Embed Size (px)
Citation preview

Strukturaufklärung von Naturstoffen aus Bakterien der
Gattung Streptomyces
vorgelegt von
Diplom-Chemiker
Jonny Nachtigall
aus Hamburg
Von der Fakultät II – Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
- Dr. rer. nat. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. Thomas Friedrich
Erster Berichter: Prof. Dr. rer. nat. Roderich D. Süßmuth
Zweiter Berichter: Prof. Dr. rer. nat. Hans-Peter Fiedler
Tag der wissenschaftlichen Aussprache: 27.01.2012
Berlin 2012
D 83

2

3

4

5
Zusammenfassung
In der vorliegenden Arbeit werden acht neue Naturstoffe aus vier verschiedenen
Bakterienstämmen der Gattung Streptomyces in ihrer chemischen Struktur
beschrieben. Zur Strukturaufklärung wurden umfangreiche massenspektrometrische
und NMR-spektroskopische Analysemethoden eingesetzt. Bei den Naturstoffen
handelt es sich zum einen um sieben Vertreter der Klasse I-Polyketide, zu anderen
um einen Vertreter der Aranciamycine, welche aus einem kondensierten,
hocharomatischen System der Klasse II-Polyketide bestehen.
Die Bakterienstämme sind ausschließlich der Gattung Streptomyces zuzuordnen,
wobei diese jedoch aus völlig unterschiedlichen Habitaten isoliert wurden. Während
die eine Gruppe von Stämmen aus einem mitteleuropäischen Nadelwald isoliert
worden ist, kommt ein weiterer Stamm aus der Atacamawüste, welche mit ihren
extremen klimatischen Bedingungen eine der trockensten Orte der Erde darstellt.
Im Anschluss an ihre strukturchemische Charakterisierung wurden die Verbindungen
in verschiedenen biologischen Testsystemen auf ihre Wirkung hin untersucht. Dabei
zeigte sich ein breites Wirkspektrum der unterschiedlichen Substanzen bei
antibakteriellen, antitumor- und enzyminhibierenden Testierungen. Die hierbei
gewonnen Erkenntnisse tragen zu einem besseren Verständnis über Struktur und
Wirkung von Naturstoffen bei.

6

7
Teile der vorliegenden Arbeit wurden bereits veröffentlicht:
D. Schulz, J. Nachtigall, J. Riedlinger, K. Schneider, K. Poralla, J.F. Imhoff, W. Beil,
G. Nicholson, H.-P. Fiedler, R.D. Süssmuth, Piceamycin and its N-acetylcysteine
adduct is produced by Streptomyces sp. GB 4-2
J. Antibiot., 2009, 62, 513-518.
J. Nachtigall, D. Schulz, W. Beil, R.D. Süssmuth, H.-P. Fiedler, Aranciamycin
anhydride, a new anthracycline-type antibiotic isolated from Streptomyces sp. Tü
6384
J. Antibiot., 2010, 63, 397-399.
J. Nachtigall, A. Kulik, S. Helaly, A.T. Bull, M. Goodfellow, J.A. Asenjo, A. Maier,
J. Wiese, J.F. Imhoff, R.D Süssmuth, H.-P. Fiedler, Atacamycins A-C, 22-membered
antitumor macrolactones produced by Streptomyces sp. C38
J. Antibiot., 2011, 64, 775 – 780.
D. Schulz, J. Nachtigall, U. Geisen, H. Kalthoff, J.F. Imhoff, H.-P. Fiedler, R.D.
Süssmuth, Silvalactam, a 24-membered macrolactam antibiotic produced by
Streptomyces sp. Tü 6392
J. Antibiot., accepted

8

9
Für meine Familie

10
Danksagung
Herrn Prof. Dr. Roderich D. Süßmuth danke ich für die Aufnahme in die
Arbeitsgruppe, die interessante Themenstellung, die sehr guten Arbeitsbedingungen
sowie für die Betreuung der Arbeit.
Herrn Prof. Dr. Hans-Peter Fiedler möchte ich herzlich für die sehr gute
Zusammenarbeit bei allen gemeinsamen Projekten und für die Übernahme des
Zweigutachtens danken. Des Weiteren möchte ich mich bei allen Mitarbeitern des
Arbeitskreises von Prof. Fiedler herzlich für die freundliche und konstruktive
Zusammenarbeit bedanken, insbesondere bei Dr. Dirk Schulz, Andreas Kulik und
Nadine Horlacher.
Herrn Prof. Dr. Thomas Friedrich danke ich für die bereitwillige Übernahme des
Prufüngsvorsitzes.
Bei Kati Winter möchte ich mich für die gute Organisation im Arbeitskreis Süßmuth
bedanken. Bei Dr. Maria Schlangen und Christine Klose vom Institut für organische
Chemie an der Technischen Universität Berlin möchte ich mich herzlich für die
Unterstützung bei den hochauflösenden Massenspektrometrie- und den IR-
Messungen bedanken. Dr. Reinhard Zeisberg und Dr. Jennifer Thuma danke ich für
die tatkräftige Unterstützung bei den NMR-Experimenten.
Prof. Dr. Johannes F. Imhoff vom Kieler Wirkstoffzentrum des IFM-GEOMAR und
seinen Mitarbeitern, insbesondere Dr. Heidi Zinecker, möchte ich für die
Durchführung der Bioaktivitätsassays und die gute Zusammenarbeit danken.
Dr. Graeme Nicholson von der Universität Tübingen möchte ich für die Durchführung
der GC-MS-Analysen und der FT-ICR-MS-Messungen danken.
Allen meinen Kollegen im Arbeitskreis Süssmuth möchte ich für die nette und
kollegiale Arbeitsatmosphäre danken, besonders Dr. Soleiman Helaly, Dr. Joanna
Krawczyk, Dr. Wolfgang Müller, Dr. Anne Hänchen, Dr. Georg Sambeth und
Alexander Denisiuk.

11
Für ihre unermessliche Geduld, antreibenden Worte und die schöne Zeit während
des Zustandekommens dieser Arbeit danke ich meiner Frau Stephanie Nachtigall.

12

Inhaltsverzeichnis
13
Inhaltsverzeichnis
1 Einleitung ................................................................................... 17
1.1 Entwicklung der Naturstoffforschung, Entdeckung der Antibiotika
und Resistenzbildung .................................................................................17
2 Grundlagen ................................................................................ 21
2.1 Chromatographische Trennmethoden........................................................21
2.1.1 Retentionsmechanismen ........................................................................22
2.1.2 Hochleistungsflüssigkeitschromatographie (HPLC) ................................24
2.2 Massenspektrometrie[15-19]..........................................................................27
2.2.1 Aufbau eines Massenspektrometers.......................................................27
2.2.2 Ionenerzeugung......................................................................................28
2.2.3 Elektrospray-Ionisation (ESI) ..................................................................30
2.2.4 Massenauftrennung[15-19].........................................................................32
2.2.5 Detektion.................................................................................................42
2.3 Kernresonanz-Spektroskopie .....................................................................42
2.3.1 Experimentelle Grundlagen ....................................................................43
3 Zielsetzung................................................................................. 52
4 Strukturaufklärung des Piceamycins und seines N-
Acetylcystein-Addukts aus Streptomyces sp. GB 4-2 ........... 53
4.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces sp.
GB 4-2........................................................................................................53
4.1.1 Chemisches Screening ...........................................................................54
4.1.2 Fermentation und Isolierung ...................................................................56
4.2 Strukturaufklärung ......................................................................................58
4.2.1 HPLC-ESI-MS.........................................................................................58
4.2.2 Bestimmung der Summenformel ............................................................58
4.2.3 Aminosäureanalytik.................................................................................59
4.2.4 NMR-Spektroskopische Untersuchungen ...............................................59
4.3 Biologische Aktivität ...................................................................................62
4.4 Diskussion..................................................................................................63
5 Strukturaufklärung des Aranciamycin-Anhydrids aus
Streptomyces TÜ 6384.............................................................. 68

Inhaltsverzeichnis
14
5.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces sp.
TÜ 6384 .....................................................................................................68
5.1.1 Chemisches Screening ...........................................................................69
5.1.2 Isolierung und Reinigung ........................................................................71
5.2 Strukturaufklärung ......................................................................................72
5.2.1 HPLC-ESI-MS.........................................................................................72
5.2.2 Bestimmung der Summenformel ............................................................72
5.2.3 Zuckeranalytik.........................................................................................73
5.2.4 NMR-spektroskopische Untersuchungen................................................74
5.2.5 Biologische Aktivität ................................................................................75
5.3 Diskussion..................................................................................................76
6 Strukturaufklärung der Atacamycine A, B und C aus
Streptomyces sp. C-38.............................................................. 80
6.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces sp.
C-38 ........................................................................................................80
6.1.1 Chemisches Screening ...........................................................................81
6.1.2 Isolierung und Reinigung ........................................................................83
6.2 Strukturaufklärung ......................................................................................84
6.2.1 HPLC-ESI-MS.........................................................................................84
6.2.2 Bestimmung der Summenformel ............................................................84
6.2.3 NMR-spektroskopische Untersuchungen................................................85
6.3 Biologische Aktivität ...................................................................................91
6.4 Diskussion..................................................................................................93
7 Strukturaufklärung von TÜ 6392 A2 und TÜ 6392 D aus
Streptomyces sp. TÜ 6392........................................................ 96
7.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces sp.
TÜ 6392 .....................................................................................................96
7.1.1 Chemisches Screening ...........................................................................97
7.1.2 Isolierung und Reinigung ......................................................................100
7.2 Strukturaufklärung von TÜ 6392 A2 .........................................................102
7.2.1 HPLC-ESI-MS.......................................................................................103
7.2.2 Bestimmung der Summenformel ..........................................................104
7.2.3 NMR-Spektroskopische Untersuchungen .............................................104
7.3 Biologische Aktivität .................................................................................112

Inhaltsverzeichnis
15
7.4 Diskussion................................................................................................112
7.5 Strukturaufklärung von TÜ 6392 D ...........................................................114
7.5.1 HPLC-ESI-MS.......................................................................................114
7.5.2 Bestimmung der Summenformel ..........................................................115
7.5.3 NMR-Spektroskopische Untersuchungen .............................................116
7.5.4 Biologische Aktivität ..............................................................................118
7.6 Diskussion................................................................................................119
8 Experimenteller Teil ................................................................ 125
8.1 Chemisches Screening mittels HPLC-DAD..............................................125
8.2 HPLC-DAD-ESI-Massenspektroskopie ....................................................125
8.3 GC-MS .....................................................................................................126
8.4 ESI-FT-ICR-Massenspektrometrie ...........................................................127
8.5 HPLC-ESI-FT-Orbitrap-Massenspektrometrie..........................................127
8.6 NMR-Spektroskopie .................................................................................128
9 Anhang ..................................................................................... 129
9.1 Abkürzungsverzeichnis ............................................................................129
9.2 Literaturverzeichnis ..................................................................................132
9.3 NMR-spektroskopische Daten der in dieser Arbeit
strukturaufgeklärten Verbindungen ..........................................................137
9.3.1 Piceamycin ...........................................................................................137
9.3.2 N-Acetyl-Piceamycin.............................................................................144
9.3.3 Aranciamycin-Anhydrid .........................................................................149
9.3.4 Atacamycin A........................................................................................155
9.3.5 Atacamycin B........................................................................................162
9.3.6 Atacamycin C........................................................................................168
9.3.7 TÜ 6392 A2...........................................................................................174
9.3.8 TÜ 6392 D ............................................................................................182

16

Einleitung
17
1 Einleitung
Naturstoffe sind aufgrund ihrer potenziellen Wirksamkeit seit jeher von großer
Bedeutung. Seit der Entdeckung des Penicillins und dem Beginn der
wissenschaftlichen Forschung auf dem Gebiet der Antibiotika sind in der Natur
vorkommende Wirkstoffe aus unserem heutigen Verständnis von Pharmazeutika
nicht mehr wegzudenken. Mehr als 75 % der in den letzten 30 Jahren eingeführten
antibakteriellen Wirkstoffe und über die Hälfte aller zugelassenen
Antitumormedikamente sind Naturstoffe oder Naturstoffderivate.[2]
1.1 Entwicklung der Naturstoffforschung, Entdeckung der
Antibiotika und Resistenzbildung
Noch vor 100 Jahren waren die Heilungschancen für die meisten aller
Infektionskrankheiten verschwindend gering, bedeutete doch eine bakterielle
Infektion meist den sicheren Tod des Patienten. Seit der Frühgeschichte sind immer
wieder verheerende Epidemien überliefert, die gerade in den eng bebauten und
hygienisch schlecht gestellten Armenvierteln der Städte in aller Welt einen Großteil
der Bevölkerung dahinrafften. Mit der Verbesserung der Hygiene und der strikten
Trennung zwischen Trink- und Brauchwasser konnten zunächst die
Überlebenschancen der erkrankten Menschen verbessert werden und die
Verminderung von Infektionskrankheiten im Allgemeinen erreicht werden. Die
stärkste Waffe gegen Infektionskrankheiten wurde jedoch im Jahr 1928 entdeckt, als
Sir Alexander Fleming aus dem Schimmelpilz Penicillium notatum das Penicillin als
erstes eingesetztes Antibiotikum isolierte.[3] Mit diesem Medikament konnten zum
ersten Mal verschiedene Arten von bakteriellen Infektionen geheilt werden, die ohne
Medikation den sicheren Tod für den Patienten bedeutet hätten. Allerdings kannte
und nutzte die Menschheit die Vielseitigkeit der Natur schon seit der Antike.[4] Meist
wurden Extrakte aus Pflanzen als Schmerzhemmer, Aphrodisiakum oder
Rauschmittel verwendet und bilden somit die antike Vorgeschichte der heutigen
Pharmazie.[5]

Einleitung
b) a)
Penicillin G Sulfamidochrysoidin
Abbildung 1: Strukturformeln von a) Penicillin G und b) Sulfamidochrysoidin.
Zwar konnte die bakterizide Wirkung des Penicillins durch mikrobiologische Versuche
rasch hinreichend bewiesen werden, die Struktur des Wirkstoffs hingegen konnte erst
1944 durch Dorothy Hodgkin mittels Röntgenstrukturanalyse aufgeklärt und somit für
weitere semisynthetische Ansätze zur Verbesserung der Pharmakologie zugänglich
gemacht werden (Abbildung 1). Dabei zeigte sich, dass die pharmakophore Gruppe
des Penicillins ein β-Lactam darstellt, wonach diese Wirkstoffgruppe β-Lactam-
Antibiotika genannt wurden.
Antibakterielle Wirkstoffe sind jedoch nicht nur ausschließlich in der Natur zu finden.
Gerhard Domagk entdeckte mit dem Sulfamidochrysoidin bereits 1935 einen gänzlich
synthetischen Arzneistoff, indem er verschiedene Azofarbstoffe an mit Streptokokken
infizierte Mäuse verabreichte.[6] Die Sulfonamide wirken jedoch im Gegensatz zu den
β-Lactam-Antibiotika nur bakteriostatisch, weswegen sich bei den Sulfonamiden
zügig eine Resistenz ausbilden kann und diese heutzutage im Gegensatz zu den
Penicillin-Derivaten seltener als Wirkstoff in der Humanmedizin zu finden sind. Diese
Meilensteine markieren den Anfang des wissenschaftlichen Forschens zur
Entdeckung und Strukturaufklärung von biologisch aktiven Naturstoffen.
Inzwischen sind insgesamt sieben große Gruppen von antibiotischen Wirkstoffen
bekannt und ihre Anzahl wächst stetig weiter.[7] Dem gegenüber steht jedoch die
ebenso steigende Zahl an resistenten bakteriellen Erregern. Schon Anfang der 50er
Jahre wurden die ersten Penicillin-resistenten Bakterienstämme entdeckt, die durch
natürliche Mutationen an den Penicillin-Wirkorten oder durch Penicillin-
Inaktivierungsprozesse entstanden.[7, 8] Im Jahr 2005 infizierten sich allein in Europa
drei Millionen Menschen an resistenten Keimen, die durch kein zugelassenes
Medikament behandelt werden konnten – 50.000 davon starben.[9]
18
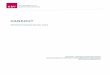
Einleitung
O
HN
O
H ONH
ONH
H
OO
O
HN
HO
NHH
OH
NH
HO2C
H
HN
O
O
Cl
H H
Cl
NH2
O
O
OHOH
CH2OH
OH
HO
CH3
O
OH3C
HOH2N
CH3
OH
b) a)
Vancomycin Oxacillin
Abbildung 2: Strukturformeln der Antibiotika a) Vancomycin und b) Oxacillin.
Zu den gefährlichsten humanpathogenen Erregern gehört der Stamm
Staphylococcus aureus, von dem manche Arten bereits Resistenzen gegen die
Antibiotika Vancomycin und Oxacillin ausgebildet haben und die vermehrt in
Krankenhäusern gefunden werden können (Abbildung 2). Vancomycin und Oxacillin
gehören zu der sogenannten Gruppe der Reserveantibiotika, welche nur gegen
Infektionen verabreicht werden, die mit der Verabreichung von anderen Antibiotika
nicht behandelt werden können. Somit kann Oxacillin als Endpunkt des
therapeutischen Erfolgs der Klasse von β-Lactam-Antibiotika angesehen werden,
sollte nicht noch ein wirksamer Nachfolger gegen multiresistente Erreger gefunden
werden.
a) b)
Linezolid Tigecyclin
Abbildung 3: Strukturformeln der Antibiotika a) Linezolid[10] und b) Tigecyclin[11].
19

Einleitung
Nach und nach verlieren ehemalige Reserveantibiotika ihre Stellung an neu
entwickelte, wirksame Vertreter wie Linezolid[10], Tigecyclin[11] (Abbildung 3) oder
Daptomycin[12] (Abbildung 4), welche heutzutage zur Ersttherapie gegen
multiresistente Bakterienstämme verwendet werden, da gegen viele Erreger kein
anderes Medikament noch Wirkung zeigt.[13]
HN
HN
NH
HN
NH
HN
NH
NH2O
HN
NHOO
NH
CONH2
O
O
CO2HO
HO
O
O
O
CO2H
O CO2H
NH
O
NH
HN
OH
O
O
CO2H
NH
O
O
NH2
10
Abbildung 4: Strukturformel von Daptomycin[12].
Somit ist ein Ziel der wissenschaftlichen Suche nach neuen Wirkstoffen auch das
Streben nach einer möglichst hohen Anzahl an wirksamen Medikamenten, um den
Wettlauf gegen die neuen multiresistenten Keime nicht zu verlieren.
20

Grundlagen
21
2 Grundlagen
2.1 Chromatographische Trennmethoden
Die Chromatographie wurde zuerst von dem russischen Botaniker Michail
Semjenowitsch Tswett im Jahr 1903 beschrieben, als er einen Chlorophyll-Extrakt
aus Pflanzen mittels einer Säule aus Calciumcarbonat in seine Bestandteile
auftrennte. Aufgrund der auftretenden farbigen Banden nannte er die neue Technik
Chromatographie, nach den griechischen Worten chroma „Farbe“ und graphein
„schreiben“. Erstaunlicherweise blieben die Ergebnisse von Tswett lange von der
Fachwelt unbeachtet. Anfang der 30er Jahre griff die Arbeitsgruppe von Richard
Kuhn am Kaiser-Wilhelm-Institut für medizinische Forschung in Heidelberg diese
Technik wieder auf, um ein Farbstoffgemisch aus Carotinoiden und Xanthophyllen
aus Pflanzenextrakten aufzutrennen. 1938 wurde Richard Kuhn der Nobelpreis für
seine allgemeine Arbeit zu den Carotinoiden und Vitaminen verliehen.
Seitdem wird die Entwicklung neuer Chromatographiemethoden stetig
vorangetrieben. Archer J.P. Martin und Richard L.M. Synge erhielten 1952 den
Chemie-Nobelpreis für die im Jahre 1941 vorhergesagte Verteilungschromatographie
zwischen einer mobilen gasförmigen und einer stationären flüssigen Phase. Diese
Entdeckung führte in den 1950er Jahren zu der Entwicklung von
Gaschromatographen.
In den 1960er Jahren erfolgte dann die Entwicklung der
Hochdruckflüssigchromatographie (HPLC). Hierzu wurden zunächst unpolare
Alkanketten über eine Etherbindung an das Kieselgel-Trägermaterial geknüpft, um
eine genügend unpolare stationäre Phase zu bilden. Durch diese Modifizierung
konnte nun ein Großteil der organischen Substanzen auf dieser Art von Säulen
retardiert werden, da für eine Wechselwirkung mit unmodifiziertem Kieselgel die
meisten Naturstoffe zu unpolar waren. Des Weiteren wurde die Druckstabilität der
Chromatographieanlagen enorm gesteigert, so dass nun eine höhere Trennleistung
möglich war. Die andauernde Entwicklung der Technik und Methodik der
Chromatographie hat diese zu einer Standardmethode für die Auftrennung von
verschiedensten Substanzen werden lassen. Durch die Nutzung verschiedener

Grundlagen
stationärer Phasen und Trenntechniken lässt sich heutzutage beinahe jedes
Stoffgemisch durch die Chromatographie in seine Bestandteile auftrennen. Auch in
der Zukunft wird daher die Chromatographie aus der Analytik – insbesondere der
Bioanalytik – nicht wegzudenken sein.
2.1.1 Retentionsmechanismen
Die Technik der chromatographischen Trennung beruht auf der unterschiedlichen
Wechselwirkung des Stoffgemischs mit der stationären Phase. Die hierdurch
hervorgerufene Retention kann anhand des dominierenden Mechanismus bei der
Trennung charakterisiert werden, jedoch handelt es sich bei den meisten Verfahren
um eine Kombination verschiedener Mechanismen. Die grundlegenden in dieser
Arbeit angewendeten Mechanismen sollen hier kurz vorgestellt werden.
2.1.1.1 Verteilungschromatographie
Die chromatographische Trennung bei der Verteilungschromatographie beruht auf
dem Gesetz der multiplen Verteilung. Ein Analyt A ist in der stationären und der
mobilen Phase unterschiedlich gut löslich, sodass sich nach dem Nernst’schen
Verteilungsgesetz ein Gleichgewicht einstellt.
AM AS
Daher kann für jede Substanz im Stoffgemisch eine Verteilungskonstante Kc
angegeben werden, welche sich aus der Konzentration eines Stoffs in der
stationären Phase cS und der mobilen Phase cM zusammensetzt.
SC
M
cK
c
Aufgrund der unterschiedlichen chemischen Struktur von Substanzen in einem
Gemisch haben diese alle unterschiedlich große Verteilungskonstanten. Genau
22

Grundlagen
23
dieser Effekt wird bei der Verteilungschromatographie ausgenutzt: Die Substanzen
verteilen sich unterschiedlich in der stationären und mobilen Phase und werden somit
aufgetrennt. Ein prominenter Vertreter dieser Trenntechnik ist die
Gaschromatographie. Hierbei wird die unterschiedliche Löslichkeit der Analyten im
Trägergas (mobile Phase) zu einem Flüssigkeitsfilm (stationäre Phase) ausgenutzt,
um die Analyten voneinander zu trennen.
2.1.1.2 Adsorptionschromatographie
Die Trennung der Analyten bei der Adsorptionschromatographie beruht auf der
unterschiedlichen Bindung der Stoffe an eine feste stationäre Phase. Bei dieser
Bindung kann es sich um Van-der-Waals-Kräfte, Wasserstoffbrückenbindungen,
Dipol-Dipol-Wechselwirkungen oder Ionenbindungen handeln, weshalb hierbei auch
häufig polare stationäre Phasen wie silikabasierende Materialien (SiO2) oder
Aluminiumoxide (Al2O3) zum Einsatz kommen. Die Auftrennung der Analyten erfolgt
über die unterschiedlich starke Bindung der Analytmoleküle an die stationäre Phase.
2.1.1.3 Größenausschlußchromatographie
Diese Trennmethode nutzt die unterschiedliche Größe der Analytmoleküle in Lösung
aus. Bei dem Säulenmaterial handelt es sich um hochvernetzte, poröse Materialien
mit einer je nach Substanzgemisch definierten Porenweite. Kleine Moleküle können
in die Poren diffundieren, während großen Molekülen weniger Porenvolumen zur
Verfügung steht und daher die Säule schneller passieren. Aufgrund dieses
Retentionsprinzips eluieren große vor kleinen Molekülen, da diesen eine kürzere
Wegstrecke in der Säule zur Verfügung steht.
2.1.1.4 Ionaustauschchromatographie
Der Trennmechanismus bei der Ionaustauschchromatographie beruht auf der
ionischen Wechselwirkung zwischen geladenen Analytmolekülen und entgegensetzt
geladenen Ionen, welche zumeist an eine polymere Matrix gebunden sind. Aufgrund

Grundlagen
24
der unterschiedlich starken Ausprägung der ionischen Wechselwirkungen zwischen
den Analyten und der stationären Phase kommt es zu unterschiedlicher Retention
der Analytmoleküle und somit zur Auftrennung.
2.1.2 Hochleistungsflüssigkeitschromatographie (HPLC)
Die Abkürzung HPLC wurde ursprünglich für high pressure liquid chromatography
gewählt, da bei der Entwicklung dieser Technologie die hohen Drücke bis 400 bar die
entscheidende Neuerung darstellten. Inzwischen hat sich die HPLC als System
weitestgehend etabliert, so dass man heutzutage von high performance liquid
chromatography spricht, da durch den Einsatz von druckstabilen Säulen und Geräten
ein wesentlich besseres Trennungsvermögen im Gegensatz zur normalen
Säulenchromatographie erreicht wird. Neuerdings werden sogar Anlagen und Säulen
bis 1200 bar eingesetzt. Hierbei spricht man von UHPLC (ultra high performance
liquid chromatography), die durch den Einsatz von kleineren Partikelgrößen der
Trennsäulen eine höhere Trennleistung und eine kürzere Analysenzeit bieten.
Aufgrund der schnellen Analysenzeiten muss auch der Detektor eine genügend
schnelle Messgeschwindigkeit besitzen, was jedoch zum jetzigen Zeitpunkt nicht von
allen Massenspektrometern erreicht werden kann.
Aufbau
Eine HPLC-Anlage besteht im Wesentlichen aus vier Teilen (Abbildung 5):
• Pumpe
• Probenaufgabesystem
• Säule
• Detektor

Grundlagen
Abbildung 5: Schematischer Aufbau einer HPLC-Anlage.
Pumpe
Die Pumpe fördert die zur Analyse benötigten Lösungsmittel aus den Vorratsgefäßen
und baut den zur Trennung nötigen Druck auf. Um Reproduzierbarkeit zu
gewährleisten, werden die Lösungsmittel zuerst von gelösten Gasen wie Stickstoff
und Sauerstoff mittels eines online-Entgasers befreit. Um die Trennung des Analyten
zu optimieren, wird in der HPLC sehr häufig mit Gradientenelution gearbeitet, d.h. die
Zusammensetzung der Lösungsmittelgemische ändert sich mit der Zeit der Messung.
Dieses resultiert in einer steigenden Elutionskraft der mobilen Phase, schärferen
Peaks und kürzeren Laufzeiten. Die Zusammenführung der Lösungsmittel findet zu
meist hinter den Pumpen in einer Mischkammer statt.
Probenaufgabe
Die Probenaufgabe erfolgt im Allgemeinen über eine Probenschleife, auf die die
flüssig vorliegende Probe aufgetragen und anschließend in die mobile Phase
eingekoppelt wird. Die mobile Phase trägt dann die Probe im Fluss mit sich durch die
Trennsäule. Im Speziellen wird dieser Vorgang entweder durch ein Sechswegeventil
oder einen Autosampler realisiert. Bei einem Sechswegeventil spritzt man die Probe
mittels einer Injektionsnadel in die Schleife, bei einem Autosampler wird die Probe in
einem separaten Gefäß vorbereitet und automatisch in die Schleife injiziert. Dieses
Prinzip ist aufgrund der hohen Automatisierbarkeit besonders gut geeignet, um hohe
Durchsatzraten zu erzielen.
25

Grundlagen
26
Säule
Grundsätzlich lassen sich in einer HPLC-Anlage Säulen von allen
Chromatographiearten betreiben, sofern die Säule selbst und das darin befindliche
Säulenmaterial den erhöhten Druck widerstehen können. Allerdings bedeutet eine
Trennung bei erhöhtem Druck nicht für jede Chromatographieart zwangsläufig einen
Vorteil, weswegen sich die die HPLC-Trennung hauptsächlich adsorptionsbasierte
Trennmechanismen eignen. Hierzu zählen zum einen die Normalphase (NP, normal
phase), und zum anderen die Umkehrphase (RP, reversed phase). Eine
Normalphasensäule enthält meist Kieselgel (SiO2), wobei diese stationäre Phase
eine polare Oberfläche besitzt und somit als Laufmittel unpolare oder wenig polare
organische Lösungsmittel benutzt werden. Zur Verbesserung der
Trenneigenschaften ist die Form der Kieselgelpartikel meist sphärisch und besitzt
eine geringe Größenverteilung. Der Trennmechanismus bei der Normalphase
basierend auf Adsorptionswechselwirkungen ist mit dem einer herkömmlichen
Kieselgelsäule vergleichbar. Aufgrund der besseren Stabilität und Permeabilität
werden neuerdings auch monolithische Kieselgelsäulen als Säulenmaterial
verwendet.[14]
Die Umkehrphase hingegen besitzt eine unpolare stationäre Phase und eine polare
mobile Phase. Das Säulenmaterial besteht aus einem oberflächenmodifizierten
Kieselgel. Dabei werden die Hydroxygruppen an der Oberfläche der Kieselgelkugeln
mit Alkylketten definierter Länge versehen. Je nach Polarität der Analyten werden mit
Octadecyl- (C18) Octyl- (C8) oder Butylresten (C4) modifizierte Säulen eingesetzt.
Aufgrund der sehr guten Trenneigenschaften für kleine Moleküle ist die
Umkehrphasechromatographie inzwischen zu der Standardtrenntechnik in der
Bioanalytik avanciert.
Detektor
Die Detektion der getrennten Komponenten erfolgt meist über einen UV/Vis-Detektor
oder einem leistungsstärkeren Diodenarraydetektor (DAD), in manchen Fällen erfolgt
eine Kopplung mit einem Massenspektrometer. Das Prinzip eines UV/Vis-Detektors
beruht auf der Lichtabsorption nach dem Lambert-Beer’schen Gesetz, nach dem die
Extinktion proportional zur Konzentration der Probe und zur Länge des
Strahlengangs ist. Aus dem kontinuierlichen, elektromagnetischen Spektrum kann

Grundlagen
27
durch einen Monochromator eine Wellenlänge herausgefiltert werden, so dass diese
in Abhängigkeit von der Retentionszeit gemessen und daraus ein Chromatogramm
erstellt werden kann. Ähnlich, jedoch leistungsfähiger, arbeiten
Diodenarraydetektoren. Bei einem DAD ist es nicht nötig, eine bestimmte
Wellenlänge herauszufiltern. Hier wird das Spektrum über einen Polychromator in
seine einzelnen Wellenlängen zerlegt und über ein Diodenfeld detektiert. Somit erhält
man alle Wellenlängen in Abhängigkeit der Retentionszeit.
Eine weitere Möglichkeit zur Detektion ist der Anschluss eines Massenspektrometers
an die HPLC. Dieses beruht heutzutage überwiegend auf dem Prinzip der
Elektrospray-Ionisation (ESI), bei dem das Lösungsmittel verdampft und die Moleküle
über eine geladene Metallkapillare ionisiert und in das Hochvakuum geleitet werden.
Das Prinzip dieser massenspektrometrischen Technik wird im folgenden Kapitel
ausführlich erklärt.
2.2 Massenspektrometrie[15-19]
2.2.1 Aufbau eines Massenspektrometers
Die Massenspektrometrie ist ein Verfahren zur Messung des Verhältnisses von
Masse zu Ladung m/z von Teilchen. Ist die Ladung z bekannt, kann aus dem
Verhältnis die Masse m des Teilchens berechnet werden. Dafür werden aus den zu
untersuchenden Molekülen zunächst über verschiedene Methoden positive oder
negative Ionen erzeugt, die anschließend nach ihrem Masse-/Ladungsverhältnis
getrennt und schließlich nachgewiesen werden (Abbildung 6).

Grundlagen
Vakuumsystem
Massen-
analysator
Ionenquelle Detektor
Einlass-
System
Daten-
system
Abbildung 6: Schematischer Aufbau eines Massenspektrometers.
2.2.2 Ionenerzeugung
Während der Entwicklung der Massenspektrometrie wurde eine Vielzahl von
Ionisationtechniken für unterschiedlich vorliegende Probenmoleküle entwickelt
(Abbildung 7). Generell lässt sich die Ionisation mit fünf verschiedene Methoden
erreichen, wovon hier die wichtigsten beschrieben sind.
Ionenquelle Analysator Detektor
- Elektronenstoß-Ionisation (EI)
- Chemische Ionisation (CI)
- Fast Atom Bombardment (FAB)
- Elektrospray-Ionisation (ESI)
- Matrix-unterstützte Laser-
desorption (MALDI)
- Atmospheric pressure chemical
Ionisation (APCI)
(Ionencyclotron, ICR)
- Quadrupol (Q)
- Magnetisches Sektorfeld
- Elektrisches Sektorfeld
- Flugzeitanalysator (TOF)
- Elektrische Ionenfalle
(ion trap, Orbitrap)
-Elektromagnetische Ionenfalle
- Vielkanalplatte
- Szintillationszähler
Vervielfacher (SEV)
- Faraday-Cup
- Sekundärelektronen-
- Felddesorption (FD)
- Feldionisation (FI)
Abbildung 7: Übersicht über Ionisations-, Analysator- und Detektortechniken.
Elektronenstoßmethoden
28

Grundlagen
29
Zu dieser Methode gehören die heute weit verbreiteten Ionisationsmethoden
Elektronenstoß- (EI) und chemische Ionisation (CI). Bei der Elektronenstoßionisation
werden aus einem Filament heraus mittels einer angelegten Spannung Elektronen
beschleunigt und durch die gasförmig vorliegende Probe geleitet. Dabei werden die
Probenmoleküle ionisiert und zerfallen spontan in kleine Molekülfragmente, welche
anschließend detektiert werden können. Bei der hiervon abgeleiteten chemischen
Ionisation wird ein Primärgas durch Elektronenstoßionisation geladen, welches
anschließend in einer Sekundärreaktion den Analyten ionisiert.
Feldionisationsmethoden
Bei der Feldionisation wird die Analyten zwischen zwei Elektroden durchgeleitet, an
denen eine hohe elektrische Spannung (10 – 20 kV) angelegt ist. Die Analytmoleküle
diffundieren in die fein verzweigte Struktur der Ionisationselektrode, wo sie durch das
gebildete elektrische Feld ionisiert und in Richtung des Analysators geleitet werden.
Liegt der Analyt gasförmig vor, so spricht man von Feldionisation (FI), liegt er flüssig
oder als Lösung vor, von Felddesorption (FD).
Teilchenbeschussmethoden
Durch den Beschuss von flüssigen oder festen Analyten mit schnellen Atomen oder
Ionen werden aus diesem Elektronen herausgeschlagen und es bilden sich
detektierbare Ionen. Bei der Verwendung von Atomen nennt sich diese Technik fast
atom bombardment (FAB), bei der Nutzung von Ionen spricht man von secondary ion
mass spectrometry (SIMS).
Photoionisationsmethoden
Die gebräuchlichste Photoionisationsmethode in der Bioanalytik ist die Matrix-
unterstützte Laser-Desorption/Ionisation (MALDI). Hierbei wird der Analyt in eine
Matrix mit definierter Anregungswellenlänge eingebettet und diese dann
kokristallisiert. Die Matrixmoleküle sind zumeist aromatische Verbindungen (wie z.B.
α-Cyano-4-hydroxyzimtsäure oder 2,5-Dihydroxybenzoesäure), da diese besonders
gut mittels Laserstrahlung angeregt werden können und relativ gut homogen
kristallisieren. Bei der anschließenden Bestrahlung der Matrix mit Laserlicht
absorbiert diese dann die Energie, was zur Verdampfung des Matrixmaterials und
schließlich zum Herauslösen des ionisierten Analyten führt.

Grundlagen
30
Sprühmethoden
Die Sprühmethoden gliedern sich in zwei unterschiedliche Methoden zur Ionisation.
Bei der Elektrospray-Ionisation (ESI) wird die Analytlösung zerstäubt, ionisiert und
die Lösungsmittelreste entfernt, so dass der Analyt als Ion vorliegt. Aufgrund der
herausragenden Bedeutung der Elektrospray-Ionisation für diese Arbeit soll diese im
nächsten Kapitel ausführlich behandelt werden. Eine weitere Sprühmethode ist die
atmospheric pressure chemical ionisation (APCI). Im Gegensatz zur ESI-Technik
wird hier zuerst die Lösung verdampft, bevor der Analyt ionisiert wird.
2.2.3 Elektrospray-Ionisation (ESI)
Elektrospray bezeichnet das Verfahren zur Zerstäubung einer Flüssigkeit mit Hilfe
eines elektrostatischen Feldes. Das hierdurch generierte Aerosol setzt sich aus polar
geladenen Tropfen mit monodisperser Größe zusammen. Zur Erzeugung wird die
Analytlösung durch eine Metallkapillare geleitet, an die eine hohe elektrische
Spannung angelegt ist. Die Analytlösung wird von dem elektrischen Feld
durchdrungen und die sich in ihr befindlichen Ionen wandern auf die Gegenelektrode
zu. Aufgrund der Anwesenheit von vielen gleichgeladenen Ionen an der
Kapillarspitze bildet sich dort ein Taylor-Kegel aus, in dem die Analytlösung nun als
Aerosol vorliegt. Durch zusätzlich eingebrachtes Trägergas wird das Lösungsmittel
verdampft, die Tröpfchenanzahl vergrößert und die Stabilität des Aerosols bis an die
Grenze geführt (Raleigh-Limit). Eine weitere Verkleinerung der Oberfläche führt zu
einer stark erhöhten Oberflächenladung und somit zum Kollaps der Tröpfchen
(Coulomb-Explosion).[16, 17]
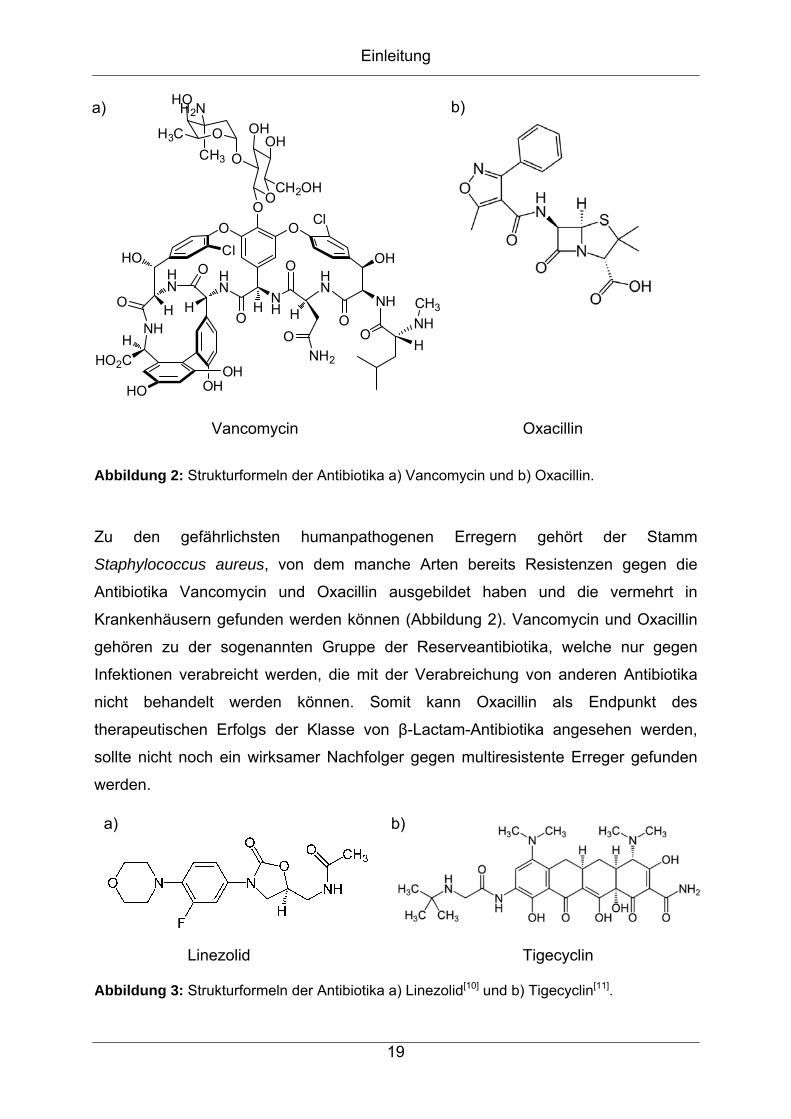
Grundlagen
Abbildung 8: Schematische Darstellung einer positiven Elektrospray-Ionisation.[15]
Durch das mehrmalige Wiederholen dieses Vorgangs entstehen Tröpfchen mit ca.
10 nm Durchmesser. Anschließend erfolgt die Bildung der freien Analytionen, wofür
es zwei verschiedene Modelle gibt. Bei dem charge residue model (CRM, Modell des
geladenen Rückstandes) geht man davon aus, dass Tröpfchen von 1 nm
Durchmesser entstehen, die nur ein ionisiertes Analytmolekül beinhalten.[16, 18] Das
ion evaporation model (IEM, Ionenevaporationsmodell) nimmt an, dass aus größeren
Tröpfchen bereits Analytionen in die Gasphase emmitiert werden.[16, 18] Liegen die
Analytmoleküle als Ionen in der Gasphase vor, werden diese durch die
Potentialdifferenz in das Massenspektrometer geführt (Abbildung 8).
Der gesamte Ionisationsprozess ist bei der ESI-Ionisation im Gegensatz zu den
vorher genannten Methoden sehr sanft. Somit können mit dieser Methode auch
empfindliche Biomoleküle praktisch ohne Quellenfragmentierung ionisiert werden,
was erstmals die massenspektrometrische Detektion von gelösten Proteinen
31

Grundlagen
ermöglichte.[15] Ein weiterer, großer Vorteil dieser Technik ist die Möglichkeit,
Analytströme online zu ionisieren, also der Ionenquelle einen konstanten Strom an
gelösten Analyten zuzuführen. Dieses ermöglicht eine Kopplung von ESI-
Massenspektrometern mit HPLC-Systemen, welche die flüssige Probe vor der
Detektion säulenchromatographisch auftrennen.
2.2.4 Massenauftrennung[15-19]
Durch ionenoptische Bauteile werden die Ionen zum Massenanalysator hin
beschleunigt und dabei durch das unterschiedliche Masse-zu-Ladungsverhältnis
(m/z) ihrer Quasimolekülmasse nach aufgetrennt. Dafür werden bei den meisten
Analysatoren elektrische oder magnetische Felder eingesetzt, um die Ionen aufgrund
ihrer Massenträgheit bzw. der damit verbundenen längeren Flugzeit massenselektiv
zu unterscheiden. Aufgrund der unterschiedlichen Anforderungen sind verschiedene
Bauarten von Massenanalysatoren entwickelt worden:
Magnet- (B)/ oder Sektorfeld (E/B)
Bestimmung der Flugzeit (TOF)
Ionencyclotronresonanz (ICR)
Ionenfalle (T)
Quadrupol (Q)
Orbitrap
Die wichtigsten Kenngrößen bei Massenanalysatoren sind Massenbereich,
Auflösungsvermögen und Scangeschwindigkeit, welche für die benutzten Geräte
verglichen werden.
Die Auflösung R eines Massenanalysators ist gegeben durch die Formel
m
Rm
wobei m die Nominalmasse und dm die Differenz zwischen zwei gerade noch
getrennten Massenpeaks darstellt. Bei den unterschiedlichen Analysatortypen
32
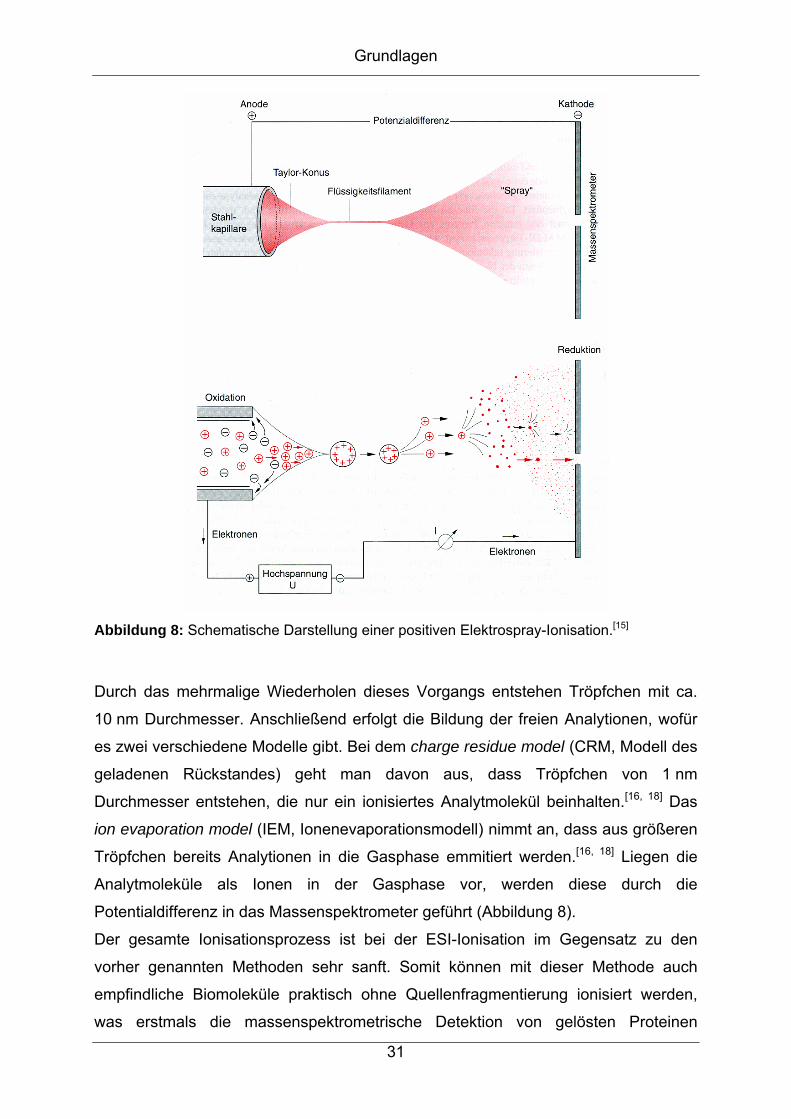
Grundlagen
wurden allerdings unterschiedliche Definitionen zur Peaktrennung verwendet, so
dass diese nicht direkt miteinander vergleichbar sind.
a) b)
Abbildung 9: Definition des Auflösungsvermögens eines Massenspektrometers, a) 10%-
bzw. 50%-Tal-Definition, b) Full Width at Half Maximum (FWHM)-Definition.[15]
Bei den sehr hochauflösenden Sektorfeld-Massenspektrometern wird die
Peaktrennung über das 10%-Tal definiert (Abbildung 9, a), bei den Quadrupol-
Massenspektrometern, die im Allgemeinen eine niedrige Auflösung bieten, gilt die
50%-Tal-Definition. Zur Messung der Auflösung eines Massenspektrometers anhand
nur eines Peaks wird die Auflösung der Halbwertsbreite (FWHM, full width at half
maximum) eines Massenpeaks m gemessen (Abbildung 9, b). Dies wird häufig bei
den TOF- und Orbitrap-Massenspektrometern angewendet und bietet den Vorteil, die
Auflösung an nur einem Peak zu bestimmen.
2.2.4.1 Quadrupol-Analysator
Der Quadrupol-Analysator arbeitet basierend auf dem Prinzip von Massenselektion
und -detektion, bestehend aus einem Bauteil zur Selektion einer definierten Masse
und dem Detektor zur Messung der Signalintensität.
Der Massenfilter besteht aus vier parallelen, auf einem Radius r angeordneten
Stäben entlang einer Achse z. An gegenüberliegenden Stäben liegt jeweils dieselbe
Polarität einer Gleichspannung U an, welche durch die gleiche Polarität einer
Wechselspannung (V cos2f t) mit der Frequenz f moduliert wird (Abbildung 10).
33

Grundlagen
Abbildung 10: Anordnung der Stabelektroden in einem Quadrupol-Massenfilter.[15]
Die nebeneinander liegenden Stäbe besitzen eine um 180° versetzte Phase mit
entgegengesetzter Polarität. Nahe der z-Achse kann das Potential daher wie folgt
beschrieben werden:
2
22
tf2cosVU),,(r
yxtyx
mit r = Radius
f = Frequenz
x,y = Raumkoordinaten
t = Zeit
U = Gleichspannung
V = Wechselspannung
Durch eine anliegende Spannung von 10-20 V erhalten die Ionen eine
Translationsenergie entlang der z-Achse. Die Bewegung in x- und y-Richtung kann
durch die beiden Gleichungen
xtfVU
)2cos(
r
ez
dx
dΦze
dt
xdmmaF
22
2
x
ytfVU
)2cos(
r
ez
dy
dΦze
dt
ydmmaF
22
2
y
34

Grundlagen
mit m = Masse des Ions
e = Elementarladung
z = Anzahl der Ladungen
a = Beschleunigung
beschrieben werden. Durch die Einführung der beiden Definitionen
2)(
2
rfm
Ueza
und 2)( rfm
Vezq
können diese Gleichungen als Mathieusche Gleichungen beschrieben werden:
0)2cos2()(d
xd2
2
xtfqatf
0)2cos2(
)(d
yd2
2
ytfqatf
Diese beiden Differentialgleichungen definieren den stabilen bzw. den instabilen
Bereich des Quadrupolfeldes für gegebene m/z-Verhältnisse, abhängig von den
Parametern a und q. Durch den Quotienten a/q ergibt sich
V
U
zeV
rfm
rfm
Uez
q
a 2)(
)(
2 2
2
dass bei gleicher Ladung z alle Massen m auf der so genannten Arbeitsgeraden
liegen.
Abbildung 11: Stabilitätsdiagramm eines Quadrupolfeldes in x- und y-Richtung.
35

Grundlagen
Nun werden Gleichspannung U und Amplitude V der Wechselspannung unter
Konstanthaltung von U/V und a/q entlang der Arbeitsgeraden so verändert, dass
nacheinander Ionen unterschiedlicher Masse im stabilen Bereich des
Quadrupolfeldes liegen. Somit kann der gesamte gewünschte Massenbereich
gescannt und die Ionen detektiert werden (Abbildung 11).
Moderne Quadrupol-Massenspektrometer bestehen häufig aus vier hintereinander
liegenden Quadrupolen, sogenannten Triple-Quadrupol- oder Tandem-
Massenspektrometern (Abbildung 12). Dieses ermöglicht die gezielte Strukturanalyse
von Ionen durch die massenselektive Filterung vorgeschalteter Quadrupole. Q0 wird
als reines ionenoptisches Bauteil ohne Gleichstrom-Komponente verwendet, um die
davor erzeugten Ionen zu stabilisieren und für Q1 zu fokussieren. Der Q1 ist der
erste Messquadrupol zum Scannen der Ionen. Q2 führt die Ionen durch eine
Kollisionskammer, die mit inertem Kollisionsgas (Stickstoff, Helium oder Argon)
gefüllt werden kann. Durch Kollision mit dem Gas fragmentieren die Ionen, wobei die
Molekülmassen der entstandenen Fragmentionen in dem nachfolgenden Quadrupol
Q3 analysiert werden können.
Abbildung 12: Schematischer Aufbau eines Triple-Quadrupol-Massenspektrometers.[15]
Mit einem Triple-Quadrupol-Massenspektrometer lassen sich nun neben dem
einfachen Trennen und Detektieren von Ionen (full scan) weitere verschiedene, auch
als MS/MS-Analysen oder Tandem-MS bezeichnete Experimente durchführen.
Bei einer Produkt-Ionen-Analyse (daughter ion scan, Abbildung 13) werden im
Quadrupol Q1 Ionen einer bestimmten Masse ausgewählt.[15] Diese Vorläuferionen
werden im Q2 fragmentiert und die generierten Tochter- oder Produktionen
36

Grundlagen
anschließend in Q3 analysiert. Aufgrund der charakteristischen Fragmentierung kann
durch die Produkt-Ionen-Analyse gezielte Strukturinformation zu dem Zielmolekül
gewonnen werden. Je nach Aufbau und Verbrückung eines Moleküls bilden sich
unterschiedliche Fragmentspektren, die durch die Abstände der Produkt-Ionen
zueinander interpretiert werden können. Aufgrund dieser Information ist die Produkt-
Ionen-Analyse die am häufigsten genutzte massenspektrometrische Analyse bei der
Strukturaufklärung von Biomolekülen.
Abbildung 13: Prinzip einer Produkt-Ionen-Analyse.[15]
Die Vorläufer-Ionen-Analyse verläuft von der Schaltung der Quadrupole genau
entgegengesetzt der Produkt-Ionen-Analyse (Abbildung 14).[15] In Q1 wird das
gesamte Massenspektrum transferiert und anschließend in Q2 sequenziell
fragmentiert. Q3 ist auf eine bestimmte, ausgewählte Fragmentmasse eingestellt.
Nun werden die durchgeleiteten Ionen in Q1 und Q3 verglichen und somit analysiert,
welche Vorläufer-Ionen dasselbe Fragment in Q3 zeigen. Mit diesem Messmodus
lassen sich Strukturanaloga aus komplexen Analyten identifizieren.
Abbildung 14: Prinzip einer Vorläufer-Ionen-Analyse.[15]
37

Grundlagen
Ähnlich funktioniert auch die Neutralverlust-Analyse (Abbildung 15).[15] Hierbei
werden Q1 und Q3 in normalen Scan-Modus gestellt, Q2 arbeitet ebenso als
Kollisionszelle. Q3 ist allerdings um die zu beobachtende Neutralverlustmasse
kleiner eingestellt als Q1. Somit lassen sich diejenigen Ionen detektieren, welche bei
Fragmentierung ein spezifisches Neutralteilchen abspalten.
konstant
Abbildung 15: Prinzip einer Produkt-Ionen-Analyse.[15]
Die neueste der Tandem-Analysen ist das multiple reaction monitoring (MRM).[15]
Es dient meist dem hochselektiven Nachweis von Analyten und nicht der
Strukturbestimmung. Dabei werden Q1 und Q3 jeweils ausschließlich auf eine
bestimmte Vorläufer- und Fragment-Masse eingestellt (Abbildung 16). Aufgrund
dieser genauen Einstellung ist das MRM hochsensitiv gegenüber dem gewünschten
Analyten. Bei der neuesten Entwicklung werden die MRM-Übergänge mit der
Retentionszeit des gewünschten Analyten verknüpft, so dass sich mit dieser Methode
praktisch unendlich viele Analyten mit einer chromatographischen Injektion
detektieren lassen (scheduled MRM, sMRM).
38

Grundlagen
Abbildung 16: Prinzip des multiple reaction monitoring (MRM).[15]
2.2.4.2 Quadrupol-Ionenfalle[16]
Als ein weiterer Massenanalysator kann die Ionenfalle genutzt werden. Diese besteht
aus einer Ringelektrode und zwei Endkappen, an die Wechselspannungen angelegt
wird und zentriert Ein- und Austrittslöcher für die Ionen eingelassen sind (Abbildung
17). Bildlich lässt sich eine Ionenfalle als einen zum Kreis gebogenen Quadrupolstab
vorstellen, dessen freie Seiten durch zwei gegenüberliegende Stäbe des Quadrupols
begrenzt sind. Für die Entwicklung der Ionenfalle wurde an Wolfgang Paul und Hans
Georg Dehmelt 1989 der Nobelpreis für Physik verliehen.
Abbildung 17: Prinzipieller Aufbau eines Ionenfallenmassenspektrometers.[15]
Das Funktionsprinzip der Ionenfalle ist vergleichbar mit dem eines Quadrupols. Die
numerischen Lösungen der Mathieuschen Differentialgleichungen definieren auch
hier den Bereich, in dem die Ionen stabile Bahnen innerhalb der Falle durchlaufen
39
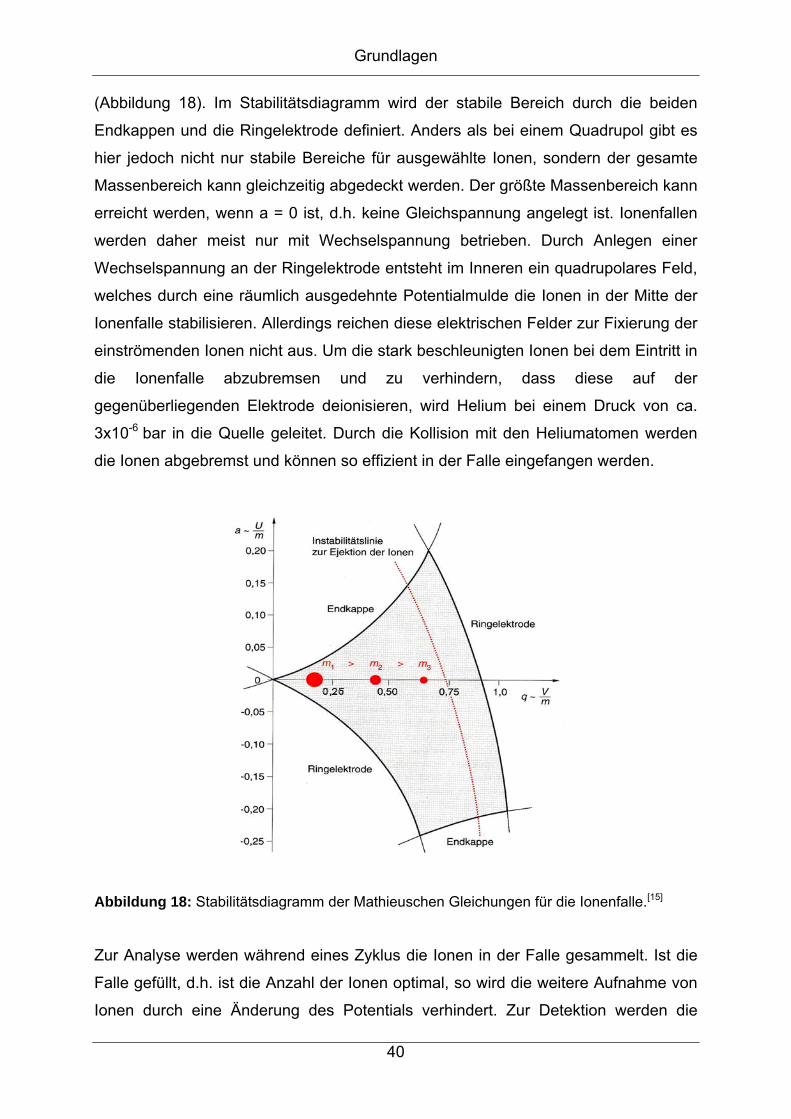
Grundlagen
(Abbildung 18). Im Stabilitätsdiagramm wird der stabile Bereich durch die beiden
Endkappen und die Ringelektrode definiert. Anders als bei einem Quadrupol gibt es
hier jedoch nicht nur stabile Bereiche für ausgewählte Ionen, sondern der gesamte
Massenbereich kann gleichzeitig abgedeckt werden. Der größte Massenbereich kann
erreicht werden, wenn a = 0 ist, d.h. keine Gleichspannung angelegt ist. Ionenfallen
werden daher meist nur mit Wechselspannung betrieben. Durch Anlegen einer
Wechselspannung an der Ringelektrode entsteht im Inneren ein quadrupolares Feld,
welches durch eine räumlich ausgedehnte Potentialmulde die Ionen in der Mitte der
Ionenfalle stabilisieren. Allerdings reichen diese elektrischen Felder zur Fixierung der
einströmenden Ionen nicht aus. Um die stark beschleunigten Ionen bei dem Eintritt in
die Ionenfalle abzubremsen und zu verhindern, dass diese auf der
gegenüberliegenden Elektrode deionisieren, wird Helium bei einem Druck von ca.
3x10-6 bar in die Quelle geleitet. Durch die Kollision mit den Heliumatomen werden
die Ionen abgebremst und können so effizient in der Falle eingefangen werden.
Abbildung 18: Stabilitätsdiagramm der Mathieuschen Gleichungen für die Ionenfalle.[15]
Zur Analyse werden während eines Zyklus die Ionen in der Falle gesammelt. Ist die
Falle gefüllt, d.h. ist die Anzahl der Ionen optimal, so wird die weitere Aufnahme von
Ionen durch eine Änderung des Potentials verhindert. Zur Detektion werden die
40

Grundlagen
41
Ionen mit aufsteigender Molekülmasse mit Hilfe von Multipolfeldern aus der
Austrittsöffnung zu einem Detektor (z.B. Sekundärelektronenvervielfältiger, SEV) hin
herausgeschleust. Die Multipolfelder (z.B. Okta-, Dekapole usw.) entstehen durch die
Kopplung des quadrupolaren Feldes an der Ringelektrode mit einem dipolaren Feld
an den speziell geformten Endkappen und induzieren mit dem Anstieg von q eine
starke Resonanz der Ionen. Dadurch können die Ionen rasch kinetische Energie
aufnehmen und schnell aus der Falle hinaus beschleunigt werden.
In modernen Ionenfallenanalysatoren mit dieser Technik können so
Scangeschwindigkeiten von bis zu 26 000 unit/s erreicht werden, 20mal schneller als
bei einem Quadrupolsystem. Des Weiteren kann das zum Abbremsen eingefüllte
Heliumgas auch als Kollisionsgas verwendet werden. Unter Nutzung der angelegten
Hochfrequenzspannung können die gewünschten Vorläufer-Ionen innerhalb der Falle
isoliert werden.
2.2.4.3 Orbitrap-Ionenfalle[16]
Die Orbitrap besteht aus einer massiven, spindelförmigen Elektrode in der Mitte und
einer ebenfalls spindelförmigen Elektrode als Umhüllung (Abbildung 19).[20] Durch
das Anlegen eines den Ionen entgegengesetzten Potentials an der inneren Elektrode
halten diese sich auf stabilen Kreisbahnen (Orbits), wenn sich Zentrifugalkraft und
Anziehungskraft gerade aufheben. Aufgrund der dezentralen Injektion der Ionen in
die Kammer oszillieren diese relativ zu ihrer Molekülmasse entlang der
Spindelelektrode. Die Frequenzen des harmonischen Oszillators sind unabhängig
von der Ionengeschwindigkeit und induzieren einen Strom, der wiederum relativ zu
ihrer Molekülmasse ist. Durch eine Fouriertransformation kann aus den überlagerten
Frequenzen aller Ionen in der Orbitrap ein Massenspektrum generiert werden.

Grundlagen
Abbildung 19: Schematischer Aufbau eines Orbitrap-Massenspektrometers.[21]
Aufgrund des Wegfalls der räumlichen Trennung von Massenselektion und –
Detektion sind diese Geräte - ähnlich wie FT-ICR-Massenspektrometer - äußerst
empfindlich und besitzen zudem eine sehr hohe Massenauflösung.[22]
2.2.5 Detektion
Die Detektion von Ionen erfolgt über die Umwandlung des Ionenstroms in einen
elektrischen Strom. Hierfür stehen verschiedene Detektoren wie Photomultiplier,
Sekundärelektronenvervielfacher oder ein Faraday-Becher zur Verfügung. Bei den
räumlich nicht getrennten Fouriertransformationsdetektoren wird der durch die Ionen
induzierte Strom gemessen und anschließend zu einem Massenspektrum
prozessiert.
2.3 Kernresonanz-Spektroskopie
Die Kernresonanzspektroskopie beruht auf dem Phänomen, dass Atomkerne in
einem homogenen Magnetfeld eine Aufspaltung ihrer Energieniveaus erfahren. Im
Jahre 1924 wurde von Wolfgang Ernst Pauli zum ersten Mal die theoretische
Grundlage für die NMR-Spektroskopie postuliert, nachdem manche Atomkerne einen
ungeraden Spin haben und somit eine Aufspaltung im magnetischen Feld erfahren
sollten.[23] Experimentell konnte die These 1933 durch das Molekularstrahlexperiment
42

Grundlagen
43
von Otto Stern nachgewiesen werden, bei dem ein Silberatom- bzw. ein
Protonenstrahl durch ein Magnetfeld in zwei Gruppen – entsprechend ihres
Spinzustandes - geteilt wird.[24] Diese Arbeit wurde 1943 mit dem Nobelpreis für
Physik ausgezeichnet. Im Jahre 1946 gelang es den beiden Arbeitsgruppen von
Bloch und Purcell unabhängig voneinander, diese Aufspaltung im Magnetfeld durch
die Energieabsorption elektromagnetischer Strahlung nachzuweisen. Für die
Entdeckung erhielten Bloch und Purcell 1952 gemeinsam den Nobelpreis für Physik.
Das von einem Atomkern erfahrene Magnetfeld ist allerdings nicht nur vom dem
angelegten homogenen Magnetfeld abhängig, sondern unterscheidet sich auch
durch die unterschiedliche chemische Anordnung eines jeden Kerns im Molekül.[25]
Dadurch lassen sich von einem Spektrum Rückschlüsse auf die Molekülstruktur der
Probe ziehen, was sich Chemiker in der Strukturbestimmung zu Nutze machen
können. 1953 wurde daher das erste kommerzielle Kernspinspektrometer der Firma
Varian Associates auf den Markt gebracht. Durch die Entwicklung von immer
stärkeren supraleitenden Magneten und die Entwicklung der
Fouriertransformationsspektrometer (FT-NMR) durch Richard Robert Ernst 1964
konnte die Empfindlichkeit stark gesteigert werden, was zu einem zusätzlichen
Bedeutungsgewinn für die Kernresonanzspektroskopie in der Strukturaufklärung
führte.[26] Zum Anfang der Technik waren die Experimente auf eindimensionale
Spektren beschränkt. Durch den steigenden Einsatz dieser Methode zur
Strukturbestimmung in den 1970er Jahren wurden zweidimensionale Experimente
entwickelt, was zu einer noch größeren Bedeutung in der analytischen Chemie
führte. Aufgrund dieser technischen Weiterentwicklungen kann heutzutage mit der
Kernresonanzspektroskopie neben der Strukturaufklärung beispielsweise die Faltung
von Proteinen gemessen werden.[27]
2.3.1 Experimentelle Grundlagen
Das einfachste aller NMR-Experimente ist das eindimensionale (1D-)Experiment,
welches sich aus den beiden Phasen Präparation und Detektion zusammensetzt
(Abbildung 20, a). Während der Präparation wird durch elektromagnetische
Manipulation das Spinsystem des Analyten in einen definierten Zustand gebracht,
anschließend misst man bei der Detektion das Ergebnis des Experiments.

Grundlagen
a) b)
Abbildung 20: a) Schematische Darstellung einer eindimensionalen NMR-Pulssequenz, b)
Wirkung eines 90°-Pulses auf z-Magnetisierung.[15]
Die Präparation des Spinsystems besteht im einfachsten Fall aus einem kurzen,
starken Anregungsimpuls auf die Gleichgewichtsmagnetisierung Mz aus der x-
Richtung. Bei geeigneter Pulsdauer verändert sich die Magnetisierung entlang der z-
Achse vollständig zur y-Achse hin (Abbildung 20, b). Nach diesem so genannten 90°-
Puls präzedieren die Kerne des Spinsystems mit ihren Lamorfrequenzen um die z-
Achse und induzieren in dem Empfangsgerät eine Spannung. Aufgrund der
transversalen Relaxation nimmt diese Spannung mit der Zeit ab, weshalb das
aufgezeichnete Signal als FID (free induction decay, freier Induktionszerfall)
bezeichnet wird. Nach der vollständigen Relaxation des Spinsystems in seinen
Grundzustand kann das Experiment beliebig oft wiederholt und das Signal addiert
werden, um die Empfindlichkeit des Gerätes zu verbessern. Anschließend wird aus
dem FID (Zeitdomäne) mittels Fourier-Transformation das aufgenommene Spektrum
(Frequenzdomäne) erzeugt.
Aufgrund der häufig auftretenden Signalüberlagerung reichen 1D-Experimente zur
Strukturaufklärung allein jedoch nicht aus. Daher wird zusätzlich die Technik der
zweidimensionalen NMR-Experimente genutzt, bei der zu den beiden
Experimentphasen Präparation und Detektion noch die indirekte Evolutionszeit t1 und
die Mischzeit hinzugefügt werden (Abbildung 21).
44

Grundlagen
45
Abbildung 21: Schematische Darstellung der Pulssequenz eines 2D-NMR Experiments.[15]
Während der Evolutionszeit t1 können die Spins frei präzedieren. Hierbei wird
gleichzeitig die Magnetisierung mit der chemischen Verschiebung des ersten Kernes
verbunden. Anschließend wird durch die Mischsequenz zunächst der Zustand der
Magnetisierung am Ende von t1 abgefragt und weiterhin die Magnetisierung vom
ersten Kern auf einen anderen Kern übertragen. Der Transfer der Magnetisierung
erfolgt über zwei verschiedene Mechanismen: die skalare Kopplung oder die dipolare
Wechselwirkung. Im Anschluss erfolgt dann die Datenakquisition, bei der die
Magnetisierung mit der chemischen Verschiebung des zweiten Kerns markiert wird.
Nach Fouriertransformation in t2-Richtung erhält man ein vom 1D-NMR-Experiment
bekanntes Bild, das eine Momentaufnahme bei der Zeit t1 darstellt. Nun werden
verschiedene Einzelexperimente mit unterschiedlicher t1-Zeit aufgenommen, so dass
die zeitliche Evolution des Spinsystems in einem Experiment abgebildet werden kann
(Abbildung 22, a). Durch eine weitere Fouriertransformation in t1-Richtung entsteht so
ein zweidimensionales Höhenkonturdiagramm. (Abbildung 22, b)

Grundlagen
b)
a)
Abbildung 22: a) Zwischen den aufeinanderfolgenden 1D-Experimenten eines 2D-
Experiments wird jeweils die t1-Zeit inkrementiert. Dadurch wird die indirekte Zeitdomäne
schrittweise abgetastet.[15] b) Nach Fouriertransformation in t2 entsteht eine Serie
eindimensionaler Spektren, die in t1 moduliert sind.[15]
Je nachdem, ob es sich hierbei um eine Kopplung zwischen gleichen
(homonuklearen) oder ungleichen (heteronuklearen) Kernen handelt, erhält man ein
zur Diagonale symmetrisches oder unsymmetrisches 2D-Spektrum.
46

Grundlagen
b)
a)
Abbildung 23: a) Schnitt durch die Daten aus Abbildung 22 parallel zu t1 durch die
jeweiligen Maxima der Signale. Dies entspricht einem free induction decay (FID) in der
indirekten Zeitdimension[15]. b) Nach Fouriertransformation auch der indirekten Dimension (t1)
entsteht eine zweidimensionale Absorptionslinie, links in dreidimensionaler Darstellung,
rechts in Aufsicht in der Darstellung als Konturplot mit Höhenlinien.[15]
Aufgrund der großen Bedeutung der verschiedenen 2D-NMR-Experimente für die
Strukturaufklärung sollen die wichtigsten ausführlich vorgestellt werden.
2.3.1.1 Das 1H,1H-COSY-Experiment
Das zweidimensionale homonukleare (1H,1H)-korrelierte NMR-Experiment liefert
NMR-Spektren, bei denen auf beiden Frequenzachsen 1H-chemische
Verschiebungen miteinander korreliert sind.[28] Hierbei erfolgt der
Magnetisierungstransfer über skalare Spin-Spin-Wechselwirkungen. Im COSY-
Experiment erhält man Information über 1H-1H-Kopplungen, d.h. über
Nachbarschaftsbeziehungen von Protonen. Allerdings sind bei diesem Experiment
nur Signale von Protonen sichtbar, die maximal drei Bindungen voneinander getrennt
sind (2J- und 3J-Kopplung). Bei dem einfachsten aller 2D-NMR-Experimente besteht
47

Grundlagen
die Pulssequenz nur aus zwei durch eine Evolutionszeit t1 getrennten 90°-Pulsen
(Abbildung 24).
Abbildung 24: Schematische Darstellung der Pulssequenz für das 1H,1H-COSY-
Experiment.[29]
Da es sich bei dem COSY-Experiment um ein homonukleares NMR-Experiment
handelt, zieht sich eine Diagonale durch das Spektrum, welche ein eindimensionales 1H-Experiment abbildet. Zusätzlich gibt es so genannte Kreuzsignale beidseitig der
Diagonalen, welche die Information aus dem COSY-Spektrum widerspiegeln. Für ein
AX-System (zwei Kerne mit großer unterschiedlicher chemischer Verschiebung)
können daher insgesamt 4 Signale beobachtet werden: die zwei Signale auf der
Diagonalen mit den Koordinaten A, A bzw. X, X und die Kreuzsignale beidseitig
der Diagonalen mit den Koordinaten A, X und X, A.
Aufgrund der enormen Bedeutung des COSY-Experiments für die Strukturaufklärung
sind eine Reihe von weiteren, ähnlichen Pulssequenzen entwickelt worden. Ein
Nachteil des COSY-Experiments ist, dass bei geringer chemischer Verschiebung der
Signale zueinander die interessanten Kreuzsignale durch die intensiveren
Diagonalsignale überlagert werden. Bei Verbindungen mit vielen, sich überlagernden
Signalen wird daher häufig das COSY-45-Experiment angewendet; hierbei wird der
zweite 90°-Puls durch einen 45°-Puls ersetzt (Abbildung 25, a). Die Magnetisierung
wird dabei so transferiert, dass die ungewünschten Diagonalsignale im Vergleich zu
den Kreuzsignalen abgeschwächt werden. Allerdings geht dieser Vorteil auch mit
einem Empfindlichkeitsverlust einher.
Bei dem long-range COSY-Experiment wird vor und nach dem zweiten 90°-Puls eine
definierte Wartezeit ∆ eingefügt. Während dieser Wartezeit können sich auch
schwache Kopplungen zwischen weit entfernten Protonen entwickeln, die im
eindimensionalen 1H-NMR-Experiment keine skalare Kopplung zueinander zeigen
(Abbildung 25, b).
48

Grundlagen
Zur Unterdrückung störender Sigulettsignale im Spektrum, z.B. des
Lösungsmittelsignals, wird häufig das DQF-COSY-Experiment (double quantum
filtered) angewendet. Hierzu wird nach dem zweiten 90°-Puls eine kurze Wartezeit ∆‘
und ein weiterer 90°-Puls eingefügt. Dadurch werden die
Magnetisierungsbestandteile von Singulettsignalen unterdrückt, während die Signale
von Spinsystemen detektiert werden können (Abbildung 25, c).
c)
b)
a)
Abbildung 25: Schematische Darstellung der Pulssequenz für das a) COSY-45-Experiment
b) Long-Range-COSY-Experiment c) DQF-COSY-Experiment[15]
2.3.1.2 Das TOCSY-Experiment
Im TOCSY-Experiment (Total Correlation Spectroscopy, vollständige Korrelations-
spektroskopie) wird die Magnetisierung durch mehrstufigen sukzessiven Transfer
über skalare J-Kopplung über das ganze Spinsystem verteilt, d.h. das TOCSY
korreliert alle Protonen eines Spinsystems miteinander.[28] Man erhält folglich
Informationen über die gesamten Spinsysteme eines Moleküls. Das TOCSY-
Experiment wird häufig bei der Strukturaufklärung von Peptiden bzw. Proteinen
eingesetzt, da beim TOCSY-Experiment charakteristische Spektrenmuster
entstehen, welche sich gut den enthaltenen Aminosäuren zuordnen lassen.
Die TOCSY-Pulssequenz ist vergleichbar mit der des COSY-Experiments, allerdings
wird hier der zweite 90°-Puls durch ein Spin-Lock ersetzt. Während der Dauer des
Spin-Locks befinden sich die Kerne nur im schwachen Hochfrequenzfeld B1, die
chemischen Verschiebungsdifferenzen der Protonen werden sehr klein und die
49

Grundlagen
50
skalaren Kopplungen überwiegen. Nun können sich die Spinzustände mischen,
Magnetisierung kann von einem zum anderen Kern übertragen werden und dieser
Transfer als Signal detektiert werden. Die Reichweite der
Magnetisierungsübertragung innerhalb eines Moleküls hängt direkt von der Dauer
des Spin-Locks ab. Durch die Verkürzung oder Verlängerung des Spin-Locks lässt
sich selektiv ein bestimmter Abschnitt eines Spinsystems anregen, wodurch die
Strukturaufklärung von Molekülen mit vielen, überlagerten Protonen ermöglicht wird.
Üblicherweise variiert die Dauer des Spin-Locks von wenigen 10 ms bis zu 300 ms.
Die eindimensionale Variante des selektiven TOCSY wird aufgrund der gezielten
Anregung von Signalen auch selTOCSY (selective Total Correlation Spectroscopy,
selektive vollständige Korrelationsspektroskopie) genannt.
2.3.1.3 Das HMQC/HSQC-Experiment
Das HMQC-Experiment[30] (Heteronuclear Multiple Quantum Coherence) verwendet
die Übertragung der Magnetisierung auf einen Heterokern und die Rückübertragung
auf den Ursprungskern.[28] Es handelt sich um ein inverses, zweidimensionales,
heteronukleares H,X-korreliertes NMR-Experiment (X = 13C, 15N). Charakteristisch für
alle inversen Verfahren ist, dass Kohärenzen im Kanal der unempfindlichen Kerne
(13C, 15N) erzeugt und dann auf die empfindlichen Kerne (im allgemeinen 1H)
übertragen werden, deren Resonanzen dann gemessen werden. Sehr ähnlich zu
dem HMQC-Experiment ist das HSQC-Experiment (Heteronuclear Single Quantum
Coherence,
Abbildung 26). Im Gegensatz zum HMQC-Experiment, bei der sich die
Magnetisierung des Heterokerns und des Protons während der Evolutionszeit
ändern, ändert sich bei dem HSQC-Experiment nur die Magnetisierung des
Heterokerns. Bei beiden Experimenten erhält man als Information die Korrelation
zwischen direkt gebundenen Kohlenstoff- und Wasserstoffatomen (1J-Kopplungen).

Grundlagen
2.3.1.4 Das HMBC-Experiment
Das HMBC-Experiment[31] (Heteronuclear Multiple Bond Correlation) unterscheidet
sich von dem HSQC-Experiment lediglich durch eine größere Wartezeit zwischen
dem ersten Protonenpuls und dem ersten Kohlenstoffpuls für die Entwicklung der
heteronuklearen Kopplung.[28] Durch die Verlängerung der Wartezeit können sich
während dieser Zeit Fernkopplungen über zwei bzw. drei Bindungen entwickeln.
Diese Korrelation von entfernt liegenden Atomen ist gerade für die Strukturaufklärung
von Naturstoffen von großer Bedeutung, da so die verschiedenen Strukturfragmente
zu einem Strukturvorschlag zusammengeführt werden können. Des Weiteren kann
mithilfe von HMBC-Experimenten auch die Lage von Heteroatomen bestimmt
werden, sofern das Molekül genug Protonen für die inverse Detektion enthält. Um
möglichst alle in einem Molekül vorhandenen Fernkopplungskonstanten (ca. 1-25 Hz)
abzudecken, wird meist ein mittlerer Wert von 60 ms (ca. 8 Hz) gewählt. Des
Weiteren verwendet man häufig einen so genannten low pass filter, um die
störenden 1JC-H-Kopplungen zu unterdrücken.[32]
Abbildung 26: Schematische Darstellung der Pulssequenz für das HSQC- bzw. HMBC-
Experiment, hier: 15N-HSQC.[15]
51

Zielsetzung
52
3 Zielsetzung
Das Ziel der vorliegenden Arbeit war die Strukturaufklärung von
Sekundärmetaboliten durch massenspektrometrische und
kernresonanzspektroskopische Analysen. Gesammelte Bakterienstämme aus
verschiedenen Habitaten wurden zuvor in der Arbeitsgruppe von Prof. H.-P. Fiedler
an der Eberhard-Karls-Universität Tübingen mittels der HPLC-DAD-Analytik
charakterisiert. Anhand der arbeitskreisinternen UV/Vis-Datenbank konnten die zu
identifizierenden Verbindungen gegen eine Vielzahl von bekannten Naturstoffen
verglichen werden.[33] Hierbei wurden zahlreiche neue, nicht in der Datenbank
enthaltene Verbindungen entdeckt, welche als uncharakterisierte Naturstoffe
klassifiziert wurden. Nach der Isolierung dieser Substanzen in Reinform sollten diese
im Folgenden strukturaufgeklärt werden, um Information über die Wirkstofffamilie und
die chemische Struktur dieser neuen Stoffe zu erhalten. Ausgehend von der
vorliegenden Struktur sollten mit Hilfe von Kooperationspartnern mit den
aufgereinigten Stoffen verschiedene antibakterielle, antitumor- und enzymatische
Testierungen durchgeführt werden, um Informationen über die Wirksamkeit zu
erlangen.

Ergebnisse und Diskussion
4 Strukturaufklärung des Piceamycins und seines N-
Acetylcystein-Addukts aus Streptomyces sp. GB 4-2
4.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces
sp. GB 4-2
Zur Untersuchung von rhizosphären Bodenbakterien wurden in der Arbeitsgruppe
von Prof. Karl Poralla an der Eberhard-Karls-Universität Tübingen Erdproben von der
Rhizosphäre junger Fichten (Schönbuch, Tübingen) genommen. Als Rhizosphäre
wird im Allgemeinen der Bereich um eine Pflanzenwurzel im Waldboden bezeichnet.
Aufgrund der starken Interaktion zwischen der Pflanze und des umliegenden
Waldbodens kommt es in diesem Bereich zu einer starken Häufung von
Mikroorganismen, die sowohl pflanzenfördernder (auxiliar, symbiotisch) als auch
pflanzenschädlicher (parasitär, pathogen) Natur sein können.[34, 35] Die hohe
Organismendichte in diesem Lebensraum zwingt die Mikroorganismen verstärkt zur
Bildung von Sekundärmetaboliten, um potentielle Konkurrenten zu bekämpfen und
sich selbst gegen andere Arten zu behaupten. Aus diesen Erdproben wurde der
Stamm Streptomyces sp. GB 4-2 isoliert und in der Arbeitsgruppe von Prof. Hans-
Peter Fiedler von Dr. Julia Riedlinger[36] taxonomisch und von Dr. Dirk Schulz[1]
bezüglich seiner Sekundärmetabolitproduktion untersucht (Abbildung 27).
Abbildung 27: Lichtmikroskopische Aufnahmen von Stamm Streptomyces sp. GB 4-2
(Kolonien auf der Agarplatte).[1]
53

Ergebnisse und Diskussion
54
4.1.1 Chemisches Screening
Zur Identifizierung von möglichen Zielkomponenten wurde in der Arbeitsgruppe von
Prof. Hans-Peter Fiedler der Bakterienstamm in unterschiedlichen Nährmedien
angezogen und die Produktion der Sekundärmetabolite in Myzel- und Kulturfiltrat
mittels HPLC-DAD-Analytik vermessen. Während des chemischen Screenings
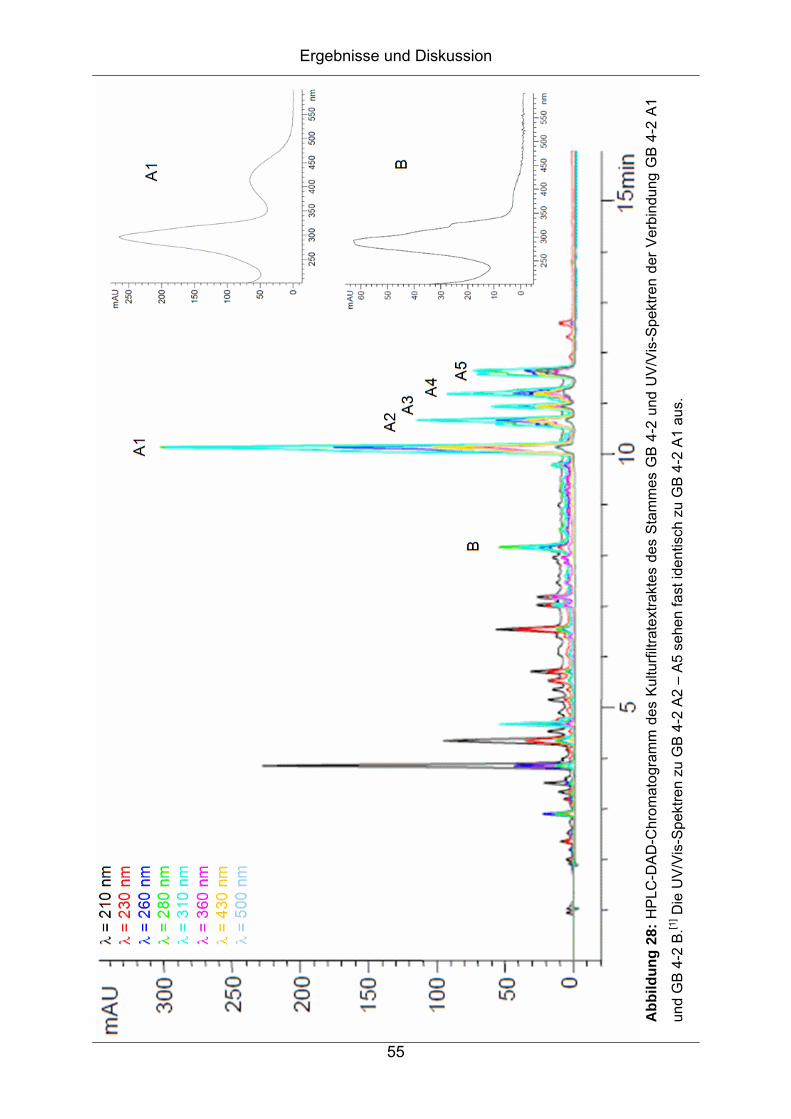
konnten insgesamt sechs Verbindungen charakterisiert werden, deren UV/Vis-
Spektren keine Übereinstimmung mit bekannten Sekundärmetaboliten in der
Datenbank zeigte.[33] Aufgrund der identischen UV/Vis-Spektren von fünf der sechs
Verbindungen handelte es sich hierbei jedoch höchstwahrscheinlich um
Stereoisomere. Diese wurden daraufhin als GB 4-2 A1 bis A5 bezeichnet, die
sechste Verbindung als GB 4-2 B (Abbildung 28). Während der Bearbeitung des
Stammes konnte beobachtet werden, dass die Verbindungen A2 bis A5 durch
Lichteinwirkung aus der Verbindung A1 entstehen. Daher wurden nur die
Komponente A1 mit der Retentionszeit von Rt = 10.1 min und die Komponente B mit
der Retentionszeit von Rt = 8.2 min als Zielkomponenten identifiziert.
Ergebnisse und Diskussion
55
Ab
bild
un
g 2
8: H
PLC
-DA
D-C
hro
mat
ogra
mm
de
s K
ultu
rfilt
rate
xtra
ktes
des
Sta
mm
es
GB
4-2
un
d U
V/V
is-S
pek
tren
der
Ver
bind
ung
GB
4-2
A1
und
GB
4-2
B.[1
] Die
UV
/Vis
-Spe
ktre
n zu
GB
4-2
A2
– A
5 se
hen
fast
iden
tisch
zu
GB
4-2
A1
aus.

Ergebnisse und Diskussion
4.1.2 Fermentation und Isolierung
Bei der von Dr. Dirk Schulz durchgeführten Produktionsoptimierung mit den drei
unterschiedlichen Nährmedien 500, SGG und OM im Schüttelkolben zeigte sich,
dass die gewünschten Metabolite in guten Ausbeuten in dem Nährmedium OM
gebildet wurden.[1] Die maximale Konzentration des Metaboliten GB 4-2 B konnte
nach 166 h mit einer Ausbeute von 2.8 mg/L erreicht werden. Zur anschließenden
Anzucht des Stammes im Bioreaktor wurden weiterhin fünf verschiedene
Bioreaktoren (New Brunswick, Biostat S, Biostat E, Biostat b20) mit dem Stamm
angeimpft und der Reaktor mit der höchsten Ausbeute bestimmt. Hierbei erwies sich
der Bioreaktor New Brunswick mit einer Ausbeute von 3.9 mg/L als bester Bioreaktor
zur Fermentation. Die Bioreaktoren Biostat S und Biostat E zeigten eine
vernachlässigbar geringere Produktion der gewünschten Verbindungen an, so dass
diese ebenfalls zur Fermentation des Stammes genutzt werden konnten.
b) a)
Schema 1: Isolierungsschema der Substanzen a) GB 4-2 A1 und b) GB 4-2 B.[1]
56

Ergebnisse und Diskussion
57
Die Isolierung der Verbindung GB 4-2 A1 erfolgte analog zu Schema 1. Nach der
Ernte wurde die Kulturbrühe zunächst mittels Druckfiltration in Kulturfiltrat und Myzel
getrennt und das Myzel verworfen. Anschließend wurde das Kulturfiltrat zunächst auf
pH 4 eingestellt und dreimal mit Ethylacetat extrahiert. Die vereinigten organischen
Phasen wurden unter Vakuum bis zur Trockne eingeengt und der erhaltene
Rohextrakt mit einem Stufengradienten von 0 %, 1 %, 2 % und 5 % an LiChroprep
Diol chromatographiert. Die Elutionsfraktionen wurden nun wiederum bis zur Trockne
eingeengt und anschließend ein 200 mg Aliquot an Sephadex LH-20 in Methanol
aufgetrennt. Der dadurch erhaltene Rohextrakt wurde schließlich mittels präparativer
RP-HPLC an C18-Material mit einem isokratischen Gradienten von 52 % Acetonitril
aufgereinigt, wodurch 5 mg GB 4-2 A1 erhalten werden konnten. Aufgrund der
starken Isomerisierung der Verbindung GB 4-2 A1 zu den isomeren Verbindungen
A2 bis A5 bei Lichteinwirkung musste der gesamte Prozess der Isolierung möglichst
unter Lichtausschluss erfolgen.
Zur Isolierung der Verbindung GB 4-2 B wurde nach der Filtration eine
Adsorptionschromatographie an Amberlite XAD-16 durchgeführt und die
Elutionsfraktion anschließend bis zum wässrigen Rückstand unter reduziertem Druck
eingeengt. Die wässrige Phase wurde nun auf pH 4 eingestellt und dreimal mit
Ethylacetat extrahiert. Hierbei ging die Verbindung in die organische Phase über, das
Lösungsmittel wurde anschließend unter reduziertem Druck entfernt. Der erhaltene
Rohextrakt wurde nun in Methanol aufgenommen und an Sephadex LH-20
chromatographiert. Der nach der Einengung der Elutionsfraktionen erhaltene
Rohextrakt II wurde anschließend in Methanol an Fractogel TSK HW-40
chromatographiert, wodurch 23 mg Rohprodukt erhalten werden konnten. Dieses
wurde dann in einem letzten Aufreinigungsschritt mittels präparativer RP-HPLC an
C18-Material mit einem linearen Gradienten von 30 - 60 % Acetonitril mit 0.1 %
Ameisensäure aufgereinigt. Hierdurch konnten 5.7 mg reines GB 4-2 B erhalten
werden.

Ergebnisse und Diskussion
58
4.2 Strukturaufklärung
4.2.1 HPLC-ESI-MS
Mit Hilfe von HPLC-ESI-MS-Untersuchungen konnten zunächst die nominalen
Molekülmassen der beiden Zielderivate bestimmt werden. Hierzu wurden Proben der
reinen Verbindungen im positiven und negativen Ionisationsmodus untersucht. In
beiden Modi war die Ionenausbeute zufriedenstellend. Daher wurde - sofern beide
Ionisationmodi ein stimmiges Ergebnis ergaben - zur vereinfachten Darstellung nur
der Positivionenmodus beschrieben. Das Derivat GB 4-2 A1 zeigte bei einer
Retentionszeit von Rt = 10.1 min ein Positiv-Molekülion [M+H]+ bei m/z 432.1, das
Derivat GB 4-2 B ein Positiv-Molekülion [M+H]+ von m/z 595.1 bei der Retentionszeit
Rt = 8.2 min. Die UV/Vis-Spektren und die Retentionszeiten stimmten mit den
experimentellen Daten aus dem chemischen Screening überein.
4.2.2 Bestimmung der Summenformel
Zur weiteren Strukturaufklärung wurde nach den HPLC-ESI-MS-Untersuchungen die
Summenformel der beiden Zielderivate GB 4-2 A1 und GB 4-2 B mittels FT-ICR-MS
bestimmt. Die Messung erfolgte durch Dipl.-Ing. Graeme J. Nicholson an der
Universität Tübingen im Positiv-Ionenmodus. Für die Berechnung der Summenformel
wurden die Elemente Kohlenstoff (C), Wasserstoff (H), Stickstoff (N), Sauerstoff (O)
und Schwefel (S) zugelassen. Eine Zusammenfassung der Ergebnisse ist in Tabelle
1 dargestellt.

Ergebnisse und Diskussion
59
Tabelle 1: Physikochemische Eigenschaften von GB 4-2 A1 und GB 4-2 B.
GB 4-2 A1 GB 4-2 B
FT-ICR-MS 432.21697 gemessen [M+H]+
432.21694 theoretisch
∆ = 0.07 ppm
595.24699 gemessen [M+H]+
595.24725 theoretisch
∆ = 0.44 ppm
Summenformel C27H29NO4 C32H38N2O7S
UV max (MeOH) 295, 410 295, 330 (sh)
4.2.3 Aminosäureanalytik
Zum Nachweis von Aminosäure-Einzelbausteinen wurde eine GC-MS-Analytik durch
Dipl.-Ing. Graeme J. Nicholson an der Universität Tübingen durchgeführt. Hierzu
wurde eine Probe beider Zielverbindungen zunächst säurekatalytisch hydrolysiert
(6 M HCl, 110 °C, 20 h), das getrocknete Hydrolysat zum N-(O-)TFA/Ethylester
derivatisiert und anschließend mittels chiraler GC-MS analysiert. Bei dem Derivat
GB 4-2 A1 konnte kein Aminosäurebaustein nachgewiesen werden, GB 4-2 B wies
hingegen die Aminosäure L-Cystein auf. Die Identifizierung erfolgte durch
Retentionszeitvergleich zu L-Cystein und der Übereinstimmung des
charakteristischen EI-Fragmentspektrums von Cystein.
4.2.4 NMR-Spektroskopische Untersuchungen
Die weiterführende Strukturaufklärung der Zielkomponenten wurde mit Hilfe von ein-
und zweidimensionalen kernresonanzspektroskopischen Experimenten durchgeführt.
Zunächst konnten die durch die FT-ICR-MS-Untersuchungen gefundenen
Summenformeln durch die 1H- und 13C-NMR-Spektren bestätigt werden. Die
detaillierte Analyse der COSY-, HSQC- und HMBC-NMR-Spektren ermöglichte die
Bestimmung aller in Abbildung 29 gezeigten 1H- und 13C-Signale. Die 1H-, 13C- und
HSQC-NMR-Spektren der Zielkomponente GB 4-2 A1 (im weiteren Verlauf als
Piceamycin bezeichnet) zeigten insgesamt 21 sp2-hybridisierte, zwei Methylen- und
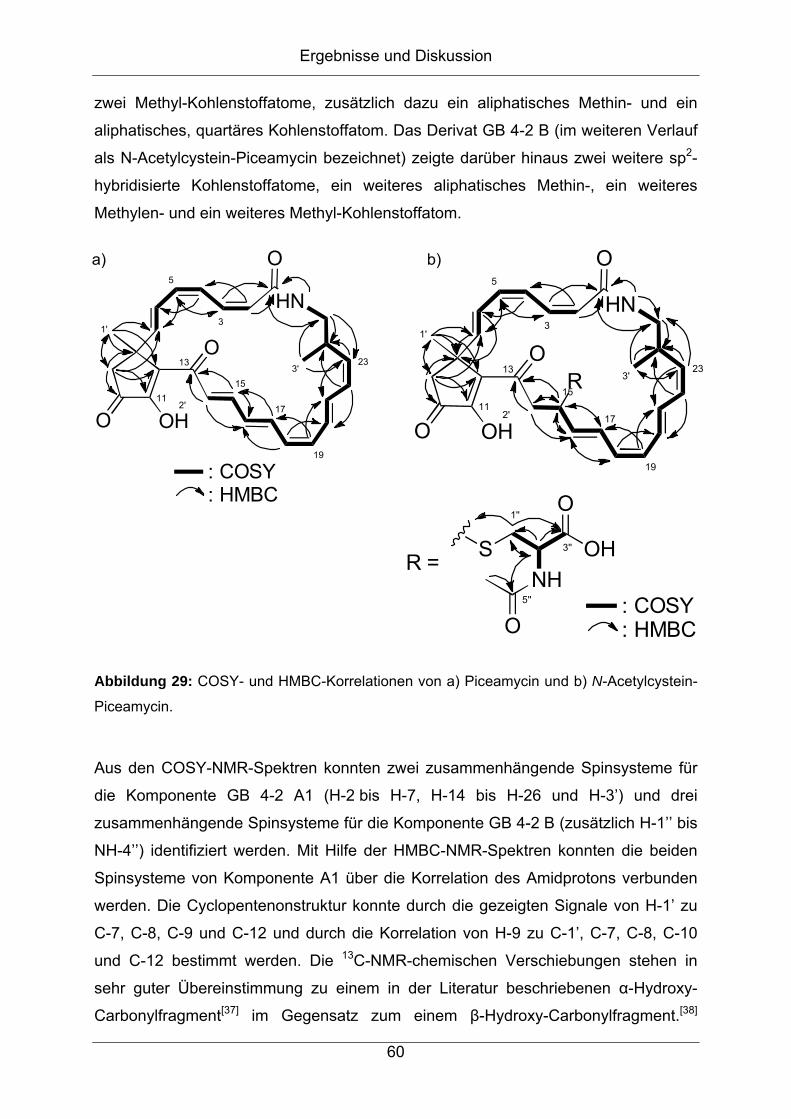
Ergebnisse und Diskussion
zwei Methyl-Kohlenstoffatome, zusätzlich dazu ein aliphatisches Methin- und ein
aliphatisches, quartäres Kohlenstoffatom. Das Derivat GB 4-2 B (im weiteren Verlauf
als N-Acetylcystein-Piceamycin bezeichnet) zeigte darüber hinaus zwei weitere sp2-
hybridisierte Kohlenstoffatome, ein weiteres aliphatisches Methin-, ein weiteres
Methylen- und ein weiteres Methyl-Kohlenstoffatom.
b)
R =S
NH
O
OH
O
HN
O
O
O OH
R
: COSY: HMBC
3
5
11
19
2313
15
17
1'
2'
3'
1''
3''
5''
a)
HN
O
O
O OH
: COSY: HMBC
3
5
11
19
2313
15
17
1'
2'
3'
Abbildung 29: COSY- und HMBC-Korrelationen von a) Piceamycin und b) N-Acetylcystein-
Piceamycin.
Aus den COSY-NMR-Spektren konnten zwei zusammenhängende Spinsysteme für
die Komponente GB 4-2 A1 (H-2 bis H-7, H-14 bis H-26 und H-3’) und drei
zusammenhängende Spinsysteme für die Komponente GB 4-2 B (zusätzlich H-1’’ bis
NH-4’’) identifiziert werden. Mit Hilfe der HMBC-NMR-Spektren konnten die beiden
Spinsysteme von Komponente A1 über die Korrelation des Amidprotons verbunden
werden. Die Cyclopentenonstruktur konnte durch die gezeigten Signale von H-1’ zu
C-7, C-8, C-9 und C-12 und durch die Korrelation von H-9 zu C-1’, C-7, C-8, C-10
und C-12 bestimmt werden. Die 13C-NMR-chemischen Verschiebungen stehen in
sehr guter Übereinstimmung zu einem in der Literatur beschriebenen α-Hydroxy-
Carbonylfragment[37] im Gegensatz zum einem β-Hydroxy-Carbonylfragment.[38]
60
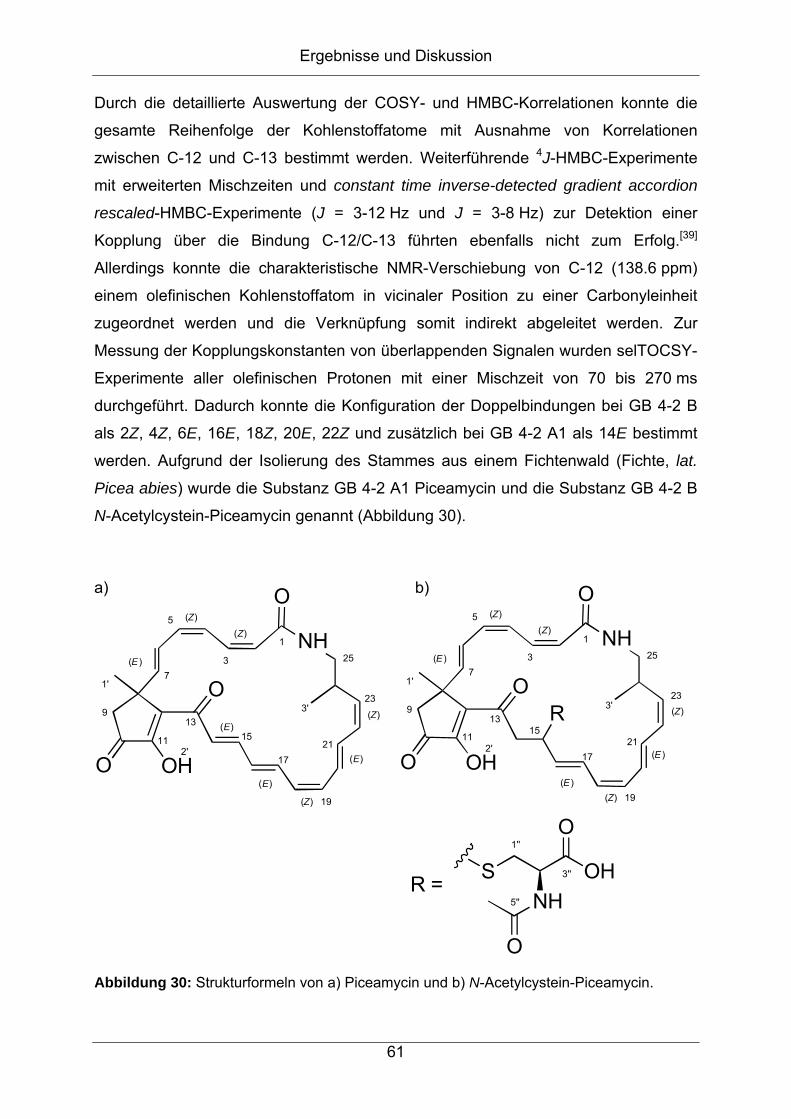
Ergebnisse und Diskussion
Durch die detaillierte Auswertung der COSY- und HMBC-Korrelationen konnte die
gesamte Reihenfolge der Kohlenstoffatome mit Ausnahme von Korrelationen
zwischen C-12 und C-13 bestimmt werden. Weiterführende 4J-HMBC-Experimente
mit erweiterten Mischzeiten und constant time inverse-detected gradient accordion
rescaled-HMBC-Experimente (J = 3-12 Hz und J = 3-8 Hz) zur Detektion einer
Kopplung über die Bindung C-12/C-13 führten ebenfalls nicht zum Erfolg.[39]
Allerdings konnte die charakteristische NMR-Verschiebung von C-12 (138.6 ppm)
einem olefinischen Kohlenstoffatom in vicinaler Position zu einer Carbonyleinheit
zugeordnet werden und die Verknüpfung somit indirekt abgeleitet werden. Zur
Messung der Kopplungskonstanten von überlappenden Signalen wurden selTOCSY-
Experimente aller olefinischen Protonen mit einer Mischzeit von 70 bis 270 ms
durchgeführt. Dadurch konnte die Konfiguration der Doppelbindungen bei GB 4-2 B
als 2Z, 4Z, 6E, 16E, 18Z, 20E, 22Z und zusätzlich bei GB 4-2 A1 als 14E bestimmt
werden. Aufgrund der Isolierung des Stammes aus einem Fichtenwald (Fichte, lat.
Picea abies) wurde die Substanz GB 4-2 A1 Piceamycin und die Substanz GB 4-2 B
N-Acetylcystein-Piceamycin genannt (Abbildung 30).
a) b)
NH
O
O
O OH
1
3
5
71'
9
11
13
15
17
19
21
23
25
3'
2'
(E)
(E)
(E)
(E)
(Z)
(Z)
(Z)
(Z)
R =S
NH
O
OH
O
1''
3''
5''
NH
O
O
O OH
1
3
5
71'
9
11
1315
17
19
21
23
25
3'
2'
(E)
(E)
(E)
(Z)
(Z)
(Z)
(Z)
R
Abbildung 30: Strukturformeln von a) Piceamycin und b) N-Acetylcystein-Piceamycin.
61

Ergebnisse und Diskussion
62
4.3 Biologische Aktivität
Piceamycin und N-Acetylcystein-Piceamycin wurden am Kieler Wirkstoffzentrum
KiWiZ am Institut für Meereswissenschaften IFM-Geomar in der Arbeitsgruppe von
Prof. Johannes F. Imhoff gegen Gram-positive und Gram-negative Bakterien sowie
gegen Eukaryoten und ausgewählte Enzyme getestet. Des Weiteren wurde in
Zusammenarbeit mit Prof. Winfried Beil an der Medizinischen Hochschule Hannover
Piceamycin gegen ausgewählte humane Krebszelllinien getestet. Dabei zeigte
Piceamycin antibakterielle Aktivität gegen die Gram-positiven Bakterien Bacillus
subtilis, Staphylococcus aureus, S. epidermidis, S. lentus und gegen das Gram-
negative Bakterium Xanthomonas campestris (Tabelle 2).
Tabelle 2: Minimale Inhibierungskonzentration von Piceamycin gegen Gram-positive
Bakterien.
Organismus MIC [µg/ml] MIC [µM]
Bacillus subtilis DSM 10 0.43 1.00
Bacillus subtilis DSM 347 0.31 0.72
Staphylococcus aureus DSM 20231 0.14 0.33
Staphylococcus lentus DSM 6672 0.45 1.05
Staphylococcus epidermidis DSM 20044 0.11 0.25
Antifungale Aktivität von Piceamycin konnte gegen die Stämme Saccharomyces
cerevisiae, Candida glabrata und Botrytis cinerea nachgewiesen werden.

Ergebnisse und Diskussion
63
Tabelle 3: Wachstumsinhibierung von Piceamycin gegen ausgewählte humane
Krebszelllinien.
Zelllinie GI50 [µg/ml] TGI [µg/ml]
AGS 0.80 3.4
HepG2 0.30 1.5
MCF 7 0.28 0.7
GI50: 50% Wachstumsinhibierung; TGI: 100% Wachstumsinhibierung
Des Weiteren zeigte das Piceamycin eine cytostatische Aktivität gegen verschiedene
humane Krebszelllinien (Tabelle 3) und eine enzymhemmende Aktivität gegen die
humane Tyrosin Phosphatase 1B (human recombinant protein tyrosine phosphatase
1B, PTP1B) mit einem IC50-Wert von 10.1 µM.[40] Kleine Moleküle zur Inhibierung der
PTP1B sind aufgrund der potentiellen therapeutischen Anwendung gegen Diabetes,
Adipositas und Krebs interessant.
Interessanterweise konnte für das N-Acetylcystein-Piceamycin weder antimikrobielle,
noch cytostatische oder enzymhemmende Aktivität nachgewiesen werden.
4.4 Diskussion
Insgesamt konnten in dem Kulturfiltrat des Stammes Streptomyces sp. GB 4-2 sechs
Verbindungen durch HPLC-DAD-Analysen nachgewiesen werden, welche aufgrund
des negativen Abgleichs mit der UV/Vis-Datenbank als potentiell neue Naturstoffe
angesehen werden konnten.[33] Während der zeitlichen Beobachtung der
Fermentation stellte sich heraus, dass die Verbindungen GB 4-2 A2 – A5 aus dem
Piceamycin entstehen. Da die Verbindungen identische UV/Vis-Spektren zu
Piceamycin zeigen, handelt es sich hierbei höchstwahrscheinlich um Stereoisomere
zu dieser Verbindung, bei denen durch Lichteinfluss einer der cis-Doppelbindungen
des Makrolactamrings in die stabilere trans-Form umgewandelt wird. Vermutlich ist
jedoch nur die Umwandlung von einer Doppelbindung möglich, da für die
Umwandlung von mehreren Doppelbindungen die Ringgröße des Makrolactams nicht
ausreichend ist. Piceamycin enthält insgesamt vier cis-Doppelbindungen, welches

Ergebnisse und Diskussion
auch das Auftreten von vier Stereoisomeren hierzu erklären könnte. Aufgrund der
nicht natürlichen Produktion der vier weiteren Isomere wurden diese nicht zur
Strukturaufklärung aufgereinigt.
Ein Literaturvergleich der aufgeklärten Struktur des Piceamycins mit anderen
makrocyclischen Polyketiden ergab, dass eine strukturelle Verwandtschaft mit dem
Hitachimycin vorliegt (Abbildung 31). Hitachimycin ist ein 19-gliedriges
Makrolactamantibiotika, welches aus dem Kulturüberstand von Streptomyces
scabrisporus isoliert wurde.[41]
HN
O
O OHOHO
24
7
10
14 1620
Abbildung 31: Strukturformel von Hitachimycin aus Streptomyces scabrisporus.[41]
Anstelle einer Methylgruppe an Position C-24 trägt das Hitachimycin jedoch einen
Phenylrest und weist weitere Unterschiede im Makrolidrückgrat auf. Zum Einen ist
ein Proton am C-8 durch eine Methyleinheit im Hitachimycin ersetzt, ebenso die
Carbonyleinheit am C-10 durch eine Methoxygruppe. Zum anderen liegt die
Kohlenstoffkette ab C-14 vollständig reduziert und an C-15 durch eine
Hydroxygruppe substituiert vor. Dieser strukturelle Unterschied führt dazu, dass das
Hitachimycin kein Michaelsystem an Position C-13 bis C-15 besitzt. Weitere
Cysteinaddukte konnten im Kulturüberstand nicht gefunden werden, was dafür
spricht, dass C-15 die am stärksten elektrophile Position des Piceamycins ist. Mit
HPLC-MS untersuchte Mikroderivatisierungsversuche von Piceamycin mit L-Cystein
zeigten, dass sich unter Standardbedingungen ein Cysteinaddukt bildet. Aufgrund
dieser Ergebnisse konnte bewiesen werden, dass das oben genannte Michael-
System für die antibakterielle und cytostatische Aktivität des Piceamycins
verantwortlich ist.
Ähnliche Beobachtungen konnten auch bei den Mycinamicinen gemacht werden.[42]
Hierbei handelt es sich wie das Piceamycin ebenfalls um ein Makrolid, welches in
seinem Kohlenstoffrückgrat ein Michaelsystem enthält (Abbildung 32). Zunächst
64
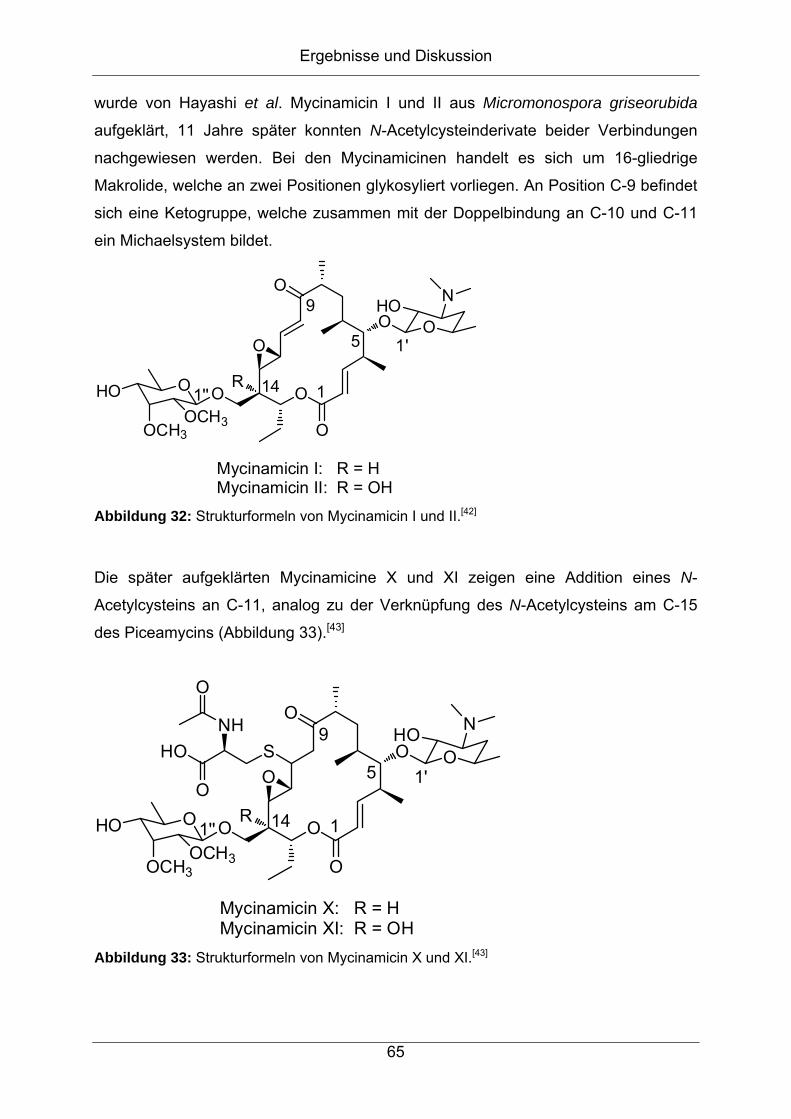
Ergebnisse und Diskussion
wurde von Hayashi et al. Mycinamicin I und II aus Micromonospora griseorubida
aufgeklärt, 11 Jahre später konnten N-Acetylcysteinderivate beider Verbindungen
nachgewiesen werden. Bei den Mycinamicinen handelt es sich um 16-gliedrige
Makrolide, welche an zwei Positionen glykosyliert vorliegen. An Position C-9 befindet
sich eine Ketogruppe, welche zusammen mit der Doppelbindung an C-10 und C-11
ein Michaelsystem bildet.
O
O
O
O
O
O O
OCH3
HO
OCH3
OHO
N
1
5
9
14
1'
1''R
Mycinamicin I: R = HMycinamicin II: R = OH
Abbildung 32: Strukturformeln von Mycinamicin I und II.[42]
Die später aufgeklärten Mycinamicine X und XI zeigen eine Addition eines N-
Acetylcysteins an C-11, analog zu der Verknüpfung des N-Acetylcysteins am C-15
des Piceamycins (Abbildung 33).[43]
O
O
O
O
O
O O
OCH3
HO
OCH3
OHO
N
1
5
9
14
1'
1''
S
NH
HO
O
O
Mycinamicin X: R = HMycinamicin XI: R = OH
R
Abbildung 33: Strukturformeln von Mycinamicin X und XI.[43]
65

Ergebnisse und Diskussion
Auch die Mycinamicine X und XI zeigen gegenüber Gram-positiven Bakterien
antibakterielle Aktivität, allerdings um ca. eine Größenordnung geringer im Vergleich
zu den Mycinamicinen I und II.[43] Der deutliche Unterschied in biologischer Aktivität
deutet auch bei den Mycinamicinen auf biologische Relevanz des Michaelsystems
hin, welches bei den Mycinamicinen X und XI durch die N-Acetylcysteinaddition
blockiert ist.
Ein weiterer Vertreter der N-Acetylcystein-Antibiotika sind die Paldimycine. Deren
Grundgerüst stellen die Paulomycine dar, welche 1982 aus Streptomyces paulus
isoliert worden sind.[44] Diese enthalten eine Isothiocyanat-Gruppe mit einem
elektrophilen Kohlenstoffatom, an das durch einen nukleophilen Angriff das
Acetylcystein angreifen kann und eine Dithiocarbamat-Gruppe bildet. Die so
entstandenen Verbindungen wurden später ebenfalls aus Streptomyces paulus
isoliert und Antibiotic 273a2α genannt.[45] Die Paldimycine entstehen durch eine
erneute Addition eines Acetylcysteins an Antibiotic 273a2α. Hierbei greift das
nukleohile Thiolat-Anion jedoch an der Didehydrobutyrineinheit des Paulomycins an
(Abbildung 34).
O
OH
RHO
O
O
NH2
COOH
O
OCH3
H3C
OH
OO
OO
O
O
N
O
O
HN
SC
S
SCOOH
HN
O
O
O
HN
S
SCOOH
HN
OS
HNHOOC
O
Paulomycin A:
R =
Antibiotic 273a2
R =
Paldimycin A:
R =
Abbildung 34: Strukturformeln von Paulomycin A, Paldimycin A und Antibiotic 273a2α.[45]
Des Weiteren sind mehrere Metabolite beschrieben, bei denen das N-Acetylcystein
mit einem Aromaten verknüpft ist.[46] Als Beispiel ist Abbildung 35 in das Phenazin
SB 212305 gezeigt, bei dem das N-Acetylcystein an das C-3 gebunden ist.
66
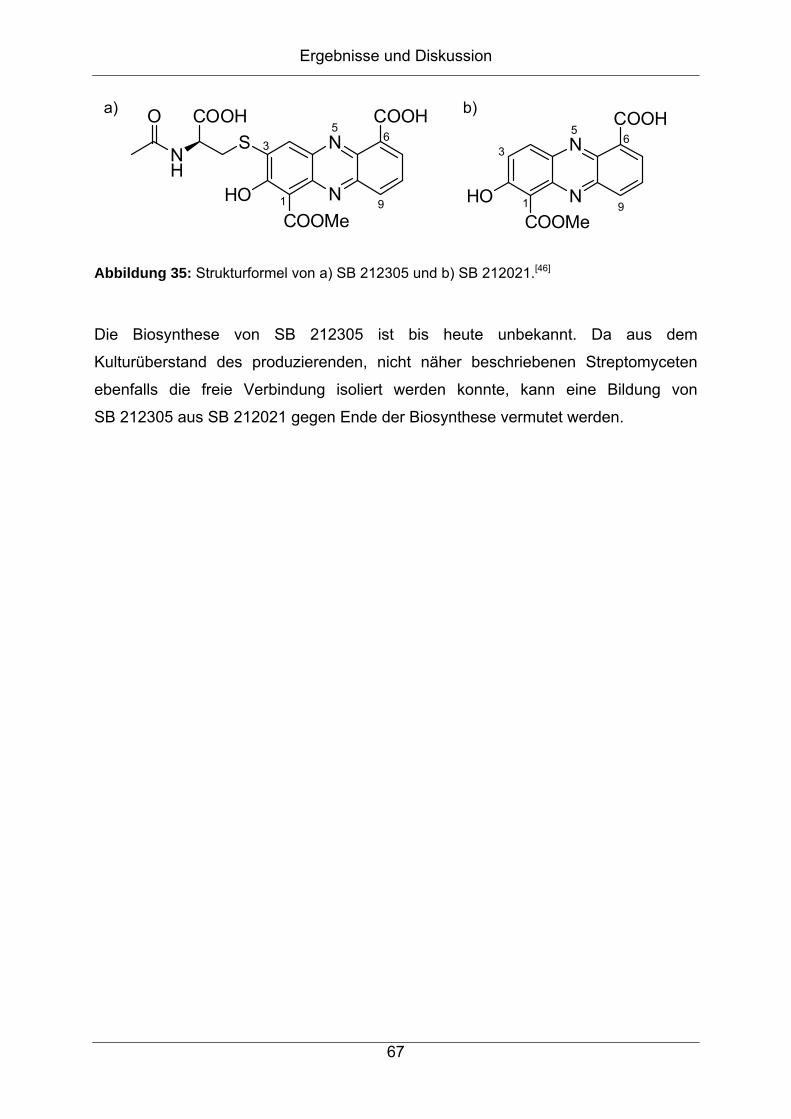
Ergebnisse und Diskussion
67
bbildung 35: Strukturformel von a) SB 212305 und b) SB 212021.[46]
nthese von SB 212305 ist bis heute unbekannt. Da aus dem
tand des produzierenden, nicht näher beschriebenen Streptomyceten
ebenfalls die freie Verbindung isoliert werden konnte, kann eine Bildung von
t t werden.
a) b)
N
N
COOH
COOMe
HO 1
3
56
9N
N
COOH
COOMe
HO
SNH
COOHO
1
3
56
9
A
Die Biosy
Kulturübers
SB 212305 aus SB 212021 gegen Ende der Biosynthese vermu e

Ergebnisse und Diskussion
5 Strukturaufklärung des Aranciamycin-Anhydrids aus
Streptomyces TÜ 6384
5.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces
sp. TÜ 6384
Der Stamm Streptomyces TÜ 6384 wurde ebenfalls in der Arbeitsgruppe von Prof.
Karl Poralla an der Eberhard-Karls-Universität Tübingen aus einer Erdprobe eines
Fichtenwaldes bei Tübingen-Bühl isoliert.[1] Die weitere taxonomische
Charakterisierung erfolgte von Dr. Julia Riedlinger[36] und die Untersuchung
hinsichtlich der Sekundärmetabolitproduktion von Dr. Dirk Schulz[1] in der
Arbeitsgruppe Arbeitsgruppe von Prof. Hans-Peter Fiedler. Die Kolonien auf der
Agarplatte bilden ein hellbeiges Substratmyzel aus, das Luftmyzel und die Sporen
sind weiß (Abbildung 36).
Abbildung 36: Lichtmikroskopische Aufnahmen von Stamm Streptomyces sp. TÜ 6384
(links: Kolonie auf der Agarplatte; rechts: Myzelpellets in der Submerskultur).[1]
Aufgrund der chemotaxonomischen und molekularbiologischen Charakteristika
konnte der Stamm der Gattung Streptomyces zugeordnet werden. Aus dem
Stoffwechsel konnte die LL-Diaminopimelinsäure, die Hauptmenachinone MK-9 und
die gesättigten iso- und anteiso-Fettsäuren mit einer C-15- und C-17-Kettenlänge
68

Ergebnisse und Diskussion
69
bestimmt werden. Durch Partialsequenzierung der 16S-rRNA konnte ebenfalls die
Gattung Streptomyces bestimmt werden.
5.1.1 Chemisches Screening
Zur Untersuchung des Stammes TÜ 6384 auf Produktion von Sekundärmetaboliten
wurde ein chemisches Screening durchgeführt. Der Stamm wurde zunächst in drei
verschiedenen Kulturmedien fermentiert und anschließend mittels HPLC-DAD-
Experimenten analysiert.[1] Bei dem Vergleich mit der UV/Vis-Stoffdatenbank ergab
sich, dass der Stamm zunächst das bekannte Antibiotika SEK 43 (Rt = 6.5 min)
produziert.[47] Des Weiteren konnte bei einer Retentionszeit von Rt = 10.4 min die
Produktion eines weiteren, unbekannten Sekundärmetaboliten nachgewiesen
werden, welcher in der Folge als TÜ 6384 A bezeichnet wurde (Abbildung 37). Das
UV/Vis-Spektrum der Substanz zeigte eine sehr gute Übereinstimmung mit dem
bekannten Metaboliten Aranciamycin, allerdings ergab die Retentionszeit von
Rt = 10.4 min keine Übereinstimmung. Aufgrund dieses Befunds wurde davon
ausgegangen, dass es sich bei der Substanz TÜ 6384 A um ein Aranciamycin-
Derivat handeln könnte.[48]
Ergebnisse und Diskussion
70
Ab
bild
un
g 37: H
PLC
-DA
D-C
hrom
atogramm
des K
ulturfiltratextrakts des Stam
mes T
Ü 6384 und U
V/V
is-Spektrum
der Verbindung T
Ü 6384
A.

Ergebnisse und Diskussion
5.1.2 Isolierung und Reinigung
Zur Isolierung der Verbindung TÜ 6384 A wurde der Stamm zunächst im 10 Liter-
Maßstab im Biostat-S Bioreaktor mit dem Kulturmedium OM fermentiert.[1] Nach einer
Inkubationszeit von 142 h wurde die Kulturbrühe zunächst mittels Druckfiltration in
Kulturüberstand und Myzel getrennt und das Myzel verworfen (Schema 2).
Schema 2: Isolierungsschema der Substanz TÜ 6384 A.
Das Kulturfiltrat wurde anschließend an Amberlite XAD-16 mit Ethanol
chromatographiert. Die Elution erfolgte nach dem Waschen mit 40 % Ethanol mit
100 % Ethanol. Der eingeengte Extrakt wurde anschließend an LiChroPrep Diol mit
einem Stufengradienten von 0 % bis 5 % DCM in Ethanol weiter aufgereinigt. Ein
50 mg Aliquot des Rohextraktes wurde anschließend mittels einer präparativen RP-
HPLC mit einem linearen Gradienten von 40 % auf 80 % Acetonitril
chromatographiert.
71

Ergebnisse und Diskussion
72
5.2 Strukturaufklärung
Augrund der starken Ähnlichkeit des UV/Vis-Spektrums mit Aranciamycin konnte
zunächst davon ausgegangen werden, dass es sich bei der Substanz TÜ 6384 A um
ein Derivat des Aranciamycins handelt (Tabelle 4). Das aufgenommene IR-Spektrum
zeigte sowohl dem Aranciamycin zugehörige, als auch weitere, nicht dem
Aranciamycin zugehörige Banden (Tabelle 4).
5.2.1 HPLC-ESI-MS
Die Auswertung von HPLC-ESI-MS-Untersuchungen ergab für das Derivat
TÜ 6384 A ein Negativmolekülion [M-H]- von m/z 709.2 und ein Positiv-Molekülion
[M+H]+ von m/z 711.2. Diese Ergebnisse zeigten ebenfalls, dass es sich bei der
Verbindung nicht um das schon literaturbekannte Aranciamycin handeln konnte, da
dies eine Molekülmasse von m/z 544.2 besitzt.
5.2.2 Bestimmung der Summenformel
Zur Bestimmung der Summenformel wurde durch Dipl.-Ing. Graeme J. Nicholson an
der Universität Tübingen eine massenspektrometrische Analyse an einem FT-ICR-
Massenspektrometer durchgeführt. Für die Berechnung der Summenformel wurden
die Elemente Kohlenstoff (C), Wasserstoff (H), Stickstoff (N) und Sauerstoff (O)
zugelassen. Eine Zusammenfassung der Ergebnisse ist in Tabelle 4 dargestellt. Im
Vergleich zum Aranciamycin besitzt TÜ 6384 A ein zusätzliches C8H6O4-Fragment.

Ergebnisse und Diskussion
Tabelle 4: Physikochemische Eigenschaften von TÜ6384 A.
TÜ 6384 A
Aussehen Rot-oranges Pulver
Drehwert 20][ Da = + 82° (c = 0.05, MeOH)
709.17728 gemessen [M-H]–
709.17741 berechnet [M-H]–
FT-ICR-MS
∆ = 0.13 ppm
C35H34O16 Summenformel
UV max (MeOH) nm (log ε) 240 (4.48), 260 (4.44), 435 (4.08)
IR νmax [cm-1] 3503, 2977, 2933, 1766, 1715, 1675, 1625, 1448,
1415, 1380, 1290, 1247, 1191, 1170, 1135, 1109,
1083, 1031, 1001, 957, 840, 758, 735
5.2.3 Zuckeranalytik
Zur Bestätigung der Stereochemie des in der Verbindung TÜ 6384 A enthaltenen
Zuckerbausteins wurde eine GC-MS-Analytik durch Dipl.-Ing. Graeme J. Nicholson
an der Universität Tübingen durchgeführt. Durch die vergleichende Analyse des
Zuckerbausteins des Aranciamycins zur Verbindung TÜ 6384 A sollte die
Stereochemie aufgeklärt werden. Hierzu wurde eine Probe von Aranciamycin und der
Verbindung TÜ 6384 A zunächst säurekatalytisch hydrolysiert (6 M HCl, 110 °C, 20
h), das getrocknete Hydrolysat zum N-(O)TFA/Ethylester derivatisiert und
anschließend mittels chiraler GC-MS-Experimente analysiert. Durch einen Vergleich
beider Analyseergebnisse konnte der in der Verbindung TÜ 6384 A enthaltene
Zucker als β-2-O-Methyl-L-Rhamnose identifiziert werden.[49]
73
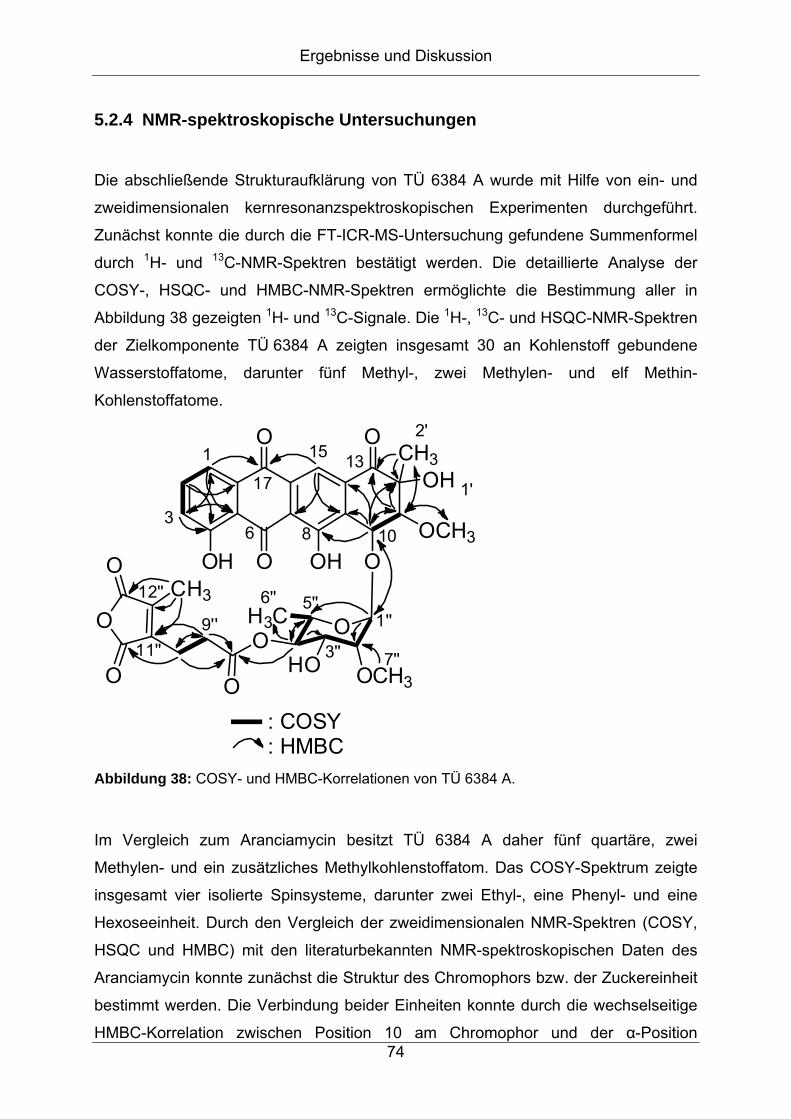
Ergebnisse und Diskussion
5.2.4 NMR-spektroskopische Untersuchungen
Die abschließende Strukturaufklärung von TÜ 6384 A wurde mit Hilfe von ein- und
zweidimensionalen kernresonanzspektroskopischen Experimenten durchgeführt.
Zunächst konnte die durch die FT-ICR-MS-Untersuchung gefundene Summenformel
durch 1H- und 13C-NMR-Spektren bestätigt werden. Die detaillierte Analyse der
COSY-, HSQC- und HMBC-NMR-Spektren ermöglichte die Bestimmung aller in
Abbildung 38 gezeigten 1H- und 13C-Signale. Die 1H-, 13C- und HSQC-NMR-Spektren
der Zielkomponente TÜ 6384 A zeigten insgesamt 30 an Kohlenstoff gebundene
Wasserstoffatome, darunter fünf Methyl-, zwei Methylen- und elf Methin-
Kohlenstoffatome.
O
OCH3HO
H3CO
O
O
O
O
CH3
O
OOH OH
O
OCH3
O
OHCH3
: COSY: HMBC
1
36 8 10
17
1513
3''
5''9''
11''
12''
1'
2'
6''
7''
1''
Abbildung 38: COSY- und HMBC-Korrelationen von TÜ 6384 A.
74
Im Vergleich zum Aranciamycin besitzt TÜ 6384 A daher fünf quartäre, zwei
Methylen- und ein zusätzliches Methylkohlenstoffatom. Das COSY-Spektrum zeigte
insgesamt vier isolierte Spinsysteme, darunter zwei Ethyl-, eine Phenyl- und eine
Hexoseeinheit. Durch den Vergleich der zweidimensionalen NMR-Spektren (COSY,
HSQC und HMBC) mit den literaturbekannten NMR-spektroskopischen Daten des
Aranciamycin konnte zunächst die Struktur des Chromophors bzw. der Zuckereinheit
bestimmt werden. Die Verbindung beider Einheiten konnte durch die wechselseitige
HMBC-Korrelation zwischen Position 10 am Chromophor und der α-Position

Ergebnisse und Diskussion
(Position 1’’) des Zuckers nachgewiesen werden. Über die COSY-Kopplung von H-9’’
zu H-10’’ und die HMBC-Kopplungen von H-9’’ zu C-8’’, C-10’’, C-11’’ und H-10’’ zu
C-8’’, C-14’’ und H-15’’ zu C-11’’, C-12’’ und C-13’’ konnte die in Abbildung 39
beschriebene Anhydrid-Struktur beschrieben werden.[50] Durch die HMBC-Korrelation
von H-4’’ zu C-8’’ konnte die Verknüpfung der Anhydrid-Einheit mit dem Zucker
nachgewiesen werden. Aufgrund der starken Ähnlichkeit mit dem Aranciamycin und
der zusätzlichen Anhydrid-Struktur wurde die Verbindung Aranciamycin-Anhydrid
genannt.
O
OCH3
HO
H3CO
O
O
O
O
CH3
O
OOH OH
O
OCH3
O
OHCH31
36 8 10
17 15 13
12
3''
5''9''
11''
12''
1'
2'
6''
7''
1''
Abbildung 39: Strukturformel von Aranciamycin-Anhydrid.
5.2.5 Biologische Aktivität
Das antimikrobielle Wirkspektrum des Aranciamycin-Anhydrids wurde in einem
Agardiffusionstest gegen die Stämme Bacillus subtilis DSM 10, Escherichia coli K12,
Saccharomyces cerevisiae ATCC 9010 und Botrytis cinerea TÜ 157 mit einer
Konzentration von 0.1 bis 1 mg/ml getestet. Ähnlich wie Aranciamycin zeigte das
Aranciamycin-Anhydrid schwache antibakterielle Aktivität nur gegen Bacillus subtilis.
Die Inhibierung des Wachstums von verschiedenen Tumorzellen wurde mit
Aranciamycin selbst verglichen. Dabei wies Aranciamycin-Anhydrid eine schlechtere
Aktivität auf (Tabelle 5).
75
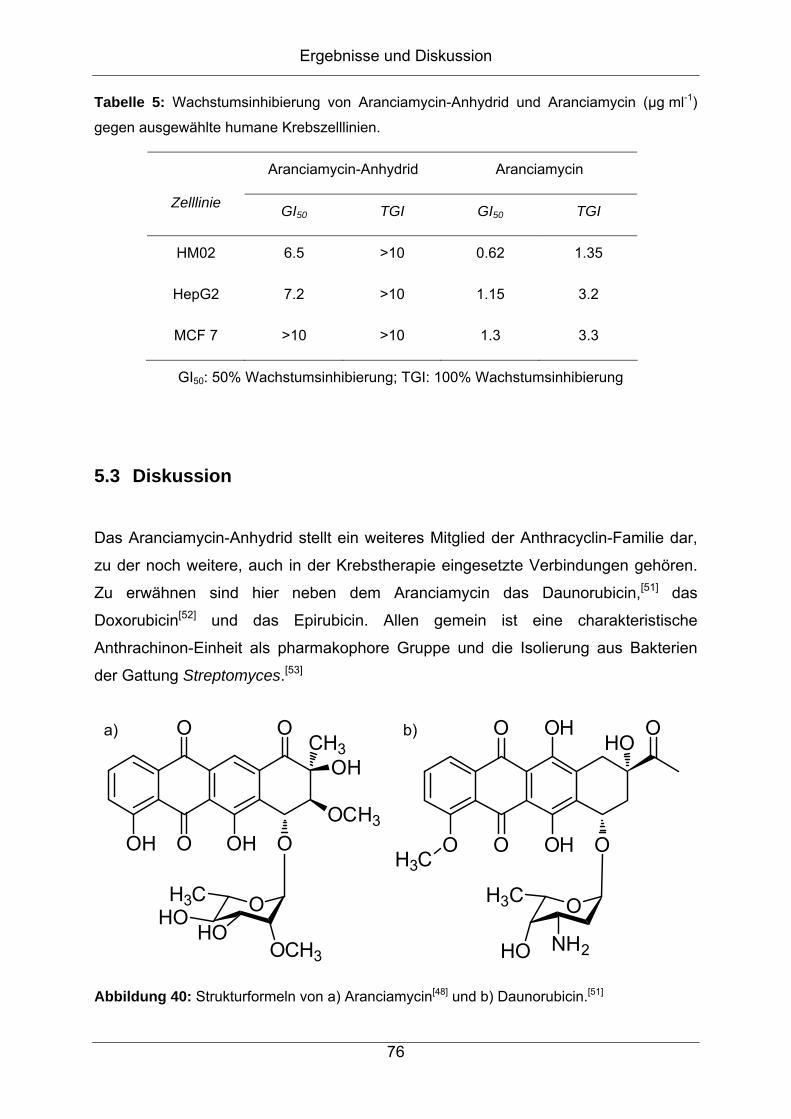
Ergebnisse und Diskussion
Tabelle 5: Wachstumsinhibierung von Aranciamycin-Anhydrid und Aranciamycin (µg ml-1)
gegen ausgewählte humane Krebszelllinien.
Aranciamycin-Anhydrid Aranciamycin
Zelllinie GI50 TGI GI50 TGI
HM02 6.5 >10 0.62 1.35
HepG2 7.2 >10 1.15 3.2
MCF 7 >10 >10 1.3 3.3
GI50: 50% Wachstumsinhibierung; TGI: 100% Wachstumsinhibierung
5.3 Diskussion
Das Aranciamycin-Anhydrid stellt ein weiteres Mitglied der Anthracyclin-Familie dar,
zu der noch weitere, auch in der Krebstherapie eingesetzte Verbindungen gehören.
Zu erwähnen sind hier neben dem Aranciamycin das Daunorubicin,[51] das
Doxorubicin[52] und das Epirubicin. Allen gemein ist eine charakteristische
Anthrachinon-Einheit als pharmakophore Gruppe und die Isolierung aus Bakterien
der Gattung Streptomyces.[53]
a) b)
O
OCH3
HO
H3CHO
O
OOH OH
O
OCH3
O
OHCH3
O
NH2
H3C
O
OO OH O
HOO
HO
H3C
OH
Abbildung 40: Strukturformeln von a) Aranciamycin[48] und b) Daunorubicin.[51]
76

Ergebnisse und Diskussion
Alle genannten Verbindungen werden in der Humanmedizin als Zytostatika
eingesetzt, ihre primäre Wirkungsweise beruht auf ihrer Interkalation in die DNA und
der damit verbundenen Inhibierung der DNA-synthetisierenden Topoisomerase IIα.
Des Weiteren werden die Wirkstoffe im Körper transformiert, was zur Bildung von
freien Radikalen und dies wiederum zur Spaltung der Doppelstrang-DNA führt.
Aufgrund dieses Wirkmechanismus sind die Anthracycline sowohl stark cytotoxisch
als auch mutagen. Durch die höhere Zellteilung von Krebszellen treten diese
Wirkungen zunächst hier auf, jedoch sind davon auch alle anderen, nicht mutierten
Zellen im Körper betroffen, was zu schweren, teils irreversiblen Beeinträchtigungen
führen kann. Aufgrund dieser starken Nebenwirkungen werden alle Verbindungen
dieser Klasse – obwohl auch antibiotisch aktiv – nicht als antibakterielle Wirkstoffe
eingesetzt. Im Vergleich zu Aranciamycin besitzt das hier aufgeklärte Aranciamycin-
Anhydrid eine ca. sechs- bis zehnfach verminderte Aktivität gegen humane
Krebszelllinien. Dies mag zunächst verwundern, ist doch der einzige Unterschied
zwischen beiden Verbindungen nur die zusätzliche Anhydrid-Einheit im
Aranciamycin-Anhydrid. Das Anthrachinon als primäres Pharmakophor ist
unverändert und in beiden Verbindungen identisch. Allerdings lässt sich die
unterschiedlich starke Bioaktivität sehr gut anhand der Strukturanaloga Doxorubicin
und Epirubicin aus derselben Wirkstoffklasse erläutern. Beide Stoffe sind konstitutiv
identisch, unterscheiden sich jedoch an einem stereogenen Zentrum am Saccharid
(Abbildung 41). Die Hydroxygruppe des Doxorubicins besitzt am Zucker eine S-, die
des Epirubicins eine R-Konfiguration.
O
OO OH O
HOO
H3C
OH
OH
(S)
O
NH2
OH
CH3
O
OO OH O
HOO
H3C
OH
OH
(R)
O
NH2
OH
CH3
b)a)
Abbildung 41: Strukturformeln von a) Doxorubicin und b) Epirubicin.
77

Ergebnisse und Diskussion
Dieser kleine strukturelle Unterschied äußert sich jedoch in einer wesentlich
gesteigerten Verträglichkeit des Epirubicins im Vergleich zum Doxorubicin. Die
Cardiotoxizität und somit die Verträglichkeit ist bei Epirubicin stark verringert. Somit
kann die Anhydrid-Einheit beim Aranciamycin-Anhydrid durchaus für die verminderte
Aktivität verantwortlich sein.
Weitere bekannte Naturstoffe mit einer Anhydrideinheit stellen das Tautomycin bzw.
sein Strukturanaloga Tautomycetin dar.[54, 55] Beide Naturstoffe wurden aus
Streptomyceten isoliert und zeigen sehr ähnliche Bioaktivitäten (Abbildung 42).
O
OH O
O
OH O OH O
O
O
O
O
O
O
OH O OH O OH
O
O
OO
a)
b)
Abbildung 42: Strukturformeln von a) Tautomycin[54] und b) Tautomycetin.[55]
Tautomycin und Tautomycetin sind nur sehr schwach aktiv gegen Bakterien, zeigen
jedoch antifungale Eigenschaften und eine cytotoxische Wirkung gegen humane
Leukämiezellen des Typs 562.[56] Der Anhydridteil beider Verbindungen ähnelt dem
des Aranciamycin-Anhydrids, beim Tautomycin bzw. Tautomycetin ist jedoch formal
ein Wassermolekül an die Doppelbindung addiert. Durch Fütterungsstudien mit
markiertem Acetat, Propionat, Methionin, Isobutyrat, Glycin und Glutamat konnte der
biosynthetische Ursprung von Tautomycin und Tautomycetin inklusive des
Anhydridteils aufgeklärt werden.[57] Dabei stellte sich heraus, dass der Anhydridteil
aus einer Propionat- und einer C5-Einheit zusammengesetzt wird. Aufgrund der
unterschiedlich hohen Einbauraten der Kohlenstoffatome in der C5-Einheit wurde α-
Ketoglutarat als Vorläuferbaustein vorgschlagen, welches aus Acetatbausteinen im
Krebszyklus entsteht. Eine direkte Fütterung mit Glutamat, dem unmittelbaren
Vorläufer von α-Ketoglutarat, konnte diese These bestätigen. Als Biosyntheseschritte
wird eine Aldolkondensation des α-Ketoglutarats an die Methyleneinheit des
Propionat-Coenzym A vorgeschlagen, gefolgt von einer Dehydratisierung zur Bildung
78

Ergebnisse und Diskussion
des Maleinsäureanhydrids. Im Falle von Aranciamycin-Anhydrid erfolgt nun eine
Dehydrierung, im Falle von Tautomycin bzw. Tautomycetin eine Hydratisierung zur
Bildung des Maleinsäureanhydrid-Bausteins. Aufgrund der Ähnlichkeit des
Anhydridteils von Aranciamycin-Anhydrid und Tautomycin bzw. Tautomycetin kann
dieser biosynthetische Ursprung und Reaktionsweg ebenfalls für das Aranciamycin-
Anhydrid postuliert werden.
Das Antibiotikum SEK 43 wurde ebenfalls im Kulturfiltratextrakt des Stammes
Streptomyces sp. TÜ 6384 gefunden. Dabei handelt es sich um ein
Benzophenonderivat, welches zuerst 1995 von Bindseil et al. aus Streptomyces sp.
P6417 isoliert worden ist.[58] Weitere Untersuchungen zur Biosynthese von
Anthracyclinen zeigten, dass SEK 43 von denselben Enzymen wie die Anthracycline
selbst produziert werden und durch eine alternative Zyklisierung des gleichen
Vorläufers entstehen, aus dem auch die Anthracycline produziert werden.[59]
OOH
HO
O
O
OH
HO
Abbildung 43: Strukturformel von SEK 43.[58]
79

Ergebnisse und Diskussion
6 Strukturaufklärung der Atacamycine A, B und C aus
Streptomyces sp. C-38
6.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces
sp. C-38
Der Stamm Streptomyces sp. C-38 wurde aus einer Bodenprobe aus der Atacama-
Wüste, Peru, isoliert.[60] Aufgrund der örtlichen Lage westlich der Anden am
Humboldtstrom gilt die Atacamawüste als eines der regenärmsten Gebiete der
Welt.[61] Diese extremen klimatischen Bedingungen bilden die Grundlage zur
Produktion von neuartigen Wirkstoffen von extremophilen Bakterien.
Die Kolonien des isolierten Stamms zeigten auf Agar-Festmedium ein beiges
Substratmycel, weißes Luftmycel und graubraune Sporen. Die Partialsequenzierung
der 16S-rRNA des Stammes ergab eine Zugehörigkeit zu der Gattung Streptomyces.
Die phylogenetische Analyse des Stamms ergab eine große Ähnlichkeit zum Stamm
Streptomyces griseosporus DSM 40562.[60]
Abbildung 44: Lichtmikroskopische Aufnahmen von Stamm Streptomyces sp. C-38 (links:
Kolonie auf der Agarplatte; rechts: Nahaufnahme einer Kolonie).[62]
80

Ergebnisse und Diskussion
81
6.1.1 Chemisches Screening
Zur Untersuchung des Stamms C-38 auf Produktion von Sekundärmetaboliten wurde
von Andreas Kulik von der Arbeitsgruppe Fiedler an der Universität Tübingen ein
chemisches Screening durchgeführt. Der Stamm wurde zunächst in drei
verschiedenen Kulturmedien (SGG, OM, 410) fermentiert und nach 168 h mittels
HPLC-DAD analysiert.[1] Hierbei zeigte sich, dass der isolierte Stamm C-38 drei
Verbindungen produziert, die keine Übereinstimmung mit bekannten Verbindungen in
der UV/Vis-Datenbank zeigten (Abbildung 45). Aufgrund der späten Retentionszeit
und den charakteristischen Signalen des UV/Vis-Spektrums wurde zunächst
angenommen, dass es sich bei den drei Verbindungen um Polyketidantibiotika
handelt. Die drei Zielverbindungen mit einer Retentionszeit von Rt = 13.1 min,
Rt = 13.7 min und Rt = 15.0 min wurden Atacamycin A, B und C benannt.
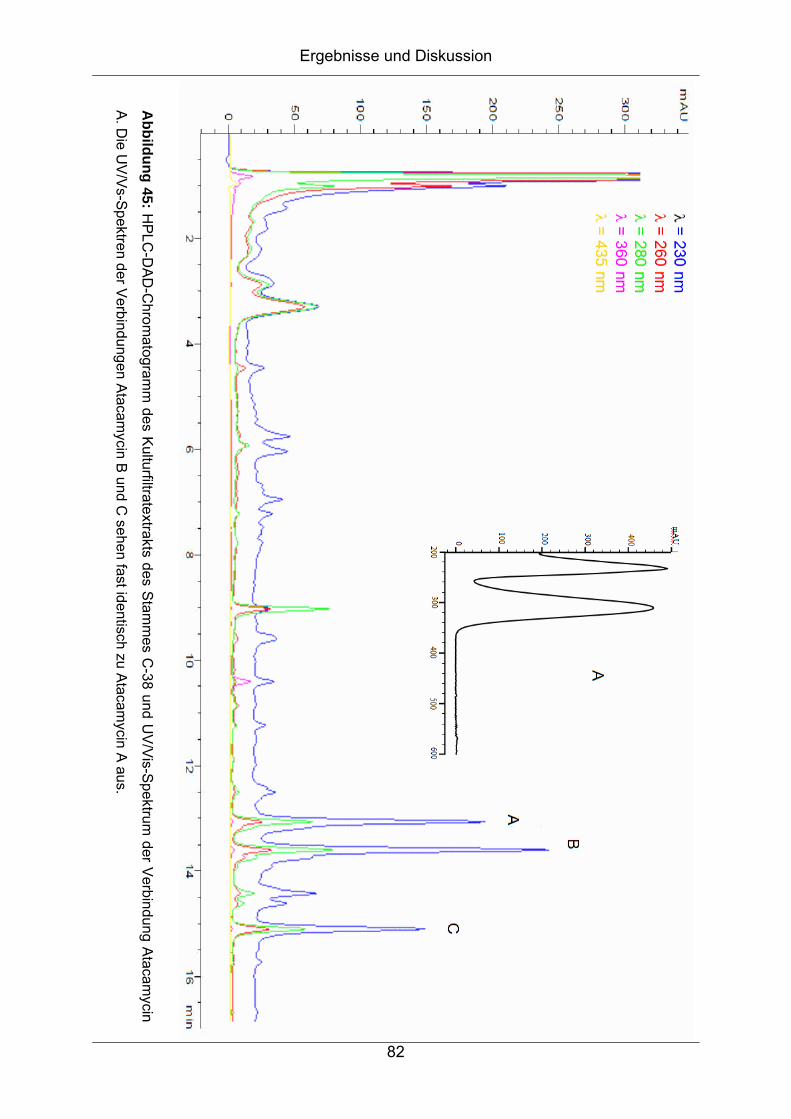
Ergebnisse und Diskussion
Ab
bild
un
g 45: H
PLC
-DA
D-C
hrom
atogramm
des K
ulturfiltratextrakts des Stam
mes C
-38 und UV
/Vis-S
pektrum der V
erbindung Atacam
ycin
A. D
ie UV
/Vs-S
pektren der Verbindungen A
tacamycin B
und C sehen fast identisch zu A
tacamycin A
aus.
82

Ergebnisse und Diskussion
6.1.2 Isolierung und Reinigung
Die Isolierung und Aufreinigung der drei Zielverbindungen wurde von Andreas Kulik
von der Arbeitsgruppe Fiedler an der Universität Tübingen durchgeführt. Zur
Isolierung der drei Zielsubstanzen wurde der Stamm im 10-Liter Biostat-S Bioreaktor
mit dem Komplexmedium NL SGG fermentiert. Nach einer Wachstumszeit von 168 h
wurde die Kulturbrühe durch Filtration in Myzel und Kulturüberstand getrennt und der
Überstand verworfen (Schema 3). Das Myzel wurde dann mit einer 1:1-Mischung
(v:v) Methanol/Aceton extrahiert, der konzentrierte Extrakt mit 3 N HCl auf pH 7
eingestellt und mit Cyclohexan extrahiert. Ein Aliquot des eingeengten
Cyclohexanextrakts wurde anschließend an Sephadex LH-20 mit Methanol als
mobile Phase chromatographiert, wobei 174 mg Rohextrakt erhalten werden
konnten. Aus diesem konnten abschließend mittels präparativer HPLC die
Atacamycine A, B und C erhalten werden.
1 g Aliquot Chromatographie an Sephadex LH-20 MeOH
174 mg Rohextrakt II
RP-PHPLC an Reprosil Pur Basic, Wasser /Methanol mit 0.1% HCOOH linearer Gradient 75-100 %
16 mg Atacamycin A 18 mg Atacamycin B 13 mg Atacamycin C
3.1 g Rohextrakt I
Konzentration unter Vakuum Einstellen auf pH 7 mit 3 N HCl 2x Extraktion mit 250 mL Cyclohexan
2.58 L Myzelextrakt
3x Extraktion mit 1 L 1:1 Methanol/Aceton
650 g Myzel
Schema 3: Isolierungsschema der Atacamycine A, B und C.
83

Ergebnisse und Diskussion
84
6.2 Strukturaufklärung
Die UV/Vis-Spektren aller drei Verbindungen zeigten zwar hohe Übereinstimmung
untereinander, jedoch keine Übereinstimmung beim Abgleich mit der UV/Vis-
Datenbank des chemischen Screenings. Aufgrund der Absorptionsmaxima bei
= 310 nm wurde zunächst ein Polyketid-Grundkörper vermutet. Die starke
Retention aller drei Verbindungen an einer C18-Phase deutet darauf hin, dass es sich
um sehr unpolare Strukturen ohne ionische oder polare funktionelle Gruppen
handelt.
6.2.1 HPLC-ESI-MS
Über HPLC-ESi-MS-Untersuchungen konnte zunächst die Molekülmasse der drei
Zielverbindungen ermittelt werden. Das MS-Spektrum von Atacamycin A wies ein
Positiv-Molekülion [M+H]+ von 501.2, Atacamycin B ein Positiv-Molekülion [M+H]+
von 471.3 und Atacamycin C ein Positiv-Molekülion [M+H]+ von 455.2 auf. Aufgrund
der charakteristischen Molekülmassendifferenzen von 30 amu und 16 amu konnte
davon ausgegangen werden, dass es sich bei Derivat B um ein Demethyl-deoxy-
atacamycin A und bei Derivat C um das Deoxy-atacamycin B handelt.
6.2.2 Bestimmung der Summenformel
Die Summenformel der drei Verbindungen wurde am Institut für organische Chemie
der TU Berlin mittels einer LTQ Orbitrap XL bestimmt. Für die Berechnung der
Summenformel wurden die Elemente Kohlenstoff (C), Wasserstoff (H), Sauerstoff (O)
und Stickstoff (N) zugelassen. Die Ergebnisse sind in Tabelle 6 dargestellt.

Ergebnisse und Diskussion
Tabelle 6: Physikochemische Eigenschaften von Atacamycin A, B und C.
Atacamycin A B C
Aussehen weißes Pulver weißes Pulver weißes Pulver
Molekülmasse 500.3 470.3 454.3
Summenformel C30H44O6 C29H42O5 C29H42O4
ESI-Orbitrap-MS
gemessen
berechnet
523.30272 [M+Na]+
523.30301
493.29240 [M+Na]+
493.29245
477.29608 [M+Na]+
477.29753
UV maxMeOH [nm] 221,310 221,308 222,307
(ε[cm2µmol-1]) -46.2 (c 0.6) +3.1 (c 0.1) -8.0 (c 0.7) 20][ Da (MeOH)
IR vmax (cm-1) 3460, 2964, 2930,
2875, 1700, 1606,
1452, 1375, 1263,
1094, 1068, 981
3406, 2963, 2927,
2876, 1710, 1635,
1457, 1378, 1259,
1171, 1050, 977
3443, 2960, 2935,
2873, 1713, 1626,
1457, 1376, 1270,
1171, 1103, 971
Die Ergebnisse der Summenformelbestimmung konnten die zunächst vermuteten
strukturellen Unterschiede der Atacamycin-Derivate bestätigen. Im Vergleich zu
Atacamycin B besitzt Atacamycin A eine weitere CH2O-Einheit und Atacamycin C ein
Sauerstoff-Atom weniger.
6.2.3 NMR-spektroskopische Untersuchungen
Die Konstitution aller drei Polyketidverbindungen konnte abschließend über ein- und
zweidimensionale NMR-Spektroskopie bestimmt werden. Durch die Auswertung der
eindimensionalen Wasserstoff- und Kohlenstoff-NMR-Spektren konnte zunächst die
über die hochauflösende Massenspektrometrie ermittelten Summenformeln bestätigt
werden. Das Wasserstoff-NMR-Spektrum der Komponente C zeigte insgesamt vier
Methyl-, vier Methylen-, fünf aliphatische Methin-, ein Methoxy-, und 13 olefinische
Wasserstoffsignale. Ein weiteres, breites Signal konnte einer Hydroxygruppe
zugeordnet werden. Im Vergleich dazu besitzt die Komponente B eine
Hydroxygruppe mehr und anstelle der vier Methylen- und fünf Methingruppen drei
85

Ergebnisse und Diskussion
Methylen- und sechs aliphatische Methineinheiten. Die Komponente A hingegen
zeigte im Vergleich zu Komponente B anstelle einer Methyl- eine Methingruppe und
eine weiteres Methoxysignal. Das eindimensionale Kohlenstoff- und das DEPT-NMR-
Spektrum von Komponente C zeigte neben fünf CH3-, vier CH2- und 18 CH-Signalen
nur ein quartäres, olefinisches und ein Carbonsäureester-Kohlenstoffatom. Daraus
ergibt sich eine ungewöhnlich niedrige Anzahl an Heteroatomen im Vergleich zur
Absolutanzahl der Kohlenstoffatome, welches ebenfalls die Vermutung bestätigt,
dass die Verbindungen zu der Gruppe der Polyketide gehören.
Das zweidimensionale COSY-NMR-Spektrum zeigte für die Komponenten B und C
insgesamt drei isolierte Spinsysteme: ein Ethylensystem (C5), ein C13-System und ein
verzweigtes C10-System (Abbildung 46). Für die Komponente A wurden jedoch
anstelle des C10-Systems zwei C5-Systeme angezeigt. Aufgrund der schon bei den
Komponenten B und C sehr schwach zu ermittelnden Kopplung zwischen den beiden
Protonen der jeweiligen C5-Systeme wurde zunächst davon ausgegangen, dass es
sich auch bei Komponente A um ein zusammenhängendes C-10-System handelt und
die Kopplung nur durch veränderte geometrische Einflüsse nicht gemessen werden
konnte.
O
O
OH
HO
MeO
OMe
27
29
11
15
16
21 25
17a)
O
O
OH
HOOMe
1H-1H COSY
HMBC
b)
OH
86
Abbildung 46: COSY- und HMBC-Korrelationen von a) Atacamycin A, b) Atacamycin B und
c) Atacamycin C.
O
O
OMe
c)

Ergebnisse und Diskussion
87
Durch die Auswertung der HMBC-Spektren der drei Verbindungen konnten die
ermittelten COSY-Spinsysteme und alle weiteren singulären Einheiten miteinander
verknüpft und die Struktur aufgeklärt werden. Beide Protonen der Ethyleneinheit (C-
2, C-3) zeigten einerseits Korrelation zu dem Carbonsäureester-Kohlenstoffatom (C-
1), andererseits konnte eine Verbindung zu dem quartären Kohlenstoffatom (C-4), zu
einer weiteren olefinischen CH-Gruppe (C-5) und zu einer singulären Methyleinheit
nachgewiesen werden. Diese zeigte ebenfalls wechselseitige Korrelation zur
Ethyleneinheit, des Weiteren auch eine Korrelation zur endständigen CH-Gruppe aus
dem C10-System und zu dem olefinischen Kohlenstoffatom. Durch diese bei allen drei
Verbindungen sichtbare Korrelation konnten die ersten beiden Fragmente
miteinander verbunden werden.
Während der weiteren HMBC-NMR-spektroskopischen Auswertung wurde der erste
Unterschied zwischen den Komponenten B, C und der Komponente A ersichtlich.
Derivat B und C zeigten beide die zusammenhängende C10-Einheit, während Derivat
A eine Kopplung weniger und dadurch formal zwei C5-Einheiten zeigte. An der
Verbindung beider Teilstücke an C-9 besitzt Derivat A anstelle eines Protons eine
Methoxygruppe, die durch die Kopplung von H-28 zu C-9 zugeordnet werden konnte.
Die Verbindung des nächsten Fragments erfolgt über die HMBC-Kopplung von H-29
zu C-13. Hierbei stellte sich heraus, dass die Kopplung zwischen H-12 und H-13
ebenfalls im COSY-NMR-Spektrum hätte erwartet werden können. Die weitere
Konstitution wurde anhand der COSY- und HMBC-NMR-Spektren aufgeklärt. Ein
weiterer struktureller Unterschied von Atacamycin A und B zu Atacamycin C war die
Präsenz einer Hydroxygruppe an C-14. Somit konnte durch NMR-Spektroskopie die
Differenz der Summenformeln auch in der Struktur vollständig aufgeklärt werden
(Abbildung 47). Durch die Kopplung von H-21 zu C-1 konnte die Ringverknüpfung
des Makrolactons nachgewiesen werden.

Ergebnisse und Diskussion
O
(E)O
(E)(E)
(E)
OH
OMe
R3
R2
(Z)
(E)
R1
(E)
14
7
11
13 15
17
20
2627
29
(E)
(E)
OMe
H
H
R1 R2 R3
A (1)
B (2)
C (3)
OH
OH
H
Abbildung 47: Strukturformeln der Atacamycine A, B und C.
Die Stereochemie der Doppelbindungen der Komponenten sollte zunächst durch die
in den 1H-NMR-Spektren gezeigten Kopplungskonstanten der olefinischen Protonen
bestimmt werden. Aufgrund der teilweisen Überlagerung mehrerer aufgespalteter
Signale im olefinischen Bereich war die Bestimmung aller
Doppelbindungskonfigurationen aus den 1H-NMR-Spektren jedoch nicht möglich.
Daher wurde zur Bestimmung der Kopplungskonstanten von überlagerten Signalen
ein selTOCSY-Experiment durchgeführt. Dazu wurde zunächst die Einstrahlfrequenz
eines Protons in räumlicher Nähe zu dem Proton gewählt, von dem die
Kopplungskonstante bestimmt werden sollte. Hierbei ist es wichtig, dass die
Einstrahlfrequenz keine Überlagerung mit weiteren Signalen zeigt, da dies sonst zu
schwer interpretierbaren Ergebnissen führt. Daraus konnten die folgenden
Kopplungskonstanten von J2,3 = 15.5 Hz, J6,7 = 15.2 Hz, J10,11 = 15.8 Hz,
J16,17 = 10.7 Hz, J18,19= 14.7 Hz und J22,23 = 15.7 Hz und die Konfiguration dieser
Doppelbindungen als 2E, 6E, 10E, 16Z, 18E und 22E bestimmt werden. Zur
Bestimmung der Stereochemie der Doppelbindung an C-4 und C-5 wurde ein
NOESY-Experiment durchgeführt. Eine Kopplung von H-3 zu H-5 zeigte dabei die
4E-Konfiguration der Doppelbindung (Abbildung 47).
Zur Aufklärung der absoluten Stereochemie der sieben stereogenen Zentren des
Atacamycin A wurde dies zunächst in verschiedenen Lösungsmitteln und -gemischen
zur Kristallisation angesetzt. Leider konnten aus diesen Ansätzen auch bei
verminderter Temperatur (4°C) keine für die Einkristallanalyse brauchbaren Kristalle
erhalten werden.
88

Ergebnisse und Diskussion
O
O
OH
OMeHO
MeO * *
*
**
**
Abbildung 48: Die drei stereochemischen Fragmente zur Berechnung der Stereochemie am
Beispiel Atacamycin A.
Experimente zur Bestimmung der relativen Stereochemie mittels NOESY-Spektren
und gleichzeitigen strukturchemischen Berechnungen wurden ebenfalls durchgeführt.
Aufgrund der großen Anzahl an Stereozentren wurde die Modellverbindung
Atacamycin A zunächst in drei Strukturfragmente aufgeteilt, da andernfalls die
strukturchemischen Berechnungen zu umfangreich gewesen wären (Abbildung 48).
Die experimentell beobachteten NOESY-Kopplungen zwischen den für die
Stereochemie relvanten Zentren waren jedoch nicht eindeutig einer Konfiguration
zuzuordnen, so dass die Bestimmung der Stereochemie mit dieser Methode keine
verlässlichen Aussagen geben konnte.
Weiterführende Experimente mit NMR-spektroskopischen Ansätzen zur Aufklärung
der absoluten Stereochemie des Atacamycin A nach Murata[63] wurden ebenfalls
durchgeführt. Diese Methode basiert darauf, dass in azyklischen Systemen die
Konformation von zwei benachbarten asymmetrischen Zentren durch gestaffelte
(„staggered“) Rotamere bestimmt wird, und dass deren relative Stereochemie durch
die Auswertung von Kohlenstoff-Proton-Spinkopplungskonstanten (2,3JC,H) und
Proton-Proton-Spinkopplungskonstanten (3JH,H) aufgeklärt werden kann. Durch
Vergleichen dieser Kopplungskonstanten kann das vorwiegende Rotamer aus den
sechs möglichen Positionen für die threo- und erythro-Form bestimmt werden.
Die Anwendbarkeit dieser Methode für zyklische Systeme hängt jedoch stark von der
Ringspannung des Moleküls ab. Ist die Ringspannung zu stark, ist eine freie
Drehbarkeit der Rotamere nicht gegeben und die erhaltenen Kopplungskonstanten
können nicht mit den in der Murata-Methode beschriebenen Kopplungen in
89

Ergebnisse und Diskussion
90
Übereinstimmung gebracht werden. Bei den hier aufgeklärten Atacamycinen handelt
es sich um 22-gliedrige Makrolactone mit einer hohen Anzahl an Doppelbindungen
und asymmetrischen Kohlenstoffatomen im Ring. Sowohl Doppelbindungen als auch
stereogene Zentren können zu einer hohen Ringspannung beitragen, weswegen die
Methode vom Murata beim Atacamycin A als eine unsichere Methode betrachtet und
nicht angewendet wurde.
Eine weitere NMR-spektroskopische Analyseverfahren zur Bestimmung der
Stereochemie ist die Methode nach Kishi[64]. Hierbei wird die zu untersuchende
Verbindung in seine stereochemischen Fragmente (Abbildung 48) unterteilt und
diese einzeln mit NMR-Spektroskopie charakterisierten, synthetischen Substanzen
verglichen. Hierbei wird angenommen, dass stereochemische Einflüsse
näherungsweise nur bis zu einem Abstand von einer Methyleneinheit beachtet
werden müssen. Sind stereogene Zentren weiter entfernt voneinander, wird der
gegenseitige Einfluss vernachlässigbar. Zum Vergleich der synthetischen
Verbindungen mit dem Naturstoff werden nur die 13C-chemischen Verschiebungen
genutzt, und aus diesem Grund ist es unabdingbar, dass die Vergleichssubstanzen
genau mit der Struktur der Fragmente übereinstimmen, da eine kleine
strukturchemische Änderung eine große Änderung der 13C-chemischen
Verschiebung bedeutet. Beim Atamcamycin A handelt es sich um die Abfolge Methyl-
Methoxy (C-8 und C-9) und Methyl-Hydroxy-Hydroxy-Methoxy (C-12, C-13, C-14 und
C-15). Leider sind für diese Abfolgen keine synthetischen Vergleichssubstanzen
erhältlich, so dass die Methode nach Kishi zur Aufklärung der Stereochemie der
Atacamycine nicht angewendet werden konnte.
Ebenfalls angewendet wurde eine relativ neue in der Strukturaufklärung von
Naturstoffen eingesetzte NMR-spektroskopische Technik, welche die Präsenz von
dipolarer Restkopplung (RDC, residual dipolar coupling) in einem asymmetrischen
Molekül zu Konfigurationsanalyse nutzt.[65] Hierbei wird während des NMR-
Experiments in dem Naturstoff eine dipolare Restkopplung erzwungen, die sich in die
Konfiguration des jeweiligen stereogenen Zentrums umrechnen lässt. Aufgrund der
Neuheit der Technik werden dafür NMR-Pulstechniken verwendet, die eine Avance-
Konsole erfordern und aufgrund unkompatibler NMR-Spektrometer nicht an dem
500-MHz NMR-Spektrometer des Instituts für Chemie der TU Berlin durchgeführt
werden konnten.
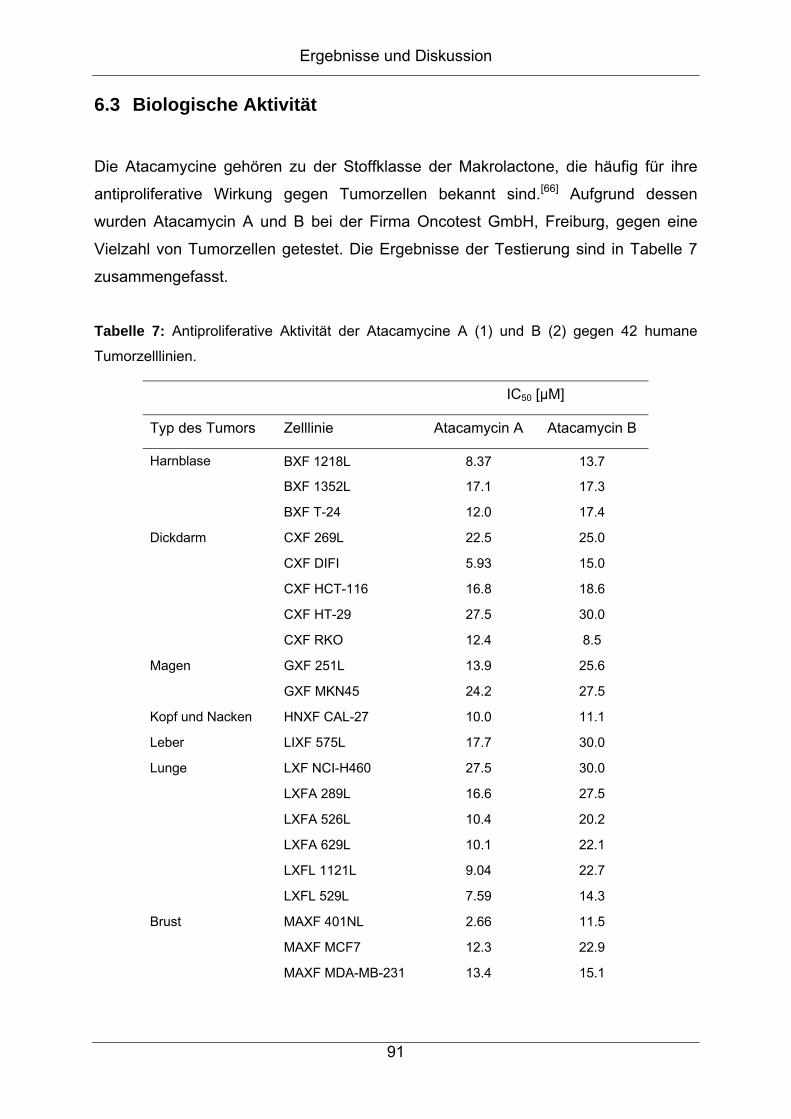
Ergebnisse und Diskussion
91
6.3 Biologische Aktivität
Die Atacamycine gehören zu der Stoffklasse der Makrolactone, die häufig für ihre
antiproliferative Wirkung gegen Tumorzellen bekannt sind.[66] Aufgrund dessen
wurden Atacamycin A und B bei der Firma Oncotest GmbH, Freiburg, gegen eine
Vielzahl von Tumorzellen getestet. Die Ergebnisse der Testierung sind in Tabelle 7
zusammengefasst.
Tabelle 7: Antiproliferative Aktivität der Atacamycine A (1) und B (2) gegen 42 humane
Tumorzelllinien.
IC50 [µM]
Typ des Tumors Zelllinie Atacamycin A Atacamycin B
Harnblase BXF 1218L 8.37 13.7
BXF 1352L 17.1 17.3
BXF T-24 12.0 17.4
Dickdarm CXF 269L 22.5 25.0
CXF DIFI 5.93 15.0
CXF HCT-116 16.8 18.6
CXF HT-29 27.5 30.0
CXF RKO 12.4 8.5
Magen GXF 251L 13.9 25.6
GXF MKN45 24.2 27.5
Kopf und Nacken HNXF CAL-27 10.0 11.1
Leber LIXF 575L 17.7 30.0
Lunge LXF NCI-H460 27.5 30.0
LXFA 289L 16.6 27.5
LXFA 526L 10.4 20.2
LXFA 629L 10.1 22.1
LXFL 1121L 9.04 22.7
LXFL 529L 7.59 14.3
Brust MAXF 401NL 2.66 11.5
MAXF MCF7 12.3 22.9
MAXF MDA-MB-231 13.4 15.1
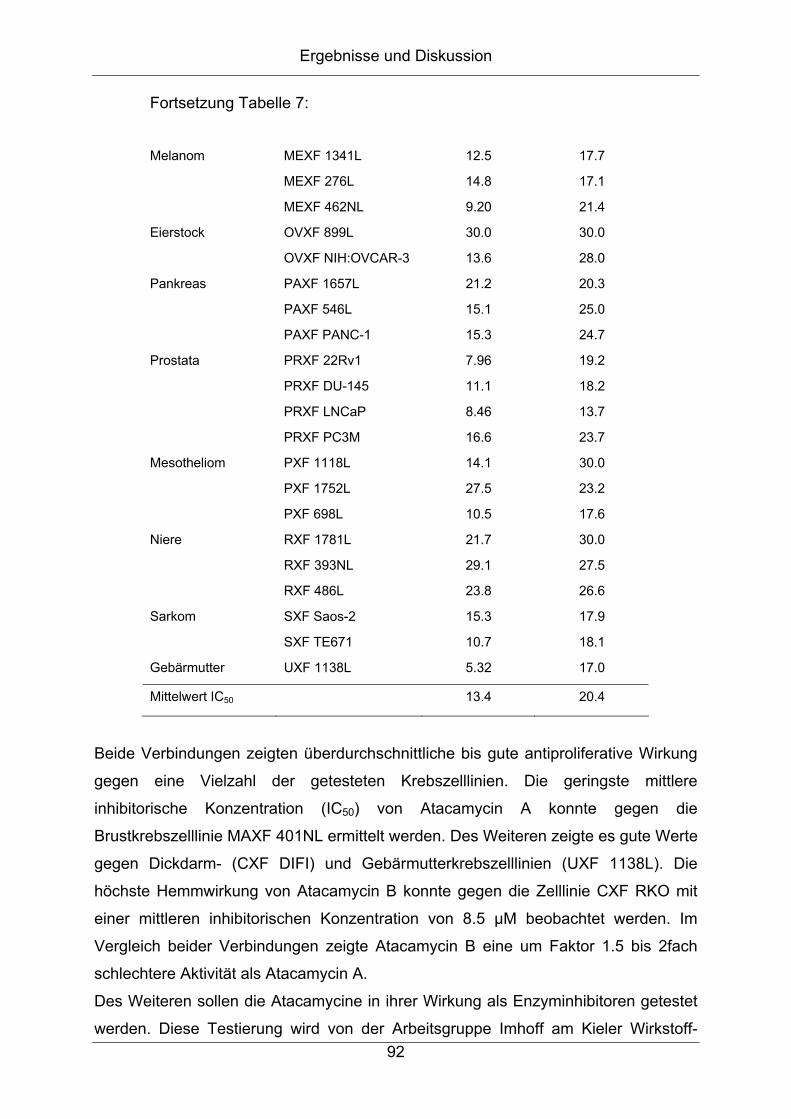
Ergebnisse und Diskussion
92
Fortsetzung Tabelle 7:
Melanom MEXF 1341L 12.5 17.7
MEXF 276L 14.8 17.1
MEXF 462NL 9.20 21.4
Eierstock OVXF 899L 30.0 30.0
OVXF NIH:OVCAR-3 13.6 28.0
Pankreas PAXF 1657L 21.2 20.3
PAXF 546L 15.1 25.0
PAXF PANC-1 15.3 24.7
Prostata PRXF 22Rv1 7.96 19.2
PRXF DU-145 11.1 18.2
PRXF LNCaP 8.46 13.7
PRXF PC3M 16.6 23.7
Mesotheliom PXF 1118L 14.1 30.0
PXF 1752L 27.5 23.2
PXF 698L 10.5 17.6
Niere RXF 1781L 21.7 30.0
RXF 393NL 29.1 27.5
RXF 486L 23.8 26.6
Sarkom SXF Saos-2 15.3 17.9
SXF TE671 10.7 18.1
Gebärmutter UXF 1138L 5.32 17.0
Mittelwert IC50 13.4 20.4
Beide Verbindungen zeigten überdurchschnittliche bis gute antiproliferative Wirkung
gegen eine Vielzahl der getesteten Krebszelllinien. Die geringste mittlere
inhibitorische Konzentration (IC50) von Atacamycin A konnte gegen die
Brustkrebszelllinie MAXF 401NL ermittelt werden. Des Weiteren zeigte es gute Werte
gegen Dickdarm- (CXF DIFI) und Gebärmutterkrebszelllinien (UXF 1138L). Die
höchste Hemmwirkung von Atacamycin B konnte gegen die Zelllinie CXF RKO mit
einer mittleren inhibitorischen Konzentration von 8.5 µM beobachtet werden. Im
Vergleich beider Verbindungen zeigte Atacamycin B eine um Faktor 1.5 bis 2fach
schlechtere Aktivität als Atacamycin A.
Des Weiteren sollen die Atacamycine in ihrer Wirkung als Enzyminhibitoren getestet
werden. Diese Testierung wird von der Arbeitsgruppe Imhoff am Kieler Wirkstoff-

Ergebnisse und Diskussion
Zentrum, Leibniz-Institut für Meereswissenschaften, IFM-GEOMAR, Kiel durchgeführt
und war zum Zeitpunkt der Drucklegung der Dissertation noch nicht abgeschlossen.
6.4 Diskussion
Die Atacamycine konnten in dieser Arbeit als neuartige Makrolidantibiotika in ihrer
Struktur aufgeklärt werden. Die Grundstruktur der Makrolide ist eine kurze (C7) bis
sehr lange (C61) Kohlenstoffkette, welche über eine Carbonsäureestereinheit zu
einem Lacton verknüpft ist.[67] Durch die unterschiedliche Anordnung von
Doppelbindungen und Methyl-, Hydroxy-, Methoxy-, Zucker- oder weiteren
dekorativen Bausteinen ist ihre strukturelle Vielfalt nahezu unbegrenzt. Die
Atacamycine zeigen hohe strukturelle Ähnlichkeit zu Dictyostatin, welches ebenfalls
einen 22-gliedrigen Ring als Grundgerüst und ein ähnliches exozyklisches Fragment
besitzt (Abbildung 49).[68]
O
(E)O
(E)(E)
(E)
OH
OMeHO
MeO
(Z)
(E)
R1
R1 =
(E)a) b)
O O
OH
OH OH
HO
Abbildung 49: a) Strukturformel von Dictyostatin aus einem Stamm der Gattung Spongia
sp., b) zum Vergleich Atacamycin A.
Dictyostatin zeigt Aktivität gegen humane Krebszelllinien im nanomolaren Bereich
und wurde anfang der 90er Jahre aus einem marinen Schwamm der Gattung
Spongia sp. isoliert.[69] Weitere Untersuchungen zeigten, dass die überaus starke
Wirkung des Dictyostatins auf eine Hemmung des Abbaus der Mikrotubuli und
dadurch verursachte Störung der Zellteilung zurückzuführen ist.[70] Dieser
Wirkmechanismus wird aufgrund der ähnlichen Struktur und biologischen
Wirksamkeit ebenfalls für die Atacamycine angenommen. Zur Klärung des genauen
Wirkortes und –mechanismus sind allerdings weiterführende experimentelle
93

Ergebnisse und Diskussion
Untersuchungen nötig. Weitere bekannte Makrolide mit einem 22-gliedrigen
Grundgerüst stellen die Wortmannilactone[71] dar, welche ebenfalls aus Schwämmen
isoliert wurden (Abbildung 50). Das Swinholide[72] wird ebenfalls als ein 22-gliedriges
Lacton synthetisiert, anschließend erfolgt die Konjugation von zwei Lactoneinheiten
zu einem Dimer. Dadurch entsteht der aussergewöhnliche Naturstoff mit einer C2-
Rotationsachse.
O
OO
O
OMe
OH OH O
O
MeO
OHOH
O
OMe
OOHHO
OHOH
OMeb)
a) HO
HO
OH
O
O
OH
Abbildung 50: Strukturformeln von a) Wortmannilacton A[71] und b) Swinholide A.[72]
Die Swinholide zeigen ebenfalls ein dem Dictyostatin ähnliches Wirkspektrum.[73]
Aufgrund der schwierigen und kostspieligen Gewinnung von Sekundärmetaboliten
aus Schwämmen ist jedoch eine pharmazeutische Anwendung des Dictyostatins
nicht abzusehen. Die ebenfalls sehr kosten- und zeitaufwendige Totalsynthese kann
aufgrund der vielen Stereozentren nicht als einfache Alternative angesehen werden.
Da es sich bei dem Produktionsstamm der Atacamycine um einen terrestrischen
Actinomycetenstamm handelt, können diese Naturstoffe relativ einfach und in großen
Mengen gewonnen werden.
Leider konnte im Rahmen dieser Arbeit die Stereochemie der Atacamycine nicht
aufgeklärt werden, obwohl unterschiedliche Ansätze hierzu verfolgt worden sind.
Aufgrund ihrer hohen strukturellen Flexibilität ist eine Einkristallzüchtung von
Makrolactonen nur schwer möglich. Zur Vermeidung von weiteren Substanzverlusten
wurden daher die Versuche zur Kristallisation ohne Ergebnis eingestellt. Generell ist
die Bestimmung der absoluten Stereochemie von Makrolactonen aufgrund der oben
94

Ergebnisse und Diskussion
genannten Probleme schwierig und wird meist erst Jahre nach der ersten
Veröffentlichung der Substanz beschrieben. Zwei der heute noch verwendeten
Vertreter dieser Gruppe, das Nystatin[74] und das Erythromycin A,[75] wurden erst 31
bzw. 12 Jahre nach der ersten Erwähnung vollständig in ihrer Stereochemie
beschrieben (Abbildung 51).[76, 77] Die erste Veröffentlichung des Dictyostatins enthält
eine Strukturformel, die in einer späteren Publikation als inkorrekt widerlegt wurde.
Letztendlich bleibt nur die Totalsynthese als finales Mittel, um sowohl die Konstitution
als auch die Konfiguration des Naturstoffes abschließend zu klären.[78]
95
b) a)
Abbildung 51: Strukturformeln von a) Nystatin und b) Erythromycin A.

Ergebnisse und Diskussion
7 Strukturaufklärung von TÜ 6392 A2 und TÜ 6392 D aus
Streptomyces sp. TÜ 6392
7.1 Herkunft und Taxonomie des Bakterienstammes Streptomyces
sp. TÜ 6392
Der Stamm Streptomyces sp. TÜ 6392 wurde ebenfalls wie der Stamm Streptomyces
sp. TÜ 6384 aus einer Erdprobe eines Fichtenwaldes bei Tübingen (Rammert, Bühl)
isoliert.[1] Abbildung 52 zeigt eine lichtmikroskopische Aufnahme des Stammes auf
der Agarplatte. Das Substratmyzel der Bakterien ist beige, das Luftmycel weiß und
die Sporen sind ebenfalls weiß gefärbt. Die Sporenketten sind spiralförmig (Spira-
Typ).
Abbildung 52: Lichtmikroskopische Aufnahmen von Stamm Streptomyces sp. TÜ 6392
(links: Kolonie auf der Agarplatte; rechts: filamentöses Myzel in der Submerskultur).[1]
Die taxonomische Charakterisierung des Stammes erfolgte durch Dr. Dirk Schulz,
AG Fiedler, an der Universität Tübingen. In der Zellwand des Stammes konnte LL-
Diaminopimelinsäure nachgewiesen und der Stamm daher chemotaxonomisch der
Gattung Streptomyces zugeordnet werden. Des Weiteren konnten die
Hauptmechaninone MK-9 (H6, H8), gesättigte und ungesättigte iso- und anteiso-
96

Ergebnisse und Diskussion
97
Fettsäuren nachgewiesen werden, welche ebenfalls charakteristisch für Stämme der
Gattung Streptomyces sind. Durch die Partialsequenzierung der 16S-rRNA konnte
die Einordnung bestätigt werden.[1]
7.1.1 Chemisches Screening
Zur Untersuchung des Stammes auf Produktion von Sekundärmetaboliten wurde von
Dr. Dirk Schulz von der Arbeitsgruppe von Prof. H.-P. Fiedler an der Universität
Tübingen ein chemisches Screening durchgeführt. Der Stamm wurde zunächst in
drei verschiedenen Kulturmedien (SGG, OM, 410) fermentiert und nach 120 h mittels
HPLC-DAD analysiert.[1] Bei der Analyse des Kulturfiltrats konnte ein Analyt mit der
Retentionszeit von Rt = 8.7 min identifiziert werden, welche keine Übereinstimmung
mit bekannten Verbindungen in der UV/Vis-Datenbank zeigte.
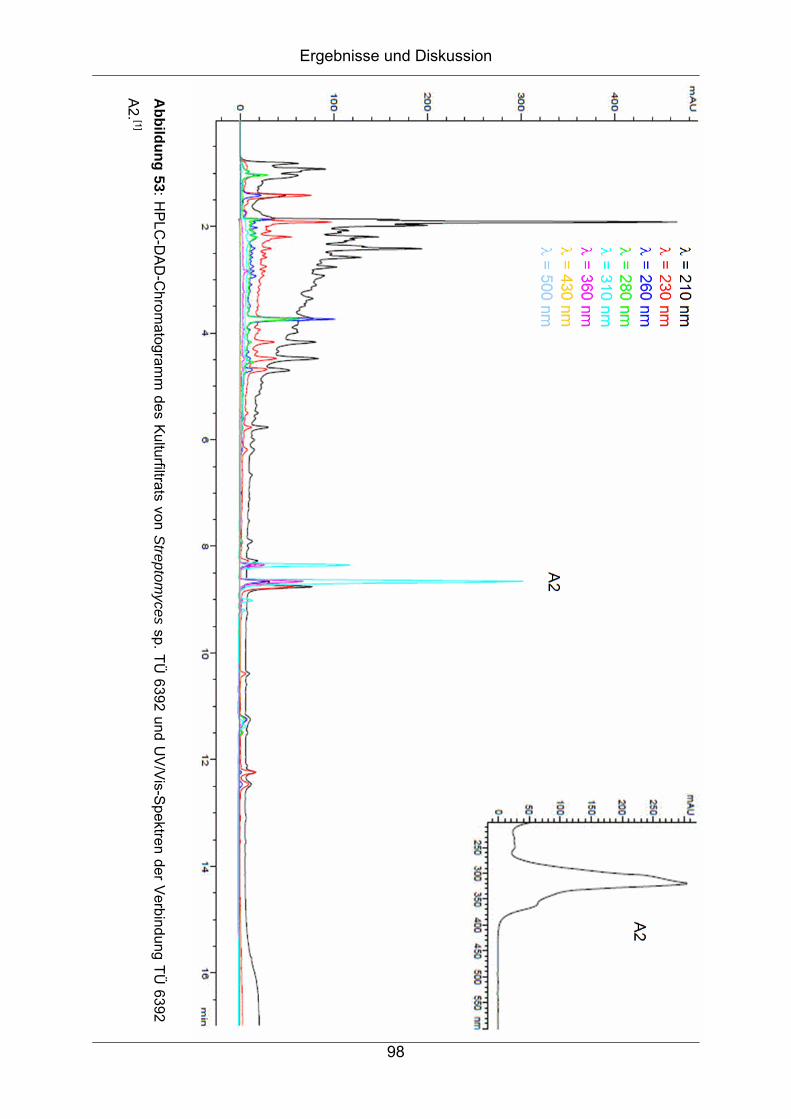
Ergebnisse und Diskussion
98
Ab
bild
un
g 53: H
PLC
-DA
D-C
hrom
atogramm
des K
ulturfiltrats von Streptom
yces sp. TÜ
6392 und UV
/Vis-S
pektren der Verbindung T
Ü 6392
A2. [1]
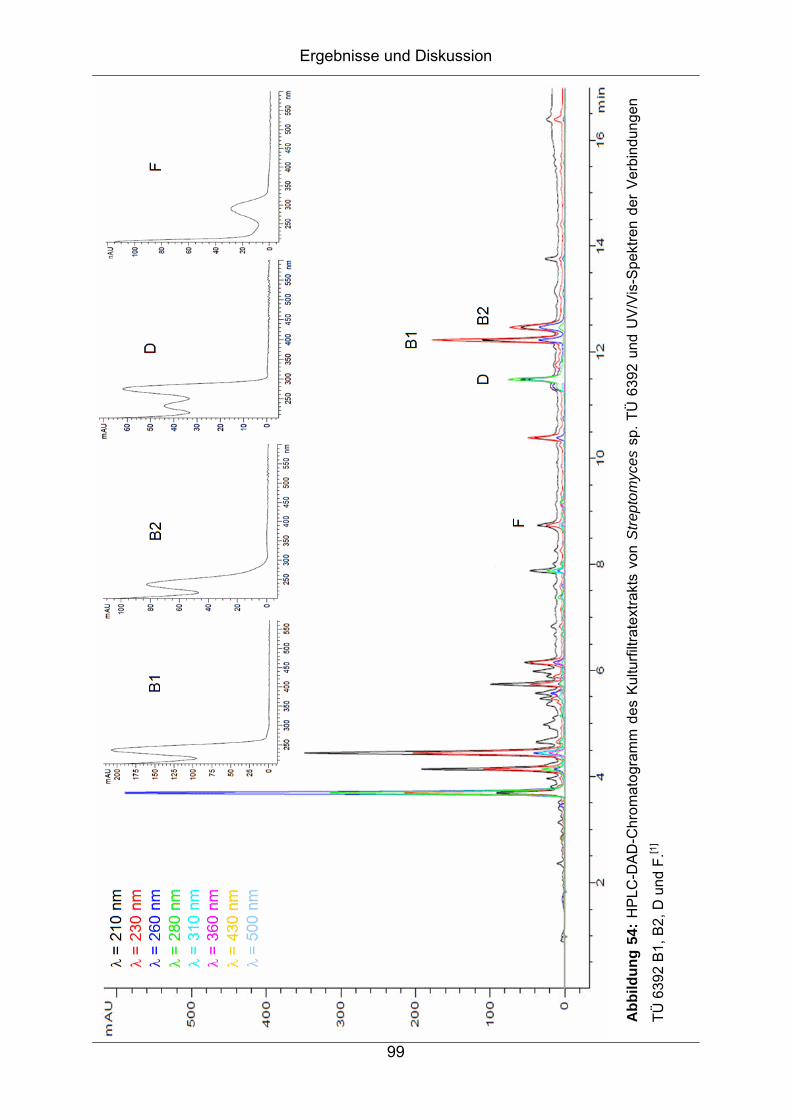
Ergebnisse und Diskussion
Ab
bild
un
g 5
4: H
PLC
-DA
D-C
hro
mat
ogra
mm
de
s K
ultu
rfilt
rate
xtra
kts
von
Str
epto
myc
es s
p. T
Ü 6
392
und
UV
/Vis
-Spe
ktre
n de
r V
erbi
ndun
gen
TÜ
639
2 B
1, B
2, D
und
F.[1
]
99

Ergebnisse und Diskussion
Bei der ebenfalls durchgeführten Analyse des Kulturfiltratextrakts konnten die
Verbindungen TÜ 6392 B1 (Rt = 12.2), B2 (Rt = 12.4), B3 (Rt = 10.4), D (Rt = 11.4)
und F (Rt = 7.8) gefunden werden, welche keine positive Übereinstimmung mit der
UV/Vis-Datenbank ergaben.[1] Die Substanz TÜ 6392 A2 konnte nur im Kulturfiltrat
nachgewiesen werden. Durch eine weiterführende Recherche in der
Naturstoffdatenbank Dictionary of Natural Products[79] konnte jedoch für alle bis auf
einen Analyten eine bereits bekannte Verbindung zugeordnet werden. Die
Verbindung TÜ 6392 B1 konnte aufgrund ihres UV/Vis-Spektrums dem Naturstoff
A88696 C,[80] die Verbindung TÜ 6392 B2 dem Naturstoff A88696 F und die
Verbindung TÜ 6392 F als Germidicin A[81] identifiziert werden. Zur
Strukturaufklärung sollten insgesamt zwei von dem Stamm Streptomyces TÜ 6392
produzierten Verbindungen (TÜ 6392 A2 und D) in Reinform isoliert werden.[1]
100
Abbildung 55: Strukturformeln von a) A88696 C,[80] b) A88696 F und c) Germicidin A.[81]
7.1.2 Isolierung und Reinigung
Die Fermentation und Isolierung der Zielverbindungen wurde von Dr. Dirk Schulz,
Arbeitsgruppe Fiedler, Universität Tübingen, durchgeführt.[1] Hierzu wurde der
Stamm Streptomyces sp. TÜ 6392 im 20 Liter Maßstab in einem Biostat b20-
Bioreaktor fermentiert. Als bestes Medium zur Produktion der beiden
Zielverbindungen im Bioreaktor zeigte sich SGG ohne Calciumcarbonat, welches als
Produktionsmedium verwendet wurde. Nach der optimalen Inkubationszeit von 168 h
wurde die Fermentation gestoppt und die Kulturbrühe mittels Druckfiltration in
Kulturfiltrat und Mycel getrennt. Das Kulturfiltrat wurde zunächst an Amberlite XAD-
16 chromatographiert, wodurch die beiden Zielverbindungen getrennt werden
b)
O
H3CCH3
CH3
OH
CH3H3C
O
OH
O
H3CCH3
CH3
OH
CH3H3Ca) c)
O
O
HO
O

Ergebnisse und Diskussion
101
konnten (Schema 4). Die Verbindung A2 adsorbierte nicht an das Material, während
die Verbindung D mit 100 % Methanol eluiert werden konnte. Zur Gewinnung von A2
wurde der Durchlauf auf pH 8 eingestellt, mit Ethylacetat extrahiert und die
organische Phase anschließend unter Vakuum eingeengt. Der so erhaltene
Rohextrakt wurde dann an LiChroPrep-Diol chromatographiert, dabei eluierte die
Verbindung A2 mit 10 % Methanol. Anschließend wurde das Rohprodukt mittels
präparativer HPLC an Nucleosil-100 C18-Material mit einem linearen Gradienten von
35-65 % Acetonitril mit 0.5 % Ameisensäure isoliert. Aufgrund der starken
Lichtempfindlichkeit der Verbindung A2 wurden alle durchgeführten Schritte unter
Lichtausschluss durchgeführt.
Die Verbindung TÜ 6392 D konnte bei der Chromatographie an Amberlite XAD-16 in
der 100 %-Methanol-Elutionsfraktion erhalten werden. Zur weiteren Aufreinigung
wurde der eingeengte Rohextrakt ebenfalls chromatographisch an LiChroPrep-Diol
aufgereinigt und der eingeengte Rohextrakt anschließend an Sephadex LH-20 in
Methanol aufgetrennt. Der so erhaltene Rohextrakt wurde zuletzt über eine
präparative C18-HPLC mit einem linearen Gradienten von 50 – 80 % Acetonitril mit
0.1 % Ameisensäure aufgereinigt, um die Verbindung TÜ 6392 D in Reinform zu
erhalten.

Ergebnisse und Diskussion
102
Schema 4: Isolierungsschema der Substanzen TÜ 6392 A2 und TÜ 6392 D.
Kulturfiltrat 14 Liter
Chromatographie an Amberlite XAD-16 Elution mit 100 % MeOH, Konzentration unter Vakuum
994 mg Rohextrakt
Chromatographie an LiChroPrep Diol Dichlormethan/MeOH Stufengradient 0 %, 2 %, 5 %, 10 % MeOH
416 mg Rohextrakt I
Chromatographie an Sephadex LH-20 MeOH
123 mg Rohextrakt II
RP-PHPLC an Nucleosil-100 C-18, Wasser /Acetonitril mit 0.1 % HCOOH linearer Gradient 50-80 %
9 mg TÜ 6392 D
7.2 Strukturaufklärung von TÜ 6392 A2
Aufgrund der stark unterschiedlichen UV/Vis-Spektren und Retentionszeiten wurde
zunächst nicht davon ausgegangen, dass es sich bei den beiden Verbindungen TÜ
6392 A2 und D um Strukturisomere handelt. Aufgrund der raschen Zersetzung der
Substanz TÜ 6392 A2 unter Lichteinwirkung wurde möglichst unter Lichtausschluss
gearbeitet. Analysen, die starke Lichtquellen als Energiequelle nutzen, konnten mit
Rücksicht auf die Substanz nicht durchgeführt werden. Des Weiteren zeigte sich,
dass die Substanz TÜ 6392 A2 bei zunehmender Aufreinigung zur Aggregation
neigte.
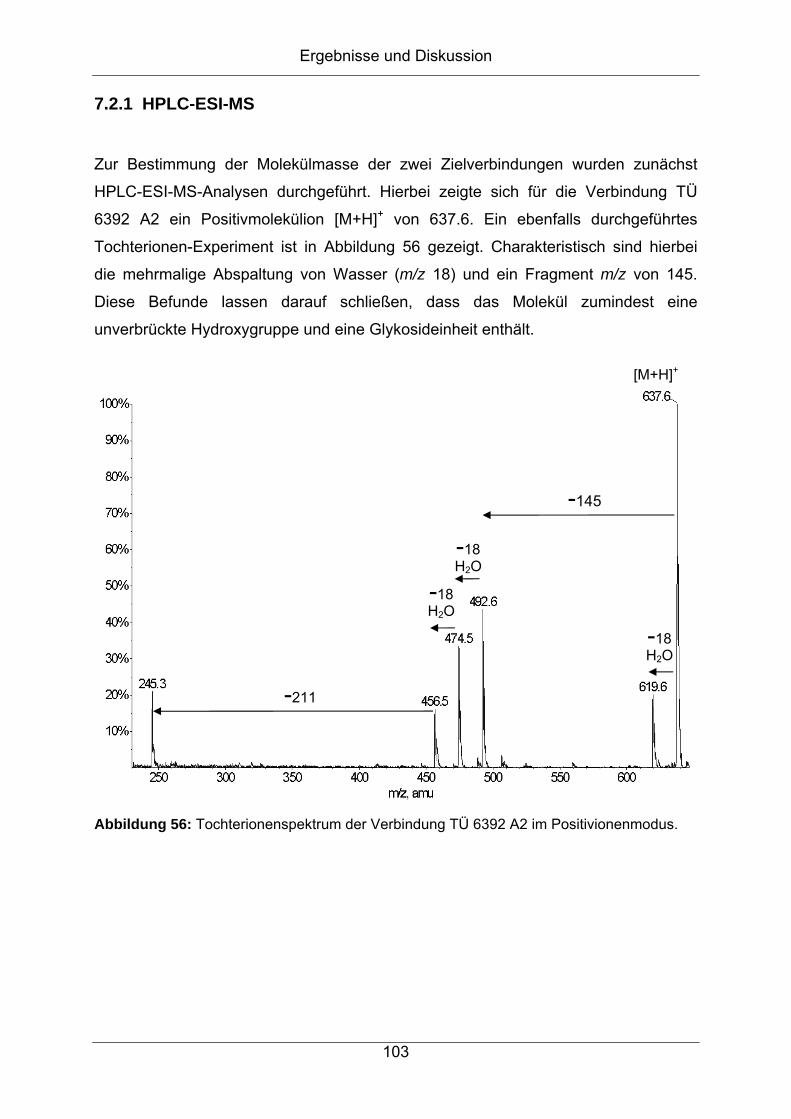
Ergebnisse und Diskussion
7.2.1 HPLC-ESI-MS
Zur Bestimmung der Molekülmasse der zwei Zielverbindungen wurden zunächst
HPLC-ESI-MS-Analysen durchgeführt. Hierbei zeigte sich für die Verbindung TÜ
6392 A2 ein Positivmolekülion [M+H]+ von 637.6. Ein ebenfalls durchgeführtes
Tochterionen-Experiment ist in Abbildung 56 gezeigt. Charakteristisch sind hierbei
die mehrmalige Abspaltung von Wasser (m/z 18) und ein Fragment m/z von 145.
Diese Befunde lassen darauf schließen, dass das Molekül zumindest eine
unverbrückte Hydroxygruppe und eine Glykosideinheit enthält.
[M+H]+
-211
-18
-18 H2O
-18 H2O
-145
H2O
Abbildung 56: Tochterionenspektrum der Verbindung TÜ 6392 A2 im Positivionenmodus.
103

Ergebnisse und Diskussion
104
7.2.2 Bestimmung der Summenformel
Die Summenformel der Verbindung TÜ 6392 A2 wurde von Dr. Dirk Schulz in der
Arbeitsgruppe von Prof. Johannes F. Imhoff im Kieler Wirkstoffzentrum KiWiZ am
Institut für Meereswissenschaften IFM-Geomar an einem MicrOTOF-II bestimmt. Für
die Berechnung wurden die Elemente Kohlenstoff (C), Wasserstoff (H), Stickstoff (N)
und Sauerstoff (O) zugelassen. Eine Zusammenfassung der Ergebnisse ist in Tabelle
8 dargestellt.
Tabelle 8: Physikochemische Eigenschaften von TÜ 6392 A2.
TÜ 6392 A2
Aussehen weißes Pulver
Molekülmasse 636.4
Summenformel C38H56N2O6
ESI-TOF-MS
gemessen
berechnet
637.4194 [M+H]+
637.4211
UV maxMeOH [nm] (ε[cm2µmol-1]) 315 (4.53)
7.2.3 NMR-Spektroskopische Untersuchungen
Zur Strukturaufklärung der beiden Zielverbindungen wurden beide zunächst in
DMSO-d6 gelöst und ein eindimensionales 1H-NMR-Spektrum bei 25 °C
aufgenommen. Von der Verbindung TÜ 6392 D konnte ein interpretierbares
Protonenspektrum und im Anschluss ein kompletter Spektrensatz in DMSO-d6
aufgenommen werden. Die Verbindung TÜ 6392 A2 allerdings koagulierte im NMR-
Röhrchen, sodass kein auswertbares 1H-NMR-Spektrum gemessen werden konnte
(Abbildung 59).
Ergebnisse und Diskussion
Ab
bild
un
g 5
7: 1H
-NM
R-S
pekt
rum
von
TÜ
639
2 A
2 in
DM
SO
-d6
bei 2
98 K
.
105

Ergebnisse und Diskussion
106
Zur Vermeidung der Koagulation wurden verschiedene NMR-Lösungsmittel (CD3OD,
ACN-d3, DCM-d2, CDCl3, Pyridin-d5, Benzol-d6) und Mischungen daraus zur NMR-
Messung verwendet, ohne die Koagulation unterbinden zu können. Ein weiterer
Ansatz zur Umgehung der Koagulation stellte das Messen bei veränderter
Temperatur dar. Hierzu wurde die Verbindung zunächst in DMSO-d6 gelöst und
anschließend bei 360 K ein eindimensionales 1H-NMR-Spektrum aufgenommen.
Dieses Spektrum konnte aufgrund der Schärfe der Signale im Gegensatz zum
Spektrum bei 298 K ausgewertet werden (Abbildung 58). Bei einem
Stabilitätsversuch bei 360 K über mehrere Stunden zeigte sich jedoch, dass die
Verbindung nicht ausreichend temperaturstabil ist, um die gesamte Messdauer von
ca. 60 h ohne Zerfall zu überstehen. Die weiteren, zur Strukturaufklärung
notwendigen ein- und zweidimensionalen Spektren konnten daher nicht bei 360 K
aufgenommen werden.
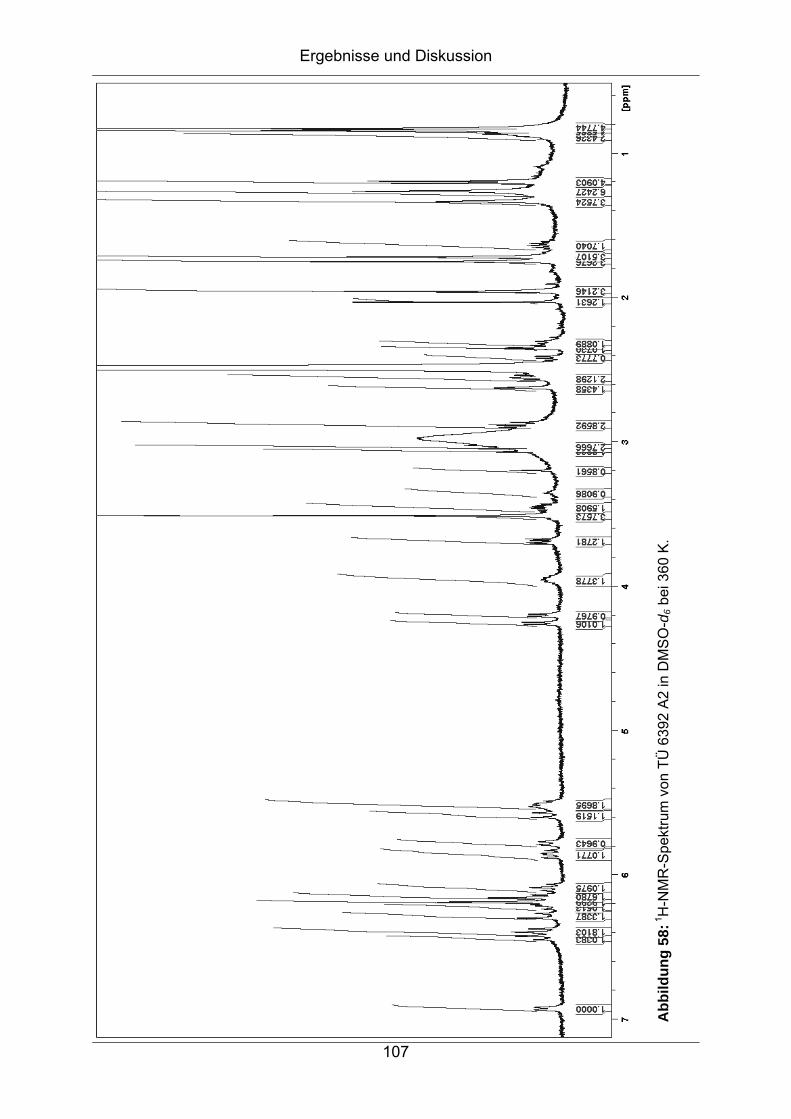
Ergebnisse und Diskussion
Ab
bild
un
g 5
8: 1H
-NM
R-S
pekt
rum
von
TÜ
639
2 A
2 in
DM
SO
-d6
bei 3
60 K
.
107

Ergebnisse und Diskussion
108
Anstelle einer Temperaturerhöhung zur Umgehung der Koagulation wurde nun
versucht, durch eine Messung bei niedriger Temperatur ein auswertbares NMR-
Spektrum aufzunehmen. Hierzu wurde die Substanz TÜ 6392 A2 in DCM-d2 gelöst
und die Probe während des NMR-Experiments auf 273 K temperiert (Abbildung 59).
Dieser Ansatz führte zu auswertbaren Spektren, so dass die zeitintensiven ein- und
zweidimensionalen Spektren auch über einen längeren Zeitraum ohne
Substanzverlust durchgeführt werden konnten.
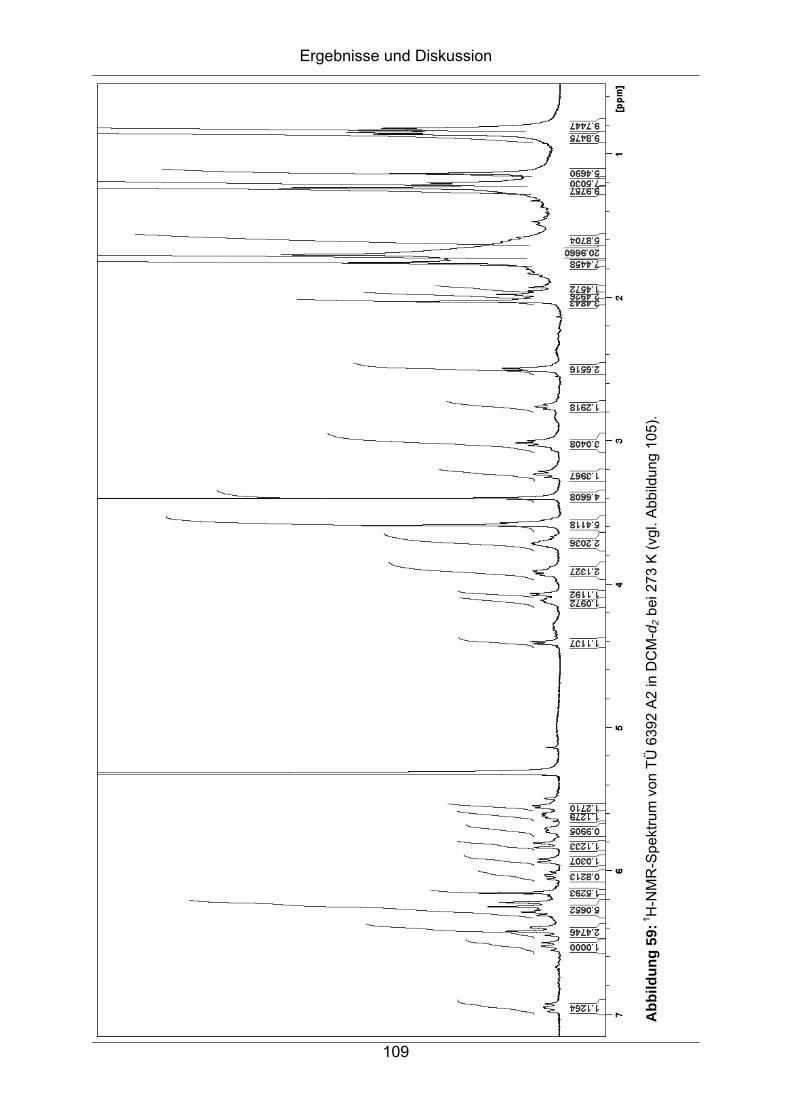
Ergebnisse und Diskussion
Ab
bild
un
g 5
9: 1H
-NM
R-S
pekt
rum
von
TÜ
639
2 A
2 in
DC
M-d
2 be
i 273
K (
vgl.
Abb
ildun
g 10
5).
109

Ergebnisse und Diskussion
Das 1H-NMR-Spektrum der Verbindung TÜ 6392 A2 zeigte insgesamt 15 Signale im
olefinischen Bereich, drei Signale zwischen 4 - 5 ppm und 17 Signale im
aliphatischen Bereich. Die Integration aller Signale des 1H-NMR-Spektrums ergab
eine Summe von 52 Protonen. Die Signale bei δH 3.59, δH 2.03, δH 1.76, δH 1.70, δH
1.21, δH 1.14, δH 0.85 und δH 0.83 ppm konnten durch eine Integralsumme von
jeweils drei Protonen zu Methylgruppen zugeordnet werden, das Signal bei δH 2.50
ppm durch die Integralsumme von zwei Protonen zu einer Methyleneinheit. Die 13C-,
DEPT- und HSQC-NMR-Spektren zeigten zwei zusätzliche Methyleneinheiten bei δC
65.6 ppm und δC 32.3 ppm. Insgesamt konnten so acht Methyl-, drei Methylen-, 21
Methin- und sechs quartäre Kohlenstoffatome zugeordnet werden. Aufgrund des
Fehlens der Kopplung im HSQC-Spektrum und der starken Verschiebung im
olefinischen Bereich wurde bei dem Signal bei δH 5.55 ppm davon ausgegangen,
dass es sich hierbei um ein Säureamidsignal handelt. Durch die detaillierte Analyse
der COSY-, HSQC-, und HMBC-Experimente konnte die Struktur von TÜ 6392 A2
vollständig beschrieben werden (Abbildung 60).
HNOHO
O
O
NH2O
HO
1H-1H COSY
HMBC
Abbildung 60: COSY- und HMBC-Kopplungen von TÜ 6392 A2.
Das COSY-NMR-Spektrum zeigt insgesamt sechs voneinander getrennte
Spinsysteme: eine Isobutyl-, zwei Pentenyl-, eine Ethenyl-, eine Isopentyl- und eine
Hexoseeinheit. (H-5 bis H-9, H-11 bis H-15, H-17 bis H-19, H-21 bis NH-1 und H-32,
H-24 bis H-27 und H-1’ bis H-5’). Fünf der isolierten Spinsysteme konnten durch die
HMBC-Korrelation der Methylsignale H-28, H-29, H-30 und H-31 verbunden werden
und bilden so das Kohlenstoffgerüst des Polyketid-Aglykons. Die Verbindung der
verbleibenden Hexoseeinheit an das Aglykon konnte durch die HMBC-Korrelation
von H-1’ zu C-11 hergestellt werden. Die chemische Verschiebung von δC 55.3 ppm
110
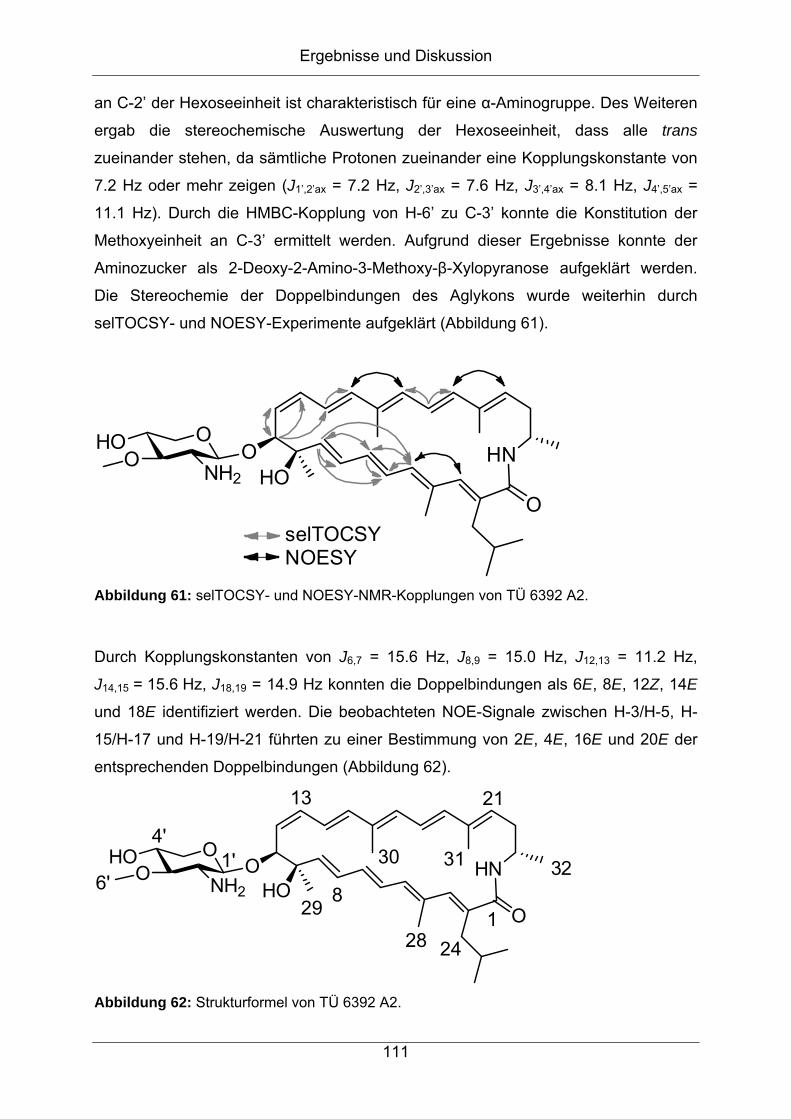
Ergebnisse und Diskussion
an C-2’ der Hexoseeinheit ist charakteristisch für eine α-Aminogruppe. Des Weiteren
ergab die stereochemische Auswertung der Hexoseeinheit, dass alle trans
zueinander stehen, da sämtliche Protonen zueinander eine Kopplungskonstante von
7.2 Hz oder mehr zeigen (J1’,2’ax = 7.2 Hz, J2’,3’ax = 7.6 Hz, J3’,4’ax = 8.1 Hz, J4’,5’ax =
11.1 Hz). Durch die HMBC-Kopplung von H-6’ zu C-3’ konnte die Konstitution der
Methoxyeinheit an C-3’ ermittelt werden. Aufgrund dieser Ergebnisse konnte der
Aminozucker als 2-Deoxy-2-Amino-3-Methoxy-β-Xylopyranose aufgeklärt werden.
Die Stereochemie der Doppelbindungen des Aglykons wurde weiterhin durch
selTOCSY- und NOESY-Experimente aufgeklärt (Abbildung 61).
HNOHO
O
O
NH2O
HO
selTOCSYNOESY
Abbildung 61: selTOCSY- und NOESY-NMR-Kopplungen von TÜ 6392 A2.
Durch Kopplungskonstanten von J6,7 = 15.6 Hz, J8,9 = 15.0 Hz, J12,13 = 11.2 Hz,
J14,15 = 15.6 Hz, J18,19 = 14.9 Hz konnten die Doppelbindungen als 6E, 8E, 12Z, 14E
und 18E identifiziert werden. Die beobachteten NOE-Signale zwischen H-3/H-5, H-
15/H-17 und H-19/H-21 führten zu einer Bestimmung von 2E, 4E, 16E und 20E der
entsprechenden Doppelbindungen (Abbildung 62).
HNOHO
O
O
NH2O
HO
18
13 21
2428
29
30 31 321'4'
6'
Abbildung 62: Strukturformel von TÜ 6392 A2.
111

Ergebnisse und Diskussion
7.3 Biologische Aktivität
Die Arbeiten zur Bestimmung der biologischen Aktivität gegen Gram-positive und
Gram-negative Bakterien, Eukaryoten, humane Krebszelllinien und ausgewählte
Enzyme werden von Dr. Dirk Schulz in der Arbeitsgruppe von Prof. Johannes F.
Imhoff am Kieler Wirkstoffzentrum KiWiZ am Institut für Meereswissenschaften IFM-
Geomar koordiniert und dauern zum Zeitpunkt der Abfassung dieser Dissertation
noch an.
7.4 Diskussion
Der Sekundärmetabolit TÜ 6392 A2 konnte im Rahmen dieser Arbeit
strukturaufgeklärt werden. Dabei handelt es sich um ein 24-gliedriges Makrolactam,
welches mit einer Xylopyranose-Zuckereinheit verknüpft ist. Vergleichbar mit den
vorher beschriebenen Atacamycinen können Makrolactame durch unterschiedliche
Anordnungen von Doppelbindungen, Kohlenwasserstoffketten oder
Zuckerbausteinen eine nahezu unbegrenzte Anzahl an möglichen Naturstoffen
bilden. Durch die aufgeklärte Struktur von TÜ 6392 A2 lassen sich die bei der
Isolierung beobachteten Schwierigkeiten erklären. Durch eine hohe Anzahl an
konjugierten Doppelbindungen (> 4) ist das Molekül empfindlich gegenüber
kurzwelligem Licht. Der starke Unterschied zwischen dem sehr hydrophoben Aglykon
und dem relativ hydrophilen Zuckerbaustein erklärt die starke Neigung zur
Koagulation bei Raumtemperatur.
Eine Literaturrecherche ergab eine Ähnlichkeit zu dem ebenfalls 24-gliedrigen
Makrolactam Incednine, welches 2008 von Futamura et al. aus Streptomyces sp.
ML694-90F3 aufgeklärt wurde (Abbildung 63).[82] Abgesehen von der Gruppe an C-2
(Methoxy- statt einer Isobutyleinheit) ist das Aglykon identisch zu TÜ 6392 A2.
HN
O
OHO
O
O
NHHOO
ONH
Abbildung 63: Strukturformel von Incednine.[82]
112

Ergebnisse und Diskussion
Die α-Zuckereinheit beider Verbindungen unterscheidet sich nur durch die
unterschiedliche Verknüpfung der Methylgruppe (Position 2’ statt Position 3’ beim TÜ
6392 A2). Des Weiteren ist beim Incednine eine Disaccharid- anstelle einer
Monosaccharid-Einheit an das Aglykon verknüpft. Für Incednine werden ebenfalls
große Probleme bei der Aufreinigung und Vermessung beschrieben.
Interessanterweise wird in der Literatur die Messung ebenfalls bei -5 °C
durchgeführt, analog zur Strukturaufklärung von TÜ 6392 A.
Incednine wurde auf seine Suppression gegen die antiapoptotische Wirkung von Bcl-
2 und Bcl-xL getestet. Bcl-xL-überexprimierende Ms-1-Zellen zeigen Resistenz zu
einer Vielzahl von Antitumorwirkstoffen, z.B. Inostamycin[83] oder Vinblastin[84]
(Abbildung 64). Diese Resistenz konnte allerdings durch Koapplikation des
Antitumorwirkstoffs mit 100 nM Incednine vermieden werden, wohingegen eine
einzelne Gabe von Incednine keine Apoptose induzieren konnte. Diese
Untersuchungen weisen darauf hin, dass Incednine die anti-apoptotische Funktion
von Bcl-2/Bcl-xL durch einen einzigartigen Wirkmechanismus inaktivieren kann.
O
HO
OH O
OH
O
OO
HO
HO
a) b)
Abbildung 64: Strukturformeln von a) Inostamycin und b) Vinblastin.
Aufgrund der starken Ähnlichkeit von TÜ 6392 A2 zu Incednine sollen dieselben
Tests ebenfalls mit TÜ 6392 A2 durchgeführt werden. Fehlende
Vergleichsexperimente zu dem Zeitpunkt dieser Arbeit lassen eine nähere
Untersuchung nicht zu.
113

Ergebnisse und Diskussion
114
7.5 Strukturaufklärung von TÜ 6392 D
Im Gegensatz zur Verbindung TÜ 6392 A2 zeigte die Verbindung TÜ 6392 D bei
ihrer Strukturaufklärung weder Lichtempfindlichkeit noch Neigung zur Koagulation.
Das charakteristische UV/Vis-Spektrum beider Verbindungen zeigte ebenfalls keine
Übereinstimmung, weswegen zunächst davon ausgegangen wurde, dass es sich bei
der Verbindung TÜ 6392 D nicht um ein Derivat der Verbindung TÜ 6392 A2 handelt.
7.5.1 HPLC-ESI-MS
Die durchgeführten HPLC-ESI-MS-Experimente zeigten für die Verbindung
TÜ 6392 D ein Positiv-Molekülion [M+H]+ von 456.3 und ein Negativ-Molekülion [M-
H]- von 454.3. Zur Strukturaufklärung wurden ebenfalls Tandem-MS-Experimente im
Positiv- und Negativ-Ionenmodus aufgenommen. Aufgrund der besseren
Fragmentierung ist im Folgenden nur das Negativ-Fragmentspektrum gezeigt
(Abbildung 65). Ausgehend von dem Pseudomolekülion mit m/z = 454.3 ist eine
Abspaltung von m/z = 44 zu sehen, was dem Verlust einer CO2-Einheit entspricht.
Ein zweites, wenn auch deutlich schwächeres Fragment von m/z = 44 ist zwischen
m/z = 191.2 und m/z = 147.1 zu sehen. Aus dieser Information lässt sich ableiten,
dass die Verbindung TÜ 6392 D eine freie Carbonsäureeinheit besitzt. Darüber
hinaus beinhaltet das Molekül eine verbrückte Carbonsäureestereinheit, die jedoch
erst durch die fortschreitende Fragmentierung des Moleküls detektiert wird. Die
weiteren beobachteten Fragmente liefern aufgrund ihrer unspezifischen Größe keine
Information zur Strukturaufklärung der Verbindung TÜ 6392 D.
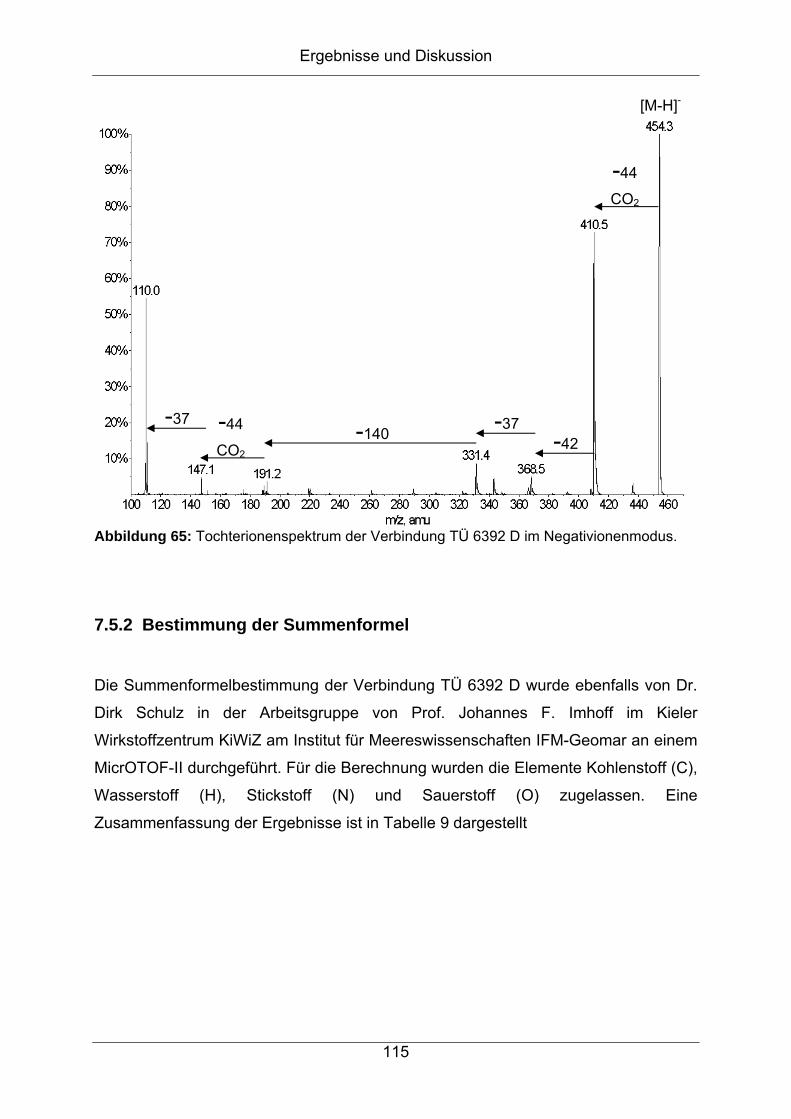
Ergebnisse und Diskussion
Abbildung 65: Tochterionenspektrum der Verbindung TÜ 6392 D im Negativionenmodus.
-42 -37 -44
CO2
-37 -140
[M-H]-
-44
CO2
7.5.2 Bestimmung der Summenformel
Die Summenformelbestimmung der Verbindung TÜ 6392 D wurde ebenfalls von Dr.
Dirk Schulz in der Arbeitsgruppe von Prof. Johannes F. Imhoff im Kieler
Wirkstoffzentrum KiWiZ am Institut für Meereswissenschaften IFM-Geomar an einem
MicrOTOF-II durchgeführt. Für die Berechnung wurden die Elemente Kohlenstoff (C),
Wasserstoff (H), Stickstoff (N) und Sauerstoff (O) zugelassen. Eine
Zusammenfassung der Ergebnisse ist in Tabelle 9 dargestellt
115

Ergebnisse und Diskussion
116
Tabelle 9: Physikochemische Eigenschaften von TÜ 6392 D.
TÜ 6392 D
Aussehen weißes Pulver
Molekülmasse 455.3
Summenformel C27H37NO5
ESI-TOF-MS
gemessen
berechnet
456.2632 [M+H]+
456.2744
UV maxMeOH [nm] (ε[cm2µmol-1]) 234 (3.40)
275 (3.53)
7.5.3 NMR-Spektroskopische Untersuchungen
Im Gegensatz zur Verbindung TÜ 6392 A2 konnten von Verbindung TÜ 6392 D
auswertbare NMR-Spektren (1H-, 13C-, DEPT-, COSY-, HSQC-, und HMBC-NMR-
Spektrum) in DMSO-d6 bei 25 °C erhalten werden. Zur Strukturaufklärung wurden
zusätzlich noch alle notwendigen zweidimensionalen NMR-Spektren in CD3OD
aufgenommen, da dieses Lösungsmittel im Vergleich zu DMSO-d6 feinere
Aufspaltungsmuster zeigte. Die gesamte, im Folgenden dargestellte
Strukturaufklärung erfolgte auf der Basis der in CD3OD aufgenommenen NMR-
Spektren (Tabelle 19). Das 1H-NMR-Spektrum der Verbindung TÜ 6392 D zeigte drei
Signale im olefinischen, ein Signal im mittleren und 19 Signale im aliphatischen
Bereich. Die Integration aller Signale ergab eine Anzahl von 37 Protonen, wobei die
Signale bei δH 0.89, δH 0.93, δH 1.15, δH 1.70 und δH 0.83 ppm zu je einer
Methylgruppe zugeordnet werden konnten. Die 13C-, DEPT- und HSQC-NMR-
Spektren zeigten insgesamt fünf Methyl-, sechs Methylen-, drei olefinische und vier
aliphatische Methin- und neun quartäre Kohlenstoffatome. Durch detaillierte
Auswertung des HSQC-Experiments konnten die Signale bei δH 1.55, δH 0.88,
δH 1.43, δH 1.18, δH 1.55, δH 1.28, δH 1.66, δH 1.43, δH 2.51 und δH 1.62 ppm zu fünf
Methyleneinheiten zugeordnet werden. Zusammen mit dem Signal bei δH 1.04 ppm,
welches eine Integralsumme von zwei Protonen zeigte, konnten somit alle
Methylenprotonen zugeordnet werden. Durch die im Folgenden beschriebene

Ergebnisse und Diskussion
detaillierte Analyse der COSY-, HSQC-, und HMBC-Experimente konnte die Struktur
von TÜ 6392 D vollständig aufgeklärt werden (Abbildung 66).
O
H3CCH3
CH3
OH
CH3H3C
O
O
OH
HN
916
1718
19
20
22
1'
3'
5'
1H-1H COSY
HMBC
Abbildung 66: COSY- und HMBC-NMR-Kopplungen von TÜ 6392 D.
Das COSY-Spektrum zeigte insgesamt drei voneinander getrennte Spinsysteme,
wobei das Spinsystem H-17 – H-5 – H-6 – H-7 – H-18 ein charakteristisches
strukturelles Merkmal aus dem Naturstoff Abyssomicin darstellt.[85] Die HMBC-
Kopplungen von H-4 zu C-2, C-3, C-16 und von H-15 zu C-1 und C-2 konnten als
Tetronsäurestruktur aufgelöst werden. Durch die intensive HMBC-Kopplung von H-
11, H-14 und H-15 zu C-1 konnte die spiroartige Verknüpfung eines sechsgliedrigen
Rings zum Tetronsäurebaustein nachgewiesen werden, wodurch ein Spirotetronat
als Grundstruktur aufgeklärt werden konnte. Durch die HMBC-Kopplungen von H-12
zu C-10, von H-10 zu C-8 und die COSY-Kopplung von H-8 zu H-3 konnte ferner die
Struktur des elfgliedrigen Hauptrings aufgeklärt werden. Dieser bis jetzt aufgeklärte
Baustein konnte als identisch zum Antibiotikum A88696 C identifiziert werden,
welches im Jahr 1993 von der Arbeitsgruppe um Bonjouklian in den Lilly Research
Laboratories aus dem Stamm Streptomyces sclerotialus aufgeklärt worden ist.[80]
Aufgrund dieser starken Ähnlichkeit kann davon ausgegangen werden, dass
A88696 C und TÜ 6392 D denselben Biosyntheseweg besitzen und damit auch die
Stereochemie der beiden Naturstoffe identisch ist. Im Vergleich zu A88696 C besitzt
TÜ 6392 D jedoch eine zusätzliche C5H3NO2-Einheit. Durch die COSY-Kopplung von
H-2’ zu H-3’, die HMBC-Kopplungen von H-2’ zu C-1’, C-3’, C-4’ und von H-3’ zu C-
117

Ergebnisse und Diskussion
1’, C-2’, C-4’ und dem charakteristischen Verlust einer CO2-Einheit im
Tochterionenspektrum konnte der Baustein als eine Tetradehydroprolin-Einheit
aufgeklärt werden. Die HMBC-Kopplung von H-4 zu C-1’ und die große chemische
Verschiebung von C-4 (δC 34.3 ppm) im Vergleich zur vergleichbaren Position in der
Verbindung A88696C (δC 28.7 ppm)[80] konnte die Verknüpfung der Einheit an C-4
bestätigen. Die Struktur von TÜ 6392 D ist in Abbildung 67 zusammenfassend
dargestellt.
O
H3CCH3
CH3
OH
CH3H3C
O
1
3
57
9
12
15
16
1718
1920
22
O
OH
HN1'
3'
5'
Abbildung 67: Strukturformel von TÜ 6392 D.
7.5.4 Biologische Aktivität
Die Arbeiten zur Bestimmung der biologischen Aktivität gegen Gram-positive und
Gram-negative Bakterien, Eukaryoten, humane Krebszelllinien und ausgewählte
Enzyme werden von Dr. Dirk Schulz in der Arbeitsgruppe von Prof. Johannes F.
Imhoff im Kieler Wirkstoffzentrum KiWiZ am Institut für Meereswissenschaften IFM-
Geomar koordiniert und dauern zum Zeitpunkt dieser Dissertation an. Bisher konnte
die molekulare Zielstruktur bzw. eine entsprechende biologische Aktivität von TÜ
6392 D nicht gefunden werden.
118
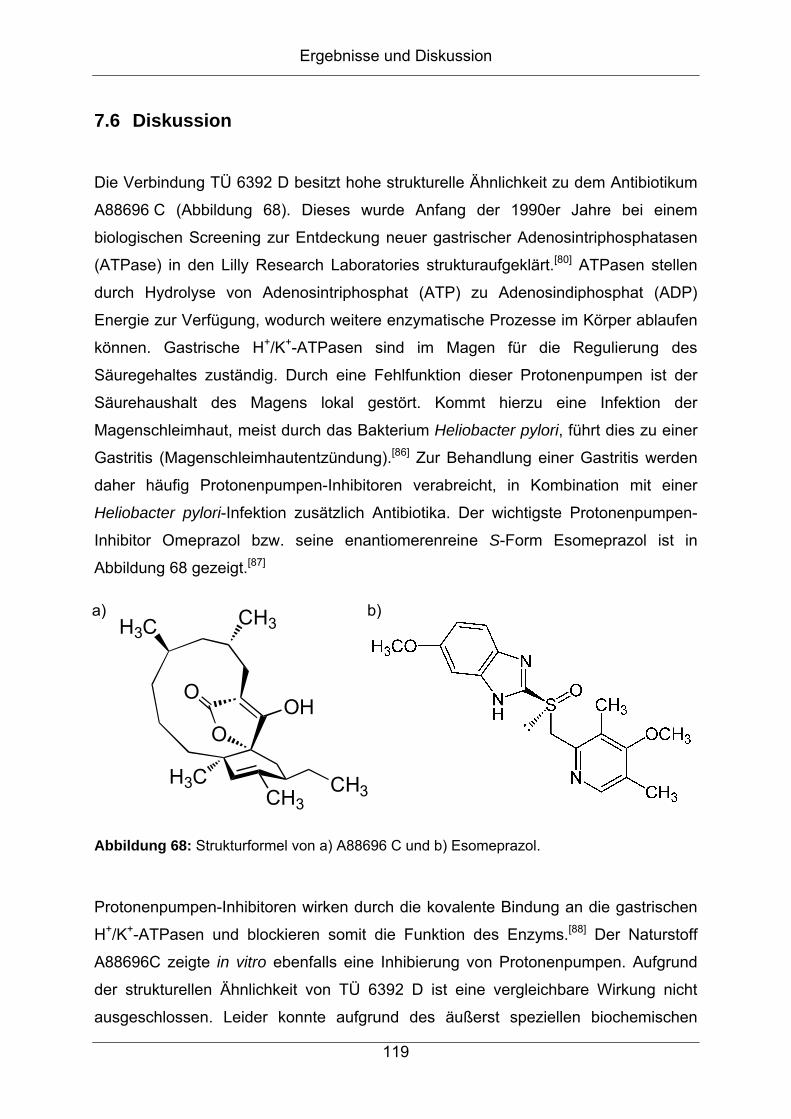
Ergebnisse und Diskussion
7.6 Diskussion
Die Verbindung TÜ 6392 D besitzt hohe strukturelle Ähnlichkeit zu dem Antibiotikum
A88696 C (Abbildung 68). Dieses wurde Anfang der 1990er Jahre bei einem
biologischen Screening zur Entdeckung neuer gastrischer Adenosintriphosphatasen
(ATPase) in den Lilly Research Laboratories strukturaufgeklärt.[80] ATPasen stellen
durch Hydrolyse von Adenosintriphosphat (ATP) zu Adenosindiphosphat (ADP)
Energie zur Verfügung, wodurch weitere enzymatische Prozesse im Körper ablaufen
können. Gastrische H+/K+-ATPasen sind im Magen für die Regulierung des
Säuregehaltes zuständig. Durch eine Fehlfunktion dieser Protonenpumpen ist der
Säurehaushalt des Magens lokal gestört. Kommt hierzu eine Infektion der
Magenschleimhaut, meist durch das Bakterium Heliobacter pylori, führt dies zu einer
Gastritis (Magenschleimhautentzündung).[86] Zur Behandlung einer Gastritis werden
daher häufig Protonenpumpen-Inhibitoren verabreicht, in Kombination mit einer
Heliobacter pylori-Infektion zusätzlich Antibiotika. Der wichtigste Protonenpumpen-
Inhibitor Omeprazol bzw. seine enantiomerenreine S-Form Esomeprazol ist in
Abbildung 68 gezeigt.[87]
a) b)
O
H3CCH3
CH3
OH
CH3H3C
O
Abbildung 68: Strukturformel von a) A88696 C und b) Esomeprazol.
Protonenpumpen-Inhibitoren wirken durch die kovalente Bindung an die gastrischen
H+/K+-ATPasen und blockieren somit die Funktion des Enzyms.[88] Der Naturstoff
A88696C zeigte in vitro ebenfalls eine Inhibierung von Protonenpumpen. Aufgrund
der strukturellen Ähnlichkeit von TÜ 6392 D ist eine vergleichbare Wirkung nicht
ausgeschlossen. Leider konnte aufgrund des äußerst speziellen biochemischen
119
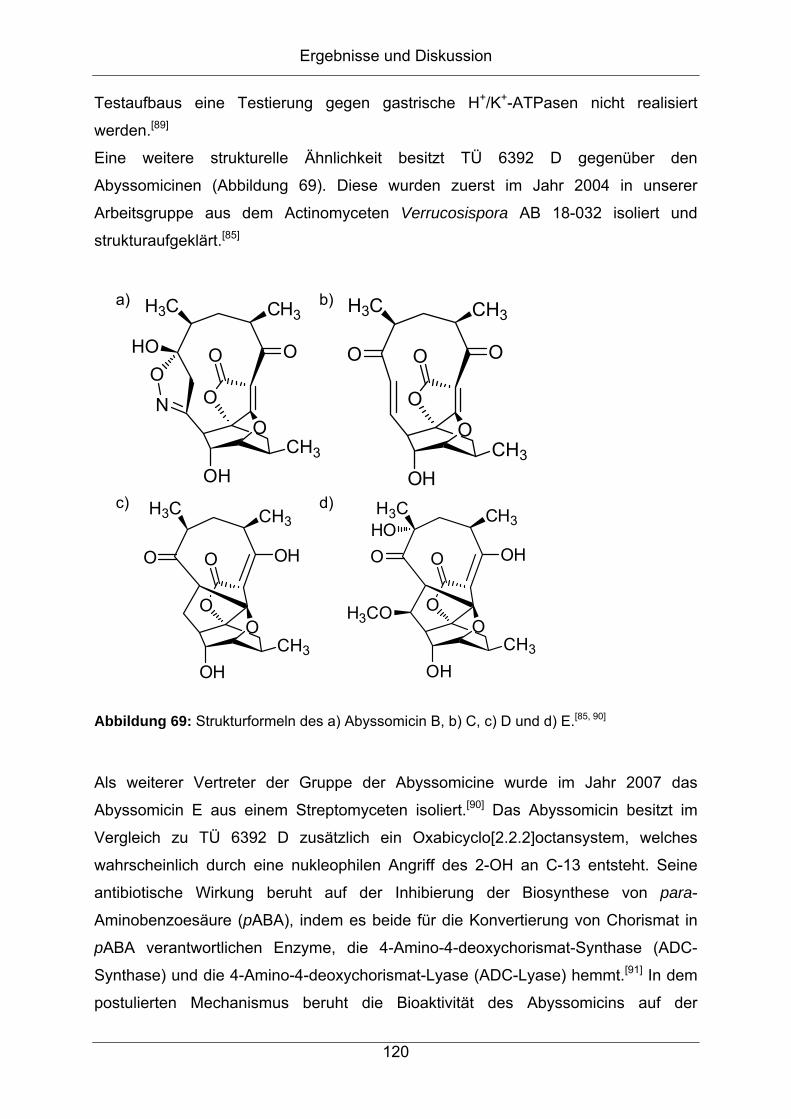
Ergebnisse und Diskussion
Testaufbaus eine Testierung gegen gastrische H+/K+-ATPasen nicht realisiert
werden.[89]
Eine weitere strukturelle Ähnlichkeit besitzt TÜ 6392 D gegenüber den
Abyssomicinen (Abbildung 69). Diese wurden zuerst im Jahr 2004 in unserer
Arbeitsgruppe aus dem Actinomyceten Verrucosispora AB 18-032 isoliert und
strukturaufgeklärt.[85]
120
a) b)
O
CH3H3C
O
O
O
OH
CH3
HO
O
N
O
CH3H3C
O
O
O
OH
CH3
O
d) c)
Abbildung 69: Strukturformeln des a) Abyssomicin B, b) C, c) D und d) E.[85, 90]
Als weiterer Vertreter der Gruppe der Abyssomicine wurde im Jahr 2007 das
Abyssomicin E aus einem Streptomyceten isoliert.[90] Das Abyssomicin besitzt im
Vergleich zu TÜ 6392 D zusätzlich ein Oxabicyclo[2.2.2]octansystem, welches
wahrscheinlich durch eine nukleophilen Angriff des 2-OH an C-13 entsteht. Seine
antibiotische Wirkung beruht auf der Inhibierung der Biosynthese von para-
Aminobenzoesäure (pABA), indem es beide für die Konvertierung von Chorismat in
pABA verantwortlichen Enzyme, die 4-Amino-4-deoxychorismat-Synthase (ADC-
Synthase) und die 4-Amino-4-deoxychorismat-Lyase (ADC-Lyase) hemmt.[91] In dem
postulierten Mechanismus beruht die Bioaktivität des Abyssomicins auf der
OH
CH3H3C
O
O
O
OH
CH3
O OH
CH3H3C
HO
O O
OO
H3CO
CH3
OH

Ergebnisse und Diskussion
offensichtlichen Ähnlichkeit des Oxabicyclooctansystems zur Konformation des
Chorismats in Lösung (Abbildung 70).
COOH
O
HO
HOOC
COOH
OH
O
COOH
=
Abbildung 70: Diaxiale Konformation von Chorismat in wässriger Lösung.
Des Weiteren scheint das Michael-System an C7 – C9 für die antibakterielle Aktivität
verantwortlich zu sein, vergleichbar mit dem Michael-System des Piceamycins. Das
antibiotisch inaktive Abyssomicin B besitzt ähnlich wie das N-Acetylcystein-
Piceamycin eine Substitution an dieser Stelle, einhergehend mit dem Verlust der
Bioaktivität.
Die Biosynthese von pABA ist essentieller Teil der Biosynthese von
Tetrahydrofolaten.[92] Aufgrund des Vorkommens dieses Biosynthesewegs in
Bakterien, Pilzen und Pflanzen, jedoch nicht in Menschen, ist die para-
Aminobenzoesäuresynthese ein bevorzugtes Ziel für Antibiotika. Aufgrund der
strukturellen Ähnlichkeit zu den Abyssomicinen sollte die Verbindung TÜ 6392 D
ebenfalls gegen die Aminodesoxychorismat-Synthase getestet werden. Die Arbeiten
hierzu dauern zum Zeitpunkt der Dissertation an.
Aufgrund der strukturellen Ähnlichkeit kann ausgehend von der Biosynthese von
Abyssomicin C die Biosynthese von TÜ 6392 D postuliert werden. Die Biosynthese
von Polyketiden wird historisch gesehen in drei Gruppen aufgeteilt, welche aufgrund
ihrer Abfolgen und der Art ihrer Enzyme charakterisiert werden. Polyketidsynthasen
des Typs I (PKS-I) sind in Modulen organisierte Enzyme, wobei jeses Modul für einen
kettenverlängerten Schritt verantwortlich ist. Bei Polyketidsynthasen des Typs II
(PKS-II) handelt es sich hingegen um Multienzymkomplexe, die aus einem einzigen
Satz von iterativ wirkenden Enzymen bestehen und meist hocharomatische
Polyketide produzieren. Polyketidsynthasen des Typs III (PKS-III) besitzen im
Vergleich zu den beiden vorher genannten PKS einen etwas anderen
Biosyntheseweg. Während bei Typ I- und Typ II-Polyketidsynthasen die Bausteine
zunächst durch eine Addition an eine Phosphopantetheineinheit eines
121

Ergebnisse und Diskussion
Acylcarrierproteins (ACP) aktiviert werden muß, können die Enzyme der Typ-III
Polyketidsynthasen (PKS-III) die Substrate direkt umsetzen.
Bei den Abyssomicinen handelt es sich um Produkte der Typ-I Polyketidsynthase.
Aufgrund von durchgeführten Fütterungsexperimenten konnte der Ursprung jedes
einzelnen Kohlenstoffatoms des Abyssomicins aufgeklät werden. Insgesamt wird
Abyssomicin C aus zwei Propionat-, einer Glucose- und fünf Acetateinheiten
zusammengesetzt. Zu der Verbindung TÜ 6392 D kann ebenfalls eine Hypothese zur
Biosynthese aufgestellt werden, ausgehend auf einem strukturchemischen Vergleich
von Abyssomicin C und TÜ 6392 D. Eine Hypothese zum Aufbau des linearen
Polyketidvorläufers von TÜ 6392 D ist in Abbildung 71 gezeigt.
O
KS AT ACP KS AT
DH KR
ACP
SO
KS
SO
AT
DH KR
ACP KS
SO
AT
DH KR
ACP KS
SO
ER
AT
DH KR
ACP KS
S
ER
O
AT
DH KR
ACP KS
S
ER
O
AT
DH KR
ACP
S
ER
BeladungModul 1
Modul 2Modul 3
Modul 4Modul 5
Modul 6
Abbildung 71: Hypothese zum Aufbau des linearen Polyketidvorläufers von TÜ 6392 D.
Das Kohlenstoffgerüst von TÜ 6392 D wird linear von einer Typ-I Polyketidsynthase
aus fünf Propionat- und zwei Acetateinheiten synthetisiert. Zunächst wird eine
Propionateinheit mit der Beladungseinheit verknüpft, anschließend erfolgt die
zweifache Kettenverlängerung mit Propionateinheiten, welche beide zum Alken
reduziert werden. Die folgende Kettenverlängerung erfolgt durch einen
Acetatbaustein, welcher vollständig zum Alkan reduziert wird. Im Anschluß erfolgt
durch Modul 4 und 5 die Verknüpfung von zwei weiteren Propionateinheiten, welche
ebenfalls beide vollständig reduziert werden. Abschließend wird eine weitere
Acetateinheit als siebter Baustein angefügt und ebenfalls vollständig reduziert.
122

Ergebnisse und Diskussion
Zuletzt wird die Enolpyruvateinheit eingebaut und dabei gleichzeitig der lineare
Baustein vom dem Multienzymkomplex getrennt. Für das Abyssomicin wurden zwei
unterschiedliche Mechanismen zur Verknüpfung vorgeschlagen, welche beide zum
gewünschten Produkt führen (Abbildung 72). Zum einen kann die Freisetzung der
linearen Polyketidkette durch den nucleophilen Angriff der Hydroxygruppe des
Enoylpyruvats stattfinden, bei der ein Ester gebildet wird. Im Anschluß findet die
Cyclisierung zur Tetronsäureeinheit statt.
O S
ACP
HO
SOACP
O O
O SACP
OO
OH
O S
ACP
HO
SOACP
O SOH
OO
OH
OH
ACP
Glykolyse Glykolyse
a) b)
Abbildung 72: Hypothese zur Biosynthese des linearen Tetronatbaustein von TÜ 6392 D
nach a) Jia et al.[93], b) nach Keller et al.[94]
Beim alternativen Mechanismus erfolgt zunächst ein nukleophiler Angriff der
Polyketidkette auf die Enolpyruvateinheit unter Ausbildung einer Kohlenstoff-
Kohlenstoff-Bindung, im Anschluß würde das Lacton gebildet werden. Zur
Ausbildung des Spirotetronats erfolgt dann zwischen der exozyklischen
Doppelbindung des Tetronatbausteins und den beiden Doppelbindungen des
Olefinrests eine Diels-Alder-Reaktion. Im letzten Schritt erfolgt die Addtion einer
formalen Tetradehydroprolineinheit an C-4 von TÜ 6392 D (Abbildung 73).
123

Ergebnisse und Diskussion
OO
HO
O
O
HO
HN
O
OH
[4+2]-Cyclo-Addition
O
O
HO
HN
O
HO
Abbildung 73: Hypothese zur Zyklisierung des linearen Vorläufers analog zur Abyssomicin-
Biosynthese.[94]
124

Experimenteller Teil
125
8 Experimenteller Teil
8.1 Chemisches Screening mittels HPLC-DAD
Die Sekundärstoffanalytik wurde mit einer Methode, die von Fiedler et al. 1993
etabliert wurde, durchgeführt.[33] Die HPLC-DAD-Experimente wurden mit einer
Agilent HP 1090M Anlage mit thermostatisiertem Autosampler und Diodenarray
Detektor (Agilent Technologies, Waldbronn) durchgeführt. Sofern nicht anders
angegeben, wurden als Detektionswellenlängen = 210, 230, 260, 280, 310, 360,
435, 500 nm und als Pilotwellenlänge = 210 nm gewählt. Die Flussrate lag bei
2 mL/min. Injiziert wurden jeweils 10 µL Analytlösung. Die verwendeten Parameter
sind in Tabelle 10 zusammengefasst.
Die unterschiedlichen Sekundärmetabolite wurden anhand ihres UV/Vis-Spektrums
und ihrer Retentionszeiten charakterisiert. Die UV/Vis-Spektren wurden mit den
Spektren der HPLC-UV/Vis-Datenbank der Arbeitsgruppe Fiedler verglichen,[33] um
auszuschließen, dass bereits bekannte Substanzen isoliert werden.
Tabelle 10: HPLC-Parameter für die Sekundärstoffanalytik.
Säulenmaterial Korn-
größe
Maße der
Hauptsäule
Maße der
Vorsäule Mobile Phase Gradient
Nucleosil 100 C-18
(Maisch,
Ammerbuch)
5 µm 125 mm x
4.6 mm I.D.
20 mm x
4.6 mm I.D.
0.1 % H3PO4
(A),
CH3CN (B)
0 % - 100 %
B in 15 min
8.2 HPLC-DAD-ESI-Massenspektroskopie
Für die massenspektrometrischen Experimente wurde ein QTRAP 2000-
Massenspektrometer (MDS Sciex/Applied Biosystems, Darmstadt), das mit
Elektrospray-Ionisation ausgestattet ist, verwendet. Das Massenspektrometer wurde
mit der Analyst-Software Version 1.4.1 gesteuert und alle Experimente mit dieser
Software ausgewertet.

Experimenteller Teil
126
Gekoppelt wurde das Massenspektrometer mit einer Kapillar 1100-HPLC-Anlage von
Agilent Technologies (Waldbronn) mit DAD-Detektor. Die Flussrate betrug 60 µL/min.
Injiziert wurden 10 µL. Eine Zusammenfassung der Parameter ist in Tabelle 11
angegeben. Ein Gradient von 0 % B bis 100 % B in 10 min wurde als
Routinegradient verwendet.
Tabelle 11: HPLC-Parameter für die Kapillar-HPLC-DAD-ESI-MS-Kopplung.
Säulenmaterial Partikel-
größe
Poren-
größe Säulemaße Mobile Phase
Luna RP C-18
(Phenomenex,
Aschaffenburg)
3 µm 100 Å 50 mm x
1 mm I.D.
H2O + 0.1 % HCOOH (A),
MeOH + 0.1 % HCOOH (B)
8.3 GC-MS
Sämtliche GC-MS-Experimente wurden von Dipl.-Ing. Graeme Nicholson am Institut
für Organische Chemie an der Eberhard-Karls-Universität Tübingen durchgeführt.
Die entsprechenden Proben (ca. je 100 µg) wurden unter Stickstoff hydrolysiert
(200 µL 6 N HCl und 5 µL Phenol, 110 °C, 24 h) und das Hydrolysat im
Stickstoffstrom zur Trockne eingeengt. Das Hydrolysat wurde mit 200 μL 15 %
Acetylchlorid in abs. Ethanol für 30 min bei 110 °C behandelt und im Stickstoffstrom
zur Trockne eingeengt. Anschließend wurde mit 100 μL Dichlormethan und 50 μL
Trifluoressigsäureanhydrid für 10 min bei 110 °C behandelt, wieder zur Trockne
gebracht und mittels eines Agilent 5975 GC-MS (Agilent Technologies, Waldbronn)
mit einer Chirasil Kapillarsäule (20 m x 0.25 mm, 0.13 μm Filmdicke) analysiert.

Experimenteller Teil
127
8.4 ESI-FT-ICR-Massenspektrometrie
Die hochauflösenden Massenspektren wurden von Dipl.-Ing. Graeme Nicholson an
der Universität Tübingen durchgeführt. Für die ESI-FT-ICR-MS-Messung wurde ein
APEX II FT-ICR-Massenspektrometer (Bruker-Daltonics, Bremen) mit Elektrospray-
Ionisierung im Positivionenmodus verwendet. Die Proben wurden in Methanol in
einer Konzentration von ca. 1 µg/mL gelöst.
8.5 HPLC-ESI-FT-Orbitrap-Massenspektrometrie
Weitere hochauflösende Massenspektren, ergänzend zur ESI-FT-ICR-
Massenspektrometrie, wurden an der Technischen Universität Berlin an einer LTQ
Orbitrap XL (Thermo Scientific, Bremen) mit Elektrospray-Ionisierung durchgeführt.
Dem Massenspektrometer wurde zur chromatographischen Trennung eine Agilent
1200 HPLC-Anlage (Agilent Technologies, Waldbronn) vorgeschaltet. Die genauen
chromatographischen Parameter sind in Tabelle 12 gezeigt. Die Flussrate betrug 1.0
mL/min. Injiziert wurden 10 µL. Ein Gradient von 5 % B bis 100 % B in 25 min wurde
als Routinegradient verwendet. Zur Auswertung der HPLC-MS-Spektren wurde die
Software Xcalibur des Geräteherstellers verwendet.
Tabelle 12: Verwendete HPLC-Parameter für die ESI-FT-Orbitrap-MS-Kopplung.
Säulenmaterial Partikel-
größe
Poren-
größe Säulemaße Mobile Phase
Eclipse XDB-C18
(Agilent, Waldbronn) 5 µm 100 Å
150 mm x
4.6 mm I.D.
H2O + 0.1 % HCOOH (A),
MeOH + 0.1 % HCOOH (B)

Experimenteller Teil
128
8.6 NMR-Spektroskopie
Für die NMR-spektroskopischen Experimente wurde ein DRX 500 NMR-
Spektrometer (Bruker, Karlsruhe) mit einem BBI-Probenkopf mit z-Gradienten
verwendet. Zur Auswertung aller ein- und zweidimensionaler Spektren wurde die
Topspin-Software verwendet. Die chemischen Verschiebungen sind in δ-Werten
(ppm) relativ zum Restsignal der undeuterierten Lösungsmittelanteile angegeben.
Die Kopplungskonstanten J sind in Hertz (Hz) angegeben. Das Lösungsmittel ist
zusammen mit den spektroskopischen Daten aufgeführt. Die Spektren wurden,
sofern nicht anders angegeben, bei 298 K aufgenommen. Die 13C-NMR-Spektren
wurden mit einer 1H-Breibandentkopplung aufgenommen.

Abkürzungen
129
9 Anhang
9.1 Abkürzungsverzeichnis
Å Ångström
ACP acyl carrier protein
AT Acyltransferase
ATP Adenosintriphosphat
ADP Adenosindiphosphat
COSY correlated spectroscopy
CRM charged residue model
Da Dalton
DAD Dioden Array Detektor
DBE Doppelbindungsäquivalente
DEPT distortionless enhancement by polarisation transfer
DH Dehydratase
DNA Desoxyribonucleinsäure
DMSO Dimethylsulfoxid
DQF-COSY COSY mit Doppelquantenfilter (double quantum filtered
COSY)
EMS enhanced MS
EPI enhanced product ion scan
ER Enoylreduktase
ESI Elektrospray-Ionisation
FID free induction decay
FT-ICR-MS Fourier-Transform-Ionen-Zyklotron-
Massenspektrometer/Massenspektrometrie

Abkürzungen
130
GC Gaschromatographie
HMBC heteronuclear multiple bond correlation
HMQC heteronuclear multiple quantum coherence
HPLC High performance liquid chromatography
(Hochleistungsflüssigkeitschromatographie)
HSQC heteronuclear single quantum coherence
Hz Hertz
IC50 mittlere inhibitorische Konzentration
I.D. Innendurchmesser
IEM ion evaporation model (Ionenevaporationsmodell)
J Kopplungskonstante
K Kelvin
KR Ketoreduktase
KS Ketosynthase
LC Flüssigkeitschromatographie
m Masse
m/z Masse zu Ladungs-Verhältnis
MALDI Matrix-assisted laser desorption/ionisation (Matrix-
unterstützte Laserdesorption/Ionisation)
mAU milliabsorbance units
MeOH Methanol
MHz Megahertz
MIC minimale Hemmkonzentration
min Minuten
mL Milliliter
MLEV Entkopplungssequenz nach Malcom Levitt
mm Millimeter

Abkürzungen
131
MS Massenspektrometer/Massenspektrometerie
nm Nanometer
NMR nuclear magnetic resonance (Kernspinresonanz)
NOESY nuclear overhauser enhancement spectroscopy (Kern-
Overhauser-Effekt-Spektroskopie)
PKS Polyketidsynthase
ppm parts per million
RNA Ribonucleinsäure
RP reversed phase (Umkehrphase)
Rt Retentionszeit
T Temperatur
TIC Totalionenchromatogramm
TOCSY total correlated spectroscopy
TOF time of flight
TPPI time proportional phase increment (zur Evolutionszeit
proportionales Phaseninkrement)
UV ultraviolettes Licht
Vis sichtbares Licht
XIC extracted ion chromatogram
Wellenlänge

Literaturverzeichnis
132
9.2 Literaturverzeichnis
[1] D. Schulz, Universität Tübingen (Tübingen), 2009. [2] D. J. Newman, G. M. Cragg, J Nat Prod 2007, 70, 461-477. [3] A. Fleming, Br J Exp Pathol 1929, 10, 226 -236. [4] H.-J. Böhm, G. Klebe, H. Kubinyi, Wirkstoffdesign, Spektrum Akademischer
Verlag, Heidelberg, Berlin, Oxford, 2002. [5] G. Habermehl, P. E. Hammann, H. C. Krebs, Naturstoffchemie, Springer
Verlag, Berlin, Heidelberg, New York, 2002. [6] G. Domagk, Deutsche Medizinische Wochenschrift 1935, 61, 250. [7] H. B. Bode, R. Muller, Angew Chem Int Ed Engl 2005, 44, 6828-6846. [8] F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand, D. Häbich, Angew
Chem Int Ed Engl 2006, 45, 5072-5129. [9] Technology Review, Vol. 4, 2007. [10] C. W. Ford, J. C. Hamel, D. M. Wilson, J. K. Moerman, D. Stapert, R. J.
Yancey, Jr., D. K. Hutchinson, M. R. Barbachyn, S. J. Brickner, Antimicrob Agents Chemother 1996, 40, 1508-1513.
[11] D. Felmingham, J Chemother 2005, 17 Suppl 1, 3-4. [12] M. Debono, B. J. Abbott, R. M. Molloy, D. S. Fukuda, A. H. Hunt, V. M.
Daupert, F. T. Counter, J. L. Ott, C. B. Carrell, L. C. Howard, et al., J Antibiot (Tokyo) 1988, 41, 1093-1105.
[13] M. S. Butler, M. A. Cooper, J Antibiot (Tokyo) 2011, 64, 413-425. [14] S. Altmaier, K. Cabrera, J Sep Sci 2008, 31, 2551-2559. [15] F. Lottspeich, J. W. Engels, Bioanalytik, Spektrum Akademischer Verlag,
München, 2006. [16] J. H. Gross, Mass Spectrometry, Springer Verlag, Berlin, Heidelberg, New
York, 2004. [17] K. Cammann, Instrumentelle Analytische Chemie, Spektrum akademischer
Verlag, Heidelberg, Berlin, 2001. [18] E. de Hoffmann, Stroobant, V., Mass Spectrometry, John Wiley & Sons,
Chichester, New York, Weinheim, Brisbane, Singapore, Toronto, 2003. [19] H. Budzikiewicz, Schäfer, M., Massenspektrometrie, Wiley-VCH Verlag,
Weinheim, 2005. [20] A. Makarov, Anal Chem 2000, 72, 1156-1162. [21] W. f. Wikimedia commons. [22] A. Makarov, E. Denisov, A. Kholomeev, W. Balschun, O. Lange, K. Strupat, S.
Horning, Anal Chem 2006, 78, 2113-2120. [23] M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen
Chemie, Thieme Verlag, 1991. [24] M. H. Lewitt, Spin Dynamics, Wiley & Sons, Chichester, 2001. [25] H. Friebolin, Ein- und zweidimensionale NMR-Spektroskopie, Wiley-VCH,
1999. [26] H. Pfeifer, Magnetic Resonance in Chemistry 1999, 37, 154 - 159. [27] H. Günther, NMR-Spektroskopie, Thieme Verlag, 1992. [28] S. Berger, S. Braun, 200 and more NMR experiments, Wiley-VCH, Weinheim,
2004. [29] R. S. Macomber, A complete introduction to modern NMR spectroscopy,
Wiley-VCH, New York, 1998. [30] L. Müller, J Am Chem Soc 1979, 101, 4481 - 4484.

Literaturverzeichnis
133
[31] A. Bax, M. F. Summers, J Am Chem Soc 1986, 108, 2093 - 2094. [32] H. Kogler, O. W. Sörensen, R. R. Bodenhausen, J Magn Reson 1983, 55, 157
- 163. [33] H. P. Fiedler, Nat Prod Lett 1993, 2, 119 - 128. [34] J. Garbaye, Experientia 1991, 47, 370 - 375. [35] J. M. Whipps, J Exp Bot 2001, 52, 487-511. [36] J. Riedlinger, Universität Tübingen (Tübingen), 2006. [37] T. Hatsui, C. Nojima, H. Takeshita, Bull Chem Soc Jpn 1989, 62, 2932 - 2938. [38] S. Bolvig, F. Duus, E. Hansen, Magn Reson Chem 1998, 36, 315 - 324. [39] C. E. Hadden, G. E. Martin, V. V. Krishnamurthy, Magn Reson Chem 2000,
38, 143 - 147. [40] R. Hooft van Huijsduijnen, A. Bombrun, D. Swinnen, Drug Discov Today 2002,
7, 1013-1019. [41] S. Omura, A. Nakagawa, K. Shibata, H. Sano, Tetrahedron Lett 1982, 23,
4713 - 4716. [42] M. Hayashi, M. Ohno, S. Satoi, J. Chem. Soc. Chem. Commun. 1908, 119 -
121. [43] K. Kinoshita, S. Takenaka, M. Hayashi, J Antibiot (Tokyo) 1991, 44, 1270-
1273. [44] A. D. Argoudelis, T. A. Brinkley, T. F. Brodasky, J. A. Buege, H. F. Meyer, S.
A. Mizsak, J Antibiot (Tokyo) 1982, 35, 285-294. [45] A. D. Argoudelis, L. Baczynskyj, J. A. Buege, V. P. Marshall, S. A. Mizsak, P.
F. Wiley, J Antibiot (Tokyo) 1987, 40, 408-418. [46] M. L. Gilpin, D. Fulston, R. Payne, R. Cramp, I. Hood, J Antibiot (Tokyo) 1995,
48, 1081 - 1085. [47] G. Meurer, M. Gerlitz, E. Wendt-Pienkowski, L. C. Vining, J. Rohr, C. R.
Hutchinson, Chem Biol 1997, 4, 433-443. [48] W. Keller-Schierlein, J. Sauerbier, U. Vogler, H. Zähner, Helv Chim Acta 1970,
53, 779 - 789. [49] W. Keller-Schierlein, A. Muller, Experientia 1970, 26, 929-930. [50] A. Naganawa, Y. Ichikawa, M. Isobe, Tetrahedron 1994, 50, 8969 - 8982. [51] M. Dubost, P. Ganter, R. Maral, L. Ninet, S. Pinnert, J. Preudhomme, G. H.
Werner, Compt Rend 1963, 257, 1813 - 1815. [52] F. Arcamone, S. Franceschi, S. Penco, A. Selva, Tetrahedron Lett 1969, 13,
1007 - 1010. [53] M. J. Piccart-Gebhart, N Engl J Med 2006, 354, 2177-2179. [54] X. C. Cheng, T. Kihara, H. Kusakabe, J. Magae, Y. Kobayashi, R. P. Fang, Z.
F. Ni, Y. C. Shen, K. Ko, I. Yamaguchi, et al., J Antibiot (Tokyo) 1987, 40, 907-909.
[55] X. C. Cheng, M. Ubukata, K. Isono, J Antibiot (Tokyo) 1990, 43, 890-896. [56] J. Magae, C. Watanabe, H. Osada, X. C. Cheng, K. Isono, J Antibiot (Tokyo)
1988, 41, 932-937. [57] M. Ubukata, Cheng, X.-Ch., Uzawa, J., Isono, K., J Chem Soc Perkin 1 1995,
2399. [58] K. U. Bindseil, P. Hug, H. H. Peter, F. Petersen, B. E. Roggo, J Antibiot
(Tokyo) 1995, 48, 457-461. [59] V. B. Rajgarhia, N. D. Priestley, W. R. Strohl, Metab Eng 2001, 3, 49-63. [60] C. K. Okoro, R. Brown, A. L. Jones, B. A. Andrews, J. A. Asenjo, M.
Goodfellow, A. T. Bull, Antonie Van Leeuwenhoek 2009, 95, 121-133. [61] J. D. A. Clark, Geomorphology 2006, 73, 101 - 114. [62] A. Kulik, Personal Communication, 2009.

Literaturverzeichnis
134
[63] N. Matsumori, D. Kaneno, M. Murata, H. Nakamura, K. Tachibana, J Org Chem 1999, 64, 866-876.
[64] N. Hayashi, Y. Kobayashi, Y. Kishi, Org Lett 2001, 3, 2249-2252. [65] M. Zweckstetter, Nat Protoc 2008, 3, 679-690. [66] S. Omura, Macrolide Antibiotics: Chemistry, Biology, and Practice, Academic
Press, Boston, 2002. [67] S. Omura, Macrolide Antibiotics - Chemistry, Biology, and Practice, Acedemic
Press, Amsterdam, Boston, New York, Oxford, 2002. [68] G. R. Pettit, Z. A. Cichacz, US Patent 5430053, 1995. [69] G. R. Pettit, Z. A. Cichacz, F. Gao, M. R. Boyd, J. M. Schmidt, J Chem Soc
Chem Comm 1994, 1111 - 1112. [70] R. A. Isbrucker, J. Cummins, S. A. Pomponi, R. E. Longley, A. E. Wright,
Biochem Pharmacol 2003, 66, 75-82. [71] Y. Dong, J. Yang, H. Zhang, J. Lin, X. Ren, M. Liu, X. Lu, J. He, J Nat Prod
2006, 69, 128-130. [72] M. Kobayashi, J. Tanaka, T. Katori, M. Matsura, I. Kitagawa, Tetrahedron Lett
1989, 30, 2963 - 2966. [73] M. R. Bubb, I. Spector, A. D. Bershadsky, E. D. Korn, J Biol Chem 1995, 270,
3463-3466. [74] E. L. Hazen, R. F. Brown, US Patent 797183, 1957. [75] R. L. Bunch, J. M. Mcguire, US Patent 2653899, 1953. [76] J. M. Lancelin, F. Paquet, J. M. Beau, Tetrahedron Lett 1988. [77] D. R. Harris, S. G. Mcgeachin, H. H. Mills, Tetrahedron Lett 1965, 11, 679 -
685. [78] M. E. Maier, Nat Prod Rep 2009, 26, 1105-1124. [79] Dictionary of Natural Products on DVD, Chapman & Hall Chemical Database,
London, 1982 - 2011. [80] R. Bonjouklian, J. S. Mynderse, A. H. Hunt, J. B. Deeter, Tetrahedron Lett
1993, 34, 7857 - 7860. [81] F. Petersen, H. Zahner, J. W. Metzger, S. Freund, R. P. Hummel, J Antibiot
(Tokyo) 1993, 46, 1126-1138. [82] Y. Futamura, R. Sawa, Y. Umezawa, M. Igarashi, H. Nakamura, K. Hasegawa,
M. Yamasaki, E. Tashiro, Y. Takahashi, Y. Akamatsu, M. Imoto, J Am Chem Soc 2008, 130, 1822-1823.
[83] M. Imoto, K. Umezawa, Y. Takahashi, H. Naganawa, Y. IItaka, H. Nakamura, Y. Koizumi, Y. Sasaki, M. Hamada, T. Sawa, T. Takeuchi, J Nat Prod 1990, 53, 825 - 829.
[84] N. Neuss, M. Gormann, W. Hargrove, N. J. Cone, K. Biemann, G. Buchi, R. E. Manning, J Am Chem Soc 1964, 86, 1440 - 1442.
[85] B. Bister, D. Bischoff, M. Ströbele, J. Riedlinger, A. Reicke, F. Wolter, A. T. Bull, H. Zähner, H. P. Fiedler, R. D. Süssmuth, Angew Chem Int Ed Engl 2004, 43, 2574-2576.
[86] https://secure.wikimedia.org/wikipedia/de/wiki/Magengeschwür. [87] J. Labenz, K. U. Petersen, W. Rösch, H. Koelz, Deutsches Ärzteblatt 2002,
37, 2425 - 2429. [88] W. Kühlbrandt, Nat Rev Mol Cell Biol 2004, 5, 282-295. [89] A. H. Dantzig, P. L. Minor, J. L. Garrigus, D. S. Fukuda, J. S. Mynderse,
Biochem Pharmacol 1991, 42, 2019-2026. [90] X. M. Niu, S. H. Li, H. Gorls, D. Schollmeyer, M. Hilliger, S. Grabley, I. Sattler,
Org Lett 2007, 9, 2437-2440.

Literaturverzeichnis
135
[91] S. Keller, H. S. Schadt, I. Ortel, R. D. Süssmuth, Angew Chem Int Ed Engl 2007, 46, 8284-8286.
[92] A. Bermingham, J. P. Derrick, Bioessays 2002, 24, 637-648. [93] X. Y. Jia, Z. H. Tian, L. Shao, X. D. Qu, Q. F. Zhao, J. Tang, G. L. Tang, W.
Liu, Chem Biol 2006, 13, 575-585. [94] S. Keller, Technische Universität Berlin (Berlin), 2007.

136
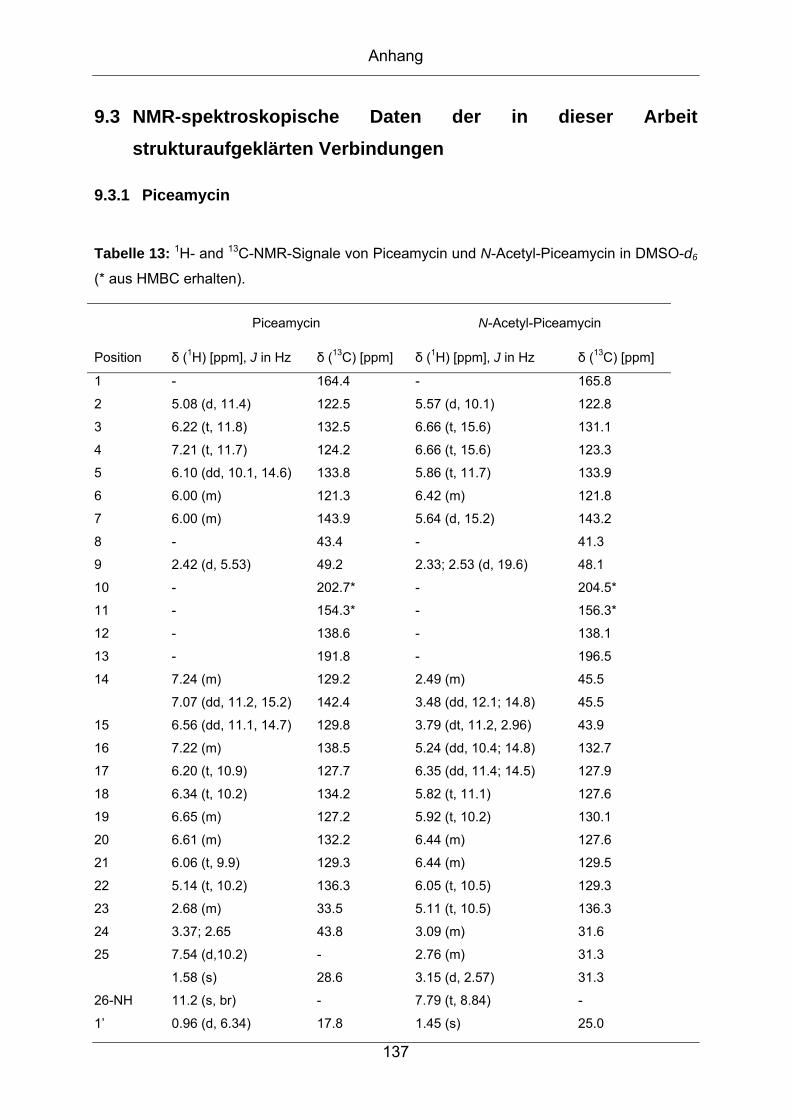
Anhang
137
9.3 NMR-spektroskopische Daten der in dieser Arbeit
strukturaufgeklärten Verbindungen
9.3.1 Piceamycin
Tabelle 13: 1H- and 13C-NMR-Signale von Piceamycin und N-Acetyl-Piceamycin in DMSO-d6
(* aus HMBC erhalten).
Piceamycin N-Acetyl-Piceamycin
Position δ (1H) [ppm], J in Hz δ (13C) [ppm] δ (1H) [ppm], J in Hz δ (13C) [ppm]
1 - 164.4 - 165.8
2 5.08 (d, 11.4) 122.5 5.57 (d, 10.1) 122.8
3 6.22 (t, 11.8) 132.5 6.66 (t, 15.6) 131.1
4 7.21 (t, 11.7) 124.2 6.66 (t, 15.6) 123.3
5 6.10 (dd, 10.1, 14.6) 133.8 5.86 (t, 11.7) 133.9
6 6.00 (m) 121.3 6.42 (m) 121.8
7 6.00 (m) 143.9 5.64 (d, 15.2) 143.2
8 - 43.4 - 41.3
9 2.42 (d, 5.53) 49.2 2.33; 2.53 (d, 19.6) 48.1
10 - 202.7* - 204.5*
11 - 154.3* - 156.3*
12 - 138.6 - 138.1
13 - 191.8 - 196.5
14 7.24 (m) 129.2 2.49 (m) 45.5
7.07 (dd, 11.2, 15.2) 142.4 3.48 (dd, 12.1; 14.8) 45.5
15 6.56 (dd, 11.1, 14.7) 129.8 3.79 (dt, 11.2, 2.96) 43.9
16 7.22 (m) 138.5 5.24 (dd, 10.4; 14.8) 132.7
17 6.20 (t, 10.9) 127.7 6.35 (dd, 11.4; 14.5) 127.9
18 6.34 (t, 10.2) 134.2 5.82 (t, 11.1) 127.6
19 6.65 (m) 127.2 5.92 (t, 10.2) 130.1
20 6.61 (m) 132.2 6.44 (m) 127.6
21 6.06 (t, 9.9) 129.3 6.44 (m) 129.5
22 5.14 (t, 10.2) 136.3 6.05 (t, 10.5) 129.3
23 2.68 (m) 33.5 5.11 (t, 10.5) 136.3
24 3.37; 2.65 43.8 3.09 (m) 31.6
25 7.54 (d,10.2) - 2.76 (m) 31.3
1.58 (s) 28.6 3.15 (d, 2.57) 31.3
26-NH 11.2 (s, br) - 7.79 (t, 8.84) -
1’ 0.96 (d, 6.34) 17.8 1.45 (s) 25.0
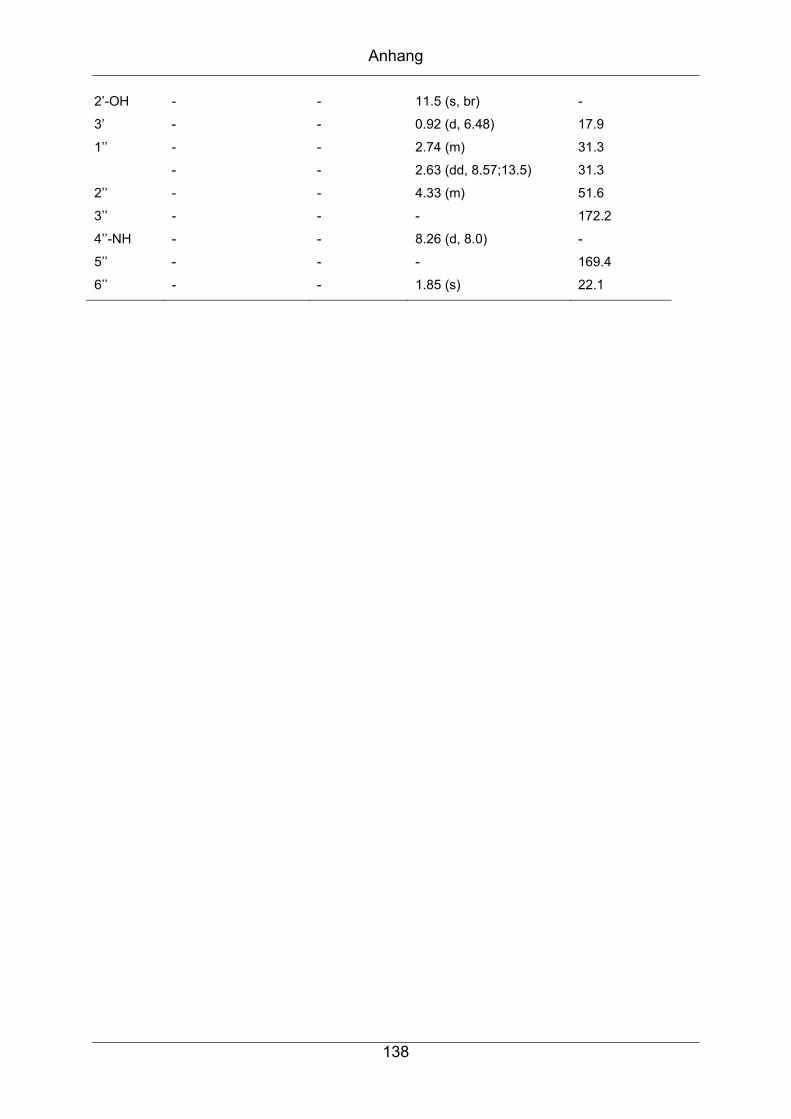
Anhang
138
2’-OH - - 11.5 (s, br) -
3’ - - 0.92 (d, 6.48) 17.9
1’’ - - 2.74 (m) 31.3
- - 2.63 (dd, 8.57;13.5) 31.3
2’’ - - 4.33 (m) 51.6
3’’ - - - 172.2
4’’-NH - - 8.26 (d, 8.0) -
5’’ - - - 169.4
6’’ - - 1.85 (s) 22.1
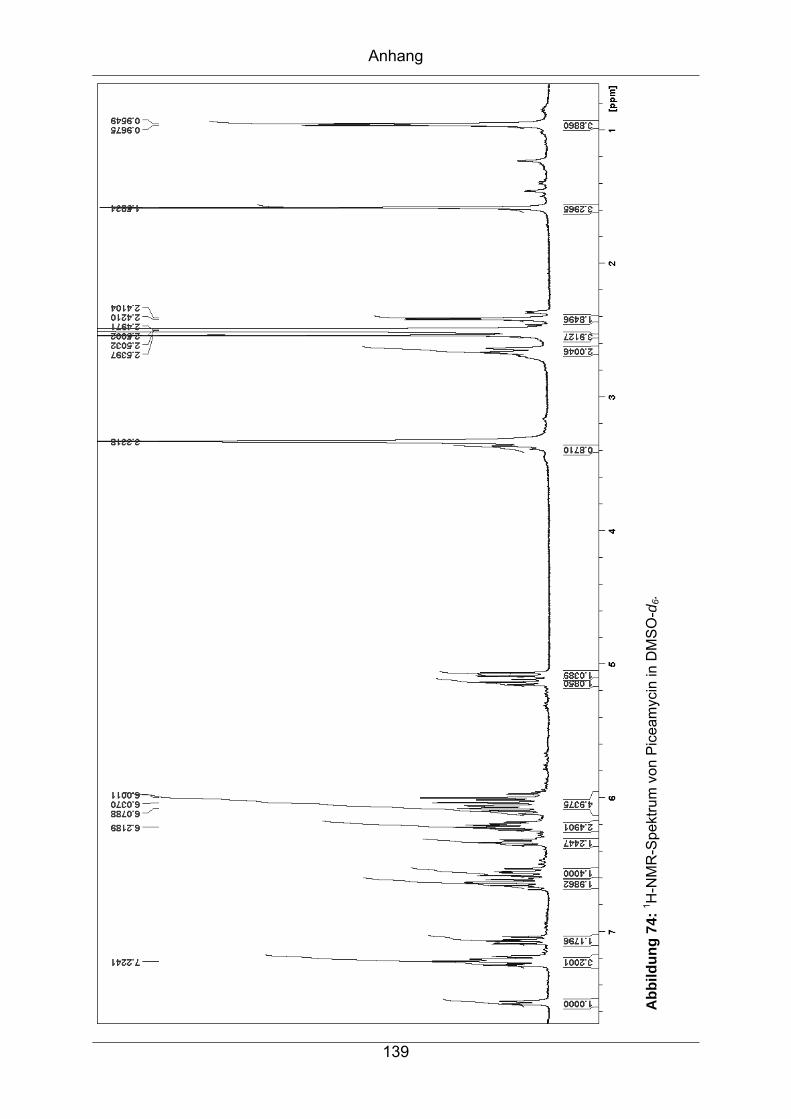
Anhang
Ab
bild
un
g 7
4: 1 H
-NM
R-S
pekt
rum
von
Pic
eam
ycin
in D
MS
O-d
6.
139
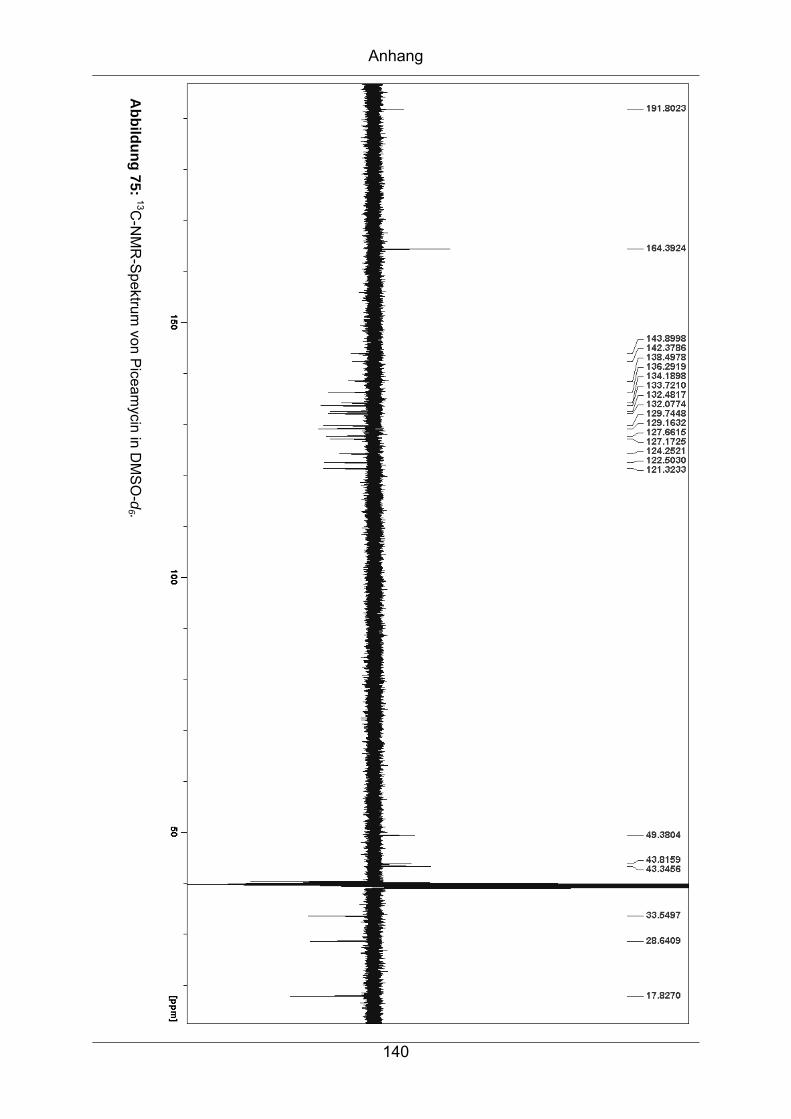
Anhang
Ab
bild
un
g 75: 13C
-NM
R-S
pektrum von P
iceamycin in D
MS
O-d
6 .
140
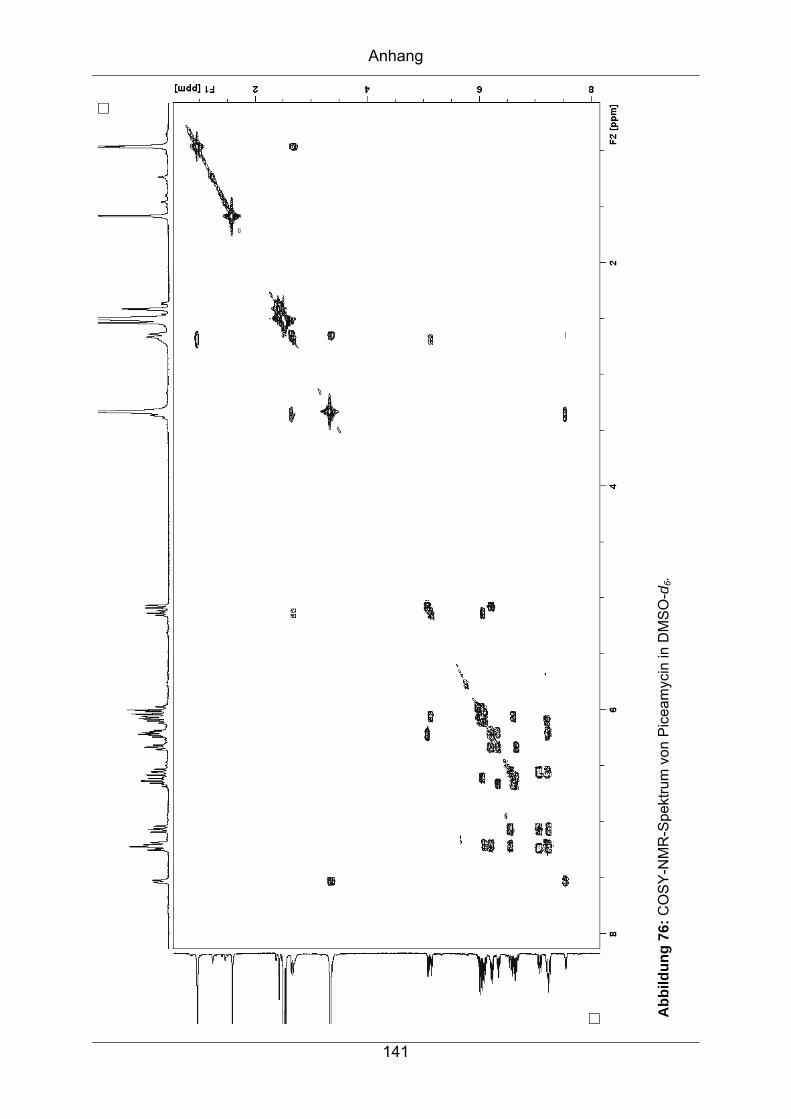
Anhang
Ab
bild
un
g 7
6: C
OS
Y-N
MR
-Spe
ktru
m v
on P
icea
myc
in in
DM
SO
-d6.
141
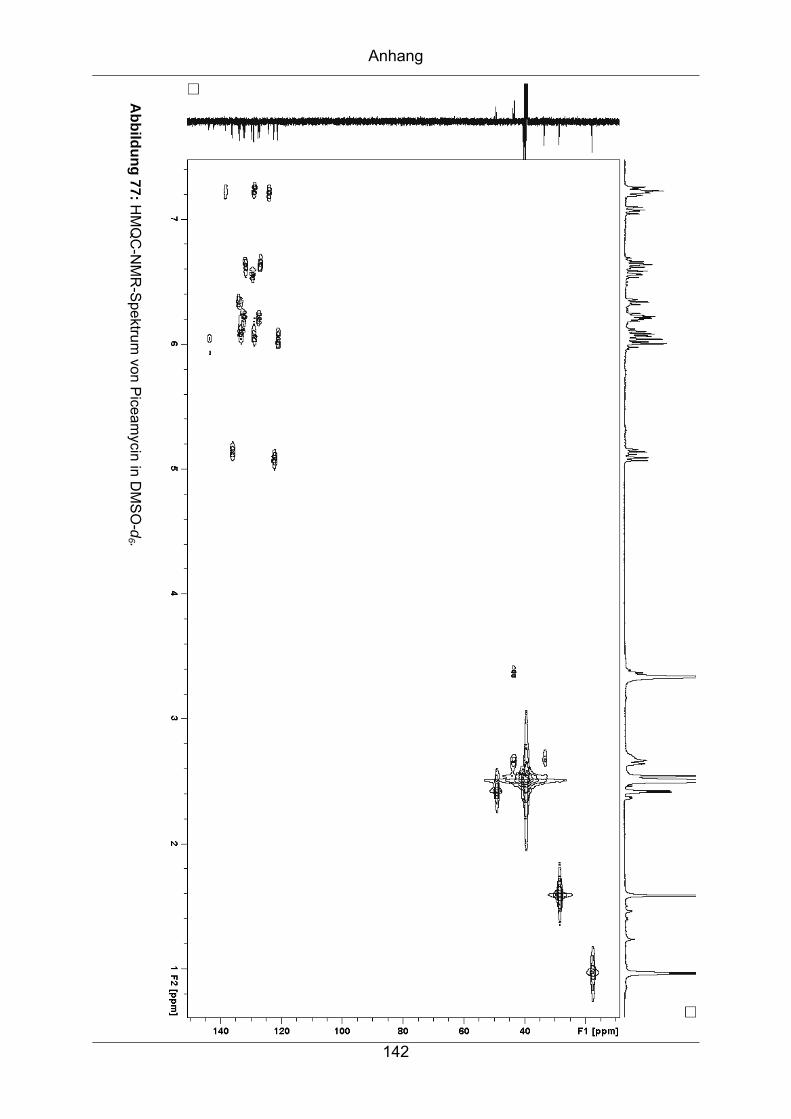
Anhang
Ab
bild
un
g 77: H
MQ
C-N
MR
-Spektrum
von Piceam
ycin in DM
SO
-d6 .
142

Anhang
Ab
bild
un
g 7
8: H
MB
C-N
MR
-Spe
ktru
m v
on P
icea
myc
in in
DM
SO
-d6.
143

Anhang
9.3.2 N-Acetyl-Piceamycin
Ab
bild
un
g 79: 1H
-NM
R-S
pektrum von N
-Acetyl-P
iceamycin in D
MS
O-d
6 .
144
Anhang
Ab
bild
un
g 8
0: 13
C-N
MR
-Spe
ktru
m v
on N
-Ace
tyl-P
icea
myc
in in
DM
SO
-d6.
145
Anhang
Ab
bild
un
g 81: C
OS
Y-N
MR
-Spektrum
von N-A
cetyl-Piceam
ycin in DM
SO
-d6 .
146
Anhang
Ab
bild
un
g 8
2: H
SQ
C-N
MR
-Spe
ktru
m v
on N
-Ace
tyl-P
icea
myc
in in
DM
SO
-d6.
147
Anhang
Ab
bild
un
g 83: H
MB
C-N
MR
-Spektrum
von N-A
cetyl-Piceam
ycin in DM
SO
-d6 .
148

Anhang
149
9.3.3 Aranciamycin-Anhydrid
Tabelle 14: 1H- and 13C-NMR-Signale von Aranciamycin-Anhydrid in CDCl3.
Position δ (1H) [ppm], J in Hz δ (13C) [ppm]
1 7.88 (d, 7.5) 120.9
2 7.75 (t, 7.9) 138.5
3 7.34 (d, 8.3) 125.3
4 - 163.2
5 - 115.9
6 - 193.2
7 - 119.1
8 - 162.5
9 - 133.3
10 5.19 (d, 2.4) 72.4
11 3.72 (d, 2.5) 85.9
12 - 76.9
13 - 198.9
14 - 136.3
15 8.41 (s) 118.0
16 - 134.0
17 - 180.6
18 - 133.6
1’ 3.54 (s) 60.3
2’ 1.54 (s) 23.1
1’’ 5.65 (s, br) 100.5
2’’ 3.53 (dd, 1.4; 3.6) 80.3
3’’ 3.59 (dd, 3.6; 9.8) 69.6
4’’ 4.87 (dd, 9.8; 9.8) 75.1
5’’ 3.92 (dq, 9.8; 6.2) 67.6
6’’ 1.23 (d, 6.2) 17.7
7’’ 3.56 (s) 59.0
8’’ - 171.8
9’’ 2.77 (m) 31.3
10’’ 2.78 (m) 19.9
11’’ - 142.0
12’’ - 142.6
13’’ - 166.1
14’’ - 165.8
15’’ 2.10 (s) 9.7
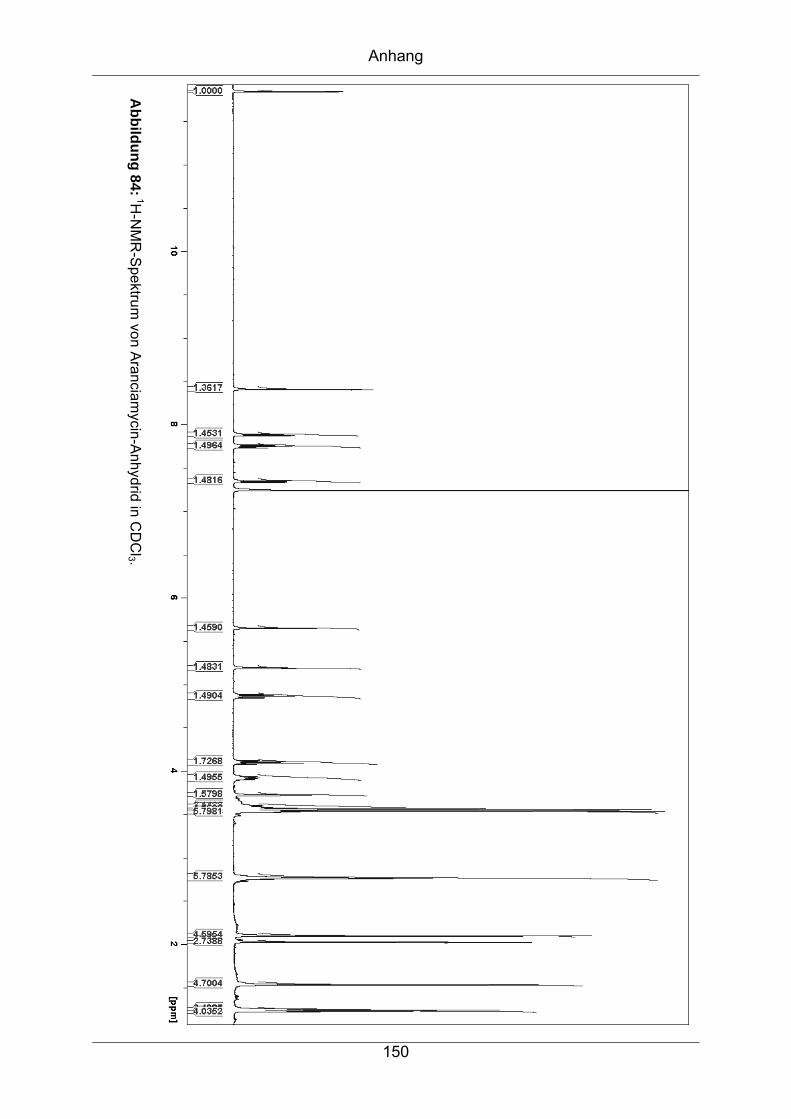
Anhang
150
Ab
bild
un
g 84: 1H
-NM
R-S
pektrum von A
ranciamycin-A
nhydrid in CD
Cl3 .
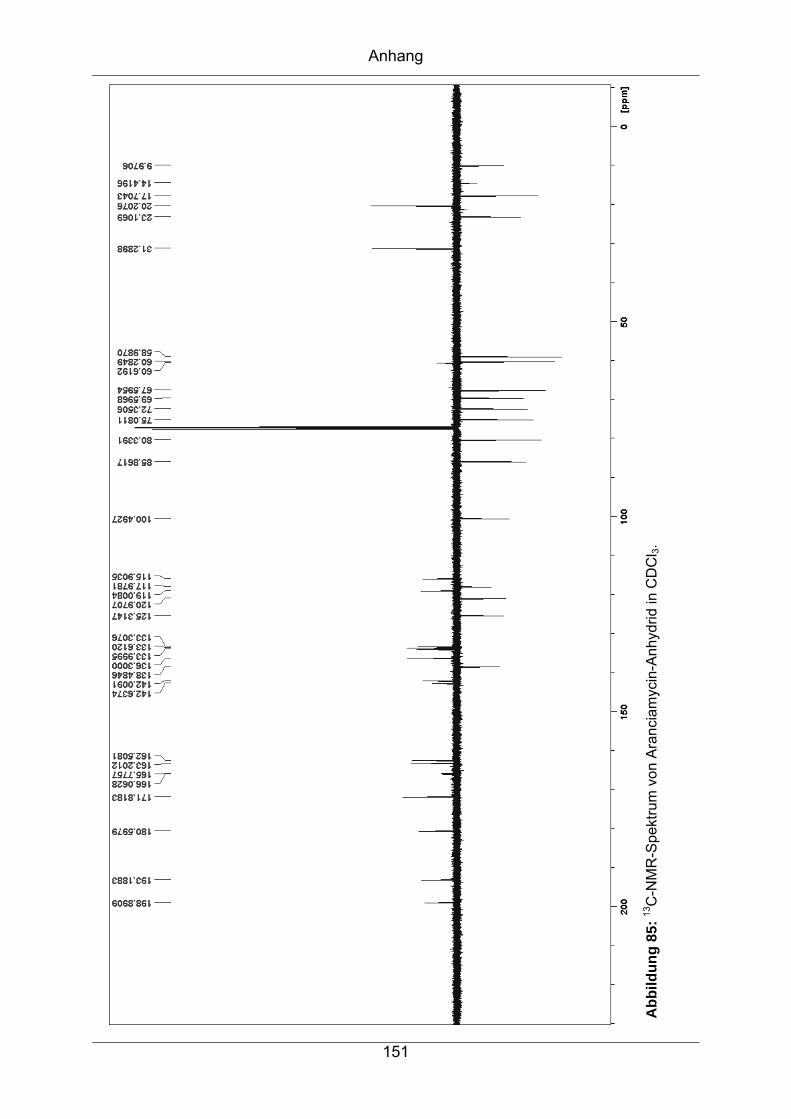
Anhang
Ab
bild
un
g 8
5: 13
C-N
MR
-Spe
ktru
m v
on A
ranc
iam
ycin
-Anh
ydrid
in C
DC
l 3.
151
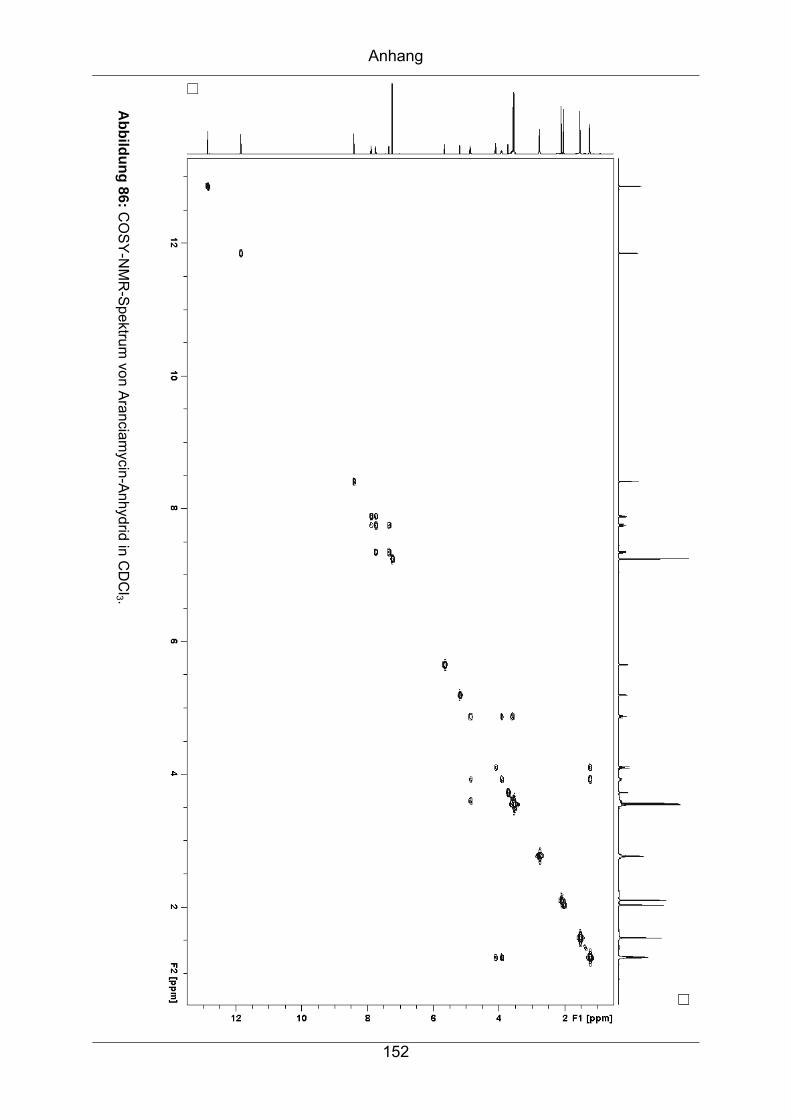
Anhang
Ab
bild
un
g 86: C
OS
Y-N
MR
-Spektrum
von Aranciam
ycin-Anhydrid in C
DC
l3 .
152
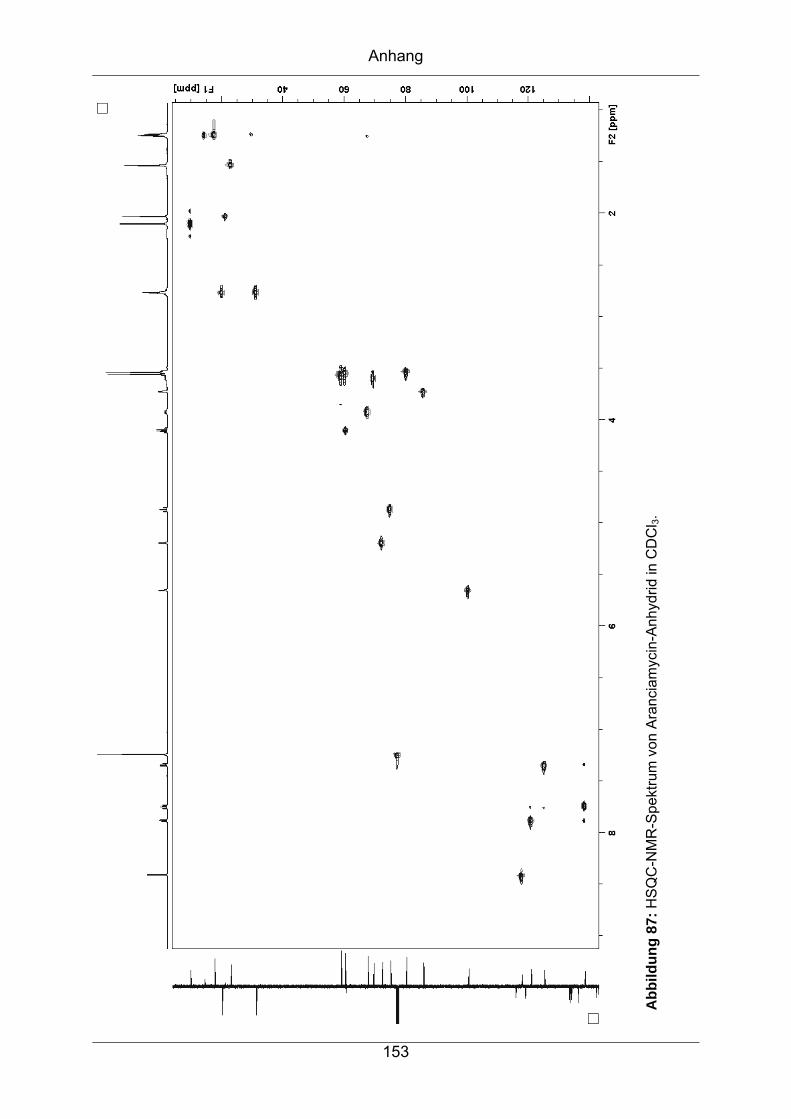
Anhang
Ab
bild
un
g 8
7: H
SQ
C-N
MR
-Spe
ktru
m v
on A
ranc
iam
ycin
-Anh
ydrid
in C
DC
l 3.
153
Anhang
154
Ab
bild
un
g 88: H
MB
C-N
MR
-Spektrum
von Aranciam
ycin-Anhydrid in C
DC
l3 .
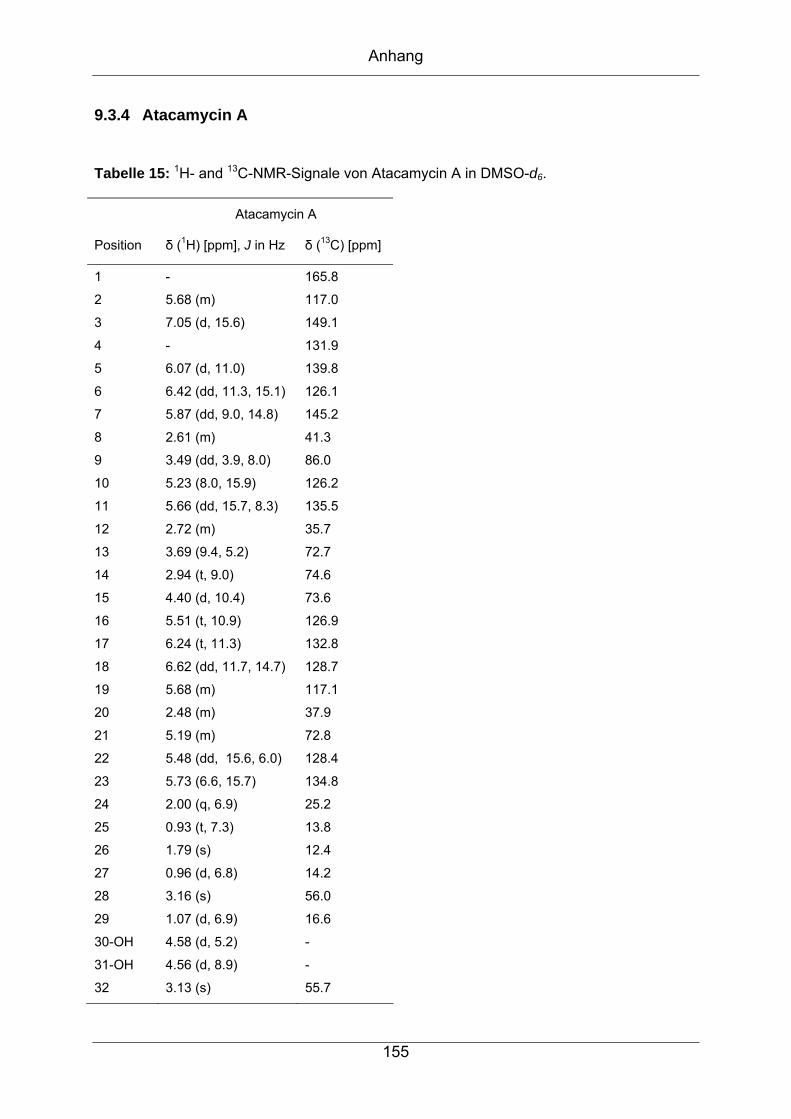
Anhang
155
9.3.4 Atacamycin A
Tabelle 15: 1H- and 13C-NMR-Signale von Atacamycin A in DMSO-d6.
Atacamycin A
Position δ (1H) [ppm], J in Hz δ (13C) [ppm]
1 - 165.8
2 5.68 (m) 117.0
3 7.05 (d, 15.6) 149.1
4 - 131.9
5 6.07 (d, 11.0) 139.8
6 6.42 (dd, 11.3, 15.1) 126.1
7 5.87 (dd, 9.0, 14.8) 145.2
8 2.61 (m) 41.3
9 3.49 (dd, 3.9, 8.0) 86.0
10 5.23 (8.0, 15.9) 126.2
11 5.66 (dd, 15.7, 8.3) 135.5
12 2.72 (m) 35.7
13 3.69 (9.4, 5.2) 72.7
14 2.94 (t, 9.0) 74.6
15 4.40 (d, 10.4) 73.6
16 5.51 (t, 10.9) 126.9
17 6.24 (t, 11.3) 132.8
18 6.62 (dd, 11.7, 14.7) 128.7
19 5.68 (m) 117.1
20 2.48 (m) 37.9
21 5.19 (m) 72.8
22 5.48 (dd, 15.6, 6.0) 128.4
23 5.73 (6.6, 15.7) 134.8
24 2.00 (q, 6.9) 25.2
25 0.93 (t, 7.3) 13.8
26 1.79 (s) 12.4
27 0.96 (d, 6.8) 14.2
28 3.16 (s) 56.0
29 1.07 (d, 6.9) 16.6
30-OH 4.58 (d, 5.2) -
31-OH 4.56 (d, 8.9) -
32 3.13 (s) 55.7

Anhang
Ab
bild
un
g 89: 1H
-NM
R-S
pektrum von A
tacamycin A
in DM
SO
-d6 .
156
Anhang
Ab
bild
un
g 9
0: 1
3 C-N
MR
-Spe
ktru
m v
on A
taca
myc
in A
in D
MS
O-d
6.
157
Anhang
Ab
bild
un
g 91: C
OS
Y-N
MR
-Spektru
m von A
tacamycin A
in DM
SO
-d6 .
158
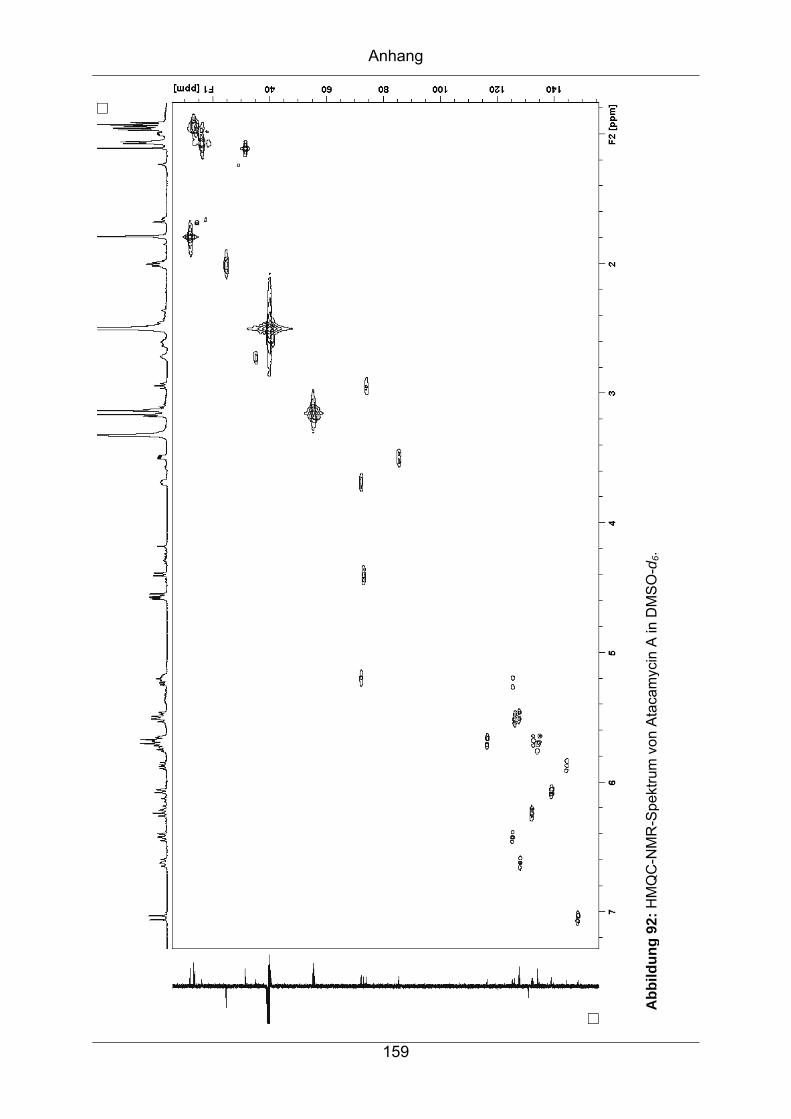
Anhang
Ab
bild
un
g 9
2: H
MQ
C-N
MR
-Spe
ktru
m v
on A
taca
myc
in A
in D
MS
O-d
6.
159
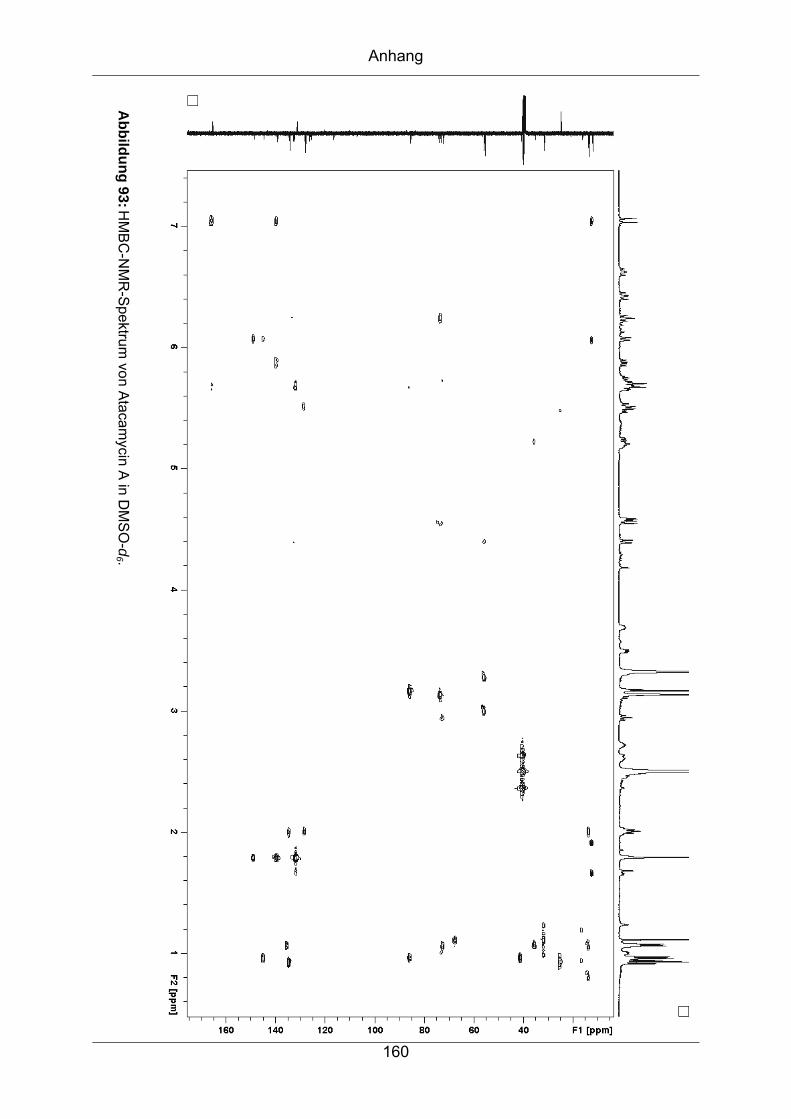
Anhang
Ab
bild
un
g 93: H
MB
C-N
MR
-Spektrum
von Atacam
ycin A in D
MS
O-d
6 .
160
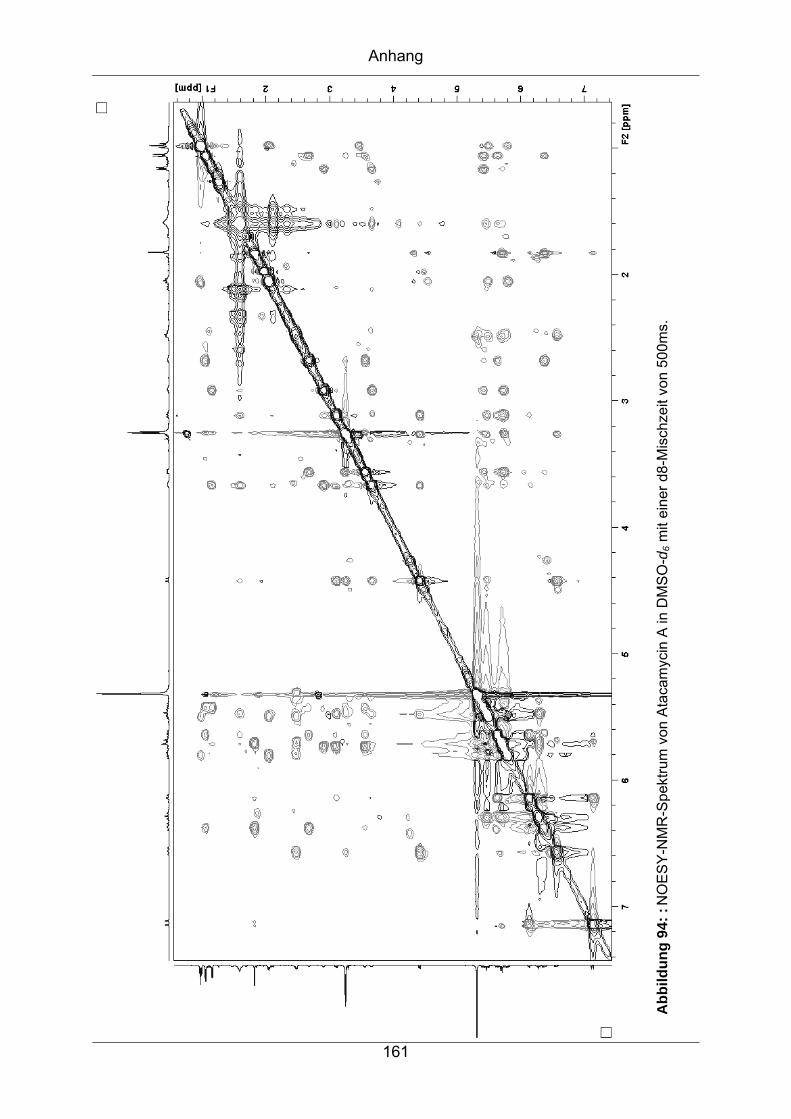
Anhang
Ab
bild
un
g 9
4: :
NO
ES
Y-N
MR
-Spe
ktru
m v
on A
taca
myc
in A
in D
MS
O-d
6 m
it ei
ner
d8-M
isch
zeit
von
500m
s.
161
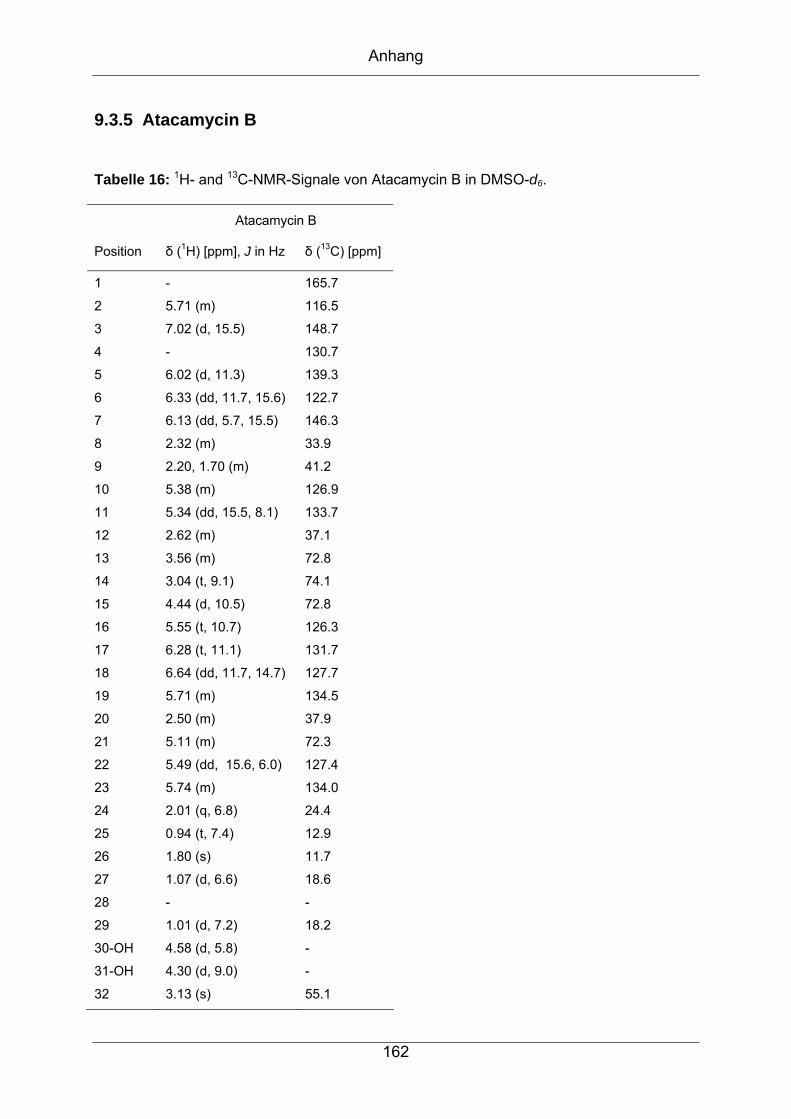
Anhang
162
9.3.5 Atacamycin B
Tabelle 16: 1H- and 13C-NMR-Signale von Atacamycin B in DMSO-d6.
Atacamycin B
Position δ (1H) [ppm], J in Hz δ (13C) [ppm]
1 - 165.7
2 5.71 (m) 116.5
3 7.02 (d, 15.5) 148.7
4 - 130.7
5 6.02 (d, 11.3) 139.3
6 6.33 (dd, 11.7, 15.6) 122.7
7 6.13 (dd, 5.7, 15.5) 146.3
8 2.32 (m) 33.9
9 2.20, 1.70 (m) 41.2
10 5.38 (m) 126.9
11 5.34 (dd, 15.5, 8.1) 133.7
12 2.62 (m) 37.1
13 3.56 (m) 72.8
14 3.04 (t, 9.1) 74.1
15 4.44 (d, 10.5) 72.8
16 5.55 (t, 10.7) 126.3
17 6.28 (t, 11.1) 131.7
18 6.64 (dd, 11.7, 14.7) 127.7
19 5.71 (m) 134.5
20 2.50 (m) 37.9
21 5.11 (m) 72.3
22 5.49 (dd, 15.6, 6.0) 127.4
23 5.74 (m) 134.0
24 2.01 (q, 6.8) 24.4
25 0.94 (t, 7.4) 12.9
26 1.80 (s) 11.7
27 1.07 (d, 6.6) 18.6
28 - -
29 1.01 (d, 7.2) 18.2
30-OH 4.58 (d, 5.8) -
31-OH 4.30 (d, 9.0) -
32 3.13 (s) 55.1

Anhang
Ab
bild
un
g 9
5: 1H
-NM
R-S
pekt
rum
von
Ata
cam
ycin
B in
DM
SO
-d6.
163
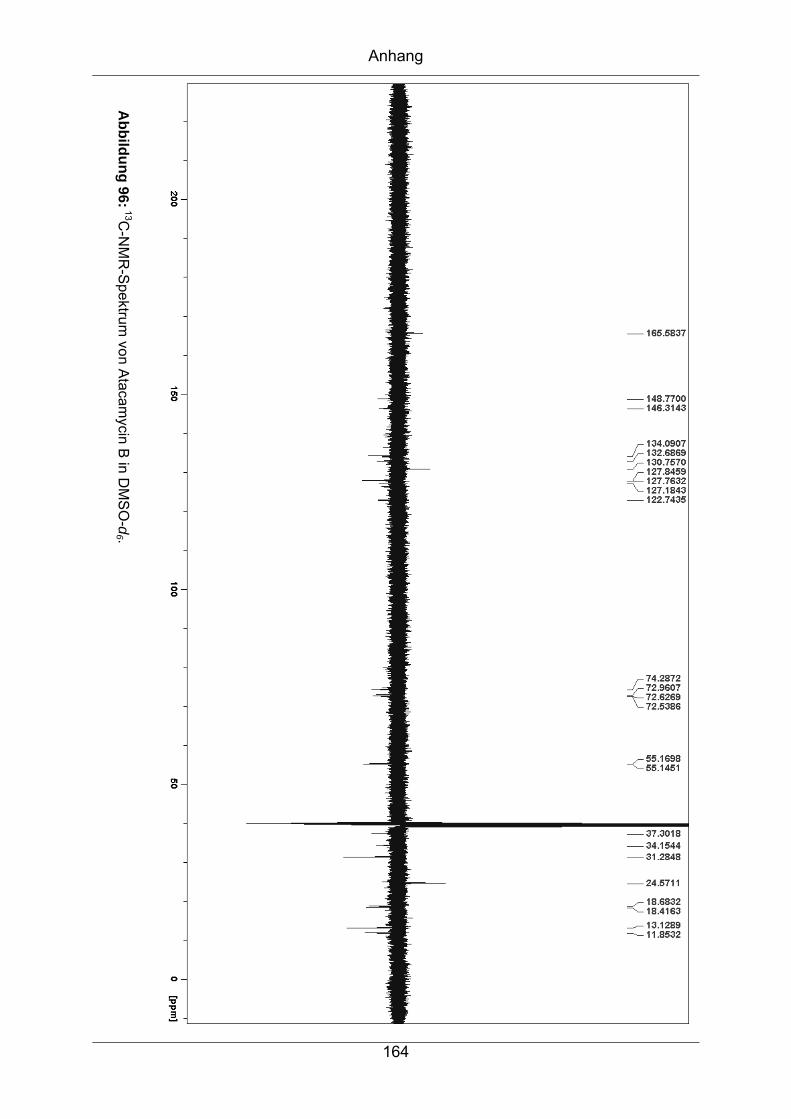
Anhang
Ab
bild
un
g 96: 13C
-NM
R-S
pektrum von A
tacamycin B
in DM
SO
-d6 .
164
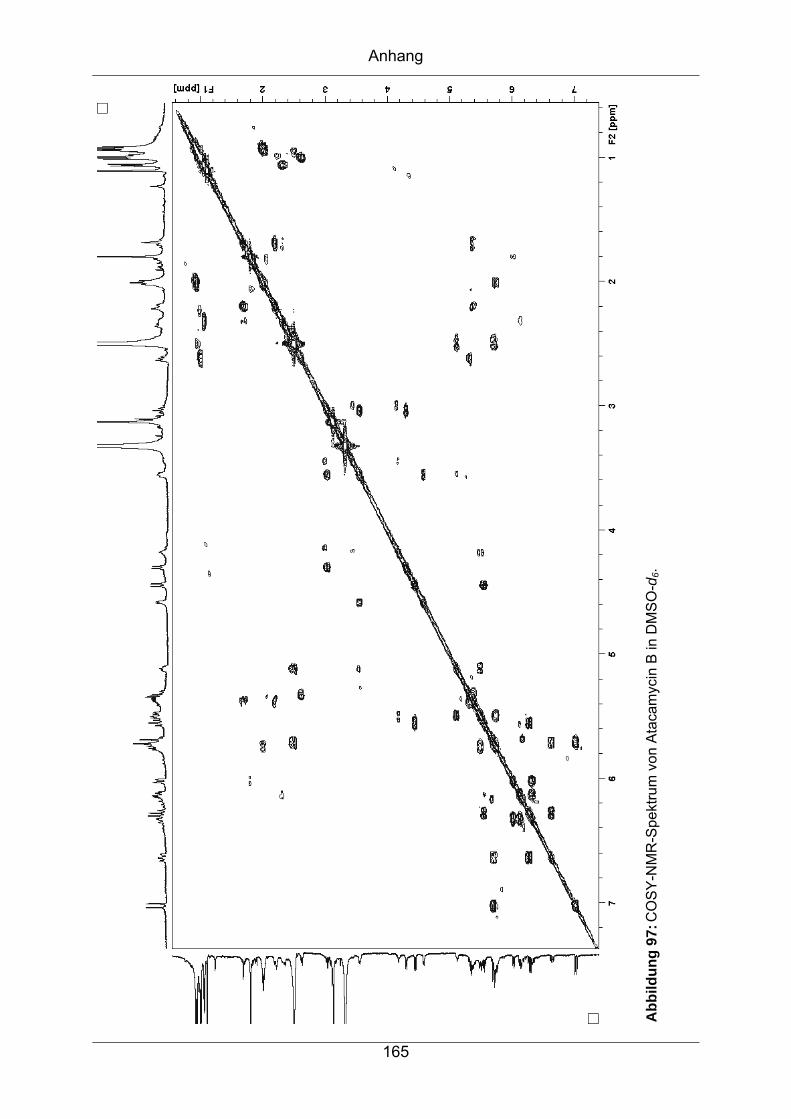
Anhang
Ab
bild
un
g 9
7: C
OS
Y-N
MR
-Spe
ktru
m v
on A
taca
myc
in B
in D
MS
O-d
6.
165
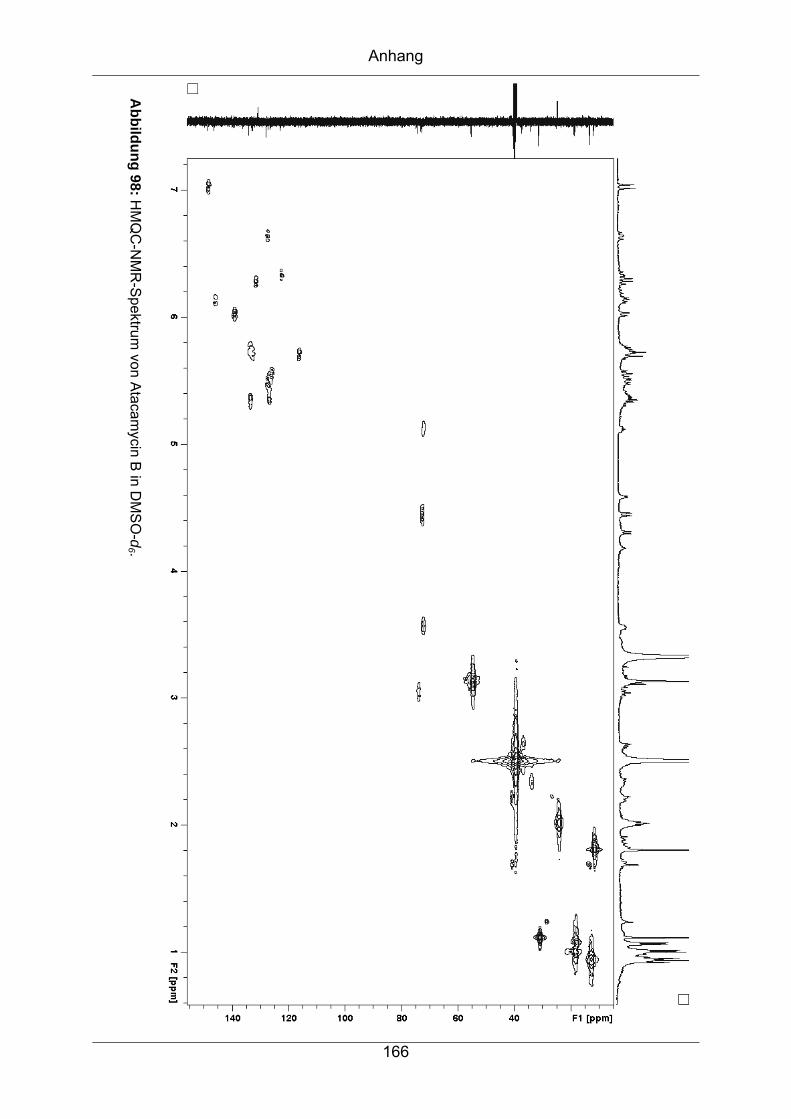
Anhang
Ab
bild
un
g 98: H
MQ
C-N
MR
-Spektrum
von Atacam
ycin B in D
MS
O-d
6 .
166
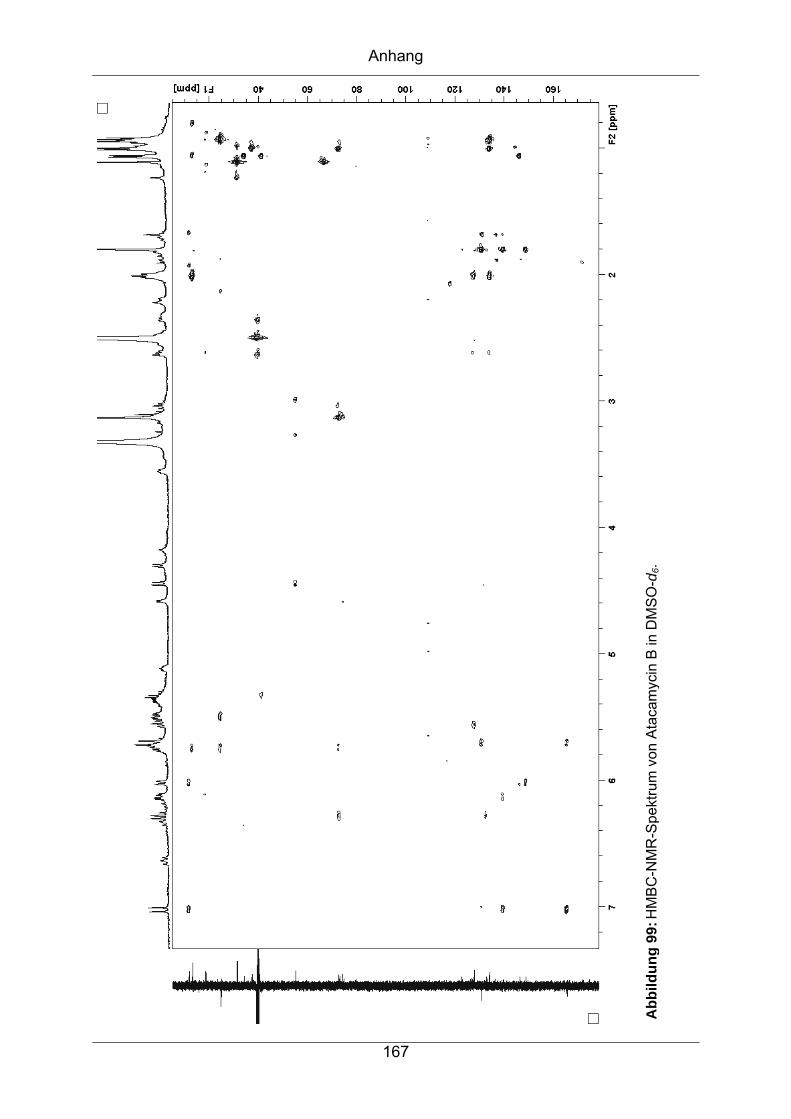
Anhang
Ab
bild
un
g 9
9: H
MB
C-N
MR
-Spe
ktru
m v
on A
taca
myc
in B
in D
MS
O-d
6.
167

Anhang
168
9.3.6 Atacamycin C
Tabelle 17: 1H- and 13C-NMR-Signale von Atacamycin C in DMSO-d6.
Atacamycin C
Position δ (1H) [ppm], J in Hz δ (13C) [ppm]
1 - 165.7
2 5.70 (d, 15.5) 133.5
3 7.02 (d, 15.5) 148.7
4 - 130.9
5 6.06 (d, 11.2) 139.3
6 6.33 (dd, 11.2, 15.2) 122.9
7 6.13 (dd, 5.3, 15.2) 145.8
8 2.32 (m) 34.3
9 2.23, 1.68 (dt, 13.0, 3.1) 41.2
10 5.33 (m) 127.7
11 5.33 (m) 134.2
12 1.97 (m) 43.4
13 3.72 (s, br) 68.9
14 1.40 (dd, 4.6, 6.8) 43.3
15 4.36 (m) 71.2
16 5.07 (t, 10.7) 129.3
17 6.18 (t, 10.7) 131.1
18 6.61 (dd, 10.7, 14.7) 127.6
19 5.68 (m) 132.8
20 2.48 (m) 37.9
21 5.12 (m) 72.6
22 5.49 (dd, 15.7, 6.0) 127.5
23 5.74 (m) 134.1
24 2.01 (q, 7.0) 24.4
25 0.93 (t, 7.4) 12.8
26 1.80 (s) 11.6
27 1.07 (d, 6.6) 18.5
28 - -
29 1.00 (d, 6.8) 19.0
30-OH 4.49 (d, 4.6) -
31-OH - -
32 3.11 (s) 54.7
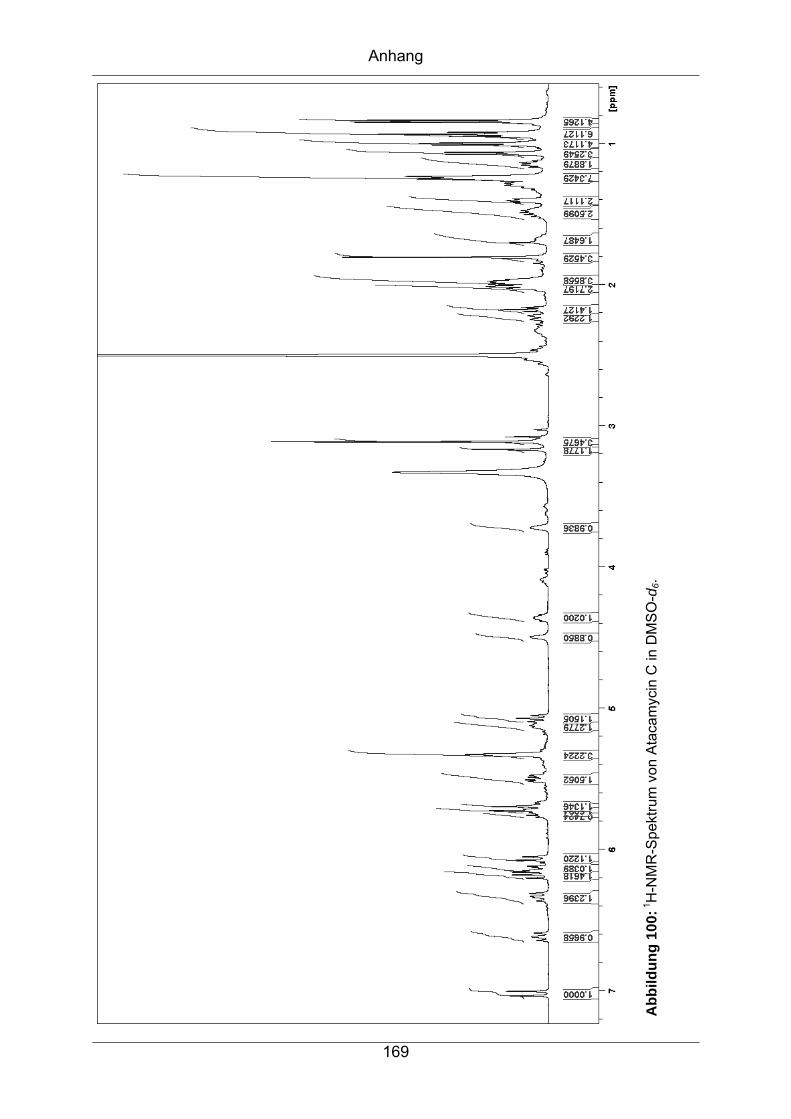
Anhang
Ab
bild
un
g 1
00: 1
H-N
MR
-Spe
ktru
m v
on A
taca
myc
in C
in D
MS
O-d
6.
169
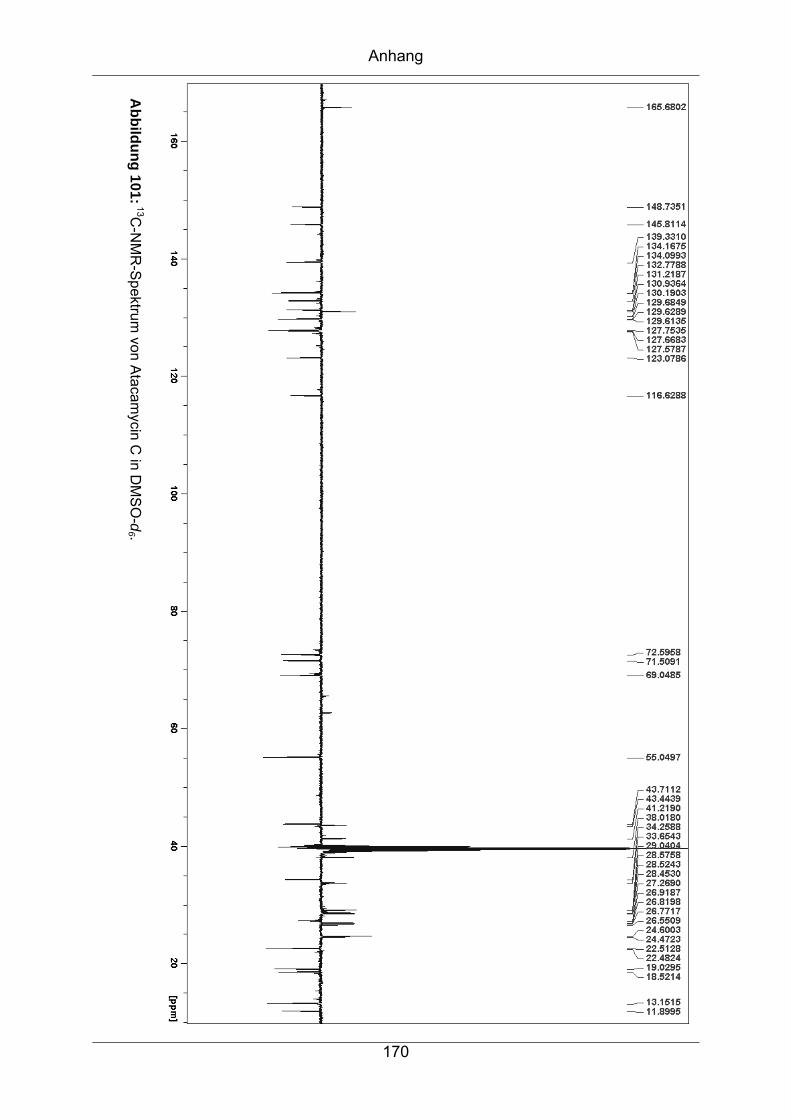
Anhang
Ab
bild
un
g 101: 13C
-NM
R-S
pektrum von A
tacamycin C
in DM
SO
-d6 .
170
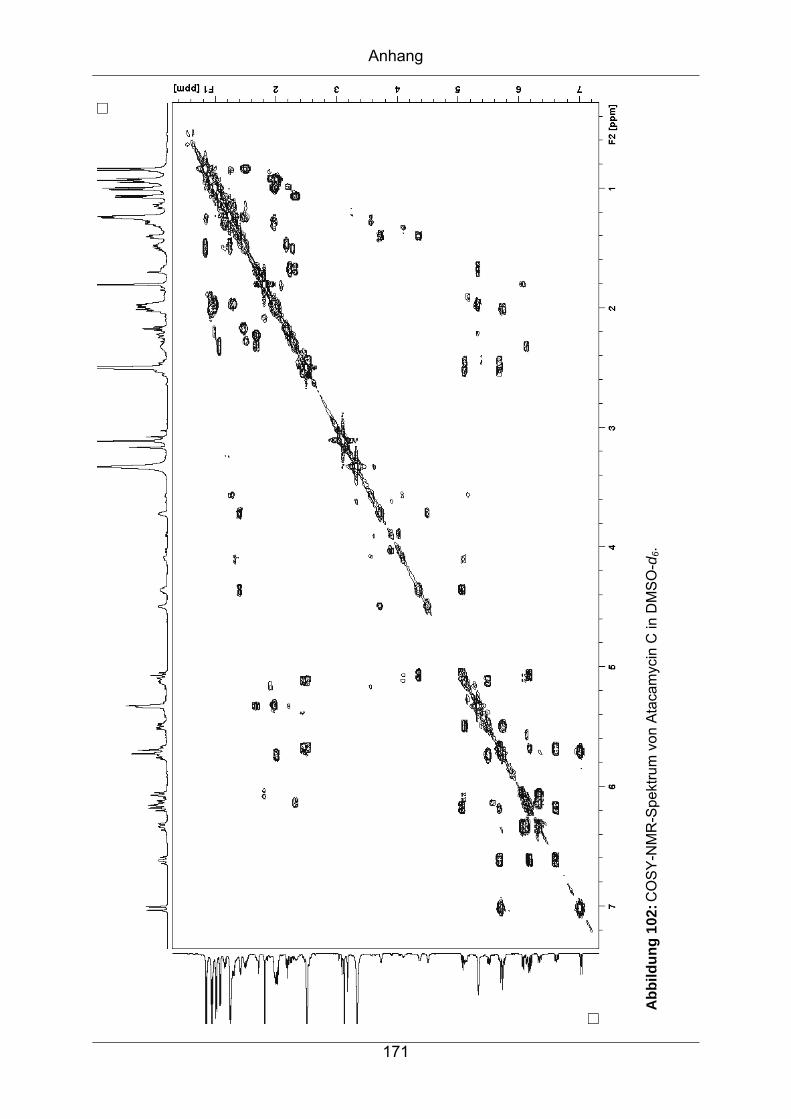
Anhang
Ab
bild
un
g 1
02: C
OS
Y-N
MR
-Spe
ktru
m v
on A
taca
myc
in C
in D
MS
O-d
6.
171
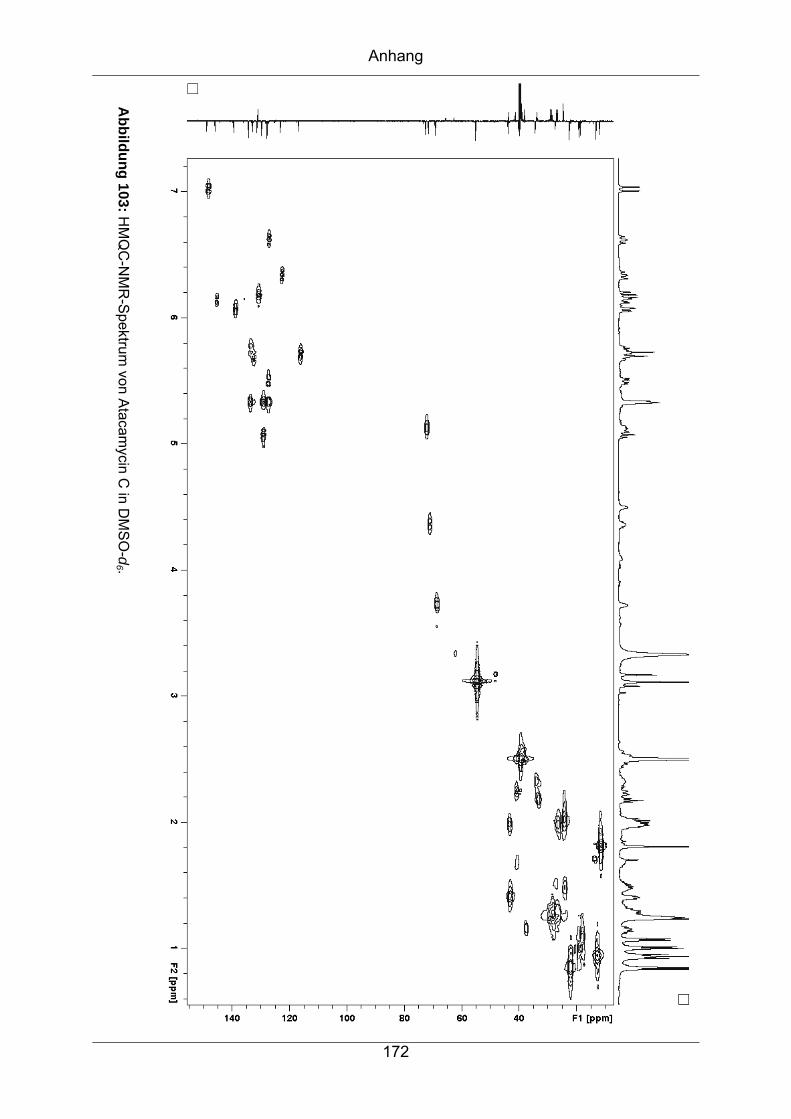
Anhang
Ab
bild
un
g 103: H
MQ
C-N
MR
-Spektrum
von Atacam
ycin C in D
MS
O-d
6 .
172
Anhang
Ab
bild
un
g 1
04: H
MB
C-N
MR
-Spe
ktru
m v
on A
taca
myc
in C
in D
MS
O-d
6.
173

Anhang
174
9.3.7 TÜ 6392 A2
Tabelle 18: 1H- and 13C-NMR Signale von TÜ 6392 A2 in DCM-d2 bei 273 K.
TÜ 6392 A2
Position δ (1H) [ppm], J in Hz δ (13C) [ppm], mult.
1 - 171.6 (s)
2 - 137.1 (s)
3 6.15 (1H, s) 135.1 (d)
4 - 134.2 (s)
5 5.93 (1H, d, 11.3) 137.2 (d)
6 6.42 (1H, dd, 11.3, 15.6) 127.0 (d)
7 6.03 (1H, dd, 11.3, 15.6) 135.9 (d)
8 6.28 (1H, dd, 11.3, 15.0) 127.7 (d)
9 5.82 (1H, d, 15.0) 141.7 (d)
10 - 77.8 (s)
11 4.07 (1H, d, 5.9) 88.8 (d)
12 5.61 (1H, dd, 5.9, 11.2) 125.7 (d)
13 6.29 (1H, t, 11.2) 133.4 (d)
14 6.95 (1H, dd, 11.2, 15.6) 128.9 (d)
15 6.24 (1H, d, 15.6) 137.0 (d)
16 - 136.5 (s)
17 6.24 (1H, d, 11.2) 131.0 (d)
18 6.53 (1H, dd, 11.2, 14.9) 124.3 (d)
19 6.40 (1H, d, 14.9) 137.1 (d)
20 - 139.2 (s)
21 5.72 (1H, dd, 4.4, 11.1) 126.7 (d)
1.98 (1H, m) 22
3.04 (1H, m) 32.3 (t)
23 4.11 (1H, m) 44.6 (d)
24 2.50 (2H, d, 6.9) 36.0 (t)
25 1.60 (1H, tqq, 6.4, 6.7, 6.9) 29.3 (d)
26 0.85 (3H, d, 6.4) 22.4 (q)
27 0.83 (3H, d, 6.7) 22.2 (q)
28 2.03 (3H, s) 15.9 (q)
29 1.14 (3H, s) 24.8 (q)
30 1.76 (3H, s) 12.9 (q)
31 1.70 (3H, s) 12.5 (q)
32 1.21 (3H, d, 6.8) 19.1 (q)
1-NH 5.55 (1H, d, 7.7)

Anhang
175
1’ 4.41 (1H, d, 7.2) 106.7 (d)
2’ 2.76 (1H, dd, 7.2, 7.6) 55.3 (d)
3’ 3.01 (1H, dd, 7.6, 8.1) 85.1 (d)
4’ 3.72 (1H, m) 69.5 (d)
3.23 (1H, dd, 11.1, 11.4) 5’
3.92 (1H, dd, 4.7, 11.4) 65.6 (t)
6’ 3.59 (3H, s) 59.9 (q)

Anhang
Ab
bild
un
g 105: 1H
-NM
R-S
pektrum von T
Ü 6392 A
2 in DC
M-d
2 bei 273 K.
176
Anhang
Ab
bild
un
g 1
06: 1
3 C-N
MR
-Spe
ktru
m v
on T
Ü 6
392
A2
in D
CM
-d2
bei 2
73 K
.
177
Anhang
Ab
bild
un
g 107: C
OS
Y-N
MR
-Spektrum
von TÜ
6392 A2 in D
CM
-d2 bei 273 K
.
178
Anhang
Ab
bild
un
g 1
08:
HS
QC
-NM
R-S
pekt
rum
von
TÜ
639
2 A
2 in
DC
M-d
2 be
i 273
K.
179
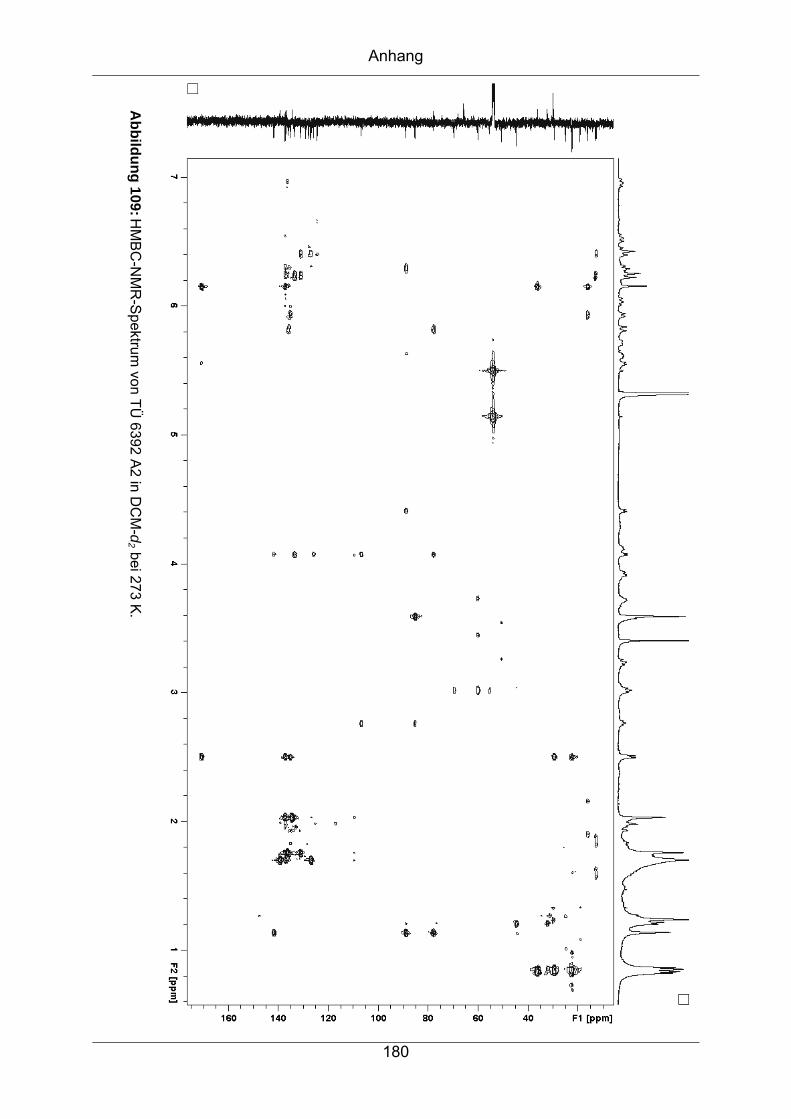
Anhang
Ab
bild
un
g 109: H
MB
C-N
MR
-Spektrum
von TÜ
6392 A2 in D
CM
-d2 bei 273 K
.
180
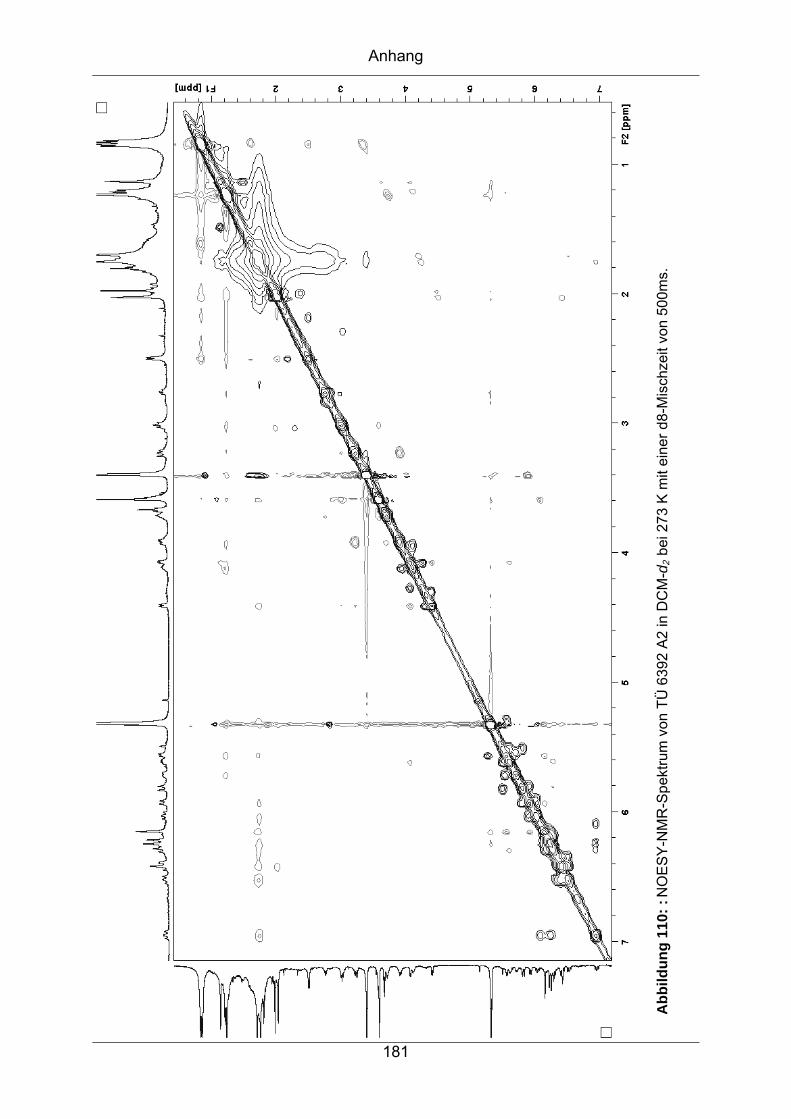
Anhang
Ab
bild
un
g 1
10:
: NO
ES
Y-N
MR
-Sp
ektr
um v
on T
Ü 6
392
A2
in D
CM
-d2
bei 2
73 K
mit
eine
r d8
-Mis
chze
it vo
n 50
0ms.
181
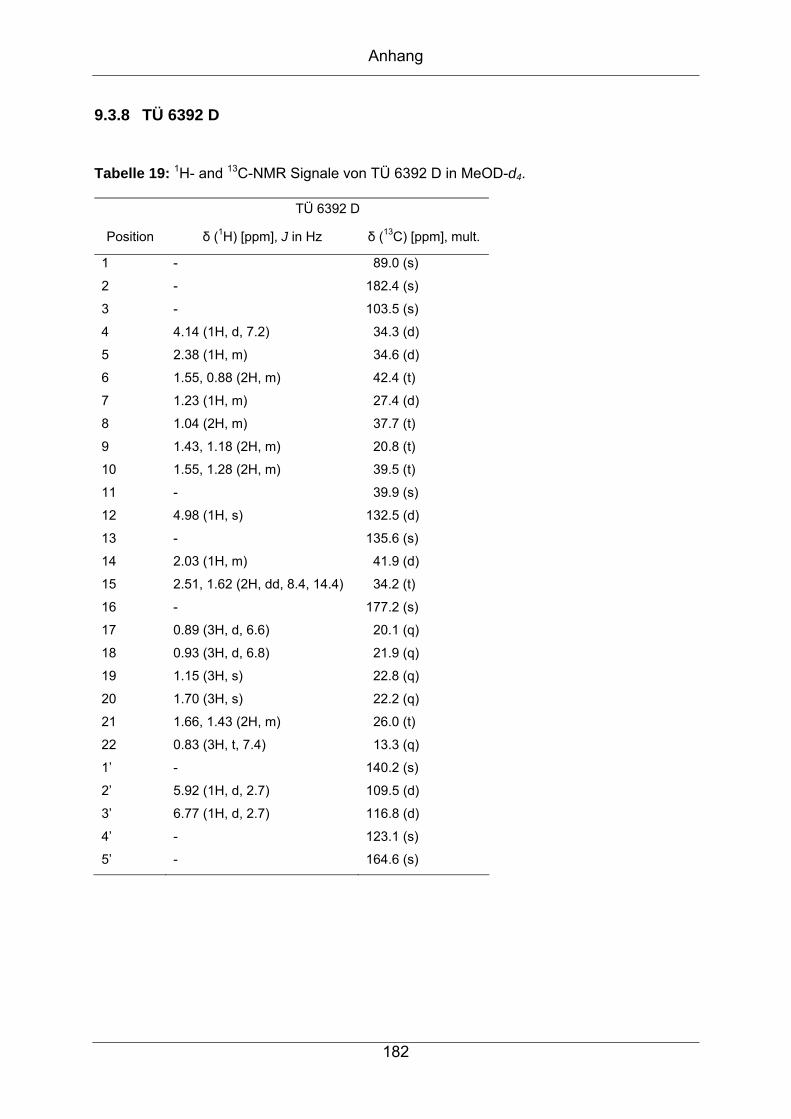
Anhang
182
9.3.8 TÜ 6392 D
Tabelle 19: 1H- and 13C-NMR Signale von TÜ 6392 D in MeOD-d4.
TÜ 6392 D
Position δ (1H) [ppm], J in Hz δ (13C) [ppm], mult.
1 - 89.0 (s)
2 - 182.4 (s)
3 - 103.5 (s)
4 4.14 (1H, d, 7.2) 34.3 (d)
5 2.38 (1H, m) 34.6 (d)
6 1.55, 0.88 (2H, m) 42.4 (t)
7 1.23 (1H, m) 27.4 (d)
8 1.04 (2H, m) 37.7 (t)
9 1.43, 1.18 (2H, m) 20.8 (t)
10 1.55, 1.28 (2H, m) 39.5 (t)
11 - 39.9 (s)
12 4.98 (1H, s) 132.5 (d)
13 - 135.6 (s)
14 2.03 (1H, m) 41.9 (d)
15 2.51, 1.62 (2H, dd, 8.4, 14.4) 34.2 (t)
16 - 177.2 (s)
17 0.89 (3H, d, 6.6) 20.1 (q)
18 0.93 (3H, d, 6.8) 21.9 (q)
19 1.15 (3H, s) 22.8 (q)
20 1.70 (3H, s) 22.2 (q)
21 1.66, 1.43 (2H, m) 26.0 (t)
22 0.83 (3H, t, 7.4) 13.3 (q)
1’ - 140.2 (s)
2’ 5.92 (1H, d, 2.7) 109.5 (d)
3’ 6.77 (1H, d, 2.7) 116.8 (d)
4’ - 123.1 (s)
5’ - 164.6 (s)
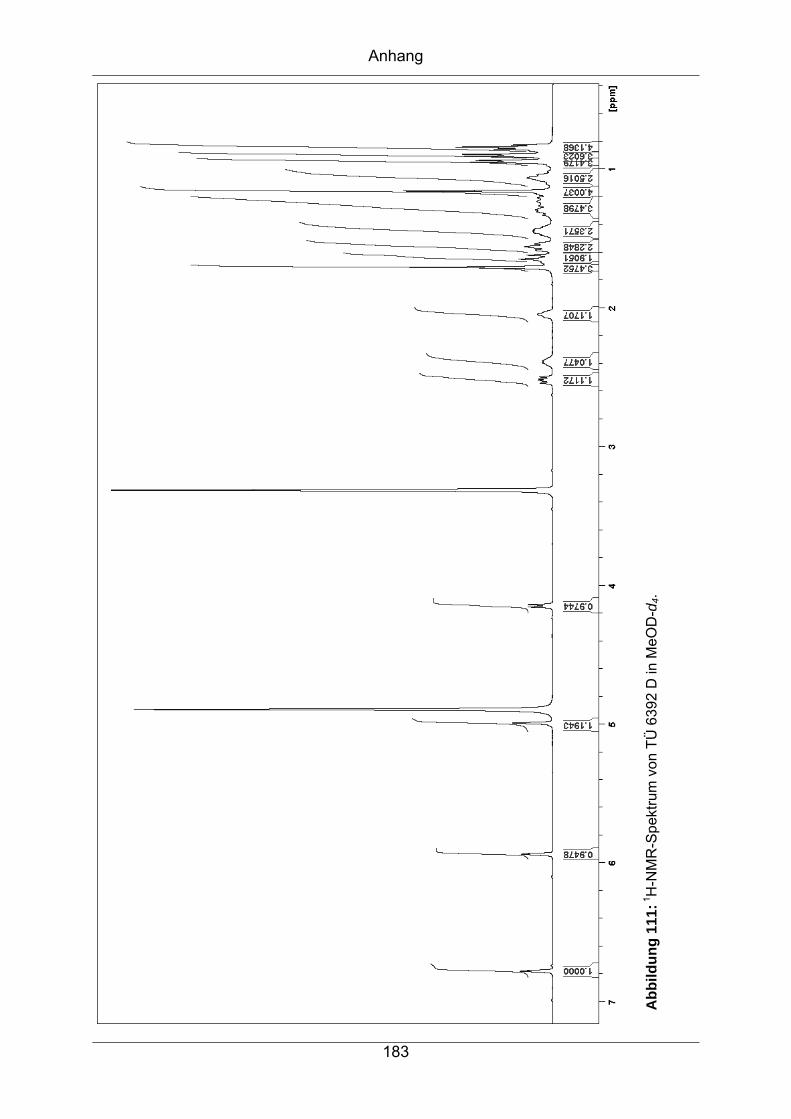
Anhang
Ab
bild
un
g 1
11: 1
H-N
MR
-Spe
ktru
m v
on T
Ü 6
392
D in
MeO
D-d
4.
183
Anhang
Ab
bild
un
g 112: 13C
-NM
R-S
pektrum von T
Ü 6392 D
in MeO
D-d
4 .
184
Anhang
Ab
bild
un
g 1
13: C
OS
Y-N
MR
-Spe
ktru
m v
on T
Ü 6
392
D in
MeO
D-d
4.
185
Anhang
Ab
bild
un
g 114: H
SQ
C-N
MR
-Spektrum
von TÜ
6392 D in M
eOD
-d4 .
186
Anhang
Ab
bild
un
g 1
15: H
MB
C-N
MR
-Spe
ktru
m v
on T
Ü 6
392
D in
MeO
D-d
4.
187














![S2k-Leitlinie: Diagnostik bei Verdacht auf eine ...€¦ · von Oralcephalosporinen anstelle von Penicillin-derivaten werden Überempfindlichkeiten ange-führt [10]. Darüber hinaus](https://img.pdfslide.org/doc/110x75/6061763e77829b193251e501/s2k-leitlinie-diagnostik-bei-verdacht-auf-eine-von-oralcephalosporinen-anstelle.jpg)