgang einer solchen Reaktion gehorcht dem Gesetz
58 E* = - lg z - Millivolt, wobei (v + 2) - = I .
12
Dabei bedeutet v den Mange1 oder UberschuB an Reagens (ge- messen in Vielfachen der Ausgleichskonzentration e,,, so daS z. B. bei einer Chlorsilbertitration fur einen SilberuberschuS von 10-5 Milli- mol.cm-3 v = 1 ist). Die Potentiale e, sind als Differenzen gegen das Ausgleichspotential e,, gemessen. n ist die Anzahl der Ladungen, die Stoff und Reagens bei der Umsetzung austauschen, also 1 fur
.... --f Fe" + Ti .... hg' + C1' -f AgCl oder Fe"' + TI 4-
Hg" + S" 72 HgS oder Sn" + J 2 -+ f- Sn"" + 2J' 3 fur
usw. Kennt man die Reaktionsgleichung, also das n, und hat man den
Potentialgang am Umschlag (den Richtungskoeffizienten der Wende- tangente) festgelegt, so kann man den weiteren Verlauf der Kurve berechnen. Umgekehrt : Kennt man die Steilheit der Kurve im Wende- punkt und den weiteren Verlauf, so ist damit eindeutig das ?z bestimmt. Praktisch macht man das am einfachsten durch Probieren, indem man fur einige Werte von n Kurven zeichnet und dann sieht, welche sich den gernessenen Punkte am besten anschmiegt. Die stark aus- gezogenen Kurven in Fig. 2 stellen den berechenbaren Potentialgang einer symmetrischen Reaktion dar, bei der Stoff und Reagens vier Ladungen tauschen, eine ganz ungewohnlich hohe Zahl. Obwohl aber die gemessenen Punkte sich vorzuglich den Kurven anpassen (die Abweichungen liegen durchweg in den Grenzen der MeBfehler) und obwohl sie sich n u r d i e sen Kurven anpassen und in keiner Weise mit den Kurven etwa fur n = 3 oder gar n = 2 zur Deckung zu bringen sind, ware es miiljig, die Formel dieser Reaktion zu suchen, denn der Divisor 4 ist mit wechselnden Versuchsbedingungen ver- anderlich und bleibt nicht einmal ganzzahlig. Die gestrichelte Kurve I11 stellt eine andere Messung dar, bei der nicht 0,01, sondern 0,05 Millimol J' unter sonst gleichen Bedingungen titriert wnrden
Z. anorg. u. allg. Chem. Bd. 195. 6

82 Zeitschrift fur anorganische und allgemeine Chemie. Band 195. 1931
(es ist dies die genaue Wiederholung des von GORBATSCHEFF und KAS- SATKINA angegebenen Versuches). Fur diese Kurve ware der Divisor 4,s ; die Potentialschritte sind noch merklich kleiner, die Steilheit der Wendetangente aber fast die gleiche.
Man ersieht daraus: Obwohl Entfarbung des Jods und zweiter Wendepunkt der Volumenpotentialkurve scharE mit dem Verbrauch von 2 Val Permanganat auf 1 Mol Jodid zusammenfallen und obwohl diese Kurve nach Symmetrie und Kriimmung durchaus berechenbar scheint, ist es unmoglich zu sagen, welches nun d i e potentialbestim- mende Reaktion ist. Es andern sich offenbar an dieser Stelle die Konzentrationen einer ganzen Reihe von Stoffen und zwar so, daB ilas Verhaltnis, in dem sie sich andern, je nach den Versuchsbedin- gungen schwankt, immer aber die Konzentrationsanderung an dieser Stelle starker ist, als an den benachbarten.
Zusammenfassung
1. Potentiometrisch kann Jodid entweder so bestimmt werden, daB man unter Zugabe von Tetrachlorkohleastoff bis zum ersten Wendepunkt beim Verbrauch von 1 Val Permanganat auf 1 Mol Jodid titriert oder ohne Zusatz bis zum zweiten Wendepunkt, der beim doppelten Verbrauch eintritt. Sind auf 1 Mol Jodid nicht mehr als 10 Mol Bromid vorhanden, so ist die erste Art unbedingt genauer ; bei hoheren Bromiduberschussen hangt es von den Eigenschaften des benutzten Potentiometers ab, melches Verfahren bessere Ergebnisse liefert .
2. Der zweite Umschlag kann auch ohne Potentiometer erfaBt werden, indem man unter Zusatz einer kleinen Menge von Tetra- chlorkohlenstoff bis zum Verschwinden der Jodfarbe titriert ; der Endpunkt ist auf wenige Tropfen 0,Ol n-Losung zu erkennen.
Frl. ANNI JACOB danke ieh auch an dieser Stelle bestens fur sorg- falt.ige und gewandte Hilfe bei den Versuchen.
Xralzkfwrt a. X., Chemisches Institut der Universitat.
Bei der Redaktion eingegangen am 4. November 1930.




![Aus der Abteilung für Nuklearmedizin Direktor: Prof. Dr ... · Pertechnetat-Anion ein dem Jodid vergleichbares Verhalten. [6, 15] Helman et al. [40] stellten im Rahmen von in-vitro](https://img.pdfslide.org/doc/110x75/5e15cd1081ba745cb54d6ef3/aus-der-abteilung-fr-nuklearmedizin-direktor-prof-dr-pertechnetat-anion.jpg)

![chemistry.mdma...Die Oxvdation $ Jahrg. 74 ist 3/194 Il N -LP- (2-Methoxy-phenyl) -äthyl]-pyridiniunj bromid (VI). Atis 2 g Bromid und 0.75 g Pyridin wie oben dargestellt. Das Produkt](https://img.pdfslide.org/doc/110x75/60dd9527f6bf256fb62c2936/-die-oxvdation-jahrg-74-ist-3194-il-n-lp-2-methoxy-phenyl-thyl-pyridiniunj.jpg)





















