Embed Size (px)
Citation preview

TECHNISCHE UNIVERSITÄT MÜNCHEN
Wacker-Lehrstuhl für Makromolekulare Chemie
Molecular Brushes of Poly(2-oxazoline)s
Ning Zhang
Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität
München zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
genehmigten Dissertation.
Vorsitzender: Univ.- Prof. Dr. Ulrich K. Heiz
Prüfer der Dissertation: 1. Univ.- Prof. Dr. Rainer Jordan, Technische Universität Dresden
2. Univ.- Prof. Dr. Sevil Weinkauf
Die Dissertation wurde am 24.08.2010 bei der Technischen Universität München eingereicht
und durch die Fakultät für Chemie am 19.10.2010 angenommen


Die vorliegende Arbeit wurde in der Zeit von November 2006 bis März 2010 unter der
Leitung von Prof. Dr. Rainer Jordan am Wacker-Lehrstuhl für Makromolekulare Chemie von
Prof. Dr. Dr. h.c. Bernhard Rieger der Technischen Universität München angefertigt.


Acknowledgements
First and foremost, I would like to gratefully and sincerely thank my supervisor, Prof. Dr.
Rainer Jordan, for giving me the opportunity to work on such an exciting project. He
introduced me to the field of polymer and interface science, broadened my perspective and
knowledge, and gave me confidence to apply myself in my graduate study. I am deeply
impressed by his wide knowledge, patient guidance as well as the professional attitude
towards scientific work. When I made the decision to stay in Munich for the last few months
of my Ph.D. study, he also showed his kind understanding of it which has been of great
importance to me. Without his constant support, guidance and encouragement this dissertation
would not have been possible.
I would also like to express my deep gratitude to Prof. Dr. Dr. h. c. Bernhard Rieger for
giving me the opportunity to work at the Wacker-Lehrstuhl für Makromolekulare Chemie.
His constructive comments and scientific work style continually impressed me and have been
of great value for my work. I wish to sincerely thank Prof. Dr. -Ing. Oskar Nuyken, whose
generous supports and encouragement always inspired me and became a large driving force
for the present study. I gratefully thank Dr. Carsten Troll and Dr. -Ing. Heidi Samarian for
their supports both technically and scientifically.
A warm and heartfelt thank goes to Dr. Marin Steenackers and Dr. Robert Luxenhofer for
their fruitful cooperation, enlightening discussion and most importantly their warm friendship
throughout my Ph.D. study. Marin taught me the valuable laboratory techniques, and put time
and energy into correcting this dissertation. „Luxi‟ always helped me with suggestion on both
my work and my life, and offered me the “warmest hotel” in Dresden. Words are not
sufficient enough to express all my appreciation for their countless help.
I am grateful to Dr. Tilo Pompe from Leibniz Institute of Polymer Research Dresden for
performing the protein adsorption and cell adhesion experiments, and Dr. Schilp Sören from
Universität Heidelberg for offering the surfaces. Without these excellent cooperations, this
dissertation would not have been completed.
Many thanks to Naïma Aurelia Hutter for her precise English-German translation, as well as
her kind help during my graduate study. At the same time, I would like to thank Anita Schulz
and Hans Koss for their kind assistance in the lab work.

I would like to give my special thank to my labmates Dr. Stephan Huber, Dr. Tianzhu Zhang,
Amir Doroodian, and Khalifah Salmeia for the harmonious lab atmosphere.
Thanks go to all the other Makros, Dr. Martin Schneider, Andreas Feigl, Dr. Liyi Chen,
Gerhard Richter, Dr. Carola Gantner, Ulrike Will, Dr. Michael Reif, Dr. Sergei Vagin, Uwe
Seemann, Alexander Schöbel, Timo Korfmann, Udo Schmidt, Paul Heinz, Monika Kellner,
Anastasia Golosova, Sabine Martinetz-Große, Timo Anselment, Pierre Göppert, Dr. Julia
Müller, Felix Schulz, Frank Deubel, Stephan Klaus, Christian Hanisch, Joachim Dengler, Dr.
Carly Anderson, Manuel Winkenstette, Philip Zehetmaier, Sanna Zimmer, Sandra
Hochwarter, Robert Reichardt, Maximilian Lehenmeier, Konrad Hindelang, Meifang Yin,
Tobias Diesner, Abdussalam Qaroush, Manfred Gunesch, Anna Lennartson, as well as other
new comers for making my stay such a pleasant and great experience. I sincerely appreciate
all the advices and skills that I have picked up in this period of time.
I would like to thank my parents and my friends for their love and help.
Finally, I would like to give my last but unique thank to my wife Hang for her endless love,
termless sacrifice and patience.

To my wife Hang


I
Table of Contents
1. Introduction············································································1
1.1 Molecular brushes··········································································2
1.1.1 Synthesis of molecular brushes··················································3
1.1.2 Stimuli-responsive molecular brushes··········································8
1.1.2.1 Stimuli-responsive polymers·········································8
1.1.2.2 Thermo-responsive molecular brushes······························9
1.1.3 Molecular brushes on surfaces·················································12
1.2 Polymer Brushes··········································································14
1.2.1 Self-initiated photografting and photopolymerization (SIPGP)···········17
1.2.2 Bottle-brush brushes (BBBs)···················································20
1.3 Bioactive surfaces·······································································21
1.4 Poly(2-oxazoline)s·········································································24
1.4.1 2-Oxazoline·······································································24
1.4.2 Living cationic ring-opening polymerization (LCROP) ···················24
1.4.3 Thermo-responsive poly(2-oxazoline)s·······································27
1.4.4 Biomedical applications of poly(2-oxazoline)s······························28
2. Objective of this work······························································30
3. Results and discussion······························································31
3.1 Monomer synthesis········································································31
3.2 Molecular brushes via free radical and cationic polymerization of
2-oxazolines·················································································33
3.2.1 Reaction of 2-isopropenyl-2-oxazoline·······································33

II
3.2.2 Polymerizations··································································34
3.2.3 Thermo-responsiveness of poly(2-oxazoline)s molecular brushes········42
3.2.4 Morphology of adsorbed molecular brushes on surfaces···················45
3.2.5 Cytotoxicity assay, MTT·······················································47
3.3 Molecular brushes via anionic and cationic ring-opening polymerization····49
3.3.1 Anionic polymerization of 2-isopropenyl-2-oxazoline·····················49
3.3.2 Kinetic studies of the living cationic ring-opening polymerization of
2-oxazolines······································································52
3.3.3 Thermo-responsive properties - impact of different parameters on the
LCST··············································································58
3.3.3.1 Side chain length effect··············································58
3.3.3.2 Concentration effect··················································64
3.3.3.3 Backbone length effect···············································65
3.4 Molecular brushes with copoly(2-oxazoline)s side chains·························68
3.4.1 Background·······································································68
3.4.2 Molecular brushes with statistical copolymer side chains·················68
3.4.2.1 Synthesis·······························································69
3.4.2.2 LCST of molecular brushes with random copolymer side chains
···········································································70
3.4.3 Molecular brushes with block copolymer side chains······················73
3.4.3.1 LCST of molecular brushes with block copolymer side chains
···········································································73
3.5 Bottle-brush brushes of poly(2-oxazoline)s··········································76
3.5.1 Background·······································································76
3.5.2 Bottle-brush brushes of poly(2-oxazoline)s on glassy carbon············77
3.5.2.1 Glassy carbon·························································77
3.5.2.2 Self-initiated photografting and photopolymerization···········78
3.5.2.3 Surface-initiated living cationic ring-opening polymerization··82

III
3.5.2.4 Wettability of the bottle-brush brushes····························86
3.5.2.5 Further functionalization of bottle-brush brushes················87
3.5.2.6 Stability test ···························································88
3.6 Bottle-brush brushes on self-assembled monolayers·······························89
3.6.1 Self-assembled monolayers····················································89
3.6.1.1 3-Aminopropyltrimethoxysilane monolayers·····················90
3.6.2 Bottle-brush brushes on 3-aminopropyltrimethoxysilane modified
oxidized silicon and glass substrates········································90
3.6.2.1 Self-initiated photografting and photopolymerization·········90
3.6.2.2 Surface-initiated living cationic ring-opening polymerization·92
3.6.3 Bottle-brush brushes on self-assembled monolayers of biphenylthiol on
gold···············································································95
3.7 Protein adsorption and cell adhesion on bottle-brush brushes of
poly(2-oxazoline)s·······································································99
3.7.1 Background·······································································99
3.7.2 Bottle-brush brushes of poly(2-oxazoline)s on oxidized silicon substrate
···················································································100
3.7.3 Protein adsorption······························································102
3.7.3.1 Variation of the BBB side chain composition···················103
3.7.3.2 Variation of the BBB side chain length··························105
3.7.3.3 Variation of the BBB pendant chain end groups················107
3.7.4 Cell adhesion····································································108
4. Summary············································································111
5. Zusammenfassung·································································120

IV
6. Experimental········································································130
6.1 Instruments···············································································130
6.2 Methods····················································································132
6.3 Materials···················································································134
6.4 Monomer synthesis······································································135
6.5 Synthesis of molecular brushes·······················································136
6.5.1 Free radical and anionic polymerizations···································136
6.5.2 Macroinitiator···································································137
6.5.3 Living cationic ring-opening polymerization·······························138
6.6 Preparation of bottle-brush brushes·················································142
6.6.1 Bottle-brush brushes on glassy carbon······································142
6.6.2 Bottle-brush brushes on 3-aminopropyltrimethoxysilane modified
oxidized silicon·································································144
6.6.3 Bottle-brush brushes on biphenyl modified gold··························146
7. References···········································································147

V
Abbreviation and Symbols
ACN acetonitrile
AFM atomic force microscopy
AIBN N,N-azobisisobutyronitrile
ATR-FTIR attenuated total reflection Fourier transform infrared
ATRP atom transfer radical polymerization
a.u. arbitrary units
-b- -block-
BBB bottle-brush brush
BDE bond dissociation energy
b.p. boiling point
BT 4-mercapto-1,1‟-biphenyl thiol
Boc N-tert-butyloxycarbonylpiperazine
BuOx 2-butyl-2-oxazoline
-co- copolymerize
DMAc dimethyl acetamide
DNA deoxyribonucleic acid
DP degree of polymerization
DRIFT diffusion reflectance Fourier transformed infrared
EBCD electron beam induced carbon deposition
EBCL electron beam chemical lithography
EtOx 2-ethyl-2-oxazoline
equiv. equivalence
-g- -graft-
GC gas chromatography
GPC gel permeation chromatography
iPrOx 2-iso-propyl-2-oxazoline
IPOx 2-isopropenyl-2-oxazoline
IR infrared
LCROP living cationic ring-opening polymerization
LCST lower critical solution temperature

VI
MDR multiple drug resistance
MeOTf methyl trifluoromethane sulfonate
MeOx 2-methyl-2-oxazoline
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
n-BuLi n-buthyl lithium
NonOx 2-n-nonyl-2-oxazoline
nPrOx 2-n-propyl-2-oxazoline
OTf Triflate (trifluoromethanesulfonate)
P poly
PBS phosphate buffered saline
PDI poly dispersity index
PEG poly(ethylene glycol)
PEO poly(ethylene oxide)
pipd piperidine
PIPOx poly(2-isopropenyl-2-oxazoline)
PIPOxA poly(2-isopropenyl-2-oxazoline) prepared by anionic polymerization
PIPOxR poly(2-isopropenyl-2-oxazoline) prepared by free radical polymerization
PMMA poly(methyl methacrylate)
PNAAm poly(N-alkyl acrylamide)
PNIPAAm poly(N-isopropyl acrylamide)
POMA poly(octadecene-alt-maleic anhydride)
POx poly(2-oxazoline)s
PS poly(styrene)
RBITC rhodamine B isothiocyanate
SAM self-assembled monolayer
SEM scanning electron microscopy
SIP surface-initiated polymerization
SIPGP self-initiated photografting and photopolymerization
SIPP surface-initiated photopolymerization
SPM scanning probe microscopy
St styrene
THF tetrahydrofuran
TFA trifluoroacetic acid
UCST upper critical solution temperature

VII
UV ultraviolet
cm centimeter
°C degree Celsius
eV electron volt
g gram
h hour
H heterotactic
hd dry polymer layer thickness
initial concentration of the initiating group
I isotactic
apparent propagation rate
kDa kilo Dalton (1000g/mol)
L liter
m meter
[M]0 initial monomer concentration
[M]t monomer concentration of the time
mbar millibar
min minute
mmol millimole
mol % molar percent
Mn number average molar mass
Mw weight average molar mass
NAv avogadro constant
ng 10-9
gram
nm 10-9
meter
Rg radius of gyration
S syndiotactic
tp polymerization time
T temperature
v/v volume ratio
w/w weight ratio
wt % weight percent

VIII
λ wavelength
μm micrometer
ρ bulk density
ζ grafting density

Chapter 1 Introduction
1
Chapter 1 Introduction
Driven by the desire to produce novel materials for new applications, polymer science as an
interdisciplinary field came into being in 17th
century. Since then, polymer has been used in a
wide range of applications, such as coatings, adhesives, plastics, elastomers, and natural
biopolymers e.g. DNA and proteins that are essential for life. A key feature of successful and
versatile polymer materials is that it is possible to build in properties by control, including the
control of the molecular architecture, and chemical functionality. For example, the progress in
living/controlled polymerization techniques has enabled the preparation of polymers with well
defined molecular architecture. Figure 1 shows different possible polymer architectures.
Figure 1. Different possible polymer architectures, linear, star, hyperbranched, brush polymer
and dendrimer.
A linear polymer is a macromolecule in which the monomers are linked in one continuous
string. A branched polymer may be a star polymer, graft/brush polymer and
hyperbranched/dendritic polymer. A star polymer is defined as a macromolecule containing
one single branch point from which linear chains (arms) emanate. A dendritic polymer, being
a perfectly hyperbranched polymer, is monodisperse macromolecule and has a completely
regular branching structure leading to a further branching. A brush polymer, also called

Introduction Chapter 1
2
molecular brush is copolymer containing a high density of side chains, which leads to
significant congestion and stretching of the backbone. Recently, branched polymer has gained
much attention due to its interesting and sometimes unexpected properties opening new areas
of applications.
1.1 Molecular brushes
Molecular brushes1,9
are unique polymeric molecules. They are also referred to as bottle brush
macromolecules because of their appearance. The conformation and physical properties of
molecular brushes are mainly controlled by the steric repulsion between grafted side chains
because of high crowding. Molecular brushes of different conformations have been prepared
in the past decade. Figure 2 presents the conformational change of molecular brushes with
the increasing grafting densities. Figure 5 shows some other factors that can also determine
the conformations, e.g. flexible or stiff side chains and backbone, homopolymers or
copolymers side chains, linear or branched in side chains and backbone. All these variables
affect the conformation and properties, but it is believed that the dense grafting of pendant
chains has the greatest effect. Dense grafting enables a formation of entropically unfavorable
elongated backbone chain, and at the same time, ensures the high density of functional groups
on the side chain along the backbone. The unique structure endows molecular brushes with
novel properties, facilitating its potential applications as sensors,2 elastomers
3,4 and template
for nanowires.5
Bottle brush structures can also be found in nature. Proteoglycans,6
for instance, are
polyelectrolyte brush-like macromolecules that consist of a protein backbone with
carbohydrate side chains (Figure 3). These bottle brush molecules widely exist in the
connective tissue and function from cell signaling and cell surface protection to joint
lubrication and lung clearance. In addition, enlarged „molecular brush‟ such as callistemon,7
also known as bottlebrush is a genus of shrubs and trees found in Australia.

Chapter 1 Introduction
3
Figure 2. Conformational change of molecular brush as a function of grafting density.
Figure 3. Structural hierarchy of proteoglycan aggregate in cartilage.8 (a) Transmission
electron microscopy (TEM) micrograph of the aggregate, (b) enlarged TEM micrograph of
protein backbone with glucosaminoglycan side chains, (c) chemical structure of the
disaccharide repeating unit of the side chains (From Ref. [9]).
1.1.1 Synthesis of molecular brushes
As comb copolymers, molecular brushes can be prepared by the grafting through approach
(polymerization of macromonomers), grafting onto approach (polymer analogue coupling of
polymers onto a backbone with pendant attachment groups), or the grafting from approach
(polymerization from a macroinitiator backbone). All the three strategies have been
successfully employed for the synthesis of defined molecular brushes. However, depending
on the functionality and polarity of the monomers, each approach has its advantages and
limitations.9
For example, the grafting through approach ensures side chain attachment to
every backbone unit, thus leads to the formation of molecular brush of high grafting density
(Figure 4). However, the synthesis of macromonomer is inevitable in this method and has
been proved to be very difficult. Moreover, the macromonomers are difficult to polymerize to

Introduction Chapter 1
4
a high degree and difficult to separate from molecular brushes. The grafting onto approach
involves the separate synthesis of the backbone and side chain by different living
polymerization techniques (Figure 4). Therefore, the resulting molecular brushes are
structurally well defined with respect to their backbones and side chains. However, a limited
grafting density could be easily found out due to the steric interaction between the grafted
side chain and the approaching side chains. The grafting from approach involves the synthesis
of a backbone polymer (macroinitiator) with initiation sites along the backbone, which is
subsequently used to initiate polymerization of the side chains (Figure 4). The macroinitiator
can be synthesized directly by the polymerization of monomers with initiating moieties or by
synthesis and conversion of a precursor to macroinitiator with initiating moieties. The gradual
growth of the side chains impairs the steric concerns which are often the limiting factors in
the grafting through and grafting onto methods. Therefore, the grafting from approach allows
for the preparation of molecular brushes with a high grafting density and a narrow molecular
weight distribution. However, compared to the grafting through approach, grafting from has
relatively less control of side chain length and grafting density.
All of the above mentioned methods aim at controlling different structural parameters,
including chemical composition, grafting density, degree of polymerization (DP) of side
chains and the backbone. Even though each of these strategies demonstrates distinct
advantages with respect to the molecular design, the grafting from strategy is the most
broadly applied method for the synthesis of densely grafted polymers.35

Chapter 1 Introduction
5
Figure 4. Pathways for synthesis of molecular brushes: „grafting through‟, „grafting onto‟
and „grafting from‟ („I‟ is an initiating group).
Within each strategy, various polymerization techniques such as anionic polymerization, ring-
opening (metathesis) polymerization, conventional and controlled radical polymerizations,
and various coupling reactions (for example „click chemistry‟) have been employed.
Therefore, brush molecules with different architectures as well as different backbone and side
chain compositions have been successfully prepared (Figure 5). In the past decade, particular
attention has been paid to develop living ionic as well as controlled radical polymerization
techniques, which have been proven particularly useful and effective for the preparation of
well-defined, functional molecular brushes. It is worth mentioning that controlled radical
polymerization techniques are suitable for the synthesis of molecular brushes due to the
employed low radical concentration, thereby intermolecular and intramolecular termination
reactions are suppressed. This is especially important for brush polymerizations because of
the high local concentration of side chains that exist in the vicinity of the backbone polymer.

Introduction Chapter 1
6
Figure 5. Various branching topologies and architectures of molecular brushes (From Ref.
[9]).
So far, such brush-like molecules have mainly been prepared via atom transfer radical
polymerization (ATRP).10,11 ATRP is an example of a controlled/living radical polymerization
where carbon-carbon bond is formed through transition metal catalyst. ATRP reactions are
tolerant to many functional groups like allyl, amino, epoxy, hydroxyl and vinyl groups.12
This
method prevails in many aspects, such as the ease of preparation, the commercial availability
of the reactant, including catalyst (usually copper complexes), pyridine based ligands, and
initiators (alkyl halides). In addition, the ease of forming pendant -bromoester groups on a

Chapter 1 Introduction
7
polymer backbone and grafting side chains from the multifunctional initiator also make ATRP
a suitable method for molecular brush synthesis. However, the steric interactions between the
initiation sites and the rather bulky ligands used in ATRP will inevitably affect the grafting
efficiency.13
Matyjaszewski and co-workers prepared molecular brushes using the grafting
from method via ATRP, and compared the initiation efficiency of brush molecules with that
of linear polymers (Figure 6).13
They found a significantly reduced initiation efficiency for
the molecular brushes which was attributed to the congested environment and high local
concentration of initiation sites. However, it should be noted, for a defined synthesis of
molecular brushes, a high initiation efficiency is needed.
Figure 6. ATRP for the synthesis of molecular brushes. Left: Mechanism of metal complex-
mediated ATRP; Right: Initiation efficiency as a function of monomer conversion for the
brush and linear polymerizations (From Ref. [13,14]).
Up to now, molecular brushes based on poly(methacrylate)s or poly(styrene) (PS) backbones
and various side chains such as poly(acrylate), poly(methacrylate), PS, poly(N-isopropyl
acrylamide) (PNIPAAm) and sugar have been prepared via ATRP. Meanwhile, molecular
brushes carrying different functionalities as well as different block copolymer side chains to
form core-shell structures have been synthesized. The topologies of these molecular brushes
are shown in Figure 5.

Introduction Chapter 1
8
1.1.2 Stimuli-responsive molecular brushes
1.1.2.1 Stimuli-responsive polymers
Stimuli-responsive polymers15
are defined as polymers that undergo large and abrupt physical
or chemical changes in response to the external environment changes. These so called
„responsive-polymers‟ have been given as stimuli-sensitive,16
intelligent,17
smart18 , 19
or
environmentally sensitive polymers.20
The stimuli could be either chemical or physical nature.
Chemical stimuli such as pH, ionic strength or another chemical will change the interactions
between polymer chains or between polymer chains and solvent molecules. Whereas, physical
stimuli refers to the alteration of molecular interactions at critical onset points with
temperature, electric or magnetic fields, and mechanical stress. These responses of polymer
systems are very useful in bio-related applications such as drug delivery21,22 ,23 ,24 ,25
and
biotechnology. Some systems have been developed to combine two or more responsive
mechanisms into one polymer system. For instance, temperature-sensitive polymers may also
respond to pH changes.26,27, 28,29
Temperature is the most widely used stimulus for responsive polymer systems. One unique
property of temperature-responsive polymers is the presence of a critical solution temperature.
Therefore, hydrophilic polymers with a lower critical solution temperature (LCST) have
gained much attention. These polymers are soluble in water through hydrogen bond with
water molecules below their LCST. Increasing the temperature impairs the formation of
hydrogen bond. When temperature is above the LCST, polymers dehydrate and become
insoluble in water as the hydrophobic interactions become entropically favorable (Figure 8).
For example, poly(N-isopropylacrylamide) (PNIPAAm) has a cloud point of around 32 °C,
across which it undergoes a reversible volume phase transition caused by the coil-to-globule
transition. However, the mixture of polymer and solution is miscible when the temperature is
below the LCST. The phase transition is favored by the entropic gain as water molecules
associated with the side chain (isopropyl moieties) are released into the bulk aqueous phase.

Chapter 1 Introduction
9
Figure 7. Thermo-responsive polymer below and above its LCST in water (From Ref.[30]).
The phase transition temperature can be adjusted by changing structural parameters such as
molecular weight,31 , 32
end groups,32, 33
and molecular structure.34
According to previous
studies, the LCST usually decreases with the increased molecular weight due to an attenuated
polymer-solvent interaction. The incorporation of hydrophobic groups in the polymer causes a
decrease of the LCST. The hydrophilic/hydrophobic balance can be additionally changed by
the end groups.
Figure 8. Illustration of reversible temperature-responsive solubility of linear polymers at
their lower critical solution temperature (LCST). Left: hydrated polymer below its LCST and
right: single chain collapse and intermolecular aggregate due to loss of water above the LCST.
1.1.2.2 Thermo-responsive molecular brushes
In solution, the length and flexibility of the molecular brush is mainly controlled by the
solvent quality, temperature, pH, light and ionic strength. The ability to trigger these
conformational changes using various stimuli will greatly enhance the significance and
practical value of these molecules. Actually, one focus of the related research of molecular
brushes is the development of stimuli-responsive adaptive systems,35
especially hydrophilic
molecular brushes with a lower critical solution temperature (LCST) in aqueous environments

Introduction Chapter 1
10
for biomedical applications such as biosensors and drug delivery systems. 36,37,38,39,40,41,42,43,
44,45
The extended conformation of molecular brushes in solution can be adjusted by incorporation
of stimuli-responsive segments. At the transition temperature, the polymer brushes not only
show the change of the contour length, they also show a transition from rod-like to globular
conformation. It is known that linear polymers become insoluble and aggregate at their LCST
due to hydrophobic interactions. Unlike linear polymers, it is very interesting that after
collapsing, the brush molecules do not aggregate and remains in solution as single
molecules.36
These thermo-responsive molecular brushes have been widely investigated in the past decade.
The first example was reported by Schmidt and coworkers in 2004.36
Molecular brushes with
thermo-responsive polymer side chains were prepared by ATRP of 2-hydroxyethyl
methacrylate (HEMA) followed by subsequent reaction with bromoisobutyric acid bromide to
form a poly-2-bromoisobutyryloxyethyl methacrylate macroinitiator followed by a second
ATRP of (N-isopropylacrylamide) (NIPAAm) to form the side chain. The resulting molecular
brushes collapsed in aqueous solution at LCST of the PNIPAAm side chains. The
conformation of the brush molecular showed a transition from a single macromolecule
cylinder to a globule within a small temperature range (collapse from a radius of gyration
from 61nm at 20 °C to 25 nm at 32 °C).
Like linear thermo-responsive polymers, the LCST of molecular brushes can be tuned by
varying the hydrophilicity of the backbone and/or side chains. The simplest way to achieve
this fine tuning is a copolymerization of hydrophilic and hydrophobic monomers of the side
chain of the molecular brushes. For instance, molecular brushes with statistical or block
copolymers of di(ethylene glycol) methyl ether methacrylate (MEO2MA) (hydrophobic) and
tri(ethylene glycol) methyl ether methacrylate (MEO3MA) (hydrophilic) in the side chain
have been prepared by grafting from a poly-2-bromoisobutyryloxyethyl methacrylate
macroinitiator by ATRP.40
In the case of brushes with statistical copolymer side chains, the
LCST increased with the mole fraction of MEO3MA in the side chain. However, very
interestingly, for the molecular brushes with block copolymer side chains, the LCST curve of
the block brushes showed two stages during heating, exhibiting both intermolecular and
intramolecular aggregation (Figure 9). This behavior was strongly dependent on the sequence
of the side chain segments. As the temperature increased, molecules consisting of collapsed
PMEO2MA (core) and PMEO3MA (shell) segments aggregated to precipitate as larger

Chapter 1 Introduction
11
aggregates. However, brush molecules with PMEO3MA-b-PMEO2MA side chains formed
vesicles consisting of aggregated PMEO2MA shell segments and soluble PMEO3MA core
segments.
Figure 9. Temperature dependence of the hydrodynamic diameter change for 0.3 wt %
aqueous solution of block molecular brushes. Solid curve: P(PMEO3MA-b-PMEO2MA) brush.
Dashed curve: P(PMEO2MA-b-PMEO3MA) (From Ref. [40]).
Another interesting phenomenon is the unusual dependence between aggregation and
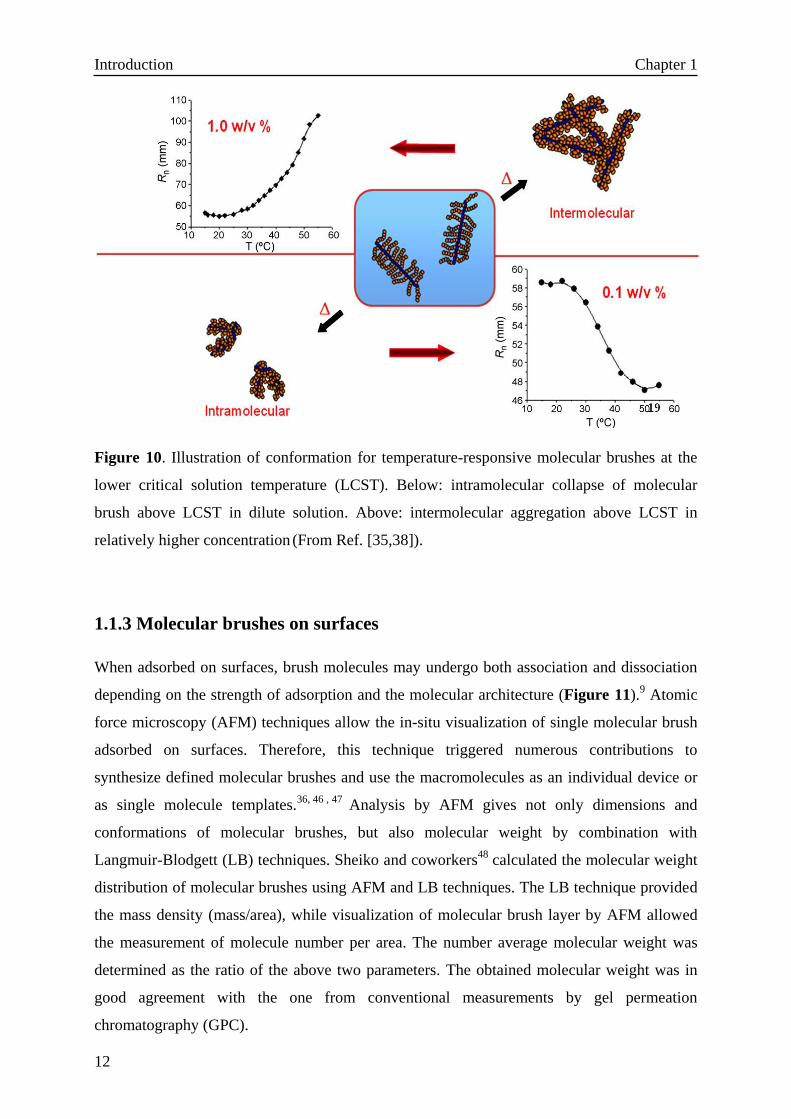
concentration. Matyjaszewski and co-workers38
prepared two thermosensitive molecular
brushes with thermo-responsive side chains. As shown in Figure 10, dynamic light scattering
(DLS) studies were performed for aqueous solutions of molecular brushes below and above
the LCST, and found an unusual concentration-dependent LCST. Due to the compact
structure of molecular brushes, intramolecular collapse occurred when the average distance
between brush molecules was much larger compared to the hydrodynamic dimensions of the
individual macromolecules. However, when the concentration of the solution of molecular
brushes was increased to a level where the separation distance was comparable to the brush
hydrodynamic dimensions, intermolecular aggregation occurred, which is typically observed
for solutions of linear polymers.
Introduction Chapter 1
12
Figure 10. Illustration of conformation for temperature-responsive molecular brushes at the
lower critical solution temperature (LCST). Below: intramolecular collapse of molecular
brush above LCST in dilute solution. Above: intermolecular aggregation above LCST in
relatively higher concentration (From Ref. [35,38]).
1.1.3 Molecular brushes on surfaces
When adsorbed on surfaces, brush molecules may undergo both association and dissociation
depending on the strength of adsorption and the molecular architecture (Figure 11).9 Atomic
force microscopy (AFM) techniques allow the in-situ visualization of single molecular brush
adsorbed on surfaces. Therefore, this technique triggered numerous contributions to
synthesize defined molecular brushes and use the macromolecules as an individual device or
as single molecule templates.36, 46 , 47
Analysis by AFM gives not only dimensions and
conformations of molecular brushes, but also molecular weight by combination with
Langmuir-Blodgett (LB) techniques. Sheiko and coworkers48
calculated the molecular weight
distribution of molecular brushes using AFM and LB techniques. The LB technique provided
the mass density (mass/area), while visualization of molecular brush layer by AFM allowed
the measurement of molecule number per area. The number average molecular weight was
determined as the ratio of the above two parameters. The obtained molecular weight was in
good agreement with the one from conventional measurements by gel permeation
chromatography (GPC).

Chapter 1 Introduction
13
Figure 11. AFM height images along with schematics demonstrate four distinct
conformations observed for molecular brushes adsorbed on different surfaces. The
conformational transformations occur due to variations in the fraction of adsorbed side chains
(From Ref. [9]).
The unusual properties of brush macromolecules on surfaces arise from their ability to change
conformation in response to the substrate of varied surface energy as well as the lateral
compression. Recently, Sheiko and co-workers49
used ATRP to prepare the polymer
backbone with more than 2000 2-hydroxyethyl methacrylate units and side chains containing
140 n-butyl acrylate units. They successively deposited the brush-like molecules on the
surfaces of various liquid and solid substrates. As the densely grafted side chains adsorbed,
they underwent steric repulsion which created tension in the backbone. This tension, was
controlled by the grafting density, the side chain length, and the extent of substrate attraction.
After adsorption of molecular brushes, the side chain adsorption-induced tension may
overcome the covalent bond strength and cause molecular fracture via C-C bond scission
(Figure 12).49,50
The scission of the backbone was verified by the observation of shortened
brush molecules.

Introduction Chapter 1
14
Figure 12. (a) AFM height images of the molecular brush with long side chains (n = 140)
were measured at different exposure times to the water/propanol (99.8/0.2 wt/wt) substrate. (b)
The molecules became shorter with greater exposure time to the substrate, which was
attributed to spontaneous scission of the polymer backbone (From Ref. [49]).
1.2 Polymer Brushes
Polymer brushes are ensembles of polymer molecules that are terminally attached to an
interface. Polymers can be anchored to a surface either by physical e.g. electrostatic
adsorption or chemical linkage. As one of the coating techniques, application of polymer
brushes has extended much beyond the conventional decoration and protection aspects and
becomes a popular technology in diverse high-tech areas ranging from computer chips51
to
biomedical surfaces.76 Investigation in polymer brushes is driven by the desire to control the
interfacial properties of the polymer layer and its compatibility with the environment.
Polymer chains of the polymer brushes exhibit different conformations on surfaces. If the
distance between two anchoring points is larger than the size of the surface-attached polymers,
the segments of the individual chains can not interact with each other and as a result polymer
chain will adopt a mushroom or pancake conformation (Figure 13). A completely different
situation is obtained if the polymer chains are attached to the surface at a short distance

Chapter 1 Introduction
15
between the anchor points, and the polymer chains overlap. In this case, the segments of the
chains try to avoid each other. At high grafting density, adsorbed polymers change their
conformation from a random coil to an extended chain (Figure 13).
Figure 13. Schematic illustration of polymer brushes with different grafting densities
prepared by „grafting to‟ and „grafting from‟ methods (From Ref. [66]).
As shown in Figure 13, polymer brushes can be prepared by the grafting to or grafting from
method (mostly referred to as surface-initiated polymerization (SIP)). In the first method, one
or more anchor groups of polymer chain can attach to the substrate. The grafting to method is
difficult to achieve high grafting densities because of the screening of reactive surface sites by
already anchored polymers. Therefore, films generated by this method are limited to
thicknesses between 1 and 20 nm. The SIP from surface-bound initiators is a powerful
alternative to control the density of polymer brushes (Figure 14). Since the diffusing species
during the SIP are small monomer molecules (instead of macromolecules in the grafting to
method), a high grafting density can be achieved. Up to now, almost all the developed
polymerization techniques in solution have been successfully adapted for the SIP, such as free,
controlled and living radical polymerization, living anionic and cationic polymerization, and
ring-opening metathesis polymerization.76

Introduction Chapter 1
16
Figure 14. Scheme for the preparation of (structured) polymer brushes by surface initiated
polymerization (SIP). Polymer brushes can be prepared on either initiator-anchored surface
(e.g. self assembled monolayer (SAM)) or initiator-free surfaces. Functionalities can be
introduced by analogue reactions. Furthermore, structured polymer brushes can be obtained
on structured SAM by chemical lithography and microcontact printing. Multi-block
copolymer brushes can be realized by a variety of polymerization methods.
Within various strategies developed for the preparation of polymer brushes, the most popular
way is to grow monomers from or attach a polymer chain to a surface-bonded initiator system.
Defined initiator systems can be prepared by the modification of the substrate with a self-
assembled monolayers (SAMs) of bi-functional molecules bearing a surface grafting function
and an initiator function for the SIP (Figure 14). SAMs can be formed on almost any surface,
as long as the anchor group is correctly chosen.52
Due to the versatility of such system,
various surface coupling strategies have been developed during the last decades. One way is
that polymer brushes were bonded to gold substrate by thiols53 , 54
or to silica by silane
functions.55,56
However, these SAM systems have a limited chemical/thermal/UV stability and
make them inappropriate for long-term applications. For instance, silane-based systems on
oxides are prone to hydrolyze and have a poor stability in saline solutions at 37 °C, thus
impairing their use for many biomedical applications.57,58
Thiol-based coatings are mainly
SIP
Structured Monolayer
SIP
SIP
Monolayer
Substrate
Polymer Brushes
with Functional Groups
Multi-component Polymer Brush
Surface
Active Group
Initiator
SIP
Structured Monolayer
SIP
SIP
Monolayer
Substrate
Polymer Brushes
with Functional Groups
Multi-component Polymer Brush
SIP
Structured Monolayer
SIP
SIP
Monolayer
Substrate
Polymer Brushes
with Functional Groups
Multi-component Polymer Brush
Surface
Active Group
Initiator

Chapter 1 Introduction
17
limited to coinage metal substrates, and their limited thermal59
and UV stability60
are well-
known. Therefore, more effective and stable systems are demanded for the preparation of
polymer brushes.
1.2.1 Self-initiated photografting and photopolymerization (SIPGP)
In the 1990s, Rånby and coworkers61
developed a photografting polymerization procedure to
prepare polymer grafts. A crosslinked polymer substrate such as poly(ethylene) (PE),
poly(ethylene terephthalate) (PET), nylon, poly(propylene) (PP) and poly(vinylchloride)
(PVC) were submerged in a solution of benzophenone in bulk vinyl monomer. Benzophenone
here acts as a photosensitizer. After irradiation by UV light of a spectral distribution between
200 and 400 nm, 2-5 μm thick polymer layers were formed on the substrates. This one-step
procedure has the advantage of e.g. formation of thick and dense polymer grafts directly on
polymer substrates without the immobilization of initiators. Later, it was shown that a broad
variety of vinyl monomers can be directly grafted onto organic substrates such as polyolefines
in the absence of benzophenone by self-initiated photografting and photopolymerization
(SIPGP).62 , 63
In this case, the substrate was immerged only in bulk monomer and then
irradiated with UV-light, resulting in the formation of polymer brush on the organic substrates.
Here, the presence of benzophenone was no more necessary and a mechanism of SIPGP of
vinyl monomer (e.g. styrene)64
was proposed and described as follows. As shown in Figure
15, styrene absorbs a photon and reaches its excited state. This excited single state can
transform through intersystem crossing (ISC) to its excited triplet state, which is in
equilibrium with a biradical form. The biradical can subsequently initiate a free radical
polymerization in solution. Besides, it can also abstract a hydrogen from the substrate. As a
consequence, the radical formed on the substrate initiates the free radical surface-initiated
polymerization. Therefore, defined, stable, and homogeneous polymer brush layers can be
prepared directly onto surface containing abstractable groups. From the above mentioned
procedure, it was found that the formation of defined, reactive interlayer such as a SAMs is
not necessary, and surface-initiated polymerization of vinyl monomers can be performed and
directly graft polymer onto surfaces. However, it should be noted that hydrogen can only be
abstracted from surfaces when the triplet state of monomer possess enough energy. Brown63
and Fang65
calculated the excited states of acrylic acid, revealing a potential energy of 112.5
kcalmol-1
for the T3 triplet state of acrylic acid (T1: 71.4 kcalmol-1
; T2: 86.9 kcalmol-1
). In
this case, the hydrogen could be abstracted from the PE substrate due to the T3-state potential

Introduction Chapter 1
18
energy of acrylic acid is higher than the C-H bond dissociated energy (BDE) on PE substrates
(the BDE of primary, secondary and tertiary hydrogen atoms for PE is 100, 96 and 93
kcalmol-1
).
Figure 15. Principle of the self-initiated photografting and photopolymerization of styrene on
a polymer substrate (From Ref. [66]).
Recently, Jordan and co-workers found that structured, extremely stable and homogenous
polymer brush of controlled morphology can be prepared directly onto carbonaceous
materials by the SIPGP of vinyl monomers.67,68,69
First, a stable ultrathin (around 1 nm)
template layer of carbonaceous material was locally deposited on an inorganic substrate by
means of a focused electron beam. Amplification of the template by surface-initiated
polymerization resulted in polymer brush layers of a controlled three dimensional shape. The
dependence between the polymer layer thickness and the amount of locally deposited carbon
allowed the preparation of complex 3D polymer architectures on surfaces. Taking advantage
of the high thermal and chemical stability of the grafting, polymer brushes were further
modified by polymer analogue reactions under drastic conditions such as nitration or
sulfonation of poly(styrene) (PS) brushes. By this general approach, stable polymer brushes
having all kinds of dimensions, architectures and chemical functionalities can be prepared on
various substrates (Figure 16).
hν ISCHC CH2
HC CH2
HC CH2
S T
HC CH2
T HC CH2
HC CH3
+ P
HC CH3
+ P
HC CH2
P + n grafted polystyrene
HC CH2
HC CH2
polystyrene in solution+ n
HC CH2
+ PH

Chapter 1 Introduction
19
Figure 16. Structured polymer brushes by carbon templating on various substrates. a) A step
pyramid of polystyrene (PS) brushes on silicon nitride created from a carbon template of
concentric squares written with different electron doses (top: irradiation scheme, below: AFM
image); b) AFM and height analysis of „STAIRS‟ templated on a silicon substrate with lines
of 100 nm width and increasing electron dose for each letter (1, 2, 4, 8, 12, 16 mC/cm2). PS
brush lines of 6, 10, 16, 35, 44 and 53 nm height, and widths were characterized between 200
and 350 nm; c) AFM image of PS brush objects of different sizes and shapes on a bare
gallium arsenide substrate (maximum structure height: 0.5 µm). The variation of the polymer
brush thickness is due to the change of the polymer grafting density, which is caused by the
different applied electron beam dosage (From Ref. [68]).

Introduction Chapter 1
20
1.2.2 Bottle-brush brushes (BBBs)
Using atom transfer radical polymerization (ATRP), Baker and co-workers70
prepared
polymer brushes of poly(methyl methacrylate) (PMMA) through the polymerization of MMA
from a surface-bound initiator on gold. It was found that only 10 % of the surface-bound
initiators actually initiated the polymerization, so that many initiator sites remained on the
surface and the produced polymer brushes only exhibited a low grafting density. The
insufficient initiation could be explained by the partial screening of initiators by the grafted
polymer chains, or the loss of initiator through termination and chain transfer reactions.71,72
This indicates that it is difficult to obtain polymer brushes with extended polymer chains
using ATRP or free radical polymerizations.
An effective strategy to generate polymer brushes with extended polymer chains is the
preparation of branched i.e. so called „bottle-brush brushes‟ (BBBs) structures from surfaces.
For instance, as shown in Figure 17, 176 nm-thick poly(hydroxyethyl methacrylate)
(PHEMA) brush was created on gold. After functionalization of the pendant hydroxyl groups,
a 358 nm-thick polymer film was obtained. The drastic increase in thickness was due to the
mass increase of repeating unit and the subsequent stretching of the main chain.
Figure 17. Extension of PHEMA brushes by functionalization of side chains to form bottle-
brush brush structures (From Ref. [76]).
Until now, only very few research groups have reported on BBB systems.73,74,75,76
However, it
is of certain importance to study on this topic in depth since it has a promising future in

Chapter 1 Introduction
21
biological applications. The complex polymer architectures intriguingly resemble the
structure of various polyglycans that is found on nearly every living cell. It has been shown
that carbohydrates in the form of polysaccharides, glycoproteins, glycolipids and other
glycoconjugates play essential roles in many biological processes. They serve as recognition
sites for the cell and contribute to the steric repulsion which prevents undesirable nonspecific
adhesion.77
Actually, highly aggregated surface-tethered carbohydrate ligands e.g. the
glycocalyx are dominated by glycosylated molecules on the cell surface, resulting in not only
the enhancement of binding strength in specific recognition of proteins but also the
minimization of nonspecific protein adsorption.73
Non-toxic BBBs structures as biomimetic
functional soft interfaces between solid semiconductors and biological systems may find
direct applications for designing advanced biomedical devices.
1.3 Bioactive surfaces
Protein adsorption78,79
at interfaces widely exists in nature ranging from food processing to
biomedical science. Preventing adsorption of proteins80,81
is a key for the performance of
medical devices and all applications including biosensors,82
cell culture,83
biomedical
implants from hip replacement to contact lenses,84
etc. As a macro fouling (attachment of
larger organisms), marine fouling (Figure 18) is a constant problem for boats, ports and
anything kept in the sea for a period of time. Marine organisms are prone to adhere to almost
any type of surface. Modern method for preventing fouling is to use paints containing metals
such as copper or tin, as well as herbicides. However, these metals are toxic and cause severe
environmental problems. Alternatives for reducing the unwanted bio fouling is to graft self-
assembled monolayer (SAM) or antifouling polymers onto surfaces.85,86,88

Introduction Chapter 1
22
Figure 18. Macro fouling i.e. organisms accumulated on a ship hull, however, no significant
adsorption was observed on antifouling painting area (From Ref. [87]).
So far oligoethylene glycol monolayers on gold79, 88
or SiO289
have been the mostly
investigated SAMs for resisting non specific protein adsorption. The protein-resistant
efficiency of these short monodisperse molecular layers was reported to increase with the
length of the oligoethylene glycol. A minimum of two ethylene glycol units is necessary for
negligible protein adsorption.78
However, the limited chemical, thermal and UV stability of
these SAMs make them inappropriate for long-term applications under tough conditions.
Polymer brushes via surface-initiated polymerization (SIP) have triggered numerous
investigations of synthetic surface-bound polymers as non-fouling coatings. Up to now, the
SIP to prepare the protein-resistant surface is regarded as one of the best methods due to the
versatility of monomers and stability.
In spite of the various reported criteria for the design of antifouling polymers, some common
parameters i.e. the presence of flexible hydrophilic groups and highly branched architecture
can lead to effective protein resistance. So far, a variety of polymers including nonionic,90
zwitterionic,91, 92
peptidomimetic93
and polysaccharides94,95,96
polymers have been developed
to resist nonspecific protein adsorption and have gained some success in in vitro and in vivo.
However, to date, very few effective non fouling surfaces meet the practical demands of a
daily application. In the following part, some promising polymers for non-fouling surfaces are
introduced.
Poly(ethylene glycol) (PEG) and PEG-based polymers are widely studied polymers to prevent
the adsorption of proteins and cells.97
However, current PEG technology has major limitations

Chapter 1 Introduction
23
for long-term applications. PEG coatings can undergo oxidative degradation and lose their
function when placed in in vivo.98,99,100
Moreover, PEG system is short of functional groups
for ligand immobilization which is required for many applications. The introduction of
additional functional groups into PEG may, however, alter its properties.101
In a word, there is
continuous need to develop new and effective system to overcome such problems.
Zwitterionic polymers are macromolecules with oppositely charged groups located at the
chain ends. Polymers incorporating zwitterionic molecules such as phosphatidylcholines102
and carboxybetaine methacrylate103
are promising for anti-biofouling surfaces. It is believed
that zwitterionic polymers resist nonspecific protein adsorption with either a mixture of
anionic and cationic terminal groups based charge interactions or a strong hydration of the
polymer through ionic salvation.
Surfaces coated with oligosaccharide grafted polymers mimic the antifouling glycocalyx,94
which directs the specific interactions such as cell-cell recognition and prevents the
undesirable non-specific adhesion of other molecules and cells. In addition, dendritic
saccharide surfactant polymers were prepared by Marchant95
as antifouling interface materials
to reduce platelet adhesion. In this case, the hexyl side chains facilitate the adsorption of the
surfactant polymers onto hydrophobic substrates, while the maltose dendron side chains
provide a dense canopy of protective glycocalyx-like layer as an antifouling interface that
suppresses platelet adhesion.
Poly(2-methyl-2-oxazoline), a peptide-like hydrophilic polymer, has showed very promising
properties in biological application since the first demonstration of its biocompatibility by
Goddard.124
Recently, Konradi and co-workers investigated a comb polymer consisting of
poly(2-oxazoline) side chains and a polycationic backbone as the protein-repellent polymer.
The comb polymer adsorbed on the negatively charged surface through static charge
interaction forming a polymer coating. Such coatings with an optimal side chain grafting
density restrict a protein adsorption as low as 2ng/cm2, which is quantitatively equal to the
protein-repellent property of PEG coatings. However, it should be noted that the electro-static
adsorbed coating is prone to desorption at certain conditions i.e. in salt water, and thus
covalent attachment is preferred.

Introduction Chapter 1
24
1.4 Poly(2-oxazoline)s
1.4.1 2-Oxazoline
Oxazolines are five-membered heterocyclic compounds. Depending on the location of the
double bond, three different oxazolines can be classified. Among them, 2-oxazoline is the
most investigated and widely used compound (Figure 19).
Figure 19. 2-Oxazoline, 3-oxazoline and 4-oxazoline (from left to right).
1.4.2 Living cationic ring-opening polymerization (LCROP)
The 2-oxazoline ring can be opened and polymerized to form a polypeptide-analog polymer,
which is known as ring-opening polymerization. In 1966, Kagiya et al. reported the first time
on the ring-opening polymerization of 2-oxazolines.104
It was shown that the polymerization
reaction of 2-oxazolines was not disturbed by chain transfer and termination under
appropriate conditions. In the following decades, many reports showed that depending on the
type of initiator, the mechanism of chain growth can be cationic, or covalent. When cationic
initiators such as methyl triflate are used, the polymerization is cationic. The cationic
propagating species of the oxazolinium salt is stable. Therefore, it was conveniently employed
in the synthesis of block copolymers and end-functionalized polymers. The living cationic
ring-opening polymerization (LCROP) behavior of 2-oxazolines having a variety of 2-
substituents has been extensively investigated by Saegus and Kobayashi et al.105,106,107
If the
monomer reacts with more nucleophilic alkyl halides, e.g. methyl iodide, the polymerization
is slow and proceeds via a different mechanism, namely the covalent mechanism.
The general mechanism of cationic polymerization for initiation and propagation of 2-
oxazolines is shown in Figure 20. The nucleophilic nitrogen of the 2-oxazoline ring can

Chapter 1 Introduction
25
convert to oxazolinium cation under the attack of electrophiles such as alkylating agents, or
Lewis and Brønsted acids. The resulting oxazolinium ring can further react with another 2-
oxazoline so that the ring is opened and a linear amide structure is formed. This process can
be repeated until a nucleophilic terminating reagent terminates the polymerization.
Figure 20. Mechanism of cationic ring-opening polymerization of 2-oxazolines.
It is generally believed that increasing the nucleophilicity of the initiator counter ion and
monomer decreases the tendency of the living cationic ring-opening polymerization (LCROP)
to be ionic. With the counter ion of non-nucleophilic trifluoromethylsulfonate (triflate, OTf),
the propagating species is the cation of any type of 2-oxazoline and the propagation rate
increases significantly.108,109
Methyl trifluoromethylsulfonate (MeOTf) as a commonly used
initiator shows many advantages in LCROP of 2-oxazolines. Due to its high reactivity,
MeOTf reacts fast with 2-oxazolines even below room temperature and the propagation starts
at temperatures above 40 °C. Fast and quantitative initiation with respect to the propagation of
polymerization is crucial when preparing well-defined polymers with narrow molar mass
distributions. Since the initiation is nearly instantaneous and the formed oxazolinium triflate
salt is sterically and electronically similar to the propagating polymer chain end, poly(2-
oxazoline)s (POx) with low polydispersity indices (i.e. Mw/Mn of 1.02~1.20) can be prepared.
However, in spite of the living character of the cationic polymerization, side reaction can
occur via chain transfer reactions especially at high temperatures (T>120 °C) and at very low
[M]0/[I]0 ratios. Hoogenboom and co-workers110
found a shoulder in the GPC traces at high
molar mass region of poly(2-ethyl-2-oxazoline)s (PEtOx). This might be due to the
occurrence of chain transfer reactions followed by chain coupling at high monomer

Introduction Chapter 1
26
conversions as already reported earlier by Litt and coworkers.111
Moreover, yellow
polymerization mixtures were obtained at low monomer concentrations ([M]<3 M), indicating
the occurrence of side reactions. An optimum monomer concentration between 4 M and 7 M
was established for the living cationic ring-opening polymerization of 2-ethyl-2-oxazoline
initiated with benzyl bromide in N,N-dimethylacetamide at 100 °C.
The living character of the polymerization of 2-substituted 2-oxazoline provides synthetic
possibilities to tailor macromolecules with a broad variety of architectures, composition,
numerous side and end functions as well as the ease of preparation of block, gradient and
random copolymers. Therefore, these polypeptide-like polyamides of various architectures
have found many early-stage or potential applications as stabilizers, compatibilizers and
thermo-settings.112
In the past decades, the biocompatibility as well as stealth
behavior113 , 114 , 115
of POx triggered the application of POx in the biomedical field.116
Moreover, water soluble POx with a lower critical solution temperature (LCST) broadened
the research area for thermo-responsive or so called „smart‟ materials. Figure 21 shows POx
of various structures and their biomedical applications.
Figure 21. Poly(2-oxazoline)s have potential use as biomaterials and thermo-responsive
materials. In addition, the easy access to hydrophilic, hydrophobic, and fluorophilic polymers
by simply changing the side chain or by the combination of click chemistry provides the
possibility to prepare a range of polymer structures and the self-assembly of copolymers
(From Ref. [117]).

Chapter 1 Introduction
27
1.4.3 Thermo-responsive poly(2-oxazoline)s
Recently, hydrophilic poly(2-oxazoline)s (POx) came into focus as temperature-responsive
polymers. Compare to the well-studied PNIPAAm and PEG systems, POx prepared by living
cationic ring-opening polymerization (LCROP) of 2-oxazolines have several advantages e.g.
ease of access of a broad variety of architectures, composition, and end functionalities.
Additionally, the lower critical solution temperature (LCST) of hydrophilic POx can be
tailored over a broad temperature range by means of the molar mass and composition
adjustment.
In 1988, Kwei and co-workers first investigated the thermo-sensitivity of aqueous solutions of
poly(2-ethyl-2-oxazoline) (PEtOx). It was shown that the LCST is dependent on the polymer
concentration, and cloud points in a range of 61°C-64°C were found for the molar mass of
500 kg/mol to 20 kg/mol.118
Since the first report of thermo-responsive POx, the LCST of
hydrogels, linear, homo- and coPOx were investigated in details as a function of the side
chain and/or end-group functionality as well as the molar mass and concentration. In 1992,
Uyama and Kobayashi161
reported that poly(2-isopropyl-2-oxazoline) (PiPrOx) exhibited a
cloud point in the range of 36-39 °C, which makes it a suitable candidate for biomedical
purposes. Recently, Park and Kataoka167
reported that poly(2-n-propyl-2-oxazoline) (PnPrOx)
also exhibited thermo-sensitivity in water, with a cloud point of 24 °C in a 1 wt% aqueous
solution. In addition, Schubert and co-workers168
finely tuned the LCST of poly(2-oxazoline)s
by varying composition and molar mass. The thermal transitions of these copolymers with
cloud points of ~34 °C showed no hysteresis or concentration dependence, making them
superior to PNIPAAm.
Besides homopolymers, the copolymerization of different 2-oxazoline monomers allows
accurate control over the LCST. Park and Katoaka171
reported the copolymerization of 2-
isopropyl-2-oxazoline (iPrOx) with the hydrophilic 2-ethyl-2-oxazoline (EtOx), which led to
a linear increase of the cloud point up to 67 °C as a function of the EtOx content of 75 mol %.
Copolymerization of iPrOx with the more hydrophobic nPrOx also resulted in a linear
dependence between cloud points and the composition. Recently, Jordan and co-workers
further decreased the cloud point of PiPrOx down to 9 °C by gradient copolymerization of
iPrOx with hydrophobic 2-n-butyl-2-oxazoline (BuOx) and 2-n-nonyl-2-oxazoline
(NonOx).119

Introduction Chapter 1
28
Furthermore, Jordan and co-workers investigated the effect of end groups with different
polarity on the cloud point of PiPrOx.33
It was found the LCST strongly depends on the
polarity of the terminal moiety especially for low molecular weight polymer. For instance, the
introduction of a hydrophobic unit e.g. n-nonyl decreased the cloud point to 28 °C as
compared to a terminal methyl group (cloud point = 47 °C). In addition, the location of the
amide groups in the POx and poly(N-alkyl acrylamide)s (PAAm) resulted in different LCSTs,
e.g. POx pendant group modification caused a stronger shift of LCST as compared to PAAm.
In addition, Schlaad‟s group tuned the LCST of copolymers of iPrOx and 2-(3-butenyl)-2-
oxazoline in a range of 5-90 °C by introducing various side chain functionalities through the
„click‟ reaction.120
Kim and co-workers121
investigated thermo-responsive micellar gels
formed by diblock PEtOx-b-Poly(ε-caprolactone) at low temperature. As temperature
increases, gel-sol transition and subsequent precipitation occured due to the collapse of PEtOx.
Another interesting phenomenon of POx is that some of them show an upper critical solution
temperature (UCST), above which a mixture is miscible in all proportions. The UCST is in
general dependent on pressure, degree of polymerization as well as the polydispersity in
polymer mixtures. The phase separation at the UCST is in general driven by an unfavorable
entropy, in particular, interactions between components favor a partially demixed state.
Schubert and co-workers reported that poly(2-phenyl-2oxazoline) (PPhOx) showed an UCST
in ethanol.122
With addition of water to the ethanol solution, the solubility of PPhOx
increased, leading to a solubility maximum in the range 6-25 wt% of water in ethanol. This
maximum solubility was due to the presence of monomeric water molecules in these solvent
mixtures. These water molecules formed hydrogen bonds with the polymeric amide groups,
and resulted in a compatibilizing hydration shell around the polymer.
1.4.4 Biomedical applications of poly(2-oxazoline)s
In the past decade, poly(2-oxazoline)s (POx) were found nontoxic/biocompatible123
and,
similar to the well established poly(ethylene oxide) (PEO). Besides, it exhibited the so-called
„stealth‟ effect. In 1989, Goddard demonstrated poly(2-ethyl-2-oxazoline) (PEtOx) as a
biocompatible polymer.124
Afterwards, 125
I-labeled POx was found to be excreted from mice
without significant accumulation in organs. Recent studies by Jordan and co-workers on the
biodistribution and excretion of radiolabeled poly(2-methyl-2-oxazoline) (PMeOx) and
PEtOx took this research to a new level.113
No accumulation in tissue and rapid clearance

Chapter 1 Introduction
29
from blood were found. Chung and co-workers125
also investigated blood compatibility of
PEtOx in in vitro. PEtOx was attached to a polyurethane film and reduced platelet adsorption
was observed.
Another interesting aspect of POx is their high chemical functionality e.g. they can be used as
drug and protein conjugates. Polymer-drug conjugation is a versatile method to increase the
circulation time and improve the solubility, moreover, the targeting group on the polymer can
deliver the drug to expected places. Since the first report of PMeOx as peptide conjugates by
Saegusa,126
a number of studies have been carried out to investigate POx in the polymer-drug
system. For example, Jordan and co-workers investigated the PMeOx and PEtOx conjugates
with 111
In radiolabel.113
In addition, Hoogenboom and co-workers demonstrated PEtOx as a
potential alternative to poly(ethylene oxide) (PEO) for protein/drug conjugates.127
Comparable results were obtained for PEtOx in regard to PEO, e.g., protein-rejecting
properties, drug-release profile, and in vitro cytotoxicity. Up to now, there have been some
polymer-drug conjugates on the market for the treatment of different diseases. However, there
are continuous demands for new polymer-drug conjugates regarding a diversity of
requirements.

Objective of this work Chaper 2
30
Chapter 2 Objective of this work
In this work, molecular brushes based on poly(2-oxazoline)s are to be synthesized for the first
time and characterized with respect to their defined architecture. Key compound of the work
is the 2-isopropenyl-2-oxazoline with two polymerizable groups: First, the vinyl group in the
2-position for living anionic or radical polymerization forming the molecular brush back bone,
second, the 2-oxazoline ring for the living cationic ring-opening polymerization forming the
pendant polymer chains of the molecular brush. Synthetic routes are to be investigated and
optimized to allow a direct synthetic access to well-defined molecular brushes in solution as
well as to well-defined bottle-brush brushes on solid surfaces. As the living cationic ring-
opening polymerization (LCROP) of 2-oxazolines allows a precise variation of the side chain
length and composition and end group functionality, the impact of such structural variations
upon the final polymer properties such as solubility, aggregation, thermo-responsiveness,
properties of bottle-brush brush coatings such as layer morphology, wetting behavior,
adsorption and adhesion characteristics are main aspects of this work.

Chapter 3 Results and discussion
31
Chapter 3 Results and discussion
3.1 Monomer synthesis
Oxazolines are five-membered heterocyclic compounds with a double bond in the ring
(shown in Figure 22). The double bond can be located in three different positions resulting in
oxazolines of different types.128
2-oxazolines are the most common and investigated
compounds. There are many ways in which 2-oxazoline can be synthesized e.g. from amino
alcohols, amides, haloamides, aziridines, epoxides, grignard reagents and reaction of SOCl2
on hydroxy amides.129,130,131,132,133,134
Figure 22. Structure of 2-substituted oxazoline.
2-oxazoline monomers are directly accessible in high yields via different synthetic routes.
One of the common used one-step synthesis is thought the addition of amino ethanol to
nitriles,132
which was developed by Seeliger and co-workers. As shown in Figure 23, through
a one-step synthesis from the corresponding nitrile compound, 2-n-propryl-, 2-isopropyl- and
2-butyl- 2-oxazoline can be prepared by a reaction with amino ethanol in the presence of
cadmium or zinc acetate in high yield. Thus, broad choices of 2-oxazolines are accessible by
changing the substituted group in nitriles.
Figure 23. Synthesis of 2-oxazolines by a reaction between amino ethanol and nitriles.
As shown in Figure 24, 2-isopropenyl-2-oxazoline (IPOx) was synthesized according to the
procedure published previously by Seeliger.135
Specifically, 2-ethyl-2-oxazoline (EtOx)

Results and discussion Chaper 3
32
reacted with paraformaldehyde in the presence of catalytic amounts of strong bases (e.g.
triethylamine) to form 2-[1-(hydroxymethyl)ethyl]-oxazoline. Water was eliminated from the
hydroxymethyl derivatives at elevated temperatures in the presence of alkali metal alkoxides
or other strong alkaline substances. However, it should be noted that the reaction with EtOx
proceeds differently when formaldehyde is present in excess: the reaction of IPOx with
formaldehyde leads to hydroxyethyl oxazoline derivatives as shown in Figure 24. Therefore,
a stochiometric amount of paraformaldehyde was used in the reaction with EtOx.
Figure 24. Synthesis of 2-isopropenyl-2-oxazoline and spontaneous side reactions.

Chapter 3 Results and discussion
33
3.2 Molecular brushes via free radical and cationic polymerization
of 2-oxazolines
3.2.1 Reaction of 2-isopropenyl-2-oxazoline
2-Isopropenyl-2-oxazoline (IPOx) allows four ways of polymerization as shown in Figure 25:
(1) olefinic polymerization via radical or anionic polymerization to produce poly((2-
oxazoline-2-yl)propylene), (2) living cationic ring-opening polymerization (LCROP) to
poly((N-methacryloyl imino)-ethylenes)), (3) cationic polymerization to give ring-preserved
polymer, and (4) spontaneous polymerization of IPOx by the N-alkylation of the 2-oxazoline
moiety with strong alkylating agents to poly[1-(3-alkyl-2-oxazolinium-2-yl)ethylene.136
Figure 25. Possible ways to polymerize IPOx. 1) Radical or anionic polymerization to
produce poly((2-oxazoline-2-yl)propylene), 2) LCROP to poly((N-methacryloyl imino)-
ethylenes)), 3) cationic polymerization to give ring-preserved polymer, and 4) spontaneous
polymerization of IPOx by the N-alkylation of the 2-oxazoline moiety with strong alkylating
agents to poly(1-(3-alkyl-2-oxazolinium-2-yl)ethylene).
We became interested in the preparation of molecular brushes of 2-oxazolines. On the one
hand, 2-oxazolines can be polymerized by living cationic ring-opening polymerization. This

Results and disccusion Chaper 3
34
allows for the incorporation of initiators with functional groups, terminal groups, and
especially broad choice of monomers, which facilitates design and construct polymers with
multifunctional architectures. On the other hand, the non-toxic peptide-like poly(2-
oxazoline)s have shown potential applications as responsive materials and biomaterials. We
choose IPOx as starting molecule for the preparation of molecular brushes i.e. free radical
polymerization of IPOx forming backbone with pendant 2-oxazoline ring. Subsequently, as
shown in Figure 26, LCROP of 2-oxazolines from the charged PIPOx backbone form side
chains.
For the preparation of POx cylindrical molecular brushes all three routes (grafting from,
grafting through or grafting onto) are possible. For example, the grafting through of POx
macromonomers were employed with comonomers to realize sufficient degrees of
polymerization.137,138,139,140
Since POx is prepared by LCROP, it is of advantage to use the
grafting from approach, analogue to the surface-initiated polymerization of 2-oxazolines on
surfaces or nanoparticles.54,141
For the grafting from approach, several synthetic strategies are
possible. Kobayashi et al.142
used hydroxyl groups of saponified poly(ethylene-co-(vinyl
acetate)) to create pendant tosylate moieties for the grafting of POx side chains and Nuyken et
al.143 , 144 , 145 , 146
used linear as well as (hyper-) branched macroinitiators containing
benzylchloride functions for the grafting of 2-oxazolines. Both approaches lead to comb
polymers with POx side chains of medium grafting density. Though some reports have been
published in the past for comb like poly(2-oxazoline)s, they are either showing low grafting
density or lacking reactivity for the LCROP of 2-oxazlines.
3.2.2 Polymerizations
The monomer IPOx has two orthogonal polymerizable groups, a vinyl group for the free
radical or living anionic polymerization and the 2-oxazoline ring for the living cationic ring-
opening polymerization (LCROP). It has been shown that the conversion of the vinyl group to
poly(2-isopropenyl-2-oxazoline) (PIPOx) by radical147 , 148 , 149 ,215
as well as anionic216
polymerization does not affect the 2-oxazoline ring. Since the oxazoline ring can be well
preserved during the vinyl polymerization, it is possible to prepare molecular brushes from
POx.
Firstly, IPOx was initiated by N,N-azobisisobutyronitrile (AIBN) and polymerized by free
radical polymerization to form the molecular brush backbone; Successively, 2-oxazolines i.e.

Chapter 3 Results and discussion
35
2-methyl-2-oxazoline (MeOx), 2-ethyl-2-oxazoline (EtOx), 2-isopropyl-2-oxazoline (iPrOx),
2-n-propyl-2-oxazoline (nPrOx) and 2-n-butyl-2-oxazoline (BuOx) were polymerized by
LCROP to form the side chains. For the latter, the PIPOx backbone has to be converted to a
macroinitiator salt with pendant oxazolinium rings by the reaction with a stochiometric
amount of methyl triflate (MeOTf). This approach has the advantage that a robust and very
effective macroinitiator 150
is formed with equal reactivities as compared to e.g. triflates or
tosylates.151
Moreover, the charge at each monomer unit, thus a polyelectrolyte induces
significant chain stretching due to the coulomb repulsion. This improves the accessibility of
the initiation sites for the monomers along the main chain, as shown in Figure 27. A fast
initiation reaction is crucial to obtain side chains of low polydispersities, especially for
grafting by the ionic living polymerization. Both aspects, macroinitiator chain stretching and
the formation of the oxazolinium salt, work in favor for a fast initiation reaction of the
LCROP for a defined side chain grafting. As outlined in the reaction pathway (Figure 26)
molecular brushes with five types of poly(2-oxazoline) homopolymer side chains were
synthesized. MeOx gives very hydrophilic brushes, BuOx gives very hydrophobic brushes,
EtOx, nPrOx and iPrOx result in slightly amphiphilic molecular brushes which are known to
show a sharp LCST.167
The results of the radical polymerization of IPOx and the grafting
from polymerization of 2-alkyl-2-oxazoliness by LCROP using the macroinitiator salt are
summarized in Table 1.
As outlined in Figure 26, the molecular brush backbone prepared by free radical
polymerization is facile but not well controlled, thus not suitable for preparation of defined
polymers. An alternative would be to use controlled radical polymerization. However,
previous studies showed it is difficult to convert IPOx by widely used atom transfer radical
polymerization (ATRP) to PIPOx. Only oligomeric products were obtained probably due to a
strong complexation of the copper by the pendant 2-oxazoline units of oligomers.

Results and disccusion Chaper 3
36
Figure 26. Synthesis of molecular brushes of 2-oxazolines via combination of radical
polymerization of 2-isopropenyl-2-oxazoline and subsequent cationic ring-opening
polymerization of 2-alkyl-2-oxazolines („R‟ = prepared by radical polymerization).
Figure 27. Schematic illustration of the formation of stretched macroinitiator (PIPOxOTf) by
methylation of methyl triflate (MeOTf) and successive cationic ring-opening polymerization
of 2-oxazlines to form side chains.

Chapter 3 Results and discussion
37
Figure 28. 1H NMR spectra of a) PIPOx
R and b) PIPOxOTf
R along with the chemical
structures and assignments. The free radical polymerization did not affect the integrity of the
pendant 2-oxazoline ring. The new signal at 3.7 ppm and the quantitative shift of the two
triplets from the pendant 2-oxazoline ring as well as the backbone signals (c,d to c‟,d‟)
indicate a quantitative conversion to the polycationic macroinitiator PIPOxOTf. The intensity
ratio of the -methyl signals (d) at 1.12 (syndiotactic, S), 1.24 (heterotactic, H) and 1.39
(isotactic, I) ppm was used to determine the polymer tacticity.
Similar to earlier reports,148,215
the free radical polymerization of IPOx using AIBN resulted in
hydrophilic PIPOxR with a molar mass distribution of 2.12. The analysis by
1H NMR
confirmed that the pendant 2-oxazoline rings were preserved (Figure 28). The integral ratio
of the ring methylene groups (a, 2H and b, 2H) and the corresponding signal integrals of the
polymer backbone (c, 2H; d 3H) is in excellent agreement and confirms the depicted polymer
structure. Closer inspection of the proton backbone signals (c, d) in Figure 28 also reveals a
fine structure due to the tacticity of PIPOx.147
Similar to PMMA,152
the tacticity of PIPOx can
be estimated from the relative intensity of the characteristic -methyl signals for syndio (S) -
(1.12 ppm), hetero (H) - (1.24 ppm) and isotactic (I) (1.39 ppm) polymer sequences. For
PIPOx, an S:H:I ratio of 57:37:6 was found. The determined S:H:I ratio for PIPOx is in
agreement with earlier findings by Kagiya et al.147

Results and disccusion Chaper 3
38
The polycationic macroinitiator salts, PIPOxOTfR, were prepared from PIPOx
R and methyl
triflate (MeOTf). The 1H NMR spectrum of PIPOxOTf
R is shown in Figure 28. Upon
methylation, a new signal of the N-methyl group at 3.7 ppm appears and the characteristic
two triplet signals of the 2-oxazoline methylene protons at 3.76 and 4.16 ppm are
quantitatively shifted to 4.54 and 5.08 ppm. The intensity ratio of the new N-methyl
oxazolinium ring and the methyl signals is again in excellent agreement with signal intensities
originating from the backbone protons. Based on 1H NMR spectral analysis, the conversion of
PIPOxR to the macroinitiator salt, PIPOxOTf
R, was quantitative.
However, in the spectrum of the macroinitiator some low molar mass impurities are
noticeable. First attempts to remove these impurities resulted in minor but noticeable
decomposition of the oxazolinium rings (by e.g. hydrolysis). Thus, we used PIPOxOTfR as
obtained after a single precipitation and washing step to initiate the side chain grafting from
polymerization of 2-methyl-, 2-ethyl-, 2-n-propyl-, 2-iso-propyl- and 2-butyl- 2-oxazolines
by LCROP. The targeted length of the side chains was set to m = 25 by the initial [M]0/[I]0
ratio.
According to our previous accounts on functionalized POx,151
N-Boc-piperazine (Boc) was
used as the terminating reagent. This terminal group is useful for 1H NMR end group analysis
and moreover, after deprotection of the secondary amine group, it allows an additional
functionalization of each side chain end.151,153,154,155,156
The obtained molecular brushes were analyzed by gel permeation chromatography (GPC).
For all five graft copolymers a significant increase of the molar mass was observed and no
remaining macroinitiator could be detected. In all cases a monomodal distribution was
obtained and no fraction of polymers having lower molar masses was observed. Because of
the side chain grafting, the number average molar mass (characterized by GPC) increased
from 9.8×103 g/mol to Mn = 7×10
4 g/mol, 8×10
4 g/mol, 5×10
4 g/mol, 5×10
4 g/mol, 6×10
4
g/mol for MeOx, EtOx, iPrOx, nPrOx, and BuOx, respectively. In Figure 29, the GPC trace
of PIPOxR (1) is compared with the traces of the resulting molecular brushes (2-4).

Chapter 3 Results and discussion
39
Figure 29. GPC traces of PIPOxR and molecular brushes P(IPOx-g-MeOx)
R, P(IPOx-g-
EtOx)R and P(IPOx-g-iPrOx)
R.
The successful side chain formation from the macroinitiators by the grafting from approach
was further confirmed by 1H NMR spectroscopy (Figure 30). For all grafting polymerization,
the signals corresponding to the oxazolinium pendant group disappeared completely. Instead,
characteristic signals for the respective side chain polymers could be unambiguously assigned.
Furthermore, a successful end functionalization with N-boc-piperazine is confirmed by the
presence of a strong signal around 1.45 ppm originating from the tert. butyl group
(overlapping with the -methyl signals of the brush backbone). Estimation of the average
degree of polymerization, m, of the POx side chains was performed by comparing the signal
integral of the tert. butyl group and the -methyl signals at 1.45 ppm of the brush backbone
versus the methylene signal of the POx side chain at 3.45 ppm. Although a degree of
polymerization of m = 25 for the side chains was targeted, in all cases slightly shorter chains
with m = 20 were determined (Table 1).
An incomplete grafting from or termination reaction would calculate to longer side chains.
Thus, this result indicates a very high grafting density (within experimental error, 100%) and
efficient end group functionalization via the termination reaction. The resulting shorter side
chains may be caused by chain transfer reactions or, more likely, residual MeOTf in the
macroinitiator salt. In fact, for the grafting of 2-oxazolines minor amounts of homopolymer
was observed that we attribute to traces of MeOTf in the macroinitiator. However, to ensure a

Results and disccusion Chaper 3
40
homogeneous and high grafting density along the polymer backbone, we decided to avoid
elaborate cleaning procedures of the macroinitiator.
Figure 30. 1H NMR spectra of the molecular brushes a) P(IPOx-g-MeOx)
R and b) P(IPOx-g-
iPrOx)R with their assignments. Besides the characteristic backbone and POx monomer unit
signals (a, b), end group signals (d, f) are detectable.

Chapter 3 Results and discussion
41
Figure 31. FTIR spectra of PIPOxR (solid line) and the molecular brush P(IPOx-g-MeOx)
R
(grey dashed line).
Further confirmation for the successful grafting of POx from the macroinitiator salts was
obtained by FTIR spectroscopy. In Figure 31, the IR spectra of PIPOxR and P(IPOx-g-
MeOx)R are compared. The strong bands at 1654 and 1118 cm
-1 are assigned to the (C=N)
and (C-O) moieties and the bands at 986 and 951 cm-1
originate from the ring skeletal
vibration of the pendant 2-oxazoline rings in PIPOxR. After ring-opening polymerization,
these bands disappeared completely, and a new sharp and intensive band appeared around
1630 cm-1
which is characteristic for the carbonyl stretching mode of the amide function
(amide I)151,212
and confirms the successful grafting by LCROP to P(IPOx-g-MeOx)R.
Moreover, the characteristic CHx stretching pattern between 2800 and 3200 cm-1
and the CHx
deformation modes for POx observed between 1365 and 1477 cm-1
as well as the carbonyl
stretching vibration of the Boc group are found in the spectrum.

Results and disccusion Chaper 3
42
Table 1. Poly(2-oxazoline) macroinitiators prepared from the PIPOx backbone by free radical
polymerization and corresponding molecular brushes of homopoly(2-oxazoline) side chains.
Polymer Yield
(%)
Mn
(kg/mol)b
PDIb
(Mw/Mn) n
b m
c m
theor.
Mntheor.
(kg/mol)d
PIPOxnR 50a 9.8 2.12 88 - - -
PIPOxnOTfR 96 - - - - - -
P(IPOxn-g-MeOxm)R 80 70.0 1.69 88 19 25 168
P(IPOxn-g-EtOxm)R 76 78.0 1.75 88 23 25 227
P(IPOxn-g-iPrOxm)R 50 53.0 1.58 88 18 25 205
P(IPOxn-g-nPrOxm)R 79 56.0 1.61 88 20 25 205
P(IPOxn-g-BuOxm)R 77 61.0 1.65 88 21 25 236
a) calculated against initial initiator feed.
b) as calculated from GPC traces.
c) average degree of polymerization, (m) calculated from end group analysis using 1H NMR
spectral data.
d) calculated using GPC determined molar mass of precursor polymer and 1H NMR
determined degree of side chain polymerization.
3.2.3 Thermo-responsiveness of poly(2-oxazoline)s molecular brushes
The hydrophilicity of poly(2-alkyl-2-oxazoline)s is determined by the length of the pendant
alkyl group. Longer alkyl chains of the monomer unit results in an increasingly stronger
amphiphilic character of the monomer unit and the polymer becomes a non-ionic
polysoap.55,153,157
As most of the water-soluble polymers,158
hydrophilic POx have a lower
critical solution temperature (LCST) above which the polymer becomes water-insoluble.
Taking advantage of the flexibility of the 2-oxazoline chemistry and the LCROP giving
access to defined (co-) polymers having various side and end groups, recent studies by
Kataoka167
and our group33,119
demonstrated the fine-tuning of the LCST of POx over a broad
temperature range. Until now, the LCST of hydrogels and linear POx homo- and copolymers
were investigated in detail as a function of the side chain and/or end group functionality as
well as the molar mass and concentration.159 ,118
Here, the temperature-dependant water
solubility of the obtained POx molecular brushes were determined. The LCST was measured
analogue to previous accounts33,146
by turbidity measurements of a 1.0 wt % aqueous solution
of P(IPOx-g-EtOx)R, P(IPOx-g-iPrOx)
R, and P(IPOx-g-nPrOx)
R. The cloud point was defined

Chapter 3 Results and discussion
43
at 10 % optical transmittance decrease of an aqueous polymer solution. The results are
summarized in Figure 32. P(IPOx-g-MeOx)R was too hydrophilic and remained soluble in
water at a temperature above 95 °C. Whereas, P(IPOx-g-BuOx)R was not water soluble in the
accessible temperature range.
For all three thermo-responsive molecular brush solutions, the LCST transition was found to
be very sharp (the complete transition occurred within a temperature range of 0.5 - 1.5 °C)
and fully reversible, with no remaining insoluble polymer fraction below the respective LCST.
The LCST of P(IPOx-g-EtOx)R was found to be about 53 °C, which is significantly lower
than the LCST between 73 °C and 69 °C found for linear PEtOx with a comparable high
molar mass as reported by Du Prez.160
For molecular brushes with PiPrOx side chains, a
transition temperature of 30 °C was determined. Also in this case, the LCST is significantly
lower than the earlier reported values for linear PiPrOx (35-45 °C)161,167
as well as crosslinked
PiPrOx hydrogels (shown in Table 2). For the molecular brush with PnPrOx side chains, a
transition temperature of 19 °C was determined. Again, the LCST is significantly lower than
the earlier reported values for linear PnPrOx (23.8 °C ~). It was reported that linear PnPrOx
homopolymer show independent sharp LCST at around 24 °C almost regardless of the molar
mass and concentration. The present findings suggest that the compact structure of a
molecular brush slightly affects the LCST, which is consistent to the previous report i.e. the
unique molecular architecture has minor effects upon the value of the LCST.36,38,40
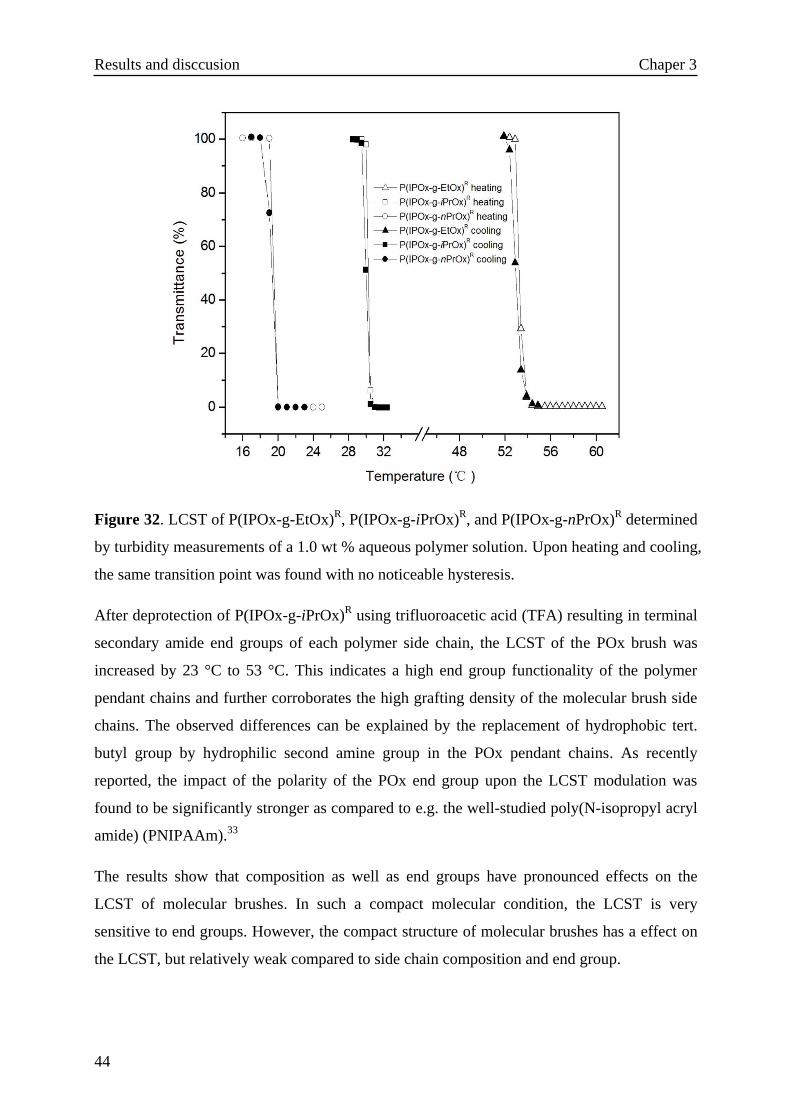
Results and disccusion Chaper 3
44
Figure 32. LCST of P(IPOx-g-EtOx)R, P(IPOx-g-iPrOx)
R, and P(IPOx-g-nPrOx)
R determined
by turbidity measurements of a 1.0 wt % aqueous polymer solution. Upon heating and cooling,
the same transition point was found with no noticeable hysteresis.
After deprotection of P(IPOx-g-iPrOx)R using trifluoroacetic acid (TFA) resulting in terminal
secondary amide end groups of each polymer side chain, the LCST of the POx brush was
increased by 23 °C to 53 °C. This indicates a high end group functionality of the polymer
pendant chains and further corroborates the high grafting density of the molecular brush side
chains. The observed differences can be explained by the replacement of hydrophobic tert.
butyl group by hydrophilic second amine group in the POx pendant chains. As recently
reported, the impact of the polarity of the POx end group upon the LCST modulation was
found to be significantly stronger as compared to e.g. the well-studied poly(N-isopropyl acryl
amide) (PNIPAAm).33
The results show that composition as well as end groups have pronounced effects on the
LCST of molecular brushes. In such a compact molecular condition, the LCST is very
sensitive to end groups. However, the compact structure of molecular brushes has a effect on
the LCST, but relatively weak compared to side chain composition and end group.

Chapter 3 Results and discussion
45
Table 2. Summary of the LCST measurements of polymers and comparison of thermal
behavior with corresponding linear polymers.
P(IPOxn-g-EtOxm)R P(IPOxn-g-iPrOxm)R P(IPOxn-g-nPrOxm)R
LCST (°C) 53 30 19
Width of
transitiona (°C) 1.5 1 1.0
LCST of linear
polymerb (°C) 69160
36c
(Mn ≈16.7 kg/mol) 161
24c
(Mn ≈ 347kg/mol)168
a temperature range between 100 % and ~ 0 % transmittance.
b of comparable molar mass.
c no references were found which studied 2-iso-propyl-2-oxazoline and 2-n-propyl-2-
oxazoline based polymers of comparable molar mass.
3.2.4 Morphology of adsorbed molecular brushes on surfaces
The visualization of molecular brushes by e.g. AFM allows for the direct determination of
their molecular dimensions, such as contour length, brush width, backbone curvature, and
persistence length, and the overall dimensions, such as radius of gyration and end-to-end
distance in real space. The 1H NMR spectroscopy data indicate that the grafting from
reactions from the macroinitiator salts are quantitative and a molecular brushes with very high
grafting densities were formed. This will result in cylindrical molecular brushes with a
strongly stretched polymer brush backbone.9,46,162
To investigate the molecular shape of the
synthesized molecular brushes, AFM was used to visualize the individual molecules deposited
on naturally oxidized silicon substrates from a dilute chloroform solution. Molecular brushes
adsorb onto silicon substrate due to the interaction between molecular side chains and the
substrate. In Figure 33 the AFM measurement of the adsorbed P(IPOx-g-EtOx)R is shown.
The individual molecular brushes could be successfully visualized and display the expected
and characteristic shape of strongly stretched chains. Clearly, the length of the polymer
brushes varies considerably because of the synthesis of the PIPOx backbone via free radical
polymerization and their broad dispersity. This, of course, is not surprising. With a
polydispersity index of 2.12 for the polymer backbone a wide variety of polymers with short
and long backbones is observed. However, as the polymer side chains were prepared by
LCROP, the width of the polymer appears to be uniform.
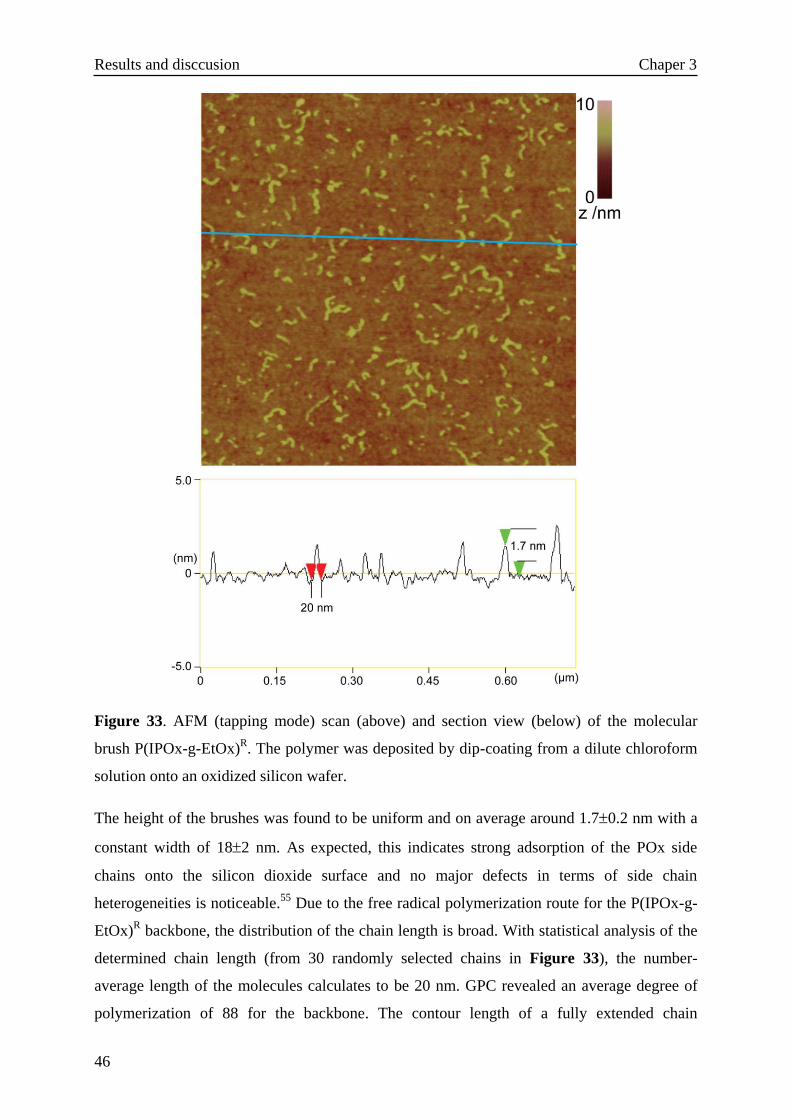
Results and disccusion Chaper 3
46
Figure 33. AFM (tapping mode) scan (above) and section view (below) of the molecular
brush P(IPOx-g-EtOx)R. The polymer was deposited by dip-coating from a dilute chloroform
solution onto an oxidized silicon wafer.
The height of the brushes was found to be uniform and on average around 1.70.2 nm with a
constant width of 182 nm. As expected, this indicates strong adsorption of the POx side
chains onto the silicon dioxide surface and no major defects in terms of side chain
heterogeneities is noticeable.55
Due to the free radical polymerization route for the P(IPOx-g-
EtOx)R backbone, the distribution of the chain length is broad. With statistical analysis of the
determined chain length (from 30 randomly selected chains in Figure 33), the number-
average length of the molecules calculates to be 20 nm. GPC revealed an average degree of
polymerization of 88 for the backbone. The contour length of a fully extended chain

Chapter 3 Results and discussion
47
calculates to be 22 nm, which further corroborates the assumption of a strongly stretched
cylindrical brush due to the high crowding of the POx side chains.49
3.2.5 Cytotoxicity assay, MTT
Toxicity test of the obtained molecular brushes was performed by Dr. Robert Luxenhofer
from the research group of Prof. Sasha Kabanov at University of Nebraska Medical Center,
Omaha. Polymers which are suitable for uses in living organisms have to fulfill requirements
concerning their biocompatibility, cytotoxicity, possibly biodegradability, and/or
immunocompatibility.163
The biocompatibility of poly(2-oxazoline)s have been known for a
period of time.110,120
Here, the toxicity test of the prepared molecular brushes were performed
according to a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
MTT assay is a standard colorimetric assay for measuring cellular proliferation (cell growth).
It can also be used to determine cytotoxicity of a given compound.164
Yellow MTT is reduced
to purple formazan in the mitochondria of living cells. This reduction takes place only when
reductase enzymes are active, and therefore conversion is often used as a measurement for
viable (living) cells.165
MCF-7/ADR cells, a human breast multiple drug resistance (MDR) cancer cell line, were
incubated with P(IPOx-g-MeOx)R for 72 h. As shown in Figure 34, P(IPOx-g-MeOx)
R
showed 100 % viability when the concentration was below 2 mg/mL. The cell viability kept
above 80 % even if the polymer concentration was increased to 10 mg/mL. This indicated that
the P(IPOx-g-MeOx)R
caused no harm to cells under the conditions used and seemed to be
suitable as a non-toxic material in living organisms.

Results and disccusion Chaper 3
48
Figure 34. Cell viability determined by the MTT assay as a function of the P(IPOx-g-
MeOxBoc)R concentration. The data are presented as mean ± SEM (standard error of the
mean).

Chapter 3 Results and discussion
49
3.3 Molecular brushes via anionic and cationic ring-opening
polymerization
In this part of work, we prepared molecular brushes by a combination of anionic vinyl
polymerization of 2-isopropenyl-2-oxazoline (IPOx) for the backbone and the living cationic
ring-opening polymerization (LCROP) of 2-oxazoline moiety for the side chain synthesis.
Two macroinitiators with different degree of polymerizations (DP) were prepared to
polymerize nPrOx or iPrOx with variable side chain DP. The kinetics of the LCROP of 2-
oxazolines in the presence of short and long macroinitiators were investigated. The results
were compared with the polymerization of 2-oxazoline initiated by methyl triflate (MeOTf)
under the same reaction condition. The thermo-responsive properties of the resulting
molecular brushes were studied to examine the effect of side chain length on the LCST values.
Furthermore, various parameters such as concentration and molar mass in determining the
LCST of brush molecules were also investigated.
3.3.1 Anionic polymerization of 2-isopropenyl-2-oxazoline
It was shown in the previous chapter that the conversion of the 2-isopropenyl-2-oxazoline
(IPOx) to poly(2-isopropenyl-2-oxazoline) (PIPOx) by free radical polymerization did not
affect the 2-oxazoline ring. However, the molar mass distribution of the PIPOx backbone was
relatively broad due to the used synthetic route. In order to synthesize a more defined
molecular brush, the anionic polymerization was adopted to prepare PIPOx backbone. It was
reported that the conversion of IPOx to PIPOx by living anionic polymerization did not affect
the 2-oxazoline ring.216
Moreover, the it gave narrow molar mass distributions of PIPOx.
Therefore, a better overall control of the resulting molecular brushes is expected. However,
early reports on anionic polymerization of IPOx showed the formation of polymers with low
molar mass and poor PDI e.g. Mn = 1447 g/mol, PDI = 1.55.216

Results and disccusion Chaper 3
50
Figure 35. Molecular brushes prepared by anionic polymerization of IPOx initiated by butyl
lithium and LCROP of 2-oxazolines to form the side chains. („A‟ = prepared by anionic
polymerization)
Table 3. PIPOx backbones prepared by living anionic polymerization.
Entry Mn(kg/mol) PDI DPa DPb
PIPOxAI 5.8 1.15 52 58
PIPOxAII 14.6 1.21 131 146
PIPOxAIII 24.0 1.20 216 225
a) degree of polymerization calculated from apparent molecular weight from GPC traces
b) degree of polymerization calculated from end group analysis from 1H NMR
In this part of work, PIPOx backbone was prepared through the anionic polymerization of
IPOx with butyl lithium (BuLi) as the initiator (Figure 35). Briefly, IPOx monomer was
added dropwise into a BuLi containting THF solution at -50 °C. Three PIPOxA polymers with
different molar mass were obtained by varying the [M]0/[I]0 ratio. There, the molar mass
weight average (Mw), polydispersity (PDI = Mw/Mn), degree of polymerization (DP) and end-
group analysis by 1H NMR are summarized in Table 3. The anionic polymerization of IPOx
resulted in PIPOxA with a PDI between 1.15 and 1.21 depending on the targeted chain length.
Compared to an early report fro Tomalia et al.,216
higher degrees of polymerization and lower

Chapter 3 Results and discussion
51
molar mass distributions are obtained here. However, the PDI of 1.21 indicates that the
anionic polymerization is accompanied, at least to some extent, by termination and chain
transfer reactions via proton abstraction.166
The estimation of the average DP, based on gel
permeation chromatography data (with PMMA standards for calibration), appears to be
accurate since it nicely fits the results calculated from 1H NMR end-group analysis. As shown
in Figure 36, the complementary end-group analysis of PIPOxA based on
1H NMR data using
the signals at 0.9 ppm (9H) of the terminal butyl group versus the signals of the monomer
units resulted in a DP which is similar to the value calculated from GPC, i.e. a DP of 52
calculated from GPC and a DP of 58 found from 1H NMR. The tacticity i.e. the S:H:I ratio of
73:25:2 for PIPOxA was calculated from
1H NMR. The living anionic polymerization of IPOx
resulted in PIPOxA which showed a significant larger syndiotactic and lower isotactic fraction
compared to results from PIPOxR. This can be explained by the steric demand of the pendant
2-oxazoline ring during the slower anionic reaction.
Figure 36. 1H NMR spectrum of PIPOx
A along with the chemical structure and assignment.
The spectrum indicates that the anionic polymerization did not affect the pendant 2-oxazoline
ring. The intensity ratio of the -methyl signals (d) at 1.12 (syndiotactic, S), 1.24 (heterotactic,
H) and 1.39 (isotactic, I) ppm was used to determine the polymer tacticity.

Results and disccusion Chaper 3
52
3.3.2 Kinetic studies of the living cationic ring-opening polymerization of 2-
oxazolines
The moderate propagation rate for the polymerization of 2-oxazoline allows for kinetic
investigations.211
Recently, our group performed kinetic measurements through gas
chromatography, revealing that multiple initiating groups (2, 3 and 4) are of equal reactivity
for initiating the polymerization of 2-oxazolines and uniform poly(2-oxazoline)s with 2, 3 and
4 arms were achieved (Figure 37).211
Analogue to these studies, gas chromatography (GC)
was used for kinetic investigations of the cationic polymerization of 2-oxazolines with
defined macroinitiator salts of different length and the obtained kinetic behavior was
compared with the one initiated by MeOTf. It should be noted that the polymerization was
carried out in a glass vial sealed with a septum, and the GC automatically collected samples at
the set time intervals.
Figure 37. Preparation of linear, multi-armed and brush-like poly(2-oxazoline)s.
To compare the kinetics of polymerization using mono- and multifunctional initiators, one
would compare the apparent polymerization rate. However, the apparent polymerization rate
for macroinitiators is inaccurate because of the intrinsic error in determining the molar mass
of the macroinitiator. Therefore, we compared the apparent polymerization rate per initiating
group i.e. single oxazolinium triflate. In the equation below,
denotes the apparent
propagation rate for the polymerization, [M] is the monomer concentration, is
concentration of the initiating group and t is the reaction time.
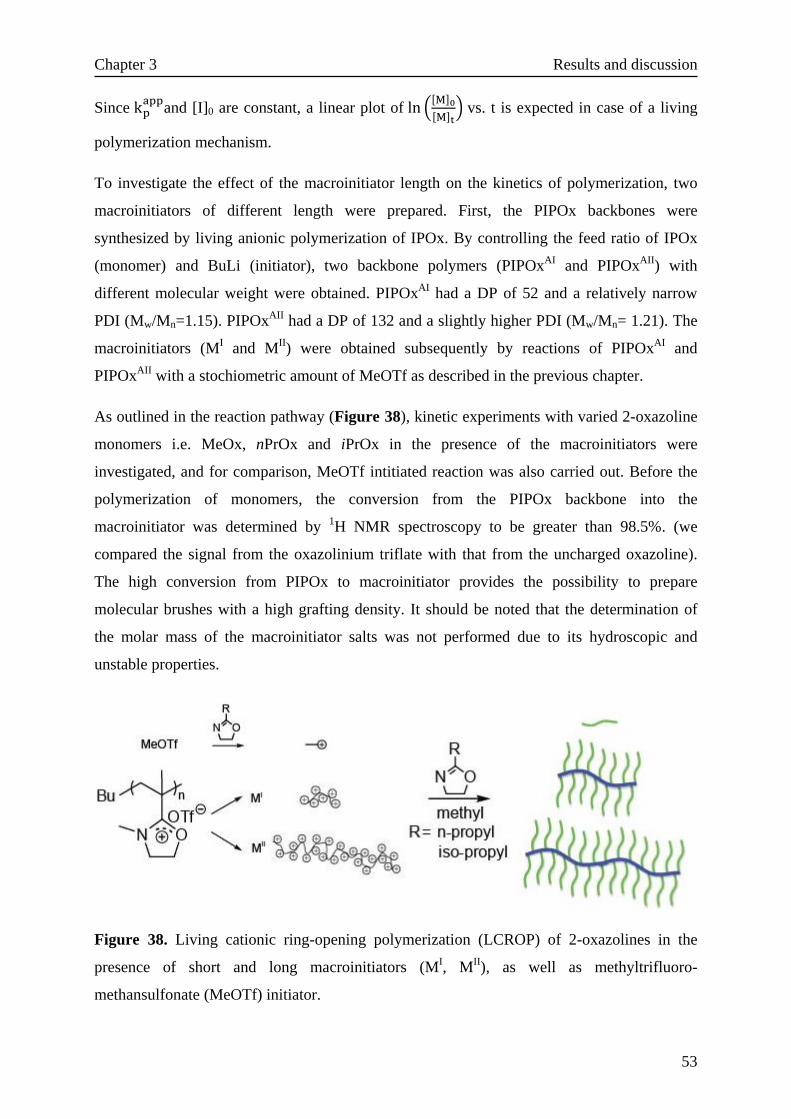
Chapter 3 Results and discussion
53
Since
and [I]0 are constant, a linear plot of
vs. t is expected in case of a living
polymerization mechanism.
To investigate the effect of the macroinitiator length on the kinetics of polymerization, two
macroinitiators of different length were prepared. First, the PIPOx backbones were
synthesized by living anionic polymerization of IPOx. By controlling the feed ratio of IPOx
(monomer) and BuLi (initiator), two backbone polymers (PIPOxAI
and PIPOxAII
) with
different molecular weight were obtained. PIPOxAI
had a DP of 52 and a relatively narrow
PDI (Mw/Mn=1.15). PIPOxAII
had a DP of 132 and a slightly higher PDI (Mw/Mn= 1.21). The
macroinitiators (MI and M
II) were obtained subsequently by reactions of PIPOx
AI and
PIPOxAII
with a stochiometric amount of MeOTf as described in the previous chapter.
As outlined in the reaction pathway (Figure 38), kinetic experiments with varied 2-oxazoline
monomers i.e. MeOx, nPrOx and iPrOx in the presence of the macroinitiators were
investigated, and for comparison, MeOTf intitiated reaction was also carried out. Before the
polymerization of monomers, the conversion from the PIPOx backbone into the
macroinitiator was determined by 1H NMR spectroscopy to be greater than 98.5%. (we
compared the signal from the oxazolinium triflate with that from the uncharged oxazoline).
The high conversion from PIPOx to macroinitiator provides the possibility to prepare
molecular brushes with a high grafting density. It should be noted that the determination of
the molar mass of the macroinitiator salts was not performed due to its hydroscopic and
unstable properties.
Figure 38. Living cationic ring-opening polymerization (LCROP) of 2-oxazolines in the
presence of short and long macroinitiators (MI, M
II), as well as methyltrifluoro-
methansulfonate (MeOTf) initiator.
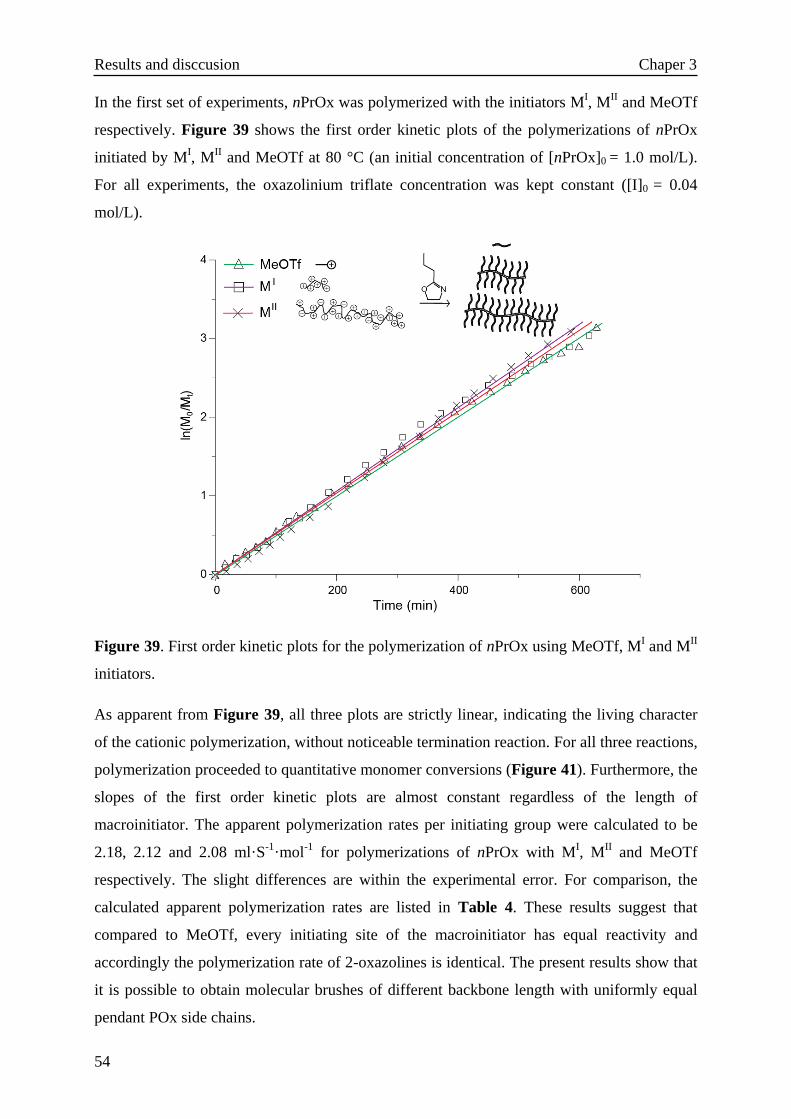
Results and disccusion Chaper 3
54
In the first set of experiments, nPrOx was polymerized with the initiators MI, M
II and MeOTf
respectively. Figure 39 shows the first order kinetic plots of the polymerizations of nPrOx
initiated by MI, M
II and MeOTf at 80 °C (an initial concentration of [nPrOx]0 = 1.0 mol/L).
For all experiments, the oxazolinium triflate concentration was kept constant ([I]0 = 0.04
mol/L).
Figure 39. First order kinetic plots for the polymerization of nPrOx using MeOTf, MI and M
II
initiators.
As apparent from Figure 39, all three plots are strictly linear, indicating the living character
of the cationic polymerization, without noticeable termination reaction. For all three reactions,
polymerization proceeded to quantitative monomer conversions (Figure 41). Furthermore, the
slopes of the first order kinetic plots are almost constant regardless of the length of
macroinitiator. The apparent polymerization rates per initiating group were calculated to be
2.18, 2.12 and 2.08 ml·S-1
·mol-1
for polymerizations of nPrOx with MI, M
II and MeOTf
respectively. The slight differences are within the experimental error. For comparison, the
calculated apparent polymerization rates are listed in Table 4. These results suggest that
compared to MeOTf, every initiating site of the macroinitiator has equal reactivity and
accordingly the polymerization rate of 2-oxazolines is identical. The present results show that
it is possible to obtain molecular brushes of different backbone length with uniformly equal
pendant POx side chains.

Chapter 3 Results and discussion
55
Table 4. Calculation of apparent rate constants per initiating group for the polymerization of
2-oxazolines using MeOTf and different macroinitiators.
Initiator Degree of
functionalizationa Monomer [M]0/[I]0 kp(ml·S-1·mol-1)
MeOTf 100 % nPrOx 25/1 2.08
MI 98 % nPrOx 25/1 2.18
MII 98.5 % nPrOx 25/1 2.12
MeOTf 100% iPrOx 25/1 1.25
MII 98.5 % iPrOx 25/1 1.28
MeOTf 100% MeOx 25/1 2.90
MII 98.5 % MeOx 25/1 2.95
a Determined by
1H NMR.
To further confirm the above results, using more steric monomer iPrOx, the kinetic behavior
of the polymerization initiated by MII was studied and compared with that initiated by MeOTf
under identical reaction conditions. The apparent polymerization rate per initiating group was
calculated to be 1.28 and 1.25 ml·S-1
·mol-1
for MII and MeOTf, respectively. Even for steric
demanding 2-oxazoline monomers such as iPrOx, the apparent polymerization rate per
initiator function is the same as MeOTf. Likewise, using MeOx as the monomer, the kinetics
of polymerizations initiated by MII and MeOTf were studied, revealing the apparent
polymerization rate per initiating group to be 2.95 and 2.90 ml·S-1
·mol-1
respectively. The
results of kinetic studies are summarized in Table 4. These results indicate that initiating sites
(oxazolinium triflate) along the macroinitiator have equal reactivity in the polymerization of
2-oxazolines, and can form side chains of equal length regardless of the type of monomer and
the length of macroinitiator. Moreover, every oxazolinium triflate in the macroinitiator has the
same reactivity as MeOTf.
For the grafting from polymerization, PIPOx has to be first converted to a macroinitiator salt
with pendant oxazolinium rings. The formed oxazolinium triflate is a very effective initiator
for polymerization of 2-oxazolines. 150
As shown in the previous chapter, the charging of each
monomer unit along the PIPOx chain to a polyelectrolyte induces significant chain stretching
which improves the accessibility of each initiation site for 2-oxazoline monomers. Both
aspects, the formation of the oxazolinium salt and macroinitiator chain stretching, work in
favor for a fast initiation reaction of the living cationic ring-opening polymerization for a
defined side chain grafting. Therefore, molecular brushes with defined side chains can be
prepared using this method.

Results and disccusion Chaper 3
56
Figure 40. Increase of the number average molar mass (Mn) of P(IPOx-g-nPrOx) versus the
conversion of nPrOx with MII as the macroinitiator.
Another evidence for the living character of the polymerizations of 2-oxazolines from the
macroinitiator was obtained from GPC measurements. Figure 40 shows that the number-
average molecular weight (Mn) of the molecular brushes increased linearly with the increasing
conversion of monomer with PnPrOx as the side chain (from MII). It should be noted that, the
Mn determined by GPC is lower than the theoretical values calculated from conversion of
nPrOx, which is consistent with a previous report by Matyjaszewski.13
This is due to the
relatively compact structure of molecular brushes compared to linear polymers, and different
hydrodynamic volumes of brush molecules as compared to the linear PMMA used for GPC
calibration. It was found at 100% conversion that the characterized molar mass is significant
lower than expected. This deviation is because of the highly compact structure of molecular
brushes and the possible termination reaction at high conversion.
The kinetics of polymerizations of MeOx, nPrOx and iPrOx in the presence of MII were also
compared. Figure 41 shows the correlation between monomer consumption and reaction time
for the polymerizations of different 2-oxazolines using MII. It was observed in each
experiment that the monomer consumption proceeded to 100%. The polymerizations of
MeOx, nPrOx and iPrOx were complete in approx. 10h, 15h and 24h, respectively. Moreover,
the first order kinetic plots were found to be strictly linear as shown in Figure 42, indicating
the living character of polymerization of 2-oxazolines.

Chapter 3 Results and discussion
57
Figure 41. Plots of MeOx, nPrOx and iPrOx consumption vs. LCROP time for the
polymerization in the presence of MII.
Figure 42. First order kinetic plots for the polymerization of MeOx, EtOx and nPrOx in the
presence of MII.

Results and disccusion Chaper 3
58
3.3.3 Thermo-responsive properties - impact of different parameters on the
LCST
The LCST of linear thermo responsive polymers, including POx, is a function of molar mass
and concentration.33,118,159
Besides, it also can be affected by the structure and polarity of side
chain and end groups.119,161,162,163,167,168
Hence, it is expected that these parameters also have a
certain influence on the LCST of molecular brushes. The influence of the side chain
composition and end group functionalities on the LCST were discussed in the previous
chapter. Here we investigated the effect of other possible factors e.g. side chain length,
concentration and backbone length on the LCST of molecular brushes of POx.
3.3.3.1 Side chain length effect
It was shown in the previous chapter that the unique molecular architecture has only minor
effects upon the LCST. Here, to examine the effect of side chain length, molecular brushes
with PnPrOx or PiPrOx pendant chains of different length were prepared with the
macroinitiators (MI, M
II). The side chain length was controlled by terminating the side chain
polymerizations after a given time, and afterwards determined from the kinetic plot of
monomer conversion vs LCROP time (Figure 41). Characterizations of macroinitiators MI,
MII and molecular brushes prepared by LCROP of nPrOx/iPrOx from macroinitiators are
summarized in Table 5. MI 1 - M
I 3 are molecular brushes prepared from macroinitiator M
I
with different PnPrOx side chain length, i.e. LCROP of nPrOx for 1, 2 and 8 h, respectively.
MII 1 - M
II 4 are brushes from macroinitiator M
II with different PnPrOx side chain length, i.e.
LCROP of nPrOx for 0.5, 1, 2 and 8 h, respectively. MII 5 - M
II 7 are brushes from M
II with
different PiPrOx length, i.e. LCROP of iPrOx for 1.3, 3 and 24 h, respectively.

Chapter 3 Results and discussion
59
Table 5. Synthesis of macroinitiators and molecular brushes with PnPrOx/ PiPrOx side chains.
Initiator Polymerizati
on time (h)a
Side
chain
Convers
ion (%)b DPc
Mn
(kg/mol)d Mw/Mn
d LCST
(°C)e
PIPOxI - 0 - 0 - 5.8 1.15 -
MI 1
MI
1
PnPrOx
22 6 16.4 1.18 19.5
MI 2 2 42 11 27.3 1.17 18.1
MI 3 8 98 24 40.7 1.14 19.2
PIPOxII - 0 - 0 - 14.6 1.21 -
MII 1
MII
0.5
PnPrOx
11 3 25.8 1.26 24
MII 2 1 22 6 46.2 1.23 19.2
MII 3 2 42 11 63 1.28 18.2
MII 4 8 97 24 102 1.30 19.2
MII 5 1.3
PiPrOx
19 4 35.1 1.24 29
MII 6 3 40 10 48.5 1.24 28.1
MII 7 24 99 25 86.9 1.24 29.1
a. 2nd
cationic grafting polymerization time
b. as calculated by GC measurement
c. degree of polymerization of side chain calculated from GC measurements
d. as calculated from GPC traces
e. determination of the cloud point by turbidity measurements of a 1.0 wt % aqueous
polymer solution (upon heating curves)
The thermo-responsive properties of the resulting brushes containing side chains with
different length were investigated by turbidity measurements. The cloud point was defined
analogue to previous reports146
at 10 % transmittance decrease of a 1.0 wt % aqueous polymer
solution. The results are summarized in Table 5. For molecular brushes with PnPrOx side
chains initiated by MII (Figure 43), the transmittance of the aqueous polymer solution
decreased sharply when the temperature reached the cloud point. In addition, the cloud point
of the resulted brush molecules with PnPrOx side chains from MII versus their side chain
length were plotted to analyze the side chain length effect upon the cloud point (Figure 44).
The cloud point for MII
1 (DP of side chain = 3) was found to be 24 °C which is identical with
the value of linear PnPrOx with comparable molar mass (23.8 °C). When the length of side
chain increased (3<DP<10), the cloud point continuously decreased to 18.2 °C for MII 3 at a
side chain DP of 10, which nicely demonstrates the increased crowding as the side chain is
growing from the macroinitiator.

Results and disccusion Chaper 3
60
Figure 43. Determination of the cloud points of molecular brushes with different lengths of
PnPrOx side chains by turbidity measurements. The transition temperatures were determined
by 10% transmittance decrease for brush molecules in 1.0 wt % of aqueous solution (only
heating curves are shown).
More interestingly, the further increase of side chain length (10<DP<25) resulted in a slight
increase of the LCST to 19.2 °C (MII 4). This subtle, but distinct dependency between the
LCST and the side chain length was reproducible and could not be neglected. As shown in
Figure 45, like linear polymer, the molecular brush with short side chains adopts a coiled
conformation. Successively, the molecular brush becomes stretched as the side chain length
increases, which results in a decrease of cloud point as discussed in the previous chapter.
However, the terminal part of side chain becomes loose as the side chain length further
increases. Given the same grafting density, the free volume per side chain is proportional to d2
(d is the apparent diameter of a molecular brush as shown in Figure 45). Therefore, the local
concentration of the side chain terminal part significantly decreases when the side chain
length increases. It is known for POx that the cloud point increases with the decrease of
concentration. As a result, the cloud point increased.

Chapter 3 Results and discussion
61
Figure 44. LCST of molecular brushes with PnPrOx side chains of different length along
with the structure of the molecular brush.
To summarize the effect of side chain length on the LCST of molecular brushes, at very short
side chain length (DP = 3) the side chain oligomers do not yet induce a steric crowding, and
the LCST is virtually identical with linear polymer.169,170
However, as the side chains grow
the crowding of side chains results in a decrease of LCST by several degrees. Furthermore, as
the side chain length increases further, the segments of side chain end experience the
increased volume/side chain i.e. a decrease of local concentration. Thus, the LCST values
increase again, albeit only slightly.

Results and disccusion Chaper 3
62
Figure 45. Illustration for the side chain length effect on the conformation of the molecular
brushes. Top: profile of a molecular brush, middle: section view of molecular brushes and
volume/side chain at different side chain length, bottom: conformation of molecular brushes
with different side chain length.
To further certify this subtle side chain length effect on the LCST, molecular brush with
PnPrOx side chain in different length from a shorter macroinitiator (MI) and brush with
poly(2-iso-propyl-2-oxazoline) (PiPrOx) side chain in different length from MII were also
compared. Similar results were found out. The LCST value of MII 5 (DP = 4) was found to be
29 °C and decreased to 28 °C for MII 6 (DP = 10) as PiPrOx side chain grew. The further
increase of the side chain length resulted in a slight increase of LCST to 29.1 °C for MII 7 (DP
= 24). On the other hand, the LCST value of MI 1 (DP = 6) decreased from 19.5 to 18.1 °C for
MI 2 (DP = 11) and increased to 19.2 °C for M
I 3 (DP = 24) as the PnPrOx side chain grew.
It was reported by Schmidt36
and Matyjaszewski40
that molecular brushes tend to aggregate
intramolecularly in dilute brush solutions as shown in Figure 46. However, linear polymers

Chapter 3 Results and discussion
63
tend to aggregate intermolecularly upon heating. In order to better understand the observed
LCST behavior, dynamic light scattering (DLS) studies were performed to investigate the side
chain length effect on aggregation behaviors of the brush molecules. The apparent diameters
of MII 1and M
II 2 were measured at different temperatures in dilute aqueous solutions. As
shown in Figure 47, the diameter of MII 2 continuously decreased to a minimum (with a
diameter decrease of 11 %) at 22 °C and suddenly increased upon further increasing of the
temperature. However, in the case of MII1, the apparent diameter kept constant below 30 °C,
and drastically increased upon further increasing of the temperature. These observations
corroborate the formation of dense brush like structure of MII 2 and the linear polymer
resemblance of MII 1, which confirms our postulation of side chain length effect on the
conformation of molecular brush. The postulated side chain length effect on the conformation
is shown in Figure 45.
Figure 46. Molecular brushes tend to aggregate intermolecularly at high concentration,
whereas intramolecular collapse is preferred at dilute solution (From Ref. [38]).

Results and disccusion Chaper 3
64
Figure 47. Temperature dependence of apparent diameters of MII 1 and M
II 2 in 0.3 wt %
aqueous solutions.
3.3.3.2 Concentration effect
The concentration effects on the LCST of the homoPOx and coPOx have been investigated
previously. For the hydrophilic PEtOx,118
a strong decrease of the cloud point temperature
was observed with increasing concentration, while the cloud point temperature of amphiphilic
PnPrOx almost kept constant regardless of the increasing concentration.167
However, the
exact nature of the dependence of polymer hydration on variations e.g. chain length and
concentration for the POx is not yet understood.
Here, the cloud point of amphiphilic P(IPOx-g-nPrOx)A at various concentrations was
investigated. As shown in Figure 48, the LCST continuously dropped from 24 °C at 0.1 wt %
to 19 °C at 1.0 wt %. However, a further increase of concentration did not affect the LCST
significantly (not shown in the picture). The significant decrease of LCST observed here is in
contrast to the results of linear PnPrOx reported before.167
Some previous reports revealed
that the unique and compact structure of molecular brushes facilitate intramolecular collapse
above the LCST when the average distance between molecules is much larger than their
hydrodynamic dimensions i.e. in dilute solution.38
However, like most thermo-responsive
linear polymers in solution, intermolecular aggregation is preferred when the intermolecular

Chapter 3 Results and discussion
65
distance was comparable to the individual molecular dimensions i.e. at high concentration.
The abnormal LCST dependence on the concentration observed here most likely ascribes to
the compact structure of molecular brushes. Figure 48 depicts the concentration effect on the
LCST of P(IPOx-g-nPrOx)A.
Figure 48. LCST of molecular brush P(IPOx-g-nPrOx)A at various concentrations (side chain
DP = 25, determined from heating curves).
3.3.3.3 Backbone length effect
In a recent report, the effect of molar mass on the cloud point of linear homoPOx has been
investigated,118
revealing a stronger influence to the more hydrophilic POx in comparison
with the more hydrophobic POx. For example, Schubert and co-workers reported that PEtOx
only exhibited a LCST when DP was larger than 100. Specifically, the LCST decreased from
94 °C at DP = 100 to 66 °C at DP > 500. In contrast, PnPrOx exhibited LCST ranging from
30 °C to 43 °C at the DP of 50-150. Only minor changes in LCST could be observed at DP >
150.
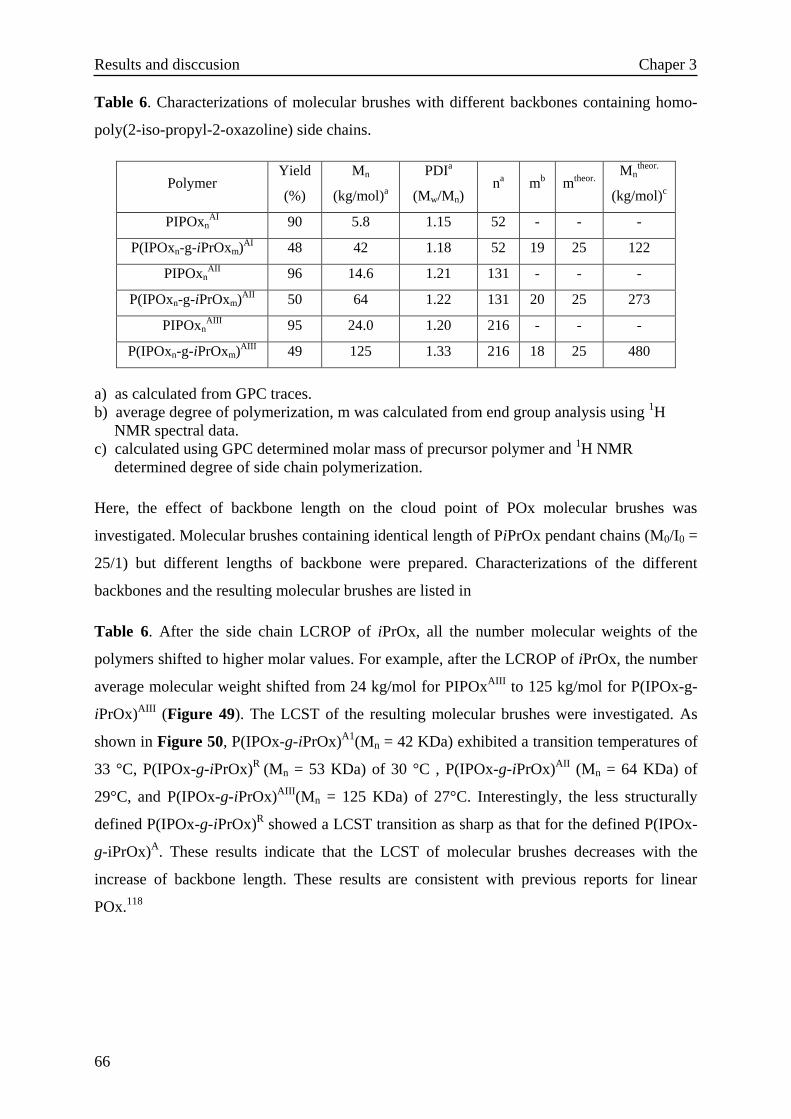
Results and disccusion Chaper 3
66
Table 6. Characterizations of molecular brushes with different backbones containing homo-
poly(2-iso-propyl-2-oxazoline) side chains.
Polymer Yield
(%)
Mn
(kg/mol)a
PDIa
(Mw/Mn) n
a m
b m
theor.
Mntheor.
(kg/mol)c
PIPOxnAI 90 5.8 1.15 52 - - -
P(IPOxn-g-iPrOxm)AI 48 42 1.18 52 19 25 122
PIPOxnAII 96 14.6 1.21 131 - - -
P(IPOxn-g-iPrOxm)AII 50 64 1.22 131 20 25 273
PIPOxnAIII 95 24.0 1.20 216 - - -
P(IPOxn-g-iPrOxm)AIII 49 125 1.33 216 18 25 480
a) as calculated from GPC traces.
b) average degree of polymerization, m was calculated from end group analysis using 1H
NMR spectral data.
c) calculated using GPC determined molar mass of precursor polymer and 1H NMR
determined degree of side chain polymerization.
Here, the effect of backbone length on the cloud point of POx molecular brushes was
investigated. Molecular brushes containing identical length of PiPrOx pendant chains (M0/I0 =
25/1) but different lengths of backbone were prepared. Characterizations of the different
backbones and the resulting molecular brushes are listed in
Table 6. After the side chain LCROP of iPrOx, all the number molecular weights of the
polymers shifted to higher molar values. For example, after the LCROP of iPrOx, the number
average molecular weight shifted from 24 kg/mol for PIPOxAIII
to 125 kg/mol for P(IPOx-g-
iPrOx)AIII
(Figure 49). The LCST of the resulting molecular brushes were investigated. As
shown in Figure 50, P(IPOx-g-iPrOx)A1
(Mn = 42 KDa) exhibited a transition temperatures of
33 °C, P(IPOx-g-iPrOx)R
(Mn = 53 KDa) of 30 °C , P(IPOx-g-iPrOx)AII
(Mn = 64 KDa) of
29°C, and P(IPOx-g-iPrOx)AIII
(Mn = 125 KDa) of 27°C. Interestingly, the less structurally
defined P(IPOx-g-iPrOx)R showed a LCST transition as sharp as that for the defined P(IPOx-
g-iPrOx)A. These results indicate that the LCST of molecular brushes decreases with the
increase of backbone length. These results are consistent with previous reports for linear
POx.118

Chapter 3 Results and discussion
67
Figure 49. GPC traces of PIPOxAIII
backbone and molecular brush P(IPOx-g-iPrOx)AIII
.
Figure 50. LCST of molecular brushes with identical PiPrOx side chains but different
backbone length in 1.0 wt % aqueous solution (side chain DP = 25, determined from heating
curves).

Results and disccusion Chaper 3
68
3.4 Molecular brushes with copoly(2-oxazoline) side chains
3.4.1 Background
A large number of reports investigated structure and composition property relationships for
POx, mainly by varying the monomer composition or end groups. For linear poly(2-
oxazoline)s (POx), changing the length of alkyl chain in the 2-position, the nature of the POx
can be changed from hydrophilic (2-methyl and 2-ethyl as substituents) to hydrophobic POx
with longer alkyl or phenyl 2-substituents. For example, Schubert and co-workers finely tuned
the LCST of poly(2-oxazoline)s by varying composition and molar mass.168
In addition, the
amphiphilicity can be adjusted by the combination of hydrophilic and hydrophobic monomers
in different degree of polymerization ratio. For instance, Kataoka171 ,167
and Jordan33,119
systematically investigated the accurate control of the thermo-sensitivity of linear POx by
copolymerization of 2-alkyl-2-oxazolines. The LCST of the copolymers decreased with an
increase of the content of hydrophobic monomer i.e. 2-n-propyl-2-oxazoline (nPrOx).
Parameters influencing the thermo sensitivity of linear POx are expected to have similar
effect on the LCST of molecular brushes of POx. Here, molecular brushes of copoly(2-
oxazoline) side chains were prepared. The thermo-responsive properties of the resulting
molecular brushes were investigated.
3.4.2 Molecular brushes with statistical copolymer side chains
The grafting from approach and living cationic ring-opening polymerization (LCROP) of 2-
oxazoline allow the efficient and accurate control of the chemical composition of the polymer
chains. In addition to molecular brushes with homopolymer side chains, here the synthesis of
molecular brushes with both, statistical- and block- copolymer side chains is described and
their thermo-responsive properties are investigated. The kinetics of the polymerization of 2-
methyl-2-oxazoline (MeOx) and nPrOx in the presence of macroinitiator MII were
investigated before the preparation of molecular brushes. The apparent polymerization rates
per initiating group were calculated to be 2.12 and 2.95 mL·S-1
·mol-1
for nPrOx and MeOx,
respectively. As shown in Figure 51, the two first order kinetic plots are strictly linear,
indicating a living character of the polymerization. Furthermore, the minor difference in the
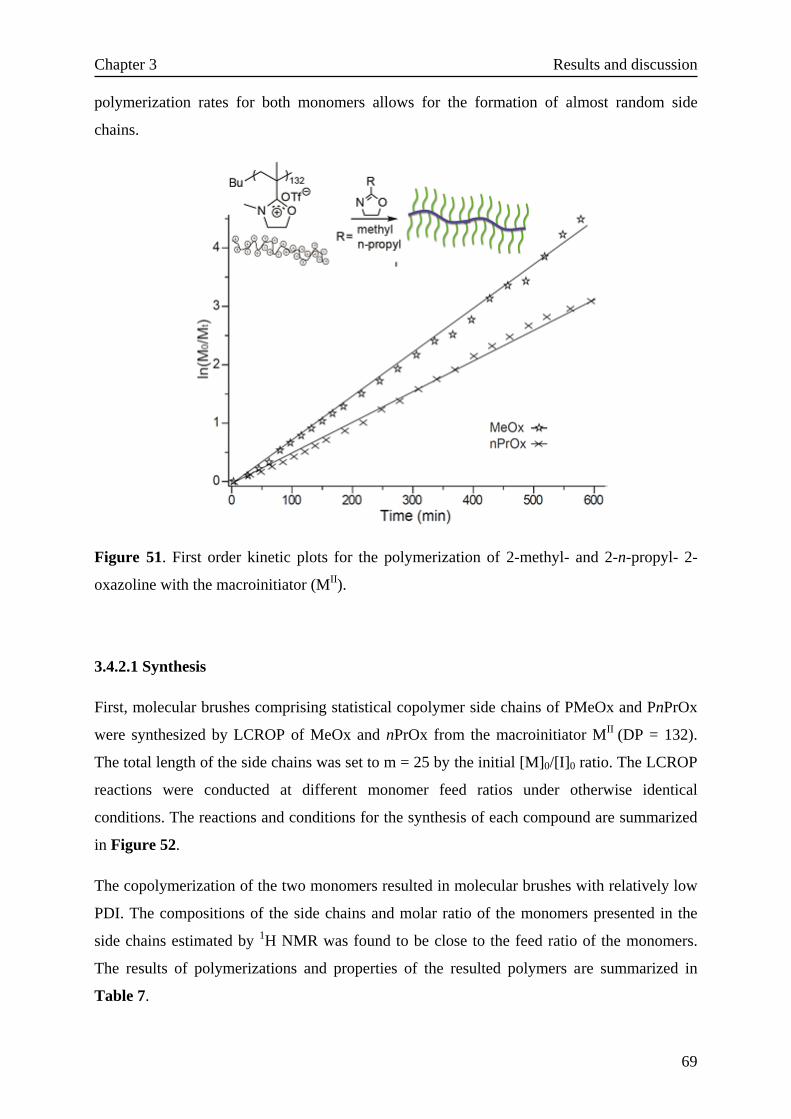
Chapter 3 Results and discussion
69
polymerization rates for both monomers allows for the formation of almost random side
chains.
Figure 51. First order kinetic plots for the polymerization of 2-methyl- and 2-n-propyl- 2-
oxazoline with the macroinitiator (MII).
3.4.2.1 Synthesis
First, molecular brushes comprising statistical copolymer side chains of PMeOx and PnPrOx
were synthesized by LCROP of MeOx and nPrOx from the macroinitiator MII
(DP = 132).
The total length of the side chains was set to m = 25 by the initial [M]0/[I]0 ratio. The LCROP
reactions were conducted at different monomer feed ratios under otherwise identical
conditions. The reactions and conditions for the synthesis of each compound are summarized
in Figure 52.
The copolymerization of the two monomers resulted in molecular brushes with relatively low
PDI. The compositions of the side chains and molar ratio of the monomers presented in the
side chains estimated by 1H NMR was found to be close to the feed ratio of the monomers.
The results of polymerizations and properties of the resulted polymers are summarized in
Table 7.

Results and disccusion Chaper 3
70
Figure 52. Reaction scheme of the synthesis of molecular brushes with statistical and block
copoly(2-oxazoline) side chains. The backbone was synthesized by anionic polymerization of
IPOx.
3.4.2.2 LCST of molecular brushes with random copolymer side chains
Some of the resulting molecular brushes (M1-M5) with statistical coPOx side chains
exhibited thermo-responsive properties i.e having LCST. The LCST was determined by
measuring transmittance of light through aqueous solutions of polymers using a UV
spectrometer. The transmittance of 1.0 wt % aqueous solution of brush molecules was
monitored at 500 nm at a heating and cooling rate of 1.0 °C 5min-1
. The results are
summarized in Table 7.
The linear homoPnPrOx and homoPMeOx have LCST at around 23.8 °C and above 100°C
respectively, and molecular brushes composed of these polymers are expected to have similar
value of LCST. Figure 53 shows the transmittance of the aqueous solution of molecular
brushes. It is seen the transmittance of the polymer solution decreased sharply when the
temperature reaches their LCST. In addition, the dependence of LCST value on MeOx

Chapter 3 Results and discussion
71
content was plotted and shown in Figure 54. The LCST values increased from 19 °C (MeOx
mol % = 0) to 79 °C (MeOx mol % = 70 %), suggesting the balance shifted towards higher
hydrophilicity due to the higher content of MeOx. Moreover, LCST values were found to
increase linearly with the increasing molar content of MeOx (Figure 54) which is consistent
with linear POx. No change in the transmittance was observed in case of MeOx content above
80 %, which is due to the too high hydrophilicity of the side chains.
Table 7. Molecular brushes with copolymer side chains from MeOx and nPrOx.
entry Feed (x:y)
(nPrOx:MeOx) Mn (KDa)a PDIa LCSTb
Composition
(x:y)c
M1 25:0 84 1.20 19 25:0
M2 20:5 92 1.21 33 19:6
M3 15:10 95 1.2 51 14:11
M4 12.5:12.5 98 1.22 57 12:13
M5 11:14 99 1.28 79 9:16
M6 10:15 102 1.26 - 8:17
M7 5:20 113 1.4 - 5:20
M8 0:25 121 1.21 - 0:25
M9 12.5:12.5 96 1.21 32 13:12
M10 12.5:12.5 121 1.6 81 14:11
a) as calculated from GPC traces
b) determined by measuring 10% transmittance decrease of light at λ = 500 nm through
aqueous solutions of the corresponding polymer using a UV spectrometer
c) calculated from 1H NMR spectroscopy

Results and disccusion Chaper 3
72
Figure 53. Determination of cloud points of molecular brushes with different MeOx content.
Only heating curves are shown here (the same transition point was found with no noticeable
hysteresis upon cooling).
Figure 54. LCST values versus MeOx molar content of the side chain of molecular brushes.

Chapter 3 Results and discussion
73
3.4.3 Molecular brushes with block copolymer side chains
Molecular brush with PnPrOx-block-PMeOx (M9) and PMeOx-block-PnPrOx (M10) block
sequences in the side chain were prepared to examine the segment sequences effect on the
LCST. M9 and M10 are molecular brushes with opposite block sequences i.e. M9 has a
PnPrOx core and a PMeOx shell, while M10 has a PMeOx core and a PnPrOx shell. The side
chain length of the molecular brushes was set to nPrOx:MeOx = 12.5:12.5. Molecular
structures of M9 and M10 are shown in Figure 55. The composition of side chain and molar
ratio of comonomers presented in the side chain were estimated by 1H NMR. The final
composition of side chains was found close to the feed ratio of the comonomers. In addition,
the GPC trace of the molecular brushes shifted to higher molecular weight after a second
LCROP of 2-oxazoline, indicating the successful introduction of the second block, however
with slightly increased PDI. The characterization of M9 and M10 with respect to 1H NMR
and GPC is listed in Table 7.
3.4.3.1 LCST of molecular brushes with block copolymer side chains
LCST values of M9 and M10 were measured and compared with the value of M4, which
consists of statistical copolymer side chain of identical nPrOx/MeOx ratio and side chain
length. The LCST were determined at 10% transmittance decrease of a 1.0 wt % aqueous
polymer solution as shown in Figure 56. The sequence of blocks in the side chain of
molecular brushes was found to have a pronounced impact on the LCST value. For M9, with
a sequence PnPrOx-b-PMeOx, the LCST was found to be 32 °C in contrast to a transition
temperature of 81 °C for M10 with a sequence of PMeOx-b-PnPrOx. A hydrophilic brush
core with a hydrophobic shell resulted in a higher LCST than a hydrophobic brush core with a
hydrophilic shell. This observation suggests that the more hydrophobic PnPrOx block inside
the molecular brush firstly aggregates at its intrinsic LCST and then drive the brush molecule
to precipitate. However, in the case of PMeOx as the inside block, the nPrOx shell starts to
aggregate forming small aggregates around the brush molecule while the inside PMeOx block
is still soluble. Due to the driving force is not strong enough, brush molecule remains soluble.
In the recent report by Matyjaszewski, 40
the impact of the block sequence in molecular brush
side chains of oligo(ethylene oxide) on the LCST value was investigated. Molecular brush

Results and disccusion Chaper 3
74
with a hydrophilic core (MEO3MA) and a hydrophobic shell (MEO2MA) has a lower LCST
as compared to molecular brushes with a hydrophobic core and a hydrophilic shell.
The results observed here seem contradict to Matyjaszewski‟s findings. However, it should be
noted the side chain length as well as the hydrophilic/ hydrophobic ratio in the side chain in
this experiment differ significantly from the ones in literature. They are not comparable
virtually. Additionally, it was discussed in the previous section that the LCST of M4, with
statistical copolymer side chains, is 57 °C, which is just in between the LCST of M9 and M10.
This also corroborates the formation of random or slightly gradient copoly(2-oxazoline) side
chains as illustrated in Figure 55.
Figure 55. Illustration of the formation of molecular brushes with statistical and block
copoly(2-oxazoline) side chains.

Chapter 3 Results and discussion
75
Figure 56. Determination of LCST values of molecular brushes with statistical and block
copolymer side chains (MeOx:nPrOx = 12.5:12.5, molar ratio) by turbidity measurements of
1.0 wt% aqueous solutions. (only heating curves are shown)
Figure 57. LCST of linear POx and molecular brushes of POx.
To sum up, the LCST of POx molecular brushes can be adjusted in a broad range from 16 to
81°C by copolymerizing 2-oxazolines, varying end groups, molar mass, side chain length and
composition (Figure 57). In addition, molecular brushes of block coPOx side chains have
been prepared forming core-shell structures.

Results and disccusion Chaper 3
76
3.5 Bottle-brush brushes of poly(2-oxazoline)s
3.5.1 Background
The preparation of polymer brushes172
on solid substrates, to tune the interfacial properties,
has been of great interest due to the potential application of polymer brushes as sensing layers,
anti-corrosion layers, protein resistant surfaces and nanostructured surfaces.76,173,174,175
Recently, our group developed the preparation of nano/micro structured polymer brushes on a
variety of surfaces by electron beam chemical lithography (EBCL)/electron beam carbon
deposition (EBCD) and successive self-initiated photografting and photopolymerization
(SIPGP).66,67,68,69
SIPGP, compared to other methods for the preparation of polymer brushes,
has gained much interest. It allows the preparation of polymer brushes on various surfaces
simply by irradiation of the substrate by UV light in the presence of bulk monomer. The only
requirement is the presence of surface functionalities on the substrate possessing abstractable
groups that can be abstracted by a photoactivated monomer. The formed surface-bound
radicals initiate a free radical polymerization. This method allows for the formation of
polymer grafts on relatively undefined organic surfaces. Specific pretreatment and
modification of surfaces with special initiator functions is strongly facilated.
In this part of work, poly(2-isopropenyl-2-oxazoline) (PIPOx) brushes were prepared by the
SIPGP of 2-isopropenyl-2-oxazoline (IPOx) on various substrates such as bare polished
glassy carbon, amino silane modified glass slide/silicon wafer and biphenylthiol self-
assembled monolayers (SAMs) modified gold. Subsequently, by following the synthetic route
for the preparation of molecular brushes of poly(2-oxazoline)s in the previous chapter, PIPOx
brushes were converted to macroinitiator brushes by the reaction with MeOTf. This
macroinitiator was then used to prepare brushes of bottle-brushes of poly(2-oxazoline)s (POx)
by the LCROP of different 2-oxazoline monomers. In the following, these structures are
referred to „bottle-brush brushes‟ (BBBs).
Molecular brushes of PMeOx side chains have been found non-toxic in the previous chapter.
In this part of work, polymer brushes of POx were introduced between solid substrates and
biosystems. Micro/nano structured polymer brushes have been realized by stencil mask or
electron beam chemical lithography. The discussion of first biomedical test i.e. non-specific

Chapter 3 Results and discussion
77
protein adsorption and cell adhesion using these BBB coatings will be shown in the last
chapter of the thesis.
3.5.2 Bottle-brush brushes of poly(2-oxazoline)s on glassy carbon
3.5.2.1 Glassy carbon
Glassy carbon,176
also called vitreous carbon, is a non-graphitizing carbon that combines
glassy and ceramic properties. Remarkable properties of glassy carbon include high
temperature and chemical resistance, and impermeability to gases and liquids.177
Therefore,
glassy carbon is widely used as electrode materials in electrochemistry, as well as for high
temperature crucibles and as a component of some prosthetic devices. Glassy carbon is
prepared by a series of heat treatments of organic precursors, such as polymeric resins at
temperatures up to 3000 oC. The structure of glassy carbon has long been a subject of debate.
Early structural models assumed that both sp2 and sp
3 -bonded atoms were present, but it is
now known that glassy carbon is composed of 100% sp2 material. Recent research suggests
that glassy carbon has a fullerene-related structure (Figure 58).178
Figure 58. Models for the structure of glassy carbon: a) The Jenkins-Kawamura model of
glassy carbon, La and Lc are the lengths of the graphitic domains perpendicular and parallel
to the graphite axis; b) at low-temperature and c) at high-temperature glassy carbon made
from the Harris-Tsang model (From Ref. [178]).

Results and disccusion Chaper 3
78
3.5.2.2 Self-initiated photografting and photopolymerization
The monomer 2-isopropenyl-2-oxazoline (IPOx) has two orthogonal polymerizable groups,
namely a vinyl group for living anionic or radical polymerization (here used for the SIPGP)
and the 2-oxazoline ring for the living cationic ring-opening polymerization (LCROP). After
thorough cleaning with ultrasound in different organic solvents, a polished glassy carbon
substrate was submerged in bulk IPOx and irradiated with UV-light of a spectral distribution
between 300 and 400 nm (λmax = 350 nm) for 40 h. In order to structure the substrate, the UV-
irradiation was performed through a stencil mask (TEM grid) with rectangular openings of
50×50 µm2. After the polymerization, the substrate was rigorously cleaned by ultrasound in
several solvents of different polarity to ensure that only chemically grafted polymer remains
on the substrate. The preparation of structured PIPOx brushes on glassy carbon is
schematically outlined in Figure 59 (for structuring) and Figure 60.
Figure 59. Scheme of the setup for SIPGP of IPOx, structures can be introduced by UV light
through a transmission electron microscopy (TEM) grid (opening: 50×50µm2).
Figure 60. Preparation of structured P(IPOx-g-2-alkyl-2-oxazoline) bottle-brush brushes on
glassy carbon. a)-b) Structured PIPOx brush was created on glassy carbon by UV-induced
SIPGP of IPOx, structures were from a stencil mask. b)-c) Living cationic ring-opening
polymerization of 2-oxazolines from the grafted PIPOx macroinitiators to form bottle-brush
brushes.

Chapter 3 Results and discussion
79
AFM measurements revealed that a polymer brush layer with a thickness of 76 ± 9 nm was
selectively formed on the irradiated surface area after 16h UV irradiation (Figure 61).
Moreover, the surface roughness of the native glassy carbon substrate (rms 4.6 nm) was
rendered by the additional polymer layer, showing a lower average roughness value of 3.0 nm
(rms).
The successful modification of the glassy carbon substrate by PIPOx brushes was further
confirmed by infrared (IR) spectroscopy. In Figure 66, the characteristic absorption bands of
PIPOx are observed. The strong bands at 1648 cm-1
and 1128 cm-1
are assigned to the (C=N)
and (C-O) stretching modes. The two bands at 987 and 951 cm-1
originate from the ring
skeletal vibration of the 2-oxazoline rings. These assignments are in agreement with bulk
PIPOx as discussed previously.
Figure 61. AFM 3D view and depth analysis of structured PIPOx brush (irradiation time =
16h). The average height was determined by depth analysis to be h = 76nm.
As shown in previous studies, using different substrates such as polyethylene,62
aromatic
SAMs on gold69
and oxidized diamond,67
the grafting reaction and the formation of polymer
brushes occurs via the SIPGP mechanism in which the monomer acts as a photosensitizer to
activate a surface functional group by hydrogen abstraction to start a free radical surface-
initiated polymerization. The only requirement for the photografting reaction is the possibility
for hydrogen abstraction by a radical mechanism. The SIPGP of IPOx also follows this
mechanism as shown in Figure 63. All the presented SIPGP experiments in the thesis were
performed at room temperature using a UV-light of a spectral distribution between 300 and
400 nm (max = 350) from Rayonet.

Results and disccusion Chaper 3
80
Various studies have investigated the surface functionalities of polished glassy carbon
substrates. Raman spectroscopy has shown that polishing severely disrupts the substrate
structure.179
Polishing does not only affect the upper monolayer but dramatically changes the
substrate microstructure within a region of 10-20 nm. Polished glassy carbon surfaces are
composed of smaller microcrystallites (compared to bulk glassy carbon) with many graphitic
edges.179
The formation of PIPOx brushes on polished glassy carbon can be explained by the
low C-H bond dissociation energy (BDE) on such graphitic edges.180
Furthermore, polishing
causes a partial oxidation of the glassy carbon substrate resulting in C-OH and C=O surface
functionalities (Figure 62).181
Collier et al. reported that polished glassy carbon substrates are
covered with up to 10 % of aromatic OH groups.182
Due to the low BDE of aromatic alcohol
groups,183
it is most likely that hydrogen atoms are also abstracted radically from surface OH
functionalities during the SIPGP process.
Figure 62. Representative surface functionalities on glassy carbon surface.
Figure 63. SIPGP reaction mechanism for the formation of PIPOx brush on polished glassy
carbon.

Chapter 3 Results and discussion
81
Figure 64. Preparation of P(IPOx-g-2-alkyl-2-oxazoline) bottle-brush brushes on glassy
carbon. a)-b) PIPOx brushes were created on the substrate by the SIPGP of IPOx. c)
Conversion of the PIPOx brush backbone to the surface bound macroinitiator PIPOxOTf. d)
LCROP of 2-alkyl-2-oxazoline from the macroinitiator. e) Termination of the side chain
polymerization with N-Boc-piperazine and deprotection of the Boc groups allows the further
functionalization of the side chain end groups with rhodamine B isothiocyanate.
Ex situ kinetic studies of the SIPGP of IPOx monomer were performed on individual glassy
carbon samples at different UV irradiation times (2 - 40 h) using the same stencil mask. In
Figure 65, the thickness of the polymer brush layer as measured by AFM under ambient
conditions is plotted as a function of the UV irradiation time. For polymerization times below
10 h, an almost constant growth rate of 6.1 nm.h-1
is observed. However, the layer thickness
growth rate decreases significantly for longer polymerization times. We observed that the
bulk monomer phase became highly viscous with longer irradiation times due to self-initiated
photopolymerization of the IPOx monomer in solution. The limited film growth can therefore
be explained by either the monomer concentration decrease and/or the limited mass transport
of the remaining monomer molecules. This kinetic behavior is in agreement with previous
reports for SIPGP for other vinyl monomers.184

Results and disccusion Chaper 3
82
Figure 65. PIPOx brush layer thickness on polished glassy carbon as a function of the
irradiation time as measured by AFM on structured polymer layer (50×50 µm2).
3.5.2.3 Surface-initiated living cationic ring-opening polymerization
We have discussed the preparation of molecular brushes from the dual-functional IPOx
monomer in the previous chapter. The PIPOx backbone was converted quantitatively with
methyl triflate to a polycationic macroinitiator for living cationic polymerization of different
2-alkyl-2-oxazolines. Here, we have adapted this synthetic route to prepare bottle-brush
brushes on glassy carbon as shown Figure 64. PIPOx brushes were converted into the
polycationic macroinitiator salt (PIPOxOTf) by submerging the PIPOx modified glassy
carbon substrate in a solution of methyl trifluoromethanesulfonate (MeOTf). Successively, the
LCROP of 2-ethyl-2-oxazoline (EtOx) was performed for 20 min at 130 °C. N-Boc-
piperazine was used as terminating reagent. This terminal group allows an additional
functionalization of each side chain end after deprotection of the secondary amine in
trifluoroacetic acid (TFA). After the LCROP and deprotection of the Boc group, the substrate
was intensively cleaned by ultrasound in different solvent to remove physisorbed material.
The successful conversion of PIPOx brushes to P(IPOx-g-EtOx) bottle-brush brushes was
confirmed by IR spectroscopy (Figure 66). The (C=N) and (C-O) stretching bands at 1648,
1128 and 987cm-1
as well as the two ring skeletal vibration bands from the pendant 2-
oxazoline ring in PIPOx brushes disappeared. A new intensive band appeared around 1627

Chapter 3 Results and discussion
83
cm-1
which is characteristic for the carbonyl stretching mode of the amide function. Moreover,
the characteristic CHx deformation modes for EtOx are observed around 1421 cm-1
. The
complete disappearance of the oxazoline ring IR bands indicates a high side chain grafting
density and quantitative reaction.
Figure 66. IR spectra of PIPOx and P(IPOx-g-EtOxBoc) brushes on glassy carbon.
AFM analysis of the resulting polymer layer revealed that the polymer brushes did not desorb
during the successive reaction steps. This demonstrates that besides the ultrasound stability,
the polymer grafts created on glassy carbon are thermally (the LCROP was performed at
130°C) as well as chemically stable under various reaction conditions. Furthermore, as shown
in Figure 67, AFM measurements showed a significant thickness increase of the polymer
structures from 159 ± 8 nm to 330 ± 10 nm after the LCROP. The layer thickness increase of
approximately 108 % is attributed to the increase of the molecular mass of the grafted chains
and the additional stretching of the backbone due to the crowding of the side chains. We also
found that the deprotection of the Boc group results in a slight thickness decrease of
approximately 5 % due to material loss.

Results and disccusion Chaper 3
84
Figure 67. AFM 3D view of polymer brushes structure on glassy carbon. a) The SIPGP of
IPOx for 40h through a stencil mask gives structured PIPOx brushes with a thickness of 159 ±
8 nm. b) The LCROP of EtOx and termination with N-Boc-piperazine results in 330 ± 10 nm
thick P(IPOx-g-EtOxBoc) bottle-brush brushes. c) Same structure after deprotection of the
side chain terminal Boc group.
Also for the second surface-initiated living cationic ring-opening polymerization (SI-LCROP),
the thickness increase was investigated as a function of the polymerization time. For this, a
PIPOx brush was prepared by SIPGP and then divided into three pieces. With each sample,
SI-LCROP was performed using MeOx as the monomer for 60, 120, and 240 minutes (Figure
68) at 80 °C. The percentage of the respective thickness increase due to the formation of
bottle-brush brushes with PMeOx side chains was found to be linear as shown in Figure 69.
The systematic thickness increase demonstrates that the sensitive SI-LCROP can be
performed in reproducible and consistent manner and individual SI-LCROP reactions can be
compared. However, these experiments do not give a detailed picture about the grafting
efficiency and the resulting polymer architecture. On the other hand, based on our experience
with SI-LCROP using initiator functionalized self-assembled monolayers on planar
substrates54
and especially on nanoparticles,141
this route to coat and functionalize a broad
variety of surfaces68
with the versatile POx has a high potential for the development of
functional surfaces for the control of protein adsorption and cell adhesion which will be
discussed in chapter 3.7.204
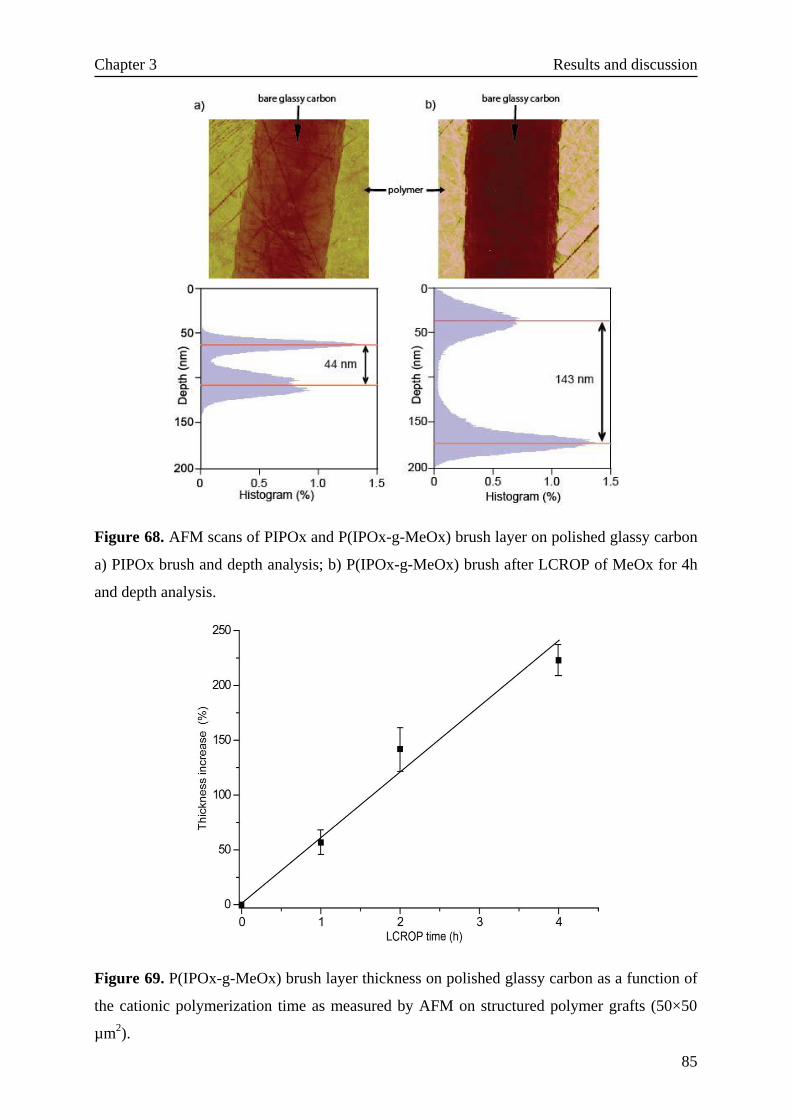
Chapter 3 Results and discussion
85
Figure 68. AFM scans of PIPOx and P(IPOx-g-MeOx) brush layer on polished glassy carbon
a) PIPOx brush and depth analysis; b) P(IPOx-g-MeOx) brush after LCROP of MeOx for 4h
and depth analysis.
Figure 69. P(IPOx-g-MeOx) brush layer thickness on polished glassy carbon as a function of
the cationic polymerization time as measured by AFM on structured polymer grafts (50×50
µm2).

Results and disccusion Chaper 3
86
3.5.2.4 Wettability of the bottle-brush brushes
The projected use of polymer brushes in the biomedical field will require a broader choice of
functional and biocompatible/bioresponsive surfaces. We have prepared molecular brushes in
solution, the LCROP from the PIPOxOTf macroinitiator could be performed with different 2-
alkyl-2-oxazolines. By changing the side chain composition, it is possible to change the
hydrophobicity of the molecular brushes and to fine-tune the LCST temperature.
In order to demonstrate that bottle-brush brushes with a variety of side chains are accessible
by an analogue approach, we have performed the LCROP from PIPOxOTf macroinitiator
brush with different 2-alkyl-2-oxazolines under identical reaction conditions. We have tested
2-methyl-, 2-ethyl, 2-n-propyl- and 2-butyl-2-oxazoline (MeOx, EtOx, nPrOx and BuOx
respectively). The influence of the bottle-brush brushes side chains on the hydrophilic/
hydrophobic character of the polymer coating was investigated by contact angle
measurements (Table 8). The LCROP of the different 2-alkyl-2-oxazolines was performed
under identical reaction conditions from homogeneous and unstructured PIPOxOTf brushes
on glassy carbon. Figure 70 shows that the hydrophilicity of the bottle-brush-brushes can be
adjusted by the side chain composition.
Figure 70. Static water contact angles of PIPOx and P(IPOx-g-2-alkyl-2-oxazoline) on glassy
carbon.

Chapter 3 Results and discussion
87
Table 8. Static water contact angles of on different homogeneous polymer brush coatings on
polished glassy carbon substrates. The PIPOx brushes were performed by the SIPGP of IPOx
over night. The LCROP was performed at 130°C by microwave heating for 20 minutes.
Substrate Static contact angle (°)
Polished glassy carbon 85 ± 2
PIPOx 41 ± 3
P(IPOx-g-MeOxBoc) 43 ± 2
P(IPOx-g-EtOxBoc) 49 ± 2
P(IPOx-g-PrOxBoc) 69 ± 3
P(IPOx-g-BuOxBoc) 95 ± 2
It is noteworthy that part of LCROP reactions in this chapter were performed at 130 °C by
microwave heating. This technique is an efficient method for performing the LCROP of 2-
oxazoline, overcoming the long reaction times when carried out under conventional heating.
Furthermore, the living character of the polymerization is retained under microwave
irradiation.185
In order to estimate the influence of the microwave irradiation, the LCROP of
EtOx was performed from structured PIPOxOTf brushes on glassy carbon at 130°C by
conventional oil bath heating and in identical reaction conditions. AFM measurements
revealed a thickness increase of approximately 97 % (from 150 nm for the PIPOx brushes to
293 nm for the P(IPOx-g-EtOxBoc)). Taking the experimental error into account, this result
shows that the microwave irradiation do not accelerate significantly the surface initiated
LCROP reaction.
3.5.2.5 Further functionalization of bottle-brush brushes
In order to demonstrate that the side chain terminal amino group (after deprotection of the
Boc group) can be further functionalized, P(IPOx-g-EtOx) bottle-brush brushes were labeled
with rhodamine B isothiocyanate (Figure 64). After intensive cleaning with ultrasound in
ethanol in order to remove all physisorbed material, a strong fluorescence signal was detected
selectively at the polymer modified surface (Figure 71), indicating that the fluorescent dye
was covalently bonded to the polymer brushes. This experiment shows that despite the high
crowding of polymer chains in such bottle-brush brushes, the side chain terminal amino group
is still accessible for further functionalization, even with steric demanding organic molecules.

Results and disccusion Chaper 3
88
The functionalization of the terminal amino group of bottle-brush brushes with biomolecules
(polysaccharides, proteins, etc.) will be the subject of future experiment.
Figure 71. Fluorescence image and section analysis of a patterned P(IPOx-g-EtOx) bottle
brushes labeled with rhodamine B isothiocyanate on glassy carbon. The bright regions are
polymer brushes coated glassy carbon.
3.5.2.6 Stability test
To test the stability of the BBB coating, prolonged ultrasonication in toluene, acetate, ethanol
and water 5minutes each was performed. These unusual drastic treatments had no significant
effect on thickness as well as morphology as confirmed by IR and AFM analysis. Therefore,
we used these cleaning procedures to remove the physisorbed polymer and clean the substrate.
In addition, BBBs coated substrates were boiled in solvents such as water, chloroform and
toluene over night. Again, no influence can be found. Moreover, the polymer brush also
showed very good stability in chemical harsh conditions such as strong acid (HCl, 1M) and
base (NaOH, 1M) at 100 °C over night. These stability tests showed that the polymer layer
was covalently attached to the glassy carbon substrate.

Chapter 3 Results and discussion
89
3.6 Bottle-brush brushes on self-assembled monolayers
3.6.1 Self-assembled monolayers
Self-assembled monolayers (SAMs) allow a defined modification of inorganic substrates such
as gold, and oxidized surfaces.186
For the preparation of SAMs, the inorganic substrate is
immersed into a dilute organic solution of reactive molecules. The molecules (covalently)
bind to the substrate and assemble into a monolayer. Typically, these reactive molecules are
composed of a reactive anchor group which binds to the substrate and a mesogen in order to
induce local order into the monomolecular layer. For example, n-alkyl thiols form S-Au
bonds on gold substrate, yielding densely packed monolayers when the n-alkyl chains are
longer than ten carbon atoms. Similarly, glass and silicon substrates can be modified with
chloro- or alkoxy silanes.
Silicon wafers are ideal substrates for the study of surface initiated polymerizations (SIP) as
they are relatively smooth (for AFM investigation), reflective (for spectroscopic
investigations) and readily available. The surface of silicon consists of a thin layer of silicon
oxide with silane functionalities. Thus, the treatment of silicon substrate with chlorosilanes or
alkoxysilanes results in a polysiloxane network that is covalently linked to the native SiO2
surface via siloxane bonds (Figure 73). Chlorosilanes are very sensitive to water, and
generally form less-ordered SAMs than alkoxysilanes. Alkoxysilanes are utilized widely since
they are less reactive and easier to handle. The preparation of SAMs on glass is identical to
that of silicon substrates. Glass is optically transparent and can be thus characterized by UV-
vis spectroscopy in the transmittance mode.
In the previous chapter, the self-initiated photografting and photopolymerization (SIPGP) of
2-isopropenyl-2-oxazoline (IPOx) on glassy carbon was described. Here, adapting this
procedure, poly(2-isopropenyl-2-oxazoline) (PIPOx) brushes and bottle-brush brushes (BBBs)
were created on aminosilane modified glass/ oxidized silicon and biphenylthiol modified gold
substrates. Before the grafting of PIPOx brush on SAM modified substrate, attempts were
made to grow PIPOx brush directly on silicon/ glass and gold substrates by SIPGP. However,
no PIPOx brushes were found after SIPGP of IPOx due to the higher bond-dissociation
energy of SiO-H bond on silicon/ glass and the absence of abstractable hydrogen on gold.

Results and disccusion Chaper 3
90
3.6.1.1 3-Aminopropyltrimethoxysilane monolayers
Silicon substrates and glass slides were first cleaned by a piranha solution (H2SO4/H2O2 (30 %
aqueous solution) (v/v) = 3/1) and successively rinsed with deionized water. The water
contact angles on oxidized silicon and glass substrates were nearly 10°, revealing the high
cleanness and uniformity of the surfaces. SAMs of 3-aminopropyltrimethoxysilane were
prepared on these substrates according to a synthetic procedure shown in Figure 73. The
water contact angle of SAM-modified substrates was found to be 55±3°. It has been reported
previously that the contact angle of aminoalkoxysilane SAMs ranges from 40° to
63°.187,188,189,190
It has also been found that a 100% amino-terminated monolayer exhibit a
contact angle of approximately 60°.191
Here, the contact angle of SAM modified samples is in
good agreement previous studies.
3.6.2 Bottle-brush brushes on 3-aminopropyltrimethoxysilane modified
oxidized silicon and glass substrates
3.6.2.1 Self-initiated photografting and photopolymerization
The 3-aminopropyltrimethoxysilane modified silicon substrate was submerged in 2-
isopropenyl-2-oxazoline (IPOx) monomer and the self-initiated photografting and
photopolymerization (SIPGP) was performed for 24 h with UV-light of a spectral distribution
between 300 and 400 nm (λmax = 350 nm). After 24 h of irradiation and thorough cleaning of
the substrate in water, ethanol and ethyl acetate, AFM analysis revealed that a polymer layer
with a thickness of 56±4 nm was formed on the silicon substrate. The NH-H bond
dissociation energy (BDE) of 3-aminopropyltrimethoxysilane was calculated to be approx. 97
kcal⋅mol-1
.192
The formation of PIPOx brush on aminosilane functionalized substrate can be
explained by the SIPGP mechanism in which surface hydrogen atoms from amino groups can
be abstracted radically by excited IPOx molecules.
Structured PIPOx brushes with dimension of 50×50 µm2 were created on aminosilane
modified glass by SIPGP of IPOx with UV irradiation through a stencil mask (TEM grid) as
shown in Figure 72. After 24 h UV irradiation, a 130 nm-thick polymer layer was formed on
the glass substrate, which is around two times the thickness of PIPOx brush on silicon

Chapter 3 Results and discussion
91
substrate (56 nm after identical SIPGP time). This might be caused by the optical
transparency of the glass substrate between 300 and 400 nm. The SIPGP experiments were
performed by placing the substrate in between two identical UV lamps. For the transparent
glass substrate, the UV intensity in the vicinity of surface is approximately two times of that
on silicon substrate. Therefore, the PIPOx brush on glass substrate is twice as thick as on
silicon substrate. This is in agreement with the results of Steenackers‟ PhD thesis.66
The successful modification of the silicon substrate by PIPOx brushes was further confirmed
by infrared (IR) spectroscopy (Figure 75). The strong adsorption bands at 1648 cm-1
and
1128 cm-1
were assigned to the (C=N) and (C-O) stretching modes of the 2-oxazoline ring.
Besides, the two bands at 987 cm-1
and 951 cm-1
originated from the ring skeletal vibration of
the 2-oxazoline rings.
Figure 72. AFM scans (60 × 60 µm2) and section analysis of the structured polymer brush on
glass. a) The SIPGP of IPOx for 24 h (λmax = 350 nm) through a stencil mask results in
structured PIPOx brushes with a thickness of 130 ± 15 nm on 3-aminopropyltrimethoxysilane
modified glass substrate. b) The LCROP of 2-methyl-2-oxazoline (MeOx) from the surface
bound macroinitiator results in bottle-brush brushes with a layer thickness of 340 ± 10 nm.
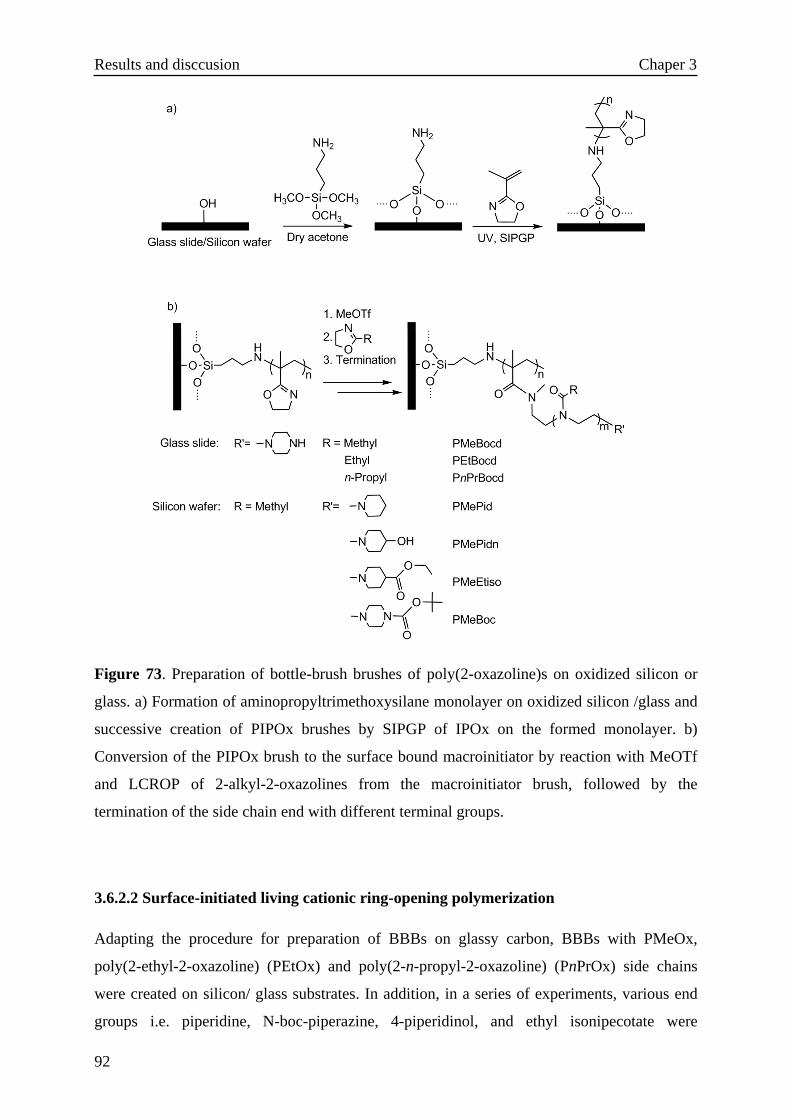
Results and disccusion Chaper 3
92
Figure 73. Preparation of bottle-brush brushes of poly(2-oxazoline)s on oxidized silicon or
glass. a) Formation of aminopropyltrimethoxysilane monolayer on oxidized silicon /glass and
successive creation of PIPOx brushes by SIPGP of IPOx on the formed monolayer. b)
Conversion of the PIPOx brush to the surface bound macroinitiator by reaction with MeOTf
and LCROP of 2-alkyl-2-oxazolines from the macroinitiator brush, followed by the
termination of the side chain end with different terminal groups.
3.6.2.2 Surface-initiated living cationic ring-opening polymerization
Adapting the procedure for preparation of BBBs on glassy carbon, BBBs with PMeOx,
poly(2-ethyl-2-oxazoline) (PEtOx) and poly(2-n-propyl-2-oxazoline) (PnPrOx) side chains
were created on silicon/ glass substrates. In addition, in a series of experiments, various end
groups i.e. piperidine, N-boc-piperazine, 4-piperidinol, and ethyl isonipecotate were

Chapter 3 Results and discussion
93
introduced into the BBBs. Here, different end groups provide a broad choice of functionalities
for further functionalization and i.e. protein immobilization. The preparation of 3-
aminopropyltrimethoxysilane self-assembled monolayer on oxidized silicon or glass
substrates, SIPGP of IPOx and the LCROP of 2-oxazoines are summarized in Figure 73.
Figure 74. AFM scans (50 × 50 µm2) and depth analysis of the polymer brush on oxidized
silicon. a) The SIPGP of IPOx for 24 h (λmax = 350 nm) results in 56 ± 3 nm thick PIPOx
brushes on 3-aminopropyltrimethoxysilane modified silicon substrate. b) The LCROP of
MeOx from the surface-bound macroinitiator results in a 140 ± 10 nm thick bottle-brush
brush (the dark region is the bare substrate, the bright area is covered by BBB).
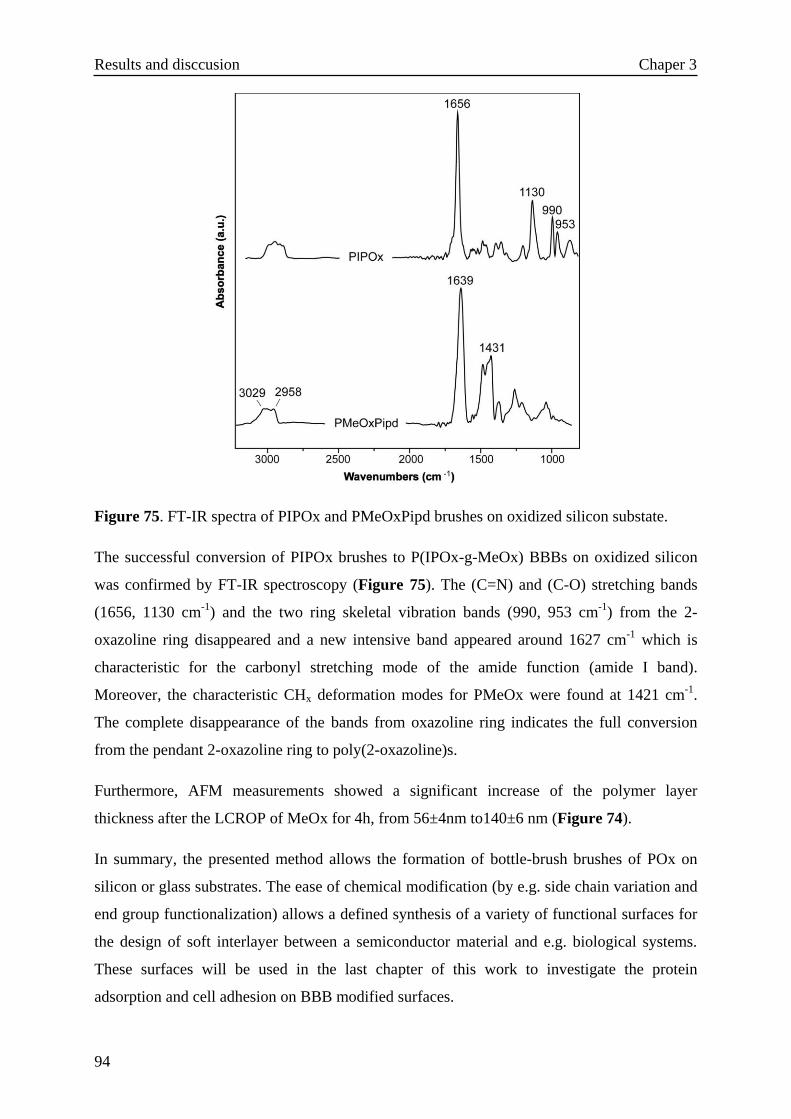
Results and disccusion Chaper 3
94
Figure 75. FT-IR spectra of PIPOx and PMeOxPipd brushes on oxidized silicon substate.
The successful conversion of PIPOx brushes to P(IPOx-g-MeOx) BBBs on oxidized silicon
was confirmed by FT-IR spectroscopy (Figure 75). The (C=N) and (C-O) stretching bands
(1656, 1130 cm-1
) and the two ring skeletal vibration bands (990, 953 cm-1
) from the 2-
oxazoline ring disappeared and a new intensive band appeared around 1627 cm-1
which is
characteristic for the carbonyl stretching mode of the amide function (amide I band).
Moreover, the characteristic CHx deformation modes for PMeOx were found at 1421 cm-1
.
The complete disappearance of the bands from oxazoline ring indicates the full conversion
from the pendant 2-oxazoline ring to poly(2-oxazoline)s.
Furthermore, AFM measurements showed a significant increase of the polymer layer
thickness after the LCROP of MeOx for 4h, from 56±4nm to140±6 nm (Figure 74).
In summary, the presented method allows the formation of bottle-brush brushes of POx on
silicon or glass substrates. The ease of chemical modification (by e.g. side chain variation and
end group functionalization) allows a defined synthesis of a variety of functional surfaces for
the design of soft interlayer between a semiconductor material and e.g. biological systems.
These surfaces will be used in the last chapter of this work to investigate the protein
adsorption and cell adhesion on BBB modified surfaces.

Chapter 3 Results and discussion
95
3.6.3 Bottle-brush brushes on self-assembled monolayers of biphenylthiol
on gold
It has been reported that the electron beam irradiation of ω-functionalized biphenylthiol self-
assembled monolayers (SAMs) causes lateral cross-linking of the biphenyl species.69,193,194
The successive self-initiated photografting and photopolymerization (SIPGP) of vinyl
monomers on such SAMs allows the formation of stable nanostructured polymer brushes on
gold substrate.69
Here, SIPGP of 2-isopropenyl-2-oxazoline (IPOx) on nanostructured ω-functionalized
biphenylthiol SAMs on gold which were selectively cross-linked and chemically modified by
electron beam chemical lithography (EBCL) was investigated. The increasing extent of cross-
linking for the SAMs caused the formation of PIPOx brushes with a gradient grafting density.
As a consequence, a PIPOx brush with a gradient polymer layer thickness was obtained.
Successive LCROP of 2-n-propyl-2-oxazoline (nPrOx) resulted in gradient bottle-brush
brushes (BBBs).
Sören Schilp from the research group of Prof. Michael Grunze at the Universität Heidelberg
prepared biphenylthiol SAMs on gold and irradiated them with a focused electron beam. As
shown in Figure 76, the 4-mercapto-1,1′-biphenyl SAM on gold was structured and cross-
linked by electron irradiation using a focused electron beam of a scanning electron
microscope (SEM) coupled to a pattern generator (direct e-beam writing). The pattern
generator allows the creation of almost any 2D structure. Furthermore, the applied electron
dosage can be controlled within each structure. For a 10×50 μm2 gradient, a write field of 100
parallel 10×0.5 μm2 lines was created. The lines were written with linearly increasing electron
dosage from 0 to 10 mC/cm2 (corresponding to 0~1 as indicated in Figure 77). The electron
beam induced lateral cross-linking of biphenylthiol SAMs, and therefore enhanced the
stability of the monolayer due to the multiple adhesion sites of the entire SAM layer.
It is known that thiol-gold interaction can be photooxidize60
and thiols desorbed from the gold
surface during UV irradiation. However, the electron induced cross-linking renders the SAM
stable at elevated temperature and UV irradiation. Depending on the applied electron dosage,
the extent of the electron-induced cross-linking can be varied, which cause the formation of
biphenylthiol monolayer with a gradual increase of cross-linking66
, thus resulting in SAM
areas of increasing UV stability. On a basis of that, a successive SIPGP reaction occurs on a
surface with increasing grafting sites, and therefore results in polymer brushes gradients.

Results and disccusion Chaper 3
96
Figure 76. Schematic illustration of bottle-brush brushes (BBBs) formed on Au. (a) Electron
beam irradiation of BT SAMs on gold, (b) Cross-linking of the biphenyl mesogen, (c) SIPGP
of IPOx monomer with UV-irradiation results in patterned PIPOx brush, (d) LCROP of 2-
oxazoline forms the BBBs structures.
Following the previously reported procedure for the SIPGP of styrene and acrylic
monomers,69
we performed photopolymerization of 2-isopropenyl-2-oxazoline (IPOx) on
biphenyl thiol SAM as illustrated in Figure 76. After 16h UV irradiation, structured and
gradient PIPOx brushes were formed on biphenyl thiol monolayer. Figure 77 (A) shows
AFM scans of the resulting polymer structure. Between 0 and 0.2 (corresponding to electron
dosage 0 and 2 mC/cm2), the thickness of PIPOx brush increases steadily and becomes
constant between 2 and 10 mC/cm2. As reported before, layer thickness of polymer brush
prepared by SIPGP on EBCL modified SAMs strongly depended on the electron dosage. The
polymer layer thickness increased with the applied electron dosage. However, after reaching
to a maximum value at about 3-4 mC/cm2, a further increase of applied electron dosage

Chapter 3 Results and discussion
97
resulted in a decrease of polymer layer thickness. In our study, the varying polymer layer
thickness is ascribed to the not complete cross-linking of biphenyl molecules. This incomplete
cross-linking decreases the number of grafting sites due to the partial desorption of the non-
crosslinked molecules during photopolymerization. As a result, the polymer layer thickness
varied. Comparing this gradient with our previous results on the morphology control of PS
brushes prepared using the same technique,69
the resulting polymer layer thicknesses of the
gradient as a function of the electron beam irradiation dose are slightly different. This may be
due to the different electron induced cross-linking of the two SAMs, different electron energy
and pressure applied during the EBCL, and different monomers used for SIPGP. Another
explanation might be the significantly enhanced stress of the sterical demanding BBB upon
the surface attachment sites, namely the thiol-gold bond. Different to linear polymer brushes
as in previous work, BBB of increasing grafting density will induce a quite significant stress
since the effective grafting density is much higher. It has been reported by Sheiko et al. that
C-C bond scission occurs when molecular brushes adsorb on surfaces and strongly interacts
with the substrate.49
The system relaxes by chain scission in the molecular brush backbone as
well as side chains. Here, the crowding induced stress of BBB attached to a crosslinked
monolayer might cause detachment of larger fractions of the coating which results in a
decrease of the total layer thickness as a critical effective grafting density is reached.
The obtained PIPOx brushes were subsequently converted into the polycationic macroinitiator
salt by submerging the modified gold substrate in a solution of methyl trifluoromethane-
sulfonate (MeOTf). Successively, the LCROP of 2-n-propyl-2-oxazoline (nPrOx) was
performed for 2h at 80 °C followed by termination by piperidine. After LCROP of nPrOx, the
polymer layer thickness increased significantly from 59 nm to 112 nm at electron dosage of
10 mC/cm2
(Figure 77). It should be noted that the shape of the gradient structure was well
preserved after the LCROP, and no significant material loss could be observed.
In previous chapter it was discussed that molecular brush of PnPrOx side chain exhibited a
lower critical solution temperature (LCST) at around 20 °C. Accordingly, surfaces coated
with BBBs of PnPrOx side chain are expected to show thermo-responsive properties.
Crossing the critical solution temperature, the layer will become hydrophobic and collapse
because of the bad solvent conditions. However, in temperature dependant measurements of
the static water contact angle no changes of the BBB layer wettability was found within a
temperature range from 15 °C to 50 °C. Our finding is in contrast to the switching behavior of
well studied thermo-responsive polymer i.e. poly-N-isopropyl acrylamide (PNIPAAm) coated

Results and disccusion Chaper 3
98
substrate. PNIPAAm in water undergoes a sharp phase transition at its LCST of 32 °C. When
grafted onto solid substrates, PNIPAAm brushes show a temperature-dependence in the
physicochemical surface properties e.g. roughness195
, thickness and contact angle.196
It was
proposed that the rearrangement of the PNIPAAm chain at its critical temperature cause the
drastic change of contact angle. However, for the BBBs of poly(2-oxazoline)s, both the
SIPGP of IPOx via grafting from method and the side chain crowding resulted in the
formation of densely packed polymer chains. This will significantly decrease the mobility of
the polymer chains for the rearrangement. Therefore, the switching behavior of such dense
polymer structure was not observed.
Figure 77. AFM images of bottle-brush brushes of POx on biphenylthiol gradient modified
gold (maximum electron dosage = 10 mC/cm2). (A) after SIPGP of IPOx for 16h, (B) after
cationic ring-opening polymerization of nPrOx for 2h and terminated by piperidine, and c)
section view of (A) and (B).
In summary, in this part of work, the preparation of structured bottle-brush brushes of poly(2-
oxazoline)s of micro/nano scale on gold substrates is described. The polymer layer thickness
can be directly controlled by the extent of electron-induced cross-linking of the SAM layer.

Chapter 3 Results and discussion
99
3.7 Protein adsorption and cell adhesion on bottle-brush brushes
of poly(2-oxazoline)s
3.7.1 Background
Protein adsorption and its induced responses including cell adhesion are able to largely
influence the biomaterial performance and are key criteria in the fields of biosensing and
bioengineering.82,84,83,197
Proteins tend to adsorb nonspecifically to most surfaces and by this
impair the performance of surface based devices.198
To cope with this problem several
strategies to suppress nonspecific protein adsorption, non fouling surfaces have been
developed and extensively investigated.199
Non fouling polymer surfaces are referred to as
surfaces which are resistant towards the adsorption of proteins.90,200
In the past decades, a
variety of polymers have been investigated to realize non fouling surfaces.90,91,92,93,94,95
However, only few were reported to meet the practical applications.
Poly(ethylene glycol) (PEG) is up to now the most studied polymer for the prevention of the
protein adsorption and unwanted cells adhesion in vitro and in vivo. 201
It has good solubility
in most solvents, enabling chemical modification on its terminal hydroxyl groups.202
However,
PEG coatings may lose their function in complex biological fluids and can undergo oxidative
degradation when used for longer time.98,99,100
Moreover, the PEG system mainly contains
primary hydroxyl terminal group which can be modified. Owing to the deficiency of
modifiable functional groups, the possibility for specific end group modifications are not
achievable, which is in most cases desirable for various applications. A potential solution is to
introduce additional functional groups into PEG system, but this has been reported to alter its
properties significantly and probably result in negative coating performance.101, 203
Consequently, there is a continuous need for the development of alternative polymers to
overcome such problems in practical applications.
In search of non fouling polymers, poly(2-oxazoline)s (POx) came into focus as a potential
alternative to the well studied PEG systems.113,116,204
It has been reported that surfaces coated
with this type of polymer have quantitatively equal properties as PEG-based coatings and are
similarly resistant toward nonspecific protein adsorption.204, 205
Additionally, the living
cationic ring-opening polymerization (LCROP) of 2-substituted-2-oxazoline provides
synthetic possibilities to tailor macromolecules with a broad selection of side and end

Results and discussion Chapter 3
100
functions, which enables the polymer with a broad variety of architectures and composition.
206,207,208,209,210,211,212
For example, Konradi et al. have investigated the protein adsorption on comb polymer
consisting of hydrophilic poly(2-methyl-2-oxazoline) (PMeOx) side chains and a polycationic
backbone.204
The positively charged comb polymer adsorbed on negatively charged surface
through coulomb interaction. Such coatings with an optimal side chain grafting density
strongly reduce protein adsorption to a value of less than 2 ng/cm2, which is quantitatively
equal to the performance of PEG coatings. However, the electrostatic adsorbed coating is
prone to desorption at certain conditions and especially in biological environment with higher
salt concentration and thus, a covalent attachment is preferable.
In this part of the work, oxidized silicon surfaces were modified with various bottle-brush
brushes (BBBs) of neutral POx. As described in previous chapters, the polymer is covalently
attached onto the surface, which prevents the desorption of polymer chains. To examine the
biocompatibility of POx coatings, protein adsorption and cell adhesion on BBBs modified
solids were investigated. Moreover, the influence of the polymer structure e.g. side chain
composition, side chain length and terminal groups on its bioadsorption properties is
discussed.
3.7.2 Bottle-brush brushes of poly(2-oxazoline)s on oxidized silicon
substrate
Bottle-brush brushes (BBBs) of poly(2-oxazoline)s (POx) with different side chains /end
groups were prepared on oxidized silicon and characterized with respect to their contact angle
and polymer layer thickness. The results are summarized in Table 9.
Sample 1-4 are BBBs of POx with a variation of the pendant chain end groups. For sample
preparation, the PIPOx coated substrate was first divided into four pieces, and converted to
the macroinitiator brush by methyl triflate (MeOTf), and successively LCROP of 2-methly-2-
oxazoline (MeOx) was allowed to proceed for 4 h. Finally, the LCROP was terminated with
piperidine, N-boc piperazine, 4-piperidinol, and ethyl isonipecotate, respectively, introducing
different end groups in the pendant chains of BBBs. Significant increase of polymer layer
thickness was observed after the formation of BBBs. For example, the polymer layer
thickness increased by 128 % from 57 ± 7nm (PIPOx) to 130 ± 5nm for sample 2 (end group

Chapter 3 Results and discussion
101
is piperidine). However, the contact angle after formation of BBBs only showed minor
changes. Contact angles of PIPOx changed from 49° to 46°, 49 to 44°, 49° to 33°, 49° to 42°
for sample 1, 2, 3 and 4, respectively. The minor effect of end groups on the contact angles is
expectable and can be interpreted by the surface rearrangement of the flexible polymer brush
layer upon change of the solvent quality/ polarity.
Sample 5-7 are BBBs of POx with a variation of the side chain composition. As above, a
PIPOx modified substrate was divided into three pieces and LCROP of MeOx, 2-ethyl-2-
oxazoline (EtOx) and 2-n-propyl-2-oxazoline (nPrOx) were performed. All these samples
were finally treated with piperidine to terminate the living polymerization. Drastic thickness
increase was also observed for these samples. In addition, the surface wettability was found to
change significantly after LCROP. Specifically, water contact angles were found to be 38°, 43°
and 56° for BBBs of PMeOx, PEtOx and PnPrOx side chains. These values are consistent
with the ones of BBBs on glassy carbon as described in Chapter 3.5.2.
Sample 8-11 are BBBs of POx with a variation of the hydrophilic side chain length. Similarly,
a substrate with a PIPOx brush was divided into four pieces. LCROP of MeOx was allowed to
proceed at 80 °C for 0h, 1h, 2h and 4h respectively. Here, the increase of BBBs thickness was
investigated as a function of the LCROP polymerization time. The percentage of the thickness
increase due to the formation of BBBs was found to be linear against reaction time. In
addition, the contact angle of BBBs varied significantly with the different length of side
chains e.g. from 71° for sample 8 (LCROP = 0h) to 43° for sample 11 (LCROP = 4h). The
results indicated that the surface wettability of BBBs increased with increasing length of the
hydrophilic side chain. However, the comparison between sample 10 and sample 11 revealed
that further increase of the side chain length only showed minor effect on the contact angle
value.

Results and discussion Chapter 3
102
Table 9. Preparation of bottle-brush brushes of 2-oxazolines on oxidized silicon and
characterization with regard to the water contact angle and the polymer layer thickness.
Sample Side chain/ CROP time
End groups (R)
CAa (PIPOx)
CAa(after
LCROP) Thicknessb
(PIPOx) Thicknessb
(after LCROP)
1.PMeBoc
MeOx/4h
N-Boc-
piperazine 49±2° 46° 49±5nm 101±6nm
2.PMePid piperidine 49±2° 44° 57±7nm 130±5nm
3.PMepidn 4-piperidinol 49±2° 33° 54±5nm 117±7nm
4.PMeetiso ethyl
isonipecotate 49±2° 42° 51±6nm 110±5nm
5.PMe MeOx/4h piperazine
51±2° 38±3° 67±8nm 245±7nm
6.PEt EtOx/4h 51±2° 43° 73±9nm 260±11nm
7.PnPr nPrOx/4h 51±2° 56±3° 78±9nm 267±10nm
8.PMe0 MeOx/0h
piperidine
50±2° 71±3° 43±9nm 54±8nm
9.PMe1 MeOx/1h 50±2° 51±2° 42±6nm 62±7nm
10.PMe2 MeOx/2h 50±2° 45±2° 51±7nm 82±6nm
11.PMe4 MeOx/4h 50±2° 43±2° 50±6nm 116±5nm
a: Static water contact angle (CA) was measured at room temperature (25 °C).
b: Polymer brush thickness was determined by AFM depth analysis.
3.7.3 Protein adsorption
The protein adsorption and cell adhesion measurements were performed by Dr. Tilo Pompe
from the research group of Prof. Carsten Werner at the Max Bergmann Center of Biomaterials,
TU Dresden/Leibniz Institute of Polymer Research Dresden.
Fibronectin is a high molar mass glycoprotein which is involved in many cellular processes.
Here, it serves as a model protein for the investigation of adsorption properties on BBBs.
Briefly, BBBs were immersed in 70 % ethanol/water to provide sterile conditions for cell
culture. Thereafter, BBBs were immersed in a 50 µg/mL fibronectin (purified from human
plasma) solution in phosphate buffered saline (PBS) at pH 7.4 and 37 °C for 1 h.
Carboxytetramethylrhodamine was used to label fibronectin prior to the experiments. Non-
adsorbed fibronectin was removed by rinsing twice with PBS. Finally, the fluorescence
microscopic analysis was performed on the protein adsorbed BBBs surfaces. Figure 78 shows
the fluorescent images of BBBs with a variation of side chain composition i.e. PnPrOx,
PEtOx and PMeOx after the fibronectin culture. Strong fluorescence could be detected on

Chapter 3 Results and discussion
103
BBBs with PnPrOx side chain. However, almost no fluorescence signal was found on BBBs
with PMeOx and PEtOx side chains. It indicates the protein adsorption is strongly affected by
the side chain composition of BBBs. Likewise, the influence of side chain length and end
group of BBBs side chains was investigated by fluorescence microscopic analysis as well.
Results confirm the significant effects of BBBs structure on the adsorption behavior of
fibronectin (Fluorescent images are not shown here).
Additionally, fibronectin adsorption was quantified using fluorescence intensity per unit area.
The amount of adsorbed fibronectin on different surfaces was plotted together with that on
poly(octadecene-alt-maleic anhydride) POMA coated surface, which was used as a reference
because it exhibits a large adsorption of fibronectin (~600 ng/cm2) via stable amide bonds. In
the following part, the quantification of protein adsorption on different BBBs surfaces as well
as the manipulation of the BBBs will be particularly analyzed and discussed.
Figure 78. Fluorescent images of BBBs with PnPrOx, PEtOx and PMeOx side chains after 1
hour fluorescent-labeled fibronectin culture.
3.7.3.1 Variation of the BBB side chain composition
The amount of adsorbed fibronectin on BBBs with a variation of side chains was investigated
and results are shown in Figure 79. BBBs with PMeOx and PEtOx pendant chains (Sample 5
and 6) efficiently suppressed protein adsorption down to a value of 6 ng/cm2, demonstrating
an excellent non fouling property of BBB surface. However, a different result was obtained

Results and discussion Chapter 3
104
for protein adsorption on BBB with PnPrOx pendant chains (Sample 7), where a high amount
of adsorbed fibronection (90 ng/cm2) was observed. This significant difference may be
ascribed to the altered hydrophilicity of POx coatings. In general, hydrophobic interaction is
expected to play an important role in the protein adsorption. A hydrophobic surface renders a
higher adsorption of proteins, whereas a hydrophilic surface repels the approaching proteins,
resulting in a sharply reduced adsorption. In this study, the BBBs surfaces are believed to be
manipulated markedly hydrophilic with PMeOx and PEtOx pendant chains (Sample 5 and 6),
which greatly impeded the adsorption of fibronectin. However, the BBB modified with
PnPrOx pendant chains (Sample 7) is slightly hydrophobic, therefore, the amount of adsorbed
fibronectin on the surface is higher.
Approving above assumption, the water contact angles of BBBs with variable side chain
composition were investigated and the results are listed in Table 9. BBBs with PMeOx and
PEtOx side chains showed water contact angles of 38° and 43° at both room temperature and
37 °C (the temperature that the protein adsorption experiments were performed). This
indicates a pronounced hydrophilic property of the surfaces. However, BBB with PnPrOx side
chains exhibited water contact angles of 56° at room temperature and 60° at 37 °C, thus a
more hydrophobic surface. These results are consistent with the protein adsorption behavior.
Additionally, the observation corroborates the criteria for designing non fouling polymers i.e.
manipulation of surface hydrophilicity is a key factor for the prevention of nonspecific protein
adsorption.

Chapter 3 Results and discussion
105
Figure 79. The amount of adsorbed fibronectin as a function of BBBs side chains i.e. PnPrOx,
PEtOx and PMeOx side chains.
3.7.3.2 Variation of the BBB side chain length
As a second parameter, side chain length of the BBBs was varied and its influence on the
protein adsorption was investigated. Briefly, PMe1 (Sample 9) which was prepared from 1 h
LCROP of MeOx, represents BBBs with short side chains; PMe2 (Sample 10) from 2 h
LCROP of MeOx represents BBBs with middle side chains; and PMe4 (Sample 11) from 4 h
LCROP of MeOx represents BBBs with long side chains. It should be noted that PMeOx is
very hydrophilic, therefore, the prepared BBBs are expected to reduce the adsorption of
proteins greatly.
Figure 80 shows the amount of adsorbed fibronectin on BBBs with a variation of side chain
length. For short side chain BBBs, a protein adsorption concentration of 45 ng/cm2 was
observed. As the side chain length increases, a significant decrease in protein adsorption was
revealed. Protein adsorption concentration of 11 ng/cm2 was observed for BBBs with both
middle side chains and long side chains. Evidenced by the above results, it seems reasonable
to conclude that protein adsorption can be influenced by side chain length of BBBs. However,
it is worth noted that the polymer layer thickness of these three BBBs surface is in fact not
comparable. As shown in Table 9, the polymer layer thicknesses were found to increase by
48 %, 61% and 132 % after LCROP of MeOx for 1 h, 2h and 4h, respectively. It suggests a

Results and discussion Chapter 3
106
large difference in polymer layer thickness between BBBs of PMe1, PMe2 and PMe4. To
determine whether the polymer layer thickness of BBBs in this case plays a key role in the
protein adsorption, the following experiments were performed.
Protein adsorption on BBBs possessing the same PMeOx side chain length but different
polymer layer thickness was compared. Here, BBBs with layer thicknesses of 130 nm
(Sample 2), 116 nm (Sample 11) and 51 nm (no shown in this study) are used as the model
BBBs for comparison. The protein adsorption on these BBBs surfaces was quantified,
revealing the adsorbed protein concentration of 12, 11 and 8 ng/cm2 for BBBs with layer
thicknesses of 130 nm, 116 nm and 51 nm, respectively. Compared with BBBs of different
side chain length, results here produced a somewhat opposite correlation between protein
adsorption and polymer thickness. However, the difference in the protein adsorption
concentration is in fact too small (within 4 ng/cm2), which is most likely caused by the
experimental error. By all means, the decreased polymer layer thickness does not show a
significant effect on protein adsorption.
Based on the discussion above, it is more reasonable to conclude that the side chain length
played the key role in the protein adsorption properties of BBBs. As the hydrophilic side
chain length increases, the surface wettablity increases accordingly. As a result, a reduced
amount of adsorbed protein was observed. Actually, water contact angles for the BBBs with
short, middle and long PMeOx side chains were found to be 51°, 45° and 43° respectively,
which was in a good agreement with the above explanation. Again, this result corroborates the
criteria for designing non fouling surfaces i.e. hydrophilic surfaces impede the nonspecific
protein adsorption.

Chapter 3 Results and discussion
107
Figure 80. The amount of adsorbed fibronectin on BBBs with PMeOx side chain as a
function of side chain length.
3.7.3.3 Variation of the BBB pendant chain end groups
In addition to the variation of side chain composition and side chain length, the
biocompatibility of BBBs can also be influenced by the pendant chain end groups. Various
chain termini (piperidine, N-boc piperazine, 4-piperidinol, and ethyl isonipecotate) were
introduced to the BBBs side chain end to examine the effect of the end groups upon the
protein adsorption behavior. Small changes in protein adsorption on the BBBs could be
observed (Figure 81) after the introduction of different end groups, which is probably
because, a) very little amount of adsorbed proteins limit the accuracy of measurement and, b)
a small portion of end groups contained in the entire polymer layer contributes little to the
protein adsorption. However, the successful introduction of different end groups provides the
possibility for further functionalization e.g. protein immobilization.
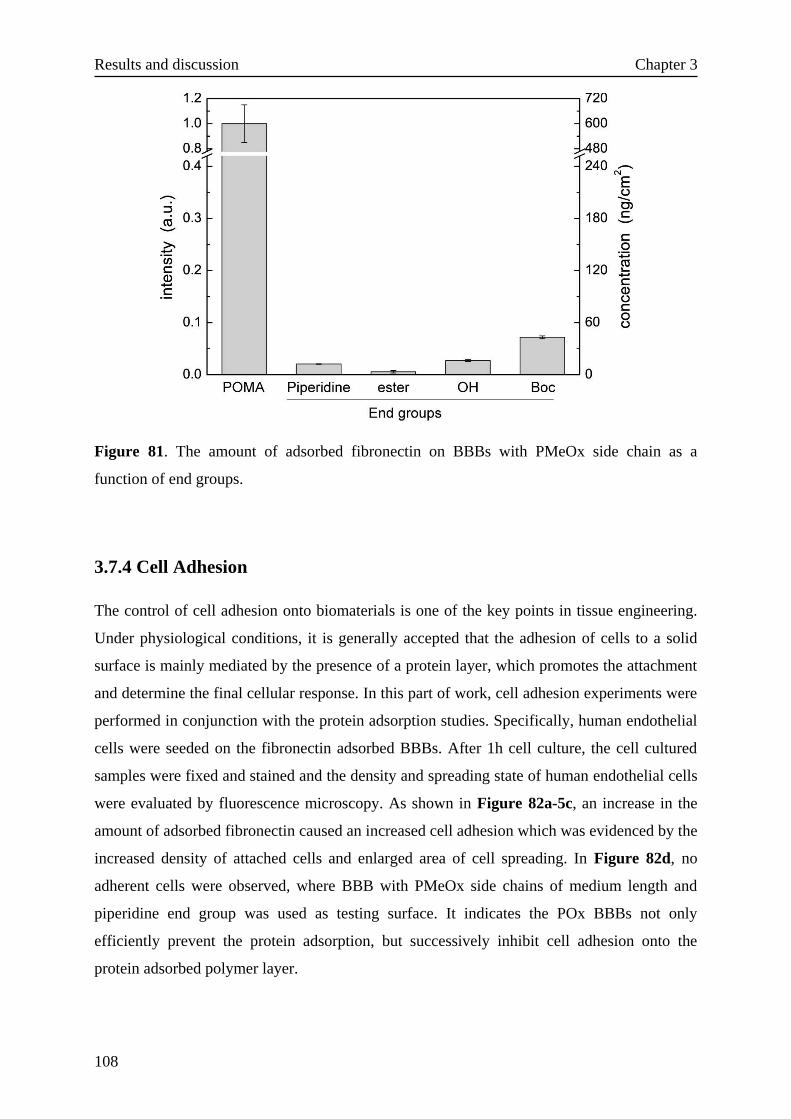
Results and discussion Chapter 3
108
Figure 81. The amount of adsorbed fibronectin on BBBs with PMeOx side chain as a
function of end groups.
3.7.4 Cell Adhesion
The control of cell adhesion onto biomaterials is one of the key points in tissue engineering.
Under physiological conditions, it is generally accepted that the adhesion of cells to a solid
surface is mainly mediated by the presence of a protein layer, which promotes the attachment
and determine the final cellular response. In this part of work, cell adhesion experiments were
performed in conjunction with the protein adsorption studies. Specifically, human endothelial
cells were seeded on the fibronectin adsorbed BBBs. After 1h cell culture, the cell cultured
samples were fixed and stained and the density and spreading state of human endothelial cells
were evaluated by fluorescence microscopy. As shown in Figure 82a-5c, an increase in the
amount of adsorbed fibronectin caused an increased cell adhesion which was evidenced by the
increased density of attached cells and enlarged area of cell spreading. In Figure 82d, no
adherent cells were observed, where BBB with PMeOx side chains of medium length and
piperidine end group was used as testing surface. It indicates the POx BBBs not only
efficiently prevent the protein adsorption, but successively inhibit cell adhesion onto the
protein adsorbed polymer layer.

Chapter 3 Results and discussion
109
These cell adhesion studies nicely support the results of the protein adsorption experiments
outlined above. Furthermore, they demonstrate the ability of the POx BBBs system to tune the
protein adsorption and cell adhesion, and additionally to provide specific chemical end groups
for further functionalization strategies.
Figure 82. Fluorescence images of cell adhesion on different fibronectin-adsorbed BBBs of
POx. In correlation to the amount of adsorbed fibronectin, different degrees of cell adhesion
can be observed on POx BBBs surfaces: (a) very good cell adhesion with highly spread cells
on BBB with PnPrOx side chains of medium length and piperidine end group, (b) good cell
adhesion with spread cells on BBB with PMeOx side chains of medium length and Boc end
groups, (c) not good cell adhesion with few and almost unspread cells on BBB with PMeOx
side chains of medium length and OH end groups, (d) no cell adhesion on BBB with PMeOx
side chain of medium length and piperidine end group. [fibronectin (red), actin (green), and
nuclei (blue). Scale bar: 50 µm.]
In summary, the protein adsorption and the successive cell adhesion on various BBBs of POx
was studied. Quantification of adsorbed protein reveals that, the manipulation of polymer
composition and architecture can greatly influence the biocompatibility of the resulting BBBs
surfaces. Specifically, (1) BBBs with very hydrophilic PMeOx and PEtOx side chains show
no or significantly reduced protein adsorption as compared to more hydrophobic BBBs with
PnPrOx side chains, (2) Given the same side chain composition, a stronger protein resistance

Results and discussion Chapter 3
110
is observed for BBBs with longer hydrophilic side chains, which is also ascribed to the
increased surface wettability of BBBs. (3) Side chain end groups influences the bioadsorption
properties as well, however, the influence is minor. The cell adhesion experiments revealed
the amount of adhered cells on protein layers, which nicely supported the quantification
results of protein adsorption. This study may open up the possibility for broadening the choice
of protein resistant surfaces in the future.

Chapter 4 Summary
111
Chapter 4 Summary
In this work, the first synthesis of well-defined molecular brushes based on poly(2-
oxazoline)s is described. Molecular brushes have been synthesized via the combination of
free radical/ as well as anionic polymerization of 2-isopropenyl-2-oxazoline (IPOx) (for the
backbone) and living cationic ring-opening polymerization (LCROP) of 2-oxazolines (for the
side chains) through the “grafting from” method (Figure 83). For the brush molecules with a
backbone made by free radical polymerization, the polydispersity index (PDI) is relatively
broad due to the uncontrolled character of the reaction. However, in the case of the backbone
prepared by living anionic polymerization, molecular brushes with relatively narrow PDI in
both the backbone and the side chains were achieved.
Figure 83. Scheme for the synthesis of molecular brushes of poly(2-oxazoline)s: (1)
backbone by free radical as well as anionic polymerization, (2) synthesis of the macroinitiator,
(3) Synthesis of pendant chains by living cationic ring-opening polymerization.
The grafting from polymerization of 2-oxazoline e.g. 2-n-propyl-2-oxazoline (nPrOx) and 2-
iso-propyl-2-oxazoline (iPrOx) from the macroinitiator follows linear first order kinetics and
the molar mass of the resulting polymers increases with the monomer conversion. It was
shown that the apparent polymerization rate for each intitiation function is independent of the
size of the macroinitiators and the initiating sites are of equal reactivity. As consequence, well

Summary Chapter 4
112
defined molecular brushes with high grafting densities of pendant chains of equal length can
be realized (Figure 84).
Figure 84. First order kinetic plots for the polymerization of nPrOx with MeOTf , MI and M
II.
The resulting molecular brushes with homopolymer side chains e.g. poly(2-ethyl-2-oxazoline)
(PEtOx), poly(2-isopropyl-2-oxazoline) (PiPrOx) and poly(2-n-propyl-2-oxazoline) (PnPrOx)
showed thermo-responsive properties i.e. having defined cloud point in aqueous solution.
Factors such as side chain length, backbone length (molar mass), side chain end groups and
concentration were investigated in detail and have significant effect on the LCST. For
example, as shown in Figure 85, the LCST values of P(IPOx-g-nPrOx) decreased from 24 °C
(side chain degree of polymerization is 3) to a minimum of 18.2 °C (side chain degree of
polymerization is 10). Further increase of the side chain length slightly increased the LCST
because of the increase of free volume per side chain (the longer the side chains, the higher
the mobility and the free volume the side chains).

Chapter 4 Summary
113
Figure 85. LCST of molecular brushes P(IPOx-g-nPrOx)A as a function of side chain length.
The LCST was determination by transmittance changes (10% decrease) for molecular brushes
in 1.0 wt % of aqueous solution (heating curves).
As shown in Figure 86, the LCST of the molecular brushes decreased steadily with the
increasing backbone length which is consistent with previous reports for linear poly(2-
oxazoline)s. As for the concentration effect, the LCST of P(IPOx-g-nPrOx)A
decreased from
24 °C to 19.1 °C as the concentration increased from 0.1 % to 1.0 % but further increase of
the concentration has only little effect on the LCST decrease (Figure 87).

Summary Chapter 4
114
Figure 86. Cloud point of molecular brush P(IPOx-g-iPrOx)A as a function of the PIPOx
backbone unit (all the molecular brushes with a side chain DP =25, determined from heating
curves).
Figure 87. Cloud point of molecular brush P(IPOx-g-nPrOx)A as a function of the
concentration. (side chain DP = 25, determined from heating curves).

Chapter 4 Summary
115
Molecular brushes with random/ gradient copolymer i.e. poly(nPrOx-co-MeOx) side chains
were also prepared. It was found that the clould point could be tuned in the range of 19-81 °C
by variation of the content of hydrophilic MeOx monomer (Figure 88). The cloud points as a
function of the percentage of MeOx in the molecular brushes are shown in Figure 89.
Furthermore, we prepared molecular brushes with block copolymer side chain and
investigated their thermo-responsiveness. Molecular brushes with a hydrophilic core of
PMeOx and a hydrophobic shell of PnPrOx resulted in a higher LCST compared to brushes
with a hydrophobic brush core and a hydrophilic shell.
Figure 88. Determination of cloud points of molecular brushes with random copolymer side
chains.

Summary Chapter 4
116
Figure 89. LCST values versus MeOx molar content of the side chain of molecular brushes.
The self-initiated photografting and photopolymerization (SIPGP) of 2-isopropenyl-2-
oxazoline on glassy carbon and aminosilane modified oxidized silicon resulted in
homogeneous and stable poly(2-isopropenyl-2-oxazoline) (PIPOx) brushes. The pendant
oxazoline ring of the PIPOx brushes allowed the conversion to macroinitiator brushes and a
successive living cationic ring-opening polymerization (LCROP) with different substituted 2-
oxazoline monomers, which resulted in the so-called bottle-brush brushes (BBBs). Moreover,
we demonstrated the possibility to functionalize the BBB side chain termini with specific
sterically demanding molecules. The reactions are summarized in Figure 90. The ease of
chemical modification allows for a precise fabrication of a broad variety of functional
surfaces for the design of a soft interlayer between solids and e.g. biological systems.

Chapter 4 Summary
117
Figure 90. Left: Reaction scheme of the preparation of well-defined bottle-brush brushes on
glassy carbon; and Right: Corresponding AFM 3D and fluorescent images of the polymer
brushes structure on glassy carbon. (a) The self-initiated photografting and
photopolymerization (SIPGP) of 2-isopropenyl-2-oxazoline (IPOx) for 40h through a stencil
mask results in structured PIPOx brushes with a thickness of 159 ± 8 nm. (b) The living

Summary Chapter 4
118
cationic ring-opening polymerization (LCROP) of EtOx and termination with N-Boc-
piperazine results in 330 ± 10 nm thick P(IPOx-g-EtOxBoc) BBBs. (c) Deprotection of the
side chain terminal Boc group results in a 313 nm thick P(IPOx-g-EtOx). (d) The side chain
end of BBB was further labelled by Rhodamine B and its fluorescent image (right).
Finally, we investigated the protein adsorption of fibronectin on BBBs of poly(2-oxazoline)s
(POx) on oxidized silicon substrates. Quantification of adsorbed protein reveals that, the
manipulation of polymer composition and architecture can greatly influence the bioadsorption
properties of the resulting BBBs surfaces. Specifically, (1) BBBs with PMeOx and PEtOx
side chains due to the strong hydrophilicity, show very much reduced protein adsorption as
compared to BBBs with PnPrOx side chains (Figure 91 a), (2) Given the same side chain
composition, a stronger protein resistance is observed for BBBs with longer hydrophilic side
chains, which is also ascribed to the increased surface wettability of BBBs (Figure 91 b). (3)
Side chain end groups influences the bioadsorption properties as well, however, the influence
is rather limited. The further cell adhesion experiments revealed the amount of adhered cells
on protein layers, which nicely supported the quantification results of protein adsorption
(Figure 92). The obtained results rearding the thermoresponsiveness of poly(2-oxazoline)
brushes and their interactions with biomolecules may open up the possibility for broadening
the choice of protein resistant surfaces in the future.
Figure 91. Protein adsorption as a function of side chain compositon and side chain length on
the BBBs of poly(2-oxazoline)s. a) BBBs with a variation of side chain composition. b) BBBs
with a variation of side chain length.

Chapter 4 Summary
119
Figure 92. Fluorescence images of cell adhesion on different fibronectin-adsorbed BBBs of
POx. In correlation to the amount of adsorbed fibronectin, different degrees of cell adhesion
can be observed on POx BBBs surfaces: (a) very good cell adhesion with highly spread cells
on BBB with PnPrOx side chains of medium length and piperidine end group, (b) good cell
adhesion with spread cells on BBB with PMeOx side chains of medium length and Boc end
groups, (c) not good cell adhesion with few and almost unspread cells on BBB with PMeOx
side chains of medium length and OH end groups, (d) no cell adhesion on BBB with PMeOx
side chain of medium length and piperidine end group. [fibronectin (red), actin (green), and
nuclei (blue). Scale bar: 50 µm.]

Zusammenfassung Chapter 5
120
Zusammenfassung
Diese Arbeit beschäftigte sich erstmals mit der Synthese wohldefinierter molekularer Bürsten
auf Basis von Poly(2-oxazolin)en. Sämtliche hier beschriebenen molekularen Bürsten wurden
durch Kombination aus freier radikalischer, sowie lebender anionischer Polymerisation von 2-
iso-Propenyl-2-oxazolin (IPOx) (zur Herstellung des Polymerrückgrates) und einer lebenden
kationischen Ringöffnungspolymerisation von 2-Oxazolinen (zur Herstellung der Seitenketten)
nach dem “grafting from” Ansatz synthetisiert (Abbildung 93). Die Bürstenpolymere, deren
Rückgrat / Polymerhauptkette über freie radikalische Polymerisation hergestellt wurde, haben
aufgrund des verhältnismäßig unkontrollierten Polymerisationsmechanismus einen relativ
hohen Polydispersitätsindex (PDI). Wenn das Polymerrückgrat über anionische
Polymerisation hergestellt wird, kann jedoch sowohl in der Hauptkette, als auch in den
Seitenketten ein niedriger PDI erzielt werden.
Abbildung 93. Herstellung von Poly(2-oxazolin) Bürstenpolymeren über freie radikalische
oder anionische Polymerisation (zur Synthese der Hauptkette), anschließende Umsetzung mit
MeOTf unter Bildung des Makroinitiators und der Aufbau der Seitenketten über eine lebende
kationische Ringöffnungspolymerisation mit 2-Oxazolinen.
Ausgehend von einem Makroinitiator erfolgt die „grafting from“ Polymerisation von 2-
Oxazolinen, wie zum Beispiel 2-n-Propyl-2-oxazolin (nPrOx) und 2-iso-Propyl-2-oxazolin
(iPrOx), einer Reaktionskinetik erster Ordnung und die Molmasse des entstehenden Polymers
steigt mit dem Monomerumsatz. Es konnte gezeigt werden, dass alle Initiatorgruppen gleiche
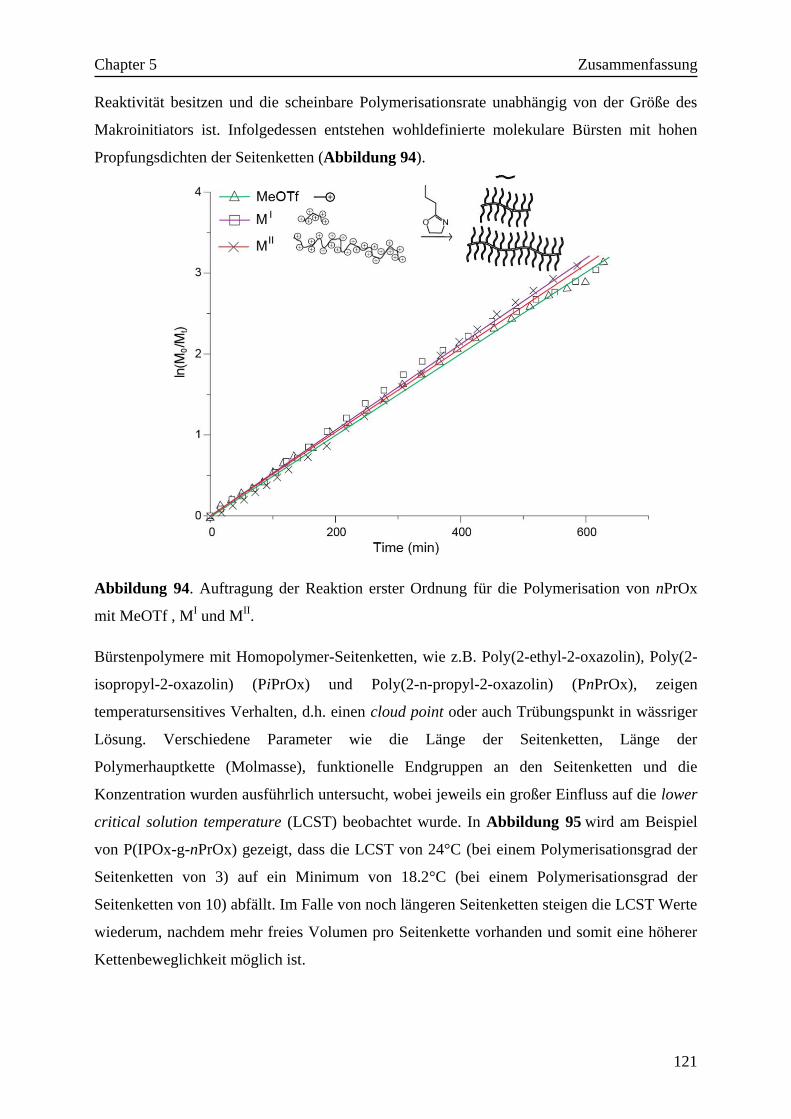
Chapter 5 Zusammenfassung
121
Reaktivität besitzen und die scheinbare Polymerisationsrate unabhängig von der Größe des
Makroinitiators ist. Infolgedessen entstehen wohldefinierte molekulare Bürsten mit hohen
Propfungsdichten der Seitenketten (Abbildung 94).
Abbildung 94. Auftragung der Reaktion erster Ordnung für die Polymerisation von nPrOx
mit MeOTf , MI und M
II.
Bürstenpolymere mit Homopolymer-Seitenketten, wie z.B. Poly(2-ethyl-2-oxazolin), Poly(2-
isopropyl-2-oxazolin) (PiPrOx) und Poly(2-n-propyl-2-oxazolin) (PnPrOx), zeigen
temperatursensitives Verhalten, d.h. einen cloud point oder auch Trübungspunkt in wässriger
Lösung. Verschiedene Parameter wie die Länge der Seitenketten, Länge der
Polymerhauptkette (Molmasse), funktionelle Endgruppen an den Seitenketten und die
Konzentration wurden ausführlich untersucht, wobei jeweils ein großer Einfluss auf die lower
critical solution temperature (LCST) beobachtet wurde. In Abbildung 95 wird am Beispiel
von P(IPOx-g-nPrOx) gezeigt, dass die LCST von 24°C (bei einem Polymerisationsgrad der
Seitenketten von 3) auf ein Minimum von 18.2°C (bei einem Polymerisationsgrad der
Seitenketten von 10) abfällt. Im Falle von noch längeren Seitenketten steigen die LCST Werte
wiederum, nachdem mehr freies Volumen pro Seitenkette vorhanden und somit eine höherer
Kettenbeweglichkeit möglich ist.

Zusammenfassung Chapter 5
122
Abbildung 95. LCST von P(IPOx-g-nPrOx)A Bürstenpolymeren als Funktion der
Seitenkettenlänge. Die LCST wurde über die Transmissionsänderung (10% Abnahme) einer
wässrigen Polymerlösung (1.0 gew.) bestimmt (Aufnahme einer Heizkurve).
In Abbildung 96 wird deutlich, dass die LCST der Bürstenpolymere mit wachsender Länge
der Hauptkette ansteigt. Dies ist in guter Übereinstimmung mit Ergebnissen aus Experimenten
mit linearen Poly(oxazolin)en. Was den Konzentrationseffekt betrifft, so sinkt die LCST von
P(IPOx-g-nPrOx)A von 24°C auf 19.1°C ab, wenn die Konzentration von 0.1% auf 1.0%
erhöht wird, wobei eine weitere Konzentrationserhöhung nur noch sehr geringen Einfluss
aufzeigt (Abbildung 97).

Chapter 5 Zusammenfassung
123
Abbildung 96. Der Cloud point von P(IPOx-g-iPrOx)A Bürsten als Funktion der Anzahl an
IPOx Einheiten in der Hauptkette (Polymerisationsgrad der Seitenketten von jeweils 25 -
Aufnahme einer Heizkurve)
Abbildung 97. Der Cloud point von P(IPOx-g-nPrOx)A als Funktion der Konzentration
(Polymerisationsgrad der Seitenketten = 25, Aufnahme einer Heizkurve).

Zusammenfassung Chapter 5
124
Im weiteren Verlauf der Arbeit wurden Bürstenpolymere mit statistisch verteilten oder
Gradientcopolymer- Seitenketten synthetisiert, z.B. Poly(nPrOx-co-MeOx). Durch Variation
des Monomeranteils an hydrophilem MeOx konnte der cloud point im Bereich zwischen 19°C
und 81°C definiert eingestellt werden kann. (Abbildung 98). Der cloud point als Funktion des
MeOx Anteils ist in Abbildung 99 dargestellt. Des Weiteren wurden Bürsten mit
Blockcopolymer Seitenketten hergestellt und ebenfalls deren Temperaturverhalten untersucht.
Polymere mit hydrophilem Kern aus PMeOx Bürsten und einer hydrophoberen PnPrOx
Schale besitzen eine höhere LCST als diejenigen mit umgekehrtem Aufbau.
Abbildung 98. Bestimmung des clound point von Bürstenpolymeren mit unterschiedlicher,
statistischer Monomerzusammensetzung in den Seitenketten.

Chapter 5 Zusammenfassung
125
Abbildung 99. Der Cloud point der Bürstenpolymere als Funktion der Anzahl an MeOx
Einheiten pro P(MeOx-co-nPrOx) Seitenkette.
Durch so genannte self-initiated photografting and photopolymerization (SIPGP) von 2-iso-
Propenyl-2-oxazolin auf glasartigem Kohlenstoff und Aminosilan-modifiziertem SiOx und
Biphenylthiol modifizierten Goldsubstraten wurden homogene und stabile Poly(2-
isopropenyl-2-oxazolin) Polymerbürsten hergestellt. Über die Oxazolingruppen der Poly(2-
isopropenyl-2-oxazolin) Bürsten konnte nach Initiierung mit MeOTf eine zweite lebende
kationische Ringöffnungspolymerisation (engl.: „living cationic ring-opening
polymerization“ LCROP) mit verschieden substituierten 2-Oxazolin Monomeren erfolgen.
Dieser Reaktionsschritt führte zur Ausbildung von pfeifenputzerförmigen Polymerbürsten
(engl.: „bottle-brush brushes“ (BBB)). Außerdem konnte gezeigt werden, dass es möglich ist
die Endgruppen der Seitenketten dieser BBBs mit sterisch anspruchsvollen Molekülen zu
funktionalisieren. Das entsprechende Reaktionsschema ist in Abbildung 100 dargestellt.
Aufgrund der vergleichsweise unkomplizierten chemischen Modifizierungsmöglichkeiten
kann eine Vielzahl an unterschiedlich funktionalisierten Oberflächen als Grenzfläche
zwischen Festkörpern und z.B. biologischen Systemen hergestellt werden.
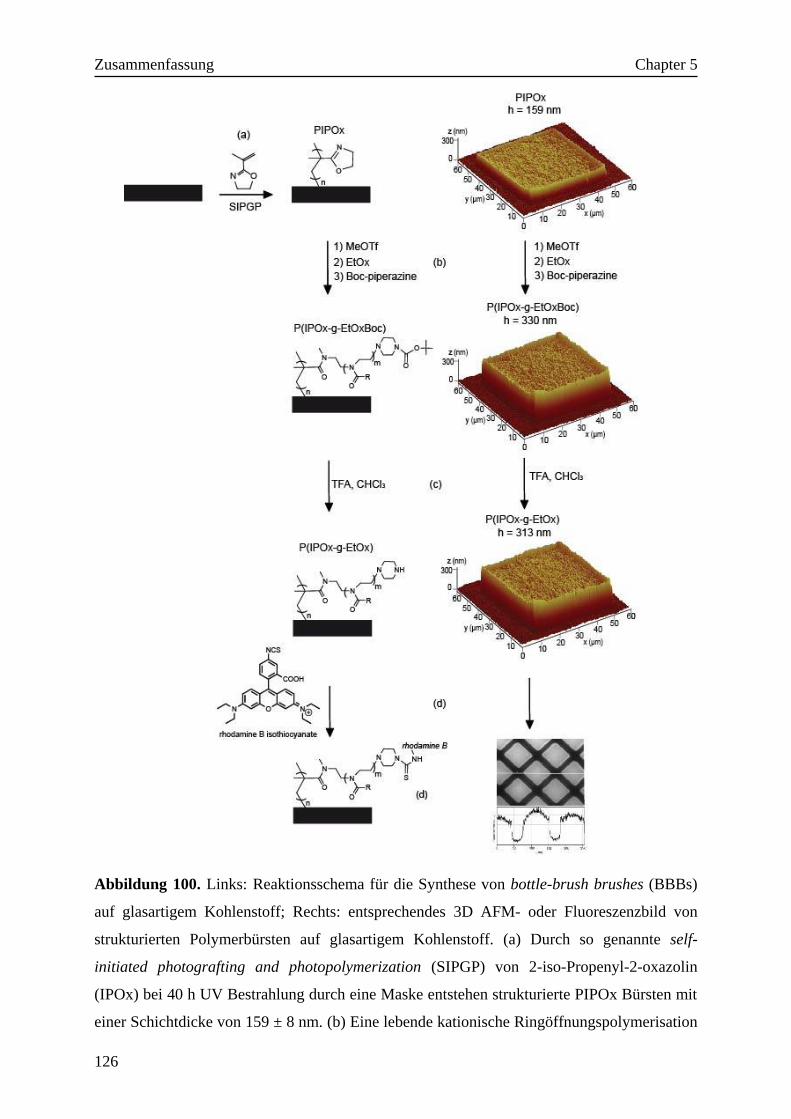
Zusammenfassung Chapter 5
126
Abbildung 100. Links: Reaktionsschema für die Synthese von bottle-brush brushes (BBBs)
auf glasartigem Kohlenstoff; Rechts: entsprechendes 3D AFM- oder Fluoreszenzbild von
strukturierten Polymerbürsten auf glasartigem Kohlenstoff. (a) Durch so genannte self-
initiated photografting and photopolymerization (SIPGP) von 2-iso-Propenyl-2-oxazolin
(IPOx) bei 40 h UV Bestrahlung durch eine Maske entstehen strukturierte PIPOx Bürsten mit
einer Schichtdicke von 159 ± 8 nm. (b) Eine lebende kationische Ringöffnungspolymerisation

Chapter 5 Zusammenfassung
127
(engl.: „living cationic ring-opening polymerization“ LCROP) von EtOx und anschließender
Kettenabbruch mit N-Boc-Piperazin ergab 330 ± 10 nm dicke P(IPOx-g-EtOxBoc) BBBs. (c)
Die Entschützen der terminalen Boc-Schutzgruppen in der Seitenketten liefert 313 nm dicke
P(IPOx-g-EtOx) BBBs. (d) Die freien Aminogruppen der BBB-Seitenketten wurde in einem
weiteren Schritt mit Rhodamin B fluoreszenzmarkiert (rechts: Fluoreszenzbild).
Schließlich wurde das Adsorptionsverhalten von Fibronektin auf POx BBBs auf SiOx
Substraten untersucht. Die Anzahl an adsorbiertem Protein lässt einen deutlichen
Zusammenhang zwischen der Polymerzusammensetzung und -architektur und dem
Adsorptionsverhalten von Biomolekülen auf den entsprechenden BBB-modifizierten
Oberflächen erkennen. Folgende Punkte konnten in dem Zusammenhang besonders
festgehalten werden: (1) BBBs mit PMeOx und PEtOx Seitenketten weisen aufgrund ihrer
Hydrophilie im Gegensatz zu BBBs mit PnPrOx Seitenketten eine geringere
Proteinadsorption auf (Abbildung 101 a). (2) Im Falle von gleicher chemischer
Zusammensetzung der Seitenketten zeichnen dich diejenigen mit längeren hydrophilen
Seitenketten durch eine stärkere Proteinresistenz aus, was ebenfalls auf eine höhere
Benetzbarkeit der Oberfläche zurückzuführen ist (Abbildung 101 b). (3) Unterschiedliche
Seitenkettenendgruppen haben ebenfalls, wenn auch in geringem Maße, Einfluss auf die
Proteinadsorption. Anschließende Zelladsorptionsexperimente auf den Protein-modifizierten
BBBs sind in guter Übereinstimmung mit den vorangegangenen Ergebnissen aus der
Untersuchung der Proteinadsorption (Abbildung 102). Die, in dieser Arbeit gewonnen
Ergebnisse im Bereich Temperaturverhalten von Poly(2-oxazolin) basierten
Bürstenpolymeren und deren Wechselwirkung mit Biomolekülen liefern wichtige
Erkenntnisse, um das Spektrum an proteinresistenten Polymeren zu erweitern.

Zusammenfassung Chapter 5
128
Abbildung 101. Proteinadsorption an Poly(2-oxazolin) BBBs als Funktion der
Seitenkettenzusammensetzung und der Seitenkettenlänge a) BBBs mit unterschiedlichen
Zusammensetzungen der Seitenketten. b) BBBs mit unterschiedlicher Seitenkettenlänge.
Abbildung 102. Fluoreszenzbilder der Zelladsorption auf Fibronektin modifizierten POx
BBBs. In Abhängigkeit der Menge an adsorbiertem Fibronektin werden unterschiedlich starke
Zelladhäsionen auf POx BBB Oberflächen beobachtet: (a) Sehr gute und weit verbreitete
Zelladhäsion auf BBBs mit PnPrOx Seitenketten, (b) gute und leicht verteilte Zelladhäsion
auf BBBs mit mittellangen PMeOx Seitenketten und Boc-geschützten Endgruppen, (c)

Chapter 5 Zusammenfassung
129
geringe Zelladhäsion und geringe Verbreitung auf BBBs mit mittellangen PMeOx
Seitenketten und Hydroxy-Endgruppen, (d) Keine Zelladhäsion auf BBBs mit mittellangen
PMeOx Seitenketten und Piperidin Endgruppen. [Die Fluoreszenzbilder der Zelladhäsion
zeigen Fibronektin (rot), Aktin (grün) und Zellkerne (blau). Die Größenordnung beträgt 50
m]

Experimental Chapter 6
130
Chapter 6 Experimental
6.1 Instruments
Atomic force microscopy (AFM)
Fa. Digital Instruments, MMAFM-2
Scanner: 5298 J and 5308 E
AFM tips: Veeco, MPP-11100
All AFM measurements were performed in tapping mode. The AFM measurements were
analyzed and visualized using the Nanoscope III-software (version 5.12r3, Digital
Instruments).
Gel permeation chromatography (GPC)
GPC was performed on a Waters system (pump mod. 510, RI-detector mod. 410) using Resi
Pore Guard (50 × 7.5 mm) and 2× Resi Pore (300 × 7.5 mm) columns as the stationary and
dimethyl acetamide (DMAc) (75 mmol/L LiBr, T = 80 °C, 1 mL/min) as the mobile phase.
The calculation of the average molar mass was performed using a calibration with
poly(methyl methacrylate) (PMMA) standards from PSS (Mainz, Germany). Prior to the
measurements, the polymer samples were dissolved in DMAc and filtered through 0.2µm
PTFE filters.
Lower critical solution temperature (LCST)
Turbidity measurements were carried out on a Cary 3 UV-vis spectrophotometer from Varian.
The cloud point was determined by spectrophotometric detection of the changes in
transmittance at λ= 500 nm of the aqueous polymer solutions (1.0 wt %). The heating/cooling

Chapter 6 Experimental
131
rate was 0.5 K min−1
followed by a 10 min period of constant temperature to ensure
equilibration. Given values for the cloud point were determined as the temperature
corresponding to a 10 % decrease in optical transmittance.
Nuclear magnetic resonance (NMR)
1H NMR spectra were recorded on a Bruker ARX 300 at 292 K. The spectra were calibrated
using the solvent signals (CDCl3 7.26 ppm, CD3CN 1.94 ppm).
Fourier transform infrared spectroscopy (FTIR)
IR spectra were obtained on a Bruker IFS 55s spectrometer with a MCT detector and a single
bounce SplitPea-ATR sampling accessory from Harrick with a diamond crystal sampling unit
at a spectral resolution of 4 cm-1
. For diffuse reflectance Fourier transform infrared (DRIFT),
for each spectrum, 500 scans were accumulated with a spectral resolution of 4 cm-1
.
Background spectra were recorded on corresponding bare substrate samples.
Gas chromatography (GC)
GC was performed on a Varian CP 3380 equipped with a CombiPal robot arm and with a
Nordion NB-54 column (25 m, 0.20 mm, 0.25 mm) and FID detector (helium carrier gas). For
the kinetic measurements, the polymerization mixture was prepared and sealed in a glove-box
under an inert and dry atmosphere. The agitator was preheated to the indicated temperature.
The CombiPal was programmed for 2 syringe wash cycles (ACN) prior and after sampling.
The sealed reaction container was introduced to the agitator immediately ( 1 s) before the
first sampling, in order to obtain a zero-time value. Per injection, 2 µL of the reaction mixture
were taken. The monomer consumption was followed by the change of the ratio of the
integrals of the monomer and the internal standard (chlorobenzene).

Experimental Chapter 6
132
Dynamic light scattering (DLS)
DLS experiments were performed on a Malvern Zetasizer Nano ZS. A He-Ne laser with a
wavelength of 632.8 nm was applied as the light source. Measurements were taken at 2.0 °C
intervals.
Fluorescence microscopy
Fluorescence microscopy images were obtained with an Axiovert 200M Zeiss AG microscope
equipped with an ORCA-ER camera (Hamamatsu Photonics Japan). The sample was
irradiated using a 00 filter set (530-585 nm). The cross section analysis was obtained by pixel
analysis of the 256 bit black and white fluorescence image using the Image J software
package.
Microwave (MW)
Microwave-assisted synthesis was performed using a CEM Discover LabMate system.
Contact angle (CA)
The water contact angles were determined with a full automated Krüss DSA 10 Mk2 contact
angle goniometer. The data were obtained with the aid of the Krüss Drop Shape Analysis v3
software package.
6.2 Methods
Micro-structuring of the substrates by electron beam lithography
The electron beam lithography was performed in the research group of Prof. Grunze. The
electron beam lithography of the ω-functionalized biphenylthiol SAMs and the preparation of
the electron beam induced carbon deposits (EBCDs) were performed under identical
irradiation conditions. No precursor molecules were introduced into the vacuum chamber
during the EBCD process.

Chapter 6 Experimental
133
A flood gun (Specs Flood Gun 15/40, electron energy: 50 eV) was used to irradiate the
samples through a stencil mask (Quantifoil Micro Tools, Jena, hole radius: 1µm, center-to-
center distance: 4µm).
Direct writing with a focused e-beam was performed with a LEO 1530 scanning electron
microscope with a Raith Elphy Plus Pattern Generator System (REPGS) software. The
electron beam energy was set at 3 keV, vacuum pressure ~ 5 x 10-6
mbar.
Cytotoxicity assay, MTT
MCF 7/ADR cells, a human breast MDR cancer cell line, were seeded in 96 well plates (104
cells per well) and were allowed to reattach for 24h. Treatment solutions were prepared from
a 1 mg/mL polymer stock solution in assay buffer (containing 122 mM NaCl, 25 mM
NaHCO3, 10 mM glucose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES), 3 mM KCl, 1.2 mM MgSO4, 1.4 mM CaCl2, and 0.4 mM K2HPO4, pH 7.4) by
appropriate dilution with media (Dulbecco‟s Modified Eagle‟s Medium) (DMEM),
supplemented with 10% FBS, 25 mM HEPES and penicillin/streptomycin) for 2 h incubation.
For 72 h incubation, 1 mg/mL stock solutions where prepared in full media. The cells were
incubated for 72 h with P(IPOx-g-MeOx)R in 200 µL of treatment solution. After discarding
the treatment solution, cells were washed thrice with PBS. FBS-free DMEM (100 µL/well) as
well as 25 µL of a 5 mg/mL solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT, Invitrogen, Eugene, OR, USA) in PBS were added and the cells incubated at
37 °C for 2 hours. The media was discarded subsequently and replaced with 100 µL of
solvent (25% v/v DMF, 20% w/v SDS in H2O). The purple formazan product was allowed to
dissolve over night and the absorbance at 570 nm was obtained using a plate reader
(SpectraMax M5, Molecular Devices). Positive controls were cells treated with media alone;
negative controls were wells without cells. Each concentration was repeated in four wells,
results are expressed as mean ± standard error of the mean(SEM).
Protein adsorption and cell adhesion
Protein adsorption and cell culture was performed similarly to procedures described
recently213
. Briefly, samples were immersed in 70% ethanol/water to provide sterile
conditions for cell culture. Thereafter polymer surfaces were immersed in a 50 µg/mL
fibronectin (purified from human plasma) solution in phosphate buffered saline (PBS) (Sigma)

Experimental Chapter 6
134
at pH 7.4 and 37 °C for 1 h. Carboxytetramethylrhodamine FluoReporter (Invitrogen,
Karlsruhe, Germany) was used to label fibronectin prior to the experiments.
Human endothelial cells from the umbilical cord vein (HUVECs) were seeded in endothelial
cell growth medium ECGM (Promocell, Heidelberg, Germany) containing 2 % fetal calf
serum at density of 1105 cells per cm² on the fibronectin-coated substrates. After 1 h of cell
culture samples were fixed with 4% paraformaldehyde for 10 min and stained with DAPI
(Sigma) and phalloidin-Alexa488 (Invitrogen) to visualize nuclei and actin cytoskeleton,
respectively, in fluorescence microscopy. The microscopic analysis was performed on inverse
epi-fluorescence microscope (DMIRE2, Leica Microsystems, Germany) with a 40x oil
immersion objective using Openlab software (Perkin Elmer). The protein amounts remaining
on the substrate surface were quantified in terms of fluorescence intensities using Openlab
software. Reference intensities were collected from fibronectin-coated poly(octadecen-alt-
maleic anhydride) surfaces and uncoated surfaces.
6.3 Materials
All chemicals were purchased from Aldrich, or Acros (American chemical society purity or
higher). Solvents of lower grade were purified and dried prior to use. The 2-oxazoline
monomers were dried with CaH2 and purified by fractionation distillation at reduced pressure.
THF was dried by refluxing over sodium and benzophenone. Dry THF was collected until the
mixture turn to blue/purple. The 2-isopropenyl-2-oxazoline was stored at -25 °C and degassed
by at least 4 freeze-thaw cycles before SIPGP.
Gold substrate pretreatment
Thin gold film (around 50 nm thick) on p-doped silicon substrates were obtained from Albert
Coatings, Heidelberg.214
Before use, the substrates were cleaned by exposing the surfaces for
2 hours in UV-light (253 nm) under inert atmosphere. The surfaces were then washed with
dimethylformamide and ethanol and dried by a jet of nitrogen.
Glass substrates:
Mechanically polished borosilicate glass (Borofloat® 33) was obtained from Schott GmbH,
Mainz. Before use, the samples were cleaned by ultrasound in toluene, ethyl acetate and
ethanol, 2 min each and dried by a jet of nitrogen.

Chapter 6 Experimental
135
Glassy carbon substrates:
Mechanically polished glassy carbon substrates (Sigradur®) were purchased from HTW
Hochtemperatur-Werkstoffe GmbH, Thierhaupten. Before use, the samples were cleaned by
ultrasound in toluene, ethyl acetate and ethanol, 2 min each and dried by a jet of nitrogen.
Silicon substrates:
P-doped Si(100) wafers were obtained from Wacker Chemie AG, Munich, Germany. Before
use, the samples were sonificated for 2 minutes in toluene, ethyl acetate and ethanol, dried by
a jet of nitrogen and cleaned in a piranha solution, rinsed with water and dried again.
6.4 Monomer synthesis
2-n-Propyl-2-oxazoline (nPrOx)
In a Schlenk flask, 15 g (0.21 mol) of n-butyronitrile, 18 g (0.26 mol, 1.2 eq) aminoethanol
and 0.9g (3.2 mmol) cadmium acetate dihydrat were stirred and heated to 130 °C. After 12 h,
the reaction was cooled to room temperature. The red raw product was purified by vacuum
distillation (boiling point (bp), 57 °C at 11 mbar) and then stored under a dry nitrogen
atmosphere prior to use (yield, 16.5 g, 68%).
1H NMR (250 MHz, CDCl3): δ=4.27 (t, 2H), δ=3.81 (t, 2H), δ=2.23 (t, 2H), δ=1.69 (st, 2H),
δ=0.95 (t, 3H).
2-n-Butyl-2-oxazoline and 2-iso-propyl-2-oxazoline were synthesized accordingly.
2-n-Butyl-2-oxazoline (BuOx)
1H NMR (250 MHz, CDCl3): δ=4.17 (t, , 2H), δ=3.77 (t, 2H), δ=2.23 (t, 2H), δ=1.60 (q, 2H),
δ=1.38 (sp, 2H), δ=0.88 (t, 3H).

Experimental Chapter 6
136
2-iso-Propyl-2-oxazoline (iPrOx)
1H NMR (250 MHz, CDCl3): δ=4.23 (t, 2H), δ=3.78 (t, 2H), δ=2.62 (sp, 3H), δ= 1.18 (d, 6H).
2-Isopropenyl-oxazoline (IPOx)
IPOx was synthesized according to the procedure reported before.135
2-ethyl-oxazoline
reacted with 1 equivalent of paraformaldehyde in the presence of catalytic amounts of
triethylamine to form the hydroxyethyl derivative. Water can be eliminated readily at elevated
temperatures when alkali metal alkoxide is used as catalysts. Finally, 2-isopropenyl-oxazoline
is distilled under reduced pressure before use.
1H NMR (250 MHz, CDCl3): δ=5.78 (s, 1H), δ=5.41 (s, 1H), δ=4.27 (t, 2H), δ= 3.93 (t,
2H),δ= 2.00 (t, 3H).
6.5 Synthesis of molecular brushes
6.5.1 Free radical and anionic polymerizations
Free radical polymerization: poly(2-isopropenyl-2-oxazoline); PIPOxR
Under a dry nitrogen 2-isopropenyl-2-oxazoline (IPOx) (1.00 g, 9.00 mmol) and AIBN (14.8
mg, 0.09 mmol) was heated to 60°C for 8 h. Afterwards, the reaction mixture was equilibrated
to room temperature, diluted with chloroform (approx. 15 mL), and precipitated into dry
diethyl ether. The product was reprecipitated twice using chloroform and ether. After
filtration and freeze drying from water, a colorless powder was obtained (0.5 g, yield = 50 %).
The obtained yield is typical for the free radical polymerization of IPOx as reported earlier by
Fréchet et al.215

Chapter 6 Experimental
137
1H NMR (CDCl3): δ (ppm) 4.16 (br, 2H), 3.76 (br, 2H), 1.98–1.76 (br, 2H) and 1.24–1.12 (br,
3H).
IR (ATR, film): 2941 (C-H str) (m), 1654 (C=N str) (vs), 1118 (C-O) (vs) (unconjugated),
986, 951(vs) (ring skeletal vibration) cm-1
. Analysis of the ratio of -methyl proton signals
calculated to a tacticity ratio of syndiotactic(S):heterotactic(H):isotactic(I) of 57.0:37.2:5.8 .
GPC: PDI = 2.12, Mn = 9800 g/mol.
Living anionic polymerization: poly(2-isopropenyl-2-oxazoline); PIPOxA
Adapting an early procedure of Tomalia et al.,216
3 mL dry THF and 0.1 mL of a 2.5 M n-
butyl lithium solution in hexane (0.236 mmol, 1 equiv) were cooled under a dry argon
atmosphere, to approx. -50 °C (acetone/dry ice). Over a period of 30 min, 1 mL (9.45 mmol,
40 equiv) IPOx was added dropwise. After stirring the mixture for another 30 min at -50 °C,
the mixture was equilibrated to room temperature and stirred again for 30 min. The polymer
was endcapped by addition of 1 mL methanol. The mixture was then precipitated using
diethyl ether. After freeze-drying (water), 1.07 g (95 % yield) of PIPOxA was obtained as a
colorless powder.
1H NMR (CDCl3): δ (ppm) 4.16 (br, 2H), 3.76 (br, 2H), 1.86–1.75 (br, 2H), 1.24–1.14 (br, 3H)
and 0.85(br, C4H9).
IR (ATR, film): 2941 (C-H str) (m), 1654 (C=N str) (vs), 1118 (C-O) (vs) (unconjugated),
986, 951(vs) (ring skeletal vibration) cm-1
. Analysis of the ratio of -methyl proton signals
calculated to a tacticity ratio of syndiotactic(S):heterotactic(H):isotactic(I) of 73.0:24.8:2.2
(Figure 1).
GPC: PDI =1.20, Mn = 24350 g/mol.
6.5.2 Macroinitiator
Macroinitiator salt: poly(2-isopropenyl-2-oxazolium triflate); PIPOxOTf

Experimental Chapter 6
138
Under dry and inert conditions, PIPOx (222 mg, 1.0 equiv of oxazoline unit) and 394 mg (2.4
mmol, 1.2 equiv) of MeOTf were added to 5 mL dry acetonitrile at approx. -35 °C. After
stirring for 5 h at 0 °C, the mixture was poured into cold and dry diethyl ether to precipitate
the oxazolinium salt. The colorless precipitate was washed twice with cold ether to yield 529
mg (1.92 mmol, 96 %).
1H NMR (CD3CN): δ (ppm) 5.08 (br, 2H, =N-CH2-CH2-O-), 4.54 (br, 2H, =N-CH2-CH2-O-),
3.70 (s, 3H, CH3-N), 2.49 (2H, -C-CH2-), 1.36 (br, 3H, -C-CH3)
6.5.3 Living cationic ring-opening polymerization
Poly(2-isopropenyl-2-oxazoline-g-2-methyl-2-oxazoline); P(IPOx-g-MeOxboc)R
At 0°C, PIPOxOTfR (110 mg, 0.4 mmol, 1.0 equiv) was dissolved in 10 mL of acetonitrile
and 850 mg of 2-methyl-2-oxazoline (MeOx) (10 mmol, 25 equiv) was added. The
polymerization solution was heated by a prepared oil bath to 85 °C and stirred for 20 h. The
mixture was cooled to 0 °C, and 298 mg (1.6 mmol, 4 equiv) of N-tert-
butyloxycarbonylpiperazine (N-Boc-pip) was added. After stirring the reaction mixture for 4
h at room temperature, an excess of finely grounded potassium carbonate (~60 mg) was added
and the mixture was allowed to stir overnight. The solvent was removed under reduced
pressure and the residual dissolved in approx. 15 mL chloroform and then precipitated three
times into dry diethyl ether. The product was freeze-dried (water) to yield a colorless powder
(860 mg, 80 % yield). Additionally, the product was purified by column chromatography
using Sephadex G100 to quantitatively separate the product from minor portions of
homopolymer side products.
1H NMR (CD3Cl): δ (ppm) 3.44 (br, 82H, -N-CH2-CH2-N-),2.13/2.07 (m, 57H, -CO-CH3),
1.98/1.92 (br, 2H, CH2-C),1.46/1.44 (br, 12H, CH3Boc
and CH3-C).
IR (ATR, film): 2939 (C-H str) (m), 1696 (C=O str, ester), 1629 (C=O str, amide I) (vs), 1419
δ(CH2-CO) (s), 731 cm-1
r(CH2) (w).
GPC: PDI =1.69, Mn= 69500 g/mol.

Chapter 6 Experimental
139
Poly(2-isopropenyl-2-oxazoline-g-2-ethyl-2oxazoline); P(IPOx-g-EtOxboc)R
As described above, P(IPOx-g-EtOx)R was obtained using 110 mg of PIPOxOTf
R, 990 mg 2-
ethyl-2-oxazoline (EtOx) and 298 mg N-Boc-pip. The polymerization time was 30h. After
column chromatography and freeze drying, a colorless powder was obtained (940 mg, 76 %
yield).
1H NMR (CD3Cl): δ (ppm) 3.44 (br, 86H, -N-CH2-CH2-N-),2.39(br, 41H, -CO-CH2CH3),
1.98/1.92 (br, 2H, -CH2-C), 1.46/1.44 (br, 12H, CH3Boc
and CH3-C), 1.10(br, 62H, -CO-
CH2CH3).
IR (ATR, film) 2937 (C-H str) (m), 1699 (C=O str, ester), 1629 (C=O str, amide I) (vs), 1428
δ(CH2-CO) (s), 731 cm-1
r(CH2) (w).
GPC: PDI =1.75, Mn = 77500 g/mol.
Poly(2-isopropenyl-2-oxazoline-g-2-isopropyl-2-oxazoline); P(IPOx-g-iPrOxboc)R
Following the procedure described above, P(IPOx-g-iPrOx)R was obtained using 110 mg
PIPOxOTfR, 1.13 g iPrOx and 298 mg N-Boc-pip. The polymerization time was 40 h. After
freeze drying (water), a colorless powder was obtained (630 mg, 50 % yield). In this case, no
homopolymer side product could be detected.
1H NMR (CD3Cl): δ (ppm) 3.45 (br, 72H, -N-CH2-CH2-N-),2.88 (br, 22H, -CO-CH(CH3)2),
1.98/1.92 ( br, 2H, CH2-C), 1.46/1.45 (br, 12H, CH3Boc
and CH3-C), 1.09 (br, 105H, -
CH(CH3)2).

Experimental Chapter 6
140
IR (ATR, film): 2937 (C-H str) (m), 1698 (C=O str, ester),1637 (C=O str, amide I) (vs),
1419 δ(CH2-CO) (s), 731 cm-1
r(CH2) (w).
GPC: PDI =1.58, Mn = 53200 g/mol.
Poly(2-isopropenyl-2-oxazoline-g-2-isopropyl-2-oxazoline) (P(IPOx-g-iPrOx))R
200 mg P(IPOx-g-iPrOxboc)R was dissolved in a solution of 1 mL trifluoroacetic acid (TFA)
and 1mL of chloroform. The mixture was stirred at RT for 2 h. Then, the mixture was dried
and redissolved in chloroform and precipitate twice in diethyl ether. After freeze drying
(water), a colorless powder was obtained (150 mg, 78% yield).
1H NMR (CD3Cl): δ (ppm) 3.45 (br, 42H, -N-CH2-CH2-N-),2.88 (br, 18H, -CO-CH(CH3)2),
1.09 (br, 67H, -CH(CH3)2).
GPC: PDI = 1.45, Mn = 50000 g/mol
Poly(2-isopropenyl-2-oxazoline-g-2-isopropyl-2-oxazoline); P(IPOx-g-iPrOx)A
Following the procedure described above, P(IPOx-g-iPrOx)A
was obtained accordingly (49 %
yield). Also in this case, no homopolymer side product could be detected.
1H NMR (CD3Cl): δ (ppm) 3.45 (br, 76H, -N-CH2-CH2-N-),2.88(br, 23H, -CO-CH(CH3)2),
1.98/1.92 ( br, 2H, CH2-C), 1.46/1.45 (br, 12H, CH3Boc
and CH3-C), 1.09 (br, 110H, -
CH(CH3)2).
IR (ATR, film): 2937 (C-H str) (m), 1698 (C=O str, ester),1637 (C=O str, amide I) (vs),
1419 δ(CH2-CO) (s), 731 cm-1
r(CH2) (w).
GPC: PDI =1.33, Mn = 125000 g/mol.

Chapter 6 Experimental
141
Poly(2-isopropenyl-2-oxazoline-g-2-npropyl-2-oxazoline) (M-I1~M-I3 and M-II1~M-II4)
gas chromatography (GC) measurement
At 0 °C, MI/M
II (44 mg, 0.16 mmol, 1.0 equiv) was dissolved in 4 mL of acetonitrile and 452
mg of nPrOx (4.0 mmol, 25 equiv) was added. The polymerization solution was performed at
80 °C. Samples were withdrawn periodically for monomer conversion (GC) and molecular
weight (GPC) measurement. The polymerization was stopped by adding excess of piperidine.
After stirring the reaction mixture for 8 h at room temperature, an excess of finely grounded
potassium carbonate (~60 mg) was added and the mixture was allowed to stir overnight. The
solvent was removed under reduced pressure and the residual dissolved in chloroform and
then precipitated three times into dry diethyl ether. The product was freeze-dried (water) to
yield a colorless powder. Additionally, the product was purified by column chromatography
using Sephadex G100 to quantitatively separate the product from minor portions of
homopolymer side products.
1H NMR (CDCl3): δ=3.44 (s, br, 93 H, N-(CH2)2-N); δ=2.33/2.20 (m, 48 H, CO-CH2-CH2-
CH3); δ=1.62 (s, br, 51 H, CO-CH2-CH2-CH3); δ=0.94 (s, br, 75 H, CO-CH2-CH2-CH3).
Poly(2-isopropenyl-2-oxazoline-g-2-isopropyl-2-oxazoline) (M-II5~M-II7) were
synthesized according to the above procedure.
1H NMR (CDCl3): δ (ppm) 3.45 (br, 76H, -N-CH2-CH2-N-) 2.88(br, 23H, -CO-CH(CH3)2),
1.98/1.92 (br, 2H, CH2-C), 1.45 (br, 3H, CH3-C), 1.09 (br, 110H, -CH(CH3)2).

Experimental Chapter 6
142
6.6 Preparation of bottle-brush brushes
6.6.1 Bottle-brush brushes on glassy carbon
Self-initiated photopolymerization and photografting (SIPGP) of IPOx on glassy carbon.
Polished glassy carbon substrates (or other surfaces) were cleaned by sequential
ultrasonification in ethanol, ethyl acetate and toluene before use. The glassy carbon substrates
were clamped with a square mesh grids (square size: 50×50 µm2) and subsequently
submerged in approximately 2 mL of freshly distilled and degassed IPOx in a glass vial.
Polymerization was allowed to complete in 2 to 40 h under constant irradiation with UV light
(λmax = 350 nm) at room temperature (RT). After photopolymerization, the samples were
immediately cleaned by sequential ultrasonification in ethanol, ethyl acetate and toluene (all
HPLC grade) for five minute each.
IR (DRIFT): 1648 cm-1
(C=N stretching), 1128 cm-1
(C-O stretching), 987-1
and 951-
1(oxazoline skeletal viberation).
Poly(2-isopropenyl-2-oxazolium triflate) on glassy carbon; PIPOxOTf
The poly(2-isopropenyl-2-oxazoline) (PIPOx) modified glassy carbon substrates were
submerged in a solution of 2 mL acetonitrile (ACN) with an excess amount of methyl
trifluoromethane sulfonate (MeOTf) (0.1 g) at approximately -35 °C under nitrogen
atmosphere. After stirring for 6 h at 0-5 °C, the mixture was allowed to equilibrate to RT and
was stirred for another 2 h.

Chapter 6 Experimental
143
Surface initiated living cationic ring-opening polymerization (SI-LCROP).
Without washing, the glassy carbon substrate coated with PIPOxOTf was taken to a
microwave reaction vial filled with a solution of 1 g 2-alkyl-2-oxazoline (alkyl = methyl,
ethyl, propyl or butyl) and several drops of MeOTf in 3mL ACN at 0 °C. Catalytic amounts
of MeOTf were added to consume the minor impurities in the liquid phase. The reaction
solution was irradiated by microwaves for 20 min with a temperature setting of 130 °C. The
solution was cooled to 0 °C, and 150 mg of N-tertbutoxycarbonylpiperazine (N-Boc-
piperazine) dissolved in 1 mL of ACN was added. Successively, the solution was stirred for
16 h at room temperature. After this, an excess of potassium carbonate (70 mg) was added to
the solution and stirred overnight. The substrate was then removed from the solution and
cleaned by sequential ultrasonication in deionized water, ethanol, ethyl acetate and toluene for
1 minute each.
IR (DRIFT) (side chain = PEtOx): 2932 cm-1
(CHx stretching), 1627 cm-1
(carbonyl stretching
of the amide), 1421cm-1
(CHx deformation).
Deprotection of poly(2-isopropenyl-2-oxazoline-g-2-ethyl-2oxazoline-Boc) (P(IPOx-g-
EtOx)).
The P(IPOx-g-EtOxBoc) coated glassy carbon substrates were submerged in a solution of 1
mL trifluoroacetic acid (TFA) and 1 mL chloroform. The mixture was stirred at RT for 3 h.
Then, the glassy carbon substrate was neutralized in a 5 % NaHCO3 aqueous solution for 2 h.
Finally, the polymer coated glassy carbon substrate was thoroughly cleaned by
ultrasonification in water, ethanol, ethyl acetate and toluene.

Experimental Chapter 6
144
Fluorescent labeling of P(IPOx-g-EtOx) grafts
A P(IPOx-g-EtOx) modified glassy carbon substrate was submerged in a 15 mM rhodamine B
isothiocyanate solution in methanol for 3 days at RT. Finally, the functionalized glassy carbon
substrate was cleaned by ultrasonification in ethanol.
6.6.2 Bottle-brush brushes on SAM of 3-aminopropyltrimethoxysilane on
oxidized silicon substrate
Aminopropyltrimethoxysilane SAM
The glass slides were covered with a 5% v/v 3-aminopropyltrimethoxysilane solution in dry
acetone and left in ultrasonication for 30 minutes at room temperature under nitrogen. After
the reaction was completed, the slides were washed with acetone, and then dried under
vacuum. (Water contact angle: 55±3°)

Chapter 6 Experimental
145
Self-initiated photopolymerization and photografting (SIPGP) of IPOx on
aminopropyltrimethoxysilane SAM
Adapt the procedure for preparation of SIPGP of IPOx on glassy carbon, PIPOx brushes on
aminopropyltrimethoxysilane monolayer modified glass slide were prepared analogously.
DRIFT-IR: 1656 cm-1
(C=N stretching), 1130 cm-1
(C-O stretching), 990-1
and 953-
1(oxazoline skeletal viberation).
Surface-initiated Living Cationic Ring-Opening Polymerization (SI-LCROP)

Experimental Chapter 6
146
Poly(2-isopropenyl-2-oxazoline-g-2-methyl-2-oxazoline) (P(IPOx-g-MeOx)), Poly(2-isopro
penyl-2-oxazoline-g-2-ethyl-2-oxazoline) (P(IPOx-g-EtOx)) and Poly(2-isopropenyl-2-
oxazoline-g-2-propyl-2-oxazoline) (P(IPOx-g-nPrOx)) were prepared according the procedure
for the preparation of Bottle-brush brushes on glassy carbon substrates. Moreover, P(IPOx-g-
MeOx) brushes with different side chain length and termini were also prepared analogously.
DRIFT-IR(side chain = PMeOx, end group = piperidine): 2958 cm-1
(CHx stretching), 1639
cm-1
(carbonyl stretching of the amide), 1431cm-1
(CHx deformation).
6.6.3 Bottle-brush brushes on SAM of biphenyl on gold
Preparation of ω-functionalized biphenylthiol SAMs on gold
The synthesis of 4-mercapto-1,1‟-biphenyl (BT), 4‟-methyl-1,1‟-biphenyl-4-thiol (MBT), 4‟-
hydroxy-1,1‟-biphenyl-4-thiol (HBT) and 4‟-nitro-1,1‟-biphenyl-4-thiol (NBT) and the
preparation of the SAMs was performed in the research group of Prof. Grunze. The SAMs
were prepared by immerging the gold substrate in a 15 mmol solution of the respective ω-
functionalized biphenylthiols in ethanol for three days. The substrates were cleaned by
sonification for 5 minutes in air and dried.
PIPOx brushes & bottle-brush brushes on SAMs on gold
PIPOx brush as well as successive LCROP of 2-oxazoline on biphenylthiol modified gold
surface was prepared according to the procedure for preparation of bottle-brush brushes on
glassy carbon.

Chapter 7 References
147
References
[1] Zhang, M.; Müller, A. H. E. J. Polym. Sci. Part A: Polym. Chem. 2005, 43, 3461-3481.
[2] Xu, H.; Sun, F. C.; Shirvanyants, D. G.; Rubinstein, M.; Shabratov, D.; Beers, K. L.;
Matyjaszewski, K.; Sheiko, S. S. Adv. Mater. 2007, 19, 2930-2934.
[3] Pakula, T.; Zhang, Y.; Matyjaszewski , K.; Lee, H.; Boerner, H.; Qin, S.; Berry, G. C.
Polymer 2006, 47, 7198-7206.
[4] Djalali, R.; Li, S. Y.; Schmidt, M. Macromolecules 2002, 35, 4282-4288.
[5] Neugebauer, D.; Zhang, Y.; Pakula, T.; Sheiko, S. S.; Matyjaszewski, K.
Macromolecules 2003, 36, 6746-6755.
[6] Bourret, R. B.; Borkovich, K. A.; Simon, M. I. Annu. Rev. Biochem. 1991, 60, 401-441.
[7] http://en.wikipedia.org/wiki/Callistemon
[8] Seog, J.; Dean, D.; Plaas, A. H. K.; Weng-Palms, S.; Grodzinsky, A. J.; Ortiz, C.
Macromolecules 2002, 35, 5601-5615.
[9] Sheiko, S. S.; Sumerlin, B. S.; Matyjaszewski, K. Prog. Polym. Sci. 2008, 33, 759-785.
[10] Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Macromolecules 1995, 28,
1721-1723.
[11] Wang, J.; Matyjaszewski, K. J. Am. Chem. Soc. 1995, 117, 5614-5615.
[12] Cowie, J. M. G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials;
CRC Press Taylor and Francis Group: Boca Raton, Fl, 3rd Ed. 2008, 82-84.
[13] Sumerlin, B. S.; Neugebauer, D.; Matyjaszewski, K. Macromolecules 2005, 38, 702-
708.
[14] Neugebauer, D.; Sumerlin, B. S.; Matyjaszewski, K.; Goodhart, B.; Sheiko, S. S.
Polymer 2004, 45, 8173-8179.
[15] Stuart, M. A. C.; Huck, W. T. S.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.;
Sukhorukov, G. B.; Szleifer, I.; Tsukruk, V. V.; Urban, M.; Winnik, F.; Zauscher, S.;
Luzinov, I.; Minko, S. Nature Materials 2010, 9, 101-113.
[16] Jeong, B.; Gutowska, A. Trends Biotechnol. 2002, 20, 305-311.
[17] Kikuchi, A.; Okano, T. Prog. Polym. Sci. 2002, 27,1165-1193.
[18] Hoffman, A. S. et al. J. Biomed. Mater. Res. 2000, 52, 577-586.
[19] Galaev, I. Y.; Mattiasson, B. Trends Biotechnol. 1999, 17, 335-340.
[20] Qiu, Y.; Park, K. Adv. Drug Deliv. Rev. 2001, 53, 321-339.
[21] Sershen, S.; West, J. Adv. Drug Deliv. Rev. 2002, 54, 1225-1235.

References Chapter 7
148
[22] Yokoyama, M. Drug Discov. Today 2002, 7, 426-432.
[23] Chilkoti, A.; Dreher, M.R.; Meyer, D. E.; Raucher, D. Adv. Drug. Deliv. Rev. 2002, 54,
613-630.
[24] Bromberg, L. E.; Ron, E. S. Adv. Drug Deliv. Rev. 1998, 31, 197-221.
[25] Weidner, J. Drug Discov. Today 2001, 6, 1239-1248.
[26] Pinkrah, V. T.; Snowden, M. J.; Mitchell, J. C.; Seidel, J.; Chowdhry, B. Z.; Fern, G. R.
Langmuir 2003, 19, 585-590.
[27] Peng, T.; Cheng, Y. L. Polymer 2001, 41, 2091-2100.
[28] Bignotti, F.; Penco, M.; Sartore, L.; Peroni, I.; Mendichi, R.; Casolaro, M.; D‟Amore,
A. Polymer 2000, 41, 8247-8256.
[29] Gan, L. H.; Gan, Y. Y.; Deen, G. R. Macromolecules 2000, 33, 7893-7897.
[30] Huber, S. Doctor thesis, TU München, 2007.
[31] Otake. K.; Inomata, H.; Konno, M.; Saito, S. Macromolecules 1990, 23, 283-289.
[32] Furyk, S.; Zhang, Y.; Ortiz-Acosta, D.; Cremer, P. S.; Bergbreiter, D. E. J. Polym. Sci.
Part A: Polym. Chem. 2006, 44, 1492-1501.
[33] Huber, S.; Hutter, N.; Jordan, R. Colloid Polym. Sci. 2008, 286, 1653-1661.
[34] Satokawa, Y.; Shikata, T.; Tanaka, F.; Qiu, X.; Winnik, F. M. Macromolecules 2009,
42, 1400-1403.
[35] Lee, H.; Pietrasik , J.; Sheiko, S. S.; Matyjaszewski, K. Prog. Polym. Sci. 2010, 35,
24-44.
[36] Li, C.; Gunari, N.; Fischer, K.; Janshoff, A.; Schmidt, M. Angew. Chem., Int. Ed. 2004,
43, 1101-1104.
[37] Yamamoto, S.; Pietrasik, J.; Matyjaszewski, K. Macromolecules 2008, 41, 7013-7020.
[38] Pietrasik, J.; Sumerlin, B. S.; Lee, R. Y.; Matyjaszewski, K. Macromol. Chem. Phys.
2007, 208, 30-36.
[39] Lutz, J. F.; Akdemir, O.; Hoth, A. J. Am. Chem. Soc. 2006, 128, 13046-13047.
[40] Yamamoto, S.; Pietrasik, J.; Matyjaszewski, K. Macromolecules 2007, 40, 9348-9353.
[41] Lutz, J. F.; Hoth, A. Macromolecules 2006, 39, 893-896.
[42] Lutz, J. F.; Weichenhan, K.; Akdemir, Ö.; Hoth, A. Macromolecules 2007, 40, 2503-
2508.
[43] Fournier, D.; Hoogenboom, R.; Thijs, H. M. L.; Paulus, R. M.; Schubert, U. S.
Macromolecules 2007, 40, 915-920.
[44] Balamurugan, S. S.; Bantchev, G. B.; Yang, Y.; McCarley, R. L. Angew. Chem. Int. Ed.

Chapter 7 References
149
2005, 44, 4872-4876.
[45] Lutz, J. F. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3459-3470.
[46] Sheiko, S. S.; Möller, M. Chem. Rev. 2001, 101, 4099-4124.
[47] Xu, H.; Shirvaniants, D.; Beers, K.; Matyjaszewski, K.; Rubinstein, M.; Sheiko, S. S.
Phys. Rev. Lett. 2004, 93, 206103/1-206103/4.
[48] Sheiko, S. S.; Silva, M. D.; Shirvaniants, D.; LaRue, I.; Prokhorova, S.; Möller, M.;
Beers, K.; Matyjaszewski, K. J. Am. Chem. Soc., 2003, 125 , 6725-6728.
[49] Sheiko, S. S.; Sun, F. C.; Randall, A.; Shirvanyants, D.; Rubinstein, M.; Lee, H.;
Matyjaszewski, K. Nature, 2006, 440, 191-194.
[50] Lebedeva, N. V.; Sun, F. C.; Lee, H. I.; Matyjaszewski, K.; Sheiko, S. S. J. Am. Chem.
Soc. 2008, 130, 4228-4229.
[51] Raynor, J. E.; Capadona, J. R.; Collard, D. M. Biointerphases 2009, 4, FA3-FA16.
[52] Yan, L.; Zhao, X. M.; Whitesides, G. M. J. Am. Chem. Soc. 1998,120, 6179-6180.
[53] Purrucker, O.; Förtig, A.; Jordan, R.; Tanaka, M. Chem. Phys. Chem. 2004, 5, 327-335.
[54] Jordan, R; Ulman, A. J. Am. Chem. Soc. 1998, 120, 243-247.
[55] Rehfeldt, F.; Tanaka, M.; Pagnoni, L.; Jordan, R. Langmuir 2002, 18, 4908-4914.
[56] Förtig, A.; Jordan, R.; Graf, K.; Schiavon, G.; Purrucker, O.; Tanaka, M. Macromol.
Symp. 2004, 210, 329-338.
[57] Wang, A.; Tang, H.; Cao, T.; Salley, S. O.; Simon Ng, K. Y. J. Colloid Interface Sci.
2005, 291, 438-447.
[58] Hernando, J.; Pourrostami, T.; Garrido, J. A.; Williams, O. A; Gruen, D. M.; Kromka,
A.; Steinmüller, D.; Stutzmann, M. Diamond Relat. Mater. 2007, 16, 138-143.
[59] Schlenoff, J. B.; Li, M.; Ly, H. J. Am. Chem. Soc. 1995, 117, 12528-12536.
[60] Brewer, N. J.; Janusz, S.; Critchley, K.; Evans, S. D.; Leggett, G. J. J. Phys. Chem. B
2005, 109, 11247-11256.
[61] Yang, W.; Rånby, B. J. Appl. Polym. Sci. 1996, 62, 533-543.
[62] Deng, J. P.; Yang, W. T.; Rånby, B. Macromol. Rapid Commun. 2001, 22, 535-538.
[63] Wang, H. L.; Brown, H. R. Macromol. Rapid Commun. 2004, 25, 1095-1099.
[64] Li, S. J.; Li, C. G.; Li, T.; Cheng, J. J.; “Polymer Photochemistry Principles and
Applications”, 1st edition, Fudan University Press, Shanghai. 1993, 110.
[65] Fang, W. H.; Liu, R. Z. J. Am. Chem. Soc. 2000, 122, 10886-10894.
[66] Steenackers, M. Doctor thesis, TU München, 2007.
[67] Steenackers, M.; Lud, S. Q.; Niedermeier, M.; Bruno, P.; Gruen, D. M.; Feulner, P.;

References Chapter 7
150
Stutzmann, M.; Garrido, J. A.; Jordan, R. J. Am. Chem. Soc. 2007, 129, 15655-15661.
[68] Steenackers, M.; Jordan, R.; Küller, A.; Grunze, M. Adv. Mater. 2009, 21, 2921-2925.
[69] Steenackers, M.; Küller, A.; Stoycheva, S.; Grunze, M.; Jordan, R. Langmuir 2009, 25,
2225-2231.
[70] Kim, J. B.; Bruening, M. L.; Baker, G. L. J. Am. Chem. Soc. 2000, 122, 7616-7617.
[71] Kim, J. B.; Huang, W. X.; Miller, M. D.; Baker, G. L.; Bruening, M. L. J. Polym. Sci.,
Part A: Polym. Chem. 2003, 41, 386-394.
[72] Xiao, D. Q.; Wirth, M. J. Macromolecules 2002, 35, 2919-2925.
[73] Yang, Q.; Tian, J.; Hu, M. X.; Xu, Z. K. Langmuir 2007, 23, 6684-6690.
[74] Lee, H. J.; Nakayama, Y.; Matsuda, T. Macromolecules 1999, 32, 6989-6995.
[75] Ma, H.; Li, D.; Sheng, X.; Zhao, B.; Chilkoti, A. Langmuir 2006, 22, 3751-3756.
[76] Advincula, R. C.; Brittain, W. J.; Caster, K. C.; Rühe, J. Polymer Brushes: Synthesis,
Characterization, Applications; Wiley-VCH: Weinheim, Germany.
[77] Dwek, R. A. Chem. Rev. 1996, 96, 683-720.
[78] Prime, K. L.; Whitesides, G. M. J. Am. Chem. Soc. 1993, 115, 10714-10721.
[79] Sigal, G. B.; Mrksich, M.; Whitesides, G. M. J. Am. Chem. Soc. 1998, 120, 3464-3473.
[80] Krishnan, S.; Weinman, C. J.; Ober, C. K. J. Mater. Chem., 2008, 18, 3405-3413.
[81] Hucknall, A.; Rangarajan, S.; Chilkoti, A. Adv. Mater. 2009, 21, 2441-2446.
[82] Herrwerth, S.; Rosendahl, T.; Feng, C.; Fick, J.; Eck, W.; Himmelhaus, M.; Dahint, R.;
Grunze, M. Langmuir 2003, 19, 1880-1887.
[83] Murthy, N.; Campbell, J.; Fausto, N.; Hoffman, A. S.; Stayton, P. S. J. Controlled
Release 2003, 89, 365-374.
[84] Werner, C.; Maitz, M. F.; Sperling, C. J. Mater. Chem. 2007, 17, 3376-3384.
[85] Nath, N.; Hyun, J.; Ma, H.; Chilkoti, A. Surf. Sci, 2004, 570, 98-110.
[86] Mrksich, M.; Whitesides, G. M. Annu. Rev. Biophys. Biomol Struct. 1996, 25, 55-78.
[87] http://www.bootschoon.com/antifouling.htm
[88] Herrwerth, S.; Eck, W.; Reinhardt, S.; Grunze, M. J. Am. Chem. Soc. 2003, 125, 9359-
9366.
[89] Lasseter, T. L.; Clare, B. H.; Abbott, N. L.; Hamers, R. J. J. Am. Chem. Soc. 2004, 126,
10220-10221.
[90] Tanaka, M.; Mochizuki, A.; Shiroya, T.; Motomura, T.; Shimura, K.; Onishi, M.;
Okahata, Y. Colloids Surf., A 2002, 203, 195-204.
[91] Cho, W. K.; Kong, B.; Choi, I. S. Langmuir 2007, 23, 5678-5682.

Chapter 7 References
151
[92] Ladd, J.; Zhang, Z.; Chen, S.; Hower, J. C.; Jiang, S. Biomacromolecules, 2008, 9,
1357-1361.
[93] Statz, A. R.; Meagher, R. J.; Barron, A. E.; Messersmith, P. B. J. Am. Chem. Soc. 2005,
127, 7972-7973.
[94] Holland, N. B.; Qiu, Y.; Ruegsegger, M.; Marchant, R. E. Nature 1998, 392, 799-801.
[95] Zhu, J.; Marchant, R. E. Biomacromolecules 2006, 7, 1036-1041.
[96] Raynor, J. E.; Petrie, T. A.; Fears, K. P.; Latour, R. A.; García A. J.; Collard, D. M.
Biomacromolecules 2009, 10, 748-755.
[97] Harris, J. M. Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical
Applications (Ed: J. M. Harris), Plenum, New York 1992, p. 1.
[98] Herold, D. A.; Keil, K.; Bruns, D. E. Biochem. Pharmacol. 1989, 38, 73-76.
[99] Shen, M.; Martinson, L.; Wagner, M. S.; Castner, D. G.; Ratner, B. D.; Horbett, T. A.
J. Biomater. Sci., Polym. Ed. 2002, 13, 367-390.
[100] Li, L.; Chen, S.; Jiang, S. J. Biomater. Sci., Polym. Ed. 2007, 18, 1415-1427.
[101] Hansen, C. B.; Kao, G. Y.; Moase, E. H.; Zalipsky, S.; Allen, T. M.; Biochim. Biophys.
Acta-Biomembranes 1995, 1239, 133-144.
[102] Hayward, J. A.; Chapman, D. Biomaterials 1984, 5, 135-142.
[103] Zhang, Z.; Chao, T.; Chen, S.; Jiang, S. Langmuir, 2006, 22, 10072-10077.
[104] Kagiya, T.; Narisawa, S.; Maeda, T.; Fukui, K. J. Polym. Sci., Polym. Lett. Ed. 1966,
4, 441-445.
[105] Saegusa, T.; Kobayashi, S. Encyclopedia of Polymer Science and Technology, John
Wiley and Sons, New York , 1976.
[106] Kobayashi, S. Prog. Polym. Sci. 1990, 15, 751-823.
[107] Chujo, Y.; Saegusa, T. Ring-Opening Polymerization, Hanser, Munich, 1993.
[108] Aoi, K.; Okada, M. Prog. Polym. Sci. 1996, 21, 151-208.
[109] Hoogenboom, R.; Fijten, M. W. M.; Schubert, U. S. J. Polym. Sci. Polym. Chem. 2004,
42, 1830-1840.
[110] Hoogenboom, R.; Paulus, R. M.; Fijten, M. W. M.; Schubert, U. S. J. Polym. Sci.
Polym. Chem. 2005, 43, 1487-1497.
[111] Litt, M.; Levy, A.; Herz, J. J. Macromol. Sci. Chem. 1975, 5, 703-727.
[112] Kobayashi, S.; Uyama, H. Polym. News 1991, 16, 70 -76.
[113] Gaertner, F. C.; Luxenhofer, R.; Blechert, B.; Jordan, R.; Essler, M. J. Contr. Release
2007, 119, 291-300.

References Chapter 7
152
[114] Woodle, M. C.; Engbers, C. M.; Zalipsky, S. Bioconjugate Chem. 1994, 5, 493-496.
[115] Zalipsky, S.; Hansen, C. B.; Oaks, J. M.; Allen, T. M. J. Pharm. Sci. 1996, 85, 133-
137.
[116] Adams, N.; Schubert, U. S. Adv. Drug Delivery Rev. 2007, 59, 1504-1520.
[117] Hoogenboom, R. Angew. Chem., Int. Ed. 2009, 48, 7978-7994.
[118] Lin, P.; Clash, C.; Pearce, E. M.; Kwei, T. K.; Aponte, M. A. J. Polym. Sci. Part B
1988, 26, 603-619.
[119] Huber, S.; Jordan, R. Colloid Polym. Sci. 2008, 286, 395-402.
[120] Diehl, C.; Schlaad, H. Macromol. Biosci.2009, 9, 157-161.
[121] Kim, C.; Lee, S. C.; Kang, S.W.; Kwon, I. C.; Jeong, S. Y. J. Polym. Sci. Part B 2000,
38, 2400-2408.
[122] Hoogenboom, R.; Thijs, H. M. L.; Wouters, D.; Hoeppener, S.; Schubert, U. S. Soft
Matter 2008, 4, 103-107.
[123] Schlaad, H.; Diehl, C.; Gress, A.; Meyer, M.; Demirel, A. L.; Nur, Y.; Bertin, A.
Macromol. Rapid Commun. 2010, 31, 511-525.
[124] Goddard, P.; Hutchinson, L. E.; Brown, J.; Brookman, L. J. J. Controlled Release
1989, 10, 5-16.
[125] Park, Y. S.; Kang, Y. S.; Chung, D. J. e-Polym. 2002, no. 016.
[126] Miyamoto, M.; Naka, K.; Shiozaki, M.; Chujo, Y.; Saegusa, T. Macromolecules 1990,
23, 3201-3205.
[127] Mero, A.; Pasut, G.; Via, L. D.; Fijten, M. W. M.; Schubert, U. S.; Hoogenboom, R.;
Veronese, F. M. J. Controlled Release 2008, 125, 87-95.
[128] Meyers, A. I.; Mihelich, E. D. Angew. Chem. Int. Ed. 1976, 15, 270-281.
[129] Gabriel, S.; Eschenbach, G. Chem. Ber. 1897, 23, 2494-2497.
[130] Zarka, M. T.; Nuyken, O.; Weberskirch, R. Chem. Eur. J. 2003, 9, 3228-3234.
[131] Cesana, S.; Auernheimer, J.; Jordan, R.; Kessler, H.; Nuyken, O. Macromol. Chem.
Phys. 2006, 207, 183-192.
[132] Witte, H.; Seeliger, W. Liebigs Ann. Chem. 1974, 6, 996-1009.
[133] Persigehl, P.; Jordan, R.; Nuyken, O. Macromolecules 2000, 33, 6977-6981.
[134] Taubmann, C.; Luxenhofer, R.; Cesana, S.; Jordan, R. Macromol. Biosci. 2005, 5,
603-612.
[135] Seeliger, W.; Aufderhaar, E.; Diepers, W.; Feinauer, R.; Nehring, R.; Thier, W.;
Hellman, H. Angew. Chem. Int. Ed. 1966, 5, 875-888.

Chapter 7 References
153
[136] Miyamoto, M.; Sano, Y.; Kimura, Y.; Saegusa, T. Macromolecules, 1985, 18, 1641-
1648.
[137] Groß, A.; Maier, G.; Nuyken, O. Macromol. Chem. Phys 1996, 197, 2811-2826.
[138] Uyama, H.; Honda, Y.; Kobayashi, S. J. Polym. Sci., Part A: Polym. Chem. 1993, 31,
123-128.
[139] Shoda, S. I.; Masuda, E.; Furukawa, M.; Kobayashi, S. J. Polym. Sci., Part A: Polym.
Chem. 1992, 30, 1489-1494.
[140] Kobayashi, S.; Kaku, M.; Sawada, S.; Saegusa, T. Polym. Bull. 1985, 13, 447-451.
[141] Jordan, R.; West, N.; Ulman, A.; Chou, Y. M.; Nuyken, O., Macromolecules 2001,
34, 1606-1611.
[142] Kobayashi, S.; Shimano, Y.; Saegusa, T. Polym. J. 1991, 23, 1307-1315.
[143] Nuyken, O.; Rueda-Sanchez, J.; Voit, B. Macromol. Rapid Commun. 1997, 18, 125-
131.
[144] Grasmüller, M.; Rueda-Sanchez, J.; Voit, B.; Nuyken, O. Macromol. Symp. 1998,
127, 109-114.
[145] Nuyken, O.; Rueda-Sanchez, J.; Voit, B. Polym. Bull. 1997, 38, 657-664.
[146] Weberskirch, R.; Hettich, R.; Nuyken, O.; Schmaljohann, D.; Voit, B. Macromol.
Chem. Phys 1999, 200, 863-873.
[147] Kagiya, T.; Matsuda, T.; Zushi, K. J. Macromol. Sci. Chem. 1972, 6, 1349-1372.
[148] Kagiya, T.; Matsuda, T.; Nakato, M.; Hirata, R. J. Macromol. Sci. Chem. 1972, 6,
1631-1652.
[149] Dibona, D. M.; Fibiger, R. F.; Gurnee, E. F.; Shuetz, J. E. J. Appl. Polym. Sci. 1986,
31, 1509-1514.
[150] Nuyken, O.; Maier, G.; Gross, A.; Fischer, H. Macromol. Chem. Phys. 1996, 197, 83-
95.
[151] Luxenhofer, R.; Jordan, R. Macromolecules 2006, 39, 3509-3516.
[152] Bovey, F. A.; Tiers, G. V. D., J. Polym. Sci. 1960, 44, 173-182.
[153] Bonne, T. B.; Lüdtke, K.; Jordan, R.; Stepanek, P.; Papadakis, C. M. Colloid Polym.
Sci. 2004, 282, 833-843.
[154] Bonne, T. B.; Papadakis, C. M.; Lüdtke, K.; Jordan, R. Colloid Polym. Sci. 2007,
285, 491-497.
[155] Ivanova, R.; Komenda, T.; Bonne, T. B.; Lüdtke, K.; Mortensen, K.; Pranzas, P. K.;
Jordan, R.; Papadakis, C. M. Macromol. Chem. Phys. 2008, 209, 2248-2258.

References Chapter 7
154
[156] Purrucker, O.; Förtig, A.; Lüdtke, K.; Jordan, R.; Tanaka, M. J. Am. Chem. Soc.
2005, 127, 1258-1264.
[157] Foreman, M. B.; Coffman, J. P.; Murcia, M. J.; Cesana, S.; Jordan, R.; Smith, G. S.;
Naumann, C. A. Langmuir 2003, 19, 326-332.
[158] Gil, E. S.; Hudson, S. M. Prog Polym. Sci. 2004, 29, 1173-1222.
[159] Diab, C.; Akiyama, Y.; Kataoka, K.; Winnik, F. M. Macromolecules 2004, 37, 2556-
2562.
[160] Christova, D.; Velichkova, R.; Loos, W.; Goethals, E. J.; du Prez, F. Polymer 2003,
44, 2255-2261.
[161] Uyama, H.; Kobayashi, S. Chem. Lett. 1992, 9, 1643-1646.
[162] Tang, C.; Dufour, B.; Kowalewski, T.; Matyjaszewski, K. Macromolecules 2007, 40,
6199-6205.
[163] Rihova, B. Adv Drug Deliv. Rev. 1996, 21, 157-176.
[164] Mosmann, T. J. Immunol Meth. 1983, 65, 55-63.
[165] http://en.wikipedia.org/wiki/MTT_assay
[166] Arest, Y.; A. A.; Litvesenko, G. I. Prog. Polym. Sci. 1996, 21, 335-398.
[167] Park, J. S.; Kataoka, K. Macromolecules 2007, 40, 3599-3609.
[168] Hoogenboom, R.; Thijs, H. M. L.; Jochems, M. J. H. C.; van Lankvelt, B. M.; Fijten,
M.W. M.; Schubert, U. Chem. Commun. 2008, 5758-5760.
[169] Zhang, B.; Gröhn, F.; Pedersen, J. S.; Fischer, K.; Schmidt, M. Macromolecules 2006,
39, 8440-8450.
[170] Saariaho, M.; Subbotin, A.; Ikkala1, O.; Brinke, G. Macromol. Rapid Commun. 2000,
21, 110-115.
[171] Park, J. S.; Kataoka, K. Macromolecules 2006, 39, 6622-6630.
[172] Nie, Z.; Kumacheva, E. Nat. Mater. 2008, 4, 277-290.
[173] Bailey, R. C.; Hupp, J. T. Anal. Chem. 2003, 75, 2392-2398.
[174] Stoykovich, M. P.; Cao, H. B.; Yoshimoto, K.; Ocola, L. E.; Nealey, P. F. Adv. Mater.
2003, 15, 1180- 1184.
[175] Nath, N.; Chilkoti, A. Adv. Mater. 2002, 14, 1243-1247.
[176] Cowlard, F. C.; Lewis, J. C. J. Mater. Sci. 1967, 2, 507-512.
[177] http://en.wikipedia.org/wiki/Glassy_carbon
[178] Harris, P. J. F. Philos. Mag. 2004, 84, 3159-3167.
[179] Rice, R. J.; McCreery, R. L. Anal. Chem. 1989, 61, 1637-1641.

Chapter 7 References
155
[180] Mehandru, S. P.; Anderson, A. B.; Angus, J. C. J. Phys. Chem. 1992, 96, 10978-
10982.
[181] Ray III, K. G.; McCreery, R. L. J. Electroanal. Chem. 1999, 469, 150-158.
[182] Collier, W. G.; Tougas, T. P. Anal. Chem. 1987, 59, 396-399.
[183] Wright, J. S.; Carpenter, D. J.; McKay, D. J.; Ingold, K. U. J. Am. Chem. Soc. 1997,
119,4245-4252.
[184] Steenackers, M.; Küller, A.; Ballav, N.; Zharnikov, M.; Grunze, M.; Jordan R. Small
2007, 3, 1764-1773.
[185] Hoogenboom, R.; Wiesbrock, F.; Huang, H.; Leenen, M. A. M.; Thijs, H. M. L.; van
Nispen, S. F. G. M.; van der Loop, M.; Fustin, C.; Jonas, A. M.; Gohy, J.; Schubert,
U. S. Macromolecules 2006, 39, 4719-4725.
[186] Ulman, A. Chem. Rev. 1996, 96, 1533-1554.
[187] Balachander, N.; Sukenik, C.N. Langmuir 1990, 6 ,1621-1627.
[188] Naciri, J.; Fang, J. Y.; Moore, M.; Shenoy, D.; Dulcey, C. S.; Shashidhar, R. Chem.
Mater. 2000, 12, 3288-3295.
[189] Heiney, P. A.; Grüneberg, K.; Fang, J.; Dulcey, C.; Shashidhar, R. Langmuir 2000, 16,
2651-2657.
[190] Sugimura, H.; Hozumi, A.; Kameyama, T.; Takai, O. Surf. Interface Anal. 2002, 34,
550-554.
[191] Heise, A.; Menzel, H.; Yim, H.; Foster, M. D.; Wieringa, R. H.; Schouten, A. J.; Erb,
V.; Stamm, M. Langmuir 1997, 13, 723-728.
[192] Gribov, L. A.; Novakov, I. A.; Pavlyuchko, A. I.; Korolkov, V. V.; Orlinson, B. S. J.
Struct. Chem. 2004, 45, 951-959.
[193] Eck, W.; Gölzhäuser, A.; Zharnikov, M.; Stadler, V.; Geyer, W.; Grunze M.
PCT/DE00/03264, 1999.
[194] Gölzhäuser, A.; Eck, W.; Geyer, W.; Stadler, V.; Weimann, T.; Hinze, P.; Grunze, M.
Adv. Mater. 2001, 13, 806-809.
[195] Jia, X.; Jiang, X.; Liu, R.; Yin, J. Macromol. Chem. Phys.2009, 210, 1876-1882.
[196] Takei, Y. G.; Aoki, T.; Sanui, K.; Ogata, N.; Sakurai, Y.; Okano, T. Macromolecules
1994, 27, 6163-6166.
[197] Boxshall, K.; Wu, M.; Cui, Z.; Cui, Z.; Watts, J. F.; Baker, M. A. Surf. Interface
Anal. 2006, 38, 198-201.
[198] Chen, S.; Jiang, S. Adv. Mater. 2008, 20, 335-338.

References Chapter 7
156
[199] Johnston, E.; Ratner, B. D. Immobilized Biomolecules in Analysis, Oxford Press, 1998.
[200] Ratner, B. D.; Hoffman, A. S.; Schoen, F. J.; Lemons, J. E. Biomaterials science: an
introduction to materials in medicine,Academic Press, 2004.
[201] Harris, J. M. Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical
Applications, Plenum, 1992.
[202] Rampal, J. B. Microarrays, 2nd Edition, Humana press, Volume 1, 2007.
[203] Woodle, M. C.; Lasic, D. D. Biochim. Biophys. Acta 1992, 1113, 171-199.
[204] Konradi, R.; Pidhatika, B.; Mühlebach, A.; Textor, M. Langmuir 2008, 24, 613-616.
[205] Pidhatikaa, B.; Möllerb, J.; Vogelb, V.; Konradi, R. CHIMIA 2008, 62, 264-269.
[206] Lüdtke, K.; Jordan, R.; Hommes, P.; Nuyken, O.; Naumann, C. A. Macromol. Biosci.
2005, 5, 384-393.
[207] Kobayashi, S.; Uyama, H. J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 192-209.
[208] Hoogenboom, R.; Wiesbrock, F.; Leenen, M. A. M.; Thijs, H. M. L.; Huang, H. Y.;
Fustin, C. A.; Guillet, P.; Gohy, J. F.; Schubert, U. S. Macromolecules 2007, 40,
2837-2843.
[209] Fijten, M. W. M.; Kranenburg, J. M.; Thijs, H. M. L.; Paulus, R. M.; van Lankvelt, B.
M.; de Hullu, J.; Springintveld, M.; Thielen, D. J. G.; Tweedie, C. A.; Hoogenboom,
R.; Van Vliet, K. J.; Schubert, U. S. Macromolecules 2007, 40, 5879-5886.
[210] Fijten, M. W. M.; Haensch, C.; van Lankvelt, B. M.; Hoogenboom, R.; Schubert, U. S.
Macromol. Chem. Phys. 2008, 209, 1887-1895.
[211] Luxenhofer, R.; Bezen, M.; Jordan, R. Macromol. Rapid Commun. 2008, 29, 1509-
1513.
[212] Jordan, R.; Martin, K.; Räder, H. J.; Unger, K. K. Macromolecules 2001, 34, 8858-
8865.
[213] Herlotz, M.; Werner, C.; Pompe, T. Biomaterials 2009, 30, 395-402.
[214] Georg Albert PVD-Coatings, Heidelberg, Germany.
[215] Havard, J. M.; Yoshida, M.; Pasini, D.; Vladimirov, N.; Frechet, J. M. J.; Medeiros, D.
R.; Patterson, K.; Yamada, S.; Willson, C. G.; Byers, J. D. J. Polym. Sci. Part A:
Polym. Chem. 1999, 37, 1225-1236.
[216] Tomalia, D. A.; Thill, B. P.; Fazio, M. J. Polym. J. 1980, 12, 661-675.



![EINE POLY-KONTEXTURALE SYSTEMTHEORIE und deren … · Eberhard von Goldammer [*] EINE POLY-KONTEXTURALE SYSTEMTHEORIE UND DEREN KONSEQUENZEN I. AusgangsSituation Das Thema des Vortrags](https://img.pdfslide.org/doc/110x75/5d4d434388c99309068b8d0b/eine-poly-kontexturale-systemtheorie-und-deren-eberhard-von-goldammer-eine.jpg)







![CHIRAL OXAZOLINE AND BIS OXAZOLINE - uni-regensburg.de...C.2.10 IIILiquid-State-Analysis of Ruthenium(III) Complexes – [Ru (7a-9)3] 71 C.2.11 Formation of Zinc(II) Complexes using](https://img.pdfslide.org/doc/110x75/611fb9ae503029547a5f8447/chiral-oxazoline-and-bis-oxazoline-uni-c210-iiiliquid-state-analysis-of.jpg)

















