Embed Size (px)
Citation preview

Eine selbstreplizierende Peptidnukleinsäure
Dissertation
zur Erlangung des Doktorgrades
der Fakultät für Chemie und Biochemie
der Ruhr-Universität Bochum
vorgelegt von
Tobias A. Plöger
aus Remscheid
Bochum 2011

1. Referent: Prof. Dr. G. von Kiedrowski, Ruhr-Universität Bochum
2. Referent: Prof. Dr. M. Feigel, Ruhr-Universität Bochum
3. Referent: Prof. Dr. R. Heumann, Ruhr-Universität Bochum
Die vorliegende Arbeit wurde in der Zeit von November 2006 bis Oktober 2011 an der
Fakultät für Chemie und Biochemie der Ruhr-Universität Bochum unter der Leitung von
Prof. Dr. Günter von Kiedrowski angefertigt.

Teile dieser Arbeit wurden veröffentlicht:
Artikel
T. A. Plöger, G. von Kiedrowski:
Improved Large Scale Liquid Phase Synthesis and High Temperature NMR
Characterization of Short (F-)PNAs
Helv. Chim. Acta 2011, 94, 1952.
Vorträge
06/06 Improved Synthesis of F-PNA Monomers
PACE Annual Review Meeting, Venedig (Italien)
06/07 Toward PNA replication analysis via kinetic titrations using 19F-NMR spectroscopy
ECLT-Workshop „Dynamic Modelling and Kinetic Data Fitting Using
SimFit”, Venedig (Italien)
10/08 Synthesis of F-PNA Monomers and Oligomers
PACE Final Review Meeting, BMZ Dortmund
Poster
09/08 F-PNA: Synthesis and Potential Application in PNA Replication Analysis
III. Nucleinsäurechemie-Treffen, Stuttgart

“Few organic chemists willingly undertake syntheses in an unsuitable solvent using
unpromising starting materials, or attempt fusion reactions between unprotected, unactivated,
multifunctional reagents at 65°C.”
L. E. Orgel, R. Lohrmann, Acc. Chem. Res. 1974, 7, 368.

Inhaltsverzeichnis
i
Inhaltsverzeichnis
Allgemeiner Teil ........................................................................................................................ 1
1 Einleitung ............................................................................................................................ 1
1.1 Grundlagen der präbiotischen Chemie......................................................................... 1
1.2 Die RNA-Welt-Hypothese ........................................................................................... 5
1.3 Die Bedeutung selbstreplizierender Systeme für die präbiotische Chemie ................. 7
1.3.1 Templatgesteuerte Synthesen................................................................................ 7
1.3.2 Selbstreplizierende Systeme auf der Basis von Nukleinsäuren ............................ 9
1.3.3 Selbstreplizierende Systeme auf der Basis von Peptiden.................................... 20
1.3.4 Artifizielle Replikatoren...................................................................................... 24
1.4 Die PNA-Welt als Modell einer Prä-RNA-Welt........................................................ 29
1.4.1 Intention des PNA-Designs................................................................................. 29
1.4.2 Das Potential einer PNA-Welt ............................................................................ 33
1.4.3 Schlüssige präbiotische Synthesewege für PNA-Bausteine................................ 35
1.4.4 Chemische Stabilität............................................................................................ 37
1.4.5 Katalytische Aktivität.......................................................................................... 41
1.4.6 PNA und der Ursprung der Homochiralität ........................................................ 42
1.4.7 Speicherung und Weitergabe genetischer Informationen ................................... 43
1.5 Kinetische 1H-NMR-Titration.................................................................................... 48
2 Aufgabenstellung .............................................................................................................. 50
3 System-Design .................................................................................................................. 52
3.1 Auswahl der NMR-Sonde .......................................................................................... 52
3.2 Wasserlöslichkeit ....................................................................................................... 54
3.3 Effektive Synthese...................................................................................................... 55
3.4 Vergleichbarkeit mit bekannten Systemen................................................................. 55
4 Synthesen und Spektroskopie............................................................................................ 57
4.1 Synthesestrategie I: Festphasensynthese.................................................................... 57
4.1.1 Retrosynthese ...................................................................................................... 57
4.1.2 Fluorlabel ............................................................................................................ 58
4.1.3 Geschützte Nukleobasen-essigsäuren ................................................................. 62
4.1.4 Rückgrat .............................................................................................................. 66

Inhaltsverzeichnis
ii
4.1.5 Monomere ........................................................................................................... 68
4.1.6 Löslichkeitsvermittler.......................................................................................... 71
4.1.7 Oligomersynthese................................................................................................ 73
4.2 Synthesestrategie II: Konvergente Flüssigphasensynthese ........................................ 81
4.2.1 Vorüberlegungen................................................................................................. 81
4.2.2 Fmoc/Bhoc-Chemie ............................................................................................ 82
4.2.3 Boc/Z-Chemie ..................................................................................................... 83
4.3 Synthesestrategie III: Zugang über ein vollständig geschütztes Rückgrat................. 91
4.3.1 Grundlagen .......................................................................................................... 91
4.3.2 Synthese des C-terminalen Trimers .................................................................... 94
4.3.3 Synthese des N-terminalen Trimers .................................................................. 102
4.4 NMR-Spektroskopie................................................................................................. 111
4.4.1 PNA und das Rotamer-Problem........................................................................ 111
4.4.2 Hochtemperatur-NMR-Spektroskopie .............................................................. 114
4.4.3 19F-NMR-Spektroskopie ................................................................................... 126
5 Ligation und Replikation................................................................................................. 130
5.1 Voruntersuchungen .................................................................................................. 130
5.2 HPLC-Kinetiken....................................................................................................... 136
5.2.1 Kalibrierung ...................................................................................................... 136
5.2.2 Auswertungsverfahren ...................................................................................... 138
5.2.3 Ligation und Replikation in Imidazol-Puffern .................................................. 140
5.2.4 Ligation und Replikation in Sulfonsäure-Puffern ............................................. 147
6 Zusammenfassung und Ausblick .................................................................................... 162
6.1 Zusammenfassung.................................................................................................... 162
6.2 Ausblick ................................................................................................................... 173
Experimenteller Teil............................................................................................................... 176
7 Geräte und Materialien.................................................................................................... 176
7.1 Magnetische Kernresonanzspektroskopie ................................................................ 176
7.2 Massenspektrometer................................................................................................. 176
7.2.1 FAB-MS............................................................................................................ 176
7.2.2 EI-MS................................................................................................................ 176
7.2.3 MALDI-TOF-MS.............................................................................................. 177
7.2.4 HR-ESI-MS....................................................................................................... 177

Inhaltsverzeichnis
iii
7.3 Chromatographische Methoden ............................................................................... 177
7.3.1 Dünnschichtchromatographie............................................................................ 177
7.3.2 Säulenchromatographie..................................................................................... 177
7.3.3 HPLC................................................................................................................. 177
7.4 UV-Spektroskopie.................................................................................................... 178
7.5 Automatisierte Peptidsynthese ................................................................................. 178
7.6 Gefriertrocknung ...................................................................................................... 178
7.7 Chemikalien ............................................................................................................. 178
8 Maschinelle Festphasensynthese..................................................................................... 180
9 Allgemeine Arbeitsvorschriften ...................................................................................... 181
9.1 Grignard-Reaktion mit Trockeneis (AAV 1) ........................................................... 181
9.2 Darstellung von Aminosäuremethylestern (AAV 2)................................................ 181
9.3 Verseifung von Aminosäuremethylestern (AAV 3)................................................. 181
9.4 Reduktive Aminierung mit elementarem Wasserstoff (AAV 4).............................. 182
9.5 Brommethylierung von Fluoraromaten (AAV 5)..................................................... 182
9.6 N9-Alkylierung bei Purin-Derivaten (AAV 6)......................................................... 182
9.7 Einführung von Carbamaten an der N6-Position von Adenin (AAV 7) .................. 183
9.8 Einführung von Carbamaten an der N4-Position von Cytosin (AAV 8).................. 183
9.9 Esterspaltung und Einführung des Carbonyls an Guanin-Derivaten (AAV 9) ........ 183
9.10 N9-Alkylierung am Guanin (AAV 10)................................................................... 184
9.11 Einführung von Carbamaten an der N2-Position von Guanin (AAV 11) .............. 184
9.12 TOTU-Kopplung (AAV 12) .................................................................................. 184
9.13 Esterspaltung mit Aluminium(III)chlorid in Ethanthiol (AAV 13) ....................... 184
9.14 EDC-Kopplung (AAV 14) ..................................................................................... 185
9.15 Abspaltung der Boc-Schutzgruppe mit Trifluoressigsäure (AAV 15)................... 185
9.16 Abspaltung der Z-Schutzgruppe mit TFMSA (AAV 16)....................................... 185
9.17 HBTU-Kopplung (AAV 17) .................................................................................. 186
9.18 DCC/HOSu-Kopplung (AAV 18).......................................................................... 186
9.19 Fmoc-Abspaltung mit Diethylamin (AAV 19) ...................................................... 186
9.20 Brop-Kopplung (AAV 20) ..................................................................................... 186
10 Synthesen ...................................................................................................................... 187
10.1 F-AcOH (70) .......................................................................................................... 187
10.2 Me2-Aeg(H)-OMe*2HCl (78)................................................................................ 188

Inhaltsverzeichnis
iv
10.3 Fmoc-Aeg(CBhoc)-OH (82) ..................................................................................... 189
10.4 Fmoc-Aeg(H)-OMe (83) ........................................................................................ 190
10.5 CBhoc-AcOH (84) .................................................................................................... 192
10.6 H-Aeg(H)-OH (85)................................................................................................. 193
10.7 H-Aeg(H)-OH (85)................................................................................................. 194
10.8 F-CH2Br (92).......................................................................................................... 195
10.9 F’-CH2Br (95) ........................................................................................................ 196
10.10 F’-AcOH (96)....................................................................................................... 197
10.11 A-AcOBn (104).................................................................................................... 198
10.12 C-AcOBn (105) .................................................................................................... 199
10.13 ABhoc-AcOBn (106) .............................................................................................. 200
10.14 CBhoc-AcOBn (107) .............................................................................................. 201
10.15 ABhoc-AcOH (108)................................................................................................ 203
10.16 ABhoc-AcOH (108)................................................................................................ 204
10.17 T-AcOH (109) ...................................................................................................... 205
10.18 GBhoc-AcOH (110)................................................................................................ 206
10.19 (2-Amino-6-chlor-purin-9-yl)essigsäure-benzylester (112)................................. 208
10.20 (2-Benzhydryloxycarbonylamino-6-chlor-purin-9-yl)essigsäure-benzylester......
(113) ..................................................................................................................... 209
10.21 A-AcOEt (120)..................................................................................................... 210
10.22 ABhoc-AcOEt (121) ............................................................................................... 211
10.23 H-Aeg(H)-OMe*2HCl (124)................................................................................ 212
10.24 Fmoc-Aeg(ABhoc)-OMe (125) .............................................................................. 213
10.25 Fmoc-Aeg(CBhoc)-OMe (126)............................................................................... 215
10.26 Fmoc-Aeg(GBhoc)-OH (127)................................................................................. 216
10.27 Fmoc-Aeg(F)-OH (128a) ..................................................................................... 217
10.28 Fmoc-Aeg(F’)-OH (128b).................................................................................... 219
10.29 Fmoc-Aeg(F4)-OH (128c) .................................................................................... 220
10.30 Fmoc-Aeg(H)-OH (129) ...................................................................................... 222
10.31 Fmoc-Aeg(T)-OH (132)....................................................................................... 223
10.32 Fmoc-Aeg(ABhoc)-OH (133)................................................................................. 225
10.33 Fmoc-Aeg(GBhoc)-OMe (134) .............................................................................. 226
10.34 Fmoc-Aeg(F)-OMe (135a)................................................................................... 227

Inhaltsverzeichnis
v
10.35 Fmoc-Aeg(F’)-OMe (135b) ................................................................................. 229
10.36 Fmoc-Aeg(F4)-OMe (135c).................................................................................. 231
10.37 N,N-Dimethyl-β-alanin Hydrochlorid (140) ........................................................ 233
10.38 Me2-Aeg(CBhoc)-OMe (141) ................................................................................. 234
10.39 (Me)2-Aeg(CBhoc)-OH*HCl (142) ........................................................................ 235
10.40 Me2-Aeg(H)-OH (145)......................................................................................... 237
10.41 H-Aeg(F)-OMe*TFA (185) ................................................................................. 238
10.42 Boc-Aeg(H)-OMe (187)....................................................................................... 239
10.43 Boc-Aeg(H)-OMe (187)....................................................................................... 240
10.44 Boc-Aeg(AZ)-OH (188) ....................................................................................... 241
10.45 AZ-AcOEt (189) ................................................................................................... 243
10.46 AZ-AcOH (190).................................................................................................... 244
10.47 Boc-Aeg(AZ)-OMe (191) ..................................................................................... 245
10.48 CZ-AcOH (192) .................................................................................................... 247
10.49 CZ-AcOH (192) .................................................................................................... 248
10.50 CZ (193)................................................................................................................ 249
10.51 CZ-AcOBn (194) .................................................................................................. 250
10.52 Boc-Aeg(CZ)-OH (195)........................................................................................ 251
10.53 CZ-AcOMe (196).................................................................................................. 252
10.54 Boc-Aeg(CZ)-OMe (197) ..................................................................................... 254
10.55 Boc-Aeg(GZ)-OH (198) ....................................................................................... 256
10.56 GZ-AcOH (199).................................................................................................... 257
10.57 (2-Amino-6-chlor-purin-9-yl)essigsäure-methylester (200) ................................ 258
10.58 (2-Amino-6-chlor-purin-9-yl)essigsäure-methylester (200) ................................ 259
10.59 (2-Benzyloxycarbonylamino-6-chlor-purin-9-yl)essigsäure-methylester (201) .. 260
10.60 Boc-Aeg(GZ)-OMe (202) ..................................................................................... 261
10.61 H-Aeg(GZ)-OMe*TFA (203) ............................................................................... 263
10.62 Boc-Aeg(F)-OMe (204) ....................................................................................... 264
10.63 2-Amino-N-(2-dimethylamino-ethyl)-acetamid Di-trifluoracetat (205) .............. 266
10.64 [(2-Dimethylamino-ethylcarbamoyl)-methyl]carbaminsäure-tert-butylester.......
(207) ..................................................................................................................... 267
10.65 H-Aeg(C)-Aeg(A)-Aeg(G)-Aem*4TFA (224) .................................................... 268
10.66 Boc-Aeg(Alloc)-OMe (225)................................................................................. 269

Inhaltsverzeichnis
vi
10.67 Boc-Aeg(Fmoc)-OMe (226) ................................................................................ 270
10.68 Z-Aeg(H)-OMe (227)........................................................................................... 272
10.69 Boc-Aeg(Alloc)-OH (228) ................................................................................... 274
10.70 H-Aeg(Fmoc)-OMe*TFA (229) .......................................................................... 275
10.71 Z-Aeg(Boc)-OMe (230) ....................................................................................... 276
10.72 Boc-Aeg(Alloc)-Aeg(Fmoc)-OMe (231)............................................................. 277
10.73 Z-Aeg(Boc)-OH (232).......................................................................................... 280
10.74 H-Aeg(Alloc)-Aeg(Fmoc)-OMe*TFA (233)....................................................... 281
10.75 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OMe (234)................................................ 282
10.76 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OH (235) .................................................. 284
10.77 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-Aem (236) ................................................ 286
10.78 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(H)-Aem (237) ...................................................... 288
10.79 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(GZ)-Aem (238)..................................................... 289
10.80 Z-Aeg(Boc)-Aeg(H)-Aeg(GZ)-Aem (239)........................................................... 290
10.81 Z-Aeg(Boc)-Aeg(AZ)-Aeg(GZ)-Aem (240) ......................................................... 292
10.82 Z-Aeg(H)-Aeg(AZ)-Aeg(GZ)-Aem*2TFA (241) ................................................. 294
10.83 Z-Aeg(CZ)-Aeg(AZ)-Aeg(GZ)-Aem*TFA (242).................................................. 295
10.84 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OH*3TFA (248) ................................... 296
10.85 Me2-Aeg(Me)-OH*2HCl (249)............................................................................ 298
10.86 Me2-Aeg(Me)-OH (249) ...................................................................................... 299
10.87 H-Aeg(H)-OBn*2pTsOH (253) ........................................................................... 300
10.88 Boc-Aeg(H)-OBn (254) ....................................................................................... 301
10.89 Boc-Aeg(Fmoc)-OBn (255) ................................................................................. 302
10.90 Boc-Aeg(Fmoc)-OBn (255) ................................................................................. 303
10.91 H-Aeg(Fmoc)-OBn (256)..................................................................................... 304
10.92 Boc-Aeg(Alloc)-Aeg(Fmoc)-OBn (257).............................................................. 305
10.93 H-Aeg(Alloc)-Aeg(Fmoc)-OBn*TFA (258)........................................................ 307
10.94 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(Fmoc)-OBn (259)............................................... 308
10.95 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(H)-OBn (260)..................................................... 309
10.96 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(GZ)-OBn (261) ................................................... 310
10.97 Boc-Aeg(CZ)-Aeg(H)-Aeg(GZ)-OBn (262) ......................................................... 312
10.98 Boc-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn (263).......................................................... 313
10.99 H-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn*TFA (264).................................................... 315

Inhaltsverzeichnis
vii
10.100 Me2-Aeg(Me)-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn*2TFA (265) ............................ 316
10.101 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-Aeg(C)-Aeg(A)-Aeg(G)-Aem*6TFA
(268) ................................................................................................................... 318
10.102 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OMe*3TFA (291)............................... 319
11 Ligations- und Replikationsexperimente ...................................................................... 320
Anhang ........................................................................................................................................I
12 Zur Nomenklatur der hier beschriebenen Verbindungen...................................................I
13 Abkürzungsverzeichnis ......................................................................................................I
14 HR-ESI-MS-Spektren von PNA 224, F-PNA 248 und F-PNA 268 ................................ V
15 SimFit-Dateien ................................................................................................................VI
15.1 SimFit-Eingabedateien .............................................................................................VI
15.1.1 A-Modell ...........................................................................................................VI
15.1.2 B-Modell ..........................................................................................................VII
15.2 Konzentrations-Zeit-Daten.................................................................................... VIII
15.2.1 Ligation in 1-Methylimidazol-Puffer ............................................................. VIII
15.2.2 Ligation in Imidazol-Puffer................................................................................ X
15.2.3 Ligation in HEPES- und MOPS-Puffer ...........................................................XII
15.2.4 Ligation in Gegenwart von Pyridin oder HOAt ............................................. XIV
15.2.5 Ligation in Anwesenheit verschiedener Salze ............................................... XVI
15.2.6 Ligation in Gegenwart von 0.4 M EDC ....................................................... XVIII
15.2.7 Ligation bei pH 6.6...................................................................................... XVIII
15.2.8 Ligation in Gegenwart von PEG .................................................................... XIX
15.2.9 Ligation bei unterschiedlichen Imidazol-Konzentrationen ............................. XX
15.2.10 Ligation bei vier unterschiedlichen intialen Hexamerkonzentrationen........ XXI
16 Danksagung.................................................................................................................XXII
17 Lebenslauf ................................................................................................................. XXIII
18 Literaturverzeichnis................................................................................................... XXIV

Allgemeiner Teil 1 Einleitung
1
Allgemeiner Teil
1 Einleitung
1.1 Grundlagen der präbiotischen Chemie
Die präbiotische Chemie versteht die Entstehung des Lebens als einen chemischen
Evolutionsprozess und versucht diesen nachzuvollziehen. Dabei ist die Frage nach der Natur
des Lebens im wissenschaftlichen Kontext weiterhin Gegenstand der Diskussion. Minimale
Anforderungen formulierte die NASA im Rahmen ihres Exobiology Programms, indem sie
Leben als ein sich selbst erhaltendes System definierte, das im Sinne Darwins evolvieren
kann. Drei fundamentale Voraussetzungen hierfür wurden von Oparin definiert:[1]
Metabolismus: Der Stoffwechsel, also der Austausch von Energie und Materie, hält
ein lebendes System im Nichtgleichgewichtszustand und bildet somit die Grundlage
für eine Weiterentwicklung;
Selbstreplikation: Das System katalysiert nicht nur seine eigene Synthese, sondern
gibt gleichzeitig Informationen über seine chemische Struktur an die nächste
Generation weiter;
Mutation: Eine gewisse Fehlerquote bei der Replikation kann zu einer Verbesserung
des bestehenden Systems und so zur Evolution im Sinne Darwins führen.
Historisch betrachtet galt die Aufmerksamkeit zunächst der Suche nach plausiblen Ent-
stehungswegen für Monomere und Submonomere der heutigen Biopolymere. Ausgangspunkt
war anfänglich die Annahme einer reduzierenden präbiotischen Atmosphäre, in der die
biologisch relevanten Elemente C, O, N und S in ihren reduzierten Formen CH4, H2O, NH3
und H2S vorlagen. Weiterhin nahm man an, dass diese Uratmosphäre verschiedenen Energie-
formen (solare Strahlung, elektrische Entladung, Hitze) ausgesetzt war, was zur Synthese
einfachster organischer Vorläufermoleküle führte, die sich in der Hydrosphäre sammelten.
Aus der sogenannten „Ursuppe“ sollen sich dann spontan die ersten Formen von Leben
gebildet haben. Diese Modellvorstellung wurde von Oparin[2, 3] und Haldane[4] unabhängig

Allgemeiner Teil 1 Einleitung
2
voneinander entwickelt und ist als Oparin-Haldane-Hypothese in die Wissenschafts-
geschichte eingegangen, wobei Haldane im Gegensatz zu Oparin von einer Atmosphäre aus
CO2 und NH3 ausging.
1953, einem Annus Mirabilis der Naturwissenschaften,[5] gelang es Miller, Aminosäuren
(insbesondere Glycin und Alanin) und einige einfache organische Verbindungen (speziell
Ameisen- und Milchsäure) aus einem „Ursuppen“-Experiment zu isolieren.[6] Miller folgte
dem Hinweis seines Doktorvaters Urey, indem er in einer Glasapparatur die Oparinsche
Uratmosphäre (CH4, NH3, H2O, H2) elektrischen Entladungen aussetzte, wobei er die
Parameter Temperatur sowie Reaktionsdauer variierte und die Produkte anschließend
analysierte. Das Miller-Urey-Experiment wird als Startpunkt der (neueren und experimen-
tellen) präbiotischen Chemie verstanden, da hier erstmalig die Frage nach dem Ursprung des
Lebens mit naturwissenschaftlichen Methoden bearbeitet wurde.
Schon 1862 gelang Butlerow die Synthese von Zuckern aus einer wässrigen Formaldehyd-
Lösung unter basischen Bedingungen.[7] Dieser komplex verlaufende Prozess wird als
Formose-Reaktion bezeichnet und liefert ein Gemisch von ca. 40 Zuckern, darunter auch
Ribose und Desoxyribose.[8] Während die Synthese des Primärprodukts Glykolaldehyd durch
anorganische Verbindungen wie Ca(OH)2[9] oder CaCO3
[10] katalysiert wird, verläuft die
Reaktionskaskade in ihrer Gesamtheit autokatalytisch.[11] Problematisch ist hierbei zum einen
die geringe Ausbeute an Zuckern, die für die Nukleinsäurechemie relevant sind (< 1 %), und
zum anderen das Fehlen eines Trennmechanismus unter präbiotischen Bedingungen. Daneben
gibt die geringe Stabilität der Ribose Anlass, an der präbiotischen Relevanz dieser Reaktion
zu zweifeln, sodass an der Synthese stabilisierter Ribose-Derivate wie Ribose-2,4-bisphos-
phat[12] und Diribose-Borat-Komplexen[13] gearbeitet wurde.
Durch das Erhitzen von Ammoniumcyanid auf 70 °C gelang Oro 1960 die Synthese der
Nukleobase Adenin.[14] Wenig später zeigte er, dass dabei fünf Moleküle HCN zu einem
Molekül Adenin reagieren.[15] Mittels Variation der Reaktionsbedingungen gelang auch die
Synthese weiterer Purinderivate wie der Nukleobase Guanin, zwei Zwischenprodukten des
Purinabbaus (Hypoxanthin, Xanthin) und Diaminopurin.[16] Den beiden Letztgenannten
spricht man auch eine Bedeutung in frühen Phasen der molekularen Erkennung zu.[17, 18]
Gerade die Synthese von Adenin ist von hoher präbiotischer Relevanz, da Adenin nicht nur
Bestandteil der Nukleinsäuren ist, sondern auch als Bestandteil von ATP eine zentrale Rolle
im Stoffwechsel der Zellen spielt. Erklären lässt sich dies durch einen Vorteil im Selektions-

Allgemeiner Teil 1 Einleitung
3
prozess der Biomoleküle: Adenin weist in der Stoffklasse der Purine die höchste Resonanz-
energie je π-Elektron auf.[19]
Während Cytosin aus Cyanoacetylen und Harnstoff bzw. Kaliumcyanat zunächst nur in
geringem Umfang hergestellt werden konnte,[20, 21] gelang die Synthese später ausgehend von
Cyanoacetaldehyd und Harnstoff in hohen Ausbeuten von bis zu 50 %.[22] Hierbei entstand
auch Uracil. Weiterhin wurde aus Cyanoacetaldehyd und Guanidinhydrochlorid 2,4-Diamino-
pyrimidin synthetisiert, das sich zu Cytosin, Isocytosin und Uracil hydrolysieren ließ.[23] Die
erfolgreichen Purin- und Pyrimidin-Synthesen erfordern allesamt hohe Konzentrationen an
Startbausteinen, die als unrealistisch für die postulierte Urerde gelten. In vielen Arbeiten
werden daher Möglichkeiten zur Konzentration von Lösungen durch Trocknungsprozesse an
Lagunen und Stränden sowie durch eutektisches Ausfrieren diskutiert.[22-24]
Die Annahme einer reduzierenden Uratmosphäre erschien zunächst folgerichtig, wenn man
bedenkt, dass Wasserstoff das am häufigsten vorkommenden Element im Universum ist.
Allerdings besteht die Atmosphäre unserer direkten Nachbarplaneten Venus und Mars zu
93-98 % aus CO2. Auch Untersuchungen an ältesten Sedimentgesteinen deuten auf eine Ur-
atmosphäre hin, die einen hohen Gehalt an CO2 und N2 aufwies. Miller selbst führte in den
80er Jahren Untersuchungen mit verschiedenen Gasmischungen aus N2, NH3, H2 und H2O
durch, denen entweder CH4, CO oder CO2 als Kohlenstoffquelle zugesetzt wurde.[25, 26] Es
zeigte sich, dass die Synthese von HCN, HCHO, NH3 und Harnstoff, ausgehend von CO oder
CO2, nur bei molaren Verhältnissen von H2 zu CO bzw. CO2 größer als 1.0 gelingt. Auch die
Aminosäuresynthese auf der Basis von CO oder CO2 erfordert hohe molare Verhältnisse von
H2 zu CO bzw. CO2, wobei aber fast ausschließlich Glycin entsteht. In neueren Experimenten
konnten allerdings größere Mengen verschiedener Aminosäuren nachgewiesen werden.[27]
Verneint man die zunächst angenommene reduzierende Uratmosphäre, so stellt sich die Frage
nach den Reduktionsäquivalenten, die für den Aufbau von Biomolekülen benötigt werden. In
Millers Experimenten wurden diese vom Wasserstoff geliefert, wobei die Flucht des
Wasserstoffs ins All gegen das Vorhandensein hoher H2-Partialdrücke spricht. Alternative
Modelle sehen Eisen als Elektronenquelle. In der Thioester-Welt[28] von de Duve liefert Fe2+
durch Bestrahlung mit dem UV-Licht der Sonne ein Elektron je Atom, während in
Wächtershäusers Theorie des Chemoautotrophen Lebensursprungs[29] die oxidative Bildung
von Pyrit aus Fe2+-haltigen Mineralien (z.B. Pyrrhotin) und Schwefelwasserstoff Ursprung
der Reduktionsäquivalente ist.

Allgemeiner Teil 1 Einleitung
4
Auch ein extraterrestrischer Ursprung organischen Materials wird diskutiert, da die frühe Erde
Ort zahlreicher Einschläge von Asteroiden, Meteoriten und Kometen war.[30] In sogenannten
kohligen Chlondriten wie dem Murchinson-Meteoriten wurden ca. 500 organische Verbin-
dungen gefunden[31] – unter ihnen präbiotisch relevante Moleküle wie Di- und Mono-
Aminosäuren, Purine, Pyrimidine und Zucker-Alkohole. Als molekularer Ursprung dieses
Materials gelten interstellare Molekülwolken, die CO, CO2, MeOH, NH3 und H2O enthielten
und im Laufe der Entwicklung des Sonnensystems akkretierten. Im Jahre 2002 gelang zwei
Forschergruppen unabhängig voneinander die Synthese von Aminosäuren bei der Herstellung
von künstlichem interstellarem Eis aus den genannten Ausgangsstoffen im Hochvakuum unter
UV-Licht-Bestrahlung.[32, 33]

Allgemeiner Teil 1 Einleitung
5
1.2 Die RNA-Welt-Hypothese
Unsere moderne DNA-RNA-Protein-Welt ist von sehr großer Komplexität geprägt, sodass
eine de novo Synthese unter präbiotischen Bedingungen ausgeschlossen scheint. Denkbarer
erscheinen evolvierbare Systeme, die nur aus einer Verbindungsklasse bestehen, welche
sowohl in der Lage ist, Informationen zu speichern und weiterzugeben als auch katalytische
Aktivität aufzuweisen. Noch bevor der spätere Nobelpreisträger Gilbert den Begriff der
RNA-Welt prägte[34], erkannten Woese[35], Crick[36] und Orgel[37] das Potential der RNA,
einen solchen autonomen „Organismus“ zu bilden. Auch Eigen[38-41] und Kuhn[42-44] wiesen in
unabhängigen Arbeiten zu Selbstorganisation und Evolution auf die besondere Rolle hin, die
die RNA bei der Entstehung des Lebens eingenommen haben könnte. Die Entdeckung der re-
versen Transkriptase,[45, 46] einer RNA-abhängigen DNA-Polymerase, und der Ribozyme,[47-50]
also enzymatisch wirkender RNA, führte nicht nur zur Erweiterung bzw. Revision molekular-
biologischer Dogmen, sondern gab auch der RNA-Welt-Hypothese Auftrieb.[51]
Die besondere Bedeutung und Flexibilität der RNA zeigt sich auch darin, dass sie in drei
verschiedenen Formen und Funktionen auftritt (tRNA, mRNA, rRNA) und wichtige
Coenzyme von ihren Nukleotiden abstammen. In den Ribosomen wird die Knüpfung der
Peptidbindungen von der RNA und nicht von den ribosomalen Proteinen katalysiert, was als
biologisches Relikt der RNA-Welt verstanden werden kann. Für das im Vergleich zur DNA
frühe Auftreten der RNA spricht, dass RNA in heutigen Zellen vor allem in entwicklungsge-
schichtlich alten Prozessen vorkommt. Darüber hinaus werden Desoxyribonukleotide durch
enzymatische Reduktion von Ribonukleotiden gebildet und Desoxythymidylsäure entsteht
durch die Methylierung der Desoxyuridylsäure. Gegen das frühere Auftreten der RNA lässt
sich anführen, dass 2’-Desoxyribose im Vergleich zu Ribose stabiler und besser löslich ist
und durch die Abwesenheit der 2’-Hydroxyfunktion keine Konkurrenz zwischen 2’-5’- und
3’-5’-Verknüpfungen besteht.[52]
Neben möglichen präbiotischen Verfahren zur Herstellung von Ribose und Nukleobasen stellt
sich im RNA-Welt-Szenario auch die Frage nach der Synthese von Nukleosiden und
Nukleotiden. Obwohl präbiotische Nukleosidsynthesen bislang nur in geringen Ausbeuten
und exklusiv für Purinbasen gelangen,[53, 54] wird die Phosphorylierung von Nukleosiden bis
heute als wahrscheinlichster Zugang zu Nukleotiden diskutiert.[55] Alternative Syntheserouten,
wie sie von Sutherland vorgeschlagen wurden, umgehen freie Ribose und freie Pyrimidin-

Allgemeiner Teil 1 Einleitung
6
Basen.[56, 57] Daneben wird die Beteiligung von Ribozymen bei der Nukleosid- bzw.
Nukleotidsynthese in Erwägung gezogen.[58]
Aufgrund der genannten Probleme wird mittlerweile von mindestens einem einfacheren und
niedriger organisierten System ausgegangen, welches der RNA-Welt als „Prä-RNA-Welt“
voranging.[55, 59-61] Die Vereinfachungen zielen dabei vor allem auf das Zucker-Phosphat-
Rückgrat der RNA ab, während die Basenpaarung als zentrales Element der Informations-
weitergabe beibehalten wird.

Allgemeiner Teil 1 Einleitung
7
1.3 Die Bedeutung selbstreplizierender Systeme für die präbiotische Chemie
1.3.1 Templatgesteuerte Synthesen
Naylor und Gilham beschrieben 1966 die erste nichtenzymatische templatgesteuerte Konden-
sation zweier kurzer DNA-Fragmente an einer komplementären Matrize.[62] Sie setzten die
Thymidin-Oligonukleotide d(pT)5 bzw. d(pT)6 in Gegenwart von Polydesoxyadenosin-
Oligonukleotiden als komplementärer Matrize mit dem wasserlöslichen Carbodiimid CMC 1
um und erhielten die entsprechenden Ligationsprodukte d(pT)10 und d(pT)12 in 3 bzw. 5 %
Ausbeute (Abbildung 1).
CNH
NH
N+
O
O
C
N
N
N+
OC
NH
NH
N+
O O
PO
O
O
OH
P
O
O
O
CNH
NH
N+
O
O
OH
PO
O
O
O
PO
O
O
OH
+
H2O
+
R2
R1 R1 R1R2
1 2 2* 5
3
4
3
R1
2
Abbildung 1. CMC-vermittelte Kondensation von Phosphat-Komponente 2 und Hydroxyl-Komponente 4 zum Phosphodiester 5. – Gezeigt ist sowohl die Hinreaktion als auch die konkurrierende Hydrolyse des aktivierten Phosphats 2*.
Für die präbiotische Chemie stellt sich die Frage, wie ihrerseits die Penta- und Hexamere ent-
standen sind. In diesem Zusammenhang untersuchten Orgel und seine Mitarbeiter ausführlich
die nichtenzymatische Polymerisation von Monomeren an komplementären Matrizen.[63]
Zunächst wurden Poly-U- bzw. Poly-C-Matrizen verwendet, da Purin-Mononukleoside
(tripel-)helicale Strukturen mit Poly-Pyrimidin-Sequenzen bilden. Pyrimidin-Mononukleoside
hingegen bilden keine stabilen Strukturen mit Poly-Purin-Sequenzen, was in ihrer geringeren

Allgemeiner Teil 1 Einleitung
8
Tendenz zur Basenstapelung begründet ist. Als Kondensationsmittel wurde mit EDC wieder-
um ein wasserlösliches Carbodiimid verwendet. Statt der natürlichen 3’-5’-Verknüpfung
beobachtete man überwiegend 2’-5’- und auch 5’-5’-Bindungen.[64, 65] Die Verwendung
präaktivierter Mononukleotide in Form ihrer 5’-Phosphoimidazolide (ImpA bzw. ImpG)
konnte die Effizienz der Bindungsbildung beschleunigen, aber nicht deren Regioselektivität
verändern.[66] Dies gelang erst durch die Verwendung zweiwertiger Metall-Ionen[67, 68] und
durch eine Variation am Imidazolring[69]. So lässt sich im ImpA/Poly-U-System mittels
Zugabe von Pb2+ nicht nur die Ausbeute an längeren Oligomeren deutlich steigern, sondern
auch die Regiochemie in einem beträchtlichen Umfang hin zur 3’-5’-Verknüpfung
umkehren.[67] Im Falle des ImpG/Poly-(C)-Systems bedingt Pb2+-Katalyse hingegen
2’-5’-Verknüpfungen, während Zn2+-Katalyse zu den gewünschten 3’-5’-Bindungen führt.[68]
Wird zur Synthese der aktivierten Nukleotide nun 2-Methylimidazol anstelle von Imidazol
verwendet (Abbildung 2), erhält man auch ohne Zusatz zweiwertiger Metall-Ionen längere
3’-5’-verknüpfte Oligomere.[69]
O
OHOH
OP
O
OH
NN
B
6
Abbildung 2. Struktur eines 5’-Phosphoro(2-methyl)imidazolids 6.
Diese drei Untersuchungen verdeutlichen, wie groß der Einfluss kleiner Änderungen des
Systems auf die Konformationen der beteiligten Komplexe sein kann und wie diese ihrerseits
die Effizienz einer templatierten Reaktion beeinflussen.
Auch die Zusammensetzung des Templats ist entscheidend. So nimmt bei der Verwendung
von Copolymeren aus Pyrimidin- und Purin-Basen als Matrize deren Wirkung mit steigendem
Purinanteil ab.[70] Als Erklärung dient einerseits die schlechte Basenstapelung von Uracil und
Cytosin, andererseits bilden Poly-G-Bereiche stabile Quadruplex-Strukturen, die Interaktio-
nen zwischen C und G unterdrücken. An einer Matrize der Sequenz CCGCC gelang 1984 die
erste nichtenzymatische Oligonukleotidsynthese mit sequenziellem Informationstransfer.[71]
Das purinreiche Produkt kann aber seinerseits nicht als Matrize wirken, was zur Selbstreplika-
tion geführt hätte.

Allgemeiner Teil 1 Einleitung
9
1.3.2 Selbstreplizierende Systeme auf der Basis von Nukleinsäuren
Das erste enzymfreie selbstreplizierende System konnte 1986 durch von Kiedrowski
vorgestellt werden.[72] Das in Abbildung 3 dargestellte Design umfasst das terminal
geschützte DNA-Hexamer d(MeCCGCGGClPh) (C/7) mit palindromischer Sequenz, das als
Templat dient, und zwei Trimere d(MeCCGp) (A/8) und d(CGGClPh) (B/9), aus denen unter
EDC-Aktivierung neues Templat C (7) gebildet werden kann.
N N
N NH
O
NH2O
O
PO OH
O
O
O
PO O
O
N
O
O
PO O
O
MeO N
N
O
NH2
N
O
NH2
N N
N NH
O
NH2
N N
N NH
O
NH2O
O
PO O
O
O
O
PO O
O
O
O
PO O
O
OH N
Cl
N
O
NH2
EDC
N N
N NH
O
NH2
N N
N NH
O
NH2O
O
PO O
O
O
O
PO O
O
O
O
PO O
O
O N
Cl
N
O
NH2
N N
N NH
O
NH2O
O
PO
O
O
O
PO O
O
N
O
O
PO O
O
MeO N
N
O
NH2
N
O
NH2
A / 8
B / 9 C / 7
Abbildung 3. Selbstreplizierendes Minimalsystem nach von Kiedrowski.[72]
Die Verwendung von DNA verhindert unerwünschte Nebenprodukte, da sie die unnatürliche
2’-5’-Verknüpfung von vornherein ausschließt, ebenso die Einführung von Schutzgruppen am
3’-Terminus von A, am 5’-Terminus von B und damit am 3’- und 5’-Ende von C. Purin- und
Pyrimidin-Basen stehen in einem ausgewogenen Verhältnis zueinander und sorgen für einen
ausreichenden Templateffekt, der außerdem durch die höhere Komplexstabilität von Trimeren
gegenüber Monomeren beeinflusst wird. Die Reaktion erfolgt durch den nukleophilen Angriff
der 5’-Hydroxylgruppe von B auf die aktivierte 3’-Phosphatgruppe von A. Nebenprodukt ist

Allgemeiner Teil 1 Einleitung
10
das 3’-3’-verknüpfte Pyrophosphat AppA. EDC muss im Überschuss (ca. 17-fach) zugesetzt
werden, da aktiviertes A* hydrolysiert und verbraucht wird.
Zum Nachweis der Autokatalyse wurde die Anfangskonzentration der Matrize C (7) c0
variiert und die Bildungsgeschwindigkeiten von C (7) und AppA wurden kinetisch
untersucht. Während letztere nicht durch initiale Templatzugabe beeinflusst wird, erhöht sich
die Reaktionsgeschwindigkeit der Produktbildung proportional zur Quadratwurzel von c0.
Daraus wurde folgende Gleichung abgeleitet, die als „Quadratwurzelgesetz der Autokatalyse“
bekannt ist.
βcαt
cv 0c
d
d (1)
Eine Auftragung der ermittelten Anfangsreaktionsgeschwindigkeiten vc gegen die Quadrat-
wurzeln der initialen Konzentrationen an C ergibt eine Gerade, deren Steigung α ein Maß für
die Autokatalyse ist. Der Ordinatenabschnitt β hingegen ist ein Maß für die Synthese von C in
Abwesenheit einer Matrize, die oft auch als Hintergrundreaktion bezeichnet wird. Das
Quadratwurzelgesetz wurde 1987 von Zielinski und Orgel an einem tetrameren System auf
Basis rückgratmodifizierter Ribonukleotide bestätigt (Abbildung 4).[73]
OH
OH
OH
OH
N
N
O
NH2
N
N
O
NH2
O
NH
O
NH
PO O
O
NOH N
NNH
O
NH2
N N
N NH
O
NH2
O
O
NH
PO O
O
PO O
O
N3
OH
OH
N
N
O
NH2
O
NH2
O
NH
PO O
O
NOH N
NNH
O
NH2
OH
N
N
O
NH2
N N
N NH
O
NH2
O
O
NH
PO O
O
O
POH O
O
N3 OH
A / 10
B / 11 C / 12
EDC
Abbildung 4. Selbstreplizierendes Minimalsystem nach Zielinski und Orgel.[73]
Abbildung 5 zeigt ein einfaches Reaktionsmodell für ein selbstreplizierendes System. Die bei-
den Eduktbausteine A und B werden hierbei reversibel vom Templat C gebunden, wobei der
termolekulare Komplex ABC entsteht. Da in diesem Komplex die reaktiven Enden von A und

Allgemeiner Teil 1 Einleitung
11
B in räumlicher Nähe preorganisiert sind, wird der folgende irreversible Ligationsschritt be-
günstigt. Es bildet sich der Templatduplex C2, der reversibel zu zwei Molekülen C
dissozieren kann, die in weiteren Cyclen als Templat zur Verfügung stehen.
Abbildung 5. Reaktionsmodell für ein autokatalytisches und selbstreplizierendes System.
Gleichung 2 gibt die Reaktionsgeschwindigkeit an, mit der C gebildet wird, und berücksich-
tigt neben dem autokatalytischen auch den nicht-autokatalytischen Reaktionsweg. Zu Beginn
der Reaktion ist die Konzentration an C noch gering und der Großteil des Produkts wird über
den nicht-autokatalytischen Weg gebildet. Erst wenn genug C gebildet wurde, endet die
Induktionsphase und die Kurve steigt steil an, um schließlich durch Verbrauch der Edukte A
und B in die Sättigung zu laufen. Auf diesem Weg kommt die sigmoide Form der
Konzentrations-Zeit-Kurve zustande.

Allgemeiner Teil 1 Einleitung
12
])()[)((d
db0a00 kcckcbca
t
c p (2)
c = zur Zeit t neu gebildete Konzentration an C
a0 = Ausgangskonzentration von A
b0 = Ausgangskonzentration von B
c0 = Ausgangskonzentration von C
ka = Geschwindigkeitskonstante des autokatalytischen Reaktionskanals
kb = Geschwindigkeitskonstante des nicht-autokatalytischen Reaktionskanals
p = autokatalytische Reaktionsordnung
Die autokatalytische Reaktionsordnung p kann, wie nachfolgend gezeigt wird, theoretisch die
Werte ½ oder 1 annehmen, während in realen Systemen auch Werte zwischen ½ und 1
möglich sind. Betrachtet man den autokatalytischen Teil von Gleichung 1 zu frühen
Reaktionszeiten, ergibt sich:
pct
c d
d (3)
Trennung der Variablen und Integration im Zeitintervall zwischen 0 und t und im
dazugehörigen Konzentrationsintervall zwischen c0 und c ergibt:
c
c
t
pt
c
c
0 0
dd (4)
Nun muss zwischen p = ½ und p = 1 unterschieden werden.
Für p = ½ gilt:
1
1
1
d
d nn xn
xx
y (5)

Allgemeiner Teil 1 Einleitung
13
Es ergibt sich die Lösung:
2
00 22
tcctcc
(6)
Für p = 1 gilt:
xxx
yln
1
d
d (7)
Somit lautet die Lösung von (4):
tcctcc elnln 00 (8)
Während Gleichung 8 eine exponentielle Wachstumsfunktion beschreibt, nimmt die Konzen-
tration in Gleichung 6 mit dem Quadrat der Zeit zu. Da die Auftragung der Konzentration c in
Abhängigkeit der Zeit t eine Parabel ergibt, spricht man hier von parabolischem Wachstum.
Die Näherung gilt nur für frühe Reaktionszeiten, weil der Verbrauch der Edukte hier nicht
betrachtet wurde. Nimmt p den Wert 0 an, gibt es keinen autokatalytischen Effekt und es
kommt gemäß Gleichung 9 zu linearem Wachstum:
tcctctc
c c
c
tc
c
t
0
000 00
dddd
(9)
Ursache des parabolischen Wachstums ist die Produktinhibition, die dadurch entsteht, dass
ein Großteil der gebildeten Templatmoleküle in inaktiven Duplexen C2 vorliegen, da die
Komplexbildungskonstante für den Templatduplex C2 (K2) in der Regel ein bis zwei Größen-
ordnungen höher ist als für den termolekularen Komplex ABC (K1). Erst ein System mit
K1 > K2 ist daher zu exponentiellem Wachstum fähig. Dieses ist jedoch, wie Szathmary 1989
theoretisch gezeigt hat, Voraussetzung für Selektion (survival of the fittest), also ein Szenario,
in dem der effizienteste Replikator seine Konkurrenten verdrängt, während parabolisches
Wachstum zu Koexistenz (survival of everybody) führt.[74] Im Falle der Koexistenz liegt der
effizienteste Replikator zwar in höheren Konzentrationen vor, kann aber seine Mitbewerber

Allgemeiner Teil 1 Einleitung
14
nicht verdrängen. Die durch parabolisches Wachstum garantierte Sequenz-Vielfalt muss dem
exponentiellen Wachstum der RNA-Welt vorausgegangen sein, da sie den Aufbau funktio-
neller Moleküle wie der Ribozyme begünstigen konnte.
In den folgenden Jahren variierte und erweiterte die Arbeitsgruppe von Kiedrowski das 1986
vorgestellte System mit dem Ziel den templatierten Reaktionskanal zu verstärken und gleich-
zeitig die Hintergrundreaktion zurückzudrängen (Abbildung 6). Die Dominanz der Hinter-
grundreaktion hatte dazu geführt, dass das theoretisch erwartete sigmoide Konzentrations-
Zeit-Profil (s.o.) nicht beobachtet werden konnte. So wurde ein katalytisches System
untersucht, bei dem die Matrize C (7) (im Folgenden C1) beibehalten wurde, während die
Eduktbausteine A2 (13) und B2 (14) so designt waren, dass im reaktionsbestimmenden Schritt
eine Pyrophosphatbindung gebildet wird.[75] Das resultierende Hexamer C2 (15) ist nicht mit
C1 (7) identisch, kann aber als chemisch markiertes Produkt angesehen werden, welches die
HPLC-Analyse des Systems erleichtert.i Streng genommen findet keine echte Selbstreplika-
tion statt. Man fand heraus, dass unter gleichen Bedingungen C2 (15) im matrizenabhängigen
Reaktionskanal 21-mal schneller gebildet wird als C1 (7), während der nicht-matrizen-
abhängige Weg nur zehnmal schneller ist. Das System profitiert daher von der größeren
Nukleophilie der angreifenden 5’-Gruppe und der damit verbundenen schnelleren Replikati-
on. Die Verwendung von Trimeren, welche nur teilweise oder gar nicht komplementär zu C
sind, zeigt erwartungsgemäß einen Rückgang in der Reaktionsgeschwindigkeit (2- bis 50-mal
langsamer), der sich am stärksten im matrizenabhängigen Reaktionskanal bemerkbar macht.
Schließlich gelang es im Jahr 1992 das erste System vorzustellen, das sigmoides (s-förmiges)
Wachstum zeigt.[76] Hierbei reagiert ein Tridesoxynukleotid-3’-phosphat A2 (13) mit einem
5’-Amino-Trimer B3 (16) zu einem selbstkomplementären hexameren 3’-5’-Phosphoamidat
C3 (17). Die Nukleophilie der angreifenden 5’-Gruppe ist erneut gesteigert. Aus Gründen der
einfacheren Analyse (s.o.) wurde erneut die Phophodiester-Matrize C1 (7) als Templat ge-
wählt, da sie einen vergleichbaren katalytischen Effekt wie die Phosphoamidat-Matrize
C4 (18) besitzt. Letztere liefert somit ein indirektes Maß für den autokatalytischen Effekt von
C3 (17). Das Verhältnis von templatierter zu nicht-templatierter Synthese, definiert durch
b
a
k
k (10)
i C1 und C2 unterscheiden sich nur durch eine Schutzgruppe am 5’-Ende, d.h. sie können als chemisch sehr ähnlich betrachtet werden. Da sie sich durch Chromatographie trennen lassen, entfällt die Differenzbildung von HPLC-Integralen, die eine mögliche Fehlerquelle darstellt.

Allgemeiner Teil 1 Einleitung
15
kann beim Übergang vom Phosphodiester-System zum Phosphoamidat-System von 25 auf
420 M-1/2 gesteigert werden. Weitere Untersuchungen zeigen, dass die Reaktivität der Kon-
densation maßgeblich von der Stapelung der beiden Nukleobasen bestimmt wird, die der
Verknüpfungsstelle benachbart sind. Dies geschieht in der Reihe G-G > G-C > C-G > C-C,
wobei beispielsweise der Reaktivitätsunterschied zwischen G-C und C-G etwa eine Größen-
ordnung beträgt. Da ka nach Arrhenius mit steigender Temperatur zunimmt, während die
Konzentration an C durch das Schmelzen der termolekularen Komplexe ABC abnimmt, ist es
zu erwarten, dass die Auftragung von ka gegen die Temperatur ein Maximum durchläuft.
Diese optimale Reaktionstemperatur liegt in der Nähe der Schmelztemperatur des Templat-
duplexes des jeweiligen Replikators.[77]
N N
N NH
O
NH2O
O
PO OH
O
O
O
PO O
O
N
O
O
PO O
O
O N
N
O
NH2
N
O
NH2
R1
N N
N NH
O
NH2
N N
N NH
O
NH2O
O
PO O
O
O
O
PO O
O
O
O
PO O
O
R2 N
Cl
N
O
NH2
N N
N NH
O
NH2
N N
N NH
O
NH2O
O
PO O
O
O
O
PO O
O
O
O
PO O
O
X N
Cl
N
O
NH2
N N
N NH
O
NH2O
O
PO
O
O
O
PO O
O
N
O
O
PO O
O
O N
N
O
NH2
N
O
NH2
R1
EDC
A1 / 8: R1 = CH3
A2 / 13: R1 = CH2SCH3
B1 / 9: R2 = HOB2 / 14: R2 = HO3PO-
B3 / 16: R2 = H2N
C1 / 7: R1 = CH3, X = OC2 / 15: R1 = CH2SCH3, X = PO4
-
C3 / 17: R1 = CH2SCH3, X = NHC4 / 18: R1 = H, X = NH
Abbildung 6. (Selbst)replizierende Minimalsysteme nach von Kiedrowski.[72, 75, 76]

Allgemeiner Teil 1 Einleitung
16
Die bislang untersuchten Replikatoren waren alle selbstkomplementär, wohingegen das
natürliche Vorbild der Nukleinsäure-Replikation mit komplementären Strängen arbeitet, die
wechselseitig ihre Synthese katalysieren. Im Falle eines solchen kreuzkatalytischen Prozesses
wird im ersten Cyclus das zu C komplementäre Produkt C’ gebildet, welches im zweiten
Cyclus die Synthese des ursprünglichen Templates C katalysiert. Parallel hierzu wird im
zweiten Cyclus das zu C’ komplementäre Produkt C gebildet, welches im ersten die Synthese
des ursprünglichen Templates C’ katalysiert. Diese beiden Reaktionswege sind über die
Bildung desselben Templatduplexes CC’ miteinander verknüpft (Abbildung 7).
Abbildung 7. Reaktionsmodell für ein kreuzkatalytisches System.
Im Jahr 1994 stellten Sievers und von Kiedrowski ein minimales kreuzkatalytisches System
ausgehend von den vier trimeren Vorstufen N3CCGp (A), MTMCGGp (A’), nCCGClPh (B) und
nCGGClPh (B’) vor.[78, 79] Da in diesem System die beiden komplementären Matrizen N3CCGpnCCGClPh (C) und MTMCGGpnCGGClPh (C’) das Produkt der gleichen Ligations-
chemie sind, kommt es gleichzeitig zur autokatalytischen Selbstreplikation der beiden selbst-
komplementären Produkte N3CCGpnCGGClPh (C’’) und MTMCGGpnCCGClPh (C’’’) gemäß:

Allgemeiner Teil 1 Einleitung
17
A’ + B + C’’ → C’’2 (11)
und
A + B’ + C’’’ → C’’’2 (12)
Weil die vier Produkte die gleiche zentrale Subsequenz G-C aufweisen (s.o.), zeigen alle
Kondensationen eine ähnliche Effizienz, unabhängig vom Pyrimidin-Gehalt und unabhängig
davon, ob die Reaktion auto- oder kreuzkatalytisch verläuft. Informationstransfer kann durch
Zugabe eines der vier Produkte in chemisch markierter Form nachgewiesen werden, da
hierdurch nur die Synthese der entsprechenden Sequenz selektiv stimuliert wird.
Schon 1993 wurden erste Anstrengungen unternommen, ein System zu realisieren, in dem
Wettbewerb um die Ressource Precursor besteht.[80] Dies gelang dadurch, dass die bereits
bekannte Sequenz CCGCGG nun aus drei Startbausteinen aufgebaut wurde, nämlich PGCCG, H2NCGp und H2NG. Das Ergebnis ist ein gekoppeltes katalytisches Netzwerk mit sechs ver-
schiedenen Rückkopplungen unterschiedlicher Effizienz. Zur Untersuchung wurde das Netz-
werk in Subsysteme zerlegt, die auf ihren jeweiligen Beitrag zum Gesamtsystem untersucht
wurden. Das Hexamer PGCCGpnCGpnG und das Pentamer PGCCGpnCGp verhalten sich als
gekoppelte Autokatalysatoren, indem sie einerseits egoistisch um die gleichen Startbausteine
konkurrieren, andererseits altruistisch ihre wechselseitige Synthese katalysieren. Das Haupt-
produkt der nicht-templatierten Synthese, PGCCGpnG, stellt hingegen einen isolierten Auto-
katalysator dar, der mit den anderen Spezies um die Vorstufen konkurriert. Die nächste Hürde
bestand nun darin, ein System zu entwerfen, das exponentielles Wachstum zeigt und so
Darwinsche Selektion möglich macht.
Das 1998 vorgestellte SPREAD-Verfahren (SPREAD = Surface Promoted Replication and
Exponential Amplification of DNA-analogues) ermöglicht erstmals eine Überwindung der
Produktinhibition.[81] Wie in Abbildung 8 dargestellt, werden zunächst Templatmoleküle über
Disulfidbrücken irreversibel auf einer Festphase immobilisiert (1). Nach Zugabe einer Lösung
komplementärer Fragmente, die über Wasserstoffbrückenbindungen reversibel an das
jeweilige Templat binden (2), kommt es durch Zugabe von EDC zur Ligation (3) und damit
zur Synthese eines zum ursprünglichen Templat komplementären Stranges. Dieser wird von
der immobilisierten Matrize getrennt (4), seinerseits immobilisiert und im nächsten
Synthesecyclus eingesetzt. Durch diese räumliche Trennung der Kopie vom Templat wird die
Reassoziation unterdrückt und die Produktinhibition umgangen. Das SPREAD-Verfahren ist
ein iterativer Prozess und erfordert daher das Eingreifen eines Experimentators.

Allgemeiner Teil 1 Einleitung
18
Eine autonome SPREAD-Variante würde ein sich selbst erhaltendes chemisches System
darstellen, welches in der Lage wäre, Darwinsche Evolution zu zeigen.
Abbildung 8. Schematische Darstellung der SPREAD-Technik. – Die Abbildung ist aus der Originalarbeit entnommen worden.[81]
Paul und Joyce präsentierten 2002 ein Ligase-Ribozym, das seine eigene Bildung aus zwei
RNA-Fragmenten katalysiert (Abbildung 9).[82] Hierbei wird das Fragment A zu einer Lösung
des vorher hergestellten Komplexes BT aus Fragment B und Templat T gegeben, wodurch
sich der reaktive termolekulare Komplex ABT bilden kann. Für frühe Reaktionszeiten wurde
eine autokatalytische Reaktionsordnung von p = 1 gefunden, die charakteristisch für exponen-
tielles Wachstum ist und sich durch einen verhältnismäßig instabilen Templat-Duplex TT
erklären lässt. Allerdings können die neu gebildeten Ribozymmoleküle kaum als Matrize
wirken, da die Edukt-Fragmente als stabile AB-Komplexe vorliegen, was die Bildung des
termolekularen Komplexes erschwert. Als Folge lässt sich kein exponentielles Wachstum
beobachten. Dieses autokatalytische System wurde nachfolgend von Lincoln und Joyce zu
einem kreuz-katalytischen System weiterentwickelt, dessen Effizienz durch in vitro Evolution
der beteiligten Ribozyme optimiert wurde.[83] Dadurch beträgt die Verdopplungszeit statt
17 Stunden nur noch eine Stunde und die Replikation bricht nicht mehr nach zwei Cyclen ab,

Allgemeiner Teil 1 Einleitung
19
sondern kann bei entsprechender Versorgung mit Edukten unendlich fortgesetzt werden. Lam
und Joyce überführten die Ribozyme in sogenannte „Adaptazyme“ und konnten so gezielt
exponentielles Wachstum durch die Zugabe von bestimmten Liganden einschalten.[84]
Abbildung 9. Selbstreplizierendes Ligase-Ribozym nach Joyce. – Die Abbildung ist aus der Originalarbeit entnommen worden.[82]

Allgemeiner Teil 1 Einleitung
20
1.3.3 Selbstreplizierende Systeme auf der Basis von Peptiden
Als das erste selbstreplizierende Peptid 1996 von Ghadiri vorgestellt wurde,[85] waren
mögliche präbiotische Szenarien für die Entstehung von Aminosäuren (s. 1.1) und die
Kondensation zu Polypeptiden schon lange bekannt. Da Peptide in heutigen Organismen als
Funktionsmoleküle wirken und nicht direkt an Replikationsprozessen beteiligt sind, wurden
neue Modelle molekularer Evolution möglich.
Während die molekulare Erkennung bei Nukleinsäuren nachvollziehbar durch die Basen-
paarung erfolgt, also durch die Sequenz gegeben ist, spielen bei peptidischen Systemen auch
höhere Strukturebenen (Sekundär-, Tertiär- und Quartärstruktur) eine wichtige Rolle. Der
Großteil der bisher beschriebenen Peptid-Replikatoren basiert auf dem Tertiär-Strukturmotiv
des Coiled-Coil (engl.: Doppelwendel). Hierbei handelt es sich um oligomere Anordnungen
von mindestens zwei amphipathischen (α)-Helices, deren Primärsequenz aus charakteristi-
schen heptameren Wiederholungseinheiten (abcdefg)n besteht (Abbildung 10).
Abbildung 10. Helixrad-Projektion einer Heptade eines dimeren Coiled-Coil. – Die molekulare Erkennung findet sowohl durch hydrophobe Wechselwirkungen zwischen den Positionen a und d statt als auch aufgrund elektrostatischer Wechselwirkungen zwischen den Positionen e und g. Die Reste in den Positionen b, c und f sind dem Lösungsmittel zugewandt und nehmen nicht direkt an der Ausbildung der Tertiärstruktur teil.
Der 1996 von Ghadiri beschriebene Replikator T ist ein Peptid aus 32 Aminosäureresten,
dessen Sequenz an die Leucin-Reißverschluss-Domäne des Transkriptionsfaktors GCN4 der
Hefe angelehnt ist und das in einem Gleichgewicht dimerer und trimerer Strukturen vorliegt.
Dieses Peptid T ist in der Lage, seine eigene Bildung aus einem elektrophilen Fragment E
(17mer) und einem nukleophilen Fragment N (15mer) zu katalysieren. Die dabei neu
entstehende Amidbindung wird unter den Bedingungen der nativen chemischen Ligation[86]
gebildet (Abbildung 11).

Allgemeiner Teil 1 Einleitung
21
S
O
NH
R1 NH2
NH
O
SH
NH2
NH
O
S
O
NH
R1
NH
O
NH
R1
SH
O
+
E N T
Abbildung 11. Native Ligation nach Kent.[86] – Der C-terminale Thiobenzylester von E wird durch den N-terminalen Cyteinrest von N unter Ausbildung eines intermediären Thioesters angegriffen, der sich nachfol-gend in die thermodynamisch stabilere Amidbindung umlagert, wobei T entsteht.
Erste kinetische Untersuchungen zeigten parabolisches Wachstum gemäß dem Quadratwur-
zelgesetz der Autokatalyse und demzufolge eine Reaktionsordnung von p = 0.5. Die autokata-
lytische Effizienz betrug in diesem Fall ca. 500 M-1/2. Aufgrund einer verdoppelten initialen
Fragmentkonzentration konnte später eine Reaktionsordnung von p = 0.63 gefunden werden,
was die Beteiligung von quartermolekularen Komplexen des Typs ENTT neben den vorher
postulierten termolekularen Komplexen vom Typ ENT wahrscheinlich macht.ii
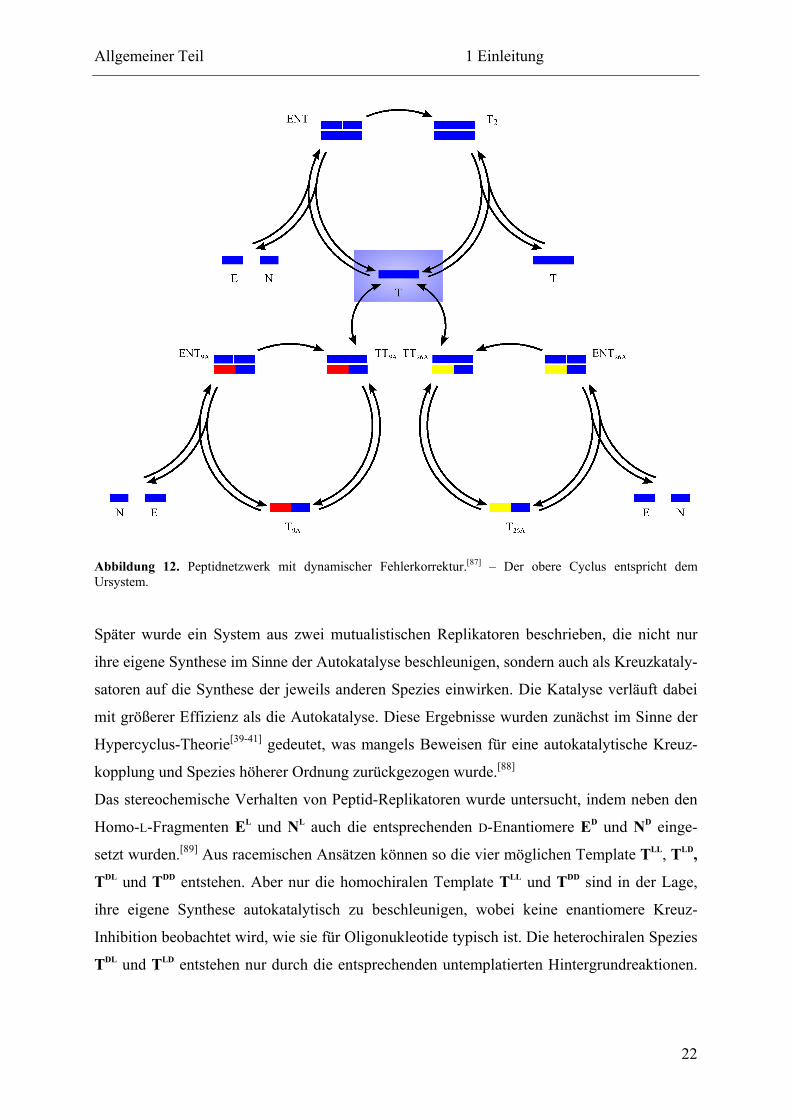
Bald darauf wurde ein Peptidnetzwerk vorgestellt, das eine dynamische Fehlerkorrektur
zeigt.[87] Hierbei werden neben den ursprünglichen Fragmenten E und N auch die beiden
Alanin-Mutanten E9A und N26A eingesetzt, sodass die vier Produkte T, T9A, T26A und T9A/26A
erwartet werden. Es zeigt sich, dass T als egoistischer Autokatalysator nur seine eigene Bil-
dung katalysiert, während T9A und T26A anstatt ihrer eigenen die Synthese von T katalysieren
(Abbildung 12). Dabei liegt die katalytische Effizienz der mutierten Peptide bei 75 % der
Aktivität von T. Das doppelt mutierte Produkt T9A/26A ist autokatalytisch sowie katalytisch
inaktiv.
ii Unter Berücksichtigung der Produktinhibition ergibt sich für ein quartermolekulares System im geschwindig-keitsbestimmenden Reaktionsschritt eine autokatalytische Reaktionsordnung von 2/3.
Allgemeiner Teil 1 Einleitung
22
Abbildung 12. Peptidnetzwerk mit dynamischer Fehlerkorrektur.[87] – Der obere Cyclus entspricht dem Ursystem.
Später wurde ein System aus zwei mutualistischen Replikatoren beschrieben, die nicht nur
ihre eigene Synthese im Sinne der Autokatalyse beschleunigen, sondern auch als Kreuzkataly-
satoren auf die Synthese der jeweils anderen Spezies einwirken. Die Katalyse verläuft dabei
mit größerer Effizienz als die Autokatalyse. Diese Ergebnisse wurden zunächst im Sinne der
Hypercyclus-Theorie[39-41] gedeutet, was mangels Beweisen für eine autokatalytische Kreuz-
kopplung und Spezies höherer Ordnung zurückgezogen wurde.[88]
Das stereochemische Verhalten von Peptid-Replikatoren wurde untersucht, indem neben den
Homo-L-Fragmenten EL und NL auch die entsprechenden D-Enantiomere ED und ND einge-
setzt wurden.[89] Aus racemischen Ansätzen können so die vier möglichen Template TLL, TLD,
TDL und TDD entstehen. Aber nur die homochiralen Template TLL und TDD sind in der Lage,
ihre eigene Synthese autokatalytisch zu beschleunigen, wobei keine enantiomere Kreuz-
Inhibition beobachtet wird, wie sie für Oligonukleotide typisch ist. Die heterochiralen Spezies
TDL und TLD entstehen nur durch die entsprechenden untemplatierten Hintergrundreaktionen.

Allgemeiner Teil 1 Einleitung
23
Erstaunlicherweise findet selbst bei Einzelmutationen in den Edukt-Fragmenten keine
Autokatalyse statt.
Auch bei peptischen Replikatoren gibt es die Bemühung, die autokatalytische Effizienz ε zu
steigern, indem zum einen die Hintergrundreaktion zurückgedrängt und zum anderen die Pro-
duktinhibition vermindert wird. Letzteres führt auch zu einer Erhöhung der autokatalytischen
Reaktionsordnung p. Die Überwindung der Produktinhibition wird wegen eines intrinsischen
Problems erschwert, da alle Maßnahmen zur Destabilisierung des Templat-Duplexes höchst-
wahrscheinlich auch zu einer Destabilisierung des termolekularen Komplexes führen werden.
Die Gruppe von Chmielewski entwickelte Peptid-Replikatoren, deren Autokatalyse durch den
Einfluss von pH-Wert[90] oder Salzkonzentration[91] moduliert werden kann. Später präsentier-
ten Issac und Chmielewski die Variante RI26 ihres ursprünglichen Replikators E1E2, welche
um eine Heptade verkürzt ist und unter vergleichbaren Bedingungen nicht als Dimer, sondern
als Tetramer vorliegt.[92] Im Vergleich zum Ursprungssytem wurde eine 90-fach langsamere
Hintergrundreaktion ermittelt (ε = 1.0 × 105 M
2.09) und durch die geringere Anzahl an Leucin-
Resten und die dadurch reduzierten hydrophoben Wechselwirkungen zwischen den Fragmen-
ten RI26a und RI26b erklärt. Die Reaktionsordnung p liegt mit 0.91 deutlich über dem Wert
von 0.75, welcher für ein produktinhibiertes tetrameres System zu erwarten ist, sodass das
System als schwach exponentiell charakterisiert wird. Als Ursache für die Reduktion der
Produktinhibition wird die verringerte Stabilität quaternärer Komplexe (RI26)4 gegenüber
dimeren (E1E2)2 Komplexen angenommen.
Auch der Austausch einzelner Aminosäure-Reste im Ursprungssystem E1E2 führt zu interes-
santen Ergebnissen. So verursacht die Destabilisierung des hydrophoben Kerns durch Aus-
tausch eines einzelnen Leucin-Rests in Position 19 gegen Alanin bzw. Glycin eine 50- bzw.
75-fach langsamere Hintergrundreaktion.[93] Der Austausch des Glutaminsäure-Rests an
Position 20 durch den Helixbrecher Prolin führt zu einer 35-fach langsameren Hintergrund-
reaktion (ε = 3.2 × 104 M2.09).[94] Wie im Falle von RI26 liegt das Produkt in Lösung als
Tetramer vor und wird durch eine ermittelte Reaktionsordnung von p = 0.91 als schwach
exponentiell charakterisiert.

Allgemeiner Teil 1 Einleitung
24
1.3.4 Artifizielle Replikatoren
Ein erstes System auf dem Weg zu artifizieller Replikation publizierten Rebek et al 1990.[95]
Das Templat C (19) enthält ein Erkennungsmotiv auf der Basis eines Adenosin-Derivats und
ein dazu komplementäres Amid-Derivat der Kempschen Trisäure (Abbildung 13). Die
Ligation der Bausteine erfolgt durch die Knüpfung einer Amidbindung zwischen einem Amin
A (20) und einem Carbonsäure-Aktivester B (21) in Chloroform. Die hier beobachtete
Autokatalyse gehorcht ebenfalls dem Quadratwurzelgesetz (ε = 80 M-1/2).[96]
NO
O
O
O
O
O
F
F F
F
F
H NN
NN
O
O
O
NH
NH2
H
NN
NN
O
O O
NH
NO
O
O
O
H
ONH
H
NN
NN
O
OO
NH
NO
O
O
O
H
ONH
H
NN
NN
O
O O
NH
NO
O
O
O
H
ONH
H
O
OO
NNN
NNH
H
NH2
N
O
O
O
O
H
O
OFF
F
FF
NO
O
O
O
NH
O
NN
NN
O
O
O
NH
HH
A / 20
B / 21
C / 19 C / 19
C / 19
C / 19
A / 20B / 21
Abbildung 13. Replikator nach Rebek et al.[95] – Reaktion des bimolekularen AB-Komplexes zum cis-konfigu-rierten Produkt (links) und des termolekularen ABC-Komplexes zum trans-konfigurierten Produkt (rechts).
In den folgenden Jahren wurde der Mechanismus des Replikators intensiv diskutiert.[97-100]
Reinhoudt et al. untersuchten daher das komplexe System genauer und identifizierten fünf
verschiedene Reaktionswege für die Bildung des Templats C: (i) durch die Hintergrund-
reaktion, (ii) über einen binären Komplex AB, (iii) über den termolekularen Komplex ABC,
(iv) über den aktivierten Aktivester und schließlich (v) über das aktivierte Amid.[101] Die
Arbeit beendete die Kontroverse, da sie sowohl Rebek als auch seinem Kontrahenten Menger
Recht gibt: In Lösungen niedriger Konzentration findet Selbstreplikation statt, in Lösungen

Allgemeiner Teil 1 Einleitung
25
hoher Konzentration auch Amid-Katalyse. Den bis dahin präferierten Reaktionskanal über
den binären Komplex konnte Rebek durch die Substitution des Naphtyl- mittels eines
Biphenyl-Spacers zurückdrängen und ein sigmoides Reaktionsprofil erhalten.[102]
Ein erster vollsynthetischer Replikator wurde 1992 von Terfort und von Kiedrowski
vorgestellt (Abbildung 14).[103] Hierbei basiert die molekulare Erkennung nicht auf Motiven
aus der Natur wie der Basenpaarung, sondern auf organischen Analoga wie in diesem Fall auf
Amidinium-Carboxylat-Salzbrücken. Die Kondensation von Amin A/22a und Aldehyd B/23a
ist autokatalytisch und wird durch Zugabe von Templat C/24a gemäß dem
Quadratwurzelgesetz beschleunigt (p = 0.5, ε = 16 M-1/2). Die Geschwindigkeit der Reaktion
von A/22b mit B/23b zu Produkt C/24b kann durch Zugabe der strukturell analogen Matrize
C/24c erhöht werden; dabei kann man allerdings eine lineare Abhängigkeit der Reaktionsrate
von der Konzentration an C/24c beobachten. Die Katalyse erster Ordnung lässt sich durch
einen ternären Komplex ABC (hier: 22b23b24c) erklären, der stabiler ist als der Templat-
Duplex CC (hier: 24c24c).
O
R1
O
O O
N
H
R3
R4
O
OONN
H H
H H
N
H
R2
R1
O
O O N N
HH
HH
N
H
R3
R4
O
OONN
H H
H H
NH2
R2
N N
HH
HH-
+
+
a: R1 = R4 = NO2, R2 = R3 = tBub: R1 = NO2, R2 = Me, R3 = tBu, R4 = Hc: R1 = R4 = H, R2 = R3 = tBu
-
+
+
A / 22 B / 23 C / 24
C / 24 C / 24
- -
Abbildung 14. Artifizielles Replikations-System nach Terfort und von Kiedrowski.[103]
Im Jahr 1997 beschrieben Wang und Sutherland ein selbstreplizierendes System auf der Basis
einer Diels-Alder-Cycloaddition zwischen dem Dien 25 und dem Maleinimid 26 (Abbildung
15).[104] Die autokatalytische Reaktionsordnung liegt mit p = 0.8 zwischen parabolischer und
exponentieller Replikation, was sich anhand einer sterisch gehinderten Templatassoziation
erklären lässt.

Allgemeiner Teil 1 Einleitung
26
NN
NH
O
NN
O
O
O
NH
C5H11
O
H
H
N
O
N N
HH
N N
H
C5H11
O
O
N
O
O
H25 26
27
Abbildung 15. Selbstreplizierendes System nach Wang und Sutherland.[104]
Zwei Varianten dieses Systems wurden 2005 vorgestellt, um seine stereochemischen Eigen-
schaften besser untersuchen zu können.[105] Während Wang und Sutherland 1997 von einer
endo-Konfiguration von 27 ausgingen, konnte im Rahmen der Untersuchungen von Kinder-
mann et al. nur das exo-Produkt nachgewiesen werden. Die Analyse der individuellen Reak-
tionen von Dien (R)-28 bzw. (S)-28 mit Dienophil 29 in Gegenwart von 10 % Templat 30
zeigt, dass es homochirale autokatalytische sowie heterochirale kreuzkatalytische Reaktions-
kanäle gibt, deren Templatduplexe vergleichbar populiert sind (Abbildung 16).
R2
N
O
O
R3 R2 N
O
O
R3
R2
N
O
O
R3 N
O
O
R3R2
N
NH
O N
O
O O
OH
R1 R1
O
O
N
O
O
H
R1 R1
N
NH
O
+
+
28 29 30
A / 28 B / 29
C / 30
Abbildung 16. Selbstreplikations-System nach Kindermann und von Kiedrowski. – Termolekularer Komplex (links) und homo-und heterochirale Reaktionskanäle (rechts).
Ein weiteres minimales Selbstreplikations-System auf der Basis einer Cycloaddition wurde
2001 von Philp vorgestellt (p = 0.9, ε = 5000 M-1/2).[106] Hier reagiert ein Nitron 31 als
1,3-Dipol mit einem Maleinimid 32 als Dipolarophil in einer Huisgen-Reaktion; dabei kann
nur eins der diastereomeren Produkte 33 als Katalysator für seine eigene Bildung dienen
(Abbildung 17). Dies lässt sich damit erklären, dass das cis-Isomer eine geschlossene Geome-
trie annimmt und daher nicht fähig ist, einen ternären Komplex auszubilden, während das

Allgemeiner Teil 1 Einleitung
27
trans-Isomer als egoistischer Replikator fungiert und die eigene Bildung exklusiv beschleu-
nigt. Dieses System wurde später durch geringe strukturelle Variationen optimiert.[107]
N NH
O
N+
ON
O
O
OO H
ON N
O
OO
O HO
NH
N
H
H
+
31 32 33
Abbildung 17. Replikations-System auf der Basis einer 1,3-dipolaren Huisgen-Cycloaddition.[106] – Gezeigt ist nur das autokatalytisch aktive trans-Isomer.
Dieckmann et al. stellten 2010 ein System vor, das auf der Cycloaddition eines Fulvens 34 mit
einem Maleinimid 35 basiert, und untersuchten es mit Hilfe von zeitaufgelöster NMR-
Spektroskopie und Computer-Simulationen (Abbildung 18).[108, 109]
N
O
H
N
O
O
ORO
N
O
H
N
O
OOR
O
N
O
N
O
OOR
O
H
+
35a: R = H 3435b: R = Me
36a: R = H: NN 36b: R = Me: NN'
36c: R = H: NX36d: R = Me: NX'
Abbildung 18. Bimolekulare Hintergrundreaktion des Replikators nach Dieckmann und von Kiedrowski.[108, 109]
Unter Blockierung der supramolekularen Erkennungsstellen werden im Laufe der Reaktion
zwei Diastereomere NN und NX mit gleichen Raten gebildet. Ist molekulare Erkennung
möglich, entsteht ein Netzwerk, in dem das Diastereomer NN als egoistischer Autokatalysator
wirkt, während das andere Isomer NX als schwacher Altruist fungiert (Abbildung 19). Daher
wird NN im Vergleich zu NX mit einer Diastereoselektivität von 16:1 gebildet.

Allgemeiner Teil 1 Einleitung
28
Abbildung 19. Fulven-Replikator-System nach Dieckmann und von Kiedrowski. – Ein Zwei-Buchstaben-Code wird zur Bezeichnung der Produkte verwendet. Der erste differenziert die Diels-Alder-Produkte (N: endo, X: exo), der zweite die Orientierung der Amidopyridin-Erkennungsstelle (N: auf der gleichen Seite wie die Brücke, X: auf der entgegengesetzten Seite der Brücke). Die Abbildung ist der Originalarbeit entnommen.[108]

Allgemeiner Teil 1 Einleitung
29
1.4 Die PNA-Welt als Modell einer Prä-RNA-Welt
1.4.1 Intention des PNA-Designs
PNA wurde ursprünglich entworfen, um mit DNA-Duplexen Triplex-Strukturen auszu-
bilden.[110] Solche Triplex-Strukturen bilden sich in der großen Furche der DNA mit
Homopurin-Regionen, indem der dritte und Triplex-bildende Strang durch Hoogsteen- statt
durch Watson-Crick-Basenpaarung bindet (Abbildung 20).
N N
N N
NHH
N
O
O
HN
N
O
N
O
H
N N
N N
O
N
H
H
H
NN
O
N
H
HN
N+
O
NH
H
H
Abbildung 20. Watson-Crick- (blau) und Hoogsteen-Basenpaarungen (rot) beim T·A-T-Triplett (links) und C+·G-C-Triplett (rechts). – Der Triplex-bildende Strang bindet an die Seiten der Purine, die nicht an der Watson-Crick-Basenpaarung teilnehmen (N7 und N6 bei Adenin, N7 und O6 bei Guanin).
Beim computergestützten Design gingen Nielsen et al. 1991 daher von einem normalen
T·A-T-Triplex aus, entfernten das Zucker-Phosphat-Rückgrat und ersetzten es durch ein
Polyamid-Rückgrat.[110] Es erwies sich, dass die sechs Bindungen einer monomeren
2-(Aminoethyl)glycin-Einheit (AEG) und die drei Bindungen eines Methylencarbonyl-
Spacers optimal für die Bindung an DNA in der B-Konformation sind. Eine unabhängige
Studie auf Basis von Molecular-Modelling im selben Jahr führte zu vergleichbaren
Ergebnissen.[111] Darüber hinaus wurde AEG schon 1987 als Baustein für ein alternatives
Rückgrat vorgeschlagen.[112] Abbildung 21 zeigt die Primärstruktur von PNA im Vergleich zu
den Strukturen der natürlichen Biopolymere.

Allgemeiner Teil 1 Einleitung
30
OH
O
O
OB
PO O
O
O
OB
PO O
O
O
OHB
PO O
n
n
NH2
NO
O
NH
N
O
B
O
NH
NO
B
O
NH2
B
OH
O
O
OB
PO O
O
O
OB
PO O
O
O
OHB
PO O
OH
OH
OH
n
NHO
NHR
R
OHO
NHR
O
O
NH2
R
n
Abbildung 21. Primärstrukturen von PNA, DNA, RNA und Peptiden (von links nach rechts).
Pyrimidinreiche PNAs bilden in der Regel keine Triplexstrukturen der Form
DNA·DNA-PNA, sondern des Typs PNA-DNA·PNA. Aus doppelsträngiger DNA verdrängen
sie den nicht-komplementären Strang (Strand-Invasion). Bei diesen Komplexen bindet ein
PNA-Strang über Watson-Crick-Basenpaare im antiparallelen Modus, der andere über
Hoogsteen-Basenpaare im parallelen Modus. Dieses und weitere Strukturmotive für die
Hybridisierung von PNA an doppelsträngige DNA zeigt Abbildung 22. PNAs mit
ausgeglichenem Pyrimidin/Purin-Verhältnis bilden mit DNA und RNA Duplexe, deren
antiparallele Orientierung ebenfalls bevorzugt ist. Die Struktur wird hierbei von der DNA
oder RNA beeinflusst, während der PNA-Strang die Struktur des Bindungspartners adaptiert.
Abbildung 22. Strukturmotive für die Hybridisierung von PNA an doppelsträngige DNA. – a) Triplex; b) Triplex-Invasion; c) Duplex-Invasion; d) Double-Duplex-Invasion.

Allgemeiner Teil 1 Einleitung
31
Untersuchungen an einem nicht-selbstkomplementären PNA-15mer mit einer zentralen
gtac-Einheit und einem ausgeglichenen Pyrimidin/Purin-Gehalt zeigten, dass die geringere
elektrostatische Abstoßung zwischen PNA und DNA bzw. RNA sowohl eine Steigerung der
Affinität als auch der Spezifität verursacht.[113] Die untersuchten PNA/DNA- und PNA/RNA-
Komplexe schmelzen in der antiparallelen Orientierung 16.2 °C bzw. 21.7 °C später als die
korrespondierenden DNA/DNA- bzw. RNA/RNA-Duplexe. In der parallelen Orientierung
betragen die Werte für ΔTm 13.4 °C bzw. 21.1 °C. Diese erhöhte Stabilität zeigt sich wie
erwartet bei niedrigen bis hohen Salzkonzentrationen, während PNA/DNA- und DNA/DNA-
Duplexe oberhalb von 1 M Na+ identische thermische Stabilitäten besitzen, da hohe Salzkon-
zentrationen die Stabilität doppelsträngiger DNA erhöhen. Detaillierte Messungen mit Hilfe
der BIAcore Technik und weitere thermische Messungen zeigen, dass PNA/RNA-Duplexe
durchschnittlich 4 °C später schmelzen als die korrespondierenden PNA/DNA-Komplexe und
schnellere Assoziationskonstanten und langsamere Dissoziationskonstanten aufweisen.[114]
Basenfehlpaarungen führen in PNA/DNA-Duplexen zu deutlicheren Destabilisierungen als in
DNA/DNA-Duplexen. Im Beispielsystem (H-tgtacgtcacaacta-NH2) wurden nacheinander die
Basen der zentralen gtac-Einheit ausgetauscht und ein durchschnittlicher Wert für ΔTm von
15 °C ermittelt. Die geringste Diskriminierung im PNA/DNA-Duplex beträg 8 °C, im
DNA/DNA-Duplex 4 °C. Komplementäre PNAs bilden unter Beachtung der Watson-Crick-
Basenpaarung helicale Duplexe, deren Stabilität gegenüber allen anderen Kombinationen von
DNA, RNA und PNA deutlich höher ist.[115, 116] Die entstehende Struktur wird P-Form
genannt und unterscheidet sich von den bisher bekannten Nukleinsäurestrukturen (Tabelle 1,
Abbildung 23). Sie wird auch im Falle von Triplexen beobachtet.

Allgemeiner Teil 1 Einleitung
32
Tabelle 1. Vergleich der A- B- und Z-Form von DNA mit der P-Form von PNA
A-DNA B-DNA Z-DNA P-PNA
Drehsinn der Helix rechts rechts links rechts und links (1:1)
Durchmesser ~ 26 Å ~ 20 Å ~ 18 Å ~ 28 Å
Basenpaare pro Windung 11 10.5 12 18
Anstieg der Helix je Basenpaar 2.6 Å 3.4 Å 3.7 Å 3.2 Å
Neigung der Basen gegenüber der Helix-Achse
20 ° 6 ° 7 ° < 1 %
Twist 31 ° 36 ° 60 °/2 19.8 °
Abbildung 23. Strukturen gelöster PNA-Komplexe. – Die Abbildung wurde aus Referenz[117] entnommen; das Original stammt aus [118].

Allgemeiner Teil 1 Einleitung
33
1.4.2 Das Potential einer PNA-Welt
Alles bisher bekannte Leben auf der Erde basiert auf Zellen, also auf geschlossenen Kompar-
timenten. In den Zellen selbst können sich, z.B. in Form vom Organellen, weitere
Kompartimente befinden, die verschiedene Reaktionsräume formen. Die Kompartimentierung
hat eine Reihe von Auswirkungen auf die Bestandteile eines chemischen Systems: Sie werden
in einem definierten Bereich lokalisiert, ihre Diffusion wird limitiert und sie werden vor
äußeren chemischen Einflüssen geschützt. Die damit verbundene Konzentrationserhöhung im
Inneren der Kompartimente begünstigt Reaktionen wie die Bildung von Biopolymeren und
stellt einen alternativen Weg zur Konzentrierung von Monomeren durch Austrocknung oder
eutektisches Ausfrieren dar. Die Frage nach der Entstehung erster Zellen und Zellvorläufer,
der sogenannten Protozellen, ist ebenfalls Gegenstand der präbiotischen Chemie; dabei muss
die Struktur einer Protozelle und die ihr zu Grunde liegende Chemie nicht zwangsläufig ein
Abbild der heutigen hoch spezialisierten Zellen sein.[119-128] Eine lebende Zelle wird in diesem
Zusammenhang durch die Kooperation von Genen, Metabolismus und Kompartimentier-
ung charakterisiert. Im Vergleich zu den von Oparin formulierten Voraussetzungen für Leben
(Selbstreplikation, Mutation, Metabolismus; vgl. 1.1) werden hier Selbstreplikation und
Mutation im Begriff Gen zusammengefasst und die Anforderungen um das Konzept der
Kompartimentierung erweitert. Ein leistungsfähiges Modell zur Beschreibung minimaler
Zellen, das Chemoton-Modell, wurde 1971 von Ganti vorgestellt und in den folgenden Jahren
weiter ausgebaut.[129-134] In diesem Modell sind ein genetisches, ein metabolisches und ein
membranformendes Subsystem stöchiometrisch gekoppelt.
Das Potential der PNA als genetisches Material in Protozellen wurde während des PACE-
Projekts (Programmable Artificial Cell Evolution) intensiv theoretisch und experimentell
untersucht.[135-138] Während die Gene moderner Zellen von Lipid-Membranen umschlossen
sind, war es Teil dieses Protozellen-Konzeptes, sie in Form von PNA an der Oberfläche eines
Lipid-Aggregats oder in diesem selbst physikalisch zu binden, um eine starke Verbindung
zwischen Geno- und Phänotyp zu garantieren. Die gewünschte Assoziation der PNA zur
Lipid-Hülle sollte durch die Konjugation mit Lipiden[139, 140] oder durch chemische Modifi-
zierung des Rückgrats[141, 142] erreicht werden (Abbildung 24, links und Mitte). Auf diese
Weise sollten PNAs synthetisiert werden, die in einem hydrophoben organischen Medium
löslich sind, wie es im Inneren von Mizellen vorherrscht. Bereits unmodifizierte PNA zeigt
unter Zugabe aprotischer organischer Solventien (DMF, 1,4-Dioxan) eine nahezu unbeein-
trächtigte Duplexstabilität, die auch in der Extrapolation hin zu 100 % organischem Solvenz

Allgemeiner Teil 1 Einleitung
34
nur geringfügig abnimmt.[143] Es war außerdem vorgesehen, das genetische Material PNA
direkt mit dem Metabolismus der Protozelle zu verbinden, um Selektion und damit Evolution
auf Basis der metabolischen Leistung zu ermöglichen. Zu diesem Zweck wurde ein
Ruthenium-Komplex mit einem PNA-Strang verknüpft (Abbildung 24, rechts). Der resul-
tierende Metallkomplex 39 soll als Photo-Sensibilisator fungieren, dem Metabolismus der
Zelle Energie zur Verfügung stellen und Nukleobasen der PNA als Elektronen-Relais nutzen.
NO
OO
NH
NO
OO
NH
NH
O
N
N
NH
O
N
OO
Ru2+
N
N
N
N
B B B
37 38 39
Abbildung 24. Strukturen von Lecine-PNA (links), Octanoly-lysin-PNA (Mitte) und eines PNA-Photo-Sensibilisator-Konjugats (rechts).

Allgemeiner Teil 1 Einleitung
35
1.4.3 Schlüssige präbiotische Synthesewege für PNA-Bausteine
Schon 1988 berichteten Ogura et al., dass Ethylendiamin neben Methylamin das Hauptpro-
dukt der Photolyse von CH4 und NH3 ist.[144] Interessanterweise wird gerade die photolytische
Instabilität dieser beiden Verbindungen oft als Gegenargument einer reduzierenden Uratmos-
phäre angeführt. Miller und Nelson gelang 1996 die Synthese von 2-(Aminoethyl)glycin
(AEG) durch eine Strecker-Synthese mit Ethylendiamin (ED), Formaldehyd und Blausäu-
re.[145] Bei Ausgangskonzentrationen von 10-1 M bis hin zu 10-4 M erreichten sie Ausbeuten
von 11-79 % an AEG, als Nebenprodukte wurden N,N’-Ethylendiamindiessigsäure und
N,N-Ethylendiamindiessigsäure gefunden. Später konnte die Ausgangskonzentration sogar
noch auf 10-5 M erniedrigt werden, bei Ausbeuten von 18 bzw. 33 %.[146] Aus Gemischen von
CH4, NH3, N2 und H2O sind unter elektrischer Entladung bei 25 und 100 °C sowohl AEG als
auch ED zugänglich, während sich bei der Polymerisation von NH4CN ohne und mit Zusatz
von Formaldehyd nur ED nachweisen lässt. Die hohe Löslichkeit von AEG*H2O und seines
Hydrochlorids in Wasser (6.4 bzw. 13.7 mol/l) ermöglicht eine Konzentrierung dieser Verbin-
dung durch trocknende Strände und Lagunen. Adenin- und Guanin-N9-essigsäure sind aus der
Polymerisation von NH4CN in der Gegenwart von Glycin zugänglich, während Cytosin- und
Uracil-N1-essigsäure aus der Umsetzung von Hydantoinsäure und Cyanoacetaldehyd
hergestellt werden können.[146]
Im Falle der PNA-Bausteine scheint auch ein extraterrestrischer Ursprung möglich. So fanden
Meierhenrich et al. bei der künstlichen Herstellung von interstellarem Eis neben 16 Amino-
säuren einige Diaminosäuren,[33] die später auch in einer Probe des Murchinson-Meteoriten
nachgewiesen wurden.[147] Bereits eine Dekade vor diesen Ergebnissen schlug Nielsen
präbiotische PNA-Varianten vor, die auf 2,4-Diaminobutansäure (40) und 2,5-Diamino-
pentansäure (41) basieren (Abbildung 25).[148]
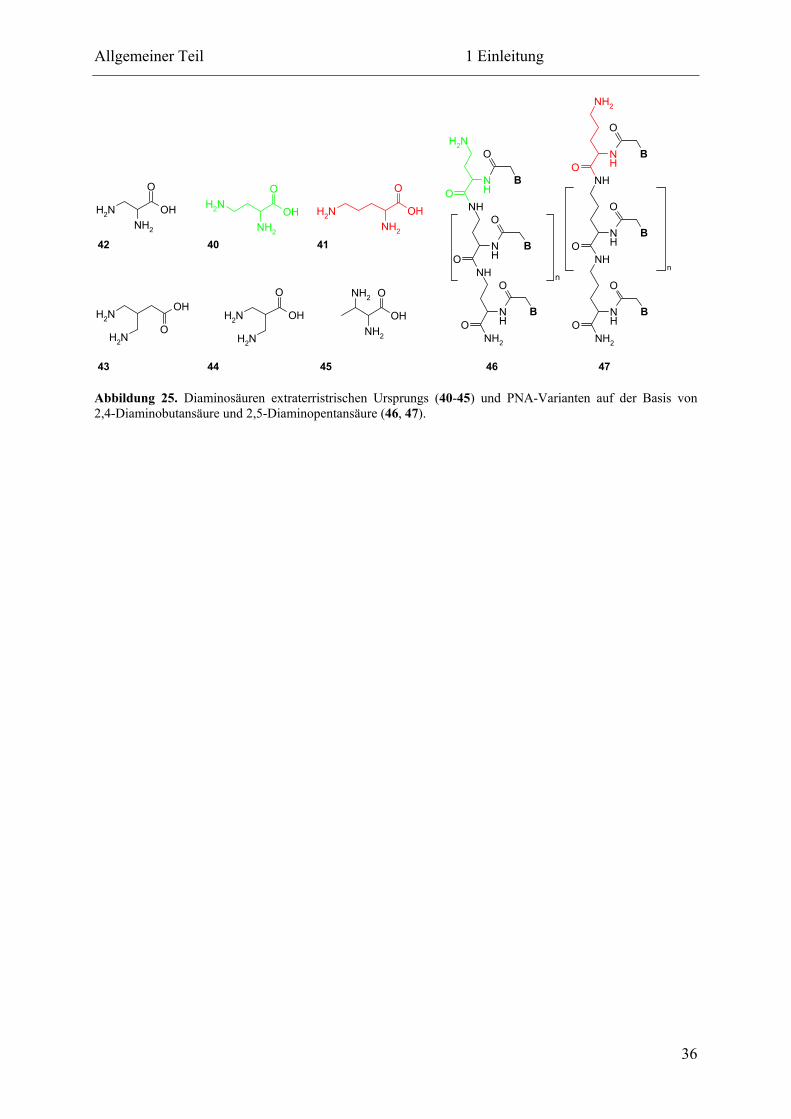
Allgemeiner Teil 1 Einleitung
36
NH2 OH
NH2
O
NH2 OH
NH2
O
NH2
NH2
OH
ONH2
NH2
OH
O
OH
O
NH2
NH2
NH2 OH
O
NH2
NH
NH2
ONH
O
B
NH
ONH
O
B
NH2
ONH
O
B
n
NH2
ONH
O
B
NHO
NH
O
B
NHO
NH
O
B
NH2
n
42 40 41
43 44 45 46 47
Abbildung 25. Diaminosäuren extraterristrischen Ursprungs (40-45) und PNA-Varianten auf der Basis von 2,4-Diaminobutansäure und 2,5-Diaminopentansäure (46, 47).

Allgemeiner Teil 1 Einleitung
37
1.4.4 Chemische Stabilität
Auf die Stabilität der PNA gegenüber biologischen Enzymen wie Proteasen und Peptidasen
ist schon früh hingewiesen worden.[149] In einem präbiotischen Kontext ist zunächst die
Langzeitstabilität der Monomere in wässrigen Lösungen von Bedeutung, damit eine kritische
Konzentration zur Oligomerisierung erreicht werden kann.
Die terminale Amino-Gruppe der PNA besitzt einen pKa-Wert von etwa 10-11 und ist sterisch
deutlich flexibler als die α-Amino-Funktion einer natürlichen Aminosäure, die darüber hinaus
nur einen pKa-Wert von 9-10 aufweist. Diese erhöhte Basizität und Reaktivität führt zu
Nebenreaktionen, die abhängig vom pH-Wert der Lösung sind. Experimentell konnte gezeigt
werden, dass das t-Monomer 48 bei 20 °C im pH-Bereich von 7.0 bis 10.0 über mehrere
Wochen chemisch stabil ist und erst bei pH 11 die in Abbildung 26 gezeigte Umlagerung zu
49 mit einer Halbwertszeit von 34 Tagen eingeht.[150] Der pH-Wert hat Einfluss auf die
Population der beiden Rotamere der tertiären Amid-Gruppe und verschiebt das Gleichgewicht
in Richtung cis-Isomer, von dem aus der Acyl-Transfer stattfinden kann.
NOH
O
N
O
NH2
NH
O
O
N
NH
O
O
NOH
O
NH2
O
NH
OH
O
NH
O
N
NH
OO
trans-48 cis-48 49
Abbildung 26. N-Acyl-Transfer am PNA-Thymin-Monomer 48 ausgehend vom cis-Rotamer.
Die Halbwertszeit und damit die Langzeitstabilität bei pH 7 und 20 °C wird auf 1000 Jahre
geschätzt, was in präbiotischen Dimensionen ein kurzer Zeitraum ist. Weiterhin scheint die
Cyclisierung des PNA-Rückgrats unter dem Einfluss erhöhter Temperatur oder durch
Aktivierung der Carboxylfunktion unvermeidbar (Abbildung 27). So cyclisiert AEG 50 bei
100 °C unter neutralen Bedingungen zum Monoketopiperazin 51,[146] ebenso wie PNA-
Monomere 52 unter den Bedingungen der EDC-Aktivierung zu 53 reagieren[151]. Auch
Monomer-Alkylester 54 cyclisieren unter neutralen oder basischen Bedingungen.[152] Daher
werden bei Ligations-Experimenten stets Dimere oder höhere Oligomere verwendet, was als
Schwachpunkt der PNA-Welt-Hypothese gilt.[153] Bei der präbiotischen Peptidsynthese ist das

Allgemeiner Teil 1 Einleitung
38
Problem der Cyclisierung auf der Stufe der aktivierten Dipeptide 55 bekannt, wobei
Diketopiperazine 56 entstehen.[154]
NH2
N
OO
O
B
NH2
NH
O
X
O
R
RNH
N
O
O
B
NH
NH
O
O R
R
NH2
NH
O
OH
NH
NH
O
NH2
N
OO
OH
B
NH
N
O
O
B
-HX
100 °C EDCa) b)
c) d)
50 51 52 53
54 53 55 56
Abbildung 27. a) – c) Cyclisierung monomerer PNA-Strukturen; d) Cyclisierung aktivierter Dipeptide.
Dessen ungeachtet können PNA-Monomere auch nach der Reaktion zum Monoketopiperazin
einer Oligomerisierung zur Verfügung stehen, wenn sie geeignet aktiviert werden. Die
koreanische Firma Panagene hat ein Verfahren entwickelt, bei dem PNA-Oligomere aus
selbstaktivierten stabilen Monomeren 57 aufgebaut werden.[155] Diese enthalten elektronenzie-
hende Schutzgruppen wie Benzothiazol-2-sulfonyl (Bts), Nitrobenzolsulfonyl oder Thiadia-
zolsulfonylderivate (Abbildung 28). Natürlich können diese Gruppen nicht unbedingt als
präbiotisch angesehen werden, was auch nicht die Intention des Designs war. Jedoch zeigt das
Verfahren einen möglichen Ausweg aus dem Monomer-Problem.

Allgemeiner Teil 1 Einleitung
39
NOH
O
B
O
NH
PG N
O
N O
B
PG
O OS
N S
O OS
NO2
O OS
N SN
Ph
O OS
N SN
O
Ph
N
O
B
O
NH2
N
O
N O
B
PG
N
O
B
O
NH
N
O
B
O
NH
PG
EDC
PG:
+
DIPEA, DMF,40 °C, 2 h
58 57
57 59 60
Abbildung 28. Selbstaktivierte Monomere der Firma Panagene (oben) und ihre Oligomerisierung ohne Zugabe von Kopplungsreagenzien (unten).
Interessanterweise berichteten vor kurzem Luisi und Mitarbeiter, dass Dimere, Trimere und
Tetramere 61a-c in geringer Ausbeute aus Monomer-Ethylestern 62 unter Katalyse des
einfachen Dipeptids Ser-His entstehen können (Abbildung 29).[156]
NH2
N
OO
O
N
NH
O
O
NN
OO
O
N
NH
O
O
H
Hn
v n = 2, 6.5 % v n = 3, 3.6 %v n = 4, 9.1 %
a: b: c:n
Ser-His
62 61
Abbildung 29. Bildung von kurzen PNA-Oligomeren. – Reagenzien und Reaktionsbedingungen: 10 mM PNA Monomer, 1.5 mM Ser-His, 100 mM Natriumborat pH 10, 6 % (v/v) DMF, 35 h, 25 °C.
Auf der Stufe der Oligomere tritt neben dem bereits besprochenen Acyl-Transfer, der zu
einem umgelagerten Produkt gleicher Masse führt, zusätzlich eine Abbaureaktion unter
Transamidierung auf, bei der die letzte monomere Einheit abgespalten wird (Abbildung 30).

Allgemeiner Teil 1 Einleitung
40
NH2
N
OO
NH
N
OO
NH O
NH
R NH
NH
O
NH
N
OO
NH
O
NH
R
O
n-1
B
NH2N
OO
NH
O
NH
R
NH
N
O
O
B B
a)
b)
a)
b)
nB
63 64
53 65
B
+n
B
Abbildung 30. Transamidierung von PNA-Oligomeren. – a) unter Wanderung der N-terminalen Nukleobase; b) unter Abspaltung des N-terminalen Monomers. Generell lassen sich chemische Veränderungen erst ab pH 9 beobachten, typische Halbwertszeiten liegen zwischen 15 und 163 Tagen bei pH 9 und 1.5 Stunden bis 21 Tagen bei pH 12.
Neben der Abhängigkeit vom pH-Wert lässt sich auch ein entscheidender Einfluss der
Sequenz beobachten, wobei vor allem die beiden N-terminalen Basen entscheidend sind.
Ursächlich könnte eine Veränderung der Rotamer-Population sein, da der Abbau vom
cis-Isomer auszugehen scheint (s.o.). Da sich die Abbaureaktionen durch Alkylierung des
N-Terminus zum sekundären Amin deutlich verlangsamen und sich nach Acylierung gar nicht
mehr beobachten lassen, scheint die Stabilität der Oligomere im präbiotischen Kontext
weniger kritisch als die der Monomere.

Allgemeiner Teil 1 Einleitung
41
1.4.5 Katalytische Aktivität
Voraussetzung für katalytisch aktive PNA ist in Analogie zu RNA die Ausbildung von
Sekundär- bzw. Tertiärstrukturen. Diese Bedingung ist zwar erfüllt, da PNA sowohl
Triplexe[157] und Quadruplexe[158, 159] als auch komplexere Strukturen[160] bilden kann, es gibt
aber noch kein Beispiel katalytischer Aktivität, wie sie für ein evolvierendes System auf
PNA-Basis nötig wäre. Nielsen äußerte 1993 die Möglichkeit einer kombinierten PNA/RNA-
Welt, in der PNA als stabiler Träger der genetischen Information fungiert, während RNA
katalytische Funktionen wie die PNA-Replikation übernimmt.[148] Dies scheint nicht abwegig
zu sein, da die Knüpfung von Peptidbindungen in den Ribosomen von der RNA katalysiert
wird. Unter dem Namen „PNAzymes“ stellten Strömberg et al. künstliche RNA-Restriktions-
Enzyme vor.[161, 162] Die katalytische Aktivität kommt hier allein durch chelatisierte Zn(II)-
bzw. Cu(II)-Ionen zustande, während die PNA zum einen als Trägermaterial für den Chelat-
Komplex dient, zum anderen durch Watson-Crick-Basenpaarung an die Zielsequenz bindet.
Diese Funktionen kann auch ein Rückgrat aus 2′-O-Methyl-oligoribonukleotiden über-
nehmen,[163] das allerdings anfälliger für eine Degradierung durch natürliche Nukleasen ist.

Allgemeiner Teil 1 Einleitung
42
1.4.6 PNA und der Ursprung der Homochiralität
Das sogenannte Homopolymer-Problem beschreibt die Schwierigkeiten, aus einer racemi-
schen Lösung von Monomeren homochirale Oligomere zu bilden und diese erfolgreich zu
replizieren. Zum einen ist die Wahrscheinlichkeit unter diesen Bedingungen ein homochirales
n-mer zu erhalten gleich (½)n, wenn die untemplatierte Primerverlängerung nicht enantio-
selektiv ist. Zum anderen führt die enantioselektive Kreuz-Inhibition zu Kettenabbrüchen
während der Replikation.[164, 165]
Die Achiralität der PNA umgeht das erste Teilproblem. Aber die enantioselektive Kreuz-
Inhibition lässt sich durch die bloße Verwendung einer achiralen PNA-Matrize nicht lösen,
wie bei der Oligomerisierung von z.B. 2-MeImG an einem PNA-C10-Templat gezeigt
wurde.[166] Einen möglichen Ausweg stellt der graduelle Übergang von einem achiralen
genetischen Material wie der PNA zu einem chiralen Homopolymer wie der RNA dar. Da das
AEG-Rückgrat von sich aus achiral ist, bildet sich in Lösung ein 1:1 Gleichgewicht zwischen
links- und rechtsgängigen Helices, das erst durch die Einführung chiraler Liganden wie
einfacher α-Aminosäuren oder auch von Didesoxynukleotiden zu einer bevorzugten Chiralität
verschoben werden kann.[115, 116, 167-169] Die Chiralität, die durch den bloßen Ersatz zweier
PNA-Einheiten durch Desoxynukleotide in einem PNA-Templatstrang entsteht, wird durch
den an sich achiralen Strang weitergeleitet, sodass es bei einer templatierten Verlängerung
eines achiralen Primers zur ferngesteuerten Enantioselektion kommt.[168] Diese Übertragung
chiraler Informationen über größere Entfernung durch kovalente und nicht-kovalente
Bindungen führt zu einem möglichen Szenario, in dem kleine homochirale DNA- oder RNA-
Segmente ihre enantioselektive Replikation durch den Einbau in eine PNA-Helix gewährleis-
ten. Evolutive Prozesse könnten im weiteren Verlauf für eine schrittweise Reduzierung des
PNA-Anteils in den Chimären sorgen. In einem von Nielsen 2007 aufgestellten Modell findet
diese Abfolge von Induktion, Replikation und Evolution in einer Population von Protozellen
statt, und führt zu einem homochiralen Genom auf der Basis von RNA.[170] Hierbei ging er
von einer Population aus, in der jede Zelle bedingt durch eine hohe Mutationsrate über ein
einzigartiges Genom verfügt. Wenn nun eine dieser Zellen einen deutlichen evolutiven
Vorteil besitzt, kann sie zum Urahn der RNA-Welt werden.

Allgemeiner Teil 1 Einleitung
43
1.4.7 Speicherung und Weitergabe genetischer Informationen
Mit eingeschränkter Effizienz und unter geeigneten Ligationsbedingungen kann ein
PNA-C10-Strang die Oligomerisierung von PNA-G-Dimeren katalysieren (Abbildung 31).[171]
Da das gleiche Templat auch die Oligomerisierung von aktiviertem Guanidinmonophosphat
und somit die Bildung von RNA katalysiert, scheint ein genetischer Übergang von einer
PNA- zu einer RNA-Welt chemisch möglich.[170, 171] In analogen Experimenten dienten DNA-
Stränge als Templat für die Oligomerisierung von aktivierten RNA-Monomeren und PNA-
Dimeren.[171, 172]
Abbildung 31. Genetischer Übergang zwischen PNA, RNA und DNA am Beispiel von Homo-Sequenzen.

Allgemeiner Teil 1 Einleitung
44
Auch ein Informationstransfer auf der Basis von Heterosequenzen ist möglich (Abbildung
32).[173] DNA-templatierte PNA-Synthesen zeigen im Falle von Polypyrimidin-Templaten
eine gute Spezifität und Effizienz, während purinhaltige Sequenzen besonders im Falle von
Adenin eine deutlich geringere Genauigkeit aufweisen.[172] Letzteres ist auch bei der Synthese
von RNA an RNA- und DNA-Templaten beobachtet worden.[174] In jedem Fall benötigen
effiziente RNA-Synthesen an einem PNA-Templat einen Primer bestehend aus wenigstens
drei Basen, da erst unter diesen Bedingungen eine Helix-Struktur entsteht, die einem
DNA/RNA-Hybrid ähnelt. Ohne diese Präorganisation der Helix kommt es durch 2’-5’-
Verknüpfungen, Cyclisierungen und Pyrophosphatbildungen zum Abbruch der Synthese.
Abbildung 32. Genetischer Übergang zwischen PNA, RNA und DNA am Beispiel von Hetero-Sequenzen. – x/X = A, C, G, T; y/Y = die zu x/X komplementäre Base.
Bei Untersuchungen zu templatgesteuerten PNA-Ligationsreaktionen benutzten Mattes und
Seitz ein heptameres PNA-Templat der Sequenz gcbacgg, wobei bei b = c ein komple-
mentäres Templat und bei b = g ein Templat mit Fehlpaarung erhalten wird.[175, 176] Wie in
Abbildung 33 dargestellt, hängt die Wirkung der Template in hohem Ausmaß von den unter-
schiedlichen Ligationsbedingungen ab. Im Falle der durch EDC und Imidazol vermittelten
Kondensation ist die untemplatierte Reaktion ineffizient, während das komplementäre
Templat die Ausbeute steigert und das fehlpaarende Templat sogar zum besten Ergebnis
führt. Dieses Ergebnis weist auf die Bedeutung von Flexibilität für Ligationsreaktionen hin.
Durch die Substitution einer PNA-Einheit durch ein isosteres Dipeptid gewinnt das System
zum einen zusätzliche Flexibilität, zum anderen wird der unspezifische kooperative Effekt der
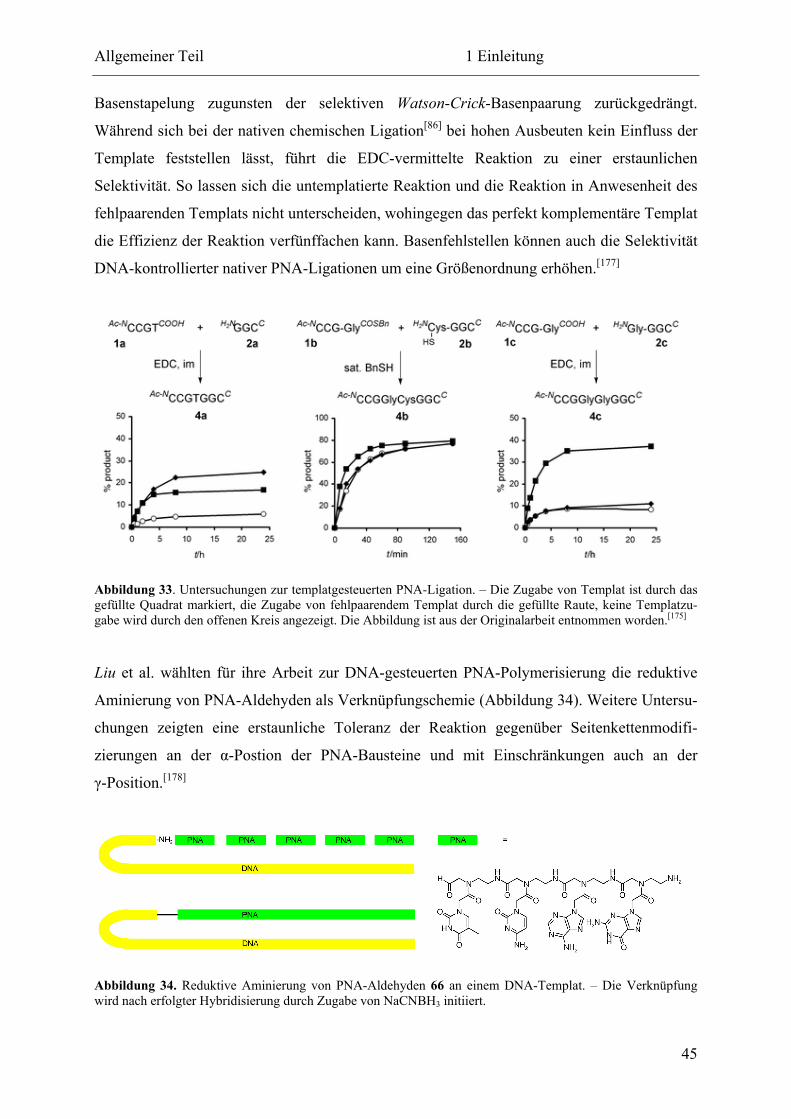
Allgemeiner Teil 1 Einleitung
45
Basenstapelung zugunsten der selektiven Watson-Crick-Basenpaarung zurückgedrängt.
Während sich bei der nativen chemischen Ligation[86] bei hohen Ausbeuten kein Einfluss der
Template feststellen lässt, führt die EDC-vermittelte Reaktion zu einer erstaunlichen
Selektivität. So lassen sich die untemplatierte Reaktion und die Reaktion in Anwesenheit des
fehlpaarenden Templats nicht unterscheiden, wohingegen das perfekt komplementäre Templat
die Effizienz der Reaktion verfünffachen kann. Basenfehlstellen können auch die Selektivität
DNA-kontrollierter nativer PNA-Ligationen um eine Größenordnung erhöhen.[177]
Abbildung 33. Untersuchungen zur templatgesteuerten PNA-Ligation. – Die Zugabe von Templat ist durch das gefüllte Quadrat markiert, die Zugabe von fehlpaarendem Templat durch die gefüllte Raute, keine Templatzu-gabe wird durch den offenen Kreis angezeigt. Die Abbildung ist aus der Originalarbeit entnommen worden.[175]
Liu et al. wählten für ihre Arbeit zur DNA-gesteuerten PNA-Polymerisierung die reduktive
Aminierung von PNA-Aldehyden als Verknüpfungschemie (Abbildung 34). Weitere Untersu-
chungen zeigten eine erstaunliche Toleranz der Reaktion gegenüber Seitenkettenmodifi-
zierungen an der α-Postion der PNA-Bausteine und mit Einschränkungen auch an der
γ-Position.[178]
Abbildung 34. Reduktive Aminierung von PNA-Aldehyden 66 an einem DNA-Templat. – Die Verknüpfung wird nach erfolgter Hybridisierung durch Zugabe von NaCNBH3 initiiert.

Allgemeiner Teil 1 Einleitung
46
Bei den bisher vorgestellten templatgesteuerten PNA-Synthesen fand die Ligation jeweils am
Rückgrat statt. Eine andere Herangehensweise stellt das sequenzspezifische Auffüllen von
Basenlücken dar. Diese Möglichkeit wurde bislang wenig in Betracht gezogen, da die Bildung
der analogen N-glycosidischen Bindungen in Wasser eine ungünstige Gleichgewichtslage
besitzt.[63] Im Jahr 2009 wurden dennoch zwei Arbeiten veröffentlich, bei denen PNA bzw.
PNA-Derivate durch das Auffüllen von Basenlücken templatgesteuert synthetisiert
wurden.[179, 180]
In dem von Heemstra und Liu vorgestellten System werden native dodecamere PNA-
Template und komplementäre Substrate mit ein oder zwei Basenlücken in der Strangmitte
oder am Strangende verwendet.[180] Hinzu kommen Nukleobasen, die an den Positionen N1
(Pyrimidine) bzw. N9 (Purine) mit einem Essigsäure- oder Acetaldehyd-Linker versehen sind
und durch Peptidkopplung beziehungsweise reduktive Aminierung mit dem Substrat
verknüpft werden. Dabei stellt sich heraus, dass die Auffüllreaktion in der Strangmitte
effizienter als am Strangende ist, was sich durch die günstigere sterische Präorganisation und
ein erhöhtes Maß an Basenstapelung erklären lässt. Weiterhin spielen die Basenpaarungen
eine Rolle, da Guanin und Cytosin bereitwilliger eingebaut werden als Adenin und Thymin.
Das Auffüllen der Basenfehlstellen gelingt durch reduktive Aminierung effektiver und
spezifischer als unter Einsatz von Kopplungsreagenzien. Als negativ erwies sich die Tatsache,
dass bei diesem Vorgang eine desoxyPNA-Einheit entsteht, die aufgrund ihrer hohen
Flexibilität zu einer starken Destabilisierung von PNA/DNA-Duplexen und (PNA)2/DNA-
Triplexen führt.[181]
Das von Ghadiri vorgestellte System basiert auf einem Rückgrat 67 aus sich wiederholenden
Dipeptideinheiten.[179] Eine der beiden Aminosäuren ist stets Cytosin, über dessen Thiol-
funktion die Nukleobasenthioester 68 durch Transthioesterbildung reversibel gebunden
werden, während sich durch die Wahl der zweiten Aminosäure Löslichkeitseigenschaften,
elektrostatische Eigenschaften und die Flexibilität des Rückgrats steuern lassen. Nach dem
Mischen eines decameren Poly-Cystein-Rückgrats 67 mit Nukleobasenthioestern 68 stellt sich
innerhalb von ca. 30 Minuten ein stabiles Produkt-Gleichgewicht aus tPNAs 69 ein, das sich
jedoch durch die Zugabe eines Überschusses von Nukleobasenthioesters 68 verschieben lässt
(Abbildung 35, oben). Die gebildeten tPNAs binden sequenzspezifisch an komplementäre
DNA-, RNA- und tPNA-Stränge. Wenn die Bildung der tPNAs 69 in Gegenwart eines DNA-
Templats stattfindet, bestimmt dieses die Zusammensetzung und Sequenz der tPNAs 69.
Verändert man im Laufe der Zeit die äußeren Bedingungen durch Zugabe von DNA-
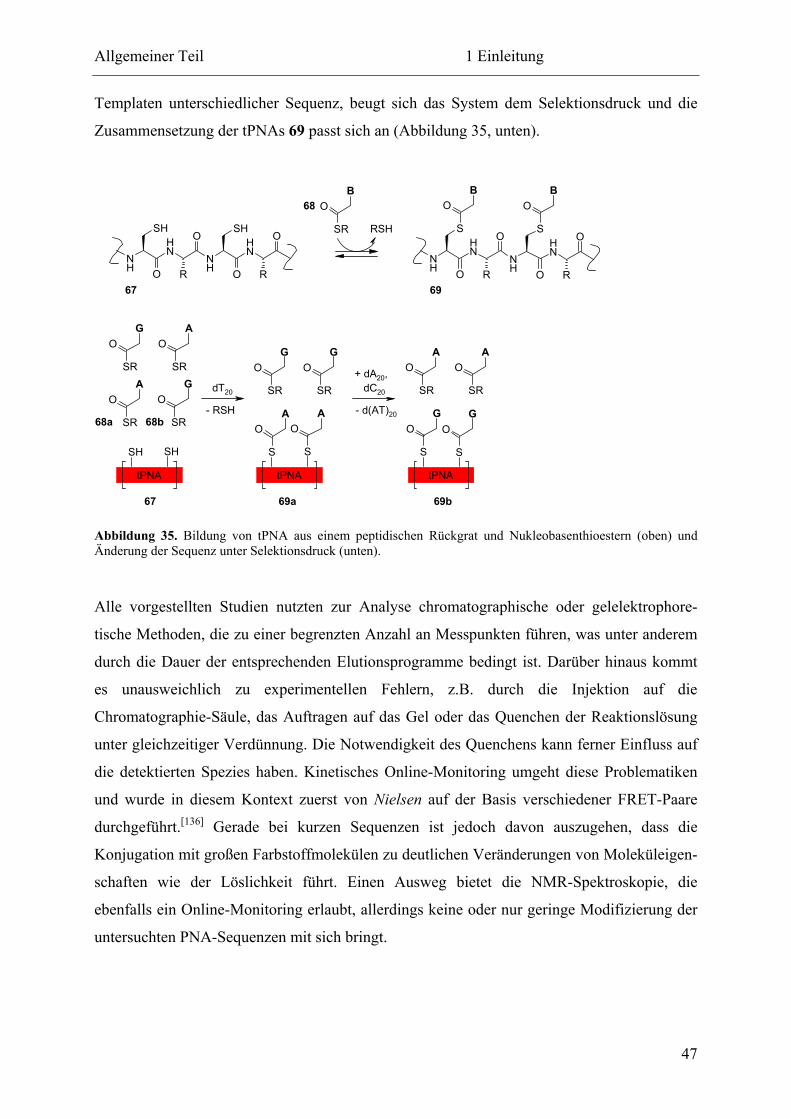
Allgemeiner Teil 1 Einleitung
47
Templaten unterschiedlicher Sequenz, beugt sich das System dem Selektionsdruck und die
Zusammensetzung der tPNAs 69 passt sich an (Abbildung 35, unten).
O
S
A
O
S
A
O
SR
A
O
SR
G
O
SR
A
O
SR
G
O
SR
G
O
SR
G
O
SR
A
O
SR
A
O
SR
B
RSH
NH
NH
NH
NH
O
O
O
OSH
R
SH
R
S
O
B
S
O
B
NH
NH
NH
NH
O
O
O
O
R R
SH
SH
O
S
G
O
S
G
tPNA
dT20
tPNA
+ dA20, dC20
tPNA
67 69
68
67 69a 69b
68a 68b- RSH - d(AT)20
Abbildung 35. Bildung von tPNA aus einem peptidischen Rückgrat und Nukleobasenthioestern (oben) und Änderung der Sequenz unter Selektionsdruck (unten).
Alle vorgestellten Studien nutzten zur Analyse chromatographische oder gelelektrophore-
tische Methoden, die zu einer begrenzten Anzahl an Messpunkten führen, was unter anderem
durch die Dauer der entsprechenden Elutionsprogramme bedingt ist. Darüber hinaus kommt
es unausweichlich zu experimentellen Fehlern, z.B. durch die Injektion auf die
Chromatographie-Säule, das Auftragen auf das Gel oder das Quenchen der Reaktionslösung
unter gleichzeitiger Verdünnung. Die Notwendigkeit des Quenchens kann ferner Einfluss auf
die detektierten Spezies haben. Kinetisches Online-Monitoring umgeht diese Problematiken
und wurde in diesem Kontext zuerst von Nielsen auf der Basis verschiedener FRET-Paare
durchgeführt.[136] Gerade bei kurzen Sequenzen ist jedoch davon auszugehen, dass die
Konjugation mit großen Farbstoffmolekülen zu deutlichen Veränderungen von Moleküleigen-
schaften wie der Löslichkeit führt. Einen Ausweg bietet die NMR-Spektroskopie, die
ebenfalls ein Online-Monitoring erlaubt, allerdings keine oder nur geringe Modifizierung der
untersuchten PNA-Sequenzen mit sich bringt.
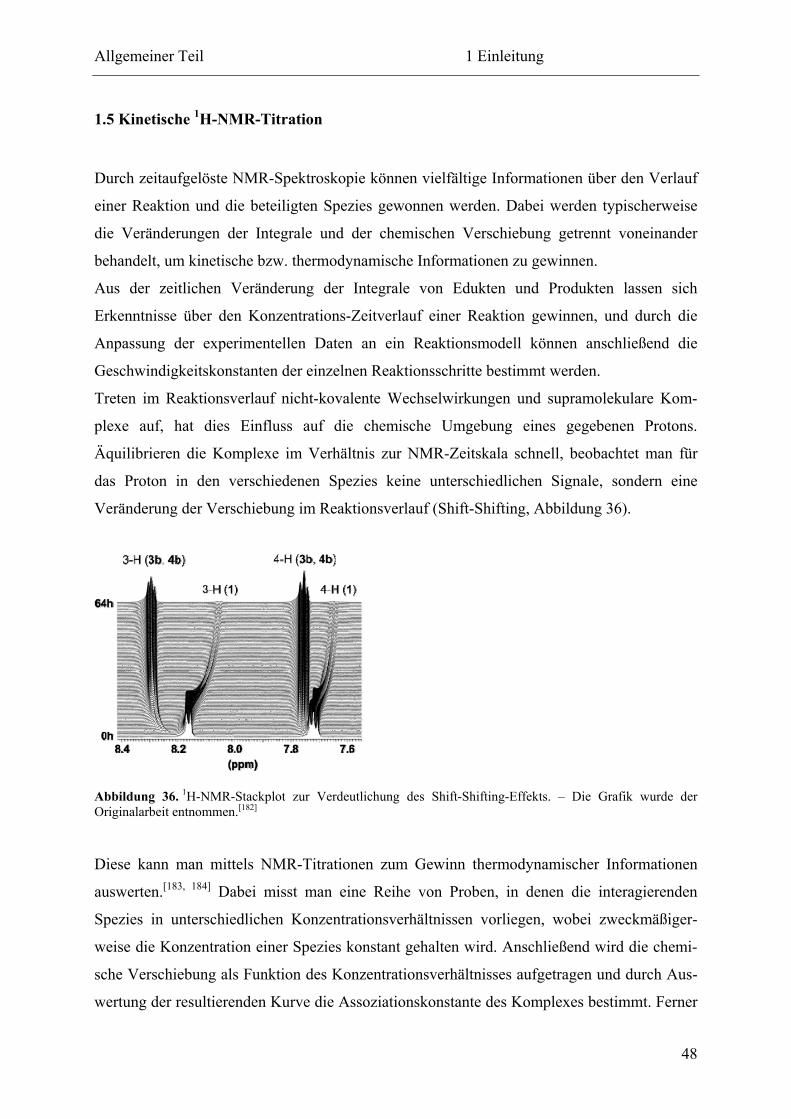
Allgemeiner Teil 1 Einleitung
48
1.5 Kinetische 1H-NMR-Titration
Durch zeitaufgelöste NMR-Spektroskopie können vielfältige Informationen über den Verlauf
einer Reaktion und die beteiligten Spezies gewonnen werden. Dabei werden typischerweise
die Veränderungen der Integrale und der chemischen Verschiebung getrennt voneinander
behandelt, um kinetische bzw. thermodynamische Informationen zu gewinnen.
Aus der zeitlichen Veränderung der Integrale von Edukten und Produkten lassen sich
Erkenntnisse über den Konzentrations-Zeitverlauf einer Reaktion gewinnen, und durch die
Anpassung der experimentellen Daten an ein Reaktionsmodell können anschließend die
Geschwindigkeitskonstanten der einzelnen Reaktionsschritte bestimmt werden.
Treten im Reaktionsverlauf nicht-kovalente Wechselwirkungen und supramolekulare Kom-
plexe auf, hat dies Einfluss auf die chemische Umgebung eines gegebenen Protons.
Äquilibrieren die Komplexe im Verhältnis zur NMR-Zeitskala schnell, beobachtet man für
das Proton in den verschiedenen Spezies keine unterschiedlichen Signale, sondern eine
Veränderung der Verschiebung im Reaktionsverlauf (Shift-Shifting, Abbildung 36).
Abbildung 36. 1H-NMR-Stackplot zur Verdeutlichung des Shift-Shifting-Effekts. – Die Grafik wurde der Originalarbeit entnommen.[182]
Diese kann man mittels NMR-Titrationen zum Gewinn thermodynamischer Informationen
auswerten.[183, 184] Dabei misst man eine Reihe von Proben, in denen die interagierenden
Spezies in unterschiedlichen Konzentrationsverhältnissen vorliegen, wobei zweckmäßiger-
weise die Konzentration einer Spezies konstant gehalten wird. Anschließend wird die chemi-
sche Verschiebung als Funktion des Konzentrationsverhältnisses aufgetragen und durch Aus-
wertung der resultierenden Kurve die Assoziationskonstante des Komplexes bestimmt. Ferner

Allgemeiner Teil 1 Einleitung
49
lassen sich die freie Energie und die Entropie der Komplexbildung durch Titrationen bei
variabler Temperatur bestimmen. Die 2006 von Stahl und von Kiedrowski vorgestellte
kinetische NMR-Titration erlaubt es erstmals, die zeitlichen Veränderungen der Integrale und
der Verschiebungen gleichzeitig durch das Programm SimFit auszuwerten.[182, 185] Anders als
bei der klassischen NMR-Titration ist es dadurch nicht mehr nötig, verschiedene Konzentrati-
onen manuell einzustellen, da dies in situ durch das Reaktionssystem selbst erfolgt. Daher
werden manuelle Fehler reduziert und ein Online-Monitoring des betrachteten Systems wird
möglich. Die Analyse der zeitabhängigen Veränderung der chemischen Verschiebung erfolgt
dabei anhand:
i
n
i i tt
1obs )()( (13)
δobs(t) = beobachtete chemische Verschiebung des betrachteten Protons zu Zeit t
γi = Molenbruch der Spezies i
δi = chemische Verschiebung des Protons in Spezies i
Der Molenbruch einer Spezies i wird aus ihrer aktuellen Konzentration und der Konzentration
aller Spezies n, die zu dem Signal des beobachteten Protons beitragen, berechnet. Die chemi-
schen Verschiebungen der reinen Spezies werden entweder durch Einzelmessungen bestimmt
oder im Rahmen des Fittings angepasst.

Allgemeiner Teil 2 Aufgabenstellung
50
2 Aufgabenstellung
Das Ziel der vorliegenden Arbeit bestand in der Entwicklung und Synthese eines
Selbstreplikations-Systems auf der Basis von PNA, das durch kinetischen NMR-Titration
untersucht werden kann. Anders als in den bisherigen Studien zur templatierten Ligation von
PNA sollte daher von selbstkomplementären Sequenzen ausgegangen werden, deren Synthese
und Reinigung als schwierig bekannt ist.[186] Beim Design des Systems sollten vier Aspekte
besonders berücksichtigt werden:
1. Auswahl einer geeigneten NMR-Sonde
Das von Stahl und von Kiedrowski entwickelte Konzept der kinetischen NMR-Titration
(siehe 1.5) wird unter Verwendung der 1H-NMR-Spektroskopie erfolgreich bei der Analyse
artifizieller Replikatoren eingesetzt.[108, 109, 182] Dabei wird die Änderung der chemischen Ver-
schiebung mindestens eines Protons über die Zeit verfolgt. Zweckmäßigerweise wählt man
Kerne aus, deren Signal keine Überlappungen aufweisen, weil diese die Auswertung der Da-
ten erheblich erschweren. Artifizielle Replikatoren erfüllen in der Regel diese Anforderung,
da sie eine überschaubare Anzahl an Protonen mit hinreichend unterschiedlicher magnetischer
Umgebung aufweisen. Die aus monomeren Einheiten aufgebauten Oligonukleotide und deren
Analoga besitzen dagegen eine große Anzahl an 1H-Kernen mit sehr ähnlicher magnetischer
Umgebung, sodass die entsprechenden Spektren durch komplexe Kopplungsmuster und
Signalüberlappungen geprägt sind. Es war demgemäß das Ziel, das Konzept der kinetischen 1H-NMR-Titration auf einen anderen NMR-aktiven Kern zu übertragen und ggf. die
notwendigen Modifizierungen an der PNA durchzuführen.
2. Wasserlöslichkeit
Die elektrische Neutralität des PNA-Rückgrats ist ursächlich für die besondere Stabilität von
Duplexen und Triplixen unter der Beteiligung von PNA, führt aber auch zu einer Reihe von
Problemen. Hierunter fallen eine reduzierte Wasserlöslichkeit,[187] eine ausgeprägte Tendenz
zur Aggregation und Selbstorganisation[116] und die limitierte Aufnahme in Zellen[188]. Um
dem entgegenzuwirken, werden typischerweise die Strangenden durch die Einführung termi-
naler, positiv geladener Gruppen modifiziert, beispielsweise durch L-Lysin[110] oder Sper-
min[189]. Alternativ wird das Rückgrat selbst durch geladene Gruppen modifiziert.[181, 190, 191]

Allgemeiner Teil 2 Aufgabenstellung
51
Darüber hinaus wurden PNA/DNA-Chimären,[192, 193], PNA-Peptid-Konjugate[139, 140] und
PNA-Konjugate mit hochmolekularem Polyethylenglykol[194] oder mit Polyethylenimin[195]
entwickelt. Diese Methoden gehen jedoch mit der Einführung zusätzlicher reaktiver Gruppen
und/oder Chiralitätszentren einher.[116] Die Synthese von Fmoc-geschützten Monomeren, die
an ihren sekundären Aminen anstelle von Nukleobasen neutrale oder positiv geladene
Löslichkeitsvermittler als Seitenketten tragen, konnte dies vermeiden.[196]
Das aufzubauende Replikations-System sollte so wenig wie möglich von der originalen PNA-
Struktur abweichen und keinesfalls Chiralitätszentren besitzen. Besondere Aufmerksamkeit
erfordert hierbei, dass gerade selbstkomplementäre Sequenzen für ihre Tendenz zur
Aggregation bekannt sind.[186]
3. Effektive Synthese
Im Vergleich zur Analytik durch HPLC, Gel-Elektrophorese oder FRET benötigen NMR-
Experimente größere Mengen der zu analysierenden Substanzen, welche im Rahmen dieser
Arbeit hergestellt werden sollten. Dabei sollte von einfachen Vorstufen ausgegangen werden,
um eine kostengünstige Synthese zu gewährleisten.
4. Vergleichbarkeit mit bekannten Systemen
Aus thermodynamischen Überlegungen basieren die meisten im Arbeitskreis von Kiedrowski
veröffentlichten Arbeiten zur (Selbst-)Replikation von Nukleinsäuren auf hexameren
Templaten.[72, 75, 76, 78-80] Dies sollte in dieser Arbeit beibehalten werden, auch um Systeme auf
Basis von DNA und PNA vergleichen zu können. Langfristig sollte es dieses Vorgehen auch
ermöglichen, komplexere Systeme mit genetischen Übergängen zwischen PNA und DNA zu
untersuchen.
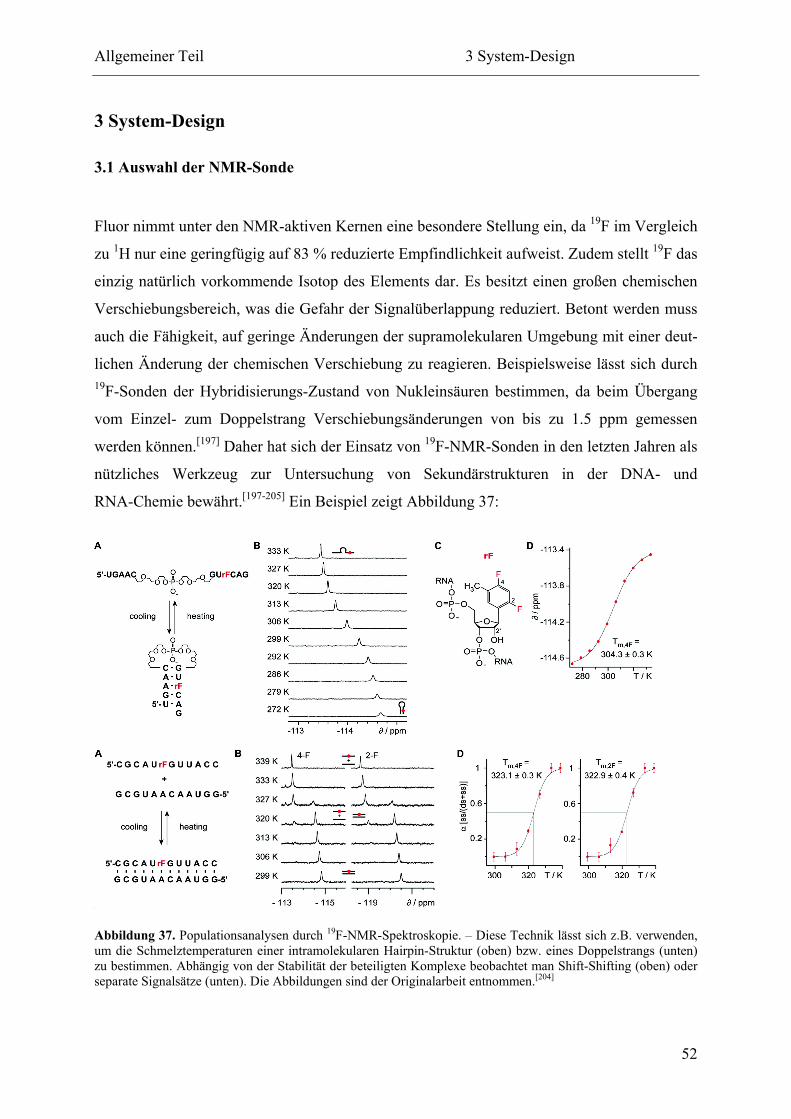
Allgemeiner Teil 3 System-Design
52
3 System-Design
3.1 Auswahl der NMR-Sonde
Fluor nimmt unter den NMR-aktiven Kernen eine besondere Stellung ein, da 19F im Vergleich
zu 1H nur eine geringfügig auf 83 % reduzierte Empfindlichkeit aufweist. Zudem stellt 19F das
einzig natürlich vorkommende Isotop des Elements dar. Es besitzt einen großen chemischen
Verschiebungsbereich, was die Gefahr der Signalüberlappung reduziert. Betont werden muss
auch die Fähigkeit, auf geringe Änderungen der supramolekularen Umgebung mit einer deut-
lichen Änderung der chemischen Verschiebung zu reagieren. Beispielsweise lässt sich durch 19F-Sonden der Hybridisierungs-Zustand von Nukleinsäuren bestimmen, da beim Übergang
vom Einzel- zum Doppelstrang Verschiebungsänderungen von bis zu 1.5 ppm gemessen
werden können.[197] Daher hat sich der Einsatz von 19F-NMR-Sonden in den letzten Jahren als
nützliches Werkzeug zur Untersuchung von Sekundärstrukturen in der DNA- und
RNA-Chemie bewährt.[197-205] Ein Beispiel zeigt Abbildung 37:
Abbildung 37. Populationsanalysen durch 19F-NMR-Spektroskopie. – Diese Technik lässt sich z.B. verwenden, um die Schmelztemperaturen einer intramolekularen Hairpin-Struktur (oben) bzw. eines Doppelstrangs (unten) zu bestimmen. Abhängig von der Stabilität der beteiligten Komplexe beobachtet man Shift-Shifting (oben) oder separate Signalsätze (unten). Die Abbildungen sind der Originalarbeit entnommen.[204]

Allgemeiner Teil 3 System-Design
53
Eine Anwendung im Bereich der PNA-Forschung gibt es noch nicht. Darüber hinaus sind
bislang nur fünf Arbeiten veröffentlicht worden, die sich mit Fluor-modifizierter PNA
befassen:
1. eine mehrstufige Synthese von (2,4-Difluor-5-methylphenyl)essigsäure 70 und dem
entsprechenden Boc-geschützten Monomer[206]
2. eine Studie zum Einfluss von Fluoraromaten auf die Hybridisierungseigenschaften[207]
3. zwei Arbeiten zur Synthese und Hybridisierung fluorierter olefinischer PNA[208, 209]
4. eine Studie zur 18F-Markierung.[210]
Betrachtet man die Studien zur DNA- und RNA-Chemie, ist ein deutlicher Trend zur Fluor-
Markierung durch modifizierte Basen festzustellen, da die Sonde in direkte räumliche Nähe
zur supramolekularen Erkennungsstelle gebracht wird. Für diese Arbeit wurde 2,4-Difluor-
toluol (F) (71) ausgewählt, weil dieses Molekül ein fast perfektes Isoster der natürlichen Base
Thymin (72) ist (Abbildung 38).[211, 212]
NH
NH
O
O F
F
72 71
Abbildung 38. Vergleich von Thymin (72) und 2,4-Difluortoluol (F) (71). – Die Carbonylfunktionen der natürlichen Base sind im Isoster durch CF-Gruppen ersetzt worden, die NH-Funktionalitäten durch CH-Gruppen.
Die entsprechende Essigsäure 70 war kommerziell nicht erhältlich und die einzig bislang
beschriebene Synthese erfordert hochtoxische Reagenzien und einen erheblichen Arbeitsauf-
wand über sechs Synthesestufen.[206] Daher bestand das erste Ziel der Arbeit in der Planung
und Durchführung einer kurzen und effektiven Synthese für dieses Molekül.

Allgemeiner Teil 3 System-Design
54
3.2 Wasserlöslichkeit
N-Methyl- und N-Ethylaminogruppen sind Bestandteile der originalen PNA-Struktur. Die aus
ihnen abgeleiteten N,N-Dimethyl- und N,N-Diethylaminogruppen liegen im physiologischen
pH-Bereich überwiegend protoniert vor und sind somit ideale Löslichkeitsvermittler. Voraus-
setzung ist allerdings, dass bei ihrer Implementierung keine Stereozentren entstehen, die eine
bevorzugte Chiralität induzieren und so das 1:1-Gleichgewicht zwischen links- und rechtsgän-
gigen Helices stören (vgl. 1.4.6).[115, 116, 167-169]
N
O
NH
O
NH2
B
N
O
NH
O
NH
B
N
OH
O
NH
N
O
NH2
O
B
N
O
N
O
B
NH
O
N O
PEG
73 76 75
74 77 78
Abbildung 39. Modifizierung der Termini. – Das primäre Amid 73 und das freie Amin 74 (jeweils orange) sollen durch vergleichbare N,N-Dimethylamino-Derivate 76 und 77 (jeweils grün) ersetzt werden. Hierzu werden ein spezieller Linker 75 und ein modifiziertes Rückgrat 78 benötigt.
Im Rahmen einer Festphasensynthese (siehe 3.3) werden PNAs wie Peptide ausgehend vom
C-Terminus synthetisiert; hierbei entsteht typischerweise C-terminal ein primäres Amid 73
und N-terminal ein freies Amin 74. Eine Modifizierung des C-Terminus kann während der
Abspaltung des Oligomers vom Harz erfolgen. Hierzu schien der HMBA-Linker 75 sehr gut
geeignet, da sich die Esterfunktion dieses Linkers durch eine Vielzahl von Nukleophilen spal-
ten lässt. Bei der Umsetzung mit Alkali entsteht die freie Säure, mit Alkohol der entsprechen-
de Ester, mit Hydrazin das Hydrazon, mit Ammoniak das primäre und mit primären Aminen
das jeweilige sekundäre Amid.[213, 214] Für die gewünschte Modifizierung bot sich daher die
Reaktion mit N,N-Dimethylaminoethylamin an (Abbildung 39, oben). Aufgrund seiner guten
Quelleigenschaften in polaren Lösungsmitteln (DMF, NMP) wurde Tentagel als
Trägermaterial gewählt. Der N-Terminus lässt sich durch den Einsatz eines modifizierten
Monomers variieren. Im konkreten Fall bot sich demzufolge die Synthese des Rückgrats 78
an, dessen terminale Aminofunktion dimethyliert ist (Abbildung 39, unten).

Allgemeiner Teil 3 System-Design
55
3.3 Effektive Synthese
Typischerweise wird PNA an der festen Phase ausgehend von Fmoc/Bhoc-[39], Boc/Z-[40],
Fmoc/Mmt- oder Fmoc/Z-[41] geschützten Monomeren dargestellt. Dies kann sowohl manuell
als auch maschinell am Peptid- und, mit Einschränkung, am DNA-Synthesizer unter Verwen-
dung modifizierter Protokolle für die Peptidsynthese geschehen. Bei der Festphasensynthese
von PNA bietet es sich prinzipiell an, die Sequenz C-terminal mit einer Aminosäure zu begin-
nen, um der Aggregation des wachsenden Oligomers an der Festphase entgegenzuwirken.
Hier wurde Glycin wegen seiner Achiralität gewählt.
Der Lehrstuhl besitzt einen Peptidsynthesizer sowie die Erfahrung mit maschineller Peptid-
synthese, sodass zunächst dieser Weg eingeschlagen wurde. Die Wahl fiel auf die
Fmoc/Bhoc-Schutzgruppenstrategie, da diese die Verwendung starker Säuren sowohl
während der Synthese als auch bei der anschließenden Abspaltung von der Festphase umgeht.
Außerdem erlaubt sie auf einfache Weise die Synthese von Peptidkonjugaten; dies war inner-
halb der Arbeitsgruppe ebenfalls angedacht. Fmoc/Bhoc-Monomere sind kommerziell erhält-
lich, aber ihre Anschaffung war im Rahmen dieses Projekts nicht finanzierbar. Des Weiteren
sollte die Synthese der Monomere zu einem grundlegenden synthetischen Verständnis der
PNA-Synthese führen, um gezielt Modifizierungen einführen zu können.
3.4 Vergleichbarkeit mit bekannten Systemen
Bereits 1997 wurde die Kristallstruktur eines PNA-Duplex gelöst, dem eine hexamere
selbstkomplementäre Sequenz (79a, NcgtacgC) zu Grunde lag.[115] Auf dieser Struktur basierte
das System-Design (Abbildung 40). Wie bereits erwähnt, wurde zunächst die natürliche
Nukleobase Thymin (72) gegen 2,4-Difluortoluol (71) ausgetauscht und die Strangenden mit
nicht-reaktiven Löslichkeitsvermittlern sowie der C-Terminus zusätzlich mit Glycin
modifiziert. Außerdem wurde die Sequenz selbst leicht variiert, um das Isoster in der
mittleren Position des N-terminalen Trimers zu positionieren und den Einfluss auf die
Ligation zu minimieren, während die Fluorsonde im gleichen Abstand von der
supramolekularen Erkennungsseite verbleibt (79b, N’cfgcagGlyC’).

Allgemeiner Teil 3 System-Design
56
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
NH ON N
O
NH2
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
N
NH
NHO
N
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
ON N
O
NH2
NH
NHO
N
NH2
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
N
OH
79b
80
81
Abbildung 40. Kristallstruktur eines PNA-Duplex (oben links),[115] Strukturformel des Einzelstrangs (oben rechts) und erstes Design eines Replikations-Systems auf der Basis von PNA (unten). – Grün: nicht-reaktive Löslichkeitsvermittler; gelb: 19F-NMR-Sonden; blau: Glycin-Einheit; rot: Ligationsstellen und daraus resul-tierende zentrale Amidbindung.
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
O
ON
NH O
N
NH
O
O
N
NH
NO
O
O
O
N
N N
O
NH2
NH2
NH2
N
N
N
NH
O
O
N
N
N
NH2
N
N
NH
O
NH2
N
O
NH2
79a

Allgemeiner Teil 4 Synthesen und Spektroskopie
57
4 Synthesen und Spektroskopie
4.1 Synthesestrategie I: Festphasensynthese
4.1.1 Retrosynthese
Die retrosynthetische Analyse von Fmoc/Bhoc-Monomeren ist in Abbildung 41 am Beispiel
des Cytosin-Monomers 82 dargestellt. Das tertiäre Amid stellt hierbei die strategische
Bindung dar, die durch die Reaktion eines sekundären Amins 83 mit einer Carbonsäure 84
unter den Bedingungen einer Peptidkopplung aufgebaut werden kann. Daher gilt es generell,
die gewünschten Nukleobasen bzw. deren Isostere mit einem Essigsäure-Linker zu versehen
und, sofern vorhanden, die reaktionsfreudigen exocyclischen Aminofunktionen orthogonal
zum Rückgrat zu schützen.
N
O
O NH
OH
OO
N
N
O
NH O
O
NH
O NH
O
OO
O
N
N
O
NH O
O
OH
NH
NH2 OH
O
NH
N
O
NH2
+
82 83
85
84
86
Abbildung 41. Retrosynthetische Analyse des Fmoc/Bhoc-Monomers 82.

Allgemeiner Teil 4 Synthesen und Spektroskopie
58
4.1.2 Fluorlabel
Eine mögliche Syntheseroute für F-Essigsäure 70 ist aus der Literatur bekannt.[206] Die
Autoren wählten einen arbeitsintensiven Weg über sechs Stufen und verwendeten eine Reihe
hochtoxischer Stoffe (Abbildung 42).
F
F
F
F
Br
F
F
CN
F
F
COOH
F
F
O
N2
F
F
O
O
F
F
OH
O
50 % 100 % 98 % 60 %
58 %
71 87 88 89 90
91 70
nichtangegeben
a b c d e
f
Abbildung 42. Synthese von F-Essigsäure 70 nach Takeuchi et al.[206] – Reagenzien und Reaktionsbedingungen: (a) Br2, Fe; (b) CuCN, DMF, 60 °C, 3 h; (c) H2SO4, H2O, Rückfluss, 1 h; (d) (1) ClCO2Et, TEA, THF, -15 °C, 15 min; (2) CH2N2, Et2O, 0 °C → RT, 3 h; (e) PhCOOAg, MeOH, -25 °C → RT, 3 h; (f) 1 M aq. LiOH, THF.
Daher bot sich die Suche nach einem überschaubareren Weg an, und dies möglichst mit
weniger toxischen Reagenzien. Ausgehend von dem gleichen Startmaterial 71 wurde eine
zweistufige Synthese entwickelt, die aus der Reaktionsfolge Brommethylierung und
Grignard-Reaktion bestand. Zunächst lieferte die Umsetzung von 2,4-Difluortoluol (71) mit
para-Formaldehyd in 33%iger essigsaurer Bromwasserstoffsäure nur geringe Mengen der
gewünschten Substanz 92. Erst durch die Zugabe der Lewis-Säure Zinkbromid wurde die
brommethylierte Verbindung 92 in guter Ausbeute erhalten. Die Notwendigkeit der
Modifizierung der ursprünglich entwickelten Vorschrift zur Brommethylierung von van der
Made und van der Made[215] dürfte dem deaktivierenden Einfluss der Fluorsubstituenten
geschuldet sein und stellt die Analogie zur Chlormethylierung nach Blanc stärker heraus.
Durch die sorgfältige Analyse der Kopplungsmuster des 1H-NMR- und besonders des 13C-
NMR-Spektrums (Abbildung 43 und Abbildung 44) konnte die Substitution an der
gewünschten Position bestätigt werden.

Allgemeiner Teil 4 Synthesen und Spektroskopie
59
C
F
C
F
C
F
C
F
1J(C,F) = 245 Hz 2J(C,F) = 21 Hz 3J(C,F) = 8 Hz 4J(C,F) = 3 Hz
Abbildung 43. Charakteristische Werte für C-F-Kopplungskonstanten.[216]
Abbildung 44. 13C-NMR-Spektrum (50 MHz, CDCl3) von Fluor-Aromat 92. – Zuordnung der Signale: δ = 161.28 (dd, 1J(C,F) = 250, 3J(C,F) = 11.9, C(4)); 159.01 (dd, 1J(C,F) = 250, 3J(C,F) = 12.3, C(2)); 133.21 (dd, 3J(C,F) = 6.90, 3J(C,F) = 4.20, C(6)); 120.91 (dd, 2J(C,F) = 29.1, 3J(C,F) = 4.20, C(5)); 121.23 (dd, 2J(C,F) = 26.1, 4J(C,F) = 4.20, C(1)); 103.89 (dd, 2J(C,F) = 24.9, 2J(C,F) = 26.5, C(3)); 25.39 (d, 3J(C,F) = 3.8, CH2); 14.01 (d, 3J(C,F) = 3.10, CH3).
Die Überführung in das entsprechende Grignard-Reagenz und die folgende Umsetzung mit
trockenem CO2 erfolgte nach der Standard-Vorschrift aus dem Lehrbuch „Organikum“.[217]
Die Zielverbindung 70 ließ sich in einer Ausbeute von 38 % über zwei Stufen darstellen
(Abbildung 45).

Allgemeiner Teil 4 Synthesen und Spektroskopie
60
F
F
F
F
Br
F
F
OH
O
NH
NH
O
O
71 92 70 72
a b
66 % 57 %
Abbildung 45. Synthese des hydrophoben Isosters 70 des Thymins (72) – Reagenzien und Reaktionsbedin-gungen: (a) (CH2O)n, ZnBr2, 33 % HBr in AcOH, 120 °C, 4 h; (b) (1) Mg, I2, Et2O, Rückfluss, 2 h; (2) CO2, 0 °C, 1 h; (3) H+, H2O.
Anschließend wurde versucht, diese Reaktions-Sequenz auch auf Derivate des 2,4-Difluor-
toluols (71) anzuwenden. Tatsächlich ließ sich 1,3-Difluorbenzol (93), ein Isoster des Uracils
(94), zunächst in die brommethylierete Verbindung 95 und dann in die entsprechende
Essigsäure 96 überführen (Abbildung 46).
F
F
F
F
Br
F
F
OH
O
NH
NH
O
O
93 95 96 94
a b
40 % 56 %
Abbildung 46. Synthese des hydrophoben Isosters 96 des Uracils (94). – Reagenzien und Reaktionsbedin-gungen: (a) (CH2O)n, ZnBr2, 33 % HBr in AcOH, 120 °C, 4 h; (b) (1) Mg, I2, Et2O, Rückfluss, 2 h; (2) CO2, 0 °C, 1 h; (3) H+, H2O.
Die Umsetzung von Pentafluor-benzol (97) zeigte hingegen unter den Bedingungen der
Brommethylierung keine Reaktion zu 98, was durch den hohen Grad der Deaktivierung
verständlich ist. Das Substitutionsmuster des 1-Fluor-3-methylbenzol (99) bedingte ein
Gemisch von Regio-Isomeren 100, das sich nicht auftrennen ließ (Abbildung 47, links).
Weitere interessante Fluor-Label sind kommerziell bereits als Essigsäuren erhältlich
(Abbildung 47, rechts). So können die beiden Verbindungen 101 und 102 als Isostere des
Uracils (94) angesehen werden. Der Ersatz des Carbonylsauerstoffs durch CF3-Gruppen ist
zwar sterisch nicht annähernd so gut wie der durch Fluor, bietet jedoch ein intensives Signal
in einem anderen Verschiebungsbereich, das außerdem ein abweichend schwächeres Shift-
Shifting erwarten lässt.

Allgemeiner Teil 4 Synthesen und Spektroskopie
61
F
F
FF
F
F
F
Br
FF
F
F F
Br
FFF
F
OH
O
F
OH
O F
F
F
97 98 101 102
99 100
a
a
Abbildung 47. Grenzen der selektiven Brommethylierung (links) und kommerziell erhältliche Isostere mit Trifluormethylgruppen (rechts). – Reagenzien und Reaktionsbedingungen: (a) (CH2O)n, ZnBr2, 33 % HBr in AcOH, 120 °C, 4 h; (b) (1) Mg, I2, Et2O, Rückfluss, 2 h; (2) CO2, 0 °C, 1 h; (3) H+, H2O.

Allgemeiner Teil 4 Synthesen und Spektroskopie
62
4.1.3 Geschützte Nukleobasen-essigsäuren
Neben dem Backbone und den Fluor-Labeln wurden geschützte Nukleobasen in Form ihrer
Essigsäuren benötigt. Ausgehend von den freien Basen bietet sich zum einen die Möglichkeit,
die exocyclischen Aminofunktionen zu schützen, um anschließend den Essigsäure-Linker ein-
zuführen. Zum anderen kann erst der Linker eingeführt werden, um anschließend die Amino-
funktionen zu schützen. Im Rahmen dieser Arbeit wurde der zweite Ansatz gewählt, wobei
auf Vorschriften aus der Literatur zurückgegriffen werden konnte (Abbildung 48).[218, 219]
Adenin (103) und Cytosin (86) wurden jeweils mit starken Basen in DMF umgesetzt und
anschließend an N9 bzw. N1 durch Bromessigsäure-benzylester alkyliert. Die beiden
Nukleobasen-benzylester 104 und 105 entstanden mit guten Ausbeuten und wurden im
folgenden Reaktionsschritt durch das Kopplungsreagenz N,N-Carbonyl-diimidazol (CDI) in
die Bhoc-geschützten Derivate 106 und 107 überführt. Bei dieser Reaktion wird CDI zunächst
durch das jeweilige Amin am zentralen Carbonyl-Kohlenstoffatom angegriffen, was zu einem
tetraedischen Übergangszustand führt. Aus diesem wird Imidazol eliminiert und es entsteht
ein N-Carbonyl-imidazol-Derivat, welches in einer weiteren Additions-Eliminierungs-
Reaktion mit Benzhydrol zur Bhoc-geschützten Verbindung 106 bzw. 107 reagiert. Die ab-
schließende basische Verseifung der Benzylester ergab die gewünschten Zielverbindungen
108 und 84 mit Ausbeuten von 38 bzw. 54 % über je drei Stufen.
Bei Thymin (72) entfällt die Schützung einer exocyclischen Funktion, sodass nach der
Alkylierung mit Bromessigsäure-methylester direkt verseift werden konnte. Dabei entstand
Thymin-1-essigsäure (109) mit einer Ausbeute von 62 %.
Die Synthese des Guanin-Derivats 110 erfolgte nicht ausgehend vom freien Guanin, da
Guanin und Guanin-Derivate in der Keto-Form bevorzugt in der Position N7 alkyliert
werden.[220, 221] Stattdessen wurde von kommerziell erhältlichem 2-Amino-6-chlor-purin (111)
ausgegangen, das als Guanin-Derivat in der Enol-Form verstanden werden kann und
bevorzugt in der Position N9 alkyliert wird. Die Alkylierung mit Bromessigsäure-benzylester
verlief unter Verwendung von Kaliumcarbonat in DMF mit einer Ausbeute von 71 % nach
Umkristallisation. Zur Einführung der Bhoc-Schutzgruppe wurde 112 zunächst mit dem
Phosgenspender Triphosgen in trockenem THF umgesetzt, anschließend wurde der dabei ent-
standene Chlorwasserstoff mit Hüning-Base neutralisiert und dann Benzhydrol zugegeben.
Nach Rühren, wässriger Aufarbeitung und Umkristallisation aus Methanol konnte der Bhoc-
geschützte Guanin-essigsäure-ester 113 mit einer Ausbeute von 67 % isoliert werden.
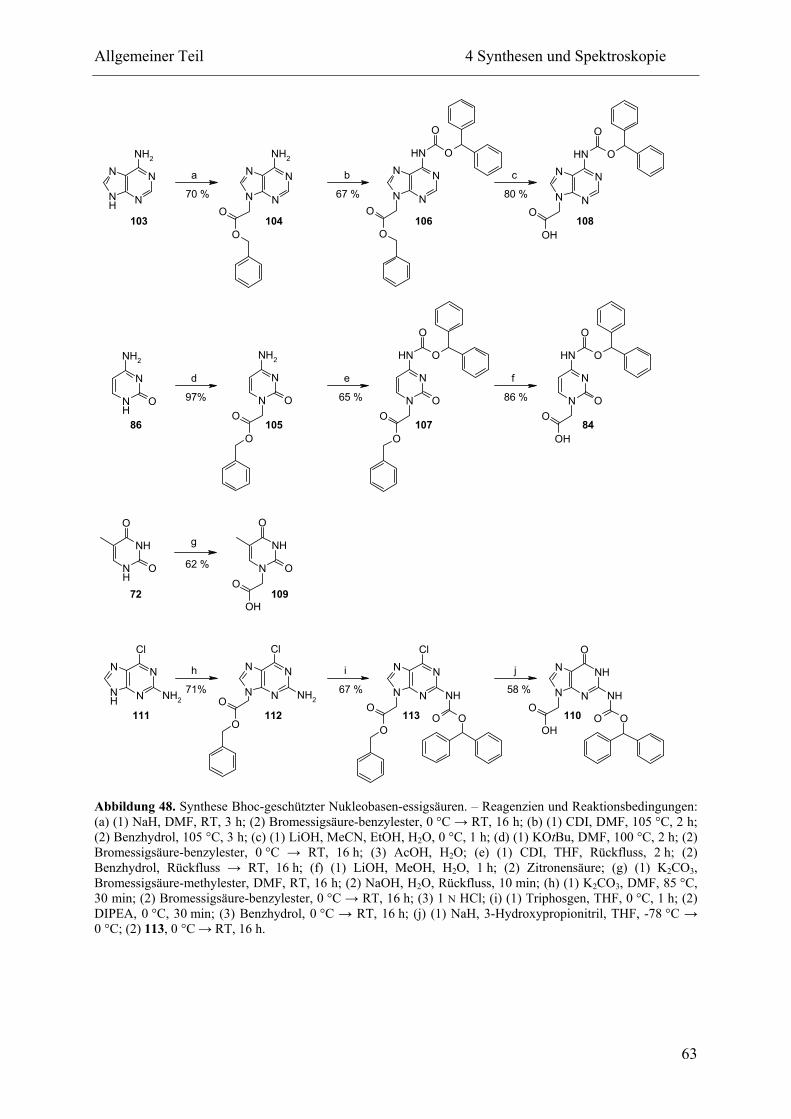
Allgemeiner Teil 4 Synthesen und Spektroskopie
63
N N
N N
NH2
O
O
NH N
N N
NH2
N N
N N
NH
O
O
O
ON N
N N
NH
O
O
OH
O
NH
N
O
NH2
N
N
O
NH2
O
O
N
N
O
NH
OH
O
O
O
N
N
O
NH
O
O
O
O
NH
NH
O
O
N
NH
O
O
OH
O
NH N
N N
Cl
NH2
O
ON N
N N
Cl
NH2
O
O
N N
N N
Cl
NH
OOOH
O
N N
N NH
O
NH
OO
103 104 106 108
a b c
70 % 67 % 80 %
d e f
97% 65 % 86 %
g
86 105 107 84
72 109
h i j
71% 67 % 58 %
111 112 113 110
62 %
Abbildung 48. Synthese Bhoc-geschützter Nukleobasen-essigsäuren. – Reagenzien und Reaktionsbedingungen: (a) (1) NaH, DMF, RT, 3 h; (2) Bromessigsäure-benzylester, 0 °C → RT, 16 h; (b) (1) CDI, DMF, 105 °C, 2 h; (2) Benzhydrol, 105 °C, 3 h; (c) (1) LiOH, MeCN, EtOH, H2O, 0 °C, 1 h; (d) (1) KOtBu, DMF, 100 °C, 2 h; (2) Bromessigsäure-benzylester, 0 °C → RT, 16 h; (3) AcOH, H2O; (e) (1) CDI, THF, Rückfluss, 2 h; (2) Benzhydrol, Rückfluss → RT, 16 h; (f) (1) LiOH, MeOH, H2O, 1 h; (2) Zitronensäure; (g) (1) K2CO3, Bromessigsäure-methylester, DMF, RT, 16 h; (2) NaOH, H2O, Rückfluss, 10 min; (h) (1) K2CO3, DMF, 85 °C, 30 min; (2) Bromessigsäure-benzylester, 0 °C → RT, 16 h; (3) 1 N HCl; (i) (1) Triphosgen, THF, 0 °C, 1 h; (2) DIPEA, 0 °C, 30 min; (3) Benzhydrol, 0 °C → RT, 16 h; (j) (1) NaH, 3-Hydroxypropionitril, THF, -78 °C → 0 °C; (2) 113, 0 °C → RT, 16 h.

Allgemeiner Teil 4 Synthesen und Spektroskopie
64
Im abschließenden Reaktionsschritt erfolgte die Abspaltung des Benzylesters parallel mit der
Einführung der Carbonylfunktion in der Position N6. Hierzu wurde bei -78 °C das Natriumal-
koholat des Hydroxypropionitrils 114 in trockenem THF dargestellt und anschließend unter
Eisbadkühlung mit dem geschützten Guanin-benzylester 113 versetzt. Aus dem Benzylester
113 entstand der 2-Cyanoethyl-ester 115, der durch ß-Eliminierung weiter zum Carboxylat
110 reagierte (Abbildung 49). Gleichzeitig wurde der Chlorsubstituent in der Position N6
durch eine 2-Cyanoethoxy-Gruppe substituiert, welche daraufhin ihr Sauerstoffatom unter
ß-Eliminierung an den Purinring abgab. Nach Ausfällen erhielt man die geschützte Guanin-
essigsäure 110 mit einer Ausbeute von 28 % über drei Stufen. Als problematisch erwies sich
die Abtrennung des Benzylalkohols (116) im letzten Schritt, da dieser sowohl Löslichkeit in
organischen Lösungsmitteln als auch in Wasser besitzt und aufgrund seines hohen
Siedepunkts auch durch Trocknung nur schwer zu entfernen war.
N N
N N
Cl
NH
OOO
O
PhPh
N
N NH
O
NH
OOOH
O
N
Ph Ph
NaO
N
N
N N
O
NH
OO
NH
O
O
N
HN
Ph Ph
ON
ON
O
N
OHN
HO
ONa
- NaCl
4
- 2
-
-
oder
oder
-
H2O
113 115 110
114
114
117 114
116
117
118
119
Abbildung 49. Mechanismus der Esterspaltung und der Einführung der Carbonylfunktion.
Im Verlauf der Arbeit konnte die Synthese der geschützten Adenin-essigsäure 108 durch die
Verwendung einer alternativen Vorschrift optimiert werden.[218] Hierzu wurde Adenin-
ethylester 103 synthetisiert, der genau wie das entsprechende Bhoc-Derivat 121 deutlich
besser zu kristallisieren war als die entsprechenden Benzyl-Analoga. Die Gesamtausbeute an
108 betrug 40 % über drei Stufen (Abbildung 50).

Allgemeiner Teil 4 Synthesen und Spektroskopie
65
N N
N N
NH2
O
O
NH N
N N
NH2
N N
N N
NH
O
O
O
ON N
N N
NH
O
O
OH
O 103 120 121 108
a b c
67% 67 % 88 %
Abbildung 50. Synthese von ABhoc-AcOH (108) über Adenin-essigsäure-ethylester 120. – Reagenzien und Reak-tionsbedingungen: (a) (1) NaH, DMF, RT, 3 h; (2) Bromessigsäure-ethylester, 0 °C → RT, 16 h; (b) (1) CDI, DMF, 105 °C, 2 h; (2) Benzhydrol, 105 °C, 3 h; (c) (1) LiOH, MeCN, EtOH, H2O, 0 °C, 1 h.

Allgemeiner Teil 4 Synthesen und Spektroskopie
66
4.1.4 Rückgrat
Des Weiteren galt es N- und C-terminal orthogonal geschütztes AEG herzustellen, welches
ausgehend von der freien Diaminosäure 85 darstellbar ist. 2-Amino-ethylglycin (85) ist durch
Alkylierung von Ethylendiamin (122) mit Halogenessigsäuren[222] oder durch reduktive
Aminierung von Glyoxylsäure (123) mit Ethylendiamin (122)[223] zugänglich. In beiden
Fällen stellt die Monofunktionalisierung des Ethylendiamins (122) die entscheidende Hürde
dar, sodass mit einem Überschuss an Diamin gearbeitet werden muss. Im Rahmen dieser
Arbeit wurde zunächst der Weg über die reduktive Aminierung gewählt, da er hohe
Ausbeuten bei einfacher Durchführung versprach. Tatsächlich konnte die Zielverbindung
durch katalytische Hydrierung des zunächst gebildeten Imins mit einer Ausbeute von 65 %
gewonnen werden (Abbildung 51). Nach Abtrennung des Katalysators durch Filtration,
Entfernen der leichtflüchtigen Komponenten und Ausfällen durch Zugabe von 2-Propanol war
keine weitere Reinigung mehr notwendig. Ethylendiamin (122) ist durch den Chelateffekt ein
guter Komplexbildner, daher konnte der Palladium-Katalysator nicht erneut eingesetzt
werden. Darüber hinaus ist eine lebhafte Durchmischung des Reaktionsgemisches, bevorzugt
unter Überdruck, unabdingbar, da ansonsten keine Reaktion einsetzt. Die nachfolgende
Schützung der Carboxylfunktion als Methylester wurde durch Umsetzung mit Thionylchlorid
in Methanol erreicht und lieferte das Dihydrochlorid 124 mit einer Ausbeute von 81 %. Die
nachfolgende Schützung des terminalen primären Amins mit der Fmoc-Gruppe erforderte
aufgrund zahlreicher Möglichkeiten zur Nebenreaktion besondere Aufmerksamkeit:
1. Das primäre Amin muss vor der Umsetzung zunächst aus seinem Hydrochlorid freige-
setzt werden. Sobald dies geschehen ist, kann es zur intramolekularen Aminolyse des
Methylesters unter Entstehung von Ketopiperazin 53 kommen. Aufgrund dieser
Tatsache empfiehlt sich eine zügige Zugabe von Fmoc-Cl.
2. Auch das sekundäre Amin lässt sich mit Fmoc-Cl schützen, sodass unter diesem
Blickwinkel das Reagenz in der Kälte langsam hinzugegeben werden sollte. Die
Verwendung eines Überschusses an Diaminosäure-methylester 124 bietet sich wegen
der teuren Darstellung nicht an.
3. Sekundäre Amine (z.B. Piperidin, Diethylamin) sind bevorzugte Reagenzien, um die
Fmoc-Gruppe abzuspalten. Daher sollte die Reaktionsmischung nicht unnötig lange
rühren, rasch gereinigt und weiter umgesetzt werden.
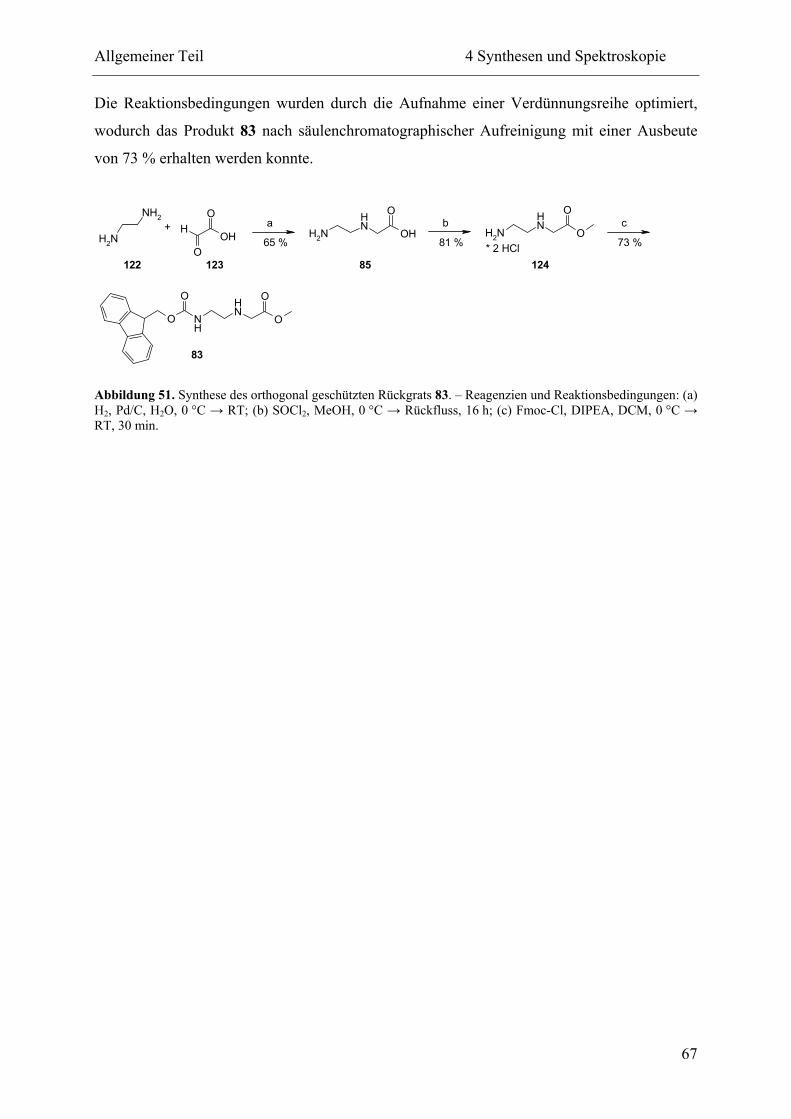
Allgemeiner Teil 4 Synthesen und Spektroskopie
67
Die Reaktionsbedingungen wurden durch die Aufnahme einer Verdünnungsreihe optimiert,
wodurch das Produkt 83 nach säulenchromatographischer Aufreinigung mit einer Ausbeute
von 73 % erhalten werden konnte.
NH
O
O
NH
O
O
NH
O
O
NH2
NH
OH
O
NH2NH2
NH2
HOH
O
O * 2 HCl
+
65 % 81 % 73 %
122 123 85 124
83
a b c
Abbildung 51. Synthese des orthogonal geschützten Rückgrats 83. – Reagenzien und Reaktionsbedingungen: (a) H2, Pd/C, H2O, 0 °C → RT; (b) SOCl2, MeOH, 0 °C → Rückfluss, 16 h; (c) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 30 min.

Allgemeiner Teil 4 Synthesen und Spektroskopie
68
4.1.5 Monomere
Die Kopplung der modifizierten Nukleobasen und der Isostere an das Rückgrat 83 erfolgte
mit dem Kopplungsreagenz TOTU in guten Ausbeuten. Während die nachfolgenden
alkalischen Verseifungen im Falle der Adenin- und Cytosin-Derivate 125 und 126 problemlos
verliefen (Abbildung 52), ließen sich das Monomer des Guanins 127 und die der Isostere
128a-c nicht isolieren (Abbildung 53).
N N
N N
NHBhoc
N
O
NH
O
OMe
N N
N N
NHBhoc
N
O
NH
O
OH
N N
N N
NHBhoc
O
OH
NH
NH
O
OMe
N
N
O
NH
OH
O
Bhoc
NH
O
OH
N
N
O
NH
N
O
Bhoc
NH
NH
O
OMe
N
N
O
NH
N
O
Bhoc
NH
O
OMe
67 % 68 %
83 125 133
Fmoc FmocFmoc
+
a b
108
48 % 94 %
83 126 82
Fmoc FmocFmoc
+
a c
84
Abbildung 52. Synthese der Monomere von Adenin und Cytosin über die Reaktionsfolge Peptidkopplung und Verseifung. – Reagenzien und Reaktionsbedingungen: (a) TOTU, DIPEA, DMF, RT, 3 h; (b) (1) LiOH (2 eq.), THF, H2O; (2) Zitronensäure; (c) (1) LiOH (1.5 eq.), THF, H2O; (2) HCl.
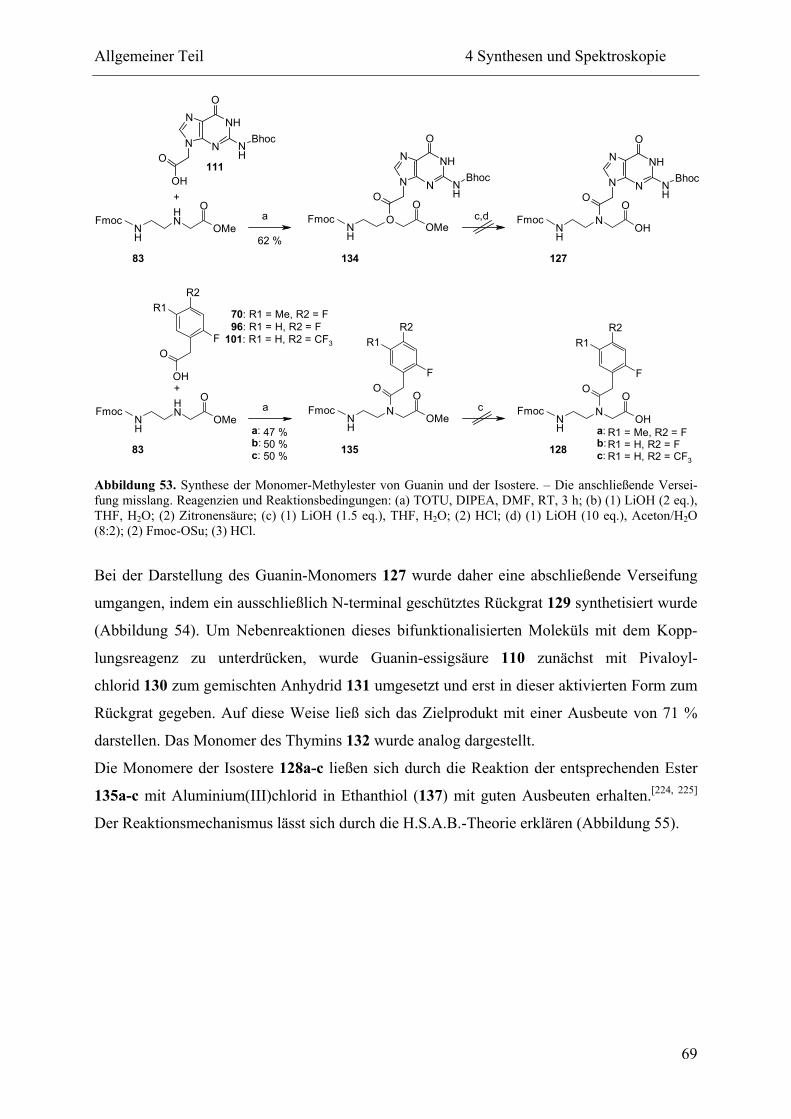
Allgemeiner Teil 4 Synthesen und Spektroskopie
69
N
N NH
O
NH
OH
O
N Bhoc
NH
O
OH
N
N NH
O
NH
N
O
N Bhoc
NH
NH
O
OMe NH
O
OMe
N
N NH
O
NH
O
O
N Bhoc
NH
NH
O
OMe NH
O
OMe
R2
F
N
O
R1
R2
F
OH
O
R1
R2
F
N
O
R1
NH
O
OH
62 %
83 134 127
Fmoc FmocFmoc
+
a c,d
111
83 135 128
Fmoc FmocFmoc
+
a c
47 %50 %50 %
70: R1 = Me, R2 = F 96: R1 = H, R2 = F101: R1 = H, R2 = CF3
a:b:c:
R1 = Me, R2 = FR1 = H, R2 = FR1 = H, R2 = CF3
a:b:c:
Abbildung 53. Synthese der Monomer-Methylester von Guanin und der Isostere. – Die anschließende Versei-fung misslang. Reagenzien und Reaktionsbedingungen: (a) TOTU, DIPEA, DMF, RT, 3 h; (b) (1) LiOH (2 eq.), THF, H2O; (2) Zitronensäure; (c) (1) LiOH (1.5 eq.), THF, H2O; (2) HCl; (d) (1) LiOH (10 eq.), Aceton/H2O (8:2); (2) Fmoc-OSu; (3) HCl.
Bei der Darstellung des Guanin-Monomers 127 wurde daher eine abschließende Verseifung
umgangen, indem ein ausschließlich N-terminal geschütztes Rückgrat 129 synthetisiert wurde
(Abbildung 54). Um Nebenreaktionen dieses bifunktionalisierten Moleküls mit dem Kopp-
lungsreagenz zu unterdrücken, wurde Guanin-essigsäure 110 zunächst mit Pivaloyl-
chlorid 130 zum gemischten Anhydrid 131 umgesetzt und erst in dieser aktivierten Form zum
Rückgrat gegeben. Auf diese Weise ließ sich das Zielprodukt mit einer Ausbeute von 71 %
darstellen. Das Monomer des Thymins 132 wurde analog dargestellt.
Die Monomere der Isostere 128a-c ließen sich durch die Reaktion der entsprechenden Ester
135a-c mit Aluminium(III)chlorid in Ethanthiol (137) mit guten Ausbeuten erhalten.[224, 225]
Der Reaktionsmechanismus lässt sich durch die H.S.A.B.-Theorie erklären (Abbildung 55).
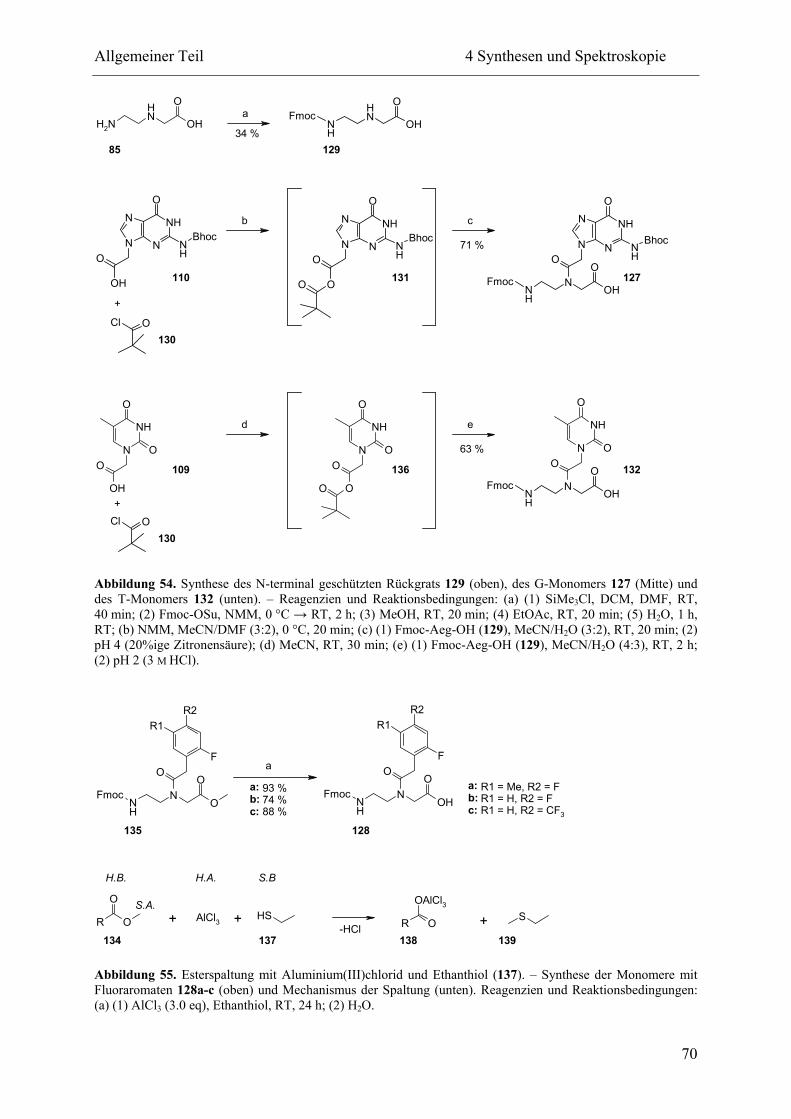
Allgemeiner Teil 4 Synthesen und Spektroskopie
70
OH
O
NBhoc
N
N NH
O
NH
OCl
O
O
NBhoc
N
N NH
O
NH
O
NH
O
OH
N
N NH
O
NH
N
O
N Bhoc
N
NH
O
O
OH
O
NH
NH2 OH
O
NH
O
OHNH
Fmoc
OCl
N
NH
O
O
N
O
NH
O
OH
N
NH
O
O
O
O
O
+
71 %
110 131 127
130
Fmoc
b c
34 %
85 129
a
+
130
63 %
Fmoc
d e
109 136 132
Abbildung 54. Synthese des N-terminal geschützten Rückgrats 129 (oben), des G-Monomers 127 (Mitte) und des T-Monomers 132 (unten). – Reagenzien und Reaktionsbedingungen: (a) (1) SiMe3Cl, DCM, DMF, RT, 40 min; (2) Fmoc-OSu, NMM, 0 °C → RT, 2 h; (3) MeOH, RT, 20 min; (4) EtOAc, RT, 20 min; (5) H2O, 1 h, RT; (b) NMM, MeCN/DMF (3:2), 0 °C, 20 min; (c) (1) Fmoc-Aeg-OH (129), MeCN/H2O (3:2), RT, 20 min; (2) pH 4 (20%ige Zitronensäure); (d) MeCN, RT, 30 min; (e) (1) Fmoc-Aeg-OH (129), MeCN/H2O (4:3), RT, 2 h; (2) pH 2 (3 M HCl).
R2
F
N
O
OH
O
NH
R1R2
F
N
O
O
O
NH
R1
R O
O
AlCl3 SH SR O
OAlCl3
FmocFmoc
135 128
+ + +-HCl
H.B. H.A. S.B
S.A.
a
134 137 138 139
R1 = Me, R2 = FR1 = H, R2 = FR1 = H, R2 = CF3
a:b:c:
93 %74 %88 %
a:b:c:
Abbildung 55. Esterspaltung mit Aluminium(III)chlorid und Ethanthiol (137). – Synthese der Monomere mit Fluoraromaten 128a-c (oben) und Mechanismus der Spaltung (unten). Reagenzien und Reaktionsbedingungen: (a) (1) AlCl3 (3.0 eq), Ethanthiol, RT, 24 h; (2) H2O.

Allgemeiner Teil 4 Synthesen und Spektroskopie
71
4.1.6 Löslichkeitsvermittler
Als N-terminale Löslichkeitsvermittler wurden sowohl zweifach methyliertes ß-Alanin 140
als auch ein terminal N-methyliertes Rückgrat 78 synthetisiert (Abbildung 56). Die letztge-
nannte Verbindung wurde anschließend mit geschützter Cytosin-essigsäure 84 gekoppelt, was
in moderater Ausbeute gelang. Bei der Synthese von 141 zeigte sich die Verwendung von
EDC als Kopplungsreagenz gegenüber der von TOTU als überlegen. Da das Produkt der
anschließenden Verseifung 142 ein inneres Salz ist, gestalteten sich die Aufarbeitung und die
Reinigung durch Säulenchromatographie schwierig.
NH
O
O
NNH
OH
O
NN
NH2
HOH
O
O
N
N
NH
O
N
O
Bhoc
N O
O
OH
O
NOH
O
NH2
N
N
NH
O
N
O
Bhoc
N OH
O
*2HCl
+95 % 98 % 54 %
144 123 145 78
*HCl
31 %143 140
a
b c d
79 %
e
141 142
Abbildung 56. Synthese zweier N-terminaler Löslichkeitsvermittler. – N,N-Dimethyl-ß-alanin (140) (oben) und ein C-Monomer 142 mit terminal methyliertem Rückgrat 78 (unten). Reagenzien und Reaktionsbedingungen: (a) (1) Formalin, Ameisensäure, Rückfluss, 16 h; (2) HCl; (b) H2, Pd/C, H2O, RT; (c) SOCl2, MeOH, 0 °C → Rückfluss, 16 h; (d) CBhoc-AcOH (84), EDC, HOBt, DIPEA, DMF, 0 °C → RT, 16 h; (e) (1) LiOH (2 eq.), THF, H2O, 0 °C → RT, 2 h; (2) HCl.
Die Modifizierung des C-Terminus sollte durch die Abspaltung des Oligomers vom
HMBA-Linker 75 durch Reaktion mit N,N-Dimethylaminoethylamin 143 erfolgen
(Abbildung 57, oben). Bei HMBA 75 und anderen Hydroxymethyl-funktionalisierten
Festphasen wird das erste Monomer typischerweise manuell gekoppelt, da hierbei eine Ester-
funktion entsteht, deren Bildung andere Bedingungen erfordert als die folgenden Kopplungen
zur Amidbindung. Das Templat und somit auch das C-terminale Trimer sollten als ersten
Baustein eine Glycin-Einheit enthalten, die zum einen der Aggregation des wachsenden

Allgemeiner Teil 4 Synthesen und Spektroskopie
72
Oligomers an der Festphase entgegenwirkt und zum anderen die Selbstabspaltung nach dem
ersten Monomer verhindert. Diese tritt auf, wenn das freie Amin 144 die Esterbindung zum
Linker unter Ausbildung eines Sechsrings 145 nukleophil angreifen kann (Abbildung 57,
unten). Bei Peptiden ist dieser Effekt auf der Stufe des Dipeptids bekannt.
N
O
NH2 O
O
B(Bhoc)O
B(Bhoc)
N
NH
O
N
O
NH2 NH
O
B(Bhoc)
O
O
NH
NNH
O
N
O
B(Bhoc)
O
NHN
H
NO
OO
B(Bhoc)
NH2
N
O
NH
OH
linker
144 145 146
PEG
PEG
+
linker
PNA PNA
143 75
147 148
Abbildung 57. Abspaltung des Oligomers von der Festphase durch Umsetzung mit N,N-Dimethylaminoethyl-amin (143) (oben) und Selbstabspaltung von der Festphase und deren Unterdrückung durch Einbau einer Glycin Einheit (unten).
Bei dem folgenden Screening verschiedener Reaktionsbedingungen erwies sich MSNT (149)
als effektivstes Reagenz zum Koppeln von Fmoc-geschütztem Glycin an den HMBA-
Linker 75 (Abbildung 58). Mit dieser aus der Oligonukleotid-Synthese bekannten Verbindung
ließ sich die Glycin-Einheit mit einer Ausbeute von 82 % an die Festphase koppeln, während
sich das anspruchsvolle Guanin-Monomer 127 immerhin noch mit moderaten 54 % koppeln
ließ.
O
NH
ONH
O
Fmoc
S N
O
O N
N
NO2
OH
O
NH
75 150
a: 20 %b: 26 %c: 65 %d: 82 %
PEG
MSNT149
PEG
Abbildung 58. Kopplung von Fmoc-Gly-OH an die HMBA-Festphase. – Reagenzien und Reaktionsbedingung-en: (a) Fmoc-Gly-OH (3.2 eq.), HBTU, DIPEA, DMF, RT, 1.5 h; (b) Fmoc-Gly-OH (5.0 eq.), TOTU, DIPEA, DMF, RT, 24 h; (c) Fmoc-Gly-OH (10 eq.), DIC, DIPEA, DMAP, DCM, RT, 24 h; (d) Fmoc-Gly-OH (5.0 eq.), MSNT, MeIm, DCM, RT, 24 h.

Allgemeiner Teil 4 Synthesen und Spektroskopie
73
4.1.7 Oligomersynthese
Die folgenden maschinellen Festphasensynthesen erfolgten jeweils im Maßstab von 10 µmol
in einem 3 ml Reaktionsgefäß. Als N-terminaler Löslichkeitsvermittler wurde zunächst
ß-Alanin 140 ausgewählt. Vom Kopplungsreagenz HBTU (151) wurden pro Kopplungsschritt
5.0 Äquivalente eingesetzt, während von den PNA-Monomeren jeweils 5.3 Äquivalente ver-
wendet wurden (Abbildung 59).
O
N
NN
NN
O
O
NNH
O
B(Bhoc)
N
NN
O
N N
O
N
N
O
ON
NH
O
B(Bhoc)
N
N
N
O
O
NH
O
B(Bhoc)
N
N
O
NH2 NH
O
B(Bhoc)
O
On
N
O
NH
NH
O
B(Bhoc)
O
O
ON
NH n
O
B(Bhoc)
PF6-
PF6
-
PF6
-+
+
-
-
Fmoc Fmoc+ +
Fmoclinker
Fmoclinker
+
152 151 153 154
156 155
157
158
Abbildung 59. Aktivierung mit HBTU(N-Form bzw. Guanidinium-Salz) und anschließende Kopplung.
Dieser leichte Überschuss an Monomeren 152 gegenüber dem Kopplungsreagenz erklärt sich
aus der in 1.4.4 besprochenen besonderen Reaktivität der Aminofunktion. Diese kann mit
nicht umgesetztem HBTU zum entsprechenden Guanidinium-Salz 159 reagieren und würde
danach einer Kettenverlängerung nicht mehr zur Verfügung stehen (Abbildung 60).
Nach der Kopplung wurden nicht umgesetzte Aminofunktionen durch eine Mischung von
Essigsäureanhydrid, Pyridin und NMP acetyliert, um Deletionsmutanten zu vermeiden und
die chromatographische Abtrennung von Nebenprodukten zu erleichtern.
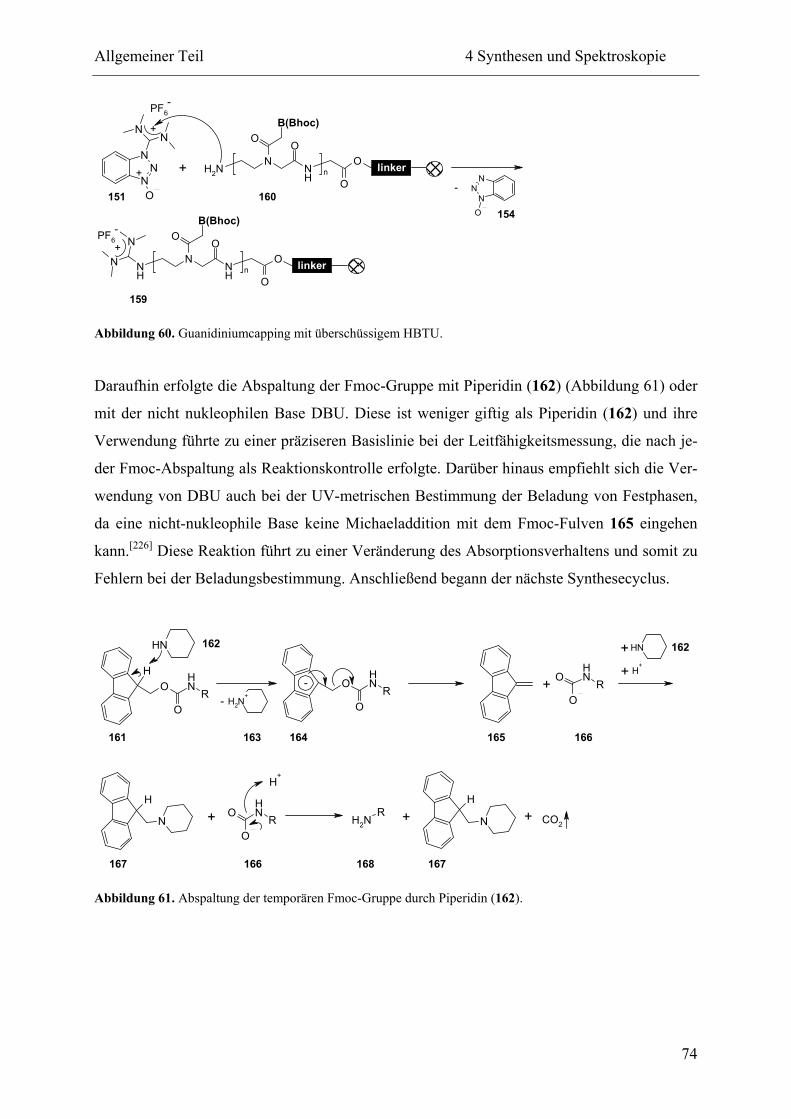
Allgemeiner Teil 4 Synthesen und Spektroskopie
74
N
NN
NN
O
N
O
NH2 NH
O
B(Bhoc)
On
O
N
O
NH
NH
O
B(Bhoc)
On
N
N
O
N
NN
O
PF6
-
PF6
-
+
+ + linker
linker
+
-
159
154
151 160
Abbildung 60. Guanidiniumcapping mit überschüssigem HBTU.
Daraufhin erfolgte die Abspaltung der Fmoc-Gruppe mit Piperidin (162) (Abbildung 61) oder
mit der nicht nukleophilen Base DBU. Diese ist weniger giftig als Piperidin (162) und ihre
Verwendung führte zu einer präziseren Basislinie bei der Leitfähigkeitsmessung, die nach je-
der Fmoc-Abspaltung als Reaktionskontrolle erfolgte. Darüber hinaus empfiehlt sich die Ver-
wendung von DBU auch bei der UV-metrischen Bestimmung der Beladung von Festphasen,
da eine nicht-nukleophile Base keine Michaeladdition mit dem Fmoc-Fulven 165 eingehen
kann.[226] Diese Reaktion führt zu einer Veränderung des Absorptionsverhaltens und somit zu
Fehlern bei der Beladungsbestimmung. Anschließend begann der nächste Synthesecyclus.
O NH
O
RO NH
O
R
H
NH
H
N
NH
NH2
+
O NH
O
R
H+
O NH
O
RH
+
CO2
H
NNH2
R
-
-
+
+
+
+
++
161 163 164 165 166
162 162
167 166 168 167
Abbildung 61. Abspaltung der temporären Fmoc-Gruppe durch Piperidin (162).

Allgemeiner Teil 4 Synthesen und Spektroskopie
75
Nach erfolgter Festphasensynthese wurden zunächst die Bhoc-Schutzgruppen durch
Umsetzung mit Trifluoressigsäure entfernt (Abbildung 62). Dabei wurden 5 % m-Cresol (169)
zugesetzt, um reaktive Carbenium-Ionen 170 abzufangen und eine unerwünschte Alkylierung
der Nukleobasen zu unterdrücken.
OHPh
Ph
OH NH
O
RC+
Ph
Ph H NH2
R CO2O
HNH
O
RPh
Ph
OH
NH
OH+
R
Ph
Ph
OH
OH
PhPh
OH Ph
Ph
+ +
TFA
oder oder
und Folgeprodukte
171 172 170 173 168
169
174 175 176
Abbildung 62. Abspaltung der permanenten Bhoc-Gruppe durch TFA.
Im Anschluss wurde das Zielmolekül 177 abgespalten, indem die Festphase über Nacht mit
einer Lösung von N,N-Dimethylaminoethylamin (143) in DMF bei Raumtemperatur behan-
delt wurde. Nachdem die flüchtigen Komponenten im Vakuum entfernt wurden, erfolgte die
Analyse des Rohprodukts durch RP-HPLC und MALDI-TOF-MS (Abbildung 63). Bei der
Analyse der ersten Templat-Synthese konnten neben dem Zielmolekül 177 auch zwei
Fragmente 178 und 179 nachgewiesen werden, deren Synthese jeweils bei der Kopplung der
Cytosin-Monomere 82 abgebrochen war (Abbildung 64). Im Folgenden wurden daher pro
Cytosin-Einheit zwei Kopplungscyclen mit jeweils frischen Reagenzien angewandt.
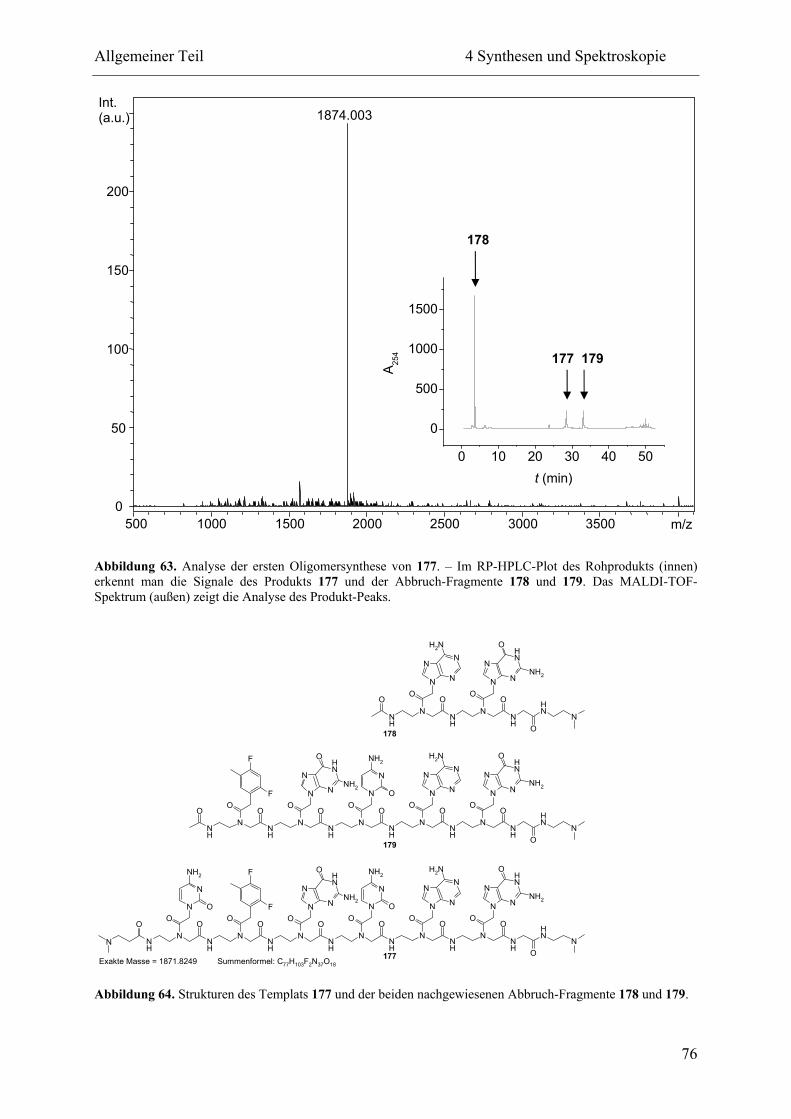
Allgemeiner Teil 4 Synthesen und Spektroskopie
76
Abbildung 63. Analyse der ersten Oligomersynthese von 177. – Im RP-HPLC-Plot des Rohprodukts (innen) erkennt man die Signale des Produkts 177 und der Abbruch-Fragmente 178 und 179. Das MALDI-TOF-Spektrum (außen) zeigt die Analyse des Produkt-Peaks.
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH
O
N
N
O
NH2
NNH
OO
N N
NNH
O
NH2
NH
N
OOO
F
F
NH
O
NH
N
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NH
O
NH
N
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH
O
N
N
O
NH2
NNH
OO
N N
NNH
O
NH2
NNH
N
OOOO
F
FN
N
O
NH2
NH
O
NH
NO
NH
N
178
179
177Exakte Masse = 1871.8249 Summenformel: C77H103F2N37O18
Abbildung 64. Strukturen des Templats 177 und der beiden nachgewiesenen Abbruch-Fragmente 178 und 179.
m/z
1874.003
0
50
100
150
200
Int. (a.u.)
500 1000 1500 2000 2500 3000 3500
0 10 20 30 40 50
0
500
1000
1500
A2
54
t (min)
177 179
178

Allgemeiner Teil 4 Synthesen und Spektroskopie
77
Nachdem das Lösungsmittel am Synthesizer von DMF auf NMP umgestellt wurde, ergaben
sich generell sauberere Rohprodukte, wie in Abbildung 65 zu sehen ist.
Abbildung 65. RP-HPLC-Analyse von zwei verschiedenen Templat-Synthesen. – Bei ansonsten identischen Be-dingungen wurde DMF (links) bzw. NMP (rechts) als Lösungsmittel verwendet.
Es wurde auch ein Templat 180 mit dem Monomer 127c hergestellt, das eine CF3-Gruppe
trägt (Abbildung 66). Die erzielten und in Abbildung 67 dargestellten Ergebnisse waren dabei
mit den vorangegangenen vergleichbar.
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH
O
N
N
O
NH2
NNH
OO
N N
NNH
O
NH2
NNH
N
OOOO
FN
N
O
NH2
NH
O
NH
NO
NH
N
FFF
Exakte Masse = 1907.8061 Summenformel: C77H101F4N37O18 Abbildung 66. Templat 180 mit CF3-Gruppe am Isoster.
Die Aufreinigung von PNA erfolgt wie bei Peptiden typischerweise durch RP-HPLC unter
Verwendung eines Wasser-Acetonitril-Gradienten, wobei den beiden Eluenten jeweils
0.1 % Trifluoressigsäure (TFA) zugesetzt werden. Allerdings wird im Falle von PNA die
Säule auf eine Temperatur von 50-60 °C geheizt, um die typische Selbstassoziation dieser
Verbindungsklasse zu unterdrücken.[227, 228] Abbildung 68 zeigt am Beispiel der Reinigung
des PNA-Trimers 81, wie drastisch sich die Säulentemperatur auf das Trennergebnis
auswirkt. Bei Raumtemperatur findet man eine Reihe zum Teil stark verbreiterter Signale,
während man bei 55 °C hauptsächlich drei definierte Signale für das Trimer und die beiden
möglichen Abbruch-Fragmente findet.
0 10 20 30 40 50 600.00
0.05
0.10
0.15
0.20
0.25
A2
54
t (min)
0 10 20 30 40 50 60 700.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
A2
54
t (min)
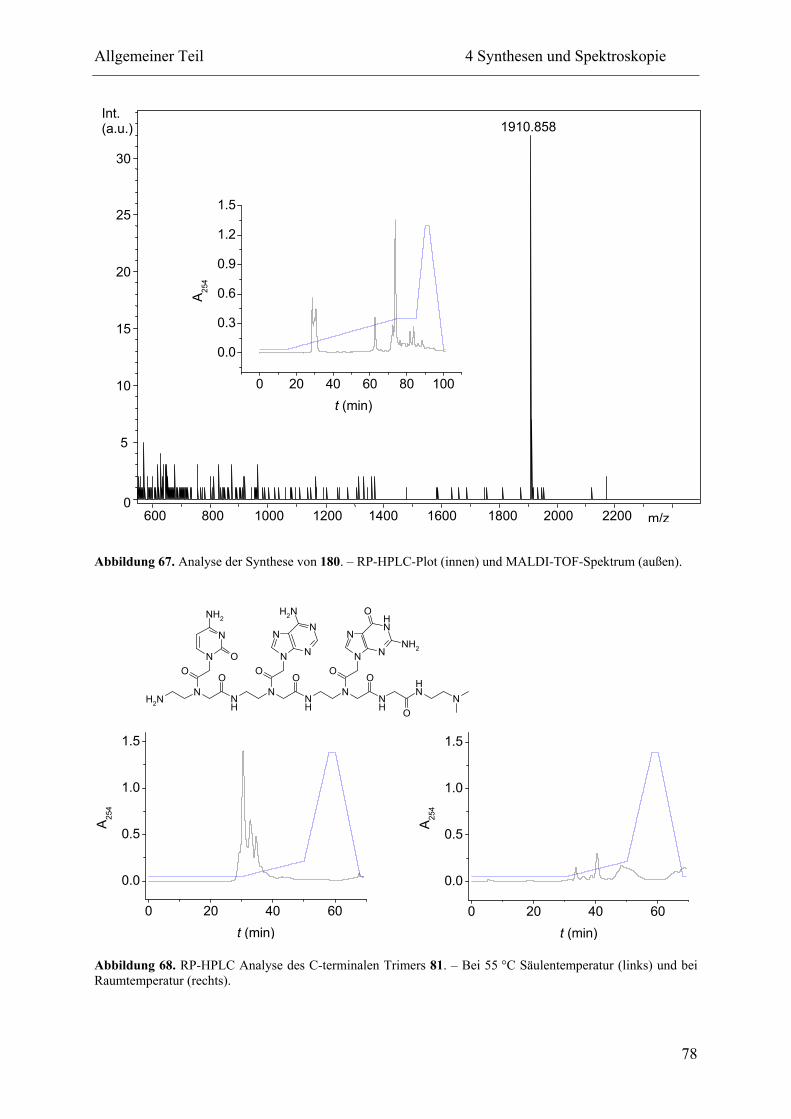
Allgemeiner Teil 4 Synthesen und Spektroskopie
78
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH2
O
N
N
O
NH2
NH
O
NH
N
Abbildung 67. Analyse der Synthese von 180. – RP-HPLC-Plot (innen) und MALDI-TOF-Spektrum (außen).
Abbildung 68. RP-HPLC Analyse des C-terminalen Trimers 81. – Bei 55 °C Säulentemperatur (links) und bei Raumtemperatur (rechts).
0 20 40 60
0.0
0.5
1.0
1.5
A2
54
t (min)
0 20 40 60
0.0
0.5
1.0
1.5
A2
54
t (min)
1910.858
0
5
10
15
20
25
30
600 800 1000 1200 1400 1600 1800 2000 2200 m/z
Int. (a.u.)
0 20 40 60 80 100
0.0
0.3
0.6
0.9
1.2
1.5A
254
t (min)

Allgemeiner Teil 4 Synthesen und Spektroskopie
79
An diesem Punkt wurde der Weg zu (F-)PNA-Bausteinen über die maschinelle Festphasen-
synthese aus drei Gründen verlassen:
1. Nach Abspaltung von der Festphase und Reinigung durch HPLC lagen die Ausbeuten
an Oligomer im Bereich von 10 %. Dies ist für PNA-Synthesen sehr gering, für die
Ausbeuten im Bereich von 26-38.5 % (15-32mere, Fmoc/Bhoc)[229] bis hin zu 61 %
(17mer, Boc/Z)[230] berichtet werden. Der Wechsel des verwendeten Lösungsmittels
und die Einführung doppelter Kopplungen führten zu sichtbaren Fortschritten. In der
Summe reichten diese Verbesserungen aber nicht aus, um die zeitnahe Synthese der
benötigten Substanzen zu gewährleisten.
2. Die maschinelle Festphasensynthese erforderte einen hohen Überschuss an den teuren
bzw. aufwendig herzustellenden Monomeren. Der damit verbundene Aufwand an Zeit,
Geld und Arbeitskraft schien nach Analyse der eben beschriebenen Ergebnisse nicht
mit einer effektiven und fristgerechten Durchführung des Projekts vereinbar.
3. Die Trennung der PNA-Trimere von ihren Abbruch-Fragmenten, dem acetylierten
Mono- und Dimer, gestaltete sich schwierig. Ursächlich war hier die geringe Reten-
tion dieser stark hydrophilen Verbindungen auf der zur Verfügung stehenden Säule.
Weder die Verwendung sehr flacher Gradienten noch der Wechsel zum Wasser-
Methanol-Gradienten brachten hier greifbare Ergebnisse.
Zunächst war nicht eindeutig zu klären, an welcher Stelle die Ausbeuten so drastisch einbra-
chen. Daher wurde in einem gemeinsamen Projekt mit Dietz sowohl die Effizienz der
Kopplungen als auch die Effektivität der Abspaltung untersucht. Aus Kostengründen geschah
dies anhand einer Modellverbindung: einem Peptid mit der Sequenz H-Lys-Lys-Pro-Tyr-Ile-
Leu-OH.[231] Zusammenfassend konnte gezeigt werden, dass die Synthese der Modellverbin-
dung am standardmäßig verwendeten Wang-Linker sowie am HMBA-Linker mit guten
Effizienzen gelang. Die Abspaltung des Peptids in Form seiner freien Säure erfolgte von
beiden Linkern durch die Behandlung mit TFA bzw. NaOH in zufriedenstellender Ausbeute.
Allerdings war es nicht möglich, das Oligomer mit N,N-Dimethylaminoethylamin (143) von
der Festphase abzuspalten. Dies deckte sich zwar mit den Erfahrungen bei den voran-
gegangenen PNA-Synthesen, erschien aber verwunderlich, da die Reaktionsbedingungen
analog zu einem Beispiel aus vertrauenswürdiger Literatur gewählt wurden.[213] In diesem
Beispiel wurde ein Peptid mit Ethanolamin von der HMBA-Festphase abgespalten.

Allgemeiner Teil 4 Synthesen und Spektroskopie
80
Leider ist die Anzahl der literaturbekannten Anwendungen des HMBA-Linkers und der
beschriebenen Variationen der Reaktionsbedingungen begrenzt. Der einzig bekannte
alternative Weg zu C-terminalen sekundären Amiden führt über die sogenannten Backbone
Amide Linker (BAL) (181a-b).[232] Hierbei handelt es sich um Linker mit einer Aldehyd-
funktion, die zunächst manuell in einer reduktiven Aminierung mit einem primären Amin
zum sekundären Amin umgesetzt wird. Bevor das gewünschte Oligomer durch maschinelle
oder manuelle Fmoc-Festphasenchemie aufgebaut werden kann, muss zunächst das erste
Monomer in einem weiteren manuellen Schritt gekoppelt werden. Die Abspaltung mit TFA in
Dichlormethan führt dann zum gewünschten C-terminalen Amid. Erste Versuche mit dem
Modellpeptid am MALDRE-Linker (181b)[233] führten zum gewünschten sekundären Amid
mit Ausbeuten von bis zu 48 %. Abbildung 69 zeigt, wie prinzipiell auch PNAs an diesem
Linker aufgebaut werden können.
O
R
O NH
ON
N
O
N
NH
O
PG1
OO
R
O NH
O
NNH2
N
NH
O
R
O NH
O
NHN
OO
NHN
O
NH
O
X
n
N
NaCNBH3
Kopplung
a: R = OMe (BAL) b: R = H (AMEBA oder MALDRE)
B
B
PNA-Monomer
181 143 182a-b
reduktive Aminierung
183a-b
184
TFA
Fmoc-SPPS
Abspaltung
B(PG2)
Abbildung 69. Potentieller Zugang zu C-terminal Amid-funktionalisierten PNAs 184 über Festphasen mit Aldehydfunktion 181.

Allgemeiner Teil 4 Synthesen und Spektroskopie
81
4.2 Synthesestrategie II: Konvergente Flüssigphasensynthese
4.2.1 Vorüberlegungen
Die Alternative zur Festphasensynthese ist die Synthese der Oligomere in Lösung, wie sie vor
den bahnbrechenden Arbeiten von Merrifield standardmäßig zum Aufbau von Peptiden be-
nutzt wurde. Auch wenn sie sich nicht automatisieren lässt, bietet sie doch eine Reihe von
Vorteilen:
1. Typischerweise wird mit nur geringen oder gar keinen Überschüssen an Reagenzien
und Monomeren bzw. Fragmenten gearbeitet, sodass weniger wertvolle Substanzen
verbraucht werden.
2. Das wachsende Oligomer kann nach jedem Reaktionsschritt isoliert und aufgereinigt
werden, wohingegen bei der SPPS nach den einzelnen Reaktionsschritten nur
gewaschen wird, was zu einer Akkumulation von Verunreinigungen bzw. Mutanten
führen kann.
3. Nach jedem Reaktionsschritt kann eine vollständige Charakterisierung erfolgen
(NMR-Spektroskopie, Massenspektrometrie, HPLC u.a.). Auf diese Weise werden
Probleme früh eingegrenzt und erkannt. Hingegen erfolgt die Kontrolle des Reaktions-
fortschritts bei der manuellen SPPS typischerweise durch Testreaktionen (z.B. Kaiser-
Test), am Synthesizer auch UV-spektroskopisch oder wie im Falle des hier
verwendeten Geräts durch Leitfähigkeitsmessung.
4. Dem Hochskalieren von Synthesen sind in flüssiger Phase deutlich weniger Grenzen
gesetzt, sodass ein rascher Zugang zu den benötigten Mengen möglich erschien.
Es war nun das Ziel, die zwei Trimere, die zum Aufbau des Replikations-Systems benötigt
wurden, in Lösung herzustellen und anschließend zum hexameren Templat zu ligieren.

Allgemeiner Teil 4 Synthesen und Spektroskopie
82
4.2.2 Fmoc/Bhoc-Chemie
Zunächst wurde auf Basis der vorhandenen Substanzen versucht, eine erste Kondensations-
reaktion zum Dimer durchzuführen. Als Säure-Komponente wurde für diesen Versuch das
bereits vorhandene Cytosin-Monommer 82 ausgewählt, während sich die Wahl der Amino-
Komponente schwieriger gestaltete. Es erscheint zunächst logisch, vom vollständig
geschützten Monomer auszugehen und anstatt der Esterverseifung eine Fmoc-Abspaltung
durchzuführen. Jedoch greift das freigesetzte Amin entweder sofort die Carbonylfunktion des
Esters unter Ringschlussbildung an oder attackiert die Carbonylfunktion des Essigsäurelinkers
unter Transamidierung (Abbildung 30). Diese Problematiken setzten sich bei der Lagerung
fort und stellen zudem in einem Kopplungsansatz Konkurrenzreaktionen dar. Die Schwierig-
keiten bei Synthese und Lagerung wurden zunächst hintangestellt, indem, ausgehend von
einem Boc-geschützten Monomer, das TFA-Salz 185 dargestellt wurde. Die Synthese des
geschützten PNA-Dimers 186 (Abbildung 70) konnte zwar durch DC- und 1H-NMR-Analyse
bestätigt werden, aber es war nicht möglich, die Zielverbindung zu reinigen, da eine
Löslichkeit nur in extrem polaren Solventien wie DMF und DMSO gegeben war, die sich
nicht zur Chromatographie eigneten. Ebenso war eine Kristallisation nicht möglich.
N
N
NH
O
N
O
Bhoc
NH
OH
O
NH2
O
F
F
N
O
OMe NH
O
F
F
N
O
N
N
NH
O
N
O
NH
O
OMe
Bhoc
*TFA+
82 185 186
Fmoc Fmoca
Abbildung 70. Konvergente Flüssigphasensynthese. – Modellreaktion mit einem Fmoc/Bhoc-Monomer. Reagenzien und Reaktionsbedingungen: (a) HATU, DIPEA, DMF, RT, 16 h.
Neben der generellen Tendenz von PNA zu aggregieren, lässt sich auch die Wahl der Schutz-
gruppen als Erklärung für die hier aufgetretene minimale Löslichkeit heranziehen. Betrachtet
man typische Peptidsynthesen an der festen Phase, so wird bei der Verwendung Fmoc-
geschützter Aminosäuren in der Regel polares DMF oder NMP als Lösungsmittel verwendet,
wohingegen bei Synthesen mit Boc-geschützten Aminosäuren weitaus unpolareres DCM
eingesetzt wird. Daraus wurde geschlossen, dass eine Synthese auf Basis von Boc-geschütz-
ten Verbindungen zu deutlich verbesserter Löslichkeit bei den jeweiligen Reaktionen und vor
allem während der folgenden Reinigungsschritte durch Chromatographie führen würde.

Allgemeiner Teil 4 Synthesen und Spektroskopie
83
4.2.3 Boc/Z-Chemie
Die Synthese der entsprechenden Boc/Z-Monomere war notwendig, da diese Verbindungen
nur zu sehr hohen Preisen erhältlich waren. Zur Darstellung von N-terminal Boc-geschütztem
Rückgrat 187 wurde der Methylester 124 unter nukleophiler DMAP-Katalyse mit Boc-
Anhydrid in DCM umgesetzt (Abbildung 71). Nach säulenchromatographischer Reinigung
ließen sich nur 54 % der Zielverbindung isolieren, sodass in einem weiteren Experiment die
Bedingungen der Fmoc-Schützung von 124 auf die Einführung der Boc-Gruppe übertragen
wurden. Hierdurch ließ sich die Ausbeute auf 63 % steigern. Anzumerken ist, dass die bei der
Synthese von Fmoc-Aeg-OMe (83) beschriebenen Konkurrenzreaktionen auch bei der
analogen Boc-Schützung auftreten.
OO NH
NH
OO
ONH2
NH
O
*2HCl
124 187
a oder b
a: 54 %b: 63 %
Abbildung 71. Synthese von Boc-Aeg-OMe (187). – Reagenzien und Reaktionsbedingungen: (a) Boc2O, DMAP, NMM, DCM, -15 °C → RT, 5 h; (b) Boc2O, TEA, DCM, 0 °C → RT, 16 h.
Die Synthese des Adenin-Monomers 188 wurde analog zu dem beschriebenen Fmoc/Bhoc-
Derivat 132 durchgeführt (Abbildung 72): Der bereits dargestellte Ethylester 120 wurde
zunächst mit CDI umgesetzt und dann wurde Benzylalkohol anstelle von Benzhydrol zugege-
ben. Nach Durchführung der schon bekannten Reaktionssequenz aus Verseifung, Kopplung
und Verseifung wurde das Boc/Z-geschützte Adenin-Monomer 188 mit einer Ausbeute von
32 % über fünf Synthesestufen erhalten.

Allgemeiner Teil 4 Synthesen und Spektroskopie
84
N N
N N
NH2
O
O
N N
N N
NH
O
O
OH
ON N
N N
NH
O
O
O
O
N N
N N
NH
O
O
OHO NH
OO
N
O
N N
N N
NH
O
O
OO NH
OO
N
O
55 % 80 % 81 %
a b c
90 %
b
120 189 190
191 188
Abbildung 72. Synthese von Boc-Aeg(AZ)-OH (188). – Reagenzien und Reaktionsbedingungen: (a) (1) CDI, DMF, 105 °C, 2 h; (2) Benzylalkohol, 105 °C, 3 h; (b) (1) LiOH, THF, H2O, 0 °C → RT, 2 h; (2) 1 M HCl; (c) Boc-Aeg-OMe (187), TOTU, DIPEA, DMF, RT, 3 h.
Die Synthese der Boc-geschützten Cytosin-essigsäure 192 wurde zum einen analog zum
Fmoc-Derivat 84 durchgeführt, zum anderen wurde ein Weg gewählt, bei dem zunächst die
exocyclische Aminofunktion geschützt wird. Bei dieser Synthese wurde Cytosin (86) unter
Schützung der Positionen N1 und N4 mit 2.2 Äquivalenten Chlorameisensäure-benzylester
versetzt. Die Schutzgruppe an N1 ist instabil und wurde durch die folgende wässrige
Aufarbeitung wieder entfernt. Anschließend wurde 193 mit Chloressigsäure-methylester
alkyliert und dann zu Essigsäure 192 verseift. Aufgrund der geringen Ausbeute des ersten
Schritts zeigte sich der bereits bekannte Zugang über 105 und 194 überlegen. Die Synthese
des Monomers 195 erfolgte über die bekannte Sequenz Peptidkopplung und Verseifung. Nach
fünf Stufen konnte das Cytosin-Monomer 195 mit einer Ausbeute von 12 bzw. 45 % erhalten
werden (Abbildung 73).
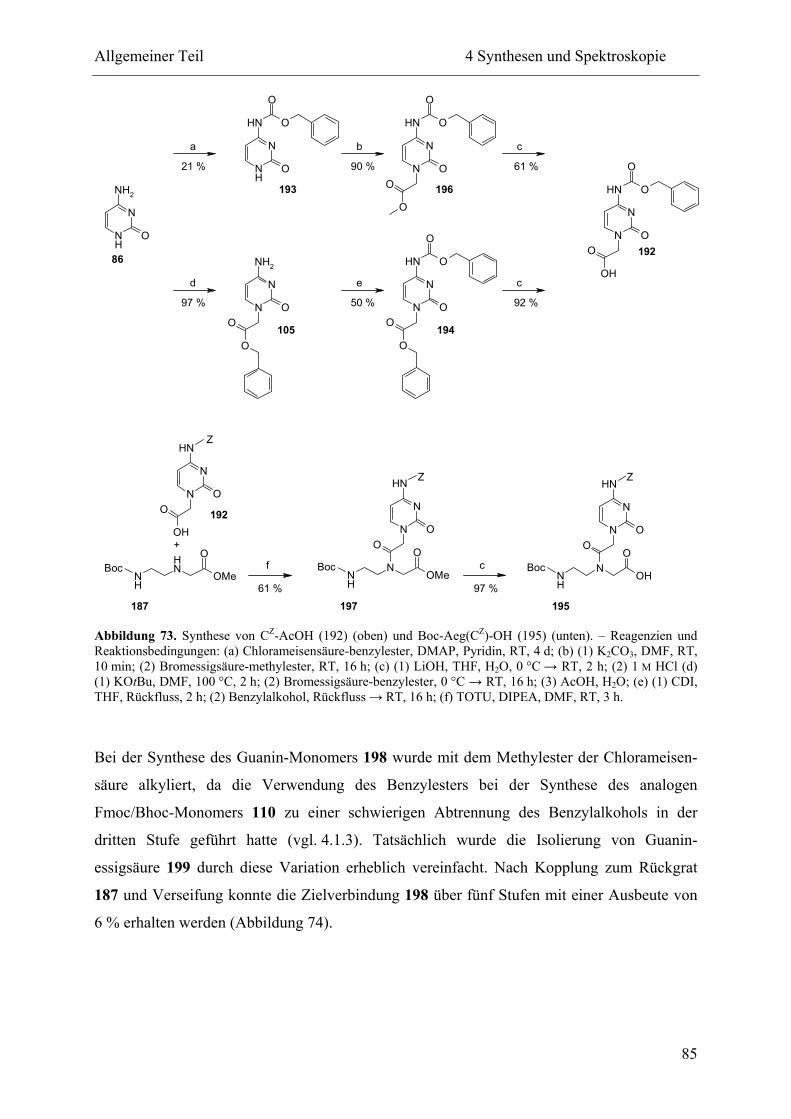
Allgemeiner Teil 4 Synthesen und Spektroskopie
85
O
O
NH
NH
N
O
O
O
NH
N
N
O
O
O
NH
N
O
NH2
N
N
O
NH2
O
O
N
N
O
NH
O
O
OH
O
N
N
O
NH
O
O
O
O
N
N
O
NH
OH
O
NH
O
OH
N
N
O
NH
N
O
NH
NH
O
OMe
N
N
O
NH
N
O
NH
O
OMe
21 % 90 % 61 %
86
193 196
105 194
192
a b c
97 % 50 % 92 %
d e c
61 % 97 %
187 197 195
Boc Boc Boc
+
f c
192
Z
Z Z
Abbildung 73. Synthese von CZ-AcOH (192) (oben) und Boc-Aeg(CZ)-OH (195) (unten). – Reagenzien und Reaktionsbedingungen: (a) Chlorameisensäure-benzylester, DMAP, Pyridin, RT, 4 d; (b) (1) K2CO3, DMF, RT, 10 min; (2) Bromessigsäure-methylester, RT, 16 h; (c) (1) LiOH, THF, H2O, 0 °C → RT, 2 h; (2) 1 M HCl (d) (1) KOtBu, DMF, 100 °C, 2 h; (2) Bromessigsäure-benzylester, 0 °C → RT, 16 h; (3) AcOH, H2O; (e) (1) CDI, THF, Rückfluss, 2 h; (2) Benzylalkohol, Rückfluss → RT, 16 h; (f) TOTU, DIPEA, DMF, RT, 3 h.
Bei der Synthese des Guanin-Monomers 198 wurde mit dem Methylester der Chlorameisen-
säure alkyliert, da die Verwendung des Benzylesters bei der Synthese des analogen
Fmoc/Bhoc-Monomers 110 zu einer schwierigen Abtrennung des Benzylalkohols in der
dritten Stufe geführt hatte (vgl. 4.1.3). Tatsächlich wurde die Isolierung von Guanin-
essigsäure 199 durch diese Variation erheblich vereinfacht. Nach Kopplung zum Rückgrat
187 und Verseifung konnte die Zielverbindung 198 über fünf Stufen mit einer Ausbeute von
6 % erhalten werden (Abbildung 74).

Allgemeiner Teil 4 Synthesen und Spektroskopie
86
NH N
N N
Cl
NH2N N
N N
Cl
NH2O
O
N N
N N
Cl
NH
O
O
N N
N NH
O
NH
OH
O
N N
NNH
O
NH
N
O
NH
O
OMe
N N
NNH
O
NH
N
O
NH
O
OH
N N
NNH
O
NH
N
O
NH2
O
OMe
a: 44 %b: 56 %
49 % 58 %
a oder b c d
59 % 62 %
Z
Boc Boc
Z
Z Z
Z
e f
g
quant.
111 200 201 199
202 198
203
Abbildung 74. Synthese von GZ-AcOH (199) (oben), Boc-Aeg(GZ)-OH (199) (Mitte) und H-Aeg(GZ)-OMe*TFA (203) (unten). – Reagenzien und Reaktionsbedingungen: (a) (1) K2CO3, DMF, 85 °C, 30 min; (2) Bromessigsäure-methylester, 0 °C → RT, 16 h; (3) 1 M HCl; (b) (1) NaH, DMF; (2) Bromessigsäure-methylester; (c) (1) Triphosgen, THF, 0 °C, 1 h; (2) DIPEA, 0 °C, 30 min; (3) Benzylalkohol, 0 °C → RT, 16 h; (d) (1) NaH, 3-Hydroxypropionitril, THF, -78 °C → RT, 3 h; (2) 201, 0 °C → RT, 16 h; (e) TOTU, DIPEA, DMF, RT, 3 h, SC; (f) (1) LiOH, THF, H2O, 0 °C → RT, 2 h; (2) 1 M HCl; (g) TFA, DCM, 0 °C → RT, 2 h.
Zur Synthese des fluoraromatischen Monomers 182 wurde die bereits dargestellte
F-Essigsäure 70 an das geschützte Rückgrat 187 gekoppelt und anschließend die Boc-Gruppe
acidolytisch entfernt (Abbildung 75).
OO NH
OO
F
F
N
O
OO NH
NH
OO
F
F
OH
O
ONH2
O
F
F
N
O+
83 % 55 %
70
187 204 182
a b
*TFA
Abbildung 75. Synthese von H-Aeg(F)-OMe*TFA (182). – Reagenzien und Reaktionsbedingungen: (a) TOTU, DIPEA, DMF, RT, 3 h; (b) TFA, DCM, 0 °C → RT, 2 h.

Allgemeiner Teil 4 Synthesen und Spektroskopie
87
Darüber hinaus wurde Verbindung 205 hergestellt, das Amid von Glycin mit N,N-Dimethyl-
amioethylamin (143), welches zur Modifizierung des C-Teminus dienen sollte (Abbildung
76). Hierzu wurde Boc-geschütztes Glycin 206 zunächst in einer durch EDC vermittelten
Kopplung mit dem Amin 143 umgesetzt und im Anschluss die Schutzgruppe entfernt.
NH
O
NNH
O
ONH2
NOH
O
NH
O
O
NH
O
NNH2+52 % 100 %
* 2TFA206 143 207 205
a b
Abbildung 76. Synthese des C-terminalen Löslichkeitsvermittlers 205 ausgehend von N-Boc-Glycin (206). –Reagenzien und Reaktionsbedingungen: (a) EDC, HOBt, TEA, Chloroform, 0 °C → RT, 16 h; (b) TFA, DCM, 0 °C → RT, 2 h.
Zuerst wurde das Dimer 208a aus Cytosin 195 und Fluoraromat 182 synthetisiert, um so die
Ergebnisse mit denen der Synthese von 186 unter Verwendung der Fmoc/Bhoc-Strategie ver-
gleichen zu können (Abbildung 77). Die Kopplung erfolgte unter Verwendung des sehr effek-
tiven Kopplungsreagenzes HATU (209) und lieferte nach Säulenchromatographie das Ziel-
molekül mit einer Ausbeute von 51 %. Der Wechsel der Schutzgruppenstrategie brachte auf
der Stufe des Dimers 208a einen greifbaren Fortschritt, indem nun das Produkt unter üblichen
Bedingungen chromatographiert werden konnte. Zudem wurde das Dimer 210 aus Adenin
188 und Guanin 203 hergestellt. Dabei wurden nacheinander eine Reihe von Reaktions-
parametern variiert und der Einfluss auf die Effizienz qualitativ durch HPLC-Analyse
bestimmt. Der Wechsel des Lösungsmittels von DMF zu NMP, die Erhöhung der Temperatur
von 25 auf 55 °C und die Zugabe von insgesamt vier Äquivalenten Basen führten zu
sichtbaren Verbesserungen, denen zufolge die Zielverbindung schließlich mit einer Ausbeute
von 40 % erhalten werden konnte.

Allgemeiner Teil 4 Synthesen und Spektroskopie
88
N
N
NH
O
N
O
NH
OH
O
NH2
O
F
F
N
O
OMe NH
O
F
F
N
O
N
N
NH
O
N
O
NH
O
OMe
OHNH
O
N
O
N N
N N
NH
N N
N NH
O
NH
NH2
O
N
O
OMe
N N
N NH
O
NH
N N
N N
NH
NH
O
N
O
NH
O
N
O
OMe
*TFA
+51 %
a
195 182 208a
Z
Boc
Z
Boc
*TFA
+Boc
Z
40 %
b
Z
Z
Z
Boc
188 203 210
Abbildung 77. Konvergente Flüssigphasensynthese I. – Synthese von Boc-Aeg(CZ)-Aeg(F)-OMe (208a) (oben) und Boc-Aeg(AZ)-Aeg(GZ)-OMe (210) (unten). Reagenzien und Reaktionsbedingungen: (a) HATU, DMF, DIPEA, RT, 16 h; (b) HBTU, DIPEA, NMP, 55 °C, 16 h.
Beachtet man, dass hierbei HBTU (151) verwendet wurde, das im Gegensatz zu HATU (209)
nicht zu intramolekularer Basenkatalyse (Abbildung 78) imstande ist, und die Tatsache, dass
purinreiche Sequenzen in besonderem Maße zur Aggregation neigen, ist die erzielte Ausbeute
an 210 durchaus zufriedenstellend.
N
N
NN
OO
N
NH
O
PG
NH
H
NO
OO
PF6
-N
N
NN
O
N
N
BPG
BPG
+
211 212 209
Abbildung 78. Intramolekulare Basenkatalyse bei der Verwendung des Kopplungsreagenzes HATU (209) (links) und seine O- bzw. Uronium-Form (rechts).
Bei der nachfolgenden Synthese des Trimers 213 der Sequenz cfg kam es zu einem
drastischen Einbruch der Kopplungsausbeute auf 19 % nach Reinigung durch HPLC
(Abbildung 79).

Allgemeiner Teil 4 Synthesen und Spektroskopie
89
OHNH
O
F
F
N
O
N
N
NH
O
N
O
NH
O
N N
N NH
O
NH
NH
O
N
O
NH
O
F
F
N
O
N
N
NH
O
N
O
NH
O
OMe
208b 213
Boc
Z
Boc
Z
Z
a
19 %
Abbildung 79. Konvergente Flüssigphasensynthese II: Boc-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OMe (213). – Reagenzien und Reaktionsbedingungen: (a) H-Aeg(GZ)-OMe*TFA (203), TOTU, DIPEA, DMF, 16 h, RT.
In dieser Versuchsreihe wurden sowohl für die analytische als auch für die präparative HPLC
Säulen mit einer Nitrophenyl-Normalphase verwendet. Dieses Material eignet sich insbeson-
dere für die Trennung von Verbindungen mit einer unterschiedlichen Anzahl von aromati-
schen Ringen, da die Nitroaromaten der stationären Phase mit den Aromaten der Analyten
wechselwirken. Interessant ist, welch starken Einfluss das Mischungsverhältnis des Laufmit-
tels hat, insbesondere da es sich bei beiden Komponenten um Alkohole handelt (Abbildung
80). Die zur Verfügung stehende präparative Säule war schon etliche Jahre alt und verlor
unter den Bedingungen der Trennung kontinuierlich geringe Mengen Substanz, sodass alle
erhaltenen Produkte leicht verunreinigt waren. Dies und der Einbruch der ohnehin mäßigen
Kopplungsausbeuten auf dem Weg zum Trimer führten zu einem erneuten Umdenken und zu
einem Wechsel der Synthesestrategie.
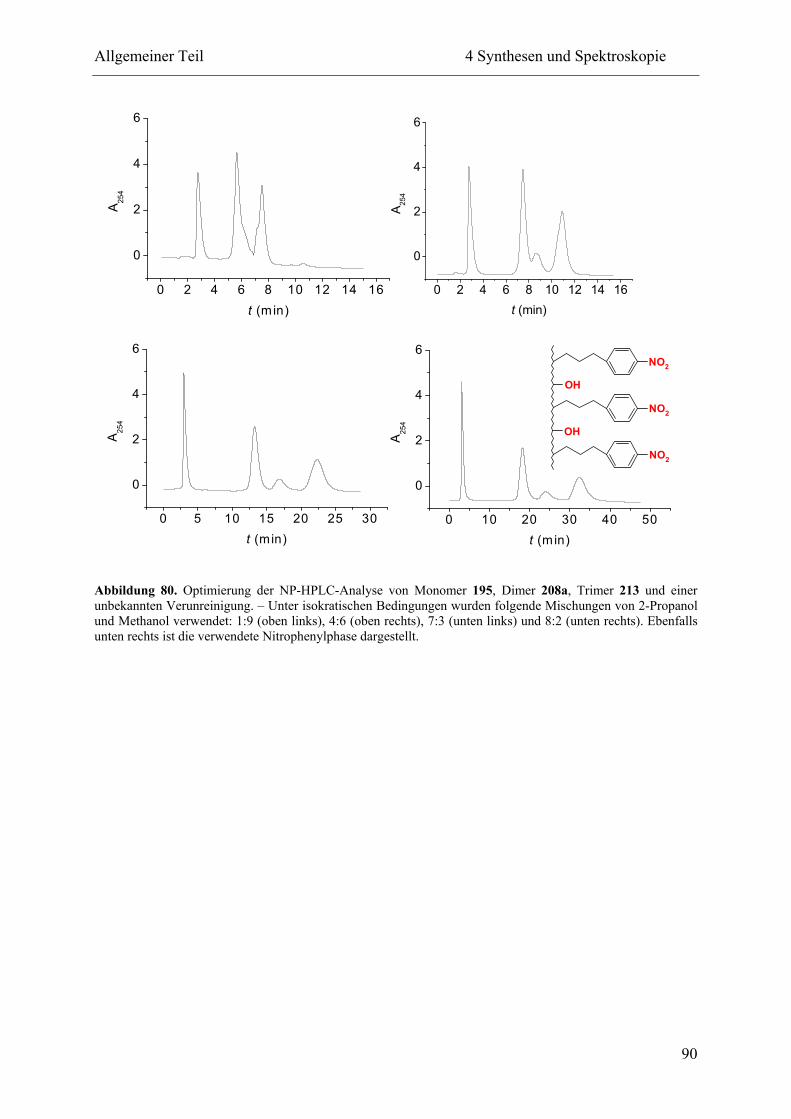
Allgemeiner Teil 4 Synthesen und Spektroskopie
90
Abbildung 80. Optimierung der NP-HPLC-Analyse von Monomer 195, Dimer 208a, Trimer 213 und einer unbekannten Verunreinigung. – Unter isokratischen Bedingungen wurden folgende Mischungen von 2-Propanol und Methanol verwendet: 1:9 (oben links), 4:6 (oben rechts), 7:3 (unten links) und 8:2 (unten rechts). Ebenfalls unten rechts ist die verwendete Nitrophenylphase dargestellt.
0 10 20 30 40 50
0
2
4
6
A2
54
t (min)
OH
OH
NO2
NO2
NO2
0 5 10 15 20 25 30
0
2
4
6
A2
54
t (min)
0 2 4 6 8 10 12 14 16
0
2
4
6A
254
t (min)
0 2 4 6 8 10 12 14 16
0
2
4
6
A25
4
t (min)

Allgemeiner Teil 4 Synthesen und Spektroskopie
91
4.3 Synthesestrategie III: Zugang über ein vollständig geschütztes Rückgrat
4.3.1 Grundlagen
Strategien zur Synthese von PNA in flüssiger Phase sind intensiv von Condom und
Mitarbeitern erarbeitet worden.[234-241] In der Literatur ist nur ein einfaches Beispiel bekannt,
das nicht aus seiner Arbeitsgruppe stammt; dabei handelt es sich um eine Dimerisierung mit
anschließender Cyclisierung.[242] In den Arbeiten von Condom lassen sich insgesamt fünf
Strategien zum Aufbau von PNAs erkennen, die je nach Syntheseproblem kombiniert und
variiert werden:
1. Der einfache konvergente Weg entspricht im Wesentlichen dem im vorherigen
Abschnitt beschriebenen Verfahren: Ein C-und ein N-terminal entschütztes Monomer
werden unter geeigneten Kopplungsbedingungen zum Dimer umgesetzt. Hierbei ist
die Ausbeute der Kopplung gering. Eine weitere Kettenverlängerung führt zu
erneuten Effizienzverlusten.
2. Beim konvergenten Ansatz wird zuerst die Carboxylform eines Monomers 214 mit
dem Alkylester 124 des Rückgrats gekoppelt (Abbildung 81). Daraufhin wird das
sekundäre Amin von 215 mit einer geschützten Nukleobasen-essigsäure 216 zum
tertiären Amid umgesetzt und nachfolgend der Alkylester verseift. Problematisch ist
hier vor allem die Diskriminierung zwischen primärem und sekundärem Amin im
ersten Kopplungsschritt. Außerdem bricht auch hier die Ausbeute mit wachsender
Kettenlänge ein, sodass diese Herangehensweise auf Di- und Trimere beschränkt ist.
3. Der Zugang über ein komplett geschütztes Rückgrat führt die geschützten
Nukleobasen möglichst spät in das Zielmolekül ein, da diese die Hauptursache für
Löslichkeitsprobleme sind (Abbildung 81). Für die Maskierung der sekundären
Amine des Rückgrats benötigt man bei dieser Strategie so viele orthogonale Schutz-
gruppen, wie es unterschiedliche Basen in der Zielverbindung gibt. Diese Schutzgrup-
pen müssen darüber hinaus orthogonal zu denen der exocyclischen Aminofunktionen
der Basen sein. Zusätzlich können die gewünschten Modifizierungen am N- und
C-Terminus den Einsatz von ein oder zwei weiteren Schutzgruppen erforderlich

Allgemeiner Teil 4 Synthesen und Spektroskopie
92
machen. Diese Herangehensweise wird von Condom „Fully Protected Polyamide
Backbone Approach“ genannt und soll im Folgenden durch FPBA abgekürzt werden.
Besonders elegant ist diese Strategie für die Synthese von Homo-Sequenzen, da hier
alle Nukleobasen in einem Schritt eingeführt werden können. Im Weiteren kann man
ausgehend von einem komplett geschützten Rückgrat zu mehreren verschiedenen
Sequenzen gelangen. Später wurde dieser Ansatz auch auf die Festphasensynthese
von PNA übertragen.[243]
4. Eine Erweiterung der letztgenannten Strategie ist der kombinierte Einsatz von ge-
schützten Rückgrat-Einheiten und geschützten PNA-Einheiten. Dies empfiehlt sich,
wenn drei oder mehr verschiedene Nukleobasen und/oder andere Funktionalitäten im
Zielmolekül vorhanden sind, da in diesen Fällen die Anforderungen an die Schutz-
gruppenstrategie enorm steigen.
5. Die Fragmentkondensation von kleinen Einheiten wie Di-, Tri- und Tetrameren zu
größeren Einheiten bietet einen eleganten Zugang zu längeren PNAs. Hierbei sind die
Fragmente selbst durch eine der anderen Herangehensweisen synthetisiert worden.

Allgemeiner Teil 4 Synthesen und Spektroskopie
93
N
O
NH
N
O
NH
N
O
NH
O
n
N
O
NH
N
O
NH
O
N
O
NH
O
n
N
O
NH
O
N
O
NH
O
N
O
NH
O
n
N
O
NH
N
O
NH
N
O
NH
n
ONH
O
NH
N
O
NH
O
NH
O
O
NH2
OHN
O
NH
O
O
OH
ON
O
NH
O
N
O
NH
O
OHN
O
NH
O
N
O
NH
O
N
O
NH
O
N
O
NH
O
N
O
NH
O
n
R1
R2
R1
R2
B(PG)1
R1
R2
B(PG)1 B(PG)2
R1
R2
B(PG)1 B(PG)2 B(PG)n
B(PG)1
B(PG)1
B(PG)1 B(PG)2
B(PG)1 B(PG)2
B(PG)2
R3
R4
B1 B2 Bn
124
214
215
217
218
219
220
221
222
216
223
PG1 PG2 PGn
PG2 PGn
PGn
R1
R1
R1
R1
Abbildung 81. Strategien zum Aufbau kurzer PNAs nach Condom et al. – Konvergenter Ansatz (links) und Zugang über ein vollständig geschütztes Rückgrat (FPBA) (rechts). R1 und R2 können sowohl weitere Schutz-gruppen als auch diverse terminale Modifizierungen sein, die während des Aufbaus der geschützten PNA inert sind. Im Zuge der Entschützung zum Zielmolekül erfahren diese Reste entweder eine Veränderung (z.B. R2 = OMe, R4 = OH) oder bleiben unberührt (z.B. R2 = R4 = NH2).

Allgemeiner Teil 4 Synthesen und Spektroskopie
94
4.3.2 Synthese des C-terminalen Trimers
Auf Basis dieser Arbeiten wurde zunächst ein Syntheseschema für das C-terminale Trimer
224 aufgestellt und dabei der Weg über ein vollständig geschütztes Rückgrat gewählt
(Abbildung 82). PNA 224 weist eine Sequenz aus drei verschiedenen Basen auf, sodass es ein
Rückgrat mit drei orthogonalen Schutzgruppen zu synthetisieren galt. Zum einen fiel die
Wahl auf Fmoc und Boc, da diese komplett orthogonal zueinander sind und ausgiebige
Erfahrungen im Umgang mit diesen Schutzgruppen vorlagen. Zum anderen wurde die Alloc-
Gruppe gewählt, da diese im Arbeitskreis bei der Synthese von Tris-Linkern bereits
erfolgreich verwendet wurde und bei geeigneter Wahl der Abspaltbedingungen komplett
orthogonal zu Boc und Fmoc ist. Im Weiteren sollte der C-Terminus des Rückgrats temporär
als Methylester geschützt werden, bis der Löslichkeitsvermittler eingeführt wird. Dieser
erfuhr eine Modifizierung hin zum Morpholin-Ring, da diese Funktionalität günstigere
Eigenschaften bei den beabsichtigten Normalphasen-Chromatographien versprach. Ort der
positiven Ladung in wässriger Lösung sollte nun das Ring-Stickstoffatom sein. Die
beabsichtigte strukturelle Nähe zur PNA ist auch hier ausreichend gegeben, da
N-Ethyleinheiten ebenfalls Teil der PNA-Struktur sind; die Etherfunktionalität und der
Ringschluss stellen allerdings eine Erweiterung dar. Die Nukleobasen und das Isoster sollten
am Ende der Synthese in Form ihrer Essigsäuren eingeführt werden und wenn nötig Z-
geschützt sein. Daher erschien es sinnvoll auch den N-Terminus auf diese Weise zu schützen,
um die Basen und die Ligationsstelle in einem Schritt entschützen zu können.

Allgemeiner Teil 4 Synthesen und Spektroskopie
95
N
O
N
O
N
O
N
O
N
O
N
O
N
O
N
O
XNH
NR2
OR1
Boc OHAlloc
Alloc
OMe
Alloc
H
Boc
OMeFmoc
Z OH
AllocZ
Boc GZ
AllocZ
Boc H
AllocZ
Boc Fmoc
HZ
Boc GZ
AZ
Z
Boc GZ
AZ
ZH GZ
AZ
ZCZ GZ
AH
C G
N
N
N
N
N
N
N
N
Alloc
ZBoc
OMe
Fmoc
Boc
OMeH
Boc OMe
Boc OMe
Alloc
H
OMe
Fmoc
OMe
H
Boc
Boc
Boc
Z OMe
HZ OMe
H
H OMe
Fmoc
Fmoc
AllocZ
BocOH
Fmoc
187
226
229
231
233
234
235
236
237
238
239
240
241
242
224
187
225124
227 228
230
232
X R2R1
=
X = H, Z, Boc
R1 = H, Boc, Alloc, Fmoc, (geschützte) Nukleobasen-essigsäuren
R2 = OH, OMe, N-CH2-CH2-N(-CH2-CH2-)2O
Abbildung 82. Syntheseschema des C-terminalen Trimers 224. – Der Kasten erläutert die Notation.
Das benötigte Rückgrat 85 wurde nun nicht mehr durch die in Abbildung 51 gezeigte
reduktive Aminierung mit elementarem Wasserstoff dargestellt, da die Kapazitäten der zur
Verfügung stehenden Hydrier-Anlagen zu gering waren und außerdem die Verwendung des
teuren Palladium-Katalysators vermieden werden sollte. Stattdessen wurde Chloressig-
säure (243) über einen Zeitraum von acht Stunden in einen Überschuss an heftig gerührtem
und gekühltem Ethylendiamin (122) eingetragen (Abbildung 83).[222] Typischerweise

Allgemeiner Teil 4 Synthesen und Spektroskopie
96
verlaufen Alkylierungen an Diaminen rasch und mit geringer Selektivität, sodass es zur
Bildung von mono-, bi-, tri- und tetra-alkylierten Produkten kommt.[244] Da sich Chlor-
essigsäure (243) nur langsam in Ethylendiamin (122) löst, kann hier der Anteil an mehrfach
alkyliertem Produkt zurückgedrängt werden. Überschüssiges Ethylendiamin wurde nach einer
Nachrührzeit von 16 Stunden destillativ entfernt und in Folgeansätzen recycelt. Durch Zugabe
von DMSO konnte die Zielverbindung 85 aus dem Rohprodukt ausgefällt werden.
NH2 NH2 OHNH
O
NH2OHCl
O
+a
122 243 8570 %
Abbildung 83. Alternativer Zugang zum PNA-Rückgrat 85. – Reagenzien und Reaktionsbedingungen: (a) (1) Eisbadkühlung, 8 h; (2) RT, 16 h.
Wie beschrieben wurde dann zum Methylester Hydrochlorid 124 umgesetzt. Anschließend
wurde ein Teil dieses Ansatzes durch Reaktion mit Benzyl-N-succinimidyl-carbonat zum
N-terminal Z-geschützten Derivat 227 umgesetzt. Zwei weitere Teile wurden durch Reaktion
mit Boc-Anhydrid in das bereits bekannte Rückgrat 185 überführt. Nachfolgend galt es die
sekundären Amine orthogonal zu schützen, was durch Umsetzung von 227 mit Boc-Anhydrid
und von 185 mit den Chlorameisensäure-estern der 9-Fluorenylmethyl- bzw. Allyl-Gruppe
gelang. Vor der Kopplung zum Di- bzw. Trimer bestand noch die Notwendigkeit, die Ester-
funktionen von 230 und 225 zu verseifen und den N-Terminus von 226 freizusetzen. Bis
hierhin konnten alle beschriebenen Reaktionsschritte in großem Maßstab bei guten bis sehr
guten Ausbeuten durchgeführt werden; eine Reinigung durch Säulenchromatographie war nur
nach der Fmoc-Schützung notwendig. Die orthogonal entschützten Rückgrat-Fragmente 228
und 229 wurden durch Umsetzung mit HBTU (151) und DIPEA in DMF zu Dimer 231
gekoppelt, das nach Chromatographie an Kieselgel in 76%iger Ausbeute als farbloser Schaum
erhalten wurde. Auf die Boc-Abspaltung folgte die Elongation zu Trimer 234 durch Reaktion
mit 232 unter den gleichen Bedingungen. Nach der Aufreinigung ließ sich das vollständig
geschützte Rückgrat 234 mit einer im Vergleich zu 231 deutlich geringeren Ausbeute von
62 % isolieren (Abbildung 84).

Allgemeiner Teil 4 Synthesen und Spektroskopie
97
Boc OHAlloc
Alloc
OMe
Alloc
H
Boc
OMeFmoc
Z OH
a b
c d e
f
(62 %)
(85 %)
g
AllocZ
BocOMe
Fmoc
Boc
OMeH
(76 %)
Boc OMe
Boc OMe
Alloc
H
OMeFmoc
OMe
HBoc
Boc
Boc
Z OMe
HZ OMe
H
H OMe
Fmoc
Fmoc
(86 %)
(63 %) (94 %) (91 %)
(96 %)
(93 %) (99 %)d e
g
187
226
229
231
233
234
187
225124
227 228
230
232
Abbildung 84. Synthese des vollständig geschützten Rückgrats 234. – Reagenzien und Reaktionsbedingungen: (a) Alloc-Cl, TEA, DCM, 0 °C → RT, 2 h; (b) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 4 h, SC; (c) Z-OSu, NMM, MeCN, -15 °C → RT, 5 h; (d) 1 M LiOH, THF, 0 °C → RT, 2 h; (e) TFA, DCM, 0 °C → RT, 2 h, Ausfällen, (f) Boc2O, TEA, DCM, RT, 16 h; (g) HBTU, DIPEA, DMF, RT, 16 h, SC.
Zur Einführung des Löslichkeitsvermittlers wurde zunächst der Methylester von 234 alkalisch
verseift. Obwohl die Fmoc-Gruppe unter den verwendeten Reaktionsbedingungen als stabil
gilt,[245] konnte in der Dünnschichtchromatographie eine quantitative Abspaltung beobachtet
werden, sodass vor der Aufarbeitung erneut geschützt werden musste. Ein vergleichbarer
Effekt ist auch von der Verseifung N-terminal Fmoc-geschützter Monomere bekannt.[246]
Einen möglichen Mechanismus soll Abbildung 85 aufzeigen.
NOH
O
NH
R
OHO
NNH
R
OO
ON
O
O
NH
R
OO
NH
OH
O
NH
R
OH
H2O
-CO2
-
244
234 245 246 247
Abbildung 85. Postulierter Mechanismus für die Fmoc-Abspaltung während der Esterverseifung von 234.
Die Kopplung von 235 mit 2-Morpholinoethylamin (143) wurde zunächst mit HBTU (151)
durchgeführt (Abbildung 86) und führte zu einer mäßigen Ausbeute von 44 %. Daher wurde
ein alternativer Weg gewählt, indem 235 zunächst durch Umsetzung mit DCC und
N-Hydroxysuccinimid in DCM in den entsprechenden N-Hydroxysuccinimidyl-ester über-
führt wurde. Dieser wurde am nächsten Tag bei -15 °C mit dem Amin 143 zum Zielmolekül

Allgemeiner Teil 4 Synthesen und Spektroskopie
98
236 umgesetzt, welches nach Säulenchromatographie mit nunmehr 84 % Ausbeute isoliert
werden konnte.
N
OAlloc
ZBoc Fmoc
Nb
(69 %)
Alloc
ZBoc
OMe
Fmoc
Alloc
ZBoc
OH
Fmoca
(84 %)
234
235
236 Abbildung 86. Vom vollständig geschützten Rückgrat 234 zum C-terminalen Amid 236. – Reagenzien und Reaktionsbedingungen: (a) (1) 1 M LiOH, THF, 0 °C → RT, 2 h; (2) Fmoc-Cl, 0 °C, 1 h, SC; (b) (1) DCC, HOSu, DCM, 0 °C → RT; (2) 4-(2-Aminoethyl)morpholin (143), -15 °C → RT, 4 h, SC.
Nun konnte damit begonnen werden, sukzessiv die sekundären Amine zu entschützen und die
Nukleobasen einzuführen. Den Startpunkt bildete die Fmoc-Gruppe, die durch Umsetzung mit
Diethylamin in DCM abgespalten wurde (Mechanismus siehe Abbildung 61). Die Kopplung
von Guanin-essigsäure 199 wurde zunächst mit HBTU (151) durchgeführt. Da die Ausbeute
mit 53 % aber deutlich hinter den Erwartungen zurückblieb, wurde anschließend das von
Condom vorgeschlagenen Reagenz Brop verwendet, wodurch sich das Ergebnis erheblich
verbessern ließ (Abbildung 87). Dieser Effekt konnte im Folgenden auch bei der Kopplung
der anderen Nukleobasen beobachtet werden.
N
O
N
O
N
O
N
O
N
O
N
O
N
O
AllocZ
Boc GZ
AllocZ
Boc H
AllocZ
Boc Fmoc
H
ZBoc GZ
AZ
ZBoc GZ
AZ
ZH GZ
AZ
ZCZ GZ
N
N
N
N
N
N
N
b
d
f
a
c
e
(79 %)
(86 %)
(61 %)
(67 %)
(47 %)
(95 %)
236
237
238
239
240
241
242
Abbildung 87. Vom komplett aufgebauten Rückgrat 236 zur komplett geschützten PNA 242. – Reagenzien und Reaktionsbedingungen: (a) DEA, DCM, RT, 1.5 h, SC; (b) GZ-AcOH (199), Brop, TEA, DCM, 0 °C → RT, 16 h, SC; (c) Pd[P(Ph)3]4, DEA, DCM, 0 °C → RT, 1 h, SC; (d) AZ-AcOH (190), Brop, TEA, DMF, 0 °C → RT, 16 h, SC; (e) TFA, HSiEt3, DCM, 0 °C → RT, 2 h, Ausfällen; (f) CZ-AcOH (192), Brop, TEA, DMF, 0 °C → RT, 16 h, Ausfällen, RP-HPLC.

Allgemeiner Teil 4 Synthesen und Spektroskopie
99
Im nächsten Schritt wurde die Alloc-Schutzgruppe Palladium-katalysiert entfernt, um darauf-
hin Adenin-essigsäure 190 an das Rückgrat von 239 zu koppeln. Während die Einführung der
ersten Nukleobase noch in DCM durchgeführt werden konnte, musste hier bereits das deutlich
polarere DMF als Lösungsmittel verwendet werden. Auch die abschließende Säulenchromato-
graphie erforderte stark polare Bedingungen, sodass ein Stufengradient ausgehend von einer
Ethylacetat/Methanol-Mischung hin zu purem Methanol verwendet wurde. Die Zielverbin-
dung 240 konnte mit einer Ausbeute von 67 % isoliert werden, die somit knapp 20 % unter
der vorangegangenen Kopplung lag.
Schließlich wurde die Boc-Gruppe durch Behandlung mit TFA in DCM abgespalten. Die
hierbei entstehenden reaktiven tert-Butyl-Kationen können durch Eliminierung zu Isobuten
reagieren oder die Nukleobasen in einer Friedel-Crafts-artigen Reaktion alkylieren
(vgl. Abbildung 62). Letzteres wurde durch die Zugabe des Opfernukleophils Triethylsilan
verhindert. Nachdem mit Cytosin die letzte der drei Nukleobasen in das Zielmolekül
eingeführt worden war, erfolgte die Aufreinigung von 242 durch RP-HPLC, da sich das
Rohprodukt auch in purem Methanol nicht mehr lösen ließ und daher eine Reinigung unter
Normalphasen-Bedingungen nicht mehr möglich war. Auch hier war die erreichte Ausbeute
im Vergleich zur vorangegangenen Kopplung um 20 % reduziert. Die finale acidolytische
Abspaltung der drei Z-Schutzgruppen erfolgte durch Umsetzung mit Trifluormethan-
sulfonsäure in TFA (Abbildung 88). Zum Abfangen der entstehenden Carbokationen wurden
Thioanisol und m-Cresol (169) zugesetzt. Nach einer Reaktionszeit von zwei Stunden wurde
das Rohprodukt durch Zugabe von Diethylether ausgefällt, zentrifugiert und mehrfach mit
Diethylether gewaschen. Damit entsprachen die Reaktionsbedingungen etwa denen, die von
Nielsen in seinen Laboratorien bei der Festphasensynthese verwendet werden. Anschließend
wurde durch RP-HPLC gereinigt und die Zielverbindung in 55%iger Ausbeute isoliert.
N
O
N
O
AZ
Z
CZ GZ
A
H
C G
N
Na
(55 %)
242
224
Abbildung 88. Finale Entschützung zum Trimer 224. – Reagenzien und Reaktionsbedingungen: (a) TFMSA, TFA, m-Cresol, Thioanisol, RT, 2 h, Ausfällen, RP-HPLC.
Eine Alternative zu Trifluormethansulfonsäure ist der ebenfalls stark saure (wasserfreie)
Fluorwasserstoff. Seine Verwendung wurde ausgeschlossen, da er schwerer zu handhaben ist
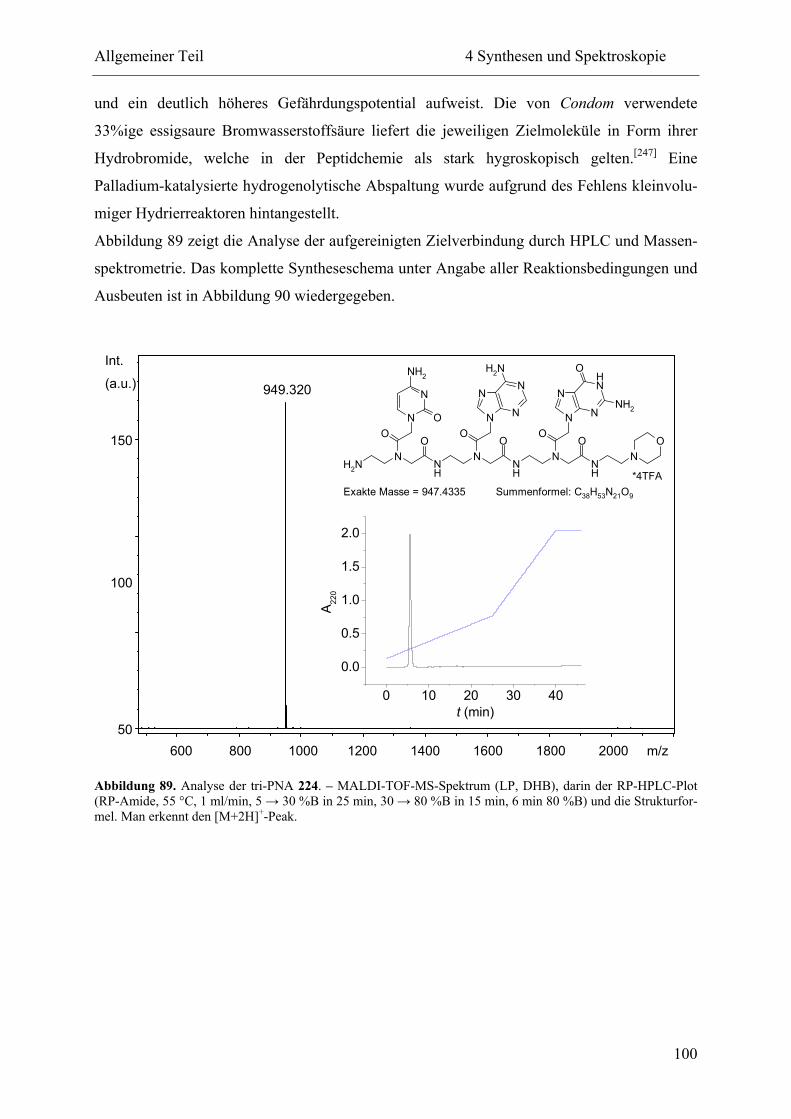
Allgemeiner Teil 4 Synthesen und Spektroskopie
100
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH2
O
N
N
O
NH2
NH
N
O
Exakte Masse = 947.4335 Summenformel: C38H53N21O9
*4TFA
0 10 20 30 40
0.0
0.5
1.0
1.5
2.0
t (min)
A2
20
und ein deutlich höheres Gefährdungspotential aufweist. Die von Condom verwendete
33%ige essigsaure Bromwasserstoffsäure liefert die jeweiligen Zielmoleküle in Form ihrer
Hydrobromide, welche in der Peptidchemie als stark hygroskopisch gelten.[247] Eine
Palladium-katalysierte hydrogenolytische Abspaltung wurde aufgrund des Fehlens kleinvolu-
miger Hydrierreaktoren hintangestellt.
Abbildung 89 zeigt die Analyse der aufgereinigten Zielverbindung durch HPLC und Massen-
spektrometrie. Das komplette Syntheseschema unter Angabe aller Reaktionsbedingungen und
Ausbeuten ist in Abbildung 90 wiedergegeben.
Abbildung 89. Analyse der tri-PNA 224. – MALDI-TOF-MS-Spektrum (LP, DHB), darin der RP-HPLC-Plot (RP-Amide, 55 °C, 1 ml/min, 5 → 30 %B in 25 min, 30 → 80 %B in 15 min, 6 min 80 %B) und die Strukturfor-mel. Man erkennt den [M+2H]+-Peak.
150
100
50
Int.
(a.u.)
600 800 1000 1200 1400 1600 1800 2000 m/z
949.320

Allgemeiner Teil 4 Synthesen und Spektroskopie
101
N
O
N
O
N
O
N
O
N
O
N
O
N
O
N
O
XNH
NR2
OR1
Boc OH
Alloc
Alloc
OMe
Alloc
H
Boc
OMeFmoc
Z OH
AllocZ
Boc GZ
AllocZ
Boc H
AllocZ
Boc Fmoc
HZ
Boc GZ
AZ
Z
Boc GZ
AZ
ZH GZ
AZ
ZCZ GZ
AH
C G
N
N
N
N
N
N
N
N
a b
c
i
d
k
m
e
f
(69 %)
(62 %)
(85 %)
o
g
Alloc
ZBoc
OMe
Fmoc
Boc
OMeH
(76 %)
Boc OMe
Boc OMe
Alloc
H
OMeFmoc
OMe
H
Boc
Boc
Boc
Z OMe
HZ OMe
HH OMe
Fmoc
Fmoc
(86 %)
(63 %) (94 %) (91 %)
(96 %)
(93 %) (99 %)d e
g
AllocZ
BocOH
Fmoch
j
l
n
p
(84 %)
(79 %)
(86 %)
(61 %)
(67 %)
(47 %)
(95 %)
(55 %)
187
226
229
231
233
234
235
236
237
238
239
240
241
242
224
187
225124
227 228
230
232
X R2R1
=
X = H, Z, Boc
R1 = H, Boc, Alloc, Fmoc, (geschützte) Nukleobasen-essigsäuren
R2 = OH, OMe, N-CH2-CH2-N(-CH2-CH2-)2O
Abbildung 90. Syntheseschema des C-terminalen Trimers 224 inklusive Reaktionsbedingungen und Ausbeuten; der Kasten erläutert die Notation. – Reagenzien und Reaktionsbedingungen: (a) Alloc-Cl, TEA, DCM, 0 °C → RT, 2 h; (b) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 4 h, SC; (c) Z-OSu, NMM, MeCN, -15 °C → RT, 5 h; (d) 1 M
LiOH, THF, 0 °C → RT, 2 h; (e) TFA, DCM, 0 °C → RT, 2 h, Ausfällen, (f) Boc2O, TEA, DCM, RT, 16 h; (g) HBTU, DIPEA, DMF, RT, 16 h, SC; (h) (1) 1 M LiOH, THF, 0 °C → RT, 2 h; (2) Fmoc-Cl, 0 °C, 1 h, SC; (i) (1) DCC, HOSu, DCM, 0 °C → RT; (2) 4-(2-Aminoethyl)morpholin (143), -15 °C → RT, 4 h, SC; (j) DEA, DCM, RT, 1.5 h, SC; (k) GZ-AcOH (199), Brop, TEA, DCM, 0 °C → RT, 16 h, SC; (l) Pd[P(Ph)3]4, DEA, DCM, 0 °C → RT, 1 h, SC; (m) AZ-AcOH (190), Brop, TEA, DMF, 0 °C → RT, 16 h, SC; (n) TFA, HSiEt3, DCM, 0 °C → RT, 2 h, Ausfällen; (o) CZ-AcOH (192), Brop, TEA, DMF, 0 °C → RT, 16 h, Ausfällen, RP-HPLC; (p) TFMSA, TFA, m-Cresol, Thioanisol, RT, 2 h, Ausfällen, RP-HPLC.

Allgemeiner Teil 4 Synthesen und Spektroskopie
102
4.3.3 Synthese des N-terminalen Trimers
Die Darstellung des N-terminalen Trimers 248 hätte im Prinzip analog zum C-terminalen
Pendant erfolgen können, da es sich auch in diesem Fall um eine Sequenz aus drei
verschiedenen Basen handelte. Ebenso galt es eine freie Ligationsstelle und einen Löslich-
keitsvermittler einzuführen.
Gerade der Löslichkeitsvermittler gab den Ausschlag ein Schema zu entwickeln, das den
Einsatz von geschützten Rückgrat-Einheiten und einer geschützten PNA-Einheit kombiniert.
Denn im Verlauf des Projekts kam die Idee auf, mehr als eine positive Ladung pro Trimer
einzuführen. Tatsächlich bot es sich gerade in diesem Fragment an, da hier die stark hydro-
phobe Fluorsonde platziert ist. Zusätzlich war es beabsichtigt, sich so nah wie möglich an der
originalen PNA-Struktur zu orientieren und somit kein Chiralitätszentrum einzuführen. Dies
gelang durch die Synthese der vollständig N-methylierten Rückgrateinheit 249, deren
N-Methylgruppen intrinsische PNA-Strukturelemente darstellen (Abbildung 91). Eine mögli-
che Variation stellt das N-ethylierte Derivat 250 dar, das die gleichen Anforderungen erfüllt.
Darüber hinaus kann dieser Ansatz auch noch erweitert werden, indem bereits
oligomerisiertes Rückgrat 251 als Startmaterial eingesetzt wird, sodass pro Einheit eine
zusätzliche Ladung zur Verfügung steht. Zuerst wurde die Methylierung von AEG 85 durch
die Umsetzung mit Formalin-Lösung und Ameisensäure nach Leuckart und Wallach erreicht,
allerdings führte die reduktive Aminierungen unter der Verwendung von elementarem
Wasserstoff zu einer deutlich höheren Ausbeute von 75 % im Vergleich zu 41 %.
NR
O
NH
O
NN n
O
NH
NH2 OH
O
NN OH
O
NN OH
NH
R
O
NH
O
NH
NH2 n
a oder b
*2HCl *2HCla: 41 %b: 75 %
85 249 250
251 252
Abbildung 91. N-terminale Löslichkeitsvermittler auf Basis von AEG (85). – Synthese von Me2-Aeg(Me)-OH (249) (oben links), Struktur der Ethyl-Variante 250 (oben rechts) und möglicher Zugang zu höheren Homologen 252 (unten). Reagenzien und Reaktionsbedingungen: (a) (1) Formalin-Lsg., Ameisensäure, Rückfluss, 16 h; (2) HCl; (b) (1) Formalin-Lsg., H2O, 0 °C → RT, 1 h; (2) H2, Pd/C.

Allgemeiner Teil 4 Synthesen und Spektroskopie
103
N
N
N
XNH
NR2
OR1
Boc OHAlloc
Alloc
OBnAlloc
H
CZ
OBnFmoc
OH
Alloc GZ
Alloc H
Alloc Fmoc
H GZ
F GZ
F GZ
FCZ GZ
FC G
OBn
OBn
OBn
OBn
OBn
OBn
OBn
OH
FmocBoc
OBnH
Boc OMe
Boc OMe
Alloc
H
OBnFmoc
OBn
H
Boc
Boc
Fmoc
OBn
H
H
OHH
H
254
255
256
257
258
259
260
261
262
263
264
265
248
187
253
228
195
85
Boc
CZ
Boc
CZ
Boc
Boc
Boc
Boc
CZ
CZ
CZ
H
Me
Me
OH
Me
OH
H
H
CZ
85
249
X R2R1
=
X = H, Boc, (Me)2
R1 = H, Me, Alloc, Fmoc, (geschützte) Nukleobasen-essigsäuren, F-Essigsäure
R2 = OH, OMe, OBn
225
Abbildung 92. Syntheseschema für die N-terminale tri-PNA 248. – Der Kasten erläutert die Notation.
Wie aus Abbildung 92 zu ersehen ist, wurde für die Synthese des N-terminalen Trimers 248
neben dem Löslichkeitsvermittler auch Cytosin-Monomer 195 gebraucht, welches bereits im
Rahmen der konvergenten Flüssigphasensynthese synthetisiert worden war. Das benötigte
Alloc-geschützte Fragment 228 war aus der Synthese des ersten Trimers bekannt, sodass es
noch ein Fmoc-geschütztes Fragment zu synthetisieren galt. Dessen Carboxylfunktion würde
im fertigen Trimer der Ligationsstelle entsprechen und sollte als Benzylester geschützt
werden. Benzylester sind ebenso wie Z-Gruppen in stark saurer Lösung instabil, daher sollte
es auch hier möglich sein, die Basen und die Ligationsstelle parallel im letzten Schritt zu ent-

Allgemeiner Teil 4 Synthesen und Spektroskopie
104
schützen. Das freie Rückgrat 85 wurde dazu säurekatalysiert mit Benzylalkohol in Toluol
verestert (Abbildung 93). Die Reaktion wurde am Wasserabscheider durchgeführt, um sowohl
das Kristallwasser der eingesetzten para-Toluolsulfonsäure als auch das durch die Reaktion
entstandene Wasser aus dem Gleichgewicht zu entfernen. Wichtig war es hierbei, nicht nur
die Siedetemperatur des Lösungsmittels von 110 °C zu überschreiten, sondern eine konstante
Temperatur von 135-140 °C zu erreichen, da andernfalls keine Reaktion erfolgte. Die
sukzessive Schützung von primärer und sekundärer Aminofunktion mit der Boc- bzw. der
Fmoc-Gruppe erlaubte es, ausgehend vom Benzylester 253, das vollständig geschützte
Fragment 255 mit einer Ausbeute von 41 % herzustellen. Dieses mäßige Ergebnis ließ sich
auf 73 % verbessern, indem das Produkt der ersten Schützung 250 nicht isoliert, sondern in
einem Eintopf-Verfahren direkt weiter zu 255 umgesetzt wurde.
O
NH
NH2 OH
O
NH
NH2 O
O
NNH
O
O
NH
NH
O
c 48 %
a b
*pTsOH
Boc
Fmoc
73 % 84 %
Boc d
73 %
85 253 255
254
Abbildung 93. Zugang zum Alloc-geschützten Fragment 255. – Reagenzien und Reaktionsbedingungen: (a) BnOH, pTsOH, Toluol, 140 °C, 16 h; (b) (1) Boc2O, TEA, DCM, 0 °C → RT, 5 h, SC; (2) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 16 h, SC; (c) Boc2O, TEA, DCM, 0 °C → RT, 16 h, SC; (d) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 16 h, SC.
Danach wurde die Boc-Gruppe abgespalten und das mit nahezu quantitativer Ausbeute
erhaltene Amino-Fragment 256 HBTU-vermittelt mit der Carboxyl-Komponente 228
gekoppelt (Abbildung 94). Nach säulenchromatographischer Aufreinigung ließ sich das
vollständig geschützte Dimer 253 in 73%iger Ausbeute isolieren. Nun wurde die temporäre
Boc-Schutzgruppe abgespalten und das entstandene Amino-Fragment 254 mit dem Cytosin-
Monomer 195 zum trimeren Produkt 259 umgesetzt. Wie im Falle des C-terminalen Trimers
234 kam es hierbei zu einem Einbruch der Kopplungsausbeute, die mit 65 % wiederum gut
10 % unter dem Wert für die Kopplung zum Dimer lag.

Allgemeiner Teil 4 Synthesen und Spektroskopie
105
Boc OHAlloc
Alloc
OBnAlloc
H
CZ
OBn
Fmoc
OH
Alloc FmocOBn
FmocBoc
OBnH
Boc OMeAlloc
OBnFmoc
Boc
Fmoc
255
256
257
258
259
228
195Boc
CZ
Boc
(86 %)(85 %)
(73 %)
(100 %)
(65 %)
a b
c
d
c
225
Abbildung 94. Aufbau von Dimer 257 und Trimer 259. – Reagenzien und Reaktionsbedingungen: (a) 1 M LiOH, THF, 0° → RT, 2 h; (b) TFA, DCM, 0° → RT, 2 h, Ausfällen; (c) HBTU, DIPEA, DMF, RT, 16 h, SC; (d) TFA, HSiEt3, DCM, 0 °C → RT, 2 h.
Hiernach wurde die Fmoc-Gruppe durch Umsetzung mit Diethylamin entfernt und die so
freigesetzte sekundäre Aminofunktion Brop-vermittelt mit Guanin-essigsäure 199 gekoppelt
(Abbildung 95). Die erzielten Ausbeuten entsprachen mit 88 bzw. 72 % den Erwartungen.
Hingegen waren die Ergebnisse der folgenden Abspaltung der Alloc-Gruppe aufgrund der
geringen Ausbeute nicht zufriedenstellend, sodass nach Alternativen zu den von Condom vor-
geschlagenen Bedingungen gesucht wurde.
Alloc GZ
Alloc H
H GZ
OBn
OBn
OBn
OBn
259
260
261
262
CZ
Boc
CZ
Boc
Boc
Boc
CZ
CZ
(86 %)
(72 %)
(57 %)
a
b
c
Alloc Fmoc
Abbildung 95. Von Trimer 259 zu 263. – Reagenzien und Reaktionsbedingungen: (a) DEA, DCM, RT 1.5 h; (b) GZ-AcOH (199), Brop, TEA, DMF, 0 °C → RT, 16 h; (c) Pd[P(Ph)3]4, DEA, DCM, RT, 1.5 h.
Hierbei half ein Blick auf den Mechanismus der Palladium-katalysierten Abspaltung.[248] Als
katalytisch aktive Spezies wird näherungsweise ein koordinativ ungesättigter Palladium(0)-
Komplex wie z.B. Pd(0)L2 angenommen, der im ersten Schritt an die Doppelbindung
koordiniert, bevor er als Nukleophil unter Ausbildung eines π-Allyl-Komplexes reagiert
(Abbildung 96). Nach dieser oxidativen Addition erfolgt durch den Angriff des Nukleophils
eine reduktive Eliminierung, bei der der Katalysator zurückgebildet wird.

Allgemeiner Teil 4 Synthesen und Spektroskopie
106
NR2
NR2
Nu
Nu -
Pd(0)
L L
NR2
-
Pd(II)
L L
Pd0L2
+
Abbildung 96. Katalysecyclus der Palladium-vermittelten Alloc-Abspaltung.
Setzt man kein externes Nukleophil als Abfangreagenz hinzu, reagiert das freigesetzte Amin
mit dem π-Allyl-Komplex und es entsteht ein Allylamin als Ergebnis einer decarboxylativen
Umlagerung (Abbildung 97). Dieser Prozess verläuft schneller bei sekundären als bei
primären Aminen. Der Einsatz des sekundären Diethylamis (DEA) als Akzeptor, wie Condom
ihn vorschlägt, führt demnach zu einer Situation, in der Substrat und DEA um die Allyl-
Gruppe konkurrieren. Daher ist es folgerichtig DEA in hohem Überschuss einzusetzen. Auch
die Wahl einer sterisch unanspruchsvollen Verbindung ist sinnvoll, um eine rasche Reaktion
zu begünstigen. Jedoch muss man hinterfragen, ob und wie weit sich die entschützte
PNA (-NR2) überhaupt vom Palladium-Komplex entfernt, bevor es zur reduktiven
Eliminierung kommt, also ob das zugesetzte DEA überhaupt ausreichende Gelegenheit
bekommt, in der gewünschten Art zu wirken.
C
O
O- Pd0L2- CO2
Pd(II)
L LR1R2N R1R2N
+
Abbildung 97. Bildung von Allylaminen in Abwesenheit eines Abfangreagenzes.
Erschwerend kommt hinzu, dass der Allyl-Abfang reversibel ist, da Allylammonium-
Verbindungen in der Lage sind, ihre Allylfunktion auf Nukleophile zu übertragen und es
daher zur Rückreaktion kommen kann (Abbildung 98). Hingegen sind C-Opfernukleophile in
der Lage, die Allylfunktion irreversibel abzufangen. Hier eignen sich CH-acide Verbindungen

Allgemeiner Teil 4 Synthesen und Spektroskopie
107
wie Dimedon (266) (pKa = 5.3) oder N,N’-Dimethylbarbitursäure (267) (pKa = 4.7), die zu
ihren entsprechenden Mono- oder Diallylderivaten reagieren.
NuH Nu
NH
O
NH
NuH
O O
N N
O
OO
EH
EH2C
O
OH EH
C
O
OHEH2
Pd(II)
L LPd
(II)
L L
EH Pd(II)
L L
COO
Pd0 Pd0
- CO2
+ + +
+
mit Nu =
mit Nu = E =
+ + -
- CO2
+ +
-
+
- Pd0L2
R1R2NCO2 R1R2N+H R1R2HN+
R1R2N
R1R2N-
R1R2N
R1R2N R1R2NH R1R2NH+EH-
Nu-
266 267
Abbildung 98. Einfluss des Opfernukleophils auf den Reaktionsverlauf der Alloc-Abspaltung. – Reversible Übertragung von Allylgruppen durch Allylamoniumsalze (oben) und irreversibler Abfang durch CH-acide Verbindungen (unten).
Für die Alloc-Entschützung wurde nachfolgend N,N’-Dimethylbarbitursäure (267) verwendet,
da Dimedon (266) dazu neigt, hydrolytisch stabile Ketoenamine zu bilden. So ließ sich die
Ausbeute in diesem Schritt von 57 auf 88 bzw. 90 % steigern (Abbildung 99). Mit der
folgenden Einführung von F-Essigsäure 70 war der Aufbau der Basensequenz komplett,
sodass die temporäre Boc-Schutzgruppe säurekatalysiert entfernt werden konnte. Das
resultierende Produkt 264 wurde mit dem Löslichkeitsvermittler 249 in Form seines
N-Hydroxysuccinimidyl-esters zum geschützten Trimer 265 umgesetzt. Nach Aufreinigung
durch präparative RP-HPLC konnte dieses mit 46 % Ausbeute isoliert werden.

Allgemeiner Teil 4 Synthesen und Spektroskopie
108
N
N
Alloc GZ
H GZ
F GZ
F GZ
FCZ GZ
OBn
OBn
OBn
OBn
OBn
261
262
263
264
265
Boc
Boc
Boc
CZ
CZ
CZ
H
Me
OHMe
OHH
H
CZ
85
249
(88 %)
(75 %)
(88 %)
(46 %)
a
b
dc
e
(75 %)
Abbildung 99. Einführung von F-Essigsäure 70 und Löslichkeitsvermittler 249. – Reagenzien und Reaktionsbe-dingungen: (a) Pd[P(Ph)3]4, NDMBA, THF, 0° → RT, 2 h; (b) F-AcOH (70), Brop, TEA, DMF, 0 °C → RT, 16 h; (c) (1) Formalin-Lsg., H2O, 0 °C → RT, 1 h; (2) H2, Pd/C; (d) TFA, HSiEt3, DCM, 0 °C → RT, 2 h; (e) (1) 249, DCC, HOSu, DCM, 0° → RT; (2) 264, -15° → RT, 4 h.
Die nachfolgende Abspaltung der Z-Gruppen und des Benzylesters erfolgte auch hier mit
Trifluormethansulfonsäure in Gegenwart von Thioanisol und m-Cresol (169) in TFA. Die
Ausbeute an F-PNA-Trimer 248 nach Reinigung durch RP-HPLC betrug 56 %; die
Analysedaten sind in Abbildung 100 gezeigt. Die PNA liegt als dreifaches TFA-Salz vor
(vgl. Abbildung 122), was einer Protonierung der beiden Aminen des Löslichkeitsvermittlers
und der Position N3 (pKa = 4.17) des Cytosins entspricht. Die Position N7 des Guanins ist
unter den gegebenen Bedingungen nicht protoniert (pKa = 3.3).[249] Abbildung 101 gibt das
komplette Syntheseschema unter Angabe aller Reagenzien und Reaktionsbedingungen
wieder.
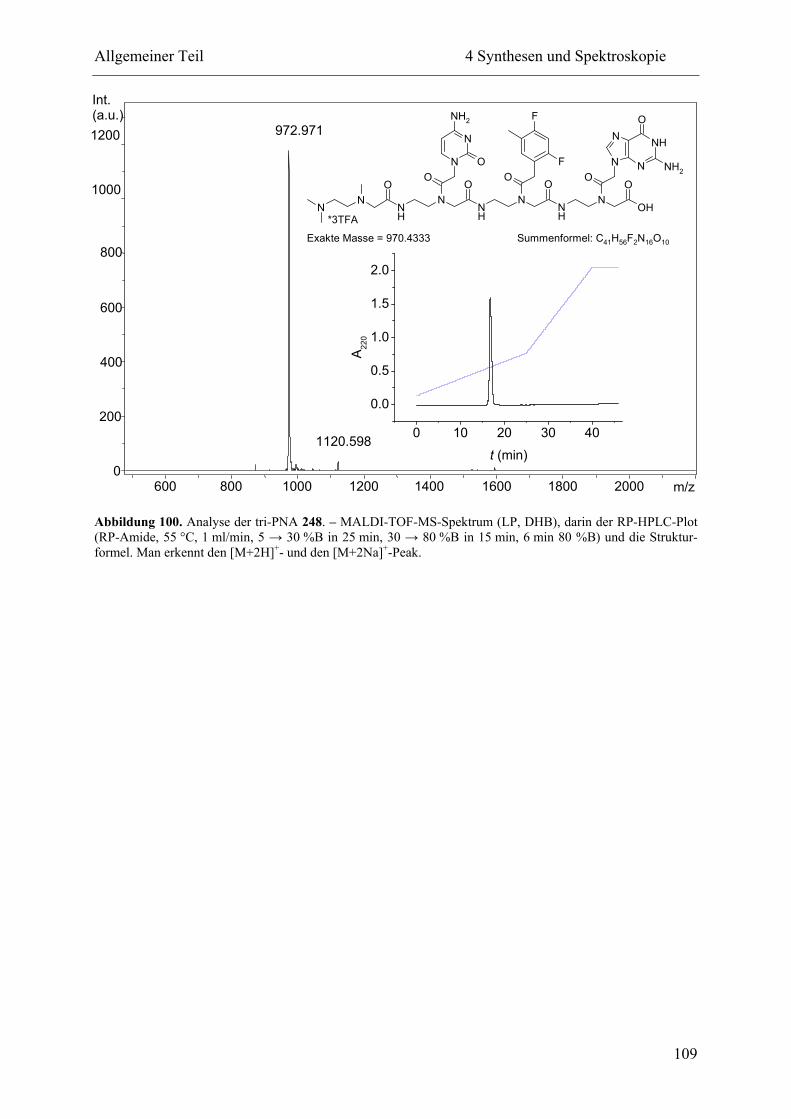
Allgemeiner Teil 4 Synthesen und Spektroskopie
109
Abbildung 100. Analyse der tri-PNA 248. – MALDI-TOF-MS-Spektrum (LP, DHB), darin der RP-HPLC-Plot (RP-Amide, 55 °C, 1 ml/min, 5 → 30 %B in 25 min, 30 → 80 %B in 15 min, 6 min 80 %B) und die Struktur-formel. Man erkennt den [M+2H]+- und den [M+2Na]+-Peak.
972.971
0
200
400
600
800
600 800 1000 1200 1400 1600 1800 2000 m/z
1120.598
N N
NNH
O
NH2
NH
N
O
NH
NNH
NOH
OOO
N
N
O
NH2
O
F
F
OO
NN
*3TFA
Exakte Masse = 970.4333 Summenformel: C41H56F2N16O10
0 10 20 30 40
0.0
0.5
1.0
1.5
2.0
A2
20
t (min)
1200
1000
Int. (a.u.)

Allgemeiner Teil 4 Synthesen und Spektroskopie
110
N
N
N
XNH
NR2
OR1
Boc OHAlloc
Alloc
OBnAlloc
H
CZ
OBnFmoc
OH
Alloc GZ
Alloc H
Alloc Fmoc
H GZ
F GZ
F GZ
FCZ GZ
FC G
OBn
OBn
OBn
OBn
OBn
OBn
OBn
OH
FmocBoc
OBnH
Boc OMe
Boc OMe
Alloc
H
OBn
Fmoc
OBn
H
Boc
Boc
Fmoc
OBn
H
H
OH
H
H
254
255
256
257
258
259
260
261
262
263
264
265
248
187
253
228
195
85
Boc
CZ
Boc
CZ
Boc
Boc
Boc
Boc
CZ
CZ
CZ
H
Me
Me
OH
Me
OHH
H
CZ
85
249
(86 %)
(86 %)
(86 %)
(85 %)
(85 %)
(73 %)
b'(48 %)
(73 %)
(100 %)
(65 %)
(86 %)
(72 %)
(88 %)
(75 %)
(88 %)
(46 %)
a
b
c d
e f
g
h
g
i
j
k
j
hl
m
n
(75 %)
(56 %)
X R2R1
=
X = H, Boc, (Me)2
R1 = H, Me, Alloc, Fmoc, (geschützte) Nukleobasen-essigsäuren, F-Essigsäure
R2 = OH, OMe, OBn
225
Abbildung 101. Syntheseschema des N-terminalen Trimers 248 inklusive Reaktionsbedingungen und Ausbeuten; der Kasten erläutert die Notation. – Reagenzien und Reaktionsbedingungen: (a) BnOH, pTsOH, Toluol, 140 °C, 16 h; (b) Boc2O, TEA, DCM, 0 °C → RT, 16 h, SC; (b’) (1) Boc2O, TEA, DCM, 0 °C → RT, 5 h, SC; (2) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 16 h, SC; c) Alloc-Cl, TEA, DCM, 0 °C → RT, 2 h; (d) Fmoc-Cl, DIPEA, DCM, 0 °C → RT, 16 h, SC; (e) 1 M LiOH, THF, 0° → RT, 2 h; (f) TFA, DCM, 0° → RT, 2 h, Ausfällen; (g) HBTU, DIPEA, DMF, RT, 16 h, SC; (h) TFA, HSiEt3, DCM, 0 °C → RT, 2 h; (i) DEA, DCM, RT 1.5 h; (j) GZ-AcOH (199) oder F-AcOH (70), Brop, TEA, DMF, 0 °C → RT, 16 h; (k) Pd[P(Ph)3]4, NDMBA, THF, 0 °C → RT, 2 h; (l) (1) Formalin-Lsg., H2O, 0 °C → RT, 1 h; (2) H2, Pd/C; (m) (1) 249, DCC, HOSu, DCM, 0 °C → RT; (2) 264, -15 °C → RT, 4 h; (n) TFMSA, TFA, m-Cresol, Thioanisol, RT, 2 h, Ausfällen, RP-HPLC.

Allgemeiner Teil 4 Synthesen und Spektroskopie
111
4.4 NMR-Spektroskopie
4.4.1 PNA und das Rotamer-Problem
Die tertiäre Amidbindung einer PNA-Einheit ist in der Lage zwei verschiedene Konformatio-
nen anzunehmen (Abbildung 102). So kann die Carbonylgruppe des Essigsäure-Linkers
entweder in Richtung des C-Terminus zeigen oder von ihm abgewandt sein. Im ersten Fall
spricht man vom cis-Rotamer, im zweiten vom trans-Rotamer. Dabei ist die Rotation um die
Amidbindung derart gehindert, dass der Austausch zwischen beiden Formen bei Raum-
temperatur langsam im Verhältnis zur NMR-Zeitskala ist. Dies gilt auch für Carbamat-
Schutzgruppen am sekundären Amin. Durch temperaturabhängige 1H-NMR-Messungen an
vier Monomeren und einem Dimer wurde für die Aktivierungsenergie der cis-trans-
Isomerisierung ein Wert von 19±2 kcal mol-1 bestimmt, die Austauschrate bei 37 °C beträgt
0.5-2 s-1.[250]
B
N
O
R
O
NH
Hn N
R
O
NH
Hn
O
BR
N+
O
RR
R
N
O
RR
Abbildung 102. Gehinderte Rotation um die tertiäre Amidbindung (links) und ihre mesomeren Grenzstrukturen.
In Komplexen mit DNA[251, 252], RNA[253] oder einem anderen PNA-Strang[115] liegen alle
tertiären Amide in der cis-Konformation vor, wohingegen Monomere und ungepaarte PNAs
in einem Gemisch aus trans- und cis-Rotameren existieren. Die Anzahl der in Lösung be-
findlichen unterschiedlichen Rotamere lässt sich durch 2n berechnen, wobei n die Anzahl der
PNA-Einheiten ist. In jedem Rotamer erfahren die Kerne eine Änderung ihrer magnetischen
Umgebung und damit ihrer chemischen Verschiebung. Weil diese Änderung abhängig von
der Entfernung des entsprechenden Kerns zum Ursprung der Rotation ist, lassen sich im
Spektrum nicht zwingend alle theoretisch vorliegenden Rotamere beobachten. Beispielsweise
erkennt man bei terminalen Gruppen wie N-Boc meist nur zwei unterschiedliche Signale, die
durch die Rotation des nächstliegenden Amids verursacht werden, obwohl aufgrund der
Sequenzlänge vier oder acht unterschiedliche Rotamere in Lösung vorliegen. Da typischer-
weise bereits diese beiden Signale überlappen, ist der Effekt weiter entfernter Rotations-

Allgemeiner Teil 4 Synthesen und Spektroskopie
112
zentren in einer Linienverbreiterung zu suchen, die oftmals die präzise Integration der
Spektren erschwert. Trotz dieser Effekte sind die NMR-Spektren von Monomeren (n = 1,
2 Rotamere) meist gut interpretierbar, wohingegen die Komplexität der Daten bereits auf der
Stufe des Trimers (n = 3, 8 Rotamere) eine detaillierte Analyse ausschließt. Des Weiteren
entstehen Probleme bei der praktischen Durchführung der Messungen, da das Auftreten von
mehrfachen Signalsätzen die Signalintensität herabsetzt. Daher muss, bei angenommener
gleichmäßiger Population aller Rotamere, entweder eine n-fach höher konzentrierte Probe
bereitgestellt oder die n-fache Messzeit aufgewandt werden. In der Regel sind die verschie-
denen Rotamere aber ungleich populiert, wodurch sich der Aufwand für ein gutes Signal-
Rausch-Verhältnis nochmals erhöht. Dadurch nimmt besonders die Aufnahme von relevanten 13C-NMR-Spektren viel Zeit in Anspruch und entzieht sich damit der Routineanalytik.
Abbildung 103 zeigt Ausschnitte eines 13C-NMR-Spektrums vom geschützten trimeren
Rückgrat 234, das mit 10835 Scans aufgenommen wurde. Trotz dieses hohen Aufwands lässt
sich in vielen Fällen nicht eindeutig zwischen Grundrauschen und Signal unterscheiden. In
den bisherigen Arbeiten zur Flüssigphasen-Synthese kurzer PNAs wurden keine 13C-NMR-
Daten angegeben[234, 235] bzw. diese nicht interpretiert[236, 239]; darüber hinaus wurden die 1H-NMR-Signale nur zum Teil zugeordnet. Im Zuge der in den vorangegangenen Abschnitten
vorgestellten Synthesen kam der Wunsch auf, sich diesem Problem zu nähern, da gerade die
zahlreichen Möglichkeiten zur genauen Analytik als großer Vorteil der Synthese in flüssiger
Phase erkannt wurden.

Allgemeiner Teil 4 Synthesen und Spektroskopie
113
Abbildung 103. 13C-NMR-Spektrum von 234. – Aufgenommen in DMSO-d6 bei 30 °C (100 MHz, 10835 Scans, 40 mg Substanz in 600 µl Lösungsmittel). Die Vergrößerungen zeigen den Bereich der Carbonyl-signale von Amid- bzw. Ester-Funktionen (links) und Carbamat-Funktionen (rechts).
NH
N
OOO
NH
NO
OOO
O
NNH
O
OOO

Allgemeiner Teil 4 Synthesen und Spektroskopie
114
4.4.2 Hochtemperatur-NMR-Spektroskopie
Eine Lösung des beschriebenen Problems versprach die Erhöhung der Messtemperatur, da
hierdurch die Rotationsbarrieren leichter überwunden werden und folglich die Rotationsraten
zunehmen. Daher wurden NMR-Experimente bei variabler Temperatur durchgeführt und die
Änderungen der Linienformen beobachtet. Typischerweise ließen sich dabei im Bereich des
langsamen Austauschs für einen Kern zwei oder mehr breite Linien beobachten, die sich bei
Temperaturerhöhung in einem Übergangsbereich weiter verbreiterten und zunehmend über-
lappten, um schließlich am Koaleszenzpunkt in ein breites flaches Signal überzugehen. Dieses
wurde in einem weiteren Übergangsbereich immer schmaler, bis es schließlich im Bereich des
schnellen Austauschs die Form einer scharfen Linie annahm. Solch ein Experiment wurde
zunächst für das vollständig geschützte Rückgrat 234 durchgeführt, bei dem alle während der
Synthese relevanten Schutzgruppen anwesend sind. Abbildung 104 zeigt Spektrenausschnitte,
die im Abstand von jeweils 10 °C aufgenommen worden sind. Die Ausschnitte zeigen
relevante Signale von Fmoc-, Alloc- und Boc-Schutzgruppe.
Abbildung 104. Ausschnitte aus 1H-NMR-Spektren (250 MHz, DMSO-d6) von 234 bei verschiedenen Temperaturen. – FmocCHarom. und Amid-NHs (links), AllocCH2CH=CH2 (Mitte) und BocCH3 (rechts).

Allgemeiner Teil 4 Synthesen und Spektroskopie
115
Tabelle 2 gibt die im Rahmen der vorliegenden Daten abgeschätzten Temperaturen für
Koaleszenz und schnellen Austausch der drei Schutzgruppen an. Die Koaleszenztemperatur
Tc ist allerdings keine Konstante, sondern eine von der Messfrequenz abhängige Größe, die
sich näherungsweise bei verdoppelter Messfrequenz um 10 °C erhöht.
Tabelle 2. Temperaturen für Koaleszenz und schnellen Austausch der Boc-, Alloc- und Fmoc-Gruppe von 234.
Schutzgruppe Koaleszenztemp. Tc (°C) schneller Austausch (°C)
Boc 40 80
Alloc 60 110
Fmoc 70 110
Man erkennt deutlich, dass die Gruppen unterschiedliche Koaleszenztemperaturen aufweisen,
die sowohl mit den jeweiligen sterischen Eigenschaften als auch mit den Positionen im
Molekül korrelieren. Wie man erwarten kann, hat die sterisch sehr anspruchsvolle Fmoc-
Gruppe mit 70 °C die höchste Koaleszenztemperatur. Betrachtet man nun die verbleibenden
beiden Gruppen isoliert, ist die Boc-Gruppe aufgrund ihrer Struktur unbeweglicher und
sterisch anspruchvoller als die Alloc-Gruppe. Dieser Effekt wird jedoch durch die mit der
Positionierung am C-Terminus gewonnenen Flexibilität derart überkompensiert, dass sie mit
40 °C den geringsten Wert für Tc aufweist. Ab 110 °C kommt es zur thermisch induzierten
Abspaltung der Fmoc-Gruppe, deren Fortgang sich durch das Erscheinen eines Singuletts bei
6.2 ppm verfolgen lässt, das den terminalen olefinischen Protonen des Fmoc-Fulvens
entspricht (vgl. Abbildung 61).
Die Entwicklungen aller Signale der Alloc-Gruppe sind in Abbildung 105 dargestellt. Aus
den zunächst undefinierten Signalen entstehen bei 110 °C genau die Kopplungsmuster, die
man erwarten kann. Auch das Signal-Rausch-Verhältnis des 13C-NMR-Spektrums verbessert
sich durch hohe Messtemperaturen merklich, sodass die Carbonylregion problemlos analysiert
werden kann (Abbildung 106). Eine Analyse des Methylenbereichs (nicht gezeigt) ist wegen
der hohen Anzahl ähnlicher Signale nicht in Gänze möglich.
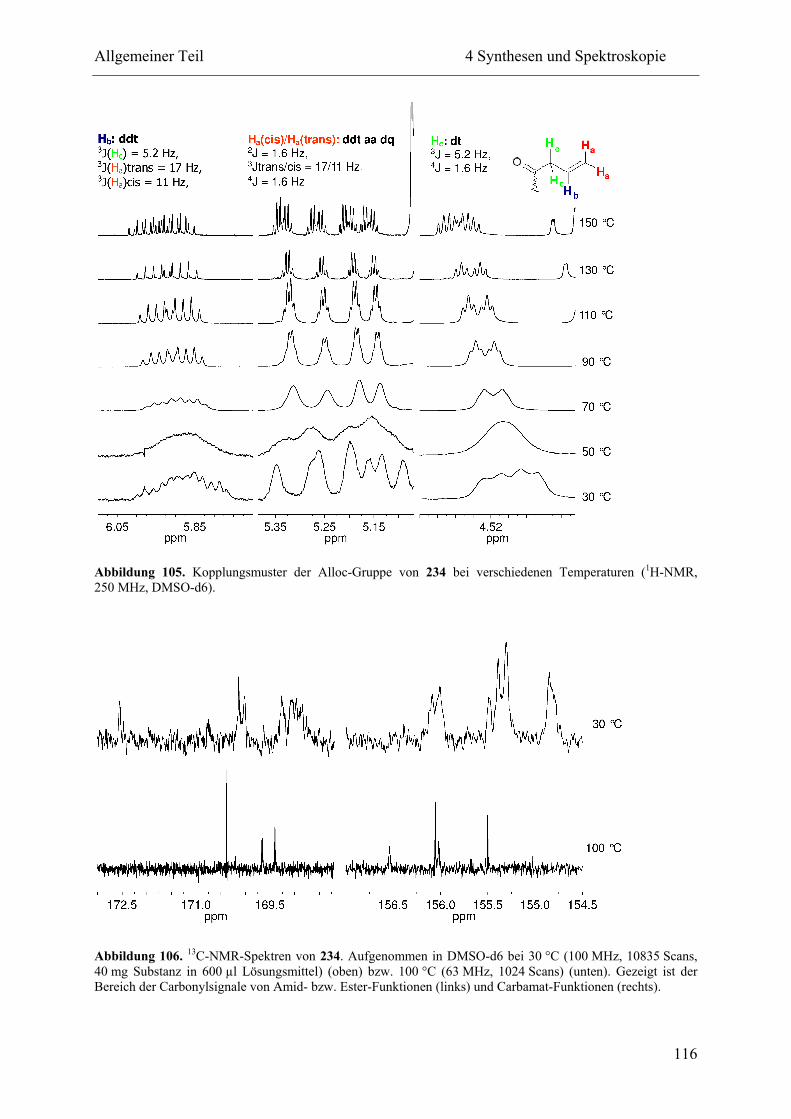
Allgemeiner Teil 4 Synthesen und Spektroskopie
116
Abbildung 105. Kopplungsmuster der Alloc-Gruppe von 234 bei verschiedenen Temperaturen (1H-NMR, 250 MHz, DMSO-d6).
Abbildung 106. 13C-NMR-Spektren von 234. Aufgenommen in DMSO-d6 bei 30 °C (100 MHz, 10835 Scans, 40 mg Substanz in 600 µl Lösungsmittel) (oben) bzw. 100 °C (63 MHz, 1024 Scans) (unten). Gezeigt ist der Bereich der Carbonylsignale von Amid- bzw. Ester-Funktionen (links) und Carbamat-Funktionen (rechts).
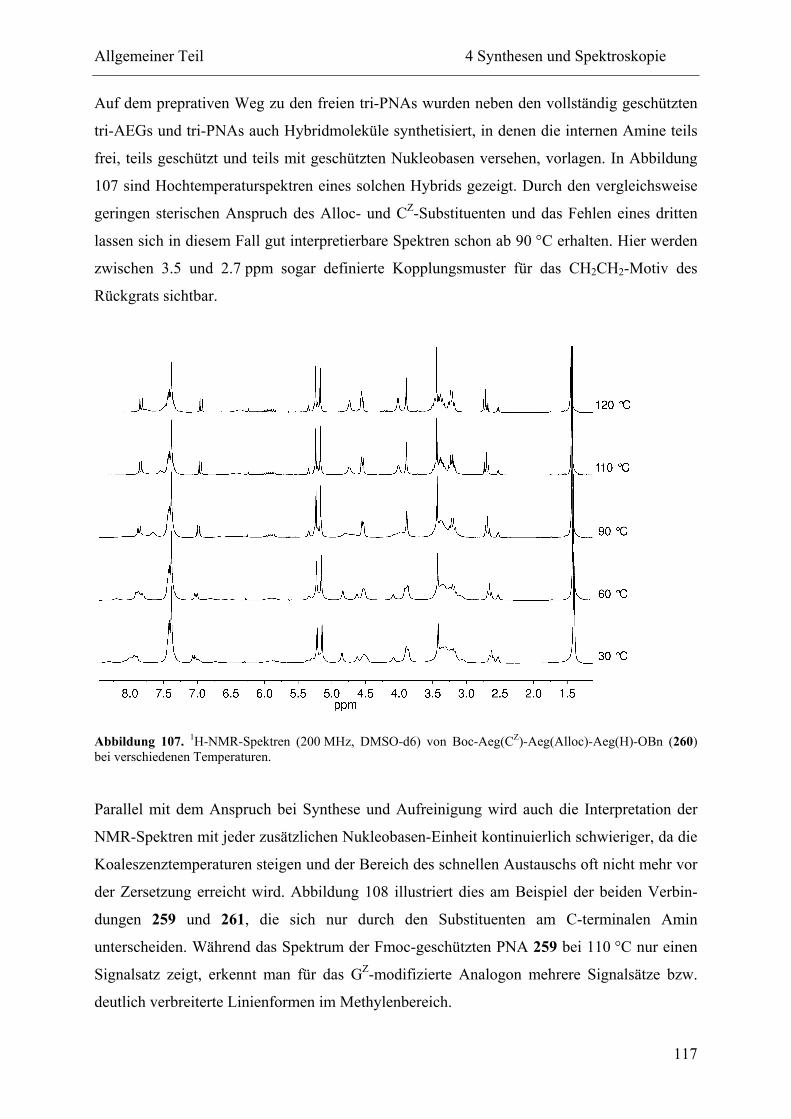
Allgemeiner Teil 4 Synthesen und Spektroskopie
117
Auf dem preprativen Weg zu den freien tri-PNAs wurden neben den vollständig geschützten
tri-AEGs und tri-PNAs auch Hybridmoleküle synthetisiert, in denen die internen Amine teils
frei, teils geschützt und teils mit geschützten Nukleobasen versehen, vorlagen. In Abbildung
107 sind Hochtemperaturspektren eines solchen Hybrids gezeigt. Durch den vergleichsweise
geringen sterischen Anspruch des Alloc- und CZ-Substituenten und das Fehlen eines dritten
lassen sich in diesem Fall gut interpretierbare Spektren schon ab 90 °C erhalten. Hier werden
zwischen 3.5 und 2.7 ppm sogar definierte Kopplungsmuster für das CH2CH2-Motiv des
Rückgrats sichtbar.
Abbildung 107. 1H-NMR-Spektren (200 MHz, DMSO-d6) von Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(H)-OBn (260) bei verschiedenen Temperaturen.
Parallel mit dem Anspruch bei Synthese und Aufreinigung wird auch die Interpretation der
NMR-Spektren mit jeder zusätzlichen Nukleobasen-Einheit kontinuierlich schwieriger, da die
Koaleszenztemperaturen steigen und der Bereich des schnellen Austauschs oft nicht mehr vor
der Zersetzung erreicht wird. Abbildung 108 illustriert dies am Beispiel der beiden Verbin-
dungen 259 und 261, die sich nur durch den Substituenten am C-terminalen Amin
unterscheiden. Während das Spektrum der Fmoc-geschützten PNA 259 bei 110 °C nur einen
Signalsatz zeigt, erkennt man für das GZ-modifizierte Analogon mehrere Signalsätze bzw.
deutlich verbreiterte Linienformen im Methylenbereich.
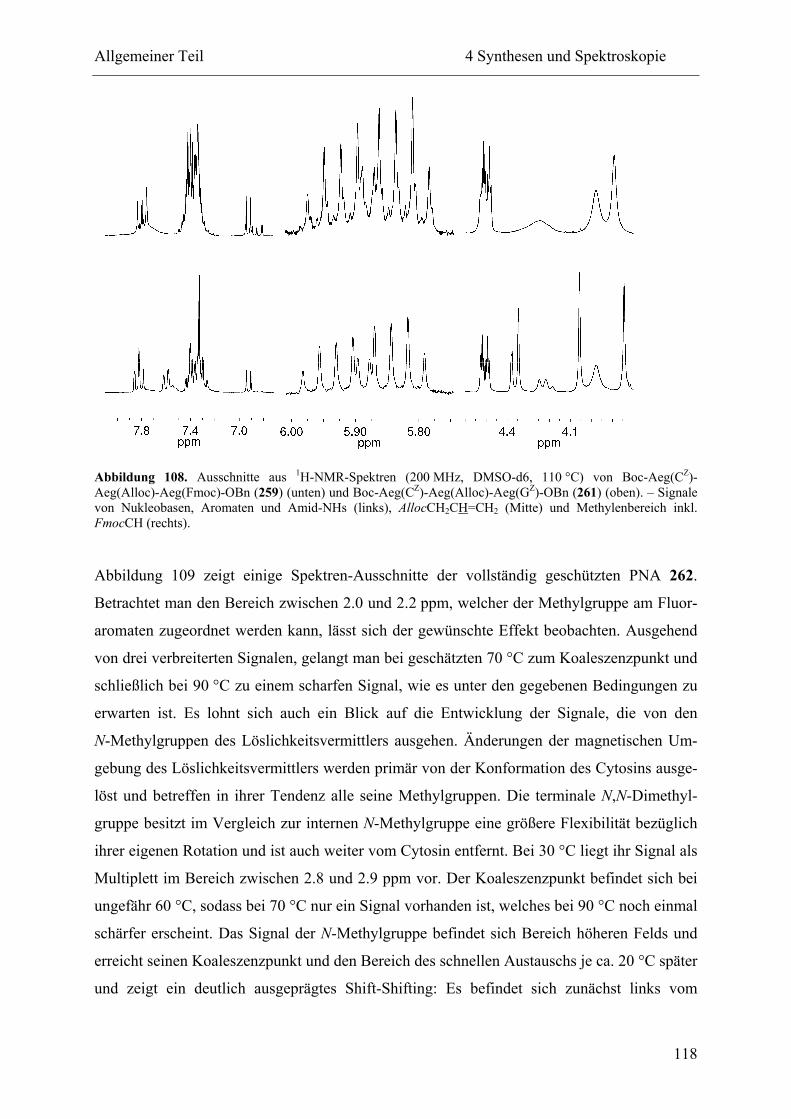
Allgemeiner Teil 4 Synthesen und Spektroskopie
118
Abbildung 108. Ausschnitte aus 1H-NMR-Spektren (200 MHz, DMSO-d6, 110 °C) von Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(Fmoc)-OBn (259) (unten) und Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(GZ)-OBn (261) (oben). – Signale von Nukleobasen, Aromaten und Amid-NHs (links), AllocCH2CH=CH2 (Mitte) und Methylenbereich inkl. FmocCH (rechts).
Abbildung 109 zeigt einige Spektren-Ausschnitte der vollständig geschützten PNA 262.
Betrachtet man den Bereich zwischen 2.0 und 2.2 ppm, welcher der Methylgruppe am Fluor-
aromaten zugeordnet werden kann, lässt sich der gewünschte Effekt beobachten. Ausgehend
von drei verbreiterten Signalen, gelangt man bei geschätzten 70 °C zum Koaleszenzpunkt und
schließlich bei 90 °C zu einem scharfen Signal, wie es unter den gegebenen Bedingungen zu
erwarten ist. Es lohnt sich auch ein Blick auf die Entwicklung der Signale, die von den
N-Methylgruppen des Löslichkeitsvermittlers ausgehen. Änderungen der magnetischen Um-
gebung des Löslichkeitsvermittlers werden primär von der Konformation des Cytosins ausge-
löst und betreffen in ihrer Tendenz alle seine Methylgruppen. Die terminale N,N-Dimethyl-
gruppe besitzt im Vergleich zur internen N-Methylgruppe eine größere Flexibilität bezüglich
ihrer eigenen Rotation und ist auch weiter vom Cytosin entfernt. Bei 30 °C liegt ihr Signal als
Multiplett im Bereich zwischen 2.8 und 2.9 ppm vor. Der Koaleszenzpunkt befindet sich bei
ungefähr 60 °C, sodass bei 70 °C nur ein Signal vorhanden ist, welches bei 90 °C noch einmal
schärfer erscheint. Das Signal der N-Methylgruppe befindet sich Bereich höheren Felds und
erreicht seinen Koaleszenzpunkt und den Bereich des schnellen Austauschs je ca. 20 °C später
und zeigt ein deutlich ausgeprägtes Shift-Shifting: Es befindet sich zunächst links vom

Allgemeiner Teil 4 Synthesen und Spektroskopie
119
DMSO-Signal, kreuzt es bei 70 °C und ist schließlich ab 90 °C rechts von ihm zu erkennen.
Indirekt lässt sich durch diese Betrachtungen die Koaleszenztemperatur des geschützten
Cytosins auf 80 °C abschätzen. Diese deckt sich mit der Analyse der Signale für CZ-C(6)H
und CZ-C(5)H bei 7.82 bzw. 6.91 ppm. Schließlich taucht ab 110 °C bei ca. 4.5 ppm ein
Signal auf, das aus einer nicht bekannten Zersetzung des Moleküls herrührt. Zu diesem Zeit-
punkt hat sich das Spektrum bereits vereinfacht, aber es liegen immer noch breite Signale für
die CH2-Gruppen der Glycin- und Linker-Einheiten (5.1-3.6 ppm) und eine benzylische CH2-
Gruppe (5.20 ppm) vor (Abbildung 110). Zusammenfassend ließ sich die Analyse der
vollständig geschützten PNA 262 vereinfachen. So konnten einige Signale zugeordnet werden
und es war möglich, erste Aussagen über die Koaleszenzpunkte der geschützten Basen bzw.
des Isosters zu treffen. Einer vollständigen Analyse stand die Zersetzung des Moleküls vor
der optimalen Messtemperatur entgegen.
Abbildung 109. Ausschnitte aus 1H-NMR-Spektren (200 MHz, DMSO-d6) von 262 bei verschiedenen Tem-peraturen. – Signale der Nukleobasen und Aromaten (links), der Methylen-Protonen (Mitte) und der Methyl-Protonen inkl. eines Methylen-Signals (rechts).

Allgemeiner Teil 4 Synthesen und Spektroskopie
120
Abbildung 110. 1H-NMR-Spektrum (200 MHz, DMSO-d6) der geschützten N-terminalen tri-PNA 262 bei 110 °C. – Zuordnung der Signale: δ = 8.62 (s(br), div. N+R3); 8.08 (s(br), 1 H, NH); 7.76 (m, 5 H, C-C(6)H, FCHarom., G-C(8)H, 2 × NH); 7.41 - 7.34 (m, 15 H, 10 × ZCHarom., 5 × BnCHarom.), 7.12 (t(br), 1 H, NH); 6.89 (m, 2 H, C-C(6)H, FCHarom.); 5.27 (s, 2 H, OCH2Ph); 5.21 (s, 2 H, OCH2Ph); 5.18 (s(br), 2 H, OCH2Ph); 5.02 (s(br), 2 H, BaseCH2); 4.69 (s(br), 2 H, BaseCH2); 4.24 (s(br), 2 H, BaseCH2); 4.02 (s(br), 4 H, 2 × GlyCH2); 3.63 - 3.20 (m(br), 18 H, 2 × GlyCH2, 3.5 × CH2CH2); darunter das Signal für Wasserreste aus dem LM; 2.90 (t, 3J = 6.0, 2 H, 0.5 × CH2CH2); 2.84 (s, 6 H, N(CH3)3); 2.40 (s, 3 H, NCH3); 2.12 (s(br), 3 H, FCH3).
Abbildung 111 zeigt das Spektrum der C-terminalen geschützten tri-PNA 242. Auch hier fällt
der scharfe Peak bei 4.5 ppm auf, der aus der beginnenden Zersetzung des Moleküls stammt.
Der mehrfache Signalsatz für die Wasserstoffatome an C(2) und C(8) des Adenins weisen auf
eine gehinderte Rotation dieser Nukleobase hin, die sich aus ihrem sterischen Anspruch und
der Position in der Sequenzmitte erklären lässt. Im Gegensatz hierzu liegt das Signal für das
Wasserstoffatom an C(5) des Cytosins als scharfes Dublett vor. Auch die Resonanzen für das
C(6)H des Cytosins und das C(8)H des Guanins lassen nur einen Signalsatz erkennen, obwohl
die Signale überlappen. Diese Situation spiegelt sich auch in der Linienform der Resonanzen
wieder, die von den an die Basen gebundenen Methylengruppen stammen. Es lassen sich drei
Linien unterschiedlicher Höhe und Breite erkennen, die auf Basis der Analyse der aromati-
schen Protonen zugeordnet werden können. Das schärfste und daher intensivste Signal lässt
sich der an das Cytosin gebundenen Methylengruppe zuordnen, das breiteste der drei Signale
sollte vom Methylencarbonyl-Linker am Adenin stammen und das Signal mittlerer Breite und
Intensität vom Linker am Guanin. Zwischen 8.1 und 7.6 und bei 6.4 ppm erkennt man sehr
N N
NNH
O
NH
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH
O
O
O
F
F
OO
NN
O
O
*2TFA
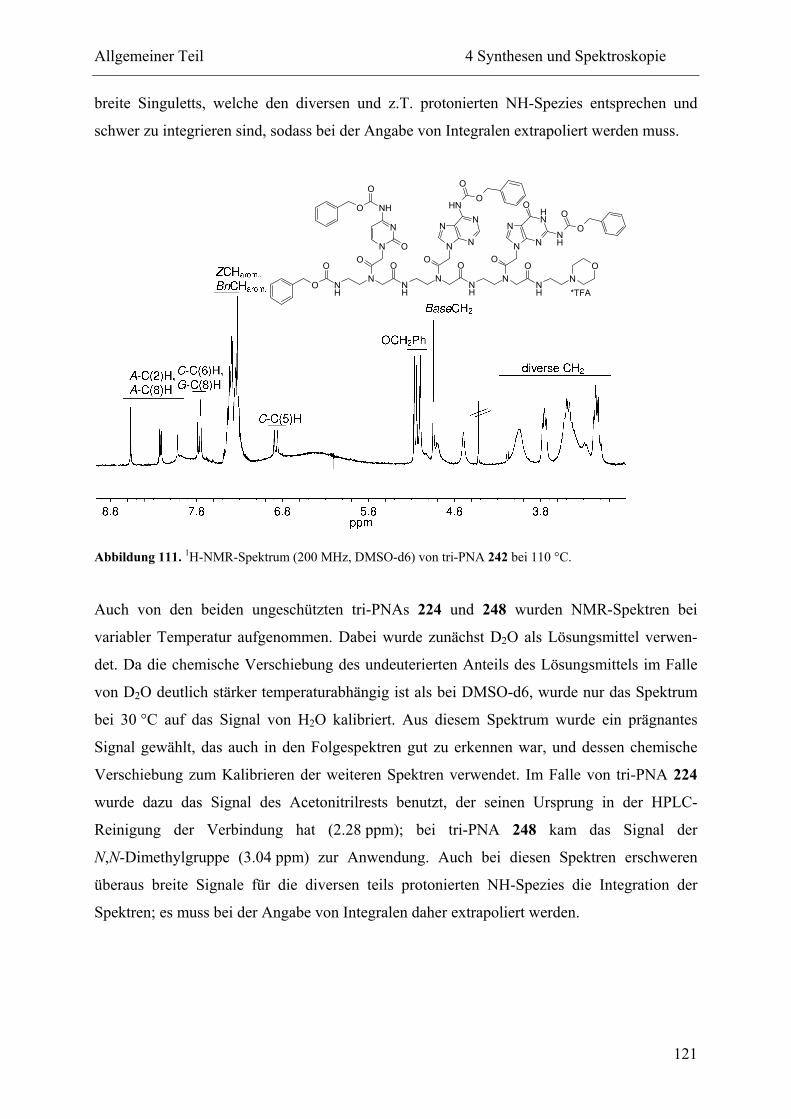
Allgemeiner Teil 4 Synthesen und Spektroskopie
121
breite Singuletts, welche den diversen und z.T. protonierten NH-Spezies entsprechen und
schwer zu integrieren sind, sodass bei der Angabe von Integralen extrapoliert werden muss.
Abbildung 111. 1H-NMR-Spektrum (200 MHz, DMSO-d6) von tri-PNA 242 bei 110 °C.
Auch von den beiden ungeschützten tri-PNAs 224 und 248 wurden NMR-Spektren bei
variabler Temperatur aufgenommen. Dabei wurde zunächst D2O als Lösungsmittel verwen-
det. Da die chemische Verschiebung des undeuterierten Anteils des Lösungsmittels im Falle
von D2O deutlich stärker temperaturabhängig ist als bei DMSO-d6, wurde nur das Spektrum
bei 30 °C auf das Signal von H2O kalibriert. Aus diesem Spektrum wurde ein prägnantes
Signal gewählt, das auch in den Folgespektren gut zu erkennen war, und dessen chemische
Verschiebung zum Kalibrieren der weiteren Spektren verwendet. Im Falle von tri-PNA 224
wurde dazu das Signal des Acetonitrilrests benutzt, der seinen Ursprung in der HPLC-
Reinigung der Verbindung hat (2.28 ppm); bei tri-PNA 248 kam das Signal der
N,N-Dimethylgruppe (3.04 ppm) zur Anwendung. Auch bei diesen Spektren erschweren
überaus breite Signale für die diversen teils protonierten NH-Spezies die Integration der
Spektren; es muss bei der Angabe von Integralen daher extrapoliert werden.
N
N
O
NH
N
O
N N
NN
NH
O
O
NH
O
NH
NNH
NNH
OOO
ON
OO
N N
NNH
O
NH
O
O
O
O
O
*TFA
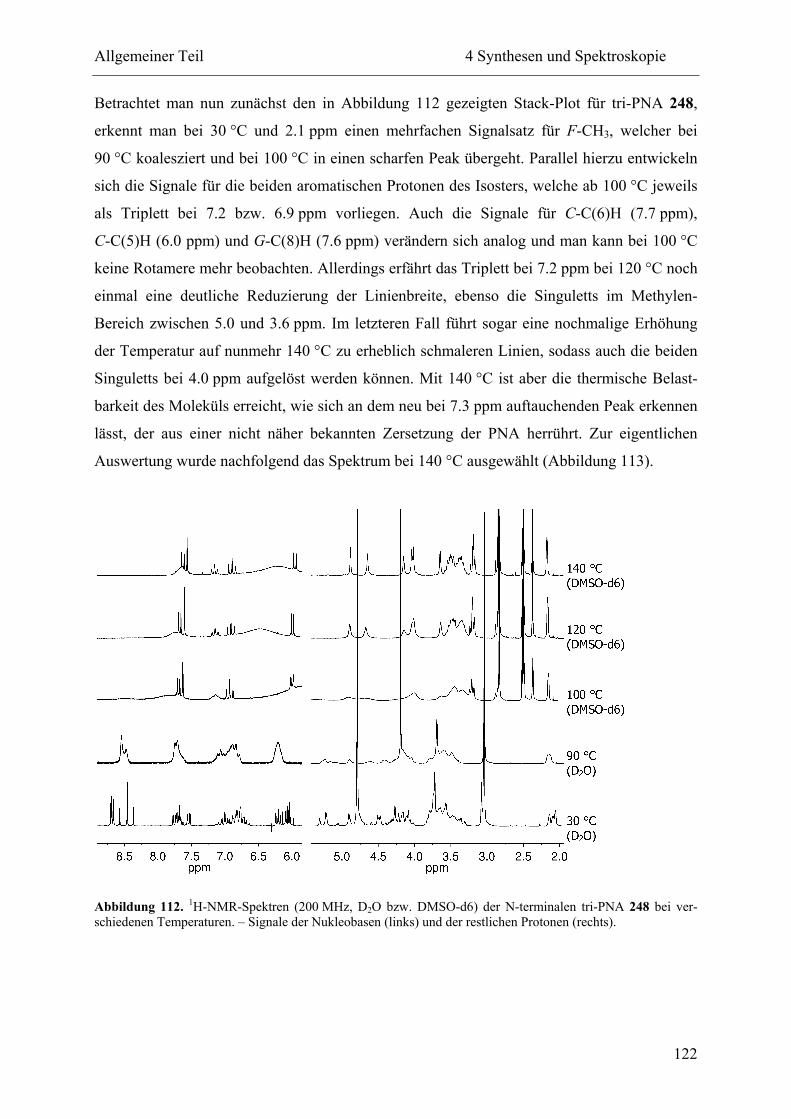
Allgemeiner Teil 4 Synthesen und Spektroskopie
122
Betrachtet man nun zunächst den in Abbildung 112 gezeigten Stack-Plot für tri-PNA 248,
erkennt man bei 30 °C und 2.1 ppm einen mehrfachen Signalsatz für F-CH3, welcher bei
90 °C koalesziert und bei 100 °C in einen scharfen Peak übergeht. Parallel hierzu entwickeln
sich die Signale für die beiden aromatischen Protonen des Isosters, welche ab 100 °C jeweils
als Triplett bei 7.2 bzw. 6.9 ppm vorliegen. Auch die Signale für C-C(6)H (7.7 ppm),
C-C(5)H (6.0 ppm) und G-C(8)H (7.6 ppm) verändern sich analog und man kann bei 100 °C
keine Rotamere mehr beobachten. Allerdings erfährt das Triplett bei 7.2 ppm bei 120 °C noch
einmal eine deutliche Reduzierung der Linienbreite, ebenso die Singuletts im Methylen-
Bereich zwischen 5.0 und 3.6 ppm. Im letzteren Fall führt sogar eine nochmalige Erhöhung
der Temperatur auf nunmehr 140 °C zu erheblich schmaleren Linien, sodass auch die beiden
Singuletts bei 4.0 ppm aufgelöst werden können. Mit 140 °C ist aber die thermische Belast-
barkeit des Moleküls erreicht, wie sich an dem neu bei 7.3 ppm auftauchenden Peak erkennen
lässt, der aus einer nicht näher bekannten Zersetzung der PNA herrührt. Zur eigentlichen
Auswertung wurde nachfolgend das Spektrum bei 140 °C ausgewählt (Abbildung 113).
Abbildung 112. 1H-NMR-Spektren (200 MHz, D2O bzw. DMSO-d6) der N-terminalen tri-PNA 248 bei ver-schiedenen Temperaturen. – Signale der Nukleobasen (links) und der restlichen Protonen (rechts).
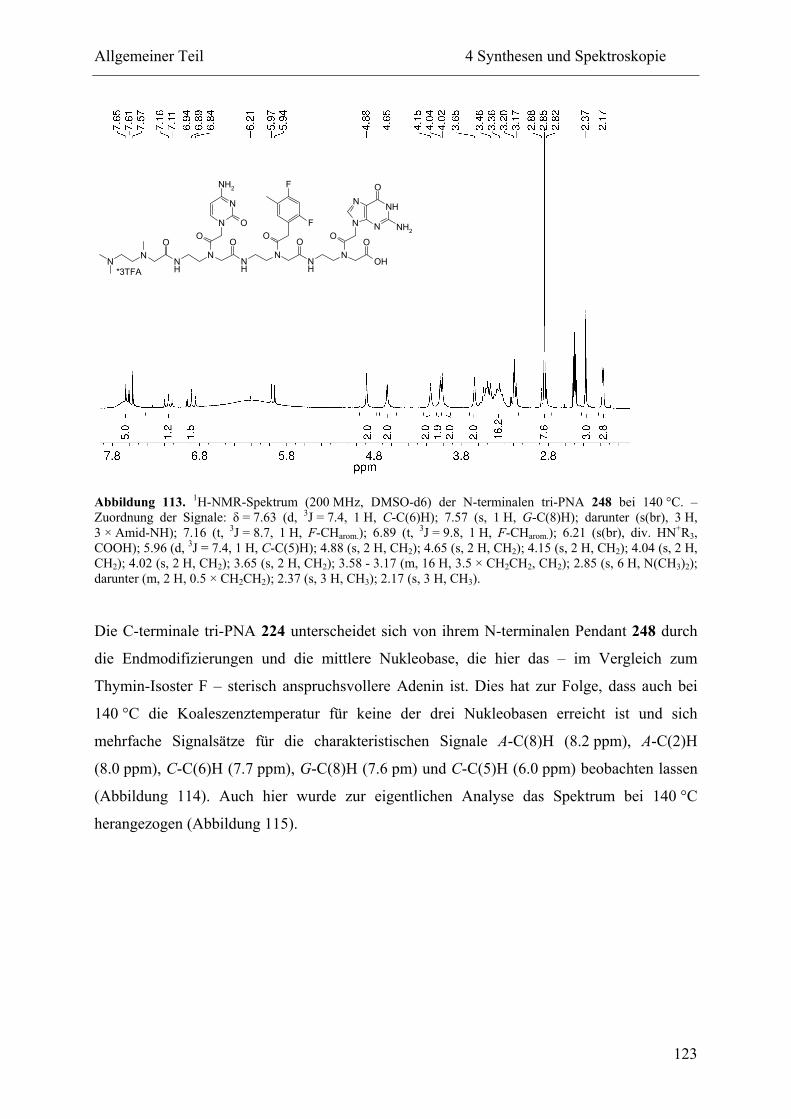
Allgemeiner Teil 4 Synthesen und Spektroskopie
123
Abbildung 113. 1H-NMR-Spektrum (200 MHz, DMSO-d6) der N-terminalen tri-PNA 248 bei 140 °C. – Zuordnung der Signale: δ = 7.63 (d, 3J = 7.4, 1 H, C-C(6)H); 7.57 (s, 1 H, G-C(8)H); darunter (s(br), 3 H, 3 × Amid-NH); 7.16 (t, 3J = 8.7, 1 H, F-CHarom.); 6.89 (t, 3J = 9.8, 1 H, F-CHarom.); 6.21 (s(br), div. HN+R3, COOH); 5.96 (d, 3J = 7.4, 1 H, C-C(5)H); 4.88 (s, 2 H, CH2); 4.65 (s, 2 H, CH2); 4.15 (s, 2 H, CH2); 4.04 (s, 2 H, CH2); 4.02 (s, 2 H, CH2); 3.65 (s, 2 H, CH2); 3.58 - 3.17 (m, 16 H, 3.5 × CH2CH2, CH2); 2.85 (s, 6 H, N(CH3)2); darunter (m, 2 H, 0.5 × CH2CH2); 2.37 (s, 3 H, CH3); 2.17 (s, 3 H, CH3).
Die C-terminale tri-PNA 224 unterscheidet sich von ihrem N-terminalen Pendant 248 durch
die Endmodifizierungen und die mittlere Nukleobase, die hier das – im Vergleich zum
Thymin-Isoster F – sterisch anspruchsvollere Adenin ist. Dies hat zur Folge, dass auch bei
140 °C die Koaleszenztemperatur für keine der drei Nukleobasen erreicht ist und sich
mehrfache Signalsätze für die charakteristischen Signale A-C(8)H (8.2 ppm), A-C(2)H
(8.0 ppm), C-C(6)H (7.7 ppm), G-C(8)H (7.6 pm) und C-C(5)H (6.0 ppm) beobachten lassen
(Abbildung 114). Auch hier wurde zur eigentlichen Analyse das Spektrum bei 140 °C
herangezogen (Abbildung 115).
N N
NNH
O
NH2
NH
N
O
NH
NNH
NOH
OOO
N
N
O
NH2
O
F
F
OO
NN
*3TFA
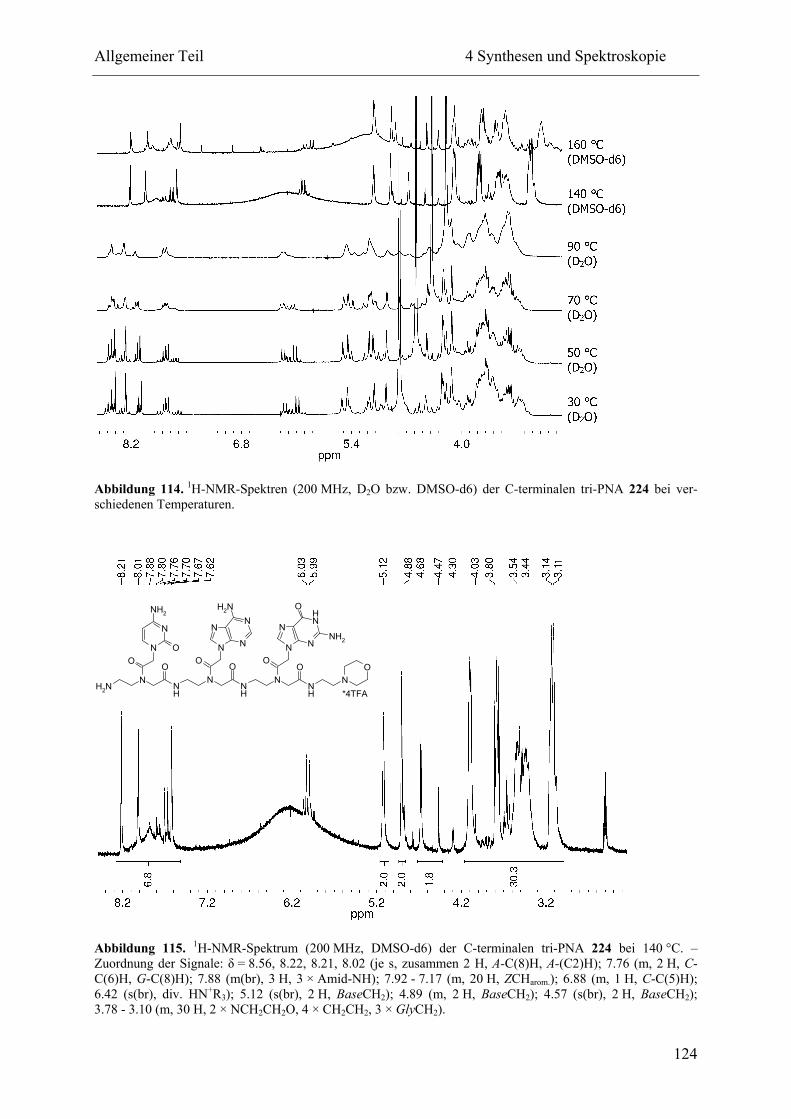
Allgemeiner Teil 4 Synthesen und Spektroskopie
124
Abbildung 114. 1H-NMR-Spektren (200 MHz, D2O bzw. DMSO-d6) der C-terminalen tri-PNA 224 bei ver-schiedenen Temperaturen.
Abbildung 115. 1H-NMR-Spektrum (200 MHz, DMSO-d6) der C-terminalen tri-PNA 224 bei 140 °C. – Zuordnung der Signale: δ = 8.56, 8.22, 8.21, 8.02 (je s, zusammen 2 H, A-C(8)H, A-(C2)H); 7.76 (m, 2 H, C-C(6)H, G-C(8)H); 7.88 (m(br), 3 H, 3 × Amid-NH); 7.92 - 7.17 (m, 20 H, ZCHarom.); 6.88 (m, 1 H, C-C(5)H); 6.42 (s(br), div. HN+R3); 5.12 (s(br), 2 H, BaseCH2); 4.89 (m, 2 H, BaseCH2); 4.57 (s(br), 2 H, BaseCH2); 3.78 - 3.10 (m, 30 H, 2 × NCH2CH2O, 4 × CH2CH2, 3 × GlyCH2).
N
N
O
NH2
N
O
N N
NN
NH2
NH2
O
NH
NNH
NNH
OO
N
OO
N N
NNH
O
NH2
O
*4TFA

Allgemeiner Teil 4 Synthesen und Spektroskopie
125
Die Grenzen der verwendeten Methode liegen in der thermischen Belastbarkeit der analysier-
ten Substanzen und der verwendeten Geräte. Denn Temperaturen oberhalb von 100 °C
schränken nicht nur die Wahl des Lösungsmittels ein, sondern auch die des zu verwendenden
Spektrometers. Von den in Bochum vorhandenen Geräten konnte zunächst das Modell
DRX-250 der Firma Bruker verwendet werden, bis dieses im Laufe der Arbeit einen moder-
nen Probenkopf mit Gradientenspulen erhielt. Die Spezifikationen erlaubten keine weiteren
Messungen oberhalb von ca. 80 °C, demzufolge wurden spätere Messungen am Modell
DPX-200 durchgeführt. Daneben kann es zu chemischen Veränderungen des Analyten
kommen – wie zur Abspaltungen von Schutzgruppen und daraus resultierenden Folgereakti-
onen. Diese chemischen Modifizierungen können zum einen bereits vor dem Erreichen des
schnellen chemischen Austauschs an allen Rotationszentren auftreten. Zum anderen können
sie im Bereich des schnellen Austauschs langsam z.B. während einer 13C-NMR-Messung
stattfinden. Besonders betroffen davon sind Moleküle mit ungeschützten funktionellen
Gruppen wie das aus 234 gewonnene Carbonsäurederivat 235. Während vom Methylester 234
sogar ein 13C-NMR-Spektrum bei 100 °C aufgenommen werden konnte (Messzeit ca. 1 h),
setzte die Abspaltung der Fmoc-Gruppe aus 235 so rasch ein, dass nicht einmal ein Protonen-
Spektrum gewonnen werden konnte. Der Mechanismus der Abspaltung folgt vermutlich dem
in Abbildung 85 dargestellten Weg.
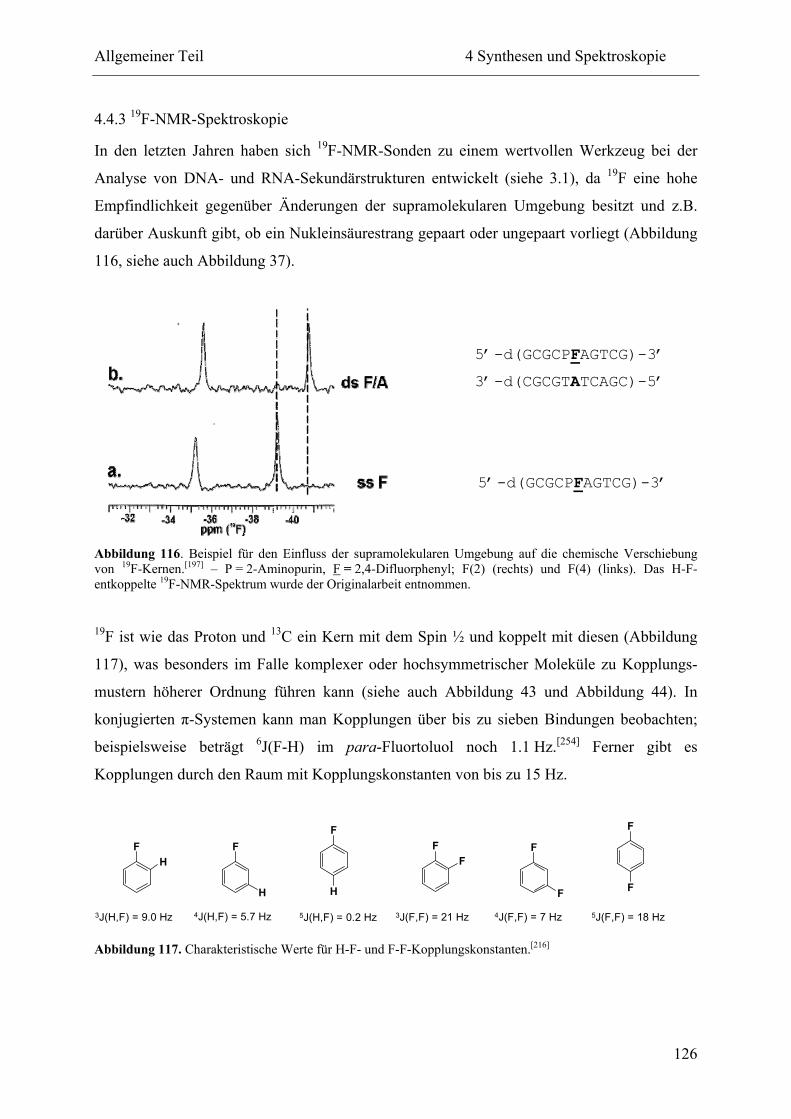
Allgemeiner Teil 4 Synthesen und Spektroskopie
126
4.4.3 19F-NMR-Spektroskopie
In den letzten Jahren haben sich 19F-NMR-Sonden zu einem wertvollen Werkzeug bei der
Analyse von DNA- und RNA-Sekundärstrukturen entwickelt (siehe 3.1), da 19F eine hohe
Empfindlichkeit gegenüber Änderungen der supramolekularen Umgebung besitzt und z.B.
darüber Auskunft gibt, ob ein Nukleinsäurestrang gepaart oder ungepaart vorliegt (Abbildung
116, siehe auch Abbildung 37).
Abbildung 116. Beispiel für den Einfluss der supramolekularen Umgebung auf die chemische Verschiebung von 19F-Kernen.[197] – P = 2-Aminopurin, F = 2,4-Difluorphenyl; F(2) (rechts) und F(4) (links). Das H-F-entkoppelte 19F-NMR-Spektrum wurde der Originalarbeit entnommen.
19F ist wie das Proton und 13C ein Kern mit dem Spin ½ und koppelt mit diesen (Abbildung
117), was besonders im Falle komplexer oder hochsymmetrischer Moleküle zu Kopplungs-
mustern höherer Ordnung führen kann (siehe auch Abbildung 43 und Abbildung 44). In
konjugierten π-Systemen kann man Kopplungen über bis zu sieben Bindungen beobachten;
beispielsweise beträgt 6J(F-H) im para-Fluortoluol noch 1.1 Hz.[254] Ferner gibt es
Kopplungen durch den Raum mit Kopplungskonstanten von bis zu 15 Hz.
F
H
F
H
F
H
F
FF
F
F
F
3J(H,F) = 9.0 Hz 4J(H,F) = 5.7 Hz 5J(H,F) = 0.2 Hz 3J(F,F) = 21 Hz 4J(F,F) = 7 Hz 5J(F,F) = 18 Hz
Abbildung 117. Charakteristische Werte für H-F- und F-F-Kopplungskonstanten.[216]
5’-d(GCGCPFAGTCG)-3’
3’-d(CGCGTATCAGC)-5’
5’-d(GCGCPFAGTCG)-3’
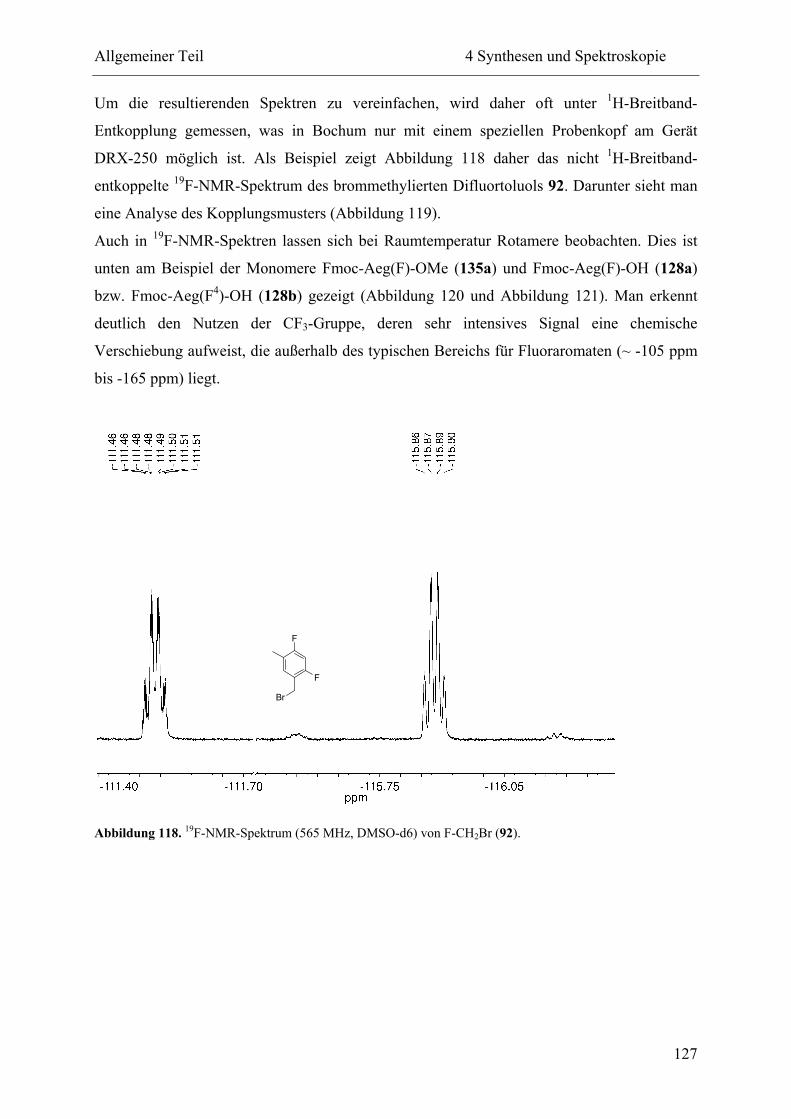
Allgemeiner Teil 4 Synthesen und Spektroskopie
127
Um die resultierenden Spektren zu vereinfachen, wird daher oft unter 1H-Breitband-
Entkopplung gemessen, was in Bochum nur mit einem speziellen Probenkopf am Gerät
DRX-250 möglich ist. Als Beispiel zeigt Abbildung 118 daher das nicht 1H-Breitband-
entkoppelte 19F-NMR-Spektrum des brommethylierten Difluortoluols 92. Darunter sieht man
eine Analyse des Kopplungsmusters (Abbildung 119).
Auch in 19F-NMR-Spektren lassen sich bei Raumtemperatur Rotamere beobachten. Dies ist
unten am Beispiel der Monomere Fmoc-Aeg(F)-OMe (135a) und Fmoc-Aeg(F)-OH (128a)
bzw. Fmoc-Aeg(F4)-OH (128b) gezeigt (Abbildung 120 und Abbildung 121). Man erkennt
deutlich den Nutzen der CF3-Gruppe, deren sehr intensives Signal eine chemische
Verschiebung aufweist, die außerhalb des typischen Bereichs für Fluoraromaten (~ -105 ppm
bis -165 ppm) liegt.
Abbildung 118. 19F-NMR-Spektrum (565 MHz, DMSO-d6) von F-CH2Br (92).
F
F
Br

Allgemeiner Teil 4 Synthesen und Spektroskopie
128
Abbildung 119. 19F-NMR-Spektrum (565 MHz, DMSO-d6) von F-CH2Br (92). – Analyse des Kopplungsmus-ters für F(4). Die Beträge der Kopplungskonstanten sind in Hz angegeben.
Abbildung 120. 19F-NMR-Spektren (565 MHz, DMSO-d6) von Fmoc-Aeg(F)-OMe (135a) (unten) und Fmoc-Aeg(F)-OH (128a) (oben).
N
O
O NH
OR
OO
F
F
135a: R = Me128a: R = H
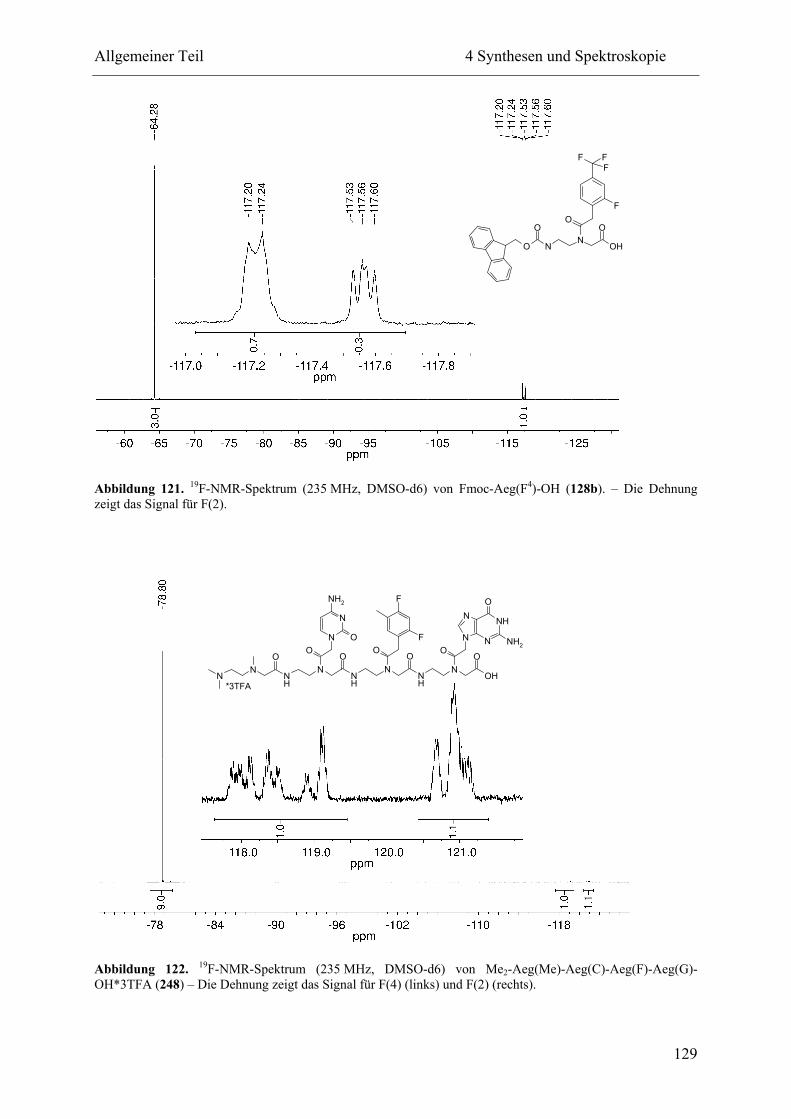
Allgemeiner Teil 4 Synthesen und Spektroskopie
129
Abbildung 121. 19F-NMR-Spektrum (235 MHz, DMSO-d6) von Fmoc-Aeg(F4)-OH (128b). – Die Dehnung zeigt das Signal für F(2).
Abbildung 122. 19F-NMR-Spektrum (235 MHz, DMSO-d6) von Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OH*3TFA (248) – Die Dehnung zeigt das Signal für F(4) (links) und F(2) (rechts).
N
O
O N OH
OO
F
FFF
N N
NNH
O
NH2
NH
N
O
NH
NNH
NOH
OOO
N
N
O
NH2
O
F
F
OO
NN
*3TFA

Allgemeiner Teil 5 Ligation und Replikation
130
5 Ligation und Replikation
5.1 Voruntersuchungen
Der nächste Schritt bestand darin, geeignete Bedingungen für die Ligation der beiden Trimere
224 (A) und 248 (B) zum hexameren Templat 268 (T) zu finden (Abbildung 123). Dabei
wurde ein besonderes Augenmerk auf die Wahl des Kopplungsreagenzes und möglicher
nukleophiler Katalysatoren gelegt.
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
NH ON N
O
NH2
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
NH
NH
NO
ON
N
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
ON N
O
NH2
NH
NH2
NO
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
NH
OH
N
N
O
268 / T
248 / A
224 / B
Abbildung 123. Potentielles Selbstreplikations-System auf der Basis der tri-PNAs 224 und 248. – Grün: nicht-reaktive Löslichkeitsvermittler; gelb: 19F-NMR-Sonden; rot: Ligationsstellen und daraus resultierende zentrale Amidbindung.
Die Autoren der ersten richtungsweisenden Arbeit zur PNA-Ligation benutzten für ihre Expe-
rimente das Kopplungsreagenz EDC und Imidazol-Puffer, nachdem sie festgestellt hatten,
dass die Verwendung von 2-Methylimidazol-Puffer die Oligomerisierung unterdrückt.[171]

Allgemeiner Teil 5 Ligation und Replikation
131
Dennoch wurde in einer folgenden Studie der gleichen Arbeitsgruppen 2-Methylimidazol-
Puffer verwendet und das fehlgeschlagene Experiment erfolgreich wiederholt.[172] Für die
Synthese von DNA/PNA-Chimären an PNA- und DNA-Templaten wurde hingegen erneut
Imidazol-Puffer verwendet.[255] Die Aufgabe der Imidazole 269 besteht nicht allein in der Re-
gulierung des pH-Werts, sondern vor allem in der nukleophilen Katalyse: Sie reagieren mit
dem zunächst aus EDC und Säure-Komponente 270 gebildeten O-Acylisoharnstoff 271 zu
den weniger hydrolyseempfindlichen Imidazoliden 272, die als effizientes Acylierungsmittel
mit der Amino-Komponente 273 zum Amid 274 reagieren (Abbildung 124).
OHR1
O
NR1
O
N
R2
NH N
R2
R3 NH2
NH N
R2
269
NH
R1
O
R3OR1
O
NH
NH
N+
H
EDC
- EDU270 272 274
269 273
-
+
271
Abbildung 124. Nukleophile Katalyse bei der EDC-vermittelten Knüpfung einer Amidbindung. – Aktivierung der Carbonsäure 270 durch Umsetzung mit EDC zum O-Acylisoharnstoff 271, Reaktion zum Imidazolid 272 und anschließende Acylierung des Amins 273.
Der Einfluss der verwendeten Imidazol-Derivate auf die Kopplungseffizienz templatgesteuer-
ter Reaktionen ist aus den Arbeiten von Orgel zur RNA-Polymerisation wohlbekannt,[69] doch
gibt es bislang keine systematische Untersuchung zum Einfluss verschiedener Imidazole auf
die Ligation von PNA. Eine entsprechende Studie wurde vor 15 Jahren angekündigt, aber bis-
lang nicht veröffentlicht,[171] sodass es sich anbot, diese Lücke durch ein Screening verschie-
dener Aktivierungsreagenzien und Imidazol-Puffer zu schließen. Daher wurde der Effekt von
Imidazol (275), 1-Methylimidazol (276), 2-Methylimidazol (277) und 4(5)-Hydroxymethyl-
imidazol (278) auf die Ligation von A und B zu T durch RP-HPLC und MALDI-TOF-MS
untersucht (Abbildung 125, Tabelle 3); die übrigen Reaktionsbedingungen orientierten sich an
den eben erwähnten Studien. Zum Vergleich wurde auch imidazolfrei mit MOPS-Puffer
gearbeitet. Neben EDC (279) kamen das ebenfalls wasserlösliche Carbodiimid CMC (1) und
das Triazin-basierte Reagenz DMT-MM (280) zum Einsatz. Durch seine beiden Sechsringe
besitzt CMC (1) einen größeren sterischen Anspruch und ist, im Gegensatz zu EDC (279),
durch das quartäre Stickstoff-Atom am Morpholin-Ring nicht in der Lage, cyclische
Strukturen auszubilden (Abbildung 126).

Allgemeiner Teil 5 Ligation und Replikation
132
N
NH
N
N
N
NH
N
NH OH
C
N
N
N+
O
C
N
N
N
O
N+
N
N
N
OO
*pTsOH *HCl275 276
277 278
1 279
280
Abbildung 125. Eingesetzte Imidazole 275-278 (links), verwendete Carbodiimide 1 und 279 (Mitte) und DMT-MM (280, rechts).
CN N N+
HCl NH N
+
NCH3CH2
NH N+
NCH2CH3
N N+
NCH3CH2 H
Cl
Cl
Cl Cl
CN N N+
HCl N N
+
NCH3CH2 H
279
279
281
281 282 283
Abbildung 126. Strukturen von EDC*HCl in D2O[256] (oben) und in DMSO-d6[257] (unten). – Auch in
gepufferten wässrigen Lösungen liegen cyclische und offenkettige Spezies nebeneinander vor; das Verhältnis ist pH-abhängig.[258]
Die mit Abstand größte Ausbeute an Ligationsprodukt 268 konnte bei der Verwendung von
1-Methylimidazol-Puffer (276) und EDC (279) erreicht werden (Tabelle 3). Erstaunlicherwei-
se war die Reaktion in MOPS-Puffer deutlich effizienter als bei Verwendung der Imidazole
275, 277 und 278, obwohl in diesem Fall keine nukleophile Katalyse möglich ist. Als Neben-
produkt ließ sich in allen Fällen das Guanidinium-Derivat 284 des C-terminalen Trimers 224
identifizieren (Abbildung 127, vgl. Abbildung 60). Beim Einsatz des sterisch anspruchs-
volleren Reagenzes CMC (1) konnte in keinem Fall die gewünschte hexa-PNA 268
nachgewiesen werden.
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH
O
N
N
O
NH2
NH
N
O
NH
NH
N
+
284
Abbildung 127. Nebenprodukt 284 der EDC vermittelten PNA-Ligation.

Allgemeiner Teil 5 Ligation und Replikation
133
Tabelle 3. Screening unterschiedlicher Reaktionsbedingungen für die Ligation der Trimere 224 und 248 zum Hexamer 268. – Reaktionsbedingungen: 5 mM PNA-Trimere, RT, 4 d. Die Ausbeuten wurden durch RP-HPLC-Analyse und UV-Detektion bei 260 nm unter Vernachlässigung von Hyperchromieeffekten bestimmt.
Nr. Puffer Kopplungsreagenz Ausbeute
1 0.4 M Im pH 7.5 0.2 M EDC 5 %
2 0.4 M 1-MeIm pH 7.5 0.2 M EDC 63 %
3 0.4 M 2-MeIm pH 7.5 0.2 M EDC 8 %
4 0.2 M 4-HOCH2-Im pH 7.2 0.2 M EDC 3 %
5 0.2 M MOPS pH 7.0 0.2 M EDC 17 %
6 0.4 M Im pH 7.5 0.2 M CMC -
7 0.4 M 1-MeIm pH 7.5 0.2 M CMC -
8 0.4 M 2-MeIm pH 7.5 0.2 M CMC -
9 0.2 M 4-HOCH2-Im pH 7.2 0.2 M CMC -
10 0.2 M MOPS pH 7.0 0.2 M CMC -
11 0.4 M MOPS pH 7.0 0.2 M DMT-MM 16 %
Die Ligation der beiden Trimere 224 und 248 wurde ferner in wasserfreiem DMF untersucht,
da die PNA-Replikation im Kontext des PACE-Projektes auch in einem hydrophoben
organischen Medium diskutiert wurde, wie es im Inneren von Mizellen vorherrscht
(vgl. 1.4.2). Weil die Zugabe aprotischer organischer Solventien (DMF, 1,4-Dioxan) die
Stabilität von PNA-Duplexen nur wenig beeinflusst,[143] sollten templatierte Reaktionen auch
unter diesen Bedingungen möglich sein. Die Uronium-Kopplungsreagenzien COMU (285)
und HBTU (151) zeigten eine vergleichbare Effizienz, während von den drei verwendeten
Carbodiimid-Reagenzien nur EDC (279) Umsatz zur hexa-PNA 268 zeigte (Abbildung 128,
Tabelle 4).
O
O
N
NC
O
N N+
O
C
N
NPF6
-N
NN
O
N
N+
285 151 286
Abbildung 128. Die Kopplungsreagenzien COMU (285), HBTU (151) und DIC (286) (von links nach rechts).

Allgemeiner Teil 5 Ligation und Replikation
134
Tabelle 4. Screening unterschiedlicher Kopplungsreagenzien für die Ligation der Trimere 224 und 248 zu Hexamer 268 in DMF. – Reaktionsbedingungen: 5 mM PNA-Trimere, 50 mM DIPEA, DMF, RT, 4 d. Die Ausbeuten wurden durch RP-HPLC-Analyse und UV-Detektion bei 260 nm unter Vernachlässigung von Hyperchromieeffekten bestimmt.
Nr. Kopplungsreagenz Ausbeute
1 1.2 eq. COMU 62 %
2 1.2 eq. HBTU 59 %
3 1.2 eq. EDC 22 %
4 1.2 eq. CMC -
5 1.2 eq. DIC -
6 1.2 eq. DIC, 1.2 eq. DMAP -
Analytische Daten für die isolierte hexa-PNA 268 zeigen Abbildung 129 und Abbildung 130,
ein HR-ESI-MS-Spektrum ist im Anhang dargestellt. Die UV-Schmelzkurve der PNA 268
wurde mit der einer analogen DNA 287 verglichen, deren Sequenz d(CTGCAG) sich nur
durch den Ersatz des hydrophoben Isosters durch Thymin unterscheidet. Der experimentell
ermittelte Schmelzpunkt der PNA 268 ist im Vergleich zu dem der DNA 287 um ca. 3 °C
niedriger, obwohl PNA-Duplexe in der Regel deutlich stabiler sind. Darüber hinaus besitzt die
UV-Schmelzkurve der hexa-PNA 268 eine deutlich geringere Steilheit als die der DNA 287
und lässt so auf ein reduziertes Maß an Kooperativität schließen. Beide Befunde lassen sich
mit dem isosteren Ersatz des Thymins durch den Fluoraromaten erklären. So zeigen Untersu-
chungen von Frey und Woski an PNA-15meren, dass der Einbau des strukturell vergleich-
baren Thymin-Isosters 4-Fluorbenzol gegenüber Adenin den Schmelzpunkt ansonsten kom-
plementärer PNA/DNA-Duplexe um 12.2 °C vermindert.[207] Auch die geringe Kooperativität
lässt sich auf den Einbau des Fluoraromaten zurückführen, der in Lösung keine Wasserstoff-
brücken ausbildet, wodurch nur vier der sechs Basenpaare im Duplex auf diese Art stabilisiert
sind. Als Nukleationskeim für die kooperative Helixbildung werden allerdings wenigstens
zwei intakte und benachbarte Basenpaare benötigt. Die Kenntnis des Schmelzpunkts ist von
Bedeutung, da die optimale Reaktionstemperatur natürlicher Replikatoren im Bereich der
Schmelztemperatur der jeweiligen Templatduplexe liegt (vgl. 1.3.2).[77]
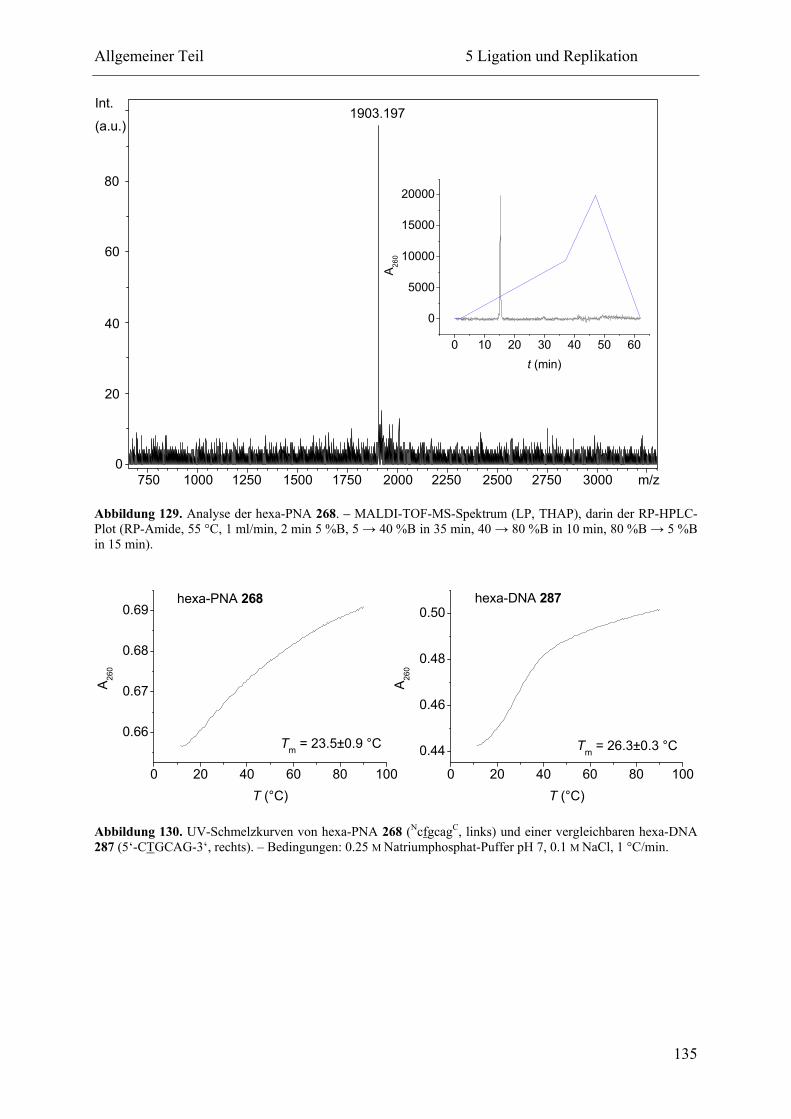
Allgemeiner Teil 5 Ligation und Replikation
135
Abbildung 129. Analyse der hexa-PNA 268. – MALDI-TOF-MS-Spektrum (LP, THAP), darin der RP-HPLC-Plot (RP-Amide, 55 °C, 1 ml/min, 2 min 5 %B, 5 → 40 %B in 35 min, 40 → 80 %B in 10 min, 80 %B → 5 %B in 15 min).
Abbildung 130. UV-Schmelzkurven von hexa-PNA 268 (NcfgcagC, links) und einer vergleichbaren hexa-DNA 287 (5‘-CTGCAG-3‘, rechts). – Bedingungen: 0.25 M Natriumphosphat-Puffer pH 7, 0.1 M NaCl, 1 °C/min.
0 20 40 60 80 100
0.66
0.67
0.68
0.69
Tm = 23.5±0.9 °C
A2
60
T (°C)
hexa-PNA 268
0 20 40 60 80 100
0.44
0.46
0.48
0.50
A2
60
T (°C)
Tm = 26.3±0.3 °C
hexa-DNA 287
0
20
40
60
80
750 1000 1250 1500 1750 2000 2250 2500 2750 3000 m/z
Int.
(a.u.) 1903.197
0 10 20 30 40 50 60
0
5000
10000
15000
20000
A2
60
t (min)

Allgemeiner Teil 5 Ligation und Replikation
136
5.2 HPLC-Kinetiken
5.2.1 Kalibrierung
Nachdem Kopplungsbedingungen für eine effektive Reaktion gefunden waren, galt es den
zeitlichen Verlauf der Ligationsreaktion zu untersuchen. Hierbei wurde mittels RP-HPLC
analysiert, um Substanz zu sparen. Die bei den Voruntersuchungen entstandene hexa-PNA
268 wurde isoliert, sodass sie weiteren Experimenten in variabler Konzentration zugesetzt
werden konnte. Ein typisches Chromatogramm ist in Abbildung 131 wiedergegeben und zeigt
deutlich den dominierenden Einfluss des Fluoraromaten auf die Retentionszeit.
0 5 10 15 20 25
0
5000
10000
15000
20000
25000
A2
60
t (h)
4.40
7.67
11.6
12.2
12.8
Abbildung 131. Typisches RP-HPLC-Chromatogramm. – Die verschiedenen Peaks wurden isoliert und durch MALDI-TOF-MS zugeordnet: tr = 4.40 min: 224; tr = 7.67 min 224+EDC (284); tr = 11.6 min: 268; tr = 12.2 min: 248; tr = 12.8 min: 248+EDC (288).
Zur Auswertung der Chromatogramme wurde das Verhältnis der Peakflächen von poten-
tiellem Templat 268 und Trimer 248 bestimmt, da beide Substanzen ähnliche Reten-
tionszeiten aufweisen und im Chromatogramm scharfe Signale zeigen. Die Verwendung
dieser internen Kalibrierung macht die Messergebnisse unabhängig von Änderungen der
Probenkonzentration und von Volumenfehlern bei Entnahme, Verdünnung und Auftragung
auf die Säule. Die größte Fehlerquelle bei diesem Vorgehen ist die Differenzbildung der
HPLC-Integrale, um zu einem gegebenen Zeitpunkt die initiale Hexamerzugabe aus der ge-
samten Hexamerkonzentration herauszurechnen. Zur Umrechnung der Peakflächenverhält-
nisse in Konzentrationsverhältnisse wurde eine Kalibrierkurve aufgenommen, indem sechs
Proben mit bekannten Konzentrationen von 248 und 268 gemessen und die Flächen-

Allgemeiner Teil 5 Ligation und Replikation
137
verhältnisse gegen die Konzentrationsverhältnisse aufgetragen wurden. Wie in Abbildung 132
dargestellt, wurde dabei ein linearer Zusammenhang gefunden.
0 2 4 6 8
0
1
2
3
4
Datenpunkte lineare RegressionK
onz
en
tra
tion
sve
rhäl
tnis
Flächenverhältnis
Abbildung 132. Kalibrierkurve zur Umrechnung von Flächen- in Konzentrationsverhältnisse. – Ergebnis der linearen Regression: f(x) = (-0.02±0.09) + (0.48±0.02) × x. Fitparameter: R2 = 0.9936, SD = 0.1417.
Unter der Annahme, dass tri-PNA A (248) entweder unverbraucht vorliegt oder zur hexa-
PNA T (268) reagiert, gilt:
00 ATAT (14)
[T] = Konzentration von T zum Zeitpunkt t
[A] = Konzentration von A zum Zeitpunkt t
[T]0 = Ausgangskonzentration von T
[A]0 = Ausgangskonzentration von A
Das Konzentrationsverhältnis κ lässt sich daher wie folgt ausdrücken:
TTA
T
A
T
00 (15)
Durch Auflösen nach [T] lässt sich aus der Kenntnis der Ausgangskonzentrationen und des
Konzentrationsverhältnisses die tatsächliche Konzentration zum Zeitpunkt t berechnen:
1
)AT(T 00
(16)

Allgemeiner Teil 5 Ligation und Replikation
138
5.2.2 Auswertungsverfahren
Die Auswertung der experimentellen Daten erfolgte mit Hilfe des Programms SimFit
(Nonlinear Fitting by Dynamic Simulation), das die Berechnung theoretischer Konzen-
trations-Zeit-Kurven durch dynamische Simulation mit der Kurvenanpassung durch nicht-
lineare Regression kombiniert.[185] Grundlage der Analyse der Reaktionskinetiken waren zwei
Reaktionsgleichungssysteme, die nachfolgend als A-Fitting (zwei Parameter) und B-Fitting
(drei Parameter) bezeichnet werden. Das A-Fitting bezieht neben einem nicht katalysierten
Reaktionskanal (k1, Gleichung 17) zwei Abklingprozesse ein, für die jeweils die gleiche
Geschwindigkeitskonstante k2 angenommen wird (Gleichung 18 und 19).
T BA 1 k (17)
X A 2k (18)
Y B 2k (19)
Das B-Fitting enthält zusätzlich einen katalysierten Reaktionskanal, in dem als Folge des
Quadratwurzelgesetzes der Autokatalyse der Koeffizient 0.5 auftritt (Gleichung 20, vgl.
1.3.2). Der Quotient von k3 und k1 entspricht hier somit dem autokatalytischen Überschuss-
Faktor ε, mit dessen Hilfe die Effizienz verschiedener Replikations-Systeme verglichen
werden kann (Tabelle 5, vgl. Gleichung 10).
T 1.5 T 0.5 BA 3 k (20)
Tabelle 5. Auswertung von Selbstreplikations-Systemen auf Nukleinsäurebasis anhand des autokatalytischen Überschuss-Faktors ε.
System [Bausteine]
(mM)
T
(°C)
Puffer
(1 M)
pH [MgCl2]
(M)
[EDC]
(M)
[T]init.
(%)
ε
(M-1/2)
v. Kiedrowski 1986[72] 10 0 MES 6.15 0.05 0.2 0-8 25
Zielinski u. Orgel 1987[73] 1 5 HEPES 7.5 - 0.06 0-100 340
v. Kiedrowski et al. 1989[75] 4 20 MES 6.15 0.05 0.2 0-40 80
v. Kiedrowski et al. 1991[76] 1 30 HEPES 7.5 - 0.2 0-32 420
Sievers u. v. Kiedrowski 1994
(ApB bzw. BpA)[78, 79]
1 30 HEPES 7.55 - 0.2 0-32 408
354

Allgemeiner Teil 5 Ligation und Replikation
139
In beiden Modellen berücksichtigen die Abklingprozesse sowohl die Bildung von Nebenpro-
dukten aus EDC und den Trimeren als auch die Hydrolyse des EDCs zum Harnstoff und die
damit einhergehende Desaktivierung der Carbonsäure-Komponente 248. Auch die durch den
entstehenden Harnstoff beeinträchtigte Bildung von Komplexen wird hier miteinbezogen. Da
die EDC-Hydrolyse durch Carbonsäuren katalysiert wird, variiert ihre Geschwindigkeit
vermutlich mit der Konzentration an PNA-Carbonsäure 248 und an Trifluoressigsäure, die
sich in allen Reaktionslösungen befand, weil die PNAs in Form ihrer mehrfachen TFA-Salze
vorlagen. Dieser Zusammenhang wird durch das verwendete Modell allerdings nicht
berücksichtigt.
Wie gut ein Modell mit den Daten übereinstimmt, kann anhand der Fehlerquadratsumme
(RMS) abgelesen werden. Ein Wert von unter 2 % wird als Hinweis auf einen guten Fit
gewertet, ein Wert von 2 % gilt als gut annehmbar und ein Wert zwischen 2 und 5 % als
annehmbar. Zwischen 5 und 10 % spricht man von einem schlechten Fit, während ein RMS-
Wert von über 10 % nicht mehr akzeptiert wird. Allerdings gibt ein Modell mit kleinem
RMS-Wert vor allem dann nicht zwangsläufig die chemische Realität wieder, wenn den
gegebenen Observablen zu viele Variablen gegenüberstehen und daher deutliche Kovarianzen
auftreten. Die Kovarianz cov(k1, k2) zweier variabler Geschwindigkeitskonstanten k1 und k2 ist
ein Maß für ihre Abhängigkeit voneinander und gibt somit Auskunft über die Verlässlichkeit
eines Modells. Bei einer Kovarianz von 0 sind die beiden Variablen völlig unabhängig
voneinander, eine Kovarianz nahe 1 bedeutet indessen, dass ein höherer Wert von k1 ebenfalls
einen höheren Wert von k2 bedingt. Wenn die Kovarianz hingegen nahe -1 ist, führt ein
höherer Wert von k1 zu einem niedrigeren Wert von k2 und umgekehrt. Ein Absolutwert der
Kovarianz von unter 0.9 soll hier als akzeptabel angesehen werden. In diesem Zusammenhang
muss außerdem erwähnt werden, dass die von SimFit angegebenen Fehlergrenzen der
berechneten Geschwindigkeitskonstanten den mathematischen Fehler bei der Simulation,
nicht aber die systematischen experimentellen Fehler berücksichtigen.

Allgemeiner Teil 5 Ligation und Replikation
140
5.2.3 Ligation und Replikation in Imidazol-Puffern
Zunächst wurden Versuchsreihen bei vier verschiedenen Temperaturen unter der Verwendung
von 1-Methylimidazol-Puffer aufgenommen. Dabei wurden jeweils Reaktionen ohne und mit
zwei verschiedenen initialen Hexamerzugaben verfolgt (Abbildung 133, Tabelle 6). Der
Beitrag des initial zugegebenen Hexamers wurde hier und in allen folgenden Experimenten
herausgerechnet.
Abbildung 133. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 in 1-Methylimidazol-Puffer. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.4 M 1-Methylimidazol-Puffer pH 7.5, 25 µl Ansatzgröße. A: cov(k1, k2) = 0.591, cov(k1, k3) = -0.628 und cov(k2, k3) = 0.205. B: cov(k1, k2) = 0.547, cov(k1, k3) = -0.553 und cov(k2, k3) = 0.337. C: cov(k1, k2) = 0.788, cov(k1, k3) = -0.260 und cov(k2, k3) = 0.335. D: cov(k1, k2) = -0.131, cov(k1, k3) = -0.793 und cov(k2, k3) = 0.814.
0 2 4 6 8 10
0.0
0.4
0.8
1.2
[T] (
mM
)
t (h)
0 % T20 % T30 % T
C 20 °C
0 2 4 6 8 10
0.0
0.5
1.0
1.5
2.0
2.5
0 % T16 % T55 % T
[T] (
mM
)
t (h)
D 30 °C
0 20 40 60 80 100
0.0
0.4
0.8
1.2
0 % T20 % T40 % T
[T] (
mM
)
t (h)
A 0 °C
0 5 10 15 20 25
0.0
0.4
0.8
1.2
[T] (
mM
)
t (h)
0 % T20 % T40 % T
B 10 °C

Allgemeiner Teil 5 Ligation und Replikation
141
Tabelle 6. Kinetische Auswertung der Ligation in 1-Methylimidazol-Puffer. – Die Modelle A’ und B’ vernachlässigen die Abklingprozesse.
Nr. T (°C) Fit RMS (%) k1 (M-1 s-1) k2 (s
-1) k3 (M-3/2 s-1) ε (M
-1/2)
1 0 A 1.17 (2.60±0.05)×10-4 (1.20±0.09)×10-6 - -
2 B 1.20 (2.50±0.07)×10-4 (1.05±0.09)×10-6 (1.7±14.4)×10-5 0.58
3 10 A 1.13 (6.9±0.2)×10-4 (9±4)×10-7 - -
4 A’ 1.21 (6.39±0.08)×10-4 - - -
5 B 0.287 (5.79±0.07)×10-4 (1.3±0.1)×10-6 (3.7± 0.2)×10-3 6.4
6 B’ 0.726 (5.3±0.1)×10-4 - (2.9±0.4)×10-3 5.5
7 20 A 1.79 (1.24±0.09)×10-3 (0.02±2.20)×10-6 - -
8 B 0.999 (9.7±0.5)×10-4 (1±1)×10-6 (9.9± 0.9)×10-3 10
9 30 A 4.27 (4.1±0.4)×10-3 (0.4±2.6)×10-6 - -
10 B 1.23 (2.4±0.1)×10-3 (0.3±1.0)×10-6 (3.9±0.5)×10-2 16
Bei 0 °C hat die initiale Templatzugabe qualitativ keinen Einfluss auf die Geschwindigkeit
oder die Ausbeute der Reaktion. Dies äußert sich bei der kinetischen Modellierung durch
vergleichbare RMS-Werte für das A- und B-Fitting, eine schlecht bestimmte Geschwindig-
keitskonstante für den autokatalytischen Reaktionskanal und einen ε-Wert von 0.58 M-1/2.
Erhöht man die Temperatur auf 10 °C, sind die Datenpunkte in besserem Einklang mit dem
B- als mit dem A-Modell und man erhält einen zehnfach höheren Wert für ε, sodass auf einen
schwachen autokatalytischen Reaktionskanal geschlossen werden kann. Werden keine
Abklingprozesse berücksichtigt, erhält man ähnliche Werte für k1, k3 und ε, während die
entsprechenden RMS-Werte ansteigen (Modell A’ und B’ in Tabelle 6). Die deutlichsten
Änderungen des Reaktionsverlaufs treten durch Zugabe der ersten 20 % Templat auf,
während die Zugabe von weiteren 20 % relativ betrachtet einen geringeren Einfluss hat.
Dieses Verhalten ist in seiner Tendenz typisch für parabolische Replikatoren: Variiert man bei
ihnen die initiale Templatzugabe [T]0 in der Reihe [T]0(1) = 0, [T]0(2) = y, [T]0(3) = 2y und
[T]0(4) = 4y (y = 2-10 %), spiegelt der Konzentrationsverlauf [T] = [T](i) - [T](1) das Ver-
hältnis 1 : 2½ : 4½ wieder.[77] Folglich ist der stärkste relative Einfluss bei der geringsten
initialen Templatzugabe zu finden; der stärkste absolute Einfluss bei der größten Zugabe. Das
Experiment bei 20 °C zeigt hingegen bei der größten Templatzugabe auch den stärksten
relativen Einfluss auf den Konzentrationsverlauf. Bei 30 °C konnte mit 16 M-1/2 der höchste
Wert für den autokatalytischen Überschuss-Faktor ε bestimmt werden. Dieser Befund ist
allerdings mit Vorsicht zu betrachten, da der Verlauf der theoretischen Kurve für das
Experiment bei 55 % Templatzugabe erkennbar von den Datenpunkten nach oben abweicht,
wodurch auch der größte RMS-Wert innerhalb dieser Serie zustande kommt. Als Erklärung

Allgemeiner Teil 5 Ligation und Replikation
142
kann zum einen die Carbonsäure-katalysierte EDC-Hydrolyse herangezogen werden. Denn
während die Konzentration der PNA-Carbonsäure 248 in allen Experimenten konstant bei
5 mM liegt, korreliert die Konzentration an Trifluoressigsäure mit der Menge des zugesetzten
Templats: Ohne Zugabe von Templat beträgt sie 35 mol/l, bei Zusatz von 16 % Templat
40 mol/l und bei 55 % Templatzugabe schon 52 mol/l. Daher kann angenommen werden, dass
die EDC-Hydrolyse mit steigender initialer Templatzugabe schneller verläuft; ein Umstand,
der im Reaktionsmodell nicht berücksichtigt wird. Zum anderen sind durch die hohe Templat-
konzentration auch größere Fehler bei der Subtraktion der HPLC-Integrale möglich. Es muss
an dieser Stelle angemerkt werden, dass Templatzugaben von mehr als 30 % nur in der frühen
Phase des Forschungsfelds üblich waren, sodass zum Vergleich nur das von Zielinski und
Orgel untersuchte tetramere System auf der Basis modifizierter Ribonukleotide herangezogen
werden kann.[73] Bei diesem wurden nach Zusatz von 25, 50 und 100 % Templat jeweils die
theoretisch erwarteten Steigerungen von Anfangsgeschwindigkeit und Ausbeute beobachtet.
Unter der Annahme, dass in keiner der vier Versuchsreihen nennenswerte Selbstreplikation zu
beobachten war, wurden die Experimente ohne initiale Templatzugabe als Reaktionen zweiter
Ordnung behandelt, um sowohl die Geschwindigkeitskonstanten für die einzelnen Reaktionen
zu ermitteln als auch Ea aus einer Arrhenius-Auftragung zu bestimmen. Hierzu wurde
zunächst die reziproke Konzentration an Trimer A (248) gegen die Zeit aufgetragen und an
die ersten, augenscheinlich linear verlaufenden Datenpunkte eine Gerade angepasst, aus deren
Steigung sich gemäß
ktAA t
0][
1
][
1 (21)
die jeweilige Geschwindigkeitskonstante ablesen ließ. Abbildung 134 zeigt dieses Vorgehen
am Beispiel der Reaktion bei 30 °C.
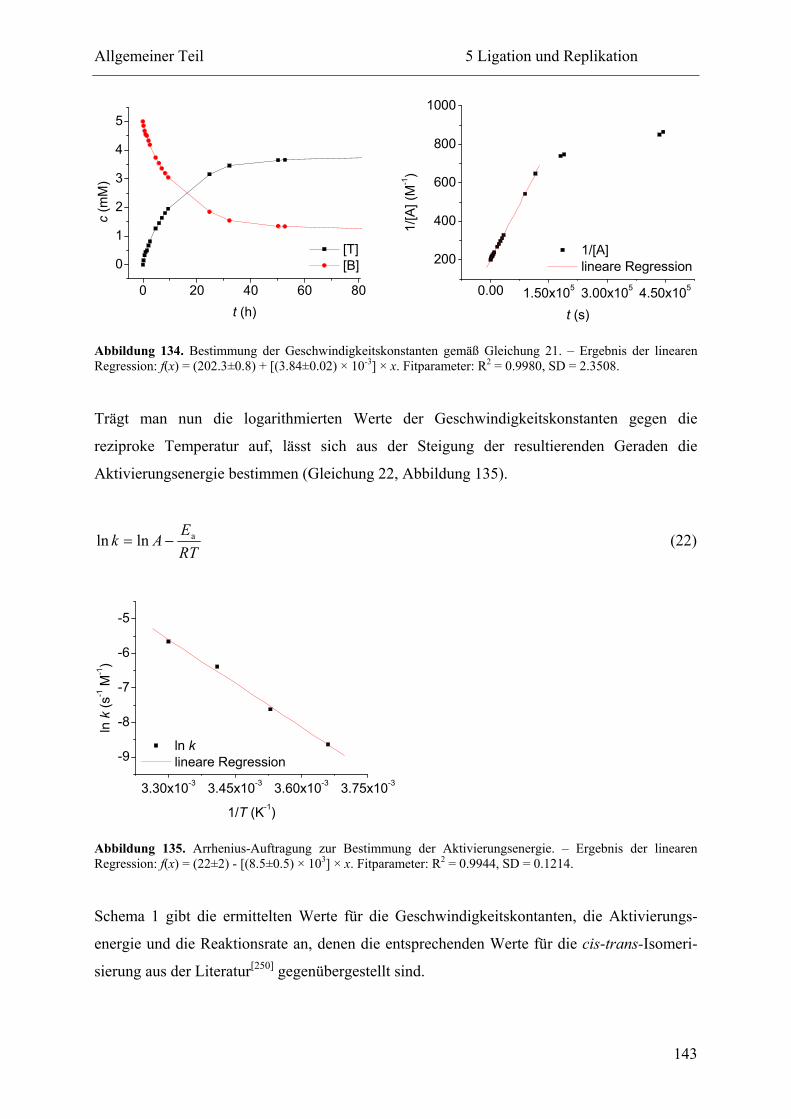
Allgemeiner Teil 5 Ligation und Replikation
143
Abbildung 134. Bestimmung der Geschwindigkeitskonstanten gemäß Gleichung 21. – Ergebnis der linearen Regression: f(x) = (202.3±0.8) + [(3.84±0.02) × 10-3] × x. Fitparameter: R2 = 0.9980, SD = 2.3508.
Trägt man nun die logarithmierten Werte der Geschwindigkeitskonstanten gegen die
reziproke Temperatur auf, lässt sich aus der Steigung der resultierenden Geraden die
Aktivierungsenergie bestimmen (Gleichung 22, Abbildung 135).
RT
EAk alnln (22)
3.30x10-3 3.45x10-3 3.60x10-3 3.75x10-3
-9
-8
-7
-6
-5
ln k lineare Regression
ln k
(s-1
M-1)
1/T (K-1)
Abbildung 135. Arrhenius-Auftragung zur Bestimmung der Aktivierungsenergie. – Ergebnis der linearen Regression: f(x) = (22±2) - [(8.5±0.5) × 103] × x. Fitparameter: R2 = 0.9944, SD = 0.1214.
Schema 1 gibt die ermittelten Werte für die Geschwindigkeitskontanten, die Aktivierungs-
energie und die Reaktionsrate an, denen die entsprechenden Werte für die cis-trans-Isomeri-
sierung aus der Literatur[250] gegenübergestellt sind.
0 20 40 60 80
0
1
2
3
4
5
[T] [B]
c (m
M)
t (h)
0.00 1.50x105 3.00x105 4.50x105
200
400
600
800
1000
1/[A
] (M
-1)
t (s)
1/[A] lineare Regression

Allgemeiner Teil 5 Ligation und Replikation
144
k (Ligation, 0 °C) = (1.78±0.06)×10-4 s-1 M-1 k (Ligation, 10 °C) = (3.19±0.02)×10-4 s-1 M-1 k (Ligation, 20 °C) = (1.68±0.02)×10-3 s-1 M-1 k (Ligation, 30 °C) = (3.84±0.02)×10-3 s-1 M-1 k (Ligation, 37 °C)b = (7.01±0.05)×10-3 s-1 M-1 k (cis-trans, 37 °C)a = 0.5-2 s-1 Ea (Ligation) = 70±4 kJ/mol
Ea (cis-trans)a = 79±8 kJ/mol
ν (Ligation) = 7.01×10-3 s-1 M-1 × (5 mM)2 = 2×10-7 M s-1 ν (cis-trans)a = 1.25 s-1 × 5 mM = 6×10-3 M s-1
Schema 1. Vergleich der experimentell bestimmten Geschwindigkeitskonstanten für die Ligation und der daraus abgeleiteten Werte für Aktivierungsenergie und Reaktionsrate mit Literaturwerten für die cis-trans-Isomerierung. – ader Literatur entnommen.[250] bextrapoliert.
Während die Aktivierungsenergie einer cis-trans-Isomerisierung und die der Ligation
dieselbe Größenordnung aufweisen, unterscheiden sich die Geschwindigkeitskonstanten bei
37 °C um drei und die entsprechenden Reaktionsraten um vier Größenordnungen zu Gunsten
der Isomerisierung. Trotz dieser Daten ist es schwierig, den Einfluss der gehinderten Rotation
auf eine mögliche templatierte Ligation abzuschätzen: In den bekannten PNA-Komplexen
liegen alle tertiären Amide in der cis-Konformation vor (vgl. 4.4.1), was daher auch für den
potentiellen ternären Komplex ABT und den resultierenden Produkt-Duplex T2 angenommen
werden kann. In beiden Fällen müssen acht oder zwölf Amidbindungen cis-orientiert sein,
abhängig davon, welche Rolle die Konformation der nicht durch H-Brücken gebundenen
Adenin- und Fluoraromat-Reste spielt. Das Ausmaß einer möglichen Präorientierung ist für
das vorliegende System nicht eindeutig bestimmt. Die NMR-Untersuchungen an den
Trimeren 224 und 248 haben gezeigt, dass diese als Rotamerengemisch vorliegen, wobei
keine Aussagen zur energetischen Bevorzugung bestimmter Konformationen gemacht werden
konnten. Aus 1D- und 2D-NMR-Untersuchungen von Chen et al. ist hingegen bekannt, dass
ein nicht-selbstkomplementäres PNA-Oktamer (NAla-gcacagcc-LysC) bevorzugt in der
cis-Konformation vorliegt.[250] Darüber hinaus ist die Kinetik der Komplexbildung nicht
aufgeklärt, z.B. inwiefern ein oder zwei intakte Basenpaare in cis-Konformation als
Nukleationskeime fungieren und Einfluss auf die Rotamerenpopulation sowie auf den
Fortgang der Komplexierung nehmen. Ist dies der Fall, kann die Aktivierungsenergie für die
komplette cis-trans-Isomerisierung eines Oligomers oder eines Komplexes nicht additiv aus
dem Wert einer einzelnen Isomerisierung berechnet werden. Um den möglichen Einfluss
einer Prääquilibrierung der Rotamerenpopulation auf die Ligation zu untersuchen, wurde eine
Lösung der Trimere und des Hexamers (10 bzw. 8 mM) in 1-Methylimidazol-Puffer (0.8 M,

Allgemeiner Teil 5 Ligation und Replikation
145
pH 7.5) auf zwei Ansätze verteilt. Ein Ansatz wurde 72 h bei -20 °C eingefroren, der andere
verblieb bei Raumtemperatur. Danach wurden beide Ansätze mit dem gleichen Volumen
einer 0.4 M wässrigen Lösung von EDC versetzt und der Reaktionsverlauf bei
Raumtemperatur durch HPLC-Analyse verfolgt und verglichen. Dabei ließen sich keine
Unterschiede feststellen.
Imidazol-Puffer wurde u.a. von Mattes und Seitz in ihren Arbeiten zur templatierten Ligation
komplementärer PNAs verwendet (vgl. 1.4.7).[175, 176] Wie bei den in Tabelle 3 vorgestellten
Screeningexperimenten konnte ohne Templatzugabe nur ca. 6 % an Ligationsprodukt nach-
gewiesen werden. Aufgrund dieser Parallele wurden auch im Rahmen dieser Arbeit
Replikationsexperimente in Imidazol-Puffer durchgeführt, wobei die Reaktionstemperaturen
mit 0 bzw. 10 °C den hier vorliegenden kürzeren Sequenzen und der Fehlpaarung angepasst
wurden (Abbildung 136, Tabelle 7). Qualitativ betrachtet weisen die Konzentrationsverläufe
mit und ohne Templatzugabe deutlichere Unterschiede auf, als es bei Verwendung von
1-Methylimidazol der Fall ist (Abbildung 133). Dies wird durch die kinetische Auswertung
bestätigt: Sowohl bei 0 als auch bei 10 °C führt das B-Modell zu einer besseren Anpassung an
die Datenpunkte als das A-Modell und für den autokatalytischen Überschuss-Faktor ε können
Werte von 12 bzw. 33 M-1/2 ermittelt werden. Den größten Effekt des Templats zeigt somit die
Versuchsreihe bei 10 °C. Generell haben niedrige Temperaturen einen positiven Einfluss auf
die Effizienz der Ligation, wie ein parallel bei Raumtemperatur durchgeführtes Experiment
zeigt. Hier erreicht die Produktbildung deutlich früher ihre Sättigung als bei 0 bzw. 10 °C, ein
Effekt, der sowohl durch das Fehlen des autokatalytischen Reaktionskanals (ε = 0.14 M-1/2) als
auch die Begünstigung der Nebenreaktionen (EDC-Hydrolyse, EDC-Addukte 284 und 288)
erklärt werden kann. Die Datenpunkte für 30 und 50 % Templatzugabe unterscheiden sich im
Rahmen der angenommenen Messungenauigkeit nicht, was sich wiederum durch die
TFA-katalysierte Hydrolyse des Kopplungsreagenzes und Fehler bei der Subtraktion der
HPLC-Integrale erklären lässt. Betrachtet man die Zeit bis zum 10%igen Umsatz, so ist die
Reaktion ohne Templatzugabe in Imidazol-Puffer zwar deutlich langsamer als in 1-Methyl-
imidazol-Puffer (16 bzw. 10 h), zeigt aber bei Zugabe von Hexamer 268 den größeren
Templateffekt (ε = 33 bzw. 6.4 M-1/2). Daher wurde zu diesem Zeitpunkt als Arbeitshypothese
angenommen, dass eine langsame Kopplung den templatierten Reaktionskanal generell
begünstigt. Aus den Arbeiten zur Selbstreplikation von DNA ist der gegenläufige Effekt be-
kannt. Hier führte die Variation der Verknüpfungschemie ausgehend von der Phosphodiester-

Allgemeiner Teil 5 Ligation und Replikation
146
über die Pyrophosphat- hin zur Phosphoamidatbindung zu schnelleren und gleichzeitig
effizienteren Replikatoren.[72, 75, 76]
Abbildung 136. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 in Imidazol-Puffer. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.4 M Imidazol-Puffer pH 7.5, 25 µl Ansatzgröße. 0 °C: cov(k1, k2) = 0.641, cov(k1, k3) = 0.229 und cov(k2, k3) = -0.558. 10 °C: cov(k1, k2) = 0.501, cov(k1, k3) = -0.153 und cov(k2, k3) = 0.740. RT: cov(k1, k2) = -0.670, cov(k1, k3) = -0.968 und cov(k2, k3) = 0.833.
Tabelle 7. Kinetische Auswertung der Ligation in Imidazol-Puffer.
Nr. T (°C) Fit RMS (%) k1 (M-1 s-1) k2 (s
-1) k3 (M-3/2 s-1) ε (M
-1/2)
1 0 A 1.75 (4.5±0.4)×10-4 (5±1)×10-6 - -
2 B 0.977 (3.3±0.2)×10-4 (5.5±0.6)×10-6 (3.7±0.5)×10-3 12
3 10 A 4.06 (7±1)×10-4 (5±2)×10-6 - -
4 B 1.35 (3.6±0.2)×10-4 (5.7±0.6)×10-6 (1.18±0.09)×10-2 33
5 RT A 0.112 (6.7±0.1)×10-4 (1.72±0.03)×10-5 - -
6 B 0.130 (6.6±0.5)×10-4 (1.69±0.07)×10-5 (0.09±4.90)×10-3 0.14
0 10 20 30
0.0
0.2
0.4
0.6
0.8
1.0
[T] (
mM
)
t (h)
0 % T, 0 °C30 % T, 0 °C50 % T, 0 °C 0 % T, RT
A
0 10 20 30
0.0
0.4
0.8
1.2
1.6B
[T] (
mM
)
t (h)
0 % T, 10 °C30 % T, 10 °C50 % T, 10 °C 0 % T, RT

Allgemeiner Teil 5 Ligation und Replikation
147
5.2.4 Ligation und Replikation in Sulfonsäure-Puffern
Die oben dargestellten Experimente zeigten einen merklichen Einfluss des verwendeten
nukleophilen Katalysators auf die Reaktionsgeschwindigkeit und die mögliche Templatierung
der Ligation. Daher wurden nun der nukleophile Katalysator und weitere Reaktionsbe-
dingungen variiert. Ziel war es einerseits, den postulierten termolekularen Komplex zu
stabilisieren, zumal schon die in Abbildung 130 gezeigte Schmelzkurve des Hexamers keinen
definierten Schmelzpunkt erkennen ließ. Andererseits sollte die Ligationsgeschwindigkeit
weiter herabgesetzt werden, da als Hypothese angenommen wurde, dass dies den
templatierten Reaktionskanal begünstigt (s.o.).
Da nicht alle potentiellen nukleophilen Katalysatoren wie Imidazol auch als Puffersubstanz
dienen können, galt es zunächst einen geeigneten Puffer auszuwählen, dem anschließend die
jeweiligen Katalysatoren zugesetzt werden sollten. Hierzu wurden Ligationen in MOPS- und
HEPES-Puffer in An- und Abwesenheit von Imidazol bzw. 1-Methylimidazol durchgeführt
(Abbildung 137, Tabelle 8). Um eine effektive Pufferung zu erreichen, wurde als pH- der
jeweilige pKa-Wert der Puffersubstanz gewählt. Ohne Zusatz eines nukleophilen Katalysators
ließ sich für die Ligation in HEPES-Puffer bei 10 °C nur ein sehr geringer Templateffekt
ausmachen (ε = 5 M-1/2), für die analoge Reaktion in MOPS-Puffer hingegen der bislang
deutlichste (ε = 91 M-1/2). In Anwesenheit von Imidazol konnten für beide Puffersysteme
ähnlich starke Templateffekte nachgewiesen werden (ε = 59 bzw. 40 M-1/2). Diese Experimen-
te zeigen, dass der Zusammenhang zwischen Reaktionsgeschwindigkeit und Templatierung
komplexer ist, als zunächst angenommen wurde. Denn trotz eines vergleichbaren
Templateffekts verläuft die Imidazol-katalysierte Reaktion in MOPS- wesentlich schneller als
in HEPES-Puffer. Da die Ligation in MOPS-Puffer außerdem zu deutlich höheren Ausbeuten
führt, wurde dieser auch für die folgenden Experimente verwendet. Bei Zusatz von 1-Methyl-
imidazol zeigte sich kein Templateffekt, dafür ergab die entsprechende Reaktion bei
Raumtemperatur mit gut 60 % eine ähnlich hohe Ausbeute wie die Verwendung von
1-Methylimidazol-Puffer (vgl. 5.1). Die kinetische Modellierung der bei Raumtemperatur
durchgeführten Experimente nach dem B-Modell ergab generell starke Kovarianzen und sehr
kleine Werten für ε. Da zudem die RMS-Werte von A- und B-Modell ähnlich sind, kann man
unter diesen Bedingungen nicht von nennenswerter Selbstreplikation ausgehen.
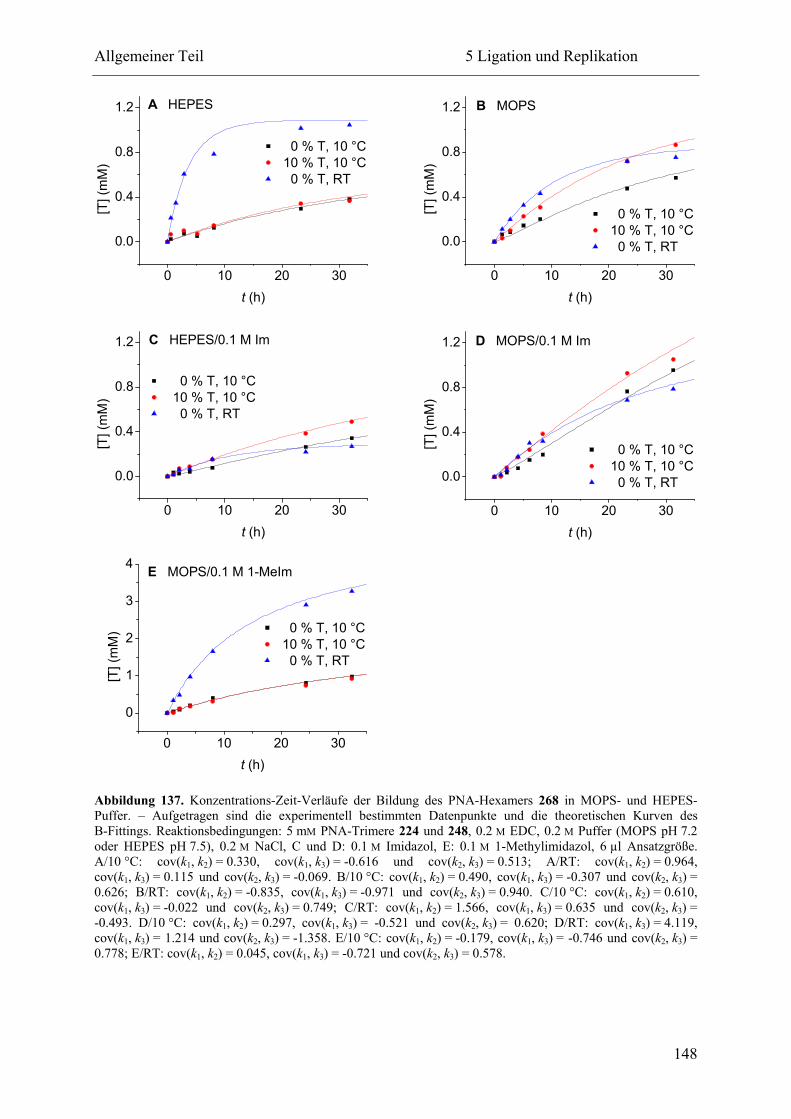
Allgemeiner Teil 5 Ligation und Replikation
148
Abbildung 137. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 in MOPS- und HEPES-Puffer. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M Puffer (MOPS pH 7.2 oder HEPES pH 7.5), 0.2 M NaCl, C und D: 0.1 M Imidazol, E: 0.1 M 1-Methylimidazol, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = 0.330, cov(k1, k3) = -0.616 und cov(k2, k3) = 0.513; A/RT: cov(k1, k2) = 0.964, cov(k1, k3) = 0.115 und cov(k2, k3) = -0.069. B/10 °C: cov(k1, k2) = 0.490, cov(k1, k3) = -0.307 und cov(k2, k3) = 0.626; B/RT: cov(k1, k2) = -0.835, cov(k1, k3) = -0.971 und cov(k2, k3) = 0.940. C/10 °C: cov(k1, k2) = 0.610, cov(k1, k3) = -0.022 und cov(k2, k3) = 0.749; C/RT: cov(k1, k2) = 1.566, cov(k1, k3) = 0.635 und cov(k2, k3) = -0.493. D/10 °C: cov(k1, k2) = 0.297, cov(k1, k3) = -0.521 und cov(k2, k3) = 0.620; D/RT: cov(k1, k3) = 4.119, cov(k1, k3) = 1.214 und cov(k2, k3) = -1.358. E/10 °C: cov(k1, k2) = -0.179, cov(k1, k3) = -0.746 und cov(k2, k3) = 0.778; E/RT: cov(k1, k2) = 0.045, cov(k1, k3) = -0.721 und cov(k2, k3) = 0.578.
0 10 20 30
0.0
0.4
0.8
1.2[T
] (m
M)
t (h)
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
A HEPES
0 10 20 30
0.0
0.4
0.8
1.2 B MOPS
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
[T] (
mM
)
t (h)
0 10 20 30
0
1
2
3
4E MOPS/0.1 M 1-MeIm
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2
0 % T, 10 °C 10 % T, 10 °C 0 % T, RT
C HEPES/0.1 M Im
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2
0 % T, 10 °C 10 % T, 10 °C 0 % T, RT
D MOPS/0.1 M Im
[T] (
mM
)
t (h)

Allgemeiner Teil 5 Ligation und Replikation
149
Tabelle 8. Kinetische Auswertung der Ligation in MOPS- und HEPES-Puffer.
Nr. Puffer Kat. T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 HEPES - 10 A 1.08 (2.2±0.2)×10-4 (3.5±0.8)×10-6 - -
2 B 1.05 (2.0±0.2)×10-4 (3.8±0.9)×10-6 (1±1)×10-3 5
3 RT A 2.12 (3.4±0.2)×10-3 (2.8±0.2)×10-5 - -
4 B 2.23 (3.2±0.2)×10-3 (3.0±0.2)×10-5 (2.0±0.2)×10-2 6.7
5 MOPS - 10 A 3.18 (4.2±0.5)×10-4 (3±1)×10-6 - -
6 B 0.864 (1.7±0.1)×10-4 (4.9±0.3)×10-6 (1.55±0.06)×10-2 91
7 RT A 0.359 (9.3±0.1)×10-4 (1.04±0.02)×10-5 - -
8 B 0.359 (9.3±0.5)×10-4 (1.04±0.07)×10-5 (0.1±4.8)×10-4 0.01
9 HEPES Im 10 A 1.70 (1.8±0.3)×10-4 (1±1)×10-6 - -
10 B 0.477 (1.04±0.05)×10-4 (2.8±0.4)×10-6 (5.9±0.3)×10-3 59
11 RT A 0.539 (2.9±0.2)×10-4 (1.1±0.1)×10-5 - -
12 B 0.539 (2.7±0.2)×10-4 (1.21±0.09)×10-5 (3±2)×10-3 11
13 MOPS Im 10 A 2.68 (4.7±0.3)×10-4 (1±6)×10-7 - -
14 B 1.47 (2.5±0.2)×10-4 (8±3)×10-7 (9.9±0.9)×10-3 40
15 RT A 1.10 (5.4±0.2)×10-4 (3.9±0.5)×10-6 - -
16 B 1.10 (5.4±0.2)×10-4 (4.1±0.3)×10-6 (1±2)×10-3 2
17 MOPS 1-MeIm 10 A 1.22 (5.6±0.2)×10-4 (2.5±0.4)×10-6 - -
18 B 1.28 (5.5±0.4)×10-4 (2.2±0.6)×10-6 (0.1±2.1)×10-3 0.2
19 RT A 2.14 (3.50±0.08)×10-3 (0.5±2.1)×10-7 - -
20 B 1.99 (3.5±0.1)×10-3 (0.3±2.5)×10-7 (2±4)×10-3 0.6
Die Produktverteilung nach zwei Tagen zeigt, dass der Zusatz von Imidazolen die Bildung
von Nebenprodukten bei 10 °C nahezu vollständig und bei Raumtemperatur um wenigstens
die Hälfte zurückdrängt (Tabelle 9). Der Fortgang der Reaktionen wird bei 10 °C oder
Imidazolzugabe nicht durch den Verbrauch des N-terminalen Trimers 248 in Nebenreaktionen
begrenzt. Auch das Guanidinium-Capping des C-terminalen Trimers 224 spielt keine
entscheidende Rolle (Daten nicht gezeigt). Wahrscheinlicher ist die Limitierung durch
Carbonsäure-katalysierte EDC-Hydrolyse.

Allgemeiner Teil 5 Ligation und Replikation
150
Tabelle 9. Produktverteilung der Ligation in MOPS- und HEPES-Puffer ohne initiale Templatzugabe nach zwei Tagen Reaktionszeit. – Das Nebenprodukt mit der Masse 1200 konnte nicht zugeordnet werden, aufgrund seiner Retentionszeit kann angenommen werden, dass es den Fluoraromaten enthält (vgl. 5.2.1).
Nr. Puffer Kat. T (°C) 268 (%) 248 (%) 248+EDC (%) M = 1200 (%)
1 HEPES - 10 11 60 20 9
2 RT 23 6 52 19
3 MOPS - 10 14 46 30 10
4 RT 18 7 55 20
5 HEPES Im 10 11 88 < 1 < 1
6 RT 6 69 18 7
7 MOPS Im 10 24 73 1 2
8 RT 18 52 23 7
9 MOPS 1-MeIm 10 28 69 1 2
10 RT 69 11 14 5
Als alternative Zusätze wurden Pyridin (289) und HOAt (290) getestet (Abbildung 139,
Tabelle 10). Beide Substanzen konnten von Richert und Mitarbeitern im Kontext ihrer
Arbeiten zur RNA- bzw. DNA-Primerverlängerung in umfangreichen Screenings als effektive
nukleophile Katalysatoren identifiziert werden.[259-265] In Gegenwart von Pyridin veläuft die
Ligation bei Raumtemperatur mit 69 % Ausbeute an hexa-PNA 268 ebenso effizient wie in
Anwesenheit von 1-Methylimidazol (s.o.). Allerdings ist der Einfluss initialer Templatzugabe
bei 10 °C auf den Reaktionsverlauf gering (ε = 0.19 M-1/2). Dieser Zusammenhang konnte
bereits mehrfach bei den vorangegangenen Experimenten beobachtet werden: Reaktionsbe-
dingungen die bei Raumtemperatur zu einer effizienten Ligation führen, bedingen bei 10 °C
keine oder eine sehr geringen Auswirkung des zugesetzten Hexamers. Ist der Templateffekt
indessen unter den gegebenen Bedingungen ausgeprägter, erhält man nach einem Tag bei
10 °C mehr Hexamer als bei Raumtemperatur. Die mit Abstand schnellsten und effizientesten
Ligationen konnten bei Zusatz des O-Nukleophils HOAt beobachtet werden. So wurden
bereits nach einer halben Stunde Reaktionszeit 30-40 % neu gebildetes Hexamer nach-
gewiesen. Als Ausnahme zum oben Gesagten war hier trotz effizienter Ligation bei Raum-
temperatur die Ausbeute nach einem Tag bei 10 °C größer. Betrachtet man die in Abbildung
138 dargestellten Messpunkte, erkennt man außerdem einen tendenziell negativen Effekt der
zugesetzten hexa-PNA auf die Reaktionsgeschwindigkeit und die Ausbeute.
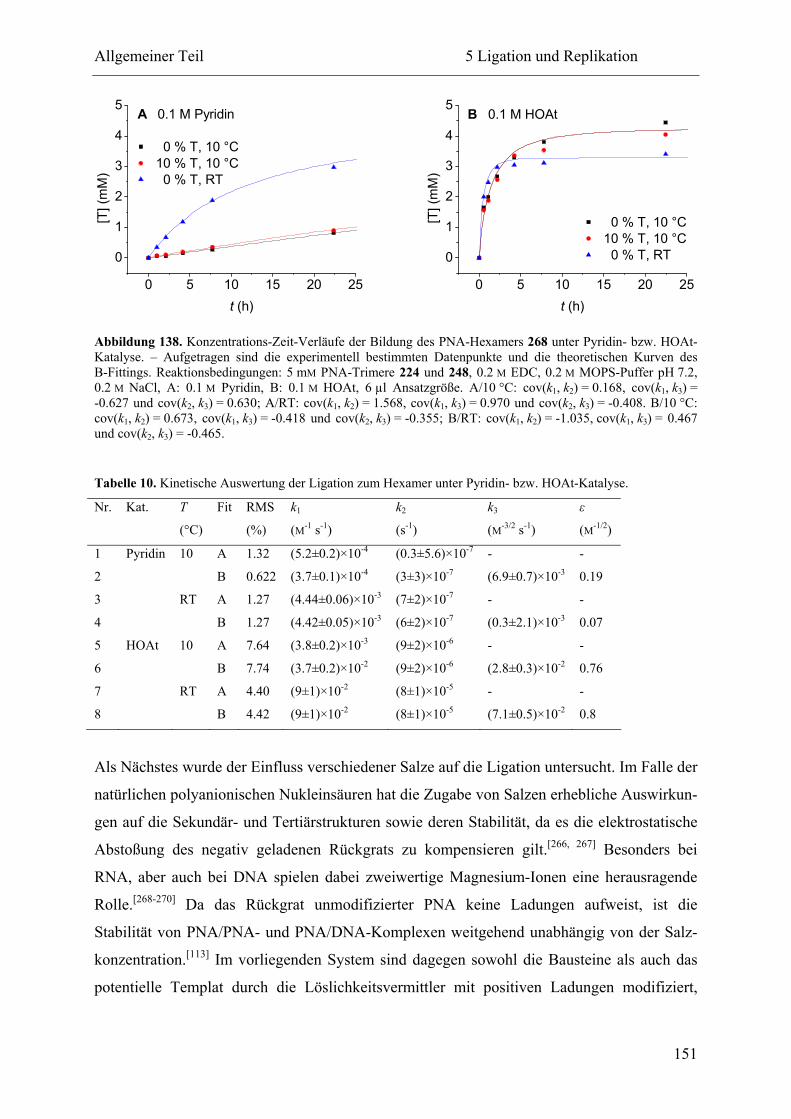
Allgemeiner Teil 5 Ligation und Replikation
151
Abbildung 138. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 unter Pyridin- bzw. HOAt-Katalyse. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.2, 0.2 M NaCl, A: 0.1 M Pyridin, B: 0.1 M HOAt, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = 0.168, cov(k1, k3) = -0.627 und cov(k2, k3) = 0.630; A/RT: cov(k1, k2) = 1.568, cov(k1, k3) = 0.970 und cov(k2, k3) = -0.408. B/10 °C: cov(k1, k2) = 0.673, cov(k1, k3) = -0.418 und cov(k2, k3) = -0.355; B/RT: cov(k1, k2) = -1.035, cov(k1, k3) = 0.467 und cov(k2, k3) = -0.465.
Tabelle 10. Kinetische Auswertung der Ligation zum Hexamer unter Pyridin- bzw. HOAt-Katalyse.
Nr. Kat. T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 Pyridin 10 A 1.32 (5.2±0.2)×10-4 (0.3±5.6)×10-7 - -
2 B 0.622 (3.7±0.1)×10-4 (3±3)×10-7 (6.9±0.7)×10-3 0.19
3 RT A 1.27 (4.44±0.06)×10-3 (7±2)×10-7 - -
4 B 1.27 (4.42±0.05)×10-3 (6±2)×10-7 (0.3±2.1)×10-3 0.07
5 HOAt 10 A 7.64 (3.8±0.2)×10-3 (9±2)×10-6 - -
6 B 7.74 (3.7±0.2)×10-2 (9±2)×10-6 (2.8±0.3)×10-2 0.76
7 RT A 4.40 (9±1)×10-2 (8±1)×10-5 - -
8 B 4.42 (9±1)×10-2 (8±1)×10-5 (7.1±0.5)×10-2 0.8
Als Nächstes wurde der Einfluss verschiedener Salze auf die Ligation untersucht. Im Falle der
natürlichen polyanionischen Nukleinsäuren hat die Zugabe von Salzen erhebliche Auswirkun-
gen auf die Sekundär- und Tertiärstrukturen sowie deren Stabilität, da es die elektrostatische
Abstoßung des negativ geladenen Rückgrats zu kompensieren gilt.[266, 267] Besonders bei
RNA, aber auch bei DNA spielen dabei zweiwertige Magnesium-Ionen eine herausragende
Rolle.[268-270] Da das Rückgrat unmodifizierter PNA keine Ladungen aufweist, ist die
Stabilität von PNA/PNA- und PNA/DNA-Komplexen weitgehend unabhängig von der Salz-
konzentration.[113] Im vorliegenden System sind dagegen sowohl die Bausteine als auch das
potentielle Templat durch die Löslichkeitsvermittler mit positiven Ladungen modifiziert,
0 5 10 15 20 25
0
1
2
3
4
5
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
A 0.1 M Pyridin[T
] (m
M)
t (h)
0 5 10 15 20 25
0
1
2
3
4
5B 0.1 M HOAt
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
[T] (
mM
)
t (h)
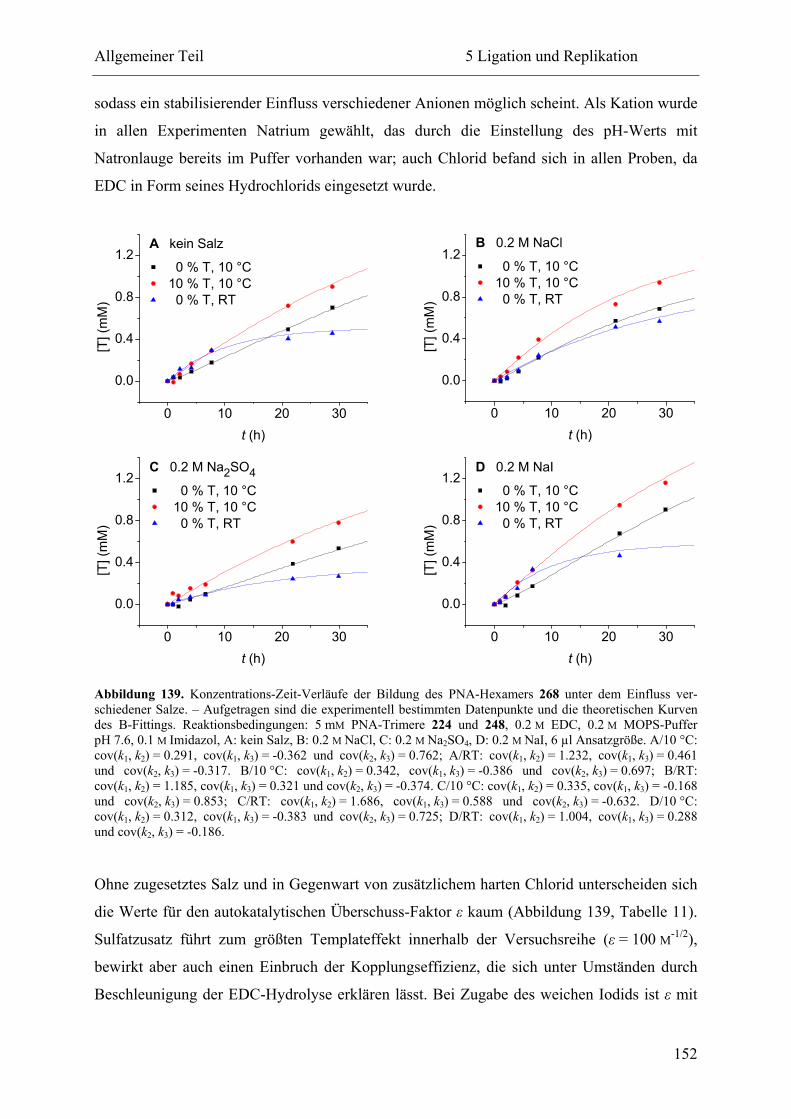
Allgemeiner Teil 5 Ligation und Replikation
152
sodass ein stabilisierender Einfluss verschiedener Anionen möglich scheint. Als Kation wurde
in allen Experimenten Natrium gewählt, das durch die Einstellung des pH-Werts mit
Natronlauge bereits im Puffer vorhanden war; auch Chlorid befand sich in allen Proben, da
EDC in Form seines Hydrochlorids eingesetzt wurde.
Abbildung 139. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 unter dem Einfluss ver-schiedener Salze. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.1 M Imidazol, A: kein Salz, B: 0.2 M NaCl, C: 0.2 M Na2SO4, D: 0.2 M NaI, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = 0.291, cov(k1, k3) = -0.362 und cov(k2, k3) = 0.762; A/RT: cov(k1, k2) = 1.232, cov(k1, k3) = 0.461 und cov(k2, k3) = -0.317. B/10 °C: cov(k1, k2) = 0.342, cov(k1, k3) = -0.386 und cov(k2, k3) = 0.697; B/RT: cov(k1, k2) = 1.185, cov(k1, k3) = 0.321 und cov(k2, k3) = -0.374. C/10 °C: cov(k1, k2) = 0.335, cov(k1, k3) = -0.168 und cov(k2, k3) = 0.853; C/RT: cov(k1, k2) = 1.686, cov(k1, k3) = 0.588 und cov(k2, k3) = -0.632. D/10 °C: cov(k1, k2) = 0.312, cov(k1, k3) = -0.383 und cov(k2, k3) = 0.725; D/RT: cov(k1, k2) = 1.004, cov(k1, k3) = 0.288 und cov(k2, k3) = -0.186.
Ohne zugesetztes Salz und in Gegenwart von zusätzlichem harten Chlorid unterscheiden sich
die Werte für den autokatalytischen Überschuss-Faktor ε kaum (Abbildung 139, Tabelle 11).
Sulfatzusatz führt zum größten Templateffekt innerhalb der Versuchsreihe (ε = 100 M-1/2),
bewirkt aber auch einen Einbruch der Kopplungseffizienz, die sich unter Umständen durch
Beschleunigung der EDC-Hydrolyse erklären lässt. Bei Zugabe des weichen Iodids ist ε mit
0 10 20 30
0.0
0.4
0.8
1.2 0 % T, 10 °C 10 % T, 10 °C 0 % T, RT
C 0.2 M Na2SO4
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2 0 % T, 10 °C10 % T, 10 °C 0 % T, RT
D 0.2 M NaI
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2 0 % T, 10 °C10 % T, 10 °C 0 % T, RT
A kein Salz
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2 0 % T, 10 °C10 % T, 10 °C 0 % T, RT
B 0.2 M NaCl
[T] (
mM
)t (h)
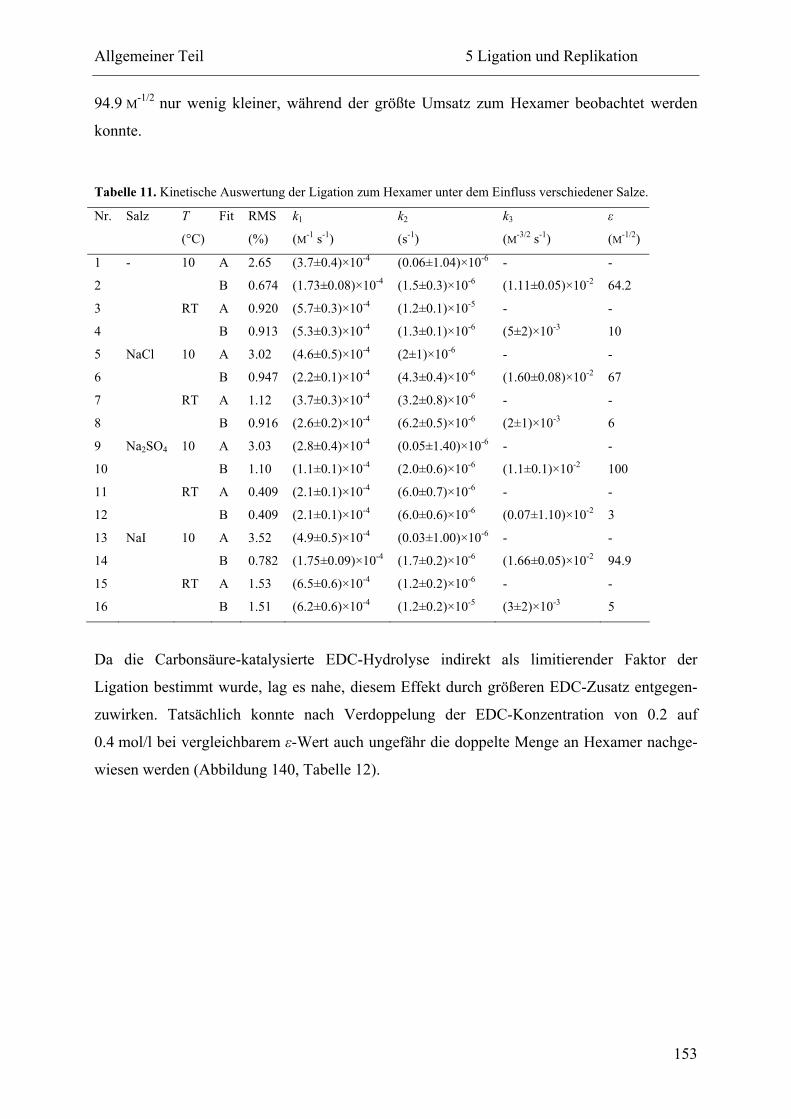
Allgemeiner Teil 5 Ligation und Replikation
153
94.9 M-1/2 nur wenig kleiner, während der größte Umsatz zum Hexamer beobachtet werden
konnte.
Tabelle 11. Kinetische Auswertung der Ligation zum Hexamer unter dem Einfluss verschiedener Salze.
Nr. Salz T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 - 10 A 2.65 (3.7±0.4)×10-4 (0.06±1.04)×10-6 - -
2 B 0.674 (1.73±0.08)×10-4 (1.5±0.3)×10-6 (1.11±0.05)×10-2 64.2
3 RT A 0.920 (5.7±0.3)×10-4 (1.2±0.1)×10-5 - -
4 B 0.913 (5.3±0.3)×10-4 (1.3±0.1)×10-6 (5±2)×10-3 10
5 NaCl 10 A 3.02 (4.6±0.5)×10-4 (2±1)×10-6 - -
6 B 0.947 (2.2±0.1)×10-4 (4.3±0.4)×10-6 (1.60±0.08)×10-2 67
7 RT A 1.12 (3.7±0.3)×10-4 (3.2±0.8)×10-6 - -
8 B 0.916 (2.6±0.2)×10-4 (6.2±0.5)×10-6 (2±1)×10-3 6
9 Na2SO4 10 A 3.03 (2.8±0.4)×10-4 (0.05±1.40)×10-6 - -
10 B 1.10 (1.1±0.1)×10-4 (2.0±0.6)×10-6 (1.1±0.1)×10-2 100
11 RT A 0.409 (2.1±0.1)×10-4 (6.0±0.7)×10-6 - -
12 B 0.409 (2.1±0.1)×10-4 (6.0±0.6)×10-6 (0.07±1.10)×10-2 3
13 NaI 10 A 3.52 (4.9±0.5)×10-4 (0.03±1.00)×10-6 - -
14 B 0.782 (1.75±0.09)×10-4 (1.7±0.2)×10-6 (1.66±0.05)×10-2 94.9
15 RT A 1.53 (6.5±0.6)×10-4 (1.2±0.2)×10-6 - -
16 B 1.51 (6.2±0.6)×10-4 (1.2±0.2)×10-5 (3±2)×10-3 5
Da die Carbonsäure-katalysierte EDC-Hydrolyse indirekt als limitierender Faktor der
Ligation bestimmt wurde, lag es nahe, diesem Effekt durch größeren EDC-Zusatz entgegen-
zuwirken. Tatsächlich konnte nach Verdoppelung der EDC-Konzentration von 0.2 auf
0.4 mol/l bei vergleichbarem ε-Wert auch ungefähr die doppelte Menge an Hexamer nachge-
wiesen werden (Abbildung 140, Tabelle 12).

Allgemeiner Teil 5 Ligation und Replikation
154
Abbildung 140. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 bei zwei verschiedenen EDC-Konzentrationen. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, A: 0.4 M EDC, B: 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.1 M Imidazol, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = -0.316, cov(k1, k3) = -0.814 und cov(k2, k3) = 0.779; A/RT: cov(k1, k2) = 1.377, cov(k1, k3) = 0.596 und cov(k2, k3) = -0.397. B/10 °C: cov(k1, k2) = 0.342, cov(k1, k3) = -0.386 und cov(k2, k3) = 0.697; B/RT: cov(k1, k2) = 1.185, cov(k1, k3) = 0.321 und cov(k2, k3) = 0.374.
Tabelle 12. Kinetische Auswertung der Ligation zum Hexamer bei zwei verschiedenen EDC-Konzentrationen.
Nr. EDC T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 0.4 10 A 3.18 (9.9±0.7)×10-4 (5±8)×10-7 - -
2 B 0.674 (4.6±0.2)×10-4 (3.9±0.2)×10-6 (3.0±0.1)×10-2 65
3 RT A 0.592 (1.01±0.02)×10-3 (8.5±0.4)×10-6 - -
4 B 0.632 (9.5±0.2)×10-4 (9.3±0.4)×10-6 (6±1)×10-3 6
5 0.2 10 A 3.02 (4.6±0.5)×10-4 (2±1)×10-6 - -
6 B 0.947 (2.2±0.1)×10-4 (4.3±0.4)×10-6 (1.60±0.08)×10-2 67
7 RT A 1.12 (3.7±0.3)×10-4 (3.2±0.8)×10-6 - -
8 B 1.09 (3.5±0.3)×10-4 (3.7±0.7)×10-6 (2±1)×10-3 6
Nakajima und Ikada zeigten 1995, dass die Aktivierung einer Carbonsäure durch EDC am
effektivsten bei pH 3.5-4.5 verläuft, während die optimale Bildung der Peptidbindung bei
pH 4-6 stattfindet.[271] Da EDC besonders schnell im sauren pH-Bereich hydrolysiert wird,
werden EDC-vermittelte Kopplungen von Peptiden und Proteinen bevorzugt bei pH 4.5-7.5
durchgeführt.[272] Bei noch höheren pH-Werten sinkt die Reaktionsgeschwindigkeit und die
Ausbeute bricht ein. Tatsächlich findet auch die PNA-Ligation im untersuchten System mit
sinkendem pH-Wert schneller und effektiver statt (Abbildung 141). Betrachtet man den
autokatalytischen Überschuss-Faktor, so tritt mit ε = 112 M-1/2 der größte Templateffekt bei
pH 6.6 auf, der kleinste mit ε = 40 M-1/2 bei pH 7.2 (Tabelle 13).
0 5 10 15 20 25
0.0
0.4
0.8
1.2
1.6
2.0
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
A 0.4 M EDC[T
] (m
M)
t (h)
0 5 10 15 20 25
0.0
0.4
0.8
1.2
1.6
2.0
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
B 0.2 M EDC
[T] (
mM
)
t (h)
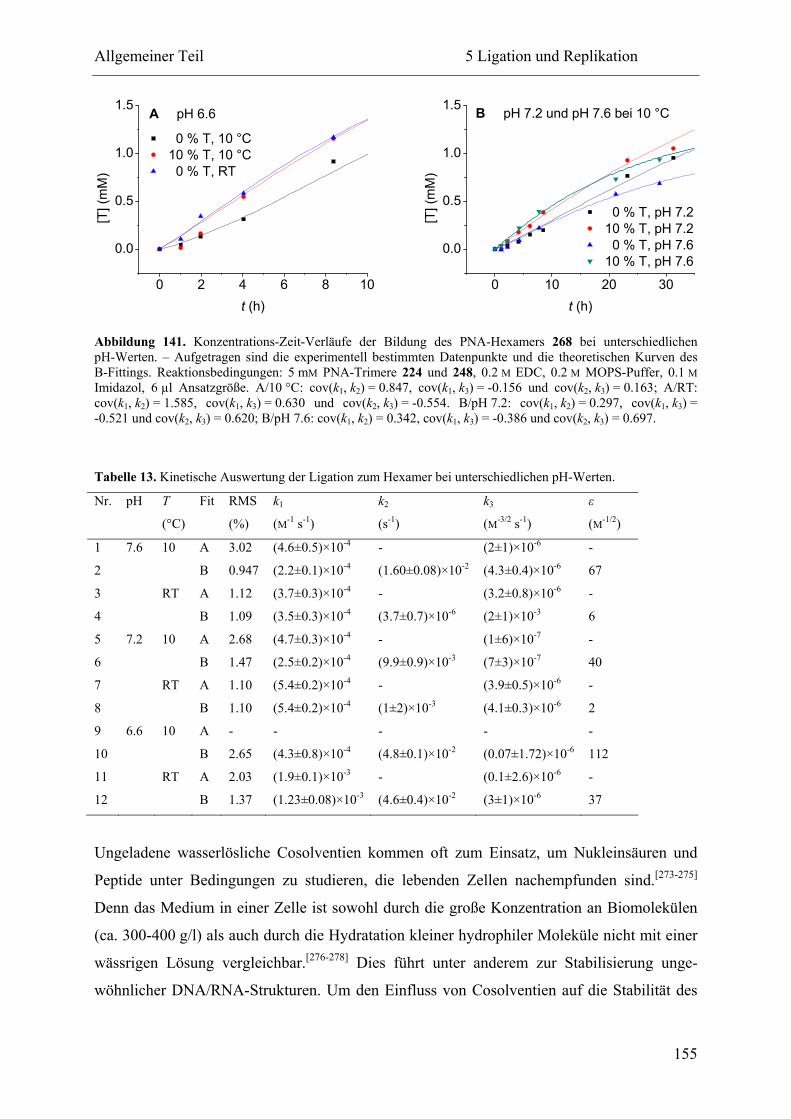
Allgemeiner Teil 5 Ligation und Replikation
155
Abbildung 141. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 bei unterschiedlichen pH-Werten. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer, 0.1 M Imidazol, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = 0.847, cov(k1, k3) = -0.156 und cov(k2, k3) = 0.163; A/RT: cov(k1, k2) = 1.585, cov(k1, k3) = 0.630 und cov(k2, k3) = -0.554. B/pH 7.2: cov(k1, k2) = 0.297, cov(k1, k3) = -0.521 und cov(k2, k3) = 0.620; B/pH 7.6: cov(k1, k2) = 0.342, cov(k1, k3) = -0.386 und cov(k2, k3) = 0.697.
Tabelle 13. Kinetische Auswertung der Ligation zum Hexamer bei unterschiedlichen pH-Werten.
Nr. pH T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 7.6 10 A 3.02 (4.6±0.5)×10-4 - (2±1)×10-6 -
2 B 0.947 (2.2±0.1)×10-4 (1.60±0.08)×10-2 (4.3±0.4)×10-6 67
3 RT A 1.12 (3.7±0.3)×10-4 - (3.2±0.8)×10-6 -
4 B 1.09 (3.5±0.3)×10-4 (3.7±0.7)×10-6 (2±1)×10-3 6
5 7.2 10 A 2.68 (4.7±0.3)×10-4 - (1±6)×10-7 -
6 B 1.47 (2.5±0.2)×10-4 (9.9±0.9)×10-3 (7±3)×10-7 40
7 RT A 1.10 (5.4±0.2)×10-4 - (3.9±0.5)×10-6 -
8 B 1.10 (5.4±0.2)×10-4 (1±2)×10-3 (4.1±0.3)×10-6 2
9 6.6 10 A - - - - -
10 B 2.65 (4.3±0.8)×10-4 (4.8±0.1)×10-2 (0.07±1.72)×10-6 112
11 RT A 2.03 (1.9±0.1)×10-3 - (0.1±2.6)×10-6 -
12 B 1.37 (1.23±0.08)×10-3 (4.6±0.4)×10-2 (3±1)×10-6 37
Ungeladene wasserlösliche Cosolventien kommen oft zum Einsatz, um Nukleinsäuren und
Peptide unter Bedingungen zu studieren, die lebenden Zellen nachempfunden sind.[273-275]
Denn das Medium in einer Zelle ist sowohl durch die große Konzentration an Biomolekülen
(ca. 300-400 g/l) als auch durch die Hydratation kleiner hydrophiler Moleküle nicht mit einer
wässrigen Lösung vergleichbar.[276-278] Dies führt unter anderem zur Stabilisierung unge-
wöhnlicher DNA/RNA-Strukturen. Um den Einfluss von Cosolventien auf die Stabilität des
0 2 4 6 8 10
0.0
0.5
1.0
1.5
0 % T, 10 °C10 % T, 10 °C 0 % T, RT
[T] (
mM
)
t (h)
A pH 6.6
0 10 20 30
0.0
0.5
1.0
1.5
0 % T, pH 7.210 % T, pH 7.2 0 % T, pH 7.610 % T, pH 7.6
[T] (
mM
)
t (h)
B pH 7.2 und pH 7.6 bei 10 °C
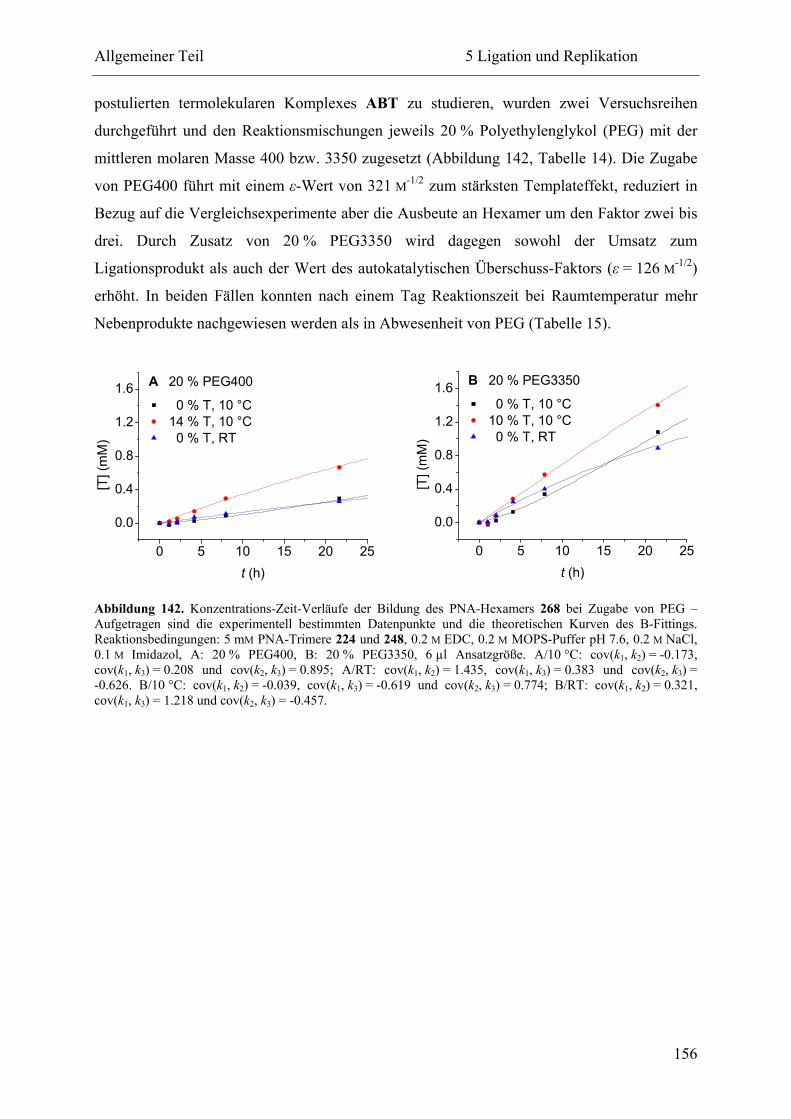
Allgemeiner Teil 5 Ligation und Replikation
156
postulierten termolekularen Komplexes ABT zu studieren, wurden zwei Versuchsreihen
durchgeführt und den Reaktionsmischungen jeweils 20 % Polyethylenglykol (PEG) mit der
mittleren molaren Masse 400 bzw. 3350 zugesetzt (Abbildung 142, Tabelle 14). Die Zugabe
von PEG400 führt mit einem ε-Wert von 321 M-1/2 zum stärksten Templateffekt, reduziert in
Bezug auf die Vergleichsexperimente aber die Ausbeute an Hexamer um den Faktor zwei bis
drei. Durch Zusatz von 20 % PEG3350 wird dagegen sowohl der Umsatz zum
Ligationsprodukt als auch der Wert des autokatalytischen Überschuss-Faktors (ε = 126 M-1/2)
erhöht. In beiden Fällen konnten nach einem Tag Reaktionszeit bei Raumtemperatur mehr
Nebenprodukte nachgewiesen werden als in Abwesenheit von PEG (Tabelle 15).
Abbildung 142. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 bei Zugabe von PEG – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.2 M NaCl, 0.1 M Imidazol, A: 20 % PEG400, B: 20 % PEG3350, 6 µl Ansatzgröße. A/10 °C: cov(k1, k2) = -0.173, cov(k1, k3) = 0.208 und cov(k2, k3) = 0.895; A/RT: cov(k1, k2) = 1.435, cov(k1, k3) = 0.383 und cov(k2, k3) = -0.626. B/10 °C: cov(k1, k2) = -0.039, cov(k1, k3) = -0.619 und cov(k2, k3) = 0.774; B/RT: cov(k1, k2) = 0.321, cov(k1, k3) = 1.218 und cov(k2, k3) = -0.457.
0 5 10 15 20 25
0.0
0.4
0.8
1.2
1.6 0 % T, 10 °C14 % T, 10 °C 0 % T, RT
A 20 % PEG400
[T] (
mM
)
t (h)
0 5 10 15 20 25
0.0
0.4
0.8
1.2
1.6 0 % T, 10 °C10 % T, 10 °C 0 % T, RT
B 20 % PEG3350
[T] (
mM
)
t (h)
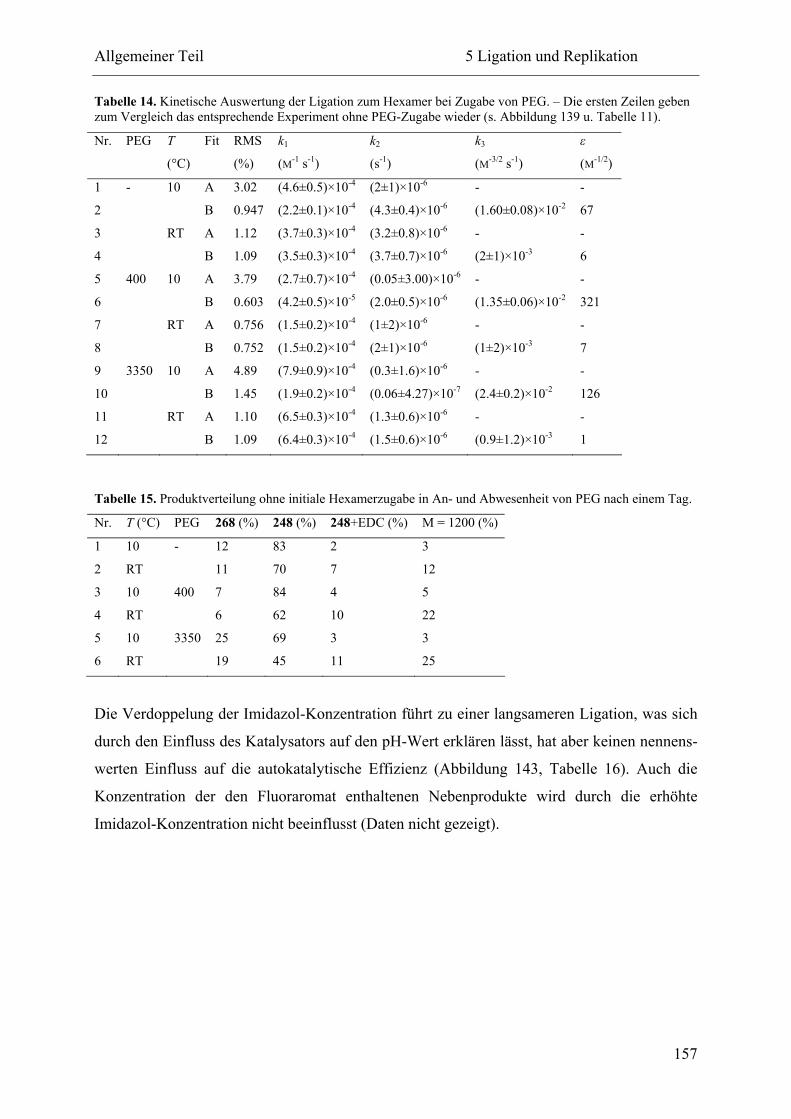
Allgemeiner Teil 5 Ligation und Replikation
157
Tabelle 14. Kinetische Auswertung der Ligation zum Hexamer bei Zugabe von PEG. – Die ersten Zeilen geben zum Vergleich das entsprechende Experiment ohne PEG-Zugabe wieder (s. Abbildung 139 u. Tabelle 11).
Nr. PEG T
(°C)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 - 10 A 3.02 (4.6±0.5)×10-4 (2±1)×10-6 - -
2 B 0.947 (2.2±0.1)×10-4 (4.3±0.4)×10-6 (1.60±0.08)×10-2 67
3 RT A 1.12 (3.7±0.3)×10-4 (3.2±0.8)×10-6 - -
4 B 1.09 (3.5±0.3)×10-4 (3.7±0.7)×10-6 (2±1)×10-3 6
5 400 10 A 3.79 (2.7±0.7)×10-4 (0.05±3.00)×10-6 - -
6 B 0.603 (4.2±0.5)×10-5 (2.0±0.5)×10-6 (1.35±0.06)×10-2 321
7 RT A 0.756 (1.5±0.2)×10-4 (1±2)×10-6 - -
8 B 0.752 (1.5±0.2)×10-4 (2±1)×10-6 (1±2)×10-3 7
9 3350 10 A 4.89 (7.9±0.9)×10-4 (0.3±1.6)×10-6 - -
10 B 1.45 (1.9±0.2)×10-4 (0.06±4.27)×10-7 (2.4±0.2)×10-2 126
11 RT A 1.10 (6.5±0.3)×10-4 (1.3±0.6)×10-6 - -
12 B 1.09 (6.4±0.3)×10-4 (1.5±0.6)×10-6 (0.9±1.2)×10-3 1
Tabelle 15. Produktverteilung ohne initiale Hexamerzugabe in An- und Abwesenheit von PEG nach einem Tag.
Nr. T (°C) PEG 268 (%) 248 (%) 248+EDC (%) M = 1200 (%)
1 10 - 12 83 2 3
2 RT 11 70 7 12
3 10 400 7 84 4 5
4 RT 6 62 10 22
5 10 3350 25 69 3 3
6 RT 19 45 11 25
Die Verdoppelung der Imidazol-Konzentration führt zu einer langsameren Ligation, was sich
durch den Einfluss des Katalysators auf den pH-Wert erklären lässt, hat aber keinen nennens-
werten Einfluss auf die autokatalytische Effizienz (Abbildung 143, Tabelle 16). Auch die
Konzentration der den Fluoraromat enthaltenen Nebenprodukte wird durch die erhöhte
Imidazol-Konzentration nicht beeinflusst (Daten nicht gezeigt).
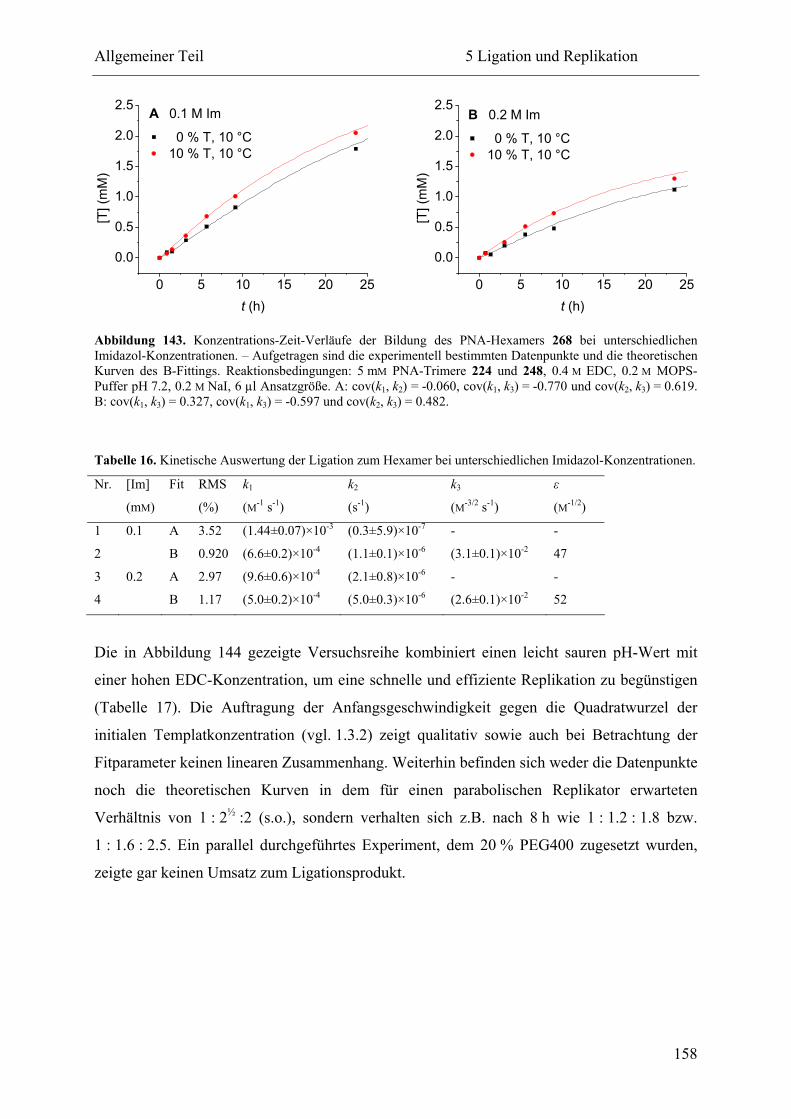
Allgemeiner Teil 5 Ligation und Replikation
158
Abbildung 143. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 bei unterschiedlichen Imidazol-Konzentrationen. – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des B-Fittings. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.4 M EDC, 0.2 M MOPS-Puffer pH 7.2, 0.2 M NaI, 6 µl Ansatzgröße. A: cov(k1, k2) = -0.060, cov(k1, k3) = -0.770 und cov(k2, k3) = 0.619. B: cov(k1, k3) = 0.327, cov(k1, k3) = -0.597 und cov(k2, k3) = 0.482.
Tabelle 16. Kinetische Auswertung der Ligation zum Hexamer bei unterschiedlichen Imidazol-Konzentrationen.
Nr. [Im]
(mM)
Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 0.1 A 3.52 (1.44±0.07)×10-3 (0.3±5.9)×10-7 - -
2 B 0.920 (6.6±0.2)×10-4 (1.1±0.1)×10-6 (3.1±0.1)×10-2 47
3 0.2 A 2.97 (9.6±0.6)×10-4 (2.1±0.8)×10-6 - -
4 B 1.17 (5.0±0.2)×10-4 (5.0±0.3)×10-6 (2.6±0.1)×10-2 52
Die in Abbildung 144 gezeigte Versuchsreihe kombiniert einen leicht sauren pH-Wert mit
einer hohen EDC-Konzentration, um eine schnelle und effiziente Replikation zu begünstigen
(Tabelle 17). Die Auftragung der Anfangsgeschwindigkeit gegen die Quadratwurzel der
initialen Templatkonzentration (vgl. 1.3.2) zeigt qualitativ sowie auch bei Betrachtung der
Fitparameter keinen linearen Zusammenhang. Weiterhin befinden sich weder die Datenpunkte
noch die theoretischen Kurven in dem für einen parabolischen Replikator erwarteten
Verhältnis von 1 : 2½ :2 (s.o.), sondern verhalten sich z.B. nach 8 h wie 1 : 1.2 : 1.8 bzw.
1 : 1.6 : 2.5. Ein parallel durchgeführtes Experiment, dem 20 % PEG400 zugesetzt wurden,
zeigte gar keinen Umsatz zum Ligationsprodukt.
0 5 10 15 20 25
0.0
0.5
1.0
1.5
2.0
2.5
0 % T, 10 °C10 % T, 10 °C
A 0.1 M Im[T
] (m
M)
t (h)
0 5 10 15 20 25
0.0
0.5
1.0
1.5
2.0
2.5
0 % T, 10 °C10 % T, 10 °C
B 0.2 M Im
[T] (
mM
)
t (h)
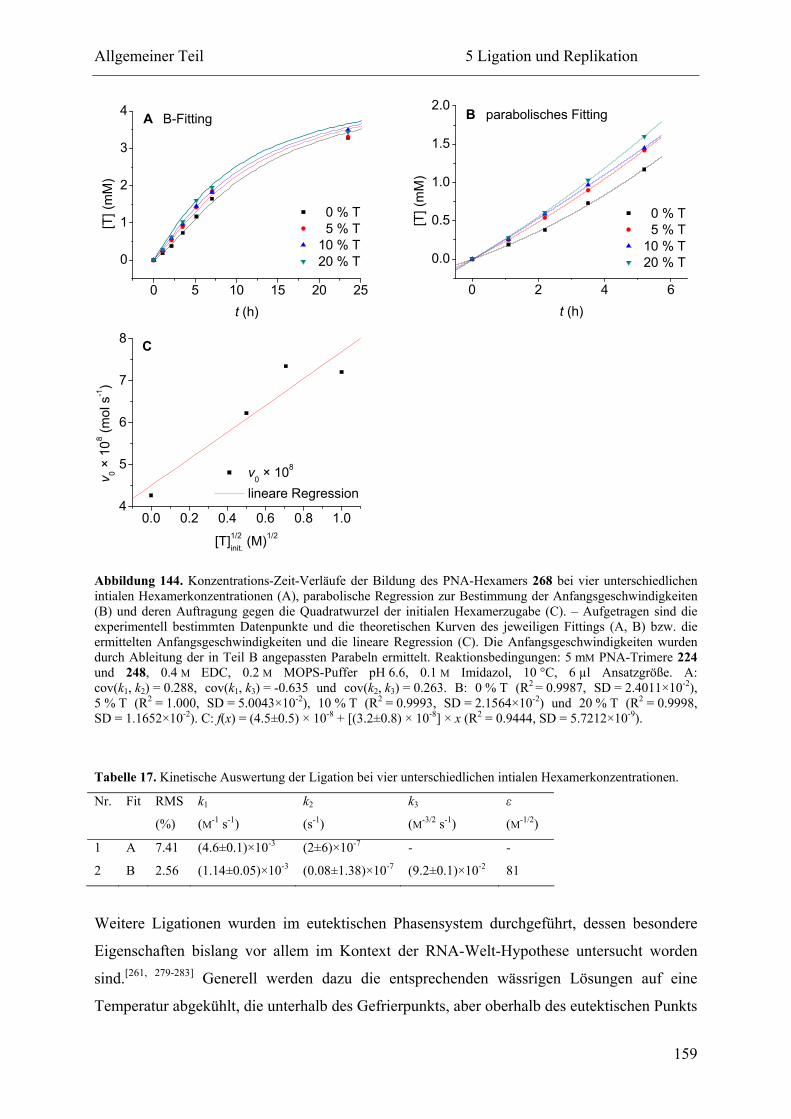
Allgemeiner Teil 5 Ligation und Replikation
159
Abbildung 144. Konzentrations-Zeit-Verläufe der Bildung des PNA-Hexamers 268 bei vier unterschiedlichen intialen Hexamerkonzentrationen (A), parabolische Regression zur Bestimmung der Anfangsgeschwindigkeiten (B) und deren Auftragung gegen die Quadratwurzel der initialen Hexamerzugabe (C). – Aufgetragen sind die experimentell bestimmten Datenpunkte und die theoretischen Kurven des jeweiligen Fittings (A, B) bzw. die ermittelten Anfangsgeschwindigkeiten und die lineare Regression (C). Die Anfangsgeschwindigkeiten wurden durch Ableitung der in Teil B angepassten Parabeln ermittelt. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.4 M EDC, 0.2 M MOPS-Puffer pH 6.6, 0.1 M Imidazol, 10 °C, 6 µl Ansatzgröße. A: cov(k1, k2) = 0.288, cov(k1, k3) = -0.635 und cov(k2, k3) = 0.263. B: 0 % T (R2 = 0.9987, SD = 2.4011×10-2), 5 % T (R2 = 1.000, SD = 5.0043×10-2), 10 % T (R2 = 0.9993, SD = 2.1564×10-2) und 20 % T (R2 = 0.9998, SD = 1.1652×10-2). C: f(x) = (4.5±0.5) × 10-8 + [(3.2±0.8) × 10-8] × x (R2 = 0.9444, SD = 5.7212×10-9).
Tabelle 17. Kinetische Auswertung der Ligation bei vier unterschiedlichen intialen Hexamerkonzentrationen.
Nr. Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 A 7.41 (4.6±0.1)×10-3 (2±6)×10-7 - -
2 B 2.56 (1.14±0.05)×10-3 (0.08±1.38)×10-7 (9.2±0.1)×10-2 81
Weitere Ligationen wurden im eutektischen Phasensystem durchgeführt, dessen besondere
Eigenschaften bislang vor allem im Kontext der RNA-Welt-Hypothese untersucht worden
sind.[261, 279-283] Generell werden dazu die entsprechenden wässrigen Lösungen auf eine
Temperatur abgekühlt, die unterhalb des Gefrierpunkts, aber oberhalb des eutektischen Punkts
0.0 0.2 0.4 0.6 0.8 1.04
5
6
7
8
v0 × 108
lineare Regression
v 0 ×
10
8 (
mol
s-1)
[T]1/2
init. (M)1/2
C
0 5 10 15 20 25
0
1
2
3
4
0 % T 5 % T10 % T20 % T
[T] (
mM
)
t (h)
A B-Fitting
0 2 4 6
0.0
0.5
1.0
1.5
2.0B parabolisches Fitting
0 % T 5 % T10 % T20 % T
[T] (
mM
)
t (h)
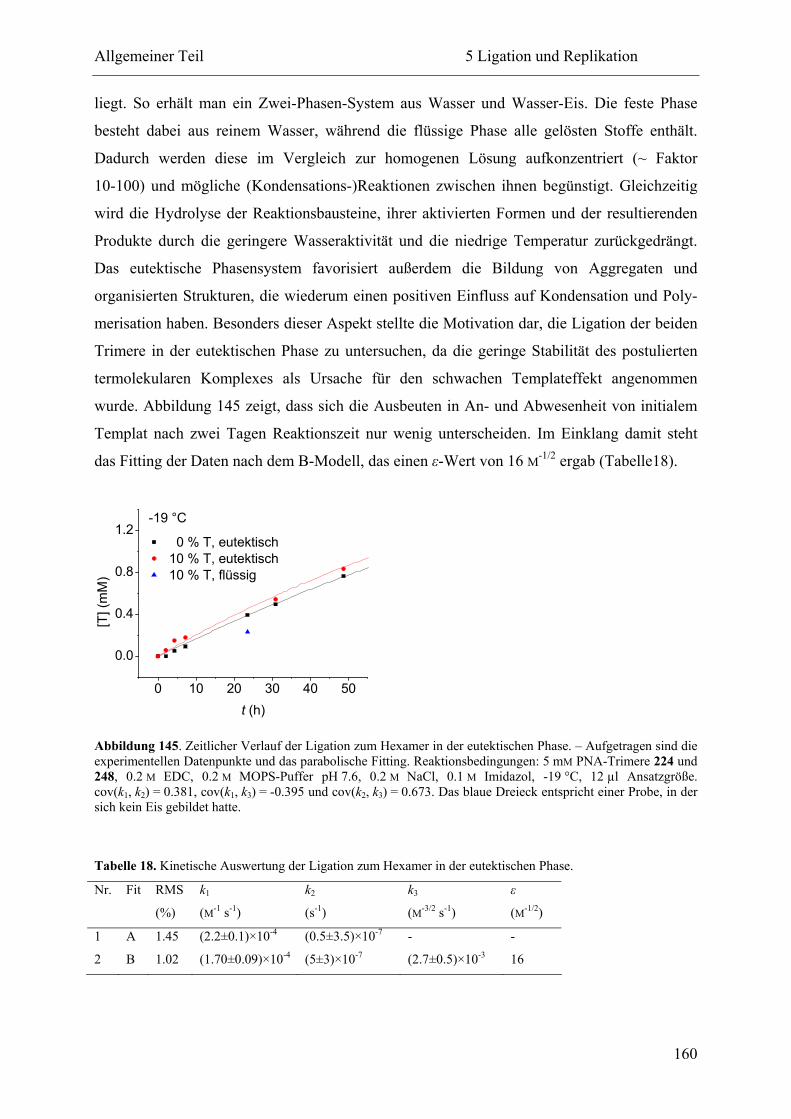
Allgemeiner Teil 5 Ligation und Replikation
160
liegt. So erhält man ein Zwei-Phasen-System aus Wasser und Wasser-Eis. Die feste Phase
besteht dabei aus reinem Wasser, während die flüssige Phase alle gelösten Stoffe enthält.
Dadurch werden diese im Vergleich zur homogenen Lösung aufkonzentriert (~ Faktor
10-100) und mögliche (Kondensations-)Reaktionen zwischen ihnen begünstigt. Gleichzeitig
wird die Hydrolyse der Reaktionsbausteine, ihrer aktivierten Formen und der resultierenden
Produkte durch die geringere Wasseraktivität und die niedrige Temperatur zurückgedrängt.
Das eutektische Phasensystem favorisiert außerdem die Bildung von Aggregaten und
organisierten Strukturen, die wiederum einen positiven Einfluss auf Kondensation und Poly-
merisation haben. Besonders dieser Aspekt stellte die Motivation dar, die Ligation der beiden
Trimere in der eutektischen Phase zu untersuchen, da die geringe Stabilität des postulierten
termolekularen Komplexes als Ursache für den schwachen Templateffekt angenommen
wurde. Abbildung 145 zeigt, dass sich die Ausbeuten in An- und Abwesenheit von initialem
Templat nach zwei Tagen Reaktionszeit nur wenig unterscheiden. Im Einklang damit steht
das Fitting der Daten nach dem B-Modell, das einen ε-Wert von 16 M-1/2 ergab (Tabelle18).
0 10 20 30 40 50
0.0
0.4
0.8
1.2 0 % T, eutektisch10 % T, eutektisch10 % T, flüssig
-19 °C
[T] (
mM
)
t (h)
Abbildung 145. Zeitlicher Verlauf der Ligation zum Hexamer in der eutektischen Phase. – Aufgetragen sind die experimentellen Datenpunkte und das parabolische Fitting. Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.2 M NaCl, 0.1 M Imidazol, -19 °C, 12 µl Ansatzgröße. cov(k1, k2) = 0.381, cov(k1, k3) = -0.395 und cov(k2, k3) = 0.673. Das blaue Dreieck entspricht einer Probe, in der sich kein Eis gebildet hatte.
Tabelle 18. Kinetische Auswertung der Ligation zum Hexamer in der eutektischen Phase.
Nr. Fit RMS
(%)
k1
(M-1 s-1)
k2
(s-1)
k3
(M-3/2 s-1)
ε
(M-1/2)
1 A 1.45 (2.2±0.1)×10-4 (0.5±3.5)×10-7 - -
2 B 1.02 (1.70±0.09)×10-4 (5±3)×10-7 (2.7±0.5)×10-3 16

Allgemeiner Teil 5 Ligation und Replikation
161
Eine Carbonsäure kann auch durch die Überführung in den entsprechenden Methylester in ge-
ringem Maße für die Reaktion mit einem Amin aktiviert werden. Die schwache Aktivierung
führt zu einer langsamen Reaktion, die im vorliegenden System den katalytischen Einfluss
des zugesetzten Templats begünstigen sollte. Als potentielle Nebenreaktion wird hier anstelle
der Carbonsäure-katalysierten Hydrolyse des EDCs die Hydrolyse des Methylesters zurück
zur Säure erwartet, die durch die HPLC-Analyse verfolgt werden kann. Methylester wurden
bereits von Luisi und Mitarbeitern bei der Ser-His-katalysierten Kondensation von PNA-
Monomeren in basischem Milieu verwendet (s. 1.4.4).[156] Durch Umsetzung von 248 mit
HATU, DIPEA und Methanol in DMF konnte der in Abbildung 146 dargestellte PNA-
Methylester 291 nach Aufreinigung mittels RP-HPLC in 15%iger Ausbeute erhalten werden.
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH2
O
F
F
OO
NN
N N
NNH
O
NH2
291
Abbildung 146. PNA-Methylester 291.
Dieser wurde dann mit dem Amino-Fragment 224 in gepufferter wässriger Lösung (0.2 M
MOPS pH 7.2, 0.2 M NaI) bei 10 °C bzw. Raumtemperatur in An- und Abwesenheit des
potentiellen Templats umgesetzt. Nach fünf Tagen Reaktionszeit konnte weder bei
Raumtemperatur noch bei 10 °C das gewünschte Ligationsprodukt nachgewiesen werden.
Eine minimale Hydrolyse des Methylesters 291 wurde nur bei Raumtemperatur festgestellt
und war unabhängig vom zugegebenen Hexamer.

Allgemeiner Teil 6 Zusammenfassung und Ausblick
162
6 Zusammenfassung und Ausblick
6.1 Zusammenfassung
Im Rahmen der vorliegenden Arbeit wurde ein Selbstreplikations-System auf der Basis von
PNA entwickelt und synthetisiert. Dieses besteht aus zwei kurzen Fragmenten, deren Ligation
zu einem selbstkomplementären Templat durch verschiedene Peptidkopplungsreagenzien
vermittelt wird. Besonderes Augenmerk wurde auf die Einführung einer Heterokern-NMR-
Sonde gelegt, um das System durch kinetische NMR-Titration analysieren zu können. Da
NMR-Experimente vergleichsweise hohe Substanz-Konzentrationen erfordern, wurde auf
eine hohe Wasserlöslichkeit der Oligomere geachtet. Außerdem wurde eine effektive und
modulare Syntheseroute gefunden, um die benötigten Substanzmengen bereitzustellen und
Sequenz-Variationen zu ermöglichen.
Als Heterokern-NMR-Sonde wurde der 19F-Kern gewählt, da dieser eine Reihe von
wünschenswerten Eigenschaften besitzt: Zum einen reagiert er auf geringe Änderungen der
supramolekularen Umgebung mit einer deutlichen Änderung der chemischen Verschiebung.
Zum anderen weist 19F einen großen chemischen Verschiebungsbereich auf, der die Gefahr
der Signalüberlappung reduziert und die Analyse der Messungen vereinfacht. Im Vergleich zu 1H besitzt er eine nur geringfügig auf 83 % reduzierte Empfindlichkeit und stellt das einzig
natürlich vorkommende Isotop des Elements dar. Dies sollte kinetische NMR-Messungen mit
einer geringen Anzahl von Scans und somit einer hohen Dichte von Messpunkten erlauben.
Das Fluor-Label kann entweder am Rückgrat oder an den Basen lokalisiert werden. Für
letztere Lokalisation sprach, dass die Sonde in direkter räumlicher Nähe zur supramolekularen
Erkennungsstelle gebracht wird. Unter den möglichen Kandidaten wurde für diese Arbeit
2,4-Difluortoluol (F) (71) ausgewählt, da dieses Molekül ein nahezu perfektes Isoster der
natürlichen Base Thymin (72) ist (Abbildung 147).[211, 212]

Allgemeiner Teil 6 Zusammenfassung und Ausblick
163
F
F
F
F
OH
O
NH
NH
O
O
72 71 70
Abbildung 147. Die natürliche Nukleobase Thymin (72) und ihr fluoraromatisches Isoster 71; daneben die für den Einbau in PNA benötigte Essigsäure 70.
Für die entsprechende Essigsäure 70 gibt es eine literaturbekannte sechsstufige Synthese bei
der eine Reihe hochtoxischer Stoffe verwendet werden (Abbildung 148).[206] Um diesen
arbeitsintensiven Weg abzukürzen, wurde von 2,4-Difluortoluol (71) ausgehend eine
zweistufige Synthese entwickelt, die aus der Reaktionsfolge Brommethylierung und
Grignard-Reaktion besteht. Dabei wurde die ursprünglich entwickelte Vorschrift zur
Brommethylierung von van der Made und van der Made durch Zugabe der Lewis-Säure Zink-
bromid modifiziert, da der durch die Fluorsubstituenten stark deaktivierte Aromat sonst nicht
zur Reaktion zu bringen ist.[215] Durch die gleiche Sequenz ließ sich auch 1,3-Difluorbenzol
(93), ein Isoster des Uracils (94), in die entsprechende Essigsäure 95 überführen.
F
F
F
F
Br
F
F
CN
F
F
OOH
F
F
O
N2
F
F
O
O
F
F
OH
O
F
F
Br
50 % 100 % 98 % 60 %
58 %
71 87 88 89 90 91
92 70
nichtangegeben
a b c d e
f
g 66 %
57 %
h
Abbildung 148. Synthese von F-Essigsäure 70 ausgehend von 2,4-Difluortoluol (71). – Nach Takeuchi et al.[206] (grün) und nach Plöger und von Kiedrowski (blau). Reagenzien und Reaktionsbedingungen: (a) Br2, Fe; (b) CuCN, DMF, 60 °C, 3 h; (c) H2SO4, H2O, Rückfluss, 1 h; (d) (1) ClCO2Et, TEA, THF, -15 °C, 15 min; (2) CH2N2, Et2O, 0 °C → RT, 3 h; (e) PhCOOAg, MeOH, -25 °C → RT, 3 h; (f) 1 M LiOH, THF; (g) (CH2O)n, ZnBr2, 33 % HBr in AcOH, 120 °C, 4 h; (h) (1) Mg, I2, Et2O, Rückfluss, 2 h; (2) CO2, 0 °C, 1 h; (3) H+, H2O.
Eine bekannte Möglichkeit, die Wasserlöslichkeit von PNA zu erhöhen und ihrer ausgepräg-
ten Tendenz zur Aggregation und Selbstorganisation entgegenzuwirken, besteht in der
Modifizierung mit positiven Ladungen. Hierbei kommt es durch die Einführung von
Chiralitätszentren und/oder zusätzlichen Funktionalitäten häufig zu Abweichungen von der

Allgemeiner Teil 6 Zusammenfassung und Ausblick
164
originalen PNA-Struktur. Dies sollte hier vermieden werden, um die Struktur der beteiligten
Komplexe nicht zu beeinflussen und Nebenreaktionen bei der EDC-Ligation auszuschließen.
Daher wurde als Strukturmotiv die PNA-inherente N,N-Dimethylaminogruppe gewählt.
Bei der Festphasensynthese lässt sich der N-Terminus mittels eines modifizierten Monomers
oder durch Anhängen einer modifizierten Aminosäure variieren, sodass sowohl zweifach
methyliertes ß-Alanin 140 als auch ein terminal N,N-dimethyliertes Rückgrat 78 synthetisiert
wurde, das anschließend mit geschützter Cytosin-essigsäure 84 gekoppelt wurde (Abbildung
149). Die Modifizierung des C-Terminus kann während der Abspaltung des Oligomers vom
Harz erfolgen. Hierzu schien der HMBA Linker 75 sehr gut geeignet, weil sich die
Esterfunktion dieses Linkers durch eine Vielzahl von Nukleophilen spalten lässt. Im
konkreten Fall sollte dies durch Reaktion mit N,N-Dimethylaminoethylamin 143 erfolgen. Die
Abspaltung eines Peptids durch Ethanolamin war aus vertrauenswürdiger Literatur
bekannt.[213]
NH
OH
O
NN
NH2
HOH
O
O
N
N
NH
O
N
O
Bhoc
N OH
O
+
144 123 145 142
b
a
95 %
98 % c
54 % d
79 %
Abbildung 149. Synthese eines C-Monomers 142 mit modifiziertem Rückgrat. – Reagenzien und Reaktionsbe-dingungen: (a) H2, Pd/C, H2O, RT; (b) SOCl2, MeOH, 0 °C → Rückfluss, 16 h; (c) CBhoc-AcOH (84), EDC, HOBt, DIPEA, DMF, 0 °C → RT, 16 h; (d) (1) LiOH (2 eq.), THF, H2O, 0 °C → RT, 2 h; (2) HCl.
Die benötigten Oligomere sollten durch maschinelle Festphasensynthese nach der
Fmoc/Bhoc-Strategie synthetisiert werden. Hierzu wurden im ersten Schritt die Fmoc/Bhoc-
geschützten Monomere der vier natürlichen Nukleobasen ausgehend von Ethylendiamin,
Glyoxylsäure und den freien Basen nach teils modifizierten literaturbekannten Vorschriften in
25 Schritten synthetisiert. Außerdem wurden drei Fluorlabel in insgesamt zehn Schritten in
ihre Fmoc-geschützten Monomere überführt (Abbildung 150).

Allgemeiner Teil 6 Zusammenfassung und Ausblick
165
NH
NH
O
OMe NH
O
OMe
R2
F
N
O
R1
R2
F
OH
O
R1
R2
F
N
O
R1
NH
O
OH
83 135 128
Fmoc FmocFmoc
+
a b
47 %50 %50 %
70: R1 = Me, R2 = F96: R1= H, R2 = F101: R1 = H, R2 = CF3
a:b:c:
93 %74 %88 %
a:b:c:
R1 = Me, R2 = FR1= H, R2 = FR1 = H, R2 = CF3
a:b:c:
Abbildung 150. Synthese von Fmoc-geschützten Monomeren verschiedener Thymin- bzw. Uracil-Isostere. – Reagenzien und Reaktionsbedingungen: (a) TOTU, DIPEA, DMF, RT, 3 h; (b) (1) AlCl3 (3.0 eq), Ethanthiol, RT, 24 h; (2) H2O.
Startpunkt für das Sequenzdesign war die Betrachtung der Kristallstruktur einer selbstkomple-
mentären hexa-PNA.[115] Die meisten im Arbeitskreis von Kiedrowski veröffentlichten
Arbeiten zur (Selbst-)Replikation von Nukleinsäuren basieren aufgrund von thermodynami-
schen Überlegungen auf hexameren Templaten und trimeren Fragmenten.[72, 75, 76, 78-80] Diese
Sequenzlängen wurden beibehalten, um Systeme auf Basis von DNA und PNA miteinander
vergleichen zu können und – auf längere Sicht – komplexere Systeme mit genetischen Über-
gängen zwischen PNA und DNA zu entwerfen. Die natürliche Nukleobase Thymin (72)
wurde durch 2,4-Difluortoluol (71) substituiert und die Strangenden mit den achiralen nicht-
reaktiven Löslichkeitsvermittlern modifiziert. Darüber hinaus wurde die Sequenz selbst leicht
variiert, um das Isoster in der mittleren Position des N-terminalen Trimers zu platzieren.
Dadurch sollte der Einfluss des Fluoraromaten auf die Ligation minimiert werden, während
die Distanz zur supramolekularen Erkennungsseite unverändert bleibt.
Die beiden Template 177 und 180 wurden durch maschinelle Festphasensynthese am Peptid-
Synthesizer hergestellt, mittels RP-HPLC gereinigt und durch MALDI-MS charakterisiert
(Abbildung 151). Der Wechsel des Lösungsmittels von DMF zu NMP und die Einführung
doppelter Kopplungen steigerten die Ausbeuten und Reinheit der Rohprodukte. Allerdings be-
trugen die Ausbeuten an gereinigtem Produkt je ca. 10 % und lagen damit in einem für
PNA-Synthesen unbefriedigenden Bereich, sodass eine zeitnahe und wirtschaftliche Synthese
der benötigten Substanzen nicht gewährleistet war. Ursächlich für die schlechten Ausbeuten
war der verwendete HMBA-Linker, der es überraschenderweise nicht gestattete, das syntheti-
sierte Oligomer quantitativ von der Festphase abzuspalten. Eine weitere Schwierigkeit be-
stand in der chromatographischen Abtrennung der Trimere von ihren jeweiligen Deletions-
mutanten.

Allgemeiner Teil 6 Zusammenfassung und Ausblick
166
NNH
N
OO
NH
O
N N
NNH
O
NH2
O
N N
NN
NH2
O
NNH
O
N
N
O
NH2
NNH
OO
N N
NNH
O
NH2
NNH
N
OOOO
FN
N
O
NH2
NH
O
NH
NO
NH
N
R2
R1
177: R1 = CH3, R2 = F; 180: R1 = H, R2 = CF3
Abbildung 151. Durch Festphasensynthese hergestellte Template 177 und 180.
Diese Probleme konnten umgangen werden, indem die beiden Trimere 224 und 248 durch
Flüssigphasensynthesen dargestellt wurden (Abbildung 152). Dazu wurden zwei von Condom
entwickelte Synthesestrategien angewandt. Beide Trimere stehen dadurch in ausreichender
Mengen zur Verfügung, um Replikationsexperimente durchführen zu können.
NH
N
O
NH
NNH
NOH
OOO
N
N
O
NH2
O
F
F
OO
NN
N N
NNH
O
NH2
NN
N
O
NH2
N
O
NH2
O
NH
NNH
NNH
OO
N
OOO
N N
NNH
O
NH2N
NN
NH2
248 224
Abbildung 152. Durch Flüssigphasensynthese dargestellte tri-PNAs 248 und 224.
Zur Darstellung des C-terminalen Trimers 224 wurde der „Fully Protected Polyamide
Backbone Approach“ adaptiert, der Löslichkeitsprobleme umgeht, indem die geschützten
Nukleobasen möglichst spät in das Zielmolekül implementiert werden. Die exocyclischen
Aminofunktionen der Nukleobasen wurden hierbei nicht mehr durch die Bhoc-Gruppe,
sondern durch die Z-Gruppe geschützt, die erst durch Behandlung mit sehr starken Säuren
abgespalten wird. Die Synthese der entsprechenden Nukleobasen-essigsäuren erfolgte
ausgehend von Adenin, Cytosin und Guanin in neun Syntheseschritten. Als Löslichkeits-
vermittler wurde anstelle des gebräuchlichen L-Lysin-amids 292 das N-Ethyl-morpholino-
amid 293 gewählt, da es die achirale PNA-Struktur aufrechterhält und eine positive Ladung
bereitstellt. Die Löslichkeit des N-terminalen Pendants 248 wurde durch das vollständig
N-methylierte Rückgrat 249 sichergestellt, das nur PNA-immanente Struktureinheiten auf-
weist und unter physiologischen Bedingungen zweifach geladen ist. Seine Synthese gelang
ausgehend von AEG (85) durch reduktive Aminierung in guter Ausbeute (Abbildung 153).

Allgemeiner Teil 6 Zusammenfassung und Ausblick
167
O
NH
NH2 OH
O
NN OH
RNH
N
OO
B
NH
n
NH2
NH2
O
RNH
N
OO
B
NH
N
O
n
a oder b
*2HCla: 41 %b: 75 %
85 249
292 293
Abbildung 153. Vergleich der Strukturen von L-Lysin-amid 292 und N-Ethyl-morpholino-amid 293 (oben) und Synthese des N-terminalen Löslichkeitsvermittlers 249 auf Basis von AEG (85) (unten). – Reagenzien und Reaktionsbedingungen: (a) (1) Formalin-Lsg., Ameisensäure, Rückfluss, 16 h; (2) HCl; (b) (1) Formalin-Lsg., H2O, 0 °C → RT, 1 h; (2) H2, Pd/C.
Zur Synthese des N-terminalen Trimers 248 wurde ein Schema entwickelt, das den sehr
polaren Löslichkeitsvermittler 249 erst zu einem späten Zeitpunkt der Synthese einführt, um
Probleme bei der Chromatographie zu vermeiden. Der Einsatz einer weiteren orthogonalen
Schutzgruppe konnte hierbei vermieden werden, indem sowohl geschützte Rückgrat-
Einheiten als auch eine geschützte PNA-Einheit kombiniert wurden. Schlüsselbausteine
beider Strategien sind vollständig orthogonal geschützte AEG-Monomere, aus denen durch
geeignete Schützung und Kopplung sukzessive Oligomere aufgebaut werden. Vier dieser
Bausteine wurden auf Basis von AEG (85) in insgesamt acht Schritten synthetisiert.
Ausgehend von den Z-geschützten Nukleobasen-essigsäuren, F-Essigsäure, dem Löslichkeits-
vermittler und den vier geschützten AEG-Monomeren wurden beide Trimere in weiteren 28
Schritten aufgebaut.
Die Synthese in flüssiger Phase ist prinzipiell auch deshalb reizvoll, weil alle Zwischen-
produkte mit Standardmethoden analysiert werden können. Im Fall der PNA-Flüssigphasen-
synthese tritt allerdings ein Problem auf, für das in der Literatur bisher keine Lösung
vorgeschlagen wurde: Bei Raumtemperatur erhält man für AEGs und ungepaarte PNAs
komplexe NMR-Spektren, da die tertiäre Amid- oder Carbamatbindung jeder AEG-Einheit
sowohl in der cis- als auch in der trans-Konformation vorliegen kann und der Austausch
zwischen den möglichen 2n Konformeren langsam im Verhältnis zur NMR-Zeitskala ist.
Daher wurden im Rahmen dieser Arbeit insgesamt 22 Monomere, Dimere und Trimere durch 1H-NMR-Spektroskopie bei erhöhten Temperaturen von bis zu 160 °C untersucht, um die
Rotationsraten zu erhöhen. Von zehn dieser Verbindungen konnten Spektren im Bereich des
schnellen Austauschs erhalten werden (Abbildung 154). Bei Spektren von weiteren neun
Substanzen konnte die Anzahl an Signalsätzen deutlich reduziert und eine beachtliche

Allgemeiner Teil 6 Zusammenfassung und Ausblick
168
Verringerung der Linienbreiten erreicht werden. Zwei Verbindungen zersetzten sich unter den
Messbedingungen und in einem Fall führte die Temperaturerhöhung zu einer Komplikation
des Spektrums. Trotz dieser Einschränkungen konnte die Strukturanalyse geschützter und
ungeschützter AEGs und PNAs deutlich verbessert werden. Außerdem lassen sich aus den
vorliegenden Spektren erstmals Erkenntnisse über das dynamische Verhalten geschützter
AEGs und PNAs gewinnen.
Abbildung 154. 1H-NMR-Spektren von Boc-Aeg(Alloc)-Aeg(Fmoc)-OBn (257) in DMSO-d6 bei 30 °C (200 MHz, oben) bzw. 100 °C (250 MHz, unten).
Anschließend wurde die Ligation der beiden Trimere 224 und 248 in wässrigen gepufferten
Lösungen mittels RP-HPLC untersucht (Abbildung 155). Ziel war es dabei, Bedingungen zu
finden, unter denen das Ligationsprodukt 268 als Templat wirkt und dadurch seine eigene
Synthese katalysiert. Hierzu wurden die Reaktionstemperatur und der pH-Wert variiert, ver-
schiedene Puffer und nukleophile Katalysatoren getestet und die Einflüsse unterschiedlicher
Anionen bestimmt. Daneben wurden Versuchsreihen unter Zugabe von PEG, in der eutek-
tischen Phase und mit dem PNA-Methylester 289 als aktivierter Komponente durchgeführt.
Zur kinetischen Modellierung der experimentellen Daten wurde das Programm SimFit
verwendet, das die Berechnung theoretischer Konzentrations-Zeit-Kurven durch dynamische
Simulation mit der Kurvenanpassung durch nichtlineare Regression verbindet. Die Messwerte

Allgemeiner Teil 6 Zusammenfassung und Ausblick
169
wurden dabei auf der Basis zweier Reaktionsgleichungssysteme ausgewertet: Das A-Modell
berücksichtigt neben einem unkatalysierten Reaktionskanal (k1) zwei Abklingprozesse (k2),
während das B-Modell zusätzlich einen autokatalytischen Reaktionskanal (k3) einbezieht. Die
Übereinstimmung eines Modells mit den Daten wurde anhand des RMS-Werts bestimmt und
die Verlässlichkeit der ermittelten Geschwindigkeitskonstanten durch deren Kovarianzen
überprüft. Stimmt unter gegebenen Reaktionsbedingungen das B-Modell besser mit den
Daten überein als das A-Modell, wird dies als Hinweis auf Selbstreplikation gewertet und
deren Effizienz durch den autokatalytischen Überschuss-Faktor ε angegeben.
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
NH ON N
O
NH2
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
NH
NH
NO
ON
N
N
NH
NO
O
NH
ON
N
N
NH
O
NH2
ON
N
N
N
NH2
ON
ON N
O
NH2
NH
NH2
NO
N
NH
O
ON
N
N
NH
O
NH2
N
NH
NO
O
O
O
F
F
N N
O
NH2
NH
OH
N
N
O
268 / T
248 / A
224 / B
Abbildung 155. Selbstreplikations-System auf der Basis der tri-PNAs 224 und 248. – Grün: nicht-reaktive Lös-lichkeitsvermittler; gelb: 19F-NMR-Sonden; rot: Ligationsstellen und daraus resultierende zentrale Amidbindung.
Die optimale Reaktionstemperatur wurde nicht aus der Schmelztemperatur des Templat-
duplexes abgeleitet, da diese aufgrund einer flachen Schmelzkurve nicht genau definiert ist.
Infolgedessen wurden 10 °C durch Experimente bei variabler Temperatur und den Vergleich
von Literaturwerten als geeignet bestimmt. Als Puffersubstanzen wurden zum einen Imidazol
und 1-Methylimidazol gewählt, die gleichzeitig als nukleophile Katalysatoren wirken, zum

Allgemeiner Teil 6 Zusammenfassung und Ausblick
170
anderen die Sulfonsäure-Puffer MOPS und HEPES, denen die jeweiligen Katalysatoren
zugesetzt wurden. Da EDC in Form seines Hydrochlorids verwendet wurde, enthielten alle
Reaktionsmischungen Chlorid-Ionen, die Ansätze in Sulfonsäure-Puffern enthielten darüber
hinaus, durch die Einstellung des pH-Werts mit Natronlauge, Natrium-Ionen. Alle verwende-
ten nukleophilen Katalysatoren konnten die Bildung von Nebenprodukten, die durch die
Addition von EDC an die Trimere entstehen, zurückdrängen. Als unerheblich erwies es sich,
ob die Katalysatoren einem Sulfonsäure-Puffer zugesetzt wurden (0.1-0.2 M) oder selbst die
Puffersubstanz darstellten (0.4 M). Pyridin, 1-Methylimidazol und vor allem HOAt konnten
als effiziente nukleophile Katalysatoren für die PNA-Ligation bei Raumtemperatur iden-
tifiziert werden: In ihrer Gegenwart wurden mit bis zu 90 % die größten Ausbeuten an
hexa-PNA 268 nachgewiesen. Hinweise auf einen nennenswerten Templateffekt gab es dabei
nicht. Ähnlich effektive Methoden zur PNA-Ligation in wässriger Phase nutzen die native
chemische Ligation nach Kent oder die daraus abgeleitete native chemische
iCys-Verknüpfung und sind daher in Bezug auf die verwendeten Sequenzen einge-
schränkt.[175-177, 284, 285] Templateffekte konnten hingegen sowohl in reinem Imidazol- und
MOPS-Puffer als auch in Imidazol-haltigem MOPS- und HEPES-Puffer beobachtet werden.
Während bei Replikatoren auf DNA-Basis die Variation der Verknüpfungschemie ausgehend
von der Phosphodiester- über die Pyrophosphat- hin zur Phosphoamidatbindung zu
schnelleren und gleichzeitig effizienteren Replikatoren geführt hat,[72, 75, 76] ist der Zusammen-
hang zwischen Reaktionsgeschwindigkeit und Templateffekt im vorliegenden System
komplexer: Vergleicht man die Ligationen in Imidazol- und 1-Methylimidazol-Puffer über
zwei Tage, so verläuft erstere langsamer und zeigt einen Templateffekt (ε = 28 M-1/2), während
letztere deutlich schneller ist und kaum Einfluss des Templats zeigt (ε = 6.7 M-1/2) (Abbildung
156). Hingegen erfolgt die Reaktion unter Imidazol-Katalyse in MOPS-Puffer fast dreimal so
schnell wie in HEPES-Puffer, während die autokatalytische Effizienz mit ε = 40 bzw. 59 M-1/2
vergleichbar ist (Abbildung 157). Zum Ausgangspunkt der weiteren Optimierungen wurde
jeweils das MOPS/Imidazol-System gewählt, da es, verglichen mit der HEPES-Variante,
einen größeren Umsatz zeigt und im Gegensatz zum Imidazol-Puffer auch die Variation des
nukleophilen Katalysators erlaubt. Der pH-Wert wurde für das Referenzexperiment von 7.2
auf 7.6 erhöht, wodurch ε von 40 auf 67 M-1/2 gesteigert werden konnte.

Allgemeiner Teil 6 Zusammenfassung und Ausblick
171
Abbildung 156. PNA-Ligation in Imidazol- und 1-Methylimidazol-Puffer bei 10 °C. – Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.4 M Puffer pH 7.5, 25 µl Ansatzgröße.
Abbildung 157. PNA-Ligation unter Imidazol-Katalyse in MOPS und HEPES-Puffer bei 10 °C. – Reaktions-bedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M Puffer pH 7.2 (MOPS) bzw. 7.5 (HEPES), 0.2 M NaCl, 6 µl Ansatzgröße.
Ohne zugesetztes Salz wich der Wert für den autokatalytischen Überschuss-Faktor ε mit
64.2 M-1/2 nur wenig vom Referenzsystem ab, während die Zugabe von NaI einen positiven
Einfluss auf die Ausbeute und Templatierung der Reaktion zeigte (ε = 94.9 M-1/2). Sulfatzusatz
führte zwar zu einem geringfügig größeren autokatalytischen Überschuss-Faktor von
ε = 100 M-1/2, bewirkte aber parallel einen Einbruch der Kopplungseffizienz. Durch die
Verdoppelung der EDC-Konzentration auf 0.4 mol/l ließ sich dessen Hydrolyse z.T.
kompensieren, sodass man die doppelte Ausbeute an Ligationsprodukt bei ähnlichem Tem-
plateffekt erhielt (ε = 65 M-1/2). Die Erniedrigung des pH-Werts von 7.6 auf 6.6 beschleunigte
die Ligation und verstärkte den beobachtbaren Einfluss des Templats beträchtlich
(ε = 112 M-1/2). Auch die Zugabe von Polyethylenglykolen zeigte einen positiven Einfluss auf
den autokatalytischen Überschuss-Faktor: In Gegenwart von 20 % PEG400 konnte mit
0 10 20 30 40 50
0.0
0.4
0.8
1.2
1.6
2.0 0 % T30 % T50 % T
Imidazol-Puffer[T
] (m
M)
t (h)
0 10 20 30 40 50
0.0
0.4
0.8
1.2
1.6
2.0 0 % T20 % T40 % T
1-Methylimidazol-Puffer
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2
0 % T10 % T
MOPS-Puffer/0.1 M Imidazol
[T] (
mM
)
t (h)
0 10 20 30
0.0
0.4
0.8
1.2
0 % T10 % T
HEPES-Puffer/0.1 M Imidazol
[T] (
mM
)
t (h)

Allgemeiner Teil 6 Zusammenfassung und Ausblick
172
321 M-1/2 der für das vorliegende System größte Wert von ε bestimmt werden, während die
Ausbeute gleichzeitig um den Faktor zwei bis drei reduziert wurde. Durch Zusatz von 20 %
PEG3350 wurde hingegen sowohl der Umsatz zum Ligationsprodukt als auch der Wert des
autokatalytischen Überschuss-Faktors (ε = 126 M-1/2) erhöht. Im eutektischen Phasensystem
zeigte die initiale Templatzugabe wenig Einfluss auf den Reaktionsverlauf, sodass für ε nur
ein Wert von 16 M-1/2 gefunden wurde. Ein erstes Experiment unter Verwendung des PNA-
Methylesters 289 als aktivierter Komponente zeigte keinen Umsatz.
Bei allen Versuchsreihen korrelierte die Abweichung der theoretischen Kurven von den
Messpunkten qualitativ mit der Menge an initial zugesetztem Templat. Dabei sagte das
Modell generell einen höheren Umsatz voraus, als tatsächlich experimentell beobachtet
werden konnte. Dies kann zum einen dadurch erklärt werden, dass bei hohen Templatkonzen-
trationen auch größere Fehler bei der Subtraktion der HPLC-Integrale möglich sind. Zum
anderen erfassen die Abklingprozesse im Reaktionsmodell nur die Carbonsäure-katalysierte
EDC-Hydrolyse an sich, während die potentielle Abhängigkeit ihrer Geschwindigkeit von der
initialen Carbonsäurekonzentration unberücksichtigt bleibt. Diese war bei den durchgeführten
Experimenten proportional zur Templatzugabe, da alle PNAs in Form ihrer mehrfachen
Trifluoracetate eingesetzt wurden.

Allgemeiner Teil 6 Zusammenfassung und Ausblick
173
6.2 Ausblick
Die vorliegende Arbeit hat die konzeptionellen und synthetischen Grundlagen geschaffen, um
die potentielle PNA-Selbstreplikation durch kinetische 19F-NMR-Titration zu untersuchen.
Erste mittels RP-HPLC ausgewertete Ligations- und Replikationsexperimente zeigen, dass
PNA-Selbstreplikation prinzipiell möglich ist. Zukünftig gilt es daher, den beobachteten, bis-
lang schwachen Templateffekt zu verstärken, um einen effektiven Replikator zu entwickeln.
Hierzu können die Reaktionsbedingungen für das untersuchte System weiter modifiziert und
optimiert werden. Vielversprechende Ansatzpunkte bietet dabei sowohl das Screening
wasserlöslicher Cosolventien und nukleophiler Katalysatoren (Abbildung 158) als auch die
Variation des pH-Werts.
N
NH
N
N
N
NH
N
NH
N
NH
N
N
N
NH
NO2
N
NH
CN
CNN
NH
OH275 276 277 294 295 296 297 298 278
NH
NN
NN
N
OHN
NN
OHCl N
N
N
O
OH
N NN
N
OH299 154 300 290 301
N NN
N
NOH N+
O
O
N
289 302 303 304 305 306
N OO
OH
NH N
N N
NH2
NH
NN
NH
N NN
307 103 308 309
Abbildung 158. Potentielle nukleophile Katalysatoren für die PNA-Ligation und Replikation auf der Basis von Imidazolen (275-278, 294-298), Benzotriazolen (154, 299-300) bzw. -trazin (301), Pyridinderivaten (289, 302-306) und weiteren Heterocyclen (103, 307-309). – Die blau dargestellten Verbindungen wurden im Rahmen der Arbeit bereits getestet.
Alternativ kann die Stabilität des postulierten termolekularen Komplexes durch den Einsatz
längerer Bausteine gesteigert werden. Dies erscheint sinnvoll, da die flache UV-Schmelz-
kurve des resultierenden Produktduplexes 268 (Abbildung 130) auf einen wenig kooperativen
Prozess hinweist und darüber hinaus nicht die zuverlässige Bestimmung der Schmelztempe-

Allgemeiner Teil 6 Zusammenfassung und Ausblick
174
ratur erlaubt, deren Kenntnis wichtig für die Wahl der optimalen Reaktionstemperatur ist. In
diesem Zusammenhang kann auch eine alternative Positionierung des Fluoraromaten in
Betracht gezogen werden. Seine jetzige Lage in der Sequenzmitte von Trimer 248 sollte den
Einfluss auf die Ligation minimieren und einen geeigneten Abstand zur molekularen
Erkennungsseite gewährleisten. Im Sinne stabiler Komplexe und eines kooperativen Schmel-
zens scheint auch ein Einbau des Isosters am N-Terminus sinnvoll, da hierdurch mehr Basen-
paare direkt benachbart wären, die an Watson-Crick-Wasserstoffbrücken beteiligt sind. Die
Carbonsäure-katalysierte EDC-Hydrolyse kann in künftigen Experimenten zurückgedrängt
werden, indem die HPLC-gereinigten PNA-Trifluoracetate freigesetzt und anschließend durch
Gel-Permeations-Chromatographie entsalzt werden. Hierdurch würde außerdem die zugege-
bene Templatmenge nicht mehr die initiale Carbonsäurekonzentration beeinflussen, die
ihrerseits Auswirkungen auf die Geschwindigkeit der EDC-Hydrolyse haben sollte. Dieser
Zusammenhang wird als Hauptursache dafür angenommen, dass die Diskrepanz zwischen den
theoretischen Kurven und den Messpunkten bei hohen Templatkonzentrationen ausgeprägter
ist als bei niedrigen. Um zudem die Fehler bei der Auswertung der HPLC-Integrale zu
minimieren, bietet sich die Synthese und Verwendung eines chemisch markierten Templats
an. Auch der Einsatz des PNA-Methylesters 291 als aktivierter Komponente bleibt weiterhin
interessant, wenn günstigere Reaktionsbedingungen gefunden werden. Ein wichtiger Aspekt
wird dabei die Wahl eines geeigneten pH-Werts sein, dessen Erhöhung gleichzeitig die Kon-
zentration an Hydroxid-Ionen und an unprotoniertem Amin erhöht, und somit die Hydrolyse
des Methylesters sowie dessen Aminolyse zum Ligationsprodukt beschleunigt. Darüber
hinaus kann der Reaktionsmischung das Dipeptid Ser-His zugesetzt werden, von dem bekannt
ist, dass es die Kondensation von PNA-Monomeren in basischem Milieu katalysiert.[156]
Sobald Bedingungen für eine effektive Replikation geschaffen worden sind, soll diese durch
zeitaufgelöste 19F-NMR-Spektroskopie verfolgt und analysiert werden. Neben einer präzisen
Temperaturkontrolle ist auch die H-F-Entkopplung von Bedeutung, da durch eine reduzierte
Signal-Aufspaltung eine höhere Intensität erreicht werden kann und eine einfachere Analyse
möglich ist. Nach der Durchführung und NMR-Analyse erster erfolgreicher Selbsteplikations-
experimente bieten sich eine Reihe interessanter chemischer Modifizierungen am System an:
Dazu zählt neben weiteren Sequenz-Variationen auch die Verwendung von Fluorsonden mit
CF3-Gruppen, da diese alternative Shift-Shifting-Informationen bieten. Auch das Design eines
kreuzkatalytischen Systems ist möglich. Ein Fernziel ist weiterhin der Einsatz eines Diamino-
Fragments 310, das zu zwei enantiomeren hexa-PNAs 311 führt (Abbildung 159). Auf diese

Allgemeiner Teil 6 Zusammenfassung und Ausblick
175
Weise entstünde ein denkbares Szenario für den Übergang von achiraler PNA zu einem
(homo-)chiralen Derivat. Außerdem erlauben die Amino-modifizierten PNAs 311 den Aufbau
verzweigter Strukturen. Für alle angesprochenen synthetischen Modifikationen des Systems
bietet die in dieser Arbeit verwendete modulare PNA-Flüssigphasensynthese einen guten
Ausgangspunkt.
NH
N
OO
B
OH NH2
N
OO
B
NH2
PNA
NH
N
OO
B
NH2
NH
N
OO
B
NH
N
OO
B
NH2
NH
N
OO
B
PNA +
PNAPNA
PNAPNA
312 310
(S)-311
(R)-311
Abbildung 159. Durch den Einsatz eines Diamino-Fragments 310 entstehen bei der Ligation zwei enantiomere Produkte 311 mit Verzweigungsstellen (durch den grünen Pfeil markiert).

Experimenteller Teil 7 Geräte und Materialien
176
Experimenteller Teil
7 Geräte und Materialien
7.1 Magnetische Kernresonanzspektroskopie
Zur Aufnahme von Kernresonanzspektren wurden die Geräte DPX-200 (200 MHz), DPX-250
(250 MHz) und DRX-400 (400 MHz) der Fa. Bruker verwendet. Das jeweilige Lösungsmittel
diente bei der Kalibrierung der 1H- und 13C-Spektren als interner Standard.[286, 287] Zur
Kalibrierung der 19F-Spektren wurde Trifluoressigsäure als externer Standard verwendet.[254]
Die chemischen Verschiebungen sind in der δ-Skala (Einheit ppm) und die Kopplungskon-
stanten in der Einheit Hz angegeben. Sofern es nicht anders angegeben ist, wurden die
Spektren bei 30 °C aufgenommen.
7.2 Massenspektrometer
7.2.1 FAB-MS
Die Aufnahme von Spektren für die Fast-Atom-Bombardment-Massenspektrometrie (FAB-
MS) erfolgte mit dem Spektrometer Autospec der Firma VG Instruments bei einer
Beschleunigungsspannung von 8 kV und Beschuss durch Cs+. Als Matrix fanden 3-Nitro-
benzylalkohol, Glycerin und Milchsäure Verwendung. Die relativen Peakintensitäten
beziehen sich auf den Basispeak (I = 100 %).
7.2.2 EI-MS
Die Aufnahme der Elektronenstoß-Ionisations-Spektren (EI-MS) erfolgte mit dem
Spektrometer Autospec der Firma VG Instruments. Auch hier beziehen sich die relativen
Peakintensitäten auf den Basispeak (I = 100 %).

Experimenteller Teil 7 Geräte und Materialien
177
7.2.3 MALDI-TOF-MS
Für die Matrix-Assisted-Laser-Desorption-Ionization-Time-Of-Flight-Massenspektrometrie
(MALDI-TOF-MS) wurde das Gerät Daltonics autoflex der Fa. Bruker verwendet. Als
Matrizes kamen 2,5-Dihydroxybenzoesäure (DHB), 2,4,6-Trihydroxyacetophenon (THAP)
und α-Cyano-4-hydroxyzimtsäure (CHCA) zum Einsatz.
7.2.4 HR-ESI-MS
Die Aufnahme der hochauflösenden Elektrospray-Ionisations-Spektren (HR-ESI-MS) erfolgte
mit dem Spektrometer LTQ-Orbitrap XL der Fa. Thermo Scientific.
7.3 Chromatographische Methoden
7.3.1 Dünnschichtchromatographie
Für die Dünnschichtchromatographie wurden Fertigfolien des Typs Kieselgel 60 F254 der Fa.
Merck (10 × 10 cm, Schichtdicke 0.25 mm Kieselgel, Fluoreszenzindikator) verwendet.
Detektiert wurde durch UV-Licht der Wellenlänge 254 nm bzw. durch Anfärben mit einer
Lösung von 4.00 g Molybdatophosphorsäure in 100 ml Methanol.
7.3.2 Säulenchromatographie
Für die Säulenchromatographie wurde Kieselgel des Typs ICN Silica 32-63 60 Å unter
Normal- oder Überdruck verwendet.
7.3.3 HPLC
Für die präparative Reinigung der PNAs wurde die VisionTM Workstation der Fa. Applied
Biosystems mit einem AFC2000 Roboter benutzt. Für die analytische HPLC wurde entweder
eine HPLC-Anlage der Firma Kontron verwendet (HPLC-Pumpe 422, Autosampler 460, UV-
Detektor 430, Mischkammer M494, Steuerungssoftware: Kroma System 2000) oder ebenfalls
die VisionTM Workstation der Fa. Applied Biosystems. Bei der Aufnahme der HPLC-
Kinetiken kam eine Anlage des Typs Beckman Gold® zum Einsatz (126 Solvent Module, 168
Detector). Alle Trennungen wurden bei einer Säulentemperatur von 55 °C durchgeführt.

Experimenteller Teil 7 Geräte und Materialien
178
Die verwendeten Säulen für die präparative und semipräparative HPLC waren:
Bischoff: Prontosil 200-5-C18-ace-EPS, 5 μm, 100 × 20 mm
Bischoff: Prontosil 200-5-C18-ace-EPS, 5 μm, 50 × 20 mm
Supelco: Discovery BIO Wide Pore C18, 5 µm, 250 × 10 mm
Die eingesetzten Säulen für die analytische HPLC waren:
Macherey-Nagel: Nucleodur 100-5 C18 ec, 250 × 4.0 mm
Supelco: Ascentis RP-Amide, 5 μm, 250 × 4.6 mm
Elutionsmittel:
A: 0.1 % TFA in ddH2O
B: 0.1 % TFA in MeCN
7.4 UV-Spektroskopie
Die UV-Spektren wurden mit dem Modell Cary 1E der Firma Varian aufgenommen.
7.5 Automatisierte Peptidsynthese
Die automatisierte Synthese der Peptide wurde am Synthesizer ABI 433A der Firma Applied
Biosystems durchgeführt.
7.6 Gefriertrocknung
Die Gefriertrocknung von wasserlöslichen Verbindungen erfolgte mit der Anlage Alpha 1-2
der Firma Christ.
7.7 Chemikalien
Die verwendeten Lösungsmittel wurden wie folgt getrocknet und aufgereinigt:
Cyclohexan wurde destilliert.
Ethylacetat wurde destilliert.
DCM für die Säulenchromatographie wurde destilliert.
DCM der Qualitätsstufe „peptide grade“ wurde von Biosolve erworben.

Experimenteller Teil 7 Geräte und Materialien
179
DMF der Qualitätsstufe „peptide grade“ wurde von Biosolve erworben.
NMP der Qualitätsstufe „peptide grade“ wurde von Applied Biosystems erworben.
Pyridinabs. wurde über Calciumhydrid refluxiert und destilliert.
Tetrahydrofuranabs. wurde über Natrium refluxiert und destilliert.
HMBA-Tentagel-Festphase wurde von Novabiochem bezogen. Alle weiteren Chemikalien
wurden in den Qualitätsstufen „p.a.“ bzw. „zur Synthese“ eingesetzt.

Experimenteller Teil 8 Maschinelle Festphasensynthese
180
8 Maschinelle Festphasensynthese
Die Synthese von PNAs nach der Fmoc/Bhoc-Chemie mit dem Synthesizer ABI 433A der Fa.
Applied Biosystems erfolgte mit modifizierten Prozeduren für die Peptidsynthese in einem
Reaktor mit einem Volumen von 3 ml. Die Synthesen wurden im Maßstab von 10 µmol
durchgeführt, wobei die verwendeten Festphasen Beladungen zwischen 0.1 und 0.2 mmol/g
aufwiesen. Pro Synthesecyclus wurden 5.3 eq. PNA-Monomere bzw. 5.0 eq. N,N-Dimethyl-β-
alanin eingesetzt. Bei schwierigen Kopplungen wurden zwei Cyclen hintereinander
durchgeführt. Als Kopplungsreagenz wurde HBTU verwendet. Zum Cappen nicht-reagierter
Aminofunktionen wurde eine Lösung von Essigsäureanhydrid, Pyridin und NMP im
Verhältnis 1:25:25 benutzt.
Während der Arbeit wurden weitere Modifizierungen bezüglich der Chemikalien und Materi-
alien vorgenommen. So wurde das Lösungsmittel von DMF auf NMP umgestellt und anstatt
Piperidin wurde eine 6%ige Lösung von DBU in NMP zum Abspalten der Fmoc-Gruppe
verwendet.
Nach der Synthese wurde die Festphase in eine Spritze mit Fritte überführt, 2 h im Ölpumpen-
vakuum getrocknet und zur Abspaltung der temporären Bhoc-Schutzgruppen mit 2 ml einer
5%igen Lösung von m-Cresol in TFA behandelt. Nach 2 h wurde mit DMF und DCM
gewaschen und 1 h im Ölpumpenvakuum getrocknet. Danach ließ man die Festphase in DMF
quellen und gab 296 mg (3.36 mmol, 367 µl) N,N-Dimethylaminoethylamin hinzu. Nach 24 h
wurde die Abspaltlösung aufgefangen und die Festsphase mit 4 ml DMF gewaschen. Die
produkthaltigen Lösungen wurden vereinigt und vom Lösungsmittel befreit. Der Rückstand
wurde mit DEE versetzt und im Ultraschallbad behandelt, wobei das Produkt als farbloser
Feststoff ausfiel. Dieser wurde durch Zentrifugieren isoliert und anschließend mehrmals mit
kaltem Diethylether gewaschen. Das so erhaltene Rohprodukt wurde anschließend mittels
RP-HPLC gereinigt und die produkthaltigen Fraktionen wurden dann gefriergetrocknet.

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
181
9 Allgemeine Arbeitsvorschriften
9.1 Grignard-Reaktion mit Trockeneis (AAV 1)
Man legt Magnesium-Späne (2 eq.) in Diethyletherabs. (0.1 ml pro mmol Substrat) vor und
tropft derart 1/20 der Gesamtmenge an Substrat (0.050 eq.) hinzu, dass die Reaktion initiiert
wird. Die restliche Menge des Substrats (0.95 eq.) wird in Diethyletherabs. (0.25 ml pro mmol
Substrat) gelöst und innerhalb von 10 min so hinzugegeben, dass der Ether leicht siedet. Es
wird 2.5 h refluxiert und anschließend auf -10 °C abgekühlt. Daraufhin wird 1 h lang ein
kräftiger Strom von trockenem CO2 eingeleitet, wobei die Temperatur 0 °C nicht übersteigen
darf. Danach wird mit Eis und 6 M Salzsäure hydrolysiert, die Etherphase abgetrennt und die
wässrige Phase mit Ether extrahiert. Die vereinigten organischen Phasen werden mit
gesättigter NaCl-Lösung gewaschen, über Magnesiumsulfat getrocknet und destillativ vom
Lösungsmittel befreit.
9.2 Darstellung von Aminosäuremethylestern (AAV 2)
Man suspendiert die Aminosäure (1 eq.) in Methanol, tropft unter Eisbadkühlung vorsichtig
Thionylchlorid (2.4 eq.) hinzu und erhitzt über Nacht unter Rückfluss. Anschließend wird die
Reaktionsmischung im Eisbad abgekühlt und mit dem doppelten Volumen Diethylether
versetzt, wobei das Produkt in Form seines (Di-)Hydrochlorids als farbloser Feststoff ausfällt.
Man lässt 30 min rühren und saugt das Produkt über eine Fritte ab. Abschließend wäscht man
das Produkt mit Diethylether und trocknet es im Ölpumpenvakuum.
9.3 Verseifung von Aminosäuremethylestern (AAV 3)
Der Methylester wird entweder in H2O/THF (1:1) gelöst, im Eisbad auf 0 °C abgekühlt und
mit LiOH*H2O versetzt oder zuerst nur in THF vorgelegt und bei 0 °C mit 1 M LiOH-Lösung
behandelt. Man lässt 1 h bei 0 °C und 1 h bei RT rühren. Daraufhin wird das Reaktionsge-
misch unter Eisbadkühlung mit 20%iger Zitronensäure-Lösung (Bhoc-geschützte Verbindun-

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
182
gen) oder 1 M HCl (Boc-geschützte Verbindungen) auf pH 2-3 eingestellt und anschließend
dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen werden mit ges.
NaCl-Lösung gewaschen und über MgSO4 getrocknet. Man erhält das Produkt nach Entfernen
des Lösungsmittels am Rotationsverdampfer.
9.4 Reduktive Aminierung mit elementarem Wasserstoff (AAV 4)
Man legt das Amin in Wasser vor (2/3 der Gesamtmenge), kühlt im Eisbad ab und tropft lang-
sam und unter heftigem Rühren eine Lösung des Aldehyds in Wasser (1/3 der Gesamtmenge)
hinzu. Es wird 1 h bei RT nachgerührt. Nach Zugabe des Palladium-Katalysators (10 % auf
Aktivkohle) wird unter leichtem Druck (1-3 bar) mit Wasserstoff hydriert. Anschließend
filtriert man den Katalysator über Kieselgur ab, engt die Reaktionsmischung ein und
coevaporiert zweimal mit je 100 ml Toluol.
9.5 Brommethylierung von Fluoraromaten (AAV 5)
Man löst Paraformaldehyd (1.0 eq.) in einer 33%igen Lösung von HBr in Essigsäure (0.5 ml
pro mmol Substrat) und gibt anschließend den Fluoraromaten (1.0 eq.) und Zinkbromid
(22-45 mol%) hinzu. Danach lässt man 4 h bei 120 °C rühren. Nachdem die Reaktionsmi-
schung abgekühlt ist, gibt man Wasser hinzu und extrahiert dreimal mit Dichlormethan. Die
vereinigten organischen Phasen werden vorsichtig mit gesättigter NaHCO3-Lösung und
NaCl-Lösung gewaschen, über MgSO4 getrocknet und destillativ vom Lösungsmittel befreit.
9.6 N9-Alkylierung bei Purin-Derivaten (AAV 6)
Das Purin-Derivat (1.0 eq.) wird in DMF suspendiert und mit Natriumhydrid (60%ig in
Mineralöl, 1.2 eq.) in zwei Portionen im Abstand von 1 h versetzt. Nach Beendigung der
Gasentwicklung (2-3 h) wird der Ansatz mit einem Eisbad gekühlt und der entsprechende
Bromessigsäureester (1.1 eq.) innerhalb von 30 min zugetropft. Der Ansatz wird über Nacht
bei RT gerührt. Das Lösungsmittel wird am Trockeneisrotationsverdampfer vollständig
entfernt. Der zähflüssige Rückstand wird mit Wasser versetzt und intensiv gerührt oder im

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
183
Ultraschallbad behandelt, bis eine gleichmäßige Suspension entsteht. Der ausgefallene Fest-
stoff wird abgesaugt, ausgiebig mit Wasser und anschließend mit Diethylether gewaschen.
9.7 Einführung von Carbamaten an der N6-Position von Adenin (AAV 7)
Der entsprechende Adenin-9-essigsäure-ester (1.0 eq.) und Carbonyldiimidazol (1.5 eq.)
werden in DMF gelöst. Die Lösung wird unter Argon auf 105 °C erwärmt und 2 h bei dieser
Temperatur gerührt. Anschließend wird um ca. 10 °C abgekühlt, mit Benzhydrol (1.5 eq.)
versetzt und weitere 3 h bei 105 °C gerührt. Die Reaktion wird durch Zugabe von Wasser
abgebrochen. Danach wird dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Phasen werden mit ges. NaCl-Lösung gewaschen, über MgSO4 getrocknet und bis zur
Trockene evaporiert.
9.8 Einführung von Carbamaten an der N4-Position von Cytosin (AAV 8)
Man erhitzt eine Suspension aus Cytosin-1-essigsäure-ester (1.0 eq.) und Carbonyldiimidazol
in THFabs. für 2 h zum Rückfluss. Im Anschluss wird die Suspension unter Argon-
Schutzgasatmosphäre etwas abgekühlt und mit Benzhydrol (1.6 eq.) versetzt. Man rührt wei-
tere 2 h bei Rückflusstemperatur und danach über Nacht bei RT. Nach der Zugabe von
Methanol wird das Lösungsmittel am Rotationsverdampfer entfernt.
9.9 Esterspaltung und Einführung des Carbonyls an Guanin-Derivaten (AAV 9)
Vorgelegt wird eine 60%ige Dispersion von NaH in Mineralöl (5.0 eq.) in absolutem THF.
Nachdem die Suspension in einem iso-Propanol/Trockeneis-Bad auf -78 °C abgekühlt wurde,
wird langsam 3-Hydroxypropionitril (5.0 eq.) zugetropft. Es wird 30 min bei -78 °C und wei-
tere 2.5 h bei 0 °C gerührt. Man gibt dann den geschützten Guanin-9-essigsäure-ester (1.0 eq.)
hinzu und lässt die Reaktionsmischung über Nacht rühren.

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
184
9.10 N9-Alkylierung am Guanin (AAV 10)
Eine Suspension von 2-Amino-6-chlor-purin (1.0 eq.) in DMF wird für 30 min auf 85 °C
erwärmt und mit K2CO3 (1.5 eq.) versetzt. Anschließend lässt man 30 min bei 85 °C rühren.
Danach wird im Eisbad auf 0 °C abgekühlt und tropfenweise mit einer Lösung des jeweiligen
Bromessigsäureesters (1.1 eq.) in DMF versetzt. Man lässt über Nacht bei RT rühren, filtriert
und versetzt das Filtrat unter Rühren mit gekühlter 1 M HCl. Nach 30 min saugt man den
entstandenen Niederschlag ab, wäscht ihn mit wenig Wasser und reichlich Diethylether und
trocknet ihn im Vakuum.
9.11 Einführung von Carbamaten an der N2-Position von Guanin (AAV 11)
Eine Suspension des entsprechenden (2-Amino-6-chlor-purin-9-yl)essigsäureesters (1.0 eq.)
in THF wird bei 0 °C mit Triphosgen (0.37 eq.) versetzt und 1 h bei dieser Temperatur ge-
rührt. Danach tropft man langsam DIPEA (2.2 eq.) hinzu, lässt 30 min rühren und gibt dann
den jeweiligen Alkohol (1.2 eq.) hinzu. Man lässt die Reaktionsmischung über Nacht rühren,
gibt Methanol hinzu und entfernt die Lösungsmittel destillativ am Rotationsverdampfer.
9.12 TOTU-Kopplung (AAV 12)
Vorgelegt werden die Amin- und die Carboxyl-Komponente (je 1.0 eq.) in DMF. Hierzu
werden nacheinander DIPEA (1.0 eq.) und TOTU (1.0 eq.) gegeben. Man läst die Reaktions-
mischung über Nacht bei RT rühren. Anschließend wird mit dem zehnfachen Volumen Ethyl-
acetat verdünnt und mit je dem gleichen Volumen ges. NaHCO3- und NaCl-Lösung
gewaschen. Nach dem Trocknen über MgSO4 wird das Lösungsmittel am Rotationsverdam-
pfer entfernt
9.13 Esterspaltung mit Aluminium(III)chlorid in Ethanthiol (AAV 13)
Man legt AlCl3 (3.0 eq) in Ethanthiol (1 ml pro mmol AlCl3) vor und gibt anschließend den
Ester (1.0 eq.) hinzu. Die Reaktionsmischung wird 24 h bei RT gerührt und anschließend auf

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
185
die dreifache Menge Wasser gegossen. Man extrahiert dreimal mit Dichlormethan, wäscht die
vereinigten organischen Phasen mit ges. NaCl-Lösung und trocknet über MgSO4.
9.14 EDC-Kopplung (AAV 14)
Vorgelegt wird eine Lösung des Amino-Fragments in Form seines Trifluoracetats bzw.
Hydrochlorids (1.0 eq.); HOBt (1.2 eq.) und TEA bzw. DIPEA (4.0 eq.) in Chloroform bzw.
DMF. Unter Eisbadkühlung wird das geschützte Carboxyl-Fragment, gelöst im gleichen
Volumen Chloroform bzw. DMF, zugegeben. Anschließend gibt man EDC*HCl (1.2 eq.)
hinzu und lässt das Reaktionsgemisch 1 h bei 0 °C und 24 h bei RT rühren. Danach wird ggf.
mit Chloroform bzw. Ethylacetat verdünnt, mit ges. NaHCO3- und NaCl-Lösung gewaschen,
über MgSO4 getrocknet und vom Lösungsmittel befreit.
9.15 Abspaltung der Boc-Schutzgruppe mit Trifluoressigsäure (AAV 15)
Man löst die Boc-geschützte Verbindung in Dichlormethan. Wenn das Substrat Nukleobasen
oder Fmoc-Gruppen enthält, wird Triethylsilan als Abfangreagenz hinzugegeben. Dann wird
unter Eisbadkühlung mit Trifluoressigsäure versetzt und die Reaktionsmischung 1 h bei 0 °C
und 1 h bei RT gerührt. Danach wird am Rotationsverdampfer eingeengt und verbleibende
Trifluoressigsäure durch viermalige Coevaporation mit Toluol entfernt.
9.16 Abspaltung der Z-Schutzgruppe mit TFMSA (AAV 16)
Die Z-geschützte Verbindung wird in TFA gelöst und mit m-Cresol und Thioanisol versetzt.
Danach gibt man tropfenweise TFMSA zu und schüttelt die Reaktionsmischung für 2 h bei
RT. Nun wird die Reaktion durch die Zugabe von 45 ml Diethylether abgebrochen und die
entstandene Suspension auf 0 °C abgekühlt. Man zentrifugiert, dekantiert und wäscht das
Produkt zweimal mit Diethylether. Das so erhaltene Rohprodukt wird durch präparative
RP-HPLC gereinigt und die produkthaltigen Fraktionen werden gefriergetrocknet.

Experimenteller Teil 9 Allgemeine Arbeitsvorschriften
186
9.17 HBTU-Kopplung (AAV 17)
Zu einer Lösung der Carboxyl-Komponente (1.0 eq.) und DIPEA (2.0 eq.) in DMF wird unter
Argon-Schutzgasatmosphäre HBTU (1.0 eq.). gegeben. Man rührt 30 min, fügt dann die
Amino-Komponente hinzu und lässt über Nacht bei RT unter Argon-Schutzgasatmosphäre
rühren. Am nächsten Tag wird mit dem zehnfachen Volumen Ethylacetat verdünnt und nach-
einander mit 1 M KHSO4-, ges. NaHCO3- und NaCl-Lösung gewaschen. Man trocknet über
MgSO4 und entfernt das Lösungsmittel am Rotationsverdampfer.
9.18 DCC/HOSu-Kopplung (AAV 18)
Die Carboxyl-Komponente (1.0 eq.) und HOSu (1.5 eq.) werden in DCM bzw. DMF
vorgelegt und unter Argon-Schutzgasatmosphäre auf 0 °C abgekühlt. Man gibt DCC (1.1 eq.)
hinzu und rührt über Nacht. Danach kühlt man die Reaktionsmischung auf -15 °C ab, versetzt
mit der Amino-Komponente (1.0 eq.) und rührt 1 h bei -15 °C und weitere 3 h bei RT. Der
entstandene Dicyclohexylharnstoff wird dann abfiltriert und das Filtrat am
(Trockeneis-)Rotationsverdampfer eingeengt. Der Rückstand wird in 500 ml EtOAc
aufgenommen und mit ges. NaHCO3- und NaCl-Lösung gewaschen. Danach trocknet man die
organische Phase über MgSO4 und entfernt das Lösungsmittel unter vermindertem Druck.
9.19 Fmoc-Abspaltung mit Diethylamin (AAV 19)
Man löst die Fmoc-geschützte Verbindung in DCM, versetzt mit Diethylamin und rührt 1.5 h
bei RT. Anschließend werden die flüchtigen Komponenten am Rotationsverdampfer entfernt.
9.20 Brop-Kopplung (AAV 20)
Man legt das Carboxyl- und das Amino-Fragment (je 1.0 eq.) in DCM bzw. DMF vor, gibt
TEA (2.2 eq.) hinzu und kühlt unter Argon-Schutzgasatmosphäre auf 0 °C ab. Nach Zugabe
von Brop (1.1-1.3 eq.) rührt man über Nacht bei RT.

Experimenteller Teil 10 Synthesen
187
10 Synthesen
10.1 F-AcOH (70)
(2,4-Difluor-5-methyl-phenyl)essigsäure
F
F
OH
O
Summenformel: C9H8F2O2 M = 186.16 g/mol
Ansatz:
22.1 g 100 mmol 1-Brommethyl-2,4-difluor-5-methyl-benzol (92)
4.87 g 200 mmol Magnesiumspäne
35 ml Diethyletherabs.
Trockeneis
Durchführung:
Die Durchführung erfolgt gemäß AAV 1. Dabei bleibt ein weißer Feststoff zurück, der
säulenchromatographisch (Silica, DCM DCM/MeOH 10:1) gereinigt wird.
Ausbeute:
10.7 g (57.5 mmol) = 57 % d.Th.
Analysedaten:
Rf (DCM): 0.20. 1H-NMR (200 MHz, CDCl3): δ = 9.86 (s(br) 1 H, COOH); 7.06 (dd aa t,
2 × J = 8.3, 1 H, CHarom.); 6.78 (dd aa t, 2 × J = 9.6, 1 H, CHarom.); 3.64 (s, 2 H, CH2); 2.22 (s,
3 H, CH3). 13C-NMR (50 MHz, CDCl3): δ = 177.12 (C=O); 160.56 (dd, 1J(C,F) = 247,
3J(C,F) = 11.5, C(4)); 159.26 (dd, 1J(C,F) = 247, 3J(C,F) = 11.9, C(2)); 133.37 (dd, 3J(C,F) = 6.50, 3J(C,F) = 5.40, C(6)); 121.10 (dd, 2J(C,F) = 17.3, 3J(C,F) = 3.80, C(5)); 116.33
(dd, 2J(C,F) = 15.7, 4J(C,F) = 3.80, C(1)); 103.54 (dd aa t, 2 × 2J(C,F) = 26.1, C(3)); 33.77 (d, 3J(C,F) = 2.70, CH2); 14.02 (d, 3J(C,F) = 3.50, CH3).
19F-NMR (565 MHz, DMSO-d6):
-115.57 (m, 1 F, F(4)); -117.17 (m, 1 F, F(2)). EI-MS: 186.0 (32) [M]+•; 141.0 (100)
[M-COOH] +. HR-EI-MS: 186.0502 (30) [M]+• (ber.: 186.0492).

Experimenteller Teil 10 Synthesen
188
10.2 Me2-Aeg(H)-OMe*2HCl (78)
(2-Dimethylamino-ethylamino)essigsäure-methylester Dihydrochlorid
O
O
NH
N
*2HCl
Summenformel: C7H16N2O2*2HCl M = 196.68 g/mol
Ansatz:
1.86 g 12.7 mmol Me2-Aeg(H)-OH (145)
2.22 ml 30.5 mmol Thionylchlorid
100 ml Methanol
Durchführung:
Die Durchführung erfolgt gemäß AAV 2. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
2.44 g (12.4 mmol) = 98 % d.Th.
Analysedaten: 1H-NMR (200 MHz, D2O): δ = 4.11 (s, 2 H, GlyCH2); 3.84 (s, 3 H, OCH3); 3.60 (s, 4 H,
CH2CH2); 2.99 (s, 6 H, (H3C)2N). 13C-NMR (50 MHz, D2O): δ = 168.00 (C=O); 53.93,
52.81, 48.16, 43.75, 42.02 (OCH3, GlyCH2, (CH3)2NCH2CH2). FAB-MS: 161.1 (100)
[M+H]+.

Experimenteller Teil 10 Synthesen
189
10.3 Fmoc-Aeg(CBhoc)-OH (82)
{[2-(4-Benzhydryloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonyl-
amino)-ethyl]-amino}essigsäure
N
N
N
O
N
O
O
O
O N OH
OO
Summenformel: C39H35N5O8 M = 701.74 g/mol
Ansatz:
500 mg 699 µmol Fmoc-Aeg(CBhoc)-OMe (126)
43.8 mg 1.05 mmol LiOH*H2O
10 ml H2O
10 ml THF
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Das Rohprodukt wird in 30 ml Acetonitril für
30 min bei Rückflusstemperatur gerührt, abgesaugt und mit Acetonitril und Ether gewaschen.
Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
457 mg (651 µmol) = 93 % d.Th.

Experimenteller Teil 10 Synthesen
190
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.87 (m, 3 H, C(6)H, FmocCHarom.);
7.68 (d, 3J = 6.8, 2 H, FmocCHarom.); 7.36 (m, 16 H, 10 × BhocCHarom., 4 × FmocCHarom.,
2 ×NH); 6.94, 6.92 (je d, 3J = 7.3, zusammen 1 H, C(5)H); 6.80 (s, 1 H, BhocCH); 4.35 - 3.99
(m, 5 H, FmocCH, FmocCH2, GlyCH2); 4.82 (s, 1.1 H, N(1)CH2); 4.63 (s, 0.9 H, N(1)CH2);
3.44 - 3.10 (m, 4 H, CH2CH2). MALDI-TOF-MS: 700.64 (116) [M-H]-.
10.4 Fmoc-Aeg(H)-OMe (83)
[2-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethylamino]essigsäure-methylester
NH
O
O
NH
O
O
Summenformel: C20H22N2O4 M = 354.41 g/mol
Ansatz:
4.00 g 19.7 mmol H-Aeg(H)-OMe*2HCl (124)
5.10 g 19.7 mmol Fmoc-Cl
12.9 ml 73.8 mmol DIPEA
500 ml DCM
Durchführung:
Man legt 4.00 g (19.7 mmol) H-Aeg(H)-OMe*2HCl (124) in 400 ml DCM vor und kühlt die
Suspension im Eisbad ab. Nach der Zugabe von 12.9 ml (73.8 mmol) DIPEA wird zügig eine
Lösung von 5.10 g (19.7 mmol) Fmoc-Cl in 100 ml DCM zugetropft. Man lässt 30 min bei
RT rühren und entfernt danach das Lösungsmittel am Rotationsverdampfer. Nach
Säulenchromatographie (Silica, DCM/MeOH 20:1) erhält man das Produkt als farbloses Öl,
das langsam erstarrt.
Ausbeute:
5.12 g (14.4 mmol) = 73 % d.Th.

Experimenteller Teil 10 Synthesen
191
Analysedaten:
Rf (DCM/MeOH 10:1): 0.60. 1H-NMR (200 MHz, CDCl3): δ = 7.78 (m, 2 H, Fmoc-
CHarom.); 7.60 (m, 2 H, FmocCHarom.); 7.35 (m, 4 H, FmocCHarom.); 5.48 (t(br), 1 H, NH); 4.44
(m, 2 H, FmocCH2); 4.21 (m, 1 H, FmocCH); 3.72 (s, 3 H, OCH3); 3.39 (s, 2 H, GlyCH2);
3.27 (dt aa q, 3J = 5.5, 2 H, FmocNHCH2); 2.75 (t, 3J = 5.5, 2 H, FmocNHCH2CH2); 1.80
(s(br), 1 H, NH). 13C-NMR (50 MHz, CDCl3): δ = 173.04 (COOMe); 156.75 (FmocC=O);
144.16 (FmocCarom.); 141.45 (FmocCarom.); 127.79 (FmocCHarom.); 127.18 (FmocCHarom.);
125.22 (FmocCHarom.); 120.09 (FmocCHarom.); 66.71 (FmocCH2); 52.01 (OCH3); 50.38,
48.79, 47.42, 40.75 (FmocCH, GlyCH2, CH2CH2). FAB-MS: 355.1 (100) [M+H] +.

Experimenteller Teil 10 Synthesen
192
10.5 CBhoc-AcOH (84)
4-N-(Benzhydryloxycarbonyl)cytosin-1-essigsäure
N
N
O
NH
OH
O
O
O
Summenformel: C20H17N3O5 M = 379.38 g/mol
Ansatz:
16.2 g 34.5 mmol CBhoc-AcOMe (107)
15.9 g 37.9 mmol LiOH*H2O
250 ml Methanol
Durchführung:
16.2 g (34.5 mmol) CBhoc-AcOMe (107) werden in 250 ml Methanol suspendiert und danach
auf 0 °C abgekühlt. Nach der Zugabe von 15.9 g (37.9 mmol) LiOH*H2O lässt man 1 h bei
0 °C rühren. Anschließend gibt man eine Lösung von 36.5 g Zitronensäure (190 mmol) in
123 ml Wasser hinzu und saugt den entstandenen Niederschlag ab. Nach dem Trocknen im
Ölpumpenvakuum erhält man das Produkt in Form eines farblosen Feststoffs.
Ausbeute:
11.3 g (29.8 mmol) = 86 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 7.91 (d, 3J = 7.3, 1 H, C(6)H); 7.46 - 7.26 (m, 10 H,
BhocCHarom.); 6.87 (d, 3J = 7.3, 1 H, C(5)H); 6.78 (s, 1 H, BhocCH); 4.27 (N(1)CH2). 13C-NMR (50 MHz, DMSO-d6): δ = 170.01 (COOH); 162.51 (C(4)), 155.28 (BhocC=O);
152.60 (C(2)); 151.22 (C(6)), 140.53 (BhocCarom.); 128.63, 127.91, 126.52 (3 × BhocCHarom.);
93.33 (C(5)); 77.43 (BhocCH); 52.29 (N(1)CH2). FAB-MS: 409 (30) [M+Na]+; 386 (85)
[M+H]+; 167.1 (100).

Experimenteller Teil 10 Synthesen
193
10.6 H-Aeg(H)-OH (85)
N-(2-Aminoethyl)glycin
NH
OH
O
NH2
Summenformel: C4H10N2O2 M = 118.14 g/mol
Ansatz:
4.60 g 50.0 mmol Glyoxylsäure Monohydrat (123)
8.97 g 149 mmol Ethylendiamin (122)
300 ml Wasser
2.00 g Pd/C (10 %)
1.12 l H2
Durchführung:
Die Durchführung erfolgt gemäß AAV 4. Das zurückbleibende Öl versetzt man mit 100 ml
iso-Propanol und behandelt es im Ultraschallbad, wobei ein weißer Feststoff ausfällt. Man
lässt die Mischung über Nacht bei -20 °C im Kühlschrank stehen, filtriert den ausgefallenen
Feststoff ab und wäscht ihn mit kaltem iso-Propanol.
Ausbeute:
3.86 g (32.7 mmol) = 65 % d.Th.
Analysedaten:
1H-NMR (200 MHz, D2O): δ = 3.24 (s, 2 H, GlyCH2); 3.04 - 2.83 (m, 4 H, CH2CH2).
13C-NMR (50 MHz, D2O): δ = 178.26 (C=O); 51.56 (GlyCH2); 46.53 (NH2CH2CH2); 38.56
(NH2CH2CH2). FAB-MS: 119.1 (100) [M+H] +.

Experimenteller Teil 10 Synthesen
194
10.7 H-Aeg(H)-OH (85)
N-(2-Aminoethyl)glycin
NH
OH
O
NH2
Summenformel: C4H10N2O2 M = 118.14 g/mol
Ansatz:
131 g 1.39 mol Chloressigsäure (243)
823 g 13.7 mol Ethylendiamin (122)
Durchführung:
Man legt 823 g (13.7 mol) Ethylendiamin (122) vor und kühlt im Eisbad. Unter heftigem
Rühren werden innerhalb von 8 h portionsweise 131 g (1.39 mol) Chloressigsäure (243) ein-
getragen. Dabei wird jeweils gewartet bis sich die vorangegangene Portion komplett gelöst
hat. Man rührt über Nacht bei RT nach und entfernt am nächsten Morgen überschüssiges
Ethylendiamin (122), das für weitere Ansätze aufgehoben wird. Der erhaltene Rückstand wird
mit 2 l DMSO versetzt und über Nacht kräftig gerührt, wodurch das Produkt als farbloser
Feststoff ausfällt. Man saugt es ab, wäscht es mit DMSO und Diethylether und trocknet es im
Ölpumpenvakuum.
Ausbeute:
116 g (980 mmol) = 70 % d.Th.
Analysedaten:
siehe 10.6

Experimenteller Teil 10 Synthesen
195
10.8 F-CH2Br (92)
1-(Brommethyl)-2,4-difluor-5-methylbenzol
F
F
Br
Summenformel: C8H7BrF2 M = 221.05 g/mol
Ansatz:
20.0 g 156 mmol 2,4-Difluortoluol (71)
4.93 g 156 mmol Paraformaldehyd
15.8 g 70.0 mmol Zinkbromid
80 ml 33% HBr in Essigsäure
Durchführung:
Die Durchführung erfolgt gemäß AAV 5. Dabei bleibt ein braunes Öl zurück, das säulenchro-
matographisch (Silica, CH) gereinigt wird. Man erhält das Produkt als farblose Flüssigkeit.
Ausbeute:
22.6 g (0.120 mol) = 66 % d.Th.
Analysedaten:
Rf (CH): 0.34. 1H-NMR (200 MHz, CDCl3): δ = 7.20 (dd aa t, 2 × J = 8.3, 1 H, CHarom.);
6.77 (dd aa t, 2 × J = 9.6, 1 H, CHarom.); 4.46 (s, 2 H, CH2); 2.23 (s, 3 H, CH3). 13C-NMR
(50 MHz, CDCl3): δ = 161.28 (dd, 1J(C,F) = 250, 3J(C,F) = 11.9, C(4)); 159.01 (dd, 1J(C,F) = 250, 3J(C,F) = 12.3, C(2)); 133.21 (dd, 3J(C,F) = 6.90, 3J(C,F) = 4.20, C(6)); 120.91
(dd, 2J(C,F) = 29.1, 3J(C,F) = 4.20, C(5)); 121.23 (dd, 2J(C,F) = 26.1, 4J(C,F) = 4.20, C(1));
103.89 (dd, 2J(C,F) = 24.9, 2J(C,F) = 26.5, C(3)); 25.39 (d, 3J(C,F) = 3.8, CH2); 14.01 (d, 3J(C,F) = 3.10, CH3).
19F-NMR (565 MHz, DMSO-d6): -111.49 (m, 1 F, F(4)); -115.88
(m, 1 F, F(2)). EI-MS: 268 (72) [M+2Na]+; 253 (100); 141.0 (78) [M-Br]+.

Experimenteller Teil 10 Synthesen
196
10.9 F’-CH2Br (95)
1-(Brommethyl)-2,4-difluor-benzol
F
F
Br
Summenformel: C7H5BrF2 M = 207.02 g/mol
Ansatz:
2.28 g 20.0 mmol 1,3-Difluorbenzol (93)
600 mg 20.0 mmol Paraformaldehyd
900 mg 4.00 mmol Zinkbromid
10 ml 33% HBr in Essigsäure
Durchführung:
Die Durchführung erfolgt gemäß AAV 5. Man erhält das Produkt nach säulenchromatogra-
phischer Reinigung (Silica, CH) als farblose Flüssigkeit.
Ausbeute:
1.66 g (8.02 mmol) = 40 % d.Th.
Analysedaten:
Rf (CH): 0.14. 1H-NMR (200 MHz, CDCl3): δ = 7.43 - 7.31 (m, 1 H, CHarom.); 6.92 - 6.77
(m, 2 H, 2 × CH); 4.48 (s, 2 H, CH2). 13C-NMR (50 MHz, CDCl3): δ = 163.51 (dd,
1J(C,F) = 251, 3J(C,F) = 11.9, C(4)); 163.74 (dd, 1J(C,F) = 253, 3J(C,F) = 12.3, C(2)); 132.48
(dd, 3J(C,F) = 9.90, 3J(C,F) = 4.60, C(6)); 121.74 (dd, 2J(C,F) = 14.6, 4J(C,F) = 4.90, C(1));
112.20 (dd, 2J(C,F) = 21.8, 4J(C,F) = 3.80, C(5)); 104.74 (dd aa t, 2 × 2J(C,F) = 25.4, C(3));
25.36 (d, 3J(C,F) = 4.20, CH2). FAB-MS: 253.3 (100) [M+2Na]+.

Experimenteller Teil 10 Synthesen
197
10.10 F’-AcOH (96)
(2,4-Difluor-phenyl)essigsäure
O
F
F
OH
Summenformel: C8H6F2O4 M = 172.13 g/mol
Ansatz:
3.00 g 14.5 mmol 1-(Brommethyl)-2,4-difluor-benzol (95)
4.87 g 26.0 mmol Magnesiumspäne
30 ml Diethyletherabs.
nach Bedarf Trockeneis
Durchführung:
Die Durchführung erfolgt gemäß AAV 2. Man erhält das Produkt nach säulenchromatogra-
phischer Reinigung (Silica, DCM/MeOH 10:1) als farblosen Feststoff.
Ausbeute:
1.40 g (8.13 mmol) = 56 % d.Th.
Analysedaten:
Rf (DCM/MeOH 10:1): 0.57. 1H-NMR (200 MHz, CDCl3): δ = 10.03 (s(br), 1 H, COOH);
7.05 (m 1 H, CHarom.); 6.71 (m, 2 H, 2 × CHarom.); 3.43 (s, 2 H, CH2). 13C-NMR (50 MHz,
CDCl3): δ = 177.57 (d, 4J = 2.6, C=O); 162.25 (dd, 1J(C,F) = 248, 3J(C,F) = 12.3, C(4));
161.09 (dd, 1J(C,F) = 248, 3J(C,F) = 11.1, C(2)); 132.23 (m, C(6)); 117.93 (m, C(1)); 111.22
(dd, 2J(C,F) = 21.1, 4J(C,F) = 2.30, C(5)); 103.78 (dd aa t, 2 × 2J(C,F) = 26.5, C(3)); 34.74 (d, 3J(C,F) = 2.70, CH2). EI-MS: 172.0 (26) [M]+•; 127.0 (100) [M+-COOH].

Experimenteller Teil 10 Synthesen
198
10.11 A-AcOBn (104)
Adenin-9-essigsäure-benzylester
N N
N N
NH2
O
O
Summenformel: C14H13N5O2 M = 283.29 g/mol
Ansatz:
6.76 g 50.0 mmol Adenin (103)
2.40 g 60.0 mmol NaH 60%ig in Mineralöl
12.6 g 55.0 mmol Bromessigsäure-benzylester
230 ml DMF
130 ml Wasser
Durchführung:
Die Durchführung erfolgt gemäß AAV 6. Nach Trocknung im Ölpumpenvakuum erhält man
das Produkt in Form eines farblosen Feststoffs.
Ausbeute:
9.90 g (34.9 mmol) = 70 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 8.14 (s, 2 H, C(2)H, C(8)H); 7.36 (s, 5 H, BnCHarom.);
7.29 (s, 2 H, NH2); 5.20 (s, 2 H, CH2); 5.15 (s, 2 H, CH2). FAB-MS: 307.0 (20) [M+Na]+;
284.0 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
199
10.12 C-AcOBn (105)
Cytosin-1-essigsäure-benzylester
N
N
O
NH2
O
O
Summenformel: C13H13N3O3 M = 259.27 g/mol
Ansatz:
20.6 g 185 mmol Cytosin (86)
23.2 g 207 mmol Kalium-tert-butoxid
46.7 g 204 mmol Bromessigsäure-benzylester
300 ml DMF
250 ml Wasser
Durchführung:
20.6 g (185 mmol) Cytosin (86) und 23.2 g (207 mmol) Kalium-tert-butoxid werden in
300 ml DMF suspendiert und anschließend unter Argon-Schutzgasatmosphäre 2 h bei 100 °C
gerührt. Anschließend wird die Suspension zunächst langsam auf RT und dann auf 0 °C
abgekühlt. Nachdem innerhalb von 30 min 46.7 g (204 mmol) Bromessigsäure-benzylester
hinzugetropft wurden, wird die Reaktionsmischung über Nacht bei RT gerührt. Die Reaktion
wird durch Zugabe von 12 ml Essigsäure gequencht und das Lösungsmittel am Trockeneis-
Rotationsverdampfer entfernt. Man versetzt den Rückstand mit 250 ml Wasser und lässt 1 h
kräftig rühren. Dann wird der Rückstand abgesaugt, ausgiebig mit Wasser gewaschen und im
Ölpumpenvakuum getrocknet.
Ausbeute:
46.5 g (179 mmol) = 97 % d.Th.

Experimenteller Teil 10 Synthesen
200
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 7.58 (d, 3J = 7.3, 2 H, C(6)H); 7.17 (d(br), 3J = 8.3,
2 H, NH2); 7.36 (s, 5 H, BnCHarom.); 5.69 (d, 3J = 7.3, 1 H, C(5)H); 5.16 (s, 2 H, GlyCH2);
4.51 (s, 2 H, N(1)CH2). FAB-MS: 260.1 (35) [M+H]+; 167.1 (100).
10.13 ABhoc-AcOBn (106)
6-N-(Benzhydryloxycarbonyl)adenin-9-essigsäure-benzylester
N N
N N
NH
O
O
O
O
Summenformel: C28H23N5O4 M = 493.53 g/mol
Ansatz:
9.90 g 34.9 mmol A-AcOBn (104)
8.49 g 52.4 mmol CDI
9.65 g 52.4 mmol Benzhydrol
50 ml DMF
200 ml Wasser
Durchführung:
Die Durchführung erfolgt gemäß AAV 7. Der Rückstand wird säulenchromatographisch
aufgereinigt (Silica, 30 % EtOAc in CH → 70 % EtOAc in CH, Gradient in Schritten von je
10 %, je ein Säulenvolumen).
Ausbeute:
11.6 g (23.5 mmol) = 67 % d.Th.

Experimenteller Teil 10 Synthesen
201
Analysedaten:
Rf (30 % EtOAc in CH): 0.53. 1H-NMR (200 MHz, DMSO-d6): δ = 10.96 (s, 1 H,
BhocNH); 8.63 (s, 1 H, C(2)H); 8.47 (s, 1 H, C(8)H); 7.56 - 7.28 (m, 15 H, BnCHarom.,
BhocCHarom.); 6.83 (s, 1 H, BhocCH); 5.29 (s, 2 H, CH2); 5.21 (s, 2 H, CH2).
10.14 CBhoc-AcOBn (107)
4-N-(Benzhydryloxycarbonyl)cytosin-1-essigsäure-benzylester
N
N
O
NH
O
O
O
O
Summenformel: C27H23N3O5 M = 469.50 g/mol
Ansatz:
13.7 g 52.8 mmol C-AcOBn (105)
13.7 g 84.5 mmol CDI
15.4 g 84.5 mmol Benzhydrol
350 ml THFabs.
50 ml Methanol
Durchführung:
Die Durchführung erfolgt gemäß AAV 8. Der so erhaltene Rückstand wird mit 500 ml
Ethanol versetzt und 30 min bei Rückflusstemperatur gerührt. Anschließend wird das Produkt
in Form eines farblosen Feststoffs abgesaugt.
Ausbeute:
16.2 g (34.5 mmol) = 65 % d.Th.

Experimenteller Teil 10 Synthesen
202
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 11.08 (s, 1 H, BhocNH); 8.07 (d, 3J = 7.3, 1 H, C(6)H);
7.49 - 7.25 (m, 15 H, 5 × BnCHarom., 10 × BhocCHarom.); 7.00 (d, 3J = 7.3, 1 H, C(5)H); 6.82
(s, 1 H, BhocCH); 5.19 (s, 2 H, BnCH2); 4.71 (s, 2 H, N(1)CH2). 13C-NMR (50 MHz,
DMSO-d6): δ = 167.89 (COOBn); 163.42 (C(4)); 154.98 (BhocC=O); 152.30 (C(2)); 150.40
(C(6)); 140.33 (BhocCarom.); 135.54 (BnCarom.); 128.55, 128.45, 128.18, 127.92, 127.87,
126.46 (3 × BnCHarom., 3 × BhocCHarom.); 94.20 (C(5)); 77.52 (BhocCH); 66.41 (BnCH2);
50.67 (N(1)CH2). FAB-MS: 492.1 (29) [M+Na]+; 470.1 (36) [M+H]+, 167.1 (100).

Experimenteller Teil 10 Synthesen
203
10.15 ABhoc-AcOH (108)
6-N-(Benzhydryloxycarbonyl)adenin-9-essigsäure
N N
N N
NH
O
O
OH
O
Summenformel: C21H17N5O4 M = 403.40 g/mol
Ansatz:
11.6 g 23.5 mmol ABhoc-AcOBn (106)
90 ml 1 M NaOH
45 ml Ethanol
45 ml Acetonitril
Durchführung:
11.6 g (23.5 mmol) ABhoc-AcOBn (106) werden in 45 ml Ethanol und 45 ml Acetonitril ge-
löst, mit einem Eisbad gekühlt und mit 90 ml 1 M NaOH-Lösung versetzt. Nach 1 h Rühren
bei 0 °C wird die Reaktionsmischung mit 20%iger Zitronensäure-Lösung auf pH-Wert 2-3
eingestellt. Der entstehende Feststoff wird abgesaugt, mit 200 ml Wasser und etwas Diethyl-
ether gewaschen und im Ölpumpenvakuum getrocknet.
Ausbeute:
7.60 g (18.8 mmol) = 80 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 10.94 (s, 1 H, BhocNH); 8.62 (s, 1 H, C(2)H); 8.45 (s,
1H, C(8)H); 7.55 - 7.28 (m, 10 H, BhocCarom.); 6.83 (s, 1 H, BhocCH); 5.09 (s, 2 H, CH2). 13C-NMR (50 MHz, DMSO-d6): δ = 169.01 (COOMe); 152.11, 151.64, 151.13 (C(2), C(4),
BhocC=O); 149.34 (C(6)); 144.81 (C(8)); 140.84 (BhocCarom.); 128.44 (Bhoc-o-CHarom.);
127.66 (Bhoc-p-CHarom.) 126.48 (Bhoc-m-CHarom.); 122.68 (C(5)); 77.24 (BhocCH2); 44.24
(N(9)CH2). FAB-MS: 426.0 (10) [M+Na]+; 404.0 (7) [M]+; 154.0 (100).

Experimenteller Teil 10 Synthesen
204
10.16 ABhoc-AcOH (108)
6-N-(Benzhydryloxycarbonyl)adenin-9-essigsäure
N N
N N
NH
O
O
OH
O
Summenformel: C21H17N5O4 M = 403.40 g/mol
Ansatz:
10.0 g 23.2 mmol ABhoc-AcOEt (121)
90 ml 1 M NaOH
45 ml Ethanol
45 ml Acetonitril
Durchführung:
s. 10.15
Ausbeute:
8.34 g (20.7 mmol) = 88 % d.Th.
Analysedaten:
s. 10.15

Experimenteller Teil 10 Synthesen
205
10.17 T-AcOH (109)
Thymin-1-essigsäure
N
NH
O
O
OH
O
Summenformel: C7H6N2O4 M = 184.15 g/mol
Ansatz:
10.0 g 79.3 mmol Thymin (72)
11.0 g 80.0 mmol K2CO3
7.60 ml 80.0 mmol Bromessigsäure-methylester
80 ml 1 M NaOH
240 ml DMF
Durchführung:
Zu einer Suspension aus 10.0 g (79.3 mmol) Thymin (72) und 11.0 g (80.0 mmol) K2CO3 in
DMF werden 12.3 g (80.0 mmol, 7.60 ml) Bromessigsäure-methylester innerhalb von 10 min
zugetropft. Die Reaktionslösung wird über Nacht bei RT gerührt, filtriert und bis zur
Trockenheit eingeengt. Der Rückstand wird mit Wasser (70 ml) und 4 M HCl (3.2 ml)
behandelt und 30 min gerührt. Der Niederschlag wird abfiltriert und mit Wasser gewaschen
(2 × 40 ml). Zu dem Feststoff werden 80 ml 1 M NaOH gegeben. Die Lösung wird für 10 min
unter Rückfluss gerührt, dann auf 0 °C abgekühlt, vorsichtig mit 30 ml 4 M HCl versetzt und
30 min gerührt. Das Produkt wird durch Filtration erhalten und dreimal mit je 50 ml Wasser
und einmal mit 40 ml Diethylether gewaschen. Abschließend wird es im Ölpumpenvakuum
getrocknet.
Ausbeute:
9.05 g (49.2 mmol) = 62 % d.Th.

Experimenteller Teil 10 Synthesen
206
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 13.11 (s(br), 1 H, COOH); 11.34 (s, 1 H, NH); 7.49 (d,
3J = 1.3, 1 H, CH); 4.36 (s, 2 H, CH2); 1.75 (s, 3 H, CH3). 13C-NMR (50 MHz, DMSO-d6):
δ = 169.71 (COOH); 164.40 (C(4)); 151.03 (C(2)); 141.84 (C(6)); 108.38 (C(5)); 48.43
(CH2); 11.94 (CH3). FAB-MS: 207.0 (5) [M+Na]+; 185.0 (36) [M]+; 154.0 (100).
10.18 GBhoc-AcOH (110)
(2-Benzhydryloxycarbonylamino-6-chlor-purin-9-yl)essigsäure
N N
N NH
O
NH
OH
O
OO
Summenformel: C21H17N5O5 M = 419.40 g/mol
Ansatz:
19.0 g 37.0 mmol (2-Benzhydryloxycarbonylamino-6-chlor-purin-9-yl)essig-
säure-benzylester (113)
7.16 g 180 mmol NaH 60%ig in Mineralöl
12.2 ml 180 mmol 3-Hydroxypropionitril
200 ml THF
Durchführung:
Die Durchführung erfolgt gemäß AAV 9. Anschließend wird das Lösungsmittel am Rotati-
onsverdampfer entfernt und der Rückstand in 150 ml Wasser aufgenommen. Man stellt den
pH-Wert der Lösung mit 20%iger Zitronensäure-Lösung auf 4 ein und kühlt auf 0 °C ab. Der
entstandene Feststoff wird abgesaugt, mit Wasser gewaschen und aus Methanol um-
kristallisiert.
Ausbeute:
5.32 g (15.5 mmol) = 58 % d.Th.

Experimenteller Teil 10 Synthesen
207
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 11.71 (s, 1 H, NH); 11.24 (s, 1 H, NH); 7.93 (s, 1 H,
C(8)H); 7.48 - 7.26 (m, 1 H, 10 × BhocCHarom.); 6.86 (s, 1 H, BhocCH); 4.87 (CH2).
13C-NMR (50 MHz, DMSO-d6): δ = 169.96 (COOH); 157.53, 155.98, 154.24, 149.46,
147.84 (C(2), C(4), C(6), C(8), BhocC=O); 140.55 (BhocCarom.); 128.05, 127.78, 126.42,
(BhocCHarom.); 118.91 (C(5)); 77.55 (BhocCH); 46.89 (CH2). FAB-MS: 464.1 (4) [M+2Na];
442.1 (14) [M+Na]+; 420.2 (3), [M+H]+; 154.0 (100).

Experimenteller Teil 10 Synthesen
208
10.19 (2-Amino-6-chlor-purin-9-yl)essigsäure-benzylester (112)
N N
N N
Cl
NH2
O
O
Summenformel: C14H12ClN5O2 M = 317.74 g/mol
Ansatz:
50.0 g 295 mmol 2-Amino-6-chlor-purin (111)
60.1 g 437 mmol K2CO3
50.3 ml 322 mol Bromessigsäure-benzylester
500 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 10. Das erhaltene Rohprodukt wird aus Acetonitril
umkristallisiert. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
65.4 g (205 mmol) = 71 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 8.13 (s, 1 H, C(8)H); 7.37 (s, 5 H, BnCHarom.); 7.01 (s,
2 H, NH2); 5.20 (s, 2 H, BnCH2); 5.07 (s, 2 H, N(9)CH2). 13C-NMR (50 MHz, DMSO-d6):
δ = 167.63 (COOBn); 159.97, 154.27, 149.51, 143.44 (C(2), C(4), C(6), C(8)); 135.31
(BnCarom.); 128.50, 128.30, 128.09 (3 × BnCHarom.); 122.94 (C(5)); 66.79 (BnCH2); 44.07
(N(9)CH2). FAB-MS: 340.0 (5) [M+Na]+; 318.0 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
209
10.20 (2-Benzhydryloxycarbonylamino-6-chlor-purin-9-yl)essigsäure-benzylester (113)
N N
N N
Cl
NH
O
O
OO
Summenformel: C28H22ClN5O4 M = 527.97 g/mol
Ansatz:
31.0 g 97.6 mmol (2-Amino-6-chlor-purin-9-yl)essigsäure-benzylester (112)
10.8 g 36.4 mmol Triphosgen
21.5 g 117 mmol Diphenylcarbinol
36.8 ml 215 mmol DIPEA
500 ml THF
Durchführung:
Die Durchführung erfolgt gemäß AAV 11. Der Rückstand wird in 500 ml DCM aufgenom-
men und mit 10%iger Zitronensäure-Lösung, ges. NaHCO3- und NaCl-Lösung gewaschen.
Nach dem Trocknen über MgSO4 wird das DCM am Rotationsverdampfer entfernt und der
Rückstand aus Methanol umkristallisiert. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
34.7 g (65.8 mmol) = 67 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 11.02 (s, 1 H, BhocNH); 8.52 (s, 1 H, C(8)H); 7.54 -
7.27 (m, 15 H, 5 × BnCHarom.., 10 × BhocCHarom.); 6.83 (s, 1 H, BhocCH); 5.22 (BnCH2); 5.19
(N(9)CH2). 13C-NMR (50 MHz, DMSO-d6): δ = 167.28 (COOBn); 153.13, 152.30, 150.91,
149.25, 146.72 (C(2), C(4), C(6), C(8), BhocC=O); 140.84 (BhocCarom.); 135.17 (BnCarom.);
128.48, 128.44, 128.23, 127.97, 127.70, 126.77, 126.45 (3 × BnCHarom., 3 × BhocCHarom.,
C(5)); 77.06 (BhocCH); 66.89 (BnCH2); 44.47 (N(9)CH2). FAB-MS: 550.1 (29) [M+Na]+;
528.1 (63) [M+H]+; 167.1 (100).

Experimenteller Teil 10 Synthesen
210
10.21 A-AcOEt (120)
Adenin-9-essigsäure-ethylester
N N
N N
NH2
O
O
Summenformel: C9H11N5O2 M = 221.22 g/mol
Ansatz:
50.0 g 370 mmol Adenin (103)
17.8 g 445 mmol NaH (60%ig in Mineralöl)
45.1 ml 408 mmol Bromessigsäure-ethylester
1 l DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 6. Der entstandene beige Feststoff wird in 500 ml
Ethanol für 45 min zum Rückfluss erhitzt. Nach dem Abkühlen auf RT saugt man das Produkt
in Form farbloser Kristalle ab, wäscht es mit Ethanol und trocknet es im Ölpumpenvakuum.
Ausbeute:
55.6 g (251 mmol) = 67 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 8.14 (s, 1 H, C(2)H); 8.12 (s, 1 H, C(8)H); 7.30 (s(br),
2 H, NH2); 5.07 (s, 2 H, N(9)CH2); 4.16 (q, 3J = 7.1, 2 H, CH2CH3); 1.20 (t, 3J = 7.1, 3 H,
CH3). 13C-NMR (50 MHz, DMSO-d6): δ = 168.00 (C=O); 155.99 (C(6)); 152.68 (C(2));
149.73 (C(4)); 141.29 (C(8)); 118.27 (C(5)); 61.37 (CH2CH3); 43.94 (N(9)CH2); 14.00 (CH3).
FAB-MS: 221.1 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
211
10.22 ABhoc-AcOEt (121)
6-N-(Benzhydryloxycarbonyl)adenin-9-essigsäure-ethylester
N N
N N
NH
O
O
O
O
Summenformel: C23H21N5O4 M = 431.45 g/mol
Ansatz:
7.72 g 34.9 mmol A-AcOEt (120)
8.49 g 52.4 mmol CDI
9.65 g 52.4 mmol Benzhydrol
50 ml DMF
200 ml Wasser
Durchführung:
Die Durchführung erfolgt gemäß AAV 7. Das Rohprodukt wird abgesaugt, mit Wasser
gewaschen und aus Ethanol umkristallisiert. Nach Trocknen im Ölpumpenvakuum erhält man
das Produkt als farblosen Feststoff.
Ausbeute:
10.1 g (23.4 mmol) = 67 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 10.96 (s, 1 H, BhocNH); 8.63 (s, 1 H, C(2)H); 8.45 (s,
1 H, C(8)H); 7.77 - 7.19 (m, 10 H, BhocCHarom.); 6.83 (s, 1 H, BhocCH); 5.20 (s, 2 H,
N(9)CH2); 4.18 (q, 3J = 7.1, 2 H, CH2CH3); 1.21 (t, 3J = 7.1, 3 H, CH3). FAB-MS: 454.0 (19)
[M+Na]+; 432.0 (23) [M]+; 388.0 (24) [M-CO2]+; 167.0 (100).

Experimenteller Teil 10 Synthesen
212
10.23 H-Aeg(H)-OMe*2HCl (124)
(2-Amino-ethylamino)essigsäure-methylester Dihydrochlorid
NH
O
O
NH2
*2HCl
Summenformel: C5H12N2O2*2HCl M = 203.06 g/mol
Ansatz:
93.0 g 787 mmol Aeg (85)
138 ml 1.90 mol Thionylchlorid
1.5 l Methanol
Durchführung:
Die Durchführung erfolgt gemäß AAV 2. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
130 g (641 mmol) = 81 % d.Th.
Analysedaten:
1H-NMR (200 MHz, D2O): δ = 4.14 (s, 2 H, GlyCH2); 3.86 (s, 3 H, OCH3); 3.56 - 3.39 (m,
4 H, CH2CH2). 13C-NMR (50 MHz, D2O): δ = 167.73 (C=O); 54.03 (OCH3); 48.03
(GlyCH2); 39.81 (NH2CH2CH2); 35.79 (NH2CH2). FAB-MS: 154.0 (100) [M+Na]+; 133.1
(70) [M]+.

Experimenteller Teil 10 Synthesen
213
10.24 Fmoc-Aeg(ABhoc)-OMe (125)
{[2-(6-Benzhydryloxycarbonylamino-purin-9-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-
amino}essigsäure-methylester
O
ONH
N
O
N N
N N
O NH
O
OO
Summenformel: C41H37N7O7 M = 739.79 g/mol
Ansatz:
1.77 g 5.00 mmol Fmoc-Aeg(H)-OMe (83)
2.02 g 5.00 mmol ABhoc-AcOH (108)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, DCM/MeOH 80:1) als farblosen Schaum.
Ausbeute:
2.72 g (3.68 mmol) = 74 % d.Th.

Experimenteller Teil 10 Synthesen
214
Analysedaten:
Rf (DCM/MeOH 30:1): 0.15. 1H-NMR (2 Rotamere) (200 MHz, DMSO-d6): δ = 10.93 (s,
1 H, BhocNH); 8.60 (s, 0.30 H, C(2)H); 8.55 (s, 0.60 H, C(2)H); 8.34 (s, 1 H, C(8)H); 7.87 (d, 3J = 7.3, 2 H, FmocCHarom.); 7.67 (m, 2 H, FmocCHarom.); 7.54 (d, 3J = 7.1, 4 H,
FmocCHarom.); 7.34 (m, 11 H, BhocCHarom., FmocNH); 6.84 (s, 1 H, BhocCH); 5.38 (s, 1.08,
N(9)CH2); 5.20 (s, 0.75, N(9)CH2); 4.47 - 4.22 (m, 4 H, FmocCH, FmocCH2, GlyCH); 4.11
(s, 1 H, GlyCH); 3.76 (s, 0.85, OCH3); 3.61 (s, 2.15 H, OCH3); 3.83 - 3.19 (m(br), CH2CH2). 13C-NMR (2 Rotamere) (50 MHz, DMSO-d6): δ = 169.89 (COOMe); 169.41 (COOMe);
167.12 (N(9)CH2CO); 166.78 (N(9)CH2CO); 156.47, 156.18, 154.03, 152.31, 151.56, 151.13,
149.28 (FmocC=O, BhocC=O, C(2), C(4), C(6)); 145.19, 143.88, 140.91, 140.77 (C(8),
BhocCarom., 2 × FmocCarom.); 128.47, 127.69, 127.62, 127.14, 127.05, 125.09, 124.95
(BhocCHarom., FmocCHarom.); 122.62 (C(5)); 120.14 (FmocCHarom.); 77.29 (BhocCH); 65.52
(FmocCH2); 54.92, 52.37, 51.82, 46.75 (GlyCH2, OCH3, N(9)CH2, CH2CH2, FmocCH). FAB-
MS: 762.3 (47) [M+Na]+; 740 (37) [M+H]+; 696.3 (40) [M-NH=C=O]+; 167.1 (100).

Experimenteller Teil 10 Synthesen
215
10.25 Fmoc-Aeg(CBhoc)-OMe (126)
{[2-(4-Benzhydryloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonyl-
amino)-ethyl]-amino}essigsäure-methylester
N
N
NH
O
N
O
O
O
O NH
O
OO
Summenformel: C40H37N5O8 M = 715.77 g/mol
Ansatz:
1.90 g 5.00 mmol Fmoc-Aeg(H)-OMe (83)
2.02 g 5.00 mmol CBhoc-AcOH (84)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man kristallisiert aus Acetonitril um und erhält das
Produkt als farblosen Schaum.
Ausbeute:
1.33 g (1.86 mmol) = 37 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 11.01 (s, 1 H, BhocNH); 7.87 (m, 3 H,
C(6)H, FmocCHarom.); 7.69 (d, 3J = 7.1, 2 H, FmocCHarom.); 7.52 (m, 16 H, 10 × BhocCHarom.,
4 × FmocCHarom., 2 × NH); 6.94 (d(br), 3J = 7.2, 1 H, C(5)H); 6.80 (s, 1 H, BhocCH); 4.83 (s,
1.2 H, N(1)CH2); 4.64 (s, 0.8 H, N(1)CH2); 4.36 - 4.08 (m, 5 H, FmocCH, FmocCH2,
GlyCH2); 3.71 (s, 0.9 H, OCH3); 3.62 (s, 2.1 H, OCH3); 3.47 - 3.10 (m, 4 H, CH2CH2).

Experimenteller Teil 10 Synthesen
216
10.26 Fmoc-Aeg(GBhoc)-OH (127)
{[2-(2-Benzhydryloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxy-
carbonylamino)-ethyl]-amino}essigsäure
N
O
N
O
ON
N NH
O
NH
O NH
OH
OO
Summenformel: C40H35N7O8 M = 741.77 g/mol
Ansatz:
200 mg 588 µmol Fmoc-Aeg(H)-OH (129)
247 mg 588 µmol GBhoc-AcOH (110)
73.8 µl 600 µmol Pivaloylchlorid (130)
130 µl 1.18 mmol NMM
nach Bedarf Triethylamin
6 ml Acetonitril
2 ml DMF
2 ml Wasser
Durchführung:
Zu einer Lösung von 247 mg (588 µmol) GBhoc-AcOH (110) und 130 µl (1.18 mmol) NMM in
5 ml MeCN/DMF (3:2) werden bei 0 °C 73.8 µl (600 µmol) Pivaloylchlorid gegeben. An-
schließend wird der Ansatz für 20 min bei 0 °C gerührt. Parallel suspendiert man
200 mg (588 µmol) Fmoc-Aeg(H)-OH (129) in 5 ml MeCN/H2O (3:2) und bringt den
Feststoff durch tropfenweise Zugabe von Triethylamin in Lösung. Danach gibt man die
Lösung zur ersten Reaktionsmischung, rührt 20 min bei RT und stellt anschließend mit
20%iger Zitronensäure-Lösung auf pH 4 ein. Die Lösungsmittel werden bei einer Badtempe-
ratur von 40 °C am Trockeneis-Rotationsverdampfer entfernt, der Rückstand in 2 ml
Methanol aufgenommen und tropfenweise zu 25 ml Eiswasser gegeben. Der resultierende
Niederschlag wird abgesaugt, mit Wasser und Diethylether gewaschen und im Vakuum
getrocknet. Man erhält das Produkt als farblosen Feststoff.

Experimenteller Teil 10 Synthesen
217
Ausbeute:
311 mg (419 µmol) = 71 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.90 (m, 2 H, FmocCHarom.); 7.81, 7.80
(je s, zusammen 1 H, C(8)H); 7.66 (m, 2 H, FmocCHarom.); 7.37 (m, 16 H, 10 × BhocCHarom.,
4 × FmocCHarom., 2 × NH); 6.86 (s, 1 H, BhocCH); 5.1 (s, 1.1 H, N(9)CH2); 4.94 (s, 0.9 H,
N(9)CH2); 4.39 - 4.01 (m, 5 H, FmocCH, FmocCH2, GlyCH2); 3.49 - 3.14 (m, 4 H, CH2CH2).
MALDI-TOF-MS: 762.34 (125) [M-H+Na]-.
10.27 Fmoc-Aeg(F)-OH (128a)
{[2-(2,4-Difluor-5-methyl-phenyl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-amino}-
essigsäure-methylester
N
O
O NH
OH
OO
F
F
Summenformel: C28H26F2N2O5 M = 508.53 g/mol
Ansatz:
565 mg 1.00 mmol Fmoc-Aeg(F)-OMe (135a)
400 mg 3.00 mmol AlCl3
3 ml Ethanthiol (137)
Durchführung:
Die Durchführung erfolgt nach AAV 13. Das Rohprodukt wird säulenchromatographisch
(Silica, DCM/MeOH 30:1) gereinigt. Man erhält das Produkt als farblosen Feststoff.

Experimenteller Teil 10 Synthesen
218
Ausbeute:
473 mg (930 µmol) = 93 % d.Th.
Analysedaten: 1H-NMR (400 MHz, DMSO-d6) (2 Rotamere):δ =7.87 (d, 3J = 7.6, 2 H, FmocCHarom.);
7.66 (d, 3J = 7.6, 2 H, FmocCHarom.); 7.50 (s(br), 1 H, FmocNH); 7.35 (m, 4 H, FmocCHarom.);
7.24 - 6.92 (m, 2 H, FCHarom.); 4.47 - 4.12 (m, 3 H, FmocCH, FmocCH2); 3.96 (s, 1.01 H,
FCH2); 3.92 (s, 0.89 H, FCH2); 3.67 (s, 0.78 H, GlyCH2); 3.53 (s, 1.22 H, GlyCH2); 3.45 (t, 3J = 6.1, 0.78 H, FmocNHCH2CH2); 3.37 (t, 3J = 6.1, 1.16 H, FmocNHCH2CH2); 3.23 (m,
0.88 H, FmocNHCH2CH2); 3.15 (m, 1.17 H, FmocNHCH2CH2); 2.13 (s, 1.5 H, CH3); 2.10 (s,
1.5 H, CH3). 13C-NMR (100 MHz, DMSO-d6) (2 Rotamere): δ = 160.00 (C=O); 159.87
(C=O); 156.26 (FmocC=O); 156.06 (FmocC=O); 143.86 (FmocCarom.); 143.79 (FmocCarom.);
140.70 (FmocCarom.); 140.64 (FmocCarom.); 133.72 (m, FC(6)); 133.43 (m, FC(6)); 127.53
(FmocCHarom.); 127.03 (FmocCHarom.); 125.19 (FmocCHarom.); 125.03 (FmocCHarom.); 119.68
(m, FC(5)); 119.18 (m, FC(1)); 102.24 (m, FC(3)); 65.47 (FmocCH2); 52.20 (FmocCH);
48.27 (s(br), FmocNHCH2CH2); 47.80 (s(br), FmocNHCH2CH2); 47.09 (GlyCH2); 46.70
(GlyCH2); 38.64 (s(br), FmocNHCH2CH2); 38.21 (s(br), FmocNHCH2CH2); 32.20 (FCH2);
31.88 (FCH2); 13.35 (CH3). Die 13C-Signale für FC(2) und FC(4) sind aufgrund der
Aufspaltung und der damit verbundenen geringen Intensität nicht zu sehen. 19F-NMR
(2 Rotamere) (545 MHz, DMSO-d6): = -115.85 (m, 0.44 F, F(4)); -116.11 (m, 0.56 F,
F(4)); -116.32 (m, 0.44 F, F(2)); -116.52 (m, 0.56 F, F(2)). FAB-MS: 154.0 (100); 509.1 (19)
[M+H]+, 531.1 (20) [M++Na]. MALDI-TOF-MS: 507.5 (55) [M-H]-.

Experimenteller Teil 10 Synthesen
219
10.28 Fmoc-Aeg(F’)-OH (128b)
{[2-(2,4-Difluor-phenyl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-amino}essigsäure-
methylester
N
O
O N OH
OO
F
F
Summenformel: C27H24F2N2O5 M = 494.50 g/mol
Ansatz:
500 mg 983 µmol Fmoc-Aeg(F’)-OMe (135b)
393 mg 2.95 mmol AlCl3
5 ml Ethanthiol
Durchführung:
Die Durchführung erfolgt nach AAV 30. Das Rohprodukt wird säulenchromatographisch
(Silica, DCM/MeOH 30:1) gereinigt. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
358 mg (724 µmol) = 74 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 12.7 (s(br), 1 H, COOH); 7.88 (d, 3J = 7.2, 2 H, FmocCHarom.); 7.68 (d, 3J = 7.1, 2 H, FmocCHarom.); 7.41 - 7.10 (m, 7 H,
4 × FmocCHarom., 2 × F’CHarom., FmocNH); 6.98 (m, 1 H, F’CHarom.); 4.00 (m, 3 H,
FmocCH2, FmocCH); 3.92 (s, 2 H, F’CH2); 3.75, 3.60 (je s, zusammen 2 H, GlyCH2); 3.49 -
3.15 (m(br), 4 H, CH2CH2). FAB-MS: 495.2 (28) [M+H]+; 517.1 (82) [M+Na]+; 179.1 (100).
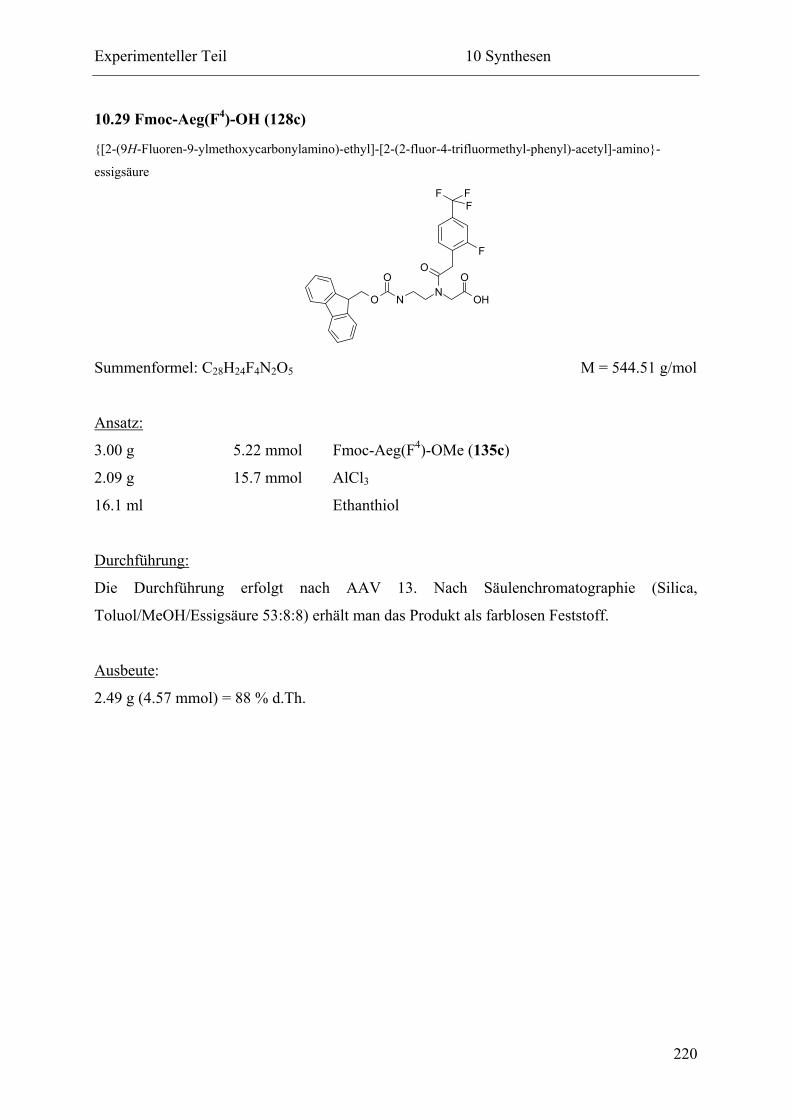
Experimenteller Teil 10 Synthesen
220
10.29 Fmoc-Aeg(F4)-OH (128c)
{[2-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethyl]-[2-(2-fluor-4-trifluormethyl-phenyl)-acetyl]-amino}-
essigsäure
N
O
O N OH
OO
F
FFF
Summenformel: C28H24F4N2O5 M = 544.51 g/mol
Ansatz:
3.00 g 5.22 mmol Fmoc-Aeg(F4)-OMe (135c)
2.09 g 15.7 mmol AlCl3
16.1 ml Ethanthiol
Durchführung:
Die Durchführung erfolgt nach AAV 13. Nach Säulenchromatographie (Silica,
Toluol/MeOH/Essigsäure 53:8:8) erhält man das Produkt als farblosen Feststoff.
Ausbeute:
2.49 g (4.57 mmol) = 88 % d.Th.

Experimenteller Teil 10 Synthesen
221
Analysedaten:
Rf (Toluol/MeOH/AcOH 53:8:8): 0.35. 1H-NMR (400 MHz, DMSO-d6) (2 Rotamere):
δ = 7.88 (d(br), 3J = 7.3, 2 H, FmocCHarom.); 7.81 (m(br), 2 H, FmocCHarom.); 7.59 - 7.29 (m,
8 H, 3 × F4CHarom., 4 × FmocCHarom., NH); 4.35 - 4.25 (m, 3 H, FmocCH2, FmocCH); 3.98,
3.89 (je s, zusammen 2 H, GlyCH2); 3.73, 3.50 (je s, zusammen 2 H, F4CH2); 3.50 - 3.14 (m,
4 H, F4CH2, FmocNHCH2CH2). 13C-NMR (100 MHz, DMSO-d6) (2 Rotamere): δ = 171.23
(COOH); 170.69 (COOH); 169.41 (GlyC=O); 168.94 (GlyC=O); 160.42 (dq aa dm, 1J(C,F) = 247, F4C(2)); 156.36 (FmocC=O); 156.13 (FmocC=O); 143.87 (FmocCarom.);
143.84 (FmocCarom.); 140.74 (FmocCarom.); 140.70 (FmocCarom.); 133.22 (d, 3J(C,F) = 4.42,
F4C(1)); 133.02 (d, 3J(C,F) = 4.56, F4C(6)); 129.24 (dq aa m, F4C(4)); 128.23 (d, 2J(C,F) = 16.9, F4C(1)); 127.57 (FmocCHarom.); 127.02 (FmocCHarom.); 125.12 (FmocCHarom.);
125.04 (FmocCHarom.); 126.98 (FmocCHarom.); 123.89 (dq, 1J(C,F) = 272, 4J(C,F) = 2.30,
F4CH3); 120.83 (dq aa m, F4C(5)); 120.07 (FmocCHarom.); 119.95 (FmocCHarom.); 112.25 (dq
aa dm, 2J(C,F) = 26.7, F4C(3)); 65.47 (FmocCH2); 65.39 (FmocCH2); 50.40, 50.38 49.82,
48.59, 46.80; 45.72; (FmocCH, GlyCH2, FmocNHCH2CH2); 38.74 (FmocNHCH2); 38.13
(FmocNHCH2); 32.82 (F4CH2); 32.45 (F4CH2). 19F-NMR (235 MHz, DMSO-d6)
(Rotamere): δ = -64.28 (s, 3 F, CF3), -117.21 (m, 0.7 F, F4C(2)F); -117.57 (m, 0.7 F,
F4C(2)F). FAB-MS: 589.1 (18) [M+2Na]+; 567.1 (100) [M+Na]+; 545.1 (20) [M+H]+.

Experimenteller Teil 10 Synthesen
222
10.30 Fmoc-Aeg(H)-OH (129)
[2-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethylamino]essigsäure
NH
O NH
OH
OO
Summenformel: C19H20N2O4 M = 340.38 g/mol
Ansatz:
3.00 g 25.4 mmol H-Aeg(H)-OH (85)
8.04 ml 63.3 mmol SiMe3Cl
8.04 g 23.8 mmol Fmoc-OSu
11.2 ml 101 mmol NMM
51 ml DCM
3 ml DMF
Durchführung:
3.00 g (25.4 mmol) H-Aeg(H)-OH (85) werden fein gemörsert und in einer Mischung aus
51 ml DCM und 3 ml DMF suspendiert. Man gibt 8.04 ml (6.88 g, 63.3 mol) SiMe3Cl hinzu
und rührt 40 min bei RT. Danach wird auf 0 °C abgekühlt und mit 8.04 g (23.8 mmol)
Fmoc-OSu versetzt. Anschließend werden innerhalb von 30 min 11.2 ml (10.3 g, 101 mmol)
NMM hinzugetropft. Man rührt 2 h bei RT, gibt 6 ml Methanol hinzu und rührt weitere
20 min. Dann werden 60 ml Ethylacetat hinzugegeben und es wird weitere 20 min gerührt.
Man saugt den Feststoff ab, wäscht ihn mit Ethylacetat und trocknet ihm im Membranpum-
penvakuum. Danach wird der Feststoff in 90 ml Wasser aufgenommen und die entstandene
Suspension 1 h bei RT gerührt. Man saugt ab, suspendiert den Feststoff in 150 ml Methanol,
rührt 30 min bei RT, saugt erneut ab und wäscht mit Methanol. Nach dem Trocknen im
Ölpumpenvakuum erhält man das Produkt als farblosen Feststoff.
Ausbeute:
2.94 g (8.64 mmol) = 34 % d.Th.

Experimenteller Teil 10 Synthesen
223
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 7.89 (d, 3J = 6.8, 2 H, FmocCHarom.); 7.68 (d, 3J = 7.2,
2 H, FmocCHarom.); 7.60 - 7.21 (m, 4 H, FmocCHarom.); 4.35 (d, 3J = 6.9, 2 H, FmocCH2); 4.23
(t, 3J = 6.9, 1 H, FmocCH); 3.88 (s, 2 H, GlyCH2); 3.45 - 3.16 (m, 2 H, FmocNHCH2CH2);
3.03 (m, 2 H, FmocNHCH2CH2). FAB-MS: 363.0 (36) [M+Na]+; 341.0 (100) [M]+.
10.31 Fmoc-Aeg(T)-OH (132)
{[2-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethyl]-[2-(5-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-yl)-ac
etyl]-amino}essigsäure
N
NH
O
O
NOH
O
NH
O
OO
Summenformel: C26H26N4O7 M = 506.52 g/mol
Ansatz:
764 mg 4.15 mmol T-OAc (109)
1.43 g 4.15 mmol Fmoc-Aeg(H)-OH (129)
536 µl 4.36 mmol Pivaloylchlorid (130)
915 µl 8.30 mmol NMM
nach Bedarf Triethylamin
15 ml Acetonitril
20 ml Acetonitril / H2O (4:3)

Experimenteller Teil 10 Synthesen
224
Durchführung:
Zu einer Lösung von 764 mg (4.15 mmol) T-AcOH (109) und 915 µl (8.30 mmol) NMM in
15 ml Acetonitril werden bei 0 °C 536 µl (4.36 mmol) Pivaloylchlorid gegeben. Anschließend
wird der Ansatz für 30 min bei 0 °C gerührt. Parallel suspendiert man 1.43 g (4.15 mmol)
Fmoc-Aeg(H)-OH (129) in 20 ml MeCN/H2O (4:3) und bringt den Feststoff durch Zugabe
von TEA in Lösung. Man gibt diese Lösung zur ersten, rührt 2 h bei RT, stellt mit 3 M HCl
auf pH 2 ein und lässt über Nacht rühren. Anschließend wird zentrifugiert, dekantiert und der
Niederschlag mit Diethylether gewaschen.
Ausbeute:
1.33 g (2.62 mmol) = 63 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): 12.75 (s(br), 1 H, COOH); 11.30, 11.29 (je
s, zusammen 1 H, N(3)H); 7.89 (d, 3J = 6.9, 2 H, FmocCHarom.); 7.68 (d, 3J = 7.1, 2 H,
FmocCHarom.); 7.34 (m, 6 H, 4 × FmocCHarom., 2 × NH); 4.65 (s, 1.1 H, N(1)CH2); 4.48 (s,
0.9 H, N(1)CH2); 4.2 (m, 5 H, FmocCH2, FmocCH, GlyCH2); 3.26 (m, 4 H, CH2CH2); 1.73
(s, 3 H, CH3). MALDI-TOF-MS: 505.37 (154) [M-H] -.

Experimenteller Teil 10 Synthesen
225
10.32 Fmoc-Aeg(ABhoc)-OH (133)
{[2-(6-Benzhydryloxycarbonylamino-purin-9-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-
amino}essigsäure
O
ONH
N
O
N N
N N
O NH
OH
OO
Summenformel: C40H35N7O7 M = 725.77 g/mol
Ansatz:
2.11 g 2.86 mmol Fmoc-Aeg(ABhoc)-OMe (125)
240 mg 5.72 mmol LiOH*H2O
120 ml H2O/THF 1:1
Durchführung:
Die Durchführung erfolgt nach AAV 3. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, DCM/MeOH 10:1) als farblosen Feststoff.
Ausbeute:
1.41 g (1.94 mmol) = 68 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 10.65 (s(br), 1 H, COOH); 8.62, 8.55 (je
s, zusammen 1 H, C(2)H); 8.36, 8.33 (je s, zusammen 1 H, C(8)H); 7.90 (m, 3 H,
2 × FmocCHarom., NH); 7.70 (m, 2 H, FmocCHarom.); 7.58 (m, 4 H, FmocCHarom.); 7.38 (m,
10 H, BhocCHarom.); 6.87 (s, 1 H, BhocCH); 5.30, 5.20 (je s, zusammen 1 H, N(9)CH2); 4.07
(m, 5 H, FmocCH2, FmocCH, GlyCH); 3.45, 3.22 (je m(br), zusammen 4 H, CH2CH2).
MALDI-TOF-MS: 724.54 (124) [M-H] -.
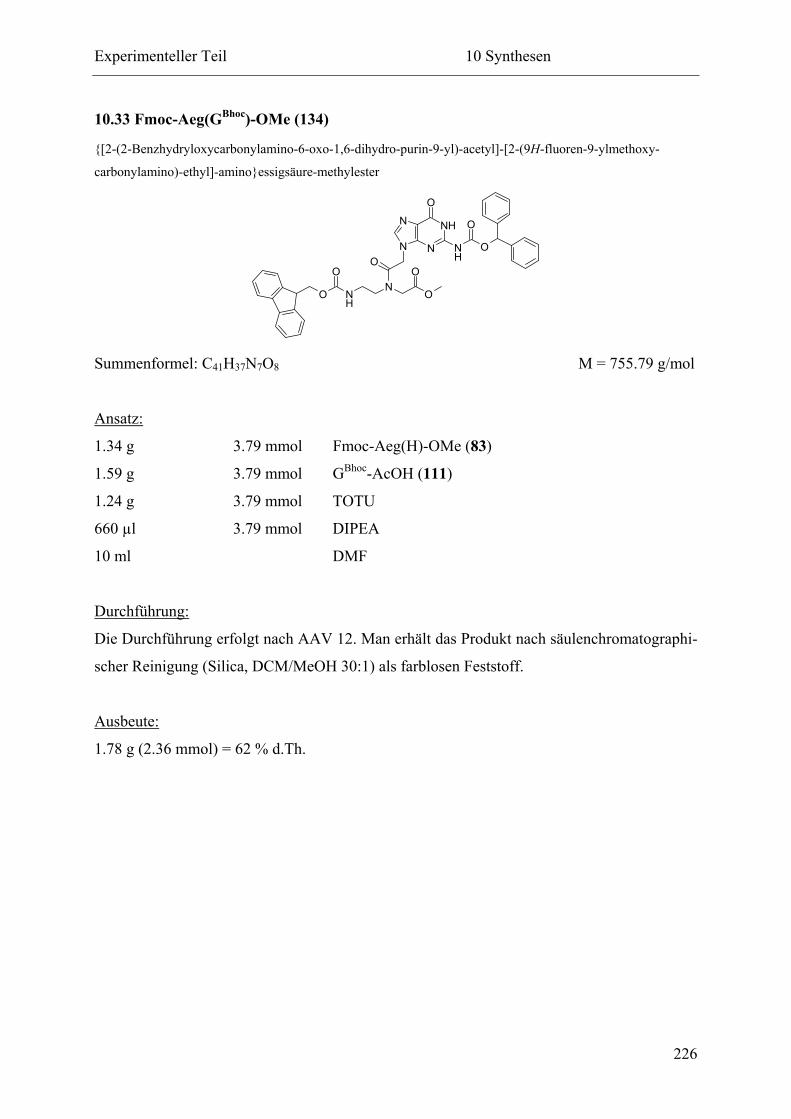
Experimenteller Teil 10 Synthesen
226
10.33 Fmoc-Aeg(GBhoc)-OMe (134)
{[2-(2-Benzhydryloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)-acetyl]-[2-(9H-fluoren-9-ylmethoxy-
carbonylamino)-ethyl]-amino}essigsäure-methylester
N
O
N
O
ON
N NH
O
NH
O NH
O
OO
Summenformel: C41H37N7O8 M = 755.79 g/mol
Ansatz:
1.34 g 3.79 mmol Fmoc-Aeg(H)-OMe (83)
1.59 g 3.79 mmol GBhoc-AcOH (111)
1.24 g 3.79 mmol TOTU
660 µl 3.79 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, DCM/MeOH 30:1) als farblosen Feststoff.
Ausbeute:
1.78 g (2.36 mmol) = 62 % d.Th.

Experimenteller Teil 10 Synthesen
227
Analysedaten:
Rf (DCM/MeOH 30:1): 0.03. 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 11.67,
11.62 (je s, zusammen 1 H, NH); 11.23 (s, 1 H, NH); 7.84 (m, 3 H, 2 × FmocCHarom., C(8)H);
7.66 (m, 2 H, FmocCHarom.); 7.38 (m, 16 H, 10 × BhocCHarom., 4 × FmocCHarom., 2 × NH);
6.86 (s, 1 H, BhocCH); 5.12 (s, 1.2 H, N(9)CH2); 4.94 (s, 0.8 H, N(9)CH2); 4.28 (m, 5 H,
FmocCH, FmocCH2, GlyCH2); 3.75 (s, 1 H, CH3); 3.75 (s, 2 H, CH3); 3.50, 3.14 (je m,
zusammen 4 H, CH2CH2). FAB-MS: 778.0 (3) [M+Na]+; 756.0 (4) [M]+; 712.0 (3)
[M-CO2]+; 167.0 (100).
10.34 Fmoc-Aeg(F)-OMe (135a)
{[2-(2,4-Difluor-5-methyl-phenyl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-amino}-
essigsäure-methylester
N
O
O NH
O
OO
F
F
Summenformel: C29H28F2N2O5 M = 522.55 g/mol
Ansatz:
1.77 g 5.00 mmol Fmoc-Aeg(H)-OMe (83)
931 mg 5.00 mmol F-AcOH (70)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, CH/EtOAc 1:4) als farblosen Feststoff.

Experimenteller Teil 10 Synthesen
228
Ausbeute:
1.66 g (4.15 mmol) = 83 % d.Th.
Analysedaten:
Rf (CH/EtOAc 1:4): 0.62. 1H-NMR (400 MHz, DMSO-d6) (2 Rotamere): δ = 7.88 (d, 3J = 7.1, 2 H, FmocCHarom.); 7.68 (d, 3J = 7.6, 2 H, FmocCHarom.); 7.41 (m u. s(br), 3 H,
FmocCHarom., FmocNH); 7.36 - 7.28 (m, 2 H, FmocCHarom.); 7.19 - 6.99 (m, 2 H, FCHarom.);
4.53 - 3.95 (m, 5 H, FmocCH, FmocCH2, FCH2); 3.72 (s, 1.1 H, GlyCH2); 3.70 (s, 0.9 H,
GlyCH2); 3.63 (s, 2.2 H, OCH3); 3.57 (s, 0.8 H, OCH3); 3.49 (t, 3J = 6.3, 1.35 H,
FmocNHCH2CH2); 3.37 (m, 0.57 H, FmocNHCH2CH2); 3.25 (m, 1.4 H, FmocNHCH2CH2);
3.14 (m, 0.6 H, FmocNHCH2CH2); 2.15 (s, 3 H, CH3). 13C-NMR (100 MHz, DMSO-d6)
(2 Rotamere): δ = 170.10 (C=O); 169.89 (C=O); 169.75 (C=O); 169.67 (C=O); 159.14 (dd, 1J(C,F) = 244, 3J(C,F) = 12.3, FC(4)); 158.55 (dd, 1J(C,F) = 244, 3J(C,F) = 12.3, FC(2));
156.29 (FmocC=O); 156.09 (FmocC=O); 143.84 (FmocCarom.); 143.79 (FmocCarom.); 140.72
(FmocCarom.); 140.69 (FmocCarom.); 133.69 (m, FC(6)); 133.40 (m, FC(6)); 127.55
(FmocCHarom.); 126.98 (FmocCHarom.); 125.06 (FmocCHarom.); 125.00 (FmocCHarom.); 120.05
(FmocCHarom.); 119.52 (dd, 4J(C,F) = 3.07, 2J(C,F) = 16.8, FC(5)); 118.62 (m, C(1)); 102.86
(dd aa t, 2 × 3J(C,F) = 26.1, FC(3)); 102.80 (dd aa t, 2 × 3J(C,F) = 26.1, FC(3)); 65.43
(FmocCH2); 65.34 (FmocCH2); 52.08 (OCH3); 51.62 (OCH3); 49.81 (FmocCH) 47.86
(FmocNHCH2CH2); 47.57 (FmocNHCH2CH2); 46.73 (GlyCH2); 46.65 (GlyCH2); 38.80
(FmocNHCH2CH2); 38.18 (FmocNHCH2CH2); 32.26 (FCH2); 31.76 (FCH2); 13.38 (m,
FCH3). 19F-NMR (546 MHz, DMSO-d6) (2 Rotamere):δ = -115.64 (m, 1 F, F(4)); -116.26
(m, 0.69 F, F(2)); -116.47 (m, 0.27 F, F(2)). FAB-MS: 545.2 (23) [M+Na] +; 523.2 (40)
[M+H]+; 178.1 (100).

Experimenteller Teil 10 Synthesen
229
10.35 Fmoc-Aeg(F’)-OMe (135b)
{[2-(2,4-Difluor-phenyl)-acetyl]-[2-(9H-fluoren-9-ylmethoxycarbonylamino)-ethyl]-amino}essigsäure-
methylester
N
O
O N O
OO
F
F
Summenformel: C28H26F2N2O5 M = 508.53 g/mol
Ansatz:
1.77 g 5.00 mmol Fmoc-Aeg(H)-OMe (83)
860 mg 5.00 mmol F’-AcOH (96)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, DCM/MeOH 80:1) als farblosen Feststoff.
Ausbeute:
1.28 g (2.52 mmol) = 50 % d.Th.

Experimenteller Teil 10 Synthesen
230
Analysedaten:
Rf (DCM/MeOH 50:1): 0.19. 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.88 (d, 3J = 7.1, 2 H, FmocCHarom.); 7.68 (d, 3J = 7.07, 2 H, FmocCHarom.); 7.61 - 7.45 (m, 7 H,
4 × FmocCHarom., 2 × F’CHarom., FmocNH); 6.99 (m, 1 H, F’CHarom.); 4.36 - 4.22 (m, 3.7 H,
FmocCH2, FmocCH, F’CH2); 4.06 (s, 1.3 H, F’CH2); 3.76 (s, 1.1 H, GlyCH2); 3.70 (s, 0.9 H,
GlyCH2); 3.62 (s, 3 H, OCH3); 3.54 - 3.09 (m(br), 4 H, CH2CH2). 13C-NMR (50 MHz,
DMSO-d6) (2 Rotamere): δ = 170.20, 169.94, 169.84, 169.69 (F’CH2CO, COOMe); 161.29
(dd, 1J(C,F) = 245, 3J(C,F) = 12.7, F’C(4)); 160.61 (dd, 1J(C,F) = 247, 3J(C,F) = 12.7,
F’C(2)); 156.40 (FmocC=O); 156.18 (FmocC=O); 143.87 (FmocCarom.); 140.79 (FmocCarom.);
132.57 (dd, 3J(C,F) = 6.14, 3J(C,F) = 9.59, F’C(6)); 127.63 (FmocCHarom.); 127.07
(FmocCHarom.); 125.08 (FmocCHarom.); 120.13 (FmocCHarom.); 119.32 (m, F’C(1)); 110.96
(dd, 2J(C,F) = 20.7, 4J(C,F) = 3.45, F’C(5)); 103.44 (dd aa t, 2 × 2J(C,F) = 26.1, F’C(3));
65.47 (FmocCH2); 51.70 (OCH3); 52.17, 49.85, 47.90, 47.61, 46.77 (CH2CH2, GlyCH2,
FmocCH); 32.37 (F’CH2); 31.09 (F’CH2). FAB-MS: 531.2 (20) [M+Na]+; 509.2 (28)
[M+H]+; 178.1 (100).

Experimenteller Teil 10 Synthesen
231
10.36 Fmoc-Aeg(F4)-OMe (135c)
{[2-(9H-Fluoren-9-ylmethoxycarbonylamino)-ethyl]-[2-(2-fluor-4-trifluormethyl-phenyl)-acetyl]-amino}-
essigsäure-methylester
N
O
O NH
O
OO
F
FFF
Summenformel: C29H26F4N2O5 M = 558.53 g/mol
Ansatz:
3.95 g 11.2 mmol Fmoc-Aeg(H)-OMe (83)
2.48 g 11.2 mmol F4-AcOH (101)
3.66 g 11.2 mmol TOTU
1.95 ml 11.2 mmol DIPEA
25 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Der Rückstand wird mit 40 ml Methanol versetzt,
15 min im Ultraschallbad behandelt und anschließend abgesaugt. Nach dem Waschen mit
Methanol und Trocknen im Vakuum erhält man das Produkt als farblosen Feststoff.
Ausbeute:
3.44 g (6.16 mmol) = 55 % d.Th.

Experimenteller Teil 10 Synthesen
232
Analysedaten: 1H-NMR (400 MHz, DMSO-d6) (2 Rotamere): δ = 7.85 (m, 2 H, FmocCHarom.); 7.69 - 7.23
(m, 10 H, 6 × FmocCHarom., 3 × F4CHarom., 1 NH); 4.34 (m, 2 H, FmocCH2); 4.22 (m, 1 H,
FmocCH); 4.06, 3.92 (je s, zusammen 2 H, GlyCH2); 3.90, 3.75 (je m, zusammen 2 H,
F4CH2); 3.71, 3.62 (je s, zusammen 3 H, CH3); 3.53 - 3.13 (m, 4 H, F4CH2, CH2CH2). 13C-
NMR (100 MHz, DMSO-d6) (2 Rotamere): δ = 170.07 (COOMe); 169.66 (COOMe);
169.40 (GlyC=O); 169.14 (GlyC=O); 160.42 (dq aa dm, 1J(C,F) = 247, F4C(2)); 156.34
(FmocC=O); 156.11 (FmocC=O); 143.85 (FmocCarom.); 143.82 (FmocCarom.); 140.72
(FmocCarom.); 140.70 (FmocCarom.); 133.22 (dq aa t, 3J(C,F) = 4.98, F4C(6)); 132.93 (dq aa d, 3J(C,F) = 4.56, F4C(6)); 128.76 (dq aa m, F4C(4)); 127.54 (FmocCHarom.); 127.09 (d, 2J(C,F) = 23.8, F4C(1)); 126.98 (FmocCHarom.); 125.06 (FmocCHarom.); 125.00 (FmocCHarom.);
123.41 (dq, 1J(C F) = 272, 4J(C,F) = 2.30, F4CH3); 120.85 (dq aa t, 4J(C,F) ~ 3J(C,F) = 3.87,
F4C(5)); 120.05 (FmocCHarom.); 119.95 (FmocCHarom.); 112.25 (dq aa dm, 2J(C,F) = 25.7,
F4C(3)); 65.43 (FmocCH2); 65.34 (FmocCH2); 52.11 (OCH3); 51.63 (OCH3); 49.84, 47.59;
46.72; (FmocCH, GlyCH2, FmocNHCH2CH2); 38.79 (FmocNHCH2); 38.12 (FmocNHCH2);
32.90 (F4CH2); 32.42 (F4CH2). 19F-NMR (235 MHz, DMSO-d6) (2 Rotamere): δ = -64.28
(s, 3 F, CF3), -117.22 (m, 0.7 F, F4C(2)F); -117.56 (m, 0.3 F, F4C(2)F). FAB-MS: 582.1 (39)
[M+Na]+; 559.1 (40) [M+H]+; 178.1 (100).

Experimenteller Teil 10 Synthesen
233
10.37 N,N-Dimethyl-β-alanin Hydrochlorid (140)
3-Dimethylamino-propionsäure Hydrochlorid
OH
O
N*HCl
Summenformel: C5H11NO2*HCl M = 153.61 g/mol
Ansatz:
3.56 g 40.0 mmol β-Alanin (143)
25 ml Ameisensäure
5 ml Formalin
5 ml HClkonz.
Durchführung:
Man erhitzt eine Mischung aus 3.56 g (40.0 mmol) β-Alanin, 25 ml Ameisensäure und 5 ml
Formalin über Nacht unter Rückfluss. Im Anschluss wird die Reaktionsmischung auf RT ab-
gekühlt und mit 5 ml konzentrierter Salzsäure versetzt. Danach werden die Lösungsmittel
destillativ entfernt und der erhaltene Rückstand abgesaugt. Nach dem Waschen mit Essig-
säure erhält man das Produkt als farblosen Feststoff.
Ausbeute:
1.90 g (12.3 mmol) = 31 % d.Th.
Analysedaten: 1H-NMR (200 MHz, D2O): δ = 3.42 (t, 3J = 6.7, 2 H, CH2); 2.91 - 2.85 (m, 8 H, 2 × CH3,
CH2). 13C-NMR (50 MHz, D2O): δ = 174.34 (C=O); 53.52 (Me2NCH2); 43.32 (CH3); 29.22
(CH2COOH). FAB-MS: 154.1 (13) [M+HCl]+; 118.1 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
234
10.38 Me2-Aeg(CBhoc)-OMe (141)
[[2-(4-Benzhydryloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-(2-dimethylamino-ethyl)-amino]-
essigsäure-methylester
N
N
NH
O
N
O
O
O
N O
O
Summenformel: C27H31N5O6 M = 521.58 g/mol
Ansatz:
983 mg 5.00 mmol Me2-Aeg(H)-OMe*HCl (78)
1.90 g 5.00 mmol CBhoc-AcOH (84)
960 mg 5.00 mmol EDC*HCl
810 mg 6.00 mmol HOBt
1.74 ml 10.0 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 14. Das Rohprodukt wird säulenchromatographisch
(Silica, DCM/MeOH/TEA 10:1:0.1) gereinigt. Man erhält das Produkt als farblosen Schaum.
Ausbeute:
1.42 g (2.72 mmol) = 54 % d.Th.

Experimenteller Teil 10 Synthesen
235
Analysedaten:
Rf (DCM/MeOH/TEA 10:1:0.1): 0.31. 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere):
δ = 10.99 (s, 1 H, BhocNH); 7.91 (m, 1 H, C(6)H); 7.48 - 7.25 (m, 10 H, BhocCHarom.); 6.93
(m, 1 H, C(5)H); 6.80 (s, 1 H, BhocCH); 4.83 (s, 1.15 H, N(1)CH2); 4.64 (s, 0.77 H,
N(1)CH2); 4.37 (s, 0.69 H, GlyCH2); 4.10 (s, 1.09 H, GlyCH2); 3.70 (s, 1.10 H, OCH3); 3.61
(s, 1.80 H, OCH3); 3.51 - 3.28 (m, 4 H, CH2CH2); 2.19 (s, 3.56 H, (H3C)2N); 2.12 (s, 2.18 H,
(H3C)2N). 13C-NMR (2 Rotamere) (50 MHz, DMSO-d6): δ = 170.15 (COOMe); 169.83
(COOMe); 167.65 (N(1)CH2C=O); 167.47 (N(1)CH2C=O); 163.40 (C(4)); 163.34 (C(4));
155.31 (BhocC=O); 152.75 (C(2)); 151.40 (C(6)); 140.75 (BhocCarom.); 128.91 (BhocCHrom.);
128.21 (BhocCHrom.); 126.80 (BhocCHrom.); 94.10 (C(5)); 77.79 (BhocCH); 57.86
(Me2NCH2); 57.19 (Me2NCH2); 52.45 (N(1)CH2); 52.05 (OCH3); 45.94, 45.73, 45.44
(Me2NCH2CH2, GlyCH2, (H3C)2N). FAB-MS: 544.2 (14) [M+Na]+; 522.2 (32) [M+H]+;
167.1 (100).
10.39 (Me)2-Aeg(CBhoc)-OH*HCl (142)
[[2-(4-Benzhydryloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-(2-dimethylamino-ethyl)-amino]-
essigsäure Hydrochlorid
N
N
NH
O
N
O
O
O
N O
O
*HCl
Summenformel: C26H29N5O6*HCl M = 544.01 g/mol

Experimenteller Teil 10 Synthesen
236
Ansatz:
1.16 g 2.22 mmol Me2-Aeg(CBhoc)-OMe (142)
186 mg 4.44 mmol LiOH*H2O
20 ml THF
20 ml H2O
Durchführung:
Die Durchführung erfolgt nach AAV 3. Man erhält das Produkt als farblosen Schaum.
Ausbeute:
960 mg (1.77 mmol) = 79 % d.Th.
Analysedaten:
1H-NMR (200 MHz, D2O-d6) (2 Rotamere): δ = 7.54 (d, 3J = 7.6, 1 H, C(6)H); 7.47 - 7.33
(m, 10 H, BhocCHarom.); 6.02 (d, 3J = 7.3, 1 H, C(5)H); 5.91 (s, 1 H, BhocCH); 5.27 (s,
0.20 H, N(1)CH2); 4.99 (s, 1.70 H, N(1)CH2); 4.09 (s, 1.80 H, GlyCH2); 3.95 (s, 0.20 H,
GlyCH2); 3.86 (t, 3J = 5.3, 2 H, Me2NCH2CH2); 3.37 (t, 3J = 5.3, 2 H, Me2NCH2CH2); 2.91 (s,
6 H, (H3C)2N). 1H-NMR (200 MHz, MeOH-d4) (2 Rotamere): = 7.82 (d, 3J = 7.3, 1 H,
C(6)); 7.31 (m, 11 H, BhocCHarom., C(5)H); 6.83 (s, 1 H, BhocCH); 4.68 (s, 2 H, N(1)CH2);
4.07 (GlyCH2); 3.84 (Me2NCH2CH2); 3.30 (Me2NCH2CH2); 2.85 (s, 6 H, CH3). 13C-NMR
(50 MHz, MeOH-d4) (2 Rotamere): = 176.16 (COOH); 170.48 (N(1)CH2C=O); 165.53
(C(4)); 161.35, 158.64 (C(2), BhocC=O); 151.70 (C(6)); 141.49 (BhocCarom.); 129.68, 129.21,
128.11 (BhocCHarom.); 96.89 (C(5)); 80.07 (BhocCH); 56.43 (Me2NCH2); 53.44 (GlyCH2);
52.39 (N(1)CH2); 44.95 (Me2NCH2CH2); 43.78 (CH3). FAB-MS: 530.2 (11) [M+Na]+; 508.2
(22) [M+H]+, 167.1 (100).

Experimenteller Teil 10 Synthesen
237
10.40 Me2-Aeg(H)-OH (145)
N-[2-(Dimethylamino)ethyl]glycin
NNH
OH
O
Summenformel: C6H14N2O2 M = 146.19 g/mol
Ansatz:
4.60 g 50.0 mmol Glyoxylsäure Monohydrat (123)
5.50 ml 50.4 mmol 2-Dimethylamino-ethlyamin (143)
300 ml Wasser
2.00 g Pd/C (10 %)
1.12 l H2
Durchführung:
Die Durchführung erfolgt gemäß AAV 4. Man erhält das Produkt als farblosen Feststoff, der
im Ölpumpenvakuum getrocknet wird.
Ausbeute:
6.96 g (47.7 mmol) = 95 % d.Th.
Analysedaten:
1H-NMR (200 MHz, D2O): δ = 3.30 (s, 2 H, GlyCH2); 2.99 (d, 3J = 3.0, 2 H,
(Me2NCH2CH2); 2.98 (d, 3J = 3.0, 2 H, (Me2NCH2CH2); 2.67 (s, 6 H (H3C)2N). 13C-NMR
(50 MHz, D2O): δ = 177.87 (C=O); 56.14 (GlyCH2); 51.50, 43.80, 43.39 ((H3C)2NCH2CH2).
FAB-MS: 169.1 (14) [M+Na] +; 147.1 (100) [M+H] +.

Experimenteller Teil 10 Synthesen
238
10.41 H-Aeg(F)-OMe*TFA (185)
{(2-Amino-ethyl)-[2-(2,4-difluor-5-methyl-phenyl)-acetyl]-amino}essigsäure-methylester Trifluoracetat
ONH2
O
F
F
N
O
*TFA
Summenformel: C14H18F2N2O3*TFA M = 414.33 g/mol
Ansatz:
936 mg 2.40 mmol Boc-Aeg(F)-OMe (204)
5 ml TFA
10 ml DCM
Durchführung:
Die Durchführung erfolgt anhand AAV 15. Man erhält das Produkt quantitativ als leicht
gelbliches Öl, das auch nach intensiver Trocknung im Ölpumpenvakuum noch 0.5 eq. Toluol
enthält.
Ausbeute:
994 mg (2.40 mmol) = 100 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 8.01, 7.81 (je s(br), zusammen 3 H,
H3N+); 7.26 - 7.04 (m, 2 H, FCHarom.); 4.40 (s, 0.79 H, C(1)CH2); 4.03 (s, 1.03 H, C(1)CH2);
3.10, 2.96 (je m(br), zusammen 2 H, H3N+CH2); 3.77 - 3.47 (m, 7 H, OCH3, GlyCH2,
H3N+CH2CH2); 2.18 (s(br), 3 H, FCH3).

Experimenteller Teil 10 Synthesen
239
10.42 Boc-Aeg(H)-OMe (187)
Methyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)glycinat
OO NH
NH
OO
Summenformel: C10H20N2O4 M = 232.28 g/mol
Ansatz:
14.8 g 69.8 mmol H-Aeg(H)-OMe*2 HCl (124)
4.30 g 69.8 mmol Boc2O
8.52 g 69.8 mmol DMAP
15.4 ml 140 mmol NMM
45 ml DCM
Durchführung:
Man legt 8.52 g (69.8 mmol) DMAP in 30 ml DCM vor und kühlt unter Argon-Schutzgas-
atmosphäre im Eis-Kochsalz-Bad auf -15 °C ab. Hierzu tropft man langsam eine Lösung von
4.30 g (69.8 mmol) Boc2O in 15 ml DCM. Anschließend gibt man 14.8 g (69.8 mmol)
H-Aeg(H)-OMe*2HCl (124) und 15.4 ml (14.1 g, 140 mmol) NMM hinzu, rührt 3 h bei
-15 °C und 2 h bei RT. Man entfernt das Lösungsmittel am Rotationsverdampfer, nimmt den
Rückstand in 80 ml 1 M KHSO4-Lösung auf und wäscht mit 80 ml EtOAc. Danach stellt man
die wässrige Phase mit NaHCO3 auf pH 8-9 ein und extrahiert dreimal mit je 100 ml EtOAc.
Die vereinigten organischen Phasen werden mit ges. NaCl-Lösung gewaschen, über MgSO4
getrocknet und vom Lösungsmittel befreit. Das so erhaltene Rohprodukt wird durch
Säulenchromatographie (Silica, EtOAc/MeOH 9:1) gereinigt. Man erhält das Produkt als
farbloses Öl.
Ausbeute:
8.72 g (37.5 mmol) = 54 % d.Th.
Analysedaten:
siehe 10.43

Experimenteller Teil 10 Synthesen
240
10.43 Boc-Aeg(H)-OMe (187)
Methyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)glycinat
OO NH
NH
OO
Summenformel: C10H20N2O4 M = 232.28 g/mol
Ansatz:
20.0 g 98.5 mmol H-Aeg(H)-OMe*2 HCl (124)
21.5 g 98.5 mmol Boc2O
27.3 ml 197 mmol TEA
1.5 l DCM
Durchführung:
Man legt 20.0 g (98.5 mmol) H-Aeg(H)-OMe*2HCl (124) in 1.2 l DCM vor und kühlt im
Eisbad auf -10 °C ab. Nach der Zugabe von 27.3 ml (197 mmol) TEA wird über einen Zeit-
raum von 30 min eine Lösung von 21.5 g (98.5 mmol) Boc2O in 300 ml DCM hinzugetropft.
Man lässt über Nacht nachrühren, wäscht mit 500 ml Wasser und 500 ml ges. NaCl-Lösung
und trocknet über MgSO4. Nachdem das Lösungsmittel am Rotationsverdampfer entfernt
wurde, wird das Rohprodukt säulenchromatographisch gereinigt (Silica, DCM/MeOH 50:1).
Alternativ kann auch durch fraktionierte Destillation im Ölpumpenvakuum aufgereinigt
werden. Man erhält das Produkt als farbloses Öl.
Ausbeute:
14.4 g (62.0 mmol) = 63 % d.Th.
Analysedaten:
Rf (DCM/MeOH 10:1): 0.55. Rf (EtOAc/MeOH 4:1): 0.45. 1H-NMR (200 MHz, CDCl3):
δ = 3.72 (s, 3 H, OCH3); 3.40 (s, 2 H, GlyCH2); 3.20 (dt aa q, 2 × 3J = 5.8, 2 H, BocNHCH2);
2.73 (t, 3J = 5.8, 2 H, BocNHCH2CH2); 1.43 (s, 9 H, BocCH3). 13C-NMR (50 MHz, CDCl3):
δ = 173.04 (GlyC=O); 156.21 (BocC=O); 79.34 (C(CH3)3); 51.97 (OCH3); 50.41 (GlyCH2);
48.90, 41.02 (CH2CH2); 28.53 (BocCH3). FAB-MS: 255.1 (19) [M+Na]+; 233.1 (100)
[M+H]+.

Experimenteller Teil 10 Synthesen
241
10.44 Boc-Aeg(AZ)-OH (188)
[[2-(6-Benzyloxycarbonylamino-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-amino]essigsäure
OHO NH
OO
N N
NN
NH
N
O
O
O
Summenformel: C24H29N7O7 M = 527.54 g/mol
Ansatz:
2.90 g 5.35 mmol Boc-Aeg(AZ)-OMe (189)
337 mg 8.03 mmol LiOH*H2O
30 ml THF
30 ml H2O
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Man erhält das Produkt als farblosen Schaum.
Ausbeute:
2.55 g (4.83 mmol) = 90 % d.Th.

Experimenteller Teil 10 Synthesen
242
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 12.81 (COOH); 10.67 (s, 1 H, NH);
8.60, 8.59 (je s, zusammen 1 H, C(2)H); 8.32, 8.31 (je s, zusammen 1 H, C(8)H); 7.49 - 7.30
(m, 5 H, ZCHarom.); 7.05 (t(br), 3J = 5.1, 0.57 H, NH); 6.77 (t(br), 3J = 5.1, 0.31 H, NH); 5.34
(s, 1.29 H, N(9)CH2); 5.22 (s, 2 H, ZCH2); 5.16 (s, 0.69 H, N(9)CH2); 4.34 (s, 0.64 H,
GlyCH2); 3.99 (s, 1.37 H, GlyCH2); 3.82 - 3.02 (m(br), 4 H, CH2CH2); 1.38 (s, 5.80 H,
BocCH3); 1.36 (s, 3.20 H, BocCH3). 13C-NMR (50 MHz, DMSO-d6) (2 Rotamere):
δ = 170.84 (GlyC=O); 170.36 (GlyC=O); 166.99 (N(9)CH2C=O); 166.54 (N(9)CH2C=O);
155.81 (BocC=O); 152.42, 152.20, 151.47 (ZC=O, C(2), C(4)); 149.39 (C(6)); 145.25 (C(8));
145.12 (C(8)); 136.40 (ZCarom.); 128.41 (Z-o-CHarom.); 127.98 (Z-p-CHarom.); 127.84 (Z-m-
CHarom.); 122.96 (C(5)); 78.13 (C(CH3)3); 77.78 (C(CH3)3); 66.25 (ZCH2); 47.55
(BocNHCH2CH2); 46.95 (BocNHCH2CH2); 44.06 (N(9)CH2); 43.81 (N(9)CH2); 28.16
BocCH3). FAB-MS: 550.4 (51) [M+Na]+; 528.4 (99) [M+H]+; 91.1 (100). Die Signale einer
Methylengruppe (BocHNCH2CH2) befinden sich unter dem Signal von DMSO. Darauf kann
man durch Vergleich mit dem Spektrum von Boc-Aeg(AZ)-OH schließen, das mit einer
Frequenz von 100 MHz aufgezeichnet wurde. Hierbei befinden sich die Signale der
entsprechenden Kohlenstoffatome bei 37.59 und 38.13 ppm.

Experimenteller Teil 10 Synthesen
243
10.45 AZ-AcOEt (189)
6-N-Benzyloxycarbonyl-adenin-9-essigsäure-ethylester
N N
N N
NH
O
O
O
O
Summenformel: C17H17N5O4 M = 355.36 g/mol
Ansatz:
50.0 g 226 mmol A-AcOEt (120)
35.7 ml 345 mmol Benzylalkohol
56.8 g 345 mmol CDI
1 l DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 7. Der Rückstand wird mit Wasser versetzt und
30 min im Ultraschallbad behandelt. Das Rohprodukt wird abgesaugt, mit Wasser gewaschen
und aus Methanol umkristallisiert. Nach Trocknen im Ölpumpenvakuum erhält man das
Produkt als farblosen Feststoff.
Ausbeute:
45.1 g (127 mmol) = 55 % d.Th.
Analysedaten: 1H-NMR (400 MHz, DMSO-d6): δ = 10.68 (s, 1 H, NH); 8.63 (s, 1 H, C(2)H); 8.44 (s, 1 H,
C(8)H); 7.47 - 7.33 (m, 5 H, ZCHarom.); 5.23 (s, 2 H, ZCH2); 5.20 (s, 2 H, N(9)CH2); 4.18 (q, 3J = 7.1, 2 H, CH2CH3); 1.21 (t, 3J = 7.1, 3 H, CH3).
13C-NMR (100 MHz, DMSO-d6): δ =
167.66 (COOMe, N(9)CH2C=O); 152.13 (C(4)); 151.72 (C(2)); unter C(2) oder C(4) liegt das
Signal für ZC=O; 149.52 (C(6)); 144.65 (C(8)); 136.34 (ZCarom.); 128.37 (Z-o-CHarom.);
127.95 (Z-p-CHarom.); 127.81 (Z-m-CHarom.); 122.93 (C(5)); 66.27 (ZCH2); 61.48 (CH2CH3);
44.20 (N(9)CH2); 13.94 (CH3). FAB-MS: 378.1 (27) [M+Na]+; 356.1 (100) [M+H]+. Die
Zuordnung der NMR-Signale ist durch HMBC- und HMQC-Spektroskopie abgesichert.

Experimenteller Teil 10 Synthesen
244
10.46 AZ-AcOH (190)
6-N-Benzyloxycarbonyl-adenin-9-essigsäure
N N
N N
NH
OH
O
O
O
Summenformel: C15H13N5O4 M = 327.30 g/mol
Ansatz:
44.0 g 124 mmol AZ-AcOEt (189)
7.79 g 186 mmol LiOH*H2O
250 ml THF
250 ml H2O
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Das erhaltene Rohprodukt wird in 220 ml Aceton
suspendiert und die Mischung 30 min bei Rückflusstemperatur gerührt. Man saugt ab, wäscht
mit Aceton und erhält das Produkt als farblosen Feststoff.
Ausbeute:
31.3 g (95.6 mmol) = 80 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 13.42 (s(br), 1 H, COOH); 10.71 (s(br), 1 H, NH);
8.62 (s, 1 H, C(2)H); 8.44 (s, 1 H, C(8)H); 7.48 - 7.33 (m, 5 H, ZCHarom.); 5.22 (s, 2 H, CH2);
5.09 (s, 2 H, CH2). 13C-NMR (50 MHz, DMSO-d6): δ = 169.11 (COOH, N(9)CH2C=O);
152.20 (C(4)); 151.65 (C(2)); 149.43 (C(6)); 144.84 (C(8)); 136.38 (ZCarom.); 128.41
(Z-o-CHarom.); 127.99 (Z-p-CHarom.); 127.87 (Z-m-CHarom.); 123.02 (C(5)); 66.29 (ZCH2);
44.29 (N(9)CH2). FAB-MS: 350.1 (24) [M+Na]+; 328 (90) [M+H]+; 154.0 (100).

Experimenteller Teil 10 Synthesen
245
10.47 Boc-Aeg(AZ)-OMe (191)
[[2-(6-Benzyloxycarbonylamino-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-amino]essigsäure-
methylester
OO NH
OO
N N
NN
NH
N
O
O
O
Summenformel: C25H31N7O7 M = 541.57 g/mol
Ansatz:
1.57 g 6.76 mmol Boc-Aeg(H)-OMe (187)
2.22 g 6.76 mmol AZ-AcOH (190)
2.22 g 6.76 mmol TOTU
873 µl 6.76 mmol DIPEA
15 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, DCM/MeOH 20:1) als farblosen Schaum.
Ausbeute:
2.98 g (5.50 mmol) = 81 % d.Th.

Experimenteller Teil 10 Synthesen
246
Analysedaten:
Rf (DCM/MeOH 20:1): 0.20. 1H-NMR (400 MHz, DMSO-d6) δ = 10.63 (s, 1 H, NH); 8.60
(s, 1 H, C(2)H); 8.30, 8.31 (je s, zusammen 1 H, C(8)H); 7.47 - 7.31 (m, 5 H, ZCHarom.); 7.03
(t(br), 0.69 H, NH); 6.73 (t(br), 0.31 H, NH); 5.35 (s, 1.4 H, N(9)CH2); 5.22 (s, 2 H, ZCH2);
5.17 (s, 0.6 H, N(9)CH2); 4.46 (s, 0.57 H, GlyCH2); 4.09 (s, 1.43 H, GlyCH2); 3.77 (s, 0.91 H,
OCH3); 3.62 (s, 2.10 H, OCH3); 3.58 - 3.04 (m(br), 4 H, CH2CH2); 1.39 (s, 5.94 H, BocCH3);
1.37 (s, 3.04 H, BocCH3). 13C-NMR (100 MHz, DMSO-d6) (2 Rotamere): δ = 169.85
(GlyC=O); 169.39 (GlyC=O); 167.01 (N(9)CH2C=O); 166.70 (N(9)CH2C=O); 155.80
(BocC=O); 155.53 (BocC=O); 152.38 (C(4)); 152.12 (ZC=O); 151.47 (C(2)); 149.35 (C(6));
149.32 (C(6)); 145.14 (C(8)); 145.04 (C(8)); 136.35 (ZCarom.); 128.35 (Z-o-CHarom.); 127.92
(Z-p-CHarom.); 127.78 (Z-m-CHarom.); 122.92 (C(5)); 122.87 (C(5)); 78.11 (C(CH3)3); 77.74
(C(CH3)3); 66.21 (ZCH2); 52.31 (OCH3); 51.78 (OCH3); 49.00 (GlyCH2); 47.57 (GlyCH2);
47.07 (BocNHCH2CH2); 46.86 (BocNHCH2CH2); 44.08 (N(9)CH2); 43.72 (N(9)CH2); 38.13
(BocNHCH2CH2); 37.59 (BocNHCH2CH2); 28.16 (BocCH3); 28.12 (BocCH3). FAB-MS:
564.4 (67) [M+Na]+; 542.5 (100) [M+H]+. Die Zuordnung der NMR-Signale ist durch
HMBC- und HMQC-Spektroskopie abgesichert.

Experimenteller Teil 10 Synthesen
247
10.48 CZ-AcOH (192)
4-N-(Benzyloxycarbonyl)cytosin-1-essigsäure
O
O
NH
N
N
O
OH
O
Summenformel: C14H13N3O5 M = 303.28 g/mol
Ansatz:
1.22 g 3.84 mmol CZ-AcOMe (196)
242 mg 5.77 mmol LiOH*H2O
20 ml THF
20 ml H2O
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
706 mg (2.33 mmol) = 61 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 8.03 (d, 3J = 7.7, 1 H, C(6)H); 7.38 (m, 5 H,
ZCHarom.); 7.02 (d, 3J = 7.7, 1 H, C(5)H); 5.19 (s, 2 H, ZCH2); 4.52 (s, 2 H, N(1)CH2). 13C-
NMR (50 MHz, DMSO-d6): δ = 169.34 (COOH); 163.31 (C(4)), 155.04 (ZC=O), 153.19
(C(2)), 150.41 (C(6)); 135.96 (ZCarom.); 128.50 (Z-o-CHarom.); 128.17 (Z-p-CHarom.); 127.94
(Z-m-CHarom.); 94.05 (C(5)); 66.54 (ZCH2); 50.54 (N(1)CH2). FAB-MS: 154.0 (100); 304.0
(30) [M+H]+, 326 (17) [M+Na]+.

Experimenteller Teil 10 Synthesen
248
10.49 CZ-AcOH (192)
4-N-(Benzyloxycarbonyl)cytosin-1-essigsäure
O
O
NH
N
N
O
OH
O
Summenformel: C14H13N3O5 M = 303.28 g/mol
Ansatz:
5.71 g 14.5 mmol CZ-AcOBn (194)
913 mg 21.8 mmol LiOH*H2O
60 ml THF
60 ml H2O
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
4.06 g (13.4 mmol) = 92 % d.Th.
Analysedaten:
siehe 10.48

Experimenteller Teil 10 Synthesen
249
10.50 CZ (193)
4-N-(Benzyloxycarbonyl)cytosin
O
O
NH
NH
N
O
Summenformel: C12H11N3O3 M = 245.24 g/mol
Ansatz:
3.00 g 27.0 mmol Cytosin (86)
7.60 ml 59.4 mmol Chlorameisensäure-benzylester
659 mg 5.39 mmol DMAP
48 ml Pyridin
Durchführung:
Zu einer Suspension von 3.00 g (27.0 mmol) Cytosin in 48 ml Pyridinabs. werden 659 mg
(5.40 mmol) DMAP gegeben und anschließend werden langsam 7.60 ml (9.04 g, 59.4 mmol)
Chlorameisensäure-benzylester zugetropft. Man lässt 4 Tage rühren, gießt die Reaktions-
mischung auf 100 ml Eiswasser und lässt 30 min rühren. Anschließend wird abgesaugt und
der erhaltene Feststoff mit Wasser gewaschen. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
1.41 g (5.75 mmol) = 21 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 11.09 (s(br), 2 H, 2 × NH); 7.80 (d, 1 H, 3J = 7.1,
C(6)H); 7.38 (m, 5 H, ZCHarom.); 6.92 (d, 1 H, 3J = 7.1, C(5)H); 5.17 (s, 2 H, CH2). 13C-NMR
(50 MHz, DMSO-d6): δ = 163.61 (C(4)); 155.75 (ZC=O); 153.40 (C(2)); 146.67 (C(6));
136.02 (ZCarom.); 128.45 (Z-o-CHarom.); 128.10 (Z-p-CHarom.); 127.88 (Z-m-CHarom.); 93.52
(C(5)), 66.38 (ZCH2). FAB-MS: 246.1 (37) [M+H]+; 163.0 (100).

Experimenteller Teil 10 Synthesen
250
10.51 CZ-AcOBn (194)
4-N-(Benzyloxycarbonyl)cytosin-1-essigsäure-benzylester
NH
N
N
O
O
O
O
O
Summenformel: C21H19N3O5 M = 393.40 g/mol
Ansatz :
7.54 g 29.1 mmol C-AcOBn (105)
7.56 g 46.6 mmol CDI
4.73 g 43.7 mmol Benzylalkohol
190 ml THFabs.
Durchführung:
Die Durchführung erfolgt gemäß AAV 8. Der erhaltene Rückstand wird mit 280 ml Ethanol
versetzt und 30 min bei Rückflusstemperatur gerührt. Anschließend wird das Produkt in Form
eines farblosen Feststoffs abgesaugt, mit Ethanol gewaschen und im Ölpumpenvakuum
getrocknet.
Ausbeute:
5.72 g (14.5 mmol) = 50 % d.Th.

Experimenteller Teil 10 Synthesen
251
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 11.06 (s, 1 H, NH); 8.06 (d, 3J = 7.3, 1 H, C(6)H);
7.29 - 7.48 (m, 10 H, GlyCH, ZCHarom.); 6.98 (d, 3J = 7.3, 1 H, C(5)H); 5.18 (s, 2 H, CH2);
4.69 (s, 2 H, CH2). 13C-NMR (50 MHz, DMSO-d6): δ = 167.82 (COOMe); 163.36 (C(4));
154.89 (ZC=O); 153.26 (C(2)); 150.32 (C(6)); 136.06 (Carom.); 135.51 (Carom.); 128.50,
128.40, 128.13, 127.82, 127.82, 126.42 (CHarom.); 94.15 (C(5)); 77.48 (BnCH2); 66.35
(ZCH2); 50.60 (N(1)CH2). FAB-MS: 426.1 (13) [M+Na]+; 394.1 (5) [M]+; 167.0 (100).
10.52 Boc-Aeg(CZ)-OH (195)
[[2-(4-Benzyloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-amino]-
essigsäure
OHO NH
N
OO
O
O
NH
N
N
O
O
Summenformel: C23H29N5O8 M = 503.52 g/mol
Ansatz:
720 mg 1.39 mmol Boc-Aeg(CZ)-OMe (197)
88.0 mg 2.09 mmol LiOH*H2O
10 ml THF
10 ml H2O
Durchführung:
Die Durchführung erfolgt gemäß AAV 3. Anschließend versetzt man das Rohprodukt mit
Diethylether, behandelt es kurz im Ultraschallbad, saugt den entstandenen Feststoff ab und
wäscht mit Diethylether. Zur weiteren Reinigung wird aus Methanol umkristallisiert. Man
erhält das Produkt als farblosen Feststoff.

Experimenteller Teil 10 Synthesen
252
Ausbeute:
676 mg (1.34 mmol) = 97 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.90, 7.88 (je d, 3J = 7.3, zusammen
1 H, C(6)H); 7.37 (m, 5 H, ZCHarom.); 7.02 u. 7.00 (je d, 3J = 7.33, 1 H, C(5)H); 6.95 (t(br), 3J = 5.1, 0.54 H, ZNH); 6.74 (t(br), 3J = 5.1, 0.23 H, ZNH); 5.19 (s, 2 H, ZCH2); 4.81 (s,
1.45 H, N(1)CH2); 4.63 (s, 0.55 H, N(1)CH2); 4.32 (s, 0.55 H, GlyCH2); 3.98 (s, 1.45 H,
GlyCH2); 3.62 - 3.00 (m(br), 4 H, CH2CH2); 1.38 (s, 9 H, BocCH3). 13C-NMR (50 MHz,
DMSO-d6) (2 Rotamere): δ = 170.82 (COOH); 170.44 (COOH); 167.47 (N(1)CH2C=O);
167.04 (N(1)CH2C=O); 163.14 (C(4)); 155.74, 154.96 (BocC=O, ZC=O); 153.21, 153.11
(C(2)); 150.78 (C(6)); 135.97 (ZCarom.); 128.47 (Z-o-CHarom.); 128.14 (Z-p-CHarom.); 127.91
(Z-m-CHarom.); 93.85 (C(5)); 78.03 (C(CH3)3); 66.49 (ZCH2); 28.15 (BocCH3). FAB-MS:
526.2 (26) [M+Na]+; 504.20 (5) [M+H]+; 91.0 (100). Die Signale für N(1)CH2, GlyCH2,
BocHNCH2CH2 und BocHNCH2 sind aufgrund der Signalintensität nicht zu erkennen bzw.
liegen unter den Signalen für DMSO.
10.53 CZ-AcOMe (196)
4-N-(Benzyloxycarbonyl)cytosin-1-essigsäure-methylester
O
O
NH
N
N
O
O
O
Summenformel: C15H15N3O5 M = 317.30 g/mol
Ansatz:
1.36 g 5.55 mmol CZ (193)
561 µl 59.4 mmol Bromessigsäure-methylester
767 mg 5.55 mmol K2CO3
30 ml DMF

Experimenteller Teil 10 Synthesen
253
Durchführung:
Vorgelegt werden 1.36 g (5.55 mmol) 4-N-(Benzyloxycarbonyl)cytosin (193) und 767 mg
(5.55 mmol) K2CO3 in 30 ml DMF. Man lässt 10 min rühren, gibt innerhalb von 1 min
561 µl (935 mg, 59.4 mmol) Bromessigsäure-methylester hinzu und lässt einen Tag rühren.
Nachfolgend wird das Lösungsmittel am Trockeneis-Rotationsverdampfer entfernt und das
Produkt durch Zugabe von Ethylacetat ausgefällt. Man saugt ab, wäscht mit Ethylacetat und
erhält das Produkt als farblosen Feststoff.
Ausbeute:
1.59 g (5.00 mmol) = 90 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 10.83 (s, 1 H, NH); 8.09 (d, 3J = 7.1, 1 H, C(6)H); 7.39
(m, 5 H, ZCHarom.); 7.04 (d, 3J = 7.1, 1 H, C(5)H); 5.19 (s, 2 H, ZCH2); 4.65 (s, 2 H,
N(1)CH2); 3.68 (s, 3 H, OCH3). 13C-NMR (50 MHz, DMSO-d6): δ = 168.44 (COOMe);
163.43 (C(4)); 155.02 (ZC=O); 153.07 (C(2)); 150.34 (C(6)); 135.88 (ZCarom.); 128.48 (Z-o-
CHarom.); 128.18 (Z-p-CHarom.); 127.98 (Z-m-CHarom.); 94.26 (C(5)); 66.78 (ZCH2); 52.26
(OCH3); 50.51 (N(1)CH2). FAB-MS: 356.2 (87) [M+K]+; 340.2 (35) [M+Na]+; 318.2 (100)
[M+H]+.

Experimenteller Teil 10 Synthesen
254
10.54 Boc-Aeg(CZ)-OMe (197)
[[2-(4-Benzyloxycarbonylamino-2-oxo-2H-pyrimidin-1-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-amino]-
essigsäure-methylester
OO NH
N
OO
O
O
NH
N
N
O
O
Summenformel: C24H31N5O8 M = 517.54 g/mol
Ansatz:
700 mg 2.31 mmol CZ-AcOH (192)
537 mg 2.31 mmol Boc-Aeg(H)-OMe (187)
758 mg 2.31 mmol TOTU
403 µl 2.31 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Anschließend versetzt man das Rohprodukt mit
Diethylether, behandelt es kurz im Ultraschallbad, saugt den entstandenen Feststoff ab und
wäscht mit Diethylether. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
728 mg (1.41 mmol) = 61 % d.Th.

Experimenteller Teil 10 Synthesen
255
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 10.79 (s(br), 1 H, C(4)NH); 7.93, 7.89
(je d, 3J = 7.3, zusammen 1 H, C(6)H); 7.37 (m, 5 H, ZCHarom.); 7.02 (d, 3J = 7.3, 1 H, C(5)H);
6.94 (t(br), 3J = 5.3, 0.63 H, ZNH); 6.75 (t(br), 3J = 5.3, 0.25 H, ZNH); 5.19 (s, 2 H, ZCH2);
4.82 (s, 1.45 H, N(1)CH2); 4.64 (s, 0.55 H, N(1)CH2); 4.35 (s, 0.55 H, GlyCH2); 4.07 (s,
1.45 H, GlyCH2); 3.72 (s, 0.79 H, OCH3); 3.62 (s, 2.21 H, OCH3); 3.54 - 3.00 (m(br), 4 H,
CH2CH2); 1.38 (s, 9 H, BocCH3). 13C-NMR (100 MHz, DMSO-d6) (2 Rotamere):
δ = 169.82 (COOMe); 169.47 (COOMe); 167.50 (N(1)CH2C=O); 167.21 (N(1)CH2C=O);
163.11 (C(4)); 163.05 (C(4)); 155.71 (BocC=O); 155.52 (BocC=O); 154.89 (ZC=O); 153.11
(C(2)); 150.65 (C(6)); 135.93 (ZCarom.); 128.41 (Z-o-CHarom.); 128.08 (Z-p-CHarom.); 127.85
(Z-m-CHarom.); 93.85 (C(5)); 78.00 (C(CH3)3); 77.71 (C(CH3)3); 66.45 (ZCH2); 52.20 (OCH3);
51.72 (OCH3); 49.53 (N(1)CH2); 49.31 (N(1)CH2); 49.04 (GlyCH2); 47.68 (GlyCH2); 47.00
(BocNHCH2CH2); 46.83 (BocNHCH2CH2); 38.17 (BocNHCH2); 37.64 (BocNHCH2); 28.16
(BocCH3); 28.11 (BocCH3). FAB-MS: 540.2 (32) [M+Na]+; 518.0 (16) [M+H]+; 91.0 (100).

Experimenteller Teil 10 Synthesen
256
10.55 Boc-Aeg(GZ)-OH (198)
[[2-(2-Benzyloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-
amino]essigsäure
OHO NH
OO
N
NN
NNH
O
NH
O
O
O
Summenformel: C24H29N7O8 M = 543.54 g/mol
Ansatz:
342 mg 613 µmol Boc-Aeg(GZ)-OH (202)
6 ml 1 M LiOH
6 ml THF
Durchführung:
Die Durchführung erfolgt nach AAV 3. Anschließend behandelt man den Rückstand mit
Methanol, saugt den entstandenen Feststoff ab und wäscht mit Diethylether. Man erhält das
Produkt als farblosen Feststoff.
Ausbeute:
205 mg (377 µmol) = 62 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 12.88 (s(br), 1 H, COOH); 11.52 (s,
0.28 H, NH); 11.59, 11.50 (je s(br), zusammen 1 H, NH); 11.37 (s(br), 1 H, NH); 7.83, 7.82
(je s, zusammen 1 H, C(8)H); 7.45 - 7.34 (m, 5 H, ZCHarom.); 7.05 (t(br), 0.63 H, NH); 6.75
(t(br), 0.37 H, NH); 5.25 (s, 2 H, ZCH2); 5.09 (s, 1.26 H, N(9)CH2); 4.93 (s, 0.76 H,
N(9)CH2); 4.32 (s, 0.76 H, GlyCH2); 4.99 (s, 1.26 H, GlyCH2); 3.48 - 2.97 (m(br), 4 H,
CH2CH2); 1.35, 1.34 (je s, zusammen 9 H, BocCH3). FAB-MS: 566.0 (9) [M+Na]+; 544.0 (4)
[M]+; 154 (100).

Experimenteller Teil 10 Synthesen
257
10.56 GZ-AcOH (199)
(2-Benzyloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)essigsäure
N N
N NH
O
NH
OH
O
OO
Summenformel: C15H13N5O5 M = 343.30 g/mol
Ansatz:
10.0 g 26.6 mmol (2-Benzyloxycarbonylamino-6-chlor-purin-9-yl)essig-
säure-methylester (201)
5.30 g 133 mmol NaH (60%ig in Mineralöl)
9.05 ml 133 mmol 3-Hydroxypropionitril
120 ml THFabs.
100 ml H2O
Durchführung:
Die Durchführung erfolgt nach AAV 9. Man saugt den ausgefallenen Feststoff ab und wäscht
zunächst mit Wasser und dann mit Ethylacetat. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
5.32 g (15.5 mmol) = 58 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 11.41 (s(br), 2 H, 2 × NH); 7.84 (s, 1 H, C(8)H);
7.45 - 7.34 (m, 5 H, ZCHarom.); 5.24 (ZCH2); 4.53 (N(9)CH2). 13C-NMR (50 MHz,
DMSO-d6): δ = 169.07 (COOH); 155.32, 154.61 (C(6); ZC=O); 149.19, 146.92 (C(2), C(4));
140.73 (C(8)); 135.59 (ZCarom.); 128.49 (Z-o-CHarom.); 128.23 (Z-p-CHarom.); 127.96
(Z-m-CHarom.); 119.10 (C(5)); 67.02 (ZCH2); 46.17 (N(9)CH2). FAB-MS: 388.0 (33)
[M+2Na]+; 366.0 (31) [M+Na]+; 344.0 (31) [M+H]+, 154.0 (100).

Experimenteller Teil 10 Synthesen
258
10.57 (2-Amino-6-chlor-purin-9-yl)essigsäure-methylester (200)
N N
N N
Cl
NH2
O
O
Summenformel: C8H8Cl1N5O2 M = 241.64 g/mol
Ansatz:
30.0 g 177 mmol 2-Amino-6-chlor-purin (111)
7.17 g 177 mmol NaH (60%ig in Mineralöl)
18.4 ml 200 mmol Bromessigsäure-methylester
400 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 6. Man kristallisiert aus THF um und erhält das
Produkt als farblosen Feststoff.
Ausbeute:
24.3 g (100 mmol) = 56 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 8.10 (s, 1 H, C(8)H); 6.99 (s, 2 H, NH2); 5.00 (CH2);
3.70 (OCH3). 13C-NMR (50 MHz, DMSO-d6): δ = 168.21 (C=O); 159.97 (C(6)); 154.24
(C(2)); 149.50 (C(4)); 143.47 (C(8)); 122.91 (C(5)); 52.46 (OCH3); 43.94 (CH2). FAB-MS:
242.0 (100) [M]+.

Experimenteller Teil 10 Synthesen
259
10.58 (2-Amino-6-chlor-purin-9-yl)essigsäure-methylester (200)
N N
N N
Cl
NH2
O
O
Summenformel: C8H8Cl1N5O2 M = 241.64 g/mol
Ansatz:
50.0 g 295 mmol 2-Amino-6-chlor-purin (111)
7.17 g 177 mmol K2CO3
29.4 ml 320 mmol Bromessigsäure-methylester
500 ml DMF
Durchführung:
Die Durchführung erfolgt anhand AAV 10. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
31.1 g (129 mmol) = 44 % d.Th.
Analysedaten:
siehe 10.57

Experimenteller Teil 10 Synthesen
260
10.59 (2-Benzyloxycarbonylamino-6-chlor-purin-9-yl)essigsäure-methylester (201)
N N
N N
Cl
NH
O
O
OO
Summenformel: C16H14Cl1N5O4 M = 375.77 g/mol
Ansatz:
18.5 g 76.6 mmol (2-Amino-6-chlor-purin-9-yl)essigsäure-methylester (200)
8.50 g 28.6 mmol Triphosgen
9.90 g 91.5 mmol Benzylalkohol
28.9 ml 168 mmol DIPEA
400 ml THFabs.
Durchführung:
Die Durchführung erfolgt anhand AAV 11. Der erhaltene Rückstand wird aus Methanol um-
kristallisiert. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
14.0 g (37.3 mmol) = 49 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6): δ = 10.84 (s, 1 H, NH); 8.48 (s, 1 H, C(8)H); 7.45 - 7.32
(m, 5 H, ZCHarom.); 5.18 (CH2); 5.13 (CH2); 3.71 (OCH3). 13C-NMR (50 MHz, DMSO-d6):
δ = 167.92 (COOMe); 153.08, 152.38 (C(6), ZC=O); 151.79, 149.23 (C(2), C(4)); 146.75
(C(8)); 136.50 (ZCarom.); 128.45 (Z-o-CHarom.); 127.99 (Z-p-CHarom.); 127.74 (Z-m-CHarom.);
126.75 (C(5)); 65.96 (ZCH2); 52.72 (OCH3); 43.94 (N(9)CH2). FAB-MS: 376.1 (40) [M+H]+;
55.0 (100).

Experimenteller Teil 10 Synthesen
261
10.60 Boc-Aeg(GZ)-OMe (202)
[[2-(2-Benzyloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)-acetyl]-(2-tert-butoxycarbonylamino-ethyl)-
amino]essigsäure-methylester
OO NH
OO
N
NN
NNH
O
NH
O
O
O
Summenformel: C25H31N7O8 M = 557.57 g/mol
Ansatz:
1.72 g 5.00 mmol GZ-AcOH (199)
1.16 g 5.00 mmol Boc-Aeg(H)-OMe (187)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Anschließend versetzt man das Rohprodukt mit
Diethylether, behandelt es kurz im Ultraschallbad, saugt den entstandenen Feststoff ab und
wäscht mit Diethylether. Zur weiteren Reinigung wird aus Methanol umkristallisiert. Man
erhält das Produkt als farblosen Feststoff.
Ausbeute:
1.65 g (2.96 mmol) = 59 % d.Th.

Experimenteller Teil 10 Synthesen
262
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 11.52 (s, 0.28 H, NH); 11.48 (s, 0.66 H,
NH); 11.35 (s, 1 H, NH); 7.82 (s, 0.70 H, C(8)H); 7.80 (s, 0.30 H, C(8)H); 7.46 - 7.31 (m,
5 H, ZCHarom.); 7.01 (t(br), 3J = 5.3, 0.60 H, NH); 6.75 (t(br), 0.30 H, NH); 5.26 (s, 2 H,
ZCH2); 5.09 (s, 1.38 H, N(9)CH2); 4.93 (s, 0.58 H, N(9)CH2); 4.42 (s, 0.57 H, GlyCH2); 4.08
(s, 1.45 H, GlyCH2); 3.75 (s, 0.88 H, OCH3); 3.62 (s, 2.14 H, OCH3); 3.50 - 3.22 (m(br), 4 H,
CH2CH2-; 1.39 (s, 5.94 H, BocCH3); 1.35 (s, 3.04 H, BocCH3). 13C-NMR (50 MHz,
DMSO-d6) (2 Rotamere): δ = 169.99 (COOMe); 169.48 (COOMe); 167.06 (N(9)CH2C=O);
166.67 (N(9)CH2C=O); 155.77, 155.12, 154.54 (BocC=O, C(6)); 149.49, 147.17 (C(2), C(4));
140.38 (C(8)); 135.45 (ZCarom.); 128.50 (Z-o-CHarom.); 128.33 (Z-p-CHarom.); 128.11 (Z-m-
CHarom.); 123.39, 119.18, 89.13 (C(CH3)3); 67.23, 51.83, 28.11 (BocCH3). FAB-MS: 580.2
(41) [M+Na]+; 558.2 (50) [M+H]+; 91 (100).

Experimenteller Teil 10 Synthesen
263
10.61 H-Aeg(GZ)-OMe*TFA (203)
[[2-(2-Benzyloxycarbonylamino-6-oxo-1,6-dihydro-purin-9-yl)-acetyl]-(2-ethylamino)]essigsäure-methylester
Trifluoracetat
ONH2
O
N
NN
NNH
O
NH
O
O
O
*TFA
Summenformel: C20H23N7O6*TFA M = 571.47 g/mol
Ansatz:
460mg 825 µmol Boc-Aeg(GZ)-OMe (202)
1.60 ml 10.0 mmol HSiEt3
3 ml TFA
6 ml DCM
Durchführung:
Die Durchführung erfolgt nach AAV 15. Man erhält das Produkt als leicht gelbliches Öl, das
auch noch intensiver Trocknung im Ölpumpenvakuum noch 1 eq. Toluol enthält.
Ausbeute:
471 mg (825 µmol) = 100 % d.Th.
Analysedaten:
1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 11.52 (s, 1 H, NH); 11.45 (s, 1 H, NH);
9.15 (s(br), 3 H, NH3+); 7.91, 7.86 (je s, zusammen 1 H, C(8)H); 7.80 (s, 0.30 H, C(8)H);
7.45 - 7.35 (m, 5 H, ZCHarom.); 5.26 (s, 2 H, ZCH2); 5.16 (s, 0.95 H, N(9)CH2); 4.96 (s,
1.05 H, N(9)CH2); 4.47 (s, 1.13 H, GlyCH2); 4.13 (s, 0.87 H, GlyCH2); 3.76 (s, 3 H, OCH3);
3.70, 3.53 (je t(br), zusammen 2 H, H3N+CH2CH2); 3.18, 2.97 (je m, zusammen 2 H,
H3N+CH2CH2).

Experimenteller Teil 10 Synthesen
264
10.62 Boc-Aeg(F)-OMe (204)
{(2-tert-Butoxycarbonylamino-ethyl)-[2-(2,4-difluor-5-methyl-phenyl)-acetyl]-amino}essigsäure-methylester
OO NH
OO
F
F
N
O
Summenformel: C19H26F2N2O5 M = 400.43 g/mol
Ansatz:
930 mg 5.00 mmol F-AcOH (70)
1.16 g 5.00 mmol Boc-Aeg(H)-OMe (187)
1.64 g 5.00 mmol TOTU
871 µl 5.00 mmol DIPEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt nach AAV 12. Nach Reinigung durch Säulenchromatographie
(Silica, DCM/MeOH 80:1) erhält man das Produkt als farbloses Öl.
Ausbeute:
1.66 g (4.15 mmol) = 83 % d.Th.

Experimenteller Teil 10 Synthesen
265
Analysedaten:
Rf (DCM/MeOH 50:1): 0.19. 1H-NMR (400 MHz, DMSO-d6) (2 Rotamere): δ = 7.17 -
7.05 (m, 2 H, FCHarom.); 6.91 (t(br), 0.76 H, NH); 6.68 (t(br), 0.24 H, NH); 4.33 (s, 0.43 H,
C(1)CH2); 4.03 (s, 1.57 H, C(1)CH2); 3.69 (s(br), 2 H, GlyCH2); 3.62, 3.55 (je s, zusammen
3 H, OCH3) (darunter befinden sich 0.56 H BocNHCH2CH2); 3.45 (t(br), 3J = 6.2, 1.44 H,
BocNHCH2CH2); 3.14 (dt aa dd(br), 3J = 6.1, 6.14, 1.44 H, BocNHCH2) 3.03 (dt aa q(br),
0.56 H, BocNHCH2); 2.17 (s(br), FCH3); 1.36 (s, 9 H, BocCH3). 13C-NMR (50 MHz,
DMSO-d6) (2 Rotamere): δ = 170.16 (C=O); 170.00 (C=O); 169.88 (C=O); 169.68 (C=O);
158.00 (FC(4)); 157.89 (FC(2)); 157.39 (FC(2)); 155.82 (BocC=O); 155.73 (BocC=O);
133.82, 133.72 u. 133.42 (FC(6)); 119.64, 119.48, 119.43, 118.74, 118.61 (FC(5)); 103.15,
102.89, 102.83, 102.63 (FC(3)); 77.94 (C(CH3)3); 77.69 (C(CH3)3); 52.10 (OCH3); 51.64
(OCH3); 49.76, 47.79, 47.35 u. 46.59 (GlyCH2, BocHNCH2CH2); 38.13 (BocHNCH2); 37.80
(BocHNCH2); 32.26 (FCH2); 31.75 (FCH2); 28.10 (BocCH3); 27.88 (BocCH3); 13.44 (FCH3).
Aufgrund der C-F-Kopplungen und dem Auftreten von 2 Rotameren, sind die
Signalintensitäten zu gering, bzw. das Spektrum zu komplex, um alle Signale vollständig zu
analysieren.

Experimenteller Teil 10 Synthesen
266
10.63 2-Amino-N-(2-dimethylamino-ethyl)-acetamid Di-trifluoracetat (205)
NH
O
NNH2
*2TFA
Summenformel: C6H15N3O*2TFA M = 373.25 g/mol
Ansatz:
1.10 g 4.48 mmol [(2-Dimethylamino-ethylcarbamoyl)-methyl]carbamin-
säure-tert-butylester (207)
5 ml TFA
10 ml DCM
Durchführung:
Die Durchführung erfolgt nach AAV 15. Man erhält das Produkt als farbloses Öl.
Ausbeute:
1.67 g (4.48 mmol) = 100 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6): δ = 8.69 (t, 3J = 5.6, 1 H, NH); 8.12 (s, 3 H, H3N
+);
3.72 - 3.40 (m, 4 H, CH2CH2); 3.25 - 3.07 (m, 2 H, GlyCH2); 2.81 (s, 6 H, N(CH3)2). 13C-NMR (50 MHz, DMSO-d6): δ = 166.85 (COOH); 158.63 (q, 3J(C,F) = 35.2,
F3CCOOH); 116.10 (q, 1J(C,F) = 293, F3CCOOH); 55.63 (CH2NMe2); 42.50 (N(CH3)2,
GlyCH2); 34.01 (CH2CH2NMe2).

Experimenteller Teil 10 Synthesen
267
10.64 [(2-Dimethylamino-ethylcarbamoyl)-methyl]carbaminsäure-tert-butylester (207)
NH
O
NNH
O
O
Summenformel: C11H23N3O3 M = 245.32 g/mol
Ansatz:
1.50 g 8.56 mmol Boc-Gly-OH (206)
755 mg 8.56 mmol N,N-Dimethylaminoethylamin (143)
1.97 g 10.3 mmol EDC
1.39 g 10.3 mmol HOBt
3.46 g 34.2 mmol TEA
50 ml Chloroform
Durchführung:
Die Durchführung erfolgt nach AAV 14. Man erhält das Produkt nach säulenchromatographi-
scher Reinigung (Silica, EtOAc/MeOH/TEA 7:3:0.1) als farbloses Öl.
Ausbeute:
1.10 g (4.48 mmol) = 52 % d.Th.
Analysedaten:
Rf (EtOAc/MeOH/TEA 7:3:0.1): 0.25. 1H-NMR (200 MHz, CDCl3): δ = 6.66 (s(br), 1 H,
NH); 5.32 (s(br), 1 H, NH); 3.77 (d, 3J = 5.8, 2 H, BocNHCH2); 3.35 (m, 2 H, CH2CH2NMe2);
2.44 (t, 3J = 5.8, 2 H, CH2CH2NMe2); 2.23 (s, 6 H, N(CH3)2); 1.42 (s, 9 H, BocCH3). 13C-
NMR (50 MHz, CDCl3): δ = 169.54 (GlyC=O); 156.03 (BocC=O); 80.14 (C(CH3)3); 57.78
(CH2NMe2); 45.13 (N(CH3)2, GlyCH2); 36.74 (CH2CH2NMe2); 28.41 (BocCH3). FAB-MS:
268.2 (14) [M+Na]+; 246.2 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
268
10.65 H-Aeg(C)-Aeg(A)-Aeg(G)-Aem*4TFA (224)
2-Amino-N-[12-(2-aminoethyl)-14-(4-amino-2-oxopyrimidin-1(2H)-yl)-6-[2-(6-amino-9H-purin-9-yl)acetyl]-
4,10,13-trioxo-3,6,9,12-tetraazatetradec-1-yl]-1,6-dihydro-N-(2-{[2-(morpholin-4-yl)ethyl]amino}-2-oxoethyl)-
6-oxo-9H-purine-9-acetamid
N
N
O
NH2
N
O
N N
NN
NH2
NH2
O
NH
NNH
NNH
OO
N
OO
N N
NNH
O
NH2
O
*4TFA
Summenformel: C38H53N21O9*4TFA M = 1403.08 g/mol
Ansatz:
491 mg 308 µmol Z-Aeg(CZ)-Aeg(AZ)-Aeg(GZ)-Aem (242)
1.0 ml 0.88 mmol TFMSA
0.50 ml 9.6 mmol m-Cresol
0.50 ml 8.5 mmol Thioanisol
3.0 ml TFA
Durchführung:
Die Durchführung erfolgt gemäß AAV 16. Nach Aufreinigung durch RP-HPLC und
anschließender Gefriertrocknung erhält man das Produkt als farbloses Pulver.
Ausbeute:
238 mg (170 µmol) = 55 % d.Th.

Experimenteller Teil 10 Synthesen
269
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 140 °C) (Rotamere): δ = 8.56, 8.22, 8.21, 8.02 (je s,
zusammen 2 H, A-C(8)H, A-C(2)H); 7.88 (m(br), 3 H, 3 × Amid-NH); 7.76 (m, 2 H, C-
C(6)H, G-C(8)H); 6.42 (s(br), div. HN+R3); 6.88 (m, 1 H, C-C(5)H); 5.12 (s(br), 2 H,
BaseCH2); 4.89 (m, 2 H, BaseCH2); 4.57 (s(br), 2 H, BaseCH2); 3.78 - 3.10 (m, 30 H, 2 ×
NCH2CH2O, 4 × CH2CH2, 3 × GlyCH2). MALDI-MS: 949.298 (169) [M+2H]+. HR-ESI-
MS: 950.4438 (13) [M+3H]+ (ber.: 950.4570); 949.4411 (51) [M+2H]+ (ber.: 949.4491);
948.4387 (100) [M+H]+ (ber.: 948.4413).
10.66 Boc-Aeg(Alloc)-OMe (225)
Methyl N-[(Allyloxy)carbonyl]-N-(2-{[(tert-butoxy)carbonyl]amino}-ethyl)glycinat
NO
O
NH
O
O
OO
Summenformel: C14H24N2O6 M = 316.36 g/mol
Ansatz:
24.0 g 103 mmol Boc-Aeg(H)-OMe (187)
14.4 ml 103 mmol Alloc-Cl
14.3 ml 134 mmol TEA
36 ml DCM
Durchführung:
Zu einer Lösung von 24.0 g (103 mmol) Boc-Aeg(H)-OMe (187) und 14.4 ml (19.8 g,
103 mmol) TEA in 360 ml DCM wird unter Eisbadkühlung eine Lösung von 14.3 ml (16.2 g,
134 mmol) Alloc-Cl getropft. Man lässt 2 h bei RT rühren, entfernt anschließend das
Lösungsmittel am Rotationsverdampfer und nimmt den Rückstand in 500 ml Ethylacetat auf.
Die organische Phase wird mit je 500 ml 1 M KHSO4-, ges. NaHCO3 und NaCl-Lösung
gewaschen und über MgSO4 getrocknet. Nach dem destillativen Entfernen des Lösungsmittels
erhält man das Produkt als farbloses Öl.

Experimenteller Teil 10 Synthesen
270
Ausbeute:
27.8 g (87.9 mmol) = 85 % d.Th.
Analysedaten: 1H-NMR (200 MHz, CDCl3) (2 Rotamere): δ = 5.90 (m, 1 H, CH2CH=CH2); 5.25 (m, 3 H,
NH und CH2CH=CH2); 4.60 (m, 2 H, CH2CH=CH); 4.00 (s, 2 H, GlyCH2); 3.75, 3.74 (2 × s,
3 H, OCH3); 3.46 (m, 2 H, BocNHCH2); 3.29 (m, 2 H, BocNHCH2CH2); 1.43 (s, 9 H,
BocCH3). 13C-NMR (50 MHz, CDCl3) (2 Rotamere): δ = 170.83 (GlyC=O); 170.56
(GlyC=O); 156.36 (BocC=O); 156.21 (AllocC=O); 156.07 (AllocC=O); 132.61
(CH2CH=CH2); 117.88 (CH2CH=CH2); 117.42 (CH2CH=CH2); 79.34 (C(CH3)3); 66.66
(CH2CH=CH2); 66.43 (CH2CH=CH2); 52.36 (OCH3); 49.94 (GlyCH2); 49.69 (GlyCH2);
49.02 (BocNHCH2CH2); 48.69 (BocNHCH2CH2); 39.19 (BocNHCH2); 28.46 (BocCH3).
FAB-MS: 655.4 (5) [2M+Na]+; 633.4 (3) [2M+H]+; 339.2 (25) [M+Na]+; 217.1 (100)
[M-Boc]+; 317.2 (23) [M+H]+.
10.67 Boc-Aeg(Fmoc)-OMe (226)
Methyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat
NO
O
NH
O
O
OO
Summenformel: C25H30N2O6 M = 454.53 g/mol
Ansatz:
26.5 g 114 mmol Boc-Aeg(H)-OMe (187)
29.4 g 114 mmol Fmoc-Cl
39.7 ml 228 mmol DIPEA
500 ml DCM

Experimenteller Teil 10 Synthesen
271
Durchführung:
Zur Vorlage von 26.5 g (114 mmol) Boc-Aeg(H)-OMe (187) und 39.7 ml (29.5 g, 228 mmol)
DIPEA in 400 ml DCM wird unter Eisbadkühlung eine Lösung von 29.4 g (114 mmol)
Fmoc-Cl in 100 ml DCM getropft. Man lässt 1 h bei Eisbadtemperatur und weitere 3 h bei RT
rühren, wäscht mit 10%iger Zitronensäure-, ges. NaHCO3- und NaCl-Lösung und entfernt
anschließend das Lösungsmittel am Rotationsverdampfer. Nach Reinigung durch Säulen-
chromatographie (Silica, DCM/MeOH 50:1) erhält man das Produkt als farbloses Öl.
Ausbeute:
44.0 g (96.9 mmol) = 86 % d.Th.
Analysedaten:
Rf (DCM/MeOH 50:1): 0.21. 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 7.85 (m, 2 H,
FmocCHarom.); 7.62 (m, 2 H, FmocCHarom.); 7.46 - 7.29 (m, 4 H, FmocCHarom.); 6.21 (m(br),
1 H, NH); 4.39 (m, 2 H, FmocCH2); 4.26 (m, 1H, FmocCH); 3.97 (s, 2 H, GlyCH2), 3.65 (s,
3 H, CH3), 3.33 (m, 2 H), 3.09 (m, 2 H) (CH2CH2); 1.39 (s, 9 H, BocCH3). 13C-NMR
(50 MHz, DMSO-d6, 110 °C): δ = 169.25 (COOMe); 154.95 (FmocC=O, BocC=O); 143.32
(FmocCarom.); 140.27 (FmocCarom.); 126.94 (FmocCHarom.); 126.40 (FmocCHarom.); 124.20
(FmocCHarom.); 119.30 (FmocCHarom.); 77.30 (C(CH3)3), 66.56 (FmocCH2); 50.96 (OCH3);
48.47, 47.31, 46.41, 38.26 (FmocCH, GlyCH2, CH2CH2); 27.65 (BocCH3). FAB-MS: 477
(15) [M+Na]+; 455 (5) [M+H]+; 355.1 (67) [M-Boc]+; 178.0 (100).

Experimenteller Teil 10 Synthesen
272
10.68 Z-Aeg(H)-OMe (227)
Methyl N-(2-{[(Benzyloxy)carbonyl]amino}ethyl)glycinat
NH
O
O
NH
O
O
Summenformel: C13H18N2O4 M = 266.30 g/mol
Ansatz:
37.1 g 150 mmol Z-OSu
30.4 g 150 mmol H-Aeg(H)-OMe*2HCl (124)
41.1 ml 373 mmol NMM
600 ml Acetonitril
Durchführung:
Zu einer Lösung von 37.1 g (150 mmol) Z-OSu in 60 ml MeCN werden bei -15 °C (NaCl-
Eisbadkühlung) zunächst 30.4 g (150 mmol) H-Aeg(H)-OMe*2HCl (124) und anschließend
41.1 ml (37.9 g, 373 mmol) NMM gegeben. Man lässt 3 h bei -15 °C und weitere 2 h bei RT
rühren und entfernt danach das Lösungsmittel am Rotationsverdampfer. Der Rückstand wird
in 100 ml 1 M KHSO4-Lösung aufgenommen und diese Lösung mit 500 ml Ethylacetat
gewaschen. Nachdem der pH-Wert der wässrigen Phase mit NaHCO3 auf 8 eingestellt wurde,
wird dreimal mit je 500 ml Ethylacetat extrahiert. Die vereinigten Extrakte werden mit 400 ml
ges. NaCl-Lösung gewaschen, über MgSO4 getrocknet und destillativ vom Lösungsmittel
befreit, woraufhin man das Produkt als farbloses Öl erhält.
Ausbeute:
25.0 g (93.9 mmol) = 63 % d.Th.

Experimenteller Teil 10 Synthesen
273
Analysedaten:
1H-NMR (200 MHz, CDCl3): δ = 7.38 - 7.13 (m, 5 H, ZCHarom.); 5.32 (s(br), 1 H, ZNH);
5.02 (s, 2 H, ZCH2); 3.64 (s, 3 H, OCH3); 3.32 (s, 2 H, GlyCH2); 3.20 (dt, 2 × 3J = 5.7, 2 H,
ZNHCH2); 2.68 (t, 3J = 5.7, 2 H, ZNHCH2CH2). 13C-NMR (50 MHz, CDCl3): δ = 172.95
(GlyC=O); 156.55 (ZC=O); 136.63 (ZCarom.); 128.51 (Z-o-CHarom.); 128.07 (2 × s, Z-p-CHarom.,
Z-m-CHarom.); 66.65 (ZCH2); 51.86 (OCH3); 50.23 (GlyCH2); 48.63 (ZNHCH2CH2); 40.63
(ZNHCH2). FAB-MS: 289.0 (11) [M+Na]+; 267.1 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
274
10.69 Boc-Aeg(Alloc)-OH (228)
N-[(Allyloxy)carbonyl-]-N-(2-{[(tert-butoxy)carbonyl]amino}ethyl)glycin
NOH
O
NH
O
O
OO
Summenformel: C13H22N2O6 M = 302.33 g/mol
Ansatz:
27.8 g 87.9 mmol Boc-Aeg(Alloc)-OMe (225)
180 ml 1 M LiOH
180 ml THF
Durchführung:
Die Durchführung erfolgt nach AAV 3. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
25.1 g (83.0 mmol) = 94 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 12.67 (s(br), 1 H, COOH); 6.75 (m(br),
1 H, NH); 6.01 - 5.73 (m, 1 H, CH2CH=CH2); 5.34 - 5.11 (m, 2 H, CH2CH=CH2); 4.51 (m,
2 H, CH2CH=CH); 3.92, 3.89 (je s, zusammen 2 H, GlyCH2); 3.33 - 3.05 (m, 4 H, CH2CH2);
1.36 (s, 9 H, BocCH3). 13C-NMR (50 MHz, DMSO-d6) (2 Rotamere): δ = 171.18 (COOH);
171.07 (COOH); 155.59, 155.46, 155.20 (BocC=O, AllocC=O); 133.22 (CH2CH=CH2);
116.70 (CH2CH=CH2); 116.34 (CH2CH=CH2); 77.67 (C(CH3)3); 65.34 (CH2CH=CH2); 65.13
(CH2CH=CH2); 48.97 (GlyCH2); 48.81 (GlyCH2); 47.78 (BocNHCH2CH2); 47.13
(BocNHCH2CH2); 38.36 (BocNHCH2); 38.06 (BocNHCH2); 28.18 (BocCH3). FAB-MS:
325.1 (100) [M+Na]+; 303 (19) [M+H]+; 203.1 (73) [M-Boc]+.

Experimenteller Teil 10 Synthesen
275
10.70 H-Aeg(Fmoc)-OMe*TFA (229)
Methyl N-(2-Aminoethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat Trifluoracetat
NO
O
NH2
OO
*TFA
Summenformel: C20H22N2O4*TFA M = 468.43 g/mol
Ansatz:
44.0 g 96.6 mmol Boc-Aeg(Fmoc)-OMe (226)
100 ml TFA
100 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 15. Man versetzt das Rohprodukt mit Diethylether,
saugt den entstandenen Niederschlag ab und trocknet im Ölpumpenvakuum. Man erhält das
Produkt als farblosen Feststoff.
Ausbeute:
41.2 g (87.8 mmol) = 91 % d.Th.
Analysedaten:
FAB-MS: 709.1 (5) [2M+H]+; 377.0 (11) [M+Na]+; 355.1 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
276
10.71 Z-Aeg(Boc)-OMe (230)
Methyl N-(2-{[(Benzyloxy)carbonyl]amino}ethyl)-N-[(tert-butoxy)carbonyl]glycinat
NO
O
NH
O
O
OO
Summenformel: C18H26N2O6 M = 366.42 g/mol
Ansatz:
24.9 g 93.8 mmol Z-Aeg(H)-OMe (227)
19.7 g 93.8 mmol Boc2O
13.1 ml 93.8 mmol TEA
500 ml DCM
Durchführung:
Man legt 24.9 g (93.8 mmol) Z-Aeg(H)-OMe (227) und 13.1 ml (9.51 g, 93.8 mmol) TEA in
410 ml DCM vor. Nach der Zugabe von 19.7 g (93.8 mmol) Boc2O in 90 ml DCM lässt man
über Nacht nachrühren, wäscht mit 1 M KHSO4-, ges. NaHCO3 und NaCl-Lösung und
trocknet über MgSO4. Nachdem das Lösungsmittel am Rotationsverdampfer entfernt wurde,
erhält man das Produkt als farbloses Öl.
Ausbeute:
32.9 g (89.9 mmol) = 96 % d.Th.

Experimenteller Teil 10 Synthesen
277
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.34 (s, 5 H, ZCHarom.); 7.18 (m, 1 H,
NH); 5.01 (s, 2 H, ZCH2); 3.93 (s, 0.90 H, GlyCH2); 3.91 (s, 1.10 H, GlyCH2); 3.65 (s,
1.30 H, OCH3); 3.63 (s, 1.70 H, OCH3); 3.29 - 3.07 (m, 4 H, CH2CH2); 1.37 (s, 4.10 H,
BocCH3); 1.32 (s, 4.90 H, BocCH3). 13C-NMR (50 MHz, DMSO-d6) (2 Rotamere):
δ = 170.61 (GlyC=O); 170.64 (GlyC=O); 156.07 (ZC=O); 154.89 (BocC=O); 154.55
(BocC=O); 137.21 (ZCarom.); 137.10 (ZCarom.); 128.34 (Z-o-CHarom.); 127.75 (Z-m-CHarom.);
127.66 (Z-p-CHarom.); 79.31 (C(CH3)3); 79.24 (C(CH3)3); 65.27 (ZCH2); 65.23 (ZCH2); 51.72
(OCH3); 49.28, 48.38, 47.24, 47.14 (2 × GlyCH2, 2 × ZNHCH2CH2); 27.88 (BocCH3); 27.80
(BocCH3). Das Signal für ZNHCH2 wird bei ca. 40 ppm vermutet und ist wahrscheinlich vom
DMSO-Signal überdeckt. FAB-MS: 389.1 (26) [M+Na]+; 367.1 (22) [M+H]+; 267.0 (100)
[M-Boc]+.
10.72 Boc-Aeg(Alloc)-Aeg(Fmoc)-OMe (231)
Methyl N-(2-{[N-(2-{[(tert-butoxy)carbonyl]amino}ethyl)-N-[(prop-2-en-1-yloxy)carbonyl]glycyl]amino}-
ethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat
O NH
N
OOOO
NH
NO
OOO
Summenformel:C33H42N4O9 M = 638.72 g/mol
Ansatz:
13.9 g 45.8 mmol Boc-(Alloc)-OH (228)
21.5 g 45.8 mmol H-Aeg(Fmoc)-OMe*TFA (229)
17.4 g 45.8 mmol HBTU
16.0 ml 91.6 mmol DIPEA
100 ml DMF

Experimenteller Teil 10 Synthesen
278
Durchführung:
Die Durchführung erfolgt gemäß AAV 17. Man erhält das Produkt nach Säulenchromatogra-
phie (Silica, EtOAc/MeOH 9:1) als farblosen Schaum.
Ausbeute:
22.4 g (35.0 mmol) = 76 % d.Th.
Analysedaten:
Rf (EtOAc/MeOH 9:1): 0.6. 1H-NMR (400 MHz, DMSO-d6, Rotamere): δ = 7.92 (t(br),
1 H, NH); 7.90 (d, 3J = 7.2, 1 H, FmocCHarom.); 7.88 (d, 3J = 7.3, 1 H, FmocCHarom.); 7.67 (d, 3J = 7.4, 1 H, FmocCHarom.); 7.57 (d, 3J = 7.4, 1 H, FmocCHarom.); 7.42 (m, 2 H,
FmocCHarom.); 7.33 (m, 2 H, FmocCHarom.); 6.76 (s(br), 0.9 H, NH); 6.42 (s(br), 0.1 H, NH);
5.87 (m, 1 H, CH2CH=CH2); 5.19 (m, 2 H, CH2CH=CH2); 4.47 (m, 2 H, CH2CH=CH2); 4.27
(m, 3 H, FmocCH2, FmocCH); 3.97 (m, 2 H, GlyCH2); 3.75 (m, 2 H, GlyCH2) 3.64, 3.61 (je s,
zusammen 3 H, OCH3); 3.29 - 3.05 (m, 8 H, 2 × CH2CH2); 1.37, 1.35 (je s, zusammen 9 H,
BocCH3). 13C-NMR (100 MHz, DMSO-d6, Rotamere): δ = 170.15 (COOMe); 170.04
(COOMe); 168.85 (GlyC=O); 168.79 (GlyC=O); 155.56, 155.50, 155.32, 155.23 (FmocC=O,
AllocC=O); 143.69 (FmocCarom.); 140.72 (FmocCarom.); 133.24 (CH2CH=CH2); 127.64
(FmocCHarom.); 127.58 (FmocCHarom.); 127.14 (FmocCHarom.); 126.99 (FmocCHarom.); 125.05
(FmocCHarom.); 124.78 (FmocCHarom.); 120.08 (FmocCHarom.); 120.04 (FmocCHarom.); 116.62
(CH2CH=CH2); 116.18 (CH2CH=CH2); 77.59 (C(CH3)3); 67.28 (FmocCH2); 66.95
(FmocCH2); 65.33 (CH2CH=CH2); 65.04 (CH2CH=CH2); 51.86 (OCH3); 51.76 (OCH3);
49.30, 48.67, 48.64, 48.13, 48.09, 47.45, 47.16, 46.55, 46.52 (FmocCH, 2 × GlyCH2,
CH2CH2N(Alloc), CH2CH2N(Fmoc)); 37.44, 36.89 (CH2CH2N(Alloc), CH2CH2N(Fmoc));
28.15 (BocCH3); 27.88 (BocCH3). 1H-NMR (250 MHz, DMSO-d6, 100 °C) (2 Rotamere,
da die Fmoc-Gruppe den Koaleszenzpunkt noch nicht ganz erreicht hat): δ = 7.87 (m,
2 H, FmocCHarom.); 7.61 (d, 3J = 7.4, 2 H, FmocCHarom.); 7.5 (m(br), 1 H, NH); 7.45 - 7.29 (m,
4 H, FmocCHarom.); 6.35 (t(br), 1 H, NH); 5.90 (ddt, 3Jtrans = 17, 3Jcis = 11, 3J = 5.2, 1 H,
CH2CH=CHcisHtrans); 5.27 (ddt aa dq, 3Jtrans = 17, 2J = 1.6, 4J = 1.6, 2 H, CH2CH=CHcisHtrans);
5.15 (ddt aa dq, 3Jcis = 11, 2J = 1.6, 4J = 1.6, 2 H, CH2CH=CHcisHtrans); 4.51 (dt, 3J = 5.2, 4J = 1.6, 2 H, CH2CH=CH2); 4.39 (m, 2 H, FmocCH2); 4.26 (m, 1 H, FmocCH2); 3.97 (s, 2 H,
N(Alloc)CH2CO); 3.82 (s, 1.6 H, N(Fmoc)CH2CO); 3.74 (s, 0.4 H, N(Fmoc)CH2CO); 3.65,
3.64 (je s, zusammen 3 H, OCH3); 3.36 - 3.08 (m, 8 H, 2 × CH2CH2); 1.39 (s, 9 H, BocCH3).

Experimenteller Teil 10 Synthesen
279
13C-NMR (62.9 MHz, DMSO-d6, 100 °C) (2 Rotamere, da die Fmoc-Gruppe den
Koaleszenzpunkt noch nicht ganz erreicht hat): δ = 169.39 (COOMe); 168.32 (GlyC=O);
155.03, 155.01 (2 × C=O); 143.34 (FmocCarom.); 140.31 (FmocCarom.); 132.79 (CH2CH=CH2);
127.01 (FmocCHarom.); 126.47 (FmocCHarom.); 124.26 (FmocCHarom.); 119.38 (FmocCHarom.);
116.08 (CH2CH=CH2); 77.24 (C(CH3)3); 66.64 (CH2CH=CH2); 64.79 (FmocCH2); 51.07
(OMe); 50.02 (CH2); 49.32 (CH2); 48.58 (CH2); 48.62 (CH2); 47.65 (CH2); 47.08 (CH2);
46.40 (FmocCH); 38.18 (CH2); 36.90 (CH2); 36.91 (CH2); 27.71 (BocCH3). FAB-MS: 678.3
(47) [M+K]+; 661.3 (97) [M+Na]+; 539.3 (33) [M-Boc]+; 178.1 (100).

Experimenteller Teil 10 Synthesen
280
10.73 Z-Aeg(Boc)-OH (232)
N-(2-{[(Benzyloxy)carbonyl]amino}ethyl)-N-[(tert-butoxy)carbonyl]glycin
NOH
O
NH
O
O
OO
Summenformel: C17H24N2O6 M = 352.39 g/mol
Ansatz:
32.9 g 89.9 mmol Z-Aeg(H)-OMe (230)
180 ml 1 M LiOH
180 ml THF
Durchführung:
Die Durchführung erfolgt nach AAV 3. Nachdem der pH-Wert auf 2-3 eingestellt ist, fällt das
Produkt als farbloser Feststoff aus. Man saugt ab, wäscht mit Wasser und Diethylether und
trocknet im Vakuum.
Ausbeute:
29.4 g (83.4 mmol) = 93 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 12.61 (s(br), 1 H, COOH); 7.34 (m, 5 H,
ZCHarom.); 7.17 (m(br), 1 H, ZNH); 5.01 (s, 2 H, ZCH2); 3.86 (s, 1 H, GlyCH2); 3.82 (s, 1 H,
GlyCH2); 3.35 - 3.11 (m, 4 H, CH2CH2); 1.37 (s, 4 H, BocCH3); 1.34 (s, 5 H, BocCH3). 13C-NMR (50 MHz, (DMSO-d6) (2 Rotamere): δ = 171.61 (GlyC=O); 171.46 (GlyC=O);
156.16 (ZC=O); 156.09 (ZC=O); 154.93 (BocC=O); 154.78 (BocC=O); 137.24 (ZCarom.);
137.13 (ZCarom.); 128.36 (Z-o-CHarom.); 127.75 (Z-m-CHarom.); 127.65 (Z-p-CHarom.); 79.09
(C(CH3)3); 79.07; (C(CH3)3); 65.28 (ZCH2); 65.22 (ZCH2); 49.30 (GlyCH2); 48.44 (GlyCH2);
47.26 (ZNHCH2CH2); 47.16 (ZNHCH2CH2); 27.93 (BocCH3); 27.87 (BocCH3). Das Signal
für ZNHCH2 ist wahrscheinlich vom DMSO-Signal überdeckt. FAB-MS: 375.1 (15)
[M+Na]+; 353.1 (24) [M+H]+; 297.1 (25) [M-tBu]+; 253.1 (81) [M-Boc]+; 91.0 (100).

Experimenteller Teil 10 Synthesen
281
10.74 H-Aeg(Alloc)-Aeg(Fmoc)-OMe*TFA (233)
Methyl N-[2-({N-[(Allyloxy)carbonyl]-N-(2-aminoethyl)-glycyl}amino)ethyl]-N-{[(9H-fluoren-9-yl)methoxy]-
carbonyl}glycinat Trifluoracetat
NH2
N
OOO
NH
NO
OOO
*TFA
Summenformel: C28H34N4O7*TFA M = 652.63 g/mol
Ansatz:
22.4 g 35.0 mmol Boc-Aeg(Alloc)-Aeg(Fmoc)-OMe (231)
40 ml TFA
40 ml DCM
Durchführung:
Die Durchführung erfolgt nach AAV 15. Man erhält das Rohprodukt als leicht gelbliches Öl,
das ohne weitere Reinigung im nächsten Reaktionsschritt eingesetzt wird.
Ausbeute:
22.8 g (34.9 mmol) = 99 % d.Th.
Analysedaten:
FAB-MS: 561.2 (17) [M+Na]+; 539.2 (100) [M+H]+.
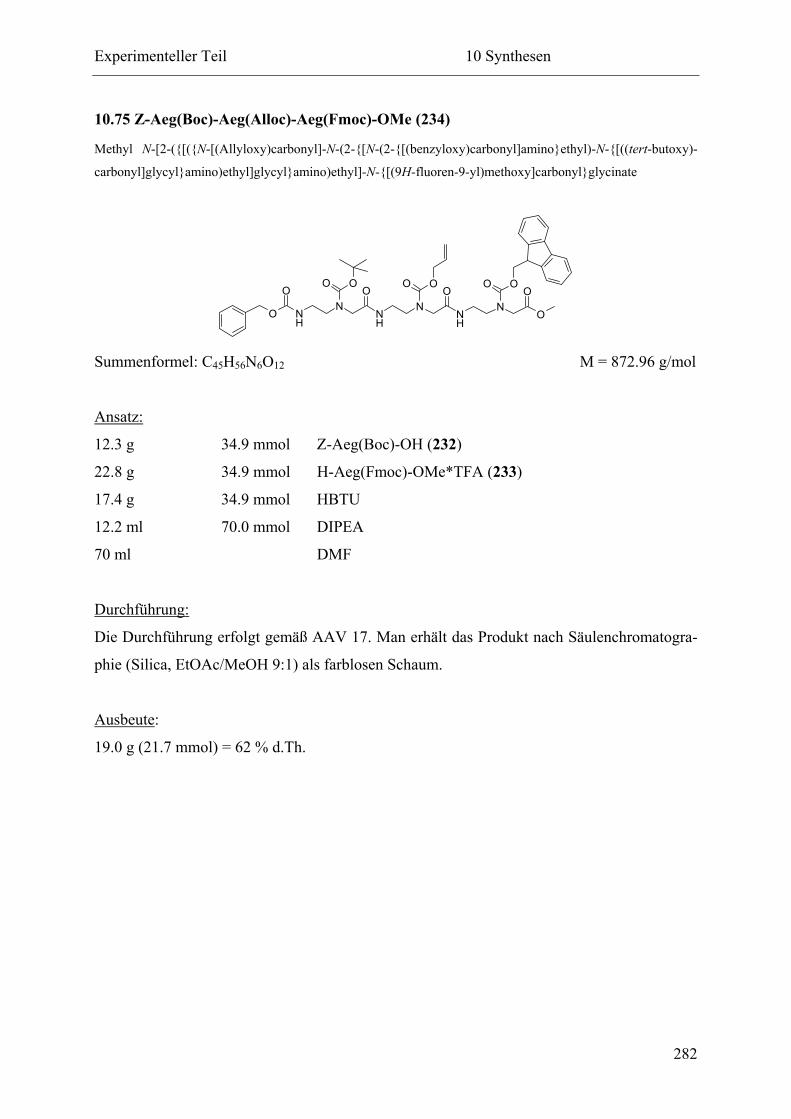
Experimenteller Teil 10 Synthesen
282
10.75 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OMe (234)
Methyl N-[2-({[({N-[(Allyloxy)carbonyl]-N-(2-{[N-(2-{[(benzyloxy)carbonyl]amino}ethyl)-N-{[((tert-butoxy)-
carbonyl]glycyl}amino)ethyl]glycyl}amino)ethyl]-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinate
NH
N
OOO
NH
NO
OOO
O
NNH
O
OOO
Summenformel: C45H56N6O12 M = 872.96 g/mol
Ansatz:
12.3 g 34.9 mmol Z-Aeg(Boc)-OH (232)
22.8 g 34.9 mmol H-Aeg(Fmoc)-OMe*TFA (233)
17.4 g 34.9 mmol HBTU
12.2 ml 70.0 mmol DIPEA
70 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 17. Man erhält das Produkt nach Säulenchromatogra-
phie (Silica, EtOAc/MeOH 9:1) als farblosen Schaum.
Ausbeute:
19.0 g (21.7 mmol) = 62 % d.Th.

Experimenteller Teil 10 Synthesen
283
Analysedaten:
Rf (EtOAc/MeOH 9:1): 0.28. 1H-NMR (250 MHz, DMSO-d6, 100 °C): δ = 7.85 (d, 3J = 7.0, 2 H, FmocCHarom.); 7.61 (d, 3J = 7.5, 2 H FmocCHarom.) darunter (s(br), 1 H, NH);
7.57 (s(br), 1 H, NH); 7.44 - 7.28 (m, 9 H, 4 × FmocCHarom., 5 × ZCHarom.); 6.81 (t(br), 1 H,
NH); 5.90 (ddt, 3J = 5.2, 3Jtrans = 17, 3Jcis = 11, 1 H, CH2CH=CHcisHtrans); 5.28 (ddt aa dq, 2J = 1.6, 3Jtrans = 17, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.15 (ddt aa dq, 2J = 1.6, 3Jcis = 11, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.04 (s, 2 H; ZCH2); 4.52 (dt, 3J = 5.2, 4J = 1.6, 2 H,
CH2CH=CH2); 4.39 (m, 2 H, FmocCH2); 4.26 (t(br), 3J = 6.2, 1 H, FmocCH); 3.97 (s, 2 H,
GlyCH2); 3.84 (s, 2 H, GlyCH2); 3.74 (s, 2 H, GlyCH2); 3.64 (s, 3 H, OCH3); 3.35 - 3.15 (m,
12 H, 3 × CH2CH2); 1.38 (s, 9 H, BocCH3). 13C-NMR (62.9 MHz, DMSO-d6, 100 °C):
169.37 (COOMe); 168.65 (GlyC=O); 168.38 (GlyC=O); 155.51, 155.04, 155.00, 154.48
(ZC=O, BocC=O, AllocC=O, FmocC=O); 143.34 (FmocCarom.); 140.32 (FmocCarom.); 136.78
(ZCarom.); 132.75 (CH2C=CH2); 127.66 (FmocCHarom.); 127.01 (Z-o-CHarom.); 126.90 (Z-m-
CHarom.); 126.47 (Z-p-CHarom.); 124.26 (FmocCHarom.); 119.38 (FmocCHarom.); 116.16
(CH2CH=CH2); 78.59 (C(CH3)3); 66.63 (CH2CH=CH2); 64.84 (FmocCH2); 51.07 (OCH3);
46.40 (FmocCH). 27.50 (BocCH3). Die Signale der übrigen CH2-Gruppen befinden sich im
Bereich 51.13 - 37.74 ppm, können jedoch z.T. unter den gegebenen Messbedingungen
(76 mM Probe, 1024 Scans) nicht sinnvoll von der Basislinie unterschieden werden. FAB-
MS: 895.3 (35) [M+Na]+; 873.3 (15) [M+H]+; 795.3 (3) [M-Boc+Na]+; 773.3 (36) [M-Boc]+;
91.0 (100).

Experimenteller Teil 10 Synthesen
284
10.76 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OH (235)
N-[2-({[{N-[(Allyloxy)carbonyl]-N-(2-{[N-(2-{[(benzyloxy)carbonyl]amino}ethyl)-N-{[(tert-butoxy)carbonyl]-
glycyl}amino)ethyl]glycyl}amino)ethyl]-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycin
OO
N
O
OHNH
OO
N
O
NH
O
N
O O
NH
O
O
Summenformel: C44H54N6O12 M = 858.95 g/mol
Ansatz:
19.0 g 21.7 mmol Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OMe (234)
7.31 g 28.2 mmol Fmoc-Cl
43.4 ml 1 M LiOH
43.4 ml THF
Durchführung:
19.0 g (21.7 mmol) Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OMe (234) (21.7 mmol) werden
43.4 ml in THF vorgelegt und auf 0 °C abgekühlt. Nach Zugabe von 43.4 ml 1 M LiOH-
Lösung wird 1 h bei 0 °C und 1 h bei RT gerührt. Die während dieser Zeit erfolgte Fmoc-
Abpaltung wird rückgängig gemacht, indem man bei 0 °C 7.31 g (28.2 mmol, 1.3 eq.) Fmoc-
Cl zugibt und eine weitere Stunde bei dieser Temperatur rührt. Danach stellt man durch
Zugabe von 1 M KHSO4-Lösung auf pH 3 ein und extrahiert dreimal mit je 150 ml
Ethylacetat. Die vereinten organischen Phasen werden mit ges. NaCl-Lösung gewaschen,
über MgSO4 getrocknet und eingeengt. Der verbleibende Rückstand wird
säulenchromatographisch gereinigt (Silica, EtOAc/MeOH 9:1 → 1:1). Man erhält das Produkt
als farblosen Schaum.
Ausbeute:
12.9 g (15.0 mmol) = 69 % d.Th.

Experimenteller Teil 10 Synthesen
285
Analysedaten:
Rf (EtOAc/MeOH 1:1): 0.46. 1H-NMR (250 MHz, DMSO-d6) (Rotamere): δ = 12.74
(s(br), 1 H, COOH); 8.00 (s(br), 2 H, 2 × NH); 7.89 (d(br), 3J = 7.4, 2 H, FmocCHarom.); 7.67
(d, 3J = 7.3, 1 H, FmocCHarom.); 7.62 (d, 3J = 7.5, 1 H, FmocCHarom.); 7.45 - 7.23 (m, 10 H,
FmocCHarom., ZCHarom, NH); 6.01 - 5.76 (m, 1 H, CH2CH=CH2); 5.20 (m, 2 H,
CH2CH=CH2); 5.00 (s, 2 H, ZCH2); 4.50 (m, 2 H, CH2CH=CH2); 4.27 (m, 3 H, FmocCH2,
FmocCH); 4.04 - 3.68 (m, 6 H, 3 × GlyCH2); 3.39 - 3.13 (m, 12 H, 3 × CH2CH2); 1.36, 1.31
(je s, zusammen 9 H, BocCH3). Erhöhte Temperaturen führen zu einer schnellen Fmoc-
Abspaltung, wahrscheinlich durch die Bildung eines cyclischen Anhydrids mit der Carboxyl-
funktion. Daher ist eine Charakterisierung oberhalb des Koaleszenzpunkts nicht möglich.
FAB-MS: 881.2 (45) [M+Na]+; 859.2 (13) [M+H]+; 759.2 (28) [M-Boc]+; 91.0 (100).
HR-ESI-MS: 861.3916 (14) [M+3H]+ (ber.: 861.4034); 860.3888 (48) [M+2H]+ (ber.:
860.3956); 859.3856 (100) [M+H]+ (ber.: 859.3878).

Experimenteller Teil 10 Synthesen
286
10.77 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-Aem (236)
N-[15-[(Allyloxy)carbonyl]-9-[(tert-butoxy)carbonyl]-3-{[(9H-fluoren-9-yl)methoxy]carbonyl}-20-(morpholin-
4-yl)-5,11,17-trioxo-3,6,9,12,15,18-hexaazaicos-1-yl]carbaminsäure-phenylmethylester
NH
N
OOO
N
O
NH
OO
N
O
NH
O
N
O O
NH
O
O
Summenformel: C50H66N8O12 M = 971.13 g/mol
Ansatz:
12.8 g 14.9 mmol Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-OH (235)
1.96 ml 14.9 mmol 4-(2-Aminoethyl)morpholin (143)
2.56 g 22.3 mmol HOSu
3.38 g 16.4 mmol DCC
60 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 18. Anschließend chromatographiert man den Rück-
stand (Silica, EtOAc/MeOH 2:1) und erhält das Produkt in Form eines farblosen Schaums.
Ausbeute:
12.1 g (12.5 mmol) = 84 % d.Th.

Experimenteller Teil 10 Synthesen
287
Analysedaten:
Rf (EtOAc/MeOH 2:1): 0.32. 1H-NMR (250 MHz, DMSO-d6, 100 °C): δ = 7.85 (d, 3J = 7.4, 2 H, FmocCHarom.); 7.72 (t(br), 1 H, NH); 7.64 (d, 3J = 7.3, 2 H, FmocCHarom.);
darunter (1 H, NH); 7.48 - 7.26 (m, 10 H, 4 × FmocCHarom., 5 × ZCHarom., NH); 6.81 (t(br),
1 H, NH); 5.90 (ddt, 3J = 5.2, 3Jtrans = 17, 3Jcis = 11, 1 H, CH2CH=CHcisHtrans); 5.27 (ddt aa dq, 2J = 1.6, 3Jtrans = 17, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.15 (ddt aa dq, 2J = 1.6, 3Jcis = 11, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.04 (s, 2 H, ZCH2); 4.52 (dt(br), 2 H, CH2CH=CH2); 4.30
(m, 3 H, FmocCH2, FmocCH); 3.88 (s, 2 H, GlyCH2); 3.85 (s, 2 H, GlyCH2); 3.74 (s, 2 H,
GlyCH2); 3.54 (m, 4 H, 2 × NCH2CH2O); 3.37 - 3.11 (m, 14 H, 3.5 × CH2CH2); 2.38 (m, 6 H,
2 × NCH2CH2O, 0.5 × CH2CH2); 1.38 (s, 9 H, BocCH3). Erhöhte Temperaturen führen zu
einer schnellen Fmoc-Abspaltung, die die Charakterisierung durch 13C-NMR-Spektroskopie
verhindert. FAB-MS: 971.1 (36) [M+H]+; 871.5 (5) [M-Boc]+; 154.1 (100). MALDI-MS:
971.917 (68) [M+H]+; 871.771 (248) [M-Boc]+. HR-ESI-MS: 973.4910 (17) [M+3H]+ (ber.:
973.5034); 972.4885 (58) [M+2H]+ (ber.: 972.4956); 971.4853 (100) [M+H]+ (ber.:
971.4878).
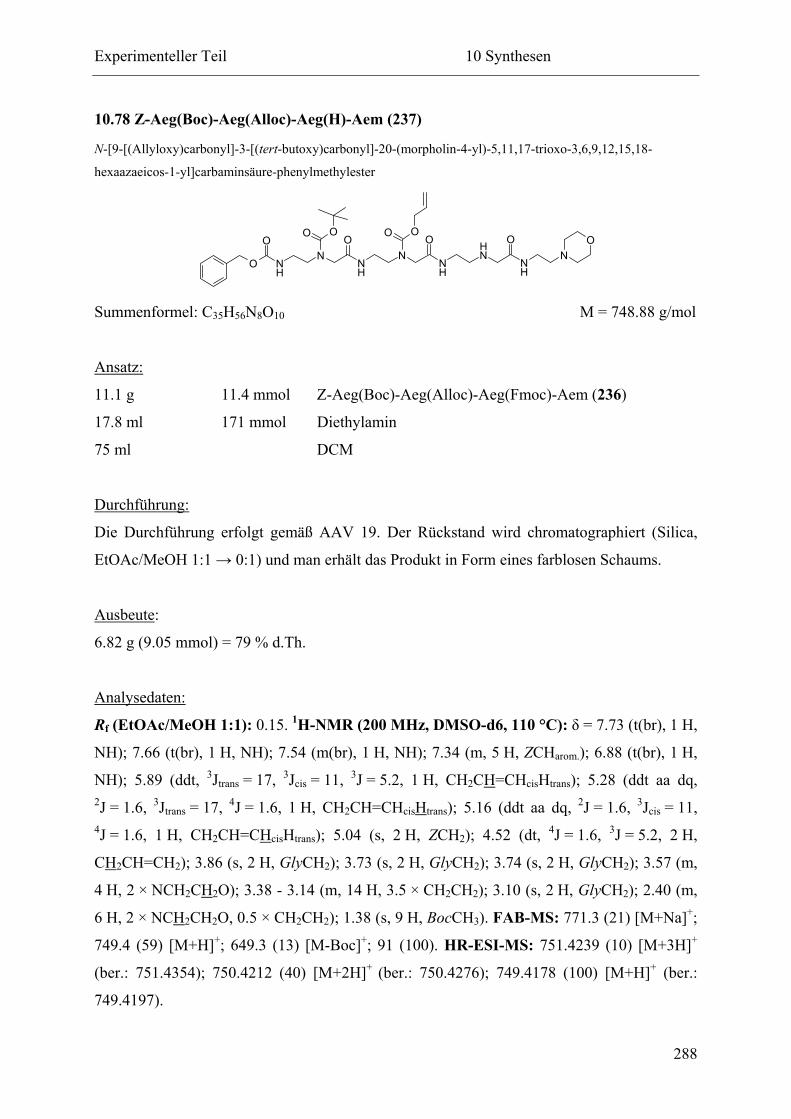
Experimenteller Teil 10 Synthesen
288
10.78 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(H)-Aem (237)
N-[9-[(Allyloxy)carbonyl]-3-[(tert-butoxy)carbonyl]-20-(morpholin-4-yl)-5,11,17-trioxo-3,6,9,12,15,18-
hexaazaeicos-1-yl]carbaminsäure-phenylmethylester
NH
N
O
NH
O
NH
OO
N
O
NH
O
N
O O
NH
O
O
Summenformel: C35H56N8O10 M = 748.88 g/mol
Ansatz:
11.1 g 11.4 mmol Z-Aeg(Boc)-Aeg(Alloc)-Aeg(Fmoc)-Aem (236)
17.8 ml 171 mmol Diethylamin
75 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 19. Der Rückstand wird chromatographiert (Silica,
EtOAc/MeOH 1:1 → 0:1) und man erhält das Produkt in Form eines farblosen Schaums.
Ausbeute:
6.82 g (9.05 mmol) = 79 % d.Th.
Analysedaten:
Rf (EtOAc/MeOH 1:1): 0.15. 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 7.73 (t(br), 1 H,
NH); 7.66 (t(br), 1 H, NH); 7.54 (m(br), 1 H, NH); 7.34 (m, 5 H, ZCHarom.); 6.88 (t(br), 1 H,
NH); 5.89 (ddt, 3Jtrans = 17, 3Jcis = 11, 3J = 5.2, 1 H, CH2CH=CHcisHtrans); 5.28 (ddt aa dq, 2J = 1.6, 3Jtrans = 17, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.16 (ddt aa dq, 2J = 1.6, 3Jcis = 11, 4J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.04 (s, 2 H, ZCH2); 4.52 (dt, 4J = 1.6, 3J = 5.2, 2 H,
CH2CH=CH2); 3.86 (s, 2 H, GlyCH2); 3.73 (s, 2 H, GlyCH2); 3.74 (s, 2 H, GlyCH2); 3.57 (m,
4 H, 2 × NCH2CH2O); 3.38 - 3.14 (m, 14 H, 3.5 × CH2CH2); 3.10 (s, 2 H, GlyCH2); 2.40 (m,
6 H, 2 × NCH2CH2O, 0.5 × CH2CH2); 1.38 (s, 9 H, BocCH3). FAB-MS: 771.3 (21) [M+Na]+;
749.4 (59) [M+H]+; 649.3 (13) [M-Boc]+; 91 (100). HR-ESI-MS: 751.4239 (10) [M+3H]+
(ber.: 751.4354); 750.4212 (40) [M+2H]+ (ber.: 750.4276); 749.4178 (100) [M+H]+ (ber.:
749.4197).

Experimenteller Teil 10 Synthesen
289
10.79 Z-Aeg(Boc)-Aeg(Alloc)-Aeg(GZ)-Aem (238)
9-[(Allyloxy)carbonyl]-N-{15-[2-(1,6-dihydro-6-oxo-2-{[(phenylmethoxy)carbonyl]amino}-9H-purin-9-
yl)acetyl]-3-[(tert-butoxy)carbonyl]-20-(morpholin-4-yl)-5,11,17-trioxo-3,6,9,12,15,18-hexaazaicos-1-yl}-
carbaminsäure-phenylmethylester
O
N N
NNH
O
NH
O
O
NH
N
O
N
O
NH
OO
N
O
NH
O
NNH
O
OOO
Summenformel: C50H67N13O14 M = 1074.17 g/mol
Ansatz:
6.78 g 9.05 mmol Z-Aeg(Boc)-Aeg(Alloc)-Aeg(H)-Aem (237)
3.88 g 11.3 mmol GZ-AcOH (199)
4.39 g 11.3 mmol Brop
2.80 ml 20.1 mmol TEA
35 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 20. Die Reaktionsmischung wird mit Chloroform
verdünnt, mit ges. NaHCO3- und NaCl-Lösung gewaschen und über MgSO4 getrocknet. Man
entfernt die Lösungsmittel am Rotationsverdampfer, chromatographiert den verbliebenen
Rückstand (Silica, EtOAc/MeOH 1:1 → 0:1) und erhält das Produkt als farblosen Schaum.
Ausbeute:
8.15 g (7.78 mmol) = 86 % d.Th.

Experimenteller Teil 10 Synthesen
290
Analysedaten:
Rf (EtOAc/MeOH 1:1): 0.10. 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 7.78 (s, 1 H,
G-C(8)H); 7.73 (t(br), 1 H, NH); 7.66 (t(br), 1 H, NH); 7.54 (m(br), 1 H, NH); 7.40 - 7.27 (m,
10 H, 10 × ZCHarom.); 6.77 (t(br), 1 H, NH); 5.89 (ddt, 3Jtrans = 17, 3Jcis = 11, 3J = 5.2, 1 H,
CH2CH=CHcisHtrans); 5.28 (s, 2 H, G-ZCH2); 5.27 (ddt aa dq, 2J = 1.6, 3Jtrans = 17, 4J = 1.6,
1 H, CH2CH=CHcisHtrans); 5.15 (ddt aa dq, 2J = 1.6, 3Jcis = 11, 4J = 1.6, 1 H,
CH2CH=CHcisHtrans); 5.04 (s, 2 H, ZCH2); 4.98 (s(br), 2 H, G-N(9)CH2); 4.51 (dt, 4J = 1.6, 3J = 5.2, 2 H, CH2CH=CH2); 4.06, 3.95 (je s(br), zusammen 2 H, GlyCH2); 3.87 (s(br), 2 H,
GlyCH2); 3.74 (s, 2 H, GlyCH2); 3.55 (m, 4 H, 2 × NCH2CH2O); 3.35 - 3.14 (m, 14 H,
3.5 × CH2CH2); 2.41 (m, 6 H, 2 × NCH2CH2O, 0.5 × CH2CH2); 1.38 (s, 9 H, BocCH3).
FAB-MS: 1096 (42) [M+Na]+; 1074.6 (31) [M+H]+; 154.0 (100). HR-ESI-MS: 1077.5062
(5) [M+4H]+ (ber.: 1077.5243); 1076.5035 (20) [M+3H]+ (ber.: 1076.5165); 1075.5010 (57)
[M+2H]+ (ber.: 1075.5087); 1074.4983 (100) [M+H]+ (ber.: 1074.5008).
10.80 Z-Aeg(Boc)-Aeg(H)-Aeg(GZ)-Aem (239)
N-{6,9-Dihydro-9-[15-[(tert-butoxy)carbonyl]-3-(2-{[2-(morpholin-4-yl)ethyl]amino}-2-oxoethyl)-2,7,13,19-
tetraoxo-21-phenyl-20-oxa-3,6,9,12,15,18-hexaazaheneicos-1-yl]-6-oxo-1H-purin-2-yl}carbaminsäure-phenyl-
methylester
O
N N
NNH
O
NH
O
O
NH
N
O
N
O
NH
NH
O
NH
O
NNH
O
OOO
Summenformel: C46H63N13O12 M = 990.09 g/mol
Ansatz:
8.98 g 8.58 mmol Z-Aeg(Boc)-Aeg(Alloc)-Aeg(GZ)-Aem (238)
991 mg 858 µmol Pd[P(Ph)3]4
26.7 ml Diethylamin
60 ml DCM

Experimenteller Teil 10 Synthesen
291
Durchführung:
Man legt 8.98 g (8.58 mmol) Z-Aeg(Boc)-Aeg(Alloc)-Aeg(GZ)-Aem (237) und 26.7 ml (18.7
g, 256 mmol, 30 eq.) Diethylamin in 60 ml DCM vor und kühlt auf 0 °C ab. Danach werden
991 mg (0.858 mmol) Pd[P(Ph)3]4 hinzugegeben und die Reaktionsmischung wird 1 h bei RT
gerührt. Anschließend werden die flüchtigen Komponenten im Vakuum entfernt und der
Rückstand durch Säulenchromatographie gereinigt (Silica, EtOAc/MeOH 1:1 → 0:1). Man
erhält das Produkt als farblosen Schaum.
Ausbeute:
5.21 g (5.27 mmol) = 61 % d.Th.
Analysedaten:
Rf (MeOH): 0.19. 1H-NMR (400 MHz, DMSO-d6) (Rotamere): δ = 8.17 (s(br), 1 H, NH);
7.86 (s(br), 2 H, 2 × NH); 7.79, 7.78 (je s, zusammen 1 H, G-C(8)H); 7.42 - 7.28 (m, 10 H,
10 × ZCHarom.); 7.24 (t(br), 1 H, NH); 5.233, 5.226 (je s, zusammen 2 H, G-ZCH2); 5.00 (s,
2 H, ZCH2); 5.05, 4.93 (je s, zusammen 2 H, G-N(9)CH2); 4.15, 3.92 (je s, zusammen 2 H,
GlyCH2); 3.75, 3.70 (je s, zusammen 2 H, GlyCH2); 3.56 - 3.06 (m, 20 H, GlyCH2,
3.5 × CH2CH2, 2 × NCH2CH2O); 2.35 (m, 6 H, 2 × NCH2CH2O, 0.5 × CH2CH2); 1.36, 1.31
(je s, zusammen 9 H, BocCH3). Bei 110 °C führen Signalverbreiterungen zu einer schlechten
Interpretierbarkeit des Spektrums. MALDI-MS: 1013.249 (210) [M+Na]+; 991.208 (120)
[M+H]+.

Experimenteller Teil 10 Synthesen
292
10.81 Z-Aeg(Boc)-Aeg(AZ)-Aeg(GZ)-Aem (240)
N-(6,9-Dihydro-9-{15-[(tert-butoxy)carbonyl]-3-(2-{[2-(morpholin-1-yl)ethyl]amino}-2-oxoethyl)-2,7,13,19-
tetraoxo-21-phenyl-9-[2-(6-{[(phenylmethoxy)carbonyl]amino}-9H-purin-9-yl)acetyl]-20-oxa-3,6,9,12,15,18-
hexaazaheneicos-1-yl}-6-oxo-1H-purin-2-yl)carbaminsäure-phenylmethylester
O
N N
NN
NH
O
O
O
N N
NNH
O
NH
O
O
NH
N
O
N
O
NH
N
O
NH
O
NNH
O
OOO
Summenformel: C61H74N18O15 M = 1299.38 g/mol
Ansatz:
6.78 g 5.26 mmol Z-Aeg(Boc)-Aeg(H)-Aeg(GZ)-Aem (239)
2.15 g 6.58 mmol AZ-AcOH (190)
4.39 g 11.3 mmol Brop
1.83 ml 20.1 mmol TEA
20 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 20. Die Reaktionsmischung wird am Trockeneis-Rota-
tionsverdampfer eingeengt und der verbleibende Rückstand durch Säulenchromatographie
gereinigt (Silica, MeOH). Man erhält das Produkt als farblosen Schaum.
Ausbeute:
4.62 g (3.56 mmol) = 67 % d.Th.

Experimenteller Teil 10 Synthesen
293
Analysedaten:
Rf (MeOH): 0.14. 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere): δ = 8.57, 8.23,
8.14, 7.96 (je m, zusammen 2 H, A-C(8)H, A-C(2)H); 7.92 - 7.17 (m, 17 H, 15 × ZCHarom.,
G-C(8)H, NH); 6.79 (t(br), 1 H, NH); 6.66 (s(br), 1 H, NH); 6.09 (s(br), 1 H, NH); 5.25 (s,
2 H, ZCH2); 5.16 (s(br), 2 H, N(9)CH2); 5.03 (s, 2 H, ZCH2); 4.87 (s(br), 2 H, N(9)CH2); 4.52
(s, 2 H, ZCH2); 4.23 (s(br), 4 H, 2 × GlyCH2); 3.81, 3.76 (je s(br), zusammen 2 H, GlyCH2);
3.57 - 3.18 (m, 18 H, 2 × NCH2CH2O, 3.5 × CH2CH2); 2.41 (m, 6 H, 2 × NCH2CH2O,
0.5 × CH2CH2); 1.38 (s, 9 H, BocCH3). MALDI-MS: 1322.861 (127) [M+Na]+; 1300.814
(171) [M+2H]+. HR-ESI-MS: 1302.5703 (8) [M+4H]+ (ber.: 1302.5894); 1301.5671 (26)
[M+3H]+ (ber.: 1301.5816); 1300.5652 (75) [M+2H]+ (ber.: 1300.5737); 1299.5621 (100)
[M+H]+ (ber.: 1299.5659).

Experimenteller Teil 10 Synthesen
294
10.82 Z-Aeg(H)-Aeg(AZ)-Aeg(GZ)-Aem*2TFA (241)
N-(6,9-Dihydro-9-{3-(2-{[2-(morpholin-4-yl)ethyl]amino}-2-oxoethyl)-2,7,13,19-tetraoxo-21-phenyl-9-[2-(6-
{[(phenylmethoxy)carbonyl]amino}-9H-purin-9-yl)acetyl]-20-oxa-3,6,9,12,15,18-hexaazaheneicos-1-yl}-6-oxo-
1H-purin-2-yl)carbaminsäure-phenylmethylester
O
N N
NN
NH
O
O
O
N N
NNH
O
NH
O
O
NH
N
O
N
O
NH
N
O
NH
O
NH
NH
O
O
*2TFA
Summenformel: C56H66N18O13*2TFA M = 1427.30 g/mol
Ansatz:
1.38 g 1.06 mmol Z-Aeg(Boc)-Aeg(AZ)-Aeg(GZ)-Aem (240)
1.00 ml 6.26 mmol HSiEt3
5 ml TFA
5 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 15 in der Gegenwart von Triethylsilan. Man nimmt
das Rohprodukt in 10 ml Ethylacetat auf, versetzt mit Diethylether, saugt den entstandenen
Feststoff ab und wäscht mit Diethylether. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
1.44 g (1.01 mmol) = 95 % d.Th.
Analysedaten:
Das Produkt zersetzt sich bei 110 °C, sodass keine HT-NMR-Charakterisierung möglich ist.
MALDI-MS: 1223.223 (355) [M+Na]+; 1200.790 (877) [M+2H]+. HR-ESI-MS: 1202.5178
(6) [M+4H]+ (ber.: 1202.5369); 1201.5152 (24) [M+3H]+ (ber.: 1201.5291); 1200.5123 (68)
[M+2H]+ (ber.: 1200.5213); 1199.5062 (100) [M+H]+ (ber.: 1199.5135).

Experimenteller Teil 10 Synthesen
295
10.83 Z-Aeg(CZ)-Aeg(AZ)-Aeg(GZ)-Aem*TFA (242)
N-(6,9-Dihydro-9-{3-(2-{[2-(morpholin-4-yl)ethyl]amino}-2-oxoethyl)-2,7,13,19-tetraoxo-15-[2-(2-oxo-4-
{[(phenylmethoxy)carbonyl]amino}pyrimidin-1(2H)-yl)acetyl]-21-phenyl-9-[2-(6-{[(phenylmethoxy)-
carbonyl]amino}-9H-purin-9-yl)acetyl]-20-oxa-3,6,9,12,15,18-hexaazaheneicos-1-yl}-6-oxo-1H-purin-2-yl)
carbaminsäure-phenylmethylester
N
N
O
NH
N
O
N N
NN
NH
O
O
NH
O
NH
NNH
NNH
OOO
ON
OO
N N
NNH
O
NH
O
O
O
O
O
*TFA
Summenformel: C70H77N21O17*TFA M = 1598.54 g/mol
Ansatz:
1.00 g 701 µmol Z-Aeg(H)-Aeg(AZ)-Aeg(GZ)-Aem (241)
266 mg 876 µmol CZ-AcOH (192)
340 mg 876 µmol Brop
440 µl 3.15 mmol TEA
5 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 20. Anschließend wird die Reaktionsmischung am
Trockeneis-Rotationsverdampfer eingeengt. Der verbleibende Rückstand wird mit
iso-Propanol versetzt und 5 min im Ultraschallbad behandelt. Man saugt den entstandenen
Niederschlag ab und wäscht mit iso-Propanol und Diethylether. Das Rohprodukt (1.08 g) wird
durch präparative RP-HPLC gereinigt. Nach Gefriertrocknung erhält man das Produkt in
Form eines farblosen Pulvers.
Ausbeute:
531 mg (332 µmol) = 47 % d.Th.

Experimenteller Teil 10 Synthesen
296
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere): δ = 8.56, 8.22, 8.21, 8.02 (je s,
zusammen 2 H, A-C(8)H, A-C(2)H); 7.76 (m, 2 H, C-C(6)H, G-C(8)H); 7.92 - 7.17 (m, 20 H,
ZCHarom.); 6.88 (m, 1 H, C-C(5)H); 5.27 (s, 2 H, ZCH2); 5.24 (s, 2 H, ZCH2); 5.20 (s, 2 H,
ZCH2); 5.19 (s, 2 H, ZCH2); 5.05 (s(br), 2 H, BaseCH2); 5.01 (s(br), 2 H, BaseCH2); 4.70
(s(br), 2 H, BaseCH2); 3.78 - 3.10 (m, 30 H, 2 × NCH2CH2O, 4 × CH2CH2, 3 × GlyCH2).
Zwischen 8.10 - 7.60 und bei 6.42 erkennt man sehr breite Singuletts, welche den diversen
NH-Spezies entsprechen und schwer zu integrieren sind. MALDI-MS: 1508.376 (34)
[M+Na]+; 1486.173 (156) [M+3H]+. HR-ESI-MS: 1487.5917 (12) [M+4H]+ (ber.:
1487.6119); 1486.5889 (37) [M+3H]+ (ber.: 1486.6041); 1485.5863 (87) [M+2H]+ (ber.:
1485.5962); 1484.5843 (100) [M+H]+ (ber.: 1484.5884).
10.84 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OH*3TFA (248)
N-[(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-N-[2-({N-[2-({N-[(4-amino-2-oxopyrimidin-1(2H)-
yl)acetyl]-N-[2-({N-[2-(dimethylamino)ethyl]-N-methylglycyl}amino)ethyl]glycyl}amino)ethyl]-N-[(2,4-
difluor-5-methylphenyl)acetyl]glycyl}amino)ethyl]glycin
N N
NNH
O
NH2
NH
N
O
NH
NNH
NOH
OOO
N
N
O
NH2
O
F
F
OO
NN
*3TFA
Summenformel: C41H56F2N16O10*3TFA M = 1313.06 g/mol
Ansatz:
305 mg 214 µmol Me2-Aeg(Me)-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn (262)
1.0 ml 0.88 mmol TFMSA
0.50 ml 9.6 mmol m-Cresol
0.50 ml 8.5 mmol Thioanisol
3.0 ml TFA

Experimenteller Teil 10 Synthesen
297
Durchführung:
Die Durchführung erfolgt gemäß AAV 16. Man erhält das Produkt in Form eines farblosen
Pulvers.
Ausbeute:
157 mg (120 µmol) = 56 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 140 °C): δ = 7.63 (d, 3J = 7.4, 1 H, C-C(6)H); 7.57 (s, 1 H,
G-C(8)H); darunter (s(br), 3 H, 3 × Amid-NH); 7.16 (t, 3J = 8.7, 1 H, FCHarom.); 6.89 (t, 3J = 9.8, 1 H, FCHarom.); 6.21 (s(br), div. HN+R3, COOH); 5.96 (d, 3J = 7.4, 1 H, C-C(5)H);
4.88 (s, 2 H, CH2); 4.65 (s, 2 H, CH2); 4.15 (s, 2 H, CH2); 4.04 (s, 2 H, CH2); 4.02 (s, 2 H,
CH2); 3.65 (s, 2 H, CH2); 3.58 - 3.17 (m, 16 H, 3.5 × CH2CH2, CH2); 2.85 (s, 6 H, N(CH3)2);
darunter (m, 2 H, 0.5 × CH2CH2); 2.37 (s, 3 H, CH3); 2.17 (s, 3 H, CH3). 19F-NMR
(235 MHz, DMSO-d6) (Rotamere): -78.80 (s, 9 F, F3CCOOH); -118.45 (m, 1 F, F(4)); -
120.99 (m, 1 F, F(2)). MALDI-MS: 972.564 (485) [M+2H]+. HR-ESI-MS: 973.4433 (13)
[M+3H]+ (ber.: 973.4568); 972.4411 (48) [M+2H]+ (ber.: 972.4490); 971.4381 (100) [M+H]+
(ber.: 971.4411).

Experimenteller Teil 10 Synthesen
298
10.85 Me2-Aeg(Me)-OH*2HCl (249)
N-[2-(Dimethylamino)ethyl]-N-methylglycin Dihydrochlorid
OH
O
NN
*2HCl
Summenformel: C7H16N16O2*2HCl M = 233.14 g/mol
Ansatz:
2.50 g 21.2 mmol Aeg (85)
3.99 ml 106 mmol Ameisensäure
5.68 ml 73.3 mmol Formalin-Lösung
Durchführung:
Man legt 2.50 g (21.2 mmol) Aeg (85) vor und gibt unter Eisbadkühlung zunächst 3.99 ml
(4.87 g, 106 mmol) Ameisensäure und anschließend 5.68 ml (6.19 g, 73.3 mmol) Formalin-
Lösung hinzu. Die Reaktionsmischung wird 12 h bei Rückflusstemperatur gerührt und danach
mit 11.6 ml konzentrierter Salzsäure versetzt. Anschließend entfernt man destillativ die
flüchtigen Komponenten und kristallisiert den verbleibenden Rückstand aus 20 ml Methanol
um. Man erhält das Produkt als farblosen Feststoff.
Ausbeute:
2.02 g (8.67 mmol) = 41 % d.Th.
Analysedaten: 1H-NMR (200 MHz, D2O): δ = 4.20 (s, 2 H, GlyCH2); 3.77 (m, 4 H, CH2CH2); 3.11 (s, 3 H,
NCH3); 3.04 (s, 6 H, N(CH3)2). 13C-NMR (50 MHz, D2O): δ = 168.26 (COOH); 57.31
(GlyCH2); 51.29 (CH2); 50.82 (CH2); 43.57 (NCH3); 42.17 (N(CH3)2). FAB-MS: 321.2 (10)
[2M+H]+; 161.1 (100) [M+H]+.

Experimenteller Teil 10 Synthesen
299
10.86 Me2-Aeg(Me)-OH (249)
N-[2-(Dimethylamino)ethyl]-N-methylglycin
OH
O
NN
Summenformel: C7H16N16O2 M = 160.22 g/mol
Ansatz:
2.50 g 21.2 mmol Aeg (85)
5.68 ml 73.3 mmol Formalin-Lösung
2.00 g Pd/C (10 %)
300 ml Wasser
Durchführung:
Die Durchführung erfolgt gemäß AAV 4. Danach kristallisiert man das Rohprodukt aus 20 ml
Methanol um und erhält das Produkt als farblosen Feststoff.
Ausbeute:
2.54 g (15.7 mmol) = 75 % d.Th.
Analysedaten:
siehe 10.85

Experimenteller Teil 10 Synthesen
300
10.87 H-Aeg(H)-OBn*2pTsOH (253)
Benzyl N-(2-Aminoethyl)glycinat Ditosylat
NH
O
O
NH2
*2pTsOH
Summenformel: C11H16N2O2*2pTsOH M = 552.66 g/mol
Ansatz:
7.56 g 64.0 mmol AEG (85)
29.4 g 154 mmol p-Toluolsulfonsäure
60 ml Benzylalkohol
500 ml Toluol
Durchführung:
Zu einer Suspension aus 7.56 g (64.0 mmol) AEG (85) in 500 ml Toluol werden 29.4 g
(154 mmol) p-Toluolsulfonsäure und 60 ml Benzylalkohol gegeben. Man erhitzt die Reakti-
onsmischung am Wasserabscheider für 6 h auf 140 °C und engt anschließend auf ca. 150 ml
ein. Nach Zugabe von 500 ml Diethylether lässt man über Nacht bei -20 °C stehen. Man saugt
den entstandenen Feststoff ab, wäscht dreimal mit Diethylether, trocknet im Ölpumpen-
vakuum und erhält so das Produkt als farblosen Feststoff.
Ausbeute:
32.2 g (54.7 mmol) = 86 % d.Th.
Analysedaten: 1H-NMR (200 MHz, D2O): δ = 7.71 (m, 4 H, pTsOHCHarom.); 7.39 (s, 5 H, BnCHarom.); 7.29
(m, 4 H, pTsOHCHarom.); 5.20 (s, 2 H, BnCH2); 4.06 (s, 2 H, GlyCH2); 3.52 (m, 4 H,
CH2CH2); 2.32 (s, 6 H, pTsOHCH3). 13C-NMR (50 MHz, D2O): δ = 166.61 (C=O); 142.32,
139.64 (2 × pTsOHCarom.); 134.43 (BnCarom.); 129.45, 128.93, 128.85, 128.49, 125.40
(3 × BnCHarom., 2 × pTsOHCHarom.); 68.52 (BnCH2); 47.71, 44.18 (H3N+CH2CH2, GlyCH2);
35.43 (H3N+CH2); 20.54 (pTsOHCH3). FAB-MS: 231 (8) [M+Na]+; 209.1 (100) [M+H]+;
119.1 (20) [M-Bn]+.

Experimenteller Teil 10 Synthesen
301
10.88 Boc-Aeg(H)-OBn (254)
Benzyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)glycinat
NH
O
O
NH
O
O
Summenformel: C16H24N2O4 M = 308.38 g/mol
Ansatz:
6.00 g 10.2 mmol H-Aeg(H)-OBn*2pTsOH (253)
2.22 g 10.2 mmol Boc2O
2.84 ml 20.4 mmol TEA
250 ml DCM
Durchführung:
Man legt 6.00 g (10.2 mmol) H-Aeg(H)-OBn*2pTsOH (253) in 250 ml DCM vor und kühlt
im Eisbad auf 0 °C ab. Nach der Zugabe von 2.84 ml (2.06 g, 20.4 mmol) TEA wird über
einen Zeitraum von 15 min eine Lösung von 2.22 g (10.2 mmol) Boc2O in 50 ml DCM hinzu
getropft. Man lässt über Nacht nachrühren, wäscht mit je 200 ml Wasser und ges. NaCl-
Lösung, trocknet über MgSO4 und entfernt das Lösungsmittel am Rotationsverdampfer. Nach
der Chromatographie des Rückstandes (Silica, EtOAc) erhält man das Produkt als farbloses
Öl.
Ausbeute:
1.52 g (4.94 mmol) = 48 % d.Th.
Analysedaten:
Rf (EtOAc): 0.26. 1H-NMR (200 MHz, DMSO-d6): δ = 7.37 (m, 5 H, BnCHarom.); 6.72
(m(br), 1 H, NH); 5.11 (s, 2 H, BnCH2); 3.37 (s, 2 H, GlyCH2); 2.97, 2.54 (je m, zusammen
4 H, CH2CH2); 1.36 (s, 9 H, BocCH3). FAB-MS: 331.2 (9) [M+Na]+; 309.2 (100) [M+H]+;
253.1 (60) [M-tBu]+.

Experimenteller Teil 10 Synthesen
302
10.89 Boc-Aeg(Fmoc)-OBn (255)
Benzyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat
NO
O
NH
O
OOO
Summenformel: C31H34N2O6 M = 530.63 g/mol
Ansatz:
1.49 g 4.83 mmol Boc-Aeg(H)-OBn (254)
1.25 g 4.83 mmol Fmoc-Cl
1.66 ml 9.66 mmol DIPEA
70 ml DCM
Durchführung:
Man legt 1.49 g (4.83 mmol) Boc-Aeg(H)-OBn (254) in 50 ml DCM vor und kühlt im Eisbad
auf 0 °C ab. Nach der Zugabe von 1.66 ml (1.23 g, 9.66 mmol) DIPEA wird über einen
Zeitraum von 20 min eine Lösung von 1.25 g (4.83 mmol) Fmoc-Cl in 20 ml DCM
hinzugetropft. Anschließend lässt man über Nacht bei RT rühren. Am darauf folgenden Tag
wäscht man mit 1 M KHSO4-, ges. NaHCO3- und NaCl-Lösung, trocknet über MgSO4 und
entfernt das Lösungsmittel destillativ am Rotationsverdampfer. Nach der Chromatographie
des Rückstandes (Silica, DCM/MeOH 50:1) erhält man das Produkt als farbloses Öl.
Ausbeute:
2.20 g (4.14 mmol) = 86 % d.Th.
Analysedaten:
siehe 10.90

Experimenteller Teil 10 Synthesen
303
10.90 Boc-Aeg(Fmoc)-OBn (255)
Benzyl N-(2-{[(tert-Butoxy)carbonyl]amino}ethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat
NO
O
NH
O
OOO
Summenformel: C31H34N2O6 M = 530.63 g/mol
Ansatz:
12.0 g 20.4 mmol H-Aeg(H)-OBn*2pTsOH (253)
4.45 g 20.4 mmol Boc2O
5.28 g 20.4 mmol Fmoc-Cl
10.5 ml 61.2 mmol DIPEA
250 ml DCM
Durchführung:
Man legt 12.0 g (20.4 mmol) H-Aeg(H)-OBn*2pTsOH (253) in 420 ml DCM vor und kühlt
im Eisbad auf 0 °C ab. Nach der Zugabe von 7.11 ml (5.28 g, 40.8 mmol) DIPEA wird über
einen Zeitraum von 20 min eine Lösung von 4.45 g (20.4 mmol) Boc2O in 120 ml DCM
hinzu getropft. Man lässt 1 h bei 0 °C und 4 h bei RT nachrühren und kühlt erneut auf 0 °C
ab. Anschließend werden 3.55 ml (2.63 g, 20.4 mmol) DIPEA und 5.28 g (20.4 mmol) Fmoc-
Cl hinzugegeben. Die Reaktionsmischung wird über Nacht bei RT gerührt. Man wäscht mit je
250 ml 1 M KHSO4-, ges. NaHCO3- und NaCl-Lösung, trocknet über MgSO4 und entfernt das
Lösungsmittel destillativ am Rotationsverdampfer. Nach der Chromatographie des Rückstan-
des (Silica, DCM/MeOH 50:1) erhält man das Produkt als farbloses Öl.
Ausbeute:
7.89 g (14.9 mmol) = 73 % d.Th.

Experimenteller Teil 10 Synthesen
304
Analysedaten:
Rf (DCM/MeOH 50:1): 0.41. 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.90 (d, 3J = 7.2, 1 H, FmocCHarom.); 7.86 (d, 3J = 7.2, 1 H, FmocCHarom.); 7.67 (d, 3J = 7.2, 1 H,
FmocCHarom.); 7.57 (d, 3J = 7.2, 1 H, FmocCHarom.); 7.33 (m, 9 H, 4 × FmocCHarom.,
5 × BnCHarom.); 6.71 (s(br), 1 H, NH); 5.15 (s, 1 H, BnCH2); 5.10 (s, 1 H, BnCH2); 4.12 (m,
5 H, FmocCH, FmocCH2, GlyCH2); 3.26, 3.06 (je m, zusammen 4 H, CH2CH2); 1.35 (s, 9 H,
BocCH3). FAB-MS: 553.3 (16) [M+Na]+; 531.3 (6) [M+H]+; 431.2 (57) [M-Boc]+; 178.1
(100).
10.91 H-Aeg(Fmoc)-OBn (256)
Benzyl N-(2-Aminoethyl)-N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinate Trifluoracetat
NO
O
NH2
OO
*TFA
Summenformel: C26H26N2O4*TFA M = 544.53 g/mol
Ansatz:
63.6 g 120 mmol Boc-Aeg(Fmoc)-OBn (255)
50 ml HSiEt3
100 ml TFA
100 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 15. Man versetzt das Rohprodukt mit Diethylether,
saugt den entstandenen Feststoff ab und wäscht mit Diethylether. Nach Trocknen im
Ölpumpenvakuum erhält man das Produkt als farblosen Feststoff.
Ausbeute:
56.5 g (104 mmol) = 86 % d.Th.

Experimenteller Teil 10 Synthesen
305
Analysedaten: 1H-NMR (200 MHz, DMSO-d6) (2 Rotamere): δ = 7.87 (m, 5 H, 2 × FmocCHarom.,
3 × H3N+); 7.61 (m, 2 H, FmocCHarom.); 7.35 (m, 9 H, 4 × FmocCHarom., 5 × BnCHarom.); 5.16
(s, 0.78 H, BnCH2); 5.12 (s, 1.22 H, BnCH2); 4.20 (m, 5 H, FmocCH, FmocCH2, GlyCH2);
3.49, 2.90 (je m, zusammen 4 H, CH2H2). 13C-NMR (50 MHz, DMSO-d6) (2 Rotamere):
δ = 169.65 (COOBn); 169.62 (COOBn); 155.68 (FmocC=O); 155.48 (FmocC=O); 143.72
(FmocCarom.); 143.62; (FmocCarom.); 140.72 (FmocCarom.); 135.68 (BnCarom.); 135.60
(BnCarom.); 128.43 (BnCHarom.); 128.16 (BnCHarom.); 127.92 (BnCHarom.); 127.74
(FmocCHarom.); 127.68 (FmocCHarom.); 127.19 (FmocCHarom.); 127.06 (FmocCHarom.); 125.02
(FmocCHarom.); 124.87 (FmocCHarom.); 120.19 (FmocCHarom.); 120.12 (FmocCHarom.); 67.62,
67.29, 66.23, 66.15 (FmocCH2, BnCH2); 49.41, 48.73, 46.40, 45.84, 37.47, 37.13 (FmocCH,
GlyCH2, CH2CH2). Das Signal für eine Methylengruppe von CH2CH2 wird bei ca. 40 ppm
vermutet und ist wahrscheinlich vom DMSO-Signal überdeckt. FAB-MS: 453.2 (10)
[M+Na]+; 431.2 (100) [M+H]+.
10.92 Boc-Aeg(Alloc)-Aeg(Fmoc)-OBn (257)
Benzyl N-(2-{[N-(2-{(tert-Butoxy)carbonyl]amino}ethyl)-N-[(prop-2-en-1-yloxy)carbonyl]glycyl]amino}ethyl)-
N-{[(9H-fluoren-9-yl)methoxy]carbonyl}glycinat
OOO
NH
NNH
NO
OOOO
O
Summenformel: C39H46N4O9 M = 714.82 g/mol
Ansatz:
13.7 g 45.3 mmol Boc-Aeg(Alloc)-OH (228)
24.7 g 45.3 mmol H-Aeg(Fmoc)-OBn*TFA (256)
17.2 g 45.3 mmol HBTU
15.8 ml 90.6 mmol DIPEA
90 ml DMF

Experimenteller Teil 10 Synthesen
306
Durchführung:
Die Durchführung erfolgt gemäß AAV 17. Man erhält das Produkt nach Säulenchromatogra-
phie (Silica, CH/EtOAc 1:2 → 1:5) als farblosen Schaum.
Ausbeute:
23.7 g (32.7 mmol) = 73 % d.Th.
Analysedaten:
Rf (EtOAc): 0.37. 1H-NMR (250 MHz, DMSO-d6, 100 °C): δ = 7.85 (d, 3J = 7.3, 2 H,
FmocCHarom.); 7.61 (d, 3J = 7.5, 2 H, FmocCHarom.); 7.56 (t(br), 1 H, NH); 7.43 - 7.27 (m, 9 H,
4 × FmocCHarom., 5 × BnCHarom.); 6.35 (t(br), 1 H, NH); 5.89 (ddt, 3Jtrans = 17, 3Jcis = 11, 3J = 5.2, 1 H, CH2CH=CHcisHtrans); 5.26 (ddt aa dq, 4J = 1.6, 3Jcis = 17, 2J = 1.6, 1 H,
CH2CH=CHcisHtrans); 5.14 (ddt aa dq, 4J = 1.6, 3Jtrans = 11, 2J = 1.6, 1 H, CH2CH=CHcisHtrans);
5.14 (s, 2 H, BnCH2); 4.52 (dt, 4J = 1.6, 3J = 5.2, 2 H, CH2CH=CH2); 4.36 (d, 3J = 6.3, 2 H,
FmocCH2); 4.23 (t(br), 3J = 6.3, 1 H, FmocCH); 4.06 (s, 2 H, GlyCH2); 3.82 (s, 2 H, GlyCH2);
3.42 - 3.06 (m, 8 H, 2 × CH2CH2); 1.38 (s, 9 H, BocCH3). 13C-NMR (63 MHz, DMSO-d6,
100 °C): δ = 168.86 (GlyC=O); 168.33 (GlyC=O); 155.03 (FmocC=O, AllocC=O); 143.32
(FmocCarom.); 140.29 (FmocCarom.); 135.30 (BnCarom.); 132.78 (CH2CH=CH2); 127.78
(FmocCHarom.); 127.42 (FmocCHarom.); 127.17 (Bn-o-CHarom.); 127.02 (Bn-m-CHarom.); 126.48
(Bn-p-CHarom.); 124.30 (FmocCHarom.); 119.38 (FmocCHarom.); 116.08 (CH2CH=CH2); 77.24
(C(CH3)3); 66.71, 65.57, 64.79 (CH2CH=CH2, BnCH2, FmocCH2); 50.03, 48.89, 47.66, 46.40,
38.19 (FmocCH, 2 × GlyCH2, 2 × CH2CH2); 27.72 (BocCH3). Die Signale der übrigen CH2-
Gruppen befinden sich im Bereich 50.84 bis 35.43 ppm, können jedoch unter den gegebenen
Messbedingungen (93 mM Probe, 734 Scans) nicht sinnvoll von der Basislinie unterschieden
werden. FAB-MS: 737 (100) [M+Na]+; 715.4 (13) [M+H]+; 615.3 (78) [M-Boc]+.

Experimenteller Teil 10 Synthesen
307
10.93 H-Aeg(Alloc)-Aeg(Fmoc)-OBn*TFA (258)
Benzyl N-[2-({N-(2-Aminoethyl)-N-[(prop-2-en-1-yloxy)-carbonyl]glycyl}amino)ethyl]-N-{[(9H-fluoren-9-yl)-
methoxy]carbonyl}glycinat Trifluoracetat
OO
NH2
NNH
NO
OOOO
*TFA
Summenformel: C34H38N4O7*TFA M = 728.72 g/mol
Ansatz:
12.8 g 17.9 mmol Boc-Aeg(Alloc)-Aeg(Fmoc)-OBn (257)
15 ml HSiEt3
30 ml TFA
30 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 15. Nach Trocknen im Ölpumpenvakuum erhält man
das Produkt als leicht gelbliches, hochviskoses Öl.
Ausbeute:
13.0 g (17.9 mmol) = 100 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 100 °C) (Rotamere): δ = 7.85 (m, 3 H, 2 × FmocCHarom.,
NH); 7.60 (m, 2 H, FmocCHarom.); 7.35 (m, 9 H, 4 × FmocCHarom., 5 × BnCHarom.); 5.88 (m,
2 H, CH2CH=CHcisHtrans, NH); 5.22 (m, 2 H, CH2CH=CH2); 5.14 (s, 2 H, BnCH2); 4.54 (m,
2 H, CH2CH=CH2); 4.37 (m, 2 H, FmocCH2); 4.24 (m, 1 H, FmocCH); 4.06, 3.96 (je s,
zusammen 2 H, GlyCH2); 3.91, 3.90 (je s, zusammen 2 H, GlyCH2); 3.55 (t(br), 3J = 6.1, 2 H),
3.30 (m, 4 H), 3.05 (t(br), 3J = 6.1, 2 H) (2 × CH2CH2). FAB-MS: 637.3 (20) [M+Na]+; 615.3
(90) [M+H]+; 154 (100). HR-ESI-MS: 617.2866 (7) [M+3H]+ (ber.: 617.2975); 616.2837
(36) [M+2H]+ (ber.: 616.2897); 615.2803 (100) [M+H]+ (ber.: 615.2819).
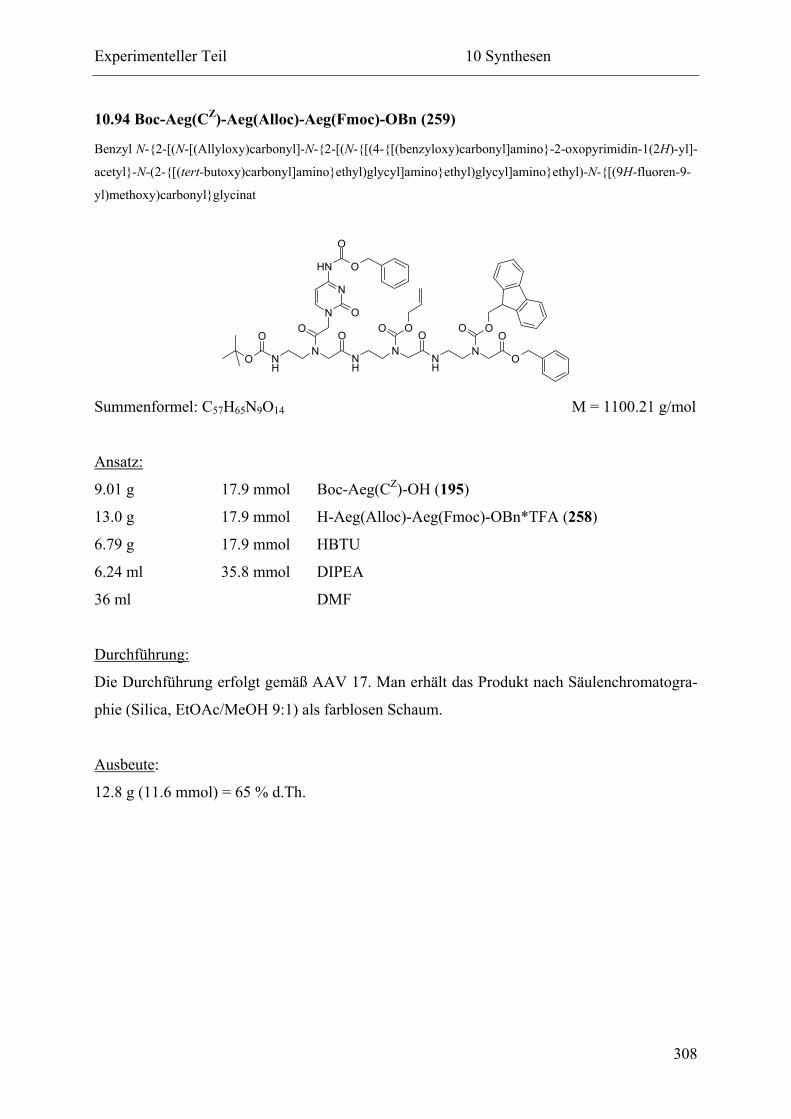
Experimenteller Teil 10 Synthesen
308
10.94 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(Fmoc)-OBn (259)
Benzyl N-{2-[(N-[(Allyloxy)carbonyl]-N-{2-[(N-{[(4-{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]-
acetyl}-N-(2-{[(tert-butoxy)carbonyl]amino}ethyl)glycyl]amino}ethyl)glycyl]amino}ethyl)-N-{[(9H-fluoren-9-
yl)methoxy)carbonyl}glycinat
N
O
NH
OO
NNH
NO
OOOOO
N
NH
O
O
N
O
NH
O
O
Summenformel: C57H65N9O14 M = 1100.21 g/mol
Ansatz:
9.01 g 17.9 mmol Boc-Aeg(CZ)-OH (195)
13.0 g 17.9 mmol H-Aeg(Alloc)-Aeg(Fmoc)-OBn*TFA (258)
6.79 g 17.9 mmol HBTU
6.24 ml 35.8 mmol DIPEA
36 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 17. Man erhält das Produkt nach Säulenchromatogra-
phie (Silica, EtOAc/MeOH 9:1) als farblosen Schaum.
Ausbeute:
12.8 g (11.6 mmol) = 65 % d.Th.

Experimenteller Teil 10 Synthesen
309
Analysedaten:
Rf (EtOAc/MeOH 9:1): 0.15. 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 7.78 (m, 4 H,
C-C(6)H, FmocCHarom., NH); 7.60 (d, 3J = 7.4, 2 H, FmocCHarom.); 7.40 (tr(br), 3J = 5.1, 1 H,
NH); 7.42 - 7.25 (m, 14 H, 5 × ZCHarom., 5 × BnCHarom., 4 × FmocCHarom.); 6.92 (d, 3J = 7.3,
1 H, C-C(5)H); 6.37 (s(br), 1 H, NH); 5.89 (ddt, 3J = 5.2, 3Jtrans = 17, 3Jcis = 11, 1 H,
CH2CH=CHcisHtrans); 5.27 (ddt aa dq, 4J = 1.6, 3Jcis = 17, 2J = 1.6, 1 H, CH2CH=CHcisHtrans);
5.14 (ddt aa dq, 4J = 1.6, 3Jtrans = 11, 2J = 1.6, 1 H, CH2CH=CHcisHtrans); 5.21 (s, 2 H,
OCH2Ph); 5.14 (s, 2 H, OCH2Ph); 4.70 (s, 2 H, C-N(1)CH2); 4.51 (dt, 4J = 1.6, 3J = 5.2, 2 H,
CH2CH=CH2); 4.36 (d, 3J = 6.0, 2 H, FmocCH2); 4.22 (t(br), 3J = 6.0, 1 H, FmocCH); 4.06 (s,
2 H, GlyCH2); 3.98 (s(br), 2 H, GlyCH2); 3.84 (s, 2 H, GlyCH2); 3.46 - 3.16 (m, 12 H,
3 × CH2CH2); 1.40 (s, 9 H, BocCH3). FAB-MS: 1122.6 (40) [M+Na]+; 91.1 (100).
10.95 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(H)-OBn (260)
Benzyl N-({[2-[(N-[(Allyloxy)carbonyl]-N-(2-{[(N-(4-{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-
yl)acetyl]-N-(2-{[(tert-butoxy)carbonyl]amino}ethyl)glycyl]amino}ethyl)glycyl]amino}ethyl)glycinat
N
O
NH
OO
NNH
NH
O
OOO
N
NH
O
O
N
O
NH
O
O
Summenformel: C42H55N9O12 M = 877.96 g/mol
Ansatz:
7.40 g 6.72 mmol Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(Fmoc)-OBn (259)
10.4 ml 101 mmol Diethylamin
45 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 19. Anschließend versetzt man mit 50 ml Ethylacetat
und 200 ml Diethylether und rührt für 10 min. Der entstandene farblose Feststoff wird

Experimenteller Teil 10 Synthesen
310
abgesaugt, mit Diethylether gewaschen und im Vakuum getrocknet. Man erhält das Produkt
in Form eines farblosen Feststoffs.
Ausbeute:
5.06 g (5.77 mmol) = 86 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 7.82 (d, 3J = 7.3, 2 H, C-C(6)H); darunter
(m(br), 1 H, NH); 7.52 (t(br), 1 H, NH); 7.40 (m, 10 H, 5 × ZCHarom., 5 × BnCHarom.); 6.93 (d, 3J = 7.3, 1 H, C-C(5)H); 6.39 (s(br), 1 H, NH); 5.90 (ddt, 3J = 5.2, 3Jtrans = 17, 3Jcis = 11, 1 H,
CH2CH=CHcisHtrans); 5.33 - 5.12 (m, 2 H, CH2CH=CH2); 5.21 (s, 2 H, OCH2Ph); 5.15 (s, 2 H,
OCH2Ph); 4.71 (s, 2 H, C-N(1)CH2); 4.52 (dt, 4J = 1.6, 3J = 5.2, 2 H, CH2CH=CH2); 3.98 (s,
2 H, GlyCH2); 3.86 (s, 2 H, GlyCH2); 3.51 - 3.15 (m, 10 H, 2.5 × CH2CH2); 3.41 (s, 2 H,
GlyCH2); 2.68 (t, 3J = 6.3, 2 H, 0.5 × CH2CH2); 1.41 (s, 9 H, BocCH3). MALDI-MS: 878.940
(128) [M+H]+. HR-ESI-MS: 880.4082 (13) [M+3H]+ (ber.: 880.4205); 879.4054 (48)
[M+2H]+ (ber.: 879.4126); 878.4023 (100) [M+H]+ (ber.: 878.4048).
10.96 Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(GZ)-OBn (261)
Benzyl N-[(Allyloxy)carbonyl]-N-[(2-{[(benzyloxy)-carbonyl]amino}-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-
N-(2-{[N-({2-[(N-{[4-{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]acetyl}-N-(2-{[(tert-butoxy)-
carbonyl]amino}ethyl)glycyl]amino}ethyl)glycyl]amino}ethyl)glycinate
N N
NNH
O
NH
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH
O
O
OO
O
OO
O
O
Summenformel: C57H66N14O16 M = 1203.25 g/mol

Experimenteller Teil 10 Synthesen
311
Ansatz:
4.84 g 5.51 mmol Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(H)-OBn (260)
2.12 g 6.17 mmol GZ-AcOH (199)
2.39 g 876 µmol Brop
1.70 ml 3.15 mmol TEA
30 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 20. Die Reaktionsmischung wird mit 300 ml Ethylace-
tat verdünnt, mit je 250 ml 1 M KHSO4- ges. NaHCO3- und NaCl-Lösung. gewaschen und
über MgSO4 getrocknet. Man entfernt das Lösungsmittel am Rotationsverdampfer, chromato-
graphiert den Rückstand (Silica, EtOAc/MeOH 4:1) und erhält das Produkt als farblosen
Schaum.
Ausbeute:
4.80 g (3.99 mmol) = 72 % d.Th.
Analysedaten:
Rf (EtOAc/MeOH 4:1): 0.08. 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere):
δ = 10.76 (s(br), 2 H, 2 × ZNH); 7.80 (m, 4 H, C-C(6)H, G-C(8)H, 2 × NH); 7.37 (m, 15 H,
10 × ZCHarom., 5 × BnCHarom.); 6.93 (m, 1 H, C-C(5)H); 6.39 (s(br), 1 H, NH); 5.88 (m, 1 H,
CH2CH=CHcisHtrans); 5.31 - 5.03 (m, 6 H, CH2CH=CH2, OCH2Ph, G-N(9)CH2); 5.27 (s, 2 H,
OCH2Ph); 5.21 (s, 2 H, OCH2Ph); 4.71 (s, 2 H, C-N(1)CH2); 4.50 (m, 2 H, CH2CH=CH2);
4.24 (s, 2 H, GlyCH2); 3.98 (s, 2 H, GlyCH2); 3.89 (s, 2 H, GlyCH2); 3.55 - 3.19 (m, 12 H,
3 × CH2CH2); 1.40 (s, 9 H, BocCH3). FAB-MS: 1203.5 (8) [M+H]+; 91.1 (100).

Experimenteller Teil 10 Synthesen
312
10.97 Boc-Aeg(CZ)-Aeg(H)-Aeg(GZ)-OBn (262)
Benzyl N-[(2-{[(Benzyloxy)carbonyl]amino}-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-N-(2-{[N-(2-[(N-{[4-
{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]acetyl}-N-{2-{[(tert-butoxy)carbony]amino}ethyl)-
glycyl]amino}ethyl)glycyl]amino}ethyl)glycinate
N N
NNH
O
NH
NH
N
O
NH
NH
NH
NO
OOO
N
N
O
NH
O
O
OO
O
O
O
Summenformel: C53H62N14O14 M = 1119.17 g/mol
Ansatz:
5.17 g 4.30 mmol Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(GZ)-OBn (261)
991 mg 858 µmol Pd[P(Ph)3]4
15.4 g 98.8 mmol N,N’-Dimethylbarbitursäure (267)
200 ml THFabs.
Durchführung:
Zu einer Lösung von 5.17 g (4.30 mmol) Boc-Aeg(CZ)-Aeg(Alloc)-Aeg(GZ)-OBn (261) in
200 ml THFabs. werden nacheinander 15.4 g (98.8 mmol) N,N’-Dimethylbarbitursäure (267)
und 991 mg (858 µmol) Pd[P(Ph)3]4 gegeben. Man lässt 2 h bei RT rühren, entfernt das
Lösungsmittel destillativ am Rotationsverdampfer und chromatographiert den Rückstand
(Silica, EtOAc/MeOH 2:1 → 1:1). Das Produkt wird als farbloser Schaum erhalten.
Ausbeute:
4.21 g (3.76 mmol) = 88 % d.Th.

Experimenteller Teil 10 Synthesen
313
Analysedaten:
Rf (EtOAc/MeOH): 0.28. 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere): δ = 7.77
(m, 4 H, C-C(6)H, G-C(8)H, 2 × NH); 7.33 (15 H, 10 × ZCHarom., 5 × BnCHarom.); 6.91 (m,
1 H, C-C(5)H); 6.39 (s(br), 1 H, NH); 5.88 (m, 1 H, CH2CH=CHcisHtrans); 5.31 - 5.03 (m, 6 H,
CH2CH=CH2, OCH2Ph, G-N(9)CH2); 5.27 (s, 2 H, OCH2Ph); 5.21 (s, 2 H, OCH2Ph); 4.71 (s,
2 H, C-N(1)CH2); 4.50 (m, 2 H, CH2CH=CH2); 4.24 (s, 2 H, GlyCH2); 3.98 (s, 2 H, GlyCH2);
3.89 (s, 2 H, GlyCH2); 3.55 - 3.19 (m, 12 H, 3 × CH2CH2); 1.40 (s, 9 H, BocCH3). MALDI-
MS: 1143.260 (317) [M+Na]+; 1120.603 (1053) [M+2H]+. HR-ESI-MS: 1122.4695 (5)
[M+4H]+ (ber.: 1122.4883); 1121.46703 (22) [M+3H]+ (ber.: 1121.4804); 1120.4646 (61)
[M+2H]+ (ber.: 1120.4726); 1119.4620 (100) [M+H]+ (ber.: 1119.4648).
10.98 Boc-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn (263)
Benzyl N-[(2-{[(Benzyloxy)carbonyl]amino}-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-N-[2-({[(N-{2-[(N-{[4-
{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]acetyl}-N-(2-{[(tert-butoxy)carbonyl]amino}ethyl)-
glycyl]amino}ethyl]-N-[(2,4-difluor-5-methylphenyl)acetyl]glycyl}amino)ethyl]glycinat
N N
NNH
O
NH
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH
O
O
OO
O
F
F
O
O
O
Summenformel: C62H68F2N14O15 M = 1287.31 g/mol
Ansatz:
1.50 g 1.34 mmol Boc-Aeg(CZ)-Aeg(H)-Aeg(GZ)-OBn (262)
300 mg 1.61 mmol F-AcOH (70)
625 mg 1.61 mmol Brop
448 µl 3.15 mmol TEA
20 ml DMF

Experimenteller Teil 10 Synthesen
314
Durchführung:
Die Durchführung erfolgt gemäß AAV 20. Die Reaktionsmischung wird mit 200 ml
Ethylacetet verdünnt, mit je 200 ml 1 M KHSO4- ges. NaHCO3- und NaCl-Lösung gewaschen
und über MgSO4 getrocknet. Man entfernt das Lösungsmittel am Rotationsverdampfer,
chromatographiert den verbliebenen Rückstand (Silica, EtOAc/MeOH 2:1) und erhält das
Produkt als farblosen Schaum.
Ausbeute:
1.29 g (1.01 mmol) = 75 % d.Th.
Analysedaten:
Rf (EtOAc/MeOH 2:1): 0.24. 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere):
δ = 7.97 - 7.72 (m, 5 H), 7.47 - 7.10 (m, 15 H), 6.92 - 6.81 (m, 2 H) (C-C(6)H, C-C(5)H,
G-C(8)H, F-C(6)H, F-C(3)H, 10 × ZCHarom., 5 × BnCHarom., 2 × NH); 6.42 (s(br), 1 H, NH);
5.25, 5.20, 5.17, 5.03, 4.92 (je s, teilweise br, zusammen 6 H, 3 × OCH2Ph); 4.73 (m, 2 H,
BaseCH2); 4.10 (m, 4 H, 2 × BaseCH2); 3.65 - 2.76 (m, 18 H, 3 × GlyCH2, 3 × CH2CH2); 1.98
(s(br), 3 H, FCH3); 1.39 (m, 9 H, BocCH3). 19F-NMR (235 MHz, DMSO-d6) (Rotamere):
δ = -119.63, -120.14 (je m, zusammen 2 F, F(2), F(4)). FAB-MS: 1309.3 (100) [M+Na]+.

Experimenteller Teil 10 Synthesen
315
10.99 H-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn*TFA (264)
Benzyl N-[2-({N-(2-{[N-(2-Aminoethyl)-N-{[4-{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]-ace-
tyl}glycyl]amino}ethyl)-N-[(2,4-difluor-5-methylphenyl)acetyl]glycyl}amino)ethyl]-N-[(2-{[(benzyloxy)-
carbonyl]amino}-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]glycinat
N N
NNH
O
NH
NH2
N
O
NH
NNH
NO
OOO
N
N
O
NH
O
O
OO
O
F
F
O
*TFA
Summenformel: C57H60F2N14O13*TFA M = 1301.22 g/mol
Ansatz:
1.43 g 1.11 mmol Boc-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn (263)
2.50 ml 15.7 mmol HSiEt3
5 ml TFA
5 ml DCM
Durchführung:
Die Durchführung erfolgt gemäß AAV 15. Man versetzt das Rohprodukt mit Diethylether,
saugt den entstandenen Feststoff ab und wäscht mit Diethylether. Nach Trocknen im
Ölpumpenvakuum erhält man das Produkt als farblosen Feststoff.
Ausbeute:
1.27 g (975 µmol) = 88 % d.Th.

Experimenteller Teil 10 Synthesen
316
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 110 °C) (Rotamere): δ = 8.08 (s(br), 1 H, NH); 7.95 - 7.68
(m, 4 H), 7.40 - 7.34 (m, 15 H), 7.11 (s(br), 1 H); 6.93 - 6.82 (m, 2 H) (C-C(6)H, C-C(5)H,
G-C(8)H, F-C(6)H, F-C(3)H, 10 × ZCHarom., 5 × BnCHarom., NH); 6.42 (s(br), 1 H, NH); 5.27,
5.21, 5.18, 5.02 (je s, teilweise br, zusammen 6 H, 3 × OCH2Ph); 4.80 - 4.60 (m, 2 H,
BaseCH2); 4.26 (s(br), 2 H), 4.08 (m(br), 4 H); 3.64 - 3.09 (m(br), 14 H) (2 × BaseCH2,
3 × GlyCH2, 3 × CH2CH2); 1.98 (s(br), 3 H, FCH3). 19F-NMR (235 MHz, DMSO-d6)
(Rotamere): δ = -77.01 (s, 3 F, F3CCOOH); -119.40, -120.12 (je m, zusammen 2 F, F(2),
F(4)). MALDI-MS: 1188.331 (70) [M+2H]+. HR-ESI-MS: 1190.4525 (8) [M+4H]+ (ber.:
1190.4745); 1189.4532 (24) [M+3H]+ (ber.: 1189.4667); 1188.4504 (67) [M+2H]+ (ber.:
1188.4589); 1187.4474 (100) [M+H]+ (ber.: 1187.4510).
10.100 Me2-Aeg(Me)-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn*2TFA (265)
Benzyl N-[(2-{[(Benzyloxy)carbonyl]amino}-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-N-{2-[(N-{2-[(N-{[4-
{[(benzyloxy)carbonyl]amino}-2-oxopyrimidin-1(2H)-yl]acetyl}-N-[2-({N-[2-(dimethylamino)ethyl]-N-methyl-
glycyl}amino)ethyl]glycyl)-N-[(2,4-difluor-5-methylphenyl)acetyl]glycyl)amino]ethyl}glycinat
N N
NNH
O
NH
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH
O
O
O
F
F
OO
NN
O
O
*2TFA
Summenformel: C64H74F2N16O14*2TFA M = 1557.44 g/mol

Experimenteller Teil 10 Synthesen
317
Ansatz:
600 mg 461 µmol H-Aeg(CZ)-Aeg(F)-Aeg(GZ)-OBn*TFA (264)
359 mg 1.54 mmol Me2-Aeg(Me)-OH*2HCl (249)
266 mg 2.31 mmol HOSu
348 mg 1.69 mmol DCC
494 µl 3.54 mmol TEA
10 ml DMF
Durchführung:
Die Durchführung erfolgt gemäß AAV 18. Nach Reinigung durch präparative RP-HPLC und
anschließender Gefriertrocknung erhält man die Zielverbindung als farbloses Pulver.
Ausbeute:
305 mg (214 mmol) = 46 % d.Th.
Analysedaten: 1H-NMR (200 MHz, DMSO-d6, 110 °C): δ = 8.62 (s(br), div. N+R3); 8.08 (s(br), 1 H, NH);
7.76 (m, 5 H, C-C(6)H, FCHarom., G-C(8)H, 2 × NH); 7.41 - 7.34 (m, 15 H, 10 × ZCHarom.,
5 × BnCHarom.); 7.12 (t(br), 1 H, NH); 6.89 (m, 2 H, C-C(5)H, FCHarom.); 5.27 (s, 2 H,
OCH2Ph); 5.21 (s, 2 H, OCH2Ph); 5.18 (s(br), 2 H, OCH2Ph); 5.02 (s(br), 2 H, BaseCH2);
4.69 (s(br), 2 H, BaseCH2); 4.24 (s(br), 2 H, BaseCH2); 4.02 (s(br), 4 H, 2 × GlyCH2);
3.63 - 3.20 (m(br), 18 H, 2 × GlyCH2, 3.5 × CH2CH2); darunter das Signal für Wasserreste
aus dem LM; 2.90 (t, 3J = 6.0, 2 H, 0.5 × CH2CH2); 2.84 (s, 6 H, N(CH3)3); 2.40 (s, 3 H,
NCH3); 2.12 (s(br), 3 H, FCH3). 19F-NMR (235 MHz, DMSO-d6) (Rotamere): δ = -77.83
(s, 6 F, F3CCOOH); -119.39, -120.09 (je m, zusammen 2 F, F(2), F(4)). MALDI-MS:
1330.842 (577) [M+2H]+. HR-ESI-MS: 1332.5663 (8) [M+4H]+ (ber.: 1332.5851);
1331.5630 (24) [M+3H]+ (ber.: 1331.5773); 1330.5610 (73) [M+2H]+ (ber.: 1330.5695);
1329.5583 (100) [M+H]+ (ber.: 1329.5616).

Experimenteller Teil 10 Synthesen
318
10.101 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-Aeg(C)-Aeg(A)-Aeg(G)-Aem*6TFA (268)
N
N
O
NH2
N
O
NH
O
NH
NNH
NNH
OOO
N N
NNH
O
NH2
OO
NN
F
F N
N
O
NH2
N
O
N N
NN
NH2
O
NH
NNH
NNH
OO
N
OO
N N
NNH
O
NH2
O
*6TFA
Summenformel: C79H107F2N37O18*6TFA M = 2585.09 g/mol
Ansatz:
6.57 mg 5.00 µmol Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OH*3TFA (248)
7.02 mg 5.00 µmol H-Aeg(C)-Aeg(A)-Aeg(G)-Aem*4TFA (224)
0.5 ml 0.8 M 1-Methylimidazol-Puffer pH 7.5
0.5 ml 0.4 M EDC*HCl
Durchführung:
Die beiden PNA-Trimere 224 (7.02 mg, 5.00 µmol) und 248 (6.57 mg, 5.00 µmol) werden in
0.5 ml 1-Methylimidazol-Puffer (0.8 M, pH 7.5) gelöst, mit dem gleichen Volumen einer
0.4 M wässrigen Lösung von EDC*HCl versetzt und dann 4 d bei RT geschüttelt. Nach
Reinigung durch präparative RP-HPLC und anschließender Gefriertrocknung erhält man die
Zielverbindung als farbloses Pulver.
Ausbeute:
7.11 mg (2.75 mmol) = 55 % d.Th.
Analysedaten:
MALDI-MS: 1903.197 (95) [M+3H]+. HR-ESI-MS: 1904.8704 (5) [M+5H]+ (ber.:
1904.8954); 1903.8673 (15) [M+4H]+ (ber.: 1903.8875); 1902.8653 (41) [M+3H]+ (ber.:
1902.8797); 1901.8629 (95) [M+2H]+ (ber.: 1901.8719); 1900.8602 (100) [M+H]+ (ber.:
1900.8641).

Experimenteller Teil 10 Synthesen
319
10.102 Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OMe*3TFA (291)
Methyl N-[(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)acetyl]-N-[2-({N-[2-({N-[(4-amino-2-oxopyrimidin-
1(2H)-yl)acetyl]-N-[2-({N-[2-(dimethylamino)ethyl]-N-methylglycyl}amino)ethyl]glycyl}amino)ethyl]-N-[(2,4-
difluor-5-methylphenyl)acetyl]glycyl}amino)ethyl]glycinat
N N
NNH
O
NH2
NH
N
O
NH
NNH
NO
OOO
N
N
O
NH2
O
F
F
OO
NN
*3TFA
Summenformel: C42H58F2N16O10*3TFA M = 1327.09 g/mol
Ansatz:
2.00 mg 1.52 µmol Me2-Aeg(Me)-Aeg(C)-Aeg(F)-Aeg(G)-OH*3TFA (248)
0.64 mg 1.7 µmol HATU
2.65 µl 15.2 µmol DIPEA
100 µl 2.53 mmol MeOH
304 µl DMFabs.
Durchführung:
2.00 mg (1.52 µmol) PNA-Trimer 248 werden mit einer Lösung von 0.64 mg (1.1 eq.) HATU
und 2.65 µl (1.96 mg, 10 eq.) DIPEA in 304 µl DMFabs. versetzt. Man lässt 15 min bei RT
schütteln, gibt 100 µl (2.53 mmol) Methanol hinzu und lässt die Reaktionsmischung über
Nacht bei RT stehen. Anschließend werden die flüchtigen Komponenten im Vakuum entfernt
(Speedvac) und der Rückstand mittels präparativer RP-HPLC gereinigt. Nach Gefriertrock-
nung erhält man die Zielverbindung als farbloses Pulver.
Ausbeute:
0.31 mg (0.23 µmol) = 15 % d.Th.
Analysedaten:
MALDI-MS: 987.495 (317) [M+2H]+.

Experimenteller Teil 11 Ligation und Replikation
320
11 Ligations- und Replikationsexperimente
Die Ligationsexperimente wurden in Reagiergefäßen (500 µl, Polypropylen) der Fa. Sarstedt
durchgeführt, die in einem Thermocycler der Fa. MJ Research (MiniCycler™ PTC-150)
temperiert wurden. Die Reaktionslösungen wurden jeweils aus einer 10 mM Stammlösung der
beiden Trimere 224 und 248 und dem gleichen Volumen einer frisch hergestellten Lösung
von EDC, Katalysator und Salz im entsprechenden Puffer angesetzt. Anschließend wurde die
Lösung gleichmäßig auf die entsprechende Anzahl Reagiergefäße aufgeteilt, die gegebenfalls
gefriergetrocknete hexa-PNA 268 enthielten. Aus diesen Ansätzen wurden in zeitlichen
Abständen je 0.5 µl entnommen und um den Faktor 100 mit einer Lösung von 5 % Acetonitril
und 0.1 % TFA in Wasser verdünnt. Die erhaltenen Proben wurden direkt mittels HPLC
analysiert oder zunächst bei RT gelagert. Die Lagerung hatte auch nach mehreren Tagen
keinen Einfluss auf die Probenzusammensetzung.

Anhang 12 Nomenklatur
I
Anhang
12 Zur Nomenklatur der hier beschriebenen Verbindungen
Die strikte Anwendung der IUPAC-Regeln führt bei vielen der größeren hier beschriebenen
Verbindungen zu sehr unübersichtlichen Namensgebilden, die damit wenig aussagekräftig
sind. Aus diesem Grund wurde hier eine Nomenklatur benutzt, die sich an die Schreibweise
von Peptidsequenzen anlehnt. Dabei bildet Aeg den Grundkörper, an dessen N-Terminus
(links) oder C-Terminus (rechts) sich weitere Aeg-Einheiten, Schutzgruppen oder Endmodifi-
zierungen befinden. Substituenten, die an das interne sekundäre Amin binden, werden wie die
Schutzgruppe einer Seitenkette notiert. PNA-Oligomere werden als poly-PNAs (z.B.
tri-PNAs) bezeichnet; handelt es sich bei den Substituenten am sekundären Amin nicht um
Nukleobasen, sondern um Schutzgruppen, wird von poly-Aegs (z.B. di-AEGs) gesprochen.
Nukleobasen und Isostere die an N1 (Pyrimidine), N9 (Purine) oder C1 mit einem Essigsäure-
Linker versehen sind, werden als Nukleobasen- bzw. Isosteren-essigsäuren bezeichnet; sind
die exocyclischen Aminofunktionen geschützt, werden sie geschützte Nukleobasen-
essigsäuren genannt. Ist die Carboxylfunktion der Essigsäure verestert, wird von
Nukleobasen-estern gesprochen.

Anhang 13 Abkürzungsverzeichnis
I
13 Abkürzungsverzeichnis
A bzw. a Adenin
aa analysiert als
abs. absolutiert
Ac Acetyl
Aeg bzw. AEG N-(2-Aminoethyl)glycin
Aem 2-Morpholinoethylamin
Alloc Allyloxycarbonyl
aq. wässrig
arom. aromatisch
B Nukleobase
Bhoc Benzhydryoxycarbonyl
ber. berechnet
Bn Benzyl
Boc tert-Butyloxycarbonyl
Brop Bromotris(dimethylamino)phosphonium-hexafluorphosphat
C bzw. c Cytosin
CDI 1,1’-Carbonyldiimidazol
CH Cyclohexan
CHCA α-Cyano-4-hydroxyzimtsäure
CMC N-Cyclohexyl-N′-(ß-[N-methylmorpholino]ethyl)carbodiimid Tosylat
d Dublett
DBU 1,8-Diazabicyclo[5.4.0]-7-undecen
DC Dünnschichtchromatographie
DCC N,N’-Dicyclohexylcarbodiimid
DCM Dichlormethan
DEA Diethylamin
DHB 2,5-Dihydroxybenzoesäure
DIPEA N,N-Diisopropylethylamin
DMAP 4-Dimethylaminopyridin
DMF N,N-Dimethylformamid

Anhang 13 Abkürzungsverzeichnis
II
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
d.Th. der Theorie
ED Ethylendiamin
EI Elektronenstoß-Ionisation
eq. Äquivalent
ESI Elektrospray-Ionisation
Et Ethyl
EDC 1-Ethyl-3-(3’-dimethylaminopropyl)carbodiimid Hydrochlorid
EtOAc Essigsäureethylester
F 2,4-Difluortoluol
FAB fast atom bombardement
Fmoc 9-Fluorenylmethoxycarbonyl
G bzw. g Guanin
ges. gesättigt
Gly Glycin
HATU 1-[Bis(dimethylamino)methylen]-1H-1,2,3-triazolo-[4,5-b]pyridinium-
hexafluorphosphat-3-oxid (N-HATU)
HBTU 1-[Bis(dimethylamino)methylen]-1H-benzotriazolium-hexafluorphosphat-
3-oxid (N-HBTU)
HEPES 2-(4-(2-Hydroxyethyl)- 1-piperazinyl)ethansulfonsäure
HMBA 4-Hydroxymethylbenzoesäure
HOAt 1-Hydroxy-7-azabenzotriazol
HOBt 1-Hydroxybenzotriazol
HOSu N-Hydroxysuccinimid
HPLC high performance liquid chromatographie
HR high resolution
Hz Hertz
konz. konzentriert
LP linear positive
Lsg. Lösung
m Multiplett
M molar

Anhang 13 Abkürzungsverzeichnis
III
MALDI matrix-assisted laser desorption ionization
Me Methyl
MeOH Methanol
MeCN Acetonitril
MES 2-(N-Morpholino)ethansulfonsäure
MHz Megahertz
min Minuten
Mmt 4-Monomethoxytrityl
MOPS 3-(N-Morpholino)propansulfonsäure
MS Massenspektrometrie
MSNT 1-(2-Mesitylensulfonyl)-3-nitro-1H-1,2,4-triazol
NDMBA N,N’-Dimethylbarbitursäure
NMM N-Methylmorpholin
NMP N-Methyl-2-pyrrolidon
NMR Kernresonanzspektroskopie (nucleic magnetic resonance)
NP normal phase
PEG Polyethylenglykol
PG protective group
Ph Phenyl
PNA Peptide Nucleic Acid / Polyamide Nucleic Acid
ppm parts per million
pTsOH p-Toluolsulfonsäure
q Quartett
R2 Bestimmtheitsmaß
RMS root mean square
RNA Ribonukleinsäure
RP reversed phase
RT Raumtemperatur
s Singulett / Sekunden
SC Säulenchromatographie
SD Standardabweichung (standard deviation)
SP solid phase
SPPS solid phase peptide synthesis

Anhang 13 Abkürzungsverzeichnis
IV
T Thymin
t Triplett / Thymin
tBu tert-Butyl
TEA Triethylamin
tert. tertiär
TFA Trifluoressigsäure
THAP 2,4,6-Trihydroxyacetophenon
THF Tetrahydrofuran
TFMSA Trifluormethansulfonsäure
TOF time of flight
TOTU O-[(Ethoxycarbonyl)cyanomethylenamino]-N,N,N′,N′-tetramethyluronium-
tetrafluorborat
UV Ultraviolett
VIS visible
Z Benzyloxycarbonyl

Anhang 14 HR-ESI-MS-Spektren
V
14 HR-ESI-MS-Spektren von PNA 224, F-PNA 248 und F-PNA 268
Abbildung 160. HR-ESI-MS-Spektren von PNA 224 (links), F-PNA 248 (Mitte) und PNA 268 (rechts).

Anhang 15 SimFit-Dateien
VI
15 SimFit-Dateien
15.1 SimFit-Eingabedateien
15.1.1 A-Modell
* MOPS-Puffer pH 7.6, 10 °C, 0.1 M Imidazol, 0.2 M NaI, Abbildung 139, Tabelle 11 define (1, t, p, 12) scale (3, 1.00000E+0) define (2, a, e, 11) scale (3, 1.00000E+0) select (t, a) read (NaI.txt) choose (exp1, exp2) reaction (A + B --> T) constant (1, 1E-4,1, 1.00000E+0, 1.00000E-10, 1.00000E+10) reaction (A --> X) constant (2, 1E-6,2, 1.00000E+0, 1.00000E-10, 1.00000E+10) reaction (B --> Y) constant (3, 1E-6,2, 1.00000E+0, 1.00000E-10, 1.00000E+10) constant (show) reaction (compile) reaction (show) int (show) assign (obs, a = A + X) assign (obs, t = T) assign (spec, A = a) assign (spec, B = # 5e-3) assign (spec, T = t) assign (spec, X = # 0) assign (spec, Y = # 0) assign (show) time (h) conc (mM) win (0, 35, 5, 0.01, 0, 6, 1, 0.01) dim (2) integ (stiff, .0000001, 4, .050000000745058, 100, 50) simplex (plot) opar (1e+25) newton (plot) plot (spec, file) make

Anhang 15 SimFit-Dateien
VII
15.1.2 B-Modell
* MOPS-Puffer pH 7.6, 10 °C, 0.1 M Imidazol, 0.2 M NaI, Abbildung 139, Tabelle 11 define (1, t, p, 12) scale (3, 1.00000E+0) define (2, a, e, 11) scale (3, 1.00000E+0) select (t, a) read (NaI.txt) choose (exp1, exp2) reaction ( A + B --> T) constant (1, 1E-4,1, 1.00000E+0, 1.00000E-10, 1.00000E+1) reaction ( A --> X) constant (2, 1E-6,2, 1.00000E+0, 1.00000E-10, 1.00000E+1) reaction ( B --> Y) constant (3, 1E-6,2, 1.00000E+0, 1.00000E-10, 1.00000E+1) reaction (A + B + 0.5 T --> 1.5 T) constant (4, 1E-2,3, 1.00000E+0, 1.00000E-10, 1.00000E+1) constant (show) reaction (compile) reaction (show) int (show) assign (obs, a = A + X) assign (obs, t = T) assign (spec, A = a) assign (spec, B = # 5e-3) assign (spec, T = t) assign (spec, X = # 0) assign (spec, Y = # 0) assign (show) time (h) conc (mM) win (0, 35, 5, 0.01, 0, 6, 1, 0.01) dim (3) integ (stiff, .0000001, 4, .05, 100, 10) simplex (plot) opar (1e+25) newton (plot) plot (spec, file) make
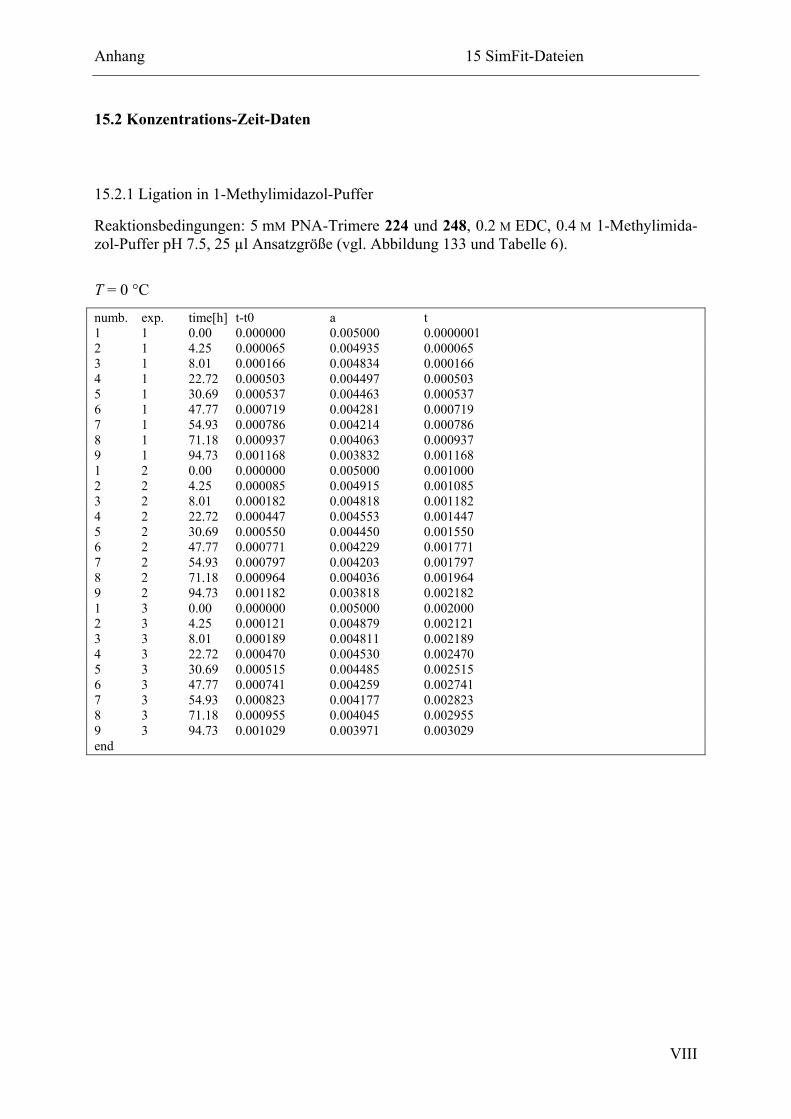
Anhang 15 SimFit-Dateien
VIII
15.2 Konzentrations-Zeit-Daten
15.2.1 Ligation in 1-Methylimidazol-Puffer
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.4 M 1-Methylimida-zol-Puffer pH 7.5, 25 µl Ansatzgröße (vgl. Abbildung 133 und Tabelle 6).
T = 0 °C
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 4.25 0.000065 0.004935 0.000065 3 1 8.01 0.000166 0.004834 0.000166 4 1 22.72 0.000503 0.004497 0.000503 5 1 30.69 0.000537 0.004463 0.000537 6 1 47.77 0.000719 0.004281 0.000719 7 1 54.93 0.000786 0.004214 0.000786 8 1 71.18 0.000937 0.004063 0.000937 9 1 94.73 0.001168 0.003832 0.001168 1 2 0.00 0.000000 0.005000 0.001000 2 2 4.25 0.000085 0.004915 0.001085 3 2 8.01 0.000182 0.004818 0.001182 4 2 22.72 0.000447 0.004553 0.001447 5 2 30.69 0.000550 0.004450 0.001550 6 2 47.77 0.000771 0.004229 0.001771 7 2 54.93 0.000797 0.004203 0.001797 8 2 71.18 0.000964 0.004036 0.001964 9 2 94.73 0.001182 0.003818 0.002182 1 3 0.00 0.000000 0.005000 0.002000 2 3 4.25 0.000121 0.004879 0.002121 3 3 8.01 0.000189 0.004811 0.002189 4 3 22.72 0.000470 0.004530 0.002470 5 3 30.69 0.000515 0.004485 0.002515 6 3 47.77 0.000741 0.004259 0.002741 7 3 54.93 0.000823 0.004177 0.002823 8 3 71.18 0.000955 0.004045 0.002955 9 3 94.73 0.001029 0.003971 0.003029 end
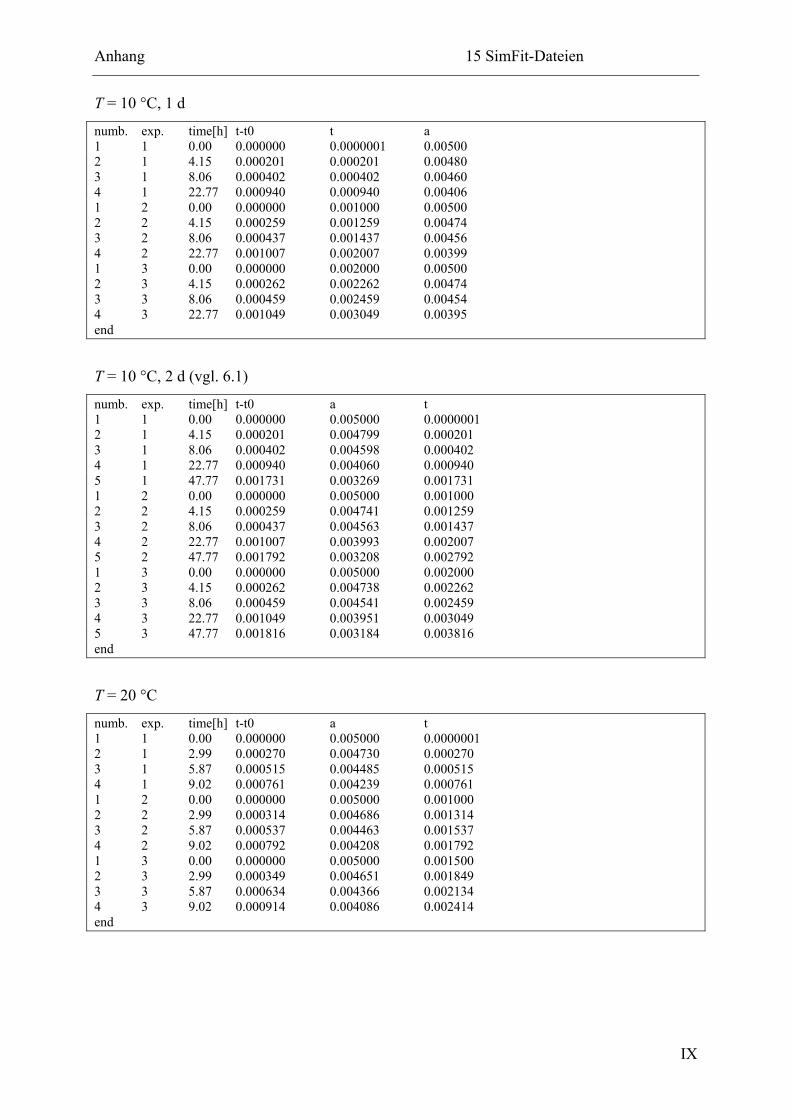
Anhang 15 SimFit-Dateien
IX
T = 10 °C, 1 d
numb. exp. time[h] t-t0 t a 1 1 0.00 0.000000 0.0000001 0.00500 2 1 4.15 0.000201 0.000201 0.00480 3 1 8.06 0.000402 0.000402 0.00460 4 1 22.77 0.000940 0.000940 0.00406 1 2 0.00 0.000000 0.001000 0.00500 2 2 4.15 0.000259 0.001259 0.00474 3 2 8.06 0.000437 0.001437 0.00456 4 2 22.77 0.001007 0.002007 0.00399 1 3 0.00 0.000000 0.002000 0.00500 2 3 4.15 0.000262 0.002262 0.00474 3 3 8.06 0.000459 0.002459 0.00454 4 3 22.77 0.001049 0.003049 0.00395 end
T = 10 °C, 2 d (vgl. 6.1)
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 4.15 0.000201 0.004799 0.000201 3 1 8.06 0.000402 0.004598 0.000402 4 1 22.77 0.000940 0.004060 0.000940 5 1 47.77 0.001731 0.003269 0.001731 1 2 0.00 0.000000 0.005000 0.001000 2 2 4.15 0.000259 0.004741 0.001259 3 2 8.06 0.000437 0.004563 0.001437 4 2 22.77 0.001007 0.003993 0.002007 5 2 47.77 0.001792 0.003208 0.002792 1 3 0.00 0.000000 0.005000 0.002000 2 3 4.15 0.000262 0.004738 0.002262 3 3 8.06 0.000459 0.004541 0.002459 4 3 22.77 0.001049 0.003951 0.003049 5 3 47.77 0.001816 0.003184 0.003816 end
T = 20 °C
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 2.99 0.000270 0.004730 0.000270 3 1 5.87 0.000515 0.004485 0.000515 4 1 9.02 0.000761 0.004239 0.000761 1 2 0.00 0.000000 0.005000 0.001000 2 2 2.99 0.000314 0.004686 0.001314 3 2 5.87 0.000537 0.004463 0.001537 4 2 9.02 0.000792 0.004208 0.001792 1 3 0.00 0.000000 0.005000 0.001500 2 3 2.99 0.000349 0.004651 0.001849 3 3 5.87 0.000634 0.004366 0.002134 4 3 9.02 0.000914 0.004086 0.002414 end

Anhang 15 SimFit-Dateien
X
T = 30 °C
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.00500 0.0000001 2 1 2.99 0.000709 0.00429 0.000709 3 1 5.87 0.001243 0.00376 0.001243 4 1 9.02 0.001795 0.00320 0.001795 1 2 0.00 0.000000 0.00500 0.000800 2 2 2.99 0.000811 0.00419 0.001611 3 2 5.87 0.001387 0.00361 0.002187 4 2 9.02 0.002009 0.00299 0.002809 1 3 0.00 0.000000 0.00500 0.002750 2 3 2.99 0.000938 0.00406 0.003688 3 3 5.87 0.001552 0.00345 0.004302 4 3 9.02 0.002134 0.00287 0.004884 end
15.2.2 Ligation in Imidazol-Puffer
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.4 M Imidazol-Puffer pH 7.5, 25 µl Ansatzgröße (vgl. Abbildung 136 und Tabelle 7).
T = 0 °C
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.00500 0.0000001 2 1 3.58 0.000097 0.00490 0.000097 3 1 6.58 0.000155 0.00485 0.000155 4 1 20.46 0.000385 0.00461 0.000385 5 1 29.77 0.000527 0.00447 0.000527 1 2 0.00 0.000000 0.00500 0.001500 2 2 3.58 0.000194 0.00481 0.001694 3 2 6.58 0.000227 0.00477 0.001727 4 2 20.46 0.000526 0.00447 0.002026 5 2 29.77 0.000673 0.00433 0.002173 1 3 0.00 0.000000 0.00500 0.002500 2 3 3.58 0.000182 0.00482 0.002682 3 3 6.58 0.000249 0.00475 0.002749 4 3 20.46 0.000509 0.00449 0.003009 5 3 29.77 0.000632 0.00437 0.003132 end
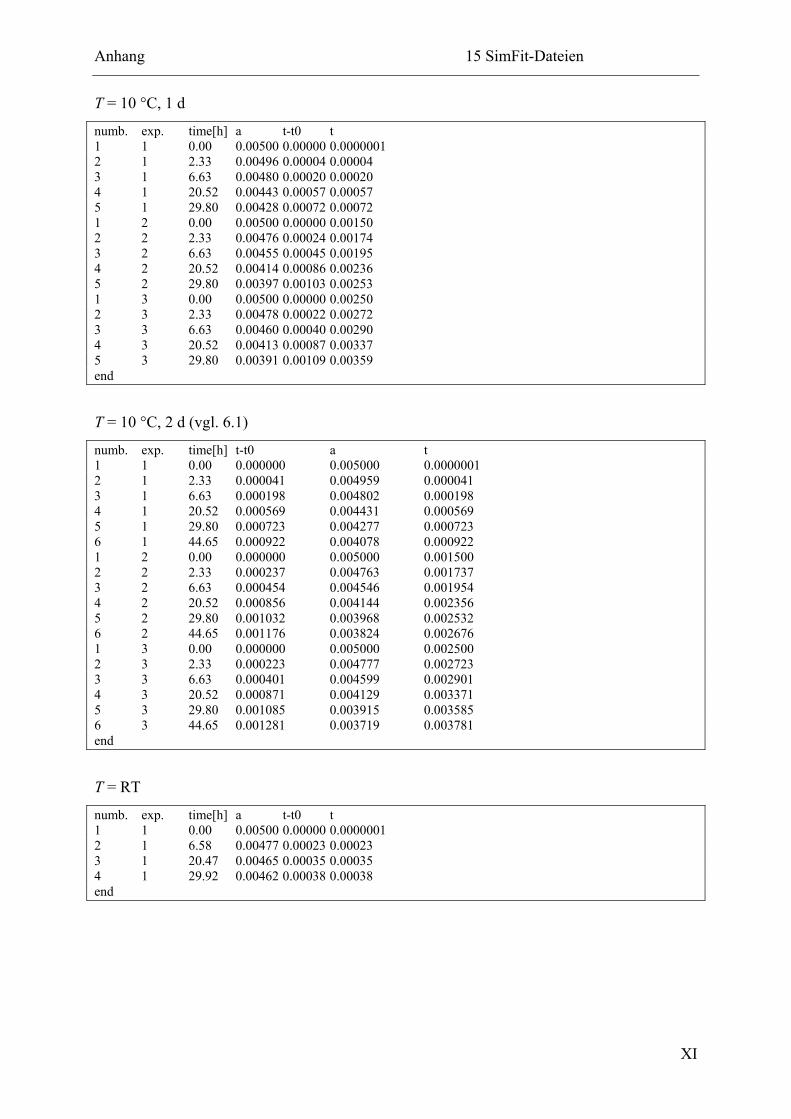
Anhang 15 SimFit-Dateien
XI
T = 10 °C, 1 d
numb. exp. time[h] a t-t0 t 1 1 0.00 0.00500 0.00000 0.0000001 2 1 2.33 0.00496 0.00004 0.00004 3 1 6.63 0.00480 0.00020 0.00020 4 1 20.52 0.00443 0.00057 0.00057 5 1 29.80 0.00428 0.00072 0.00072 1 2 0.00 0.00500 0.00000 0.00150 2 2 2.33 0.00476 0.00024 0.00174 3 2 6.63 0.00455 0.00045 0.00195 4 2 20.52 0.00414 0.00086 0.00236 5 2 29.80 0.00397 0.00103 0.00253 1 3 0.00 0.00500 0.00000 0.00250 2 3 2.33 0.00478 0.00022 0.00272 3 3 6.63 0.00460 0.00040 0.00290 4 3 20.52 0.00413 0.00087 0.00337 5 3 29.80 0.00391 0.00109 0.00359 end
T = 10 °C, 2 d (vgl. 6.1)
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 2.33 0.000041 0.004959 0.000041 3 1 6.63 0.000198 0.004802 0.000198 4 1 20.52 0.000569 0.004431 0.000569 5 1 29.80 0.000723 0.004277 0.000723 6 1 44.65 0.000922 0.004078 0.000922 1 2 0.00 0.000000 0.005000 0.001500 2 2 2.33 0.000237 0.004763 0.001737 3 2 6.63 0.000454 0.004546 0.001954 4 2 20.52 0.000856 0.004144 0.002356 5 2 29.80 0.001032 0.003968 0.002532 6 2 44.65 0.001176 0.003824 0.002676 1 3 0.00 0.000000 0.005000 0.002500 2 3 2.33 0.000223 0.004777 0.002723 3 3 6.63 0.000401 0.004599 0.002901 4 3 20.52 0.000871 0.004129 0.003371 5 3 29.80 0.001085 0.003915 0.003585 6 3 44.65 0.001281 0.003719 0.003781 end
T = RT
numb. exp. time[h] a t-t0 t 1 1 0.00 0.00500 0.00000 0.0000001 2 1 6.58 0.00477 0.00023 0.00023 3 1 20.47 0.00465 0.00035 0.00035 4 1 29.92 0.00462 0.00038 0.00038 end

Anhang 15 SimFit-Dateien
XII
15.2.3 Ligation in HEPES- und MOPS-Puffer
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M Puffer (MOPS pH 7.2 oder HEPES pH 7.5), 0.2 M NaCl, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 137 und Tabelle 8).
HEPES
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.00500 0.0000001 2 1 0.57 0.000024 0.00498 0.000024 3 1 2.87 0.000073 0.00493 0.000073 4 1 5.18 0.000050 0.00495 0.000050 5 1 8.10 0.000125 0.00487 0.000125 6 1 23.32 0.000296 0.00470 0.000296 7 1 31.82 0.000382 0.00462 0.000382 1 2 0.00 0.000000 0.00500 0.000500 2 2 0.57 0.000067 0.00493 0.000567 3 2 2.87 0.000100 0.00490 0.000600 4 2 5.18 0.000070 0.00493 0.000570 5 2 8.10 0.000146 0.00485 0.000646 6 2 23.32 0.000342 0.00466 0.000842 7 2 31.82 0.000365 0.00463 0.000865 1 3 0.00 0.000000 0.00500 0.0000001 2 3 0.57 0.000213 0.00479 0.000213 3 3 1.45 0.000347 0.00465 0.000347 4 3 2.87 0.000607 0.00439 0.000607 5 3 8.10 0.000785 0.00421 0.000785 6 3 23.32 0.001015 0.00398 0.001015 7 3 31.82 0.001046 0.00395 0.001046 end
MOPS
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.35 0.000065 0.004935 0.000065 3 1 2.77 0.000084 0.004916 0.000084 4 1 5.09 0.000145 0.004855 0.000145 5 1 8.00 0.000203 0.004797 0.000203 6 1 23.22 0.000476 0.004524 0.000476 7 1 31.72 0.000572 0.004428 0.000572 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.35 0.000031 0.004969 0.000531 3 2 2.77 0.000099 0.004901 0.000599 4 2 5.09 0.000227 0.004773 0.000727 5 2 8.00 0.000308 0.004692 0.000808 6 2 23.22 0.000721 0.004279 0.001221 7 2 31.72 0.000866 0.004134 0.001366 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.35 0.000112 0.004888 0.000112 3 3 2.77 0.000199 0.004801 0.000199 4 3 5.09 0.000327 0.004673 0.000327 5 3 8.00 0.000431 0.004569 0.000431 6 3 23.22 0.000719 0.004281 0.000719 7 3 31.72 0.000754 0.004246 0.000754 end
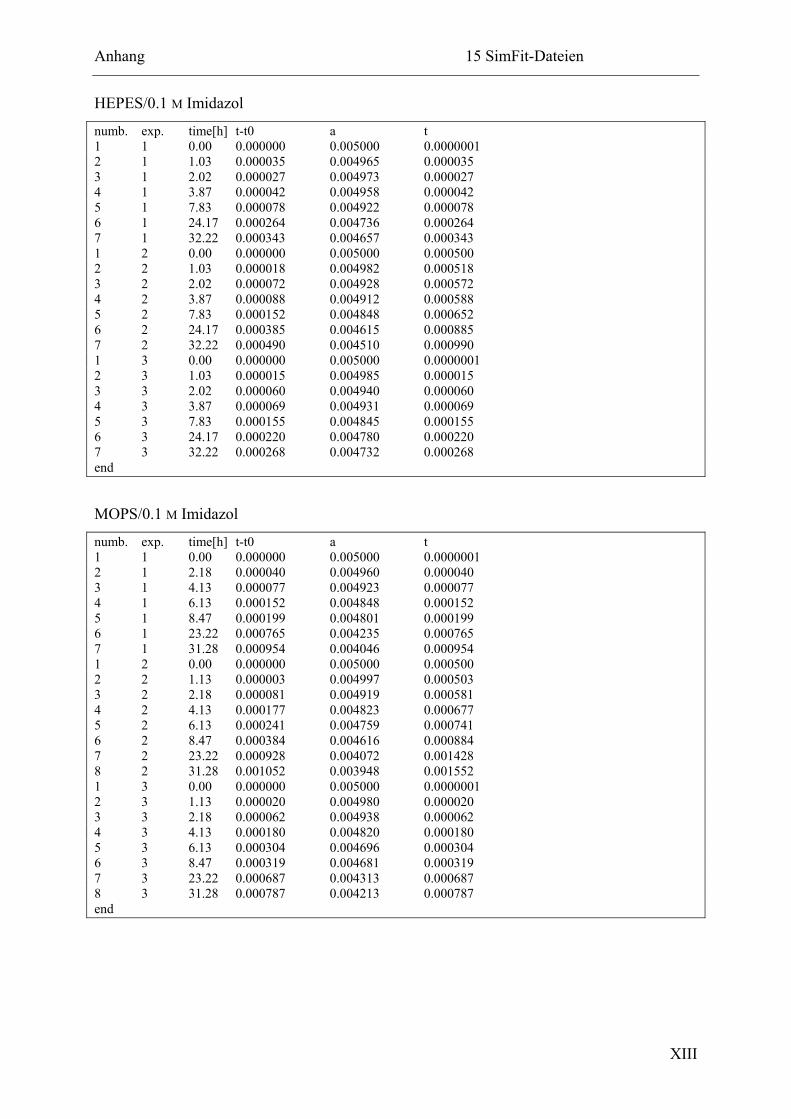
Anhang 15 SimFit-Dateien
XIII
HEPES/0.1 M Imidazol
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.03 0.000035 0.004965 0.000035 3 1 2.02 0.000027 0.004973 0.000027 4 1 3.87 0.000042 0.004958 0.000042 5 1 7.83 0.000078 0.004922 0.000078 6 1 24.17 0.000264 0.004736 0.000264 7 1 32.22 0.000343 0.004657 0.000343 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.03 0.000018 0.004982 0.000518 3 2 2.02 0.000072 0.004928 0.000572 4 2 3.87 0.000088 0.004912 0.000588 5 2 7.83 0.000152 0.004848 0.000652 6 2 24.17 0.000385 0.004615 0.000885 7 2 32.22 0.000490 0.004510 0.000990 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.03 0.000015 0.004985 0.000015 3 3 2.02 0.000060 0.004940 0.000060 4 3 3.87 0.000069 0.004931 0.000069 5 3 7.83 0.000155 0.004845 0.000155 6 3 24.17 0.000220 0.004780 0.000220 7 3 32.22 0.000268 0.004732 0.000268 end
MOPS/0.1 M Imidazol
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 2.18 0.000040 0.004960 0.000040 3 1 4.13 0.000077 0.004923 0.000077 4 1 6.13 0.000152 0.004848 0.000152 5 1 8.47 0.000199 0.004801 0.000199 6 1 23.22 0.000765 0.004235 0.000765 7 1 31.28 0.000954 0.004046 0.000954 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.13 0.000003 0.004997 0.000503 3 2 2.18 0.000081 0.004919 0.000581 4 2 4.13 0.000177 0.004823 0.000677 5 2 6.13 0.000241 0.004759 0.000741 6 2 8.47 0.000384 0.004616 0.000884 7 2 23.22 0.000928 0.004072 0.001428 8 2 31.28 0.001052 0.003948 0.001552 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.13 0.000020 0.004980 0.000020 3 3 2.18 0.000062 0.004938 0.000062 4 3 4.13 0.000180 0.004820 0.000180 5 3 6.13 0.000304 0.004696 0.000304 6 3 8.47 0.000319 0.004681 0.000319 7 3 23.22 0.000687 0.004313 0.000687 8 3 31.28 0.000787 0.004213 0.000787 end

Anhang 15 SimFit-Dateien
XIV
MOPS/0.1 M 1- Methylimidazol
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.11 0.000038 0.004962 0.000038 3 1 2.21 0.000093 0.004907 0.000093 4 1 4.05 0.000201 0.004799 0.000201 5 1 8.02 0.000404 0.004596 0.000404 6 1 24.35 0.000810 0.004190 0.000810 7 1 32.40 0.000975 0.004025 0.000975 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.11 0.000005 0.004995 0.000505 3 2 2.21 0.000120 0.004880 0.000620 4 2 4.05 0.000182 0.004818 0.000682 5 2 8.02 0.000314 0.004686 0.000814 6 2 24.35 0.000738 0.004262 0.001238 7 2 32.40 0.000919 0.004081 0.001419 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.11 0.000335 0.004665 0.000335 3 3 2.21 0.000485 0.004515 0.000485 4 3 4.05 0.000975 0.004025 0.000975 5 3 8.02 0.001651 0.003349 0.001651 6 3 24.35 0.002905 0.002095 0.002905 7 3 32.40 0.003270 0.001730 0.003270 end
15.2.4 Ligation in Gegenwart von Pyridin oder HOAt
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.2, 0.2 M NaCl, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 138 und Tabelle 10).
0.1 M Pyridin
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.02 0.000062 0.004938 0.000062 3 1 2.10 0.000056 0.004944 0.000056 4 1 4.15 0.000156 0.004844 0.000156 5 1 7.72 0.000270 0.004730 0.000270 6 1 22.37 0.000820 0.004180 0.000820 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.02 0.000078 0.004922 0.000578 3 2 2.10 0.000102 0.004898 0.000602 4 2 4.15 0.000194 0.004806 0.000694 5 2 7.72 0.000356 0.004644 0.000856 6 2 22.37 0.000900 0.004100 0.001400 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.02 0.000343 0.004657 0.000343 3 3 2.10 0.000669 0.004331 0.000669 4 3 4.15 0.001184 0.003816 0.001184 5 3 7.72 0.001882 0.003118 0.001882 6 3 22.37 0.002968 0.002032 0.002968 end
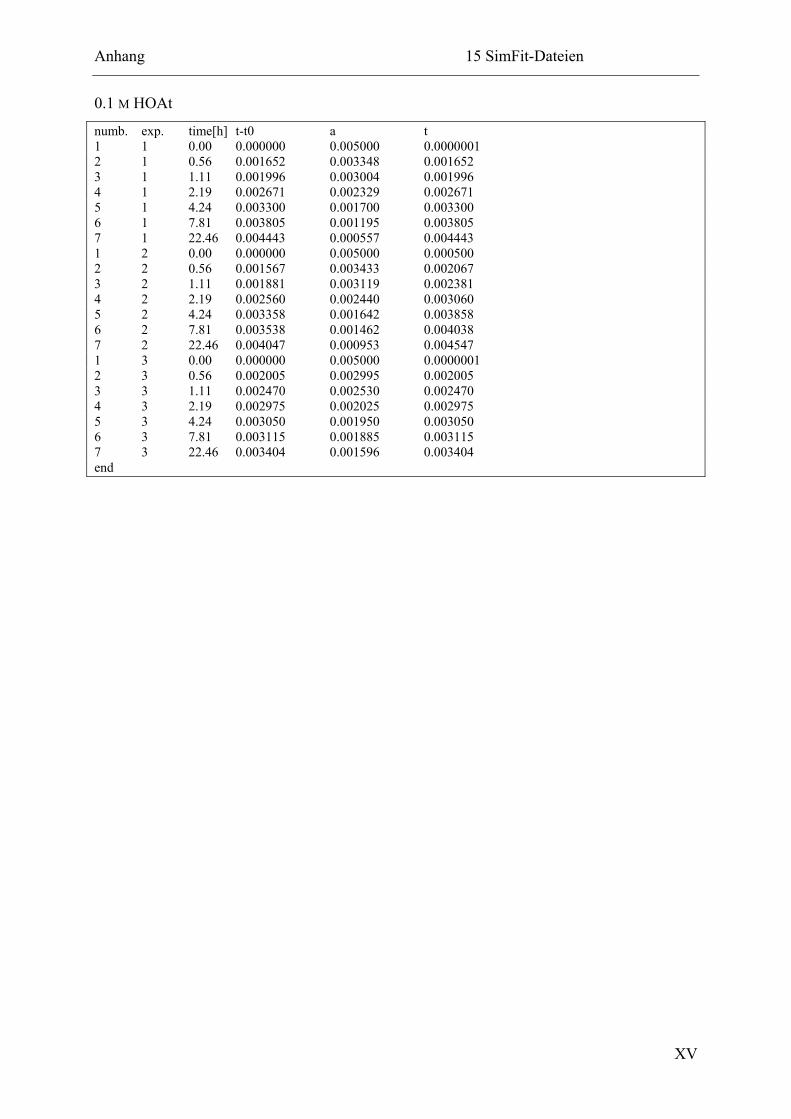
Anhang 15 SimFit-Dateien
XV
0.1 M HOAt
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 0.56 0.001652 0.003348 0.001652 3 1 1.11 0.001996 0.003004 0.001996 4 1 2.19 0.002671 0.002329 0.002671 5 1 4.24 0.003300 0.001700 0.003300 6 1 7.81 0.003805 0.001195 0.003805 7 1 22.46 0.004443 0.000557 0.004443 1 2 0.00 0.000000 0.005000 0.000500 2 2 0.56 0.001567 0.003433 0.002067 3 2 1.11 0.001881 0.003119 0.002381 4 2 2.19 0.002560 0.002440 0.003060 5 2 4.24 0.003358 0.001642 0.003858 6 2 7.81 0.003538 0.001462 0.004038 7 2 22.46 0.004047 0.000953 0.004547 1 3 0.00 0.000000 0.005000 0.0000001 2 3 0.56 0.002005 0.002995 0.002005 3 3 1.11 0.002470 0.002530 0.002470 4 3 2.19 0.002975 0.002025 0.002975 5 3 4.24 0.003050 0.001950 0.003050 6 3 7.81 0.003115 0.001885 0.003115 7 3 22.46 0.003404 0.001596 0.003404 end
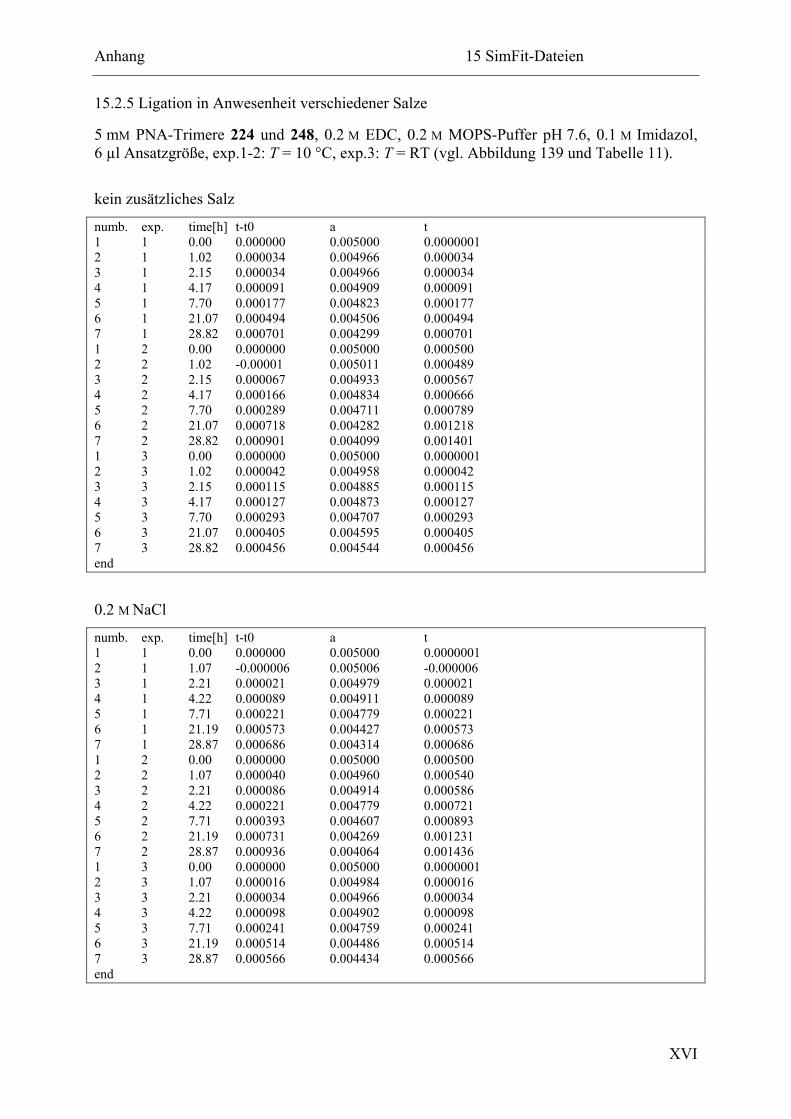
Anhang 15 SimFit-Dateien
XVI
15.2.5 Ligation in Anwesenheit verschiedener Salze
5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.1 M Imidazol, 6 µl Ansatzgröße, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 139 und Tabelle 11).
kein zusätzliches Salz
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.02 0.000034 0.004966 0.000034 3 1 2.15 0.000034 0.004966 0.000034 4 1 4.17 0.000091 0.004909 0.000091 5 1 7.70 0.000177 0.004823 0.000177 6 1 21.07 0.000494 0.004506 0.000494 7 1 28.82 0.000701 0.004299 0.000701 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.02 -0.00001 0.005011 0.000489 3 2 2.15 0.000067 0.004933 0.000567 4 2 4.17 0.000166 0.004834 0.000666 5 2 7.70 0.000289 0.004711 0.000789 6 2 21.07 0.000718 0.004282 0.001218 7 2 28.82 0.000901 0.004099 0.001401 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.02 0.000042 0.004958 0.000042 3 3 2.15 0.000115 0.004885 0.000115 4 3 4.17 0.000127 0.004873 0.000127 5 3 7.70 0.000293 0.004707 0.000293 6 3 21.07 0.000405 0.004595 0.000405 7 3 28.82 0.000456 0.004544 0.000456 end
0.2 M NaCl
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.07 -0.000006 0.005006 -0.000006 3 1 2.21 0.000021 0.004979 0.000021 4 1 4.22 0.000089 0.004911 0.000089 5 1 7.71 0.000221 0.004779 0.000221 6 1 21.19 0.000573 0.004427 0.000573 7 1 28.87 0.000686 0.004314 0.000686 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.07 0.000040 0.004960 0.000540 3 2 2.21 0.000086 0.004914 0.000586 4 2 4.22 0.000221 0.004779 0.000721 5 2 7.71 0.000393 0.004607 0.000893 6 2 21.19 0.000731 0.004269 0.001231 7 2 28.87 0.000936 0.004064 0.001436 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.07 0.000016 0.004984 0.000016 3 3 2.21 0.000034 0.004966 0.000034 4 3 4.22 0.000098 0.004902 0.000098 5 3 7.71 0.000241 0.004759 0.000241 6 3 21.19 0.000514 0.004486 0.000514 7 3 28.87 0.000566 0.004434 0.000566 end
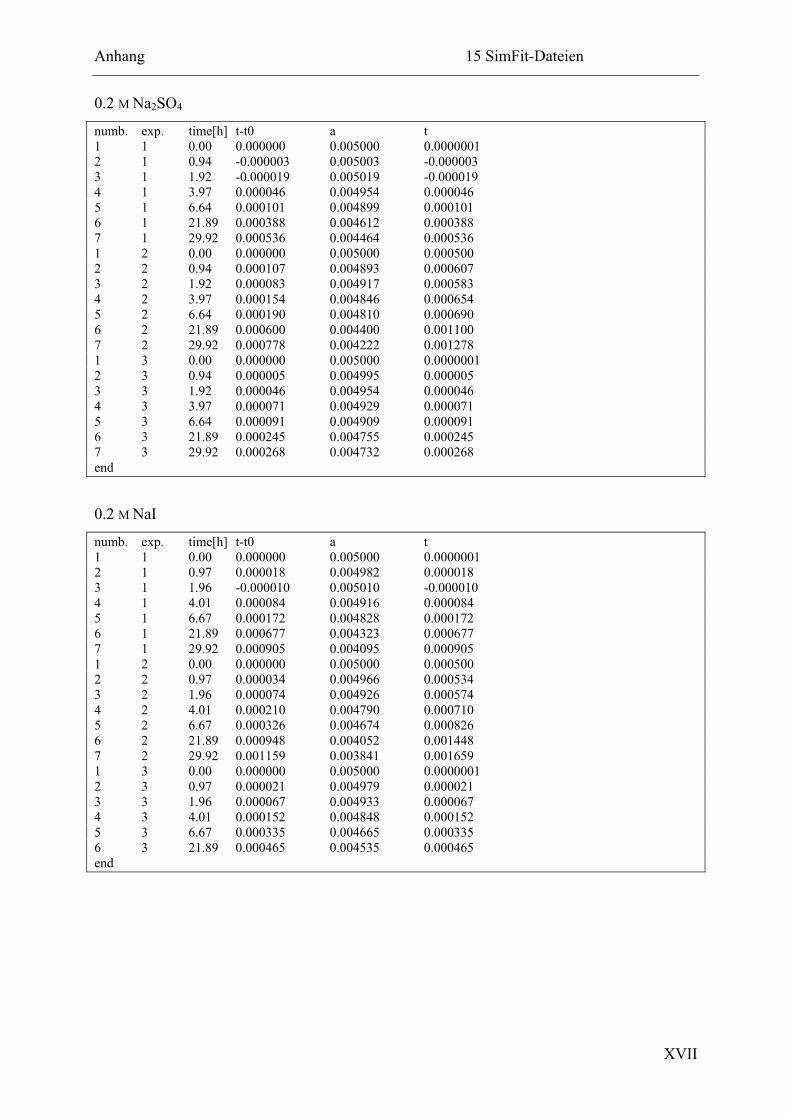
Anhang 15 SimFit-Dateien
XVII
0.2 M Na2SO4
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 0.94 -0.000003 0.005003 -0.000003 3 1 1.92 -0.000019 0.005019 -0.000019 4 1 3.97 0.000046 0.004954 0.000046 5 1 6.64 0.000101 0.004899 0.000101 6 1 21.89 0.000388 0.004612 0.000388 7 1 29.92 0.000536 0.004464 0.000536 1 2 0.00 0.000000 0.005000 0.000500 2 2 0.94 0.000107 0.004893 0.000607 3 2 1.92 0.000083 0.004917 0.000583 4 2 3.97 0.000154 0.004846 0.000654 5 2 6.64 0.000190 0.004810 0.000690 6 2 21.89 0.000600 0.004400 0.001100 7 2 29.92 0.000778 0.004222 0.001278 1 3 0.00 0.000000 0.005000 0.0000001 2 3 0.94 0.000005 0.004995 0.000005 3 3 1.92 0.000046 0.004954 0.000046 4 3 3.97 0.000071 0.004929 0.000071 5 3 6.64 0.000091 0.004909 0.000091 6 3 21.89 0.000245 0.004755 0.000245 7 3 29.92 0.000268 0.004732 0.000268 end
0.2 M NaI
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 0.97 0.000018 0.004982 0.000018 3 1 1.96 -0.000010 0.005010 -0.000010 4 1 4.01 0.000084 0.004916 0.000084 5 1 6.67 0.000172 0.004828 0.000172 6 1 21.89 0.000677 0.004323 0.000677 7 1 29.92 0.000905 0.004095 0.000905 1 2 0.00 0.000000 0.005000 0.000500 2 2 0.97 0.000034 0.004966 0.000534 3 2 1.96 0.000074 0.004926 0.000574 4 2 4.01 0.000210 0.004790 0.000710 5 2 6.67 0.000326 0.004674 0.000826 6 2 21.89 0.000948 0.004052 0.001448 7 2 29.92 0.001159 0.003841 0.001659 1 3 0.00 0.000000 0.005000 0.0000001 2 3 0.97 0.000021 0.004979 0.000021 3 3 1.96 0.000067 0.004933 0.000067 4 3 4.01 0.000152 0.004848 0.000152 5 3 6.67 0.000335 0.004665 0.000335 6 3 21.89 0.000465 0.004535 0.000465 end

Anhang 15 SimFit-Dateien
XVIII
15.2.6 Ligation in Gegenwart von 0.4 M EDC
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.4 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.1 M Imidazol, 6 µl Ansatzgröße, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 140 und Tabelle 12).
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.12 0.000080 0.004920 0.000080 3 1 2.07 0.000108 0.004892 0.000108 4 1 4.14 0.000224 0.004776 0.000224 5 1 8.49 0.000574 0.004426 0.000574 6 1 22.27 0.001209 0.003791 0.001209 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.12 0.000118 0.004882 0.000618 3 2 2.07 0.000226 0.004774 0.000726 4 2 4.14 0.000412 0.004588 0.000912 5 2 8.49 0.000746 0.004254 0.001246 6 2 22.27 0.001444 0.003556 0.001944 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.12 0.000101 0.004899 0.000101 3 3 2.07 0.000197 0.004803 0.000197 4 3 4.14 0.000290 0.004710 0.000290 5 3 8.49 0.000515 0.004485 0.000515 6 3 22.27 0.000828 0.004172 0.000828 end
15.2.7 Ligation bei pH 6.6
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.2 M EDC, 0.2 M MOPS-Puffer pH 6.6, 0.1 M Imidazol, 6 µl Ansatzgröße, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 141 und Tabelle 13).
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.02 0.000046 0.004954 0.000046 3 1 1.98 0.000131 0.004869 0.000131 4 1 4.05 0.000312 0.004688 0.000312 5 1 8.38 0.000916 0.004084 0.000916 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.02 0.000012 0.004988 0.000512 3 2 1.98 0.000163 0.004837 0.000663 4 2 4.05 0.000546 0.004454 0.001046 5 2 8.38 0.001153 0.003847 0.001653 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.02 0.000105 0.004895 0.000105 3 3 1.98 0.000342 0.004658 0.000342 4 3 4.05 0.000583 0.004417 0.000583 5 3 8.38 0.001168 0.003832 0.001168 end
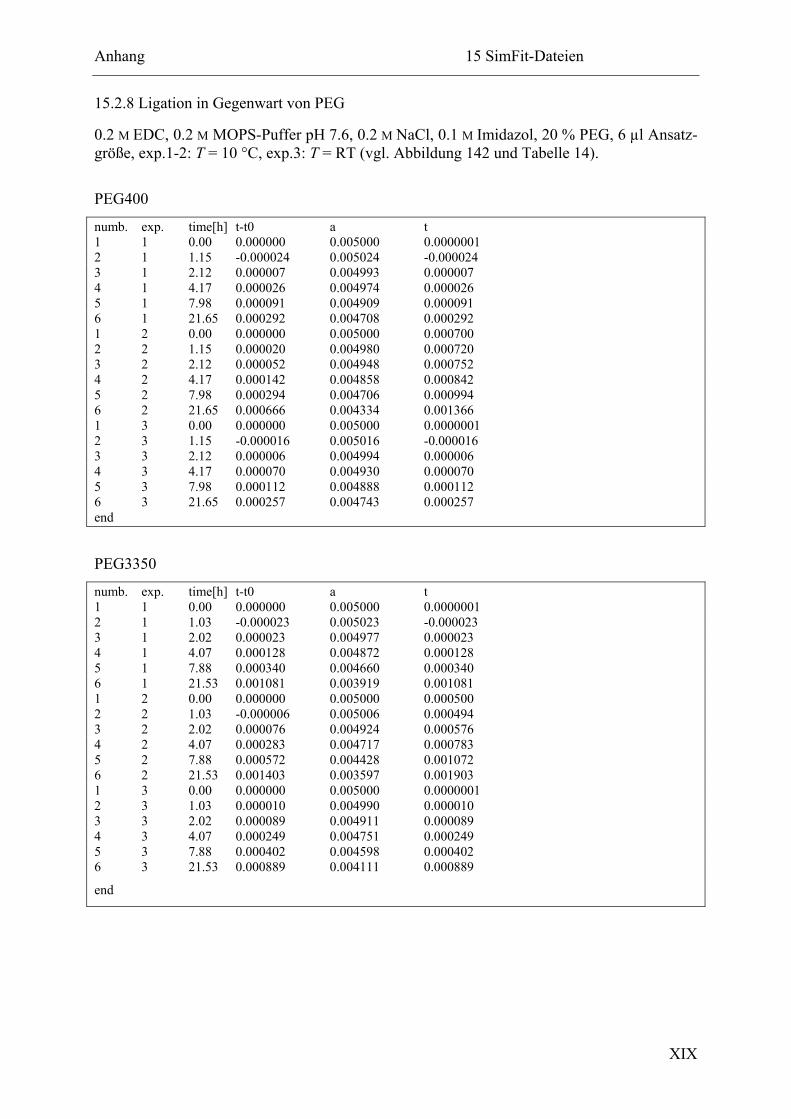
Anhang 15 SimFit-Dateien
XIX
15.2.8 Ligation in Gegenwart von PEG
0.2 M EDC, 0.2 M MOPS-Puffer pH 7.6, 0.2 M NaCl, 0.1 M Imidazol, 20 % PEG, 6 µl Ansatz-größe, exp.1-2: T = 10 °C, exp.3: T = RT (vgl. Abbildung 142 und Tabelle 14).
PEG400
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.15 -0.000024 0.005024 -0.000024 3 1 2.12 0.000007 0.004993 0.000007 4 1 4.17 0.000026 0.004974 0.000026 5 1 7.98 0.000091 0.004909 0.000091 6 1 21.65 0.000292 0.004708 0.000292 1 2 0.00 0.000000 0.005000 0.000700 2 2 1.15 0.000020 0.004980 0.000720 3 2 2.12 0.000052 0.004948 0.000752 4 2 4.17 0.000142 0.004858 0.000842 5 2 7.98 0.000294 0.004706 0.000994 6 2 21.65 0.000666 0.004334 0.001366 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.15 -0.000016 0.005016 -0.000016 3 3 2.12 0.000006 0.004994 0.000006 4 3 4.17 0.000070 0.004930 0.000070 5 3 7.98 0.000112 0.004888 0.000112 6 3 21.65 0.000257 0.004743 0.000257 end
PEG3350
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.03 -0.000023 0.005023 -0.000023 3 1 2.02 0.000023 0.004977 0.000023 4 1 4.07 0.000128 0.004872 0.000128 5 1 7.88 0.000340 0.004660 0.000340 6 1 21.53 0.001081 0.003919 0.001081 1 2 0.00 0.000000 0.005000 0.000500 2 2 1.03 -0.000006 0.005006 0.000494 3 2 2.02 0.000076 0.004924 0.000576 4 2 4.07 0.000283 0.004717 0.000783 5 2 7.88 0.000572 0.004428 0.001072 6 2 21.53 0.001403 0.003597 0.001903 1 3 0.00 0.000000 0.005000 0.0000001 2 3 1.03 0.000010 0.004990 0.000010 3 3 2.02 0.000089 0.004911 0.000089 4 3 4.07 0.000249 0.004751 0.000249 5 3 7.88 0.000402 0.004598 0.000402 6 3 21.53 0.000889 0.004111 0.000889
end
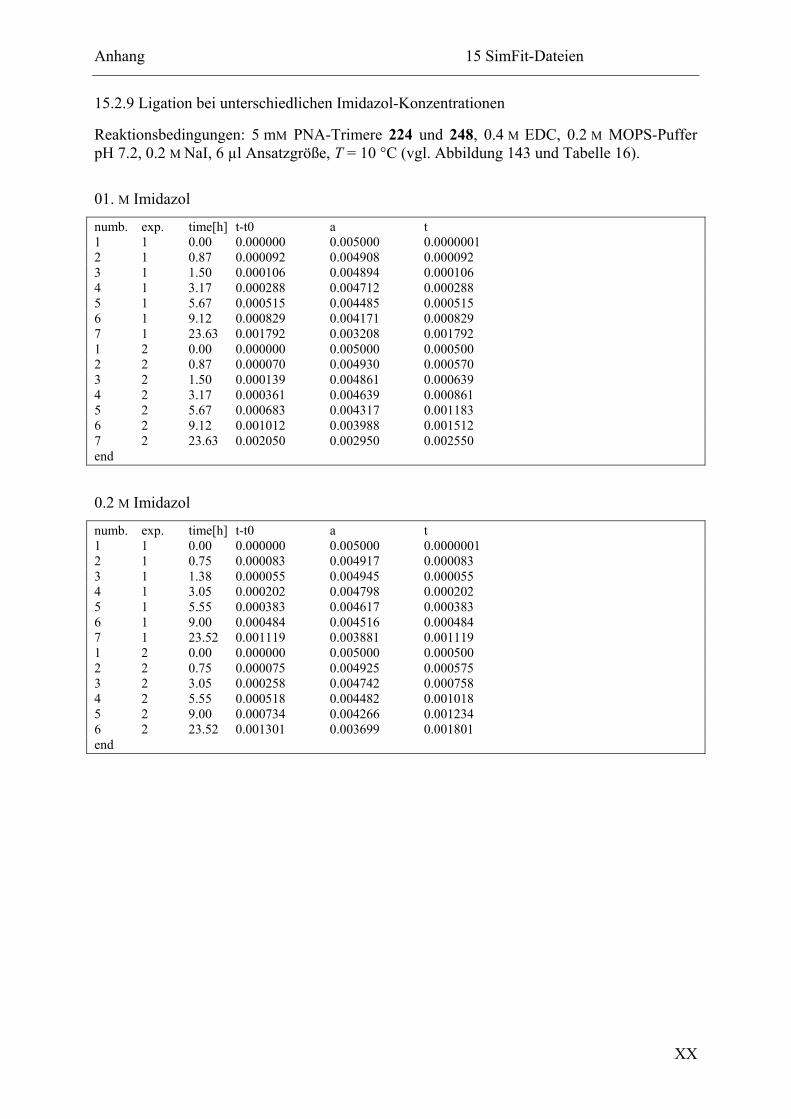
Anhang 15 SimFit-Dateien
XX
15.2.9 Ligation bei unterschiedlichen Imidazol-Konzentrationen
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.4 M EDC, 0.2 M MOPS-Puffer pH 7.2, 0.2 M NaI, 6 µl Ansatzgröße, T = 10 °C (vgl. Abbildung 143 und Tabelle 16).
01. M Imidazol
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 0.87 0.000092 0.004908 0.000092 3 1 1.50 0.000106 0.004894 0.000106 4 1 3.17 0.000288 0.004712 0.000288 5 1 5.67 0.000515 0.004485 0.000515 6 1 9.12 0.000829 0.004171 0.000829 7 1 23.63 0.001792 0.003208 0.001792 1 2 0.00 0.000000 0.005000 0.000500 2 2 0.87 0.000070 0.004930 0.000570 3 2 1.50 0.000139 0.004861 0.000639 4 2 3.17 0.000361 0.004639 0.000861 5 2 5.67 0.000683 0.004317 0.001183 6 2 9.12 0.001012 0.003988 0.001512 7 2 23.63 0.002050 0.002950 0.002550 end
0.2 M Imidazol
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 0.75 0.000083 0.004917 0.000083 3 1 1.38 0.000055 0.004945 0.000055 4 1 3.05 0.000202 0.004798 0.000202 5 1 5.55 0.000383 0.004617 0.000383 6 1 9.00 0.000484 0.004516 0.000484 7 1 23.52 0.001119 0.003881 0.001119 1 2 0.00 0.000000 0.005000 0.000500 2 2 0.75 0.000075 0.004925 0.000575 3 2 3.05 0.000258 0.004742 0.000758 4 2 5.55 0.000518 0.004482 0.001018 5 2 9.00 0.000734 0.004266 0.001234 6 2 23.52 0.001301 0.003699 0.001801 end

Anhang 15 SimFit-Dateien
XXI
15.2.10 Ligation bei vier unterschiedlichen intialen Hexamerkonzentrationen
Reaktionsbedingungen: 5 mM PNA-Trimere 224 und 248, 0.4 M EDC, 0.2 M MOPS-Puffer pH 6.6, 0.1 M Imidazol, 10 °C, 6 µl Ansatzgröße (vgl. Abbildung 144 und Tabelle 17).
numb. exp. time[h] t-t0 a t 1 1 0.00 0.000000 0.005000 0.0000001 2 1 1.09 0.000185 0.004815 0.000185 3 1 2.16 0.000378 0.004622 0.000378 4 1 3.52 0.000730 0.004270 0.000730 5 1 5.16 0.001171 0.003829 0.001171 6 1 7.05 0.001647 0.003353 0.001647 7 1 23.47 0.003277 0.001723 0.003277 1 2 0.00 0.000000 0.005000 0.000250 2 2 1.09 0.000246 0.004754 0.000496 3 2 2.16 0.000537 0.004463 0.000787 4 2 3.52 0.000895 0.004105 0.001145 5 2 5.16 0.001421 0.003579 0.001671 6 2 7.05 0.001851 0.003149 0.002101 7 2 23.47 0.003310 0.001690 0.003560 1 3 0.00 0.000000 0.005000 0.000500 2 3 1.09 0.000255 0.004745 0.000755 3 3 2.16 0.000593 0.004407 0.001093 4 3 3.52 0.000965 0.004035 0.001465 5 3 5.16 0.001450 0.003550 0.001950 6 3 7.05 0.001820 0.003180 0.002320 7 3 23.47 0.003498 0.001502 0.003998 1 4 0.00 0.000000 0.005000 0.001000 2 4 1.09 0.000277 0.004723 0.001277 3 4 2.16 0.000614 0.004386 0.001614 4 4 3.52 0.001033 0.003967 0.002033 5 4 5.16 0.001603 0.003397 0.002603 6 4 7.05 0.001951 0.003049 0.002951 7 4 23.47 0.003441 0.001559 0.004441 end

Anhang 16 Danksagung
XXII
16 Danksagung
Ich bedanke mich bei Herrn Prof. Dr. Günter von Kiedrowski dafür, dass ich in einem sehr
interessanten Bereich forschen konnte und die wissenschaftliche Freiheit bei der Durch-
führung selbstverständlich war. Ferner möchte ich mich für seinen wissenschaftlichen Rat,
seine Unterstützung und das mir entgegengebrachte Vertrauen bedanken.
Herrn Prof. Dr. Martin Feigel danke ich für die freundliche Übernahme des Korreferates.
Bei Herrn Dr. Wolf Matthias Pankau möchte ich mich für seine wertvolle wissenschaftliche
und moralische Unterstützung bedanken. Dr. Arne Dieckmann danke ich für die freundschaft-
liche Atmosphäre in „unserer Box“ und die ständige Diskussionsbereitschaft zu wissenschaft-
lichen und anderen interessanten Themen.
Hans-Jochen Hauswald (RUBiospek) danke ich für die angenehme Zusammenarbeit bei der
Aufnahme der Hochtemperatur-NMR-Spektren. Dr. Benjamin Fraenzel (AG Dr. Dirk Wol-
ters) sei für die Aufnahme der HR-ESI-Spektren gedankt.
Den Vertiefern Alexandra Aniol, Michel Beißwenger, Christian Dietz, Mei Kappels, Daniel
Krah und Kirill Sliozberg danke ich für ihren jeweiligen Anteil an der vorliegenden Arbeit.
Der gesamten Arbeitsgruppe von Kiedrowski danke ich für das positive Arbeitsklima und die
stete Bereitschaft zur Diskussion. Besonders danke ich:
Florian Kaschuba für die Unterstützung mit IT-Knowhow;
Carsten Lodwig für die technische Hilfe bei NP-HPLC-Trennungen;
Dr. Sven Mönninghoff für das Interesse an dieser Arbeit und den interessanten Aus-
tausch zu Fragen der Peptidchemie;
Jana Pyka für die angenehme „Box-Nachbarschaft“;
Tim Wilmsen für die vetrauensvolle Zusammenarbeit im Labor;
Michael Wüstefeld für seine wichtige technische Unterstützung und sein Interesse am
Fortgang meiner Arbeit.
Ganz persönlicher Dank gebührt meiner Frau Nadine und meinen Eltern Sigrid und Fred
Plöger für ihre vielfältige Unterstützung.

Anhang 17 Lebenslauf
XXIII
17 Lebenslauf
Persönliches 13.06.1982 geboren in Remscheid, verheiratet Schule 08/1988 - 07/1992 Gemeinschafts-Grundschule Bergerhof, Radevormwald 08/1992 - 06/2001 Theodor-Heuss-Gymnasium, Radevormwald 06/2001 Abschluss: Abitur (Note 1.4) Studium 10/2001 Beginn des Chemie-Studiums, Ruhr-Universität Bochum 06/2004 Abschluss: Bachelor of Science (Note 1.8)
Bachelor-Arbeit in Organischer Chemie: Beiträge zur Synthese neuartiger mehrfachfunktionalisierter Tris-Thioether-Liganden für Goldcluster vom Au55-Typ
09/2006 Abschluss: Master of Science (Note 1.1)
Master-Arbeit in Organischer Chemie: Synthese von tripodalen Tris-Thioether-Liganden auf Peptidbasis
11/2006 - 12/2011 Promotion am Lehrstuhl für Organische Chemie I Tag der mündlichen Prüfung: 02.12.2011
Studienbegleitende Tätigkeiten 06/2004 - 10/2006 studentische Hilfskraft, Lehrstuhl für Organische Chemie I,
Ruhr-Universität Bochum 11/2006 bis heute wissenschaftlicher Mitarbeiter, Lehrstuhl für Organische Chemie I,
Ruhr-Universität Bochum

Anhang 18 Literaturverzeichnis
XXIV
18 Literaturverzeichnis
[1] A. I. Oparin, Life: its Nature, Origin and Development, Academic Press, New York, 1961.
[2] A. I. Oparin, Proiskhozhdenie Zhizny [The Origin of Life], Izd. Moskoviskiy Rabochiy, Moscow, 1924.
[3] A. L. Oparin, in The origin of life. Translated from Russian by S. Morgulis., Macmillan Co., 1938.
[4] J. B. S. Haldane, Rationalist Annual 1929, 148. [5] G. von Kiedrowski, Nachrichten aus der Chemie 1993, 51, 666. [6] S. L. Miller, Science 1953, 117, 528. [7] A. Butlerow, C. R. Acad. Sci. Paris 1861, 53, 145. [8] P. Decker, H. Schweer, R. Pohlmann, J. Chromatogr. 1982, 244, 281. [9] C. Reid, L. E. Orgel, Nature 1967, 216, 455. [10] R. Mayer, L. Jaschke, Angew. Chem. Int. Ed. Engl. 1960, 72, 635. [11] T. Mizuno, A. H. Weiss, Adv. Carbohyd. Chem. Biochem. 1974, 29, 173. [12] D. Muller, S. Pitsch, A. Kittaka, E. Wagner, C. E. Wintner, A. Eschenmoser, Helv.
Chim. Acta 1990, 73, 1410. [13] A. Ricardo, M. A. Carrigan, A. N. Olcott, S. A. Benner, Science 2004, 303, 196. [14] J. Oro, Biochem. Biophys. Res. Commun. 1960, 2, 407. [15] J. Oro, Nature 1961, 191, 1193. [16] R. Sanchez, J. Ferris, L. E. Orgel, Science 1966, 153, 72. [17] G. Wächtershauser, Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 1134. [18] J. A. Piccirilli, T. Krauch, S. E. Moroney, S. A. Benner, Nature 1990, 343, 33. [19] B. Pullman, in Exobiology (Ed.: C. Ponnamperuma), North Holland Publishing
Company, Amsterdam, London, 1972, pp. 136. [20] R. A. Sanchez, J. P. Ferris, L. E. Orgel, Science 1966, 154, 784. [21] J. P. Ferris, R. A. Sanchez, L. E. Orgel, J. Mol. Biol. 1968, 33, 693. [22] M. P. Robertson, S. L. Miller, Nature 1995, 375, 772. [23] M. P. Robertson, M. Levy, S. L. Miller, J. Mol. Evol. 1996, 43, 543. [24] K. E. Nelson, M. P. Robertson, M. Levy, S. L. Miller, Origins Life Evol. Biosphere
2001, 31, 221. [25] G. Schlesinger, S. L. Miller, J. Mol. Evol. 1983, 19, 376. [26] G. Schlesinger, S. L. Miller, J. Mol. Evol. 1983, 19, 383. [27] H. Cleaves, J. Chalmers, A. Lazcano, S. Miller, J. Bada, Origins Life Evol. Biosphere
2008, 38, 105. [28] C. de Duve, Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 8253. [29] G. Wächtershauser, Microbiol. Rev. 1988, 52, 452. [30] J. Oro, Nature 1961, 190, 389. [31] K. Kvenvold, J. Lawless, K. Pering, E. Peterson, J. Flores, C. Ponnampe, I. R. Kaplan,
Nature 1970, 228, 923. [32] M. P. Bernstein, J. P. Dworkin, S. A. Sandford, G. W. Cooper, L. J. Allamandola,
Nature 2002, 416, 401. [33] G. M. M. Caro, U. J. Meierhenrich, W. A. Schutte, B. Barbier, A. A. Segovia, H.
Rosenbauer, W. H. P. Thiemann, A. Brack, J. M. Greenberg, Nature 2002, 416, 403. [34] W. Gilbert, Nature 1986, 319, 618.

Anhang 18 Literaturverzeichnis
XXV
[35] C. R. Woese, The genetic code. The molecular basis for genetic expression., Harper & Row, New York, 1967, pp. 200.
[36] F. H. C. Crick, J. Mol. Biol. 1968, 38, 367. [37] L. E. Orgel, J. Mol. Biol. 1968, 38, 381. [38] M. Eigen, Naturwissenschaften 1971, 58, 465. [39] M. Eigen, P. Schuster, Naturwissenschaften 1977, 64, 541. [40] M. Eigen, P. Schuster, Naturwissenschaften 1978, 65, 7. [41] M. Eigen, P. Schuster, Naturwissenschaften 1978, 65, 341. [42] H.-D. Försterling, H. Kuhn, K.-H. Tews, Angew. Chem. Int. Ed. Engl. 1972, 11, 821. [43] H. Kuhn, Angew. Chem. Int. Ed. Engl. 1972, 11, 798. [44] H. Kuhn, J. Waser, Angew. Chem. Int. Ed. Engl. 1981, 20, 500. [45] D. Baltimore, Nature 1970, 226, 1209. [46] H. M. Temin, S. Mizutani, Nature 1970, 226, 1211. [47] A. J. Zaug, T. R. Cech, Cell 1980, 19, 331. [48] T. R. Cech, A. J. Zaug, P. J. Grabowski, Cell 1981, 27, 487. [49] K. Kruger, P. J. Grabowski, A. J. Zaug, J. Sands, D. E. Gottschling, T. R. Cech, Cell
1982, 31, 147. [50] C. Guerriertakada, K. Gardiner, T. Marsh, N. Pace, S. Altman, Cell 1983, 35, 849. [51] T. R. Cech, Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 4360. [52] J. Dworkin, S. Miller, Origins Life Evol. Biosphere 1996, 26, 412. [53] W. D. Fuller, R. A. Sanchez, L. E. Orgel, J. Mol. Biol. 1972, 67, 25. [54] W. D. Fuller, L. E. Orgel, R. A. Sanchez, J. Mol. Evol. 1972, 1, 249. [55] L. E. Orgel, Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99. [56] C. Anastasi, F. F. Buchet, M. A. Crowe, A. L. Parkes, M. W. Powner, J. M. Smith, J.
D. Sutherland, Chem. Biodiversity 2007, 4, 721. [57] J. D. Sutherland, Angew. Chem. Int. Ed. Engl. 2007, 46, 2354. [58] P. J. Unrau, D. P. Bartel, Nature 1998, 395, 260. [59] A. Eschenmoser, E. Loewenthal, Chem. Soc. Rev. 1992, 21, 1. [60] L. E. Orgel, Nature 1992, 358, 203. [61] A. Eschenmoser, Tetrahedron 2007, 63, 12821. [62] R. Naylor, P. T. Gilham, Biochemistry 1966, 5, 2722. [63] L. E. Orgel, R. Lohrmann, Acc. Chem. Res. 1974, 7, 368. [64] J. Sulston, R. Lohrmann, L. E. Orgel, H. T. Miles, Proc. Natl. Acad. Sci. U.S.A. 1968,
59, 726. [65] J. Sulston, R. Lohrmann, L. E. Orgel, H. Schneider-Bernloehr, B. J. Weimann, H. T.
Miles, J. Mol. Biol. 1969, 40, 227. [66] B. J. Weimann, R. Lohrmann, L. E. Orgel, H. Schneider-Bernloehr, J. E. Sulston,
Science 1968, 161, 387. [67] H. L. Sleeper, R. Lohrmann, L. E. Orgel, J. Mol. Evol. 1979, 13, 203. [68] R. Lohrmann, P. K. Bridson, L. E. Orgel, Science 1980, 208, 1464. [69] T. Inoue, L. E. Orgel, J. Am. Chem. Soc. 1981, 103, 7666. [70] T. Inoue, L. E. Orgel, Science 1983, 219, 859. [71] T. Inoue, G. F. Joyce, K. Grzeskowiak, L. E. Orgel, J. M. Brown, C. B. Reese, J. Mol.
Biol. 1984, 178, 669. [72] G. von Kiedrowski, Angew. Chem. Int. Ed. Engl. 1986, 25, 932. [73] W. S. Zielinski, L. E. Orgel, Nature 1987, 327, 346. [74] E. Szathmary, I. Gladkih, J. Theor. Biol. 1989, 138, 55. [75] G. von Kiedrowski, B. Wlotzka, J. Helbing, Angew. Chem. Int. Ed. Engl. 1989, 28,
1235.

Anhang 18 Literaturverzeichnis
XXVI
[76] G. von Kiedrowski, B. Wlotzka, J. Helbing, M. Matzen, S. Jordan, Angew. Chem. Int. Ed. Engl. 1991, 30, 423.
[77] G. von Kiedrowski, Bioorg. Chem. Front. 1993, 3, 113. [78] D. Sievers, G. von Kiedrowski, Nature 1994, 369, 221. [79] D. Sievers, G. von Kiedrowski, Chem. Eur. J. 1998, 4, 629. [80] T. Achilles, G. von Kiedrowski, Angew. Chem. Int. Ed. Engl. 1993, 32, 1198. [81] A. Luther, R. Brandsch, G. von Kiedrowski, Nature 1998, 396, 245. [82] N. Paul, G. F. Joyce, Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12733. [83] T. A. Lincoln, G. F. Joyce, Science 2009, 323, 1229. [84] B. J. Lam, G. F. Joyce, Nat. Biotechnol. 2009, 27, 288. [85] D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin, M. R. Ghadiri, Nature 1996, 382,
525. [86] P. E. Dawson, T. W. Muir, I. Clarklewis, S. B. H. Kent, Science 1994, 266, 776. [87] K. Severin, D. H. Lee, J. A. Martinez, M. Vieth, M. R. Ghadiri, Angew. Chem. Int. Ed.
Engl. 1998, 37, 126. [88] D. H. Lee, K. Severin, Y. Yokobayashi, M. R. Ghadiri, Nature 1997, 390, 591. [89] A. Saghatelian, Y. Yokobayashi, K. Soltani, M. R. Ghadiri, Nature 2001, 409, 797. [90] S. Yao, I. Ghosh, R. Zutshi, J. Chmielewski, J. Am. Chem. Soc. 1997, 119, 10559. [91] S. Yao, I. Ghosh, R. Zutshi, J. Chmielewski, Angew. Chem. Int. Ed. Engl. 1998, 37,
478. [92] R. Issac, J. Chmielewski, J. Am. Chem. Soc. 2002, 124, 6808. [93] X. Q. Li, J. Chmielewski, Org. Biomol. Chem 2003, 1, 901. [94] X. Q. Li, J. Chmielewski, J. Am. Chem. Soc. 2003, 125, 11820. [95] T. Tjivikua, P. Ballester, J. Rebek, J. Am. Chem. Soc. 1990, 112, 1249. [96] J. S. Nowick, Q. Feng, T. Tjivikua, P. Ballester, J. Rebek, J. Am. Chem. Soc. 1991,
113, 8831. [97] F. M. Menger, A. V. Eliseev, N. A. Khanjin, J. Am. Chem. Soc. 1994, 116, 3613. [98] M. M. Conn, E. A. Wintner, J. Rebek, J. Am. Chem. Soc. 1994, 116, 8823. [99] F. M. Menger, A. V. Eliseev, N. A. Khanjin, M. J. Sherrod, J. Org. Chem. 1995, 60,
2870. [100] E. A. Wintner, B. Tsao, J. Rebek, J. Org. Chem. 1995, 60, 7997. [101] D. N. Reinhoudt, D. M. Rudkevich, F. de Jong, J. Am. Chem. Soc. 1996, 118, 6880. [102] V. Rotello, J. I. Hong, J. Rebek, J. Am. Chem. Soc. 1991, 113, 9422. [103] A. Terfort, G. von Kiedrowski, Angew. Chem. Int. Ed. Engl. 1992, 31, 654. [104] B. Wang, I. O. Sutherland, Chem. Commun. 1997, 16, 1495. [105] M. Kindermann, I. Stahl, M. Reimold, W. M. Pankau, G. von Kiedrowski, Angew.
Chem. Int. Ed. Engl. 2005, 44, 6750. [106] V. C. Allen, D. Philp, N. Spencer, Org. Lett. 2001, 3, 777. [107] E. Kassianidis, D. Philp, Angew. Chem. Int. Ed. Engl. 2006, 45, 6344. [108] A. Dieckmann, S. Beniken, C. Lorenz, N. Doltsinis, G. von Kiedrowski, J. Syst.
Chem., 1, 10. [109] A. Dieckmann, S. Beniken, C. D. Lorenz, N. L. Doltsinis, G. von Kiedrowski, Chem.
Eur. J. 2011, 17, 468. [110] P. E. Nielsen, M. Egholm, R. H. Berg, O. Buchardt, Science 1991, 254, 1497. [111] D. D. Weller, D. T. Daly, W. K. Olson, J. E. Summerton, J. Org. Chem. 1991, 56,
6000. [112] F. H. Westheimer, Science 1987, 235, 1173. [113] M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H.
Berg, S. K. Kim, B. Norden, P. E. Nielsen, Nature 1993, 365, 566. [114] K. K. Jensen, H. Orum, P. E. Nielsen, B. Norden, Biochemistry 1997, 36, 5072.

Anhang 18 Literaturverzeichnis
XXVII
[115] H. Rasmussen, J. Sandholm, Nat. Structural Biol. 1997, 4, 98. [116] P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm, B. Norden, Nature 1994, 368,
561. [117] P. E. Nielsen, Acc. Chem. Res. 1999, 32, 624. [118] M. Eriksson, P. E. Nielsen, Q. Rev. Biophys. 1996, 29, 369. [119] H. Okihana, C. Ponnamperuma, Nature 1982, 299, 347. [120] D. W. Deamer, Nature 1985, 317, 792. [121] D. W. Deamer, R. M. Pashley, Origins Life Evol. Biosphere 1989, 19, 21. [122] J. W. Szostak, D. P. Bartel, P. L. Luisi, Nature 2001, 409, 387. [123] P. L. Luisi, Anat. Rec. 2002, 268, 208. [124] P. L. Luisi, T. Oberholzer, A. Lazcano, Helv. Chim. Acta 2002, 85, 1759. [125] M. M. Hanczyc, S. M. Fujikawa, J. W. Szostak, Science 2003, 302, 618. [126] P. L. Luisi, The Emergence of Life – From Chemical Origins to Synthetic Biology,
Cambridge University Press, New York, 2006. [127] S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobe, D. A. Treco, J. W. Szostak,
Nature 2008, 454, 122. [128] S. S. Mansy, J. W. Szostak, Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13351. [129] T. Ganti, Az elet principuma (The Principles of Life), Gondolat, Budapest, 1971. [130] T. Ganti, BioSystems 1975, 7, 15. [131] T. Ganti, Az elet principuma (The Principles of Life), 2nd rev. ed., Gondolat,
Budapest, 1978. [132] T. Ganti, The Principles of Life, with a commentary by James Griesemer and Eörs
Szathmary, Oxford University Press, Oxford, 2003. [133] T. Ganti, Chemoton Theory, Volume I: Theory of Fluid Machineries, Kluwer
Academic/Plenum, New York, 2003. [134] T. Ganti, Chemoton Theory, Volume II: Theory of Living Systems, Kluwer
Academic/Plenum, New York, 2003. [135] Protocells – Bridging Nonliving and Living Matter, Eds. S. Rasmussen, M. A. Bedau,
L. Chen, D. Deamer, D. C. Krakauer, N. H. Packard, P. F. Stadler, MIT Press, Cambridge, London, 2009.
[136] www.istpace.org, Stand 10.10.2011. [137] S. Rasmussen, L. Chen, D. Deamer, D. C. Krakauer, N. H. Packard, P. F. Stadler, M.
A. Bedau, Science 2004, 303, 963. [138] T. Rocheleau, S. Rasmussen, P. E. Nielsen, M. N. Jacobi, H. Ziock, Phil. Trans. R.
Soc. B 2007, 362, 1841. [139] C. G. Simmons, A. E. Pitts, L. D. Mayfield, J. W. Shay, D. R. Corey, Bioorg. Med.
Chem. Lett. 1997, 7, 3001. [140] L. Good, S. K. Awasthi, R. Dryselius, O. Larsson, P. E. Nielsen, Nat. Biotechnol.
2001, 19, 360. [141] A. Püschl, S. Sforza, G. Haaima, O. Dahl, P. E. Nielsen, Tetrahedron Lett. 1998, 39,
4707. [142] L. D. Fader, M. Boyd, Y. S. Tsantrizos, J. Org. Chem. 2001, 66, 3372. [143] A. Sen, P. E. Nielsen, Nucleic Acids Res. 2007, 35, 3367. [144] K. Ogura, C. T. Migita, T. Yamada, Chem. Lett. 1988, 1563. [145] K. Nelson, S. Miller, Origins Life Evol. Biosphere 1996, 26, 345. [146] K. E. Nelson, M. Levy, S. L. Miller, Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 3868. [147] U. J. Meierhenrich, G. M. M. Caro, J. H. Bredehoft, E. K. Jessberger, W. H. P.
Thiemann, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9182. [148] P. E. Nielsen, Origins Life Evol. Biosphere 1993, 23, 323.

Anhang 18 Literaturverzeichnis
XXVIII
[149] V. V. Demidov, V. N. Potaman, M. D. Frankkamenetskii, M. Egholm, O. Buchard, S. H. Sonnichsen, P. E. Nielsen, Biochem. Pharmacol. 1994, 48, 1310.
[150] M. Eriksson, L. Christensen, J. Schmidt, G. Haaima, L. Orgel, P. E. Nielsen, New J. Chem. 1998, 22, 1055.
[151] P. E. Nielsen, Chem. Biodiversity 2007, 4, 1996. [152] T. Plöger, 2006-2010. [153] L. E. Orgel, TIBS 1998, 23, 491. [154] L. E. Orgel, J. Mol. Evol. 1989, 29, 465. [155] www.panagene.com, Stand 10.11.2010. [156] M. Gorlero, R. Wieczorek, K. Adamala, A. Giorgi, M. E. Schininà, P. Stano, P. L.
Luisi, FEBS Letters 2009, 583, 153. [157] P. Wittung, P. Nielsen, B. Norden, J. Am. Chem. Soc. 1997, 119, 3189. [158] Y. Krishnan-Ghosh, E. Stephens, S. Balasubramanian, J. Am. Chem. Soc. 2004, 126,
5944. [159] B. Datta, M. E. Bier, S. Roy, B. A. Armitage, J. Am. Chem. Soc. 2005, 127, 4199. [160] B. Petersson, B. B. Nielsen, H. Rasmussen, I. K. Larsen, M. Gajhede, P. E. Nielsen, J.
S. Kastrup, J. Am. Chem. Soc. 2005, 127, 1424. [161] M. Murtola, R. Strömberg, Org. Biomol. Chem. 2008, 6, 3837. [162] M. Murtola, M. Wenska, R. Strömberg, J. Am. Chem. Soc. 2010, 132, 8984. [163] M. Murtola, R. Strömberg, ARKIVOC 2009, 4. [164] G. Joyce, L. Orgel, S. Miller, Origins Life Evol. Biospheres 1986, 16, 445. [165] G. F. Joyce, G. M. Visser, C. A. A. van Boeckel, J. H. van Boom, L. E. Orgel, J. van
Aestrenen, Nature 1984, 310, 602. [166] J. G. Schmidt, P. E. Nielsen, L. E. Orgel, J. Am. Chem. Soc. 1997, 119, 1494. [167] P. Wittung, M. Eriksson, R. Lyng, P. E. Nielsen, B. Norden, J. Am. Chem. Soc. 1995,
117, 10167. [168] I. A. Kozlov, L. E. Orgel, P. E. Nielsen, Angew. Chem. Int. Ed. Engl. 2000, 39, 4292. [169] S. Sforza, G. Haaima, R. Marchelli, P. E. Nielsen, Eur. J. Org. Chem. 1999, 197. [170] P. E. Nielsen, Origins Life Evol. Biosphere 2007, 37, 323. [171] C. Bohler, P. E. Nielsen, L. E. Orgel, Nature 1995, 376, 578. [172] J. G. Schmidt, L. Christensen, P. E. Nielsen, L. E. Orgel, Nucleic Acids Res. 1997, 25,
4792. [173] J. G. Schmidt, P. E. Nielsen, L. E. Orgel, Nucleic Acids Res. 1997, 25, 4797. [174] A. R. Hill, L. E. Orgel, T. F. Wu, Origins Life Evol. Biosphere 1993, 23, 285. [175] A. Mattes, O. Seitz, Chem. Commun. 2001, 2050. [176] A. Mattes, O. Seitz, Angew. Chem. Int. Ed. Engl. 2001, 40, 3178. [177] S. Ficht, C. Dose, O. Seitz, ChemBioChem 2005, 6, 2098. [178] R. E. Kleiner, Y. Brudno, M. E. Birnbaum, D. R. Liu, J. Am. Chem. Soc. 2008, 130,
4646. [179] Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel, M. R. Ghadiri, Science 2009, 325, 73. [180] J. M. Heemstra, D. R. Liu, J. Am. Chem. Soc. 2009, 131, 11347. [181] B. Hyrup, M. Egholm, O. Buchardt, P. E. Nielsen, Bioorg. Med. Chem. Lett. 1996, 6,
1083. [182] I. Stahl, G. von Kiedrowski, J. Am. Chem. Soc. 2006, 128, 14014. [183] M. A. Santos, A. Afonso, M. M. Marques, C. Wilcox, Supramol. Chem. 2000, 11,
201. [184] C. S. Wilcox, Frontiers in Supramolecular Organic Chemistry and Photochemistry,
VCH, Weinheim, New York, Basel, Cambridge, 1991. [185] G. von Kiedrowski, 1990-2003.

Anhang 18 Literaturverzeichnis
XXIX
[186] P. E. Nielsen, Peptide Nucleic Acids: Protocols and Applications, Horizon Bioscience, Wymondham, 2004.
[187] S. A. Noble, M. A. Bonham, J. E. Bisi, D. A. Bruckenstein, P. H. Brown, S. C. Brown, R. Cadilla, M. D. Gaul, J. C. Hanvey, C. F. Hassman, J. A. Josey, M. J. Luzzio, P. M. Myers, A. J. Pipe, D. J. Ricca, C. W. Su, C. L. Stevenson, S. A. Thomson, R. W. Wiethe, L. E. Babiss, Drug Develop. Res. 1995, 34, 184.
[188] P. Sazani, S. H. Kang, M. A. Maier, C. F. Wei, J. Dillman, J. Summerton, M. Manoharan, R. Kole, Nucleic Acids Res. 2001, 29, 3965.
[189] B. P. Gangamani, V. A. Kumar, K. N. Ganesh, Biochem. Biophys. Res. Commun. 1997, 240, 778.
[190] P. E. Nielsen, G. Haaima, A. Lohse, O. Buchardt, Angew. Chem. Int. Ed. Engl. 1996, 35, 1939.
[191] E. Lesnik, F. Hassman, J. Barbeau, K. Teng, K. Weiler, R. Griffey, S. Freier, Nucleosides and Nucleotides 1997, 16, 1775
[192] F. Bergmann, W. Bannwarth, S. Tam, Tetrahedron Lett. 1995, 36, 6823. [193] E. Uhlmann, D. W. Will, G. Breipohl, D. Langner, A. Ryte, Angew. Chem. Int. Ed.
Engl. 1996, 35, 2632. [194] G. M. Bonora, S. Drioli, M. Ballico, A. Faccini, R. Corradini, S. Cogoi, L. Xodo,
Nucleosides, Nucleotides Nucleic Acids 2007, 26, 661 [195] P. R. Berthold, T. Shiraishi, P. E. Nielsen, Bioconjugate Chem. 2010, 21, 1933. [196] B. D. Gildea, S. Casey, J. MacNeill, H. Perry-O'Keefe, D. Sørensen, J. M. Coull,
Tetrahedron Lett. 1998, 39, 7255. [197] Y. L. Jiang, L. M. McDowell, B. Poliks, D. R. Studelska, C. Cao, G. S. Potter, J.
Schaefer, F. Song, J. T. Stivers, Biochemistry 2004, 43, 15429. [198] C. Kreutz, H. Kählig, R. Konrat, R. Micura, J. Am. Chem. Soc. 2005, 127, 11558. [199] G. L. Olsen, T. E. Edwards, P. Deka, G. Varani, S. T. Sigurdsson, G. P. Drobny,
Nucleic Acids Res. 2005, 33, 3447. [200] M. Hennig, M. L. Munzarova, W. Bermel, L. G. Scott, V. Sklenar, J. R. Williamson,
J. Am. Chem. Soc. 2006, 128, 5851. [201] C. Kreutz, H. Kählig, R. Konrat, R. Micura, Angew. Chem. Int. Ed. Engl. 2006, 45,
3450. [202] M. Hennig, L. G. Scott, E. Sperling, W. Bermel, J. R. Williamson, J. Am. Chem. Soc.
2007, 129, 14911. [203] N. B. Barhate, R. N. Barhate, P. Cekan, G. Drobny, S. T. Sigurdsson, Org. Lett. 2008,
10, 2745. [204] D. Graber, H. Moroder, R. Micura, J. Am. Chem. Soc. 2008, 130, 17230. [205] B. Puffer, C. Kreutz, U. Rieder, M.-O. Ebert, R. Konrat, R. Micura, Nucleic Acids Res.
2009, 37, 7728. [206] N. Shibata, B. K. Das, H. Honjo, Y. Takeuchi, J. Chem. Soc. Perkin Trans. 1 2001,
1605. [207] K. A. Frey, S. A. Woski, Chem. Commun. 2002, 2206. [208] M. Hollenstein, C. J. Leumann, Org. Lett. 2003, 5, 1987. [209] M. Hollenstein, C. J. Leumann, J. Org. Chem. 2005, 70, 3205. [210] B. Kuhnast, F. Dolle, B. Tavitian, J. Labelled Cpd. Radiopharm. 2002, 45, 1. [211] B. A. Schweitzer, E. T. Kool, J. Org. Chem. 1994, 59, 7238. [212] E. T. Kool, H. O. Sintim, Chem. Commun. 2006, 3665. [213] E. Atherton, R. C. Sheppard, Solid Phase Peptide Synthesis: A Practical Approach,
IRL Press at Oxford University Press, Oxford, 1989.

Anhang 18 Literaturverzeichnis
XXX
[214] S. L. Mellor, D. A. Wellings, J.-A. Fehrentz, M. Paris, J. Martinez, N. J. Ede, A. M. Bray, D. J. Evans, G. B. Bloomberg, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press, Oxford, 2000.
[215] A. W. van der Made, R. H. van der Made, J. Org. Chem. 1993, 58, 1262. [216] M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen Chemie,
6th ed., Thieme, Stuttgart, New York, 2002. [217] H. G. O. Becker, W. Berger, G. Domschke, E. Fanghänel, J. Faust, M. Fischer, F.
Gentz, K. Gewald, R. Gluch, R. Mayer, K. Müller, D. Pavel, H. Schmidt, K. Schollberg, K. Schwetlick, E. Seiler, G. Zeppenfeld, Organikum, 21st ed., Wiley-VCH, Weinheim, 2001.
[218] J. M. Coull, M. Egholm, R. P. Hodge, M. Ismail, S. B. Rajur, PCT Int. Appl. WO 96/40685 1996.
[219] F. Debaene, N. Winssinger, Org. Lett. 2003, 5, 4445. [220] M. Ashwell, C. Bleasdale, B. T. Golding, I. K. O'Neill, Chem. Soc., Chem. Commun.
1990, 955. [221] J. Kjellberg, N. G. Johansson, Nucleosides, Nucleotides Nucleic Acids 1989, 8, 225 [222] E. P. Heimer, H. E. Gallotorres, A. M. Felix, M. Ahmad, T. J. Lambros, F. Scheidl, J.
Meienhofer, Int. J. Peptide Protein Res. 1984, 23, 203. [223] G. Breipohl, EP0672647 1995. [224] M. Node, K. Nishide, M. Sai, E. Fujita, Tetrahedron Lett. 1978, 52, 5211. [225] M. Node, K. Nishide, M. Sai, K. Fuji, E. Fujita, J. Org. Chem. 1981, 46, 1991. [226] M. Gude, J. Ryf, P. White, Lett. Pept. Sci. 2002, 9, 203. [227] R. Hoffmann, G. Bril, L. Otvos, J. Chromatogr., A 1998, 814, 111. [228] Y. Wei, M. Marino, B. Thompson, J. E. Girard, J. Chromatogr., A 1999, 864, 49. [229] L. D. Mayfield, D. R. Corey, Anal. Biochem. 1999, 268, 401. [230] L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M.
Egholm, O. Buchardt, P. E. Nielsen, J. Coull, R. H. Berg, J. Pept. Sci. 1995, 1, 175. [231] S. I. Kirin, F. Noor, N. Metzler-Nolte, J. Chem. Educ. 2007, 84, 108. [232] U. Boas, J. Brask, K. J. Jensen, Chem. Rev. 2009, 109, 2092. [233] D. Sarantakis, J. J. Bicksler, Tetrahedron Lett. 1997, 38, 7325. [234] A. Farese, N. Patino, R. Condom, S. Dalleu, R. Guedj, Tetrahedron Lett. 1996, 37,
1413. [235] A. Farese, S. Pairot, N. Patino, V. Ravily, R. Condom, A. Aumelas, R. Guedj,
Nucleosides and Nucleotides 1997, 16, 1893. [236] C. Di Giorgio, S. Pairot, C. Schwergold, N. Patino, R. Condom, A. Farese-Di Giorgio,
R. Guedj, Tetrahedron 1999, 55, 1937. [237] G. Depecker, C. Schwergold, C. Di Giorgio, N. Patino, R. Condom, Tetrahedron Lett.
2001, 42, 8303. [238] M. L. Leroux, C. Di Giorgio, N. Patino, R. Condom, Tetrahedron Lett. 2002, 43,
1641. [239] C. Schwergold, G. Depecker, C. Di Giorgio, N. Patino, F. Jossinet, B. Ehresmann, R.
Terreux, D. Cabrol-Bass, R. Condom, Tetrahedron 2002, 58, 5675. [240] G. Depecker, N. Patino, C. Di Giorgio, R. Terreux, D. Cabrol-Bass, C. Bailly, A. M.
Aubertin, R. Condom, Org. Biomol. Chem. 2004, 2, 74. [241] S. Caldarelli, G. Depecker, N. Patino, A. Di Giorgio, T. Barouillet, A. Doglio, R.
Condom, Bioorg. Med. Chem. Lett. 2004, 14, 4435. [242] J. C. Verheijen, G. M. Grotenbreg, L. H. de Ruyter, P. A. M. van der Klein, G. A. van
der Marel, J. H. van Boom, Tetrahedron Lett. 2000, 41, 3991. [243] G. Upert, M. Mehiri, M.-L. Goddard, A. Di Giorgio, R. Benhida, R. Condom, N.
Patino, Tetrahedron Lett. 2005, 46, 4081.

Anhang 18 Literaturverzeichnis
XXXI
[244] F. Z. Dörwald, Side Reactions in Organic Synthesis, 1st ed., Wiley-VCH, Weinheim, 2005.
[245] T. W. Greene, P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd ed., Wiley-Interscience, New York, 1999.
[246] G. Breipohl, J. Knolle, D. Langner, G. O'Malley, E. Uhlmann, Bioorg. Med. Chem. Lett. 1996, 6, 665.
[247] M. Bodanszky, A. Bodanszky, The Practice of Peptide Synthesis, 2nd ed., Springer-Verlag, Berlin, Heidelberg, 1995.
[248] F. Guibé, Tetrahedron 1998, 54, 2967. [249] D. W. Dodd, R. H. E. Hudson, Electrophoresis 2007, 28, 3884. [250] S.-M. Chen, V. Mohan, J. S. Kiely, M. C. Griffith, R. H. Griffey, Tetrahedron Lett.
1994, 35, 5105. [251] M. Leijon, A. Graslund, P. E. Nielsen, O. Buchardt, B. Norden, S. M. Kristensen, M.
Eriksson, Biochemistry 1994, 33, 9820. [252] L. Betts, J. A. Josey, J. M. Veal, S. R. Jordan, Science 1995, 270, 1838. [253] S. C. Brown, S. A. Thomson, J. M. Veal, D. G. Davis, Science 1994, 265, 777. [254] S. Berger, S. Braun, H. O. Kalinowski, NMR-Spektroskopie von Nichtmetallen, Vol. 4,
Thieme, Stuttgart, 1994. [255] M. Koppitz, P. E. Nielsen, L. E. Orgel, J. Am. Chem. Soc. 1998, 120, 4563. [256] T. Tenforde, R. A. Fawwaz, N. K. Freeman, N. Castagnoli, J. Org. Chem. 1972, 37,
3372. [257] I. Yavari, J. D. Roberts, J. Org. Chem. 1978, 43, 4689. [258] A. Williams, I. T. Ibrahim, J. Am. Chem. Soc. 1981, 103, 7090. [259] P. Hagenbuch, E. Kervio, A. Hochgesand, U. Plutowski, C. Richert, Angew. Chem.
Int. Ed. Engl. 2005, 44, 6588. [260] J. A. R. Stütz, E. Kervio, C. Deck, C. Richert, Chem. Biodiversity 2007, 4, 784. [261] S. R. Vogel, C. Richert, Chemical Commun. 2007, 1896. [262] M. Röthlingshöfer, E. Kervio, T. Lommel, U. Plutowski, A. Hochgesand, C. Richert,
Angew. Chem. Int. Ed. Engl. 2008, 47, 6065. [263] K. Gießler, H. Griesser, D. Göhringer, T. Sabirov, C. Richert, Eur. J. Org. Chem.
2010, 2010, 3611. [264] E. Kervio, A. Hochgesand, U. E. Steiner, C. Richert, Proc. Natl. Acad. Sci. U.S.A.
2010, 107, 12074. [265] M. Röthlingshöfer, C. Richert, J. Org. Chem. 2010, 75, 3945. [266] C. F. Anderson, M. T. Record, Annu. Rev. Phys. Chem. 1995, 46, 657. [267] S.-i. Nakano, M. Fujimoto, H. Hara, N. Sugimoto, Nucleic Acids Res. 1999, 27, 2957. [268] M. T. Record, Biopolymers 1975, 14, 2137. [269] V. K. Misra, D. E. Draper, Biopolymers 1998, 48, 113. [270] M. J. Serra, J. D. Baird, T. Dale, B. L. Fey, K. Retatagos, E. Westhof, RNA 2002, 8,
307. [271] N. Nakajima, Y. Ikada, Bioconjugate Chem. 1995, 6, 123. [272] G. T. Hermanson, Bioconjugate Techniques, 2nd ed., Academic Press, London,
Amsterdam, Burlington, San Diego, 2008. [273] S.-i. Nakano, L. Wu, H. Oka, H. T. Karimata, T. Kirihata, Y. Sato, S. Fujii, H. Sakai,
M. Kuwahara, H. Sawai, N. Sugimoto, Mol. BioSyst. 2008, 4, 579. [274] S.-i. Nakano, H. T. Karimata, Y. Kitagawa, N. Sugimoto, J. Am. Chem. Soc. 2009,
131, 16881. [275] M. Z. Markarian, J. B. Schlenoff, J. Phys. Chem. B 2010, 114, 10620. [276] S. B. Zimmerman, A. P. Minton, Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27. [277] K. Luby-Phelps, Int. Rev. Cytol., 2000, 192, 189.

Anhang 18 Literaturverzeichnis
XXXII
[278] R. J. Ellis, Curr. Opin. Struct. Biol. 2001, 11, 114. [279] P. A. Monnard, A. Kanavarioti, D. W. Deamer, J. Am. Chem. Soc. 2003, 125, 13734. [280] H. Trinks, W. Schröder, C. K. Biebricher, Origins Life Evol. Biosphere 2005, 35, 429. [281] P. A. Monnard, J. W. Szostak, J. Inorg. Bio. 2008, 102, 1104. [282] P. A. Monnard, H. Ziock, Chem. Biodiversity 2008, 5, 1521. [283] J. Attwater, A. Wochner, V. B. Pinheiro, A. Coulson, P. Holliger, Nat. Commun.
2010, 1, 9. [284] C. Dose, O. Seitz, Org. Lett. 2005, 7, 4365. [285] C. Dose, S. Ficht, O. Seitz, Angew. Chem. Int. Ed. Engl. 2006, 45, 5369. [286] G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M.
Stoltz, J. E. Bercaw, K. I. Goldberg, Organometallics 2010, 29, 2176. [287] H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512.





























