Embed Size (px)
Citation preview

Intronunabhängige Expression des Glykoproteins G des Vesicular-Stomatitis-Virus
zur Pseudotypisierung lentiviraler Vektoren
Dissertation
zur Erlangung des Grades eines Doktors der Naturwissenschaften
der Fakultät für Chemie der Ruhr-Universiät Bochum
angefertigt in der
Abteilung für Molekulare und Medizinische Virologie,
Institut für Hygiene und Mikrobiologie, Medizinische Fakultät,
Ruhr-Universität Bochum
vorgelegt von
Daniela Stefanou
aus Hüls
Bochum, 2006

Die in der vorliegenden Dissertation präsentierten Experimente wurden in der Zeit von April
2001 bis März 2004 unter der Anleitung von Prof. Dr. Klaus Überla in der Abteilung für Mo-
lekulare und Medizinische Virologie des Instituts für Hygiene und Mikrobiologie der
Medizinischen Fakultät der Ruhr-Universität Bochum angefertigt.

Abkürzungen
AB Akut-Box AIDS erworbenes Immundefizienz-Syndrom (acquired immunodeficieny syndrome) APS Ammoniumperoxodisulfat b Basen BGH Rinder-Wachstumshormon (bovine growth hormone) bp Basenpaare C. elegans Caenorhabditis elegans cm Zentimeter cm2 Quadratzentimeter dATP 2’-Desoxyadenosin-5’-triphosphat DEPC Diethylpyrocarbonat DIG Digoxygenin DMEM Dulbecco’s Modified-Eagle-Medium DMSO Dimethylsulfoxid DNA Desoxyribonukleinsäure (deoxyribonucleic acid) E. coli Escherichia coli EB Elutionspuffer (elution buffer) EDTA Ethylendiaminotetraessigsäure EJC exon-junction-complex FBS Fötales Rinderserum (fetal bovine serum) g Gramm GFP grün fluoreszierendes Protein GFU grüne-Fluoreszenz-bildende Einheit (green fluorescence forming unit) °C Grad Celsius h Stunde HBS HEPES gepufferte Salzlösung (HEPES buffered saline) HCMV humanes Cytomegalievirus HEPES 4-(2-Hydroxyethyl)-1Piperazin-Ethansulfonsäure HIV humanes Immundefizienzvirus HRP Meerrettichperoxidase (horseradish peroxidase) Hsp Hitzeschockprotein IRE Iron-responsive-Element IRES interne Ribosomeneintrittsstelle IVS Intron (intervening sequence) kJ Kilojoule kb Kilobasenpaare kD Kilo-Dalton l Liter LB lysogeny broth

Abkürzungen iii
LMO LIM-domain only LTR long terminal repeat M molar MBS Maleinsäure gepufferte Salzlösung (maleic acid buffered saline) MBS-T Maleinsäure gepufferte Salzlösung mit Tween 20 MIE major immediate early min Minute µg Mikrogramm µl Mikroliter ml Milliliter MLV Maus-Leukämievirus mM millimolar mol Mol MOPS Morpholinopropansulfonsäure mRNA Messenger-Ribonukleinsäure NF Nukleärer Faktor ng Nanogramm PAGE Polyacrylamid-Gelelektrophorese PBS Phosphat gepufferte Salzlösung (phosphate buffered saline) PCR Polymerase-Kettenreaktion (polymerase chain reaction) pH potentia Hydrogenii pmol Pikomol RNA Ribonukleinsäure (Ribonucleic acid) Rpl32 Ribosomales Protein L32 RRE Rev-responsive-Element RT Reverse Transkriptase s Sekunde s. siehe s. o. siehe oben s. u. siehe unten S.O.C super optimal catabolite repression SCID-X1 X-chromosomal vererbte schwere kombinierte Immundefizienz
(X-linked severe combined Immunodeficiency) SDS Natriumdodecylsulfat (sodiumdodecylsulfate) SEAP Sekretierte Alkalische Phosphatase SIN-Vektor sich selbst inaktivierender Vektor SIV Affen- (Simian) Immundefizienzvirus SSC Natriumchlorid-Natriumcitrat-Puffer (sodium chloride sodium citrate buffer) ssRNA einzelsträngige RNA (single stranded RNA) SV40 Simian Virus 40 TAE Tris-Acetat-EDTA-Puffer

Abkürzungen iv
TBE Tris-Acetat-Borat-Puffer TEMED N,N,N’,N’-Tetramethylethylendiamin Tris Tris-(hydroxymethyl)-aminomethan tRNA Transfer-Ribonukleinsäure U Einheit (unit) Upm Umdrehungen pro Minute UsnRNP Uridinreiches kleines nukleäres Ribonukleoprotein
(uridinrich small nuclear ribonucleoprotein) usw. und so weiter UTR nicht translatierte Region (untranslated region) V Volt v/v Volumen pro Volumen VSV Vesicular-Stomatitis-Virus VSV-G Glykoprotein G des Vesicular-Stomatitis-Virus w/v Masse (weight) pro Volumen WB Westernblot z.B. zum Beispiel
Aminosäuren wurden mit den üblichen Drei- und Einbuchstabenabkürzungen, Nukleinsäure-
basen mit den gängigen Einbuchstabenabkürzungen bezeichnet.

Inhalt
1 EINLEITUNG...................................................................................................................1
1.1 GENTHERAPIE MITTELS PSEUDOTYPISIERTER LENTIVIRALER VEKTOREN ....................1
1.1.1 Viraler Gentransfer in der Gentherapie.................................................................1
1.1.2 Drei-Plasmid-Verpackungssystem .........................................................................2
1.1.3 Pseudotypisierung mit VSV-G ................................................................................3
1.2 OPTIMIERUNG DER PSEUDOTYPISIERUNGSEFFIZIENZ...................................................5
1.2.1 HCMV-MIE-5’-UTR...............................................................................................5
1.2.2 Codonoptimierung..................................................................................................8
2 MATERIAL UND METHODEN..................................................................................10
2.1 PLASMIDE..................................................................................................................10
2.1.1 VSV-G-Expressionsplasmide ................................................................................10
2.1.2 gag-pol-Expressionsplasmide...............................................................................14
2.1.3 Vektor-Konstrukte.................................................................................................15
2.1.4 Weitere Plasmide..................................................................................................15
2.2 OLIGONUKLEOTIDE UND WEITERE NUKLEINSÄUREN .................................................16
2.3 EUKARYONTEN UND PROKARYONTEN .......................................................................17
2.4 ENZYME, ANTIKÖRPER UND ANDERE PROTEINE ........................................................18
2.5 CHEMIKALIEN, KITS, SONSTIGE VERBRAUCHSMATERIALIEN......................................19
2.6 KUNSTSTOFF-EINMALARTIKEL..................................................................................20
2.7 PUFFER, MEDIEN UND LÖSUNGEN .............................................................................21
2.8 TECHNISCHE AUSSTATTUNG .....................................................................................24
2.9 ARBEITEN MIT ZELLKULTUREN UND LENTIVIREN .....................................................25
2.9.1 Kultivierung von 293T- und 293A-Zellen.............................................................25
2.9.2 Kryokonservierung von Zellen .............................................................................25
2.9.3 Auftauen kryokonservierter Zellen .......................................................................25
2.9.4 Transiente Transfektion von 293T-Zellen mittels Calcium-Phosphat..................26
2.9.5 Ernte lentiviraler Vektoren...................................................................................27
2.9.6 Infektion von 293A-Zellen mit lentiviralen Vektoren ...........................................27
2.9.7 Titerbestimmung ...................................................................................................27
2.10 ARBEITEN MIT ESCHERICHIA COLI..............................................................................28
2.10.1 Transformation von E. coli...............................................................................28

Inhalt vi
2.10.2 Kultivierung von E. coli....................................................................................28
2.11 ARBEITEN MIT PROTEINEN ........................................................................................29
2.11.1 SEAP-Bestimmung............................................................................................29
2.11.2 Westernblot-Analyse.........................................................................................29
2.12 ARBEITEN MIT NUKLEINSÄUREN ...............................................................................30
2.12.1 Fotometrische Bestimmung von Nukleinsäure-Konzentrationen .....................30
2.12.2 mRNA-Präparation...........................................................................................31
2.12.3 Northernblot-Analyse .......................................................................................31
2.12.4 Total-RNA Präparation mit anschließender DNase-Spaltung .........................32
2.12.5 RNA-Struktur-Vorhersage ................................................................................33
2.12.6 Plasmidpräparationen......................................................................................33
2.12.7 Polymerase-Kettenreaktion (PCR) ...................................................................33
2.12.8 Spaltung von DNA mittels Restriktionsendonukleasen.....................................36
2.12.9 Auffüllen von Einzelstrang-Enden....................................................................37
2.12.10 Reinigung von DNA..........................................................................................37
2.12.11 Agarose-Gelelektrophorese ..............................................................................37
2.12.12 DNA-Extraktion aus Agarose ...........................................................................38
2.12.13 Phosphorylierung und komplementäre Anlagerung von Oligonukleotiden .....38
2.12.14 Ligation.............................................................................................................38
2.12.15 TA-Klonierung..................................................................................................39
2.12.16 DNA-Sequenzierung .........................................................................................39
3 ERGEBNISSE.................................................................................................................41
3.1 EXPRESSION VON VSV-G MITTELS PCDNA3.1(+)-BASIERTER PLASMIDE ................41
3.2 EXAKTE DELETION DES ERSTEN HCMV-MIE-INTRONS............................................43
3.2.1 Einfluss des Spleiß-Prozesses und der Exon-Sequenzen auf die Expression .......43
3.2.2 Nachweis der Spleißprodukte ...............................................................................46
3.2.3 Deletion weiterer HCMV-Sequenzen aus der VSV-G-Expressionskassette .........48
3.3 EINFLUSS VON CODONOPTIMIERUNG AUF DIE EXPRESSION .......................................50
3.3.1 Erste Konstrukte mit codonoptimierter Sequenz für VSV-G ................................50
3.3.2 VSV-Gsyn-G-Fusions-RNA ....................................................................................52
3.3.3 Sequenz-Analyse der Wildtyp-VSV-G-DNA aus pCI-G........................................53
3.3.4 Korrektur der codonoptimierten VSV-G-Sequenz ................................................54
3.3.5 Kombination von HCMV-MIE-5’-UTR mit codonoptimierter VSV-G-Sequenz...58

Inhalt vii
4 DISKUSSION .................................................................................................................60
4.1 INTRONUNABHÄNGIGE EXPRESSION VON VSV-G .....................................................60
4.2 DAS HCMV-MIE-INTRON UND SPLEIßEN ALS EXPRESSIONSVERSTÄRKER ...............60
4.3 EXONISCHE HCMV-SEQUENZEN IN DER 5’-UTR ALS EXPRESSIONSVERSTÄRKER ....62
4.4 EXPRESSION EINES CODONOPTIMIERTEN, MUTIERTEN VSV-G ..................................64
4.5 PSEUDOTYPISIERUNG MIT OPTIMIERTER FÜR VSV-G CODIERENDER SEQUENZ..........66
4.6 CODONOPTIMIERUNG IN VERBINDUNG MIT DER HCMV-5’-UTR..............................67
5 ZUSAMMENFASSUNG................................................................................................69
6 LITERATUR ..................................................................................................................70
DISSERTATIONSBEZOGENE PUBLIKATION..............................................................80
DANKSAGUNG .....................................................................................................................81
LEBENSLAUF .......................................................................................................................82

1 Einleitung
1.1 Gentherapie mittels pseudotypisierter lentiviraler Vektoren
1.1.1 Viraler Gentransfer in der Gentherapie
Durch Einbringen therapeutischer Gene in Körperzellen bietet die Gentherapie neue Möglich-
keiten in der Behandlung vieler verschiedener ererbter und erworbener Krankheiten wie Mu-
koviszidose, Duchenne-Muskeldystrophie, Ornithin-Transcarbamoylase-Mangel, schwere
kombinierte Immundefizienzen, Hämophilie, AIDS, Tumorleiden, Bluthochdruck und neuro-
degenerative Erkrankungen [1-9]. Ein Kernpunkt der Gentherapie-Forschung ist die Entwick-
lung effizienter Methoden für den Transfer des therapeutischen Gens in die zu behandelnden
Zellen. Der Gentransfer kann nicht-viral (z.B. durch Gene-Gun, Elektroporation, kationische
Lipide, Nanopartikel) oder viral (z. B. durch auf Adenoviren, Adeno-assoziierte Viren, Her-
pesviren oder Retroviren basierenden Vektoren) vermittelt werden [10,11]. Die Evolution hat
die Mechanismen des viralen Gentransfers, der Teil des Vermehrungszyklus der Viren ist,
perfektioniert, so sind virale Vektoren den nicht-viralen in ihrer Effizienz überlegen [11].
Die viralen Vektoren lassen sich danach einteilen, entweder eine vorübergehende oder eine
langfristige Expression des therapeutischen Gens zu vermitteln; je nach Anwendung ist das
eine oder das andere vorzuziehen [11]. Eine langfristige Expression des Transgens ermögli-
chen z. B. die retroviralen Vektoren, die dieses in das Genom der Zielzelle integrieren. Die
beiden identischen Kopien des einzelsträngigen RNA-Genoms, das das therapeutische Gen
enthält, werden dazu nach Eintritt in das Cytoplasma von der viralen Reversen Transkriptase
in doppelsträngige DNA umgeschrieben; diese wird in den Zellkern transportiert und von der
viralen Integrase in das Wirtszellgenom eingefügt [12]. Neben dem Vorteil der dauerhaften
Transgen-Expression und der Übertragung des Transgens auf die Tochterzellen birgt die In-
tegration allerdings auch die Gefahr einer Insertionsmutagenese: Drei von zehn Patienten
entwickelten fast drei Jahre nach erfolgreicher Gentherapie ihrer X-chromosomal vererbten
schweren kombinierten Immundefizienz (SCID-X1) eine unkontrollierte exponentielle klona-
le T-Zell-Proliferation durch die Integration des retroviralen Vektors in die Nähe des Promo-

1 Einleitung 2
tors des Proto-Onkogens LMO2 [13]. Das reale Risiko der Insertions-Mutagenese bedeutet
jedoch nicht die grundsätzliche Abkehr von retroviralen Vektoren in der Gentherapie [14].
Zu den retroviralen Vektoren gehören u. a. die onkoretroviralen und die lentiviralen Vektoren.
Vertreter der Lentiviren sind z.B. das humane Immundefizienzvirus und das Immundefizienz-
virus der Affen (HIV bzw. SIV), die das erworbene Immundefizienz-Syndrom (AIDS) bei
Menschen bzw. bei Affen verursachen können [15-17]. Im Gegensatz zu den onkoretroviralen
transduzieren lentivirale Vektoren nicht nur sich teilende sondern auch sich nicht teilende
Zellen [18,19]. Lentivirale Vektoren sind somit auch für die Behandlung terminal differen-
zierter Zellen wie Neuronen oder Muskelzellen geeignet [20,21]. Für die Gentherapie postmi-
totischer Zellen bieten sich auch integrationsdefiziente lentivirale Vektoren an, da mit diesen
ein deutlich reduziertes Integrations-Risiko verbunden ist; sie sind vielen nichtintegrierenden
viralen Vektoren in ihrer höheren Klonierungskapazität und niedrigeren Immunogenität über-
legen [22].
1.1.2 Drei-Plasmid-Verpackungssystem
Die in der vorliegenden Arbeit eingesetzten integrierenden lentiviralen Vektoren sind nicht
replikationsfähig und durchlaufen den lentiviralen Replikationszyklus nur bis zur Integration
[23,24]. Zur Herstellung der Vektoren werden mindestens drei verschiedene Plasmide tran-
sient in 293T-Zellen transfiziert, die daraufhin die Vektoren produzieren. Ein Plasmid codiert
für das Vektorgenom, ein weiteres für die zur Verpackung des Virus nötigen Proteine und ein
drittes für das Hüll-Glykoprotein.
Sowohl das verwendete SIV- als auch das HIV-1-Vektorkonstrukt enthalten als Reportergen
das Gen für das grün fluoreszierende Protein (GFP) unter der Transkriptions-Kontrolle eines
HCMV-MIE-Promotor/Enhancers sowie die für die Transkription, Verpackung, reverse
Transkription und Integration notwendigen cis-aktiven lentiviralen Sequenzen [23,24]. Das
SIV-Vektorkonstrukt enthält darüber hinaus die intakten codierenden Sequenzen für die Re-
gulatorproteine Tat und Rev. Die Transkription des SIV-Vektors erfolgt durch die Promotor-
aktivität der U3-Region des 5’-LTRs (long terminal repeat) und wird durch das virale Protein
Tat verstärkt [24]. Die LTRs der Lentiviren bilden die Flanken des Vektorgenoms und beste-
hen beide aus der U3-, R- und U5-Region [25]. Die Transkription des verwendeten HIV-1-
Vektors erfolgt Tat-unabhängig durch einen HCMV-MIE-Promotor, den das Konstrukt an
Stelle der U3-Region im 5’-LTR trägt [23]. Beide Vektorkonstrukte weisen Deletionen im

1 Einleitung 3
Bereich der U3-Region des 3’-LTRs auf, die bei der reversen Transkription als Matrize für die
U3-Region des 5’-LTRs dient. Die ins Genom der Zielzelle integrierte provirale DNA weist
folglich Deletionen im lentiviralen Promotor auf und kann durch diesen nicht transkribiert
werden. Beide Vektoren werden daher als sich selbst inaktivierende Vektoren bezeichnet
[23,24].
Die Transduktion der Zielzellen durch lentivirale Vektoren ist nicht von der Neusynthese vi-
raler Proteine abhängig, alle nötigen Proteine sind bereits im Viruspartikel verpackt. Daher
können die viralen Proteine bei der Produktion der Vektoren in trans von gesonderten Plas-
miden exprimiert werden. Die in der vorliegenden Arbeit verwendeten SIV- und HIV-1-gag-
pol-Expressionsplasmide codieren für die Matrixproteine, die im Viruspartikel der Hüll-
membran von innen anliegen, die Capsidproteine, die die beiden Genom-Kopien mit der
Capsidstruktur umgeben, und die Nukleocapsidproteine, die mit der genomischen RNA kom-
plexiert sind, sowie für die Enzyme Protease, die das Gag- und Gag-Pol-Vorläuferprotein
spaltet, Reverse Transkriptase und Integrase [24,26,27]. Ferner enthält das SIV-gag-pol-
Expressionsplasmid intakte Leserahmen für die Regulatorproteine Tat und Rev sowie die ak-
zessorischen Gene vif, vpx und vpr; als Promotor dient die U3-Region im 5’-LTR [24]. Im
Gegensatz dazu enthält die Expressionskassette des verwendeten HIV-1-gag-pol-
Expressionsplasmids einen HCMV-MIE-Promotor/Enhancer und eine codonoptimierte gag-
pol-Sequenz, aber keine weiteren HIV-1-Sequenzen [26].
Werden zur Vektorproduktion ein Vektorkonstrukt und ein Verpackungsplasmid cotransfi-
ziert, die beide keine tat- und rev-Gene enthalten, so sind nötigenfalls zusätzlich tat- und rev-
Expressionsplasmide zu transfizieren. Tat verstärkt die LTR-getriebene Expression, Rev ver-
mittelt durch Wechselwirkung mit dem RNA-Element Rev-responsive-Element (RRE) den
Export ungespleißter und einfach gespleißter lentiviraler RNA aus dem Zellkern ins Cy-
toplasma [28,29].
1.1.3 Pseudotypisierung mit VSV-G
Bei der Produktion lentiviraler Vektoren wird das Glykoprotein ebenfalls in trans von einem
gesonderten Plasmid exprimiert. Natürliche SI- und HI-Viren weisen als Produkte des env-
Gens das Transmembranprotein gp41 und das externe Glykoprotein gp120 als Komplex in
ihrer Hüllmembran auf und können nur Zellen infizieren, die den CD4-Rezeptor tragen [30].
Diesem beschränkten Tropismus liegt die spezifische Wechselwirkung des Glykoprotein-

1 Einleitung 4
Komplexes mit dem CD4-Rezeptor in der Cytoplasmamembran der Wirtszelle zu Grunde.
Das gp120 interagiert zusätzlich mit einem Chemokin-Corezeptor. Nach mehrfach aufeinan-
der folgenden Konformationsänderungen der Glykoproteine und des Rezeptors vermittelt das
gp41 die Fusion der viralen mit der Wirtszellmembran, der virale Kern gelangt ins Cyto-
plasma [12].
Zur Veränderung des Tropismus eines umhüllten viralen Vektors bedient man sich der Pseu-
dotypisierung. Es werden dabei Vektoren produziert, die statt des natürlichen Glykoproteins
das eines anderen umhüllten Virus tragen und so dessen Wirtszellspezifität übernehmen.
Schon lange vor der Entwicklung viraler Vektoren waren Pseudotypen bekannt, sie entstehen
beispielsweise in Zellen, die von zwei oder mehr nicht verwandten Viren infiziert sind [31].
Von großer Bedeutung für die Erforschung viraler Vektoren und für die Gentherapie sind
Pseudotypen mit dem Glykoprotein G aus der Hülle des Vesicular-Stomatitis-Virus, da sich
mit ihnen eine sehr große Vielfalt von Zelltypen infizieren lassen.
Das umhüllte Rhabdovirus VSV mit einem einzelsträngigen kontinuierlichen RNA-Genom in
Negativstrang-Orientierung wird in die VSV-Serotypen Indiana und New Jersey unterteilt und
ist der Erreger der vesikulären Stomatitis bei Pferden, Rindern und Schweinen mit Sympto-
men, die denen der Maul- und Klauenseuche ähneln [32-35]. Die 67-kD-Monomere des tri-
meren Glykoproteins G (VSV-G) sind in der Hülle des Virus über 20 Aminosäuren lange
Transmembran-Domänen verankert, die daran angrenzenden 29 C-terminalen Aminosäuren
bilden die basische cytoplasmatische Domäne [36,37]. Die aus 446 Aminosäuren bestehende
Ectodomäne ist an zwei Stellen glycosyliert; die beiden Kohlenhydratketten sind identisch,
dreifach verzweigt und enden in jeder der drei Verzweigungen in einem Sialinsäurerest [38].
Die terminalen Sialinsäurereste sind essentiell für die Adsorption des VSV an die Wirtszelle
[39,40]. Das unreife Protein enthält N-terminal ein 16 Aminosäuren umfassendes Signalpep-
tid, das bei Eintritt des entstehenden Proteins ins endoplasmatische Retikulum abgespalten
wird [41]. Das Protein wird über den Golgi-Apparat zur Cytoplasmamembran transportiert,
für dieses Ziel enthält es das YTDIE-Exportmotiv in seiner cytoplasmatischen Domäne [42].
VSV-G stellt bei der Infektion den ersten Kontakt mit der Zielzelle her, indem es mit einem
bisher unbekannten ubiquitären zellulären Rezeptor interagiert; bei diesem Rezeptor handelt
es sich nicht – wie früher einmal angenommen – um das Membranlipid Phosphatidylserin,
letzteres spielt erst bei der Einleitung der Membranenfusion eine Rolle [43,44]. Im Gegensatz
zu natürlichen Lentiviren, deren Hülle direkt mit der Cytoplasmamembran fusioniert, gelan-
gen VSV und Pseudotypen mit dem Glykoprotein VSV-G durch clathrinabhängige rezeptor-

1 Einleitung 5
vermittelte Endocytose in die Wirtszelle [45]. Nach Ansäuerung des Endosoms führen die
elektrostatischen Wechselwirkungen zwischen dem VSV-G in der viralen und dem Phospha-
tidylserin in der endosomalen Membran zu einer drastischen Konformationsänderung des
VSV-G sowie zur Fusion der beiden Membranen, in Folge dessen das Nukleocapsid in das
Cytoplasma entlassen wird [44,46,47].
Neben dem breiten Tropismus von VSV-G-Pseudotypen haben diese auch den Vorteil, die
Virushülle so zu stabilisieren, dass die Vektoren durch Zentrifugation konzentrierbar sind und
so hochtitrige Vektorpräparate erhalten werden können [48]. Diese beiden Vorzüge sind be-
deutend für ex vivo und lokale in vivo Gentherapie-Anwendungen. Bei systemischer Verab-
reichung pseudotypisierter Vektoren sind Glykoproteine besser geeignet, die für den zu be-
handelnden Zelltyp bzw. das zu behandelnde Organ spezifisch sind, um an diesen Orten wirk-
same Konzentrationen zu erreichen und um Nebenwirkungen, die durch Transduktion nicht
zu behandelnder Zellen entstehen, zu vermindern.
1.2 Optimierung der Pseudotypisierungseffizienz
1.2.1 HCMV-MIE-5’-UTR
Für die Produktion lentiviraler Vektoren in größerem Maßstab sind stabile Verpackungs-
Zelllinien geeigneter als transient transfizierte Zellen. Bei der Herstellung solcher Verpa-
ckungs-Zelllinien für lentivirale Pseudotypen mit dem Glykoprotein VSV-G klonierte Dr.
Seraphin Kuate die für VSV-G codierende Sequenz in eine einfache induzierbare Expressi-
onskassette, da konstitutiv exprimiertes VSV-G toxisch für die Zellen ist [48]. Die VSV-G-
Expression gelang aber erst, nachdem er auch die 5’ nicht translatierte Region (5’-UTR) in-
klusive des ersten Introns der major immediate early (MIE) Gene des humanen Cytomegalie-
virus (HCMV) aus dem zu Grunde gelegten VSV-G-Expressionsplasmid zwischen Promotor
und codierender Sequenz kloniert hatte [49,50].
Das β-Herpesvirus HCMV (systematischer Name: humanes Herpesvirus 5) trägt ein dop-
pelsträngiges DNA-Genom und ist eine Ursache für mild verlaufende infektiöse Mononukleo-
sen bei älteren Kindern und Erwachsenen, für Leberentzündungen bei jüngeren Kindern, für
schwere Lungenentzündungen bei Kleinkindern und für Geburtsfehler bei Neugeborenen; bei

1 Einleitung 6
Immunbeeinträchtigten, wie Transplantations- oder AIDS-Patienten, ist der Verlauf einer
HCMV-Infektion schwerer und endet häufiger mit dem Tod [32,51]. Für die Gentherapie und
andere biotechnologische Anwendungen ist HCMV in sofern von großer Bedeutung, als dass
ihm eines der stärksten bekannten Promotor/Enhancer-Elemente entstammt, das Verwendung
in vielen eukaryotischen Expressions-Vektoren findet und auch Bestandteil der meisten in der
vorliegenden Arbeit verwendeten Expressionsplasmide ist [52-59]. Es handelt sich um die
Promotor/Enhancer-Region der Major-immediate-early-(MIE)-Gene. Die HCMV-Gene wer-
den zeitlich reguliert exprimiert – beginnend mit den Immediate-early-Genen, zu denen die
MIE-Gene gehören. Die codierenden Sequenzen beginnen im zweiten Exon. Exon 1 und die
ersten Nukleotide des Exons 2 bilden die 5’-UTR der durch alternatives Spleißen entstehen-
den mRNAs [60]. Auch diese 5’-UTR und das Intron aus dieser Region haben eine Bedeu-
tung für die heterologe Genexpression: Diese Elemente können die Expression verstär-
ken [61,62].
Der positive Effekt von Introns und des Spleißens auf die Genexpression ist auch für viele
andere Introns und Organismen beschrieben, z.B. ist (1) für die Produktion der RNA für
β-Globin durch das rekombinante SV40-Kaninchen-β-Globin-Virus eines der β-Globin-
Introns IVS1 oder IVS2 nötig [63], wird (2) die Expression des unc-54-Gens für eine Isoform
der schweren Kette des Myosins des Fadenwurms C. elegans durch eines der vier Introns na-
he dem 5’-Ende stimuliert [64], wird (3) das Gen Rpl32 für das Ribosomen-Protein L32 der
Maus ohne ein spleißbares Intron gar nicht, bei Vorhandensein eines der Introns 2 oder 3 oder
sogar eines fremden Introns aus dem Gen für die Immunglobulin schwere Kette µ der Maus
zu 10 bis 20 % und bei Vorhandensein des Introns 1 zu 100 % exprimiert [65], wird (4) das
Reportergen für die Chloramphenicol-Acetyltransferase unter der Kontrolle des Promotors
des Mais-alkohol-dehydrogenase1-Gens in Mais 100-mal stärker exprimiert, wenn sich zwi-
schen Promotor und codierender Sequenz das Intron 1 des Mais-alkohol-dehydrogenase1-
Gens befindet [66].
Als Prä-mRNA-Spleißen wird der Prozess bezeichnet, bei dem das Intron entfernt und die
flankierenden Exons ligiert werden. Das geschieht über zwei Umesterungsreaktionen. Zuerst
greift die 2’-Hydroxylgruppe eines Adenosinrestes der Verzweigungsstelle im Intron die
Phosphodiesterbindung der 5’-Spleißstelle an und es entsteht eine Lasso-Struktur; die freige-
wordene 3’-Hydroxylgruppe des 5’-Exons greift daraufhin die Phosphodiesterbindung der 3’-
Spleißstelle an, die Exons werden so verbunden und das Intron als Lasso-Struktur freigesetzt
[67]. Das Erkennen der Spleiß- und Verzweigungsstellen sowie die Katalyse der Umeste-

1 Einleitung 7
rungsreaktionen werden durch das Spleißosom vermittelt. Das Spleißosom ist ein Komplex,
der aus fünf uridinreichen kleinen nukleären Ribonukleoproteinen (UsnRNP) und zahlreichen
Nicht-snRNP-Spleißfaktoren besteht; es formiert sich schrittweise in bestimmter Reihenfolge
an der zu prozessierenden Prä-mRNA aus seinen Komponenten [68]. Im Laufe dieses Prozes-
ses werden nach und nach bestimmte Proteine 20 bis 24 Nukleotide stromaufwärts der Exon-
Exon-Verbindungsstelle angeheftet, die auch nach Beenden des Spleißens dort verbleiben,
diese werden exon-junction-complex (EJC) genannt. Der EJC ist das Bindeglied zwischen
dem Spleiß-Vorgang und nachfolgenden Schritten des mRNA-Metabolismus, er besteht aus
wenigen festen Komponenten im Inneren und einigen peripheren – zum Teil transienten –
Faktoren [69].
Es werden verschiedene Wirkungsebenen und Ursachen für die Verstärkung der Genexpressi-
on durch Introns diskutiert, für viele Introns ist jedoch noch nicht bekannt, welche die vor-
herrschende Wirkung ist [70]. So kann ein Intron beispielsweise unerlässlich sein, um den
Transport der mRNA aus dem Zellkern ins Cytoplasma zu gewährleisten, da Komponenten
des EJC durch ihre Interaktion mit Exportfaktoren die Effizienz des Exports erhöhen [71,72].
Darüber hinaus haben Komponenten des EJC und des Spleißosoms direkten Kontakt zur Po-
lyadenylierungs-Maschinerie, dies führt zu einer effizienteren Polyadenylierung und somit zu
höheren mRNA-Niveaus im Zellkern und im Cytoplasma [73-75]. Spleißen verstärkt zudem
die Translation, dies geht einher mit einer vermehrten Assoziation von gespleißter mRNA mit
Polysomen und ist wiederum durch Komponenten des EJC vermittelt [76]. Introns können
aber auch bereits auf die Transkription einwirken: als DNA, wenn sie enhancer enthalten
[77,78], oder nach der Transkription, da Komponenten der Spleißmaschinerie mit Transkrip-
tions-Initiations- bzw. –Elongations-Faktoren interagieren und auf diese Weise sowohl die
Initiation als auch die Elongation der Transkription stimulieren [79,80]. Das erste HCMV-
MIE-Intron enthält eine Bindestelle für den Transkriptionsfaktor NF1, die aber nicht so be-
deutend für den expressionsfördernden Effekt des Introns ist wie eine ebenso enthaltene Re-
gion, die dem enhancer im ersten Intron des Gens für Troponin I der Wachtel homolog ist;
diese Region wirkt in vivo muskelspezifisch, in vitro aber auch in Nicht-Muskelzellen [61,81].
Nicht nur für das erste Intron der HCMV-MIE-Gene wurde ein expressionsfördernder Effekt
nachgewiesen, sondern auch für die 5’ nicht translatierten HCMV-MIE-Exon-Sequenzen. Aus
den Untersuchungen von Ghazal und Nelson geht hervor, dass einerseits die konservierte Box
5’-TGACCTCCATAGAAGACA-3’ eine Expressionsverstärkung bewirkt – dabei spielt die
Bindung zweier im Zellkern lokalisierter Proteine an diese Box eine Rolle – andererseits je-

1 Einleitung 8
doch auch weitere Sequenzabschnitte der 5’-UTR einen Beitrag zur Expressionsverstärkung
leisten [62]. Die Bedeutung dieser weiteren Sequenzabschnitte als regulatorische Elemente
wurde bisher nicht näher charakterisiert.
Ziele der vorliegenden Arbeit waren es also, (1) zu klären, ob im Falle der VSV-G-Expression
für die Pseudotypisierung lentiviraler Vektoren das Intron oder die nicht translatierten Exon-
Sequenzen den entscheidenden positiven Effekt liefern, (2) bisher nicht beschriebene regula-
torische Elemente zu finden und (3) die HCMV-Sequenzen in der 5’-UTR auf das unerläss-
lichste einzugrenzen.
1.2.2 Codonoptimierung
Die Verstärkung der VSV-G-Expression durch die HCMV-MIE-5’-UTR zeigt, dass die
VSV-G-Expression prinzipiell gesteigert werden kann. Das bedeutet aber nicht zwangsläufig,
dass diese Maßnahme die einzig mögliche ist, noch dass sie die beste VSV-G-Expression und
die höchsten Titer der mit VSV-G pseudotypisierten Vektoren hervorbringt. Außerdem könn-
te der Verzicht auf die zusätzlichen HCMV-Sequenzen entscheidend sein, wenn in einer Ex-
pressionskassette nur ein beschränkter Platz zur Verfügung steht.
Eine Technik zur Steigerung der Expression heterologer Gene und insbesondere der Expressi-
on viraler Gene in Säugerzellen ist die Codonoptimierung: Dazu wird die codierende Sequenz
neu synthetisiert und nur jene Codone benutzt, die in stark exprimierten Genen des Wirtes am
häufigsten auftreten [82]. Dieser Technik liegt zu Grunde, dass viele Aminosäuren durch zwei
bis sechs synonyme Tripletts codiert werden und es abhängig von der Spezies und vom Ex-
pressions-Niveau eines Proteins unterschiedliche Vorlieben für die Synonyme gibt [83-85].
Die Ursache für den Einfluss der Codonoptimierung auf die Expression wird kontrovers dis-
kutiert. Der ausschlaggebende Einfluss der Codonoptimierung wird zum einen auf Translati-
ons-Ebene angenommen, da bei nicht codonoptimierten Genen auch seltenere tRNAs benötigt
werden und infolgedessen „Versorgungs-Engpässe“ vorstellbar sind und die Häufigkeit der
Codon-Synonyme in stark exprimierten Genen mit der Häufigkeit der entsprechenden Isoak-
zeptor-tRNAs korreliert [86,87]. Robinson et al. konnten jedoch in E. coli zeigen, dass die
Verfügbarkeit beladener tRNAs, die seltene Codone erkennen, erst unter extremen Bedingun-
gen, d. h. bei hohen Transkriptionsraten und mehrfacher Verwendung des suboptimalen Co-
dons in dem Gen, zum geschwindigkeitslimitierenden Faktor der Protein-Biosynthese
wird [88].

1 Einleitung 9
Ein Effekt der Codonoptimierung auf Vorgänge, die der Translation vorausgehen, ist für co-
donoptimierte gag-pol-Gene des HIV-1 und des SIV beschrieben. Diese codonoptimierten
Gene ermöglichen die von der HIV-1-5’-UTR, dem Rev-responsive-Element (RRE) und dem
Rev-Protein unabhängige Expression des Gag-Proteins aufgrund einer Erhöhung der nukleä-
ren mRNA-Stabilität und des konstitutiven Exports aus dem Zellkern [26]. Zudem führt die
cytoplasmatische Transkription von Wildtyp-HIV-1-gag ebenso zur Expression des Proteins
wie die von codonoptimiertem gag, während die Gag-Expression bei nukleärer Transkription
nur unter Verwendung des codonoptimierten gag-Gens gelingt; somit können es in dem Fall
nur prätranslationale Mechanismen wie mRNA-Stabilisierung und -Prozessierung sowie
nukleocytoplasmatischer Export sein, die entscheidend zur Expression codonoptimierter Gene
beitragen [89]. Eine mRNA-Stabilisierung wird schon dadurch erreicht, dass die Codonopti-
mierung gleichzeitig zu einer Steigerung des GC-Gehalts der Sequenz führt, was eine Steige-
rung der Halbwertszeit der mRNA zur Folge hat [90].
Weitere Ziele der vorliegenden Arbeit waren es folglich, (4) die HCMV-MIE-5’-UTR-
unabhängige VSV-G-Expression durch eine codonoptimierte für VSV-G codierende Sequenz
zu untersuchen, (5) Hinweise auf die Wirkungsebene der Codonoptimierung als Expressions-
verstärkung zu finden und (6) zu überprüfen, ob durch Codonoptimierung alleine oder in
Kombination mit der HCMV-MIE-5’-UTR noch eine Steigerung der Titer der mit VSV-G
pseudotypisierten lentiviralen Vektoren möglich ist.

2 Material und Methoden
2.1 Plasmide
2.1.1 VSV-G-Expressionsplasmide
a) pHIT-G
pHIT-G enthält die für VSV-G codierende Sequenz [91], leitet sich von dem MLV-env-
Expressionsplasmid pHIT 123 ab und trägt somit das SV40 ori, die HCMV-major-immediate-
early-(MIE)- Promotor/Enhancer-Region sowie die 5’-UTR mitsamt des ersten Introns der
HCMV-MIE-Gene [92].
b) pCD-G
pCD-G wurde von Prof. Dr. Ralf Wagner erhalten, der die für VSV-G codierende Sequenz
aus pHIT-G in das kommerziell erhältliche pcDNA3.1(+) (Invitrogen, Karlsruhe) eingesetzt
hat.
c) pCD-Gsyn
pCD-Gsyn wurde von Prof. Dr. Ralf Wagner erhalten, der die codonoptimierte DNA für VSV-
G entsprechend der im Genbankeintrag J02428 veröffentlichten Aminosäure-Sequenz des
VSV-G synthetisiert und in pcDNA3.1(+) eingesetzt hat.
d) pCI-G
Manuela Günther fügte das erste HCMV-MIE-Intron in pCD-G ein, indem sie das 1,2-kb-
Fragment des durch die Restriktionsendonukleasen SnaBI und HindIII gespaltenen pHIT-G in
durch dieselben Restriktionsendonukleasen gespaltenes pCD-G klonierte.
Die für VSV-G codierende Sequenz wurde im Rahmen der vorliegenden Arbeit in Zusam-
menarbeit mit Dr. Dennis Hoffmann sequenziert (s. 2.12.16).

2 Material und Methoden 11
e) pCI-Gsyn
Manuela Günther fügte das erste HCMV-MIE-Intron in pCD-Gsyn ein, indem sie das 1,2-kb-
Fragment des durch die Restriktionsendonukleasen SnaBI und HindIII gespaltenen pHIT-G in
durch dieselben Enzyme gespaltenes pCD-Gsyn klonierte.
f) pCI-GΔI
Zur Deletion des HCMV-MIE-Introns aus pCI-G wurde mit den Restriktionsendonukleasen
SacII und HindIII zunächst ein 893 bp großer, das Intron umfassender Sequenz-Abschnitt aus
pCI-G (s. Abb. 2-1) entfernt und anschließend durch Ligation einer Verbindungs-Sequenz, die
durch Phosphorylierung und komplementäre Aneinanderlagerung der Oligonukleotide ΔIs
und ΔIa gewonnen wurde, die herausgeschnittene Nicht-Intron-Sequenz wieder ergänzt. Nach
der Klonierung wurde pCI-GΔI zur Kontrolle von der SacII- bis zur HindIII-Schnittstelle se-
quenziert (s. 2.12.16).
Abb. 2-1. Plasmidkarten. – Schematische Darstellung der Plasmide pCI-G, pCI-Gsyn und pCD-G und Lage der Erkennungssequenzen der eingesetzten Restriktionsendonukleasen. HCMV-P: Promo-tor/Enhancer-Region der Major-immediate-early-Gene des humanen Cytomegalievirus; HCMV-Intron: erstes Intron der HCMV-major-immediate-early-Gene; VSV-G: codierende Sequenz für das VSV-G-Protein; VSV-Gsyn: nach Genbankeintrag J02428 codonoptimierte codierende Sequenz für das VSV-G-Protein; Ori: Replikationsursprung des SV40-Virus; Neomycin: Neomycin-Resistenzgen; Amp: Ampicillin-Resistenz-Gen.
g) pCI-GΔIΔSL
Aus pCI-G (s. Abb. 2-1) wurden das HCMV-MIE-Intron und die vermutlich stemloop bilden-
de HCMV-MIE-Exon-Sequenz entfernt. Dazu wurde pCI-G mit den Restriktionsendonuklea-
sen HindIII und SacII gespalten und zuviel herausgeschnittene Sequenzen durch Einfügen

2 Material und Methoden 12
einer Verbindungs-Sequenz, die durch Phosphorylierung und komplementäre Aneinanderla-
gerung der Oligonukleotide ΔSLs und ΔSLa gewonnen wurde, wieder ergänzt. Nach der Klo-
nierung wurde pCI-GΔIΔSL zur Identifizierung von der SacII- bis zur HindIII-Schnittstelle
sequenziert (s. 2.12.16).
h) pCSL-G
Aus pCI-G (s. Abb. 2-1) wurden das HCMV-MIE-Intron und die HCMV-MIE-Exon-Sequenz
5’ der stemloop bildenden Exon-Sequenz entfernt. Dazu wurde pCI-G mit den Restriktion-
sendonukleasen BamHI und SacI gespalten und zuviel herausgeschnittene Sequenzen durch
Einfügen einer Verbindungs-Sequenz, die durch Phosphorylierung und komplementäre Anei-
nanderlagerung der Oligonukleotide ΔSLs und ΔSLa gewonnen wurde, wieder ergänzt. Nach
der Klonierung wurde pCSL-G zur Identifizierung von der ersten SacI- bis zur BamHI-
Schnittstelle sequenziert (s. 2.12.16).
i) pCI-Gsyn-G
Durch PCR wurde das Start-Codon in der Wildtyp-DNA-Sequenz für VSV-G von ATG zu
ATA mutiert, 5’ der codierenden Sequenz eine EcoRI-Schnittstelle und unmittelbar 3’ des
Stop-Codons eine XhoI-Schnittstelle eingefügt. Das Amplifikat wurde mit den Restriktion-
sendonukleasen EcoRI und XhoI gespalten und in mit den gleichen Restriktionsendonuklea-
sen gespaltenes pCI-Gsyn (s. Abb. 2-1) kloniert.
j) pCI-GsynΔBam
pCI-GsynΔBam wurde zur Konstruktion von pCI-GsynMut benötigt. pCI-Gsyn (s. Abb. 2-1)
wurde mit der Restriktionsendonuklease BamHI gespalten, die entstandenen Einzelstrang-
Enden zu vollständigen Doppelsträngen aufgefüllt und intramolekular miteinander ligiert.
Nach Denaturierung der Ligase wurde vor der Klonierung eventuell noch vorhandenes unver-
ändertes pCI-Gsyn mit der Restriktionsendonuklease BamHI gespalten.
k) pCI-GsynMut
Die vsv-gsyn-Sequenz wurde an den Positionen 169 (A C), 171 (C G) und 288 (G C)
mittels PCR mutiert (s. 2.12.7). Dabei wurde die Sequenz zwischen der HindIII- und PmlI-
Schnittstelle unter Verwendung von Mutagenese-Primern in drei überlappenden Abschnitten

2 Material und Methoden 13
amplifiziert und die Abschnitte durch Overlap-Extension-PCR wieder zur Sequenz vollstän-
diger Länge zusammengefügt (s. 2.12.7). Nach Sequenzierung des Overlap-Extension-PCR-
Produkts wurde dieses mit den Restriktionsendonukleasen HindIII und PmlI gespalten und in
ebenso gespaltenes pCI-GsynΔBam kloniert. Plasmide mit BamHI-Schnittstelle wurden von
der HindIII- bis zur PmlI-Schnittstelle sequenziert (s. 2.12.16).
Wäre das mutierte Fragment in pCI-Gsyn statt in pCI-GsynΔBam kloniert worden, hätte nicht
durch Spaltung mittels Restriktionsendonukleasen zwischen dem mutierten Plasmid und ei-
nem als Verunreinigung mitgeführten, ungespaltenen bzw. einfach gespaltenen und religierten
pCI-Gsyn unterschieden werden können, da die Mutagenese keine Schnittstellen für verfügbare
Restriktionsendonukleasen betraf.
l) pCI-Gopt
Da unter den untersuchten pCI-GsynMut-Klonen, die alle mindestens eine zusätzliche, uner-
wünschte Mutation aufwiesen, ein Klon war, bei dem die unerwünschte Mutation nahe genug
an einer Restriktionsschnittstelle (PmlI-Schnittstelle) lag, wurde nicht weiter nach einem Klon
ohne zusätzliche Mutation gesucht, da dies wenig effizient schien, sondern die unerwünschte
Mutation durch eine einfache Mutagenese-PCR revertiert.
Dazu wurde die Sequenz von der HindIII- bis zur PmlI-Schnittstelle unter Verwendung eines
Mutagenese-Primers amplifiziert (s. 2.12.7), mit den Restriktionsendonukleasen PmlI und
HindIII gespalten und in ebenso gespaltenes pCI-GsynΔBam kloniert. Plasmide mit BamHI-
Schnittstelle wurden von der HindIII- bis zur PmlI-Schnittstelle sequenziert (s. 2.12.16).
Das so erhaltene pCI-Gopt trägt eine Sequenz, die für das gleiche VSV-G codiert wie pCI-G,
im Unterschied dazu allerdings codonoptimiert ist (s. auch Tab. 2-1).
2 Material und Methoden 14
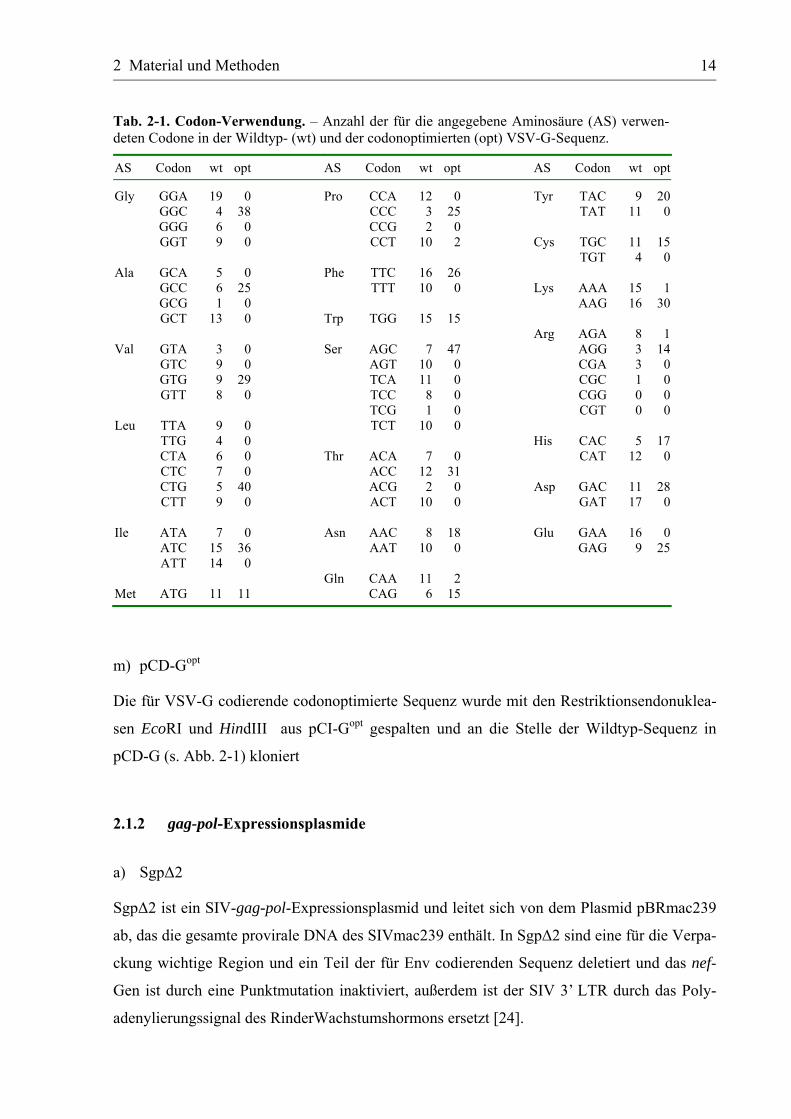
Tab. 2-1. Codon-Verwendung. – Anzahl der für die angegebene Aminosäure (AS) verwen-deten Codone in der Wildtyp- (wt) und der codonoptimierten (opt) VSV-G-Sequenz.
AS Codon wt opt AS Codon wt opt AS Codon wt opt
Gly GGA 19 0 Pro CCA 12 0 Tyr TAC 9 20 GGC 4 38 CCC 3 25 TAT 11 0 GGG 6 0 CCG 2 0 GGT 9 0 CCT 10 2 Cys TGC 11 15 TGT 4 0Ala GCA 5 0 Phe TTC 16 26 GCC 6 25 TTT 10 0 Lys AAA 15 1 GCG 1 0 AAG 16 30 GCT 13 0 Trp TGG 15 15 Arg AGA 8 1Val GTA 3 0 Ser AGC 7 47 AGG 3 14 GTC 9 0 AGT 10 0 CGA 3 0 GTG 9 29 TCA 11 0 CGC 1 0 GTT 8 0 TCC 8 0 CGG 0 0 TCG 1 0 CGT 0 0Leu TTA 9 0 TCT 10 0 TTG 4 0 His CAC 5 17 CTA 6 0 Thr ACA 7 0 CAT 12 0 CTC 7 0 ACC 12 31 CTG 5 40 ACG 2 0 Asp GAC 11 28 CTT 9 0 ACT 10 0 GAT 17 0 Ile ATA 7 0 Asn AAC 8 18 Glu GAA 16 0 ATC 15 36 AAT 10 0 GAG 9 25 ATT 14 0 Gln CAA 11 2 Met ATG 11 11 CAG 6 15
m) pCD-Gopt
Die für VSV-G codierende codonoptimierte Sequenz wurde mit den Restriktionsendonuklea-
sen EcoRI und HindIII aus pCI-Gopt gespalten und an die Stelle der Wildtyp-Sequenz in
pCD-G (s. Abb. 2-1) kloniert
2.1.2 gag-pol-Expressionsplasmide
a) SgpΔ2
SgpΔ2 ist ein SIV-gag-pol-Expressionsplasmid und leitet sich von dem Plasmid pBRmac239
ab, das die gesamte provirale DNA des SIVmac239 enthält. In SgpΔ2 sind eine für die Verpa-
ckung wichtige Region und ein Teil der für Env codierenden Sequenz deletiert und das nef-
Gen ist durch eine Punktmutation inaktiviert, außerdem ist der SIV 3’ LTR durch das Poly-
adenylierungssignal des RinderWachstumshormons ersetzt [24].

2 Material und Methoden 15
b) Hgpsyn
Dieses HIV-1-gag-pol-Expressionsplasmid wurde von Prof. Dr. Ralf Wagner erhalten. Es
basiert auf pcDNA3.1(+) und enthält unter der Kontrolle eines CMV-Promotors das bloße
synthetische, codonoptimierte HIV-1-gag-pol-Gen – ohne 5’ UTR und ohne Rev-responsive-
Element [26].
2.1.3 Vektor-Konstrukte
a) ViCGΔBH
Katrin Bräutigam ersetzte in dem SIV-Vektor-Konstrukt ViGΔBH [24], das sich von dem die
gesamte provirale SIVmac239-DNA enthaltenden Plasmid pBRmac239 ableitet, das an Posi-
tion des nef-Genes befindliche GFP-Gen mit stromaufwärts liegender U3-Region des Spleen
Focus Forming Virus durch das GFP-Gen aus HIV-CS-CG mit stromaufwärts liegendem
HCMV-MIE-Promotor und erhielt so ViCGΔBH. Die gag-pol-, vif-, vpx-, vpr-, env- und nef-
Gene sind durch Punktmutationen und Deletionen inaktiviert. Die U3-Region des 3’ LTR ist
fast vollständig deletiert, daher handelt es sich um einen sich selbst inaktivierenden Vektor
(SIN-Vektor).
b) HIV-CS-CG
Die U3-Region des 5’ LTR dieses von Ulrike Blömer erhaltenen HIV-1-Vektor-Konstrukts ist
durch einen HCMV-MIE-Promotor ersetzt und in der U3-Region des 3’ LTR TATA-Box
sowie Bindungsstellen für die Transkriptionsfaktoren Sp1 und NF-κB deletiert (SIN-Vektor).
Als Reportergen dient GFP mit einem internen HCMV-MIE-Promotor [23].
2.1.4 Weitere Plasmide
a) pcTat
Das von Joachim Hauber erhaltene HIV-1-tat-Expressionsplasmid enthält das tat-Gen eines
HIV-1 cDNA-Klons unter transkriptioneller Kontrolle eines HCMV-MIE-Promotors [93].

2 Material und Methoden 16
b) pcRev
Das von Joachim Hauber erhaltene HIV-1-rev-Expressionsplasmid enthält das rev-Gen eines
HIV-1 cDNA-Klons unter transkriptioneller Kontrolle eines HCMV-MIE-Promotors [93].
c) pSEAP
pSEAP-Control (BD Biosciences, Heidelberg; hier als „pSEAP“ bezeichnet) dient der Ex-
pression der Sekretierten Alkalischen Phosphatase in eukaryotischen Zellen.
2.2 Oligonukleotide und weitere Nukleinsäuren
Tab. 2-2. Primer für Polymerase-Kettenreaktionen und Sequenzierungen.
Bezeichnung Sequenz (5’-3’)
3’XhoI-VSV-Ga1 CTGCACTCGAGTTATTACTTTCCAAGTCGGT 5’EcoRI-VSV-Gs1 CCGCCGAATTCATAAAGTGCCTTTTGTACTTAG Exa1 GTACTCGTCCACCAGCACGT Exs1 TCCTTGACACGAAGCTTGGT ILa1 CTTCACTTGCAGGGCGGTGCCGATCAGGT ILs1 GCACCGCCCTGCAAGTGAAGATGCCCAA Krypta2 TGAGCTTGACGGGCAATAATGG Krypts2 TCAGATCGCCTGGAGACGCCAT M13 Forward (-20)3 GTAAAACGACGGCCAG
M13 Reverse3 CAGGAAACAGCTATGAC MutVSV-GsynMuta1 TCCACCAGCACGTGGTGAGGGGTCACTTGCACGATCACGGCCTCGGCGTCGGTCACG
GTGGCGTA pCI-GIa2 GTACAAAAGGCACTTCATGGTGG pCI-GIs2 CACGCTGTTTTGACCTCCATAGA QHa1 GATGCTGTGGGTGATGTACTTGGGGCCGTA QHs1 AGTACATCACCCACAGCATCAGGAGCTTCA SEQpCI-G11 CCTTTCCATGGGTCTTTTCTGC SEQpCI-G21 CGAAGCAGTGATTGTCCAGGTG SEQpCI-G31 GGGTCTTCCAATCTCTCCAGTGG SEQpCI-G41 TGTAGAGTTATGGACAGTGGGGCA SEQpCI-G51 CTGATCATTCCGACCATTCTTGAG SEQpCI-G61 ATGGCTGGCAACTAGAAGGCAC SEQpCI-GsynMuta1 CTTCACTTTGTAGTCGCTGT SEQpCI-GsynMuts1 CCACCAGACATAATAGCTGA
1 Bezugsquelle: SIGMA-ARK, Darmstadt 2 Bezugsquelle: biomers.net, Ulm 3 Bezugsquelle: Invitrogen, Karlsruhe

2 Material und Methoden 17
Tab. 2-3. Oligonukleotide für die komplementäre Anlagerung zu Verbindungssequenzen. Die Nukleinsäuren wurden von biomers.net, Ulm bezogen.
Bezeichnung Sequenz (5’-3’)
SLa GATCCGAGCTCGGTACCAAGCTTCGTGTCAAGGACGGTGAGTCACTCTTGGCACGCGGTTCACTAAACGAGCT
SLs CGTTTAGTGAACCGCGTGCCAAGAGTGACTCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCG ΔIa AGCTTCGTGTCAAGGACGGTGAGTCACTCTTGGCACGGGGAATCCGCGTTCCAATGCACCGTTCCC
GGCCGC ΔIs GGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGACTCACCGTCCTTGACACGA
ΔSLa AGCTTGGGAATCCGCGTTCCAATGCACCGTTCCCGGCCGC ΔSLs GGCCGGGAACGGTGCATTGGAACGCGGATTCCCA
Tab. 2-4. Weitere Nukleinsäuren und Nukleotide.
Bezeichnung Bezugsquelle
Fischsperma-DNA „DNA, MB grade“ Roche, Mannheim
ssRNA-Leiter NEB, Frankfurt/Main
100 bp DNA Leiter Invitrogen, Karlsruhe
1 kb DNA Leiter Invitrogen, Karlsruhe
dATP, dCTP, dGTP, dTTP Amersham, Freiburg
ATP Amersham, Freiburg
2.3 Eukaryonten und Prokaryonten
Tab. 2-5. Zelllinien.
Zelllinie Beschreibung ATCC-Nr.
293A Humane Nierenepithel-Zellen, mit Adenovirus-5-DNA transformiert CRL-1573 293T 293A-Zellen, in die das Gen für das große T-Antigen des SV40-Virus eingefügt wurde CRL-11268
Tab. 2-6. Bakterienstämme.
Bakterienstamm Genotyp Bezugsquelle
DH5α F-, φ80dlacZΔM15, Δ(lacZYA-argF)U169, deoR, recA1, endA1, hsdR17 (rk-, mk+), phoA, supE44, λ-, thi-1, gyrA96, relA1
Invitrogen, Karlsruhe
XL2-Blue recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F´ proAB lacIqZΔM15 Tn10 (Tetr) Amy Camr]
Stratagene, Amsterdam, NL

2 Material und Methoden 18
2.4 Enzyme, Antikörper und andere Proteine
Tab. 2-7. Polymerasen und DNA-/RNA-modifizierende Enzyme. Geeignete Reaktionspuffer wurden von den angegebenen Herstellern mitgeliefert.
Bezeichnung Produktname Bezugsquelle
DNase I DNA-free Ambion, Huntingdon, UKReverse Transkriptase SuperScript II RT Invitrogen, Karlsruhe RNase A Applichem, Darmstadt T4-DNA-Ligase TaKaRa DNA-Ligation-Kit Ver.2.1 MoBiTec, Göttingen T4-DNA-Ligase NEB, Frankfurt/Main T4-DNA-Polymerase NEB, Frankfurt/Main T4-Polynukleotid-Kinase NEB, Frankfurt/Main Taq- und Tfu-DNA-Polymerase-Mischung Arrow-Taq-Polymerase Qbiogene, Heidelberg Taq-DNA-Polymerase Amersham, Freiburg
Tab. 2-8. Restriktionsendonukleasen. Die roten Linien symbolisieren die Schnittstellen. Die Enzyme wurden mit Reaktionspuffern von NEB in Frankfurt am Main bezogen.
Bezeichnung Erkennungssequenz Bezeichnung Erkennungssequenz
BamHI G|G A T C C C C T A G|G SacI G A G C T|C
C|T C G A G
EcoRI G|A A T T C C T T A A|G SacII C C G C|G G
G G|C G C C
HindIII A|A G C T T T T C G A|A XhoI C|T C G A G
G A G C T|C
PmlI C A C|G T G G T G|C A C
Tab. 2-9. Antikörper und Protein-Größenstandards.
Bezeichnung Bezugsquelle Proteingrößenstandard „Precision Plus“ Biorad, München Monoklonaler Antikörper gegen HIV-1 p24 (AG3.0), produziert in Balb/c Milzzellen × SSP2/10 Myelomzellen, Zellkulturüberstand
AIDS Research and Reference Reagent Program, Dr. Jonathan Allan [94]
Monoklonaler Antikörper gegen VSV-G, produziert in Maus, Klon P5D4, Ascites-Flüssigkeit
Sigma-Aldrich, München
Polyklonale Kaninchen-Anti-Maus-Immunglobuline, meerrettichper-oxidasekonjugiert (HRP)
DakoCytomation, Hamburg

2 Material und Methoden 19
2.5 Chemikalien, kits, sonstige Verbrauchsmaterialien
Tab. 2-10. Chemikalien.
Chemikalie Bezugsquelle
4-(2-Hydroxyethyl)-1Piperazin-Ethansulfonsäure (HEPES), freie Säure Biomol, Hamburg Acrylamid-Bisacrylamid-Lösung 40 % (37,5:1) Carl Roth, Karlsruhe Agar Applichem, Darmstadt Agarose Invitrogen, Karlsruhe Ammoniumperoxodisulfat (APS) Carl Roth, Karlsruhe Ampicillin Applichem, Darmstadt Aqua B. Braun B. Braun, Melsungen Bromphenolblau Sigma-Aldrich, München Calciumchlorid (CaCl2) J. T. Baker, Griesheim Diethylpyrocarbonat (DEPC) Applichem, Darmstadt Dimethylsulfoxid (DMSO) Applichem, Darmstadt Dinatriumhydrogenphosphat (Na2HPO4) J. T. Baker, Griesheim Eisessig J. T. Baker, Griesheim Ethanol J. T. Baker, Griesheim Ethidiumbromidlösung 1% Carl-Roth, Karlsruhe Ethylendiaminotetraessigsäure (EDTA) VWR, Darmstadt Formaldehyd 37 % J. T. Baker, Griesheim Formamid (deionisiert) Sigma-Aldrich, München Glucose J. T. Baker, Griesheim Glycerin J. T. Baker, Griesheim Hefeextrakt Applichem, Darmstadt Isopropanol J. T. Baker, Griesheim Kaliumacetat J. T. Baker, Griesheim Kaliumchlorid J. T. Baker, Griesheim Magermilchpulver Heirler Cenovis, RadolfzellMagnesiumchlorid J. T. Baker, Griesheim Maleinsäure J. T. Baker, Griesheim β-Mercaptoethanol Sigma-Aldrich, München
Morpholinopropansulfonsäure (MOPS) Applichem, Darmstadt N,N,N’,N’-Tetramethylethylendiamin (TEMED) Carl Roth, Karlsruhe Natriumacetat J. T. Baker, Griesheim Natriumchlorid (NaCl) J. T. Baker, Griesheim Natriumcitrat J. T. Baker, Griesheim Natriumdodecylsulfat (SDS) Applichem, Darmstadt Natriumhydroxid (NaOH) J. T. Baker, Griesheim Saccharose Sigma-Aldrich, München Salzsäure (HCl) J. T. Baker, Griesheim Tris-(hydroxymethyl)-aminomethan (Tris) Applichem, Darmstadt Trypton Applichem, Darmstadt Tween 20 Applichem, Darmstadt

2 Material und Methoden 20
Tab. 2-11. Kits.
Bezeichnung Bezugsquelle
Alpha Innotech Chemiglow West Biozym, Hess. Oldendorf DIG High Prime DNA Labelling and Detection Starter Kit II Roche, Mannheim DIG PCR Probe Synthesis Kit Roche, Mannheim DNA-free Ambion, Huntingdon, UK Fast Track 2.0 Kit Invitrogen, Karlsruhe Jetquick DNA Clean-Up Spin Kit Genomed, Löhne Jetquick Gel Extraction Spin Kit Genomed, Löhne Jetquick PCR Purification Spin Kit Genomed, Löhne Jetstar Maxi Plasmid Purification Kit Genomed, Löhne Phospha-Light-SEAP-Reporter-Gene-Assay-System Applied Biosystems, Darmstadt Qiagen Plasmid Maxi Kit Qiagen, Hilden Qiaprep Spin Miniprep Kit Qiagen, Hilden Qiaquick Gel Extraction Kit Qiagen, Hilden Qiaquick Nucleotide Removal Kit Qiagen, Hilden Qiaquick PCR Purification Kit Qiagen, Hilden RNeasy Mini Kit Qiagen, Hilden Superscript First-Strand Synthesis System for RT-PCR Invitrogen, Karlsruhe TaKaRa DNA-Ligation-Kit Ver.2.1 MoBiTec, Göttingen Topo TA Cloning Invitrogen, Karlsruhe
Tab. 2-12. Sonstige Verbrauchsmaterialien.
Bezeichnung Bezugsquelle
0,2-µm-Spritzenvorsatzfilter Sarstedt 0,45-µm-Spritzenvorsatzfilter Sarstedt Filterpapier Whatman Schleicher & Schuell, Dassel Filterpapier für „Mini Trans-Blot Cell“ Bio-Rad, München Nytran Nylon-Membran Whatman Schleicher & Schuell, Dassel Protran Nitrocellulose-Membran Whatman Schleicher & Schuell, Dassel Zellstoff B. Braun, Melsungen Zentrifugenröhrchen Beckman, Krefeld
2.6 Kunststoff-Einmalartikel
Einmalartikel aus Kunststoff (z.B. Zellkulturflaschen, Pipetten, Pipettenspitzen, Reaktionsge-
fäße usw.) wurden von den Firmen Nunc (Wiesbaden), Sarstedt (Nümbrecht), Biozym (Hes-
sisch Oldendorf), Starlab (Ahrensburg) und Greiner (Frickenhausen) bezogen.

2 Material und Methoden 21
2.7 Puffer, Medien und Lösungen
Tab. 2-13. Medien, Puffer, Lösungen für Arbeiten mit Zellkulturen und Lentiviren. In „Aqua B. Braun“. Für Fertigprodukte ist die Bezugsquelle angegeben.
Medium Zusammensetzung Bezugsquelle
Kulturmedium DMEM 10 % (v/v) FBS 1 % (v/v) Penicillin-Streptomycin
Einfriermedium Kulturmedium 10 % (v/v) DMSO frisch angesetzt, auf Eis gekühlt
Infektionsmedium DMEM 5 % (v/v) FBS 1 % (v/v) Penicillin-Streptomycin
Dulbecco’s Modified Eagle Medium (DMEM)
mit L-Glutamin 4,5 g/l D-Glucose ohne Pyruvat
Invitrogen, Karlsruhe
Fötales Rinderserum (FBS) 30 min bei 56 °C inaktiviert Biochrom, Berlin Penicillin-Streptomycin-Lösung 10000 U/ml Penicillin G
10000 µg/ml Streptomycin in 0,85 % NaCl-Lösung
Invitrogen, Karlsruhe
10 × PBS 1,37 M NaCl 27 mM KCl 43 mM Na2HPO4 14 mM KH2PO4 pH 7,4
Invitrogen, Karlsruhe
2 M CaCl2 2 M CaCl2 steril filtriert
10 × HBS 250 mM HEPES (freie Säure) 1,4 M NaCl 14 mM Na2HPO4
2 × HBS 20 % (v/v) 10 × HBS, Aliquote mit pH 7,05 bis 7,15 Transfektionseffizienzen ermit-telt, ungeeignete Aliquote verworfen

2 Material und Methoden 22
Tab. 2-14. Bakterienmedien. In Reinstwasser.
Bezeichnung Zusammensetzung
S.O.C.-Medium 2 % (w/v) Trypton 0,5 % (w/v) Hefe-Extrakt 8,6 mM NaCl 2,5 mM KCl 10 mM MgCl2 20 mM Glucose pH 7,0
LB-Ampicillin-Agar-Platten 1,5 % (w/v) Agar in LB-Medium 0,01 % (w/v) Ampicillinin Petrischalen gegossen
LB-Medium 1 % (w/v) Trypton 0,5 % (w/v) Hefeextrakt 0,17 M NaCl pH 7,0
LB-Ampicillin-Medium LB-Medium 0,01 % (w/v) Ampicillin
Tab. 2-15. Puffer und Lösungen für RNA-Agarose-Gelelektrophorese und Northernblots.
Bezeichnung Zusammensetzung
RNA-Probenpuffer 50 % (v/v) Formamid (deionisiert) 16,6 % (v/v) Formaldehyd 37 % 10 % (v/v) 10 × MOPS 0,2 ‰ (w/v) Bromphenolblau 1,5 ‰ (v/v) Ethidiumbromidlösung 1 % in DEPC-Wasser
DEPC-Wasser Reinstwasser mit 1 ‰ DEPC geschüttelt über Nacht unter dem Abzug inkubiert DEPC durch Autoklavieren inaktiviert
10 × MOPS 200 mM MOPS 50 mM Natriumacetat 20 mM EDTA in DEPC-Wasser pH 7,0
20 × SSC 3 M NaCl 300 mM Natriumcitrat in Reinstwasser pH 7,0
DIG Easy Hyb, Anti-Digoxigenin-Antikörper, CSPD
Bestandteile des „DIG High Prime DNA Labelling and Detection Starter Kit II“
Tab. 2-16. Puffer für die Agarose-Gelelektrophorese.
Puffer Bezugsquelle
50 × TAE Applichem, Darmstadt10 × TBE Applichem, Darmstadt

2 Material und Methoden 23
Tab. 2-17. Puffer und Lösungen für SDS-PAGE, Westernblots und Probenaufarbeitung. In Reinstwasser, falls nicht anders angegeben.
Bezeichung Zusammensetzung
WB-Probenpuffer 1,1 % (w/v) SDS 11 % (v/v) Glycerin 11 % (v/v) β-Mercaptoethanol 8,9 % (v/v) 0,5 M Tris-Puffer, pH 6,8 1,1 ‰ (w/v) Bromphenolblau
20 % Saccharose-Lösung 20 % (w/v) Saccharose 10 % (v/v) 10 × PBS in „Aqua B. Braun“ steril filtriert
10 % Trenngel 25 % (v/v) Acrylamid-Bisacrylamid-Lösung 40 % (37,5:1) 33 % (v/v) 1,5 M Tris-Puffer, pH 8,8 0,1 % (w/v) SDS 0,1 % (w/v) APS 0,8 ‰ (v/v) TEMED
Sammelgel 12,5 % (v/v) Acrylamid-Bisacrylamid-Lösung 40 % (37,5:1)14 % (v/v) 0,5 M Tris-Puffer, pH 6,8 0,1 % (w/v) SDS 0,1 % (w/v) APS 0,8 ‰ (v/v) TEMED
10 × MBS 1 M Maleinsäure 1,5 M NaCl pH 7,5
MBS-T 10 % (v/v) 10 × MBS 0,3 % (v/v) Tween 20
5 % Magermilch 5 % (w/v) Magermilchpulver in MBS-T
Anti-HIV-1-p24-Lösung 0,5 ‰ (v/v) Monoklonaler Antikörper gegen HIV-1 p24 in 5 % Magermilch
Anti-VSV-G-Lösung 0,01 ‰ (v/v) Monoklonaler Antikörper gegen VSV-G in 5 % Magermilch
Anti-Maus/HRP-Lösung 0,5 ‰ (v/v) Kaninchen-Anti-Maus-Immunglobuline/HRP in 5 % Magermilch
Chemiglow-West-Lösung 50 % (v/v) Chemiglow-West-Luminol/Enhancer 50 % (v/v) Chemiglow-West stabile Peroxid-Lösung
Tab. 2-18. Puffer für Mini-Plasmidpräparationen. In Reinstwasser.
Puffer Zusammensetzung
P1 50 mM Tris 10 mM EDTA 0,1 ‰ RNase A pH 8,0
P2 200 mM NaOH 1 % (w/v) SDS
P3 3 M KaliumacetatpH 5,5
1/10 × EB 1 mM Tris pH 8,5

2 Material und Methoden 24
2.8 Technische Ausstattung
Tab. 2-19. Geräte und Apparaturen.
Gerät Bezugsquelle
Analysenwaage “Scaltec SPB63” Denver, Göttingen Bilddokumentations-System “ChemiImager 4400” Biozym, Hess. Oldendorf CO2-Inkubator „Heraeus HERAcell 240“ Thermo Electron, Langenselbold Elektroblot-Apparatur “Mini Trans-Blot Cell” Bio-Rad, München Elektrophorese-Apparatur (horizontal) “PerfectBlue” Peqlab, Erlangen Elektrophorese-Apparatur (vertikal) “Mini Protean 3” Bio-Rad, München Folienschweißgerät Krups, Solingen Fotometer „BioPhotometer“ Eppendorf, Hamburg Flüssigstickstoff-Behälter „MVE Cryosystem 6000“ Chart, Marietta, GA, USA Gefrierschrank (-20 °C) Siemens, München Gefrierschrank (-80 °C) „-86C ULT Freezer“ Thermo Electron, Dreieich Gefrierschrank (-80 °C) „MDF-U71V“ Sanyo, München Heißluftsterilisator “Heraeus T6420” Thermo Electron, Langenselbold Hochsensitives Bilddokumentations-System Hamamatsu, Herrsching am Ammersee Inverses Fluoreszenzmikroskop “Axiovert 100” Carl Zeiss, Oberkochen Inverses Mikroskop “TMS” Nikon, Düsseldorf Kreisschüttler „Certomat SII“ Sartorius, Göttingen Magnetrührer “IKA-Combimag RCT” IKA-Werke, Staufen Mikropipetten “Pipetman P” Gilson, Bad Camberg Mikrotiterplatten-Luminometer “Orion” Berthold, Pforzheim Mikrowellengerät “R-220A” Sharp, Hamburg Mikrozentrifuge „5417 C“ Eppendorf, Hamburg Neubauer-Zählkammer Carl Roth, Karlsruhe pH-Meter “GPHR 1400A” Greisinger Electronic, Regenstauf Pipettierhilfe “Pipetus-akku” Hirschmann, Eberstadt Präzisionswaage “Scaltec SBA31” Denver, Göttingen Reinstwasseranlage “Seralpur Pro 90 CN” Elga, Celle Schüttelgerät “Scientific Industries Vortex-Genie 2” Carl Roth, Karlsruhe Schüttelinkubator “Thermomixer comfort” Eppendorf, Hamburg Schüttelwasserbad “1086” GFL, Burgwedel Schüttler (wippend) „Rockomat“ Tecnomara, Zürich, CH Sicherheitswerkbank „Heraeus Herasafe“ Thermo Electron, Langenselbold Spannungsgeber “Consort E835” Peqlab, Erlangen Spannungsgeber „Power Pack P25” Biometra, Göttingen Thermocycler “MJ Research PTC100” Bio-Rad, München Tischzentrifuge „6K15“ mit Winkelrotor „12500“ Sigma, Osterode am Harz Tischzentrifuge „GPR“ mit Ausschwingrotor Beckman Coulter, Krefeld Ultrazentrifuge „Optima L-70 K“ mit Ausschwingrotor „SW 41 Ti“ Beckman Coulter, Krefeld Wasserbad Memmert, Schwabach Wasserbad Breda Scientific, Breda, NL
Zur Ausstattung gehörten ferner ein 37°C- und zwei 4°C-Räume.

2 Material und Methoden 25
2.9 Arbeiten mit Zellkulturen und Lentiviren
2.9.1 Kultivierung von 293T- und 293A-Zellen
Die Zellen wurden zur Erhaltung in 75 cm2 Zellkulturflaschen in 20 ml Kulturmedium kulti-
viert. Die Kulturen wurden dreimal wöchentlich passagiert; dazu wurde das Kulturmedium
verworfen, der Zellrasen mit 10 ml 1 x PBS abgespült, die Zellen mit 2 ml Trypsinlösung
abgelöst und nach Zugabe von 8 ml frischem Kulturmedium resuspendiert. 1 ml der Suspen-
sion wurde zu 19 ml Kulturmedium in eine neue Zellkulturflasche übertragen, der Rest ent-
weder verworfen oder für weitere Experimente verwendet. Die Kulturen wurden bei 37 °C
unter 5 % CO2 und gesättigter Luftfeuchtigkeit inkubiert; diese Bedingungen wurden bei allen
Zellkultur-Inkubationen in den im Folgenden beschriebenen Methoden eingehalten.
2.9.2 Kryokonservierung von Zellen
Um den Bestand kryokonservierter Kulturen zu erhalten, wurden frische Kulturen in 175 cm2
Zellkulturflaschen expandiert. Bei Erreichen von 80 % Konfluenz wurde der Zellkultur-
Überstand verworfen, der Zellrasen mit 20 ml PBS gespült, die Zellen mit 4 ml Trypsinlösung
abgelöst, nach Zugabe von 46 ml Kulturmedium resuspendiert, mit Hilfe einer Neubauer-
Zählkammer gezählt und durch Zentrifugation bei 1000 Upm (Tischzentrifuge „GPR“) sedi-
mentiert. Mit eiskaltem Einfriermedium wurden die Zellen auf Eis in einer Dichte von 5 × 106
bis 1 × 107 Zellen/ml resuspendiert und in 1-ml-Portionen auf Kryogefäße verteilt. Die Kultu-
ren wurden in einem isolierten Gefäß bei – 80 °C langsam eingefroren. Nach zwei Tagen
wurden die Kulturen in einen mit Flüssigstickstoff gefüllten Behälter überführt und dort gela-
gert.
2.9.3 Auftauen kryokonservierter Zellen
Die Eigenschaften der verwendeten 293T- und 293A-Zellen veränderten sich nach einigen
Passagen, daher mußten alle zwei bis drei Monate die Kulturen verworfen und kryokonser-
vierte Zellen niedriger Passage frisch in Kultur genommen werden. Ein Kryogefäß mit 1 ml
Kultur wurde aus dem Flüssigstickstoff-Behälter direkt in ein 37 °C warmes Wasserbad über-
führt und dort unter Schwenken aufgetaut. Unmitteltbar nach dem Auftauen wurde die Zell-

2 Material und Methoden 26
suspension zu 20 ml Kulturmedium gegeben und die Zellen dann gleich durch Zentrifugation
bei 1000 Upm (Tischzentrifuge „GPR“) sedimentiert. Das Sediment wurde in 5,5 ml Kultur-
medium resuspendiert, 5 ml der Suspension wurden in eine 25 cm2 Zellkulturflasche gegeben,
die restlichen 0,5 ml wurden zu 4,5 ml Kulturmedium in eine weitere 25 cm2 Zellkulturfla-
sche gegeben. Nach etwa acht Stunden Inkubation wurde das Kulturmedium in beiden Kultu-
ren gegen frisches ausgetauscht. War die dünner ausgesäte Kultur nach zwei bis drei Tagen
konfluent, so wurde diese expandiert, die andere verworfen, andernfalls wurde die dichter
ausgesäte Kultur expandiert.
2.9.4 Transiente Transfektion von 293T-Zellen mittels Calcium-Phosphat
a) Zur Vektor-Produktion, für die Westernblot-Analyse und für die Total-RNA-Präparation
Am Tag vor der Transfektion wurden 1,2 × 106 Zellen in eine 25 cm2 Zellkulturflasche aus-
plattiert. Unmittelbar vor der Transfektion wurde das Medium (5 ml pro Flasche) gewechselt,
dazu wurde stets frisch angesetztes Medium verwendet. Die zu transfizierende DNA (s. Ab-
bildungsunterschriften im Kapitel 3 Ergebnisse) wurde gegebenenfalls mit Fischsperma-DNA
auf 15 µg aufgefüllt und auf Eis zu 31 µl 2 M CaCl2 gegeben. Der Ansatz wurde mit „Aqua
B. Braun“ auf 250 µl aufgefüllt, kurz gevortext und dazu unter Vortexen 250 µl eisgekühlter
2 × HBS-Puffer zugetropft. Der gesamte Ansatz wurde dann zum Überstand der zu transfizie-
renden Zellen getropft und die Flasche in waagerechter Position vorsichtig geschwenkt, um
eine gleichmäßige Verteilung der Calcium-Phosphat-DNA-Präzipitate zu gewährleisten. Acht
bis zwölf Stunden nach der Transfektion wurde das Medium gewechselt.
b) Für die mRNA-Präparation
Am Tag vor der Transfektion wurden 7 × 106 293T-Zellen in eine 175 cm2 Zellkulturflasche
ausgesät. Unmittelbar vor der Transfektion wurde der Zellkulturüberstand durch 35 ml frisch
angesetztes Kulturmedium ersetzt.
Auf Eis wurden zu 217 µl 2M CaCl2 35 µg des SIV-Vektor-Plasmids ViCGΔBH, 35 µg des
HIV-1-gag-pol-Expressions-Plasmids Hgpsyn, 14 µg VSV-G-Expressionsplasmid und 21 µg
Fischsperma-DNA gegeben, mit „Aqua B. Braun“ auf 3,5 ml aufgefüllt und kurz gevortext.
Dazu wurden unter vortexen 3,5 ml eiskalter 2 × HBS-Puffer getropft und der gesamte Ansatz
anschließend zum Überstand der zu transfizierenden Zellen getropft. Durch vorsichtiges
Schwenken der Flasche in waagerechter Position wurde für eine gleichmäßige Verteilung der

2 Material und Methoden 27
Calcium-Phosphat-DNA-Präzipitate gesorgt. Acht bis zwölf Stunden nach der Transfektion
wurde das Kulturmedium gewechselt. Zwei Tage nach Transfektion wurde die mRNA präpa-
riert
2.9.5 Ernte lentiviraler Vektoren
Zwei Tage nach der Transfektion wurde der Zellkulturüberstand 5 bis 10 min bei 1200 Upm
(Tischzentrifuge „GPR“) zentrifugiert und der Überstand anschließend durch einen 0,45 µm
Spritzenvorsatz-Filter filtriert. Das Filtrat wurde sofort in Infektions-Experimenten eingesetzt,
nicht benötigte Mengen wurden in Portionen von 1 ml bei – 80 °C aufbewahrt.
2.9.6 Infektion von 293A-Zellen mit lentiviralen Vektoren
Am Tag vor der Infektion wurden unter Verwendung von Infektionsmedium 1 × 105 293A-
Zellen pro Kavität in eine 24-Kavitäten-Platte ausgesät. Zur Infektion wurden frisch geerntete
oder im Wasserbad bei 37 °C aufgetaute Vektor-Präparate 1:10 und 1:100 in Infektionsmedi-
um verdünnt. Von den Zellen wurde der Überstand entfernt und 200 µl unverdünntes, 1:10-
bzw. 1:100 verdünntes Vektorpräparat zugegeben. Nach einer Inkubation von 4 h wurden die
Kavitäten mit Infektionsmedium auf je 1 ml aufgefüllt.
2.9.7 Titerbestimmung
Da alle eingesetzten Vektorkonstrukte das Gen für das grün fluoreszierende Protein (GFP)
enthalten, konnte die grüne Fluoreszenz für die Titerbestimmung ausgenutzt werden. Mit Hil-
fe eines inversen Fluoreszenzmikroskops wurden die grün fluoreszierenden Zellen in einer der
angesetzten Verdünnungsstufen in 7 von 196 Gesichtsfeldern einer Kavität zwei bis drei Tage
nach der Infektion ausgezählt. Der Titer ergab sich dann durch Multiplikation mit dem Ver-
dünnungsfaktor, anschließender Multiplikation mit dem Faktor 196/7, um die Zahl der grünen
Zellen in der gesamten Kavität zu errechnen, sowie Multiplikation mit dem Faktor 5, um die
Zahl der grüne-Fluoreszenz-bildenden Einheiten in 1 ml unverdünntem Vektorpräparat
(GFU/ml) zu erhalten.

2 Material und Methoden 28
2.10 Arbeiten mit Escherichia coli
2.10.1 Transformation von E. coli
Plasmide wurden in E. coli transformiert, um durch anschließende Vermehrung der transfor-
mierten E. coli eine Vermehrung der Plasmide zu erreichen. Es wurden kommerziell erwor-
bene, chemisch kompetente E. coli XL2-Blue sowie von Dr. Susann Lucke und Dr. Hella
Monse hergestellte chemisch kompetente E. coli DH5α transformiert.
Dazu wurden 1 bis 2 µl einer Plasmidlösung (100 ng/µl), eines Ligationsproduktes oder eines
TA-Klonierungs-Produktes zu 10 bis 25 µl E. coli XL2-Blue- bzw. 50 µl E. coli DH5α-
Suspension gegeben, behutsam gemischt und 30 min auf Eis inkubiert. Nach einem Hitze-
schock von 45 s bei 42 °C wurde für weitere 2 min auf Eis inkubiert und das 9fache des Vo-
lumens der Bakterien-Suspension an S.O.C.-Medium zugegeben. Es wurde 1 h unter Schüt-
teln bei 37 °C inkubiert. Zuletzt wurde der gesamte Ansatz oder nur ein Teil davon mit einem
Drigalski-Spatel auf LB-Ampicillin-Agar-Platten ausplattiert und über Nacht bei 37 °C inku-
biert.
2.10.2 Kultivierung von E. coli
a) Für Mini-Plasmidpräparationen
Eine Kolonie auf einer LB-Ampicillin-Agar-Platte mit transformierten E. coli wurde mit einer
Pipettenspitze durchstoßen und letztere in ein vorbereitetes Kultur-Röhrchen mit 2 bis 3 ml
LB-Ampicillin-Medium gegeben. Alternativ wurden 2 bis 3 ml LB-Ampicillin-Medium mit
50 µl LB-Ampicillin-Medium angeimpft, das seinerseits beim Ansatz einer Kolonie-PCR (s.
2.12.7) angeimpft wurde. Die Kulturen wurden über Nacht bei 37 °C geschüttelt.
b) Für Maxi-Plasmidpräparationen
100 bis 500 ml LB-Ampicillin-Medium wurden mit 1/1000 Volumen Vorkultur (z. B. Rest
einer Kultur für Mini-Plasmidpräparation) oder 1/1000 Volumen Glycerinkultur angeimpft.
Die Kultur wurde über Nacht bei 37 °C geschüttelt.

2 Material und Methoden 29
c) Glycerinkultur
Zur Kryokonservierung wurden 500 µl einer frischen E. coli Kultur vorsichtig mit 500 µl
Glycerin vermischt und bei – 20 °C eingefroren.
2.11 Arbeiten mit Proteinen
2.11.1 SEAP-Bestimmung
Zwei Tage nach Cotransfektion des Expressionsplasmids für die Sekretierte Alkalische
Phosphatase pSEAP (100 ng auf 15 µg Gesamt-DNA) wurde das Enzym im Zellkulturüber-
stand mit Hilfe des „Phospha-Light-SEAP-Reporter-Gene-Assay-System“ quantifiziert. Dabei
erfolgte die Bestimmung im Chemilumineszenz-Verfahren mit CSPD als Substrat. Die An-
satzgröße wurde halbiert, ansonsten wurden die Angaben des Herstellers genau befolgt. Die
Chemilumineszenz wurde mittels Mikrotiterplatten-Luminometer gemessen.
2.11.2 Westernblot-Analyse
a) Lyse transfizierter Zellen für den Westernblot
Zwei Tage nach Transfektion wurde der Zellkultur-Überstand dekantiert und der Zellrasen
vorsichtig mit PBS gespült. Anschließend wurden 500 µl WB-Probenpuffer auf den Zellrasen
pipettiert. Nach kurzer Inkubation bei Raumtemperatur wurde das visköse Lysat in ein 1,5 ml
Reaktionsgefäß überführt und bis zur Verwendung bei – 20 °C aufbewahrt.
b) Aufarbeitung der Viruspartikel für den Westernblot
Die Viruspartikel wurden aus dem Vektorpräparat mittels Ultrazentrifugation durch ein Sac-
charosekissen sedimentiert. Dazu wurden 2 ml 20 % Saccharose-Lösung in ein Zentrifugen-
röhrchen vorgelegt, das Vektorpräparat darüber geschichtet und das Röhrchen mit Infekti-
onsmedium aufgefüllt. Es wurde mit 28000 Upm für 2 h bei 4 °C in der Ultrazentrifuge
zentrifugiert und anschließend der gesamte flüssige Überstand abgehoben. Das (unsichtbare)
Sediment wurde durch wiederholtes Abspülen des Bodens des Zentrifugenröhrchens mit
100 µl WB-Probenpuffer in selbigem gelöst.

2 Material und Methoden 30
c) Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Das Gießen des vertikalen Gels und die Vorbereitung der Elektrophoresekammer erfolgten
nach Angaben des Herstellers der Apparatur „Mini Protean 3“.
Das Gel bestand zu etwa ¾ aus Trenngel und zu etwa ¼ aus Sammelgel.
Es wurden 5 µl des Größenstandards „Precision Plus“ und je 20 µl der Zell-Lysate bzw. Vi-
ruspartikel-Proben aufgetragen. Die Zell-Lysate und die Viruspartikel-Proben sind zuvor für
5 min bei 100 °C denaturiert und für 2 min bei 14000 Upm in der Mikrozentrifuge zentrifu-
giert worden. Die Elektrophorese erfolgte für 45 min bei 200 V.
d) Transfer und Immunodetektion
Nach SDS-PAGE wurden die Proteine aus dem Trenngel auf eine Nitrocellulose-Membran im
Elektroblot-Verfahren in 1 h bei 100 V übertragen. Die Apparatur „Mini Trans-Blot Cell“
wurde dabei nach Angaben des Herstellers zusammengesetzt.
Die Membran wurde dann 2 min in MBS-T gewaschen, über Nacht in 5 % Magermilch ge-
blockt und anschließend unterhalb der 50 kD Bande des Größenstandards durchtrennt. Der
Abschnitt mit den Proteinen kleiner als 50 kD wurde für 3 h in Anti-HIV-1-p24-Lösung inku-
biert. Der Abschnitt mit den Proteinen größer als 50 kD wurde für 3 h in Anti-VSV-G-Lösung
inkubiert. Die Membranen wurden dann fünfmal für 5 min in MBS-T gewaschen, für 2 h in
Anti-Maus/HRP-Lösung inkubiert und wieder fünfmal für 5 min in MBS-T gewaschen.
Zur Detektion wurden insgesamt 0,5 ml Chemiglow-West-Lösung auf die Membranen geträu-
felt, diese dann 5 min zwischen zwei Folien inkubiert und nach Herauswalzen der überschüs-
sigen Flüssigkeit eingeschweißt. Die Chemilumineszenz wurde mit einer hochsensitiven Ka-
mera dokumentiert.
2.12 Arbeiten mit Nukleinsäuren
2.12.1 Fotometrische Bestimmung von Nukleinsäure-Konzentrationen
Mit einem Fotometer wurde die Extinktion der DNA (RNA) in Wasser bei 260 nm gemessen.
Einer Extinktion von 1 entsprechen 50 µg DNA (40 µg RNA) pro ml Wasser.

2 Material und Methoden 31
2.12.2 mRNA-Präparation
Zur Lyse der Zellen und zur mRNA-Präparation wurde das „Fast-Track 2.0 Kit“ nach den
Angaben des Herstellers benutzt. Dabei wurde die Lyse durch SDS vermittelt und zur Bin-
dung und Isolation der PolyA+-RNA Oligo-dT-Zellulose-Pulver eingesetzt. Das gewonnene
RNA-Sediment wurde in 25 µl Fast-Track-2.0-Elutions-Puffer gelöst.
2.12.3 Northernblot-Analyse
a) Synthese der Sonde
Die Sonde wurde mit dem „PCR DIG Probe Synthesis Kit“ – aus dem Digoxigenin (DIG)
Markierungs- und Detektionssystem von Roche – durch Einbau von DIG-11-dUTP mittels
Polymerase-Ketten-Reaktion generiert. Sie war gegen die 130 b 5’ des AAUAAA-Motivs des
Rinder-Polyadenylierungssignals im hgpsyn-Transkript und in den Transkripten der
pcDNA3.1(+) basierten VSV-G-Expressionsplasmide gerichtet.
b) Denaturierung der Proben
Die mRNA-Proben und die RNA-Leiter wurden mit je 2 Volumen RNA-Probenpuffer ver-
setzt, 10 min bei 65 °C denaturiert und sofort auf Eis gekühlt. Es wurden je 2 µg mRNA bzw.
5 µl RNA-Leiter eingesetzt.
c) RNA-Agarose-Gelelektrophorese
Die mRNA-Proben wurden in einem 1,5 %igen Agarose-Gel getrennt; dazu wurden 0,75 g
Agarose in 47,3 ml 1 × MOPS aufgekocht, 2,7 ml Formaldehyd (37 %) zugefügt und das Gel
gegossen. Die horizontale Elektrophorese-Apparatur wurde so hergerichtet, dass die Puffer-
kammern nur so weit mit 1 × MOPS gefüllt wurden, dass das Gel nicht überschichtet wurde.
Die Proben wurden in die trockenen Taschen gefüllt und sind zunächst bei 100 V für ca.
10 min ins Gel eingelaufen. Dann wurde das Gel mit 1 × MOPS überschichtet und die Proben
bei 80 V für 2 h getrennt.
d) Transfer der RNA
Die aufgetrennten Proben wurden durch Kapillar-Transfer auf die positiv geladene Nylon-
membran übertragen. Dazu wurde ein rechteckiger Plastik-Deckel von der Größe des Gels in

2 Material und Methoden 32
ein 20 × SSC Reservoir gestellt, ein mit 20 × SSC durchtränktes, dickes Filterpapier als Brü-
cke so über den Deckel gelegt, dass beide Enden in das 20 × SSC ragten. Darauf wurde das
Gel gelegt, dann folgte die Membran, anschließend ein trockenes Filterpapier von der Größe
des Gels. Passgenau um das Gel wurde eine Maske gelegt, die aus einer Klarsichthülle zuge-
schnitten worden war, um den direkten Kontakt der über dem Gel befindlichen trockenen La-
gen mit der unter dem Gel befindlichen nassen Lage zu verhindern. Auf das trockene Filter-
papier wurde ein etwa 10 cm hoher Stapel Zellstoff gelegt, das sandwich wurde mit einer
Glasplatte und einem Gewicht abgeschlossen. Der Transfer erfolgte über Nacht, die Membran
wurde anschließend kurz in 2 × SSC gewaschen und für 30 min bei 120 °C im Heißluftsterili-
sator gebacken.
e) Hybridisierung und Detektion der Sonde
Prähybridisierung und Hybridisierung mit „DIG Easy Hyb“ sowie nachfolgende Wasch-
Schritte erfolgten nach den Angaben des Herstellers des Digoxigenin (DIG) Markierungs- und
Detektionssystems unter Verwendung der dort aufgeführten Puffer und Lösungen; dabei ent-
hielt die Hybridisierungslösung 4 µl Sonde/ml „DIG Easy Hyb“ und für die Prähybridisie-
rung, die Hybridisierung sowie den hochstringenten Wasch-Schritt wurde eine Temperatur
von 50 °C gewählt.
Die hybridisierte Sonde wurde mit einem Anti-Digoxigenin-Antikörper, der mit Alkalischer
Phosphatase konjugiert war, unter Verwendung des Substrats CSPD im Chemilumineszenz-
Verfahren nach Angaben des Herstellers detektiert. Die Chemilumineszenz wurde mit einer
hochsensitiven Kamera dokumentiert.
2.12.4 Total-RNA Präparation mit anschließender DNase-Spaltung
Zwei Tage nach Transfektion wurden mit Trypsin abgelöste 293T-Zellen durch Zentrifugati-
on bei 1200 Upm (Tischzentrifuge „GPR“) sedimentiert, mit eiskaltem PBS gewaschen, er-
neut sedimentiert, die Sedimente in flüssigem Stickstoff schockgefroren und bis zur RNA-
Präparation bei -80 °C gelagert.
Die Total-RNA wurde mit Hilfe des „RNeasy Mini Kits“ nach dem Protokoll des Herstellers
gewonnen. Unter Verwendung von „DNA-free“ wurden anschließend eventuelle DNA-
Verunreinigungen in der Total-RNA-Präparation mittels DNase gespalten und die DNase an-
schließend inaktiviert, dabei wurden die Anweisungen des Herstellers genau befolgt.

2 Material und Methoden 33
2.12.5 RNA-Struktur-Vorhersage
Zur Vorhersage von RNA-Sekundär-Strukturen wurde das Programm „RNAfold“ mit den
Standard-Einstellungen über den Vienna-RNA-Web-Server (http://rna.tbi.univie.ac.at/) be-
nutzt [95].
2.12.6 Plasmidpräparationen
Plasmide wurden aus E. coli im kleinen (Mini-) und großen (Maxi-) Maßstab unter Verwen-
dung von kits präpariert. Bei geringeren Anforderungen an die Reinheit des Präparats wurden
Mini-Plasmidpräparationen nach der folgenden kostengünstigeren Methode durchgeführt:
1,5 ml Kultur wurden 1 min bei 14000 Upm in der Mikrozentrifuge zentrifugiert und das Se-
diment anschließend in 250 µl Puffer P1 resuspendiert. Mit 250 µl Puffer P2 wurden
die Bakterien dann unter Schwenken lysiert. Durch Zugabe von 250 µl Puffer P3 und unter
erneutem Schwenken wurden genomische Bakterien-DNA, Proteine, Zelltrümmer und SDS
gefällt. Nach Zentrifugation für 10 min bei 14000 Upm in der Mikrozentrifuge wurde der Ü-
berstand in ein neues Reaktionsgefäß überführt. Durch Zugabe von 0,7 Volumen Isopropanol
wurde die Plasmid-DNA gefällt. Nach Zentrifugation für 1 h bei 4 °C und 14000 Upm in der
Mikrozentrifuge wurde das DNA-Sediment mit 70 % (v/v) Ethanol gewaschen, dann wurde
für 10 min bei 4 °C und 14000 Upm in der Mikrozentrifuge zentrifugiert. Der Überstand wur-
de dekantiert, das DNA-Sediment an der Luft getrocknet und in 30 µl 1/10 × EB gelöst.
2.12.7 Polymerase-Kettenreaktion (PCR)
a) Mutagenese-PCR
Die zur Konstruktion des Plasmids pCI-Gsyn-G benötigte PCR erfolgte unter Verwendung von
Arrow-Taq-DNA-Polymerase, einer Mischung aus Taq-DNA-Polymerase und korrekturle-
sender Tfu-DNA-Polymerase. Der PCR-Ansatz wurde dem angegebenen Programm gemäß
im Thermocycler inkubiert.

2 Material und Methoden 34
Ansatz: Programm:
1 µl pCI-G (10 pg/µl) 2 min 94 °C 1 µl primer 5’EcoRI-VSV-Gs (10 pmol/µl) 30 s 94 °C 1 µl primer 3’XhoI-VSV-Ga (10 pmol/µl) 30 s 58 °C × 35 5 µl 10 × T. Pol Puffer 100 s 72 °C 0,2 µl dNTPs (je 10 mM) 7 min 72 °C 0,1 µl Arrow-Taq-DNA-Polymerase (5 U/µl) 42 µl „Aqua B. Braun“
Für die Konstruktion des Plasmids pCI-GsynMut wurde die Sequenz zwischen der HindIII-
und PmlI-Schnittstelle in drei überlappende zu amplifizierende Abschnitte unterteilt und drei
Mutagenes-PCRs durchgeführt. Die Ansätze wurden nach dem unten angegebenen Programm
im Thermocycler inkubiert.
Ansätze: Programm: Ansatz 1: Ansatz 2: Ansatz 3:
pCI-Gsyn (10 pg/µl) 2 µl 2 µl 2 µl 2 min 94 °C primer Exs (10 pmol/µl) 1 µl 30 s 94 °C primer ILa (10 pmol/µl) 1 µl 30 s 55 °C × 35 primer ILs (10 pmol/µl) 1 µl 45 s 72 °C primer QHa (10 pmol/µl) 1 µl 7 min 72 °C primer QHs (10 pmol/µl) 1 µl primer Exa (10 pmol/µl) 1 µl 10 × T. Pol Puffer 5 µl 5 µl 5 µl dNTPs (je 10 mM) 0,2 µl 0,2 µl 0,2 µl Arrow-Taq-Polymerase (5 U/µl) 0,1 µl 0,1 µl 0,1 µl „Aqua B. Braun“ 41 µl 41 µl 41 µl
Für die Konstruktion des Plasmids pCI-Gopt wurde zur Reversion der unerwünschten Mutati-
on folgender PCR-Ansatz nach dem angegebenen Programm im Thermocycler inkubiert:
Ansatz: Programm:
1 µl pCI-GsynMut 2 min 94 °C 1 µl primer MutVSV-GsynMuta (10 pmol/µl) 30 s 94 °C 1 µl primer Exs (10 pmol/µl) 30 s 55 °C × 35 5 µl 10 × T. Pol Puffer 1 min 72 °C 0,2 µl dNTPs (je 10 mM) 7 min 72 °C 0,1 µl Arrow-Taq-Polymerase (5 U/µl) 42 µl „Aqua B. Braun“

2 Material und Methoden 35
b) Overlap-Extension-PCR
Bei einer Overlap-Extension-PCR lagern sich in den ersten Zyklen die denaturierten Teil-
Sequenzen an den komplementären Überlappungs-Stellen aneinander und werden verlängert,
in den späteren Zyklen wird die so entstandene Sequenz vollständiger Länge mit den äußeren
primern amplifiziert (s. auch Abb. 2-1).
Abb. 2-2. Overlap-Extension-PCR-Produkt. – primer sind durch blaue Pfeile, Restriktionsschnittstellen durch senkrechte Balken und VSV-Gsyn-Sequenzabschnitte in rosa dargestellt.
Die Exs-ILa- (224 bp), ILs-QHa- (134 bp) und QHs-Exa-Amplifikate (191 bp) (s. o.) wurden
im Rahmen der Konstruktion des Plasmids pCI-GsynMut durch eine Overlap-Extension-PCR
zur Sequenz vollständiger Länge zusammengefügt. Der PCR-Ansatz wurde dem unten ange-
gebenen Programm gemäß im Thermocycler inkubiert.
Ansatz: Programm:
1 µl Exs-ILa-Amplifikat (Gelextrakt) 2 min 94 °C 1 µl ILs-QHa-Amplifikat (Gelextrakt) 30 s 94 °C 1 µl QHs-Exa-Amplifikat (Gelextrakt) 30 s 55 °C × 35 1 µl primer Exs (10 pmol/µl) 45 s 72 °C 1 µl primer Exa (10 pmol/µl) 7 min 72 °C 5 µl 10 × T. Pol Puffer 0,2 µl dNTPs (je 10 mM) 0,1 µl Arrow-Taq-Polymerase (5 U/µl) 40 µl „Aqua B. Braun“

2 Material und Methoden 36
c) Kolonie-PCR
Alternativ zur Spaltung mittels Restriktionsendonukleasen wurden TA-Klone durch Kolonie-
PCR analysiert. Jede zu untersuchende Kolonie transformierter E. colis wurde mit je einer
Pipettenspitze durchstoßen, diese zuerst in 50 µl LB-Ampicillin-Medium und dann in 15 µl
Grundmischung ausgespült. Die PCR-Ansätze wurden nach dem unten angegebenen Pro-
gramm im Thermocycler inkubiert. Die angeimpften Medium-Aliquote wurden bei 4 °C gela-
gert, mit diesen konnten erforderlichenfalls später größere Kulturen angeimpft werden.
Programm: Grundmischung (pro Ansatz):
5 min 95 °C 1,5 µl 10 x PCR-Puffer 20 s 95 °C 0,3 µl dNTPs (je 10 mM) 20 s 55 °C × 40 0,3 µl primer M13 Forward (-20) (10 pmol/µl) 90 s 72 °C 0,3 µl primer M13 Reverse (10 pmol/µl) 12,5 µl „Aqua B. Braun“ 0,15 µl Taq-Polymerase
d) Reverse-Transkriptase-PCR
Je 5 µg Total-RNA wurden mit 2 pmol primer Krypta unter Verwendung des kits „Super-
script First-Strand Synthesis System for RT-PCR“ nach Angaben des Herstellers revers
transkribiert und wie folgt amplifiziert:
Ansatz: Programm:
2,5 µl 10 × PCR-Puffer 2 min 95 °C 0,5 µl dNTPs (je 10 mM) 20 s 95 °C 0,5 µl primer Krypts (10 pmol/µl) 20 s 57 °C × 35 0,5 µl primer Krypta (10 pmol/µl) 80 s 72 °C 1 µl cDNA 7 min 72 °C 0,5 µl Taq-Polymerase 19,5 µl „Aqua B. Braun“
2.12.8 Spaltung von DNA mittels Restriktionsendonukleasen
Maxi-Plasmidpräparationen wurden zur Kontrolle und Mini-Plasmidpräparationen in Reihen-
untersuchungen zur Identifizierung oder zum Treffen einer Vorauswahl für eine anschließen-
de Sequenzierung durch Spaltung mittels Restriktionsendonukleasen analysiert. 1 bis 2 µg

2 Material und Methoden 37
DNA wurde mit 1 µl des angegebenen Enzyms für 8 bis 12 h unter den vom Hersteller ange-
gebenen Pufferbedingungen bei der für das Enzym optimalen Temperatur inkubiert.
2.12.9 Auffüllen von Einzelstrang-Enden
Bei der Spaltung von DNA durch Restriktionsendonukleasen entstandene überhängende Ein-
zelstrang-Enden konnten mit Hilfe des Enzyms T4-DNA-Polymerase zu doppelsträngigen
Enden aufgefüllt werden. Diese Fill-in-Reaktion wurde nach den Anweisungen des Herstel-
lers des Enzyms durchgeführt.
2.12.10 Reinigung von DNA
a) PCR-Produkte und mittels Restriktionsendonukleasen gespaltene DNA
Erforderlichenfalls wurden PCR-Produkte und durch Spaltung mittels Restriktionsendo-
nukleasen entstandene Fragmente mit Hilfe von PCR-Purification-Kits oder DNA-Clean-Up-
Kits nach Angaben der Hersteller gereinigt. Alternativ erfolgte die Reinigung durch Agarose-
Gelelektrophorese mit anschließender Extraktion der DNA aus der entsprechenden Gel-
Bande, wenn unerwünschte DNA-Moleküle entfernt werden sollten (s. u.).
b) DNA < 100 bp
Nach komplementärer Anlagerung von Oligonukleotiden wurden die so gewonnenen Doppel-
stränge mit Hilfe des „Qiaquick Nucleotide Removal Kits“ nach Anweisungen des Herstellers
gereinigt.
2.12.11 Agarose-Gelelektrophorese
DNA-Moleküle wurden der Größe nach durch Agarose-Gelelektrophorese getrennt. Die Aga-
rose-Konzentration (0,8 bis 1,5 % (w/v)) und der Lauf-Puffer (1 × TAE- oder 0,5 × TBE-
Puffer) wurden abhängig von der Größe der zu trennenden DNA-Moleküle nach den Angaben
in „Molecular Cloning“ [96] gewählt. Ebenfalls in Abhängigkeit von der Größe der zu tren-
nenden DNA-Moleküle wurde die 100 bp DNA-Leiter oder die 1 kb DNA-Leiter als Größen-

2 Material und Methoden 38
standard aufgetragen. Die horizontale Apparatur wurde gemäß der Anleitung des Herstellers
benutzt.
2.12.12 DNA-Extraktion aus Agarose
DNA wurde aus Agarosegel-Banden mit Hilfe von Gel-Extraction-Kits unter Beachtung der
Anweisungen des jeweiligen Herstellers extrahiert.
2.12.13 Phosphorylierung und komplementäre Anlagerung von Oligonukleotiden
Die anzulagernden Oligonukleotide wurden zunächst mittels T4-Polynukleotid-Kinase
phosphoryliert, um eine nachfolgende Ligation zu ermöglichen. Der Phosphorylierungs-
Ansatz (s. u.) wurde 1 h bei 37 °C inkubiert. Anschließend wurden 7,5 µl 20 × SSC zugefügt,
um nichtkomplementäre Anlagerungen zu reduzieren, und 10 min bei 70 °C denaturiert. Die
komplementäre Anlagerung wurde durch langsames (0,3 °C pro min) Abkühlen auf 21 °C
erreicht.
Phosphorylierungsansatz:
2,5 µl Oligonukleotid 1 2,5 µl Oligonukleotid 2 2 µl ATP (10 mM) 4 µl 10 × Kinase-Puffer 26 µl „Aqua B. Braun“ 3 µl T4-Poynukleotid-Kinase (10 U/µl)
2.12.14 Ligation
DNA-Moleküle wurden bei 16 °C mittels T4-DNA-Ligase in einem 10-µl-Ansatz über Nacht
ligiert, dabei wurden der mitgelieferte Puffer und 400 U T4-DNA-Ligase eingesetzt.
Alternativ erfolgte die Ligation unter Verwendung des „DNA Ligation Kit, Version 2.1“
(auch T4-DNA-Ligase-basierend) bei 16 °C in 30 min unter genauer Befolgung der Anleitung
des Herstellers.

2 Material und Methoden 39
2.12.15 TA-Klonierung
Die Taq-Polymerase hängt ein einzelnes dATP an die 3’-Enden von PCR-Produkten. Über
diese 3’-A-Überhänge können PCR-Produkte in spezielle TA-Klonierungs-Plasmide einge-
fügt werden, welche linearisiert sind und einzelne 3’-T-Überhänge aufweisen. Das benutzte
Plasmid pCR-2.1-TOPO aus dem „TOPO-TA-Cloning-Kit“ besitzt außerdem eine kovalent
gebundene Topoisomerase I, die die Ligation des komplementär angelagerten PCR-Produkts
mit dem Plasmid bewirkt. Nach TA-Klonierung gemäß den Anweisungen des Herstellers
wurden die Plasmide in E. coli transformiert.
2.12.16 DNA-Sequenzierung
a) Im Hause
Sowohl Sinn- als auch Gegensinnstrang der für VSV-G codierenden Sequenz in pCI-G wur-
den in drei Abschnitten sequenziert. Für die Sinnstrang-Sequenzierung wurden die primer
SEQpCI-G1, SEQpCI-G2 und SEQpCI-G3 und für die Gegensinnstrang-Sequenzierung die
primer SEQpCI-G4, SEQpCI-G5 und SEQpCI-G6 eingesetzt. Die Ansätze für die Didesoxy-
Kettenabbruch-Reaktion wurden wie unten angegeben im Thermocycler inkubiert.
Danach wurden die Reaktionsprodukte gefällt. Dazu wurden die Ansätze jeweils mit 80 µl
„Aqua B. Braun“, 10 µl 3 M Natriumacetat, pH 5,2 und 250 µl Ethanol versetzt, 30 min bei
4 °C und 14000 Upm in einer Mikrozentrifuge zentrifugiert, die Überstände verworfen,
350 µl 70 % (v/v) Ethanol zugegeben, weitere 10 min bei 4 °C und 14000 Upm zentrifugiert,
die Überstände erneut verworfen und die unsichtbaren Sedimente an der Luft getrocknet.
Die Sequenzierung wurde von Dr. Dennis Hoffmann an Geräten der Abteilung für Humange-
netik durchgeführt.
Reaktionsansätze Programm:
3 µl pCI-G (0,1 µg/µl) 2 min 95 °C 1 µl primer (10 pmol/µl) 20 s 95 °C 4 µl ABI-Ready-Reaction-Mix 20 s 57 °C × 35 4 min 60 °C

2 Material und Methoden 40
b) Seqlab
Die Tab. 2-20 zeigt die Plasmide und PCR-Produkte, die mit dem jeweils angegebenen pri-
mer von der Firma Seqlab in Göttingen sequenziert wurden.
Tab. 2-20. Sequenzierungen durch Seqlab.
Matrizen-DNA primer Querverweis
100 ng OE-Amplifikat 20 pmol Exa 2.1.1 k) 600 ng pCI-GsynMut 20 pmol pCI-GsynMuts 2.1.1 k) 600 ng pCI-GsynMut 20 pmol pCI-GsynMuta 2.1.1 k) 600 ng pCI-Gopt 20 pmol pCI-GsynMuts 2.1.1 l) 600 ng pCI-Gopt 20 pmol pCI-GsynMuta 2.1.1 l)
c) Genterprise
Die Tab. 2-21 zeigt die Plasmide und PCR-Produkte, die mit dem jeweils angegebenen pri-
mer von der Firma Genterprise in Mainz sequenziert wurden.
Tab. 2-21. Sequenzierungen durch Genterprise.
Matrizen-DNA primer Querverweis
300 ng pCI-GΔI 60 pmol pCI-GIs 2.1.1 f)
300 ng pCI-GΔI 60 pmol pCI-GIa 2.1.1 f)
35 ng 0,3-kb-pCI-G-Amplifikat 60 pmol Krypts 2.12.4
35 ng 0,3-kb-pCI-G-Amplifikat 60 pmol Krypta 2.12.4
35 ng 0,3-kb-pCI-GΔI-Amplifikat 60 pmol Krypts 2.12.4
35 ng 0,3-kb-pCI-GΔI-Amplifikat 60 pmol Krypta 2.12.4
35 ng pCR-2.1(0,7-kb-Amplifikat) 60 pmol M13 Forward (-20) 2.12.4
35 ng pCR-2.1(0,7-kb-Amplifikat) 60 pmol M13 Reverse 2.12.4
35 ng pCR-2.1(1,15-kb-Amplifikat) 60 pmol M13 Forward (-20) 2.12.4
35 ng pCR-2.1(1,15-kb-Amplifikat) 60 pmol M13 Reverse 2.12.4

3 Ergebnisse
3.1 Expression von VSV-G mittels pCDNA3.1(+)-basierter Plasmide
VSV-G-Expressionsplasmide werden zur Produktion pseudotypisierter Viren eingesetzt. Dazu
hat sich das Plasmid pHIT-G bewährt [24,91,97,98], in welchem die VSV-G-Transkription
durch den HCMV-major-immediate-early-Promotor (HCMV-MIE-Promotor) angetrieben
wird und sich zwischen Promotor und der für VSV-G codierenden Sequenz das erste Intron
und die flankierenden, nicht translatierten Exon-Sequenzen der HCMV-MIE-Gene befinden
(s. Abb. 3-1). Dr. Seraphin Kuate brachte das VSV-G-Gen unter die Kontrolle eines durch
Ponasteron induzierbaren Promotors [50] und stellte dabei fest, dass die Expression nur ge-
lang, wenn nicht nur die für VSV-G codierende Sequenz, sondern auch die HCMV-MIE-
Intron- und -Exon-Sequenzen in die induzierbare Expressionskassette kloniert wurden [49].
Dieses Ergebnis wurde zunächst in der vorliegenden Arbeit mit pCDNA3.1(+)-basierten
Plasmiden nachvollzogen.
Abb. 3-1. Expressionskassetten der VSV-G-Expressionsplasmide pHIT-G, pCD-G und pCI-G (schematische Darstellung). – HCMV-Promotor: Promotor/Enhancer-Region der Major-immediate-early-Gene (MIE-Gene) des humanen Cytomegalievirus; Ex 1: HCMV-MIE-Exon 1; Ex 2: nicht translatiertes 5’-Ende des HCMV-MIE-Exons 2; Intron: erstes HCMV-MIE-Intron; VSV-G: codieren-de Sequenz für das Glykoprotein G des Vesicular-Stomatitis-Virus; VSV 3’-UTR: nicht translatierte Wildtypsequenz am 3’-Ende des VSV-G-Gens; SV40 pAd: Polyadenylierungssignal des SV40-Virus; BGH pAd: Polyadenylierungssignal des Rinder-Wachstumshormons.
Dazu wurden die Pseudotypisierungseffizienz und das VSV-G-Expressionsvermögen der
pCDNA3.1(+)-Konstrukte pCD-G und pCI-G untersucht, welche nur die für VSV-G codie-

3 Ergebnisse 42
rende Sequenz bzw. sowohl die für VSV-G codierende Sequenz und als auch das HCMV-
MIE-Intron mit flankierenden, nicht translatierten Exon-Sequenzen enthielten (s. Abb. 3-1),
und diese denen des pHIT-G gegenübergestellt.
Durch Cotransfektion der VSV-G-Expressionsplasmide wurden SIV-Vektoren pseudotypi-
siert und deren Titer anschließend auf 293A-Zellen als grüne-Fluoreszenz-bildende Einheiten
pro ml (GFU/ml) bestimmt. Mit pHIT-G wurden Titer von (5,7 ± 2,1) × 105 GFU/ml erzielt;
die unter Verwendung von pCD-G erhaltenen Titer waren etwa 7000fach niedriger
((82 ± 13) GFU/ml); mittels pCI-G wurden wieder Titer in der ursprünglichen Größenord-
nung erreicht ((6,8 ± 1) × 105 GFU/ml) (s. Abb. 3-2).
Abb. 3-2. VSV-G-Expression mittels pHIT-G, pCIG und pCD-G. - Zur Produktion lentiviraler Vektoren wurden 293T-Zellen mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Nach zwei Tagen wurden die Vektoren geerntet und die Zellen lysiert. A: Die Titer der lentiviralen Vekto-ren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Experimenten. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt). B: Die Zelllysate wurden unter Verwendung monoklonaler Antikörper gegen VSV-G und HIV-1 p24 einer Westernblot-Analyse unterzogen. CA: Capsidprotein.
Es bestätigte sich in der Westernblot-Analyse von Lysaten entsprechend transfizierter SIV-
Partikel produzierender 293T-Zellen, dass sich VSV-G mit pHIT-G und pCI-G stark expri-
mieren ließ, die Expression von VSV-G mittels pCD-G jedoch unterhalb der Nachweisgrenze
blieb (s. Abb. 3-2). Ein damit übereinstimmendes Bild zeigte sich auch in der Northernblot-
Analyse von mRNA-Präparationen aus 293T-Zellen, die SIV-Vektoren produzierten, die mit-
tels eines HIV-1-gag-pol-Expressionsplasmids verpackt worden waren: Sogar bei – im-

3 Ergebnisse 43
Vergleich zur pCI-G-Probe – stärkerem gag-pol-Signal, das zur Normierung herangezogen
wurde, ließ sich keine von pCD-G transkribierte VSV-G-mRNA nachweisen (s. Abb. 3-3).
Abb. 3-3. Northernblot-Analyse der von pCI-G und pCD-G transkribierten mRNA aus transfizierten Zellen. – 293T-Zellen wurden mit ViCGΔBH (SIV-Vektorkonstrukt), Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und dem angegebenen VSV-G-Expressionsplasmid cotransfiziert. Die nach zwei Ta-gen aus den transfizierten Zellen isolierte mRNA wurde einer Northernblot-Analyse unterzogen. Die DNA-Sonde war gegen die 130 b 5’ des AAUAAA-Motivs des Rinder-Polyadenylierungssignals in den gag-pol- und VSV-G-Transkripten gerichtet.
3.2 Exakte Deletion des ersten HCMV-MIE-Introns
3.2.1 Einfluss des Spleiß-Prozesses und der Exon-Sequenzen auf die Expression
Da das Plasmid pCI-G im Vergleich zum Plasmid pCD-G sowohl das erste Intron als auch
das Exon 1 und das nicht translatierte 5’-Ende des Exons 2 der HCMV-major-immediate-
early-Gene (HCMV-MIE-Gene) enthielt, wurde überprüft, ob der beobachtete positive Effekt
auf die Expression durch das Intron bzw. das Spleißen, oder durch die nicht translatierten E-
xon-Sequenzen bewirkt wird.
Dazu wurde das VSV-G-Expressionsplasmid pCI-GΔI (s. Abb. 3-4) konstruiert, das als pri-
märes Transkript, also ohne Spleiß-Prozesse, eine mRNA liefert, die identisch ist mit der rei-
fen, d. h. gespleißten, mRNA aus pCI-G, also auch die HCMV-MIE-Exon-Sequenzen enthält.

3 Ergebnisse 44
Abb. 3-4. Expressionskassetten der VSV-G-Expressionsplasmide pCI-G, pCI-GΔI und pCD-G (schematische Darstellung). – HCMV-Promotor: Promotor/Enhancer-Region der Major-immediate-early-Gene (MIE-Gene) des humanen Cytomegalievirus; Ex 1: HCMV-MIE-Exon 1; Ex 2: nicht translatiertes 5’-Ende des HCMV-MIE-Exons 2; Intron: erstes HCMV-MIE-Intron; VSV-G: codieren-de Sequenz für das Glykoprotein G des Vesicular-Stomatitis-Virus; BGH pAd: Polyadenylierungs-signal des Rinder-Wachstumshormons.
Die Pseudotypisierungseffizienz und das VSV-G-Expressionsvermögen der Plasmide pCI-G,
pCI-GΔI und pCD-G wurden wie in 3-1 anhand lentiviraler Vektoren durch Titerbestimmung
sowie Western- und Northernblot-Analyse miteinander verglichen.
Abb. 3-5. VSV-G-Expression mittels pCI-GΔI. – A: Zur Produktion lentiviraler Vektoren wurden 293T-Zellen mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg Hgpsyn (HIV-1-gag-pol-Expressions-plasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die Titer der nach zwei Tagen geernteten lentiviralen Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Ex-perimenten. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt). B: 293T-Zellen wurden mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die Zellen wurden zwei Tage nach Transfektion lysiert und einer Westernblot-Analyse unter Verwen-dung monoklonaler Antikörper gegen VSV-G und HIV-1 p24 unterzogen. CA: Capsidprotein.
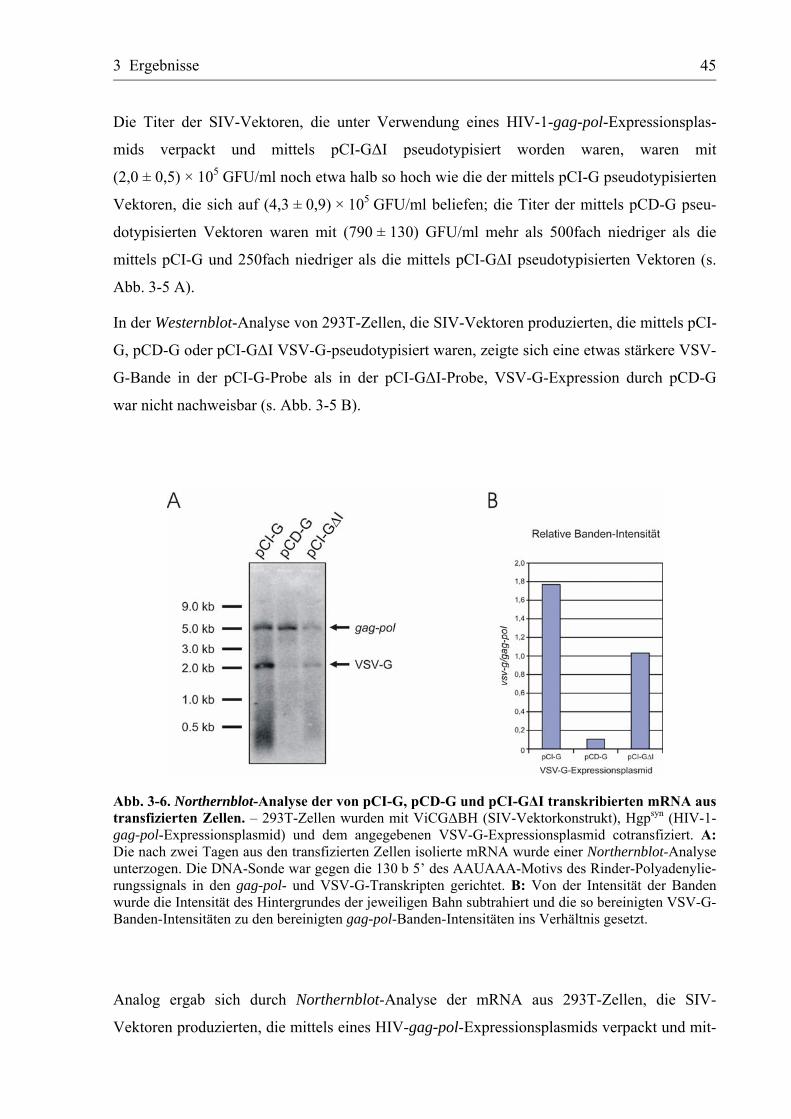
3 Ergebnisse 45
Die Titer der SIV-Vektoren, die unter Verwendung eines HIV-1-gag-pol-Expressionsplas-
mids verpackt und mittels pCI-GΔI pseudotypisiert worden waren, waren mit
(2,0 ± 0,5) × 105 GFU/ml noch etwa halb so hoch wie die der mittels pCI-G pseudotypisierten
Vektoren, die sich auf (4,3 ± 0,9) × 105 GFU/ml beliefen; die Titer der mittels pCD-G pseu-
dotypisierten Vektoren waren mit (790 ± 130) GFU/ml mehr als 500fach niedriger als die
mittels pCI-G und 250fach niedriger als die mittels pCI-GΔI pseudotypisierten Vektoren (s.
Abb. 3-5 A).
In der Westernblot-Analyse von 293T-Zellen, die SIV-Vektoren produzierten, die mittels pCI-
G, pCD-G oder pCI-GΔI VSV-G-pseudotypisiert waren, zeigte sich eine etwas stärkere VSV-
G-Bande in der pCI-G-Probe als in der pCI-GΔI-Probe, VSV-G-Expression durch pCD-G
war nicht nachweisbar (s. Abb. 3-5 B).
Abb. 3-6. Northernblot-Analyse der von pCI-G, pCD-G und pCI-GΔI transkribierten mRNA aus transfizierten Zellen. – 293T-Zellen wurden mit ViCGΔBH (SIV-Vektorkonstrukt), Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und dem angegebenen VSV-G-Expressionsplasmid cotransfiziert. A: Die nach zwei Tagen aus den transfizierten Zellen isolierte mRNA wurde einer Northernblot-Analyse unterzogen. Die DNA-Sonde war gegen die 130 b 5’ des AAUAAA-Motivs des Rinder-Polyadenylie-rungssignals in den gag-pol- und VSV-G-Transkripten gerichtet. B: Von der Intensität der Banden wurde die Intensität des Hintergrundes der jeweiligen Bahn subtrahiert und die so bereinigten VSV-G-Banden-Intensitäten zu den bereinigten gag-pol-Banden-Intensitäten ins Verhältnis gesetzt.
Analog ergab sich durch Northernblot-Analyse der mRNA aus 293T-Zellen, die SIV-
Vektoren produzierten, die mittels eines HIV-gag-pol-Expressionsplasmids verpackt und mit-

3 Ergebnisse 46
tels pCI-G, pCD-G oder pCI-GΔI pseudotypisiert waren, dass das Verhältnis von VSV-G- zu
gag-pol-mRNA in Lentiviren produzierenden 293T-Zellen, die transfiziertes pCI-GΔI enthiel-
ten, nur 1,7fach geringer war als in solchen, die pCI-G enthielten, aber 10,1fach höher als in
solchen, die pCD-G enthielten (s. Abb. 3-6).
3.2.2 Nachweis der Spleißprodukte
Um zu beweisen, dass die VSV-G-Expression mit pCI-GΔI nicht durch das Spleißen der
mRNA, sondern durch das Vorliegen der fusionierten Exon-Sequenzen so stark war, wurde
gezeigt, dass bei der Konstruktion dieses Plasmids nicht eventuell vorhandene kryptische
Spleißstellen aktiviert wurden. Dazu wurde Total-RNA aus transfizierten 293T-Zellen isoliert
und die 5’-Enden der primären VSV-G-Transkripte und der Spleißprodukte mittels RT-PCR
amplifiziert. Die Abb. 3-7 zeigt das Resultat der Agarose-Gelelektrophorese der Amplifikate:
Die mit Abstand intensivste Bande repräsentiert ein etwa 300 bp großes Amplifikat in der
pCI-G- und in der pCI-GΔI-Probe. In der pCI-G Probe zeigen sich noch zwei weitere Banden:
Eine zarte bei etwas mehr als 1,1 kb und eine nicht erwartete bei etwa 700 bp.
Abb. 3-7. Transkripte und Spleißprodukte: Agarose-Gelelektrophorese von RT-PCR-Produkten. – 293T-Zellen wurden mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des an-gegebenen VSV-G-Ex-pressionsplasmid cotransfiziert. Aus den transfizierten Zellen wurde Total-RNA präpariert und von den in dieser enthaltenen VSV-G-RNAs die gesamten 5’-Enden einschließlich der ersten 132 b der für VSV-G codie-renden Sequenz mit dem primer Krypta revers transkribiert, zur Kontrolle wurde die Reaktion auch ohne Reverse Transkriptase (RT) angesetzt. Dann wurden die Einzelstränge mit dem Primer-Paar Krypts und Krypta ausgehend vom Transkriptionsstart amplifiziert und die Amplifikate einer Agarose-Gelelektrophorese unterzogen

3 Ergebnisse 47
1 130 1130-bp-pCI-G TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGACGTAAGTACC 713-bp-pCI-G TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGAC--------- 303-bp-pCI-G TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGAC--------- 303-bp-pCI-GΔI TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGAC--------- pCI-GΔI TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGAC--------- pCI-G TCAGATCGCCTGGAGACGCCATCCACGCTGTTTTGACCTCCATAGAAGACACCGGGACCGATCCAGCCTCCGCGGCCGGGAACGGTGCATTGGAACGCGGATTCCCCGTGCCAAGAGTGACGTAAGTACC ————————— 131 260 1130-bp-pCI-G GCCTATAGAGTCTATAGGCCCACCCCCTTGGCTTCTTATGCATGCTATACTGTTTTTGGCTTGGGGTCTATACACCCCCGCTTCCTCATGTTATAGGTGATGGTATAGCTTAGCCTATAGGTGTGGGTTA 713-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G GCCTATAGAGTCTATAGGCCCACCCCCTTGGCTTCTTATGCATGCTATACTGTTTTTGGCTTGGGGTCTATACACCCCCGCTTCCTCATGTTATAGGTGATGGTATAGCTTAGCCTATAGGTGTGGGTTA 261 390 1130-bp-pCI-G TTGACCATTATTGACCACTCCCCTATTGGTGACGATACTTTCCATTACTAATCCATAACATGGCTCTTTGCCACAACTCTCTTTATTGGCTATATGCCAATACACTGTCCTTCAGAGACTGACACGGACT 713-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G TTGACCATTATTGACCACTCCCCTATTGGTGACGATACTTTCCATTACTAATCCATAACATGGCTCTTTGCCACAACTCTCTTTATTGGCTATATGCCAATACACTGTCCTTCAGAGACTGACACGGACT ——————— ————— 391 520 1130-bp-pCI-G CTGTATTTTTACAGGATGGGGTCTCATTTATTATTTACAAATTCACATATACAACACCACCGTCCCCAGTGCCCGCAGTTTTTATTAAACATAACGTGGGATCTCCACGCGAATCTCGGGTACGTGTTCC 713-bp-pCI-G --------------GATGGGGTCTCATTTATTATTTACAAATTCACATATACAACACCACCGTCCCCAGTGCCCGCAGTTTTTATTAAACATAACGTGGGATCTCCACGCGAATCTCGGGTACGTGTTCC 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G CTGTATTTTTACAGGATGGGGTCTCATTTATTATTTACAAATTCACATATACAACACCACCGTCCCCAGTGCCCGCAGTTTTTATTAAACATAACGTGGGATCTCCACGCGAATCTCGGGTACGTGTTCC ——————————————— 521 650 1130-bp-pCI-G GGACATGGGCTCTTCTCCGGTAGCGGCGGAGCTTCTACATCCGAGCCCTGCTCCCATGCCTCCAGCGACTCATGGTCGCTCGGCAGCTCCTTGCTCCTAACAGTGGAGGCCAGACTTAGGCACAGCACGA 713-bp-pCI-G GGACATGGGCTCTTCTCCGGTAGCGGCGGAGCTTCTACATCCGAGCCCTGCTCCCATGCCTCCAGCGACTCATGGTCGCTCGGCAGCTCCTTGCTCCTAACAGTGGAGGCCAGACTTAGGCACAGCACGA 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G GGACATGGGCTCTTCTCCGGTAGCGGCGGAGCTTCTACATCCGAGCCCTGCTCCCATGCCTCCAGCGACTCATGGTCGCTCGGCAGCTCCTTGCTCCTAACAGTGGAGGCCAGACTTAGGCACAGCACGA 651 780 1130-bp-pCI-G TGCCCACCACCACCAGTGTGCCGCACAAGGCCGTGGCGGTAGGGTATGTGTCTGAAAATGAGCTCGGGGAGCGGGCTTGCACCGCTGACGCATTTGGAAGACTTAAGGCAGCGGCAGAAGAAGATGCAGG 713-bp-pCI-G TGCCCACCACCACCAGTGTGCCGCACAAGGCCGTGGCGGTAGGGTATGTGTCTGAAAATGAGCTCGGGGAGCGGGCTTGCACCGCTGACGCATTTGGAAGACTTAAGGCAGCGGCAGAAGAAGATGCAGG 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G TGCCCACCACCACCAGTGTGCCGCACAAGGCCGTGGCGGTAGGGTATGTGTCTGAAAATGAGCTCGGGGAGCGGGCTTGCACCGCTGACGCATTTGGAAGACTTAAGGCAGCGGCAGAAGAAGATGCAGG 781 910 1130-bp-pCI-G CAGCTGAGTTGTTGTGTTCTGATAAGAGTCAGAGGTAACTCCCGTTGCGGTGCTGTTAACGGTGGAGGGCAGTGTAGTCTGAGCAGTACTCGTTGCTGCCGCGCGCGCCACCAGACATAATAGCTGACAG 713-bp-pCI-G CAGCTGAGTTGTTGTGTTCTGATAAGAGTCAGAG------------------------------------------------------------------------------------------------ 303-bp-pCI-G ---------------------------------------------------------------------------------------------------------------------------------- 303-bp-pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-GΔI ---------------------------------------------------------------------------------------------------------------------------------- pCI-G CAGCTGAGTTGTTGTGTTCTGATAAGAGTCAG AGCTGACAG
AGGTAACTCCCGTTGCGGTGCTGTTAACGGTGGAGGGCAGTGTAGTCTGAGCAGTACTCGTTGCTGCCGCGCGCGCCACCAGACATAAT ————————— ———————
911 1040 1130-bp-pCI-G ACTAACAGACTGTTCCTTTCCATGGGTCTTTTCTGCAGTCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG 713-bp-pCI-G --------------------------------------TCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG 303-bp-pCI-G --------------------------------------TCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG 303-bp-pCI-GΔI --------------------------------------TCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG pCI-GΔI --------------------------------------TCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG pCI-G ACTAACAGACTGTTCCTTTCCATGGGTCTTTTCTGCAGTCACCGTCCTTGACACGAAGCTTGGTACCGAGCTCGGATCCGCCGCCACCATGAAGTGCCTTTTGTACTTAGCCTTTTTATTCATTGGGGTG ———————————————————— 1041 1130 1130-bp-pCI-G AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA 713-bp-pCI-G AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA 303-bp-pCI-G AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA 303-bp-pCI-GΔI AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA pCI-GΔI AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA pCI-G AATTGCAAGTTCACCATAGTTTTTCCACACAACCAAAAAGGAAACTGGAAAAATGTTCCTTCTAATTACCATTATTGCCCGTCAAGCTCA
Abb. 3-8. Transkripte und Spleiß-Produkte: Alignment der RT-PCR-Produkt-Sequenzen. – Nach präparativer Agarose-Gelelektrophorese wurden die 0,3 kb großen Amplifikate aus der pCI-G- und pCI-GΔI-Probe direkt sequenziert, die 0,7 kb und 1,1 kb großen Amplifikate aus der pCI-G-Probe zunächst in pCR2.1-TOPO TA-kloniert und dann als inserts dieses Plasmids sequenziert. (Alle Se-quenzierungen wurden von der Firma GENterprise durchgeführt). Übereinstimmungen mit HCMV-MIE-Exon-Sequenzen sind blau, mit dem ersten HCMV-MIE-Intron orange und mit der für VSV-G codierenden Sequenz grün unterlegt. Die Sequenz-Motive für die 5’-Spleißstellen sind rot, für die 3’-Spleißstellen schwarz und für die Verzweigungsstellen grün unterstrichen [99].
Zur eindeutigen Identifizierung der Banden in Abb. 3-7 wurden die 0,3 kb großen Amplifikate
direkt nach Isolation aus einem Agarose-Gel und die 0,7 und 1,1 kb großen Amplifikate nach
TA-Klonierung sequenziert und die Sequenzen hinsichtlich Spleiß- und Verzweigungsstellen
analysiert (s. Abb. 3-8)..

3 Ergebnisse 48
3.2.3 Deletion weiterer HCMV-Sequenzen aus der VSV-G-Expressionskassette
Von pCI-GΔI transkribierte VSV-G-mRNA enthält in der 5’UTR eine 138 Basen umfassende
aus HCMV-MIE-Exonen stammende Sequenz, die nicht notwendigerweise in ihrer ganzen
Länge für die positive Wirkung auf die VSV-G Expression unerlässlich ist. Verzichtbare Se-
quenzanteile sollten in einem nächsten Schritt herausgefiltert und entfernt werden.
Dazu wurde zunächst die Sekundär-Struktur der HCMV-MIE-5’UTR mit Hilfe des Pro-
gramms RNA-Fold vorhergesagt (s. Abb. 3-9) [100,101]. Ein Bereich von 32 Nukleotiden
zeigte bei der Berechnungen eine Stemloop-Struktur, an deren Spitze sich die Spleißstelle
befindet; diese Struktur mit einer Änderung der freien Enthalpie von ΔG = - 42 kJ/mol bildet
sich laut Berechnung nicht aus, wenn das Intron noch enthalten ist.
Abb. 3-9. Sekundärstruktur-Vorhersage der HCMV-MIE-5’-UTR. – A: 5’-Nicht-translatierte-Region (UTR) ohne Intron. B: 5’-UTR mit HCMV-Intron. Türkis: Exon 1 5’ des stemloops, Violett: Exon 1 im stemloop, Oran-ge: HCMV-Intron, Blau: Exon 2. Die Strukturen wurden mit dem Programm „RNAfold“ berech-net [100,101].
Zur Überprüfung der Entbehrlichkeit dieser Struktur für die VSV-G-Expression wurden die
Plasmide pCI-GΔIΔSL und pCSL-G konstruiert, die im Unterschied zu pCI-GΔI von den
HCMV-Sequenzen in der 5’UTR gerade die stemloop bildende Sequenz nicht (pCI-GΔIΔSL)
bzw. nur noch die stemloop bildende Sequenz (pCSL-G) enthalten (s. Abb. 3-10 A).

3 Ergebnisse 49
Die Deletion der stemloop bildenden Sequenz aus pCI-GΔI reduzierte den Titer der entspre-
chend pseudotypisierten SIV-Vektoren von (2,3 ± 0,3) ×106 GFU/ml auf etwa die Hälfte
((1,2 ± 0,1) × 106 GFU/ml), während die Deletion der Sequenz 5’ der stemloop bildenden
Sequenz den Titer um den Faktor 23 auf (1,0 ± 0,1) × 105 GFU/ml reduzierten; dieser mittels
pCSL-G erzielte Titer war allerdings immer noch 10fach höher als der mittels pCD-G erhalte-
ne ((1,0 ± 0,4) × 104 GFU/ml) (s. Abb. 3-10 B).
Abb. 3-10. Pseudotypisierungseffizienz nach Deletion von HCMV Sequenzen aus der 5’UTR. – A: Schematische Darstellung der 5’UTR der angegebenen VSV-G-Expressionsplasmide. Türkis: HCMV-MIE-Exon-1-Sequenzen 5’ des vorhergesagten stemloops, violett: HCMV-MIE-Exon-1-Sequenzen im stemloop, blau: HCMV-MIE-Exon-2-Sequenzen, gelb: pCDNA3.1(+)-Sequenzen, grau: KOZAK-Sequenz. B: Zur Produktion lentiviraler Vektoren wurden 293T-Zellen mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die Titer der lentiviralen Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Experimenten. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt).
In Übereinstimmung damit konnten durch Westernblot-Analyse der Lysate von 293T-Zellen,
die pseudotypisierte SIV-Vektoren produzierten, und Northernblot-Analyse der mRNA aus
293T-Zellen, die pseudotypisierte SIV-Vektoren produzierten, die mittels eines HIV-gag-pol-
Expressionsplasmids verpackt worden waren, in der pCSL-G Probe kein VSV-G-Protein bzw.
keine VSV-G-mRNA nachgewiesen werden, während die Nachweise für die pCI-GΔI- und
pCI-GΔIΔSL-Proben gelangen (s. Abb. 3-11).

3 Ergebnisse 50
Abb. 3-11. Western- und Northernblot-Analyse nach Deletion von HCMV-Sequenzen aus der 5’UTR. – A: Westernblot-Analyse. 293T-Zellen wurden mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die Lysate dieser Zellen wurden einer Westernblot-Analyse unter Verwendung monoklonaler Antikörper gegen VSV-G und HIV-1 p24 unterzogen. CA: Capsidprotein. B: Northernblot-Analyse. 293T-Zellen wurden mit ViCGΔBH (SIV-Vektorkonstrukt), Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und dem angegebenen VSV-G-Expressionsplasmid cotransfiziert. Die nach zwei Tagen aus den transfizierten Zellen isolierte mRNA wurde einer Northernblot-Analyse unterzogen. Die DNA-Sonde war gegen die 130 b 5’ des AAUAAA-Motivs des Rinder-Polyadenylierungssignals in den gag-pol- und VSV-G-Transkripten gerichtet.
3.3 Einfluss von Codonoptimierung auf die Expression
3.3.1 Erste Konstrukte mit codonoptimierter Sequenz für VSV-G
Es wurde untersucht, ob durch Codonoptimierung der für VSV-G codierenden Sequenz auch
unter völligem Verzicht von HCMV-Sequenzen in der 5’ nicht translatierten Region der
VSV-G-mRNA eine gute Expression des VSV-G-Proteins möglich ist.
Dazu wurde von Prof. Dr. Ralf Wagner die codonoptimierte DNA für VSV-G entsprechend
der im Genbankeintrag J02428 veröffentlichten Aminosäure-Sequenz des VSV-G syntheti-
siert und zur Konstruktion des Plasmids pCD-Gsyn in pcDNA3.1(+) kloniert (s. Abb. 3-12).

3 Ergebnisse 51
Für Kontroll-Experimente wurden das erste Intron und die flankierenden nicht translatierten
Exon-Sequenzen der HCMV-major-immediate-early-Gene aus pHIT-G in pCD-Gsyn zwischen
HCMV-Promotor und der für VSV-G codierenden Sequenz kloniert und so das Plasmid pCI-
Gsyn erzeugt (s. Abb. 3-12).
Abb. 3-12. Expressionskassetten der VSV-G-Expressionsplasmide pCD-G, pCD-Gsyn, pCI-G und pCI-Gsyn (schematische Darstellung). – HCMV-Promotor: Promotor/Enhancer-Region der Major-immediate-early-Gene (MIE-Gene) des humanen Cytomegalievirus; Ex 1: HCMV-MIE-Exon 1; Ex 2: nicht translatiertes 5’-Ende des HCMV-MIE-Exons 2; Intron: erstes HCMV-MIE-Intron; VSV-G: codierende Sequenz für das Glykoprotein G des Vesicular-Stomatitis-Virus; VSV-Gsyn: nach Gen-bankeintrag J02428 codonoptimierte codierende Sequenz für das VSV-G-Protein; BGH pAd: Polya-denylierungssignal des Rinder-Wachstumshormons.
Es wurden SIV-Vektoren hergestellt, bei deren VSV-G-Pseudotypisierung eines der Plasmide
pCD-G, pCD-Gsyn, pCI-G oder pCI-Gsyn eingesetzt wurde. Die Abb. 3-13 A zeigt, dass die
Titer der mittels pCD-G (220 GFU/ml), pCD-Gsyn (1,1 × 103 GFU/ml) und pCI-Gsyn
(2,0 × 103 GFU/ml) pseudotypisierten Vektoren alle um zwei bis drei Zehnerpotenzen niedri-
ger lagen als die Titer der mittels pCI-G (5,7 × 105 GFU/ml) pseudotypisierten Vektoren. Da
sogar pCI-Gsyn keine hohen Titer lieferte, wurde die VSV-Gsyn-Expression durch Westernblot-
Analyse von Zell-Lysaten und Virus-Partikeln untersucht (s. Abb. 3-13 B). So konnte in Lysa-
ten von SIV-Vektoren produzierenden 293T-Zellen, sowohl durch pCI-G als auch durch pCI-
Gsyn exprimiertes VSV-G nachgewiesen werden. In den produzierten Vektoren konnte VSV-
G nur dann nachgewiesen werden, wenn sie mittels pCI-G nicht aber mittels pCI-Gsyn pseudo-
typisiert waren.
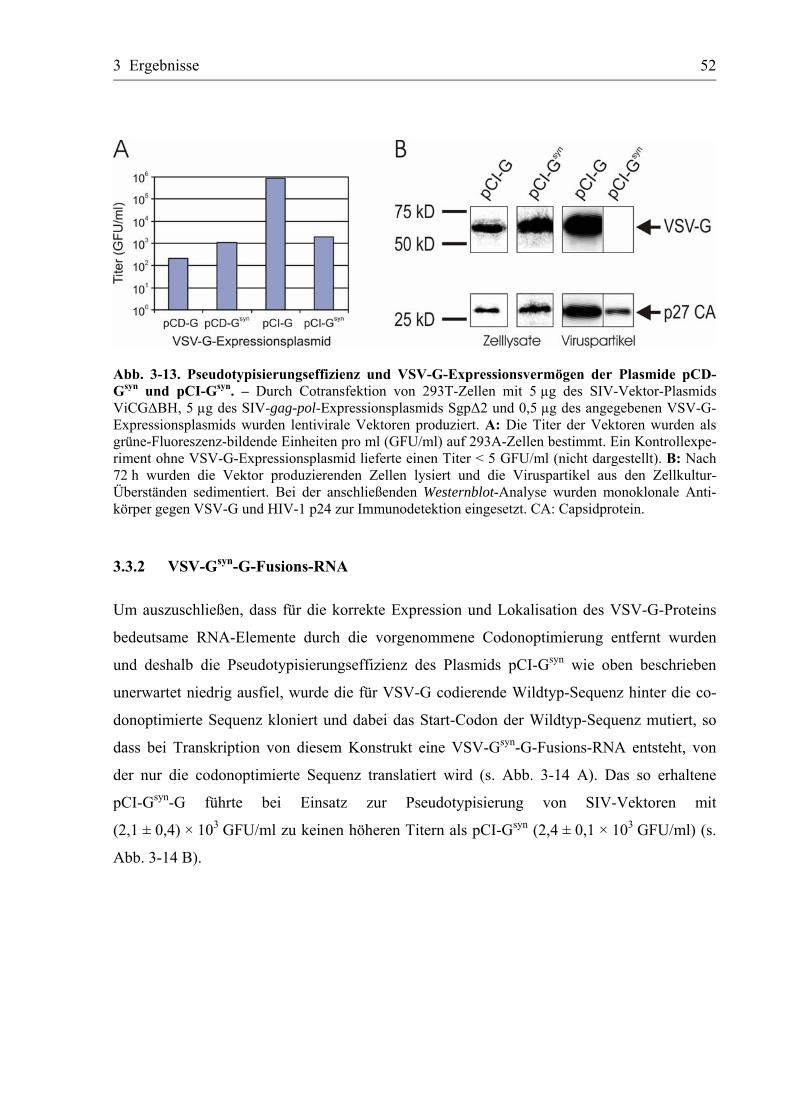
3 Ergebnisse 52
Abb. 3-13. Pseudotypisierungseffizienz und VSV-G-Expressionsvermögen der Plasmide pCD-Gsyn und pCI-Gsyn. – Durch Cotransfektion von 293T-Zellen mit 5 µg des SIV-Vektor-Plasmids ViCGΔBH, 5 µg des SIV-gag-pol-Expressionsplasmids SgpΔ2 und 0,5 µg des angegebenen VSV-G-Expressionsplasmids wurden lentivirale Vektoren produziert. A: Die Titer der Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Ein Kontrollexpe-riment ohne VSV-G-Expressionsplasmid lieferte einen Titer < 5 GFU/ml (nicht dargestellt). B: Nach 72 h wurden die Vektor produzierenden Zellen lysiert und die Viruspartikel aus den Zellkultur-Überständen sedimentiert. Bei der anschließenden Westernblot-Analyse wurden monoklonale Anti-körper gegen VSV-G und HIV-1 p24 zur Immunodetektion eingesetzt. CA: Capsidprotein.
3.3.2 VSV-Gsyn-G-Fusions-RNA
Um auszuschließen, dass für die korrekte Expression und Lokalisation des VSV-G-Proteins
bedeutsame RNA-Elemente durch die vorgenommene Codonoptimierung entfernt wurden
und deshalb die Pseudotypisierungseffizienz des Plasmids pCI-Gsyn wie oben beschrieben
unerwartet niedrig ausfiel, wurde die für VSV-G codierende Wildtyp-Sequenz hinter die co-
donoptimierte Sequenz kloniert und dabei das Start-Codon der Wildtyp-Sequenz mutiert, so
dass bei Transkription von diesem Konstrukt eine VSV-Gsyn-G-Fusions-RNA entsteht, von
der nur die codonoptimierte Sequenz translatiert wird (s. Abb. 3-14 A). Das so erhaltene
pCI-Gsyn-G führte bei Einsatz zur Pseudotypisierung von SIV-Vektoren mit
(2,1 ± 0,4) × 103 GFU/ml zu keinen höheren Titern als pCI-Gsyn (2,4 ± 0,1 × 103 GFU/ml) (s.
Abb. 3-14 B).

3 Ergebnisse 53
Abb. 3-14. Einfluss regulatorischer Elemente innerhalb der für VSV-G codierenden Sequenz auf die Pseudotypisierungseffizienz. – A: Schematische Darstellung der Expressionskassette des pCI-Gsyn-G. B: Durch Cotransfektion von 293T-Zellen mit 5 µg des SIV-Vektor-Plasmids ViCGΔBH, 5 µg des SIV-gag-pol-Expressionsplasmids SgpΔ2 und 1 µg des angegebenen VSV-G-Expressions-plasmids wurden lentivirale Vektoren produziert und deren Titer als grüne-Fluoreszenz-bildende Ein-heiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Experimenten. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt).
3.3.3 Sequenz-Analyse der Wildtyp-VSV-G-DNA aus pCI-G
Um die Ursache für das Mißlingen der Pseudotypisierung mittels pCI-Gsyn zu finden, wurde
die Aminosäure-Sequenz des VSV-Gsyn mit der des verwendeten Wildtyp-VSV-G verglichen.
Dazu wurde die Wildtyp-VSV-G-DNA aus pCI-G in Zusammenarbeit mit Dr. Dennis Hoff-
mann sequenziert und die erhaltene Sequenz in die entsprechende Aminosäure-Sequenz über-
setzt. Der Vergleich dieser Sequenz mit der, auf der die Codonoptimierung beruhte, zeigt
zwei Unterschiede auf (s. Abb. 3-15): An Position 57 befindet sich in VSV-Gsyn ein Isoleucin
statt eines Leucins und an Position 96 ein Glutamin statt eines Histidins.

3 Ergebnisse 54
1 60 VSV-G MKCLLYLAFLFIGVNCKFTIVFPHNQKGNWKNVPSNYHYCPSSSDLNWHNDLIGTALQVK VSV-Gsyn MKCLLYLAFLFIGVNCKFTIVFPHNQKGNWKNVPSNYHYCPSSSDLNWHNDLIGTAIQVK
61 120 VSV-G MPKSHKAIQADGWMCHASKWVTTCDFRWYGPKYITHSIRSFTPSVEQCKESIEQTKQGTW VSV-Gsyn MPKSHKAIQADGWMCHASKWVTTCDFRWYGPKYITQSIRSFTPSVEQCKESIEQTKQGTW
121 180 VSV-G LNPGFPPQSCGYATVTDAEAVIVQVTPHHVLVDEYTGEWVDSQFINGKCSNYICPTVHNS VSV-Gsyn LNPGFPPQSCGYATVTDAEAVIVQVTPHHVLVDEYTGEWVDSQFINGKCSNYICPTVHNS
181 240 VSV-G TTWHSDYKVKGLCDSNLISMDITFFSEDGELSSLGKEGTGFRSNYFAYETGGKACKMQYC VSV-Gsyn TTWHSDYKVKGLCDSNLISMDITFFSEDGELSSLGKEGTGFRSNYFAYETGGKACKMQYC
241 300 VSV-G KHWGVRLPSGVWFEMADKDLFAAARFPECPEGSSISAPSQTSVDVSLIQDVERILDYSLC VSV-Gsyn KHWGVRLPSGVWFEMADKDLFAAARFPECPEGSSISAPSQTSVDVSLIQDVERILDYSLC
301 360 VSV-G QETWSKIRAGLPISPVDLSYLAPKNPGTGPAFTIINGTLKYFETRYIRVDIAAPILSRMV VSV-Gsyn QETWSKIRAGLPISPVDLSYLAPKNPGTGPAFTIINGTLKYFETRYIRVDIAAPILSRMV
361 420 VSV-G GMISGTTTERELWDDWAPYEDVEIGPNGVLRTSSGYKFPLYMIGHGMLDSDLHLSSKAQV VSV-Gsyn GMISGTTTERELWDDWAPYEDVEIGPNGVLRTSSGYKFPLYMIGHGMLDSDLHLSSKAQV
421 480 VSV-G FEHPHIQDAASQLPDDESLFFGDTGLSKNPIELVEGWFSSWKSSIASFFFIIGLIIGLFL VSV-Gsyn FEHPHIQDAASQLPDDESLFFGDTGLSKNPIELVEGWFSSWKSSIASFFFIIGLIIGLFL
481 511 VSV-G VLRVGIHLCIKLKHTKKRQIYTDIEMNRLGK VSV-Gsyn VLRVGIHLCIKLKHTKKRQIYTDIEMNRLGK
Abb. 3-15. Aminosäure-Sequenzvergleich zwischen VSV-G und VSV-Gsyn. – Die angegebene A-minosäuresequenz des VSV-G ist eine Übersetzung des DNA-Sequenzierungs-Resultats. Die Amino-säuresequenz des VSV-Gsyn entspricht dem Genbankeintrag J02428 und wurde so bei der Codonopti-mierung zugrunde gelegt. Unterschiede der Sequenzen sind orange markiert.
3.3.4 Korrektur der codonoptimierten VSV-G-Sequenz
Die codonoptimierte VSV-G-Sequenz wurde so verändert, dass sie für ein Protein mit tatsäch-
lich identischer Sequenz zum verwendetetn Wildtyp-VSV-G codiert. Zunächst wurde unter-
sucht, ob diese Veränderung der codonoptimierten VSV-G-Sequenz eine Pseudotypisierung
wieder möglich macht. Folglich wurde mittels PCR-Mutagenese-Methoden in pCI-Gsyn das
Codon ATC für Isoleucin an Position 169 der VSV-Gsyn-Sequenz gegen CTG für Leucin und
das Codon CAG für Glutamin an Position 286 der VSV-Gsyn-Sequenz gegen CAC für Hisiti-
din ausgetauscht und so pCI-Gopt konstruiert (s. Abb. 3-16).

3 Ergebnisse 55
Abb. 3-16. Expressionskassetten der VSV-G-Expressionsplasmide pCI-Gsyn, pCI-Gopt und pCD-Gopt (schematische Darstellung). – HCMV-Promotor: Promotor/Enhancer-Region der Major-immediate-early-Gene (MIE-Gene) des humanen Cytomegalievirus; Ex 1: HCMV-MIE-Exon 1; Ex 2: nicht translatiertes 5’-Ende des HCMV-MIE-Exons 2; Intron: erstes HCMV-MIE-Intron; VSV-Gsyn: nach Genbankeintrag J02428 codonoptimierte codierende Sequenz für das VSV-G-Protein; VSV-Gopt: mutierte VSV-Gsyn-DNA-Sequenz (rote Sternchen symbolisieren Mutationen); BGH pAd: Polyadeny-lierungssignal des Rinder-Wachstumshormons.
Im Gegensatz zu mittels pCI-Gsyn pseudotypisierten SIV-Vektoren ((3,6 ± 2,3) × 103 GFU/ml)
konnten die Vektoren mittels pCI-Gopt genau so effizient pseudotypisiert werden wie mittels
pCI-G ((3,2 ± 1,3) × 105 GFU/ml bzw. (4,3 ± 1,3) × 105 GFU/ml) (s. Abb. 3-17 A).
Abb. 3-17. VSV-G-Expression mittels pCI-Gopt. – Zur Produktion lentiviraler Vektoren wurden 293T-Zellen mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 1 µg (A) bzw. 2 µg (B) des angegebenen VSV-G-Expressionsplasmids cotransfiziert. A: Die Titer der Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Ex-perimenten. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt). B: Die transfizierten Zellen wurden lysiert und einer Westernblot-Analyse unter Verwen-dung monoklonaler Antikörper gegen VSV-G und HIV-1 p24 unterzogen. CA: Capsidprotein.
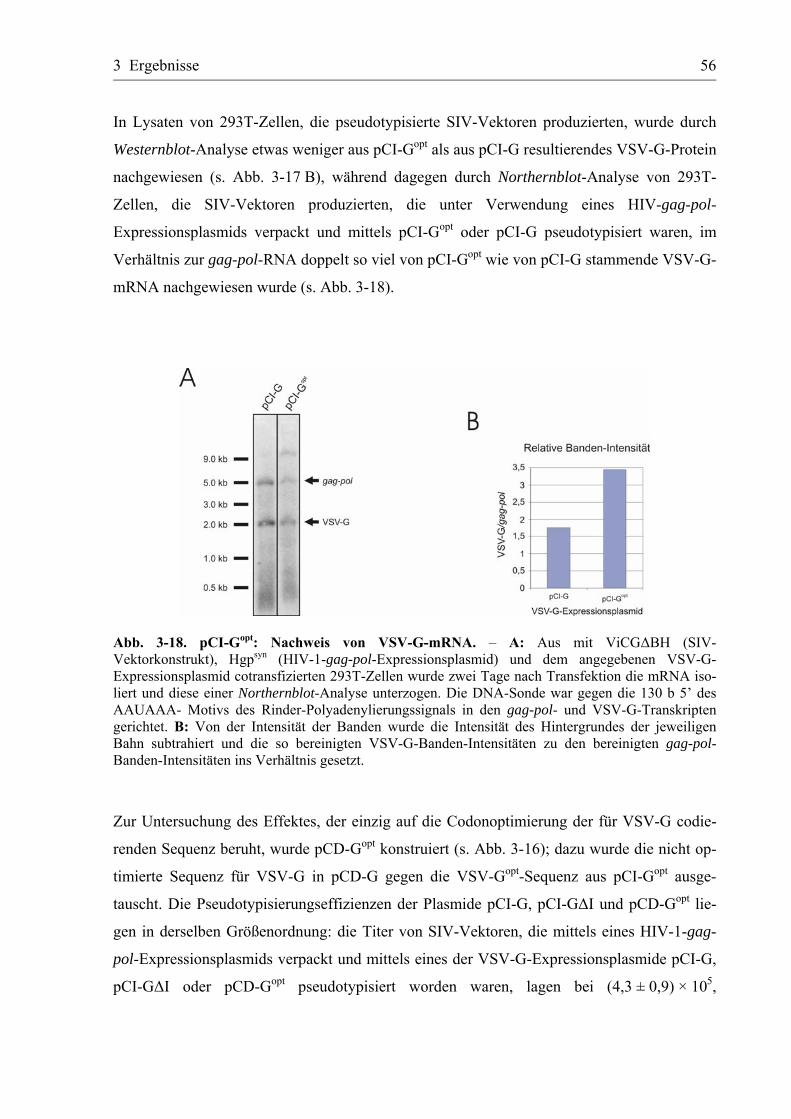
3 Ergebnisse 56
In Lysaten von 293T-Zellen, die pseudotypisierte SIV-Vektoren produzierten, wurde durch
Westernblot-Analyse etwas weniger aus pCI-Gopt als aus pCI-G resultierendes VSV-G-Protein
nachgewiesen (s. Abb. 3-17 B), während dagegen durch Northernblot-Analyse von 293T-
Zellen, die SIV-Vektoren produzierten, die unter Verwendung eines HIV-gag-pol-
Expressionsplasmids verpackt und mittels pCI-Gopt oder pCI-G pseudotypisiert waren, im
Verhältnis zur gag-pol-RNA doppelt so viel von pCI-Gopt wie von pCI-G stammende VSV-G-
mRNA nachgewiesen wurde (s. Abb. 3-18).
Abb. 3-18. pCI-Gopt: Nachweis von VSV-G-mRNA. – A: Aus mit ViCGΔBH (SIV-Vektorkonstrukt), Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und dem angegebenen VSV-G-Expressionsplasmid cotransfizierten 293T-Zellen wurde zwei Tage nach Transfektion die mRNA iso-liert und diese einer Northernblot-Analyse unterzogen. Die DNA-Sonde war gegen die 130 b 5’ des AAUAAA- Motivs des Rinder-Polyadenylierungssignals in den gag-pol- und VSV-G-Transkripten gerichtet. B: Von der Intensität der Banden wurde die Intensität des Hintergrundes der jeweiligen Bahn subtrahiert und die so bereinigten VSV-G-Banden-Intensitäten zu den bereinigten gag-pol-Banden-Intensitäten ins Verhältnis gesetzt.
Zur Untersuchung des Effektes, der einzig auf die Codonoptimierung der für VSV-G codie-
renden Sequenz beruht, wurde pCD-Gopt konstruiert (s. Abb. 3-16); dazu wurde die nicht op-
timierte Sequenz für VSV-G in pCD-G gegen die VSV-Gopt-Sequenz aus pCI-Gopt ausge-
tauscht. Die Pseudotypisierungseffizienzen der Plasmide pCI-G, pCI-GΔI und pCD-Gopt lie-
gen in derselben Größenordnung: die Titer von SIV-Vektoren, die mittels eines HIV-1-gag-
pol-Expressionsplasmids verpackt und mittels eines der VSV-G-Expressionsplasmide pCI-G,
pCI-GΔI oder pCD-Gopt pseudotypisiert worden waren, lagen bei (4,3 ± 0,9) × 105,

3 Ergebnisse 57
(2,0 ± 0,5) × 105 und (1,3 ± 0,2) × 105 GFU/ml, während die mittels pCD-G pseudotypisierter
Vektoren bei (790 ± 130) GFU/ml lagen (s. Abb. 3-19 A).
Abb. 3-19: VSV-G-Expression mittels pCD-Gopt. – A: Zur Produktion lentiviraler Vektoren wurden 293T-Zellen mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die Titer der Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Gezeigt sind Mittelwert und Standardabweichung aus drei Experimenten. Kontrollexperi-mente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt). B: 293T-Zellen wurden mit 5 µg ViCGΔBH (SIV-Vektor-Konstrukt), 5 µg SgpΔ2 (SIV-gag-pol-Expressionsplasmid) und 2 µg des angegebenen VSV-G-Expressionsplasmids cotransfiziert. Die transfizierten Zellen wurden lysiert und einer Westernblot-Analyse unter Verwendung monoklonaler Antikörper gegen VSV-G und HIV-1 p24 unterzogen. CA: Capsidprotein. C: 293T-Zellen wurden mit ViCGΔBH (SIV-Vektorkonstrukt), Hgpsyn (HIV-1-gag-pol-Expressionsplasmid) und dem angegebe-nen VSV-G-Expressionsplasmid cotransfiziert. Die nach zwei Tagen aus den transfizierten Zellen isolierte mRNA wurde einer Northernblot-Analyse unterzogen. Die DNA-Sonde war gegen die 130 b 5’ des AAUAAA- Motivs des Rinder-Polyadenylierungssignals in den gag-pol- und VSV-G-Transkripten gerichtet.

3 Ergebnisse 58
Außerdem konnte in Lysaten von 293T-Zellen, die SIV-Vektoren produzierten, welche mit-
tels pCD-Gopt oder pCI-G pseudotypisiert worden waren, bzw. in der mRNA von 293T-
Zellen, die SIV-Vektoren produzierten, die mittels eines HIV-1-gag-pol-Expressionsplasmids
verpackt und mittels pCD-Gopt oder pCI-G pseudotypisiert worden waren, durch Western-
bzw. Northernblot-Analyse VSV-G-Protein bzw. -mRNA nachgewiesen werden – aus pCD-G
resultierende VSV-G-mRNA bzw. resultierendes -Protein konnte im Gegensatz dazu nicht
nachgewiesen werden (s. Abb. 3-19).
3.3.5 Kombination von HCMV-MIE-5’-UTR mit codonoptimierter VSV-G-Sequenz
Zuletzt wurde geprüft, ob codonoptimierte Konstrukte, die zusätzlich die unter 3.1 und 3.2
beschriebenen HCMV-Sequenzen enthalten, besser zur Pseudotypisierung geeignet sind als
Konstrukte, die entweder nur die codonoptimierte VSV-G-Sequenz ohne HCMV-Sequenzen
in der 5’-UTR oder diese HCMV-Sequenzen in Verbindung mit der Wildtyp-VSV-G-Sequenz
enthalten. Dazu wurden pCI-G, pCD-Gopt und pCI-Gopt miteinander verglichen.
Da nur geringe Unterschiede in der Pseudotypisierungseffizienz erwartet wurden, wurde diese
in Abhängigkeit unterschiedlicher transfizierter Mengen dieser Konstrukte betrachtet. Es
wurden die Titer von HIV-1-Vektoren bestimmt, die unter Verwendung von 0,03, 0,1, 0,3
oder 1 µg der Plasmide pCI-G, pCD-Gopt oder pCI-Gopt pseudotypisiert worden waren
(s. Abb. 3-20 A).
Bei der Produktion der HIV-1-Vektoren wurde auch das Expressionsplasmid für die Sekre-
tierte Alkalische Phosphatase (SEAP) cotransfiziert, welches üblicherweise zum Vergleich
von Transfektionseffizienzen eingesetzt wird, und die SEAP in den Vektorpräparaten durch
eine Chemilumineszenz-Reaktion quantifiziert. Die dabei gemessene relative Licht-Einheit
pro Sekunde (RLU/s) ist ein Maß für die SEAP-Aktivität im Vektorpräparat (s. Abb. 3-20 B).
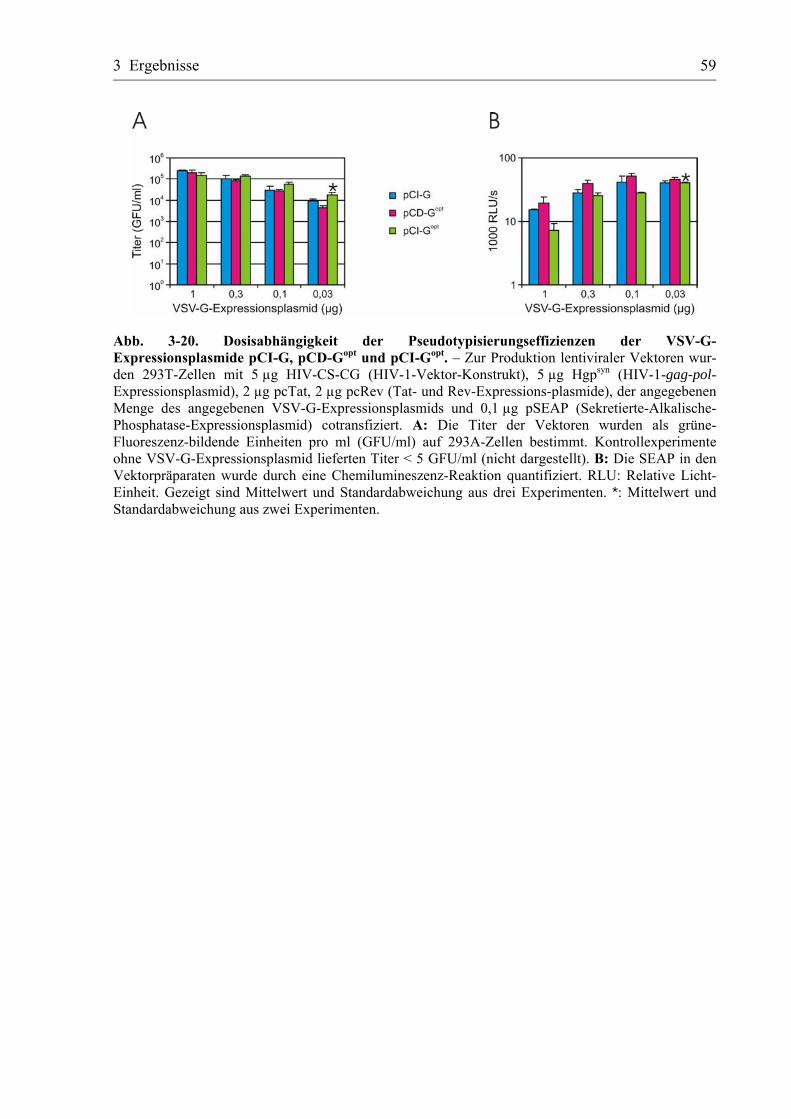
3 Ergebnisse 59
Abb. 3-20. Dosisabhängigkeit der Pseudotypisierungseffizienzen der VSV-G-Expressionsplasmide pCI-G, pCD-Gopt und pCI-Gopt. – Zur Produktion lentiviraler Vektoren wur-den 293T-Zellen mit 5 µg HIV-CS-CG (HIV-1-Vektor-Konstrukt), 5 µg Hgpsyn (HIV-1-gag-pol-Expressionsplasmid), 2 µg pcTat, 2 µg pcRev (Tat- und Rev-Expressions-plasmide), der angegebenen Menge des angegebenen VSV-G-Expressionsplasmids und 0,1 µg pSEAP (Sekretierte-Alkalische-Phosphatase-Expressionsplasmid) cotransfiziert. A: Die Titer der Vektoren wurden als grüne-Fluoreszenz-bildende Einheiten pro ml (GFU/ml) auf 293A-Zellen bestimmt. Kontrollexperimente ohne VSV-G-Expressionsplasmid lieferten Titer < 5 GFU/ml (nicht dargestellt). B: Die SEAP in den Vektorpräparaten wurde durch eine Chemilumineszenz-Reaktion quantifiziert. RLU: Relative Licht-Einheit. Gezeigt sind Mittelwert und Standardabweichung aus drei Experimenten. *: Mittelwert und Standardabweichung aus zwei Experimenten.

4 Diskussion
4.1 Intronunabhängige Expression von VSV-G
Die Ergebnisse der vorliegenden Arbeit zeigen, dass es möglich ist, VSV-G unter Verwen-
dung des HCMV-major-immediate-early-(MIE)-Promotors auch ohne das erste Intron der
HCMV-MIE-Gene effizient zu exprimieren und zur Pseudotypisierung von SIV-, HIV-1- so-
wie SIV-Vektoren, die mit Hilfe eines HIV-1 gag-pol-Expressionsplasmids verpackt wurden,
einzusetzen. Das Intron mit flankierenden nicht translatierten exonischen HCMV Sequenzen –
insgesamt 965 Nukleotide – kann auf die ersten 106 Nukleotide des ersten Exons der HCMV-
MIE-Gene reduziert werden, um einen vergleichbaren Effekt zu erzielen. Auf HCMV-
Sequenzen in der 5’-UTR kann völlig verzichtet werden, wenn statt der Wildtyp-VSV-G-
Sequenz eine codonoptimierte Sequenz eingesetzt wird.
4.2 Das HCMV-MIE-Intron und Spleißen als Expressionsverstärker
In der vorliegenden Arbeit wurde der Effekt des ersten HCMV-MIE-Introns auf die HCMV-
MIE-Promotor/Enhancer gesteuerte Expression des Glykoproteins G des Vesicular-Stoma-
titis-Virus und auf die Effizienz, mit der lentivirale Vektoren VSV-G-pseudotypisiert werden,
untersucht. Diese Untersuchungen wurden ausgehend vom Plasmid pHIT-G mit
pCDNA3.1(+)-basierten VSV-G-Expressionsplasmiden durchgeführt.
Dem pHIT-G entspricht das pCDNA3.1(+)-basierte Plasmid pCI-G: Bis auf die 3’-UTR und
das Polyadenylierungssignal stimmen die Expressionskassetten überein (s. Abb. 3-1), die
Westernblot-Analyse der Lysate entsprechend transfizierter 293T-Zellen zeigt keine Unter-
schiede im VSV-G-Expressionsvermögen (s. Abb. 3-2 B), und Lentiviren können mit beiden
Plasmiden gleich gut pseudotypisiert werden (s. Abb. 3-2 A). pHIT-G enthält die 5’-UTR der
HCMV-MIE-Gene mitsamt des ersten Introns [91,92], da gezeigt wurde, dass dieses Intron
die heterologe Expression in Säugerzellen positiv regulieren kann: Chapman et al. haben die
HCMV-Promotor getriebene Expression des Gewebe-Plasminogenaktivators, des humanen
Gerinnungsfaktors VIII und des HIV-gp120 jeweils mit und ohne HCMV-Intron in der

4 Diskussion 61
5’-UTR verglichen, dabei wurde für die beiden letztgenannten Glykoproteine ohne das Intron
eine Expressionsstärke von 74 % bzw. 2 % der Expressionsstärke mit Intron erhalten, für das
erstgenannte Glykoprotein ergab sich kein Unterschied in den Expressionsstärken [61].
Innerhalb dieser Spanne liegen auch die hier präsentierten Ergebnisse: Die Pseudotypisie-
rungseffizienz des pCI-GΔI (ohne Intron, s. Abb. 3-4) beträgt 46 % der des pCI-G (mit Intron,
s. Abb. 3-4) und es wird laut Westernblot-Analyse durch pCI-GΔI etwas weniger VSV-G
exprimiert als durch pCI-G (s. Abb. 3-5). Dieser Unterschied zeigt sich bereits prätranslatio-
nal: Aus pCI-GΔI resultieren im Verhältnis zur gag-pol-RNA 58 % der VSV-G-mRNA, die
aus pCI-G resultieren (s. Abb. 3-6). Es wird also auch für das hier verwendete Expressions-
system bestätigt, dass das erste HCMV-MIE-Intron die durch den HCMV-MIE-
Promotor/Enhancer gesteuerte Expression positiv reguliert und dass diese Regulation vor der
Translation erfolgt. Diese Wirkung des Introns beruht auf einer in dem Intron enthaltenen
Enhancer-Sequenz, die der Enhancer-Sequenz im ersten Intron des Gens für Troponin I der
Wachtel homolog ist [61,81].
Die Verwendung des ersten HCMV-MIE-Introns in VSV-G-Expressionsplasmiden, die zur
Pseudotypisierung lentiviraler Vektoren eingesetzt werden, wird an dieser Stelle als durchaus
entbehrlich eingestuft. Ein Vektortiter, der um die Hälfte reduziert ist und sich noch in dersel-
ben Größendordnung befindet, ist immer noch sehr gut. Ist es etwa. erforderlich, die VSV-G-
Expressionskassette in ein Gefüge zu klonieren, in dem der Platz beschränkt ist, so kann ohne
große Verluste in der Pseudotypisierungseffizienz auf die 827 bp umfassende Intron-Sequenz
verzichtet werden.
Im Gegensatz dazu führt der Verzicht auf die verbleibenden exonischen Sequenzen der
HCMV-MIE-5’-UTR in VSV-G-Expressionsplasmiden zu einer drastischen Reduktion des
Titers der lentiviralen Vektoren, die mittels dieser Plasmide pseudotypisiert wurden. Die
Pseudotypisierungseffizienz des pCD-G (keine HCMV-MIE-Sequenzen in der 5’-UTR, s.
Abb. 3-4) beträgt nur 0,4 % der des pCI-GΔI (s. Abb. 3-5 A). Weniger drastisch ist der Unter-
schied zwar auf mRNA-Ebene – im Verhältnis zur gag-pol-RNA ergeben sich aus pCD-G
10 % der VSV-G-mRNA, die sich aus pCI-GΔI ergeben (s. Abb. 3-6). Durch pCD-G expri-
miertes VSV-G-Protein konnte jedoch durch Westernblot-Analyse nicht nachgewiesen wer-
den (s. Abb. 3-5 B).
Nun wäre denkbar, dass in pCI-GΔI kryptische Spleißstellen aktiviert wurden [102]. Dann
würden der herausragenden VSV-G-Expression mittels pCI-GΔI die gleichen Mechanismen
zugrunde liegen wie der mittels pCI-G. Daher wurde die gesamte RNA entsprechend transfi-

4 Diskussion 62
zierter Zellen hinsichtlich VSV-G-Primär-Transkripte und Spleißprodukte untersucht. Die
RT-PCR vom Transkriptionsstart bis zur 132. Base der für VSV-G codierenden Sequenz er-
bringt für die pCI-GΔI-Probe nur ein einziges 302 bp umfassendes Amplifikat, das sich auch
in der pCI-G-Probe wieder findet (s. Abb 3-7). Dies ist der Nachweis für das aus pCI-GΔI
resultierende Primärtranskript und die aus pCI-G resultierende regulär gespleißte VSV-G-
mRNA und wird auch durch die Sequenzanalyse der Amplifikate bestätigt (s. Abb. 3-8). Da
die RT-PCR der pCI-GΔI-Probe keine weiteren Amplifikate erzeugt, läßt sich folgern, dass
das aus pCI-GΔI resultierende Transkript tatsächlich nicht gespleißt wird. Es sind also andere
Mechanismen für die im Vergleich zu pCD-G überragende VSV-G-Expression durch pCI-
GΔI verantwortlich als durch Introns und Spleißen bewirkte.
Somit ist auch die expressionsfördernde Wirkung der vollständigen, aus Exon- und Intronse-
quenzen bestehenden HCMV-MIE-5’-UTR – wie sie im Plasmid pCI-G vorliegt – überwie-
gend auf die Exons und nur mäßig auf das Intron zurückzuführen.
Für pCI-G wurde neben dem regulären Spleißprodukt durch ein 1,13 kb grosses Amplifikat
das Primärtranskript und durch ein 713 bp grosses Amplifikat in geringen Mengen eine weite-
re, nicht erwartete RNA nachgewiesen (s. Abb. 3-7). Aus der erhaltenen Sequenz des uner-
warteten Amplifikats (s. Abb. 3-8) läßt sich schließen, dass es sich um ein – bisher noch nicht
beschriebenes – alternatives Spleißprodukt handelt und das erste HCMV-MIE-Intron demzu-
folge ein optionales Exon enthält [103]. Diese Schlußfolgerung wird auch dadurch gestützt,
dass die Sequenz an den entsprechenden Stellen die von Yeo et al. [99] beschriebenen Kon-
sensussequenzen für die 5’- und 3’-Spleiß- sowie für die Verzweigungsstelle aufweist
(s. Abb. 3-8).
4.3 Exonische HCMV-Sequenzen in der 5’-UTR als Expressionsverstärker
Die HCMV-MIE-Exon-Sequenzen in der 5’-UTR der VSV-G-Expressionskassette, die die
oben beschriebene positive Regulation der Expression bewirken, können weiter eingeschränkt
und aufgegliedert werden. Die Sekundärstruktur-Vorhersage für die 5’-UTR in der mRNA
zeigt eine Stemloop-Struktur, die von 15 Nukleotiden des 3’-Endes des Exons 1 und von den
gesamten 17 Nukleotiden der nichttranslatierten Sequenz des Exons 2 gebildet wird; fast an
der Spitze dieser Struktur steht also die Fusionsstelle der beiden Exone (s. Abb. 3-9). Das
VSV-G-Expressionsplasmid pCSL-G enthält aus der HCMV-MIE-5’-UTR nur diese stemloop

4 Diskussion 63
bildende Sequenz (s. Abb. 3-10 A). Auch wenn der Nachweis der VSV-G-mRNA und des
Proteins nicht gelang (s. Abb. 3-11) und die Titer der lentiviralen Vektoren, die mit Hilfe die-
ses Plasmids pseudotypisiert wurden, nur 4 % der Titer der mit pCI-GΔI pseudotypisierten
Vektoren erreichen, so sind diese Titer immer noch 10mal höher als die der mit pCD-G pseu-
dotypisierten Vektoren (s. Abb. 3-10 B). Das VSV-G-Expressionsplasmid pCI-GΔIΔSL ent-
hält aus der HCMV-MIE-5’-UTR nur die Sequenz des Exons 1 5’ der stemloop bildenden
Sequenz (s. Abb. 3-10 A). Die aus diesem Plasmid resultierende VSV-G-mRNA und das Pro-
tein konnten nachgewiesen werden, die Titer der mit diesem Plasmid pseudotypisierten Vek-
toren erreichen 52 % der Titer der mit pCI-GΔI pseudotypisierten Vektoren.
Die stemloop bildende Sequenz hat folglich einen positiven Einfluss auf die VSV-G-
Expression, dieser ist aber vergleichsweise gering, so dass die Reduktion der HCMV-
Sequenzen in der 5’-UTR auf die bloße stemloop bildende Sequenz für die effiziente Pseudo-
typisierung lentiviraler Vektoren nicht ausreicht. Da diese Sequenz eine Spleißstelle um-
spannt, resultiert sie aus pCI-G erst nach dem Spleißen der prä-mRNA, ihre Funktion entfaltet
sie also auch erst als gespleißte RNA. Regulatorische Elemente in nicht translatierten Regio-
nen von mRNAs, wie z.B. (1) das Iron-responsive-Element (IRE) in der 5’- bzw. 3’-UTR der
mRNA der Ferritin-H- und -L-Kette bzw. des Transferrinrezeptors [104], (2) die Akut-Box
(AB) in der 5’-UTR der mRNA der Ferritin-H- und -L-Kette und des Amyloid Vorläuferpro-
teins [105,106], (3) ein Translationsverstärker-Element in der 5’-UTR der humanen Hsp70
mRNA [107] und (4) die interne Ribosomeneintrittsstelle (IRES) in einigen viralen RNAs und
eukaryotischen mRNAs [108] sind bereits bekannt. Die positive Regulation der Expression
durch die in der vorliegenden Arbeit angeführte stemloop bildenden Sequenz sowie zugrunde
liegende Mechanismen sind jedoch bisher noch nicht beschrieben. Die vorhergesagte Stem-
loop-Struktur ist mit ΔG = - 42 kJ/mol etwas weniger stabil als die Stemloop-Struktur der A-
kut-Box der mRNA der Ferritin-L-Kette mit ΔG = - 67 kJ/mol [105]. Es sind weitere Experi-
mente nötig, um zu klären, ob die Sekundärstruktur der RNA in diesem Sequenzabschnitt
tatsächlich von Bedeutung ist, sowie ob und welche zellulären Proteine an die Stemloop-
Struktur in der 5’-UTR der mRNA binden.
Beinahe der gesamte VSV-G-Expressions-fördernde Effekt der exonischen nichttranslatierten
HCMV-MIE-Exon-Sequenzen kann mithin allein der Sequenz des Exons 1 5’ der stemloop
bildenden Sequenz zugeschrieben werden. In diesem Abschnitt liegt auch die von Ghazal und
Nelson beschriebene positive Transkriptions-Kontroll-Domäne, die wiederum eine Sequenz

4 Diskussion 64
enthält, die in Cytomegalieviren verschiedener Spezies konserviert ist und eine Bindestelle für
zwei Zellkernproteine darstellt [62].
Es fällt auf, dass die in der vorliegenden Arbeit durchgeführten Western- und Northernblots
bei der Untersuchung der unter der Verwendung der Plasmide pCD-G und pCSL-G gewonne-
nen Proben an ihre untere Nachweisgrenze gestoßen sind. Das erklärt die folgenden scheinba-
ren Widersprüche: (1) Obwohl für die genannten Proben kein VSV-G im Westernblot nach-
gewiesen wird, werden infektiöse, pseudotypisierte lentivirale Vektoren nachgewiesen
(s. Abb. 3-2, Abb. 3-5, Abb. 3-10 B, Abb. 3-11 A). (2) In Abb. 3-6 wird VSV-G-mRNA, die
sich aus pCD-G ergibt, nachgewiesen, in Abb. 3-3 dagegen nicht. (3) Obwohl die Verwen-
dung von pCD-G zu 10fach niedrigeren Titern führt als die Verwendung von pCSL-G
(s. Abb. 3-10 B), kann aus pCI-G resultierende VSV-G-mRNA nachgewiesen werden
(s. Abb. 3-6), die aus pCSL-G resultierende jedoch nicht (s. Abb. 3-11 B).
4.4 Expression eines codonoptimierten, mutierten VSV-G
Das VSV-G-Protein, das durch die Sequenz codiert wird, die nach dem Genbankeintrag
J02428 codonoptimiert wurde, erwies sich als eine VSV-G-Mutante, bei der im Vergleich zur
Wildtyp-Aminosäuresequenz an der Position 57 Leucin gegen Isoleucin und an der Position
96 Histidin gegen Glutamin ausgetauscht ist (s. Abb. 3-15). Es wird zwar intrazellulär expri-
miert, ist aber nicht für die Pseudotypisierung von lentiviralen Vektoren geeignet, weil es
nicht in ausreichender Menge in die Virushülle gelangt: Lentivirale Vektoren, die mit Hilfe
des Plasmids pCI-Gsyn pseudotypisiert sind, erreichen kaum 0,4 % der Titer, die mittels pCI-G
pseudotypisierte lentivirale Vektoren erreichen (s. Abb. 3-13 A). In Lysaten entsprechend
transfizierter 293T-Zellen kann sowohl das von pCI-G stammende Wildtyp- als auch das von
pCI-Gsyn stammende mutierte VSV-G durch Westernblot-Analyse nachgewiesen werden – das
mutierte VSV-G scheint sogar akkumulierter zu sein als das Wildtyp-VSV-G. In den unter-
suchten Viruspartikeln bleibt das mutierte VSV-G jedoch im Gegensatz zum Wildtyp-VSV-G
unter der Nachweisgrenze (s. Abb. 3-13 B). Das mutierte Protein wird also offensichtlich
exprimiert, aber wahrscheinlich nicht in dem Maße wie das Wildtyp-Protein zur Cytoplas-
mamembran transportiert und infolgedessen auch nur unzureichend in die Virushülle einge-
baut.

4 Diskussion 65
Die Aminosäuresequenzen von mutiertem VSV-G und Wildtyp-VSV-G unterscheiden sich an
zwei Positionen, die Nukleinsäuresequenzen unterscheiden sich im vorliegenden Fall jedoch
im Mittel an jeder vierten Position, da für das Wildtyp-VSV-G eine Wildtyp-DNA codiert
und für das mutierte VSV-G eine codonoptimierte DNA. Führen nun die beiden Mutationen
der Aminosäuresequenz oder die vielen (stillen) Mutationen auf Nukleinsäureebene zu dem
mangelhaften Transport des VSV-G zur Plasmamembran?
Es wäre z. B. möglich, dass die Wildtyp-VSV-G-mRNA Signale für die Lokalisation der
mRNA enthält, ähnlich wie die zip codes, die bei lokalisierten mRNAs meist in der 3’-UTR
enthalten sind, und die Lokalisation der mRNA Voraussetzung ist für die richtige Lokalisati-
on des resultierenden Proteins [109]. Dann müssten bei der Pseudotypisierung mit dem Kon-
strukt pCI-Gsyn-G ähnlich hohe lentivirale Vektor-Titer erzielt werden wie bei der Pseudotypi-
sierung mit pCI-G bzw. deutlich höhere Titer als bei der Verwendung von pCI-Gsyn. In pCI-
Gsyn-G ist die Wildtyp-VSV-G-Sequenz mit deletiertem Startcodon hinter die codonoptimierte
Sequenz – unter Erhalt des Stop-Codons der codonoptimierten Sequenz – eingefügt, so dass
aus dieser Fusions-DNA eine Fusions-RNA, aber weder ein Fusions-Protein noch ein Wild-
typ-Protein entsteht. Die Titer der mit pCI-Gsyn-G pseudotypisierten Vektoren waren jedoch
genauso niedrig wie die der mit pCI-Gsyn pseudotypisierten.
Einen weiteren Befund, der gegen die Mutationen auf Nukleinsäureebene als Ursache für den
gestörten VSV-G-Transport zur Plasmamembran spricht, liefern Experimente mit dem
VSV-G-Expressionsplasmid pCI-Gopt, bei dem die VSV-Gsyn-Sequenz so verändert wurde,
dass es sich nach wie vor um eine codonoptimierte Sequenz handelt, die aber tatsächlich für
dieselbe Aminosäuresequenz codiert wie die Wildtyp-VSV-G-Sequenz: Mittels pCI-Gopt
pseudotypisierte lentivirale Vektoren erreichen im Gegensatz zu denen, die mittels pCI-Gsyn
pseudotypisiert sind, gleichhohe Titer wie mittels pCI-G pseudotypisierte Vektoren (s. Abb.
3-17 A). Außerdem zeigt die Westernblot-Analyse kein angereicherteres von pCI-Gopt als von
pCI-G stammendes VSV-G in den Zellen (s. Abb. 3-17 B). Es ist also mindestens eine der
beiden Mutationen in der Aminosäuresequenz des VSV-G, das aus pCI-Gsyn resultiert, die
eine effiziente Pseudotypisierung lentiviraler Vektoren mittels dieses Plasmids verhindert.
Die Aminosäuresequenz der VSV-G-Mutante entspricht der im Genbankeintrag J02428 wie-
dergegebenen Sequenz, die Rose und Gallione 1981 publiziert haben. Diese Sequenz war je-
doch noch mit Unsicherheiten behaftet [110]. In einer Veröffentlichung derselben Autoren
aus dem Jahre 1985 findet sich eine korrigierte Sequenz für VSV-G desselben Virusstammes
(San Juan): hier ist nun an Position 96 die Aminosäure Histidin und nicht mehr Glutamin an-

4 Diskussion 66
gegeben, an Position 57 ist nach wie vor die Aminosäure Isoleucin genannt, andererseits sind
die VSV-G-Sequenzen anderer Virusstämme aufgeführt, die an der Position ein Leucin auf-
weisen, an anderen Stellen aber weiter abweichen [111]. Im Unterschied zu Rose und Gallio-
ne und in Übereinstimmung mit den Daten in der vorliegenden Arbeit fanden Bilsel und Ni-
chol an der Position 57 der Aminosäuresequenz des VSV-G des Stammes San Juan die Ami-
nosäure Leucin, allerdings weicht deren Sequenz wiederum an zwei anderen Stellen von der
von Rose und Gallione sowie von der Wildtyp-Sequenz in der vorliegenden Arbeit ab [33].
Da es sowohl VSV-Stämme gibt, deren Glykoprotein G an der Position 57 ein Leucin auf-
weist, als auch solche die eben dort ein Isoleucin tragen, aber keine mit einem Glutamin statt
eines Histidins an Position 96, ist die Akkumulation der VSV-G-Mutante in der Zelle auf die
Mutation an Position 96 zurückzuführen. Es ist bekannt, dass VSV-G-Mutanten, die gar nicht
oder wenig effektiv zur Plasmamembran transportiert werden und die Mutation wie die Mut-
ante in der vorliegenden Arbeit in der Ectodomäne tragen, sich initial nicht richtig falten, in-
folgedessen nicht trimerisieren, im Endoplasmatischen Retikulum akkumulieren und aggre-
gieren [112].
4.5 Pseudotypisierung mit optimierter für VSV-G codierender Sequenz
Mit Hilfe des HCMV-MIE-Promotors läßt sich VSV-G von der Wildtyp-DNA-Sequenz nur
dann ausreichend exprimieren, wenn die 5’-UTR mindestens die oben beschriebene Region
aus dem HCMV-MIE-Exon 1 enthält, die sich 5’ der stemloop bildende Sequenz befindet.
Erfolgt die Expression dagegen von einer codonoptimierten Sequenz, kann auf HCMV-
Sequenzen in der 5’-UTR gänzlich verzichtet werden. So beträgt die Pseudotypisierungseffi-
zienz des VSV-G-Expressionsplasmids pCD-Gopt (codonoptimierte VSV-G-Sequenz, keine
HCMV-Sequenzen in der 5’ UTR, s. Abb. 3-16) immerhin 30 % der Pseudotypisierungseffi-
zienz des pCI-G (Wildtyp-VSV-G-Sequenz, enthält Exon 1, erstes Intron, nichttranslatierte
Region des Exons 2 der HCMV-MIE-Gene, s. Abb. 3-4) und 65 % der Pseudotypisierungsef-
fizienz des pCI-GΔI (wie pCI-G, aber ohne das erste HCMV-MIE-Intron, s. Abb. 3-4); dage-
gen beträgt die Pseudotypisierungseffizienz des pCD-G nur 0,6 % der des pCD-Gopt (s. Abb.
3-19 A). Im Gegensatz zur VSV-G-Expression mittels pCD-G ist die VSV-G-Expression mit-
tels pCD-Gopt durch Westernblot-Analyse ebenso gut nachweisbar wie die mittels pCI-G

4 Diskussion 67
(s. Abb. 3-19 B). Auch die aus pCD-Gopt resultierende mRNA zeigt sich wie die aus pCI-G
resultierende mRNA deutlich in der Northernblot-Analyse, während der Nachweis einer aus
pCD-G resultierenden mRNA im selben Experiment nicht gelingt (s. Abb. 3-19 C).
Die Wirkungsebene der Codonoptimierung wird in der Literatur kontrovers diskutiert [86-88].
Die Ergebnisse der vorliegenden Arbeit zeigen einen deutlichen Effekt auf Prätranslations-
ebene: Bei jeweils gleicher 5’-UTR resultiert – im Vergleich zur gag-pol-RNA – deutlich
mehr mRNA aus den codonoptimierten VSV-G-Sequenzen als aus den Wildtyp-Sequenzen
(s. Abb. 3-18, Abb. 3-19 C). Durch die Codonoptimierung hat sich der GC-Gehalt der für
VSV-G codierenden Sequenz von 43,8 % auf 62,9 % erhöht, damit ist auch die codonopti-
mierte VSV-G-mRNA stabiler als die Wildtyp-mRNA [90].
4.6 Codonoptimierung in Verbindung mit der HCMV-5’-UTR
Das Plasmid pCI-Gopt, das als Kombination der expressionsfördernden Maßnahmen die nicht
translatierten HCMV-Sequenzen und die codonoptimierte VSV-G-Sequenz enthält (s. Abb.
3-16), kann nicht eingesetzt werden, um eine absolute Steigerung der Titer der pseudotypi-
sierten Vektoren zu erreichen. Sollte – was im Allgemeinen nicht von Bedeutung ist – für
bestimmte Experimente die Reduktion der Plasmid-Dosis essentiell sein, so könnten dann bei
Verwendung des pCI-Gopt höhere Titer erzielt werden als bei Verwendung des pCI-G (nicht
translatierte HCMV-Sequenzen in der 5’-UTR, Wildtyp-VSV-G-Sequenz, s. Abb. 3-12) oder
pCD-Gopt (nur codonoptimierte VSV-G-Sequenz, s. Abb. 3-16).
Bei Variation der Menge des VSV-G-Expressionsplasmids, das zur Produktion von Lentivi-
ren transfiziert wird, steigt der Titer der pseudotypisierten Lentiviren mit zunehmender Men-
ge des VSV-G-Expressionsplasmids bis zum Erreichen einer Obergrenze an. Beim Vergleich
dieser Dosisabhängigkeit der Pseudotypisierungseffizienz der Plasmide pCI-G, pCD-Gopt und
pCI-Gopt, wird mit pCI-Gopt die Obergrenze schon mit geringerer Menge des Plasmids erreicht
als mit pCI-G oder pCD-Gopt. Bei Transfektion hoher Mengen VSV-G-Expressionsplasmids
führt die Verwendung des Plasmids pCI-Gopt zu etwas geringeren Pseudotypisierungseffizien-
zen als die der Plasmide pCI-G oder pCD-Gopt, bei Transfektion niedriger Mengen VSV-G-
Expressionsplasmids dagegen zu deutlich höheren Pseudotypisierungseffizienzen (s. Abb.
3-20 A).

4 Diskussion 68
Darüber hinaus gehen höhere Titer einher mit geringeren Aktivitäten der Sekretierten Alkali-
schen Phosphatase (SEAP), die von einem cotransfizierten Plasmid exprimiert wird (s. Abb.
3-20 B). In der Regel wird die SEAP benutzt, um Transfektionseffizienzen zu beurteilen. Im
vorliegenden Fall sind dagegen niedrigere SEAP-Aktivitäten nicht mit niedrigeren Transfek-
tionseffizienzen gleich zu setzen, die Korrelation sinkender SEAP-Aktivitäten mit steigenden
Mengen VSV-G-Expressionsplasmids kann durch die Toxizität des VSV-G erklärt werden
[48]. Die Toxizität des VSV-G ist wahrscheinlich auch die Ursache dafür, dass die Titer
pseudotypisierter Lentiviren unabhängig vom eingesetzten VSV-G-Expressionsplasmid im-
mer ein annähernd gleiches Maximum erreichen.
Folglich wird die Additivität der expressionsfördernden Effekte der Codonoptimierung und
der nicht translatierten HCMV-Sequenzen durch die Toxizität des VSV-G maskiert. Erst bei
Transfektion niedrigerer Plasmid-Dosen und submaximalen Titern wird ein Vorteil des pCI-
Gopt gegenüber den Plasmiden pCD-Gopt und pCI-G deutlich.

5 Zusammenfassung
Lentivirale Vektoren werden zur Erweiterung ihres Wirtszellspektrums pseudotypisiert, d. h. mit einem heterologen Glykoprotein in der Hüllmembran ausgestattet. Bei der Herstellung pseudotypisierter lentiviraler Vektoren mit Hilfe eines 3-Plasmid-Verpackungssystems wird das heterologe Hüllprotein mittels eines Plasmids exprimiert. Ein hohes Expressionsvermögen und damit eine hohe Pseudotypisierungseffizienz dieses Plasmids sind eine Voraussetzung für hochtitrige Vektorpräparate, die von großer Bedeutung in der Gentherapie sind.
Gegenstand der vorliegenden Arbeit ist die Pseudotypisierung lentiviraler Vektoren mit VSV-G. Ausgangspunkt war die Notwendigkeit einer 965 bp langen aus dem nicht translatier-ten Exon 1, dem ersten Intron und dem nicht translatiertern 5’-Ende des Exons 2 der Major-immediate-early-Gene des humanen Cytomegalievirus (HCMV-MIE-Gene) bestehenden Se-quenz zwischen HCMV-MIE-Promotor und der für VSV-G codierenden Sequenz in der Ex-pressionskassette des VSV-G-Expressionsplasmids für die Pseudotypisierung lentiviraler Vektoren.
Die vorliegende Arbeit belegt, dass das 827 bp umfassende Intron, dessen positiver Effekt auf die heterologe Genexpression in der Literatur beschrieben ist, eine vergleichsweise geringe Wirkung auf die Pseudotypisierungseffizienz der untersuchten VSV-G-Expressionsplasmide ausübt und daher verzichtbar ist. Erstmals beschrieben wird hier eine Sequenz, die als RNA vermutlich eine Stemloop-Struktur bildet und aus dem 3’-Ende des HCMV-MIE-Exons 1 und dem 5’-Ende des Exons 2 besteht, aber auch nur geringfügig zur Steigerung der VSV-G-Expression und der Pseudotypisierungseffizienz beiträgt. Den entscheidenden positiven Effekt liefert ein Sequenzabschnitt des Exons 1, der auch die in der Literatur beschriebene konser-vierte Box umfasst. Die HCMV-Sequenz, die für die Expression der für VSV-G codierenden Wildtyp-Sequenz in der 5’-nicht-translatierten-Region (5’-UTR) unverzichtbar ist, wurde auf die 106 bp 5’ der Stemloop-Struktur eingegrenzt. Darüber hinaus ist ein bisher nicht beschrie-benes optionales Exon im ersten HCMV-MIE-Exon aufgefallen.
Eine weitere Technik zur Verstärkung der Expression viraler Gene ist die Codonoptimierung. Die Expression einer codonoptimierten VSV-G-Sequenz zur Pseudotypisierung lentiviraler Vektoren führt in der vorliegenden Arbeit zur völligen Entbehrlichkeit von HCMV-Sequenzen in der 5’-UTR. Die Codonoptimierung entfaltet hier ihre Wirkung auf Prätransla-tionsebene, dies wird in der Literatur kontrovers diskutiert.
Eine absolute Steigerung der Titer VSV-G-pseudotypisierter lentiviraler Vektoren kann auf-grund der Cytotoxizität des VSV-G weder durch die Codonoptimierung der VSV-G-Sequenz noch durch die Kombination der Codonoptimierungs- mit der HCMV-MIE-5’-UTR-Strategie erreicht werden.

6 Literatur
1. Lee, T. W., Matthews, D. A. und Blair, G. E. 2005. Novel molecular approaches to cystic fibrosis gene therapy. Biochem. J. 387: S. 1-15
2. Chakkalakal, J. V., Thompson, J., Parks, R. J. und Jasmin, B. J. 2005. Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB J. 19: S. 880-91
3. Mian, A., McCormack, W. M., Jr., Mane, V., Kleppe, S., Ng, P., Finegold, M., O'Brien, W. E., Rodgers, J. R., Beaudet, A. L. und Lee, B. 2004. Long-term cor-rection of ornithine transcarbamylase deficiency by WPRE-mediated overexpression using a helper-dependent adenovirus. Mol. Ther. 10: S. 492-9
4. Cavazzana-Calvo, M., Lagresle, C., Hacein-Bey-Abina, S. und Fischer, A. 2005. Gene therapy for severe combined immunodeficiency. Annu. Rev. Med. 56: S. 585-602
5. Graw, J., Brackmann, H. H., Oldenburg, J., Schneppenheim, R., Spannagl, M. und Schwaab, R. 2005. Haemophilia A: from mutation analysis to new therapies. Nat. Rev. Genet. 6: S. 488-501
6. Strayer, D. S., Akkina, R., Bunnell, B. A., Dropulic, B., Planelles, V., Pomerantz, R. J., Rossi, J. J. und Zaia, J. A. 2005. Current status of gene therapy strategies to treat HIV/AIDS. Mol. Ther. 11: S. 823-42
7. Sutter, A. P. und Fechner, H. 2006. Gene therapy for gastric cancer: is it promising? World J. Gastroenterol. 12: S. 380-7
8. Raizada, M. K. und Der, S. S. 2006. Potential of gene therapy strategy for the treat-ment of hypertension. Hypertension 47: S. 6-9
9. Chen, Q., He, Y. und Yang, K. 2005. Gene therapy for Parkinson's disease: progress and challenges. Curr. Gene Ther. 5: S. 71-80
10. Gardlik, R., Palffy, R., Hodosy, J., Lukacs, J., Turna, J. und Celec, P. 2005. Vec-tors and delivery systems in gene therapy. Med. Sci. Monit. 11: S. RA110-RA121
11. Young, L. S., Searle, P. F., Onion, D. und Mautner, V. 2006. Viral gene therapy strategies: from basic science to clinical application. J. Pathol. 208: S. 299-318
12. Nisole, S. und Saib, A. 2004. Early steps of retrovirus replicative cycle. Retrovirolo-gy. 1: S. 9

6 Literatur 71
13. Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M., McCormack, M. P., Wulffraat, N., Leboulch, P., Lim, A., Osborne, C. S., Pawliuk, R., Morillon, E., Sorensen, R., Forster, A., Fraser, P., Cohen, J. I., de Saint, B. G., Alexander, I., Wintergerst, U., Frebourg, T., Aurias, A., Stoppa-Lyonnet, D., Romana, S., Rad-ford-Weiss, I., Gross, F., Valensi, F., Delabesse, E., Macintyre, E., Sigaux, F., Soulier, J., Leiva, L. E., Wissler, M., Prinz, C., Rabbitts, T. H., Le Deist, F., Fi-scher, A. und Cavazzana-Calvo, M. 2003. LMO2-associated clonal T cell prolifera-tion in two patients after gene therapy for SCID-X1. Science 302: S. 415-9
14. Ginn, S. L., Curtin, J. A., Kramer, B., Smyth, C. M., Wong, M., Kakakios, A., McCowage, G. B., Watson, D., Alexander, S. I., Latham, M., Cunningham, S. C., Zheng, M., Hobson, L., Rowe, P. B., Fischer, A., Cavazzana-Calvo, M., Hacein-Bey-Abina, S. und Alexander, I. E. 2005. Treatment of an infant with X-linked se-vere combined immunodeficiency (SCID-X1) by gene therapy in Australia. Med. J. Aust. 182: S. 458-63
15. Gallo, R. C. 2002. Historical essay. The early years of HIV/AIDS. Science 298: S. 1728-30
16. Hahn, B. H., Shaw, G. M., De Cock, K. M. und Sharp, P. M. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287: S. 607-14
17. Montagnier, L. 2002. Historical essay. A history of HIV discovery. Science 298: S. 1727-8
18. Miller, D. G., Adam, M. A. und Miller, A. D. 1990. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell Biol. 10: S. 4239-42
19. Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., Verma, I. M. und Trono, D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: S. 263-7
20. Gregory, L. G., Waddington, S. N., Holder, M. V., Mitrophanous, K. A., Buckley, S. M., Mosley, K. L., Bigger, B. W., Ellard, F. M., Walmsley, L. E., Lawrence, L., Al Allaf, F., Kingsman, S., Coutelle, C. und Themis, M. 2004. Highly efficient EIAV-mediated in utero gene transfer and expression in the major muscle groups af-fected by Duchenne muscular dystrophy. Gene Ther. 11: S. 1117-25
21. Azzouz, M., Kingsman, S. M. und Mazarakis, N. D. 2004. Lentiviral vectors for treating and modeling human CNS disorders. J. Gene Med. 6: S. 951-62
22. Yanez-Munoz, R. J., Balaggan, K. S., Macneil, A., Howe, S. J., Schmidt, M., Smith, A. J., Buch, P., Maclaren, R. E., Anderson, P. N., Barker, S. E., Duran, Y., Bartholomae, C., Von Kalle, C., Heckenlively, J. R., Kinnon, C., Ali, R. R. und Thrasher, A. J. 2006. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 12: S. 348-53

6 Literatur 72
23. Miyoshi, H., Blomer, U., Takahashi, M., Gage, F. H. und Verma, I. M. 1998. De-velopment of a self-inactivating lentivirus vector. J. Virol. 72: S. 8150-7
24. Schnell, T., Foley, P., Wirth, M., Munch, J. und Uberla, K. 2000. Development of a self-inactivating, minimal lentivirus vector based on simian immunodeficiency virus. Hum. Gene Ther. 11: S. 439-47
25. Pereira, L. A., Bentley, K., Peeters, A., Churchill, M. J. und Deacon, N. J. 2000. A compilation of cellular transcription factor interactions with the HIV-1 LTR promo-ter. Nucleic Acids Res. 28: S. 663-8
26. Wagner, R., Graf, M., Bieler, K., Wolf, H., Grunwald, T., Foley, P. und Uberla, K. 2000. Rev-independent expression of synthetic gag-pol genes of human immuno-deficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum. Gene Ther. 11: S. 2403-13
27. Scarlata, S. und Carter, C. 2003. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 1614: S. 62-72
28. Brigati, C., Giacca, M., Noonan, D. M. und Albini, A. 2003. HIV Tat, its TARgets and the control of viral gene expression. FEMS Microbiol. Lett. 220: S. 57-65
29. Hope, T. J. 1999. The ins and outs of HIV Rev. Arch. Biochem. Biophys. 365: S. 186-91
30. Doms, R. W., Earl, P. L., Chakrabarti, S. und Moss, B. 1990. Human immunode-ficiency virus types 1 and 2 and simian immunodeficiency virus env proteins possess a functionally conserved assembly domain. J. Virol. 64: S. 3537-40
31. Cronin, J., Zhang, X. Y. und Reiser, J. 2005. Altering the tropism of lentiviral vec-tors through pseudotyping. Curr. Gene Ther. 5: S. 387-98
32. Modrow, S., Falke, D. und Truyen, U. 2003. Molekulare Virologie. Spektrum A-kademischer Verlag, Heidelberg, Berlin:
33. Bilsel, P. A. und Nichol, S. T. 1990. Polymerase errors accumulating during natural evolution of the glycoprotein gene of vesicular stomatitis virus Indiana serotype isola-tes. J. Virol. 64: S. 4873-83
34. Kelley, J. M., Emerson, S. U. und Wagner, R. R. 1972. The glycoprotein of vesicu-lar stomatitis virus is the antigen that gives rise to and reacts with neutralizing antibo-dy. J. Virol. 10: S. 1231-5
35. Letchworth, G. J., Rodriguez, L. L. und Del cbarrera, J. 1999. Vesicular stomati-tis. Vet. J. 157: S. 239-60

6 Literatur 73
36. Rose, J. K., Welch, W. J., Sefton, B. M., Esch, F. S. und Ling, N. C. 1980. Vesicu-lar stomatitis virus glycoprotein is anchored in the viral membrane by a hydrophobic domain near the COOH terminus. Proc. Natl. Acad. Sci. U. S. A 77: S. 3884-8
37. Doms, R. W., Keller, D. S., Helenius, A. und Balch, W. E. 1987. Role for adenosi-ne triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J. Cell Biol. 105: S. 1957-69
38. Reading, C. L., Penhoet, E. E. und Ballou, C. E. 1978. Carbohydrate structure of vesicular stomatitis virus glycoprotein. J. Biol. Chem. 253: S. 5600-12
39. Schloemer, R. H. und Wagner, R. R. 1974. Sialoglycoprotein of vesicular stomatitis virus: role of the neuraminic acid in infection. J. Virol. 14: S. 270-81
40. Schloemer, R. H. und Wagner, R. R. 1975. Cellular adsorption function of the sia-loglycoprotein of vesicular stomatitis virus and its neuraminic acid. J. Virol. 15: S. 882-93
41. Irving, R. A., Toneguzzo, F., Rhee, S. H., Hofmann, T. und Ghosh, H. P. 1979. Synthesis and assembly of membrane glycoproteins: presence of leader peptide in nonglycosylated precursor of membrane glycoprotein of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A 76: S. 570-4
42. Nishimura, N., Plutner, H., Hahn, K. und Balch, W. E. 2002. The delta subunit of AP-3 is required for efficient transport of VSV-G from the trans-Golgi network to the cell surface. Proc. Natl. Acad. Sci. U. S. A 99: S. 6755-60
43. Schlegel, R., Tralka, T. S., Willingham, M. C. und Pastan, I. 1983. Inhibition of VSV binding and infectivity by phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell 32: S. 639-46
44. Coil, D. A. und Miller, A. D. 2004. Phosphatidylserine is not the cell surface recep-tor for vesicular stomatitis virus. J. Virol. 78: S. 10920-6
45. Sun, X., Yau, V. K., Briggs, B. J. und Whittaker, G. R. 2005. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 338: S. 53-60
46. Carneiro, F. A., Ferradosa, A. S. und Da Poian, A. T. 2001. Low pH-induced con-formational changes in vesicular stomatitis virus glycoprotein involve dramatic struc-ture reorganization. J. Biol. Chem. 276: S. 62-7
47. Carneiro, F. A., Bianconi, M. L., Weissmuller, G., Stauffer, F. und Da Poian, A. T. 2002. Membrane recognition by vesicular stomatitis virus involves enthalpy-driven protein-lipid interactions. J. Virol. 76: S. 3756-64

6 Literatur 74
48. Burns, J. C., Friedmann, T., Driever, W., Burrascano, M. und Yee, J. K. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentrati-on to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U. S. A 90: S. 8033-7
49. Kuate, S. 2001. Persönliche Mitteilung.
50. Kuate, S., Wagner, R. und Uberla, K. 2002. Development and characterization of a minimal inducible packaging cell line for simian immunodeficiency virus-based lenti-viral vectors. J. Gene Med. 4: S. 347-55
51. Alford, C. A. und Britt, W. J. 1990. Cytomegalovirus, S. 1981-2003. Fields, B. N. und Knipe, D. M. (Hrsg.) Virology. Raven Press, New York.
52. Boshart, M., Weber, F., Jahn, G., Dorsch-Hasler, K., Fleckenstein, B. und Schaffner, W. 1985. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41: S. 521-30
53. Thomsen, D. R., Stenberg, R. M., Goins, W. F. und Stinski, M. F. 1984. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A 81: S. 659-63
54. Ghazal, P., Lubon, H., Fleckenstein, B. und Hennighausen, L. 1987. Binding of transcription factors and creation of a large nucleoprotein complex on the human cy-tomegalovirus enhancer. Proc. Natl. Acad. Sci. U. S. A 84: S. 3658-62
55. Sinclair, J. H. 1987. The human cytomegalovirus immediate early gene promoter is a strong promoter in cultured Drosophila melanogaster cells. Nucleic Acids Res. 15: S. 2392
56. Sabbioni, S., Negrini, M., Rimessi, P., Manservigi, R. und Barbanti-Brodano, G. 1995. A BK virus episomal vector for constitutive high expression of exogenous cDNAs in human cells. Arch. Virol. 140: S. 335-9
57. Norderhaug, L., Olafsen, T., Michaelsen, T. E. und Sandlie, I. 1997. Versatile vectors for transient and stable expression of recombinant antibody molecules in mammalian cells. J. Immunol. Methods 204: S. 77-87
58. Gerolami, R., Uch, R., Jordier, F., Chapel, S., Bagnis, C., Brechot, C. und Man-noni, P. 2000. Gene transfer to hepatocellular carcinoma: transduction efficacy and transgene expression kinetics by using retroviral and lentiviral vectors. Cancer Gene Ther. 7: S. 1286-92
59. Hallauer, P. L. und Hastings, K. E. 2000. Human cytomegalovirus IE1 promo-ter/enhancer drives variable gene expression in all fiber types in transgenic mouse ske-letal muscle. BMC. Genet. 1: S. 1

6 Literatur 75
60. Castillo, J. P. und Kowalik, T. F. 2002. Human cytomegalovirus immediate early proteins and cell growth control. Gene 290: S. 19-34
61. Chapman, B. S., Thayer, R. M., Vincent, K. A. und Haigwood, N. L. 1991. Effect of intron A from human cytomegalovirus (Towne) immediate-early gene on heterolo-gous expression in mammalian cells. Nucleic Acids Res. 19: S. 3979-86
62. Ghazal, P. und Nelson, J. A. 1991. Enhancement of RNA polymerase II initiation complexes by a novel DNA control domain downstream from the cap site of the cy-tomegalovirus major immediate-early promoter. J. Virol. 65: S. 2299-307
63. Buchman, A. R. und Berg, P. 1988. Comparison of intron-dependent and intron-independent gene expression. Mol. Cell Biol. 8: S. 4395-405
64. Okkema, P. G., Harrison, S. W., Plunger, V., Aryana, A. und Fire, A. 1993. Se-quence requirements for myosin gene expression and regulation in Caenorhabditis e-legans. Genetics 135: S. 385-404
65. Chung, S. und Perry, R. P. 1989. Importance of introns for expression of mouse ribosomal protein gene rpL32. Mol. Cell Biol. 9: S. 2075-82
66. Callis, J., Fromm, M. und Walbot, V. 1987. Introns increase gene expression in cultured maize cells. Genes Dev. 1: S. 1183-200
67. Umen, J. G. und Guthrie, C. 1995. The second catalytic step of pre-mRNA splicing. RNA. 1: S. 869-85
68. Will, C. L. und Luhrmann, R. 2001. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13: S. 290-301
69. Tange, T. O., Nott, A. und Moore, M. J. 2004. The ever-increasing complexities of the exon junction complex. Curr. Opin. Cell Biol. 16: S. 279-84
70. Le Hir, H., Nott, A. und Moore, M. J. 2003. How introns influence and enhance eukaryotic gene expression. Trends Biochem. Sci. 28: S. 215-20
71. Rafiq, M., Suen, C. K., Choudhury, N., Joannou, C. L., White, K. N. und Evans, R. W. 1997. Expression of recombinant human ceruloplasmin--an absolute require-ment for splicing signals in the expression cassette. FEBS Lett. 407: S. 132-6
72. Le Hir, H., Gatfield, D., Izaurralde, E. und Moore, M. J. 2001. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 20: S. 4987-97
73. Lutz, C. S., Murthy, K. G., Schek, N., O'Connor, J. P., Manley, J. L. und Alwine, J. C. 1996. Interaction between the U1 snRNP-A protein and the 160-kD subunit of

6 Literatur 76
cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev. 10: S. 325-37
74. McCracken, S., Lambermon, M. und Blencowe, B. J. 2002. SRm160 splicing co-activator promotes transcript 3'-end cleavage. Mol. Cell Biol. 22: S. 148-60
75. Lu, S. und Cullen, B. R. 2003. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA. 9: S. 618-30
76. Nott, A., Le Hir, H. und Moore, M. J. 2004. Splicing enhances translation in mam-malian cells: an additional function of the exon junction complex. Genes Dev. 18: S. 210-22
77. Brooks, A. R., Blackhart, B. D., Haubold, K. und Levy-Wilson, B. 1991. Charac-terization of tissue-specific enhancer elements in the second intron of the human apo-lipoprotein B gene. J. Biol. Chem. 266: S. 7848-59
78. Rossi, P. und de Crombrugghe, B. 1987. Identification of a cell-specific transcripti-onal enhancer in the first intron of the mouse alpha 2 (type I) collagen gene. Proc. Natl. Acad. Sci. U. S. A 84: S. 5590-4
79. Fong, Y. W. und Zhou, Q. 2001. Stimulatory effect of splicing factors on transcrip-tional elongation. Nature 414: S. 929-33
80. Kwek, K. Y., Murphy, S., Furger, A., Thomas, B., O'Gorman, W., Kimura, H., Proudfoot, N. J. und Akoulitchev, A. 2002. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat. Struct. Biol. 9: S. 800-5
81. Yutzey, K. E., Kline, R. L. und Konieczny, S. F. 1989. An internal regulatory ele-ment controls troponin I gene expression. Mol. Cell Biol. 9: S. 1397-405
82. Gustafsson, C., Govindarajan, S. und Minshull, J. 2004. Codon bias and heterolo-gous protein expression. Trends Biotechnol. 22: S. 346-53
83. Gouy, M. und Gautier, C. 1982. Codon usage in bacteria: correlation with gene ex-pressivity. Nucleic Acids Res. 10: S. 7055-74
84. Wada, K., Aota, S., Tsuchiya, R., Ishibashi, F., Gojobori, T. und Ikemura, T. 1990. Codon usage tabulated from the GenBank genetic sequence data. Nucleic Acids Res. 18 Suppl: S. 2367-411
85. Crick, F. H., Barnett, L., Brenner, S. und Watts-Tobin, R. J. 1961. General nature of the genetic code for proteins. Nature 192: S. 1227-32
86. Ikemura, T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for

6 Literatur 77
a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 151: S. 389-409
87. Sharp, P. M. und Devine, K. M. 1989. Codon usage and gene expression level in Dictyostelium discoideum: highly expressed genes do 'prefer' optimal codons. Nucleic Acids Res. 17: S. 5029-39
88. Robinson, M., Lilley, R., Little, S., Emtage, J. S., Yarranton, G., Stephens, P., Millican, A., Eaton, M. und Humphreys, G. 1984. Codon usage can affect efficien-cy of translation of genes in Escherichia coli. Nucleic Acids Res. 12: S. 6663-71
89. Kofman, A., Graf, M., Deml, L., Wolf, H. und Wagner, R. 2003. Codon usage-mediated inhibition of HIV-1 gag expression in mammalian cells occurs independent-ly of translation. Tsitologiia 45: S. 94-100
90. Graf, M., Deml, L. und Wagner, R. 2004. Codon-optimized genes that enable inc-reased heterologous expression in mammalian cells and elicit efficient immune res-ponses in mice after vaccination of naked DNA. Methods Mol. Med. 94: S. 197-210
91. Fouchier, R. A., Meyer, B. E., Simon, J. H., Fischer, U. und Malim, M. H. 1997. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuc-lear import. EMBO J. 16: S. 4531-9
92. Soneoka, Y., Cannon, P. M., Ramsdale, E. E., Griffiths, J. C., Romano, G., Kingsman, S. M. und Kingsman, A. J. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 23: S. 628-33
93. Malim, M. H., Hauber, J., Fenrick, R. und Cullen, B. R. 1988. Immunodeficiency virus rev trans-activator modulates the expression of the viral regulatory genes. Nature 335: S. 181-3
94. Simm, M., Shahabuddin, M., Chao, W., Allan, J. S. und Volsky, D. J. 1995. Aber-rant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J. Virol. 69: S. 4582-6
95. Hofacker, I. L. 2003. Vienna RNA secondary structure server. Nucleic Acids Res. 31: S. 3429-31
96. Sambrook, J. und Russell, D. W. 2001. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor: S. -2100
97. Lee, M. T., Coburn, G. A., McClure, M. O. und Cullen, B. R. 2003. Inhibition of human immunodeficiency virus type 1 replication in primary macrophages by using Tat- or CCR5-specific small interfering RNAs expressed from a lentivirus vector. J. Virol. 77: S. 11964-72

6 Literatur 78
98. Richardson, R. M., Tokunaga, K., Marjoram, R., Sata, T. und Snyderman, R. 2003. Interleukin-8-mediated heterologous receptor internalization provides resistan-ce to HIV-1 infectivity. Role of signal strength and receptor desensitization. J. Biol. Chem. 278: S. 15867-73
99. Yeo, G., Hoon, S., Venkatesh, B. und Burge, C. B. 2004. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl. Acad. Sci. U. S. A 101: S. 15700-5
100. McCaskill, J. S. 1990. The equilibrium partition function and base pair binding pro-babilities for RNA secondary structure. Biopolymers 29: S. 1105-19
101. Zuker, M. und Stiegler, P. 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9: S. 133-48
102. Nelson, K. K. und Green, M. R. 1990. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc. Natl. Acad. Sci. U. S. A 87: S. 6253-7
103. Norton, P. A. 1994. Alternative pre-mRNA splicing: factors involved in splice site selection. J. Cell Sci. 107 ( Pt 1): S. 1-7
104. Hentze, M. W. und Kuhn, L. C. 1996. Molecular control of vertebrate iron metabo-lism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. U. S. A 93: S. 8175-82
105. Rogers, J. T., Leiter, L. M., McPhee, J., Cahill, C. M., Zhan, S. S., Potter, H. und Nilsson, L. N. 1999. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5'-untranslated region sequences. J. Biol. Chem. 274: S. 6421-31
106. Thomson, A. M., Cahill, C. M., Cho, H. H., Kassachau, K. D., Epis, M. R., Bridges, K. R., Leedman, P. J. und Rogers, J. T. 2005. The acute box cis-element in human heavy ferritin mRNA 5'-untranslated region is a unique translation enhancer that binds poly(C)-binding proteins. J. Biol. Chem. 280: S. 30032-45
107. Vivinus, S., Baulande, S., van Zanten, M., Campbell, F., Topley, P., Ellis, J. H., Dessen, P. und Coste, H. 2001. An element within the 5' untranslated region of hu-man Hsp70 mRNA which acts as a general enhancer of mRNA translation. Eur. J. Bi-ochem. 268: S. 1908-17
108. Mokrejs, M., Vopalensky, V., Kolenaty, O., Masek, T., Feketova, Z., Sekyrova, P., Skaloudova, B., Kriz, V. und Pospisek, M. 2006. IRESite: the database of expe-rimentally verified IRES structures (www.iresite.org). Nucleic Acids Res. 34: S. D125-D130
109. Jansen, R. P. 2001. mRNA localization: message on the move. Nat. Rev. Mol. Cell Biol. 2: S. 247-56

6 Literatur 79
110. Rose, J. K. und Gallione, C. J. 1981. Nucleotide sequences of the mRNA's encoding the vesicular stomatitis virus G and M proteins determined from cDNA clones contai-ning the complete coding regions. J. Virol. 39: S. 519-28
111. Gallione, C. J. und Rose, J. K. 1985. A single amino acid substitution in a hydrophobic domain causes temperature-sensitive cell-surface transport of a mutant viral glycoprotein. J. Virol. 54: S. 374-82
112. Doms, R. W., Ruusala, A., Machamer, C., Helenius, J., Helenius, A. und Rose, J. K. 1988. Differential effects of mutations in three domains on folding, quaternary structure, and intracellular transport of vesicular stomatitis virus G protein. J. Cell Bi-ol. 107: S. 89-99

Dissertationsbezogene Publikation
Kuate, S., Stefanou, D., Hoffmann, D., Wildner, O. und Überla, K. 2004. Production of lentiviral vectors by transient expression of minimal packaging genes from recombi-nant adenoviruses. J. Gene Med. 6: S. 1197-205.

Danksagung
Ich danke Herrn Prof. Dr. Klaus Überla sehr herzlich für die Aufnahme in seine Arbeitsgrup-pe, für die Möglichkeit, an verschiedenen Projekten mitwirken und so vielfältige Methoden erlernen zu dürfen, und für die unzähligen Diskussionen, aus denen ich immer neue Motivati-on schöpfte und die mir halfen, das Ziel nicht aus den Augen zu verlieren.
Herrn Prof. Dr. Bernd-Joachim Benecke und Herrn Prof. Dr. William S. Sheldrick danke ich für die Bereitschaft, als Koreferent bzw. dritter Prüfer der Promotionskommission beizutreten.
Dr. Susann Lucke teilte drei Jahre mit mir die Laborbank, vielen Dank für die tolle Team-Arbeit!
Dr. Thomas Grunwald möchte ich meinen Dank für seine ständige Hilfsbereitschaft und für die Einführung in wesentliche Methoden aussprechen.
Dr. Dennis Hoffmann danke ich für die Hilfe beim Sequenzieren für die vielen wertvollen Ratschläge und interessanten Diskussionen.
Dr. Alexander Stang gilt mein Dank für viele fachliche Diskussionen und lustige Hörspieldia-loge.
Das Ende meiner Labortätigkeit überschnitt sich mit dem Anfang meiner Schwangerschaft, in dieser Zeit war ich auf die selbstlose technische Assistenz meiner Kollegen angewiesen. Be-sonders häufig (aber nicht ausschließlich) halfen mir Dr. Hella Monse, Matthias Tenbusch und Dr. Susann Lucke. Herzlichen Dank!
Ich möchte die Gelegenheit nutzen, jener kleinen Gruppe zu danken, ohne die kein Labor wirklich funktioniert. Ich spreche von den Kollegen, die nicht nur ihren eigenen Kram aufge-räumt haben, sondern darüber hinaus tatkräftig dem Bestreben des Universums nach Unord-nung entgegengewirkt sowie organisatorische Verantwortung übernommen haben. Dafür dan-ke ich Bettina Tippler, Dr. Dennis Hoffmann, Dr. Susann Lucke, Dr. Hella Monse und Dr. Thomas Grunwald.
Für das Engagement bei dem für das Arbeitsklima so wichtigen „Drumherum“ (Weihnachts-feier, Betriebsausflug usw..) danke ich allen damaligen Mitarbeitern der Abteilung für Mole-kulare und Medizinische Virologie. Hierzu gehören auch: Andreia Fernandes, Annika Müh-lenkamp, Bärbel Rölleke, Ph.D. Charles Esimone, Claudius Grossmann, Dr. Godwin Nchin-da, Heike Seidenstücker, Klaus Sure, Manuela Günther, Nicola Ternette, Regina Bütermann, Rosemarie Bohr, Sabine Brandt, Dr. Seraphin Kuate, Dr. Uli Heindel, Uli Schumacher, Ulla Vogel, Vera Siegmund und Yvonne Klingen.
Ich bedanke mich bei Susi, Vera, Dennis und Alexios für das Korrekturlesen meiner Arbeit.
Meinen Eltern danke ich für die Ermöglichung meiner Ausbildung und vor allem für die lie-bevolle Betreuung meines Sohnes Elias.
Ich danke meinem Mann Alexios für seine Unterstützung und meinem Sohn Elias für die vie-len fröhlichen Stunden.

Lebenslauf
Name: Daniela Stefanou
Geburtsdatum: 13. April 1975
Geburtsort: Rheinberg
Familienstand: verheiratet
Kinder: ein Sohn (21 Monate)
1981 – 1985 Städt. Gemeinschaftsgrundschule St. Hubert
1985 – 1994 Städt. Gymnasium Thomaeum, Kempen Abitur im Mai 1994
WS 1994 – SS 1999 Studium der Biochemie an der Universität Leipzig Diplom im September 1999
März 1999 – September 1999 Diplomarbeit: „Studien zur Profilin-Polyprolin-Interaktion mittels Fluoreszenz-Korrelations-Spektroskopie“ am Institut für Biochemie (Prof. Dr. U. Hahn), Fakultät für Biowissenschaften, Pharmazie und Psychologie, Universität Leipzig
November 1999 – März 2000 Wissenschaftliche Hilfskraft am Institut für Biochemie (Prof. Dr. U. Hahn), Fakultät für Biowissenschaften, Pharmazie und Psychologie, Universität Leipzig
April 2000 – Dezember 2000 Wissenschaftliche Hilfskraft am Institut für Virologie (Prof. Dr. U. G. Liebert, Dr. J. Hofmann), Medizinische Fakultät, Universität Leipzig
Seit April 2001 Promotionsstudiengang Chemie, Ruhr-Universität Bochum
April 2001 – März 2004 Wissenschaftliche Mitarbeiterin in der Abteilung für Molekulare und Medizinische Virologie (Prof. Dr. K. Überla), Medizinische Fakultät, Ruhr-Universität Bochum, mit Möglichkeit zur Promotion
Seit April 2004 Schreiben der Dissertation
31.05.2004 – 15.09.2004 Mutterschutz





























