Embed Size (px)
Citation preview

ICD in O2-Dimeren nach resonanterAuger-Anregung
Bachelorarbeitam Institut für Kernphysik Frankfurt
MiriamWeller
November 2012

Erklärung nach §30 Abs. 11 der Ordnung für den Bachelor- und d en Masterstu-diengang Physik der Johann Wolfgang Goethe-Universität vo m 26.02.2008
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne Benutzunganderer als der angegebenen Quellen und Hilfsmittel verfasst habe. Alle Stellen der Ar-beit, die ich wörtlich oder sinngemäß aus Veröffentlichungen oder aus anderen frem-den Texten entnommen habe, sind von mir als solche kenntlichgemacht worden. Ichhabe die Arbeit nicht, auch nicht auszugsweise, für eine andere Prüfung verwendet.
Frankfurt am Main, den 14.11.2012, Miriam Weller

Inhaltsverzeichnis
1. Einleitung 5
2. Theoretische Grundlagen 72.1. Atome und Moleküle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72.2. Anregungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102.3. Photoionisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102.4. Auger-Effekt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112.5. ICD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.5.1. Vorangegangene Experimente . . . . . . . . . . . . . . . . . . . . 142.5.2. ICD nach resonanter Auger-Anregung . . . . . . . . . . . . . . . 142.5.3. Knock-Off . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.6. RA-ICD in (O2)2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.6.1. Das (O2)2-Molekül . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.6.2. Die Reaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
3. Messung 193.1. Synchrotron . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 193.2. COLTRIMS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3.2.1. Gasjet . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.2.2. Spektrometer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 233.2.3. Detektoren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 253.2.4. Aufnahme der Rohdaten . . . . . . . . . . . . . . . . . . . . . . . 27
4. Analyse 294.1. Datenverarbeitung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.1.1. ROOT und lmf2root . . . . . . . . . . . . . . . . . . . . . . . . . . 294.1.2. Zeiten und Orte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294.1.3. Vorsortieren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.2. Auswertung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334.2.1. Impulse und Energien . . . . . . . . . . . . . . . . . . . . . . . . . 334.2.2. Kalibrierung der Auftrefforte . . . . . . . . . . . . . . . . . . . . . 354.2.3. Energie-Eichung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 354.2.4. Auswählen von Impulsbereichen . . . . . . . . . . . . . . . . . . . 384.2.5. Korrigieren der Impulsverteilungen . . . . . . . . . . . . . . . . . 404.2.6. Eingrenzung der Elektronenflugzeit . . . . . . . . . . . . . . . . . 41
4.3. Fehlerbetrachtung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3

Inhaltsverzeichnis
5. Ergebnisse 43
6. Ausblick 47
A. Atomare Einheiten 49
B. Parameter zum Experiment 51B.1. Spektrometerdaten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51B.2. Daten zum Experiment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Abbildungsverzeichnis 53
Literaturverzeichnis 55
4

1. Einleitung
In der nun etwa einhundertjährigen Geschichte der Atomphysik und damit verbun-den der Quantenmechanik wurden viele bahnbrechende Entdeckungen gemacht. Inrasantem Tempo wurden Einblicke in grundlegende Prozesse der Atomhülle gewon-nen,mit denen verschiedenste technische Geräte entwickelt werden konnten (eines derwichtigsten Beispiele ist hier der Laser). Die Atomphysik ist nicht nur von zentralerBedeutung für viele Forschungsbereiche, zum Beispiel die Laser-, Plasma- und Astro-physik sowie die Meteorologie sondern sie steht auch an der Grenze zwischen Physikund Chemie. Doch immer noch sind viele Fragen zur Dynamik der Atomhülle undder Wechselwirkung von Atomen untereinander, aber auch mit elektromagnetischerStrahlung offen. Selbst mit Hochleistungsrechnern können heutzutage viele quanten-mechanische Rechnungen noch immer nicht durchgeführt werden. Die experimentel-len Methoden zur Analyse von Prozessen auf atomarer Skala werden immer genauer,sodass es mittlerweile sogar möglich ist, sie zeitabhängig zu betrachten (vgl. [Dem10]).
Ein grundlegender Abregungsprozess von Atomen und Molekülen ist der Interato-mare Coulombische Zerfall (ICD). Er wurde 1997 von L. Cederbaum et al. erstmals vorge-schlagen [CZT97]. Obwohl der Prozess in vielen Systemen der bevorzugte Zerfallska-nal ist, blieb er über lange Zeit unentdeckt, da aufgrund der besseren experimentellenKontrollierbarkeit vor allem isolierte Atome untersucht wurden. ICD tritt allerdingsin Molekülen oder in Clustern, die durch Van-der-Waals- und Wasserstoffbrückenbin-dungen interagieren, auf. Bei dieser Zerfallsart kann die Energie, die bei der Abregungeines Atoms oder Moleküls frei wird, strahlungslos an ein benachbartes Teilchen über-tragen werden und dieses ionisieren.Vor etwa 10 Jahren wurde der ICD erstmals an Ne2-Molekülen experimentell beob-
achtet [MKHM03, JCS+04] und seitdem an verschiedenen anderen atomaren und mo-lekularen Systemen nachgewiesen [UFL+08, JSH+10, HJK+10, TSK+12]. Diesem Zer-fallsprozess können verschiedene Anregungsprozesse vorangehen. 2012 wurde die re-sonante Auger Anregung theoretisch vorgeschlagen [GKKC12]. Diese bietet die Mög-lichkeit, den angeregten Zustand, der durch ICD zerfällt, genau zu kontrollieren.
In der vorliegenden Arbeit wurde der Intermolekulare Coulombische Zerfall in (O2)2-Molekülen untersucht. Eintreffende Photonen einer Energie von 539,4 eV führen zu-nächst zu einer Innerschalenanregung eines O2-Moleküls: 1s → 3σu. Anschließendfindet der Auger-Zerfall statt. Danach befindet sich das Molekül noch immer in einemangeregten Zustand und ist außerdem einfach ionisiert. Die verbleibende Anregungs-energie reicht nun nicht aus, um das Doppelionisationspotential des O2-Moleküls zuüberwinden. Sie wird auf ein weiteres O2-Molekül, das sich in Van-der-Waals-Reich-weite befindet, übertragen. Dieses wird ebenfalls ionisiert, die Reaktionsprodukte sind
5

1. Einleitung
das primäre schnelle Auger-Elektron, das langsame ICD-Elektron und zwei O+2 -Mole-
külionen. Aufgrund der Coulomb-Kraft stoßen sie sich gegenseitig ab und fliegen inentgegengesetzten Richtungen auseinander ("Coulomb-Explosion"):
hν + (O2)2Anregung−−−−−→ (O2)
∗
2
Auger−−−−→Zerfall
(O2)+ ∗
2 + e−Auger
ICD−−→ (O2)++2 + e−Auger + e−ICD
Coulomb−−−−−→Explosion
O+2 +O+
2 + e−Auger + e−ICD
Das Experiment wurde mit der COLd Target Recoil Ion Momentum Spectroscopy-Technik (COLTRIMS) am berliner Elektronensynchrotron BESSY II durchgeführt. Da-beiwird ein kalter Sauerstoff-Gasjet mit einemRöntgenphotonenstrahl gekreuzt. Nach-dem die Reaktion stattgefunden hat, werden die Recoil-Ionen und das ICD-Elektrondurch ein elektrisches Feld zu den Detektoren geführt und registriert. Anschließendwerden die aufgenommenen Daten digital verarbeitet und analysiert.
6

2. Theoretische Grundlagen
2.1. Atome und Moleküle
n=1
n=2
Abbildung 2.1.: Sauerstoff imBohrschen Atommodell
Nach dem Bohr-Sommerfeldschen Atommodell [Boh13]besteht ein Atom aus dem Atomkern, der aus Protonenund Neutronen gebildet wird, und der Atomhülle, in dersich die Elektronen auf elliptischen Bahnen bewegen. Soherrscht ein Gleichgewicht zwischen der elektrostatischenAnziehungskraft zwischen Elektronen und Kern und derZentripetalkraft, die aus der Bewegung der Elektronen aufder gekrümmten Bahn resultiert. Diese Elektronenbahnenwerden auch als Energieniveaus bezeichnet.Würde man dies klassisch erklären wollen, müsste den
Elektronen, da sie auf der Kreisbahn um den Kern eine be-schleunigte Bewegung ausführen, ständig Energie zugeführt werden. Diese Energiewürden die Elektronen dann nach der klassischen Elektrodynamik wieder in Formvon Strahlung emittieren. Eine solche Strahlungwird allerdings nicht beobachtet. Klas-sisch betrachtet wäre für ein Elektron der Masse me in der Atomhülle außerdem jedekinetische Energie und damit auch jeder Kernabstand r möglich.Betrachtet man nun aber beispielsweise die Absorptionsspektren des Wasserstoffs
(Abbildung 2.2), so sind dort diskrete Linien zu erkennen. Da diese Beobachtungennicht mit dem klassischen Modell übereinstimmten, wurden in der Quantenmechanikdiskrete Energieniveaus für die Elektronen eingeführt (vgl. [Dem10], S. 106ff). DamitNiels Bohr erklären konnte, dass die Elektronen auf ihren Bahnen den Kern stabil um-laufen, musste er zwei Postulate aufstellen (vgl. [Mes06], S. 732ff; [Boh13], § 2f):
Übergänge nur zwischen bestimmten Energieniveaus hνij = ∆Eij = Ei − Ej
Erlaubte Energieniveaus 2 · π · rn = n · λ = n · hmeve
Hier steht ν für die Frequenz und λ für die Wellenlänge eines absorbierten oderemittierten Photons.Die Energieniveaus Ei werden mit Hilfe von Quantenzahlen beschrieben. Mit der
Zeit verbesserten sich die experimentellen Möglichkeiten und immer feinere Aufspal-tungen wurden in den elektronischen Energieniveaus gefunden1. Je nachdem, wie sehrdas Modell zur Erklärung dieser Befunde erweitert wurde, wurden weitere Quanten-zahlen eingeführt, um die möglichen elektronischen Zustände genauer zu spezifizie-ren.1Feinstrukturaufspaltung, Lamb-Shift, Zeeman-Effekt, Hyperfeinstruktur
7

2. Theoretische Grundlagen
Abbildung 2.2.: Absorptionsspektrum des Wasserstoffs: Es lassen sich diskrete, scharfe schwarze Lini-en erkennen, die den Übergängen zwischen den Energieniveaus in der Atomhülle desWasserstoffs entsprechen (Quelle: pst-spectramit Daten von cdsweb.u-strasbg.fr).
Hauptquantenzahl Im ursprünglichen Modell von Niels Bohr reichte eine Quanten-zahl aus, um die Energieniveaus, aus denen sich die damals beobachtbaren Spek-tren des Wasserstoffs ergeben, zu beschreiben: Die Hauptquantenzahl n = 1, 2, . . .(oder auch n = K,L,M, · · · ).
Drehimpulsquantenzahl Arnold Sommerfeld führte eine weitere Quantenzahll = 0, · · · , l, · · · , n− 1 (oder auch l = s, p, d, f, g, h, · · · ) ein, um die Elliptizität derElektronenbahn zu einer bestimmten Hauptquantenzahl zu beschreiben. Heu-te wird l als Drehimpulsquantenzahl bezeichnet und beschreibt als Eigenwert desDrehimpulsoperators den Bahndrehimpuls des Elektrons.
Magnetische Quantenzahl Die magnetische Quantenzahl m = −l, · · · , m, · · · , l drücktdie räumliche Orientierung des Bahndrehimpulses des Elektrons aus.
Spinquantenzahl Der Eigendrehimpuls des Elektrons wird durch die Spinquantenzahls = ±1 dargestellt.
In aktuellen Modellen zur Beschreibung der Atome werden die elektrostatischenKräfte im und um das Atom oft durch Coulomb-ähnliche Potentialkurven dargestellt.Auch die Elektronenbewegung wird nicht mehr als feste Ellipse angenommen, viel-mehr werden im Orbitalmodell Aufenthaltswahrscheinlichkeiten für die Elektronenberechnet, aus denen sich unterschiedlich geformte Orbitale für Elektronen in ver-schiedenen Energiezuständen ergeben. Doch auch diese Orbitale werden noch im-mer durch Quantenzahlen kategorisiert. Die Aufenthaltswahrscheinlichkeiten werdenmathematisch mit orts- und zeitabhängigen Wellenfunktionen beschrieben, deren Be-tragsquadrat die Wahrscheinlichkeit angibt, das Elektron zu einem Zeitpunkt in einembestimmten Raumvolumen zu finden. Diese Wellenfunktionen ψ folgen aus der Schrö-dingergleichung, die die Grundlage der theoretischen Beschreibung der Quantenme-chanik bildet (vgl. [Dem10], S. 124ff, S. 155). Die zeitunabhängige, stationäre Schrödin-gergleichung für ein freies Teilchen der Massem lautet:
− h̄2
2m∆ψ(~r, t) = ih̄
∂ψ(~r, t)
∂t(2.1)
Hier werden mit ~r die Raumkoordinaten angegeben, in denen das Teilchen zum Zeit-punkt t zu finden ist. Für wasserstoffähnliche Atome2 haben die Wellenfunktionen(Orbitale) ψ in Polarkooridinaten r, ϑ, und ϕ die Form:
ψn,l,m(r, ϑ, ϕ) = Rn,l(r) · Y ml (ϑ, ϕ) (2.2)
2Ein Elektron, Coulomb-Potential.
8

2.1. Atome und Moleküle
x
y
z
1s: n=1, l=0,m=0
x
y
z
2px: n=2, l=1,m=±1
y
x
z
3dz2 : n=3, l=2,m=0
x
y
z
3dxz : n=3, l=2,m=±1
x
y
z
4fxyz : n=4, l=3,m=±2
Abbildung 2.3.: Beispiele einiger Orbitale in wasserstoffähnlichen Atomen
Sie hängen von den Laguerre-PolynomenRn,l(r) und von den KugelflächenfunktionenY ml (ϑ, ϕ) ab. Diese sind wiederum abhängig von den Quantenzahlen n, l undm. Setzt
man diese Funktionen ein, so ergibt sich für die Wellenfunktion des 1s-Orbitals:
ψ1,0,0(r, ϑ, ϕ) =1√π
(
Z
a0
)3
2
e−Z r
a0 (2.3)
Hierbei bezeichnet Z die Kernladung und a0 den Bohrschen Radius. Dieses 1s-Orbitalund vier weitere der ersten Orbitale sind in Abbildung 2.3 graphisch dargestellt.
Moleküle
Mit dem Begriff Molekül bezeichnet man mehrere Atome, die durch verschiedenechemische Bindungen zusammengehalten werden. Diese Bindungen kommen durchÜberlappen der Wellenfunktionen der Elektronen der beteiligten Atome zustande. Esgibt zwei Ansätze, diesenÜberlappmathematisch darzustellen: Zum Einen die (LC)AO-Methode3: hier werden die Orbitale der einzelnen im Molekül enthaltenen Atomedurch lineare Kombination zu Molekülorbitalen erweitert. Dem entgegen versuchtman bei der MO-Methode4, die Gesamt-Elektronenverteilungen bezogen auf alle imMolekül enthaltenen Kerne zu errechnen.In den Molekülen werden die Orbitale nach ihrem Drehimpuls und damit der Sym-
metrie ihrer Wellenfunktionen ψ eingeordnet: So gibt es in Molekülen σ-Orbitale (Dre-himpuls l = 0), die rotationssymmetrische Bindungen zwischen atomaren s- und/oderp-Orbitalen beschreiben und π-Orbitale (Drehimpuls l = 1), die eine nicht rotations-symmetrische Bindung darstellen. Handelt es sich um Atome mit vielen Elektronen,ergeben sich für ein Molekül auch Bindungen höheren Drehimpulses, zum Beispiel δ-und ϕ-Orbitale. Diese hohen Drehimpulse können durch Bindungen zwischen atoma-ren d-, f - und höheren Orbitalen zu Stande kommen (vgl. [Mes06], S. 876ff).Für besondere Atom- und Molekülanordnungen werden spezielle Bezeichnungen
verwendet: Im Folgenden wird mit dem Begriff Dimer ein Molekül, das aus zwei glei-chenUntereinheiten, denMonomeren besteht, bezeichnet. Sammeln sichmehr undmehr
3„(linear) combination of atomic orbitals“4„molecular orbitals“
9

2. Theoretische Grundlagen
Atome zu einem sehr großen Molekül an, so spricht man von einem Cluster. Die Clus-tergröße wird allerdings auch als allgemeiner Begriff verwendet, um die Anzahl derKonstituenten in einemMolekülverbund anzugeben.
2.2. Anregungen
Wird einem Atom Energie, zum Beispiel in Form eines Photons, zugeführt, kann esdiese aufnehmen, indem ein Elektron auf ein höheres Energieniveau angehoben wird(vgl. Bohrsche Postulate). Man spricht hier von Anregung, da sich das Atom danach ineinem höheren Energiezustand befindet. Die Anregung ist am effektivsten, wenn daseinfallende Photon genau die Energiedifferenz zwischen zwei elektronischen Zustän-den trägt. Dadurch ergibt sich die Möglichkeit, durch Variieren der Photonenenergiebestimmte Anregungszustände gezielt auszuwählen.DaMoleküle ausmehreren Atomen bestehen, die durch eine chemische Bindung zu-
sammengehalten werden, gibt es innerhalb eines Moleküls mehr Freiheitsgrade als ineinem Atom. Das Molekül kann zum Beispiel um die Bindung verdreht und bezüglichderen Länge gestreckt und gestaucht werden. Im Gegensatz zu einem Atom kann alsoein Molekül nicht nur elektronisch angeregt werden, vielmehr stehen auch Rotations-undVibrationsmoden zur Verfügung, umAnregungsenergie aufzunehmen. Daraus er-geben sich viele Energieniveaus, die besetzt werden können und im entsprechendenEnergiespektrum auftauchen. Diese Rotations- und Vibrationszustände können für be-stimmte Moleküle berechnet und auch im Energiespektrum unterschieden werden.
2.3. Photoionisation
n=1
n=2
Abbildung 2.4.: Photoionisation:Ein Photon löst ein Elektronaus der Atomhülle heraus.
Jedes Energieniveau, das ein Elektron in der Atomhül-le einnehmen kann, entspricht einer bestimmten Bin-dungsenergie zwischen Elektron und Atomkern. Legtman das Bohrsche Atommodell zu Grunde, kann mansich vorstellen, dass das Elektron, je weiter es vomAtomkern entfernt ist, weniger elektrostatische Anzie-hung durch die positiven Ladungen dort erfährt. Die„äußersten“ Elektronen sind also am schwächsten anden Atomkern gebunden. Sie werden auch Valenzelek-tronen5 genannt, da sie für die chemischen Eigenschaf-ten des Atoms verantwortlich sind.Wird dem Elektron nun mindestens diese Bindungs-
energie, zum Beispiel in Form eines Photons, zugeführt, kann es aus dem Atom her-ausgelöst werden. Das freie Elektron hat dann eine Geschwindigkeit, die der übrigenkinetischen Energie entspricht:
Ekin,e = h · ν − EB (2.4)
5Von lat. valere = vermögen
10
2.4. Auger-Effekt
Beträgt die Photonenenergie hν genau den Wert der Bindungsenergie EB , so erhältman ein freies Elektron, das keine kinetische Energie und damit auch keinen Impulsträgt. Dieser sogenannte Photoeffekt wurde 1905 von Albert Einstein für Oberflächenhergeleitet ([Ein05], § 8) und später von Niels Bohr in seinem Modell der Photoionisa-tion auf einzelne Atome übertragen ([Boh13], § 4).
10-1 100
Wah
rsch
einlichk
eit
Photoa
bsorption
Photonenenergie [keV]
Absorptionskanten
M5
M4
M3
M2
M1
L3 L2 L1
K
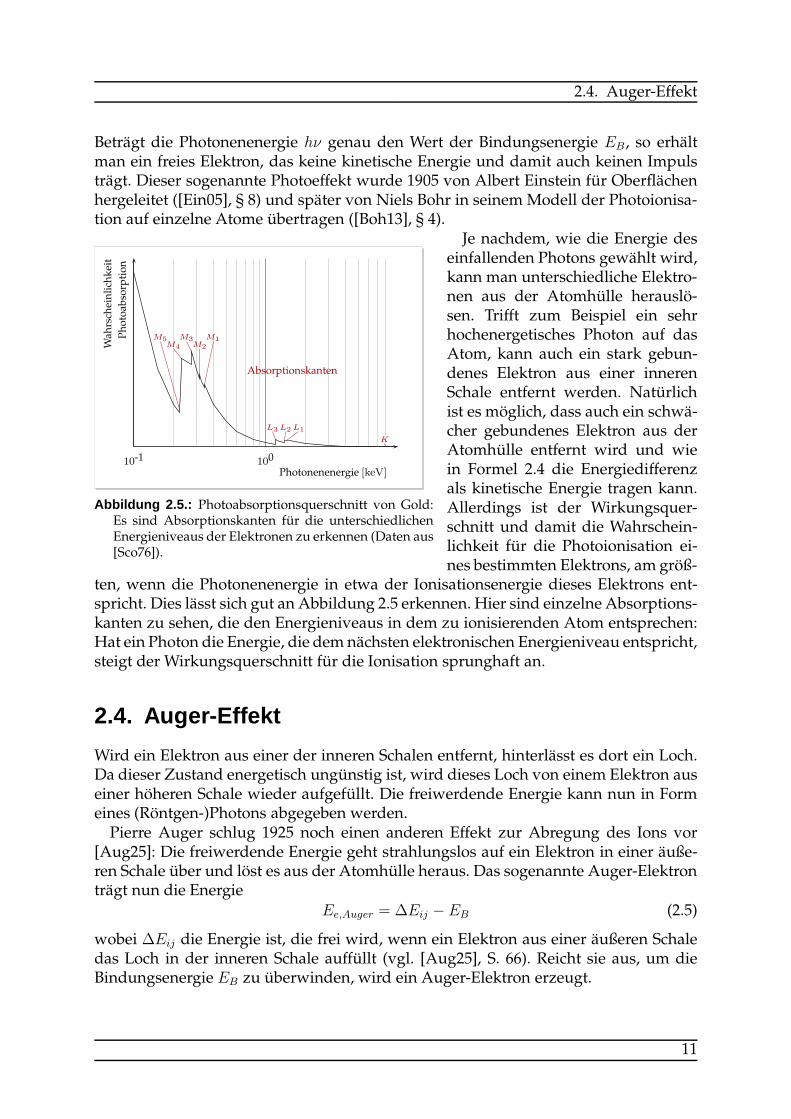
Abbildung 2.5.: Photoabsorptionsquerschnitt von Gold:Es sind Absorptionskanten für die unterschiedlichenEnergieniveaus der Elektronen zu erkennen (Daten aus[Sco76]).
Je nachdem, wie die Energie deseinfallenden Photons gewählt wird,kann man unterschiedliche Elektro-nen aus der Atomhülle herauslö-sen. Trifft zum Beispiel ein sehrhochenergetisches Photon auf dasAtom, kann auch ein stark gebun-denes Elektron aus einer innerenSchale entfernt werden. Natürlichist es möglich, dass auch ein schwä-cher gebundenes Elektron aus derAtomhülle entfernt wird und wiein Formel 2.4 die Energiedifferenzals kinetische Energie tragen kann.Allerdings ist der Wirkungsquer-schnitt und damit die Wahrschein-lichkeit für die Photoionisation ei-nes bestimmten Elektrons, am größ-
ten, wenn die Photonenenergie in etwa der Ionisationsenergie dieses Elektrons ent-spricht. Dies lässt sich gut an Abbildung 2.5 erkennen. Hier sind einzelne Absorptions-kanten zu sehen, die den Energieniveaus in dem zu ionisierenden Atom entsprechen:Hat ein Photon die Energie, die demnächsten elektronischen Energieniveau entspricht,steigt der Wirkungsquerschnitt für die Ionisation sprunghaft an.
2.4. Auger-Effekt
Wird ein Elektron aus einer der inneren Schalen entfernt, hinterlässt es dort ein Loch.Da dieser Zustand energetisch ungünstig ist, wird dieses Loch von einem Elektron auseiner höheren Schale wieder aufgefüllt. Die freiwerdende Energie kann nun in Formeines (Röntgen-)Photons abgegeben werden.Pierre Auger schlug 1925 noch einen anderen Effekt zur Abregung des Ions vor
[Aug25]: Die freiwerdende Energie geht strahlungslos auf ein Elektron in einer äuße-ren Schale über und löst es aus der Atomhülle heraus. Das sogenannte Auger-Elektronträgt nun die Energie
Ee,Auger = ∆Eij − EB (2.5)
wobei ∆Eij die Energie ist, die frei wird, wenn ein Elektron aus einer äußeren Schaledas Loch in der inneren Schale auffüllt (vgl. [Aug25], S. 66). Reicht sie aus, um dieBindungsenergie EB zu überwinden, wird ein Auger-Elektron erzeugt.
11

2. Theoretische Grundlagen
n=1
n=2
a)
n=1
n=2
b)
Abbildung 2.6.: Auger-Effekt: Nachdem ein Elektron aus einerinneren Schale entfernt wurde (a), wird das Loch wiederaufgefüllt. Die freiwerdende Energie reicht aus, um dasAtom doppelt zu ionisieren (b).
DerAuger-Effekt kann nichtnur nach einer Ionisation statt-finden, sondern auch, wennein Elektron aus einer inne-ren Schale auf ein hohes Ener-gieniveau angeregt wird, kannauf diese Weise die Abregungerfolgen6. Füllt nicht das Elek-tron auf dem höchsten besetz-ten Energieniveau, sondern ei-nes aus einem mittleren Ni-veau das kernnahe Loch auf,so verbleibt das Atom in ei-nem angeregten Zustand. Dieser kann nun wiederum durch den Auger-Effekt zer-fallen. So können sich sogenannte Auger-Kaskaden bilden, wenn mehrere Elektronennacheinander in niedrigere Energiezustände übergehen.
Auch in angeregtenMolekülen steht der Auger-Zerfallskanal offen. Ein Loch in einerkernnahen Schale kann dann von einem Elektron aus einem Molekülorbital aufgefülltwerden.
2.5. ICD
Interatomic Coulombic Decay, kurz ICD, ist ein grundlegender Zerfallsprozess fürschwach gebundene, also durch Van-der-Waals- oder Wasserstoffbrückenbindungenzusammengehaltene Moleküle und Cluster, der im Jahr 1997 theoretisch vorhergesagtwurde [CZT97]. Der experimentelle Nachweis erfolgte einige Jahre später zunächst fürNe-Dimere [JCS+04, MKHM03] und später für eine Vielzahl von anderen Systemen.
Der Prozess läuft wie folgt ab: Nachdem ein Atom elektronisch angeregt wurde,kann die Abregung auch stattfinden, indem die Anregungsenergie in Form eines vir-tuellen Photons, also strahlungslos, auf ein benachbartes Atom übertragen wird unddieses ionisiert. Wie wahrscheinlich diese Form der Abregung ist, hängt unter ande-rem davon ab, welche anderen Zerfallskanäle zur Verfügung stehen. Ist das Atom nurangeregt, steht auch der Auger-Zerfallskanal als Konkurrenzprozess zur Verfügung.Wurde das erste Atom jedoch vorher schon ionisiert, ist es möglich, dass die Anre-gungsenergie nicht für die zweite Ionisation ausreicht, aber doch groß genug ist, umdas benachbarte Atom einfach zu ionisieren. In solchen Systemen ist der interatoma-re Coulombische Zerfall (ICD) der bevorzugte Zerfallskanal. Dieser Prozess findet aufeiner sehr kurzen Zeitskala von wenigen Femtosekunden statt [KC07], da die Energienur übertragen werden kann, solange die Atome oder Moleküle, zwischen denen dieEnergie übertragen wird, noch nicht dissoziiert sind. Ihm können verschiedene Anre-gungsprozesse vorangehen:
6Man nennt diesen Effekt bei resonanter Anregung auch Resonanter Auger-Effekt.
12

2.5. ICD
a)
n=1
n=2
A+
n=1
n=2
B
A+ −B
b)
n=1
n=2
A+
n=1
n=2
B+
A+ −B+
Abbildung 2.7.: ICD: Durch Photoionisation ensteht ein einfach geladenes, angeregtes Ion (a). Die An-regungsenergie wird an ein in der Nähe befindliches Atom übertragen und löst auchaus dessen Hülle ein Elektron heraus (b).
ICD nach Photoionisation Ein Elektron aus einer mittleren Schale wird entfernt. DieEnergie, die beim Auffüllen des resultierenden Lochs frei wird, reicht nicht füreine weitere Ionisation aus und wird daher auf ein benachbartes Atom oder Mo-lekül übertragen und ionisiert dieses.
Resonanter ICD (RICD) Ein Atom oderMolekül wird resonant elektronisch angeregt.Die freie Stelle in der ursprünglichen Schale des angeregten Elektrons wird voneinem weiteren Elektron aufgefüllt. Nun kann einerseits eine Autoionisation desangeregten Zustandes stattfinden, andererseits wurde experimentell [BJM+05]und theoretisch [KGCA09] bestätigt, dass die freiwerdende Energie auch aufein benachbartes System übertragen werden und dieses ionisieren kann. Nimmthier das ursprünglich angeregte Elektron direkt am ICD teil, spricht man von p-RICD7, von den beiden Molekülfragmenten ist anschließend eines ionisiert undeines imGrundzustand. Füllt ein anderes Elektron das Loch in der inneren Schalewieder auf, bezeichnet man den Vorgang als s-RICD8. Hier sind nach dem Zerfallein angeregtes und ein ionisiertes Fragment vorzufinden.
Auger-ICD Nach dem Entfernen eines inneren Elektrons findet Auger-Zerfall in demAtom oder Molekül statt. Reicht die freiwerdende Energie nach einem elektro-nischen Übergang an einem Punkt der Auger-Kaskade nicht mehr aus, um dasAtom oder Molekül weiter zu ionisieren, findet ICD statt und ein benachbartesAtom oder Molekül wird ionisiert [MLS+06].
Resonanter Auger-ICD Ähnlich wie beim Auger-ICD, allerdings startet der Auger-Zerfall nicht nach einer Ionisation, sondern nach einer resonanten Anregung (sie-he 2.5.2 und Abbildung 2.9).
7Von engl. to participate = teilnehmen8Von engl. to spectate = zuschauen
13
2. Theoretische Grundlagen
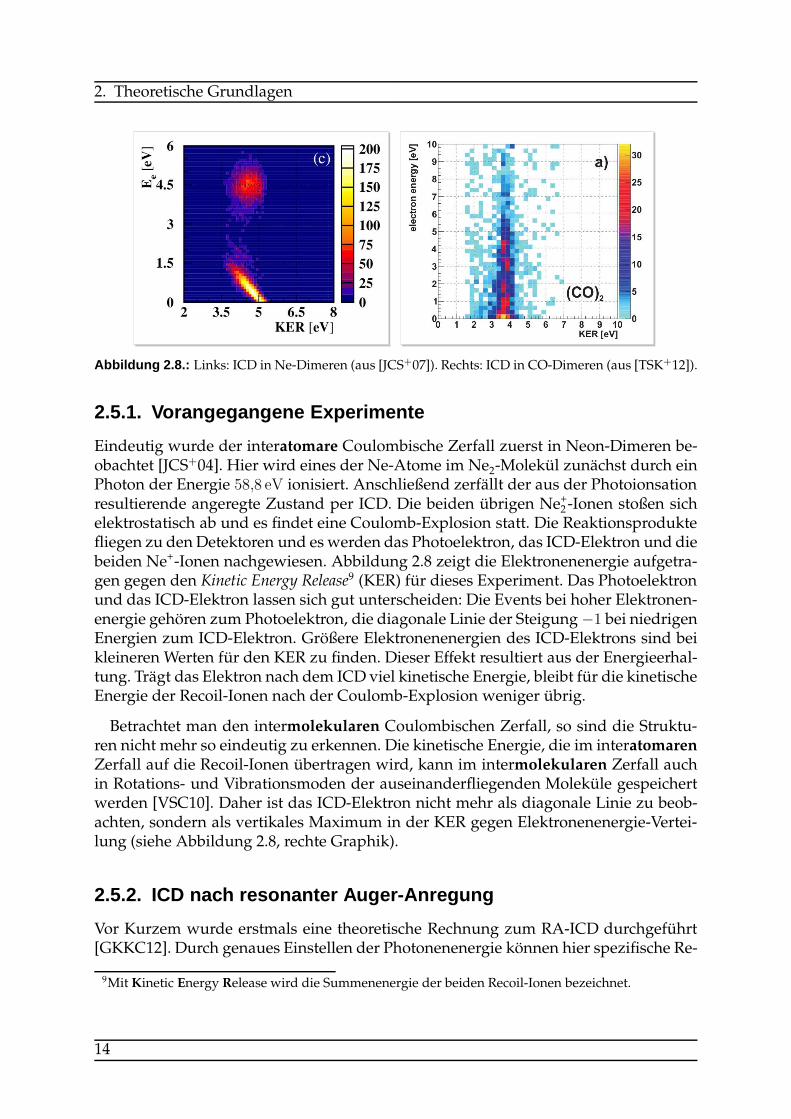
Abbildung 2.8.: Links: ICD in Ne-Dimeren (aus [JCS+07]). Rechts: ICD in CO-Dimeren (aus [TSK+12]).
2.5.1. Vorangegangene Experimente
Eindeutig wurde der interatomare Coulombische Zerfall zuerst in Neon-Dimeren be-obachtet [JCS+04]. Hier wird eines der Ne-Atome im Ne2-Molekül zunächst durch einPhoton der Energie 58,8 eV ionisiert. Anschließend zerfällt der aus der Photoionsationresultierende angeregte Zustand per ICD. Die beiden übrigen Ne+2 -Ionen stoßen sichelektrostatisch ab und es findet eine Coulomb-Explosion statt. Die Reaktionsproduktefliegen zu den Detektoren und es werden das Photoelektron, das ICD-Elektron und diebeiden Ne+-Ionen nachgewiesen. Abbildung 2.8 zeigt die Elektronenenergie aufgetra-gen gegen den Kinetic Energy Release9 (KER) für dieses Experiment. Das Photoelektronund das ICD-Elektron lassen sich gut unterscheiden: Die Events bei hoher Elektronen-energie gehören zum Photoelektron, die diagonale Linie der Steigung−1 bei niedrigenEnergien zum ICD-Elektron. Größere Elektronenenergien des ICD-Elektrons sind beikleineren Werten für den KER zu finden. Dieser Effekt resultiert aus der Energieerhal-tung. Trägt das Elektron nach dem ICD viel kinetische Energie, bleibt für die kinetischeEnergie der Recoil-Ionen nach der Coulomb-Explosion weniger übrig.
Betrachtet man den intermolekularen Coulombischen Zerfall, so sind die Struktu-ren nicht mehr so eindeutig zu erkennen. Die kinetische Energie, die im interatomarenZerfall auf die Recoil-Ionen übertragen wird, kann im intermolekularen Zerfall auchin Rotations- und Vibrationsmoden der auseinanderfliegenden Moleküle gespeichertwerden [VSC10]. Daher ist das ICD-Elektron nicht mehr als diagonale Linie zu beob-achten, sondern als vertikales Maximum in der KER gegen Elektronenenergie-Vertei-lung (siehe Abbildung 2.8, rechte Graphik).
2.5.2. ICD nach resonanter Auger-Anregung
Vor Kurzem wurde erstmals eine theoretische Rechnung zum RA-ICD durchgeführt[GKKC12]. Durch genaues Einstellen der Photonenenergie können hier spezifische Re-
9Mit Kinetic Energy Release wird die Summenenergie der beiden Recoil-Ionen bezeichnet.
14

2.5. ICD
a)
A∗ B
A∗ −B
b)
A+∗ B
A+∗ −Bc)
A+ B+
A+ −B+
d)
A+ B+
Abbildung 2.9.: RA-ICD: Mit einem Photon wird ein Atom angeregt (a). Es findet Auger-Zerfall in demAtom statt: Ein Elektron wird aus der Hülle entfernt und es bleibt ein einfach gela-denes, angeregtes Ion zurück (b). Die Anregungsenergie wird an ein in der Nähe be-findliches Atom übertragen und löst auch aus dessen Hülle ein Elektron heraus (c).Aufgrund der elektrostatischen Kräfte zwischen den beiden einfach geladenen Ionenfindet eine Coulomb-Explosion statt (d).
15
2. Theoretische Grundlagen
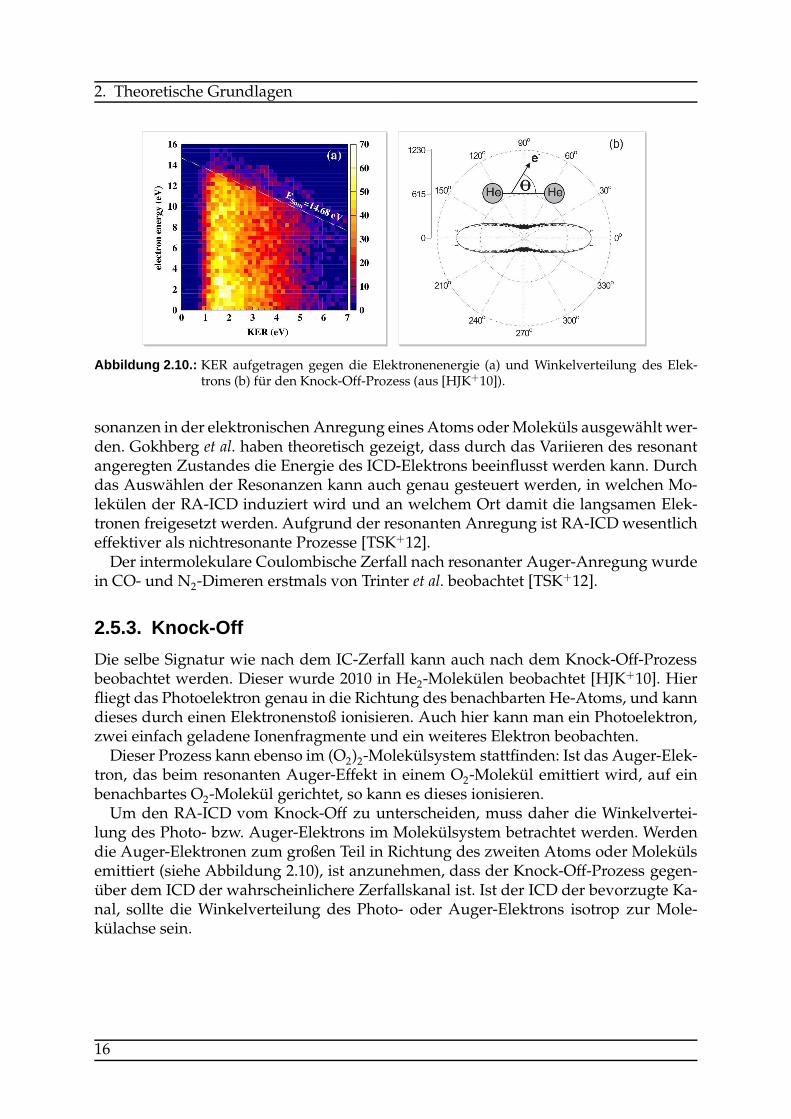
Abbildung 2.10.: KER aufgetragen gegen die Elektronenenergie (a) und Winkelverteilung des Elek-trons (b) für den Knock-Off-Prozess (aus [HJK+10]).
sonanzen in der elektronischen Anregung eines Atoms oderMoleküls ausgewählt wer-den. Gokhberg et al. haben theoretisch gezeigt, dass durch das Variieren des resonantangeregten Zustandes die Energie des ICD-Elektrons beeinflusst werden kann. Durchdas Auswählen der Resonanzen kann auch genau gesteuert werden, in welchen Mo-lekülen der RA-ICD induziert wird und an welchem Ort damit die langsamen Elek-tronen freigesetzt werden. Aufgrund der resonanten Anregung ist RA-ICD wesentlicheffektiver als nichtresonante Prozesse [TSK+12].Der intermolekulare Coulombische Zerfall nach resonanter Auger-Anregung wurde
in CO- und N2-Dimeren erstmals von Trinter et al. beobachtet [TSK+12].
2.5.3. Knock-Off
Die selbe Signatur wie nach dem IC-Zerfall kann auch nach dem Knock-Off-Prozessbeobachtet werden. Dieser wurde 2010 in He2-Molekülen beobachtet [HJK+10]. Hierfliegt das Photoelektron genau in die Richtung des benachbarten He-Atoms, und kanndieses durch einen Elektronenstoß ionisieren. Auch hier kann man ein Photoelektron,zwei einfach geladene Ionenfragmente und ein weiteres Elektron beobachten.Dieser Prozess kann ebenso im (O2)2-Molekülsystem stattfinden: Ist das Auger-Elek-
tron, das beim resonanten Auger-Effekt in einem O2-Molekül emittiert wird, auf einbenachbartes O2-Molekül gerichtet, so kann es dieses ionisieren.Um den RA-ICD vom Knock-Off zu unterscheiden, muss daher die Winkelvertei-
lung des Photo- bzw. Auger-Elektrons im Molekülsystem betrachtet werden. Werdendie Auger-Elektronen zum großen Teil in Richtung des zweiten Atoms oder Molekülsemittiert (siehe Abbildung 2.10), ist anzunehmen, dass der Knock-Off-Prozess gegen-über dem ICD der wahrscheinlichere Zerfallskanal ist. Ist der ICD der bevorzugte Ka-nal, sollte die Winkelverteilung des Photo- oder Auger-Elektrons isotrop zur Mole-külachse sein.
16

2.6. RA-ICD in (O2)2
2.6. ICD nach resonanter Auger-Anregung in (O 2)2
2.6.1. Das (O2)2-Molekül
H L
TX
Abbildung 2.11.: Die verschiedenen möglichen Ori-entierungen der beiden O2-Moleküle im (O2)2[AAB+99]: Mit unterschiedlichen Orientierungenvariiert die Bindungslänge.
Das (O2)2-Molekül besteht aus zweiO2-Molekülen, die durch Van-der-Waals-Kräfte interagieren [AAB+99].Die sogenannte Van-der-Waals-Bin-dung zwischen den O2-Molekülenist keine Molekülbindung im zu-vor genannten Sinne (siehe Kapi-tel 2.1), da hier die Wellenfunktio-nen der Elektronen der beiden ein-zelnen O2-Moleküle nicht überlap-pen. Die Elektronen sind weiterhinstark an den O2-Molekülen lokali-siert. Da sie allerdings in den Atom-hüllen beweglich sind, ändert sichdie Ladungsverteilung in einem ein-zelnen, insgesamt neutralen Molekülständig; die Ladungsschwerpunkte verschieben sich und es kann sich ein temporärerDipol ausbilden. Befindet sich in dessen Nähe nun ein zweites Molekül, so verschiebtsich aufgrund des Dipolfeldes von Molekül 1 auch die Ladungsverteilung in Molekül2. Die beiden resultierenden Dipolmoleküle ziehen sich elektrostatisch an, die zunächsttemporären Dipolverteilungen werden durch die gegenseitige Wechselwirkung stabi-ler und es kann von einer Bindung gesprochen werden (vgl. [Dem10], S. 317f).Der Bindungsabstand variiert mit der Orientierung der beiden O2-Moleküle zuein-
ander [AAB+99, NTVP11] (vgl. Abb. 2.11). Die Gleichgewichtsabstände sind in folgen-der Tabelle zusammengestellt:
H X L TRO
2−O
2[Å] 3,56 3,63 3,74 4,26
Die Sauerstoff-Atome im O2 sind durch σ- und π-Molekülorbitale verbunden. Daherkann innerhalb der O2-Moleküle erst ein resonanter Auger-Zerfall stattfinden. Das Io-nisationspotential für O2 liegt bei 12,07 eV10. Die verbleibende Anregungsenergie wirdzu dem benachbarten O2-Molekül transferiert und reicht aus, um dieses zu ionisieren.
10http://physics.nist.gov/cgi-bin/Ionization/table.pl?ionization=O2
17

2. Theoretische Grundlagen
2.6.2. Die Reaktion
Die Reaktionsgleichung für das beobachtete Experiment ist gegeben durch:
Anregung: hν(539,4 eV) + (O2)2 → (O2)∗
2
Auger-Zerfall: (O2)∗
2 → (O2)+ ∗
2 + eAuger
ICD: (O2)+ ∗
2 + eAuger → (O2)++2 + eAuger + eICD
Coulomb-Explosion: (O2)++2 + eAuger + eICD → O+
2 +O+2 + eAuger + eICD
(O2)2*
Grundzustand (O2)2
Potential V
hν = 539,4eV
Auger-Elektron
~ 500eV
ICD-Elektron
~ 1eV
(O2)2+*
O2+ + O2
+
Abbildung 2.12.: Schematischer Ablauf der Reaktion
Im ersten Schritt wird ein O2-Molekül durch ein Photon derEnergie 539, 4 eV resonant an-geregt [GSS10]:
1s→ 3σu
DieAbregung erfolgt zunächstdurch einenAuger-Zerfall. Da-bei wird ein Auger-Elektronmit der kinetischen Energievon etwa 500 eV emittiert.Übrig bleibt ein einfach io-nisiertes, angeregtes O+
2∗-Mo-
lekülion mit einem zweitenO2-Molekül im Grundzustandin Van-der-Waals-Reichweite.Die übrige Anregungsenergiereicht nun nicht mehr aus,um das erste Molekül zwei-fach zu ionisieren, deshalb fin-det ICD statt. Die Energie wirdauf das benachbarte O2 über-tragen und ionisiert dieses.Daraus resultiert ein langsa-mes ICD-Elektron von weni-gen eV und zwei O+
2 -Molekül-ionen. Aufgrund ihrer Ladungen stoßen sich die Ionen elektrostatisch ab, es findet eineCoulomb-Explosion statt und das Molekül dissoziiert. In Abbildung 2.12 ist der zu un-tersuchende Prozess graphisch dargestellt.
18

3. Messung
3.1. Synchrotron
Die Messung wurde am Elektronensynchrotron BESSY II1 in Berlin durchgeführt.
~vevc≈ 0
0 < vc< 1
vc≈ 1
Abbildung 3.1.: Abstrahlcharakteristikder Elektronen im Synchrotron bei Be-schleunigung transversal zu ve
Synchrotrons sind in der Lage, die hochener-getischen Photonen zur Anregung der Sauer-stoff-Moleküle bereitzustellen. Sie funktionierennach dem Prinzip der klassischen Elektrodyna-mik, dass beschleunigte Ladungen Energie inForm von elektromagnetischer Strahlung emittie-ren. Werden nun Elektronen auf ihrer Bahn ab-gelenkt, also beschleunigt, geht Energie in Formvon elektromagnetischer Strahlung (Photonen)verloren (siehe Abbildung 3.1). Diese Photonenwerden nach der klassischen Elektrodynamik beieiner transversalen Beschleunigung nicht nur inFlugrichtung, sondern auch zu einem großen Teilsenkrecht zur Elektronenbahn abgestrahlt2. In einem Synchrotron bewegen sich dieElektronen allerdings mit nahezu Lichtgeschwindigkeit, so dass die Abstrahlung rela-tivistisch betrachtet werden muss. Im Bezugssystem des Elektrons bleibt die Abstrahl-charakteristik natürlich gleich, doch transformiert man nun in das Laborsystem, brei-tet sich die sogenannte Synchrotronstrahlung in einem Vorwärtskegel aus ([Mes06], S.862). Der Öffnungswinkel ϑ dieses Kegels hängt von der Geschwindigkeit v der Elek-tronen ab:
sin(ϑ) =
√
1− v2
c2(3.1)
Der Energieverlust eines Elektrons in einem Umlauf in einem Speicherring bei ultrare-lativistischer Betrachtung3 ist gegeben durch [PRSZ09]:
∆E =e2 · E4
3 · ε0 ·R · (mec2)4(3.2)
Möchte man nun hochenergetische Synchrotronstrahlung erzeugen, muss also beigleichbleibender hoher Teilchenenergie E der Ablenkungsradius R möglichst kleinwerden.
1www.helmholtz-berlin.de2vgl. Hertzscher Dipol3v ≈ c
19

3. Messung
(a) BESSY II (b) Beamline Layout
Abbildung 3.2.: (a) : Übersicht BESSY II (Quelle: BESSY).(b): Das optische Layout der Beamline U49/2-PGM1 am BESSY II (Quelle: BESSY).
Um hochenergetische Elektronen zu erhalten, werden diese am BESSY II nach demVerlassen der Quelle zunächst durch einen Linearbeschleuniger auf Energien im Bereichvon MeV beschleunigt, bevor sie im Synchrotron auf 1,7GeV gebracht werden können.Haben die Elektronen diese Energie erreicht, findet die Injektion in den Speicherringstatt, in den Undulatoren eingebaut sind. Undulatoren werden in modernen Synchro-tronstrahlungsquellen zur Erzeugung von sehr brillanter, hochenergetischer Strahlungeingesetzt. Eine hohe Brillanz bedeutet, dass eine hohe Zahl an Photon eines schma-lenWellenlängenbereiches pro Fläche, Raumwinkelbereich und Zeit erreicht wird (vgl.[Her08], S. 424). Ein Undulator ist eine einige Meter lange Anordnung unterschiedlichgepolter Magnete, die die Elektronen auf eine Wellenbahn mit einer Periodenlängevon einigen Millimetern zwingen. An jedem Umkehrpunkt wird Synchrotronstrah-lung emittiert, die sich addiert. Am Ende des Undulators werden nun die Elektronenweiter durch den Speicherring geleitet, während sich die Synchrotronstrahlung gerad-linig weiter durch die Beamline ausbreitet.Da die Synchrotronstrahlung noch ein Spektrum an Wellenlängen enthält, ist ein
wichtiges Bauteil in der Beamline der Monochromator: Dieser besteht aus Spiegelnund Gittern, die die Strahlung spektral aufspalten, so dass mit einem Schlitz die ge-wünschte Wellenlänge herausgegriffen werden kann (siehe Abbildung 3.2b).
Magnete
Elektronenbahn
Synchrotron-lichtkegel
Abbildung 3.3.: Undulator: Die Elektronenbahnverläuft sinusförmig, die Elektronen emittie-ren Synchrotronstrahlung.
Anschließend wird der Photonenstrahldurch röntgenoptische Systeme fokussiert,bevor er dem Experiment zur Verfügungsteht.Damit zeitaufgelöste Messungen wie
im vorliegenden Experiment erst möglichwerden, muss eine bestimmte Zeitspannezwischen zwei aufeinanderfolgenden Er-eignissen liegen. Nur so können die zu ei-ner Reaktion gehörigen Teilchen einanderkoinzident zugeordnet werden. Aus die-
20

3.2. COLTRIMS
Foto: M. Schöffler
DetektorenPhotonenstrahl
Magnetfeldspulen
Gasjet
Gasjet
yPhotonenstrahl
x
Flugzeitrichtung
z
Detektor
ϕϑ
Teilchenbahn
Abbildung 3.4.: Messaufbau am BESSY II und das durch den Aufbau definierte Koordinatensystem.
sem Grund wurde die Messung während der Single Bunch Operation des BESSY IIdurchgeführt. Während dieser ist der Speicherring nicht komplett mit Elektronen ge-füllt, sondern es befindet sich nur ein einziges Elektronenpaket im Ring. Ein Umlaufdieses Pakets dauert 800,5515 ns und pro Umlauf erreicht ein Lichtblitz das Experi-ment.
3.2. COLTRIMS
Die COLd Target Recoil Ion Momentum Spectroscopy – kurz: COLTRIMS-Methodestellt eine sehr ausgereifte Möglichkeit dar, kinematisch vollständige Messungen inder Vielteilchen-Spektroskopie durchzuführen. Mit diesem Verfahren ist es möglich,die Fragmente nach einer Teilchenkollission im gesamten Raumwinkelbereich von 4πdreidimensional fokussiert zu detektieren. Im Zentrum der COLTRIMS-Kammer be-findet sich die Reaktionszone, in der ein überschallschneller Gasjet mit dem vom Un-dulator kommenden Photonenstrahl gekreuzt wird. Dort findet die Reaktion statt, eswerden Atome oder auch Moleküle ionisiert und Ionen und Elektronen anschließenddurch ein elektrisches Feld zu den Detektoren geführt. Das ganze Spektrometer sowiedie Detektoren sind in einer Vakuumkammer angebracht, um Untergrund durch Ioni-sation von Restgasteilchen zu verringern. In der Targetzone herrscht daher ein Druckim Bereich von 10−9mbar.
Durch die Orientierung des Spektrometers und der Detektoren wird auch das Koor-dinatensystem, in demdie Teilchenimpulse betrachtet werden, festgelegt. Die Flugzeit-richtung, also von der Reaktionszone auf den Detektor zeigend, legt die z-Achse fest,die Ortsrichtungen auf dem Detektor die x- und y-Achse. Dabei wird die y-Richtungdurch die Jetrichtung vorgegeben, die x-Richtung durch den Photonenstrahl.
Im Folgenden werden die einzelnen Komponenten genauer erklärt:
21

3. Messung
3.2.1. Gasjet
Düse
Expansion 1Skimmer 1
Expansion 2 Skimmer 2
Experimentier-kammer
Abbildung 3.5.: Schema des Gasjetszur Targetpräparation
Das gewünschte Target aus (O2)2-Molekülen wirdals Gasjet präpariert. Um die Impulse, die die Teil-chen durch die Reaktion erhalten, genau messenzu können, muss eine Möglichkeit gefunden wer-den, ein Target mit einer möglichst homogenen Im-pulsverteilung zu generieren. Dies wird mit einemÜberschall-Gasjet (Schema in Abb. 3.5) realisiert:Sauerstoff strömt mit starkem Überdruck von
10 bar durch eine 30µm feine Düse in die Ex-pansionskammer, in der Unterdruck (∼ 10−7 bar)herrscht. Aufgrund des Druckunterschiedes findeteine Überschallexpansion statt, wobei die thermi-sche Bewegung der Gasteilchen in eine gerichteteumgewandelt wird. Nun bewegt sich das Gas wei-ter in Richtung Skimmer, eine 300µm feine Öffnung,mit der ein kleiner Bereich gekühlter (sich mit dergleichen Geschwindigkeit bewegender) Gasteilchen ausgewählt wird. Um die Ge-schwindigkeitsverteilung weiter zu verbessern, wird diese Prozedur noch einmal wie-derholt: Mit einem zweiten Skimmer wird wiederum ein kleiner Jetkegel ausgeschnit-ten.Hinter der Reaktionszone, also gegenüber der Gasdüse, befindet sich der Jetdump,
ein schmales Röhrchen, in das die Gasteilchen, die nicht reagiert haben, hineinfliegenund hinter dem sie anschließend abgepumpt werden. Zur weiteren Analyse der im Jetenthaltenen Teilchen kann am Jetdump ein Massenspektrometer angebracht werden.Bei ausgeschaltetem Jet kann damit auch das in der Kammer enthaltene Restgas ana-lysiert werden. Der Jetdump ist wichtig, um das Vakuum innerhalb der Kammer aufeinem hohen Niveau zu halten, da er den Rückstrom der Teilchen in die Vakuumkam-mer verhindert.
Partialdruck 2. Dumpstufe, m = 64 u [x10
bar]
0
77
Vordruck [bar]
Abbildung 3.6.: Jetkurven für O2 (schwarz) und (O2)2(rot) bei 300K. Zu beachten ist, dass der Partial-druck für (O2)2 gegenüber dem vonO2 umdrei Grö-ßenordnungen hochskaliert wurde.
Um den Vordruck des Gases einstel-len zu können, ist an der Gasflasche einDruckminderer angebracht. Die Tem-peratur des Targetgases kann an der Jet-düse mit Hilfe eines Kryostaten und ei-ner Heizung kontrolliert werden. DasVariieren von Druck und Temperaturdes Gases vor dem Einlassen in dieKammer ermöglicht ein Optimieren derClustergröße im Jet auf das gewünsch-te Molekül. Zu diesem Zweck wer-den zunächst bei verschiedenen Düsen-und damit Gastemperaturen sogenann-te Jetkurven aufgenommen, um denAnteil an (O2)2 im Jet zu bestimmen.
22

3.2. COLTRIMS
Abbildung 3.6 zeigt eine solche Testkurve für 300K. Darstellungsbedingt wurde diey-Achse für (O2)2 um drei Größenordnungen hochskaliert. Wie leicht zu sehen ist, isterst bei hohen Vordrucken eine größere Anzahl an (O2)2-Dimeren im Jet zu finden.
Die mittlere kinetische Energie der Teilchen im Jet ergibt sich aus dem Energieerhal-tungssatz für die Moleküle im Targetgas. Da der Jet haupsächlich aus O2-Molekülenbesteht, deren Geschwindigkeitsverteilung durch die Überschallexpansion und dasAusschneiden durch die beiden Skimmer sehr schmal ist, haben auch alle anderenTeilchen im Jet die Geschwindigkeit dieser O2-Moleküle. Sie unterscheiden sich auf-grund ihrer unterschiedlichen Massen aber im Impuls. Um die thermische Energie derTeilchen in einem Gas zu berechnen, benötigt man die Anzahl der Freiheitsgrade f .Ein O2-Molekül besitzt drei Translations-, zwei Rotations- und Vibrationsfreiheitsgra-de. Da es sich um einen kalten Jet handelt, findet sich die innere Energie des Gasesaber vor allem in den Translations- und Rotationsmoden wieder. Die Vibrationsmodendes Sauerstoffs werden erst bei viel höheren Temperatuten angeregt und können daherals Freiheitsgrad vernachlässigt werden. Insgesamt ergibt sich also f = 5. Nun kanndie Energie und damit die Geschwindigkeit eines O2-Moleküls im Gas ausgerechnetwerden ([Mes06], S. 212):
Ekin,O2=
1
2mO
2v2jet =
f
2· kBTjet =
5kBTjet2
(3.3)
vjet =
√
5kBTjetmO
2
=⇒ pjet,(O2)2= m(O
2)2vjet =
√
20kBTjetmO2
(3.4)
Mit einer Jettemperatur von Tjet = 300K, der Teilchenmasse mO2= 32 u und der
Boltzmann-Konstante kB = 1,381 · 10−23 JK4 ergibt sich für den Impuls eines (O2)2-
Teilchens in Jetrichtung pjet,(O2)2= 33,33 a.u..
3.2.2. Spektrometer
Das Spektrometer besteht aus parallelen Kupferplatten, an die ein elektrisches Potenti-al angelegt wird. Dadurchwird ein elektrisches Feld erzeugt. Die Geometrie des Feldeslässt sich dadurch regeln, dass die einzelnen Kupferplatten nicht alle auf dem selbenPotential liegen. Mittels zwischen die Platten geschalteter Widerstände lässt sich einPotentialverlauf erzeugen. Das resultierende elektrische Feld lenkt die Elektronen undIonen in entgegengesetzten Richtungen auf die Detektoren. Außerdem beschleunigtdas elektrische Feld, das innerhalb des Spektrometers vorliegt, die Teilchen von derReaktionszone aus in Richtung der Detektoren. Zur Fokussierung in Flugzeitrichtungist zwischen Beschleunigungsstrecke und Detektor eine feldfreie Driftstrecke vorgese-hen. Durch Einstellen des Verhältnisses dieser beiden Strecken kann das Spektrome-ter so eingestellt werden, dass alle Teilchen, die in der Reaktionszone mit dem glei-chen Impulsbetrag in die gleiche Richtung, aber aufgrund der Ausdehnung von Gas-jet (ØJet ∼ 1mm) und Photonenstrahl (Øhν ∼ 100µm) an unterschiedlichen Orten inder Flugzeitrichtung z starten, gleichzeitig auf dem Detektor auftreffen. Die genaue4http://physics.nist.gov/cgi-bin/cuu/Value?k
23
3. Messung
Detektorebene
Drift Beschleunigung
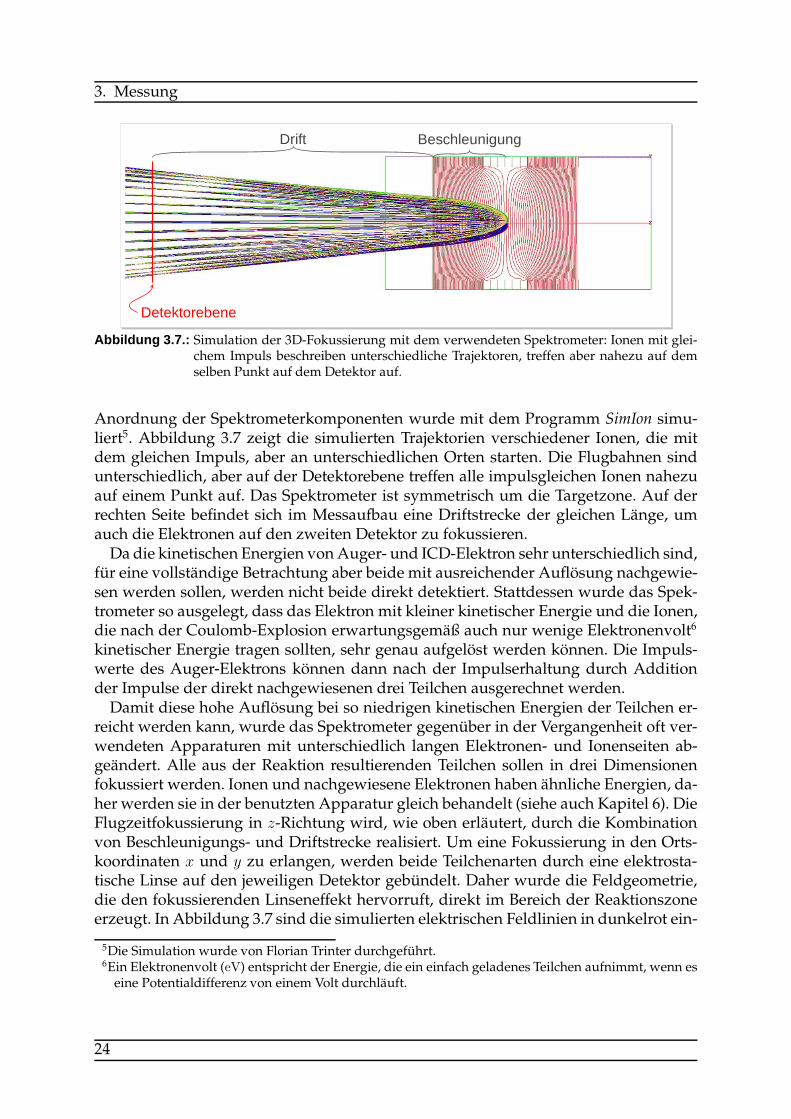
Abbildung 3.7.: Simulation der 3D-Fokussierung mit dem verwendeten Spektrometer: Ionen mit glei-chem Impuls beschreiben unterschiedliche Trajektoren, treffen aber nahezu auf demselben Punkt auf dem Detektor auf.
Anordnung der Spektrometerkomponenten wurde mit dem Programm SimIon simu-liert5. Abbildung 3.7 zeigt die simulierten Trajektorien verschiedener Ionen, die mitdem gleichen Impuls, aber an unterschiedlichen Orten starten. Die Flugbahnen sindunterschiedlich, aber auf der Detektorebene treffen alle impulsgleichen Ionen nahezuauf einem Punkt auf. Das Spektrometer ist symmetrisch um die Targetzone. Auf derrechten Seite befindet sich im Messaufbau eine Driftstrecke der gleichen Länge, umauch die Elektronen auf den zweiten Detektor zu fokussieren.Da die kinetischen Energien vonAuger- und ICD-Elektron sehr unterschiedlich sind,
für eine vollständige Betrachtung aber beide mit ausreichender Auflösung nachgewie-sen werden sollen, werden nicht beide direkt detektiert. Stattdessen wurde das Spek-trometer so ausgelegt, dass das Elektron mit kleiner kinetischer Energie und die Ionen,die nach der Coulomb-Explosion erwartungsgemäß auch nur wenige Elektronenvolt6
kinetischer Energie tragen sollten, sehr genau aufgelöst werden können. Die Impuls-werte des Auger-Elektrons können dann nach der Impulserhaltung durch Additionder Impulse der direkt nachgewiesenen drei Teilchen ausgerechnet werden.Damit diese hohe Auflösung bei so niedrigen kinetischen Energien der Teilchen er-
reicht werden kann, wurde das Spektrometer gegenüber in der Vergangenheit oft ver-wendeten Apparaturen mit unterschiedlich langen Elektronen- und Ionenseiten ab-geändert. Alle aus der Reaktion resultierenden Teilchen sollen in drei Dimensionenfokussiert werden. Ionen und nachgewiesene Elektronen haben ähnliche Energien, da-her werden sie in der benutzten Apparatur gleich behandelt (siehe auch Kapitel 6). DieFlugzeitfokussierung in z-Richtung wird, wie oben erläutert, durch die Kombinationvon Beschleunigungs- und Driftstrecke realisiert. Um eine Fokussierung in den Orts-koordinaten x und y zu erlangen, werden beide Teilchenarten durch eine elektrosta-tische Linse auf den jeweiligen Detektor gebündelt. Daher wurde die Feldgeometrie,die den fokussierenden Linseneffekt hervorruft, direkt im Bereich der Reaktionszoneerzeugt. In Abbildung 3.7 sind die simulierten elektrischen Feldlinien in dunkelrot ein-
5Die Simulation wurde von Florian Trinter durchgeführt.6Ein Elektronenvolt (eV) entspricht der Energie, die ein einfach geladenes Teilchen aufnimmt, wenn eseine Potentialdifferenz von einem Volt durchläuft.
24

3.2. COLTRIMS
gezeichnet. Zu beachten ist, dass in der Nähe der Reaktionszone ein hoher Feldgradi-ent herrscht. Aufgrunddessen kann es leicht zu Verzerrungen in der Impulsverteilungkommen, wenn Moleküle nicht genau in der Mitte der Reaktionszone, sondern wei-ter rechts oder links ionisiert werden. Diese Fehler müssen später in der Analyse derDaten korrigiert werden (siehe Kapitel 4.2.5).Aufgrund der komplizierten Feldgeometrie in der Reaktionszone muss sicherge-
stellt werden, dass im kompletten Spektrometer kein magnetisches Feld herrscht. Diezuvor genannte Simulation ergab, dass schon ein Feld von wenigen µT7 zu Abwei-chungen in den Teilchentrajektorien führt, die so stark sind, dass eine eindeutige Re-konstruktion der Anfangsimpulse der Ionen und Elektronen rechnerisch nicht mehrmöglich ist. Um äußere Magnetfelder zu eliminieren, wurden um die gesamte COL-TRIMS-Kammer herum drei Spulen installiert, die einen Würfel der Kantenlänge 2mbilden (siehe Abbildung 3.4). Mit diesen kann das magnetische Feld so gegengeregeltwerden, dass im Bereich des Spektrometers die magnetischen Feldkomponenten in al-len drei Raumrichtungen gleich null sind.Wie in Kapitel 3.2.1 erwähnt, tragen die Targetteilchen im Gasjet durch die Über-
schallexpansion schon einen Impuls in y-Richtung. Sie treffen also nicht mittig auf demDetektor auf, sondern das Bild wird in y-Richtung verschoben. Diese Verschiebungkann so stark sein, dass bei großem Jetoffset nicht alle nachzuweisenden Teilchen denDetektor treffen. Um diesem Effekt entgegenzuwirken, wurden die Spektrometerplat-ten leicht schräg gestellt, so dass die ganze Detektorfläche ausgenutzt wird. Da dieseKompensation allerdings nicht ausreichend genau sein kann, muss der Jetoffset spä-ter in den Daten durch eine Korrektur der y-Koordinate der Ionen genau ausgeglichenwerden. Die Elektronen betrifft dies aufgrund ihrer geringen Masse nicht.Alle wichtigen Daten, die aus der Simulation zum Spektrometer gewonnen wurden,
sind in Anhang B.1 zusammengestellt.
3.2.3. Detektoren
Der Nachweis der Teilchen erfolgt durch ein Detektorsystem, das es ermöglicht, dieFlugzeit und die Auftrefforte der Ionen und Elektronen auf dem Detektor zu rekon-struieren. Die Detektoren sind aufgebaut aus einem Microchannel Plate (MCP) undeiner Delay-Line-Anode, deren Funktion im Folgenden erläutert wird:
Microchannel Plate
Das Microchannel Plate dient dazu, die schwachen elektrischen Signale einzelner Teil-chen zu verstärken. In der 1,5mm dicken Glasplatte befinden sich tausende schrägerPoren mit jeweils einem Durchmesser von etwa 25µm [Roe10]. Die Ober- und Unter-seite des MCPs sind metallbeschichtet, zwischen ihnen liegt eine Spannung im Bereichvon einigen kV an. Trifft nun ein Teilchen auf das MCP, schlägt es aus der Wand ei-ner der Poren Sekundärelektronen heraus. Diese treffen wiederum auf die Wand, umdort weitere Elektronen auslösen und sich so weiter zu vervielfachen. Währenddessen7Größenordnung des Erdmagnetfeldes.
25

3. Messung
bewirkt die an den Oberflächen angelegte Spannung, dass sich die Teilchen auch indie richtige Richtung, zur Delay-Line-Anode hin, bewegen. Um zusätzlich zu verhin-dern, dass sich die Sekundärteilchen zurück in Richtung Spektrometer bewegen, sindzwei dieser MCPs in einer „Chevron-Anordnung“ angebracht. Die Kanäle, durch diesich die Teilchen bewegen, bilden also eine V-Förmige Struktur (Siehe Abbildung 3.8).Außerdem bewirkt diese Anordnung eine weitere Verstärkung, da auch Teilchen, dieinnerhalb des ersten MCPs keine Wand berührt haben, nun einschlagen. Letztendlichkann das Signal eines einzelnen Elektrons oder Ions so um über sieben Größenordun-gen verstärkt werden [Roe10].
Porenauftreffendes
Teilchen
zurAnode
Hoch-spannung
MCP 1
MCP 2
Abbildung 3.8.: Schematische Darstel-lung einer MCP-Chevronanordnung.
Die MCP-Anordnung dient, außer zur Verstär-kung der Teilchensignale, zur Flugzeitmessung: Ver-lässt nach der Verstärkung eine Elektronenwolkeund damit eine große Anzahl Ladungen das MCP,so schwankt die zwischen Ober- und Unterseite an-gelegte Spannung. Dieser Spannungspuls wird ka-pazitiv ausgekoppelt, registriert und als Zeitsignalweiterverarbeitet. In Abschnitt 4.1.2 wird erläutert,wie aus diesem Signal die Flugzeit des Teilchens er-mittelt werden kann.
Delay-Line-Anode
Abbildung 3.9.: Hex-Anode (Quelle:RoentDek)
Die Lawine von Sekundärelektronen erreicht an-schließend die Delay-Line-Anode. Eine Delay-Line-Anode besteht aus meist zwei (Quad-Anode) oderdrei (Hex-Anode) Ebenen gespannten Kupferdrah-tes. Auf jeder Ebene sind zwei Drähte parallel mehr-mals gewickelt: ein Signal- und ein Referenzdraht.Das Signal ist räumlich so gut lokalisiert, dass essich nur auf einem der beiden ausbreitet, Störsigna-le und Rauschen jedoch auf beiden. So kann durchDifferenzbildung ein großer Teil des Rauschens be-reinigt werden. Hier wurden sowohl auf der Ionen-als auch auf der Elektronenseite so genannte Hex-Anoden verwendet, welche aus drei Draht-Ebenenbestehen, die jeweils in einem Winkel von 60◦ zu-einander angeordnet sind (vgl. Abb. 3.9).Die Ortsmessung mit einer Delay-Line-Anode funktioniert über das Messen von
Zeitdifferenzen. Trifft eine Elektronenwolke auf demDraht auf, bewegt sich das Signalin beiden Richtungen auf ihm weiter und wird an beiden Enden gemessen. Da dieDrahtlänge und damit auch die gesamte Laufzeit bekannt ist, kann schon mit einemSignal der Auftreffort auf dem Draht ermittelt werden. Die Messung der Laufzeitenbeider Richtungen bietet eine zusätzliche Redundanz. Außerdem kann so die Funk-tion und richtige Einstellung des Detektors überprüft werden, da die Zeitsumme der
26

3.2. COLTRIMS
Signale an einem Draht konstant bleiben muss. Dadurch, dass drei Drahtlagen genutztwerden, kann man den zweidimensionalen Auftreffort des Teilchens durch Überlage-rung der drei eindimensionalen Orte mit großer Genauigkeit rekonstruieren.
3.2.4. Aufnahme der Rohdaten
Die vom MCP und den Delay-Line-Anoden gemessenen Pulse werden nun elektro-nisch aufbereitet, um anschließend vom Computer verarbeitet werden zu können.Zunächst werden sie nochmals elektronisch durch einen Fast Amplifier verstärkt,
anschließend wird mit Hilfe eines Constant Fraction Discriminators (CFD) die Zeit-präzision der Signale erhöht [Roe09]. Der CFD wandelt die analogen Pulse, die un-terschiedliche Höhen und Breiten haben können, in digitale Signale um. Jetzt werdendiese Signale mit Hilfe eines Time-to-Digital-Converters (TDC) zurWeiterverarbeitungan den PC übergeben.
Hier werden sie von der Daten-Aufnahme und -Analysesoftware CoboldPC [Roe06]aufgenommen und im ListMode-Format abgespeichert. Zu jedem gemessenen Eventwerden in der ListMode-Datei alle verfügbaren Informationen zugeordnet und stehenso zur weiteren Analyse bereit.Die Signale, die aufgenommen werden, sind: der Bunchmarker, der angibt, wann
das Elektronenpaket einen Umlauf im Speicherring beendet hat, die MCP-Treffer desElektronen- und des Ionendetektors und für jeden Detektor und jede Detektorlage diezwei Zeitsignale, wann der elektrische Puls das jeweilige Drahtende erreicht hat. Einvollständiger Rohdatensatz zu einem aufgenommenen Elektron oder Ion enthält alsosieben Zeitsignale plus den Bunchmarker.
27


4. Analyse
4.1. Datenverarbeitung
4.1.1. ROOT und lmf2root
Das Softwarepaket ROOT ist ein am CERN entwickeltes, auf die Programmierspra-che C++ aufgesetztes Analysetool für Experimente der Teilchenphysik1. Seine Stärkeliegt darin, große Mengen von Daten effizient und schnell zu verarbeiten. Die Daten,die in einer speziellen ROOT-Datei gespeichert werden, können auf verschiedensteWeise sortiert, ausgewählt und visualisiert werden. Es steht eine große Anzahl anFunktionen zur Verfügung, die angewendet werden können und auch unterschiedli-che Histogramm-Typen, um die Daten darzustellen. Außerdem lässt sich ROOT durcheigene Makros beliebig erweitern.
4.1.2. Berechnung von Flugzeiten und Auftrefforten der Tei lchen
Aus den aufgenommenen Zeitsignalen müssen nun zunächst die Auftrefforte und dieFlugzeiten der Teilchen berechnet werden.
Die Flugzeiten lassen sich aus denMCP-Signalen (te,MCP und trec,MCP ), dem Bunch-marker-Signal (tbm) und dem bekannten Abstand zwischen zwei Bunchmarkern amBESSY II (800,5515 ns) ermitteln. Der Bunchabstand wird gebraucht, um die Zeitsigna-le, die vom TDC erzeugt werden, in Relation zu setzen. Die Elektronen bewegen sichaufgrund ihrer geringen Masse viel schneller zum Detektor als die Recoil-Ionen. IhreFlugzeit beträgt in dem hier durchgeführten Experiment um die 30 ns (vgl. Abbildung4.4) und ist damitmehr als eine Größenordnung kleiner als der Abstand zwischen zweiLichtblitzen. Daher kann sie wie folgt berechnet werden:
tofe = mod ((te,MCP − tbm) , 800.5515 ns) (4.1)
Die Flugzeit der untersuchten O+2 Recoil-Ionen ist, wie auch in Abbildung 4.3 zu erken-
nen, mit über 9000 ns über eine Größenordnung länger als der Bunchmarker-Abstand.Deshalb werden die Ionen mit Hilfe der Elektronen dem korrekten Bunchmarker zu-geordnet:
tofrec = trec,MCP − te,MCP + tofe (4.2)
1http://root.cern.ch/
29
4. Analyse
v-layer rec [ns]-100 -80 -60 -40 -20 0 20 40 60 80 100
Tim
e su
m r
ec v
[ns]
-5
-4
-3
-2
-1
0
1
2
3
4
5
0
1000
2000
3000
4000
5000
6000
7000
v-layer rec [ns]-100 -80 -60 -40 -20 0 20 40 60 80 100
Tim
e su
m r
ec v
[ns]
-5
-4
-3
-2
-1
0
1
2
3
4
5
0
5000
10000
15000
20000
25000
30000
35000
Abbildung 4.1.: Zeitsummenkorrektur: Links: Zeitsumme einer der drei Detektorlagen gegen den Auf-treffort auf dieser Lage in ns vor der Autokalibrierung, Rechts: nachher.
Die Auftrefforte der Teilchen werden aus den Zeitsignalen der jeweils drei Detek-torlagen rekonstruiert. Die Laufzeit eines elektrischen Signals über die gesamte Draht-länge ist bekannt und es wird angenommen, dass sich das Signal linear auf dem An-odendraht bewegt. So kann das gemessene Signal in ns direkt einer Strecke in mm aufdem Detektor zugeordnet werden.
4.1.3. Vorsortieren
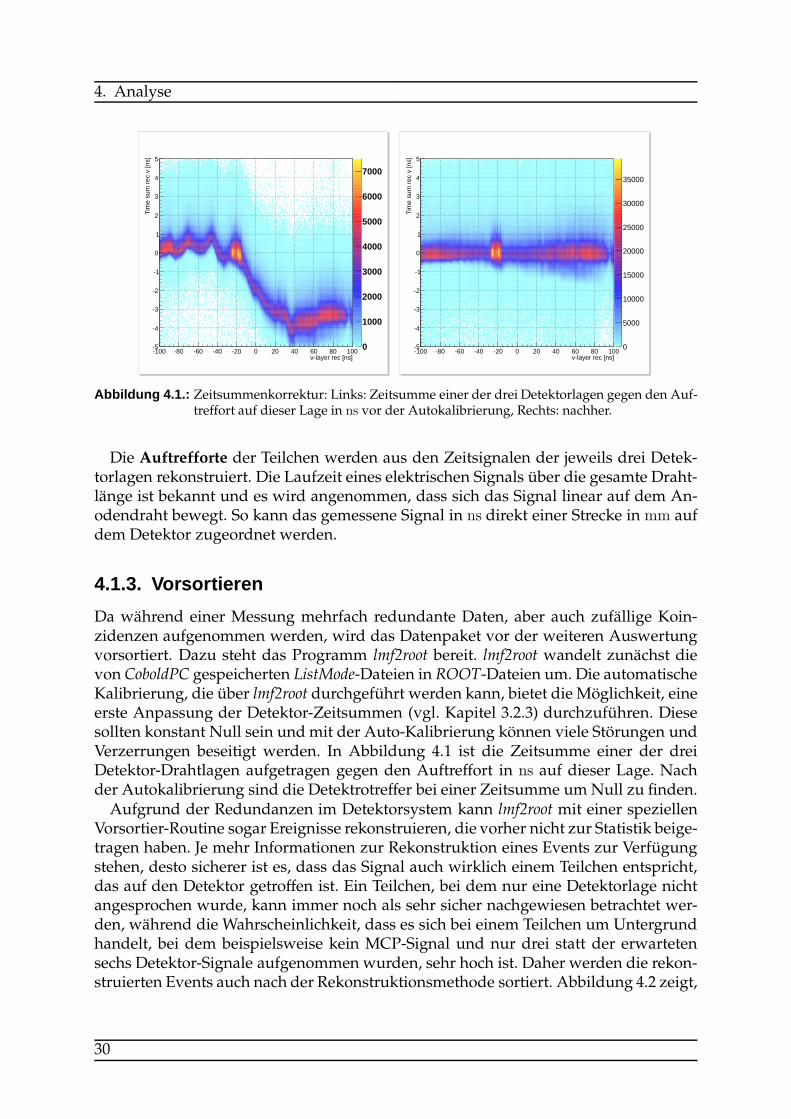
Da während einer Messung mehrfach redundante Daten, aber auch zufällige Koin-zidenzen aufgenommen werden, wird das Datenpaket vor der weiteren Auswertungvorsortiert. Dazu steht das Programm lmf2root bereit. lmf2root wandelt zunächst dievon CoboldPC gespeicherten ListMode-Dateien in ROOT-Dateien um. Die automatischeKalibrierung, die über lmf2root durchgeführt werden kann, bietet die Möglichkeit, eineerste Anpassung der Detektor-Zeitsummen (vgl. Kapitel 3.2.3) durchzuführen. Diesesollten konstant Null sein und mit der Auto-Kalibrierung können viele Störungen undVerzerrungen beseitigt werden. In Abbildung 4.1 ist die Zeitsumme einer der dreiDetektor-Drahtlagen aufgetragen gegen den Auftreffort in ns auf dieser Lage. Nachder Autokalibrierung sind die Detektrotreffer bei einer Zeitsumme umNull zu finden.Aufgrund der Redundanzen im Detektorsystem kann lmf2root mit einer speziellen
Vorsortier-Routine sogar Ereignisse rekonstruieren, die vorher nicht zur Statistik beige-tragen haben. Je mehr Informationen zur Rekonstruktion eines Events zur Verfügungstehen, desto sicherer ist es, dass das Signal auch wirklich einem Teilchen entspricht,das auf den Detektor getroffen ist. Ein Teilchen, bei dem nur eine Detektorlage nichtangesprochen wurde, kann immer noch als sehr sicher nachgewiesen betrachtet wer-den, während die Wahrscheinlichkeit, dass es sich bei einem Teilchen um Untergrundhandelt, bei dem beispielsweise kein MCP-Signal und nur drei statt der erwartetensechs Detektor-Signale aufgenommen wurden, sehr hoch ist. Daher werden die rekon-struierten Events auch nach der Rekonstruktionsmethode sortiert. Abbildung 4.2 zeigt,
30
4.1. Datenverarbeitung
Methode0 2 4 6 8 10 12 14 16 18 200
100
200
300
400
500
310×Hits Recoil 1
Methode0 2 4 6 8 10 12 14 16 18 200
50
100
150
200
250
310×Hits Recoil 2
Abbildung 4.2.: Anzahl der Ionendetektortreffer nach Rekonstruktion mit unterschiedlichenMethoden
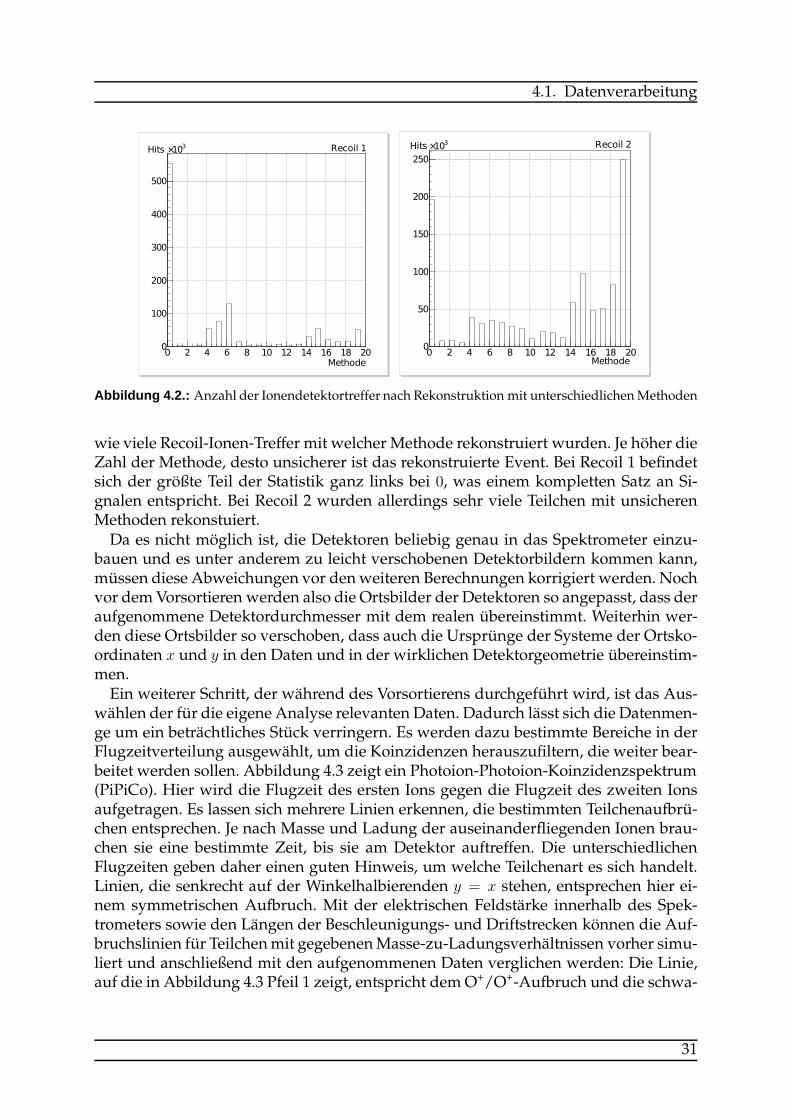
wie viele Recoil-Ionen-Treffer mit welcher Methode rekonstruiert wurden. Je höher dieZahl der Methode, desto unsicherer ist das rekonstruierte Event. Bei Recoil 1 befindetsich der größte Teil der Statistik ganz links bei 0, was einem kompletten Satz an Si-gnalen entspricht. Bei Recoil 2 wurden allerdings sehr viele Teilchen mit unsicherenMethoden rekonstuiert.Da es nicht möglich ist, die Detektoren beliebig genau in das Spektrometer einzu-
bauen und es unter anderem zu leicht verschobenen Detektorbildern kommen kann,müssen diese Abweichungen vor denweiteren Berechnungen korrigiert werden. Nochvor demVorsortieren werden also die Ortsbilder der Detektoren so angepasst, dass deraufgenommene Detektordurchmesser mit dem realen übereinstimmt. Weiterhin wer-den diese Ortsbilder so verschoben, dass auch die Ursprünge der Systeme der Ortsko-ordinaten x und y in den Daten und in der wirklichen Detektorgeometrie übereinstim-men.Ein weiterer Schritt, der während des Vorsortierens durchgeführt wird, ist das Aus-
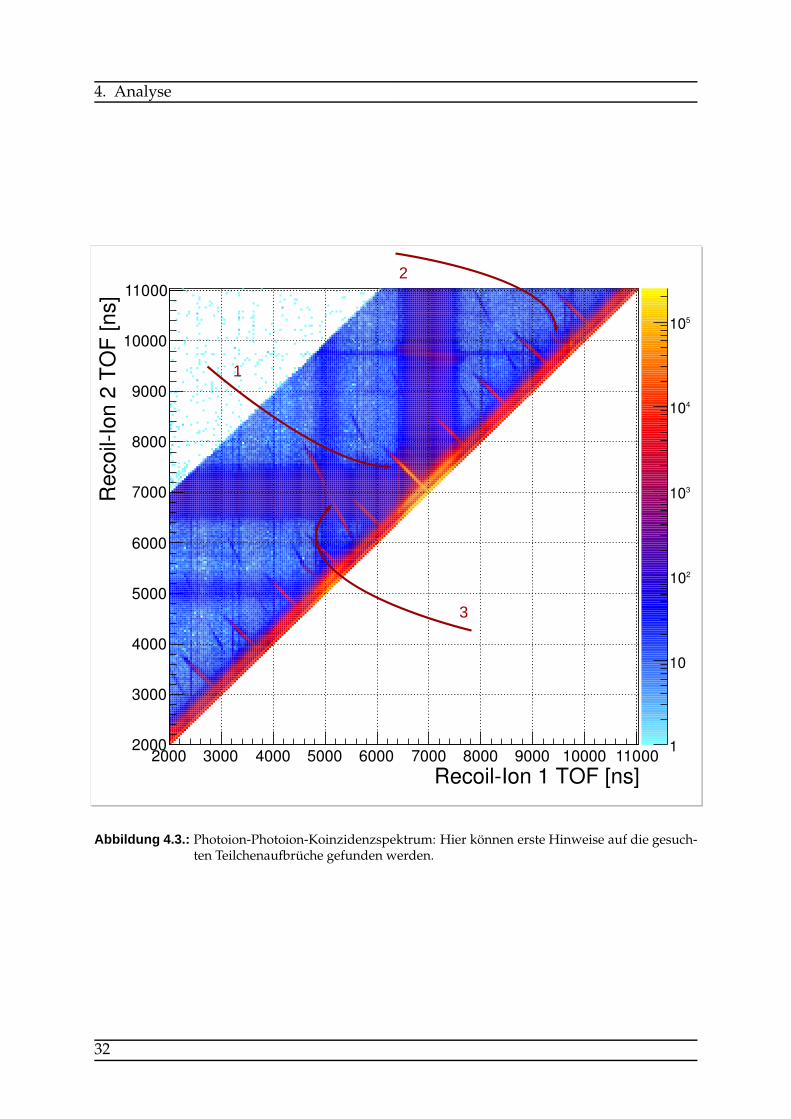
wählen der für die eigene Analyse relevanten Daten. Dadurch lässt sich die Datenmen-ge um ein beträchtliches Stück verringern. Es werden dazu bestimmte Bereiche in derFlugzeitverteilung ausgewählt, um die Koinzidenzen herauszufiltern, die weiter bear-beitet werden sollen. Abbildung 4.3 zeigt ein Photoion-Photoion-Koinzidenzspektrum(PiPiCo). Hier wird die Flugzeit des ersten Ions gegen die Flugzeit des zweiten Ionsaufgetragen. Es lassen sich mehrere Linien erkennen, die bestimmten Teilchenaufbrü-chen entsprechen. Je nach Masse und Ladung der auseinanderfliegenden Ionen brau-chen sie eine bestimmte Zeit, bis sie am Detektor auftreffen. Die unterschiedlichenFlugzeiten geben daher einen guten Hinweis, um welche Teilchenart es sich handelt.Linien, die senkrecht auf der Winkelhalbierenden y = x stehen, entsprechen hier ei-nem symmetrischen Aufbruch. Mit der elektrischen Feldstärke innerhalb des Spek-trometers sowie den Längen der Beschleunigungs- und Driftstrecken können die Auf-bruchslinien für Teilchenmit gegebenenMasse-zu-Ladungsverhältnissen vorher simu-liert und anschließend mit den aufgenommenen Daten verglichen werden: Die Linie,auf die in Abbildung 4.3 Pfeil 1 zeigt, entspricht demO+/O+-Aufbruch und die schwa-
31
4. Analyse
Recoil-Ion 1 TOF [ns]2000 3000 4000 5000 6000 7000 8000 9000 10000 11000
Reco
il-Io
n 2
TO
F [n
s]
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
102
103
104
105
10
1
2
1
3
Abbildung 4.3.: Photoion-Photoion-Koinzidenzspektrum: Hier können erste Hinweise auf die gesuch-ten Teilchenaufbrüche gefunden werden.
32

4.2. Auswertung
che Linie am Ende von Pfeil 2 dem O+2/O
+2 -Aufbruch. Linien mit einemWinkel zu Dia-
gonalen, der von 90◦ abweicht, zeigen unsymmetrische Aufbrüche wie etwa O++/O+
(Pfeil 3). Parallele Linien zu den erwähnten Aufbrüchen sind falsche Koinzidenzen.Die Linien sind um den Bunchabstand von ≈ 800 ns verschoben, hier wurden alsoRecoil-Ionen dem falschen Bunchmarker zugeordnet.Wie oben beschrieben kann man nun während des Vorsortierens den so gefundenen
Flugzeitbereich in den Ionendaten auswählen und in die ROOT-Datei übernehmen.
4.2. Auswertung
4.2.1. Berechnung von Impulsen und Energien
Impulse
Anders als in den vorangegangenen Experimenten benutzten COLTRIMS-Spektrome-tern wurde, wie in Abschnitt 3.2.2 erklärt, in dieser Messung eine Spektrometergeome-trie gewählt, in der Ionen und Elektronen gleich behandelt werden.
Die Impulse in den beiden Ortsrichtungen x und y werden nach der aus der New-tonschen Mechanik bekannten Formel
p = m · v = m · st
(4.3)
berechnet. Die Strecke s berechnet sich aus dem Ortsnullpunkt auf dem Detektor x0und dem Auftreffort x des jeweiligen Teilchens.Damit ergibt sich für die Recoil-Ionen:
pr,x = mO2· x− x0
tofO+2
(4.4)
und analog für die y-Richtung:
pr,y = mO2· y − y0
tofO+2
(4.5)
In der Flugzeitrichtung werden die Teilchen durch das elektrische Feld im Spektro-meter beeinflusst. Dieses beschleunigt die Ionen und Elektronen zu den Detektorenhin, bewirkt also eine Steigerung ihrer kinetischen Energie und muss in der Berech-nung mit berücksichtigt werden. Aufgrund der elektrostatischen Linse im Bereich derReaktionszone wird die Feldgeometrie nicht vernachlässigbar verzerrt und es kannkein ein homogenes Feld angenommen werden. Allerdings ist die aus der Reaktionresultierende kinetische Energie der Teilchen viel kleiner als die Energie, die sie auf-grund der Beschleunigung im Feld erhalten. Deshalb kann in der linearen Näherungangenommen werden, dass unterschiedliche Startorte der Teilchen keinen messbarenUnterschied in der Energieaufnahme im Feld bewirken. Die Coulomb-Kraft auf einTeilchen mit der Ladung q in einem elektrischen Feld ~E ist gegeben durch:
~FC = ~E · q (4.6)
33

4. Analyse
Elektron 1 TOF [ns]-400 -300 -200 -100 0 100 200 300 400
0
10000
20000
30000
40000
50000
Abbildung 4.4.: Verteilung der Elektronenflug-zeit: Der scharfe Peak liegt bei etwa 33 ns undrepräsentiert die Flugzeitmitte t0. Die niede-renergetischen Elektronen mit kleinen Impul-sen führen hier zu einer schmalen Flugzeitver-teilung.
Damit ergibt sich für den Impuls diesesTeilchens in die vom Feld beeinflusste Flug-zeitrichtung z:
pr,z = Fz ·∆t = | ~FC | ·∆t = | ~E| · q ·∆t (4.7)
Je nachdem, in welche Richtung sich dasTeilchen nach der Reaktion bewegt, ist des-sen Flugzeit unterschiedlich lang. Hat einElektron etwa einen Impuls in Richtung Io-nendetektor, dauert es länger, bis es vomelektrischen Feld zum Elektronendetektorgeführt wird, als wenn es nach der Reakti-on direkt in diese Richtung fliegt. Um dieaufgenommenen Zeiten in Beziehung zu-einander zu setzen, wird ∆t wie folgt be-rechnet: ∆t = (tofrec − t0). Hierbei be-zeichnet t0 die Flugzeitmitte, also die Zeit,die das Elektron bis zum Detektor braucht,wenn es mit dem Impuls pz = 0 in der Re-aktionszone startet. (tofrec) entspricht der gemessenen Flugzeit. Die Zeit t0 wurdedurch Mitteln über die gesamte Flugzeitverteilung der Teilchen gefunden. Da die Zeit-differenz ∆t die einzige Variable in der Gleichung zur Berechnung von pz ist, wurde,um die Berechnung in der Auswerteroutine zu vereinfachen, die Formel 4.7 im Pro-grammcode durch Zusammenfassen der Konstanten zu einem Feldparameter verein-facht.
Da das Spektrometer symmetrisch aufgebaut und kein Magnetfeld angelegt ist, wer-den die Impulse für Elektronen und Ionen analog berechnet.
Energien
Aus den Impulsen können nun auch die Energien der Teilchen berechnet werden:
Für die Recoil-Ionen wird der Kinetic Energy Release (KER) berechnet. Dieser be-schreibt die Aufbruchsenergie der beiden O2-Moleküle und wird aus dem Relativim-puls ~prel der Ionen und der reduzierten Masse µ des Systems errechnet:
~prel =~pr1 − ~pr2
2(4.8)
µ =mr1 ·mr2
mr1 +mr2
=mO
22
2 ·mO2
=mO
2
2(4.9)
KER =|~prel|22 · µ (4.10)
34

4.2. Auswertung
Die Elektronenenergie des detektierten ICD-Elektrons wird aus dem Elektronenimpuls~pe = (pe,x, pe,y, pe,z) wie folgt berechnet:
Ee =|~pe|22 ·me
(4.11)
4.2.2. Kalibrierung der Auftrefforte
Nach dem Vorsortieren müssen zunächst die Detektoren kalibriert werden. Dazu wer-den auf die vorhandenen Datensätze konstante Parameter addiert, um die physikali-schen Nullpunkte mit denen auf dem Detektor in Übereinstimmung zu bringen, umweitere Berechnungen durchführen zu können.
4.2.3. Energie-Eichung
Da Messaufbauten nicht beliebig genau sind und viele systematische Fehlerquellenbeinhalten, die die aufgenommenen Werte beeinflussen, ist ein wichtiger Schritt dieEnergie-Eichung der aufgenommenen Daten. Um am Ende der Auswertung korrek-te Absolutwerte zu erhalten, müssen die aufgenommenen Daten mit Werten aus derLiteratur verglichen und eventuell angepasst, also geeicht werden.
Eichung der Photonen-Energien
Beamline-Offset
Abbildung 4.5.: Eichung der Photonenenergie: In derRate der auf den Detektor treffenden Recoil-Ionen(schwarz) sind Maxima erkennbar, die mit Litera-turwerten aus [TFK+08] (blau) verglichen werden.
Noch vor der Messung wird dieEnergie der Photonen geeicht. Da-zu wird ein Photonenenergiebe-reich von 537 eV bis 545 eV in 0,1 eV-Schritten durchgescannt. Zu jedemWert der Photonenenergie wird dieRate der auf den Detektoren auf-treffenden Elektronen und Recoil-Ionen aufgenommen. Trägt mannun die Recoil-Rate gegen die Pho-tonenenergie auf, so lassen sich Re-sonanzen erkennen, die mit den Li-teraturwerten aus [TFK+08] vergli-chen werden. Es lässt sich ablesen,um wie viele eV die Spektren gegeneinander verschoben sind. So ergibt sich ein Offsetder Beamline in der Photonenenergie von 0,95 eVUmdie gewünschte Anregungsener-gie von 539,4 eV zu erhalten, müssen also an der Beamline 540,35 eV Photonenenergieeingestellt werden. Das Beamline-Offset ist eine Eigenschaft von Undulator und Mo-nochromator der Beamline. Daher muss die Photonenenergie-Eichung nachdem Syn-chrotron und Beamline neu eingestellt wurden, erneut durchgeführt werden. In derletzten Woche der Strahlzeit nach der Maschinenschicht war so das Beamline-Offsetdeutlich kleiner, nämlich bei 0,45 eV.
35
4. Analyse
Eichung der Ionen-Energien
Photon Energy: 412 eV
Abbildung 4.6.: Eichung des KER: Die schwarze Kurveist das Spektrum aus [LEBW96], in rot sind die inBerlin gemessenen Werte dargestellt.
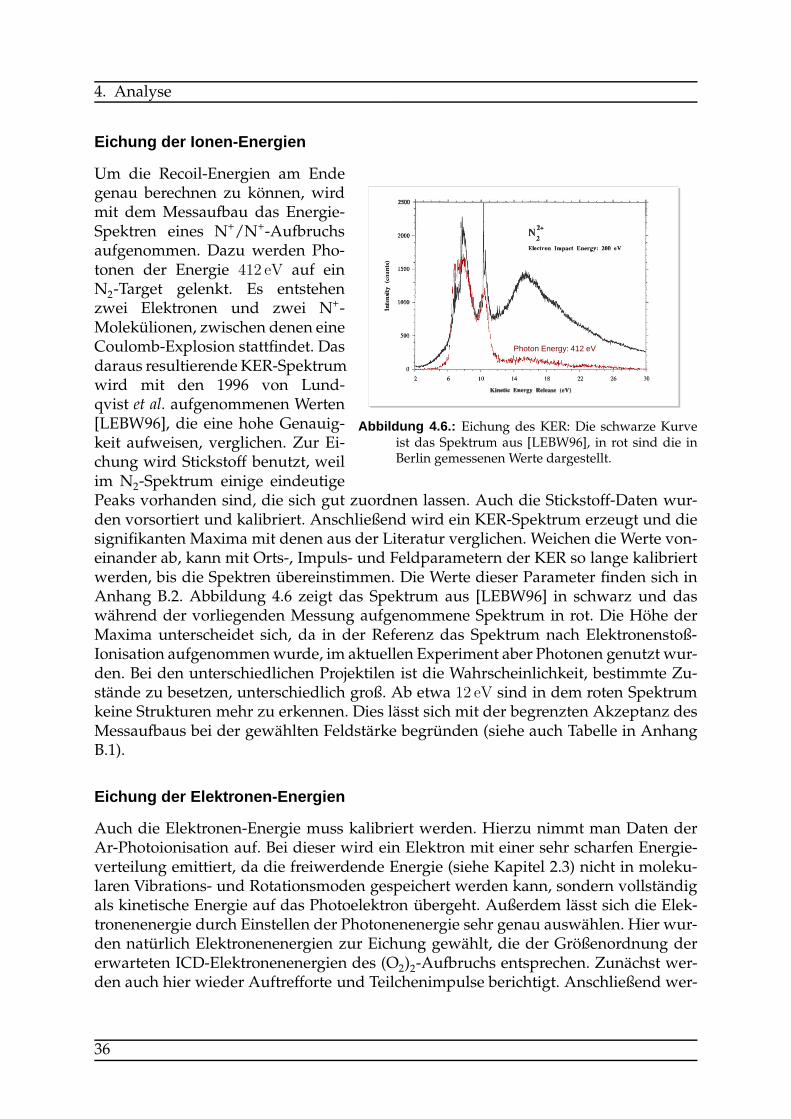
Um die Recoil-Energien am Endegenau berechnen zu können, wirdmit dem Messaufbau das Energie-Spektren eines N+/N+-Aufbruchsaufgenommen. Dazu werden Pho-tonen der Energie 412 eV auf einN2-Target gelenkt. Es entstehenzwei Elektronen und zwei N+-Molekülionen, zwischen denen eineCoulomb-Explosion stattfindet. Dasdaraus resultierende KER-Spektrumwird mit den 1996 von Lund-qvist et al. aufgenommenen Werten[LEBW96], die eine hohe Genauig-keit aufweisen, verglichen. Zur Ei-chung wird Stickstoff benutzt, weilim N2-Spektrum einige eindeutigePeaks vorhanden sind, die sich gut zuordnen lassen. Auch die Stickstoff-Daten wur-den vorsortiert und kalibriert. Anschließend wird ein KER-Spektrum erzeugt und diesignifikanten Maxima mit denen aus der Literatur verglichen. Weichen die Werte von-einander ab, kann mit Orts-, Impuls- und Feldparametern der KER so lange kalibriertwerden, bis die Spektren übereinstimmen. Die Werte dieser Parameter finden sich inAnhang B.2. Abbildung 4.6 zeigt das Spektrum aus [LEBW96] in schwarz und daswährend der vorliegenden Messung aufgenommene Spektrum in rot. Die Höhe derMaxima unterscheidet sich, da in der Referenz das Spektrum nach Elektronenstoß-Ionisation aufgenommenwurde, im aktuellen Experiment aber Photonen genutzt wur-den. Bei den unterschiedlichen Projektilen ist die Wahrscheinlichkeit, bestimmte Zu-stände zu besetzen, unterschiedlich groß. Ab etwa 12 eV sind in dem roten Spektrumkeine Strukturen mehr zu erkennen. Dies lässt sich mit der begrenzten Akzeptanz desMessaufbaus bei der gewählten Feldstärke begründen (siehe auch Tabelle in AnhangB.1).
Eichung der Elektronen-Energien
Auch die Elektronen-Energie muss kalibriert werden. Hierzu nimmt man Daten derAr-Photoionisation auf. Bei dieser wird ein Elektron mit einer sehr scharfen Energie-verteilung emittiert, da die freiwerdende Energie (siehe Kapitel 2.3) nicht in moleku-laren Vibrations- und Rotationsmoden gespeichert werden kann, sondern vollständigals kinetische Energie auf das Photoelektron übergeht. Außerdem lässt sich die Elek-tronenenergie durch Einstellen der Photonenenergie sehr genau auswählen. Hier wur-den natürlich Elektronenenergien zur Eichung gewählt, die der Größenordnung dererwarteten ICD-Elektronenenergien des (O2)2-Aufbruchs entsprechen. Zunächst wer-den auch hier wieder Auftrefforte und Teilchenimpulse berichtigt. Anschließend wer-
36

4.2. Auswertung
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8 10
0.5
1
1.5
2
2.5
3
0
5
10
15
20
25EE
lektr
on [e
V]
Kosinus (ϑ)
a)
φ [°]-150 -100 -50 0 50 100 150
0
0.5
1
1.5
2
2.5
3
0
2
4
6
8
10
12
14
16
18
EE
lektr
on [e
V]
b)
EElektron [eV]0 1 2 3 4 5 6 7 8 9 10
0
10
20
30
40
50
60
70
80
co
un
ts
c)
Kosinus (ϑ)
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8 1
EE
lektr
on
1 [e
V]
0
0.5
1
1.5
2
2.5
3
0
5
10
15
20
25
30
d)
-150 -100 -50 0 50 100 150
EE
lektr
on [e
V]
0
0.5
1
1.5
2
2.5
3
0
2
4
6
8
10
12
14
16
18
20
φ [°]
e)
EElektron [eV]0 1 2 3 4 5 6 7 8 9 10
0
20
40
60
80
100
co
un
ts
f)
Abbildung 4.7.: Eichung der Elektronen-Energie: Die obere Reihe zeigt die Daten vor der Eichung, dieuntere nachher. In der ersten Spalte ist die Elektronenenergie gegen cos(ϑ) aufgetragen,in der zweiten gegen ϕ und in der dritten als 1D-Histogramm.
37

4. Analyse
den die Elektronenenergien berechnet und gegen ϕ und cos(ϑ) (siehe Abbildung 3.4auf Seite 21) aufgetragen. Die Energie des Elektrons sollte eine schmale Verteilung auf-weisen und sich in Abhängigkeit dieser Winkel nicht ändern. Durch Anpassen derParameter für Ortsnullpunkt, elektrisches Feld und die Flugzeitmitte kann die Ver-teilung so angepasst werden, dass dies auch in den aufgenommenen Daten stimmt.Abbildung 4.7 zeigt cos(ϑ), ϕ und die Energie des Elektrons vor (a, b, c) und nach (d,e, f) der Eichung.
Überprüfung der Energie-Eichung
Wie in Kapitel 4.2.3 kann nun auch das KER-Spektrum des O+/O+-Aufbruchs mit Li-teraturwerten [LEB+96] verglichen werden, um die KER-Eichung zu überprüfen. DieEichung wurde nicht direkt mit dem O2-KER durchgeführt, da in dem mit dem an-gelegten elektrischen Feld messbaren niedrigen KER-Bereich nur wenige eindeutigeEnergie-Peaks zu finden sind. Der Vergleich zeigt allerdings, dass die Eichung gelun-gen ist, die Verteilungen weisen deutliche Übereinstimmungen auf.
4.2.4. Auswählen von Impulsbereichen
Wie man in Abbildung 4.3 sehen kann, sind die O+2/O
+2 -Aufbrüche von Rauschen un-
terlegt. Damit die ICD-Signatur gefunden werden kann, müssen die wirklichen Ereig-nisse von falschen Koinzidenzen und nicht relevanten Signalen getrennt werden. Dieslässt sich durch Auswählen von sinnvollen Bereichen in den Impulsverteilungen errei-chen.Für die Summe der Recoil-Impulse ist aufgrund der Impulserhaltung bei jedem ein-
zelnen Aufbruch nur ein bestimmter Wertebereich erlaubt. Die bei der Reaktion frei-werdende Energie teilt sich auf die Recoil-Ionen und die Elektronen auf. Da die Re-coilionen genau entgegengesetzt voneinander wegfliegen, wäre ihre Impulssummegleich Null, wenn die Elektronen keinen Impuls tragen würden. Die Recoil-Impulsekönnen sich also nur höchstens um den Elektronenimpuls unterscheiden. Das ICD-Elektron ist sehr langsam und trägt daher nur einen vernachlässigbaren Impuls, dasAugerelektron allerdings führt zu einer Verbreiterung der Impulssumme der Recoi-lionen. Außerdem führt auch die begrenzte experimentelle Auflösung zu einer Ver-breiterung der Impulssummenverteilung. Die Impulsdifferenz ist proportional zumKER (vgl. Formel 4.10). Die Bereiche, die die Impulssummen in x-, y- und z-Richtungannehmen dürfen, können also bis auf die durch Augerelektronen-Impuls und Auf-lösung gegebene Breite begrenzt werden. Bei Abbildung 4.8 ist zu beachten, dass diedargestellten Histogramme aus Gründen der besseren Vergleichbarkeit nur einen Aus-schnitt aus den Impulsverteilungen darstellen. In den Bildern a) und c) befindet sichein Großteil der Statistik außerhalb des dargestellten Bereichs, die relevanten Datenliegen allerdings innerhalb dieser Ausschnitte. Auf der Horizontalen, bei etwa einerImpulsdifferenz von etwa 0, und auf der Diagonalen, die in dem dargestellten Aus-schnitt zu erkennen ist und über den Rand hinaus weiterverläuft befinden sich diemeisten nicht relevanten Daten. Diese werden durch Anwenden der Bedingungen
38
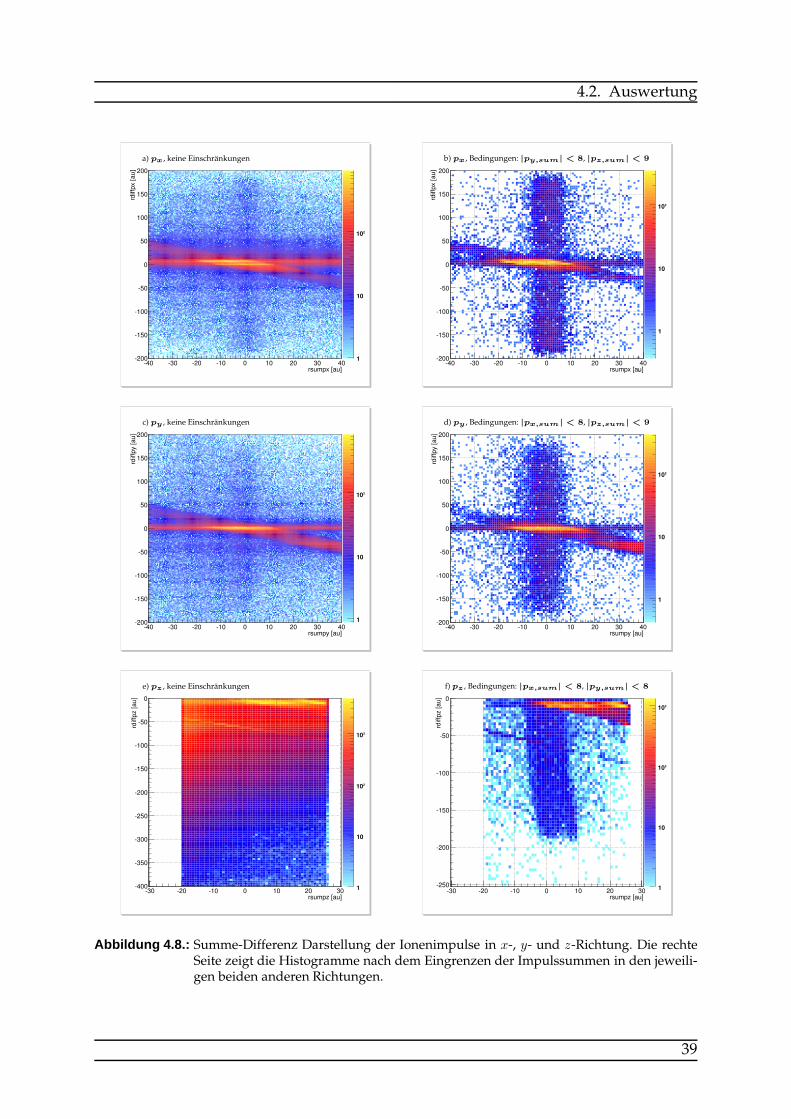
4.2. Auswertung
rsumpx [au]-40 -30 -20 -10 0 10 20 30 40
rdiffp
x[a
u]
-200
-150
-100
-50
0
50
100
150
200
1
10
102
rsumpy [au]-40 -30 -20 -10 0 10 20 30 40
rdiffp
y[a
u]
-200
-150
-100
-50
0
50
100
150
200
1
10
102
rsumpz [au]-30 -20 -10 0 10 20 30
rdiffp
z[a
u]
-400
-350
-300
-250
-200
-150
-100
-50
0
1
10
102
103
rsumpx [au.. ]-40 -30 -20 -10 0 10 20 30 40
rdiffp
x[a
u]
-200
-150
-100
-50
0
50
100
150
200
1
10
102
rsumpy [au]-40 -30 -20 -10 0 10 20 30 40
rdiffp
y[a
u]
-200
-150
-100
-50
0
50
100
150
200
1
10
102
rsumpz [au]-30 -20 -10 0 10 20 30
rdiffp
z[a
u]
-250
-200
-150
-100
-50
0
1
10
102
103
a) px , keine Einschränkungen b) px , Bedingungen: |py,sum| < 8, |pz,sum| < 9
c) py , keine Einschränkungen d) py , Bedingungen: |px,sum| < 8, |pz,sum| < 9
e) pz , keine Einschränkungen f) pz , Bedingungen: |px,sum| < 8, |py,sum| < 8
Abbildung 4.8.: Summe-Differenz Darstellung der Ionenimpulse in x-, y- und z-Richtung. Die rechteSeite zeigt die Histogramme nach dem Eingrenzen der Impulssummen in den jeweili-gen beiden anderen Richtungen.
39

4. Analyse
rsumpr [au]0 5 10 15 20 25 30 35 40 45 50
KE
R [e
V]
0
5
10
15
20
25
1
10
102
103
Abbildung 4.9.: Werte für die Relativimpulssum-me, die größer als 30 a.u. sind, wer-den nicht berücksichtigt.
nicht weiter berücksichtigt, was zu einersehr großen Veränderung von e) nach f)durch die Anwendung der Summenim-pulsbedingungen in x- und y-Richtungführt. Die scharfen Begrenzungen derFlugzeitimpulssumme in negativer undpositiver Richtung in den Abbildungene) und f) ist auf das Ausschneiden derrelevanten Ereignisse während des Vor-sortierens zurückzuführen (vgl. Kapitel4.1.3).
Trägt man die Summe der Relativim-pulse gegen den KER auf (Abbildung4.9), so erkennt man, dass im Bereichhoher Relativimpulssummen und kleinerKERs viele Ereignisse liegen. Diese nichtzum ICD-Prozess gehörigen Events wer-den mit einer Einschränkung der Relati-vimpulssumme auf Werte ≤ 30 a.u. abge-schnitten.
4.2.5. Korrigieren der Impulsverteilungen
Da ein Spektrometer im wirklichen Messaufbau nie so genau realisiert werden kannwie in der Simulation berechnet, kommt es auch zu geringen Abweichungen in derFeldgeometrie. Diese Verzerrungen der Feldlinien, vor allem in dem Bereich um dieReaktionszone, in der große Potentialdifferenzen auf kleinen Abständen herrschen(vgl. Kapitel 3.2.2), führen zu leicht veränderten Teilchenbahnen. Da die Teilchen nunnach einer verkürzten oder verlängerten Flugzeit auf einem anderen Ort auf dem De-tektor auftreffen, ergeben sich Fehler in den anschließenden Impulsberechnungen. Dasich das elektrische Feld im Spektrometer über die Messung aber nicht ändert, kön-nen diese Abweichungen in der Analyse korrigiert werden. Erwartungsgemäß solltendie Impulssummen der Recoil-Ionen in der Summe-Differenz-Darstellung (Abbildung4.10) auf einem vertikalen Streifen mit dem Mittelpunkt bei der Impulssumme 0 ab-gebildet sein. Dies ergibt sich aus der Impulserhaltung der Reaktionsprodukte. DieAusdehnung des Streifens auf der Impulssummen-Achse ergibt sich aus dem Auger-elektronenimpuls und der experimentellen Auflösung (siehe Abschnitt 4.2.4). In Abbil-dung 4.10 sieht man, dass dieser Streifen bei steigendem Impulsdifferenzbetrag zu grö-ßeren Impulssummen gebogen ist. Dieser Abbildungsfehler lässt sich durch Addiereneiner Korrekturfunktion auf den Summenimpuls der Ionen eliminieren. Die verwen-dete Korrekturfunktion ist linear:
pz,sum,corr = −0,0357 · pz,diff − 2,2801 a.u. (4.12)
40
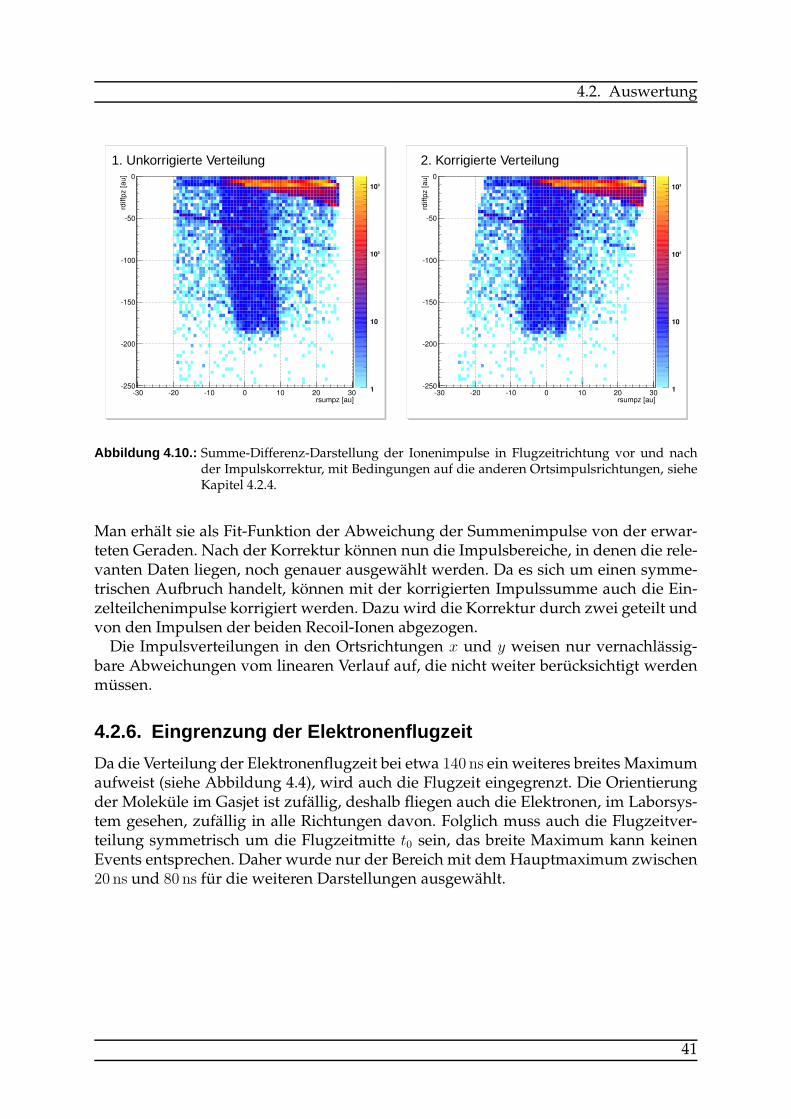
4.2. Auswertung
rsumpz [au]-30 -20 -10 0 10 20 30
rdiffp
z[a
u]
-250
-200
-150
-100
-50
0
1
10
102
103
rsumpz [au]-30 -20 -10 0 10 20 30
rdiffp
z[a
u]
-250
-200
-150
-100
-50
0
1
10
102
103
1. Unkorrigierte Verteilung 2. Korrigierte Verteilung
Abbildung 4.10.: Summe-Differenz-Darstellung der Ionenimpulse in Flugzeitrichtung vor und nachder Impulskorrektur, mit Bedingungen auf die anderen Ortsimpulsrichtungen, sieheKapitel 4.2.4.
Man erhält sie als Fit-Funktion der Abweichung der Summenimpulse von der erwar-teten Geraden. Nach der Korrektur können nun die Impulsbereiche, in denen die rele-vanten Daten liegen, noch genauer ausgewählt werden. Da es sich um einen symme-trischen Aufbruch handelt, können mit der korrigierten Impulssumme auch die Ein-zelteilchenimpulse korrigiert werden. Dazu wird die Korrektur durch zwei geteilt undvon den Impulsen der beiden Recoil-Ionen abgezogen.Die Impulsverteilungen in den Ortsrichtungen x und y weisen nur vernachlässig-
bare Abweichungen vom linearen Verlauf auf, die nicht weiter berücksichtigt werdenmüssen.
4.2.6. Eingrenzung der Elektronenflugzeit
Da die Verteilung der Elektronenflugzeit bei etwa 140 ns ein weiteres breites Maximumaufweist (siehe Abbildung 4.4), wird auch die Flugzeit eingegrenzt. Die Orientierungder Moleküle im Gasjet ist zufällig, deshalb fliegen auch die Elektronen, im Laborsys-tem gesehen, zufällig in alle Richtungen davon. Folglich muss auch die Flugzeitver-teilung symmetrisch um die Flugzeitmitte t0 sein, das breite Maximum kann keinenEvents entsprechen. Daher wurde nur der Bereich mit dem Hauptmaximum zwischen20 ns und 80 ns für die weiteren Darstellungen ausgewählt.
41

4. Analyse
4.3. Fehlerbetrachtung
Schon während der Simulation des Spektrometers wurde eine Abschätzung der zuerreichenden experimentellen Auflösung erstellt (vgl. Anhang B.1). Da allerdings beikeiner theoretischen Überlegung alle Fehlerquellenmit einbezogenwerden können, istdie erreichte experimentelle Auflösung etwas schlechter als die durch die Simulationabgeschätzte. Während der Durchführung und auch bei der anschließenden Auswer-tung der Daten wurden viele Schritte durchgeführt, um möglichst genaue Messergeb-nisse zu erzielen:Schon vor der eigentlichen Messung wurde die Photonenenergie kalibriert. Das Jet-
system mit Expansion und Skimmern wird verwendet, um die Geschwindigkeitsver-teilung der Gasmoleküle soweit wie möglich zu verkleinern. Von den Detektoren wer-den redundante Signale aufgenommen, um zusätzliche Absicherungen zu erhaltenund die nachgeschaltete Elektronik hat unter anderem den Zweck, die analogen Si-gnale so genau wie möglich zu erfassen.Während des Vorsortierens und in der Analyse der Daten werden die Detektor-
Zeitsummen kalibriert, Ortsverteilungen korrigiert und verschoben. Die Impulse inFlugzeitrichtung wurden korrigiert und relevante Daten wurden durch Bedingungenbezüglich der Flugzeit und der Impulserhaltung ausgewählt. Außerdem fand eine Ei-chung der Ionen- und Elektronenenergien statt.Betrachtet man nun 1D-Histogramme wie etwa das des KERs in Abbildung 4.6, so
kann man aus der Breite der Maxima in der Verteilung und dem Vergleich mit denhochaufgelösten Literaturwerten die experimentelle Auflösung zum Beispiel für denKER abschätzen: Es können Maxima der Breite von etwa 0,2 eV aufgelöst werden.Zu beachten ist dabei aber, dass die Verbreiterung von Linien im Energiespektrum
allerdings nicht nur auf die experimentelle Auflösung zurückzuführen ist. Auch diebegrenzte Lebensdauer der Zustände kannmit der HeisenbergschenUnschärferelationin eine Verbreiterung der Maxima übersetzt werden (vgl. [Dem10], S. 102, 106):
∆x ·∆p ≥ h̄ und ∆E ·∆t ≥ h̄
In denMolekülspektren wie z. B. Abbildung 5.2 (KER gegen Elektronenenergie) füh-ren auch molekulare Rotations- und Vibrationsmoden zu einer weniger diskreten, un-schärferen Verteilung.
42
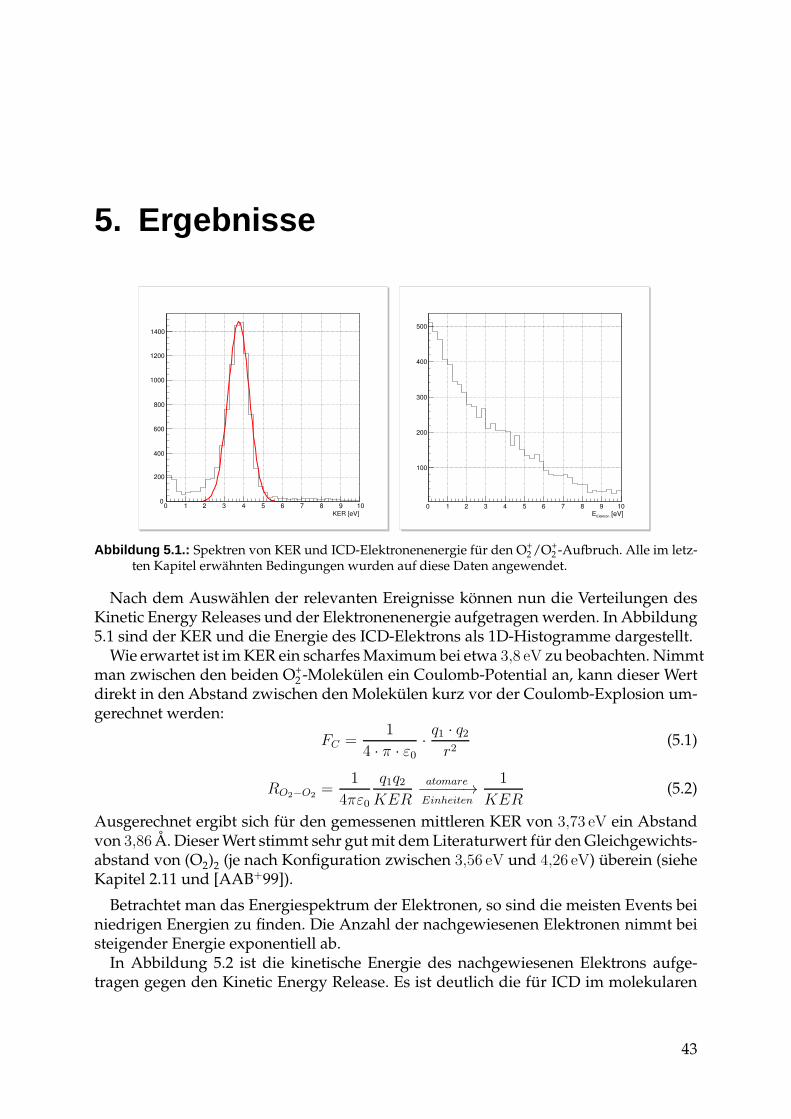
5. Ergebnisse
0 1 2 3 4 5 6 7 8 9 10
200
400
600
800
1000
1200
1400
0
KER [eV] EElektron [eV]0 1 2 3 4 5 6 7 8 9 10
100
200
300
400
500
Abbildung 5.1.: Spektren von KER und ICD-Elektronenenergie für den O+2/O
+2 -Aufbruch. Alle im letz-
ten Kapitel erwähnten Bedingungen wurden auf diese Daten angewendet.
Nach dem Auswählen der relevanten Ereignisse können nun die Verteilungen desKinetic Energy Releases und der Elektronenenergie aufgetragen werden. In Abbildung5.1 sind der KER und die Energie des ICD-Elektrons als 1D-Histogramme dargestellt.Wie erwartet ist imKER ein scharfesMaximum bei etwa 3,8 eV zu beobachten. Nimmt
man zwischen den beiden O+2 -Molekülen ein Coulomb-Potential an, kann dieser Wert
direkt in den Abstand zwischen den Molekülen kurz vor der Coulomb-Explosion um-gerechnet werden:
FC =1
4 · π · ε0· q1 · q2
r2(5.1)
RO2−O2=
1
4πε0
q1q2
KER
atomare−−−−−→Einheiten
1
KER(5.2)
Ausgerechnet ergibt sich für den gemessenen mittleren KER von 3,73 eV ein Abstandvon 3,86Å.DieserWert stimmt sehr gutmit demLiteraturwert für denGleichgewichts-abstand von (O2)2 (je nach Konfiguration zwischen 3,56 eV und 4,26 eV) überein (sieheKapitel 2.11 und [AAB+99]).
Betrachtet man das Energiespektrum der Elektronen, so sind die meisten Events beiniedrigen Energien zu finden. Die Anzahl der nachgewiesenen Elektronen nimmt beisteigender Energie exponentiell ab.In Abbildung 5.2 ist die kinetische Energie des nachgewiesenen Elektrons aufge-
tragen gegen den Kinetic Energy Release. Es ist deutlich die für ICD im molekularen
43
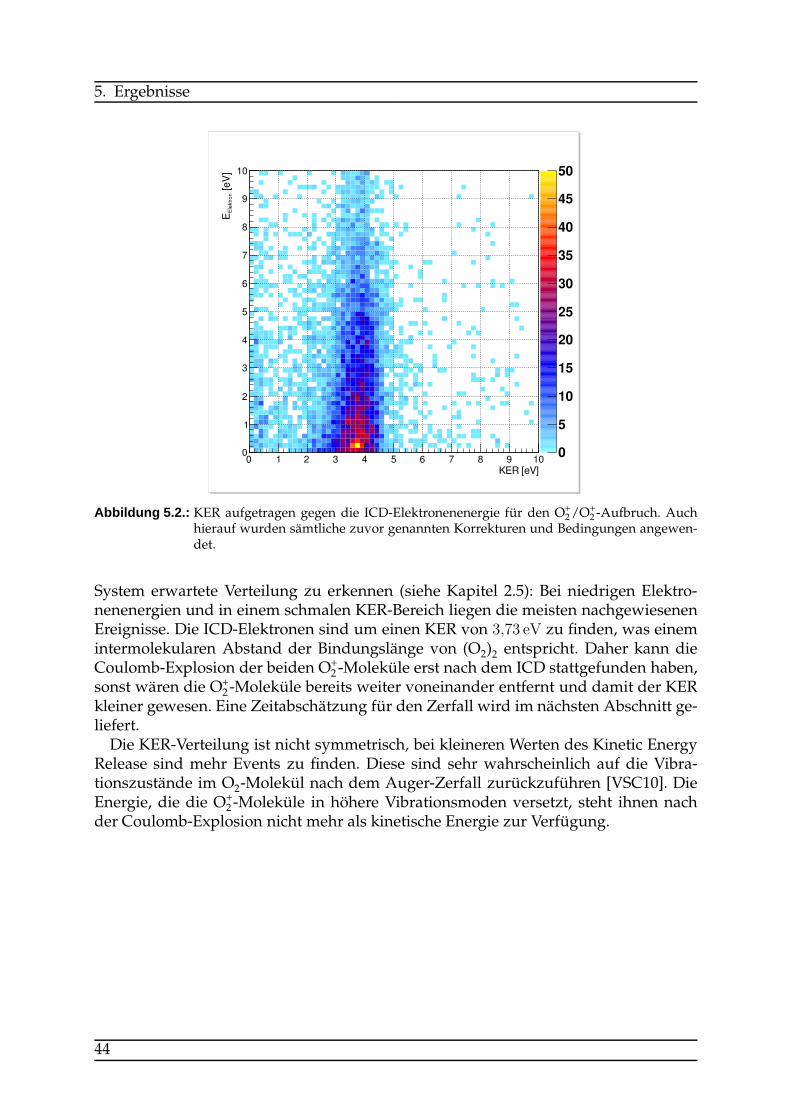
5. Ergebnisse
KER [eV]0 1 2 3 4 5 6 7 8 9 10
EE
lektr
on [e
V]
0
1
2
3
4
5
6
7
8
9
10
0
5
10
15
20
25
30
35
40
45
50
Abbildung 5.2.: KER aufgetragen gegen die ICD-Elektronenenergie für den O+2/O
+2 -Aufbruch. Auch
hierauf wurden sämtliche zuvor genannten Korrekturen und Bedingungen angewen-det.
System erwartete Verteilung zu erkennen (siehe Kapitel 2.5): Bei niedrigen Elektro-nenenergien und in einem schmalen KER-Bereich liegen die meisten nachgewiesenenEreignisse. Die ICD-Elektronen sind um einen KER von 3,73 eV zu finden, was einemintermolekularen Abstand der Bindungslänge von (O2)2 entspricht. Daher kann dieCoulomb-Explosion der beiden O+
2 -Moleküle erst nach dem ICD stattgefunden haben,sonst wären die O+
2 -Moleküle bereits weiter voneinander entfernt und damit der KERkleiner gewesen. Eine Zeitabschätzung für den Zerfall wird im nächsten Abschnitt ge-liefert.Die KER-Verteilung ist nicht symmetrisch, bei kleineren Werten des Kinetic Energy
Release sind mehr Events zu finden. Diese sind sehr wahrscheinlich auf die Vibra-tionszustände im O2-Molekül nach dem Auger-Zerfall zurückzuführen [VSC10]. DieEnergie, die die O+
2 -Moleküle in höhere Vibrationsmoden versetzt, steht ihnen nachder Coulomb-Explosion nicht mehr als kinetische Energie zur Verfügung.
44
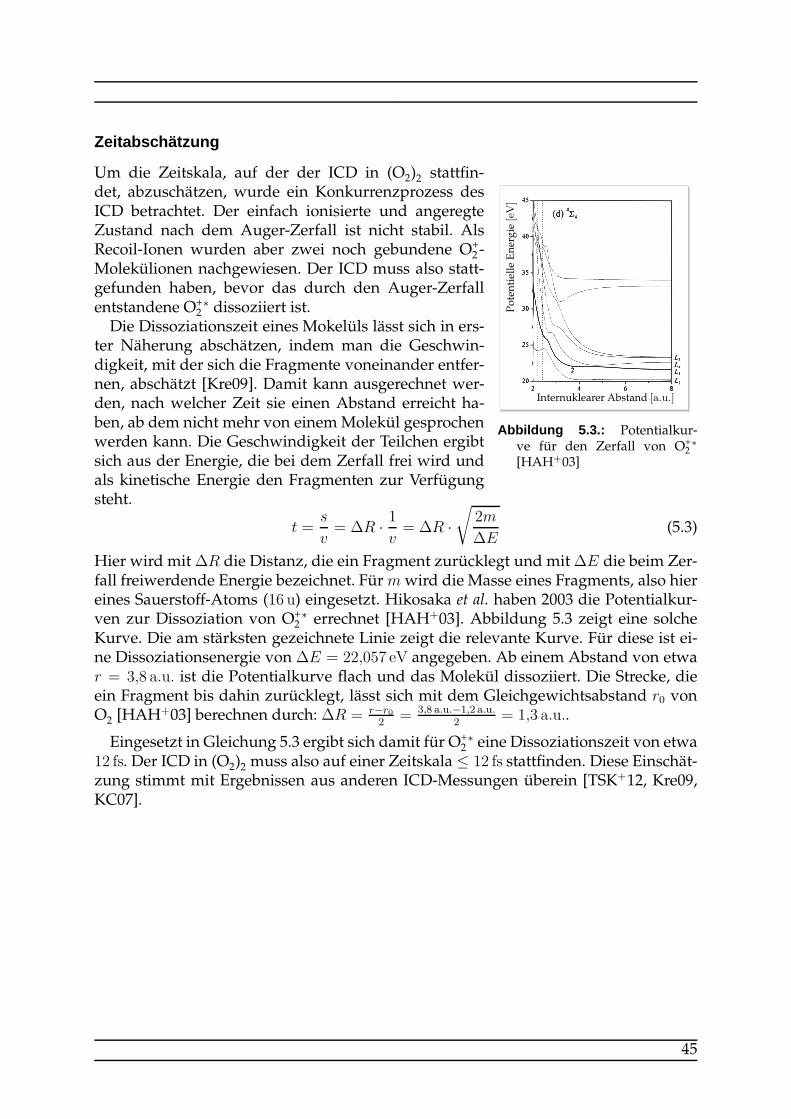
Zeitabschätzung
Internuklearer Abstand [a.u.]
Potentielle
Ene
rgie
[eV]
Abbildung 5.3.: Potentialkur-ve für den Zerfall von O+
2∗
[HAH+03]
Um die Zeitskala, auf der der ICD in (O2)2 stattfin-det, abzuschätzen, wurde ein Konkurrenzprozess desICD betrachtet. Der einfach ionisierte und angeregteZustand nach dem Auger-Zerfall ist nicht stabil. AlsRecoil-Ionen wurden aber zwei noch gebundene O+
2 -Molekülionen nachgewiesen. Der ICD muss also statt-gefunden haben, bevor das durch den Auger-Zerfallentstandene O+
2∗ dissoziiert ist.
Die Dissoziationszeit eines Mokelüls lässt sich in ers-ter Näherung abschätzen, indem man die Geschwin-digkeit, mit der sich die Fragmente voneinander entfer-nen, abschätzt [Kre09]. Damit kann ausgerechnet wer-den, nach welcher Zeit sie einen Abstand erreicht ha-ben, ab dem nicht mehr von einemMolekül gesprochenwerden kann. Die Geschwindigkeit der Teilchen ergibtsich aus der Energie, die bei dem Zerfall frei wird undals kinetische Energie den Fragmenten zur Verfügungsteht.
t =s
v= ∆R · 1
v= ∆R ·
√
2m
∆E(5.3)
Hier wird mit ∆R die Distanz, die ein Fragment zurücklegt und mit∆E die beim Zer-fall freiwerdende Energie bezeichnet. Fürmwird die Masse eines Fragments, also hiereines Sauerstoff-Atoms (16 u) eingesetzt. Hikosaka et al. haben 2003 die Potentialkur-ven zur Dissoziation von O+
2∗ errechnet [HAH+03]. Abbildung 5.3 zeigt eine solche
Kurve. Die am stärksten gezeichnete Linie zeigt die relevante Kurve. Für diese ist ei-ne Dissoziationsenergie von ∆E = 22,057 eV angegeben. Ab einem Abstand von etwar = 3,8 a.u. ist die Potentialkurve flach und das Molekül dissoziiert. Die Strecke, dieein Fragment bis dahin zurücklegt, lässt sich mit dem Gleichgewichtsabstand r0 vonO2 [HAH+03] berechnen durch: ∆R = r−r0
2= 3,8 a.u.−1,2 a.u.
2= 1,3 a.u..
Eingesetzt in Gleichung 5.3 ergibt sich damit für O+2∗ eine Dissoziationszeit von etwa
12 fs. Der ICD in (O2)2 muss also auf einer Zeitskala ≤ 12 fs stattfinden. Diese Einschät-zung stimmt mit Ergebnissen aus anderen ICD-Messungen überein [TSK+12, Kre09,KC07].
45

5. Ergebnisse
EElektron [eV]
100
200
300
400
500
600
0 2 4 6 8 10 12 14 16 18 200
100
200
300
400
500
600
700
e1_ke,e2_ke 0
counts
Abbildung 5.4.: Vergleich der Energien der registrierten Elektronen für RA-ICD (schwarz) und ICDnach Photoionisation (rot) bei einer Photonenenergie von 46 eV.
Ähnliche Messungen
An der Advanced Light Source in Berkeley wurden von F. Sturm et al. Messungen zumICD in (O2)2 nach Photoionisation durchgeführt. Hier wurden die O2-Moleküle mitPhotonen der Energie von 46 eV innerschalenionisiert, die Abregung erfolgte über ICD.Im Gegensatz zumAuger-Elektron beim ICD nach resonanter Auger-Anregung ist dasPhotoelektron bei diesem Prozess niederenergetisch. Es trägt eine kinetische Energie inder Größenordnung einiger eV, ähnlich wie das ICD-Elektron.In das 1D-Histogramm in Abbildung 5.4 sind die Energiespektren der aufgenomme-
nen Elektronen von denMessungen in Berlin (schwarzer Graph) und in Berkeley (roterGraph) eingezeichnet. Bei dem ICD nach Photoionisation (rot) lassen sich Photo- undICD-Elektron im Energiespektrum nicht unterscheiden. Vergleicht man die Daten abermit dem schwarzen Graph für den RA-ICD, bei dem nur das ICD-Elektron nachgewie-sen wurde, so kann man die Daten ab etwa 5 eV Elektronenenergie dem Photoelektronzuordnen.
46

6. Ausblick
Ausschluss des Knock-Off-Prozesses
Um den intermolekularen coulombischen Zerfall eindeutig identifizieren zu können,muss der in Kapitel 2.5.3 schon angeführte Knock-Off-Prozess ausgeschlossen wer-den. Dazu ist es nötig, die Impulsdaten des Auger-Elektrons auszurechnen und des-sen Winkelverteilung zu betrachten. Ist sie isotrop zur Molekülachse der Recoil-Ionen,so ist ICD der wahrscheinlichste Zerfallskanal. Zeigt sie allerdings Ausprägungen inRichtung der O2-Moleküle, ist es wahrscheinlicher, dass das Auger-Elektron ein Se-kundärelektron aus dem benachbarten Molekül herausschlägt.
Spektrometer
Um in Zukunft auch größere Ionenimpulse und -energien nachweisen zu können, sindnoch weitere Anpassungen am Spektrometer durchzuführen. Es wäre wünschenswert,die Feldgeometrie im Spektrometer asymmetrisch zu gestalten, also das Feld auf derIonenseite zu verstärken. Bisher war es allerdings nicht möglich, das Feld auf der Io-nenseite so zu verändern, dass die Elektronenseite nicht beeinflusst wird. Um die guteFokussierung auf der Elektronenseite zu erhalten, wurde daher im vorliegenden Ex-periment die symmetrische Feldgeometrie gewählt.
Anwendungen
In der vorliegenden Arbeit wurde ICD im Modellsystem (O2)2 nachgewiesen. Bei die-sem sehr effizienten und schnellen Prozess werden langsame Elektronen erzeugt. Mitder gewählten Photonenenergie konnte eine schmale, molekülspezifische Resonanzzur Anregung ausgewählt werden, nach der der ICD-Prozess abläuft.Eswurde bereits nachgewiesen, dass langsame Elektronen, wie sie beim ICD-Prozess
erzeugtwerden, DNA-Einfach- undDoppelstrangbrüche induzieren können [BCH+00,HBC+03]. Mittels ICD nach resonanter Auger-Anregung kann gesteuert werden, inwelchen Molekülen genau diese Elektronen erzeugt werden, da die Resonanz mole-külspezifisch ist. So könnte etwa in einem größeren molekularen System genau eineStelle für die Produktion der langsamen Elektronen und auch deren Energie ausge-wählt werden [GKKC12]. Dies bietet die Möglichkeit der Anwendung beispielsweisein der Krebstherapie [SS12].
47


A. Atomare Einheiten
Beim Rechnen in atomaren Maßstäben ist es sinnvoll, die verwendeten Einheiten derGrößenordnung anzupassen. Dazu werden die bekannten Größen in SI-Einheiten aufatomare Größen (a.u.) normiert, etwa den Bohrschen Radius a0 oder die Elementarla-dung e. Die folgende Tabelle gibt einen Überblick über grundlegende und abgeleiteteatomare Größen und ihre Umrechnungsfaktoren in SI-Einheiten (vgl. [Mes06], S. 734f).Für die Konstanten wurden die aktuellen CODATA-Werte1 verwendet.
Name a.u.-Wert Formel SI-Wert
Länge 1 a0 0,5292 · 10−10m
Masse 1 me 9,1094 · 10−31 kg
Lichtgeschwindigkeit 137 c = 1α
2,9979 · 108 ms
Geschwindigkeit 1 v0 =a0Eh
h̄2,1877 · 106 m
s
Impuls 1 p = mev0 1,9929 · 10−24 kg·ms
Drehimpuls 1 h̄ 1,0546 · 10−34 J · s
Zeit 1 t = h̄Eh
2,4189 · 10−17 s
Energie 1 Eh = h̄2
a20me
4,3597 · 10−18 J
Ladung 1 e 1,6022 · 10−19C
1http://physics.nist.gov/cuu/Constants/index.html
49

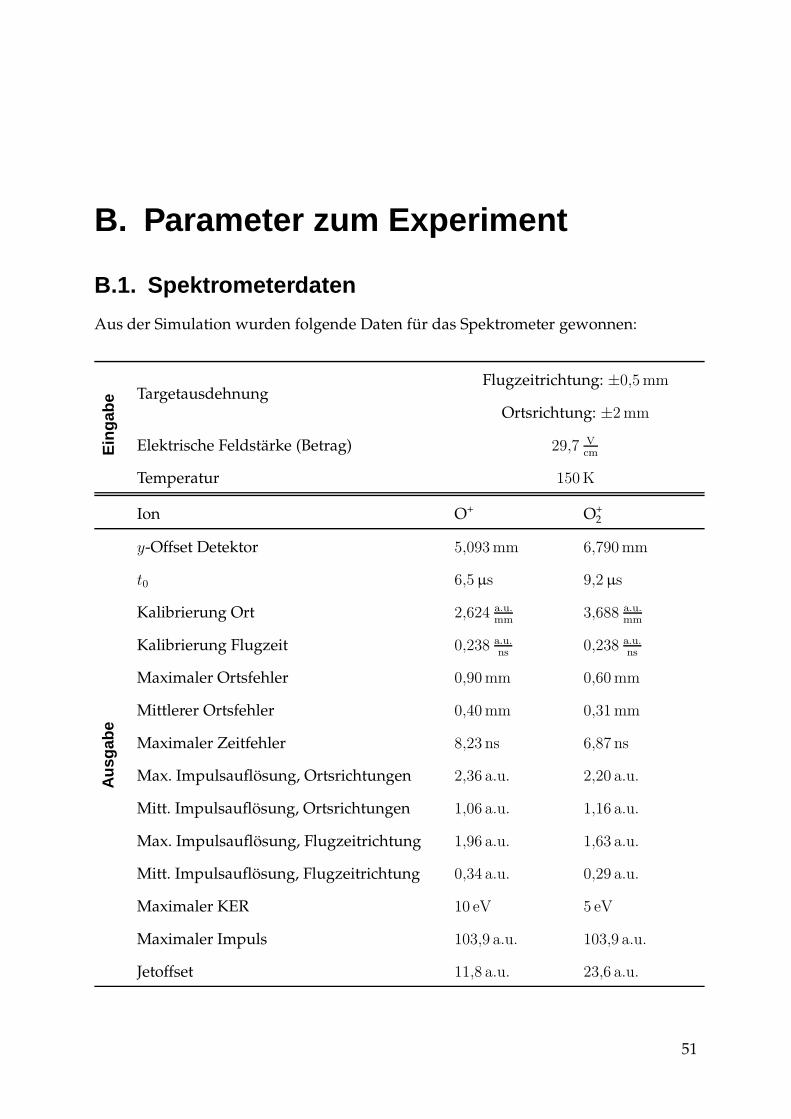
B. Parameter zum Experiment
B.1. Spektrometerdaten
Aus der Simulation wurden folgende Daten für das Spektrometer gewonnen:
Ein
gabe
TargetausdehnungFlugzeitrichtung: ±0,5mm
Ortsrichtung: ±2mm
Elektrische Feldstärke (Betrag) 29,7 Vcm
Temperatur 150K
Ion O+ O+2
Aus
gabe
y-Offset Detektor 5,093mm 6,790mm
t0 6,5µs 9,2µs
Kalibrierung Ort 2,624 a.u.mm
3,688 a.u.mm
Kalibrierung Flugzeit 0,238 a.u.ns
0,238 a.u.ns
Maximaler Ortsfehler 0,90mm 0,60mm
Mittlerer Ortsfehler 0,40mm 0,31mm
Maximaler Zeitfehler 8,23 ns 6,87 ns
Max. Impulsauflösung, Ortsrichtungen 2,36 a.u. 2,20 a.u.
Mitt. Impulsauflösung, Ortsrichtungen 1,06 a.u. 1,16 a.u.
Max. Impulsauflösung, Flugzeitrichtung 1,96 a.u. 1,63 a.u.
Mitt. Impulsauflösung, Flugzeitrichtung 0,34 a.u. 0,29 a.u.
Maximaler KER 10 eV 5 eV
Maximaler Impuls 103,9 a.u. 103,9 a.u.
Jetoffset 11,8 a.u. 23,6 a.u.
51

B. Parameter zum Experiment
B.2. Daten zum Experiment
Pho
tone
nstr
ahl Polarisation linear
Beamline-Offset −0,95 eV
Beamline-Offset ab dem 20.02.12 −0,45 eV
Strahlstrom der Elektronen im Ring ∼ 23,5mA – ∼ 5mA
Gas
-Jet
Temperatur 300K
Druck ∼ 10 bar
Verhältnis O+2/O
+2 :O
+/O+ ∼ 1%
Vak
uum
Expansionskammer Größenordnung 10−4mbar
Zweite Jetstufe Größenordnung 10−5mbar
Targetzone Größenordnung 10−9mbar
Jetdump Größenordnung 10−8mbar
Ana
lyse
para
met
er
Flugzeitgrenzen O+2/O
+2 -Aufbruch tof -Summe: 19250 ns, 19750 ns
tof -Differenz: 0 ns, −2000 ns
Rec
oilio
nen
x-Verschiebung 0,96mm
y-Verschiebung (Jetoffset) 8,28mm
Flugzeitmittelpunkt 9333 ns
Impulsfaktor Ortsrichtungen x, y 1,34
Feldparameter Eichung 0,277 a.u.ns
Ele
ktro
nen
x-Verschiebung −1,32mm
y-Verschiebung −2,622mm
Rotation Detektor −50 ◦
Flugzeitmittelpunkt 33,18 ns
Impulsfaktor Ortsrichtungen x, y 1,32
Feldparameter Eichung 0,27 a.u.ns
52

Abbildungsverzeichnis
2.1. Sauerstoff im Bohrschen Atommodell . . . . . . . . . . . . . . . . . . . . 72.2. Absorptionsspektrum des Wasserstoffs . . . . . . . . . . . . . . . . . . . 82.3. Orbitale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92.4. Photoionisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102.5. Photoabsorptionsquerschnitt Gold . . . . . . . . . . . . . . . . . . . . . . 112.6. Auger-Effekt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.7. ICD-Schema . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132.8. ICD in Ne- und in CO-Dimeren . . . . . . . . . . . . . . . . . . . . . . . . 142.9. ICD nach Resonanter Auger-Anregung . . . . . . . . . . . . . . . . . . . 152.10. Knock-Off . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162.11. Das (O2)2-Molekül . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172.12. Schematischer Ablauf der Reaktion . . . . . . . . . . . . . . . . . . . . . . 18
3.1. Abstrahlcharakteristik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 193.2. BESSY II . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.3. Undulator . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.4. Messaufbau, Koordinatensystem . . . . . . . . . . . . . . . . . . . . . . . 213.5. Gasjet . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.6. Jetkurve . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.7. Simulation der 3D-Fokussierung . . . . . . . . . . . . . . . . . . . . . . . 243.8. Microchannel Plate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263.9. Delay-Line-Anode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
4.1. Zeitsummenkorrektur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 304.2. Reconstruction flags . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314.3. PiPiCo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 324.4. Verteilung der Elektronenflugzeit . . . . . . . . . . . . . . . . . . . . . . . 344.5. Eichung der Photonenenergien . . . . . . . . . . . . . . . . . . . . . . . . 354.6. KER-Eichung mit N2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364.7. Eichung der Elektronen-Energie . . . . . . . . . . . . . . . . . . . . . . . . 374.8. Summe-Differenz-Darstellung der Recoil-Impulse . . . . . . . . . . . . . 394.9. Beschränkung der Relativimpulssumme . . . . . . . . . . . . . . . . . . . 404.10. Recoil Summe-Differenz-Darstellung . . . . . . . . . . . . . . . . . . . . . 41
5.1. KER und Elektronenenergie . . . . . . . . . . . . . . . . . . . . . . . . . . 435.2. KER gegen Elektronenenergie . . . . . . . . . . . . . . . . . . . . . . . . . 445.3. Potentialkurve O+
2∗ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
5.4. Elektronenenergiespektren . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
53


Literaturverzeichnis
[AAB+99] AQUILANTI, V. ; ASCENZI, D. ; BARTOLOMEI, M. ; CAPPALLETTI, D. ; CA-VALLI, S. ; CASTRO VITORES, M. de ; PIRANI, F.: Molecular Beam Scatte-ring of Aligned Oxygen Molecules. The Nature of the Bond in the O2−O2
Dimer. In: J. Am. Chem. Soc. 121 (1999), Nr. 46
[Aug25] AUGER, P.: Sur les rayons β secondaires produits dans un gaz par desrayons X. In: Comptes rendus 180 (1925), S. 65 – 68
[BCH+00] BOUDAÏFFA, B. ; COULTIER, P. ; HUNTING, D. ; HUELS, M. A. ; SANCHE,L.: Resonant formation of DNA strand breaks by low-energy (3 to 20 eV)electrons. In: Science 287 (2000), Nr. 1658
[BJM+05] BARTH, S. ; JOSHI, S. ; MARBURGER, S. ; ULRICH, V. ; LINDBLAD, A. ; ÖHR-WALL, G. ; BJÖRNEHOLM, O. ; HERGENHAHN, U.: Observation of resonantInteratomic Coulombic Decay in Ne clusters. In: J. Chem. Phys. 122 (2005),Nr. 241102
[Boh13] BOHR, N.: On the Constitution of Atoms and Molecules. In: PhilosophicalMagazine 26 (1913), Nr. 1
[CZT97] CEDERBAUM, L. S. ; ZOBELEY, J. ; TARANTELLI, F.: Giant IntermolecularDecay and Fragmentation of Clusters. In: Phys. Rev. Lett. 79 (1997), Nr. 24
[Dem10] DEMTRÖDER, W.: Experimentalphysik 3 - Atome, Moleküle und Festkörper. 4.Heidelberg : Springer, 2010. – ISBN 978–3–642–03910–2
[Ein05] EINSTEIN, A.: Über einen die Erzeugung und Verwandlung des Lichtesbetreffenden heuristischen Gesichtspunkt. In:Annalen der Physik 17 (1905),S. 132 – 148
[GKKC12] GOKHBERG, K. ; KOLORENC, P. ; KULEFF, A. ; CEDERBAUM, L. S.: A newintermolecular mechanism to selectively drive photoinduced damages.In: Nature, submitted (2012)
[GSS10] GUILLEMIN, R. ; SIMON, M. ; SHIGEMASA, E.: Doppler effect in fragmentautoionization following core-to-valence excitation in O2. In: Phys. Rev. A82 (2010), Nr. 051401
[HAH+03] HIKOSAKA, Y. ; AOTO, T. ; HALL, R. I. ; ITO, K. ; HIRAYAMA, R.: Inner-valence states of O2+ and dissociation dynamics studied by threshold
55

Literaturverzeichnis
photoelectron spectroscopy and a configuration interaction calculation.In: J. Chem. Phys. 119 (2003), Nr. 7693
[HBC+03] HUELS, M. A. ; BOUDAÏFFA, B. ; COULTIER, P. ; HUNTING, D. ; SANCHE,L.: Single, Double, and Multiple Double Strand Breaks Induced in DNAby (3 – 1000 eV) Electrons. In: J. Am. Chem. Soc. 125 (2003), Nr. 15
[Her08] HERTEL, I. V.: Atome, Moleküle und optische Physik 1 - Atomphysik undGrundlagen der Spektroskopie. Heidelberg : Springer, 2008. – ISBN 978–3–540–30613–9
[HJK+10] HAVERMEIER, T. ; JAHNKE, T. ; KREIDI, K. ; WALLAUER, R. ; VOSS, S. ;SCHÖFFLER, M. ; SCHÖSSLER, S. ; FOUCAR, L. ; NEUMANN, N. ; TITZE, J.; SANN, H. ; KÜHNEL, M. ; VOIGTSBERGER, J. ; MALAKZADEH, A. ; SI-SOURAT, N. ; SCHÖLLKOPF, W. ; SCHMIDT-BÖCKING, H. ; GRISENTI, R. E.; DÖRNER, R.: Single Photon Double Ionization of the Helium Dimer. In:Phys. Rev. Lett. 104 (2010), Nr. 153401
[JCS+04] JAHNKE, T. ; CZASCH, A. ; SCHÖFFLER, M. S. ; SCHÖSSLER, S. ; KNAPP,A. ; KÄSZ, M. ; TITZE, J. ; WIMMER, C. ; KREIDI, K. ; GRISENTI, R. E. ;STAUDTE, A. ; JAGUTZKI, O. ; HERGENHAHN, U. ; SCHMIDT-BÖCKING, H.; DÖRNER, R.: Experimental Observation of Interatomic Coulombic Decayin Neon Dimers. In: Phys. Rev. Lett. 93 (2004), Nr. 16
[JCS+07] JAHNKE, T. ; CZACH, A. ; SCHÖFFLER, M. ; SCHÖSSLER, S. ; KÄSZ, M. ;TITZE, J. ; KREIDI, K. ; GRISENTI, R. E. ; STAUDTE, A. ; JAGUTZKI, O. ;SCHMIDT, L. Ph. H. ; SEMENOV, S. K. ; CHEREPKOV, N. A. ; SCHMIDT-BÖCKING, H. ; DÖRNER, R.: Photoelectron and ICD electron angular dis-tributions from fixed-in-space neon dimers. In: J. Phys. B 40 (2007), S. 2597
[JSH+10] JAHNKE, T. ; SANN, H. ; HAVERMEIER, T. ; KREIDI, K. ; STUCK, C. ; ME-CKEL, M. ; SCHÖFFLER, M. ; NEUMANN, N. ; WALLAUER, R. ; VOSS, S. ;CZASCH, A. ; JAGUTZKI, O. ; MALAKZADEH, A. ; AFANEH, F. ; WEBER,Th. ; SCHMIDT-BÖCKING, H. ; DÖRNER, R.: Ultrafast energy transfer bet-ween water molecules. In: Nature Physics 6 (2010), S. 139 – 142
[KC07] KULEFF, A. I. ; CEDERBAUM, L. S.: Tracing Ultrafast Interatomic ElectronicDecay Processes in Real Time and Space. In: Phys. Rev. Lett. 98 (2007), Nr.083201
[KGCA09] KOPELKE, S. ; GOKHBERG, K. ; CEDERBAUM, L. S. ; AVERBUKH, V.: Calcu-lation of resonant interatomic Coulombic decay widths of inner-valence-excited states delocalized due to inversion symmetry. In: J. Chem. Phys.130 (2009), Nr. 144103
[Kre09] KREIDI, Katharina: Untersuchung der Zerfallsmechanismen und der Lokalisie-rung von Vakanzen in Ne2, Fachbereich Physik, Goethe-Universität Frank-furt am Main, Diss., 2009
56

Literaturverzeichnis
[LEB+96] LUNDQVIST, M. ; EDVARDSSON, D. ; BALTZER, P. ; LARSSON, M. ; WANN-BERG, B.: Observation of pedissociation and tunnelling processes inO2+
2 : astudy using Doppler free kinetic energy release spectroscopy and ab initioCI calculations. In: J. Phys. B 29 (1996), Nr. 499
[LEBW96] LUNDQVIST, M. ; EDVARDSSON, D. ; BALTZER, P. ; WANNBERG, B.:Doppler-free kinetic energy release spectrum of N2+
2 . In: J. Phys. B 29(1996), Nr. 1489
[Mes06] MESCHEDE, D.: Gerthsen Physik. 23. Heidelberg : Springer, 2006. – ISBN3–540–25421–8
[MKHM03] MARBURGER, S. ; KUGELER, O. ; HERGENHAHN, U. ; MÖLLER, T.: Expe-rimental Evidence for Interatomic Coulombic Decay in Ne Clusters. In:Phys. Rev. Lett. 90 (2003), Nr. 20
[MLS+06] MORISHITA, Y. ; LIU, X.-J. ; SAITO, N. ; LISCHKE, T. ; KATO, M. ; PRÜMPER,G. ; OURA, M. ; YAMAOKA, H. ; TAMENORI, Y. ; SUZUKI, I. H. ; UEDA, K.:Experimental Evidence of Interatomic Coulombic Decay from the AugerFinal States in Argon Dimers. In: Phys. Rev. Lett. 96 (2006), Nr. 243402
[NTVP11] NGUYEN, T. ; TIMERGHAZIN, Q. ; VACH, H. ; PESLHERBE, G.: Mechanical-ly induced generation of highly reactive excited-state oxygen moleculesin cluster scattering. In: J. Chem. Phys. 134 (2011), Nr. 064035
[PRSZ09] POVH, B. ; RITH, K. ; SCHOLZ, C. ; ZETSCHE, F.: Teilchen und Kerne. 8.Heidelberg : Springer, 2009. – ISBN 978–3–540–68075–8
[Roe06] ROENTDEK HANDELS GMBH (Hrsg.): CoboldPC user manual. 6.2.90.2. ImVogelshaag 8, 65579 Kelkheim-Ruppertshain: RoentDek Handels GmbH,Mai 2006
[Roe09] ROENTDEK HANDELS GMBH (Hrsg.): The RoentDek Constant Fraction Dis-criminators CFD8b, CFD4b and CFD1b. 9.1.904.1. Im Vogelshaag 8, 65579Kelkheim-Ruppertshain: RoentDek Handels GmbH, April 2009
[Roe10] ROENTDEK HANDELS GMBH (Hrsg.): MCP Delay Line Detector Manual.9.22.1003.1. Im Vogelshaag 8, 65579 Kelkheim-Ruppertshain: RoentDekHandels GmbH, März 2010
[Sco76] SCOFIELD, J. H.: Theoretical Photoionization Cross Sections from 1 to 1500keV. In: Lawrence Livermore National Laboratory Report. LLNL, 1976 (UCRL-51326)
[SS12] SURDUTOVICH, E. ; SOLOV’YOV, A. V.: Double strand breaks in DNAresulting from double ionization events. In: Eur. Phys. J. D 66 (2012), Nr.206
57

Literaturverzeichnis
[TFK+08] TANAKA, T. ; FEIFEL, R. ; KITAJIMA, M. ; TANAKA, H. ; SORENSEN, S. L. ;SANKARI, R. ; FANIS, A. D. ; PIANCASTELLI, M.-N. ; KARLSSON, L. ; UE-DA, K.: Symmetry-resolved x-ray absorption fine structure and resonantAuger-spectator-electron decay study of O 1s → Rydberg resonances inO2. In: Phys. Rev. A 78 (2008), Nr. 022516
[TSK+12] TRINTER, F. ; SCHÖFFLER, M. S. ; KIM, H.-K. ; STURM, F. ; COLE, K. ; NEU-MANN, N. ; VREDENBORG, A. ; WILLIAMS, J. ; BOCHAROVA, I. ; GUIL-LEMIN, R. ; SIMON, M. ; BELKACEM, A. ; LANDERS, A. L. ; WEBER, Th. ;SCHMIDT-BÖCKING, H. ; DÖRNER, R. ; JAHNKE, T.: Experimental Proofof Resonant Auger Decay Driven Intermolecular Coulombic Decay. In:Nature, submitted (2012)
[UFL+08] UEDA, K. ; FUKUZAWA, H. ; LIU, X.-J. ; SAKAI, K. ; PRÜMPER, G. ; MORIS-HITA, Y. ; SAITO, N. ; SUZUKI, I. H. ; NAGAYA, K. ; IWAYAMA, H. ; YAO, M.; KREIDI, K. ; SCHÖFFLER, M. ; JAHNKE, T. ; SCHÖSSLER, S. ; DÖRNER, R.; WEBER, Th. ; HARRIES, J. ; TAMENORI, Y.: Interatomic Coulombic decayfollowing Auger decay: Experimental evidence in rare-gas dimers. In: J.Electron Spectrosc. Relat. Phenom. 166-167 (2008), Nr. 3-10
[VSC10] VENDRELL, O. ; STOYCHEV, S. D. ; CEDERBAUM, L. S.: Generation of High-ly DamagingH2O
+ Radicals by Inner Valence Shell Ionization ofWater. In:Chem. Phys. Chem. 11 (2010), S. 1006 – 1009
58

Danksagung
Ganz am Ende meiner Arbeit möchte ich die Möglichkeit nutzen, einigen PersonenDanke zu sagen, von denen mich viele nicht nur während der Bachelorarbeit unter-stützt haben:
An erster Stelle steht Prof. Dr. Reinhard Dörner: Danke für die freundliche Aufnahmein die Arbeitsgruppe und das sehr interessante Thema für meine Arbeit. Außerdemdanke dafür, dass ich auch als Bachelorstudentin bereits an zwei Konferenzen teilneh-men durfte. Vielen Dank für die große Unterstützung!
Ein ganz großes Dankeschön geht an meinen direkten Betreuer Florian Trinter. Dankefür die viele Zeit, die Du aufgewendet hast, um mir alle meine Fragen ausführlich zubeantworten, aber auch für die vielen Hinweise und Tipps und Tricks, die Du an michweitergegeben hast. Außerdem vielen Dank für das Korrekturlesen meiner Arbeit.Auch ein großer Dank geht an Dr. Markus Schöffler, der nicht nur immer ein offenesOhr für mich hatte und mir ganz spontan noch wertvolle Hinweise zu meiner Arbeitgegeben, sondern außerdem während der Strahlzeit und der Tagungen stets daraufgeachtet hat, dass ich nicht untergehe :-)
Ichmöchte meinen ehemaligen und aktuellen Bürokollegen Dr. Jasmin Titze, Ute Lenz,Stefan Zeller, Martin Richter, Melanie Salomon und Maximilian Schütt herzlich für dienette Arbeitsatmosphäre danken. Bei Fragen hat immer mindestens eine oder einervon euch sofort alles stehen- und liegengelassen und mir weiterhelfen können.Außerdem Danke an die Leute im Raum 01.322, dass ich bei euch immer eine Aus-zeit nehmen konnte: Neben Florian sind das Markus Waitz (Danke, Markus, für denDetektor-Verkabelungskurs in Berlin und dafür, dass ich immer genug zu essen be-kommen habe ;-)), Jörg Voigtsberger und Hong-Keun Kim – ich hoffe, ich habe euchalle nicht zu sehr von eurer eigenen Arbeit abgehalten ;-)In der Arbeitsgruppe Atomphysik hatte ich von Anfang an das Gefühl, willkommenzu sein. Vielen Dank euch allen für die angenehme Atmosphäre!
Ein herzlicher Dank geht an meine Korrekturleser Ole Hinrichs und Christian Stuck.Christian, Dir möchte ich nicht nur für das Korrekturlesen, sondern auch für den im-mer zuverlässigen IT-Support und vor allem fürs Aufmuntern, wenn ich mal wiedernicht an mich selbst glauben wollte, danken.
Danke an meine Freunde und an meine Kolleginnen bei HengelerMueller – vor allemdafür, dass Ihr mich auch mal von meiner Arbeit abgelenkt und damit verhindert habt,dass ich während meiner Bachelorarbeitsphase nichts anderes als die Uni sehe.
Am Ende stehen die Personen, die mich am längsten begleiten: Meine Familie, vorallem meine Eltern und Schwestern. Vielen Dank Euch, dass ihr mich bis jetzt immer
59

Literaturverzeichnis
unterstützt und anmich geglaubt habt. Ohne das Vertrauen darauf, dass ich nicht allei-ne bin und immer auf Euch zählen kann, hätte ich sicherlich nicht das Selbstvertrauengehabt, ein Physikstudium in Frankfurt anzufangen und bis zu diesem Punkt zu brin-gen.
60





















![Optimierung 1...(b) ϕ heißt Zielfunktion [engl.: objective function], F heißt zul¨assiger Bereich [engl.: feasible region] der gegebenen Aufgabe. Jeder Punkt x∈ F heißt zul¨assig](https://img.pdfslide.org/doc/110x75/5febfab46df6691b384d66ad/optimierung-1-b-heit-zielfunktion-engl-objective-function-f-heit.jpg)

![BIBLID [1699-3225 (2007) 11, 83-104]Reg Berol vidi” (sic Magnus) ϕ De : facesque • 779 quod non ego punior ipsa M2c UP ς De, in mg. add. ponor in ignem Dec • 780 monitumque](https://img.pdfslide.org/doc/110x75/6099936da8dd3e49fb4b83dc/biblid-1699-3225-2007-11-83-104-reg-berol-vidia-sic-magnus-de-facesque.jpg)





