Embed Size (px)
Citation preview

Thermische Analyse
Grundlagen und Praktikumsanleitung
Otto-Schott-Institut für Glaschemie
Materialkundliches Praktikum Teil 1
Marek Patschger

Inhalt
1 Einleitung 1
2 Methoden der thermischen Analyse 1
2.1 Thermodynamische Grundlagen 2
2.2 Messmethodische Grundlagen: die Temperaturerfassung 4
2.3 Überblick über die Methoden der thermischen Analyse 6
2.4 Differenzthermoanalyse (DTA) 7
2.4.1 Das Grundprinzip der DTA 8
2.4.2 Beeinflussung von thermischen Effekten: apparative Faktoren 9
2.4.3 Beeinflussung von thermischen Effekten: Probenbehandlung 11
2.4.4 Auswertung thermischer Effekte 12
2.5 Differential Scanning Calorimetry (DSC) 14
2.6 Dilatometrie (DIL) 17
2.7 Erhitzungsmikroskop (EHM) 21
2.8 Simultane Thermoanalyse (STA) 22
2.9 Fazit 24
3 Durchführung des Praktikums 25
4 Quellen 26

1
1 Einleitung
Die thermische Analyse ist eine der grundlegenden Methoden der Festkörperanalytik und damit für die
Natur- und Ingenieurswissenschaften von großer Relevanz. Die Vorteile dieser Methode liegen vor
allem in dem geringen technischen Aufwand und den niedrigen Anforderungen an die Beschaffenheit
der Probe (z.B. Korngröße, Kornverteilung, Kristallinität, Textur). Trotzdem stellt die Thermoanalyse
eine leistungsfähige und ökonomische Meßmethode dar, mit der sich vielfältige Aussagen über einen
Werkstoff erzielen lassen. Dazu zählen unter anderem Analysen zu Phasenbestand, Reinheit,
Synthesebedingungen, physikalischen Stoffkonstanten und Reaktionskinetik. Auch die Erstellung von
Zustandsdiagrammen ist mit Hilfe der Thermischen Analyse möglich [1]. Dem gegenüber stehen die
mit Hilfe der Thermoanalyse nicht allgemeingültig zu beantwortenden Fragen nach dem zugrunde-
liegende physikalische Effekt einer detektierten Eigenschaftsänderung und welche Methode für die
Lösung eines spezifischen Messproblems am geeignetsten ist.
Der Begriff der Thermoanalyse ist sehr weit gefasst. Eine mögliche Definition lautet: „Die thermische
Analyse ist eine Gruppe von Methoden, bei denen physikalische und chemische Eigenschaften einer
Substanz bzw. eines Substanz-und/oder Reaktionsgemisches als Funktion der Temperatur oder Zeit
gemessen werden, während die Substanz einem geregelten Temperaturprogramm unterworfen wird“
[2]. Die thermische Analyse ist somit eine Methode zur Charakterisierung eines Stoffes als Funktion
von Temperatur und Zeit. Da die meisten Substanzen ihre chemische Zusammensetzung und/oder ihre
Struktur bei Variation der Temperatur ändern und solche Vorgänge mit Veränderungen der
spezifischen Wärme oder auch der Masse verbunden sind, können während einer Temperatur-
behandlung thermische Effekte detektiert werden. Je nach zu untersuchender Eigenschaften werden
dabei besondere Anforderungen an den apparativen Aufbau gestellt. Somit wurden im Laufe der Zeit
sehr diffizile experimentelle Aufbauten zur thermischen Analyse entwickelt: die Methoden der
Thermoanalyse.
2 Methoden der thermischen Analyse
Zur thermischen Analyse sind im Allgemeinen alle physikalischen Eigenschaften geeignet, die sich
temperaturabhängig signifikant ändern, ebenso wie chemische Reaktionen, die sich als Funktion der
Temperatur detektieren lassen. Hier sind sowohl Änderungen der spezifischen Wärme als auch
Masseänderungen geeignete Eigenschaften, mit deren Hilfe man mögliche temperaturabhängige
Reaktionen und Reaktionsketten erfassen und deuten kann.
Für die Auswertung derartiger Messungen ist das Grundwissen des Chemikers, Physikers oder
Materialwissenschaftlers normalerweise ausreichend. Dem Anwender muss jedoch bewusst sein, dass

2
bei der thermischen Analyse neben den reinen Probeneigenschaften eine Vielzahl von Faktoren wie
z.B. der apparativer Aufbau des Analysegerätes oder der Ablauf des Messvorganges eine nicht
vernachlässigbare Rolle spielen. Kenntnisse über thermodynamische und messmethodische
Grundlagen werden also vor allem im Hinblick auf die Vergleichbarkeit von Messungen an
unterschiedlichen Geräten und die Reproduzierbarkeit von Einzelmessungen wichtig. Im gesteigerten
Maße gilt dies bei der Deutung von Messergebnissen, denen komplexe Vorgänge zugrunde liegen. An
dieser Stelle sind vor allem dynamisch-mechanische Prozesse anzuführen, die neben einem
komplizierten mathematischen Rüstzeug umfangreiche Spezialkenntnisse zum rheologischen
Verhalten von viskoelastischen Körpern und den möglichen Wechselwirkungen aller Einflussgrößen
untereinander erfordern.
2.1 Thermodynamische Grundlagen
Im Rahmen dieser Versuchsanleitung sollen lediglich die entscheidenden Zusammenhänge der
thermodynamischen Grundlagen der Thermoanalyse dargestellt werden. Für eine detailliertere
Charakterisierung sei auf die folgende Literatur verwiesen:
- ATKINS, P.W., DE PAULA , J. (2006): Physikalische Chemie, 4. vollständig überarbeitete
Auflage. Wiley-VCH Verlag Weinheim.
- HEIDE, K. (1979): Dynamische thermische Analysemethoden, 1. Auflage. VEB Deutscher
Verlag für Grundstoffindustrie Leipzig.
- HEMMINGER, W.F., CAMMENGA, H.K. (1989): Methoden der thermischen Analyse, 1.
Auflage. Springer-Verlag Berlin, Heidelberg, New York, London, Paris, Tokyo.
Das grundlegende Ziel der Thermoanalyse besteht darin, die Eigenschaften eines Stoffes bzw. deren
Änderungen während einer vorgegebenen Temperaturänderung zu analysieren. Es gilt also, eine
bestimmte Menge einer Substanz zu erwärmen, wofür eine stoffspezifische Wärmemenge zugeführt
werden muss, die Wärmekapazität C:
� = ����
(2-1) C - Wärmekapazität dQ - zugeführte Wärmemenge dT - Temperaturänderung
Zwar lässt sich diese Wärmemenge experimentell relativ leicht bestimmen, die erhaltenen Messwerte
sind jedoch wesentlich von der Versuchsdurchführung und von Zustandsvariablen wie Druck und
Temperatur abhängig [1]. Für die Charakterisierung von Stoffsystemen im thermodynamischen
Gleichgewicht nutzt man aus diesem Grund thermodynamische Potentiale, die sich aus der
GIBBS‘schen Fundamentalgleichung und den daraus abgeleiteten Differentialgleichungen ergeben.
3
Hier bietet sich vor allem die freie Enthalpie g an, da im Gleichgewicht die Variablen Druck und
Temperatur für das gesamte System gleich sind und ihre Messung relativ leicht möglich ist [1]:
���, , �� = − ��
(2-2)
G - freie Enthalpie T - Temperatur p - Druck ni - Stoffmenge H - Enthalpie S - Entropie
Über die Ableitung der kalorischen und thermischen Zustandsgleichung, die sich für die speziellen
Messprobleme der thermischen Analyse besonders eignen, lässt sich schließlich die molare
Wärmekapazität bei isobarer Prozessführung cp formulieren:
�� = �� ����
(2-3)
cp - molare Wärmekapazität eines isobaren Prozesses H - Enthalpie T - Temperatur p - Druck
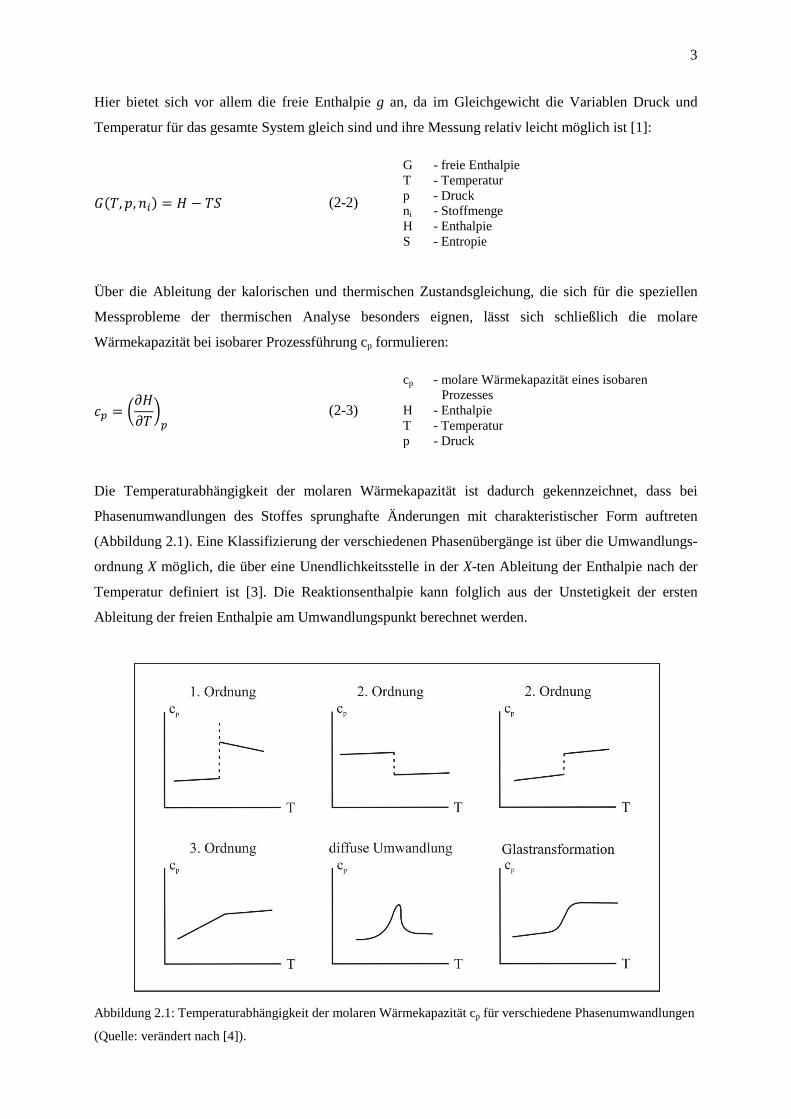
Die Temperaturabhängigkeit der molaren Wärmekapazität ist dadurch gekennzeichnet, dass bei
Phasenumwandlungen des Stoffes sprunghafte Änderungen mit charakteristischer Form auftreten
(Abbildung 2.1). Eine Klassifizierung der verschiedenen Phasenübergänge ist über die Umwandlungs-
ordnung X möglich, die über eine Unendlichkeitsstelle in der X-ten Ableitung der Enthalpie nach der
Temperatur definiert ist [3]. Die Reaktionsenthalpie kann folglich aus der Unstetigkeit der ersten
Ableitung der freien Enthalpie am Umwandlungspunkt berechnet werden.
Abbildung 2.1: Temperaturabhängigkeit der molaren Wärmekapazität cp für verschiedene Phasenumwandlungen
(Quelle: verändert nach [4]).

4
Typische Phasenumwandlungen erster Ordnung sind z.B. Kristallisations- und Schmelzprozesse,
Übergänge zweiter Ordnungen sind dagegen häufig Ordnungs-Unordnungs-Umwandlungen in
Mischkristallen wie z.B. das Verhalten ferromagnetischer Stoffe im Bereich der Curie-Temperatur.
Auch die Glastransformation stellt eine Umwandlung 2. Ordnung dar, wobei hier die kinetische
Komponente (Abkühlgeschwindigkeit) häufig die entscheidende Rolle spielt. Theoretisch sind auch
Umwandlungen mit dritter und höherer Ordnungen denkbar, aufgrund der sich schwierig gestaltenden
Messung von kleinen ∆H-Werten sind hier jedoch oft keine eindeutigen Zuordnungen möglich.
Die Thermodynamik liefert an dieser Stelle eine grundlegende Erkenntnis: aus der Temperatur-
abhängigkeit der spezifischen Wärme können wichtige Informationen über einen Phasenumwand-
lungsvorgang gewonnen werden. Um dies experimentell detektieren zu können, ist zunächst erst
einmal eine möglichst genaue Temperaturerfassung nötig.
2.2 Messmethodische Grundlagen: die Temperaturerfassung
Im Bereich bis 2000 °C werden für die Temperaturerfassung im Rahmen der thermischen Analyse
üblicherweise Thermoelemente verwendet. Diese bestehen aus zwei unterschiedlichen Metalldrähten,
die an einem Ende z.B. durch eine Schweißperle elektrisch leitend miteinander verbunden sind
(Thermopaar). Aufgrund des SEEBECK-Effektes, der das Auftreten einer Spannung zwischen zwei
Stellen unterschiedlicher Temperatur eines Leiters beschreibt, kommt es an den freien Enden zu einer
elektrischen Spannung, wenn sich Mess- und Vergleichstemperatur voneinander unterscheiden
(Abbildung 2.2). Eine Kontaktstelle des Thermopaares wird auf konstanter Temperatur gehalten
(Vergleichstemperatur), mit der anderen wird gemessen (Messtemperatur). Die aufgezeichnete
Potentialdifferenz ist dann annähernd proportional zur Temperaturdifferenz.
Die Auswahl einer geeigneten Metallpaarung für das Thermoelement muss an das spezifische
Analyseproblem angepasst werden, wobei verschiedene Faktoren eine Rolle spielen. Grundsätzlich
strebt man in dem für die Messung entscheidenden Temperaturbereich eine möglichst hohe
Thermospannung an, da bei hohen Messwerten der absolute Fehler des Thermoelements nur eine
untergeordnete Rolle spielt. Dieser liegt bei handelsüblichen Thermoelementen im Bereich von 2 bis 8
K. Daneben ist ein hoher Grad an Linearität im Verhältnis von Temperatur und Thermospannung vor
allem für die dynamischen Methoden der thermischen Analyse (vgl. Kapitel 2.3) nötig, die mit einer
konstanten Aufheizrate arbeiten.
Zusätzlich spielen insbesondere im Hochtemperaturbereich die chemische Beständigkeit (Korrosions-
festigkeit, Oxidationsverhalten) und das Rekristallisationsverhalten der Metalle eine wichtige Rolle.
Beide Aspekte können zur Veränderung des Drahtwiderstandes führen und damit zu einer
Verfälschung der aufgezeichneten Werte der Thermospannung. Ferner sollte man sich vor dem Einsatz

5
vergewissern, ob im angestrebten Temperaturbereich der Messung einzelne Bestandteile der Probe
verdampfen und diese Eutektika mit den Metallen des Thermoelementes bilden können. Bei Platin und
Phosphor kann das bereits ab 590 °C der Fall sein.
Abbildung 2.2: Schematische Darstellung eines Thermoelementes (Quelle: verändert nach [5]).
Bezüglich des spezifischen Widerstandes der Metalldrähte spielt neben der Technologie der
Drahtherstellung (Grad an Verunreinigungen, Texturen) auch das Eindiffundieren von Verunreini-
gungen während der Anwendung eine wichtige Rolle. Dieses stellt einen irreversiblen Prozess dar und
ist maßgeblich für die Lebensdauer des Thermoelementes verantwortlich, was sich wiederum auf die
Kostenstruktur des Einsatzes auswirkt.
Diese Vielzahl an Faktoren ist für unterschiedliche Analyseprobleme naturgemäß nicht in einer
einzigen Metallkombination zu vereinigen. Dementsprechend werden je nach Einsatzbereich
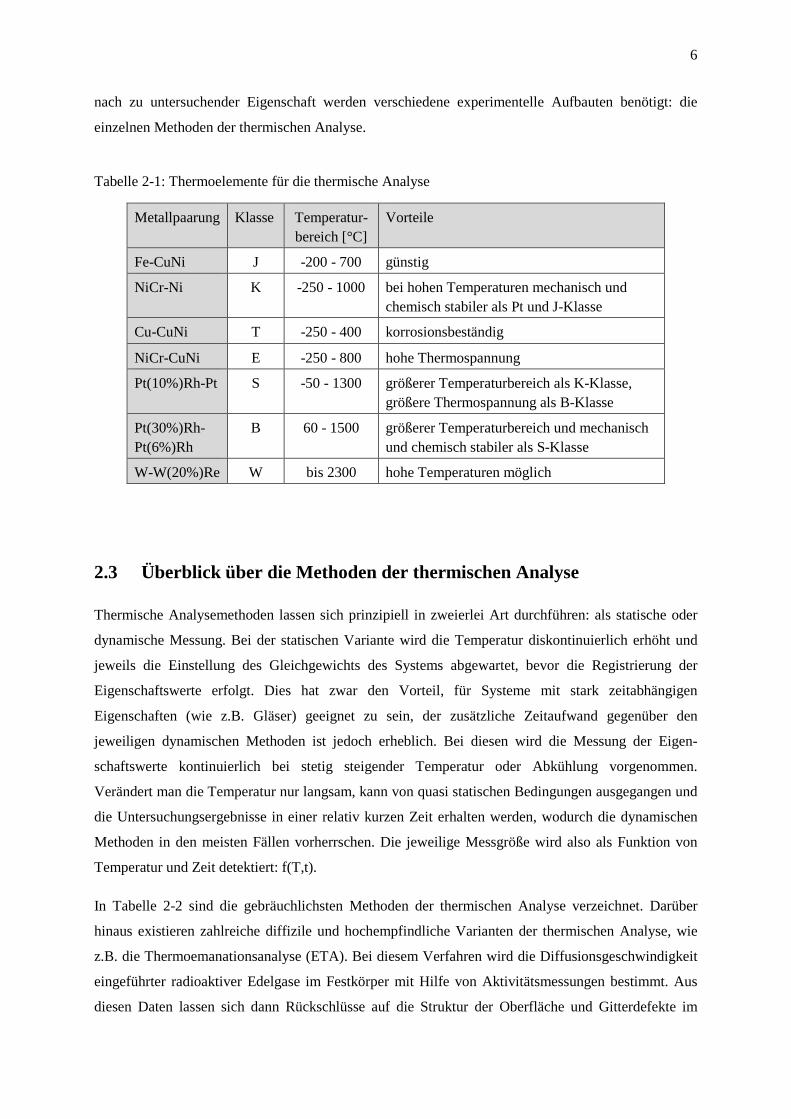
unterschiedliche Materialpaarungen verwendet, von denen die gebräuchlichsten in Tabelle 2-1
aufgeführt sind. Eine Systematisierung der verschiedenen Metallpaarungen findet über die sogenannte
Klasse statt, die neben entsprechenden Grundwerten auch zulässige Abweichungen definiert.
Die Problematik einer korrekten Temperaturerfassung im Rahmen der thermischen Analyse ist mit der
Auswahl eines geeigneten Thermoelementes nicht abgeschlossen. Vor allem apparative Einflüsse auf
die Differenz zwischen den realen Temperaturverhältnissen innerhalb der Probe und den vom
Thermoelement aufgezeichneten Temperaturen spielen eine wichtige Rolle. Diese werden am Beispiel
der Differenzthermoanalyse in Abschnitt 2.4 erläutert.
Kann die Temperatur zuverlässig bestimmt werden, ist ein kontrolliertes Aufheizprogramm
realisierbar und damit auch die temperaturabhängige Beobachtung von Eigenschaftsänderungen. Je
6
nach zu untersuchender Eigenschaft werden verschiedene experimentelle Aufbauten benötigt: die
einzelnen Methoden der thermischen Analyse.
Tabelle 2-1: Thermoelemente für die thermische Analyse
Metallpaarung
Klasse
Temperatur-bereich [°C]
Vorteile
Fe-CuNi
J
-200 - 700
günstig
NiCr-Ni
K
-250 - 1000
bei hohen Temperaturen mechanisch und chemisch stabiler als Pt und J-Klasse
Cu-CuNi
T
-250 - 400
korrosionsbeständig
NiCr-CuNi
E
-250 - 800
hohe Thermospannung
Pt(10%)Rh-Pt
S
-50 - 1300
größerer Temperaturbereich als K-Klasse, größere Thermospannung als B-Klasse
Pt(30%)Rh-Pt(6%)Rh
B
60 - 1500
größerer Temperaturbereich und mechanisch und chemisch stabiler als S-Klasse
W-W(20%)Re
W
bis 2300
hohe Temperaturen möglich
2.3 Überblick über die Methoden der thermischen Analyse
Thermische Analysemethoden lassen sich prinzipiell in zweierlei Art durchführen: als statische oder
dynamische Messung. Bei der statischen Variante wird die Temperatur diskontinuierlich erhöht und
jeweils die Einstellung des Gleichgewichts des Systems abgewartet, bevor die Registrierung der
Eigenschaftswerte erfolgt. Dies hat zwar den Vorteil, für Systeme mit stark zeitabhängigen
Eigenschaften (wie z.B. Gläser) geeignet zu sein, der zusätzliche Zeitaufwand gegenüber den
jeweiligen dynamischen Methoden ist jedoch erheblich. Bei diesen wird die Messung der Eigen-
schaftswerte kontinuierlich bei stetig steigender Temperatur oder Abkühlung vorgenommen.
Verändert man die Temperatur nur langsam, kann von quasi statischen Bedingungen ausgegangen und
die Untersuchungsergebnisse in einer relativ kurzen Zeit erhalten werden, wodurch die dynamischen
Methoden in den meisten Fällen vorherrschen. Die jeweilige Messgröße wird also als Funktion von
Temperatur und Zeit detektiert: f(T,t).
In Tabelle 2-2 sind die gebräuchlichsten Methoden der thermischen Analyse verzeichnet. Darüber
hinaus existieren zahlreiche diffizile und hochempfindliche Varianten der thermischen Analyse, wie
z.B. die Thermoemanationsanalyse (ETA). Bei diesem Verfahren wird die Diffusionsgeschwindigkeit
eingeführter radioaktiver Edelgase im Festkörper mit Hilfe von Aktivitätsmessungen bestimmt. Aus
diesen Daten lassen sich dann Rückschlüsse auf die Struktur der Oberfläche und Gitterdefekte im

7
Volumen des Probenkörpers ziehen, die unter anderem einen Einfluss auf die Geschwindigkeit von
Festkörperreaktionen haben.
Tabelle 2-2: Die wichtigsten Methoden der thermischen Analyse
Bezeichnung
Abkürzung
Messgrößen als f(T,t)
Thermogravimetrie
TGA
Masseänderung und deren Ableitung nach der Temperatur
Differenzthermoanalyse
DTA
Wärmetönung
Differential Scanning Calorimetrie
DSC
Wärmefluss
Thermooptische Analyse
TOA
optische Veränderung
Dilatometrie
DIL
Ausdehnung
Viskosimetrie
-
Viskosität
Erhitzungsmikroskop
EHM
Gestaltänderung
Thermisch simulierter Strom
TSC
Relaxationsspektrum
Thermisch-mechanische Analyse
TMA
Längenänderung bei Belastung (z.B. Zug- und Scherbelastung)
Simultane Thermoanalyse
STA
mehrere Messgrößen
Thermo-Gasanalyse
EGA
Gasabspaltungsreaktionen
Dynamisch-mechanische-thermische Spektroskopie
DMTS
Elastizitäts-, Scher-, Verlust- und
Speichermodul, tanδ
Hochtemperatur-Röntgenbeugung
-
Gitterparameter, Symmetrie, Atompositionen
Thermoakustische Analyse
TSA
Schallgeschwindigkeit
Im Rahmen des Praktikums soll der Fokus auf die im Otto-Schott-Institut vorhandenen Methoden der
thermischen Analyse gerichtet werden: die Differenzthermoanalyse, die Differential-Scanning-
Calometry, die Dilatometrie, die Erhitzungsmikroskopie und eine Anlage zur simultanen Thermo-
analyse. Im Folgenden werden die Besonderheiten des jeweiligen experimentellen Aufbaus, die
Auswertung der Messkurven und dabei zu beachtende Faktoren erläutert.
2.4 Differenzthermoanalyse (DTA)
Die gebräuchlichste Methode der thermischen Analyse ist die Differenzthermoanalyse (DTA). Dies
begründet sich zum einen durch die vielfältigen Anwendungsmöglichkeiten. Es sind z.B. quantitative
Aussagen über die relative Änderung von Energie, Masse, Gitterparametern, optischen Parametern,
Dampfdruck, Volumen, Leitfähigkeit, magnetischen Eigenschaften und Diffusionsgeschwindigkeiten

8
möglich. Zum anderen wirkt die DTA dem messmethodischen Problem der klassischen thermischen
Analyse, dem Nachweis geringer Differenzen großer Messwerte, entgegen, indem keine absoluten
Temperaturen gemessen werden, sondern nur Differenzen.
2.4.1 Das Grundprinzip der DTA
Die Messanordnung der DTA besteht aus zwei Tiegeln (meist aus Platin), in deren unmittelbarer Nähe
zwei Thermoelemente angebracht sind (Abbildung 2.3). Ein Tiegel enthält die Analysesubstanz, der
andere eine Inertsubstanz, die in dem zu untersuchenden Temperaturbereich keinerlei thermische
Effekte zeigt. Wie in Abschnitt 2.2 erläutert, produzieren beide Thermoelemente eine temperatur-
abhängige Spannung, die den jeweiligen Temperaturen entspricht. Da die Thermoelemente gegen-
einander geschaltet sind, kann eine Differenzspannung abgegriffen werden, die mit der Differenz der
Temperaturen übereinstimmt. Diese Apparatur befindet sich auf einer isothermen Fläche innerhalb des
Ofens, der mit kontrollierten Aufheizprogrammen betrieben werden kann.
Abbildung 2.3: Schematische Darstellung einer Differenzthermoanalyse (Quelle: verändert nach [1]).
Wird dem Ofen eine lineare Temperatur-Zeit-Funktion vorgegeben, zeigt die Inertsubstanz
entsprechend eine lineare Aufheizkurve TI (t) (Abbildung 2.4). Diese wird mit der Aufheizkurve der
Analysesubstanz TPR (t) verglichen, der die gleiche Wärmemenge zugeführt wird. Findet in der Probe
ein thermischer Effekt wie z.B. eine chemische Reaktion oder eine Kristallisation statt, der im
dargestellten Fall Energie verbraucht (endotherm), kann die zugeführte Wärmemenge nicht vollständig
zur Erhöhung der Temperatur genutzt werden. Die Aufheizkurve der Probe verzögert sich also im
Vergleich zur linearen Temperatur-Zeit-Kurve der Inertsubstanz, woraus sich die aufgezeichnete
Differenzkurve ∆T (t) ergibt. Mit Hilfe der DTA lässt sich also die sog. Wärmetönung registrieren, die
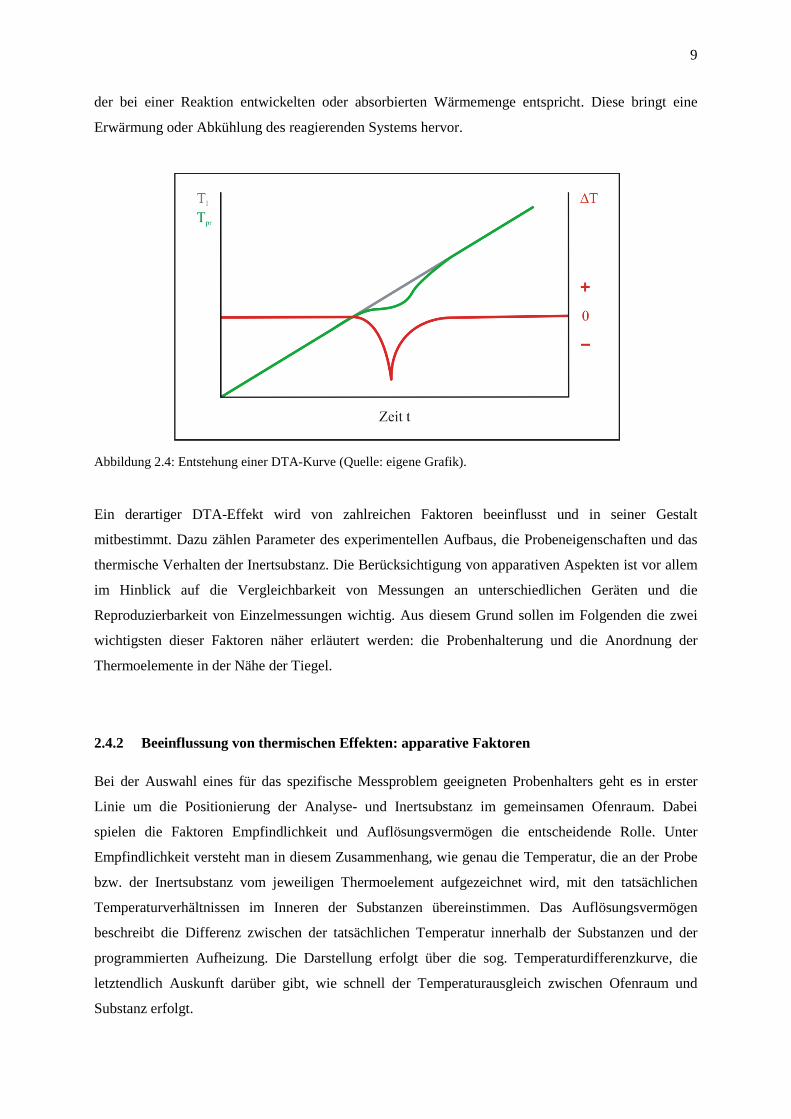
9
der bei einer Reaktion entwickelten oder absorbierten Wärmemenge entspricht. Diese bringt eine
Erwärmung oder Abkühlung des reagierenden Systems hervor.
Abbildung 2.4: Entstehung einer DTA-Kurve (Quelle: eigene Grafik).
Ein derartiger DTA-Effekt wird von zahlreichen Faktoren beeinflusst und in seiner Gestalt
mitbestimmt. Dazu zählen Parameter des experimentellen Aufbaus, die Probeneigenschaften und das
thermische Verhalten der Inertsubstanz. Die Berücksichtigung von apparativen Aspekten ist vor allem
im Hinblick auf die Vergleichbarkeit von Messungen an unterschiedlichen Geräten und die
Reproduzierbarkeit von Einzelmessungen wichtig. Aus diesem Grund sollen im Folgenden die zwei
wichtigsten dieser Faktoren näher erläutert werden: die Probenhalterung und die Anordnung der
Thermoelemente in der Nähe der Tiegel.
2.4.2 Beeinflussung von thermischen Effekten: apparative Faktoren
Bei der Auswahl eines für das spezifische Messproblem geeigneten Probenhalters geht es in erster
Linie um die Positionierung der Analyse- und Inertsubstanz im gemeinsamen Ofenraum. Dabei
spielen die Faktoren Empfindlichkeit und Auflösungsvermögen die entscheidende Rolle. Unter
Empfindlichkeit versteht man in diesem Zusammenhang, wie genau die Temperatur, die an der Probe
bzw. der Inertsubstanz vom jeweiligen Thermoelement aufgezeichnet wird, mit den tatsächlichen
Temperaturverhältnissen im Inneren der Substanzen übereinstimmen. Das Auflösungsvermögen
beschreibt die Differenz zwischen der tatsächlichen Temperatur innerhalb der Substanzen und der
programmierten Aufheizung. Die Darstellung erfolgt über die sog. Temperaturdifferenzkurve, die
letztendlich Auskunft darüber gibt, wie schnell der Temperaturausgleich zwischen Ofenraum und
Substanz erfolgt.

10
Abbildung 2.5 zeigt zwei Realisierungsmöglichkeiten einer DTA-Probenhalterung. Variante 1 besteht
aus einem Metallblock mit zwei Bohrungen, in die Probe und Inertsubstanz eingeführt werden. Das
Metall zwischen beiden Tiegeln führt zu einem schnellen Temperaturausgleich und sorgt für ein
entsprechend hohes Auflösungsvermögen. Mit einer solchen Probenanordnung können zwar
benachbarte thermische Effekte gut voneinander getrennt werden, die Empfindlichkeit ist jedoch
relativ gering. Variante 2 besteht dagegen aus einem Metallblock mit nur einer Bohrung. Der
Temperaturausgleich zwischen den Tiegeln ist hier im Wesentlichen auf Konvektion beschränkt und
entsprechend verlangsamt. Der hohen Empfindlichkeit steht ein relativ schlechtes Auflösungs-
vermögen gegenüber.
Abbildung 2.5: Verschiedene Probenhalter für die DTA (Quelle: verändert nach [1]).
Die Verwendung eines Probenhalters sollte stets an das konkrete Messproblem angepasst werden, je
nachdem ob das Hauptaugenmerk auf dem Auflösungsvermögen oder der Empfindlichkeit liegt.
Dennoch hat sich für die meisten DTA-Analysen die Variante 1 durchgesetzt, wobei man den Nachteil
des geringen Auflösungsvermögens durch eine kleine Einwaage (wenig Probensubstanz) und einer
Verbesserung der Wärmeleitung zwischen den Tiegeln zu kompensieren versucht.
Die Anordnung des Temperaturmessfühlers wird zwar zwangsläufig durch den Aufbau der Proben-
halterung mitbestimmt, sie kann aber in gewissen Grenzen variiert werden. Vor allem hinsichtlich der
Optimierung der Empfindlichkeit der Meßanordnung stellt sie einen wichtigen Aspekt dar. Auch hier
existieren wieder verschiedene Faktoren, die zu beachten und gegeneinander abzuwägen sind: die
geometrische Anordnung der Heizelemente im Ofenraum, die Art der verwendeten Thermoelemente
und die Reproduzierbarkeit der räumlichen Ausrichtung der Thermoelemente. Abbildung 2.6 zeigt
verschiedene Varianten, wobei für die meisten Messungen Variante 2 zu bevorzugen ist, da diese die
höchste Reproduzierbarkeit aufweist. Die Bedeutung dieses Faktors wird deutlich, wenn man bedenkt,
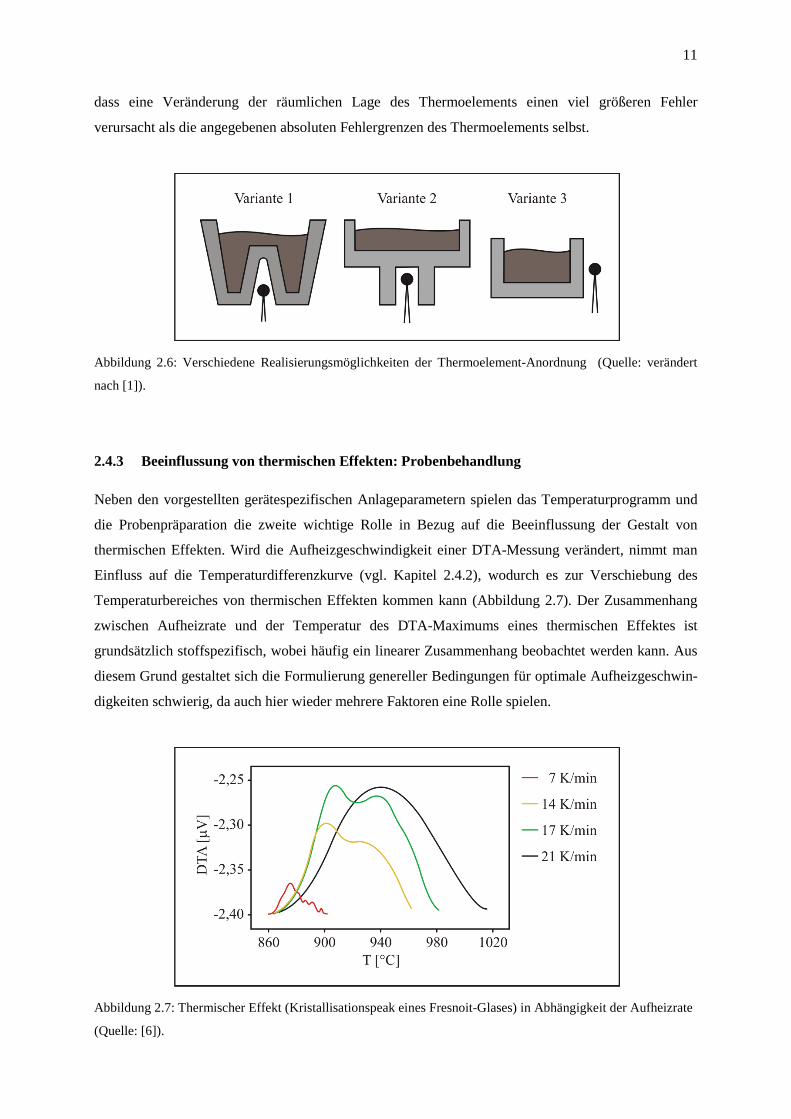
11
dass eine Veränderung der räumlichen Lage des Thermoelements einen viel größeren Fehler
verursacht als die angegebenen absoluten Fehlergrenzen des Thermoelements selbst.
Abbildung 2.6: Verschiedene Realisierungsmöglichkeiten der Thermoelement-Anordnung (Quelle: verändert
nach [1]).
2.4.3 Beeinflussung von thermischen Effekten: Probenbehandlung
Neben den vorgestellten gerätespezifischen Anlageparametern spielen das Temperaturprogramm und
die Probenpräparation die zweite wichtige Rolle in Bezug auf die Beeinflussung der Gestalt von
thermischen Effekten. Wird die Aufheizgeschwindigkeit einer DTA-Messung verändert, nimmt man
Einfluss auf die Temperaturdifferenzkurve (vgl. Kapitel 2.4.2), wodurch es zur Verschiebung des
Temperaturbereiches von thermischen Effekten kommen kann (Abbildung 2.7). Der Zusammenhang
zwischen Aufheizrate und der Temperatur des DTA-Maximums eines thermischen Effektes ist
grundsätzlich stoffspezifisch, wobei häufig ein linearer Zusammenhang beobachtet werden kann. Aus
diesem Grund gestaltet sich die Formulierung genereller Bedingungen für optimale Aufheizgeschwin-
digkeiten schwierig, da auch hier wieder mehrere Faktoren eine Rolle spielen.
Abbildung 2.7: Thermischer Effekt (Kristallisationspeak eines Fresnoit-Glases) in Abhängigkeit der Aufheizrate
(Quelle: [6]).
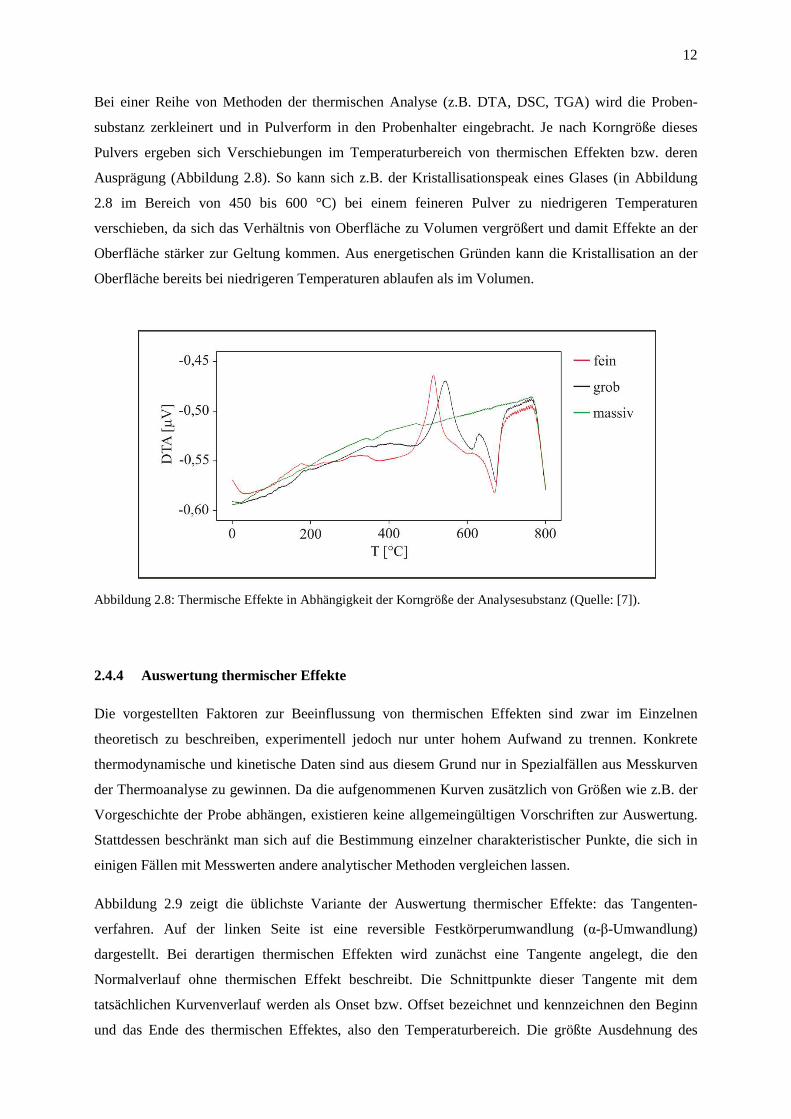
12
Bei einer Reihe von Methoden der thermischen Analyse (z.B. DTA, DSC, TGA) wird die Proben-
substanz zerkleinert und in Pulverform in den Probenhalter eingebracht. Je nach Korngröße dieses
Pulvers ergeben sich Verschiebungen im Temperaturbereich von thermischen Effekten bzw. deren
Ausprägung (Abbildung 2.8). So kann sich z.B. der Kristallisationspeak eines Glases (in Abbildung
2.8 im Bereich von 450 bis 600 °C) bei einem feineren Pulver zu niedrigeren Temperaturen
verschieben, da sich das Verhältnis von Oberfläche zu Volumen vergrößert und damit Effekte an der
Oberfläche stärker zur Geltung kommen. Aus energetischen Gründen kann die Kristallisation an der
Oberfläche bereits bei niedrigeren Temperaturen ablaufen als im Volumen.
Abbildung 2.8: Thermische Effekte in Abhängigkeit der Korngröße der Analysesubstanz (Quelle: [7]).
2.4.4 Auswertung thermischer Effekte
Die vorgestellten Faktoren zur Beeinflussung von thermischen Effekten sind zwar im Einzelnen
theoretisch zu beschreiben, experimentell jedoch nur unter hohem Aufwand zu trennen. Konkrete
thermodynamische und kinetische Daten sind aus diesem Grund nur in Spezialfällen aus Messkurven
der Thermoanalyse zu gewinnen. Da die aufgenommenen Kurven zusätzlich von Größen wie z.B. der
Vorgeschichte der Probe abhängen, existieren keine allgemeingültigen Vorschriften zur Auswertung.
Stattdessen beschränkt man sich auf die Bestimmung einzelner charakteristischer Punkte, die sich in
einigen Fällen mit Messwerten andere analytischer Methoden vergleichen lassen.
Abbildung 2.9 zeigt die üblichste Variante der Auswertung thermischer Effekte: das Tangenten-
verfahren. Auf der linken Seite ist eine reversible Festkörperumwandlung (α-β-Umwandlung)
dargestellt. Bei derartigen thermischen Effekten wird zunächst eine Tangente angelegt, die den
Normalverlauf ohne thermischen Effekt beschreibt. Die Schnittpunkte dieser Tangente mit dem
tatsächlichen Kurvenverlauf werden als Onset bzw. Offset bezeichnet und kennzeichnen den Beginn
und das Ende des thermischen Effektes, also den Temperaturbereich. Die größte Ausdehnung des
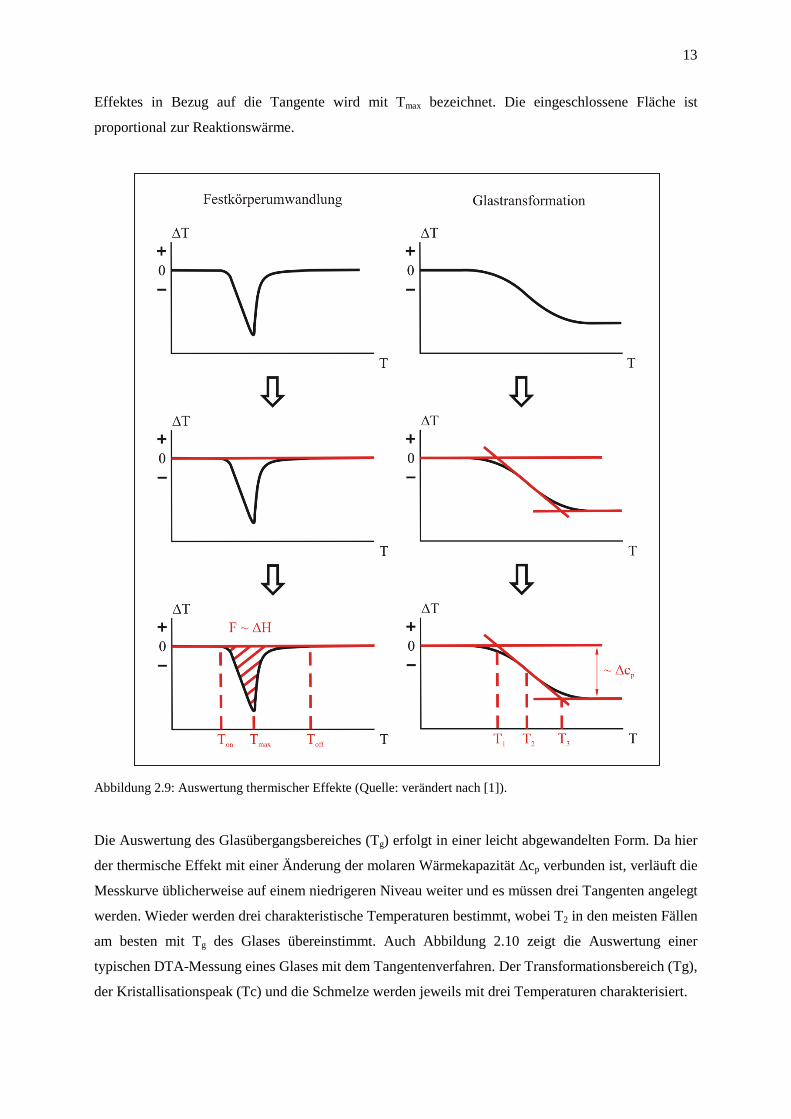
13
Effektes in Bezug auf die Tangente wird mit Tmax bezeichnet. Die eingeschlossene Fläche ist
proportional zur Reaktionswärme.
Abbildung 2.9: Auswertung thermischer Effekte (Quelle: verändert nach [1]).
Die Auswertung des Glasübergangsbereiches (Tg) erfolgt in einer leicht abgewandelten Form. Da hier
der thermische Effekt mit einer Änderung der molaren Wärmekapazität ∆cp verbunden ist, verläuft die
Messkurve üblicherweise auf einem niedrigeren Niveau weiter und es müssen drei Tangenten angelegt
werden. Wieder werden drei charakteristische Temperaturen bestimmt, wobei T2 in den meisten Fällen
am besten mit Tg des Glases übereinstimmt. Auch Abbildung 2.10 zeigt die Auswertung einer
typischen DTA-Messung eines Glases mit dem Tangentenverfahren. Der Transformationsbereich (Tg),
der Kristallisationspeak (Tc) und die Schmelze werden jeweils mit drei Temperaturen charakterisiert.
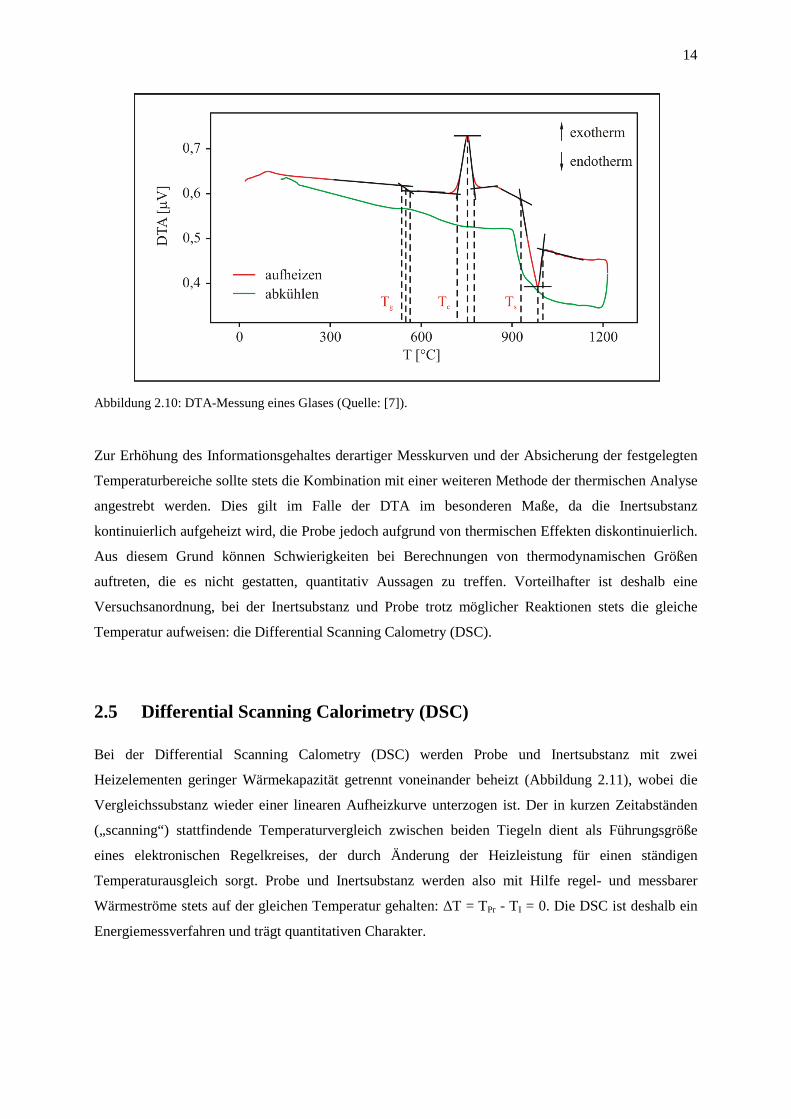
14
Abbildung 2.10: DTA-Messung eines Glases (Quelle: [7]).
Zur Erhöhung des Informationsgehaltes derartiger Messkurven und der Absicherung der festgelegten
Temperaturbereiche sollte stets die Kombination mit einer weiteren Methode der thermischen Analyse
angestrebt werden. Dies gilt im Falle der DTA im besonderen Maße, da die Inertsubstanz
kontinuierlich aufgeheizt wird, die Probe jedoch aufgrund von thermischen Effekten diskontinuierlich.
Aus diesem Grund können Schwierigkeiten bei Berechnungen von thermodynamischen Größen
auftreten, die es nicht gestatten, quantitativ Aussagen zu treffen. Vorteilhafter ist deshalb eine
Versuchsanordnung, bei der Inertsubstanz und Probe trotz möglicher Reaktionen stets die gleiche
Temperatur aufweisen: die Differential Scanning Calometry (DSC).
2.5 Differential Scanning Calorimetry (DSC)
Bei der Differential Scanning Calometry (DSC) werden Probe und Inertsubstanz mit zwei
Heizelementen geringer Wärmekapazität getrennt voneinander beheizt (Abbildung 2.11), wobei die
Vergleichssubstanz wieder einer linearen Aufheizkurve unterzogen ist. Der in kurzen Zeitabständen
(„scanning“) stattfindende Temperaturvergleich zwischen beiden Tiegeln dient als Führungsgröße
eines elektronischen Regelkreises, der durch Änderung der Heizleistung für einen ständigen
Temperaturausgleich sorgt. Probe und Inertsubstanz werden also mit Hilfe regel- und messbarer
Wärmeströme stets auf der gleichen Temperatur gehalten: ∆T = TPr - TI = 0. Die DSC ist deshalb ein
Energiemessverfahren und trägt quantitativen Charakter.
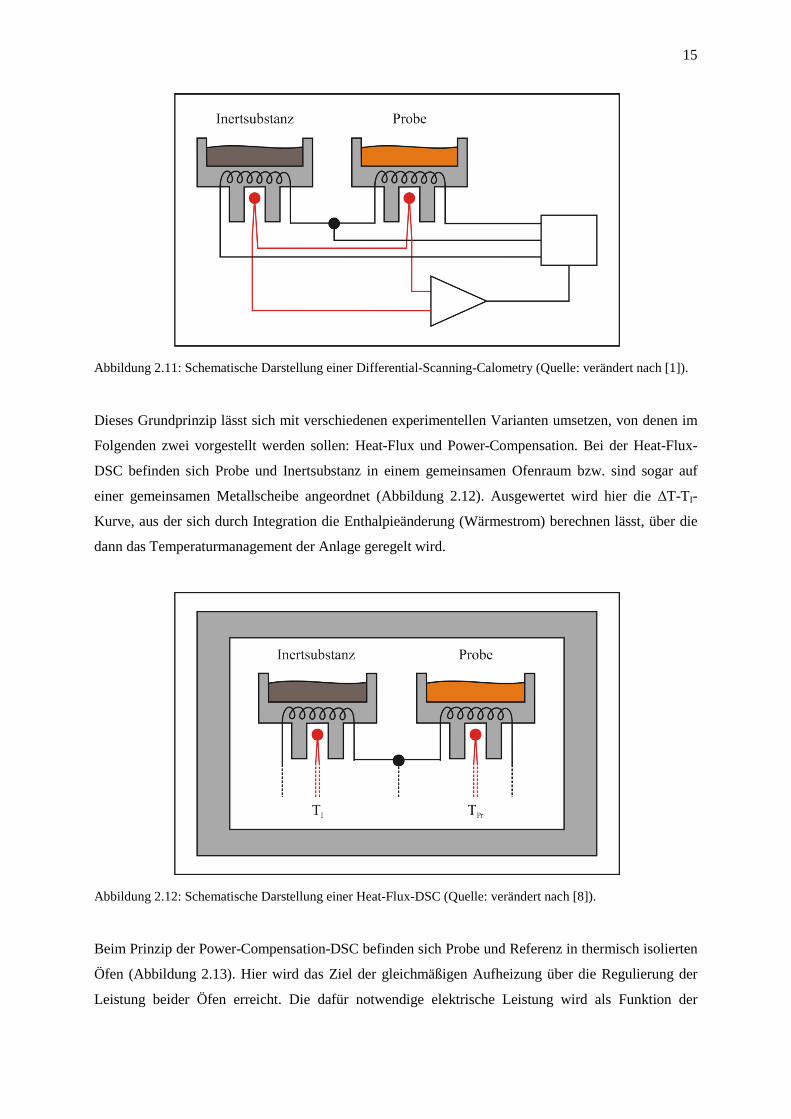
15
Abbildung 2.11: Schematische Darstellung einer Differential-Scanning-Calometry (Quelle: verändert nach [1]).
Dieses Grundprinzip lässt sich mit verschiedenen experimentellen Varianten umsetzen, von denen im
Folgenden zwei vorgestellt werden sollen: Heat-Flux und Power-Compensation. Bei der Heat-Flux-
DSC befinden sich Probe und Inertsubstanz in einem gemeinsamen Ofenraum bzw. sind sogar auf
einer gemeinsamen Metallscheibe angeordnet (Abbildung 2.12). Ausgewertet wird hier die ∆T-TI-
Kurve, aus der sich durch Integration die Enthalpieänderung (Wärmestrom) berechnen lässt, über die
dann das Temperaturmanagement der Anlage geregelt wird.
Abbildung 2.12: Schematische Darstellung einer Heat-Flux-DSC (Quelle: verändert nach [8]).
Beim Prinzip der Power-Compensation-DSC befinden sich Probe und Referenz in thermisch isolierten
Öfen (Abbildung 2.13). Hier wird das Ziel der gleichmäßigen Aufheizung über die Regulierung der
Leistung beider Öfen erreicht. Die dafür notwendige elektrische Leistung wird als Funktion der
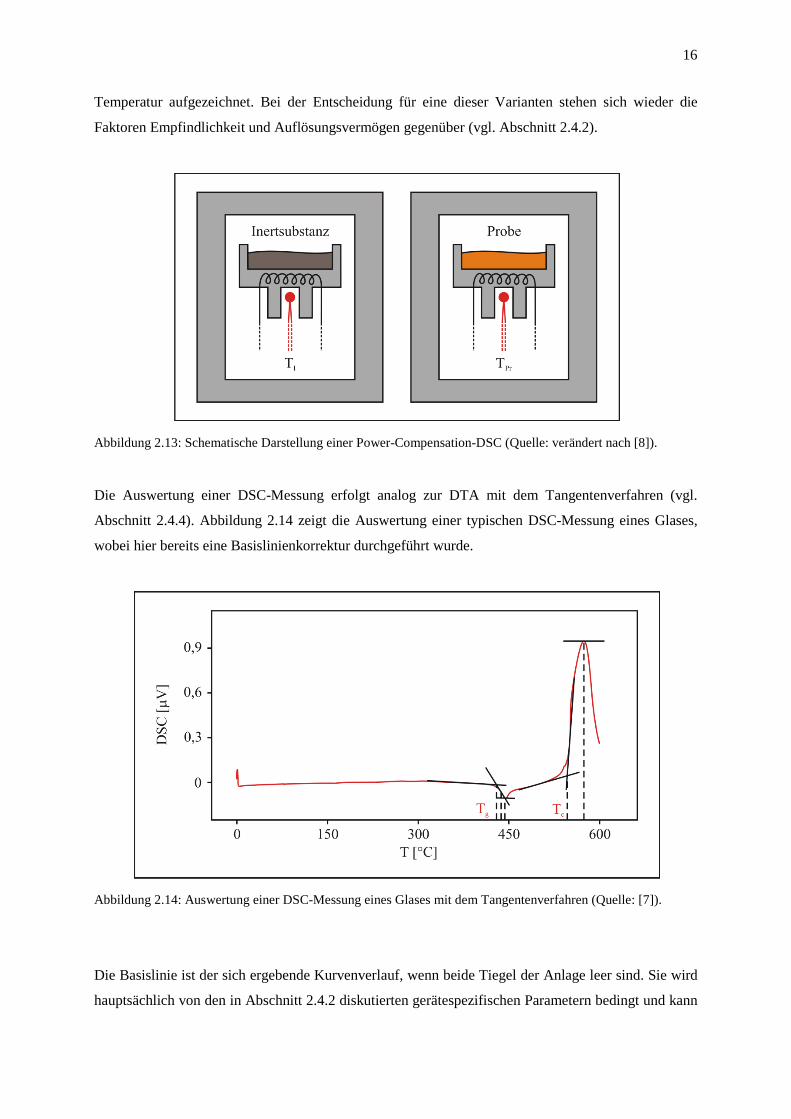
16
Temperatur aufgezeichnet. Bei der Entscheidung für eine dieser Varianten stehen sich wieder die
Faktoren Empfindlichkeit und Auflösungsvermögen gegenüber (vgl. Abschnitt 2.4.2).
Abbildung 2.13: Schematische Darstellung einer Power-Compensation-DSC (Quelle: verändert nach [8]).
Die Auswertung einer DSC-Messung erfolgt analog zur DTA mit dem Tangentenverfahren (vgl.
Abschnitt 2.4.4). Abbildung 2.14 zeigt die Auswertung einer typischen DSC-Messung eines Glases,
wobei hier bereits eine Basislinienkorrektur durchgeführt wurde.
Abbildung 2.14: Auswertung einer DSC-Messung eines Glases mit dem Tangentenverfahren (Quelle: [7]).
Die Basislinie ist der sich ergebende Kurvenverlauf, wenn beide Tiegel der Anlage leer sind. Sie wird
hauptsächlich von den in Abschnitt 2.4.2 diskutierten gerätespezifischen Parametern bedingt und kann

17
zur Minimierung derartiger Einflüsse von der Messkurve abgezogen werden. Die Gefahr einer
Fehlinterpretation von gerätespezifischen Kurvenverläufen kann somit reduziert werden.
2.6 Dilatometrie (DIL)
Die Längenänderung eines Festkörpers bei Temperaturänderung wird über den thermischen
Ausdehnungskoeffizienten charakterisiert. Dieser ist ein relativer Wert, der angibt, um den wievielten
Teil seiner Länge sich ein Stoff ändert, wenn die Temperatur von einem beliebigen Ausgangswert um
einen Kelvin steigt:
� = 1∆� ∗
∆���
(2-4)
α - Längenausdehnungskoeffizient ∆T - Temperaturdifferenz ∆l - Längenänderung l0 - Ausgangslänge
Unter dem Volumenausdehnungskoeffizient ß versteht man (bei Annahme von isotropen
Eigenschaften) den dreifachen Wert des Längenausdehnungskoeffizienten (ß = 3α). Eine anisotrope
Ausdehnung kann jedoch nur als Tensor beschrieben werden [9].
Die thermische Ausdehnung von festen Körpern ist bei gleicher Temperaturerhöhung weitaus geringer
als bei einer Flüssigkeit oder eines Gases gleicher Zusammensetzung, da die Teilchen (Atome, Ionen,
Molekülverbände) an feste Gitterplätze gebunden sind. Die Absolutwerte der thermischen
Ausdehnung von Feststoffen liegen daher in aller Regel lediglich bei 10-5 bis 10-7 K-1. Wird die
Auslenkung in einem Festkörper größer als die maximale Bindungslänge, werden die Teilchen von
ihrer Gleichgewichtslage entfernt und der Stoff schmilzt.
Die Messung der thermischen Ausdehnung erfolgt mit Dilatometern. Da sich die Erfassung des
Volumens von Festkörpern in Abhängigkeit von der Temperatur sehr aufwendig gestaltet, wird unter
Annahme isotroper Eigenschaften nur die Längenänderung gemessen und als repräsentativ angesehen.
Handelsübliches Kalknatronsilikatglas hat einen thermischen Ausdehnungskoeffizienten im Bereich
von 7 ∗ 10-6 K-1. Bei einer gängigen Probenlänge von 25 mm und einer Temperaturdifferenz von 10 K
müssen also Längenänderungen von 1,75 µm registriert werden. Dies kann mit mechanischen,
optischen, elektrischen oder röntgenographischen Methoden erfolgen. Bei der Auswahl der Mess-
technik spielen die Faktoren Probenbeschaffenheit, Messgenauigkeit und Temperaturbereich die
wichtigste Rolle.
In einem Dilatometer wird eine stabförmige Probe in ein Messystem bekannter thermischer
Ausdehnung eingespannt und in einem Ofen aufgeheizt (Abbildung 2.15). Die Aufzeichnung der
Längenänderung als Funktion der Temperatur f(T) erfolgt bei modernen Dilatometern über die
Einkopplung in ein mechanisches Sensorsystem (engl. Linear Variable Differential Transformer
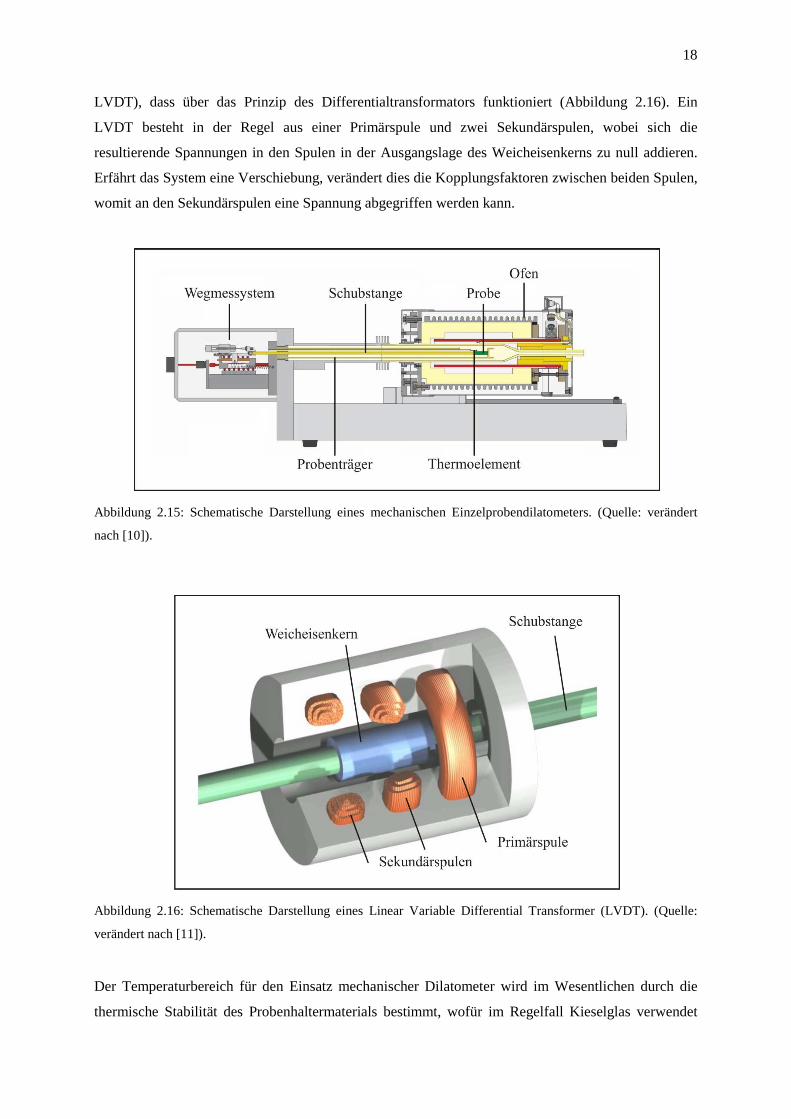
18
LVDT), dass über das Prinzip des Differentialtransformators funktioniert (Abbildung 2.16). Ein
LVDT besteht in der Regel aus einer Primärspule und zwei Sekundärspulen, wobei sich die
resultierende Spannungen in den Spulen in der Ausgangslage des Weicheisenkerns zu null addieren.
Erfährt das System eine Verschiebung, verändert dies die Kopplungsfaktoren zwischen beiden Spulen,
womit an den Sekundärspulen eine Spannung abgegriffen werden kann.
Abbildung 2.15: Schematische Darstellung eines mechanischen Einzelprobendilatometers. (Quelle: verändert
nach [10]).
Abbildung 2.16: Schematische Darstellung eines Linear Variable Differential Transformer (LVDT). (Quelle:
verändert nach [11]).
Der Temperaturbereich für den Einsatz mechanischer Dilatometer wird im Wesentlichen durch die
thermische Stabilität des Probenhaltermaterials bestimmt, wofür im Regelfall Kieselglas verwendet
19
wird (bis ca. 1000 °C temperaturstabil). Zusätzlich geht die (nicht völlig lineare) Temperaturabhängig-
keit der Ausdehnung des Probenhaltermaterials als zu beachtende Größe ein. Aus diesen Gründen
verwendet man im Bereich von Präzisions- und Hochtemperaturmessungen optische Interferenz-
längenmessyteme (ca. 30 nm Genauigkeit).
Die Methode der Interferenzdilatometrie nutzt das Prinzip des Interferenzkomparators. Hierbei werden
Lichtwegdifferenzen gemessen, die sich durch Interferenz des Lichtes zwischen einer definierten
Wegstrecke (Referenz) und der zu messenden Strecke ergeben. Diese entsprechen dem Abstand bzw.
der Dicke der Probe in der Messanordnung. Das Licht kann dabei sowohl weiß (polychromatisch) als
auch monochromatisch sein (Laser).
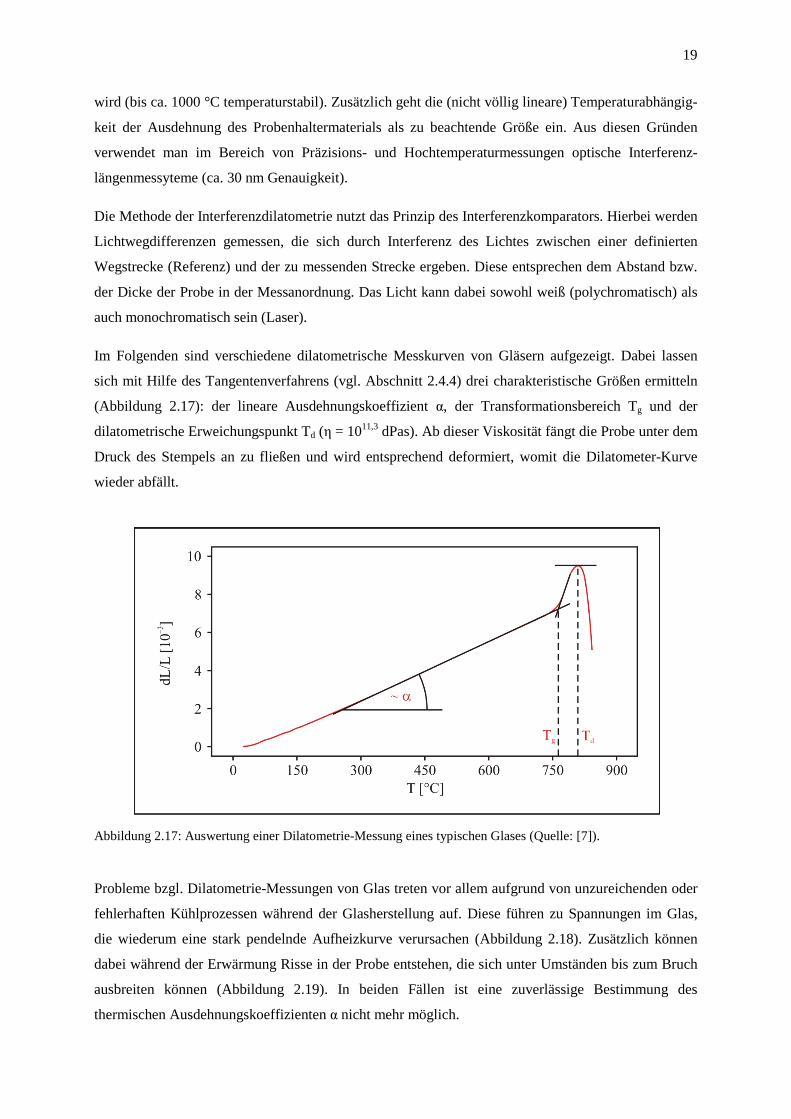
Im Folgenden sind verschiedene dilatometrische Messkurven von Gläsern aufgezeigt. Dabei lassen
sich mit Hilfe des Tangentenverfahrens (vgl. Abschnitt 2.4.4) drei charakteristische Größen ermitteln
(Abbildung 2.17): der lineare Ausdehnungskoeffizient α, der Transformationsbereich Tg und der
dilatometrische Erweichungspunkt Td (η = 1011,3 dPas). Ab dieser Viskosität fängt die Probe unter dem
Druck des Stempels an zu fließen und wird entsprechend deformiert, womit die Dilatometer-Kurve
wieder abfällt.
Abbildung 2.17: Auswertung einer Dilatometrie-Messung eines typischen Glases (Quelle: [7]).
Probleme bzgl. Dilatometrie-Messungen von Glas treten vor allem aufgrund von unzureichenden oder
fehlerhaften Kühlprozessen während der Glasherstellung auf. Diese führen zu Spannungen im Glas,
die wiederum eine stark pendelnde Aufheizkurve verursachen (Abbildung 2.18). Zusätzlich können
dabei während der Erwärmung Risse in der Probe entstehen, die sich unter Umständen bis zum Bruch
ausbreiten können (Abbildung 2.19). In beiden Fällen ist eine zuverlässige Bestimmung des
thermischen Ausdehnungskoeffizienten α nicht mehr möglich.
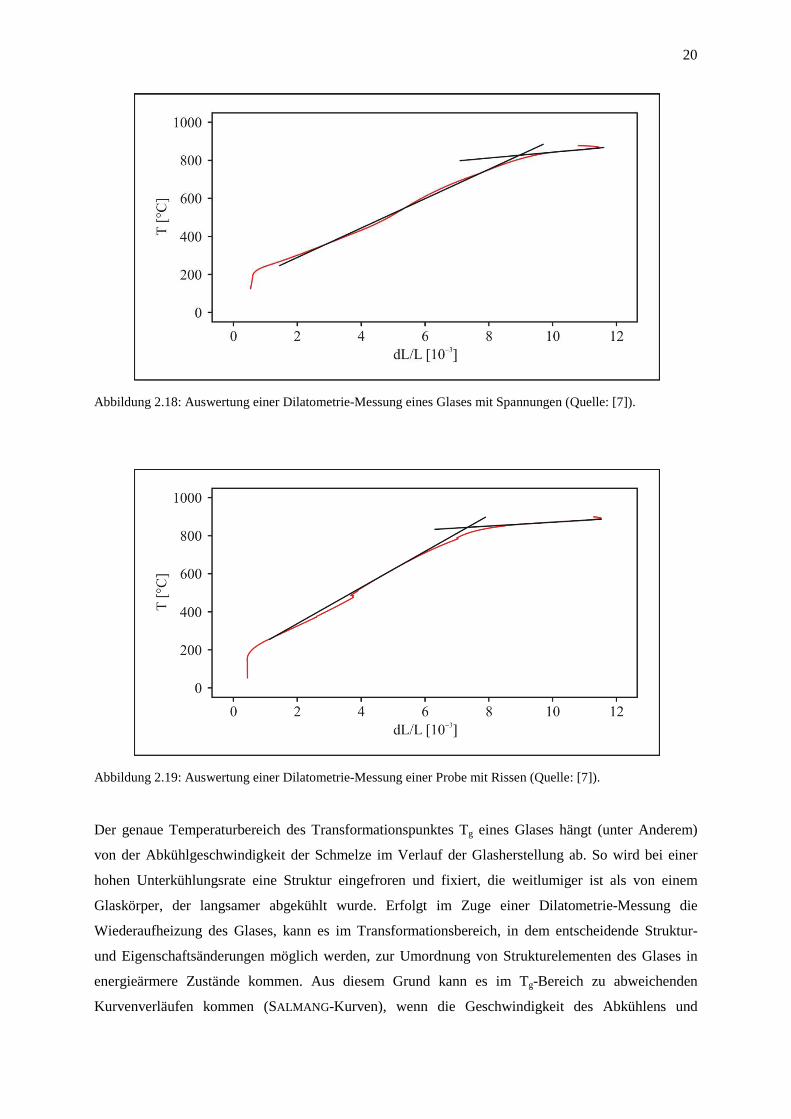
20
Abbildung 2.18: Auswertung einer Dilatometrie-Messung eines Glases mit Spannungen (Quelle: [7]).
Abbildung 2.19: Auswertung einer Dilatometrie-Messung einer Probe mit Rissen (Quelle: [7]).
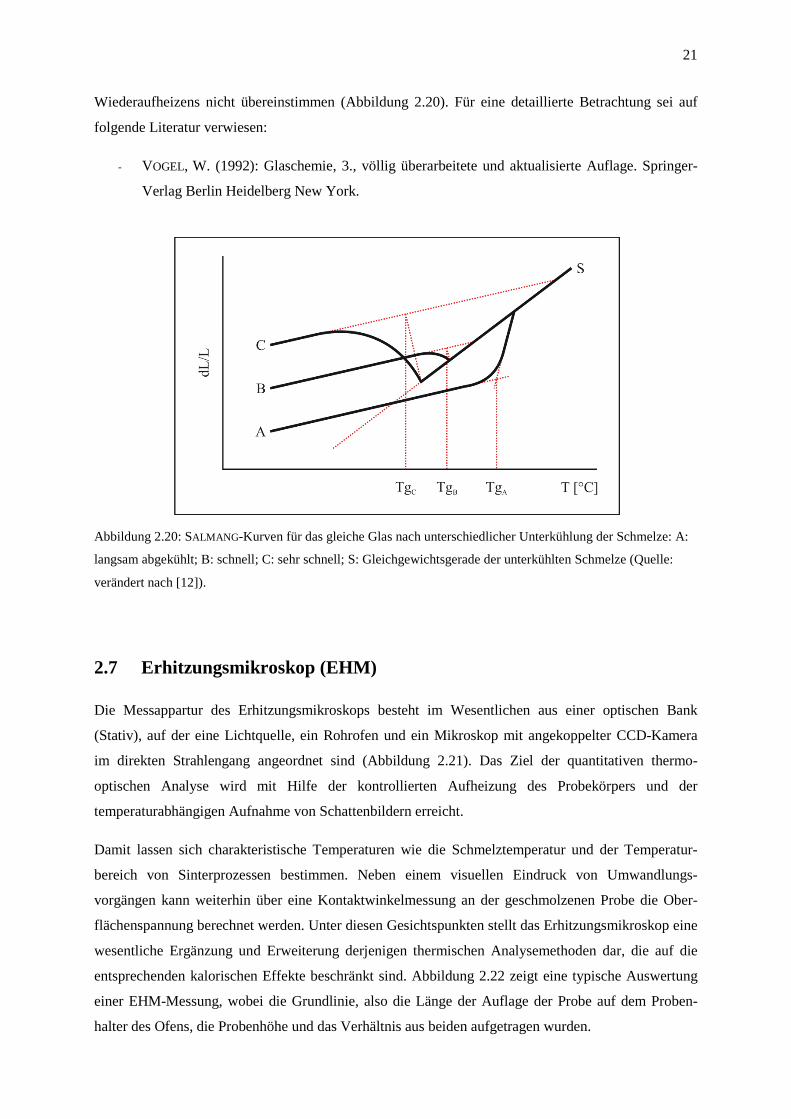
Der genaue Temperaturbereich des Transformationspunktes Tg eines Glases hängt (unter Anderem)
von der Abkühlgeschwindigkeit der Schmelze im Verlauf der Glasherstellung ab. So wird bei einer
hohen Unterkühlungsrate eine Struktur eingefroren und fixiert, die weitlumiger ist als von einem
Glaskörper, der langsamer abgekühlt wurde. Erfolgt im Zuge einer Dilatometrie-Messung die
Wiederaufheizung des Glases, kann es im Transformationsbereich, in dem entscheidende Struktur-
und Eigenschaftsänderungen möglich werden, zur Umordnung von Strukturelementen des Glases in
energieärmere Zustände kommen. Aus diesem Grund kann es im Tg-Bereich zu abweichenden
Kurvenverläufen kommen (SALMANG -Kurven), wenn die Geschwindigkeit des Abkühlens und
21
Wiederaufheizens nicht übereinstimmen (Abbildung 2.20). Für eine detaillierte Betrachtung sei auf
folgende Literatur verwiesen:
- VOGEL, W. (1992): Glaschemie, 3., völlig überarbeitete und aktualisierte Auflage. Springer-
Verlag Berlin Heidelberg New York.
Abbildung 2.20: SALMANG -Kurven für das gleiche Glas nach unterschiedlicher Unterkühlung der Schmelze: A:
langsam abgekühlt; B: schnell; C: sehr schnell; S: Gleichgewichtsgerade der unterkühlten Schmelze (Quelle:
verändert nach [12]).
2.7 Erhitzungsmikroskop (EHM)
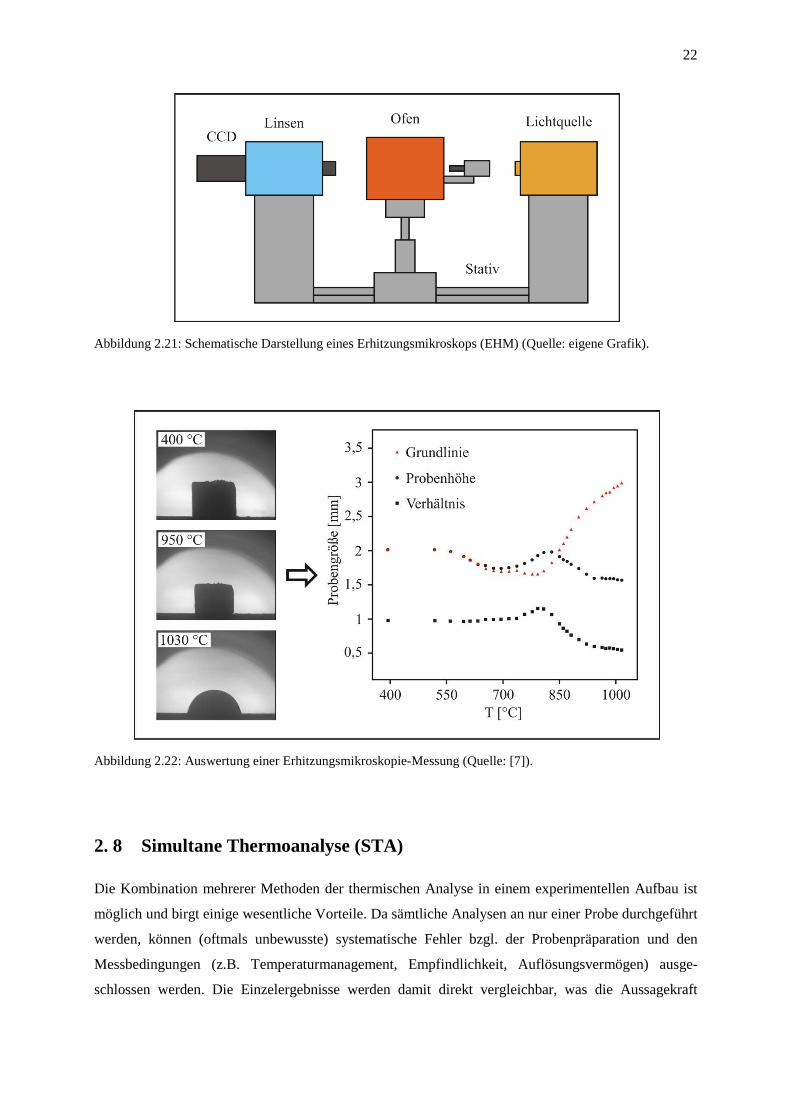
Die Messappartur des Erhitzungsmikroskops besteht im Wesentlichen aus einer optischen Bank
(Stativ), auf der eine Lichtquelle, ein Rohrofen und ein Mikroskop mit angekoppelter CCD-Kamera
im direkten Strahlengang angeordnet sind (Abbildung 2.21). Das Ziel der quantitativen thermo-
optischen Analyse wird mit Hilfe der kontrollierten Aufheizung des Probekörpers und der
temperaturabhängigen Aufnahme von Schattenbildern erreicht.
Damit lassen sich charakteristische Temperaturen wie die Schmelztemperatur und der Temperatur-
bereich von Sinterprozessen bestimmen. Neben einem visuellen Eindruck von Umwandlungs-
vorgängen kann weiterhin über eine Kontaktwinkelmessung an der geschmolzenen Probe die Ober-
flächenspannung berechnet werden. Unter diesen Gesichtspunkten stellt das Erhitzungsmikroskop eine
wesentliche Ergänzung und Erweiterung derjenigen thermischen Analysemethoden dar, die auf die
entsprechenden kalorischen Effekte beschränkt sind. Abbildung 2.22 zeigt eine typische Auswertung
einer EHM-Messung, wobei die Grundlinie, also die Länge der Auflage der Probe auf dem Proben-
halter des Ofens, die Probenhöhe und das Verhältnis aus beiden aufgetragen wurden.
22
Abbildung 2.21: Schematische Darstellung eines Erhitzungsmikroskops (EHM) (Quelle: eigene Grafik).
Abbildung 2.22: Auswertung einer Erhitzungsmikroskopie-Messung (Quelle: [7]).
2. 8 Simultane Thermoanalyse (STA)
Die Kombination mehrerer Methoden der thermischen Analyse in einem experimentellen Aufbau ist
möglich und birgt einige wesentliche Vorteile. Da sämtliche Analysen an nur einer Probe durchgeführt
werden, können (oftmals unbewusste) systematische Fehler bzgl. der Probenpräparation und den
Messbedingungen (z.B. Temperaturmanagement, Empfindlichkeit, Auflösungsvermögen) ausge-
schlossen werden. Die Einzelergebnisse werden damit direkt vergleichbar, was die Aussagekraft

23
einzelner thermischer Effekte deutlich erhöht. Zusätzlich kann Probenmaterial eingespart und der
zeitliche Aufwand der Messung entsprechend reduziert werden.
Das üblichste Prinzip der STA ist die Verknüpfung von Thermogravimetrie (TGA) und DSC (vgl.
Abschnitt 2.5), da für eine genaue Enthalpiebestimmung mittels DSC auch immer die zugehörige
Probenmasse erforderlich ist. Das Grundprinzip der TGA besteht in der kontinuierlichen temperatur-
abhängigen Bestimmung der Probenmasse, wobei die messtechnische Ausführung sehr komplex ist.
Zum Einen sind bei der Auswahl einer dem spezifischen Messproblem angepassten Präzisionswaage
(hauptsächlich Balken-, Torsions, oder Federwaagen) die Faktoren Wägebereich, Genauigkeit und
Empfindlichkeit zu berücksichtigen. Zum Anderen spielen weitere Gesichtspunkte wie durch die
Temperaturabhängigkeit der Gasdichte verursachte Auftriebseffekte und die Temperaturmessung am
beweglichen Probentiegel eine wichtige Rolle.
Abbildung 2.23 zeigt die schematische Darstellung einer STA-Anlage, die TGA und DSC kombiniert.
Die Spülung des Probenraumes mit Inertgas verhindert dabei die Korrosion von Tiegel- und
Waagematerial, wenn während der Temperaturbehandlung aggressive Gase freigesetzt werden. Neben
dem hier dargestellten Prinzip sind im Bereich der STA vor allem speziell entwickelte Laboraufbauten
verbreitet, die auf bestimmte Messprobleme zugeschnitten sind. Die STA am Otto-Schott-Institut stellt
z.B. eine Kombination aus DTA, Dilatometrie und der Messung der Ultraschallaufzeit an einer
kompakten Glasprobe dar. Zusätzlich kann über eine Punktmasse eine mechanische Anregung
(Zugkraft) des Prüfkörpers erfolgen, die eine Bestimmung von viskoelastischen Stoffkonstanten
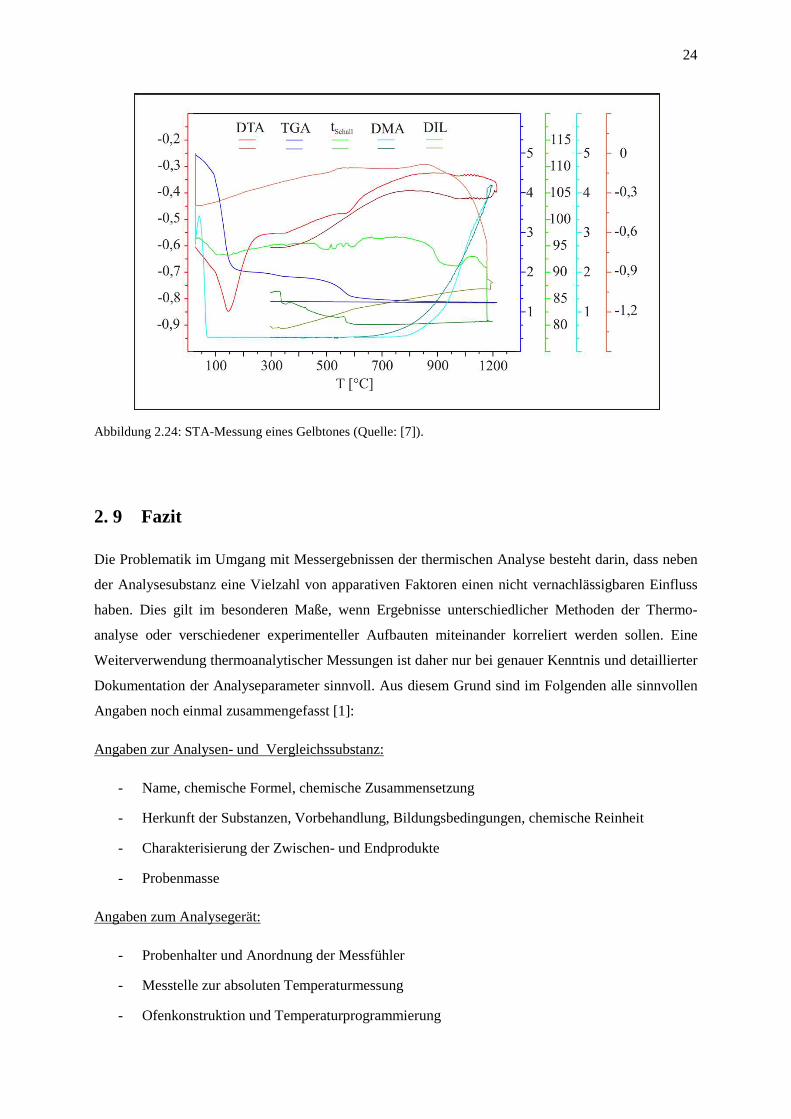
ermöglicht. Eine typische STA-Messung ist in Abbildung 2.24 dargestellt.
Abbildung 2.23: Schematische Darstellung einer STA (TGA + DSC) (Quelle: verändert nach [13]).
24
Abbildung 2.24: STA-Messung eines Gelbtones (Quelle: [7]).
2. 9 Fazit
Die Problematik im Umgang mit Messergebnissen der thermischen Analyse besteht darin, dass neben
der Analysesubstanz eine Vielzahl von apparativen Faktoren einen nicht vernachlässigbaren Einfluss
haben. Dies gilt im besonderen Maße, wenn Ergebnisse unterschiedlicher Methoden der Thermo-
analyse oder verschiedener experimenteller Aufbauten miteinander korreliert werden sollen. Eine
Weiterverwendung thermoanalytischer Messungen ist daher nur bei genauer Kenntnis und detaillierter
Dokumentation der Analyseparameter sinnvoll. Aus diesem Grund sind im Folgenden alle sinnvollen
Angaben noch einmal zusammengefasst [1]:
Angaben zur Analysen- und Vergleichssubstanz:
- Name, chemische Formel, chemische Zusammensetzung
- Herkunft der Substanzen, Vorbehandlung, Bildungsbedingungen, chemische Reinheit
- Charakterisierung der Zwischen- und Endprodukte
- Probenmasse
Angaben zum Analysegerät:
- Probenhalter und Anordnung der Messfühler
- Messtelle zur absoluten Temperaturmessung
- Ofenkonstruktion und Temperaturprogrammierung

25
- Art und Eichung der Thermoelemente
- Form und Material der Tiegel
- Empfindlichkeit des elektronischen ∆T-Verstärkers
Angaben zum Messvorgang:
- Aufheiz- oder Abkühlgeschwindigkeit
- Beschreibung der Gasatmosphäre (Druck, Zusammensetzung; statisch oder dynamisch)
- Packungsdichte
Angaben zur Auswertung der Messungen:
- Methoden zur Identifizierung eines thermischen Effektes
- Maßstab von Abszisse und Ordinate
- Nullinie des Analysengerätes
- Registriermethode (Originaldiagramm)
3 Durchführung des Praktikums
Zum Praktikumstermin werden die im Otto-Schott-Institut vorhanden Geräte zur thermischen Analyse
vorgestellt und Besonderheiten der experimentellen Aufbauten erläutert. Anschließend bekommt jede
Praktikumsgruppe Messkurven, die mit den vorhandenen Geräten aufgenommen wurden. Diese sind
mit Hilfe des erläuterten Tangentenverfahrens auszuwerten. Neben der Bestimmung charakteristischer
Temperaturen ist der Kurvenverlauf hinsichtlich ablaufender Reaktionen zu interpretieren. Die Diskus-
sion sollte weiterhin mögliche Abweichungen vom theoretischen Kurvenverlauf und anderweitige
gerätespezifische Fehlereinflüsse beinhalten. Das Protokoll ist auf die Auswertung und Diskussion zu
beschränken.
Das Praktikum zur thermischen Analyse endet mit einem Abtestat. Neben einer Diskussion über das
vorgelegte Protokoll sollen messmethodische Grundlagen und zu berücksichtigende Faktoren bei der
Kurvenauswertung erörtert werden. Eine Terminvereinbarung findet nach Protokollabgabe statt.

26
4 Quellen
[1] HEIDE, K. (1979): Dynamische thermische Analysemethoden, 1. Auflage. VEB Deutscher
Verlag für Grundstoffindustrie Leipzig.
[2] DIN EN 51005-08 (2005): Thermische Analyse (TA) - Begriffe. Beuth Verlag GmbH, Berlin.
[3] RÜSSEL, C. (2009): Skript zur Vorlesung Glas - Grundlagen.
[4] MÜNSTER, A. (1956): Statistische Thermodynamik, 1. Auflage. Springer-Verlag Berlin,
Heidelberg, New York.
[5] JÄNSCH, D. (2009): Thermoelektrik - Eine Chance für die Automobilindustrie, 1. Auflage.
expert-Verlag, Renningen.
[6] NAGEL, M. (2011): Phasen- und Texturanalyse gerichteter durch elektrochemisch induzierte
Keimbildung hergestellter Fresnoit-Glaskeramiken. Dissertation, Friedrich-Schiller-Universi-
tät, Jena.
[7] ohne Autor (2010): Messungen am Otto-Schott-Institut, Friedrich-Schiller-Universi-tät, Jena.
[8] HÖHNE, G., HEMMINGER, W., FLAMMERSHEIM, H.-J. (1996): Differential Scanning
Calorimetry - An Introduction for Practitioners, 1. Auflage. Springer-Verlag Berlin, Heidel-
berg, New York, Barcelona, Budapest, Hong Kong, London, Mailand, Paris, Santa Clara,
Singapur, Tokyo.
[9] KLEBER, W., BAUTSCH, H.J., BOHM, J. (1998): Einführung in die Kristallographie, 18., stark
bearbeitete Auflage. Verlag Technik GmbH, Berlin.
[10] ohne Autor (2011): Produktprospekt Dilatometer DIL 402 PC. Netzsch-Gerätebau GmbH,
Selb.
[11] http://de.wikipedia.org/wiki/Differentialtransformator; aufgerufen am 18.11.2011, Stand:
10.11.2011.
[12] VOGEL, W. (1992): Glaschemie, 3., völlig überarbeitete und aktualisierte Auflage. Springer-
Verlag Berlin Heidelberg New York.
[13] ohne Autor (2011): Produktprospekt STA 409. Netzsch-Gerätebau GmbH, Selb.





























