Embed Size (px)
Citation preview

Verfahrenstechnik für IngenieureLehr- und Übungsbuch
Mit CDKarl SchwisterVolker Leven

Schwister/LevenVerfahrenstechnik
für Ingenieure


Karl Schwister Volker Leven
Verfahrenstechnik für IngenieureLehr- und Übungsbuch
Mit 317 Bildern und 51 Tabellen
Fachbuchverlag Leipzigim Carl Hanser Verlag

Autoren:Prof. Dr. rer. nat. Karl SchwisterDipl.-Ing. Volker Leven
Fachhochschule DüsseldorfFachbereich Maschinenbau und VerfahrenstechnikLehr- und Forschungsgebiet Chemie und Bioverfahrenstechnik
Bibliografische Information der Deutschen NationalbibliothekDie Deutsche Nationalbibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie;detaillierte bibliografische Daten sind im Internet über http://dnb.d-nb.de abrufbar.
ISBN 978-3-446-43070-9E-Book-ISBN 978-3-446-43136-2
Einbandbild und Bild Seite 2: LyondellBasell Industries, Basell Polyolefine GmbH Wesseling
Die Wiedergabe von Gebrauchsnamen, Handelsnamen, Warenbezeichnungen usw. in diesem Werk berechtigt auch ohne besondere Kennzeichnung nicht zu der Annahme, dass solche Namen im Sinne der Warenzeichen- und Marken-schutz-Gesetzgebung als frei zu betrachten wären und daher von jedermann benutzt werden dürften.
Dieses Werk ist urheberrechtlich geschützt.Alle Rechte, auch die der Übersetzung, des Nachdrucks und der Vervielfältigung des Buches oder Teilen daraus, vorbehalten. Kein Teil des Werkes darf ohne schriftliche Genehmigung des Verlages in irgendeiner Form (Fotokopie, Mikrofilm oder ein anderes Verfahren), auch nicht für Zwecke der Unterrichtsgestaltung, reproduziert oder unter Verwendung elektronischer Systeme verarbeitet, vervielfältigt oder verbreitet werden.
Fachbuchverlag Leipzig im Carl Hanser Verlag© 2013 Carl Hanser Verlag München www.hanser-fachbuch.deLektorat: Jochen HornHerstellung: Katrin WulstEinbandrealisierung: Stephan RönigkSatz, Druck und Bindung: Kösel, KrugzellPrinted in Germany

Vorwort
Das Lehr- und Übungsbuch Verfahrenstechnik für In-genieure bietet eine kompakte, verständliche und an den Bedürfnissen der Praxis ausgerichtete Gesamt-darstellung über den vielfältigen und weit verzweig-ten Bereich der Verfahrens technik. Es gibt eine ers-te Einführung in die notwendigen Grundlagen wie Thermodynamik, Kinetik, Katalyse, Strömungstech-nik sowie Statistik und behandelt natürlich die Grundverfahren der Mechanischen und Thermi-schen Verfahrenstechnik sowie die Chemische Re-ak tionstechnik. Neben den zahlreichen Übungsauf-gaben und Exkursen wird auch dem Fachfremden ein Einstieg in diese wichtige Ingenieurdisziplin er-möglicht.
Die Abgrenzung der vier Teilbereiche erfolgt nach traditionellem Verständnis. Danach basieren die Grundoperationen der Mechanischen Verfahrens-technik im Wesentlichen auf den Grundgesetzen der Mechanik. Hinzu kommen die in der Thermischen Verfahrenstechnik zusammengefassten Gesetzmäßig-keiten des Stoff- und Wärmetransports. Das kom-plexe Gebiet der Chemischen Reaktionstechnik be-schäftigt sich mit chemischen Umsetzungen, für deren quantitative Beschreibung sowohl die Ther-modynamik als auch die Kinetik benötigt wird. Bei der Anwendung der dargelegten Theorien und Be-rechnungsmethoden stoßen Studenten und Absol-venten häufig auf Probleme, die den Einstieg in Lehrveranstaltungen, Seminare oder Praktika er-schweren.
Die Zielsetzung des vorliegenden Lehr- und Übungsbuches besteht darin, den Studenten der Fach- und Vertiefungsrichtungen Verfahrenstechnik, Biotechnologie, Lebensmitteltechnologie, Pharma- und Kosmetikindustrie, Kunststoffindustrie, Metall-verarbeitung, Bergbau und Hüttenwesen sowie einer Reihe von Industrie- und Umweltbereichen das nö-tige Grundwissen einerseits, aber auch eine Auf-gaben samm lung von Berechnungsbeispielen und Stoffdaten andererseits, zur schnellen und erfolgrei-chen Einarbeitung an die Hand zu geben.
Besonders möchten wir uns bei Frau Antje Kim Fraederich bedanken, die neben ihrem Studium die beiliegende CD konzipiert und erstellt hat. Für das Korrekturlesen bedanken wir uns bei Frau Janine Mater und Herrn Klaus Vogelsang. Unseren Frauen Karin und Regine möchten wir auf diesem Wege unseren herzlichen Dank für ihr Verständnis und ihre Geduld während der Erarbeitung des Manu-skriptes ausdrücken.
Für die in diesem Buch enthaltenen Fehler und Mängel sind wir alleine verantwortlich.
Düsseldorf, im Oktober 2012 Volker Leven und Karl Schwister


Inhalt
Vorwort . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
VTG – Verfahrenstechnische Grundlagen . . 13
1 Physikalische Größen und Einheitensysteme . . . . . . . . . . . . . . . . 14
1.1 Größen und Größenarten . . . . . . . . . . . . . 141.2 Größen- und Zahlen wertgleichungen . . . 161.3 Zustandsgrößen und Prozessgrößen . . . . 171.4 Zustandsfunktionen . . . . . . . . . . . . . . . . 181.5 Gehalts- und Konzentra tionsangaben . . . 19
1.5.1 Massenanteil . . . . . . . . . . . . . . 201.5.2 Stoffmengenanteil . . . . . . . . . . 201.5.3 Volumenanteil . . . . . . . . . . . . . 211.5.4 Massenkonzentration . . . . . . . 211.5.5 Stoffmengenkonzentration . . . 211.5.6 Volumenkonzentration . . . . . . 211.5.7 Molalität . . . . . . . . . . . . . . . . . . 221.5.8 Aktivität . . . . . . . . . . . . . . . . . . 22
1.6 Umrechnungen und Mischungs- rechnung . . . . . . . . . . . . . . . . . . . . . . . . 22
2 Statistische Grundlagen . . . . . . . . . . . 252.1 Fehlerarten . . . . . . . . . . . . . . . . . . . . . . . 25
2.1.1 Grobe Abweichung von Messwerten . . . . . . . . . . . . . . . 25
2.1.2 Systematische Abweichung von Messwerten . . . . . . . . . . . 25
2.1.3 Zufällige Abweichung von Messwerten . . . . . . . . . . . . . . . 26
2.2 Darstellung von Messreihen . . . . . . . . . . 262.3 Erfassung der Messwert abweichung . . . . 29
2.3.1 Normalverteilung nach Gauss . . . . . . . . . . . . . . . . . . . . 30
2.3.2 Standardabweichung . . . . . . . 302.3.3 Vertrauensbereich . . . . . . . . . . 31
2.4 Fehlerfortpflanzung . . . . . . . . . . . . . . . . 322.4.1 Methode der oberen und
unteren Grenze . . . . . . . . . . . . 322.4.2 Gausssche Fehler-
fortpflanzung . . . . . . . . . . . . . . 332.4.3 Lineare Fehlerfortpflanzung . 33
2.5 Grafische Auswertung von Messdaten . . 342.5.1 Lineare und nichtlineare
Skalen . . . . . . . . . . . . . . . . . . . . 342.5.2 Anfertigung einer grafischen
Darstellung . . . . . . . . . . . . . . . 352.5.3 Grafische Auswertung linearer
Zusammenhänge . . . . . . . . . . . 36
3 Aggregatzustände und Phasenlehre . 383.1 Gasförmiger Zustand . . . . . . . . . . . . . . . 38
3.1.1 Ideales Gas . . . . . . . . . . . . . . . . 383.1.2 Gasgemische . . . . . . . . . . . . . . 403.1.3 Reale Gase . . . . . . . . . . . . . . . . 42
3.2 Flüssiger Zustand . . . . . . . . . . . . . . . . . . 453.2.1 Dichte und Volumen-
ausdehnung . . . . . . . . . . . . . . . 453.2.2 Viskosität von Flüssigkeiten . 473.2.3 Oberflächenspannung . . . . . . 48
3.3 Fester Zustand . . . . . . . . . . . . . . . . . . . . 493.3.1 Kristallgitter und Kristall-
systeme . . . . . . . . . . . . . . . . . . 493.3.2 Methoden zur Ermittlung
der Festkörperstruktur . . . . . . 513.4 Phasenumwandlung von Reinstoffen . . . 52
3.4.1 Druck-Temperatur-Phasen-diagramm . . . . . . . . . . . . . . . . . 52
3.4.2 Clausius-Clapeyron- Gleichung . . . . . . . . . . . . . . . . . 54
3.4.3 Regel von Trouton . . . . . . . . . 553.5 Binäre Phasen gleich gewichte . . . . . . . . . 553.6 Ternäre Phasen gleich gewichte . . . . . . . . 59

8 Inhalt
3.7 Verdünnte Lösungen . . . . . . . . . . . . . . . . 603.7.1 Kolligative Eigenschaften . . . . 603.7.2 Löslichkeit . . . . . . . . . . . . . . . . 62
4 Strömungstechnische Grundbegriffe 654.1 Allgemeine Grundlagen . . . . . . . . . . . . . 654.2 Kontinuitätsgleichung . . . . . . . . . . . . . . 664.3 Strömung ohne Reibung . . . . . . . . . . . . . 67
4.3.1 Gleichung von Bernoulli . . . . 674.3.2 Gleichung von Torricelli . . . 69
4.4 Strömung mit Reibung . . . . . . . . . . . . . . 704.4.1 Viskosität . . . . . . . . . . . . . . . . . 704.4.2 Widerstandsbeiwert . . . . . . . . 71
4.5 Rohrströmung mit Reibung . . . . . . . . . . 724.5.1 Laminare Strömung . . . . . . . . 724.5.2 Turbulente Strömung . . . . . . . 734.5.3 Druckverlust in Rohrleitungen 734.5.4 Druckverlust in Formstücken
und Armaturen . . . . . . . . . . . . 75
5 Produktionstechnische Grundbegriffe . . . . . . . . . . . . . . . . . . . . 76
5.1 Verfahrensentwicklung . . . . . . . . . . . . . . 765.2 Verfahrensinformationen . . . . . . . . . . . . . 775.3 Fließschemata von Anlagen . . . . . . . . . . 78
5.3.1 Grundfließschema . . . . . . . . . . 795.3.2 Verfahrensfließschema . . . . . . 795.3.3 Rohrleitungs- und
Instrumenten fließschema . . . 805.3.4 Mess- und Regelschema . . . . . 82
5.4 Stoffdaten und Verfahrensablauf . . . . . . 825.4.1 Stoffdaten . . . . . . . . . . . . . . . . . 825.4.2 Sicherheitstechnische Daten . 835.4.3 Toxikologische Daten . . . . . . . 84
5.5 Scale-up – Probleme . . . . . . . . . . . . . . . . 84
CRT – Chemische Reaktionstechnik . . . . . . . 87
6 Grundlagen der Reaktionstechnik . . . 886.1 Einführung und Grundbegriffe . . . . . . . . 88
6.1.1 Klassifizierung chemischer Reaktionen . . . . . . . . . . . . . . . . 88
6.1.2 Beurteilungsgrößen und Definitionen . . . . . . . . . . . . . . . 89
6.2 Chemische Thermo dynamik . . . . . . . . . . 926.2.1 Systeme und Zustandsgrößen 92
6.2.2 Erster Hauptsatz . . . . . . . . . . . 926.2.3 Standardenthalpien . . . . . . . . 936.2.4 Zweiter Hauptsatz . . . . . . . . . . 946.2.5 Chemisches Gleichgewicht . . 96
6.3 Stoff- und Wärme bilanzen . . . . . . . . . . . 986.3.1 Transportprozesse . . . . . . . . . 996.3.2 Erhaltungssätze . . . . . . . . . . . . 100
7 Kinetik chemischer Reaktionen . . . . . 1027.1 Reaktions geschwindigkeit . . . . . . . . . . . 1027.2 Gesetze der Reaktions kinetik . . . . . . . . . 103
7.2.1 Differenzialgleichungen . . . . . 1047.2.2 Reaktionen nullter Ordnung . 1057.2.3 Reaktionen erster Ordnung . . 1057.2.4 Reaktionen zweiter Ordnung 1077.2.5 Reaktionen dritter Ordnung . . 1087.2.6 Molekularität einer Reaktion 109
7.3 Bestimmung von Reaktionsordnungen . . 1097.3.1 Differenzialmethode . . . . . . . . 1107.3.2 Methode der Anfangs-
geschwindigkeiten . . . . . . . . . 1107.3.3 Integrationsmethode . . . . . . . . 1117.3.4 Halbwertszeitmethode . . . . . . 1117.3.5 Konzentrationsabhängige
Messgrößen . . . . . . . . . . . . . . . 1117.3.6 Experimentelle Bestimmungs-
methoden . . . . . . . . . . . . . . . . . 1127.4 Kinetik komplexer Reaktionen . . . . . . . . 113
7.4.1 Gleichgewichtsreaktionen . . . 1147.4.2 Parallelreaktionen . . . . . . . . . . 1157.4.3 Folgereaktionen . . . . . . . . . . . . 116
7.5 Theorie der Reaktions geschwindigkeit . . 1177.5.1 Temperaturabhängigkeit der
Reaktionsgeschwindigkeit . . . 1187.5.2 Theorie des aktivierten
Komplexes . . . . . . . . . . . . . . . . 120
8 Aktivierung von Reaktionen und Katalyse . . . . . . . . . . . . . . . . . . . . . 122
8.1 Aktivierung von Reaktionsprozessen . . . 1238.1.1 Thermische Aktivierung . . . . 1238.1.2 Katalytische Aktivierung . . . . 1248.1.3 Aktivierung durch Initiator-
zerfall . . . . . . . . . . . . . . . . . . . . 1268.1.4 Biokatalytische Aktivierung . 1268.1.5 Fotochemische Aktivierung . . 128
8.2 Homogene und heterogene Systeme . . . . 128

9Inhalt
8.3 Heterogene Katalyse . . . . . . . . . . . . . . . . 1288.3.1 Heterogene Reaktionen mit
Feststoffen . . . . . . . . . . . . . . . . 1288.3.2 Heterogene Reaktionen mit
Fluiden . . . . . . . . . . . . . . . . . . . 1348.3.3 Reaktionsablauf . . . . . . . . . . . . 135
8.4 Homogene Katalyse . . . . . . . . . . . . . . . . 1368.4.1 Einphasige Reaktionssysteme 1378.4.2 Säure- und Basenkatalyse . . . 1388.4.3 Enzymkatalytische Reaktionen 1408.4.4 Reversible Hemmung von
Enzymen . . . . . . . . . . . . . . . . . 143
9 Ideale Reaktoren . . . . . . . . . . . . . . . . . 1469.1 Klassifizierung von Reaktoren . . . . . . . . 146
9.1.1 Allgemeine Betriebsformen . . 1469.1.2 Vermischung im Reaktor . . . . 1479.1.3 Wärmetechnische Betriebs-
formen . . . . . . . . . . . . . . . . . . . 1489.1.4 Grundtypen chemischer
Reaktoren . . . . . . . . . . . . . . . . . 1509.1.5 Stoff- und Wärmebilanzen . . . 151
9.2 Diskontinuierlich betriebener Rührkessel 1529.2.1 Isotherm betriebener
Rührkessel . . . . . . . . . . . . . . . . 1539.2.2 Adiabat betriebener
Rührkessel . . . . . . . . . . . . . . . . 1549.2.3 Polytrop betriebener
Rührkessel . . . . . . . . . . . . . . . . 1559.3 Kontinuierliche Betriebs führung ohne
Rückvermischung der Reaktionsmasse . 1569.4 Kontinuierliche Betriebsführung mit
Rückvermischung der Reaktionsmasse . 1599.5 Rührkesselkaskade . . . . . . . . . . . . . . . . . 161
9.5.1 Gestaltung und stoffliche Bilanzierung . . . . . . . . . . . . . . 162
9.5.2 Berechnung von Rührkessel-kaskaden . . . . . . . . . . . . . . . . . 163
9.6 Vergleichende Betrachtung der Reaktoren . . . . . . . . . . . . . . . . . . . . . . . . 165
10 Reale Reaktoren und Verweilzeitverteilungen . . . . . . . . . . . . . . . . . . . . . 168
10.1 Abweichungen vom idealen Verhalten . . 16810.2 Verweilzeit unter suchungen zur
Charakterisierung des Vermischungs-verhaltens . . . . . . . . . . . . . . . . . . . . . . . . 169
10.2.1 Verweilzeitspektrum und Verweilzeit-Summenfunktion 170
10.2.2 Messung der Verweilzeit-verteilungen . . . . . . . . . . . . . . . 171
10.3 Berechnung und Aus wertung von Verweil zeit verteilungen . . . . . . . . . . . . . . 17210.3.1 Idealer kontinuierlicher
Rührreaktor . . . . . . . . . . . . . . . 17210.3.2 Kaskade von kontinuierlich
betriebenen idealen Rührreaktoren . . . . . . . . . . . . . 173
10.3.3 Laminar durchströmter Rohrreaktor . . . . . . . . . . . . . . . 174
10.4 Reaktoren mit realem Verhalten . . . . . . . 17510.4.1 Dispersionsmodell . . . . . . . . . 17510.4.2 Kaskadenmodell . . . . . . . . . . . 17810.4.3 Berechnungsbeispiele . . . . . . . 179
MVT – Mechanische Verfahrenstechnik – Grundoperationen . . . . . . . . . . . . . . . . 183
11 Charakterisierung von Partikeln und dispersen Systemen . . . . . . . . . . . . . . 184
11.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 18411.2 Partikelgrößen und Merkmale . . . . . . . . 18511.3 Kenngrößen einer Verteilung . . . . . . . . . 187
11.3.1 Verteilungssumme . . . . . . . . . 18711.3.2 Verteilungsdichte . . . . . . . . . . 188
11.4 Verteilungsgesetze . . . . . . . . . . . . . . . . . . 19011.4.1 Potenzverteilung nach Gates-
Gaudin-Schumann . . . . . . . . . 19011.4.2 Gausssche Normalverteilungs-
funktion . . . . . . . . . . . . . . . . . . 19111.4.3 Logarithmische Normal-
verteilung . . . . . . . . . . . . . . . . . 19111.4.4 RRSB-Verteilung . . . . . . . . . . . 19211.4.5 Vergleich der Verteilungen
und Kennwerte . . . . . . . . . . . . 19311.5 Messen einer Partikel größenverteilung . . 194
12 Zerteilung von Feststoffen, Flüssigkeiten und Gasen . . . . . . . . . . . 197
12.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 19712.2 Zerkleinerung . . . . . . . . . . . . . . . . . . . . . 197
12.2.1 Näherungsformeln . . . . . . . . . 19912.2.2 Zerkleinerungsgrad . . . . . . . . 200

10 Inhalt
12.2.3 Bruchvorgang . . . . . . . . . . . . . 20012.2.4 Zerkleinerungsmaschinen . . . 201
12.3 Flüssigkeitszerteilung . . . . . . . . . . . . . . . 20312.3.1 Berieselung . . . . . . . . . . . . . . . 20312.3.2 Zerstäubung . . . . . . . . . . . . . . . 20312.3.3 Zerspritzung . . . . . . . . . . . . . . 207
12.4 Begasung . . . . . . . . . . . . . . . . . . . . . . . . 207
13 Trennen disperser Systeme . . . . . . . . 21013.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 21013.2 Absetzprozesse . . . . . . . . . . . . . . . . . . . . 210
13.2.1 Sedimenter . . . . . . . . . . . . . . . . 21413.2.2 Trennschärfe und
Abscheidegrad . . . . . . . . . . . . . 21613.2.3 Zentrifuge . . . . . . . . . . . . . . . . 21813.2.4 Zyklone . . . . . . . . . . . . . . . . . . . 22313.2.5 Koagulation und Flokkulation 22513.2.6 Flotation . . . . . . . . . . . . . . . . . . 226
13.3 Filtrationsprozesse . . . . . . . . . . . . . . . . . 22713.3.1 Kuchenfiltration . . . . . . . . . . . 22713.3.2 Querstromfiltration . . . . . . . . . 23213.3.3 Tiefenfiltration . . . . . . . . . . . . . 234
14 Mischen . . . . . . . . . . . . . . . . . . . . . . . . . 23614.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 23614.2 Mischen von Feststoffen . . . . . . . . . . . . . 23814.3 Statisches Mischen von Fluiden . . . . . . . 24114.4 Dynamisches Mischen von Flüssigkeiten 242
14.4.1 Laminarer Bereich . . . . . . . . . 24614.4.2 Turbulenter Bereich . . . . . . . . 24614.4.3 Übergangsbereich . . . . . . . . . . 24714.4.4 Rühren von nicht-Newton-
schen Flüssigkeiten . . . . . . . . 24814.4.5 Scale-up – Maßstabs-
übertragung . . . . . . . . . . . . . . . 24814.4.6 Weitere Anwendungsgebiete 249
15 Agglomerieren . . . . . . . . . . . . . . . . . . . 25215.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 25215.2 Einteilung der Agglomeration . . . . . . . . . 253
15.2.1 Aufbauagglomeration (Pelletieren) . . . . . . . . . . . . . . . 253
15.2.2 Pressagglomeration (Formpressen) . . . . . . . . . . . . . 255
16 Transport von Stoffen . . . . . . . . . . . . . 25816.1 Arten der Förderung . . . . . . . . . . . . . . . . 258
16.2 Transport von Flüssigkeiten . . . . . . . . . . 25816.2.1 Verdrängungspumpen . . . . . . 25916.2.2 Zentrifugalpumpen . . . . . . . . . 26016.2.3 Strahlpumpen . . . . . . . . . . . . . 26116.2.4 Berechnungen . . . . . . . . . . . . . 262
16.3 Transport von Gasen . . . . . . . . . . . . . . . 26616.3.1 Lüfter und Gebläse . . . . . . . . . 26616.3.2 Verdichter . . . . . . . . . . . . . . . . 269
16.4 Feststoffförderung . . . . . . . . . . . . . . . . . . 27116.4.1 Gurt-, Gliederbandförderer
und Becherwerke . . . . . . . . . . 27116.4.2 Schnecken- und
Spiralförderer . . . . . . . . . . . . . 27216.4.3 Pneumatische Förderung . . . . 273
TVT – Thermische Verfahrenstechnik – Grundoperationen . . . . . . . . . . . . . . . . 277
17 Verdampfen und Kondensieren . . . . . 27817.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 278
17.1.1 Dampf . . . . . . . . . . . . . . . . . . . . 28017.1.2 Wärmeübertragung . . . . . . . . 28117.1.3 Wärmeaustauscher . . . . . . . . . 283
17.2 Verdampfen und Eindampfen . . . . . . . . . 284
18 Kristallisation . . . . . . . . . . . . . . . . . . . . 28818.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 28818.2 Berechnungen zur Kristallisation . . . . . . 28918.3 Technische Anwendung . . . . . . . . . . . . . . 291
19 Trocknen . . . . . . . . . . . . . . . . . . . . . . . . 29319.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 29319.2 Trocknungsarten und Trocknungskurven 29719.3 Bauarten von Trocknern . . . . . . . . . . . . . . 299
20 Destillation und Rektifikation . . . . . . . . . . . . . . . . . . . . . 300
20.1 Grundlagen . . . . . . . . . . . . . . . . . . . . . . 30020.1.1 Ideales Zweistoffgemisch . . . . 30020.1.2 Reales Zweistoffgemisch . . . . 30620.1.3 Mischungslücken . . . . . . . . . . 308
20.2 Destillation . . . . . . . . . . . . . . . . . . . . . . . 31020.2.1 Absatzweise (einfache)
Destillation . . . . . . . . . . . . . . . . 31020.2.2 Fraktionierte Destillation . . . . 31420.2.3 Kontinuierliche Destillation . . 315

11Inhalt
20.2.4 Trägerdampfdestillation . . . . . 31620.2.5 Vakuumdestillation . . . . . . . . . 317
20.3 Rektifikation . . . . . . . . . . . . . . . . . . . . . . 31720.3.1 Grundlagen der Rektifikation 31820.3.2 Bilanzen an einer Rekti-
fikationskolonne . . . . . . . . . . . 32120.3.3 Wärmebedarf und Heiz-
leistung . . . . . . . . . . . . . . . . . . 33220.3.4 Füllkörper- und Packungs-
kolonnen . . . . . . . . . . . . . . . . . . 33220.3.5 Rektifikationsverfahren . . . . . 334
21 Sorption . . . . . . . . . . . . . . . . . . . . . . . . 33921.1 Absorption . . . . . . . . . . . . . . . . . . . . . . . 339
21.1.1 Grundlagen der Absorption . . 33921.1.2 Bilanzierung und Berechnung 34421.1.3 NTU/HTU-Konzept für die
Absorption . . . . . . . . . . . . . . . . 35021.1.4 Kenngrößen eines Absorbers 354
21.1.5 Wärmebilanz bei der Absorption . . . . . . . . . . . . . . . . 355
21.1.6 Anwendung der Absorption . . 35721.2 Adsorption . . . . . . . . . . . . . . . . . . . . . . . 359
21.2.1 Grundlagen der Adsorption . . 35921.2.2 Adsorptionsmittel . . . . . . . . . . 36121.2.3 Beispiele einiger Adsorptions-
mittel . . . . . . . . . . . . . . . . . . . . 36221.2.4 Mechanismen der Adsorption 36421.2.5 Bilanzierung von Adsorbern . 37121.2.6 Wärmebilanz an einem
Festbettadsorber . . . . . . . . . . . 37421.2.7 Technische Anwendungen
und Bauformen . . . . . . . . . . . . 376
Hinweise zur beigefügten CD . . . . . . . . . . . . . 378
Sachwortverzeichnis . . . . . . . . . . . . . . . . . . . 379


VTG Verfahrenstechnische Grundlagen
1 Physikalische Größen und Einheitensysteme2 Statistische Grundlagen3 Aggregatzustände und Phasen4 Strömungstechnische Grundbegriffe5 Produktionstechnische Grundbegriffe
einer Strömung, einer Wärmeübertragung oder des Stofftransports ist in allen Fällen mit der inneren und äußeren Reibung zu begründen. Bei der Über-tragung der Wärme werden Teilchen mit hoher ther mischer Bewegung Energie an benachbarte Teil-chen abgeben. Strömungsimpulse entstehen, wenn schnelle auf langsame Teilchen treffen. Der Stoff-transport entsteht, wenn Teilchen aufgrund ihrer kinetischen Bewegungsenergie in andere Bereiche vordringen. Alle Vorgänge, die auf eine submikros-kopische Bewegung von Teilchen zurückzuführen sind, lassen sich mit mathematischen Gleichungen beschreiben.
In diesem Teil des Buches werden neben grund-legenden naturwissenschaftlichen Gesetzmäßigkei-ten die verfahrenstechnischen Grundlagen bespro-chen. Es geht um Definitionen von Begriffen, um das Vergrößern von Laborverfahren in den Produktions-maßstab (Scale-up) und um chemische Stoffum-wandlungsprozesse in verschiedenen Reaktionsap-paraten. Daneben werden Transportvorgänge von Fluiden, Wärme und Stoffen in ihren physikalischen Grundprinzipien dargestellt. Bei diesen drei Er-scheinungsformen handelt es sich um Ausgleichs-vorgänge auf submikroskopischer Ebene, bei denen Volumenelemente sich gegeneinander verschieben oder aneinander reiben können. Der Widerstand

1 Physikalische Größen und Einheitensysteme
1 .1 Größen und GrößenartenEin wesentliches Ziel der naturwissenschaftlichen und technischen Forschung ist die Beschreibung der in der Natur ablaufenden Vorgänge bzw. der technischen Prozesse durch mathematische Glei-chungen. Diese werden entweder durch Experimen-te oder durch theoretische Überlegungen erhalten. Diese Gleichungen stellen einen funktionalen Zu-sammenhang zwischen den für den betrachteten Prozess maßgeblichen erfassbaren Eigenschaften oder Erscheinungen des Systems her, die auch allge-mein Einflussgrößen genannt werden. Solche Grö-ßen sind z. B. Länge, Masse, Zeit, Stromstärke, Kon-zentration, Arbeit oder Energie. Jede dieser Größen G lässt sich aufspalten in ein Produkt aus dem Zah-lenwert {G} und der dazugehörigen Einheit [G]:
G = {G} · [G] (1-1)
Die Einheit ist eine willkürlich wählbare, aber ver-einbarte Größe der gleichen Art wie die betrachtete Größe. Die physikalische Größe der Zeit t = 60 s be-steht beispielsweise aus dem Zahlenwert {t} = 60 und der Einheit [t] = s. Statt der Einheit „Sekunde“ kann auch eine andere Zeiteinheit verwendet wer-den, z. B. „Minute“ oder „Stunde“.
Eine Gleichung zwischen verschiedenen Ein-flussgrößen (Größengleichung) beinhaltet immer die Arten (Einheiten) dieser Größe und deren Zah-lenwerte. Größengleichungen sind daher im Unter-schied zu den reinen Zahlenwertgleichungen (z. B.: 4 · 2 = 8) auch Einheitengleichungen. Eine Größen-gleichung ist demzufolge auch nur dann erfüllt, wenn Zahlenwert und Einheit auf beiden Seiten übereinstimmen.
Gleichartige Größen werden unter dem Begriff Größenarten zusammengefasst. So stellen die Grö-ßen Arbeit und Wärme etwas grundsätzlich anderes dar, gehören jedoch beide der gemeinsamen Grö-ßenart Energie an. Der überwiegende Teil der physi-kalischen und chemischen Größenarten ist durch Naturgesetze miteinander verknüpft. Einige müssen jedoch unabhängig voneinander festgelegt werden. Sie werden als Grundgrößenarten oder Basisgrö-ßen bezeichnet. Aus diesen Basisgrößen werden die abgeleiteten Größen definiert.
Bisher existierte eine Vielzahl von Einheitensys-temen, z. B. das physikalische und das technische Einheitensystem u. v. a.; daneben kommen noch die britischen und US-Einheitensysteme. Die Gremien der Meterkonvention haben das sog. Internationa-le Einheitensystem (Système International d’Uni-tés = SI) empfohlen. Durch das „Gesetz über Einhei-ten im Meßwesen“ vom 2. Juli 1969 wurde das Internationale Einheitensystem für die Bundesrepu-blik Deutsch land gesetzlich vorgeschrieben Die Ba-sisgrößen, Basiseinheiten und Einheitenzeichen sind in Tabelle 1.1 gezeigt.
Tabelle 1 .1 SI-Basisgrößen und Basiseinheiten
Basisgröße BasiseinheitName
Länge
Masse
Zeit
Stromstärke
Temperatur
Stoffmenge
Lichtstärke
Meter
Kilogramm
Sekunde
Ampere
Kelvin
Mol
Candela
Einheiten-zeichen
m
kg
s
A
K
mol
cd

1.1 Größen und Größenarten 15
1Die Basiseinheiten des Internationalen Einhei-tensystems sind gegenwärtig wie folgt definiert:
1 Meter ist gleich der Länge der Strecke, die Licht im Vakuum während der Dauer von 1/299 792 458 Sekunden durchläuft.
1 Kilogramm ist die Masse des Internationalen Kilogrammprototyps in Paris, einem Zylinder aus einer Pt-Ir-Legierung von 39 mm Höhe und glei-chem Durchmesser.
1 Sekunde ist die Zeitdauer von 9 192 631 770 Schwingungsperioden der Strahlung des 133-Cä-sium isotops.
1 Ampere ist die Stärke eines zeitlich unverän-derlichen Stromes, der durch zwei im Vakuum par-allel im Abstand von 1 m voneinander angeordnete, geradlinige, unendlich lange Leiter von vernachläs-sigbar kleinem Querschnitt fließend zwischen die-sen Leitern elektrodynamisch eine längenbezogene Kraft von 2 · 10–7 Newton je 1 m Leiterlänge hervor-rufen würde.
1 Kelvin ist der 273,16te Teil der thermodyna-mischen Temperatur des Tripelpunktes von Wasser genau definierter Isotopenzusammensetzung.
1 Candela ist die Lichtstärke einer monochro-matischen Strahlungsquelle mit einer Frequenz von exakt 540 · 1012 Hz, deren Strahlstärke in die her-ausgegriffene Richtung 1/683 W/sr beträgt.
1 Mol ist die Stoffmenge eines Systems, das so viele Teilchen enthält, wie Atome in 0,012 kg des Kohlenstoffisotops 12C enthalten sind. (Diese Zahl
NA = 6,022 141 · 1023 Atome/Mol heißt Avogadro-Konstante.)
Dezimale Vielfache und Teile von Einheiten wer-den durch Voransetzen von Präfixen ausgedrückt (vgl. Tabelle 1.2).
Die Vielzahl möglicher Größen lässt sich auf die sieben Basisgrößen zurückführen. Eine Basisgröße kann nicht weiter auf andere Größen reduziert wer-den. Daher gibt es für eine Basisgröße keine Defini-tion, sondern nur eine Messvorschrift, mit der ihre Einheit festgelegt wird.
Exkurs 1 .1 Vom Urmeter bis zur Neudefinition der Längen-einheit Meter
Die ist seit Ende des 18. Jahrhundertsin Gebrauch. Der Ursprung ist ein Beschluss derfranzösischen Nationalversammlung, ein einheitlichesLängenmaß zu schaffen. Das Königreich Bayern trat 1870,noch vor der Reichsgründung, der InternationalenMeterkonvention bei und erhielt als einer der damals 27beteiligten Staaten eine offizielle Kopie des Prototyps von1889 aus einer Platin-Iridium-Legierung. Während des 3.Reiches musste Bayern ein Exemplar an die Physikalisch-Technische Reichsanstalt Berlin abgeben.
Der wurde erst 1960 abgelöst, als dieGeneralkonferenz für Maß und Gewicht das Meter als das1 650 763,73-Fache der Wellenlänge der von Atomen desNuklids Krypton-86 im Vakuum ausgesandten Strahlungdefinierte. Damit wurde eine etwas höhere Genauigkeitdefiniert.
Da die früheren Definitionen des Urmeters auf der Basis desinternationalen Prototyps bzw. einer bestimmten Wellenlängeim Vergleich zur mit Atomuhren gemessenen SI-BasiseinheitSekunde relativ ungenau waren, entschloss man sich, dasMeter neu zu definieren. Seit 1983 ist die
wie folgt festgelegt:
ist die Strecke, die das Licht im Vakuum in einer Zeitvon 1 / 299 792 458 Sekunden durchläuft.
Längeneinheit Meter
Meterprototyp
SI-BasiseinheitMeter
1 Meter
Alle anderen Größen sind abgeleitete Größen und können entsprechend ihrer Definition als solche dargestellt werden. Als Beispiel einer abgeleiteten Größe soll die Kraft betrachtet werden. Für sie gilt das physikalische Gesetz:
Kraft = Masse · BeschleunigungF = m · a
Mit der Masse m = 1 kg als Basisgröße und der Be-schleunigung a = 1 m/s2 als bereits abgeleitete Grö-ßenart ergibt sich:
Tabelle 1 .2 Präfixe für dezimale Vielfache und Teile von Ein-heiten (DIN 1301, Auszug)
Präfixe Kurz-zeichen
Bedeutung der Präfixe
ExaPetaTeraGigaMegaKiloHektoDeka
DeziZentiMilliMikroNanoPikoFemtoAtto
EPTGMkhda
dcmµnpfa
10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit
18
15
12
9
6
3
2
1
10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit10 -Faches der Einheit
–1
–2
–3
–6
–9
–12
–15
–18

1 Physikalische Größen und Einheitensysteme16
F = m · a = 1 kg · 1 m/s2 = 1 kg · m/s2 = 1 N
Die abgeleitete Einheit kg · m · s−2 hat die neue Be-zeichnung Newton erhalten. 1 N ist daher die Kraft F, die der Masse m = 1 kg die Beschleunigung a = 1 m/s2 erteilt.
Tabelle 1 .3 Einige abgeleitete Größenarten und Einheiten des Internationalen Einheitensystems
Größenart Einheit physikal.Gleichung
N (Newton)
Pa (Pascal)
J (Joule)
W (Watt)
Hz (Herz)
Kraft
Druck
Energie
Leistung
Frequenz
F = m a
p = F / A
W = F s
P = W / t
f = / t
�
�
1
Einheiten-gleichung
N = kg m / s
Pa = kg / (m s )
J = kg m / s
W = kg m / s
Hz = 1/s
�
�
�
�
2
2
2 2
2 3
Viele der häufig verwendeten Einheiten sind keine SI-Einheiten. Sie sind jedoch Vielfache von SI-Ein-heiten, wie z. B. die Einheit Liter (1 l = 10–3 m3). In der folgenden Tabelle 1.4 sind einige gebräuchliche Umrechnungsfaktoren für übliche Einheiten zusam-mengestellt.
Tabelle 1 .4 Umrechnungsfaktoren häufig verwendeter Einheiten
Basisgröße BasiseinheitName
Einheiten-zeichen
Minute (min)
Stunde (h)
Tag (d)
Jahr (a)
1 min = 60 s
1 h = 3 600 s
1 d = 86 400 s
1 a = 31 536 000 s
Zeit ( )t
Länge ( )l
Volumen ( )V
Druck ( )p
Energie ( )E
Liter (l)
Bar (bar)
Elektronvolt (eV)
1 l = 10 m–3 3
1 bar = 100 000 Pa1 eV = 1,602 · 10 J–19
1 A = 10 m–10Angström (A)
1 .2 Größen und Zahlenwertgleichungen
Größengleichungen sind im Unterschied zu den frü-her häufig verwendeten Zahlenwertgleichungen un-abhängig von den verwendeten Einheiten und ent-halten daher auch keine Umrechnungsfaktoren. Es
ist daher zweckmäßig, Definitionen und Gesetze stets als Größengleichungen anzugeben.
Größengleichungen sind Gleichungen zwischen physikalischen Größen. Sie enthalten nur die Sym-bole der physikalischen Größen und Zahlenwerte, die aus mathematischen Operationen entstanden sind. Andere Zahlenwerte oder Zeichen, die aus der Umrechnung unterschiedlicher Einheiten stammen, enthalten sie nicht.
In Größengleichungen ist die physikalische Grö-ße vollständig angegeben, also als Produkt aus Zah-lenwert und Einheit. Folgendes Beispiel soll diesen Sachverhalt verdeutlichen:
Kraft = Masse · BeschleunigungF = m · g
Bei einer Masse von m = 90 kg sind der Zahlenwert {m} = 90 und die Einheit [m] = kg in die Größenglei-chung einzusetzen. Mit der Fallbeschleunigung von g = 9,81 m/s2 ist ebenso zu verfahren. Im Inter natio-nalen Einheitensystem ergibt sich:
F m g= ⋅ = ⋅ = ⋅90 9 81 882 9kg , m/s , kg m/ s2 2
Mit 1 kg · m/s2 = 1 N ist die Kraft F = 882,9 N.Im Technischen Einheitensystem wäre
m Fg
= = = ⋅909 81
9 17kp
, m/s, kp s
m2
2
Wird die Kraft F = 882,9 N in die Krafteinheit des Technischen Einheitensystems umgerechnet, so er-gibt sich mit 1 kp = 9,81 N:
F = 882,9 N = 882,9 N · (1/9,81) · kp/N = 90 kp
Mathematische Beziehungen zwischen reinen Zah-len werden Zahlenwertgleichungen genannt. Sie werden nur in Sonderfällen verwendet, z. B. bei der Umrechnung verschiedener Temperatureinheiten. Die gebräuchlichste Einheit ist Grad Celsius. In den USA ist auch die Einheit degree Fahrenheit in Ge-brauch. Die meistverwendete Einheit für wissen-schaftliche Zwecke ist das Kelvin.
Die Zahlenwerte der unterschiedlichen Einhei-ten sind durch Zahlenwertgleichungen verknüpft. Die Umrechnung einer Temperaturangabe von Fah-

1.3 Zustandsgrößen und Prozessgrößen 17
1renheit in Celsius ist in dem folgenden Beispiel ge-zeigt:
{ϑC} = 0,5556 · ({ϑF} − 32) (1-2)
Nach Gleichung (1-2) lässt sich der Zahlenwert der Celsius-Temperatur errechnen. Für die Tempe-ratur nach der Fahrenheit-Skala ist der reine Zah-lenwert einzusetzen. Für 140 °F ergibt sich bei-spielsweise:
{ϑC} = 0,5556 · (140 − 32) = 60
Weitere Temperaturumrechnungsformeln sind in Tabelle 1.5 zusammengestellt.
Tabelle 1 .5 Temperaturumrechnungsformeln häufig verwen-dete Einheiten wie Grad Celsius (°C), Grad Fahrenheit (°F), Kelvin (K), Grad Rankine (°Ra) und Grad Réaumur (°Re)
Umrechnungsformeln
{ } = 0,5556 · ({ } – 32){ } = (1,80 · { }) + 32
C F
F C
{ } = 0,5556 · ({ } – 491,67){ } = (1,80 · { }) + 491,67
C Ra
Ra C
{ } = { } – 273,15{ } = { } + 273,15
C
C
TT
{ } = 1,25 · { }{ } = 0,80 · { }
C Re
Re C
Grad Celsius Grad Farenheit
Grad Celsius Grad Réaumur
Grad Celsius Kelvin
Grad Celsius Grad Rankine
Neben diesen wichtigsten Temperaturskalen gibt es noch eine Reihe veralteter Skalen, wie die nach Delisle, Newton oder RØmer.
1 .3 Zustandsgrößen und Prozessgrößen
Eine Zustandsgröße (Zustandsvariable) ist eine physikalische Größe oder ein Parameter in einer Zustands gleichung, die nur vom aktuellen Zustand eines betrachteten Systems abhängt. Der Weg, auf dem dieser Zustand erreicht wurde, ist daher nicht von Interesse. Eine Zustandsgröße beschreibt nur eine Eigenschaft des Systems in diesem Zustand.
Temperatur, Druck, Masse, Dichte, Energie und En-tropie sind Beispiele von Z ustandsgrößen.
In der Thermodynamik wird ein System eindeu-tig beschrieben, beispielsweise durch Angabe der Zustandsgrößen Druck p, Temperatur T, Volumen V, Stoffmenge n bzw. Masse m, Enthalpie H und Entro-pie S. Diese Zustandsgrößen bleiben konstant, wenn sich das System im thermodynamischen Gleichge-wicht befindet.
Physikalische Größen, die den Zustand eines thermodynamischen Systems beschreiben, werden thermodynamische Zustandsgrößen genannt. Es wird unterschieden:
Thermische Zustandsgrößen: Temperatur T, Volumen V und Druck p.
Kalorische Zustandsgrößen: Innere Energie U, Enthalpie H, Entropie S und weitere.
Spezifische Zustandsgrößen: Physikalische Größen, die in der Regel auf die Masse eines Stoffes oder Körpers oder auf Raumdimensionen eines Sys-tems (Volumen, Flächeninhalt, Länge) bezogen sind. Nach DIN-Norm ist der Begriff spezifisch jedoch nur für den Massenbezug reserviert. Spezifische Größen werden mit Kleinbuchstaben bezeichnet (Ausnah-men: Masse m und Stoffmenge n). Beispiel: Spezifi-sches Volumen v = V /m.
Molare Zustandsgrößen: Auf die Stoffmenge n (Substanzmenge, Molmenge) bezogene Zustands-größen, auch stoffmengenbezogene Zustandsgrößen genannt. Sie werden durch den Index m gekenn-zeichnet. Beispiel: Molares Volumen Vm = V / n.
Extensive Zustandsgrößen: Physikalische Grö-ßen, die zur Teilchenzahl proportional sind. Der Wert einer solchen Zustandsgröße ändert sich mit der Größe des betrachteten Systems. Beispiele sind Masse m, Stoffmenge n, Volumen V, Enthalpie H und Entropie S. Das Pendant der extensiven Größe ist die intensive Größe.
Intensive Zustandsgrößen: Physikalische Grö-ßen, die sich bei unterschiedlicher Größe des be-trachteten Systems nicht ändern. Es werden sys-tem eigene intensive Größen wie beispielsweise Temperatur T und Druck p und stoffeigene inten-sive Größen wie alle spezifischen und molaren Grö-ßen unterschieden.
Es ist natürlich auch möglich, extensive in inten-sive Größen umzuwandeln, indem diese auf eine

1 Physikalische Größen und Einheitensysteme18
bestimmte Masse (spezifische Größe) oder auf eine bestimmte Stoffmenge (molare Größe) bezogen wer-den. Das Volumen ist daher eine extensive Größe, während das molare Volumen im Unterschied hier-zu eine intensive Größe darstellt.
Im Unterschied zu Zustandsgrößen beschreiben Prozessgrößen einen Prozessschritt zwischen zwei Zuständen. Sie stellen keine Eigenschaften des Sys-tems dar, sondern beschreiben einen Austauschpro-zess zwischen zwei Systemen oder zwischen einem System mit seiner Umgebung. Prozessgrößen sind wegabhängig, also abhängig davon, wie der Prozess geführt wird.
1 .4 ZustandsfunktionenZustandsgleichungen stellen einen funktionalen Zu-sammenhang zwischen thermodynamischen Zu-standsgrößen her, mit deren Hilfe sich der Zustand eines thermodynamischen Systems beschreiben lässt. Eine der Zustandsgrößen wird als Zustands funk-tion gewählt und die anderen von ihr abhängigen Zustandsgrößen als Zustandsvariablen. Mit Zu-standsgleichungen lassen sich Eigenschaften von Gasen, Flüssigkeiten, Fluidgemischen und Feststof-fen beschreiben.
Die bekanntesten Zustandsgleichungen dienen der Beschreibung von Gasen und Flüssigkeiten. Die wichtigste und zugleich auch einfachste Zustands-gleichung dieser Art ist die allgemeine Gasglei-chung.
p · V = n · R · T (1-3)
p Druck, V Volumen, n Stoffmenge, T Temperatur, R allgemeine Gaskonstante
Es konnte experimentell gezeigt werden, dass diese Gleichung (1-3) auf viele Gase näherungsweise bei geringen Drücken und hohen Temperaturen an-wendbar ist. Bei kleinen spezifischen Volumina und hohen Drücken treten zu große Abweichungen von der durch die thermische Zustandsgleichung des idealen Gases gegebene Gesetzmäßigkeit auf. Diese können dann nicht mehr vernachlässigt werden.
Eine Möglichkeit zur Berücksichtigung der Ab-weichungen besteht darin, die thermische Zustands-gleichung des idealen Gases durch einen Realgas-faktor Z zu korrigieren:
p · V = Z · n · R · T (1-4)
p Druck, V Volumen, Z Realgasfaktor, n Stoffmenge, T Tempera-tur, R allgemeine Gaskonstante
Für ideale Gase ist Z = 1, für reale Gase werden Vi-rialkoeffizienten (Kräfte zwischen den Molekülen) angehängt, die experimentell, in manchen Fällen auch rechnerisch, zu ermitteln sind:
Z p Vn R T
B TV
C TV
D TVm
= ⋅⋅ ⋅
= + + +1 2
( ) ( ) ( )
m m3
(1-5)
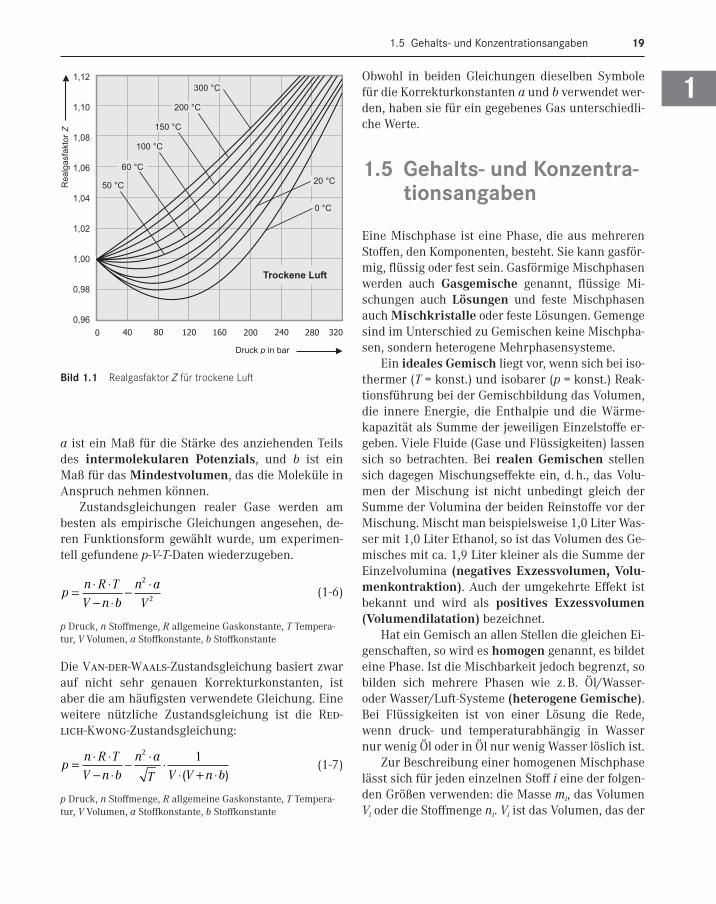
Der Realgasfaktor ist vom physikalischen Zustand abhängig. B(T), C(T), D(T) usw. sind die Virialkoeffi-zienten. In Bild 1.1 ist die Abhängigkeit des Realgas-faktors Z für trockene Luft vom Druck p und der Temperatur T gezeigt.
Der holländische Physiker van der Waals ent-wickelte eine historisch interessante Gleichung, welche die Kräfte zwischen den Molekülen als Ursa-che für die Abweichung berücksichtigte. Die Zu-standsgleichung enthält außer den bekannten Grö-ßen zwei Parameter a und b, die man experimentell für ein gegebenes Gas bestimmen muss. Parameter
Exkurs 1 .2 Beispiele von wegunabhängigen Zustandsgrößen
Das Volumen einer definierten Gasmenge Luft wirddurch Änderung des Druckes und der Temperaturvon einem bestimmten Anfangswert zu einembestimmten Endwert gebracht. Experimentelllässt sich feststellen, dass die Reihenfolge, in derdie Druck- und Temperaturänderung vorgenommenwurde, keinen Einfluss auf die Volumenänderung
hat. Die Änderung des Volumens ist immer gleichgroß und unabhängig vom Weg.
Ein Bergsteiger hat auf einem Berggipfel einebestimmte, von der Höhe des Berges abhängigepotenzielle Energie. Es ist gleichgültig, auf welchemWeg der Bergsteiger den Gipfel erreicht hat. DieArbeit hingegen, die der Bersteiger aufzuwendenhat, um auf den Gipfel zu gelangen (Energie), hängtnatürlich vom zurückgelegten Weg ab.
VV
V
A
E
∆
1.5 Gehalts- und Konzentra tionsangaben 19
1
a ist ein Maß für die Stärke des anziehenden Teils des intermolekularen Potenzials, und b ist ein Maß für das Mindestvolumen, das die Moleküle in Anspruch nehmen können.
Zustandsgleichungen realer Gase werden am besten als empirische Gleichungen angesehen, de-ren Funktionsform gewählt wurde, um experimen-tell gefundene p-V-T-Daten wiederzugeben.
p n R TV n b
n aV
= ⋅ ⋅− ⋅
− ⋅2
2 (1-6)
p Druck, n Stoffmenge, R allgemeine Gaskonstante, T Tempera-tur, V Volumen, a Stoffkonstante, b Stoffkonstante
Die Van-der-Waals-Zustandsgleichung basiert zwar auf nicht sehr genauen Korrekturkonstanten, ist aber die am häufigsten verwendete Gleichung. Eine weitere nützliche Zustandsgleichung ist die Red-lich-Kwong-Zustandsgleichung:
p n R TV n b
n aT V V n b
= ⋅ ⋅− ⋅
− ⋅ ⋅⋅ + ⋅
2 1( )
(1-7)
p Druck, n Stoffmenge, R allgemeine Gaskonstante, T Tempera-tur, V Volumen, a Stoffkonstante, b Stoffkonstante
Obwohl in beiden Gleichungen dieselben Sym bole für die Korrekturkonstanten a und b verwendet wer-den, haben sie für ein gegebenes Gas unterschiedli-che Werte.
1 .5 Gehalts und Konzentrationsangaben
Eine Mischphase ist eine Phase, die aus mehreren Stoffen, den Komponenten, besteht. Sie kann gasför-mig, flüssig oder fest sein. Gasförmige Mischphasen werden auch Gasgemische genannt, flüssige Mi-schungen auch Lösungen und feste Mischphasen auch Mischkristalle oder feste Lösungen. Gemenge sind im Unterschied zu Gemischen keine Mischpha-sen, sondern heterogene Mehrphasensysteme.
Ein ideales Gemisch liegt vor, wenn sich bei iso-thermer (T = konst.) und isobarer (p = konst.) Reak-tionsführung bei der Gemischbildung das Volumen, die innere Energie, die Enthalpie und die Wärme-kapazität als Summe der jeweiligen Einzelstoffe er-geben. Viele Fluide (Gase und Flüssigkeiten) lassen sich so betrachten. Bei realen Gemischen stellen sich dagegen Mischungseffekte ein, d. h., das Volu-men der Mischung ist nicht unbedingt gleich der Summe der Volumina der beiden Reinstoffe vor der Mischung. Mischt man beispielsweise 1,0 Liter Was-ser mit 1,0 Liter Ethanol, so ist das Volumen des Ge-misches mit ca. 1,9 Liter kleiner als die Summe der Einzelvolumina (negatives Exzessvolumen, Volu-menkontraktion). Auch der umgekehrte Effekt ist bekannt und wird als positives Exzessvolumen (Volumendilatation) bezeichnet.
Hat ein Gemisch an allen Stellen die gleichen Ei-genschaften, so wird es homogen genannt, es bildet eine Phase. Ist die Mischbarkeit jedoch begrenzt, so bilden sich mehrere Phasen wie z. B. Öl/Wasser- oder Wasser/Luft-Systeme (heterogene Gemische). Bei Flüssigkeiten ist von einer Lösung die Rede, wenn druck- und temperaturabhängig in Wasser nur wenig Öl oder in Öl nur wenig Wasser löslich ist.
Zur Beschreibung einer homogenen Mischphase lässt sich für jeden einzelnen Stoff i eine der folgen-den Größen verwenden: die Masse mi, das Volumen Vi oder die Stoffmenge ni. Vi ist das Volumen, das der
1,12
1,10
1,08
1,06
1,04
1,02
1,00
0,98
0,9640 80 120 160 200 240 280 3200
Druck in barp
Rea
lgas
fakt
orZ
100 °C
60 °C
50 °C
150 °C
200 °C
300 °C
20 °C
0 °C
Trockene Luft
Bild 1 .1 Realgasfaktor Z für trockene Luft

1 Physikalische Größen und Einheitensysteme20
Stoff i allein bei gegebener Temperatur und Druck und vorliegendem Aggregatzustand einnehmen würde.
Im Internationalen Einheitensystem ist die Stoff-menge eine Basisgröße, deren Basiseinheit das Mol ist (vgl. Abschn. 1.1). Die Stoffmenge von 1 mol eines Stoffes besteht aus ebenso vielen Einzelteil-chen (Atome, Moleküle oder Ionen), wie Atome in 12 · 10–3 kg des Kohlenstoffnuklids 12C enthalten sind. Bei der Verwendung der Basisgröße Stoffmen-ge müssen daher die einzelnen Teilchen des Sys-tems genau spezifiziert sein. Es darf nicht heißen 1 mol Sauerstoff, sondern es muss zum Ausdruck gebracht werden, ob es sich um Sauerstoffatome (O), Sauerstoffmoleküle (O2) oder Ozon (O3) handelt. Es muss daher geschrieben werden: 1 mol O =̂ 16 g, 1 mol O2 =̂ 32 g bzw. 1 mol O3 =̂ 48 g.
Eine Mischphase wird durch die Materiemenge (Masse, Stoffmenge oder Molvolumen) jeder Kom-ponente beschrieben. Von Interesse ist im Allgemei-nen nur der intensive, d. h. von der Größe des Sys-tems unabhängige Zustand. Bei den Einzelmengen wird auf eine extensive Zustandsgröße bezogen, d. h. eine zur Materiemenge proportionalen Größe, wie Masse, Stoffmenge oder Molvolumen (vgl. Ab-schn. 1.3). Die Einzelmengen bestimmen die Zusam-mensetzung der Mischphase. Zusätzlich werden zwei voneinander unabhängige intensive Zustand-größen benötigt, z. B. Druck und Temperatur.
Um die Menge des gelösten Stoffes im Lösungs-mittel zu charakterisieren, gibt es verschiedene Möglichkeiten: Angabe des Massenanteils w Angabe des Stoffmengenanteils x Angabe des Volumenanteils ϕ Angabe der Massenkonzentration ß Angabe der Stoffmengenkonzentration c Angabe der Volumenkonzentration σ
Soll die Anteilsgröße angegeben werden, so werden stets Quotienten gleicher Größen wie Masse, Volu-men oder Stoffmenge verwendet. Bei der Angabe ei-ner Konzentration wird die Menge des gelösten Stof-fes als Masse, Volumen oder Stoffmenge auf das Volumen der Flüssigkeit bezogen. Konzentrationsan-gaben gelten immer nur für eine gegebene Tempera-tur.
1 .5 .1 Massenanteil
Der Massenanteil (Formelzeichen w) einer Kompo-nente an einem Stoffgemisch ist die anteilige Masse dieser Komponente an der Gesamtmasse des Stoff-gemisches. Berechnet wird der Massenanteil der Komponente i als Quotient aus der Masse der Kom-ponente mj und der Summe aller Massen des Stoff-gemisches aus k Komponenten.
wm
mi
i
jj
k=
=∑
1
(1-8)
Der Massenanteil einer Komponente des Gemisches liegt zwischen 0 und 1:
0 ≤ wi ≤ 1 (1-9)
Die Massenanteile aller k Bestandteile eines Stoff-gemisches addieren sich zu 1.
wii
k
==∑ 1
1
(1-10)
Multipliziert man den Massenanteil mit 100 %, so kann man ihn auch als prozentuale Größe angeben, also Gewichtsprozent (Gew.-%) bzw. Massenprozent. Dies sollte nach DIN 1 310 jedoch vermieden werden.
1 .5 .2 Stoffmengenanteil
Der Stoffmengenanteil (Formelzeichen x) einer Komponente an einem Stoffgemisch ist die relative Anzahl der Teilchen (Atome, Moleküle, Ionen) die-ser Komponente an der Gesamtteilchenzahl des Stoffgemisches. Im Unterschied dazu beschreibt das Stoffmengenverhältnis die relative Anzahl an Teil-chen der Komponenten zueinander.
Berechnet wird der Stoffmengenanteil der Kom-ponente i als Quotient aus der Stoffmenge der Kom-ponente ni und der Summe aller Stoffmengen des Stoffgemisches aus k Komponenten.
xn
ni
i
jj
k=
=∑
1
(1-11)

1.5 Gehalts- und Konzentra tionsangaben 21
1Der Stoffmengenanteil einer Komponente des Gemi-sches liegt zwischen 0 und 1:
0 ≤ xi ≤ 1 (1-12)
Die Stoffmengenanteile aller k Bestandteile eines Stoffgemisches addieren sich zu 1.
xii
k
==∑ 1
1
(1-13)
Multipliziert man den Stoffmengenanteil mit 100 %, so kann man ihn als prozentuale Größe angeben, also Stoffmengenprozent oder Molprozent (Mol-%).
1 .5 .3 Volumenanteil
Der Volumenanteil (Formelzeichen ϕ) einer Kompo-nente an einem Stoffgemisch ist das anteilige Volu-men dieser Komponente an der Summe der Volu-mina aller Komponenten des Stoffgemisches. Be-rechnet wird der Volumenanteil der Komponente i als Quotient aus dem Volumen der Komponente Vi und der Summe aller Volumina des Stoffgemisches aus k Komponenten.
ϕii
jj
k
V
V=
=∑
1
(1-14)
Der Volumenanteil einer Komponente des Gemi-sches liegt zwischen 0 und 1:
0 ≤ ϕi ≤ 1 (1-15)
Die Volumenanteile aller k Bestandteile eines Stoff-gemisches addieren sich zu 1.
ϕii
k
==∑ 1
1
(1-16)
Multipliziert man den Volumenanteil mit 100 %, so kann man ihn auch als prozentuale Größe angeben, also Volumenprozent (Vol.-%). Bei der Zusammen-setzung von Gasen oder bei der Festlegung von Ex-plosionsgrenzen ist diese Angabe üblich.
1 .5 .4 Massenkonzentration
Die Massenkonzentration (Formelzeichen ß) ist eine Gehaltsangabe, bei der die Masse mi eines Stoffes i auf das Volumen V eines Stoffgemisches oder einer Lösung bezogen angegeben wird.
iim
ßV
= (1-17)
Die SI-Einheit lautet [ß] = kg/m3. Bei Arbeiten im Labor wird meistens die Einheit g/l verwendet.
1 .5 .5 Stoffmengenkonzentration
Die Stoffmengenkonzentration (Formelzeichen c) ist eine Gehaltsangabe, bei der die Stoffmenge ni eines Stoffes i, bezogen auf das Volumen V eines Stoffge-misches oder einer Lösung, angegeben wird.
cnVi
i= (1-18)
Die SI-Einheit lautet [c] = mol/m3. Bei Arbeiten im Laboratorium wird meistens die Einheit mol/l ver-wendet.
1 .5 .6 Volumenkonzentration
Die Volumenkonzentration (Formelzeichen s) ist eine Gehaltsangabe für Mischphasen, insbesondere von Lösungen, bei der das Volumen Vi eines Stoffes i, bezogen auf das Gesamtvolumen V eines Stoffge-misches oder einer Lösung, angegeben wird.
s iiV
V= (1-19)
Die SI-Einheit lautet [s] = m3/m3. Bei Arbeiten im Laboratorium wird meistens die Einheit l/l verwen-det. Die Volumenkonzentration unterscheidet sich von der Gehaltsangabe Volumenanteil, da bei der Volumenkonzentration eine mögliche Volumenkon-traktion mit berücksichtigt ist.

1 Physikalische Größen und Einheitensysteme22
1 .5 .7 Molalität
Die Molalität (Formelzeichen b) ist der Quotient aus der Stoffmenge ni des gelösten Stoffes i und der Masse mLsgm des Lösungsmittels.
bn
mi=
Lsgm.
(1-20)
Die SI-Einheit lautet [b] = mol/kg.
1 .5 .8 Aktivität
Aufgrund interionischer Wechselwirkungen in kon-zentrierten Lösungen ist die wirksame Konzentra-tion oder Aktivität der Lösung kleiner als die tat-sächliche Konzentration. Die Abweichungen sind umso größer, je höher die Konzentrationen der Stof-fe sind.
a f cc
= ⋅°
(1-21)
a Aktivität, f Aktivitätskoeffizient, c Stoffmengenkonzentration, c° Standard-Stoffmengenkonzentration (1 mol/l)
1 .6 Umrechnungen und Mischungsrechnung
Im Laboratorium werden mitunter Lösungen herge-stellt, indem zwei Lösungen unterschiedlichen Mas-senanteils zu einer neuen Lösung vermischt wer-den. Manchmal wird auch eine Lösung hergestellt, indem eine höherkonzentrierte Lösung mit Lösungs-mittel (z. B. Wasser) verdünnt wird. Die Mengen bzw. die Massenanteile werden mit der Mischungs-gleichung berechnet:
m1 w1 + m2 w2 + . . . = (m1 + m2 + . . .) wMischung (1-22)
m1 Masse der Lösung 1, m2 Masse der Lösung 2, w1 Massen anteil der Lösung 1, w2 Massenanteil der Lösung 2, wMischung Massen anteil der entstandenen Mischung
Benutzt man zum Verdünnen einer Lösung reines Lösungsmittel, so beträgt der Massenanteil des rei-nen Lösungsmittels w2 = 0.
Beispiel 1 .1: Es sollen 2,5 kg Schwefelsäure mit w (H2SO4) = 15 % aus einer konzentrierten Schwefelsäure (w = 95 %) mit Wasser hergestellt werden. Welche Masse an konz. Schwefelsäure und welche Masse an Wasser sind abzuwiegen?1. Zunächst wird die Mischungsgleichung aufgestellt und
werden die Daten der Aufgabe definiert:m1 · w1 + m2 · w2 = (m1 + m2) · wMischung
m1 = Schwefelsäure, w(H2SO4) = 95 %m2 = Wasser
Beide Massen ergeben zusammen nach dem Mischen 2 500 g Lösung (m1 + m2 = 2 500 g)
m2 = 2 500 g – m1
w1 = 95 % w2 = 0 % wMischung = 15 %2. Die Daten werden in die Mischungsgleichung einge-
setzt: m1 · 95 % + (2 500 g − m1) · 0 % = 2 500 g · 15 % Für m1 ergibt sich nach Umstellung der Gleichung: m1 = 395 g Schwefelsäure mit w = 95 %.3. Die Menge an Wasser kann aus der Gesamtmenge her-
zustellender Schwefelsäure berechnet werden. Masse der 95 %-Schwefelsäure und Masse Wasser ergeben zusammen 2 500 g.
m2 = 2 500 g – m1 = 2 500 g − 395 g = 2 105 g Wasser4. Es werden 2 105 g Wasser vorgelegt und langsam darin
395 g Schwefelsäure mit w = 95 % eingerührt. Man er-hält 2,5 kg Schwefelsäure mit einer Konzentration von 15 %.
In der Praxis muss schnell gerechnet werden. Formt man die Mischungsgleichung (1-22) für zwei Kom-ponenten um, dann erhält man:
m1 w1 + m2 w2 = (m1 + m2) wMischung
m1 w1 − m1 wMischung = m2 wMischung – m2 w2
m1 (w1 − wMischung) = m2 (wMischung. − w2)
mm
w ww w
1
2
2
1
=−
−Mischung
Mischung
(1-23)

1.6 Umrechnungen und Mischungsrechnung 23
1wMischung
w2 . . . m2
w1 . . . m1w wMischung 2–
w w1 Mischung–
Die Differenzen (wMischung – w2) zu (w1 – wMischung) ver-halten sich wie die Massen der beiden Komponenten. Dieses Verhältnis lässt sich in Form eines Kreuzes, dem Mischungskreuz (Andreaskreuz), darstellen und damit das Massenverhältnis berechnen:
Masse m
Stoffmenge nVolumen V
Teilchen-anzahl N
M =
mAtom =
Vm =
= NA =m
m
V
m Nn
N
n
V nρ
Bild 1 .2 Umrechnungsformeln von Masse m in Teilchen-anzahl N, Volumen V und Stoffmenge n mit der Dichte ρ und der molaren Masse M
Zur schnellen Umrechnung von Stoff- und Gehalts-größen können die Gleichungen in Bild 1.2 genutzt werden.
Beispiel 1 .2: Welche Masse an Natriumchloridlösung mit einem Massenanteil von w (NaCl) = 18 % muss zu 500 g einer Lösung mit einem Massenanteil von w (NaCl) = 8 % gegeben werden, damit eine Mischung mit w (NaCl) = 10 % entsteht?1. Zunächst werden die Daten der Aufgabe definiert: w1 = 18 %, w2 = 8 % und wMischung = 10 % und entsprechend dem Mischungskreuz eingesetzt.2. Aus der Differenzbildung ergibt sich: wMischung – w2 = 10 % − 8 % = 2 % ⇒ m1 = 2 w1 − wMischung = 18 % − 10 % = 8 % ⇒ m2 = 8 Die beiden Kochsalzlösungen sind daher im Verhältnis
2 zu 8 bzw. 1 zu 4 miteinander zu mischen.
3. Da 500 g einer 8 %-NaCl-Lösung mit einer 18 %-NaCl-Lösung gemischt werden sollen, erfolgt der nächste Rechenschritt als Dreisatz:
4 Teile entsprechen 500 g NaCl (w = 8 %) 1 Teil entspricht x g NaCl (w = 18 %) x = 1/4 · 500 g = 125 g NaCl (w = 18 %)4. Es werden 125 g der 18 %-Kochsalzlösung benötigt, um
mit 500 g der 8 %-Kochsalzlösung eine Mischkonzentra-tion von 10 %-NaCl zu erhalten.
Massenanteil in Stoffmengenanteil
wx M
x Mi
i i
i ii
k=⋅
⋅=∑
1
(1-24)
xw
M wM
ii
ii
ii
k=⋅
=∑
1
(1-25)
wi Massenanteil der Komponente i in %, xi Stoffmengenanteil der Komponente i in %, Mi molare Masse der Komponente i in g/mol
Massenanteil in Stoffmengenkonzentration
wc M
ii i=⋅⋅r 10
(1-26)
cwMi
i
i
=⋅ ⋅r 10
(1-27)
wi Massenanteil der Komponente i in %, ci Stoffmengenkonzent-ration der Komponente i in mol/l, r Dichte des Lösung in g/ml, Mi molare Masse der Komponente i in g/mol
Massenanteil in Volumenanteil
wii i=⋅σ ρρ
(1-28)
σρ
ρii
i
w=
⋅ (1-29)
wi Massenanteil der Komponente i in %, si Volumenanteil der Komponente i in %, ri Dichte des reinen Stoffes in g/ml, r Dichte der Lösung in g/ml

1 Physikalische Größen und Einheitensysteme24
Massenanteil in Massenkonzentration
wii=
⋅ρ 10ß (1-30)
wßi i= ⋅ ⋅10 ρ (1-31)
wi Massenanteil der Komponente i in %, ßi Massenkonzentration in g/l, r Dichte des Lösung in g/ml
Massen-konzentration ß
Stoffmengen-konzentration c
Volumen-konzentration c =
=
·
ß
M
ß c M= ·
Bild 1 .3 Umrechnungsformeln von der Massenkonzentration ß in die Stoffmengenkonzentration c und in die Volumenkonzen-tration s mit der Dichte r und der molaren Masse M

2 Statistische Grundlagen
2 .1 FehlerartenUnter einem Fehler verstand man lange Zeit die Ab-weichung von einem festgelegten Zustand oder Ver-fahren in einem bezüglich seiner Funktionen deter-minierten System. Mittlerweile wurde jedoch diese Definition verändert. Das Deutsche Institut für Nor-mung (DIN) definiert Fehler als einen „Merkmals-wert, der die vorgegebenen Forderungen nicht er-füllt“ und als „Nichterfüllung einer Anforderung“. Die Anforderung wird dabei definiert als „Erforder-nis oder Erwartung, die festgelegt, üblicherweise vorausgesetzt oder verpflichtend ist“.
Grundsätzlich ist jede Messung einer physika-lischen Größe mit Fehlern behaftet, wobei man nach DIN 1 319 nicht mehr von Fehlern, sondern von Abwei chungen der Messwerte und von Un sicher-heiten bei den Messergebnissen sprechen sollte. Die Empfehlung unterstreicht, dass mit dem Wort Fehler im Zusammenhang mit Messergebnissen nicht ein falsches Ergebnis gemeint ist, sondern die Streuung der Messwerte um den „wahren“ Wert der Messgröße, der in der Regel unbekannt ist.
In den üblichen Konventionen wird ein Mess-ergebnis für eine Größe x wie folgt angegeben:
x = xw ± Δx (2-1)
Dabei ist xw der „wahrscheinlichste“ oder „beste“ Schätzwert für das Messergebnis und Δx die Mess-unsicherheit. Die Angabe bedeutet, dass das Mess-ergebnis mit einer gewissen Sicherheit im Intervall xw – Δx ≤ x ≤ xw + Δx liegt. Δx wird bei dieser Schreib-weise auch als absolute Messunsicherheit be-zeichnet.
Es sind drei Arten verschiedener Abweichungen von Messwerten (Fehlern) zu unterscheiden, die in
den folgenden Abschnitten näher beschrieben wer-den: grobe Abweichung von Messwerten, systematische Abweichung von Messwerten, zufällige Abweichung von Messwerten.
2 .1 .1 Grobe Abweichung von Messwerten
Grobe Abweichungen sind vermeidbare Abweichun-gen und sollten durch sorgfältiges Arbeiten, kriti-sches Überprüfen und Kontrollieren der Ergebnisse vermieden werden. Mögliche Ursachen sind: Unachtsamkeit ungeeignetes Messverfahren falsche Bedienung der Messgeräte Messbedingungen sind ungeeignet Fehler bei der Protokollierung und Auswertung
der Messwerte
Treffen eine oder mehrere Ursachen zu, sind die Messungen oder Auswertungen falsch und müssen wiederholt werden. Grobe Abweichungen von Mess-werten werden durch keine Fehlertheorie erfasst.
2 .1 .2 Systematische Abweichung von Messwerten
Bei einer systematischen Abweichung von Messwer-ten wird das Messergebnis unter gleichen Mess-bedingungen stets in gleichem Sinne beeinflusst. Bei der Wiederholung der Messung unter gleichen Bedingungen sind die Abweichungen gleich und können somit weder erkannt noch verhindert wer-den. Beispiele für systematische Abweichungsquel-len sind:

2 Statistische Grundlagen26
Verwendung falscher Messgeräte Alterung der Messgeräte ungültige physikalische Gesetze Einfluss äußerer Parameter (z. B. Druck, Tempe-
ratur, Störfelder)
Systematische Abweichungen lassen sich ganz oder teilweise ausschalten durch Veränderung der Mess-bedingungen oder Kalibrierung der Messgeräte. Eine Berücksichtigung der systematischen Fehler in der Fehlerrechnung oder der zahlenmäßigen An-gabe im Ergebnis (vgl. Gl. 2-1) findet jedoch nicht statt.
2 .1 .3 Zufällige Abweichung von Messwerten
Selbst bei vollständiger Eliminierung aller systema-tischen Abweichungen erhält man bei wiederholter Messung der gleichen physikalischen Größe selten übereinstimmende Messergebnisse. Die Messwerte werden immer um den „wahren“ Wert streuen. Diese Abweichung wird als zufällig bezeichnet und lässt sich mit den Gesetzen der Statistik beschrei-ben. Einige Beispiele für mögliche Ursachen sind im Folgenden gezeigt. Messgröße hat einen Zufallscharakter (z. B. radio-
aktiver Zerfall) zufällige und nicht vorhersehbare Einflüsse Ableseabweichungen (Parallaxenfehler) Schätzungen und Interpolationen auf Mess-Ska-
len unterschiedliche Reaktionszeiten des Experimen-
tators (Stoppuhr)
Zufällige Abweichungen von Messwerten sind nicht vermeidbar. Sie lassen sich jedoch durch Wiederho-lungsmessungen verringern.
2 .2 Darstellung von Messreihen
Bei einer Befragung von 150 Hörern einer Verfah-renstechnik-Vorlesung wurden folgende Daten er-mittelt:(1) Familienstand(2) Studienrichtung(3) Interesse an der Vorlesung(4) Anzahl der Geschwister(5) Anzahl der Hochschulsemester(6) Körpergröße(7) Entfernung von der Uni zur Wohnung
Die Gesamtheit aller Hörer der Vorlesung nennt man Beobachtungsmenge, den einzelnen Hörer Beobachtungseinheit. Die in dem Beispiel erfrag-ten Eigenschaften oder Sachverhalte heißen Beob-achtungsmerkmale. Bei der Beobachtung werden folgende Merkmalstypen unterschieden: qualitative Merkmale (vgl. 1 und 2) Rangmerkmale (vgl. 3) quantitativ diskrete Merkmale (vgl. 4 und 5) quantitativ stetige Merkmale (vgl. 6 und 7)
Im Folgenden werden wir uns hauptsächlich mit quantitativen Merkmalen befassen, da in den Natur-wissenschaften und der Technik Daten in erster Linie durch Messen oder Zählen gewonnen werden. Es ist daher zunächst davon auszugehen, dass zu jeder Beobachtungseinheit ein quantitatives Merk-mal gehört oder dass die verschiedenen Merkmale getrennt voneinander untersucht werden sollen.
Beispiel 2 .1: Häufigkeitsverteilungen einer Stichprobe30 Studenten in einem Vorkurs Mathematik erhielten in der Reihenfolge ihrer Matrikelnummer folgende Noten für die Abschlussklausur: 3, 2, 5, 3, 4, 3, 5, 4, 3, 2, 3, 4, 2, 3, 3, 2, 3, 4, 5, 1, 3, 2, 1, 4, 4, 3, 2, 1, 4, 3.
In Tabelle 2.1 sind der Zahlenwerte dieser zunächst völ-lig ungeordneten Liste als Strichliste oder Häufigkeitstabel-le dargestellt. Die Anzahl der einzelnen Striche ergibt die absoluten Häufigkeiten der jeweiligen Noten.

2.2 Darstellung von Messreihen 27
2Tabelle 2 .1 Strichliste einer Häufigkeitstabelle
Note absoluteHäufigkeit
Strichliste relativeHäufigkeit
3
6
11
7
3
= 30
–
n
1
2
3
4
5
6
0,100
0,200
0,367
0,233
0,100
–
= 1,000�
Anteilin %
10,0
20,0
36,7
23,3
10,0
–
= 100�
Eine grafische Darstellung kann die Übersichtlichkeit noch weiter erhöhen. Es kommen Stabdiagramme, Häufigkeits-polygone oder Histogramme infrage. Ein Histogramm be-steht aus Rechtecken, deren Grundseiten die verschiede-nen Noten als Mittelpunkt besitzen. Die Größen der Balken bilden die absoluten Häufigkeiten der entsprechenden No-ten ab, da bei der Bewertung einer Arbeit die Gesamtnote im Allgemeinen aus mehreren Einzelbewertungen ermittelt wird. Liegt dieser Durchschnitt zum Beispiel zwischen 1,5 und 2,5, so erhält der Student die Note 2. Es findet somit bereits hier eine Klasseneinteilung statt, d. h., mehrere Werte werden zu einer Klasse zusammengefasst.
1 2 3 4 65
8
4
12
absolu
teH
äufigkeit
Note
Bild 2 .1 Histogramm-Darstellung
Dividiert man die absoluten Häufigkeiten durch die Anzahl der Studenten (Messwerte), so erhält man die relativen Häufigkeiten (vgl. 4. Spalte in Tabel-le 2.1), deren Gesamtsumme den Wert 1,000 ergibt. Die Multiplikation der relativen Häufigkeit mit 100 ergibt die prozentualen Anteile (vgl. 5. Spalte in Ta-belle 2.1). Die grafischen Darstellungen der absolu-ten Häufigkeiten haben den Nachteil, dass die ent-
sprechenden Höhen der Balken mit der Anzahl der Beobachtungswerte steigen. Im Unterschied zu den absoluten Häufigkeiten können die relativen Häufig-keiten nie größer als eins werden und bieten sich somit bei immer demselben Maßstab zur grafischen Auswertung an.
Gegeben seien n Beobachtungswerte (Zahlen) x1, x2, . . . , xn, dann wird das n-Tupel x = x1, x2, . . . , xn eine Stichprobe vom Umfang n genannt. Die einzel-nen Zahlen xi nennt man Stichprobenwerte. Die in der Stichprobe vorkommenden verschiedenen Werte heißen Merkmalswerte; sie werden mit x*1, x*2, . . . x*N bezeichnet. Die Anzahl des Auftretens von x*k in einer Stichprobe wird absolute Häufigkeit von x*k genannt und wird mit hk = h(x*k) bezeichnet. Den Quotienten rk = hk / n nennt man relative Häufig-keit von x*k in der Stichprobe für k = 1, 2, . . . N.
Für die absoluten und relativen Häufigkeiten gelten folgende Eigenschaften:
h nkk
N
==
∑1
0 1 11
≤ ≤ ==
∑r rk kk
N
f r alle k; für alle k; 0 1 11
≤ ≤ ==
∑r rk kk
N
f r alle k;
Die Summe der absoluten Häufigkeiten der Merk-malswerte, die nicht größer als x*i sind, werden abso lute Summenhäufigkeit des Merkmalswertes x*i genannt und mit Hi bezeichnet. Es gilt daher
H hi kk
i
==
∑1
(2-2)
Die absoluten Summenhäufigkeiten aus Beispiel 2.2 sind in der 3. Spalte der Tabelle 2.2 dargestellt. Mit den relativen Häufigkeiten ri erhält man als relative Summenhäufigkeit
R ri kk
i
==
∑1
(2-3)
den relativen Anteil der Merkmalswerte, die nicht größer als x*i sind. In Tabelle 2.2 sind die relativen Summenhäufigkeiten für das Beispiel 2.2 in der letzten Spalte berechnet. In Bild 2.2 (unten) sind über den Merkmalswerten die relativen Summen-
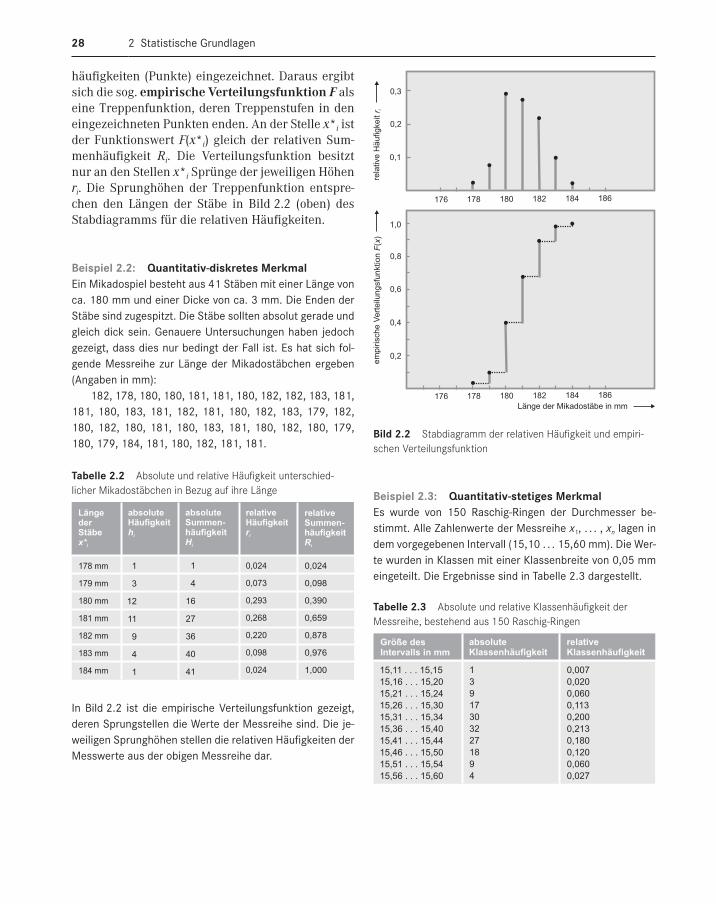
2 Statistische Grundlagen28
häufigkeiten (Punkte) eingezeichnet. Daraus ergibt sich die sog. empirische Verteilungsfunktion F als eine Treppenfunktion, deren Treppenstufen in den eingezeichneten Punkten enden. An der Stelle x*i ist der Funktionswert F(x*i) gleich der relativen Sum-menhäufigkeit Ri. Die Verteilungsfunktion besitzt nur an den Stellen x*i Sprünge der jeweiligen Höhen ri. Die Sprunghöhen der Treppenfunktion entspre-chen den Längen der Stäbe in Bild 2.2 (oben) des Stabdiagramms für die relativen Häufigkeiten.
Beispiel 2 .2: Quantitativdiskretes MerkmalEin Mikadospiel besteht aus 41 Stäben mit einer Länge von ca. 180 mm und einer Dicke von ca. 3 mm. Die Enden der Stäbe sind zugespitzt. Die Stäbe sollten absolut gerade und gleich dick sein. Genauere Untersuchungen haben jedoch gezeigt, dass dies nur bedingt der Fall ist. Es hat sich fol-gende Messreihe zur Länge der Mikadostäbchen ergeben (Angaben in mm):
182, 178, 180, 180, 181, 181, 180, 182, 182, 183, 181, 181, 180, 183, 181, 182, 181, 180, 182, 183, 179, 182, 180, 182, 180, 181, 180, 183, 181, 180, 182, 180, 179, 180, 179, 184, 181, 180, 182, 181, 181.
Tabelle 2 .2 Absolute und relative Häufigkeit unterschied-licher Mikadostäbchen in Bezug auf ihre Länge
LängederStäbex*i
absoluteSummen-häufigkeitHi
absoluteHäufigkeithi
relativeHäufigkeitri
1
4
16
27
36
40
41
1
3
12
11
9
4
1
178 mm
179 mm
180 mm
181 mm
182 mm
183 mm
184 mm
0,024
0,073
0,293
0,268
0,220
0,098
0,024
relativeSummen-häufigkeitRi
0,024
0,098
0,390
0,659
0,878
0,976
1,000
In Bild 2.2 ist die empirische Verteilungsfunktion gezeigt, deren Sprungstellen die Werte der Messreihe sind. Die je-weiligen Sprunghöhen stellen die relativen Häufigkeiten der Messwerte aus der obigen Messreihe dar.
176
176
178
178
180
180
182
182
184
184
186
186
0,1
0,2
0,2
0,3
0,4
0,6
0,8
1,0
em
piris
ch
eV
ert
eilu
ng
sfu
nktio
n(
)F
xre
lative
Hä
ufig
ke
itr i
Länge der Mikadostäbe in mm
Bild 2 .2 Stabdiagramm der relativen Häufigkeit und empiri-schen Verteilungsfunktion
Beispiel 2 .3: Quantitativstetiges MerkmalEs wurde von 150 Raschig-Ringen der Durchmesser be-stimmt. Alle Zahlenwerte der Messreihe x1, . . . , xn lagen in dem vorgegebenen Intervall (15,10 . . . 15,60 mm). Die Wer-te wurden in Klassen mit einer Klassenbreite von 0,05 mm eingeteilt. Die Ergebnisse sind in Tabelle 2.3 dargestellt.
Tabelle 2 .3 Absolute und relative Klassenhäufigkeit der Messreihe, bestehend aus 150 Raschig-Ringen
Größe desIntervalls in mm
absoluteKlassenhäufigkeit
relativeKlassenhäufigkeit
1
3
9
17
30
32
27
18
9
4
0,007
0,020
0,060
0,113
0,200
0,213
0,180
0,120
0,060
0,027
15,11 . . . 15,15
15,16 . . . 15,20
15,21 . . . 15,24
15,26 . . . 15,30
15,31 . . . 15,34
15,36 . . . 15,40
15,41 . . . 15,44
15,46 . . . 15,50
15,51 . . . 15,54
15,56 . . . 15,60

2.3 Erfassung der Messwert abweichung 29
2
In Bild 2.3 ist das dazugehörige Histogramm in der oberen Hälfte gezeigt. Bei der Darstellung der doppelten Klassen-breite (vgl. Bild 2.3, untere Hälfte) ist darauf zu achten, dass der Maßstab (Ordinate) entsprechend geändert wur-de. Die relativen Klassenhäufigkeiten sind dann: 0,027, 0,173, 0,413, 0,300 und 0,087.
15,0
15,0
15,2
15,2
15,4
15,4
15,6
15,6
15,8
15,8
0,10
0,20
0,20
0,40
0,30
0,15
0,10
0,05
0,25
0,50
absolu
teH
äufigkeit
()
hx
absolu
teH
äufigkeit
()
hx
Höhe der Raschig-Ringe in mm
Bild 2 .3 Histogramme der Durchmesserverteilung von Ra-schig-Ringen bei unterschiedlicher Klassenbreite
2 .3 Erfassung der Messwertabweichung
Wird eine Messgröße x nur einmal gemessen, so lässt sich aufgrund statistischer Überlegungen kei-ne Aussage über die Abweichung von Messwerten machen. In einem solchen Fall ist man auf die An-gabe der geschätzten größtmöglichen Ungenauig-keit angewiesen. Sie ergibt sich aus der Ablese-genauigkeit auf der verwendeten Skala, aus der Genauigkeitsklasse oder aus anderen Informatio-nen.
Beim Ablesen einer Skala vergleicht man die Lage eines Messpunktes oder eines Zeigers mit den
Teilstrichen der Skala. Handelt es sich um eine fein unterteilte Skala (Millimeterunterteilung eines Li-neals), so nimmt man den nächstgelegenen Teil-strich als Bestwert und schätzt die Unsicherheit mit ± (halbe Intervallbreite) ab. Bei gröber geteilten Skalen kann man die Lage des Zeigers zwischen zwei Teilstrichen interpolieren und als Dezimal-stelle angeben. Die Unsicherheit wird dann zu ± (1 Zehntelbruchteil) angenommen. Bei Digital-anzeigen beträgt die Unsicherheit ± 1 in der letzten Stelle der Anzeige. Bei Messungen, bei denen Unsi-cherheiten schwieriger zu schätzen sind, lässt sich durch Wiederholung der Messungen die Messunsi-cherheit bestimmen.
Häufig ist man an einer knappen Beschreibung einer Messreihe interessiert, beispielsweise durch Angabe einer typischen Eigenschaft der Häufig-keitsverteilung. Diese soll Auskunft darüber geben, wo sich die Mitte der beobachteten Messwerte be-findet und wie groß der Bereich ist, über den sich die Werte erstrecken. Die Statistik unterscheidet da-her zwischen der Lage und den Streuungsmaßzah-len.
Betrachtet man eine Messreihe x1, . . . , xn, so wird
xn
x xn
xn ii
n
= + + ==∑1 1
11
( . . . ) (2-4)
arithmetisches Mittel (oder empirischer Mittel-wert) der Messreihe genannt.
Werden die Zahlen der Messreihe x1, . . . , xn je-doch der Größe nach geordnet, so lautet die entstan-dene Messreihe:
x1, . . . , xn (2-5)
mit x1 ≤ xi ≤ xn. Die Zahl
x x nx x n
n
n
=
=+( ) /
( / )
falls ungerade
falls gerade1 2
2
(2-6)
wird (empirischer) Median der Messreihe x1, . . , xn genannt. Der Median stellt eine Grenze zwischen zwei Hälften von Beobachtungen dar. Er hat gegen-über dem arithmetischen Mittelwert den Vorteil, un-empfindlicher gegenüber Ausreißern zu sein (vgl. Beispiel 2.4).
























![Curriculum für das Fach Deutsch als Zweitsprache am ... DEUTSCH[1].pdfDreyer, Hilke und Richard Schmitt: Lehr – und Übungsbuch der deutschen Grammatik . Hueber: Ismaning, 2000](https://img.pdfslide.org/doc/110x75/5af7cc937f8b9a44658b8aae/curriculum-fr-das-fach-deutsch-als-zweitsprache-am-deutsch1pdfdreyer-hilke.jpg)



