Embed Size (px)
Citation preview

Neurologische Klinik
des St.-Josef-Hospitals Bochum
- Universitätsklinik -
der Ruhr-Universität Bochum
Direktor: Prof. Dr. med. H. Przuntek
_______________________________________
Laktat- und Pyruvatspiegel bei Patienten mit degenerativer Ataxie
in Ruhe und während Fahrradbelastung
Inaugural-Dissertation
zur
Erlangung des Doktorgrades der Medizin
einer
Hohen Medizinischen Fakultät
der Ruhr-Universität Bochum
vorgelegt von
Jochen B. Müller
aus Gronau/Westfalen
Bochum 1999

Dekan: Prof. Dr. Eysel
Referent: Prof. Dr. Kuhn
Koreferent: Prof. Dr. Hanstein
Tag der mündlichen Prüfung: 16. Mai 2002

Für Stefanie.

Inhaltsverzeichnis
Kapitel Seite
1 Einleitung und Fragestellung ............................................................................... 1
1.1 Einteilung und Symptomatik der degenerativen Ataxien ................................. 1
1.2 Genetik und Pathogenese...................................................................................... 6
1.3 Fragestellung der Arbeit ....................................................................................... 8
2 Materialien und Methoden ................................................................................. 13
2.1 Materialien ........................................................................................................... 13
2.1.1 Geräte..................................................................................................................... 13
2.1.2 Verbrauchsartikel................................................................................................... 13
2.1.3 Chemikalien........................................................................................................... 14
2.2 Versuchsablauf und Entnahme der Proben...................................................... 14
2.3 Bestimmung von Pyruvat.................................................................................... 17
2.4 Bestimmung von Laktat...................................................................................... 18
2.5 Einfluß von Fehlerquellen................................................................................... 19
2.6 Patientengut ......................................................................................................... 21
2.7 Statistische Auswertung ...................................................................................... 21
3 Hauptteil mit Darstellung der Ergebnisse......................................................... 22
3.1 Charakterisierung der Patienten und Darstellung der Einzelergebnisse....... 22
3.1.1 Patient C5-UL, männlich, 51 Jahre ....................................................................... 23
3.1.2 Patient C1-AK, männlich, 38 Jahre ....................................................................... 25
3.1.3 Patientin C4-GN, weiblich, 49 Jahre ..................................................................... 27
3.1.4 Patient C10-MR, männlich, 34 Jahre..................................................................... 28
3.1.5 Patient C11-KR, männlich, 69 Jahre ..................................................................... 30
3.1.6 Patient C12-KSG, männlich, 59 Jahre................................................................... 31
3.1.7 Patient C6-GH, männlich, 65 Jahre ....................................................................... 33
3.1.8 Patient C2-HK, männlich, 58 Jahre ....................................................................... 35
3.1.9 Patient C7-KSR, männlich, 61 Jahre ..................................................................... 37

3.1.10 Patient C9-SW, männlich 52 Jahre........................................................................ 39
3.1.11 Patient C8-GK, weiblich, 51 Jahre ........................................................................ 40
3.2 Statistische Auswertung des gewonnenen Datenmaterials .............................. 42
3.2.1 Deskriptive Darstellung der Daten ........................................................................ 42
3.2.2 Statistische Auswertung mittels t-Test .................................................................. 44
4 Diskussion............................................................................................................. 46
4.1 Kommentierung der Ergebnisse......................................................................... 47
4.1.1 Laktat ..................................................................................................................... 47
4.1.2 Pyruvat................................................................................................................... 51
4.1.3 Laktat-Pyruvat-Quotient........................................................................................ 52
4.2 Diskussion der Ergebnisse .................................................................................. 52
5 Zusammenfassung ............................................................................................... 58
6 Literaturverzeichnis ............................................................................................ 60

Verzeichnis der Abbildungen Abbildung 1: Laktat u. Pyruvat in [mg/dl] für Patient C5-UL u. Kontrollperson .................................. 25
Abbildung 2: Laktat u. Pyruvat in[mg/dl] für Patient C1-AK u. Kontrollperson................................... 27
Abbildung 3: Laktat u. Pyruvat in [mg/dl] für Patientin C4-GN u. Kontrollperson .............................. 28
Abbildung 4: Laktat u. Pyruvat in [mg/dl] für Patient C10-MR u. Kontrollperson............................... 30
Abbildung 5: Laktat u. Pyruvat in [mg/dl] für Patient C11-KR u. Kontrollperson................................ 31
Abbildung 6: Laktat u. Pyruvat in [mg/dl] für Patient C12-KSG u. Kontrollperson ............................. 33
Abbildung 7: Laktat u. Pyruvat in [mg/dl] für Patient C6-GH u. Kontrollperson ................................. 35
Abbildung 8: Laktat u. Pyruvat in [mg/dl] für Patient C2-HK u. Kontrollperson ................................. 37
Abbildung 9: Laktat u. Pyruvat in [mg/dl] für Patient C7-KSR u. Kontrollperson ............................... 38
Abbildung 10: Laktat u. Pyruvat in [mg/dl] für Patient C9-SW u. Kontrollperson ............................... 40
Abbildung 11: Laktat u. Pyruvat in [mg/dl] für Patientin C8-GK u. Kontrollperson ............................ 42
Abbildung 12: Zeitliche Entwicklung der Laktatkonzentration ............................................................... 43
Abbildung 13: Zeitliche Entwicklung der Pyruvatkonzentration............................................................. 43

Verzeichnis der Tabellen Tabelle 1: Datendokumentation ................................................................................................................... 15
Tabelle 2: Versuchsprotokoll........................................................................................................................ 16
Tabelle 3: Diagnose, Alter und Geschlecht der Probanden ....................................................................... 23
Tabelle 4: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C5-UL u. Kontrollperson............................. 24
Tabelle 5: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C1-AK u. Kontrollperson ............................ 26
Tabelle 6: Laktat- u. Pyruvatwerte in [mg/dl] für Patientin C4-GN u. Kontrollperson ......................... 28
Tabelle 7: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C10-MR u. Kontrollperson.......................... 29
Tabelle 8: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C11-KR u. Kontrollperson .......................... 31
Tabelle 9: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C12-KSG u. Kontrollperson........................ 32
Tabelle 10: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C6-GH u. Kontrollperson .......................... 34
Tabelle 11: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C2-HK u. Kontrollperson .......................... 36
Tabelle 12: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C7-KSR u. Kontrollperson ........................ 38
Tabelle 13: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C9-SW u. Kontrollperson .......................... 40
Tabelle 14: Laktat- u. Pyruvat in [mg/dl] für Patientin C8-GK u. Kontrollperson ................................ 41
Tabelle 15: Vergleich der Patienten und Kontrollen hinsichtlich der Differenz der logarithmierten
Werte zum Zeitpunkt t4 und t0 .......................................................................................................... 45
Tabelle 16: Kilometerleistung der Probanden ............................................................................................ 53

Verzeichnis der verwendeten Abkürzungen ADCA .................................... autosomal dominant zerebelläre Ataxie
DNA ...................................... Desoxyribonukleinsäure
FADH2 .................................. Flavinadenindinucleotid (reduzierte Form)
GPT ....................................... Glutamat-Pyruvat-Transaminase
IDCA ..................................... idiopathisch sporadisch zerebelläre Ataxie
LDH ...................................... Laktatdehydrogenase
NAD+ ..................................... Nicotinamidadenindinucleotid
OPCA ..................................... olivopontozerebelläre Atrophie
SCA ........................................ spinozerebelläre Ataxie

Einleitung und Fragestellung 1
1 Einleitung und Fragestellung
1.1 Einteilung und Symptomatik der degenerativen Ataxien
Die Übersetzung des griechischen Wortes „ataxia“ bedeutet soviel wie „Unordnung“ oder
„Verwirrung“. Nicht nur die Krankheitssymptomatik sondern auch die Einteilung der
Ataxien scheint dieser Bedeutung zu entsprechen, wenn man einerseits die Vielfältigkeit
der Symptome und andererseits die zahlreichen Einteilungsversuche betrachtet. Der
Begriff „Ataxie“ bezeichnet eine Störung des geordneten Ablaufs und der Koordination
von Muskelbewegungen, was sich durch Störungen der Okulomotorik und
Diadochokinese, Dysarthrie, Intentionstremor, Rumpfataxie sowie Ataxie beim Stehen
oder Gehen bemerkbar machen kann. Die nachfolgende Übersicht soll einen kurzen
Überblick über die heterogene Gruppe der Ataxien geben.
Frühe Einteilungen orientierten sich an Krankheitserscheinungen und wurden nach
Erstbeschreibern benannt (z.B. Friedreich 1863, Menzel 1891). Wenige Jahre später
wurden neue Einteilungen auf der Grundlage neuropathologischer Veränderungen
erarbeitet (Holmes 1907, Greenfield 1954). Nicht zuletzt wegen ihres geringen Nutzens für
den klinischen Gebrauch wurde die letztgenannte Einteilung zugunsten einer
Klassifikation nach klinischen und genetischen Gesichtspunkten verlassen (Harding 1983).
Harding teilte die Ataxien in früh, d. h. in der Regel vor dem 25. Lebensjahr beginnende
(z.B. Friedreich-Ataxie) und spät beginnende, also nach dem 25. Lebensjahr beginnende
Formen ein. Bei den spät beginnenden Formen wurden die idiopathischen zerebellären
Atrophien (IDCA) den autosomal dominant erblichen zerebellären Ataxien (ADCA) mit 3
Unterformen gegenübergestellt: ADCA I ist ein klinisch und genetisch sehr heterogener
Subtyp, dem ca. ¾ der Patienten zugeordnet werden können. Klinisch stehen eine
progressive Ataxie, kombiniert mit zusätzlichen Symptomen wie Sakkadenverlangsamung,
unterschiedlich ausgeprägter supranukleärer Ophthalmoplegie, Pyramidenbahnzeichen,
Muskelatrophien, Basalgangliensymptomen und Sensibilitätsstörungen im Vordergrund;
seltener kommen extrapyramidalmotorische Zeichen, Inkontinenz und Entwicklung einer

Einleitung und Fragestellung 2
Demenz vor. Während Subtyp II Erkrankungen mit begleitender pigmentärer
Retinadegeneration umfaßt, grenzt sich Subtyp III durch eine Ataxie mit reiner
Kleinhirnsymptomatik ohne zusätzliche Symptome ab.
Die von Harding vorgenommene Einteilung hat sich jedoch als unbefriedigend erwiesen, u.
a. weil es hinsichtlich des klinisch manifesten Erkrankungsbeginns bei den autosomal
dominanten Ataxien eine hohe Varianz vom jungen bis zum höheren Lebensalter gibt
(Übersicht von Klockgether u. Mitarb. 1995). Auch hinsichtlich der Symptomatik besteht
eine große Variabilität, so daß neben einer rein zerebellären Symptomatik auch
Mischformen existent sind, die eine exakte Zuordnung unmöglich machen (Harding 1984).
Heute werden die degenerativen Ataxien grundsätzlich in erbliche und nichterbliche
Erkrankungen unterschieden, wobei beiden Krankheitsgruppen das Hauptsymptom der
progressiven Ataxie gemeinsam ist.
Die erblichen Formen werden entsprechend ihrem formalgenetischen Erbgang in
autosomal rezessive, autosomal dominante und die sehr seltenen, hier nicht weiter
beschriebenen, X-chromosomal vererbten Ataxien unterteilt. Die nichterblichen Ataxien
unterliegen einer Einteilung in idiopathische Erkrankungen ohne bekannte Ursache und
symptomatische Ataxien mit bekannter, d.h. beispielsweise entzündlicher, toxischer,
paraneoplastischer oder physikalischer Genese:
Erbliche Ataxien:
• autosomal rezessive Ataxien
• autosomal dominante Ataxien
• X-chromosomal vererbte Ataxien
Nichterbliche Ataxien:
• idiopathisch zerebelläre Ataxien
• symptomatische Ataxien
Die häufigste der autosomal rezessiven Ataxien ist die Friedreich-Ataxie mit einer
progredienten Ataxie, Areflexie der unteren Extremität, Auftreten von

Einleitung und Fragestellung 3
Pyramidenbahnzeichen trotz schlaffer Paraparese sowie Entwicklung einer Dysarthrie
wenige Jahre nach Erkrankungsmanifestation. Zusätzlich bestehen häufig eine
Kardiomyopathie, Glukosestoffwechselstörungen und Skelettdeformitäten wie Skoliose
und Friedreich-Hohlfuß mit Krallenzehen. Außerdem gehören zu der Gruppe der
autosomal rezessiven Ataxien die selteneren Formen wie die Abetalipoproteinämie, der
Morbus Refsum, die Ataxia teleangiectasia, die Vitamin-E-Mangelataxie, die früh
beginnende zerebelläre Ataxie mit erhaltenen Muskeleigenreflexen (Fickler-Winkler) und
andere früh beginnende zerebelläre Ataxien.
Die jüngsten molekulargenetischen Identifikationen von Genloci und die teilweisen
Entdeckungen zugrundeliegender Mutationen der autosomal dominanten zerebellären
Ataxien machen eine Neuordnung der autosomal dominant vererbten Ataxien anhand ihrer
genetischen Grundlagen erforderlich. Diese Neuordnung unterliegt einer ständigen
Anpassung an die jeweils neuen Erkenntnisse auf dem Gebiet der Molekulargenetik
(Übersicht von Schöls u. Mitarb. 1997).
Die ADCA haben die Bezeichnung „spinozerebelläre Ataxie (SCA)“ erhalten und werden
nach ihren Genorten in der Reihenfolge ihrer Beschreibung durchnumeriert. Bis 1999
waren 7 Unterformen bekannt; bis 2001 wurden 14 Subtypen der ADCA unterschieden:
SCA 1 bis 8 und 10 bis 14, zuzüglich der dentatorubropallidoluysianen Atrophie
(Übersicht von Riess u. Mitarb. 2001).
Im Vergleich zu der von Harding vorgenommenen Klassifikation entsprechen die SCA-
Typen 1 bis 4, 8, 12 und 13 dem ADCA-Typ I, die SCA 7 dem ADCA-Subtyp II und die
SCA 5, 6, 10, 11 und 14 dem ADCA-Typ III.
Mit Einschränkung der klinisch schwierigen Differenzierbarkeit einiger Subtypen lassen
sich die SCA wie folgt unterscheiden:
• Die SCA 1 stellt eine Erkrankung mit verschiedenartigen Mischbildern aus
peripherer Polyneuropathie, Pyramidenbahnschädigung, externer Ophthalmoplegie,
bulbärer Zungenatrophie und Paresen der fazialen Muskulatur dar; gemeinsam ist

Einleitung und Fragestellung 4
den Patienten lediglich die zerebelläre Ataxie und im weiteren Verlauf der
Erkrankung eine Schluckstörung. Die Feststellung, daß die Erkrankung in
nachfolgenden Generationen dazuneigt, früher auszubrechen, wurde als
„Antizipation“ bezeichnet, was für alle SCA bis auf die SCA 6 zutrifft (Komure u.
Mitarb. 1995).
• Das Krankheitsbild der SCA 2 unterscheidet sich von dem der SCA 1 durch das
vermehrte Auftreten von verlangsamten Blicksakkaden, Reflexabschwächung und
geringerer Spastik.
• Die SCA 3 wurde genetisch in derselben Region wie die Machado-Joseph-
Erkrankung, eine ebenfalls autosomal dominant vererbte, subakut verlaufende
Kleinhirnatrophie, lokalisiert, unterscheidet sich aber klinisch von ihr und besitzt
ähnliche Symptome wie die SCA 1 (Übersicht von Schöls u. Mitarb. 1997). Neben
Ataxie, Dysarthrie und ausgeprägtem Nystagmus werden Pseudoexophthalmus,
Restless-Legs-Syndrom und je nach Erkrankungsbeginn Spastik bzw. periphere
Polyneuropathie beobachtet.
• Die Symptomatik der SCA 4 ist gekennzeichnet durch eine zerebelläre Ataxie mit
normaler Augenbeweglichkeit und neuropathischen Beschwerden bei
Pyramidenbahnschädigung.
• Die SCA 5 ist auf rein zerebelläre Zeichen beschränkt und zeigt keine
Einschränkung der Lebenserwartung.
• Patienten, die an der SCA 6 leiden, zeigen rein zerebelläre Symptome mit meist
episodischem Verlauf bei ebenfalls normaler Lebenserwartung.
• Die SCA 7 ist charakterisiert durch eine Ataxie mit Retinadegeneration, welche zu
einem Visusverlust bis zur totalen Erblindung führt. Zusätzliche
Krankheitssymptome sind verlangsamte Blicksakkaden, externe Ophthalmoplegie
und Pyramidenbahnzeichen.
• Charakteristische Symptome der SCA 8 sind Ataxie, Dysarthrie und Nystagmus.
• Bei der SCA 10 kann zur Ataxie-Symptomatik mit Dysarthrie und Nystagmus auch
eine Epilepsie treten.
• Die SCA 11 ist neben gesteigerten Muskeleigenreflexen ebenfalls durch die Trias
Ataxie, Dysarthrie und Nystagmus gekennzeichnet.

Einleitung und Fragestellung 5
• Bei der SCA 12 finden sich Ataxie, Nystagmus und Tremor.
• Die SCA 13 kann - bei sehr langsamen Fortschreiten - u. U. bereits in frühester
Kindheit ausbrechen und weist neben motorischer und geistiger Retardierung
Ataxie, Dysarthrie, Nystagmus und Reflexsteigerung auf.
• Die SCA 14 ist ebenfalls langsam progredient. Charakteristika sind neben der
Ataxie bei frühem Ausbrechen auch Kopftremor- und myoklonus.
• Die dentatorubropallidoluysiane Atrophie (DRPLA) ist durch Ataxie, Dysarthrie
und Demenz gekennzeichnet. Bei frühem Beginn tritt eine Myoklonusepilepsie, bei
spätem Beginn eine Choreoathetose hinzu.
Unter dem Oberbegriff der idiopathisch zerebellären Ataxie (IDCA) wird eine heterogene
Gruppe neurodegenerativer Erkrankungen zusammengefaßt, die einerseits durch eine
fortschreitende Ataxie mit Beginn im Erwachsenenalter und andererseits durch das
sporadische Auftreten gekennzeichnet ist (Harding 1981). Man unterscheidet die rein
zerebelläre Form mit zerebellärer kortikaler Atrophie von einer Form mit zusätzlichen
nicht zerebellären Symptomen, der eine olivopontozerebelläre Atrophie (OPCA) zugrunde
liegt (Klockgether u. Mitarb. 1990). Die klinischen Diagnosekriterien für eine
idiopathische zerebelläre Ataxie sind eine progressive, anders nicht erklärbare Ataxie mit
Krankheitsbeginn nach dem 25. Lebensjahr und eine leere Familienanamnese bei negativer
Blutsverwandtschaft der Eltern (Übersicht von Klockgether u. Mitarb. 1995).
Ist die idiopathische zerebelläre Ataxie mit weiteren Symptomen vergesellschaftet, kann es
sich um eine Multisystematrophie (MSA) handeln. Mit dem Begriff MSA wird eine
progressive idiopathische Neurodegeneration des Erwachsenenalters umschrieben, wobei
in unterschiedlichem Ausmaß zerebelläre Dysfunktion, autonomes Versagen und
Parkinson-Symptomatik mit schlechtem Ansprechen auf L-Dopa-Therapie kombiniert sein
können.
Neben den idiopathischen zerebellären Ataxien gehören noch die symptomatischen
Ataxien zur Gruppe der nichterblichen Ataxien. Im einzelnen sind dies Ataxien bei
chronischem Alkoholismus, der zu einer zerebellären kortikalen Degeneration führen kann.

Einleitung und Fragestellung 6
Andere Noxen sind z.B. Antiepileptika, bestimmte Zytostatika, Schwermetalle oder
Lösungsmittel. Eine Ataxie kann auch die seltene neurologische Komplikation einer
Hypothyreose sein, die bei entsprechender Behandlung meist reversibel ist. Eine Ataxie
kann auch bei chronischem Fettmalabsorptionssyndrom und konsekutivem Vitamin-E-
Mangel auftreten. Auch bei Bronchial-, Ovarial-, und Mammakarzinomen oder
Lymphomen im Rahmen eines paraneoplastischen Geschehens kann sich eine Ataxie
entwickeln. Schließlich muß noch die Ataxie mit physikalischer Genese, d.h. Erhöhung der
Körperkerntemperatur auf > 41° Celsius, Erwähnung finden.
1.2 Genetik und Pathogenese
Molekulargenetische Untersuchungen haben gezeigt, daß die Expansion von
Trinukleotidsequenzen sowohl bei autosomal rezessiv als auch bei autosomal dominant
vererbten Ataxien eine Rolle spielen.
Als Prototyp der autosomal rezessiven Ataxien wird im folgenden auf die Friedreich-
Ataxie eingegangen: Ende der achtziger Jahre konnte das Gen der Friedreich-Ataxie in der
Zentromer-Region des Chromosoms 9 lokalisiert werden (Chamberlain u. Mitarb. 1988).
Wenige Jahre später gelang es, die für die Mehrzahl der Fälle ursächliche Mutation, eine
Verlängerung der GAA-Trinukleotidsequenz, nachzuweisen (Campuzano u. Mitarb. 1996).
Studien an Hefe-Spezies, die das vom Friedreich-Gen kodierte Protein, das Frataxin, in
homologen Formen enthalten, konnten zeigen, daß der Verlust des Frataxins nicht nur zu
oxidativem Stress bei Exposition gegenüber Wasserstoffperoxid und Eisen führt, sondern
auch eine Überladung der Hefezellen mit mitochondrialem Eisen verursacht (Foury 1997).
Außerdem konnten an Muskelbiopsien von Patienten mit Morbus Friedreich
mitochondriale Defekte, besonders des Komplexes I der Atmungskette, nachgewiesen
werden (Schöls u. Mitarb. 1996). Die Friedreich-Ataxie ist nach heutiger Erkenntnis eine
mitochondriale Erkrankung auf genetischer Basis, bei der das Fehlen des Frataxins eine
Eisenanhäufung in den Mitochondrien verursacht. Die damit verbundene mitochondriale

Einleitung und Fragestellung 7
Anfälligkeit gegenüber oxidativem Streß bedingt über eine Störung der mitochondrialen
Energiegewinnung den Zelltod.
Auch bei den ADCA spielen repetitive Trinukleotidsequenzen eine Rolle. Fast alle bisher
bekannten Subtypen werden durch die Expansion von sich wiederholenden Trinukleotid-
Sequenzen in den entsprechenden Genen oder in deren Nähe hervorgerufen (Übersicht von
Riess u. Mitarb. 2001). So sind die für die hier untersuchten Ataxie-Subtypen SCA 1, SCA
3 und SCA 6 verantwortlichen Mutationen sogenannte CAG-“repeats“. Die entsprechend
erweiterten Gensequenzen kodieren für verlängerte Polyglutaminstränge („Ataxine“). Die
entsprechenden Genorte wurden wie folgt lokalisiert: Der betreffende Genort für die SCA
1 auf dem kurzen Arm von Chromosom 6 (Orr u. Mitarb. 1993), für SCA 3 auf dem langen
Arm von Chromsom 14 (Takiyama u. Mitarb. 1993; Stevanin u. Mitarb. 1994) und für
SCA 6 auf dem kurzen Arm von Chromosom 19 (Zhuchenko u. Mitarb. 1997).
Es wird angenommen, daß die Krankheitserscheinungen weniger durch einen Verlust der
ursprünglich kodierten Proteininformation, als vielmehr durch eine bisher nicht näher
bekannte zerstörerische Funktion des verändert kodierten Polyglutaminproteins
hervorgerufen werden (Klockgether u. Mitarb. 1998). Möglicherweise stehen die
verlängerten Polyglutaminketten im Zusammenhang mit neuronalen Einschlusskörperchen
in den Zellkernen betroffener Gehirnareale, wobei deren definitive Bedeutung für die
Neurodegeneration noch unbekannt ist. Es gibt Hinweise, daß die Einschlußkörperchen
Teil eines zellulären Abwehrmechanismus gegen die wahrscheinlich toxisch wirksamen
Polyglutaminproteine sein könnten (Übersicht von Riess u. Mitarb. 2001).
Dazu paßt die Entdeckung, daß bestimmte, offenbar der Ubiquitinierung zuführende und
somit schützend wirkende, Chaperone, nämlich Hitzeschockproteine (Hsp 70), in den
Einschlußkörperchen gefunden wurden (Chai u. Mitarb. 1999). Auch die Entdeckung, daß
in Zellkultur eine Überexpression von Chaperonen zu einem verminderten Absterben von
Zellen führte, unterstützt diese Hypothese (Cummings u. Mitarb. 1998).

Einleitung und Fragestellung 8
Die Mutation der SCA 6 unterscheidet sich von den übrigen autosomal dominant vererbten
Ataxien dadurch, daß sie einen spannungsabhängigen Kalziumkanal betrifft und seine
physiologische Funktion beeinträchtigt (Ludwig u. Mitarb. 1997).
Trotz dieser Fortschritte hinsichtlich der molekulargenetischen Grundlagen autosomal
dominant vererbter Ataxien ist ihr genauer Pathomechanismus noch immer Gegenstand der
Forschung: Die Funktion der verschiedenen Ataxine ist derzeit ebenso unbekannt wie die
Interaktion der jeweiligen Ataxine auf zellulärer Ebene. Im Hinblick auf die Fragestellung
dieser Arbeit bleibt festzustellen, daß bei Patienten mit autosomal dominant zerebellärer
Ataxie mitochondriale Enzymdefekte gefunden wurden: Bei einer Untersuchung von 30
Patienten mit autosomal dominant zerebellärer Ataxie wurden bei fast der Hälfte der
Patienten mitochondriale Abnormalitäten festgestellt (Schöls u. Mitarb. 1996).
In der gleichen Untersuchung wurden in der Gruppe der nichterblichen Ataxien Störungen
der Mitochondrienenzyme bei 3 von 6 Patienten mit idiopathisch zerebellärer Ataxie und
bei keinem von 6 Patienten mit symptomatischer, d. h. sprue-induzierter, alkoholtoxischer
bzw. paraneoplastisch bedingter Ataxie beschrieben (Schöls u. Mitarb. 1996).
1.3 Fragestellung der Arbeit
1962 prägten Luft u. Mitarb. den Begriff der „mitochondrialen Myopathie“, für deren
Diagnose neben dem Nachweis morphologisch veränderter Mitochondrien und einem
spezifischen biochemischen Defekt an Mitochondrien außerdem ein durch den
mitochondrialen Stoffwechseldefekt erklärbares klinisches Erscheinungsbild wie z. B.
Muskelschwäche, Myalgien oder Intoleranz gegenüber Ausdauerleistungen gegeben sein
muß (Luft u. Mitarb. 1962). Diagnostisch wegweisend für diese Erkrankungen ist eine
Erhöhung des Laktatspiegels im venösen Blut in Ruhe oder bei Belastung (Reichmann u.
Mitarb. 1988/2). 15 Jahre später prägten Shapira u. Mitarb. den Begriff der
„mitochondrialen Enzephalomyopathie“ (Shapira u. Mitarb. 1977). Die mitochondrialen
Enzephalomyopathien zeichnen sich durch in ihrer Struktur bzw. Funktion veränderte
Mitochondrien im Gehirn und in der Muskulatur aus, wie dies beispielsweise bei dem

Einleitung und Fragestellung 9
Kearns-Sayre-Syndrom (Retinitis pigmentosa, Opthalmoplegie, Ataxie, Ptosis, kardiale
Komplikationen) oder den Krankheitsbildern „myoclonus epilepsy with ragged red fibers“
(= MERRF) und „mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like
episodes“ (= MELAS) der Fall ist (Kearns u. Mitarb. 1958, Fukuhara u. Mitarb. 1980,
Pavlakis u. Mitarb. 1984). Für diese Erkrankungen wird ebenfalls eine häufige bis sehr
häufige Erhöhung des Laktatspiegels und eine weniger häufige bis häufige Erhöhung des
Pyruvatspiegels angegeben (Zenner u. Mitarb. 1990).
Anfang der neunziger Jahre berichteten verschiedene Autoren über Autopsien von
Patienten mit olivopontozerebellärer Atrophie und beschrieben einen Zusammenhang
zwischen zerebellärer Degeneration und mitochondrialen Abnormalitäten: Truong u.
Mitarb. berichteten 1990 über die Autopsie einer Patientin, die klinisch an zerebellärer
Dysarthrie, Muskelschwäche, Bradykinesie und Gangataxie litt. In-vitro-Untersuchungen
des mitochondrialen Stoffwechsels dieser Patientin hatten einen Defekt des Komplexes I
der Atmungskette aufgedeckt. Der pathologische Befund ergab striatoniagrale
Degeneration und olivopontozerebelläre Atrophie (Truong u. Mitarb. 1990).
1991 beschrieben Kageyama u. Mitarb. den Fall einer Patientin, die klinisch an
progressiver Ataxie, Muskelschwäche und Hörverlust litt. Laborchemisch wurden bei
dieser Patientin erhöhte Laktat- und Pyruvatspiegel in Ruhe und bei Belastung gefunden;
die Untersuchung einer Muskelbiopsie ergab eine Erniedrigung der Komplex-I-Aktivität.
Bei der Autopsie fand sich eine deutliche Degeneration des olivopontozerebellären
Systems (Kageyama u. Mitarb. 1991).
Über mitochondriale Dysfunktion war zuvor nicht nur im Zusammenhang mit
olivopontozerebellärer Atrophie, sondern auch in Verbindung mit verschiedenen anderen
neurodegenerativen Erkrankungen wie Morbus Huntington (Brennan u. Mitarb. 1985),
Morbus Parkinson (Schapira u. Mitarb. 1989) oder Morbus Alzheimer (Reichmann u.
Mitarb. 1993) berichtet worden.

Einleitung und Fragestellung 10
Um zu verdeutlichen, wie es bei einer mitochondrialen Störung zu einer Akkumulation von
Laktat und Pyruvat im Stoffwechsel kommt, folgt ein kurzer, vereinfachter Exkurs über
das normale Prinzip der biologischen Oxidation in den Mitochondrien:
In einem System von vier Multienzymkomplexen läuft in der mitochondrialen Atmungskette über mehrere Teilschritte letztlich eine Knallgasreaktion ab, d. h. aus Wasserstoff und Sauerstoff wird Wasser und Energie gebildet. Hervorzuheben ist die Tatsache, daß die oxidative Phosphorylierung an der inneren Mitochondrienmembran lokalisiert ist. Der Citratzyklus und die Fettsäure-Oxidation, die NADH und FADH2 als Reduktionsäquivalente für die mitochondriale Energiegewinnung liefern, laufen in der benachbarten mitochondrialen Matrix ab. Die stufenweise Elektronen-Übertragung vom NADH und FADH2 zum Sauerstoff bedingt einen Protonenfluß aus der mitochondrialen Matrix heraus. So entsteht eine elektrische Potentialdifferenz, bei deren Ausgleich durch Rückfluß von Protonen in die mitochondriale Matrix Energie in Form von ATP gebildet wird. Die innere Mitochondrienmembran stellt also (nach Löffler 1993) eine „energetisierte Membran“ dar. Der größte der vier Enzymkomplexe der Atmungskette ist der Komplex I, die NADH-Ubichinonreduktase, die Ubichinon mit Hilfe von NADH reduzieren kann. Komplex I enthält bis zu 30 Untereinheiten, von denen einige durch das mitochondriale Genom kodiert sind. Neben dem Coenzym Flavinmononucleotid (FMN) spielen auch Eisen-Schwefel-Komplexe eine Rolle. Vom NADH werden Elektronen über FMN und die Eisen-Schwefel-Komplexe auf das Ubichinon übertragen, es entsteht Ubichinol. Komplex II ist wesentlich kleiner und als einziger Komplex nicht in der Membran selber, sondern an der Innenseite der Membran lokalisiert. Der Komplex II ist eine Succinat-Ubichinonreduktase, die FAD bzw. FADH2 als prosthetische Gruppe enthält. Durch diesen Komplex werden die Reduktionsäquivalente von Succinat, die im innermitochondrial lokalisierten Citratzyklus generiert werden, auf Ubichinon übertragen. Komplex III beinhaltet die Ubichinol-Cytochrom-c-Reduktase. Vom Ubichinol fließen Elektronen über die Cytochrom-c-Reduktase zum Cytochrom c gemäß folgender Gleichung: Ubichinol + 2 Cytochrom cox → Ubichinon + 2 Cytochrom cred

Einleitung und Fragestellung 11
Elektronen des reduzierten Cytochrom c werden durch den Komplex IV, die Cytochrom-c-Oxidase auf Sauerstoff übertragen, der mit freien Wasserstoff-Protonen unter Bildung von Wasser reagiert: 4 Cytochrom c (+2) + 4 H+ + O2 → 4 Cytochrom c (+3) + 2 H2O
Zunächst muß festgestellt werden, daß - unabhängig von der mitochondrialen
Stoffwechselsituation - bereits ein erhöhter zytosolischer NADH/NAD+-Quotient einen
erhöhten zytosolischen Laktat-Pyruvat-Quotienten bedingt, da die Laktat-Dehydrogenase
Pyruvat zu Laktat unter Oxidation von NADH zu NAD+ reduziert. Dies ist zum Beispiel
physiologischerweise im kontrahierenden Muskel der Fall.
Die zytosolische Laktat- und Pyruvat-Konzentration steigt analog, wenn es aufgrund eines
Defektes der mitochondrialen NADH-Oxidationskapazität zunächst zu einem Anstieg des
mitochondrialen und über Shuttle-Mechanismen schließlich auch zu einem Anstieg des
zytosolischen NADH/NAD+-Quotienten kommt. Ein erhöhter mitochondrialer NADH-
Gehalt bedingt eine allosterische Hemmung der mitochondrialen Pyruvat-Dehydrogenase,
was zu einem Anstieg von Pyruvat im Mitochondrium führt; die Verstoffwechselung des
aus dem Zytosol übertretenden Pyruvat bleibt aus. Dadurch kommt es zu einem Rückstau
von Pyruvat im Zytosol, welches dann größtenteils durch die zytosolische Laktat-
Dehydrogenase in Laktat umgewandelt wird. Die Abhängigkeit des Laktat-Pyruvat-
Gleichgewichtes von der Konzentration des NADH und NAD+ führt letztlich zu einer
Verschiebung des Gleichgewichtes zugunsten von Laktat, so daß nicht nur der jeweilige
Wert von Laktat und Pyruvat im Serum, sondern auch der Laktat-Pyruvat-Quotient
ansteigt (Zierz u. Mitarb. 1989).
Um Defekte der Atmungskette zu diagnostizieren, ist eine einfache Bestimmung des
Laktatspiegels im Serum sinnvoll (Jackson u. Mitarb. 1995). So war bei einer
Untersuchung von 30 Patienten mit mitochondrialer Enzephalomyopathie der
Laktatspiegel bei 19 Patienten (= 63 %) pathologisch erhöht; der Pyruvatspiegel hingegen
war in Ruhe nur bei 14 Patienten (= 47 %) erhöht (Zierz u. Mitarb. 1989). Da die
Bestimmung der Laktat- und Pyruvatwerte in Ruhe bei mitochondrialen Erkrankungen

Einleitung und Fragestellung 12
manchmal keine eindeutige Erhöhung ergibt, reicht eine einfache Bestimmung dieser
Werte selten aus. Bei einer nicht richtungsweisenden Erhöhung der Laktat- und
Pyruvatwerte empfiehlt sich daher der Fahrradergometer-Belastungstest, durch den ein
Anstieg beider Werte, vor allem aber ein starker Anstieg des Laktatwertes beobachtet
werden kann (Reichmann u. Mitarb. 1988/2, Reichmann u. Mitarb. 1992). Zierz u. Mitarb.
fanden in ihrer Untersuchung entsprechend einen Anstieg des Laktatwertes sowie des
Laktat-Pyruvat-Quotienten bei einem Großteil (83 % bzw. 80 %) der von ihnen
untersuchten 30 Patienten bei einer Belastung von 30 Watt für 15 Minuten auf einem
Fahrradergometer. Die Pyruvat-Werte blieben bei beinahe 2/3 der Patienten normwertig
(Zierz u. Mitarb. 1989).
In dieser Arbeit wird eine Untersuchungsreihe vorgestellt, bei der die peripher-venösen
Laktat- und Pyruvatwerte von 11 Patienten mit degenerativer Ataxie in Ruhe und unter 15-
minütiger Belastung auf einem Fahrradtrainer untersucht wurden, um festzustellen, ob eine
Erhöhung dieser Werte im peripheren Blut bzw. eine Erhöhung des Laktat-Pyruvat-
Quotienten meßbar ist.

Materialien und Methode 13
2 Materialien und Methoden
2.1 Materialien
2.1.1 Geräte
Es standen folgende Geräte zur Verfügung:
Fahrradtrainer „Golf“ (Firma Kettler)
Blutdruckmanschette (Firma Boso)
Stethoskop (Firma Littmann)
Stauschlauch (Firma Bayer)
Kühlschrank (Firma Siemens)
Pipetten in verschiedenen Größen (Firma Eppendorf)
Präparative Kühlzentrifuge Modell J2-21 (Firma Beckman)
GPR Tischzentrifuge (Firma Beckman)
Photometer für die Klinische Chemie ECOM 6122 (Firma Eppendorf)
Eisakkus für Wasserbad
2.1.2 Verbrauchsartikel
An Verbrauchsartikeln wurden benutzt:
Vasofix-Braunülen® 1,1 x 33 mm (Firma Braun Melsungen AG)
Vasofix-Braunülen® 1,3 x 45 mm (Firma Braun Melsungen AG)
Mandrins 18 G x 45 mm (Firma Braun Melsungen AG)
Mandrins 20 G x 45 mm (Firma Braun Melsungen AG)
Serum-Monovetten, ohne Gerinnungskügelchen (Firma Kabe Labortechnik)
Eppendorf-Cups (Firma Eppendorf)
15 ml Polysteren-Zentrifugierröhrchen (Firma Corning)
Pipettierspitzen, Größe „blau“ und „gelb“ (Firma Greiner)
Plastik-Reagenzröhrchen
Deckel für Plastik-Reagenzröhrchen
Küvetten bzw. Halbküvetten mit 1 cm Schichtdicke

Materialien und Methode 14
2.1.3 Chemikalien
Folgende Chemikalien fanden zur Laktat- und Pyruvatbestimmung Verwendung:
Perchlorsäure/Perchlorat 0,165 mol/l (Zentralapotheke der St. Elisabeth-Stiftung, Bochum)
Perchlorsäure 1,0 mol/l (Zentralapotheke der St. Elisabeth-Stiftung, Bochum)
„Lactat für die Sportmedizin“ (Firma Boehringer Mannheim, Best.-Nr. 1178750),
enthaltend:
Lösung 1 = 1 x 30 ml Puffer
Lösung 2 = 1 x NAD (lyoph.)
Lösung 3 = 1 x 0,7 ml GPT
Lösung 4 = 1 x 0,7 ml LDH
Set „Pyruvat“ (Firma Boehringer Mannheim, Best.-Nr. 124982), enthaltend:
Phosphat für 1 x 35 ml destilliertes Wasser
NADH für 1 x 4,0 ml destilliertes Wasser
1 x 0,5 ml gebrauchsfertige LDH
Aqua dest. (Firma Waldeck)
2.2 Versuchsablauf und Entnahme der Proben
Am Vortag des Versuchs sowie unmittelbar vor Versuchsbeginn wurden die Probanden
eingehend über Ablauf und Ziel des Versuchs aufgeklärt.
Vor Versuchsbeginn erfolgte außerdem die Rücksprache mit dem behandelnden Arzt, um
Risikopatienten (Patienten mit Herz-Kreislauferkrankungen wie Rhythmusstörungen,
koronarer Herzerkrankung, Zustand nach Myokardinfarkt und Hypertonie) zu erfassen
bzw. Ausschlußkriterien zu erkennen (Schmerzen, postoperative Patienten, Nüchternzeit
kürzer als 1,5 h, Ruhepause kürzer als 1 h).
Anhand eines standardisierten Fragebogens wurden Personalien, Familien- und
Eigenanamnese, Symptomatik und Medikation dokumentiert (Tabelle 1).

Materialien und Methode 15
Patientenname / Anrede
Kürzel
Geburtsdatum /Alter
Straße / Wohnort / Telefon
Beruf
Gewicht / Größe
Sport / Trainingszustand
Nikotin / Alkohol / Coffein
Ernährungsgewohnheiten- &
besonderheiten
Geburten / Menses
Vater
Mutter
Geschwister
Sozialanamnese
Neurologische Diagnose
Symptome / Krankengeschichte /
Familienanamnese
weitere Erkrankungen (renale /
hepatische Dysfunktion / Muskel-
erkrankung / kardiovaskuläre Risiken ?)
aktuelle Medikation
Tabelle 1: Datendokumentation

Materialien und Methode 16
Puls, Blutdruck und Zeitpunkt der letzten Nahrungs- und Medikamenteneinnahme wurden
ebenfalls erfaßt (Tabelle 2):
Datum, Beginn der Untersuchung
Name
Puls zu Beginn
RR zu Beginn
Puls zu Ende
RR zu Ende
letzte Medikation
letztes Essen
Braunülenlage; Zeitpunkt des
Legens
Bemerkungen
Tabelle 2: Versuchsprotokoll
Nach Legen einer Vasofix-Braunüle in der Größe 1,3 x 45 mm bzw. 1,1 x 33 mm in die
Ellenbeuge wurde ungestautes Blut für die Untersuchung vor Belastungsbeginn (t0) in
einer 10-ml-Serummonovette entnommen, aus der zuvor die Gerinnungskügelchen entfernt
worden waren.
Danach begann der Belastungstest auf dem Kettler-Fahrradtrainer „Golf“ in der
Belastungsstufe 1. Die Probanden hielten eine Belastung von ca. 25 bis 30 Watt während
15 Minuten ein.

Materialien und Methode 17
Zu den Zeitpunkten
t1 = 1 Minute nach Belastunsbeginn,
t2 = 5 Minuten nach Belastungsbeginn,
t3 = 10 Minuten nach Belastungsbeginn,
t4 = 15 Minuten nach Belastungsbeginn,
t5 = 1 Minute nach Belastungsende und
t6 = 5 Minuten nach Belastungsende
wurden weitere Blutproben ungestaut über die liegende Braunüle entnommen.
Vor, während und nach Versuchsende erfolgten Puls- und Blutdruckkontrollen; als
Abbruchkriterien waren ein Pulsanstieg größer 150/Minute, ein Blutdruckanstieg auf
größer 180 mm Hg systolisch und jegliches subjektives Unwohlsein (Schwäche, Übelkeit,
Schwindel etc.) des Patienten festgelegt. Die gewonnen Proben wurden sofort verarbeitet.
2.3 Bestimmung von Pyruvat
Unmittelbar nach der Blutentnahme zu den Zeitpunkten t0 bis t6 wurden jeweils 3 ml Blut
zu je 3 ml, in einem 15-ml-Zentrifugierröhrchen vorgelegter, kalter Perchlorsäure gegeben,
vorsichtig durchmischt und bis zur weiteren Verarbeitung im Labor kalt gestellt.
Nach der Enteiweißung des Blutes mit der Perchlorsäure erfolgte die weitere Aufbereitung
im Labor erfolgt gemäß der Packungsbeilage des Pyruvat-Sets (Anlage):
Im Labor wurden die Proben 10 Minuten bei 3.000 Umdrehungen/Minute zentrifugiert, 2
ml des Überstandes wurden abpipettiert und zu 1 ml Phosphatpuffer (Lösung 1 des
Pyruvat-Sets) in ein Reagenzröhrchen gegeben.
Dann wurden die Röhrchen gut durchmischt, 15 Minuten auf Eis gelegt und anschließend
erneut für 5 Minuten bei 3.000 Umdrehungen/Minute zentrifugiert.
Bei jeder neu angebrochenen Packung des Pyruvat-Sets mußte vor der weiteren
Bearbeitung die Extinktionsabnahme, die durch die Verdünnung des Bestimmungsansatzes
bei Zusatz des Enzyms LDH (Lösung 3 des Pyruvat-Sets) hervorgerufen wurde, einmal bei
Wellenlänge Hg 340 nm mit einem Photometer bestimmt werden. Dazu wurde die
Bestimmung mit Wasser statt mit Filtrat durchgeführt und ∆EWasser von ∆EFiltrat abgezogen.

Materialien und Methode 18
Außerdem wurde vor der Pyruvatbestimmung der Küvettenleerwert gemessen und in das
Photometer eingespeichert.
In eine Küvette mit 1 cm Schichtdicke wurden 0,20 ml NADH (Lösung 2 des Pyruvat-
Sets) zusammen mit 2,00 ml des auf Raumtemperatur (25° C) erwärmten Filtrats gegeben
und vorsichtig gemischt. Nach Bestimmung der Extinktion E1 im Photometer wurde das
Enzym LDH zugegeben (Lösung 3 des Pyruvat-Sets) und vorsichtig gemischt.
Die Extinktion E2 konnte nach etwa 10 Minuten, also nach Stillstand der Reaktion
LDH
Pyruvat + NADH + H+ ⇔ L-Lactat + NAD+
gemessen werden.
Die Konzentration des Pyruvats in der Probe errechnete sich anhand der Formel
cPyruvat [mg/100ml] = 4,30 x (E1 - E2).
Nach Herstellerangaben beträgt der Normbereich im venösen Nüchternblut 0,36-0,59
mg/100 ml bei einem Meßbereich von ca. 0,04-4,0 mg/dl.
Bei 9 von 11 Patienten und bei 9 von 11 Kontrollpersonen konnten Pyruvat-Doppelt-
bestimmungen durchgeführt werden, um Meßfehler zu minimieren.
2.4 Bestimmung von Laktat
Unmittelbar nach der Blutentnahme zu den Zeitpunkten t0 bis t6 wurden jeweils 20 µl Blut
mit je 200 µl (in einem Eppendorf-Gefäß vorgelegter) eiskalter 0,165 mol/l
Perchlorsäure/Perchlorat enteiweißt, gut durchmischt und bis zur weiteren Verarbeitung im
Labor gekühlt aufbewahrt. Im Labor wurde das Blut-Enteiweißungslösungs-Gemisch 2
Minuten bei 12.000 Umdrehungen/Minute zentrifugiert und der Überstand zur weiteren
Verarbeitung abpipettiert.

Materialien und Methode 19
Für jede Laktat-Meßreihe mußte ein Reagenzien-Leerwert bestimmt werden, dazu wurden
500 µl Reagenzlösung mit 50 µl eiskalter Enteiweißungslösung versetzt und bei 20° bis
25° Celsius Raumtemperatur im Eppendorf-Photometer bei Wellenlänge Hg 340 nm
bestimmt. Außerdem wurde vor der Bestimmung der Küvettenleerwert gemessen und in
das Photometer eingespeichert.
Das Testprinzip beruhte auf folgenden Reaktionsgleichungen:
LDH
L-Laktat + NAD+ ⇔ Pyruvat + NADH + H+
GPT
Pyruvat + L-Glutamat ⇔ L-Alanin + α-Ketoglutarat
Für die Laktatbestimmung wurden 500 µl Reagenzlösung und 50 µl Probenüberstand
gemischt, in eine Küvette überführt und nach 30 Minuten bei Wellenlänge Hg 340 nm im
Photometer gegen den Reagenzien-Leerwert gemessen und wie folgt berechnet:
EProbe - EReagenzienleerwert = ∆E
Die Berechnung der Laktat-Konzentration (cLactat) im Blut erfolgte dann mittels der Formel
cLactat [mg/dl] = 170,7 x ∆E
Die Normalwerte im venösen Nüchternblut betragen nach Herstellerangaben 9-16 mg/dl.
Bei 7 von 11 Patienten und bei 7 von 11 Kontrollpersonen wurden Laktat-
Doppeltbestimmungen durchgeführt, um Meßfehler zu vermeiden.
2.5 Einfluß von Fehlerquellen
Fehler bei der Bestimmung von Laktat und Pyruvat kann man in systematische und nicht-
systematische Fehler unterteilen.
Um systematische Fehler zu vermeiden, wurden die von Zierz u. Mitarb. gemachten
Vorgaben für die Bestimmung von Laktat und Pyruvat beachtet (Zierz u. Mitarb. 1989):

Materialien und Methode 20
Da körperliche Aktivität vor dem Fahrradbelastungstest zu einem Anstieg der Laktatwerte
führen kann, wurden Anamnese und körperliche Untersuchung der Probanden unmittelbar
vor dem Belastungstest durchgeführt, um so eine ca. 60-minütige Ruhepause einhalten zu
können. Teilweise mußten jedoch die Kontrollpersonen, die sich aus nichtneurologischen
Abteilungen rekrutierten, weite Wege zurücklegen und zahlreiche Treppen steigen, bis sie
den Untersuchungsraum in der Neurologischen Klinik erreichten, so daß bei ihnen
eventuell falsch hohe Laktatspiegel bestimmt wurden.
Die Anamnese suchte mögliche Einflußgrößen auf die Funktion der Atmungskette zu
erfassen, im einzelnen wurden Trainingszustand (Hollmann u. Mitarb. 1990), Rauchen
(Smith u. Mitarb. 1993), Muskelerkrankungen und hepatische Dysfunktion (Robinson
1989) sowie die aktuelle Medikation erfragt und dokumentiert (vgl. Tabelle 1).
Um falsch positive Laktatanstiege durch zu lange Stauungszeiten zu vermeiden, wurde das
Blut bei allen Probanden ungestaut aus einer möglichst großlumigen Braunüle (1,3 x 45
mm) entnommen. Allerdings mußte aufgrund der lokalen Venenverhältnisse bei einzelnen
Probanden eine etwas kleinlumigere Braunüle (1,1 x 33 mm) gewählt werden, so daß es
eventuell hämolysebedingt zur Bestimmung von zu hohen Laktatwerten kommen konnte.
Dieser nichtsystematische Fehler dürfte insgesamt jedoch vernachlässigbar sein.
Falls die Gewinnung von Blut durch eine Braunüle zu falsch hohen Laktatwerten führen
sollte, wäre dies ein systematischer Fehler, der Patienten und Kontrollen beträfe und somit
die Aussagekraft der Ergebnisse nicht stören würde.
Da für die Untersuchungsreihe kein Fahrradergometer zur Verfügung stand, sondern nur
ein privat beschaffter Fahrradtrainer, war die Standardisierung dieses Tests nur
mittelmäßig: Das Einhalten der Belastungsstärke konnte nicht durch eine digital gesteuerte
Wirbelstrombremse wie bei einem Ergometer, sondern nur durch ein analoges
Rundinstrument, das die Umdrehungs- und Kilometerzahl wiedergab, überwacht werden.
Nichtsystematische Fehler könnten sich bei der laborchemischen Bestimmung von Laktat
und Pyruvat ereignet haben. Die Liste reicht von unterschiedlich temperierter

Materialien und Methode 21
Perchlorsäure (bzw. Perchlorsäure/Perchlorat), Pipettierfehlern, nicht ordnungsgemäßem
Abpipettieren des Überstandes, Verwechslung von Proben über nicht gewechselte,
verunreinigte Pipettierspitzen bis zu falscher Kalibrierung des Photometers und läßt sich
sicher fortsetzen. Diese Fehlermöglichkeiten können für die Erklärung einzelner
„Ausreißer“-Werte herangezogen werden. Um die genannten Fehler zu minimieren, wurde
auf Chargengleichheit der verwendeten Laktat- und Pyruvatsets sowie auf Gleichheit der
verwendeten Chemikalien und Gerätschaften geachtet. Außerdem wurde die Bestimmung
stets eigenhändig durchgeführt, also nicht anderen Personen überlassen. Um „Ausreißer“
besser erkennen zu können, wurden nicht nur 7 Werte (t0 bis t6), teilweise in 1-minütigen
Abstand bestimmt, sondern auch Doppelt- und teilweise Dreifachbestimmungen
durchgeführt.
2.6 Patientengut
Es wurden insgesamt 11 Patienten im Alter von 34 bis 69 Jahren aus der Neurologischen
Universitätsklinik des St.-Josef-Hospitals untersucht, davon waren 9 männlich und 2
weiblich (vgl. Tabelle 3, Kapitel 3.1). Das durchschnittliche Alter der Patienten betrug
53,4 Jahre. Die alters- und geschlechtsentsprechenden Kontrollprobanden (37 bis 70 Jahre)
rekrutierten sich aus der orthopädischen, inneren und dermatologischen Abteilung des St.-
Josef-Hospitals. Das Durchschnittsalter der Kontrollpersonen betrug 54,9 Jahre.
2.7 Statistische Auswertung
Die Erfassung der Daten erfolgte mit dem Programm Excel 95. Die statistische Aus-
wertung wurde mit dem Programm Statistica für Windows vorgenommen. Bei dem
durchgeführten statistischen Testverfahren, dem t-Test, werden die Mittelwerte zweier
unverbundener Stichproben derselben intervallskalierten Variablen auf Unterschiedlichkeit
getestet.

Hauptteil mit Darstellung der Ergebnisse 22
3 Hauptteil mit Darstellung der Ergebnisse
Das Problem der statistischen Betrachtung des gewonnenen Datenmaterials besteht darin,
daß die zum Zeitpunkt der Fahrradergometrie gültige Diagnose „olivopontozerebelläre
Atrophie“ der 11 Patienten nach 1996 aufgrund molekulargenetischer Forschungen
modifiziert wurde. So ist die ursprünglich homogene Gruppe der 11 OPCA-Patienten nun
in 6 Subgruppen (vgl. Tabelle 3) zerfallen, was die statistische Auswertung erheblich
erschwert und die Ergebnisse weniger reliabel macht.
Für die eigentliche statistische Auswertung interessant sind die Laktat- und Pyruvatwerte
zu den Zeitpunkten t0 (Ruhewert) und t4 (maximale Belastung nach 15 Minuten). Die
übrigen Werte wurden als Zwischenwerte zur Verlaufskontrolle dokumentiert und
graphisch aufgetragen.
3.1 Charakterisierung der Patienten und Darstellung der Einzelergebnisse
Es wurden 22 Probanden untersucht, die sich in 9 männliche und 2 weibliche Patienten
sowie in 9 männliche und 2 weibliche Kontrollpersonen aufteilen:
Patient Kontrollperson
C5-UL, männlich, 51 Jahre
Diagnose: spinozerebelläre Ataxie Typ 1
XVII-HC, männlich, 51 Jahre
C1-AK, männlich, 38 Jahre,
Diagnose: spinozerebelläre Ataxie Typ 3
XXIV-HK, männlich, 41 Jahre
C4-GN, weiblich, 49 Jahre
Diagnose: spinozerebelläre Ataxie Typ 3
IX-IW, weiblich, 52 Jahre
C10-MR, männlich, 34 Jahre
Diagnose: spinozerebelläre Ataxie Typ 3
XII-UP, männlich, 37 Jahre
C11-KR, männlich, 69 Jahre
Diagnose: spinozerebelläre Ataxie Typ 3
VI-AR, männlich, 70 Jahre

Hauptteil mit Darstellung der Ergebnisse 23
C12-KSG, männlich, 59 Jahre
Diagnose: spinozerebelläre Ataxie Typ 6
XXIX-EP, männlich, 57 Jahre
C6-GH, männlich, 65 Jahre
Diagnose: autosomal dominante zerebelläre Ataxie
XIX-GN, männlich, 64 Jahre
C2-HK, männlich, 58 Jahre
Diagnose: autosomal dominante zerebelläre Ataxie
XXII-AS, männlich, 58 Jahre
C7-KSR, männlich, 61 Jahre
Diagnose: Idiopathische sporadische zerebelläre Ataxie
VIII-FH, männlich, 59 Jahre
C9-SW, männlich, 52 Jahre
Diagnose: Idiopathische sporadische zerebelläre Ataxie
XIII-AL, männlich, 53 Jahre
C8-GK, weiblich, 51 Jahre
Diagnose: Multisystematrophie
XI-ER, weiblich, 62 Jahre
Tabelle 3: Diagnose, Alter und Geschlecht der Probanden
Es folgt eine Beschreibung der einzelnen Patienten mit Diagnosen, ggf.
Ausschlußdiagnosen, aktueller Anamnese, Familienanamnese, neurologischen Unter-
suchungsbefunden, aktueller Medikation und evtl. relevanten fachfremden Diagnosen.
Sofern nicht anders vermerkt, beziehen sich die Angaben auf den Untersuchungszeitpunkt
während der Studie im Frühjahr bzw. Herbst 1996. Ggf. haben einzelne Ergebnisse und
Erkenntnisse der Zeit nach 1996 Eingang in die Beschreibung gefunden, hierzu siehe die
jeweiligen Hinweise. Außerdem sind für jeden Patienten die Laktat- und Pyruvatwerte zu
den Zeitpunkten t0 (Ruhewert) bis t6 (5 Minuten nach Belastungsende) als Tabelle und als
Grafik wiedergegeben. Da überwiegend Mehrfachbestimmungen durchgeführt wurden,
haben die errechneten Mittelwerte Eingang in die Tabelle gefunden.
3.1.1 Patient C5-UL, männlich, 51 Jahre
Herr UL. hatte eine molekulargenetisch gesicherte spinozerebelläre Ataxie vom Typ 1, die
seit etwa 1987 progredient war. Es handelte sich um ein autosomal dominant vererbtes
Leiden, an dem auch Herrn UL.s Mutter, eine Schwester, ein Bruder und drei Tanten
mütterlicherseits erkrankt waren.

Hauptteil mit Darstellung der Ergebnisse 24
Die neurologische Untersuchung ergab eine langsame, sakkadierte Blickfolge,
Schluckerschwernis und dysarthrisches langsames Sprechen. Das Gangbild war bei
langsamen Gehtempo und geringer Schrittgröße breitbasig und spastisch-ataktisch, die
Arme wurden gut mitbewegt. Der insgesamt spastisch erhöhte Muskeltonus war vor allem
am linken Arm ausgeprägt. Die Feinmotorik war eingeschränkt, wie der dysmetrische
Fingernaseversuch, die ataktisch-hypermetrisch durchgeführten Zeigeversuche und die
Bradydysdiachokinese deutlich machten. Die Muskeleigenreflexe waren bei negativem
Achillessehnenreflex und nicht erhältlichen Bauchhautreflexen seitengleich lebhaft
auslösbar. Herr UL. gab eine bimalleoläre Pallanaesthesie von 6/8 über beiden Processus
styloidei radii an. Nebenbefundlich war eine latente Hyperthyreose bei Struma
multinodosa mit kaltem Schilddrüsenknoten im linken unteren Schilddrüsendrittel bekannt.
Die aktuelle Medikation bestand aus Branigen 2-1-1 und Biomagnesin 2x1.
Herr UL. zeigte während der körperlichen Belastung auf dem Fahrradtrainer eine
durchgängig konstante Belastbarkeit (vgl. Tabelle 17, Kap. 4). Laborchemisch konnten
folgende Laktat- und Pyruvatwerte im venösen Blut bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 12,461 13,059 17,924 17,924 18,521 19,033 15,96
LaktatKontrolle 12,461 12,461 18,948 17,924 23,898 24,154 19,545
PyruvatPatient 0,819 0,673 0,841 1,02 0,993 1,002 1,06
PyruvatKontroll
e
1,122 1,211 1,293 1,223 1,378 1,352 1,413
Tabelle 4: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C5-UL u. Kontrollperson
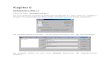
Wie Abbildung 1 zeigt, entsprechen sich die Laktatwerte von Herrn UL. und der alters-
und geschlechtsentsprechenden Kontrollperson zu den Zeitpunkten t0 (Ruhewert), t1 (1.
Belastungsminute), t2 (5. Belastungsminute) und t3 (10. Belastungsminute). Auffällig ist,
daß die Laktatwerte der Kontrollperson zu den Zeitpunkten t4 (15. Belastungsminute), t5
(1 Minute nach Belastungsende) und t6 (5 Minuten nach Belastungsende) sowie die
gesamten Pyruvatwerte der Kontrollperson sichtbar über denen von Herrn UL. liegen. Die
Laktat-Ruhewerte von Patient bzw. Kontrolle steigen während der Belastung um die Hälfte

Hauptteil mit Darstellung der Ergebnisse 25
bzw. das Zweifache an. Die Pyruvatwerte steigen jeweils nur wenig, und zwar von ca. 0,8
mg/dl auf 1,0 mg/dl bei Herrn UL. bzw. von ca. 1,1 mg/dl auf 1,4 mg/dl bei der
Kontrollperson.
0
5
10
15
20
25
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,6
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 1: Laktat u. Pyruvat in [mg/dl] für Patient C5-UL u. Kontrollperson
3.1.2 Patient C1-AK, männlich, 38 Jahre
Bei Herrn AK. war durch Gen-Analyse eine spinozerebelläre Ataxie vom Typ 3 gesichert
worden. Die Familienanamnese war positiv hinsichtlich der Machado-Joseph-Krankheit
bei Ururgroßmutter, Urgroßmutter und Großvater mütterlicherseits, auch war bei der
Mutter, deren Zwillingsschwester und einem Neffen dieselbe Diagnose gesichert worden.
Bei Herrn AK. handelte es sich klinisch-anamnestisch um ein langsam schleichend-
progredientes Krankheitsgeschehen. Herr AK. berichtete von morgendlicher Fallneigung;
es wäre durch seine verlangsamten Reaktionen und die Unfähigkeit, Gleichgewichts-
verschiebungen nicht schnell genug ausgleichen zu können, zu täglichen Sturzereignissen
gekommen.
Die neurologische Untersuchung des Herrn AK. ergab eine sakkadierte Blickfolge,
geringen horizontalen Nystagmus sowie eine zerebelläre Dysarthrie. Bei insgesamt
verlangsamten Bewegungen fand sich eine leichte, linksbetonte Abduktionsschwäche der
Arme und eine Tonuserhöhung der Extremitäten, besonders der Beine, ohne sichere
Zuordnung bezüglich Spastik oder Rigor. Herr AK. zeigte im Romberg-Stehversuch nur
geringes Schwanken und in den Zeigevesuchen Ataxie und Dysmetrie, in den Beinen auch
Intentionstremor. Die Muskeleigenreflexe ließen sich beinbetont sehr lebhaft auslösen, der

Hauptteil mit Darstellung der Ergebnisse 26
Babinski-Reflex war nicht sicher positiv. Im Knöchelbereich fand sich eine
Pallhypästhesie von 6/8. Herr AK. hatte eine mäßige Hohlfußdeformität bei leichter
Supinationsfehlstellung, jedoch ohne Paresen in der Fußpronation.
Die aktuelle Medikation bestand aus 3x125 mg Madopar und PK Merz 3x1; hierunter
war es zu einer leichten Verbesserung der Beweglichkeit gekommen.
Herr AK. zeigte beim Fahrradversuch ein normales Leistungsvermögen (vgl. Tab. 17); im
peripher-venösen Blut konnten folgende Laktat- und Pyruvatwerte bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 8,876 11,347 19,631 33,799 41,651 43,187 44,894
LaktatKontrolle 11,01 12,802 20,996 27,91 42,59 49,589 46,516
PyruvatPatient 0,447 0,838 0,894 1,114 1,651 1,457 1,578
PyruvatKontroll
e
0,993 0,525 1,176 1,361 1,808 1,813 2,122
Tabelle 5: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C1-AK u. Kontrollperson
Die grafische Auswertung zeigt sich ungefähr entsprechende Laktat- und Pyruvatwerte von
Kontrollperson und Patient. Bei den Pyruvatwerten scheint ein Trend zu höheren Werten
der Kontrollperson vorzuliegen. Die Laktatwerte von Patient und Kontrollperson steigen
unter der Belastung auf jeweils mehr als das Vierfache des jeweiligen Ruhewertes, bei den
Pyruvatwerten ist ein Anstieg um mehr als das Dreifache (Patient) bzw. Doppelte
(Kontrollperson) des jeweiligen Ruhewertes festzustellen.
05
101520253035404550
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
0
0,5
1
1,5
2
2,5
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.

Hauptteil mit Darstellung der Ergebnisse 27
Abbildung 2: Laktat u. Pyruvat in[mg/dl] für Patient C1-AK u. Kontrollperson
3.1.3 Patientin C4-GN, weiblich, 49 Jahre
Bei Frau GN. war durch Gen-Analyse eine spinozerebelläre Ataxie vom Typ 3 gesichert
worden. Familienanamnestisch erwähnenswert waren die an Ataxie leidende Mutter, eine
Großtante, der Großvater, die Schwester des Großvaters und der Urgroßvater.
Vor 10 Jahren wäre erstmals Gangunsicherheit, einhergehend mit starker Fallneigung,
aufgetreten. Gelegentlich hätte Frau GN. Doppelbilder bemerkt, seit 2 Jahren hätte sie eine
Sprachstörung.
Bei der neurologischen Untersuchung fanden sich eine vertikale Blickeinschränkung nach
oben, eine leichte Ptosis rechts, spontaner Nystagmus und beidseitiger
Blickrichtungsnystagmus sowie eine Abducens- und Blickheberschwäche. Frau GN.s
Dysarthrie war ausgeprägt, sie artikulierte schlecht und unverständlich. Das Gangbild war
langsam, ataktisch und breitbasig mit ausgleichenden Mitbewegungen der Arme. Es fand
sich eine diskrete Absinktendenz im Beinhalteversuch ohne isolierte Paresen. Im
Fingernase- und Kniehackeversuch ließ sich eine ausgeprägte rechtsbetonte Dysmetrie
beobachten, während die Dysdiachokinese eher linksbetont erschien. Die
Muskeleigenreflexe der Arme waren seitengleich und prompt; der Patellarsehnenreflex war
beidseits gut auslösbar, der Achillessehnenreflex war nicht auslösbar, Bauchhautreflexe
waren in allen Etagen erhältlich. Rechts ließ sich ein fraglicher Spontanbabinski auslösen.
Frau GN. gab eine Pallhypästhesie von 6/8 an beiden Knöcheln an, die übrige
Sensibititätsprüfung war unauffällig.
Die aktuelle Medikation war PK-Merz, je morgens und abends 2 Dragees, wodurch
initial Sprache und Gangbild gebessert worden waren.
Frau GN.s Leistung muß als unterdurchschnittlich bewertet werden, da sie den
Fahrradbelastungstest nur mit vielen kleinen Pausen durchhalten konnte (vgl. Tab. 17).
Außerdem mußte die Belastungsstufe ab ca. der Hälfte der Zeit im Vergleich zu den
anderen Patienten auf Belastungsstufe 0 gestellt werden, was eine Erklärung für die sich
entsprechenden Laktatwerte von Patient und Kontrollperson während der Erholungsphase
(t5 und t6 = 1 bzw. 5 Minuten nach Belastungsende) sein könnte; die vor diesen

Hauptteil mit Darstellung der Ergebnisse 28
Zeitpunkten gemessenen Laktatwerte steigen nämlich stärker an als die der
Kontrollperson. Bei den Pyruvatwerten sind kaum nennenswerte Unterschiede zu
vermerken, außer zum Zeitpunkt t6 (5 Minuten nach Belastungsende), wo der Wert der
Kontrollperson um 0,5 mg/dl höher liegt als der von Frau GN.
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 12,29 14,083 33,97 50,186 48,82 44,041 43,528
LaktatKontrolle 14,509 11,949 22,02 36,018 37,895 44,382 43,528
PyruvatPatient 0,767 0,864 0,882 1,372 1,212 1,247 1,374
PyruvatKontroll
e
0,778 0,868 0,72 1,367 1,312 1,225 1,84
Tabelle 6: Laktat- u. Pyruvatwerte in [mg/dl] für Patientin C4-GN u. Kontrollperson
0
10
20
30
40
50
60
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,61,8
2
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 3: Laktat u. Pyruvat in [mg/dl] für Patientin C4-GN u. Kontrollperson
3.1.4 Patient C10-MR, männlich, 34 Jahre
Bei Herrn MR. wurde 1994 die Diagnose einer autosomal dominant vererbten zerebellären
Ataxie vom genetischen Typ der spinozerebellären Ataxie Typ 3 gestellt. Der Vater von
Herrn MR. hat als Patient C11-KR ebenfalls an dieser Untersuchung teilgenommen.
Die neurologische Untersuchung ergab einen horizontalen Blickrichtungsnystagmus und
eine sakkadierte Blickfolge. Die Sprache war zerebellär dysarthrisch. Bei geringer
Rumpfataxie war das Gangbild ataktisch mit mäßiger spastischer Komponente. Der
Seilgang war nur angedeutet, ausdauerndes Stehen nur mit offenen Augen möglich. In den

Hauptteil mit Darstellung der Ergebnisse 29
Zeigeversuchen an Armen und Beinen war eine geringe Ataxie und Dysmetrie
nachweisbar. Ferner ließ sich eine leichte Bradydysdiadochokinese beobachten. Der
Patellarsehnenreflex war beidseits sehr lebhaft, jedoch nicht verbreitert auslösbar. Der
Achillessehnenreflex war rechts nicht und links nur schwach auslösbar. Die Untersuchung
ergab keine Hinweise auf Atrophien und Sensibilitätsausfälle. Herr MR. gab eine 3-malige
Nykturie an, war aber nicht inkontinent.
Zum Untersuchungszeitpunkt erhielt Herr MR. die Studienmedikation Trimethoprim bzw.
Cotrimoxazol, die jedoch, wie inzwischen nachgewiesen wurde, keinen Effekt auf die
Erkrankung hat.
Herr MR. zeigte eine gute bis überdurchschnittliche Leistung während des
Fahrradbelastungstests (vgl. Tab. 17). Es konnten folgende Blutwerte bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 6,997 7,681 17,839 30,128 51,296 50,783 48,394
LaktatKontrolle 2,476 2,646 11,351 18,008 23,556 27,824 26,715
PyruvatPatient 0,407 0,299 0,692 1,032 1,498 1,045 2,187
PyruvatKontroll
e
0,205 0,262 0,331 0,754 0,66 0,673 0,811
Tabelle 7: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C10-MR u. Kontrollperson
Die grafische Auswertung zeigt deutlich, daß die Laktatwerte von Herrn MR. vor, während
und nach der Belastung jeweils um fast das Doppelte höher sind als die der entsprechenden
Kontrollperson. Die Pyruvatwerte des Patienten liegen ebenfalls, außer zum Zeitpunkt t1
(1. Belastungsminute) und t2 (5. Belastungsminute), ebenfalls deutlich über denen der
Kontrollperson.

Hauptteil mit Darstellung der Ergebnisse 30
0
10
20
30
40
50
60
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
0
0,5
1
1,5
2
2,5
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 4: Laktat u. Pyruvat in [mg/dl] für Patient C10-MR u. Kontrollperson
3.1.5 Patient C11-KR, männlich, 69 Jahre
Die molekulargenetisch gesicherte spinozerebelläre Ataxie vom Typ 3 des Herrn KR.
wurde 1994 diagnostiziert (vgl. die Einzelfallbeschreibung des Sohnes C10-MR, Abschnitt
3.1.4).
Die neurologische Untersuchung ergab bei verlangsamten Blicksakkaden eine nur leicht
sakkadierte Blickfolge und geringen horizontalen Blickrichtungsnystagmus. Der
optokinetische Nystagmus war in horizontaler wie vertikaler Richtung gestört. Die
Dysarthrie war eher diskret. Das Gangbild ließ sich als spastisch-ataktisch und hölzern-
breitbasig beschreiben. Im Romberg-Stehversuch zeigte sich eine ungerichtete
Fallneigung, bei eng gestellten Füßen war der Gang auch bei offenen Augen schwankend.
Die im Fingernaseversuch nachweisbare Dysmetrie und Ataxie war nur leichtgradig, der
Kniehackeversuch war ataktisch und dysmetrisch; außerdem fiel eine
Bradydysdiadochokinese auf. Der Muskeltonus war nicht alteriert. Die kleinen
Fußmuskeln und die kleinen Handmuskeln waren beidseits atrophisch, insbesondere im
Bereich des Interosseus dorsalis I. Herr KR. gab an beiden Knöcheln eine Pallanästhesie
und im Bereich beider Handgelenke eine Pallhypästhesie von 6/8 an. Die
Muskeleigenreflexe waren an den Armen seitengleich schwach auslösbar und an den
Beinen erloschen. Die Pyramidenbahnzeichen waren negativ.
Zum Untersuchungszeitpunkt erhielt der Patient die Studienmedikation Trimethoprim bzw.
Cotrimoxazol, die jedoch, wie inzwischen nachgewiesen wurde, keinen Effekt auf die
Erkrankung hat.

Hauptteil mit Darstellung der Ergebnisse 31
Herr KR. zeigte eine gutes, ausdauerndes Leistungsvermögen (vgl. Tab. 17).
Laborchemisch konnten folgende Werte aus dem peripher-venösen Blut bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 8,194 14,253 35,335 61,282 67,597 75,45 66,996
LaktatKontrolle 22,191 24,751 26,117 22,02 17,241 12,29 12,461
PyruvatPatient 0,256 0,643 0,651 0,63 1,599 1,619 2,448
PyruvatKontroll
e
0,629 0,591 0,762 0,798 1,139 1,15 0,893
Tabelle 8: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C11-KR u. Kontrollperson
Wie Abbildung 5 zeigt, liegen die Laktatwerte der entsprechenden Kontrollperson zu den
Zeitpunkten t0 (Ruhewert) und t1 (1. Belastungsminute) über denen des Patienten. Mit
andauernder Belastung steigen jedoch die Laktatwerte des Patienten auf über das
Neunfache des Ruhewertes (t0), während die der gesunden Kontrollperson in der
Erholungsphase auf fast die Hälfte des Ruheniveaus sinken. Die Pyruvatwerte des
Patienten liegen erst ab dem Belastungsgipfel (t4) über denen der Kontrollperson.
01020304050607080
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
0
0,5
1
1,5
2
2,5
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 5: Laktat u. Pyruvat in [mg/dl] für Patient C11-KR u. Kontrollperson
3.1.6 Patient C12-KSG, männlich, 59 Jahre
Die spinozerebelläre Ataxie Typ 6 des Herrn KSG. konnte molekulargenetisch gesichert
werden. Die Erkrankung war gekennzeichnet durch Tagesschwankungen bezüglich der
Gangsicherheit, schien aber nicht eindeutig progredient zu sein.

Hauptteil mit Darstellung der Ergebnisse 32
Der neurologische Befund beinhaltete eine sakkadierte Blickfolge und einen leichten
horizontalen Blickrichtungsnystagmus. Die zerebelläre Dysarthrie war eher diskret. Das
Gangbild war ataktisch, beim Romberg-Stehversuch geriet Herr KSG. in leichtes
Schwanken. Im Fingernaseversuch war eine diskrete Ataxie zu verzeichnen, auch der
Kniehackeversuch erschien dysmetrisch. Es war eine Eudiadochokinese zu dokumentieren.
Es fanden sich keine Paresen oder Atrophien. Im Knöchelbereich gab Herr KSG. eine
Pallhypästhesie von 5-6/8 an. Die Temperaturdiskrimination im Fuß- und
Unterschenkelbereich war auf beiden Seiten reduziert, sonst bestanden keine weiteren
Sensibilitätsausfälle. Der Reflexstatus war unauffällig. Herr KSG. nahm keine
Medikamente ein.
Herr KSG. zeigte eine gutes Ausdauervermögen bei überdurchschnittlicher Leistung auf
dem Fahrradtrainer (vgl. Tab. 17). Im Blut wurden folgende Werte bestimmt:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 12,29 21,423 41,053 65,293 96,189 99,262 99,433
LaktatKontrolle 5,719 5,377 11,267 14,424 23,215 26,885 20,313
PyruvatPatient 0,752 1,002 1,32 1,829 1,72 1,941 2,625
PyruvatKontroll
e
0,451 0,521 0,656 0,705 0,762 0,815 1,268
Tabelle 9: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C12-KSG u. Kontrollperson
Sowohl in Ruhe als auch während und nach der Belastung liegen die Laktat- und
Pyruvatwerte des Patienten deutlich über dem der Kontrollperson. Die Laktatwerte von
Herrn KSG. steigen auf Maximalwerte, die bei ungefähr dem Achtfachen des Ruhewertes
(t0) liegen. Die Laktatwerte der Kontrollperson steigen auf Maximalwerte, die bei deutlich
über dem Vierfachen des Ruhewertes (t0) liegen. Die Pyruvatwerte des Patienten sind
ebenfalls deutlich höher, nämlich fast doppelt so hoch wie die der Kontrollperson und
steigen während der Belastungsphase (t1 bis t4) und anschließenden Erholungsphase (t5
und t6) kontinuierlich an.

Hauptteil mit Darstellung der Ergebnisse 33
0102030405060708090
100
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
0
0,5
1
1,5
2
2,5
3
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 6: Laktat u. Pyruvat in [mg/dl] für Patient C12-KSG u. Kontrollperson
3.1.7 Patient C6-GH, männlich, 65 Jahre
Herr GH. litt an einer autosomal dominant vererbten zerebellären Ataxie, durch direkte
Analyse der Desoxyribonukleinsäure (DNA) waren die Subtypen 1 und 3 der
spinozerebellären Ataxie ausgeschlossen worden. Herr GH. gab an, seit 1991 an einer
zunehmenden Gangunsicherheit zu leiden und seit Frühjahr 1995 eine Gehstütze zu
benötigen, da er oft nach vorne und hinten stürze. Seit einigen Jahren verschlechterte sich
die Sprache, die Funktion der Hand beim Schreiben sowie das Knöpfen und Schlucken.
Aus der Familienanamnese war zu erfahren, daß der Vater hohe Schuhe habe tragen
müssen, später Gehstöcke benutzt habe und Probleme hatte, das Eßbesteck zu halten. Die
Probleme des Vaters hätten mit 55 Jahren begonnen, er wäre mit 82 Jahren verstorben. Die
Schwester des Patienten wäre 72 Jahre alt und liefe ebenfalls schlecht, was jedoch auf
Kniebeschwerden zurückgeführt würde; sie hätte auch Blasenprobleme.
Die neurologische Untersuchung des Herrn GH. ergab eine sakkadierte Blickfolge, einen
horizontalen Blickrichtungsnystagmus und leichten vertikalen Nystagmus sowie
dysmetrische Blicksakkaden. Es bestanden dysmetrische Blicksakkaden und eine Störung
des optokinetischen Nystagmus in vertikalen Richtung. Die Sprache war mäßig zerebellär-
dysarthrisch. Das Gangbild war ataktisch bei vorgebeugtem Oberkörper. Im Romberg-
Stehversuch zeigte sich eine nach wenigen Sekunden eine ungerichtete Fallneigung. Bei
den Zeigeversuchen war eine deutliche Hypermetrie, Ataxie und Intentionstremor
nachweisbar; die Bradydysdiadochokinese war eher leichtgradig und linksbetont. Der
Muskeltonus war nicht sicher alteriert. Bei seitengleichen, mittellebhaften

Hauptteil mit Darstellung der Ergebnisse 34
Muskeleigenreflexen an den Armen und beidseits lebhaften Patellarsehnenreflex war der
Achillessehnenreflex rechts mit einzelnen Kloni auffällig. Die Babinski-Reaktion war
nicht sicher positiv. Es fand sich eine beidseitige Pallhypästhesie von 5-6/8 gegenüber 8/8
an den Handgelenken. Das Temperaturempfinden im Fuß- und Handbereich war etwas
reduziert.
Die aktuelle Medikation bestand aus Acerbon 2,5 1x1 bei Hypertonus und Dridase 2x1.
Krankengymnastik 2x/Woche.
Herr GH. zeigte eine unterdurchschnittliche, aber konstante Leistung (vgl. Tab. 17).
Folgende Laktat- und Pyruvatwerte konnten bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 7,511 8,705 17,326 12,974 20,399 19,033 13,656
LaktatKontrolle 11,181 11,266 25,008 30,299 38,834 37,639 32,604
PyruvatPatient 0,759 0,815 0,965 0,97 1,169 1,195 1,268
PyruvatKontroll
e
0,8 0,971 1,043 1,498 1,543 1,606 1,89
Tabelle 10: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C6-GH u. Kontrollperson
Die Auswertung der Laktatwerte zeigt während der gesamten Belastungszeit (t1 bis t4) und
anschließenden Ruhephase (t5 und t6) jeweils niedrigere Laktatwerte des Patienten als der
entsprechenden Kontrollperson; auch die Pyruvatwerte des Herrn GH. liegen durchweg
niedriger als die der Kontrollperson. Die Laktatwerte sowohl von Patient als auch von
Kontrollperson steigen während der Belastung an und fallen danach wieder ab; die
Pyruvatwerte von Patient und Kontrolle steigen jeweils bis zum Ende des beobachteten
Zeitraumes an.

Hauptteil mit Darstellung der Ergebnisse 35
05
10152025303540
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,61,8
2
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 7: Laktat u. Pyruvat in [mg/dl] für Patient C6-GH u. Kontrollperson
3.1.8 Patient C2-HK, männlich, 58 Jahre
Herr HK. litt an einer autosomal dominant vererbten zerebellären Ataxie mit Ausschluß
der SCA-Subtypen 1 und 3 durch direkte DNA-Analyse.
Die Symptomatik hätte vor etwa 10 Jahren mit Gleichgewichtsstörungen beim schnellen
Laufen begonnen und wäre seitdem langsam progredient. Ein im Januar 1996
durchgeführtes NMR wäre unauffällig gewesen. Familienanamnestisch wären im höheren
Lebensalter aufgetretene Gehstörungen des Vaters, der mit 85 Jahren verstorben wäre,
bekannt. Eine Tante und eine Onkel der väterlichen Linie sowie der Großvater hätten
ebenfalls Gehstörungen gehabt, der Großvater wäre im Alter von 55 Jahren verstorben.
Im Rahmen der klinisch-neurologischen Untersuchung fand sich beim Blick nach rechts
ein horizontaler Nystagmus nach links. Die Sprache war dysarthrisch und skandierend. Bei
langsamem Gehtempo und ungleicher Schrittgröße erschien das Gangbild spastisch-
ataktisch, die Wende war unauffällig. Blind- und Seilgang waren unmöglich. Es fanden
sich keine Atrophien und Faszikulationen. Der Muskeltonus an den Beinen war spastisch
erhöht, kein Zahnradphänomen. Der Klonus in beiden Fußgelenken war nicht erschöpflich.
Die weitere Untersuchung ergab keinen Tremor, keine Hyperkinesen und eine nur leichte
Einschränkung der Feinmotorik. Die Dysmetrie war im Fingernaseversuch linksbetont, im
Kniehackeversuch war sie rechtsbetont. Der Romberg-Stehversuch war pathologisch, der
Arm- und Beinhalteversuch war unauffällig. Die Muskeleigenreflexe waren an beiden
Armen rechtsbetont sehr lebhaft. Der Patellarsehnenreflex war beidseits pathologisch ohne
Erweiterung der reflexogenen Zone, der Achillessehnenreflex zeigte beidseits einen

Hauptteil mit Darstellung der Ergebnisse 36
unerschöpflichen Klonus. Die Bauchhautreflexe waren gut und seitengleich auslösbar. Der
Babinski war an den Fußsohlen beidseits positiv. Die Sensibilitätserkennung war
unauffällig.
Die Medikation bestand aus 1x1 Jodthyrox 100 µg. Ein Therapieversuch mit Carnitin i.v.
im Rahmen des weiteren stationären Aufenthaltes führte zur Symptomverschlechterung.
Im peripher-venösen Blut des Patienten HK., der eine durchschnittliche Leistung bei
konstantem Leistungsvermögen zeigte (vgl. Tab. 17), wurden folgende Werte bestimmt:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 7,34 10,754 5,975 5,292 5,121 5,975 8,194
LaktatKontrolle 8,791 11,01 26,288 43,102 55,989 72,889 60,598
PyruvatPatient 0,361 0,417 0,439 0,589 0,404 0,28 0,456
PyruvatKontroll
e
0,623 0,761 0,952 1,128 1,342 1,385 2,231
Tabelle 11: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C2-HK u. Kontrollperson
Der Vergleich der Laktatwerte von Patient und Kontrollperson ergibt auffällig niedrige
Laktatwerte für Patient HK., die mit zunehmender Belastungsdauer unter den Ruhewert
(t0) fallen. Die Laktatwert der Kontrollperson dagegen steigen während der Belastung auf
über das Achtfache des Ruhewertes (t0). Die Pyruvatwerte des Patienten bleiben in etwa
konstant bei 0,4 mg/dl, die Pyruvatwerte der Kontrollperson steigen stetig bis zum Ende
des beobachteten Zeitraumes auf über das Dreifache des Ruhewertes (t0).
01020304050607080
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
0
0,5
1
1,5
2
2,5
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.

Hauptteil mit Darstellung der Ergebnisse 37
Abbildung 8: Laktat u. Pyruvat in [mg/dl] für Patient C2-HK u. Kontrollperson
3.1.9 Patient C7-KSR, männlich, 61 Jahre
Herr SR. litt an einer idiopathisch sporadischen zerebellären Ataxie, die sich vor etwa 3
Jahren durch eine langsam progrediente Gangstörung mit schwer ausgleichbarem
Schwanken sowie mit Störungen der Feinmotorik und der Sprache erstmalig bemerkbar
gemacht hatte. Durch molekulargenetische Untersuchungen war ausgeschlossen worden,
daß Herr SR. Genträger für eine spinozerebelläre Ataxie vom Typ 1 oder Typ 3 war.
Aus der Familienanamnese ergaben sich keine Hinweise für eine erbliche Disposition der
Erkrankung des Herrn SR.
Der neurologische Untersuchung zeigte runde, mittelweite Pupillen, eine regelrechte
Konvergenzreaktion und freie Beweglichkeit der Augen in alle Richtungen. Ein
Nystagmus war nicht nachweisbar. Bei Durchführung der Stand- und Gangversuche
imponierte die ataktische Gangstörung, bei Augenschluß die spontane Fallneigung. Der
Unterberger-Tretversuch und Blindgang waren nicht durchführbar, das Liniengehen war
nur mit Mühe und ausfahrenden Bewegungen möglich. Die Schrittgröße war klein und die
Armmitbewegung war reduziert. Die Bradydysdiadochokinese war beidseits nachweisbar.
Im Fingernase- und Kniehackeversuch war eine deutliche Intentionsataxie- und dysmetrie
nachweisbar. Die Kraftentwicklung war seitengleich ohne Hinweise für Paresen. Bei
fehlenden pathologischen Reflexen ließen sich die Muskeleigenreflexe seitengleich
mittellebhaft auslösen. Die Sensibilität war bis auf herabgesetztes bimalleoläres
Vibrationsempfinden von 5/8 nicht gemindert.
Die aktuelle Medikation beinhaltete PK-Merz 2x1, Levothym 3x1, Dihydergot retard
1x1, Harzol 1x1 bei obstruktivem Prostataadenom und Vitamin-B-Komplex 1x1 bei
Zustand nach Billroth-II-Resektion mit konsekutiver Vitamin-B12-Mangel-Resorption. Ein
Therapieversuch mit L-Carnitin über 2 Wochen im Verlauf des stationären Aufenthaltes
hatte zu keiner Besserung der Symptomatik geführt.
Herr KSR. zeigte eine durchschnittliche Leistung (vgl. Tab. 17). Aus der Braunüle der
Kontrollperson konnte zu den Zeitpunkten t5 und t6 (1 bzw. 5 Minuten nach

Hauptteil mit Darstellung der Ergebnisse 38
Belastungsende) kein Blut mehr zur laborchemischen Bestimmung von Laktat und Pyruvat
gewonnen werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 12,974 15,107 31,836 43,016 56,246 58,208 52,063
LaktatKontrolle 8,194 20,655 33,628 35,676 20,313 Abbruch Abbruch
PyruvatPatient 0,785 0,663 0,637 1,199 1,447 1,224 1,789
PyruvatKontroll
e
0,679 1,2 1,544 1,604 1,423 Abbruch Abbruch
Tabelle 12: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C7-KSR u. Kontrollperson
Zwar fehlen die Laktat- und Pyruvatwerte für die Kontrollperson zu den Zeitpunkten t5
und t6 (1 bzw. 5 Minuten nach Belastungsende); es ist jedoch ersichtlich, daß die
Laktatwerte des Patienten nach anfänglicher Entsprechung mit zunehmender
Belastungsdauer stärker ansteigen als die der Kontrollperson. Die Pyruvatwerte von
Patient und Kontrollperson unterscheiden sich durch Maxima zu verschiedenen
Zeitpunkten; das Maximum der Pyruvatwerte des Patienten liegen am Ende des
beobachteten Zeitraumes, während das Maximum der Kontroll-Pyruvatwerte zum
Zeitpunkt t3 (10. Belastungsminute) zu verzeichnen ist.
0
10
20
30
40
50
60
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,61,8
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 9: Laktat u. Pyruvat in [mg/dl] für Patient C7-KSR u. Kontrollperson

Hauptteil mit Darstellung der Ergebnisse 39
3.1.10 Patient C9-SW, männlich 52 Jahre
Herr SW. litt an einer idiopathischen sporadischen zerebellären Ataxie. Die
molekulargenetische Untersuchung der bekannten Mutationen für zerebelläre Ataxien
ergab einen negativen Befund, so daß bei Herrn SW. ein vererbtes Leiden
unwahrscheinlich erschienen war. Ein initial festgestellter Carnitinmangel von 0,6 mg/dl
i.S. (bei Normalwerten von 0,8 bis 1,5 mg/dl) spielte ursächlich anscheinend keine Rolle,
da eine Therapie mit 3 mal täglich einer Trinkampulle L-Carnitin über ein halbes Jahr
keinen überzeugend positiven Einfluß auf die Krankheitssymptomatik hatte, so daß die
Carnitin-Substitution schließlich eingestellt worden war.
Der Vater von Herrn SW. wäre mit 72 Jahren an einer Lungenerkrankung verstorben, hätte
aber an einem Parkinson-Syndrom mit Tremor gelitten und in den letzten Jahren nicht
selbständig gehen können. Die Mutter wäre 43-jährig an einer Bluterkrankung verstorben.
Die Geschwister wären gesund.
Die neurologische Untersuchung ergab eine leicht sakkadierte Blickfolge, dysmetrische
Blicksakkaden und einen in vertikaler Richtung gestörten optokinetischen Nystagmus.
Hypakusis rechtsseitig. Die Sprache war zerebellär dysarthrisch, das Gangbild breitbasig
ataktisch mit geringer Unsicherheit im Seilgang. Im Romberg-Stehversuch und im
Einbeinstand zeigte Herr SW. ungerichtetes Schwanken. Die Motorik der Hände und Beine
war dysmetrisch-ataktisch. An den Beinen ließ sich zusätzlich Intentionstremor mit
rechtsseitiger Betonung feststellen. Der Muskeltonus war eher reduziert, Paresen oder
Atrophien waren nicht vorhanden. Die Muskeleigenreflexe waren seitengleich lebhaft
auslösbar. Der Babinski-Reflex war links fraglich positiv, rechts negativ. Im
Knöchelbereich fand sich eine Pallhypästhesie von 7/8 beidseits. Die Medikation bestand
aus Isoptin 1x1.
Herr SW. zeigte gute Belastbarkeit und ein überdurchschnittliches Leistungsvermögen
während des Fahrradbelastungstests (vgl. Tab. 17), und es konnten folgende Blutwerte
bestimmt werden:
t0 t1 t2 t3 t4 t5 t6
LaktatPatient 9,986 14,168 25,349 34,055 60,598 49,503 38,405

Hauptteil mit Darstellung der Ergebnisse 40
LaktatKontrolle 7,34 6,999 16,558 17,753 24,324 21,85 19,034
PyruvatPatient 0,567 0,14 0,617 0,761 0,714 1,419 1,935
PyruvatKontroll
e
0,739 0,578 0,964 0,877 1,217 1,266 1,094
Tabelle 13: Laktat- u. Pyruvatwerte in [mg/dl] für Patient C9-SW u. Kontrollperson
Abbildung 10 zeigt, daß die Laktatwerte des Patienten kontinuierlich bis zum Maximum
zum Zeitpunkt t4 (15. Belastungsminute) ansteigen und jeweils deutlich über denen der
Kontrollperson liegen. Die Pyruvatwerte des Patienten liegen unter denen der
Kontrollperson, außer zu den Zeitpunkten t5 und t6 (= 1 bzw. 5 Minuten nach
Belastungsende), wo sie die Pyruvatwerte der Kontrollperson übersteigen.
010203040506070
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,61,8
2
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 10: Laktat u. Pyruvat in [mg/dl] für Patient C9-SW u. Kontrollperson
3.1.11 Patient C8-GK, weiblich, 51 Jahre
Frau GK. litt an einer Multisystematrophie mit rasch progredientem Verlauf kombiniert
mit einer ausgeprägten Blutdruckhypotonieneigung bei vegetativer Dysregulation. Die
aktuelle stationäre Aufnahme erfolgte bei zunehmender Sturzneigung.
Der neurologische Untersuchungsbefund ergab eine Hypomimie bei unauffälligem
Hirnnervenstatus. Frau GK. hatte eine deutliche Sprachartikulationsstörung bei
Hypophonie. Die Körperhaltung war vornüber gebeugt, die Retropulsionsneigung war
ausgeprägt. Das Gehen war nur mit Hilfe möglich, dabei zeigte Frau GK. starke
Schwankneigung. Das Gangtempo war verlangsamt und die Schrittgröße vermindert, die
Armmitbewegung beim Gehen war reduziert. Weiterhin fand sich ein rechtsbetonter

Hauptteil mit Darstellung der Ergebnisse 41
Extremitätenrigor und beidseitige Bradydysdiadochokinese. Der Fingernaseversuch wurde
dysmetrisch ausgeführt, der Kniehackeversuch war unauffällig. Bei fehlenden Paresen und
fehlenden Pyramidenbahnzeichen waren die Muskeleigenreflexe seitengleich mittellebhaft
auslösbar. Bimalleolär war eine Pallhypästhesie von 5/8 nachweisbar, ansonsten war keine
Sensibilitätsstörung vorhanden.
Die Medikation zum Untersuchungszeitpunkt bestand aus Madopar disp. 6 x 1, Antiparkin
1-½-0, Madopar HBS 1x1 zur Nacht, Budipin 10 mg 4x1, Dridase 1x1 zur Nacht, DOPS
100 4x1, Dogmatil 1x1, Astonin H 1x½, Gutron-Tropfen 4x15.
Frau GK. zeigte eine unterdurchschnittliche Leistung (vgl. Tab. 17), für sie mußte von
Anfang an eine kleinere Belastungsstufe als für die übrigen Probanden gewählt werden.
Frau GK.s Werte:
t0 t1 t2 t3 T4 t5 t6
LaktatPatient 14,253 11,608 15,363 16,814 12,545 13,998 12,120
LaktatKontrolle 5,292 6,999 24,069 35,506 36,53 44,211 45,577
PyruvatPatient 0,957 1,045 0,972 0,899 0,998 0,948 0,955
PyruvatKontroll
e
0,534 0,486 0,851 0,8 1,161 1,329 1,785
Tabelle 14: Laktat- u. Pyruvat in [mg/dl] für Patientin C8-GK u. Kontrollperson
Die Laktatwerte der Patientin liegen durchgängig über den auffällig niedrigen Werten der
Kontrollperson. Die Pyruvatruhewerte (t0) gleichen sich, während der Belastung (t1 bis t4)
steigen die Pyruvatwerte der Patientin jeweils unter dem Niveau der Kontrollwerte, bis sie
die Werte der Kontrollperson in der Erholungsphase (t5 und t6) leicht übersteigen.

Hauptteil mit Darstellung der Ergebnisse 42
02468
1012141618
t0 t1 t2 t3 t4 t5 t6
Laktat Pat.Laktat Kontr.
00,20,40,60,8
11,21,41,61,8
2
t0 t1 t2 t3 t4 t5 t6
Pyruvat Pat.Pyruvat Kontr.
Abbildung 11: Laktat u. Pyruvat in [mg/dl] f. Patientin C8-GK u. Kontrollperson
3.2 Statistische Auswertung des gewonnenen Datenmaterials
3.2.1 Deskriptive Darstellung der Daten
Für Laktat und Pyruvat wird die zeitliche Entwicklung der Meßwert-Konzentrationen bei
Patienten und Kontrollpersonen zusammenfassend graphisch dargestellt:
[mg/dl]

Hauptteil mit Darstellung der Ergebnisse 43
Zeitpunkt der Messung
Lakt
atM
edia
n; 2
5%-,
75%
-Per
zent
ile; M
inim
um, M
axim
um
����������������������
����������������������
���������������������������������
������������������������������������������������
��������������������������������������������
�������������������������������������������������������
������������������������������������������������������������������
������������������������
����������������������
��������������������������������������������
�����������������������������������������������������������������������������
���������������������������������������������������������������������������������������������������
���������������������������������������������������������������������������������������������������
����������������������������������������������������������������������������������������
0
20
40
60
80
100
Ruhe 1 Min. 5 Min. 10 Min. 15 Min. EB 1 Min. EB 5 Min.
���������Patienten���������Kontrollen
Abbildung 12: Zeitliche Entwicklung der Laktatkonzentration. Dargestellt sind der Median (schwarzer bzw. weißer Punkt), 25%- bzw. 75%-Perzentile (karierte Kästchen) sowie Minimum und Maximum (Striche) der Laktatwerte.

Hauptteil mit Darstellung der Ergebnisse 44
Zeitpunkt der Messung
Pyru
vat
Med
ian;
25%
-, 75
%-P
erze
ntile
; Min
imum
, Max
imum
���������������������������������
��������������������������������������������
��������������������������������������������
�������������������������������������������������������
���������������������������������
������������������������������������
������������������������������������������������������������������������
��������������������������������������������
��������������������������������������������
��������������������������������������������
��������������������������������������������
�������������������������������������������������������
��������������������������������������������
��������������������������������������������������������������������������������������������������������������
0,0
0,5
1,0
1,5
2,0
2,5
Ruhe 1 Min. 5 Min. 10 Min. 15 Min. EB 1 Min. EB 5 Min.
���������Patienten���������Kontrollen
[mg/dl]
Abbildung 13: Zeitliche Entwicklung der Pyruvatkonzentration. Dargestellt sind der Median (schwarzer bzw. weißer Punkt), 25%- bzw. 75%-Perzentile (karierte Kästchen) sowie Minimum und Maximum (Striche) der Laktatwerte.
3.2.2 Statistische Auswertung mittels t-Test
Wie zu Beginn dieses Kapitels ausgeführt, ist für die statistische Auswertung der
Vergleich der Ataxie-Patienten und der Kontrollpersonen hinsichtlich Laktat und Pyruvat
zum Zeitpunkt der Maximalbelastung (t4) in Bezug auf den Ruhewert (t0) bedeutsam. (Die
übrigen Meßwerte (t1, t2, t3, t5, t6) dienen der deskriptiven Verlaufsbeurteilung und sind
im Hinblick auf zukünftige Meßreihen aufgrund zu geringer zeitlicher Trennschärfe
anteilig vernachlässigbar.)
Das interessierende Unterschiedsmaß ist der Quotient der Meßwert-Konzentration zum
Zeitpunkt t4 geteilt durch die Meßwert-Konzentration zum Zeitpunkt t0. Da es sinnvoll ist,
relative Veränderungen zu betrachten, wird der t-Test für die logarithmierten Werte
gerechnet und zwar als Quotient log [t4/t0].

Hauptteil mit Darstellung der Ergebnisse 45
Variable (log t4 – log t0)
Mittelwert Patienten
Mittel-wert Kontroll-personen
t-Wert p Anzahl der Patienten bzw. der Kontroll-personen
Standard-abwei-chung Patienten
Standard-abwei-chung Kontroll- personen
Laktat 0,5236 0,533 - 0,06434 0,9493 je 11 0,3801 0,2971 Pyruvat 0,2901 0,2785 0,1407 0,8895 je 11 0,2536 0,1032
Tabelle 15: Vergleich der Patienten und Kontrollen hinsichtlich der Differenz der
logarithmierten Werte zum Zeitpunkt t4 und t0
Aus den vorliegenden Daten sind aufgrund des t–Tests keine Unterschiede zwischen den
Ataxie-Patienten und Kontrollpersonen für Laktat und Pyruvat abzuleiten, da p > 0,05 ist.
In beiden Gruppen liegt der Mittelwert der Differenzen der logarithmierten Werte des
Laktat bei etwa 0,52 (Patienten) bzw. etwa 0,53 (Kontrollpersonen). Für Pyruvat liegt der
Mittelwert der Differenzen der logarithmierten Werte zwischen 0,28 (Kontrollpersonen)
und 0,29 (Patienten).

Diskussion 46
4 Diskussion
1996 konnten Schöls und Mitarbeiter in einer systematischen Studie durch die
Untersuchung von Muskelbiopsien nachweisen, daß Störungen der mitochondrialen
Funktion ein häufig zu beobachtendes Phänomen bei Patienten mit degenerativer Ataxie
sind (Schöls u. Mitarb. 1996).
Ziel dieser Studie war daher, herauszufinden, ob der Fahrradbelastungstest ein mögliches
Screening-Verfahren zur Diagnosefindung und -sicherung bei Ataxie-Patienten ist. Dazu
müßte sich der mitochondriale Defekt im peripher-venösen Blut in Ruhe und/oder während
der Belastung einem Kontrollkollektiv gegenüber nachweisen lassen. Nach dem Vorbild
des von Zierz beschriebenen Fahrradbelastungstest wurden 11 Patienten mit degenerativer
Ataxie einer 15-minütigen Fahrradbelastung ausgesetzt und dabei Blutproben zu
verschiedenen Zeitpunkten vor, während und nach der Belastung gewonnen (Zierz u.
Mitarb. 1989).
Unter der Annahme, daß den degenerativen Ataxien ein Defekt der Atmungskette
zugrunde liegt und aufgrund der Erkenntnis, daß dieser Defekt sich nicht auf das zentrale
Nervensystem beschränkt, sondern sich auch in nichtneuronalen, „peripheren“ Geweben
wie Muskelgewebe nachweisen läßt, wurde peripher-venöses Blut von Ataxie-Patienten
untersucht. Sollte die Dysfunktion der Atmungskette also ein systemischer biochemischer
Defekt sein, müßte sich dieser auch im peripher-venösen Blut darstellen, welches ein im
klinischen Alltag leichter zu gewinnendes und mit geringerem Aufwand zu untersuchendes
Medium als Muskelgewebe darstellt.

Diskussion 47
4.1 Kommentierung der Ergebnisse
4.1.1 Laktat
Wie zu Beginn von Kapitel 3 angeführt, muß die Interpretation der Daten die Tatsache
berücksichtigen, daß die 11 Patienten des Gesamtkollektivs zwar alle Ataxie-Patienten
sind, im Endeffekt aber 6 verschiedene Diagnosen haben, die von der SCA 1, SCA 3, SCA
6 und autosomal dominant zerebellärer Ataxie (ADCA) über die idiopathische zerebelläre
Ataxie (IDCA) bis zur Multisystematrophie (MSA) reichen: Durch den Zerfall des
ursprünglich homogenen Patientenkollektivs mit der Diagnose „olivopontozerebelläre
Atrophie“ (OPCA) in die genannten Subgruppen ergibt sich das Problem der
Undurchführbarkeit statistischer Berechnungen innerhalb der Subgruppen. Die statistische
Auswertung des Gesamtkollektivs wiederum ist zu grob, da sie z. B. keine Unterschiede
zwischen erblichen und nichterblichen Ataxien macht. Zugunsten einer möglichst hohen
Fallzahl (n = 11) für die statistische Auswertung wurde dennoch eine Berechnung mit dem
Gesamtkollektiv aller Ataxie-Patienten durchgeführt.
Die statistisch-rechnerische Auswertung der Laktatwerte zum Zeitpunkt der
Maximalbelastung aller 11 Patienten und deren Kontrollpersonen mit dem t-Test ergibt
keinen signifikanten Unterschied (vgl. Tab. 15, Kapitel 3). Da die Patientengruppe klein
und inhomogen ist, bedarf das Ergebnis einer Überprüfung in einer größeren und
einheitlicheren Stichprobe.
Wie die deskriptive Auswertung der Daten (vgl. Abbildung 12, Kapitel 3) zeigt, liegen die
Patienten-Laktatmediane unter Belastung überwiegend über denen der Kontrollpersonen.
Ferner fällt bei der Betrachtung von Abbildung 12 auf, daß die Laktatspiegel der Patienten
und Kontrollpersonen zwar von einem fast identischen Ruhemedian ausgehen, sich aber
mit zunehmender Belastungsdauer zunehmend stärker voneinander unterscheiden. Der
größte Abstand zwischen den Laktatmedianen der 11 Patienten und Kontrollpersonen ist
zum Zeitpunkt t4, also zum Zeitpunkt der längsten Belastung (15. Belastungsminute),
erreicht.

Diskussion 48
Aufgrund sich kaum unterscheidender Ruhewerte erscheint es richtig, zu postulieren, daß
es nicht genügt, bei Verdacht auf eine mitochondriale Erkrankung lediglich die Ruhewerte
von Laktat (und Pyruvat) zu erfassen. Die Probanden müssen einer körperlichen Belastung
ausgesetzt werden, um einen Stoffwechseldefekt anhand des dann stark steigenden
Laktatspiegels diagnostizieren zu können. Diese Überlegung deckt sich mit den
Ergebnissen von Zierz, der unter Ruhebedingungen bei nur 63 %, aber unter Belastung auf
dem Fahrradergometer bei 83 % der von ihm untersuchten 30 Patienten mit
mitochondrialer Enzephalomyopathie eine Erhöhung des Laktatspiegels fand (Zierz u.
Mitarb. 1989). (Es stellt sich dabei die Frage, warum „nur“ bei 83 % und nicht bei allen
Patienten erhöhte Laktatspiegel gefunden wurden.)
Die unter Maximalbelastung höheren Laktatmediane der Patienten sind zumindest
auffällig. Sollte sich dieses Ergebnis in einer größeren Studie mit einheitlichen
Krankheitsbildern mit statistischer Signifikanz nachweisen lassen, könnte man schließen,
daß ein Zusammenhang zwischen mitochondrialer Störung und zerebellärer Degeneration
gegeben ist, der sich im peripher-venösen Blut unter körperlichen Belastung meßbar
nachweisen läßt. Dabei wäre auch interessant zu untersuchen, ob es innerhalb der Gruppe
der erblichen Ataxien sowie zwischen erblichen und nichterblichen Ataxien Unterschiede
hinsichtlich der Laktatwerte in Ruhe und unter Belastung gibt.
Vergleicht man beispielsweise die Grafiken einiger SCA-3-Patienten (C10-MR, C11-KR
und C4-GN: siehe Abbildungen 3 bis 5, Kapitel 3) sowie des Patienten C12-KSG mit einer
SCA-6 (Abbildung 6, Kapitel 3) mit der Grafik des SCA-1-Patienten C5-UL (Abbildung 1,
Kapitel 3), so fallen deutliche Unterschiede auf: Während die Laktatwerte des Patienten
C5-UL auf bzw. unter dem Niveau der Laktatwerte der Kontrollperson liegen, steigen die
Laktatwerte der anderen Patienten unter Belastung deutlich über das Niveau der jeweiligen
Kontrollperson. Da hier jedoch Einzelfälle untersucht werden, kann nicht auf das gesamte
Krankenkollektiv geschlossen werden.
Abbildung 2 zeigt dagegen, daß die Laktatwerte des Patienten C1-AK etwa gleich hoch
wie die der Kontrollperson sind.

Diskussion 49
Über die Ursache beinah gleicher Laktatwerte bei Patient und Kontrollperson kann man
spekulieren: Vielleicht hat Patient C1-AK eine besonders gute körperliche Kondition. Dies
wird zwar durch die im Fahrradbelastungstest gezeigte durchschnittliche Kilometerleistung
bestätigt; Herr AK. gab aber an, nicht regelmäßig Sport zu treiben.
Rauchen (vgl. Abschnitt 4.2) entfällt als mögliche Ursache, da sowohl Patient als auch
Kontrollperson angaben, 30 bis 40 Zigaretten pro Tag zu rauchen und z. B. Patient C10-
MR ebenfalls ca. 30 Zigaretten pro Tag rauchte.
Vielleicht ist die Mitochondriopathie des Patienten C1-AK auch noch nicht manifest,
sondern erst in ihren Anfängen begriffen. (Dem steht allerdings die bereits in 1990 erfolgte
klinische Manifestation bzw. Diagnosestellung der Ataxie entgegen). Man kann auch
vermuten, daß die Mitochondriopathie einem Schwelleneffekt unterliegt (Wallace 1988),
d. h. der verbleibende funktionsfähige Teil der Mitochondrien kompensiert den Ausfall der
defekten Mitochondrien.
Denkbar ist auch, daß Herr AK. eine Spontanmutation der mitochondrialen DNA hat, die
sich kompensatorisch auf einen möglichen Atmungskettendefekt ausgewirkt hat. Vielleicht
hat aber die Kontrollperson eine klinisch noch nicht manifeste Mitochondriopathie, die
sich anhand der Laktatspiegel in Ruhe und unter Belastung bereits zum
Untersuchungszeitpunkt vermuten lassen könnte.
Auch muß z. B. ein möglicher Leberschaden der Kontrollperson Berücksichtigung finden,
da durch den Cori-Zyklus ein Teil der Stoffwechsellast von der Muskulatur zur Leber
verlagert wird:
Im gesunden Organismus wird das in der aktiven Muskulatur glykolyse-bedingt anfallende
Pyruvat durch die Laktat-Dehydrogenase in Laktat umgewandelt, gelangt über die
Blutbahn zur Leber, wo es abermals durch die Laktat-Dehydrogenase in Pyruvat
umgewandelt wird. Aus Pyruvat entsteht dann im Rahmen der hepatischen
Gluconeogenese Glukose, welches von der kontrahierenden Muskulatur utilisiert werden
kann. Dieser Zyklus ist in Form des erhöhten Laktatspiegels im Blut nachvollziehbar, da
aufgrund des hohen NADH/NAD+-Verhältnisses im tätigen Muskel vor allem Laktat
entsteht. Ein Leberschaden oder auch ein Defekt der verschiedenen Laktat-
Dehydrogenase-Typen der jeweiligen Gewebe könnte hier also zu Veränderungen führen.

Diskussion 50
Zusammenfassend läßt sich sagen, daß ein nichtpathologisches Ergebnis beim
Fahrradbelastungstest eine Mitochondriopathie nicht mit Sicherheit ausschließen kann, und
daß die geäußerten hypothetischen Überlegungen der Überprüfung in Studien mit größeren
Fallzahlen bedürfen.
Die eigenen Ergebnisse zeigen, daß die körperliche Belastung der Probanden in jedem Fall
lang genug gewählt werden muß, um eine mögliche vorhandene Mitochondriopathie zu
bestätigen, da die Laktatspiegel nicht unmittelbar nach Beginn der körperlichen Belastung,
sondern erst mit einer Latenz von 10 bzw. 15 Minuten Fahrradbelastung steigen. (Es stellt
sich die Frage, ob eine längere Belastungsdauer, z. B. 20 oder 25 Minuten ein deutlicheres
Ergebnis zur Folge hätte.) Die Forderung nach einer möglichst langen körperlichen
Belastung und somit möglichst aussagekräftigen laborchemischen Ergebnissen kollidiert
allerdings häufig mit dem Leistungsvermögen und Trainingszustand der Patienten: Gerade
für ältere Patientinnen und Patienten sowie Versuchspersonen in fortgeschrittenen
Krankheitsstadien erscheint der Fahrradbelastungstest ungeeignet.
Als Alternative scheint sich hier eine Laufbandergometrie auf der Grundlage der all-
täglichen Ausdauerbelastung des Gehens anzubieten (Schmidt u. Mitarb. 1997). Bei
nominell gleicher Belastung wie bei der Fahrradergometrie kommt es bei der Lauf-
bandergometrie nicht nur zu einer weniger starken Belastung des M. rectus femoris mit
entsprechend geringeren Ermüdungserscheinungen sondern auch zu schwächeren Blut-
druckanstiegen und geringerem myokardialen Sauerstoffverbrauch (Zervazi 1987, Schmidt
u. Mitarb. 1997). Bei Ataxiepatienten, die meist ein unsicheres Gangbild - manchmal
verbunden mit einer Fallneigung - haben, erscheint die Laufbandbelastung verglichen mit
der Fahrradbelastung allerdings als wenig zumutbar: Bei der Fahr-radbelastung ist
aufgrund des sitzend oder halbliegend ausgeübten Radfahrens und der Stützmöglichkeit
der Arme auf dem Lenker bzw. neben dem Körper weniger Balance nötig als bei der Lauf-
bandbelastung. Auch unter praktischen Gesichtspunkten erscheint der Fahr-
radbelastungstest günstiger: Sämtliche am Arm durchgeführten diagnostischen
Maßnahmen wie Blutentnahmen und Blutdruck- oder Pulsmessungen sind an ruhig

Diskussion 51
gehaltenen Armen (Fixation am Lenker bzw. auf der Liegefläche) einfacher
durchzuführen.
Da die Ataxie-Patienten zum Teil ältere Patienten mit entsprechend schlechtem
Trainingszustand und schweren Begleiterkrankungen sind, erscheint eine Belastung von
maximal 15 Minuten als empfehlenswert (vgl. z. B. die Fallbeschreibung der Patientin C8-
GK, Abschnitt 3.1.11). Eine 15 Minuten überschreitende Belastungsdauer ist aufgrund
kardiopulmonaler Risiken nur noch unter EKG-Kontrolle und intensiver ärztlicher
Beobachtung anzuraten. Mit längerer Belastungsdauer dürfte sich also nicht nur der
diagnostische Aufwand, sondern auch die Zahl der Komplikationen und die Zahl der
Versuchsabbrüche aufgrund von muskulärer Erschöpfung multiplizieren. Schließlich
gehen die bisher publizierten Arbeiten ebenfalls von einer 15-minütiger Belastung aus, so
daß bei Wahl dieser Belastungsdauer eine optimale Vergleichbarkeit gegeben ist (Zierz u.
Mitarb. 1989, Schmidt u. Mitarb. 1997)
4.1.2 Pyruvat
Die Auswertung der Pyruvat-Blutwerte aller 11 Ataxie-Patienten ergibt, bezogen auf den
auf den Ruhewert und den Meßwert zum Zeitpunkt der Maximalbelastung, keinen
statistisch signifikanten Unterschied bei der Berechnung mit dem t-Test. Die
Pyruvatmediane des Gesamtkollektivs aller 11 Patienten und der entsprechenden
Kontrollpersonen haben weitgehend die gleiche Größe (vgl. Abbildung 13, Kapitel 3).
Die - sich zumindest weniger als die Laktatspiegel unterscheidenden - Pyruvatspiegel
lassen sich durch die Aktivität der zytosolischen Laktat-Dehydrogenase erklären, die das
anfallende Pyruvat in Laktat umwandelt. Wie in der Einleitung erörtert, ist das Verhältnis
von Laktat und Pyruvat von der NADH bzw. NAD+-Konzentration abhängig und führt zu
einer Verschiebung zugunsten des Laktats.
Die Überlegung, daß die Laktatspiegel wesentlich aussagekräftiger als die Pyruvatspiegel
sind, deckt sich mit den von Zierz gemachten Angaben: So war der Pyruvatspiegel bei der
Untersuchung des venösen Blutes von 30 Patienten mit mitochondrialen
Enzephalomyopathien in Ruhe bei nur 14 (47 %) und unter Belastung bei nur 11 (37 %)
der Patienten erhöht (Zierz u. Mitarb. 1989).

Diskussion 52
Der Gipfel der Pyruvat-Blutspiegel liegt bei den meisten der Patienten (vgl. Abbildungen 1
bis 11, Kapitel 3) am Ende des Beobachtungszeitraumes. Für zukünftige
Untersuchungsreihen empfiehlt sich (auch im Hinblick auf die Laktatwerte) eine weitere
Blutentnahme etwa 10 Minuten nach Ende der Belastung; fakultativ könnte dafür die
Blutentnahme zum Zeitpunkt t5 (1 Minute nach Ende der Belastung) aufgrund eher
geringer Aussagekraft und fehlender Trennschärfe im Vergleich zum Laborwert t4 (15.
und letzte Belastungsminute) entfallen.
4.1.3 Laktat-Pyruvat-Quotient
Aus den gemessenen Werten für Laktat und Pyruvat kann man den Laktat-Pyruvat-
Quotienten berechnen. Da die Werte für Pyruvat der Patienten mit denen der Kontrollen
auffallend identisch sind, erscheint die Berechnung eines Quotienten mit Pyruvat als
Nennergröße überflüssig.
Während Zierz und Mitarbeiter bei einer etwa vergleichbaren Anzahl von Patienten
erhöhte Laktatspiegel bzw. Laktat-Pyruvat-Quotienten fanden (83 % bzw. 80 %), bewerten
Jackson und Mitarbeiter den Laktat-Pyruvat-Quotienten im Vergleich mit Blut-
Lakatwerten als weniger aussagekräftig (Zierz u. Mitarb. 1989, Jackson u. Mitarb. 1995).
Insgesamt dürften die Unterschiede zwischen Laktatwerten und Laktat-Pyruvat-Quotienten
wenig ausgeprägt sein. Angesichts der allgemein defizitären Finanzlage erscheint es nicht
notwendig, neben Laktat auch noch Pyruvat laborchemisch zu bestimmen, um dann einen
Quotienten zu berechnen, wo bereits der Laktatwert und sein Verhalten unter Belastung
ausreichend Interpretationsansatz bieten.
4.2 Diskussion der Ergebnisse
Unter körperlicher Belastung liegen die Laktatmediane der Ataxie-Patienten zwar
überwiegend über denen der Kontrollpersonen, jedoch ergibt sich in der statistischen
Berechnung kein signifikanter Unterschied. Somit ist das Ergebnis des Fahrrad-
belastungstests in diesem Fall nicht pathologisch. Vor dem Hintergrund der von Schöls
beschriebenen Tatsache, daß Störungen der mitochondrialen Funktion häufig bei Ataxie-
Patienten zu beobachten sind (Schöls u. Mitarb. 1996), können die tendenziell erhöhten

Diskussion 53
Laktatspiegel des untersuchten inhomogenen Patientenkollektivs möglicherweise als
Hinweis auf eine mitochondriale Dysfunktion aufgefaßt werden:
Eine zu vermutende Ursache einer verminderten mitochondrialen Oxidationsfähigkeit ist
die Abhängigkeit der mitochondrialen Enzymkapazität vom individuellen
Trainingszustand (Hollmann u. Mitarb. 1990). Störungen der aeroben Enzymleistung
finden sich z. B. bei Patienten mit nichtmitochondrialen Myopathien bzw. starker
muskulärer Inaktivitätsatrophie (Schmidt u. Mitarb. 1997).
Zwar ließen sich bei den 11 untersuchten Patienten Unterschiede im Trainingszustand
feststellen - nennenswert vor allem die Konditionsschwäche der beiden Patientinnen C4-
GN und C8-GK - es war jedoch bei keinem der 11 untersuchten Patienten eine Myopathie
oder eine höhergradige Muskelatrophie objektivierbar. Um die Unterschiede hinsichtlich
des Trainingszustandes für die Auswertung zu minimieren, wurden den Patienten ungefähr
leistungsentsprechende Kontrollpersonen zugeordnet, wie die nachfolgende Tabelle
verdeutlicht. Es ist jeweils die Kilometerleistung, die während der 15 Minuten
Fahrradbelastung erbracht wurde, angegeben:
Patient C5-UL 5,0 km Kontrollperson 4,7 km
Patient C1-AK 6,2 km Kontrollperson 7,0 km
Patientin C4-GN 3,8 km Kontrollperson 4,8 km
Patient C10-MR 7,4 km Kontrollperson 6,9 km
Patient C11-KR 5,9 km Kontrollperson 5,9 km
Patient C12-KSG 6,8 km Kontrollperson 7,0 km
Patient C6-GH 4,0 km Kontrollperson 6,4 km
Patient C7-KSR 5,1 km Kontrollperson 6,3 km
Patient C2-HK 5,7 km Kontrollperson 6,0 km
Patientin C8-GK 3,7 km Kontrollperson 5,5 km
Patient C9-SW 7,0 km Kontrollperson 6,4 km
Tabelle 16: Kilometerleistung der Probanden

Diskussion 54
Wie zu Beginn dieses Kapitels erwähnt, zeigte Schöls 1996 an Muskelbiopsien bei
insgesamt 22 von 61 Patienten mit degenerativer Ataxie mitochondriale Defekte, die
insbesondere den Komplex I der Atmungskette betrafen. Bei 31 % der untersuchten
Friedreich-Patienten und bei 47 % der Patienten mit autosomal dominant vererbter Ataxie
sowie bei 50 % der Patienten mit idiopathisch sporadisch zerebellärer Ataxie wurden
mitochondriale Enzymdefekte festgestellt (Schöls u. Mitarb. 1996).
Inzwischen konnte gesichert werden, daß die Friedreich-Ataxie eine mitochondriale
Krankheit auf genetischer Basis ist, bei der das Fehlen des vom Friedreich-Gen kodierten
Frataxins durch Eisenanhäufung in den Mitochondrien zu einer Störung der Atmungskette
führt (vgl. Kap. 1). Ein mit einer entsprechend großen Anzahl von Friedreich-Patienten
durchgeführter Fahrradbelastungstest könnte Hinweise für die Sensitivität des
Fahrradbelastungstest bei neurodegenerativen Mitochondriopathien geben. Die Friedreich-
Ataxie könnte gewissermaßen als „Musterkrankheit“ dienen. So bietet sich z. B. auch ein
Vergleich der Laktat- und Pyruvatspiegel von Friedreich-Patienten mit den Laktat- und
Pyruvatspiegeln der Patienten mit autosomal dominant vererbten Ataxien an.
Das pathogenetische Modell der Friedreich-Ataxie könnte auch für die spinozerebellären
Ataxien zutreffen: Ein durch repetitive CAG-Sequenzen erweitertes Gen kodiert
möglicherweise ein verändertes Protein, welches (ähnlich wie das Frataxin bei der
Friedreich-Ataxie) negativen Einfluß auf die mitochondriale Oxidationsfähigkeit ausübt.
Jüngere Veröffentlichungen stellen allerdings die Rolle nukleärer Einschlußkörperchen als
Auslöser oder Folge der Neurodegeneration in den Vordergrund (Übersicht von Riess u.
Mitarb. 2001). Die Rolle der von den SCA-Genen verändert kodierten Proteine in der
Pathogenese der Ataxien ist insgesamt noch weitgehend unklar.
Neben den Veränderungen in der nukleären DNA sind Veränderungen in der
mitochondrialen DNA (mtDNA) eine weitere mögliche Erklärung für die vermutete
Dysfunktion der oxidativen Phosphorylierung der Ataxie-Patienten: Die Atmungskette
steht am Kreuzungspunkt zweier verschiedenartiger genetischer Systeme (Zeviani u.
Mitarb. 1998), dem nukleären Genom, das den Mendelschen Vererbungsgesetzen folgt und

Diskussion 55
dem mitochondrialen Genom, das maternal vererbt wird. Trotz ihrer Komplexität und
genetischen Besonderheiten, auf die noch eingegangen wird, ist die mtDNA bei der
Synthese von Proteinen und bei der Vervielfältigung ihrer Erbinformation von der
nukleären DNA direkt und indirekt abhängig: Ein großer Teil der mitochondrialen Enzyme
wird vom Zellkern kodiert, an zytosolischen Ribosomen kodiert und in die Mitochondrien
transportiert. Zudem benötigen die Mitochondrien für die Replikation, Transkription und
Translation Faktoren, die von der nukleären DNA kodiert werden (Übersicht von Clayton
1991). Die Abhängigkeit der Funktionsfähigkeit der Mitochondrien (und damit der
Atmungskette) von nukleärer und mitochondrialer DNA könnte eine Brücke zwischen
repititiven CAG-Sequenzen in der nukleären DNA und Störungen der mitochondrialen
Funktion sein. Die durch die verlängerten Trinukleotidsequenzen verändert kodierten
Proteine könnten die Atmungskette direkt oder auf dem Umweg über das mitochondriale
Genom in ihrer Funktion beeinträchtigen.
Das mitochondriale Genom ist durch eine Reihe genetischer Besonderheiten
gekennzeichnet, wie nachfolgend kurz erläutert:
Die ausschließlich maternal vererbte ringförmige mitochondriale DNA kodiert für 13
Strukturproteine der Atmungskette sowie entsprechende Transfer-Ribonukleinsäure
(tRNA) und ribosomale Ribonukleinsäure (rRNA).
Menschliche Zellen enthalten viele hundert Mitochondrien, welche bei einer Zellteilung
nach dem Zufallsprinzip auf die neu entstehenden Zellen verteilt werden. Aufgrund der
hohen Mutationsrate der mtDNA kann es vorkommen, daß neben der ursprünglichen
mtDNA auch mutante mtDNA in die Tochterzellen übertragen wird. Das Vorhandensein
zweier verschiedener Kopien derselben mitochondrialen Gene in derselben Zelle oder
demselben Gewebe wird als Heteroplasmie bezeichnet. Die als mitotische Segregation
bezeichnete Tendenz der Trennung der 2 verschiedenen mitochondrialen Genome bewirkt
mitunter, daß schließlich zwei Zellreihen vorliegen, von denen jede Mitochondrien nur
einen Typs enthält. Entsprechend kann der eigentlich maternale Verbungsmodus
mitochondrialer Krankheiten als sporadisch erscheinen (nach Parker 1989).
Die mtDNA ist durch eine hohe Rate von Spontanmutationen und mangelnde
Reparaturmechanismen gekennzeichnet. In der Akkumulation dieser Mutationen mit

Diskussion 56
zunehmendem Alter (DiMauro u. Mitarb. 1993) und der damit verbundenen Abnahme der
mitochondrialen Enzymkapazität wird eine mögliche Ursache für altersabhängige
neurodegenerative Krankheiten gesehen (Schulz u. Mitarb. 1994).
Störungen der mitochondrialen Genetik wie häufige Spontanmutationen und fehlende
Reparaturmechanismen (zumal im Zusammenhang mit Heteroplasmie und mitotischer
Segregation) können sicher krankheitsverursachend im Rahmen von Neurodegeneration
sein, stellen aber im Fall der erblichen Ataxien, insbesondere im Fall der autosomal
dominant zerebellären Ataxien wahrscheinlich keine primäre Krankheitsursache dar: Die
hier untersuchten ADCA-Patienten haben z. T. eindeutige Familienanamnesen (siehe
Kapitel 3), d. h. ihre Erkrankungen treten nicht sporadisch bzw. gemäß maternalem
Vererbungsmuster auf. (Anders verhält es sich bei den Patienten mit nichterblichen
Ataxien, wo Störungen des mitochondrialen Erbgutes primär krankheitsverursachend sein
könnten.)
Es ist daher nicht davon auszugehen, daß eine mögliche Ursache für die mitochondriale
Dysfunktion der hier untersuchten Ataxie-Patienten ausschließlich im Bereich des
mitochondrialen Genoms liegt. Vielmehr ist - ähnlich wie bei der Friedreich-Ataxie - eine
Störung im Bereich des nukleären Genoms mit entsprechendem Einfluß auf die
mitochondriale Funktion zu vermuten. Mitochondriale Mutationen treten im übrigen auch
im Zusammenhang mit physiologischem Altern auf und sind für sich genommen nicht
beweisend für einen neurodegenerativen Prozess (Kadenbach 1995).
Der Einfluß des Rauchens auf die mitochondriale Leistungsfähigkeit ist eine weitere
potentielle Ursache für pathologische Laktat- und Pyruvatspiegel: An Thrombozyten von
Rauchern stellten Smith und Mitarbeiter eine 24%-ige Abnahme der Komplex-I-Aktivität
fest (Smith u. Mitarb. 1993).
Eine eindeutige Aussage über den generellen Einfluß des Rauchens läßt sich aufgrund der
Heterogenität des untersuchten Kollektivs jedoch nicht treffen: Es waren nicht nur 6 der
Patienten ehemals aktive oder jetzt aktive Raucher oder Gelegenheitsraucher, sondern auch
5 der Kontrollpersonen ehemals aktive oder jetzt aktive Raucher oder Gelegenheitsraucher.
(Der Einfluß des eventuellen Passivrauchens aller Probanden bleibt dabei unbetrachtet.) Es

Diskussion 57
ist jedoch anzunehmen, daß Rauchen die mitochondriale Funktion einzelner hier
untersuchter Probanden negativ beeinflußt und damit zu einer Erhöhung der Laktatspiegel
geführt hat.
Insgesamt betrachtet, stellt der Fahrradbelastungstest einen gering invasiven Suchtest zur
Klärung des Verdachts einer Mitochondriopathie dar. Trotz der vermuteten 75 bis 80%-
igen Sensitivität des Fahrradbelastungstest (Schmidt u. Mitarb. 1997) gibt nur die
histologisch-histochemische, immunhistologische und biochemische Aufarbeitung einer
Muskelbiopsie abschließende diagnostische Sicherheit (Reichmann u. Mitarb. 1988/1):
Zwar ist die Muskelbiopsie (verglichen mit dem Fahrradbelastungstest) ein invasives
Verfahren, die Untersuchung von Muskelbiopsien bleibt aber bei der Klärung von
Mitochondriopathien gleichsam der diagnostische „Goldstandard“.
Im Rahmen von Therapiestrategien steht dem Fahrradbelastungstest möglicherweise eine
Renaissance bevor: Aufgrund der jüngsten Erkenntnis, daß die Friedreich-Ataxie eine vom
nukleären Genom kodierte Mitochondriopathie ist, kann der Fahrradbelastungstest zur
Verlaufsbeurteilung von Therapieansätzen bei dieser Erkrankung dienen. Die Wirksamkeit
des Einsatzes von Antioxidantien, die Gabe von Coenzym Q10 und die Gabe von Carnitin
zur Förderung alternativer Wege der mitochondrialen Energiegewinnung (Schöls u.
Mitarb. 1999) oder die Verabreichung von Chelatoren (Wong u. Mitarb. 1999) dürfte sich
mittels des Fahrradbelastungstests laborchemisch überprüfen lassen.
Trotz in der deskriptiven Auswertung auffallender leichter Erhöhung der Laktatwerte der
untersuchten 11 Ataxie-Patienten konnte in der Gesamtschau der gewonnenen
Erkenntnisse hinsichtlich der Frage, ob tatsächlich eine mitochondriale Dysfunktion
vorliegt, und ob es sich hierbei um ein primäres oder sekundäres Phänomen handelt, keine
eindeutige Klarheit erzielt werden: Möglicherweise lassen sich eindeutig pathologische
Laktatspiegel im peripher-venösen Blut bei einem einheitlicheren und größeren
Patientenkollektiv als Ausdruck einer mitochondrialen Störung finden.

Zusammenfassung 58
5 Zusammenfassung
Nachdem Schöls und Mitarbeiter in einer systematischen Studie an Muskelgewebe
nachweisen konnten, daß Defekte der Atmungskette bei Patienten mit degenerativer Ataxie
ein häufiges Phänomen sind (Schöls u. Mitarb. 1996), wurden 11 Ataxie-Patienten mit dem
von Zierz und Mitarbeitern beschriebenen Fahrradbelastungstest untersucht (Zierz u.
Mitarb. 1989). Dazu wurden 7 Proben peripher-venösen Blutes vor, während und nach
einer Fahrradbelastung entnommen, um laborchemisch Laktat und Pyruvat als mögliche
periphere Indikatoren einer Mitochondriopathie bei Patienten mit degenerativer Ataxie
messen zu können.
Die untersuchten Patienten hatten ein Durchschnittsalter von 53,4 Jahren und teilten sich in
9 männliche und 2 weibliche Patienten auf, wobei die Diagnosen sowohl die
spinozerebelläre Ataxie (SCA) vom Typ 1, 3 und 6 als auch weitere autosomal dominant
zerebelläre Ataxien mit SCA-Mutationsausschluß sowie die idiopathisch sporadisch
zerebelläre Ataxie umfaßten. 1 Patientin hatte eine Multisystematrophie. Die
Kontrollpersonen hatten ein Durchschnittsalter von 54,9 Jahren.
Die deskriptive Auswertung zeigte zum Zeitpunkt der Maximalbelastung einen leicht
erhöhten Laktatmedian des Gesamtkollektivs der Patienten im Vergleich zum Median der
Kontrollpersonen. Trotz der Heterogenität des Patientenkollektivs hinsichtlich der Ataxie-
Diagnosen wurden die Laktatwerte aller Patienten zum Zeitpunkt der Maximalbelastung in
Relation zum Ruhewert mit denen der alters- und geschlechtsentsprechenden
Kontrollpersonen mit dem t-Test statistisch verglichen. Es konnte kein signifikanter
Unterschied festgestellt werden. Die Berechnung des Signifikanzniveaus des
Pyruvatspiegels zum Zeitpunkt der Maximalbelastung ergab in Bezug auf den Ruhewert
ebenfalls keine Signfikanz. Die deskriptive Auswertung der Pyruvatwerte zeigte nur
geringe Unterschiede zwischen Patienten und Kontrollpersonen, so daß die Aussagekraft
der Pyruvatspiegel hinsichtlich der Klärung einer Mitochondriopathie vernachlässigbar
erscheint.

Zusammenfassung 59
Da sich die Ruhe-Laktatmediane von Patienten und Kontrollen kaum unterscheiden, ist
anscheinend eine ausreichend lang andauernde körperliche Belastung mit entsprechend
zunehmenden Laktatspiegeln notwendig, um eine potentielle Mitochondriopathie abklären
zu können. Für Ataxie-Patienten erscheint aufgrund etwaiger Gangstörungen eine
Fahrradergometrie geeigneter als eine Laufbandergometrie. Lassen sich bei einem
größeren und einheitlicheren Patientenkollektiv pathologische Laktatspiegel finden, muß
eine mitochondriale Störung bei Ataxie-Patienten grundsätzlich vermutet werden.
Nach der jüngsten Entdeckung, daß die Friedreich-Ataxie eine vom nukleären Genom
kodierte mitochondriale Erkrankung ist, dürfte dem Fahrradbelastungstest als gering
invasive Untersuchungsmethode im Vergleich zum „Goldstandard“ Muskelbiopsie neue
Bedeutung zukommen: Einerseits kann die Friedreich-Ataxie als „Musterkrankheit“ für die
degenerativen Ataxien und andere neurodegenerative Erkrankungen dienen. Andererseits
könnten mittels des Fahrradbelastungstests mögliche Therapieansätze des M. Friedreich
mit Coenzym Q10, Vitamin C, Carnitin oder Chelatoren hinsichtlich ihrer Wirksamkeit
besser beurteilt werden.

Literaturverzeichnis 60
6 Literaturverzeichnis
1. Brennan, W. Jr., Bird E. D. and Aprille J. R.: Regional mitochondrial respiratory activity in Huntington’s disease brain. Journal of Neurochemistry (1985) 44, 1948-1950
2. Campuzano, V., L. Montermini, M. D. Moltò u. Mitarb.: Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271 (1996) 1423-1427
3. Chai, Y., Koppenhafer, S. L., Bonini, N. M., Paulson, H. L.: Analysis of the role of heat shock protein (Hsp) moleculare chaperones in polyglutamine disease. Journal of the Neurological Science 19 (1999): 10338-10347
4. Chamberlain, S., J. Shaw, A. Rowland u. Mitarb.: Mapping of mutation causing Friedreich’s ataxia to human chromosome 9. Nature 334 (1988) 248-250
5. Clayton, D. A.: Replication and transcription of vertebrate mitochondrial DNA. Annuals of Revised Cellular Biology 7 (1991) 453-478
6. Cummings, C. J., Mancini, M. A., Antalffy B., DeFranco D., Orr, H. T., Zoghbi, H. Y.: Chaperone suppresion of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA 1. Nat. Genet. 19 (1998) 148-154
7. DiMauro, S., C. Moraes: Mitochondrial Encephalomyopathies (Review). Archives of Neurology 50 (1993) 1197-1208
8. Foury F., O. Cazzalino: Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Letters 411 (1997) 373-377
9. Friedreich, N.: Über degenerative Atrophie der spinalen Hinterstränge. Virchow’s Archive of Pathology and Anatomy 26 (1863) 391-419
10. Fukuhara, N., S. Tokiguchi, S. Shirakawa, T. Tsubaki: Myoclonus epilepsy associated with ragged-red-fibers (mitochondrial abnormalities): disease entity or syndrome ? Light and electromicroscopic studies of two cases and review of literature. Journal of the Neurological Science 47 (1980) 117-133
11. Greenfield, J. G.: The spino-cerebellar degenerations. Blackwell, Oxford 1954

Literaturverzeichnis 61
12. Harding, A. E. Idiopathic late onset cerebellar ataxia. A clinical and genetic study of 36 cases. Journal of Neurology and Science 51 (1981) 259-271
13. Harding, A. E.:Classification of the hereditary disorders and paraplegias. Lancet (1983) 1151-1155
14. Harding, A. E.: The hereditary ataxias and paraplegias. Churchill Livingstone. Edinburgh 1984
15. Hollmann, W., T. Hettinger: Sportmedizin, Arbeits- und Trainingsgrundlagen. 3. Auflage, Schattauer, Stuttgart, New York (1990) S. 317
16. Holmes, G.: An attempt to classify cerebellar disease, with a note on Marie’s hereditary cerebellar ataxia. Brain 30 (1907) 545-567
17. Jackson, M., J. Schaefer, M. Johnson, A. Morris, D. Turnbull, L. Bindoff: Presentation and clinical investigation of mitochondrial respiratory chain disease. A study of 51 patients. Brain 118 (1995) 339-357
18. Kadenbach, B.: Nachlassende Leistungsfähigkeit im Alter. Ursache: stochastische Mutationen der Mitochondrien-DNA ? Fortschritte der Medizin 35-36 (1995) 32
19. Kageyama, Y., Ichikawa K., Fujioka A., Tstusumi A., Yorifuji S. and Miyoshi K.: An autopsy case of mitochondrial encephalomyopathy with prominent degeneration in olivo-ponto-cerebellar system. Acta neuropathologica 83 (1991) 83 99-103
20. Kearns, T., G. Sayre: Retinitis pigmentosa, external ophthalmoplegia and complete heart-block. Unusual syndrome with histologic study in one of two cases. Archive of Ophthalmology 60 (1958) 280-289
21. Klockgether, T., Schroth, G., Diener H. C., Dichgans, J.: Idiopathic cerebellar ataxia of late onset: natural history and MRI morphology. Journal of Neurology, Neurosurgery and Psychiatry 53 (1990) 297-305
22. Klockgether, T., Bürk, K., Auburger G., Dichgans, J.: Klassifikation und Diagnostik der degenerativen Ataxien. Nervenarzt 66 (1995) 571-581
23. Klockgether, T., B. Evert: Genes involved in hereditary ataxias. Trends in Neurosciences 21 (1998) 413-418

Literaturverzeichnis 62
24. Komure, O., A. Sano, N. Nishino u. Mitarb.: DNA analysis in hereditary dentatorubral-pallidoluysian atrophy: Correlation between CAG repeat length and phenotypic variation and the molecular basis of anticipation. Neurology 45 (1995) 143-149
25. Löffler, G., Petrides, P.: Biochemie und Pathobiochemie. 6. Auflage 1998. Springer-Verlag. S. 495 ff
26. Löffler, G.: Funktionelle Biochemie. 1. Auflage 1993. Springer-Verlag. S. 163 ff
27. Ludwig, A., V. Flockerzi, F. Hoffmann: Returned expression and cellular localisation of the alpha-1 and beta-subunit of high-voltage-activated calcium channel in rat brain. Journal of Neuroscience 17 (1997) 1339-1349
28. Luft, R., D. Ikkos, G. Palmieri, L. Ernster, N. Afzelius: A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control. A correlated clinical, biochemical and morphological study. Journal of clinical investigation 41 (1962) 1776
29. Menzel, P.: Beitrag zur Kenntnis der hereditären Ataxie und Kleinhirnatrophie. Archiv für Psychatrie und Nervenkrankheiten 22 (1891) 160-190
30. Orr, H. T., M. Chung, S. Banfi u. Mitarb.: Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nature Genetics 4 (1993) 221-226
31. Parker, W.D. Jr.: Sporadic neurologic diseases and the electron transport chain: a hypothesis. In Pascuzzi (Ed.), Proceedings of the 1989 scientific meeting of the American Society for Neurological Investigation: new developments in neuromuscular disease. Bloomingtom: Indiana University Printing Services 1990. (1989) 59-94
32. Pavlakis S., P. Phillips, S. DiMauro, D. DeVivo, L. Rowland: Mitochondrial myopathy, encephalomyopathy, lactic acidosis and stroke-like epidsodes: A distinctive clinical syndrome. Annuals of Neurology 16 (1984) 481-488
33. Reichmann, H., H. Mertens: Moderne Diagnostik von Myopathien. Deutsches Ärzteblatt 85 (1988/1) A-3358-3366 (Heft 47)
34. Reichmann, H., R. Rohkamm, K. Richer, H. Mertens: Mitochondriale Myopathien, Deutsche medizinische Wochenschrift 113 (1988/2) 106-113
35. Reichmann, H., R. Gold: Metabolische Myopathien und Enzephalomyopathien. Deutsches Ärzteblatt 89 (1992) A 1862-1866 [Heft 20]

Literaturverzeichnis 63
36. Reichmann, H., Flörke, S., Hebenstreit, G., Schrubar, H., Riederer, P.: Analyses of energy metabolism and mitochondrial genome in post-mortem brain from patients with Alzheimer’s disease. Journal of Neurology 240 (1993) 377-380
37. Riess, O., Schmidt, Th., Schöls, L.: Autosomal dominant vererbte spinozerebellare Ataxien. Deutsches Ärzteblatt 98 (2001) A 1319-1326 [Heft 23]
38. Robinson, B. H.: Lactic acidemia. In: C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle, (editors): The metabolic basis of inherited disease. Volume 1, 6th edition. New York. Mc Graw-Hill (1989) 869-888
39. Schapira, A.H.V., Cooper, J. M., Dexter, D., Jenner, P., Clark J. B., Marsden C. D.: Mitochondrial complex I deficiency in Parkinson’s disease. Lancet I (1989) 1269
40. Schmidt, M., M. Kunkel, P. Schuff-Werner, M. Naumann, H. Reichmann, C. Reimers: Standardisierte aerobe Gehbelastung auf dem Laufband bei Gesunden sowie Patienten mit mitochondrialen und nicht-mitochondrialen Myopathien. Der Nervenarzt 68 (1997) 831-835
41. Schöls, L., H. Reichmann, G. Amoiridis, P. Seibel, S. Wagener, S. Seufert, H. Przuntek: Mitochondrial disorders in degenerative ataxias. European Journal of Neurology 3 (1996) 55-60
42. Schöls, L., O. Rieß, G. Amoiridis, A. Rieß, H. Przuntek, J. T. Epplen: Genetische Diagnostik, Klassifikation und klinische Krankheitsentitäten hereditärer Ataxien. Fortschritte der Neurologie und Psychiatrie 65 (1997) 79-89
43. Schöls, L., C. Hardt: Friedreich-Ataxie. Psycho 25 (1999)
44. Schulz, J. B., M. F. Beal: Mitochondrial dysfunction in movement disorders. Current Opinion in Neurology 7 (1994) 333-339
45. Shapira, Y., S. Harel, A. Russell: Mitochondiral Encephalomyopathy. A group of neuromuscular disorders with defects in oxidative metabolism. Israelian Journal of Medical Science 13 (1977) 161-164
46. Smith, P. R., J. M. Cooper, G. G. Govan, A. E. Harding, A. H. V. Schapira: Smoking and Mitochondrial Function: A Model for Environmental Toxins. Quarterly Journal of Medicine 86 (1993) 657-660

Literaturverzeichnis 64
47. Stevanin, G,. E. Le Guern, N. Ravise u. Mitarb.: A third locus for autosomal dominant cerebellar ataxias type I maps to chromosome 14q24.3-qter: Evidence for the existence of a fourth locus. American Journal of Human Genetics 54 (1994) 11-20
48. Stryer, L.: Biochemie. Korr. Nachdruck 1991. Spektrum-Verlag. S. 413 ff
49. Takiyama, Y., M. Nishiwaza, H. Tanaka u. Mitarb. : The gene for Machado-Joseph-Disease maps to human chromsome 14q. Nature Genetics 4 (1993) 300-303
50. Truong, D.D., Harding, A. E., Scaravilli F., Smith, S. J. M., Morgan-Hughes, J. A., Marsden C. D.: Movement Disorders in Mitochondrial Myopathies. Movement Disorders Volume 5, No. 2 (1990) 109-117
51. Wallace, D. C., X. Zheng, M. T. Lott: Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological and biochemical characterization of a mitochondrial DNA disease. Cell. 55 (1988) 601-610
52. Wong, A., J. Yang, P. Cavadini, C. Gellera, B. Lonnerdal, F. Taroni, G. Cortopassi: The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inibitors of apoptosis. Human Molecular Genetics 8 (1999) 425-430
53. Zenner, K., R. Gold, B. Meurers, H. Reichmann: Die mitochondrialen Enzephalomyo-pathien Kearns-Sayre-Syndrom, MELAS und MERRF im Vergleich. Der Nervenarzt 61 (1990) 597-603
54. Zervazi, R.: Hämodynamische Reaktion unter verschiedenen Belastungsformen. In: Rost, R., F. Weber (Hrsg.): Kardiologie im Sport. Deutscher Ärzteverlag, Köln.
55. Zeviani, M., V. Tiranti, C. Piantadosi: Mitochondrial disorders. Medicine 77 (1998) 59-72
56. Zhuchenko, O., J. Bailey, P. Bonnen u. Mitarb.: Autosomal dominant cerebellar ataxia (SCA 6) associated with small polyglutamine expansions in the alpha1a-voltage-dependent calcium channel. Nat. Genet. 15 (1997), 62-69
57. Zierz, St., S. Meeßen, F. Jerusalem: Lactat- und Pyruvatspiegl in der Diagnostik mitochondrialer Myopathien. Nervenarzt 60 (1989) 545-548

DANKSAGUNG
Ohne die engagierte Mitarbeit zahlreicher Mitarbeiter der Ruhr-Universität Bochum hätte
diese Arbeit nicht erstellt, überarbeitet und eingereicht werden können. Herzlichen Dank
allen, die mitgeholfen haben.
Mein Dank geht insbesondere an Herrn Prof. Dr. W. Kuhn, der das Thema bereitgestellt
hat und die Arbeit routiniert begleitet hat. Namentlich danken möchte ich auch Herrn PD
Dr. L. Schöls, der für meine Fragen immer ein offenes Ohr hatte.
Meiner Frau Stefanie danke ich für ihre liebevolle Unterstützung, fachlich kompetente
Ratschläge und große Hilfe bei der kritischen Revision des Textes.
Außerdem möchte ich
den Patienten und Probanden für ihre Aufgeschlossenheit und Mitarbeit,
den MTAs des Liquorlabors für zahlreiche Hilfen und Tips,
Thorsten Schulte für seine kritischen Meinungsäußerungen,
Ralf Wegener für EDV-Unterstützung,
Inge und Hans Weiberg für die Leihgabe des Kettler-Heimtrainers,
Eltern und Schwiegereltern für emotionalen Beistand,
dem Pflegepersonal des St.-Josef-Hospitals,
dem ärztlichen Personal der Neurologischen und Orthopädischen Klinik sowie
dem Personal der Neurologischen Ambulanz danken.

LEBENSLAUF
Personalien Name und Vorname: Jochen B. Müller Geburtsdatum: 20.11.1972 Geburtsort: Gronau in Westfalen Nationalität: deutsch Familienstand: verheiratet, 2 Kinder Konfession: römisch-katholisch Eltern: Wolfgang Müller, Diplom-Volkswirt Gisela Müller, M. A. Schule, Studium und beruflicher Werdegang 1978-1982 Pestalozzi-Grundschule, Ahaus 1982-1991 Alexander-Hegius-Gymnasium, Ahaus 01.07.1991 Eintritt in die Deutsche Marine als Marinesänitätsoffizieranwärter, bis 01.09.1992 militärische Grundausbildung und Seekadettenlehrgang an der
Marineschule Mürwik, seemännische Grundausbildung auf SSS „Gorch Fock“, Flottenpraktikum auf FGS „Augsburg“,
Offizierlehrgang an der Sanitätsakademie der Bundeswehr, militärfachlicher Lehrgang an der Marineversorgungsschule, Pflegepraktikum im Bundeswehrkrankenhaus Hamm
10.09.92 Immatrikulation für das Studium der Humanmedizin an der Ruhr-Unversität Bochum
09.09.1994 Ärztliche Vorprüfung 29.08.1995 1. Abschnitt der ärztlichen Prüfung 10.09.1997 2. Abschnitt der ärztlichen Prüfung 14.10.1997 Beginn des Praktischen Jahres im Marienhospital Herne,
Universitätsklinik der Ruhr-Universität Bochum mit Wahlfach Orthopädie (St.-Josef-Hospital Bochum, Universitätsklinik)
17.11.1998 3. Abschnitt der ärztlichen Prüfung 30.11.1998 Erlangung der Erlaubnis zur vorübergehenden Ausübung des
ärztlichen Berufes 01.12.1998 Beginn der Tätigkeit als Arzt im Praktikum im
Bundeswehrkrankenhaus Hamm (Chefarzt: Dr. med. V. Gedicke) 28.12.2000 Erlangung der Vollapprobation; Ernennung zum Stabsarzt 01.09.2001 Beginn der Tätigkeit als Weiterbildungsassistent (Fachrichtung
Allgemeinmedizin) in der Praxis Dr. med. H. Reineke, Soest

Anlage: Arbeitsanweisung „Lactat für die Sportmedizin“ und Arbeitsanweisung „Pyruvat“