Embed Size (px)
Citation preview

Untersuchungen zur Modulation der
Ionenleitfähigkeit von P2X3- und P2X7-Rezeptoren
durch extrazelluläre Phosphorylierung und
Regulation der Rezeptorexpression in
HEK 293-Zellen
Dissertation
zur
Erlangung des Doktorgrades
der Naturwissenschaften
(Dr. rer. nat.)
dem
Fachbereich Pharmazie
der Phillipps-Universität Marburg
vorgelegt von
Doychin Toshev Stanchev aus Bulgarien
Marburg/Lahn 2006

Danksagung
Diese Doktorarbeit entstand am Rudolf-Boehm-Institut für Pharmakologie und Toxikologie
der Universität Leipzig unter der Leitung von Herrn Prof. Dr. Dr. Peter Illes. Für seine
Unterstützung, die wertvollen Anregungen und seine stetige Diskussionbereitschaft möchte
ich meinen herzlichsten Dank aussprechen.
Gleichermaßen danke ich Herrn Prof. Dr. Dr. Josef Krieglstein, der mir die Dissertation an
der Universität in Marburg ermöglicht hat.
Für seine großzügige Unterstützung möchte ich mich im besonderen Maße bei Herrn Prof.
Dr. Günter Heidemann bedanken. Als Vertrauensdozent der Friedrich Naumann Stiftung
half er mir nicht nur, meinen Platz in einem qualifizierten Team einnehmen zu können,
sondern sorgte darüber hinaus durch seine Fürsorge dafür, mich mit Hilfe eines
Stipendiums der Friedrich-Naumann-Stiftung während der dreijährigen Promotionszeit
finanziell abzusichern.
Weiterhin danke ich Frau PD Dr. Kerstin Wirkner für ihre stetige und intensive fachliche
Betreuung, sowie die hervorragende praxisnahe Unterstützung mit der Patch-Clamp
Technik.
Frau Dipl. Biol. Doreen Milius spreche ich meinen Dank für die Zusammenarbeit bei der
Untersuchung P2X7-transfizierter HEK293-Zellen aus.
Mein Dank gilt auch Frau Monika Henschke und Frau Helga Sobottka für die Kultivierung
von HEK293-Zellen und DRG-Neuronen.
Ganz besonderen Dank verdient das gesamte Kollegium des Instituts für Pharmakologie
und Toxikologie, welches mir meine Arbeit in einem außerordentlich guten Arbeitsklima
ermöglicht hat.

1. Einleitung und Fragestellung..................................................... 7
1.1. Purine und ATP.....................................................................................7
1.2. P2-Rezeptoren........................................................................................9
1.2.1. P2Y-Rezeptorfamilie ......................................................................................... 9
1.2.2. P2X-Rezeptorfamilie ....................................................................................... 11
1.2.2.1. Lokalisation und Distribution ................................................................... 12
1.2.2.2. Struktur der P2X-Rezeptoren .................................................................... 12
1.2.2.3. Physiologische Eigenschaften ................................................................... 14
1.2.3. P2X3-Rezeptor .................................................................................................. 16
1.2.3.1. Lokalisation ................................................................................................ 16
1.2.3.2. Struktur ...................................................................................................... 16
1.2.3.3. Physiologische Eigenschaften ................................................................... 17
1.2.4. P2X7-Rezeptor .................................................................................................. 20
1.2.4.1. Lokalisation ................................................................................................ 20
1.2.4.2. Struktur ...................................................................................................... 20
1.2.4.3. Physiologische Eigenschaften ................................................................... 21
1.2.4.4. Membranporenbildung, morphologische Änderungen und Einfluss von
Hypoxie auf den P2X7-Rezeptor ............................................................................. 23
1.3. Aufgaben und Zielstellungen..............................................................24
2. Materialen und Methoden ....................................................... 26
2.1. Chemikalien .........................................................................................26
2.2. Material und Methoden zu den P2X7-Rezeptorexperimenten........28
2.2.1. Material............................................................................................................. 28
2.2.2. Methoden .......................................................................................................... 29

4
2.2.2.1. Nukleinsäure-Methoden ............................................................................ 29
2.2.2.2. Transiente Transfektion von HEK 293-Zellen ......................................... 33
2.3. Material und Methoden zu den P2X3-Rezeptorexperimenten........34
2.3.1 . Kulturmedium für hHEK293-P2X3-Zellen: ................................................. 34
2.3.2. Extrazelluläre Lösung (EC) für HEK293- und Hinterwurzelganglien-
(DRG) Zellen: ............................................................................................................ 34
2.3.3. Intrazelluläre Lösung (IC) für HEK293- und Hinterwurzelganglien -
Zellen: ......................................................................................................................... 35
2.3.4 Präparationen und Kultivierungen ................................................................. 35
2.3.4.1. Präparation der DRG Zellen und ihre Kultivierung ................................ 35
2.3.4.2. Kultivierung von HEK293-hP2X3-Zellen................................................. 36
2.3.5. Patch-Clamp Technik ...................................................................................... 36
2.3.6. Aufbau und Ausstattung des Messplatzes ..................................................... 38
2.3.7. Die praktische Durchführung von Patch-Clamp-Experimenten ................ 41
2.3.8. Datenanalyse und Statistik.............................................................................. 43
3. Ergebnisse.................................................................................. 45
3.1. Potenzierung der α,ß-meATP-induzierten Ströme durch UTP. ....45
3.2. Interaktionen mit PKC Aktivatoren und Inhibitoren .....................50
3.3. Einfluss von Mutationen auf die UTP-induzierte Potenzierung der
α,ß-meATP-induzierten Ströme an mit P2X3-Rezeptoren transient
transfizierten HEK293-Zellen ...................................................................53

5
3.4. Hemmung der maximalen Stromantwort von α,ß-meATP durch
Inhibitoren der Proteinkinase C oder durch Mutationen im P2X3-
Rezeptor.......................................................................................................58
3.5. Effekt von Mutationen des P2X3-Rezeptors auf die Kinetik der
Rezeptorantwort. ........................................................................................60
3.6. Potenzierung der α,ß-meATP-induzierten Ströme durch UTP bei
DRG-Neuronen und Interaktionen mit PKC-Inhibitoren. ....................65
3.7. P2X7-EGFP Klonierung und Eigenschaften des
Rezeptorkonstruktes ..................................................................................68
4. Diskussion.................................................................................. 74
4.1. Regulation des humanen rekombinanten P2X3-Rezeptors durch
ekto-Proteinkinase C. .................................................................................74
4.2. Sauerstoff- und Glukose-Mangel erhöhen die Expressionsrate des
P2X7-Rezeptors in der HEK 293-Zellmembran. .....................................78
5. Zusammenfassung .................................................................... 81
6. Eigene Publikationen................................................................ 84
6.1. Tagungsbeiträge ..................................................................................84
6.2. Originalarbeiten ..................................................................................85

6
7. Literatur .................................................................................... 86
8. Abkürzungsverzeichnis .......................................................... 100
9. Lebenslauf ............................................................................... 103

1. Einleitung und Fragestellung
1.1. Purine und ATP
Die Purine wurden erstmalig 1884 von dem deutschen Chemiker Emil Fischer isoliert und
in eine eigene chemische Familie eingeordnet. Purine sind organische, heterozyklische,
aromatische Stoffe, die aus einem Pyrimidinring und einem Imidazolring bestehen (Abb.1).
Abb. 1. Chemische Grundstruktur der Purine.
Mit A, B und C wurde die Strukturformel der Purine auf drei unterschiedliche Arten
dargestellt. In D ist ein dreidimensionales Modell der Purinstruktur abgebildet. Mit den
blauen Kugeln wurden die Stickstoffatome markiert, mit den grauen Kugeln die
Kohlenstoffatome und mit den weissen Kugeln die Wasserstoffatome.
Für die Funktion lebender Organismen spielt das Purinnukleotid Adenosintriphosphat
(ATP) eine essentielle Rolle. Es ermöglicht einerseits die Energieübertragung in der Zelle,
fungiert andererseits aber auch als wichtiger Neurotransmitter bzw. Kotransmitter (Hoyle
und Burnstock, 1986). ATP besteht aus Adenosin, an das drei Phosphatreste gebunden sind
(Abb.2).

8
Abb. 2. Chemische Grundstruktur des ATP
Im menschlichen Organismus wird ATP als ein Produkt der oxidativen Phosphorylierung
in den Mitochondrien produziert (Belitser und Tsibakova, 1939). Beim Abbau eines
Phosphatrestes wird Energie freigesetzt. So ist ATP der Hauptenergieträger im
menschlichen Organismus (Lipmann, 1949). Außer dieser Funktion spielt ATP eine Rolle
als universales Signalmolekül (Burnstock, 2001).
Neben ATP können auch andere Purine (Adenosin, ADP, AMP, GDP, GTP) und
Pyrimidine (UDP, UTP) eine Signalfunktion im Organismus ausüben (Starke, 2005). Die
extrazellulären Purine und Pyrimidine beeinflussen viele biologische Prozesse, wie z.B. die
Kontraktilität der glatten Muskeln, die Erregungsübertragung zwischen den Nervenzellen,
die exogene und endogene Sekretion, die Immunantwort, die Erregung, die
Blutplättchenaggregation, den Schmerz u.a. (Ralevic und Burnstock 1998). Die Wirkung
von Purinen ist zum ersten Mal von Drury und Szent-Györgyi 1929 beschrieben worden.
Diese Autoren konnten zeigen, dass aus Herzmuskulatur extrahiertes Adenosin und 5’-
Adenosinmonophosphat (AMP) einen biologischen Effekt haben. Wurden die extrahierten
Purine einem Versuchstier injiziert, resultierten Herzstillstand, arterielle Dilatation,
Erniedrigung des Blutdruckes und eine Inhibition der Darmkontraktion. Gillespie (1934)
beschrieb bei Katzen und Kaninchen zwei entgegen gesetzte Effekte: Während
Vasodilatation und Blutdrucksenkung infolge Adenosin- und AMP-Gabe auftraten, führte
ATP zur Erhöhung des Blutdruckes. Das war der erste Anhaltspunkt für die Existenz
mehrerer Purin-Rezeptoren. 1972 vermutete Burnstock eine Beteiligung von ATP bei der
Erregungsübertragung zwischen Neuronen oder Neuronen und Glattmuskelzellen. 1978
postulierte er, dass spezifische, extrazellulär lokalisierte Rezeptoren für Adenosin und ATP
existieren. Mit der ersten Klonierung eines ATP-spezifischen Rezeptors 1990 (s. Kennedy,

9
2000) nahm die diesbezügliche Forschung einen deutlichen Aufschwung. Mittlerweile
wurden Purin- und Pyrimidin-Rezeptoren, die heute als P2-Rezeptoren bezeichnet werden
(siehe Kapitel 1.2.), in fast allen Zellarten gefunden und deren Beteiligung an vielen
physiologischen und pathologischen Prozessen nachgewiesen.
1.2. P2-Rezeptoren
P2-Rezeptoren sind auf der Oberfläche fast aller Säugetierzellen zu finden (von Kügelgen,
2005). Den Begriff „purinerge Rezeptoren“ führte Burnstock 1978 ein. Er unterteilte sie in
zwei große Rezeptorfamilien, die P1- und P2-Purinorezeptoren. Die P1-Rezeptoren
klassifizierte er als Rezeptoren für Adenosin. Durch ihre molekularen, biochemischen und
pharmakologischen Eigenschaften unterteilte er Adenosinrezeptoren in vier Subtypen: A1-,
A2A-, A2B-, und A3-Rezeptoren. Alle P1-Rezeptoren sind alle an G-Proteine gekoppelt
(Ralevic und Burnstock, 1998). Zur P2-Rezeptorfamilie gehören die so genannten P2X-
Rezeptoren, welche Liganden-aktivierte Kationenkanäle sind, und die G-Protein-
gekoppelten P2Y-Rezeptoren.
1.2.1. P2Y-Rezeptorfamilie
Die P2Y-Rezeptorfamilie gehört zu den evolutionär sehr früh entstandenen
Rezeptorfamilien. Dranoff et al., (2000) behaupteten, dass purinerge Signalmechanismen
wahrscheinlich die phylogenetisch ältesten sind. Diese Rezeptorfamilie gehört zur
Rhodopsinfamilie G-Protein-gekoppelter Rezeptoren (von Kügelgen, 2005). Bis jetzt
wurden bei Säugetieren 8 verschiedene P2Y-Rezeptorsubtypen (P2Y1, 2, 4, 6, 11, 12, 13, 14)
kloniert (North und Barnard, 1997; Ralevic und Burnstock, 1998; von Kügelgen und
Wetter, 2000; Jacobson et al., 2002) und funktionell als P2Y-Rezeptoren definiert.
Die P2Y-Rezeptoren besitzen die typischen Merkmale G-Protein-gekoppelter Rezeptoren,
einschließlich der 7 hydrophoben transmembranären Regionen, welche durch 3
intrazelluläre und 3 extrazelluläre Schleifen verbunden sind. Der N-Terminus befindet sich
extrazellulär und der C-Terminus intrazellulär (Abb. 3) (von Kügelgen, 2005).
Alle bis jetzt bekannten P2Y-Subtypen besitzen auf ihrer extrazellulären Domaine 4
Cystein-Reste, die zwei Disulfidbrücken bilden. Eine Disulfidbrücke befindet sich
zwischen dem N-Terminus und der dritten extrazellulären Schleife; die zweite
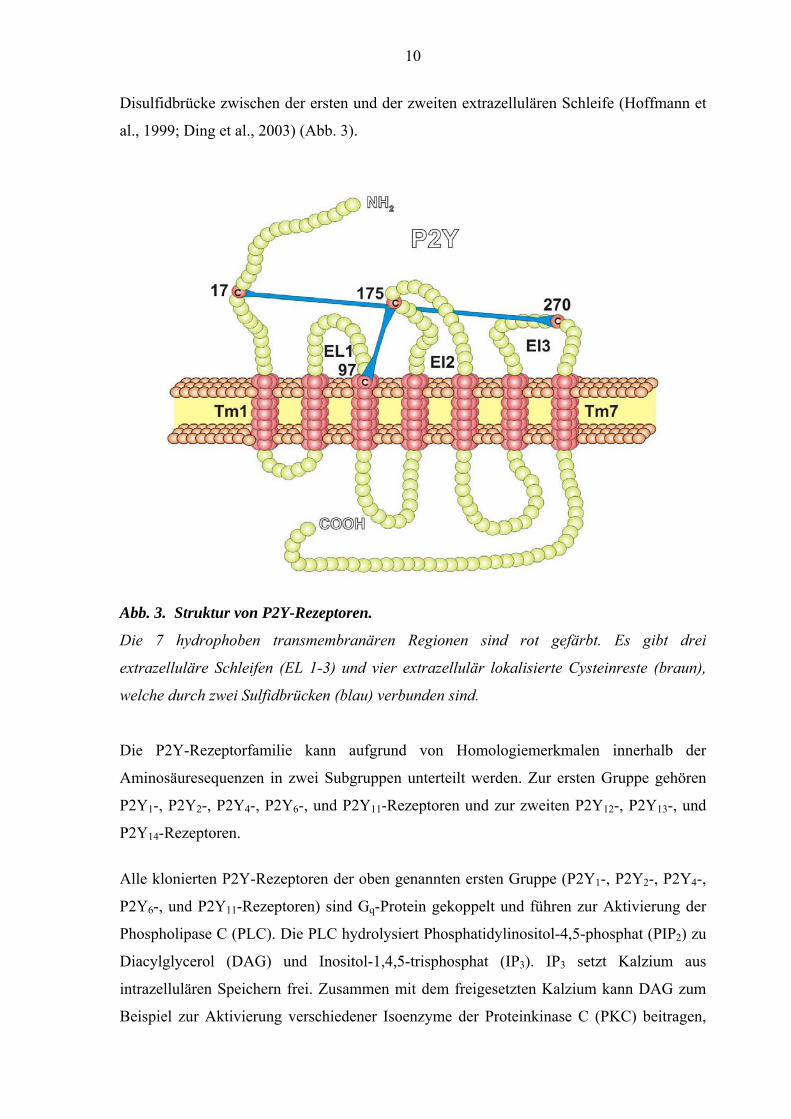
10
Disulfidbrücke zwischen der ersten und der zweiten extrazellulären Schleife (Hoffmann et
al., 1999; Ding et al., 2003) (Abb. 3).
Abb. 3. Struktur von P2Y-Rezeptoren.
Die 7 hydrophoben transmembranären Regionen sind rot gefärbt. Es gibt drei
extrazelluläre Schleifen (EL 1-3) und vier extrazellulär lokalisierte Cysteinreste (braun),
welche durch zwei Sulfidbrücken (blau) verbunden sind.
Die P2Y-Rezeptorfamilie kann aufgrund von Homologiemerkmalen innerhalb der
Aminosäuresequenzen in zwei Subgruppen unterteilt werden. Zur ersten Gruppe gehören
P2Y1-, P2Y2-, P2Y4-, P2Y6-, und P2Y11-Rezeptoren und zur zweiten P2Y12-, P2Y13-, und
P2Y14-Rezeptoren.
Alle klonierten P2Y-Rezeptoren der oben genannten ersten Gruppe (P2Y1-, P2Y2-, P2Y4-,
P2Y6-, und P2Y11-Rezeptoren) sind Gq-Protein gekoppelt und führen zur Aktivierung der
Phospholipase C (PLC). Die PLC hydrolysiert Phosphatidylinositol-4,5-phosphat (PIP2) zu
Diacylglycerol (DAG) und Inositol-1,4,5-trisphosphat (IP3). IP3 setzt Kalzium aus
intrazellulären Speichern frei. Zusammen mit dem freigesetzten Kalzium kann DAG zum
Beispiel zur Aktivierung verschiedener Isoenzyme der Proteinkinase C (PKC) beitragen,
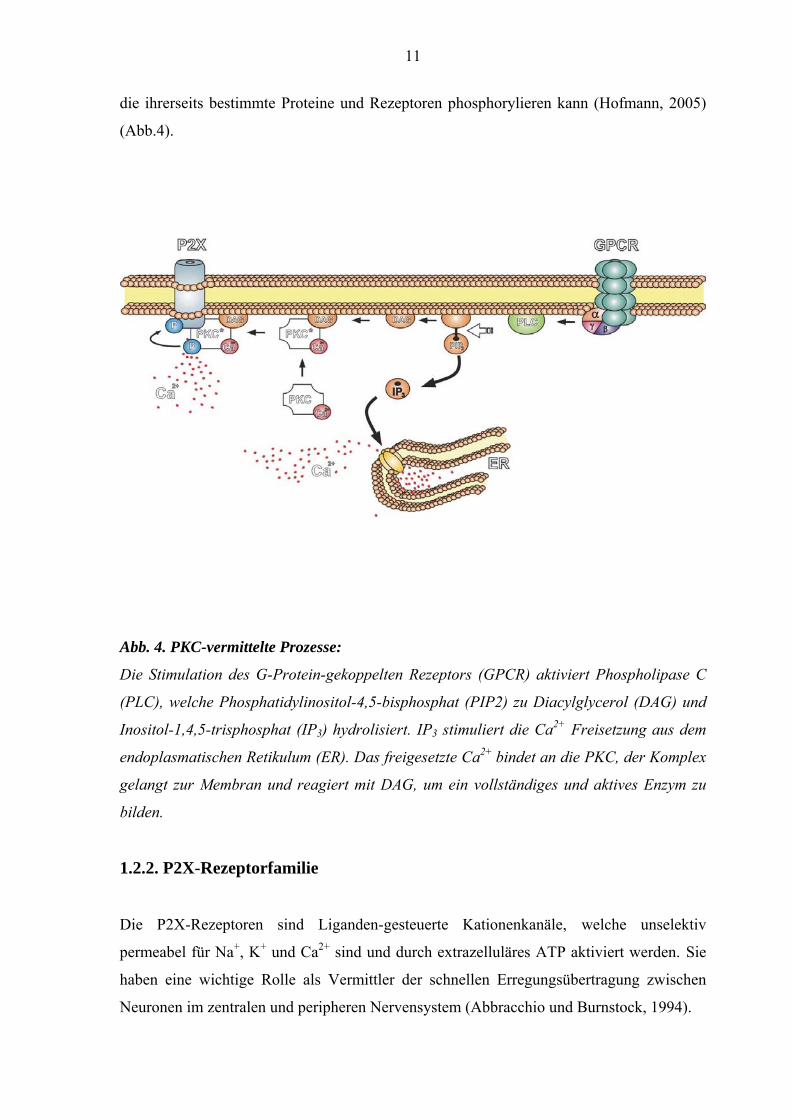
11
die ihrerseits bestimmte Proteine und Rezeptoren phosphorylieren kann (Hofmann, 2005)
(Abb.4).
Abb. 4. PKC-vermittelte Prozesse:
Die Stimulation des G-Protein-gekoppelten Rezeptors (GPCR) aktiviert Phospholipase C
(PLC), welche Phosphatidylinositol-4,5-bisphosphat (PIP2) zu Diacylglycerol (DAG) und
Inositol-1,4,5-trisphosphat (IP3) hydrolisiert. IP3 stimuliert die Ca2+ Freisetzung aus dem
endoplasmatischen Retikulum (ER). Das freigesetzte Ca2+ bindet an die PKC, der Komplex
gelangt zur Membran und reagiert mit DAG, um ein vollständiges und aktives Enzym zu
bilden.
1.2.2. P2X-Rezeptorfamilie
Die P2X-Rezeptoren sind Liganden-gesteuerte Kationenkanäle, welche unselektiv
permeabel für Na+, K+ und Ca2+ sind und durch extrazelluläres ATP aktiviert werden. Sie
haben eine wichtige Rolle als Vermittler der schnellen Erregungsübertragung zwischen
Neuronen im zentralen und peripheren Nervensystem (Abbracchio und Burnstock, 1994).

1.2.2.1. Lokalisation und Distribution
P2X-Rezeptoren haben eine relativ breite Verteilung in verschiedenen nativen Zell- und
Gewebetypen. Nörenberg und Illes (2000) berichteten, dass P2X-Rezeptoren in Neuronen
fast aller Gehirn- und Rückenmarkregionen zu finden sind. Sie können außerdem in vielen
Retinazellarten sowie autonomen und primär-sensorischen Neuronen lokalisiert werden
(Brandle et al., 1998; Jabs et al., 2000; Wheeler-Schilling et al., 2000; Dunn et al., 2001).
Zahlreiche Gliazelltypen exprimieren ebenfalls P2X-Rezeptoren, z.B. Müller-Zellen der
Retina (Pannicke et al., 2000; Neal et al., 1998; Liu et al., 1998) und Schwannzellen
peripherer Neurone (Verkhratsky et al., 2000). Sie werden außerdem noch in verschiedenen
Arten von Epithel- und Endothelzellen, im Skelettmuskel und im hämatopoetischen
Gewebe exprimiert (North, 2002).
1.2.2.2. Struktur der P2X-Rezeptoren
Sieben Subtypen von P2X-Rezeptoren (P2X1 bis P2X7) wurden bis jetzt bei Säugetieren
kloniert (Buell et al., 1996). Zusätzlich wurde noch ein anderes Mitglied dieser
Rezeptorfamilie identifiziert. Es wurde im Huhn nachgewiesen und P2X8-Rezeptor genannt
(Bo et al., 2000). Alle Subtypen haben allgemein ähnliche Hauptstrukturen (Abb. 5.).
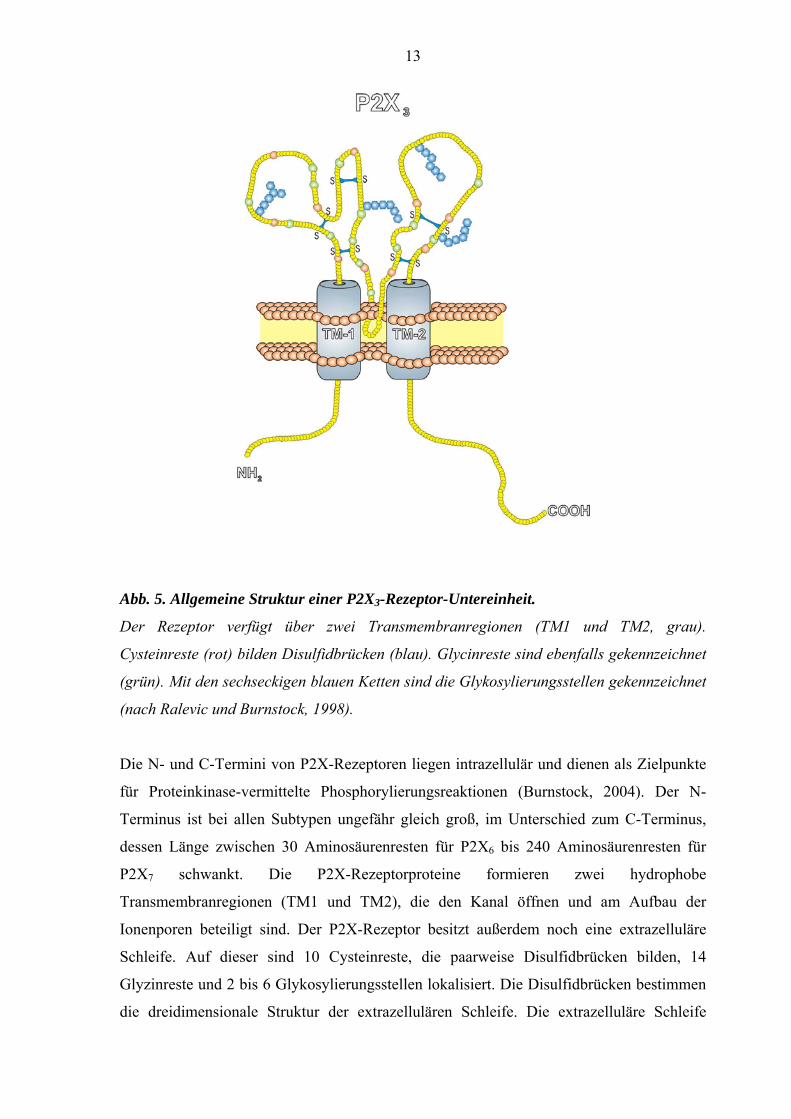
13
Abb. 5. Allgemeine Struktur einer P2X3-Rezeptor-Untereinheit.
Der Rezeptor verfügt über zwei Transmembranregionen (TM1 und TM2, grau).
Cysteinreste (rot) bilden Disulfidbrücken (blau). Glycinreste sind ebenfalls gekennzeichnet
(grün). Mit den sechseckigen blauen Ketten sind die Glykosylierungsstellen gekennzeichnet
(nach Ralevic und Burnstock, 1998).
Die N- und C-Termini von P2X-Rezeptoren liegen intrazellulär und dienen als Zielpunkte
für Proteinkinase-vermittelte Phosphorylierungsreaktionen (Burnstock, 2004). Der N-
Terminus ist bei allen Subtypen ungefähr gleich groß, im Unterschied zum C-Terminus,
dessen Länge zwischen 30 Aminosäurenresten für P2X6 bis 240 Aminosäurenresten für
P2X7 schwankt. Die P2X-Rezeptorproteine formieren zwei hydrophobe
Transmembranregionen (TM1 und TM2), die den Kanal öffnen und am Aufbau der
Ionenporen beteiligt sind. Der P2X-Rezeptor besitzt außerdem noch eine extrazelluläre
Schleife. Auf dieser sind 10 Cysteinreste, die paarweise Disulfidbrücken bilden, 14
Glyzinreste und 2 bis 6 Glykosylierungsstellen lokalisiert. Die Disulfidbrücken bestimmen
die dreidimensionale Struktur der extrazellulären Schleife. Die extrazelluläre Schleife

14
bildet noch eine hydrophobe H5-Region nahe am Porenvorhof. Diese Region ist für eine
mögliche Rezeptorregulierung durch Kationen (Magnesium, Kalzium, Zink und Kupfer-
Ionen sowie Protonen) verantwortlich. Die ATP- Bindungsstellen der extrazellulären
Schleife in der Nähe der TM1 und TM2 sind für die verschiedenen Subtypen
unterschiedlich. Die P2X1-7 Rezeptoren haben eine 30-50% identische Aminosäuresequenz
(Burnstock, 2004). Für die Stöchiometrie der P2X-Rezeptoren wurde ein Protein aus drei
Untereinheiten, die die Form eines gestreckten Trimers haben, vorgeschlagen (Khakh,
2001).
Native P2X-Rezeptoren können dabei aus gleichen (Homomere) oder aus verschiedenen
Untereinheiten (Heteromere) aufgebaut sein. Die heteromeren P2X-Rezeptoren
unterscheiden sich von den homomeren P2X-Rezeptoren in ihren pharmakologischen
Eigenschaften.
1.2.2.3. Physiologische Eigenschaften
Universaler Agonist für die P2X-Rezeptoren ist ATP. Mehr oder weniger spezielle
Agonisten sind α,β-Methylen ATP (α,ß-meATP), UTP, ADP, 2-Methylthio ATP
(2MeSATP), 2',3'-(Benzoyl-4-benzoyl)-ATP (BzATP) und CTP. Die verschiedenen
Agonisten besitzen eine unterschiedlich starke Wirkpotenz an den einzelnen P2X-
Rezeptortypen.
Native P2X-Rezeptoren können über die Reihenfolge der Agonistenwirksamkeit, durch
selektive Antagonisten und anhand ihrer Zink- und Protonen-Empfindlichkeit
charakterisiert werden. Allerdings stehen subtypselektive Antagonisten von P2X-
Rezeptoren immer noch nicht in ausreichendem Ausmaß zur Verfügung; Suramin und
Pyridoxal-5’-phosphat-6-azophenyl-2’,4’-disulfonsäure (PPADS) sind unselektive
Antagonisten.
Wie schon erwähnt, sind die P2X-Rezeptoren durchlässig für positiv geladene monovalente
und bivalente Ionen. Manche Rezeptoren sind auch für größere Moleküle (wie Propidium
Iodid) permeabel (z.B. P2X7; Di Virgilio, 1995).
P2X-Rezeptortypen desensibilisieren in unterschiedlichem Maße. Bei manchen Rezeptoren
entwickelt sich eine vollständige Desensibilisierung innerhalb einiger Millisekunden (P2X1
und P2X3), bei anderen Rezeptoren ist dieser Vorgang 100-1000-mal langsamer (P2X2 und
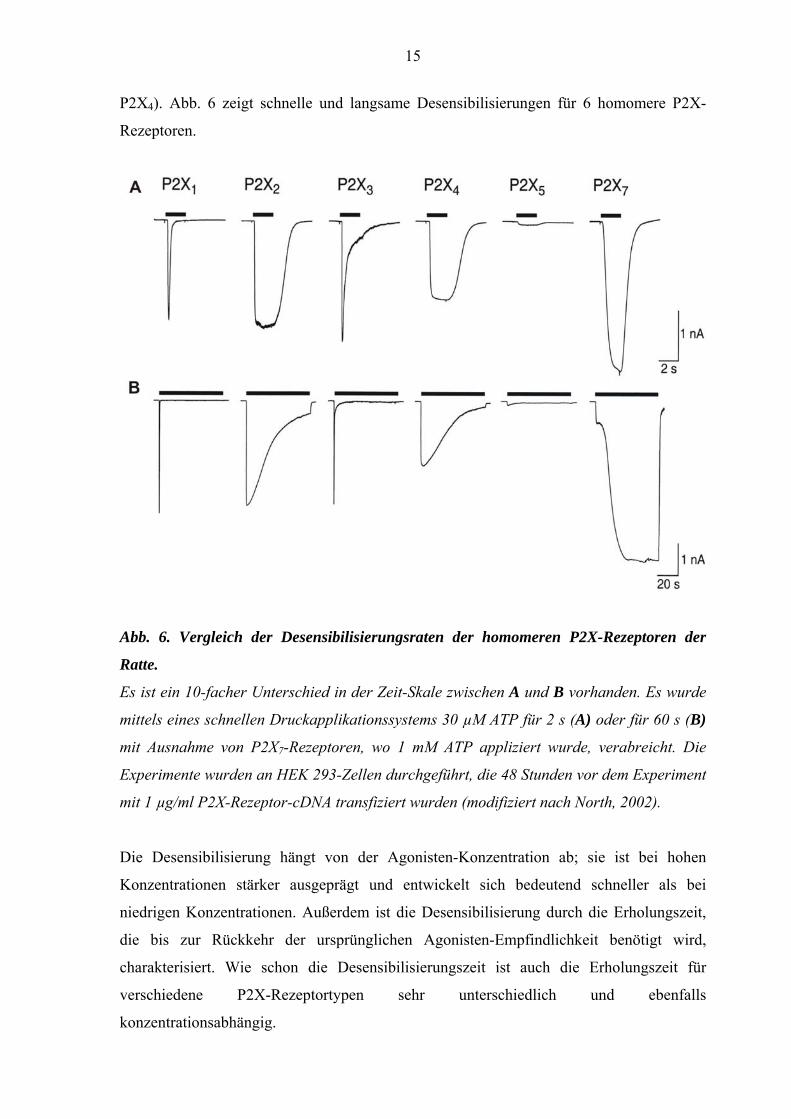
15
P2X4). Abb. 6 zeigt schnelle und langsame Desensibilisierungen für 6 homomere P2X-
Rezeptoren.
Abb. 6. Vergleich der Desensibilisierungsraten der homomeren P2X-Rezeptoren der
Ratte.
Es ist ein 10-facher Unterschied in der Zeit-Skale zwischen A und B vorhanden. Es wurde
mittels eines schnellen Druckapplikationssystems 30 µM ATP für 2 s (A) oder für 60 s (B)
mit Ausnahme von P2X7-Rezeptoren, wo 1 mM ATP appliziert wurde, verabreicht. Die
Experimente wurden an HEK 293-Zellen durchgeführt, die 48 Stunden vor dem Experiment
mit 1 µg/ml P2X-Rezeptor-cDNA transfiziert wurden (modifiziert nach North, 2002).
Die Desensibilisierung hängt von der Agonisten-Konzentration ab; sie ist bei hohen
Konzentrationen stärker ausgeprägt und entwickelt sich bedeutend schneller als bei
niedrigen Konzentrationen. Außerdem ist die Desensibilisierung durch die Erholungszeit,
die bis zur Rückkehr der ursprünglichen Agonisten-Empfindlichkeit benötigt wird,
charakterisiert. Wie schon die Desensibilisierungszeit ist auch die Erholungszeit für
verschiedene P2X-Rezeptortypen sehr unterschiedlich und ebenfalls
konzentrationsabhängig.

1.2.3. P2X3-Rezeptor
1.2.3.1. Lokalisation
Die Lokalisation des P2X3-Rezeptors in den sensorischen Neuronen erzeugte erhebliches
Interesse für die Rolle dieses Rezeptors im Schmerzgeschehen (Burnstock, 2000; Kowaluk
und Jarvis, 2001). Der P2X3-Rezeptor wurde zum ersten Mal aus Hinterwurzelganglien
(„dorsal root ganglion“, DRG) der Ratte kloniert (Chen et al., 1995; Lewis, 1995). Die
P2X3-cDNA weiterer Spezies wurde noch aus dem menschlichen Herzen (Garcia-Guzman
et al., 1997) und aus dem Zebrafisch (Boué-Grabot et al., 2000; Egan et al., 2000) isoliert.
Im DRG werden P2X3-Rezeptoren in hoher Anzahl in kleinen und mittleren Neuronen,
welche in die Schmerzperzeption involviert sind, exprimiert. Die nozizeptiven Neuronen
besitzen sowohl homomere P2X3-Rezeptoren als auch heteromere P2X2/3-Rezeptoren
(Lewis et al., 1995; Burgard et al., 1999). Es wurde beschrieben, dass der homomere P2X3-
Rezeptor vorwiegend in kleineren sensorischen Neuronen exprimiert wird, während der
heteromere P2X2/3-Rezeptor in sensorischen Neuronen mittlerer Größe vorkommt (Ueno et
al., 1999; Cockayne et al., 2000). Außerdem gibt es Beweise für das Vorhandensein von
P2X3-Rezeptoren in Rückenmarkneuronen, sympathischen und parasympathischen
Neuronen und enterischen Neuronen.
1.2.3.2. Struktur
Die Aminosäuresequenz des P2X3-Rezeptors besitzt 43% Homologie mit der des P2X1-
Rezeptors und 47% Homologie mit der des P2X2-Rezeptors (Ralevic und Burnstock,
1998). Die räumliche Struktur des P2X3-Rezeptors ist noch nicht genau bekannt, es wird
jedoch vermutet, dass er wie alle anderen P2X-Rezeptoren aus drei Untereinheiten besteht
(North, 2002). Das Modell einer Untereinheit des humanen P2X3-Rezeptors ist kürzlich
von unserer Arbeitsgruppe publiziert worden (siehe Abb. 7, Wirkner et al., 2005).
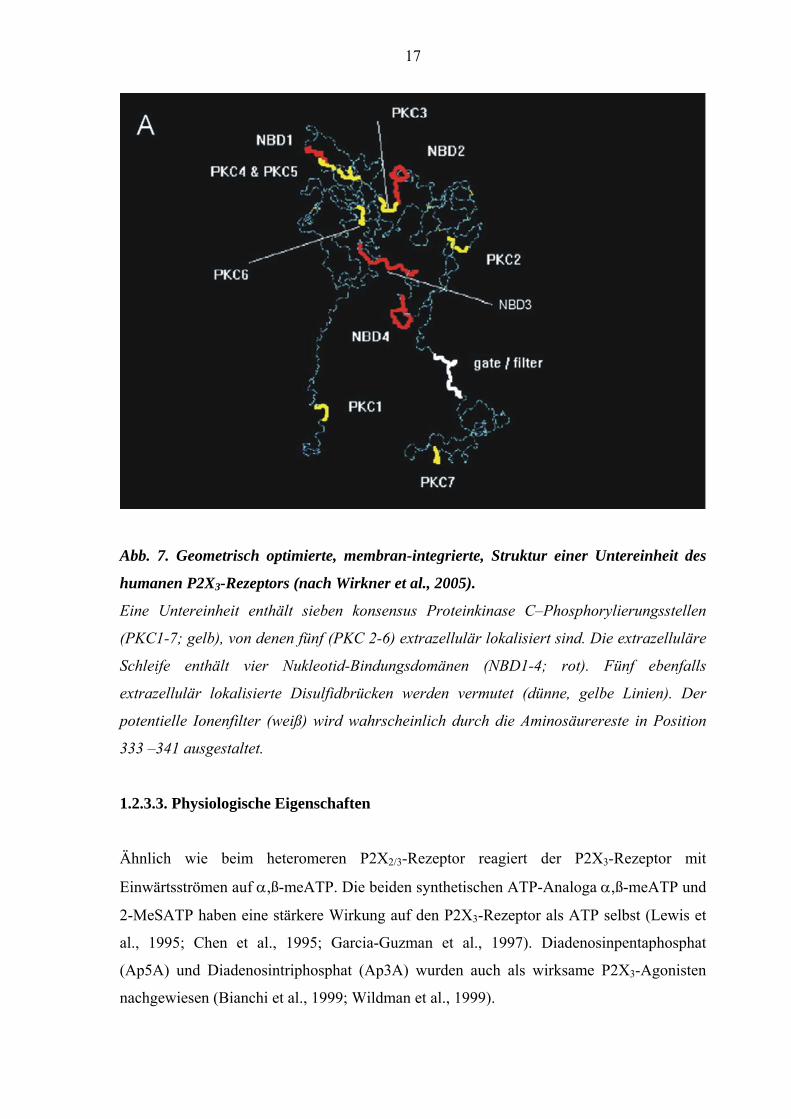
17
Abb. 7. Geometrisch optimierte, membran-integrierte, Struktur einer Untereinheit des
humanen P2X3-Rezeptors (nach Wirkner et al., 2005).
Eine Untereinheit enthält sieben konsensus Proteinkinase C–Phosphorylierungsstellen
(PKC1-7; gelb), von denen fünf (PKC 2-6) extrazellulär lokalisiert sind. Die extrazelluläre
Schleife enthält vier Nukleotid-Bindungsdomänen (NBD1-4; rot). Fünf ebenfalls
extrazellulär lokalisierte Disulfidbrücken werden vermutet (dünne, gelbe Linien). Der
potentielle Ionenfilter (weiß) wird wahrscheinlich durch die Aminosäurereste in Position
333 –341 ausgestaltet.
1.2.3.3. Physiologische Eigenschaften
Ähnlich wie beim heteromeren P2X2/3-Rezeptor reagiert der P2X3-Rezeptor mit
Einwärtsströmen auf α,ß-meATP. Die beiden synthetischen ATP-Analoga α,ß-meATP und
2-MeSATP haben eine stärkere Wirkung auf den P2X3-Rezeptor als ATP selbst (Lewis et
al., 1995; Chen et al., 1995; Garcia-Guzman et al., 1997). Diadenosinpentaphosphat
(Ap5A) und Diadenosintriphosphat (Ap3A) wurden auch als wirksame P2X3-Agonisten
nachgewiesen (Bianchi et al., 1999; Wildman et al., 1999).

18
Unspezifische Antagonisten für den P2X3-Rezeptor sind Suramin, PPADS und TNP-ATP
(North et al., 2000). A-317491 ist ein wirksamer und selektiver Antagonist für P2X3- und
P2X2/3-Rezeptoren (Torben et al., 2003). Der heteromere P2X2/3-Rezeptor wurde besser
durch TNP-ATP blockiert als durch Suramin und PPADS (Burgard et al., 2000; Thomas et
al., 1998; Virginio et al. 1998; Spelta et al., 2002). Di-Inosinpentaphosphat (Ip5I) blockiert
viel stärker P2X3-Rezeptoren als heteromere P2X2/3-Rezeptoren, was nützlich sein kann
beim Unterscheiden zwischen diesen beiden Rezeptoren (Dowd et al., 1998; Liu et al.,
2001).
Homomere P2X3- und heteromere P2X2/3-Rezeptoren werden außerdem noch durch
verschiedene Ionen gesteuert, was für das Bestimmen des Rezeptorentyps wichtig ist. Die
H+-Ionen inhibieren die agonisteninduzierten Ströme am P2X3-Rezeptor und potenzieren
dieselben Ströme am heteromeren P2X2/3-Rezeptor (Liu et al., 2001; Stoop et al., 1999).
Höhere Konzentrationen an Ca2+-Ionen inhibieren den heteromeren P2X2/3-Rezeptor und
haben keinen Einfluss auf den homomeren P2X3-Rezeptor (Virginio et al., 1998).
Der homomere P2X3-Rezeptor ist selektiv durchlässig für Na+-, K+- und Ca2+-Ionen. Die
relative Permeabilität gegenüber Ca2+- im Verhältnis zu der gegenüber Na+-Ionen (PCa/PNa)
beträgt ~ 1,2 (Virginio et al., 1998). Die Kalzium-Permeabilität von P2X2/3- und P2X3-
Rezeptoren ist vergleichbar. Eine zeitabhängige Leitfähigkeitszunahme für N-methyl-D-
glukamin (NMDG) wurde nur am heteromeren P2X2/3-Rezeptor beobachtet (Khakh et al.,
1998).
Die Desensibilisierungseigenschaften des schnell aktivierenden und schnell
desensibilisierenden P2X3-Rezeptors sind von großer physiologischer Bedeutung, da sie
das Empfinden eines kurzandauernden, schmerzhaften Stimulus ermöglichen, ohne den
Organismus durch intensive, langandauernde Schmerzen zu belasten.
Kleinere Konzentrationen (30-300 nM) von ATP induzieren Ströme, die über einige
Sekunden persistieren können, bei höheren ATP Konzentrationen (≥30 µM) beobachtet
man jedoch eine schnelle Desensibilisierung (Lewis et al., 1995). Im Gegensatz zum
homomeren P2X3-Rezeptor desensibilisiert der heteromere P2X2/3-Rezeptor nicht (Collo et
al., 1996; Burgard et al., 1999). Insofern ähneln sich P2X2/3- und P2X2-Rezeptoren trotzt
unterschiedlicher pharmakologischer Eigenschaften. Die verzögerte Erholungszeit von
P2X3 sollte bei repetitiven Agonisten-Applikationen berücksichtigt werden; der
Applikationsabstand von α,β-meATP (3 μM) muss mindestens 4-5 min betragen, um

19
stabile Stromantworten erhalten zu können (Wirkner et al., 2005). Die Rückkehr des P2X3-
Rezeptors aus der Desensibilisierung konnte mit einer steigenden Konzentration von
extrazellulärem Kalzium deutlich beschleunigt werden. Bei 1 mM Kalzium ist die
vollständige Erholungszeit 7 min und bei 10 mM Kalzium verringerte sich diese Zeit auf
3,5 min (North, 2002).
Interaktionen zwischen P2X3-Rezeptor und anderen Rezeptoren, die sich an DRG-
Neuronen befinden und dort im Schmerzgeschehen involviert sind, wurden in den letzten
Jahren wiederholt beschrieben. Piper und Docherty (2000) berichteten über eine negative
Interaktion zwischen dem heteromeren P2X2/3-Rezeptor und dem Capsaicin-, Hitze- und
pH-sensitiven Vanilloidrezeptor (TRPV1). Eine ähnlich geartete negative Interaktion
wurde von Sokolova et al. (2001) für P2X3- und GABAA-Rezeptoren beschrieben. Eine
Wechselwirkung zwischen P2X3- und P2Y1-Rezeptoren wurde von Gerevich et al. (2005)
beobachtet. Die P2Y1-Rezeptoragonisten ADP-ß-S und 2-meSADP hemmten die α,ß-
meATP-induzierten Ströme in DRG Zellen. Dieser Effekt von α,ß-meATP wird durch
intrazelluläre Applikation des G-Protein-Antagonisten GDP-ß-S aufgehoben, als Hinweis
dafür, dass G-Proteine am Transduktionsmechanismus des P2Y-Rezeptors beteiligt sind.
Zur Aufklärung der Rolle des P2X3-Rezeptors an der Nozizeption wurden selektive
Rezeptorantagonisten, Antisense-Oligonukleotide und kleine interferierende RNA bzw.
P2X3-Rezeptor-defiziente Mäuse verwendet.
Aus dem Hinweis, dass ATP bei Entzündungen freigesetzt wird, stellt sich die Frage, ob
ATP mit anderen Faktoren der Entzündung interagiert und wie sich dies auf die DRG-
Zellen auswirkt. Xu und Huang (2002) zeigten, dass die ATP-induzierten Antworten von
DRG Zellen, die aus Ratten eines Entzündungsmodells entnommen wurden, zwei- bis
dreimal größer sind als die ATP-induzierten Ströme von DRG Zellen, die von
unbehandelten Ratten stammen. Das deutet auf eine Hochregulierung der P2X3- und
P2X2/3-Proteine hin, die durch einen Western-Blot tatsächlich nachgewiesen werden
konnte. Substanz P und Bradykinin sind potentielle Entzündungsmediatoren. Die durch
diese Substanzen aktivierten Rezeptoren können eine Steigerung von ATP-induzierten
Strömen in Oozyten, die P2X3- und P2X2/3-Rezeptoren exprimieren, bewirken. Ursache
war wahrscheinlich eine Phosphorylierung am N-Terminus des Rezeptors durch PKC
(Paukert et al., 2001). Es ist schon seit langer Zeit bekannt, dass die Leitfähigkeit mancher
ionotroper Aminosäurerezeptoren durch verschiedene Proteinkinasen moduliert wird (Moss

20
und Smart, 1996; Swope et al., 1999; Köles et al., 2001). Obwohl diese
Phosphorylierungseffekte am häufigsten an der intrazellulären Seite der Rezeptoren
beobachtet wurden, wurde der Teilnahme von Ektoproteinkinasen an einer
Phosphorylierung der extrazellulären Rezeptorseite ebenfalls mehrmals diskutiert
(Wieraszko und Ehrlich, 1994; Ehrlich und Kornecki, 1999). So könnte insbesondere
intrazelluläres aber auch extrazelluläres ATP, seine klassische Funktion als Phosphatdonor
ausüben.
1.2.4. P2X7-Rezeptor
1.2.4.1. Lokalisation
Die cDNA, die den Ratten-P2X7-Rezeptor kodiert, wurde zum ersten Mal aus
überdeckenden Fragmenten des oberen Halsganglions und der medialen Habenula des
Gehirns hergestellt; die komplette cDNA wurde danach aus Rattenhirn isoliert (Surprenant
et al., 1996). Menschliche (Rassendren et al., 1997) und Mäuse (Chessell et al., 1998)
cDNA wurde aus Monozyten bzw. aus mikroglialen Zellen kloniert. Der P2X7-Rezeptor
wird darüber hinaus in humanen Müllerzellen (Pannicke et al., 2000), Astrozyten (John et
al., 2001), Epithelzellen der Atemwege (Korngreen et al., 1998), azinösen
Ohrenspeicheldrüsenzellen (Alzola et al., 1998; Soltoff et al., 1993), Duktuszellen der
exokrinen Pankreas (Hede et al., 1999; Luo et al., 1999), Hypophysenzellen (Chung et al.,
2000), humanen Hautfibroblasten (Solini et al., 1999), Osteoklasten (Gartland et al., 2001),
Vena saphena-Zellen (Cario-Toumaniantz et al., 1998; Loirand et al., 1995),
Glattmuskelzellen (Ugur et al., 1997; Zou et al., 2001), Makrophagen (North, 2002) und
Lymphozyten (Sluyter et al., 2001) exprimiert.
1.2.4.2. Struktur
Der P2X7-Rezeptor ist strukturell den anderen P2X-Untereinheiten ähnlich, jedoch
unterscheidet ihn sein längerer C-Terminus (Abb. 8.).
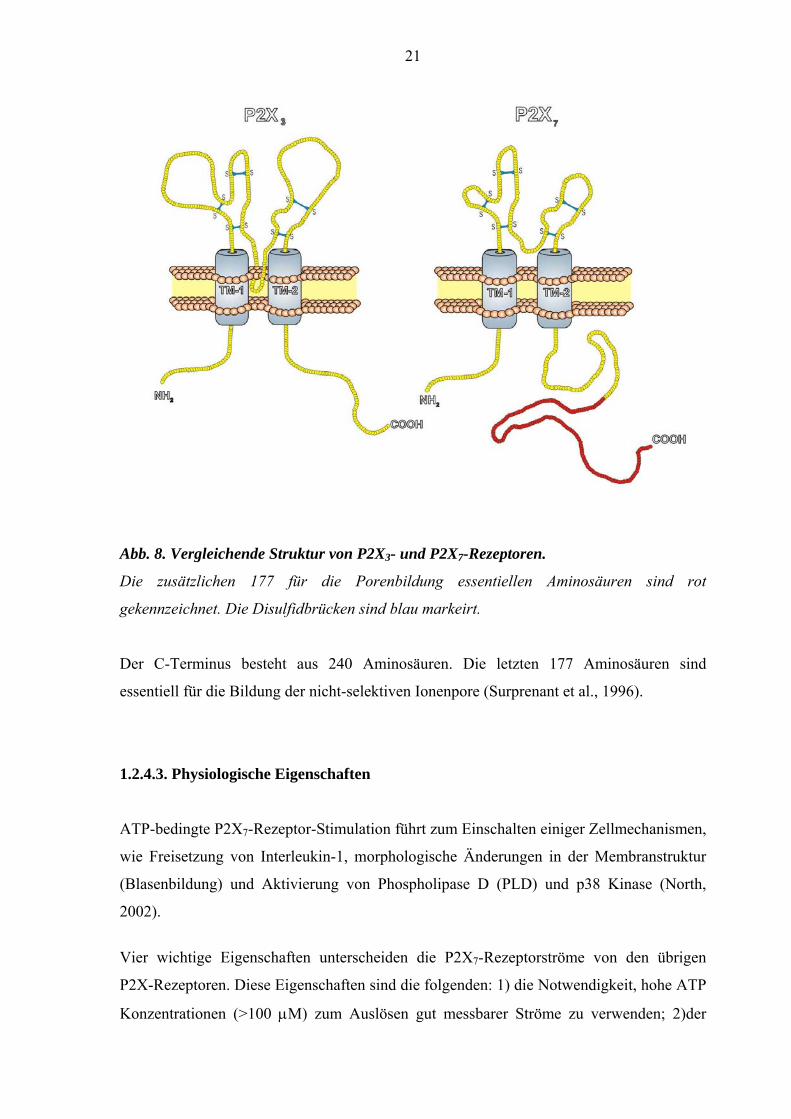
21
Abb. 8. Vergleichende Struktur von P2X3- und P2X7-Rezeptoren.
Die zusätzlichen 177 für die Porenbildung essentiellen Aminosäuren sind rot
gekennzeichnet. Die Disulfidbrücken sind blau markeirt.
Der C-Terminus besteht aus 240 Aminosäuren. Die letzten 177 Aminosäuren sind
essentiell für die Bildung der nicht-selektiven Ionenpore (Surprenant et al., 1996).
1.2.4.3. Physiologische Eigenschaften
ATP-bedingte P2X7-Rezeptor-Stimulation führt zum Einschalten einiger Zellmechanismen,
wie Freisetzung von Interleukin-1, morphologische Änderungen in der Membranstruktur
(Blasenbildung) und Aktivierung von Phospholipase D (PLD) und p38 Kinase (North,
2002).
Vier wichtige Eigenschaften unterscheiden die P2X7-Rezeptorströme von den übrigen
P2X-Rezeptoren. Diese Eigenschaften sind die folgenden: 1) die Notwendigkeit, hohe ATP
Konzentrationen (>100 μM) zum Auslösen gut messbarer Ströme zu verwenden; 2)der

22
Befund, dass BzATP eine 10- bis 30-mal stärkere Wirkung als ATP besitzt; 3) die
Tatsache, dass der Effekt von ATP und BzATP durch Reduzierung der extrazellulären
Konzentration von Ca2+ und Mg2+ verstärkt wird (Surprenant et al., 1996); 4) der Befund,
dass die Ströme auffällige Änderungen in ihrer Kinetik und Amplitude nach wiederholter
Applikationen desselben Agonisten aufweisen können.
Die Antagonisten können in vier verschiedene Gruppen gegliedert werden: 1) Ionen, wie
Kalzium, Magnesium, Zink, Kupfer und Protonen inhibieren P2X7-Rezeptorströme. 2) Die
Ströme sind gegenüber den üblichen, unselektiven P2X-Rezeptorantagonisten Suramin und
PPADS unempfindlich (Surprenant et al., 1996). Das Suraminanalogon NF279 ist
wirkungsvoller als Suramin und PPADS (Klapperstuck et al., 2000). Der humane P2X7-
Rezeptor zeigt eine größere Empfindlichkeit gegenüber PPADS (Michel et al., 2000) und
oxidiertem ATP als der entsprechende Rezeptor der Ratte. Der effektivste Antagonist
dieser Substanzgruppe ist jedoch Brilliant Blue G, das am Ratten P2X7-Rezeptor mit einer
hohen Affinität und Wirksamkeit bindet (Jiang et al., 2000). 3). Die dritte Gruppe der
P2X7-Inhibitoren enthält zwei große organische Kationen, Kalmidazolium und KN-62. 4)
Zur vierten Gruppe der P2X7-Antagonisten zählt 17ß-Estradiol.
Bei kurzdauernder Agonistenapplikation zeigt der P2X7-Rezeptorkanal eine schwache
NMDG Permeabilität, die bei längeren Applikationszeiten jedoch markant zunimmt
(Surprenant et al., 1996; Virginio et al., 1999). Die Zeitkonstante der
Permeabilitätssteigerung (PX/PNa) steigt von ~1 s für Dimethylamin, 4 s für Tris, bis 10 s
für NMDG an. Sogar wenn die Pore NMDG-permeabel ist, bleibt ihre Selektivität für
Kationen erhalten (Virginio et al., 1999). Die BzATP Konzentrationen, die für das Öffnen
des Kanals notwendig sind, entsprechen denen, die für die Porenöffnung benötigt werden.
Die Konzentrations-Wirkungskurve von BzATP bzgl. der Porenöffnung, ist im Bereich von
0,3 bis 30 µM BzATP sehr steil (Virginio et al., 1999). Die Permeabilitätsmessungen
wurden unter Abwesenheit von zweiwertigen Ionen durchgeführt, ohne extrazelluläres
Kalzium oder Magnesium dem Inkubationsmedium hinzuzufügen. Divalente Kationen (1
mM Magnesium, 2 mM Kalzium) beeinflussen die Zuname der Permeabilität für NMDG
negativ (Virginio et al., 1999). Die Permeabilität konnte auch durch genetische
Veränderungen modifiziert werden. Durch eine Deletion der Aminosäurenreste 419 bis 595
wurde die Aufnahme des Fluoreszenzfarbstoffes YO-PRO-1 stark reduziert (Surprenant et
al., 1996).

23
Die Einwärtsströme in HEK 293-Zellen, die durch ATP oder BzATP induziert wurden,
zeigten keine Desensibilisierung während der mehrere Minuten andauernden Applikation
(Abb. 6).
1.2.4.4. Membranporenbildung, morphologische Änderungen und Einfluss von
Hypoxie auf den P2X7-Rezeptor
ATP oder BzATP induzierten erhebliche Veränderungen in mit dem P2X7-Rezeptor
transfizierten HEK 293-Zellen (Mackenzie et al., 2001; Virginio et al., 1999). Nach ~30 s
kontinuierlicher BzATP (30 µM)-Applikation begann die Zellmembran große Blasen zu
bilden. Nach 1-2 Minuten entwickelten sich mehr Blasen, die sich auch teilweise wieder
vereinigen. Eine wichtige Frage war, ob diese Eigenschaften P2X7-spezifisch sind oder ob
sie zusätzliche Moleküle von der Gast/Mutter-Zelle benötigen.
Die Membranblasenbildung, die bei der Aktivierung des P2X7-Rezeptors auftritt, wurde bei
keinem anderen Mitglied der Familie beobachtet und scheint auf abwärts gerichtete
Signaltransduktionsmechanismen hinzudeuten, die keinen Einfluss auf die Bewegung der
Ionen durch die Membran haben. Punktmutationen, die selektiv den Ionenfluß und die
Membranblasenbildung inhibieren, sollten in Zukunft verstärkt untersucht werden.
Folge einer P2X7-Rezeptoraktivierung kann der Zelltod sein. Die Literatur ist hier, wegen
der unterschiedlichsten methodischen Ansätze, die verwendet werden, extrem
unübersichtlich. Zum Beispiel wird die Aufnahme von YO-PRO-1, als Nachweis für den
Zelltod verwendet, da angenommen wird, dass über die sich ausbildenden großen
Membranporen Zellbestandteile nach außen strömen und den Zelluntergang einleiten
könnten. Es ist aber anzumerken, dass Zellen die den P2X7-Rezeptor exprimieren, YO-
PRO-1 wiederholt aufnehmen können und, dass die Zellen zu diesem Zeitpunkt noch
lebensfähig sind (Virginio et al., 1999). Das Ausmaß der Laktatdehydrogenaseaktivität im
Zellmedium kann als verlässlicherer Marker für den Zelltod verwendet werden. Der Zelltod
von mit P2X7-Rezeptor transfizierten HEK 293-Zellen tritt aber erst nach minutenlanger
Applikation von BzATP auf (Mackenzie et al., 2001; Virginio et al., 1999).
Sauerstoff- und Glukosemangel verursachen in vitro und in vivo eine Überempfindlichkeit
der neuronalen P2X7-Rezeptoren (Cavaliere et al., 2002; Wirkner et al., 2005) und eine

24
Verstärkung der entsprechenden Rezeptorexpression (Cavaliere et al., 2004; Franke et al.,
2004).
1.3. Aufgaben und Zielstellungen
Ziel dieser Arbeit war es, das Wissen über zwei der wichtigsten Rezeptoren der P2X-
Rezeptorfamilie, P2X3- und P2X7-Rezeptoren, zu erweitern. Vorhergehende
Untersuchungen unserer Arbeitsgruppe zeigten, dass die Leitfähigkeit schmerzrelevanter
P2X3-Rezeptoren durch niedrige Konzentrationen von UTP, welche jedoch selbst noch
nicht zur Aktivierung von P2X3-Rezeptoren führte, gesteigert werden konnte. Dieser Effekt
wurde nur dann beobachtet, wenn endogen auf HEK293-Zellen lokalisierte P2Y-
Rezeptoren pharmakologisch ausgeschaltet wurden.
In der vorliegenden Arbeit sollte mit Hilfe der Patch-Clamp-Technik der Mechanismus, der
dieser Interaktion zugrunde liegt, aufgeklärt werden. Es interessierte natürlich auch, ob
neben UTP, andere Nukleotide ebenfalls P2X3-Rezeptor-vermittelte Ströme potenzieren
können.
Ausgehend vom Erkenntnisstand, dass die Leitfähigkeit verschiedener Rezeptoren vor
allem durch intrazelluläre Phosphorylierungsreaktionen moduliert werden kann, sollten
unselektive und selektive Proteinkinaseaktivatoren und -inhibitoren untersucht werden.
Darüber hinaus kamen Rezeptormutanten zum Einsatz, bei denen konsensus PKC-Stellen
durch Punktmutationen eliminiert wurden. Um eine mögliche physiologische Relevanz
dieser im Expressionsmodell der HEK293-Zelle beobachteten Befunde zu testen, wurden
auch Versuche an Hinterwurzelganglienzellen der Ratte, welche ebenfalls P2X3-
Rezeptoren exprimieren, durchgeführt.
Ergebnisse unserer Arbeitsgruppe an primären kortikalen Neuronen wiesen auf eine
verstärkte Expression des endogenen P2X7-Rezeptors nach in-vitro-Ischämie hin (Wirkner
et al., 2005). Ausgehend von diesen Befunden sollte an einem Zellmodell der Einfluss
ischämischer Bedingungen auf den intrazellulären Transport bzw. auf die
Membranexpression des P2X7-Rezeptors sowie ausgewählter P2X7-Rezeptormutanten
aufgeklärt werden. Dazu sollte zunächst ein C-terminal mit EGFP gekoppelter P2X7-
Rezeptor kloniert und ein daraus resultierender Fusionsprotein-kodierender-Vektor (P2X7-
EGFP) hergestellt werden, welcher transient in HEK293-Zellen eingebracht werden

25
konnte. Dieses heterologe Expressionssystem ermöglichte die Lokalisierung des P2X7-
Rezeptors innerhalb der Zelle, sowie die Verfolgung des Rezeptorprotein-Transportes
(„Rezeptor-Trafficking“) innerhalb der Zelle. In der vorliegenden Arbeit sollte
elektrophysiologisch geklärt werden, ob die hergestellten Mutanten funktionell aktive
Rezeptoren bilden und inwieweit ischämische Bedingungen die Funktionsfähigkeit dieser
Rezeptoren verändern.
2. Materialen und Methoden
2.1. Chemikalien
α,ß-Methylen-adenosin-5´-triphosphat α,ß-meATP Biotrend
12-(2-Cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-
5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole
Gö 6976 Calbiochem
2′-3′-O-(4-Benzoylbenzoyl)adenosin 5′-triphosphat
triethylammoniumsalz
BzATP Sigma
6-N,N-Diethyl-b-g-dibromomethylen-D-adenosin-5-
triphosphat
ARL 67156 Sigma
7-Deaza-7-carbamoyladenosinhydrat Sangivamycin Sigma
Adenosin-5´-triphosphat ATP Sigma
Agarose Gibco
Chloroform
Diacylglycerollacton DAG-Lakton Merck
Dimethylsulfoxid DMSO Calchemie
DNA Größenstandard (1kb) Sigma
DNA - Ladepuffer Sigma
Desoxynucleotidtriphosphat dNTPs Sigma
Dulbecco’s Modifiziertes Eagle Medium DMEM Sigma
Essigsäure (100%) Gibco
Ethanol (100%) Merck
Ethidiumbromid (EtBr, 10mg/ml) EtBr Merck
Ethylendiaminotetraessigsäure EDTA Sigma
Ethylenglycol-bis(2-aminoethylether)-N,N,N´,N´-
tetraessigsäure
EGTA Sigma
Fötales Kälberserum FKS Sigma
Geneticin Seromed
Gentamycin Gibco
Glycerol (100%) Gibco
Guanosin 5´O-(2-thiodiphosphat) GDP-ß-S Sigma
Isopropanol (100%) Sigma
27
Kanamycin Merck
LB Agar Gibco
L-Glutamin Gibco
N-2-hydroxyethylpiperazin-N´-ethansulfonsäure HEPES Sigma
Natriumacetat Sigma
Nichtessentielle Aminosäurelösung NEAA Sigma
Phorbol 12-myristat 13-acetat PMA Gibco
Physiologische Salzlösung nach Hank HBSS Sigma
Polyfect (4 x 1ml) Gibco
QIAGEN Plasmid Kits QIAGEN
Rinderserumalbumin BSA Sigma
Restriktionsenzyme + -puffer QIAGEN
T4 DNA Ligase BioLabs
T4 DNA Puffer Promega
Tetrodotoxin TTX Promega
Tetraethylamonium TAE Sigma
Tris-HCl Biotrend
Uridin-5´-triphosphat UTP Sigma
Q-Solution QIAGEN
pEGFP-N1 Clonetech
Ethidium Bromid EtBr Sigma
Taq Polymerase Eppendorf
Ligase Puffer Roche
Trypton Sigma
Natriumchlorid NaCl Merck
Hefeextrakt Sigma
Salzsäure HCl Merck
LB-Medium Gibco
Polymerase Puffer Eppendorf
Oligonukleotide Roche
D-Glucose Merck
Magnesiumchlorid MgCl2 Merck
Kalziumchlorid CaCl2 Merck

28
Cesiumchlorid CsCl Sigma
Cesiumhydroxid CsOH Sigma
2.2. Material und Methoden zu den P2X7-Rezeptorexperimenten
2.2.1. Material
Zelllinien
HEK293 (Human Embryonic Kidney Cells)
Oligonukleotide
X7GFP-for: 5’-CTA TAC TGC AGA ATT CAT GCC GGC CTG CTG CAG C-3’
X7GFP-rev: 5’-ATG TAG GTA CCC AGT AAG GAC TCT TGA AGC C-3’
Vektoren
pEGFP-N1 (Clonetech, Heidelberg), DVektor zur Expression von C-terminal gekoppelten -
EGFP Fusionsproteinen in eukaryotischen Zellen. Bei EGFP handelt es sich um eine
Variante von GFP, deren Emissionsmaximum in den längerwelligen Bereich verschoben ist
und die für hellere Fluoreszenz optimiert wurde.
Geräte
Brutschrank für Zellkultur Heraeus Kelvitron, Hanau, D
Brutschrank für Klonierung Heraeus Kelvitron, Hanau, D
Schüttler Gerhardt, Bonn, D
Elektrophoresekammer Amersham Pharmacia Biotech AB,
USA
Geldokumentationssystem, Raytest, Straubenhardt, D
Thermomixer Eppendorf, Hamburg, D
Thermocycler, PTC 200 Biozym Diagnostik GMBH, Oldendorf,
D
Photospektrometer Eppendorf, Hamburg, D
pH-Messgerät inoLab, Wellheim, D

29
Wasserbad,GFL Burgwedel, D
Minizentrifuge Eppendorf, Hamburg, D
Magnetrührer, Heidolph MR 3002 Kelheim, D
Neubauer-Zählkammer, 0,0025mm2 Laborcenter, Nürnberg
Spannungsquelle Amersham Pharmacia Biotech AB,
USA
Vortexer: Heidolph, Kelheim D
Waagen, Mettler AT 261 Delta range Toledo, Schweiz
CO2-Gasflasche Linde AG, Höllriegelskreuth, D
Deionisierungsanlage: MilliQ Millipore, Eschborn, D
2.2.2. Methoden
2.2.2.1. Nukleinsäure-Methoden
Isolierung von Plasmid-DNA
Die Präparation von Plasmid-DNA erfolgte nach den Protokollen der Plasmid Miniprep
oder Plasmid Midi Kits von QIAGEN (QIAGEN; Hilden; D). Basierend auf der alkalischen
Lyse der Bakterien wird die Plasmid-DNA an einer siliziumdioxidhaltigen Membran unter
Hochsalzbedingungen adsorbiert und anschließend unter Niedrigsalzbedingungen eluiert.
DNA-Sequenzierung
Alle Sequenzierreaktionen wurden durch die Firma Medigenomix (Martinsried, D) nach
dem Protokoll des DNA Sequencing Kit BigDyeTM Terminator Cycle Sequencing Ready
Reaction von Perkin Elmer (Applied Biosystems; Foster City; U.S.A.) durchgeführt. Die
Reaktionen wurden am ABI 310 Sequenziergerät analysiert.
Restriktionsanalyse
Für den Verdau von 250-500 ng Plasmid-DNA wurden 2 µl 10 x Restriktionspuffer
(passend zum schneidenden Enzym), 2 µl 10 x BSA (wenn nötig für die Enzymaktivität),
0,5-1 µl Restriktionsenzym (5-10 U) und H2O in einem Endvolumen von 20 µl für zwei

30
Stunden bei der für das Enzym optimalen Temperatur inkubiert. Die Analyse der verdauten
DNA erfolgte auf einem 1%igem Agarose-Gel. Für den präparativen Restriktionsverdau
wurden 10-50 μg DNA und 10-100 U des Restriktionsenzyms eingesetzt.
Agarosegelelektrophorese
Lösungen und Reagenzien:
10 x DNA-Ladepuffer: Sigma, (Deisenhofen, D)
50 x TAE-Laufpuffer: 2M Tris-HCl
1M Natriumacetat
50mM EDTA
pH mit HCl auf 8,5 einstellen
Zur Größenbestimmung von DNA-Fragmenten wurden 1%ige Agarose-Gele verwendet,
denen 5 µl EtBr (10 mg/ml) je 100 ml 1 x TAE zugefügt wurde. Die elektrophoretische
Auftrennung der DNA-Fragmente erfolgte in horizontalen Laufkammern bei 100 V in 1 x
TAE-Laufpuffer. Den Proben wurde vor dem Auftragen 1/10 des Gesamtvolumens an 10 x
DNA-Ladepuffer hinzugefügt. Als Größenstandard wurde ein 1 kb bp-DNA-Leiter
mitgeführt.
Isolierung von DNA-Fragmenten aus Agarosegelen
Die Isolierung von DNA-Fragmenten wurde nach dem Protokoll des QiaEx extraction kit
(QIAGEN, Hilden, D) durchgeführt.
DNA-Fragment Ligation
Lösungen und Reagenzien:
T4 DNA Ligase
10 x Ligase Puffer

31
Protokoll:
Folgenden Ansatz in ein Eppendorf-Gefäß pipettieren:
50 ng verdaute Vektor-DNA
x ng DNA Fragment, welches in Vektor einkloniert werden soll (molares Verhältnis
von Insert:Vektor ist 5:1)
1 μl 10 x Ligase Puffer
1 μl T4 DNA Ligase
mit H2O auf ein Endvolumen von 10 μl auffüllen
Reaktion über Nacht bei 15°C inkubieren.
Herstellung von Kanamycin-Agarplatten
LB Agar Platten: 1 % Trypton
0,5 % Hefe Extrakt
0,17 M Natriumchlorid
1,5 % Agar
pH auf 7,0 mit HCl einstellen, anschließend für 30 min autoklavieren, auf ca 50 °C
abkühlen lassen, 50 µg / ml Kanamycin hinzufügen und Platten unter Sterilbank gießen.
Diese sind bei 4 °C ca 6 Monate haltbar.
Transformation von Plasmiden mittels Thermoschock
Lösungen und Reagenzien:
Tg2-kompetente Zellen
Autoklavieren des LB-Medium
LB-Medium: 1 % Trypton
0,5 % Hefe Extrakt
0,17 M Natriumchlorid
pH mit HCl auf 7,0 einstellen und für 30 min autoklavieren
Protokoll:
3 μl des Ligationsansatzes in ein Eppendorf-Gefäß überführen.
Kompetente Zellen auf Eis auftauen und 50 μl zu Ligationsansatz pipettieren.

32
Inkubation der Transformationsansätze auf Eis für 30 min.
LB-Medium auf 42°C vorwärmen.
Ansätze für 90 s bei 42°C inkubieren und anschließend für 3 min auf Eis stellen.
Zugabe von 500 μl des vorgewärmten LB-Mediums bei RT.
Inkubation der Ansätze bei 37°C für 1 h.
Ausplattieren der Ansätze auf die Kanamycin-Agarplatten und Inkubation über Nacht bei
37°C im Brutschrank.
Einzelne Kolonien picken und 5 ml LB Medium animpfen.
Bakterien über Nacht bei 37°C im Schüttler wachsen lassen.
Plasmid-DNA Isolation per Plasmid Miniprep Kit (QIAGEN, Hilden, D) und
anschließende Überprüfung der Klone mittels Restriktionsanalyse.
Amplifikation von DNA mittels PCR
Lösungen und Reagenzien:
Taq Polymerase (5 U/μl)
10 x Polymerase Puffer
10 x dNTPs
Q-Solution
Oligonukleotide
Protokoll:
Folgenden Reaktionsansatz in ein 0,5 ml PCR-Gefäß pipettieren:
x μl DNA (100-150 ng Plasmid-DNA)
5 μl 10 x Polymerase-Puffer
10 µl Q-Solution
1,25 μl 10 x dNTPs
1,25 μl Primer 1 (10 pmol/µl)
1,25 μl Primer 2 (10pmol/µl)
x μl ddH2O (auffüllen auf 49,75 μl)
0,5 μl Taq Polymerase (1 Einheit)
PCR-Ansatz in die PCR-Maschine stellen und Programm auswählen.

33
Schritt 1 95 °C 5 min
Schritt 2 95 °C 30 sec
Schritt 3 56 °C 30 sec
Schritt 4 72 °C 2 min
Schritt 5 72 °C 7 min
Schritt 6 4 °C ∞
Schritte 1, 5 und 6 werden einmalig, Schritte 2-4 36 mal durchgeführt.
Analyse der PCR-Produkte auf einem Agarose-Gel.
2.2.2.2. Transiente Transfektion von HEK 293-Zellen
Die transiente Transfektion erfolgte mit Hilfe des Polyfect Transfektionsreagenz von
QIAGEN nach dem Protokoll des Herstellers.
Zellkultur
Lösungen und Reagenzien:
Zellkulturmedium: 500 ml DMEM (Dulbecco’s Minimal Essential Medium)
10 % Fötales Kälberserum
1 mM L-Glutamin
Kultivierung von HEK 293-Zellen
Die Kultivierung der Zellen erfolgte im Brutschrank bei 37°C und 10 % CO2 . Die Zellen
wurden 2 x wöchentlich bei einer Dichte von ungefähr 2 x 105 Zellen/ml gesplittet. Sie
wurden mit einem Zellschaber vom Boden der Zellkulturflasche abgelöst und etwa 1/5 der
Kultur in eine neue Zellkulturflasche mit frischem Zellkulturmedium überführt.

34
Kryokonservierung von Zellen
5 x 105 - 1 x 106 Zellen wurden geerntet, in 1,5 ml Einfrier-Medium resuspendiert, in
Einfrierröhrchen überführt und sofort bei -70°C eingefroren. Nach einigen Tagen wurden
die Zellen zur längeren Aufbewahrung in flüssigen Stickstoff überführt.
Auftauen kryokonservierter Zellen
Die Zellen wurden bei 37°C im Wasserbad aufgetaut, mit 15 ml eiskaltem
Zellkulturmedium verdünnt und 5 min bei 1.200 rpm und 4°C abzentrifugiert. Nach
Abkippen des Überstandes wurden die Zellen in Zellkulturmedium resuspendiert und in
Zellkulturflaschen überführt.
2.3. Material und Methoden zu den P2X3-Rezeptorexperimenten
2.3.1 . Kulturmedium für hHEK293-P2X3-Zellen
Dulbecco´s Modifiziertes Eagle Medium (DMEM):
500 ml; Kat.Nr. 22320-022 Gibco BRL (Invitrogen, D), bestehend aus Natriumpyruvat
(110 mg/l),D-Glucose (1000 mg/l), L-Glutamin (580 mg/l) und HEPES (5958 mg/l). 500 µl
Geneticin, 5 ml NEAA (Nichtessentielle Aminosäuren) und 50 ml FKS wurden
hinzugefügt.
2.3.2. Extrazelluläre Lösung (EC) für HEK293- und Hinterwurzelganglien-
(DRG) Zellen
Die EC bestand aus (in mM):
NaCl, 135; KCl, 4,5; CaCl2, 2; MgCl2, 1; HEPES, 10; Glukose, 10; Einstellung des pH
Wertes auf pH=7,4 erfolgte mit NaOH
Manche Versuche sind in Mg2+ freier EC durchgeführt, bei den DRG Zellen wurde noch
0,5 µM Tetrodotoxin (TTX) hinzugefügt.

35
2.3.3. Intrazelluläre Lösung (IC) für HEK293- und Hinterwurzelganglien -
Zellen
Die IC bestand aus (in mM):
CsCl, 140; MgCl2, 2; CaCl2, 1; HEPES, 10; und EGTA, 11. Die Einstellung des pH-Wertes
auf pH=7,4 erfolgte mit CsOH.
Für manche Versuche wurde 0,3 mM GDP-ß-S hinzugefügt.
2.3.4 Präparationen und Kultivierungen
2.3.4.1. Präparation der DRG-Zellen und ihre Kultivierung
Die DRG-Zellen wurden von 1 bis 2 Tage alten Ratten gewonnen. Zuerst wurde die Ratte
dekapitiert. Dann wurden alle Organe entfernt und die Wirbelsäule eröffnet. Nach
Entnahme des Rückenmarks wurden die Ganglien entnommen und in HBSS auf Eis
gesammelt. Die Ganglien müssen vor der weiteren Verarbeitung noch von möglichen
Bindegewebsresten gesäubert werden.
Die in HBSS befindlichen Ganglien wurden im Greiner-Röhrchen bei 1000 U/Min für 5
Minuten zentrifugiert. Danach wurde der HBSS Überstand bis auf 1ml abpipettiert. Dazu
wurden anschließend 1ml Kollagenase, 1ml Dispase und 150 µl DNase gegeben. Dann
erfolgte die Inkubation im Wasserbad bei 37°C für 12 Minuten. Gelegentlich sollte das
Greiner-Röhrchen geschüttelt werden um die enzymatische Dissoziation anzuregen.
Schließlich wurde noch 1 ml Trypsin zugegeben und die Inkubation für weitere 7 Minuten
bei 37°C im Wasserbad fortgesetzt. Um die Enzymreaktion zu stoppen, wurden 2 ml
Medium in die Suspension gegeben. Mit einer Glasspipette (die Pipette soll keine scharfen
Kanten haben) wurde (ca 20x) trituriert. So wurden die noch erhaltenen Zellverbände
gelöst. Die Suspension, die zunehmend trüber wurde, wurde durch ein steriles Zellsieb
(Porengröße 70µm) filtriert und dann bei 1000 U/Min für 10 Minuten ohne Bremse
zentrifugiert. Es wurde ein Pellet gebildet. Der verbleibende Überstand wurde vorsichtig
entfernt. Nach Schätzung der erwarteten Zellzahl wurde das Pellet mit Medium (ca 1 ml)
resuspendiert.
Die Zellzahl wurde mittels Neubauer Zählkammer bestimmt. 30 000 Zellen wurden in einer
Petrischale (3,5 cm Durchmesser) ausgesät bei einer empfohlenen Tropfengröße von etwa

36
70 µl. Die verwendeten Petrischalen sind vorher mit Poly-L-Lysin beschichtet und 2 x mit
HBSS und Wasser gespült worden. Die ausgesäten Zellen wurden im Brutschrank (37°C,
5-7% CO2) für 1 bis 2 Stunden inkubiert. Anschließend wurden noch 2 ml Medium
hinzugegeben. Nach 3 Tagen können an den Kulturen elektrophysiologische Messungen
vorgenommen werden.
2.3.4.2. Kultivierung von HEK293-hP2X3-Zellen
Die erste humane embryonale Nierenzelllinie HEK293 (human embryonic kidney;
European Collection of Cell Cultures, Porton Down, U.K.) wurde 1977 entwickelt und
beschrieben (Graham et. al., 1977). Diese Zelllinie wurde auch für unsere Experimente
verwendet. Uns wurde eine stabil transfizierte HEK293-Zellinie im Rahmen einer
Forschungskooperation von der Grünental GmbH (Aachen, D) zur Verfügung gestellt.
Die HEK293-hP2X3-Zellen wurden einmal pro Woche gesplittet. Die Zellen wurden mit
einem Zellschaber vom Boden der Zellkulturflasche abgelöst, mechanisch trituriert und
etwa 1/5 der Kultur in eine neue Zellkulturflasche mit frischem Zellkulturmedium
überführt. Die Einsaatdichte sollte bei 5 x 105 Zellen in 10-15 ml Medium liegen. Nach 2
bis 7 Tagen wurden die Zellen für die Messung verwendet.
2.3.5. Patch-Clamp Technik
Die Patch-Clamp Technik ist heute eine der wichtigsten elektrophysiologischen
Arbeitsmethoden. Sie wurde von Erwin Neher und Bert Sakmann 1976 entwickelt. Mit
Hilfe dieser Methode wurde es ermöglicht, die elektrische Aktivität von Ionenkanälen an
einzelnen Zellen zu messen. Die Öffnung von Ionenkanälen und die Aktivität von
Ionentransportern verändern die Leitfähigkeit der Membran, was zur Änderung des
Membranpotentials führt. Zur Konstanthaltung des Membranpotentials ist in den dreißiger
Jahren die Spannungsklemme (Voltage-Clamp) entwickelt worden. Ausgangspunkt der
Spannungsklemme ist ein vom Verstärker erzeugter Kompensationsstrom, der im Whole-
cell-Modus über die Pipette in die Zelle fließt und Änderungen des Membranpotentials der
untersuchten Zelle ausgleicht.
Grundlage dieses Verfahrens ist eine mit Elektrolytlösung gefüllte Glasmikropipette, die
auf der Membran einer Zelle aufgesetzt wurde (Abb. 9A). Zwischen die Glaspipette und

37
die Zellmembran soll eine Abdichtung, deren elektrischer Widerstand im Gigaohm-Bereich
liegt, entstehen. Das erreicht man durch leichten Unterdruckaufbau. Diese Abdichtung
(engl. Seal) zeigt, dass die Zelle und die Pipette elektrisch von der Umgebung isoliert sind.
Die Pipettenspitze hat einen Durchmesser von 1-2 µm und im gefüllten Zustand einen
Widerstand von 4-9 MΩ. Durch den Unterdruck wird die Zellmembran in diese Spitze
hineingezogen, zwischen Glas und Membran bildet sich ein enger Kontakt aus, der zu
einem Abdichtungswiderstand von 5-7 GΩ führt. So entsteht die Cell-attached
Konfiguration (Abb. 9B). Das ist die Ausgangskonfiguration bei dieser Methode. Ein
Vorteil dieser Konfiguration ist, dass intrazelluläre Mechanismen nicht beeinflusst werden.
Das Ruhemembranpotential der Zelle bleibt ebenfalls unverändert. Nur in dem
Membranstück unter der Pipette, dem sogenannten Patch, wird im Spannungsklemm-
Modus das Membranpotential vom Verstärker kontrolliert.
Wenn man die Pipette in einer Cell-attached Konfiguration schnell von der Zelle wegzieht,
kann sich ein Membranstück von der Zelle lösen und an der Pipettenspitze bleiben, ohne
dass der Sealwiderstand zerstört wird. Diesen Messmodus nennt man Inside-out-
Konfiguration (Abb. 9D). Diese Konfiguration ist gut geeignet für Einzelkanalmessung.
Die innere (zytoplasmatische) Seite der Membran ist der Badlösung zugewandt.
Stärkerer Unterdruck im Cell-attached-Modus kann zum Platzen des Patches führen,
wodurch man zur Whole-Cell-Konfiguration (Abb. 9C) oder Ganzzellableitung übergeht.
Diese Konfiguration ist durch einen relativ niederohmigen Zugang von der Pipette zur
gesamten Zelle charakterisiert. Dies ermöglicht die Gesamtheit der Ionenströme der
untersuchten Zelle zu messen. Durch die Öffnung in der Zellmembran können
verschiedene Substanzen von der Pipettenlösung in die Zelle diffundieren und so
intrazelluläre Mechanismen beeinflussen. Das Membranpotential der gesamten Zelle kann
im Spannungsklemm-Modus kontrolliert werden. Die Whole-Cell-Messkonfiguration dient
als Ausgangskonfiguration für die Outside-out-Konfiguration (Abb. 9E). Dabei wird ein
Stück Membran aus der Zelle herausgelöst und schließt sich an der Pipettenspitze zu einer
Art Halbvesikel. So hat man an der Pipettenmündung ein Stück Zellmembran, das der
Außenseite der Badlösung zugewandt ist. Diese Konfiguration ist auch sehr gut für die
Untersuchung von ligandengesteuerten Ionenkanälen geeignet, weil man leicht Substanzen
von der extrazellulären Seite applizieren kann.

38
Im Rahmen dieser Arbeit wurden die Experimente nur in der Whole-cell-Konfiguration im
Voltage-clamp-Modus an Hinterwurzelganglienzellen (Dorsalwurzelganglienzellen, DRG-
Zellen) der Ratte und HEK-293 Zellen durchgeführt.
Abb. 9. Schematische Durchführung eines Patch-Clamp-Experimentes durch die vier
wichtigsten Konfigurationen der Patch-Clamp-Technik:
A. Annäherung der Pipette zur Membranoberfläche; B. Cell-attached; C. Whole-Cell; D.
Inside-out; E. Outside-out (modifieziert nach http://www.science-
display.com/patchclamp.html)
2.3.6. Aufbau und Ausstattung des Messplatzes
Die wichtigsten Teile eines Patch-Clamp-Arbeitsplatzes sind: Mikroskop mit Kamera,
Vorverstärker, Verstärker, Applikationssystem, Mikromanipulatoren, Computer. Die Patch-
Clamp-Messungen werden mit einer sehr empfindlichen Technik durchgeführt. Der
Kontakt zwischen Zelle und Patch-Pipette kann durch leichte Erschütterungen zerstört
werden. Deswegen befinden sich das Mikroskop, die Mikromanipulatoren und der
Vorverstärker auf einem schwingungsgedämpften Tisch (TMC, Peabidy, USA) (Abb. 10-

39
1). Die Ströme, die gemessen wurden, liegen im Pikoampere-Bereich, was auch eine sehr
gute Abschirmung gegen elektromagnetische Störungen mittels eines Faraday’schen Käfigs
(Abb. 10-2) erfordert. Alle Metallelemente, welche sich innerhalb des Käfigs befanden,
wurden mit einem zentralen Erdungsblock verbunden, um von außen eingetragenes
Rauschen zu vermeiden. Auf dem schwingungsgedämpften Tisch steht ein inverses
Mikroskop (Axiovert 100, Zeiss, Jena, D) (Abb. 10-3), das für Zellkulturen gut geeignet ist.
Auf dem Mikroskoptisch ist eine Kammer für 35 mm Schalen (Abb. 10-4) eingebaut. Dort
wurden die Zellkulturen für die elektrophysiologischen Ableitungen platziert. Zur visuellen
Identifizierung und besseren Auswahl der Zellen wurde eine Kamera (Oscar Color Camera,
World Precision Instruments, USA) (Abb. 10-5) montiert. Außer der normalen Belichtung
verfügt das Mikroskop über eine UV-Lampe (HBO-50) (Abb. 10-6). Der Verlauf der
Patch-Clamp Experimente kann über ein Oszilloskop (Oscilloscope Type HM305, Hameg
GmbH, Frankfurt, D) (Abb. 10-7) beobachtet werden. Auf dem schwingungsgedämpften
Tisch sind zwei motorische Mikromanipulatoren (Luigs & Neumann, Ratingen, D) (Abb.
10-8) befestigt. Die Applikationssystemspitze (Abb. 10-9), die mit der 13-fachen Spindel
des Applikationssystems verbunden ist, ist am rechten Manipulator befestigt. Die lokale
Applikation der Pharmaka an die Zellen erfolgt über ein rechnergestütztes schnelles
Druckapplikationssystem (DAD-12, ALA Scientific Instruments Inc. Westbury, USA)
(Abb. 10-10). Das Applikationssystem wurde mittels eines Laptops (Compaq; LTE Lite
4/25C, Compaq Computer Corporation, Japan) (Abb. 10-11) gesteuert. Am linken
Manipulator ist der Patch-Clamp-Vorverstärker (CV 203BU HEADSTAGE, Axon
Instruments) (Abb. 10-12) befestigt. Er ist mit dem Axopatch 200B (Axon Instruments,
Foster City, USA) Verstärker (Abb. 10-13) verbunden. Der Verstärker ermöglicht das
eingegebene Membranpotential konstant zu halten und den dazu notwendigen
Kompensationsstrom zu messen. Die Potential- und Stromausgänge des Verstärkers
Axopatch 200B wurden mit einem Interface (Analog-Digital-Umwandler, Digidata 1200,
Axon Instruments) (Abb. 10-14), einem Laserdruckschreiber (Laserjet 5MP, Hewlett
Packard) (Abb. 10-15) und dem Oszilloskop verbunden. Das Interface wandelt die
Analogsignale in Digitalsignale um und ist mit dem Computer (Peacock PC+Genuintel
Pentium III Processor; System: Microsoft Windows 98) (Abb. 10-16), verbunden. Die
Aufnahme der digitalen Daten wurde mit dem Programm Clampex (Version 8.0.0.81,
1984-1999 Axon Instruments, New South Wales, USA) durchgeführt. Die Aufnahmen
wurden mit Hilfe des Programms Clampfit (Version 8.2.0.235, 1984-2001 Axon
Instruments, New South Wales, USA) analysiert.

40
In den Pipettenhalter (Abb. 10-17) am Vorverstärker wurde eine mit ca. 10 µl
Pipettenlösung gefüllte Glasmikropipette (3-5 MΩ) gespannt. Über silberdrahtchlorierte
Bad- und Pipetten-Elektroden besteht eine elektrische Verbindung zum Vorverstärker. Die
Glasmikropipetten wurden aus Borosilikatglas (GB150F-8P, Science Products GmbH,
Hofheim, D) hergestellt. Sie wurden mit einem horizontalen Pipettenziehgerät (Modell P-
97, Sutter Instruments, Novato, USA) (Abb. 10-18) vor der Messung frisch gezogen.
Wichtig ist, dass die gefüllten Pipetten blasenfrei sind, um einen ungehinderten Stromfluss
zu ermöglichen.
Abb. 10. Ausstattung eines Patch-Clamp-Messplätzes.
Die Erläuterungen zu den Zahlen sind im Text (oben) dargestellt.

2.3.7. Die praktische Durchführung von Patch-Clamp-Experimenten
Damit man einen guten Seal herstellen kann, soll man eine neugezogene und saubere
Pipette benutzen. Die Pipette wurde blasenfrei mit intrazellulärer Lösung aufgefüllt und in
den Pipettenhalter am Vorverstärker eingespannt, so dass alles luftdicht wird. Die
aufgefüllte Pipette wurde mit der Hilfe des Mikromanipulators in die Badlösung
eingetaucht. Dann wurde der Nullwert für das Potential eingestellt. Die chlorierte
Badelektrode wurde mittels einer Elektrolytbrücke mit der Badlösung verbunden. Der
Strom soll ungehindert zwischen Pipette und Badelektrode fließen. Nach dem Abgleich
beider Ströme wurde auf dem Oszilloskop ein rechteckförmiger Kommandospannungspuls
beobachtet, der bei unseren Voreinstellungen etwa 10 mV betrug. Mit Hilfe des
Mikromanipulators wurde die Pipettenspitze nah an die Zelloberfläche herangeführt. Eine
Berührung der Membranoberfläche durch die Pipette konnte als eine geringe Erhöhung des
Pipettenwiderstandes am Oszilloskop verfolgt werden. Nach der Berührung der
Zelloberfläche durch die Pipette erfolgte ein leichtes Saugen an der Pipette. Dadurch wurde
der Widerstand zwischen Pipette und Zellmembran immer höher und es bildete sich ein
Seal. Dann wurde durch den Verstärker das gewünschte Haltepotential (-70 mV)
eingestellt. Nach der Sealbildung wurde der Unterdruck entfernt und der Seal überprüft.
Das auf dem Oszilloskop beobachtete Signal sollte bis auf kleine kapazitive Artefakte am
Anfang und Ende des Spannungspulses fast vollständig flach sein. Diese beiden Artefakte
resultieren von der Pipettenkapazität. Diese kleinen kapazitiven Artefakte wurden mittels
der Pipettenkapazitäts-Kompensationsfunktion des Verstärkers abgeglichen. Jetzt befindet
sich die Zelle in der Cell-Attached Konfiguration (Abb.9B). Dann wurde die Membran
unter der Pipettenspitze durch stärkeres Ansaugen, oder betätigen der „Zap-Modus“-Taste
(eine hochfrequente und hochamplitudige Wechselspannung) durchbrochen und die
Whole-Cell-Konfiguration hergestellt. Das Bild im Oszilloskop ändert sich wie in Abb. 9C
dargestellt. Die kapazitiven Artefakte werden wesentlich größer, denn statt des kleinen
Stücks Membran muss jetzt bei jeder Spannungsänderung die gesamte Fläche der
Zellmembran umgeladen werden. Dann wurde die Ableitung optimiert, indem der
Serienwiderstand reduziert und die Kompensationen für Kapazität und Serienwiderstand
aktiviert wird
Nach dem Erreichen der Whole-Cell-Konfiguration, wurde die Applikationsspitze des
DAD-Systems (ALA, Scientific Instruments Inc. Westbury, USA) mit der Hilfe des
zweiten Mikromanipulators in die Nähe (ungefähr 100 µm) der Zelle geführt, von welcher

42
die Ableitung erfolgte; so konnten die verschiedenen Lösungen in die unmittelbare
Umgebung dieser Zelle appliziert werden. Die lokale Verabreichung der Substanzen
erfolgte über ein schnelles Druckapplikationssystem, welches bis zu 12 verschiedene
Substanzen oder Konzentrationen nacheinander applizieren kann. Außerdem verfügt das
System über ein weiteres druckunabhängiges Gefäß, über das die Zelle kontinuierlich mit
extrazellulärer Lösung umspült wird. Die Austauschzeit der verwendeten Lösungen für das
DAD-System wurde durch die Applikation von destilliertem Wasser an eine offene Patch-
Pipette bestimmt. Der Potentialsprung von 10 auf 90 Prozent der maximalen
Peakamplitude erfolgte in 15,9 ± 0,9 ms (n = 7).
Die verwendeten Applikationsschemas werden in Abbildung 11 gezeigt.
Abb. 11. Applikationsschema für die elektrophysiologischen Untersuchungen.
A. Applikationsschema für die Applikation von UTP, UDP, Uridin, GTP, ATP und
PMA. Die Applikationsdauer dieser Substanzen ist mit grün gekennzeichnet. α,ß-
meATP wird in den angegebenen Abständen für jeweils 2 s appliziert (rot
gekennzeichnet).
B. Applikationsschema für DAG-Lakton. Die Farben sind analog wie im Punkt A.

43
C. Applikationsschema für Konzentrations-Wirkungskurven mit α,ß-meATP. Mit rot ist
die Applikation von α,ß-meATP dargestellt.
D. Applikationsschema für Konzentrations-Wirkungskurven mit BzATP. Mit rot ist die
Applikation von BzATP gekennzeichnet.
2.3.8. Datenanalyse und Statistik
Die Datenaufnahme wurde mit dem Verstärker Axopatch 200B (Axon Instruments)
durchgeführt und mit dem Computer und den entsprechenden Programmen (siehe oben)
ausgewertet.
Die Digitalisierungsfrequenz wurde auf 5 kHz eingestellt und sie sollte nach dem Nyquist-
Kriterium mindestens doppelt so hoch sein wie die höchste im Signal vorkommende
Frequenz. Die hochfrequenten Signale, die in der Regel nur dem Hintergrundrauschen
entsprechen, wurden mittels eines Tiefpassfilters bis zu einer Grenzfrequenz von 2 kHz
eliminiert.
Die relativen Leitfähigkeiten der passierenden Ionen und ihre jeweiligen
Gleichgewichtspotentiale bestimmen das Umkehrpotential eines Membranstroms. Am
Umkehrpotential ist kein Nettostrom messbar. Wenn der Strom nur von einer Ionenart
getragen wird, entspricht das Umkehrpotential dem Gleichgewichtspotential des
entsprechenden Ions und kann anhand der Nernst Gleichung berechnet werden:
EIon = RT/zF x ln([Ion]a/[Ion]i)
EIon: Gleichgewichts-(Nernst-) Potential des jeweiligen Ions
R: allgemeine Gaskonstante z: Wertigkeit des Ions
T: absolute Temperatur in Kelvin F: Faraday-Konstante
([Ion]a/[Ion]i): Ionenkonzentration außerhalb bzw. innerhalb der Zelle
Für diese Arbeit wurden die angegebenen Umkehrpotentiale empirisch aus Stromantworten
ermittelt, indem derjenige Wert bestimmt wurde, bei dem der untersuchte Strom seine
Orientierung änderte, also den Betrag Null hatte.

44
Die Grundparameter der durch Agonisten induzierten Ströme wurden mittels der folgenden
mathematischen Gleichung bestimmt:
I=Imax/[1+(EC50/Agonist)n]
Hier steht „I“ für den durch den Agonisten induzierten Gleichgewichts-Strom
(Plateaustrom), „Imax“ für den maximalen Strom bei unendlicher Agonisten-Konzentration,
„n“ für den Hill-Koeffizienten und EC50 steht für jene Konzentration des Agonisten, bei der
50 % von Imax erreicht werden. Die Konzentrations-Wirkungs-Kurven wurden mit dem
Programm SigmaPlot und SigmaStat (Jandel Scientific, Erkrath, D) erstellt. Die graphische
Darstellung erfolgte mit Hilfe der Programme CorelDraw (Version 11) und Microsoft
Office (Microsoft, Richmond, USA).
Alle Ergebnisse werden als arithmetische Mittelwerte mit ihrem Standardfehler von „n“
Zellen vorgestellt. Mehrere Vergleiche mit einer Kontrollgruppe wurden durchgeführt mit
einer einseitig gerichteten Analyse der Varianzen (one-way-ANOVA), der entweder ein
nicht parametrischer Dunn’s-Test oder der parametrische Bonferroni’s t-Test folgte. Zwei
Messwertreihen wurden, wenn sie normal verteilt sind und eine vergleichbare
Standardabweichung hatten, mit dem parametrischen Student’s t-Test verglichen. Wo der
Student’s t-Test nicht verwendet werden konnte, kam der nicht-parametrische Mann-
Wilcoxon-Test zur Anwendung. Unterschiede mit einer Irrtumswahrscheinlichkeit von p
kleiner als 0,05 wurden als statistisch signifikant bezeichnet.
Zum Auswerten der Abfallphasen der Ströme während der Desensibilisierung von P2X3-
Rezeptoren wurde 20 s lang α,ß-meATP appliziert. Die Aufnahmen wurden mit Faktor 5
durch die pClamp 8.0 Software reduziert und die Kurvenabfallphasen wurden mittels
Origin Software (OriginLab, Northampton, MA, USA) so angepaßt, dass die
Zeitkonstanten (Offsetkonstanten τoff1 und τoff2 und Onsetkonstante τon(10-90) ) ausgerechnet
werden konnten.
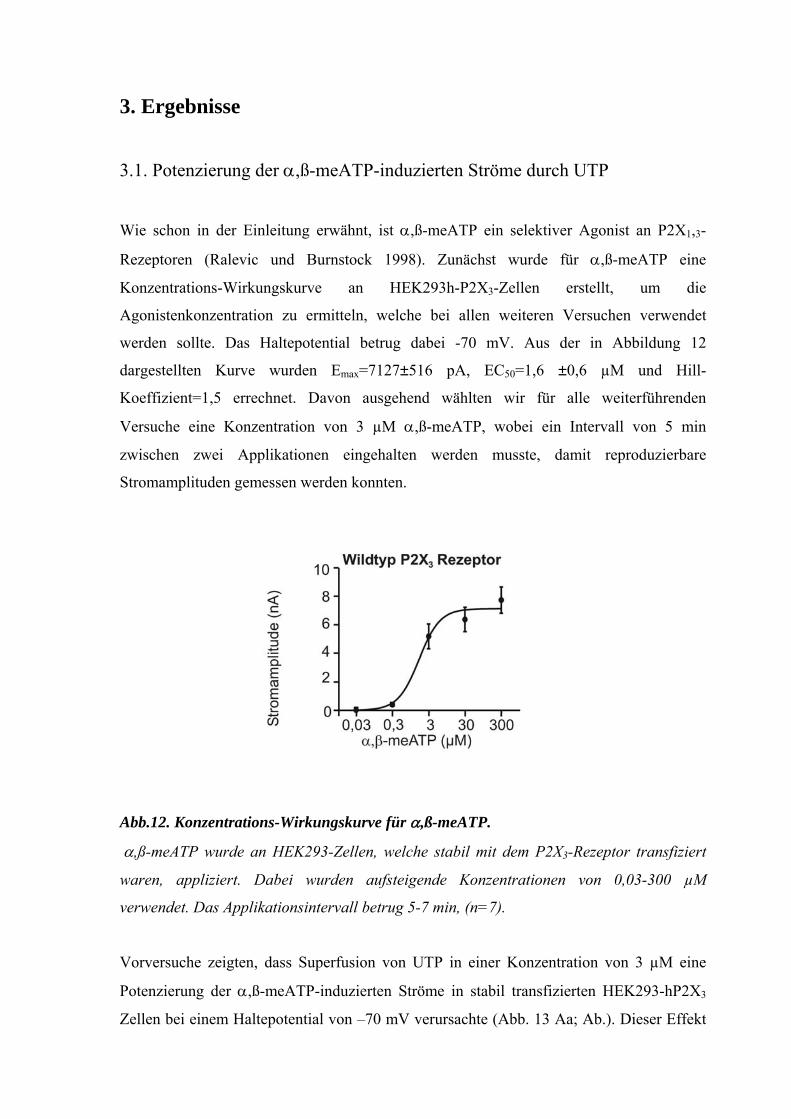
3. Ergebnisse
3.1. Potenzierung der α,ß-meATP-induzierten Ströme durch UTP
Wie schon in der Einleitung erwähnt, ist α,ß-meATP ein selektiver Agonist an P2X1,3-
Rezeptoren (Ralevic und Burnstock 1998). Zunächst wurde für α,ß-meATP eine
Konzentrations-Wirkungskurve an HEK293h-P2X3-Zellen erstellt, um die
Agonistenkonzentration zu ermitteln, welche bei allen weiteren Versuchen verwendet
werden sollte. Das Haltepotential betrug dabei -70 mV. Aus der in Abbildung 12
dargestellten Kurve wurden Emax=7127±516 pA, EC50=1,6 ±0,6 µM und Hill-
Koeffizient=1,5 errechnet. Davon ausgehend wählten wir für alle weiterführenden
Versuche eine Konzentration von 3 µM α,ß-meATP, wobei ein Intervall von 5 min
zwischen zwei Applikationen eingehalten werden musste, damit reproduzierbare
Stromamplituden gemessen werden konnten.
Abb.12. Konzentrations-Wirkungskurve für α,ß-meATP.
α,ß-meATP wurde an HEK293-Zellen, welche stabil mit dem P2X3-Rezeptor transfiziert
waren, appliziert. Dabei wurden aufsteigende Konzentrationen von 0,03-300 µM
verwendet. Das Applikationsintervall betrug 5-7 min, (n=7).
Vorversuche zeigten, dass Superfusion von UTP in einer Konzentration von 3 µM eine
Potenzierung der α,ß-meATP-induzierten Ströme in stabil transfizierten HEK293-hP2X3
Zellen bei einem Haltepotential von –70 mV verursachte (Abb. 13 Aa; Ab.). Dieser Effekt
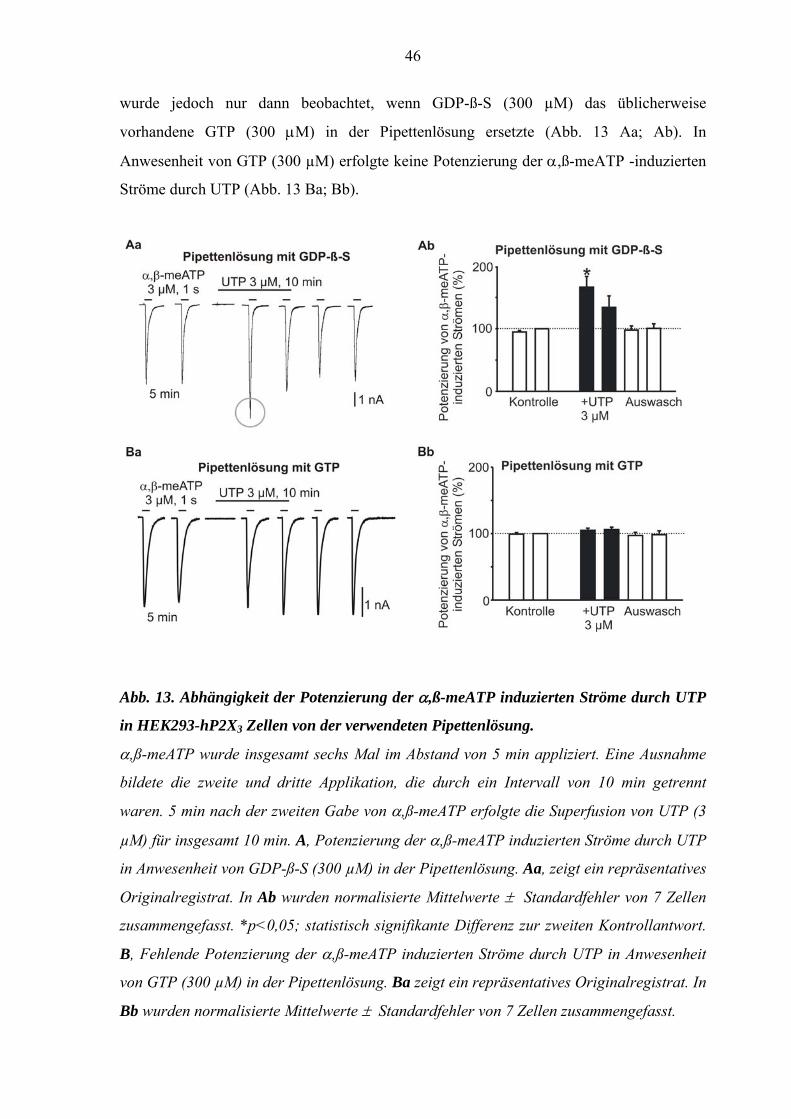
46
wurde jedoch nur dann beobachtet, wenn GDP-ß-S (300 µM) das üblicherweise
vorhandene GTP (300 μM) in der Pipettenlösung ersetzte (Abb. 13 Aa; Ab). In
Anwesenheit von GTP (300 µM) erfolgte keine Potenzierung der α,ß-meATP -induzierten
Ströme durch UTP (Abb. 13 Ba; Bb).
Abb. 13. Abhängigkeit der Potenzierung der α,ß-meATP induzierten Ströme durch UTP
in HEK293-hP2X3 Zellen von der verwendeten Pipettenlösung.
α,ß-meATP wurde insgesamt sechs Mal im Abstand von 5 min appliziert. Eine Ausnahme
bildete die zweite und dritte Applikation, die durch ein Intervall von 10 min getrennt
waren. 5 min nach der zweiten Gabe von α,ß-meATP erfolgte die Superfusion von UTP (3
µM) für insgesamt 10 min. A, Potenzierung der α,ß-meATP induzierten Ströme durch UTP
in Anwesenheit von GDP-ß-S (300 µM) in der Pipettenlösung. Aa, zeigt ein repräsentatives
Originalregistrat. In Ab wurden normalisierte Mittelwerte ± Standardfehler von 7 Zellen
zusammengefasst. *p<0,05; statistisch signifikante Differenz zur zweiten Kontrollantwort.
B, Fehlende Potenzierung der α,ß-meATP induzierten Ströme durch UTP in Anwesenheit
von GTP (300 µM) in der Pipettenlösung. Ba zeigt ein repräsentatives Originalregistrat. In
Bb wurden normalisierte Mittelwerte ± Standardfehler von 7 Zellen zusammengefasst.

47
Es interessierte, ob der Effekt von UTP konzentrationsabhängig ist. Deshalb wurden 0,3-
3000 nM UTP im oben beschriebenen Versuchsablauf getestet (Abb. 14A). Höhere UTP-
Konzentrationen konnten nicht untersucht werden, da bereits 30 µM UTP die P2X3-
Rezeptoren aktivierte, selbst Einwärtsströme induzierte und bei längerdauernder
Applikation schließlich eine Desensibilisierung des P2X3-Rezeptors hervorrief (Abb. 14
C). Während 0,3 und 3 nM UTP keine signifikante Potenzierung der α,β-meATP-Ströme
bewirkte, erfolgte eine Steigerung der Stromantworten bei Konzentrationen ab 30 nM,
wobei Maximaleffekte bei 3 µM UTP registriert wurden.
Weiterhin wurde untersucht, welchen Einfluss die Dauer der Applikation von 3 µM UTP
auf die α,ß-meATP-induzierten Ströme hat. Dazu wurde die Dauer der UTP-Gabe vor der
gemeinsamen α,β-meATP-Applikation von 1 min bis auf 5 min gesteigert. Eine
mindestens vierminütige UTP-Applikation war vor Gabe von α,ß-meATP nötig, um eine
signifikante Steigerung zu erzielen (Abb. 14 B). Folglich schien der Zeitverlauf des UTP-
Effektes die klassischen P2X/P2Y-rezeptorvermittelten Reaktionen, die normalerweise
innerhalb von Millisekunden oder Sekunden anstatt Minuten stattfinden, auszuschließen.
Das deutete darauf hin, dass die UTP-Wirkung durch einen anderen Mechanismus als eine
direkte Rezeptoraktivierung zustande kommt.
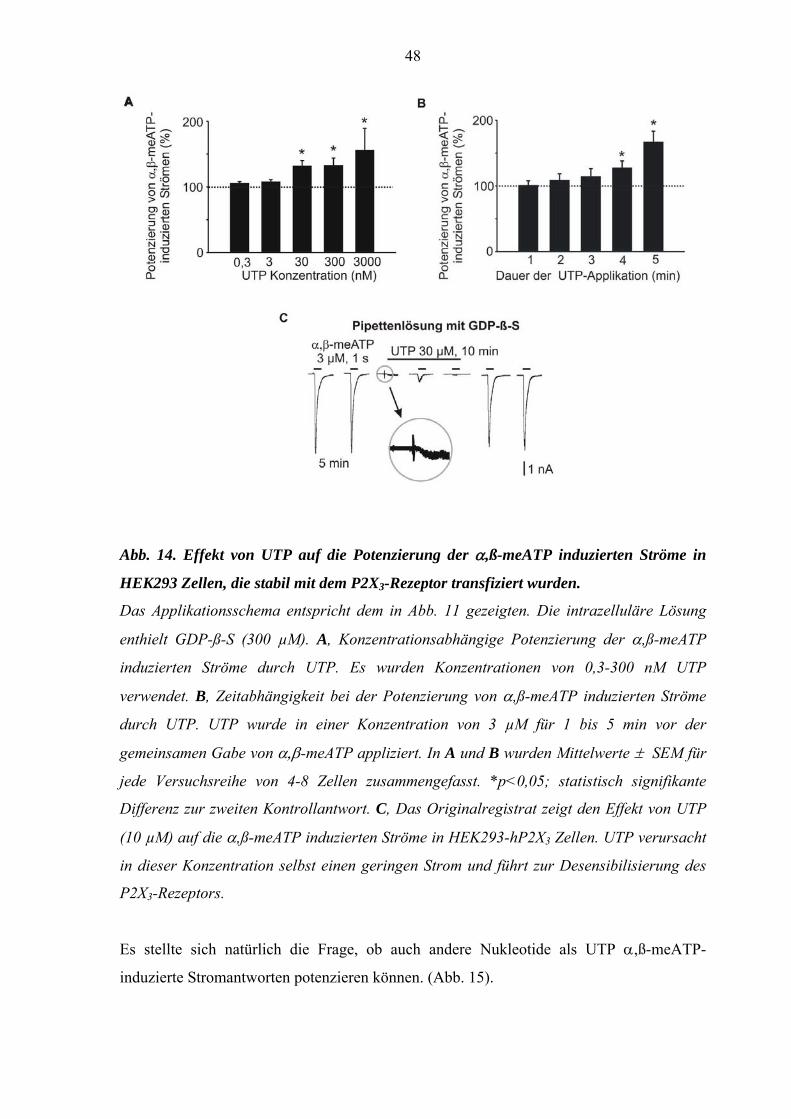
48
Abb. 14. Effekt von UTP auf die Potenzierung der α,ß-meATP induzierten Ströme in
HEK293 Zellen, die stabil mit dem P2X3-Rezeptor transfiziert wurden.
Das Applikationsschema entspricht dem in Abb. 11 gezeigten. Die intrazelluläre Lösung
enthielt GDP-ß-S (300 µM). A, Konzentrationsabhängige Potenzierung der α,ß-meATP
induzierten Ströme durch UTP. Es wurden Konzentrationen von 0,3-300 nM UTP
verwendet. B, Zeitabhängigkeit bei der Potenzierung von α,ß-meATP induzierten Ströme
durch UTP. UTP wurde in einer Konzentration von 3 µM für 1 bis 5 min vor der
gemeinsamen Gabe von α,β-meATP appliziert. In A und B wurden Mittelwerte ± SEM für
jede Versuchsreihe von 4-8 Zellen zusammengefasst. *p<0,05; statistisch signifikante
Differenz zur zweiten Kontrollantwort. C, Das Originalregistrat zeigt den Effekt von UTP
(10 µM) auf die α,ß-meATP induzierten Ströme in HEK293-hP2X3 Zellen. UTP verursacht
in dieser Konzentration selbst einen geringen Strom und führt zur Desensibilisierung des
P2X3-Rezeptors.
Es stellte sich natürlich die Frage, ob auch andere Nukleotide als UTP α,ß-meATP-
induzierte Stromantworten potenzieren können. (Abb. 15).
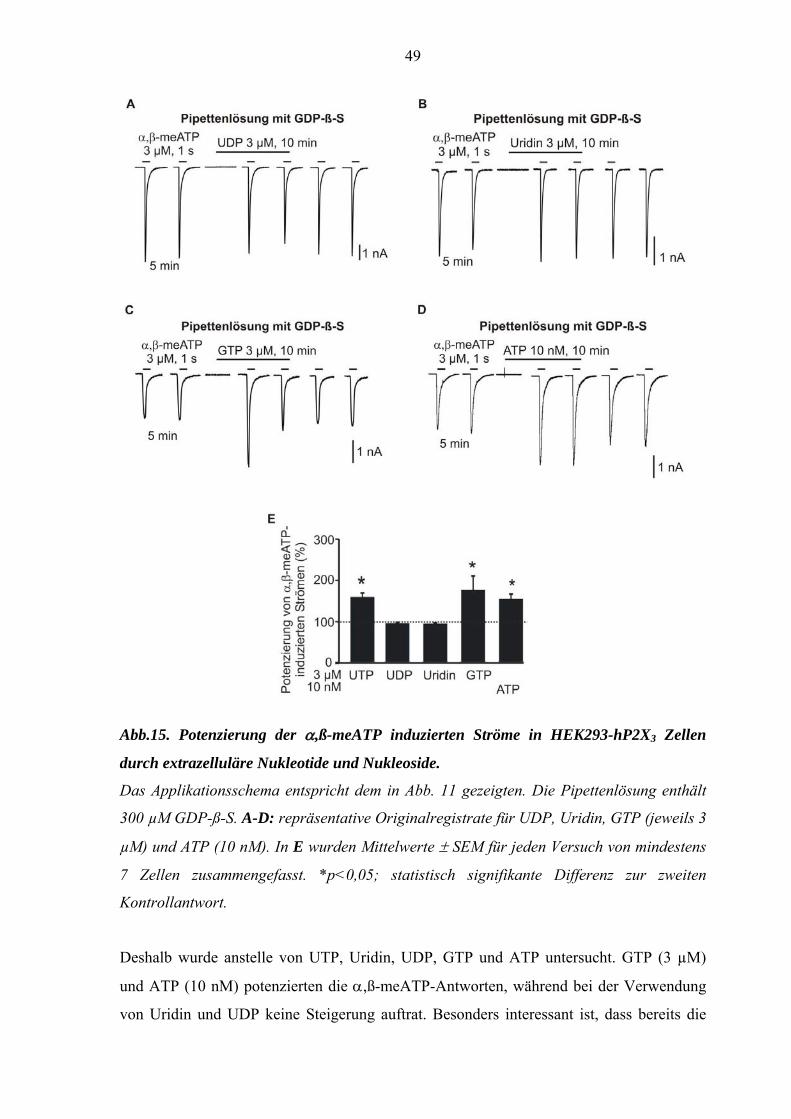
49
Abb.15. Potenzierung der α,ß-meATP induzierten Ströme in HEK293-hP2X3 Zellen
durch extrazelluläre Nukleotide und Nukleoside.
Das Applikationsschema entspricht dem in Abb. 11 gezeigten. Die Pipettenlösung enthält
300 µM GDP-ß-S. A-D: repräsentative Originalregistrate für UDP, Uridin, GTP (jeweils 3
µM) und ATP (10 nM). In E wurden Mittelwerte ± SEM für jeden Versuch von mindestens
7 Zellen zusammengefasst. *p<0,05; statistisch signifikante Differenz zur zweiten
Kontrollantwort.
Deshalb wurde anstelle von UTP, Uridin, UDP, GTP und ATP untersucht. GTP (3 µM)
und ATP (10 nM) potenzierten die α,ß-meATP-Antworten, während bei der Verwendung
von Uridin und UDP keine Steigerung auftrat. Besonders interessant ist, dass bereits die

50
geringe ATP-Konzentration von 10 nM potenzierend wirkte. ATP-Konzentrationen >10
nM konnten nicht getestet werden, da dadurch eine Aktivierung bzw. Desensibilisierung
von P2X3-Rezeptoren erfolgte Abb. 15D).
3.2. Interaktionen mit PKC Aktivatoren und Inhibitoren
Infolge der Anwesenheit von konsensus Phosphorylierungsstellen in der extrazellulären
Schleife des P2X3-Rezeptors (Mager et al., 2004), wurde eine funktionale Bedeutung dieser
strukturellen Merkmale als potenzielles Ziel für ekto-PKC-induzierte Phosphorylierung
angenommen.
Um diese Frage genauer zu untersuchen, wurden die selektiven PKC-Aktivatoren
Diazylglyzerollakton (DAG-Lakton) und Phorbol-12-myristat 13-acetat (PMA) ausgewählt
und gemäß dem üblichen Protokoll, das für die Nukleotidapplikation verwendet wurde,
superfundiert. Die Superfusion von DAG-Lakton wurde von 10 auf 6 min verkürzt und
begann 1 min vor der ersten Applikation von α,β−meATP. Sowohl DAG-Lakton (1 µM)
als auch PMA (0.1 µM) vergrößerten die α,ß-meATP-induzierten Ströme (Abb. 16).
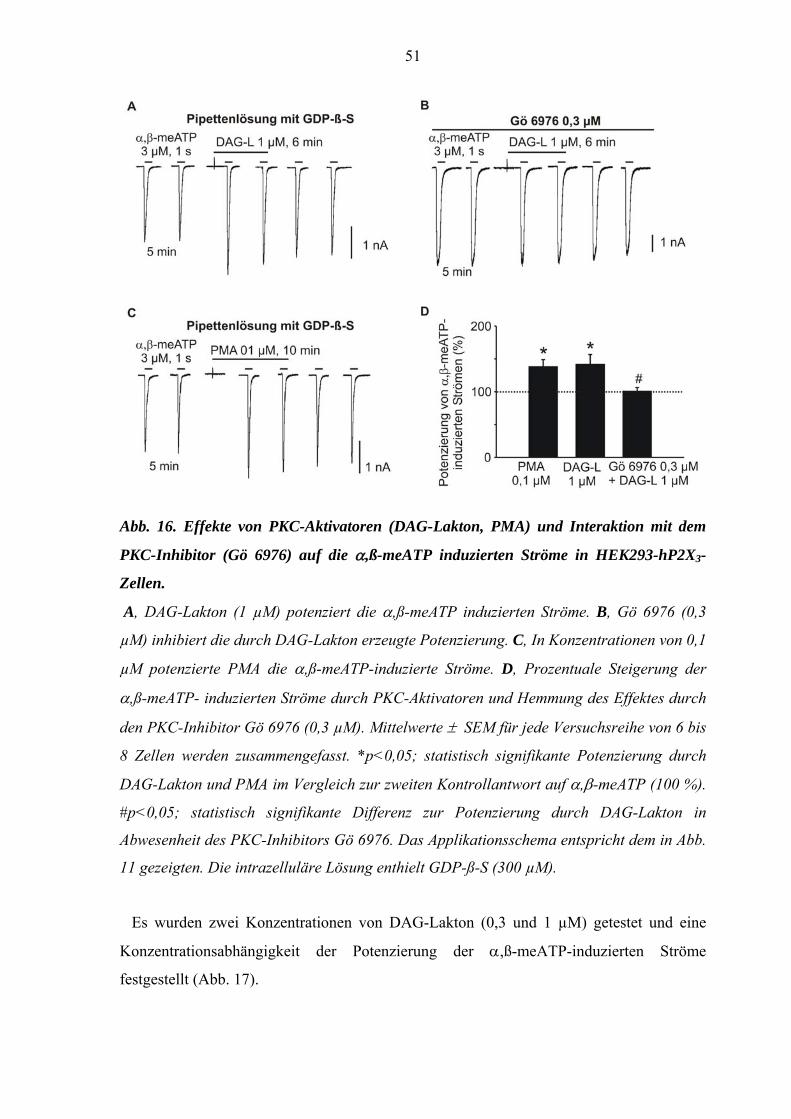
51
Abb. 16. Effekte von PKC-Aktivatoren (DAG-Lakton, PMA) und Interaktion mit dem
PKC-Inhibitor (Gö 6976) auf die α,ß-meATP induzierten Ströme in HEK293-hP2X3-
Zellen.
A, DAG-Lakton (1 µM) potenziert die α,ß-meATP induzierten Ströme. B, Gö 6976 (0,3
µM) inhibiert die durch DAG-Lakton erzeugte Potenzierung. C, In Konzentrationen von 0,1
µM potenzierte PMA die α,ß-meATP-induzierte Ströme. D, Prozentuale Steigerung der
α,ß-meATP- induzierten Ströme durch PKC-Aktivatoren und Hemmung des Effektes durch
den PKC-Inhibitor Gö 6976 (0,3 µM). Mittelwerte ± SEM für jede Versuchsreihe von 6 bis
8 Zellen werden zusammengefasst. *p<0,05; statistisch signifikante Potenzierung durch
DAG-Lakton und PMA im Vergleich zur zweiten Kontrollantwort auf α,β-meATP (100 %).
#p<0,05; statistisch signifikante Differenz zur Potenzierung durch DAG-Lakton in
Abwesenheit des PKC-Inhibitors Gö 6976. Das Applikationsschema entspricht dem in Abb.
11 gezeigten. Die intrazelluläre Lösung enthielt GDP-ß-S (300 µM).
Es wurden zwei Konzentrationen von DAG-Lakton (0,3 und 1 µM) getestet und eine
Konzentrationsabhängigkeit der Potenzierung der α,ß-meATP-induzierten Ströme
festgestellt (Abb. 17).
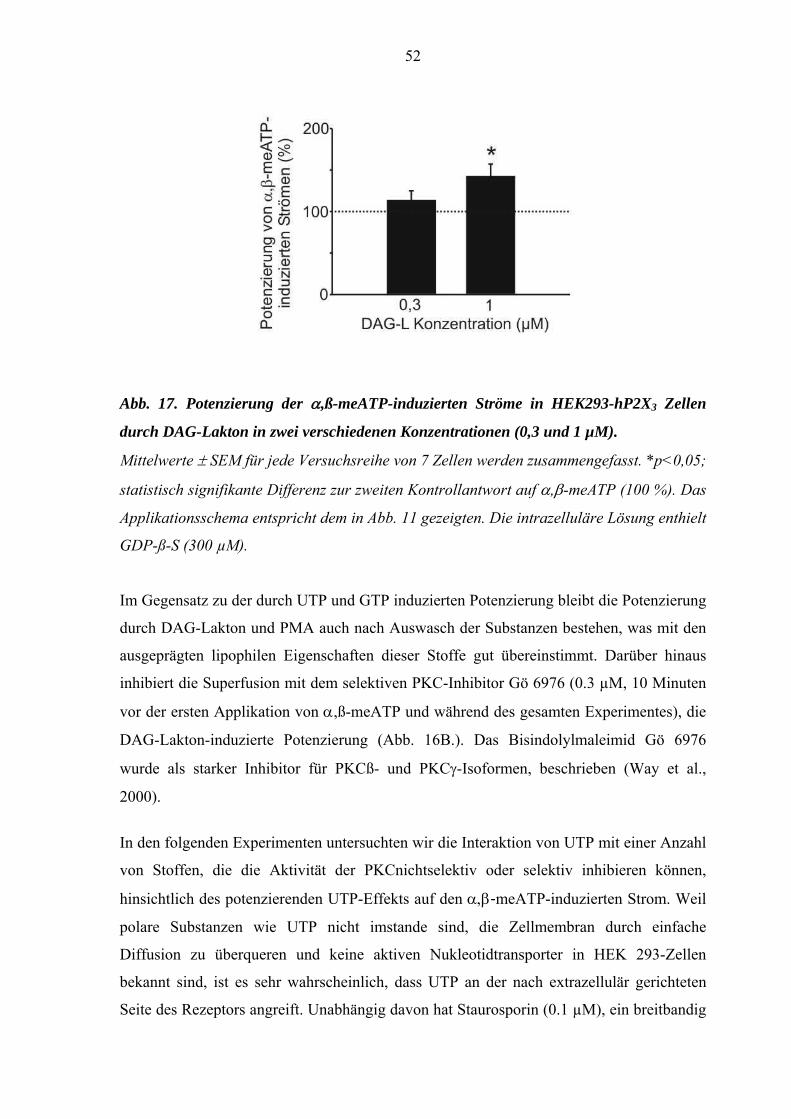
52
Abb. 17. Potenzierung der α,ß-meATP-induzierten Ströme in HEK293-hP2X3 Zellen
durch DAG-Lakton in zwei verschiedenen Konzentrationen (0,3 und 1 µM).
Mittelwerte ± SEM für jede Versuchsreihe von 7 Zellen werden zusammengefasst. *p<0,05;
statistisch signifikante Differenz zur zweiten Kontrollantwort auf α,β-meATP (100 %). Das
Applikationsschema entspricht dem in Abb. 11 gezeigten. Die intrazelluläre Lösung enthielt
GDP-ß-S (300 µM).
Im Gegensatz zu der durch UTP und GTP induzierten Potenzierung bleibt die Potenzierung
durch DAG-Lakton und PMA auch nach Auswasch der Substanzen bestehen, was mit den
ausgeprägten lipophilen Eigenschaften dieser Stoffe gut übereinstimmt. Darüber hinaus
inhibiert die Superfusion mit dem selektiven PKC-Inhibitor Gö 6976 (0.3 µM, 10 Minuten
vor der ersten Applikation von α,ß-meATP und während des gesamten Experimentes), die
DAG-Lakton-induzierte Potenzierung (Abb. 16B.). Das Bisindolylmaleimid Gö 6976
wurde als starker Inhibitor für PKCß- und PKCγ-Isoformen, beschrieben (Way et al.,
2000).
In den folgenden Experimenten untersuchten wir die Interaktion von UTP mit einer Anzahl
von Stoffen, die die Aktivität der PKCnichtselektiv oder selektiv inhibieren können,
hinsichtlich des potenzierenden UTP-Effekts auf den α,β-meATP-induzierten Strom. Weil
polare Substanzen wie UTP nicht imstande sind, die Zellmembran durch einfache
Diffusion zu überqueren und keine aktiven Nukleotidtransporter in HEK 293-Zellen
bekannt sind, ist es sehr wahrscheinlich, dass UTP an der nach extrazellulär gerichteten
Seite des Rezeptors angreift. Unabhängig davon hat Staurosporin (0.1 µM), ein breitbandig

53
wirkender Inhibitor von verschiedenen Proteinkinasen, und ein (nominell) Mg2+-freies
Medium, welches typischerweise Proteinkinase-abhängige Reaktionen inhibiert (Christie et
al., 1992), den Potenzierungseffekt durch UTP unterdrückt (Abb. 18.). Der selektive,
membranpermeable PKC-Inhibitor Gö 6976, der nichtselektive Ekto-PKC-Inhibitor K252b
(0.2 µM) (Kase et al., 1987) und das membranundurchlässige PKC-Inhibitorpeptid (10
µM) inhibierten alle die UTP-induzierte Potenzierung der α,ß-meATP-Ströme (Abb. 18).
Zusammenfassend kann gesagt werden, dass UTP durch ekto-PKC-vermittelte Übertragung
der terminalen Phosphatgruppen von Nukleotiden die extrazellulären
Phosphorylierungstellen des P2X3-Rezeptors zu beeinflussen scheint.
Abb. 18. Effekt von verschiedenen PKC-Inhibitoren auf die α,ß-meATP-induzierten
Ströme in HEK293-hP2X3 Zellen.
Das Applikationsschema entspricht dem in Abb. 11. dargestellten. Mittelwerte ± SEM für
jede Versuchsreihe von mindesten 6 bis 9 Zellen werden zusammengefasst. *p<0,05;
statistisch signifikante Differenz zur zweiten Kontrollantwort (100 %).
3.3. Einfluss von Mutationen auf die UTP-induzierte Potenzierung der α,ß-
meATP-induzierten Ströme an mit P2X3-Rezeptoren transient transfizierten
HEK293-Zellen
Im Gegensatz zu den bisher beschriebenen Experimenten waren die im folgenden
verwendeten HEK 293-Zellen hier nicht stabil sondern mit dem P2X3-Rezeptor oder einer

54
seiner Mutanten transient transfiziert. Um die funktionell relevanten PKC-
Phosphorylierungsstellen am P2X3-Rezeptor zu indentifizieren, wurden die Ser- und Thr-
Reste innerhalb von fünf der sieben Konsensus-Phosphorylierungsstellen nacheinander
durch die neutrale Aminosäure Ala ersetzt. Eine intrazelluläre PKC Stelle, die in allen
P2X-Rezeptoren hochkonserviert ist, befindet sich N-terminal (PKC1, 12-14), während die
zusätzlichen Stellen extrazellulär lokalisiert sind (PKC2, 134-136; PKC3, 178-180; PKC4,
196-198; PKC6, 269-271) (Mager et al., 2004). Die PKC5-Stelle wurde in unseren
Experimenten nicht verändert. Zusätzlich wurden noch zwei Ser-Reste (S110, S267), die
im humanen P2X3-Rezeptor, aber nicht in dem der Ratte vorhanden sind, zu Ala mutiert.
Keine der Mutationen verhinderte die Membranexpression des P2X3-Rezeptors oder seine
Empfindlichkeit gegenüber extrazellulären Nukleotiden in den HEK 293-Zellen. In allen
Fällen verursachten α,ß-meATP-Applikationen reproduzierbare Stromamplituden (für die
zweite Stromantwort im üblichen Applikationsschema, siehe Abb. 19).
Abb. 19. Einfluss der Mutationen von Ser- und Thr-Resten zu Ala in den PKC
Konsensus-Phosphorylierungsstellen des Wildtyp-P2X3-Rezeptors auf seine
elektrophysiologischen Eigenschaften.
Die intrazelluläre Lösung enthielt GDP-ß-S 300 µM. Das Applikationsschema entspricht
dem in Abb. 11 beschriebenen. A, Es ist keine potenzierende Wirkung von UTP auf den
α,β-meATP-Strom an der Mutante T134A zu beobachten (Inaktivierung der PKC2-Stelle;
s. Abb. 16). B. Die Potenzierung an der Mutante S267A bleibt erhalten (keine Mutationen
in einer der Konsensus-PKC-Stellen).
Trotz der T13A- (PKC1-Stelle), S267A- und S269A-Mutationen (PKC4-Stelle und ihre
unmittelbare Nachbarschaft) beobachteten wir eine unveränderte Potenzierung der α,ß-

55
meATP-induzierten Ströme durch UTP, während die Mutationen T134A (PKC2-Stelle),
S178A und N177V (PKC3-Stelle) die Steigerung derselben Ströme durch UTP
verhinderten (Abb. 20). Bereits aus dieser Abbildung ist ersichtlich, dass die Leitfähigkeit
der Mutante T134A nach der Applikation von 3 μM α,β-meATP bedeutend geringer war
als die der Mutante S267A (s. die unterschiedliche Stromeichung in Abb. 19A und B; Abb.
20). Diese Änderungen sollten jedoch nicht überinterpretiert werden, da infolge der
unterschiedlichen Expression des Rezeptorproteins nach transienter Transfektion sehr
variable Stromgrößen beobachtet werden können. Es fällt auf, dass nach der Substitution
von Thr in Position-12 mit Ala, nach α,ß-meATP-Applikation kein messbarer Strom
hervorgerufen werden konnte. Dieses Ergebnis publizierten bereits Paukert et al. (2001).
Die Autoren vermuteten eine mutationsverursachte Störung der P2X3-Kanalexpression in
der Zellmembran. Deswegen veränderten wir Thr nur in Position-13 auf Ala; diese
Mutation hemmte nicht die Stromamplitude, sondern schien sie sogar zu vergrößern (Abb.
20).
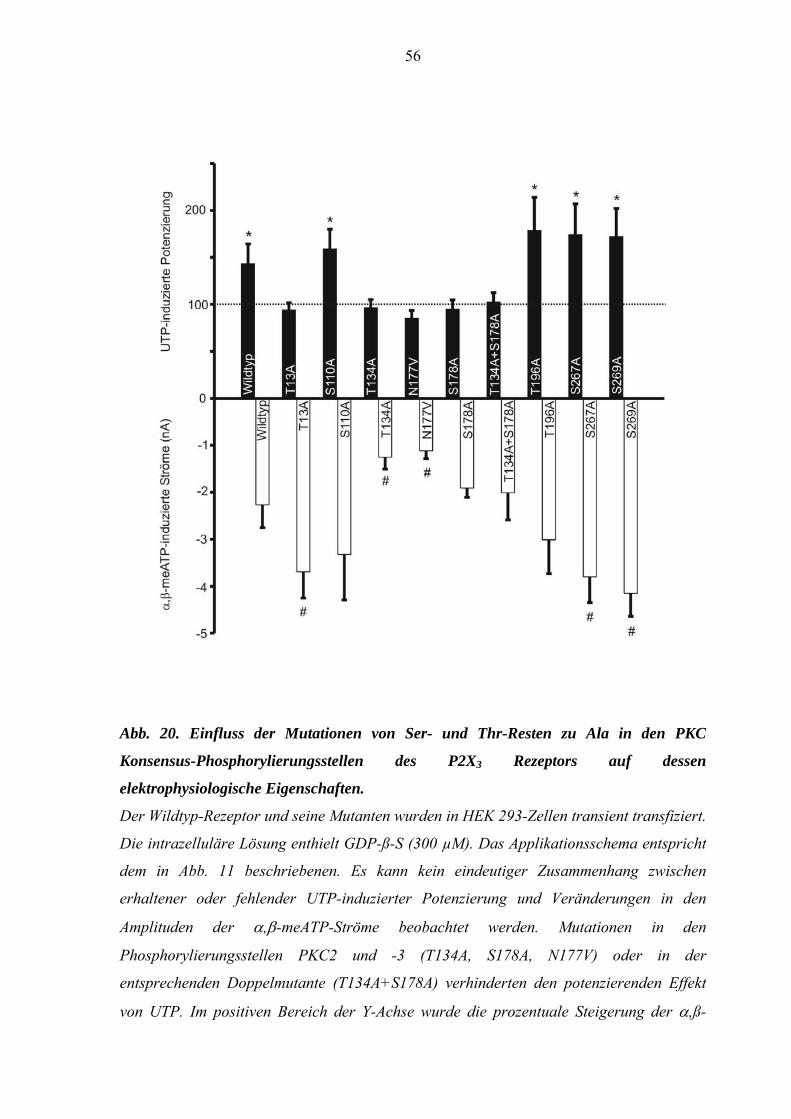
56
Abb. 20. Einfluss der Mutationen von Ser- und Thr-Resten zu Ala in den PKC
Konsensus-Phosphorylierungsstellen des P2X3 Rezeptors auf dessen
elektrophysiologische Eigenschaften.
Der Wildtyp-Rezeptor und seine Mutanten wurden in HEK 293-Zellen transient transfiziert.
Die intrazelluläre Lösung enthielt GDP-ß-S (300 µM). Das Applikationsschema entspricht
dem in Abb. 11 beschriebenen. Es kann kein eindeutiger Zusammenhang zwischen
erhaltener oder fehlender UTP-induzierter Potenzierung und Veränderungen in den
Amplituden der α,β-meATP-Ströme beobachtet werden. Mutationen in den
Phosphorylierungsstellen PKC2 und -3 (T134A, S178A, N177V) oder in der
entsprechenden Doppelmutante (T134A+S178A) verhinderten den potenzierenden Effekt
von UTP. Im positiven Bereich der Y-Achse wurde die prozentuale Steigerung der α,ß-

57
meATP-induzierten Ströme durch UTP dargestellt. Mittelwert ± SEM von mindestens 7
Zellen; *p<0,05; statistisch signifikanter Unterschied im Vergleich zur zweiten
Stromamplitude, die auf 100% gesetzt wurde. Im negativen Bereich der Y-Achse wurden
Mittelwerte ± SEM der Stromamplituden aufgetragen. #p>0,05; statistisch signifikanter
Unterschied des Mittelwerts der Stromamplitude im Vergleich zu dem des Wildtyp-
Rezeptors. Das Applikationsschema entspricht dem in Abb. 20 beschriebenen. Die
intrazelluläre Lösung enthielt GDP-ß-S 300 µM in allen Versuchen.
Wenn α,β-meATP (3 µM) sechsmal im Abstand von 5 min und für jeweils 1 s mit dem
üblichen Applikationsschema auf transient mit dem Wildtyp P2X3-Rezeptor oder seinen
Mutanten transfizierte HEK 293-Zellen appliziert wurde, induzierte UTP nach einer
Applikationszeit von 5 min eine kleinere Potenzierung der α,ß-meATP-induzierten Ströme
(43.6 ± 21.1%; n=7) (Abb. 20) als in stabil transfizierten HEK 293-hP2X3-Zellen (66.4 ±
17.1%; n=7) (Abb. 20). Dies führen wir auf die geringere Expressionsdichte des P2X3-
Rezeptors in der HEK293-Zellmembran bei transienter als bei permamenter Transfektion
zurück.
Die Steigerung der α,ß-meATP-Ströme durch UTP blieb in den Mutanten S110A, T196A,
S267A und S269A erhalten (Abb. 20). Daraus kann gefolgert werden, dass Mutationen in
den Ser-Resten, die beim P2X3-Rezeptor des Menschen nicht aber bei dem der Ratte (S110,
S267) vorkommen, die potenzierende Wirkung von UTP nicht beeinflussen, ebenso wie
Mutationen in den Thr und Ser-Resten, welche innerhalb der extrazellulären PKC-
Phosphorylierungsstellen PKC4 und PKC6 (T196A, S269A) lokalisiert sind. Im Gegensatz
dazu, gelang es uns mit Mutationen in den Thr- und Ser-Resten, die in den extrazellulären
PKC-Phosphorylierungsstellen PKC2 und PKC3 (T134A, S178A) lokalisiert waren, den
UTP-Effekt zu unterbinden.
Sowohl die gleichzeitige Substitution von Thr-134 und Ser-178 durch Ala (T134A+S178A)
als auch die Substitution von Asn-177 neben dem Ser-178 durch Val (N177V) führten zu
UTP-resistenten Mutanten (Abb. 20).
Überraschenderweise verhinderte die Mutation von Thr zu Ala im intrazellulären N-
Terminus die potenzierende Wirkung von UTP ebenfalls, obwohl UTP als stark polare
Verbindung die Zellmembran nicht durchdringen dürfte und deshalb keine
Phosphorylierung an der PKC1-Stelle durch UTP zu erwarten ist (Abb. 20). Gründe hierfür
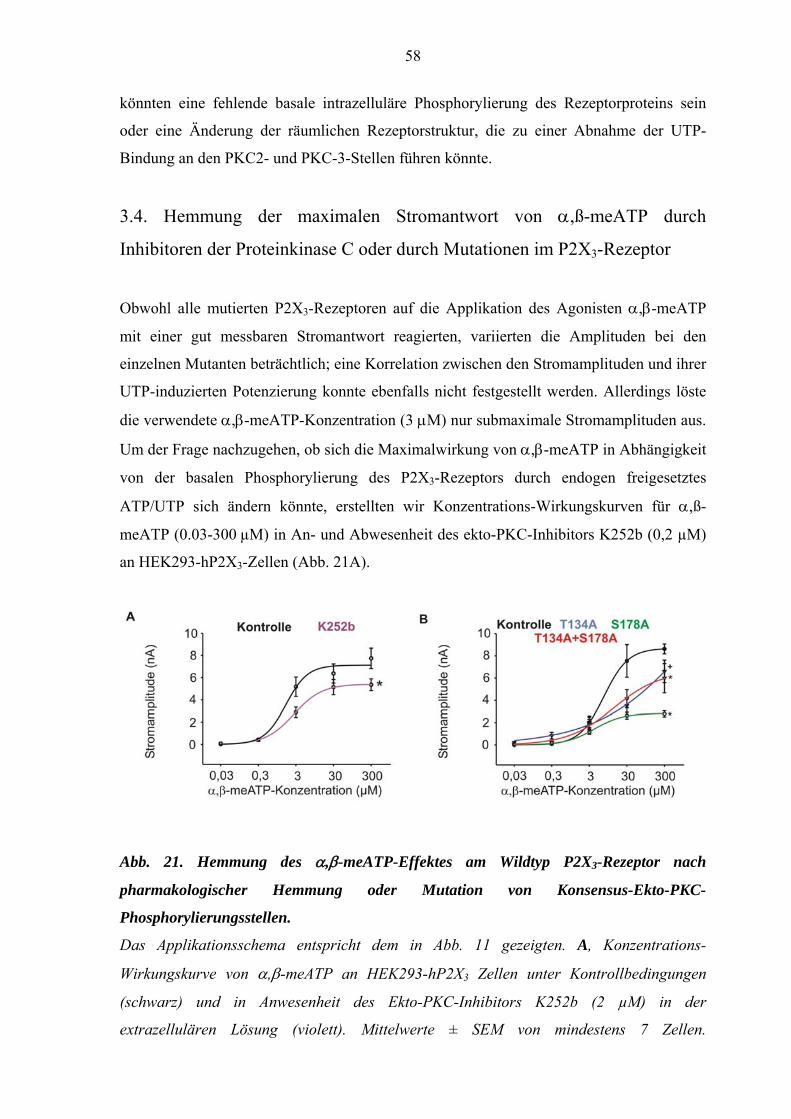
58
könnten eine fehlende basale intrazelluläre Phosphorylierung des Rezeptorproteins sein
oder eine Änderung der räumlichen Rezeptorstruktur, die zu einer Abnahme der UTP-
Bindung an den PKC2- und PKC-3-Stellen führen könnte.
3.4. Hemmung der maximalen Stromantwort von α,ß-meATP durch
Inhibitoren der Proteinkinase C oder durch Mutationen im P2X3-Rezeptor
Obwohl alle mutierten P2X3-Rezeptoren auf die Applikation des Agonisten α,β-meATP
mit einer gut messbaren Stromantwort reagierten, variierten die Amplituden bei den
einzelnen Mutanten beträchtlich; eine Korrelation zwischen den Stromamplituden und ihrer
UTP-induzierten Potenzierung konnte ebenfalls nicht festgestellt werden. Allerdings löste
die verwendete α,β-meATP-Konzentration (3 μM) nur submaximale Stromamplituden aus.
Um der Frage nachzugehen, ob sich die Maximalwirkung von α,β-meATP in Abhängigkeit
von der basalen Phosphorylierung des P2X3-Rezeptors durch endogen freigesetztes
ATP/UTP sich ändern könnte, erstellten wir Konzentrations-Wirkungskurven für α,ß-
meATP (0.03-300 µM) in An- und Abwesenheit des ekto-PKC-Inhibitors K252b (0,2 µM)
an HEK293-hP2X3-Zellen (Abb. 21A).
Abb. 21. Hemmung des α,β-meATP-Effektes am Wildtyp P2X3-Rezeptor nach
pharmakologischer Hemmung oder Mutation von Konsensus-Ekto-PKC-
Phosphorylierungsstellen.
Das Applikationsschema entspricht dem in Abb. 11 gezeigten. A, Konzentrations-
Wirkungskurve von α,β-meATP an HEK293-hP2X3 Zellen unter Kontrollbedingungen
(schwarz) und in Anwesenheit des Ekto-PKC-Inhibitors K252b (2 µM) in der
extrazellulären Lösung (violett). Mittelwerte ± SEM von mindestens 7 Zellen.

59
*p<0,05;statistisch signifikanter Unterschied zum Effekt der höchsten α,β-meATP-
Konzentration unter Kontrollbedingungen. B, Konzentrations-Wirkungskurven von α,β-
meATP an HEK293 Zellen, die mit dem Wildtyp P2X3-Rezeptor (schwarz) seinen Mutanten
T134A (blau), S178A (grün) und T134A + S178A (rot) transient transfiziert wurden.
Mittelwerte ± SEM von mindestens 6 Zellen. *p<0,05; statistisch signifikanter Unterschied
zum Effekt der höchsten α,β-meATP-Konzentration am Wildtyp P2X3-Rezeptor (schwarz).
Der membranunpermeable PKC-Inhibitor K252b verringerte die maximale Stromantwort
auf α,ß-meATP, beeinflusste aber den EC50-Wert nicht. Daher scheint, trotz vergleichbarer
Wirkpotenz des Agonisten, die Entwicklung der vollen intrinsischen Aktivität eine basale
Ekto-Phosphorylierung des P2X3-Rezeptors vorauszusetzen.
Diese Annahme wird durch weitere Experimente gestützt, in denen Konzentrations-
Wirkungskurven von α,ß-meATP an mit dem Wildtyp P2X3-Rezeptor transient
transfizierten HEK 293-Zellen erstellt wurden; zusätzlich wurden auch die Mutanten
T134A, S178A und T134A+S178A in diese Untersuchungen mit einbezogen (Abb. 21 B).
Aminosäure-Substitutionen in den relevanten Ekto-PKC-Phosphorylierungsstellen des
Rezeptors führten ähnlich wie die pharmakologische Ausschaltung der Ekto-PKC durch
K252b, zu einer Abnahme der maximalen Stromantworten auf α,β-meATP. Die EC50
Werte blieben auch in dieser experimentellen Serie unverändert.
Es wurde nicht untersucht, ob der Austausch der Ser- oder Thr-Reste durch Ala in den
relevanten Ekto-PKC-Phosphorylierungsstellen PKC2 und -3 die Synthese des Rezeptors
oder seinen Einbau in die Zellmembran beeinflussen können. Da jedoch die verwendete
submaximale Konzentration von α,β-meATP (3 μM) an jeder einzelnen Mutanten gut
messbare Stromantworten auslöste, können gravierende Modifikationen der
Rezeptorsynthese oder des Rezeptortraffickings ausgeschlossen werden. Die potenzierende
Wirkung von UTP wäre durch solche modulatorischen Effekte sowieso nicht beeinträchtigt
gewesen.

3.5. Effekt von Mutationen des P2X3-Rezeptors auf die Kinetik der
Rezeptorantwort
In den vorhergehenden Experimenten führte die Substitution von Thr-134 und Ser-178
durch Ala (bei unveränderten Thr-196- und Ser-269-Resten) dazu, dass die potenzierende
Wirkung von UTP unterbunden wurde. Daraus lässt sich schließen, dass die PKC2- und
PKC3-Phosphorylierungstellen T134 und S178 für die Regulierung der P2X3-
Rezeptorströme essentiell sind. In den folgenden Experimenten wurde an den Ekto-PKC-
Phosphorylierungsstellen PKC2, -3, -4 und -6 des P2X3-Rezeptors die neutrale Aminosäure
Ala stets durch die negativ geladene Aminosäure Asp ersetzt. Dadurch sollte die
Phosphorylierungsreaktion an den einzelnen Konsensus-Phosphorylierungsstellen durch
UTP nachgeahmt und ihre Daueraktivierung verursacht werden. Tatsächlich verhinderte
die Asp-Substitution an allen vier PKC-Stellen die durch UTP (3 μM) verursachte
Potenzierung der α,ß-meATP-induzierten Ströme und beeinflusste sowohl die
Stromamplituden als auch ihre Aktivierungs- und Desensibilisierungs-Kinetik. Die Ala-
Substitution an denselben PKC-Phosphorylierungsstellen hatte keine nennenswerte
Auswirkung auf die Stromamplitude und ihr Desensibilisierungsverhalten.
Konzentrations-Wirkungskurven von α,β-meATP an HEK293-Zellen, die mit dem Wildtyp
P2X3-Rezeptor oder seinen Mutanten T134D oder S178D transfiziert wurden um die PKC2
und -3-Stellen zu daueraktivieren und gleichzeitig auszuschalten, werden in Abb. 22A
dargestellt.
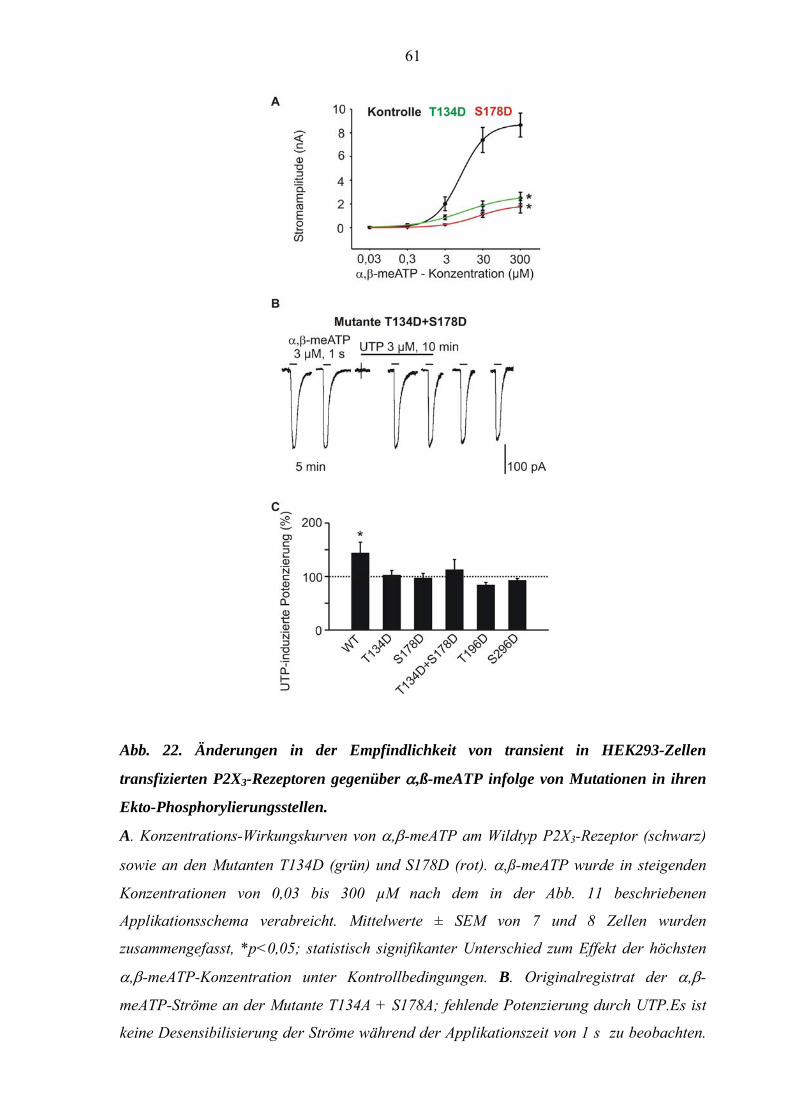
61
Abb. 22. Änderungen in der Empfindlichkeit von transient in HEK293-Zellen
transfizierten P2X3-Rezeptoren gegenüber α,ß-meATP infolge von Mutationen in ihren
Ekto-Phosphorylierungsstellen.
A. Konzentrations-Wirkungskurven von α,β-meATP am Wildtyp P2X3-Rezeptor (schwarz)
sowie an den Mutanten T134D (grün) und S178D (rot). α,ß-meATP wurde in steigenden
Konzentrationen von 0,03 bis 300 µM nach dem in der Abb. 11 beschriebenen
Applikationsschema verabreicht. Mittelwerte ± SEM von 7 und 8 Zellen wurden
zusammengefasst, *p<0,05; statistisch signifikanter Unterschied zum Effekt der höchsten
α,β-meATP-Konzentration unter Kontrollbedingungen. B. Originalregistrat der α,β-
meATP-Ströme an der Mutante T134A + S178A; fehlende Potenzierung durch UTP.Es ist
keine Desensibilisierung der Ströme während der Applikationszeit von 1 s zu beobachten.
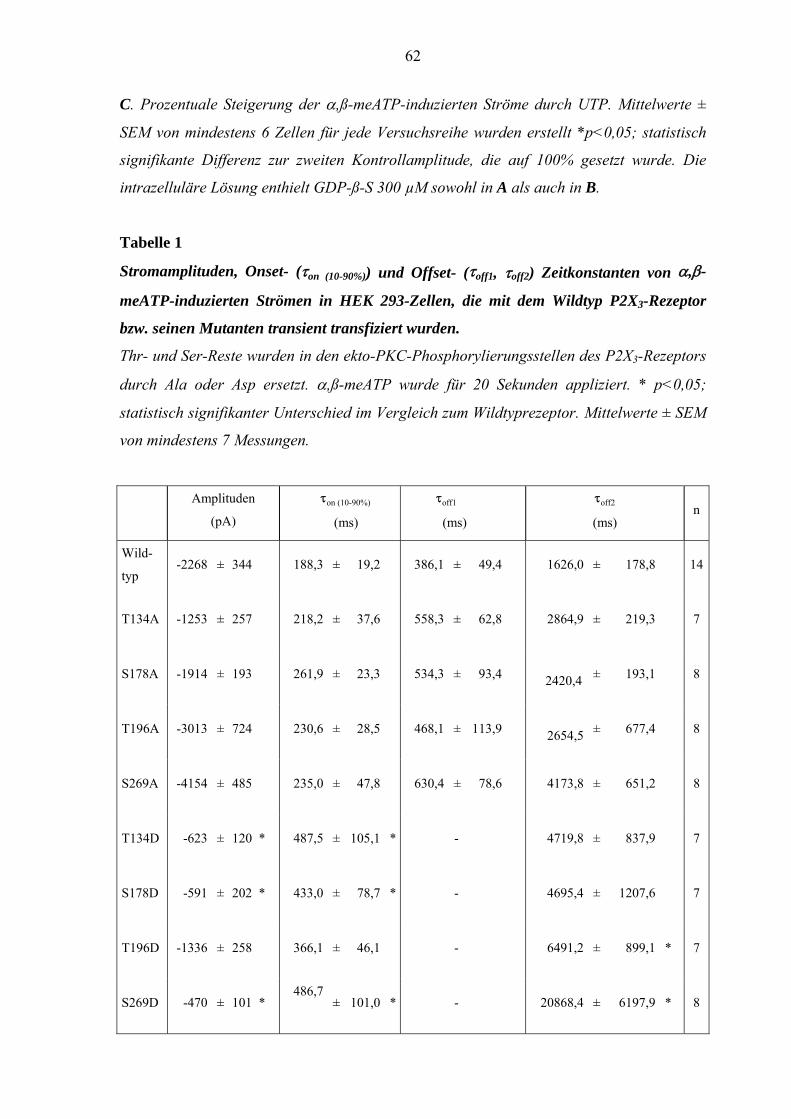
62
C. Prozentuale Steigerung der α,ß-meATP-induzierten Ströme durch UTP. Mittelwerte ±
SEM von mindestens 6 Zellen für jede Versuchsreihe wurden erstellt *p<0,05; statistisch
signifikante Differenz zur zweiten Kontrollamplitude, die auf 100% gesetzt wurde. Die
intrazelluläre Lösung enthielt GDP-ß-S 300 µM sowohl in A als auch in B.
Tabelle 1
Stromamplituden, Onset- (τon (10-90%)) und Offset- (τoff1, τoff2) Zeitkonstanten von α,β-
meATP-induzierten Strömen in HEK 293-Zellen, die mit dem Wildtyp P2X3-Rezeptor
bzw. seinen Mutanten transient transfiziert wurden.
Thr- und Ser-Reste wurden in den ekto-PKC-Phosphorylierungsstellen des P2X3-Rezeptors
durch Ala oder Asp ersetzt. α,ß-meATP wurde für 20 Sekunden appliziert. * p<0,05;
statistisch signifikanter Unterschied im Vergleich zum Wildtyprezeptor. Mittelwerte ± SEM
von mindestens 7 Messungen. Amplituden
(pA)
τon (10-90%)
(ms)
τoff1
(ms)
τoff2
(ms) n
Wild-
typ -2268 ± 344 188,3 ± 19,2 386,1 ± 49,4 1626,0 ± 178,8 14
T134A -1253 ± 257 218,2 ± 37,6 558,3 ± 62,8 2864,9 ± 219,3 7
S178A -1914 ± 193 261,9 ± 23,3 534,3 ± 93,4
2420,4 ± 193,1 8
T196A -3013 ± 724 230,6 ± 28,5 468,1 ± 113,9
2654,5 ± 677,4 8
S269A -4154 ± 485 235,0 ± 47,8 630,4 ± 78,6 4173,8 ± 651,2 8
T134D -623 ± 120 * 487,5 ± 105,1 * - 4719,8 ± 837,9 7
S178D -591 ± 202 * 433,0 ± 78,7 * - 4695,4 ± 1207,6 7
T196D -1336 ± 258 366,1 ± 46,1 - 6491,2 ± 899,1 * 7
S269D -470 ± 101 * 486,7
± 101,0 * -
20868,4 ± 6197,9 * 8

63
Diese Experimente demonstrieren, dass die Mutation T134D die frühe und schnelle
Desensibilisierungskomponente der α,β-meATP-induzierten Ströme unterdrückt. Es wurde
vermutet, dass die dauerhafte Aktivierung und Desensibilisierung der P2X3 Rezeptor-
Mutanten jede weitere Modulation durch UTP verhindert (Abb. 22B, C; Tabelle 1). Keine
der Mutationen unterdrückte jedoch die Membranexpression des P2X3-Rezeptors in den
HEK 293-Zellen oder verhinderte die Empfindlichkeit gegenüber extrazellulären
Nukleotiden, da die α,β-meATP-Applikation (3 μM) immer reproduzierbare
Stromamplituden auslöste.
Die eingehende Prüfung der Ergebnisse dokumentiert, dass die Ala-Substitutionen α,β-
meATP-induzierte Stromamplituden nicht wesentlich veränderten, während die Asp-
Substitutionen zu einer ausgeprägten Verminderung dieser Amplituden führten (Abb. 22 B;
Table 1).
Die Amplitude der schnell desensibilisierenden P2X1-Rezeptorströme wird durch zwei
gegenläufige Mechanismen bestimmt; einerseits durch die agonistische Rezeptorbesetzung
(Rettinger und Schmalzing, 2003) und zum anderen durch die begleitende
Desensibilisierung (Rettinger und Schmalzing, 2004). Um die Desensibilisierung des P2X3-
Wildtyprezeptors und seiner Mutanten quantifizieren zu können, bestimmten wir die
Zeitkonstante des Stromabfalls zur Ausgangslinie während einer 20 s dauernden
Applikation von α,β-meATP (3 μM).
Die Offsetzeitkonstante wurde als biexponentiell, mit schneller Kurzzeitphase und
langsamer Langzeitphase, berechnet (τoff1, τoff2; Abb. 23) (Fabbretti et al., 2004). Zusätzlich
wurden noch von denselben Registraten die Onsetzeitkonstanten (τon(10–90%)) bestimmt.
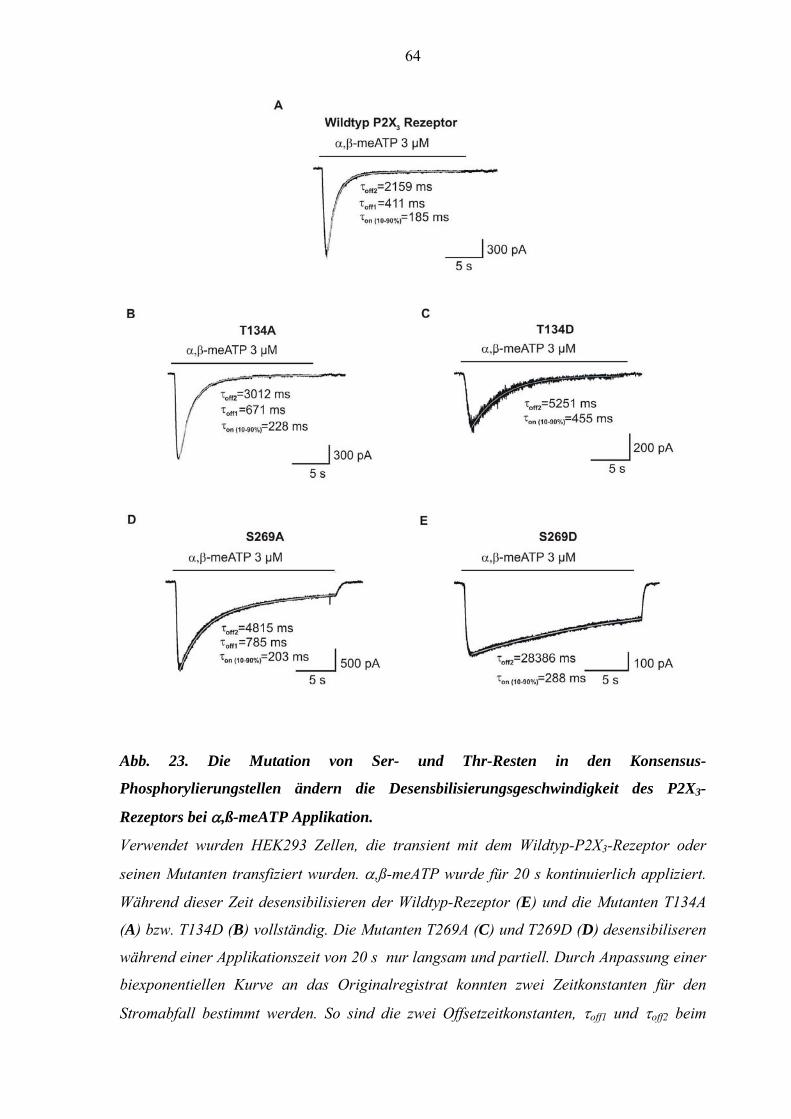
64
Abb. 23. Die Mutation von Ser- und Thr-Resten in den Konsensus-
Phosphorylierungstellen ändern die Desensbilisierungsgeschwindigkeit des P2X3-
Rezeptors bei α,ß-meATP Applikation.
Verwendet wurden HEK293 Zellen, die transient mit dem Wildtyp-P2X3-Rezeptor oder
seinen Mutanten transfiziert wurden. α,ß-meATP wurde für 20 s kontinuierlich appliziert.
Während dieser Zeit desensibilisieren der Wildtyp-Rezeptor (E) und die Mutanten T134A
(A) bzw. T134D (B) vollständig. Die Mutanten T269A (C) und T269D (D) desensibiliseren
während einer Applikationszeit von 20 s nur langsam und partiell. Durch Anpassung einer
biexponentiellen Kurve an das Originalregistrat konnten zwei Zeitkonstanten für den
Stromabfall bestimmt werden. So sind die zwei Offsetzeitkonstanten, τoff1 und τoff2 beim

65
Wildtyprezeptor und den Mutanten T134A (A) und T269A (C) vorhanden. Die
Offsetzeitkonstanten der Mutanten T134D (B) und T269D (D) können mit
monoexponentiellen Kurven und mit nur einer Zeitkonstante τoff2 gefitted werden. Die
Onsetzeitkonstanten τon(10-90) wurden ebenfalls berechnet.
Diese Experimente wurden durchgeführt, um festzustellen, ob strukturelle Änderungen der
PKC-Phosphorylierungsstellen (Ala- und Asp-Substitutionen) einerseits die endogene
Phosphorylierung verhindern und andererseits die Desensibilisierungskinetik ändern
können. Es wurde erwartet, dass eine Ala-Substitution den Potenzierungseffekt des
endogenen ATP ausschalten wird, während die Asp-Substitution die Wirkung des
endogenen ATP (oder exogenen UTP) nachahmen könnte. Wenn allerdings die vier PKC-
Phosphorylierungsstellen auf der extrazellulären Schleife des Rezeptors durch Ersatz von
Ser oder Thr durch Ala (T134A, S178A, T196A, S269A) der Reihe nach mutiert wurden,
blieben die beiden Onsetzeitkonstanten τoff1 und τoff2 sowie die Onsetzeitkonstante τon(10-90),
mit Ausnahme von S269A, wo τoff1 und τoff2, nicht aber τon(10-90) erhöht sind, unverändert
(Abb. 23; Tabelle 1). Die Größe der Stromamplituden war relativ stabil. Die einzige
Ausnahme bildet wieder die S269A-Mutante, wo die Ströme angestiegen sind (Abb. 23;
Tabelle 1). Substitution von Ser oder Thr durch Asp innerhalb der oben genannten PKC-
Phosphorylierungsstellen, veränderte den biexponentiellen Abfall auf monoexponentiell,
vermutlich durch Eliminierung der Konstante τoff1 und gleichzeitiger Steigerung der
Konstante τoff2 (Abb. 23; Tabelle 1). Außerdem wurde τon(10–90%) in allen Asp-Mutanten
Mutanten erhöht, obwohl für T196D diese Veränderung die statistische Signifikanzgrenze
nicht erreichte (Tabelle 1). Die langsame Onsetzeitkonstante der Stromantwort in
Verbindung mit dem Verschwinden der schnellen τoff1 des Abfalls resultiert in einem sehr
viel langsameren Strom bei den Asp-Mutanten als beimWildtyprezeptor (Abb. 23; Tabelle
1). Außerdem stieg die späte Konstante τoff2 durch Asp-Substitutionen an den PKC-Stellen
2 bis 6 graduell an (Abb. 23; Tabelle 1).
3.6. Potenzierung der α,β-meATP-induzierten Ströme durch UTP bei DRG-
Neuronen und Interaktionen mit PKC-Inhibitoren
Für die folgenden Experimente wurden nozizeptive Hinterwurzel (DRG)-Neurone mit
kleinem Durchmesser genutzt. Obwohl diese Neurone sowohl mit dem schnell
desensibilisierenden homomeren P2X3-Rezeptor als auch dem heteromeren P2X2/3-
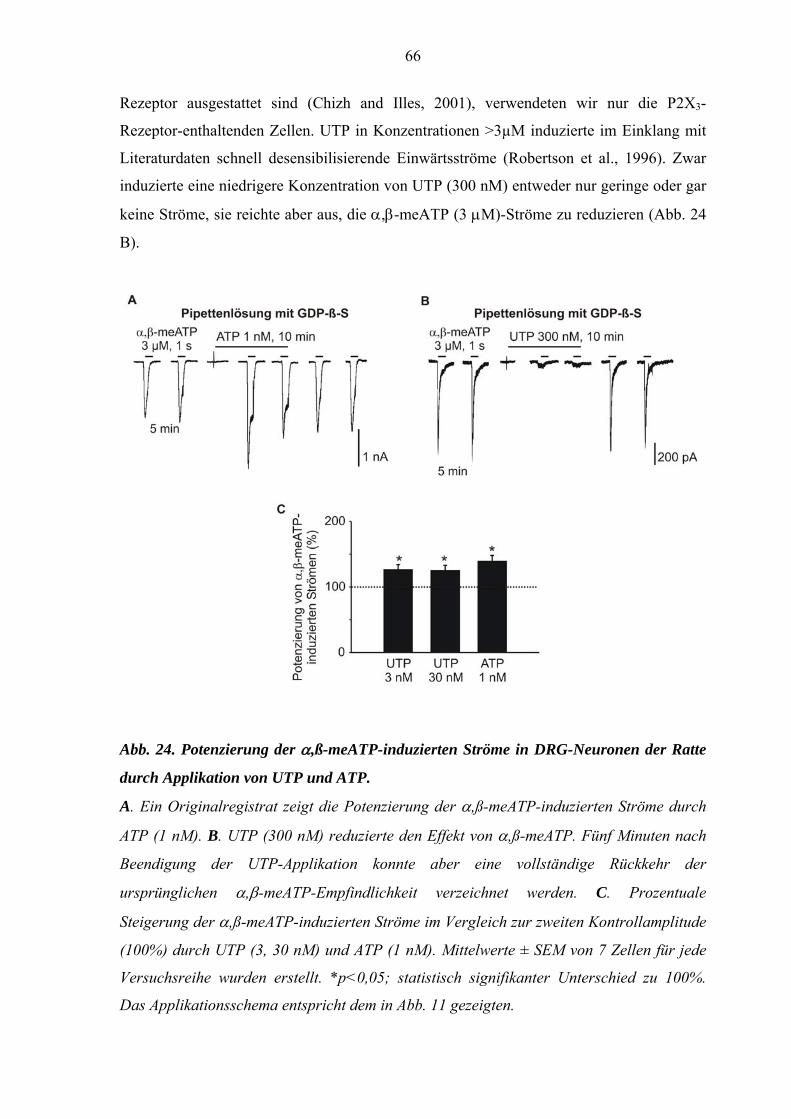
66
Rezeptor ausgestattet sind (Chizh and Illes, 2001), verwendeten wir nur die P2X3-
Rezeptor-enthaltenden Zellen. UTP in Konzentrationen >3µM induzierte im Einklang mit
Literaturdaten schnell desensibilisierende Einwärtsströme (Robertson et al., 1996). Zwar
induzierte eine niedrigere Konzentration von UTP (300 nM) entweder nur geringe oder gar
keine Ströme, sie reichte aber aus, die α,β-meATP (3 μM)-Ströme zu reduzieren (Abb. 24
B).
Abb. 24. Potenzierung der α,ß-meATP-induzierten Ströme in DRG-Neuronen der Ratte
durch Applikation von UTP und ATP.
A. Ein Originalregistrat zeigt die Potenzierung der α,ß-meATP-induzierten Ströme durch
ATP (1 nM). B. UTP (300 nM) reduzierte den Effekt von α,ß-meATP. Fünf Minuten nach
Beendigung der UTP-Applikation konnte aber eine vollständige Rückkehr der
ursprünglichen α,β-meATP-Empfindlichkeit verzeichnet werden. C. Prozentuale
Steigerung der α,ß-meATP-induzierten Ströme im Vergleich zur zweiten Kontrollamplitude
(100%) durch UTP (3, 30 nM) und ATP (1 nM). Mittelwerte ± SEM von 7 Zellen für jede
Versuchsreihe wurden erstellt. *p<0,05; statistisch signifikanter Unterschied zu 100%.
Das Applikationsschema entspricht dem in Abb. 11 gezeigten.
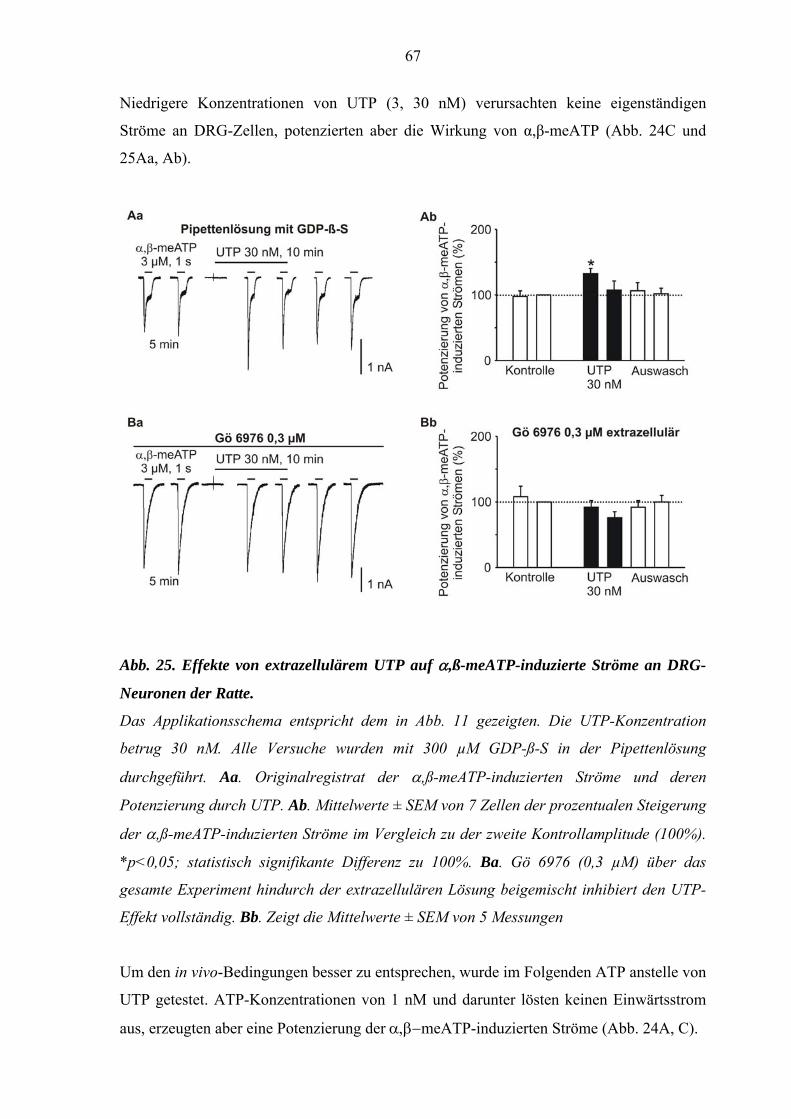
67
Niedrigere Konzentrationen von UTP (3, 30 nM) verursachten keine eigenständigen
Ströme an DRG-Zellen, potenzierten aber die Wirkung von α,β-meATP (Abb. 24C und
25Aa, Ab).
Abb. 25. Effekte von extrazellulärem UTP auf α,ß-meATP-induzierte Ströme an DRG-
Neuronen der Ratte.
Das Applikationsschema entspricht dem in Abb. 11 gezeigten. Die UTP-Konzentration
betrug 30 nM. Alle Versuche wurden mit 300 µM GDP-ß-S in der Pipettenlösung
durchgeführt. Aa. Originalregistrat der α,ß-meATP-induzierten Ströme und deren
Potenzierung durch UTP. Ab. Mittelwerte ± SEM von 7 Zellen der prozentualen Steigerung
der α,ß-meATP-induzierten Ströme im Vergleich zu der zweite Kontrollamplitude (100%).
*p<0,05; statistisch signifikante Differenz zu 100%. Ba. Gö 6976 (0,3 µM) über das
gesamte Experiment hindurch der extrazellulären Lösung beigemischt inhibiert den UTP-
Effekt vollständig. Bb. Zeigt die Mittelwerte ± SEM von 5 Messungen
Um den in vivo-Bedingungen besser zu entsprechen, wurde im Folgenden ATP anstelle von
UTP getestet. ATP-Konzentrationen von 1 nM und darunter lösten keinen Einwärtsstrom
aus, erzeugten aber eine Potenzierung der α,β−meATP-induzierten Ströme (Abb. 24A, C).

68
In Anwesenheit des PKC-Inhibitors Gö 6976 (0.3 µM) wurde die UTP-Potenzierung der
α,β-meATP-induzierten Ströme nicht mehr beobachtet (Abb. 25 Ba, Bb). Daher kann
gefolgert werden, dass niedrige Konzentrationen von ATP und UTP den nativen P2X3-
Rezeptor von DRG-Zellen gleichermaßen phosphorylieren.
Es fiel auf, dass relativ wenige DRG-Neurone mit P2X2/3-Rezeptoren ausgestattet waren.
Sie wurden nicht in die oben dargestellten Auswertungen aufgenommen, reagierten aber
trotzdem auf UTP mit einer Potenzierung der α,β-meATP-induzierten Ströme.
3.7. P2X7-EGFP Klonierung und Eigenschaften des Rezeptorkonstruktes
Um einen DNA-Vektor, der für ein P2X7-EGFP Fusionsprotein kodiert, herstellen zu
können, wurde der humane P2X7-Rezeptor in die multiple Klonierungsregion des pEGFP-
N1 Vektors von Clontech eingefügt. Die P2X7-DNA Sequenz wurde mit Hilfe einer PCR
amplifiziert, wobei durch überhängende Enden der P2X7-EGFP-for und P2X7-EGFP-rev
Primer jeweils eine Sse8387I bzw. KpnI Restriktionsschnittstelle eingeführt wurde. Der
P2X7-EGFP-rev Primer deletierte außerdem das Stopcodon der P2X7-Sequenz. Das
amplifizierte P2X7-Insert wurde anschließend mit den Restriktionsenzymen Sse8387I und
KpnI verdaut und über kompatible Enden in den mit PstI und KpnI verdauten pEGFP-N1
Vektor ligiert (Abb.26).

69
Abb. 26. Restriktionskarte und Klonierungsstelle des P2X7-Rezeptors im pEGFP-N1
Vektor.
Die Restriktionsschnittstellen für Kpn I und Pst I sind mit roten Pfeilen markiert. Das
EGFP-Gen ist durch den grünen Pfeil dargestellt. Die Lage des „SV40 early mRNA
polyadenylation“- Signals ist blau markiert. Das Kanamycin/Neomycin-Resistenzgen ist
durch einen blauen Pfeil dargestellt. Der pUK–Plasmid-Replikationsbeginn (pUK plasmid
replication origin) ist in hellblau, der menschliche Zytomegalovirus (CMV) immediate
early Promotor ist mit orangenem Pfeil dargestellt.
Die Klonierung erfolgte so, dass die offenen Leserahmen der P2X7- und EGFP-Sequenz
miteinander fusioniert wurden. Mit diesem Konstrukt konnte die Expression eines P2X7-
Rezeptors, an dessen C-Terminus sich ein EGFP Protein anschließt, in HEK Zellen erzielt
werden.
Der humane P2X7-Rezeptor (C-terminal mit EGFP (P2X7-EGFP) gekoppelt), wurde
kloniert, um damit die zelluläre Lokalisation und das intrazelluläre Wandern (Trafficking)
bei ischämischen Bedingungen in vitro beobachten zu können. Die P2X7-E496A und P2X7-
I568N-Rezeptormutanten verhindern das Traffickingverhalten (Wiley et al., 2003) und die
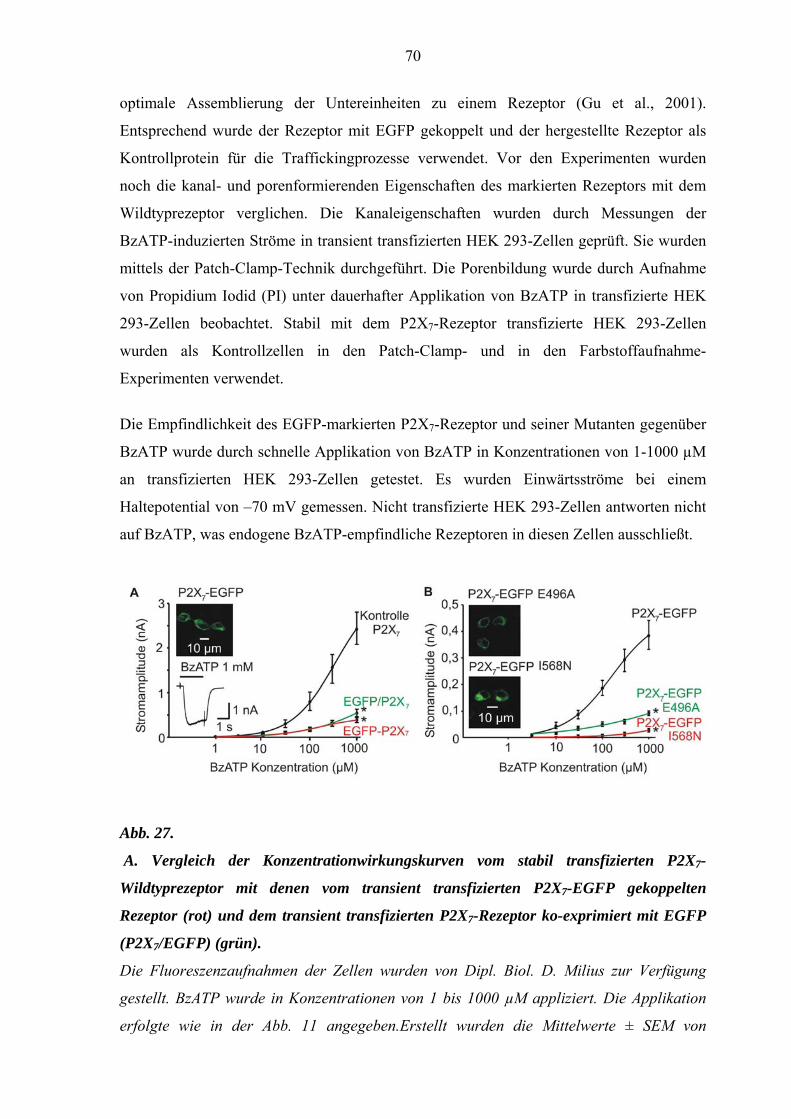
70
optimale Assemblierung der Untereinheiten zu einem Rezeptor (Gu et al., 2001).
Entsprechend wurde der Rezeptor mit EGFP gekoppelt und der hergestellte Rezeptor als
Kontrollprotein für die Traffickingprozesse verwendet. Vor den Experimenten wurden
noch die kanal- und porenformierenden Eigenschaften des markierten Rezeptors mit dem
Wildtyprezeptor verglichen. Die Kanaleigenschaften wurden durch Messungen der
BzATP-induzierten Ströme in transient transfizierten HEK 293-Zellen geprüft. Sie wurden
mittels der Patch-Clamp-Technik durchgeführt. Die Porenbildung wurde durch Aufnahme
von Propidium Iodid (PI) unter dauerhafter Applikation von BzATP in transfizierte HEK
293-Zellen beobachtet. Stabil mit dem P2X7-Rezeptor transfizierte HEK 293-Zellen
wurden als Kontrollzellen in den Patch-Clamp- und in den Farbstoffaufnahme-
Experimenten verwendet.
Die Empfindlichkeit des EGFP-markierten P2X7-Rezeptor und seiner Mutanten gegenüber
BzATP wurde durch schnelle Applikation von BzATP in Konzentrationen von 1-1000 µM
an transfizierten HEK 293-Zellen getestet. Es wurden Einwärtsströme bei einem
Haltepotential von –70 mV gemessen. Nicht transfizierte HEK 293-Zellen antworten nicht
auf BzATP, was endogene BzATP-empfindliche Rezeptoren in diesen Zellen ausschließt.
Abb. 27.
A. Vergleich der Konzentrationwirkungskurven vom stabil transfizierten P2X7-
Wildtyprezeptor mit denen vom transient transfizierten P2X7-EGFP gekoppelten
Rezeptor (rot) und dem transient transfizierten P2X7-Rezeptor ko-exprimiert mit EGFP
(P2X7/EGFP) (grün).
Die Fluoreszenzaufnahmen der Zellen wurden von Dipl. Biol. D. Milius zur Verfügung
gestellt. BzATP wurde in Konzentrationen von 1 bis 1000 µM appliziert. Die Applikation
erfolgte wie in der Abb. 11 angegeben.Erstellt wurden die Mittelwerte ± SEM von

71
mindesten 6 Messungen proVersuchsreihe . *p<0,05; statistisch signifikanter Unterschied
zum Mittelwert der Stromamplituden beim Wildtyprezeptor.
B. Konzentrationswirkungsverhältnisse des transient transfizierten P2X7-EGF-
gekoppelten Rezeptors und seiner Mutanten P2X7-EGFP-E496A (grün) und -I568N
(rot).
Die Mutanten und Fluoreszenzaufnahmen der Zellen wurden von Dipl. Biol. D. Milius zur
Verfügung gestellt. BzATP wurde in Konzentrationen von 3 bis 1000 µM appliziert. Die
Applikation erfolgte wie in der Abb. 11 angegeben. Mittelwerte ± SEM. von 6-8 Zellen pro
Versuchsreihe wurden erstellt; *p<0,05: statistisch signifikanter Unterschied zu den durch
maximale Agonistenkonzentrationen ausgelösten Stromamplituden beim transient
transfizierten P2X7-EGFP gekoppelten Rezeptor.
Abbildung 27 präsentiert Konzentrationswirkungsverhältnisse des transient transfizierten
P2X7-EGFP gekoppelten Rezeptors verglichen mit dem P2X7-Wildtyprezeptor, wobei
beide entweder transient transfiziert und ko-exprimiert mit EGFP (P2X7/EGFP) wurden
oder der P2X7-Rezeptor stabil in HEK 293-Zellen transfiziert wurde. Diese
Konzentrations-Wirkungskurven zeigen ähnliche Sensibilität des P2X7-EGFP gekoppelten
Rezeptors auf BzATP im Vergleich zum P2X7-Wildtyp (transient transfiziert und ko-
exprimiert mit EGFP) in HEK 293-Zellen. Eine um einiges höhere Sensibilität gegenüber
allen BzATP-Konzentrationen zeigte der stabil in HEK 293-Zellen transfizierte P2X7-
Rezeptor im Vergleich mit den beiden anderen Transfektionsarten. Dieser Unterschied
könnte an der höheren Anzahl von in der Zellmembran exprimierten Rezeptorproteinen in
den stabil transfizierten Zellen im Vergleich zu den transient transfizierten Zellen liegen.
Wie erwartet, zeigten die beiden Mutanten (P2X7-EGFP-E496A und -I568N) eine sehr
schwache Empfindlichkeit auf BzATP im Vergleich zum Wildtyprezeptor. Die zelluläre
Verteilung von EGFP-Fluoreszenz und eine typische durch BzATP induzierte
Stromamplitude werden in Abb. 27 gezeigt.
In den folgenden Experimenten wurden die HEK293-Zellen entweder in normoxischem
und glukosehaltigem Medium oder in ischämischem und glukosefreiem Medium für 4-12
h vorinkubiert. Die normoxischen Zellen wurden in einer Atmosphäre versetzt mit 5% CO2
bei 37oC im üblichen Labor-Brutschrank gehalten. Die ischämischen Zellen wurden
hingegen in einem N2-Inkubator mit 5% CO2 und 1,1% O2 ebenfalls bei 37oC inkubiert.
Die Dauer der Vorinkubation betrug 4-12 h. Ischämische Inkubation von P2X7-EGFP
transient transfizierten Zellen für 4 Stunden konnte die BzATP (1-1000 µM)-induzierten

72
Ströme nicht ändern (Daten nicht gezeigt). Leider führte eine Verlängerung dieser Zeit auf
12 Stunden zusammen mit den latenten durch die Zelltransfektionsmethode verursachten
Zellschäden der HEK 293-Zellen zu einer irreversiblen Schädigung und machte eine
weitere Untersuchung unmöglich. Deswegen wurde der Effekt einer zwölfstündigen
ischämischen Simulation an stabil transfizierten Wildtyp P2X7-Rezeptor HEK 293-Zellen
untersucht (Abb. 28).
Abb. 28. Konzentrations-Wirkungskurven für den Wildtyp P2X7-Rezeptor, stabil
transfiziert in HEK 293-Zellen unter normalen (Kontrolle, schwarz), normoxischen
(grün) und hypoxischen Bedingungen (rot).
Die Fluoreszenzaufnahmen unter Hypoxie und Normoxie wurden von Dipl. Biol. D. Milius
gemacht. BzATP wurde in Konzentrationen von 1 bis 1000 µM appliziert. Die Applikation
erfolgte wie in der Abb. 11 angegeben. Mittelwerte ± SEM von 7 Zellen pro Versuchsreihe
wurden zusammengefasst; *p<0,05: statistisch signifikanter Unterschied zu den
Kontrollapplikationen von BzATP bei 100, 300 und 1000 µM.
Die erste Serie von Experimenten verglich die BzATP induzierten Ströme in Zellen, die in
normalem Zellmedium inkubiert wurden mit den gleichen Zellen, die 12 Stunden in einem
glukosehaltigen und serumfreien extrazellulären Medium inkubiert wurden
(Normoxiezellen). Obwohl die normoxischen Zellen empfindlicher als die Kontrollzellen
auf Agonistenkonzentrationen von 100, 300 µM als die Kontrollzellen schienen, waren sie
bei 1000 µM fast gleich empfindlich. Jedoch waren die BzATP-induzierten Ströme von
HEK 293-Zellen nach der Inkubation in glukose- und serum-freiem extrazellulärem

73
Medium bei Sauerstoffentzug für 12 Stunden deutlich erhöht im Vergleich zu Strömen
unter Kontrollbedingungen (Abb. 28). Deswegen unterstützt die Serumentnahme und in
vitro Ischämie wahrscheinlich den Einbau des Rezeptors in die Membran oder die
Stabilisation des Rezeptors darin.

4. Diskussion
4.1. Regulation des humanen rekombinanten P2X3-Rezeptors durch ekto-
Proteinkinase C
Das wichtigste Ergebnis dieser Arbeit ist die Identifikation einer neuen regulatorischen
Wirkung von Nukleotiden auf die P2X3-Rezeptorfunktion durch Phosphorylierung der
PKC-Phosphorylierungsstellen auf der extrazellulären Schleife des Rezeptors. Es wird
vermutet, dass eine Ektoproteinkinase und die gleichzeitige Abgabe von ATP und
Glutamat (Chen et al., 1996) am Geschehen der hippokampalen synaptischen Plastizität
[Langzeitpotenzierung (LTP)] beteiligt sind (Wieraszko und Ehrlich, 1994; Ehrlich und
Kornecki, 1999; Fujii, 2004). So konnte durch nieder (aber nicht hoch)-frequente
elektrische Stimulation der Schafferkollateralen-kommissuralen Faserbündel in
Pyramidenzellen der CA1-Region des Hippocampus nur dann eine LTP induziert werden,
wenn exogenes ATP vorhanden war. Deshalb wurde gefolgert, dass dieses Nukleotid eine
Schlüsselrolle bei der Entstehung von LTP spielt. ATP könnte unter diesen Bedingungen
eher ein Oberflächenprotein phosphorylieren als P2-Rezeptoren aktivieren. Enzymatisch
stabile strukturelle Analoga von ATP oder ATP in Anwesenheit des Ektoproteinkinase
Inhibitors K252b induzierten keine LTP (Wieraszko und Ehrlich, 1994). Es gibt keine
Information über die Herkunft der Ektoproteinkinase im Extrazellulärraum. Wir vermuten,
dass dieses intrazellulär hergestellte Enzym mittels Transporter oder durch die geschädigte
Zellmembran nach aussen gelangt. Dadurch könnte die Empfindlichkeit von Endo- und
Ektoproteinkinase C gegenüber seinen pharmakologischen Inhibitoren praktisch identisch
sein.
G-Protein-gekoppelte Rezeptoren, wie solche für Substanz P, Bradykinin (Paukert et al.,
2001), Glutamat (metabotrope Glutamatrezeptoren; Vial et al., 2004) und ATP/UTP [P2Y2
(Molliver et al., 2002); P2Y4 (Fischer et al., 2003)] können die P2X-Rezeptorfunktion
durch intrazelluläre Proteinkinase mit ATP als Substrat verändern. Die Endoproteinkinase
könnte entweder ein assoziiertes Protein (Adinolfi et al., 2003; Vial et al., 2004) oder den
Rezeptorkanal selbst phosphorylieren (Paukert et al., 2001) und dadurch die
Ionenleitfähigkeit steuern. Die Phosphorylierung einer großen Anzahl verschiedener
interagierender Proteine, die den P2X-Rezeptorumsatz kontrollieren, könnte den Transport
dieser Rezeptoren zur Zellmembran fördern und hierdurch zu einer Sensibilisierung im

75
Verletzungszustand (Xu und Huang, 2004) oder zu einer Zunahme der synaptischen
Aktivität führen (Khakh, 2001; Bobanovic et al., 2002).
In den vorliegenden Experimenten wurden UTP und GTP auf HEK293-P2X3 Zellen
appliziert, weil diese Nukleotide erst in viel höheren Konzentrationen als ATP (Fischer et
al., 2003) den P2X3-Rezeptor aktivieren und weil sie in ihrer Mg2+-assoziierten Form
Substrate für die Ektoproteinkinase sind. Der Potenzierungseffekt von UTP wurde nur für
den Fall beobachtet, bei dem die negative Interaktion zwischen dem UTP-empfindlichen
P2Y4-Rezeptor (Fischer et al., 2003) und dem transfizierten P2X3-Rezeptor durch die
intrazelluläre Applikation von GDP-ß-S (Sternweis und Pang, 1990) gehemmt wurde.
Unter diesen Bedingungen verursachten niedrigere UTP-Konzentrationen, die den Rezeptor
nicht aktivieren, eine Steigerung der Rezeptorantwort auf α,β-meATP.
Die UTP-induzierte Potenzierung zeigte eine Konzentrations- und Zeitabhängigkeit.
Konzentrationen über einem Grenzwert von 3 µM aktivierten und desensibilisierten den
Rezeptor. Dadurch wurde es unmöglich weitere α,β-meATP-induzierte Ströme zu
beobachten. Für die Potenzierung der α,β-meATP-induzierten Ströme spielt die
Nukleotidstruktur der Stoffe eine essentielle Rolle; die entsprechenden Nukleoside bzw.
ihre Mono- oder Diphosphate sind unwirksam. Die durch UTP induzierte Potenzierung ist
dosisabhängig. Interessant ist, dass ATP in viel niedrigerer Konzentration (10 nM) die
gleiche Potenzierung wie UTP induziert. Die Ursache für die Potenzierung bei einer
niedrigen ATP-Konzentration ist, dass ATP vermutlich die optimale Struktur als Substrat
für die PKC besitzt. Der Fakt, dass ATP selber diese Potenzierung verursacht, ist ein
Nachweis, dass dieser Prozess unter physiologischen Bedingungen eine Rolle spielt.
Logischerweise sollte die UTP-induzierte Potenzierung auch bei einem Haltepotential von
+40 mV und bei der Outside-out Konfiguration auftreten. Leider konnten wir durch den
starken Rundown-Effekt die von uns gewünschte Potenzierung nicht sehen (nicht
präsentiert).
Ausgangspunkt unserer Überlegungen war, dass die Ekto-PKC für die potenzierende
Wirkung von UTP auf die α,β-meATP-induzierten Ströme verantwortlich ist. Deshalb
verwendeten wir eine Reihe von PKC-Aktivatoren (PMA, DAG-Lakton), die einen
ähnlichen Effekt wie UTP verursachten. Die Potenzierung unterschied sich von der UTP-
induzierten Potenzierung insofern, dass sie nicht mehr auswaschbar war, was gut mit der
hohen Lipidlöslichkeit der verwendeten Substanzen übereinstimmt. Außerdem war der

76
Effekt dosisabhängig. Die fehlende Potenzierung der α,β-meATP-induzierten Ströme
sowohl durch UTP als auch durch PMA oder DAG-Lakton in Anwesenheit von Pharmaka
(Staurosporin, K252b, PKC-Inhibitor-Peptid und Gö 6976) (Kase et al., 1987; Way et al.,
2000) oder nach Manipulationen (Mg2+-freies extrazelluläres Medium) (Christie et al.,
1992), für die bekannt ist, dass sie die PKC-Aktivität inhibieren, belegt den gemeinsamen
Wirkmechanismus. Daraus kann geschlussfolgert werden, dass entweder ein
Oberflächenprotein auf der Zellmembran oder Phosphorylierungsstellen auf der
extrazellulären Schleife des P2X3-Rezeptors Ziele der Ekto-PKC sind.
Punktmutationen in Ekto-PKC-Phosphorylierungsstellen des P2X3-Rezeptors bestätigen,
dass UTP ein Substrat für die Ekto-PKC ist. Durch das Ersetzen der entscheidenden Ser-
und Thr-Reste durch Ala wurde die Beteiligung der Phosphorylierungsstellen PKC2 und
PKC3 (T134A, S178A) deutlich. Die Substitution dieser wichtigen Ser- und Thr-Reste
durch Asp unterstreicht die Bedeutung der zwei anderen Phosphorylierungsstellen PKC4
und PKC6 (Thr-196, Ser-269) ebenfalls.
Offensichtlich wird durch die negativ geladene Aminosäure Asp die PKC-induzierte
Phosphorylierung durch UTP, welches ein Substrat von Ekto-PKC ist, nachgeahmt. Die
Asp-Mutationen interagieren mit dem Effekt von UTP, reduzieren darüber hinaus die
Amplituden der α,β-meATP-induzierten Ströme und verlangsamen deren
Desensibiliserungskinetik. Wir nehmen an, dass dies durch die übermäßige Aktivierung der
Ekto-PKC Stelle mittels der in den Rezeptor eingefügten Asp-Reste bedingt ist. Eine
konstitutive Desensibilisierung des Rezeptors führt zwangsläufig zu verminderten und
verlangsamten Stromamplituden. Im Gegensatz zu den Asp-Mutationen, veränderten die
Ala-Mutationen in PKC2 und -3 nur die potenzierende Wirkung von UTP, nicht aber die
Amplitude oder Kinetik der α,β-meATP-Ströme
Alle P2X-Rezeptoren enthalten positiv geladene Aminosäuren (Lys, Arg) in der
extrazellulären Schleife, die ATP durch Anziehen der negativ geladenen Phosphatgruppe
binden können, außerdem koordiniert Phe die Bindung des Adeninringes von ATP (Jiang et
al., 2000; North, 2002; Roberts und Evans, 2004). An P2X1- oder P2X2-Rezeptoren wurden
diese Agonisten-Bindungsstellen vorwiegend im Übergang zwischen der extrazellulären
Schleife und dem transmembranen Bereich identifiziert. Eine Substitution dieser basischen
Aminosäurereste durch das neutrale Ala verhinderte die Bindung von ATP, ohne mit dem
Einbau der P2X-Rezeptoren in die Zellmembran zu interferieren. Diese Agonisten-

77
Bindungsstellen sind jedoch mit den von uns untersuchten Ekto-PKC-
Phosphorylierungsstellen nicht identisch. Im letzteren Fall wurden die für die PKC-Stellen
typischen Ser- und Thr-Reste durch Ala oder Asp ersetzt. Durch diese Änderungen erfolgte
keine Modifikation der Agonisten-Bindung, sehr wohl aber die der basalen Aktivierung des
P2X3-Rezeptors durch eine Phosphorylierungsreaktion. Die molekulare Simulation des
Rezeptors zeigte, dass die Agonisten-Bindung- und Ektoproteinkinase C-Stellen in der
extrazellulären Schleife räumlich unterschiedlich untergebracht sind. Letzten Endes wurden
in Mutationsanalysen von konservierten Cysteinresten, welche Disulfidbrücken im
extrazellulären Bereich des P2X3-Rezeptors bilden, festgestellt, dass beim Ersatz von Cys
durch Ala die Agonistenempfindlichkeit reduziert wird (Clyne et al., 2002; Ennion und
Evans, 2002; Nakazawa et al., 2004). Die Ekto-PKC-Stellen wiesen jedoch keine
Überlappung mit den für die Bildung der Disulfidbrückenbildung notwendigen Cys-Resten
auf.
Obwohl die Phosphorylierung des P2X1-Rezeptorkanals durch PMA Markierung mit
[32P]Orthophosphat und folgender Immunpräzipitation demonstriert wurde (Vial et al.,
2004), konnten wir in unseren Experimenten wegen des zu niedrigen Expressionsniveaus
der P2X3-Rezeptoren in HEK 293-Zelllinien den Grad der Kanalphosphorylierung nicht
messen (C. Vial und R. J. Evans, nichtpublizierte Beobachtung). Allerdings deuten unsere
Experimente stark darauf hin, dass niedrige UTP-Konzentrationen PKC-Stellen im
extrazellulären Bereich des P2X3-Rezeptors phosphorylieren können.
Zum Schluss wurden noch native P2X3-Rezeptoren von DRG-Neuronen, die an der
Schmerzübertragung beteiligt sind (Robertson et al., 1996; Dunn et al., 2001), darauf hin
untersucht, ob sie durch extrazelluläre Nukleotide moduliert werden. Gemäß vorherigen
Ergebnissen induziert UTP in Konzentrationen >0,3 µM schnell inaktivierende
Einwärtsströme (Robertson et al., 1996). Allerdings potenzierte UTP in niedrigeren
Konzentrationen von 3-30 nM ausnahmslos die α,β-meATP-induzierten Ströme infolge
eines Mechanismus, der von der PKC-Aktivität abhängig ist, da der PKC-Inhibitor Gö
6976 die UTP-induzierte Potenzierung verhinderte. Die nativen P2X3-Rezeptoren von
DRG-Neuronen der Ratte und die in HEK 293-Zellen exprimierten rekombinanten
humanen P2X3-Rezeptoren reagierten sehr ähnlich auf UTP.
Schließlich wurde vorgeschlagen, dass unter physiologischen Bedingungen niedrige
extrazelluläre Konzentrationen von endogenem ATP (Lazarowski et al., 2000) die

78
Empfindlichkeit des P2X3-Rezeptors gegenüber diesem Agonisten erhöhen können. Solch
eine Aktivierung könnte eine Voraussetzung für die volle Funktionalität der
schmerzübermittelnden P2X3-Rezeptoren in peripheren sowie zentralen Prozessen der
Hinterwurzelganglienzellen sein. Allerdings sei hier erwähnt, dass höhere Konzentrationen
von endogenem ATP den Rezeptor desensibilisieren können, wie dies durch Inhibitoren der
ATP-abbauenden Phosphohydrolase gezeigt werden konnte.
Die agonistische Funktion von ATP oder seiner Strukturanaloga könnte somit durch das
Zusammenspiel mehrerer teilweise gegenläufiger Prozesse bestimmt werden: 1)
Phosphorylierung der Ekto-PKC-Stellen und somit eine Funktionssteigerung; 2)
Desensibiliserung der Agonisten-Bindungsstelle des Rezeptors und somit eine
Funktionsabnahme; 3) Aktivierung des Rezeptors durch Besetzung der Agonisten-
Bindungsstellen durch ATP und somit Induktion der nicht-selektiven kationischen
Leitfähigkeit; und 4) schließlich eine Hemmung der P2X3-Rezeptoraktivierung durch eine
gleichzeitige Stimulation von inhibitorischen P2Y-Rezeptoren.
4.2. Sauerstoff- und Glukose-Mangel erhöhen die Expressionsrate des P2X7-
Rezeptors in der HEK 293-Zellmembran
Die metabotropen und ionotropen Rezeptoren werden möglicherweise durch die Clathrin-
und Dynamin-abhängigen endozytotischen Mechanismen internalisiert, worauf sie dann
zwischen der Zellmembran und den endosomalen Kompartimenten ständig ausgetauscht
werden (Carroll und Zukin, 2002; Marchese et al., 2003). Es wurde gezeigt, dass der
Transport von P2X-Rezeptoren spezifisch für einzelne Untereinheiten ist. P2X4-Rezeptoren
haben ein zyklisches Verhalten und gehen rasch in die und aus der Zellmembran, im
Gegensatz zu den P2X2-Rezeptoren, welche stabil auf der neuronalen Oberfläche
exprimiert werden (Bobanovic et al., 2002). Die Internalisierung der P2X-Rezeptoren,
deren Passage zwischen den endosomalen und lysosomalen Kompartimenten, die
Integration in die Zellmembran und die Stabilisierung an der Zelloberfläche scheinen durch
Transportstrukturen in der Rezeptoruntereinheit gesteuert zu sein (Chaumont et al., 2004).
Diese Transportstrukturen könnten Disulfidbrücken oder glykosylierte Asparaginreste in
der extrazellulären Schleife enthalten. Beide sind sehr wichtig für die Rezeptorstruktur und
die folgende Qualitätskontrolle, die normalerweise im endoplasmatischen Retikulum

79
stattfindet (P2X1, Ennion and Evans, 2002; P2X2, Torres et al., 1999). Bestimmte
Aminosäuresequenzen auf dem C-Terminus aller P2X-Untereinheiten (P2X4; Royle et al.,
2002; Chaumont et al., 2004) scheinen das Rezeptorenrecycling zu regulieren und den
Rezeptor in der Zellmembran zu stabilisieren.
Die Veränderung einer zweibasischen Aminosäure (Adriouch et al., 2002) und der
Polymorphismus I568N (Ile-568 zu Asn) hat normale Funktion und Trafficking-
Eigenschaften beim menschlichen P2X7 Rezeptor unterbunden (Wiley et al., 2003).
Ausserdem scheint der E496A (Glu-496 zu Ala)-Polymorphismus den optimalen Aufbau
des P2X7 Rezeptors zu behindern, wodurch sowohl die Ethidium Iodid-Aufnahme als auch
die Apoptose in Lymphozyten von homozygoten Objekten/Organismen unterdrückt wird
(Gu et al., 2001; Barden et al., 2003). In unseren Experimenten zeigten stabil mit dem
menschlichen P2X7-Rezeptor transfizierte HEK293 Zellen einen stabilen Einwärtsstrom
und eine ausgeprägte Propidium Iodid-Aufnahme nach Zugabe von BzATP. Im Gegensatz
dazu haben transiente Expressionsprozeduren entweder mit zwei Plasmiden, die beide den
P2X7-Rezeptor und EGFP kodieren (P2X7/EGFP) oder mit einem Plasmid, das den am C-
Terminus mit EGFP markierten P2X7 kodiert, in HEK 293-Zellen viel niedrigere
Stromantworten auf BzATP gezeigt. Der Grund hierfür könnte eine geringere
Rezeptordichte wegen der transienten Transfektion sein. Allerdings konnten beide
Mutanten (E496A, I568N) nicht die Kationenkanäle und die hochpermeablen Poren öffnen,
was eine gestörte Funktionalität oder fehlende Integrierung in der Zellmembran als Ursache
haben könnte. Durch konfokale Laserscanning-Mikroskopiebilder und deren quantitative
Evaluierung konnte kein Unterschied zwischen der Intensität des Fluoreszenzsignals der
EGFP-markierten Wildtyprezeptoren und den beiden Mutanten in der Zellmembran
festgestellt werden. Die optische Untersuchung der Mikroskopiebilder deutet darauf hin,
dass I568N relativ gleichmäßig im Zytoplasma der HEK 293-Zellen verteilt ist, während
der Wildtyp P2X7-Rezeptor und seine Mutante E496A bevorzugte Orte in der Zellmembran
hat.
In vitro Ischämie verursachte in ZNS-Neuronen mittels der Beteiligung von P2-Rezeptoren
zytotoxische Effekte wie zelluläre Schwellung, die Freisetzung von Laktatdehydrogenase
und Kernfragmentation, die zum Zelltod führen (Amadio et al., 2002; Cavaliere et al.,
2002). Eine Hochregulierung und/oder Überempfindlichkeit des P2X7-Rezeptorsubtyps
scheint sehr wichtig für ischämische und entzündliche Prozesse im Gehirn zu sein
(Sperlagh et al., 2002; Cavaliere et al., 2004; Wirkner et al., 2005). Die gezeigten

80
Experimente unterstützen diese Idee, indem sie gezeigt haben, dass der Glukose- und
Sauerstoffmangel die Expression des P2X7-EGFP in der Zellmembran der HEK 293-Zellen
erhöht, entweder durch die Zunahme der Integrierung oder die Abnahme der
Internalisierung der P2X7-Rezeptoren. Gleichzeitig werden die Stromantworten auf BzATP
nach Ischämie im Vergleich mit der normoxischen Kontrolle potenziert. Schließlich zeigen
die Mutanten E396A und I568N keine höhere Expression in der Zellmembran nach
Ischämie im Vergleich mit dem P2X7-Wildtyprezeptor.
Die Notwendigkeit einer langandauernden ischämischen Anregung zur Betrachtung einer
starken Antwort auf die in vitro Ischämie zu bekommen, liegt sicherlich am Widerstand der
epithelialen HEK 293-Zelllinie gegenüber Einschränkungen des Stoffwechsels. Primäre
neuronale Kulturen und besonders Neuronen unter in situ Bedingungen sind in dieser
Hinsicht vermutlich bedeutend empfindlicher. Schließlich könnten P2X7-Rezeptoren ihre
Trafficking-Eigenschaften während der Ischämie ändern und zur ATP-verursachten
Beschädigung der verschiedenen Zellarten einschließlich Gehirnneuronen beitragen.

5. Zusammenfassung
In der vorliegenden Arbeit wurden an zwei Typen von ionotropen ATP-empfindlichen
P2X-Rezeptoren (P2X3, P2X7) Experimente zu den folgenden zwei Fragestellungen
durchgeführt.
A. Regulation der Ionenleitfähigkeit des P2X3-Rezeptors durch Phosphorylierung
einzelner Aminosäuren der extrazellulären Schleife
1. Nukleotide wie UTP, GTP und ATP steigerten den α,β-meATP-induzierten Strom an
mit humanen P2X3-Rezeptoren transfizierten HEK293-Zellen und an nativen P2X3-
Rezeptoren von Hinterwurzelganglienzellen (DRG-Neurone) der Ratte. Allerdings musste
die Interaktion zwischen den endogenen P2Y- und P2X-Rezeptoren durch die Blockade
von G-Proteinen mittels intrazellulärem GDP-β-S unterbunden werden, um den
potenzierenden Effekt der Nukleotide beobachten zu können.
2. Der Nukleotid-Effekt entwickelte sich konzentrations- und zeitabhängig. Der lange
Zeitabstand zwischen Nukleotid-Applikation und maximaler Wirkung (Minutenbereich)
lässt die Vermutung zu, dass es sich weder um ionotrope (Millisekundenbereich) noch um
klassische G-Protein-vermittelte (Sekundenbereich) Effekte handelt.
3. Deshalb haben wir angenommen, dass eine Übertragung der terminalen Phosphatreste
der Nukleotide auf Konsensus-Phosphorylierungsstellen der extrazellulären Schleife des
hP2X3-Rezeptors die Grundlage für den beobachteten Effekt sein könnte. Anhand einer
Computer-Modellierung kommen Proteinkinase C (PKC)-Phosphorylierungsstellen als
Zielorte in Frage. Tatsächlich konnten PKC-Aktivatoren (DAG-Lakton, PMA) eine dem
UTP ähnliche Steigerung der α,β-meATP-induzierten-Ströme auslösen. Die Wirkung der
PKC-Aktivatoren und die des UTP konnten gleichermaßen durch PKC-Inhibitoren
(Staurosporin, Gö 6976, PKC-inhibitorisches Peptid, K252b) aufgehoben werden. Da
weder UTP noch das PKC-inhibitorische Peptid oder K252b in das Zellinnere diffundieren,
scheint erwiesen zu sein, dass die Phosphorylierung an extrazellulären PKC-Stellen des
P2X3-Rezeptors entsteht.
4. P2X3-Rezeptormutanten, in deren Ekto-PKC-Phosphorylierungsstellen (PKC-2 und -3;

82
nicht aber PKC-4 und -6); die relevanten Ser- oder Thr-Reste durch Ala ersetzt wurden,
reagierten nicht mehr auf die Applikation von UTP mit einer Erhöhung der α,β-meATP-
induzierten Ströme. Die Stromamplituden der Mutanten waren kleiner als die des Wild-
Typ-Rezeptors, die Desensibilisierungskinetik war jedoch nicht wesentlich verändert.
Hingegen führte die Substitution von Ser oder Thr in allen vier Ekto-PKC-
Phosphorylierungsstellen durch Asp zu einer signifikanten Verlangsamung der
Stromantworten und einer Hemmung ihrer Amplituden. Darüber hinaus entfiel die
Wirkung von α,β-meATP vollständig.
5. Die Potenzierung der α,β-meATP-induzierten Ströme durch UTP und geringe
Konzentrationen von ATP konnte an DRG-Neuronen der Ratte ebenfalls nachgewiesen
werden. Dieser Effekt wurde durch PKC-Hemmer verhindert.
B. Regulation der Ionenleitfähigkeit des P2X7-Rezeptors durch eine vermehrte Expression
des Rezeptorproteins in der Zellmembran nach ischämischen Stimuli
1. Es wurden EGFP-gekoppelte P2X7-Rezeptoren und zwei Mutanten hergestellt. Diese
Mutanten (E496A, I568N) weisen ein unvollständiges Traffickingverhalten und
demzufolge einen reduzierten Einbau in die Zellmembran auf bzw. werden infolge der
nicht-optimalen Assemblierung der Untereinheiten nicht zum funktionsfähigen Rezeptor
zusammengebaut.
2. Elektrophysiologische und Dye-Uptake- (Propidium Iodid-Aufnahme) Experimente
belegten die Funktionsfähigkeit des Wildtyp-Rezeptors und die fehlende Funktionalität der
beiden Mutanten.
3. Eine in vitro Ischämie erzeugte ein vermehrtes Erscheinen des Wild-Typ-Rezeptors in
der Zellmembran; die funktionsuntüchtigen Mutanten wurden jedoch unverändert nur in
geringem Maße an dieser Stelle exprimiert. Diese Aussage konnte durch
immunhistochemische Experimente mittels konfokaler Laserscanning-Mikroskopie belegt
werden. Elektrophysiologische Experimente bestätigten, dass ein vermehrtes Erscheinen
des P2X7-Rezeptors in der Zellmembran nach Ischämie mit höheren Amplituden der α,β-
meATP-induzierten Ströme einhergeht.

83
C. Nukleotid- und Ischämie-bedingte Potenzierung ionotroper P2X-Rezeptorströme
Exemplarisch wurde an zwei prototypischen P2X (P2X3 und P2X7)-Rezeptoren gezeigt,
dass gewebsschädigende Vorgänge (z.B. Schmerz oder Ischämie) über eine vermehrte
Nukleotidabgabe in den Extrazellulärraum bzw. durch z.Z. unbekannte Transduktionswege
die Funktion der o.g. Rezeptoren steigern können. Die jeweiligen Mechanismen sind die
Phosphorylierung von Ekto-PKC-Stellen und ein erhöhtes Erscheinen des Rezeptors in der
Zellmembran (Förderung des Einbaus in die Membran oder Hemmung der Aufnahme in
das Zellinnere).

6. Eigene Publikationen
6.1. Tagungsbeiträge
Stanchev D, Klebingat M, Wirkner K, Fleming G, Eschrich K, Illes P (2004) UTP
potentiates P2X3 receptor-mediated currents in HEK 293-hP2X3 cells by
phosphorylation of the extracellular loop (3rd Leipzig Research Festival for Life
Sciences 2004)
Stanchev D, Klebingat M, Köles L, Dihazi S, Fleming G, Eschrich K, Illes P, Wirkner K
(2005) Potenzierung von P2X3 Rezeptor-vermittelten Strömen durch
Phosphorylierung der extrazellulären Schleife in HEK 293-hP2X3 Zellen (46.
Frühjahrstagung der DGTP 2005)
Stanchev D, Millius D, Lange-Dohna Ch, Roßner S, Wirkner K, Illes P (2005)
Investigation of functional properties and intracellular distribution of hP2X7-EGFP
receptors and their mutants in HEK 293 cells during normoxia and in vitro ischemia
(First Joint Italian-German Purine Club Meeting 2005)
Wirkner K, Stanchev D, Klebingat M, Köles L, Dihazi H, Flemming G, Eschrich K, Illes P
(2005) Facilitation of P2X3 receptor-mediated currents by phosphorylation of the
extracellular loop in HEK 293-hP2X3 cells and in rat dorsal root ganglion neurons.
(First Joint Italian-German Purine Club Meeting 2005)
Millius D, Stanchev D, Lange-Dohna Ch, Roßner S, Wirkner K, Illes P (2005)
Investigation of funktional properties an intracellular distribution of hP2X7-EGFP
receptors and their mutants in HEK 293 cells during normoxia and ischemia. (4rd
Leipzig Research Festival for Life Sciences 2005)

6.2. Originalarbeiten
Wirkner K, Stanchev D, Koles L, Klebingat M, Dihazi H, Flehmig G, Vial C, Evans RJ,
Furst S, Mager PP, Eschrich K, Illes P (2005) Regulation of human recombinant
P2X3 receptors by ecto-protein kinase C. J. Neurosci. 25:7734-7742.
Stanchev D, Flehmig G, Gerevich Z, Norenberg W, Dihazi H, Furst S, Eschrich K, Illes P,
Wirkner K. (2006) Decrease of current responses at human recombinant P2X3
receptors after substitution by Asp of Ser/Thr residues in protein kinase C
phosphorylation sites of their ecto-domains. Neurosci Lett. 393:78-83.
Milius D, Stanchev D, Lange-Dohna C, Rossner S, Illes P. Oxygen- and glucose-
deprivation increase the expression of recombinant P2X7 receptors in the plasma
membrane of HEK293 cells. Eingereicht bei FEBS Letters.

7. Literatur
Abbracchio MP, Burnstock G (1994) Purinoceptors: Are there families of P2X and P2Y
purinoceptors? Pharmacol. Ther. 64:445–475.
Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A (2003) Tyrosine
phosphorylation of HSP90 within the P2X7 receptor complex negatively regulates
P2X7 receptors. J. Biol. Chem. 278:37344-37351.
Adriouch S, Dox C, Welge V, Seman M, Koch-Nolte F, Haag F (2002) Cutting edge: a
natural P451L mutation in the cytoplasmic domain impairs the function of the mouse
P2X7 receptor. J. Immunol. 168:4108-4112.
Alzola E, Perez-Etxebarria A, Kabre E, Fogarty DJ, Metioui M, Chaib N, Macarulla JM,
Matute C, Dehaye JP, Marino A (1998) Activation by P2X7 agonists of two
phospholipases A2 (PLA2) in ductal cells of rat submandibular gland. Coupling of
the calcium-independent PLA2 with kallikrein secretion. J. Biol. Chem. 273:30208-
30217.
Amadio S, D'Ambrosi N, Cavaliere F, Murra B, Sancesario G, Bernardi G, Burnstock G,
Volonte C (2002) P2 receptor modulation and cytotoxic function in cultured CNS
neurons. Neuropharmacology. 42:489-501.
Barden JA, Sluyter R, Gu BJ, Wiley JS (2003) Specific detection of non-functional human
P2X7 receptors in HEK293 cells and B-lymphocytes. FEBS Lett. 538:159-162.
Barnard EA, Simon J, Webb TE (1997) Nucleotide receptors in the nervous system. An
abundant component using diverse transduction mechanisms. Mol. Neurobiol.
15:103–129.
Belitser VA, Tsibakova ET (1939) Oxidative Phosphorylierung. Biokhimiia 4:516-535
Bianchi BR, Lynch KJ, Touma E, Niforatos W, Burgard EC, Alexander KM, Park HS, Yu
H, Metzger R, Kowaluk E, Jarvis MF, van Biesen T (1999) Pharmacological

87
characterization of recombinant human and rat P2X receptor subtypes. Eur. J.
Pharmacol. 376:127-138.
Bo X, Schoepfer R, Burnstock G (2000) Molecular cloning and characterization of a novel
ATP P2X receptor subtype from embryonic chick skeletal muscle. J. Biol. Chem.
275:14401–14407.
Bobanovic LK, Royle SJ, Murrell-Lagnado RD (2002) P2X receptor trafficking in neurons
is subunit specific. J. Neurosci. 22:4814-4824.
Boue-Grabot E, Akimenko MA, Seguela P (2000) Unique functional properties of a
sensory neuronal P2X ATP-gated channel from zebrafish. J. Neurochem. 75:1600-
1607.
Brandle U, Kohler K, Wheeler-Schilling TH (1998) Expression of the P2X7-receptor
subunit in neurons of the rat retina. Brain Res. Mol. Brain Res. 62:106-109.
Buell G, Collo G, Rassendren F (1996) P2X receptors: an emerging channel family. Eur J.
Neurosci. 8:2221-2228.
Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Kage KL, Touma E, Kowaluk EA,
Jarvis MF (2000) Competitive antagonism of recombinant P2X2/3 receptors by 2', 3'-
O-(2,4,6-trinitrophenyl) adenosine 5'-triphosphate (TNP-ATP). Mol. Pharmacol.
58:1502-1510.
Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Touma E, Metzger RE, Kowaluk EA,
Jarvis MF (1999) P2X receptor-mediated ionic currents in dorsal root ganglion
neurons. J. Neurophysiol. 82:1590-1598.
Burnstock G (2000) P2X receptors in sensory neurones. Br. J. Anaesth. 84:476-488.
Burnstock G (2001) Purine-mediated signalling in pain and visceral perception. Trends
Pharmacol. Sci. 22:182-188.

88
Burnstock G (2004) Introduction: P2 receptors. Curr. Top. Med. Chem. 4:793-803.
Cario-Toumaniantz C, Loirand G, Ferrier L, Pacaud P (1998) Non-genomic inhibition of
human P2X7 purinoceptor by 17beta-oestradiol. J. Physiol. 508:659-666.
Carroll RC, Zukin RS (2002) NMDA-receptor trafficking and targeting: implications for
synaptic transmission and plasticity. Trends Neurosci. 25:571-577.
Cavaliere F, Sancesario G, Bernardi G, Volonte C (2002) Extracellular ATP and nerve
growth factor intensify hypoglycemia-induced cell death in primary neurons: role of
P2 and NGFRp75 receptors. J. Neurochem. 83:1129-1138.
Chaumont S, Jiang LH, Penna A, North RA, Rassendren F (2004) Identification of a
trafficking motif involved in the stabilization and polarization of P2X receptors. J.
Biol. Chem. 279:29628-29638.
Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN (1995) A P2X
purinoceptor expressed by a subset of sensory neurons. Nature 377:428-431.
Chen CC, Chen WC (1996) Increased protein kinase C isoform gamma in the hippocampus
of pentylenetetrazol-induced chemoshocked mouse.Brain Res. 725:75-80.
Chessell IP, Simon J, Hibell AD, Michel AD, Barnard EA, Humphrey PP (1998) Cloning
and functional characterisation of the mouse P2X7 receptor. FEBS Lett. 439:26-30.
Chizh BA, Illes P (2001) P2X receptors and nociception. Pharmacol. Rev. 53:553-568.
Christie A, Sharma VK, Sheu SS (1992) Mechanism of extracellular ATP-induced increase
of cytosolic Ca2+ concentration in isolated rat ventricular myocytes. J. Physiol.
445:369-388.
Chung HS, Park KS, Cha SK, Kong ID, Lee JW (2000) ATP-induced [Ca2+](i) changes and
depolarization in GH3 cells. Br. J. Pharmacol. 130:1843-1852.

89
Clyne JD, Wang LF, Hume RI (2002) Mutational analysis of the conserved cysteines of the
rat P2X2 purinoceptor. J. Neurosci. 22:3873-3880.
Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S, Malmberg AB,
Cain G, Berson A, Kassotakis L, Hedley L, Lachnit WG, Burnstock G, McMahon
SB, Ford AP (2000) Urinary bladder hyporeflexia and reduced pain-related behaviour
in P2X3-deficient mice. Nature 407:1011-1015.
Collo G, North RA, Kawashima E, Merlo-Pich E, Neidhart S, Surprenant A, Buell G
(1996) Cloning OF P2X5 and P2X6 receptors and the distribution and properties of an
extended family of ATP-gated ion channels. J. Neurosci. 16:2495-2507.
Di Virgilio F (1995) The P2Z purinoceptor: an intriguing role in immunity, inflammation
and cell death. Immunol. Today 16:524-528
Ding Z, Kim S, Dorsam RT, Jin J, Kunapuli SP (2003) Inactivation of the human P2Y12
receptor by thiol reagents requires interaction with both extracellular cysteine residues,
Cys17 and Cys270, Blood 101:3908–3914.
Dowd E, McQueen DS, Chessell IP, Humphrey PP (1998) P2X receptor-mediated
excitation of nociceptive afferents in the normal and arthritic rat knee joint. Br. J.
Pharmacol. 125:341–346.
Dranoff JA, O'Neill AF, Franco AM, Cai SY, Connolly GC, Ballatori N (2000) A primitive
ATP receptor from the little skate Raja erinacea. J. Biol. Chem. 275:30701–30706.
Drury, Szent-Gyorgyi (1929) A historical perspective on the discovery of adenyl purines.
Biol. Res. Nurs. 2000; 1:265-275.
Dunn PM, Zhong Y, Burnstock G (2001) P2X receptors in peripheral neurons. Prog.
Neurobiol. 65:107-134.
Egan TM, Cox JA, Voigt MM (2000) Molecular cloning and functional characterization of
the zebrafish ATP-gated ionotropic receptor P2X3 subunit. FEBS Lett. 475:287-290.

90
Ehrlich YH, Kornecki E (1999) Ecto-protein kinases as mediators for the action of secreted
ATP in the brain. Prog. Brain Res. 120:411-426.
Ennion SJ, Evans RJ (2002) Conserved cysteine residues in the extracellular loop of the
human P2X1 receptor form disulfide bonds and are involved in receptor trafficking to
the cell surface. Mol. Pharmacol. 61:303-311.
Fabbretti E, Sokolova E, Masten L, D'Arco M, Fabbro A, Nistri A, Giniatullin R (2004)
Identification of negative residues in the P2X3 ATP receptor ectodomain as structural
determinants for desensitization and the Ca2+-sensing modulatory sites. J. Biol. Chem.
279:53109-53115.
Fischer W, Wirkner K, Weber M, Eberts C, Köles L, Reinhardt R, Franke H, Allgaier C,
Gillen C, Illes P (2003) Characterization of P2X3, P2Y1 and P2Y4 receptors in cultured
HEK293-hP2X3 cells and their inhibition by ethanol and trichloroethanol. J.
Neurochem. 85: 779-790.
Franke H, Gunther A, Grosche J, Schmidt R, Rossner S, Reinhardt R, Faber-Zuschratter H,
Schneider D, Illes P (2004) P2X7 receptor expression after ischemia in the cerebral
cortex of rats. J. Neuropathol. Exp. Neurol. 63:686-699.
Fujii S (2004) ATP- and adenosine-mediated signaling in the central nervous system: the
role of extracellular ATP in hippocampal long-term potentiation. J. Pharmacol. Sci.
94:103-106.
Garcia-Guzman M, Stuhmer W, Soto F (1997) Molecular characterization and
pharmacological properties of the human P2X3 purinoceptor. Brain Res. Mol. Brain
Res. 47:59-66.
Gerevich Z, Muller C, Illes P (2005) Metabotropic P2Y1 receptors inhibit P2X3 receptor-
channels in rat dorsal root ganglion neurons. Eur. J. Pharmacol. 521:34-38.

91
Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line
transformed by DNA from human adenovirus type 5. Gen Virol. 36:59-74.
Gu BJ, Zhang WY, Bendall LJ, Chessell IP, Buell GN, Wiley JS (2000) Expression of
P2X7 purinoceptors on human lymphocytes and monocytes: evidence for
nonfunctional P2X7 receptors. Am J. Physiol. Cell Physiol. 279:C1189-1197.
Hede SE, Amstrup J, Christoffersen BC, Novak I (1999) Purinoceptors evoke different
electrophysiological responses in pancreatic ducts. P2Y inhibits K+ conductance, and
P2X stimulates cation conductance. J. Biol. Chem. 274:31784-31791.
Hoffmann F, Moro S, Nicholas RA, Harden TK, Jacobson KA (1999) The role of amino
acids in extracellular loops of the human P2Y1 receptor in surface expression and
activation processes. J. Biol. Chem. 274: 14639–14647.
Hoyle CH, Burnstock G (1986) Evidence that ATP is a neurotransmitter in the frog heart.
Eur. J. Pharmacol. 124:285-289.
Jabs R, Guenther E, Marquordt K, Wheeler-Schilling TH (2000) Evidence for P2X3, P2X4,
P2X5 but not for P2X7 containing purinergic receptors in Muller cells of the rat retina.
Brain Res. Mol. Brain Res. 76:205-210.
Jacobson KA, Jarvis MF, Williams M (2002) Purine and pyrimidine (P2) receptors as drug
targets, J. Med. Chem. 45:4057–4093.
Jiang LH, Mackenzie AB, North RA, Surprenant A (2000) Brilliant blue G selectively
blocks ATP-gated rat P2X7 receptors. Mol. Pharmacol. 58:82-88.
John GR, Simpson JE, Woodroofe MN, Lee SC, Brosnan CF (2001) Extracellular
nucleotides differentially regulate interleukin-1beta signaling in primary human
astrocytes: implications for inflammatory gene expression. J. Neurosci. 21:4134-
4142.

92
Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato
A, Kaneko M (1987) K-252 compounds, novel and potent inhibitors of protein kinase
C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res.
Commun. 142:436-440.
Kennedy C (2000) The discovery and development of P2 receptor subtypes. J. Auton.
Nerv. Syst. 81:158-163.
Khakh BS (2001) Molecular physiology of P2X receptors and ATP signalling at synapses.
Nat. Rev. Neurosci. 2:165-174.
Khakh BS, Kennedy C (1998) Adenosine and ATP: progress in their receptors' structures
and functions. Trends Pharmacol. Sci. 19:39-41.
Klapperstuck M, Buttner C, Nickel P, Schmalzing G, Lambrecht G, Markwardt F (2000)
Antagonism by the suramin analogue NF279 on human P2X1 and P2X7 receptors. Eur.
J. Pharmacol. 387:245-252.
Korngreen A, Ma W, Priel Z, Silberberg SD (1998) Extracellular ATP directly gates a
cation-selective channel in rabbit airway ciliated epithelial cells. J. Physiol. 508:703-
720.
Lazarowski ER, Boucher RC, Harden TK (2000) Constitutive release of ATP and evidence
for major contribution of ecto-nucleotide pyrophosphatase and nucleoside
diphosphokinase to extracellular nucleotide concentrations. J. Biol. Chem. 275:31061-
31068.
Lewis C, Neidhart S, Holy C, North RA, Buell G, Surprenant A. (1995) Coexpression of
P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory
neurons. Nature 377:432-435.
Lipmann F (1948-1949) Biosynthetic mechanisms. Harvey Lect. 44:99-123.

93
Liu Y, Wakakura M (1998) P1-/P2-purinergic receptors on cultured rabbit retinal Müller
cells. Jpn. J. Ophthalmol. 42:33-40.
Loirand G, Pacaud P (1995) Mechanism of the ATP-induced rise in cytosolic Ca2+ in
freshly isolated smooth muscle cells from human saphenous vein. Pflugers Arch.
430:429-436.
Luo X, Zheng W, Yan M, Lee MG, Muallem S (1999) Multiple functional P2X and P2Y
receptors in the luminal and basolateral membranes of pancreatic duct cells. Am. J.
Physiol. 277:205-215.
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A (2001) Rapid
secretion of interleukin-1beta by microvesicle shedding. Immunity 15:825-835.
Mager PP, Weber A, Illes P (2004) Bridging the gap between structural bioinformatics and
receptor research: the membrane-embedded, ligand-gated, P2X glycoprotein receptor.
Curr. Top. Med. Chem. 4:1657-1705.
Marchese C, Visco V, Aimati L, Cardinali G, Kovacs D, Buttari B, Bellocci M, Torrisi
MR, Picardo M (2003) Nickel-induced keratinocyte proliferation and up-modulation
of the keratinocyte growth factor receptor expression. Exp. Dermatol. 12:497-505.
Michel AD, Kaur R, Chessell IP, Humphrey PP (2000) Antagonist effects on human P2X7
receptor-mediated cellular accumulation of YO-PRO-1. Br. J. Pharmacol. 130:513-
520.
Molliver DC, Cook SP, Carlsten JA, Wright DE, McCleskey EW (2002) ATP and UTP
excite sensory neurons and induce CREB phosphorylation through the metabotropic
receptor, P2Y2. Eur. J. Neurosci. 16:1850-1860.
Nakazawa K, Ojima H, Ishii-Nozawa R, Takeuchi K, Ohno Y (2004) Amino acid
substitutions from an indispensable disulfide bond affect P2X2 receptor activation. Eur.
J. Pharmacol. 483:29-35.

94
Neal MJ, Cunningham JR, Dent Z (1998) Modulation of extracellular GABA levels in the
retina by activation of glial P2X-purinoceptors. Br. J. Pharmacol. 124:317-322.
North RA (2002) Molecular physiology of P2X receptors. Physiol. Rev. 82:1013-1067.
North RA, Barnard EA (1997) Nucleotide receptors. Curr. Opin. Neurobiol. 7: 346–357.
North RA, Surprenant A (2000) Pharmacology of cloned P2X receptors. Annu. Rev.
Pharmacol. Toxicol. 40:563-580.
Nörenberg W, Illes P (2000) Neuronal P2X receptors: localisation and functional
properties. Naunyn Schmiedebergs Arch. Pharmacol. 362:324-339.
Kowaluk EA, Jarvis MF (2000) Therapeutic potential of adenosine kinase inhibitors.
Expert Opin. Investig Drugs. 9:551-564.
Pannicke T, Fischer W, Biedermann B, Schadlich H, Grosche J, Faude F, Wiedemann P,
Allgaier C, Illes P, Burnstock G, Reichenbach A (2000) P2X7 receptors in Muller glial
cells from the human retina. J. Neurosci. 20:5965-5972.
Paukert M, Osteroth R, Geisler HS, Brändle U, Glowatzki E, Ruppersberg JP, Grunder S
(2001) Inflammatory mediators potentiate ATP-gated channels through the P2X3
subunit. J. Biol. Chem. 276: 21077-21082
Piper AS, Docherty RJ (2000) One-way cross-desensitization between P2X purinoceptors
and vanilloid receptors in adult rat dorsal root ganglion neurones. J. Physiol. 523: 685-
696.
Ralevic V, Brunstock G (1998) Receptors for purines and pyrimidines. Pharmacol. Rev.
50:413-492.
Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A (1997) The
permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J.
Biol. Chem. 272:5482-5486.

95
Rettinger J, Schmalzing G (2003) Activation and desensitization of the recombinant P2X1
receptor at nanomolar ATP concentrations. J. Gen. Physiol. 121:451-461.
Rettinger J, Schmalzing G (2004) Desensitization masks nanomolar potency of ATP for the
P2X1 receptor. J. Biol. Chem. 279:6426-6433.
Roberts JA, Evans RJ (2004) ATP binding at human P2X1 receptors. Contribution of
aromatic and basic amino acids revealed using mutagenesis and partial agonists. J.
Biol. Chem. 279:9043-9055.
Robertson SJ, Rae MG, Rowan EG, Kennedy C (1996) Characterization of a P2X-
purinoceptor in cultured neurones of the rat dorsal root ganglia. Br. J. Pharmacol.
118:951-956.
Royle SJ, Bobanovic LK, Murrell-Lagnado RD (2002) Identification of a non-canonical
tyrosine-based endocytic motif in an ionotropic receptor. J. Biol. Chem. 277:35378-
35385.
Sluyter R, Barden JA, Wiley JS (2001) Detection of P2X purinergic receptors on human B
lymphocytes. Cell Tissue Res. 304:231-236.
Sokolova E, Nistri A, Giniatullin R (2001) Negative cross talk between anionic GABAA
and cationic P2X ionotropic receptors of rat dorsal root ganglion neurons. J. Neurosci.
21:4958-4968.
Soltoff SP, McMillian MK, Talamo BR, Cantley LC (1993) Blockade of ATP binding site
of P2 purinoceptors in rat parotid acinar cells by isothiocyanate compounds. Biochem.
Pharmacol. 45:1936-1940.
Spelta V, Jiang LH, Surprenant A, North RA (2002) Kinetics of antagonist actions at rat
P2X2/3 heteromeric receptors. Br. J. Pharmacol. 135:1524-1530.

96
Sperlagh B, Kofalvi A, Deuchars J, Atkinson L, Milligan CJ, Buckley NJ, Vizi ES (2002)
Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat
hippocampus. J. Neurochem. 81:1196-1211.
Starke K (2005)Grundlagen der Pharmakologie des Nervensystems. In: Aktories:
Förstermann, Hofmann, Starke; Editors: Elsevier GmbH, München,; Allgemeine
Pharmakologie und Toxikologie; S.137
Sternweis PC, Pang IH (1990) The G protein-channel connection. Trends Neurosci.
13:122-126.
Stoop R, Thomas S, Rassendren F, Kawashima E, Buell G, Surprenant A, North RA (1999)
Contribution of individual subunits to the multimeric P2X2 receptor: estimates based
on methanethiosulfonate block at T336C. Mol. Pharmacol. 56:973-981.
Surprenant A (1996) Functional properties of native and cloned P2X receptors. Ciba Found
Symp. 198:208-219
Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. (1996) The cytolytic P2Z
receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272:735-
738.
Swope SL, Moss SJ, Raymond LA, Huganir RL. (1999) Regulation of ligand-gated ion
channels by protein phosphorylation. Adv. Second Messenger Phosphoprotein Res.
33:49-78.
Thomas S, Virginio C, North RA, Surprenant A (1998) The antagonist trinitrophenyl-ATP
reveals co-existence of distinct P2X receptor channels in rat nodose neurones. J.
Physiol. 509:411-417.
Torres G.E, Egan T.M, Voigt M.M, (1999) Hetero-oligomeric assembly of P2X receptor
subunits. Specificities exist with regard to possible partners. J. Biol. Chem. 274:6653–
6659.

97
Ueno S, Tsuda M, Iwanaga T, Inoue K (1999) Cell type-specific ATP-activated responses
in rat dorsal root ganglion neurons. Br. J. Pharmacol. 126:429-436.
Ugur M, Drummond RM, Zou H, Sheng P, Singer JJ, Walsh JV Jr (1997) An ATP-gated
cation channel with some P2Z-like characteristics in gastric smooth muscle cells of
toad. J. Physiol. 498:427-442.
Verkhratsky A, Steinhauser C (2000) Ion channels in glial cells. Brain Res. Brain Res. Rev.
32:380-412.
Vial C, Roberts JA, Evans RJ (2004) Molecular properties of ATP-gated P2X receptor ion
channels. Trends Pharmacol. Sci. 25:487-493.
Virginio C, MacKenzie A, North RA, Surprenant A (1999) Kinetics of cell lysis, dye
uptake and permeability changes in cells expressing the rat P2X7 receptor. J. Physiol.
519:335-346.
Virginio C, MacKenzie A, Rassendren FA, North RA, Surprenant A (1999) Pore dilation of
neuronal P2X receptor channels. Nat. Neurosci. 2:315-321.
Virginio C, North RA, Surprenant A (1998) Calcium permeability and block at homomeric
and heteromeric P2X2 and P2X3 receptors, and P2X receptors in rat nodose neurones.
J. Physiol. 510:27-35.
Virginio C, Robertson G, Surprenant A, North RA (1998) Trinitrophenyl-substituted
nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3
receptors. Mol. Pharmacol. 53:969-973.
von Kugelgen I (2005) Pharmacological profiles of cloned mammalian P2Y-receptor
subtypes. Pharmacol. Ther. 110:415-432.
von Kügelgen I, Wetter A (2000) Molecular pharmacology of P2Y-receptors. Naunyn-
Schmiedeberg's Arch. Pharmacol. 362:310–323.

98
Way KJ, Chou E, King GL (2000) Identification of PKC-isoform-specific biological
actions using pharmacological approaches. Trends Pharmacol. Sci. 21:181-187.
Wheeler-Schilling TH, Marquordt K, Kohler K, Jabs R, Guenther E (2000) Expression of
purinergic receptors in bipolar cells of the rat retina. Brain Res. Mol. Brain Res.
76:415-418.
Wieraszko A, Ehrlich YH (1994) On the role of extracellular ATP in the induction of long-
term potentiation in the hippocampus. J. Neurochem. 63:1731-1738.
Wildman SS, Brown SG, King BF, Burnstock G (1999) Selectivity of diadenosine
polyphosphates for rat P2X receptor subunits. Eur. J. Pharmacol. 367:119-123.
Wiley JS, Dao-Ung LP, Li C, Shemon AN, Gu BJ, Smart ML, Fuller SJ, Barden JA, Petrou
S, Sluyter R. (2003) An Ile-568 to Asn polymorphism prevents normal trafficking and
function of the human P2X7 receptor. J. Biol. Chem. 278:17108-17113.
Wirkner K, Franke H, Inoue K, Illes P (1998) Differential age-dependent expression of
alpha2 adrenoceptor- and P2 purinoceptor-functions in rat locus coeruleus neurons.
Naunyn Schmiedebergs Arch. Pharmacol. 357:186-189.
Wirkner K, Stanchev D, Koles L, Klebingat M, Dihazi H, Flehmig G, Vial C, Evans RJ,
Furst S, Mager PP, Eschrich K, Illes P (2005) Regulation of human recombinant P2X3
receptors by ecto-protein kinase C. J. Neurosci. 25:7734-7742.
Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C und Barsoumian EL (2004)
Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-
coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br. J.
Pharmacol. 141:141–151.
Xu GY, Huang LY (2002) Peripheral inflammation sensitizes P2X receptor-mediated
responses in rat dorsal root ganglion neurons. J. Neurosci. 22: 93-102.

99
Xu GY, Huang LY (2004) Ca2+/calmodulin-dependent protein kinase II potentiates ATP
responses by promoting trafficking of P2X receptors. Proc. Natl. Acad. Sci. USA.
101:11868-11873.
Zou H, Ugur M, Drummond RM, Singer JJ (2001) Coupling of a P2Z-like purinoceptor to
a fatty acid-activated K+ channel in toad gastric smooth muscle cells. J. Physiol.
534:59-70.

100
8. Abkürzungsverzeichnis
α,ß-meATP -α,ß-Methylen-Adenosin-5'-triphosphat
2-meSADP -2-Methylthio-Adenosin-5'-diphosphat
2MeSATP -2-Methylthio-Adenosin-5'-triphosphat
ADP -Adenosin-5'-diphosphat
AMP -Adenosin-5'-monophosphat
Ap3A -Di(adenosin5')triphosphat
Ap5A -Di(adenosin-5')pentaphosphat
ATP - Adenosin-5'-triphosphat
BSA -Rinderserum Albumin (Bovine Serum Albumin)
BzATP -2',3'-(Benzoyl-4-benzoyl)-Adenosin-5'-triphosphat
CaCl2 -Kalziumchlorid
cDNA -zyklische Desoxyribonukleinsäure
CMV -Zytomegalovirus
CsCl -Cesiumchlorid
CsOH -Cesiumhydroxid
CTP -Cytidin-5'-triphosphat
DAG - Diacylglycerol
DMEM -Dulbecco’s Modifiziertes Eagle Medium
DMSO -Dimethylsulfoxid
DNA -Desoxyribonukleinsäure
dNTPs -Desoxynucleotid Triphosphat
DRG -Hinterwurzelganglien Zellen (Dorsal Root Ganglion)
EC -extrazelluläre Lösung
EC50 -halbmaximale effektive Konzentration
EDTA -Ethylendiaminotetraessigsäure
EGFP -verstärktes grünfluoreszierendes Protein (Enhanced Green
Fluorescent Protein)
EGTA -Ethylenglycol-bis(2-aminoethylether)-N,N,N´,N´-tetraessigsäure
FKS -Fötales Kälberserum
EL -extrazelluläre Schleife (Extracellular Loop)
ER -endoplasmatisches Retikulum
EtBr -Ethidiumbromid

101
GABA -γ-Aminobuttersäure
GDP -Guanosin-5'-diphosphat
GDP-ß-S -Guanosin-5'-O-(2'-thiodiphosphat)
GPRC -G-Protein gekoppelter Rezeptor
GTP -Guanosin-5'-triphosphat
HBSS -Physiologische Salzlösung nach Hank (Hanks' Balanced Salt
Solution)
HCl -Salzsäure
HEK -humane, embryonale Nierenzellen (Human Embryonic Kidney)
HEPES -N-2-Hydroxyethylpiperazin-N´-ethansulfonsäure
IC -intrazelluläre Lösung
IP3 -Inositol-1,4,5-trisphosphat
Ip5I -Di(inosin-5')pentaphosphat
LTP -Langzeitpotenzierung (Long-Term Potentiation)
MgCl2 -Magnesiumchlorid
NaCl -Natrumchlorid
NBD -Nukleotid-Bindungsdomäne
NEAA -Nichtessentielle Aminosäure Lösung (Nonessential Amino Acids)
NMDG -N-Methyl-D-glukamine
Pca -relative Permeabilität für Kalzium
PCR -Polymerase-Kettenreaktion
PIP2 -Phosphatidylinositol-4,5-phosphat
PKC -Proteinkinase C
PLC -Phospholipase C
PLD -Phospholipase D
PMA -Phorbol 12-myristat 13-acetat
PPADS -Pyridoxal-5’-phosphat-6-azophenyl-2’,4’-disufonsäure
RT -Raumtemperatur
RNA -Ribonukleinsäure
SEM -Standardfehler der Mittelwerte (Standard Error of the Mean)
TEA -Tetraethylammonium
TM -transmembraner Bereich
TNP-ATP -2',3'-O-(2,4,6-trinitrophenyl)adenosin-5'-triphosphat
TRPV1 -Trancient Receptor Potential Vanilloid 1
102
TTX -Tetrodotoxin
UDP -Uridin-5'-triphosphat
ZNS -zentrales Nervensystem

9. Lebenslauf
Name: Doychin Toshev Stanchev
Anschrift: Köbisstr.6
04317 Leipzig
Telefon: 0179/7526123
E-mail: [email protected]
Geburtsdatum: 26.07.1977
Geburtsort: Russe, Bulgarien
Staatsangehörigkeit: bulgarisch
Familienstand: ledig
Führerschein: Klasse M und B
Ausbildung
1984-1991 Grundschule „Otetz Paisiy“ Sandrovo
1991-1996 Allgemeinbildende Oberschule „Sv. Konstantin –Kiril
Philosoph“ Russe für europäische Sprachen mit
intensivem Erlernen der deutschen Sprache
1996-2002 Medizinische Universität Sofia, Fakultät für Pharmazie
13.03.2001- Deutschkurs am Herder-Institut in Leipzig
12.07.2001 Studienabschluss mit dem Titel „Diplompharmazeut“
Berufserfahrung
2000-2002 Praktika in bulgarischen Apotheken in Sofia
3.10.2000 Teilnahme am Internationalen Symposium für 7.10.2000
Pharmaziestudenten (Second IPSF Scientific

104
Symposium, Albena, Bulgarien)
22.07.2001- Arbeit in der Anlage für schwerbehinderte Kinder
11.09.2001 „Fasanenhof“ in Stuttgart
01.01.2002-
10.07.2002 Arbeit als Apotheker in bulgarischen Apotheken
01.08.2002- Promotion an der Universität Leipzig in der
30.06.2003 Abteilung „Pharmakologie für Naturwissenschaftler“ des
Institutes für Pharmazie der Fakultät für
Biowissenschaften, Pharmazie und Psychologie
01.04.2003- Stipendiat der Friedrich-Naumann-Stiftung
31.03.2006
ab 01.07.2003 Promotion an der Universität Leipzig am
Rudolf-Boehm-Institut für Pharmakologie und
Toxikologie
seit 1.04.2006 Wissenschaftlicher Mitarbeiter an der
Universität Leipzig
Preise und Drittmittel-Einwerbung
10.12.2004 Posterpreis beim 3. Leipziger Research Festival
01.01.2007-
31.12.2007 Projektförderung im Rahmen des formel.1 -Programmes der
Medizinischen Fakultät der Universität Leipzig (Summe: 41.290 €)
Kongreßteilnahmen
15.03.2005- Teilnahme an der 46. Frühjahrstagung der
17.03.2005 Deutschen Gesellschaft für Experimentelle und
Klinische Pharmakologie und Toxikologie (DGPT)
18.09.2005- Teilnahme am „First Joint Italian-German Purine
20.09.2005 Club Meeting“

105
Fremdsprachen
Deutsch sehr gute Kenntnisse (abgelegte Deutsche
Sprachprüfung für den Hochschulzugang ausländischer
Studienbewerber (DSH)-Prüfung – Zeugnis der
Universität Leipzig vom 12.07.2001)
Englisch gute Kenntnisse (Schulkenntnisse)
Russisch gute Kenntnisse (Schulkenntnisse)
Weitere Kenntnisse
Computerkenntnisse: MS.Office, Internet, CorelDRAW
Hobbys
Freistilringen, Radfahren

Erklärung Ich versichere, dass ich meine Dissertation
„Untersuchungen zur Modulation der Ionenleitfähigkeit von P2X3- und P2X7-Rezeptoren durch extrazelluläre Phosphorylierung und Regulation
der Rezeptorexpression in HEK 293-Zellen“ selbstständig und ohne unerlaubte Hilfe angefertigt habe und mich dabei keiner anderen als
der von mir ausdrücklich bezeichneten Quellen bedient habe.
Die Dissertation wurde in der jetzigen oder einer ähnlichen Form noch bei keiner anderen
Hochschule eingereicht und hat noch keinen sonstigen Prüfungszwecken gedient.
Marburg, den 11.01.2007
Doychin Stanchev





























