Embed Size (px)
Citation preview

358
Mage. Es zeigt sich also, dag sich die K o l - l o i d t e i l c h e n g r S g e a u f d i e K r i s t a l l i t - g r 6 g e d e r Z e l l u l o s e z u r f i c k f f i h r e n l~if~t.
Mit HiIfe der S c h e r r e r 'schen Beziehung 1/igt sich - - in gfinstigen FNlen - - auch die Kristallitform bzw. ihre AchsenverhNtnisse be- stimmen.
D. Kristallitanordnung.
Als letztes Problem wurde die Bestimmung der Kristallitanordnung angefiihrt, wie sie mit Hilfe der von P o l a n y i und Weii3enberg(a) angegebenen Methoden erfolgen kann. Ebenso wie die Untersuchung an Metallen gezeigt hat, dag die Kenntnis dieser Strukturen yon grot~er Bedeutung ftir das Verst~indnis ihrer Festigkeits- eigenschaften ist, darf man annehmen, daf~ ~ihn- liches fiir den Feinbau der Gele gilt, die aus Kryptokristallen aufgebaut sind. Wir haben in den letzten Jahren die r6ntgenspektrographische Methode auch wesentlich zu~.Untersuchungen dieser Art an natfirlichen Zellulosefasern und Kunstfasern sowie an tierischen Geweben- be- nutzt. Dabei haben wir die Erfahrung gemacht, dag hier vielleicht noch mehr als bei den Me-
1~) P o l a n y i u. W e i B e n b e r g , Naturw. 1925, Heft 18; Zeitschr. f. Phys. 7, 149 (1921); 8, 20 (1921).
tallen die z w e i p h a s i g e N a t u r der G e b i l d e ffir ihr physikalisches Verhalten entscheidend ist 14).
Man findet die einfache Faserstruktur - - die Kristallite mit e in e r Kristallachse parallel der Faserachse - - bei manchen Zellulosefasern und bei natfirlicher Seide; andere biologische Oe- bilde zeigen komplizierteren Bau; so liegen bei der sog. Walzstruktur - - wie sie gewalzte Folien zeigen - - alle Kristallite untereinander parallel oder sie zerfallen in wenige parallel ge- ordnete Oruppen usw. Dieselben Typen wie bei den organischen linden sich bei anorganischen biologischen Kristallaggregaten 15) und ebenso bei den k/instlich geformten Metallen, Kunst- fasern und anderen mikro- und kryptokristalli- nischen Geffigen.
Ffir das Verstiindnis ihrer Entstehung ist die Kenntnis der Kristallwachstumsbedingungen n/Stig. Die Entstehung der Faserstruktur ist zu- meist auf Dehnung und Zug zurfickzuffihren15). Hier sind auch die neuen Beobachtungen am Kautschuk zu erw~ihnen, die J. R. K a t z 16) mit- geteilt hat.
14) Ber. d. Deutsch. chem. Ges. 58, 1254 (1925). 15) Siehe 14); ferner A. Fr ey, Naturw. 13, 403
(1925); F. R inne , Naturw. 18, 690 (1925). 16) j. R. Ka tz , Chem.-Ztg. 49, 353 (1925).
Die Methoden zur Bestimmung der Ladungsgri fle kolloider Teilchen. Yon H. R. K r U yt (Utrecht). (Eingegangen a m 24. Oktober 1925.')
(Zusammenfassender Vortrag, gehalten in der IV. Hauptversammlung der Kolloid-Oesellschaft in Nfirnberg.)
Zwecks Berechnung der Ladungsgr6t~e kol- loider Teilchen kann man die Messung ver- schiedener Or6gen verwenden. Mehrere dieser Gr6fSen stehen indes mit der zu bestimmenden Ladung in einem Zusammenhang, der uns bis- her quantitativ nicht bekannt ist. So k6nnte man z. B. denken an den Zusammenhang zwischen der Ausflockungskonzentration und elektrischer Ladung bzw. an den elektrovisk6sen Effekt kolloider L6sungen. Von der ErBrterung derartiger Verfahren soll indes in dieser Mit- teilung abgesehen werden. Mehr direkt geben uns nur Messungen der kataphoretischen Ge- schwindigkeit bzw. diejenige der elektrischen Ueberffihrung fiber die Teilchengr6t~e Aufschlug.
I. M a k r o s k o p i s c h e K a t a p h o r e s e b e i g e f ~ t r b t e n S o l e n .
Bekanntlich haben S. E. L i n d e r und H. Pic- ton 1) zum ersten Male die Kataphorese unter-
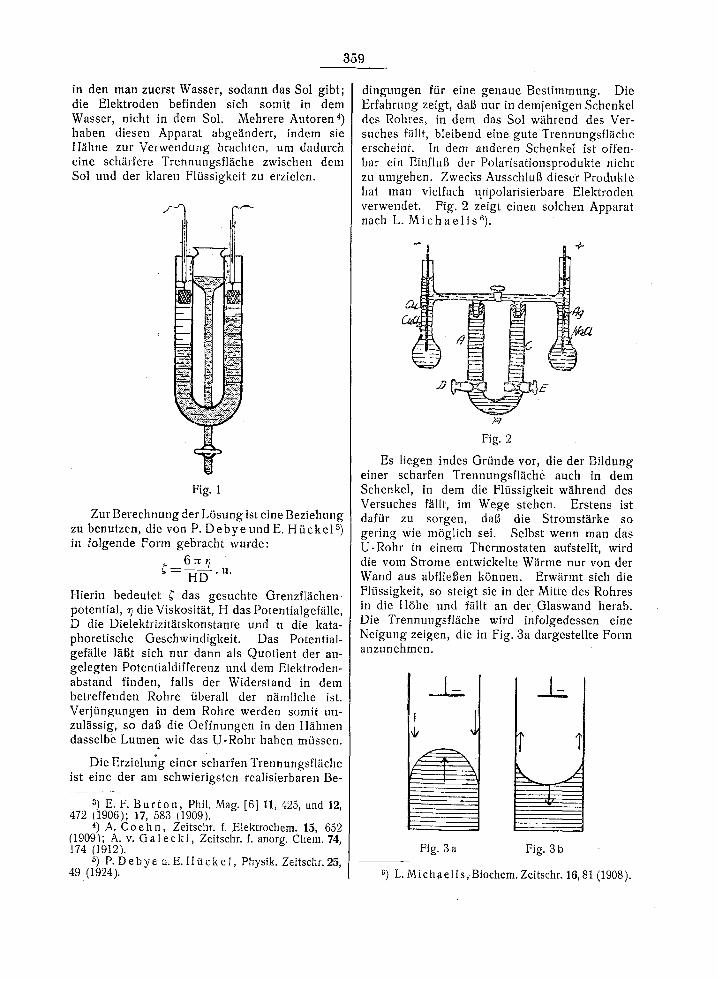
sucht, indem sie ein Sol in ein U-Rohr brachten' welches die beiden Elektroden enthielt. Diese Forscher sowie andere benutzten auch wohl ein gerades Rohr2), welches vertikal aufgestellt war. Am oberen bzw. unteren Ende war je eine Elektrode aufgestellt. Diese Methode hat den Nachteil, dai~ die Elektroden sich in dem Sol selbst befinden, dag sich somit die Trennungs- fl~iche zwischen dem gef~irbten Sol und der klaren Plfissigkeit an einer der Elektroden bildel. Infolgedessen wird die Trennungsfl~iche sofort durch die Polarisationsprodukte der be- treffenden Elektrode verunreinigt.
E, F. B u r t o n 3) hat diese Schwierigkeit um- gangen in seinem bekannten Apparat (Fig. 1),
1) H. P i c ton u. S.E. L inde r , Joum. Chem. Soc. 61, 148 (1892); 71, 568 (1897).
3) W. R. W h i t n e y u. J.C. B l a k e , Joum. Amer. Chem. Soc. 26, 1339 (1904).
359
in den man zuerst Wasser, sodann das Sol gibt; die Elektroden befinden sich somit in dem Wasser, nicht in dem Sol. Mehrere Autoren4) haben diesen Apparat abge~.ndert, indem sie H~ihne zur Verwendung brachten, urn dadurch eine seh~irfere Trennungsfl~iche zwischen dem Sol und der klaren F1/issigkeit zu erzielen.
F
Fig. 1
Zur Berechnung der L6sung ist eine Beziehung zu benutzen, die yon P. D e b y e und E. Hf icke l s) in folgende Form gebraeht wurde:
6 ~ ~ ' = ~ - f f . u.
Hierin bedeutet ~" das gesuchte Grenzfl/ichen- potential, ~) die Viskosit~it, H das Potentialgeffille, D die Dielektrizit/~tskonstante und u die kata- phoretische Geschwindigkeit. Das Potential- gef~ille l~igt sich nur dann als Quotient der an- gelegten Potentialdifferenz und dem Elektroden- abstand finden, falls der Widerstand in dem betreffenden Rohre fiberall der n~imtiche ist. Verjfingungen in dem Rohre werden somit un- zul~issig, so dag die Oeffnungen in den H~ihnen dasselbe Lumen wie das U-Rohr haben mfissen.
Die Erzielung einer scharfen Trennungsfl~iche ist eine der am schwierigsten realisierbaren Be-
~) E. F. B u r t o n , Phil. Mag. [6] 11, 425, und 12, 472 (1906); 17, 588 {1909).
4) A. C o e h n, Zeitschr. f. Elektrochem. 15, 652 (1909~; A. v. G a l e c k i , Zeitschr. f. anorg. Chem. 74, 174 (1912).
5) p. D e bye u. E. H/i e k e 1, Physik. Zeitschr. 25, 49 (1924),
dingungen ffir eine genaue Bestimmung. Die Erfahrung zeigt, dag nut in demjenigen Schenkel des Rohres~ in dem das Sol wfihrend des Ver- suches fNlt, bleibend eine gute Trennungsfl~iehe erscheint. In dem anderen Schenkel ist often- bar ein Einflug der Polarisationsprodukte nicht zu umgehen. Zwecks Ausschlufi diesel" Produkte hat man vielfach unpolarisierbare Elektroden verwendet. Fig. 2 zeigt einen solchen Apparat nach L. M i c h a e l i s 6 ) .
Fig. 2
Es liegen indes Grfinde vor, die der Bildung einer scharfen Trennungsfl~iche auch in dem Schenkel, in dem die Flfissigkeit w~thrend des Versuches fNlt, im Wege stehen. Erstens ist dafiir zu sorgen, da6 die Stromstfirke so gering wie mSglich sei. Selbst wenn man das U-Rohr in einem Thermostaten aufsteltt, wird die vom Strome entwickelte W/~rme nur yon der Wand aus abfliegen k6nnen. Erw~rmt sich die Flfissigkeit, so steigt sie in der Mitre des Rohres in die H6he und fiillt an der Glaswand herab. Die Trenmmgsfl~iche wird infolgedessen eine Neigung zeigen, die in Fig. 3a dargestellte Form anzunehmen.
Fig. 3 a Fig. 8 b
6) L. IViichaelis, Bi0chem. Zeitschr. 16, 81 (1908).

360
Anderseits abet tritt ein elektroendosmotischer Wasserstrom der Glaswand entlang aufT). Bei einem negativ geladenen Sol f~illt das Sol in dem Kathodenrohr; der elektroendosmotische Transport ist der Kathode zu gerichtet, so dab diese Verh~iltnisse eine Formiinderung der Trennungs.fl~iche bedingen, wie sie in Fig. 3b zum Ausdruck kommt.
Tritt somit eine scharfe Trennungsfl~iche auf, so liegt die M/Sglichkeit vor, dab die beiden betreffenden Fehler sich gerade kompensieren. In diesem Fall entspricht der gemessene u-Weft keineswegs dem theoretischen u der oben ge- gebenen Formel. Jedenfalls sind solche Mes- sungen, die mit Spann'ungen von 110 bzw. 220 Volt ausgeffihrt werden, auf Grund der dabei auftretenden Stromw~irme grogen Bedenken unterworfen. Unserer Erfahrung nach a~beitet man am besten mit etwa 16 Volt; zwecks Er- zielung einer gr6Beren Genauigkeit ist dann die lange Dauer des Versuches mit in den Kauf zu nehmen.
Arbeitet man mit Solen,. die nur wenig oder gar keinen Elektrolyt enthalten, so treten die Komplikationen, welche eine Folge der Strom- w~irme und der Polarisationsprodukte sind, nut wenig in den Vordergrund. Schwierigkeiten machen sich erst dann geltend, falls in dem Sol Elektrolyte vorhanden sind in Konzentrationen von einigen Millimol pro Liter. Nun ist aber h~iufig die Untersuchung reiner Sole vom kol- loidchemischen Standpunkte weniger wichtig, als das Studium des Einflusses, deft zugesetzte Elektrolyte au[ die elektrische Ladung aus/iben.
Aui~erdem aber treten aus folgenden Qrtinden noch-andere Schwierigkeiten auf: Wir betonten soeben, dab es ftir die Berechnung des Potential- gef~lles erwtinscht ist, dab der Widerstand im Rohre fiberall tier gleiche sei. Das spezifische LeitvermSgen des Soles und der fiber demselben stehenden Flfissigkeit soll somit das n~imliche sein. Da die Solteilchen ihren Beitrag zu dem Leitvermt~gen liefern, ist es somit nicht zul~issig, das Sol mit einer Flfissigkeit zu fiberschichten, die die n~imliche Elektrolytkonzentration wie das Sol besitzt. Diese Konzentration soll gr6Ber sein. Anderseits ist es logisch, zu versuchen, der fiber dem Sol geschichteten Flfissigkeit die- selbe Konzentration zu erteilen, als die inter- mizellare Fltissigkeit des Sols aufweist. Wird hierftir Sorge getragen, so bleiben, ungeachtet
7) M. v. S m o l u c h o w s k i , Oraetz' Handbuch der Elektr. u. Magn. II (Leipzig 1914), 366--428.
des elektrolytischen Transports, die Kolloid- teilchen stets in demselben Medium.
Es h~ilt aber schwer, gerade jene Kon- zentration anzuwenden, da man h~iufig das Adsorptionsgleichgewicht nicht kennt, das sich be[ der Verteilung des zugesetzten Elektrolyten zwischen den Kolloidteilchen und der inter- mizellaren Flfissigkeit einstellt. M6glich wird es, dab Ultrafiltration hier ein Mittel w~ire zur Herstellung der intermizellaren Flfissigkeit bzw. zur Ermittelung ihrerZusammensetzung.
Ist abet die tiber das Sol g'eschichtete Fliissig- keit nicht identisch mit der intermizellaren Flfissigkeit, so wird sich gerade in der Trennungs- fliiche zwischen dem Sol und der fiberstehenden klaren Flfissigkeit ein Diffusionspotential bilden, so dal~ man dann die betreffenden Messungen gerade an der ungtinstigsten Stelle des Rohres ausffihrt. Gelegentlich der Untersuchungen, die T e n d e l o o im verflossenen Winter in meinem Laboratorium ausgeffihrt hat, haben wit in Erfahrung gebracht, wie sehr die Ergeb- nisse auseinandergehen b e i wechselnder Zu- sammensetzung der fiberstehenden Flfissigkeit.
Die Messungen wurden mittels eines sogleich zu beschreibenden Apparates an Gelatinesolen mit verschiedenen P~-Werten ausgeffihrt bei Zusatz neutraler Salze. Es stellte sich dabei heraus, dab der Wert der kataphoretischen Ge- schwindigkeit ein sehr verschiedener war, je nachdem man eine Flfissigkeit benutzte, der so- viel Siiure zugesetzt war, d.af~ sie denselben Pa- Weft hatte als das Sol, oder eine solche, die auBerdem gerade soviel Neutralsalz enthielt als das Sol, oder schlieBlich eine solche, die soviel Salz enthielt, dab sie dasselbe elektrische Leit- verm6gen als das Sol aufwies. Es h~ilt schwer, festzustellen, welche der betreffenden Messungen nun tats~ichlich die wahre kataphoretische Ge- schwindigkeit ergibt.
Aus obigem ersehen wir, dab dem Verfahren mit dem U-Rohr mehrere Schwierigkeiten an- h~ingen infolge der wenig geeigneten Stelle, an der man die Messungen ausffihrt, und der Form- ~inderung, die die Trennungsflfiche erleidet.
II. M a k r o s k o p i s c h e K a t h p h o r e s e an u n g e f ~ r b t e n S o l e n .
Falls sich das zu untersuchende Sol nicht ohne weiteres vonder darfiberstehenden Fliissig- keit unterscheiden l~iBt, hat man ein Hilfsmittel zu suchen, das uns in den Stand setzt, die Trennungsflfiche sichtbar zu machen. Wir ver- wendeten zu diesem Zweck das T yn d a 11-Licht. Dies ist m6glich, da die fiber dem Sol befind-

Fig, 4
36I
liche Fliissigkeit diese Erscheirmng nicht auf- weist, das Sol dagegen wohl.
Anfangs warfen wir einen Lichtstrahl auf das U-Rohr , und zwar dorthin, wo sich die Trennungsfl~iche befand. Alsbald stellte sich heraus, dat~ sich die Messung besser ausftihren l~ifat, wenn man dieses Lichtbfischel von oben in den Schenkel, in dem sich die fallende Trennungsfl~tche befindet, eintreten liit~t. Wir gelangten so zu der Versuchsanordnung, die in den Fig. 4 und 5 dargestellt i s t .
An den Schenkeln des U-Rohres sind seitlich R6hrchen angebracht, die umkehrbare Elektroden enthalten. Der ganze Apparat ist in einem Thermostaten aufgehangt. Der Hahn l~il~t sich manipulieren mittels eines l~ingeren Hebels, welcher aus dem Wasser des Thermostaten heraustritt. Aus unserer Fig. 5 ist ersichtlich, dab fiber den Schenkel ein Blechzylinder ge- schoben ist, der an der Stelle, wo sich die Trennungsfl~.che befindet, einen Schlitz hat. An der AuBenseite des Zylinders l~iBt sich mittels einer Mikrometerschraube ein Index auf- und abbewegen. Die Verschiebung des Index later sich an der Schraubenmutter ablesen. In dem zweiten horizontalen Zylinder befindet sich eine kleine 4-Voltlampe, deren Licht senkrecht in den betreffenden Schenkel des U-Rohres ge- worfen wird.
Es stellte sich heraus, dat~ speziell bei Solen, die nur ein schwaches Tyndall-Licht aufweisen, die Benutzung des Zylinders und das Ablesen mittels des beschriebenen Index sehr wich t ig sind ftir die Genauigkeit der Beobachtungen.
Fig. 6 zeigt einen Thermostaten, in dem sich ein solches Instrument befindet.
Fig. 5

362
~ C
Fig. 7
T h e S v e d b e r g s) verwandte eine andere physikalische Erscheinung zum Sichtbarmachen der Trennungsfl~iche. Er bestrahlt das U-Rohr seitlich rnit ultraviolettem Licht (Fig. 7). Die Fluoreszenz, welche hierdurch bei Eiweigl6sungen auftritt, lfiBt sich auf photographischen Wege fixieren. Fig. 8 zeigt die Abbildung der beiden Apparatenschenkel. Man ersieht daraus wiederum deutlich, dat3 die Trennungsfl~.che in dem die fallende Trennungsfl~.che enthNtenden Schenkel scMrfer zutage tritt als in dem anderen. Aut3er- dern aber ergibt sich gleichfalls, dal~ aueh die erstgenannte Trennungsflgche nicht ideal ist. Auch bei unserem Verfahren l~,gt ihre Schg.rfe h~iufig zu wiinschen tibrig.
Es ist fibrigens selbstredend, dag s/imtliche Bedenken, die wir ira vorhergehenden Para- graphen nannten, auch fiir den in diesem Para- graphen er6rterten Apparat gelten.
III. B e o b a c h t u n g an E i n z e l t e i l c h e n .
Die Messung der kataphoretischen Oe- schwindigkeit mittels des Ultramikroskopes wurde zu.m ersten Male yon A. C o t t o n und H. M o u t o n 0 ) ausgeftihrt. T h e S v e d b e r g 1~ konstruierte zu diesem Zwecke eine Kfivette, die in Fig. 9 dargestellt ist. Dieselbe erm~Sglicht, Messungen mittels des Spalt-Ultramikroskopes auszufiihren, wobei das elektrische Feld scnk- recht zur Richtung des einfallenden Strahles steht. Handelt es sich urn die Untersuchung
s) T h e S v e d b e r g, Journ. Amer. Chem. Soc. 45, 954 (1923).
9) A. C o t t o n u. H. Mou ton , Les ultramicro- scopes (Paris 1906).
10) The S v e d b e r g , Nova Acta Upsaliensis [4] 2, Nr. 1 (1907).
Fig. 8
von Solen des V~Q-Typus, sorist es erwiinscht, auch MessurJgen anstellen zu~'k6nnen, wobei die Richtung des Lichtes und des elektrischen Feldes einander parallel sind. H.R. K r u y t ll) konstruierte 1916 eine ffir diese Zwecke ge- eignete Kiivette, die in Fig. 10 abgebildel ist.
Q T v v ' O
Fig. 9
-Z ) /
Fig. 10
Dieser Kfivettentypus eignet sich ffir Sole, die fiugerst geringe Elektrolytmengen enthalten. I~t dieser Bedingung nicht Geniige geleistet, so entsteht eine sehr stOrende Gasentwicklung an den blanken Platinelektroden. Die Qas- blasen erzeugen eine so starke Bewegung der Flfissigkeit, dag das Beobachten unm6glich wird. Uebrigens tragt die Stromw~_rme das ihrige zu den St6rungen bei. H .R. K r u y t und A. E. v a n Ar, ke l ~2) ist es gelungen eine Ktivette zu
n) H. R. Kruyt , Proc. Acad. Amsterdam 18, 1625 (1916); KolI.-Zeitschr. 19, 161 (1916).
12) H. R. Kruyt u. A.E, van Arkel , Koi1.-Zeit- schr. 82, 91 (1923).

363
konstruieren, bei deren Verwendung sich die Schwierigkeiten, welche bei Kfivetten des so- eben genannten Typus auftreten, umgehen lassen. Dieselbe soll in Kombinationen mit dem Paraboloidmikroskop verwendet werden. Fig. 11 stellt dieselbe in der Ansicht und Aufsicht dar, w~thrend in Fig. 12 eine vollst~indigere Abbildung
~: z ) , c c
z J t A t 3 ,I IH 0 ; @ x ~ 6r C
Kataphorese-Kfivette von oben gesehen.
~---Y .~ H _. /1"_.]-;
i i
Kataphorese-Kfivette, wobei das Abschluflglas weg- genommen ist, yon der Seite ges%hen.
Fig. 11
~ / / / / 2 2 " / / / / / / X / / / / ~ ~
Fig. 12
gegebeff ist. Ffir Einzelheiten sei auf die betreffende Abhandlung verwiesen, Die hier gezeichnete Vorrichtung ermSglicht ungest6rte Beobachtungen bei Elektrolytgehalten bis etwa 20 Millimol pro Liter. Ferner ist es mSglich, mittels derselben Messungen anzustellen in allen Schichten zwischen dem Boden und demDeck- glase der'Kfivette. Dies ist absolut notwendig, da, wie oben betont wurde, an den Glasw/inden ein elektroendosmotischer Transport stattfindet.
M. v. S m o l u c h o w s k i la) berechnete seiner- zeit, wie man aus einer in verschiedenen Schichten angestellten Anzahl Beobachtungen den Fehler, welcher infolge des elektroend- osmotischen Transports auftritt, v611ig eliminieren k a n n .
In F!g. 13a ist eine Reihe yon negativ ge- ladenen Teilchen in verschiedenen Schichten
1~) M.v, S m o l u c h o w s k i . loc. cit.
Fig. 13 a Fig. 13b
§
abgebildet; in Fig. 13b die Lage derselben, wenn der Strom w/ihrend einiger Augenblicke durch- gegangen ist. Die Teilchen an der Wand haben eine unver~nderliche Lage, darauf folgen die Teilchen, welche von dem elektroendosmotischen Wasserstrom zur Kathode geftihrt werden, w/ihrend weiter v o n d e r Wand enffernt die Teilchen kataphoretisch zur Anode gefiihrt werden. Zwischen ]enen Schichten befindet sich somit eine solche, in welcher die Teilchen stillstehen. Dies alles lftgt sich in der K r u y t - v a n Arkel ' schen Kfivette scharf beobachten, indem man das Mikroskop auf verschiedene Schichttiefen einstellt.
-4, .4
Fig. 14
T h e S v e d b e r g 14) bediente sich eines Wech sel- stromes zur Beobachtung kataphoretischer Ge- schwindigkeiten im Ultramikroskop. Fig. 1 4 stellt seine sehr einfache Kfivette dar. Das Verfahren erfordert indes einen Wechselstrom bekannter Frequenz. Die Teilchen werden von diesem Strome hin- und herbewegt, so dag dieselben eine leuchtende Linie bilden. Man miBt die L~inge derselben und erh~ilt somit auf Orund der bekannten Frequenz des Stromes die nStigen Daten zur Berechnung. Wir glauben aber, dab diese Kfivette, die yon S v e d b e r g auch ffir Oieichstrom verwendet wird, etwas zu untief ist. Infolgedessen wird d i e soeben genannte Kfivette nach M. v. S m o l u c h o w s k i (Fig. 12) wohl sehr stark deformiert. Ferner treten in der v511ig geschlossenen Kiivette leicht StSrungen ein infolge der Bildung yon Gasblasen. Dies wird in dem Apparat yon H. R. K r u y t und A. E. v a n A r k e l infol'ge der vorhandenen seit- lichen Oeffnungen (Fig. 11) vSllig vermieden. Nicht allein mittels des Ultramikroskops, sondern auch mit dem gew5hnlichen Mikroskop hat man-
14) The Sv edberg, Koll.-Zeitschr. 24, 156 (1919).

364
Beobachtungen ausgeffihrt, dann aber selbst- verst~indlich an Systemen geringerer Dispersit/it, als die Kollo[de es sin& So haben R. Elf"is 15) nnd F. P o w i s 16) Messungen an Oelemulsionen angestellt. Dabei kam ein Apparat zur Ver- wendung, der aus einem Objektglas besteht, um welches zwei Metalldr~ihte gelegt sind, w/ihrend ein Deckglas auf diesen beiden Elek- troden ruht. Nach unserer Erfahrung lassen sich mittels dieses Apparates jedoch reproduzier- bare Ergebnisse nicht erzielen. Herr H u i z i n g hat in meinem Laboratorium einen verbesse:ten Apparat angefertigt; sobald wir darfiber Ntiheres in Erfahrung gebracht haben werden, hoffen wir darfiber zu berichten.
IV. U e b e r f f i h r u n g s v e r f a h r e n .
Die bisher er6rterten Messungen der Kata- phorese setzten uns nur in den Stand, das P o t e n t i a l in der Doppelschicht kennen zu lernen. N~ihere Kenntnisse fiber die elektrischen Verhtiltnisse in der Doppelschicht liefert uns eine Messung der Ueberf/ihrung. Die ersten Untersuchungen naeh dieser Richtung rfihren yon J. D u e l a u x 17) her, abet noch 1919 be- zeichnete T h e S v e d b e r g dieses Verfahren als wenig aussichtsvoll. Seitdem sind aber die interessanten Untersuchungen yon (3. Va r g a18) und die yon R. W i n t g e n 19) und seiner Mit- arbeiter bekannt geworden. In diesen Unter- suchungen wurde neben der Kataphorese (z. B. die des Zinnoxydsols) das elektrische Leitver- mSgen sowohl des Sols als auch der inter- mizellaren Flfissigkeit ermittelt, wtihrend schlieg- lich gemessen wurde, eine wie groge Menge des Kolloids yon einer bestimmten Anzahl Coulombs transportiert wurde.
Aus diesen Messungen l~igt s ich ableiten, wie grog die Aequivalentaggregation ist. Hier~ unter versteht R. Win t g e n die Anzahl von Mole- kfilen Zinnoxyd, die einer elem entare n elektrischen Ladung entsprieht.
Untersuchungen dieser Art erweitern unsere Kenntnisse fiber die Art und Weise der Ladung
1~) R. E l l i s , Zeitschr. f. physik. Chem. 78, 321 (1911).
16) F. Powis , Zeitschr. f. physik. Chem. 89, 91 (1915).
17) j. Duc laux , Joum. chimie physique 7, 405 (1909).
is) G. Varga, Kolloidchem. Beih. II, 1 (1919). 19) R. W i n t g e n , Zeitschr. physik, f. Chem. 103,
238 (1922); 107, 403 (1923); 109, 378 (1924).
der untersuchten Kolloidteilchen ganz ungemein. Nicht nur wird deren Or6ge dadurch bekannt, sondern wir erfahren dadurch ii~iheres zur Be- antwortung der Frage. weIche Ionen sich bei der Bildung der Doppelschicht beteiligen.
Fig. 15
Fig. 16
Die Untersuchung l~gt sich mittels eines Apparates messen, der aus drei BechergI~isern zusammengesetzt ist (Fig. 15). Eines derselben enthNt die Kathode, ein zweites die Anode, schlieglich das mittlere die Mittenflfissigkeit. Im allgemeinen ltifit sich jeder Apparat, mit welchem man direkt aus Konzentrations- ~nderungen Ueberffihrungszahlen bestimmt, zu diesem Zwecke verwenden. In Fig. 16' ist ein Apparat abgebildet, der yon R. W i n t g e n in seinen sp~.teren Versuchen verwendet wurde.

365
Wie die Figur zeigt, lassen sich der Kathoden- Und Anodenraum voneinander trennen durch Verwendung eines Schliffes.
Utrecht, September 1925.
van't Hoff - Laboratorium.
D i s k u s s i o n s b e m e r k u n g . H o c k (Giet~en) : Ist wohi schon der Versuch
gemacht worden, bei der Untersuchung narnent- lich der farblosen Sole nach dern U-Rohr-Ver- fahren zwecks scharfer Bestimmung der Tren- nungslinie die Schtierenrnethode anzuwenden, die sich hierftir doch gut eiguen rn/ii3te?
Die Methoden zur Bestimmung der Teilchengr6fie, Von A l f r e d K u h n (Leipzig).
(Vortrag auf der IV. Hauptversammlung der Kolloid-Oesellschaft in Ntirnberg 1925.)
Um eine gegebene Zerteilung als kolloid de- finieren zu k6nnen, ist es n6tig, den Zerteilungs- grad dieses Systems zu kennen. Innerhalb eines kolloiden Systems variieren die Eigenschaften wiederurn rnit der Teilchengr6t~e und der Hfiufig- keitsverteilung der verschiedenen Gr6i~enklassen, wodurch eine genauere Bestimrnung der Teil- chengr6i~e efforderlich wird. Die Bestimmung der Teilchengr61~e gestaltet sich damit zu einer grunds~ttzlichen Aufgabe der Kolloidchernie.
Ich beginne 'mit den Methoden, die auf mechanischen Eigenschaften beruhen. Diesen schliefien sich die optischen Methoden an. Die Auswahl der Methode richtet sich nach dem Zweck der Untersuchung, je nachdem es sich u r n die Eingrenzung eines Systems als kolloid innerhalb der dispersen Systerne fiberhaupt handelt oder aber urn die genauere Bestimrnung der vorhandenen Teilchengr6l~en und deren H~iufigkeitsver teilung.
A. Bestimmung der TeilchengrOfie dutch kinetische und mechanische A~ethoden.
I. B e s t i m m u n g d e r T e i l e h e n g r 6 t ~ e aus d e r F a l l g e s c h w i n d i g k e i t .
Ein Teilehen, welches sich unier dern Einflul~ der Sehwerkraft in einer Flfissigkeit befindet, bewegt sich nach S t o k e s dann rnit konstanter Geschwindigkeit v, wenn die Reibungskraft 6 n r/r v gleieh der Erdbeschleunigung
4 ~, r:' (Sp-- sin) g
ist, wobei r den Radius des Teilchens, "J7 die Viskositiit des Dispersionsmittels, sp die Dichte der dispersen Phase, Sm die des Dispersions- mittels und g die Erdbeschleunigung ist. Daraus ergibt sich ftir r
// 9 ~ . v r = 2 ( s ~ s ~ ) g (1)
Nennen wir den in der Zeit t zur/ickgelegten Weg x, so erhalten wir die Forrnel in ihrer praktischen Anwendungsforrn
9~x~ ' r = 2 (sp--sm) t g ' (2)
oder fassen wir die konstanten GrSl3en in
]/ k = 2 (Sp--Sm)g
zusammen, so ergibt sich / -
U r n eine Vorstellung von den zu erwartenden Geschwindigkeiten zu geben, sei an ein Beispiel von E. H a t s e h e k ~) erinnert. Es handle sich um ein Goldteilchen von 2 # Durchrnesser, also r = l . t 0 - l c r n , sp=19,3 , s m = l , g = 9 0 0 , ~ = 0 , 0 1 (20~ Daraus ergibt sich eine Oe- schwindigkeit yon 0,04 turn in der Sekunde oder 2,4 rnnrn in der Minute. '~, ~Eine Goldsuspension yon diesem Dispersitiitsgrade wfirde sich also in einer Stunde in einer H6he yon 14 cm abkl~iren. Gehen wit zu einern kolloiden Qoldsol fiber rnit Teilehen von 10 .u!,, so erhalten wir I/i0000 der berechnete~ Geschwindigkeit, also 0,014 rnrn in der Stunde oder 10 mrn in einem Monat. Ist die Dichte der Substanz kleiner, so wird der Sedirnentationsvorgang noch langsamer. F f i r die Dichte 3 errechnet sich etwa 1,3 rnm irn Monat. Man kann etwa 50 t,f, irn Falle eines Schwermetalls wie Gold als die praktische Grenze betraehten.
Urn diese Methode auf kleinere Teilehen anwenden zu kSnnen, mut~ g vergr6i~ert werden, wozu s ich die Zentrifugalbesehleunigung gut eignet. Wit haben dann wieder das Gleieh- gewicht zwischen der Reibungskraft
1) E. H a t s c h e k , Introduction to the Physics and Chemistry of Colloid~ (London 1922), 32;





























