Embed Size (px)
Citation preview

JOURNALTUMORZENTRUM ERFURT
INHALT
Seite 3�� Neuroendokrine Tumoren
der Lunge
Seite 8�� Schlafende Stammzellen und
ihre Erweckung
Seite 9�� LymphoplasmozytischesLymphom (einschließlichMorbus Waldenström, WM)
Seite 13�� Die Bedeutung des Ki-67-Antigens in der Tumor-pathologie
Seite 16�� Hochleistungsmedizin und Menschlichkeit –ein Widerspruch?
Seite 19�� Wünsche eines Krebspatienten
Seite 20�� Auffangen – Informieren –
BegleitenFrauenselbsthilfe nach Krebs –längst auch für Männer
Seite 21�� Wichtige Fortschritte bei
Diagnose und Behandlungvon Brustkrebs in denletzten Jahren erreicht
Seite 23�� Gemeinsames
Veranstaltungsverzeichnis
Seite 24�� Angebote des
Tumorzentrum Erfurt e.V.
02/2009
ISSN 1868-291X
MMeehhrr zzuumm TThheemmaa KKrreebbssssttaammmmzzeell lleenn lleesseenn aauuff SSeeiittee 88
Oberstes Ziel der Tumorzentren istdie ständige Verbesserung der Be-treuung aller Tumorpatienten imEinzugsgebiet. Neben der Vermitt-lung aktuellen Wissens kommt da-bei der Förderung von Interdiszipli-narität, Kooperation und Kommu-nikation aller in die Patientenver-sorgung eingebundenen Bereicheeine herausragende Bedeutung zu. Die jährlich stattfindenden Onkolo-gischen Konferenzen sind dieserAufgabe in besonderer Weise ver-pflichtet. Seit 22 Jahren organisiertdas Erfurter Tumorzentrum insbe-sondere für die Region Mittel- undWestthüringen im Herbst einemehrtägige Tagung. Bis auf wenigeAusnahmen fand sie auf dem Hain-stein in Eisenach statt, in diesemJahr am 20. und 21. November. Die
Themenauswahl berücksichtigt so-wohl Innovationen in der Onkolo-gie als auch Probleme der prakti-schen Umsetzung neuer Diagnose-und Therapieverfahren und ist glei-chermaßen an niedergelasseneund in der Klinik tätige Ärzte ge-richtet. Die 22. Onkologische Konferenzwidmete sich den vier Themen-schwerpunkten – Prognosefaktoren in der Onko-
logie,– Diagnostische und therapeuti-
sche Standards bei Intrakraniel-len Tumoren und bei Hodentu-moren,
– Innovative Behandlungskon-zepte bei weit fortgeschritte-nen Tumorerkrankungen,
– Krebsstammzellen.
22. Onkologische Konferenz des Tumorzentrum Erfurt
Prof. Dr. Andreas Tumpp vom Deutschen Krebsforschungszentrum Heidelberg ist einer der renommiertesten Forscher auf dem Gebiet der Krebsstammzellen.

Dr. Hubert GöbelGeschäftsführer
Wir wünschen allen Mitgliedern, Partnern, Freunden und Förderern des Tumorzentrum Erfurt e.V.
ein frohes Weihnachtsfest und ein gesundes neues Jahr.
Wir danken Ihnen herzlich für Ihr Engagement und hoffen auf eine
weitere gute Zusammenarbeit.
Prof. Dr. Berthold UlshöferVorsitzender des Vorstandes
Prof. Dr. Hartwig KosmehlVorsitzender
des Wissenschaftlichen Beirates

�� SSee iittee 33 ��JJOOUURRNNAALL 0022//22000099
�� Neuroendokrine Tumoren der Lunge
Sayegh Y., R. BonnetKlinik für Pneumologie, Zentralklinik Bad Berka
EinführungBronchiale Karzinoide gehören zu den seltenen pulmona-len Neubildungen. Sie zeichnen sich durch neuroendokri-ne Differenzierung der Zellen und ein relativ indolentesklinisches Verhalten aus.Sie wurden früher zu den bronchialen Adenomen gezählt.Wegen ihres Potentials zu metastasieren, werden sie je-doch heutzutage als maligne Neubildungen betrachtet.Wie Karzinoide an anderen Stellen des Körpers leiten sichauch die bronchialen Karzinoide von diffus im Körper ver-teilten neuroendokrinen Zellen ab, welche in der Embryo-genese aus dem Neuralrohr in den Körper eingewandertsind. Diese erstmals 1938 von Feyrter in der Mukosa des Tra-cheobronchialsystems beschriebenen „hellen Zellen“ wur-den 1949 von Froelich auch im Lungenparenchym gefun-den. Ungefähr 20 Jahre später wurden von Pearse dieseZellen entsprechend ihrer histochemischen Eigenschaftendem APUD-Zellsystem (amin-precursor-uptake-and-de-carboxylation) zugeordnet, da die Zellen die Fähigkeit be-sitzen, Vorstufen biogener Amine aufzunehmen und zudecarboxylieren. Die daraus entstehenden Tumoren wur-den als Apudome bezeichnet. Oberndorfer fand 1907 beisechs Patienten ebenfalls multiple ileale Tumoren, die imUnterschied zu Karzinomen kein infiltratives Wachstumund keine Metastasierung aufwiesen. Daher prägte er denBegriff „Karzinoid“ (Karzinom-ähnlich).Erspamer und Asero wiesen 5-Hydroxytryptamin (Seroto-nin) in Mucosaextrakten des Gastrointestinaltraktes nach,und ein Jahr später, 1953, konnte Lambeck chromatogra-phisch Serotonin bei einem Appendixkarzinoid darstellen. Anhand histogenetischer Gesichtspunkte und je nach ih-rer Lokalisation unterteilten Williams und Sander die Kar-zinoide in • Vorderdarm- (Lunge, Magen, Duodenum, oberes Jeju-
num und Pankreas), • Mitteldarm- (hinteres Jejunum, Ileum, Appendix, Zö-
kum) und • Enddarmkarzinoide (Kolon und Rektum). Lungenkarzinoide stehen nach den Karzinoiden des Ga-strointestinaltrakts an zweiter Stelle in der Häufigkeit(Tab.1). Andere typische Lokalisationen von Karzinoidensind der Thymus und die Ovarien.
Arrigoni et al. hatten bereits 1972 die aggressiver wach-senden und früher metastasierenden Karzinoide als ,,aty-pisch“ von den „typischen Karzinoiden“ abgegrenzt. Neuroendokrine Tumoren der Lunge treten meist spora-disch auf, jedoch in bis zu 15% im Rahmen eines MEN-I-Syndroms.
EpidemiologieBronchiale Karzinoide machen etwa 1-2% aller Lungentu-moren im Erwachsenenalter aus. Bei Kindern zählen sie zuden häufigsten malignen Lungenerkrankungen. Sie prä-sentieren sich typischerweise erst im späten Jugendalter.
Dabei beträgt das Verhältnis von typischen zu atypischenKarzinoiden etwa 4:1.Die globale Inzidenzrate liegt zwischen 0,2 bis 2 Fälle auf100.000 Einwohner pro Jahr. In den meisten Untersu-chungsserien sind Frauen häufiger betroffen als Männer.Die ansteigende Inzidenz in den letzten Jahrzehnten istweniger auf eine Zunahme der Häufigkeit zurückzufüh-ren, sondern auf die bessere bildgebende Diagnostik, wel-che heutzutage auch viele asymptomatische Tumoren de-tektiert. Das Durchschnittsalter, in dem die Diagnose beitypischen Karzinoiden gestellt wird, liegt bei 45 Jahren.Atypische Karzinoide werden im Durchschnitt erst 10 Jah-re später manifest.
Tabelle 1 Lungenkarzinoide
1-2 % aller Lungentumoren25 % aller Karzinoide80-90 % typische Karzinoide0-20 % atypische Karzinoide70-80 % proximal, 20-30% peripher61 % liegen rechtsseitig, v. a. im Mittellappen
RisikofaktorenEine Assoziation von Karzinoiden und Nikotinkonsum istnicht bewiesen. In vielen epidemiologischen Studien wa-ren 1/3 bis 2/3 aller Patienten mit bronchialen KarzinoidenExraucher. Einige dieser Untersuchungen ergeben auch ei-ne höhere Prävalenz von atypischen Karzinoiden bei Rau-chern. Eine eindeutige Kausalität ließ sich durch dieseStudien jedoch nicht belegen. Weitere Umweltfaktorenoder Karzinogene als Risikofaktor bei Karzinoiden sindnicht bekannt.
Genetische PrädispositionObwohl selten, gibt es familiär ein gehäuftes Auftretenvon Karzinoiden. Patienten mit dem autosomal dominan-ten Syndrom der multiplen endokrinen Neoplasie Typ I(MEN I) weisen eine hohe Inzidenz von malignen endokri-nen Neubildungen und Vorderdarm-Karzinoiden (d.h. vonThymus, Lunge, Magen, Dünndarm) auf. Jedoch wurdenauch familiäre pulmonale Karzinoide beschrieben, dienicht mit dem MEN-I-Syndrom assoziiert sind.
KlassifikationHistologisch sind bronchiale Karzinoide Teil eines Spek-trums von endokrinen Tumoren der Lunge, welche sicheindrucksvoll durch ein unterschiedliches biologischesVerhalten auszeichnen. An dem einen Ende des Spek-trums finden sich die typischen Karzinoide. Diese sindhoch differenziert, langsam wachsend und selten in extra-thorakale Strukturen metastasierend. Am anderen Endedes Spektrums sind schlecht differenzierte neuroendokri-ne Tumoren anzutreffen. Typifizierend dafür steht daskleinzellige Bronchialkarzinom (SLCL) mit hochaggressi-vem Verhalten und damit schnellem Wachstum und frü-her Metastasierung. Das biologische Verhalten von atypi-schen Karzinoiden liegt intermediär zwischen beiden.Ungeachtet ihres heterogenen klinischen Verhaltens tei-len Lungentumoren mit neuroendokriner Differenzierungeinige morphologische und biochemische Charakteristika:1. Die Fähigkeit, Neuropeptide zu synthetisieren.2. Das Vorhandensein von submikroskopisch zytoplas-
matischen dichten (neuroendokrinen) Granula, wel-che nur elektronenmikroskopisch nachgewiesen wer-den können.

�� SSeeiittee 44 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
WHO-KlassifikationNeuroendokrine Tumoren der Lunge waren in der Vergan-genheit Gegenstand einer beträchtlichen Kontroverse.Hieraus resultierten multiple, teils unübersichtliche undverwirrende Klassifikationen. Die letzte, allgemein akzeptierte WHO-Klassifikation istvon 2004. In ihr werden neuroendokrine Tumoren derLunge in ein klinisch-pathologisches Spektrum eingeteilt,welches von der diffusen idiopathischen neuroendokrinenZellhyperplasie (DIPNECH) bis hin zum niedrig differen-zierten kleinzelligen Bronchialkarzinom und zu den groß-zelligen neuroendokrinen Tumoren reicht (Tab. 2).
Tabelle 2 WHO-Kriterien (2004) für die Diagnose der neu-roendokrinen Tumoren
TUMORTYP KRITERIEN
Typische Karzinoidmorphologie mit <2 Mitosen/2mm2
Karzinoidtumoren (10 HPFs), keine Nekrose und >0,5cm
Atypische Karzinoidmorphologie mit 2-10 Mito-Karzinoidtumoren sen/2mm2 (10 HPFs) oder Nekrose (oft
punktuell angeordnet)
Großzellige neuroendo- Neuroendokrine Morphologie (organoide krine Karzinome Strukturen mit trabekulären, rosettenförmigen
oder palisadenartigen Strukturen)
Hohe Mitoserate >10/2mm2 (10 HPFs), im Median 70/2mm2
Nekrose (oft große Regionen)
Zytologische Eigenschaften von NSCLC: große Zellen, kleines Verhältnis von Zellkern zum Zy-toplasma, einige Tumoren weisen feines nu-kleäres Chromatin und fehlende Nukleoli auf, zählen jedoch aufgrund ihrer Zellgröße und ih-res reichlichen Zytoplasmas zu den NSCLC
Positive immunhistochemische Markierung für ein oder mehrere NE-Marker (andere als NSE) und/oder neuroendokrine Granulae im Elek-tronenemissionsmikroskop
Kleinzellige neuroendo- Kleine Zellen (normalerweise kleiner als 3 Lym-krine Karzinome phozyten
Geringes Zytoplasma
Nuklei: feines granuläres Chromatin, keine oder matte Nukleoli
Hohe Mitoserate: >11 Mitosen/2mm2
(10 HPFs), im Median 80/2mm2 (10 HPFs) oft Nekrosen, sehr häufig große Zonen
( HPF - high-power field; NSCLC - non-small cell lung carcinoma)
TNM- KlassifikationDas Staging pulmonaler Karzinoide ist denen von Lungen-tumoren gleichgestellt. Typische Karzinoide gehören mei-stens zu den Stadium-I-Tumoren, während mehr als dieHälfte der atypischen Karzinoide zu den Tumoren der Sta-dien II und III gehören.
Klinische MerkmaleDie Mehrzahl der Tumoren (80%) entsteht in den proxima-len Atemwegen. Die Beschwerden sind meistens durchdie Stenosierung der Atemwege infolge der Tumormassebedingt. Patienten leiden demzufolge unter Husten, Gie-
men, Luftnot oder wiederkehrenden Infektionen durchRetentionspneumonien im gleichen Lungensegment oderLungenlappen. Zudem können aufgrund der typischenHypervaskularisation der Tumoren Blutungen mit Hämop-tysen entstehen. Gelegentlich treten Brustschmerzen auf.Die Diagnose wird meist sehr spät gestellt, häufig werdenPatienten längere Zeit symptomatisch therapiert oder beirezidivierenden Infekten mit verschiedenen Antibiotika.Im Gegensatz hierzu sind die Patienten mit peripherenLungenkarzinoiden (20%) asymptomatisch. Diese Tumo-ren werden häufig als Zufallsbefund bei Röntgen-Thorax-aufnahmen gefunden.
Peptid Produktion und Paraneoplastische SyndromeIm Gegensatz zu den Karzinoiden mit Ursprung aus demprimitiven Mitteldarm haben Karzinoide des primitivenVorderdarms, zu denen auch die Lungenkarzinoide gehö-ren, im Allgemeinen einen niedrigeren Gehalt an Seroto-nin und verursachen daher üblicherweise kein Karzinoid-syndrom. Der Grund hierfür ist, dass Vorderdarm-Karzi-noide häufig einen Mangel an aromatischen Aminode-carboxylasen haben und Serotonin und seine Metabolitenicht selbst herstellen können.Obwohl sie eine große Vielfalt anderer Peptide und Hor-mone innerhalb der Zelle synthetisieren (Gastrin releasingPeptid, 5-Hydroxytryptophan und Chromogranine), se-zernieren bronchiale Karzinoide nur gelegentlich bioakti-ve Amine. Das Resultat hieraus ist, dass der Hormonspie-gel im Plasma bzw. im Urin sehr niedrig ist und die Karzi-noide so kaum entdeckt werden können. Nur ein geringerAnteil der Patienten entwickelt klinisch ein paraneoplasti-sches Syndrom bedingt durch die Peptidsekretion.Karzinoidsyndrome sind sehr selten mit einer Tumorgrö-ße von mehr als 5cm vergesellschaftet. Jedoch tritt dasKarzinoidsyndrom in mehr als 80 % der Fälle bei Patien-ten mit Lungenkarzinoid und Lebermetastasen auf.
Karzinoide KriseBiopsien oder Manipulationen an einem aktiv sezernieren-den bronchialen Karzinoid können in sehr seltenen Fällenzu karzinoiden Krisen durch massive systemische Aus-schüttung von bioaktiven Mediatoren führen. Die Patien-ten können akut einen Flush, Durchfall, ein Bronchospas-mus, Azidose, Hypo- oder Hypertensionen, Tachykardieoder einen Myokardinfarkt entwickeln.
Cushing SyndromDurch die ektope Produktion von ACTH (Adrenocortico-tropes Hormon) können sowohl typische als auch atypi-sche Karzinoide ein Cushing-Syndrom verursachen. Dabeisind bronchiale Karzinoide die häufigste Ursache für ek-tope ACTH-Produktion, andere maligne Ursachen stellendas SCLC und CUP dar.In 1-2% der Fälle entwickeln Patienten mit einem bronchia-len Karzinoid ein Cushing-Syndrom. Das Cushing-Syndromführt die Patienten meist erst zum Arzt. Der Beginn ist oftakut und es liegt häufig eine Hypokaliämie vor. Die Mehr-zahl der ACTH-produzierenden bronchialen Karzinoide istkleiner als 2cm, was das Stellen der richtigen Diagnose er-schwert. Einige Studien gehen davon aus, dass ACTH-pro-duzierende Karzinoide aggressiver sind als hormonstilleKarzinoide. Andere jedoch gehen von einem ähnlichenOutcome aus, wenn bei den Patienten eine Resektion mitmediastinaler Lymphadenektomie durchgeführt wird.
�� SSee iittee 55 ��JJOOUURRNNAALL 0022//22000099
AkromegalieEine Akromegalie infolge von ektoper Produktion vonGHRH (Growth-hormone-releasing hormone) durch einbronchiales Karzinoid ist extrem selten, wobei die Lungen-karzinoide die häufigste Ursache für eine ektopischeGHRH-Sekretion sind.
Diagnostik1. LaborDie Bestimmung des Serotonin-Metaboliten 5-Hydroxyin-dolessigsäure (5-HIES) als Screening-Nachweismethodegehört zur diagnostischen Aufarbeitung, obwohl diesesneuroendokrine Tumorprodukt nur in seltenen Fällen im24-h-Urin erhöht ist.Ein Merkmal der NET der Lunge als sogenannte Vorder-darm-Karzinoide ist ein Defizit des Enzyms Dopa-Decarb-oxylase, weswegen diese Tumoren vermehrt 5-Hydroxy-tryptophan und nicht 5-Hydroxytryptamin (Serotonin) indas Gefäßsystem sezernieren. Dies erklärt die relativ ho-hen Urinspiegel an 5-Hydroxytryptophan und nur seltennachweisbare 5-HIES im 24-h-Urin dieser Patienten. DerNachweis einer erhöhten 5-HIES-Konzentration im 24-h-Urin ist spezifisch und sensitiv für NET des Mitteldarms,selten jedoch für NET des Vorder- und Mitteldarms. DieBestimmung von Serotonin im Blut spielt aufgrund zu ho-her interindividueller tageszeitlicher Schwankungen eineabsolut untergeordnete Rolle in der klinischen Diagnostik.Jedoch zeigt die Serotonin-Bestimmung aus den Throm-bozyten eine höhere Sensitivität insbesondere bei Tumo-ren, die durch eine niedrige Serotoninproduktion charak-terisiert sind (NET des Vorderdarms und Residualtumo-ren), als die 5-HIES-Bestimmung aus dem Urin. Die Sero-toninkonzentration der Thrombozyten wird nicht durcheine serotoninreiche Diät beeinflusst. Die Bestimmung von Chromogranin A (CgA) im Serum alsBreitspektrummarker für neuroendokrine Tumoren stelltein relativ sensitives Verfahren dar. Als Bestandteil derMembran der Sekretgranula von neuroendokrinen Zellenwird CgA im Rahmen der Hypersekretion mit Peptid- undPolypeptidhormonen kosezerniert. Erhöhte Chromogra-nin-A-Blutspiegel finden sich bei fast allen metastasiertenNET und sind in Abhängigkeit einzelner Hypersekretions-syndrome z. B. mit erhöhten Werten von 5-HIES im 24-h-Urin wie beim Karzinoidsyndrom assoziiert. Neuronenspezifische Enolase (NSE) kann bei neuroendo-krinen Tumoren pathologisch erhöht sein. Gemeinsam weisen diese eine höhere Sensitivität auf alseinzeln bestimmt. In der Routinediagnostik eines Lungen-karzinoids ist bei Abwesenheit einer entsprechenden Sym-ptomatik die Bestimmung der Serotonin-Metabolitennicht notwendig.
2. Röntgen-Thorax75% der Patienten mit einem bronchialen Karzinoid ha-ben ein suspektes Röntgen-Thorax. Die meisten Raumfor-derungen sind rund bis oval (2-5 cm im Durchschnitt) undhilär bzw. perihilär gelegen. Eine Kavernenbildung ist sel-ten. Pleurale Beteiligung ist unüblich, kann aber mit einerpostobstruktiven Pneumonie vergesellschaftet sein.
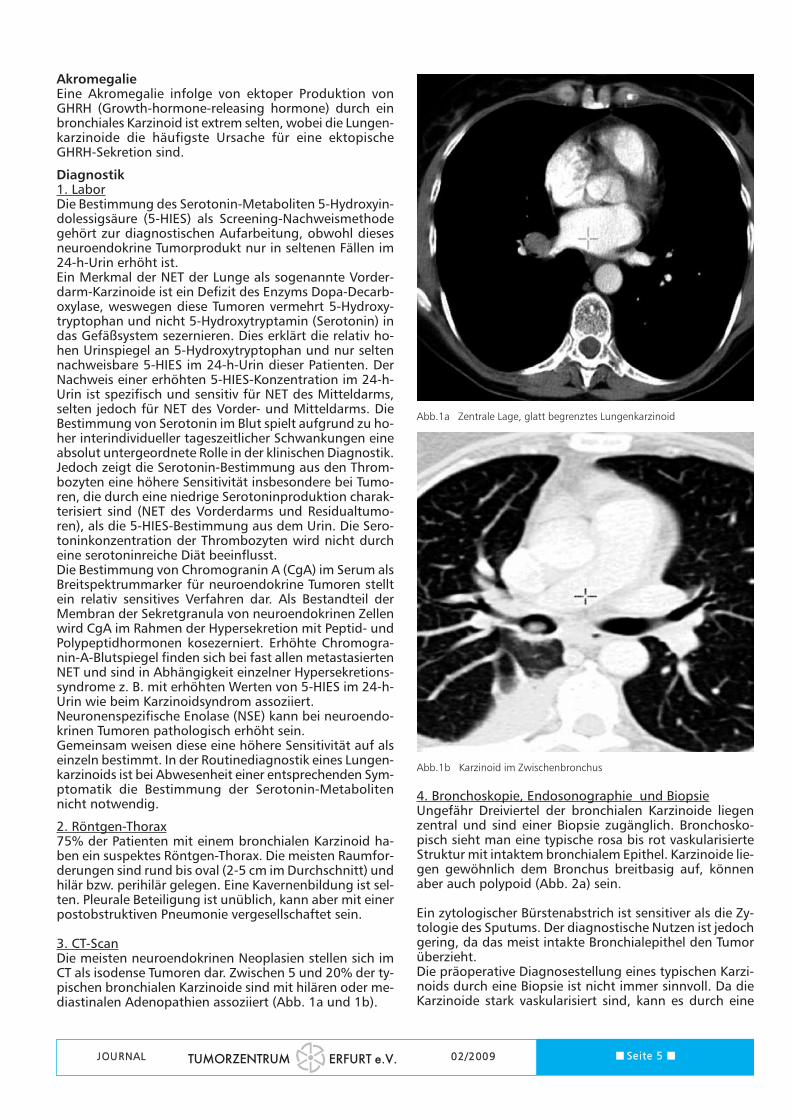
3. CT-ScanDie meisten neuroendokrinen Neoplasien stellen sich imCT als isodense Tumoren dar. Zwischen 5 und 20% der ty-pischen bronchialen Karzinoide sind mit hilären oder me-diastinalen Adenopathien assoziiert (Abb. 1a und 1b).
Abb.1a Zentrale Lage, glatt begrenztes Lungenkarzinoid
Abb.1b Karzinoid im Zwischenbronchus
4. Bronchoskopie, Endosonographie und BiopsieUngefähr Dreiviertel der bronchialen Karzinoide liegenzentral und sind einer Biopsie zugänglich. Bronchosko-pisch sieht man eine typische rosa bis rot vaskularisierteStruktur mit intaktem bronchialem Epithel. Karzinoide lie-gen gewöhnlich dem Bronchus breitbasig auf, könnenaber auch polypoid (Abb. 2a) sein.
Ein zytologischer Bürstenabstrich ist sensitiver als die Zy-tologie des Sputums. Der diagnostische Nutzen ist jedochgering, da das meist intakte Bronchialepithel den Tumorüberzieht.Die präoperative Diagnosestellung eines typischen Karzi-noids durch eine Biopsie ist nicht immer sinnvoll. Da dieKarzinoide stark vaskularisiert sind, kann es durch eine
�� SSeeiittee 66 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
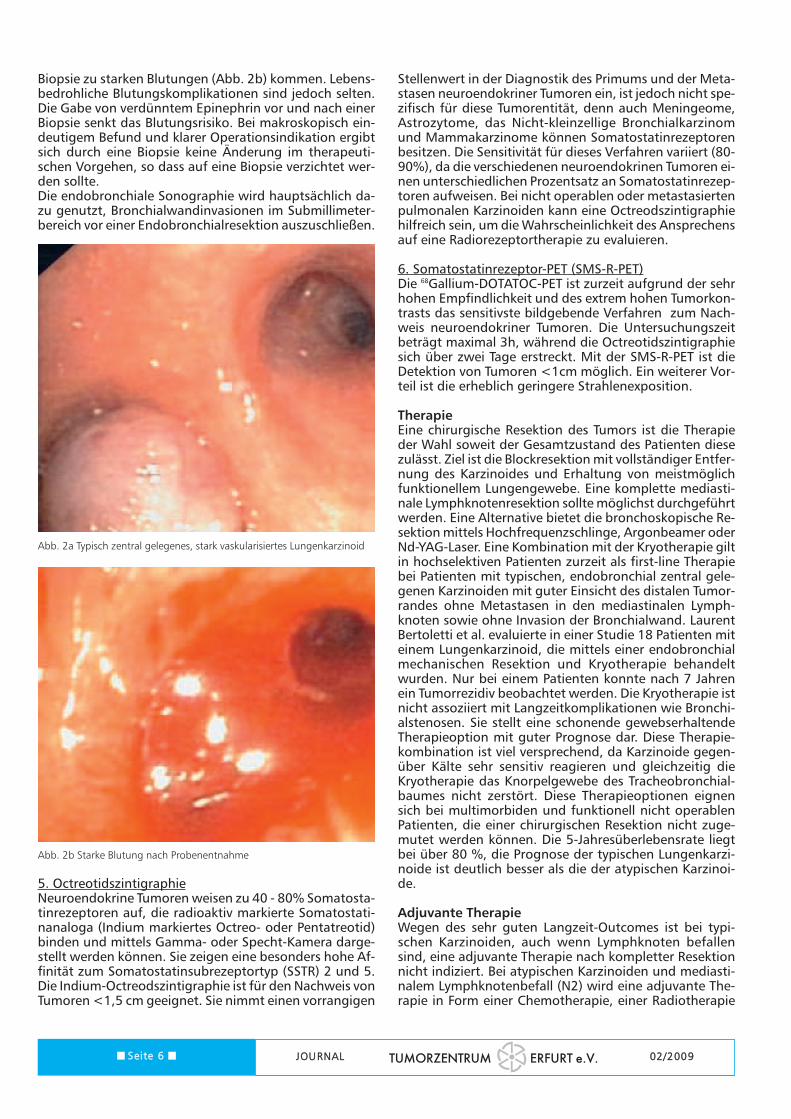
Biopsie zu starken Blutungen (Abb. 2b) kommen. Lebens-bedrohliche Blutungskomplikationen sind jedoch selten.Die Gabe von verdünntem Epinephrin vor und nach einerBiopsie senkt das Blutungsrisiko. Bei makroskopisch ein-deutigem Befund und klarer Operationsindikation ergibtsich durch eine Biopsie keine Änderung im therapeuti-schen Vorgehen, so dass auf eine Biopsie verzichtet wer-den sollte.Die endobronchiale Sonographie wird hauptsächlich da-zu genutzt, Bronchialwandinvasionen im Submillimeter-bereich vor einer Endobronchialresektion auszuschließen.
Abb. 2a Typisch zentral gelegenes, stark vaskularisiertes Lungenkarzinoid
Abb. 2b Starke Blutung nach Probenentnahme
5. OctreotidszintigraphieNeuroendokrine Tumoren weisen zu 40 - 80% Somatosta-tinrezeptoren auf, die radioaktiv markierte Somatostati-nanaloga (Indium markiertes Octreo- oder Pentatreotid)binden und mittels Gamma- oder Specht-Kamera darge-stellt werden können. Sie zeigen eine besonders hohe Af-finität zum Somatostatinsubrezeptortyp (SSTR) 2 und 5.Die Indium-Octreodszintigraphie ist für den Nachweis vonTumoren <1,5 cm geeignet. Sie nimmt einen vorrangigen
Stellenwert in der Diagnostik des Primums und der Meta-stasen neuroendokriner Tumoren ein, ist jedoch nicht spe-zifisch für diese Tumorentität, denn auch Meningeome,Astrozytome, das Nicht-kleinzellige Bronchialkarzinomund Mammakarzinome können Somatostatinrezeptorenbesitzen. Die Sensitivität für dieses Verfahren variiert (80-90%), da die verschiedenen neuroendokrinen Tumoren ei-nen unterschiedlichen Prozentsatz an Somatostatinrezep-toren aufweisen. Bei nicht operablen oder metastasiertenpulmonalen Karzinoiden kann eine Octreodszintigraphiehilfreich sein, um die Wahrscheinlichkeit des Ansprechensauf eine Radiorezeptortherapie zu evaluieren.
6. Somatostatinrezeptor-PET (SMS-R-PET)Die 68Gallium-DOTATOC-PET ist zurzeit aufgrund der sehrhohen Empfindlichkeit und des extrem hohen Tumorkon-trasts das sensitivste bildgebende Verfahren zum Nach-weis neuroendokriner Tumoren. Die Untersuchungszeitbeträgt maximal 3h, während die Octreotidszintigraphiesich über zwei Tage erstreckt. Mit der SMS-R-PET ist dieDetektion von Tumoren <1cm möglich. Ein weiterer Vor-teil ist die erheblich geringere Strahlenexposition.
TherapieEine chirurgische Resektion des Tumors ist die Therapieder Wahl soweit der Gesamtzustand des Patienten diesezulässt. Ziel ist die Blockresektion mit vollständiger Entfer-nung des Karzinoides und Erhaltung von meistmöglichfunktionellem Lungengewebe. Eine komplette mediasti-nale Lymphknotenresektion sollte möglichst durchgeführtwerden. Eine Alternative bietet die bronchoskopische Re-sektion mittels Hochfrequenzschlinge, Argonbeamer oderNd-YAG-Laser. Eine Kombination mit der Kryotherapie giltin hochselektiven Patienten zurzeit als first-line Therapiebei Patienten mit typischen, endobronchial zentral gele-genen Karzinoiden mit guter Einsicht des distalen Tumor-randes ohne Metastasen in den mediastinalen Lymph-knoten sowie ohne Invasion der Bronchialwand. LaurentBertoletti et al. evaluierte in einer Studie 18 Patienten miteinem Lungenkarzinoid, die mittels einer endobronchialmechanischen Resektion und Kryotherapie behandeltwurden. Nur bei einem Patienten konnte nach 7 Jahrenein Tumorrezidiv beobachtet werden. Die Kryotherapie istnicht assoziiert mit Langzeitkomplikationen wie Bronchi-alstenosen. Sie stellt eine schonende gewebserhaltendeTherapieoption mit guter Prognose dar. Diese Therapie-kombination ist viel versprechend, da Karzinoide gegen-über Kälte sehr sensitiv reagieren und gleichzeitig die Kryotherapie das Knorpelgewebe des Tracheobronchial-baumes nicht zerstört. Diese Therapieoptionen eignensich bei multimorbiden und funktionell nicht operablenPatienten, die einer chirurgischen Resektion nicht zuge-mutet werden können. Die 5-Jahresüberlebensrate liegtbei über 80 %, die Prognose der typischen Lungenkarzi-noide ist deutlich besser als die der atypischen Karzinoi-de.
Adjuvante TherapieWegen des sehr guten Langzeit-Outcomes ist bei typi-schen Karzinoiden, auch wenn Lymphknoten befallensind, eine adjuvante Therapie nach kompletter Resektionnicht indiziert. Bei atypischen Karzinoiden und mediasti-nalem Lymphknotenbefall (N2) wird eine adjuvante The-rapie in Form einer Chemotherapie, einer Radiotherapie

�� SSee iittee 77 ��JJOOUURRNNAALL 0022//22000099
oder einer Kombination aus beiden Therapieoptionen an-geraten. Es gibt jedoch keine prospektiven Studien, dieden Erfolg adjuvanter Therapieformen für Patienten mitbronchialen Karzinoiden (typisch oder atypisch) doku-mentieren, zudem gibt es nur sehr wenige Erfahrungeneinzelner Institutionen.Obwohl sich Karzinoide als relativ radioresistent erwiesenhaben, ist die Radiotherapie bei lokal fortgeschrittenennicht resezierbaren Primärtumoren als palliative Maßnah-me sinnvoll. Zudem wird empfohlen, dass dieses Patien-tenkollektiv eine Kombination aus Radio- und Chemothe-rapie bekommt, in ähnlicher Form wie bei Patienten mitSCLC. Jedoch fallen die Ansprechraten bei Patienten mitSCLC im Vergleich besser aus.
ChemotherapieDer Anteil der Patienten mit Metastasen liegt zwischen 5und 70%. Metastasen können sehr spät auftreten, auchnoch Jahrzehnte nach Diagnosestellung. Aufgrund des seltenen Auftretens dieses Tumors gibtkaum prospektive Studien und nur wenig Erfahrung mitder Chemotherapie. Patienten mit metastasierendenBronchialkarzinoiden wurden oft mit einem Chemothera-pieprotokoll, ähnlich dem des SCLC, behandelt (z.B. Cis-platin in Kombination mit Etoposid oder Cisplatin in Kom-bination mit Paclitaxel). Weitere zum Einsatz kommende Substanzen sind Interfe-ron-Alpha, 5-Fluorouracil, Streptozocin, Doxorubicin, In-hibitoren von Angiogenese (z. B. Bevacizumab) und Tyro-sinkinaseinhibitoren (z.B. Sunitinib). In der Regel sind dieAnsprechraten jedoch gering:• In einer Studie aus Uppsala, Schweden, wurden acht
Patienten mit Cisplatin-Etoposid behandelt. Zwei Pa-tienten mit einem typischen Lungenkarzinoid sprachen über einen Zeitraum von sechs bzw. acht Monaten auf die Therapie an. Bei einem Patienten mit atypischen Karzinoid zeigte sich ein stabiler Krankheitsverlauf über sieben Monate. Fünf Patienten entwickelten eine Progression nach 3 bis 4 Monaten. Patienten mit ei-nem Karzinoidsyndrom sprachen seltener auf diese Kombination an als Patienten ohne Karzinoidsyndrom. Es gab keine Korrelation zwischen dem Ansprechen auf die Behandlung und der Ki-67-Expression.
• Von 7 Patienten, die mit einer Kombination aus Strep-tozotocin und 5-Fluorouracil behandelt wurden, wies lediglich ein Patient einen stabilen Krankheitsverlauf über einen Zeitraum von acht Monaten auf. Die restli-chen sechs Patienten zeigten eine Progression.
• Zwei Patienten mit einer Kombinationstherapie 5-Fluo-rouracil/Interferon-Alpha zeigten einen progressiven Verlauf nach drei bzw. sechs Monaten.
• Bei zwei mit einer Kombination aus Streptozotocin-Do-xorubicin behandelten Patienten konnte ein radiolo-gisch stabiler Krankheitsverlauf über acht bzw. zehn Monate erreicht werden.
• Von zwei Patienten, die mit Paclitaxel und Doxorubicin behandelt wurden, erreichte einer für acht Monate ei-nen stabilen Krankheitsverlauf, der andere für elf Mo-nate.
• Von zwei Patienten mit Paclitaxel-Monotherapie ent-wickelte einer nach einem Monat eine Progression. Der andere zeigte radiologisch eine stabile Tumorgrö-ße, biochemisch jedoch eine Progression nach sieben Monaten.
Interessanterweise sprechen Patienten mit neuroendokri-nen Tumoren anderer Organe (z.B. Pankreas) besser an alsLungenkarzinoide. Sogar die Kombination aus Cisplatinund Etoposid, die sehr effektiv bei SCLC oder aggressivenatypischen Karzinoiden anderer Organe ist, hat eine limi-tierte Effektivität bei typischen und atypischen Karzinoi-den der Lunge.
BiotherapieIn der Studie von Granberg et al. wurde Interferon-Alphaallein oder in Kombination mit anderen Biotherapeutikaevaluiert. 27 Patienten erhielten Interferon-Alpha alleinoder in Kombination mit Octreotid. Bei 21 dieser Patien-ten kam es sowohl radiologisch als auch biochemisch zueinem progressiven Krankheitsverlauf. Vier Patienten mittypischem Karzinoid zeigten einen stabilen Krankheits-verlauf, im Median über 15 Monate. Es gab keinen Un-terschied im Ansprechen bei Patienten mit oder ohne kar-zinoidem Syndrom. Es konnte auch keine Korrelation zwi-schen dem Ansprechen und jeglichen immunhistochemi-schen Analysen der Ki-67-Expression gefunden werden.Ebenso gab es keine Unterschiede hinsichtlich des Über-lebens bei Patienten, die eine Therapie mit Interferon-Al-pha allein oder in Kombination mit Octreotid bekommenhatten.Die Wirksamkeit von Interferon-Alpha und Octreotid beiLungenkarzinoiden ist somit begrenzt. Nur sehr wenigePatienten weisen einen stabilen Krankheitsverlauf auf. DerStellenwert dieser Therapien liegt überwiegend beim klas-sischen Karzinoidsyndrom in einer Symptombesserung.
RadiorezeptortherapieIm Falle eines nicht-operablen, metastasierten Somatosta-tinrezeptor-positiven Karzinoids führt eine rezeptorver-mittelte Radiotherapie mit an Octreotid gekoppeltem Yttrium90 (DOTATOC) oder Indium111 (In-Pentetreotid) beimehr als der Hälfte der Patienten zur Symptombesserung.
PrognoseTypische bronchiale Karzinoide haben eine gute Progno-se; nur 1-2% der Karzinoide rezidivieren. Die 5-Jahres-überlebensrate liegt zwischen 75 und 100%:Eine unvollständige Resektion ist mit einer signifikantschlechteren Prognose assoziiert. Atypische Karzinoidehaben eine große Tendenz zu metastasieren. Die 5-Jahres-überlebensrate variiert zwischen 30 und 65%.Die Prognose verschlechtert sich, wenn Lymphknoten be-fallen sind. Eine Prognoseverschlechterung bei Befall vonLymphknoten bei typischen Karzinoiden ist allerdingsnicht sicher beschrieben.
Korrespondenzadresse:
Sayegh Y.Adjunct Professor of Medicine Dr. med. R. Bonnet M.D. Zentralklinik Bad Berka Klinik für PneumologieRobert-Koch-Allee 999437 Bad BerkaTelefon 0364585-1501e-Mail [email protected]

�� SSeeiittee 88 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
�� Schlafende Stammzellen und ihreErweckung
Dr. Ernst-Dieter JaraschBioRegion Rhein-Neckar-Dreieck e.V., Heidelberg
Mit einer Serie neuer Veröffentlichungen in hochkarätigenFachzeitschriften hat Prof. Dr. Andreas Trumpp, der 2008von Lausanne nach Heidelberg gewechselt ist, seinen Rufals einer der weltweit führenden Stammzellforscher be-kräftigt. Zusätzlich zu seiner Professur und Leitung derAbteilung für Zellbiologie am Deutschen Krebsfor-schungszentrum (DKFZ) übernahm er die wissenschaftli-che Leitung des neu gegründeten „Heidelberg Institutefor Stem Cell Technology and Experimental Medicine“ (HI-STEM gGmbH), um in enger Kooperation mit dem DKFZ,dem Universitätsklinikum und der Universität Heidelbergdie Erforschung von Stammzellen voranzutreiben und ih-re Ergebnisse therapeutisch nutzbar zu machen. DerSchwerpunkt liegt dabei auf der Identifizierung und Be-kämpfung von Krebsstammzellen.
Andreas Trumpp, Jahrgang 1964, hatte an der UniversitätFreiburg i. Br. Biologie studiert und seine Doktorarbeit aufdem Gebiet der molekularen Entwicklungsbiologie beiRolf Zeller und Thomas Graf am EMBL in Heidelbergdurchgeführt. 1994 ging er für sechs Jahre an die Univer-sität von Kalifornien in San Francisco, wo er in den Labo-ratorien von J. Michael Bishop (Nobelpreisträger 1989)und Gail R. Martin Mausmodelle für die Erforschung des„fibroblast growth factor 8 (Fgf8) und des Myc-Onkogensentwickelte. Im April 2000 wurde er Leiter des Genetik-und Stammzelllabors am Schweizerischen Institut für Ex-perimentelle Krebsforschung (ISREC) in Epalinges bei Lau-sanne und wurde dort 2005 zum Professor für Molekula-re Onkologie und Stammzellbiologie der École Polytech-nique Féderale de Lausanne (EPFL; Eidgenössische Techni-sche Hochschule Lausanne) berufen. 2008 folgte er einemRuf nach Heidelberg auf die W3-Professur und Leitung derAbteilung Stammzellen und Krebs am Deutschen Krebs-forschungszentrum.
Tiefschlaf als Schutz von StammzellenAn der EPFL hatten Trumpp und seine Mitarbeiter, darun-ter Dr. Anne Wilson, am Ludwig Institute for Cancer Re-search in Lausanne im Knochenmark der Maus eine klei-ne Population von Stammzellen entdeckt, die in Nischendes Knochenmarks versteckt nahezu lebenslang in einerArt Winterschlaf verharren und nur in einer Notfallsitua-tion aktiviert werden. Während es bei Blutstammzellender Maus normalerweise etwa jeden Monat zu einer Zell-teilung kommt, teilen sich die „schlafenden“ Stammzel-len, die etwa 15 Prozent der gesamten Stammzellpopula-tion ausmachen, während des gesamten Lebens der Mausnur fünfmal. Auf den Menschen übertragen entsprächedas einer Zellteilung alle 18 Jahre. Werden diese Zellen je-doch geweckt – etwa bei einer Verletzung des Knochen-marks oder durch Ausschüttung von Botenstoffen – zei-gen sie das höchste Regenerationspotenzial aller Stamm-zellen. Sie können das gesamte blutbildende System beiMäusen, deren Knochenmark durch Bestrahlung zerstörtworden war, wieder erneuern. Wenn das Knochenmark
repariert und der gesunde Zustand wieder hergestellt ist,fallen diese Zellen erneut in den Tiefschlaf, während diegroße Mehrzahl der „aktiven“ Stammzellen für die Auf-rechterhaltung des physiologischen Gleichgewichts derBlutzellen verantwortlich ist.
Krebsstammzelle im Knochenmark einer Maus (Bildquelle: DKFZ)
Der Schlafzustand kann als ein Schutzmechanismus derZellen aufgefasst werden, da die meisten Chemothera-peutika und anderen Zellgifte nur auf sich teilende Zelleneinwirken. Auch Mutationen im Erbgut erfolgen in der Re-gel nur während der Zellteilung. Trumpp und seine Mitar-beiter haben maßgeblich zur Charakterisierung der Kno-chenmarks-Nische bei Blutstammzellen beigetragen undentdeckt, dass das Krebsprotein c-Myc den Eintritt undAustritt dieser hämatopoetischen Stammzellen aus ihrerNische kontrolliert. In einer gerade veröffentlichten Publi-kation (Marieke Essers et al., Nature, February 11, 2009)zeigten die Forscher, dass der Immunbotenstoff Interfe-ron alpha die schlafenden Blutstammzellen im Knochen-mark aktiviert und dadurch für die Wirkung von Zellgiftenwie zum Beispiel 5-Fluorouracil angreifbar macht.
Für Andreas Trumpp stellen diese Ergebnisse auch einenSchlüssel zum Verständnis der Krebsstammzellen dar, de-ren Rolle bei der Initiierung und dem Wachstum von Tu-moren heute im Brennpunkt der onkologischen For-schung steht. Am DKFZ wird unter seinem Vorstandsvor-sitzenden und Wissenschaftlichen Vorstand Prof. Dr. Ot-mar D. Wiestler der Erforschung der Krebsstammzellenund darauf aufbauend der Entwicklung neuer Strategiender Krebstherapie besonders Rechnung getragen. In ei-nem umfassenden Review haben Trumpp und Wiestlerden gegenwärtigen Forschungsstand und die neuen The-rapiekonzepte zusammengefasst (Nature Clinical PracticeOncology, April 22, 2008).
Das HI-STEM InstituteAm „Heidelberg Institute for Stem Cell Technology and Ex-perimental Medicine“ (HI-STEM gGmbH) mit Trumpp alswissenschaftlichem Leiter wird die anwendungsorientier-te Forschung an den Krebsstammzellen gebündelt. Ge-gründet wurde das HI-STEM von der privaten Dietmar-Hopp-Stiftung als Mehrheitsgesellschafter sowie demDKFZ, an dem das Institut auch angesiedelt sein wird. Esstellt das Kernstück des Stammzell-Netzwerks dar, das imRahmen des Spitzencluster-Wettbewerbs des Bundesfor-schungsministeriums als einer der Schwerpunkte zur Wei-

�� SSee iittee 99 ��JJOOUURRNNAALL 0022//22000099
terentwicklung des BioRN-Clusters „Zellbasierte und Mo-lekulare Biologie in der Metropolregion Rhein-Neckar"identifiziert worden ist. Mit dem Gewinn des Wettbe-werbs 2008 erhalten Trumpp und seine neu am HI-STEMangesiedelten Arbeitsgruppen zusätzliche Förderungdurch das BMBF. Durch ihre Verbindung von Grundlagen-forschung, translationaler Medizin und wirtschaftlichemPotenzial auf einem hochaktuellen, zukunftsträchtigenGebiet im Kampf gegen Krebs haben die Forschungspro-jekte am HI-STEM zentrale Bedeutung für die Entwicklungdes BioRN Clusters.
Metastasen induzierende KrebsstammzellenDie zuerst in Leukämien entdeckten Krebsstammzellen(„cancer stem cells“, CSC) haben die Theorie der Krebsent-stehung und der Metastasierung in den letzten Jahren re-volutioniert. Nicht jede vom Primärtumor abgelöste (dis-seminierte) Krebszelle hat die Fähigkeit, Fernmetastasenzu bilden. Wahrscheinlich kann das nur eine winzig klei-ne Subpopulation, die sogenannten „Metastasen induzie-renden Krebsstammzellen“ (Metastasis inducing cancerstem cells, MICs). Diese Zellen befinden sich, wie andereStammzellen auch, normalerweise in einem Ruhezustandund teilen sich nur sehr selten; wie diese haben sie auchein unbegrenztes Selbsterneuerungspotenzial und die Ka-pazität, verschiedenartige Zelltypen zu erzeugen. Beson-ders wichtig ist, dass Krebsstammzellen gegenüber derBehandlung mit gebräuchlichen Chemotherapeutika resi-stent sind. Sie verstecken sich in einer speziellen als„Stammzell-Nische“ bezeichneten Mikroumgebung, inder sie im Ruhezustand verharren und für Therapien nurschwer erreichbar sind. Das erklärt, warum nach einerChemotherapie der Krebs zunächst oft verschwindet,denn die Tumorzellen, welche die Masse des Primärtu-mors ausmachen, sind nicht oder kaum resistent. Oftmalsviele Jahre später kommt es dann zu einem von den che-motherapieresistenten MICs gebildeten Rezidiv. Eine di-rekt auf Krebsstammzellen zielende Therapie könnte bes-sere Heilung bringen. Die Forschungsgruppe von Trumpphat nun ein spezielles Xenograft-Modell in der Maus ent-wickelt, mit dem es möglich ist, MICs aus menschlichemBlut oder Knochenmark zu identifizieren. Das Modell er-möglicht die gezielte Suche nach Wirkstoffen, die sichspezifisch gegen Zielmoleküle auf den MICs richten. Da-mit würden sich ganz neue Behandlungsmöglichkeitenfür Patienten mit metastasierenden Tumoren ergeben, diefür die überwiegende Zahl von Krebstodesfällen verant-wortlich sind.
© BIOPRO Baden-Würtemberg GmbH (Erstveröffentlichung auf www.bio-pro.de)
Korrespondenzadresse:
Dr. Ernst-Dieter JaraschBioRegion Rhein-Neckar-Dreieck e.V.Im Neuenheimer Feld 58269120 HeidelbergTelefon 06221-64922-0e-Mail [email protected]
�� Lymphoplasmozytisches Lymphom(einschließlich Morbus Waldenström,WM)
Michael Herold4. Medizinische Klinik, HELIOS Klinikum Erfurt
Definition / Pathologie / GenetikDas lymphoplasmozytische Lymphom (LPL) ist eine reifzel-lige B-Zell-Neoplasie vom kleinzelligen Typ nach der WHO-Klassifikation. Von einem Morbus Waldenström bzw. ei-ner Makroglobulinämie Waldenström (WM) spricht manbei Nachweis eines monoklonalen IgM jeglicher Konzen-tration und einer Knochenmarkinfiltration, was bei ca.30 % der LPL der Fall ist. Nach der aktuellen WHO-Klassi-fikation von 2008 handelt es sich dabei um eine eigen-ständige Krankheitsentität [1]. Die Erstbeschreibung dieses Erkrankungssyndroms durchJ. Waldenström datiert aus dem Jahre 1944. Er beschriebzwei Patienten mit Anämie, Lymphknotenschwellungen,Hepatosplenomegalie und einer Hyperproteinämie mitZeichen der Hyperviskosität, im Knochenmark der Patien-ten fanden sich plasmazellähnliche Zellen [2]. Morpholo-gisch ist das LPL gekennzeichnet durch eine Proliferationkleiner B-Zellen, plasmazytoider Zellen (mit breitem Zyto-plasmasaum und einem kompakten lymphozyten-ähnli-chen Kern) und teilweise auch Plasmazellen. Das Befalls-muster im Lymphknoten ist meist interfollikulär, das fürdie B-CLL typische pseudofollikuläre Wachstumsbild fehlt.Locker eingestreut finden sich Immunoblasten und reak-tiv vermehrt Mastzellen und Epitheloidzellen. PAS-positi-ve Einschlüsse in den Zellkernen und im Zytoplasma (Dut-scher- und Russell-Körperchen) sind häufig nachweisbarund ein Indiz für das Vorliegen eines Lymphoms [3]. Be-fallen sind in der Regel das Knochenmark (~90 % bei LPLund per definitionem immer bei WM), Lymphknoten unddie Milz. Leukämische Verläufe kommen vor, die Zellzah-len sind jedoch meist deutlich niedriger als bei der B-CLL.Immunphänotypisch ist das LPL eindeutig von anderenkleinzelligen NHL, insbesondere von der B-CLL abzugren-zen. Typischerweise werden die Pan-B-Zell-Marker CD 19,CD 20, CD 22 und CD 79a exprimiert, dagegen fehlen CD 5, CD 10 und CD 23. Die Ursprungszelle des LPL ist ei-ne Postkeimzentrums-Gedächtnis-B-Zelle mit plasmazel-lulärer Differenzierung. Die klonalen Zellen weisen IgH-und IgL-Umlagerungen sowie somatische Mutationen inden IgH-Regionen auf. Etwa 50 % der LPL-Patienten zei-gen eine t(9;14) und ein Rearrangement des PAX5-Gens,welches eine wichtige Rolle in der B-Zell-Entwicklungspielt.Nach der WHO-Klassifikation wird eine klare Abgrenzungzum CD 5+ lymphoplasmozytoiden Immunozytom derKiel-Klassifikation vorgenommen; alle diese Fälle werdenheute der B-CLL zugerechnet. Klinisch handelt es sich je-doch bei diesen Patienten um eine prognostisch ungün-stigere CLL-Subgruppe; man spricht von der Untergruppeder lymphoplasmozytoid differenzierten B-CLL. Transfor-mationen von LPL in diffus großzellige B-Zell-Lymphomewerden beobachtet.
Epidemiologie und klinisches ErscheinungsbildDas lymphoplasmozytische Lymphom ist eine seltene En-

�� SSeeiittee 1100 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
tität der kleinzelligen Non-Hodgkin-Lymphome. Es machtnur etwas mehr als 1 % der NHL aus [4], d.h. es ist mitca.1-2 Neuerkrankungen pro Jahr/1 Mio. Einwohner zurechnen. Betroffen sind Patienten in höherem Lebensalter,das mediane Erkrankungsalter liegt zwischen 60 und 65Jahren und Männer erkranken häufiger als Frauen. DerVerlauf der Erkrankung ist meist indolent und das media-ne Überleben liegt 5 bis 6 Jahren [4]. Beim M. Walden-ström wird immer wieder über eine familiäre Häufung be-richtet. Bei bis zu 20% der Betroffenen weist ein Ver-wandter ersten Grades eine B-Zell-Neoplasie auf. [5]Das klinische Bild ist beim LPL gekennzeichnet durch einegeneralisierte Lymphadenopathie, häufig besteht eine He-patosplenomegalie. Während eine diffuse oder noduläreInfiltration des Knochenmarks fast immer vorliegt, sindleukämische Verläufe eher selten. Die Laborparameter zei-gen eine maximale Senkungsbeschleunigung, meist eineAnämie und im Falle des M. Waldenström ein monoklo-nales IgM; bei hohen Paraproteinkonzentrationen (ab>30 g/l) kann es zum Auftreten eines Hyperviskositätssyn-droms kommen. Zeichen dafür sind Schleimhautblutun-gen, neurologische Symptome wie Kopfschmerz, Schwin-del, Hör- und Sehstörungen, Somnolenz bis zum Komaund kardiovaskulären Symptomen (Herzinsuffizienz). Hierkann in der Akutphase der Einsatz der Plasmapherese er-forderlich werden und noch vor Einleitung einer Chemo-therapie rasch Abhilfe schaffen. In seltenen Fällen sindKryoglobulinämien oder auch IgM-Kälteagglutinine mitentsprechender klinischer Symptomatik zu finden, AL-Amyloidosen können auftreten. Auch über Assoziationenmit einer Hepatitis C-Infektion wurde berichtet [6].Zur Diagnosesicherung sollte, wenn irgend möglich, eineLymphknotenexstirpation erfolgen. Nur wenn keine peri-pheren Lymphome vorhanden sind, sollte eine bioptischeMaterialgewinnung (CT- oder Sonografie-gestützt) erfol-gen. Gelegentlich muss man sich auch mit einer alleinigenKnochenmarkhistologie begnügen. Die Stadieneinteilungdes lymphoplasmozytischen Lymphoms erfolgt nach derAnn-Arbor-Klassifikation. Diese Einteilung spielt jedochbei dieser Entität eine eher untergeordnete Rolle, da, wiebereits erwähnt, mehr als 90 % der betroffenen Patien-ten bei Diagnosestellung einen Knochenmarkbefall habenund damit dem Stadium IV zuzuordnen sind. Die klinischeStadiendiagnostik beim LPL umfasst neben Anamneseund klinischer Untersuchung auch den Einsatz der bildge-benden Diagnostik: zu fordern sind eine Thorax-Röntgen-aufnahme in 2 Ebenen, eine Sonografie von Hals undOberbauch, sowie CT von Thorax und Abdomen. Eine wei-terführende Diagnostik (Endoskopien, MRT und nuklear-medizinische Untersuchungsmethoden inkl. PET) solltevom klinischen Bild abhängig gemacht werden. Als Unter-suchung zur pathologischen Stadiendiagnostik ist dieKnochenmarkbiopsie obligat.
Therapie
StrahlentherapieEinen kurativen Therapieanspruch beim LPL gibt es nur beiden wenigen Patienten mit einem lokalisierten Erkran-kungsstadium (Ann-Arbor-Stadium I und II); dabei han-delt es sich jedoch nur um ca. 5 % der Patienten. Hierkommt die Bestrahlung zum Einsatz, die in Anlehnung andas Vorgehen bei den follikulären Lymphomen durchge-führt wird: bei Patienten bis 75 Jahre als extended field
oder auch total nodale Bestrahlung mit einer Dosis im Re-ferenzpunkt von 40 Gy, bei Patienten über 75 Jahre als ei-ne involved field Bestrahlung mit der gleichen Dosis. Au-ßerdem hat die Strahlentherapie einen Stellenwert als ad-ditive Maßnahme zur Chemotherapie, z.B. bei großen,verdrängenden oder Beschwerden verursachendenLymphknotenpaketen, bei Restbefunden nach Chemothe-rapie oder auch als symptomatische Maßnahme beikrankheitsdominanter Milz.An dieser Stelle ist auch die Splenektomie zu erwähnen;die Milzgröße kann im Einzelfall extreme Ausmaße anneh-men und ist oft mit einer Zytopenie vergesellschaftet. Hierkann eine Splenektomie dem Patienten Erleichterung ver-schaffen und oft bessert sich auch die Zytopenie.
Chemotherapie/ImmunchemotherapieBeim generalisierten LPL (95 %) besteht bis heute kein ku-rativer Therapieanspruch. Aus diesem Grund wird die Ein-leitung einer systemischen Therapie vom Beschwerdebilddes Patienten, von den hämatologischen Parametern unddem Progressionverhalten des Lymphoms abhängig ge-macht. Ähnlich wie bei den generalisierten follikulärenLymphomen oder auch der B-CLL gelten als Indikation füreine Chemotherapie/Immunchemotherapie:• das Vorhandensein von B-Symptomen (Nachtschweiß, Fieber und/oder Gewichtsverlust) bzw. ein ausgepräg-tes Fatigue-Syndrom,
• eine hämatopoetische Insuffizienz mit Anämie und/oder Thrombozytopenie (Hb < 6,25 mmol/l; Thrombozyten < 100 Gpt/l),
• ein rasches Wachstum von Lymphknoten und/ oder Milz und
• das Auftreten eines Hyperviskositäts-Syndroms.Nicht immer ist es leicht dem betroffenen Patienten einsolches Vorgehen plausibel zu machen, es gibt jedochauch beim LPL, wie bei den anderen indolenten NHL, kei-ne Studiendaten, die zeigen konnten, dass eine frühzeiti-ge Einleitung einer Chemotherapie einen Überlebensvor-teil erzielt. Das abwartend exspektative Verhalten (watchand wait) ist deshalb bei den asymptomatischen Patien-ten mit lymphoplasmozytischem Lymphom weiterhin derStandard.Speziell für den M. Waldenström wurde ein prognosere-levanter Risikoscore entwickelt, der für eine Therapieent-scheidung hilfreich ist (Tabelle 1) [7].
Tabelle 1Internationaler Prognostischer Index WM (ISSWM)
Risiko-Score WMParameter Niedrig Intermediär Hoch
Alter > 65 Jahre o + +
Hb < 11.5 g/dlThrombozyten < 100 x 109/l < 1 Faktor 2 Faktoren > 3 Faktorenß2 M > 3 mg/l Alter AlterIgM > 70 g/l < 65 Jahre > 65 Jahre
5 - Jahresüberleben 87 % 68 % 36 %

�� SSeeiittee 1111 ��JJOOUURRNNAALL 0022//22000099
Für die therapiebedürftigen Patienten mit LPL/WM steheneine Reihe wirksamer Zytostatika und Zytostatikakombi-nationen zur Verfügung (Tabelle 2). In der Primärtherapiekönnen Ansprechraten (CR + PR) um 60-80 % erreichtwerden, der Anteil von kompletten Remissionen ist aller-dings gering (10-20 %), im Median überleben die Patien-ten etwas mehr als 5 Jahre. Auf Grund der Seltenheit die-ser Lymphomentität liegen keine Daten von größeren Pha-se III-Studien vor. In vielen Studien wurden allgemein in-dolente Lymphome eingeschlossen, darunter auch miteinem Anteil von 10-20 % LPL. In den Studien vor der Ärader WHO/REAL-Klassifikation handelte es sich dabei zu-dem oft noch um CD 5+-Erkrankungen, die man heuteder B-CLL mit plasmazytoider Differenzierung zuordnet.Daraus resultiert eine ausgesprochene Heterogenität undeine sichere evidenzbasierte Therapieempfehlung für dasLPL kann heute noch nicht gegeben werden.Chlorambucil hat seit langem einen festen Platz in derChemotherapie des LPL, dabei gibt es keinen Unterschiedzwischen einer niedrig dosierten kontinuierlichen Gabeund einer intermittierenden höheren Dosis. Auch dieKombination mit Glucocortikoiden scheint keinen Einflußauf die Effektivität zu haben. Die kumulative monatlicheDosis liegt um ca. 1,5 mg/kg KG. Vergleichbare Ergebnis-se können mit anderen Alkylanzien wie Cyclophosphamid,Trofosfamid oder Bendamustin erzielt werden. Aggressi-vere Therapieprogramme wie COP oder CHOP wurdennicht in Phase III-Studien gegen Chlorambucil geprüft,sind aber offenbar nicht effektiver bei einer deutlich hö-heren Toxizität. Die Nukleosidanaloga stellen eine neuereBehandlungsoption auch bei LPL dar. Cladribin (2-Chloro-deoxyadenosin, 2-CdA) und Fludarabin sind auch bei mitAlkylanzien vorbehandelten Patienten hoch wirksam; eskonnten Ansprechraten von 30-50 % erzielt werden [8; 9; 10]. In der Primärtherapie kann bei 55-90 % der Pa-tienten eine Remission induziert werden [11; 12]. Eine neuere therapeutische Option ist der Einsatz des mo-noklonalen Antikörpers Rituximab (Anti-CD20-Antikör-per); aus Phase II-Studien gibt es Hinweise für eine hoheEffektivität bei rezidivierten Patienten und auch in der Pri-märtherapie. Insbesondere in Kombination mit verschie-denen Chemotherapieprotokollen konnten gute Ergeb-nisse erzielt werden (Dexamethason, Cyclophosphamid,Rituximab [13], R-CHOP [14]). In einer ersten prospektivenrandomisierten Studie der GLSG konnte die Überlegenheiteiner kombinierten Immunchemotherapie (R-CHOP) ge-genüber alleiniger CHOP-Therapie in der Primärtherapievon LPL gezeigt werden [14]. Hier ist allerdings anzumer-ken, dass es für den Einsatz von Rituximab bei LPL/WMkeine explizite Zulassung gibt. Beim Einsatz von Rituximabist der sog. IgM-Flare zu beachten, ein krisenhafter An-stieg des IgM mit der Gefahr des Hyperviskositätssyn-droms; dieses Phänomen tritt jedoch bei der Kombinati-on von Rituximab mit Chemotherapie in der Regel nichtauf.Im Rezidiv nach oder bei Refraktärität gegenüber der Pri-märtherapie sind die Nukleosidanaloga die Substanzender Wahl; beim Einsatz von Cladribin und Fludarabin istderen Kreuzresistenz zu beachten; ohnehin ist vor dem se-quentiellen Einsatz von Nukleosidanaloga wegen ihrerausgeprägten T-Zell-Toxizität zu warnen. Kombinationenvon Fludarabin/Cyclophosphamid (FC), Fludarabin/ Cyclo-phosphamid/Mitoxantron (FCM) oder Cladribin/Mitoxan-tron haben sowohl bei der B-CLL als auch bei follikulären
und Mantelzell-Lymphomen eine hohe Wirksamkeit ge-zeigt und es kann davon ausgegangen werden, dass die-se auch beim LPL effektiv sind. Die erneute Kombinationmit Rituximab ist möglich, sie muss letztendlich von deraktuellen Situation (refraktär, Früh- oder Spätrezidiv) ab-hängig gemacht werden.Neue therapeutische Optionen eröffnen sich durch denEinsatz neuer Substanzen wie dem ProteasomeninhibitorBortezomib, den sog. Imids (Thalidomid, Revlimid) oderauch mTOR-Inhibitoren (Everolimus), die Ergebnisse vonderzeit laufenden Studien sind abzuwarten [15].
Tabelle 2Therapieschemata bei lymphoplasmozytischem Lymphom (Auswahl)
Schema/Substanz Dosierung Bemerkung
Chlorambucil 0,4-0,8 mg/kg KG q 2 Wo. Bis zum maxima-6-8 mg/m2 KOF x len Ansprechen10d q 4 Wo.20 mg x 3 d+/- Prednisolon q 2 Wo.50 mg
Bendamustin 70-100 mg/m2 d 1 + 2 6-8 ZyklenKOF q 4 Wo. + Rituximab
375 mg/m2 inf.d 0 oder 1
COP 6-8 ZyklenCyclophosphamid 400 mg/m2 KOF d 1-5 + RituximabVincristin 2 mg d 1 375 mg/m2
Prednisolon 100 mg/m2 KOF d 1-5 d 0 oder 1q 4 Wo.
BOP 6-8 ZyklenBendamustin 60 mg/m2 KOF d 1-5 + RituximabVincristin 2 mg d 1 375 mg/m2
Prednisolon 100 mg/m2 KOF d 1-5 d 0 oder 1q 4 Wo.
MCP 6-8 ZyklenMitoxantron 8 mg/m2 KOF d 1 + 2 + RituximabChlorambucil 3 x 3 mg/m2 KOF d 1-5 375 mg/m2
Prednisolon 25 mg/m2 KOF d 1-5 d 0 oder 1q 4 Wo.
CHOP 6-8 ZyklenCyclophosphamid 750 mg/m2 KOF d 1 + RituximabAdriblastin 50 mg/m2 KOF d 1 375 mg/m2
Vincristin 2 mg d 1 d 0 oder 1100 mg d 1
Prednisolon q 3 Wo.Fludarabin 25 mg/m2 KOF d 1-5 4-6 Zyklen
q 4 Wo. Second-line+ Rituximab
FC 4-6 ZyklenFludarabin 30 mg/m2 KOF d 1-3 Second-lineCyclophosphamid 250 mg/m2 KOF d 1-3 + Rituximab
FB 4-6 ZyklenFludarabin 30 mg/m2 KOF d 1-3 Second-lineBendamustin 30 mg/m2 KOF d 1-3 + Rituximab
q 4 Wo.FCM 4-6 ZyklenFludarabin 25 mg/m2 KOF d 1-3 Second-lineCyclophosphamid 200 mg/m2 KOF d 1-3 + RituximabMitoxandron 6-8 mg/m2 KOF d 1
q 4 Wo.
Cladribin 0,07 mg/kg KG d 1-7 civi bis 4 Zyklenq 4 Wo. Second-line0,1 mg/kg KG d 1-5 + Rituximab
2h-inf.q 4 Wo.
Die Remissionsbeurteilung bei LPL erfolgt analog den an-deren nodalen Lymphomen, beim M. Waldenström spieltzusätzlich die Reduktion des monoklonalen IgM eine we-

�� SSeeiittee 1122 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
sentliche Rolle bei der Evaluierung des Ansprechens aufdie Therapie (Tabelle 3).
Tabelle 3Remissionskriterien bei M. Waldenström
Ansprechen Kriterien
Komplette Komplette Rückbildung aller objektivenRemission Krankheitsbefunde zum Zeitpunkt des (CR) Restaging mit völliger Rückbildung vor-
bestehender Lymphknotenschwellungen sowie einer vorbestehenden Hepatome-galie und Splenomegalie für mindestens 6 Wochen. Ausschluss einer weiter beste-henden Lymphominfiltration des Kno-chenmarks durch Knochenmark-Biopsie, fehlender Nachweis des monoklonalen Proteins in der Immunfixation. Der CR-Status muss erneut unter anderem durch eine Immunfixation frühestens 6 Wochen nach initialem Restaging bestätigt wer-den.
Partielle Mindestens 50%ige Reduktion der mono-Remission klonalen IgM-Serumkonzentration in der (PR) Serumelektrophorese und mindestens
50% Rückgang der Adenopathie/Orga-nomegalie im CT. Keine neuen Krank-heitsmanifestationen.
„Minimales Mindestens 25%ige, aber weniger als Ansprechen“ 50%ige Reduktion der monoklonalen (MR) IgM-Serumkonzentration in der Serum-
elektrophorese. Keine neuen Krankheits-manifestationen.
„Stable Weniger als eine 25%ige Reduktion und disease“ (SD) weniger als ein 25%iger Anstieg der mo-
noklonalen IgM-Serumkonzentration in der Serumelektrophorese ohne Progress der Adenopathie/Organomegalie, Zyto-penien oder klinisch relevanter Sympto-me.
Progress (PD) Mindestens 25%iger Anstieg der mono-klonalen IgM-Serumkonzentration in der Serumelektrophorese, bestätigt in einer zweiten Messung, oder neu aufgetretene klinisch relevante Krankheitssymptome oder -befunde.
Die Hochdosistherapie (mit oder ohne Ganzkörperbe-strahlung) mit nachfolgender autologer peripherer Blut-stammzelltransplantation (ABSCT) ist eine auch heutenoch als experimentell anzusehende Behandlungsvarian-te für jüngere Patienten (< 60 Jahre) und sollte vorerstausschließlich im Rahmen von klinischen Studien zum Ein-satz kommen. Noch mehr trifft dies zu für die allogeneTransplantation, hier ist bei konventionellem Vorgehenmit einer sehr hohen transplantationsassoziierten Morta-lität zu rechnen; größere Erfahrungen mit einer dosisre-duzierten Konditionierung vor allogener Transplantationliegen bisher nicht vor.
Literatur
1. Swerdlow SH, Berger F, Pileri SA, Harris NL, Jaffe ES, Stein H (2008): Lymphoplasmozytic lymphoma. In: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues; Eds.: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW; WHO Press Geneva; 194 – 195
2. Waldenström J (1944): Incipient myelomatosis or ”essential” hyperglo-bulinemia with fibrinogenopenia: a new syndrom ? Acta Med Scand 117: 216-222
3. Coupland SE, Dallenbach FE, Stein H (2000): Kleinzellige B-Zell-Lympho-me: Differentialdiagnostische Leitlinien. Pathologe 21: 147 - 161
4. The Non-Hodgkin’s Lymphoma Classifikation Project (1997): A clinical evaluation of the international lymphoma study group classification of Non-Hodgkin’s Lymphoma. Blood 89: 3909 – 3918
5. Kristinsson SY, Bjorkholm M, Goldin LR, McMaster ML, Turesson I, Land-gren O (2008). Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenstrom macroglobuli-nemia patients: a population-based study in Sweden. Blood; 112(8): 3052 - 3056.
6. Lai R, Weiss LM (1998): Hepatitis C virus and non-Hodgkin’s lymphoma [editorial; comment]. Am J Pathol 109: 508 - 510
7. Kantarjian HM, Alexanian R, Koller CA, Kurzrock R, Keating MJ (1990): Fludarabine therapy in macroglobulinemic lymphoma. Blood 75: 1928 – 1931
8. Zinzani PL, Gherlinzoni F, Bendandi M, Zaccaria A, Aitini E, Salvucci M, Tura S (1995): Fludarabine treatment in resistant Waldenstrom’s macro-globulinemia. Eur J Haematol 54: 49 – 52
9. Leblond V, Ben-Othman T, Deconinck E, et al. (1998): Activity in previously treated Waldenstrom’s macroglobulinemia: a report on 71 cases. J Clin Oncol 16: 2060 – 2064
10. Dimopoulos MA, Kantarjian HM, Weber D, et al. (1994): Primary thera-py of Waldenstrom’s macroglobulinemia with 2-chlorodeoxyadenosine. J Clin Oncol 12: 2694 - 2698
11. Foran JM, Rohatiner AZS, Coiffier B et al. (1999) : Multicenter phase II study of fludarabine phosphate for patients with newly diagnosed lym-phoplasmozytoid lymphoma, Waldenstroms macroglobulinemia and mantle cell lymphoma. J ClinOncol 17: 546 – 553
12. Dimopoulos M, Anagnostopoulus A, Kyrtsonis MC, et al. (2006) : Pri-mary treatment of Waldenstrom’s macroglobulinemia (WM) with dea-xamethason, rituximab and cyclophosphamide. Blood 108: abstract128
13. Treon SP, Gertz MA, Dimopoulos M, et al. (2006): Update on treatment recommendations from the Third International Workshop on Walden-strom’s macroglobulinemia. Blood 107: 3442 - 3446
14. Buske C, Hoster E, Dreyling M, Eimermacher H, Wandt H, Metzner B, et al.(2009): The addition of rituximab to front-line therapy with CHOP (R-CHOP) results in a higher response rate and longer time to treatment failure in patients with lymphoplasmacytic lymphoma: results of a ran-domized trial of the German Low-Grade Lymphoma Study Group (GLSG). Leukemia.23(1):153 - 161.
15. Treon SP (2009): How I treat Waldenstrom’s macroglobulinemia. Blood 114: 2375 - 2385
Korrespondenzadresse:
Prof. Dr. med. Michael Herold4. Medizinische KlinikHämatologie, internistische Onkologie, HämostaseologieHELIOS Klinikum ErfurtNordhäuser Str. 7499089 ErfurtTelefon 0361-7812473e-Mail [email protected]

�� SSeeiittee 1133 ��JJOOUURRNNAALL 0022//22000099
�� Die Bedeutung des Ki-67-Antigensin der Tumorpathologie
Barbara Henkel, Hartwig KosmehlInstitut für Pathologie, HELIOS Klinikum Erfurt, undGemeinschaftspraxis für Pathologie
Zellbiologie des Ki-67-AntigensDas Ki-67-Antigen wurde 1983 von J. Gerdes an der Uni-versität Kiel bei der Herstellung spezifischer Marker gegenHodgkin- und Reed-Sternberg-Zellen entdeckt. Das Ki-67-Protein ist ein Proteinase-sensibles Zellprotein, das für dieAufrechterhaltung des Zellzyklus absolut erforderlich ist.Seine Expression ist eng an die Zellproliferation gekoppelt.Das Ki-67-Protein akkumuliert in der G1-, S- und G2-Pha-se sowie während der Mitose, wo es seine maximale Kon-zentration erreicht. Nach Abschluss der Mitose fällt derSpiegel des Ki-67-Proteins und Ki-67 ist in der ruhendenZelle (G0) nicht nachweisbar. Das Ki-67-Protein hat einekurze biologische Halbwertszeit, die durch die hohe Pro-teinase-Sensitivität erklärt werden kann. Offensichtlichwird es zu jeder Zellteilung de novo synthetisiert.Das Ki-67-Protein kann als Kernprotein immunhistoche-misch dargestellt werden. Zwischenzeitlich sind zahlrei-che spezifische monoklonale Antikörper gegen das Ki-67-Antigen beschrieben (z.B. der Klon MIB-1). Der ursprüng-liche Ki-67-Antikörper erkennt das Protein im Kontrast zuden neuen Klonen nur im unfixierten Gewebe. Bei stan-dardisierter Fixierung und bei standardisiertem Antigen-Retrival erfolgt die Demaskierung des Antigens proportio-nal zur Zeit, so dass in standardisierten immunhistoche-mischen Verfahren die Ki-67-Spiegel differenter Probenuntereinander vergleichbar sind.
Die konzeptionelle Bedeutung des Ki-67-Index bei derTumorklassifikationDie Benennung von Tumoren erfolgt prinzipiell nach 2 Ge-sichtspunkten:
1. Nach der Differenzierung (welcher normale Zelltyp wird von der Tumorzelle nachgeahmt).
2. Nach der Dignität (gutartig oder bösartig).Für maligne Tumoren ist ein schnelles Tumorwachstum charakteristisch und ein hoher Ki-67-Index kann folg-lich die hohe Proliferationsrate bösartiger Tumoren schnell und einfach im histologischen Präparat anzei-gen.In der Synopsis: Das Ki-67-Antigen ist ein Prolifera-tionsmarker. Die Proliferation ist ein von der Differen-zierung unabhängiges Phänomen. Der Ki-67-Index leistet keinen Beitrag zur Differenzierungsbestim-mung. Der Ki-67-Index ist eng verknüpft mit der Digni-tätsbestimmung von Tumoren. Gutartige Tumoren ha-ben in der Regel einen niedrigeren Ki-67-Index als bös-artige Tumoren. Das schließt nicht aus, dass auch gut-artige Tumoren oder entzündliche und reparative Ver-änderungen einen Ki-67-Index wie maligne Tumoren erreichen können.
Das Ki-67-Antigen in malignen LymphomenZunächst leistet bereits die Verteilung der proliferativ-ak-tiven Zellen im Lymphknoten einen Beitrag zur Diagnostik.
Finden sich die Ki-67-markierten Zellen in follikulärenStrukturen und ist anhand der Verteilung der proliferativ-aktiven Zellen eine erhaltene Gliederung in B- und T-Zoneerkennbar, spricht dieses Befundmuster eher für eine re-aktive Lymphadenopathie bzw. für ein follikuläres Lym-phom.Eine diffuse ungeordnete Verteilung der Ki-67-markiertenLymphozyten in einem Lymphknoten ist ein diagnosti-scher Hinweis auf ein malignes Lymphom mit diffusemWuchsbild. Bei einem Ki-67-Index von ca. 1 bis 20% ist inerster Linie ein indolentes (niedriggradiges) Lymphom inBetracht zu ziehen. Ab einen Ki-67-Index von 45% müs-sen maligne Lymphome mit einem gesteigerten Blasten-gehalt differenzialdiagnostisch bedacht werden.Die noch gut bekannte REAL-Klassifikation, einem Vorläu-fer der aktuellen WHO-Klassifikation, unterschied in hoch-maligne, aggressive Lymphome mit einem Ki-67-Indexvon 45 bis 65% und in hochmaligne, hochaggressive Lym-phome mit einem Ki-67-Index über 65%. So zeigt das Bur-kitt-Lymphom eine Wachstumsfraktion von über 90%.Der Ki-67-Index in einem Lymphknoten muss immer vordem histologischen Gesamtbild bewertet werden. DieLymphozyten im Follikelzentrum immunologisch stimu-lierter Lymphknoten haben oft einen höheren Ki-67-Indexals die Lymphozyten in den follikulären Lymphomen Grad I und Grad II. Die Graduierung der follikulären Lym-phome in den Grad I bis III erfolgt auch nicht anhand derproliferativ-aktiven Zellen sondern anhand der Blastenz-ahl innerhalb der neoplastischen follikulären Strukturen!Der Ki-67-Index in follikulären Strukturen ist somit einsehr relativer Parameter, der nur vor dem Gesamtbild derErkrankung eingeordnet werden kann (Abb. 1 bis 6 und7 bis 8).
Der Ki-67-Index als Hilfe zur Klassifikation von Weich-gewebstumorenFür die Dignitätsbestimmung von Weichgewebstumorenkann der Ki-67-Index nur mit großem Vorbehalt herange-zogen werden. Charakteristisch ist, dass pseudosarkoma-töse Läsionen wie die Fasciitis nodularis oder die Fasciitisproliferans einen hohen Ki-67-Index haben können.Ist die Differenzierungsrichtung und die Dignität einesWeichgewebstumors durch das Aufdecken von zytologi-schen Atypien, pathologischen Mitosen, das Gefäßmusterund durch andere immunhistochemisch dargestellte An-tigene oder durch chromosomale Aberrationen aufge-deckt, kann der Ki-67-Index einen Beitrag zum histopa-thologischen Grading leisten. Das histopathologischeGrading ist für Sarkome des Weichgewebes ein unab-dingbarer diagnostischer Parameter, da er in einem direk-ten Bezug zum 5-Jahres-Überleben und zur Wahl der The-rapie steht. In Europa ist das FNCLCC-Grading-System dasam weitesten verbreitete. Es wird auch von der WHO ak-zeptiert.
Das FNCLCC-Grading umfasst 3 Parameter:
• DifferenzierungsgradScore-Wert 1 - Sarkome mit hoher Ähnlichkeit zur ent-sprechenden mesenchymalen Normalzelle; Score-Wert 2- Sarkome mit definierbaren histologischen Tumor-typ; Score-Wert 3 - embryonale undifferenzierte Sarko-me, Synovialsarkome, Osteosarkome und PNET.
�� SSeeiittee 1144 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
• Mitosezahl Wird ebenfalls in einem Score-Wert 1 bis 3 erfasst. 0 bis 10 Mitosen pro 10 hochauflösende Gesichtsfel-der werden mit dem Score-Wert 1 belegt, 10 bis 19 Mi-tosen mit den Score-Wert 2 und mehr als 20 Mitosen pro 10 hochauflösende Gesichtsfelder werden mit dem Score-Wert 3 belegt.
• NekroseIst das Sarkom frei von Nekrosen, ergibt sich ein Score-Wert 0, bei weniger als 50% Nekrosen der Score-Wert 1 und bei mehr als 50% Nekrosen der Score-Wert 3.
Die Summe der Score-Werte ergibt den histopathologi-schen Grad. Der Summen-Score 2 oder 3 Punkte ist demGrad I verbunden, Grad II-Sarkome haben einen Sum-menscore von 4 oder 5 und Grad III-Sarkome (hochmali-gne) haben einen Summen-Score-Wert 6, 7 oder 8.Der Ki-67-Index steht in einem direkten Bezug zur Mito-sezahl und kann somit einen Beitrag zur Graduierung derWeichgewebssarkome leisten.
Abb. 1 Abb. 2
Abb. 3 Abb. 4
Abb. 5 Abb. 6
Abbildungen 1-6 In den Abb. 1 und 2 sind ein reaktiver und ein neopla-stischer Lymphfollikel eines follikulären Lymphoms Grad 2 gegenüber gestellt(HE-Färbung). Die Immunreaktion für bcl2 unterscheidet den reaktivenLymphfollikel (Abb. 3) und das follikuläre Lymphom (Abb. 4). In dem folli-kulären Lymphom verhindert die Überexpression von bcl2 den programmier-ten Zelltod der Zellen der Follikel. In reaktiven Follikeln (Abb. 5) ist die Proli-ferationsrate der Follikelzentrums-Zellen oft höher als in den neoplastischenFollikeln (Abb. 6) von follikulären Lymphomen, bei denen das Tumorwachs-tum auch durch die Verhinderung des programmierten Zelltods (bcl2-Über-expression) eintritt.
Der Ki-67-Index bei der Klassifikation von Lungentu-morenDas Karzinom mit der höchsten Proliferationsrate in derLunge ist das kleinzellige Karzinom. Bei einem Ki-67-Indexvon über 50% muss die Frage nach einem kleinzelligenKarzinom mit weiterer Methodik beantwortet werden.Die Mitoserate ist für die Einordnung der pulmonalenKarzinoide ganz entscheidend. Die WHO-Klassifikationunterscheidet das typische Karzinoid und das atypischeKarzinoid der Lunge. Typische Karzinoide haben wenigerals 2 Mitosefiguren pro 2 mm² und Nekrosen werdennicht gefunden. Atypische Karzinoide habe 2 bis 10 Mito-sen pro 2 mm² und/oder Herde von Nekrosen. Bei allenKarzinoiden mit einem gesteigerten Ki-67-Index bestehtsomit die Differenzialdiagnose eines atypischen Karzi-noids. Während das typische Karzinoid eine exzellentePrognose hat, ist beim atypischen Karzinoid bereits zumZeitpunkt der Diagnosestellung mit einer Lymphknoten-metastasierung zu rechnen. Für das atypische Karzinoidwird eine 5-Jahres-Überlebensrate von 61 bis 73% ange-geben.
Abb. 7 Abb. 8
Abbildungen 7, 8 Diffuse großzellige B-Zell-Lymphome sind durch einenblastären Zellphänotyp (Abb. 7, HE) und durch einen Ki-67-Index über 80%gekennzeichnet (Abb.8; Ki-67).
Der Ki-67-Index in der Klassifikation von HirntumorenPrognose und Tumorbiologie von Geschwülsten des ZNSund der Hirnhäute werden mit den WHO-Graden I-IV be-schrieben. Ab WHO Grad III ist das Verhalten der Tumoremaligne. Für die Einteilung der Hirntumoren in die WHO-Grade ist der Ki-67-Index eine große Hilfe. Im Falle der Me-ningiome unterscheidet der Ki-67-Index wesentlich zwi-schen dem WHO Grad I und II. Ab einem Ki-67-Index von3% besteht der dringende Verdacht auf ein atypischesMeningeom (WHO Grad II) (siehe Abb. 9 und 10).Bei der biologisch und therapeutisch wichtigen Unter-scheidung zwischen Astrozytomen des WHO Grad II undIII ist ab einen Ki-67-Index von 5% mit einem Grad III-Tu-mor (maligne) zu rechnen. Ab einen Ki-67-Index größer als10% besteht die Differenzialdiagnose eines Glioblastoms(WHO Grad IV) .
Der Ki-67-Index in Nierenzellkarzinomen, oralen Plat-tenepithelkarzinomen, sinonasalen und nasopharyn-gealen KarzinomenAuch in den vorgenannten Karzinomtypen ist das Ki-67-Antigen ein wichtiges Hilfsmittel zur Graduierung und Ty-pisierung, wenngleich für diese Entitäten keine klarenProzentgrenzen des Ki-67-Index vorgegeben sind.Dass ein hoher Ki-67-Index nicht in jedem Fall mit einerungünstigen Prognose einhergehen muss, zeigen die na-sopharyngealen Karzinome vom Plattenepitheltyp. In die-

�� SSeeiittee 1155 ��JJOOUURRNNAALL 0022//22000099
ser WHO-Entität verbirgt sich das lymphoepitheliale Kar-zinom vom Schmincke-Regaud-Typ, das durch einen Ki-67-Index oft von mehr als 50% gekennzeichnet ist und ei-ne günstige Prognose aufgrund der Strahlen- und Chemo-therapiesensitivität hat.
Abb. 9 Abb. 10
Abbildungen 9, 10 Die Abb. 9 demonstriert den niedrigen Ki-67-Index ei-nes Meningeoms WHO Grad I im Kontrast zur gesteigerten mitotischen Ak-tivität im Meningeom WHO Grad II (Abb. 10). Die Meningeome unterschei-den sich durch ihre Rezidivrate.
Der Ki-67-Index in der Diagnostik von Mammatumo-renZunächst gibt es Mammakarzinomtypen, die mit einemhohen Ki-67-Index verbunden sind. Ein Beispiel für einesolche Entität ist das medulläre Karzinom der Mamma.Das häufigste Mammakarzinom ist das invasiv-duktaleKarzinom. Insbesondere für diesen Karzinomtyp ist dasGrading nach Elston und Elllis ein entscheidender progno-stischer und therapieentscheidender Parameter. Das Gra-ding umfasst das Ausmaß der Tubulusbildung (1 bis 3Punkte), der Kernpleomorphie (1 bis 3 Punkte) und der Mi-tosezahl. Auch die Mitosezahl wird mit 1 bis 3 Punkten be-legt. Der histopathologische Grad 1, hochdifferenziert, wirdbei einem Punkte-Score von 3 bis 5 Punkten vergeben, derGrad 2, moderat differenziert, bei einem Summen-Scorevon 6 bis 7 Punkten und der Grad 3, schlecht differenziert,bei einem Punktwert 8 bis 9.Mitosen sind im Gewebe oft schwer zu identifizieren, ins-besondere wenn die Fixierung des Tumorgewebes nichtoptimal ist. Hier kann der Ki-67-Index zur Überprüfungdes ermittelten Mitosewertes herangezogen werden.
Der Ki-67 Index im ProstatakarzinomProstatakarzinome weisen oft auch unbehandelt ein lang-jähriges, langsames Wuchsverhalten auf. Folglich reflek-tiert die Proliferationsrate bzw. der Ki-67-Index nur sehrbedingt das biologische Verhalten.Die Erfassung der mitotischen Aktivität ist zur Einschät-zung der Biologie des Prostatakarzinoms nicht akzeptiert.Das modifizierte histopathologische Grading nach Glea-son beschreibt vielmehr die architektonischen Parameterdes Tumors (tubulär, kribriform, strangförmig, solid, dis-soziativ).Das Beispiel der Prostatakarzinome zeigt, dass die Inter-pretation des Ki-67-Index in Relation zu der Prognose en-titätsspezifisch ist. Es ist nicht ohne weiteres möglich, voneinem Organ und von einem Karzinomtyp auf den ande-ren zu schließen.
In der Synopsis ist das Ki-67-Antigen und die Bestimmungdes Ki-67-Index eine einfache und gute Methode, einen
wichtigen tumorbiologischen Parameter zu beschreiben.Der Ki-67-Index ist hilfreich zur Dignitätseinordnung vonNeubildungen. Allgemein gilt, dass ein hoher Ki-67-Indexmit einem aggressiven Verhalten des Tumors verbundenist. Auch gutartige Tumoren können schnell wachsen, sodass im Einzelfall der Ki-67-Index keine absolute Größe fürdie Bewertung der Dignität darstellt.In vielen Entitäten wie malignen Lymphomen, Weichge-webssarkomen und Hirntumoren ist der Ki-67-Index eineentscheidende Größe für die Festlegung des histopatho-logischen Grades.Jenseits der großen Bedeutung des Ki-67-Index für Digni-tätsbewertung und Graduierung gilt: Eine Bewertung desKi-67-Index kann nur im histologischen Gesamtkontext er-folgen!
Korrespondenzadresse:
Dr. med. Barbara HenkelUniv. Prof. Dr. med. H. KosmehlHELIOS Klinikum Erfurt Institut für Pathologie und Gemeinschaftspraxis für PathologieNordhäuser Str. 7499089 ErfurtTelefon 0361-7812751e-Mail [email protected]

�� SSeeiittee 1166 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
�� Hochleistungsmedizin und Mensch-lichkeit – ein Widerspruch?
Vortrag auf dem Symposium „Zwischen Evi-denz und Empathie – Dilemma oder Chancefür die Palliativmedizin“ am 20. Juni 2009 inBad Berka
Wolfgang HiddemannMedizinische Klinik und Poliklinik III, Klinikum derLudwig-Maximilians-Universität München, CampusGroßhadern
Einleitung
Die bahnbrechenden Errungenschaften, die die Medizinim letzten Jahrhundert erleben durfte, der zunehmendeEinfluss naturwissenschaftlicher Erkenntnisse und nichtzuletzt die Verschiebung allgemeiner gesellschaftlicherWerte haben dazu geführt, die Ganzheitlichkeit des kran-ken Menschen aus dem Auge zu verlieren. Diese Tendenzwird durch die aktuellen gesetzgeberischen und finanziel-len Rahmenbedingungen erheblich verstärkt. Um dieserEntwicklung entgegen zu treten, erscheint es notwendig,uns auf die traditionellen ärztlichen Aufgaben und Wertezu besinnen. Wir dürfen unsere Verpflichtung dem kran-ken Menschen gegenüber nicht vernachlässigen und müs-sen die Betrachtung des ganzen Menschen als zentralesElement unserer Tätigkeit bewahren, um nicht den Berufdes Arztes mit der Tätigkeit eines Mediziners oder hoch-qualifizierten Facharbeiters einzutauschen und damit dieessentiellen Elemente unserer ethischen und ärztlichenEinstellung aufzugeben. Es muss daher der Anspruch anuns selber sein und bleiben, Hochleistungsmedizin undMenschlichkeit miteinander zu verbinden.
Hochleistungsmedizin
Der Begriff „Hochleistungsmedizin“ löst fast immer diespontane Assoziation mit der Apparatemedizin aus, mithochspezialisierter Technologie, mit Computer-gestützterDiagnostik und Therapie, mit einer Medizin, der der Pa-tient hilflos ausgeliefert ist und in der der Arzt allenfallsals kundiger Techniker erscheint. Dieses Bild ist nicht grundsätzlich falsch. Die Hochlei-stungsmedizin schließt die hoch-technisierte Apparate-medizin zweifellos ein. Ohne entsprechende technischeAusrüstungen wären viele Leistungen der modernen Me-dizin undenkbar. Wie sollte beispielsweise eine Operationam Herzen ohne Apparate erfolgen? Wie sollte der Aus-fall der Niere ohne Dialysegeräte kompensiert werden?Wie sollte ein Atemversagen ohne Beatmungsmaschineund Intensivstation überbrückt werden?Hochleistungsmedizin ist aber weit mehr als eine Medizinmit Apparaten. Hochleistungsmedizin ist zuallererst undunverzichtbar eine auf hohen Leistungen und eine auf ho-hen fachlichen Qualifikationen von Ärzten und Pflege-kräften aufbauende Medizin. Ein hohes Sachwissen und
die fortwährende Bereitschaft zur Fort- und Weiterbil-dung sind ein entscheidendes Element der Hochleistungs-medizin. Ohne adäquates Fachwissen können auch nochso weit entwickelte Apparate nicht sachgerecht zum Wohldes Patienten eingesetzt werden und verfehlen hoch-technisierte diagnostische und therapeutische Verfahrenihr Ziel. Hohes Fachwissen ist somit unverzichtbare Grund-lage jeden ärztlichen Handelns. Fachwissen alleine reicht jedoch nicht aus, um wirklich„Arzt“ zu sein. Arztsein beinhaltet als zweites ebenso un-verzichtbares Grundelement die Menschlichkeit, d.h. dasBewusstsein für den kranken Menschen in seinen körper-lichen und seelischen Dimensionen und das Bewusstseinfür die Verantwortung, die ein Arzt übernimmt, wenn eres sich zur Aufgabe macht, kranken Menschen zu helfen.
Menschlichkeit
Was bedeutet Menschlichkeit im ärztlichen Beruf konkretund wie wird sie in der täglichen Praxis erkennbar? Wennman ein Lexikon zur Hilfe nimmt und nach dem Begriff„Menschlichkeit“ sucht, so findet man als Definition:„Menschlichkeit bedeutet ein Denken und Handeln basie-rend auf Selbstlosigkeit, Mitgefühl, Warmherzigkeit, Her-zensgüte, Ehrlichkeit, Verständnis, Toleranz, Einfachheit,Gewissenhaftigkeit, Zufriedenheit, Achtsamkeit, gegen-seitigem Verantwortungsgefühl und Respekt“. Mensch-lichkeit definiert sich somit über das Denken und Handeln,d.h. eine persönliche Grundeinstellung. Menschlichkeitist in dieser Form allerdings als ein Ideal definiert, das inseiner Vollkommenheit nicht erreicht werden kann.Diese Definition von Menschlichkeit ist für den Anspruchdes Arztes, Hochleistungsmedizin und Menschlichkeitmiteinander zu verbinden, in vieler Hinsicht aufschluss-reich. Zum einen wird deutlich, dass Menschlichkeit eineallgemein gütige und erstrebenswerte Tugend ist, dienicht speziell auf den Arztberuf abzielt. Auch ein Juristoder Kaufmann, ein Bäcker oder Bankangestellter solltemenschlich sein. Natürlich wird von einem Beruf, der essich als Hauptaufgabe macht, Menschen und vor allemkranken Menschen zu helfen, in besonderer WeiseMenschlichkeit erwartet und sogar vorausgesetzt. Damitist der Arztberuf von vorn herein mit einem besonderenAnspruch verbunden, der sowohl aus seinem eigenenSelbstverständnis heraus resultiert, sich aber in gleicherWeise auch aus den Erwartungen der Patienten und derGesellschaft im allgemeinen ergibt. Nicht zuletzt aus die-sem Grunde genossen Ärzte zu allen Zeiten ein hohes An-sehen. Sie hatten und haben es sich zur Aufgabe ge-macht, Wissen und Können zu erwerben und es zumWohle kranker Menschen zu verwenden. Mit dieser Wahr-nehmung ist verbunden, dass von Ärzten ein höheresMaß an moralischem und ethischen Handeln und Denkenerwartet wird. Dies impliziert, dass Ärzte mit anderenMaßstäben gemessen werden und dass sie, um es plaka-tiv zu formulieren, „bessere Menschen“ sein sollen.
Wenn man derzeit zur Kenntnis nehmen muss, dass dashohe Ansehen des Arztberufs deutlich abgenommen hat,so liegt dies sicherlich zum Teil in der Ärzteschaft selbst be-gründet. Das Gewinnstreben, die Geltungssucht und dieMissachtung der Menschlichkeit durch einige wenige Kol-legen haben den gesamten Berufsstand in Misskredit ge-

�� SSeeiittee 1177 ��JJOOUURRNNAALL 0022//22000099
bracht. Darüber hinaus findet eine fast systematisch zunennende Demontage des ärztlichen Ansehens durch diePolitik statt, die die Ärzte zu den Hauptverantwortlichenfür die derzeitige finanzielle Misere des Gesundheitssy-stems macht und eine dem Wohl der Allgemeinheit zuwi-derhandelnde Allianz zwischen pharmazeutischer Indu-strie und Ärzteschaft unterstellt. Nur vor diesem Hinter-grund ist das Ausmaß der Kontrollinstanzen zu verstehen,die in den letzten Jahren etabliert wurden, um die Ärztezu überwachen. Beispielhaft genannt seien nur der Medi-zinische Dienst der Krankenkassen, das Institut für Quali-tätssicherung und Wirtschaftlichkeit im Gesundheitswe-sen und nicht zuletzt die Pauschalvergütung ärztlicherLeistungen. Mit derartigen Maßnahmen wird eine auf denindividuellen Patienten ausgerichtete Medizin zumindesterschwert und rangieren ökonomische Vorgaben vor me-dizinischen Erfordernissen.
Ein ganz anderer, sicherlich aber mindestens gleichbe-deutender Grund für den Ansehensverlust der Ärzte undihre Abwertung zu Medizinern im Sinne von Gesundheits-managern liegt darin, dass in unserer modernen Gesell-schaft die Menschlichkeit kaum noch als eine allgemeinerstrebenswerte Tugend angesehen wird. Obwohl keinZweifel daran bestehen kann, dass eine große Sehnsuchtnach Menschlichkeit besteht, wird das tägliche Lebendoch eher von materiellen Werten und Leistungsdenkendominiert. Menschlichkeit wird in unserem derzeitigenGesellschaftssystem nicht anerkannt und honoriert undist allenfalls zu einem Luxusgut geworden.
Wir müssen uns als Ärzte mit diesen allgemeinen Tenden-zen arrangieren. Dabei gilt es aber um so mehr, die wirk-lichen Inhalte unseres Berufs zu bewahren und Mensch-lichkeit auszuüben wo immer wir es können, vor allem inder Betreuung unserer Patienten. Dazu bedarf es gar nichtviel! Es bedarf einer Grundeinstellung, die von der Ach-tung des Menschen, und des kranken Menschen im be-sonderen, ausgeht. Es bedarf der Betrachtung des ganzenMenschen in seinen körperlichen und seelischen Dimen-sionen und der Vermeidung eines „Tunnelblicks“, der nurauf die bestehende Krankheit ausgerichtet ist. Es bedarfdes Verständnisses den Sorgen und Ängsten des krankenPatienten gegenüber und des Bemühens, diese Sorgenund Ängste zu lindern. Es bedarf schlicht und einfach„nur“ der Wahrnehmung eines anderen, kranken Men-schen.
Über diesen wesentlichen Grundeinstellungen ärztlichenHandelns darf aber nicht außer Acht gelassen werden,dass auch Ärzte nur Menschen sind. Dies bedeutet, dasses nicht bei jedem Patienten möglich sein wird, dieseGrundeinstellungen in der Praxis zu erfüllen. Dies bedeu-tet auch, dass Ärzte das Bewusstsein für Grenzen habenoder entwickeln müssen, für die Grenzen der Medizin imallgemeinen aber auch für ihre ganz persönlichen Gren-zen.
Die großen Erfolge der Medizin in den letzten Jahrzehn-ten, die der Entwicklung neuer Techniken und bahnbre-chender Erkenntnisse der Naturwissenschaften zu verdan-ken sind, verleiten dazu, den Blick für die Grenzen der Me-dizin zu verschleiern. Medizin ist jedoch keine Technik undsie ist auch keine Naturwissenschaft. Trotz aller Fortschrit-
te muss uns bewusst sein, dass Leben und Tod letztlich un-erklärbare und unverstehbare Phänomene sind und blei-ben. Leben und Tod sind jedoch die Eckpunkte innerhalbderer sich die Medizin bewegt und die die Grenzen unse-rer Möglichkeiten bestimmen. Die Achtung dieser Gren-zen und die Einsicht, dass auch die moderne Medizinnicht vor Krankheit und Tod bewahren kann, muss uns inunserem Handeln bestimmen. Dies bedeutet, dass verant-wortliches Arztsein darin bestehen kann, technisch undmedizinisch mögliche Maßnahmen zu unterlassen, wennsie dem Wohl des Patienten nicht mehr dienen und mög-licherweise nur Leiden und Sterben verlängern, und es be-deutet für diese Entscheidung die Verantwortung zuübernehmen.
Die Anerkennung der eigenen Grenzen ist manchmalnoch schwieriger umzusetzen. Sie ist aber von elementa-rer Wichtigkeit. So ist es in dem Bewusstein der Verant-wortung einem Patienten gegenüber notwendig, dieGrenze des eigenen Fachwissens zu kennen und gegebe-nenfalls zusätzlichen speziellen Rat einzuholen oder denPatienten an eine andere Institution weiterzuleiten. Fastnoch wichtiger ist die Anerkennung der eigenen physi-schen und psychischen Grenzen, die bei einem engagier-ten Arbeiten leicht aus dem Blickfeld geraten. Trotz deshohen Anspruchs an den Arzt und seiner Bereitschaft zurHilfeleistung besteht keine Verpflichtung zur Selbstaufga-be und zur Selbstaufopferung. Auch ein Arzt hat wie je-der Mensch ein Recht auf Leben, auf Familie, auf Freude,Freizeit und Genuss. Wie wichtig dieser Hinweis ist, wirddurch die hohe Zahl von Ärzten, aber auch von Pflegekräf-ten unterstrichen, die im Laufe ihres Berufslebens ein„Burn-out“-Syndrom oder eine schwere Depression erlei-den. Es steht zu befürchten, dass diese Zahl angesichts derkontinuierlich erschwerten Rahmenbedingungen für einemenschliche Medizin eher noch steigen wird. Davor gilt esuns selbst zu schützen. Wir sind als Ärzte nicht für die Rah-menbedingungen verantwortlich, unter denen wir zurZeit arbeiten müssen. Wir sind auch nicht dafür verant-wortlich, wenn medizinisch sinnvolle und notwendigeMaßnahmen von den Krankenkassen nicht anerkanntwerden. So sehr es uns schmerzt, unseren Patienten indiesen Situationen nicht die aus unserer Sicht beste Be-handlung zu kommen zu lassen, so sehr sollten wir unsdavor schützen, dafür die Verantwortung zu überneh-men. Seit der Gründung der Deutschen Gesellschaft fürInnere Medizin, eigentlich sogar seit Beginn des ärztlichenBerufsstandes ist viel über die moralischen und ethischenVerpflichtungen der Ärzte gesagt und geschrieben wor-den. Es erscheint heute an der Zeit, auch die Bedürfnissedes Arztes als Mensch zu formulieren und das Recht aufeigene Menschlichkeit einzufordern. Dies steht keinesfallsim Widerspruch zum ärztlichen Auftrag, dem krankenMenschen gegenüber, aber es definiert die Grenzen die-ser Aufgabe und unterstreicht die Achtung vor uns selbst.
Menschlichkeit ist daher ein unverzichtbares, aber kom-plexes Grundelement der ärztlichen Tätigkeit. Mensch-lichkeit macht den Unterschied aus zwischen einem Arztund einem Mediziner und definiert die Einstellung mit derwir unseren Beruf ausüben. Menschlichkeit dem Patientengegenüber darf jedoch nicht bedeuten, von uns selbst Un-menschliches zu verlangen.

�� SSeeiittee 1188 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
Hochleistungsmedizin und Menschlichkeit
Hochleistungsmedizin und Menschlichkeit sind kein Wi-derspruch! Sie sind vielmehr die beiden Grundelementeder ärztlichen Tätigkeit und daher Richtschnur unseresHandelns und Denkens. Hochleistungsmedizin erfordertintellektuelle Leistungsfähigkeit und die fortwährende Be-reitschaft zu Lernen. Menschlichkeit erfordert Wärme undZuwendung, Achtung und Respekt vor der Persönlichkeitdes kranken Menschen, aber auch vor uns selbst.
Korrespondenzadresse:
Prof. Dr. Wolfgang HiddemannMedizinische Klinik und Poliklinik IIIKlinikum der Ludwig-Maximilians-Universität MünchenCampus GroßhadernMarchioninistr. 1581377 MünchenTelefon 089-70952550e-Mail [email protected]
2. Erfurter Symposium für Pädiatrische Neuroonkologie und Neurochirurgie23. Januar 2010, 8.30 – 15.30 Uhr · Hotel Pullman Erfurt am Dom
Wissenschaftliches Programm
8.30 Uhr Begrüßung
8.45 Uhr Pathologische Einteilung kindlicher Hirntumoren und die Bedeutung molekularer Marker in der Therapiestratifizierung Prof. Dr. med. Th. Pietsch, Institut für Neuropathologie, Universitätsklinikum Bonn
9.15 Uhr Neuroradiologische Diagnostik bei ZNS-Tumoren Prof. Dr. med. Monika Warmuth-Metz, Abteilung für Neuroradiologie, Universitätsklinikum Würzburg
9.45 Uhr Pause
10.15 Uhr Klinik und elektrophysiologische DiagnostikProf. Dr. med. U. Brandl, Abteilung für Neuropädiatrie, Klinik für Kinder- und Jugendmedizin, Universitätsklinikum Jena
10.40 Uhr Indikation und chirurgische Behandlungsstrategien von sellären und parasellärenTumoren Prof. Dr. med. N. Soerensen, Klinik für Neurochirurgie, Evangelisches Krankenhaus Oldenburg
11.05 Uhr Entwicklung in der operativen Therapie hirneigener supra- und infratentorieller HirntumoreProf. Dr. med. R. Gerlach, Abteilung für vaskuläre, funktionelle und pädiatrische Neurochirurgie, Klinik für Neurochirurgie, HELIOS Klinikum Erfurt
11.30 Uhr Stellenwert der Brachytherapie bei pädiatrischen Hirntumoren Priv.-Doz. Dr. med. Aurelia Peraud, Neurochirurgische Klinik und Poliklinik, Universitätsklinikum Großhadern, München
11.55 Uhr Therapie spinaler Tumore bei KindernProf. Dr. med. Vera van Velthoven-Wurster, Abteilung für Allgemeine Neurochirurgie, Universitätsklinikum Freiburg
12.20 Uhr Mittagspause
13.20 Uhr NF2 – Neurochirurgisches Management im Kinder- und Jugendalter Prof. Dr. med. St. Rosahl, Klinik für Neurochirurgie, HELIOS Klinikum Erfurt
13.50 Uhr Aktuelle Konzepte der Chemotherapie maligner Hirntumoren des Kindes- und JugendaltersDr. med. A. von Büren, Klinik für Pädiatrische Hämatologie und Onkologie, Universitätsklinik Hamburg-Eppendorf
14.20 Uhr State of the Art der Strahlentherapie von Hirntumoren im Kindes- und JugendalterProf. Dr. med. R. D. Kortmann, Klinik für Strahlentherapie und Radioonkologie, Universitätsklinikum Leipzig
14.50 Uhr Protonentherapie – Indikationen und Möglichkeiten einer neuen Methode zur Behandlung von Hirntumoren Priv.-Doz. Dr. med. Beate Timmermann, Westdeutsches Protonentherapiezentrum Essen, Universitätsklinikum Essen
15.20 Uhr Schlusswort
Veranstalter: Tumorzentrum Erfurt e.V.
in Zusammenarbeit mit der Klinik für Kinder- und Jugendmedizin und der Klinik für Neurochirurgie, HELIOS Klinikum Erfurt
Fortbildungszertifikat der Landesärztekammer Thüringen: 7 Punkte, Kategorie A

��Wünsche eines KrebspatientenVortrag auf dem Symposium „Zwischen Evi-denz und Empathie – Dilemma oder Chancefür die Palliativmedizin“ am 20. Juni 2009 inBad Berka
Manfred HörschelmannGraitschen b. Bürgel
Wünsche sind Schecks, ausgestellt auf eine Bank, bei derwir kein Konto haben. (nach Oscar Wilde)
VorbemerkungIch zähle zu den Menschen, denen die Einweisung in eineKlinik als Horrortrip erscheint. Der Rucksack, den ich tra-ge, wird mit jedem Schritt in der Klinik schwerer.Fast alle, wie Sie hier versammelt sind, haben eine ande-re Beziehung zu einer Klinik. Fast immer ist es Ihr Arbeits-platz, sie üben dort Ihren Beruf aus. Bis auf wenige Aus-nahmen gilt für die hier Beschäftigten, dass sie Beruf alsBerufung auffassen. Darin besteht ein großer Unter-schied.Als Patient ist man eigentlich immer von einem mehr oderweniger gut organisierten System absolut abhängig.Wichtig ist mir: Ich komme nicht in die Palliativabteilung,weil ich hier sterben möchte. Ich komme in diese Klinik,weil ich Hilfe bekomme.
Der erste Wunsch: Hoffnung Wenn man sich einen Fußknochen bricht, humpelt man ineine Chirurgische Abteilung und wird geröntgt. Wurdeder Bruch festgestellt, darf man nicht mehr humpeln.Man muss zwingend in einen Krankenstuhl, bekommt jenach Schwere des Bruchs eine Behandlung und gilt nach3 bis x Wochen als geheilt. Nun humpelt man nur nochaus Pietät, aber der Knochen benimmt sich in der Regelso, wie er soll. Vor etwa 25 Jahren stellten sich bei mir erste Probleme mitder Prostata ein. Die Diagnose war erschwert; es gab inJena nur 4 Ultraschallgeräte. Die Wartezeiten waren ent-sprechend lang. Es nötigt mir noch heute Hochachtungab, dass die Ärzte auf solchen Geräten - grau in grau - et-was diagnostizieren konnten. Mit der Wende gab es reichlich Geräte und beim Urolo-gen konnte man so etwa alle 14 Tage einen rektalen Ul-traschall absolvieren, wobei nicht zwingend etwas er-kannt wurde. Biopsien gab es erst blind durch den Darm,später mit Ultraschall auf elegantere Weise. Ich habe solange gewartet, bis ich dort einen Untersuchungsterminbekam. Sechs Proben - sechs mal positiv. Etwas radikal Schlechtes als positiv zu kennzeichnen, istja eigentlich für den Patienten heimtückisch. Das war er-schütternd, aber von mir in der Tragweite nicht begriffen. 14 Tage nach der Untersuchung kam eine Radikaloperati-on in einer Klinik mit „indischer“ Toilette. „Indisch“ - siebefand sich jenseits des Ganges. Die Inkontinenz-Probleme wurden durch ein nettes Auf-klärungsgespräch ergänzt. Der Krebs war aggressiv, mangab mir zwei Wochen bis zur Orchiektomie usw. Nach 5Jahren hatte ich die Sache halbwegs überwunden undgalt nach den bestehenden Leitlinien als geheilt. Der PSA-
Wert hatte das nicht so mitbekommen und stieg langsam,aber monoton, an. Irgendwann wurde die behandelndeUrologin stutzig und ließ ein Szintigramm und ein MRTanfertigen. Die Auswertung war ernüchternd im doppel-ten Sinne: - Es gab Knochenmetastasen.- Es gab den netten Hinweis: „Heilen können wir sie nicht, aber das Leben verlängern.“
Vita brevis. - Das ganze Leben ist ja eigentlich nur Verlän-gerung desselben.
Nun folgten im Wechsel Bestrahlung, Chemo, Bestrah-lung, Chemo usw. bis die Blutwerte durch Spritzen wie-der etwas aufgemöbelt werden mussten. Die Schmerzenim Lendenbereich wurden so stark, dass ich nur noch ge-krümmt liegen konnten und förmlich auf allen Vieren vor-wärts kroch. Bitte beachten Sie, dass weder das Szinti-gramm noch die MRT-Auswertung die Ursache aufzeigenkonnten. Verstehen Sie, warum mein erster Wunsch Hoff-nung war? Ich wurde nach Bad Berka in die Palliativabteilung über-wiesen. Wieder Klinik! Wer weiß, was für Überraschungendort kommen?
Lebensqualität vs. Lebensverlängerung Mühsam mussten die Ärzte dort meine Aversion gegenMorphium abbauen. Es gelang. - Ein Hoffnungsschimmer.Die Schmerzen wurden erträglich. Der wichtigste Satz in den Gesprächen mit dem Arzt war:„Wir wollen ihnen helfen, Lebensqualität zu erhalten undzu verbessern.“ Nun kommt das Wesentliche: Man gabsich nicht mit dem MRT und dem Szintigramm zufrieden.Nach wenigen Untersuchungen stand fest, dass ein Wir-bel von den Metastasen zersetzt war und ersetzt werdensollte. Wenn Sie die Zustimmungserklärung lesen, in der dieOperation erläutert wird, kommt kaum Hoffnung auf. Ein Arzt kam und erläuterte mir nochmals, wie es tech-nisch abläuft. Die Klinikpsychologin und der Pfarrerschafften es, bei mir Hoffnung aufzubauen. Die Angst vorder Narkose war weg. Über das, was da chirurgisch ablief,denke ich lieber nicht nach. Es klappte aber. Nach einigenTagen konnte ich aufstehen, mich bewegen und dieSchmerzen nahmen ab. Hoffnung baute sich auf. Aller-dings wirkt der Krebs ja immer noch.
Der zweite Wunsch: Sprecht deutsch und menschlichmit uns!Unsere Zeit klebt an Zahlen und Messwerten. Man glaubt,man sei wissenschaftlich, wenn man in Prozenten redet.Was teilt der Arzt mit, wenn er sagt: „Die Heilungsquoteliegt bei 10%.“? Er könnte ja auch sagen: „Statistisch ge-sehen sterben 90 von 100 Patienten mit diesem Krebs“. Bitte machen Sie sich das klar, bevor sie mit Werten an denPatienten herantreten!10% heißt: zehn Anwesende von 100 dürfen gehen, dierestlichen dürfen sich auf das Sterben vorbereiten. Man-cher Politiker ist ja stolz, wenn 10% der Bevölkerung ihngewählt haben. Ärzte sind in der Regel seriöser. Zu viele Werte verunsichern, geben kaum Lebensqualitätund schaffen Frust. Selbst als Antwort auf die wenig sinn-volle Frage „Wie lange kann ich noch mit dem Krebs le-ben?“ ist das keine Antwort.
�� SSeeiittee 1199 ��JJOOUURRNNAALL 0022//22000099

�� SSeeiittee 2200 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
Ich kenne das Ende meines Lebens nicht. Kein Arzt kenntes. Er kann lediglich auf der Basis bisheriger Erfahrungenfolgern. Bitte beachten Sie: Das sind Plausibilitätsschlüs-se und die Prozentangaben machen sie logisch gesehennicht besser. Was habe ich als Patient von einem Fachchinesisch? Wenig! Alle Spezialbegriffe, die Namen der Medikamen-te sind für einen Laien, selbst wenn er etwas Latein kennt,Fremdsprache. Wenn man solche Fachbegriffe nicht alsRenommee beim Kaffeeklatsch braucht, ist der Nutzen ge-ring. Ich habe in den sechs Wochen Klinikaufenthalt oftgewünscht, dass die Damen und Herren ihren Informati-onsaustausch vor der Tür vollziehen und mit mir im gutenDeutsch über die Fragen reden, welche mich bewegen.
Der dritte Wunsch: Helft uns gegen die Mafia!Wenn Sie sich im Internet umsehen, bietet man bei KrebsMisteltherapien, Thymuspräparate, Hormon-Ersatzthera-pie sowie das Essen von bitteren Aprikosenkernen oderHimbeeren usw. an. Irgendwann geht es einem soschlecht, dass man diesem Druck erliegt. Wenn ich danneinen Arzt, bei dem ich schon fast 2 Jahre in Behandlungbin, frage, ob denn mit Ernährung, etwa die von Himbee-ren und bitteren Mandeln, etwas Förderliches gegen denKrebs getan werden kann, erwarte ich keine dumme Ant-wort, wie: „Woher soll die Krebszelle wissen, dass dieStoffe aus der Himbeere gegen sie sind?“. Bei der Chemo-therapie soll sie es aber wissen - oder? Sinnvolle Hinweise für die Ernährung können helfen. Ichbrauche keine Kochkurse, aber ich denke darüber nach,warum die einzelnen Kasten in Indien eine für die Kastetypische Ernährung haben. Mitunter wäre wohl auch beim Klinikessen selbst etwasmehr Phantasie angebracht. Wie sagte ein Patient: „Beidem Flugzeugfraß schnürt sich mir sofort der Magen zu.“Sicher etwas drastisch, aber das Essen in der touristic classist wohl nicht immer so wundervoll.
Obwohl ich schon über 20 Jahre mit dem Krebs lebe, binich erst seit März 2009 Patient in Bad Berka und in der Sta-tion für Palliativmedizin in guter Betreuung. Ich hätte mirgewünscht, so eine Betreuung schon zeitiger zu erfahren.Manche Angst und Sorge auch meiner Familie, wäre sovermieden worden. Es war Gottes Wille, dass mir dieserLichtblick nun vergönnt ist.
Wir - auch der Arzt - können nicht wissen, was morgen ist.Wir leben aus Routine oder Hoffnung und brauchen ei-gentlich ein Gefüge von Motiven.
Dazu fällt mir am Schluss Zarathustra (gemeint ist der Be-gründer einer Religion vor etwa 2700 Jahren) ein: Gute Gedanken bewirken gute Worte und gute Worte be-wirken gute Taten. Das Böse enthält immer einen Kern des Guten und das Gu-te wird siegen.
Korrespondenzadresse:
Dr. sc. nat. Manfred Hörschelmann Am Bahndamm 307616 Graitschen b. BürgelTelefon 036692-20699
�� Auffangen – Informieren – BegleitenFrauenselbsthilfe nach Krebs – längst auchfür Männer
Die Frauenselbsthilfe nach Krebs e.V. wurde 1976 vonbrustamputierten Frauen, die sich mit dem Schock derDiagnose Krebs allein gelassen fühlten, gegründet. Sie istdamit eine der ältesten und mit insgesamt rund 50.000Teilnehmern in derzeit 420 Gruppen, die in 12 Landesver-bänden organisiert sind, die größte Krebsselbsthilfeorga-nisation in Deutschland. Der Verband steht unter derSchirmherrschaft der Deutschen Krebshilfe e.V. und wirdvon ihr auch finanziell gefördert.
Die Mitglieder der Frauenselbsthilfe sind selbst an Krebserkrankt. Innerhalb der verschiedenen Verbandsebenensind sie ehrenamtlich tätig und stehen für Beratungen zurVerfügung. Die Beratung kann als Einzel- oder Gruppen-gespräch mit den Betroffenen und/oder Angehörigen beiBesuchen im Krankenhaus oder auch zu Hause durchge-führt werden Darüber hinaus gibt es telefonische und On-line-Beratungsangebote sowie verschiedene, ständig ak-tualisierte Informationsbroschüren.
Ziel der Arbeit ist es, Betroffene nach der Diagnose Krebsaufzufangen, sie über Hilfen zur Krankheitsbewältigungzu informieren und in ein Leben mit oder nach Krebs zubegleiten. Inzwischen ist der Verband auch ein wichtiger Interessen-vertreter der Krebskranken in gesundheits- und sozialpo-litischen Gremien.
Aus dem ehemaligen Selbsthilfeverband von Frauen mitBrustkrebs ist längst eine offene Selbsthilfeorganisationfür alle an einem Tumor Erkrankte geworden, in der sichauch immer mehr Männer aufgefangen und angenom-men fühlen. So ist es nicht verwunderlich, dass der Lan-desverband Thüringen der Frauenselbsthilfe nach Krebsinzwischen von einem Mann geleitet wird (siehe unten).
Kontaktdaten:
BundesgeschäftsstelleFrauenselbsthilfe nach Krebs Bundesverband e.V.Haus der KrebsselbsthilfeThomas-Mann-Str. 40 · 53111 BonnTelefon 0228-33889400Telefax 0228-33889401e-Mail [email protected]
Landesverband Thüringen e.V.Vorsitzender Hans-Jürgen MayerHelenenweg 15e · 98574 SchmalkaldenTelefon 03683-600545Telefax 03683-407460e-Mail [email protected]
In Mittel- und Westthüringen gibt es Gruppen in Bad Ber-ka, Bad Langensalza, Bad Salzungen, Eisenach, Erfurt, Go-tha, Mühlhausen, Schlotheim, Sömmerda, Tabarz, Wei-mar und Wölfis.
Ansprechpartner vermitteln auch die Kontakt- und Infor-mationsstellen für Selbsthilfegruppen (KISS) der Gesund-heitsämter und das Tumorzentrum Erfurt e.V.(www.tumorzentrum-erfurt.de)

�� SSeeiittee 2211 ��JJOOUURRNNAALL 0022//22000099
6-Punkte-Programm der Frauenselbsthilfe nach Krebs
Wir wollen:1. Krebskranke psycho-sozial begleitendurch menschliche Zuwendung in Einzelgesprächen und Aussprache in Selbsthilfegruppen gemeinsam ler-nen, mit Krebs zu leben
2. helfen, die Angst vor weiteren Untersuchungen und Behandlungen zu überwindenVermitteln von Hoffnung durch persönliche Erfahrung und eigenes Erleben
3. Vorschläge zur Stärkung der Widerstandskraft gebenaktuelle Vorträge von Fachleuten aus den verschiede-nen Bereichen des Gesundheitswesens u.a. Ernährung,Bewegung
4. die Lebensqualität verbessern helfenHilfe zur Selbsthilfe, Überwindung von Isolation, För-derung der Kreativität
5. informieren über soziale Hilfen, Versicherungs- undSchwerbehindertenrechtAnschlussheilbehandlung, Rehabilitation, Pfelgever-sicherung, Renten u.a.m.
6. die Interessen Krebskranker sozial- und gesund-heitspolitisch vertreten
��Wichtige Fortschritte bei Diagnoseund Behandlung von Brustkrebs inden letzten Jahren erreicht
M. Domrös, A. FindekleeLandesvertretung Thüringen, Verband der Ersatzkas-sen e.V. (vdek)
Brustkrebs ist bei Frauen die häufigste Todesursache un-ter den bösartigen Tumoren. Jährlich sterben in Deutsch-land ca. 46.000 Frauen an dieser Erkrankung, davon sind17.000 unter 60 Jahre. Brustkrebs verursacht bei Frauenden höchsten krebsbedingten Verlust an Lebensjahren.Diese wenigen Zahlen zeigen, dass gerade bei dieser Er-krankung Handlungsbedarf in unserem Gesundheitssy-stem besteht. Bereits 2001 stellte der Sachverständigen-rat in allen Bereichen der Versorgung (Screening, Diagno-stik, Therapie, Nachsorge) eine große Varianz fest, die sichin Über-, Unter- und Fehlversorgung manifestierte.
Ein großer Fortschritt zur Überwindung dieser unbefriedi-genden Situation war bereits die bundesweite Einführungdes Mammographie-Screenings als Leistung der Gesetzli-chen Krankenversicherung für alle Patientinnen im Alterzwischen 50 und 69 Jahren. Dazu wurde 2002 beschlos-sen, in Deutschland ein qualitätsgesichertes Mammogra-phie-Screening-Programm zu etablieren. Damit wurde
auch auf die Erfahrungen anderer Länder reagiert, die be-reits zuvor derartige Screening-Programme aufgelegt hat-ten. So lag die Rate der Todesfälle bei Brustkrebs bspw. inSchweden signifikant unter der in Deutschland. Das Mammographie-Screening ist mittlerweile flächen-deckend in der gesamten Bundesrepublik eingeführt.Auch in Thüringen ist nach einigen Anlaufschwierigkeitenseit Anfang d. J. die Flächendeckung sichergestellt. Nach-dem bereits 2007 die erste Screening-Einheit in Jena (Dr. Wurdinger) ihre Tätigkeit aufnahm, ist es zwischen-zeitlich auch im Westthüringer Bereich durch die Scree-ning-Einheit in Bad Langensalza (Dr. Buse) möglich, für al-le anspruchsberechtigten Frauen das Mammographie-Screening anzubieten.
Auf der Grundlage eines zielgerichteten Einladungsver-fahrens werden alle Frauen zwischen 50 und 69 Jahren –auch wenn sie nicht in der GKV versichert sind - zumMammographie-Screening eingeladen. Die Auswertungaller Aufnahmen durch zwei voneinander unabhängigeBefunde ist dabei sichergestellt. Die Teilnahmequote inThüringen hat sich mittlerweile bei ca. 55 % eingepegelt.Dieses Ergebnis kann nicht befriedigen, da es unseren An-sprüchen an ein umfassendes und hoch effizientes Versor-gungsangebot noch nicht entspricht. Zielgröße muss ei-ne Teilnahmequote von mind. 70 % aller Anspruchsbe-rechtigten und eingeladenen Frauen sein. Dabei werden bereits vielfältige Anstrengungen unter-nommen, um auch in der Praxis eine hohe Akzeptanz beiden Versicherten zu organisieren. In der GeschäftsstelleMammographie-Screening ist es möglich, Termine indivi-duell zu gestalten. Mehrere Mitarbeiterinnen im Callcen-ter stehen dafür bis in die Abendstunden für die Klientin-nen zur Verfügung. Außerdem gibt es in Thüringen nebenden stationären Mammographie-Einheiten in Altenburg,Eisenach, Erfurt, Gera, Jena, Meiningen und Bad Langen-salza derzeit vier mobile Mammographie-Einheiten(Mammobile), die in der Fläche vor Ort ein Screening-An-gebot unterbreiten.
Dennoch sind schon erste positive Resultate des Scree-nings vorweisbar. Bereits mit der Einführung hat der Ge-setzgeber die Evaluierung des Mammographie-Screening-Programms verfügt. Die hierzu erhobenen Daten sollenAufschluss über den Verlauf und die Ergebnisse des Scree-ning-Programms liefern.
Die Kooperationsgemeinschaft Mammographie-Scree-ning auf der Bundesebene erstellt über die Ergebnisse re-gelmäßig einen Evaluationsbericht. Der erste Evaluations-bericht wurde im September 2009 vorgestellt. In dem Be-richt sind die Auswertungen des Mammographie-Scree-nings von bundesweit 77 Screening-Einheiten aus denersten 3 Jahren des Programms von 2005 bis 2007 zusam-mengefasst. Die Ergebnisse im Bericht belegen, dass diestrengen Qualitätsvorgaben im Programm auch erfülltwerden. So werden durch das Mammographie-Screeningwesentlich häufiger kleine Tumoren aufgespürt. Der An-

�� SSeeiittee 2222 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
teil der invasiven Karzinome von einer max. Größe bis 10 mm liegt im Screening bei gut 30 %. Vor Einführungdes Mammographie-Screenings waren es nur rd. 14 %. Bei 76,6 % aller im Rahmen des Screening-Programmsentdeckten invasiven Karzinome waren die Lymphknotennoch nicht befallen. Der Ausgangswert vor Beginn desScreenings lag mit 49 % deutlich darunter. Somit konntedie Kooperationsgemeinschaft zu dem Ergebnis gelan-gen, dass für Frauen mit kleinen Tumoren ohne Metasta-sen sehr gute Chancen bestehen, vollständig geheilt zuwerden. Die betroffenen Frauen profitieren außerdemvon einer schonenderen und meistens brusterhaltendenTherapie. Trotz aller vor Einführung des Mammographie-Screenings geführten Diskussionen über Vor- und Nach-teile des neuartigen Programms überwiegen nach unse-rer Überzeugung die Vorteile deutlich gegenüber den Ri-siken.
Fortschritte hat es in den letzten Jahren aber nicht nur beider Früherkennung von Brustkrebs gegeben. Durch dieEinführung des Disease-Management-Programms (DMP)Brustkrebs konnten ebenso bei der kurativen Versorgungzum Wohle der Patientinnen gemacht werden.
Auch bei der Etablierung dieses DMP musste zunächst ei-nige Skepsis überwunden werden, schließlich weist die Er-krankung Brustkrebs deutliche Spezifika gegenüber denansonsten im DMP gemanagten klassischen chronischenErkrankungen, wie Diabetes mellitus, KHK bzw. Asthma/COPD auf. Erkannte Defizite in der Patientenversorgungbelegten aber auch hier die Handlungsnotwendigkeiten:- die Zugangsmöglichkeit zu optimaler medizinischer
Versorgung und psychosozialer Betreuung war oft-mals zufallsabhängig,
- regional uneinheitliche Strukturen der Versorgung mit unterschiedlicher Behandlungsqualität,
- in Praxen und Kliniken fehlte es an einheitlichen Dia-gnose- und Therapievorgaben, verbindlicher Qualitäts-kontrolle und anerkannten Versorgungskonzepten,
- Mängel in der Koordination der Versorgung und in der Kommunikation zwischen verschiedenen Disziplinen und Versorgungsstufen,
- unzureichende Informationen über medizinische Be-handlungsschritte sowie psychosoziale Betreuungsan-gebote.
Nach Einschätzung der Deutschen Krebsgesellschaft wur-de prognostiziert, dass allein durch eine bessere Organi-sation und eine interdisziplinär ganzheitliche Betreuungin Deutschland jährlich bis zu 10 % mehr Brustkrebspa-tientinnen gerettet werden könnten.
Das DMP Brustkrebs ist darauf angelegt, durch die Bereit-stellung evidenzbasierter Leitlinien, die Empfehlung vali-der diagnostischer Maßnahmen und Nutzen belegterWirkstoffe, vorzeitige und effektive Fokussierung auf Be-gleit- bzw. Folgeerkrankungen, Transparenz durch Doku-mentationen sowie aktive Mitarbeit der Patientinnen dengenannten Versorgungsdefiziten entgegen zu wirken. Da-
für wurden klare Schnittstellen zwischen den Versor-gungsbereichen definiert, deren optimierte Zusammenar-beit gefördert und der Informationsfluss über Sektoren-grenzen hinweg organisiert.
Der „Behandlungspfad“ von der histologischen Diagnose-sicherung über die Primärtherapie und Nachsorge ist re-gelhaft auf 5 ½ Jahre angelegt, wobei Operation und Ein-leitung der adjuvanten Therapie im Rahmen der Primär-therapie im Krankenhaus erfolgt. Daran anschließendwird unter Federführung des ProgrammverantwortlichenDMP-Arztes zur Koordination der erforderlichen Diagno-stik und Therapie auf das Zusammenwirken von Gynäko-logen, Radiologen, Onkologen, Klinik und sonstige Lei-stungserbringer abgestellt.
Der entsprechende DMP-Vertrag wurde in Thüringen zwi-schen Kassenverbänden und Kassenärztlicher Vereinigung2004 geschlossen und mittlerweile bis 2014 verlängert. Inder Anlage zum DMP-Vertrag sind die Versorgungsinhal-te zur Behandlung von Patientinnen mit Brustkrebs detail-liert definiert. Diese nehmen Bezug auf die gesetzlichenRegelungen der so genannten Risikostrukturausgleichs-verordnung (RSAV) in ihrer aktuellen Fassung. Vorange-stellt ist dem der zentrale Anspruch, die Behandlung nachdem aktuellen Stand der medizinischen Wissenschaft un-ter Berücksichtigung von evidenzbasierten Leitlinien odernach der jeweils besten verfügbaren Evidenz sowie unterBerücksichtigung des jeweiligen Versorgungssektors zuorganisieren.
Konkrete Vorgaben werden dann zur Diagnose, Therapie,Nachsorge, Palliativtherapie, Rehabilitation und Koopera-tion der Versorgungssektoren gemacht. Das zuständigeBundesversicherungsamt hat die entsprechende Zulas-sung des Programms erteilt.
In das DMP Brustkrebs haben sich in Thüringen derzeitüber 3.500 Patientinnen eingeschrieben, deren Teilnahmeam Programm freiwillig ist. Sie werden von etwa 200DMP-Ärzten betreut, in der Regel Fachärzte für Frauen-heilkunde und Geburtshilfe mit der Anerkennung „onko-logisch verantwortlicher Arzt“ oder der Teilnahme amFortbildungscurriculum „Brustkrebs“ und jährlicher brust-krebsspezifischer Fortbildung. Zusätzlich sind aus demstationären Bereich die hoch spezialisierten Brustzentrenam Waldklinikum Gera, dem Universitätsklinikum Jena,des Südharzkrankenhauses Nordhausen, des HELIOS Kli-nikums Erfurt und des SRH Zentralklinikums Suhl sekto-renübergreifend beteiligt.
Damit sind die strukturellen Voraussetzungen für einehochwertige evidenzbasierte Behandlung der Brustpa-tientinnen geschaffen. Spannend ist jedoch auch, wie sichdie neuen Rahmenbedingungen auf die Prozess- und Er-gebnisqualität ausgewirkt haben.
Instrumentarien der ärztlichen Qualitätssicherung sind fe-ster Bestandteil des DMP-Vertrages. Die wesentlichen

�� SSeeiittee 2233 ��JJOOUURRNNAALL 0022//22000099
�� Gemeinsames Veranstaltungsver-zeichnis
von Medizinisch-wissenschaftlicher Gesellschaft Erfurt e.V.HELIOS Klinikum Erfurt GmbH und Tumorzentrum Erfurt e.V.
Liebe Kolleginnen und Kollegen,
wir möchten Ihre gezielten und konzentrierten Fortbildungsaktivitätenmit einem gemeinsamen Veranstaltungsverzeichnis unterstützen und Ih-nen ein breites Spektrum zertifizierter und hoffentlich für Sie interessan-ter Fort- und Weiterbildungen anbieten. Die nachstehende Kurzfassung kann weder vollständig sein, noch um-fassend informieren. Sie soll als Orientierungshilfe dienen und Sie ani-mieren, alle weiteren Informationen und die laufenden Aktualisie-run-gen auf der Internetseite www.mwg-erfurt.de nachzulesen und / oderdirekt bei den Organisatoren zu erfragen. Über eine zahlreiche Teilnahme an den Veranstaltungen, rege Diskussio-nen sowie die Vertiefung und Ausweitung persönlicher Kontakte freuenwir uns besonders.
PD Dr. med. Prof. Dr. med. Prof. Dr. med.K. Hamm B. Ulshöfer D. EßerVorsitzender Vorsitzender Ärztlicher DirektorMWG e.V. Tumorzentrum Erfurt e.V. HELIOS Klinikum Erfurt
Januar 2010
13.01.2010, 16.00 UhrHELIOS Klinikum Erfurt, Seminarraum der Klinik für NeurologieNeurochirurgie-Primer für NeurologenProf. Dr. med. St. RosahlHELIOS Klinikum Erfurt, Klinik für Neurochirurgie
15. – 17.01.2010Leonardo Hotel Weimar (ehem. Hilton), Belvederer Allee 25, Weimar20. Gemeinsame Arbeitstagung „Angiologie interdisziplinär“HELIOS Klinikum Erfurt, Institut für diagnostische und interventionelleRadiologie und Neuroradiologie, Gefäßzentrum
16.01.2010, 9.00 bis 16.00 UhrUniversität Erfurt, AudimaxPolytraumtagHELIOS Klinikum Erfurt, Klinik für Unfall-, Hand- und Wiederherstel-lungschirurgie, Klinik für Anästhesie, Intensivmedizin und Schmerzthe-rapie
23.01.2010, 8.00 bis 15.30 UhrHotel Pullmann Erfurt am Dom2. Erfurter Symposium für Padiatrische Neuroonkologie undNeurochirurgieTumorzentrum Erfurt e.V. in Zusammenarbeit mit der Klinik fürKinder- und Jugendmedizin und der Klinik für Neurochirurgie,HELIOS Klinikum ErfurtFortbildungszertifikat LÄK Thüringen, 7 Punkte, Kategorie A
Februar 2010
27.02.2010, 9.00 bis 14.00 Uhr2. Gothaer GynäkologentagTumorzentrum Erfurt e.V. in Zusammenarbeit mit der Klinik fürGynäkologie und Geburtshilfe, HELIOS Brustzentrum Gotha,HELIOS Kreiskrankenhaus Gotha-Ohrdruf
Maßnahmen sind neben den jährlichen Fortbildungsver-anstaltungen die jährlich zu erstellenden Feedbackberich-te für die Ärzte und der Qualitätsbericht der gemeinsa-men Einrichtung von Krankenkassen und KV. Mittlerweileliegen drei Qualitätsberichte vor, der letzte für das Jahr2008. Hierbei ergibt sich ein differenziertes – aber auchdurchaus positives – Bild. Nicht alle Ziele konnten in Thü-ringen bereits erreicht werden.
Das sehr ambitionierte Ziel, nach 1 Jahr DMP-Laufzeit min-destens 70 %, nach 3 Jahren mindestens 80 % und nach5 Jahren mindestens 85 % brusterhaltende Operationendurchzuführen, wurde verfehlt. Im Jahr 2008 gelang esbei 77,2 % der neu eingeschriebenen Patientinnen; seitBeginn des DMP bei 76,6 % aller derzeit eingeschriebenenPatientinnen. Gleichwohl ist eine positive Tendenz erkenn-bar, lagen die entsprechenden Werte in den voran gegan-genen Berichten bei 72,6 % bzw. 75,8 %. Ebenfalls knappverfehlt mit aufsteigender Tendenz wurde das Ziel der Ent-fernung von mindestens 10 Lymphknoten auf alle Patien-tinnen mit invasivem Tumor (82,2 % vs. 90 %). Erreichtwurden die Qualitätsziele Hormonrezeptoranalyse, Nach-bestrahlung nach brusterhaltender Therapie und adju-vante endokrine Therapie.
Insgesamt zeigen diese Daten, dass das DMP Brustkrebspositive Ergebnisse vorweisen kann. Die Versorgung derBrustkrebspatientinnen hat sich verbessert und das inve-stierte Geld der Krankenkassen scheint gut angelegt zusein. Dabei verstehen wir den Kampf gegen den Brust-krebs als kontinuierlichen Prozess. Mammographie-Scree-ning und DMP sind wichtige Elemente. Entscheidend wirdaber auch sein, die Versorgung im Zusammenwirken vonÄrzte, Krankenhäusern und Krankenkassen weiter zu op-timieren, um zum Wohl der Patienten unser Gesundheits-system leistungsfähig und finanzierbar zu erhalten.
Korrespondenzadresse:
Michael DomrösLandesvertretung ThüringenVerband der Ersatzkassen e.V. (vdek)Lucas-Cranach-Platz 299099 ErfurtTelefon 0361-442520e-Mail [email protected]

�� SSeeiittee 2244 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
März 2010
03.03.2010, 17.00 bis 20.00 UhrKleine Synagoge ErfurtSymposium „Kolorektale Karzinome“Darmzentrum Erfurt am Katholischen Krankenhaus Erfurt in Zusam-menarbeit mit dem Tumorzentrum Erfurt e.V.
April 2010
24.04.2010, 16.00 bis 20.00 UhrHELIOS Klinikum Erfurt, Auditorium2. Erfurter Frühjahrstagung „Interdisziplinäre Hauttumor-therapie“HELIOS Klinikum Erfurt, Klinik für Hautkrankheiten und Allergologie,HELIOS Klinikum Erfurt in Zusammenarbeit mit dem TumorzentrumErfurt e.V.
Mai 2010
26.05.2010, 19.00 bis 20.30 UhrHELIOS Klinikum Erfurt, Auditorium35. Erfurter Fortbildung Hämatologie und Onkologie fürKrankenschwestern und -pflegerThema: Das Maligne MelanomReferent: Prof. Dr. med. R. Herbst, Chefarzt, Klinik für Hautkrank-heiten und Allergologie, HELIOS Hauttumorzentrum Erfurt, HELIOS Klinikum ErfurtTumorzentrum Erfurt e.V.
28.-29.05.2010Augustinerkloster ErfurtSymposium Neuromedizin 2010 (Neurochirurgie/ Neurologie/Neuroradiologie/ Radiochirurgie)28.05. 10 Jahre Novalis-Radiochirurgie in Erfurt
PD Dr. med. K. Hamm, HELIOS Klinikum Erfurt, Abteilungfür Stereotaktische Neurochirurgie und Radiochirurgie
29.05. Entzündungen und Infektionen des NervensystemsPD Dr. med. A. Steinbrecher, HELIOS Klinikum Erfurt,Klinik für Neurologie
Juni 2010
12.06.2010, 9.00 bis 14.00 UhrZentralklinik Bad Berka, Station 33, Robert-Koch-Allee 9, Bad BerkaSymposium „Palliativmedizin“Klinik für Palliativmedizin, Zentralklinik Bad Berka, in Zusammenarbeitmit dem Tumorzentrum Erfurt e.V.
16.06.2010, 17.00 bis 20.00 UhrVictor’s Residenz-Hotel Erfurt20. Erfurter Fortbildung Hämatologie und OnkologieTumorzentrum Erfurt e.V. in Zusammenarbeit mit der 4. Medizini-schen Klinik und der Klinik für Kinder- und Jugendmedizin,HELIOS Klinikum Erfurt
Oktober 2010
06.10.2010, 17.00 bis 20.00 UhrVictor’s Residenz-Hotel ErfurtSymposium „Gynäkologische Onkologie“Tumorzentrum Erfurt e.V. in Zusammenarbeit mit der Klinik fürFrauenheilkunde und Geburtshilfe, HELIOS Klinikum Erfurt
November 2010
05.-06.11.2010Haus Hainstein Eisenach23. Onkologische KonferenzTumorzentrum Erfurt e.V. in Kooperation mit der Medizinisch-Wissenschaftlichen Gesellschaft Erfurt e.V.
Dezember 2010
04.12.2010, 9.00 bis 18.00 UhrHELIOS Klinikum Erfurt, Auditorium12. Jahrestagung der Südostdeutschen Dermatologen /2. Erfurter HerbsttagungHELIOS Klinikum Erfurt, Klinik für Hautkrankheiten und Allergologie,HELIOS Hauttumorzentrum Erfurt in Zusammenarbeit mit dem Tumorzentrum Erfurt e.V.
KONTAKTADRESSEN:
Medizinisch-wissenschaftliche Gesellschaft Erfurt e.V.Vorsitzender Priv.-Doz. Dr. med. Klaus HammNordhäuser Straße 74 · 99089 ErfurtTelefon: 03 61 / 7 81-67 18Telefax: 03 61 / 7 81-67 19e-Mail: [email protected]
HELIOS Klinikum ErfurtPressesprecherin Brigitte KohlbergNordhäuser Straße 74 · 99089 ErfurtTelefon: 03 61 / 7 81-10 31Telefax: 03 61 / 7 81-10 32e-Mail: [email protected]/erfurt
Tumorzentrum Erfurt e.V.Geschäftsführer Dr. Hubert GöbelNordhäuser Straße 74 · 99089 ErfurtTelefon: 03 61 / 7 81-48 06Telefax: 03 61 / 7 81-48 03e-Mail: [email protected]
Sie können die Arbeit des Tumorzentrum Erfurt e.V.
durch Ihre Spende unterstützen!
Sparkasse Mittelthüringen
BLZ 820 510 00 · Konto-Nr. 130 123 609(Spenden sind steuerlich begünstigt!

�� SSeeiittee 2255 ��JJOOUURRNNAALL 0022//22000099
�� ANGEBOTE DES
TUMORZENTRUM ERFURT e.V.
KONSILARDIENSTE• Interdisziplinäres onkologisches KonsilJeden Mittwoch, 7.30 Uhr, Demo-Raum C 1.400 des Insti-
tuts für bildgebende Diagnostik, Hauptgebäude 1. OG,
HELIOS Klinikum Erfurt, Nordhäuser Straße 74
Anmeldungen über Telefon 03 61 / 7 81-48 02
Leitung: Prof. Dr. Herold / Prof. Dr. Ulshöfer
Jeder Arzt kann seine onkologischen Fälle persönlich ei-
nem Gremium von Experten aller Fachdisziplinen vorstel-
len. Am Ende der (kostenfreien) Beratung erhält er eine
konkrete Therapieempfehlung. Zu jeder Fallbesprechung
wird ein Protokoll angefertigt, das dem vorstellenden Arzt
und eventuellen mitbehandelnden Ärzten zugeht.
• Telefonischer KonsilardienstUnkompliziertes Vermitteln von Kontakten zu den
speziellen onkologischen Ansprechpartnern aller Fachge-
biete
f www.tumorzentrum.de
ONKOLOGISCHE LEITLINIENHilfestellung bei der Umsetzung der aktuellen Diagno-
se-, Therapie- und Nachsorgeleitlinien der Deutschen
Krebsgesellschaft und der medizinischen Fachgesellschaf-
ten.
In Ergänzung und zur praktischen Durchführung werden
diese wo nötig für die speziellen regionalen Bedingungen
adaptiert.
KONTAKTE ZU SELBSTHILFEGRUPPEN UNDHOSPIZDIENSTEN IN DER REGION
PSYCHOLOGISCHE BETREUUNGBetreuungsangebote für stationäre Patienten des HELIOS
Klinikum Erfurt sowie für Ärzte und Pflegepersonal.
FORT- UND WEITERBILDUNG• Ärzte
• Krankenschwestern und -pfleger
• Sozialdienste
DOKUMENTATION• Klinische TumordokumentationIn Erfüllung des Qualitätssicherungsauftrages des Sozial-gesetzbuches (SGB V) wird für jeden Patienten der gesam-te Krankheitsverlauf nach anerkannten Regeln (Tumorba-sisdokumentation) dokumentiert. Die Unterlagen stehendem Patienten und ihren behandelnden Ärzten zur Verfü-gung. Im Einzelfall (bei Umzug, Arztwechsel, Verlust vonOriginalunterlagen) sind sie für den Arzt eine unschätzba-re Hilfe.
• Gemeinsames Krebsregister der neuen Bundesländer
Epidemiologisch relevante Daten werden entsprechendgeltender Gesetze an das Gemeinsame Krebsregister derneuen Bundesländer weitergegeben.Mehr als 95 % der Meldungen des Einzugsgebietes kom-men vom Tumorzentrum. Diese Daten werden regelmä-ßig mit den amtlichen Sterbedaten abgeglichen und ste-hen dem meldenden Einrichtungen zur Verfügung.
SERVICE• Unterstützung der Nachbetreuung, Erinnerungsfunktion
Auf persönlichen Wunsch werden Patienten (und ihre be-treuenden Ärzte) an vereinbarte bzw. vergessene Nach-sorgetermine erinnert.
• Statistiken für Krankenhäuser und PraxenErstellung von Übersichten, Leistungsstatistiken undÜberlebenszeitanalysen für die von der jeweiligen Ein-richtung betreuten Patienten.
• InformationenKostenlose Bereitstellung von Tumor-Nachsorgepässenund Informationsmaterialien für Patienten, Ärzte, Pflege-
personal und Sozialdienste
�� HIER ERREICHEN SIE UNS
HELIOS Klinikum Erfurt GmbH
Haus 8, Nordhäuser Straße 74, 99089 Erfurt
Telefon: 03 61 / 7 81-48 02
Telefax: 03 61 / 7 81-48 03
E-Mail: [email protected]
Homepage: http://www.tumorzentrum-erfurt.de
Geschäftsführer: Dr. rer. nat. Hubert Göbel

��WISSENSCHAFTLICHER BEIRAT
Prof. Dr. med. Hartwig Kosmehl (Vorsitzender)Chefarzt, Institut für Pathologie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-27 50
Dr. med. Joachim BechlerChefarzt, Abteilung Gynäkologie und Geburtshilfe, HELIOS Kreiskrankenhaus Gotha / OhrdrufTelefon: 0 36 21 / 2 20-2 49
Adjunct Professor Dr. med. Rainer Bonnet M.D. Dpt. of Medicine, Loma Linda Univ., CaliforniaChefarzt, Klinik für Pneumologie, Zentralklinik Bad BerkaTelefon: 03 64 58 / 5 15 00
Michael DomrösLeiter der Landesvertretung ThüringenVerband der Ersatzkassen e.V. (vdek)Lucas-Cranach-Platz 2, 99099 ErfurtTelefon: 03 61 / 4 42 52 11
Dr. med. Alexander FichteUrologe, Geschwister-Scholl-Straße 6, 99085 ErfurtTelefon: 03 61 / 6 43 73 03
Dipl.-Med. Susanne KöhlerOberärztin, Innere Medizin I, HELIOS KreiskrankenhausGotha / OhrdrufTelefon: 0 36 21 / 2 20-1 30
Priv.-Doz. Dr. med. Klaus HammLeiter der Abteilung Stereotaktische Neurochirurgie undRadiochirurgie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-67 18
Prof. Dr. med. Udo B. HoymeDirektor, Klinik für Frauenheilkunde und Geburtshilfe,HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-40 00
Prof. Dr. med. Ruthild Linseem. Chefärztin, Klinik für Hautkrankheiten, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-43 00
Dr. med. André NematChefarzt, Klinik für Thoraxchirurgie und Thorakale Endo-skopie, Thoraxzentrum, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-25 90
Priv.-Doz. Dr. med. Günter OrtmannOberarzt, Klinik für Chirurgie, Hufeland Klinikum, Stand-ort Bad LangensalzaTelefon: 0 36 03 / 8 55-0
Prof. Dr. med. Dr. med. dent. Hans PistnerChefarzt, Klinik für Mund-, Kiefer- und Gesichtschirur-gie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-22 30
Dr. med. Stefan ReinschOberarzt, Klinik für Hals-, Nasen- und Ohrenheilkunde,HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-63 01
Prof. Dr. med. Steffen RosahlChefarzt, Klinik für Neurochirurgie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-22 60
Prof. Dr. med. Axel SauerbreyChefarzt, Klinik für Kinder- und Jugendmedizin, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-45 00
�� VORSTANDProf. Dr. med. Berthold Ulshöfer (Vorsitzender)Chefarzt, Klinik für Urologie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-22 00
Prof. Dr. med. Michel Herold (Stellvertr. Vorsitzender)Chefarzt, 4. Medizinische Klinik, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-25 66
Prof. Dr. med. Dirk EßerChefarzt, Klinik für Hals-, Nasen- und Ohrenheilkunde,HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-21 00
Prof. Dr. med. Hartwig KosmehlChefarzt, Institut für Pathologie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-27 50
Dr. med. Christina MüllerChefärztin, Klinik für Palliativmedizin, Zentralklinik Bad BerkaTelefon: 03 64 58 / 5 19 00
Priv.-Doz. Dr. med. Ulrike SchalldachChefärztin, Klinik für Strahlentherapie und Radioonkologie, HELIOS Klinikum ErfurtTelefon: 03 61 / 7 81-24 00
Dr. med. Jörg WenigerHämatologe und internistischer Onkologe,Geschwister-Scholl-Straße 6, 99085 ErfurtTelefon: 03 61 / 5 66 78 19
�� SSeeiittee 2266 �� JJOOUURRNNAALL 0011//22000055JJOOUURRNNAALL 0022//22000099
IMPRESSUM
ISSN 1868-291X (Print-Ausgabe)ISSN 1868-2928 (Internet)
�� Herausgeber: Tumorzentrum Erfurt e.V.
�� Redaktion: Prof. Dr. med. Hartwig Kosmehl · Dr. rer. nat. Hubert Göbel
�� Redaktionsbüro und Versand:Tumorzentrum Erfurt e.V.
Nordhäuser Straße 74 · 99089 ErfurtTelefon: 03 61 / 7 81-48 02 · Telefax: 03 61 / 7 81-48 03
E-Mail: [email protected]
�� Layout, Satz und Druck: Handmann Werbung GmbH Erfurt
�� Hinweis: Das Tumorzentrum Erfurt erstellt die Artikel nach bestem
Wissen und Gewissen. Die Verantwortung für den Inhalt der medizinischen und wissenschaftlichen Beiträge obliegt den Autoren. Sie stellen keine Handlungsempfehlungen für den
individuellen Fall dar.

Perspektiven eröffnen – SUTENT® bei mRCC und GIST *
Überleben mit SUTENT®
26,4 Monate in der First-Line bei mRCC 1
17,2 Monate bei GIST nach Imatinibversagen/-unverträglichkeit 2
1. Figlin RA, Hutson TE, Tomczak P, et al. Overall survival with sunitinib versus IFN-alpha as fi rst-line treatment of mRCC. J Clin Oncol. 2008 May; 26(15SI):256s, Abstract 5024.
2. Demetri GD, Huang X, Garrett CR, et al. Novel statistical analysis of long-term survival to account for crossover in a phase III trial of Sunitinib versus placebo in advanced GIST after imatinib failure. J Clin Oncol. 2008 May; 26(15SI):559s, Abstract 10524.
* SUTENT® ist zugelassen zur Behandlung von fortgeschrittenem/metastasiertem RCC sowie zur Behandlung nicht resezierbarer und/oder metastasierter maligner gastrointestinaler Stromatumoren (GIST), wenn eine Behandlung mit Imatinibmesylat wegen Resistenz oder Unverträglichkeit fehlgeschlagen ist.
Sutent® 12,5 mg/25 mg/37,5 mg/50 mg Hartkapseln. Wirkstoff: Sunitinib. Zusammensetzung: Wirkstoff: Eine Hartkapsel enthält Sunitinibmalat, entsprechend 12,5 mg/25 mg/50 mg Sunitinib. Sonstige Bestandteile: Mannitol (Ph.Eur.), Croscarmellose-Natrium, Povidon (K 25), Magnesiumstearat (Ph.Eur.), Gelatine, Eisen(III)-oxid (E 172), Titandioxid (E 171), Schellack, Propylenglycol, Natriumhydroxid; -25 mg/-37,5 mg/-50 mg zusätzl.: Eisen(III)-hydroxid-oxid x H2O (E 172), Eisen(II,III)-oxid (E 172). Anwendungsgebiete: Zur Behandlung nicht resezierbarer u./od. metastasierter maligner gastrointestinaler Stromatumoren (GIST), wenn eine Behandlung mit Imatinibmesylat wegen Resistenz oder Unverträglichkeit fehlgeschlagen ist. Zur Behandl. fortgeschrittener metastasierter Nierenzellkarzinome (MRCC). Gegenanzeigen: Überempfi ndlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Die wichtigsten schweren Nebenwirkungen bei der Behandlung von Patienten mit soliden Tumoren waren Lungenembolie (1 %), Thrombozytopenie (1 %), Tumor-Hämorrhagie (0,9 %), fi ebrige Neutropenie (0,4 %) und Hypertonie (0,4 %). Die häufi gsten (bei mind. 20 % der Patienten) Nebenwirkungen aller Schweregrade umfassen u. A. Erschöpfung; gastrointestinale Beschwerden wie etwa Durchfall, Übelkeit, Stomatitis, Oberbauchbeschwerden und Erbrechen; Verfärbung der Haut; Dysgeusie und Anorexie. Bei Patienten mit soliden Tumoren waren Erschöpfung, Hypertonie und Neutropenie die häufi gsten Nebenwirkungen im Schweregrad 3 und eine erhöhte Lipase das häufi gste Nebenwirkung im Schweregrad 4. Hepatitis und Leberversagen traten bei weniger als 1 % der Patienten auf und eine Verlängerung des QT-Intervalls bei weniger als 0,1 %. Ereignisse mit tödlichem Ausgang umfassten u. a. Multiorganversagen, disseminierte intravaskuläre Koagulation, peritoneale Blutungen, Rhabdomyolyse, Apoplex, Dehydrierung, Nebenniereninsuffi zienz, Nierenversagen, akute respiratorische Insuffi zienz, Pleuraerguss, Pneumo thorax, Schock und plötzlicher Tod. In GIST-Studien: Sehr häufi g: Anämie, Neutropenie, Thrombozytopenie; Hypothyreose; Appetitlosigkeit; Beeinträchtigung des Geschmackssinns, Kopfschmerzen; Hypertonie; Durchfall, Übelkeit, Stomatitis, Erbrechen, Oberbauchbeschwerden, Bauchschmerzen/aufgeblähter Bauch, Blähungen, Schmerzen im Mundbereich; Gelbfärbung/Verfärbung der Haut, palmar-plantares Erythrodysästhesie-Syndrom, Veränderung der Haar farbe, Hautausschlag; Schmerzen in den Extremitäten; Erschöpfung/Kraftlosigkeit, Schleimhautentzündung, Ödeme. Häufi g: Leukopenie, Lymphopenie; Schlafl osigkeit; Parästhesie, Schwindel, periphere Neuropathie, Hypästhesie; Nasenbluten, Dyspnoe; Chromaturie; Verstopfung, Zungenschmerzen, Mundtrockenheit, gastroösophagealer Refl ux, Ulzerationen/Beschwerden im Mundbereich; trockene Haut, Haarausfall, Dermatitis, periorbitale Ödeme, Hautreaktionen, Erythem, Ekzem,Juckreiz, Hyperpigmentierung der Haut, Abschälen der Haut, Blasenbildung, Hautschäden; Gelenkschmerzen, Muskelschmerzen/-spasmen, Rückenschmerzen, Muskelschwäche; Pyrexie; Lipase erhöht, weißes Blutbild erniedrigt, Ejektionsfraktion verringert, Hämoglobin erniedrigt, Kreatininphosphokinase erhöht, Thrombozytenzahl erniedrigt, Gewichtsabnahme, Erhöhung von Amylase, Aspartataminotransferase und Alaninaminotransferase. In der zytokinrefraktären und der nicht vorbehandelten MRCC-Studie: Sehr häufi g: Neutropenie, Thrombozytopenie, Anämie; Hypothyreose; verringerter Appetit; Beeinträchtigung des Geschmackssinns, Kopfschmerzen; Hypertonie; Nasenbluten; Durchfall, Übelkeit, Oberbauchbeschwerden, Stomatitis, Erbrechen, Bauchschmerzen/aufgeblähter Bauch, Verstopfung, Zungenschmerzen, Blähungen, Schmerzen im Mund, Mund trocken heit; palmar-plantares Erythrodysästhesie-Syndrom, Gelbfärbung/Verfärbung der Haut, Hautausschlag, trockene Haut, Veränderung der Haarfarbe, Haarausfall; Schmerzen in den Extremitäten; Erschöpfung/Kraftlosigkeit, Schleimhautentzündung, Ödeme; Ejektionsfraktion verringert, Gewichtsverlust. Häufi g: Leukopenie, Lymphopenie; verstärkter Tränenfl uss, Lidödem; Dehydratation; Schwindel, Parästhesie, periphere Neuropathie, Hypästhesie, Hyperästhesie; Hitzegefühl/-wallung; Dyspnoe, pharyngolaryngeale Schmerzen, Husten, Dysphonie, trockene/verstopfte Nase, Belastungsdyspnoe, Pleuraerguss; gastroösophagealer Refl ux, Dysphagie, Cheilitis, Zahnfl eischbluten, Hämorrhoiden, Proktalgie, Ulzerationen im Mundbereich, Magenbeschwerden, Rektalblutungen; Hautrötung, Abschälen der Haut, Juckreiz, periorbitale Ödeme, Dermatitis, Hautschäden, Schädigung/Verfärbung der Nägel, Blasenbildung, Hautreaktionen, Hyperkeratose, Akne; Gelenkschmerzen, Muskelschmerzen/-spasmen, Rückenschmerzen; Fieber, Schüttelfrost, Schmerzen, Brustschmerzen; Schlafl osigkeit, Depressionen; Thrombozytenzahl erniedrigt, Leukozytenzahl erniedrigt, Lipase erhöht, Hämoglobin erniedrigt, Kreatininphosphokinase erhöht, Aspartataminotransferase erhöht, Amylase erhöht, Kreatininwert erhöht, Blutdruck erhöht, Alaninaminotransferase erhöht. Weitere Nebenwirkungen nach Markteinführung: Häufi g: erhöhtes schilddrüsenstimulierendes Hormon (TSH). Gelegentlich: Herzinsuffi zienz, dekompensierte Herzinsuffi zienz, Linksherzversagen; Pankreatitis; Leberversagen. Selten: Verlängerung des QT-Intervalls, Torsade de pointes; gastrointestinale Perforationen. Häufi gkeit unbekannt: Infektionen (mit oder ohne Neutropenie) einschl. Pneumonien; thrombotische Mikroangiopathie; Hyperthyreose; Myopathie u./od. Rhabdomyolyse; Pleuraerguss, Lungenembolie, akute respiratorische Insuffi zienz; Proteinurie, nephrotisches Syndrom. Warnhinweis: Enthält Mannitol und Propylenglycol. Packungsgrößen: Sutent 12,5 mg/25 mg/37,5 mg/50 mg Hartkapseln: 30 Hartkapseln (N1). Bitte beachten Sie außerdem die Fachinfor-mation. Abgabestatus: Verschreibungspfl ichtig. Pharmazeutischer Unternehmer: Pfi zer Limited, Sandwich, Kent CT13 9NJ, Vereinigtes Königreich. Repräsentant in Deutschland: PFIZER PHARMA GmbH, 10785 Berlin. Stand: Juni 2009.
b-9v
8su-
hk-0
www.pfi zer.de

AFINITOR® – Jetzt weiterkommen nach TKI-Therapie*
Signifikante Verlängerung des progressionsfreien Überlebens auf 4,9 Monate.1
Oraler mTOR-Inhibitor mit Antitumor- und Antiangiogenese-Wirkung.1 – 3
Gut beherrschbares Sicherheitsprofil.1
1. Afinitor®, Fachinformation, Stand August 2009. 2. Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771-780. 3. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-484.
* VEGFR-Tyrosinkinase-Inhibitoren Sunitinib und Sorafenib, die zu den gegen VEGF gerichteten Therapien beim fortgeschrittenen Nierenzellkarzinom zählen.
Afinitor® 5 mg/10 mg Tabletten Wirkstoff: Everolimus. Zus.: 1 Tablette enth.: Arzneilich wirksamer Bestandt.: 5 mg /10 mg Everolimus. Sonst. Bestandt.: Butylhydroxytoluol, Magnesiumstearat, Lactose-Monohydrat, Hypromellose, Crospovidon Typ A, Lactose, wasserfrei. Anw.: Behandlung von Patienten mit fort geschrittenem Nierenzellkarzinom, bei denen es während od. nach einer gegen VEGF gerichteten Therapie zu einer Krankheitsprogression kommt. Gegenanz.: Überempfindlichkeit gegen den Wirkstoff, andere Rapamycin-Derivate oder einen der sonst. Bestandt. Nebenw.: Sehr häufig: Stomatitis, Hautausschlag, Erschöpfung, Asthenie, Durchfall, Appetitlosigkeit, Übelkeit, Mukositis, Infektionen, Erbrechen, Husten, Peripheres Ödem, Hauttrockenheit, Pruritus, Epistaxis, Pneumonitis, Dyspnoe, Geschmacksstörung, Anämie, Hypercholesterinämie, Hyper triglyzerid ämie, Hyperglykämie, Lymphopenie, Erhöhung Kreatinin, Hypophospha-tämie, Erhöhung ALAT/ASAT, Thrombozytopenie, Neutropenie. Häufig: Kopfschmerzen, Mundtrockenheit, Pyrexie, Gewichtsverlust, Nagelveränderungen, Hand-Fuß-Syndrom, Abdominalschmerzen, Erythem, Ödeme der Augenlieder, Schlaf-losigkeit, akneförmige Dermatitis, Brüchigwerden der Nägel, Dysphagie, Dyspepsie, Exfoliation, Hypertonie, Dehydra ta tion, Konjunktivitis, Brustschmerzen, Bluthusten, Erhöhung Bilirubin. Geglegentl.: Verzögerte Wundheilung, Angioödem, Entwicklung eines Diabetes mellitus, Herzinsuffizienz. Nicht bek.: Überempfindlichkeit, Blutungen. Warnhinweis: Enthält Lactose. Weitere Angaben: Siehe Fach info. Verschreibungspflichtig. Handelsformen: Afinitor 5 mg: 30 Tbl. (N1); Afinitor 10 mg: 30 Tbl. (N1), 90 Tbl. (N3), Klinikpackung mit 10 Tbl. Stand: November 2009 (MS 11/9.2). Novartis Pharma GmbH, 90327 Nürnberg. Tel.: (09 11) 273-0, Fax: (09 11) 273-12 653. www.novartis.dewww.afinitor.de und Tel.: 0800 - AFINITOR (0800 - 234 64 86)
Bei fortgeschrittenem Nierenzellkarzinom:
1 x 1Tablette täglich





























