Embed Size (px)
Citation preview

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Forschungsvorhaben SAFIRA
Teilprojekt B 3.2:
Zeolith-gestützte Katalysatoren zur Hydrodehalogenierung und Hydrierung von Schadstoffen im Grundwasser
Endbericht
Projektleiter: Dr. Christoph Schüth Mitarbeiter: Dipl. Chem. Nikolai-Alexeji Kummer Laufzeit des Vorhabens: 01.07.99 – 30.06.02
Das diesem Bericht zugrundeliegende Vorhaben wurde mit Mitteln des Bundesministeriums
für Bildung und Forschung unter dem Förderkennzeichen 02WT9941/1 gefördert. Die Verantwortung für den Inhalt dieser Veröffentlichung liegt beim Autor.
Tübingen, den 30.01.2003

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Inhaltsverzeichnis
1. Einleitung und Aufgabenstellung 4
2. Voraussetzungen, Planung und Ablauf des Vorhabens 4
3. Wissenschaftlicher und technischer Stand zu Projektbeginn 5
4. Zusammenarbeit mit anderen Stellen 6
5. Grundlagen 6 5.1 Katalyse in wässriger Phase 7 5.2 Zeolithe als Trägermaterial 8
5.2.1 Faujasite 9 5.2.2 Herstellung zeolithgestützter Katalysatoren 10
6. Ergebnisse 12 6.1 Pilotanlage 12
6.1.1 Einlasskonzentrationen 12 6.1.2 Versuchsaufbau 14 6.1.3 Versuche in der Pilotanlage 16
6.1.3.1 Versuch 1 16 6.1.3.2 Versuch 2 19 6.1.3.3 Regeneration deaktivierter Katalysatoren 20
6.2 Begleitende Untersuchungen 23 6.2.1 Versuchsaufbauten 23 6.2.2 Analytik 24 6.2.3 Referenzprobe 25 6.2.4 Einfluss verbreiteter ionischer Wasserinhaltsstoffen 26
6.2.4.1 Natruimsulfat 26 6.2.4.2 Natriumsulfid 27 6.2.4.3 Kupfersulfat 27 6.2.4.4 Eisen(II)sulfat 28 6.2.4.5 Natriumnitrat 29 6.2.4.6 Ionenmischungen 30 6.2.4.7 Schlussfolgerungen 31
6.2.5 Untersuchungen zur speziellen Problematik in Bitterfeld 32 6.2.5.1 Schwefelwasserstofffracht des Grundwassers 32 6.2.5.2 Schwefelwasserstoffbildung in den Reaktoren 34 6.2.5.3 Schwefelorganika 36
6.2.6 Palladiumfreisetzung 38
2

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
7. Zusammenfassende Bewertung 41
8. Ausblick 45
9. Verwertbarkeit der Ergebnisse 49
10. Fortschritte anderer Arbeitsgruppen 50
11. Publikationen, Vorträge und Patente 52
12. Literaturverzeichnis 53
3

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
1. Einleitung und Aufgabenstellung Im folgenden Abschlussbericht werden die Aktivitäten im Rahmen des vom Bundesministerium für Bildung und Forschung geförderten SAFIRA-Teilprojekts B 3.2: „Zeolith-gestützte Katalysatoren zur Hydrodehalogenierung und Hydrierung von Schadstoffen im Grundwasser“ (Förderkenn-zeichen: 02WT9941/1) über die Projektlaufzeit vom 01.07.1999 bis zum 31.06.2002 zusammenge-fasst. Der Abschlußbericht stützt sich dabei auf die während der Projektlaufzeit abgefassten Zwi-schenberichte, darüber hinaus werden auch die bisher unveröffentlichten Ergebnisse der dreijährigen Projektlaufzeit eingearbeitet. Der Ergebnisteil dieses Bericht gliedert sich in zwei Teile. Im ersten Teil werden Ergebnisse aus den Laboruntersuchungen dargestellt. Im zweiten Teil wer-den die Befunde aus dem Betrieb der Pilotanlage in Bitterfeld diskutiert.
Aufgabenstellung und Ziel dieses Teilprojekts war es, in Laborversuchen das Potenzial sowie die Limitierungen einer reduktiven katalytischen Dehalogenierung und Hydrierung halogenierter aro-matischer und aliphatischer CKW-Gemische in wässriger Phase zu ermitteln. Darüber hinaus sollte die Langzeitstabilität der entwickelten Edelmetallkatalysatoren beim Einsatz in der in Bitterfeld er-richteten Pilotanlage unter Atmosphärendruck und Grundwassertemperaturen überprüft werden.
2. Voraussetzungen, Planung und Ablauf des Vorhabens Dieses SAFIRA-Teilprojekt war eines der drei abiotisch katalytisch ausgerichteten Projekte, die im Rahmen des Projektverbundes SAFIRA durchgeführt wurden. Thematisch baute dieses Projekt auf den Vorarbeiten des Antragstellers zum Abbau von chlorierten organischen Schadstoffen mittels Edelmetallkatalysatoren, vorwiegend auf Palladiumbasis, auf (Schüth und Reinhard, 1998). Nach-dem in diesen Untersuchungen Laborversuche unter Idealbedingungen in deionisiertem Wasser durchgeführt wurden, war das Hauptziel des anschließenden SAFIRA-Teilprojektes, die Erkennt-nisse der Laborversuche auf den Feldmaßstab zu übertragen.
Im Vorlauf zur Antragstellung wurden in der mobilen Testeinheit des Umweltforschungszentrums Leipzig/Halle (UFZ) am späteren Standort der SAFIRA Pilotanlage on-site Tests zur Anwendung des Verfahrens in der Praxis durchgeführt. In diesen Tests konnte prinzipiell die Eignung des Ver-fahrens zur Behandlung der Grundwasserkontamination an diesem Standort nachgewiesen wer-den (Schüth, 1999; in UFZ Bericht 17/99). Daraufhin wurde ein Projektantrag für eine dreijährige Projektlaufzeit gestellt, in der das Verfahren als Sanierungsvariante zum kommerziellen Einsatz weiterentwickelt werden sollte. Grundkonzept des Antrages war die Aufteilung der Arbeiten in ei-nen Laborteil, der im Hydrogeochemischen Labor des Lehrstuhls für Angewandte Geologie in Tü-bingen abgearbeitet werden sollte, und in einen Geländeteil, für den ein Versuchsstand in der zu errichtenden Pilotanlage in Bitterfeld eingerichtet werden sollte. Der Laborteil war so angelegt, dass, neben einem vorab planbaren Versuchsprogramm, flexibel auf Problemstellungen, die sich aus dem Anlagenbetrieb in der Pilotanlage ergeben könnten, reagiert werden konnte.
4

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Mit Beginn der Projektförderung zum 01.07.1999 konnte das Laborversuchsprogramm in Tübingen begonnen werden. Die Inbetriebnahme des Versuchsstandes in der Bitterfelder Pilotanlage verzö-gerte sich, auf Grund von Problemen bei der Ansteuerung der Förderpumpen für die zwei installier-ten Reaktormodule, bis zum Dezember 2000. Mit dem Ende der Förderung zum 30. 06. 2002 wurde das Projekt abgeschlossen.
3. Wissenschaftlicher und technischer Stand zu Projektbeginn Bei den klassischen abiotischen Sanierungstechniken für kontaminierte Grundwässer erfolgt in der Regel nur eine Verlagerung und Aufkonzentrierung von Schadstoffen in einer zweiten Phase (z.B. Aktivkohle), die eine Nachbehandlung verlangt. Ein grundlegend anderer Sanierungsansatz beruht auf Methoden die von vornherein schadstoffzerstörenden Charakter aufweisen, also keinen weite-ren Aufbereitungsschritt erfordern. Dazu gehören z.B. thermische, oxidative oder biologische Me-thoden. Anfang der 90er Jahre wurde das Potential von 0-wertigem Eisen zur reduktiven Dehalogenierung von chlorierten Schadstoffen erkannt (Gillham und O’Hannesin, 1994; Matheson und Tratnyek, 1994) und es wird seit einigen Jahren mit guten Erfolgen in sog. Reaktiven Wänden zur in situ-Grundwassersanierung eingesetzt (Gillham, 1993). Die Einsatzmöglichkeiten dieser Me-thode beschränken sich jedoch im wesentlichen auf Schadensfälle mit chlorierten aliphatischen Schadstoffen wie Trichlorethylen (TCE) und Perchlorethylen (PCE) da eine Reaktivität gegenüber anderen relevanten Schadstoffgruppen, z.B. aromatische Verbindungen wie Chlorbenzole oder chlorierte Biphenyle, nicht gegeben ist. Selbst für TCE oder PCE muss mit Halbwertszeiten im Stundenbereich gerechnet werden (Arnold und Roberts 2000).
Auf Edelmetallen beruhende katalytische Verfahren haben den Vorteil, dass eine breite Reaktivität bei aromatischen und aliphatischen halogenierten Verbindungen gegeben ist (Hoke et al., 1992; Kovenklioglu et al. 1992; Schreier und Reinhard, 1995; Sclimm und Heitz, 1996; Schüth und Rein-hard, 1998). Außerdem sind schnelle Reaktionsraten mit Halbwertszeiten im Bereich von Sekun-den bis wenigen Minuten erreichbar (Lowry und Reinhard, 1999). Idealerweise wird der Katalysator in der Reaktion nicht verbraucht. Damit sind theoretisch sehr lange Standzeiten der Katalysatoren möglich. Eine Gruppe der Engelhard Corp. meldete bereits 1992 und 1993 in den USA 2 Patente zur Hydrodehalogenierung halogenierter organischer Verbindungen in wässriger Phase mit Palla-dium auf Aktivkohle und unterschiedlichen Reduktionsmitteln unter Umgebungsbedingungen oder bei leicht erhöhten Druck/Temperaturbedingungen an (US-Pat. No. 5,177,268, Balko et al. 1992, und 5,196,617, Kovenklioglu et al. 1993). Unter anderem wird die Dechlorierung von Chlorbenzo-len beschrieben.
Das Hauptproblem, welches bisher eine Anwendung dieses Verfahrens in der Praxis verhindert, ist die schnelle Deaktivierung von Edelmetallkatalysatoren durch die oft komplexe Grundwasserche-mie. Besonders die Sorption schwefelhaltiger ionischer Wasserinhaltsstoffe auf den katalytisch ak-tiven Metalloberflächen kann zum vollständigen Verlust der Abbaueigenschaften führen (Siantar et
5

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
al., 1996). Die Langzeitstabilität ist jedoch Voraussetzung für einen wirtschaftlichen und damit kon-kurrenzfähigen Einsatz dieser Methode.
Die einzige uns bekannte Feldanwendung eines auf Palladium basierenden katalytischen Verfah-rens zur Grundwassersanierung wurde auf dem Gelände des Lawrence Livermore National Lab (LLNL) durchgeführt. Hier wurde ein Katalysator (Palladium auf Aluminiumoxid) direkt in einer Brunnenbohrung eingebaut und elektrochemisch mit Wasserstoff versorgt (McNab et al., 2000). In diesem Fall musste der Katalysator täglich mehrere Stunden mit atmosphärischer Luft regeneriert werden.
4. Zusammenarbeit mit anderen Stellen Während der gesamten Projektlaufzeit gab es eine ausgezeichnete Zusammenarbeit mit der Arbeitsgruppe von Prof. Dr. F.-D. Kopinke (Teilprojekt B 3.1), die einen ähnlichen Ansatz zur katalytischen Behandlung des Grundwassers im Bereich der Pilotanlage verfolgte. Diese Zusammenarbeit wurde zum Ende der Projektlaufzeit auf die Entwicklung eines verbesserten katalytischen Ansatzes, der die Erfahrungen aus dem Betrieb der Pilotanlage berücksichtigt, ausgeweitet.
Die weltweit mitführende Arbeitsgruppe im Bereich der Grundwassersanierung mittels reduktiv katalytischer Verfahren ist die von Prof. Dr. M. Reinhard an der Stanford University in den USA. Mit Prof. Reinhard bestand ein reger Informationsaustausch, der auch wechselseitige Besuche einschloss.
Spezialanalytik zur Katalysatorcharakterisierung (Röntgen-Photoelektronspektroskopie (XPS)) wurde am Max-Planck Institut für Kohlenforschung im Mülheim (Prof. Dr. F. Schüth) durchgeführt. Dieses Institut ist ausgewiesenermassen eine der führenden Institutionen im Bereich der heterogenen Katalyse.
5. Grundlagen Palladium ist bei Anwesenheit von Wasserstoff in der Lage, eine reduktive Dehalogenierung von chlorierten Kohlenwasserstoffen in wässriger Phase bei Umgebungsbedingungen zu katalysieren. Dazu wird das Palladium auf einen geeigneten Träger in Pelletform aufgebracht um ein körniges, schüttfähiges Material zu erhalten. Dieser Katalysator kann dann, z.B. in Säulen eingebaut, im Durchfluss betrieben werden. Über ein geeignetes Dosiersystem muss dem zu behandelnden Wasser Wasserstoff in stöchiometrisch ausreichender Menge zugeführt werden. Endprodukt der
6

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Dehalogenierung von aliphatischen Kohlenwasserstoffen, wie z.B. TCE oder VC, ist Ethan. Das frei werdende Chlor wird in ionischer Form als Chlorid abgeführt.
In diesem Projekt wurden trägergestützte Katalysatoren eingesetzt, die von der Degussa AG nach Spezifikation angefertigt wurden. Darüber hinaus wurden für spezielle Fragestellungen auch eigene Katalysatoren präpariert. Im Folgenden wird deshalb auch kurz auf die Katalysatorpräpa-ration eingegangen.
5.1 Katalyse in wässriger Phase
Katalytische Hydrierungen oder Hydrodehalogenierungen von organischen Verbindungen werden in der chemischen Synthese häufig eingesetzt, typischerweise jedoch bei hohen Wasserstoffdrü-cken und Temperaturen sowie in der Gasphase oder organischen Lösemitteln. Das Potential die-ser Methode für den Einsatz im Grundwasserbereich ist jedoch enorm. Abb. 5.1 zeigt einige Beispielreaktionen für typische aromatische Schadstoffe in wasserstoffgesättigter wässriger Phase
Das Hauptproblem, welches bisher eine Anwendung dieses Ve
mit einem kommerziellen Palladiumkatalysator (1% Pd auf Al2O3).
ens in der Praxis verhindert, ist jedoch die schnelle Deaktivierung von Edelmetallkatalysatoren durch die oft komplexe Grundwas-
rfahr
C l
C 6 H 4 C l 2 C 6 H 5 C l C 6 H 6
C l C l R 1 R 2
R 3
C12H9Cl C12H10 C1 2 H 1 6 C12H22
Cl
R7
R6R5
Cl
C l Cl
C l C l
C 6 H6C l 6 C 6 H 6
C l R 8
C 1 0 H 1 2C 1 0 H 8 R 1 0 R 9
C14H18
C 1 4 H 1 8 C14H14
C14H10 C12H22
C14H12?
I
III
II I V
VI
V
Abb. 5.1: Beispielreaktionen aromatischer (bzw. cyclischer) Verbindungen mit einem Palldiumkatalysator in was-serstoffgesättigter wässriger Phase unter Umgebungsbedingungen (Schüth und Reinhard, 1998).
serchemie. Besonders die Sorption schwefelhaltiger ionischer Wasserinhaltsstoffe auf den kataly-tisch aktiven Metalloberflächen kann zum vollständigen Verlust der Abbaueigenschaften führen. Aus diesem Grund wurde in Vorstudien zu diesem Projekt ein Ansatz entwickelt, der längere Standzeiten der Katalysatoren durch den Einsatz stark hydrophober Trägermaterialien für die kata-lytisch aktive Komponente Palladium ermöglichen sollte.
7

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
5.2 Zeolithe als Trägermaterialien
Zeolithe sind mikroporöse, kristalline Aluminosil0,25 nm bis 1nm. Die Hohlraumdimensionen li
ikate mit einem Poreninnendurchmesser zwischen egen damit im Bereich von Durchmessern organi-
rdalkali-Gruppe) kompensiert, die sich in den Kanälen
l-Verhältnisses durch Desaluminierung werden Zeolithe unpolarer. Gleich-
scher Moleküle. Es gibt sowohl natürliche Zeolithe als auch synthetisch hergestellte Zeolithe, de-ren Struktur oft auf den natürlichen Kristallgittern beruhen. Die Gitter werden in allen Fällen aus SiO4 und AlO4
- Tetraedern aufgebaut, die über gemeinsame Sauerstoffatome zu einer dreidimen-sionalen Raumnetzstruktur verknüpft sind.
Die aufgrund der AlO4- - Einheiten hervorgerufene negative Ladung im Aluminosilikatgerüst wird
durch Kationen (meist aus der Alkali- und Eund Hohlräumen des Gitters befinden. Sie haben keine stützende Funktion und lassen sich auch aufgrund der hohen Porosität der Zeolithe leicht substituieren; Zeolithe werden deshalb häufig auch als Ionenaustauscher eingesetzt. Zu beachten ist hierbei allerdings, dass bei Änderung der Kationengröße oder bei Ersatz der einwertigen durch mehrwertige Kationen die Querschnitte der Porenöffnungen beeinflusst werden. Damit ist es möglich, die Zugänge zu den Porenöffnungen durch den Ionenaustausch zu erweitern oder zu verengen, wodurch spezifische Molekültrennun-gen möglich werden.
Durch Variation des Si/Al-Verhältnisses lassen sich Zeolithe unterschiedlicher Polarität herstellen. Bei Erhöhung des Si/Azeitig wird das Eindringen von (hydrophoben) Kohlenwasserstoffen in die wasserfreien Gerüste er-leichtert - zum einen aufgrund der geringeren Zahl austauschbarer Kationen, zum anderen aber auch wegen der mit fallendem Aluminiumgehalt abnehmenden negativen Ladung und der damit verbundenen steigenden Hydrophobie (Oleophilie) des Gerüstes. Der Übergang von einer eher hydrophilen Struktur zu einer eher hydrophoben Struktur wird bei einem Si/Al-Verhältnis von ca. 7 erreicht (Flanigen, 1991; Weitkamp et al., 1992).
Abb. 5.2: Kubooktaeder als Zeolithgrundkörper. Links: Sodalithkäfig (β-Käfig), ausgefüllte Punkte stellen Si- bzw. Al-Atome dar, nicht ausgefüllte Punkte O-Atome. Rechts: Sodalithkäfig (schematisch)
8

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Das Bauprinzip zeolithischer Strukturen erschließt sich am einfachsten, wenn man von einem Ku-booktaeder (auch abgestumpfter Oktaeder, Tetrakaidekaeder (Vierzehnflächner) bezeichnet) aus-geht. Besetzt man die Eckpunkte dieses Kubooktaeders abwechselnd mit SiO4 und AlO4
- Tetra-edern (Primärbaugruppen), so erhält man ein aus Vier- und Sechs- Ringen zusammengesetztes Aluminosilikatgerüst. Dieses aus insgesamt 24 Tetraedern bestehende Grundgerüst wird als Soda-lith oder β-Käfig bezeichnet (Abb. 5.2). Aus dieser Sekundärbaugruppe lassen sich wichtige Zeo-lithgrundstrukturen wie Sodalith, Zeolith A und Faujasit entwickeln.
5.2.1 Faujasite (Zeolith X bzw. Y)
Durch Verknüpfung der Sodalith-Käfige über hexagonale Prismen (Doppel-Sechsringe) zu einem der Diamantstruktur analogen Gitter ergibt sich die Struktur von Faujasit (Zeolith X bei niedrigen Si/Al-Verhältnissen, Zeolith Y bei hohen Si/Al-Verhältnissen). Die Sodalitheinheiten sind hier in tetraedrischer Anordnung jeweils über sechs Sauerstoffatome verbunden. Der Ausschnitt der Struktur in Abb. 5.3 zeigt einen durch zehn Sodalitheinheiten gebildeten α-Käfig. Dieser ist durch vier tetraedrisch angeordnete, aus zwölf Sauerstoffatomen gebildeten Öffnungen mit benachbarten α-Käfigen verbunden. Der Durchmesser des α-Käfigs beträgt 0,92 nm. Entscheidender als der Durchmesser des α-Käfigs ist allerdings die Porenöffnung des dreidimensionalen Porensystems, die 0,74 nm beträgt (weitporiger Zeolith). Die kristallographische Elementarzelle wird aus einer An-ordnung von acht α-Käfigen gebildet.
Abb. 5.3: Faujasit-Struktur (Zeolith X, Zeolith Y): Verknüpfung der Sodalithkäfige über hexagonale Prismen (Doppel-Sechsringe). Der Durchmesser der Porenöffnungen beträgt 0.74 nm.
Die Poren in einem Zeolith sind zwar sehr regelmäßig, doch muss das Gitter als in Grenzen flexi-bel betrachtet werden. Durch Gitterschwingungen können z.B. beim Zeolithen Y auch Moleküle mit einem Durchmesser von bis zu 0,95 nm in die Poren eindringen.
9

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
In dem hier vorgestelltem Projekt wurde (neben Aluminiumoxid) speziell der Zeolith Y200 als Trä-germaterial verwendet. Die Zahl 200 gibt hierbei das Si/Al- Verhältnis an, womit es sich bei diesem Zeolith also um einen sehr hydrophoben, unpolaren Zeolith handelt. Durch Verwendung dieses hydrophoben, kationenarmen Zeolithen als Trägermaterial sollte das Eindringen von polaren Ver-bindungen (insbesondere von Ionen) verhindert werden, während unpolare organische Schadstof-fe weiterhin (ungehindert) in die Poren gelangen können, sofern ein Eindringen der Schadstoffe nicht durch ihre Größe bzw. sterische Anordnung behindert wird. Damit sollte es möglich sein, das katalytisch aktive Metall (zumindest in den Poren) vor einem Angriff von potentiell deaktivierenden Ionen zu schützen, auch wenn deaktivierende Ionen im Gewässer vorhanden sind. Dieser Effekt konnte in Modellsystemen für Sulfit nachgewiesen werden (Schüth et al. 2000).
5.2.2 Herstellung zeolithgestützter Katalysatoren
Zur Einbringung des katalytisch aktiven Edelmetalls (z.B. Palladium) auf ein Trägermaterial (z.B. Zeolithe) stehen verschiedene Verfahren zur Verfügung, z.B. Tränkimprägnierung, In situ Synthese und Ionenaustausch. In dieser Projekt wurde ausschließlich das Verfahren der Tränkimprägnie-rung angewandt: Bei dieser Präparationstechnik erhält man die höchste Dispersion des eingesetz-ten Edelmetalls so dass die so hergestellten Katalysatoren eine sehr gute katalytische Aktivität zeigen. Abb. 5.4 zeigt mittels Tränkimprägnierung hergestellte Katalysatoren mit Zeolithen als Trä-gern. Die Katalysatorpräparation mittels Tränkimprägnierung kann in verschiedene Teilschritte aufgeteilt werden:
1. Tränkimprägnierung: Bei einem pulverförmigen Trägermaterial („Incipient-Wetness“- Impräg-nierung) wird zunächst die Wasseraufnahmekapazität des Trägermaterials bestimmt. Hierzu wird eine bestimmte Menge Trägermaterial eingewogen und im Mörser langsam soviel Wasser zuge-geben, bis das Trägermaterial gut durchfeuchtet ist, aber kein Wasser übersteht. In die so ermittel-
100 nm100 nm
Abb. 5.4: Links: Pulverförmige Zeolithe, pelletiert und mit Palladium imprägniert. Rechts: Transmissionselektronenmik-roskopische Aufnahme (TEM) von einem Zeolith Y200 tränkimprägniert mit 1 % Palladium.
10

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
te Menge Wasser wird die gewünschte Menge Komplexsalz (z.B. Palladium(II)-tetramminchlorid, [Pd(NH3)4Cl2)]) gelöst und diese Lösung wird unter mörsern tropfenweise zu einer entsprechenden Menge trockenem Trägermaterials zugegeben. Liegt das Trägermaterial in Pelletform vor wird das Verfahren etwas modifiziert. Die Metallsalzlösung wird in einem Überschuss an Wasser gelöst und dem Trägermaterial zugegeben, so dass Trägermaterial vollständig mit Wasser bedeckt ist. Unter vorsichtigem Rühren lässt man die Salzlösung (über Nacht) eintrocknen.
2. Trocknung: Das durchfeuchtete, imprägnierte Trägermaterial wird in einem vorgeheizten Ofen für 2 Stunden bei 400°C getrocknet.
3. Calcinierung: Das nach der Trocknung erhaltene Material wird nun einer ca. fünfstündige Be-handlung an Luft im Muffelofen bei 550°C unterzogen, nachdem der Ofen mit einer Aufheizrate von 1 C/min auf diese Temperatur gebracht worden ist.
4. Reduktion: Nach der Calcinierung erfolgt die Reduktion des Metallsalzes in einem Röhrenofen im Wasserstoffstrom. Das zu reduzierende Pulver oder die Pellets werden dazu in das Quarzrohr des Röhrenofens eingebracht. Der Röhrenofen wird daraufhin im Stickstoffstrom (80 ml/min) mit einer Heizrate von 10 C/min auf 400°C aufgeheizt. Die Reduktion erfolgt nun im Wasserstrom (50 ml/min) bei 400°C über 4 Stunden. Nun lässt man den trägergebundenen Katalysator im leichten Stickstoffstrom (ca. 5 ml/min) abkühlen.
In diesem Projekt wurden sowohl Palladium- als auch Rhodiumkatalysatoren auf dem als Träger-material eingesetzten Zeolithen Y200 mit einem Edelmetallgehalt von 0,5 % synthetisiert. Als was-serlösliche Metallsalze wurden Palladium(II)-tetramminchlorid (Pd(NH3)4Cl2)) bzw. Rhodium(III)-chlorid (RhCl3 x H2O, Rh-Gehalt 38-41 %) verwendet. Das Mikroporenvolumen Vmicro des verwen-deten Trägermaterials beträgt nach Herstellerangaben (Degussa AG) 0,3 cm3/g. Die Trocken-raumdichte der Zeolithe beträgt ca. 0.5 g/cm³ bei einer Größe der Pellets von ca. 3x2mm. Die Porosität einer Schüttung beträgt ca. 50%.
11

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
6. Ergebnisse Der Ergebnisteil dieses Berichts gliedert sich in zwei Teile. Im ersten Teil werden die Ergebnisse aus dem Betrieb in der Bitterfelder Pilotanlage zusammengefasst. Im zweiten Teil werden die be-gleitenden Untersuchungen, im wesentlichen Laborversuche sowie ein weiterer Feldversuche in Backnang bei Stuttgart, dargestellt.
6.1 Pilotanlage
Abb. 6.1: Bitterfelder Pilotanlage mit Schachtbau-werken und Laborgebäude.
Auf Grund von Schwierigkeiten bei der Ansteuerung der Grundwasserförderpumpen für die Reaktoren dieses Teilprojekts wurde die endgültige Inbetrieb-nahme der Pilotanlage (Abb. 6.1) erst Anfang Dezember 1999 realisiert. Danach wurden die Tests zur Langzeitstabilität der entwickelten Edelmetall-katalysatoren beim Einsatz unter Atmosphärendruck und Grundwassertemperaturen durchgeführt. Dabei konnte die vorhandene Infrastruktur in der Pilot-anlage, etwa das Standortlabor zur Durchführung der chemischen Analytik, für die Einstellung und Überwachung der Prozessabläufe genutzt werden.
6.1.1 Einlasskonzentrationen
Die Hauptkontaminante im Grundwasser an der Pilotanlage ist eindeutig Chlorbenzol mit einem Anteil von ca. 95 % an dem Gesamtgehalt der identifizierten organischen Schadstoffe. Darüber-hinaus sind 1,4-Dichlorbenzol und 1,2-Dichlorbenzol sowie Benzol über den gesamten Versuchs-zeitraum nachweisbar. Chlorierte Ethene (VC, TCE und PCE) sowie Ethane (1,1,2,2 TCA) werden nicht regelmäßig nachgewiesen und spielen im Bereich der Pilotanlage eine untergeordnete Rolle.
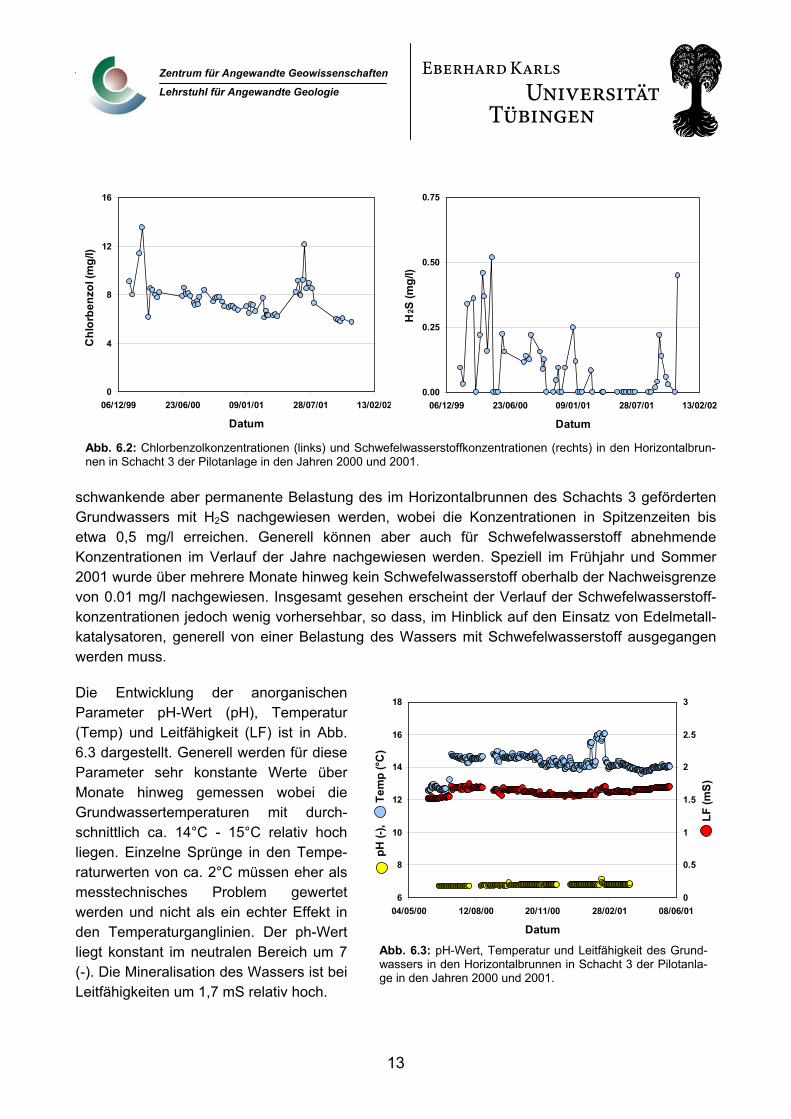
Neben den Chlorbenzolkonzentrationen sind die Schwefelwasserstoffkonzentrationen (H2S) im gefördertem Grundwasser für dieses Teilprojekt von besonderer Bedeutung, da diese Verbindung ein stark deaktivierendes Potential für Edelmetallkatalysatoren aufweist. Deshalb sind in Abb. 6.2 die Konzentrationsverläufe für Chlorbenzol (links) und Schwefelwasserstoff (rechts) im Horizontalbrunnen des Schachtes 3 in den Jahren 2000 und 2001 dargestellt. Die Chlorbenzol-konzentrationen zeigen einen leichten Rückgang im Verlauf der Jahre und liegen zum Ende des Beobachtungszeitraumes bei ca. 6 mg/l. Als Spitzenwerte werden Konzentrationen über 12 mg/l gemessen.
Grundsätzlich stellt die Schwefelwasserstoffproblematik eines der Hauptprobleme im Routinebetrieb der Reaktoren in der Pilotanlage dar. So kann über weite Zeiträume eine zwar stark
12
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
schwankende aber permanente Belastung des im Horizontalbrunnen des Schachts 3 geförderten Grundwassers mit H2S nachgewiesen werden, wobei die Konzentrationen in Spitzenzeiten bis etwa 0,5 mg/l erreichen. Generell können aber auch für Schwefelwasserstoff abnehmende Konzentrationen im Verlauf der Jahre nachgewiesen werden. Speziell im Frühjahr und Sommer 2001 wurde über mehrere Monate hinweg kein Schwefelwasserstoff oberhalb der Nachweisgrenze von 0.01 mg/l nachgewiesen. Insgesamt gesehen erscheint der Verlauf der Schwefelwasserstoff-konzentrationen jedoch wenig vorhersehbar, so dass, im Hinblick auf den Einsatz von Edelmetall-katalysatoren, generell von einer Belastung des Wassers mit Schwefelwasserstoff ausgegangen werden muss.
Die Entwicklung der anorganischen Parameter pH-Wert (pH), Temperatur (Temp) und Leitfähigkeit (LF) ist in Abb. 6.3 dargestellt. Generell werden für diese Parameter sehr konstante Werte über Monate hinweg gemessen wobei die Grundwassertemperaturen mit durch-schnittlich ca. 14°C - 15°C relativ hoch liegen. Einzelne Sprünge in den Tempe-raturwerten von ca. 2°C müssen eher als messtechnisches Problem gewertet werden und nicht als ein echter Effekt in den Temperaturganglinien. Der ph-Wert liegt konstant im neutralen Bereich um 7 (-). Die Mineralisation des Wassers ist bei Leitfähigkeiten um 1,7 mS relativ hoch.
18 3
0
4
8
12
06/12/99 23/06/00 09/01/01 28/07/01 13/02/02
Datum
Chl
orbe
nzol
(mg/
l)
0.00
0.25
0.50
06/12/99 23/06/00 09/01/01 28/07/01 13/02/02
DatumH
2S (m
g/l)
Abb. 6.2: Chlorbenzolkonzentrationen (links) und Schwefelwasserstoffkonzentrationen (rechts) in den Horizontalbrun-nen in Schacht 3 der Pilotanlage in den Jahren 2000 und 2001.
0.7516
6
8
10
12
14
16
04/05/00 12/08/00 20/11/00 28/02/01 08/06/01
Datum
pH (-
),
T
emp
(°C)
0
0.5
1
1.5
2
2.5
LF (m
S)
Abb. 6.3: pH-Wert, Temperatur und Leitfähigkeit des Grund-wassers in den Horizontalbrunnen in Schacht 3 der Pilotanla-ge in den Jahren 2000 und 2001.
13
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
0
50
100
150
200
250
01/02/00 11/05/00 19/08/00 27/11/00
Datum
Kon
zent
ratio
n (m
g/l)
Ca2+
K+
Na+
Mg2+
0
200
400
600
800
1000
01/01/00 07/03/00 12/05/00 17/07/00
DatumK
onze
ntra
tion
(mg/
l) SO42-
Cl-
Abb. 6.4: Hauptkationen (links) und Hauptanionen (rechts, ohne HCO3-) in den Horizontalbrunnen in Schacht 3 der
Pilotanlage im Jahr 2000.
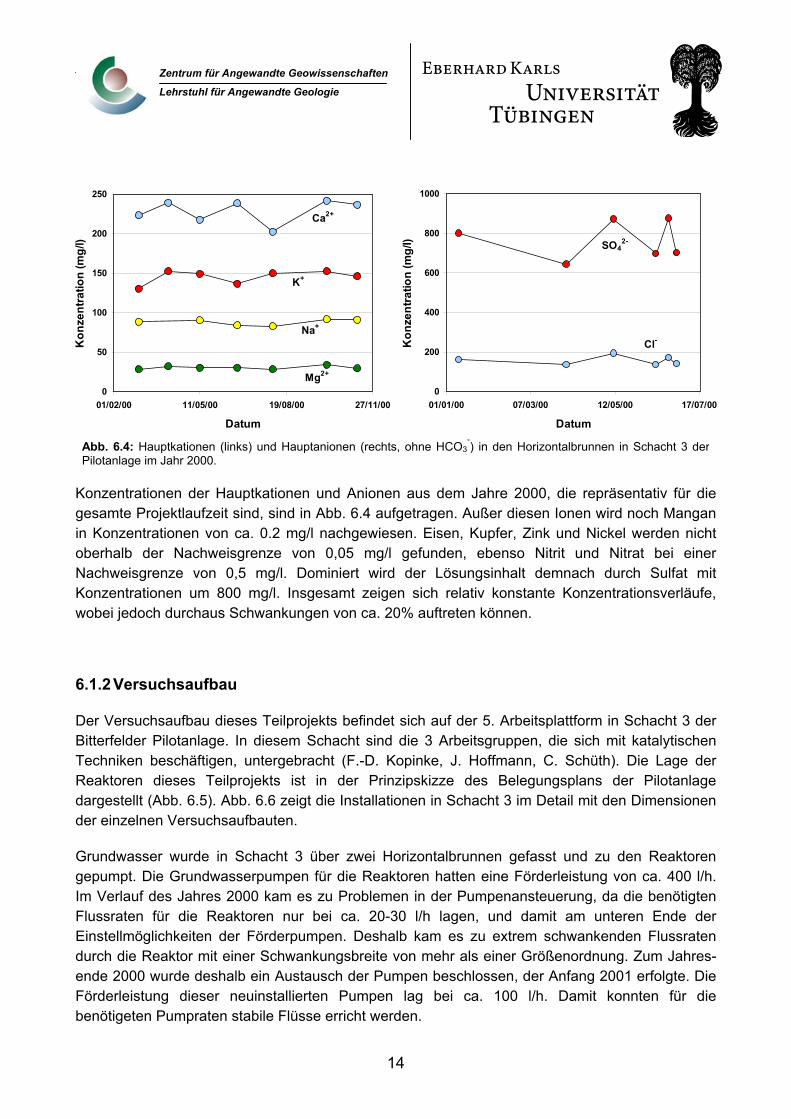
Konzentrationen der Hauptkationen und Anionen aus dem Jahre 2000, die repräsentativ für die gesamte Projektlaufzeit sind, sind in Abb. 6.4 aufgetragen. Außer diesen Ionen wird noch Mangan in Konzentrationen von ca. 0.2 mg/l nachgewiesen. Eisen, Kupfer, Zink und Nickel werden nicht oberhalb der Nachweisgrenze von 0,05 mg/l gefunden, ebenso Nitrit und Nitrat bei einer Nachweisgrenze von 0,5 mg/l. Dominiert wird der Lösungsinhalt demnach durch Sulfat mit Konzentrationen um 800 mg/l. Insgesamt zeigen sich relativ konstante Konzentrationsverläufe, wobei jedoch durchaus Schwankungen von ca. 20% auftreten können.
6.1.2 Versuchsaufbau
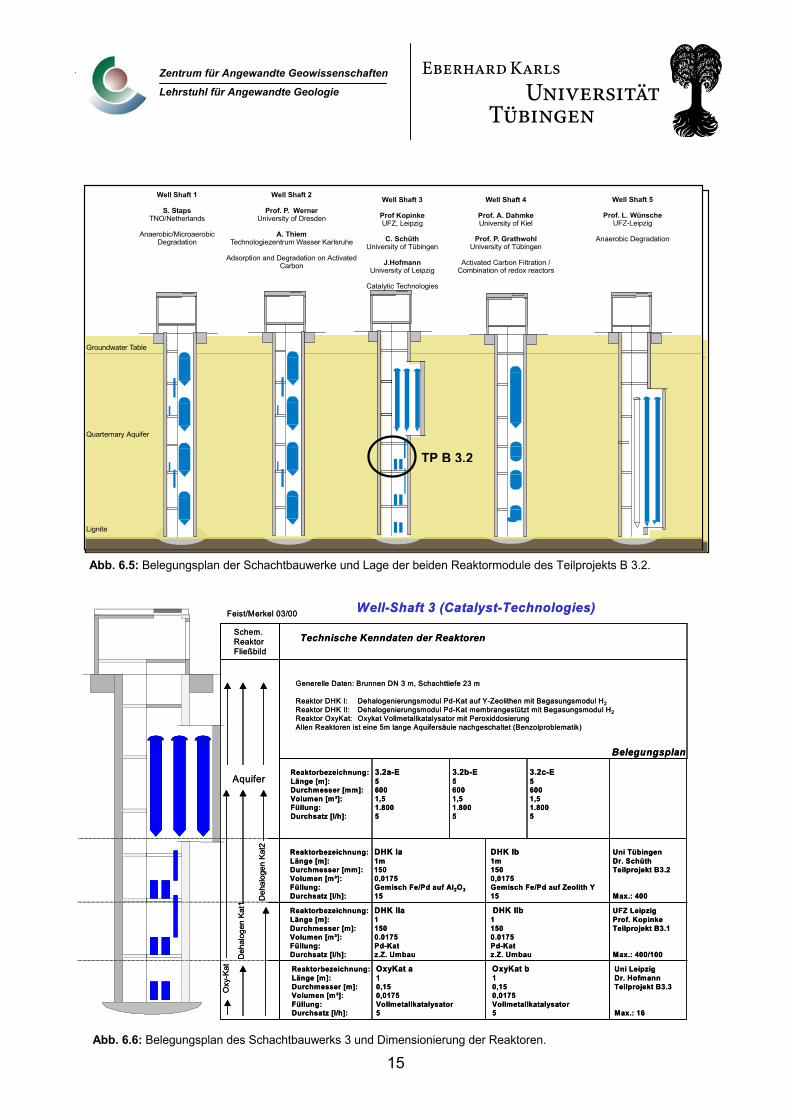
Der Versuchsaufbau dieses Teilprojekts befindet sich auf der 5. Arbeitsplattform in Schacht 3 der Bitterfelder Pilotanlage. In diesem Schacht sind die 3 Arbeitsgruppen, die sich mit katalytischen Techniken beschäftigen, untergebracht (F.-D. Kopinke, J. Hoffmann, C. Schüth). Die Lage der Reaktoren dieses Teilprojekts ist in der Prinzipskizze des Belegungsplans der Pilotanlage dargestellt (Abb. 6.5). Abb. 6.6 zeigt die Installationen in Schacht 3 im Detail mit den Dimensionen der einzelnen Versuchsaufbauten.
Grundwasser wurde in Schacht 3 über zwei Horizontalbrunnen gefasst und zu den Reaktoren gepumpt. Die Grundwasserpumpen für die Reaktoren hatten eine Förderleistung von ca. 400 l/h. Im Verlauf des Jahres 2000 kam es zu Problemen in der Pumpenansteuerung, da die benötigten Flussraten für die Reaktoren nur bei ca. 20-30 l/h lagen, und damit am unteren Ende der Einstellmöglichkeiten der Förderpumpen. Deshalb kam es zu extrem schwankenden Flussraten durch die Reaktor mit einer Schwankungsbreite von mehr als einer Größenordnung. Zum Jahres-ende 2000 wurde deshalb ein Austausch der Pumpen beschlossen, der Anfang 2001 erfolgte. Die Förderleistung dieser neuinstallierten Pumpen lag bei ca. 100 l/h. Damit konnten für die benötigeten Pumpraten stabile Flüsse erricht werden.
14
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
15
Well Shaft 3
Prof Kopinke
C. Schüth
J.Hofmann
UFZ, Leipzig
University of Tübingen
University of Leipzig
Catalytic Technologies
Well Shaft 4
Prof. A. Dahmke
Prof. P. Grathwohl
University of Kiel
University of Tübingen
Activated Carbon Filtration /Combination of redox reactors
Well Shaft 5
Prof. L. WünscheUFZ-Leipzig
Anaerobic Degradation
Groundwater Table
Quarternary Aquifer
Lignite
Well Shaft 2
Prof. P. Werner
A. Thiem
University of Dresden
Technologiezentrum Wasser Karlsruhe
Adsorption and Degradation on Activated Carbon
Well Shaft 1
S. StapsTNO/Netherlands
Anaerobic/Microaerobic Degradation
Well Shaft 3
Prof Kopinke
C. Schüth
J.Hofmann
UFZ, Leipzig
University of Tübingen
University of Leipzig
Catalytic Technologies
Well Shaft 4
Prof. A. Dahmke
Prof. P. Grathwohl
University of Kiel
University of Tübingen
Activated Carbon Filtration /Combination of redox reactors
Well Shaft 5
Prof. L. WünscheUFZ-Leipzig
Anaerobic Degradation
Groundwater Table
Quarternary Aquifer
Lignite
Well Shaft 2
Prof. P. Werner
A. Thiem
University of Dresden
Technologiezentrum Wasser Karlsruhe
Adsorption and Degradation on Activated Carbon
Well Shaft 1
S. StapsTNO/Netherlands
Anaerobic/Microaerobic Degradation
TP B 3.2
Abb. 6.5: Belegungsplan der Schachtbauwerke und Lage der beiden Reaktormodule des Teilprojekts B 3.2.
Well-Shaft 3 (Catalyst-Technologies)
Technische Kenndaten der Reaktoren
Belegungsplan
Reaktorbezeichnung: 3.2a-E 3.2b-E 3.2c-ELänge [m]: 5 5 5Durchmesser [mm]: 600 600 600Volumen [m³]: 1,5 1,5 1,5Füllung: 1.800 1.800 1.800Durchsatz [l/h]: 5 5 5
Reaktorbezeichnung: DHK Ia DHK Ib Uni TübingenLänge [m]: 1m 1m Dr. SchüthDurchmesser [mm]: 150 150 Teilprojekt B3.2Volumen [m³]: 0,0175 0,0175 Füllung: Gemisch Fe/Pd auf Al2O3 Gemisch Fe/Pd auf Zeolith YDurchsatz [l/h]: 15 15 Max.: 400
Reaktorbezeichnung: DHK IIa DHK IIb UFZ LeipzigLänge [m]: 1 1 Prof. KopinkeDurchmesser [m]: 150 150 Teilprojekt B3.1Volumen [m³]: 0.0175 0.0175Füllung: Pd-Kat Pd-KatDurchsatz [l/h]: z.Z. Umbau z.Z. Umbau Max.: 400/100
Reaktorbezeichnung: OxyKat a OxyKat b Uni LeipzigLänge [m]: 1 1 Dr. HofmannDurchmesser [m]: 0,15 0,15 Teilprojekt B3.3Volumen [m³]: 0,0175 0,0175Füllung: Vollmetallkatalysator VollmetallkatalysatorDurchsatz [l/h]: 5 5 Max.: 16
Generelle Daten: Brunnen DN 3 m, Schachttiefe 23 m
Reaktor DHK I: Dehalogenierungsmodul Pd-Kat auf Y-Zeolithen mit Begasungsmodul H2Reaktor DHK II: Dehalogenierungsmodul Pd-Kat membrangestützt mit Begasungsmodul H2Reaktor OxyKat: Oxykat Vollmetallkatalysator mit PeroxiddosierungAllen Reaktoren ist eine 5m lange Aquifersäule nachgeschaltet (Benzolproblematik)
Schem. Reaktor Fließbild
Aquifer
Oxy
-Kat
Deh
alog
enK
at1 D
ehal
ogen
Kat
2
Feist/Merkel 03/00 Well-Shaft 3 (Catalyst-Technologies)
Technische Kenndaten der Reaktoren
Belegungsplan
Reaktorbezeichnung: 3.2a-E 3.2b-E 3.2c-ELänge [m]: 5 5 5Durchmesser [mm]: 600 600 600Volumen [m³]: 1,5 1,5 1,5Füllung: 1.800 1.800 1.800Durchsatz [l/h]: 5 5 5
Reaktorbezeichnung: DHK Ia DHK Ib Uni TübingenLänge [m]: 1m 1m Dr. SchüthDurchmesser [mm]: 150 150 Teilprojekt B3.2Volumen [m³]: 0,0175 0,0175 Füllung: Gemisch Fe/Pd auf Al2O3 Gemisch Fe/Pd auf Zeolith YDurchsatz [l/h]: 15 15 Max.: 400
Reaktorbezeichnung: DHK IIa DHK IIb UFZ LeipzigLänge [m]: 1 1 Prof. KopinkeDurchmesser [m]: 150 150 Teilprojekt B3.1Volumen [m³]: 0.0175 0.0175Füllung: Pd-Kat Pd-KatDurchsatz [l/h]: z.Z. Umbau z.Z. Umbau Max.: 400/100
Reaktorbezeichnung: OxyKat a OxyKat b Uni LeipzigLänge [m]: 1 1 Dr. HofmannDurchmesser [m]: 0,15 0,15 Teilprojekt B3.3Volumen [m³]: 0,0175 0,0175Füllung: Vollmetallkatalysator VollmetallkatalysatorDurchsatz [l/h]: 5 5 Max.: 16
Generelle Daten: Brunnen DN 3 m, Schachttiefe 23 m
Reaktor DHK I: Dehalogenierungsmodul Pd-Kat auf Y-Zeolithen mit Begasungsmodul H2Reaktor DHK II: Dehalogenierungsmodul Pd-Kat membrangestützt mit Begasungsmodul H2Reaktor OxyKat: Oxykat Vollmetallkatalysator mit PeroxiddosierungAllen Reaktoren ist eine 5m lange Aquifersäule nachgeschaltet (Benzolproblematik)
Schem. Reaktor Fließbild
Aquifer
Oxy
-Kat
Deh
alog
enK
at1 D
ehal
ogen
Kat
2
Feist/Merkel 03/00
Abb. 6.6: Belegungsplan des Schachtbauwerks 3 und Dimensionierung der Reaktoren.

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Im Rahmen des Projekts wurden zwei segmentierte Reaktormodu-le installiert, die prinzipiell unab-hängig voneinander betrieben werden können. Jeder Reaktor besteht aus 5 in Edelstahl ausge-führten Einzelsegmenten mit ei- ner Länge von jeweils 20 cm und einem Durchmesser von 15 cm (Abb. 6.7). Nach jedem Segment bestand über einen Probe-nahmeport die Möglichkeit zur Probenahme. Auslass- und Ein-lassproben konnten automati-siert direkt im Analytikgebäude gezogen werden, Proben aus den einzelnen Reaktorsegmenten mußten im Schacht genommen werden.
Abb. 6.7: Segmentierter Reaktor, Detailaufnahme der Einzelsegmente(rechts). In blau ein Druckausgleichsbehälter zur Kompensation vonFlussschwankungen durch die Pulsationen der Grundwasserförderpum-pen.
6.1.3 Versuche in der Pilotanlage
Im November 1999 wurden die Reaktoren erstmalig mit Katalysatormaterial befüllt und daraufhin in Betrieb genommen. In der folgenden Projektlaufzeit wurden zahlreiche Versuche mit unterschied-lichen Ansätzen begonnen. Im folgenden werden einzelne Versuche vorgestellt, die die Problematiken des Anlagenbetriebs verdeutlichen und für die Gesamtbeurteilung des Teilprojekts wichtig sind.
6.1.3.1 Versuch 1
In folgendem Versuchsaufbau wurde zwei Problematiken Rechnung getragen, die in der Pilotanlage angetroffen werden, nämlich Sulfatreduktion im Aquifer sowie Sulfatreduktion in den Reaktoren. Die mikrobielle Sulfatreduktion im Aquifer kann bei Annahme einer Kohlenstoffquelle mit folgender Reaktionsgleichung beschrieben werden:
SHOHCOeSOCOOHCH 22243 228 ++=++ −−−
Es kann darüber hinaus aber auch durch die aktive Zugabe von elementarem Wasserstoff in der Katalysatorsäule zur Sulfatreduktion nach folgender Reaktionsgleichung kommen:
SHOHOHeSOH 22242 228 ++=++ −−−
16

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Dazu wird angenommen, dass eine sulfatreduzierende Bakterienpopulation mit dem gefördertem Grundwasser in den Reaktor gelangt und dort festgelegt wird. Durch den permanent zudosierten Wasserstoff wird dann die Sulfatreduktion angeregt.
Diese unterschiedlichen Problematiken wurden bei diesen Versuchen in der Pilotanlage durch zwei unterschiedliche Lösungsansätze berücksichtigt.
• Die H2S Gehalte des Grundwassers sollen durch Vorschalten einer Eisenschüttung vor die Katalysatorschüttung metallisch gefällt werden.
• Mikrobielle Sulfatreduktion in der Katalysatorsäule soll durch periodische Spülungen (täglich für 15 Minuten) des Reaktors mit einer ca. 5 g/l H2O2 Lösung verhindert werden.
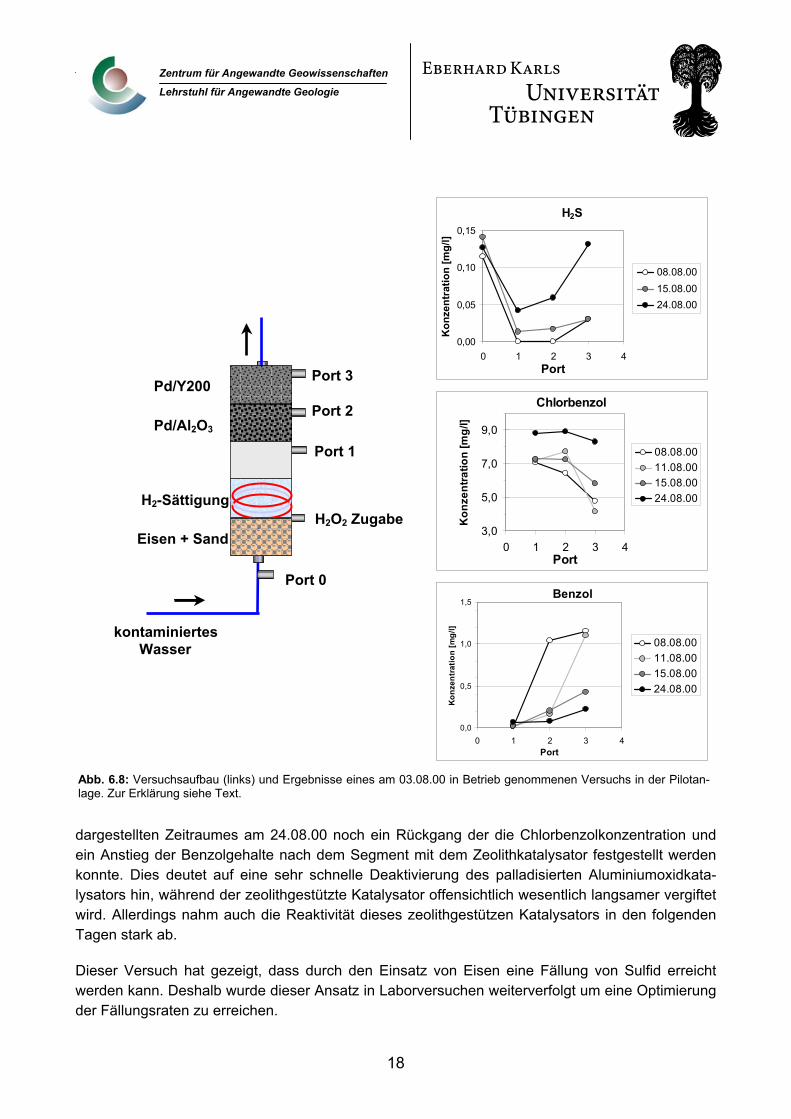
Ein am 03.08.00 in Betrieb genommer Versuchsaufbau in der Pilotanlage zeigt exemplarisch die beiden Problematiken und die Lösungsmöglichkeiten. Der Versuchsaufbau ist schematisch in Abb. 3.5 (links) dargestellt. Im Untersten der 5 Reaktorsegmente wurde eine Eisen/Sand-Mischung aus 500 g gekörntem Eisengranulat (Graugussgranulat, Gotthart Maier GmbH, Rheinfelden) und 2000 g Quarzsand zur metallischen Sulfidfällung eingebaut. Im 2. Segment wird über ein Modul (ein am Ende geschlossener Silikonschlauch der mit Wasserstoff beaufschlagt wird) Wasserstoff zugegeben. Das 3. Segment blieb leer um eine Durchmischung des mit Wasserstoff angereicherten Wassers zu erreichen. In den Segmenten 4 und 5 wurden die Katalysatoren eingebaut, wobei im 4. Segment ein palladisierter Aluminiumoxid Katalysator eingesetzt wurde, während im 5. Segment ein zeolithgestützter Katalysator (Pd auf Zeolith Y200) zum Einsatz kam. Zwischen Segment 1 und 2 erfolgte täglich für 15 Minuten die Zugabe einer konzentrierten H2O2 Lösung mit einer resultierenden Konzentration von 5g/l im gefördertem Grundwasser.
Abb. 6.8 (rechts) zeigt die Entwicklung der H2S-, Chlorbenzol- und Benzolgehalte im Säulenprofil während der ersten 3 Wochen des Versuchs, dokumentiert durch die Probenahmen an den einzelnen Probenahmeports. Es konnte gezeigt werden, dass die H2S Fracht des Grundwassers zumindest temporär durch das Vorschalten der Eisenschüttung gefällt wurde (kein H2S an Port 1). Es kommt jedoch bereits nach 12 Tagen (Beprobung am 15.08.00) zu einem Teildurchbruch des H2S durch die Eisenschüttung. Außerdem kommt es auch nach der Wasserstoffzugabe und in der Katalysator-Schüttung zur Sulfatreduktion, so dass die H2S Konzentrationen im Säulenprofil wieder ansteigen (Port 1, 2, 3). Dieser Effekt belegt, dass die tägliche Zugabe des H2O2 biologische Aktivität im Reaktor nicht verhindern kann. Es wird angenommen, dass durch die punktförmige Zugabe des H2O2 in der Reaktorsäule keine ausreichende Durchmischung im Säulenquerschnitt erreicht wird, so dass Teilbereichen der Säule nicht von der H2O2 Spülung erreicht werden.
Prinzipiell konnte jedoch auch in diesem Versuch die Dehalogenierung des Chlorbenzol und der resultierende Anstieg der Benzolgehalte im Säulenauslauf gezeigt werden. 5 Tage nach Versuchs-beginn am 08.08.00 nahm die Konzentration an Chlorbenzol von Port 1 (vor dem 1. Katalysator-segment) über Port 2 bis hin zum Port 3 (nach dem 2. Katalysatorsegment) stark ab. Ab der Beprobung am 11.08.00 konnte jedoch von Port 1 bis Port 2 (Aluminiumoxidkatalysator) kein Rückgang der Chlorbenzolkonzentration mehr festgestellt werden, während bis zum Ende des
17
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Chlorbenzol
3,0
5,0
7,0
9,0
0 1 2 3 4Port
Kon
zent
ratio
n [m
g/l]
08.08.0011.08.0015.08.0024.08.00
Benzol
0,0
0,5
1,0
1,5
0 1 2 3 4Port
Konz
entr
atio
n [m
g/l]
08.08.0011.08.0015.08.0024.08.00
H2S
0,00
0,05
0,10
0,15
0 1 2 3 4Port
Kon
zent
ratio
n [m
g/l]
08.08.0015.08.0024.08.00
Abb. 6.8: Versuchsaufbau (links) und Ergebnisse eines am 03.08.00 in Betrieb genommenen Versuchs in der Pilotan-lage. Zur Erklärung siehe Text.
Eisen + Sand
kontaminiertes Wasser
Pd/Y200
Pd/Al2O3
H2O2 ZugabeH2-Sättigung
Port 0
Port 3
Port 2
Port 1
dargestellten Zeitraumes am 24.08.00 noch ein Rückgang der die Chlorbenzolkonzentration und ein Anstieg der Benzolgehalte nach dem Segment mit dem Zeolithkatalysator festgestellt werden konnte. Dies deutet auf eine sehr schnelle Deaktivierung des palladisierten Aluminiumoxidkata-lysators hin, während der zeolithgestützte Katalysator offensichtlich wesentlich langsamer vergiftet wird. Allerdings nahm auch die Reaktivität dieses zeolithgestützen Katalysators in den folgenden Tagen stark ab.
Dieser Versuch hat gezeigt, dass durch den Einsatz von Eisen eine Fällung von Sulfid erreicht werden kann. Deshalb wurde dieser Ansatz in Laborversuchen weiterverfolgt um eine Optimierung der Fällungsraten zu erreichen.
18

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
6.1.3.2 Versuch 2
Aufgrund der zurückgehenden Schwefelwasserstoffkonzentrationen im über die Horizontalbrunnen gefördertem Grundwassers ab Frühjahr 2001 (Abb. 6.2) und der damit günstigeren Rahmen-bedingungen für die Stabilität der Katalysatoren wurden ab März 2001 weitere Neustarts der Anlage mit frischem Katalysatormaterial durchgeführt. Dabei wurden auch die Ergebnisse der Laboruntersuchungen berücksichtigt die zeigten, dass das in der Arbeitsgruppe Prof. Dahmke eingesetzte Eisen die besten Fällungseigenschaften für Sulfid aufweist (Kap. 6.2.5.1).
Im folgendem werden exemplarisch die Ergebnisse eines am 27.03.2001 begonnenen Versuchs dargestellt. Der Aufbau der Säule ist in Abbildung 6.9 dargestellt. Die unteren beiden Säulensegmente wurden mit einer Mischung aus Eisengranulat (das von der Gruppe Dahmke in ihren Reaktoren eingesetzte Eisen) und Quarzsand in einem Volumenverhältnis von 1:2 befüllt. Damit sollte verhindert werden, dass etwaige H2S Restgehalte im zufließenden Grundwasser den Katalysator erreichen. Darüber wurde das Modul zur Wasserstoffsättigung eingebaut, in dem ein unter H2-Druck gehaltener Silikonschlauch verlegt ist. Die beiden letzten Module wurden mit dem Zeolith-Katalysator befüllt. Die Eisen/Sand Mischung sowie der Katalysator wurden in Nylonnetze verpackt um ein einfacheres Befüllen und Entleeren der Säulensegmente zu ermöglichen. Im Anlagenbetrieb wird zwischen dem zweiten Eisensegment und dem Segment zur Wasserstoff-sättigung die periodische Zugabe von H2O2 zur Verhinderung einer sulfatreduzierenden Biologie durchgeführt (tägliche Spülungen mit H2O2 für 15 Minuten mit einer Konzentration von ca. 5 g/l). Die Flussrate in diesem Versuch wurde auf 20 Liter pro Stunde eingestellt, so dass die Kontaktzeit in jedem Katalysatorsegment etwa 3 Minuten betrug (jeweils 1000 g Katalysator mit einer Schüttdichte von ca. 0,5 kg/l, entsprechend einem Porenvolumen von ca. 1 Liter).
19
Abb. 6.9: Säulenaufbau für den am 27.03.2001 neugestarteten Versuch. Die linken beiden Segmente sind mit einer Mischung aus Eisen und Sand gefüllt. Darüber wird das Modul zur Wasserstoffsättigung und zwei Module gefüllt mitKatalysator aufgebaut. Die Eisen/Sand Mischung und der Katalysator wurden in einem Nylonnetz verpackt.
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
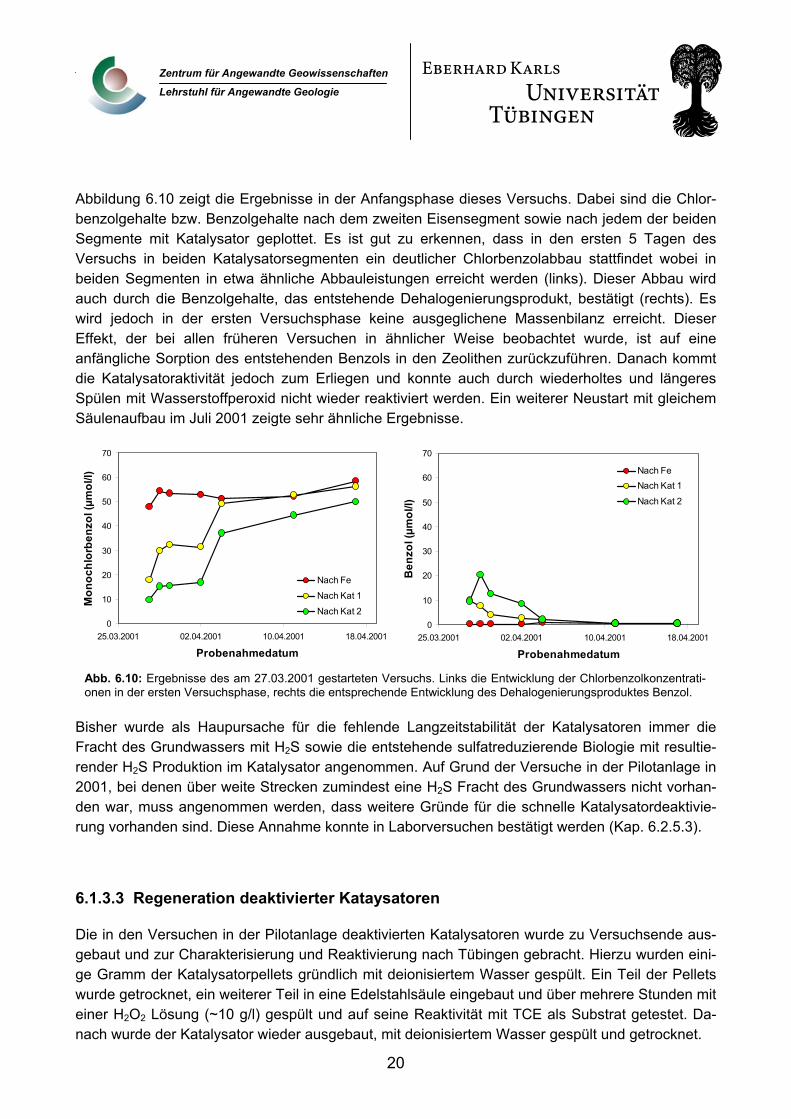
Abbildung 6.10 zeigt die Ergebnisse in der Anfangsphase dieses Versuchs. Dabei sind die Chlor-benzolgehalte bzw. Benzolgehalte nach dem zweiten Eisensegment sowie nach jedem der beiden Segmente mit Katalysator geplottet. Es ist gut zu erkennen, dass in den ersten 5 Tagen des Versuchs in beiden Katalysatorsegmenten ein deutlicher Chlorbenzolabbau stattfindet wobei in beiden Segmenten in etwa ähnliche Abbauleistungen erreicht werden (links). Dieser Abbau wird auch durch die Benzolgehalte, das entstehende Dehalogenierungsprodukt, bestätigt (rechts). Es wird jedoch in der ersten Versuchsphase keine ausgeglichene Massenbilanz erreicht. Dieser Effekt, der bei allen früheren Versuchen in ähnlicher Weise beobachtet wurde, ist auf eine anfängliche Sorption des entstehenden Benzols in den Zeolithen zurückzuführen. Danach kommt die Katalysatoraktivität jedoch zum Erliegen und konnte auch durch wiederholtes und längeres Spülen mit Wasserstoffperoxid nicht wieder reaktiviert werden. Ein weiterer Neustart mit gleichem
Bisher wurde als Haupursache für die fehlende Langzeits
Säulenaufbau im Juli 2001 zeigte sehr ähnliche Ergebnisse.
tabilität der Katalysatoren immer die Fracht des Grundwassers mit H2S sowie die entstehende sulfatreduzierende Biologie mit resultie-
.1.3.3 Regeneration deaktivierter Kataysatoren
alysatoren wurde zu Versuchsende aus-gebaut und zur Charakterisierung und Reaktivierung nach Tübingen gebracht. Hierzu wurden eini-
0
10
20
30
40
50
60
70
25.03.2001 02.04.2001 10.04.2001 18.04.2001
Probenahmedatum
Mon
ochl
orbe
nzol
(µm
ol/l)
Nach FeNach Kat 1Nach Kat 2
0
10
20
30
40
50
60
70
25.03.2001 02.04.2001 10.04.2001 18.04.2001
Probenahmedatum
Ben
zol (
µmol
/l)
Nach FeNach Kat 1Nach Kat 2
Abb. 6.10: Ergebnisse des am 27.03.2001 gestarteten Versuchs. Links die Entwicklung der Chlorbenzolkonzentrati-onen in der ersten Versuchsphase, rechts die entsprechende Entwicklung des Dehalogenierungsproduktes Benzol.
render H2S Produktion im Katalysator angenommen. Auf Grund der Versuche in der Pilotanlage in 2001, bei denen über weite Strecken zumindest eine H2S Fracht des Grundwassers nicht vorhan-den war, muss angenommen werden, dass weitere Gründe für die schnelle Katalysatordeaktivie-rung vorhanden sind. Diese Annahme konnte in Laborversuchen bestätigt werden (Kap. 6.2.5.3).
6
Die in den Versuchen in der Pilotanlage deaktivierten Kat
ge Gramm der Katalysatorpellets gründlich mit deionisiertem Wasser gespült. Ein Teil der Pellets wurde getrocknet, ein weiterer Teil in eine Edelstahlsäule eingebaut und über mehrere Stunden mit einer H2O2 Lösung (~10 g/l) gespült und auf seine Reaktivität mit TCE als Substrat getestet. Da-nach wurde der Katalysator wieder ausgebaut, mit deionisiertem Wasser gespült und getrocknet.
20
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
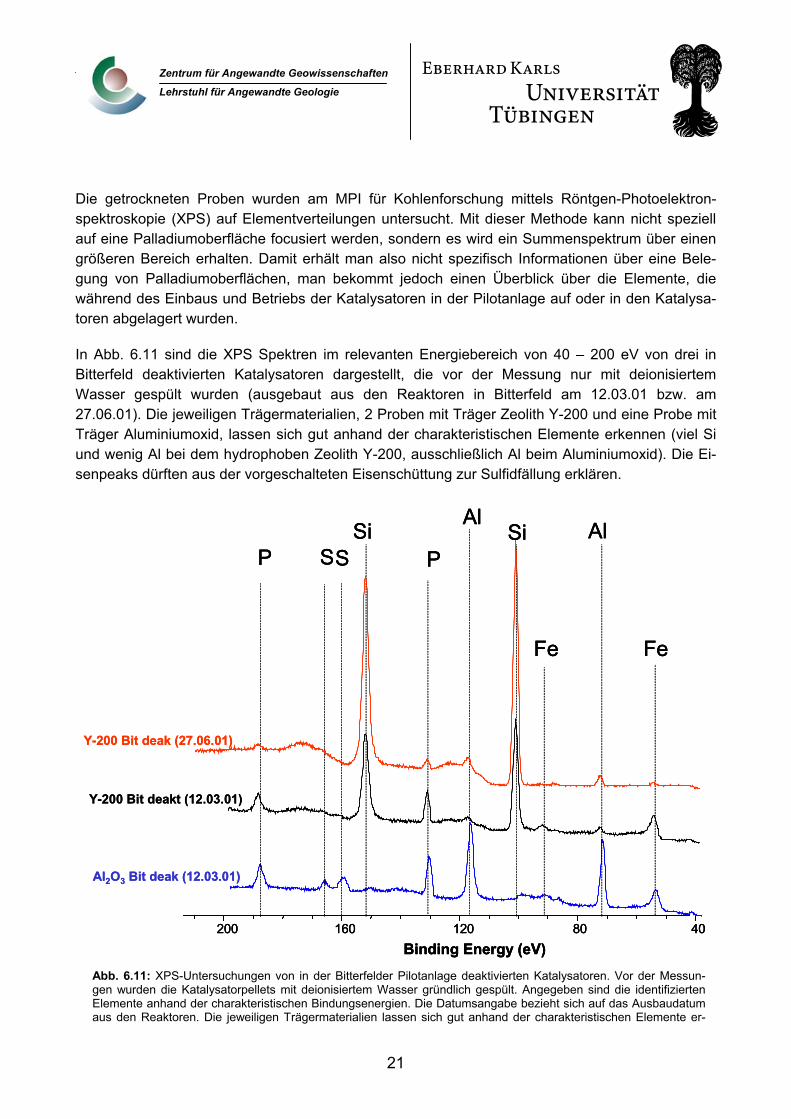
Die getrockneten Proben wurden am MPI für Kohlenforschung mittels Röntgen-Photoelektron-spektroskopie (XPS) auf Elementverteilungen untersucht. Mit dieser Methode kann nicht speziell auf eine Palladiumoberfläche focusiert werden, sondern es wird ein Summenspektrum über einen
talysatoren dargestellt, die vor der Messung nur mit deionisiertem Wasser gespült wurden (ausgebaut aus den Reaktoren in Bitterfeld am 12.03.01 bzw. am
größeren Bereich erhalten. Damit erhält man also nicht spezifisch Informationen über eine Bele-gung von Palladiumoberflächen, man bekommt jedoch einen Überblick über die Elemente, die während des Einbaus und Betriebs der Katalysatoren in der Pilotanlage auf oder in den Katalysa-toren abgelagert wurden.
In Abb. 6.11 sind die XPS Spektren im relevanten Energiebereich von 40 – 200 eV von drei in Bitterfeld deaktivierten Ka
27.06.01). Die jeweiligen Trägermaterialien, 2 Proben mit Träger Zeolith Y-200 und eine Probe mit Träger Aluminiumoxid, lassen sich gut anhand der charakteristischen Elemente erkennen (viel Si und wenig Al bei dem hydrophoben Zeolith Y-200, ausschließlich Al beim Aluminiumoxid). Die Ei-senpeaks dürften aus der vorgeschalteten Eisenschüttung zur Sulfidfällung erklären.
AlAlAl
21
kennen (viel Si und wenig Al bei dem hydrophoben Zeolith Y-200, ausschliesslich Al beim Aluminiumoxid).
4080120160200Binding Energy (eV)
Y-200 Bit deakt (12.03.01)
Al2O3 Bit deak (12.03.01)
Si Si AlSSP P
FeFe
Y-200 Bit deak (27.06.01)
4080120160200Binding Energy (eV)
4080120160200Binding Energy (eV)
Y-200 Bit deakt (12.03.01)
Al2O3 Bit deak (12.03.01)
Si Si AlSi Si AlSSP P
FeFe
SSP P
FeFe
Y-200 Bit deak (27.06.01)
Abb. 6.11: XPS-Untersuchungen von in der Bitterfelder Pilotanlage deaktivierten Katalysatoren. Vor der Messun-gen wurden die Katalysatorpellets mit deionisiertem Wasser gründlich gespült. Angegeben sind die identifizierten Elemente anhand der charakteristischen Bindungsenergien. Die Datumsangabe bezieht sich auf das Ausbaudatum aus den Reaktoren. Die jeweiligen Trägermaterialien lassen sich gut anhand der charakteristischen Elemente er-
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
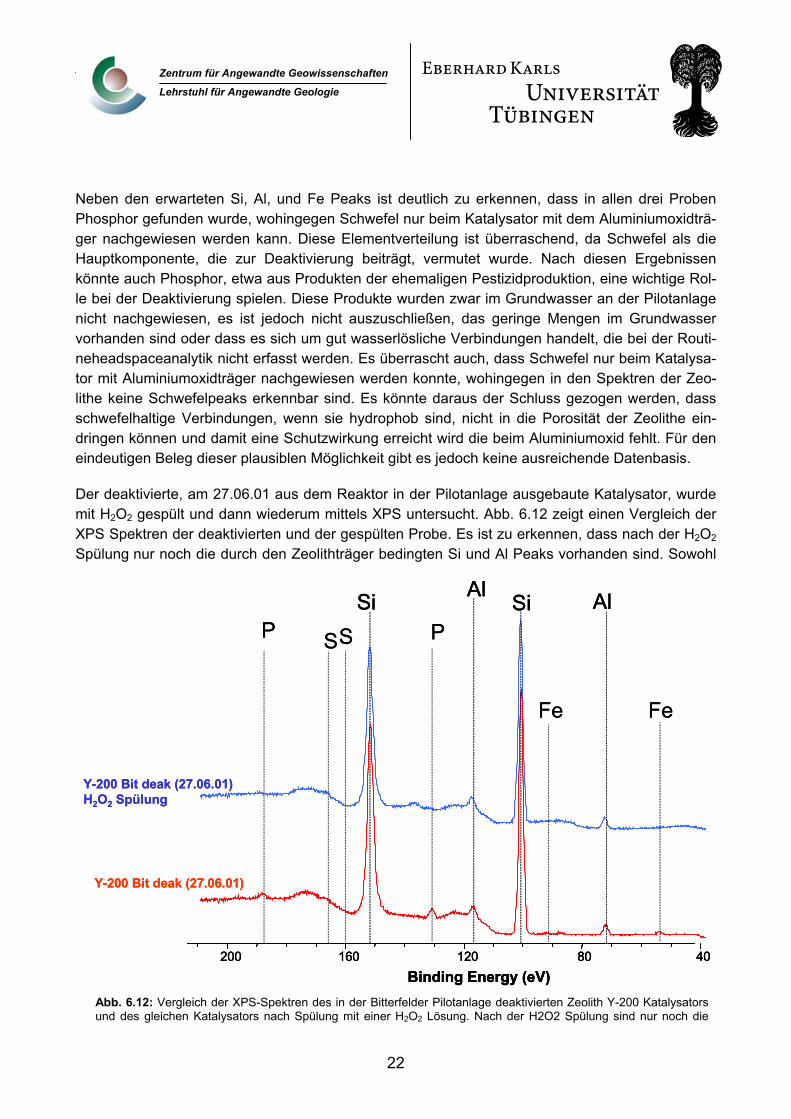
Neben den erwarteten Si, Al, und Fe Peaks ist deutlich zu erkennen, dass in allen drei Proben Phosphor gefunden wurde, wohingegen Schwefel nur beim Katalysator mit dem Aluminiumoxidträ-
de mit H2O2 gespült und dann wiederum mittels XPS untersucht. Abb. 6.12 zeigt einen Vergleich der
ger nachgewiesen werden kann. Diese Elementverteilung ist überraschend, da Schwefel als die Hauptkomponente, die zur Deaktivierung beiträgt, vermutet wurde. Nach diesen Ergebnissen könnte auch Phosphor, etwa aus Produkten der ehemaligen Pestizidproduktion, eine wichtige Rol-le bei der Deaktivierung spielen. Diese Produkte wurden zwar im Grundwasser an der Pilotanlage nicht nachgewiesen, es ist jedoch nicht auszuschließen, das geringe Mengen im Grundwasser vorhanden sind oder dass es sich um gut wasserlösliche Verbindungen handelt, die bei der Routi-neheadspaceanalytik nicht erfasst werden. Es überrascht auch, dass Schwefel nur beim Katalysa-tor mit Aluminiumoxidträger nachgewiesen werden konnte, wohingegen in den Spektren der Zeo-lithe keine Schwefelpeaks erkennbar sind. Es könnte daraus der Schluss gezogen werden, dass schwefelhaltige Verbindungen, wenn sie hydrophob sind, nicht in die Porosität der Zeolithe ein-dringen können und damit eine Schutzwirkung erreicht wird die beim Aluminiumoxid fehlt. Für den eindeutigen Beleg dieser plausiblen Möglichkeit gibt es jedoch keine ausreichende Datenbasis.
Der deaktivierte, am 27.06.01 aus dem Reaktor in der Pilotanlage ausgebaute Katalysator, wur
XPS Spektren der deaktivierten und der gespülten Probe. Es ist zu erkennen, dass nach der H2O2 Spülung nur noch die durch den Zeolithträger bedingten Si und Al Peaks vorhanden sind. Sowohl
AlAlAl
22
durch den Zeolithträger bedingten Si und Al Peaks vorhanden.
Abb. 6.12: Vergleich der XPS-Spektren des in der Bitterfelder Pilotanlage deaktivierten Zeolith Y-200 Katalysatorsund des gleichen Katalysators nach Spülung mit einer H2O2 Lösung. Nach der H2O2 Spülung sind nur noch die
Y-200 Bit deak (27.06.01)H2O2 Spülung
Y-200 Bit deak (27.06.01)
4080120160200Binding Energy (eV)
Si Si Al
SSP P
FeFe
Y-200 Bit deak (27.06.01)H2O2 Spülung
Y-200 Bit deak (27.06.01)
4080120160200Binding Energy (eV)
4080120160200Binding Energy (eV)
Si Si AlSi Si Al
SSP P
FeFe

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
die Eisen, als auch die Phosphorpeaks sind in der gespülten Probe nicht mehr nachweisbar. In ei-nem Aktivitätstest der gespülten Probe mit TCE als Testsubstrat konnte gezeigt werden, dass die Reaktivität des Katalysators nahezu vollständig wiederhergestellt war.
Die Befunde aus den Reaktivierungsversuchen und den XPS Messungen haben gezeigt, dass zumindest im Labor die in Bitterfeld deaktivierten Katalysatoren wieder regeneriert werden konn-ten. Es kann außerdem vermutet werden, dass Phosphorverbindungen bei der Deaktivierung eine Rolle spielen. Das ist jedoch kein zwingender Schluss, da durchaus andere Elemente an der De-aktivierung beteiligt sind, die mit der Nachweisgrenze der XPS Methode nicht nachweisbar sind. Diese Nachweisgrenze liegt bei ca. 0,2 – 2 Atom-%. Auch bei der Eindringtiefe sind der Methode Grenzen gesetzt, die bei ca. 1-5 nm liegen. Warum in der Bitterfelder Pilotanlage keine Reaktivie-rung bei den auch dort stattfindenden H2O2 Spülungen gelingt ist unklar. Ein Grund könnte in einer permanenten Fracht des Grundwassers mit deaktivierenden Verbindungen liegen, die nach jedem Reaktivierungsversuch eine sofortige erneute Deaktivierung auslöst.
6.2 Begleitende Untersuchungen Die begleitenden Untersuchungen wurden im wesentlichen als Laborversuche im Hydrogeochemi-schen Labor des Lehrstuhls für Angewandte Geologie in Tübingen durchgeführt. Dabei standen zunächst die Optimierung der Katalysatoren in Bezug auf Abbauraten, Abbauwege und Endpro-dukte im Vordergrund. Die Erfahrungen aus der Bitterfelder Pilotanlage, in der keine stabilen Ab-bauraten über längere Zeiten erreicht werden konnten, machten jedoch eine Erweiterung der Zielsetzung der Laborversuche nötig. Zentraler Punkt wurde eine intensivere Untersuchung der Deaktivierungsprozesse mit dem Ziel, Lösungsansätze zur Implementierung in der Pilotanlage zu entwickeln. Dazu wurden unterschiedlichen Versuche mit Wässern, die eine Lösungsfracht enthal-ten durchgeführt, um den Einfluss der unterschiedlichen Stoffe auf die Langzeitstabilität des Kata-lysators zu ermitteln.
Darüber hinaus wurde ein weiterer Geländeversuch in Backnang bei Stuttgart durchgeführt, bei dem speziell die Problematik des Aufwachsens einer sulfatreduzierenden Bakterienpopulation in den Katalysatorsäulen untersucht wurde. In diesem Versuch sollten wirksame Methoden zur Un-terdrückung mikrobiologischer Aktivität, die durch die Zugabe des Reduktionsmittels Wasserstoff induziert wurde, gefunden werden.
6.2.1 Versuchsaufbauten
In den Laborversuchen wurden im wesentlichen zwei unterschiedliche Versuchsaufbauten einge-setzt, Säulenversuche und Batchversuche (Abb. 6.13) wobei die Säulenversuche in der Regel für Langzeitexperimente mit pelletiertem Katalysator und die Bachversuche bei kürzeren Versuchszei-ten mit pulverförmigem Katalysator eingesetzt wurden.
23

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Magnetrührer
wässrige Lösung
Mininertventil
Rührfisch
Gasphase
Katalysator-säule
Pumpe
Einlassprobe
Auslassprobe
H2-Säulemit Silikonschlauch
unter H2 Druck
Teflonbeutel mit kontaminiertem
Wasser
H2 Tank
Konzentrierte wässrige Lösung mit ionischer Verbindung
Katalysator-säule
Pumpe
Einlassprobe
Auslassprobe
H2-Säulemit Silikonschlauch
unter H2 Druck
Teflonbeutel mit kontaminiertem
Wasser
H2 Tank
Konzentrierte wässrige Lösung mit ionischer Verbindung
Magnetrührer
wässrige Lösung
Mininertventil
Rührfisch
Gasphase
Magnetrührer
wässrige Lösung
Mininertventil
Rührfisch
Gasphase
Katalysator-säule
Pumpe
Einlassprobe
Auslassprobe
H2-Säulemit Silikonschlauch
unter H2 Druck
Teflonbeutel mit kontaminiertem
Wasser
H2 Tank
Konzentrierte wässrige Lösung mit ionischer Verbindung
Katalysator-säule
Pumpe
Einlassprobe
Auslassprobe
H2-Säulemit Silikonschlauch
unter H2 Druck
Teflonbeutel mit kontaminiertem
Wasser
H2 Tank
Konzentrierte wässrige Lösung mit ionischer Verbindung
Abb. 6.13: Standard-Versuchsaufbauten für die Laborversuche. Links: Säulenversuche. Rechts: Batchversuche.
Bei den Säulenversuchen (Abb. 6.13 links) wurde in einem Teflonbeutel eine zu untersuchende wässrige Testlösung angesetzt, welche mittels einer Kolbenpumpe durch die Katalysatorsäule ge-pumpt wurde. Um dem System den für die Reaktion nötigen Wasserstoff zuzuführen wurde das Wasser, bevor es die Katalysatorsäule erreichte, durch eine Säule geleitet in der ein mit Wasser-stoff unter Druck (ca. 0,8 bar) gehaltener Silikonschlauch verlegt wurde. Wasserstoff gelangt so diffusiv durch den Silikonschlauch in das vorbeiströmende Wasser. Die Katalysatorsäule hatte in der Regel eine Dimension von ca. 6 cm Länge bei einem Durchmesser von 1 cm und wurde mit ca. 2g pelletiertem Katalysator befüllt. Die Bestimmung der Schadstoffkonzentrationen am Kataly-satoreingang- und Ausgang sowie anderer relevanter Parameter (z.B. Wasserstoff oder die Haupt-anionen und -Kationen) erfolgte durch Probenahmen an Ports direkt vor und nach der Katalysatorsäule. Alle Teile des Versuchsaufbaus die in Kontakt mit der wässrigen Phase standen waren in Edelstahl oder Teflon ausgeführt.
Bei den Batchversuchen wurde ein mit einem Mininertventil verschlossenes 120 ml Glasgefäß als Reaktionsgefäß eingesetzt. In dieses Gefäß wurde wasserstoffgesättigtes Wasser eingefüllt, dem bei Bedarf Zusätze zugegeben wurden. Danach wurde der pulverförmige Katalysator und die ab-zubauende Testverbindung über das Mininertventil eingespritzt. Die Reaktionsgefäße in den Batchversuchen wurden in der Regel nur etwa zu ¾ mit Wasser gefüllt, so dass die Probennahme über die verbleibende Gasphase erfolgen konnte.
Auf Abänderungen der Versuchsaufbauten oder spezielle Aufbauten wird bei Darstellung der betreffenden Versuchsansätze im Ergebnisteil eingegangen.
6.2.2 Analytik
Für die Analytik der organischen Schadstoffe sowie der anorganischen Parameter in den Labor-versuchen wurde im wesentlichen Standardanalytik eingesetzt. Dazu gehören für die Organik GC-ECD/FID sowie GC-MS (HP 6890/5972). Bei leichtflüchtigen Verbindungen wurde in der Regel
24
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Analytik aus der Gasphase betrieben. Die Hauptanionen und Kationen wurden mittels Ionenchro-matographie analysiert (zwei Dionex DX 120). Wasserstoff wurde mit einem reduktivem Gasanaly-sator gemessen (Trace Analytical RGD 2). Im folgendem Ergebnisteil wird bei Einsatz spezieller analytischer Methoden gesondert darauf hingewiesen.
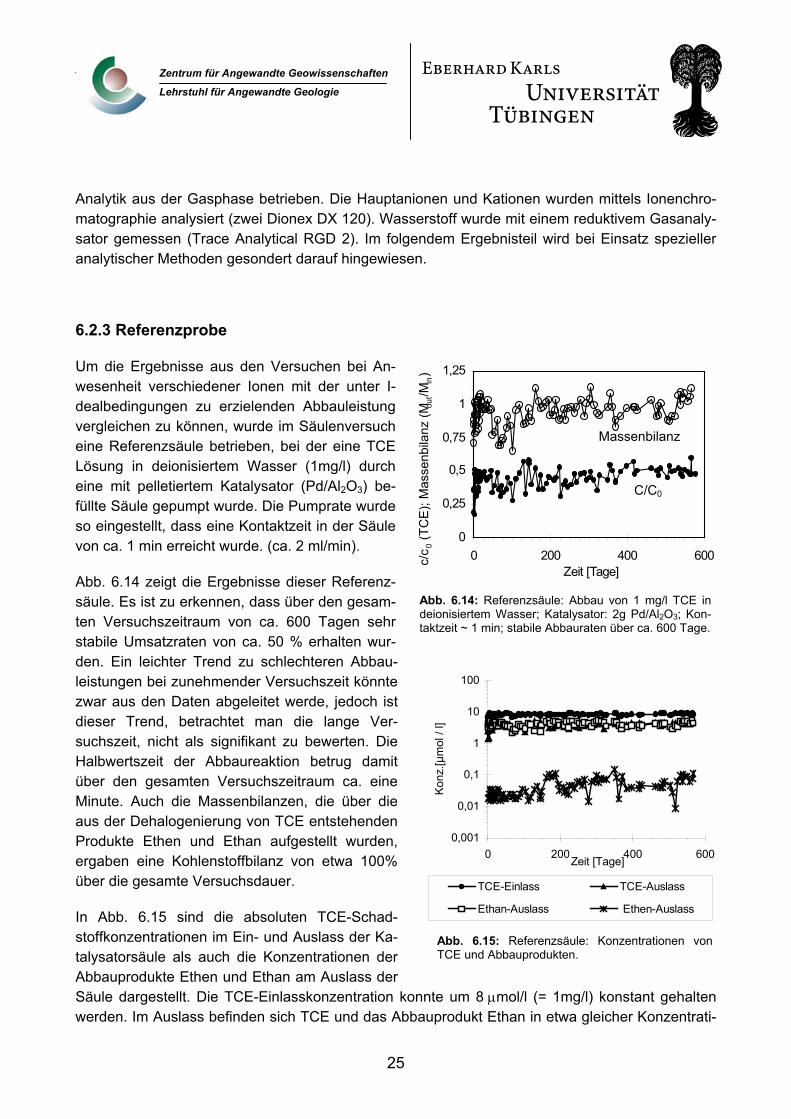
6.2.3 Referenzprobe
Um die Ergebnisse aus den Versuchen bei An-wesenheit verschiedener Ionen mit der unter I-dealbedingungen zu erzielenden Abbauleistung vergleichen zu können, wurde im Säulenversuch eine Referenzsäule betrieben, bei der eine TCE Lösung in deionisiertem Wasser (1mg/l) durch eine mit pelletiertem Katalysator (Pd/Al2O3) be-füllte Säule gepumpt wurde. Die Pumprate wurde so eingestellt, dass eine Kontaktzeit in der Säule von ca. 1 min erreicht wurde. (ca. 2 ml/min). 0
0,25
0,5
0,75
1
1,25
0 200 400 600Zeit [Tage]
c/c 0
(TC
E); M
asse
nbila
nz (M
out/M
in)
Abb. 6.14: Referenzsäule: Abbau von 1 mg/l TCE indeionisiertem Wasser; Katalysator: 2g Pd/Al2O3; Kon-taktzeit ~ 1 min; stabile Abbauraten über ca. 600 Tage.
Massenbilanz
C/C0
Abb. 6.14 zeigt die Ergebnisse dieser Referenz-säule. Es ist zu erkennen, dass über den gesam-ten Versuchszeitraum von ca. 600 Tagen sehr stabile Umsatzraten von ca. 50 % erhalten wur-den. Ein leichter Trend zu schlechteren Abbau-leistungen bei zunehmender Versuchszeit könnte zwar aus den Daten abgeleitet werde, jedoch ist dieser Trend, betrachtet man die lange Ver-suchszeit, nicht als signifikant zu bewerten. Die Halbwertszeit der Abbaureaktion betrug damit über den gesamten Versuchszeitraum ca. eine Minute. Auch die Massenbilanzen, die über die aus der Dehalogenierung von TCE entstehenden Produkte Ethen und Ethan aufgestellt wurden, ergaben eine Kohlenstoffbilanz von etwa 100% über die gesamte Versuchsdauer.
In Abb. 6.15 sind die absoluten TCE-Schad-stoffkonzentrationen im Ein- und Auslass der Ka-talysatorsäule als auch die Konzentrationen der Abbauprodukte Ethen und Ethan am Auslass der Säule dargestellt. Die TCE-Einlasskonzentration konnte um 8 µmol/l (= 1mg/l) konstant gehalten werden. Im Auslass befinden sich TCE und das Abbauprodukt Ethan in etwa gleicher Konzentrati-
0,001
0,01
0,1
1
10
100
0 200 400 600Zeit [Tage]
Konz
.[µm
ol /
l]
TCE-Einlass TCE-Auslass
Ethan-Auslass Ethen-Auslass
Abb. 6.15: Referenzsäule: Konzentrationen vonTCE und Abbauprodukten.
25

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
on, während das Zwischenprodukt Ethen nur in sehr geringer Konzentration vorliegt. Weitere Zwi-schen- oder Abbauprodukte wurden bei dieser Schadstoffkonzentration nicht nachgewiesen.
Neben der Analyse der organischen Stoffe wurde auch der Wasserstoffgehalt am Ein- und Auslass der Katalysatorsäule gemessen. Die über den Silikonschlauch (siehe auch Versuchsaufbau Abb. 6.13) eingebrachte Wasserstoffkonzentration betrug am Einlass der Katalysatorsäule kon-stant zwischen 700 und 800 µmol/l und lag damit im Bereich der Sättigungskonzentration von Wasserstoff in Wasser.
6.2.4 Einfluss verbreiteter ionischen Wasserinhaltsstoffen
Im Folgendem werden Langzeitsäulenversuche vorgestellt, bei denen gezielt der Einfluss von in vielen Grundwässern verbreiteten ionischen Verbindungen auf die Langzeitstabilität von Palladi-umkatalysatoren untersucht wurde. Der Versuchsaufbau entspricht dem des Referenzversuchs, (Kap. 6.2.1) wobei wiederum TCE als Testsubstrat in einer Konzentration von 1 mg/l eingesetzt wurde. Die Kontaktzeit in der Säule wurde entsprechend wieder über die Flussrate auf ca. 1 ml/min eingestellt. Einziger Unterschied zu dem Referenzversuch ist, dass dem deionisiertem Wasser verschiedene ionische Verbindungen zugesetzt wurden. Über den Vergleich der Einlass- und Aus-lassproben wurde wiederum die Abbauleistung des Katalysators überprüft. Die einzelnen Versuche wurden jeweils über einen Zeitraum von mehreren 10er Tagen gefahren um auch eine schleichen-de Deaktivierung des Katalysators beobachten zu können.
6.2.4.1 Natriumsulfat
0
0.5
1
0 20 40 60 80 10
Zeit (Tage)
CA
usla
ss /
C E
inla
ss [-
]
Massenbilanz
C/C0 (TCE)
1
0
Abb. 6.16: Abbau von TCE (1 mg/l) in deionisiertem Was-ser mit einem Pd/Al2O3 Katalysator im Säulenversuch. Kon-taktzeit 1 min. 1 Ab dem 25 Tag Zugabe von ~ 500 mg/lNa2SO4.
In Abb. 6.16 ist ein Säulenversuch darge-stellt, bei dem der wässrigen Phase ab ca. dem 25 Versuchstag Natriumsulfat in einer Konzentration von ca. 500 mg/l (SO4
2- Konz.: ca. 340 mg/l, Na+-Konz: ca. 81 mg/l) zugegeben wurde. Über die Versuchsdauer von ca. 80 Tagen konnte kein signifikanter Rückgang der Abbauleistung beobachtet werden. Die etwas höheren Abbauleistun-gen zu Versuchsbeginn könnten eine Folge der schlechteren Massenbilanz sein und müssen nicht auf eine Deaktivierung hinwei-sen, da die Kurven von Massenbilanz und Abbauleistung über die gesamte Versuchs-dauer in etwa äquidistant verlaufen.
26
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Dieser Versuch lässt darauf schließen, dass sowohl Na+ Ionen als auch SO42- Ionen keinen negati-
ven Einfluss auf die Abbauleistung haben. Wie bei dem Referenzversuch werden nach ca. 80 Ta-gen Umsatzraten von ca. 50% erreicht.
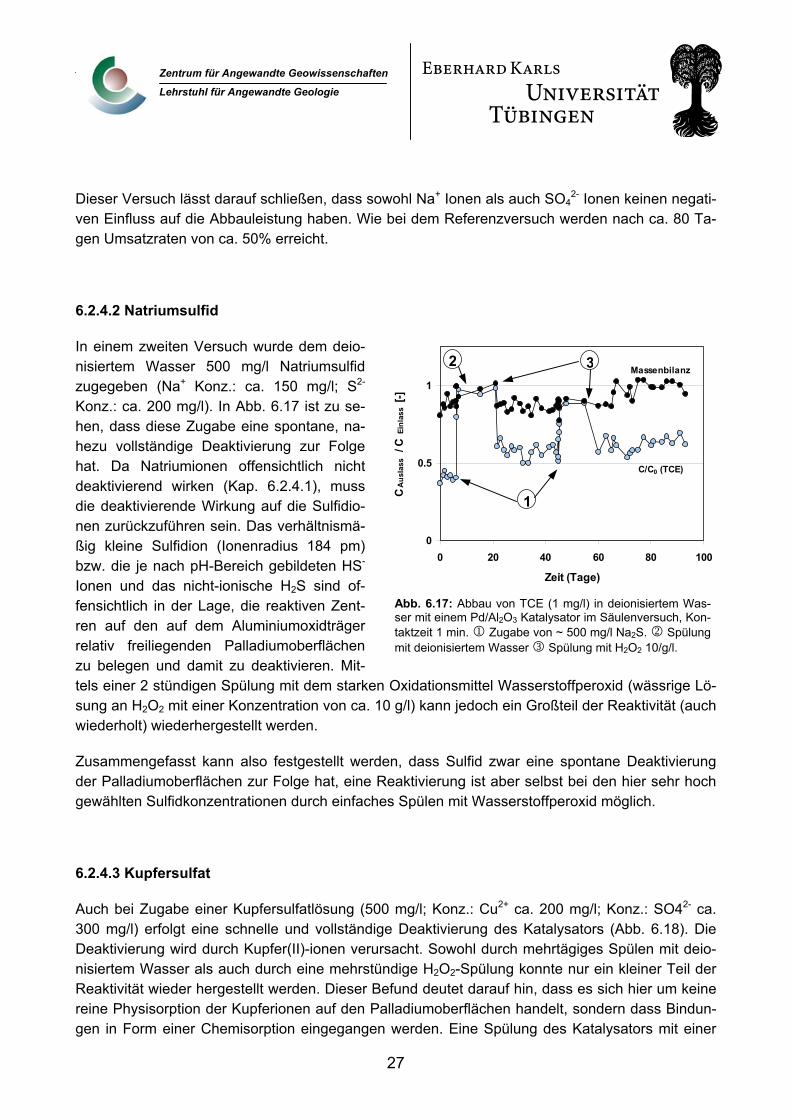
6.2.4.2 Natriumsulfid
In einem zweiten Versuch wurde dem deio-nisiertem Wasser 500 mg/l Natriumsulfid zugegeben (Na+ Konz.: ca. 150 mg/l; S2- Konz.: ca. 200 mg/l). In Abb. 6.17 ist zu se-hen, dass diese Zugabe eine spontane, na-hezu vollständige Deaktivierung zur Folge hat. Da Natriumionen offensichtlich nicht deaktivierend wirken (Kap. 6.2.4.1), muss die deaktivierende Wirkung auf die Sulfidio-nen zurückzuführen sein. Das verhältnismä-ßig kleine Sulfidion (Ionenradius 184 pm) bzw. die je nach pH-Bereich gebildeten HS- Ionen und das nicht-ionische H2S sind of-fensichtlich in der Lage, die reaktiven Zent-ren auf den auf dem Aluminiumoxidträger relativ freiliegenden Palladiumoberflächen zu belegen und damit zu deaktivieren. Mit-tels einer 2 stündigen Spülung mit dem starken Oxidationsmittel Wasserstoffperoxid (wässrige Lö-sung an H2O2 mit einer Konzentration von ca. 10 g/l) kann jedoch ein Großteil der Reaktivität (auch wiederholt) wiederhergestellt werden.
0
0.5
1
0 20 40 60 80 10
Zeit (Tage)
CA
usla
ss /
C E
inla
ss [-
]
C/C0 (TCE)
Abb. 6.17: Abbau von TCE (1 mg/l) in deionisiertem Was-ser mit einem Pd/Al2O3 Katalysator im Säulenversuch, Kon-taktzeit 1 min. 1 Zugabe von ~ 500 mg/l Na2S. 2 Spülungmit deionisiertem Wasser 3 Spülung mit H2O2 10/g/l.
2
1
0
Massenbilanz3
Zusammengefasst kann also festgestellt werden, dass Sulfid zwar eine spontane Deaktivierung der Palladiumoberflächen zur Folge hat, eine Reaktivierung ist aber selbst bei den hier sehr hoch gewählten Sulfidkonzentrationen durch einfaches Spülen mit Wasserstoffperoxid möglich.
6.2.4.3 Kupfersulfat
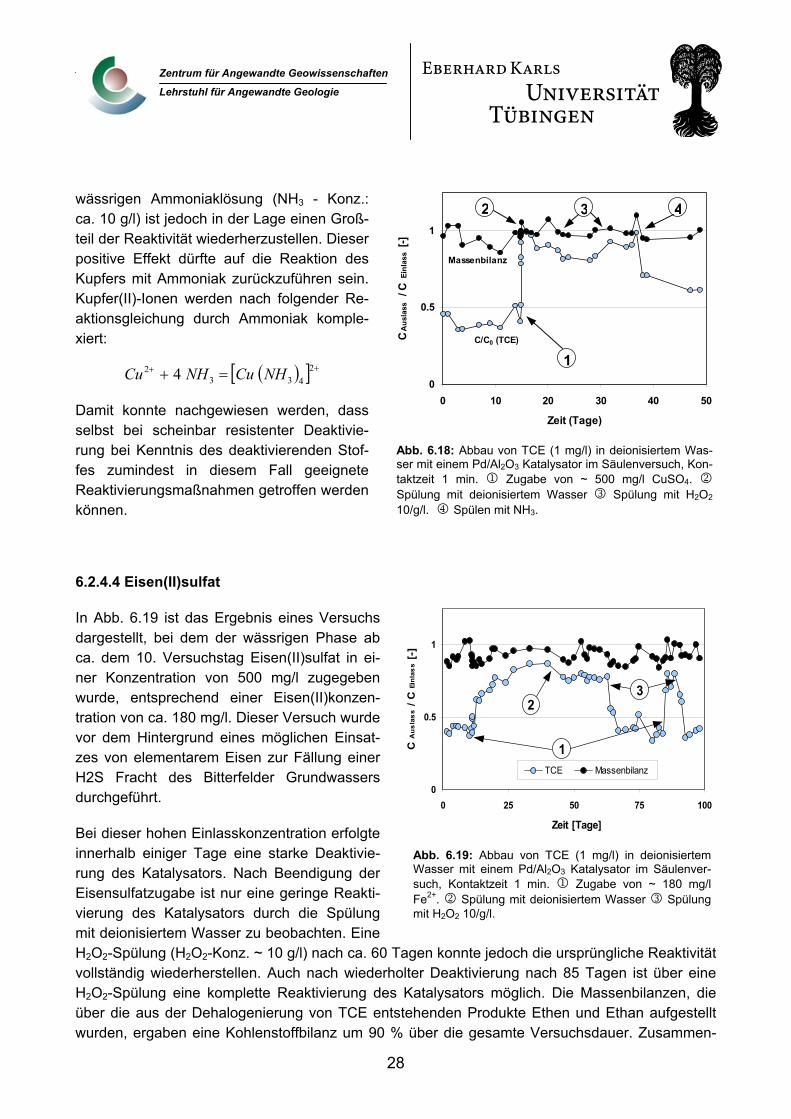
Auch bei Zugabe einer Kupfersulfatlösung (500 mg/l; Konz.: Cu2+ ca. 200 mg/l; Konz.: SO42- ca. 300 mg/l) erfolgt eine schnelle und vollständige Deaktivierung des Katalysators (Abb. 6.18). Die Deaktivierung wird durch Kupfer(II)-ionen verursacht. Sowohl durch mehrtägiges Spülen mit deio-nisiertem Wasser als auch durch eine mehrstündige H2O2-Spülung konnte nur ein kleiner Teil der Reaktivität wieder hergestellt werden. Dieser Befund deutet darauf hin, dass es sich hier um keine reine Physisorption der Kupferionen auf den Palladiumoberflächen handelt, sondern dass Bindun-gen in Form einer Chemisorption eingegangen werden. Eine Spülung des Katalysators mit einer
27
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
0
0.5
1
0 10 20 30 40 5
Zeit (Tage)C
Aus
lass
/ C
Ein
lass
[-]
Massenbilanz
C/C0 (TCE)
Abb. 6.18: Abbau von TCE (1 mg/l) in deionisiertem Was-ser mit einem Pd/Al2O3 Katalysator im Säulenversuch, Kon-taktzeit 1 min. 1 Zugabe von ~ 500 mg/l CuSO4. 2Spülung mit deionisiertem Wasser 3 Spülung mit H2O2
10/g/l. 4 Spülen mit NH3.
42
1
3
0
wässrigen Ammoniaklösung (NH3 - Konz.: ca. 10 g/l) ist jedoch in der Lage einen Groß-teil der Reaktivität wiederherzustellen. Dieser positive Effekt dürfte auf die Reaktion des Kupfers mit Ammoniak zurückzuführen sein. Kupfer(II)-Ionen werden nach folgender Re-aktionsgleichung durch Ammoniak komple-xiert:
( )[ ] +24 + =+ 33
2 4 NHCuNHCu
Damit konnte nachgewiesen werden, dass selbst bei scheinbar resistenter Deaktivie-rung bei Kenntnis des deaktivierenden Stof-fes zumindest in diesem Fall geeignete Reaktivierungsmaßnahmen getroffen werden können.
6.2.4.4 Eisen(II)sulfat
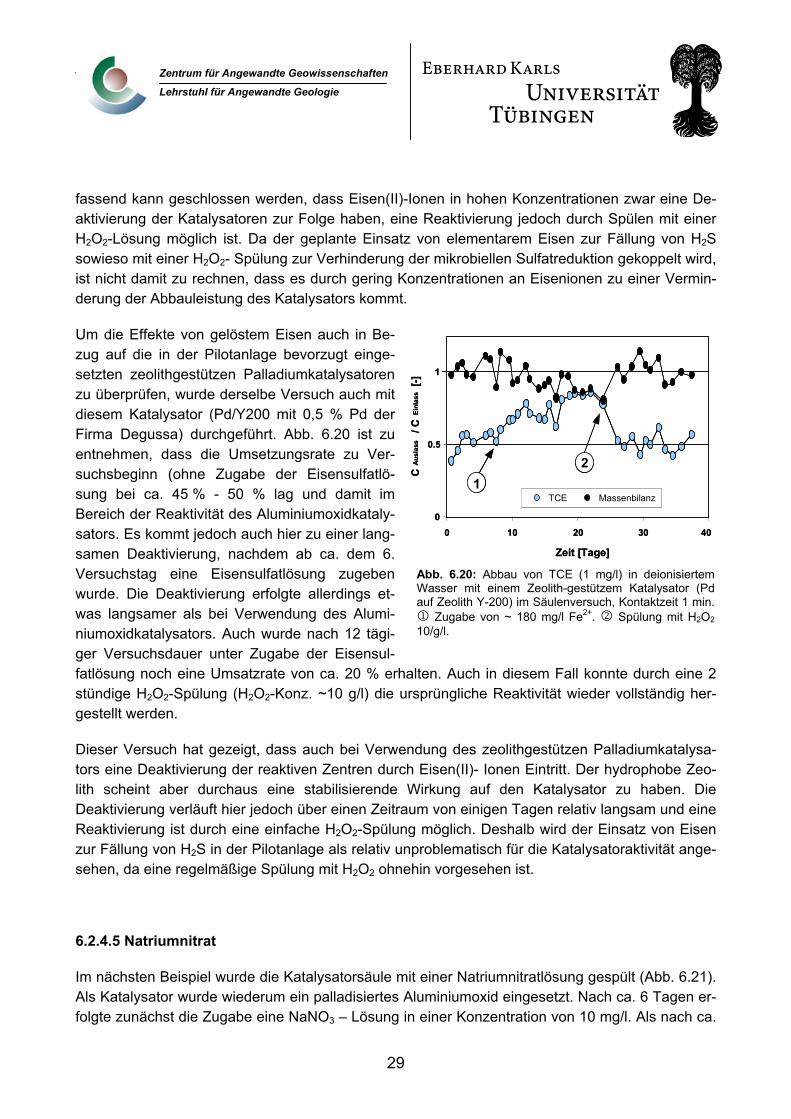
In Abb. 6.19 ist das Ergebnis eines Versuchs dargestellt, bei dem der wässrigen Phase ab ca. dem 10. Versuchstag Eisen(II)sulfat in ei-ner Konzentration von 500 mg/l zugegeben wurde, entsprechend einer Eisen(II)konzen-tration von ca. 180 mg/l. Dieser Versuch wurde vor dem Hintergrund eines möglichen Einsat-zes von elementarem Eisen zur Fällung einer H2S Fracht des Bitterfelder Grundwassers durchgeführt.
Bei dieser hohen Einlasskonzentration erfolgte innerhalb einiger Tage eine starke Deaktivie-rung des Katalysators. Nach Beendigung der Eisensulfatzugabe ist nur eine geringe Reakti-vierung des Katalysators durch die Spülung mit deionisiertem Wasser zu beobachten. Eine H2O2-Spülung (H2O2-Konz. ~ 10 g/l) nach ca. 60 Tagen konnte jedoch die ursprüngliche Reaktivität vollständig wiederherstellen. Auch nach wiederholter Deaktivierung nach 85 Tagen ist über eine H2O2-Spülung eine komplette Reaktivierung des Katalysators möglich. Die Massenbilanzen, die über die aus der Dehalogenierung von TCE entstehenden Produkte Ethen und Ethan aufgestellt wurden, ergaben eine Kohlenstoffbilanz um 90 % über die gesamte Versuchsdauer. Zusammen-
0
0.5
1
C A
usla
ss /
C Ei
nlas
s [-
]
TCE Massenbilanz
1
23
0 25 50 75 100
Zeit [Tage]
Abb. 6.19: Abbau von TCE (1 mg/l) in deionisiertemWasser mit einem Pd/Al2O3 Katalysator im Säulenver-such, Kontaktzeit 1 min. 1 Zugabe von ~ 180 mg/lFe2+. 2 Spülung mit deionisiertem Wasser 3 Spülungmit H2O2 10/g/l.
28
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
fassend kann geschlossen werden, dass Eisen(II)-Ionen in hohen Konzentrationen zwar eine De-aktivierung der Katalysatoren zur Folge haben, eine Reaktivierung jedoch durch Spülen mit einer H2O2-Lösung möglich ist. Da der geplante Einsatz von elementarem Eisen zur Fällung von H2S sowieso mit einer H2O2- Spülung zur Verhinderung der mikrobiellen Sulfatreduktion gekoppelt wird, ist nicht damit zu rechnen, dass es durch gering Konzentrationen an Eisenionen zu einer Vermin-derung der Abbauleistung des Katalysators kommt.
Um die Effekte von gelöstem Eisen auch in Be-zug auf die in der Pilotanlage bevorzugt einge-setzten zeolithgestützen Palladiumkatalysatoren zu überprüfen, wurde derselbe Versuch auch mit diesem Katalysator (Pd/Y200 mit 0,5 % Pd der Firma Degussa) durchgeführt. Abb. 6.20 ist zu entnehmen, dass die Umsetzungsrate zu Ver-suchsbeginn (ohne Zugabe der Eisensulfatlö-sung bei ca. 45 % - 50 % lag und damit im Bereich der Reaktivität des Aluminiumoxidkataly-sators. Es kommt jedoch auch hier zu einer lang-samen Deaktivierung, nachdem ab ca. dem 6. Versuchstag eine Eisensulfatlösung zugeben wurde. Die Deaktivierung erfolgte allerdings et-was langsamer als bei Verwendung des Alumi-niumoxidkatalysators. Auch wurde nach 12 tägi-ger Versuchsdauer unter Zugabe der Eisensul-fatlösung noch eine Umsatzrate von ca. 20 % erhalten. Auch in diesem Fall konnte durch eine 2 stündige H2O2-Spülung (H2O2-Konz. ~10 g/l) die ursprüngliche Reaktivität wieder vollständig her-gestellt werden.
0
0.5
1
0 10 20 30
Zeit [Tage]
C A
usla
ss/ C
Ein
lass
[-]
TCE Massenbilanz 1
2
0
0.5
1
0 10 20 30
Zeit [Tage]
C A
usla
ss/ C
Ein
lass
[-]
TCE Massenbilanz 11
22
Abb. 6.20: Abbau von TCE (1 mg/l) in deionisiertemWasser mit einem Zeolith-gestützem Katalysator (Pdauf Zeolith Y-200) im Säulenversuch, Kontaktzeit 1 min.1 Zugabe von ~ 180 mg/l Fe2+. 2 Spülung mit H2O210/g/l.
4040
Dieser Versuch hat gezeigt, dass auch bei Verwendung des zeolithgestützen Palladiumkatalysa-tors eine Deaktivierung der reaktiven Zentren durch Eisen(II)- Ionen Eintritt. Der hydrophobe Zeo-lith scheint aber durchaus eine stabilisierende Wirkung auf den Katalysator zu haben. Die Deaktivierung verläuft hier jedoch über einen Zeitraum von einigen Tagen relativ langsam und eine Reaktivierung ist durch eine einfache H2O2-Spülung möglich. Deshalb wird der Einsatz von Eisen zur Fällung von H2S in der Pilotanlage als relativ unproblematisch für die Katalysatoraktivität ange-sehen, da eine regelmäßige Spülung mit H2O2 ohnehin vorgesehen ist.
6.2.4.5 Natriumnitrat
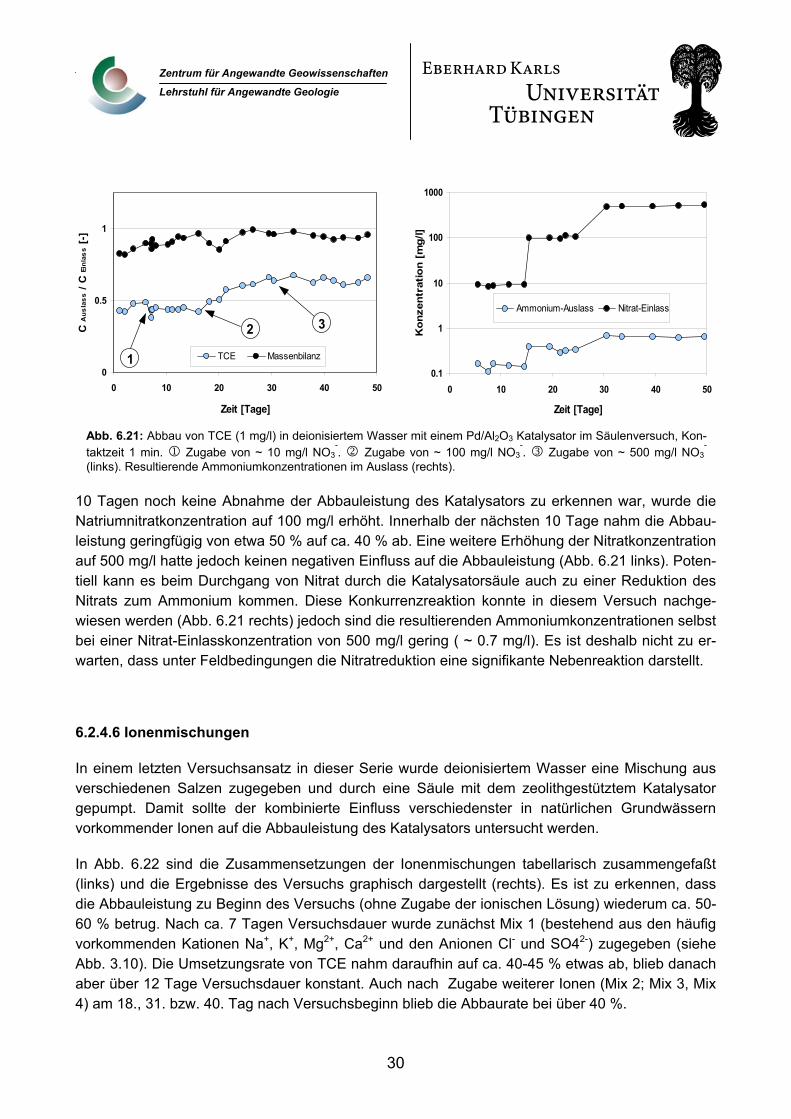
Im nächsten Beispiel wurde die Katalysatorsäule mit einer Natriumnitratlösung gespült (Abb. 6.21). Als Katalysator wurde wiederum ein palladisiertes Aluminiumoxid eingesetzt. Nach ca. 6 Tagen er-folgte zunächst die Zugabe eine NaNO3 – Lösung in einer Konzentration von 10 mg/l. Als nach ca.
29
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
0
0.5
1
0 10 20 30 40 50
Zeit [Tage]
C A
usla
ss /
C Ei
nlas
s [-
]
TCE Massenbilanz1
2 3
0.1
1
10
100
1000
0 10 20 30 40 5
Zeit [Tage]K
onze
ntra
tion
[mg/
l]
Ammonium-Auslass Nitrat-Einlass
0
Abb. 6.21: Abbau von TCE (1 mg/l) in deionisiertem Wasser mit einem Pd/Al2O3 Katalysator im Säulenversuch, Kon-taktzeit 1 min. 1 Zugabe von ~ 10 mg/l NO3
-. 2 Zugabe von ~ 100 mg/l NO3
-. 3 Zugabe von ~ 500 mg/l NO3
-
(links). Resultierende Ammoniumkonzentrationen im Auslass (rechts).
10 Tagen noch keine Abnahme der Abbauleistung des Katalysators zu erkennen war, wurde die Natriumnitratkonzentration auf 100 mg/l erhöht. Innerhalb der nächsten 10 Tage nahm die Abbau-leistung geringfügig von etwa 50 % auf ca. 40 % ab. Eine weitere Erhöhung der Nitratkonzentration auf 500 mg/l hatte jedoch keinen negativen Einfluss auf die Abbauleistung (Abb. 6.21 links). Poten-tiell kann es beim Durchgang von Nitrat durch die Katalysatorsäule auch zu einer Reduktion des Nitrats zum Ammonium kommen. Diese Konkurrenzreaktion konnte in diesem Versuch nachge-wiesen werden (Abb. 6.21 rechts) jedoch sind die resultierenden Ammoniumkonzentrationen selbst bei einer Nitrat-Einlasskonzentration von 500 mg/l gering ( ~ 0.7 mg/l). Es ist deshalb nicht zu er-warten, dass unter Feldbedingungen die Nitratreduktion eine signifikante Nebenreaktion darstellt.
6.2.4.6 Ionenmischungen
In einem letzten Versuchsansatz in dieser Serie wurde deionisiertem Wasser eine Mischung aus verschiedenen Salzen zugegeben und durch eine Säule mit dem zeolithgestütztem Katalysator gepumpt. Damit sollte der kombinierte Einfluss verschiedenster in natürlichen Grundwässern vorkommender Ionen auf die Abbauleistung des Katalysators untersucht werden.
In Abb. 6.22 sind die Zusammensetzungen der Ionenmischungen tabellarisch zusammengefaßt (links) und die Ergebnisse des Versuchs graphisch dargestellt (rechts). Es ist zu erkennen, dass die Abbauleistung zu Beginn des Versuchs (ohne Zugabe der ionischen Lösung) wiederum ca. 50-60 % betrug. Nach ca. 7 Tagen Versuchsdauer wurde zunächst Mix 1 (bestehend aus den häufig vorkommenden Kationen Na+, K+, Mg2+, Ca2+ und den Anionen Cl- und SO42-) zugegeben (siehe Abb. 3.10). Die Umsetzungsrate von TCE nahm daraufhin auf ca. 40-45 % etwas ab, blieb danach aber über 12 Tage Versuchsdauer konstant. Auch nach Zugabe weiterer Ionen (Mix 2; Mix 3, Mix 4) am 18., 31. bzw. 40. Tag nach Versuchsbeginn blieb die Abbaurate bei über 40 %.
30

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Kationen-Konz. [mg/l]
Anionen-Konz. [mg/l]
Mix 1 Na+ ~ 100 K+ ~ 50 Ca2+ ~ 100 Mg2+ ~ 50
Cl- ~ 380 SO4
2- ~ 300
Mix 2
Na+ ~ 260 K+ ~ 50 Ca2+ ~ 100 Mg2+ ~ 50
Cl- ~ 380 SO4
2- ~ 300 HCO3
- ~ 430
Mix 3
Na+ ~ 100 K+ ~ 50 Ca2+ ~ 100 Mg2+ ~ 50
Cl- ~ 380 SO4
2- ~ 300 Br- ~ 8
Mix 4 Na+ ~ 150 K+ ~ 50 Ca2+ ~ 100 Mg2+ ~ 50
Cl- ~ 380 SO4
2- ~ 300 Br- ~ 80 HPO4
2- ~ 70
0
0,25
0,5
0,75
1
1,25
0 10 20 30 40 5Zeit [Tage]
TCE Massenbilanzc/
c 0 (TC
E); M
asse
nbila
nz (M
out/M
in) 31 42
0
Abb. 6.22: Links: Anionen- und Kationenkonzentrationen in den Versuchen mit Ionenmischungen. Rechts: Ergeb-nisse, 1 mg/l TCE in deionisiertem Wasser; Katalysator: 2g Pd/Y-200; Kontaktzeit ~ 1 min; 1 Zugabe von Mix 1; 2Zugabe von Mix 2; 3 Zugabe von Mix 3; 4 Zugabe von Mix 4
6.2.4.7 Schlussfolgerungen
In den in diesem Kapitel vorgestellten Langzeitsäulenversuche konnte gezeigt werden, daß der Einfluss von vielen in Grundwässern verbreiteten ionischen Verbindungen auf die Langzeitstabilität von Palladiumkatalysatoren gering ist (z.B. Na+, K+, Mg2+, Ca2+ Cl- und SO42-). Einige ander Ionen, wie z.B. Fe2+, H2S oder S2- haben einen zum Teil stark deaktivierenden Effekt. In allen Fällen jedoch konnte eine in der Regel komplette Reaktivierung der Katalysatoren durch einfaches Spülen mit H2O2 erreicht werden.
In einem Fall, beim Einsatz von Kupferionen (Cu2+) konnte eine Reaktivierung mit H2O2 nicht erreicht werden. Das läßt auf einen anderen Deaktivierungsmechanismus, möglicherweise auf eine Chemisorption mit dem Eingehen von Bindungen zwischen dem Palladium und den Kupferionen schließen. Hier konnte durch eine Spülung des Katalysators mit einer wässrigen Ammoniaklösung ein Großteil der Reaktivität des Katalysators wiederhergestellt werden. Dieser Effekt beruht vermutlich auf einer Komplexbildung zwischen Kupfer und Ammoniak. Damit ist eine wichtige Erfahrung dieser Versuche, daß bei Kenntnis des deaktivierenden Stoffes gezielt nach Reaktivierungsmöglichkeiten gesucht werden kann.
Eine erfolgreiche Reaktivierung ist jedoch in einigen Fällen noch keine Lösung für einen erfolgreichen Langzeitbetrieb der Katalysatoren im Grundwasser. Bei einer permanenten Belastung des Grundwassers mit z.B. H2S ist eine permanente Deaktivierung zu befürchten, so daß eine Reaktivierung nur einen sehr kurzfristigen Effekt bedeuten würde. In solchen Fällen müßte eine geeignete Vorbehandlung des Wassers erfolgen, um diese Stoffe vor Erreichen des Katalysators aus dem Grundwasser zu entfernen.
31

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
6.2.5 Untersuchungen zur speziellen Problematik in Bitterfeld
Die Langzeitstabilität von Katalysatoren beim Betrieb in der wässrigen Phase ist im wesentlichen von der Zusammensetzung der wässrigen Phase abhängig. Im Bitterfelder Raum weist das Grundwasser, bedingt durch die lange Geschichte der chemischen Industrie in diesem Bereich, ein ausgesprochen komplexes organisches Schadstoffspektrum auf. Darüber hinaus ist auch auf an-organischer Seite ein hoher und heterogener Lösungsinhalt gegeben. Diese Rahmenbedingungen führen zu einer Vielzahl von potentiellen Möglichkeiten, die zu einer Katalysatordeaktivierung füh-ren können. Die Erfahrungen aus der Bitterfelder Pilotanlage haben jedoch im wesentlichen zu drei wahrscheinlichen Szenarien geführt, die zu den dortigen Stabilitätsproblemen der Katalysatoren beitragen:
• Die Schwefelwasserstofffracht (H2S) des geförderten Grundwassers in Schacht 3, induziert durch eine mikrobielle Sulfatreduktion im Aquifer,
• eine einsetzende mikrobiellen Sulfatreduktion in den Reaktorbereichen nach der Wasser-stoffzugabe, die ebenfalls zu einer H2S Bildung führt, sowie,
• eine komplexe Belastung des Grundwassers mit teilweise nicht identifizierten Schwefel- und Phosphororganika, wahrscheinlich aus der ehemaligen Pestizidproduktion.
In den begleitenden Laborversuchen wurde versucht, diese unterschiedlichen Problembereiche mit unterschiedlichen experimentellen Ansätzen anzugehen, um geeignete präventive Maßnahmen in der Pilotanlage treffen zu können. Im folgenden wird auf die Laborversuche zu den drei Problem-feldern eingegangen.
6.2.5.1 Schwefelwasserstofffracht des Grundwassers
Wie in Abb. 6.2 gezeigt wird, ist in dem im Schacht 3 der Pilotanlage geförderte Grundwasser über lange Zeiten eine mehr oder weniger starke H2S Belastung nachweisbar. Da H2S ein starkes Kata-lysatorgift ist, erfolgt deshalb eine permanente Deaktivierung des Katalysators. Deshalb ist eine Entfernung des Sulfids aus der wässrigen Phase unbedingt notwendig, bevor das Grundwasser den Katalysator erreicht.
Prinzipiell ist eine metallische Fällung dieser Sulfidfracht möglich, die z.B. durch Vorschalten einer metallischen Schüttung vor die Katalysatorsäule erreicht werden kann. Dieser Ansatz wurde des-halb in Laborversuchen auf seine Wirksamkeit untersucht. Hierzu wurden zunächst in Kurzzeitver-suchen unterschiedliche Materialien auf ihre Fällungseigenschaften getestet. Zum Einsatz kamen zwei verschiedene Eisensorten (Eisengranulat der Reaktoren des Teilprojektes 1.3, Arbeitsgruppe Prof. Dahmke, und ein Eisenschwamm), sowie ein kommerziell erhältliches, pelletiertes Zinkoxid der Degussa Hüls AG, welches verwendet wird um Gase von H2S abzureinigen. Die Versuche wurden als Säulenversuche durchgeführt. Dazu wurde in einem Teflonbeutel zunächst eine
32
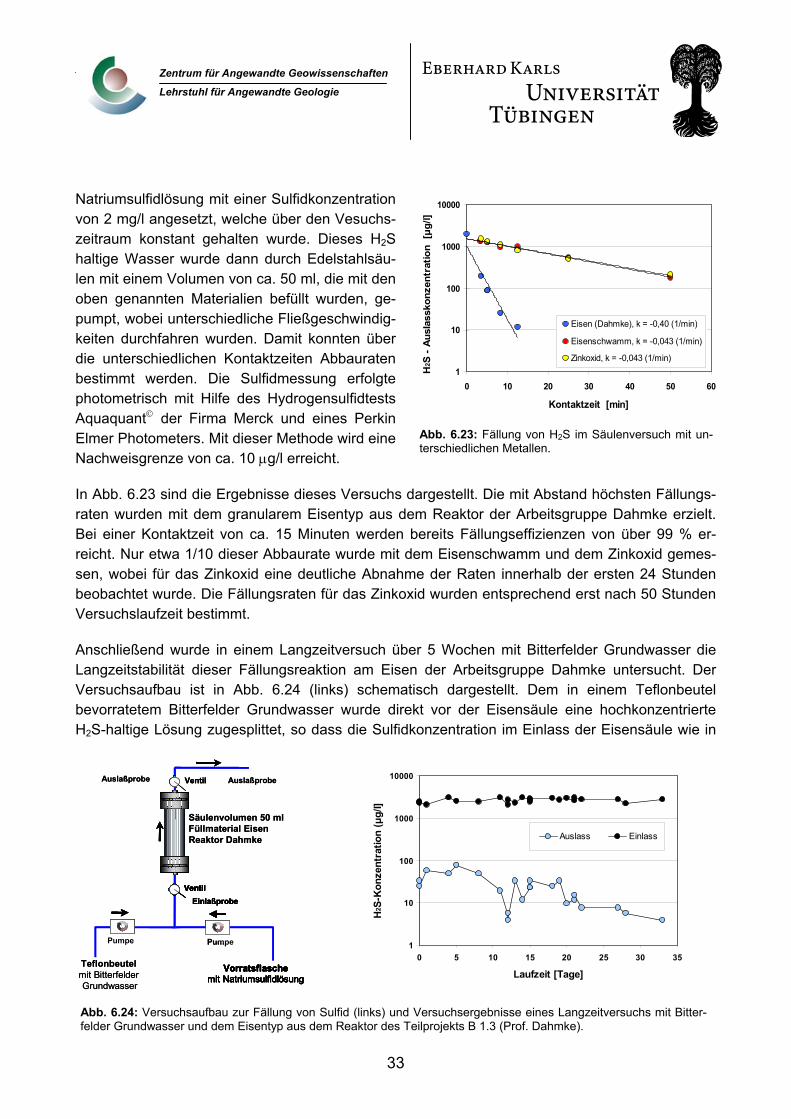
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
1
10
100
1000
10000
0 10 20 30 40 50 60
Kontaktzeit [min]
H2 S
- Au
slas
skon
zent
ratio
n [µ
g/l]
Eisen (Dahmke), k = -0,40 (1/min)
Eisenschwamm, k = -0,043 (1/min)
Zinkoxid, k = -0,043 (1/min)
Abb. 6.23: Fällung von H2S im Säulenversuch mit un-terschiedlichen Metallen.
Natriumsulfidlösung mit einer Sulfidkonzentration von 2 mg/l angesetzt, welche über den Vesuchs-zeitraum konstant gehalten wurde. Dieses H2S haltige Wasser wurde dann durch Edelstahlsäu-len mit einem Volumen von ca. 50 ml, die mit den oben genannten Materialien befüllt wurden, ge-pumpt, wobei unterschiedliche Fließgeschwindig- keiten durchfahren wurden. Damit konnten über die unterschiedlichen Kontaktzeiten Abbauraten bestimmt werden. Die Sulfidmessung erfolgte photometrisch mit Hilfe des Hydrogensulfidtests Aquaquant der Firma Merck und eines Perkin Elmer Photometers. Mit dieser Methode wird eine Nachweisgrenze von ca. 10 µg/l erreicht.
In Abb. 6.23 sind die Ergebnisse dieses Versuchs dargestellt. Die mit Abstand höchsten Fällungs-raten wurden mit dem granularem Eisentyp aus dem Reaktor der Arbeitsgruppe Dahmke erzielt. Bei einer Kontaktzeit von ca. 15 Minuten werden bereits Fällungseffizienzen von über 99 % er-reicht. Nur etwa 1/10 dieser Abbaurate wurde mit dem Eisenschwamm und dem Zinkoxid gemes-sen, wobei für das Zinkoxid eine deutliche Abnahme der Raten innerhalb der ersten 24 Stunden beobachtet wurde. Die Fällungsraten für das Zinkoxid wurden entsprechend erst nach 50 Stunden Versuchslaufzeit bestimmt.
Anschließend wurde in einem Langzeitversuch über 5 Wochen mit Bitterfelder Grundwasser die
Auslaßprobe Ventil
Ventil
Auslaßprobe
VentilVentil
Säulenvolumen 50 ml Füllmaterial Eisen Reaktor Dahmke
Auslaßprobe VentilVentil
VentilVentil
Auslaßprobe
VentilVentil
Säulenvolumen 50 ml Füllmaterial Eisen Reaktor Dahmke
100
1000
10000
-Kon
zent
ratio
n (µ
g/l]
Auslass Einlass
Langzeitstabilität dieser Fällungsreaktion am Eisen der Arbeitsgruppe Dahmke untersucht. Der Versuchsaufbau ist in Abb. 6.24 (links) schematisch dargestellt. Dem in einem Teflonbeutel bevorratetem Bitterfelder Grundwasser wurde direkt vor der Eisensäule eine hochkonzentrierte H2S-haltige Lösung zugesplittet, so dass die Sulfidkonzentration im Einlass der Eisensäule wie in
Einlaßprobe
Teflonbeutelmit BitterfelderGrundwasser
Pumpe
Einlaßprobe
Vorratsflasche mit Natriumsulfidlösung
Vorratsflasche mit Natriumsulfidlösung
Pumpe
Einlaßprobe
Teflonbeutelmit BitterfelderGrundwasser
Pumpe
Einlaßprobe
Vorratsflasche mit Natriumsulfidlösung
Vorratsflasche mit Natriumsulfidlösung
Vorratsflasche mit Natriumsulfidlösung
Vorratsflasche mit Natriumsulfidlösung
Pumpe 1
10
0 5 10 15 20 25 30 35
Laufzeit [Tage]
H2S
Abb. 6.24: Versuchsaufbau zur Fällung von Sulfid (links) und Versuchsergebnisse eines Langzeitversuchs mit Bitter-felder Grundwasser und dem Eisentyp aus dem Reaktor des Teilprojekts B 1.3 (Prof. Dahmke).
33
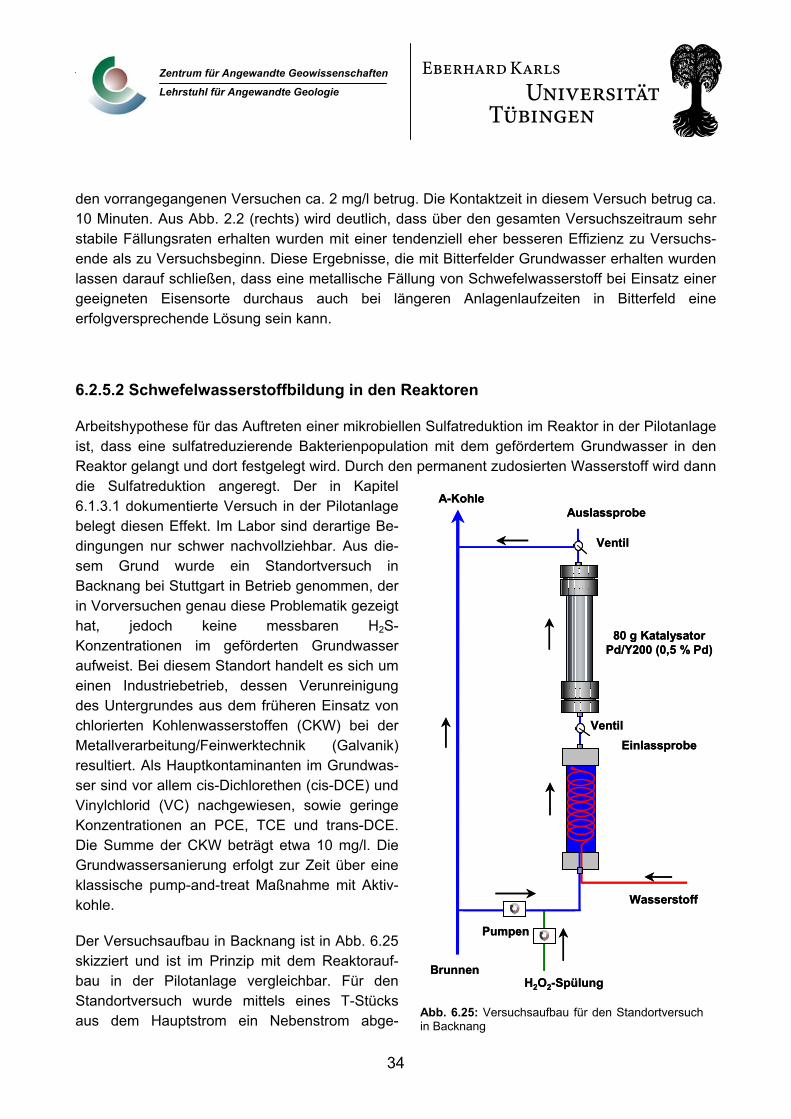
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
den vorrangegangenen Versuchen ca. 2 mg/l betrug. Die Kontaktzeit in diesem Versuch betrug ca. 10 Minuten. Aus Abb. 2.2 (rechts) wird deutlich, dass über den gesamten Versuchszeitraum sehr stabile Fällungsraten erhalten wurden mit einer tendenziell eher besseren Effizienz zu Versuchs-ende als zu Versuchsbeginn. Diese Ergebnisse, die mit Bitterfelder Grundwasser erhalten wurden lassen darauf schließen, dass eine metallische Fällung von Schwefelwasserstoff bei Einsatz einer geeigneten Eisensorte durchaus auch bei längeren Anlagenlaufzeiten in Bitterfeld eine erfolgversprechende Lösung sein kann.
6.2.5.2 Schwefelwasserstoffbildung in den Reaktoren
Arbeitshypothese für das Auftreten einer mikrobiellen Sulfatreduktion im Reaktor in der Pilotanlage ist, dass eine sulfatreduzierende Bakterienpopulation mit dem gefördertem Grundwasser in den Reaktor gelangt und dort festgelegt wird. Durch den permanent zudosierten Wasserstoff wird dann die Sulfatreduktion angeregt. Der in Kapitel 6.1.3.1 dokumentierte Versuch in der Pilotanlage belegt diesen Effekt. Im Labor sind derartige Be-dingungen nur schwer nachvollziehbar. Aus die-sem Grund wurde ein Standortversuch in Backnang bei Stuttgart in Betrieb genommen, der in Vorversuchen genau diese Problematik gezeigt hat, jedoch keine messbaren H2S-Konzentrationen im geförderten Grundwasser aufweist. Bei diesem Standort handelt es sich um einen Industriebetrieb, dessen Verunreinigung des Untergrundes aus dem früheren Einsatz von chlorierten Kohlenwasserstoffen (CKW) bei der Metallverarbeitung/Feinwerktechnik (Galvanik) resultiert. Als Hauptkontaminanten im Grundwas-ser sind vor allem cis-Dichlorethen (cis-DCE) und Vinylchlorid (VC) nachgewiesen, sowie geringe Konzentrationen an PCE, TCE und trans-DCE. Die Summe der CKW beträgt etwa 10 mg/l. Die Grundwassersanierung erfolgt zur Zeit über eine klassische pump-and-treat Maßnahme mit Aktiv-kohle.
Pumpen
Einlassprobe
Auslassprobe
Ventil
Ventil
Brunnen
A-Kohle
H2O2-Spülung
Wasserstoff
80 g KatalysatorPd/Y200 (0,5 % Pd)
Pumpen
Einlassprobe
Auslassprobe
Ventil
Ventil
Brunnen
A-Kohle
H2O2-Spülung
Wasserstoff
80 g KatalysatorPd/Y200 (0,5 % Pd)
Abb. 6.25: Versuchsaufbau für den Standortversuchin Backnang
Der Versuchsaufbau in Backnang ist in Abb. 6.25 skizziert und ist im Prinzip mit dem Reaktorauf-bau in der Pilotanlage vergleichbar. Für den Standortversuch wurde mittels eines T-Stücks aus dem Hauptstrom ein Nebenstrom abge-
34
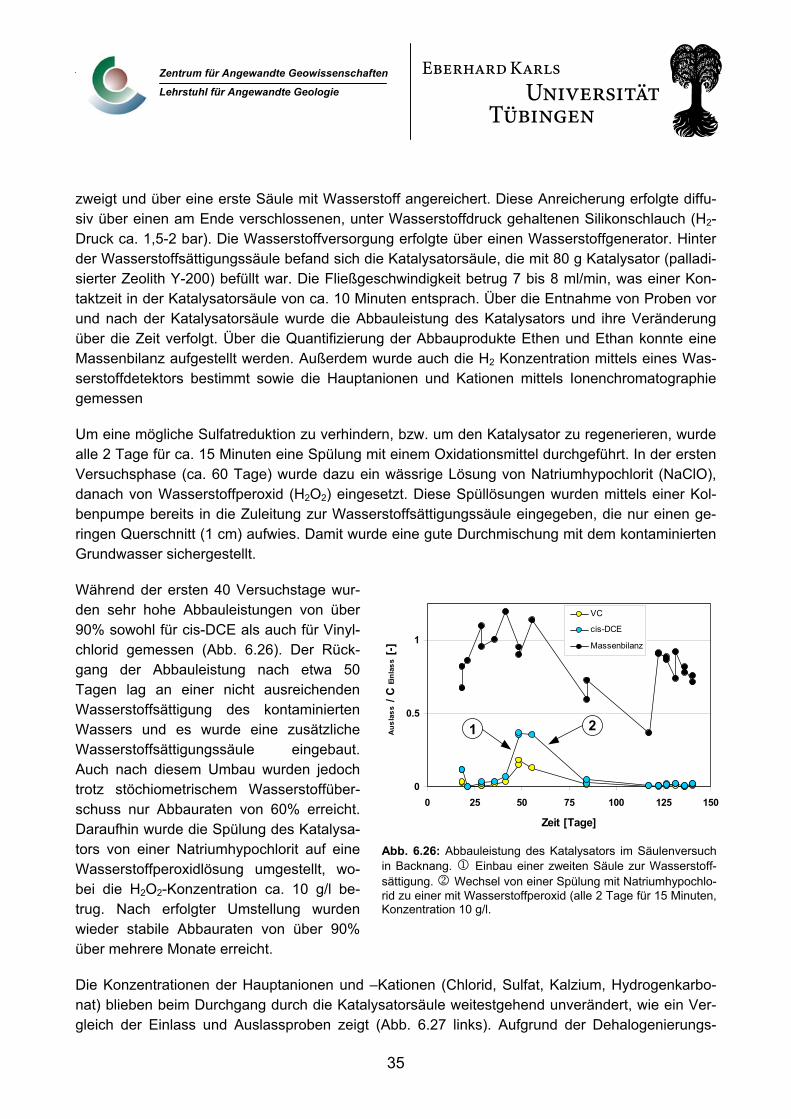
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
zweigt und über eine erste Säule mit Wasserstoff angereichert. Diese Anreicherung erfolgte diffu-siv über einen am Ende verschlossenen, unter Wasserstoffdruck gehaltenen Silikonschlauch (H2-Druck ca. 1,5-2 bar). Die Wasserstoffversorgung erfolgte über einen Wasserstoffgenerator. Hinter der Wasserstoffsättigungssäule befand sich die Katalysatorsäule, die mit 80 g Katalysator (palladi-sierter Zeolith Y-200) befüllt war. Die Fließgeschwindigkeit betrug 7 bis 8 ml/min, was einer Kon-taktzeit in der Katalysatorsäule von ca. 10 Minuten entsprach. Über die Entnahme von Proben vor und nach der Katalysatorsäule wurde die Abbauleistung des Katalysators und ihre Veränderung über die Zeit verfolgt. Über die Quantifizierung der Abbauprodukte Ethen und Ethan konnte eine Massenbilanz aufgestellt werden. Außerdem wurde auch die H2 Konzentration mittels eines Was-serstoffdetektors bestimmt sowie die Hauptanionen und Kationen mittels Ionenchromatographie gemessen
Um eine mögliche Sulfatreduktion zu verhindern, bzw. um den Katalysator zu regenerieren, wurde alle 2 Tage für ca. 15 Minuten eine Spülung mit einem Oxidationsmittel durchgeführt. In der ersten Versuchsphase (ca. 60 Tage) wurde dazu ein wässrige Lösung von Natriumhypochlorit (NaClO), danach von Wasserstoffperoxid (H2O2) eingesetzt. Diese Spüllösungen wurden mittels einer Kol-benpumpe bereits in die Zuleitung zur Wasserstoffsättigungssäule eingegeben, die nur einen ge-ringen Querschnitt (1 cm) aufwies. Damit wurde eine gute Durchmischung mit dem kontaminierten Grundwasser sichergestellt.
Während der ersten 40 Versuchstage wur-den sehr hohe Abbauleistungen von über 90% sowohl für cis-DCE als auch für Vinyl-chlorid gemessen (Abb. 6.26). Der Rück-gang der Abbauleistung nach etwa 50 Tagen lag an einer nicht ausreichenden Wasserstoffsättigung des kontaminierten Wassers und es wurde eine zusätzliche Wasserstoffsättigungssäule eingebaut. Auch nach diesem Umbau wurden jedoch trotz stöchiometrischem Wasserstoffüber-schuss nur Abbauraten von 60% erreicht. Daraufhin wurde die Spülung des Katalysa-tors von einer Natriumhypochlorit auf eine Wasserstoffperoxidlösung umgestellt, wo-bei die H2O2-Konzentration ca. 10 g/l be-trug. Nach erfolgter Umstellung wurden wieder stabile Abbauraten von über 90% über mehrere Monate erreicht.
C
0
0.5
1
0 25 50 75 100 125 150
Zeit [Tage]
Aus
lass
/ C
Einl
ass [
-]
VC
cis-DCE
Massenbilanz
1 2
Abb. 6.26: Abbauleistung des Katalysators im Säulenversuchin Backnang. 1 Einbau einer zweiten Säule zur Wasserstoff-sättigung. 2 Wechsel von einer Spülung mit Natriumhypochlo-rid zu einer mit Wasserstoffperoxid (alle 2 Tage für 15 Minuten,Konzentration 10 g/l.
Die Konzentrationen der Hauptanionen und –Kationen (Chlorid, Sulfat, Kalzium, Hydrogenkarbo-nat) blieben beim Durchgang durch die Katalysatorsäule weitestgehend unverändert, wie ein Ver-gleich der Einlass und Auslassproben zeigt (Abb. 6.27 links). Aufgrund der Dehalogenierungs-
35
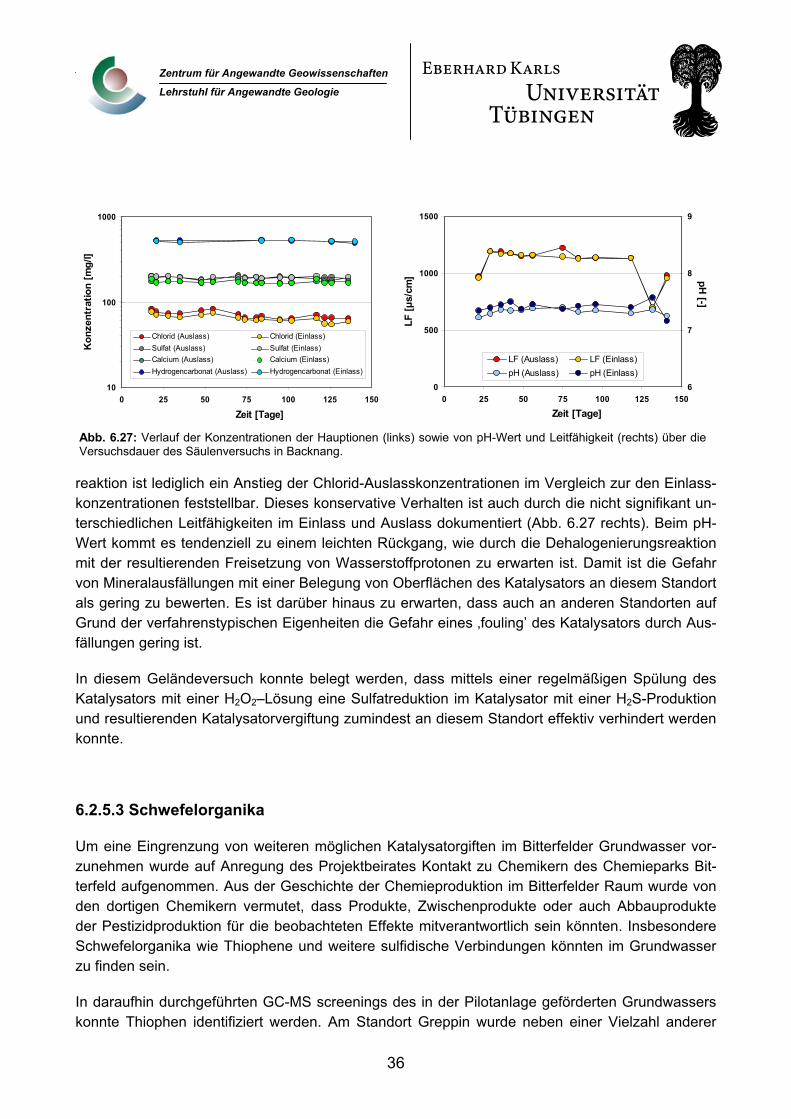
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
10
100
1000
0 25 50 75 100 125 150
Zeit [Tage]
Kon
zent
ratio
n [m
g/l]
Chlorid (Auslass) Chlorid (Einlass)Sulfat (Auslass) Sulfat (Einlass)Calcium (Auslass) Calcium (Einlass)Hydrogencarbonat (Auslass) Hydrogencarbonat (Einlass)
0
500
1000
1500
0 25 50 75 100 125 150
Zeit [Tage]LF
[µs/
cm]
6
7
8
9
pH [-]
LF (Auslass) LF (Einlass)pH (Auslass) pH (Einlass)
Abb. 6.27: Verlauf der Konzentrationen der Hauptionen (links) sowie von pH-Wert und Leitfähigkeit (rechts) über dieVersuchsdauer des Säulenversuchs in Backnang.
reaktion ist lediglich ein Anstieg der Chlorid-Auslasskonzentrationen im Vergleich zur den Einlass-konzentrationen feststellbar. Dieses konservative Verhalten ist auch durch die nicht signifikant un-terschiedlichen Leitfähigkeiten im Einlass und Auslass dokumentiert (Abb. 6.27 rechts). Beim pH-Wert kommt es tendenziell zu einem leichten Rückgang, wie durch die Dehalogenierungsreaktion mit der resultierenden Freisetzung von Wasserstoffprotonen zu erwarten ist. Damit ist die Gefahr von Mineralausfällungen mit einer Belegung von Oberflächen des Katalysators an diesem Standort als gering zu bewerten. Es ist darüber hinaus zu erwarten, dass auch an anderen Standorten auf Grund der verfahrenstypischen Eigenheiten die Gefahr eines ‚fouling’ des Katalysators durch Aus-fällungen gering ist.
In diesem Geländeversuch konnte belegt werden, dass mittels einer regelmäßigen Spülung des Katalysators mit einer H2O2–Lösung eine Sulfatreduktion im Katalysator mit einer H2S-Produktion und resultierenden Katalysatorvergiftung zumindest an diesem Standort effektiv verhindert werden konnte.
6.2.5.3 Schwefelorganika
Um eine Eingrenzung von weiteren möglichen Katalysatorgiften im Bitterfelder Grundwasser vor-zunehmen wurde auf Anregung des Projektbeirates Kontakt zu Chemikern des Chemieparks Bit-terfeld aufgenommen. Aus der Geschichte der Chemieproduktion im Bitterfelder Raum wurde von den dortigen Chemikern vermutet, dass Produkte, Zwischenprodukte oder auch Abbauprodukte der Pestizidproduktion für die beobachteten Effekte mitverantwortlich sein könnten. Insbesondere Schwefelorganika wie Thiophene und weitere sulfidische Verbindungen könnten im Grundwasser zu finden sein.
In daraufhin durchgeführten GC-MS screenings des in der Pilotanlage geförderten Grundwassers konnte Thiophen identifiziert werden. Am Standort Greppin wurde neben einer Vielzahl anderer
36
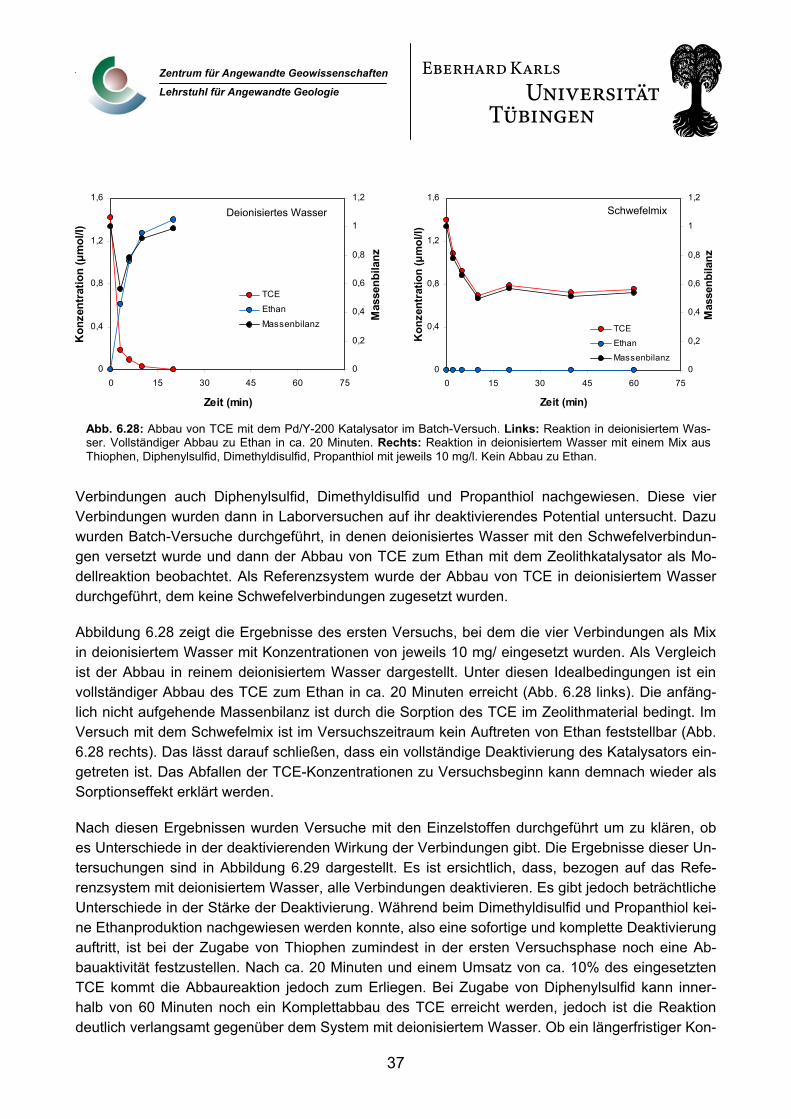
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Abb. 6.28: Abbau von TCE mit dem Pd/Y-200 Katalysator im Batch-Versuch. Links: Reaktion in deionisiertem Was-ser. Vollständiger Abbau zu Ethan in ca. 20 Minuten. Rechts: Reaktion in deionisiertem Wasser mit einem Mix aus Thiophen, Diphenylsulfid, Dimethyldisulfid, Propanthiol mit jeweils 10 mg/l. Kein Abbau zu Ethan.
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)K
onze
ntra
tion
(µm
ol/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)
Kon
zent
ratio
n (µ
mol
/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
SchwefelmixDeionisiertes Wasser
Verbindungen auch Diphenylsulfid, Dimethyldisulfid und Propanthiol nachgewiesen. Diese vier Verbindungen wurden dann in Laborversuchen auf ihr deaktivierendes Potential untersucht. Dazu wurden Batch-Versuche durchgeführt, in denen deionisiertes Wasser mit den Schwefelverbindun-gen versetzt wurde und dann der Abbau von TCE zum Ethan mit dem Zeolithkatalysator als Mo-dellreaktion beobachtet. Als Referenzsystem wurde der Abbau von TCE in deionisiertem Wasser durchgeführt, dem keine Schwefelverbindungen zugesetzt wurden.
Abbildung 6.28 zeigt die Ergebnisse des ersten Versuchs, bei dem die vier Verbindungen als Mix in deionisiertem Wasser mit Konzentrationen von jeweils 10 mg/ eingesetzt wurden. Als Vergleich ist der Abbau in reinem deionisiertem Wasser dargestellt. Unter diesen Idealbedingungen ist ein vollständiger Abbau des TCE zum Ethan in ca. 20 Minuten erreicht (Abb. 6.28 links). Die anfäng-lich nicht aufgehende Massenbilanz ist durch die Sorption des TCE im Zeolithmaterial bedingt. Im Versuch mit dem Schwefelmix ist im Versuchszeitraum kein Auftreten von Ethan feststellbar (Abb. 6.28 rechts). Das lässt darauf schließen, dass ein vollständige Deaktivierung des Katalysators ein-getreten ist. Das Abfallen der TCE-Konzentrationen zu Versuchsbeginn kann demnach wieder als Sorptionseffekt erklärt werden.
Nach diesen Ergebnissen wurden Versuche mit den Einzelstoffen durchgeführt um zu klären, ob es Unterschiede in der deaktivierenden Wirkung der Verbindungen gibt. Die Ergebnisse dieser Un-tersuchungen sind in Abbildung 6.29 dargestellt. Es ist ersichtlich, dass, bezogen auf das Refe-renzsystem mit deionisiertem Wasser, alle Verbindungen deaktivieren. Es gibt jedoch beträchtliche Unterschiede in der Stärke der Deaktivierung. Während beim Dimethyldisulfid und Propanthiol kei-ne Ethanproduktion nachgewiesen werden konnte, also eine sofortige und komplette Deaktivierung auftritt, ist bei der Zugabe von Thiophen zumindest in der ersten Versuchsphase noch eine Ab-bauaktivität festzustellen. Nach ca. 20 Minuten und einem Umsatz von ca. 10% des eingesetzten TCE kommt die Abbaureaktion jedoch zum Erliegen. Bei Zugabe von Diphenylsulfid kann inner-halb von 60 Minuten noch ein Komplettabbau des TCE erreicht werden, jedoch ist die Reaktion deutlich verlangsamt gegenüber dem System mit deionisiertem Wasser. Ob ein längerfristiger Kon-
37
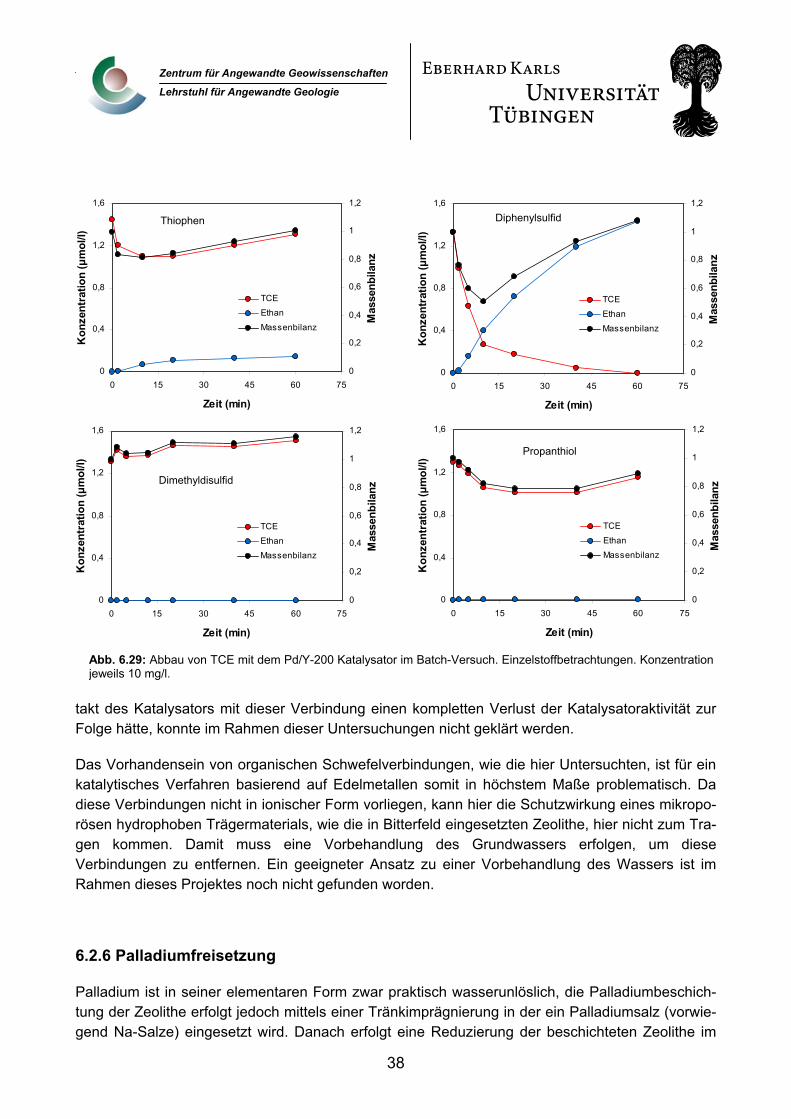
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)
Kon
zent
ratio
n (µ
mol
/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)K
onze
ntra
tion
(µm
ol/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)
Kon
zent
ratio
n (µ
mol
/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
0
0,4
0,8
1,2
1,6
0 15 30 45 60 75
Zeit (min)
Kon
zent
ratio
n (µ
mol
/l)
0
0,2
0,4
0,6
0,8
1
1,2
Mas
senb
ilanz
TCEEthanMassenbilanz
Dimethyldisulfid
Propanthiol
Diphenylsulfid Thiophen
Abb. 6.29: Abbau von TCE mit dem Pd/Y-200 Katalysator im Batch-Versuch. Einzelstoffbetrachtungen. Konzentration jeweils 10 mg/l.
takt des Katalysators mit dieser Verbindung einen kompletten Verlust der Katalysatoraktivität zur Folge hätte, konnte im Rahmen dieser Untersuchungen nicht geklärt werden.
Das Vorhandensein von organischen Schwefelverbindungen, wie die hier Untersuchten, ist für ein katalytisches Verfahren basierend auf Edelmetallen somit in höchstem Maße problematisch. Da diese Verbindungen nicht in ionischer Form vorliegen, kann hier die Schutzwirkung eines mikropo-rösen hydrophoben Trägermaterials, wie die in Bitterfeld eingesetzten Zeolithe, hier nicht zum Tra-gen kommen. Damit muss eine Vorbehandlung des Grundwassers erfolgen, um diese Verbindungen zu entfernen. Ein geeigneter Ansatz zu einer Vorbehandlung des Wassers ist im Rahmen dieses Projektes noch nicht gefunden worden.
6.2.6 Palladiumfreisetzung
Palladium ist in seiner elementaren Form zwar praktisch wasserunlöslich, die Palladiumbeschich-tung der Zeolithe erfolgt jedoch mittels einer Tränkimprägnierung in der ein Palladiumsalz (vorwie-gend Na-Salze) eingesetzt wird. Danach erfolgt eine Reduzierung der beschichteten Zeolithe im
38

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Wasserstoffstrom bei erhöhten Temperaturen. Etwaige nichtreduzierte Palladiumsalze könnten theoretisch bei einem Einsatz der Zeolithe im wässrigen Medium zu einer Palladiumfreisetzung führen. Darüber hinaus ist durch Abrieb von Feinstpartikeln vom Katalysatormaterial bei einer Re-aktorbefüllung eine Freisetzung partikelgebundenen Palladiums in der Anfangsphase einer Reak-torbetriebs möglich.
Deshalb wurden Laborversuche zur Palladiumfreisetzung durchgeführt. Dazu wurden insgesamt drei Säulen mit drei unterschiedlichen Katalysatormaterialien gefüllt und über einen Zeitraum von 40 Tagen mit Wässern unterschiedlicher pH-Werte durchströmt. Die Eluate wurden aufgefangen und auf ihren Palladiumgehalt mittels AAS (Atomadsorptionsspektroskopie) untersucht. Dazu wur-den die wässrigen Eluate im Rotationsverdampfer maximal auf 1 % des Gesamtvolumens einge-engt. Die Nachweisgrenze nach der Einengung lag bei ca. 0,1 µg/l.
Als pH-Werte wurden für die drei Säulen 5, 7 und 9 gewählt, die den in Grundwässern zu erwar-tenden pH-Bereich abdecken. Die pH-Werte wurden mittels kommerzieller pH-Pufferlösungen ein-gestellt. Die Säulenvolumina lagen bei jeweils 50 ml, die Katalysatormengen bei jeweils ca. 27 g. Bei Porositäten der Katalysatorschüttungen von ca. 50 % und Flussraten von 1 ml/min wurden Kontaktzeiten in den Säulen von ca. 25 min erreicht.
Als Katalysatormaterialien kamen zum Einsatz:
• 0,5 % Pd auf Zeolith Y-200, Degussa AG, eingesetzt wie erhalten
• 0,5 % Pd auf Zeolith Y-200, Degussa AG, nachreduziert im H2-Strom bei 400°C
• 0,5 % Pd auf Al2O3, Degussa AG
Neben diesen beschichteten Katalysatormaterialien wurde in eine vierte Säule gleicher Dimension der pelletierte Y-200 Zeolith ohne Palladium eingebaut. Diese Probe diente als Referenzprobe und wurde nur bei pH 7 durchströmt.
Die Ergebnisse dieser Versuche sind in Abbildung 6.30 dargestellt. In allen Versuchen konnte Pal-ladium im Eluat aus den Säulen nachgewiesen werden. Das gilt auch für die Referenzsäule, in der nur ein unbeschichteter Zeolith eingebaut wurde. Die Konzentrationen lagen hier bei Maximalwer-ten um 1 µg/l – 2 µg/l (entsprechend Austragsfrachten von ca. 0,001µg/min). Diese Konzentration werden als Hintergrundwerte interpretiert, bei denen unklar bleibt ob es sich um eine tatsächliche Belastung des Wassers mit Palladium oder um Artefakte in der Analytik durch die starke Einen-gung der wässrigen Eluate handelt.
Bei den palladiumhaltigen Katalysatoren werden für alle drei Materialien sehr ähnliche Austrags-muster gefunden, wobei eine deutliche Abhängigkeit der Austragsraten vom pH-Wert besteht. Ge-nerell kann geschlossen werden, dass tiefe pH-Werte einen höheren Palladiumaustrag zur Folge haben. Bei pH 9 fallen die Austräge sehr schnell in den Bereich der Hintergrundwerte und nach ei-nigen Tagen unter die Nachweisgrenze. Bei pH 5 werden für alle Materialien zu Versuchsende
39
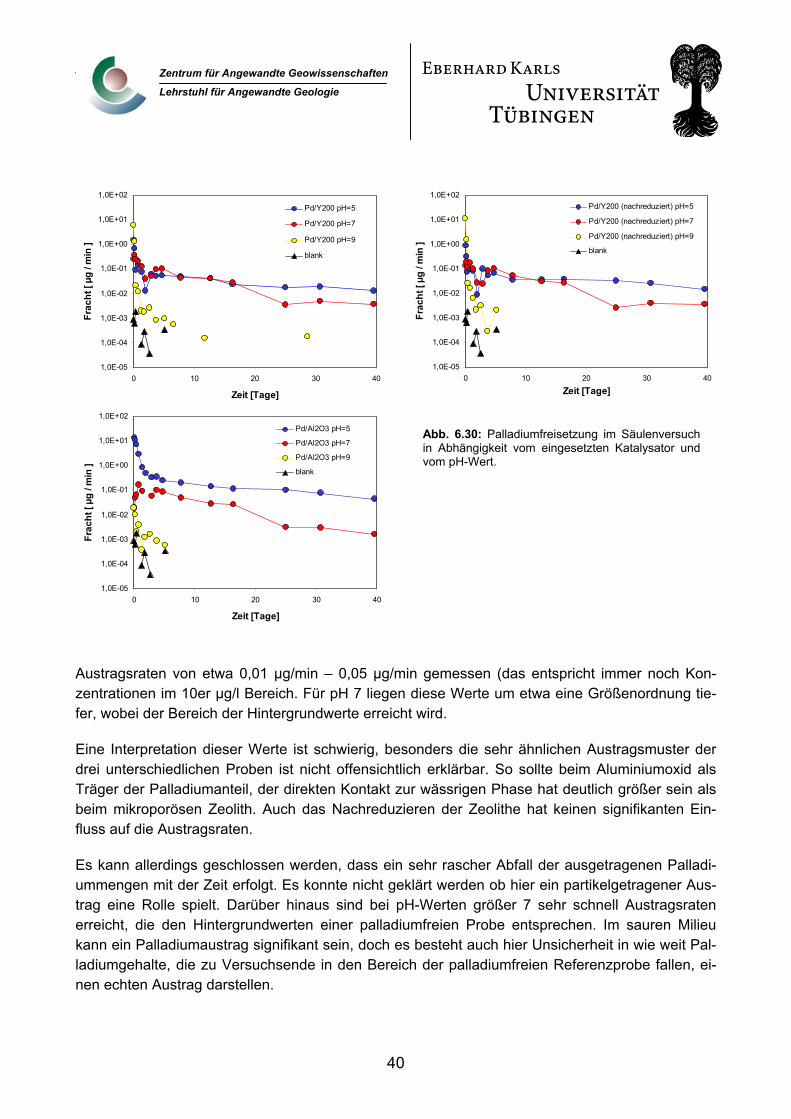
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Abb. 6.30: Palladiumfreisetzung im Säulenversuch in Abhängigkeit vom eingesetzten Katalysator und vom pH-Wert.
1,0E-05
1,0E-04
1,0E-03
1,0E-02
1,0E-01
1,0E+00
1,0E+01
1,0E+02
0 10 20 30 40
Zeit [Tage]
Frac
ht [
µg /
min
]
Pd/Y200 (nachreduziert) pH=5
Pd/Y200 (nachreduziert) pH=7
Pd/Y200 (nachreduziert) pH=9
blank
1,0E-05
1,0E-04
1,0E-03
1,0E-02
1,0E-01
1,0E+00
1,0E+01
1,0E+02
0 10 20 30 40
Zeit [Tage]
Frac
ht [
µg /
min
]
Pd/Al2O3 pH=5
Pd/Al2O3 pH=7
Pd/Al2O3 pH=9
blank
1,0E-05
1,0E-04
1,0E-03
1,0E-02
1,0E-01
1,0E+00
1,0E+01
1,0E+02
0 10 20 30 40
Zeit [Tage]
Frac
ht [
µg /
min
]
Pd/Y200 pH=5
Pd/Y200 pH=7
Pd/Y200 pH=9
blank
Austragsraten von etwa 0,01 µg/min – 0,05 µg/min gemessen (das entspricht immer noch Kon-zentrationen im 10er µg/l Bereich. Für pH 7 liegen diese Werte um etwa eine Größenordnung tie-fer, wobei der Bereich der Hintergrundwerte erreicht wird.
Eine Interpretation dieser Werte ist schwierig, besonders die sehr ähnlichen Austragsmuster der drei unterschiedlichen Proben ist nicht offensichtlich erklärbar. So sollte beim Aluminiumoxid als Träger der Palladiumanteil, der direkten Kontakt zur wässrigen Phase hat deutlich größer sein als beim mikroporösen Zeolith. Auch das Nachreduzieren der Zeolithe hat keinen signifikanten Ein-fluss auf die Austragsraten.
Es kann allerdings geschlossen werden, dass ein sehr rascher Abfall der ausgetragenen Palladi-ummengen mit der Zeit erfolgt. Es konnte nicht geklärt werden ob hier ein partikelgetragener Aus-trag eine Rolle spielt. Darüber hinaus sind bei pH-Werten größer 7 sehr schnell Austragsraten erreicht, die den Hintergrundwerten einer palladiumfreien Probe entsprechen. Im sauren Milieu kann ein Palladiumaustrag signifikant sein, doch es besteht auch hier Unsicherheit in wie weit Pal-ladiumgehalte, die zu Versuchsende in den Bereich der palladiumfreien Referenzprobe fallen, ei-nen echten Austrag darstellen.
40

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Würde man eine Fracht von 0,001 µg/min als Dauerfracht annehmen, der Bereich, der zu Ver-suchsende bei pH 7 in etwa erreicht wurde, so kann man auf dieser Basis Austragszeiten rechnen. Bei einer Kontaktzeit von 25 Minuten, die charakteristisch für Katalysereaktoren wäre, würde pro Jahr ca. 0,3 % der Palladiummasse ausgetragen. Theoretisch wäre nach ca. 250 Jahren Betriebs-zeit alles Palladium ausgetragen. Diese Annahme wäre außerdem als sehr pessimistisch zu be-zeichnen, da mit großer Wahrscheinlichkeit ein weiterer Rückgang der Austragsraten mit der Zeit zu verzeichnen wäre. Es kann deshalb auf Grund dieser Daten geschlossen werden, dass zumin-dest bei pH Werten im neutralen und basischen Bereich Palladiumfreisetzung aus den Katalysato-ren ein kurzfristiges Problem sein kann, jedoch langfristig nur eine eher untergeordnete Rolle spielen sollte.
7. Zusammenfassende Bewertung Die Erfahrungen aus diesem Projekt in der Bitterfelder Pilotanlage sowie aus den begleitenden Laborversuchen haben vor allem die Risiken katalytischer Technologien in Bezug auf die Standfestigkeit der eingesetzten Katalysatoren deutlich gemacht. In der Bewertung der ersten Projektphase sind vor dem Hintergrund der vorliegenden Ergebnisse die folgenden Schlußfolgerungen zu ziehen:
• Der katalytische Abbau von chlorierten Verbindungen in wässriger Phase ist prinzipiell möglich und wurde in-situ in der Bitterfelder Pilotanlage nachgwiesen.
• Gerade unter den hydrogeochemischen Bedingungen im Bitterfelder Raum ist jedoch eine Langzeitstabilität der eingesetzten Katalysatoren bisher nicht erreicht worden.
• Diese fehlende Langzeitstabilität resultiert mit grosser Wahrscheinlichkeit aus einer Katalysatorvergiftung durch mikrobiologisch im Grundwasser gebildeten Schwefelwasserstoff sowie durch im Grundwasser vorhandene Organoschwefel und/oder Organophosphor-verbindungen (Produktionsrückstände der ehemaligen Chemiekombinate).
• Bisher wurde keine geeignete Möglichkeit gefunden, die Katalysatoren in der wässrigen Phase vollständig vor diesen Verbindungen zu schützen.
Diese Ergebnisse stellen jedoch die Katalyse in wässriger Phase als Sanierungsvariante nicht grundsätzlich in Frage. So wurden an Standorten mit weniger komplexer Grundwasserchemie (Backnang und Denkendorf bei Stuttgart) stabile Prozessabläufe in der wässerigen Phase über Monate hinweg erreicht. Darüberhinaus konnte wiederholt gezeigt werden, dass auch die in Bitterfeld deaktivierten Katalysatoren mit einer oxidativen Behandlung (H2O2) erfolgreich regeneriert werden konnten. Diese Befunde wurden auch durch oberflächenspektroskopische Untersuchungen (XPS) belegt.
41
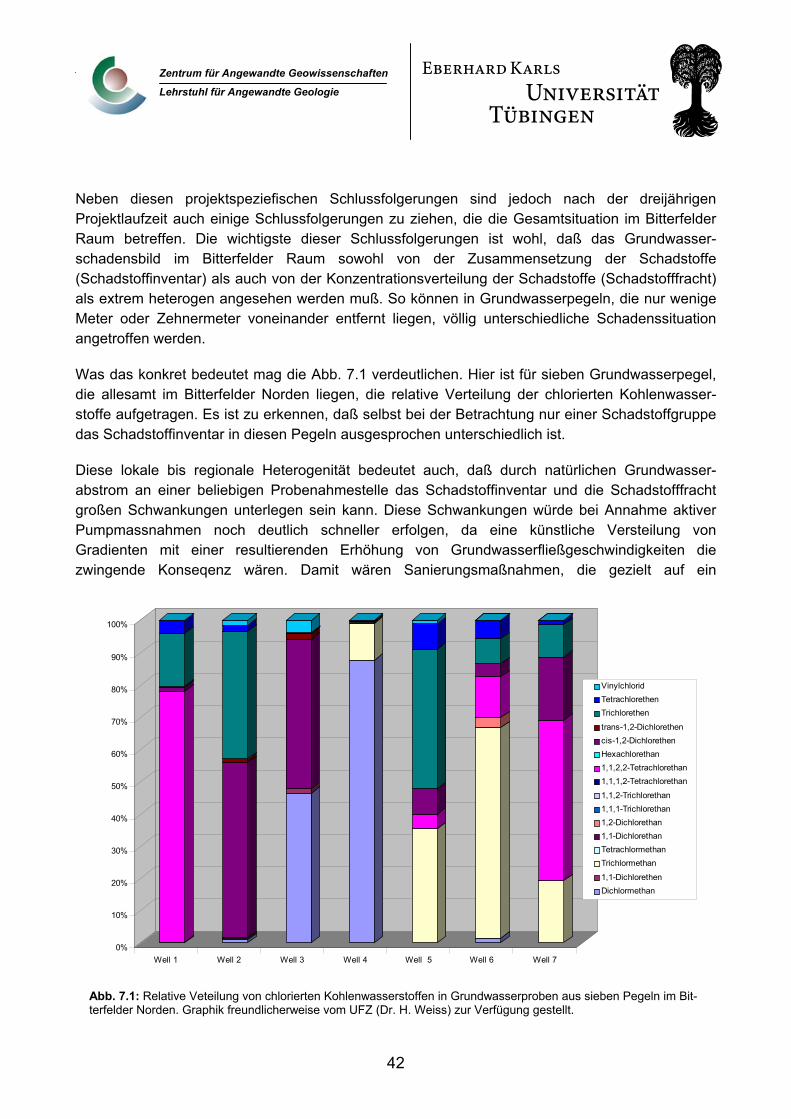
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Neben diesen projektspeziefischen Schlussfolgerungen sind jedoch nach der dreijährigen Projektlaufzeit auch einige Schlussfolgerungen zu ziehen, die die Gesamtsituation im Bitterfelder Raum betreffen. Die wichtigste dieser Schlussfolgerungen ist wohl, daß das Grundwasser-schadensbild im Bitterfelder Raum sowohl von der Zusammensetzung der Schadstoffe (Schadstoffinventar) als auch von der Konzentrationsverteilung der Schadstoffe (Schadstofffracht) als extrem heterogen angesehen werden muß. So können in Grundwasserpegeln, die nur wenige Meter oder Zehnermeter voneinander entfernt liegen, völlig unterschiedliche Schadenssituation angetroffen werden.
Was das konkret bedeutet mag die Abb. 7.1 verdeutlichen. Hier ist für sieben Grundwasserpegel, die allesamt im Bitterfelder Norden liegen, die relative Verteilung der chlorierten Kohlenwasser-stoffe aufgetragen. Es ist zu erkennen, daß selbst bei der Betrachtung nur einer Schadstoffgruppe das Schadstoffinventar in diesen Pegeln ausgesprochen unterschiedlich ist.
Diese lokale bis regionale Heterogenität bedeutet auch, daß durch natürlichen Grundwasser-abstrom an einer beliebigen Probenahmestelle das Schadstoffinventar und die Schadstofffracht großen Schwankungen unterlegen sein kann. Diese Schwankungen würde bei Annahme aktiver Pumpmassnahmen noch deutlich schneller erfolgen, da eine künstliche Versteilung von Gradienten mit einer resultierenden Erhöhung von Grundwasserfließgeschwindigkeiten die zwingende Konseqenz wären. Damit wären Sanierungsmaßnahmen, die gezielt auf ein
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
Well 1 Well 2 Well 3 Well 4 Well 5 Well 6 Well 7
VinylchloridTetrachlorethenTrichlorethentrans-1,2-Dichlorethencis-1,2-DichlorethenHexachlorethan1,1,2,2-Tetrachlorethan1,1,1,2-Tetrachlorethan1,1,2-Trichlorethan1,1,1-Trichlorethan1,2-Dichlorethan1,1-DichlorethanTetrachlormethanTrichlormethan1,1-DichlorethenDichlormethan
Abb. 7.1: Relative Veteilung von chlorierten Kohlenwasserstoffen in Grundwasserproben aus sieben Pegeln im Bit-terfelder Norden. Graphik freundlicherweise vom UFZ (Dr. H. Weiss) zur Verfügung gestellt.
42

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
bestimmtes Schadstoffspektrum abgestellt sind, nur zeitlich sehr begrenzt einsetzbar, da von einer Änderung des Schadstoffspektrums mit fortschreitender Sanierungsdauer ausgegangen werden muss. Jede Technologie, die mit hohen Kosten z.B. in-situ implemetiert wird, wäre damit mit großen Risiken bezüglich eines langfristigen Einsatzes behaftet.
Neben dieser räumlich stark heterogenen Schadstoffverteilung wird die Situation zusätzlich durch ein extrem breites Schadstoffspektrum erschwert. Im Bereich der Pilotanlage ist die Grundwasser-kontamination zwar vor allem durch Monochlorbenzol dominiert wobei untergeordnet auch andere chlorierte Kohlenwasserstoffe nachgewiesen werden können. Damit ist jedoch nicht die gesamte Kontamination erfaßt denn mittels Massenspektroskopie lassen sich viele weitere Verbindungen in geringen Konzentrationen nachweisen jedoch oft nicht exakt bestimmen. Regional gesehen können aber an anderen Grundwassermessstellen komplexeste Schadstoffmischungen nachgewiesen werden. In der Abb. 7.2 ist eine Aufstellung abgebildet, die die an einer Grundwassermessstelle imBitterfelder Norden analysierten und quantifizierten Schadstoffe zusammenfasst. Die Aufstellung umfaßt organische Verbindungen unterschiedlichster Gruppen und mit unterschiedlichsten chemisch-physikalischen Eigenschaften. So werden extrem gut wasserlösliche hydrophile Verbindungen wie Phenole neben stark hydrophoben schlecht wasserlöslichen Verbindungen wie hochchlorierte Benzole nachgewiesen.
Group Compound µg/l Group Compound µg/l
Anilines Aniline 23 Cl-Phenols Monochlorphenols 440Methylbenzamine 15 2,4-Dichlorphenol 9Chloroanilines 1181 2,4,6-Trichlorphenol 9Chloro-N-methylbenzenamine 19 Pentachlorphenol 17Dichlorobenzenamine 415 4-Chloro-3-Methylphenol 68
BTEX Benzene 26054 Phenols Phenol 93Toluene 331 Methlyphenols 328Ethylbenzene 19 Dimethylphenols 17Xylenes 147 Ethylphenol 16
CVOC Dichloromethane 111 Diethylphenol 29Trichloromethane 4449 Ethylmethylphenols 42trans-1,2-Dichloroethene 25 Trimethylphenols 12cis-1,2-Dichloroethene 69 4-Nitrophenol 168Trichloroethene 28 Pesticides Trimethylthiophosphat 53
Cl-Benzenes Chlorobenzene 37140 HCHs 1Chlormethylbenzene 241 S-Organics Trithiolan 259Bromochlorobenzene 24 Tetrachlorthiophene 662-Chlorotoluene 350 Pentachlorthiocane 191,3- /1,4-Dichlorobenzene 14073 Others Sulfur 381,2-Dichlorobenzene 9655 Bromphenylhydrazine 76Dichloromethylbenzene 64 Dimethylphthalate 761,2,4 Trichlorobenzene 1010 Ethylmandelate 2231,2,3 Trichlorbenzene 43 Benzylbutylphthalate 1221,2,4,5 Tetrachlorobenzene 42 Bromobenzene 581,2,3,4 Tetrachlorobenzene 344 Cyclohexane 5Pentachlorobenzene 7 Naphthalin 25Hexachlorobenzene 0,2 2-Naphthol 43
Abb. 7.2: Mittels Gaschromatographie und Massenspektroskopie nachgewiesene organische Verbindungen in einerGrundwasserprobe aus dem Bitterfelder Norden. Tabelle freundlicherweise vom UFZ (Dr. H. Weiss) zur Verfügunggestellt.
43

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Bewertet man ein solch komplexes Schadstoffspektrum in Bezug auf eine Behandelbarkeit durch klassische Technologien und auch durch in SAFIRA entwickelte Technologien so wird klar, dass in der Regel nur ein Bruchteil der Verbindungen durch eine einzige Technologie zu behandeln sind. Abb. 7.3 zeigt als Beispiel die gleiche Aufstellung wie in Abb. 5.2 wobei das Schadstoffspektrum auf eine Behandelbarkeit durch eine reduktiv katalytische Methode analysiert wird. In grün sind die Stoffe hervorgehoben, bei denen durch diese Methode ein vollständiger Abbau zu unbedenklichen Endprodukten angenommen werden kann. In blau sind Verbindungen markiert, bei denen ein teilweiser Abbau möglich ist aber nicht mit unbedenkichen Endprodukten gerechnet werden kann. Bei grau ausgeschriebenen Verbindungen ist keine Reaktivität zu erwarten bzw. Reaktionen könnten möglich sein, sind aber unbekannt. Schließlich sind Verbindungen in rot gehalten (Schwefelorganika), die für ein vollständiges Versagen der katalytischen Methode durch Deaktivierung der Katalysatoren sorgen könnten.
Ähnliche Bewertungen dieses Schadstoffspektrums wären wohl auch für die anderen SAFIRA Technologien abzugeben, sowohl für die abiotischen als auch für die biologischen Methoden. Damit wird klar, dass nicht eine einzelne Methode als Problemlösung angesehen werden kann. Vielmehr müßte durch intelligente Kombination mehrerer Technologien eine möglichst vollständige
Group Compound µg/l Group Compound µg/l
Anilines Aniline 23 Cl-Phenols Monochlorphenols 440Methylbenzamine 15 2,4-Dichlorphenol 9Chloroanilines 1181 2,4,6-Trichlorphenol 9Chloro-N-methylbenzenamine 19 Pentachlorphenol 17Dichlorobenzenamine 415 4-Chloro-3-Methylphenol 68
BTEX Benzene 26054 Phenols Phenol 93Toluene 331 Methlyphenols 328Ethylbenzene 19 Dimethylphenols 17Xylenes 147 Ethylphenol 16
CVOC Dichloromethane 111 Diethylphenol 29Trichloromethane 4449 Ethylmethylphenols 42trans-1,2-Dichloroethene 25 Trimethylphenols 12cis-1,2-Dichloroethene 69 4-Nitrophenol 168Trichloroethene 28 Pesticides Trimethylthiophosphat 53
Cl-Benzenes Chlorobenzene 37140 HCHs 1Chlormethylbenzene 241 S-Organics Trithiolan 259Bromochlorobenzene 24 Tetrachlorthiophene 662-Chlorotoluene 350 Pentachlorthiocane 191,3- /1,4-Dichlorobenzene 14073 Others Sulfur 381,2-Dichlorobenzene 9655 Bromphenylhydrazine 76Dichloromethylbenzene 64 Dimethylphthalate 761,2,4 Trichlorobenzene 1010 Ethylmandelate 2231,2,3 Trichlorbenzene 43 Benzylbutylphthalate 1221,2,4,5 Tetrachlorobenzene 42 Bromobenzene 581,2,3,4 Tetrachlorobenzene 344 Cyclohexane 5Pentachlorobenzene 7 Naphthalin 25Hexachlorobenzene 0,2 2-Naphthol 43
Abb. 7.3: Bewertung des Schadstoffspektrums für eine Behandelbarkeit durch eine reduktiv katalytische Methode.Grün: abbaubar zu unbedenklichen Endprodukten; Blau: Abbaubar aber in der Regel nicht zu unbedenklichen End-produkten; Grau: Kein Abbau, bzw nicht bekannt; Rot: Potentielle Katalysatorgifte. Tabelle freundlicherweise vomUFZ (Dr. H. Weiss) zur Verfügung gestellt.
44

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Behandelbarkeit des Schadstoffspektrums sichergestellt werden. Wie bereits erwähnt könnte auch in Verbindung mit der räumlichen Heterogenität der Kontamination der Einsatz von in-situ Technologien als problematisch angsehen werden. Damit wäre z.B. der Einsatz von skalierbaren und kombinierbaren Technologien zur Behandlung von ‚Hot-Spots’ durch lokale pump and treat Maßnahmen durchaus als Option in Erwägung zu ziehen. Als Verdienst von SAFIRA könnte angesehen werden, dass in diesem Verbundprojekt eine Vielzahl von Technologien entwickelt worden sind, die als einzelne zwar keine Problemlösung bieten aber in Kombination durchaus einsetzbar sein könnten.
8. Ausblick Einer der Erkentnisse dieses Projektes war, dass die katalytischen Verfahren in der wässrigen Phase außerordentlich abhängig sind vom Chemismus des Grundwassers. Bei ungünstigen Vorraussetzungen, wie sie in Bitterfeld gegeben sind, kann der Einsatz dieser Technik problema-tisch oder sogar unmöglich sein. Deshalb wurde in der letzten Phase des SAFIRA Projekts zusammen mit der Arbeitsgruppe von Prof. Dr. Kopinke vom UFZ eine alternative Technologie entwickelt, die diese Problematik umgeht. Um eine Deaktivierung der Katalysatoren durch die komplexe Grundwassermatrix zu verhindern, wird die katalytische Umsetzung der Schadstoffe in die Gasphase verlegt, wobei ein Membranfiltermodul zur Phasentrennung eingesetzt wird. Diese Trennung der Stoffflüsse, in eine von Schadstoffen abgereicherte wässrige Phase und eine mit Schadstoffen angereicherte Gasphase, bringt erhebliche Vorteile mit sich. Zum einen sind in der Gasphase auf Grund der sehr viel höheren Diffusionskoeffizienten (ca. 3 Größenordnungen) extrem hohe Reaktionsraten zu erzielen. Darüberhinaus ist in der Gasphase eine bessere Prozesskontrolle zu erreichen. So sind für die Gasphase Standardkatalysatoren erhältlich, etwa Zinkoxid (ZnO), die vorgeschaltet vor den Dehalogenierungskatalysator eine effektive Entfernung etwaiger leichtflüchtiger Schwefelverbindungen gewährleisten. Der Prozessablauf eines solchen Verfahrens läßt sich grundsätzlich in drei Teile aufteilen:
• Transfer der Schadstoffe aus der wässrigen Phase in die Gasphase mittels eines Membranfiltermoduls. Dazu muss eine ausreichende Flüchtigkeit der Verbindungen gegeben sein. Flüssigphase und Gasphase sind dabei durch poröse hydrophobe Polymermembranen getrennt, wobei auf der Gasseite Sickstoff als Spülgas eingesetzt wird. Damit wird der Strippvorgang unter Sauerstoffausschluss durchgeführt.
• Behandlung etwaiger in der wässrigen Phase vorhandener Schadstoffe mit geringer Flüchtigkeit. Das kann zum einen vor dem Membranfiltermodul, um sie in eine leichter flüchtige Spezies zu überführen, oder nach dem Membranfiltermodul in Form einer z.B. sorptiven Entfernung der nicht strippbaren Verbindungen aus der wässrigen Phase erfolgen.
45

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
• Behandlung der Gasphase sowohl in Bezug auf eine etwaige Schwefelfracht als auch in Bezug auf den katalytischen Schadstoffabbau.
Dieses Konzept wurde bereits unter Beteiligung der Arbeitsgruppen Kopinke, Grathwohl und Schüth mit sehr ermutigenden Ergebnissen sowohl in Laborversuchen, als auch in einer on-site Pilotanlage in Greppin sowie an einem Standort auf Sardinien getestet.
Die Arbeitsgruppe Schüth war dabei für die Membranfiltration verantwortlich. Die verwendeten Hohlfasermembranmodule werden von der Firma Celgard Inc. (USA) produziert und werden in der Regel zur Erzeugung sauerstofffreien Wassers in der Halbleiterindustrie genutzt. In Abb. 8.1 ist der Laborversuchsaufbau dargestellt, mit dem die Effizienz des Strippvorgangs für unterschiedlichste Wässer und Schadstoffe getestet werden kann.
Zentraler Teil des Aufbaus ist ein Celgard Hohlfasermembranmodul [ ] welches im Detail in Abb. 8.2 dargestellt ist. Dieses Modul enthält tausende mikroporöser Polypropylen Hohlfasern, die miteinender verflochten sind und um einen zentralen Kanal gelegt werden. Der Wasserfluss ergibt sich aus Abb. 8.2. Das gezeiget Modul hat eine Länge von 8” und einen Durchmensser von 2.5”
9
Abb. 8.1: Laborversuchsaufbau zum Membranstrippverfahren. 1 Wasserpumpe, 2 Gasflussregler, 3 Vacuum-pumpe mit Druckregler, 4 Hohlfasermembranmodul, 5 Wassereinlass, 6 Wasserauslass, 7 Strippgaseinlass, 8 Strippgasauslass, 9 Probenahmeports Wasser, Probenahmeport Gas.
46

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
mit einer Membranoberfläche von ca. 1.25 m2. Im Betrieb fließt das kontaminierte Wasser von unten in das Modul [ ] und fließt dann außen (shellside) an den Hohlfasern vorbei zum Auslass [ ]. Stickstoff wird als Strippgas eingesetzt und fließt im Gegenstrom durch die Hohlfasern (lumenside) (Gas ein bei , Gas aus bei ). Durch den Einsatz von Stickstoff als Strippgas soll das Eindringen von Sauerstoff verhindert werden um so eine Minimierung von möglichen Ausfällungen zu erreichen. Es wird außerdem ein Vacuum in den Hohlfasern angelegt, um die Strippeffizienzen zu erhöhen. Sowohl der Sickstofffluss als auch das Vacuum können unabhängig voneinander durch einen Gasflussregler [ ] bzw. eine Vacuumpumpe mit Regler [ ] eingestellt werden. Die Effizienz des Systems kann über Probenahmen der wässrigen Phase [ ] als auch der
Die Abb. 8.3 zeigt exemplarisch das
Gasphase [ ] kontrolliert werden.
Schadstoffspektrum in einer Grundwasserprobe vom Standort einer chemischen Fabrik, mit der die Membranfiltration im Labor mit dem in Abb. 8.2 dargestellten
Abb. 8.2: Celgard Hohlfasermembranmodul im Schnitt. © Celgard Inc..
MW11 Formula MW (g/mol) # Henry (-) * Conc. (mg/l) Vinyl Chloride C2H3Cl 62.5 2.29 n.b. Ethyl Chloride C2H5Cl 64.5 0.35 n.b. 1,1-Dichloroethene C2H2Cl2 96.9 1.10 n.b. Methylene Chloride CH2Cl2 84.9 0.09 n.b. trans-1,2-Dichloroethene C2H2Cl2 96.9 0.40 8.8 1,1-Dichloroethane C2H4Cl2 99.0 0.23 7.5 cis-1,2-Dichloroethene C2H2Cl2 96.9 0.17 7.4 Chloroform CHCl3 99.0 0.15 39.5 1,2-Dichloroethane C2H4Cl2 99.0 0.04 222.3 Carbon Tetrachloride CCl4 153.8 1.20 2.3 Trichloroethene C2HCl3 131.4 0.40 3.2 1,1,2-Trichloroethane C2H3Cl3 133.4 0.04 2.6 Tetrachloroethene C2Cl4 165.8 0.70 0.5 1,1,1,2-Tetrachloroethane C2H2Cl4 167.8 n.a. n.b
Tab. 8.3: Analyse eines Grundwassers vom Standort einer chemischen Fabrik. Konzentrationen für Verbindungenmit geringen Gehalten wurden nicht quantifiziert (n.b.).d
47
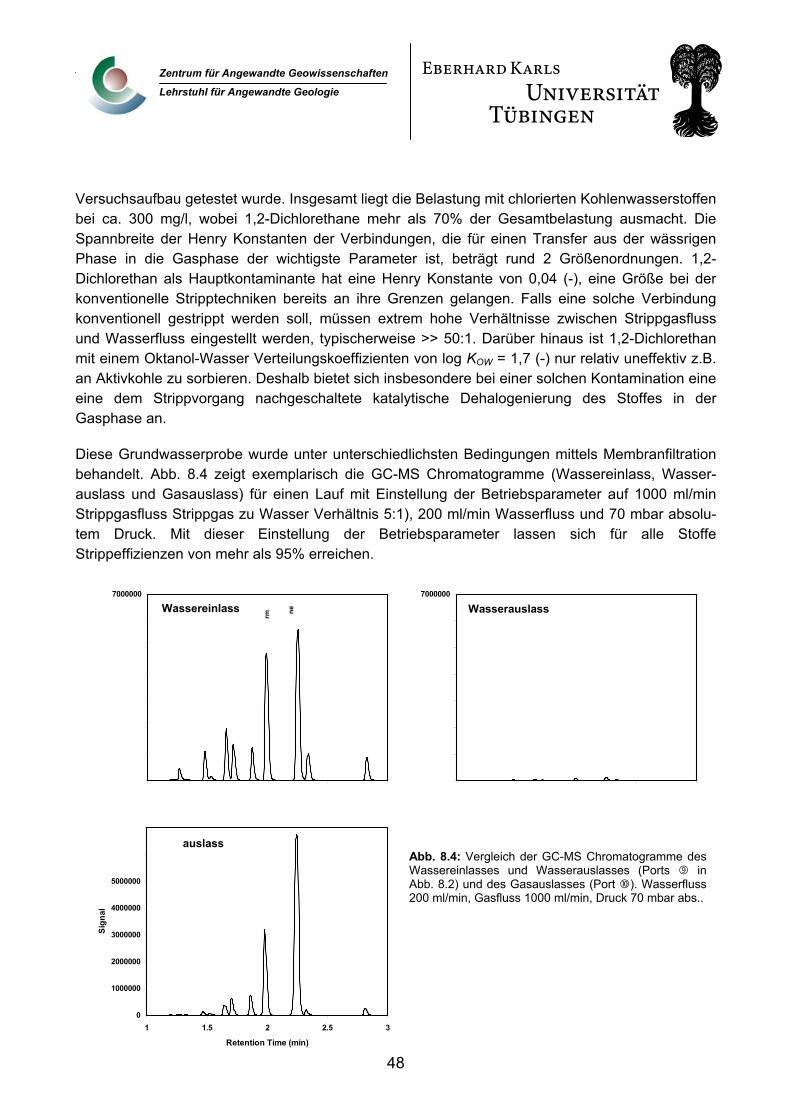
Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Versuchsaufbau getestet wurde. Insgesamt liegt die Belastung mit chlorierten Kohlenwasserstoffen bei ca. 300 mg/l, wobei 1,2-Dichlorethane mehr als 70% der Gesamtbelastung ausmacht. Die
0
1000000
2000000
3000000
4000000
5000000
6000000
1 1.5 2 2.5 3
Retention Time (min)
Sign
al
6000000
7000000
0
1000000
2000000
3000000
4000000
5000000
6000000
1 1.5 2 2.5 3
Retention Time (min)
Sign
al
Viny
l Chl
orid
e
Tric
hlor
oeth
ene
Carb
on T
etra
chlo
ride
1,2-
Dich
loro
etha
Chlo
rofo
cis-
1,2-
Dich
loro
ethe
ne
1,1-
Dich
loro
etha
netra
ns-1
,2-D
ichl
oroe
then
eM
ethy
lene
Chl
orid
e1,
1-Di
chlo
roet
hene
Ethy
l Chl
orid
e
Gas
48
Spannbreite der Henry Konstanten der Verbindungen, die für einen Transfer aus der wässrigen
P die Gasphase der wic ra äg öße 1,2-Dichlorethan als Hauptkontaminante hat eine Henry Konstante von 0,04 (-), eine Größe bei der konventionelle Stripptechniken bereits an ihre Grenzen gelangen. Falls eine solche Verbindung ko t werden so en extrem e Verhä e zwischen pgasflussund Wasserfluss eingestellt werden, typischerweise >> 50:1. Darüber hinaus ist 1,2-Dichlorethan mit einem Oktanol-Wasser Verteilungskoeffizienten von log KOW = 1,7 (-) nur relativ uneffektiv z.B. an Deshalb te sich insbesondere bei einer solchen Kontamination eineeine dem Strippvorgang nachgeschaltete katalytische Dehalogenierung des Stoffes in der G
Diese Grundwasserprobe wurde unter unterschiedlichsten Bedingungen mittels Membranfiltration be igt exempla die GC-M omatogra (Wasserein , Wasser-au uslass) für eine it Einst der Betr arameter au 00 ml/min Strippgasfluss Strippgas zu Wasser Verhältnis 5:1), 200 ml/min Wasserfluss und 70 mbar absolu-te ieser Einstell er Betr ameter n sich fü lle Stoffe Strippeffizienzen von mehr als 95% erreichen.
nrm
Wasserauslass
0
1000000
2000000
3000000
4000000
5000000
1 1.5 2 2.5 3
Retention Time (min)
Sign
al
auslass Abb. 8.4: Vergleich der GC-MS Chromatogramme desWassereinlasses und Wasserauslasses (Ports inAbb. 8.2) und des Gasauslasses (Port ). Wasserfluss200 ml/min, Gasfluss 1000 ml/min, Druck 70 mbar abs..
70000007000000
eWassereinlass
hase in htigste Pa meter ist, betr t rund 2 Gr nordnungen.
nventionell gestripp ll, müss hoh ltniss Strip
Aktivkohle zu sorbieren. bie t
asphase an.
handelt. Abb. 8.4 ze risch S Chr mme lassslass und Gasa n Lauf m ellung iebsp f 10
m Druck. Mit d ung d iebspar lasse r a

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Diese Effizienzen sind vor dem Hintergrund der geringen Henry Konstanten und des geringen Strippgas-Wasser Verhältnisses außerordentlich ermutigend Die anschließende katalytische Ab-reinigung der Gasphase, die von der Arbeitsgruppe von Prof. Kopinke im UFZ eingehend unter-sucht wurde, hat ähnlich gute Ergebnisse erbracht.
katalytische Technologien idealerweise auch k
Für den Einsatz von Membranfiltermodulen in der Grundwassersanierung im Feld liegen unseres Wissens und nach Auskunft der Herstellerfirma (Celgard Inc.) bisher keine Erfahrungen vor. Eine
eine sekundären Schadstoffströme die weiterbe-handelt werden müssen. Vielmehr wurden in diesem Projekt die Risiken dieser Technologie aufge-
ie Untersuchungen einbezogene Standort in Backnang. Hier wurde nach ca. einjährigem Betrieb einer Pilotanlage, in der spezielle Fragestel-
Publikation von Keller und Bierwagen (2001) hat aber das Potential dieser Methode zur Entfernung von MTBE aus kontaminierten Wässern in Laborversuchen untersucht. Deshalb sind hier mög-lichst auch im Gelände Langzeiterfahrungen zu sammeln, um die Stabilität der Stoffübergänge vor dem Hintergrund eines möglichen Fouling (durch anorganische Präzipitate oder als Biofouling durch Biofilme) zu untersuchen. Darüber hinaus ist das Zusammenspiel von Wasserdurchsatz, Un-terdruck und Spülgasfluss sowie eine etwaige Verschaltung mehrerer Module zu untersuchen, um für ein Spektrum von Schadstoffen optimale Strippbedingungen zu erreichen. Als Designkriterium für eine spätere Umsetzung der Technologie in den technischen Maßstab sind hier etwa Stoffflüs-se pro m2 Membranoberfläche zu quantifizieren.
9. Verwertbarkeit der ErgebnissIn dem fortgeschriebenem Verwertungsplan dieses Teilprojekts vom 14.08.2001 wird betont, dass eine erfolgreiche Umsetzung der hier untersuchten Technologie in die Praxis stark standortbezo-gen erfolgen muss. Das hat sich in der Projektlaufzeit bestätigt, wobei am Standort in Bitterfeld diese Technologie nicht ohne weitere Forschungsanstrengungen implementierbar ist.
Die Ergebnisse aus der Pilotanlage und den begleitenden Laborversuchen stellen aber nicht die Katalyse in wässriger Phase als Sanierungsvariante grundsätzlich in Frage. Nach wie vor gilt, dass besonders die schnellen Reaktionsraten und die damit möglichen kompakten Reaktordimensionen kostengünstige Lösungen zulassen sollten. Im Gegensatz zu Konkurrenztechnologien erzeugen
e
zeigt, die für jeden Standort individuell bewertet werden müssen.
Das durchaus standortbezogene Entscheidungen für einen Einsatz dieser Technologie fallen kön-nen belegt der während dieses Projekts auch in d
lungen mit Bezug zur Bitterfeldproblematik untersucht wurden, eine kommerzielle full-scale Anlage mit zeolithgestützten Katalysatoren in wässriger Phase aufgebaut, die von einem Ingenieurbüro betreut wird. Diese Anlage ist zwar nur für relativ geringe Flüsse bis ca. 300 l/h dimensioniert, konnte aber im Vergleich zu klassischen Technologien, etwa Aktivkohlefiltration, die wirtschaftlich bessere Lösung bieten. Damit ist diese Anlage unserer Kenntnis nach weltweit die erste kommer-
49

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
zielle Anlage, die auf einer reduktiven Katalyse in wässriger Phase zur Dehalogenierung chlorierter Schadstoffe beruht.
Potentiell ist auch ein Nutzerkreis dieser Technologie außerhalb der Grundwassersanierung zu sehen, etwa in der Abwasseraufbereitung. Auch hier gilt jedoch, dass die Risiken dieser Technolo-gie im Einzelfall für den jeweiligen Verwendungszweck abgeschätzt werden müssen.
ie Arbeiten in der Endphase von dieses SAFIRA Projekts in Zusammenarbeit mit Prof. Kopinke lassen sich in einem ähnlichem Zusammenhang sehen. Hier wird individuell und direkt abgeleitet
us den Arbeiten in SAFIRA, ein Lösungsweg für eine problematische Grundwasserchemie aufge-t. ls so erfolgversprechend eingeschätzt, dass Patentschrift eingereicht wurde (Kopinke et al, 2001). Eine Pilotanlage basierend auf dieser
Technologie ist vom UFZ aufgebaut worden und an einem Standort in Greppin über mehrere Mo-
ngstechno-logie entwickelt wurde, die zwar nicht universell einsetzbar ist, aber in speziellen Fällen eine wirt-
Die große Mehrzahl der Arbeiten beruhen jedoch auf oxidativen Techniken, obwohl anerkannt
torvergiftung durch Schwefelverbindungen, Hydrodechlorierungen bei Umgebungsbedingungen im wässriger
D
azeig Dieser Lösungsweg wurde von den Beteiligten aeine
nate betrieben worden. Die Ergebnisse lassen eine konkurrenzfähige Sanierungsvariante mit die-ser Technologie an diesem Standort möglich erscheinen.
Der Nutzen dieses SAFIRA Projektes liegt zusammengefasst darin, dass eine Sanieru
schaftliche Alternativoption zu klassischen Ansätzen bieten kann. Eine breite Markterschließung ist deshalb kurzfristig nicht zu erwarten, es ist jedoch gerade in der Endphase von SAFIRA ein neues Potential katalytischer Methoden aufgezeigt worden, die weitere Forschungsanstrengungen in die-ser Richtung lohnenswert machen sollte.
10. Fortschritte anderer Arbeitsgruppen
Ende 2002 erschien in der Zeitschrift Chemosphere ein ausführlicher Review-Artikel mit dem Titel ‚Heterogeneous water phase catalysis as an environmental application’ (Pirkanniemi und Sillan-pää, 2002), in dem die aktuelle Literatur zu diesem Thema ausgewertet wurde. Dabei werden so-wohl oxidative als auch reduktive Ansätze berücksichtigt. Aus dieser Auswertung von 120 Literaturstellen vorwiegend der letzten 5 Jahre wird deutlich, dass das wissenschaftliche Interesse an dieser Thematik stark zugenommen hat.
wird, dass reduktive Katalyse effizienter ist, und dass, im Gegensatz zu oxidativen Techniken, kei-ne Gefahr für eine Bildung von Dioxinen und Furanen besteht. Die Autoren kommen nach Durch-sicht der Literatur zu dem Schluss, dass jedoch auf Grund der Gefahr einer Katalysa
Phase nicht einsetzbar sind. Daraus folgt, dass zumindest in der Fachliteratur keine wesentlichen neuen Erkenntnisse publiziert wurden, die das Problem einer Katalysatorvergiftung effektiv lösen würden.
50

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Es sind nur 5 oder 6 Arbeitsgruppen in diesem Review aufgeführt, die am Thema der reduktiven Katalyse in wässriger Phase arbeiten, darunter die in SAFIRA beteiligten Gruppen Kopinke und Schüth, sowie die Arbeitsgruppe von Martin Reinhard in Stanford. Zwischen den SAFIRA Arbeits-gruppen und der Gruppe von Prof. Reinhard besteht, wie bereits angesprochen, ein intensiver Er-fahrungsaustausch. Aus der Gruppe Reinhard wurden in den letzten Jahren einige Artikel zum Thema veröffentlicht (Lowry und Reinhard 1999, 2000, 2001), die jedoch allesamt Laborbefunde
darstellen. Dazu kommen Artikel von Felis et al. (1999 a,b) Liu et al. (2001), Tundo et al. (2001) und Engelmann et al (2001), wobei Tundo und Engelmann in Isooctan/Wasser- bzw. Ace-ton/Wasser-Mischungen arbeiten.
51

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
11. Publikationen, Vorträge und Patente
ie Ergebnisse dieses Teilprojekts wurden in mehreren Veröffentlichungen in international aner-kannten Zeitschriften publiziert. Dabei sind sowohl Publikationen mit direktem Bezug zu SAFIRA [2,4,5], die zum Teil in Zusammenarbeit mit der Arbeitsgruppe von Prof. Kopinke (Teilprojekt B 3.1) entstanden sind, als auch Publikationen, bei denen Teilaspekte der SAFIRA Ergebnisse in ande-ren Forschungsbereichen eingearbeitet wurden [1,3]. Es ist außerdem noch ein weiteres Manu-skript zur Feldanwendung in Backnang in Bearbeitung, welches in Kürze bei Applied Catalysis B eingereicht werden soll.
[1] Kleineidam, S.; Schüth, C.; Grathwohl, P. (2002): Solubility-Normalized Combined Pore-Filling-Partitioning Sorption Isotherms for Organic Pollutants. Environ. Sci. Technol., 36, 4689-4697.
[2] Kopinke, F.-D.; Köhler, R.; Mackenzie, K.; Borsdorf, H.; Schüth, C. (2002): Katalytische Dechlorierung von Chlorkohlenwasserstoffen aus kontaminierten Grundwässern. Grundwas-ser, 3, 140-145.
[3] Bill, M.; Schüth, C.; Barth, J.A.C.; Kalin, R.M. (2001): Carbon Isotope Fractionation during Abiotic Reductive Dehalogenation of Trichloroethene (TCE). Chemosphere, 44, 5, 1281-1286.
[4] Kopinke, F.-D.; Köhler, R.; Mackenzie, K.; Weiss, H.; Schüth, C. (2000): In-situ-Dechlorierung von CKW im Grundwasser. Wasserwirtschaft Wassertechnik, 7, 58-63.
[5] Schüth, C.; Disser, S.; Schüth, F.; Reinhard, M. (2000): Tailoring Catalysts for Hydrode-chlorinating Chlorinated Hydrocarbon Contaminants in Groundwater. Appl. Cat. B: Environ., 28 (3-4), 147-152.
Außerdem wurden die Ergebnisse dieses Teilprojekts und des Teilprojekt B 3.1 (Prof. Kopinke) in gemeinsamen Tagungsbeiträgen vorgestellt.
[6] Kopinke, F.-D.; Mackenzie, K.; Köhler, R.; Weiß, H.; Grathwohl, P.; Schüth, C. (2002): Groundwater Decontamination by Stripping and Hydrodechlorination in the Gas Phase. 3rd International Conference on Remediation of Chlorinated and Recalcitrant Compounds, Mon-terey, May 2002, in press.
[7] Schüth, C.; Kummer, S.; Köhler, R.; Kopinke, F.-D. (2000): Palladium Catalyst Technolo-gies. Consoil, Leipzig, September 18-22.
Mit den Arbeitsgruppen von Prof. Kopinke und Prof. Grathwohl sowie Herrn Dr. Weiß aus der SA-FIRA Leitung wurde darüberhinaus ein Patent angemeldet, welches weitgehend aus den Erfah-rungen des SAFIRA Projektes resultiert.
[8] Kopinke, F.-D.; Mackenzie, K.; Köhler, R.; Weiss, H.; Schüth, C.; Grathwohl, P. (2001): Verfahren und Vorrichtung zur Dekontamination von Wässern, insbesondere von Grundwäs-sern, die stark und komplex mit organischen Halogenverbindungen (HKW) belastet sind. Pa-tentanmeldung, Deutsches Patentamt P115101DE.
D
52

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
12. Literaturverzeichnis
Arnold, W. A.; Roberts, A. L., (2000): Pathways and kinetics of chlorinated ethylene and chlorin-ated acetylene reaction with Fe(O) particles. Environ. Sci. Technol., 34: 1794-1805.
Balko, E.N., Hoke, J.B.; Gramiccioni, G.A. (1992): United States Patent No. 5,177,268.
Engelmann, M.D.; Doyle, J.G.; Cheng, I.F. (2001): The complete dechlorination of DDT by mag-nesium/palladium bimetallic particles. Chemosphere 43, 2, 195–198.
Felis, V.; De Bellefon, C.; Fouilloux, C.; Schweich, D (1999a): Hydrodechlorination and hy-
iron., 20, (2), 91–100.
modeling issues. Ind. Eng. Chem. Res. , (11), 4213–4219.
FlaniC. Naccache, Eds. Martinius Nijhoff, The Hague, (1985) p. 237.
Gillha
KoveStates Patent, No. 5,196,617.
Kovenklioglu, S.; Cao, Z; Shah, D.; Farrauto, R.J.; Balko, E.N. (1992): Direct catalytic hydro-dechlorination of toxic organics in wastwater. AIChE J., 38, 7, 1003-1012.
Lowry; G.V., Reinhard, M. (2000): echlorination in groundwater: solute ef-fects, biological control, and oxidative catalyst regeneration. Environ. Sci. Technol., 34, 13217–3223.
drodearomatization of monoaromatic chlorophenols into cyclohexanol on Ru/C catalysts ap-plied to water depollution. Influence of the basic solvent and kinetics of the reactions. Appl. Catal. B, Env
Felis, V.; Fouilloux, C.; De Bellefon, C.; Schweich, D (1999b): Three-step catalytic detoxification process of wastewater containing chlorinated aromatic compounds: experimental results and
38
gen, E. M. In: Zeolites: Science and Technology, F.R. Ribeiro, A.E. Rodrigues, L.D. Roll-mann,
Gilham, R.W.; O`Hannesin, S.F. (1994): Enhanced Degradation of Halogenated Aliphatics by Zero-Valent Iron. Ground Water, 32, 958-967.
m, R. W. (1993): Cleaning halogenated compounds from groundwater. US Patent 5,266,213.
Hoke, J.B., Gramiccioni G.A.; Balko E.N. (1992): Catalytic hydrodechlorination of chlorophenols. Appl. Catal. B, Environ. 1, 285-296.
Kopinke, F.-D.; Köhler, R.; Mackenzie, K.; Weiss, H.; Schüth, C. (2000): In-situ-Dechlorierung von CKW im Grundwasser. Wasserwirtschaft Wassertechnik, 7, 58-63.
nklioglu, S. ; Balko, E.N.; Hoke, J.B.; Farrauto, R.J.; Gramiccioni, G.A. (1993): United
Liu, Y.; Yang, F.; Yue, P.L.; Chen, G. (2001): Catalytic dechlorination of chlorophenols in water by palladium/iron. Water Res. 35 (8), 1887–1890.
Lowry; G.V., Reinhard, M. (1999): Hydrodehalogenation of 1- to 3-carbon halogenated organic compounds in water using a palladium catalyst and hydrogen gas. Environ. Sci. Technol. 33, 1905–1910.
Pd-catalyzed TCE d
53

Zentrum für Angewandte Geowissenschaften
Lehrstuhl für Angewandte Geologie
Lowry; G.V., Reinhard, M. (2001): Pd-catalyzed TCE Dechlorination in water: effect of [H2](aq) and H2-utilizing competitive solutes on the TCE dechlorination rate and product distribution.
Matheson, L. J., Tratnyek, P. G. (1994): Reductive dehalogenation of chlorinated methanes by
tive Well: Testing and
Schli
Schreier, C.G., Reinhard, M. (1995): Catalytic hydrodehalogenation of chlorinated ethylenes us-
Schüth, C. (1999): Zeolith-gestützte Katalysatoren – Ergebnisse aus der mobilen Testeinheit. In:
ert.-132
-saturated Water. Appl. Catal. B: Environ. 18,
Schü er, S.; Schüth, F.; Reinhard, M. (2000): Tailoring Catalysts for Hydrode-
hydrogen/palladium catalysts, Water Res. 30, (10), 2315-2322.
Tundo, P.; Perosa, A.; Selva, M.; Zinovyev, S.S. (2001): A mild catalytic detoxification method for PCDDs and PCDFs. Appl. Cat. B: Environ, 32, (1-2), L1–L7.
Weitk e Conference; von Ballmoos, R., Higgins, J. B., Treacy, M. M. J., Eds.; Butterworth-Heinemann, Boston: Mon-
Environ. Sci. Technol., 35, 696–702.
iron metal. Environ. Sci. Technol., 28: 2045-2053.
Mcnab, W. W. Jr., Ruiz, R., Reinhard, M. (2000): In-Situ Destruction of Chlorinated Hydrocarbons in Groundwater Using Catalytic Reductive Dehalogenation in a ReacOperational Experiences. Environ. Sci. Technol., 34: 149-153.
mm, C., Heitz, E. (1996): Development of a wastewater treatment process: reductive deha-logenation of chlorinated hydrocarbons by metals. Environ. Progress, 15, 1, 38-47.
ing palladium and hydrogen for the treatment of contaminated water. Chemosphere, 31, 6, 3475-3487.
2. Statusbericht SAFIRA - Modellstandort, Mobile Testeinheit, Pilotanlage. H. Weiß, B. Daus, G. Teutsch (Hrsg.), UFZ-Bericht 17/99, Fehler! Textmarke nicht defini
Schüth, C., Reinhard, M. (1998): Hydrodechlorination and Hydrogenation of Aromatic Com-pounds over Palladium on Alumina in Hydrogen3-4, 215-221.
th, C.; Disschlorinating Chlorinated Hydrocarbon Contaminants in Groundwater. Appl. Cat. B: Environ., 28 (3-4), 147-152.
Siantar, D.P; Schreier, C.G.; Chou, C.S. Reinhard, M. (1996): Treatment of 1,2-dibromo-3-chloropropane and nitrate-contaminated water with zero-valent iron or
amp, J.; Kleinschmit, P.; Kiss, A.; Berke, C.H In 9th international Zeolit
treal, 1992; Vol. 2, 79-87.
54





























