Embed Size (px)
Citation preview

64 W. WALTER und K.-D. BODE Bd. 681
u ber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 1)
OXYDATIONSREAKTIONEN AN THIONURETHANEN U N D
DITH IOURETHAN EN
von WOLFGANG WALTER und KLAUS-DIETER BODE
Aus dem Chemischen Staatsinstitut, lnstitut fur Organische Chemie der Universitat Hamburg Eingegangen am 21. April I964
Unsubstituierte und rnonosubstituierte Thionurethane und Dithiourethane bilden mit Wasserstoffperoxid Thionurethan- bzw. Dithiourethan-S-oxide:
Durch Substituenten R mit gronern Raumbedarf wird die Stabilitat der S-Oxide so weit erhoht, daR die Isolierung gelingt. Fehlen derartige Substituenten, so erhalt man bei der Oxydation der Thionurethane in vielen Fallen Formimido- esterdisulfide: RlN=C(OR) -S-S -C(OR)=NRI. Bei der Oxydation disub- stituierter Thionurethane und Dithiourethane lieBen sich keine S-Oxide
nachweisen.
RX-C(=SO)NHRl (X = 0, S ) .
Im Gegensatz zum S-Oxid des Rhodanins 1) waren S-Oxide offenkettiger Thioure- thane bisher nicht bekannt.
Thiourethane sind wegen ihrer biologischen Wirkungen in den letzten Jahren ausfuhrlich untersucht worden2-6), wobei ihre Oxydationsempfindlichkeit aufgefallen ist, die auch tech- nisch genutzt wird 7). DEBUS erhielt 1852 aus 0-Athyl-thionurethan mit Kupfer(I1)-chlorid oder salpetriger Saure neben anderen Produkten ein substituiertes 1.3.5-Thiadiazols). Auch HOLMBERG und ROsfN9), die 0-L-Menthyl-thionurethan mit H202 oxydierten, erhielten neben Schwefel und Schwefelsaure das entsprechende 1.3.5-Thiadiazol, was spater bestatigt wurdelo). BILLETER 1 1 ) beobachtete bei der Darstellung des N.N-Dimethyl-thionurethans eine Autoxyda- tion desselben, wobei er als Zwischenprodukt ein nicht faBbares ,,Superoxyd" annahm, das in das entschwefelte Urethan und ,,Schwefelmonoxyd" ubergehen sollte.
1) W. WALTER und G. RANDAU, Liebigs Ann. Chem. 681, 55 (1965). voranstehend. 2) N. KREUTZKAMP, Dtsch. Apotheker-Ztg. 102, 1286 (1962). 3 ) G. A. CARTER, J . L. GARRAWAY, D. M. SPENCER und R. L. WAIN, Ann. appl. Biol. 51,
4) 0 . - E . SCHULTZ und K. GLEIXNER, Arch. Pharmaz. 295, 879 (1962). 5 ) A. RIECHE, D. MARTIN und W. SCHADE, Arch. Pharmaz. 296, 770 (1963). 6 ) K.-H. RISSE, U. HORLEIN, W. WIRTH und R. GOSSWALD, Medizin und Chemie, Bd. VII,
7) R. C. MORRIS und W. J. SULLIVAN, Amer. Pat. 3081 335 v. 19.9. 1960, Shell Oil Comp.
8) H. DEBUS, Liebigs Ann. Chem. 82, 253 (1852). 9) B. HOLMBERG und W. ROS~N, Ber. dtsch. chem. Ges. 58, 1834 (1925).
10) J. V. DUSK? und J. TRTILEK, Chem. Obzor 8, 1 (1933) [C. 1933 I, 20891. 1 1 ) 0. BILLETER, Ber. dtsch. chern. Ges. 43, 1835 (1910).
135 (1963).
S. 171, Verlag Chemie GmbH, Weinheim/Bergstr. 1963.
[C. A. 59, 9886 (1963)].

1965 tjfber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 65
Zur Darstellung der fur eine systematische Untersuchung des Oxydationsverhaltens
A. Umserzung von Alkoholen mit Senfolenl2) :
notwendigen Thionurethane bedienten wir uns der folgenden Verfahren.
ROH + R’N=C=S + RO-CS-NHR’ Diese Reaktion wird durch Triathylamin katalysiert 13.14).
B. Acylierung von Aminen mit Chlorrhionameisensaureestern 15) :
RONa + CI-CS-CI -- + RO-CS-CI + NaCl, RO-CS-Cl + 2RNHz ---A RO-CS-NHR + RNHz.HCI
C. Thiolyse von Cyansuureestern: Die Darstellung einiger Vertreter der bis dahin unbe- kannten Cyansaureester gelang STROH und GERBER 16). Die Umsetzung des 12.6-Di-tert.- butyl-4-allyl-phenyl]-cyansaureesters mit Schwefelwasserstoff in Gegenwart von Triathyl- arnin ergab in guter Ausbeute das entsprechende Thionurethan :
RO-C-N + H2S ~ + RO-CS-NH2 Diese Synthese entspricht der Thiolyse.) von Nitrilen zu Thiocarbonsaureamiden 17).
RO-CS-SK + ClCH2--COzNa -- - RO --CS-S-CHz -COZNa, D. Acylierung von Aminen mil 0-Alkyl-S-carboxymethyl-xanthaten 18) :
RO-CS-S-CH2-CO2Na + R’NH2 -- + RO-CS-NHR’ + HSCH2-COzNa E. Umsetzung von Xanthogensaureesrern mit Glycin 19) :
RO-CS-SR’ + HzN-CHz-COzH -+ RO-CS-NH-CHZ-CO~H + R‘-SH F. Umsetzung von Ammoniumrhodanid mir Chlorameisensaureathylester 20) :
NH4S-C - N + CIC02CrH5 - C2H50 -CO -NH --CS-OC~HS G . Acylierung von Phenolen mit Thiocarbamidsaurechloriden 15.21) :
ArONa + CI-CS-NR’R” - ArO-CS-NR’R” -1 NaCI
* ) Annierkung bei der Korrektur (30. 1 1 . 1964): lnzwischen sind uns zwei weitere Arbeiten uber die Thiolyse von Cyansaureestern zuganglich geworden. K. A. JENSEN und A. HOLM, Acta chern. scand. 18, 826 (1964), erhielten das 0-Athyl-thionurethan aus Athylcyanat durch Thiolyse. E.GRIGAT und R. PUTTER, Chem. Ber. 97, 3022 (1964), stellten zahlreiche Thionurethane auf diesem Wege her.
12) A. W. HOFMANN, Ber. dtsch. chem. Ges. 2, 452 (1869); E. BAMBERGER, Ber. dtsch. chem. Ges. 15, 2164 (1882); A. E. DIXON und R. E. DORAN, J. chem. SOC. [London] 67, 565 ( 1 895).
13) J. F. HARRIS, J. Amer. chem. SOC. 82, 155 (1960). 14) C. N. R. RAO und R. VENKATARAGHAVAN, Tetrahedron [London] 18,531 (1962). 15a)H. RIVIER, Bull. SOC. chim. France [3] 35, 837 (1906). - 15b) D. L. GARMAISE, A. UCHI-
16) R. STROH und H. GERBER, Angew. Chem. 72, lo00 (1960); vgl. auch D. MARTIN, Angew.
171 C. VOLCKEL, Liebigs Ann. Chem. 38, 314 (1841); A. WEDDIGE, J. prakt. Chem. [2] 7, 79
18) E. BIILMANN, Liebigs Ann. Chem. 339, 351 (1905), und zwar S. 355; B. HOLMBERG, J.
19) E. B. KNOTT, Amer. Pat. 2691581 v. 12. 10. 1954, Eastman Kodak Co. [C. A. 49, 83
2 0 ) G. DELITSCH, J. prakt. Chem. [2] 10, 116 (1874). 21) M. DEL~PINE, Bull. SOC. chim. France 141 7, 404 (1910).
YAMA und A. F. MCKAY, J. org. Chemistry 27, 4509 (1962).
Chem. 76, 303 (1964); Angew. Chem. internal. Edit. 3, 311 (1964).
(1873).
prakt. Chem. [2] 71, 264 (1905).
(1955)l.
Liebigs Ann. Chem. Bd. 681 5

66 W. WALTER und K.-D. BODE Bd. 681
Thionurethane * ) der allgemeinen Formel RR'N-CS-XR (X = 0, S) sollten analog den Thiocarbonsaureamiden und Thioharnstoffen mit Oxydationsmitteln, wie Wasserstoffperoxid oder Percarbonaten bzw. Perboraten 22), S-Oxide ergeben.
Bei den nach A leicht zuganglichen monosubstituierten Thionurethanen, vgl. Tabelle 1, fiihrte die Oxydation bei Raumtemperatur oder wenig erhohter Temperatur zu Thionurethan-S-oxiden, die mit Eisen(II1)-chlorid nachgewiesen wurden. Die Oxydation verlief langsamer als bei Thioamiden, und mit Eisen(Il1)-chlorid traten stets blauviolette Farbungen auf, wahrend bei den Thioamiden eine vie1 reichhaltigere Farbskala zu beobachten war **). Die Reaktion mit Fe(I1I)-Ionen erwies sich als so empfindlich, da13 bereits Spuren von Eisen(IlI)-salzen im wasserfreien Natriumsulfat, mit dem Losungen der Thionurethan-S-oxide getrocknet wurden, eine leichte Blau- farbung hervorriefen.
Bei der Isolierung von Thionurethan-S-oxiden traten haufig Formimidoesterdisulfide auf 24) :
2 CeHs-NH-$-OR + CeHs-N=C-OR RO-C=N-C~HB 0s s 4
Die Bildung von Disulfiden durch Oxydation ist sowohl bei Thioharnstoffen25) als auch bei Thiocarbonsaureamiden bekannt 26). Wahrend die Formamidindisulfide nur als Salze dargestellt werden konnten, kennt man von den Disulfiden der Thioamide die freien Basen.
Zur Charakterisierung der Formimidoesterdisulfde genugten weder Elementar- analysen noch Molekulargewichtsbestimmungen, da sich die Disulfide von den Aus- gangsverbindungen (2 Mol) nur urn zwei H-Atome unterscheiden und die Thion- urethane zur Bildung von Assoziaten neigen 27). Zuverlassiger sind die IR-Spektren, in denen die N =C-Bande bei 6-6.2 p deutlich hervortritt, sowie die Cyanidspaltung, bei der das Thionurethan chromatographisch nachgewiesen und durch Oxydation in das ebenfalls chromatographisch nachweisbare S-Oxid iibergefiihrt wurde. Durch Schmelzpunkte und chromatographisches Verhalten konnten die Formimidoesterdi- sulfide eindeutig von den Thionurethanen unterschieden werden.
* ) Zur Nomenklatur vgl. Lit. 23). **)Die fur Thiourethan-S-oxide charakteristische Farbung gibt auch das in der VIII. Mitt.
22)Belg. Pat. 618276 v. 29. I I . 1962, RhBne-Poulenc S. A. [C. A. 59, 5142 (1963)l; eigene
23)W. HAAS und K. I R G O L I ~ : , Z. analyt. Chem. 193, 248 (1963). 24) W. WALTER und K.-D. BODE, Angew. Chem. 75, 208 (1963); Angew. Chem. internat. Edit.
2, I54 ( 1 963). 25) F. KURZER und PH. M. SANDERSON, J. chem. SOC. [London] 1957, 4461 ; H. BEYER und
R. GIEBELMANN, Z. Chem. 2, 84 (1962). 26)H. WUYTS und A. LACOURT, Bull. SOC. chim. Belgique 42, 1 (1933) [C. 1933 I, 30761;
F. HODOSAN, Bull. SOC. chim. France 1957, 633; M. A. MCCALL, J. org. Chemistry 27, 2433 ( I 962).
beschriebene Rhodanin-S-oxid-Anion.
unveroffentlichte Untersuchungen.
27)A. A. BURROWS und L. HUNTER, J. chem. SOC. [London] 1952, 4118.

1965 uber die Oxydationsprodukte von Thiocarbonsaureamiden, 1X 67
Die chromatographische Verfolgung der Oxydationen zeigte, daR die Thionurethan- S-oxide immer nur in geringen Konzentrationen neben Sulfat, Schwefel, Schwefel- wasserstoff, Disulfid und Ausgangsprodukt auftraten.
Nachdem aromatisch substituierte Thiobenzamide S-Oxide hervorstechender Stabilitat ergeben hatten 2*), wurde die ,,Sauerstoff-Seite" des Thionurethans ebenso wie die ,,Stickstoff-Seite" aromatisch substituiert (vgl. Tab. 1). D a beiderseits aroma- tisch substituierte Thionurethane nur schlecht nach A zu erhalten waren29), wurde in diesem Fall die Methode B vorgezogen.
Die aromatisch substituierten Thionurethane zerfallen bei Raumtemperatur langsam in Senfole und Phenole und sind nur im Kiihlschrank langere Zeit haltbar. Die Oxyda- tion zeigte, daR das gewiinschte Thionurethan-S-oxid gebildet wurde, a u k r d e m traten jedoch zahlreiche Nebenprodukte auf. Als Zersetzungsprodukte konnten in alkohol- freien Losungen Senfole chromatographisch nachgewiesen werden.
Die Erfahrungen aus der Thioamidreihe iiber die Stabilitat der S-Oxide lassen sich also nicht auf die Thionurethane ubertragen. Wir griffen daher auf Beobachtungen an Thioharnstoffen zuriick. Dort hatte sich am Beispiel des N.N-Di-tert.-butyl-thioharn- stoffs gezeigt30), daB durch sterische Effekte die Stabilitat der Monooxydationspro- dukte so weit erhoht werden kann, da8 ein S-Oxid isolierbar wird. Daher wurde das N-tert.-Butyl-0-tert.-butyl-thionurethan dargestellt ; die Oxydation fiihrte jedoch auch bei dieser Substanz zu einer groRen Zahl von Oxydationsprodukten *).
Am Beispiel des nach A dargestellten N-Phenyl-O-[2.6-di-tert.-butyl-phenyl]-thion- urethans sollte der gleichzeitige Einfrup sterischer Effekte und aromatischer Substituenten gepriift werden. Das in wa8rigen Alkalien32) unlosliche 2.6-Di-tert.-butyl-phenol wurde in Gegenwart von Natronlauge mit Phenylsenfol umgesetzt, wobei in geringer Ausbeute ein Produkt anfiel, dessen Elementarzusammensetzung dem gewiinschten Thionurethan entsprach und in recht guter Ausbeute ein S-Oxid ergab. Das IR- Spektrum der Substanz zeigte jedoch eine OH-Bande, so daR man in Analogie zu anderen Reaktionen mit sterisch gehinderten Phenolen eine Addition des Senfoles in
*) Die leichte Oxydierbarkeit von Carbathoxythioformarniden31) fiihrte zum Versuch, N-Carbathoxy-0-athyl-thionurethan 20) zu oxydieren. Hierbei verlief die Reaktion jedoch zu leicht in Richtung der Entschwefelung, wobei das S-Oxid ebenfalls nur nachgewiesen und nicht isoliert werden konnte. Ein Lhnliches Ergebnis zeigte das N-Carboxymethyl- 0-tetradecyl-thionurethan 19), das auf Grund seines giinstigen Loslichkeitsverhaltens erfolg- versprechend schien.
28) W. WALTER und J. CURTS, Chem. Ber. 93, 151 1 (1960). 2 9 ) W. SCHNEIDER und F. WREDE, Ber. dtsch. chem. Ges. 47, 2038 (1914). 30) W. WALTER, Habilitationsschrift Univ. Hamburg 1958. 31) W. WALTER und K.-D. BODE, Liebigs Ann. Chern. 660, 74 (1962). 32) R. STROH J. EBERSBERGER, H. HABERLAND und W. HAHN, Angew. Chem. 69, 124 (1957);
R. STROH, J. EBERSBERGER, H. HABERLAND und W. HAHN, in W. FOERST, Neuere Merhodzn der praparariven orgrinischen Chemie, Bd. 2, S. 155 , Verlag Chemie GrnbH, Weinheim/ Bergstr. 1960.
5.
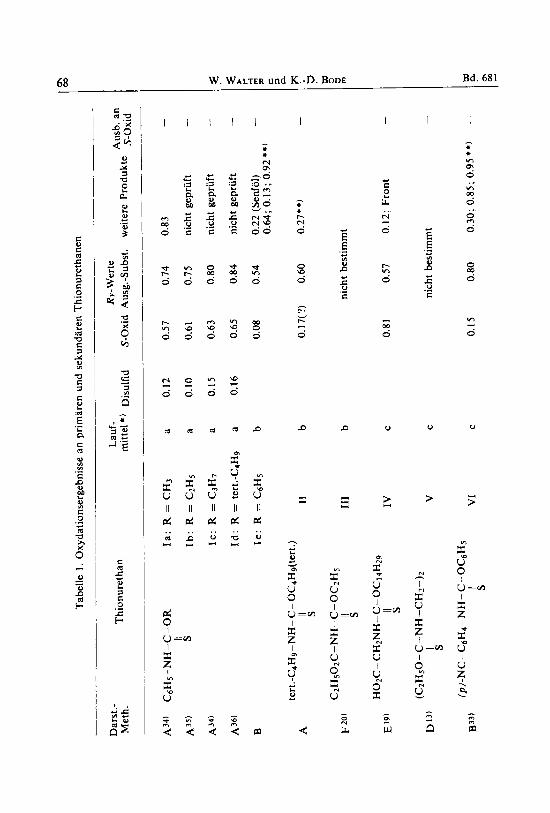
1965 uber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 69
I
In
0
N
01 . ”
2
x 0
0 0
U
B u II d d I - >
u u =m
z
9 I
I
0 x
2 0
00
0 09
U
B u II d d - - - >
v)
F 9 u
a
X
m m m m
Tab
elle
1 (
Fort
serz
ung)
-
- .-
-
Dar
st.-
Met
h.
Thi
onur
etha
n La
uf-
mit
tel*
)
h
C~
HS
O-C
-NH
~
B IS
) II S
XI1
XII
I: R =
H
R=
CH
3
RF-
Wer
te
Aus
b. a
n S-
Oxi
d A
usg-
Subs
t.
wei
tere
Pro
dukt
e S-
Oxi
d
0.56
0.
88
0.57
0.
65
0.38
; 0.
40;
-
0.42
; 0
.69
**)
0.78
0.
82
0.10
; 0.3
6 -
0.40
: 0.
42
B XIV: R
= R’= C
H3
I 0.
60
0.71
*’D
iinns
chic
htch
rom
atog
raph
ie a
n K
iese
lgel
G: L
aufm
ittel
: a
= B
enzo
llAth
anol
(3:
I),
b =
Ben
rol/C
hlor
ofor
m (
I: I)
. c
= C
hlor
ofor
mIM
etha
nol
(2: I
). C
hlor
ofor
m.
e =
Ben
zolI
Petr
olat
her
(3: I
). f
= B
enzo
lIM
ethy
lenc
hlor
id (
3: I)
, g
= B
enzo
IlC
hlor
ofor
m (
I : 3
). h
= B
enzo
lIM
etha
nol (
3: 1)
. i
= B
enzo
ll d M
etha
nol (
I : 1
). **
I Fl
eck
am S
tart
punk
t.
33)
R. P
. MU
LL
, J. A
mer
. che
m. S
OC
. 77,
581
(195
5).
34)
W. R
. OR
ND
OR
FF
und
F. A
. RIC
HM
ON
D,
Am
er. c
hem
. J. 2
2, 4
58 (
1899
). 35
) H
. L. W
HE
EL
ER
un
d B
. BA
RN
ES,
Am
er. c
hem
. J. 2
4. 6
0 (1
900)
. 36
) R
. W.
BOST
und
E. R
. AN
DR
EW
S,
J. A
mer
. che
m. S
OC
. 65, 9
00 (
1943
).

1965 uber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 71
p-Stellung annehrnen kann. Die haufig beobachtete Wanderung der Alkylgruppen32) kann hier ausgeschlossen werden, so daR wahrscheinlich folgende Reaktion abgelaufen ist :
Fur das Vorliegen eines Thioarnids sprachen bereits die gelbe Farbe der Substanz (Thionurethane ahnlicher Konstitution sind farblos) und die Farbung des S-Oxids rnit Eisen(II1)-chlorid, die nicht blau, wie bei allen Thionurethanen, sondern griin ausfiel.
Zur Prufung der Arbeitshypothese, daR die Stabilitat der Thionurethan-S-oxide durch sterische Einfliisse und gleichzeitige arornatische Substitution erhoht werden kann, wurden die in Tabelle 1 aufgefuhrten Thionurethane VIIa -VIIc dargestellt und der Oxydation unterworfen. Die Reaktionen verliefen ubersichtlicher, die Iso- lierung eines S-Oxids war jedoch auch hier nicht rnoglich. Der nachste Schritt fiihrte zu VIIIa und VIIIb, die bei der Oxydation recht einheitlich S-Oxide bildeten (Diinn- schichtchromatograrnme), bei Urnkristallisationsversuchen - selbst unter schonend- sten Bedingungen - jedoch stets die Disulfide ergaben.
Durch ortho-Substitution des Phenjdrestes an der JauerstofflSrite'' der Thion- urethane schien eine weitere Moglichkeit zur sterischen Beeinflussung der Thiocarbonyl- Gruppe gegeben. Die Oxydation von I X a ergab in 54-proz. Ausbeute eine Substanz, die alle Eigenschaften eines S-Oxids zeigt und auch im 1R-Spektrurn die bei allen Thioamid-S-oxiden beobachtete zusatzliche Bande irn Bereich von 9- 10 p enthalt. Als Nebenprodukt wurde auch hier das entsprechende Disulfid erhalten.
Wenn die Hypothese von der Erhohung der Stabilitat des Thionurethan-S-oxides durch sterische Hinderung richtig war, sollten sterisch wirksarne Methylgruppen am Phenylrest der ,,Stickstoff-Seite" die Stabilitat des S-Oxids weiter erhohen. Die Oxydation von IX b ergab ein Rohprodukt, das nach Elernentaranalyse und chrornato- graphischern Verhalten als S-Oxid anzusehen ist. Dieses Rohprodukt wandelte sich jedoch sehr leicht in das Disulfid urn, so daR statt einer Erhohung der Stabilitat eine Stabilitatsverrninderung eintrat.
Nach den bisher beschriebenen Ergebnissen war in der Thionurethanreihe ein stabiles S-Oxid zu erwarten, wenn der Sauerstoff'einen Substituenten rnit groJem Raum- bedarf tragt urid der Stickstoff hochstens monosubstituiert ist. Zur Prufung dieser Aus- sage wurden die Thionurethane XI1 -XIV oxydiert. Obgleich bisher hier kein stabiles S-Oxid zu erhalten war, zeigen die Diinnschichtchrornatograrnme (Abb. I ) deutlich den stabilisierenden EinfluR der Disubstitution an der ,,Sauerstoff-Seite" der Thion- urethane:

72 W. WALTER und K.-D. BODE Bd. 681
- - - S-Oxid
*--.-.-.
0-Phenyl- 0-[2-Methyl-phenyl]- 0-[2.6-Dimethyl-phenyI]- thionurethan (XII) thionurethan (XIII) thionurethan (XIV)
Abbildung I . Diinnschichtchrornatographische Verfolgung der Oxydation
Irn Falle der Verbindung XI reichte die Stabilisierung durch sterische Einflusse aus, urn das S-Oxid in 87-proz. Ausbeute zu isolieren:
Die fur Thioarnid-S-oxide charakteristische Eigenschaft, rnit Schwefelwasserstoff die Struktur -C(==S)NHR- zuriickzuliefern, findet sich auch in der Klasse der Thionurethan-S-oxide.
N.N-disubstituierte Thionurethane, die nach D oder G dargestellt wurden, bildeten keine S-Oxide. Gelegentlich farbten sich rnit FeC13 versetzte Oxydationsansatze schwach rot. Irn Dunnschichtchrornatograrnrn wurden jedoch entsprechende Jod/ Azid-positive Substanzen nicht erhalten. Bei Iangerer Reaktionsdauer trat oxydative Entschwefelung ein.
Als Spezialfall eines disubstituierten Thionurethans wurde XV oxydiert, das wie folgt erhalten wurde37):
Auch hier konnte rnit FeC13 kein Oxydationsprodukt nachgewiesen werden. Unsubstituierte und rnonosubstituierte Dithiourethane sind zersetzlich und zer-
fallen leicht in Mercaptan und Senfol. Daher haben wir in dieser Reihe nur wenige Versuche unternornrnen. Das N-Phenyl-S-phenyl-dithiourethan38) lieferte ein instabiles S-Oxid.
37) 0. BILLETER, Ber. dtsch. chern. Ges. 20, 1629 (1887). 38) H. RIVIER, Bull. SOC. chirn. France [4] 1, 733 (1907).

1965 uber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 73
Unter konsequenter Verfolgung der bei den Thionurethanen gewonnenen Vor- stellung, wie die Stabilitat der S-Oxide durch sterische Einflusse erhoht werden kann, oxydierten wir das S-terr.-Butyl-dithiourethan (XIX) und isolierten in 40-proz. Aus- beute das Dithiourethan-S-oxid:
( 7 3 a CHS-C-S-C-NHz I I1
CH3 S CH, SO
Dieses S-Oxid gibt wie alle anderen Thiourethan-S-oxide mit FeC13 in Methanol eine Blaufarbung, in Benzol dagegen ist die Farbung rotbraun. Die Verringerung der sterischen Beeinflussung fiihrte beirn S-Isopropyl-dithiourethan noch in 23-proz. Aus- beute zurn S-Oxid, wahrend es beim S-Athyl-dithiourethan nicht mehr gelang, ein stabiles Oxydationsprodukt zu fassen.
Disubstituierte Dithiourethane wurden aus NNdisubstituierten Dithiocarbarnaten mit Alkylhalogeniden erhalten39) :
Sie ergaben - entsprechend dem Verhalten der N.N-disubstituierten Thionurethane - keine S-Oxide, es trat vielrnehr Zersetzung ein, wobei Mercaptane, Sulfat und Schwefel gebildet wurden.
Abschlieknd wurde das Verhalten eines gemischteti Dithiourethan-thioamids gepruft, das auf folgendern Wege dargestellt wurde40) :
Das Dithiourethan-thioamid XVI lieferte ein Mono-S-oxid. Es konnte die Thio- carbonylgruppe im ,,DithiourethanteiI" oder irn ,,Thioarnidteil" des Molekuls oxydiert worden sein. Das S-Oxid gab rnit FeC13 eine rotviolette Farbung, die nach Verdunnen rnit Methanol in Blau umschlug. Die Entscheidung zugunsten der Oxydation irn ,,Thioamidteil" konnte dadurch getroffen werden, daB bei der therrnischen Zersetzung des S-Oxids das Dithiourethan XVII entstand, das mit der aus N.N-Dirnethyl-dithio- carbamat und Chloracetamid erhaltenen Substanz41) identisch war. Die Reduktion mit Schwefelwasserstoff fuhrte zu XVI zuruck.
39) M . KULKA, Canad. J. Chem. 34, 1093 (1956) [C. A. 51, 3481 (1957)]. 40) H. G. SCHICKE und G. SCHRADER, Dtsch. Bundes-Pat. I129949 v. 24. 5. 1962, Farben-
41) Amer. Pat. 2247917 (1934), Wingfoot Corp., Wilmington [C. 1942 11, 10641. fabriken Bayer AG [C. A. 57, I 3 626 (1962)l.

74 W. WALTER und K.-D. BODE Bd. 681
XVIX
Bei dieser konkurrierenden Oxydation erweist sich also der Schwefel der Thioamid- gruppe als der reaktivere.
DISKUSSION
Die Stabilitat der Thionurethan- und Dithiourethan-S-oxide konnten wir durch sperrige Substituenten am Sauerstoff bzw. Schwefel erhohen. Zur Deutung erscheint
10 uns folgende Vorstellung geeignet. Nach KING und Du~s1.42) ist die Gruppe >C=S gewinkelt, so daB cis- und trans-Lagen des Sauerstoffs bezuglich der an Kohlenstoff gebundenen Gruppen moglich sind. Fur das Thiobenzanilid-S-oxid schlieBen KING und DURST auf Grund von IR-Messungen auf eine Wasserstoffbriicke, welche die Struktur XX stabilisiert43). Unsere Befunde lassen sich erklaren, wenn man diese Arischauung dahingehend weiterentwickelt, daB im Falle der Thionurethane zur Ausbildung einer solchen Beziehung auf das C = SO-System ein einseitiger ,,Druck" ausgeubt werden mu& der die beteiligten Atorne einander nahert *) (XXI).
Die Tatsache, dab NNdisubstituierte Thionurethane keine S-Oxide ergeben, spricht ebenfalls fur diese Vorstellung, da sich in diesem Falle keine Wasserstoffbrucke ausbilden kann. Zum Verhalten der tertiaren Thionurethane steht die Fahigkeit der tertiaren Thioamide, S-Oxide zu bilden40), in auffallendem Gegensatz, der ein Aus- druck fur die Empfindlichkeit der Thioamidgruppe gegenuber elektronischen Ein- fliissen ist. Das Ausbleiben der S-Oxidbildung bei den N,N-disubstituierten Thion- urethanen konnte auch noch darauf beruhen, daB sich diese Verbindungen in die entsprechenden Thiolurethane umgelagert hattens).
* ) Anmerkung bei der Korrekrur (10. 10. 1964): Durch IR-spektroskopische Untersuchungen konnten wir inzwischen das Vorliegen einer Wasserstoffbriicke in XX nachweisen, wo- riiber an anderer Steile ausfiihrlich berichtet wird.
42) J . F. KING und T. DURST, J . Amer. chem. SOC. 85, 2676 (1963). 43) J . F. KING und T. DURST, Tetrahedron Letters [London] 1963, 585.

1965 ube r die Oxydationsprodukte von Thiocarbonsaureamiden, IX 75
RZN-$-O-CHz-CH2-NRz + RzN- -S-CHz-CHZ-NRa S 8
Es ist aber nicht wahrscheinlich, daD diese Umlagerung bereits wahrend der Oxydation eintritt, da hier wesentlich mildere Reaktionsbedingungen angewendet werden. Auch die Beobachtung, da8 I1 kein stabiles S-Oxid gibt, paBt zwanglos zu dieser Hypothese; denn offenbar verhindert die raumbeanspruchende N-tert.- Butylgruppe die Ausbildung der Wasserstoff briicke. In der gleichen Weise IaBt sich die geringere Stabilitat des S-Oxids von I X b erklaren. Diese Ansicht wird weiter gestiitzt durch die glatte Oxydation von XIX zum S-Oxid.
Die Unfahigkeit der tertiaren Thiourethane, Sauerstoff anzulagern, ist so ausgepragt, da8 sie sich zur qualitativen Unterscheidung derselben von priniaren und sekundaren Thiourethanen ausnutzen IaDt, die nach Oxydation rnit Wasserstoffperoxid rnit Eisen(II1)-chlorid in Methanol charakteristische Farbungen geben, w;ihrend bei den tertiaren Thiourethanen keine Farbreaktion auftritt.
Den FARBENFABRIKEN BAYER danken wir fur die Uberlassung von Chemikalien und dem FONDS DER CHEMISCHEN INDUSTRIE fur Unterstiitzung unserer Arbeit.
B E S C H R E I B U N G D E R V E R S U C H E Alle RP- Werre beziehen sich auf Diinnschichtchromatogramme (Triiger : Kieselgel G) rnit
dem angegebenen Laufmittel.
Dorsrellung N-nionosubsriruierrer Thionirrerhane durch Umsetzung von Senfolen mir Alkoholen nach Li t . 12) (Methode A )
N-[err.-Butyl-0-[err.-butyl-rhionurethan (11). - 2 g Natriiim wurden in 200 ccm tea.- Butylalkohol gelost, rnit 6.5 g rert.-Bufyl-senfol versetzt und 10 Min. unter Riihren zum Riick- RuR erhitzt. Die erkaltete Losung wurde in 500 ccm Wasser gegossen, mit verd. Salzsaure angesauert und 2mal rnit Chloroform extrahiert. Nach dem Trocknen iiber Natriumsulfat wurde abgedampft und aus Benzol/Petrofather umkristallisiert. Ausbeute 800 mg (7.5 %) vom Schmp. 120--121° (nach Sintern ab 112").
C9H19NOS (189.3) Ber. C 57.10 H 10.12 N 7.40 S 16.94 Gef. 57.48 10.05 7.69 16.57
Darstellung von Chlorrhionkohlensaurephenylestern
Chlorthionkohlensaure-O-~Z.6-dimerhyl-phenyll-ester. - 12.2 g (0.1 Mol) 2.6-Dimethyl-phenol wurden in 50 ccm 2n NaOH gelost und unter Riihren im Verlauf von 30 Min. zu 7.7 ccm (0.1 MoI) Thiophosgen in 50 ccm Chloroform gegeben. Dabei wurde durch Eiskiihlung die Temperatur der Mischung auf 10- 15" gehalten. Nach beendeter Zugabe wurde weitere 60 Min. geriihrt. Die organische Phase wurde dann abgetrennt, einmal rnit Wasser gewaschen und iiber Natriumsulfat getrocknet. Nachdem das Losungsmittel i. Vak. abdestilliert war, wurde i. Vak. bei einer albadtemperatur von zunachst 140- 150" und schliefllich 160- 170"

76 W. WALTER und K.-D. BODE Bd. 681
destilliert. Ausbeute 15.1 g(71.5%), Sdp.12 106-1 12", n'," = 1.5605. Das gelbe 81 kristallisiert im Tiefkiihlschrank.
C9H9CIOS (200.7) Ber. C 53.86 H 4.52 C1 17.67 S 15.98 Gef. 54.44 4.64 16.88 15.73
Analog wurden folgende Ester dargestellt :
112". Chlorfhionkohlensaure-0-[2-mefhyl-phenyll-esfer. - Ausbeute 10.0 g (54 %), Sdp.15 108 bis
CgH7CIOS (186.7) Ber. C 51.47 H 3.78 S 17.18 Gef. C 51.34 H 3.84 S 17.16
Chlorfhio1rkohlensaure-O-[2-brom-phenyll-esfer. - Ausbeute 14.7 g (58.8 %), Sdp.15 131-134".
C7HdBrCIOS (251.5) Ber. C 33.42 H 1.60 S 12.76 Gef. C 32.79 H 1.57 S 11.68
Chlorfhionkohlensaure-O-[2-chlor-phenyll-esfer. - Ausbeute 10. I g (49 %), Sdp.15 1 I8 - 123".
C7H4ClrOS (207.1) Ber. C 40.62 H 1.95 S 15.52 Gef. C 40.73 H 2.37 S 15.52
Dars fellung N-monosubs fifuierfer Thionurethane
durch Acylierung von Aminen nach Lit. 15) (Methode B )
N-[2.6-Dimefhyl-phenyll-O-phenyl-thionurefhon (VIII a). - 24.2 g (0.2 Mol) 2.6-Dimethyl- anilin wurden in 50 ccm trockenem Chloroform gelost und unter Wasserkiihlung mit 16.9 g (0.1 Mol) Chlorrhionkohlensaurephenylester in 100 ccm Chloroform versetzt. Nach 24 Stdn. wurde vom ausgefallenen Aminhydrochlorid abgesaugt und i. Vak. eingedampft. Es hinter- blieb eine gelbliche Kristallmasse (36 g). Nach Umkristallisation aus 150 ccm Benzol und 100 ccm Petrolather wurden 18 g (61 %) farblose Nadeln vom Schmp. 102.5- 103" erhalten.
C I ~ H ~ ~ N O S (257.4) Ber. C 70.02 H 5.88 N 5.44 S 12.46 Gef. 70.33 6.05 5.39 12.37
Analog wurden folgende Thionurethane dargestellt :
N-Phenyl-0-phenyl-fhionurefhan. - Ausbeute 46% d. Th., Schmp. 141 - 142" (aus Benzol/ Petrollther), Lit. 29) 142".
N-[2-Methyl-phenyl]-O-phenyl-fhionurefhan (VII a). - Ausbeute 8.8 g (73 %) vom Schmp. 109.5 - 1 11.5" (aus Benzol).
C14H13NOS (243.3) Ber. C 69.1 I H 5.39 N 5.76 S 13.18 Gef. 69.57 5.57 5.57 12.65
N-[2-Chlor-phenyl]-O-phenyl-thionurethan (VII b). - Ausbeute 3.0 g (36 %), Schmp. I19--121" (aus Benzol).
C13HloCINOS (263.8) Ber. C 59.20 H 3.82 N 5.31 S 12.16 Gef. 60.09 3.87 5.29 12.27
N-i2-Brom-phenyll-O-phenyl-rhionurefhan (VIIc). - Schmp. 128 - 128.5" (aus Benzol/ Petrolather), Ausbeute 5.3 g (34%).
Cl3HloBrNOS (308.2) Ber. C 50.66 H 3.27 N 4.55 S 10.40 Gef. 50.52 3.60 4.46 10.13

1965 uber die Oxydationsprodukte von Thiocarbonslureamiden, IX 77
N-[2.6-Diathyl-phenyl?-O-phenyl-thionurethan (VIII b). - Schmp. 141 - 143" (aus Benzol/ Methanol), Ausbeute 11.4 g (80%).
C17H19NOS (285.4) Ber. C 71.55 H 6.71 N 4.91 S 11.24 Gef. 72.31 7.02 5.13 10.90
N-Phenyl-O-[2.6-dimethyl-phenylj-thionurethan (1X a). - Schmp. 169 - 169.5" (aus Benzol), Ausbeute 7.7 g (65 %).
C15H15NOS (257.4) Ber. C 70.01 H 5.87 N 5.44 S 12.46 Gef. 70.52 5.99 5.24 12.24
N-[2-Methyl-phe1iyl]-O-[2.6-dimethyl-phenylj-~hionurethan (IX b). - Schmp. I6 I .5 - 162.5" (aus Benzol/Petrolather), Ausbeute 6.7 g (49.5 %).
C16H17NOS (271.4) Ber. C 70.82 H 6.31 N 5.16 S 11.82 Gef. 71.28 6.30 5.68 11.80
[2.6-Di-tert.-butyl-4-allyl-phenylJ-thionurethan (XI) (Methode C). - 5 g [2.6-Di-terr.-bufyl- 4-allyl-phenylj-cyansaureester wurden in 80 ccm trockenem Benzol gelost. Nach Zugabe von 0.1 ccrn Triathylamin wurde 50 Min. lang durch die Losung ein kraftiger H2S-Strom geleitet und iiber Nacht bei Raumtemperatur aufbewahrt. Nach dem Abdampfen i. Vak. wurde aus 20 ccm Benzol umkristallisiert. Ausbeute 4.4 g (78 %) farblose Nadeln. Schmp. 190- 190.5".
ClgHzsNOS (303.5) Ber. C 71.24 H 8.30 N 4.62 S 10.57 Gef. 70.83 8.54 4.79 10.95
N. N-Dirnefhyl-S-[4-cyan-benzyl]-dithiourethan (Methode D). - 18 g Natriumdimethyldithio- carbornat wurden in 250ccm Aceton gelost, mit I I g 4-Cyan-benzylchlorid versetzt und 20 Min. unter RiickfluB erhitzt. Dann wurde abfiltriert, i. Vak. auf 25 ccm eingedampft und tropfen- weise mit Wasser bis zur Triibung versetzt. Im Kiihlschrank fie1 ein farbloses Produkt an, das aus Athanol umkristallisiert wurde. Ausbeute 12.3 g (72 %, bez. auf Cyanbenzylchlorid).
CllH12N2Sz (236.4) Ber. C 55.88 H 5.11 N 11.85 S 27.13 Gef. 55.53 5.14 11.75 27.91
N. N-Dimethyl-S-carbamoylmethyl-dithiourethan (XVII). - 14.3 g Natriumdimethyldithio- carbamat wurden in 50ccm Wasser gelost, auf 50" erwarmt und unter Riihren mit l o g Chloracetamid in 200 ccm Aceton versetzt. Nach 90 Min. wurde in 500 ccm Wasser gegossen. Nach Abdampfen des Acetons i. Vak. schieden sich farblose Kristalle ab, die Fallung wurde durch Kiihlen vervollstandigt. Umkristallisieren aus Aceton ergab 10 g (56 %) farblose Nadeln, Schmp. 123-124". Lit.41) 122".
CsHloN2OSz (178.3) Ber. C 33.68 H 5.65 N 15.71 S 35.97 Gef. 33.81 5.74 15.71 35.72
N-Carboxymethyl-0-tetradecyl-thionurethan (1V) (Methode E). - Nach der Vorschrift von KNOT TI^) wurde eine Substanz erhalten, die nicht wie angegeben bei 67", sondern bei 79.5 bis 80.5" schmolz. Es handelte sich nach der Analyse jedoch um die gewiinschte Verbindung.
C17H33N03S (331.5) Ber. C 61.59 H 10.33 N 4.23 S 9.67 Gef. 61.09 9.92 4.92 9.59
Morpholino-thiocarbonsaureathylester (Methode E). - 19 g 0-Athyl-S-carboxymerhyl- xanthat wurden in 200 ccm Wasser gelast und mit 8.7 g Morpholin versetzt. Es fie1 sofort ein

78 W. WALTER und K.-D. BODE Bd. 681
Niederschlag am, der aus Methanol/Wasser umkristallisiert wurde. Ausbeute 16.3 g (93 %). Farblose Nadeln, Schmp. 56.5 - 57".
C ~ H I ~ N O ~ S (175.3) Ber. C47.96 H 7.47 N 7.99 S 18.29 Gef. 48.10 7.54 7.99 18.14
N.N-Dimethyl-O-[2-nitro-phenyl]-thionurethan (Methode G). - 8.7 g Natrium-o-nitro- phenolat wurden in 80 ccm Wasser gelost und mit 10.0 g (-I .5 Aquivv.) N.N-Dimethyl-thio- carbamidchlorid in 50 ccm Chloroform versetzt. Die Mischung wurde krhftig geschiittelt, bis die dunkelrote Farbung verschwand und eine hellgelbe organische Phase erhalten wurde. Diese wurde abgetrennt, rnit NaHCO3-Losung und Wasser gewaschen und iiber Natriumsulfat getrocknet. Nach dem Abdampfen i. Vak. wurde ein gelbes Kristallisat erhalten, das nach Umkristallisation aus Benzol/Petrolather bei 126.5 - 128" schmolz (Lit.44) 124"). Ausbeute 6.9 g (57%. bez. auf das Phenolat).
CgHloNzO3S (226.3) Ber. C 50.02 H 4.45 N 12.38 Gef. C49.57 H 4.62 N 11.96
Phenylfhionurethan (XII, Methode B). - 10 g Chlorthionameisensaure-phenylester wurden in 100 ccm Chloroform gelost. In die Losung wurde langsam Ammoniak eingeleitet, bis die gelbe Farbe verschwunden war. In einem verschlossenen Gefaf3 wurde iiber Nacht aufbewahrt, vom ausgeschiedenen Ammoniumchlorid abfiltriert, i. Vak. eingedampft und aus Benzol um- kristallisiert. Ausbeute 4.5 g (51 %), Schmp. 130.5- 132" (Lit. 15) 132.5").
Analog wurden folgende Thionurethane erhalten :
[Z-Me/hyl-phenyl]-thionurefhan (XIII). - Schmp. 155.5 - 156.5" (aus Benzol), Ausbeute 4.4 g (49 %).
CsHqNOS (167.2) Ber. C 57.47 H 5.43 N 8.38 S 19.18 Gef. 58.15 5.46 8.16 18.81
[2.6-Dimethyl-phenylI-thionurethan (XIV). - Schmp. 171 - 172" (aus Benzol), Ausbeute 3.0 g (31 %).
C9HllNOS (181.3) Ber. C 59.63 H 6.12 N 7.73 S 17.69 Gef. 59.17 6.10 7.62 16.98
3.5-Di-tert.-butyl-l-hydroxy-thiobenzanilid. - 10 g 2.6-Di-tert.-butyl-phenol wurden in 100 ccm Aceton gelost und mit 7.8 g Phenylsenfolsowie 4 g NaOH 2 Stdn. auf dem siedenden Wasserbad erhitzt. Die braungriine Losung wurde in 500 ccm Wasser gegossen und mehrfach mit Chloroform extrahiert. Nach dem Trocknen iiber Natriumsulfat wurde i. Vak. abdestilliert und aus Benzol umkristallisiert. Ausbeute 0.8 g zitronengelbe Nadeln, Schmp. 219-2220",
C21H27NOS (341 3) Ber. C 73.86 H 7.97 N 4.10 S 9.41 Gef. 73.46 7.82 4.66 9.95
S-tert.-Butyl-dithiourefhan (XIX). - Analog der von DELkPINE45) angegebenen Arbeitsweise wurde das Dithiourethan in 15-proz. Ausbeute dargestellt. Feine weiBe Nadeln, Schmp. 102-103".
CsHllNSz (149.3) Ber. C 40.22 H 7.43 N 9.38 S 42.96 Gef. 40.47 7.53 9.34 42.97
44) J. CLAUDIN, Franz. Pat. 789500 (1935), SOC. An. des Matibres Colorantes et Produits Chimiques de Saint-Denis [C. 1936 I, 28281.
45) M. DEL~PINE, Bull. SOC. chim. France [3] 27, 585 (1902).

1965 uber die Oxydarionsprodukte von Thiocarbonsaureamiden, 1X 79
Oxydution monosubstituierter Thionurethane
Allgemrine Vurschrift (vgl. Tab. I ) : 100 mg des Thionurethans wurden in 50 ccm Methylen- chlorid/Methanol ( I : I ) gelost und rnit 3-4 Aquivv. Perhydrdversetzt. Die Reaktion wurde bei 40" ausgefiihrt und rnit Hilfe der Diinnschichtchromatographie verfolgt. Der Nachweis der Oxydationsprodukte erfolgte rnit methanol. FeCI3-Losung und mit Jod/Natriumazid-Losung.
Formimidoesterdisulfide
N- Phenyl-0-arhyl-formimidoester-disulfid. - 5.4 g N-Phenyl-0-athyl-thionurethan (1 b) wurden in 25 ccm Methanol + 10 ccm Chloroform gelost und rnit 8.0 ccm (2.5 Aquivv.) Perhydrol versetzt. Die Losung wurde 40 Min. auf 28-30" gehalten, bis eine rasch starker werdende Triibung auftrat. Die Reaktion wurde durch EingieDen in 100 ccm NaCI-Losung beendet. Nach Abtrennen der organischen Phase wurde 3 ma1 mit Chloroform extrahiert, die organischen Ausziige wurden mit Wasser gewaschen und iiber Natriumsulfat getrocknet. Nach dem Eindampfen blieben 5 . I g farbloses bI zuriick, das riach Anreiben Iangsam durch- kristallisierte und mit FeCI3 eine starke Blaufarbung ergab. Das Diinnschichtchromatogramm zeigte drei Flecke rnit Jod/Azid, die dem Ausgangsprodukt, dem Thionurethan-S-oxid und dem Formimidoester-disulfid zugeordnet werden konnten. Der kristalline Riickstand wurde unter 20 ccrn Methanol fein zerkleinert; dabei blieben I .9 g farblose Substanz zuriick, die bei 85 -94" schmolz. Aus Benzol/Petrolather wurden I .6 g farblose Nadeln vom Schmp. 98" erhalten. Aus der methanolischen Losung wurden 3.4 g Ausgangssubstanz zuriickerhalten, so daR die Ausbeute an Formimidoester-disulfid 80 :< (bez. auf umgesetztes Thionurethan) betrug.
C ~ ~ H ~ O N Z O Z S Z (360.5) Ber. C 59.97 H 5.59 N 7.77 S 17.79 Gef. 60.26 5.73 7.65 17.81
N- Phenyl-0-propyl-jormiinidoester-disulfid. - I .O g N-Phenyl-0-propyl-thionurethan (I c) wurde in 20 ccm Benzol f 20 ccm Methanol gelost und mit 2.0 g Perhydrol versetzt. Nach 2 Stdn. bei 30" wurde in 100 ccm Wasser gegossen. Die organische Phase wurde abgetrennt, das Wasser 2mal mit Chloroform extrahiert, die vereinigten Ausziige wurden wieder rnit Wasser gewaschen und iiber Natriumsulfat getrocknet. Nach dem Abdampfen des Losungs- mittels i.Vak. blieben 0.9 g gelbliches bl zuriick, das im Kiihlschrank kristallisierte. Ein Diinn- schichtchromatogramm (Laufmittel: Benzol/Athanol = 3: I ) zeigte drei Flecke rnit den RF- Werten 0.3 (Disulfid), 0.6 g (Thionurethan-S-oxid) und 0.9 (Ausgangssubstanz). Umkristalli- sation aus Petrolather ergab 94 mg farblose Kristalle vom Schmp. 88-90" sowie 0.6 g Aus- gangssubstanz. Ausbeute 24 % (bez. auf umgesetztes Thionurethan).
CzoH24N2OzSz (384.5) Ber. C 61.82 H 6.23 N 7.21 S 16.51 Gef. 61.81 6.44 7.41 16.35
N-[2.6-Dimethyl-phet~ylJ-O-phenyl-formimidoester-disul~d. - 2.6 g N-12.6- Dimethyl-phenyll- 0-phenyl-thiunurethan (VlfI a ) wurden in 30 ccm Methylenchlorid + 25 ccm Methanol gelost, mit 2.0 ccm (2 Aquivv.) Perhydrol versetzt und 30 Min. bei genau 40" gehalten. Nach beendeter Reaktion wurde in 100 ccm Wasser gegossen, die organische Phase abgetrennt und die waBr. Phase einmal rnit Methylenchlorid extrahiert. Nach dem Trocknen iiber Natrium- sulfat wurde i. Vak. bei 18" Wasserbadtemperatur eingedampft. Es blieben 2.6 g farbloses

80 W. WALTER und K.-D. BODE Bd. 681
0 1 zuriick, aus dem durch Umkristallisation rnit 10 ccm Benzol und 6 ccm Petrolather 1.3 g (49 %) farblose Kristalle vom Schmp. 156- 157" erhalten wurden.
C,oH2sN201S2 (512.7) Ber. C 70.28 H 5.63 N 5.47 S 12.51 Gef. 70.78 6.05 5.29 12.44 Mol.-Gew. 468 (in Benzol)
Analog wurden dargestellt:
N-[2.6-Diathyl-phenylJ-O-phenyl-formimidoester-dis~1l~d. .- Aus 3 .O g N-[2.6-Diathyl- phenyl~-O-phenyI-thionurethan (VIII b) wurden 2.8 g (99 %) Disulfid erhalten. Reaktionsdauer 5 Stdn., Schmp. 79-80" (aus n-Heptan).
C J ~ H ~ ~ N Z O ~ S ~ (568.8) Ber. C 71.80 H 6.38 N 4.93 S 11.27 Gef. 71.45 6.28 4.78 11.33 Mol.-Gew. 564 (in Benzol)
N-[2-Methyl-phenylJ-0-[2.6-dimethyl-phenyl]-formimidoester-disulfd. - Aus 1.4 g N- ~2-Methyl-phenylj-0-[2.6-dimethyl-~henylJ-thionuretha1i (IX b) wurden 0.33 g (24 %) Disulfd erhalten. Reaktionsdauer 4 Stdn., Schmp. 142- 143" (aus Benzol/Methanol).
C ~ Z H ~ ~ N Z O ~ S ~ (540.8) Ber. C 71.08 H 5.97 N 5.18 S 11.86 71.26 5.97 5.09 11.76
Oxydation von N-Phenyl-O-[2.6-dimethyl-phenylJ-thionurethan: 2.5 g IXa wurden in 60 ccm Methylenchlorid + 40 ccm Methanol gelost und rnit 2.0 ccm Perhydrol (ca. 2 Aquiw.) bei genau 40' 4 Stdn. oxydiert. Die Losung wurde dann in NaHCO3-Losung gegossen, die orga- nische Phase abgetrennt, die waBr. Phase einmal rnit Methylenchlorid extrahiert, die ver- einigten Ausziige wurden einmal mit Wasser gewaschen. Nach demTrocknen iiber Natriumsulfat wurde i.Vak. abdestilliert; dabei verblieb ein 81, das nach dem Anreiben kristallisierte. Um- kristallisation aus Methanol/Benzol ergab 1.45 g N-Phenyl-O-[2.6-dimethyl-phenylJ-thion- urethan-S-oxid. Schmp. 124" - 125". Die Substanz gab rnit methanol. FeCI3 eine starke Blau- farbung. Im Diinnschichtchromatogramm zeigte sich nur ein Fleck rnit dem Rp-Wert 0.8 (RF-Wert der Ausgangssubstanz0.9; Laufmittel: Athanol/Benzol = 3 : 1). Aus der Mutterlauge wurden 800 mg farblose Kristalle vom Schmp. 138- 139" (Disulfid) erhalten.
N- Phenyl-0-12.6-dimethyl-pher1ylj-1hionurethan-S-oxid. - Ausbeute 54
C l sHl~N0zS (273.4)
d. Th.
Ber. C 65.91 H 5.53 N 5.12 S 11.73 Gef. 65.84 5.40 5.14 12.05 Mol.-Gew. 278 (in Benzol)
N-Phenyl-O-l2.6-dimethyl-phenyl~-formimidoester-disu/fid. - Ausbeute 800 mg (32 %). C3oHzsNzOzS2 (512.7) Ber. C 70.28 H 5.63 N 5.47 S 12.51
Gef. 69.70 5.35 4.82 12.66
Oxydation von N-[2-Methyl-phenylJ-0-[2.6-dimethyl-phe1i~lj-thionurethan ( I X b ) : Analog der Oxydation zum Disulfid. Rohausbeute 1.2 g (81 %), Schmp. 126- 130" (unscharf).
C ~ ~ H I ~ N O ~ S (287.4) Ber. C 66.87 H 5.96 N 4.87 S 11.16 Gef. 65.77 5.74 5.43 10.56
Allgemeine Vorschrift zur Oxydation primarer Thionurethane: 0.0 I Mol des primaren Thion- urethans wurde in 20-3Occm Methanol gelost und mit einem, zwei oder drei Aquivalenten Perhydrol bei 20, 30 oder 40" oxydiert. Den Losungen wurden in Abstanden von 30 Min.

1965 ube r die OxvdationsDrodukte von Thiocarbonsaureamiden. IX 81
Proben zur chromatographischen Verfolgung der Reaktion entnommen. Die Ergebnisse sind in Tab. 1 (S. 68) und in den Abbildungen der Diinnschichtchromatogramme von X11, XITI und XIV ( S . 72) wiedergegcben. i2.6-Di-tert.-butyl-4-allyl-phenyl]-thionurethan-S-oxid. - 0.7 g XI wurden in 50 ccm Methy-
lenchlorid + 30 ccm Methanol gelost und rnit l .O ccm (ca. 4 Aquivv.) Perhydrol I20 Min. bei genau 40" oxydiert. Nach beendeter Reaktion wurde i. Vak. auf 40 ccm eingedampft und im Tiefkiihlschrank iiber Nacht aufbewahrt. Die schneeweiBen Kristalle, die ausfielen, wurden abgesaugt und rnit Petrolather gewaschen. Ausbeute 650 mg (87%), Schmp. 128- 132". Mit FeCI3: rotviolett. Die Substanz war im Diinnschichtchromatogramm (Benzol/Methanol = 3 : I ) einheitlich. Sie war, wie alle Thionurethan-S-oxide, nur im Tiefkiihlschrank haltbar.
ClaH2~N02S (319.5) Ber. C67.67 H 7.89 N4.29 S 10.04 Gef. 67.53 8.32 4.82 10.01
3.5-Di-rert.-butyl-4-hydroxy-thiobenzanilid-S-oxid. - 500 mg Thioamid wurden in 50 ccm Methylenchlorid + 50 ccm Methanol gelost und mit 1.0 ccm Perhydrol 2 Stdn. bei 30-34" oxydiert. Dann wurde in 100 ccm gesattigte NaC1-Losung gegossen und m;t Methylenchlorid extrahiert. Die organischen Extrdkte wurden einmal mit Wasser gewaschen und iiber Natrium- sulfat getrocknet. Nach dem Eindampfen i . Vak. wurde aus Methanol umkristallisiert. Ausbeute 368 mg (71 %) zitronengelbe Nadeln, die rnit methanol. FeCI3 eine olivgrune Farbung gaben. Schmp. 170- 174" (nach raschem Erhitzen, Schmelzpunktmikroskop LEITZ).
C ~ I H ~ ~ N O ~ S (356.5) Ber. C 70.74 H 7.35 N 3.93 S 9.00 Gef. 71.45 7.34 3.82 8.81
Oxydation disubstituierter Thionurethane Analog der fur monosubstituierte Thionurethane angegebenen Arbeitsweise wurden die in
Tabelle 2 aufgefiihrten Substanzen oxydiert. Der Verlauf der Reaktion wurde rnit Hilfe der Diinnschichtchromatographie gepriift.
Tabelle 2. Oxydationsergebnisse an tertiaren Thionurethanen (Laufmittel: Benzol/Chloroform = 3 : I )
Thionurethan Ergebnis - -
(CH3)zN- C- O-C6H4-NO2 (0)
0 T - N - C-OC2H5 Entschwefelung
mit Jod/Azid nur 1 Fleck, RP 0.61 (Ausgangs- substanz), rnit FeCI3 I Fleck, R~0.22(rotbraun) I1
S
ll S
C6H5 ,C6H5
C2H5 ' S '1 4 'C2Hs 'N-C-0-C-N xv 37)
s
keine Reaktion
zahlreiche Jod/Azid-positive Zersetzungspro- dukte
46) H. A. STAAB und G. WALTHER, Liebigs Ann. Chem. 657, 98 (1962).
Liebigs Ann. Chern. Bd. 681 6

82 W. WALTER und K.-D. BODE Bd. 681
N.N-Dimethyl-S-thiocarbamoylmerhyl-dithiourethan-S-oxid. -- 2.0 g Dithiourethan X V I wurden in 15 ccm Dimethylformamid gelost und mit 2.0ccm Perhydrolversetzt. Dabei stieg die Temperatur auf iiber 40". Nach 30 Min. wurde tropfenweise bis zurTriibung mit Wasser versetzt und iiber Nacht im Tiefkiihlschrank aufbewahrt. Der ausgefallene Kristallbrei wurde in wenig Dimethylformamid gelost und mit Wasser bis zur Triibung versetzt. Im Kiihlschrank kristalli- sierten 1.1 g (51 %) farblose Kristalle aus, die mit FeCI3 eine rotviolette Farbung gaben. Schmp. 96-98".
C5HloN20Sj (210.3) Ber. C 28.55 H 4.79 N 13.32 S 45.73 Gef. 28.57 4.74 13.13 45.43
Oxydation von Dithiourethanen Analog der bei primaren und sekundaren Thionurethanen angegebenen Arbeitsweise wurden
die in Tabelle 3 aufgefuhrten Dithiourethane oxydiert :
Tabelle 3. Oxydationsergebnisse an Dithiourethanen - -. - -~ - .- -. - - - -~ -.
Dithiourethan RF-Werte S-Oxid [Laufmittel] *) Ausbeute 1x1 ____ __ - - - - _ _ - ~ - - - -~ -
- a) (CH3)2N - C - S-CHz-CsHs 39) nicht bestimmt [A] I / S
II 0.75 (Ausg.-Verbdg.) S
II S
ll S
(CH3)zN-C- S - C H ~ - C ~ H ~ - C N I J I ) 0.36 (nur mit FeCI3) [A] -
[(CH3)2N-C-CH2]2 47) - [A1 - b)
(CH3)2N - C - S -CH2-C -NH2 0.30 (unbekannt) [B] - bl II XVII 0.50 (Ausg.-Verbdg.) 0
(CH3)2N-C- S -CHz-C-NHl - I1 II XVI 40) S S
51
tert.-CdHg- S -C-NH2 0.50 (S-Oxid) [D] 40 II XIX 0.66 (Ausg.-Verbdg.) S
iso-C3H,-S -C-NH2 /I S
C2H5- S-C-NH2 I1 S
0.47 (S-Oxid) ID] 23 0.60 (Ausg.-Verbdg.)
0.45 (S-Oxid) [DI 0.59 (Ausg.-Verbdg.)
- d)
*) Laufmittel: A = Benzol/Athanol ( 3 : I ) ; B = Methanol; C = Benzol/Chloroform ( 3 : l ) ; D = Benzol/Metha-
a) Kein S-Oxid, nach ltingerer Dauer Zersclzg. - bJ Rotbraunfarbung mit FeCI, nur im Reagcnzglas. - C ) Zahl- no1 (3: I ) .
reiche Flecke neben dem S-Oxid. - d) 0.14; 0.75, Front.
47) J. C . THOMAS, Amer. Pat. 2384577 v. 11. 9. 1945, E. 1. du Pont de Nemours & Co. [C. A. 40, I77 ( I 946)l.

1965 Uber die Oxydationsprodukte von Thiocarbonsaureamiden, IX 83
Oxydative Entschwefelung eines Thionurethans: 1 .O g N-Carboxymethyl-0-retrodecyl-thiori- urerhan (IV) wurde in 20 ccm Methanol -t 10ccm Chloroform gelost und mit 1.0ccm Perhydrol versetzt; dabei stieg die Temperatur auf 45-50". Nach 90 Min. wurde in gesattigte NaCI-Losung gegossen, die organische Phase abgetrennt und die waBrige Phase einmal rnit Chloroform ausgeschiittelt. Nach dem Trocknen iiber Natriumsulfat wurde i. Vak. abdestil- liert und 2 ma1 aus Benzol umkrktallisiert. Ausbeute: 440 mg N-Carboxymethyl-0-rerradecyl- urethan, Schmp. 99.5- 100.5".
C I ~ H ~ ~ N O ~ (315.4) Ber. C 64.73 H 10.53 N 4.44 Gef. C 64.81 H 10.33 N 4.39
S-[err.-Bury/-dirhiourethan-S-oxid. - I50 mg XIX wurden in 15 ccm Methanol gelost und mit 0.5 ccm Perhydrol (etwa 5 Aquivv.) bei 40" oxydiert. Nach 10 Min. triibte sich die Losung und wurde auf Raumtemperatur abgekiihlt. Nach insgesamt 70 Min. wurde in 50 ccm geslttigte NaCI-Losung gegossen und mit 2mal 10 ccm Methylenchlorid extrahiert. Nach dem Trocknen und Abdestillieren des Losungsmittels i. Vak. verblieben 100 mg farblose Substanz, die aus 25 ccm Benzol umkristallisiert wurden. Ausbeute 66 mg (40%), Schmp. 141.5- 142". Feinc Nadeln; mit FeCI3 in Athanol Blau-, in Benzol Rotbraunfarbung.
CSHllNOS2 (165.3) Ber. C 36.33 H 6.74 N 8.47 S 38.80 Gef. 37.09 6.77 8.76 38.18
S-lsapropyl-dithiorwethan-S-oxid. - Darstellung analog voranstehender Verbindung. Aus- beute 23 x, Schmp. 100 - 103" (aus Benzol).
C4H9NOS2 (151.3) Ber. C 31.76 H 6.00 N 9.26 S 42.39 Gef. 31.83 5.85 9.26 42.75
Reduktion von [2.6-Di-terr.-buryl-4-allyI-phenyl~-rhiotiurethan-S-oxid: I 0 0 mg S-Oxid wur- den in 20 ccm Methanol gelost. In diese Losung wurde langsam Schwefelwasserstoff ein- geleitet. In kurzen Abstanden wurden Proben entnommen. Nach 25 Min. war chromato- graphisch kein S-Oxid mehr nachweisbar. Die Losung wurde i. Vak. eingedampft, der Riick- stand aus Benzol/Petrolather umkristallisiert. Es wurden 60 mg [2.6-Di-rerr.-butyl-4-allyl- phenylj-thionurethan isoliert. Schmp. 190- 190.5". Die Substanz gab nach Oxydation mit Perhydrol mit FeCI3 wieder eine Blaufarbung.
Spaltiitrg eities Formimidoesrerdisulfids rnit Natriumcyariid: 180 mg N- Phenyl-0-athyl-form- imidoester-disulfd wurden in 10 ccm Methanol gelost und mit 50 mg Natriumcyanid in 3 ccm Wasser 10 Min. gekocht. Die Losung wurde dann im Diinnschichtchromatogramm (Benzol/ Methanol = 3 : 1) rnit IV-Pheiryl-O-uthyl-t/~ionur~~tha~r verglichen und zeigte einen Fleck mit dem gleichen RF-Wert wie dieses. AuBerdem wurde ein Teil der Losung mit 2 Tropfen Perhydrol versetzt und chromatographiert; dabei lien sich N-Phenyl-0-athyl-thionurethan-S-oxid sowohl rnit Jod/Azid als auch mit FeCI3 nachweisen.
Reduktion von N. N- Dimethyl-S-rhiocarbamoylmethyl-dithiourethan-S-oxid: I 00 mg S-Oxid wurden in 20 ccm Dimethylformamid gelost und mit H2S behandelt. Nach 30 Min. war kein S-Oxid mehr nachzuweisen. Es wurde i. Vak. eingedampft und aus Aceton umkristallisiert. Ausbeute 65 mg N. N-Dimethyl-S-corbamoylmethyl-dithiourethan, Schmp. 153" (Mischprobe).
Thermische Zersetzung von N.N-Dimethyl-S-rhiocorbamoylmethyl-di~hioure~han-S-oxid: 100 mg S-Oxid wurden in 20 ccrn Dimethylformamid geltist und 15 Min. unter RiickfluO
6.
84 H. REINSHAGEN Bd. 681
erhitzt, bis kein S-Oxid mehr nachzuweisen war. Nach dem Eindampfen verblieb ein braun- licher Ruckstand, der mehrfach aus Aceton umkristallisiert wurde. Dabei wurden 40 rng N. N- Dimerhyl-S-carbamoylmethyl-dirhiourethan erhalten, Schmp. 1 24".
UBER NEUE HEXAHYDROCHINOLINE von HELLMUTH REINSHAGEN
Herrn Pro/. Dr., Dr. h. c . C . Schopf in Verehrung zum 65. Geburrstag gewidmer
Aus dem lnstitut fur Organische Chemie der Technischen Hochschule Darmstadt
Eingegangen am 13. Marz 1964
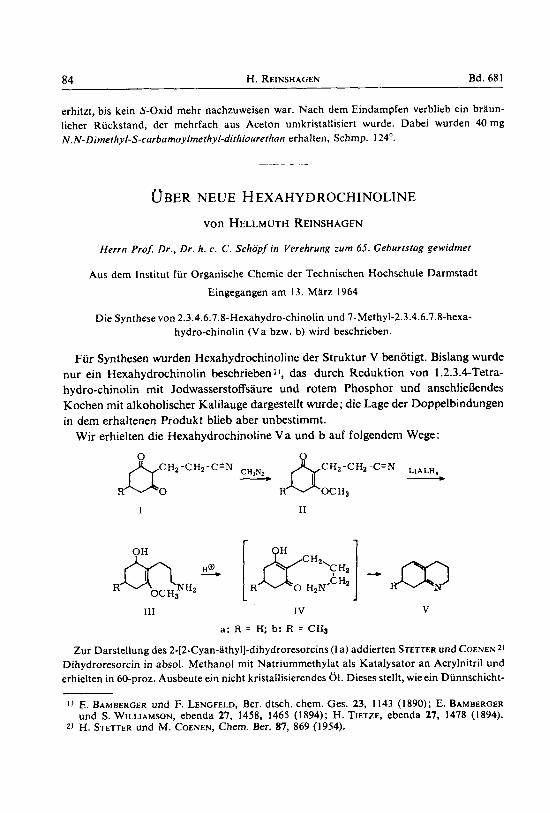
Die Synthese von 2.3.4.6.7.8-Hexahydro-chinolin und 7- Methyl-2.3.4.6.7.8-hexa- hydro-chinolin ( V a bzw. b) wird beschrieben.
Fur Synthesen wurden Hexahydrochinoline der Struktur V benotigt. Bislang wurde nur ein Hexahydrochinolin beschrieben I), das durch Reduktion von 1.2.3.4-Tetra- hydro-chinolin mit Jodwasserstoffsaure und rotem Phosphor und anschliekndes Kochen mit alkoholischer Kalilauge dargestellt wurde; die Lage der Doppelbindungen in dern erhaltenen Produkt blieb aber unbestimmt.
Wir erhielten die Hexahydrochinoline V a und b auf folgendern Wege:
C Hz -CHz-C 'N - CH,NI ~ c H z - c H z - LIALH,
R OC H3 bo I1
R I
I11 IV
a: R = H; b: R = CHI
V
Zur Darstellung des 2-[2-Cyan-athyl]-dihydroresorcins (I a) addierten STETTER und COENEN 2)
Dihydroresorcin in absol. Methanol mit Natriurnmethylat als Katalysator an Acrylnitril und erhielten in 60-proz. Ausbeute ein nicht kristallisierendes 01. Dieses stellt, wieein Dunnschicht-
1 ) E. BAMBERGER und F. LENGFELD, Ber. dtsch. chern. Ges. 23, 1143 (1890); E. BAMBERGER und S. WILLIAMSON, ebenda 27, 1458, 1465 (1894); H. TIETZE, ebenda 27, 1478 (1894).
2) H. STETTER und M. COENEN, Chern. Ber. 87, 869 (1954).













![w - keralapareekshabhavan.inkeralapareekshabhavan.in/images/sslc2015/ded_2015... · 3 t{KUnwKv kvsIbn t{KUv kvtImÀ ]cn[n A+ 90 iX-am \w apX 100 iX-am \w hsc A 80 iX-am \w apXÂ](https://img.pdfslide.org/doc/110x75/6081d422255bc151340a4c4a/w-k-3-tkunwkv-kvsibn-tkuv-kvtim-cnn-a-90-ix-am-w-apx-100-ix-am-w.jpg)















