Embed Size (px)
Citation preview

81
Methode zur quantitativen Bestimmung yon auf Diinnschichten getrennten Substanzen *
GI~NTER L:EHI~ANN, I~ANs-GEI~T ]~[AHN u n d PAeLo MA_I~TINOD**
Institut fiir Organische Chemie der Universit~t des Saarlandes, Saarbriicken
Eingegangen am 21. September 1966
Summary. A generally applicable method is presented for the quantitative deter- ruination of compounds separable by thin-layer chromatography, where the separated substance is removed from the thin-layer material by elution in a micro- chromatography column. The preparation of a suitable micro-pipette and of the column for micro-chromatography is described. The data obtained show that the procedure works only with a slight error and the results are well reproducible.
Fiir die 5~kroanalytik ist die quanti tat ive Auswertung yon Dfinnsehicht. ehromatogrammen yon groi~er Bedeutung and es ist verwunderlich, wie relativ wenig bisher in der analytischen Praxis yon dieser MSglichkeit Gebrauch gemacht wird. Das mag damit zusammenhi~ngen, dal~ die bekannten Methoden zur quanti tat iven Bestimmung diinnschiehtchro- matographisch getrennter Substanzen nur fiir Sonderfalle zur Analytik ausgewi~hlter Substanzklassen entwickelt worden sind. Allgemeiner an- wendbare Methoden sind nun in letzter Zeit yon MXLL~r U. Mitarb. [8], S C H I L C ~ [12] sowie yon D~AW~nT u. Mitarb. [3] beschrieben worden, die aber naeh unserer Ansieht das Problem der quanti tat iven Bestim- mung mit Hilfe der Diinnsehiehtehromatographie fiir die routinema~ige Laboratoriumspraxis noeh nicht gelSst haben. Allgemein zeichnen sieh bei den bisher ausgefibten Verfahren zur quanti- ta t iven Auswertung der Dfinnsehichtehromatographie zwei Richtungen ab, denen beiden gemeinsam die Entfernung des Sorptionsmittels mit der zu best immenden Substanz yon der Glasplatte ist, wobei dieses dutch Absehaben mit dem Spatel o.~. oder mit einem Sauggeri~t ge- sehieht. Ansehliel]end wird die Substanz aus dem Sorptionsmittel extra- hiert und spektralphotometrisch oder colorimetriseh gemessen [10,11,14]. Eine quanti tat ive Auswertung kann aber auch durch direkte lViessung auf der Diinnschicht dureh Planimetrieren der FleckengrSl~e, dutch Densitometrie, durch Reflektometrie, Fluorimetrie oder dureh Aus- wiegen der die Substanz enthaltenden, yon der Glasplatte entfernten Sorptionssehicht vorgenommen ~erden [11]. I)iese Methoden lassen
* 1. Mitteilung fiber Beitr~ge zur quantitativen Diinnschichtchromatographie. ** Central-Universit~t von Ecuador in Quito, seiner Zeit Humboldt-Stipendiat am Institut ffir 0rganlsche Chemie der Universit~t des Saarlandes.
6 Z. Anal, Chem., Bd. 227

82 G. LEHNIANI~, H.-G. HAHN U. P. ~At~TINOD :
jedooh sehon erkennen, re_it welchen Fehlerm6glichkeiten sie behaftet siud und in welchem MaBe mit rel0roduzierbaren Ergebnissen gerechnet werden kann. Nach unseren Erfahrungen lassen sich diinnschicht- chromatographisch getrennte Stoffe im allgemeinen nut schwierig veto Sorptionsmittel ablSsen und analytiseh auswerten, da die Extraktion yon der aktiven Oberfl&che dureh alleiniges, einfaches Behandeln mit einem entsl0rechenden LSsungsmittel nieht quantitativ erfolgt. Unsere Ergeb- nisse werden dureh die Beobachtung einiger anderer Antoren gest/itzt. So stellte SCHILO~ [12] bei der quantitativen Isolierung dfinnschicht- chromatographisch getrennter Snbstanzen fest, dal3 dutch Erws der ExtraktionslSsung die Stoffe ,,quantitativer" aus der Trggerschicht herausgelSst werden. BLAZ~J~WICZ [1] berichtet, dab naeh den bei der sgulenchromatographisehen Trennung gewonnenen Erkenntnissen eine 8ubstanz sich aus dem Kieselgel der D/innsehicht ohne Sehwierigkeiten mit dem gleiehen LSsungsmittel eluieren lassen sollte. Einschlggige Ver- suche ergaben aber, dal~ dutch Extraktion im Soxhlet-Apparat in dem geschilderten Falle gar keine und dureh Heil~extraktion nur etwa die H/~lfte der Substanz extrahiert werden konnte. Der Verwendung zu stark polarer Extraktionsmittel sind dureh das Kieselgel Grenzen gesetzt, weft dieses mit steigender Polaritgt des Extraktionsmittels zunehmend stark hydratisierte Kieselss abgibt. Dieses ist eine Ursache ffir die bei der spektralphotometrischen Gehaltsbestimmung anftretende Schwierigkeit, die aueh nicht dutch BlindwertlSsungen restlos kompensiert werden kann, da die Menge des extrahierten und Streulicht verursaehenden Gels yon Extraktion zu Extraktion variieren kann. Auch BOLLI~G~ n. K6~IG [2] sind der Meinung, dab das Sammeln des Kieselgels im Mel~kolben, Sehfit- teln mit dem LSsungsmittel sowie Filtrieren des Extr~ktes mit Fehlern behaftet ist, die dureh Verdunstungen des LSsungsmittels und dureh das Volumen des Kieselgels bedingt sind. Gv~Iss u. Mitarb. [4] weisen schliel~- lieh noeh dar~uf bin, dab beim Absaugen des Iiieselgels yon der Dfinn- s chiehtplatte Verunreinigungen bus den verwendeten S ehl/~uchen und der Extraktionshiilse eingeschleppt werden k6nnen. Die vorliegende ArbeR hatte sich die Auffindung eines m6glichst ein- fachen Analysenverfahrens zum Ziele gesetzt, das es erlauben sollte, die bisher beschriebenen, mitunter aufwendigen Methoden zur quantitativen Auswertung yon Dfinnschiehtchromatogrammen einfacher und durehAus- sehaltung vermeidbarer Fehlerm6glichkeiten besser zu gestalten. Zur Herabsetzung der m6glichen Fehler haben wit es als notwendig erachtet, eine Auftragepipette selbst herzustellen, da die k&uflichen Mikropipetten (z. B. Blutzuckerpipetten mit einem Volumen yon 10 ~1) bei der Herstel- lung ungenau ausfallen k6nnen [13] und Untersehiede im Volumen yon 7--12~ aufweisen [6]. Andererseits semen uns die Verwendung yon Mikrometerpipetten (Agla-Spritze, Hamilton Dosiervorriehtung) ffir ein

Quantitative Bestimmung von auf Dfinnschiehten getrennten Substanzen 83
Verfahren, das in jedem analytischen Laboratorium leieht durchffibxbar sein sell, zu aufwendig und der Gebrauch yon ,,Microcaps" zu teuer. Wit benutzten fiir die ~erstellung der Pipette eine Injektionskanfile, in die seitlich ein Schlitz eingefeilt und deren Spitze plangesehliffen wnrde. Beim oberflichliehen Eintauchen der Kaniile in eine Fltissigkeit steigt diese infolge der Capitlaritat bis zum Schlitz empor. Wit konnten uns iiberzeugen, dal~ bei der Verwendung des gleiehen L5sungsmittels die Fliissigkeitssaule stets in der gteichen HShe abril~ und somit eine ge- nauere Dosierung erfolgt, als mit den iibliehen Glaspipetten, bei denen die Schwierigkeiten des genanen Einstellens der Fliissigkeitssaule auf die Striehmarke noch hinzukommt. Um die yon den verschiedenen Autoren aufgezeigten Fehler bei tier Elu- tlion des Sorptionsmlttels auszuschalten, ba~iert unsere Arbeit nun darauf, das yon der Glasplatte abgeschabte Sorptionsmittel in ein Mikrochroma- tographierrohr zu iiberfiihren, zur S~ule zu sehiehten und zu eluieren, um dadureh eine quantitative AblSsung der zu besthnmenden Substanz zu erreiehen. Dureh den stet~en Zulauf yon frischem LSsungsmittel kommt es in der Mikrochromatographiersiule zu einer Verteilungsehromato- graphie und weft dgs fI~sch zufiieBende LSsungsmittel die Einstellung eines LSsungsgleiehgewichtes verhindert, zur vo]]kommenen AblSsung der Substanz vom Sorptionsmittet. iV[umber u. Mitarb. [8] eluierten gleieh- falls die Substanz aus dem zur S~ule geschichteten Sorptionsmittel, das Prinzip des Chromatographierrohres ist jedoch mit dem unserer Anord- nung nicht vergleiehbar. Falls erforderlieh, kann die Elution des Sorptionsmittels in dem yon mls entwickelten Mikrochromatographierrohr auch durch Druck besehleu. nigt werden. Die Apparatur zur Druekelution besteht aus einem Glas- reehen als Ver~efler, (lessen Abzweigungen fiber Gummisehl~uehe mit den Chromatographierrohren verbunden werden. Als Uberdruekventil bzw. Windkessel z~un Druekausgleich client eine Gummiblase (Luft- balIon). Das Abschaben des Sorptionsmitte]s yon der Glasplatte erfotgt am einfaehsten nach Lokalisierung der Substanz mit einem 0bjekttr~ger [5] in Form eines Reehteekes [1t] und daneben (zur Besthnmung des BlindweI~es) eines Rechteekes gleicher Gr61~e des chromatographisch gleich behandelten Sorptionsmittels auf einer freigebtiebenen Bahn der Diinnschicht. Die Verwendung eines Spatels zum Abheben des Sorptions- mittels, ~de hiufig beschrieben wtu'de, hat sieh als naehteilJg erwiesen, da die Sorptionssehicht nicht in einem Zuge abgeschabt werden kann, was einen Zeitverlust bedeutet, wobei die MSgtiehkeit nieht ausgesehlossen wird, dal3 kleinste tq.este des Sorptionsmittels auf der Glasoberfliche zurfiekbleiben. M_it dem seharfkantigen Glasstreifen kann die Schieht mit einem Zuge abgelSst nnd dadureh ein Substanzverlust weitgehend ver- mieden werden.
6*

84
Wir fiihrten unsere Versuche au f Diinnschichten aus Kieselgel und vor
allem aus Cellulosepulvcr durch, da wir davon ausgingen, daG Cellulose- sehichten meehaniseh widerstandsfKhiger sind und sick besonders fiir die Trennung hydrophfler Stoffe bewAhrt haben [7, 9]. Aul~crdem lasscn sick die Ergebnisse der Papierchroma~ographie in vielen FAllen auf die Dfinnschichtehromatographie an Celluloseschiehten tibertragen [13]. Zur Erzielung einer guten Fleekenausbildung, die gerade ffir eine quant i ta t ive Auswer~ung des Chromatogrammes yon grol~er Bedcu tung ist., wurde mi t 0,5 m m starken Schichten gearbeitet , denn die BeladungskapazitAt tier Sorp~ionsschicht sell nieht/iberschri~ten werden, was bei 0,25 m m starken D/innschiehten leieh~ der Fall sein kSnnte, da fiir die Bes t immung eine rela~iv grSgere Subsganzmenge als zum qualitagiven Nachweis benStigt Mrd.
Durchfi ihrung der Versuche
Sorlgtionsmittel
Cellulosepulver MN 300, lVIN 300F254 (1Yiacherey, Nagel & Co.), KieselgeI G und Kieselgel GF 254 (E. Merck AG) zur Diinnsehichtchromatographie, je nach Eigen- schaft der zu bestimmenden Verbindung. Zur Herstellung yon 5 Triigerplatten 20• cm mit einer Schichtdicke yon ca. 0,5 mm wird wie tblgt verfab2en: 1. CelluZoseschicI#en. 25 g Cellulosepulver werden mlt 150ml dest. ~*asser auf- geschl~,mmt, in einem elektrischen ]Hixger~t ca. 1--2 rain homogenisiert, oder mit 115 ml 90~ J~hylMkohol, verg~ll~ mit 1 ~ Petrol~ther (Merck), in einem versehlossenen Erlenmeyer-Kolben 30 sec l~ng geschfittelt [7]. Die Suspensionen werden wie fiblieh in ein Streichger~t gef'fillt und die Platten damit beschichtet. Wiihrend die mit der wii{~rigen Suspension bereiteten Schichten einer l~ngeren Trocknungszeit bedfirfen, sind die mit der Mkoholischcn bereitcten schon nach ca. 2 Std bei Zimmertempcratur ohne weitere Trockmmg verwendbar. 2. Kieselgelschichten. 50 g Kieselgel werden mit 100 ml dest. W~sser in einem ver- schlosscnen Erlenmeyer-Kolben 30 sec lang geschiittelt, in ein Streichger~t ein- geffillt, auf den Platten ausgestrichen und wie iiblich weiterbehandelt.
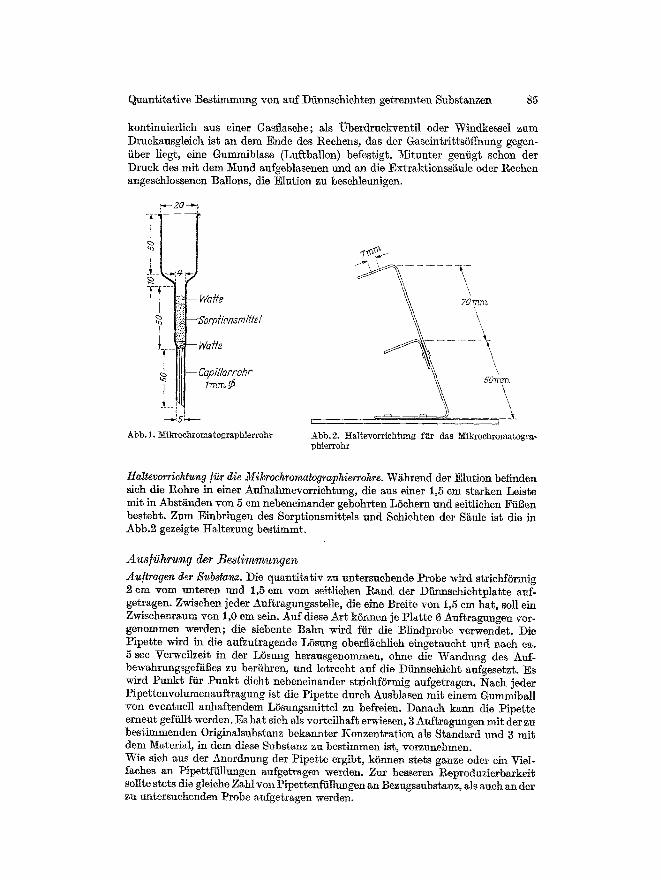
Gergte An]ertigung einer Pipette. Die Spitze einer Injek%ionskantile (080/40) wird p]an- geschliffen; in einer Entfernung yon ca. 13 mm vom Kaniilenausgang wird mit einer 1M:esserfeile ein Schlitz bis zur Kaniilenmitte eingefeilt. Nachdem tier Feilgrat beseitigt worden ist, ist die Pipette einsatzbereit. Zum Gebraueh wird diese auf einen Halter oder eine Injektionsspritze gese~zt. An]ertigung des Mikrochromatographierrohres. Ffir die Elution der zu bestimmenden Substanz aus dem Sorptionsmittel wird das in Abb. 1 skizzierte Chromatographier- rohr verwendet. Durch die Capill~re am Ende des Rohres flieBt das Elutions- mittel tropfenweisc ab. Einrichtung ]i~r eine Elutlon unter Druck. Ein Glasrohr mit angeschmolzenen I4~hnen in Form eiues Rechens dicnt zur Verteilung des Gasdruckes auf die ein- zelnen Mikroehromatographiers~ulen, die durch Gummisc}fl~tuche mit dem Rechen verbunden sind. Die Aus des Gasdruckes erfolgt kontinuierlich oder dis-
Quantitative Bestimmung von auf Diinnschiehten getrerm~en Substanzen 85
kontinuierlich aus einer Gasflasehe; als ~berdruckventil oder Windkessel zum Druekausgleich ist an dem Ende des Rechens, das der GaseintrittsSffnung gegen- fiber liegt, eine Gummiblase (Luftballon) befestigt, l~Iitunter geniigt sehon der Druck des mit den] Mund aufgeblasenen und an die Extraktionssgu]e oder Reehen angesehlossenen Ballons, die Elution zu beschleunigen.
llll--C~P ;H~176 f _ _ ~ t ~ m ~ r
Abb. L ~ ikrochromatographie r rohr
F . l
Abb.2. IIal te~'orrichtung f i i r das ~Iikrochroma~ogra- phierrohr
Haltevorrichtung/i~r die Mik~rochromatographierrohre. W~hrend der Elution befinden sich die Rohre in einer Aufnahmevorriehtung, die aus einer 1,5 em starken Leiste mit in Abstgnden yon 5 em nebeneinander gebohrten L6chern und seiflichen FiiBen besteht. Zum Einbringen des Sorptionsmittels und Sehieht~n der S~lfle ist die in Abb.2 gezeigte Halterung bestimmt.
A usli~hrung der Bestimmungen Au]tragen der Substanz. Die qu~ntit.ativ zu untersuchende Probe whxt striehfSrmig 2 cm yore unteren trod 1,5 em yore seifliehen Rand der Diinnschichtplatte auf- getragen. Zwischen jeder Auftragungsst~le, die eine Breite yon 1,5 cm hat, sou ein Zwischem'aum yon 1,0 em sein. Auf diese Art kSnnen je Platte 6 Auftragtmgen vor- genommen werden; die siebente Balm wird ffir die Blindprobe verwendet. Die Pipette wird in die aufzutragende LOsung oberfl~chlieh eingetaucht und nach cu. 5 see Verweilzeit in der L6sung herausgenommen, ohne die Wandmlg des Auf- bewahrungsgef~l]es zu berfihren, trod lotrecht auf die Diinnsehicht aufgesetzt. Es wird Punkt ffir Punkt dieht nebeneinander striehfSrmig aufgetragen. :Nach jeder Pipettenvolumenauftragung ist die Pipette durch Ausbl~sen mit einem GummibM1 yon eventuell anhaftendem LSsungsmittd zu befreien. Danach kann die Pipette erneut geffillt werden. Es b~t sieh als vorteilhaft erwiesen, 3 Auftragungen mit der zu bestimmenden Originalsubstanz bekarmter Konzentration als Standard und 3 mit dem Material, in dem diese Substa.~z zu bestimmen ist, vorztmehmen. Wie sieh aus der Anordmmg der Pipette ergibt, kS~men stets ganze oder ehl VieL laches an Pipet~tFfilllmgen aufgetl'agen werden. Zur besseren Reproduzierbaxkei~ soltte stets die gleiche Zaht yon PipettenFfillungen an Bezugssubstanz, als auch an der zu untersuehenden Probe aufgetragen werden.

86 G. LERMtN~, tt.-G. Hi~N u. P. MiRTINOD :
Entwicklung des Chromatogrammes. Das Chromatogramm wird wie fibllch mit dem fiir die Trennung der zu bestimmenden Substanz angegebenen Flle~mittel in der Kammer entwickclt. Sichtbarmachung der Substanz. Nach Erreichen einer Steigh(~he yon 10--15 em wird die Dtinnschichtplatte aus tier Trennkammer herausgenommen und das in dem Sorptionsmittel haftende Flie$mittel im Luftstrom oder im Troekenschrank ent- fernt. Zur Erkermung und zum Vergleich mit der Bezugssubstanz wird das Chro- matogramm farbloser Verbindungen, die im UV-Licht absorbieren, sowohl im iang- weIligen (366 nm) als aueh im kurzweHigen (254 nm) Ultraviolett-Lieht betrachtet. Bei der Verwendung fluorescierender Sorptionsmittel l~Bt sieh die Verbindung als dunkler Fleck auf ituoreseierendem Grunde erkennen, gleichfalls finder man auf nor- malem Sorptionsmittel solche Verbindungen, die selbst fiuorescieren. Farblose, im UV-Licht nicht absorbierende Stoffe miissen dureh geeignete Reagentien zerstS- rungsfrei siehtbar gemacht werden. Die Lage der zu bestimmenden Substanz wird nun rechteckig markiert und zur Bestimmung des Blindwertextraktes ein gleich groSes Feld auf der freien, chromatographiseh gleieh behandelten Bahn in gleicher HShe angezeiehnet. Abschaben des Sorptio~smittels yon der Tr~igerplatte. Das Absehaben des Sorptions- mittels zur Gewinnung der zu bestimmenden Verbindung aus der lok~lisierten Zone erfolgt mit der Kante eines Objekttr~gers, wobei dieser parallel zur Startlinie Feld fiir Feld abhebt. Der Vorteil bei der Verwendung eines Objekttr~gers oder G]as- streifens liegt darin, da$ stets gleieh breite Zonen der Diinnschicht abgeschabt werden und das Sorptionsmittel in einem Zuge entfernt wird. Das abgesehabte Sorlo- tionsmittel wird auf glattem l%rgamentpapier gesammelt und ver]ustlos in das Chromatographierrohr eingebracht. Ffir die Blindwertbcstimmung wird das dafiir angezeiehnete Feld ebenso behande]t. Fiillen des Mikrochromatographierrohres. Das Rohr wird un~en mit Watte oder Glas- wolle verschlossen und das zu eluierende Sorptionsmittel mit ttilfe des Pergament- papieres daraufgegeben. Das Chromatogralohierrohr befinde~ sich wi~hrend dieses Arbeitsganges in der I:faltevorrichtung (Abb.2). Mit einem dfinnen Glasstab wird das Sorptionsmittel im Rohr zur S~ule gesehiehtet, die Glasoberfl~ehe unter der abgesehabten Diinnschieht mit einem Wattebausch, der mit dem Elutionsmittel leicht getri~nkt ist, abgerieben und auf das zur Si~ule gep~ekte Sorptionsmittel gebraeht. Nach dem ~)berffihren des RShrchens in die Aufnahmevorrichtung werden ca. 9 ml des Elutionsmittels auf die S~ule gegeben und in einen 10 ml fassenden MeB- kolben eintropfen gelassen. Soll eine Substanz yore Kieselgel elulert werden, wird auf den W~tte- oder Gl~swolleverschluB eine 2--3 mm hohe Sehicht CeHuloseloulver eingebracht. Das Celinlosepulver mug mit Wasser angertihrt sein, da das unbehandelte Pulver nicht geeigne~ ist. Gfinstig l i~t sieh das obere Material einer gebrauchten Diinnschicht dazu verwenden, das nicht mit dem Fliei3mittel in Berfih- rung gekommen ist. Auf diese Cellulose wird dann das Kieselgel gesehichtet. Die Cellulose hat die Aufgabe, als Filter zu wirken und Kieselgel bei der Eintion zurfick- zuhalten. Elution der zu bestimmenden Substanz. Zur quantitutiven Elution hat sich in vielen F~llen 70~ Methy]alkohol bew~hrt, der Vorgang dauert ca. 2--6 Std. Die Ein- tion kann wesentlieh beschleunigt werden, wenn sic unter Druck erfolgt. Dazu wird ein mit einem rech~winklig gebogenen Glasrohr versehener Gummistopfen auf das Chromatographierohr gesetzt, mit einem Schlaueh an den Druckverteiler (Reehen) angeschlossen und eluiert. Schon der blol3e Druck einer aufgeblasenen Gummiblase kaim die Elution besehleunigen. Mit bekannten Gehalten an Standardsubstanz soUte die Druekelution auf Vollst~ndigkeit und Reproduzierbarkeit vorher aus- getestet und die Tropfenfolge, die yon der Diehte der geschichteten S~ule abh~ngig

Quantitative Bestimmung yon auf Diinnschichten getrennten Substanzen 87
ist, festgelegt werden. ~ffach den yon uns gemachten Erf~hrtmgen befindet sieh die zu eluierende Verbindung bereits in den ersten Millillt~rn des Elu~ts fast quantitativ. Nach der Elution wird der Mel3kolben zur ~ r k e aufgefiillt, umgeschiittelt und wmm einige Verunreinigungen in der LSsung schweben solRen, 20 rain lang zentri- fugiert. Messung der Liehtabsorption. Die Messung zur quantit~tiven Bestimmung erfolgt gegen den Blindwertextrakt dutch Lichtabsorption im sichtbaren oder im ultra- violetten Licht. Die Menge der zu bestimmenden Substanz errechnet sich aus der Extinktion bei 2raax dieser Verbindung, der Extinktion einer auf dieselbe Weise chromatographierten und eluierten Standardmenge und der bekannten/r des Standards.
Ex X = S t - -
Est
X Mengo der zu bestimmenden Substanz St Eingewogene ~enge Bezugssubstanz (Standard) E , Gemessene Extinktion der zu bestimmenden Substanz E~t Gemessene Extinktion der Bezugssubstanz (Standard). Die Formeln gelten, wenn gldohe Mengen (gleiche Anzahl yon Pipett~nftillung~n) dot Bezugssubstanz als auch der zu bestimmenden Verbindung auf die Diinnschicht aufgetragen werden. Au/stellung einer Ei&kurve zur Prii/ung, ob gas Bouguer-Lambert-Beersche Gesetz erliillt ist. Die der gefundenen Extinktion entsprechende Substanzmenge kann auch einer Eiohkurve entnommen werden. Zur Aufstelhmg dieser Eichkurve wird yon der Bezugssubstanz eine steigende Anzahl yon Pipettenfiillungen auf die Diinnschicht aufgetragen und nach der vorstehend angegebenen Arbeitsvorschrift weiter- behandelt.
Reproduzierbarkeit und t 'ehler
Direkte Reproduzierbarkeit und Volumes der Pipette. Zur Feststellung des Volumens der hergestellten Pipette wurden verschiedene L5sungen mit bekarmten Geh~lten an Farbstoffen in 70~ Methyl~lkohol hergestellt und yon diesen LSsungen je 1 Pipettenfiillung direkt in 10 ml fassende Me•kolben iiberffihrt, zttr Marke aufgeftillt und gegen das reine LSsungsmittel spektralphotometriert. Die nachstehende Auf- stellung gibt die Ergebnisse der Messungen wieder: 1. Lebensmittelrot Nr. 1 (Azorubin S), 74,5i mg in 10,0 ml 70~ ~ethylalko- hol, 2max 510 nm, Smax 10630 in 70~ Methylalkohol; 2. Lebensmittelgelb Hr. 2 (Tartr~zin 0), 55,46 mg in 10,0 ml 70~ ~ethylalko- hol, ~ a x 423 nm, Smax 18190 in 70~ Methylalkohol; 3. Lebensmittelgelb Nr. 2 (Tartrazin 0), 82,80 mg in 10,0 ml 70~ ~ethyl- alkohol.
zu i. E = 0,096 zu 2. E = 0,110 0,092 0,108 0,095 0,112 0,092 0,109 0,091 0,110
0,110 = 0,093 da 0,003 0,111
zu 3. E = 0,168 0,166 0,165
= 0,166 :]: 0,002
= 0,1t0 ::t: 0,002

88 Lrm~A~r e~ al.: Quantitative Bestimmung yon getrennten Substanzen
Abweichung vom Mittelwert: 1. = 3,20/0 2. = 1,8~ 3. = 1,2~
Das Pipettenvolumen wurde aus dem molaren Extinktionskoeffizienten der Lebens- mittelfarbstoffe erreehnet. Danaeh betr~gt das Volumen nach: 1. 5,86 tzl
2. 5,91 ~zl 3. 5,91 ~.1
Mittelwert 5,89 g.1 • 0,03 ----- =k 0,50/0 �9
Relgroduzierbarkeit der Bestlmmungs~nethode. Zur i)berprfifung der Reproduzierbar- keit der vorstehend besehriebenen Methode wxlrden auf eine Dfinnschicht (Cellulose
300) 3mal je 2 Pipettenvolumina der FarbstofflSsung Lebensmittelgelb I~r. 2 anfgetragen, chromatographiert, das Sorp~ionsmittel abgeschabt, eluiert und photo- metriert.
Ergebnis der Messungen: E = 0,221 i 0,003 = 1,3~ Abweichung vom theoretisehen Wert
0,221 • 0,003 = 1,3~ 0,218 • 0,006 = 2,70/0
.E = 0,220
Urn den Einflul~ der Zeit auf die Elution zu untersuchen, wurden 4 real je 2 Pipetten- volumina der gleichen FarbstofflSsung Lebensmittelgelb Nr. 2 auf eine Cellulose- dfinnschicht aufgetragen, chromatographier$, die Cellulose abgeschabt, unter Druck eluierb und gemessen. Die Elutionszeit betrug nur ~/4 der Zeit, die ffir eine Normal- elution benStigt wurde.
Ergebnis der Messungen: E = 0,219 • 0,005 ---- 2,20/0 Abweichung vom theoretisehen Wert
0,220 • 0,004 ---- 1,8~ 0,221 • 0,003 = 1,3~ 0,221 • 0,003 = 1,3~
----- 0,220
Zusammenfassung Es wi rd ein a l lgemein anwendbares Verfahren zur qua n t i t a t i ve n Be- s t i m m u n g df innsehich~chromatographisch t r e nnba re r Subs tanzen emp- fohlen, bei dem die Abl6sung der ge t r enn ten Subs tanzen yon dem D/ innsch ich tmate r i a l durch E lu t ion in e inem MLkroehromatographier rohr erfolgt. Die Hers te l lung einer geeigneten Mikrop ipe t t e und die Anfer t i - gung der Mikrochromatograph ie r rohre wird besckrieben. Aus den erhal- t enen Mel]werten erg ib t sieh, dab das Verfahren nur m i t e inem kleinen ]~ehler beha f t e t i s t und sieh die Ergebnisse sehr gu t reproduz ie ren lassen.
Den Herren Prof. B. EIST~T und O. NEV~U~OEFF~ danken wir Fdr ihr Interesse an dieser Arbeit und Fdr wer~volle Diskussionen.

M. Qu~ESRI and I. AK]tTAR : Paper Chromatographiy of Metal Ions 89
Li tera tur
[1] BLXZEJEWICZ, L. : diese Z. 216, 327 (1966). -- [2] BOLLINGE~, It . R., u. A. K6- NKG: diese Z. 214, 1 (1965). -- [3] DaAWE~T, F., W. tIEnvKAI~ U. A. ZIEGLE~: diese Z. 217, 22 (1966). -- [4] GEIss, F., A. KLOSE U. A. COPET: diese Z. 211, 37 (1965). -- [5] HO~EGGER, C. G. : Helv. Chim. Acta 45, 1409 {1962). -- [6] Jo~c, H. : Privat- mitteilung. - - [7] LEtKMAI~lV, G., U. I ). MARTINOD : Z. Lebensm.-Untersuch. -Forsch. 180, 269 (1966). -- [8] MILLETT, M. A., W. E. MOO~E, and J. F. SAE~AN: Anal. Chem. 36, 491 (1964) ; vgl. diese Z. 208, 441 (1965). - - [9] NY~OM, N. : Fruehtsaft- industrie 8, 205 (1963). - - [10] PATAKI, G.: Diinnschiehtctlromatographie in der Aminos~ure- und Peptidchemie. Berlin: W. de Gruyter & Co. 1966. -- [11] I~ANDE- RAT~,K.: Dfinnsehicht-Chromatographie, 2. Aufl. Weinheim: Verlag Chemie G.m.b.H. 1965. -- [12] Scn~c~n~, H. : diese Z. 199, 335 (1964). - - [13] S c ~ o ~ , P. J. : a) Dissertation Saarbrfieken 1963; b) diese Z. 205, 303 (1964). -- [14] STARL, E.: Diinnschlcht-Chromatographie. Berlin, GSttingen, Heidelberg: Springer 1962.
Dr. G. LE~A~N Institut ffir Organisehe Chemie der Universit~t des Saarlandes 66 Saarbriicken 15
Paper Chromatography of Metal Ions in Solvents Containing Methylamine MoHsn~ QU~ESHI and Miss IQ~AL AKHTA~
Department of Chemistry, Aligarh Muslim University, Aligarh (India)
Received August 7, 1965
Summary. The effect of methylamine and methylamine hydrochloride has been systematically investigated in the paper chromatography of various metal ions. I t has been found that methylamine hydrochloride like EDTA helps in preventing tailing. A new separation of Fe, Mn, iNi, and Co has been developed. The Rr-values are 0.97, 0.49, 0.08, 0.79 respectively and the solvent system used is ethylaceto- acetate/methylamine hydroehloride/conc, hydrochloric acid (20:2 : 7). The values are equally spaced, the spots are compact and the separation is probably the best yet recorded.
Me thy lamine is k n o w n to form numerous complexes wi th m e t a l ions which usua l ly form complexes wi th ammonia . I t s presence in t he so lvent sys t em is therefore a p t to mod i fy the p roper t i e s of such a sys tem. As far as we are aware sys tems conta in ing m e t h y l a m i n e have no t y e t been inves t iga t ed sy s t ema t i ca l l y for the separa t ion of m e t a l ions. W e have, therefore , s tud ied the ch romatograph ic behav iour of numerous m e t a l ions in sys tems conta in ing m e t h y l a m i n e or m e t h y l a m i n e hydrochlor ide . As a resu l t we have deve loped a new solvent sys t em which gives p r o b a b l y the bes t s epa ra t ion of Fe , Mn, Ni, and Co y e t recorded [ l , 5, 7, 9 - - 11] and we have also found t h a t m e t h y l a m i n e hydroch lo r ide is a v e r y use fu l subs tance which l ike E D T A helps to p reven t t a i l ing and should therefore prove useful in m a n y separa t ions of m e t a l ions.