Embed Size (px)
Citation preview

Preisträger
81
Die chemische Physik der Oberflächen ist einwichtiges Teilgebiet der Oberflächenphysik. IhrZiel ist ein tiefgehendes Verständnis der Verän-derungen, die in Atomen und Molekülen durchderen Bindung an eine Oberfläche verursachtwerden. Diese Veränderungen sind die Basisvon so wichtigen Effekten wie der heterogenenKatalyse, Schichtwachstum, Reibung undSchmierung, Halbleiterstrukturierung und vie-len anderen. In diesem Artikel beschreibe ichzwei neue Experimente, in denen wir die elek-tronische Anregung von Molekülen auf Ober-flächen dazu ausnutzen, die chemischen Bin-dungskräfte zu studieren und zu manipulieren.Durch Anregung und Messung mit schmalban-diger Synchrotronstrahlung können wir nunchemische Reaktionen gezielt steuern.
D ie vergangenen 35 Jahre haben eine rasante Ent-wicklung der Oberflächenphysik gebracht undsie von der frühen Physik der Dreckeffekte zu
einem reichen und reifen Teil der Physik gemacht. EineVielzahl von oberflächenspezifischen Methoden wurdeentwickelt und wird routinemäßig angewandt, was zueiner großen Fülle von Wissen über Oberflächensys-teme geführt hat [1]. In der chemischen Physik derOberflächen, einem Teilgebiet der Oberflächenphysik,ist es heute möglich, Adsorbat- und Koadsorbatsystemewohldefiniert in ihrer Zusammensetzung herzustellenund ihre exakte Geometrie, Bindungsverhältnisse,elektronische und vibratorische Zustände (einschließ-lich niederdimensionaler Bandstrukturen), ihre elek-tronischen Anregungen und deren Dynamik sowie dieDynamik und Kinetik von Oberflächen-Elementarreak-tionen (wie Haften und thermische Desorption, Diffu-sion auf der Oberfläche, Zerfall und Bildung vonOberflächenmolekülen) detailliert zu untersuchen. DerPhysiker wird bei solchen Untersuchungen immer dieprinzipiellen Elementarschritte und -mechanismen imAuge haben und verstehen wollen. Wegen der starkenEinflüsse der Präparation auf die Eigenschaften vonAdsorptionsschichten ist es wichtig, dass solche auf dasVerständnis grundlegender Mechanismen zielendenUntersuchungen stets an wohldefinierten Systemendurchgeführt werden, was am besten erreichbar ist,wenn prinzipielle, systemorientierte und methodischeArbeiten parallel betrieben werden. Dies war dieGrundidee der Forschungsanstrengungen meiner
Münchner Arbeitsgruppe in den vergangenen dreiDekaden. Unsere Arbeiten zur Geometrie von Adsorp-tionsschichten [2], zu ihren Schwingungseigenschaften [3, 4], ihrer elektronischen Spektroskopie im Valenz-[3, 5] und Rumpfbereich [6, 7], zur Dynamik elektroni-scher Anregungen [8, 9] sowie zu durch sie induziertenReaktionen [10] und zu Dynamik und Kinetik vonAdsorption [11, 12], Desorption [12, 13] und Reaktion[14] sind aufeinander bezogen und stützen sich inihren Befunden gegenseitig. Die enge Kooperation mittheoretischen Kollegen war und ist dabei entscheidendfür ein tiefergehendes Verständnis [4, 5, 11, 12].
Es ist unmöglich, über dieses breite Feld hier einenÜberblick zu geben; ich werde daher in diesem Artikeleine wichtige grundlegende Fragestellung herausgrei-fen. Es handelt sich um die quantitative Charakterisie-rung der Kopplung von Molekülen an Oberflächen, diezur Oberflächenbindung, zur Änderung der Reaktivitätadsorbierter Moleküle (Basis der heterogenen Kataly-se), zu starker Modifikation ihrer Photochemie und zuanderen interessanten Effekten führt. Zu diesem Ge-biet sind mir schon frühzeitig fruchtbare Beiträge ge-lungen [15]. Ich beschreibe zwei neue Experimente,von denen eines diese Kopplung anhand der Messungder Zeitkonstanten des Transfers elektronischer Anre-gungen quantitativ erfassbar macht und das andereeine Konsequenz dieser Vorgänge zur Selektion einesvon zwei konkurrierenden Reaktionswegen verwendet.Beide Experimente arbeiten mit Anregung und Zerfallvon Rumpfelektronen, wobei die Verwendung vonschmalbandiger Anregung und Messung – wie siedurch neue Quellen für Synchrotronstrahlung (SR) ho-her Qualität möglich geworden sind – entscheidend ist.
Unsere anderen Arbeiten auf dem Gebiet der Che-mischen Physik der Oberflächen sind auch in diesemZusammenhang wichtig, da sie die genaue Definitionder Adsorbatschichten ermöglichen, ohne die eine de-taillierte Untersuchung so subtiler physikalischer Ef-fekte, wie sie im Folgenden beschrieben werden, nichtsinnvoll wäre.
Ladungstransferzeiten zwischen Adsorbat und SubstratIn einem isolierten Molekül kann eine elektronische
Anregung zum Bruch von Bindungen führen. Dies wirddann der Fall sein, wenn der erzeugte Molekülzustandrepulsiv für eine oder mehrere Bindungen ist, undwenn die Bindung schneller bricht als eine eventuelle
Robert-Wichard-Pohl-Preis
Ultraschneller Ladungstransfer und lokalisierter Bindungsbruch
Die Kopplung von Molekülen an Oberflächen lässt sich durch die gezielte Anregungder Elektronen im Molekül charakterisieren und beeinflussen
Dietrich Menzel
Prof. Dr. DietrichMenzel, Physik-De-partment E20, Tech-nische UniversitätMünchen, D-85747Garching b. Mün-chen – Festvortraganlässlich der Ver-leihung des Robert-Wichard-Pohl-Prei-ses auf der 64. Phy-sikertagung inDresden
Physikalische Blätter56 (2000) Nr. 7/80031-9279/00/0707-81$17.50+50/0© WILEY-VCH Verlag GmbH,D-69451 Weinheim, 2000

Physikalische Blätter56 (2000) Nr. 7/882
Preisträger
Umverteilung der Energie auf andere Freiheitsgrade.Adsorbiert man das Molekül auf einer Oberfläche, soführt die Kopplung von Adsorbat und Substrat zu einerneuen Bindung, der Adsorptionsbindung, und zurMöglichkeit neuer, sehr schneller Umverteilungspro-zesse. Dies führt zum Beispiel dazu, dass durch elek-tronische Anregungen induzierte Dissoziationsprozessein Adsorbaten auf Metalloberflächen häufig stark undselektiv unterdrückt sind. Die Erforschung solcher Mo-
difikationen bildet ein eigenes, meist „DIET“ (Desorp-tion Induced by Electronic Transitions; Desorption istder zur Dissoziation analoge Oberflächenprozess) ge-nanntes Gebiet [10, 16], dessen Etablierung in nichtunwesentlichem Umfang durch meine frühen Arbeiten[15] beeinflusst wurde und das seit über 30 Jahrenwichtige Beiträge zum Verständnis von Oberflächen-prozessen geliefert hat. Als bis heute konzeptionellsehr fruchtbar erwies sich ein einfaches Zweistufenmo-dell von Anregung und deren Evolution [15], das alswesentlichen Parameter die Effektivität des Energie-und Ladungsaustauschs zwischen Adsorbat und Ober-fläche enthält. Die experimentellen Ergebnisse zuDIET [10, 16] lassen den Schluss zu, dass solche Trans-ferprozesse an Metall- und Halbleiteroberflächen sehrschnell sein müssen, d. h. im Bereich von wenigenFemtosekunden oder sogar schneller stattfinden. DieZeitkonstanten so schneller Prozesse direkt zu messenist erst in jüngster Zeit möglich geworden. Wie so oftzeigt sich hier, dass neue Methodik das Angehen langbestehender Fragen ermöglicht.
Im einfachsten Fall handelt es sich bei der zu unter-suchenden elektronischen Anregung um eine Einteil-chenanregung zu einem Ein-Loch-Ein-Elektron-Zu-stand (1h-1e-Zustand). Das Elektron wird dadurch inein im Grundzustand unbesetztes Niveau des Adsor-bats angeregt; die für seinen Transfer in das Substratnötige Zeit dient zur Charakterisierung der Kopplung.Für die genaue Definition dieser zeitlichen Evolutionist es günstig, die primäre Anregung dem System mög-lichst lokalisiert aufzuprägen. Dies lässt sich durch dieAnregung eines Rumpfelektrons in ein gebundenes, imGrundzustand unbesetztes Orbital des Systems erzie-len; d. h. das durch die Primäranregung erzeugte Lochbefindet sich in einem Rumpfniveau eines Adsorbat-atoms. Mit weichen Röntgenquanten können bestimm-te Atome durch Energieselektion und Dipolauswahl-regeln (adsorbierte Moleküle sind i. A. definiert orien-
tiert) ganz gezielt angeregt werden. Weitere Vorteilevon Rumpfanregungen sind ihre gut bekannten Lebens-dauern im Bereich von einigen Femtosekunden für diehier interessierenden Atome und Rumpfniveaus, sowiedie Tatsache, dass sich der Zustand des rumpfangereg-ten Atoms sehr gut durch ein Atom mit um 1 erhöhterKernladung („equivalent core“ oder „Z+1“ Näherung)beschreiben lässt. So ist ein rumpfangeregtes Argon-atom einem Kaliumatom äquivalent, und ein rumpfan-geregtes N2 einem NO-Molekül. Diese Näherung er-leichtert das qualitative Verständnis, ist aber zu ge-nauerer Interpretation nicht nötig.
Wir stellen uns also die Frage, wie lange das durchRumpfanregung eines Adsorbatatoms erzeugte Elek-tron in einem im Grundzustand leeren Niveau – einemResonanzniveau - auf diesem Atom lokalisiert bleibt.Die durch das Wiederauffüllen des Rumpflochs erzeug-ten Autoionisationsspektren (auch resonante Auger-Spektren oder allgemein Zerfallsspektren genannt) lie-fern die Antwort (Abb. 1). Wenn nämlich das angeregteElektron zum Zeitpunkt des Auffüllens des Rumpflochsnoch auf dem gleichen Atom lokalisiert ist, so werdendie kinetischen Energien der zur Energieerhaltung aus-gestoßenen Elektronen durch die Coulomb-Wechsel-wirkung des angeregten Elektrons mit den Valenzelek-tronen des Systems anders (nämlich um die „Specta-torshift“ höher) sein, als wenn zu diesem Zeitpunktdas angeregte Elektron bereits in das Substrat gesprun-gen ist (daneben können auch Zerfallsprozesse unterBeteiligung des angeregten Elektrons ablaufen; sieführen zu „Participant“ Linien, die energetisch norma-len Photoemissionslinien entsprechen). Ist das ange-regte Elektron zum Zeitpunkt des Rumpfloch-Zerfallsbereits ins Substrat delokalisiert, so macht sich dasdurch energetisch verschobene, „normale“ Auger-Lini-en bemerkbar.
Wenn die Zeitskalen von Rumpfloch-Füllung undElektronentransfer vergleichbar sind, werden also zweiArten von Spektrallinien entstehen, die den beiden Si-tuationen – Rumpfloch-Zerfall vor oder nach Transferdes lokal angeregten Elektrons – entsprechen, die alsoAutoionisations- bzw. normale Auger-Spektren darstel-len. Da die entsprechenden Endzustände durch zweiverschiedene, miteinander konkurrierende Ratenpro-zesse erreicht werden (Ladungstransfer bzw. Rumpf-loch-Füllung), ist das Intensitätsverhältnis der beidenSpektrenarten nach einem einfachen Ratenmodelldirekt mit dem Verhältnis der beiden Zeitskalen gekop-pelt. Auf dieser Basis wurde de facto schon vor fasteiner Dekade (durch Vergleich von resonanten undnormalen Auger-Spektren von Adsorbaten) geschlos-sen, dass bei chemisch an die Oberfläche gebundenen(„chemisorbierten“) Molekülen der Ladungstransferwesentlich schneller stattfindet als der Rumpfloch-Zer-fall [17], während bei lediglich van-der-Waals-gebunde-nen („physisorbierten“) Teilchen die Zeitskalen ver-gleichbar sind [18]. Die komplexen, sich überlappen-den Spektren machten genauere Aussagen schwer, imersten Fall sogar unmöglich. Die Verwendung vonschmalbandiger Strahlung hoher Intensität, wie sie beiso genannten SR-Quellen der dritten Generation ver-fügbar ist, hat eine neue Messmethode ermöglicht, dieeine wesentliche Verbesserung brachte und recht ge-naue, systematische Messungen möglich gemacht hat.
Hierzu wird die resonante Rumpfanregung unter sogenannten Resonant-Auger-Raman-Bedingungen vor-genommen. Dies heißt, dass die Bandbreite der Strah-
Abb. 1:a) Ein resonant und lokalisiert von einemAdsorbat-Rumpfniveau in ein unbesetz-tes gebundenes Valenzniveau des Adsor-bats angeregtes Elektron kann in dasSubstrat transferiert werden (zur Verein-fachung ist Letzteres nicht gezeichnet).Der Zerfall des Rumpflochs (b, c) findetauf vergleichbarer Zeitskala statt. DieEnergien der Zerfallselektronenspektrenhängen davon ab, ob der Rumpfloch-Zer-fall vor (b) oder nach (c) dem Ladungs-transfer stattfindet. Die (bekannte)Rumpfloch-Lebensdauer kann daher alsUhr zur Messung der Ladungstransferzeitverwendet werden.
Physikalische Blätter56 (2000) Nr. 7/8
Preisträger
83
lung deutlich unter der intrinsischen Lebensdauerbreiteder Rumpfanregung liegt und die Auger-Spektren eben-so gut aufgelöst gemessen werden. In einem isoliertenMolekül erfordert dann die Energieerhaltung, dass dieZerfallsspektren ebenso schmal sind wie die Primäran-regung („line narrowing“) und dass ihre kinetischeEnergie sich bei variierter (insbesondere durch die Re-sonanzlinie hindurch gestimmter) Anregungsenergielinear mit dieser verändert („linear dispersion“). Sieverhalten sich also wie Photoemissionslinien, deren„Bindungsenergie“ von der Anregungsenergie unabhän-gig ist, und nicht wie Auger-Linien, die unabhängigvon der Anregungsenergie konstante kinetische Ener-gie besitzen – obwohl der Prozess ein Auger-Prozess istund durch das entsprechende Coulomb-Matrixelementbeschrieben wird. In der Atom- und Molekülspektro-skopie lässt sich damit die Auflösung wesentlich ver-bessern [19]. Um eine anschauliche Vorstellung vonden zugrunde liegenden Vorgängen zu erreichen, istein explizit zeitabhängiges Bild hilfreich [20]. Wegender kleinen Anregungs-Bandbreite im Verhältnis zurLebensdauer des Zwischenzustands dauert die Anre-gung wesentlich länger als der Zerfall, sodass der ge-samte Anregungs-Zerfalls-Prozess als ein kohärenter,einstufiger Prozess beschrieben werden muss. Für iso-lierte Moleküle hat dies eine Reihe von sehr interes-santen Folgen, wenn die Zeitskalen des Rumpfloch-Zerfalls und der Bewegungen der Atome im veränder-ten Potential vergleichbar werden. Hier können dazunur die Stichpunkte „lifetime-vibrational interference“und „vibrational collapse“ bei gebundenen rumpfange-regten Zwischenzuständen sowie „atomic vs. molecularlines“ und „Doppler effect“ bei dissoziativen Zwi-schenzuständen genannt werden [19 – 20].
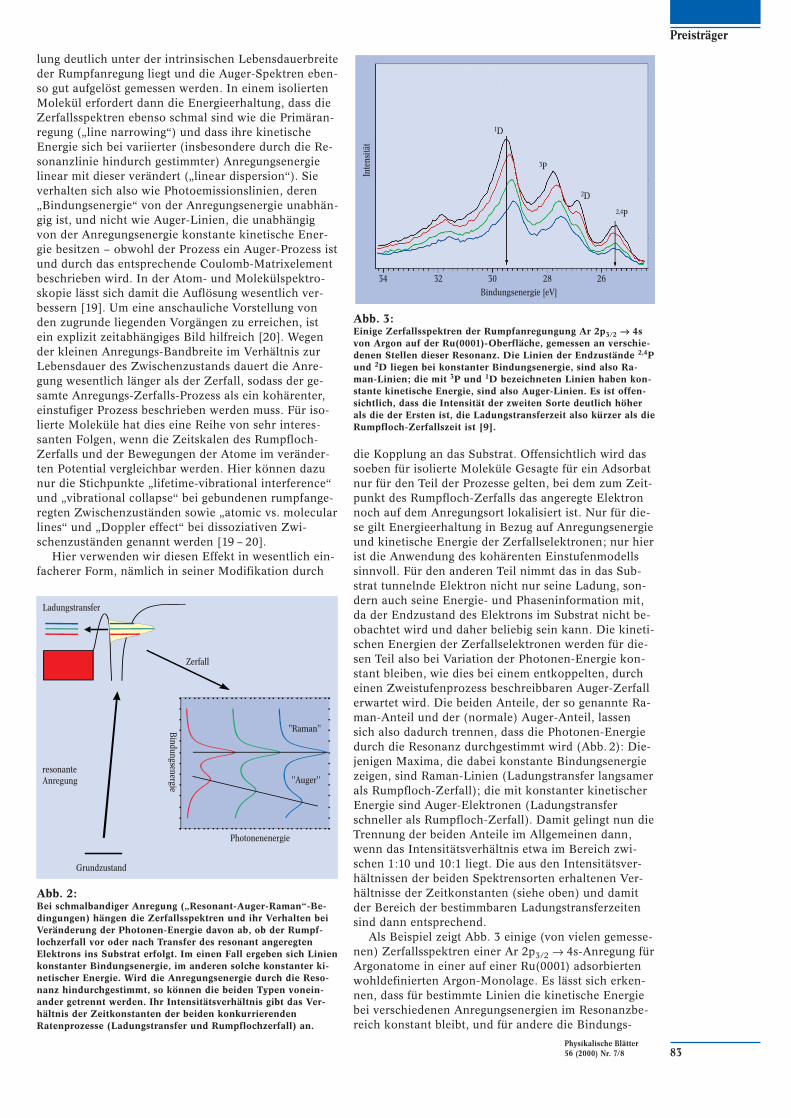
Hier verwenden wir diesen Effekt in wesentlich ein-facherer Form, nämlich in seiner Modifikation durch
die Kopplung an das Substrat. Offensichtlich wird dassoeben für isolierte Moleküle Gesagte für ein Adsorbatnur für den Teil der Prozesse gelten, bei dem zum Zeit-punkt des Rumpfloch-Zerfalls das angeregte Elektronnoch auf dem Anregungsort lokalisiert ist. Nur für die-se gilt Energieerhaltung in Bezug auf Anregungsenergieund kinetische Energie der Zerfallselektronen; nur hierist die Anwendung des kohärenten Einstufenmodellssinnvoll. Für den anderen Teil nimmt das in das Sub-strat tunnelnde Elektron nicht nur seine Ladung, son-dern auch seine Energie- und Phaseninformation mit,da der Endzustand des Elektrons im Substrat nicht be-obachtet wird und daher beliebig sein kann. Die kineti-schen Energien der Zerfallselektronen werden für die-sen Teil also bei Variation der Photonen-Energie kon-stant bleiben, wie dies bei einem entkoppelten, durcheinen Zweistufenprozess beschreibbaren Auger-Zerfallerwartet wird. Die beiden Anteile, der so genannte Ra-man-Anteil und der (normale) Auger-Anteil, lassensich also dadurch trennen, dass die Photonen-Energiedurch die Resonanz durchgestimmt wird (Abb. 2): Die-jenigen Maxima, die dabei konstante Bindungsenergiezeigen, sind Raman-Linien (Ladungstransfer langsamerals Rumpfloch-Zerfall); die mit konstanter kinetischerEnergie sind Auger-Elektronen (Ladungstransferschneller als Rumpfloch-Zerfall). Damit gelingt nun dieTrennung der beiden Anteile im Allgemeinen dann,wenn das Intensitätsverhältnis etwa im Bereich zwi-schen 1:10 und 10:1 liegt. Die aus den Intensitätsver-hältnissen der beiden Spektrensorten erhaltenen Ver-hältnisse der Zeitkonstanten (siehe oben) und damitder Bereich der bestimmbaren Ladungstransferzeitensind dann entsprechend.
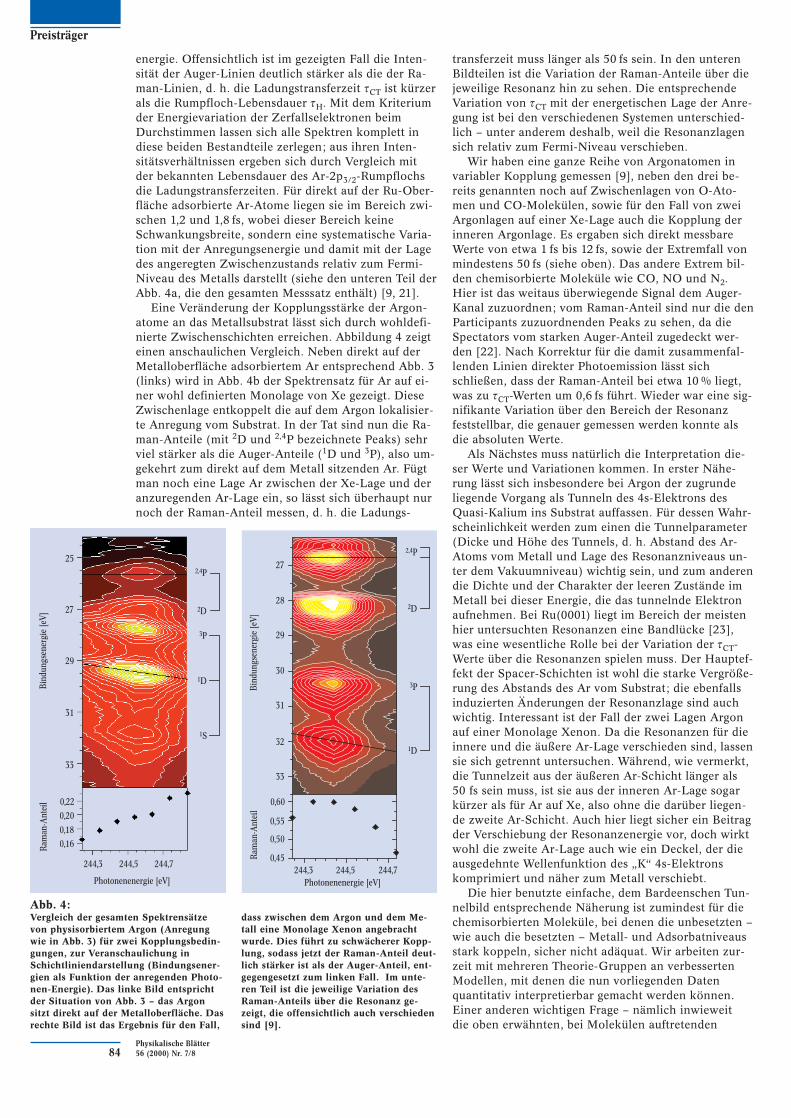
Als Beispiel zeigt Abb. 3 einige (von vielen gemesse-nen) Zerfallsspektren einer Ar 2p3/2 → 4s-Anregung fürArgonatome in einer auf einer Ru(0001) adsorbiertenwohldefinierten Argon-Monolage. Es lässt sich erken-nen, dass für bestimmte Linien die kinetische Energiebei verschiedenen Anregungsenergien im Resonanzbe-reich konstant bleibt, und für andere die Bindungs-
Abb. 2:Bei schmalbandiger Anregung („Resonant-Auger-Raman“-Be-dingungen) hängen die Zerfallsspektren und ihr Verhalten beiVeränderung der Photonen-Energie davon ab, ob der Rumpf-lochzerfall vor oder nach Transfer des resonant angeregtenElektrons ins Substrat erfolgt. Im einen Fall ergeben sich Linienkonstanter Bindungsenergie, im anderen solche konstanter ki-netischer Energie. Wird die Anregungsenergie durch die Reso-nanz hindurchgestimmt, so können die beiden Typen vonein-ander getrennt werden. Ihr Intensitätsverhältnis gibt das Ver-hältnis der Zeitkonstanten der beiden konkurrierendenRatenprozesse (Ladungstransfer und Rumpflochzerfall) an.
Abb. 3:Einige Zerfallsspektren der Rumpfanregungung Ar 2p3/2 →→ 4svon Argon auf der Ru(0001)-Oberfläche, gemessen an verschie-denen Stellen dieser Resonanz. Die Linien der Endzustände 2,4Pund 2D liegen bei konstanter Bindungsenergie, sind also Ra-man-Linien; die mit 3P und 1D bezeichneten Linien haben kon-stante kinetische Energie, sind also Auger-Linien. Es ist offen-sichtlich, dass die Intensität der zweiten Sorte deutlich höherals die der Ersten ist, die Ladungstransferzeit also kürzer als dieRumpfloch-Zerfallszeit ist [9].
Physikalische Blätter56 (2000) Nr. 7/884
Preisträger
energie. Offensichtlich ist im gezeigten Fall die Inten-sität der Auger-Linien deutlich stärker als die der Ra-man-Linien, d. h. die Ladungstransferzeit tCT ist kürzerals die Rumpfloch-Lebensdauer tH. Mit dem Kriteriumder Energievariation der Zerfallselektronen beimDurchstimmen lassen sich alle Spektren komplett indiese beiden Bestandteile zerlegen; aus ihren Inten-sitätsverhältnissen ergeben sich durch Vergleich mitder bekannten Lebensdauer des Ar-2p3/2-Rumpflochsdie Ladungstransferzeiten. Für direkt auf der Ru-Ober-fläche adsorbierte Ar-Atome liegen sie im Bereich zwi-schen 1,2 und 1,8 fs, wobei dieser Bereich keineSchwankungsbreite, sondern eine systematische Varia-tion mit der Anregungsenergie und damit mit der Lagedes angeregten Zwischenzustands relativ zum Fermi-Niveau des Metalls darstellt (siehe den unteren Teil derAbb. 4a, die den gesamten Messsatz enthält) [9, 21].
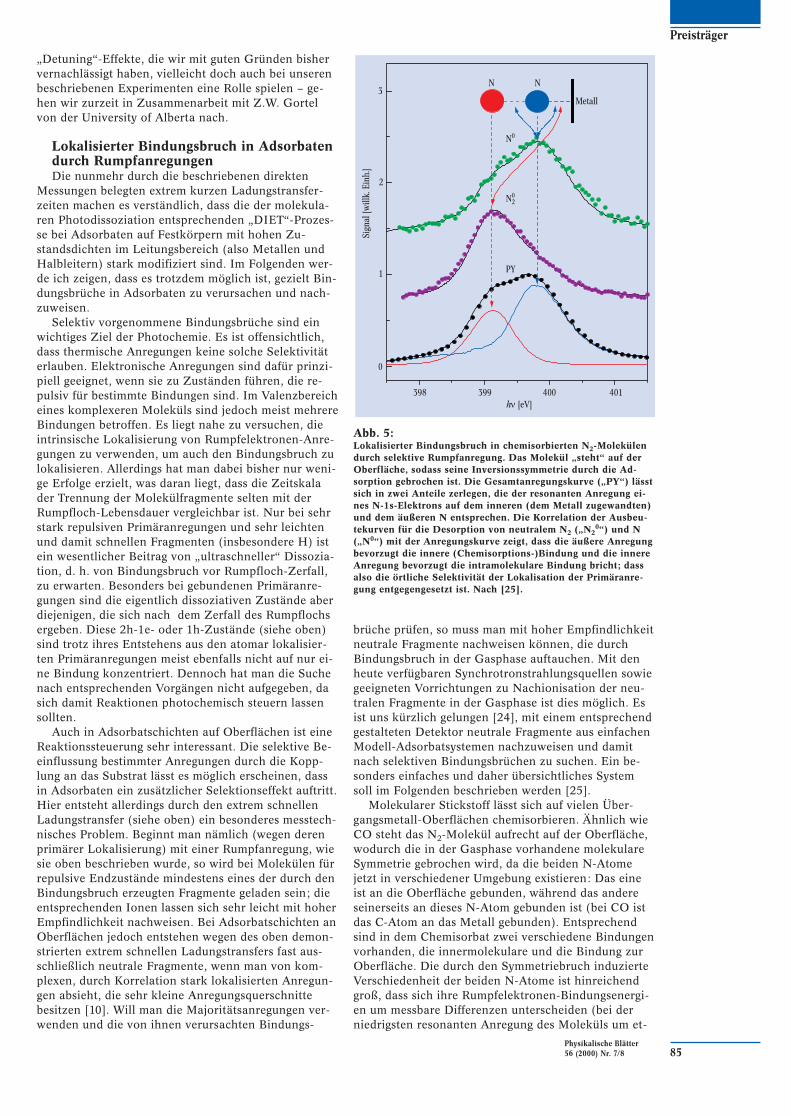
Eine Veränderung der Kopplungsstärke der Argon-atome an das Metallsubstrat lässt sich durch wohldefi-nierte Zwischenschichten erreichen. Abbildung 4 zeigteinen anschaulichen Vergleich. Neben direkt auf derMetalloberfläche adsorbiertem Ar entsprechend Abb. 3(links) wird in Abb. 4b der Spektrensatz für Ar auf ei-ner wohl definierten Monolage von Xe gezeigt. DieseZwischenlage entkoppelt die auf dem Argon lokalisier-te Anregung vom Substrat. In der Tat sind nun die Ra-man-Anteile (mit 2D und 2,4P bezeichnete Peaks) sehrviel stärker als die Auger-Anteile (1D und 3P), also um-gekehrt zum direkt auf dem Metall sitzenden Ar. Fügtman noch eine Lage Ar zwischen der Xe-Lage und deranzuregenden Ar-Lage ein, so lässt sich überhaupt nurnoch der Raman-Anteil messen, d. h. die Ladungs-
transferzeit muss länger als 50 fs sein. In den unterenBildteilen ist die Variation der Raman-Anteile über diejeweilige Resonanz hin zu sehen. Die entsprechendeVariation von tCT mit der energetischen Lage der Anre-gung ist bei den verschiedenen Systemen unterschied-lich – unter anderem deshalb, weil die Resonanzlagensich relativ zum Fermi-Niveau verschieben.
Wir haben eine ganze Reihe von Argonatomen invariabler Kopplung gemessen [9], neben den drei be-reits genannten noch auf Zwischenlagen von O-Ato-men und CO-Molekülen, sowie für den Fall von zweiArgonlagen auf einer Xe-Lage auch die Kopplung derinneren Argonlage. Es ergaben sich direkt messbareWerte von etwa 1 fs bis 12 fs, sowie der Extremfall vonmindestens 50 fs (siehe oben). Das andere Extrem bil-den chemisorbierte Moleküle wie CO, NO und N2.Hier ist das weitaus überwiegende Signal dem Auger-Kanal zuzuordnen; vom Raman-Anteil sind nur die denParticipants zuzuordnenden Peaks zu sehen, da dieSpectators vom starken Auger-Anteil zugedeckt wer-den [22]. Nach Korrektur für die damit zusammenfal-lenden Linien direkter Photoemission lässt sichschließen, dass der Raman-Anteil bei etwa 10 % liegt,was zu tCT-Werten um 0,6 fs führt. Wieder war eine sig-nifikante Variation über den Bereich der Resonanzfeststellbar, die genauer gemessen werden konnte alsdie absoluten Werte.
Als Nächstes muss natürlich die Interpretation die-ser Werte und Variationen kommen. In erster Nähe-rung lässt sich insbesondere bei Argon der zugrundeliegende Vorgang als Tunneln des 4s-Elektrons desQuasi-Kalium ins Substrat auffassen. Für dessen Wahr-scheinlichkeit werden zum einen die Tunnelparameter(Dicke und Höhe des Tunnels, d. h. Abstand des Ar-Atoms vom Metall und Lage des Resonanzniveaus un-ter dem Vakuumniveau) wichtig sein, und zum anderendie Dichte und der Charakter der leeren Zustände imMetall bei dieser Energie, die das tunnelnde Elektronaufnehmen. Bei Ru(0001) liegt im Bereich der meistenhier untersuchten Resonanzen eine Bandlücke [23],was eine wesentliche Rolle bei der Variation der tCT-Werte über die Resonanzen spielen muss. Der Hauptef-fekt der Spacer-Schichten ist wohl die starke Vergröße-rung des Abstands des Ar vom Substrat; die ebenfallsinduzierten Änderungen der Resonanzlage sind auchwichtig. Interessant ist der Fall der zwei Lagen Argonauf einer Monolage Xenon. Da die Resonanzen für dieinnere und die äußere Ar-Lage verschieden sind, lassensie sich getrennt untersuchen. Während, wie vermerkt,die Tunnelzeit aus der äußeren Ar-Schicht länger als50 fs sein muss, ist sie aus der inneren Ar-Lage sogarkürzer als für Ar auf Xe, also ohne die darüber liegen-de zweite Ar-Schicht. Auch hier liegt sicher ein Beitragder Verschiebung der Resonanzenergie vor, doch wirktwohl die zweite Ar-Lage auch wie ein Deckel, der dieausgedehnte Wellenfunktion des „K“ 4s-Elektronskomprimiert und näher zum Metall verschiebt.
Die hier benutzte einfache, dem Bardeenschen Tun-nelbild entsprechende Näherung ist zumindest für diechemisorbierten Moleküle, bei denen die unbesetzten –wie auch die besetzten – Metall- und Adsorbatniveausstark koppeln, sicher nicht adäquat. Wir arbeiten zur-zeit mit mehreren Theorie-Gruppen an verbessertenModellen, mit denen die nun vorliegenden Datenquantitativ interpretierbar gemacht werden können.Einer anderen wichtigen Frage – nämlich inwieweit die oben erwähnten, bei Molekülen auftretenden
Abb. 4:Vergleich der gesamten Spektrensätzevon physisorbiertem Argon (Anregungwie in Abb. 3) für zwei Kopplungsbedin-gungen, zur Veranschaulichung inSchichtliniendarstellung (Bindungsener-gien als Funktion der anregenden Photo-nen-Energie). Das linke Bild entsprichtder Situation von Abb. 3 – das Argonsitzt direkt auf der Metalloberfläche. Dasrechte Bild ist das Ergebnis für den Fall,
dass zwischen dem Argon und dem Me-tall eine Monolage Xenon angebrachtwurde. Dies führt zu schwächerer Kopp-lung, sodass jetzt der Raman-Anteil deut-lich stärker ist als der Auger-Anteil, ent-gegengesetzt zum linken Fall. Im unte-ren Teil ist die jeweilige Variation desRaman-Anteils über die Resonanz ge-zeigt, die offensichtlich auch verschiedensind [9].
Physikalische Blätter56 (2000) Nr. 7/8
Preisträger
85
„Detuning“-Effekte, die wir mit guten Gründen bishervernachlässigt haben, vielleicht doch auch bei unserenbeschriebenen Experimenten eine Rolle spielen – ge-hen wir zurzeit in Zusammenarbeit mit Z.W. Gortelvon der University of Alberta nach.
Lokalisierter Bindungsbruch in Adsorbatendurch RumpfanregungenDie nunmehr durch die beschriebenen direkten
Messungen belegten extrem kurzen Ladungstransfer-zeiten machen es verständlich, dass die der molekula-ren Photodissoziation entsprechenden „DIET“-Prozes-se bei Adsorbaten auf Festkörpern mit hohen Zu-standsdichten im Leitungsbereich (also Metallen undHalbleitern) stark modifiziert sind. Im Folgenden wer-de ich zeigen, dass es trotzdem möglich ist, gezielt Bin-dungsbrüche in Adsorbaten zu verursachen und nach-zuweisen.
Selektiv vorgenommene Bindungsbrüche sind einwichtiges Ziel der Photochemie. Es ist offensichtlich,dass thermische Anregungen keine solche Selektivitäterlauben. Elektronische Anregungen sind dafür prinzi-piell geeignet, wenn sie zu Zuständen führen, die re-pulsiv für bestimmte Bindungen sind. Im Valenzbereicheines komplexeren Moleküls sind jedoch meist mehrereBindungen betroffen. Es liegt nahe zu versuchen, dieintrinsische Lokalisierung von Rumpfelektronen-Anre-gungen zu verwenden, um auch den Bindungsbruch zulokalisieren. Allerdings hat man dabei bisher nur weni-ge Erfolge erzielt, was daran liegt, dass die Zeitskalader Trennung der Molekülfragmente selten mit derRumpfloch-Lebensdauer vergleichbar ist. Nur bei sehrstark repulsiven Primäranregungen und sehr leichtenund damit schnellen Fragmenten (insbesondere H) istein wesentlicher Beitrag von „ultraschneller“ Dissozia-tion, d. h. von Bindungsbruch vor Rumpfloch-Zerfall,zu erwarten. Besonders bei gebundenen Primäranre-gungen sind die eigentlich dissoziativen Zustände aberdiejenigen, die sich nach dem Zerfall des Rumpflochsergeben. Diese 2h-1e- oder 1h-Zustände (siehe oben)sind trotz ihres Entstehens aus den atomar lokalisier-ten Primäranregungen meist ebenfalls nicht auf nur ei-ne Bindung konzentriert. Dennoch hat man die Suchenach entsprechenden Vorgängen nicht aufgegeben, dasich damit Reaktionen photochemisch steuern lassensollten.
Auch in Adsorbatschichten auf Oberflächen ist eineReaktionssteuerung sehr interessant. Die selektive Be-einflussung bestimmter Anregungen durch die Kopp-lung an das Substrat lässt es möglich erscheinen, dassin Adsorbaten ein zusätzlicher Selektionseffekt auftritt.Hier entsteht allerdings durch den extrem schnellenLadungstransfer (siehe oben) ein besonderes messtech-nisches Problem. Beginnt man nämlich (wegen derenprimärer Lokalisierung) mit einer Rumpfanregung, wiesie oben beschrieben wurde, so wird bei Molekülen fürrepulsive Endzustände mindestens eines der durch denBindungsbruch erzeugten Fragmente geladen sein; dieentsprechenden Ionen lassen sich sehr leicht mit hoherEmpfindlichkeit nachweisen. Bei Adsorbatschichten anOberflächen jedoch entstehen wegen des oben demon-strierten extrem schnellen Ladungstransfers fast aus-schließlich neutrale Fragmente, wenn man von kom-plexen, durch Korrelation stark lokalisierten Anregun-gen absieht, die sehr kleine Anregungsquerschnittebesitzen [10]. Will man die Majoritätsanregungen ver-wenden und die von ihnen verursachten Bindungs-
brüche prüfen, so muss man mit hoher Empfindlichkeitneutrale Fragmente nachweisen können, die durchBindungsbruch in der Gasphase auftauchen. Mit denheute verfügbaren Synchrotronstrahlungsquellen sowiegeeigneten Vorrichtungen zu Nachionisation der neu-tralen Fragmente in der Gasphase ist dies möglich. Esist uns kürzlich gelungen [24], mit einem entsprechendgestalteten Detektor neutrale Fragmente aus einfachenModell-Adsorbatsystemen nachzuweisen und damitnach selektiven Bindungsbrüchen zu suchen. Ein be-sonders einfaches und daher übersichtliches Systemsoll im Folgenden beschrieben werden [25].
Molekularer Stickstoff lässt sich auf vielen Über-gangsmetall-Oberflächen chemisorbieren. Ähnlich wieCO steht das N2-Molekül aufrecht auf der Oberfläche,wodurch die in der Gasphase vorhandene molekulareSymmetrie gebrochen wird, da die beiden N-Atomejetzt in verschiedener Umgebung existieren: Das eineist an die Oberfläche gebunden, während das andereseinerseits an dieses N-Atom gebunden ist (bei CO istdas C-Atom an das Metall gebunden). Entsprechendsind in dem Chemisorbat zwei verschiedene Bindungenvorhanden, die innermolekulare und die Bindung zurOberfläche. Die durch den Symmetriebruch induzierteVerschiedenheit der beiden N-Atome ist hinreichendgroß, dass sich ihre Rumpfelektronen-Bindungsenergi-en um messbare Differenzen unterscheiden (bei derniedrigsten resonanten Anregung des Moleküls um et-
Abb. 5:Lokalisierter Bindungsbruch in chemisorbierten N2-Molekülendurch selektive Rumpfanregung. Das Molekül „steht“ auf derOberfläche, sodass seine Inversionssymmetrie durch die Ad-sorption gebrochen ist. Die Gesamtanregungskurve („PY“) lässtsich in zwei Anteile zerlegen, die der resonanten Anregung ei-nes N-1s-Elektrons auf dem inneren (dem Metall zugewandten)und dem äußeren N entsprechen. Die Korrelation der Ausbeu-tekurven für die Desorption von neutralem N2 („N2
0“) und N(„N0“) mit der Anregungskurve zeigt, dass die äußere Anregungbevorzugt die innere (Chemisorptions-)Bindung und die innereAnregung bevorzugt die intramolekulare Bindung bricht; dassalso die örtliche Selektivität der Lokalisation der Primäranre-gung entgegengesetzt ist. Nach [25].

Physikalische Blätter56 (2000) Nr. 7/886
Preisträger
wa 0,7 eV; für die in XPS beobachtete Ionisation insKontinuum sogar um 1,5 eV). Es ist bekannt, dass dieSignale mit niedrigerer Bindungs- bzw. Anregungsener-gie dem „inneren“, dem Metall benachbarten N-Atomentsprechen [26].
Damit lässt sich durch hinreichend schmalbandigeRöntgenstrahlung die (Rumpf-)Anregung des äußerenund des inneren N-Atoms weitgehend trennen. Beob-achtet man nun die durch Photodesorption in der Gas-phase erscheinenden neutralen N-Atome und N2-Mo-leküle, so lässt sich feststellen, ob es einen selektivenBindungsbruch gibt und in welchem Umfang welcheAnregung vorzugsweise die „äußere“ – zu N-Desorpti-on führende – oder die „innere“ – zu N2-Desorptionführende Bindung bricht. Abbildung 5 zeigt, dass in derTat eine erhebliche – wenn auch nicht völlige – Selekti-vität des Bindungsbruchs vorliegt: Das N-Signal ist se-lektiv verstärkt bei Anregung des inneren N, und dasN2-Signal bei Anregung des äußeren N. Interessanter-weise ist dies umgekehrt als nach einem naiven Lokali-sierungsmodell erwartet. Denn wenn die Lokalisierungder Primäranregung die Selektivität des Bindungs-bruchs direkt verursachen würde, also jeweils dienächstliegende Bindung gebrochen würde, sollte esumgekehrt sein: Anregung des inneren N sollte die in-nere Bindung bevorzugt brechen, was durch Desorpti-on von N2 angezeigt würde, und Anregung des äußerenN die näher liegende N-N Bindung, mit dem Ergebnisvon N-Desorption. Es sei hier angemerkt, dass vorläu-fige Messungen nahe legen, dass die Verhältnisse beiadsorbiertem CO tatsächlich so sind, d. h. dass die An-regung des (inneren) C-Atoms in der Tat vorzugsweisezu CO-Freisetzung, also Bruch der inneren Bindung,führt, und die des (äußeren) zu Bruch der „äußeren“,intramolekularen Bindung.
Diese fehlende Korrelation der Selektion des Bin-dungsbruchs zur Lokalisierung der primären Anregungim Fall des N2 ist nach dem, was wir – zur Erklärungder bei Molekülen meist fehlenden Selektivität – obenbereits über die relevanten Zeitskalen gesagt haben,leicht verständlich. Selbst wenn die Primäranregung imFranck-Condon-Bereich repulsiv ist, wird wegen derkurzen Rumpfloch-Lebensdauern der wesentliche Bin-dungsbruch erst nach Zerfall des Rumpflochs, also imAuger-Endzustand, erfolgen. Er hängt also davon ab,welche Auger-Endzustände vorzugsweise durch Zerfalldes jeweils atomar selektiv angeregten Rumpf-Niveauserzeugt werden, und für welche Bindungen sie – even-tuell bevorzugt – repulsiv wirken. Im Fall von N2 undCO haben wir darüber durch Messung der Zerfalls-spektren detaillierte Information [27]. Ohne auf dieEinzelheiten einzugehen, soll hier nur vermerkt wer-den, dass in der Tat die durch Rumpfloch-Zerfall aufdem inneren N (bei N2) und auf dem (äußeren) O (beiCO) gebildeten Zweiloch-Endzustände vorzugsweisedie intramolekulare Bindung brechen, und die auf demäußeren N und dem (inneren) C die (innere) Adsorpti-onsbindung. Wir vermerken, dass wir bei Auswahl desN2-Systems nicht nur insofern Glück hatten, dass die-ses System überhaupt selektiven Bindungsbruch auf-weist, sondern auch darin, dass sein kontraintuitivesVerhalten gleich auf den Grund der Selektivität hin-wies; bei CO wäre dies nicht so offensichtlich gewesen.Die Zerfallsspektren erklären das verschiedene Verhal-ten der beiden Systeme, das mit der unterschiedlichenAdsorbat-Substrat-Wechselwirkung zusammenhängt.
Eine andere Frage ist, warum wir mit diesen Ober-
flächensystemen mehr Glück hatten beim Nachweis se-lektiven Bindungsbruchs als andere Gruppen bei freienMolekülen wie N2O. Wir sind zurzeit wegen der nochschmalen Datenbasis nicht in der Lage zu sagen, obdies eine generelle Eigenschaft von Adsorbaten ist (wiedas oben als vorstellbar bezeichnet wurde). Denkbarwäre immerhin, dass die starke Anisotropie von Ab-schirmung und Ladungstransfer in Adsorbaten solcheSelektivitäten begünstigt. Dies ist eine interessante Fra-ge für die Zukunft. Zur Frage, ob der Effekt des selek-tiven Bindungsbruchs von praktischer Bedeutung seinkann, sollte man noch Zurückhaltung üben. Einerseitsist klar, dass ein Effekt, der zu seiner Realisierungschmalbandige Synchrotronstrahlung erfordert, nichtfür praktische Chemie wie die gezielte Synthese vonMolekülen Bedeutung haben kann. Andererseits istSynchrotronstrahlung fein fokussierbar und für dieStrukturierung von Schichtsystemen für die Halbleiter-technologie und andere Nano-Techniken verwendbar.In diesem Zusammenhang ist sicherlich ein Bindungs-bruch, der – wie in unseren Messungen – zur Entfer-nung von Fragmenten von der Oberfläche (Desorption)führt, zwar verwendbar (z. B. zu strukturiertem Ätzen),aber nicht sehr aufregend. Jedoch lässt sich leicht vor-stellen, dass dadurch gebildete freie Bindungen in derAdsorbatschicht mit anderen Adsorbaten rekombinie-ren, oder die abgespaltenen Fragmente ihrerseits mitanderen Teilchen auf der Oberfläche selektiv reagierenkönnen. Dann wäre es möglich, lokale Reaktionen so-wohl im Sinne der Nanostrukturierung als auch imSinne der atomaren Lokalisierung durchzuführen undzu verwenden. Eine andere „Anwendung“ wäre, wennsolche selektiven Reaktionen in bzw. an interplaneta-ren und interstellaren Körnern und Ähnlichem ablau-fen könnten, was vielleicht manche der erstaunlichkomplexen Weltraum-Moleküle erklären könnte. Füruns steht im Augenblick der Gewinn an Verständnisder in Adsorbaten möglichen Prozesse im Vordergrund;das schließt aber Anwendungen in der Zukunft nichtaus.
SchlussbemerkungDiese Übersicht über jüngste Ergebnisse in einem
Teilgebiet der Forschung meiner Arbeitsgruppe hat denZweck zu zeigen, dass die Chemische Physik der Ober-flächen ein reifer, aber weiterhin sehr aktiver und in-novativer Zweig der Physik ist. Wie in allen Gebietenunserer Wissenschaft ergeben sich aus wesentlichenFortschritten bei der Experimentiertechnik Möglichkei-ten, neue – aber auch altehrwürdige, bisher der direk-ten Beantwortung nicht zugängliche – Fragestellungenerschöpfender zu beantworten, wodurch neue Fragenaufgeworfen werden, was die Schönheit unserer Wis-senschaft und den Spaß an ihr ausmacht.
Wie in allen Erfolgsberichten über die Aktivitäteneiner Arbeitsgruppe muss betont werden, dass diese Er-folge auf dem Zusammenwirken einer großen Zahl voneng kooperierenden Personen beruhen und der Berich-tende sich vor allem als Exponent und Repräsentantdieser Gruppe fühlt. Diesen Umstand zu betonen istumso wichtiger, als der erfreuliche Anlass dieses Be-richts auf den Berichtenden personalisiert ist. Ichmöchte sehr nachdrücklich meine tiefe Dankbarkeitgegenüber meinen Mitarbeitern, jetzigen und früheren,und den vielen mit uns eng zusammenarbeitenden Kol-legen, vor allem in der Theorie, aber auch bei den vonuns verwendeten Synchrotron-Strahlungsquellen, beto-

Physikalische Blätter56 (2000) Nr. 7/8
Preisträger
87
nen. Im Zusammenhang mit diesem Bericht gilt diesganz besonders meinen langjährigen Mitstreitern Wil-fried Wurth und Peter Feulner, die jeweils für die hierskizzierten beiden Entwicklungen primär verantwort-lich waren und sind, sowie den direkt beteiligten Dok-toranden und Postdocs Christian Keller und MarkusStichler, sowie Silvano Lizzit und Giovanni Comellivon ELETTRA für den ersten Komplex, und für denzweiten Ralf Romberg, Sean Frigo, Alexander Ogurtsovund unseren Techniker Norbert Heckmair, der leidersehr jung verstorben ist. Beim theoretischen Verständ-nis ist hier zuvörderst Zbyszek Gortel von der Univer-sity of Alberta als wesentlicher Beiträger zu nennen.Meine jetzigen selbstständigen Mitarbeiter Peter Jakobund Wolf Widdra sowie viele frühere Mitarbeiter, ins-besondere Eberhard Umbach, Herbert Pfnür, Hans-Pe-ter Steinrück und Georg Held, sowie viele Doktoran-den und Postdocs haben in anderen Bereichen unseresgemeinsamen Forschungsprogramms den Erfolg meinerArbeitsgruppe ermöglicht. Unter meinen Kollegen ausder Theorie möchte ich in diesem Zusammenhang Wil-helm Brenig, Hans Jürgen Kreuzer und Notker Röschhervorheben.
Gefördert wurden diese Arbeiten vor allem von derDFG und in früheren Zeiten auch vom BMBF (als die-ses noch Grundlagenforschung förderte). Unsere Mes-sungen bei ELETTRA in Trieste wurden und werdenvon der EU unterstützt. Zusätzliche Flexibilität wurdeverfügbar durch den Max-Planck-Preis von MPG undAlexander-von Humboldt-Stiftung und durch denFonds der Chemischen Industrie. Für alle diese Förde-rungen sage ich hier meinen Dank.
Literatur[1] „Surface Science – The first thirty years“, C. B.
Duke (Hrsg.); Surf. Sci. 299/300 (1994); Chemical Physics – Molecules at Surfaces: Electro-nic Structure and Dynamics, D. Menzel und N.Rösch (Hrsg.); Chem. Physics 177 321 (1993).
[2] D. Menzel, Surf. Rev. Lett. 4, 1283 (1997) und 6,835 (1999) sowie Zitate darin.
[3] D. Menzel und W. Widdra, Proc. Internat. Sympos.„Physics of Lowdimensional Systems“, Oaxaca,Mexico (im Druck) und Zitate darin.
[4] M. Staufer, U. Birkenheuer, T. Belling et al.; J.Chem. Phys. 112, 2498 (2000).
[5] W. Widdra, A. Fink, S. Gokhale et al.; Phys. Rev.Letters 80, 4269 (1998).
[6] D. Menzel, Surface Sci. 299/300, 170 (1994) undZitate darin.
[7] M. Stichler, C. Keller, C. Heske et al.; Surf. Sci.448, 164 (2000).
[8] D. Menzel, J. Electron Spectrosc. Rel. Phen. 72, 19(1995) und 76, 73 (1995); W. Wurth, Appl. Phys. A65, 155 und 597 (1997).
[9] W. Wurth und D. Menzel, Chem. Physics 251, 141(2000) und Zitate darin.
[10] D. Menzel, Nucl. Instrum. Methods B 101, 1(1995); P. Feulner und D. Menzel, in: „Laser Spec-troscopy and Photochemistry on Metal Surfaces“,H. L. Dai and W. Ho (Hrsg.); World Scientific Pu-blishing Co., Singapore 1995, Kap. 16, S. 627 undZitate darin.
[11] H. Schlichting, D. Menzel, T. Brunner et al.; J.Chem. Phys. 97, 4453 (1992).
[12] H. J. Kreuzer, S. H. Payne, A. Drozdowski et al.; J.Chem. Phys. 110, 6982 (1999).
[13] H. Schlichting und D. Menzel, Surf. Sci. 272, 27(1992); K.-H. Allers, H. Pfnür, P. Feulner et al.; J.Chem. Physics 100, 3985 (1994).
[14] K. L. Kostov, H. Rauscher und D. Menzel, Surf.Sci. 287/ 288, 283 (1993); S. H. Payne, H. J. Kreu-zer, P. Jakob et al.; Surf. Sci. 424, 36 (1999) undZitate darin.
[15] D. Menzel und R. Gomer, J. Chem. Phys. 40, 1164(1964) und 41, 3311 (1964).
[16] Die Tagungsbände „DIET – Desorption Induced byElectronic Transitions“ (I-V in Springer Series onSurface Science; ab VI in Surf. Sci).
[17] D. Menzel, P. Feulner, R. Treichler et al.; PhysicaScripta T 17, 166 (1987) und Zitate darin.
[18] W. Wurth, P. Feulner und D. Menzel, PhysicaScripta T 41, 213 (1992); O. Björneholm, A. San-dell, A. Nilsson et al.; Physica Scripta. T 41, 217(1992).
[19] F. Gel’mukhanov und H. Agren, Physics Reports312, 87 (1999) und darin angegebene Literatur.
[20] E. Pahl, H.-D. Meyer und L. S. Cederbaum, Z.Phys. D 38, 215 (1996); Z. W. Gortel, R. Teshimaund D. Menzel, Phys. Rev. A 58, 1225 und 3699(1998), und ibid. A 60, 2139 (1999).
[21] C. Keller, M. Stichler, G. Comelli et al., Phys. Rev.B 57, 11951 (1998).
[22] C. Keller, M. Stichler, G. Comelli et al., Phys. Rev.Lett. 80, 1774 (1998).
[23] M. Lindroos, H. Pfnür und D. Menzel, Phys. Rev.B 33, 6684 (1986).
[24] S. P. Frigo, P. Feulner, B. Kassühlke et al., Phys.Rev. Lett. 80, 2813 (1998).
[25] R. Romberg, N. Heckmair, S. Frigo et al., Phys.Rev. Lett. 84, 374 (2000); Phys.Rev. Focus, Jan. 10(2000); Chem. & Engin. News, 31, 36 (2000).
[26] A. Nilsson, H. Tilborg und N. Martensson, Phys.Rev. Lett. 67, 1015 (1993); O. Sandell, O. Björ-neholm, A. Nilsson et al., Phys. Rev. Lett. 70, 2000(1993).
[27] Ch. Keller, Dissertation, München 1998, H. UtzVerlag, München, S. 82 (1998).





























