Embed Size (px)
Citation preview

Kolloidchemische U ntersuchungen Ober die Sulfitablauge III.
Charakteristik der elektrodialytisch erhaltenen Fraktionen.
Voa M. S a m e c und I. R i b a r i 6 . Aus dem chemischen Institut der Universitiit in Laibach.
(Mit 3 Figuren.) (gingegangen am 20. Dezember 19~6. )
Durch Dialyse wcitgchend gereinigtc Sulfitablaugc l~.t3t sich clcktro- dialytisch unter weitercr Befreiung yon den Ascheastoffc~l in zwci Antcile zerlcgen, yon denen sich der eine in dcr Mittelzcllc dcr Elcktro- dialysiervorrichtung ansammclt , der andcrc abcr in der Anodenzelle auftr i t t 1).
Dicse Festste|lung reiht sich eng an die Bcobachtungen verschic- dencr Autoren an, welchc in dcr Sulfitablaugc cbenfalls mehrerc Lignin- substanzen annehmcn.
So nannte z. B. P. Klason"-) den durch Kalziumchlorid fiillbaren Ligninanteil a-Lignin, dc11 nicht aussalzbaren Anteil abet /5-Lignin. K. A. H. M c l a n d c r konstatierte einc vcrschicdenc F~illbarkeit dcr Li- gtlinsubsta1]zen dutch NaCla). Nach H. v. E u l c r 4) wii.re dic a-Lignin- sulfos~urc ein Aldchyd, die /~-Ligninsulfos~urc abcr eine Kurbonsiiure, welche in bcst immter Bezichung zu dcn Gcrbstoffcn stehcn dtirftc. In Ubercinstimmung damit konnte P a s c h k e 5) tatsiichlich bcobachten, dai3 cin durch Natronkochung dargesre~cs ~ g n l n , we~hcS~-E Karbonsiiurc aufzufassen ist, ausgesprochcn gcrbcnde Eigcnschaften hat, wlihrend dic eingcdicktc Sulfitablaugc wohl cin gutes Ftillmaterial darstellt, das Lcdcr aber brtichig macht.
1) M. Samec u. M. Rebek , Kolloidchem. Beih. 19, 106 (1923). 2) Berl. Ber. fi3, 1862 (1920). 3) Zellulosechemie 2, 41 (1921). 4) ZeUulosechemie 3, 1 {1922). a) Zellulosechemie 4, 32 (1923).

158 KOLLOIDCHEM1SCHE BEIHEFTE BAND XXIV, HEFT 1- -4
Mit Rticksicht auf das LaugenbindungsvermSgen scheint die yon M. S a m e e und M. R e b e k elektrodialytiseh 1) erhaltene Mittelzellen- substanz mit der Gruppe des a-Lignins, die Anodenzellensubstanz aber mit dem fl-Lignin in Beziehung zu stehen, welch letztere fibrigens nach noch nicht verSffentliehten Versuehen yon I. R i b a rid wesentlich besser gerberisch wirksam ist als die Mittelzellensubstanz.
In der vorliegenden Arbeit wurde nun eine nlihere Charakteristik dieser beiden Fraktionen der Sulfitablauge versucht, welche die Be- ziehung der geschilderten Produkte zu den von anderen Autoren er- haltenen Ligninsubstanzen festlegen und eine n~here Kenntnis dieser nunmehr sehr reinen Produkte vermitteln soll.
I~
Die Darstellung der beiden Sulii tablaugen-Komponenten erfolgte im engen Anschlui3 an die yon M. S a m e c und M. R e b e k beschriebene Arbeitsweise. Die durch Dialyse erhaltene Flfissigkeit, welche wegen anders geffihrter Dialyse wesentlich verdfinnter war als bei M. S a m e c und M. R e b e k , wurde zunAeh.st elektrodialytisch konzentriert, dann elektrodialytisch welter gereinigt, und zwar so lange, bis die Leit- f~higkeit des Kuthodenzelleninhaltes nicht mehr wesentlich anstieg und etwa das Doppelte der Leitf~higkeit destillierten Wassers betrug. Hier- dutch konnte ein Gehalt yon 2,82 Proz. Trockensubstanz erreicht wer- den, w~hrend die genannten Autoren in der endgtiltig erhaltenen Mittel- zellenflfissigkeit nur 0,73 g Troekensubstanz p r o 100 cem Flfissigkeit
bekamen. Das Anodendialysat wurde im Vakuum bei 45 o konzentriert und
abermals elektrodialysiert. Bei kurz geffihrter Flektrodialyse bleibt ein groi3er Teil der organischen Substanz in der Mittelzelle, in den Anoden- raum wandern vor allem die freie Schwefels~ure und die niedriger- molekularen Anteile. Diese aus dem ursprtinglichen Anodenzelleninhalt durch abermalige Elektrodialyse erhaltene Anodenzellensubstanz wurde in vier Fraktionen yon etwa 200 bis 300 ccm geteilt und jede Fraktion ftir sich untersueht. Auf diese Weise versuchten wir nghere Aufsehlfisse tiber die Einheitliehkeit der ursprfingliehen Anodenzellensubstanz zu
erhalten. Der Kfirze halber wollen wir das bei der Elektrodialyse in der Mitten
zelle angehAufte Produkt als ,,kolloider Rest", den neben ihm im Anoden-
~) Durchgeffihrt mit dem DreizeUenapparat nach Wo. Pauli. Vgl, M. Spiegel u. Adolf A b d e r h a l d e n s Handbuch der Biochem. Arbeits- methoden IIIb.

SAMEC U.RIBARIC, KOLLOIDCHEM. UNTERSUCHUNGEN OB. SULFITABLAUGE III 159
raume erhaltenen Anteil als prim~tres Anodendialysat, die aus diesem gewonnene Mittelzellenfltissigkeit als Anodenkolloid und die daneben auftretenden anodischen Fliissigkeiten als sekund~ire Anodendialysate 1 - 2 - 3 - 4 bezeichnen. Das nachstehende Schema soll die gegen- seitige Beziehung klarer vor Augen fUhren.
S u l f i t a b l a u g e .
Sechsw6chentliche Dialyse in Faltendialysatoren 1) gegen flieflende Wasser und Konzentrieren durch kurze Elektrodialyse.
D i a l y s i e r r t i c k s t a n d .
Elektrodialytisch weiter konzentriert und bis zur Konstanz der elektro- chemischen Eigenschaffen elektrodialysiert.
/ \ K o l l o i d e r R e s t P r i m S ~ r e s A n o d e n d i a l y s a t
Im Vakuum konzentriert und abermals elektrodialysiert
/ \ A n o d e n k o l l o i d S e k u n d ~ i r e s A n o d e n d i a l y s a t
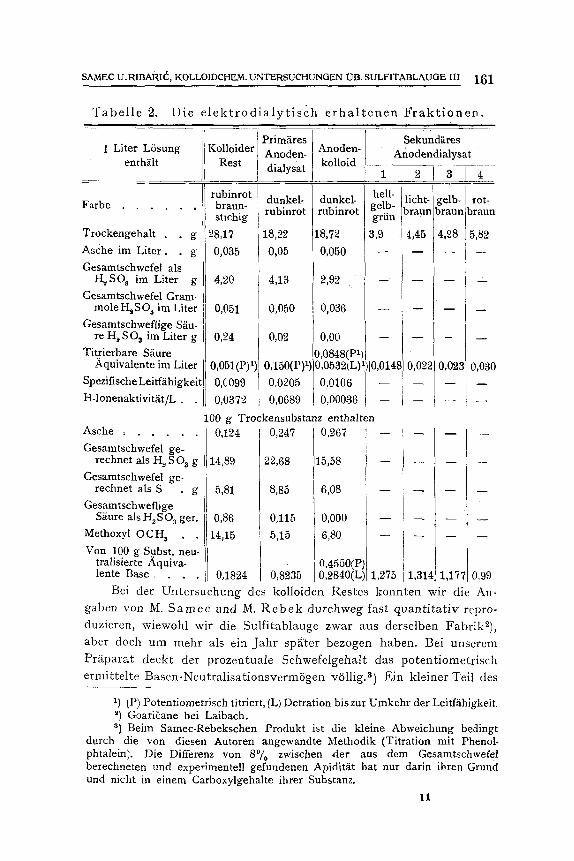
Die so erhaltenen Fraktionen wurden einerseits durch Ermitt lung des Aschengehaltes und des Gehaltes an den verschiedenen Formen yon Schwefel andererseits elektrochemisch und dutch Bestimmung der typi- sehen organischen Gruppen nliher charakterisiert. Das erhaltene Zahlen- material enthalten die Tabellen 1 und 2.
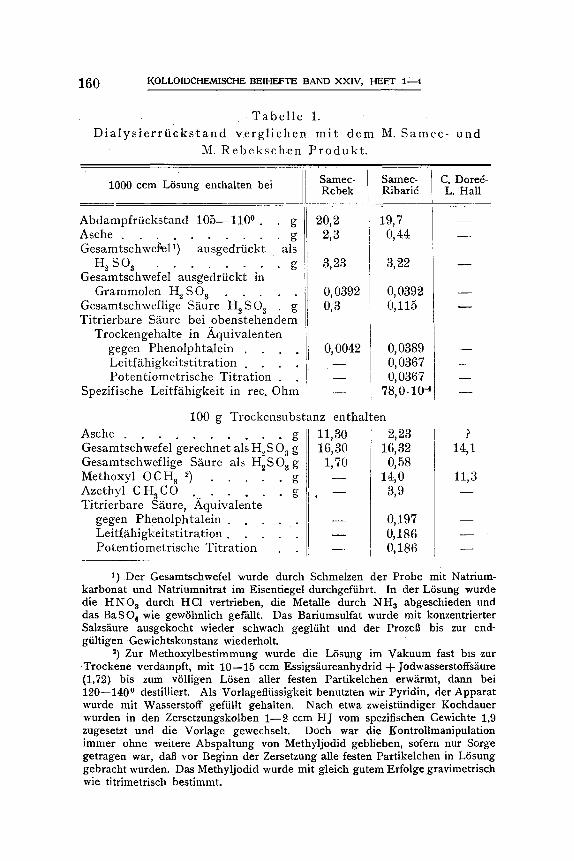
TabeUe 1 vermit tel t zuniiehst den AnschluB der vorliegenden Unter- suchung an die Experimente yon M. S a m e e und M. R e b e k sowie C. D o r e e und L. HallS) .
Der zur Elektrodialyse gelangende Dialysierrtickstand zeigt in u n - serem FaUe denselben Gehalt an Ttockensubstanz wie bei S a m e c - R e b e k , doch ist er bei dem gleichen Gehalte an Gesamtschwefel asche- ~irmer. Im Zusammenhang damit ist die ti trierbare Siiure in unserem jetzigen Falle fast zehnmal so groB und bleibt nur um ein geringes hinter dem aus dem Gesamtschwefelgehalte berechneten Basen-Neutra- tisationsverm6gen zurtick, z)
1) Im Anschlul~ an Wo. Pauli. 2) Journ. Soc. Chem. Ind. 18, 257 (1924); Zellulosechemie 5, 71 (1924). ~) 1 Grammatom S = 1 Grammatom ersetzbarer Wasserstoff.
160 KOLLOIDCHEMISCHE B E I H E F T E B A N D X X l V , H E F T 1~4
T a b e l l e 1.
D i a l y s i e r r f i c k s t a n d v e r g l i c h e n m i t d e m M. S a m e c - u n d
M. R e b e k s c h e n P r o d u k t .
1000 ccm L6sung enthalten bei C. Dored- L. Hall
Abdampfrf icks tand 105--1100 . g Asche . . . . . . . . . . g Gesamtschwefel ') ausgedrfickt als
H ~ S O 3 . . . . . . . . g Gesamtschwefel ausgedrtickt in
Grammolen H~ $O 3 . . . . . Gesamtschweflige S~ture H~SO 3 . g Ti t r ie rbare S~ture bei obens tehendem
Trockengehal te in Aquiva len ten gegen Phenolphta le in . . . . Leitf~ihigkeitst i tration . . . . Po ten t iomet r i sche T i t r a t ion .
Spezifische Leitf~higkeit in rec. Ohm
Samec- Samec- Rebek Ribarid
20,2 19,7 2,3 0,44
3,23 3,22
O, 0392 O, 0392 0,3 0,115
O, 0042 O, 0389 - - 0,0367 - - 0, 0367
i
_ _ 78,0.10 .4
100 g Trockensubs tanz entha l ten
Asche . . . . . . . . . . . g Gesamtschwcfel gerechnet als H~ S 03 g Gesamtschweflige S~ure als H~SO 3 g Methoxyl 0 C H.~ ~) . . . . . g Azethyl CHACO . . . . . . . . g Ti t r ie rbare S/~ure, Aquivalente
gegen P h e n o l p h t a l e i n . . . . . L e i t f ~ h i g k e i t s t i t r a t i o n . . . . . Potent iometr ische Ti t ra t ion
11,30 2,23 ,16,30 16,32
1,70 0,58 - - 14,0 - - 3,9
- - 0,197 - - 0,186 --- 0,186
m
f
?
14,1
11,3
1) Der Gesamtschwefel wurde durch Schmeizen der Probe mit Natrium- karbonat und Natriumnitrat im Eisentiegel durchgefiihrt. In der L6sung wurde die H NO 8 dutch H C1 vertrieben, die Metalle durch N H 3 abgeschieden und das Ba S 04 wie gew6hnlich geffillt. Das Bariumsulfat wurde mit konzentrierter Salzs/iure ausgekocht wieder schwach geglfiht und der ProzeB bis zur end- giiltigen Gewichtskonstanz wiederholt.
~) Zur Methoxylbestimmung wurde die L6sung im Vakuum fast bis zur Trockene verdampft, mit 10--15 ccm Essigs~iureanhydrid +Jodwasserstoffs~iure (1,72) bls zum v611igen L6sen aller festen Partikelchen erw~irmt, dann bei 120--140 o destilliert. Als Vorlagefliissigkeit benutzten wir Pyridin, der Apparat wurde mit Wasserstoff gefiillt gehalten. Nach etwa zweistiindiger Kochdauer wurden in den Zersetzungskolben 1--2 c c m HJ vom spezifischen Gewichte 1,9 zugesetzt und die Vorlage gewechselt. Doch war die Kontrollmanipulation immer ohne wekere Abspaltung von Methyljodid geblieben, sofern nur Sorge getragen war, dab vor Beginn der Zersetzung alle festen Partikelchen in L6sung gebracht wurden. Das Methyljodid wurde mit gleich gutem Erfolge gravimetrisch wie titrimetrisch bestimmt.
SAMEC U.RIBAIRI~, KOLLOIDCHEM.UNTERSUCHUNGEN OB. SULFITABLAUGE III 161
T a b e l l e 2. D ie e l e k t r o d i a l y t i s c h e r h a l t e n e n F r a k t i o n e n .
1 Liter LSsung enthttlt
Farbe . . . . . .
Trockengehalt . . g Asche im Liter. . g Gesamtschwefel als
H, SO s irn Liter g Gesamtschwefel Gram-
mole H, SOs im Liter Gesamtschweflige S~iu-
I Prim~ires Kolloider ] Anoden-
Rest [ dialysat
Sekund~ires Anoden- Anodendialysat kolloid
1 1 2 1 3 rubinrot dunkel- braun- rubinrot stichig
~8,17 18,22 0,035 0,05
4,20 4,13
0,051 0,050
dunkel- rubinrot
18,72
0,050
2,92
0,036
hell- licht-gelb- rot- gelb- braun braun braun griin 5 / 3,9 4,4 4,~~ 5,82
m _ _
t e l l , SO a imLiter g 0,24 0,02 0,00 -- 072 ] i 2 Titrierbare Saure 0,0848(Pl/
3 0,030 0,150(P) 1) 0,0532tL)1)[0,0148 2 0 Aquivalente im Liter 0,051(P)1)1 Spezifische Leitfiihigkeit 0,C099 0,0205 0,0106 -- H-Ionenaktivit~t/L. 0,0372 0,0689 0,00036 J - - -- --
100 g Trockensubstanz enthalten
Gesamtschwefel ge- rechnet als H o SO a g 14,89 22,68 15,58 --
Gesamtschwefelrechnet als S ge-. g 5,81 8,85 6,08 - -
Gesamtschweflige S/iure als H~SO 3 get.
0,86 0,115 0 , 0 0 0 , - - , , ~7 0,99 Methoxy! OCH s 14,15 5,15 6,80 ] - - Von 100 g Subst. neu-
tralisierte Aquiva- 0,4550(P) 0,1824 0,8235 0,2840(L)[ 1,275 1314' 1 7 lente Base . . . .
Bei der Untersuehung des kolloiden Restes konnten wir die An- gaben von M. S a m e c und M. R e b e k durchweg fast quan t i t a t iv repro- duzieren, wiewohl wir die Sulfi tablauge zwar aus derselben FabrikZ), aber doch um mehr als ein J ah r spgter bezogen haben. Bei unserem Prgpara t deckt der prozentuale Schwefelgehalt das potent iometr isch ermittel te Basen-Neutral isat ionsvermSgen vSllig, a) Ein kleiner Tell des
1) (p) Potentiometrisch titriert, (L) Detration biszur Umkehr der Leitftihigkeit. 9) Goari~ane bei Laibach. a) Beim Samec-Rebekschen Produkt ist die kleine Abweichung bedingt
durch die yon diesen Autoren angewandte Methodik (Titration mit Phenol- phtalein). Die Differenz von 80/o zwischen tier aus dem Gesamtschwefel berechneten und experimentell gefundenen Apidit~it hat nur darin ihren Grund und nicht in einem Carboxylgehalte ihrer Substanz.
11

169, KOLLOIDCHEMISCHE BEIHEFTIE BAND XXIV, HEFT 1--4
Schwefels findet sich hier noch als ,,gesamtschweflige S~ure" vor, w~h- rend bei M. S a m e c und M. R e b e k der Gehalt an dieser Schwefelform unter 0,00 Proz. lag. Die Ursache hiervon mag in der gr6f3eren Konzen- tration unserer L6sung liegen, da ja der Komplexzerfall der aldehyd- schwefligen S~ure wesentlich v o n d e r Verdtinnung abh/ingig ist.
Das prim. Anodendialysat ist dutch eine viel intensivere und reinere rubinrote Farbe ausgezeichnet als das Mittelzellenkolloid, der Schwefel- gehalt (Gesamtschwefel) ist um fund 3 Proz. h6her. Der Hauptunter- schied zwischen diesen beiden Fraktionen liegt aber in ihrem Basen, Neutralisationsverm6gen. Die t i t r ierbare S~ure betr/~gt n~mlich beim prim/~ren Anodendialysat etwa dreimal so viel, wie dem Gesamtschwefel entspricht. In diesem Anteile findet sich zwar die freie anorganische S/~ure infolge anodischer Oxydation wohl als Schwefels~ure vor, doch wtirde, selbst wenn der ganze Schwefel dieser Fraktion als H 2SO~ vor- l~ge, noch immer ein Drittel der titrierbaren S/~ure ungedeckt blciben, so dab wit auf die Gegenwart organischer S/~uren schlieBen mtissen. Ein Teil von diesen kanu durch anodische Oxydation aus dem Aldehyd (Lignin) entstanden sein, doch begegnet es mit Rticksicht auf die vielen Feststellungen anderer Autoren keiner Schwierigkeit, auch in unserem Falle an die Existenz yon vorgebildeten Karbons~uren in der Suflitablauge zu denken, welche durch Elektroosmose yon der Aldehyd- sulfos/~ure abgetrennt werdeu konnten.
Neben dieser Gegenwart yon Karbonsfiuren im Anodendialysat ist noch sein relativ niedriger Gehalt an Methoxylgruppen auffallend. Es ist sehr wahrscheinlich, daft auch ftir den Verlust der OCI-I~-Gruppen die anodische Oxydation verantwortlich zu machen ist, wobei neue OH-Gruppen auftreten wtirden und - - falls sie phenolischer Natur w~ren - - zu einer Erh6hung des Basen-Neutralisationsverm6gens bei- tragen k6nnten. Der Verlust der CHO3-Gruppe und die im sekund{iren Anodendialysat nachgewiesene Oxydatign der organischen Substanz bis zur Kohlens/~ure bedingen eine Verkleinerung der lV[olektUgr6Be und dadurch indirekt einen scheiubaren Anstieg im S-Gehalt.
Die Abtren~ung der freien SchwefelsSure aus diesem Sulfitablaugen- anteil versuchten wir zu~/ichst durch Uberftihrung in das Bariumsalz zu erreichen, doch bietet die Abscheidung des BaSO 4 wegen der sehr unangenehmen Form des Niederschlages grot3e Schwierigkeiten.
Wie erwS.hnt, enthS.lt das prim{ire Anodendialysat auf je 100 g Sub- stanz etwa um 3 g Schwefel mehr als der kolloide Rest der Mittelzelle. Fafit man diesen Schwefeltiberschufi als freie SchwefelsS.ure auf und setzt gerade so viel Ba(OH)2 Z u, als zu ihrer Neutralisation n6tig wgre,

SAMEC U. RIBARIC, I(OLLOIDCHEM. UNTERSUCHUNGEN CB. SULFITABLAUGE III 1 6 ~
so erh~tlt man einen gelblichen, flockigen Niederschlag, welcher sicl~ innerhalb einiger Tage absetzt. Die tibrigbleibende Fltissigkeit ist dun- kelrot. Betr~igt die zugesetzte Ba(OH)2-Menge so viel, dab die freie Schwefelstiure [1 S - 2 H ] sowie die dem fibrigbleibenden Sehwefel [1 S = 1 H] entsprechende S5.ure neutralisiert werden kann, so erfolgt die Ab- scheidung des Niederschlages viel langsamer und ist erst innerhalb einer Woche beendet; die Yltissigkeit ist wesentlich heller geworden.
Bei v611iger Neutralisation der L6sung, etwa gegen Phenolphta- lein oder unter potentiometrischer Messung, erfolgt selbst nach mel~reren Monarch keine scharfe Tlennung der Phasen; die Yliissifikeit ist stark triib und hellgelb, der Niederschlag besteht aus mehreren in der Farbe und tier Kornar t unterschiedenen Schichten, welche wohl durch das verschie- den rasche Absetzen der gebildeten Bariumsalze bedingt sind.
Viel glatter geht die Abtrennung der freien Schwefels~ure aus dem prim~iren Anodendialysat durch Elektrodialyse.
Im Gegensatz zur Ligninsulfosiiure l~il3t sieh nun die vermutliche Sulfoligninkarbonsiiure elektrodialytisch nicht in Form einer separaten Phase abscheiden, wohl aber finder bin starker Abtransport yon Wasser in die Elektrodenrliume statt.
Den Verlauf dieser zweiten Elektrodialyse haben wir durch Be- st immung der Trockensubstanz und der titrierbaren SSure des Anoden- zelleninhaltes kontrolliert. Wir unterbrachen den Prozef3 im Zeitpunkt, in welehem insgesamt etwa doppelt so viel S~iure ausgewandert war (genau die 2,16fache ~Ienge), wie dem l~berschul3 des Sehwefelgehaltes des prim~iren Anodendialysates fiber den Schwefelgehalt des kolloiden Restes entspricht. Wie man sieh leicht tiberzeugen ka,m, enttfiilt (tie erste Fraktion reichlich SO4-lonen , in der vierten Fraktion fanden wit nur mehr Spuren davon, so daft die gestellte Aufgabe: Trennung der SchwefelsS.ure yon der Gruppe der Sulfolignins~turen, erreicht war. Allerdings diosmiert neben der Schwefels~iure aueh eine betr~eht- liche Menge organischer Substanz zur Anode, und zwar relativ um so mehr, je weiter die Reinigung fortgeschritten ist. Aus diesem Orunde sinkt bei den aufeinanderfolgenden Portionen des sekund~ren Anoden- dialysates das Verh~iltnis zwischen der titrierbaren S~ure und dem Ab- dampfrfickstand. Die immer dunkler werdende Farbe und das steigencte TiterS.quivalent der einander folgenden Fraktionen des Anodendialy- sates sprechen daffir, daft mit zunehmender Dialysierdauer immer gr6flermolatige Substanzenteile auswandern.
Durch die neuerliche Elektrodialyse ist --- wie zu e r w a r t e n - der Schwefclgehalt des primS.ren Anodendialysates wesentlich gefallen, doch
11"

164 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1--4
enth~ilt das Anodenkolloid auch nach dieser Reinigung etwas mehr Schwefel als das Mittelzellenkolloid. M6glicherweise ist hierftir die t e i l - weise Molekiilzerkleinerung, wie sie oben bei der Besprechung der Ande- rung des 0 CH3-Gehaltes an gedeutet wurde, verantwortlich zu machen. Die titrimetrisch bestimmbare Azidit~it fibersteigt auch nach der zweiten Elektrodialyse ganz bedeutend den dem Schwefelgehalt entsprechenden S~iuregrad und best~itigt so die bereits dargelegte Ansicht, daft w i re s mit organischen S~uren zu tun haben. Der potentiometrischen Titra- tion zufolge k~ime in dieser L6sung auf jede SOa H-Gruppe etwas mehr als eine COOH- oder OH-Gruppe.
Verg le ich tman nun die zur zweiten Elektrodialyse verwendete Substanzmenge mit der im Anodenkolloid und im sekund~iren Anoden- dialysat wiedergefundenen, so ergibt sich ein Abgang, welcher grSBer ist, als die Fehlerbreite der Bestimmungen ausmacht.
Bei einem solchen Versuche kam zur Anwendung
330 ccm prim. Anodendialysat mit 5,470 g Trockensubstanz und 0,045 Aqu. S~iure. Hieraus erhielten wir
175 ccm Anodenkolloid mit 3,280 g Trockensubstanz und 0,0148 .~qu. S~ure
und in den einzelnen Fraktionen des sekund~iren Anodendialysates zu- sammen
1,915 g Trockensubstanz u. 0,0224 Aqu. S~iure; daraus ergibt sich ein Verlust yon
0,275g Trockensubstanz u. 0,0077 Aqu. S~iure.
Es bleibt zur Erkl~irung dieser Erscheinung nur die Annahme tibrig, dag Bruchstticke der organischen Molekeln bei der Elektrodialyse gas- I6rmig etwa als CO 2 abgehenl). Das S~ure~iquivalentgewicht des abge- gangenen Anteiles betrfigt 35, bewe~t sich also in der Gr6t3enordnung des S~iure~iquivalentes der Karboxylgruppe (45).
Auger dem Schwefel bleibt selbst nach der energischesten Reinigung durch Elektrodialyse sowohl im kolloiden Rest als auch im Anoden- dialysat eine kleine Menge nicht fitichtiger Aschenbestandteile zurtick; tier kolloide Rest enth~lt hiervon 0,124 Proz., das Anodenkolloid 0,26 Proz. Wie wir uns tiberzeugen konnten, besteht dieser Aschenrest vor- wiegend aus Kiesels~ure, welche also die Ligninsubstanzen bei ihrem kolloidchemischen Wandel stets begleitet. Dessenungeachtet aber ist der Reinheitsgrad unserer Produkte auch in bezug auf die schwer zu
1) Vgl. F. K6nig, Zellulosechemie 2, 100 (1921).

SAMEC U.RIBARIC, KOLLOIDCHEM.UNTERSUCHUNOEN I~B.SULFITABLAUOE III 165
entfernende Si 02 sehr viel gr6Ber als bei den entsprechenden Substanzen
yon C. D o r 6 e und B. Ha l l , deren gereinigte SSmre 48malsovielSiO 2 enthielt als unser Pr~parat.
II.
Die n~ichste Frage war nun nach der Einheitlichkeit der erhaltene~ Fraktionen gerichtet.
Wir versuchten sie durch fraktionierte F~llung der Bariumsalze
und teilweise auch der/5-Naphthylaminsalze der erhaltenen Sulfitab- laugenanteile zu klSxen.
Wie bereits angedeutet, fSAlt ein betrS~ehtlicher Teil der Lignin- substanzen bei der Neutralisation mit Bariumhydroxyd allmS.hlich aus
der L6sung aus; viel rascher erfolgt diese Abscheidung naeh Zusatz von Alkohol .
Um diese Tatsache zur weiteren Fraktionierung der Ligninsub- stanzen auszuntttzen, versetzten wir eine Portion des Dialysierrtiek-
standes mit etwas mehr Ba(OH)2 als zur v611igen Neutralisation (bei potentriometrischer Messung) n6tig war, f~illten das frei gebliebene Bariumhydroxyd in der WSxme durch Einleiten yon CO 2 aus und lieflen das Bariumkarbonat in hohen Standzylindern absitzen. Die
klare, 1,39prozentige L6sung wurde im Vakuum bei 45 o auf rund 3 Proz. Trockensubstanz gebracht, der hierbei ausgefallene geringftigige Nieder- schlag abfiltriert und zu der nun klaren Fl~s.sigkeit so viel Alkohol zu-
gesetzt, dab die Mischung in bezug auf Alkohol 33prozentig war. Der ausgeschiedene Niederschlag wurde auf einem gewogenen Filter gesam- melt, das Filtrat dureh Zusatz yon Alkohol auf 50 Proz. und nach dem Entfernen der ausgefallenen Fraktion auf 75 Proz. Alkohol gebracht.
Die schlieglich iibrigbleibende L6sung wurde auf dem Wasserbade zur Trockene verdampft. Um die Entfernung des tiberschtissigen Bariums aus der L6sung zu umgehen, setzten wit zur Gewinnung der Barium-
salze des kolloiden Restes nur so viel Ba(OH)2 zu, als zur Einstellung der potentiometrischen Neutralit~t notwendig war. So entfiel dann die FSAlung des Bariumhydroxyds mit CO 2 und alle damit verbundenen Manipulationen.
Interessant ist das Verhalter~ der Bariumsalze des kolloiden tZestes bei der nun folgenden Fraktionierung mit Alkohol.
Die 50 Proz. Alkohol und 3,93 g Bariumsalz in 100 ccm enthaltende Mischung ist gleich nach Zusatz yon Alkohol dunkelrotbraun, schwach
opaleszent, gesteht aber tib'er Nacht zu einer reeht festen Gallerte, welche alle verftigbare Fltissigkeit aufgenommen hat. Erst nach mecha-

1 6 6 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1--~-
nischer Zerteilung durch Reiben und Schtitteln gelingt es, eine kleine Menge Fltissigkeit abzuscheiden, doch liet3 sich die gallertige Phase nicht recht abfiltrieren. Aus diesem Grunde wurde die Fraktionierung mi t einer verdtinnteren L/Ssung durchgeftihrt (1,96 g Ba-Salz in 100 ccm), welche beim Stehen fiber Nacbt einen durch Filtration entfernbaren Niederschlag abschied. Um Fehler bei der Methoxylbestimmung zu vermeiden, sorgten wir daftir, dab die NiederschlSge nicht austrock- neten. Es wurden vielmehr die erhaltenen Ba-Fraktionen in 100 ccm Wasser gel6st und diese L6sung ftir die Untersuchungen weiter ver-
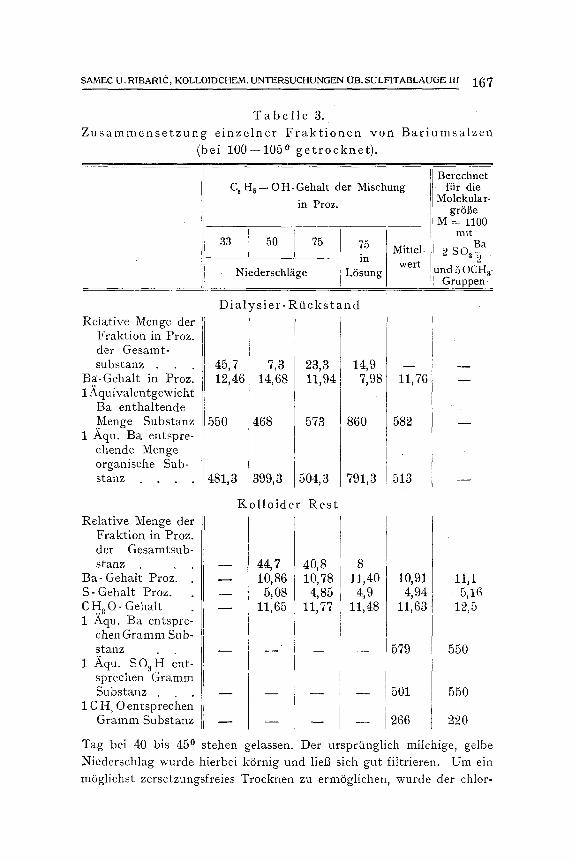
wendet. Wir erhielten so vier Fraktionen yon Bariumsalzen, deren Zusam-
mensetzung die Tabelle 3 angibt. Der Bariumgehalt bewegt sich bei den einzelnen Anteilen um 11,8
Proz. Im Dialysierrtickstand fallen in die alkoholarmen Mischungen die bariumreicheren Substanzenanteile; bei diesen kommen auf ein Aqui- valentgewicht Barium 4=81--504 g organischer Substanz, nur im Ab- dampfriickstand erreicht das Bariumgquivalent den Wert von 791; es dtirften hier wohl die Aschenbestandteile des Lignins das Resultat trti- ben, welche nach dem Abdampfen mit der organischen Substanz zu- gleich gewogen werden, und die auf ein Ba entfallende Substanzmenge
vermenren. Wenn somit im Hinblick auf die Zusammensetzung der Barium-
salze schon der Dialysierrtickstand verh~iltnism~tBig einheitlich zusam- mengesetzt erscheint, so ist das in viel h6herem Mage beim kolloiden Rest der Fall. Hier schwanken der Barium- sowie der Schwefelgehalt der einzelnen Fraktionen in sehr engen Grenzen und schlieBen sich fast v611ig an die unter Zugrundelegung der Molekulargr6t3e 1100, welche spS.ter ausftihrlich begrtindet wird und 2 S Q H- und 5CH a O-Gruppen ent- hS.lt, berechneten Werte an. Auch der Methoxylgehalt der einzelnen Fraktionen ist nahezu konstant, nur ist er durehweg etwas niedriger, als der Berechnung entsprechen wtirde; der Grund hierftir dtirfte wohl in der Bestimmungsmethode des Methoxyls liegen.
Zum gleichen Ergebnis, wenn auch nicht mit gleicher Sch~trfe, ftihrte die Untersuchung des Verhaltens unserer Sulfitablaugenfraktionen zu
/~-Naphthylamin. Wir gingen vom Merkschen /5-Naphthylamin aus, welches wir mit
tiberschtissiger HC1 in das Chlorhydrat tiberfiihrten und nach grtind- lichem Waschen und in gesS.ttigter L6sung weiter verwendeten. Die zu untersuchende Fltissigkeit wurde mit dem gleichen Volum der fi-Naphthylaminchlorhydratl6sung versetzt, gut durchgemischt und einen
SAMEC U.RIBARIC, KOLLOIDCHEM. UNTERSUCHUNGEN OB.SULF1TABLAUGE III 167
T a b e l l e 3. Z u s a m m e n s e t z u n g e i n z e l n e r F r a k t i o n e n y o n B a r i u m s a l z e n
(bei 100--105 0 g e t r o c k n e t ) .
C, H~ -- O H- Gehalt der Mischung in Proz.
33 50 75 75 Mind= in wert
Niederschliige L6sung
Berechnet fiir die
Molekular- grSfle
M = 11oo mit O Ba 2S ~ 2
und 50CHa- Gruppen _
Relative Menge der Fraktion in Proz. der Gesamt- substanz
Ba-Gehalt in Prozl 1 fi~quivalentgewicht
Ba enthaltende Menge Substanz
1 Aqu. Ba entspre- chende Menge organische Sub- stanz . . . .
Relative Menge der Fraktion in Proz. der Gesamtsub- stanz
Ba- Gehalt Proz'. S- Gehalt Proz. C H a O- Gehalt 1 Aqu. Ba entspre-
chenGramm Sub- stanz
1 Aqu. SO a~I ent- spreehen Gramm Substanz .
1 C H a O entsprecimn Gramm Substanz
Tag bei 40 bis 450
D i a l , T s i e r - R t i c k s t a n d
45, 7 12,46
7,3 14,68
550 468
481,3 399,3
23,3 11,94
573 !
504,3
K o l l o i d e r R e s t
14,9 7,98
86O
791,3
11,76
582
513
w
44, 7 10,86
5j08 11,65
40 8 10, 78
4,85 11,77
8 11,40
4,9 11,48
10,91 4,94
11,63
579
501
1266
11,1 5 , 1 6 12,5
550
550
220
stehen gelassen. Der urspriinglich milchige, gelbe Niederschlag wurde hierbei k6rnig und lieB sich gut filtrieren. Um ein m6glichst zersetzungsfreies Trockrlen zu erm6glichen, wurde der chlor-

168 KOLLOIDCHEMISCHE BEIHEFTE BAND XXlV, HEFT 1--4
frei gewaschene Niederschlag auf dem Filter durch Absaugen so welt als m~glich vom Wasser befreit und erst dann bei 45 o his zur Gewichts- konstanz getrocknet. Das erhaltene Produkt war bei dieser Arbeitsweise sandig und eigelb, wlthrend es, mit einem grSt3erenWassergehalt getrock- net, leicht zu dunklen Klumpen zusammenballt.
In den Niederschl~igen wurde tier Schwefel in der tiblichen Weise durch Schmelzen mit Soda und Salpeter, der Stickstoff nach K j e l d a h l unter Verwendung yon konzentrierter Bors~iure als Vorlages~ture und Kongorot als Indikator ermittelt.
Der beobachtete Stickstoffgehalt erm~glicht nun die Berechnung des in den Niederschliigen enthaltenen ~-Naphthylamins [1 N ~ 1 0 , 2 g /5-Naphthylamin], und aus demVergleiche mit dem Gewichte des Nieder- schlages ergibt sich die als/5-Naphthylaminsalzausgeschiedene Substanz- menge.
Um zur Kontrolle noch die in der L6sung verbliebenen Lignin- mengen zu bestimmen, wurde die /~-naphthylaminhaltige Fltissigkeit mit NaOH alkalisch gemacht, das ausgefallene/~/-Naphthylamin abfiI- triert, das Filtrat mit Ather ausgezogen und hierauf eingedampft, ge- trocknet, gewogen und verascht. Der Gewichtsverlust beim Veraschen wurde als die nach der /5-Naphthylaminf~llung in L6sung verbliebene Ligninmenge in Rechnung gesetzt.
Diese Methode gibt nattirlicherweise nur ein ann~herndes Bild der Substanzverteilung, doch stimmt die auf dem Stickstoffgehalt des Ko- agulums basierte Berechnung des gefiillten Sulfitablaugenanteiles mit dern aus dem in L6sung befindlichen Anteile einigermaflen iiberein.
BeimDialysierrtickstand fanden wir zum Beispiel nach demStickstoff- gehalt 95,3 Proz. der aschehaltigen und 97,5 Proz. der aschefreien Sub- stanz als f~illbar durch fi-Naphthylamin. Die direkte Bestimmung des in L~sung gebliebenenAnteils aber sprach ftir 98 Proz. gef~llte organische Substanz. Nach beiden Berechnungen verbleibt demnach ein kleinerTeil des Dialysierrtickstandes durch /5-Naphthylamin ungef~illt und deutet auf eine gewisse Inhomogenit~t dieser Fraktion lain.
Die einzelnen, bei verschiedenen Versuchen erhaltenen Nieder- schl~Lge stimmen in der Zusammensetzung gut miteinander tiberein. Ihr Schwefelgehalt betr~igt im Mittel 5,01 Proz. und der Stickstoff- gehalt 3,05 Proz.
Der elektrodialytisch gereinigte kolloide Rest wird durch fl-Naph- thylamin g~inzlich gef~illt, was im Vereine mit der Gleichartigkeit der einzelnen Bariumfraktionen auf eine weitgehende Homogenitlit dieses Produktes hindeutet.

SAMEC U.RIBARIC, KOLLOIDCHEM.UNTERSUCHUNGF_.aN OB. SULFITABLAUOE 1II 169
I I I .
Die Reinheit und gute Einheitlichkeit der hier beschriebenen Frakt ionen der Sulfitablauge erlaubt uns, die Resultate der physiko2
chemischen Messungen in theoretiseher Hinsicht etwas weiter aus- zuwerten. Das Mittelzellenkolloid stellt uns wie erw~ihnt die freie Ligninsulfosiiure vor, welehe in 2,8prozentiger LSsung bei Zimmer- tempera tur eine potentiometriseh feststellbare t-LIonenaktivit i i t vor~ 3,72 �9 10 -2 n aufweist. Rechnen wir die dieser Wasserstoff ionenaktivi tat
entsprechende Leitfahigkeit, so kommen wir nach dem Ansatze 3 ,72 ' 3 1 5 . 1 0 "2-a zu k = 117,1 �9 10 .4 rec. Ohm [18 o (2]. Die direkte Bestim-
mung der elektrischen Leitf~ihigkeit ftihrt zu dem Werte k = 99,75 �9 10 .4 rec. Ohm [25 o C], welcher demnach zwar sehr nahe, aber t rotz der h6he-
ren Beobachtungs tempera tur doch etwas niedriger ist, als der gemes- senen It-Ionenaktivit~it entspricht.
Diese Divergenz dtirfte auf ~ihnliche Ursachen zurtickzuftihren seir~
wie die Beobachtung yon M i c h a e l i s und M. M i z u t a n i , derzufolge z. B. eine 1/100n HC1 durch Zusatz yon 2,7n KC1 eine H- Ionenak t iv i tg t von
16/100n erlangtl). Vergleicht man nun die elektrometrisch ermittelte H- Ionenak t iv i tg t
mit dem durch Titrat ion ermittel ten Siiuregrad (bzw. mit dem S-Gehalt)
der LigninsulfosSmrel6sung, so wtirde nach den Werten der Tabelle 2: als Dissoziationsgrad a = 3;72/5,13 = 0,725 Iolgen. Im Hinblick auf die
�9 eben angedeuteten Verhgltnisse dtirfen wir darin zwar nur einen oberen Grenzwert sehen, immerhin erweist sich die Ligninsulfos~iure als recht weitgehend ionisiert.
Beim Vergleich dieses Ergebnisses mit anderen Beobachtungen abet stofien wir auf ghnlich verwickelte VerhS.ltnisse, wie sie z. B. yon W. B il t z
an Kongoro t und an anderen sulfurierten hochmolekularen Produkten beobachte t wurden.
Unsere analytischen Daten ermSglichen zun~ehst die Fest legung
des Aquivalentgewichtes der Ligninsulfos~ure, welches bei der guten Ubere ins t immung der einzelnen Resul tate recht gut fund ie r t erscheint.
Aus dem Schwefelgehalte folgt das Aquivalentgewieht ~_ = 551 aus dem titrierbaren SS.uregrade A = 549 aus dem Methoxylgehalte A = 219 und aus dem Azetylgehalte )k = 1096.
Da auf ein Schwefelfiquivalent 2,5 C HaO-Gruppen entfallen, mug als Minimatmolekulargewicht M = 1100 angenommen werden.
1) Zeitschr. t. phys. Chem. 112, 68 (1924).

1 7 0 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1--4
Die kryoskopische Molekulargewichtsbestimmung ergab bei einer 2,906prozentigen LSsung der Ligninsulfos~iure gegen reinstes Wasser eine
Gefrierpunktdepression von 0,049~ und Itihrt zu dem Molekulargewichte M = 1136, also zu dem aus dem Azetyl- und Methoxylgehalte deduzierten
Werte. Wtirde man nun den aus der H-IonenaktivitS.t gescn~ttzten Disso-
ziationsgrad a = 7 2 Proz. der Molekulargewicntsbestimmung zugrunde
legen, so Mime man auf Grund der Gefrierpunktsdepression bei Annahme, dab die LigninsulfosSure einbasisch ist, zu dem Werte M =- 1960 und bei Annahme einer zweibasischen S~iure zu M = 2780. Diese beiden Zahlen
entfernen sich aber sehr weir von den auf analytischer Basis ermittelten. Da auf 1100 Gramm unseres PrS~parates zwei Gramm-Atome Schwe-
fel, also zwei S03H-Gruppen kommen, w~re die Ligninsulfos~ure bei
einem Molekulargewiehte von 1136 zweibasiseh, bei einem Molekular- gewichte 1960 aber naheza vierbasisch Es wS~re daher sehr verlockend, zur Entscheidung der MolekulargrSfJe die Os twa ld sche Valenzregel heranzuziehen. Doch dtirfen wir nicht tibersehen, daft nach F. K S n i g 3)
das Bariumligninsulfonat sowie aueh das yon K. M e l a n d e r studierte Natriumsalz mit steigender Verdtinnung anfangs einen viel geringeren Anstieg derAquivalentf~higkeit zeigen, als dies bei Salzen sonst beobach-
tet wird, und erst in grofien Verdtinnungen eine starke Zunahme d e s .Aquivalentleitverm6gens aufweist, so dab F. K5 ni g zur Annahme aus- giebiger Assoziation seines Ligninsulfonats gezwungen war und die Va-
lenzregel ablehnen mut3te. Der Weg, auf welchem wir die freie Ligninsulfos~iure erhalten haben,
ist jedoch yon den Darstellungsmethoden K. H. A. lVielanders 4) und
F. KSni gs wesentlich verschieden, so daft wir, wenn schon keine wesent- lichen chemischen Verschiedenheiten, so doch sicherlich kolloide Zu- standsdifferenzen zwischen unserem Produkt und den Pr~paraten der
erwS~hnten Forscher anzunehmen haben. K. A. H. Mela n d e r erhielt sein Pdiparat durch Fiillung der Sulfitablauge mit Natriumchlorid, F. K S n i g durch F~illung mit Natriumbisulfat bzw. starker Schwefel-
sSmre, bei unserer Darstellungsmethode aber passierte die Ligninsulfo- s~Lure den festen Aggregatzusatz gar nicht. Ferner ist es sieher, dab in unserem Falle der weitaus grSt3te Teil der aldehydschwetligen SSmre
aus dem Ligninkomplex entfernt worden war, wS~hrend dies beim Barium-
1) Teknisk Tidskr. (1918), 176, zitiert nach F. KSnig. 3) F. KSnig, Zellulosechemie 2, 100 (1921). 3) Zellulosechemie 2, 93 (1921). ~ Teknisk Tidskr. (1918), 176, zitiert nach F. KSnig.
SAMEC U.RIBARIC, KOLLOIDCHEM. UNTERSUCHUNOEN OlB. SULFITABLAUGE III 171
l ign insu l fona t F. K 6 n i g s nicht mit Sicherheit angenommen werden kann, da nach Untersuchungen yon M. S a m e e und M. R e b e k die Bin- dung~der Schwefligen SS.ure an die Aldehydgruppe in saurer L/Ssung
relativ bestS~ndig ist. ~ i ~;Um einen besseren Einblick in die obwaltenden Verh~iltnisse zu gewinnen, untersuchten wir das Aquivalent lei tverm6gen unserer Lignin-
2~
110
l,,0
~1 's~
~0
,Y allcr~
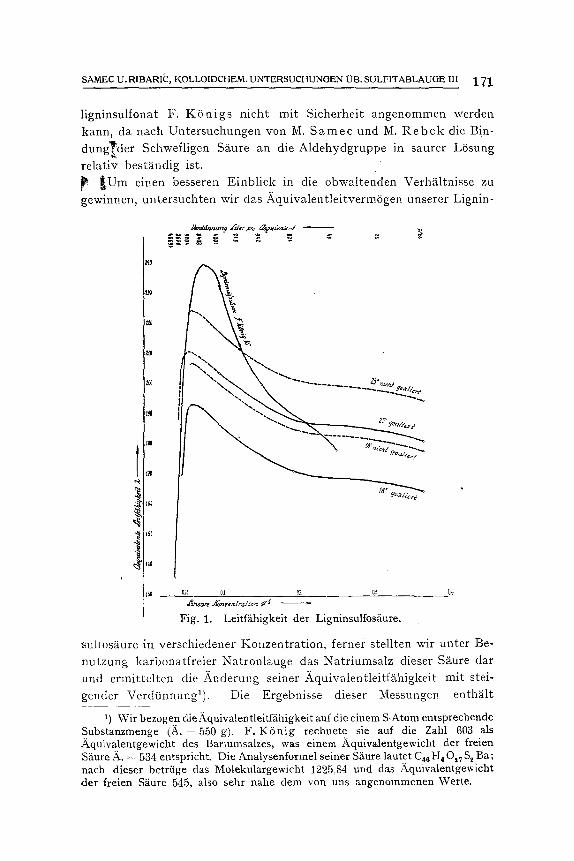
Fig. 1. Leitfiihigkeit der Ligninsulfos{iure.
sultosfiure in verschiedener Konzentrat ion, ferner stellten wir unter Be- nu t zung karbonatfreier Natronlauge das Natr iumsalz dieser S~ture dar und ermittelte,~ die Anderung seiner _~quivalentleitfghigkeit mit stei-
gender Verdtinnungl). Die Ergebnisse dieser Messungen enthglt
1) Wir bezogen die 5-quivalentleitfiihigkeit auf die einem S-Atom entsprechende Substanzmenge (5-. = 550 g). F. K6nig rechnete sie auf die Zahl 603 als Aquivalentgewicht des Bariumsalzes, was einem 5-quivalentgewicht der freien S~iure 5_. = 534 entspricht. Die Analysenformel seiner S~iure lautet C~H~O~TS 2 Ba; nach dieser betrtige das Molekulargewicht 1225,84 und das 5-quivalentgewicht tier freien S~iure 5z15, also sehr nahe dem von uns angenommenen Werte.
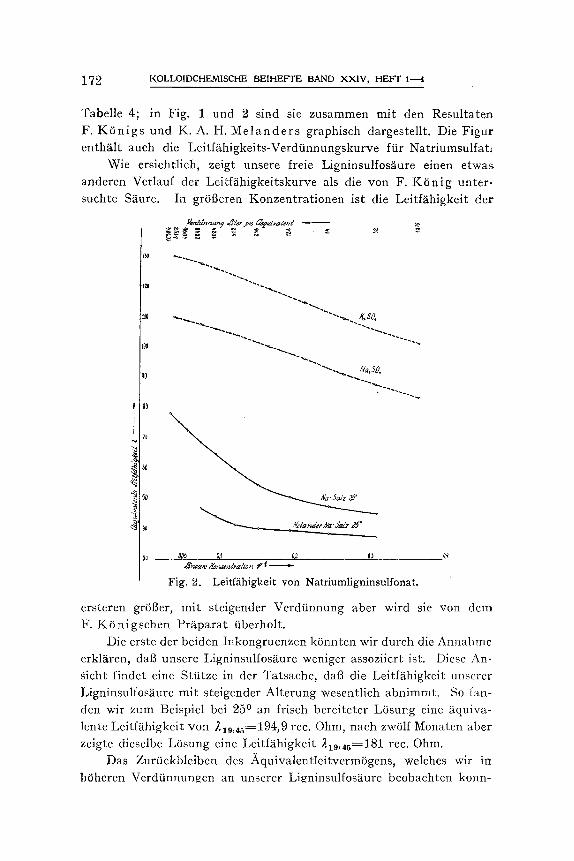
1 7 ~ N, OLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1-.-.4
Tabelle 4; in Fig. 1 und 2 sind sie zusammen mit den Resultaten F. K 6 n i g s und K. A. H. M e l a n d e r s graphiseh dargestellt. Die Figur enthS.It aueh die Leiffghigkeits-Verd~nnungskurve ftir Natriumsulfat:
Wie ersichtlich, zeigt unsere freie Ligninsulfos~iure einen etwas anderen Verlauf der Leitffihigkeitskurve als die yon F. K 6 n i g unter- suchte S~ure. In gr6t3eren Konzentrationen ist die Leitf~ihigkeit der
&so~
90 " ' " - - . . ~ /Va,50.
70 >,
~ 50 Ha- 3o&. N"
~o
Fig. 2. Leitf/ihigkeit von Natriumligninsulfonat.
e r s t e r e n gr6t3er, mit steigender Verdtinnung aber wird sie yon dem F. K6n igschen PrS.parat tiberholt.
Die erste der beiden Inkongruenzen k6nnten wir durch die Annahme erkl~tren, dab unsere Ligninsulfosgure weniger assoziiert ist. Diese An- Sicht findet eine Stiitze in der Tatsache, daft die Leiffghigkeit unserer Ligninsulfosfiure mit steigender Alterung wesentlieh abnimmt. So fan- den wir zum Beispiel bei 250 an frisch bereiteter L6sung eine /~quiva- lente Leifffihigkeit yon 210,~5=194,9 rec. Ohm, nach zwSlf Monaten abet zeigte dieselbe L6sung eine LeitfS.higkeit 219,45=181 rec. Ohm.
Das Zurtickbleiben des Aquivalentleitverm6gens, welches wir in h6heren Verdtinnun~en an unserer Li~ninsulfos~iure beobachten konn-

SAMEC U.RIBARIC, KOLLOIDCHEM.UNTERSUCHUNGEN OB.SULFITABLAUGE IlI ].~3
ten, wtirde mit der schon angedeuteten Annahme in guter Oberein- s t immung stehen, da~ die F. K S n i g s c h e Ligninsulfos~iure lloch an die Aldehydgruppen gebulldene Schweflige S~ure euth~lt, welche mit stei- gender Verdiinnung abgespalten wird und freie H 2 SO 3 in LSsung liefert.
Das typische Maximum im Verlaufe der fi~quivalentleitfiihigkeit konnten auctl wir beobachten, allerdings in wesentlich niedrigeren Kon- zentratiouen als F. K 6 n i g .
Das Natriumsalz unserer Lignillsulfosw zeigt, gleichgtiltig ob aus frischer oder gealterter L6sung bereitet, eine vSllig identische elek- trische Leitf~higkeit, es scheillt demnach die zur Neutralisatioll der Siiure benutzte Natronlauge in beiden F~illen zu demseIben Peptisationsgrade des Salzes zu ftihren. Ausnahmslos ist das AquivalentleitvermSgen unseres Natriumligninsulfonates grSt]er als bci K. A. H. M e 1 a n d e r ~) und steigt noch im Gebiete gr6ilerer Konzentration mit zunehmender Ver- dtinnullg viel steiler an. Trotzdem aber bleiben deutliche Abweichungen im u der Leitf/thigkeit-Verdtinnungskurve unseres Salzes etwa yon dem Verlaufe der Leiff~higkeit-Verd~illnungskurve yon Natriumsulfat und yon anderen anorganschen Salzen bestehen. Versuchen wir mit dem ausdriicklichen Hinweise auf diese Anomalien dennoch die O s t - w a ldsche Valenzregel zwecks allgemeiner Orientierung auf unsere Lig- ninsulfos~iure anzuwenden, so k~men wir, da ~32=45,5 und i10~4=64,75 rec. Ohm ausmacht, nach dem Ansatze B -~ (64,75--45,5) : 10zur Basizitfit yon fast genau zwei. Wir dtirfen bei Wertung dieses Ergebnisses nicht iibersehen, dab aueh bei den typisch kristalloiden Salzen die Valenz- regel nur sehr auniihernd stimmt. Unter Benutzung der Tabellen y o n L a l l d o l t - B S r n s t e i n findet man ja z. B. die Basizit~it yon Kalium- sulfat B=2,48, fiir Kaliumsulfit B=2,42, ftir Natr iumhydroarseniat B = l , 8 0 .
Die so gesch~itzte Basizit/it der Ligninsulfosliure wtirde demnaeh auf eine zwei SO3H-Gruppell entsprechende Formel als Molekularformel hindeuten, wie ja auch der Azetylgehalt zur Zahl 1100 als Molekular- gewicht fiihrt. Von dieser Gewichtsmenge w~ren durch die ana- lytisch ermittelten Atomgruppen [1 Azetyl-, 5 Methoxyl ~-) und 2 Sulfo- s/iuregruppen] 360 Einheiten gedeckt.
Ill der GrSi3enordnung st immen unsere Molekulargewichtsbestim- mungen mit den Resultaten anderer Forscher gut tibereill. P. K l a s o l l a)
t) Miilanders Substanzen diirften auch yon unserem Priiparat abweichen, da sie ja nur zu ~'l Proz. durch Naphtylamin fiillbar sind.
2) Oder eine Gruppe, die sich bei der angewandten Besdmmungsmethode wie eine CHsCO verhiilt.
a) Ark. L Kemi 6, 13 (19t7).

174 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1----4
fend nach der ebullioskopischen Methode ffir das Kalziumligninsulfonat
unter anderen auch den Molekularwert M = 1000, F. K 6 n i g kryosko- pisch ftir das Bariumsalz bei Berficksichtigung des aus der Leitf~higkeit berechneten Dissoziationsgrades M = 1108 bis 11101) und ohne Bertick-
sichtigung der Dissoziation M ~ 890. Ffir das freie Lignin aus Winter- roggenstroh fanden E. B e c k m a n n und O. L i e s c h e ~) nach der Siede- sowie Gefrierpanktsmethode mit Eisessig und mit Phenol als L6sungs- mitre 1 das Molekulargewicht 764.
Wir mfissen uns mit diesem Hinweis auf die Inkongruenz der elektro- metrischen Messungsresultate einerseits und dem kryoskopischen Ver- halten der Ligninsulfos~ure andererseits begnfigen. Eine Erklfirung
der ganzen Sachlage steht in engstem Zusammenhange mit dem all- gemeinen Problem des osmotischen Verhaltens der den Kolloidionen entsprechenden Gegenionen und dem Weehsel der Aggregation mit der
Anderung der Temperatur und der Ionisation. Wollten wir die Ansieht freier Beteiligung der elektrometrisch feststellbaren aktiven H-Ionen am osmotischen Druek beibehalten, so mfiflten wir bei der scharfen LTberein-
stimmung der kryoskopisch ermittelten und ohne Rficksieht auf die Ionisation gerechneten Molekulargr6Be mit dem aus dem chemischen Verhalten deduzierten Werte annehmen, dat3 so viel Teilehen dutch
Assoziation aus der L6sung versehwinden als durch die Ionisation neu gebildet worden sind, daft also Assoziation und Ionisation in einem
strengen gegenseitigen Abh~ngigkeitsverh~ltnis stehen wfirden. Extrapolieren wir die Leitf~higkeitskurve unseres Natriumligain-
sulfonates auf 9~ = oo, so kommen wirzu einem Werte, welcher zwisehen
86 und 90 liegt. Nach Abzug der Wanderungsgesehwindigkeit fiir das Natriumion bei 25~ verbleibt als Wanderungsgeschwindigkeit ftir das Ligninsulfos~iure-Ion 35 bis 39, also ein wesentlich h6herer Weft als
ihn F. K 6 n i g berechnet hat (auf Grund seiner Messungen kam F. K 6 n i g zu 19 rec. Ohm, w~hrend M e l a n d e r 9,3 als Wanderungsgesehwindig- keit des Ligninsulfos~ure-Ions bei 250 C annimmt).
1) F. K6nig rechnete das Molekulargewicht nach der Formel
M = s. (1 +[n- - I I ' )E" 100 V . d
in welcher s die Substanzmenge in Gramm, n die Anzahl der Ionen bedeutet, in welche ein Molekiil zerfiillt, a den Dissoziationsgrad, E die molekulare Ge- frierpunktsdepression bzw. die SiedepunktserhShung, V die Menge des L6sungs- mittels in Gramm, d die beobachtete Gefrierpunktsdepression bzw. Siede- punktserh6hung.
2) Zeitschr. L angew. Chem. 84, 285 (1921); Biochem. Zeitschr. 1~1, 293 (1921).
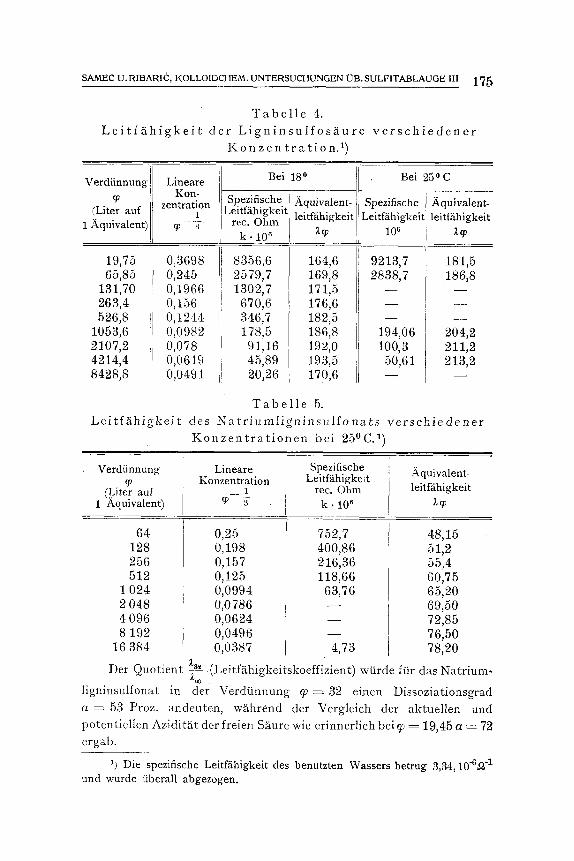
SAMEC U.RIBARIC, KOLLOIDCHEM.UNTERSUCHUNGEN I]B.SULF1TABLAUGE 1II 175
L e i t f ~ h i g k e i t T a b e l l e 4 .
d e r L i g n i n s u l f o s X u r e v e r s c h i e d e n e r K o n z e n t r a t i o n . 1)
Verd;nnung LineareKon_
(Liter auf I] zentrationl 1 ~6_quivalent) qo- i~-
19,75 65,85
131,70 263,4 526,8
1053,6 2107,2 4214,4 8428,8
0~3698 0,245 0,1966 0,156 0,1244 0,0982 0,078 0,0619 0,0491
Bei 18 0
Aquivalent- leitfiihigkeit
Zq~
Spezifische Leitfiihigkeit
rec. Ohm k �9 106
Spezifische LeitfS.higkeit
106
Bei 250 C
Aquivalent- leitfS.higkeit
8356,6 164,6 2579,7 169,8 1302,7 171,5
670,6 176,6 346,7 182,5 178,5 186,8
91,16 192,0 45,89 193,5 20,26 170,6
9213,7 2838,7
194,06 100,3
50,61
181,5 186,8
204,2 211,2
1213,2 I i
Le i t f~ th igke i t T a b e l l e 5.
des N a t r i u m l i g n i n s u l f o n a t s v e r s c h i e d e n e r K o n z e n t r a t i o n e n be i 25~ 1)
Verdiinnung Lineare Spezifische Aquivalent- rp Konzentration Leitf~ihigke~t
(Liter aut _ 1 rec. Ohm leitf~ihigkeit 1 Aquivalent) cp ~- k �9 106 Zcp
64 128 256 512
1 024 2 048 4096 8 192
16 384
0,25 0,198 0,157 0,125 0,0994 0,0786 0,0624 0,0496 0,0387
752,7 400,86 216,36 118,66
63,76
4,73
48,15 51,2 55,4 60,75 65,20 69,50 72,85 76,50 78,20
Der Quotient z3~ (LeitfS~higkeitskoeffizient) wtirde ffir das Natrium- U
ligninsulfonat in der Verdtinnung q ) = 32 einen Dissoziationsgrad a = 53 Proz. andeuten, wS.hrend der Vergleich der aktuellen und potentiellen Azidit~tt der freien SSmre wie erinnerlich bei q) = 19,45 a = 72 ergab.
1) Die spezifische Leitffihigkeit des benutzten Wassers betrug 3,34,10"6tT 1 und wurde iiberall abgezogen.

176 : KOLLO1DCHEMISCHE B E I H E F T E BAND X X l V , H E F T 1 - - 4
Wir stehen demnach hier vor sehr komplizierten Verh~iltniscen, deren Kl~rnng erst die weitere experimentelle Arbeit bringen soll.
IV.
Noch verwickelter liegen die Dinge bei unserem Anodenkolloid; doch k6nnen wir uns durch synoptische Behandlung der einzelnen Messungsresultate auch hier ein gewisses Bild fiber diese Ligninfraktion schaffen.
Die Tatsache, dab diese Substanzen durch Etektroosmose v o n d e r Hauptfrakt ion des Lignins abgetrennt werden konnten, spricht a priori ffir das Vorhandensein kleinerer Teilchen. Ob diese mit dem Gros der Substanz in einem Assoziationsgleichgewiehte stehen und beim Aus- wandern aus der Mittelzelle immer wieder nachgebildet werden oder ob es sich um andersgeartete Spaltungsstficke handelt, ist vorlS~ufig nicht aufgekl~irt, jedenfalls aber ist der weit gr6t3ere Teil der Spaltungspro- dukte erst sekund/ir durch anodische Oxydation entstanden.
Im allgemeinen physikochemischen Verhalten ist unser Anoden- kolloid der untersuchten LigninsulfosXure qualitativ ziemlich verwandt, ein wesentlicher Unterschied ergab sich nur im Verhalten bei der Titra- tion und in dem viel geringeren C H30-Gehalte. Potentiometrisch durch- geffihrt verl/iuft die Titration ganz normal, und es 15~Bt sich ein genfigend scharfer {]bergang aus dem sauren in das alkalische Gebiet verfolgen, sofern die Messungen genfigend rasch durchgeftihrt werden. {)berschfis- sige Lauge wird zeitlich nachgebunden, wobei allem Anschein nach eine Zersetzung der organischen Substanz erfogt; die LSsung bekommt einen scharfen, an Formaldehyd erinnernden Geruch.
Ein Li ter unserer 1,87prozentigen LSsung neutralisiert 8 ,48.10 "~ .~quivalente Alkali, so dab dieser Titration gemS.13 als Titer~iquivalent des Anodenkolloides A = 221 folgt.
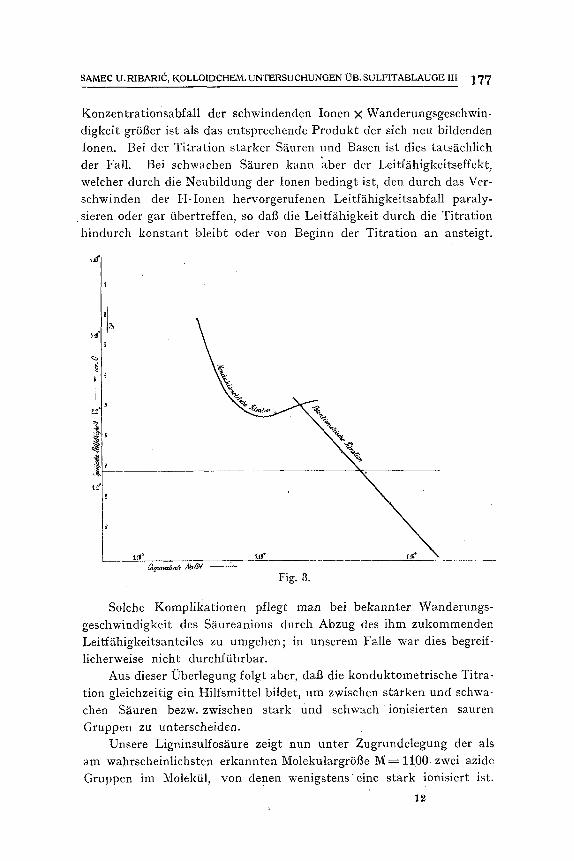
Bei der konduktometrischen Titration1) zeigt sich aber das Mini- mum der LeitfS.higkeit in viel niedrigeren Laugenzus~itzen (Fig. 3). Es bindet 1 Liter unserer LSsung bis zur Umkehr der elektrischen Leit- fghigkeit nur 5 ,32-10 .2 e% Lauge entsprechend einem Titerfiquivalent A=352.
Diese auf den ersten Blick befremdende Erscheinung wird klar, wenn wir fiberlegen, dab der Verlauf der Leitffihigkeit w~ihrend der Titration durch die VerS.nderung der Produkte: Konzentration der Ionen X Wanderungsgeschwindigkeit bes t immt ist. Die Leitf~ihigkeit wird bei der Titration demnach nur dann abnehmen, wenn das Produkt
1~ Zur LSsung wurden wachsende Laugenmengen zugesetzt.
SAMEC U.RIBARIC, KOLLOIDCHEM.UNTERSUCHUNGEN liB. SULFITABLAU(IE III 177
Konzentrationsabfall der schwindenden Ionen • Wanderungsgeschwin- digkeit gr6t3er ist als das entsprechende Produkt der sich neu bildenden Ionen. Bei der Titration starker SS.uren und Basen ist dies tatsS~chlich der Fall. Bei schwachen Sguren kann aber der Leitf~ihigkeitseffekt, welcher durch die Neubildung der Ionen bedingt ist, den dureh das Ver- schwinden der H-Ionen hervorgerufenen LeitfS~higkeitsabfall paraly- sieren oder gar tibertreffen, so dab die Leitffihigkeit durch die Titration hindurch konstant bleibt oder yon Beginn der Titration an ansteigt.
il !i i
%
Fig. 3.
Solche Komplikationen pflegt man bei bekannter Wanderungs- geschwindigkeit des SSureanions durch Abzug des ihm zukommenden LeiffSohigkeitsanteiles zu umgehen; in unserem Falle war dies begreif-
licherweise nicht durchftihrbar. Aus dieser {3berlegung folgt aber, dab die konduktometrisehe Titra-
tion gleichzeitig ein Hilfsmittel bildet, um zwischen starken und schwa- chen Sguren bezw. zwischen stark Und schwach ionisierten sauren Gruppen zu unterscheiden.
Unsere Ligninsulfosgure zeigt nun unter Zugrundelegung der als am wahrscheintichsten erkannten MolekulargrSfle M---= 1100 zwei azide Gruppen im Molektil, von denen wenigstens eine stark ionisiert ist.

178 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1 --4
Legen wir die Substanzmenge 1100 auch ffir die Diskussion der Titrations- erfolge am Anodenkolloid zugrunde, so ftihrt der LeitfShigkeitsabfall
zu drei, die potenfiometrische Titration zu insgesamt ffinf sauren
Gruppen, so dab wit also auf je 1100 g Substanz drei stark und zwei sehr schwach ionisierte Gruppen anzunehmen h~tten. 1)
Diese ffinf aziden Gruppen dfirfen wir aber wohl kaum in einer Molekel yon der Gr6Be M ----- 1100 unterbringen. Denn, wie erw~hnt, dios-
miert das Anodenkolloid viel leiehter als die LigninsulfosXure. Ferner ffihrt der Schwefelgehalt des Pr~parates zum Aquivalentgewicht A = 532
und der Methoxylgehalt (OCH 8 = 6,8 Proz.) zum Aquivalentgewicht = 456, so dab wir in 1100 g Substanz wohl fast genau zwei S-Atome,
jedoch nur 2,41 OCH3-Gruppen anzunehmen hgtten. Nach der kryoskopischen Methode ermittelt, ergab sich ffir das
Anodenkolloid die Molekulargr6Be M-----522, falls man den elektrolyti- schen Zerfallsgrad [a = 42 Proz.] nicht in Recllnung setzt. Nimmt man
abet eine freie Beteiligung der H-Ionen am osmotisehen Druck an, so
ergibt sich bei Annahme, daft die S~ure ffinfbasisch 1st, der Wert M----- 1618, und in der Annahme, dab sie dreibasiseh ist, IV[ = 1180.
Wir versuchten analog wie bei der Ligninsulfos~ure die Basizitgt nach der Valenzregel zu schgtzen und kamen, nachdem die S~ure poten-
tiometrisch neutralisiert worden war, zu den Werten B = 2,86--2,92, welche mit dem Molekularwerte M = 522 noch am besten in Einklang
zu bringen sind. Die hier klar gewordenen Unterschiede zwischen der Ligninsulfo-
s~ure und dem Anodenkolloid sind demnaeh dutch nachstehende An- gaben festgelegt:
1. Die Molekulargr6Be des Anodenkolloids ist kleiner als die Mole- kulargrSt3e der Ligninsulfos~ure; 2. der Methoxylgehalt ist geringer und 3. die Zahl der aziden Gruppen ist grSfler. Ob letztere als Karboxyl
oder als Phenolgruppen zu formulieren sind, steht nicht vollkommen
fest. Drticken wir sie als Karboxyle aus, so lieBe sich die quan- titative Beziehung zwischen dem kolloiden Rest (Ligninsulfos~.ure) und
demAnodenzellenkolloid (Sulfolignin-Karbonsgure) dureh nachstehendes Schema klarlegen, bei welehem nattirlieh die Verteilung der SO3H- ,
COOH- und OCHs-Gruppen v611ig willkfirlich ist. SOaH SOaH SOsH SOaH SO3H SO3H
I I f I I I 2 R1 R~ -- )- RI--(COOH), +R~--COOHq-R3--(COOH),q-Ra--COOH
I I ] | " I (OCH3) 3 ~OCHs) ~ OCHa OCH 8 OCHa (OCH~),
L Die Dissoziation dieser Gruppen ist ungef~ihr die gleiche wie bei H C O O C B 3 in 1/10 n-L6sung.

SAMEC U. RIBARIC, KOLLOIDCHEM. UNTERSUCHUNGEN I~IB. SULFITABLAUGE llI 179
Die potentriometriseh feststellbare H-Ionenaktivit~t betrtigt im Anodenkolloid (1,87 Proz. Trockensubstanz) bei Zimmertemperatur 3,55 �9 10 -2. Aueh diese Aktivit~it ist grSt3er, als der beobachteten elek- trischen LeitfS~higkeit entspricht. Letztere betr~gt ja 106,1 �9 104 rec. Ohm, w~ihrend die potentiometrisch gemessenen H-Ionen all ein, also ohne Beteiligung der Anionen bei 250 C eine Leitf~higkeit 124,3.10 .4 tee. Ohm bedingen w/irden. Bei dem dem kristalloiden Ztlstande nSher stehenden prim~iren Anodendialysat n~ihern sich die beobachteten und gereehneten Werte noch mehr, was fiir die frtiher erw~hnte Hydratation des kolloiden Anteiles eine weitere Stiitze w~ire.
Der Quotient H-IonenaktivitSs GesamtaziditS~t gibt uns wieder die obere Grenze ftir den elektrolytischen Zerfallsgrad (ira Sinne der klas- sisehen Theorie) an und berechnet sich zu = 0,42.
Die bier besehriebenen Fraktionen der Sulfitablauge bilden trotz gewisser Komplikationen ein sehr reines Material, an welchem das Studium der Ligninsubstanzen fortgeftihrt werden soil. Eine weitere theoretische Auswertung der hier mitgeteilten Ergebnisse im Bezug auf Farbe, relative St~rke der sauren Gruppen, Vorhandensein yon Karboxyl- und Phenolgruppen sowie yon chromophoren Gruppen, ferner weitere Konsequenzen aus dem elektrochemischen Verhalten folgt in der nS.ehsten Mitteilung.
Z u s a m m e n f a s s u n g .
1. Die yon M. S a m e c und M. R e b e k durchgefiihrte elektrodialy- tische und elektroosmotische Zerlegung der Sulfitablauge wurde ein- gehender untersucht und die Beobachtungen dieser Autoren reproduziert.
2. Der erhaltene kolloide Rest erwies sich nach Untersuchung durch fraktionierte F~llung der Bariumsalze und der Naphtylaminsalze als einheitlieh, so dab es gelungen ist, eine sehr reine und gut definierte Li- gninsulfos~ure zu gewinnen.
3. Wir ermittelten aus dem S-Gehalte, dem Methoxylgehall{e der titrierbaren Azidit~t und dem Azetylgehalte das fi~quivalentgewicht und das Minimalmolekulargewicht; letzteres wurde aucn kryoskopiscla bestimmt.
4. Das Verhalten der fi~quivalentf~higkeit der freien S~ure und des Natriumsalzes bei steigender Verdtinnuog wurde untersucht und nach de r Valenzregel versuehsweise die Basizit~t der LigninsulfosOure gesch~tzt.
5. Durch Elektroosmose werden vom Hauptanteil kleinermolatige Anteile abgetrennt. Es scheint hierbei wonl infolge anodischer Oxyda- tion ungef~ihr eine Halbierung der Molekel einzutreten.

] 8 0 KOLLOIDCHEMISCHE BEIHEFTE BAND XXIV, HEFT 1 - - 4
6. Der Methoxylgehalt des Anodenkolloids ist gering, die Azidit~it gr613er als im kolloiden Rest.
7. Die einzelnen sauren Gruppen erweisen sich 31s verschieden stark ionisiert.
8. Ein Tell der Substanz wird w~ihrend der Elektroosmose bis zu gasf6rmigen Produkten oxydiert.
9. Die O CH3-Gruppen erweisen sich bei der Elektrodialyse in bezug auf Oxydations- und Diffusionsf~ihigkeit als verschiedenwertig.
10. Die freie Ligninsulfos~iure unterliegt in w~sseriger L6sung zeit- lichen )i, nderungen, bei welchen unter anderem die elektrische Leitf~ihig- keit abnimmt.





























