Embed Size (px)
Citation preview

Synthese und Charakterisierung von potenziellen Inhibitoren
des „Macrophage infectivity potentiator“ (Mip) Proteins
von Legionella pneumophila
-
Ein neuer Ansatz in der Legionellose-Therapie
Dissertation zur Erlangung des
Naturwissenschaftlichen Doktorgrades
der Julius-Maximilians-Universität Würzburg
Vorgelegt von
Christina Juli
aus Würzburg
Würzburg, 2012

Eingereicht am:……………………………………..
bei der Fakultät für Chemie und Pharmazie
1. Gutachter:………………………………………..
2. Gutachter:………………………………………..
der Dissertation
1. Prüfer:……………………………………………
2. Prüfer:……………………………………………
3. Prüfer:……………………………………………
des Öffentlichen Promotionskolloquiums
Tag des Öffentlichen Promotionskolloquiums:……………….
Doktorurkunde ausgehändigt am:……………………………

Die vorliegende Arbeit wurde in der Zeit von Januar 2008 bis Januar 2012 am Institut für Pharmazie
und Lebensmittelchemie der Bayerischen Julius-Maximilians-Universität Würzburg unter der
Anleitung von Frau Professor Dr. Ulrike Holzgrabe mit finanzieller Unterstützung des
Sonderforschungsbereiches 630 angefertigt.


Teile dieser Arbeit wurden bereits in folgender Form veröffentlicht:
Originalarbeiten:
Juli, C.; Sippel, M.; Jäger, J.; Thiele, A.; Weiwad, M.; Schweimer, K.; Rösch, P.; Steinert,
M.; Sotriffer, C.A.; Holzgrabe, U.
Pipecolic Acid Derivatives As Small-Molecule Inhibitors of the Legionella MIP Protein
J. Med. Chem. 2011, 54(1), 277
Vorträge:
Juli, C.
Potential inhibitors of the Macrophage-infectivity-potentiator (MIP) protein of Legionella
pneumophila
Seminar im Rahmen des SFB 630, Würzburg 2012
Abstrakta und Kongreßmitteilungen:
Juli, C.; Sippel, M.; Sotriffer, C.A.; Faber, C.; Steinert, M.; Holzgrabe, U.
Design and Synthesis of novel MIP Inhibitors as Potential Compounds against Legionnaires
Disease
XXth International Symposium on Medicinal Chemistry, Wien 2008
Juli, C.; Sippel, M.; Faber, C.; Steinert, M.; Sotriffer, C.A.; Holzgrabe, U.
Pipecolic Acid Derivatives – Promising Agents against Legionnaires Disease
DPhG – Jahrestagung, Bonn 2008
Juli, C.; Sippel, M.; Faber, C.; Steinert, M.; Sotriffer, C.A.; Holzgrabe, U.
Analogues of Rapamycin as MIP Inhibitors of Legionella pneumophila
SFB 630 – Doktorandentagung, Bronnbach 2008
Chemie – Symposium der Studierenden Mainfrankens, Würzburg 2008
Juli, C.; Sippel, M.; Thiele, A.; Steinert, M.; Sotriffer, C.A.; Holzgrabe, U.
Design and Synthesis of Novel Lead Structures against MIP, a Target of Legionella
pneumophila
DPhG – Jahrestagung, Jena 2009
SFB 544 – Doktorandentagung, Heidelberg 2009
Sippel, M.; Juli, C.; Holzgrabe, U.; Sotriffer, C.A.
Legionnaires´ Disease and the Role of Legionella MIP: Fragment-Based Design and
Synthesis of the first Small-Molecule MIP Inhibitor
International Symposium of the Collaborative Research Center 630, Würzburg 2009
Juli, C.; Sippel, M.; Thiele, A.; Weiwad, M.; Jäger, J.; Steinert, M.; Schweimer, K.; Rösch,
P.; Sotriffer, C.A.; Holzgrabe, U.
The Importance of Inhibiting Infectivity Protein MIP for the Treatment of Legionellosis
DPhG – Jahrestagung, Braunschweig 2010
Chemie – Symposium der Studierenden Mainfrankens, Würzburg 2010

Juli, C.; Sippel, M.; Thiele, A.; Weiwad, M.; Jäger, J.; Steinert, M.; Schweimer, K.; Rösch,
P.; Sotriffer, C.A.; Holzgrabe, U.
MIP Inhibitors of the Pipecolic Acid Type as Potential Drugs against Legionellosis
Frontiers in Medicinal Chemistry, Saarbrücken 2011
Juli, C.; Weiwad, M.; Steinert, M.; Schwarzinger, S.; Rösch, P.; Holzgrabe, U.
Development of novel Inhibitors of MIP, a Target of Legionella pneumophila
Gemeinsame ÖPhG - DPhG – Jahrestagung, Innsbruck 2011


Danksagung
An dieser Stelle möchte ich mich recht herzlich bei allen Personen und Freunden (die ich im
Einzelnen nicht nennen kann) bedanken, die mich während der Erstellung dieser Arbeit in den
verschiedensten Bereichen unterstützt haben.
Mein besonderer Dank gilt Frau Prof. Dr. Ulrike Holzgrabe für die freundliche Aufnahme in ihren
Arbeitskreis, die äußerst interessante und abwechslungsreiche Aufgabenstellung, ihre Unterstützung
und immerwährende Diskussionsbereitschaft während der Promotion sowie das in mich gesetzte
Vertrauen, das mir selbstständiges und eigenverantwortliches Arbeiten und damit die erfolgreiche
Erstellung dieser Arbeit ermöglichte.
Weiterhin möchte ich all meinen Kooperationspartnern danken:
Prof. Dr. Christoph A. Sotriffer vom Institut für Pharmazie in Würzburg und seinen Mit-
arbeitern Dr. Martin Sippel und Michael Hein für die gute und freundschaftliche Zusammen-
arbeit sowie ihre Untersuchungen im Bereich des computergestützten Wirkstoffdesigns.
Dr. Matthias Weiwad und Dr. Alexandra Thiele sowie ihren Kollegen an der Max-Planck
Forschungsstelle für Enzymologie der Proteinfaltung in Halle für die mir entgegengebrachte
Gastfreundschaft und die Durchführung der enzymatischen Testungen.
Prof. Dr. Michael Steinert vom Institut für Mikrobiologie in Braunschweig sowie seinem
Mitarbeiter Jens Jäger für die Durchführung der In-vivo-Untersuchungen.
Prof. Dr. Paul Rösch vom Department of Biopolymers in Bayreuth sowie seinen Mitarbeitern
Dr. Kristian Schweimer und PD Dr. Stephan Schwarzinger für die Durchführung der NMR-
Bindungsstudien.
Prof. Dr. Thomas Rudel und Dr. Vera Kozjak-Pavlovic vom Institut für Mikrobiologie in
Würzburg für die Substanztestungen an Chlamydien und Neisserien.
Dr. Isobel Norville vom Defence Science and Technology Laboratory in Salisbury (UK) für
die freundliche Zusammenarbeit sowie die Untersuchungen an Burkholderien.
Dem SFB 630 - Teilprojekt Z1 für die Durchführung weiterer antimikrobieller Testungen.
Claudia Steinert und Alexander Hörst für ihre aufopferungsvollen Bemühungen bei der
Bestimmung der Enantiomerenreinheit.
Frau Ebner und Frau Möhler möchte ich recht herzlich für die freundliche Unterstützung in allen
bürokratischen Belangen sowie für die überaus erfrischenden Gespräche zwischendurch danken.

Dr. Sabine Niedermeier, Jessica Klöckner, Dr. Jens Schmitz, Dr. Tim Göbel und Dr. Eberhard Heller
danke ich für die nette Einführung in die chemische Synthese und die gute Zusammenarbeit im
„alten“ Großraumlabor 112.
Dr. Eberhard Heller danke ich für die zahlreichen Tipps und Ratschläge im Laboralltag sowie für die
Unterstützung bei diversen Synthese- bzw. NMR-Fragestellungen, ohne die man als Pharmazeut
manchmal recht hilflos gewesen wäre.
Für jegliche Unterstützung im Labor, sei es bei verschiedenen Synthesen oder der Beseitigung der
allgemeinen Unordnung, sowie für die netten Unterhaltungen bedanke ich mich bei Liana Pogorelaja.
Benjamin Schaefer, Christine Topf, Dr. Eva Kugelmann, Katja Heinig, Michaela Prinz und Dr. Petra
Kapkova danke ich für die angenehmen Gespräche während der Betreuung des Studentenpraktikums,
die die langen Nachmittage immer wieder schnell vergingen ließen.
Ein überaus großer Dank gilt Dr. Jens Schmitz für seine Mühe und Geduld beim Korrekturlesen.
Meinen Kollegen und Freunden Christine Topf, Diana Kesetovic, Georg Hiltensperger, Ines Schmidt,
Maximilian Tischer und Michaela Prinz möchte ich für die tolle Atmosphäre im „neuen“ 6er-
Syntheselabor danken. Die zahlreichen lustigen „Labormomente“ haben sehr zur allgemeinen
Aufhellung des Alltags beigetragen. Des Weiteren danke ich Georg, Max und Andi besonders für
ihre Diskussionsbereitschaft bei allen auftretenden synthetischen Problemen.
Christine, Georg, Max, Michi, Jens, Tim und Julia gilt ein großes Dankeschön für die vielen, auch
außeruniversitären fröhlichen Stunden (v.a. bei German) und eine unvergessliche Zeit in Würzburg.
Christine möchte ich besonders für die vielen Stunden v.a. im Rahmen des SFB 630 (wie bei der
Organisation der Bronnbach-Tagung, diversen Vorträgen und Tagungen) danken. Ohne Dich wäre
diese Zeit nur halb so angenehm und erheiternd geworden.
Den Randersackerern Max und Michi danke ich für die vielen wundervollen, gemeinsamen Abende
auf der Couch.
Ein ganz besonderer Dank geht an meine Eltern, Großeltern und meine Schwester für ihre großartige
Unterstützung während dieser Zeit sowie in allen Lebensbereichen und -lagen. Danke, dass Ihr
immer für mich da seid und mich einfach so nehmt wie ich bin!
Zu guter Letzt, danke ich meinem Schatz André. Danke, dass Du immer für mich da warst und bist.
Dass Du mich bei allem unterstützt und mir oftmals dabei geholfen hast, nicht den Boden unter den
Füßen zu verlieren. Ich freue mich auf eine wunderbare Zeit mit Dir!


„Darin besteht das Wesen der Wissenschaft:
Zuerst denkt man an etwas, das wahr sein könnte.
Dann sieht man nach, ob es der Fall
ist und im Allgemeinen ist es nicht der Fall.“
Bertrand Russell (1872-1970), brit. Philosoph u. Mathematiker


Meinen lieben Eltern und
meiner wunderbaren Schwester


INHALTSVERZEICHNIS I
1 EINLEITUNG ........................................................................................ 1
1.1 Legionellose .......................................................................................................... 1
1.1.1 Historischer Rückblick ..................................................................................................... 1
1.1.2 Erreger – Legionella pneumophila ................................................................................... 1
1.1.2.1 Vorkommen und parasitäre Lebensform .......................................................................... 2
1.1.2.2 Pathogenese und Epidemiologie ....................................................................................... 4
1.1.2.3 Therapie, Präventionsmaßnahmen und Diagnostik .......................................................... 5
1.1.2.4 Aufbau, Eigenschaften und Bestandteile der Zellwand .................................................... 7
1.1.3 Macrophage-Infectivity-Potentiator Protein ..................................................................... 9
1.1.3.1 Strukturelle und enzymatische Eigenschaften .................................................................. 9
1.1.3.2 Biochemische Analyse: Mip als Virulenzfaktor? ........................................................... 11
1.2 Immunsystem und immunsupprimierende Substanzen ................................. 14
1.2.1 Immunophiline ................................................................................................................ 15
1.2.1.1 FKBP12 – Strukturelle und zelluläre Eigenschaften ...................................................... 15
1.2.2 Makrozyklische Immunsuppressiva ............................................................................... 17
1.2.2.1 Immunologische Wirkung .............................................................................................. 17
1.2.2.2 Struktur-Wirkungsbeziehung .......................................................................................... 18
1.3 Strukturen der beiden PPIasen: Mip und FKBP12 ....................................... 20
1.3.1 Mip-Strukturen ............................................................................................................... 21
1.3.2 FKBP12-Strukturen ........................................................................................................ 23
1.3.3 Struktureller Vergleich der PPIasen ............................................................................... 24
1.4 Mip-ähnliche Strukturen anderer Mikroorganismen .................................... 27
1.4.1 Chlamydien ..................................................................................................................... 27
1.4.2 Trypanosomen ................................................................................................................ 28
1.4.3 Neisserien ....................................................................................................................... 29
1.4.4 Burkholderien ................................................................................................................. 30
1.5 Zielsetzung .......................................................................................................... 31
1.5.1 Chemische Synthese ....................................................................................................... 31
1.5.2 Pharmakologische Testung ............................................................................................. 33

II INHALTSVERZEICHNIS
2 DESIGN UND SYNTHESE DER ZIELVERBINDUNGEN .......................... 35
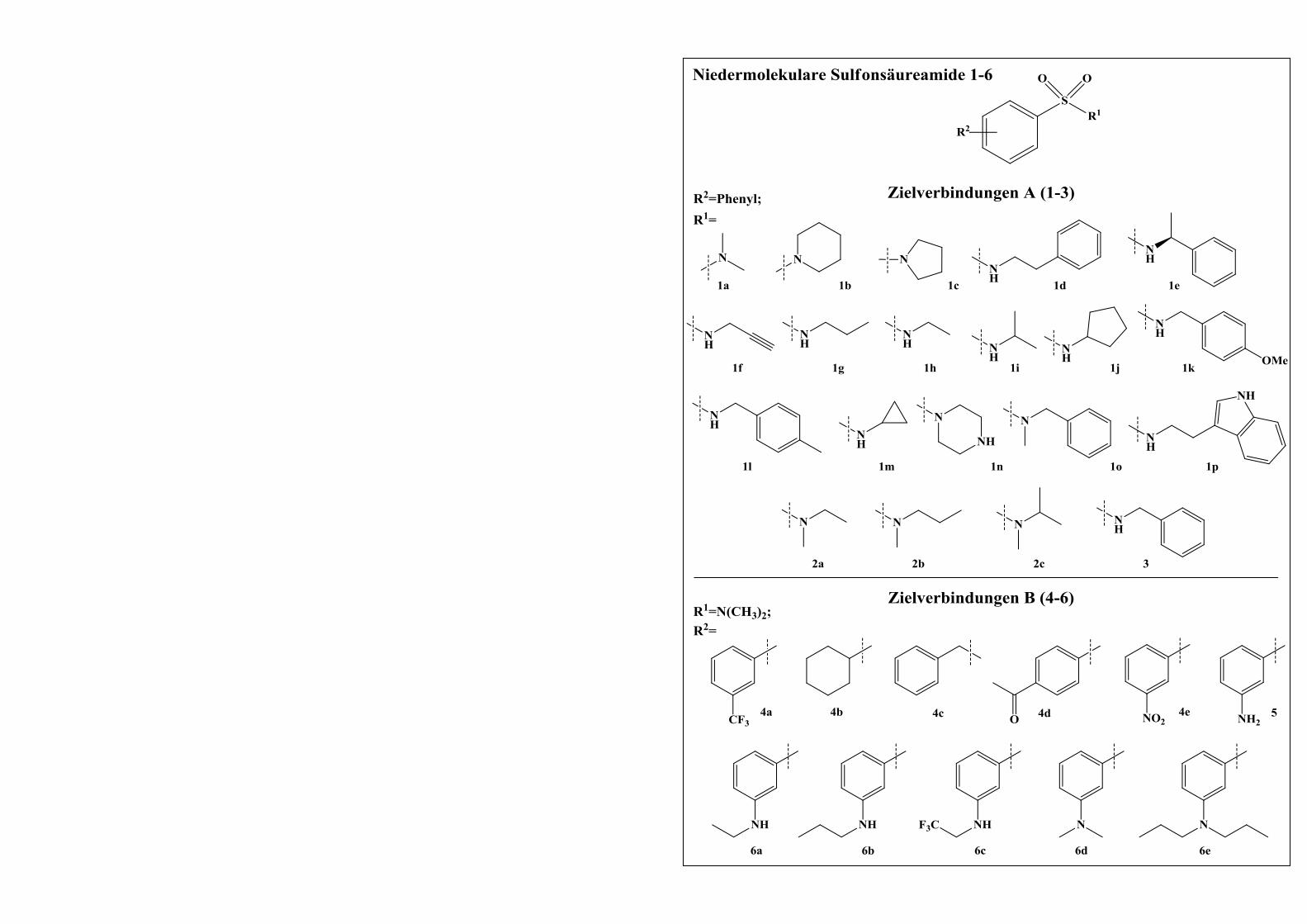
2.1 „Kleine“ Sulfonsäureamide .............................................................................. 35
2.1.1 Computergestützte Entwicklung neuartiger Mip-Inhibitoren ......................................... 35
2.1.2 Synthese der designten Sulfonsäureamid-Derivate ......................................................... 39
2.1.2.1 Darstellung von Derivaten mit der Grundstruktur A ...................................................... 40
2.1.2.1.1 Syntheseweg zur Herstellung der Phenylsulfonsäureamide 1a-1p ................................. 40
2.1.2.1.2 Syntheseweg zur Herstellung der Phenylsulfonsäureamide 2a-2c ................................. 42
2.1.2.1.3 Versuche der Synthese von N-((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid 42
2.1.2.2 Darstellung von Derivaten mit der Grundstruktur B....................................................... 45
2.1.2.2.1 Syntheseweg zur Herstellung der Verbindungen 4a-4e .................................................. 45
2.1.2.2.2 Syntheseweg zur Herstellung von Verbindung 5 ............................................................ 46
2.1.2.2.3 Syntheseweg zur Herstellung der Verbindungen 6a-6e .................................................. 46
2.1.2.3 Versuche der Synthese von Aminomethylphenylsulfon ................................................. 48
2.1.3 Diskussion des 1H-NMR-Spektrums von Verbindung 1k .............................................. 49
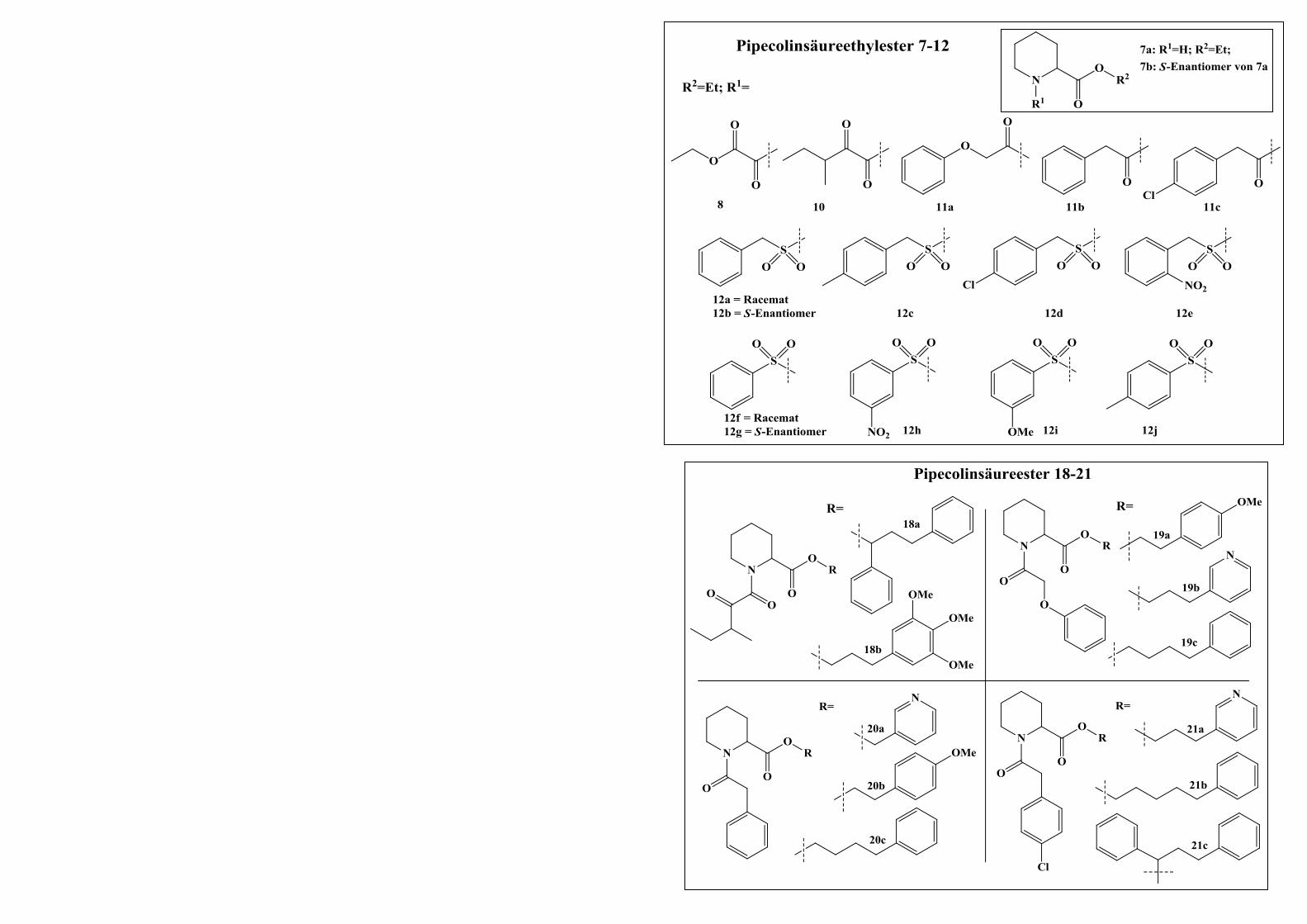
2.2 Pipecolinsäure-Derivate .................................................................................... 50
2.2.1 Docking-Untersuchungen von bekannten FKBP12-Inhibitoren ..................................... 50
2.2.2 Darstellung der Pipecolinsäure-Derivate ........................................................................ 52
2.2.2.1 Allgemeine Synthesestrategie zur Darstellung der Zielverbindung A ............................ 52
2.2.2.1.1 Syntheseweg zur Herstellung von Verbindung 18a ........................................................ 52
2.2.2.1.2 Syntheseweg zur Herstellung von Verbindung 18b ....................................................... 56
2.2.2.2 Allgemeine Synthesestrategie zur Darstellung der Zielverbindung B ............................ 57
2.2.2.2.1 Syntheseweg zur Herstellung von Verbindung 22a ........................................................ 57
2.2.2.2.2 Syntheseweg zur Herstellung von Verbindung 22b ....................................................... 58
2.2.2.2.3 Bestimmung der Enantiomerenreinheit von Verbindung 22b ........................................ 58
2.2.2.3 Allgemeine Synthesestrategie zur Darstellung von Pipecolinsäure-Derivaten mit der
Grundstruktur B .............................................................................................................. 60
2.2.2.3.1 Syntheseweg zur Herstellung der Verbindungen 19-21.................................................. 61
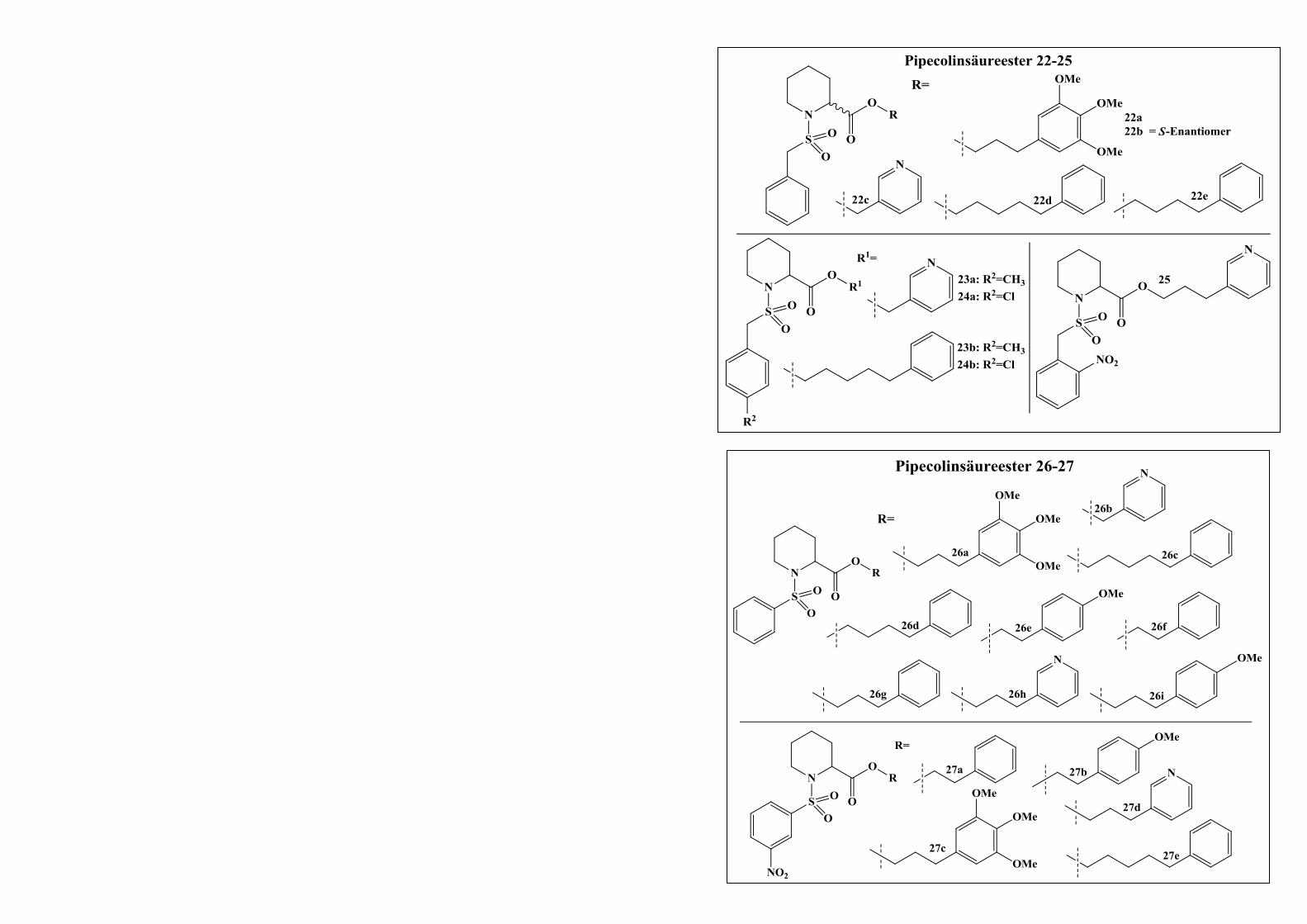
2.2.2.3.2 Syntheseweg zur Herstellung der Verbindungen 22-29.................................................. 62
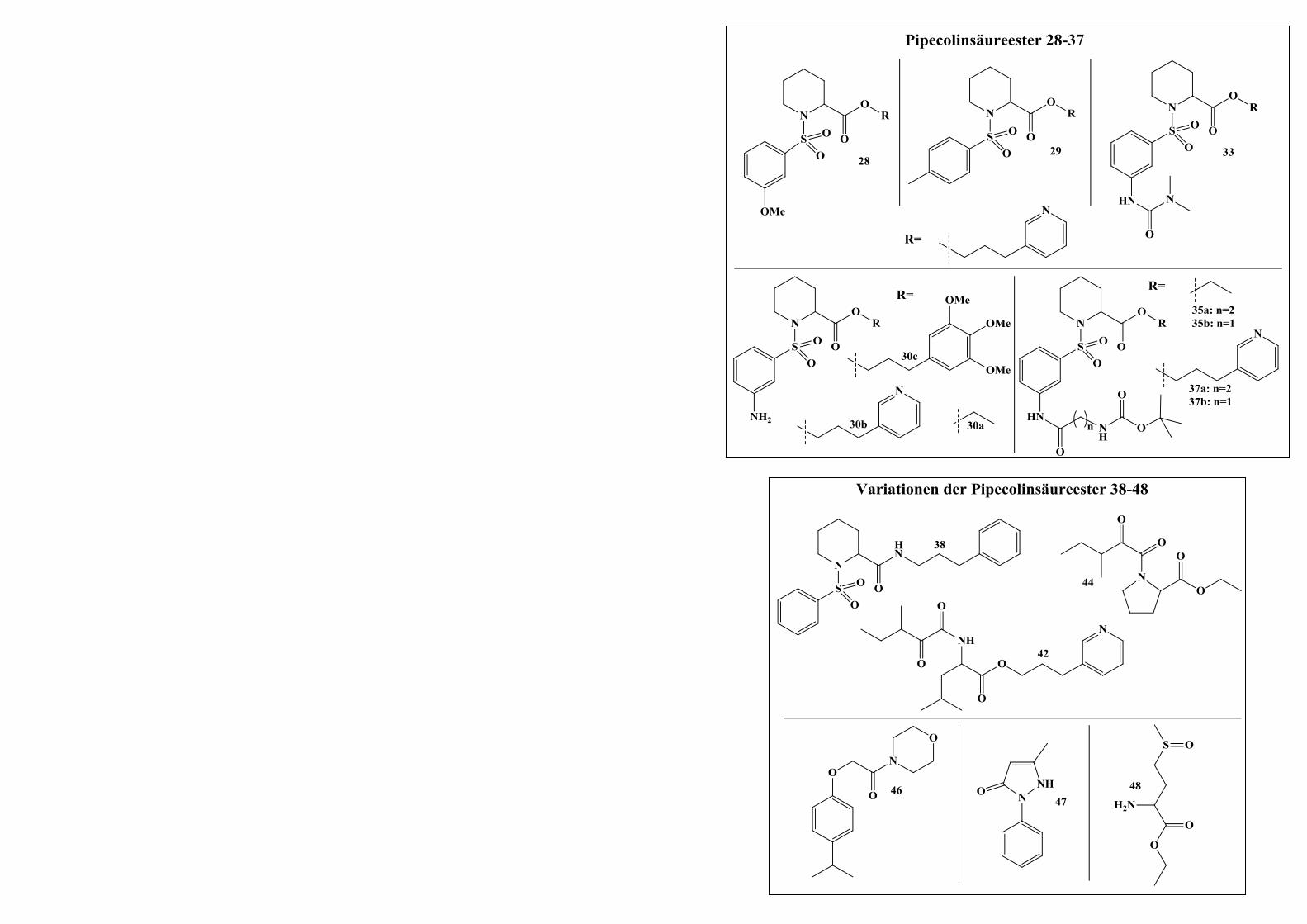
2.2.2.4 Derivatisierungen am Aromaten der Sulfonsäureamidgruppe ........................................ 64

INHALTSVERZEICHNIS III
2.2.2.4.1 Syntheseweg zur Herstellung der Verbindungen 30a-30c ............................................. 64
2.2.2.4.2 Syntheseweg zur Herstellung von Verbindung 33 ......................................................... 65
2.2.2.4.3 Syntheseweg zur Herstellung der Verbindungen 37a und 37b ...................................... 66
2.2.2.5 Variationen des Grundgerüsts der Zielverbindungen ..................................................... 69
2.2.2.5.1 Syntheseweg zur Herstellung von Verbindung 38 ......................................................... 69
2.2.2.5.2 Syntheseweg zur Herstellung von Verbindung 42 ......................................................... 70
2.2.2.5.3 Syntheseweg zur Herstellung von Verbindung 44 ......................................................... 71
2.2.3 Diskussion des 1H-NMR-Spektrums von Verbindung 22a ............................................ 71
2.3 Weitere neu-„designte” Derivate ...................................................................... 75
2.3.1 Syntheseweg zur Herstellung von Verbindung 46 ......................................................... 75
2.3.2 Syntheseweg zur Herstellung von Verbindung 47 ......................................................... 76
2.3.3 Syntheseweg zur Herstellung von Verbindung 48 ......................................................... 77
3 PHARMAKOLOGISCHE TESTUNGEN UND ERGEBNISSE .................... 79
3.1 In-vitro-Testsystem ............................................................................................ 79
3.1.1 Expression von Mip ........................................................................................................ 79
3.1.1.1 Klonierung und Transformation ..................................................................................... 79
3.1.1.2 Anzucht von E. coli-Bakterien ....................................................................................... 79
3.1.1.3 Zellaufschluss mittels mechanischer Lyse ...................................................................... 80
3.1.1.4 Reinigung von Mip ......................................................................................................... 80
3.1.2 Durchführung und Auswertung des PPIase-Assays ....................................................... 82
3.1.3 Ergebnisse ....................................................................................................................... 86
3.2 NMR-Bindungsstudien ...................................................................................... 90
3.2.1 Prinzip und Durchführung der 15
N-HSQC-NMR-Experimente ...................................... 90
3.2.2 Ergebnisse ....................................................................................................................... 91
3.3 In-vivo-Testsystem ............................................................................................. 93
3.3.1 Transwell-Assay ............................................................................................................. 93
3.3.2 Infektionsstudien ............................................................................................................ 93
3.3.2.1 Durchführung .................................................................................................................. 93
3.3.2.2 Ergebnisse ....................................................................................................................... 93

IV INHALTSVERZEICHNIS
3.4 Testungen an Mip-ähnlichen Proteinen .......................................................... 95
3.4.1 Pharmakologische Testungen an Neisserien und Chlamydien ....................................... 95
3.4.1.1 Infektionsstudien an N. gonorrhoeae .............................................................................. 95
3.4.1.2 Infektionsstudien an C. trachomatis ............................................................................... 96
3.4.2 Pharmakologische Testungen an Burkholderien ............................................................. 97
3.5 Antimikrobielle Aktivitäten gegen Protozoen ................................................ 97
4 ZUSAMMENFASSUNG ....................................................................... 101
5 SUMMARY ........................................................................................ 107
6 EXPERIMENTELLER TEIL ............................................................... 113
6.1 Allgemeine Angaben ....................................................................................... 113
6.1.1 Verwendete Geräte ........................................................................................................ 113
6.1.2 Chromatographie........................................................................................................... 114
6.1.3 Chemikalien und Lösungsmittel ................................................................................... 115
6.1.4 Abkürzungen ................................................................................................................. 116
6.2 Synthesevorschriften und analytische Daten ................................................ 117
6.2.1 Darstellung der Sulfonsäureamid-Derivate 1-6 ............................................................ 117
6.2.1.1 Synthese der Phenylsulfonsäureamide 1a-1p ............................................................... 117
6.2.1.2 Synthese der N,N-disubstituierten Phenylsulfonsäureamide 2a-2c............................... 121
6.2.1.3 Synthese von N-Benzylphenylsulfonsäureamid 3 ......................................................... 123
6.2.1.4 Synthese der N,N-Dimethylsulfonsäureamide 4a-4e .................................................... 124
6.2.1.5 Synthese von 3-Amino-N,N-dimethylphenylsulfonsäureamid 5 .................................. 126
6.2.1.6 Synthese der N-alkylierten 3-Amino-N,N-dimethylphenylsulfonsäureamide 6a-6e .... 127
6.2.2 Synthese der Piperidin-2-carbonsäureethylester 7a und 7b .......................................... 129
6.2.3 Synthese von 1-(2-Ethoxy-2-oxoacetyl)piperidin-2-carbonsäureethylester 8 .............. 130
6.2.4 Synthese von 3-Methyl-2-oxopentansäure 9 ................................................................. 131
6.2.5 Synthese von 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäureethylester 10 ...... 132
6.2.6 Darstellung der Pipecolinsäureethylester-Amide 11 und 12 ......................................... 133
6.2.6.1 Synthese der Carbonsäureamide 11a-11c aus den entsprechenden Säurechloriden ..... 133

INHALTSVERZEICHNIS V
6.2.6.2 Synthese der Sulfonsäureamide 12a-12j aus den entsprechenden Sulfonsäurechloriden
...................................................................................................................................... 136
6.2.7 Synthese der Pipecolinsäure-Amide 13-15 ................................................................... 140
6.2.8 Synthese von 3-(3,4,5-Trimethoxyphenyl)propan-1-ol 16 ........................................... 144
6.2.9 Synthese von 1,3-Diphenylpropan-1-ol 17 ................................................................... 145
6.2.10 Darstellung der Piperidinamid-2-carbonsäureester-Derivate 18-29 ............................. 146
6.2.10.1 Synthese der 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäureester 18a und 18b
..................................................................................................................................... .147
6.2.10.2 Synthese der 1-(2-Phenoxyacetyl)piperidin-2-carbonsäureester 19a-19c .................... 148
6.2.10.3 Synthese der 1-(2-Phenylacetyl)piperidin-2-carbonsäureester 20a-20c ....................... 151
6.2.10.4 Synthese der 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäureester 21a-21c ........ 153
6.2.10.5 Synthese der 1-(Benzylsulfonyl)piperidin-2-carbonsäureester 22a-22e ...................... 156
6.2.10.6 Synthese der 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäureester 23a und 23b
..................................................................................................................................... .158
6.2.10.7 Synthese der 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäureester 24a und 24b . 160
6.2.10.8 Synthese von 1-((2-Nitrobenzyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)-
propyl)ester 25 .............................................................................................................. 161
6.2.10.9 Synthese der 1-(Phenylsulfonyl)piperidin-2-carbonsäureester 26a-26i ....................... 162
6.2.10.10 Synthese der 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäureester 27a-27e......... 166
6.2.10.11 Synthese von 1-(3-Methoxyphenylsulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)-
propyl)ester 28 .............................................................................................................. 168
6.2.10.12 Synthese von 1-Tosylpiperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester 29 ......... 169
6.2.11 Synthese der 1-((3-Aminophenyl)sulfonyl)piperidin-2-carbonsäureester 30a-30c ...... 170
6.2.12 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäureethyl-
ester 31 .......................................................................................................................... 172
6.2.13 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäure 32
..................................................................................................................................... .173
6.2.14 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäure-(3-
(pyridin-3-yl)propyl)ester 33 ........................................................................................ 174
6.2.15 Darstellung von BOC-geschützten Aminosäuren 34 .................................................... 175

VI INHALTSVERZEICHNIS
6.2.15.1 Synthese von 4-((tert.-Butoxycarbonyl)amino)buttersäure 34a und 3-((tert.-Butoxy-
carbonyl)amino)propionsäure 34b ................................................................................ 175
6.2.15.2 Synthese von 2-((tert.-Butoxycarbonyl)amino)essigsäure 34c ..................................... 176
6.2.16 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)-
piperidin-2-carbonsäureethylester 35a und 1-((3-(2-((tert.-Butoxy-carbonyl)amino)-
acetamido)phenyl)sulfonyl)piperidin-2-carbonsäureethylester 35b ............................. 177
6.2.17 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)-
piperidin-2-carbonsäure 36a und 1-((3-(2-((tert.-Butoxycarbonyl)amino)acetamido)-
phenyl)sulfonyl)piperidin-2-carbonsäure 36b .............................................................. 179
6.2.18 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)-
piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester 37a und 1-((3-(2-((tert.-Butoxy-
carbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)-
propyl)ester 37b ............................................................................................................ 180
6.2.19 Synthese von N-(3-Phenylpropyl)-1-(phenylsulfonyl)piperidin-2-carbonsäureamid 38
...................................................................................................................................... 182
6.2.20 Synthese von 2-Amino-4-methylpentansäureethylester 39 .......................................... 183
6.2.21 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäureethylester 40 ..... 183
6.2.22 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure 41 .................... 184
6.2.23 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure-(3-(pyridin-3-yl)-
propyl)ester 42 .............................................................................................................. 185
6.2.24 Synthese von Pyrrolidin-2-carbonsäureethylester 43.................................................... 186
6.2.25 Synthese von 1-(3-Methyl-2-oxopentanoyl)pyrrolidin-2-carbonsäureethylester 44 ..... 187
6.2.26 Synthese von 2-(4-Isopropylphenoxy)essigsäure 45 .................................................... 188
6.2.27 Synthese von 2-(4-Isopropylphenoxy)-1-morpholinessigsäureamid 46 ....................... 189
6.2.28 Synthese von 5-Methyl-2-phenyl-1H-pyrazol-3(2H)-on 47 ......................................... 190
6.2.29 Synthese von 2-Amino-4-(methylsulfinyl)buttersäureethylester 48 ............................. 191
6.3 Expression von Mip ......................................................................................... 192
6.3.1 Verwendete Geräte ........................................................................................................ 192
6.3.2 Klonierung und Transformation des Mip-Gens ............................................................ 192
6.3.3 Anzucht von E. coli-Bakterien ...................................................................................... 192
6.3.4 Aufschluss der E. coli-K12-HB101-Zellkulturen ......................................................... 193

INHALTSVERZEICHNIS VII
6.3.5 Säulenchromatographische Reinigung von Mip ........................................................... 193
6.3.6 Dialyse von gereinigtem Mip ....................................................................................... 195
6.4 Enzymatischer Assay ....................................................................................... 195
6.4.1 Verwendete Geräte ....................................................................................................... 195
6.4.2 Herstellung der Puffer- bzw. Stammlösungen .............................................................. 196
6.4.3 Protease-gekoppelter PPIase-Assay .............................................................................. 196
7 ANHANG .......................................................................................... 199
7.1 Überblick synthetisierter Verbindungen ....................................................... 199
7.2 logP-Werte synthetisierter Verbindungen .................................................... 202
8 LITERATURVERZEICHNIS................................................................ 205


EINLEITUNG 1
1 EINLEITUNG
1.1 Legionellose
1.1.1 Historischer Rückblick
Am 01.01.2001 trat in der Bundesrepublik Deutschland das Gesetz zur Verhütung und Bekämpfung
von Infektionskrankheiten beim Menschen (Infektionsschutzgesetz - IfSG) in Kraft. Ziel des
Gesetzes ist es, „übertragbaren Krankheiten beim Menschen vorzubeugen, Infektionen frühzeitig zu
erkennen und ihre Weiterverbreitung zu verhindern“ (§1 IfSG).1
Aus diesem Grund wird eine Meldepflicht über den Krankheitsverdacht, die eigentliche Erkrankung
oder den durch die Krankheit bedingten Tod für bestimmte Infektionskrankheiten durch §7 des
Gesetzes vorgeschrieben. Neben Infektionskrankheiten wie Tuberkulose, Masern oder Hepatitis zählt
auch die Legionellose zu diesen meldepflichtigen Erkrankungen.
Erstmals trat die Legionellen-Infektion im Jahr 1976 in Philadelphia/USA auf. Bei einem
Veteranentreffen der American Legion im Bellevue Stratford Hotel erkrankten 182 Teilnehmer an
einer schweren Lungenentzündung, wobei in 29 Fällen die Erkrankung, für die bis zu diesem
Zeitpunkt noch kein Erreger bekannt war, tödlich verlief.2 Nach kurzer Zeit, in der zahlreiche
biologische Untersuchungen durchgeführt wurden, gelang es den Mikrobiologen des Center of
Disease Control Joseph McDade und Charles Shepard im Januar 1977 ein gramnegatives Bakterium
als ursächlichen Erreger dieses Ausbruchs zu identifizieren.3 Da alle Erkrankten Mitglieder der
American Legion waren, wurde das Bakterium Legionella pneumophila genannt und die Krankheit
als Legionärskrankheit bezeichnet. In darauf folgenden retrospektiven Studien konnte mittels
Antikörper-Titer-Bestimmung von ehemals Erkrankten gezeigt werden, dass bereits vergangene
Pneumonie-Epidemien wie ein Ausbruch 1957 in Austin4, 1965 im St Elizabeth´s Hospital in
Washington5 und auch 1968 in Pontiac
6 ebenfalls auf das Bakterium L. pneumophila zurückgeführt
werden können. Damals konnte auf Grund der eingeschränkten diagnostischen Möglichkeiten kein
ursächlicher Erreger identifiziert werden.
1.1.2 Erreger – Legionella pneumophila
Da L. pneumophila zunächst keiner Bakteriengattung zugeordnet werden konnte, wurde ein Jahr nach
der Entdeckung die Gattung der Legionellen (Legionella) erschaffen. Neben L. pneumophila wurden
mit Hilfe neuer Methoden im Bereich der Bakterienisolation und Kultivierung nun auch Bakterien,
die in den 40er Jahren irrtümlicher Weise zu der Gattung der Rickettsien gezählt wurden, als

2 EINLEITUNG
Legionella ssp. identifiziert. Bis heute sind mehr als fünfzig gramnegative, stäbchenförmige Vertreter
innerhalb der Familie der Legionellaceae bekannt. Nahezu alle diese Arten kommen ubiquitär in
natürlichem Süßwasser vor. Als einzige Ausnahme ist L. longbeachae zu nennen, welches im
Erdboden seinen natürlichen Lebensraum findet. Fast die Hälfte aller Legionellen-Arten ist potenziell
humanpathogen und kann nach der Aufnahme in den menschlichen Körper schwere
Lungenentzündungen auslösen. In 90 % all dieser Pneumonie-Fälle heißt der Erreger L.
pneumophila. Von dieser Art sind bis heute 15 verschiedene Serotypen bekannt.7
1.1.2.1 Vorkommen und parasitäre Lebensform
L. pneumophila tritt ubiquitär in natürlichen Süßwassersystemen wie Seen und Flüssen und auch in
künstlich geschaffenen, wie z.B. Kühltürmen, Schwimmbädern oder Klimaanlagen auf. Für
optimales Wachstum und Vermehrung benötigen die Bakterien ein zusätzliches Wirtssystem. In der
natürlichen Umgebung parasitiert L. pneumophila hauptsächlich in Protozoen wie Acanthamoeba,
Hartmanella und Naegleria. Während die Legionellen-Konzentration in diesem natürlichen Habitat
jedoch eher gering ist, kann sie in den künstlich geschaffenen Süßwassersystemen relativ hohe Werte
erreichen. Grund dafür ist, dass der Erreger dort meist in Form von komplexen Biofilmen gefunden
wird. Solche Biofilme stellen eine Ansammlung der Mikroorganismen und ihrer Ausscheidungen dar
und entstehen hauptsächlich an Grenzflächen. Eine Vielzahl von äußeren Einflüssen kann die
Bakterienzahl in einem solchen Komplex zusätzlich steigern. Zu den optimalen Wachstums-
bedingungen zählen u.a. Temperaturen zwischen 30 und 40 °C, eine regelmäßige Zufuhr von
Frischwasser und eine lange Verweildauer der Bakterien. Auch in einem solchen Verband sind das
Wachstum und die Vermehrung von L. pneumophila von der Anwesenheit eines Wirtssystems
abhängig.8
Während eines parasitären Lebenszyklus durchläuft das Bakterium zwei verschiedene Phasen: Die
nicht-replikative bzw. stationäre und die replikative Phase. Der erste Kontakt zwischen Bakterium
und Wirtszelle findet in der nicht-replikativen Phase statt, in der der Mikroorganismus in seiner
infektiösen, begeißelten Form vorliegt. In dieser Zeit zeichnet sich das Bakterium vor allem durch
seine Natriumsensitivität und seine Stressresistenz aus. Durch eine Anlagerung an die Wirtszelle
gelangt L. pneumophila zuerst durch unterschiedliche Transportmechanismen in die Zelle. Dort wird
das Bakterium sofort in einem Phagosom eingeschlossen. Die Phagosomenmembran wird zunächst
von Mitochondrien und Wirtszellvesikeln, später von Teilen des rauen Endoplasmatischen
Retikulums umgeben.9 Auf Grund dieser besonderen Ummantelung kann das bakterielle Phagosom
nicht mehr mit Lysosomen fusionieren, weshalb die Legionellen vor schädlichen äußeren Einflüssen
und intrazellulärem Abbau geschützt sind.10
Auf die Bildung des Phagosoms folgt die replikative

EINLEITUNG 3
Phase, in der sich das Bakterium vermehrt. Dabei befindet es sich in einem nicht begeißelten, nicht
infektiösen und weniger Stress resistenten Zustand. Nach vollständiger Replikation wird zuerst die
Phagosomenmembran aufgelöst und anschließend die bakteriellen Nachkommen durch speziell
gebildete Poren oder in Vesikeln eingelagert an die wässrige Umgebung abgegeben. Von dort werden
die Bakterien entweder erneut von Protozoen phagozytiert oder bilden komplexe Biofilme auf
natürlichem Untergrund, um dort zu persistieren.11
In einem solchen Verbund sind die Legionellen
zwar überlebensfähig, jedoch nicht kultivierbar. In eben diesem Zustand gelangen sie zuerst in unser
Trinkwassersystem und von dort über technische Vektoren wie Klimaanlagen, Schwimmbäder oder
Duschköpfe in den menschlichen Organismus. Durch die Inhalation kontaminierter Aerosole sind die
Bakterien in der Lage die menschliche Lunge zu befallen und eine pulmonale Infektion auszulösen.12
Die Fähigkeit, die menschliche Lunge zu besiedeln und sich gleichzeitig dort zu vermehren, lässt auf
einen humanen Lebenszyklus von L. pneumophila schließen, der dem natürlichen Parasitismus
ähnlich ist. Anstelle von Protozoen werden im menschlichen Körper jedoch hauptsächlich alveoläre
Makrophagen als Wirtssystem herangezogen.
Legionellen Legionellen-tragende Vakuole
ER Mitochondrien
Abb. 1: Parasitärer Lebenszyklus von L. pneumophila in einer Makrophagenzelle: (1) Phagozytose; (2)
Speziell ummanteltes Phagosom; (3) Replikation; (4) Ausschleusen der bakteriellen Nachkommen (modifiziert
nach 13
)
Der Phagozytosemechanismus (siehe (1) in Abb. 1) in der menschlichen Lunge ist bis heute noch
nicht vollkommen aufgeklärt. In der Literatur werden mehrere sehr unterschiedliche
Transportmechanismen beschrieben, weshalb in diesem Kapitel nicht näher darauf eingegangen
werden soll.7 Nach der Aufnahme in die Makrophagenzelle befinden sich die Legionellen zunächst
eingeschlossen in einer Vakuole (siehe (2) in Abb. 1), die wie ihr natürliches Vorbild nicht mit
Makrophagenzelle
Nukleus
1
1
1
2
3
4

4 EINLEITUNG
Lysosomen fusionieren kann. Somit werden die Bakterien auch im Inneren einer humanen
Makrophagenzelle nicht abgebaut. Nach dieser stationären Phase folgt, wie oben beschrieben, die
replikative, in der sich die Bakterien zuerst vermehren (siehe (3) in Abb. 1) und anschließend die
Vakuole auflösen. Um die Makrophagenzelle danach wieder verlassen zu können, bilden die
Legionellen Poren in der Wirtszellwand (siehe (4) in Abb. 1) und verursachen damit den Untergang
der Zelle.7 Wie erwähnt, persistieren die Legionellen nun solange in der menschlichen Lunge bis der
parasitäre Lebenszyklus von Neuem beginnt. In dieser Zustandsform sind die Bakterien in der Lage
auch auf andere Organe wie die Milz überzugehen.
Der Übergang aus einem nicht kultivierbaren in einen kultivierbaren, infektiösen Zustand und die
parasitäre Lebensform im Allgemeinen bestätigen auch die Tatsache, dass eine Übertragung dieser
Bakterien von Mensch zu Mensch bisher nie stattgefunden hat.
1.1.2.2 Pathogenese und Epidemiologie
Nach der Inhalation kontaminierter Aerosole und der damit verbundenen Besiedelung der
menschlichen Lunge ist L. pneumophila in der Lage eine pulmonale Infektion hervorzurufen. Hierbei
muss zwischen zwei verschiedenen Krankheitsverlaufsformen differenziert werden: Dem Pontiac-
Fieber und der Legionärskrankheit. Die Verwendung des Begriffs „Legionellose“ steht in der
nachfolgenden Arbeit für die Zusammenfassung der beiden Verlaufsformen.
Das Pontiac-Fieber, benannt nach dem Ort des ersten Ausbruchs,6 ist eine milde Atemwegs-
erkrankung, die in ihren Symptomen einer gewöhnlichen Influenza ähnelt. Die Krankheit zeichnet
sich durch leichtes Fieber, Kopfschmerzen, Myalgien und allgemeines Unwohlsein aus. Obwohl es
sich um eine Atemwegserkrankung handelt, können weder eine Entzündung noch Infiltrationen der
Lunge beobachtet werden. Nach einer Inkubationszeit von ca. 36 h limitiert sich die Krankheit in
einem Zeitraum von zwei bis fünf Tagen selbst.6 Die Inzidenzrate bei dieser Form der Legionellose
liegt bei ca. 95 %.14
Die Legionärskrankheit ist eine starke, zum Teil tödlich verlaufende Lungenentzündung. Nach einer
relativ langen Inkubationszeit, die bis zu zehn Tage andauern kann, klagen die Erkrankten über
Symptome wie Kopfschmerzen, hohes Fieber, Myalgien, Husten, Dyspnoen, Schmerzen beim Atmen
und Diarrhoen. Nicht selten wird auch ein Deliriums-artiger Zustand beschrieben. Im Gegensatz zum
Pontiac-Fieber manifestiert sich in fast allen Fällen eine schwere Lungenentzündung mit
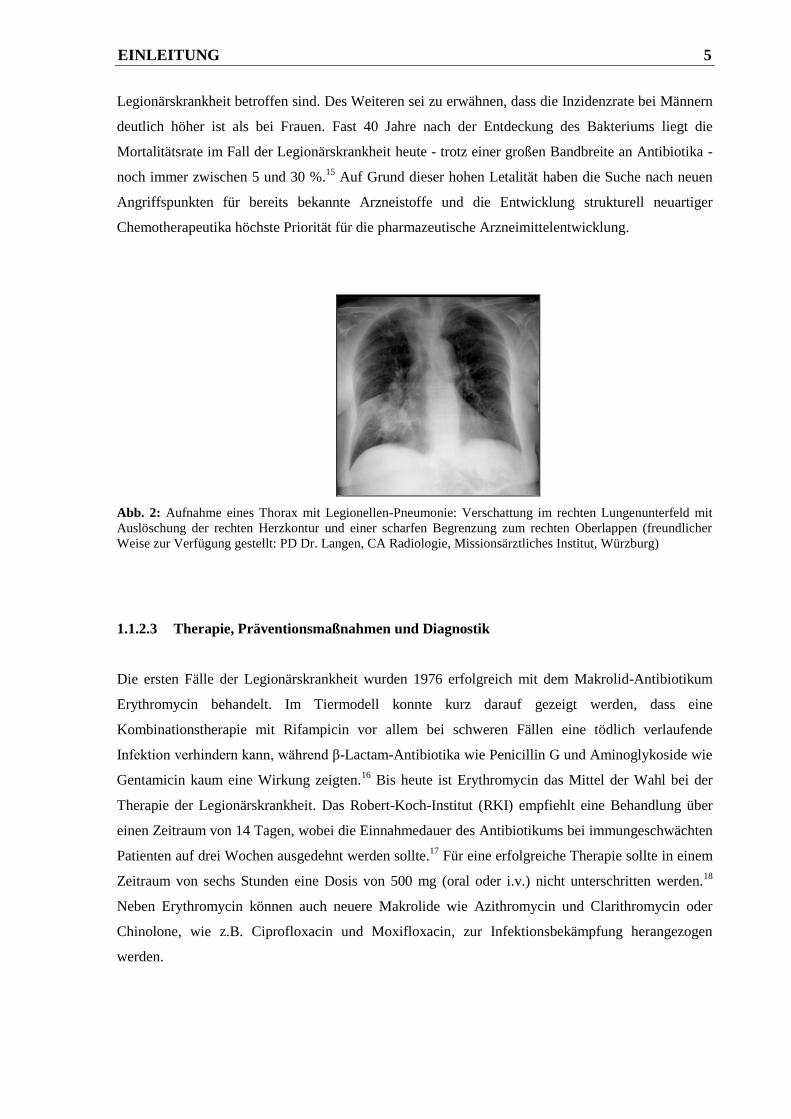
Infiltrationen von Fibrin, Makrophagen und Neutrophilen (siehe Abb. 2), die unbehandelt zum Tod
führen kann. Im Fall der schweren Verlaufsform überschreitet die Zahl der Neuerkrankungen die 5 %
nicht.14
Dieser eher niedrige Wert bestätigt die Hypothese, dass hauptsächlich ältere,
immunsupprimierte Personen, Raucher und Patienten mit Herz-Kreislaufproblemen von der
EINLEITUNG 5
Legionärskrankheit betroffen sind. Des Weiteren sei zu erwähnen, dass die Inzidenzrate bei Männern
deutlich höher ist als bei Frauen. Fast 40 Jahre nach der Entdeckung des Bakteriums liegt die
Mortalitätsrate im Fall der Legionärskrankheit heute - trotz einer großen Bandbreite an Antibiotika -
noch immer zwischen 5 und 30 %.15
Auf Grund dieser hohen Letalität haben die Suche nach neuen
Angriffspunkten für bereits bekannte Arzneistoffe und die Entwicklung strukturell neuartiger
Chemotherapeutika höchste Priorität für die pharmazeutische Arzneimittelentwicklung.
Abb. 2: Aufnahme eines Thorax mit Legionellen-Pneumonie: Verschattung im rechten Lungenunterfeld mit
Auslöschung der rechten Herzkontur und einer scharfen Begrenzung zum rechten Oberlappen (freundlicher
Weise zur Verfügung gestellt: PD Dr. Langen, CA Radiologie, Missionsärztliches Institut, Würzburg)
1.1.2.3 Therapie, Präventionsmaßnahmen und Diagnostik
Die ersten Fälle der Legionärskrankheit wurden 1976 erfolgreich mit dem Makrolid-Antibiotikum
Erythromycin behandelt. Im Tiermodell konnte kurz darauf gezeigt werden, dass eine
Kombinationstherapie mit Rifampicin vor allem bei schweren Fällen eine tödlich verlaufende
Infektion verhindern kann, während β-Lactam-Antibiotika wie Penicillin G und Aminoglykoside wie
Gentamicin kaum eine Wirkung zeigten.16
Bis heute ist Erythromycin das Mittel der Wahl bei der
Therapie der Legionärskrankheit. Das Robert-Koch-Institut (RKI) empfiehlt eine Behandlung über
einen Zeitraum von 14 Tagen, wobei die Einnahmedauer des Antibiotikums bei immungeschwächten
Patienten auf drei Wochen ausgedehnt werden sollte.17
Für eine erfolgreiche Therapie sollte in einem
Zeitraum von sechs Stunden eine Dosis von 500 mg (oral oder i.v.) nicht unterschritten werden.18
Neben Erythromycin können auch neuere Makrolide wie Azithromycin und Clarithromycin oder
Chinolone, wie z.B. Ciprofloxacin und Moxifloxacin, zur Infektionsbekämpfung herangezogen
werden.

6 EINLEITUNG
Im Gegensatz zur Therapie der Legionärskrankheit, ist der Einsatz von Antiinfektiva im Fall des
Pontiac-Fiebers nicht erforderlich. In den Therapieleitlinien des RKI für Ärzte wird lediglich eine
symptomatische Behandlung empfohlen.17
Sobald dem RKI ein Legionellosefall vorliegt, versucht man mittels Standardmethoden wie der
Bakterienkultivierung oder serologischen Methoden, wie z.B. der Pulsed-Field-Elektrophorese oder
der Polymerase-Ketten-Reaktion (PCR), den bakteriellen Vektor zu ermitteln. Ist der Erreger sowie
sein Aufenthaltsort eindeutig identifiziert, wird sofort die dort auftretende bakterielle Konzentration
bestimmt. Als quantitative Messgröße dient hierbei die Angabe von Koloniebildenden Einheiten
(KbE). Steigt dieser Parameter über einen Wert von 1 KbE/ml, sollten desinfizierende Maßnahmen
eingeleitet werden.12
In der Vergangenheit zeigten der Einsatz von heißem Wasser, ultraviolettem
Licht, Silberionen oder Chlor den größten Erfolg.19
Um das Risiko einer Legionellen-Epidemie jedoch schon vorher weitgehend zu minimieren,
verpflichtet die am 01.11.2011 in Kraft getretene Änderung der Trinkwasserverordnung (TrinkwV)20
Eigentümer von Mehrfamilienhäusern mit Großanlagen zur Trinkwassererwärmung (Inhalt ≥ 40 L)
einerseits zur Anzeige der Anlage, andererseits zu einer jährlichen Untersuchung auf Legionellen.
Generell sollte bei der Trinkwassererwärmung beachtet werden, dass die Zirkulationstemperatur des
Wassers 55 °C nicht unterschreitet und dass das in Boilern stehende Wasser mindestens einmal am
Tag auf 60 °C erhitzt wird. Desweitern sind lange und stagnierende Leitungsabschnitte sowie
Toträume generell zu vermeiden.
Vor der Einführung dieser Richtlinien kam es auch in der heutigen Zeit immer wieder zu vor allem
Reise-assoziierten oder nosokomialen Epidemien. Der letzte große Ausbruch in Deutschland
ereignete sich im Januar 2010 in einer Firma in Ulm. In diesem Fall wurde wahrscheinlich eine
unzureichende Menge an Biozid zur Verhinderung der Trinkwasserverkeimung verwendet, so dass
sich der Erreger sehr leicht über das Kühlwassersystem ausbreiten konnte. Infolgedessen wurden 64
Legionellosefälle verzeichnet, von denen fünf tödlich verliefen.21,22
Im gleichen Jahr wurden dem
RKI insgesamt 690 Fälle einer klinisch manifestierten Legionellose gemeldet.23
Diese Zahl stellt
jedoch keinesfalls die tatsächliche Rate an Neuinfektionen innerhalb eines Jahres dar.
Hochrechnungen einer Pneumonie-Studie des Kompetenznetzwerks „Community acquired
Pneumonia“ (CAPNETZ) zufolge, liegt die Zahl der Neuerkrankungen in Deutschland bei 15.000-
20.000 pro Jahr.24
Der Grund der unverhältnismäßig niedrigen Anzahl an Meldungen im Vergleich
zu den tatsächlich auftretenden Krankheitsfällen ist vermutlich die Tatsache, dass eine Legionellose
nur durch spezielle mikrobiologische Untersuchungen von anderen eher harmlosen Pneumoniearten
abgegrenzt werden kann. In der bakteriellen Diagnostik finden diese teilweise aufwendigen
Methoden aber auf Grund der hohen Kosten kaum Anwendung.
Den Goldstandard der diagnostischen Verfahren stellt die Anzucht von Legionellen dar. Als
Untersuchungsquelle kann entweder Sputum, Lungengewebe oder Blut herangezogen werden, wobei
die Anzucht aus Lungengewebe am besten gelingt. Nach erfolgreicher Kultivierung folgt bei dieser

EINLEITUNG 7
diagnostischen Methode eine serologische Typisierung der Bakterien, meist mit Hilfe monoklonaler
Antikörper. Diese Typisierung ist von großer Bedeutung, da im Fall von L. pneumophila 15
verschiedene Serogruppen bekannt sind. Nicht alle dieser Unterarten sind jedoch in der Lage eine
pulmonale Erkrankung hervorzurufen. Da Serogruppe 1 in 84 % aller Infektionen die Hauptursache
darstellt, kann bei einem solchen positiven Nachweis eine gezielte Therapie erfolgen. Da die
Kultivierung von Bakterien jedoch oft mit hohem Aufwand und nicht selten auch mit schlechtem
Erfolg verbunden ist, soll der Nachweis von Nukleinsäuren aus respiratorischem Material mittels
PCR in Zukunft die Diagnostik der Legionellose verbessern.
Als weitere diagnostische Methode sei der Antigennachweis aus diversen Untersuchungsmaterialien
zu nennen. Am häufigsten wird die Bestimmung aus Urin durchgeführt. Im Urin können bereits ein
bis drei Tage nach Ausbruch der Infektion spezifische Lipopolysaccharide von L. pneumophila
identifiziert und mit Hilfe eines speziellen Enzymimmunoassays (ELISA) detektiert werden. Bei
diesem Nachweisverfahren wird jedoch nur eine Serogruppe erfasst, während beim Antigennachweis
aus respiratorischem Material alle Serogruppen des Bakteriums nachgewiesen werden können. Als
Antigen fungiert dabei ein äußeres Membranprotein von L. pneumophila. Dieses Protein bindet
während eines direkten Immunofluoreszenzassays (DFA) an einen fluoreszenzmarkierten, Spezies-
spezifischen monoklonalen Antikörper und kann somit detektiert werden.
Seit Entdeckung des Erregers bis heute wird als Standardmethode jedoch der Antikörpernachweis in
Form eines indirekten Immunofluoreszenztests (IFA) durchgeführt. Als Testantigen sollten mehrere
Serogruppen von L. pneumophila, nicht nur die infektiöse Serogruppe 1, herangezogen werden. Da
es bei einigen Patienten jedoch zu einer zeitlich verzögerten Antikörperbildung kommen oder diese
in 30 % aller Infektionen sogar ganz ausbleiben kann, sollte der Enzymimmunoassay aus Urin dem
Antikörpernachweis vorgezogen werden.25
1.1.2.4 Aufbau, Eigenschaften und Bestandteile der Zellwand
Um für die Legionellosetherapie neue Antiinfektiva zu entwickeln, müssen zum einen die
Mechanismen der bakteriellen Infektion, zum anderen der genaue Aufbau der Legionellen bekannt
sein. Eine wichtige Rolle spielt hier die Beschaffenheit und die Zusammensetzung der bakteriellen
Zellwand. Wie bei anderen gramnegativen Bakterien setzt sich diese aus drei verschiedenen
Schichten zusammen (siehe Abb. 3).

8 EINLEITUNG
Abb. 3: Schematischer Aufbau der Zellwand von L. pneumophila (modifiziert nach Shevchuk et al.26
)
Das Zellinnere, auch Zytoplasma genannt, wird von einer inneren Membran (IM) umgeben, die auch
als zytoplasmatische oder Plasmamembran bezeichnet werden kann. Sie besteht aus einer
Phospholipiddoppelschicht, in die viele verschiedene Proteine eingelagert sind, die eine wichtige
Rolle im bakteriellen Metabolismus spielen. Zu ihnen zählen Eisen- bzw. Peptidtransporter,
Adenylatzyklasen und Methyltransferasen. Direkt an die IM schließt sich das Periplasma (PP) an,
welches sich aus Peptidoglykanen und größtenteils detoxifizierenden Proteinen zusammensetzt. Ihre
Hauptaufgabe besteht darin, radikalische Sauerstoffspezies abzufangen und abzubauen. An das PP
grenzt die Außenmembran (AM), die das Bakterium von seiner Umwelt abgrenzt und somit eine
erste Kontaktstelle für andere Zellen, Substanzen und auch für Arzneistoffe darstellt. Wie die
Innenmembran besteht die äußere Membran aus zwei Schichten, die sich jedoch in ihrer
Zusammensetzung unterscheiden: Während die nach innen gewandte Seite zum Großteil aus
Phospholipiden besteht, sind Lipopolysaccharide (LPS) die Hauptbestandteile der äußeren Schicht.
Diese LPS fungieren hauptsächlich als immundominante Antigene der Legionellen und können somit
während eines diagnostischen Verfahrens mit Hilfe von monoklonalen Antikörpern detektiert werden
(siehe Kapitel 1.1.2.3). Neben den LPS beinhaltet die äußere Membran noch ungefähr 250 weitere
Proteine, deren gesamte Funktionen noch nicht vollständig aufgeklärt sind. In Tabelle 1 sind einige
wichtige Vertreter dieser AM-Proteine und ihre Aufgaben aufgelistet.
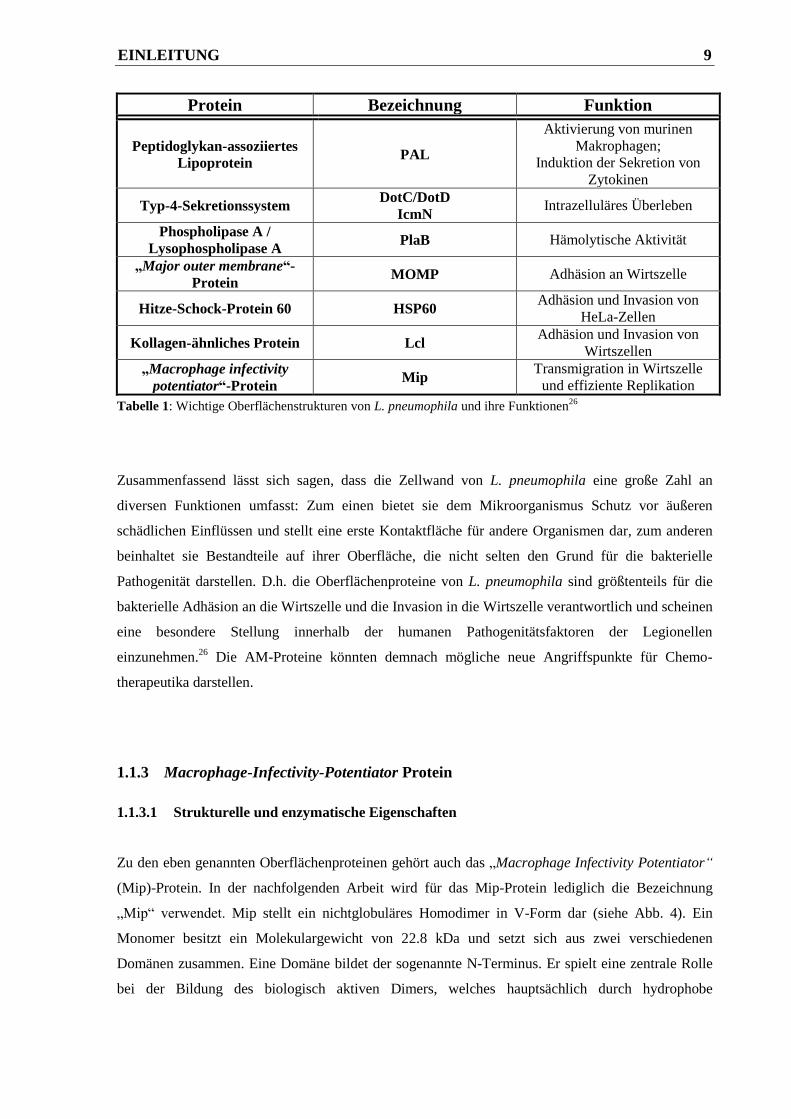
EINLEITUNG 9
Protein Bezeichnung Funktion
Peptidoglykan-assoziiertes
Lipoprotein PAL
Aktivierung von murinen
Makrophagen;
Induktion der Sekretion von
Zytokinen
Typ-4-Sekretionssystem DotC/DotD
IcmN Intrazelluläres Überleben
Phospholipase A /
Lysophospholipase A PlaB Hämolytische Aktivität
„Major outer membrane“-
Protein MOMP Adhäsion an Wirtszelle
Hitze-Schock-Protein 60 HSP60 Adhäsion und Invasion von
HeLa-Zellen
Kollagen-ähnliches Protein Lcl Adhäsion und Invasion von
Wirtszellen
„Macrophage infectivity
potentiator“-Protein Mip
Transmigration in Wirtszelle
und effiziente Replikation
Tabelle 1: Wichtige Oberflächenstrukturen von L. pneumophila und ihre Funktionen26
Zusammenfassend lässt sich sagen, dass die Zellwand von L. pneumophila eine große Zahl an
diversen Funktionen umfasst: Zum einen bietet sie dem Mikroorganismus Schutz vor äußeren
schädlichen Einflüssen und stellt eine erste Kontaktfläche für andere Organismen dar, zum anderen
beinhaltet sie Bestandteile auf ihrer Oberfläche, die nicht selten den Grund für die bakterielle
Pathogenität darstellen. D.h. die Oberflächenproteine von L. pneumophila sind größtenteils für die
bakterielle Adhäsion an die Wirtszelle und die Invasion in die Wirtszelle verantwortlich und scheinen
eine besondere Stellung innerhalb der humanen Pathogenitätsfaktoren der Legionellen
einzunehmen.26
Die AM-Proteine könnten demnach mögliche neue Angriffspunkte für Chemo-
therapeutika darstellen.
1.1.3 Macrophage-Infectivity-Potentiator Protein
1.1.3.1 Strukturelle und enzymatische Eigenschaften
Zu den eben genannten Oberflächenproteinen gehört auch das „Macrophage Infectivity Potentiator“
(Mip)-Protein. In der nachfolgenden Arbeit wird für das Mip-Protein lediglich die Bezeichnung
„Mip“ verwendet. Mip stellt ein nichtglobuläres Homodimer in V-Form dar (siehe Abb. 4). Ein
Monomer besitzt ein Molekulargewicht von 22.8 kDa und setzt sich aus zwei verschiedenen
Domänen zusammen. Eine Domäne bildet der sogenannte N-Terminus. Er spielt eine zentrale Rolle
bei der Bildung des biologisch aktiven Dimers, welches hauptsächlich durch hydrophobe
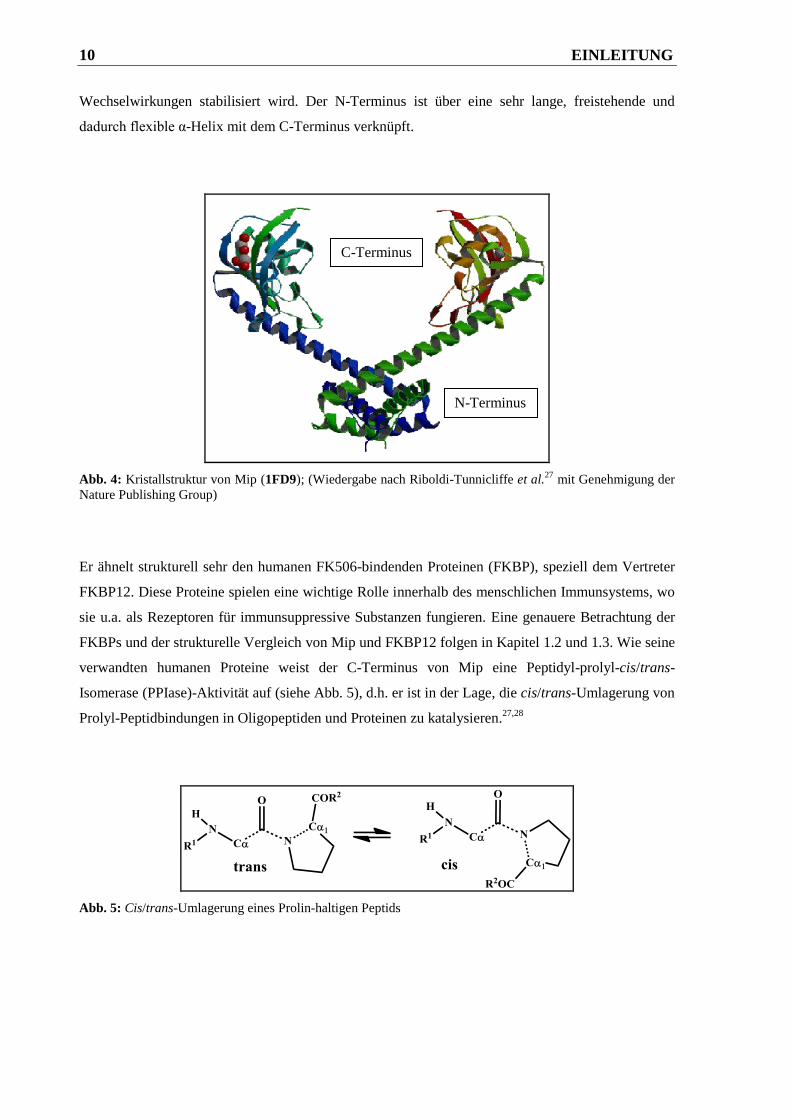
10 EINLEITUNG
Wechselwirkungen stabilisiert wird. Der N-Terminus ist über eine sehr lange, freistehende und
dadurch flexible α-Helix mit dem C-Terminus verknüpft.
Abb. 4: Kristallstruktur von Mip (1FD9); (Wiedergabe nach Riboldi-Tunnicliffe et al.27
mit Genehmigung der
Nature Publishing Group)
Er ähnelt strukturell sehr den humanen FK506-bindenden Proteinen (FKBP), speziell dem Vertreter
FKBP12. Diese Proteine spielen eine wichtige Rolle innerhalb des menschlichen Immunsystems, wo
sie u.a. als Rezeptoren für immunsuppressive Substanzen fungieren. Eine genauere Betrachtung der
FKBPs und der strukturelle Vergleich von Mip und FKBP12 folgen in Kapitel 1.2 und 1.3. Wie seine
verwandten humanen Proteine weist der C-Terminus von Mip eine Peptidyl-prolyl-cis/trans-
Isomerase (PPIase)-Aktivität auf (siehe Abb. 5), d.h. er ist in der Lage, die cis/trans-Umlagerung von
Prolyl-Peptidbindungen in Oligopeptiden und Proteinen zu katalysieren.27,28
Abb. 5: Cis/trans-Umlagerung eines Prolin-haltigen Peptids
N-Terminus
C-Terminus

EINLEITUNG 11
Während gewöhnlich die Rotationen um die Winkel Cα-(C=O) und N-Cα1 von Peptidbindungen im
gesamten Bereich von 0 – 360 ° sehr schnell verlaufen, ist die freie Drehbarkeit des (C=O)-N-
Winkels durch den partiellen Doppelbindungscharakter stark eingeschränkt - d.h. sie bewegt sich nur
im Bereich von 0 – 180 °. Von einem Peptid können daher zwei Konformationsisomere (0 ° = cis;
180 ° = trans) existieren, wobei der natürliche Anteil des cis-Isomers im Fall von normalen
sekundären Peptidbindungen ((C=O)-NH) gewöhnlich unter einem Prozent liegt. Eine Ausnahme
hiervon stellen jedoch die tertiären Peptidbindungen zwischen einem N-terminalen Prolin und einer
weiteren Aminosäure dar (Xaa-Pro) (siehe Abb. 5). In diesem speziellen Fall kann das cis-Konformer
einen viel höheren Wert (10 - 40 %) im konformeren Gleichgewicht einnehmen. Die cis/trans-
Isomerisierung von solchen Xaa-Pro-Peptiden findet auf Grund der recht hohen Aktivierungsenergie
(ca. 80 kJ/mol) nur langsam statt.29
Dies hat nachfolgend einen erheblichen Einfluss auf die
Proteinfaltung, da die Ausbildung der Tertiärstruktur solcher Peptide teilweise gehemmt ist und nur
in Anwesenheit von PPIasen stattfinden kann. Die Isomerasen agieren in diesem Fall als
„Faltungshelfer“, sogenannte Chaperone, und wandeln die Konformere in die Form um, die für eine
korrekte Faltung benötigt wird. Bei dieser Reaktion handelt es sich oftmals um den
geschwindigkeitsbestimmenden Schritt der Proteinfaltung.
1.1.3.2 Biochemische Analyse: Mip als Virulenzfaktor?
Im ersten Jahrzehnt nach der Entdeckung von L. pneumophila wurden viele biochemische
Untersuchungen durchgeführt, um die Gene, die Informationen über die intrazelluläre Infektiösität
von Makrophagen tragen, zu identifizieren. Cianciotto et al. entwickelten 1989 eine Mutante von L.
pneumophila, deren Gen, das für die Expression eines 24 kDa Oberflächenproteins codiert,
ausgeschaltet wurde.30
Nach der Behandlung einer humanen, Makrophagen-ähnlichen Zelllinie
(U937-Zellen) und humanen alveolären Makrophagen mit dieser Mutante konnten im Zellinneren
verglichen mit dem Inneren der Zellen, die zuvor mit L. pneumophila-Wildtyp inkubiert wurden,
kaum Bakterien gefunden werden. Eine nachträgliche Aktivierung des ausgeschalteten Gens
resultierte im Fall der Mutante in einer spontanen Infektion der Zellen. Diese Beobachtung bestätigte
die Notwendigkeit der Anwesenheit dieses Proteins für eine vollständige Infektion. Um ferner
aufzuklären, welcher Schritt im Infektionsprozess durch das Oberflächenprotein beeinflusst wird,
wurde die Dauer dieser Experimente auf einen Zeitraum von drei Tagen ausgedehnt. In dieser Zeit
wurde die Bakterienkonzentration im Inneren der Zellen mehrfach bestimmt. Dadurch konnte des
Weiteren gezeigt werden, dass das Oberflächenprotein keinen direkten Einfluss auf die anfängliche
bakterielle Replikation hat, sondern erst 40 h nach der Inkubation mit einem späten Schritt der

12 EINLEITUNG
Aktivierung der zellulären Infektion interagiert. Auf Grund dieser intrazellulären Eigenschaften
wurde das Protein Macrophage-Infectivity-Potentiator-Protein genannt.
Zur weiteren strukturellen Aufklärung des Virulenzmechanismus von Mip untersuchten Wintermeyer
et al. mittels ähnlicher Invasionsstudien eine Mip-Mutante mit stark reduzierter PPIase-Aktivität und
fanden keinen Einfluss dieser Enzymaktivität auf die intrazelluläre Wachstumsrate von L.
pneumophila in einzelligen Wirtssystemen wie Acanthamoeba oder U937-Zellen.31
Der genaue
Virulenzmechanismus von L. pneumophila blieb deshalb zunächst bis auf Weiteres ungeklärt,
weshalb Köhler et al. mit Hilfe verschiedener Genmutationen die genaue Funktion der beiden vorher
beschriebenen Domänen bezüglich der bakteriellen Infektiösität zu klären versuchten.32
Mit Hilfe
einer monomeren Mip-Variante sowie einer Mutante mit abgeschwächter PPIase-Aktivität konnte im
Rahmen von Infektionsstudien gezeigt werden, dass der N-Terminus, jedoch nicht die C-terminale
PPIase für das intrazelluläre Wachstum in einzelligen Wirtssystemen verantwortlich ist - d.h.
während bei der Behandlung der Wirtszellen mit der monomeren Mip-Variante die
Legionellenanzahl im Inneren abnahm, war nach der Inkubation mit der PPIase-reduzierten Mutante
keine verminderte Bakterienanzahl im Zellinneren zu verzeichnen. Dieses Resultat bestätigt auch die
früheren Beobachtungen von Wintermeyer et al.31
Im Rahmen dieser Studien wurden In-vivo-
Testungen mit denselben Mutanten durchgeführt. Da Meerschweinchen dem Menschen sehr ähnliche
Symptome einer Legionellen-Infektion zeigen, wurden sie als Tiermodell für alle weiteren In-vivo-
Untersuchungen herangezogen. Im Gegensatz zu den eben beschriebenen In-vitro-Ergebnissen
konnte in vivo eine abgeschwächte Infektion bei denjenigen Meerschweinchen beobachtet werden,
die mit der PPIase-Knock-down-Mutante infiziert worden waren. Dies lässt einerseits auf
unterschiedliche Funktionen der beiden Domänen im einzelligen Wirtssystem sowie im Tiermodell
schließen. Andererseits scheint Mip eine extrazelluläre Rolle in der Pathogenese von L. pneumophila
zu spielen.33
Die menschliche Lunge ist von einer Epithelzellschicht sowie einer extrazellulären Matrix (ECM)
umgeben. Dahinter befinden sich die alveolären Makrophagenzellen (siehe (A) in Abb. 6). Um eine
Makrophagenzelle zu infizieren, muss der Mikroorganismus zuerst diese alveoläre Barriere
überwinden. Dieser Vorgang wird als Transmigration bezeichnet. Zur Bestätigung der Hypothese
einer extrazellulären Funktion von Mip wurde von Wagner et al. ein sogenannter Transwell-Assay
entwickelt (siehe (B) in Abb. 6).33
Für diesen Test wurde in eine Zellkulturkammer eine zweite
Kammer mit einer 3 µm porösen Membran eingesetzt. Auf diese Membran wurden Lungen-
epithelzellen (NCI-H292) gesetzt, die in der Lage sind, ECM zu sekretieren, und damit die natürliche
Barriere der Lunge nachahmen. Mip-positive und Mip-negative Mutanten sowie Wildtyp-
Legionellen wurden auf diese Zellschicht gegeben und nach einer Inkubationszeit von fünf Stunden
die Bakterienkonzentration in der unteren Kammer bestimmt. Als Ergebnis dieses Transwell-Assays
konnte eine Mip-abhängige Transmigration beobachtet werden. Bei weiteren Untersuchungen stellte
sich jedoch heraus, dass Mip nicht - wie angenommen - direkt an die alveolären Epithelzellen,

EINLEITUNG 13
sondern wohl eher an einen Bestandteil der ECM bindet. Mit Hilfe von monomeren Mip-Mutanten
wurde eine Bindung zwischen dem Kollagen der ECM und der C-Domäne von Mip identifiziert.
Diese Beobachtung konnte durch eine von PPIase-Inhibitoren vermittelte Transmigrationshemmung
weiter bestätigt werden. Unter allen Kollagenbindungen erwies sich diejenige zu Kollagen IV,
welches den Hauptvertreter der Kollagene in der menschlichen Lunge darstellt, als die Stabilste.
In Anbetracht dieser Ergebnisse, scheint eine Inhibition von Mip - genauer gesagt die Hemmung
dessen PPIase-Funktion - die Transmigration der Legionellen und damit einen Befall der
menschlichen Lunge zu verhindern. Ein Ziel dieser Arbeit war, diese Hypothese zu überprüfen.
A) B)
NCI-H292 Zellen
Legionellen
Abb. 6: Transmigrationsschema: A) Ausschnitt aus der menschlichen Lunge; B) Transwell-System
Mip
Makrophagenzelle
L. pneumophila

14 EINLEITUNG
1.2 Immunsystem und immunsupprimierende Substanzen
Eine strukturelle Ähnlichkeit zwischen Mip und den humanen FKBPs wurde bereits in Kapitel
1.1.3.1 angedeutet. Aus diesem Grund darf eine Betrachtung des menschlichen Immunsystems in
dieser Arbeit nicht fehlen.
Im Jahr 1796 entdeckte der englische Arzt Edward Jenner, dass diejenigen Patienten, die zuvor mit
für den Menschen harmlosen Kuhpocken in Berührung kamen, von einer humanen Pockeninfektion
weitgehend verschont blieben. Mit der erfolgreichen Durchführung eben solcher Impfungen legte er
kurze Zeit später (1798) den Grundstein für unser heutiges immunologisches Verständnis und gilt
damit als Begründer der Immunologie.34
Die Hauptaufgabe unseres Immunsystems ist die Abwehr von für den menschlichen Organismus
potenziell schädlichen Stoffen oder Mikroorganismen. Dafür stehen ihm mehrere spezifische und
unspezifische Mechanismen zur Verfügung, durch die eine Erkrankung häufig auch ohne zusätzliche
Arzneimitteltherapie geheilt werden kann. In bestimmten Fällen, z.B. nach einer
Organtransplantation, kann eine solche körpereigene Immunreaktion jedoch zu erheblichen
Gesundheitsproblemen führen. In diesen Fällen werden in der Therapie immunsupprimierende
Substanzen eingesetzt, die eine vom menschlichen Organismus eingeleitete Immunantwort
unterdrücken sollten. Heute existieren vier verschiedene Arzneimittelgruppen von solchen
immunsupprimierenden Substanzen: Glucocorticoide, Zytostatika, Antikörper und makrozyklische
Naturstoffe. Als Hauptvertreter der Naturstoffe seien Ciclosporin, Tacrolimus und Sirolimus genannt,
welche in Kapitel 1.2.2 noch genauer betrachtet werden sollen (siehe Abb. 7).
Abb. 7: Immunsuppressiva aus der Gruppe der makrozyklischen Naturstoffe

EINLEITUNG 15
1.2.1 Immunophiline
Immunophiline stellen einen wichtigen Bestandteil des menschlichen Immunsystems dar (siehe
Kapitel 1.2.2.1). Sie zählen zu den intrazellulären Proteinen und wurden bereits in einer Vielzahl von
prokaryotischen und eukaryotischen Zellen identifiziert. Im menschlichen Körper sind sie in vielen
Organen lokalisiert, besonders hohe Konzentrationen findet man im Gehirn.35
Bis heute sind drei verschiedene Immunophilin-Familien bekannt: Cyclophiline, Parvuline und
FK506-bindende Proteine (FKBPs).
1.2.1.1 FKBP12 – Strukturelle und zelluläre Eigenschaften
In dieser Arbeit soll der Fokus auf die Klasse der FKBPs gerichtet werden. Bis heute wurden bereits
15 verschiedene Vertreter der FKBPs in unterschiedlichen Organismen beschrieben. Unter all diesen
weist das humane FKBP12 die größten strukturellen Gemeinsamkeiten mit dem bakteriellen Mip auf.
FKBP12 ist der kleinste Vertreter seiner Proteinfamilie und wurde bis heute am ausführlichsten
charakterisiert - oft wird FKBP12 auch als Prototyp aller FKBPs bezeichnet. Es besitzt ein
Molekulargewicht von 12 kDa und setzt sich aus 108 Aminosäuren zusammen (siehe Abb. 8). Diese
Peptidkette formt eine einzelne Domäne, die als Rezeptor für die makrozyklischen Naturstoffe
FK506 und Rapamycin (siehe Abb. 7) fungieren kann. Diese Rezeptoreigenschaft verhalf dem
Protein auch zu seiner Bezeichnung FK506-bindendes Protein. Die Domäne weist außerdem eine
PPIase-Aktivität auf - eine genaue Beschreibung dieser Enzymaktivität befindet sich in Kapitel
1.1.3.1. Inwieweit die beiden PPIase-Funktionen von Mip und FKBP12 miteinander in Verbindung
stehen, soll zu einem späteren Zeitpunkt dieser Arbeit geklärt werden.
Abb. 8: Kristallstruktur von FKBP12 (2PPN); (Wiedergabe nach Szep et al.36
mit Genehmigung von John
Wiley and Sons)

16 EINLEITUNG
Auf zellulärer Ebene kommt es zu unterschiedlichen Wechselwirkungen zwischen FKBP12 und
seinen natürlichen Bindungspartnern. Da diese bis heute noch nicht vollständig aufgeklärt werden
konnten, sollen nur einige Wichtige davon zum besseren Verständnis im Folgenden kurz dargestellt
werden (siehe Abb. 9).
Im sakroplasmatischen Retikulum kann das Immunophilin FKBP12 an zwei verschiedene
membranständige Calcium-freisetzende Kanäle binden: Zum einen an den Ryanodin-Rezeptor
(RyR1), zum anderen an den Inositol-1,4,5-Triphosphat-Rezeptor (IP3R). Die Komplexbildung von
FKBP12 mit RyR1 im menschlichen Körper führt zu einer Stabilisierung des Ca2+
-Kanals und einer
Modulation in der Kanalsteuerung. Durch diese Veränderung wird nach der Bindung die Anzahl der
RyR1-Rezeptoren auf höchster Leitfähigkeitsstufe erhöht und gleichzeitig die mittlere
Kanalöffnungszeit verlängert. Diese Tatsache bedingt einen erhöhten, zytosolischen Ca2+
-Einstrom,
der daraufhin einen Kontraktionsreiz in der quergestreiften Muskulatur auslöst.37,38
Der IP3-Rezeptor wird durch eine Proteinkinase-A-vermittelte Phosphorylierung aktiviert und durch
die Phosphatase Calcineurin dephosphoryliert und damit inaktiviert. Die Assoziation von FKBP12
mit IP3R führt dazu, dass das Immunophilin nun in der Lage ist, mit Calcineurin einen ternären
Komplex zu bilden. Dies beeinflusst das Phosphorylierungsmuster des IP3-Rezeptors, wodurch
wiederum der Ca2+
-Einstrom des Kanals erhöht und ein Kontraktionsreiz ausgelöst wird.39,40
Eine dritte zelluläre Funktion von FKBP12 wird durch eine Wechselwirkung von FKBP12 mit dem
inaktiven Wachstumsfaktor TGF-ß-R1 (transforming growth factor-ß-Rezeptor Typ 1) beschrieben.
Dieser Rezeptor weist eine Serin/Threonin-Kinaseaktivität auf und beeinflusst verschiedene Prozesse
wie z.B. die Proliferation oder Differenzierung von Zellen. Durch Bindung von FKBP12 kann TGF-
ß-R1 nur noch begrenzt phosphoryliert werden. Diese Downregulation der Phosphorylierung bedingt
anschließend eine Stabilisierung der inaktiven Rezeptorkonformation von TGF-ß-R1 und dadurch
eine Inhibition der TGF-Signaltransduktion.41,42
Abb. 9: Zelluläre Funktionen von FKBP12 (modifiziert nach Breiman et al.39
)

EINLEITUNG 17
1.2.2 Makrozyklische Immunsuppressiva
1.2.2.1 Immunologische Wirkung
Die natürlichen Funktionen der FKBPs sind bis heute noch nicht vollständig aufgeklärt. Eine
Inhibition der FKBPs durch die makrozyklischen Naturstoffe, wie z.B. Rapamycin oder FK506, führt
im menschlichen Körper zu einer Immunsuppression. Eine Hemmung von FKBP12 darf jedoch nicht
als Antagonismus der natürlichen Wirkung angesehen werden. Bei einer solchen Inhibition scheint es
sich vielmehr um eine Modulation des Immunophilins zu handeln, bei der die makrozyklischen
Antibiotika wohl auch eher als „Prodrug“ fungieren. Erst nach der Komplexierung des Arzneistoffs
mit dem entsprechenden Immunophilin kommt es zu einer vollständigen Entfaltung der Wirkung.
In der heutigen Transplantationsmedizin werden hauptsächlich die drei obengenannten
makrozyklischen Naturstoffe verwendet. Ciclosporin A (CsA) ist ein zyklischer Peptidmetabolit aus
der Pilzfamilie Trichoderma polysporum (LINK ex PERS.) Rifai.43
Nach der Verabreichung von CsA
bindet es in den T-Zellen des humanen Immunsystems an die Immunophilingruppe der Cyclophiline
und führt zur Immunsuppression.44
FK506 (Tacrolimus) und Rapamycin (Sirolimus) (siehe Abb. 7) sind bakteriellen Ursprungs und
entstammen der Gattung Streptomyces. FK506 wird aus Streptomyces tsukubaenis, Rapamycin aus
dem Boden der Osterinsel Rapa Nui, genauer aus dem dort lebenden Mikroorganismus Streptomyces
hygroscopius isoliert. Beide Immunsuppressiva binden zwar an das humane Immunophilin FKBP12,
weisen jedoch unterschiedliche Wirkmechanismen auf (siehe Abb. 10). Die Wirkungsweise von
FK506 ist mit der von CsA vergleichbar, - wobei FK506 weitaus potenter als CsA ist.45,46
D.h., trotz
Bindung an unterschiedliche Immunophiline, besitzen CsA und FK506 einen ähnlichen
Wirkmechanismus. Daraus folgt, dass ein dritter, wahrscheinlich identischer Bindungspartner in den
Mechanismus der Immunsuppression involviert ist.
Im Fall von FK506 und CsA ist der dritte Bindungspartner die Calcium/Calmodulin-abhängige
Phosphatase Calcineurin.47
Durch diese Wechselwirkung wird die Enzymaktivität von Calcineurin
gehemmt. Unter physiologischen Bedingungen bewirkt Calcineurin - während einer körpereigenen
Immunreaktion - zunächst eine Dephosphorylierung von nuklearen Transkriptionsfaktoren der
aktivierten T-Zellen (NF-AT) und unterstützt damit die Transkription von T-Zell-Aktivierungsgenen.
Zu diesen Aktivierungsgenen zählt auch Interleukin-2 (IL-2), das wiederum für die Bildung von
zytotoxischen Zellen verantwortlich ist. Eine Inhibition von Calcineurin, die damit verbundene
Hemmung der IL-2-Produktion sowie der Bildung von zytotoxischen Zellen führen somit am Ende
einer solchen Reaktionskaskade zu der gewünschten Immunsuppression.48
Das mit FK506 strukturverwandte Rapamycin bildet im menschlichen Immunsystem nach der
Bindung an das Immunophilin FKBP12 ebenfalls einen ternären Komplex aus. Trotz der
strukturellen Ähnlichkeit zu FK506/FKBP12 interagiert die Rapamycin/FKBP12-Assoziation jedoch

18 EINLEITUNG
nicht mit Calcineurin, sondern mit einem anderen Protein, das auf Grund dieser Bindung
mammalian-target-of-Rapamycin (mTOR) genannt wird. Das Protein mTOR weist eine
Sequenzhomologie zur Familie der Phosphatidyl-Inositolkinasen auf und wird deshalb selbst zu der
Familie der Kinasen gezählt. In IL-2 stimulierten T-Zellen hemmt der Komplex
Rapamycin/FKBP12/mTOR während eines Zellzyklus den Übergang von der G1- zur S-Phase.49
Diese Inhibition führt dann in einem späteren Schritt zu einer Unterdrückung der IL-2-Signal-
Transduktion, was sich letztendlich auf das Wachstum und die Differenzierung von T-Zellen
auswirkt.50
Kommt es nach der Arzneimittelgabe nicht zu einer vollständigen Ausbildung der T-
Zellen, werden, wie oben beschrieben, auch keine zytotoxischen Zellen mehr gebildet und das Ziel
der Immunsuppression ist erreicht.
Abb. 10: Immunologische Wirkung des FKBP12/FK506 bzw. FKBP12/Rapamycin-Komplexes (modifiziert
nach Breiman et al.39
)
1.2.2.2 Struktur-Wirkungsbeziehung
Im folgenden Kapitel werden die strukturellen Besonderheiten der beiden Immunsuppressiva, die für
die Bindung an das entsprechende Immunophilin von Bedeutung sind, näher beleuchtet.
Die Fähigkeit eines chemischen Stoffs einen ternären Komplex zu bilden, setzt voraus, dass er
mindestens zwei Bindestellen aufweist. Im Fall von FK506 und Rapamycin konnte diese „Zwei-
Domänen-Natur“ durch Schreiber et al. im Jahr 1991 mit Hilfe von Domäneanalysen bewiesen
werden.48
EINLEITUNG 19
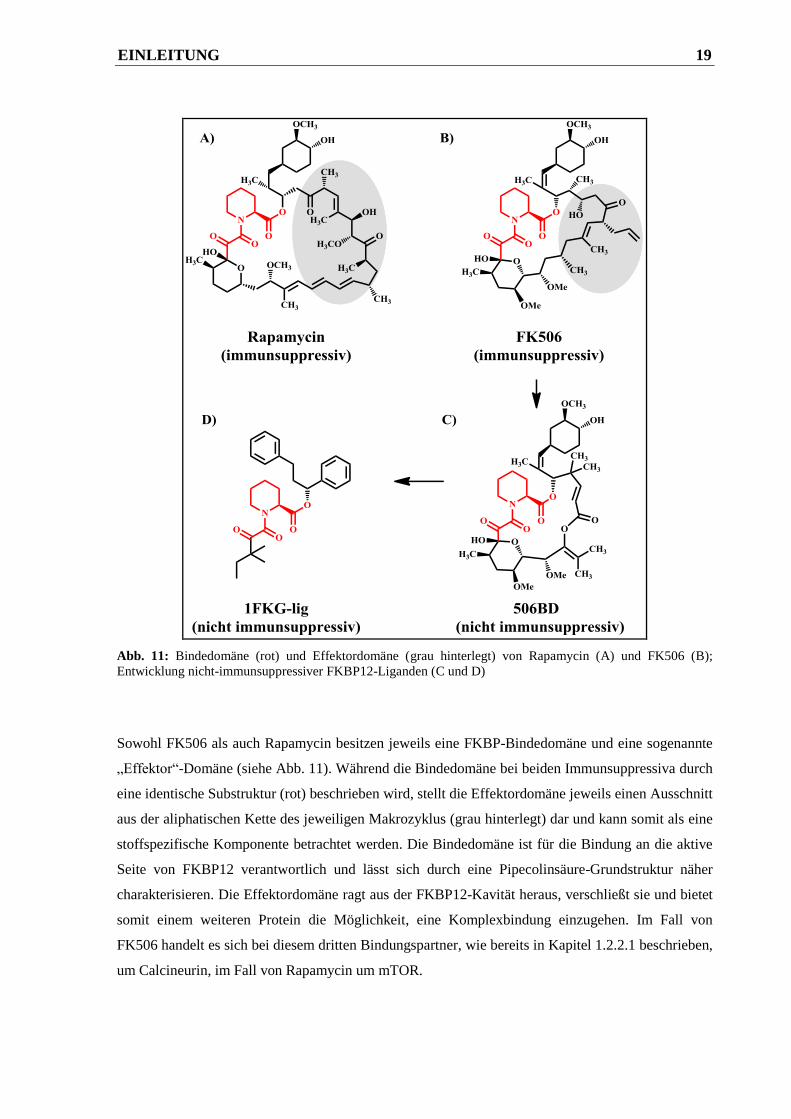
Abb. 11: Bindedomäne (rot) und Effektordomäne (grau hinterlegt) von Rapamycin (A) und FK506 (B);
Entwicklung nicht-immunsuppressiver FKBP12-Liganden (C und D)
Sowohl FK506 als auch Rapamycin besitzen jeweils eine FKBP-Bindedomäne und eine sogenannte
„Effektor“-Domäne (siehe Abb. 11). Während die Bindedomäne bei beiden Immunsuppressiva durch
eine identische Substruktur (rot) beschrieben wird, stellt die Effektordomäne jeweils einen Ausschnitt
aus der aliphatischen Kette des jeweiligen Makrozyklus (grau hinterlegt) dar und kann somit als eine
stoffspezifische Komponente betrachtet werden. Die Bindedomäne ist für die Bindung an die aktive
Seite von FKBP12 verantwortlich und lässt sich durch eine Pipecolinsäure-Grundstruktur näher
charakterisieren. Die Effektordomäne ragt aus der FKBP12-Kavität heraus, verschließt sie und bietet
somit einem weiteren Protein die Möglichkeit, eine Komplexbindung einzugehen. Im Fall von
FK506 handelt es sich bei diesem dritten Bindungspartner, wie bereits in Kapitel 1.2.2.1 beschrieben,
um Calcineurin, im Fall von Rapamycin um mTOR.

20 EINLEITUNG
Bereits im Vorfeld der Domäneanalysen wurden zahlreiche systematische Studien durchgeführt, um
die molekularen Wechselwirkungen, die die Immunsuppressiva mit den humanen FKBPs eingehen,
näher zu definieren. Zu diesem Zweck entwickelten Bierer et al. im Jahr 1990 das erste
totalsynthetische FK506-Derivat 506BD (siehe Abb. 11).51
Da FK506 und Rapamycin an dasselbe
Immunophilin binden, wurde das neue Molekül nur aus den gemeinsamen Strukturelementen
zusammengestellt. Die Verbindung wurde jedoch trotzdem auf eine mögliche Immunsuppression
untersucht. In Kompetitionsassays konnte keine Auswirkung von 506BD auf die T-Zellaktivierung
beobachtet werden. Vielmehr wurden eine Verdrängung der beiden natürlichen Makrozyklen und
damit eine Blockade ihrer immunsupprimierenden Effekte nachgewiesen. Mit diesem Ergebnis
wurde zwar bestätigt, dass die Effektordomäne allein für die Immunsuppression verantwortlich ist,
die Wirkung des Moleküls auf FKBP12 galt es jedoch noch zu klären. Da sich die FKBP12-Domäne
vor allem durch ihre PPIase-Aktivität auszeichnet und eine Hemmung dieser Enzymaktivität durch
FK506 oder Rapamycin bereits früher nachgewiesen werden konnte,52
wurde der nicht-
immunsuppressive FKBP12-Ligand auf eine mögliche PPIase-Inhibition untersucht. Mit Hilfe eines
Protease-gekoppelten PPIase-Assays53,54
konnte daraufhin durch 506BD ebenfalls eine Hemmung der
PPIase beobachtet werden. Somit war mit dem totalsynthetisierten Molekül ein erster hochaffiner,
jedoch nicht-immunsuppressiver FKBP12-Inhibitor identifiziert worden. In den darauffolgenden
Jahren wurde eine Vielzahl solcher nicht-immunsuppressiver FKBP12-Inhibitoren entwickelt, unter
denen sich 1FKG-lig (siehe Abb. 11) als besonders potent erwies (Ki = 10 nM).55
In der heutigen
medizinischen Forschung gelten diese FKBP12-Inhibitoren u.a. als vielversprechende Kandidaten in
der Therapie von neurodegenerativen Erkrankungen wie z.B. Alzheimer oder Parkinson.56
Die
Mechanismen dieser neuroprotektiven Aktivität sind jedoch noch nicht vollständig geklärt.
1.3 Strukturen der beiden PPIasen: Mip und FKBP12
Der C-Terminus von Mip setzt sich aus ungefähr 100 Aminosäuren zusammen und weist eine
Sequenzhomologie von 35 % zur FKBP12-Domäne auf.57
Auf die strukturelle Ähnlichkeit von Mip
und FKBP12 und auf die gemeinsame Zugehörigkeit zur Enzymklasse der PPIasen wurde bereits in
einem früheren Kapitel dieser Arbeit hingewiesen (siehe Kapitel 1.1.3.1). Betrachtet man nun diese
Gemeinsamkeiten, so ergibt sich als Schlussfolgerung, dass bereits bekannte FKBP12-Inhibitoren
ebenso an Mip binden und damit neben der humanen auch die bakterielle PPIase hemmen sollten. Im
Jahr 1992 konnten Fischer et al. erstmals mit Hilfe von Inhibitionsstudien eine Hemmung der Mip-
PPIase durch FK506 nachweisen.28
Inwieweit auch kleinere, nicht-immunsuppressive Verbindungen
eine Inhibition der Mip-PPIase ausüben, soll in dieser Arbeit geklärt werden.

EINLEITUNG 21
1.3.1 Mip-Strukturen
Im Jahr 2001 konnten Riboldi-Tunnicliffe et al. die erste Röntgenkristallstruktur (1FD9)27
von Mip
auflösen und damit zeigen, dass Mip als Dimer vorliegt. Ein Mip-Monomer besteht aus 213
Aminosäuren und setzt sich, wie oben beschrieben, aus zwei verschiedenen Domänen zusammen.
Die N-terminale Domäne (Aminosäuren 9-48) wird aus zwei antiparallel verlaufenden α-Helices
geformt und ist für die Dimerisierung des Proteins verantwortlich. Dabei bildet sich ein untypisches
„Vier-Helix-Bündel“ mit einem Inklinationswinkel von ca. 45 ° zwischen den beiden Helices aus.
Eine weitere Besonderheit dieser Dimerverbindung ist ein sogenannter „Methionin-Zipper“, bei dem
die Methioninreste Met38 und Met42 eines Monomers mit Met42 und Met38 des anderen in
Interaktion treten.
Die C-terminale Domäne (Aminosäuren 77-213) zeigt die typische Faltung der FKBPs und beinhaltet
die PPIase-Funktion (siehe Abb. 12). Im folgenden Kapitel soll sich die Bezeichnung „Mip“ nur noch
auf diese C-terminale Domäne beziehen (d.h. die Aminosäuren der C-Domäne werden von 1 bis 137
nummeriert). Sechs β-Stränge formen am C-terminalen Ende ein antiparalleles Faltblatt mit der
topologischen Reihenfolge β1-β2-β5-β6-β3-β4. Während alle β-Stränge über kurze Schleifen (sog.
Haarnadelschlaufen, hair-pins) miteinander verbunden sind, besteht die Haarnadelschlaufe zwischen
β5 und β6 aus 22 Aminosäuren. Dieses β-Faltblatt wird von einer kurzen α-Helix so bedeckt, dass
zwischen der Innenseite der β-Struktur und der α-Struktur eine Kavität entsteht, die sich besonders
durch ihre Hydrophobizität auszeichnet.
Abb. 12: Proteinstruktur (2VCD) der C-Domäne von Mip: Zwischen der α-Helix und dem β-Faltblatt (siehe
Pfeil) formt sich die hydrophobe Bindetasche (erstellt mit PyMOL®)
Diese Bindetasche setzt sich aus folgenden Aminosäuren zusammen: Tyr55, Phe65, Asp66, Ala75,
Phe77, Val82, Ile83, Trp86, Tyr109 und Phe126 (siehe Abb. 13). Der Indolring des Trp86 bildet den
Grund der Kavität und wird von den Resten Tyr55, Ala75, Phe77, Val82, Ile83 und Phe126
umgeben. Phe65, Asp66 und Tyr109 bilden den äußeren Rand der Bindetasche, wobei die

22 EINLEITUNG
letztgenannte Aminosäure einen Bestandteil der langen Schleife zwischen β5 und β6 darstellt, welche
an das aktive Zentrum angrenzt.27,58
Abb. 13: Sekundärstruktur (2VCD) der C-terminalen Domäne von Mip mit der Aminosäurebezeichnung im
aktiven Zentrum; (Wiedergabe nach Sippel58
mit Genehmigung des Autors)
Neben dieser Kristallstruktur existieren mittlerweile zwei weitere Proteinstrukturen von Mip. Mit
Hilfe verschiedener NMR-Analysen lösten Ceymann et al. 2008 eine Apo-Struktur von Mip
(2UZ5)59
, sowie eine Komplexstruktur (2VCD)59
, in der Rapamycin im aktiven Zentrum gebunden
vorliegt, auf. Ein Vergleich der beiden Apo-Strukturen (1FD9 bzw. 2UZ5) zeigt, dass sie sich
erheblich voneinander unterscheiden. Während die Aminosäure Tyr109 in der Kristallstruktur
(1FD9) die Bindetasche am oberen Ende abschließt und somit einen Teil des aktiven Zentrums
darstellt, ist derselbe Rest in der NMR-gelösten Struktur (2UZ5) kein Bestandteil der Kavität; er ragt
vielmehr aus der Bindetasche heraus. Überlagert man jedoch die Mip/Rapamycin-Komplexstruktur
(2VCD) mit beiden Apo-Strukturen, so zeigt sich eine größere Kongruenz mit der Kristallstruktur
(1FD9) als mit der nicht komplexierten NMR-gelösten Struktur (2UZ5) (siehe Abb. 14).
Abb. 14: Überlagerung der Komplexstruktur 2VCD (beige) mit den Apo-Strukturen 2UZ5 (blau, links) und
1FD9 (cyan, rechts); (Wiedergabe nach Sippel58
mit Genehmigung des Autors)
Val82 Ile83 Tyr109
Phe65 Asp66
Phe126 Tyr55
Trp86 Ala75
Phe77

EINLEITUNG 23
1.3.2 FKBP12-Strukturen
Während sich ein Mip-Monomer aus zwei verschiedenen Domänen zusammensetzt, zeichnet sich die
Struktur von FKBP12 nur durch eine einzelne Domäne aus (siehe Abb. 15). 108 Aminosäuren bilden
aus fünf β-Strängen eine antiparallele ß-Faltblattstruktur, die sich rechtsdrehend um eine α-Helix
lokalisiert. Damit wird die α-Helix so umhüllt, dass sich eine hydrophobe Bindetasche zwischen ihr
und dem ß-Faltblatt ausbildet.60
Abb. 15: Proteinstruktur (2PPN) von FKBP12: Zwischen der α-Helix und dem β-Faltblatt (siehe Pfeil) formt
sich die hydrophobe Bindetasche (erstellt mit PyMOL®)
Die FKBP12-Kavität setzt sich aus folgenden Aminosäuren zusammen: Tyr26, Phe36, Asp37,
Phe46, Phe48, Val55, Ile56, Trp59, Tyr82 und Phe99.58
Der Indolring des Trp59 bildet den Grund
der Kavität und wird von den restlichen Aminosäuren umgeben.59
Im Fall von FKBP12 ist in den Proteindatenbanken eine weitaus größere Anzahl an Kristall- sowie
NMR-gelösten Strukturen verfügbar. Als Vertreter der Komplexstrukturen wurde in einer früheren
Arbeit die kristallographische Struktur 1FKB61
herangezogen, während 2PPN36
eine nicht
komplexierte Kristallstruktur und 1FKT62
eine ungebundene NMR-gelöste Struktur darstellen
sollten.58
Der Vergleich der Orientierung der Aminosäuren der gewählten Strukturen zeigte kaum
Abweichungen im aktiven Zentrum. Die Überlagerung der beiden Apo-Strukturen (2PPN bzw.
1FKT, siehe Abb. 16, links), sowie die Überlagerung der Kristallstrukturen (1FKB bzw. 2PPN,
siehe Abb. 16, rechts) deutete lediglich auf kleine Unterschiede in der Stellung der Aminosäure
Asp37 hin.58
Auf Grund der Geringfügigkeit wurde diese Diskrepanz im nachfolgenden
Proteinstruktur-Vergleich vernachlässigt.

24 EINLEITUNG
Abb. 16: Überlagerung der Apo-Strukturen 2PPN (blau) und 1FKT (beige, links), sowie Überlagerung der
Kristallstrukturen 2PPN (blau) und 1FKB (grün, rechts); (Wiedergabe nach Sippel58
mit Genehmigung des
Autors)
1.3.3 Struktureller Vergleich der PPIasen
Für eine Entwicklung selektiver Mip-Inhibitoren müssen die strukturellen Gemeinsamkeiten bzw.
Unterschiede der beiden PPIasen genau erfasst werden. Aus diesem Grund wurden nun zuerst die
Aminosäurereste in beiden aktiven Zentren und anschließend die dort vorherrschende geometrische
Struktur genau miteinander verglichen. Tabelle 2 zeigt die Gegenüberstellung aller relevanten
Aminosäuren im aktiven Zentrum der beiden Proteine:
MIP FKBP12
Tyr55 Tyr26
Phe65 Phe36
Asp66 Asp37
Ala75 Phe46
Phe77 Phe48
Val82 Val55
Ile83 Ile56
Trp86 Trp59
Tyr109 Tyr82
Phe126 Phe99
Tabelle 2: Vergleich der Aminosäuren im aktiven Zentrum von Mip und FKBP1258
Val55 Ile56 Tyr82
Phe36
Asp37
Phe99
Tyr26 Phe46
Phe48 Trp59

EINLEITUNG 25
Fast alle Aminosäuren in Mip entsprechen ihrem jeweiligen Gegenstück in FKBP12. Eine Ausnahme
bildet jedoch die Aminosäure Ala75 des bakteriellen Mips, welche dem Phe46 in der FKBP12-
Bindetasche gegenübersteht. Trotz dieser Inkongruenz weist die Umgebung dieser Aminosäuren in
beiden Proteinen eine ähnlich starke Hydrophobizität auf, so dass dieser kleine Unterschied
vernachlässigbar ist. Vergleicht man jedoch die Geometrie der Aminosäuren im aktiven Zentrum der
bakteriellen sowie der humanen PPIase miteinander, so zeigen sich bereits deutlichere Unterschiede
in der räumlichen Orientierung einzelner Aminosäuren. Die Überlagerung der unkomplexierten
Kristallstrukturen (1FD9, Mip bzw. 2PPN, FKBP12) weist die größte Homologie zwischen dem
bakteriellen und dem humanen Protein auf. Erstaunlicherweise ist bei dieser Überlagerung eine
deutlich höhere strukturelle Kongruenz zu finden als bei der Überlagerung der beiden ungebundenen
Mip-Strukturen (1FD9 bzw. 2UZ5).
Betrachtet man nun die Überlagerung der beiden Rapamycin-Komplexstrukturen (2VCD, Mip und
1FKB, FKBP12, siehe Abb. 17), so zeigen sich ähnlich starke strukturelle Unterschiede. Das
bakterielle Tyr109 ist viel stärker in Richtung der hydrophoben Bindetasche orientiert als sein
humanes Gegenstück Tyr82. Die Position von Tyr109 in der Rapamycin-Komplexstruktur führt
somit zu einer erheblichen Verkleinerung der Mip-Bindetasche, was sich letztendlich auch auf die
Bindung möglicher Liganden auswirken könnte.
Abb. 17: Überlagerung der Rapamycin-Komplexstrukturen (2VCD, Mip (beige; mit Aminosäurebezeichnung)
und 1FKB, FKBP12 (grün)); Tyr109 ragt tiefer in die hydrophobe Mip-Bindetasche, als Tyr82 in die FKBP12-
Bindetasche. (Wiedergabe nach Juli et al.63
mit Genehmigung der American Chemical Society)
Als Komplexpartner wurde in allen beschriebenen Proteinstrukturen das Immunsuppressivum
Rapamycin gewählt. Seine Bindedomäne besteht aus einem Pipecolinsäure-Grundgerüst (siehe
Kapitel 1.2.2.2), das im Fall beider PPIasen starke Wechselwirkungen mit den Aminosäuren im
aktiven Zentrum eingeht und deshalb tief in die jeweilige Bindetasche gebunden wird. Der größere
Teil des Makrozyklus (= Effektordomäne) ragt aus der Kavität heraus und bietet damit eine
Tyr109 / Tyr82
Ala75 / Phe46
Tyr109
Ile83 Val82
Phe77
Ala75
Tyr55 Asp66
Phe65
Phe126 Trp86

26 EINLEITUNG
strukturelle Oberfläche für die Bindung an ein weiteres Molekül. Im Fall von Rapamycin handelt es
sich dabei um die Kinase mTOR (siehe Kapitel 1.2.2.1).
Die feste Verankerung des Pipecolinrings in der FKBP12-Bindetasche ist einerseits durch
hydrophobe Wechselwirkungen des Piperidins, andererseits durch die Bildung von
Wasserstoffbrücken zwischen Ile56 und der Esterfunktion (C=O) bzw. Tyr82 und der Amidfunktion
(C=O) bedingt. Die letztgenannte Bindung kann sich auf Grund der divergierenden räumlichen
Orientierung von Tyr109 in der Mip-Kavität jedoch nicht ausbilden. Vielmehr kommt es hier zu
einer „Kollision“ beider Funktionen. Diese Abstoßung bedingt, dass das Pipecolinsäure-Grundgerüst
in der bakteriellen Bindetasche eine andere Stellung als in der humanen einnehmen muss. Tatsächlich
lagert sich der Piperidinzyklus - im Gegensatz zur FKBP12-Kavität - aufrecht in die Mip-Kavität ein.
Aus diesem Grund können die Aminosäure Tyr109, ebenso wie die gegenüberliegende Aminosäure
Tyr55, in der Mip-Bindetasche auch nicht mehr als Wasserstoffbrücken-Donoren fungieren. Tyr55
wendet sich im Gegensatz zum humanen Tyr26 sogar von der Diketostruktur des Rapamycins ab.
Trotz dieser strukturellen Divergenzen scheinen beide Immunsuppressiva aber über dieselben
funktionellen Gruppen in die Kavitäten von FKBP12 und Mip zu binden (siehe Abb. 18).58
Abb. 18: Rapamycin/FKBP12-Komplex: Wasserstoffbrücken können sich zu Ile56 und Tyr82 ausbilden
(links). Rapamycin/Mip-Komplex: Abstoßung zwischen Tyr109 und dem Amid (C=O); Tyr55 wendet sich ab
(rechts). (Wiedergabe nach Sippel58
mit Genehmigung des Autors)
FKBP12 Mip

EINLEITUNG 27
1.4 Mip-ähnliche Strukturen anderer Mikroorganismen
In den Jahren nach der Entdeckung von Legionella-Mip (Lp-Mip) und der vollständigen Aufklärung
seiner Gensequenz wurden auch in anderen Bakterienarten Mip-ähnliche Proteine identifiziert.
Mittels eines Protease-gekoppelten PPIase-Assays53
konnte nachgewiesen werden, dass diese alle
eine PPIase-Aktivität aufweisen. Die Mip-ähnlichen Proteine greifen jedoch an unterschiedlichen
Stellen des bakteriellen Infektionsprozesses ein. Im folgenden Kapitel sollen die Mikroorganismen
mit ihrem Mip-Protein kurz vorgestellt werden.
1.4.1 Chlamydien
Chlamydia trachomatis gehört neben vielen weiteren Arten zur Gattung der Chlamydien und stellt
ein sehr bedeutendes Humanpathogen dar. Das Bakterium wird in mehrere Serogruppen unterteilt,
die je nach Typ verschiedene Krankheiten verursachen können. Während die Serotypen A bis C vor
allem in Entwicklungsländern schwere Infektionen am Auge (z.B. Trachom) hervorrufen können,
lässt sich der Großteil der Chlamydieninfektionen (Serotyp D-L) über sexuell übertragbare
Erkrankungen im Urogenitalbereich definieren. Bei Frauen kann dies zu schweren Entzündungen der
Gebärmutter oder der Eileiter führen. Bei Männern sind vor allem die Nebenhoden und die Prostata
betroffen. Außer den genannten Krankheiten kann C. trachomatis (Serotyp L1, L2 und L3) auch das
sexuell übertragbare Lymphogranuloma venereum verursachen. Bei dieser Erkrankung kommt es
hauptsächlich zu Lymphknotenschwellungen bzw. Geschwüren in der Leistengegend. C. trachomatis
zählt damit zu den häufigsten sexuell übertragenen Erregern weltweit. Zur Behandlung einer
Chlamydieninfektion eignen sich Antibiotika wie Tetrazykline oder Makrolide. Des Weiteren werden
auch Chinolone erfolgreich angewendet. Die antibiotische Behandlungsdauer sollte einen Zeitraum
von zwei Wochen nicht unterschreiten.64
Chlamydien sind wie Legionellen obligat intrazelluläre Bakterien, die sich vor allem durch ihren
zweiphasigen Lebenszyklus in humanen Schleimhautzellen auszeichnen. Eine Phase wird durch die
sogenannten extrazellulären Elementarkörper (EB) dargestellt. Die EBs sind zwar infektiös, aber
metabolisch völlig inaktiv. In dieser Phase können die Bakterien an eine geeignete humane
Wirtszelle binden. Nach ihrer Aufnahme, die entweder durch Phagozytose oder durch Pinozytose
erfolgt, fusionieren die EBs mit dem Wirtsphagosom und wandeln sich nach 8-10 Stunden in
Retikularkörper (RB) um, die nun nicht mehr durch Lysosome abgebaut werden können. In diesem
zweiten Stadium verlieren die Teilchen ihre Infektiösität und nach weiteren 10-20 Stunden beginnen

28 EINLEITUNG
sie sich zu vermehren. Nach der bakteriellen Replikation nehmen die Retikularkörper wieder die
ursprüngliche Form der EBs ein und zerstören die Wirtszelle, um sich weiter zu verbreiten.65
Lange Zeit war man sich über die Mechanismen der bakteriellen Adhäsion und Infektion im
Unklaren. Im Rahmen der Aufklärung dieser Prozesse und daran beteiligter Proteine, wurde von
Lundemose et al. u.a. ein spezielles Membranprotein identifiziert, das in den Infektionsverlauf
involviert zu sein scheint.65
Die DNA-Strukturaufklärung dieses Proteins zeigte, dass es eine
Molekülmasse von 27 kDa besitzt. Die C-terminale Domäne des Proteins besteht aus 175
Aminosäuren und weist eine Homologie von 37 % zu Lp-Mip auf. Zwei Jahre später konnte gezeigt
werden, dass dieses Mip-ähnliche Protein (Ct-Mip) auch eine PPIase-Aktivität besitzt.66
Mit Hilfe der
bekannten PPIase-Inhibitoren Rapamycin und FK506 wurde durch Inhibitionsstudien im Folgenden
die Rolle des Mip-ähnlichen Proteins während der bakteriellen Infektion aufgeklärt. Es konnte
gezeigt werden, dass die beiden Immunsuppressiva nur bei Gabe in einer frühen Phase des
Infektionsprozesses die Anzahl an Chlamydien im Zellinneren reduzieren können. Eine
Verabreichung der Arzneistoffe zu einem späten Zeitpunkt wirkt sich nicht auf die bakterielle
Vermehrung aus.
1.4.2 Trypanosomen
Die Infektionskrankheit Trypanosomiasis kann in zwei Unterarten eingeteilt werden: Die
afrikanische Form, die oft auch als Schlafkrankheit bezeichnet wird, und die amerikanische Form, die
Chagas-Krankheit. Der Erreger der Schlafkrankheit ist Trypanosoma brucei. T. brucei wird v.a. im
tropischen Afrika über Tsetsefliegen auf den Menschen übertragen. Die Chagas-Krankheit hingegen
wird v.a. im mittel- bis südamerikanischen Raum über Raubwanzen (Reduviidae) durch
Trypanosoma cruzi übertragen. Da im Rahmen dieser Arbeit lediglich die Art T. cruzi von
Bedeutung ist, soll auch nur diese im folgenden Kapitel betrachtet werden.
Bei T. cruzi handelt es sich um Protozoen, die während eines Lebenszyklus in verschiedenen Formen
auftreten können. Nach der Blutmahlzeit eines infizierten Insekts werden sogenannte
Trypomastigoten, die infektiöse Form des Mikroorganismus, im Kot neben der Einbissstelle auf der
menschlichen Haut hinterlassen. Von dort können sie entweder direkt über die Wunde oder über
Schleimhäute in den menschlichen Körper gelangen und nahe gelegene Zellen befallen. Im
Zellinneren transformieren sie zu Amastigoten, vermehren sich und verwandeln sich wieder zurück
in Trypomastigoten. Um in den Blutkreislauf zu gelangen, zerstören sie ihre Wirtszelle und befallen
danach entweder erneut Zellen oder persistieren in einer nicht infektiösen Form. Kommt es nun zu
einem weiteren Kontakt zwischen Insekt und Mensch, gelangen die persistenten Trypomastigoten
zurück in den Mitteldarm der Raubwanze, wo sie sich sofort in Epimastigoten differenzieren und

EINLEITUNG 29
weiter vermehren. Nach der Transformation zurück zur Trypomastigot-Form beginnt der Zyklus
erneut.67
Der Verlauf einer Chagas-Erkrankung kann in vier Stadien gegliedert werden: Nach ödematösen
Entzündungsreaktionen an der Bissstelle leidet der Patient zunächst an Fieber, Luftnot, Durchfall und
Bauchschmerzen. Diese Phase klingt rasch ab (ca. 30 Tage) und auf eine lange Latenzzeit (bis zu
mehreren Jahren) ohne Symptome folgt die chronische Phase der Erkrankung. Diese zeichnet sich
einerseits durch Herzprobleme, andererseits durch Zerstörung von Darmnervenzellen aus.
Unbehandelt kann dies bis hin zum Tode führen. Zum heutigen Zeitpunkt existieren nur zwei
Antibiotika für die Behandlung der Chagas-Krankheit: Nifurtimox und Benznidazol. Beide
Arzneistoffe müssen zu Beginn der akuten Krankheitsphase eingesetzt und die Therapiedauer von
zwei Monaten nicht unterschritten werden.68
Bei Untersuchungen der einzelnen Zustandsformen von T. cruzi zeigten sich viele verschiedene
bakterielle Virulenzfaktoren, u.a. ein Mip-ähnliches Protein (Tc-Mip), das jedoch nur von der
infektiösen Form der Trypomastigoten sekretiert wird.69
Die Gensequenzanalyse von Tc-Mip ergab
ein Monomer aus 167 Aminosäuren, das eine Homologie von 29.6 % zu Lp-Mip aufweist.70
Wie bei
dem verwandten Protein konnte mittels Protease-gekoppelten Enzymassays eine PPIase-Aktivität
nachgewiesen werden. In einem Invasionsassay wurde ferner festgestellt, dass Tc-Mip wie auch Lp-
Mip einen Einfluss auf die Invasion der Bakterien in die Wirtszelle hat. Die darauffolgende exogene
Behandlung der Wirtszellen mit Tc-Mip zeigte, dass das Protein eine Wechselwirkung mit der
Wirtszelle, aber nicht mit der Parasitenzelle eingeht. Dies verdeutlicht, dass das Target der Isomerase
auf der Oberfläche der Wirtszelle liegen muss.69
1.4.3 Neisserien
Neben der Chlamydieninfektion zählt auch die Gonorrhoe (Tripper) zu den häufigsten sexuell
übertragenen Erkrankungen. Auslöser einer solchen Infektion ist das intrazellulär lebende Bakterium
Neisseria gonorrhoeae, das neben Neisseria meningitis zu den bedeutendsten Vertretern der Gattung
Neisseria gehört. Der Lebensraum von N. gonorrhoeae ist hauptsächlich im menschlichen
Genitalbereich zu finden. Hier binden die Bakterien bei einer Infektion zunächst an mukosale
Epithelzellen und werden anschließend von diesen phagozytiert. Der Mechanismus der Phagozytose
wird entweder durch Pili oder sogenannte „opacity outer membrane“-Proteine vermittelt. Im
Zellinneren werden die Bakterien in eine Vakuole eingeschlossen und darin gebunden in
subepitheliales Gewebe transportiert. Dort können sie lokale Entzündungsreaktionen hervorrufen
oder/und sich über den Blutkreislauf auf den gesamten Körper ausbreiten.71
Während eine
Erkrankung bei Frauen eher asymptomatisch verläuft (nur 25 % der Betroffenen entwickeln diverse

30 EINLEITUNG
Unterleibsentzündungen), manifestiert sich beim Mann typischerweise eine akute Urethritis.72
Das
RKI empfiehlt zur Therapie der Gonorrhoe eine zweimalige Gabe von Cefixim in einem Zeitraum
von drei Tagen, oder eine einmalige intramuskuläre Verabreichung von Ceftriaxon.73
Wie bei den vorher beschriebenen Mikroorganismen existieren auch in N. gonorrhoeae zahlreiche
Virulenzfaktoren, die mit unterschiedlichen Prozessen während der humanen Infektion
wechselwirken. Die Anzahl sowie die Funktion dieser Faktoren sind noch nicht vollständig
aufgeklärt. 2005 konnten Leuzzi et al. jedoch ein Oberflächenprotein von N. gonorrhoeae mit
PPIase-Aktivität identifizieren.72
Das dimere Protein besteht aus 272 Aminosäuren und weist eine
Sequenzhomologie von 43.8 % zu Lp-Mip auf. Diese Eigenschaften und eine nachgewiesene
Inhibition durch Rapamycin ordneten das Neisserienprotein (Ng-Mip) in die Klasse der Mips ein. Im
Rahmen dieser Studie konnte auch die Funktion von Ng-Mip geklärt werden. Während alle
beschriebenen Mips in die Invasion des Mikroorganismus bzw. einen frühen Schritt der Infektion
involviert sind, spielt Ng-Mip eine bedeutende Rolle bei der Persistenz des Bakteriums in
Makrophagenzellen. Es konnte gezeigt werden, dass Mip-negative Stämme durch eine Makrophagen-
vermittelte Immunreaktion getötet werden, während Mip-positive Stämme dagegen resistent sind.
1.4.4 Burkholderien
Burkholderia pseudomallei ist der Erreger der Infektionskrankheit Melioidose, die hauptsächlich in
Regionen wie Südostasien und Nordaustralien auftritt. Klinisch manifestiert sich eine solche
Erkrankung in einer akuten Sepsis bis hin zu lokal auftretenden chronischen Infektionen. In 60 %
aller akuten Erkrankungen kann sich zusätzlich eine schwere Pneumonie entwickeln.74
In der
Therapie der akuten Phase gilt die intravenöse Gabe von Ceftazidim als Mittel der Wahl, das aber
auch durch die Reserveantibiotika Meropenem oder Imipenem ersetzt werden kann. Dieser
zweiwöchigen Initialtherapie schließt sich eine Eradikationsphase an, in der Mittel wie Cotrimoxazol
oder Doxyzyklin über mindestens 20 Wochen eingesetzt werden müssen.
B. pseudomallei zählt zu den beweglichen, gramnegativen Bakterien, die ihren Lebensraum sowohl
in der Erde als auch im Wasser finden. Aus ihrer natürlichen Umgebung gelangen sie über
verschiedene Transportmechanismen in den menschlichen Organismus. Typischerweise wird der
Mikroorganismus über die Haut, seltener auch über Inhalation oder über die Nahrung aufgenommen.
Im menschlichen Körper kann B. pseudomallei verschiedene Zellen, zu denen auch Makrophagen
gehören, befallen. Nach der Phagozytose werden die Bakterien im Zellinneren, ähnlich wie bei einer
Legionellen-Infektion, zuerst in ein Phagosom, das vor dem Abbau durch Lysosomen geschützt ist,
eingeschlossen. Anschließend findet die bakterielle Vermehrung statt. Nach der Replikation zerstören
die Bakterien die Phagosomenmembran und breiten sich direkt von Zelle zu Zelle auf den gesamten

EINLEITUNG 31
Organismus aus.75
Auf Grund dieser relativ leichten Art der Verbreitung wird B. pseudomallei auch
als biologischer Kampfstoff eingesetzt. Das Centers for Disease Control and Prevention der USA
teilte die Burkholderien deshalb in die Kategorie B der sogenannten B-Waffen ein.
Obwohl in den letzten Jahren zahlreiche Studien zu den Virulenzmechanismen dieses Bakteriums
durchgeführt wurden, bleiben weiterhin viele Fragen im Bereich der diversen Infektionsprozesse
offen. Kürzlich (2011) konnte jedoch von Norville et al. ein Mip-ähnliches Protein (Bp-Mip) als
Hauptvirulenzfaktor von B. pseudomallei identifiziert werden.74
Dieses Polypeptid setzt sich aus 113
Aminosäuren zusammen, die eine Homologie von 40 %, 45 % und 42 % zu Lp-Mip, Ng-Mip und Ct-
Mip aufweisen. Analog zu seinen verwandten Proteinen besitzt Bp-Mip im Bereich der C-Domäne
eine PPIase-Aktivität und spielt ähnlich zur Funktion von Ng-Mip eine wichtige Rolle im Bereich
des intrazellulären Überlebens und der Vermehrung des Bakteriums. Die Adhäsion an die Wirtszelle
wie im Fall von Lp-Mip und Tc-Mip wird von Bp-Mip jedoch nicht beeinflusst.
1.5 Zielsetzung
1.5.1 Chemische Synthese
Im Zentrum dieser Arbeit stand das Oberflächenprotein Mip des gramnegativen Bakteriums L.
pneumophila. Da bis dato weder eine gezielte Behandlung der Legionellen möglich ist, noch die
Mortalitätsrate der Legionellose durch eine Antibiotikagabe reduziert werden konnte, sollten im
Rahmen dieser Arbeit neue Leitstrukturen für selektive Legionellen-Chemotherapeutika entwickelt
werden. Mip stellt hierfür ein vielversprechendes neues Target dar, da es zum einen durch seine
Position auf der Oberfläche der Legionellen leicht zugänglich ist, zum anderen bislang nicht von den
eingesetzten Antiinfektiva inhibiert wird, d.h. es existieren noch keine Resistenzmechanismen.
Bereits 2008 wurde an der Universität Würzburg mittels NMR-Strukturanalyse eine Mip/Rapamycin-
Komplexstruktur (2VCD) aufgeklärt.59
Da Mip eine große Sequenzhomologie zum humanen
FKBP12 aufweist, wurde in Kooperation mit der Molecular-Modelling-Arbeitsgruppe von Prof. Dr.
Christoph Sotriffer 2VCD mit einer literaturbekannten FKBP12/Rapamycin-Kristallstruktur
verglichen und mit Hilfe verschiedener Docking-Studien nach neuen Leitstrukturen für Mip-
Inhibitoren gesucht. Dabei wurde N,N-Dimethylphenylsulfonsäureamid als ein vielversprechender
Mip-Bindepartner identifiziert, woraufhin diese Verbindung im Rahmen dieser Arbeit zunächst
hergestellt werden sollte. Um anschließend eindeutige Struktur-Wirkungsbeziehungen zwischen dem
Sulfonsäureamid und Mip aufstellen zu können, sollte N,N-Dimethylphenylsulfonsäureamid
zusätzlich strukturell verändert und somit eine kleine Substanzbibliothek von Sulfonsäureamid-
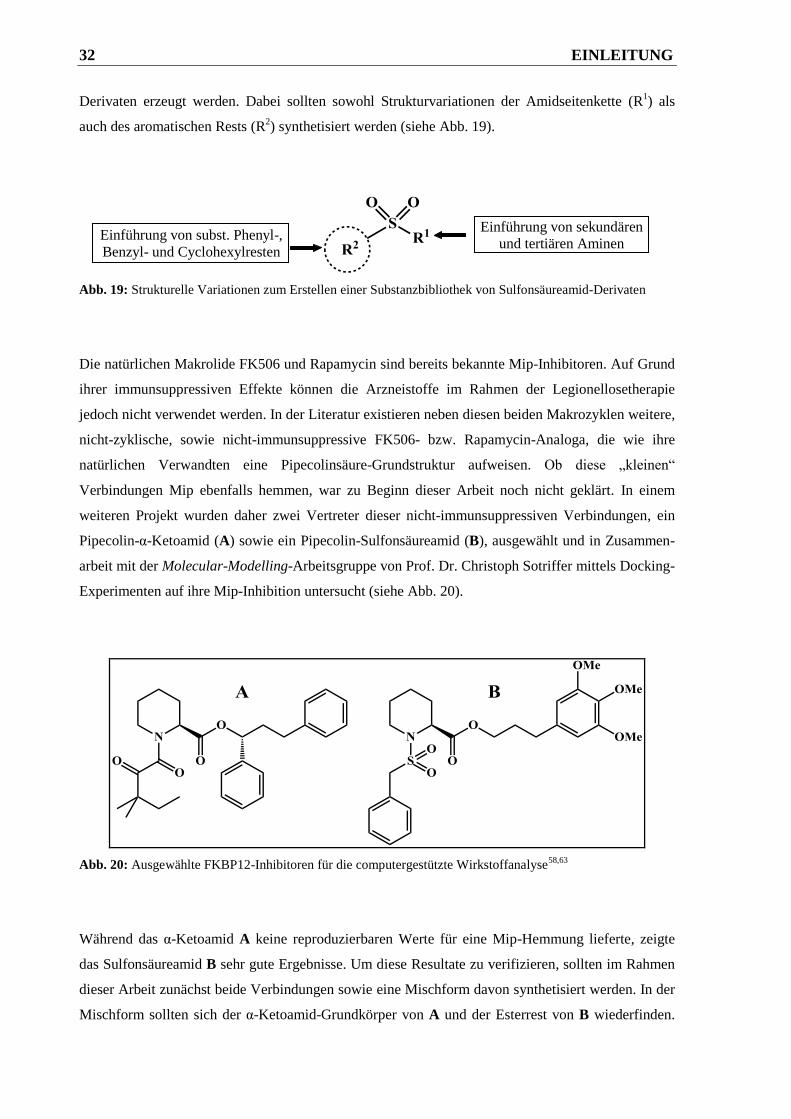
32 EINLEITUNG
Derivaten erzeugt werden. Dabei sollten sowohl Strukturvariationen der Amidseitenkette (R1) als
auch des aromatischen Rests (R2) synthetisiert werden (siehe Abb. 19).
Abb. 19: Strukturelle Variationen zum Erstellen einer Substanzbibliothek von Sulfonsäureamid-Derivaten
Die natürlichen Makrolide FK506 und Rapamycin sind bereits bekannte Mip-Inhibitoren. Auf Grund
ihrer immunsuppressiven Effekte können die Arzneistoffe im Rahmen der Legionellosetherapie
jedoch nicht verwendet werden. In der Literatur existieren neben diesen beiden Makrozyklen weitere,
nicht-zyklische, sowie nicht-immunsuppressive FK506- bzw. Rapamycin-Analoga, die wie ihre
natürlichen Verwandten eine Pipecolinsäure-Grundstruktur aufweisen. Ob diese „kleinen“
Verbindungen Mip ebenfalls hemmen, war zu Beginn dieser Arbeit noch nicht geklärt. In einem
weiteren Projekt wurden daher zwei Vertreter dieser nicht-immunsuppressiven Verbindungen, ein
Pipecolin-α-Ketoamid (A) sowie ein Pipecolin-Sulfonsäureamid (B), ausgewählt und in Zusammen-
arbeit mit der Molecular-Modelling-Arbeitsgruppe von Prof. Dr. Christoph Sotriffer mittels Docking-
Experimenten auf ihre Mip-Inhibition untersucht (siehe Abb. 20).
Abb. 20: Ausgewählte FKBP12-Inhibitoren für die computergestützte Wirkstoffanalyse58,63
Während das α-Ketoamid A keine reproduzierbaren Werte für eine Mip-Hemmung lieferte, zeigte
das Sulfonsäureamid B sehr gute Ergebnisse. Um diese Resultate zu verifizieren, sollten im Rahmen
dieser Arbeit zunächst beide Verbindungen sowie eine Mischform davon synthetisiert werden. In der
Mischform sollten sich der α-Ketoamid-Grundkörper von A und der Esterrest von B wiederfinden.
Einführung von subst. Phenyl-,
Benzyl- und Cyclohexylresten
Einführung von sekundären
und tertiären Aminen

EINLEITUNG 33
Um anschließend die für die Mip-Inhibition essenziellen Strukturmerkmale zu identifizieren, sollten
des Weiteren verschiedene Derivate der Sulfonsäureamid-Struktur B hergestellt werden (siehe Abb.
21). Dabei sollte sowohl der Benzylrest der Sulfonsäureamidseitenkette durch substituierte Benzyl-
und Phenylreste als auch das Sulfonsäureamid durch Carbonsäureamide ersetzt werden. Ferner
sollten auch Variationen der Esterseitenkette synthetisiert werden.
Die beiden Substanzbibliotheken, die der niedermolekularen Sulfonsäureamide und die der
Pipecolinsäure-Derivate, sollten schließlich mit Hilfe verschiedener pharmakologischer Testsysteme
auf ihre Mip-Wirkung untersucht und basierend auf den erhaltenen Erkenntnissen Struktur-
Wirkungsbeziehungen aufgestellt werden.
Abb. 21: Variationen am Pipecolinsäure-Grundgerüst zum Erstellen einer Substanzbibliothek
1.5.2 Pharmakologische Testung
Für die enzymatischen In-Vitro-Untersuchungen zur Bestimmung der Mip-Inhibition der neu
synthetisierten Verbindungen eignet sich ein Protease-gekoppelter PPIase-Assay.53
Dieser wurde zu
Beginn dieser Arbeit in Kooperation mit der Arbeitsgruppe von Dr. Matthias Weiwad am Max-
Planck-Institut für Enzymologie der Proteinfaltung in Halle durchgeführt und sollte am Institut für
Pharmazie der Universität Würzburg etabliert werden. Dazu sollte Mip im Rahmen dieser Arbeit
zunächst in Halle exprimiert und gereinigt werden.
Da sich die Existenz von Mip nicht nur auf die Familie der Legionellen beschränkt, sollten in
verschiedenen Kooperationsprojekten außerdem die Aktivitäten der hergestellten Verbindungen an
N. gonorrhoeae, C. trachomatis sowie an B. pseudomallei bestimmt werden.
Einführung von
aromatischen Esterseitenketten
mit Alkylspacern n = 1-5
Einführung von subst.
Benzyl- (m = 1) und
Phenylresten (m = 0)
Ersetzen der
Sulfongruppe durch
eine Carbonylfunktion

34 EINLEITUNG

ALLGEMEINER TEIL 35
2 DESIGN UND SYNTHESE DER ZIELVERBINDUNGEN
2.1 „Kleine“ Sulfonsäureamide
2.1.1 Computergestützte Entwicklung neuartiger Mip-Inhibitoren
Das bakterielle Mip sowie das humane FKBP12 gehören zur Enzymklasse der Isomerasen (PPIasen)
(siehe Kapitel 1.1.3.1). Die beiden PPIase-Domänen von Mip und FKBP12 weisen eine große
Sequenzhomologie zueinander auf. Aus diesem Grund können beide Enzyme auch durch identische
Arzneimittel, wie z.B. die makrozyklischen Immunsuppressiva, inhibiert werden. Auf Grund ihrer
immunsuppressiven Effekte dürfen diese Arzneistoffe jedoch nicht in der Therapie der bakteriellen
Legionellose verwendet werden. Daher galt es im Rahmen dieser Arbeit zunächst die strukturellen
Unterschiede zwischen beiden Proteinen aufzuzeigen und anschließend Stoffe zu entwickeln, die
selektiv das bakterielle Mip hemmen. Auf der Suche nach solchen potenziellen Mip-Inhibitoren
wurden die charakteristischen Aminosäuren im aktiven Zentrum näher betrachtet (siehe Kapitel
1.3.3). Die Überlagerung der Mip/Rapamycin-Komplexstruktur mit derjenigen des homologen
Proteins FKBP12 zeigte, dass die Bindetaschen insgesamt ähnlich sind, sich jedoch in einem
wichtigen Punkt unterscheiden: Die Mip-Aminosäure Tyr109 ragt tiefer ins aktive Zentrum der Mip-
Bindetasche hinein als ihr Gegenstück Tyr82 in die FKBP12-Kavität und verkleinert dadurch die
Mip-Bindetasche im Vergleich zur FKBP12-Kavität erheblich. Diese räumliche Diskrepanz wurde
als Ausgangspunkt für die computergestützte Entwicklung selektiver Mip-Inhibitoren genutzt. Da
dem Aminosäurerest Tyr109 eine weitere Tyrosingruppe (Tyr55) gegenüberliegt, wurde der Abstand
zwischen den beiden Tyrosin-Hydroxygruppen (Tyr-OH) berechnet und ergab einen Wert von 7.5 Å.
Unter Berücksichtigung dieses Abstands wurde nach funktionellen Gruppen gesucht, die optimal
diesen Raum ausfüllen und als Wasserstoffbrücken-Akzeptoren für die Tyr-OH-Gruppen fungieren
könnten. Als geeignete Kandidaten hierfür wurden Verbindungen mit einer Sulfon-Teilstruktur
identifiziert: Die beiden Sulfon-Sauerstoff-Atome können sich zwischen den beiden Tyr-OH-
Gruppen so orientieren, dass sich eine günstige Wasserstoffbrücken-Geometrie ausbilden kann (siehe
Abb. 22).
Die gesamten Molecular-Modelling-Untersuchungen dieser Arbeit wurden im Arbeitskreis von Prof. Dr.
Christoph Sotriffer an der Universität Würzburg hauptsächlich von Dr. Martin Sippel im Rahmen seiner
Dissertation58
durchgeführt.

36 ALLGEMEINER TEIL
Abb. 22: Abstand zwischen den Tyr-OH-Gruppen; günstige Wasserstoffbrücken-Geometrie des optimalen
Bindungspartners Sulfon
Im nächsten Schritt wurde versucht, die beiden Reste an der Sulfongruppe ebenfalls an die Mip-
Bindetasche anzupassen. Auf Grund der starken Hydrophobizität der Bindetasche sollten sich weitere
kleinere hydrophobe Gruppen als geeignet erweisen. Zur Bestätigung dieser Hypothese wurde N,N-
Dimethylphenylsulfonsäureamid (siehe Abb. 23) für weitere computergestützte Untersuchungen
ausgewählt.
Abb. 23: Struktur von N,N-Dimethylphenylsulfonsäureamid
Das Molekül ist durch eine Kopplung von Phenylsulfonsäurechlorid mit Dimethylamin synthetisch
leicht zugänglich und sollte sowohl hydrophobe Wechselwirkungen als auch Wasserstoff-
brückenbindungen mit den Aminosäuren der Mip-Kavität eingehen. Die Docking-Untersuchungen
wurden mit dem Programm GOLD 4.0 durchgeführt und zeigten für dieses Molekül sehr gute
Ergebnisse. Während das Sulfon sich, wie vorhergesagt, tatsächlich zwischen die beiden Tyr-OH-
Gruppen platzierte, lag der hydrophobe Phenylring tief in der Bindetasche verankert vor (siehe
Abb. 24).
2.5 Å
7.5 Å
2.5 Å 2.5 Å

ALLGEMEINER TEIL 37
Abb. 24: Docking von N,N-Dimethylphenylsulfonsäureamid in die Mip-Bindetasche (Proteinstruktur 2VCD)
Mit demselben Programm wurde daraufhin zuerst die Lage zahlreicher Phenylsulfonsäureamid-
Derivate, die sich lediglich in ihren hydrophoben Amidseitenketten unterschieden (siehe Abb. 25,
Strukturvariation I), in der Mip-Kavität bestimmt. Die vielversprechendsten Ergebnisse sollten
anschließend synthetisiert werden (siehe Kapitel 2.1.2.1.1 und 2.1.2.1.2).
Abb. 25: Docking-Ergebnisse: Variationen I und II an der Struktur von N,N-Dimethylphenylsulfonsäureamid
Neben den Variationen auf der Seite des Amins, wurden anschließend auch strukturelle
Veränderungen des aromatischen Teils des Moleküls untersucht (siehe Abb. 25, Strukturvariation II).
Während der Docking-Studien zeigten der Austausch des unsubstituierten Phenylrests durch
substituierte aromatische Reste sowie einen Cyclohexylrest sehr gute Ergebnisse, weshalb die
synthetische Herstellung dieser Substanzen ebenfalls angestrebt wurde (siehe Kapitel 2.1.2.2.1).
Des Weiteren fielen bei einer genauen Betrachtung der Bindetasche sowie dem darin gebundenen
N,N-Dimethylphenylsulfonsäureamid zwei Positionen auf, an denen das Molekül durch noch größere
Substituenten als den bereits untersuchten erweitert werden könnte. Einerseits sollte sich die
Einführung einer längeren Alkylseitenkette in meta-Position des Phenylrings (siehe Abb. 24,
Bindetasche A) positiv auf die Verankerung des Moleküls in der Bindetasche erweisen. Deshalb
Tyr55
Tyr109
A B
Strukturvariation I Strukturvariation II

38 ALLGEMEINER TEIL
wurde beabsichtigt, zusätzlich solche meta-substituierten Derivate zu synthetisieren (siehe Abb. 26,
Strukturvariation III / Kapitel 2.1.2.2.2 und 2.1.2.2.3).
Abb. 26: Docking-Ergebnisse: Strukturvariationen III-V von N,N-Dimethylphenylsulfonsäureamid
Andererseits könnte durch die Vergrößerung einer Amidseitenkette eine zusätzliche kleine
Seitentasche optimal adressiert werden (siehe Abb. 24, Bindetasche B). Um hierfür geeignete
Derivate zu finden, wurde ein Pharmakophor-basiertes virtuelles Screening durchgeführt. Basierend
auf N,N-Dimethylphenylsulfonsäureamid wurde mit Hilfe des Programms MOE folgendes
Pharmakophormodell erstellt: Der Phenylring des Ausgangsmoleküls wurde dafür durch ein
hydrophobes aromatisches Grundgerüst (siehe Abb. 27: brauner Polyeder), die beiden Sauerstoff-
gruppen des Sulfons durch zwei Protonen-Akzeptor-Gerüste dargestellt (siehe Abb. 27: blaue
Polyeder). Da das Schwefelatom für eine Mip-Interaktion eine eher unbedeutende Rolle spielt, wurde
es in diesem Modell vernachlässigt. Um einen räumlichen Bezug zum Protein herzustellen, wurden
die beiden Mip-Tyr-OH-Gruppen durch entsprechende Protonen-Donor-Gruppen angezeigt (siehe
Abb. 27: „kleine“ blaue Polyeder). Die Fragment-Datenbank des Programms wurde mit Hilfe dieses
Pharmakophormodells durchsucht und die passenden Fragmente anschließend in die Bindetasche
gedockt. Vielversprechende Bindemodi wurden bei N-((2-Oxo-oxazolidin-5-yl)methylbenzyl-
sulfonsäureamid identifiziert, weshalb die Synthese dieses Moleküls ebenfalls geplant wurde (siehe
Abb. 26, Strukturvariation IV / Kapitel 2.1.2.1.3). Außerdem wies aber auch Aminomethyl-
phenylsulfon recht gute Werte im Bereich der Bindemodi auf und sollte ebenfalls synthetisiert
werden (siehe Abb. 26, Strukturvariation V / Kapitel 2.1.2.3).

ALLGEMEINER TEIL 39
Abb. 27: Pharmakophormodell: Hydrophile Funktionalitäten (blau: Mip-Tyr-OH- (klein), sowie Sulfon-
Sauerstoffgruppen (groß)); hydrophobe Funktionalität (braun: aromatischer Phenylring)
Zusammenfassend lässt sich sagen, dass die Sulfongruppe ein fester Bestandteil aller neu zu
entwickelnden Moleküle sein sollte.
2.1.2 Synthese der designten Sulfonsäureamid-Derivate
Ausgehend von den Ergebnissen der Molecular-Modelling-Untersuchungen wurden zahlreiche
Sulfonsäureamid-Derivate hergestellt. Diese Moleküle lassen sich strukturell in zwei Gruppen
unterteilen (siehe Abb. 28):
Abb. 28: Grundstrukturen A und B der Sulfonsäureamid-Derivate
Das Grundgerüst der beiden Zielverbindungen ist N,N-Dimethylphenylsulfonsäureamid. In
Zielverbindung A bleibt der aromatische Bereich unverändert und der sulfonsäureamidische
Stickstoff wird unterschiedlich substituiert: Neben Alkylaminen wie Ethylamin, Propylamin,
Mip-Tyr55-OH
Mip-Tyr109-OH
Phenylrest
Sulfon-Sauer-
stoffgruppen

40 ALLGEMEINER TEIL
Propargylamin, Isopropylamin, Pyrrolidin, Piperidin, Piperazin, Cyclopropylamin, Cyclopentylamin
wurden aromatische Amine wie Benzylamin, 2-Phenylethylamin, S-1-Phenylethylamin, 4-
Methoxyamin, 4-Methylamin sowie Tryptamin anstelle des Dimethylamins an das Sulfon gekoppelt.
In Zielverbindung B stellt der aromatische Teil von N,N-Dimethylphenylsulfonsäureamid den
variablen Bereich des Moleküls dar. Der unsubstituierte Phenylring wurde durch meta- und para-
substituierte Phenylringe, einen Benzylring sowie einen Cyclohexylring ersetzt.
2.1.2.1 Darstellung von Derivaten mit der Grundstruktur A
2.1.2.1.1 Syntheseweg zur Herstellung der Phenylsulfonsäureamide 1a-1p*
Für die Synthese von Verbindungen mit der Grundstruktur A eignete sich die Kopplung von
Phenylsulfonsäurechlorid mit dem entsprechenden Amin nach einer allgemeinen Vorschrift.76
Diese
„kleinen“ Sulfonsäureamide wurden auf klassische Weise in einer nukleophilen Substitutionsreaktion
(SN2-artig) hergestellt.76
Bei dieser Reaktion greift das freie Elektronenpaar des Amins als
Nukleophil das elektrophile Schwefelatom an, wobei unter Abspaltung von Salzsäure das
gewünschte Sulfonsäureamid entsteht. Die entstehende Salzsäure kann das Edukt (Amin) jedoch
bereits während der Reaktion unter Salzbildung ausfällen. Dies hat zur Folge, dass der gewünschten
Reaktion das nukleophile Amin entzogen und damit die Ausbeute des Produkts verringert wird. Aus
diesem Grund muss eine zusätzliche Base als Hilfsbase eingesetzt werden. Die Hilfsbase geht nun
anstelle des Amins die Salzbildung mit der Salzsäure ein und unterdrückt somit die unerwünschte
Nebenreaktion. Zur Darstellung der Zielverbindungen A wurde als Hilfsbase Triethylamin
verwendet. Die Synthese verlief nach folgendem Schema (siehe Abb. 29):
Abb. 29: Syntheseweg zur Herstellung der Verbindungen 1a-1p
* Alle gekennzeichneten Verbindungen im Allgemeinen Teil sind bereits in der Literatur beschrieben (Verweise
siehe Experimenteller Teil).
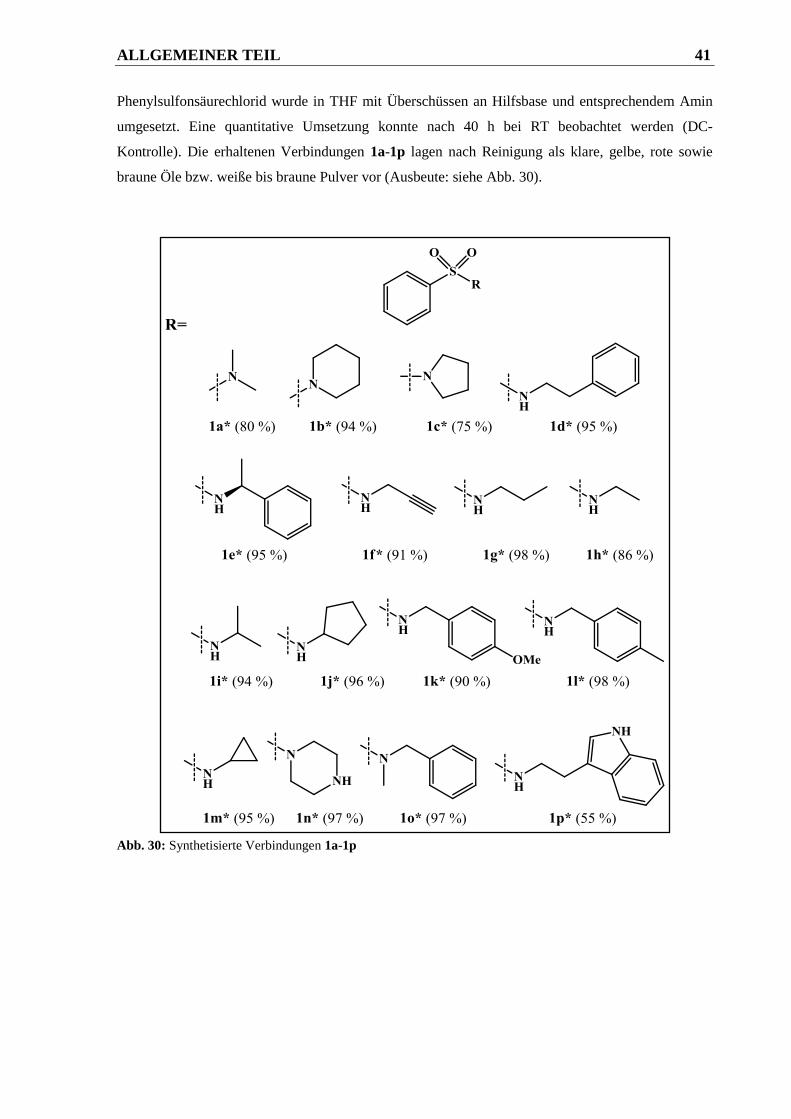
ALLGEMEINER TEIL 41
Phenylsulfonsäurechlorid wurde in THF mit Überschüssen an Hilfsbase und entsprechendem Amin
umgesetzt. Eine quantitative Umsetzung konnte nach 40 h bei RT beobachtet werden (DC-
Kontrolle). Die erhaltenen Verbindungen 1a-1p lagen nach Reinigung als klare, gelbe, rote sowie
braune Öle bzw. weiße bis braune Pulver vor (Ausbeute: siehe Abb. 30).
Abb. 30: Synthetisierte Verbindungen 1a-1p

42 ALLGEMEINER TEIL
2.1.2.1.2 Syntheseweg zur Herstellung der Phenylsulfonsäureamide 2a-2c
Die Ergebnisse der Docking-Untersuchungen zeigten, dass sich tertiäre Phenylsulfonsäureamide
vermutlich besser in die Mip-Kavität einlagern als sekundäre Amide. Zu diesem Zweck wurden
einige bereits hergestellte sekundäre Amide (Verbindungen 1g-1i) in einer klassischen nukleophilen
Substitutionsreaktion (SN2-artig) zum entsprechenden tertiären Amid umgesetzt (siehe Abb. 31).76
Das sekundäre Amid wurde dafür zuerst mit einer Base deprotoniert und anschließend mit
Methyliodid unter Säurefreisetzung (HI) gekoppelt. Bei dieser Reaktion musste keine weitere
Hilfsbase eingesetzt werden, da die entstandene Säure während der Reaktion durch die bereits
eingesetzte Base Kalium-t-butylat neutralisiert wurde.
Zur Synthese der Verbindungen 2a-2c wurden Verbindung 1g-1i jeweils in DMF mit Überschüssen
an Kalium-t-butylat und Methyliodid umgesetzt. Eine quantitative Umsetzung zum gewünschten
Produkt konnte nach 3 h bei RT beobachtet werden (DC-Kontrolle). Nach säulenchromato-
graphischer Reinigung lagen die erhaltenen Verbindungen 2a-2c als klare Öle vor (Ausbeute: siehe
Abb. 31).
Abb. 31: Syntheseweg zur Herstellung der Verbindungen 2a-2c
2.1.2.1.3 Versuche der Synthese von N-((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid
Neben den Verbindungen 1a-1p sowie 2a-2c lieferte das computergestützte Wirkstoffdesign auch N-
((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid (siehe Abb. 32), das auf Grund seiner
Oxazolidinonsubstruktur optimal den Raum der kleinen Mip-Seitentasche ausfüllen und mit den dort
vorliegenden Aminosäuren in Wechselwirkung treten können sollte.

ALLGEMEINER TEIL 43
Abb. 32: Strukturformel von N-((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid
Da lediglich 5-Chlormethyl-2-oxazolidinon kommerziell erworben werden konnte, war in diesem
Fall eine einstufige Synthese ausgehend von Phenylsulfonsäurechlorid nicht möglich. Deshalb wurde
eine andere Synthesestrategie entwickelt (siehe Abb. 33). Als Edukt wurde Phenylsulfonsäureamid
verwendet, das mit Hilfe einer geeigneten Base zuerst deprotoniert und anschließend über eine SN2-
artige Substitutionsreaktion mit 5-Chlormethyl-2-oxazolidinon unter Säureabspaltung gekoppelt
werden sollte.
Abb. 33: Syntheseweg zur Darstellung von N-((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid
Zur Deprotonierung wurden verschiedene Basen (siehe Tabelle 3) eingesetzt.
Syntheseweg Base Lösungsmittel Temperatur Molverhältnis [Sulfonamid/Chlorid/Base]
1 K2CO3 / KI DMF 100 °C 1 : 3 : 5
2 Kalium-t-butylat DMF RT 1 : 5 : 5
3 NaH THF RT 3 : 1 : 4
4 K2CO3 / Tetrabutyl-
ammoniumbromid Toluol 100 °C 1 : 1 : 1
Tabelle 3: Edukte und Reaktionsbedingungen für die Darstellung von N-((2-Oxo-oxazolidin-5-yl)-
methyl)benzylsulfonsäureamid
Phenylsulfonsäureamid wurde im entsprechenden Lösungsmittel vorgelegt und anschließend bei
einer definierten Temperatur mit der entsprechenden Base versetzt (siehe Tabelle 3). Nach 1 h wurde
5-Chlormethyl-2-oxazolidinon zugegeben und der Ansatz mehrere Stunden gerührt. Unter keiner der
angegebenen Reaktionsbedingungen fand eine Umsetzung zum gewünschten Endprodukt statt. Mit

44 ALLGEMEINER TEIL
Hilfe von DC- sowie NMR-Kontrollen konnte lediglich das Edukt Phenylsulfonsäureamid
identifiziert werden.
Auf Grund dieser „Nicht-Umsetzung“ sowie aus Kostengründen wurde Benzylchlorid anstelle von 5-
Chlormethyl-2-oxazolidinon für weitere Syntheseversuche herangezogen (siehe Abb. 34).
Abb. 34: Syntheseweg zur Herstellung von Verbindung 3
Zuerst wurde n-Butyllithium als sehr starke Base für die Deprotonierung von Phenylsulfonsäureamid
verwendet. Phenylsulfonsäureamid wurde dafür in THF gelöst und auf – 70 °C abgekühlt. Bei dieser
Temperatur wurde n-Butyllithium zugegeben und 10 min gerührt. Danach wurde der Ansatz auf RT
erwärmt und Benzylchlorid zugegeben. Eine chemische Umsetzung konnte nach 6 h bei RT
beobachtet werden (DC-Kontrolle). Um anschließend überschüssiges n-Butyllithium zu entfernen,
wurde H2O zugegeben und mit CHCl3 extrahiert. Nach säulenchromatographischer Reinigung lag
Verbindung 3 als weißes Öl vor (Ausbeute: 1 %).
Auf Grund der schlechten Ausbeute wurde die klassische Reaktion anschließend in einer
Synthesemikrowelle durchgeführt. Bei dieser Synthesevariante wurde anstelle von n-Butyllithium als
basische Komponente K2CO3 verwendet. Zusammen mit den beiden anderen Ausgangsstoffen wurde
die Base in Methanol gelöst und 3 h in der Mikrowelle erhitzt. Nach säulenchromatographischer
Reinigung konnte Verbindung 3 als weißes Pulver in einer Ausbeute von 22 % erhalten werden.
Diese zweite Variante wurde nun auf die Synthese von N-((2-Oxo-oxazolidin-5-yl)methyl)benzyl-
sulfonsäureamid übertragen, zeigte aber auch nach mehreren Tagen keinerlei Stoffumsatz (DC-
sowie NMR-Kontrolle).
Um die Verbindung N-((2-Oxo-oxazolidin-5-yl)methyl)benzylsulfonsäureamid auf dieselbe Weise
wie die Verbindungen 1a-1p, d.h. ausgehend von Phenylsulfonsäurechlorid, herzustellen, müsste 5-
Chlormethyl-2-oxazolidinon zunächst zum entsprechenden Amin umfunktionalisiert werden. Als
weitere Synthesestrategie zur Darstellung der Zielverbindung wäre deshalb eine Umfunktiona-
lisierung mit Kaliumphtalimid in Form einer Gabriel-Synthese und eine anschließende
Substitutionsreaktion von Phenylsulfonsäurechlorid und dem entstandenen 5-Aminomethyl-2-
oxazolidinon denkbar. Da für diese Reaktion jedoch nicht genügend 5-Chlormethyl-2-oxazolidinon
vorhanden war, wurde diese Strategie aus den bereits genannten Kostengründen nicht weiter verfolgt.
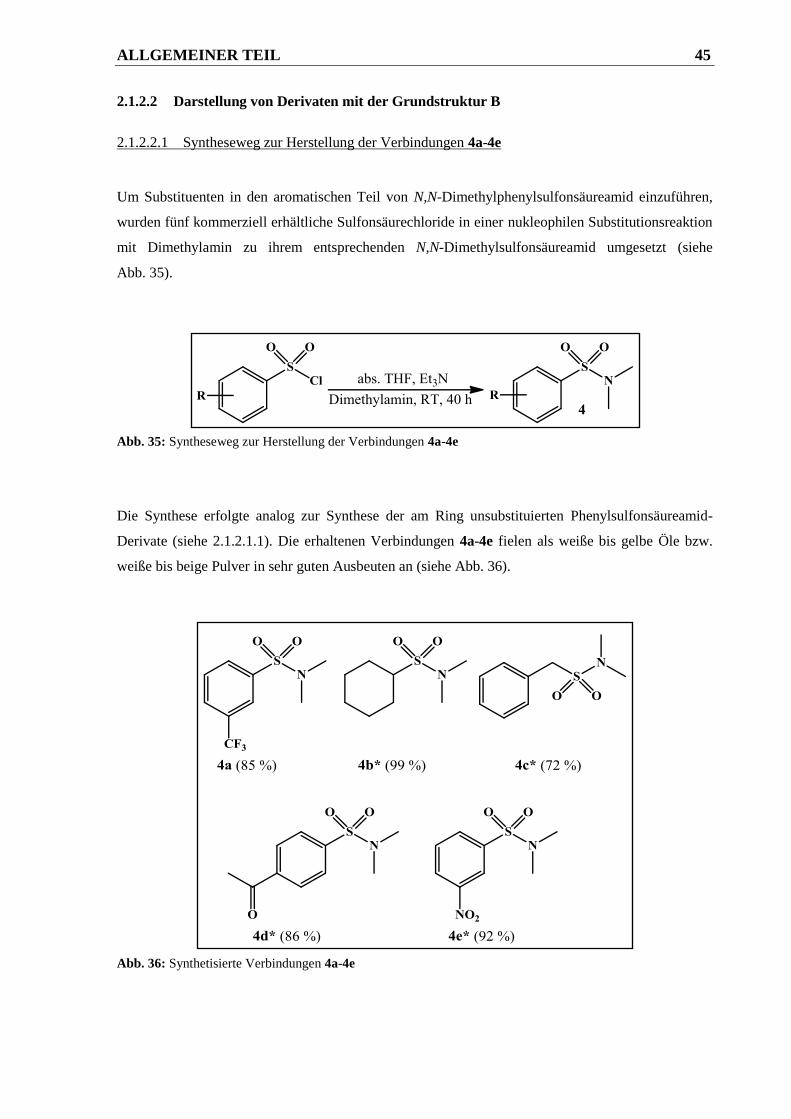
ALLGEMEINER TEIL 45
2.1.2.2 Darstellung von Derivaten mit der Grundstruktur B
2.1.2.2.1 Syntheseweg zur Herstellung der Verbindungen 4a-4e
Um Substituenten in den aromatischen Teil von N,N-Dimethylphenylsulfonsäureamid einzuführen,
wurden fünf kommerziell erhältliche Sulfonsäurechloride in einer nukleophilen Substitutionsreaktion
mit Dimethylamin zu ihrem entsprechenden N,N-Dimethylsulfonsäureamid umgesetzt (siehe
Abb. 35).
Abb. 35: Syntheseweg zur Herstellung der Verbindungen 4a-4e
Die Synthese erfolgte analog zur Synthese der am Ring unsubstituierten Phenylsulfonsäureamid-
Derivate (siehe 2.1.2.1.1). Die erhaltenen Verbindungen 4a-4e fielen als weiße bis gelbe Öle bzw.
weiße bis beige Pulver in sehr guten Ausbeuten an (siehe Abb. 36).
Abb. 36: Synthetisierte Verbindungen 4a-4e

46 ALLGEMEINER TEIL
2.1.2.2.2 Syntheseweg zur Herstellung von Verbindung 5
Für die Darstellung der meta-substituierten Phenylsulfonsäureamide wurde die Nitroverbindung 4e in
einem ersten Reaktionsschritt zur Aminoverbindung 5 reduziert, um anschließend alkyliert werden zu
können (siehe Abb. 37).
Abb. 37: Syntheseweg zur Herstellung von Verbindung 5
Die Reduktion wurde nach der Vorschrift von Bellamy et al. durchgeführt.77
Die Nitroverbindung 4e
wurde in Ethanol mit einem Überschuss an Zinn(II)chlorid x 2 H2O unter Rückfluss erhitzt. Eine
quantitative Umsetzung konnte nach 2 h beobachtet werden (DC-Kontrolle). Danach wurde H2O
zugegeben, bis sich das überschüssige Zinn(II)chlorid vollständig gelöst hatte, mit ges. NaHCO3-
Lösung auf pH 8 eingestellt und das primäre aromatische Amin mit Essigsäureethylester extrahiert.
Das beige Pulver (Verbindung 5) wurde in einer Ausbeute von 97 % erhalten.
2.1.2.2.3 Syntheseweg zur Herstellung der Verbindungen 6a-6e
Die reduktive Aminierung wurde nach der Vorschrift von Gribble et al. durchgeführt.78
Die
Kombination des Reduktionsmittels Natriumborhydrid mit einer organischen Säure ermöglicht die
einfache Alkylierung eines primären oder sekundären aromatischen Amins. Nach Gribble et al. kann
der Reaktionsverlauf durch eine Temperaturkontrolle zusätzlich in die Richtung der Mono- oder
Dialkylderivate gesteuert werden.78
Im Rahmen dieser Arbeit sollten daher nach folgendem Schema
(siehe Abb. 38) einfach alkylierte Phenylsulfonsäureamide hergestellt werden.
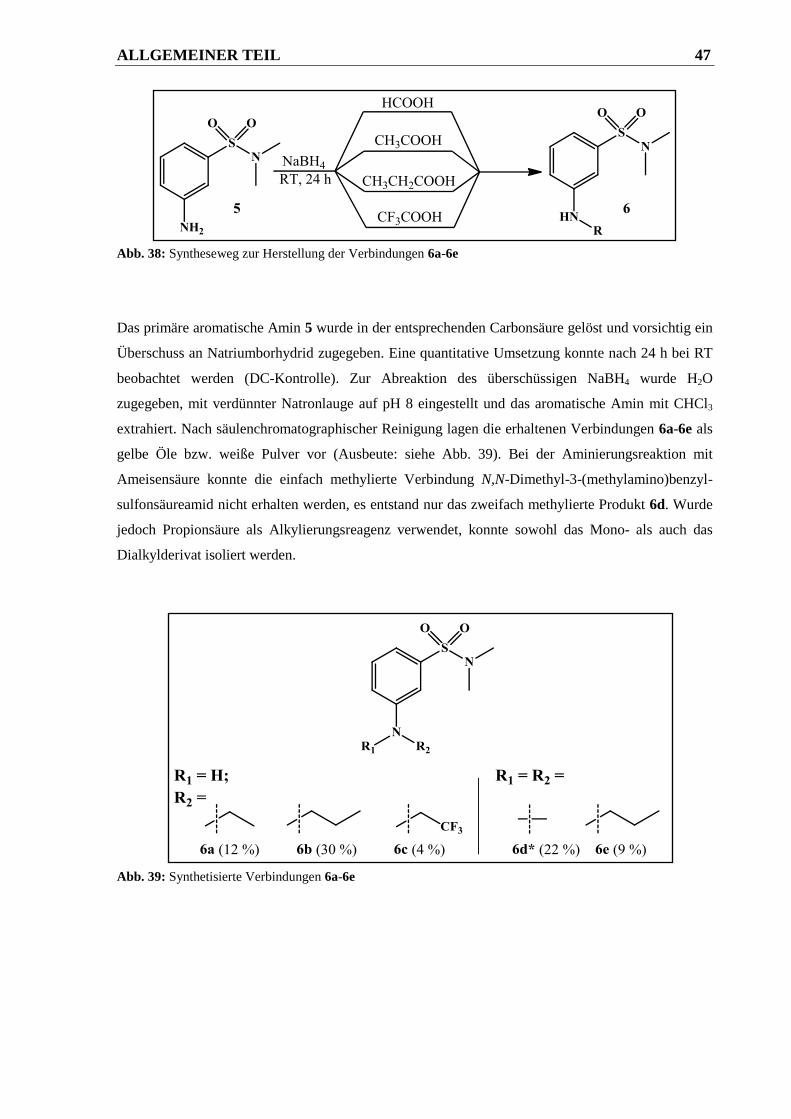
ALLGEMEINER TEIL 47
Abb. 38: Syntheseweg zur Herstellung der Verbindungen 6a-6e
Das primäre aromatische Amin 5 wurde in der entsprechenden Carbonsäure gelöst und vorsichtig ein
Überschuss an Natriumborhydrid zugegeben. Eine quantitative Umsetzung konnte nach 24 h bei RT
beobachtet werden (DC-Kontrolle). Zur Abreaktion des überschüssigen NaBH4 wurde H2O
zugegeben, mit verdünnter Natronlauge auf pH 8 eingestellt und das aromatische Amin mit CHCl3
extrahiert. Nach säulenchromatographischer Reinigung lagen die erhaltenen Verbindungen 6a-6e als
gelbe Öle bzw. weiße Pulver vor (Ausbeute: siehe Abb. 39). Bei der Aminierungsreaktion mit
Ameisensäure konnte die einfach methylierte Verbindung N,N-Dimethyl-3-(methylamino)benzyl-
sulfonsäureamid nicht erhalten werden, es entstand nur das zweifach methylierte Produkt 6d. Wurde
jedoch Propionsäure als Alkylierungsreagenz verwendet, konnte sowohl das Mono- als auch das
Dialkylderivat isoliert werden.
Abb. 39: Synthetisierte Verbindungen 6a-6e

48 ALLGEMEINER TEIL
2.1.2.3 Versuche der Synthese von Aminomethylphenylsulfon
Aminomethylphenylsulfon war ebenfalls ein Ergebnis des virtuellen Screenings. Ausschlaggebend
für die chemische Herstellung von Aminomethylphenylsulfon war die anschließend synthetisch leicht
zugängliche Vergrößerung des Moleküls über eine Kopplungsreaktion des primären Amins. Zuerst
sollte deshalb ausgehend von Phenylsulfonsäurechlorid und Nitromethan Aminomethylphenylsulfon
in einer SN2-artigen Substitutionsreaktion dargestellt werden. Dazu sollte Nitromethan mit einer
geeigneten Base zuerst deprotoniert und anschließend mit Phenylsulfonsäurechlorid unter
Säureabspaltung gekoppelt werden (siehe Abb. 40).
Abb. 40: Syntheseweg zur Darstellung von Aminomethylphenylsulfon
Zu diesem Zweck wurde Nitromethan zusammen mit einer starken Base bei RT im entsprechenden
Lösungsmittel gelöst und nach 15 min ein Äquivalent Phenylsulfonsäurechlorid zugegeben. Dieser
Ansatz wurde mehrere Stunden gerührt, wobei der Reaktionsverlauf dünnschichtchromatographisch
verfolgt wurde. Unter keiner der angegebenen Reaktionsbedingungen (siehe Tabelle 4) fand eine
Umsetzung zum gewünschten Endprodukt statt, so dass die anschließend geplante Reduktion mit
Zinn(II)chlorid gar nicht erst durchgeführt werden konnte.
Syntheseweg Base Lösungsmittel Molverhältnis [Chlorid/Nitromethan/Base]
1 Kalium-t-butylat DMF 1 : 10 : 10
2 KOH DMF 1 : 10 : 10
3 n-Butyllithium DMF 1 : 5 : 5
4 K2CO3 / KI DMF 1 : 5 : 10
5 NaH THF 1 : 3: 4
Tabelle 4: Edukte und Reaktionsbedingungen für die Darstellung von Nitromethylphenylsulfon
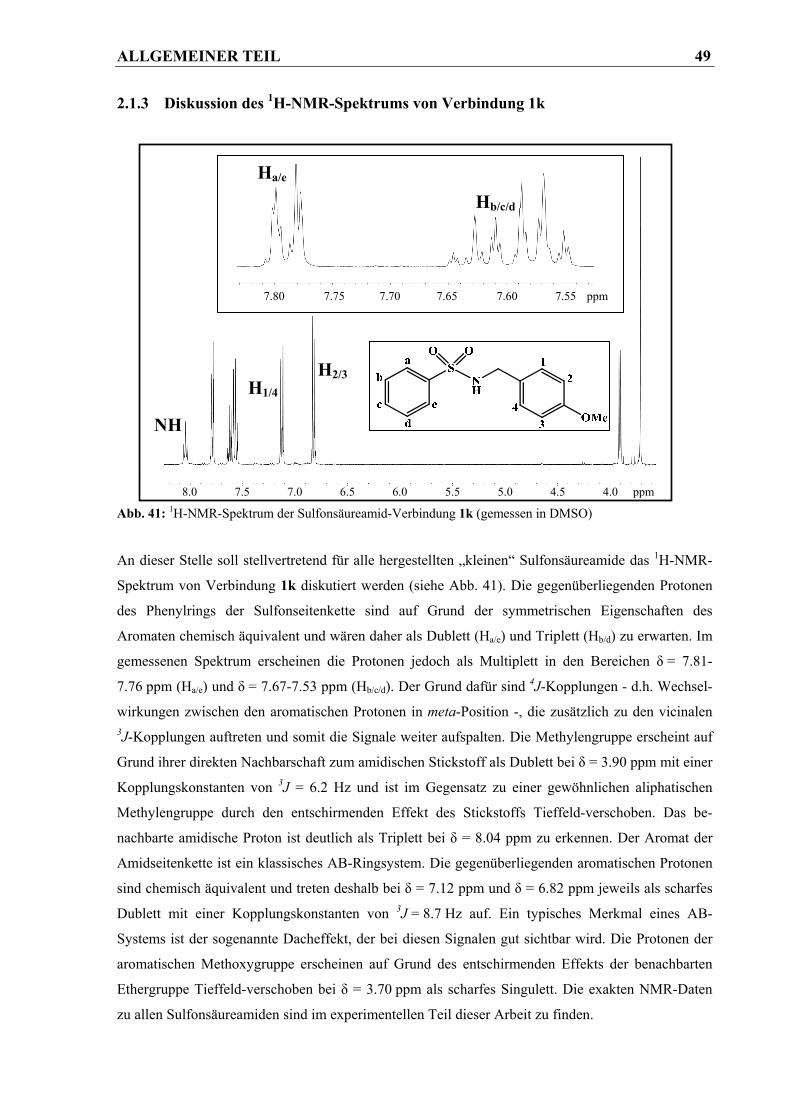
ALLGEMEINER TEIL 49
2.1.3 Diskussion des 1H-NMR-Spektrums von Verbindung 1k
Abb. 41: 1H-NMR-Spektrum der Sulfonsäureamid-Verbindung 1k (gemessen in DMSO)
An dieser Stelle soll stellvertretend für alle hergestellten „kleinen“ Sulfonsäureamide das 1H-NMR-
Spektrum von Verbindung 1k diskutiert werden (siehe Abb. 41). Die gegenüberliegenden Protonen
des Phenylrings der Sulfonseitenkette sind auf Grund der symmetrischen Eigenschaften des
Aromaten chemisch äquivalent und wären daher als Dublett (Ha/e) und Triplett (Hb/d) zu erwarten. Im
gemessenen Spektrum erscheinen die Protonen jedoch als Multiplett in den Bereichen δ = 7.81-
7.76 ppm (Ha/e) und δ = 7.67-7.53 ppm (Hb/c/d). Der Grund dafür sind 4J-Kopplungen - d.h. Wechsel-
wirkungen zwischen den aromatischen Protonen in meta-Position -, die zusätzlich zu den vicinalen 3J-Kopplungen auftreten und somit die Signale weiter aufspalten. Die Methylengruppe erscheint auf
Grund ihrer direkten Nachbarschaft zum amidischen Stickstoff als Dublett bei δ = 3.90 ppm mit einer
Kopplungskonstanten von 3J = 6.2 Hz und ist im Gegensatz zu einer gewöhnlichen aliphatischen
Methylengruppe durch den entschirmenden Effekt des Stickstoffs Tieffeld-verschoben. Das be-
nachbarte amidische Proton ist deutlich als Triplett bei δ = 8.04 ppm zu erkennen. Der Aromat der
Amidseitenkette ist ein klassisches AB-Ringsystem. Die gegenüberliegenden aromatischen Protonen
sind chemisch äquivalent und treten deshalb bei δ = 7.12 ppm und δ = 6.82 ppm jeweils als scharfes
Dublett mit einer Kopplungskonstanten von 3J = 8.7 Hz auf. Ein typisches Merkmal eines AB-
Systems ist der sogenannte Dacheffekt, der bei diesen Signalen gut sichtbar wird. Die Protonen der
aromatischen Methoxygruppe erscheinen auf Grund des entschirmenden Effekts der benachbarten
Ethergruppe Tieffeld-verschoben bei δ = 3.70 ppm als scharfes Singulett. Die exakten NMR-Daten
zu allen Sulfonsäureamiden sind im experimentellen Teil dieser Arbeit zu finden.
NH
H1/4
H2/3
7.8 0 7.75 7.70 7.65 7.60 7.55 ppm
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 ppm
Hb/c/d
Ha/e

50 ALLGEMEINER TEIL
2.2 Pipecolinsäure-Derivate
2.2.1 Docking-Untersuchungen von bekannten FKBP12-Inhibitoren
Die PPIase-Domänen der beiden Proteine (Mip und FKBP12) weisen sehr ähnliche hydrophobe
Bindetaschen auf. Auf Grund dessen kann die PPIase-Aktivität der beiden Proteine durch identische
Wirkstoffe gehemmt werden; potente Inhibitoren sind die zyklischen Makrolide Rapamycin und
FK506. Die Pipecolinsäure-Grundstruktur beider Wirkstoffe ist über dieselben Funktionalitäten tief
in der FKBP12- sowie der Mip-Bindetasche verankert (siehe Kapitel 1.3.3). Trotz dieser identischen
Wechselwirkungen bedingt die unterschiedliche geometrische Ausrichtung einzelner Aminosäuren -
Tyr109 in Mip bzw. Tyr82 in FKBP12 - im aktiven Zentrum der hydrophoben Bindetaschen, dass die
Pipecolinsäure-Grundstruktur eine andere Stellung in Mip als in FKBP12 einnimmt. Daher sollte
zunächst mit Hilfe verschiedener Docking-Studien die Lage der Pipecolinsäure-Struktur in Mip näher
untersucht und durch die so gewonnenen neuen Erkenntnisse neuartige, selektive Mip-Inhibitoren
vom Pipecolinsäure-Typ entwickelt werden.
Da für eine Mip-Inhibition die zusätzliche immunsuppressive Wirkung von Rapamycin bzw. FK506
unerwünscht ist, wurden für alle weiteren computergestützten Versuche nur Verbindungen
verwendet, die die Funktionalitäten der Bindedomäne - d.h. der Pipecolinsäure-Struktur -, aber nicht
die der Effektordomäne - d.h. der aliphatischen Kette des Makrozyklus - der Immunsuppressiva
enthalten (siehe Kapitel 1.2.2.2). In der Literatur sind bereits sehr viele solcher sogenannten nicht-
immunsuppressiven FKBP12-Inhibitoren beschrieben, von denen für die computergestützte
Wirkstoffanalyse zwei Moleküle ausgewählt wurden, die FKBP12 im unteren mikromolaren Bereich
inhibieren (1FKG-lig: Ki = 0.010 µM und MI-1: Ki = 0.23 µM) (siehe Abb. 42).
Abb. 42: Ausgewählte FKBP12-Inhibitoren für die computergestützte Wirkstoffanalyse58,63

ALLGEMEINER TEIL 51
Während Verbindung 1FKG-lig (A) die gleichen Strukturmerkmale der Bindedomäne von
Rapamycin beinhaltet, wurde die α-Keto-Amidstruktur in MI-1 (B) durch ein Sulfonsäureamid
ersetzt. Von Verbindung 1FKG-lig existierte bereits eine Struktur (1FKG)55
komplexiert mit
FKBP12. Der Vergleich der Bindemodi von 1FKG bzw. von Rapamycin im Komplex mit FKBP12
(1FKB, siehe Kapitel 1.3.2) zeigte bzgl. der Position der Pipecolinsubstruktur eine Übereinstimmung
von nahezu 100 %. Überträgt man diese FKBP12-Daten nun auf die Mip-Kavität, sollte 1FKG-lig in
ähnlicher Weise gebunden vorliegen wie Rapamycin im entsprechenden Mip-Komplex. Das Docking
dieser nicht-immunsuppressiven Struktur in Mip wurde mit dem Programm AutoDock 3.0 unter
Verwendung der Proteinstruktur 2VCD (siehe Kapitel 1.3.1) durchgeführt. Das in 2VCD gebundene
Rapamycin wurde zu diesem Zweck aus der Bindetasche entfernt und durch 1FKG-lig ersetzt. Die
Docking-Ergebnisse ergaben letztendlich zwar einen sehr guten Bindemodus für das Molekül, jedoch
stimmte er nur in einem geringen Maß mit dem Bindemodus von Rapamycin im Mip-Komplex
überein. Außerdem konnte dieser günstige Bindemodus auch nur in zwei von hundert Docking-
Versuchen reproduziert werden. Zusammenfassend lässt sich daher sagen, dass eine Bindung von
1FKG-lig an Mip zwar potenziell möglich, aber wohl eher unwahrscheinlich ist. Um dieses Ergebnis
zusätzlich noch durch einen pharmakologischen Assay zu bestätigen, wurde das Molekül
anschließend synthetisiert (siehe Kapitel 2.2.2.1).
Die Vorgehensweise bei den Docking-Studien von MI-1 verlief identisch zu den oben be-
schriebenen. Die Pipecolin-Sulfonsäureamidstruktur platzierte sich deutlich besser in die Bindetasche
von Mip als die α-Ketoamidstruktur von 1FKG-lig (siehe Abb. 43). Des Weiteren konnte sogar ein
besserer Bindemodus von MI-1 in der bakteriellen Bindetasche als in der von FKBP12 erzielt
werden. Dies gilt aber lediglich für das S-Enantiomer von MI-1. Das S-Isomer scheint sich bevorzugt
in die Kavität von Mip einzulagern, während das R-Enantiomer zwar auch, jedoch in einer anderen
Position gebunden, in der Bindetasche vorliegen kann. Zur Überprüfung dieser Ergebnisse sollte
MI-1 nun synthetisch als Racemat und als S-Enantiomer hergestellt werden (siehe Kapitel 2.2.2.2).
Abb. 43: Docking von MI-1 in die Mip-Bindetasche (2VCD); bestes Ergebnis aller möglichen Bindemodi;
(Wiedergabe nach Sippel58
bzw. nach Juli et al.63
mit Genehmigung des Autors bzw. der American Chemical
Society)

52 ALLGEMEINER TEIL
2.2.2 Darstellung der Pipecolinsäure-Derivate
2.2.2.1 Allgemeine Synthesestrategie zur Darstellung der Zielverbindung A
2.2.2.1.1 Syntheseweg zur Herstellung von Verbindung 18a
Die Synthese von Zielverbindung A wurde bereits 1993 von Holt et al. beschrieben und sollte nach
diesen Angaben durchgeführt werden.55
Diese Vorschrift startet mit der Umsetzung von 1-(2-Ethoxy-
2-oxoacetyl)piperidin-2-carbonsäureethylester 8 und 1,1-Dimethylpropylmagnesiumchlorid in Form
einer klassischen Grignard-Reaktion (siehe Abb. 44, rechtes Schema). Hierbei wird das elektrophile
Carbonyl-C-Atom des Ethylesters der Amidseitenkette von dem Grignard-Reagenz nukleophil
angegriffen. Unter Ethanolabspaltung entsteht nach Holt et al. anschließend der gewünschte α-
Ketoamid-Pipecolinsäureethylester.55
Um zu verhindern, dass die Reaktion möglicherweise über eine
nukleophile Addition bis zum tertiären Alkohol weiterläuft, wird das Grignard-Reagenz nur in einem
kleinen Überschuss eingesetzt.
Das von Holt et al. verwendete Edukt 8 war kommerziell nicht verfügbar und wurde zunächst über
eine zweistufige Synthese hergestellt (siehe Abb. 44).55
Als Ausgangsstoff für Verbindung 8 diente
die kommerziell erhältliche, racemische Pipecolinsäure, die nach Aktivierung mit Thionylchlorid
zum Pipecolinsäureethylester 7a umgesetzt wurde. Dazu wurde das Edukt in Ethanol mit einem
Überschuss an Thionylchlorid 4 h erhitzt, bis die Umsetzung quantitativ war (DC-Kontrolle) und der
Pipecolinsäureethylester 7a in Form eines gelben Öls erhalten wurde (Ausbeute: 90 %).
Dann folgte eine Amidierung von Verbindung 7a mit Monoethyloxalylchlorid in Gegenwart einer
Hilfsbase mittels einer SN2-artigen Substitutionsreaktion. Nach Vereinigung der beiden
Reaktionslösungen, bestehend aus Verbindung 7a in Dioxan sowie Monoethyloxalylchlorid und
Hilfsbase in Dioxan, konnte eine quantitative Umsetzung bei RT nach 6 h beobachtet werden (DC-
Kontrolle). Verbindung 8 lag als braunes Öl vor (Ausbeute: 94 %).
Nun sollte die von Holt et al. beschriebene Grignard-Reaktion durchgeführt werden.55
Bei der In-
situ-Herstellung des Grignard-Reagenzes 1,1-Dimethylpropylmagnesiumchlorid wurde das von Holt
verwendete Chlorid durch die bessere Abgangsgruppe Bromid ersetzt. Daher wurde 1,1-
Dimethylpropylbromid mit äquivalenten Mengen Magnesiumspänen in abs. THF suspendiert. Der
Ansatz wurde mit Iod versetzt und mehrere Stunden unter Rückfluss erhitzt, jedoch keine Umsetzung
beobachtet (DC-Kontrolle). Alternativ sollte deshalb das Grignard-Reagenz nach Mutule et al. durch
eine Mikrowellen-unterstützte Synthese hergestellt werden.79
Hierfür wurde ein Äquivalent der
Halogenkomponente mit 1.1 Äquivalenten Magnesiumspänen in abs. THF bei 80 °C umgesetzt.
Jedoch erfolgte auch nach einstündigem Erhitzen und einer zusätzlichen Zugabe von Jod in die
Reaktionslösung kein Stoffumsatz (DC-Kontrolle).
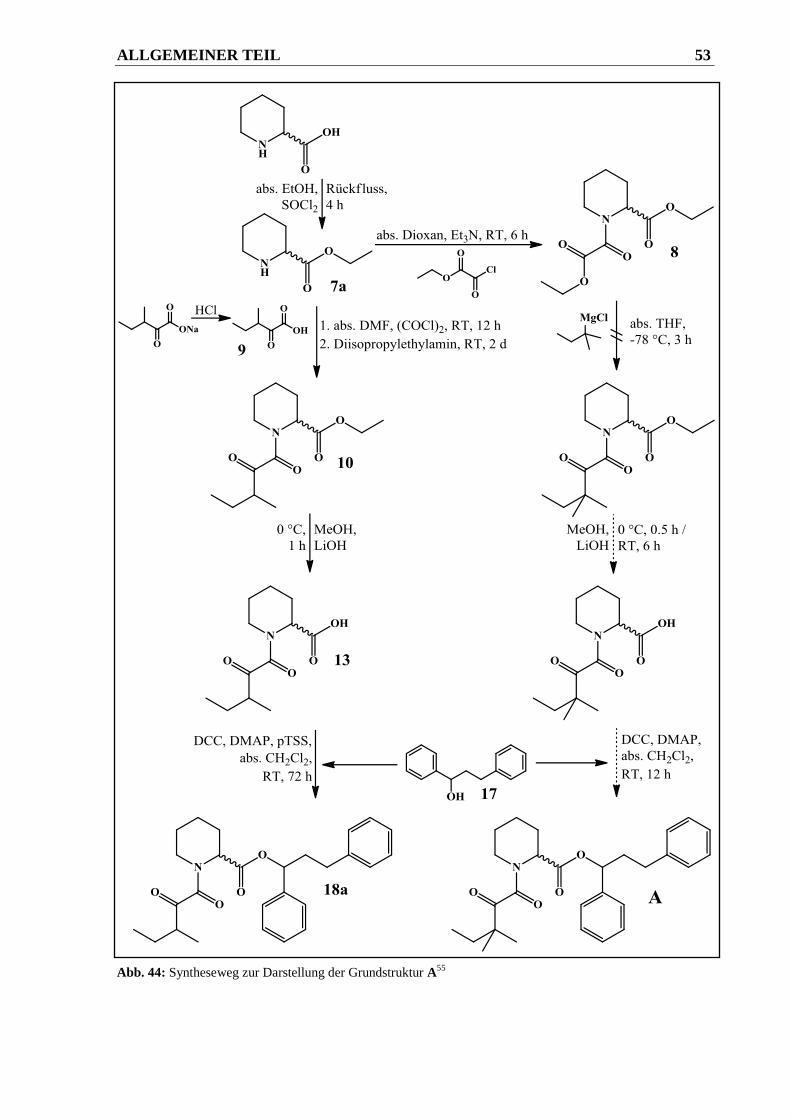
ALLGEMEINER TEIL 53
Abb. 44: Syntheseweg zur Darstellung der Grundstruktur A55

54 ALLGEMEINER TEIL
Die zweite Synthesestrategie war daher, die gewünschte Seitenkette am Pipecolin-Stickstoff
direkt durch eine Amidierung von Verbindung 7a mit 3,3-Dimethyl-2-oxovaleriansäure einzuführen.
Jedoch ist nur das Natriumsalz der 3-Methyl-2-oxovaleriansäure kommerziell erhältlich; dieses Salz
unterscheidet sich von 3,3-Dimethyl-2-oxovaleriansäure lediglich durch das Fehlen einer
Methylgruppe. Laut Rücksprache mit der Docking-Arbeitsgruppe sollte dieser geringe strukturelle
Unterschied keine Auswirkungen auf die Lage des Moleküls in der Mip-Bindetasche haben. Da das
Natriumsalz einer Säure unter normalen Bedingungen nicht direkt mit einem Amin umgesetzt werden
kann, musste zuerst die entsprechende Säure freigesetzt werden. Dafür wurde 3-Methyl-2-
oxovaleriansäure-Natriumsalz in H2O gelöst, konzentrierte Salzsäure bis pH 1 zugegeben und
anschließend mehrfach mit CHCl3 extrahiert (DC-Kontrolle). Die freigesetzte Säure 9 lag bei 4 °C in
Form weißer Nadeln vor (Ausbeute: 86 %).
Anschließend wurde Verbindung 9 zunächst mit Oxalylchlorid aktiviert und danach in Form einer
nukleophilen Substitutionsreaktion mit Verbindung 7a zum Amid umgesetzt. Dazu wurde
Verbindung 9 in DMF mit Oxalylchlorid 12 h bei RT gerührt. Danach wurde zuerst ein kleiner
Überschuss an N,N-Diisopropylethylamin und nach 5 min Verbindung 7a zugegeben. Eine
chemische Umsetzung konnte nach 1-2 d beobachtet werden (DC-Kontrolle). Nach säulen-
chromatographischer Reinigung wurde Verbindung 10 in Form eines gelben Öls mit einer Ausbeute
von 20 % erhalten.
Abgesehen von der fehlenden Methylgruppe in der Amidseitenkette entspricht Verbindung 10 dem
Zwischenprodukt 1-(3,3-Dimethyl-2-oxopentanoyl)piperidin-2-carbonsäureethylester der anfangs
angestrebten Synthese nach Holt, so dass die literaturbekannte Strategie ab diesem Zeitpunkt
weiterverfolgt werden konnte. Zunächst wurde Verbindung 10 in Methanol auf 0 °C abgekühlt und
mit einer LiOH-Lösung versetzt, bis die Umsetzung nach 1 h quantitativ war (DC-Kontrolle). Nach
Zugabe von verdünnter Salzsäure bis pH 1, wurde mehrfach mit CH2Cl2 extrahiert und schließlich
Verbindung 13 in Form eines gelben Öls erhalten (Ausbeute: 67 %).
Im nächsten Schritt wurde Verbindung 13 nach Steglich mit der gewünschten Alkoholkomponente
verestert.80
Als Reagenzien werden hierbei Dicyclohexylcarbodiimid (DCC) in geringem Überschuss
sowie Dimethylaminopyridin (DMAP) in katalytischer Menge verwendet. DCC wird im ersten
Schritt durch die eingesetzte Carbonsäure protoniert und anschließend durch eine nukleophile
Addition des entstandenen Carboxylat-Anions zum O-Acylisoharnstoff umgesetzt. Daraufhin greift
das freie Elektronenpaar der Hydroxygruppe der Alkoholkomponente am elektrophilen Carbonyl-C-
Atom des O-Acylisoharnstoffs an und es entsteht der gewünschte Ester sowie Dicyclohexylharnstoff.
Da Alkohole im Gegensatz zu Aminen schwache Nukleophile sind, können bei Reaktionen mit
Alkoholen N-Acylharnstoffe als Nebenprodukt entstehen. Um diese unerwünschte Nebenreaktion zu
unterdrücken, verwendet Steglich zusätzlich DMAP als O-Acyltransfer-Reagenz. Anstelle des
Alkohols greift dann das freie Elektronenpaar von DMAP die elektrophile Carbonylverbindung des
O-Acylisoharnstoff an und bildet unter Abspaltung von Dicyclohexylharnstoff ein reaktives Amid,

ALLGEMEINER TEIL 55
das wiederum nukleophil vom Alkohol angegriffen werden kann. Unter Abspaltung von DMAP
entsteht schließlich der gewünschte Ester.
Wie bei Standard-Veresterungsmethoden wurde bei der hier durchgeführten Steglich-Veresterung als
saurer Katalysator die organische p-Toluolsulfonsäure verwendet. Dazu wurden Verbindung 13, ein
kleiner Überschuss an 1,3-Diphenylpropan-1-ol 17 sowie die Reagenzien DCC, DMAP und p-
Toluolsulfonsäure in CH2Cl2 umgesetzt. Eine quantitative Umsetzung wurde nach 72 h bei RT
beobachtet (DC-Kontrolle). Anschließend wurde der Niederschlag von Dicyclohexylharnstoff
abfiltriert und das Lösungsmittel des Filtrats im Vakuum entfernt. Das Rohprodukt wurde in
Essigsäureethylester suspendiert, wobei sich erneut ein Niederschlag bildete, der wiederum abfiltriert
wurde. Der Vorgang des Fällens und Filtrierens wurde sooft wiederholt, bis sich nach
Essigsäureethylester-Zugabe kein Niederschlag mehr bildete. Nach säulenchromatographischer
Reinigung erhielt man Verbindung 18a in Form eines gelben Öls (Ausbeute: 30 %).
Aus Kostengründen wurde der Alkohol 17 für die Veresterung durch eine klassische Grignard-
Synthese selbst hergestellt (siehe Abb. 45).76
Abb. 45: Syntheseweg zur Herstellung von Verbindung 17
Hierzu wurden Magnesiumspäne in Diethylether mit 2-Phenylethylbromid solange zum Rückfluss
erhitzt, bis alle Magnesiumspäne abreagiert waren. Anschließend wurde auf RT erwärmt und
Benzaldehyd in Diethylether vorsichtig zugetropft. Danach wurde weitere 2 h unter Rückfluss erhitzt
und anschließend mit Eis hydrolysiert. Der entstandene Niederschlag wurde in verdünnter Salzsäure
gelöst und die organische Phase abgetrennt. Die wässrige Phase wurde mit Diethylether extrahiert
und die organischen Phasen vereinigt, um anschließend mehrmals mit NaHCO3-, NaHSO3-Lösung
und H2O gewaschen zu werden. Nach säulenchromatographischer Reinigung entstand Verbindung 17
in Form eines klaren Öls (Ausbeute: 61 %).

56 ALLGEMEINER TEIL
2.2.2.1.2 Syntheseweg zur Herstellung von Verbindung 18b
Neben den für die Docking-Untersuchungen ausgewählten Verbindungen A und B sollte auch eine
Mischform der beiden Grundstrukturen hergestellt werden, um mögliche Struktur-Wirkungs-
beziehungen aufstellen zu können. Aus diesem Grund sollte der Esterrest von Zielverbindung B mit
dem Grundkörper von Zielverbindung A gekoppelt werden (siehe Abb. 46).
Abb. 46: Strukturformel von 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäure-3-(3,4,5-trimethoxy-
phenyl)propylester
Zur Darstellung der Verbindung 18b wurde zuerst die Alkoholkomponente 16 aus der
entsprechenden Carbonsäure durch eine klassische Reduktion mit Lithiumaluminiumhydrid in
64 %iger Ausbeute hergestellt (siehe Abb. 47).76
Abb. 47: Syntheseweg zur Herstellung von Verbindung 16
Die Synthese von Verbindung 18b verlief, mit Ausnahme der eingesetzten Alkoholkomponente im
letzten Schritt, analog zu der von Verbindung 18a. Die Mischform 18b wurde in Form eines weißen
Öls in Ausbeuten von 37 % erhalten.

ALLGEMEINER TEIL 57
2.2.2.2 Allgemeine Synthesestrategie zur Darstellung der Zielverbindung B
2.2.2.2.1 Syntheseweg zur Herstellung von Verbindung 22a
Im Gegensatz zu Zielverbindung A existiert in der Literatur keine allgemeine Vorschrift für die
Herstellung der Grundstruktur B. Aus diesem Grund wurde folgendes Schema entwickelt (siehe Abb.
48). Ausgangsstoff war auch in diesem Fall die racemische Pipecolinsäure, die, wie oben beschrieben
(siehe Kapitel 2.2.2.1.1), im ersten Reaktionsschritt zum Pipecolinsäureethylester 7a umgesetzt
wurde. Danach wurde der Piperidin-Stickstoff in einer SN2-artigen Reaktion mit
Benzylsulfonsäurechlorid in Gegenwart einer Hilfsbase amidiert. Dazu wurde Verbindung 7a in
Dioxan zunächst mit einem Überschuss an N,N-Diisopropylethylamin sowie nach 10 min mit
Benzylsulfonsäurechlorid umgesetzt. Eine quantitative Umsetzung konnte nach 4.5 h bei RT
beobachtet werden (DC-Kontrolle). Nach säulenchromatographischer Reinigung entstand
Verbindung 12a in Form eines gelben Öls (Ausbeute: 77 %).
Durch basische Esterhydrolyse mit LiOH entstand im Anschluss 1-(Benzylsulfonyl)piperidin-2-
carbonsäure 15a als gelbes Öl (Ausbeute: 92 %) und durch Steglich-Veresterung schließlich die
Zielstruktur 22a als gelbes Wachs (Ausbeute: 48 %). Die genauen Synthesevorschriften wurden
bereits in Kapitel 2.2.2.1.1 beschrieben.
Abb. 48: Syntheseweg zur Darstellung der Grundstruktur B

58 ALLGEMEINER TEIL
2.2.2.2.2 Syntheseweg zur Herstellung von Verbindung 22b
Da die Docking-Studien eindeutig das S-Enantiomer (MI-1) von Grundstruktur B als optimalen
Bindungspartner der Mip-Bindetasche identifizierten, sollte dieses Enantiomer ebenfalls hergestellt
werden. Die Synthese wurde in Analogie zur Herstellung von Verbindung 22a durchgeführt. Als
Ausgangspunkt wurde in diesem Fall die enantiomerenreine, kommerziell erhältliche S-Pipecolin-
säure verwendet. Der erste Schritt lieferte Verbindung 7b als gelbes Öl mit einer Ausbeute von 91 %.
Die anschließende Amidierung mit N,N-Diisopropylethylamin als Hilfsbase zeigte im Fall des S-
Pipecolinsäureethylesters 7b jedoch auch nach mehreren Versuchen nicht die gewünschte Umsetzung
(DC- bzw. NMR-Kontrolle). Deshalb wurden sowohl das Lösungsmittel als auch die zugesetzte
Hilfsbase variiert. Bei Verwendung von Kaliumcarbonat und DMF konnte schließlich eine
quantitative Umsetzung nach 24 h bei RT erzielt (DC-Kontrolle) und Verbindung 12b in Form eines
gelben Öls mit einer Ausbeute von 63 % erhalten werden. Die anschließende basische Esterhydrolyse
sowie die Steglich-Veresterung lieferten die Zwischenstufe 15b als gelbes Öl in sehr guter Ausbeute
(99 %) sowie die Endstufe 22b in Form eines gelben Wachses (Ausbeute: 21 %).
2.2.2.2.3 Bestimmung der Enantiomerenreinheit von Verbindung 22b
Im Verlauf einer chemischen Reaktion mit reinen Enantiomeren chiraler Verbindungen kann es unter
bestimmten Bedingungen zu einer Racemisierung kommen. Auch im Fall der Herstellung des
enantiomerenreinen Sulfonsäureamid-Derivats 22b ist es möglich, dass eine solche Racemisierungs-
reaktion stattgefunden hat. Das Stereozentrum des Pipecolinsäure-Derivats 22b ist durch die
Aktivierung des Esters und der Amin-Funktion in α-Position so stark acidifiziert, dass durch
Deprotonierung und Reprotonierung eine Isomerisierung erfolgen kann. Diese Racemisierung könnte
an zwei verschiedenen Stellen während der beschriebenen Synthese eintreten: Zum einen bei der
Veresterung mit Thionylchlorid und zum anderen bei der basischen Esterhydrolyse mit LiOH.
Während eine Deprotonierung des aciden Protons bei der Aktivierung mit Thionylchlorid auf Grund
der fehlenden Base eher unwahrscheinlich ist,81
findet die Racemisierung mit hoher
Wahrscheinlichkeit im basischen Milieu während der Esterhydrolyse statt. Um diese Hypothese zu
überprüfen bzw. zu widerlegen, wurde daher die Enantiomerenreinheit von Verbindung 22b im
Arbeitskreis von Prof. Dr. Dr. Gerhard Bringmann von Claudia Steinert ermittelt.
Zur Bestimmung des Enantiomerenverhältnisses eignen sich neben spektrometrischen Methoden wie
der Polarimetrie oder elektrophoretischen Techniken wie der Kapillarelektrophorese, besonders
chromatographische Methoden wie die Hochleistungsflüssigkeitschromatographie (HPLC). Bei
letzterem gibt es drei verschiedene Trennprinzipien: 1. die Herstellung diastereomerer Derivate durch
Verwendung optisch aktiver enantiomerenreiner Reagenzien und anschließende achirale Trennung,
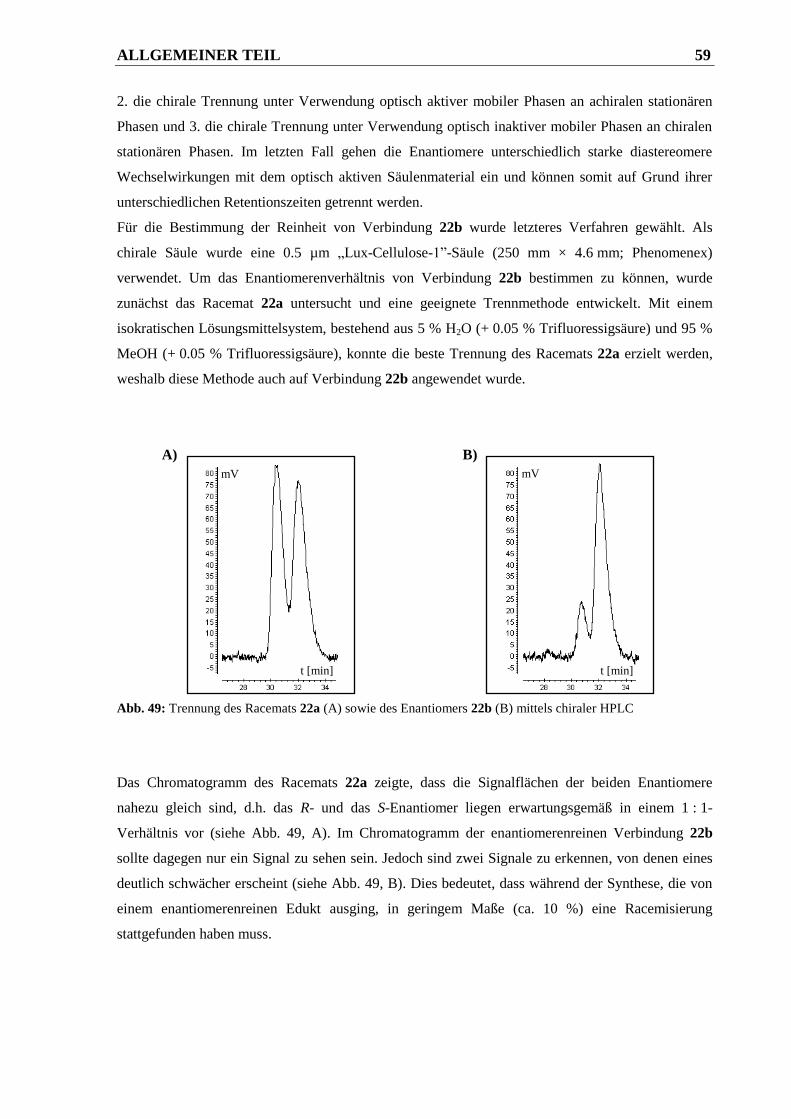
ALLGEMEINER TEIL 59
2. die chirale Trennung unter Verwendung optisch aktiver mobiler Phasen an achiralen stationären
Phasen und 3. die chirale Trennung unter Verwendung optisch inaktiver mobiler Phasen an chiralen
stationären Phasen. Im letzten Fall gehen die Enantiomere unterschiedlich starke diastereomere
Wechselwirkungen mit dem optisch aktiven Säulenmaterial ein und können somit auf Grund ihrer
unterschiedlichen Retentionszeiten getrennt werden.
Für die Bestimmung der Reinheit von Verbindung 22b wurde letzteres Verfahren gewählt. Als
chirale Säule wurde eine 0.5 µm „Lux-Cellulose-1”-Säule (250 mm × 4.6 mm; Phenomenex)
verwendet. Um das Enantiomerenverhältnis von Verbindung 22b bestimmen zu können, wurde
zunächst das Racemat 22a untersucht und eine geeignete Trennmethode entwickelt. Mit einem
isokratischen Lösungsmittelsystem, bestehend aus 5 % H2O (+ 0.05 % Trifluoressigsäure) und 95 %
MeOH (+ 0.05 % Trifluoressigsäure), konnte die beste Trennung des Racemats 22a erzielt werden,
weshalb diese Methode auch auf Verbindung 22b angewendet wurde.
A) B)
Abb. 49: Trennung des Racemats 22a (A) sowie des Enantiomers 22b (B) mittels chiraler HPLC
Das Chromatogramm des Racemats 22a zeigte, dass die Signalflächen der beiden Enantiomere
nahezu gleich sind, d.h. das R- und das S-Enantiomer liegen erwartungsgemäß in einem 1 : 1-
Verhältnis vor (siehe Abb. 49, A). Im Chromatogramm der enantiomerenreinen Verbindung 22b
sollte dagegen nur ein Signal zu sehen sein. Jedoch sind zwei Signale zu erkennen, von denen eines
deutlich schwächer erscheint (siehe Abb. 49, B). Dies bedeutet, dass während der Synthese, die von
einem enantiomerenreinen Edukt ausging, in geringem Maße (ca. 10 %) eine Racemisierung
stattgefunden haben muss.
mV mV
t [min] t [min]

60 ALLGEMEINER TEIL
2.2.2.3 Allgemeine Synthesestrategie zur Darstellung von Pipecolinsäure-Derivaten mit der
Grundstruktur B
Die Verbindungen 18a und 18b sowie die Verbindungen 22a und 22b wurden zur Bestätigung der
Docking-Ergebnisse zunächst in einem pharmakologischen Enzym-Assay (siehe Kapitel 3.1) auf ihre
potenzielle PPIase-Inhibition untersucht. Da die Resultate dieser pharmakologischen Testungen mit
den Ergebnissen der computergestützten Wirkstoffanalyse übereinstimmten, sollten anschließend
weitere Derivate mit der Grundstruktur B hergestellt werden (siehe Abb. 50).
Abb. 50: Grundstruktur der Pipecolinsäure-Derivate
Strukturelle Variationen sollten sowohl über die Ester-, als auch über die Sulfonsäureamid-
seitenkette in den Grundkörper eingeführt werden. Aus diesem Grund wurde zuerst der
Benzylsulfonsäurerest des Pipecolinsäureethylesters durch substituierte Benzylsulfonsäure- (m = 1),
substituierte sowie die unsubstituierte Phenylsulfonsäure- (m = 0) und diverse Benzylcarbonsäure-
Gruppen (m = 1) ersetzt. Diese verschiedenen Pipecolinsäureethylester wurden anschließend
willkürlich mit Alkoholkomponenten versetzt, die über einen Alkylspacer unterschiedlicher Länge an
ein aromatisches System gebunden waren. Durch die Einführung dieser verschiedenen Alkoholreste
sollte überprüft werden, welche Rolle der Abstand zwischen Piperidinring und aromatischem System
für eine Mip-Inhibition spielt.
Die Synthesen aller nachfolgenden Verbindungen verliefen nach demselben Schema wie die
Darstellung von Verbindung 22a (siehe Abb. 48 / Kapitel 2.2.2.2.1). Lediglich die Hilfsbasen,
Lösungsmittel oder die Reaktionsdauer variierten im zweiten Reaktionsschritt dieser Vorschrift
(siehe Experimenteller Teil). Nach der vierstufigen Synthese entstanden die Verbindungen 19-29
(siehe Tabelle 5, Tabelle 6 und Tabelle 7) in guten bis sehr guten Ausbeuten.
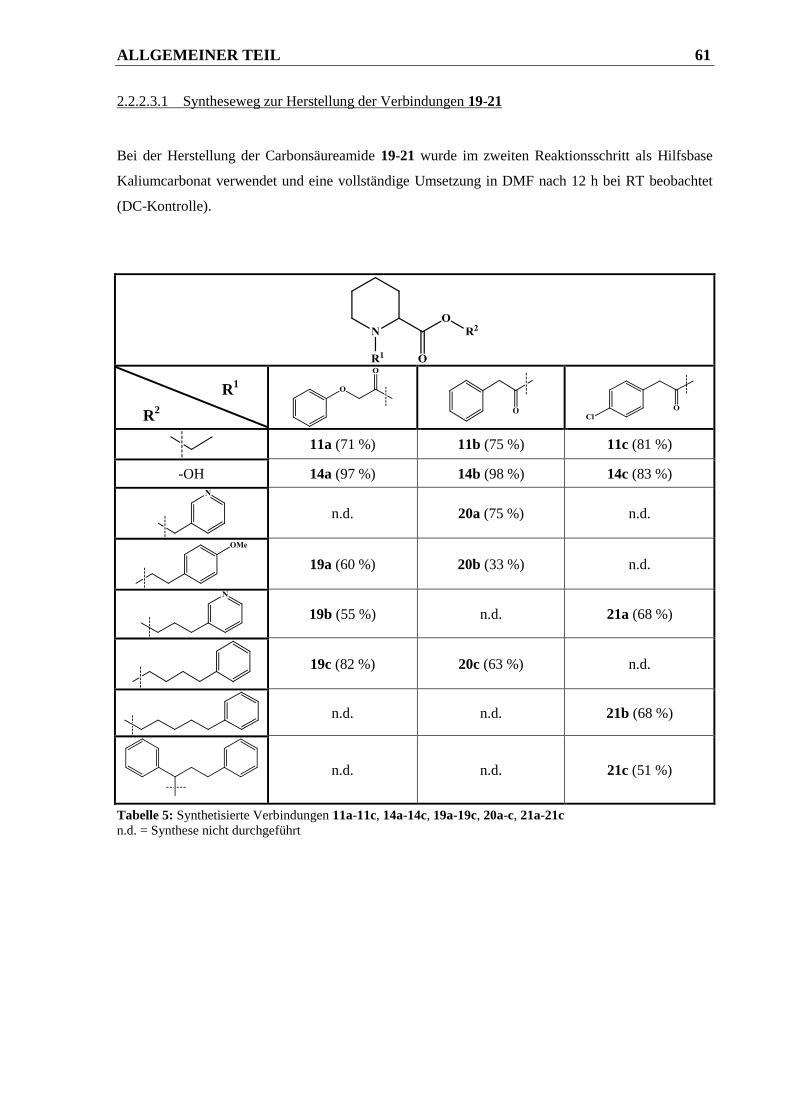
ALLGEMEINER TEIL 61
2.2.2.3.1 Syntheseweg zur Herstellung der Verbindungen 19-21
Bei der Herstellung der Carbonsäureamide 19-21 wurde im zweiten Reaktionsschritt als Hilfsbase
Kaliumcarbonat verwendet und eine vollständige Umsetzung in DMF nach 12 h bei RT beobachtet
(DC-Kontrolle).
11a (71 %) 11b (75 %) 11c (81 %)
-OH 14a (97 %) 14b (98 %) 14c (83 %)
n.d. 20a (75 %) n.d.
19a (60 %) 20b (33 %) n.d.
19b (55 %) n.d. 21a (68 %)
19c (82 %) 20c (63 %) n.d.
n.d. n.d. 21b (68 %)
n.d. n.d. 21c (51 %)
Tabelle 5: Synthetisierte Verbindungen 11a-11c, 14a-14c, 19a-19c, 20a-c, 21a-21c
n.d. = Synthese nicht durchgeführt
R1
R
2
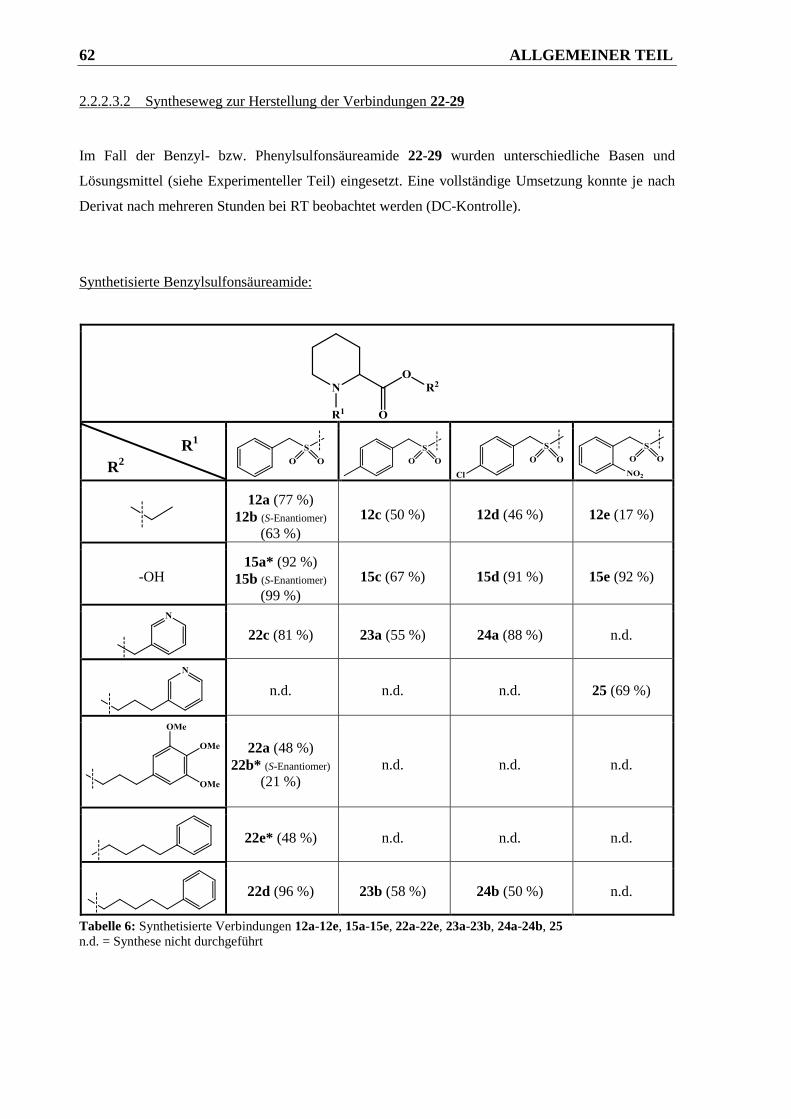
62 ALLGEMEINER TEIL
2.2.2.3.2 Syntheseweg zur Herstellung der Verbindungen 22-29
Im Fall der Benzyl- bzw. Phenylsulfonsäureamide 22-29 wurden unterschiedliche Basen und
Lösungsmittel (siehe Experimenteller Teil) eingesetzt. Eine vollständige Umsetzung konnte je nach
Derivat nach mehreren Stunden bei RT beobachtet werden (DC-Kontrolle).
Synthetisierte Benzylsulfonsäureamide:
12a (77 %)
12b (S-Enantiomer) (63 %)
12c (50 %) 12d (46 %) 12e (17 %)
-OH 15a* (92 %)
15b (S-Enantiomer) (99 %)
15c (67 %) 15d (91 %) 15e (92 %)
22c (81 %) 23a (55 %) 24a (88 %) n.d.
n.d. n.d. n.d. 25 (69 %)
22a (48 %)
22b* (S-Enantiomer) (21 %)
n.d. n.d. n.d.
22e* (48 %) n.d. n.d. n.d.
22d (96 %) 23b (58 %) 24b (50 %) n.d.
Tabelle 6: Synthetisierte Verbindungen 12a-12e, 15a-15e, 22a-22e, 23a-23b, 24a-24b, 25
n.d. = Synthese nicht durchgeführt
R1
R2
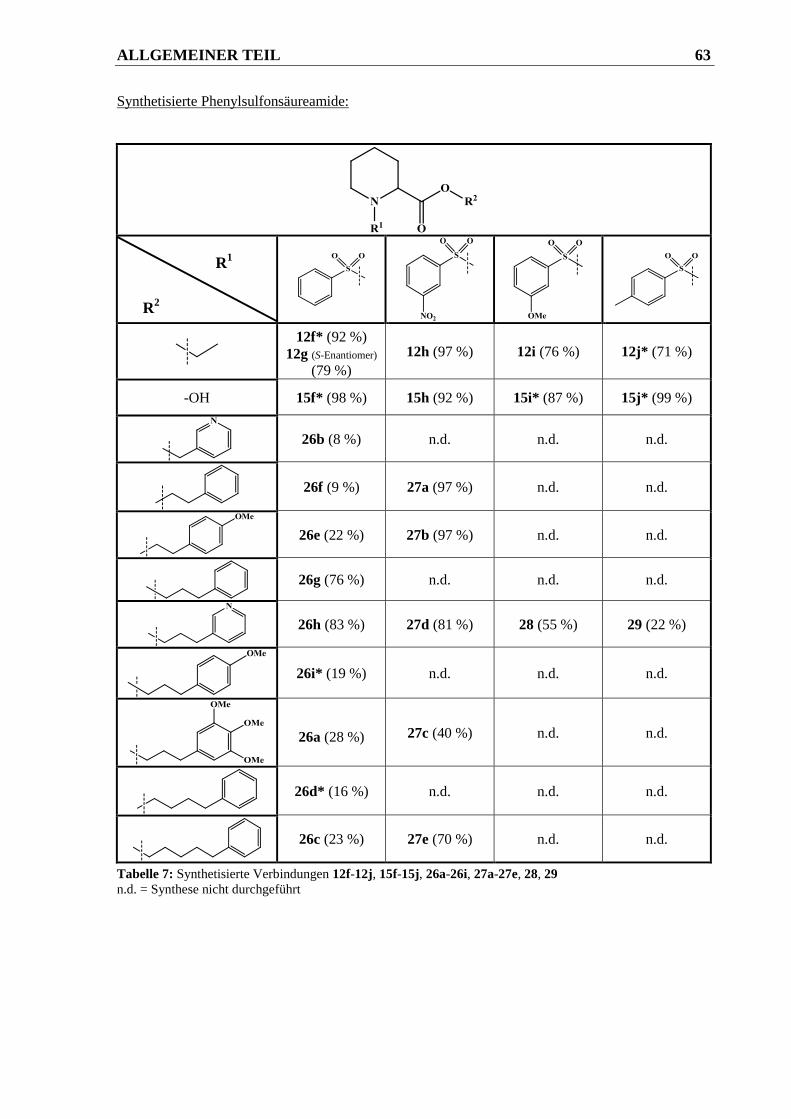
ALLGEMEINER TEIL 63
Synthetisierte Phenylsulfonsäureamide:
12f* (92 %)
12g (S-Enantiomer) (79 %)
12h (97 %) 12i (76 %) 12j* (71 %)
-OH 15f* (98 %) 15h (92 %) 15i* (87 %) 15j* (99 %)
26b (8 %) n.d. n.d. n.d.
26f (9 %) 27a (97 %) n.d. n.d.
26e (22 %) 27b (97 %) n.d. n.d.
26g (76 %) n.d. n.d. n.d.
26h (83 %) 27d (81 %) 28 (55 %) 29 (22 %)
26i* (19 %) n.d. n.d. n.d.
26a (28 %) 27c (40 %) n.d. n.d.
26d* (16 %) n.d. n.d. n.d.
26c (23 %) 27e (70 %) n.d. n.d.
Tabelle 7: Synthetisierte Verbindungen 12f-12j, 15f-15j, 26a-26i, 27a-27e, 28, 29
n.d. = Synthese nicht durchgeführt
R1
R2

64 ALLGEMEINER TEIL
2.2.2.4 Derivatisierungen am Aromaten der Sulfonsäureamidgruppe
Ein weiteres Ergebnis der Docking-Studien der Grundstruktur B
war, dass sich innerhalb der Mip-Kavität eine kleine Seitentasche
befindet, die eventuell durch große Wasserstoffbrücken-ausbildende
Substituenten der Amidseitenkette der Pipecolinsäure-Sulfonamide
adressiert werden kann, und zwar durch einen Wasserstoff-Akzeptor
in meta-Position des Benzylrings (siehe Abb. 51). Zur Einführung
geeigneter Substituenten, sollten die bereits hergestellten meta-
Nitro-Derivate 12h, 27c und 27d zuerst zum primären aromatischen
Amin reduziert und anschließend diese Aminofunktionen mit
verschiedenen Wasserstoff-Akzeptoren funktionalisiert werden.
2.2.2.4.1 Syntheseweg zur Herstellung der Verbindungen 30a-30c
Die Reduktion von primären aromatischen Nitrogruppen mit Zinn(II)chlorid wurde bereits in Kapitel
2.1.2.2.2 beschrieben und daher in Analogie zur Synthese von 3-Amino-N,N-dimethylphenyl-
sulfonsäureamid 5 durchgeführt (siehe Abb. 52). Die Verbindungen 30a-30c wurden als braunes Öl
bzw. gelbe Wachse in sehr guten Ausbeuten erhalten (siehe Abb. 52).
Abb. 52: Syntheseweg zur Darstellung der Verbindungen 30a-30c
Abb. 51: Komplex von
Grundstruktur B in der Mip-
Kavität mit zusätzlich zu
adressierender Seitentasche58,63
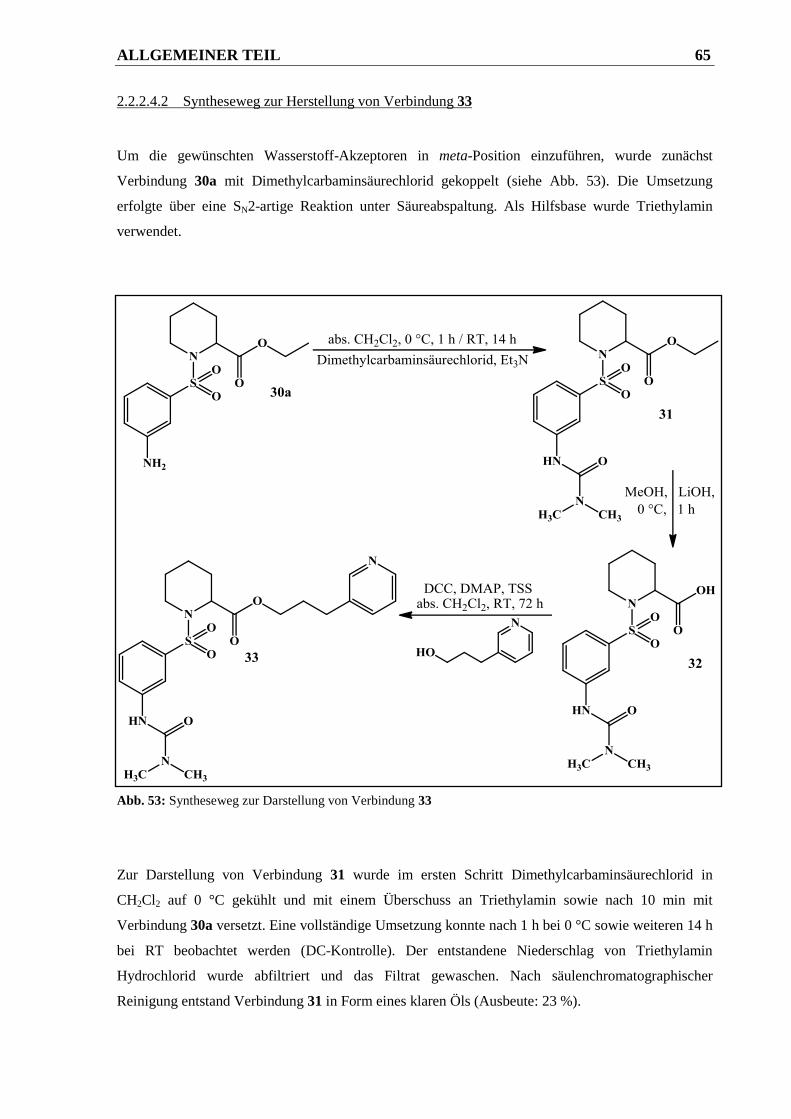
ALLGEMEINER TEIL 65
2.2.2.4.2 Syntheseweg zur Herstellung von Verbindung 33
Um die gewünschten Wasserstoff-Akzeptoren in meta-Position einzuführen, wurde zunächst
Verbindung 30a mit Dimethylcarbaminsäurechlorid gekoppelt (siehe Abb. 53). Die Umsetzung
erfolgte über eine SN2-artige Reaktion unter Säureabspaltung. Als Hilfsbase wurde Triethylamin
verwendet.
Abb. 53: Syntheseweg zur Darstellung von Verbindung 33
Zur Darstellung von Verbindung 31 wurde im ersten Schritt Dimethylcarbaminsäurechlorid in
CH2Cl2 auf 0 °C gekühlt und mit einem Überschuss an Triethylamin sowie nach 10 min mit
Verbindung 30a versetzt. Eine vollständige Umsetzung konnte nach 1 h bei 0 °C sowie weiteren 14 h
bei RT beobachtet werden (DC-Kontrolle). Der entstandene Niederschlag von Triethylamin
Hydrochlorid wurde abfiltriert und das Filtrat gewaschen. Nach säulenchromatographischer
Reinigung entstand Verbindung 31 in Form eines klaren Öls (Ausbeute: 23 %).

66 ALLGEMEINER TEIL
Diese Synthese sollte daraufhin auf die Verbindungen 30b und 30c übertragen werden. Jedoch zeigte
sich im Fall dieser „größeren“ Pipecolinsäureester 30b und 30c keine Umsetzung zum gewünschten
Endprodukt, so dass der Pipecolinsäureethylester 31 zunächst mit LiOH basisch zu seiner
entsprechenden Carbonsäure 32 hydrolysiert wurde (Ausbeute: 97 %). Anschließend lieferte die
Steglich-Veresterung von Verbindung 32 mit 3-(Pyridin-3-yl)propan-1-ol das gewünschte End-
produkt 33 als weißes Öl in einer Ausbeute von 97 %.
2.2.2.4.3 Syntheseweg zur Herstellung der Verbindungen 37a und 37b
Herstellung der Verbindungen 34a-34c
Eine weitere Strategie zur Einführung von Wasserstoff-Akzeptoren in meta-Position des Benzylrings
war die Einführung von diversen Aminosäuren. Um ein primäres aromatisches Amin mit einer
Aminosäure ohne unerwünschte Nebenreaktionen zum Amid umzusetzen, muss die NH2-Gruppe der
Aminosäure zuvor chemisch geschützt werden. In der Peptidsynthese werden zu diesem Zweck
gewöhnlich Säureanhydride, wie z.B. Di-tert-butyldicarbonat (BOC) bzw. Säurechloride, wie z.B. 9-
Fluorenylmethoxycarbonylchlorid (FMOC) verwendet. Diese bilden mit der Aminofunktion über
eine SN2-artige Reaktion Carbamate, die sich leicht unter Bildung von Kohlendioxid wieder spalten
lassen. Im Rahmen dieser Arbeit sollten 4-Aminobuttersäure, 3-Aminopropionsäure und Glycin mit
der BOC-Gruppe nach dem oberen Syntheseschema (siehe Abb. 54) geschützt werden.
Abb. 54: Syntheseweg zur Darstellung der Verbindungen 34a-34c
Die Einführung der Schutzgruppe wurde nach der Vorschrift von Ponnusamy et al. durchgeführt.82
Dazu wurde die jeweilige Aminosäure in Methanol mit einer methanolischen Triethylamin-Lösung
10 min unter Rückfluss erhitzt. Anschließend wurde das Schutzgruppen-Reagenz (BOC)2O

ALLGEMEINER TEIL 67
zugegeben und solange unter Rückfluss erhitzt, bis sich alle Bestandteile komplett gelöst hatten.
Nach Entfernen des Lösungsmittels im Vakuum wurde der ölige Rückstand in verdünnter Salzsäure
(pH 2) bei 0 °C gelöst, 10 min gerührt und anschließend diese wässrige Phase mehrfach mit
Essigsäureethylester extrahiert. Die Verbindungen 34a und 34b entstanden als klare Öle in guten bis
sehr guten Ausbeuten (siehe Abb. 55). Bei dieser Reaktion konnten lediglich die geschützte 3-
Aminopropionsäure 34a sowie die geschützte 4-Aminobuttersäure 34b isoliert werden, im Fall von
Glycin war keine Umsetzung mit (BOC)2O zu erkennen (DC- bzw. NMR-Kontrolle).
Abb. 55: Synthetisierte Verbindungen 34a-34b
Aus diesem Grund wurde eine neue Synthesestrategie eingeschlagen. Die Umsetzung von Glycin mit
(BOC)2O erfolgte in einer Synthesemikrowelle. Dazu wurde die Aminosäure in H2O mit einem
kleinen Überschuss an NaOH-Plätzchen sowie einer Lösung von (BOC)2O in THF versetzt. Eine
quantitative Umsetzung konnte nach 80 min bei 70 °C beobachtet werden (DC-Kontrolle).
Anschließend wurde verdünnte Salzsäure bis pH 2.5 zugegeben, weitere 30 min bei RT gerührt und
danach mehrfach mit CHCl3 extrahiert. Nach Umkristallisation aus Cyclohexan wurde Verbindung
34c als weißer Feststoff in Ausbeuten von 74 % erhalten.
Herstellung der Verbindungen 37a und 37b
Die geschützten Aminosäuren 34 sollten im nächsten Schritt in einer Amidierungsreaktion mit dem
primären aromatischen Amin (Verbindungen 30) gekoppelt werden. Als Methode wurde eine
Umsetzung mit den Reagenzien DCC und N-Hydroxysuccinimid gewählt.83
Dabei wird die
Carbonsäure zuerst durch DCC aktiviert und anschließend ein sogenannter Aktivester mit N-
Hydroxysuccinimid gebildet. Der Mechanismus dieser Reaktion ist analog zu dem der Steglich-
Veresterung. Durch die Bildung des Aktivesters steigt die Reaktivität der Carbonsäure, so dass diese
anschließend zum Amid umgesetzt werden kann (siehe Abb. 56).
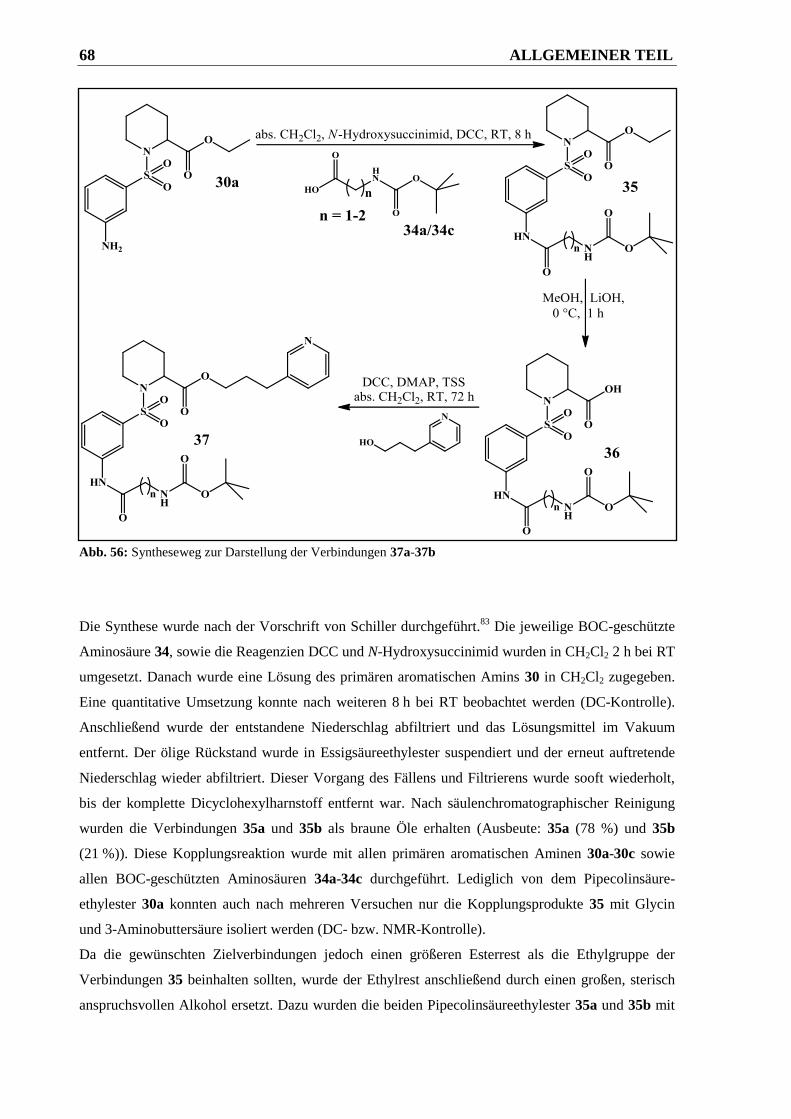
68 ALLGEMEINER TEIL
Abb. 56: Syntheseweg zur Darstellung der Verbindungen 37a-37b
Die Synthese wurde nach der Vorschrift von Schiller durchgeführt.83
Die jeweilige BOC-geschützte
Aminosäure 34, sowie die Reagenzien DCC und N-Hydroxysuccinimid wurden in CH2Cl2 2 h bei RT
umgesetzt. Danach wurde eine Lösung des primären aromatischen Amins 30 in CH2Cl2 zugegeben.
Eine quantitative Umsetzung konnte nach weiteren 8 h bei RT beobachtet werden (DC-Kontrolle).
Anschließend wurde der entstandene Niederschlag abfiltriert und das Lösungsmittel im Vakuum
entfernt. Der ölige Rückstand wurde in Essigsäureethylester suspendiert und der erneut auftretende
Niederschlag wieder abfiltriert. Dieser Vorgang des Fällens und Filtrierens wurde sooft wiederholt,
bis der komplette Dicyclohexylharnstoff entfernt war. Nach säulenchromatographischer Reinigung
wurden die Verbindungen 35a und 35b als braune Öle erhalten (Ausbeute: 35a (78 %) und 35b
(21 %)). Diese Kopplungsreaktion wurde mit allen primären aromatischen Aminen 30a-30c sowie
allen BOC-geschützten Aminosäuren 34a-34c durchgeführt. Lediglich von dem Pipecolinsäure-
ethylester 30a konnten auch nach mehreren Versuchen nur die Kopplungsprodukte 35 mit Glycin
und 3-Aminobuttersäure isoliert werden (DC- bzw. NMR-Kontrolle).
Da die gewünschten Zielverbindungen jedoch einen größeren Esterrest als die Ethylgruppe der
Verbindungen 35 beinhalten sollten, wurde der Ethylrest anschließend durch einen großen, sterisch
anspruchsvollen Alkohol ersetzt. Dazu wurden die beiden Pipecolinsäureethylester 35a und 35b mit

ALLGEMEINER TEIL 69
LiOH basisch zu den Carbonsäuren 36a und 36b hydrolysiert, wobei die Verbindungen als weiße bis
braune Wachse in sehr guten Ausbeuten anfielen (36a: 89 % und 36b: 80 %). Durch die
anschließende Steglich-Veresterung mit 3-(Pyridin-3-yl)propan-1-ol wurden die gewünschten
Endprodukte 37a und 37b als weißes Öl bzw. Wachs (Ausbeute: siehe Abb. 57) erhalten.
Abb. 57: Synthetisierte Verbindungen 37a-37b
2.2.2.5 Variationen des Grundgerüsts der Zielverbindungen
Um die Notwendigkeit der Pipecolinsäure-Esterfunktion für eine Bindung in die Mip-Kavität zu
überprüfen, sollte diese gegen eine Amidfunktion ausgetauscht werden (siehe Kapitel 2.2.2.5.1). Des
Weiteren sollte auch der Cyclohexylcharakter des Pipecolinsäure-Grundgerüsts bzgl. der Mip-
Inhibition näher untersucht werden. Dafür sollte der komplette Piperidinring durch eine offenkettige
Form (siehe Kapitel 2.2.2.5.2) und einen Pyrrolidinring (siehe Kapitel 2.2.2.5.3) ersetzt werden.
2.2.2.5.1 Syntheseweg zur Herstellung von Verbindung 38
Zur Einführung der Amidfunktion wurde im letzten Schritt der bereits bekannten vierstufigen
Synthese der Pipecolinsäure-Derivate der raumeinnehmende Alkohol durch ein strukturell ähnliches
Amin ersetzt. Um die Pipecolinsäure 15f mit dem primären Amin umzusetzen, wurde die Aktivester-
Methode mit den Reagenzien DCC und N-Hydroxysuccinimid gewählt (siehe Abb. 58). Die
Synthese verlief analog zur Darstellung der Verbindungen 35a und 35b (siehe Kapitel 2.2.2.4.3).
Verbindung 38 wurde in Form eines braunen Öls in Ausbeuten von 70 % erhalten.
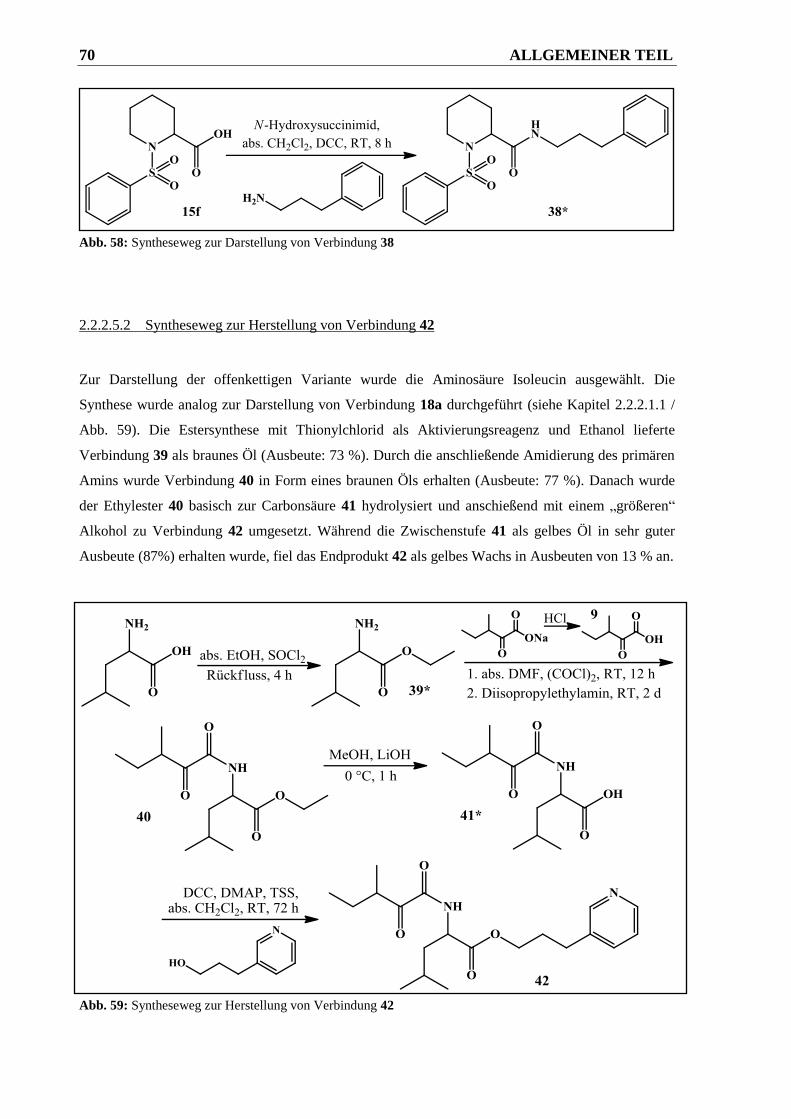
70 ALLGEMEINER TEIL
Abb. 58: Syntheseweg zur Darstellung von Verbindung 38
2.2.2.5.2 Syntheseweg zur Herstellung von Verbindung 42
Zur Darstellung der offenkettigen Variante wurde die Aminosäure Isoleucin ausgewählt. Die
Synthese wurde analog zur Darstellung von Verbindung 18a durchgeführt (siehe Kapitel 2.2.2.1.1 /
Abb. 59). Die Estersynthese mit Thionylchlorid als Aktivierungsreagenz und Ethanol lieferte
Verbindung 39 als braunes Öl (Ausbeute: 73 %). Durch die anschließende Amidierung des primären
Amins wurde Verbindung 40 in Form eines braunen Öls erhalten (Ausbeute: 77 %). Danach wurde
der Ethylester 40 basisch zur Carbonsäure 41 hydrolysiert und anschießend mit einem „größeren“
Alkohol zu Verbindung 42 umgesetzt. Während die Zwischenstufe 41 als gelbes Öl in sehr guter
Ausbeute (87%) erhalten wurde, fiel das Endprodukt 42 als gelbes Wachs in Ausbeuten von 13 % an.
Abb. 59: Syntheseweg zur Herstellung von Verbindung 42
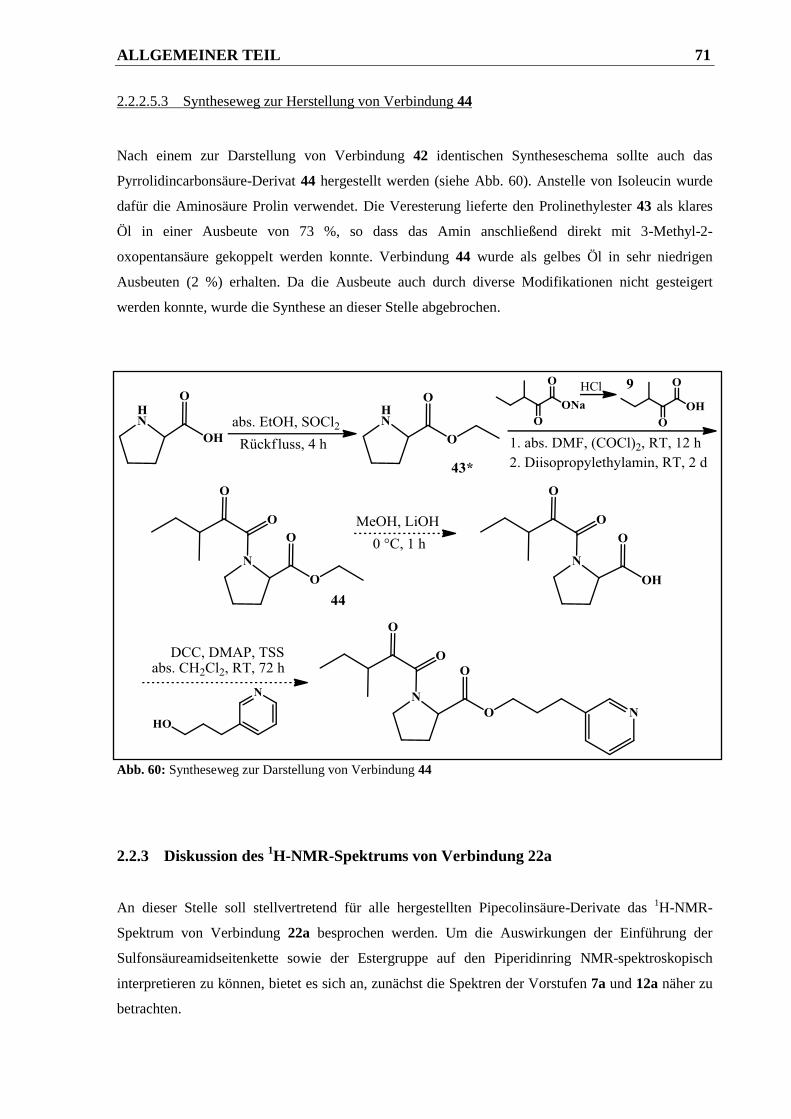
ALLGEMEINER TEIL 71
2.2.2.5.3 Syntheseweg zur Herstellung von Verbindung 44
Nach einem zur Darstellung von Verbindung 42 identischen Syntheseschema sollte auch das
Pyrrolidincarbonsäure-Derivat 44 hergestellt werden (siehe Abb. 60). Anstelle von Isoleucin wurde
dafür die Aminosäure Prolin verwendet. Die Veresterung lieferte den Prolinethylester 43 als klares
Öl in einer Ausbeute von 73 %, so dass das Amin anschließend direkt mit 3-Methyl-2-
oxopentansäure gekoppelt werden konnte. Verbindung 44 wurde als gelbes Öl in sehr niedrigen
Ausbeuten (2 %) erhalten. Da die Ausbeute auch durch diverse Modifikationen nicht gesteigert
werden konnte, wurde die Synthese an dieser Stelle abgebrochen.
Abb. 60: Syntheseweg zur Darstellung von Verbindung 44
2.2.3 Diskussion des 1H-NMR-Spektrums von Verbindung 22a
An dieser Stelle soll stellvertretend für alle hergestellten Pipecolinsäure-Derivate das 1H-NMR-
Spektrum von Verbindung 22a besprochen werden. Um die Auswirkungen der Einführung der
Sulfonsäureamidseitenkette sowie der Estergruppe auf den Piperidinring NMR-spektroskopisch
interpretieren zu können, bietet es sich an, zunächst die Spektren der Vorstufen 7a und 12a näher zu
betrachten.
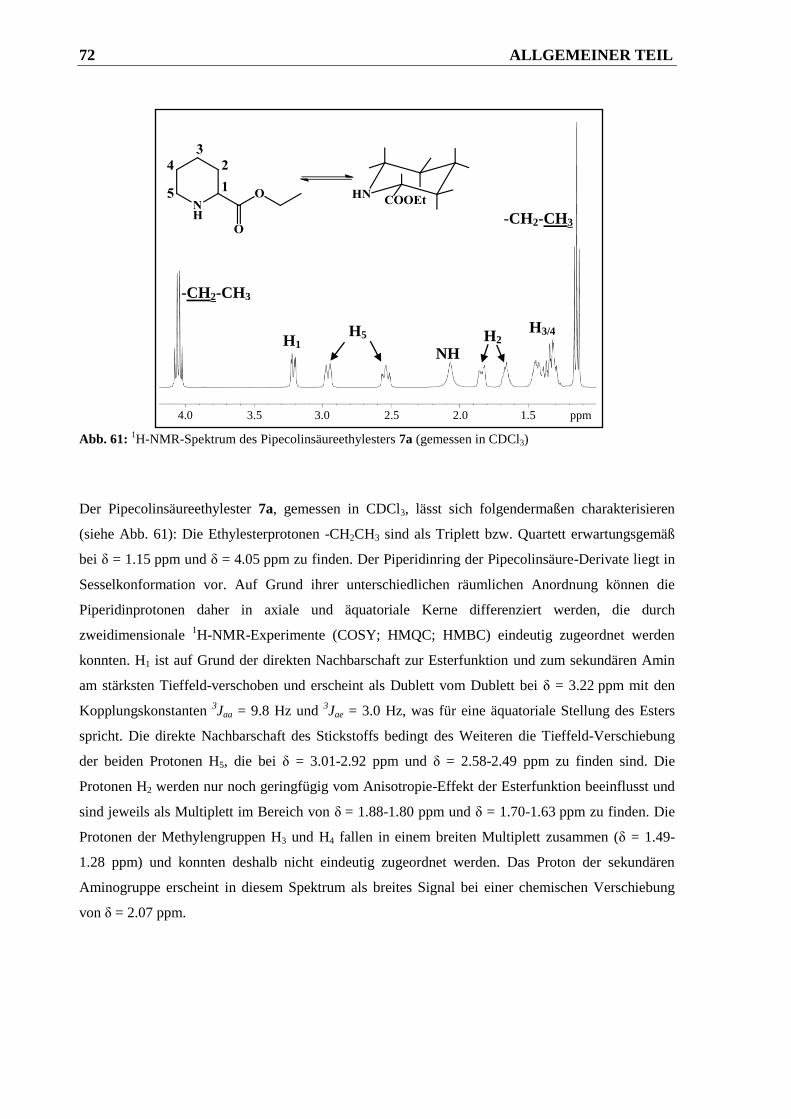
72 ALLGEMEINER TEIL
Abb. 61: 1H-NMR-Spektrum des Pipecolinsäureethylesters 7a (gemessen in CDCl3)
Der Pipecolinsäureethylester 7a, gemessen in CDCl3, lässt sich folgendermaßen charakterisieren
(siehe Abb. 61): Die Ethylesterprotonen -CH2CH3 sind als Triplett bzw. Quartett erwartungsgemäß
bei δ = 1.15 ppm und δ = 4.05 ppm zu finden. Der Piperidinring der Pipecolinsäure-Derivate liegt in
Sesselkonformation vor. Auf Grund ihrer unterschiedlichen räumlichen Anordnung können die
Piperidinprotonen daher in axiale und äquatoriale Kerne differenziert werden, die durch
zweidimensionale 1H-NMR-Experimente (COSY; HMQC; HMBC) eindeutig zugeordnet werden
konnten. H1 ist auf Grund der direkten Nachbarschaft zur Esterfunktion und zum sekundären Amin
am stärksten Tieffeld-verschoben und erscheint als Dublett vom Dublett bei δ = 3.22 ppm mit den
Kopplungskonstanten 3Jaa = 9.8 Hz und
3Jae = 3.0 Hz, was für eine äquatoriale Stellung des Esters
spricht. Die direkte Nachbarschaft des Stickstoffs bedingt des Weiteren die Tieffeld-Verschiebung
der beiden Protonen H5, die bei δ = 3.01-2.92 ppm und δ = 2.58-2.49 ppm zu finden sind. Die
Protonen H2 werden nur noch geringfügig vom Anisotropie-Effekt der Esterfunktion beeinflusst und
sind jeweils als Multiplett im Bereich von δ = 1.88-1.80 ppm und δ = 1.70-1.63 ppm zu finden. Die
Protonen der Methylengruppen H3 und H4 fallen in einem breiten Multiplett zusammen (δ = 1.49-
1.28 ppm) und konnten deshalb nicht eindeutig zugeordnet werden. Das Proton der sekundären
Aminogruppe erscheint in diesem Spektrum als breites Signal bei einer chemischen Verschiebung
von δ = 2.07 ppm.
-CH2-CH3
H5 H1
NH
H3/4 H2
4.0 3.5 3.0 2.5 2.0 1.5 ppm
-CH2-CH3

ALLGEMEINER TEIL 73
Abb. 62: 1H-NMR-Spektrum des Pipecolinsäureethylesters 12a (gemessen in CDCl3)
In Verbindung 12a wurde der sekundäre Stickstoff des Piperidinrings mit einem Benzyl-
sulfonylsubstituenten verknüpft (siehe Abb. 62). Dieser sterisch anspruchsvolle Substituent bedingt
in der Esterseitenkette die Entstehung einer Rotationsbarriere, so dass der Ethylrest nicht mehr frei
rotieren kann. Die beiden, dem Ester benachbarten Protonen der Methylengruppe -CH2-CH3 verlieren
dadurch ihre chemische Äquivalenz und sind in der Lage, auch untereinander zu koppeln. Sie
erscheinen im Spektrum daher auch nicht, wie in Verbindung 7a, als scharfes Quartett, sondern
vielmehr als ein Oktett. Die eingeführte Sulfonsäuregruppe wirkt sich des Weiteren auch teilweise
auf die Verschiebung der einzelnen Protonen des Piperidinrings aus. Während die Signale der
Protonen H2-4, kaum verschoben, als Multiplett bei δ= 2.22-2.11 ppm, δ = 1.72-1.54 ppm und
δ = 1.51-1.38 ppm auftreten, sind die Protonen H1 und H5 durch den entschirmenden Effekt des
benachbarten Sulfonsäureamids weiter Tieffeld-verschoben zu finden. Das Proton H1 ist um 1.4 ppm
am stärksten verschoben und erscheint bei δ = 4.57 ppm als Dublett mit einer Kopplungskonstanten
von 3J = 4.0 Hz. Die Protonen H5 sind im Vergleich zu Verbindung 7a um ca. 0.5 ppm Tieffeld-
verschoben und als Multiplett im Bereich von δ = 3.50-3.41 ppm sowie als Dublett vom Triplett bei
δ = 3.15 ppm zu erkennen. Die Kopplungskonstante 2J = 12.8 Hz lässt auf eine geminale, die von
3J = 3.1 Hz auf eine vicinale Kopplung der Protonen schließen. Die Methylengruppe der
Sulfonseitenkette ist auf Grund des Anisotropie-Effekts der benachbarten Sulfongruppe und des
Phenylrings Tieffeld-verschoben und erscheint als scharfes Singulett bei δ = 4.2 ppm. Die
H1
H2 H5
H2/3/4
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 ppm
1.8 1.6 1.4 1.2 ppm
7.5 7.3 ppm
4.3 4.2 ppm
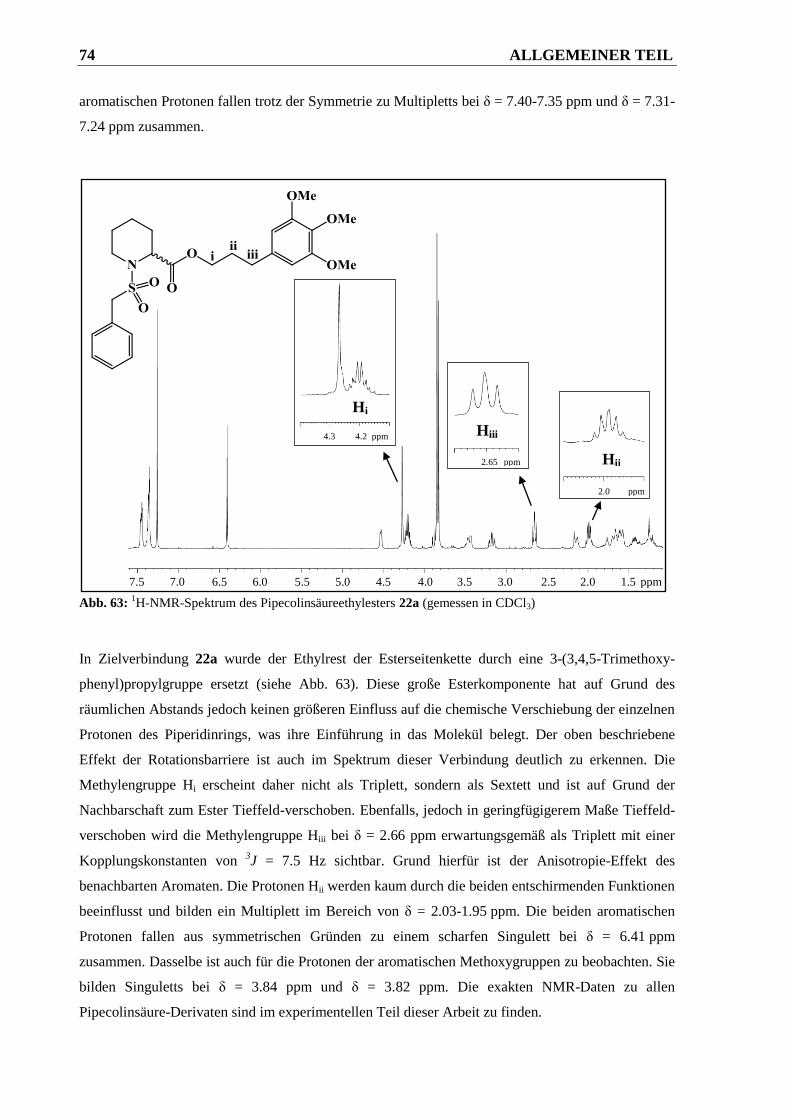
74 ALLGEMEINER TEIL
aromatischen Protonen fallen trotz der Symmetrie zu Multipletts bei δ = 7.40-7.35 ppm und δ = 7.31-
7.24 ppm zusammen.
Abb. 63: 1H-NMR-Spektrum des Pipecolinsäureethylesters 22a (gemessen in CDCl3)
In Zielverbindung 22a wurde der Ethylrest der Esterseitenkette durch eine 3-(3,4,5-Trimethoxy-
phenyl)propylgruppe ersetzt (siehe Abb. 63). Diese große Esterkomponente hat auf Grund des
räumlichen Abstands jedoch keinen größeren Einfluss auf die chemische Verschiebung der einzelnen
Protonen des Piperidinrings, was ihre Einführung in das Molekül belegt. Der oben beschriebene
Effekt der Rotationsbarriere ist auch im Spektrum dieser Verbindung deutlich zu erkennen. Die
Methylengruppe Hi erscheint daher nicht als Triplett, sondern als Sextett und ist auf Grund der
Nachbarschaft zum Ester Tieffeld-verschoben. Ebenfalls, jedoch in geringfügigerem Maße Tieffeld-
verschoben wird die Methylengruppe Hiii bei δ = 2.66 ppm erwartungsgemäß als Triplett mit einer
Kopplungskonstanten von 3J = 7.5 Hz sichtbar. Grund hierfür ist der Anisotropie-Effekt des
benachbarten Aromaten. Die Protonen Hii werden kaum durch die beiden entschirmenden Funktionen
beeinflusst und bilden ein Multiplett im Bereich von δ = 2.03-1.95 ppm. Die beiden aromatischen
Protonen fallen aus symmetrischen Gründen zu einem scharfen Singulett bei δ = 6.41 ppm
zusammen. Dasselbe ist auch für die Protonen der aromatischen Methoxygruppen zu beobachten. Sie
bilden Singuletts bei δ = 3.84 ppm und δ = 3.82 ppm. Die exakten NMR-Daten zu allen
Pipecolinsäure-Derivaten sind im experimentellen Teil dieser Arbeit zu finden.
2.65 ppm
2.0 ppm
4.2 ppm 4.3
Hi
Hiii
Hii
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 ppm

ALLGEMEINER TEIL 75
2.3 Weitere neu-„designte” Derivate
Das Pharmakophor-basierte, virtuelle Screening lieferte neben den niedermolekularen Sulfonsäure-
amiden und den Pipecolinsäure-Derivaten weitere Verbindungen, die auf Grund ihrer molekularen
Eigenschaften mit den Aminosäuren der Mip-Bindetasche wechselwirken sollten. Dazu gehörten: 2-
(4-Isopropylphenoxy)essigsäuremorpholinamid, 5-Methyl-2-phenyl-1H-pyrazol-3(2H)-on sowie 2-
Amino-4-(methylsulfinyl)buttersäureethylester.
2.3.1 Syntheseweg zur Herstellung von Verbindung 46
2-(4-Isopropylphenoxy)essigsäuremorpholinamid 46 sollte über eine zweistufige Synthese hergestellt
werden (siehe Abb. 64). Im ersten Schritt sollte p-Isopropylphenol in Form einer klassischen
Ethersynthese zu 2-(4-Isopropylphenoxy)essigsäure 45 umgesetzt und im zweiten Schritt die
entstandene Carbonsäure 45 mit Morpholin zum Endprodukt 46 amidiert werden.
Abb. 64: Syntheseweg zur Darstellung von Verbindung 46
Zur Synthese von Verbindung 45 wurde p-Isopropylphenol in methanolischer Natronlauge auf 80 °C
erhitzt und anschließend mit Chloressigsäure versetzt. Eine quantitative Umsetzung wurde nach 2 h
bei 80 °C und weiteren 12 h bei RT beobachtet (DC-Kontrolle). Danach wurden H2O und verdünnte
Salzsäure bis pH 2 zugegeben und anschließend die entstandene freie Säure mit Essigsäureethylester
extrahiert. Verbindung 45 wurde in Form eines braunen Öls in sehr guten Ausbeuten von 98 %
erhalten.
Für die Amidierung musste im nächsten Schritt die Säure 45 zuerst aktiviert werden. Hierzu sollte
mit den Reagenzien 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid (EDC) und 1-Hydroxy-
benzotriazol (HOBt) ein Aktivester hergestellt werden. Der Mechanismus kann mit der oben

76 ALLGEMEINER TEIL
beschriebenen Steglich-Veresterung gleichgesetzt werden; EDC entspricht hierbei DCC und HOBt
der alkoholischen Komponente.
Zur Darstellung von Verbindung 46 wurden ein Äquivalent Verbindung 45, 1.2 Äquivalente EDC
und HOBt sowie 3 Äquivalente Triethylamin in abs. DMF gelöst und 30 min bei RT gerührt. Danach
wurde ein Äquivalent Morpholin zugegeben und weitere 12 h bei RT gerührt. Der Reaktionsverlauf
wurde dünnschichtchromatographisch verfolgt (DC-Kontrolle: Kieselgel; Hexan : EtOAc = 7 : 3),
zeigte aber eine Vielzahl an Produkten. Nach Entfernen des Lösungsmittels im Vakuum und der
säulenchromatographischen Reinigung (Kieselgel; Hexan : EtOAc = 7 : 3) konnte lediglich das
Edukt isoliert werden (DC- bzw. NMR-Kontrolle).
Als weitere Synthesestrategie wurde daher nach einer Vorschrift von Holzgrabe und Heller ein
Äquivalent Verbindung 45 in abs. THF gelöst und 1.1 Äquivalente Pyridin, 1.05 Äquivalente
(BOC)2O sowie 0.05 Äquivalente DMAP zugegeben.84
Anschließend wurde 30 min bei RT gerührt,
danach ein Äquivalent Morpholin zugegeben und weitere 48 h bei RT gerührt. Der Reaktionsverlauf
wurde dünnschichtchromatographisch verfolgt (DC-Kontrolle: Kieselgel; Cyclohexan : EtOAc =
7 : 3), zeigte aber auch in diesem Fall eine Vielzahl an Produkten. Nach Entfernen des
Lösungsmittels im Vakuum und der säulenchromatographischen Reinigung (Kieselgel; Cyclohexan :
EtOAc = 7 : 3) konnte wiederum lediglich das Edukt isoliert werden (DC- bzw. NMR-Kontrolle).
Nach einer Vorschrift von Burkhart et al. wurde anschließend eine Kopplungsvariante mit den
Reagenzien N-Methylmorpholin und n-Propylphosphonsäureanhydrid verwendet.85
Dafür wurde
Verbindung 45 in DMF mit Morpholin, Überschüssen an N-Methylmorpholin sowie n-Propyl-
phosphonsäureanhydrid versetzt und der Reaktionsverlauf dünnschichtchromatographisch verfolgt.
Da nach 48 h keine Umsetzung sichtbar war, wurde ein weiterer Überschuss an n-
Propylphosphonsäureanhydrid zugeben. Dieser Vorgang wurde nach 7 d, da immer noch keine
Umsetzung zu erkennen war, noch einmal wiederholt. Nach weiteren 7 d wurde das Lösungsmittel
im Vakuum entfernt und nach säulenchromatographischer Reinigung Verbindung 46 in Form eines
braunen Öls isoliert (Ausbeute: 36 %).
2.3.2 Syntheseweg zur Herstellung von Verbindung 47
5-Methyl-2-phenyl-1H-pyrazol-3(2H)-on war ein weiteres Ergebnis der computergestützten Wirk-
stofffindung. Pyrazole sind in der heutigen Medizin weit verbreitet und werden hauptsächlich als
nicht-steroidale Antiphlogistika verwendet. Bereits 1883 gelang es dem Chemiker Ludwig Knorr 1,5-
Dimethyl-2-phenyl-1H-pyrazol-3(2H)-on (Phenazon) chemisch herzustellen.86
Die damals
beschriebene Synthese konnte bis heute mehrfach verbessert und weiterentwickelt werden. Der
Mechanismus einer Pyrazol-Synthese verläuft meistens über die 1,3-Cyclisierung von Acetessig-

ALLGEMEINER TEIL 77
säureethylester mit Hydrazin-Derivaten. Der Acetessigsäureethylester kann sowohl in seiner Keto-
als auch Enolform vorliegen. In Anwesenheit eines starken Nukleophils kann dieses - in diesem Fall
handelt es sich um das primäre Amin des Hydrazin-Derivats - am elektrophilen C-Atom des Enols
angreifen, wobei unter Wasserabspaltung zunächst ein Enamin entsteht. Im nächsten Schritt greift
das zweite nukleophile Amin des Hydrazins intramolekular das elektrophile Carbonyl-C-Atom an
und unter Abspaltung von Ethanol entsteht das gewünschte Pyrazol (siehe Abb. 65).
In dieser Arbeit wurde die Synthese des Pyrazols 47 nach einer Vorschrift von Lehmann et al.
durchgeführt.87
Dazu wurde Acetessigsäureethylester in Ethanol mit Phenylhydrazin 3 h zum
Rückfluss erhitzt und anschließend 12 h bei RT stehen gelassen, bis eine quantitative Umsetzung
beobachtet werden konnte (DC-Kontrolle). Nach Umkristallisieren aus n-Pentan wurde Verbindung
47 in Form eines gelben Pulvers erhalten (Ausbeute: 65 %).
Abb. 65: Syntheseweg zur Herstellung von Verbindung 47
2.3.3 Syntheseweg zur Herstellung von Verbindung 48
Zur Synthese von 2-Amino-4-(methylsulfinyl)buttersäureethylester 48 wurde die entsprechende
Säure in Form einer klassischen Veresterung mit Thionylchlorid als Aktivierungsreagenz und
Ethanol umgesetzt (siehe Abb. 66). Die Zielverbindung 48 fiel dabei als gelbes Öl in Ausbeuten von
35 % an.
Abb. 66: Syntheseweg zur Herstellung von Verbindung 48

78 ALLGEMEINER TEIL

ALLGEMEINER TEIL 79
3 PHARMAKOLOGISCHE TESTUNGEN UND ERGEBNISSE
3.1 In-vitro-Testsystem
Das Oberflächenprotein Mip stellt, wie in Kapitel 1.1.3.2 beschrieben, einen Hauptvirulenzfaktor von
L. pneumophila dar. Seine C-Domäne weist eine enzymatische PPIase-Aktivität auf, die durch
Pipecolinsäure-Derivate inhibiert werden kann. Diese Enzymhemmung kann durch den IC50-Wert
(IC50 = mittlere inhibitorische Konzentration) beschrieben werden, für dessen Bestimmung sich der
bereits 1984 entwickelte Protease-gekoppelte Enzymassay eignet.53
Die dazu notwendigen
biologischen Vorarbeiten sowie die pharmakologische Testung der synthetisierten Verbindungen
erfolgten hauptsächlich in der Forschungsgruppe von Dr. Matthias Weiwad am Max-Planck-Institut
für Enzymologie der Proteinfaltung in Halle.
3.1.1 Expression von Mip
3.1.1.1 Klonierung und Transformation
Für eine optimale Expression des Lp-Mip-Gens erwiesen sich in früheren Studien Escherichia coli
K-12-Bakterienstämme als besonders geeignet.88
Vor der Expression in E. coli musste die
gewünschte DNA-Sequenz zunächst aus L. pneumophila isoliert und anschließend in einen
geeigneten Vektor kloniert werden. Zu diesem Zweck wurde eine früher angelegte Genbibliothek des
L. pneumophila-Stamms Philadelphia I88,89
durchsucht, wobei 21 rekombinante Klone identifiziert
wurden, die für verschiedene Proteine mit einer Masse von 24 kDa codierten. Alle Klone wurden mit
Hilfe der Cosmid-Klonierungstechnik in E. coli-K-12-Bakterienstämme transformiert und
anschließend auf ihre exprimierten Proteine untersucht. Da der E. coli-K-12-Stamm HB101 eine
Expression des gewünschten Mips zeigte, wurde er für die weitere Kultivierung verwendet.28
Diese Klonierungs- bzw. Transformationsarbeiten waren bereits abgeschlossen, so dass die
praktische Durchführung in dieser Arbeit mit der Anzucht der Bakterien startete (siehe Kapitel 6.3.3).
3.1.1.2 Anzucht von E. coli-Bakterien
Für die Kultivierung des E. coli-K-12-Stamms HB101 wurde das für dieses Bakterium am häufigsten
eingesetzte LB-Nährmedium („lysogeny broth“) verwendet, da es alle essenziellen Bestandteile, die

80 ALLGEMEINER TEIL
für ein optimales Wachstum von E. coli von Bedeutung sind, enthält. Zur Herstellung des Mediums
wurden ein Gemisch aus verschiedenen Peptiden und Aminosäuren (Pepton), Hefeextrakt und
Natriumchlorid in dem. H2O gelöst, auf pH 7 eingestellt und autoklaviert. Zur Überprüfung des
optimalen Wachstums der Bakterien wurde aus dem LB-Medium eine Probe entnommen und mit
Ampicillin (100 µg/ml) versetzt. Der klonierte Vektor enthält neben dem Mip-Gen ein zusätzliches
Ampicillin-Resistenzgen; somit können in Anwesenheit des Antibiotikums selektiv nur diejenigen
Bakterien überleben, die dieses Resistenzgen enthalten - d.h. alle anderen, v.a. diejenigen, die bei der
Expression stören könnten, sterben ab. Des Weiteren wurde die Probe mit einer klonierten E. coli-K-
12-HB101-Kultur versetzt und über Nacht bei 37 °C inkubiert. Zeigte diese sogenannte Vorkultur
nach 12 h ausreichend Bakterienwachstum, wurde sie zusammen mit Ampicillin (100 µg/ml) in eine
größere Menge LB-Medium überführt und weitere 24 h bei 37 °C inkubiert. Anschließend wurden
die gewachsenen Zellen mittels Zentrifugation pelletiert, der flüssige Überstand verworfen und die
Zellpellets für die nachfolgende mechanische Lyse auf Eis abgekühlt.
3.1.1.3 Zellaufschluss mittels mechanischer Lyse
Die vorbereiteten Zellpellets wurden unter Eiskühlung in 2 mM Trizin-Puffer (N-Tris-
(hydroxymethyl)methylglycin) pH 8.5 gelöst und anschließend zu dieser Zellsuspension
verschiedene Protease-Inhibitoren (siehe Kapitel 6.3.4) gegeben, um während der Lyse einen
unerwünschten Proteinabbau durch anwesende Proteasen zu verhindern. Nach Überführung der
Suspension in den Stahlzylinder der hydraulischen Zellaufschluss (French-press)-Anlage wurden die
Bakterienzellen unter Druck aufgeschlossen und das dabei anfallende Lysat ultrazentrifugiert. Die
entstandenen Zellpellets wurden für die folgende Reinigung gesammelt und das Lysat verworfen.
3.1.1.4 Reinigung von Mip
Bei der anschließenden Proteinaufreinigung wurden als Elutionsmittel der bereits hergestellte Trizin-
Puffer und eine 35 %ige wässrige Ammoniumsulfat-Lösung verwendet. Der gesamte Reinigungs-
prozess beinhaltete vier hintereinandergeschaltete chromatographische Trennschritte und wurde bei
einer Temperatur von 4 °C durchgeführt.
Im ersten Schritt wurden die Zellpellets der Ultrazentrifugation in Trizin-Puffer suspendiert und nach
Überprüfung des pH-Werts auf eine mit diesem Puffer equilibrierte Diethylaminoethyl (DEAE)-
Säule (Fractogel®) aufgetragen. Bei dieser Trennmethode handelt es sich um das Prinzip der
schwachen Anionenaustausch-Chromatographie. Dabei geht Mip im Gegensatz zu den ebenfalls
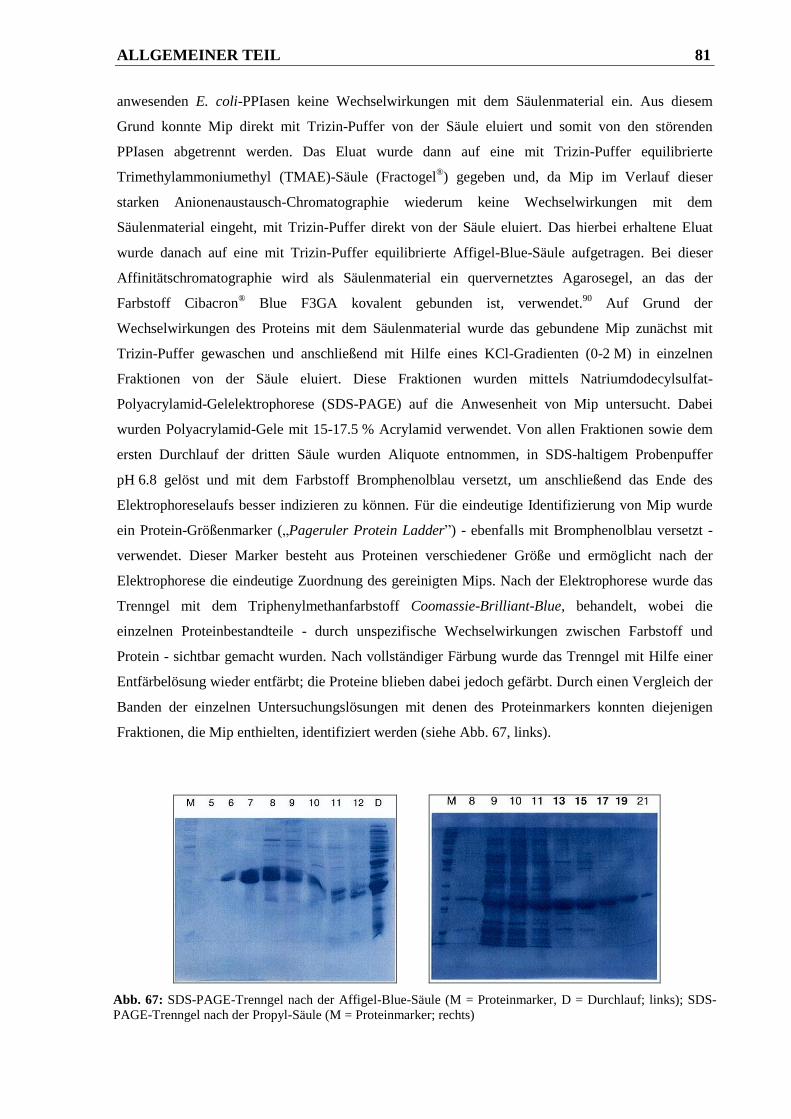
ALLGEMEINER TEIL 81
anwesenden E. coli-PPIasen keine Wechselwirkungen mit dem Säulenmaterial ein. Aus diesem
Grund konnte Mip direkt mit Trizin-Puffer von der Säule eluiert und somit von den störenden
PPIasen abgetrennt werden. Das Eluat wurde dann auf eine mit Trizin-Puffer equilibrierte
Trimethylammoniumethyl (TMAE)-Säule (Fractogel®) gegeben und, da Mip im Verlauf dieser
starken Anionenaustausch-Chromatographie wiederum keine Wechselwirkungen mit dem
Säulenmaterial eingeht, mit Trizin-Puffer direkt von der Säule eluiert. Das hierbei erhaltene Eluat
wurde danach auf eine mit Trizin-Puffer equilibrierte Affigel-Blue-Säule aufgetragen. Bei dieser
Affinitätschromatographie wird als Säulenmaterial ein quervernetztes Agarosegel, an das der
Farbstoff Cibacron® Blue F3GA kovalent gebunden ist, verwendet.
90 Auf Grund der
Wechselwirkungen des Proteins mit dem Säulenmaterial wurde das gebundene Mip zunächst mit
Trizin-Puffer gewaschen und anschließend mit Hilfe eines KCl-Gradienten (0-2 M) in einzelnen
Fraktionen von der Säule eluiert. Diese Fraktionen wurden mittels Natriumdodecylsulfat-
Polyacrylamid-Gelelektrophorese (SDS-PAGE) auf die Anwesenheit von Mip untersucht. Dabei
wurden Polyacrylamid-Gele mit 15-17.5 % Acrylamid verwendet. Von allen Fraktionen sowie dem
ersten Durchlauf der dritten Säule wurden Aliquote entnommen, in SDS-haltigem Probenpuffer
pH 6.8 gelöst und mit dem Farbstoff Bromphenolblau versetzt, um anschließend das Ende des
Elektrophoreselaufs besser indizieren zu können. Für die eindeutige Identifizierung von Mip wurde
ein Protein-Größenmarker („Pageruler Protein Ladder”) - ebenfalls mit Bromphenolblau versetzt -
verwendet. Dieser Marker besteht aus Proteinen verschiedener Größe und ermöglicht nach der
Elektrophorese die eindeutige Zuordnung des gereinigten Mips. Nach der Elektrophorese wurde das
Trenngel mit dem Triphenylmethanfarbstoff Coomassie-Brilliant-Blue, behandelt, wobei die
einzelnen Proteinbestandteile - durch unspezifische Wechselwirkungen zwischen Farbstoff und
Protein - sichtbar gemacht wurden. Nach vollständiger Färbung wurde das Trenngel mit Hilfe einer
Entfärbelösung wieder entfärbt; die Proteine blieben dabei jedoch gefärbt. Durch einen Vergleich der
Banden der einzelnen Untersuchungslösungen mit denen des Proteinmarkers konnten diejenigen
Fraktionen, die Mip enthielten, identifiziert werden (siehe Abb. 67, links).
Abb. 67: SDS-PAGE-Trenngel nach der Affigel-Blue-Säule (M = Proteinmarker, D = Durchlauf; links); SDS-
PAGE-Trenngel nach der Propyl-Säule (M = Proteinmarker; rechts)

82 ALLGEMEINER TEIL
Die Mip-enthaltenen Fraktionen wurden daraufhin vereinigt, in Ammoniumsulfat-Lösung suspendiert
und einem weiteren chromatographischen Reinigungsschritt unterzogen. Dazu wurde die
Proteinsuspension auf eine mit Trizin-Puffer und Ammoniumsulfat-Lösung equilibrierte Propyl-Säule
aufgetragen und nach dem Prinzip der hydrophoben Interaktionschromatographie mit einem
Ammoniumsulfat-Gradienten (0-35 %) in Fraktionen eluiert. Im Anschluss wurden die einzelnen
Untersuchungslösungen wiederum mittels SDS-PAGE-Elektrophorese (siehe oben) analysiert (siehe
Abb. 67, rechts). Alle Fraktionen, die reines Mip enthielten, wurden vereinigt und für die Dialyse
vorbereitet.
Im letzten Schritt der Reinigung von Mip wurden mittels Dialyse enthaltene Fremdionen, z.B.
Ammoniumsulfat, durch Diffusionsprozesse aus der Proteinlösung entfernt. Die Dialyse erfolgte unter
Verwendung von HEPES (2-(4-(2-Hydroxyethyl)-1-piperazinyl)ethansulfonsäure)-Puffer pH 7.8 und
einem dünnwandigen Dialyseschlauch für 12 h Stunden. Während der Dialyse wandern nur Ionen und
kleine Moleküle durch die semipermeable Membran des Dialyseschlauchs in Richtung des
Konzentrationsgefälles; die größeren Proteine bleiben im Inneren des Dialyseschlauchs. Auf diese
Weise konnte Mip bis auf eine vernachlässigbare Menge an Ammoniumsulfat aufkonzentriert werden.
3.1.2 Durchführung und Auswertung des PPIase-Assays
Die Untersuchungen der Mip-PPIase-Inhibition durch die hergestellten Verbindungen und die
Bestimmung der entsprechenden IC50-Werte wurden am Max-Planck-Institut durchgeführt (siehe
Kapitel 3.1). Der verwendete Enzymassay sollte während der Promotionszeit an das Institut für
Pharmazie in Würzburg übertragen und dort etabliert werden. Dies scheiterte jedoch daran, dass das
vorhandene UV-Spektrometer im gewünschten Absorptionsbereich zu große Schwankungen aufwies
und somit keine verwertbaren, konstanten Werte gemessen werden konnten. Der In-vitro-Test beruht
auf einer Protease-gekoppelten enzymatischen Reaktion. Das Prinzip dabei ist, dass die
Peptidbindungen des eingesetzten Substrats Succinyl-Ala-Phe-Pro-Phe-p-Nitroanilin durch die
ebenfalls anwesende Hilfsprotease α-Chymotrypsin nur dann gespalten werden können, wenn die
Peptidbindung zwischen Phenylalanin und Prolin in trans-Stellung vorliegt (siehe A). Die Menge an
gespaltenem trans-Substrat kann dann durch eine Absorptionsmessung des Spaltprodukts p-
Nitroanilin bei 390 nm gemessen werden. Bei pH 7.8 stellt sich zwischen den beiden Konformeren
trans und cis des Substrats ein Gleichgewicht von 80-95 : 5-20 ein.28
Dies bedingt zu Beginn der
Reaktion - d.h. innerhalb der ersten 30 Sekunden - eine schnelle Spaltung des trans-Substrats durch
die Hilfsprotease und damit verbunden einen steilen Anstieg der Absorption (siehe A).
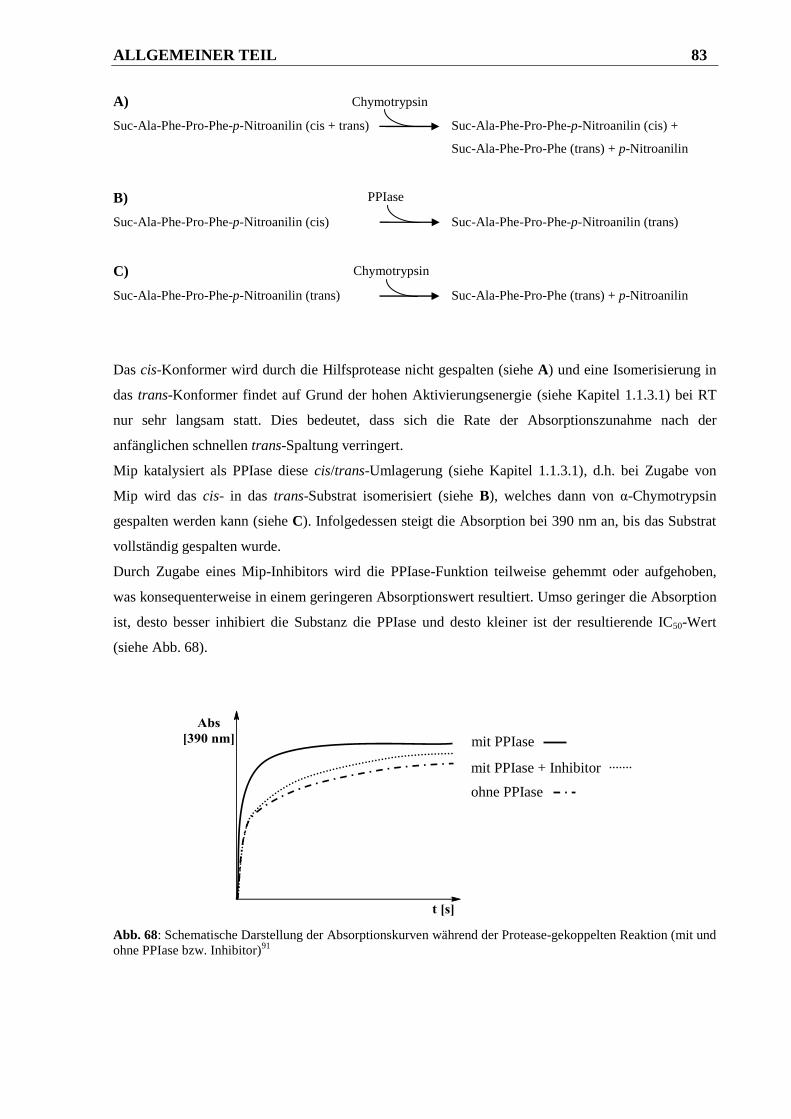
ALLGEMEINER TEIL 83
A)
Suc-Ala-Phe-Pro-Phe-p-Nitroanilin (cis + trans) Suc-Ala-Phe-Pro-Phe-p-Nitroanilin (cis) +
Suc-Ala-Phe-Pro-Phe (trans) + p-Nitroanilin
B)
Suc-Ala-Phe-Pro-Phe-p-Nitroanilin (cis) Suc-Ala-Phe-Pro-Phe-p-Nitroanilin (trans)
C)
Suc-Ala-Phe-Pro-Phe-p-Nitroanilin (trans) Suc-Ala-Phe-Pro-Phe (trans) + p-Nitroanilin
Das cis-Konformer wird durch die Hilfsprotease nicht gespalten (siehe A) und eine Isomerisierung in
das trans-Konformer findet auf Grund der hohen Aktivierungsenergie (siehe Kapitel 1.1.3.1) bei RT
nur sehr langsam statt. Dies bedeutet, dass sich die Rate der Absorptionszunahme nach der
anfänglichen schnellen trans-Spaltung verringert.
Mip katalysiert als PPIase diese cis/trans-Umlagerung (siehe Kapitel 1.1.3.1), d.h. bei Zugabe von
Mip wird das cis- in das trans-Substrat isomerisiert (siehe B), welches dann von α-Chymotrypsin
gespalten werden kann (siehe C). Infolgedessen steigt die Absorption bei 390 nm an, bis das Substrat
vollständig gespalten wurde.
Durch Zugabe eines Mip-Inhibitors wird die PPIase-Funktion teilweise gehemmt oder aufgehoben,
was konsequenterweise in einem geringeren Absorptionswert resultiert. Umso geringer die Absorption
ist, desto besser inhibiert die Substanz die PPIase und desto kleiner ist der resultierende IC50-Wert
(siehe Abb. 68).
Abb. 68: Schematische Darstellung der Absorptionskurven während der Protease-gekoppelten Reaktion (mit und
ohne PPIase bzw. Inhibitor)91
mit PPIase
mit PPIase + Inhibitor
ohne PPIase
PPIase
Chymotrypsin
Chymotrypsin

84 ALLGEMEINER TEIL
Der Enzymassay wurde UV-spektrometrisch in einem 96-Well-Plate-Format durchgeführt. Im ersten
Schritt wurden jeweils Stammlösungen von Substrat, α-Chymotrypsin und Mip in HEPES/NaCl-
Puffer pH 7.8 sowie Stammlösungen von den zu testenden Inhibitoren in Ethanol hergestellt. In jeder
Messreihe wurden neben den Messwerten der Inhibitorlösungen, bestehend aus Substrat, Mip,
Inhibitor und α-Chymotrypsin, auch ein Nullwert (Substrat und α-Chymotrypsin) sowie ein
Kontrollwert (Substrat, Mip und α-Chymotrypsin) bestimmt. Die Bestimmung der Absorptionswerte
aller Untersuchungslösungen wurde dreimal durchgeführt. Bei allen Messungen sollte ein
Temperaturbereich von 9-11 °C zwingend eingehalten werden, da nur in diesem Bereich die
Geschwindigkeit der cis/trans-Umlagerung über eine Absorptionszunahme beobachtet werden kann.
Bei höheren Temperaturen ist die Reaktion bereits nach einigen Sekunden abgeschlossen, bei
niedrigeren ist eine Isomerisierung nicht zu erkennen. Sobald die Reaktion durch die α-Chymotrypsin-
Zugabe gestartet worden war, wurde über einen Zeitraum von 200 s alle drei Sekunden die Absorption
bei 390 nm bestimmt. Die erhaltenen Absorptionswerte wurden als Funktion gegen die Zeit
aufgetragen, wobei die Absorptionswerte im Intervall von 0-30 s bei der anschließenden Auswertung
nicht berücksichtigt wurden. In dieser Zeit findet die Spaltung des trans-Konformers unabhängig von
der Mip-Aktivität statt. Um die Aktivität der PPIase, d.h. die Geschwindigkeit der cis/trans-
Isomerisierung erfassen zu können, werden diese Absorptionswerte vernachlässigt.
Die Umlagerung der Konformere beschreibt eine Reaktion erster Ordnung, d.h. die
Geschwindigkeitskonstanten (k-Werte) der einzelnen Reaktionen lassen sich nach dem Modell von
Michaelis-Menten berechnen.92
Dieses Modell gilt für Reaktionen mit folgendem prinzipiellen
Mechanismus:
Das Enzym E bindet an das Substrat S unter Bildung eines Enzym/Substrat-Komplexes mit der
Geschwindigkeitskonstanten k1. Der Komplex kann danach entweder wieder in die Ausgangsstoffe E
und S zerfallen (k-1) oder in das Produkt P umgesetzt werden (k2).
Die Geschwindigkeitskonstante der PPIase während der einzelnen Reaktionen wurde über die
nachfolgende Funktionsgleichung (1) mit Hilfe eines geeigneten Analyseprogramms berechnet:
)(0 kxeayy (Gleichung 1)
Aus den Werten der Nullwert-Messung erhielt man nach der Auswertung den nicht-enzymatischen k-
Wert (kn.enz), und aus denen der Kontrolle sowie der einzelnen Inhibitoren den jeweiligen kobs-Wert

ALLGEMEINER TEIL 85
(obs = observed). Der kn.enz-Wert, d.h. der Nullwert, wurde anschließend von allen kobs-Werten
abgezogen und man erhielt die enzymatischen k-Werte (kenz):
kenz = kobs -kn.enz (Gleichung 2)
Anschließend wurde aus den einzelnen kenz-Werten der kcat/KM-Parameter des Enzyms bei den
verschiedenen Inhibitorkonzentrationen nach folgender Formel berechnet:
][MIP
k
K
k enzcat
M
(Gleichung 3)
Der kcat/KM-Wert ist eine Geschwindigkeitskonstante zweiter Ordnung und beschreibt die katalytische
Effizienz oder Spezifität des Enzyms. Während kcat als katalytische Konstante erster Ordnung oder
auch „turnover-number“ die maximale Zahl der umgesetzten Substrat-Moleküle pro Enzym und
Sekunde darstellt, gibt KM, die sogenannte Michaelis-Menten-Konstante, die Substratkonzentration bei
halbmaximaler Reaktionsgeschwindigkeit an.
Im Fall des PPIase-Assays wurde der kcat/KM-Wert des ungebundenen Enzyms (ohne Inhibitor;
entspricht der Kontrolllösung) daher als 100 % der Enzymaktivität angenommen und alle anderen
Effizienz-Werte (mit Inhibitor) dazu ins Verhältnis gesetzt. Somit konnte die Restaktivität des Enzyms
bei verschiedenen Inhibitorkonzentrationen erhalten werden. Diese beiden Parameter gegeneinander
aufgetragen (Restaktivität gegen Inhibitorkonzentration), ergeben eine exponentiell abfallende Kurve
(siehe Abb. 69), aus der mit Hilfe geeigneter Analyseprogramme der IC50-Wert für jeden einzelnen
Inhibitor bestimmt werden konnte.
Abb. 69: Restaktivitätskurve von Verbindung 22b (IC50 = 6 µM)
[CJ168] (µM)
0 50 100 150 200
Lp
Mip
PP
Iase
activity (
% o
f co
ntr
ol)
0
20
40
60
80
100
Inhibitorkonzentration
(Verbindung 22b)
Lp-P
PIa
se-A
kti
vit
ät
[% d
er K
ontr
oll
e]
0 50 100 150 200
0
20
40
60
80
100
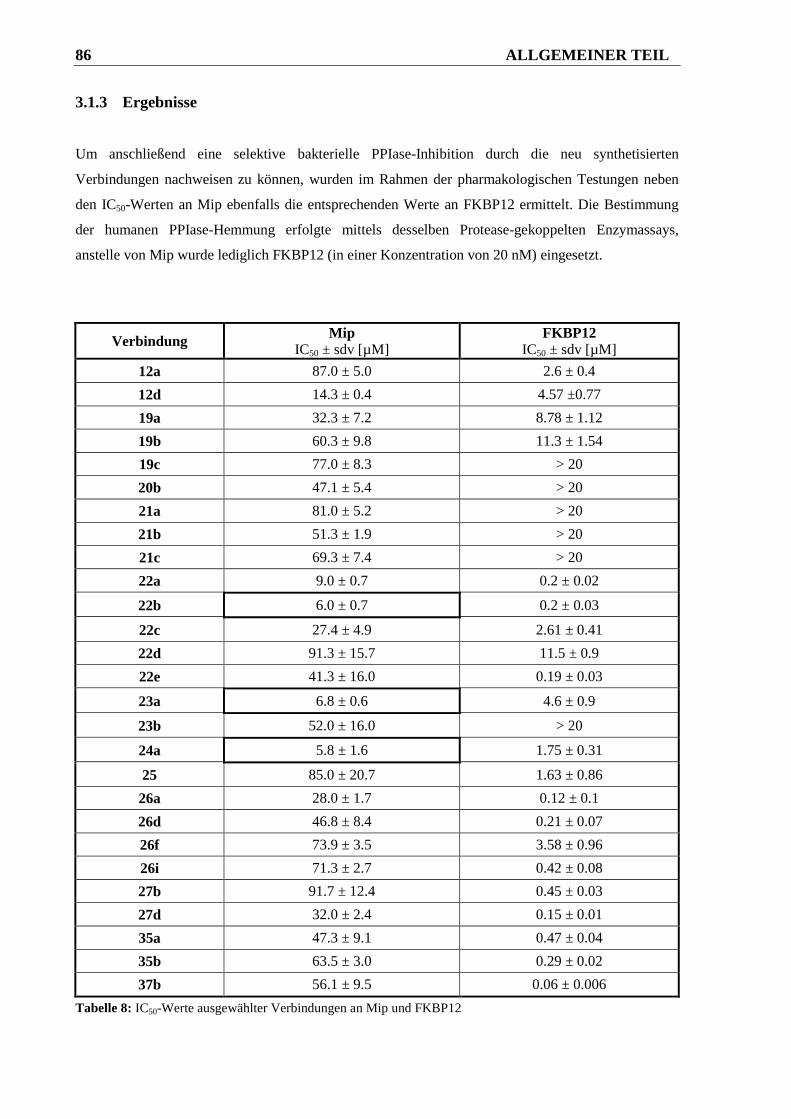
86 ALLGEMEINER TEIL
3.1.3 Ergebnisse
Um anschließend eine selektive bakterielle PPIase-Inhibition durch die neu synthetisierten
Verbindungen nachweisen zu können, wurden im Rahmen der pharmakologischen Testungen neben
den IC50-Werten an Mip ebenfalls die entsprechenden Werte an FKBP12 ermittelt. Die Bestimmung
der humanen PPIase-Hemmung erfolgte mittels desselben Protease-gekoppelten Enzymassays,
anstelle von Mip wurde lediglich FKBP12 (in einer Konzentration von 20 nM) eingesetzt.
Verbindung Mip
IC50 ± sdv [µM] FKBP12
IC50 ± sdv [µM]
12a 87.0 ± 5.0 2.6 ± 0.4
12d 14.3 ± 0.4 4.57 ±0.77
19a 32.3 ± 7.2 8.78 ± 1.12
19b 60.3 ± 9.8 11.3 ± 1.54
19c 77.0 ± 8.3 > 20
20b 47.1 ± 5.4 > 20
21a 81.0 ± 5.2 > 20
21b 51.3 ± 1.9 > 20
21c 69.3 ± 7.4 > 20
22a 9.0 ± 0.7 0.2 ± 0.02
22b 6.0 ± 0.7 0.2 ± 0.03
22c 27.4 ± 4.9 2.61 ± 0.41
22d 91.3 ± 15.7 11.5 ± 0.9
22e 41.3 ± 16.0 0.19 ± 0.03
23a 6.8 ± 0.6 4.6 ± 0.9
23b 52.0 ± 16.0 > 20
24a 5.8 ± 1.6 1.75 ± 0.31
25 85.0 ± 20.7 1.63 ± 0.86
26a 28.0 ± 1.7 0.12 ± 0.1
26d 46.8 ± 8.4 0.21 ± 0.07
26f 73.9 ± 3.5 3.58 ± 0.96
26i 71.3 ± 2.7 0.42 ± 0.08
27b 91.7 ± 12.4 0.45 ± 0.03
27d 32.0 ± 2.4 0.15 ± 0.01
35a 47.3 ± 9.1 0.47 ± 0.04
35b 63.5 ± 3.0 0.29 ± 0.02
37b 56.1 ± 9.5 0.06 ± 0.006
Tabelle 8: IC50-Werte ausgewählter Verbindungen an Mip und FKBP12
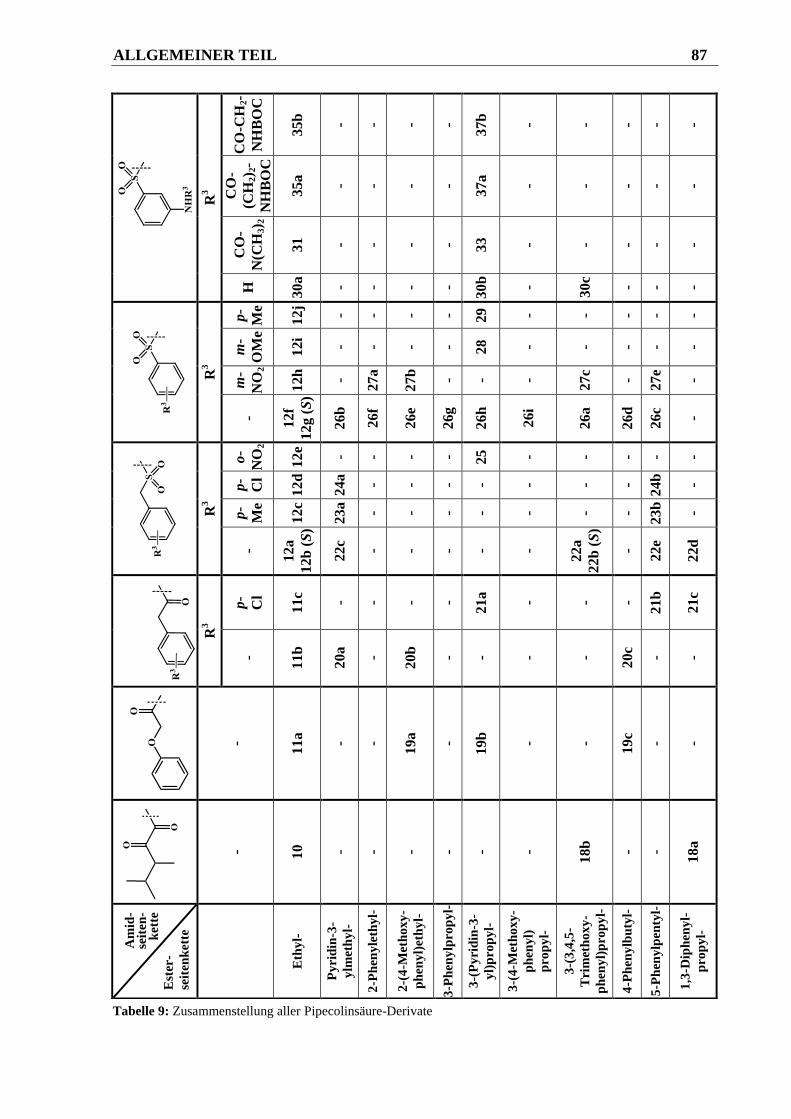
ALLGEMEINER TEIL 87
R
3
CO
-CH
2-
NH
BO
C
35
b
- - - -
37
b
- - - - -
CO
-
(CH
2) 2
-
NH
BO
C
35
a
- - - -
37
a
- - - - -
CO
-
N(C
H3) 2
31
- - - - 33
- - - - -
H
30
a
- - - -
30
b
-
30
c
- - -
R3
p-
Me
12
j
- - - - 29
- - - - -
m-
OM
e
12
i
- - - - 28
- - - - -
m-
NO
2
12
h
-
27
a
27
b
- - -
27
c
-
27
e
-
-
12
f
12
g (
S)
26
b
26
f
26
e
26
g
26
h
26
i
26
a
26
d
26
c -
R3
o-
NO
2
12e - - - - 2
5
- - - - -
p-
Cl
12d
24a
- - - - - - -
24b
-
p-
Me
12c
23a
- - - - - - -
23b
-
-
12a
12b
(S
)
22c - - - - -
22a
22b
(S
)
-
22e
22d
R3
p-
Cl
11c - - - -
21a
- - -
21b
21c
-
11b
20a
-
20b
- - - -
20c
- -
-
11a
- -
19a
-
19b
- -
19c
- -
- 10
- - - - - -
18
b
- -
18
a
A
mid
- se
iten
- k
ette
Est
er-
seit
enk
ette
Eth
yl-
Py
rid
in-3
-
ylm
eth
yl-
2-P
hen
yle
thy
l-
2-(
4-M
eth
oxy
-
ph
eny
l)et
hy
l-
3-P
hen
ylp
rop
yl-
3-(
Py
rid
in-3
-
yl)
pro
py
l-
3-(
4-M
eth
oxy
-
ph
eny
l)
pro
py
l-
3-(
3,4
,5-
Tri
met
ho
xy
-
ph
eny
l)p
rop
yl-
4-P
hen
ylb
uty
l-
5-P
hen
ylp
enty
l-
1,3
-Dip
hen
yl-
pro
py
l-
Tabelle 9: Zusammenstellung aller Pipecolinsäure-Derivate

88 ALLGEMEINER TEIL
In Tabelle 8 sind die IC50-Werte der Substanzbibliotheken an Mip bzw. FKBP12 zusammengestellt. Es
werden nur diejenigen Verbindungen dargestellt, die eine Inhibition der bakteriellen PPIase zeigten.
Bei allen anderen - wie den niedermolekularen Sulfonsäureamiden 1-6, nahezu allen Pipecolinsäure-
ethylestern 10-12, den α-Ketoamiden 18a und 18b, den meta- bzw. para-substituierten Phenyl-
sulfonsäureamid-Derivaten 28-31 und 33, den Pipecolinsäure-Variationen 38-44 sowie den neu-
designten Verbindungen 46-48 - lag der Mip-IC50-Wert deutlich über einem Wert von 100 µM, so dass
eine effiziente PPIase-Hemmung durch diese Verbindungen ausgeschlossen werden kann.
Die niedermolekularen Sulfonsäureamide 1-6 hatten keine Wirkung auf die bakterielle PPIase; eine
Hemmung der FKBP12-PPIase durch diese Verbindungen war ebenfalls nicht zu beobachten. Ein
Grund hierfür könnte sein, dass die Verbindungen 1-6 zwar in die hydrophobe Bindetasche von Mip
bzw. FKBP12 einlagern, aber nicht genügend Aktivität für die PPIase-Inhibition aufweisen, da sie den
Raum der Kavität nicht vollständig ausnutzen. In einem solchen Fall sollten höhere Inhibitor-
konzentrationen eine gewünschte PPIase-Hemmung herbeiführen. Um diese Hypothese zu überprüfen,
wurden daher Konzentrationen bis 100 µM eingesetzt; eine Messung in diesem Bereich war jedoch
nicht möglich, da alle Verbindungen ab einer Konzentration von 60 µM präzipitierten. Um dennoch
die Bindungsverhältnisse zwischen den „kleinen“ Sulfonsäureamiden und Mip zu untersuchen,
wurden NMR-Bindungsstudien durchgeführt (siehe Kapitel 3.2.2).
Bei den Pipecolinsäure-Derivaten konnte zunächst ebenfalls keine Inhibition der enzymatischen
Aktivität von Mip durch die Derivate mit der Grundstruktur A (d.h. der α- Ketoamid-Derivate (siehe
Kapitel 2.2.1); Verbindungen 10, 18a und 18b) beobachtet werden; es zeigte sich aber eine geringe
FKBP12-PPIase-Hemmung (IC50-Wert von Verbindung 18a = 4.2 µM). Auch durch strukturelle
Variationen wie z.B. ein Ersetzen des Piperidinrings in Zielverbindung A durch eine offenkettige
Struktur (Verbindung 42) oder einen Fünfring (Verbindung 44) konnte keine Inhibition von Mip
erzielt werden.
Im Gegensatz dazu wurde im Fall der Verbindungen mit der Grundstruktur B (d.h. der
Sulfonsäureamid-Derivate (siehe Kapitel 2.2.1)) eine Inhibition der bakteriellen PPIase im
mikromolaren Bereich beobachtet, jedoch nur bei „größeren“ Pipecolinsäureestern. „Kleinere“
Pipecolinsäureethylester inhibierten das bakterielle Enzym kaum, mit Ausnahme der Verbindung 12d
(IC50-Wert = 14.3 µM), welche eine Vorstufe von Verbindung 24a darstellt.
Eine sehr gute Aktivität unter den „größeren“ Pipecolinsäureestern zeigten das unsubstituierte
Benzylsulfonsäureamid-Derivat 22b (Zielstruktur B; IC50 = 6.0 µM) und das para-Methyl-
substituierte Benzylsulfonsäureamid-Derivat 23a (IC50 = 6.8 µM); der vielversprechendste Inhibitor
war das para-Chlor-substituierte Benzylsulfonsäureamid-Derivat 24a mit einem IC50-Wert von
5.8 µM. Alle anderen Strukturvariationen der Sulfonsäureamidseitenkette - wie z.B. der Austausch des
Benzylrings durch Phenylreste (Verbindungen 26-30), der Austausch der Sulfonsäureamid-Funktion
durch ein Carbonsäureamid (Verbindungen 19-21) oder die Einführung von längeren meta-
Substituenten (Verbindungen 31, 33, 35 und 37) - zeigten zwar in Einzelfällen eine Hemmung der

ALLGEMEINER TEIL 89
bakteriellen PPIase, der entsprechende IC50-Wert lag aber deutlich höher. Hierbei muss jedoch noch
erwähnt werden, dass nur im Fall der Carbonsäureamide 19c, 20b, 21a-c eine gewünschte bevorzugte
Hemmung von Mip erzielt werden konnte; alle anderen aktiven Sulfonsäureamid-Derivate zeigten
neben der Mip-Inhibition eine zusätzliche FKBP12-Hemmung.
Die Tatsache dieser „Unselektivität“ soll aber bei der weiteren Wirkstoffsuche zunächst nicht
berücksichtigt werden, da davon auszugehen ist, dass die FKBP12-Inhibition in diesen Fällen keine
unerwünschte Immunsuppression nach sich zieht, da die Sulfonsäureamid-Derivate nicht über die
Effektordomäne von Rapamycin bzw. FK506 verfügen, welche für die Wechselwirkungen mit mTOR
bzw. Calcineurin und damit für die Immunsuppression verantwortlich ist. Diese Hypothese gilt es in
Nachfolgearbeiten jedoch noch zu beweisen.
Bei der Betrachtung der inhibitorischen Werte fällt ferner auf, dass - im Widerspruch zur
ursprünglichen Annahme - die Propylkette des Esterrests nicht essenziell für die PPIase-Hemmung ist,
da bei allen Estern mit Alkylketten von n = 1-5 (kombiniert mit unterschiedlichen Sulfonsäureamiden)
eine inhibitorische Wirkung beobachtet werden konnte.
Abb. 71: Mögliche Variationen am Grundgerüst von aktiven Pipecolinsäure-Derivaten
Zusammenfassend lässt sich sagen, dass die Ergebnisse der Docking-Studien der Pipecolinsäure-
Derivate (siehe Kapitel 2.2.1) im pharmakologischen Test vollkommen bestätigt werden konnten.
Während Zielverbindungen mit der Grundstruktur A keine Mip-Hemmung zeigten, wiesen diejenigen
mit der Grundstruktur B am bakteriellen Enzym IC50-Werte im mikromolaren Bereich auf. Mit einer
Benzylsulfonsäureamidstruktur konnten die besten inhibitorischen Werte erzielt werden; an dieser
Struktur ist zwar sowohl der Austausch der Sulfongruppe durch eine Carbonylfunktion als auch der
des Benzylrings durch einen Phenylring möglich, beides führt aber zu einer Verschlechterung der
inhibitorischen Aktivität. Im Gegensatz dazu verringert die para-Chlor-Substitution am Benzylring
Bester IC50-Wert bei m = 1;
m = 0 führte zu einer Verringerung
der inhibitorischen Aktivität.
Die Variation der Kettenlänge (n = 1-5)
führte zu keiner Änderung des IC50-Werts.
Die Einführung von p-Cl zeigte keine
Veränderung des IC50-Werts und ist daher möglich.

90 ALLGEMEINER TEIL
den IC50-Wert nicht. Ferner ist der große Esterrest der Verbindungen zwar, wie angenommen, für eine
Inhibition notwendig, sein Alkylspacer, der den aromatischen Teil mit der Pipecolinsäure verbindet,
ist jedoch nicht auf die Propylkette beschränkt, sondern kann zwischen den Kettenlängen n = 1-5
variieren (siehe Abb. 71).
3.2 NMR-Bindungsstudien
3.2.1 Prinzip und Durchführung der 15
N-HSQC-NMR-Experimente
Um die exakten Bindungsverhältnisse zwischen einem Protein und einer chemischen Verbindung zu
bestimmen, eignen sich sogenannte 15
N-HSQC (Heteronuclear Single Quantum Coherence)-NMR-
Experimente. Dabei werden Wechselwirkungen, die zwischen den Kernen von direkt gebundenen
Protonen und Stickstoff-Atomen entstehen, gemessen. Als Ergebnis erhält man ein zweidimensionales
Spektrum, in dem auf der x-Achse die 1H- und auf der y-Achse die
15N-Verschiebungen aufgetragen
sind. Bei der Bindung eines Inhibitors an ein Protein kommt es u.a. zu Wechselwirkungen mit den
Stickstoff-Atomen des Proteins und damit zu einer Veränderung in der chemischen Verschiebung der
betroffenen Kerne. Misst man zunächst das ungebundene Protein und anschließend dieses im
Komplex mit einem möglichen Bindungspartner, kann man in der Überlagerung der beiden Spektren -
im Fall einer Bindung - deutlich die Veränderung in der chemischen Verschiebung der betroffenen
Atome erkennen und damit die Bindetasche identifizieren.
Um die Ergebnisse des Enzymassays (siehe Kapitel 3.1.3) stichprobenartig zu verifizieren, wurden
ausgewählte Verbindungen mittels solcher NMR-Bindungsstudien untersucht. Diese Experimente
wurden an einem 800 MHz NMR-Spektrometer im Arbeitskreis von Prof. Dr. Paul Rösch am Institut
für Biopolymere an der Universität Bayreuth durchgeführt. Für die Messungen wurde die kürzlich
beschriebene 15
N-markierte C-Domäne von Mip verwendet.59
Die zu testenden Verbindungen wurden
in Acetonitril-d3 in einer Konzentration von 23 mM gelöst und dreimal - jeweils in einem Protein-
Ligand-Verhältnis von 1 : 5; 1 : 10 und 1 : 20 - vermessen. Als Referenz wurde bei der Messung des
ungebundenen Proteins die Menge an Inhibitor durch Acetonitril-d3 ersetzt. Die standardisierten
chemischen Verschiebungen wurden als Durchschnitt der Veränderungen der chemischen
Verschiebungen (1H und
15N) durch folgende Gleichung dargestellt. Hierbei wurde ein Wert von
0.03 ppm als signifikant angesehen.63
normshift = (δ(1H)
2 + (0.1δ(
15N))
2)
1/2 (Gleichung 4)
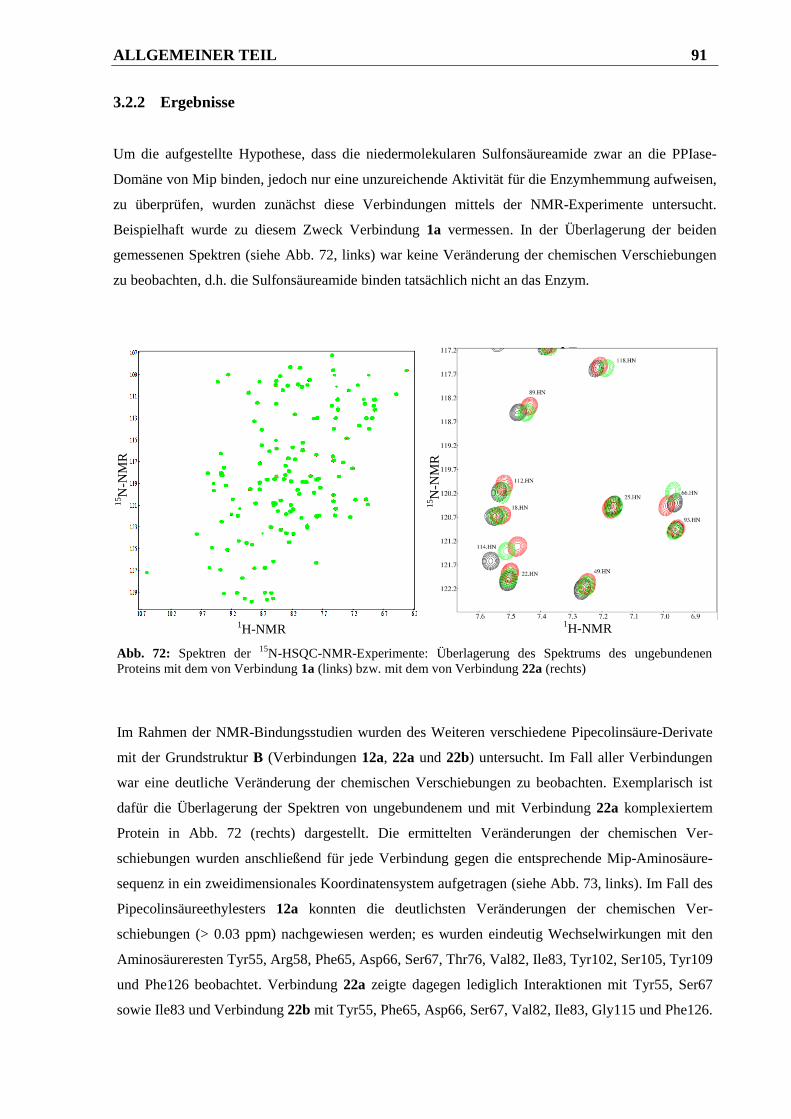
ALLGEMEINER TEIL 91
3.2.2 Ergebnisse
Um die aufgestellte Hypothese, dass die niedermolekularen Sulfonsäureamide zwar an die PPIase-
Domäne von Mip binden, jedoch nur eine unzureichende Aktivität für die Enzymhemmung aufweisen,
zu überprüfen, wurden zunächst diese Verbindungen mittels der NMR-Experimente untersucht.
Beispielhaft wurde zu diesem Zweck Verbindung 1a vermessen. In der Überlagerung der beiden
gemessenen Spektren (siehe Abb. 72, links) war keine Veränderung der chemischen Verschiebungen
zu beobachten, d.h. die Sulfonsäureamide binden tatsächlich nicht an das Enzym.
Abb. 72: Spektren der 15
N-HSQC-NMR-Experimente: Überlagerung des Spektrums des ungebundenen
Proteins mit dem von Verbindung 1a (links) bzw. mit dem von Verbindung 22a (rechts)
Im Rahmen der NMR-Bindungsstudien wurden des Weiteren verschiedene Pipecolinsäure-Derivate
mit der Grundstruktur B (Verbindungen 12a, 22a und 22b) untersucht. Im Fall aller Verbindungen
war eine deutliche Veränderung der chemischen Verschiebungen zu beobachten. Exemplarisch ist
dafür die Überlagerung der Spektren von ungebundenem und mit Verbindung 22a komplexiertem
Protein in Abb. 72 (rechts) dargestellt. Die ermittelten Veränderungen der chemischen Ver-
schiebungen wurden anschließend für jede Verbindung gegen die entsprechende Mip-Aminosäure-
sequenz in ein zweidimensionales Koordinatensystem aufgetragen (siehe Abb. 73, links). Im Fall des
Pipecolinsäureethylesters 12a konnten die deutlichsten Veränderungen der chemischen Ver-
schiebungen (> 0.03 ppm) nachgewiesen werden; es wurden eindeutig Wechselwirkungen mit den
Aminosäureresten Tyr55, Arg58, Phe65, Asp66, Ser67, Thr76, Val82, Ile83, Tyr102, Ser105, Tyr109
und Phe126 beobachtet. Verbindung 22a zeigte dagegen lediglich Interaktionen mit Tyr55, Ser67
sowie Ile83 und Verbindung 22b mit Tyr55, Phe65, Asp66, Ser67, Val82, Ile83, Gly115 und Phe126.
1H-NMR 1
H-NMR
15N
-NM
R
15N
-NM
R
92 ALLGEMEINER TEIL
Werden die so ermittelten Aminosäuren anschließend auf die Oberfläche von Mip projiziert, können
die Binderegionen, an denen der Inhibitor Wechselwirkungen mit dem Protein eingeht, eindeutig
identifiziert werden (siehe Abb. 73, rechts).
Abb. 73: Veränderungen der chemischen Verschiebungen der Aminosäurereste der Mip-PPIase-Domäne bei
Bindung der Verbindungen 12a, 22a und 22b (links); Mapping der signifikanten Veränderungen der
chemischen Verschiebung auf die Mip-Oberfläche (rechts; orange = Binderegionen von Verbindung 22b)63
Zusammenfassend lässt sich sagen, dass die Ergebnisse des Enzymassays durch die NMR-
Bindungsstudien ohne Ausnahme bestätigt werden konnten. Sowohl die „Nicht-Bindung“ der
niedermolekularen Sulfonsäureamide als auch die Bindung der Pipecolinsäure-Derivate konnten
mittels dieser spektrometrischen Methode nachgewiesen werden.
Verbindung
22b
Ver
änder
ung d
er c
hem
isch
en V
ersc
hie
bu
ng [
ppm
]
Verbindung
22a
Verbindung
12a
Aminosäuresequenz
20 40 60 80 100 120
20
20
40 100 60 80
40 100 80 60
120
120
0.08
0.03
0
0.08
0.03
0
0.08
0.03
0

ALLGEMEINER TEIL 93
3.3 In-vivo-Testsystem
3.3.1 Transwell-Assay
Als In-vivo-Testsystem für Lp-Mip eignet sich ein Transwell-Assay, der kürzlich im Arbeitskreis von
Prof. Dr. Michael Steinert an der Universität Würzburg entwickelt und etabliert wurde.33
Mit Hilfe
von Transwell-Systemen kann die Fähigkeit von Mikroorganismen, biologische Barrieren zu
überwinden, untersucht werden. Im Fall von Mip sollte auf diese Weise die Transmigration der
Legionellen durch die Lungenbarriere beobachtet (siehe Kapitel 1.1.3.2) und somit die In-vivo-
Aktivität der in vitro wirksamen Pipecolinsäure-Derivate bestimmt werden. Da bereits zu Beginn der
Studien große Probleme bei der Bildung der Epithelzellschicht auf der porösen Transwell-Membran
auftraten, konnte diese In-vivo-Untersuchung nicht durchgeführt werden. Um dennoch die Aktivität
der Pipecolinsäure-Derivate an lebenden Zellen zu bestimmen, wurde schließlich der Gentamicin-
Infektionsassay verwendet.
3.3.2 Infektionsstudien
3.3.2.1 Durchführung
Mit Hilfe von Infektionsstudien sollte die Aktivität der synthetisierten Verbindungen in vivo
bestimmt werden. Dabei sollte der Einfluss von Verbindung 22b auf die Invasion und die
intrazelluläre Replikation von L. pneumophila in Makrophagen-ähnlichen Zellen (U937) ermittelt
werden. Zu diesem Zweck wurden die Zellen entweder mit dem Legionellen-Wildtyp, einer Mip-
negativen Legionellen-Mutante oder dem E. coli-Stamm HB101 infiziert sowie mit der zu
untersuchenden Verbindung 22b (50 µM) für mehrere Stunden inkubiert. Als Positiv-Kontrolle
wurde der bekannte Inhibitor Rapamycin verwendet. Nach jeweils 2 h, 24 h und 48 h wurde die
Bakterienzahl im Inneren der U937-Zellen bestimmt.
3.3.2.2 Ergebnisse
Das Diagramm (siehe Abb. 74) zeigt die Ergebnisse des Infektionsassays. Es ist in drei Teil-
diagramme untergliedert. Ein Teildiagramm gibt dabei die Anzahl der Bakterien im Zellinneren
(KbE/ml) nach 2 h, 24 h bzw. 48 h wider.
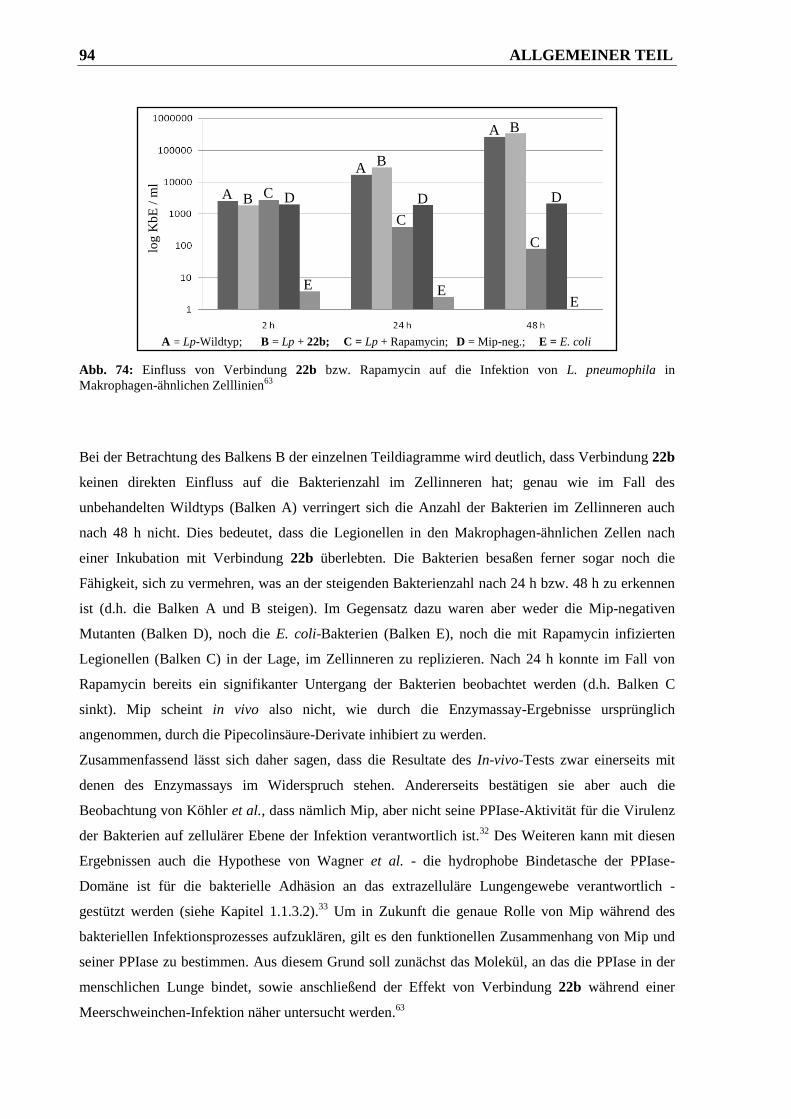
94 ALLGEMEINER TEIL
Abb. 74: Einfluss von Verbindung 22b bzw. Rapamycin auf die Infektion von L. pneumophila in
Makrophagen-ähnlichen Zelllinien63
Bei der Betrachtung des Balkens B der einzelnen Teildiagramme wird deutlich, dass Verbindung 22b
keinen direkten Einfluss auf die Bakterienzahl im Zellinneren hat; genau wie im Fall des
unbehandelten Wildtyps (Balken A) verringert sich die Anzahl der Bakterien im Zellinneren auch
nach 48 h nicht. Dies bedeutet, dass die Legionellen in den Makrophagen-ähnlichen Zellen nach
einer Inkubation mit Verbindung 22b überlebten. Die Bakterien besaßen ferner sogar noch die
Fähigkeit, sich zu vermehren, was an der steigenden Bakterienzahl nach 24 h bzw. 48 h zu erkennen
ist (d.h. die Balken A und B steigen). Im Gegensatz dazu waren aber weder die Mip-negativen
Mutanten (Balken D), noch die E. coli-Bakterien (Balken E), noch die mit Rapamycin infizierten
Legionellen (Balken C) in der Lage, im Zellinneren zu replizieren. Nach 24 h konnte im Fall von
Rapamycin bereits ein signifikanter Untergang der Bakterien beobachtet werden (d.h. Balken C
sinkt). Mip scheint in vivo also nicht, wie durch die Enzymassay-Ergebnisse ursprünglich
angenommen, durch die Pipecolinsäure-Derivate inhibiert zu werden.
Zusammenfassend lässt sich daher sagen, dass die Resultate des In-vivo-Tests zwar einerseits mit
denen des Enzymassays im Widerspruch stehen. Andererseits bestätigen sie aber auch die
Beobachtung von Köhler et al., dass nämlich Mip, aber nicht seine PPIase-Aktivität für die Virulenz
der Bakterien auf zellulärer Ebene der Infektion verantwortlich ist.32
Des Weiteren kann mit diesen
Ergebnissen auch die Hypothese von Wagner et al. - die hydrophobe Bindetasche der PPIase-
Domäne ist für die bakterielle Adhäsion an das extrazelluläre Lungengewebe verantwortlich -
gestützt werden (siehe Kapitel 1.1.3.2).33
Um in Zukunft die genaue Rolle von Mip während des
bakteriellen Infektionsprozesses aufzuklären, gilt es den funktionellen Zusammenhang von Mip und
seiner PPIase zu bestimmen. Aus diesem Grund soll zunächst das Molekül, an das die PPIase in der
menschlichen Lunge bindet, sowie anschließend der Effekt von Verbindung 22b während einer
Meerschweinchen-Infektion näher untersucht werden.63
A = Lp-Wildtyp; B = Lp + 22b; C = Lp + Rapamycin; D = Mip-neg.; E = E. coli
log
Kb
E /
ml
E
D C A B
E
D
C
A B
E
D
C
A B

ALLGEMEINER TEIL 95
3.4 Testungen an Mip-ähnlichen Proteinen
3.4.1 Pharmakologische Testungen an Neisserien und Chlamydien
Alle pharmakologischen Untersuchungen an N. gonorrhoeae und C. trachomatis fanden in der
Arbeitsgruppe von Prof. Dr. Thomas Rudel und Dr. Vera Kozjak-Pavlovic am Institut für
Mikrobiologie der Universität Würzburg statt. Für beide Bakterienarten wurden zur Bestimmung der
inhibitorischen Aktivität der hergestellten Pipecolinsäure-Derivate In-vivo-Infektionsstudien (siehe
Kapitel 3.3.2.1) durchgeführt. Zu diesem Zweck wurden exemplarisch die strukturell
unterschiedlichen Verbindungen 18b, 22a, 27d, 30b sowie 38 ausgewählt.
3.4.1.1 Infektionsstudien an N. gonorrhoeae
Zur Bestimmung der Wachstumshemmung von N. gonorrhoeae wurden die Verbindungen in drei
verschiedenen Konzentrationen (1.6 µM, 8 µM und 40 µM) mit Epithelzellen inkubiert. Als Positiv-
Kontrolle wurde der bekannte Inhibitor Fucoidin, ein Polysaccharid aus Seetang, verwendet.
Anschließend wurden die inkubierten Zellen mit Neisserien, die ein zusätzliches Gen zur Expression
von grün-fluoreszierendem Protein (GFP) in sich trugen, infiziert. Nach einer Stunde wurden die
Mikroorganismen, die weder an die Zellen adhärierten noch in diese invadiert waren, entfernt und die
Epithelzellkerne mit dem Fluoreszenzfarbstoff DAPI (4´,6-Diamin-2-phenylindol) gefärbt. Dadurch
konnte anschließend die Anzahl der grün-fluoreszierenden Neisserien, deren Adhäsions- bzw.
Invasionsfähigkeiten durch die Pipecolinsäure-Derivate nicht gehemmt worden waren, UV-
spektroskopisch bestimmt werden.
Während die Verbindungen 18b und 22a lediglich in der höchsten Konzentration (40 µM) einen
hemmenden Effekt auf die Bakterien zeigten, konnte im Fall von Verbindung 27d bei allen Inhibitor-
konzentrationen eine verminderte Bakterienzahl im Zellinneren verzeichnet werden. Verbindungen
30b und 38 zeigten weder eine Auswirkung auf die Adhäsion noch auf die Invasion der Neisserien.
Diese Ergebnisse spiegeln jedoch nur eine Tendenz bzgl. der Neisserien-Inhibition durch die
Pipecolinsäure-Derivate wider und können nicht als repräsentativ für alle Verbindungen angesehen
werden. Daher sollen in Zukunft weitere Testungen folgen, um schließlich auch mögliche Struktur-
Wirkungsbeziehungen aufstellen zu können. Ferner sollte bei diesem In-vivo-Testsystem zwingend
bedacht werden, dass ein positives Ergebnis - d.h. eine Inhibition der Neisserien-Adhäsion bzw.
Invasion - nicht automatisch auch eine Hemmung von Ng-Mip darstellt. Aus diesem Grund sollen die
positiv getesteten Verbindungen des Weiteren auch bzgl. ihres Mip-Effekts in vitro untersucht
werden.

96 ALLGEMEINER TEIL
3.4.1.2 Infektionsstudien an C. trachomatis
Im Rahmen der Infektionsstudien der obligat intrazellulären Chlamydien wurde die extrazelluläre
Form der Mikroorganismen, die sogenannten Elementarkörper (EB, siehe Kapitel 1.4.1), verwendet.
Da diese EBs metabolisch vollkommen inaktiv sind und sich nicht vermehren können, werden die
Bakterien in diesem Stadium durch einen Inhibitor nur in seltenen Fällen gehemmt. Aus diesem
Grund müssen zusätzliche Infektionsuntersuchungen durchgeführt werden, nachdem die EBs in die
Wirtszelle eingedrungen sind und sich in metabolisch aktive, vermehrungsfähige Retikularkörper
(RB) umgewandelt haben. Eine Inhibition der RBs bewirkt, dass die Replikation der Bakterien zwar
nicht gehemmt wird, aber Nachkommen mit verminderter Infektiösität gebildet werden. Um die
Aktivität potenzieller Inhibitoren auf die Chlamydien-Infektion zu bestimmen, muss deshalb neben
der anfänglichen Infektiösität der EBs auch die der Nachkommen untersucht werden.
In zwei Versuchsreihen wurden Epithelzellen mit den Pipecolinsäure-Derivaten in drei
Konzentrationen (10 µM, 20 µM und 40 µM) inkubiert und mit Chlamydia-EBs infiziert. Als
Positiv-Kontrolle wurde Wortmannin, ein Mykotoxin aus verschiedenen Schimmelpilzarten, das in
der Lage ist, die Adhäsion der Chlamydien-EBs an die Wirtszelle zu hemmen, verwendet. Die
infizierten Epithelzellen eines Untersuchungsansatzes wurden nach 24 Stunden von nicht
gebundenen Bakterien getrennt, fixiert und die Zellkerne mit dem Fluoreszenzfarbstoff Hoechst
33258 gefärbt; die eingedrungenen Bakterien wurden mit dem Cyaninfarbstoff Cy3 gefärbt.
Im zweiten Ansatz wurden die bakteriellen Nachkommen isoliert und mit frischen Epithelzellen
weitere 24 Stunden inkubiert. Danach wurde analog zur EB-Infektion vorgegangen. Auf diese Weise
konnte durch die unterschiedliche Färbung von Mikroorganismen und Epithelzellen die Anzahl an
Bakterien, die in die Wirtszelle eingedrungen waren, ermittelt werden.
Die Ergebnisse der Infektionsstudien zeigten, dass lediglich Verbindung 22a einen inhibitorischen
Effekt auf die Chlamydien-Infektion hat. Während im Fall der Nachkommen-Infektion bei einer
Inhibitorkonzentration von 40 µM eine verminderte Bakterienanzahl im Zellinneren bestimmt
werden konnte, war, wie erwartet, keine Auswirkung von Verbindung 22a auf die anfängliche EB-
Infektion zu erkennen. Bei den anderen Pipecolinsäure-Derivaten 18b, 27d, 30b und 38 wurde weder
ein Einfluss auf die EBs noch auf die Nachkommen beobachtet.
Jedoch kann auch hier auf Grund dieser Resultate nicht direkt auf eine Chlamydien-Hemmung durch
alle Pipecolinsäure-Derivate vom Benzylsulfonsäureamid-Typ (Verbindung 22a) geschlossen
werden. Die Ergebnisse des Infektionstests spiegeln lediglich einen dahingehenden Trend wider. Um
aussagekräftigere Informationen für eine Struktur-Wirkungsbeziehung zu bekommen, müssen in
Zukunft weitere Derivate vom Pipecolinsäure-Typ 22a untersucht werden. Außerdem sollen die in
vivo aktiven Substanzen ebenfalls in vitro bzgl. ihrer Wechselwirkungen mit Mip analysiert werden.
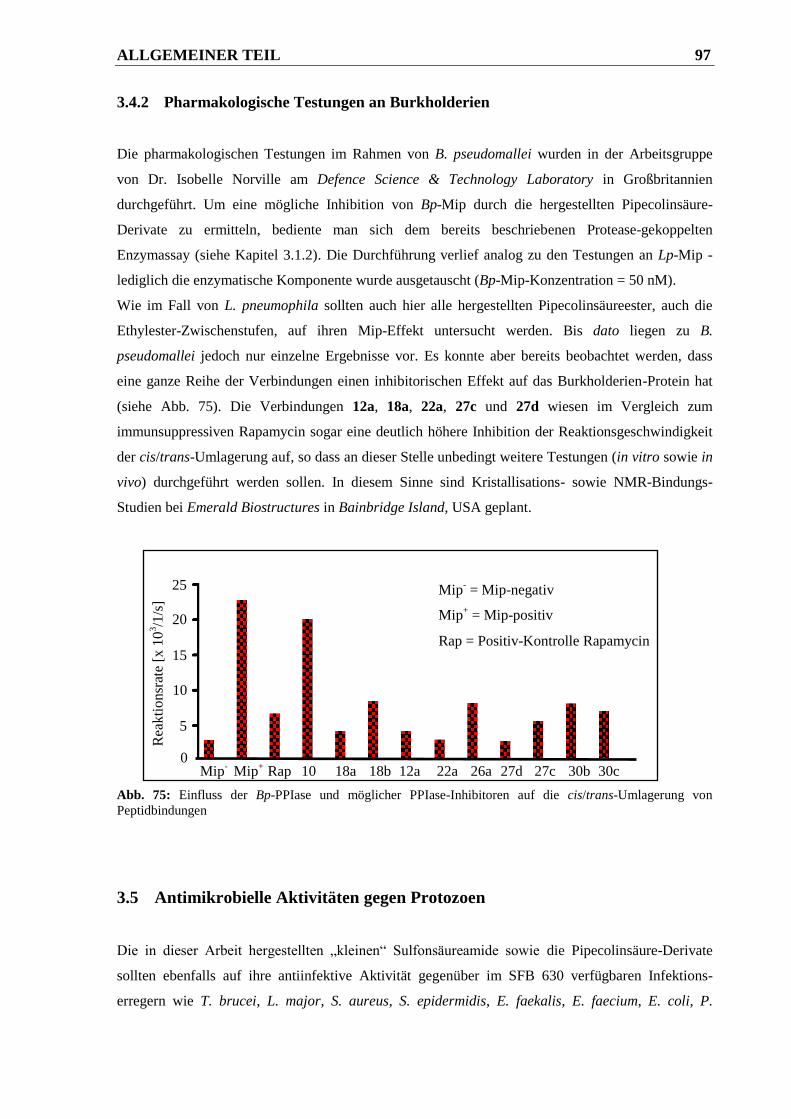
ALLGEMEINER TEIL 97
3.4.2 Pharmakologische Testungen an Burkholderien
Die pharmakologischen Testungen im Rahmen von B. pseudomallei wurden in der Arbeitsgruppe
von Dr. Isobelle Norville am Defence Science & Technology Laboratory in Großbritannien
durchgeführt. Um eine mögliche Inhibition von Bp-Mip durch die hergestellten Pipecolinsäure-
Derivate zu ermitteln, bediente man sich dem bereits beschriebenen Protease-gekoppelten
Enzymassay (siehe Kapitel 3.1.2). Die Durchführung verlief analog zu den Testungen an Lp-Mip -
lediglich die enzymatische Komponente wurde ausgetauscht (Bp-Mip-Konzentration = 50 nM).
Wie im Fall von L. pneumophila sollten auch hier alle hergestellten Pipecolinsäureester, auch die
Ethylester-Zwischenstufen, auf ihren Mip-Effekt untersucht werden. Bis dato liegen zu B.
pseudomallei jedoch nur einzelne Ergebnisse vor. Es konnte aber bereits beobachtet werden, dass
eine ganze Reihe der Verbindungen einen inhibitorischen Effekt auf das Burkholderien-Protein hat
(siehe Abb. 75). Die Verbindungen 12a, 18a, 22a, 27c und 27d wiesen im Vergleich zum
immunsuppressiven Rapamycin sogar eine deutlich höhere Inhibition der Reaktionsgeschwindigkeit
der cis/trans-Umlagerung auf, so dass an dieser Stelle unbedingt weitere Testungen (in vitro sowie in
vivo) durchgeführt werden sollen. In diesem Sinne sind Kristallisations- sowie NMR-Bindungs-
Studien bei Emerald Biostructures in Bainbridge Island, USA geplant.
Abb. 75: Einfluss der Bp-PPIase und möglicher PPIase-Inhibitoren auf die cis/trans-Umlagerung von
Peptidbindungen
3.5 Antimikrobielle Aktivitäten gegen Protozoen
Die in dieser Arbeit hergestellten „kleinen“ Sulfonsäureamide sowie die Pipecolinsäure-Derivate
sollten ebenfalls auf ihre antiinfektive Aktivität gegenüber im SFB 630 verfügbaren Infektions-
erregern wie T. brucei, L. major, S. aureus, S. epidermidis, E. faekalis, E. faecium, E. coli, P.
25
20
15
10
5
0
Mip- = Mip-negativ
Mip+ = Mip-positiv
Rap = Positiv-Kontrolle Rapamycin
Mip- Mip
+ Rap 10 18a 18b 12a 22a 26a 27d 27c 30b 30c
Rea
kti
onsr
ate
[x 1
03/1
/s]
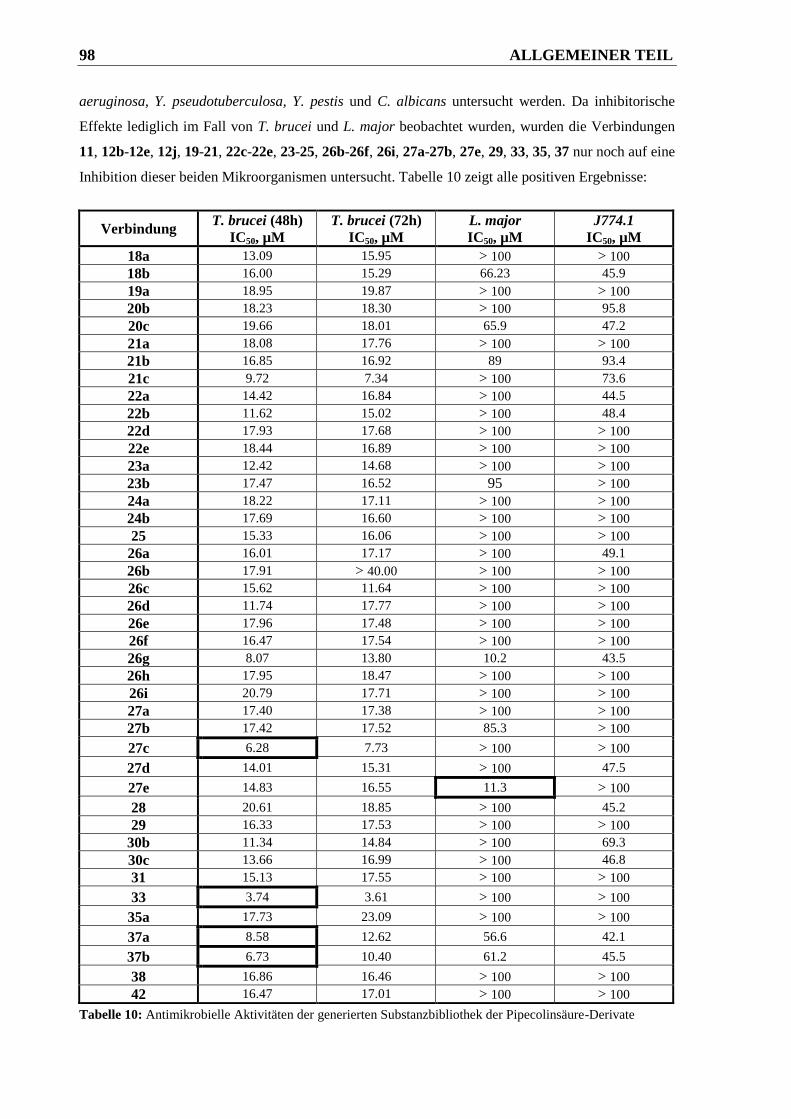
98 ALLGEMEINER TEIL
aeruginosa, Y. pseudotuberculosa, Y. pestis und C. albicans untersucht werden. Da inhibitorische
Effekte lediglich im Fall von T. brucei und L. major beobachtet wurden, wurden die Verbindungen
11, 12b-12e, 12j, 19-21, 22c-22e, 23-25, 26b-26f, 26i, 27a-27b, 27e, 29, 33, 35, 37 nur noch auf eine
Inhibition dieser beiden Mikroorganismen untersucht. Tabelle 10 zeigt alle positiven Ergebnisse:
Verbindung T. brucei (48h)
IC50, µM
T. brucei (72h)
IC50, µM
L. major
IC50, µM
J774.1
IC50, µM
18a 13.09 15.95 > 100 > 100
18b 16.00 15.29 66.23 45.9
19a 18.95 19.87 > 100 > 100
20b 18.23 18.30 > 100 95.8
20c 19.66 18.01 65.9 47.2
21a 18.08 17.76 > 100 > 100
21b 16.85 16.92 89 93.4
21c 9.72 7.34 > 100 73.6
22a 14.42 16.84 > 100 44.5
22b 11.62 15.02 > 100 48.4
22d 17.93 17.68 > 100 > 100
22e 18.44 16.89 > 100 > 100
23a 12.42 14.68 > 100 > 100
23b 17.47 16.52 95 > 100
24a 18.22 17.11 > 100 > 100
24b 17.69 16.60 > 100 > 100
25 15.33 16.06 > 100 > 100
26a 16.01 17.17 > 100 49.1
26b 17.91 > 40.00 > 100 > 100
26c 15.62 11.64 > 100 > 100
26d 11.74 17.77 > 100 > 100
26e 17.96 17.48 > 100 > 100
26f 16.47 17.54 > 100 > 100
26g 8.07 13.80 10.2 43.5
26h 17.95 18.47 > 100 > 100
26i 20.79 17.71 > 100 > 100
27a 17.40 17.38 > 100 > 100
27b 17.42 17.52 85.3 > 100
27c 6.28 7.73 > 100 > 100
27d 14.01 15.31 > 100 47.5
27e 14.83 16.55 11.3 > 100
28 20.61 18.85 > 100 45.2
29 16.33 17.53 > 100 > 100
30b 11.34 14.84 > 100 69.3
30c 13.66 16.99 > 100 46.8
31 15.13 17.55 > 100 > 100
33 3.74 3.61 > 100 > 100
35a 17.73 23.09 > 100 > 100
37a 8.58 12.62 56.6 42.1
37b 6.73 10.40 61.2 45.5
38 16.86 16.46 > 100 > 100
42 16.47 17.01 > 100 > 100
Tabelle 10: Antimikrobielle Aktivitäten der generierten Substanzbibliothek der Pipecolinsäure-Derivate
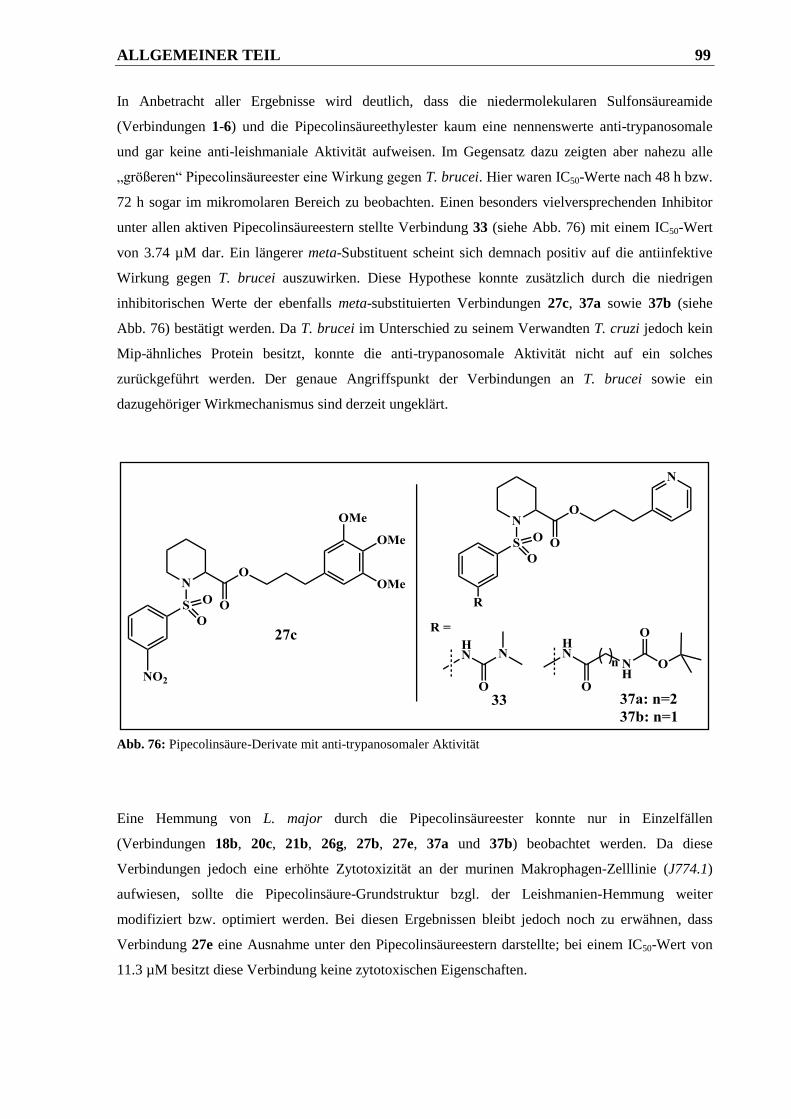
ALLGEMEINER TEIL 99
In Anbetracht aller Ergebnisse wird deutlich, dass die niedermolekularen Sulfonsäureamide
(Verbindungen 1-6) und die Pipecolinsäureethylester kaum eine nennenswerte anti-trypanosomale
und gar keine anti-leishmaniale Aktivität aufweisen. Im Gegensatz dazu zeigten aber nahezu alle
„größeren“ Pipecolinsäureester eine Wirkung gegen T. brucei. Hier waren IC50-Werte nach 48 h bzw.
72 h sogar im mikromolaren Bereich zu beobachten. Einen besonders vielversprechenden Inhibitor
unter allen aktiven Pipecolinsäureestern stellte Verbindung 33 (siehe Abb. 76) mit einem IC50-Wert
von 3.74 µM dar. Ein längerer meta-Substituent scheint sich demnach positiv auf die antiinfektive
Wirkung gegen T. brucei auszuwirken. Diese Hypothese konnte zusätzlich durch die niedrigen
inhibitorischen Werte der ebenfalls meta-substituierten Verbindungen 27c, 37a sowie 37b (siehe
Abb. 76) bestätigt werden. Da T. brucei im Unterschied zu seinem Verwandten T. cruzi jedoch kein
Mip-ähnliches Protein besitzt, konnte die anti-trypanosomale Aktivität nicht auf ein solches
zurückgeführt werden. Der genaue Angriffspunkt der Verbindungen an T. brucei sowie ein
dazugehöriger Wirkmechanismus sind derzeit ungeklärt.
Abb. 76: Pipecolinsäure-Derivate mit anti-trypanosomaler Aktivität
Eine Hemmung von L. major durch die Pipecolinsäureester konnte nur in Einzelfällen
(Verbindungen 18b, 20c, 21b, 26g, 27b, 27e, 37a und 37b) beobachtet werden. Da diese
Verbindungen jedoch eine erhöhte Zytotoxizität an der murinen Makrophagen-Zelllinie (J774.1)
aufwiesen, sollte die Pipecolinsäure-Grundstruktur bzgl. der Leishmanien-Hemmung weiter
modifiziert bzw. optimiert werden. Bei diesen Ergebnissen bleibt jedoch noch zu erwähnen, dass
Verbindung 27e eine Ausnahme unter den Pipecolinsäureestern darstellte; bei einem IC50-Wert von
11.3 µM besitzt diese Verbindung keine zytotoxischen Eigenschaften.

100 ALLGEMEINER TEIL

ALLGEMEINER TEIL 101
4 ZUSAMMENFASSUNG
Die vorliegende Arbeit beschäftigt sich mit der Synthese und Charakterisierung potenzieller
Inhibitoren des Oberflächenproteins Mip von Legionella pneumophila. Der gramnegative
Mikroorganismus ist der ursächliche Erreger der Legionellose. Die Erkrankung kann in zwei
verschiedenen Formen auftreten, dem Pontiac-Fieber, einer Grippe-ähnlichen Atemwegserkrankung,
und der Legionärskrankheit, einer schweren Lungenentzündung mit einer Mortalitätsrate von bis zu
30 %. Natürliche und künstlich geschaffene Süßwassersysteme bilden den biologischen Lebensraum
der Bakterien. Aus dieser Umgebung werden sie über technische Vektoren wie Duschen oder
Klimaanlagen durch Inhalation kontaminierter Aerosole auf den Menschen übertragen, wo sie
alveoläre Makrophagen besiedeln und eine pulmonale Infektion hervorrufen. Um die Zellen in der
Lunge zu erreichen, müssen die Mikroorganismen jedoch zuerst die alveoläre Barriere, bestehend aus
einer Epithelzellschicht und extrazellulärer Matrix, überwinden. Dafür ist das Oberflächenprotein
Mip, der Hauptvirulenzfaktor, verantwortlich. Mip ist ein Homodimer, das sich aus einer C- und
einer N-Domäne zusammensetzt. Während der N-Terminus für die Dimerisierung des Proteins
verantwortlich ist, weist der C-Terminus die typische Faltung einer Peptidyl-Prolyl-cis/trans-
Isomerase (PPIase) auf. Der Vergleich der Aminosäuresequenz der Mip-C-Domäne mit der Domäne
verschiedener humaner PPIasen zeigte eine besonders große Homologie zu FKBP12, welches zur
Familie der FK506-bindenden Proteine gehört und eine wichtige Rolle innerhalb des menschlichen
Immunsystems spielt. Bemerkenswerterweise hemmen bekannte immunsuppressive FKBP12-
Inhibitoren wie FK506 und Rapamycin neben der humanen PPIase ebenfalls das bakterielle Mip.
Außerdem wurde beobachtet, dass die C-terminale Mip-PPIase an Kollagen IV, den Hauptbestandteil
in der menschlichen Lunge, bindet und somit für die Transmigration der Legionellen in die Lunge
verantwortlich ist. Mip-Inhibitoren sollten demnach eine Legionellen-Infektion verhindern können.
Zur Verifizierung der Hypothese sollten daher im Rahmen dieser Arbeit neue Leitstrukturen für Mip-
PPIase-Inhibitoren entwickelt werden. Mit Hilfe von Molecular-Modelling-Untersuchungen
basierend auf der NMR-Struktur 2VCD wurde als eine mögliche Leitstruktur N,N-Dimethylphenyl-
sulfonsäureamid identifiziert. Deshalb sollten diese Verbindung sowie Analoga, die diverse
strukturelle Variationen in der Sulfonsäureamidseitenkette oder im aromatischen Rest enthielten,
hergestellt werden (siehe Abb. 77).

102 ALLGEMEINER TEIL
Abb. 77: Synthetisierte Verbindungen 1-6 (basierend auf der Leitstruktur von N,N-Dimethylphenyl-
sulfonsäureamid)
Obwohl die Immunsuppressiva FK506 und Rapamycin Mip-Inhibitoren darstellen, können sie auf
Grund ihrer immunsuppressiven Eigenschaften nicht in der Legionellosetherapie eingesetzt werden.
Die makrozyklischen Immunsuppressiva setzen sich im Gegensatz zu N,N-Dimethylphenyl-
sulfonsäureamid allerdings aus zwei strukturellen Einheiten, einer Binde- sowie einer Effektor-
domäne, zusammen. Die Bindedomäne mit dem Pipecolinsäure-Grundgerüst ist für die Wechsel-
wirkungen mit Mip und FKBP12 verantwortlich. Die Effektordomäne hingegen ist der aliphatische
Teil der Makrozyklen, der erst durch die Bildung eines ternären Komplexes eine Immunsuppression
hervorruft. Somit können Verbindungen vom Pipecolinsäure-Typ keine immunsuppressive Wirkung
haben und stellen demnach optimale, neue Leitstrukturen für Mip-Inhibitoren dar.
Aus diesem Grund wurden die zwei literaturbekannten, nicht-immunsuppressiven FKBP12-
Inhibitoren A und B ausgewählt und in verschiedenen Docking-Studien untersucht. Das Molecular-
Modelling zeigte, dass nur Verbindung B reproduzierbare Interaktionen mit Mip eingehen kann und
demnach ein potenzieller Inhibitor ist. Um dies zu überprüfen, sollten beide Verbindungen sowie
eine Mischform, die die strukturellen Merkmale von A und B kombiniert, hergestellt werden (siehe
Abb. 78).
Da die literaturbekannte Synthese der Grundstruktur A auch durch diverse Optimierungsversuche
nicht reproduzierbar war, wurde eine neue Strategie entwickelt. Diese lieferte als Produkt zwar nicht
die exakte Struktur von A, die hergestellte Verbindung 18a unterschied sich jedoch lediglich durch
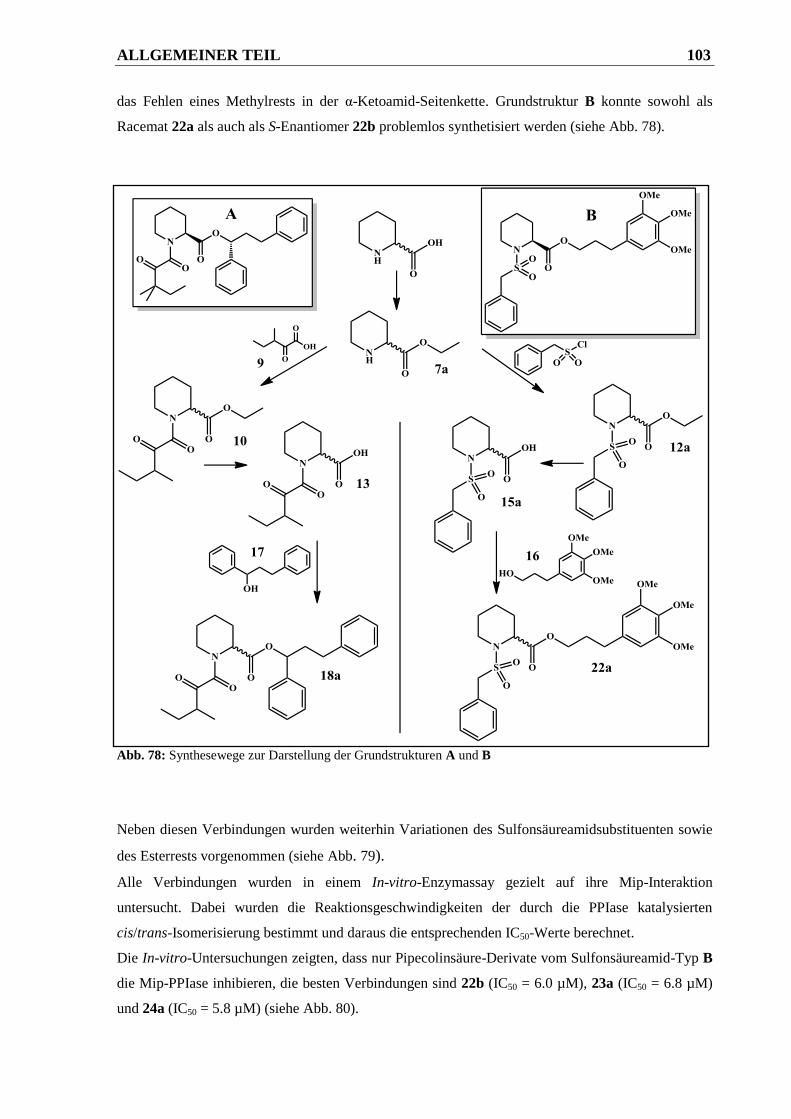
ALLGEMEINER TEIL 103
das Fehlen eines Methylrests in der α-Ketoamid-Seitenkette. Grundstruktur B konnte sowohl als
Racemat 22a als auch als S-Enantiomer 22b problemlos synthetisiert werden (siehe Abb. 78).
Abb. 78: Synthesewege zur Darstellung der Grundstrukturen A und B
Neben diesen Verbindungen wurden weiterhin Variationen des Sulfonsäureamidsubstituenten sowie
des Esterrests vorgenommen (siehe Abb. 79).
Alle Verbindungen wurden in einem In-vitro-Enzymassay gezielt auf ihre Mip-Interaktion
untersucht. Dabei wurden die Reaktionsgeschwindigkeiten der durch die PPIase katalysierten
cis/trans-Isomerisierung bestimmt und daraus die entsprechenden IC50-Werte berechnet.
Die In-vitro-Untersuchungen zeigten, dass nur Pipecolinsäure-Derivate vom Sulfonsäureamid-Typ B
die Mip-PPIase inhibieren, die besten Verbindungen sind 22b (IC50 = 6.0 µM), 23a (IC50 = 6.8 µM)
und 24a (IC50 = 5.8 µM) (siehe Abb. 80).
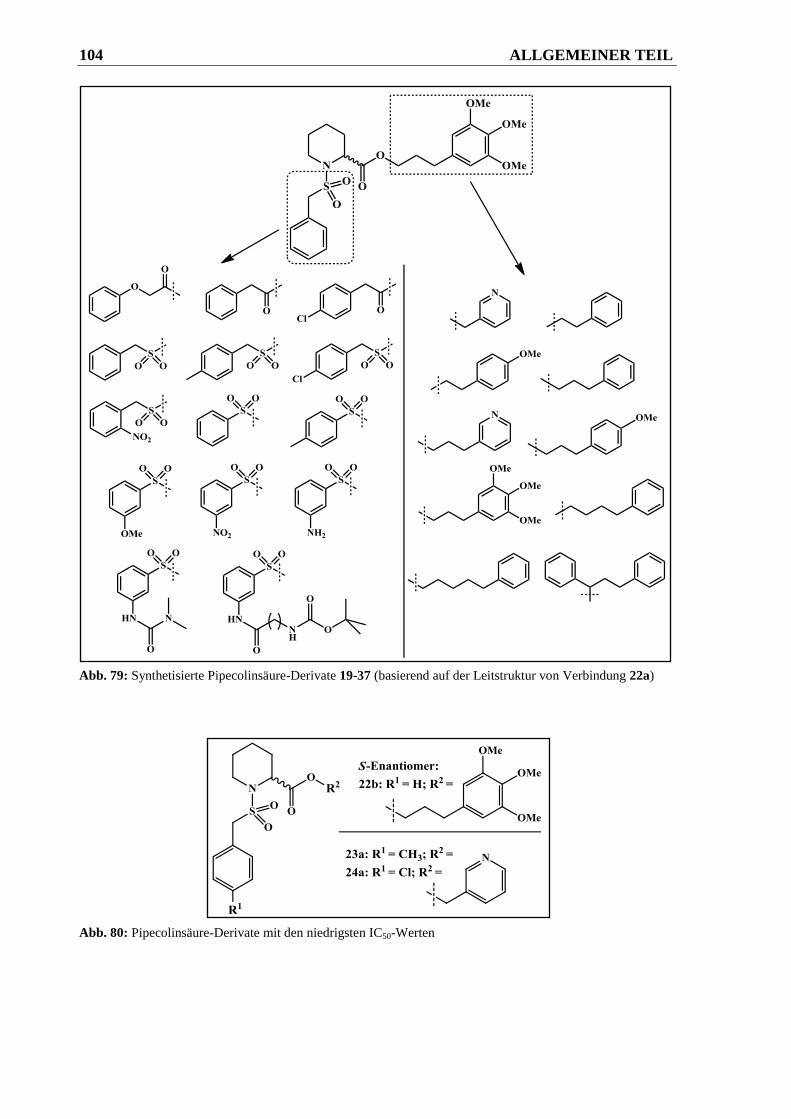
104 ALLGEMEINER TEIL
Abb. 79: Synthetisierte Pipecolinsäure-Derivate 19-37 (basierend auf der Leitstruktur von Verbindung 22a)
Abb. 80: Pipecolinsäure-Derivate mit den niedrigsten IC50-Werten

ALLGEMEINER TEIL 105
Der strukturelle Vergleich dieser drei Verbindungen mit den restlichen Pipecolinsäure-Derivaten
zeigte, dass der Benzylrest der Sulfonsäureamidseitenkette essenziell für eine Mip-Inhibition ist. Der
Aromat kann entweder unsubstituiert oder para-Chlor-substituiert vorliegen. Der Austausch mit
einem Phenylsubstituenten ist prinzipiell möglich, führt aber zu weniger aktiven Verbindungen. Der
Austausch der Sulfonsäureamid- durch eine Carbonsäureamid-Struktur führt ebenfalls zu einem
Aktivitätsverlust. Die Länge der Esterkette spielt keine entscheidende Rolle für eine Mip-Inhibition.
Der Esterrest muss aus einem endständigen aromatischen Teil bestehen, der über einen Alkyllinker
(n = 1-5) mit dem Piperidinring verknüpft ist.
Neben den enzymatischen In-vitro-Testungen wurden exemplarisch für die Verbindungen 1a, 12a,
22a und 22b HSQC-NMR-Experimente zur Bestimmung der Inhibitor-Proteinbindung durchgeführt.
Hier zeigte das „kleine“ Sulfonsäureamid 1a keine Wechselwirkungen mit Mip, weshalb
angenommen wurde, dass die Verbindungen 1-6 die Mip-Kavität nicht ausreichend ausfüllen und
daher nicht genügend Aktivität für eine Inhibition besitzen. Dagegen wurden bei allen drei
Pipecolinsäure-Derivaten 12a, 22a und 22b Interaktionen zwischen Inhibitor und Mip beobachtet,
wodurch die Ergebnisse des Enzymassays bestätigt wurden.
Für Verbindung 22b wurde zusätzlich die In-vivo-Wachstumshemmung der Legionellen mittels
Gentamicin-Infektionsstudien ermittelt. Nach der Inkubation von Makrophagenzellen mit
Mikroorganismen und Inhibitor 22b konnte jedoch keine verminderte Bakterienanzahl im Zell-
inneren beobachtet werden, d.h. das Pipecolinsäure-Derivat 22b hat keine signifikanten
Auswirkungen auf die Legionellen-Infektion in vivo. Eine Erklärung für die gute In-vitro-, aber
schlechte In-vivo-Aktivität ist, dass Mip als Gesamtmolekül, jedoch nicht seine PPIase-Aktivität für
die bakterielle Infektion verantwortlich ist. In Zukunft sollen daher weitere Studien den funktionalen
Zusammenhang von Mip und seiner PPIase aufklären.
Mip-ähnliche Proteine wurden auch in Bakterienarten wie Chlamydia trachomatis, Neisseria
gonorrhoeae und Burkholderia pseudomallei identifiziert. In vorläufigen In-vivo-Untersuchungen
konnte für Verbindung 22a eine Inhibition des Chlamydienwachstums und für die Verbindungen
18b, 22a und 27d eine Inhibition des Neisserienwachstums beobachtet werden. Es bleibt allerdings
offen, ob dies auf eine Mip-Inhibition zurückzuführen ist. Eine mögliche Inhibition der Mip-PPIase
von B. pseudomallei wurde für alle Pipecolinsäure-Derivate 18-38, 42 und 44 untersucht. Eine
Enzymhemmung, die größer war als die von Rapamycin, wurde für die Verbindungen 12a, 18a, 22a,
27c und 27d beobachtet.

106 ALLGEMEINER TEIL

ALLGEMEINER TEIL 107
5 SUMMARY
The present work focused on the synthesis and characterization of potential inhibitors of the surface
protein Mip of Legionella pneumophila. The gram-negative microorganism is the causative agent of
Legionellosis, which may occur in two different progressive forms, the Pontiac fever, a flu-like
respiratory disease, and the Legionnaires disease, a severe pneumonia with mortality rate of up to
30 %. Natural and man-made fresh water systems provide a biological habitat to the bacteria. From
this environment they were transmitted into humans via technical vectors such as showers or air
conditioners by inhaling the contaminated aerosols. Within the human lung, they affect alveolar
macrophages and cause a pulmonary infection. Though, to affect the alveolar macrophages the
microorganisms have first to cross the alveolar barrier, which consists of epithelial cells and an
extracellular matrix. For this, the surface protein Mip, which represents the main virulence factor of
the bacterium, is responsible. Mip forms a stable homodimer, which consists of two domains, a C-
and an N-domain. While the N-terminus is responsible for the dimerization of the protein, the C-
terminus exhibits the typical folding of a peptidyl-prolyl-cis/trans-isomerase (PPIase). The
comparison of the amino acid sequence of the Mip-C-domain and the domain of different human
PPIases shows a particularly high homology to FKBP12. This human PPIase belongs to the family of
the FK506 binding proteins and plays an important role within the human immune system.
Remarkably, known immunosuppressive FKBP12-inhibitors such as FK506 and Rapamycin are also
able to inhibit the bacterial Mip. Furthermore, it was observed that the C-terminal Mip-PPIase binds
to collagen IV, the main component of the human lung, and therefore, Mip is responsible for the
transmigration of Legionella. Due to this fact, Mip-inhibitors should prevent a Legionellosis.
To verify this hypothesis, the intention of this work was to develop novel lead structures for Mip-
PPIase-inhibitors. By means of molecular modeling studies based on the NMR structure 2VCD N,N-
dimethylbenzenesulfonamide was identified as a potential lead structure. Therefore, this compound
as well as analogues, containing structural variations of the sulfonamide side chain or the aromatic
residue, were aimed to prepare (see Fig. 1).
Although the macrolides FK506 and Rapamycin inhibit Mip, they cannot be used for the treatment of
Legionellosis due to their immunosuppressive effects. Compared to N,N-dimethylbenzene-
sulfonamide the macro cyclic, immunosuppressive drugs consist of two structural domains, a binding
domain and an effector domain. The binding domain with the pipecolic acid feature is responsible for
the interactions with Mip and FKBP12, respectively, whereas the effector domain, the aliphatic part
of the molecule, causes the immunosuppression by forming a ternary complex. Therefore,
compounds of the pipecolic acid type cannot affect an immunosuppression and thus, represent
optimal, novel lead structures.

108 ALLGEMEINER TEIL
Due to this fact, the two common, non-immunosuppressive FKBP12-inhibitors A and B were ana-
lyzed by means of different docking studies with the result that only compound B is able to interact
with Mip. Therefore, it was assumed that B must be a potential Mip-inhibitor. To verify this hypo-
thesis, both compounds A and B as well as a hybrid of them should be prepared (see Fig. 2).
Fig. 1: Synthesized compounds 1-6 (based on the lead structure of N,N-dimethylbenzenesulfonamide)
Despite different optimizing strategies, the reported synthetic route for compound A could not be
reproduced. Therefore, a novel strategy has been developed, but did not result in the exact structure
of A. However, compared to structure A the synthesized compound 18a was missing only one
methylene residue in the α-ketoamide side chain. Compound B could be prepared in the racemic
form 22a as well as in the enantiopure form 22b (see Fig. 2).
In addition to these compounds, further variations of the sulfonamide substituent or the ester residue
were prepared (see Fig. 3).
To determine the interaction with Mip, all synthesized compounds were analyzed by means of an in
vitro enzyme assay. For this, the reaction rates of the PPIase catalyzed cis/trans-isomerization were
determined and the corresponding IC50 values calculated. During the in vitro studies, it was observed
that only pipecolic acid compounds of the sulfonamide type B inhibit the Mip-PPIase, the most
promising compounds are 22b (IC50 = 6.0 µM), 23a (IC50 = 6.8 µM) und 24a (IC50 = 5.8 µM) (see
Fig. 4).
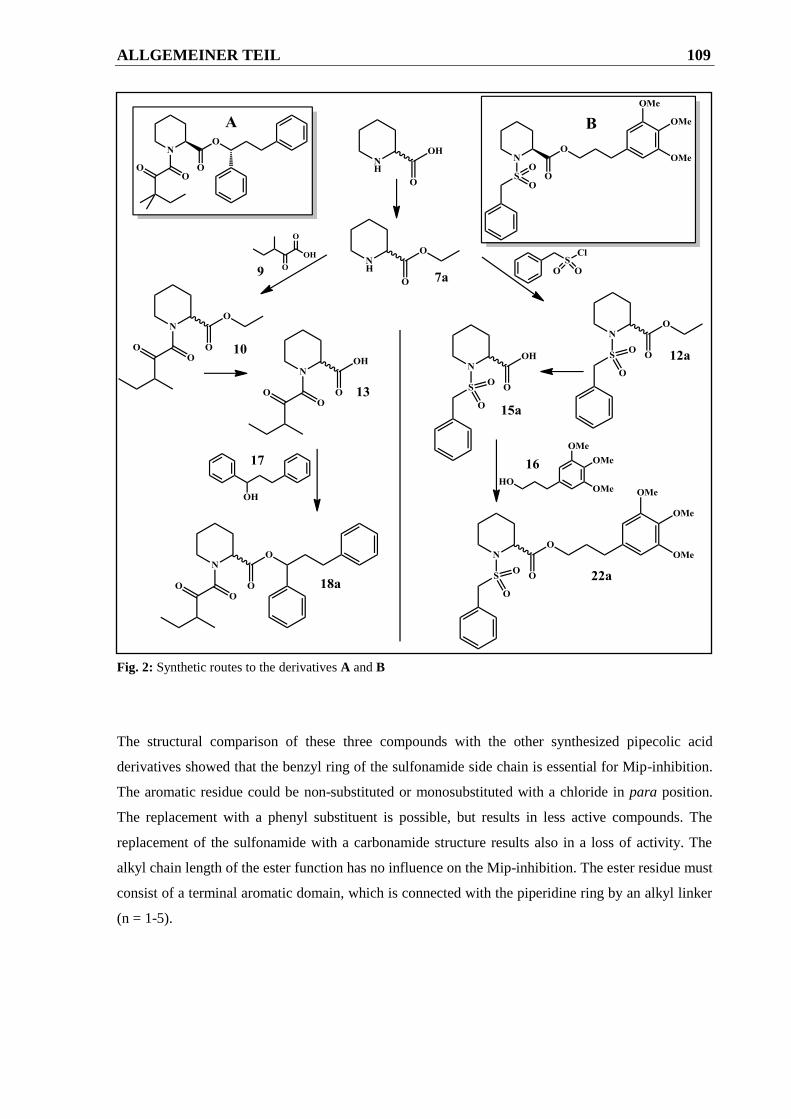
ALLGEMEINER TEIL 109
Fig. 2: Synthetic routes to the derivatives A and B
The structural comparison of these three compounds with the other synthesized pipecolic acid
derivatives showed that the benzyl ring of the sulfonamide side chain is essential for Mip-inhibition.
The aromatic residue could be non-substituted or monosubstituted with a chloride in para position.
The replacement with a phenyl substituent is possible, but results in less active compounds. The
replacement of the sulfonamide with a carbonamide structure results also in a loss of activity. The
alkyl chain length of the ester function has no influence on the Mip-inhibition. The ester residue must
consist of a terminal aromatic domain, which is connected with the piperidine ring by an alkyl linker
(n = 1-5).
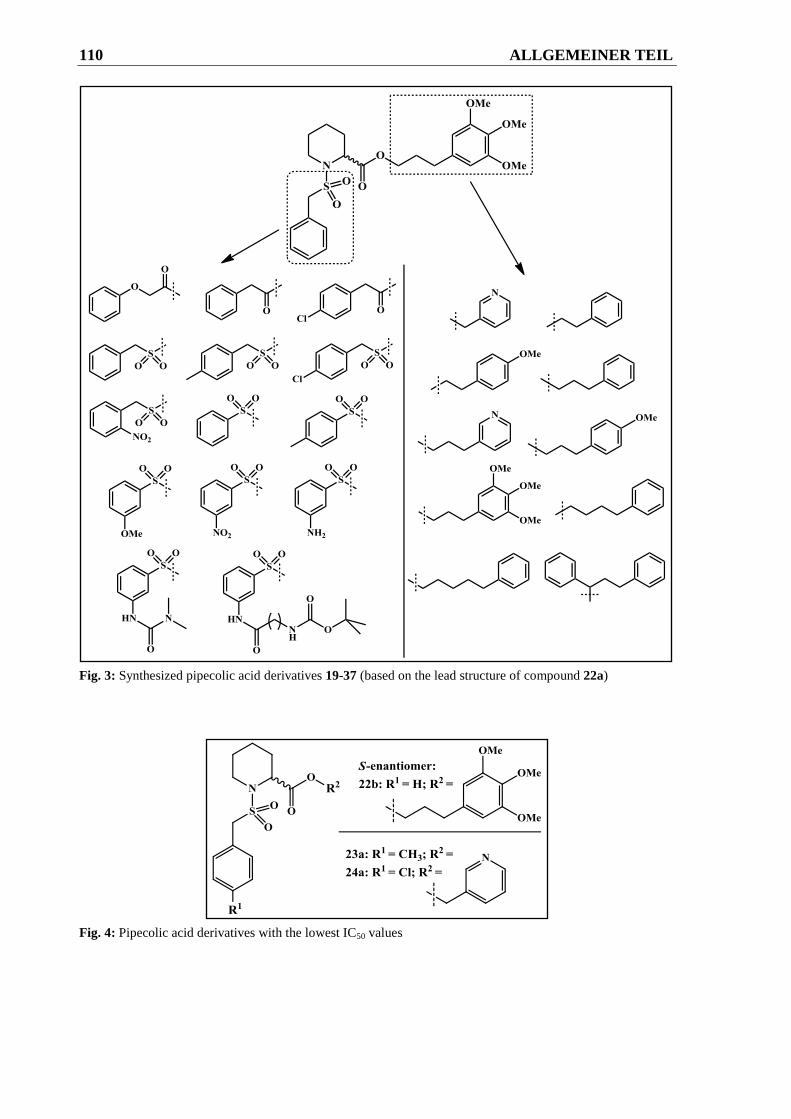
110 ALLGEMEINER TEIL
Fig. 3: Synthesized pipecolic acid derivatives 19-37 (based on the lead structure of compound 22a)
Fig. 4: Pipecolic acid derivatives with the lowest IC50 values

ALLGEMEINER TEIL 111
To determine the inhibitor-protein binding, compounds 1a, 12a, 22a and 22b were analyzed by
means of HSQC-NMR experiments. During these studies, the small sulfonamide 1a showed no
interaction with Mip, and it was therefore assumed that the compounds 1-6 do not fill sufficient the
Mip-cavity and thus, exhibit not enough activity for inhibition. In contrast, all three pipecolic acid
derivatives 12a, 22a and 22b interact with Mip, which verifies the results of the enzyme assay.
Furthermore, for compound 22b an in vivo determination of growth inhibition of Legionella by
means of a Gentamicin infection assay was carried out. After incubation of macrophages with
microorganisms and compound 22b no reduced number of bacteria in the cellular interior was
observed, which means the pipecolic acid derivative 22b has no significant influence on the
Legionella infection in vivo. One reason for the excellent in vitro and the deficient in vivo activity is
that Mip as an entire molecule, but not its PPIase-activity is responsible for inhibition. As a result,
further studies are directed to elucidate the functional correlation between Mip and its PPIase.
Mip-like proteins are also identified in species of bacteria such as Chlamydia trachomatis, Neisseria
gonorrhoeae and Burkholderia pseudomallei. Preliminary in vivo studies showed an inhibition of the
Chlamydia growth mediated by compound 22a and of the Neisseria growth mediated by compounds
18b, 22a, und 27d. However, it remains to be elucidated, whether these results are related to a Mip-
inhibition. For all pipecolic acid compounds 18-38, 42 and 44 enzymatic studies were carried out to
evaluate the inhibition of the Mip-PPIase of B. pseudomallei. An enzymatic inhibition, which was
greater than that of Rapamycin, was shown for the compounds 12a, 18a, 22a, 27c and 27d.

112 ALLGEMEINER TEIL

EXPERIMENTELLER TEIL 113
6 EXPERIMENTELLER TEIL
6.1 Allgemeine Angaben
6.1.1 Verwendete Geräte
Schmelzpunkte: Schmelzpunktapparatur MPD350:BM 3.5
Fa. Sanyo Gallenkamp B.V., Etten-Leur, Holland
Alle angegebenen Schmelzpunkte sind unkorrigiert.
IR-Spektren: Jasco FT-IR-Spektrometer
Fa. Jasco Labor- und Datentechnik GmbH, Gross-Umstadt, Deutschland
Alle Spektren wurden mit einer Diamant-ATR-Einheit aufgenommen.
1H-NMR-Spektren: Bruker Kernresonanzspektrometer Avance 400 (400.132 MHz)
Fa. Bruker BioSpin GmbH, Rheinstetten, Deutschland
Als interner Standard werden die Mittelpunkte der Signale der deuterierten Lösungsmittel verwendet
(CDCl3: 7.26 ppm; DMSO-d6: 2.50; CD3CN: 1.94 ppm).
13C-NMR-Spektren: Bruker Kernresonanzspektrometer Avance 400 (100.613 MHz)
Fa. Bruker BioSpin GmbH, Rheinstetten, Deutschland
Als interner Standard werden die Mittelpunkte der Signale der deuterierten Lösungsmittel verwendet
(CDCl3: 77.16 ppm; DMSO-d6: 39.52; CD3CN: 1.32 ppm).
Folgende Abkürzungen werden für die Beschreibung der Multiplizitäten der 1H-, bzw.
13C-Spektren
verwendet:
H-, bzw. C-aromat. = aromatisches Proton bzw. C-Atom, s = Singulett, d = Dublett, dd = Dublett
vom Dublett, ddd = Dublett vom Dublett vom Dublett, dt = Dublett vom Triplett, dq = Dublett vom
Quartett, t = Triplett, td = Triplett vom Dublett, q = Quartett, p = Pentett, sext. = Sextett, sept. =
Septett, m = Multiplett, br = breites Signal.
Die chemischen Verschiebungen sind in ppm, die Kopplungskonstanten J in Hz angegeben.
Mikrowelle: Milestone MLS-Ethos 1600
Fa. Mikrowellen-Laborsysteme, Leutkirch, Deutschland

114 EXPERIMENTELLER TEIL
Reaktionen im offenen System wurden in einem Dreihalskolben und Reaktionen im geschlossenen
System in einem Bombenrohr aus Polytetrafluorethylen (PTFE, 3.0 cm Innendurchmesser, 18 cm
Länge) ummantelt mit einem Polyetheretherketon (PEEK) durchgeführt.
Zur Absorptionssteigerung von Mikrowellenstrahlen wurden bei Reaktionen mit apolaren
Lösungsmitteln Weflon-Scheiben (PTFE mit 100 % Graphit) oder Siliciumcarbid hinzugefügt.
6.1.2 Chromatographie
Dünnschichtchromatographie (DC): DC-Alufolien Kieselgel 60 F254, Fa. Merck, Darmstadt,
Deutschland
DC-Fertigplatten SIL G-25 UV254, Fa. Machery-Nagel,
Düren, Deutschland
DC-Alufolie ALOX N/UV254, Fa. Machery-Nagel, Düren,
Deutschland
Säulenchromatographie (SC): Kieselgel 60, 0.063-0.2 mm (70-320 mesh), Fa. Merck,
Darmstadt, Deutschland
MN-Aluminiumoxid, neutral, Fa. Machery-Nagel, Düren,
Deutschland
Sprühreagenzien: 1. Dimethylaminobenzaldehyd-Lösung R 2 (Ehrlichs-
Reagenz): 0.2 g 4-Dimethylaminobenzaldehyd R werden
ohne Erwärmen in einer Mischung von 4.5 ml Wasser R und
5.5 ml Salzsäure R gelöst.
2. Kaliumpermanganat-Lösung R: 0.3 g Kalium-
permanganat R werden in 100 ml Wasser gelöst.
3. Ninhydrin-Lösung R: 0.2 g Ninhydrin R werden in
100 ml einer Lösung von 5 Volumenteilen verdünnter
Essigsäure R und 95 Volumenteilen 1-Butanol R gelöst.
Alle Sprühreagenzien wurden nach den allgemeinen Vorschriften des Ph. Eur. 5.0 hergestellt.

EXPERIMENTELLER TEIL 115
6.1.3 Chemikalien und Lösungsmittel
Chemikalien und Lösungsmittel wurden von folgenden Firmen bezogen:
Acros Organics, Geel, Belgien
Bachem Distribution Services GmbH, Weil am Rhein, Deutschland
Fisher Scientific GmbH, Schwerte, Deutschland
Sigma-Aldrich, Deisenhofen, Deutschland
VWR International, Darmstadt, Deutschland
Bei allen Reaktionen wurde ausschließlich demineralisiertes Wasser verwendet.
CH2Cl2 wurde 24 h über CaCl2 chemisch getrocknet, anschließend über P2O5 zum Sieden erhitzt und
destilliert. Et2O wurde mehrere Tage über CaCl2 chemisch getrocknet, anschließend über NaH zum
Sieden erhitzt und destilliert. Dioxan wurde 2 h über Na/K-Legierung unter Rückfluss erhitzt und
anschließend destilliert. DMF wurde über P2O5 bei 40 mbar destilliert. EtOH wurde über
Magnesiumspänen zum Sieden erhitzt und danach destilliert. THF wurde 2 h über Na/K-Legierung
unter Rückfluss erhitzt, anschließend destilliert und über frisch gepresstem Na-Draht aufbewahrt.

116 EXPERIMENTELLER TEIL
6.1.4 Abkürzungen
Äq.: Äquivalente
äq.: äquatorial
abs.: absolutiert
ax.: axial
aromat.: aromatisch
(BOC)2O: Di-tert-butyldicarbonat
DCC: Dicyclohexylcarbodiimid
dem.: demineralisiert
DMAP: Dimethylaminopyridin
DMF: Dimethylformamid
DMSO: Dimethylsulfoxid
Et3N: Triethylamin
Et2O: Diethylether
EtOAc: Essigsäureethylester
EtOH: Ethanol
FS: Feststoff
ges.: gesättigt
Lit.: Literatur
konz.: konzentriert
MeI: Methyliodid
MeOH: Methanol
NMM: N-Methylmorpholin
PE: Petrolether 40-60
PPA: n-Propylphosphonsäureanhydrid
RT: Raumtemperatur
Smp.: Schmelzpunkt
THF: Tetrahydrofuran
Verb.: Verbindung
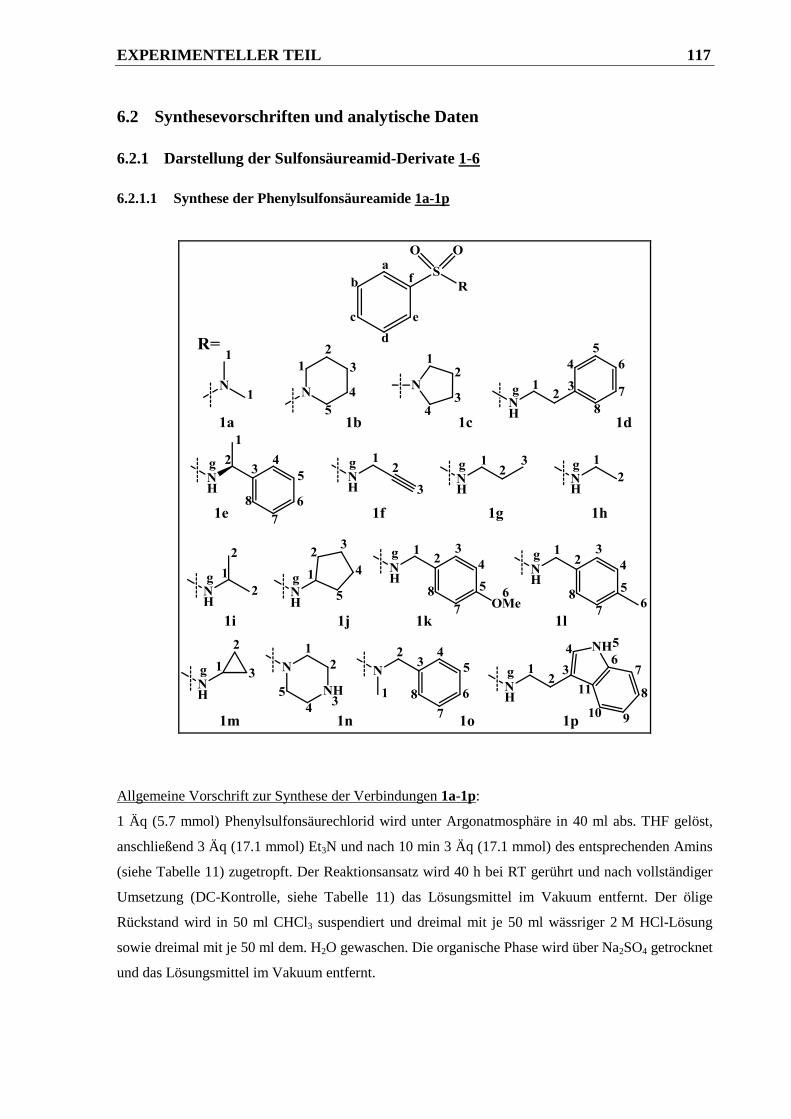
EXPERIMENTELLER TEIL 117
6.2 Synthesevorschriften und analytische Daten
6.2.1 Darstellung der Sulfonsäureamid-Derivate 1-6
6.2.1.1 Synthese der Phenylsulfonsäureamide 1a-1p
Allgemeine Vorschrift zur Synthese der Verbindungen 1a-1p:
1 Äq (5.7 mmol) Phenylsulfonsäurechlorid wird unter Argonatmosphäre in 40 ml abs. THF gelöst,
anschließend 3 Äq (17.1 mmol) Et3N und nach 10 min 3 Äq (17.1 mmol) des entsprechenden Amins
(siehe Tabelle 11) zugetropft. Der Reaktionsansatz wird 40 h bei RT gerührt und nach vollständiger
Umsetzung (DC-Kontrolle, siehe Tabelle 11) das Lösungsmittel im Vakuum entfernt. Der ölige
Rückstand wird in 50 ml CHCl3 suspendiert und dreimal mit je 50 ml wässriger 2 M HCl-Lösung
sowie dreimal mit je 50 ml dem. H2O gewaschen. Die organische Phase wird über Na2SO4 getrocknet
und das Lösungsmittel im Vakuum entfernt.
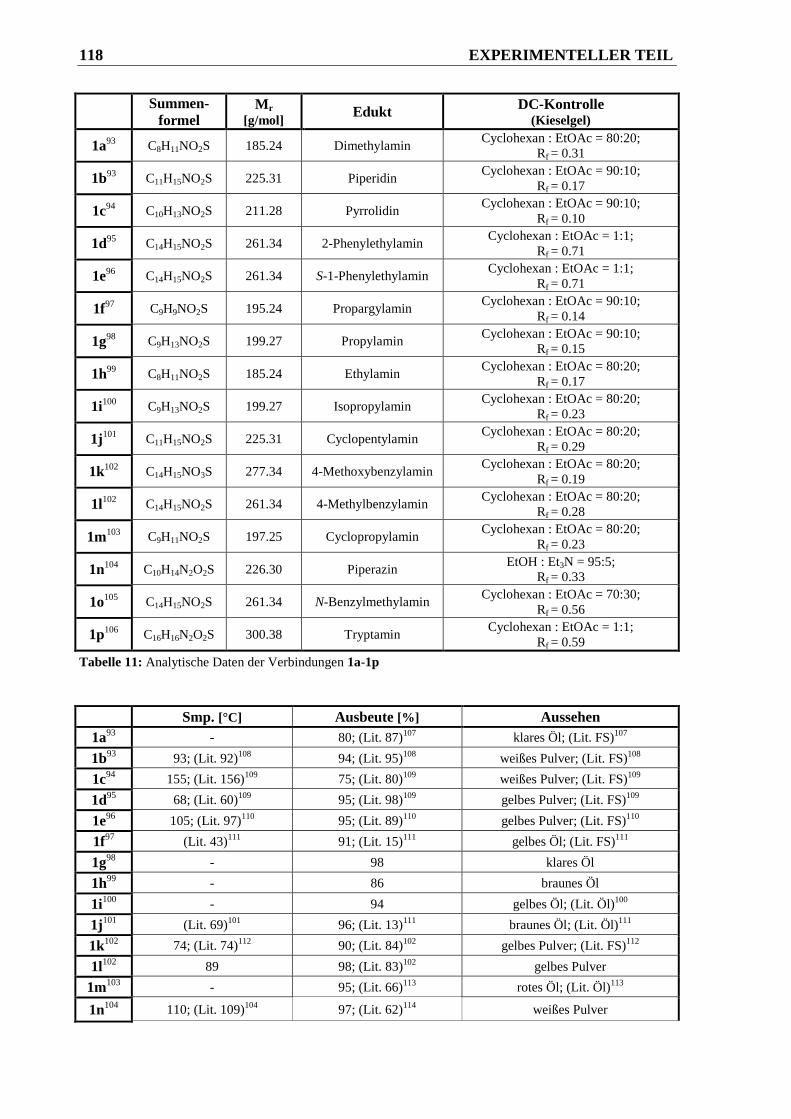
118 EXPERIMENTELLER TEIL
Summen-
formel Mr
[g/mol] Edukt
DC-Kontrolle (Kieselgel)
1a93
C8H11NO2S 185.24 Dimethylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.31
1b93
C11H15NO2S 225.31 Piperidin Cyclohexan : EtOAc = 90:10;
Rf = 0.17
1c94
C10H13NO2S 211.28 Pyrrolidin Cyclohexan : EtOAc = 90:10;
Rf = 0.10
1d95
C14H15NO2S 261.34 2-Phenylethylamin Cyclohexan : EtOAc = 1:1;
Rf = 0.71
1e96
C14H15NO2S 261.34 S-1-Phenylethylamin Cyclohexan : EtOAc = 1:1;
Rf = 0.71
1f97
C9H9NO2S 195.24 Propargylamin Cyclohexan : EtOAc = 90:10;
Rf = 0.14
1g98
C9H13NO2S 199.27 Propylamin Cyclohexan : EtOAc = 90:10;
Rf = 0.15
1h99
C8H11NO2S 185.24 Ethylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.17
1i100
C9H13NO2S 199.27 Isopropylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.23
1j101
C11H15NO2S 225.31 Cyclopentylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.29
1k102
C14H15NO3S 277.34 4-Methoxybenzylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.19
1l102
C14H15NO2S 261.34 4-Methylbenzylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.28
1m103
C9H11NO2S 197.25 Cyclopropylamin Cyclohexan : EtOAc = 80:20;
Rf = 0.23
1n104
C10H14N2O2S 226.30 Piperazin EtOH : Et3N = 95:5;
Rf = 0.33
1o105
C14H15NO2S 261.34 N-Benzylmethylamin Cyclohexan : EtOAc = 70:30;
Rf = 0.56
1p106
C16H16N2O2S 300.38 Tryptamin Cyclohexan : EtOAc = 1:1;
Rf = 0.59
Tabelle 11: Analytische Daten der Verbindungen 1a-1p
Smp. [°C] Ausbeute [%] Aussehen
1a93
- 80; (Lit. 87)107
klares Öl; (Lit. FS)107
1b93
93; (Lit. 92)108
94; (Lit. 95)108
weißes Pulver; (Lit. FS)108
1c94
155; (Lit. 156)109
75; (Lit. 80)109
weißes Pulver; (Lit. FS)109
1d95
68; (Lit. 60)109
95; (Lit. 98)109
gelbes Pulver; (Lit. FS)109
1e96
105; (Lit. 97)110
95; (Lit. 89)110
gelbes Pulver; (Lit. FS)110
1f97
(Lit. 43)111
91; (Lit. 15)111
gelbes Öl; (Lit. FS)111
1g98
- 98 klares Öl
1h99
- 86 braunes Öl
1i100
- 94 gelbes Öl; (Lit. Öl)100
1j101
(Lit. 69)101
96; (Lit. 13)111
braunes Öl; (Lit. Öl)111
1k102
74; (Lit. 74)112
90; (Lit. 84)102
gelbes Pulver; (Lit. FS)112
1l102
89 98; (Lit. 83)102
gelbes Pulver
1m103
- 95; (Lit. 66)113
rotes Öl; (Lit. Öl)113
1n104
110; (Lit. 109)104
97; (Lit. 62)114
weißes Pulver

EXPERIMENTELLER TEIL 119
Smp. [°C] Ausbeute [%] Aussehen
1o105
93; (Lit. 93)105
97; (Lit. 84)105
weißes Pulver; (Lit. FS)105
1p106
87; (Lit. 89)106
55; (Lit. 90)106
braunes Pulver
Tabelle 12: Analytische Daten der Verbindungen 1a-1p
Die IR-, 1H-NMR-, und
13C-NMR-Daten zu 1a, 1b, 1c, 1d, 1e, 1f, 1h, 1j, 1k, 1l, 1m, 1n, 1o
entsprechen den Literaturstellen107, 108, 109, 110, 111, 115, 112, 116, 113, 114, 106
.
IR [cm-1
]:
1a: 682; 732; 950; 1090; 1162; 1255; 1332; 1445; 2961; 3029;
1b: 685; 737; 1030; 1092; 1157; 1252; 1315; 1444; 2837; 3060;
1c: 645; 751; 1011; 1092; 1155; 1333; 1448; 2858; 2978; 3074;
1d: 685; 727; 1030; 1092; 1155; 1252; 1315; 1444; 2835; 3060; 3277;
1e: 685; 727; 1030; 1092; 1157; 1254; 1315; 1444; 2839; 3060; 3275;
1f: 687; 728; 1030; 1056; 1092; 1154; 1320; 1429; 2923; 2960; 3252;
1g: 688; 753; 998; 1072; 1093; 1155; 1320; 1446; 2967; 3065; 3280;
1h: 690; 758; 943; 1054; 1089; 1156; 1321; 1447; 2978; 3062; 3247;
1i: 689; 755; 995; 1072; 1092; 1156; 1322; 1446; 2975; 3066; 3277;
1j: 686; 753; 908; 1024; 1089; 1147; 1309; 1446; 2963; 3060; 3250;
1k: 690; 758; 1030; 1092; 1155; 1252; 1315; 1444; 2966; 3060; 3277;
1l: 685; 725; 1039; 1092; 1155; 1253; 1317; 1444; 2926; 3035; 3265;
1m: 687; 716; 954; 1093; 1154; 1323; 1446; 1595; 2962; 3035; 3243;
1n: 687; 739; 945; 1093; 1157; 1320; 1444; 1597; 2929; 3060; 3238;
1o: 695; 737; 940; 1089; 1157; 1336; 1448; 1585; 2945; 3035;
1p: 680; 735; 940; 1100; 1160; 1303; 1446; 1636; 3054; 3245; 3406;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
1a: (CDCl3): 7.82-7.72 (m, 2H, H-a, H-e); 7.63-7.49 (m, 3H, H-b, H-c, H-d); 2.69 (s, 6H, H-1);
1b: 7.76-7.58 (m, 5H, H-a, H-b, H-c, H-d, H-e); 2.90-2.84 (m, 4H, H-1, H-5); 1.57-1.48 (m, 4H,
H-2, H-4); 1.38-1.31 (m, 2H, H-3);
1c: 7.87-7.76 (m, 2H, H-a, H-e); 7.74-7.68 (m, 1H, H-c); 7.66-7.61 (m, 2H, H-b, H-d); 3.16-
3.12 (m, 4H, H-1, H-4); 1.65-1.61 (m, 4H, H-2, H-3);
1d: 7.80-7.76 (m, 2H, H-a, H-e); 7.72-7.68 (m, 1H, H-g); 7.66-7.52 (m, 3H, H-b, H-c, H-d);
7.29-7.10 (m, 5H, H-aromat.); 2.99-2.91 (m, 2H, H-1); 2.66 (t, 2H, 3J = 7.5, H-2);
1e: 8.19 (d, 1H, 3
J = 7.5, H-g); 7.70-7.66 (m, 2H, H-a, H-e); 7.57-7.42 (m, 3H, H-b, H-c, H-d);
7.22-7.10 (m, 5H, H-aromat.); 4.35 (p, 1H, 3J = 7.5, H-2); 1.16 (d, 3H,
3J = 7.5, H-1);

120 EXPERIMENTELLER TEIL
1f: 8.15-8.10 (m, 1H, H-g); 7.86-7.76 (m, 2H, H-a, H-e); 7.68-7.54 (m, 3H, H-b, H-c, H-d);
3.72-3.67 (m, 2H, H-1); 3.03-3.00 (m, 1H, H-3);
1g: 8.07-7.71 (m, 2H, H-a, H-e); 7.66-7.52 (m, 4H, H-b, H-c, H-d, H-g); 2.72-2.65 (m, 2H,
H-1); 1.36 (sext., 2H, 3J = 7.3, H-2); 0.78 (t, 3H,
3J = 7.3, H-3);
1h: 7.89-7.73 (m, 2H, H-a, H-e); 7.66-7.52 (m, 4H, H-b, H-c, H-d, H-g); 2.82-2.73 (m, 2H,
H-1); 0.96 (t, 3H, 3J = 7.2, H-2);
1i: 7.93-7.68 (m, 2H, H-a, H-e); 7.63-7.51 (m, 4H, H-b, H-c, H-d, H-g); 3.27-3.14 (m, 1H,
H-1); 0.91 (d, 6H, 3J = 6.5, H-2);
1j: 7.84-7.76 (m, 2H, H-a, H-e); 7.65-7.54 (m, 4H, H-b, H-c, H-d, H-g); 3.39 (sext., 1H,
3J = 6.8, H-1); 1.62-1.46 (m, 4H, äq., H-2, H-3, H-4; H-5); 1.43-1.21 (m, 4H, ax., H-2, H-3,
H-4; H-5);
1k: 8.04 (t, 1H, 3J = 6.2, H-g); 7.81-7.76 (m, 2H, H-a, H-e); 7.67-7.53 (m, 3H, H-b, H-c, H-d);
7.12 (d, 2H, 3J = 8.7, H-3, H-8); 6.82 (d, 2H,
3J = 8.7, H-4, H-7); 3.90 (d, 2H,
3J = 6.2, H-1);
3.70 (s, 3H, H-6);
1l: 8.08 (t, 1H, 3J = 6.0, H-g); 7.82-7.76 (m, 2H, H-a, H-e); 7.67-7.53 (m, 3H, H-b, H-c, H-d);
7.13-7.05 (m, 4H, H-3, H-4, H-7, H-8); 3.92 (d, 2H, 3J = 6.0, H-1); 2.25 (s, 3H, H-6);
1m: 7.90-7.88 (m, 1H, H-g); 7.84-7.79 (m, 2H, H-a, H-e); 7.69-7.57 (m, 3H, H-b, H-c, H-d);
2.13-2.05 (m, 1H, H-1); 0.51-0.43 (m, 2H, H-2, H-3); 0.34-0.38 (m, 2H, H-2, H-3);
1n: 7.76-7.56 (m, 5H, H-a, H-b, H-c, H-d, H-e); 2.79-2.74 (m, 4H, H-1, H-5); 2.73-2.68 (m, 4H,
H-2, H-4); 2.18 (br, 1H, H-3);
1o: 7.91-7.82 (m, 2H, H-a, H-e); 7.77-7.63 (m, 3H, H-b, H-c, H-d); 7.40-7.27 (m, 5H,
H-aromat.); 4.14 (s, 2H, H-2); 2.54 (s, 3H, H-1);
1p: 10.80 (s, 1H, H-5); 7.84-7.78 (m, 2H, H-a, H-c); 7.74 (t, 1H, 3J = 5.8, H-g); 7.65-7.54 (m,
3H, H-b, H-c, H-d); 7.37 (d, 1H, 3
J = 8.0, H-10); 7.32 (d, 1H, 3J = 8.0, H-7); 7.12-7.10 (m,
1H, H-4); 7.04 (t, 1H, 3J = 8.0, H-9); 6.95 (t, 1H,
3J = 8.0, H-8); 3.06-2.96 (m, 2H, H-1);
2.82-2.76 (m, 2H, H-2);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
1a: (CDCl3): 135.5 (C-f); 132.6 (C-c); 129.0 (C-b, C-d); 127.6 (C-a, C-e); 37.9 (C-1);
1b: 135.3 (C-f); 132.7 (C-c); 129.0 (C-b, C-d); 127.1 (C-a, C-e); 46.3 (C-1, C-5); 24.4 (C-2,
C-4); 22.5 (C-3);
1c: 135.7 (C-f); 132.5 (C-c); 128.9 (C-b, C-d); 126.7 (C-a, C-e); 47.3 (C-1, C-4); 24.2 (C-2,
C-3);
1d: 140.3 (C-f); 138.6 (C-3); 132.3 (C-c); 129.1 (C-b, C-d); 128.5 (C-5, C-7); 128.2 (C-4, C-8);
126.4 (C-a, C-e); 126.1 (C-6); 44.0 (C-1); 35.2 (C-2);
1e: 142.8 (C-f); 141.0 (C-3); 131.5 (C-c); 128.3 (C-b, C-d); 127.5 (C-5, C-7); 126.2 (C-6);
125.8 (C-a, C-e); 125.5 (C-4, C-8); 52.3 (C-2); 23.0 (C-1);

EXPERIMENTELLER TEIL 121
1f: 139.5 (C-f); 132.0 (C-c); 128.6 (C-b, C-d); 126.1 (C-a, C-e); 78.8 (C-2); 74.1 (C-3); 31.4
(C-1);
1g: 140.1 (C-f); 131.7 (C-c); 128.7 (C-b, C-d); 125.9 (C-a, C-e); 43.8 (C-1); 21.9 (C-2); 10.6
(C-3);
1h: 140.5 (C-f); 132.2 (C-c); 129.1 (C-b, C-d); 126.3 (C-a, C-e); 37.4 (C-1); 14.6 (C-2);
1i: 141.4 (C-f); 131.6 (C-c); 128.6 (C-b, C-d); 125.8 (C-a, C-e); 44.7 (C-1); 22.6 (C-2);
1j: 141.5 (C-f); 132.1 (C-c); 129.0 (C-b, C-d); 126.3 (C-a, C-e); 54.3 (C-1); 32.3 (C-2, C-5);
22.7 (C-3, C-4);
1k: 158.3 (C-5); 140.7 (C-f); 132.2 (C-c); 129.3 (C-2); 129.0 (C-b, C-d); 128.8 (C-3, C-8);
126.3 (C-a, C-e); 113.5 (C-4, C-7); 55.0 (C-1); 45.6 (C-6);
1l: 140.8 (C-f); 136.2 (C-2); 134.5 (C-5); 132.3 (C-c); 129.1 (C-b, C-d); 128.7 (C-3, C-8);
127.5 (C-4, C-7); 126.4 (C-a, C-e); 45.9 (C-1); 20.6 (C-6);
1m: 140.3 (C-f); 132.4 (C-c); 129.1 (C-b, C-d); 126.7 (C-a, C-e); 24.1 (C-1); 5.1 (C-2, C-3);
1n: 134.3 (C-f); 132.6 (C-c); 128.8 (C-b, C-d); 127.0 (C-a, C-e); 46.2 (C-1, C-5); 44.2 (C-2,
C-4);
1o: 136.5 (C-f); 135.5 (C-3); 132.5 (C-c); 129.0 (C-b, C-d); 128.0 (C-5, C-7); 127.6 (C-4, C-8);
127.2 (C-6); 126.6 (C-a, C-e); 52.8 (C-2); 33.9 (C-1);
1p: 140.5 (C-f); 136.1 (C-6), 132.2 (C-c); 129.1 (C-b, C-d); 126.8 (C-11); 126.3 (C-a, C-e);
122.8 (C-4); 120.8 (C-9); 118.2 (C-8); 117.8 (C-10); 111.3 (C-7); 110.8 (C-3); 43.3 (C-1);
25.0 (C-2);
6.2.1.2 Synthese der N,N-disubstituierten Phenylsulfonsäureamide 2a-2c
Allgemeine Vorschrift zur Synthese der Verbindungen 2a-2c:
1 Äq (4.0 mmol) des sekundären Amins (Verbindungen 1, siehe Tabelle 13) wird unter
Argonatmosphäre in 30 ml abs. DMF gelöst, anschließend 6 Äq (24 mmol) Kalium-t-butylat und
nach 10 min langsam 10 Äq (40 mmol) MeI zugegeben. Dieser Reaktionsansatz wird 3 h bei RT
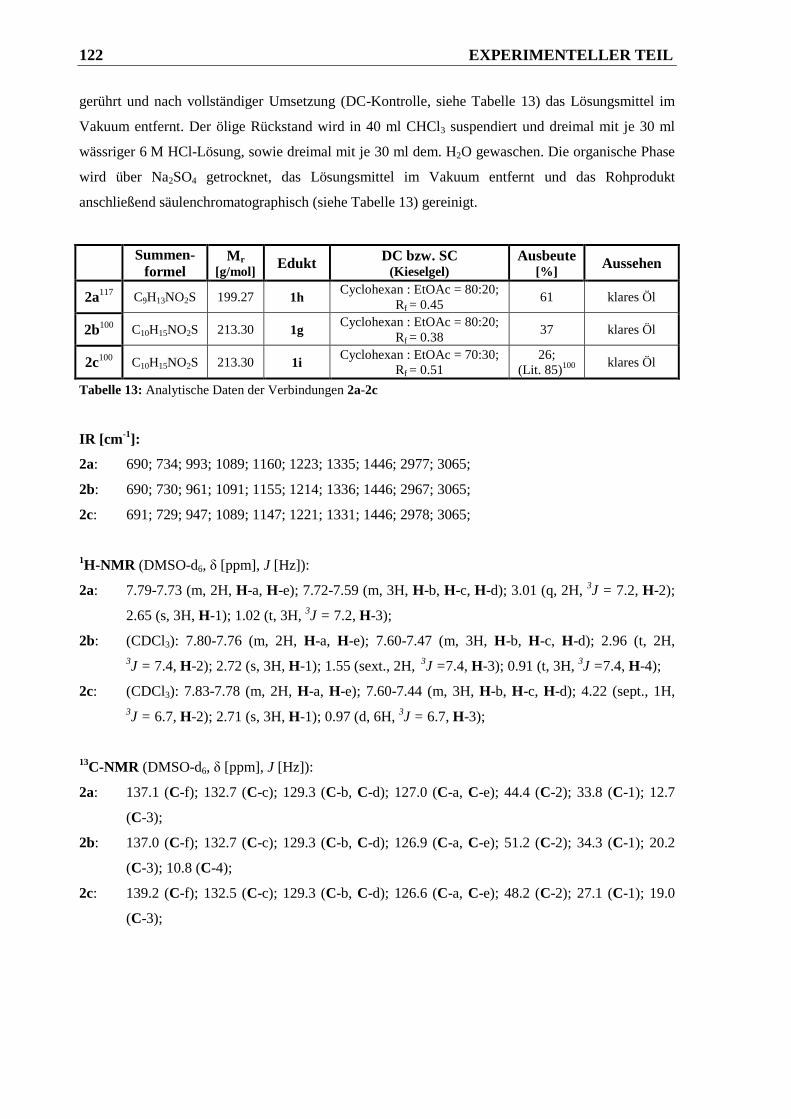
122 EXPERIMENTELLER TEIL
gerührt und nach vollständiger Umsetzung (DC-Kontrolle, siehe Tabelle 13) das Lösungsmittel im
Vakuum entfernt. Der ölige Rückstand wird in 40 ml CHCl3 suspendiert und dreimal mit je 30 ml
wässriger 6 M HCl-Lösung, sowie dreimal mit je 30 ml dem. H2O gewaschen. Die organische Phase
wird über Na2SO4 getrocknet, das Lösungsmittel im Vakuum entfernt und das Rohprodukt
anschließend säulenchromatographisch (siehe Tabelle 13) gereinigt.
Summen-
formel Mr
[g/mol] Edukt
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aussehen
2a117
C9H13NO2S 199.27 1h Cyclohexan : EtOAc = 80:20;
Rf = 0.45 61 klares Öl
2b100
C10H15NO2S 213.30 1g Cyclohexan : EtOAc = 80:20;
Rf = 0.38 37 klares Öl
2c100
C10H15NO2S 213.30 1i Cyclohexan : EtOAc = 70:30;
Rf = 0.51
26;
(Lit. 85)100
klares Öl
Tabelle 13: Analytische Daten der Verbindungen 2a-2c
IR [cm-1
]:
2a: 690; 734; 993; 1089; 1160; 1223; 1335; 1446; 2977; 3065;
2b: 690; 730; 961; 1091; 1155; 1214; 1336; 1446; 2967; 3065;
2c: 691; 729; 947; 1089; 1147; 1221; 1331; 1446; 2978; 3065;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
2a: 7.79-7.73 (m, 2H, H-a, H-e); 7.72-7.59 (m, 3H, H-b, H-c, H-d); 3.01 (q, 2H, 3J = 7.2, H-2);
2.65 (s, 3H, H-1); 1.02 (t, 3H, 3J = 7.2, H-3);
2b: (CDCl3): 7.80-7.76 (m, 2H, H-a, H-e); 7.60-7.47 (m, 3H, H-b, H-c, H-d); 2.96 (t, 2H,
3J = 7.4, H-2); 2.72 (s, 3H, H-1); 1.55 (sext., 2H,
3J =7.4, H-3); 0.91 (t, 3H,
3J =7.4, H-4);
2c: (CDCl3): 7.83-7.78 (m, 2H, H-a, H-e); 7.60-7.44 (m, 3H, H-b, H-c, H-d); 4.22 (sept., 1H,
3J = 6.7, H-2); 2.71 (s, 3H, H-1); 0.97 (d, 6H,
3J = 6.7, H-3);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
2a: 137.1 (C-f); 132.7 (C-c); 129.3 (C-b, C-d); 127.0 (C-a, C-e); 44.4 (C-2); 33.8 (C-1); 12.7
(C-3);
2b: 137.0 (C-f); 132.7 (C-c); 129.3 (C-b, C-d); 126.9 (C-a, C-e); 51.2 (C-2); 34.3 (C-1); 20.2
(C-3); 10.8 (C-4);
2c: 139.2 (C-f); 132.5 (C-c); 129.3 (C-b, C-d); 126.6 (C-a, C-e); 48.2 (C-2); 27.1 (C-1); 19.0
(C-3);

EXPERIMENTELLER TEIL 123
6.2.1.3 Synthese von N-Benzylphenylsulfonsäureamid 3118
Methode 1:
2.0 g (12.7 mmol) Phenylsulfonsäureamid werden unter Argonatmosphäre in 50 ml abs. THF gelöst,
mit Trockeneis auf -70 °C abgekühlt und langsam 0.8 g (12.7 mmol) n-Butyl-Lithium zugetropft.
Anschließend wird die Mischung 10 min bei -70 °C gerührt und danach auf RT erwärmt. 1.6 g
(12.7 mmol) Benzylchlorid werden bei RT zugetropft und 6 h bei RT gerührt. Danach werden 50 ml
dem. H2O zugegeben und das Lösungsmittel im Vakuum entfernt. Der organische Rückstand wird in
30 ml dem. H2O suspendiert und dreimal mit je 30 ml CH2Cl2 extrahiert. Die vereinigten organischen
Phasen werden über Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Das Rohprodukt
wird anschließend säulenchromatographisch gereinigt (Kieselgel; Cyclohexan : EtOAc = 1 : 1;
Rf = 0.71) und man erhält 0.03 g (0.1 mmol) von Verbindung 3 in Form eines weißen Öls.
Summenformel: C13H13NO2S
Mr = 247.31 g/mol
Ausbeute: 1 %
Aussehen: weißes Öl
Methode 2:
0.5 g (3.2 mmol) Phenylsulfonsäureamid, 0.44 g (3.2 mmol) K2CO3 und 0.4 g (3.2 mmol)
Benzylchlorid werden in einem Bombenrohr in 40 ml MeOH gelöst, langsam in 9 min auf 100 °C in
der Mikrowelle erhitzt und 3 h bei 100 °C gerührt. Danach wird das Lösungsmittel im Vakuum
entfernt und das Rohprodukt säulenchromatographisch gereinigt (Kieselgel; Cyclohexan : EtOAc =
80 : 20; Rf = 0.28). Man erhält 0.17 g (0.7 mmol) von Verbindung 3 in Form eines weißen Pulvers.
Smp. = 89 °C (Lit. 87 °C)118
Ausbeute: 22 % (Lit. 33 %)109
Aussehen: weißes Pulver (Lit. FS)109
Die 1H-NMR-, und
13C-NMR-Daten zu 3 entsprechen der Literaturstelle
109.

124 EXPERIMENTELLER TEIL
IR [cm-1
]: 687; 749; 837; 1059; 1093; 1149; 1324; 1416; 1447; 2930; 3055; 3327;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
8.14 (br, 1H, H-g); 7.81-7.77 (m, 2H, H-a, H-e); 7.67-7.52 (m, 3H, H-b, H-c, H-d); 7.31-7.17 (m,
5H, H-aromat.); 3.98 (s, 2H, H-1);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
140.2 (C-f); 137.1 (C-2); 131.8 (C-c); 128.6 (C-b, C-d); 127.7 (C-4, C-6); 127.0 (C-3, C-7); 126.6
(C-5); 125.9 (C-a, C-e); 45.6 (C-1);
6.2.1.4 Synthese der N,N-Dimethylsulfonsäureamide 4a-4e
Allgemeine Vorschrift zur Synthese der Verbindungen 4a-4e:
1 Äq (Stoffmenge siehe Tabelle 14) des entsprechenden Sulfonsäurechlorids wird unter
Argonatmosphäre in 40 ml abs. THF gelöst, anschließend 3 Äq (12.3-16.5 mmol) Et3N und nach 10
min 3 Äq (12.3-16.5 mmol) Dimethylamin zugetropft. Der Reaktionsansatz wird 40 h bei RT gerührt
und nach vollständiger Umsetzung (DC-Kontrolle, siehe Tabelle 14) das Lösungsmittel im Vakuum
entfernt. Der ölige Rückstand wird in 50 ml CHCl3 suspendiert, dreimal mit je 50 ml wässriger
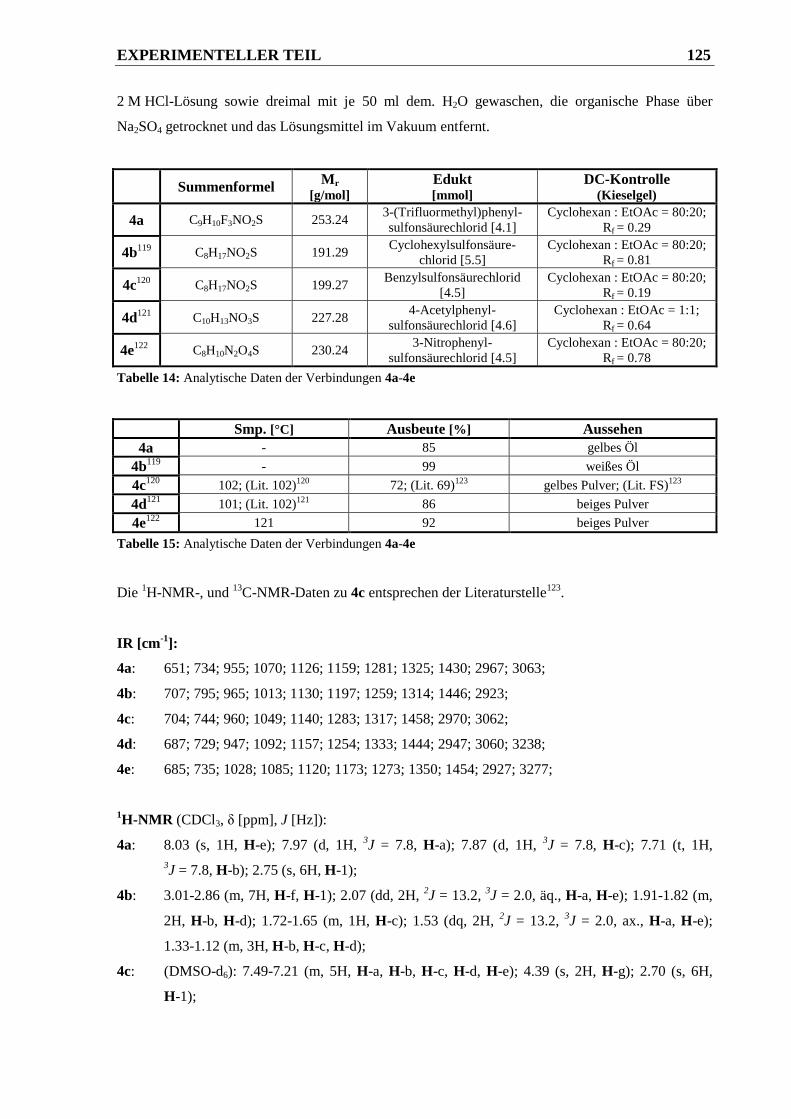
EXPERIMENTELLER TEIL 125
2 M HCl-Lösung sowie dreimal mit je 50 ml dem. H2O gewaschen, die organische Phase über
Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt.
Summenformel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
4a C9H10F3NO2S 253.24 3-(Trifluormethyl)phenyl-
sulfonsäurechlorid [4.1]
Cyclohexan : EtOAc = 80:20;
Rf = 0.29
4b119
C8H17NO2S 191.29 Cyclohexylsulfonsäure-
chlorid [5.5]
Cyclohexan : EtOAc = 80:20;
Rf = 0.81
4c120
C8H17NO2S 199.27 Benzylsulfonsäurechlorid
[4.5]
Cyclohexan : EtOAc = 80:20;
Rf = 0.19
4d121
C10H13NO3S 227.28 4-Acetylphenyl-
sulfonsäurechlorid [4.6]
Cyclohexan : EtOAc = 1:1;
Rf = 0.64
4e122
C8H10N2O4S 230.24 3-Nitrophenyl-
sulfonsäurechlorid [4.5]
Cyclohexan : EtOAc = 80:20;
Rf = 0.78
Tabelle 14: Analytische Daten der Verbindungen 4a-4e
Smp. [°C] Ausbeute [%] Aussehen
4a - 85 gelbes Öl
4b119
- 99 weißes Öl
4c120
102; (Lit. 102)120
72; (Lit. 69)123
gelbes Pulver; (Lit. FS)123
4d121
101; (Lit. 102)121
86 beiges Pulver
4e122
121 92 beiges Pulver
Tabelle 15: Analytische Daten der Verbindungen 4a-4e
Die 1H-NMR-, und
13C-NMR-Daten zu 4c entsprechen der Literaturstelle
123.
IR [cm-1
]:
4a: 651; 734; 955; 1070; 1126; 1159; 1281; 1325; 1430; 2967; 3063;
4b: 707; 795; 965; 1013; 1130; 1197; 1259; 1314; 1446; 2923;
4c: 704; 744; 960; 1049; 1140; 1283; 1317; 1458; 2970; 3062;
4d: 687; 729; 947; 1092; 1157; 1254; 1333; 1444; 2947; 3060; 3238;
4e: 685; 735; 1028; 1085; 1120; 1173; 1273; 1350; 1454; 2927; 3277;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
4a: 8.03 (s, 1H, H-e); 7.97 (d, 1H, 3J = 7.8, H-a); 7.87 (d, 1H,
3J = 7.8, H-c); 7.71 (t, 1H,
3J = 7.8, H-b); 2.75 (s, 6H, H-1);
4b: 3.01-2.86 (m, 7H, H-f, H-1); 2.07 (dd, 2H, 2J = 13.2,
3J = 2.0, äq., H-a, H-e); 1.91-1.82 (m,
2H, H-b, H-d); 1.72-1.65 (m, 1H, H-c); 1.53 (dq, 2H, 2J = 13.2,
3J = 2.0, ax., H-a, H-e);
1.33-1.12 (m, 3H, H-b, H-c, H-d);
4c: (DMSO-d6): 7.49-7.21 (m, 5H, H-a, H-b, H-c, H-d, H-e); 4.39 (s, 2H, H-g); 2.70 (s, 6H,
H-1);

126 EXPERIMENTELLER TEIL
4d: (DMSO-d6): 8.18 (d, 2H, 3J = 8.5, H-b, H-d); 7.89 (d, 2H,
3J = 8.5, H-a, H-e); 2.65 (s, 3H,
H-h); 2.64 (s, 6H, H-1);
4e: (DMSO-d6): 8.55 (ddd, 1H, 3J = 8.0,
4J = 2.2,
4J = 1.0, H-c); 8.39-8.37 (m, 1H, H-e); 8.22-
8.15 (m, 1H, H-a); 7.95 (t, 1H, 3J = 8.0, H-b); 2.68 (s, 6H, H-1);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
4a: 137.2 (C-f); 131.8 (q, 2J = 33.4, C-d); 130.8 (C-a); 129.9 (C-b); 129.3 (q,
3J = 4.0, C-c);
124.6 (q, 3J = 4.0, C-e); 124.3 (q,
1J = 235.0, C-g); 37.8 (C-1);
4b: 60.7 (C-f); 37.8 (C-1); 26.6 (C-a, C-e); 25.2 (C-b, C-d); 25.1 (C-c);
4c: (DMSO-d6): 130.8 (C-a, C-e); 129.5 (C-f); 128.3 (C-b, C-d); 128.0 (C-c); 53.0 (C-g); 37.3
(C-1);
4d: (DMSO-d6): 197.2 (C-g); 139.7 (C-f); 138.4 (C-c); 128.9 (C-b, C-d); 127.7 (C-a, C-e); 37.7
(C-1); 26.7 (C-h);
4e: (DMSO-d6): 147.8 (C-d); 136.3 (C-f); 133.1 (C-a); 131.2 (C-b); 127.4 (C-c); 121.7 (C-e);
37.2 (C-1);
6.2.1.5 Synthese von 3-Amino-N,N-dimethylphenylsulfonsäureamid 5124
1 Äq (4.0 mmol) der Verbindung 4e und 5 Äq (20.0 mmol) SnCl2 x 2 H2O werden unter
Argonatmosphäre in 40 ml abs. EtOH suspendiert und 2 h unter Rückfluss erhitzt. Nach vollständiger
Umsetzung (DC-Kontrolle: Kieselgel; EtOH : Et3N = 95 : 5; Rf = 0.65) werden 30 ml dem. H2O und
danach ges. wässrige NaHCO3-Lösung bis pH 8 zugegeben. Danach wird dreimal mit je 50 ml
EtOAc extrahiert, die vereinigten organischen Phasen über Na2SO4 getrocknet und das Lösungsmittel
im Vakuum entfernt. Man erhält 0.78 g (3.9 mmol) von Verbindung 5 in Form eines beigen Pulvers.
Summenformel: C8H12N2O2S
Mr = 200.26 g/mol
Smp. = 155 °C (Lit. 155 °C)124

EXPERIMENTELLER TEIL 127
Ausbeute: 97 % (Lit. 91 %)124
Aussehen: beiges Pulver
IR [cm-1
]: 687; 724; 953; 1080; 1149; 1311; 1450; 1593; 1628; 2972; 3069; 3374; 3468;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
7.24 (t, 1H, 3J = 7.9, H-b); 6.92 (s, 1H, H-e); 6.84-6.77 (m, 2H, H-a, H-c); 5.61 (br, 2H, H-g); 2.58
(s, 6H, H-1);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
149.2 (C-d); 134.7 (C-f); 129.3 (C-b); 117.4 (C-c); 113.8 (C-a); 111.5 (C-e); 37.3 (C-1);
6.2.1.6 Synthese der N-alkylierten 3-Amino-N,N-dimethylphenylsulfonsäureamide
6a-6e
Allgemeine Vorschrift zur Synthese der Verbindungen 6a-6e:
1 Äq (3.0 mmol) der Verbindung 5 wird in 30 ml der entsprechenden Säure (siehe Tabelle 16) gelöst,
5 Äq (15.0 mmol) NaBH4 portionsweise zugegeben und 24 h bei RT gerührt. Nach vollständiger
Umsetzung (DC-Kontrolle, siehe Tabelle 16) werden langsam 30 ml dem. H2O und anschließend
eine wässrige 10 M NaOH-Lösung bis pH 8 zugetropft. Die wässrige Phase wird viermal mit je
30 ml CHCl3 extrahiert, die vereinigten organischen Phasen über Na2SO4 getrocknet und das
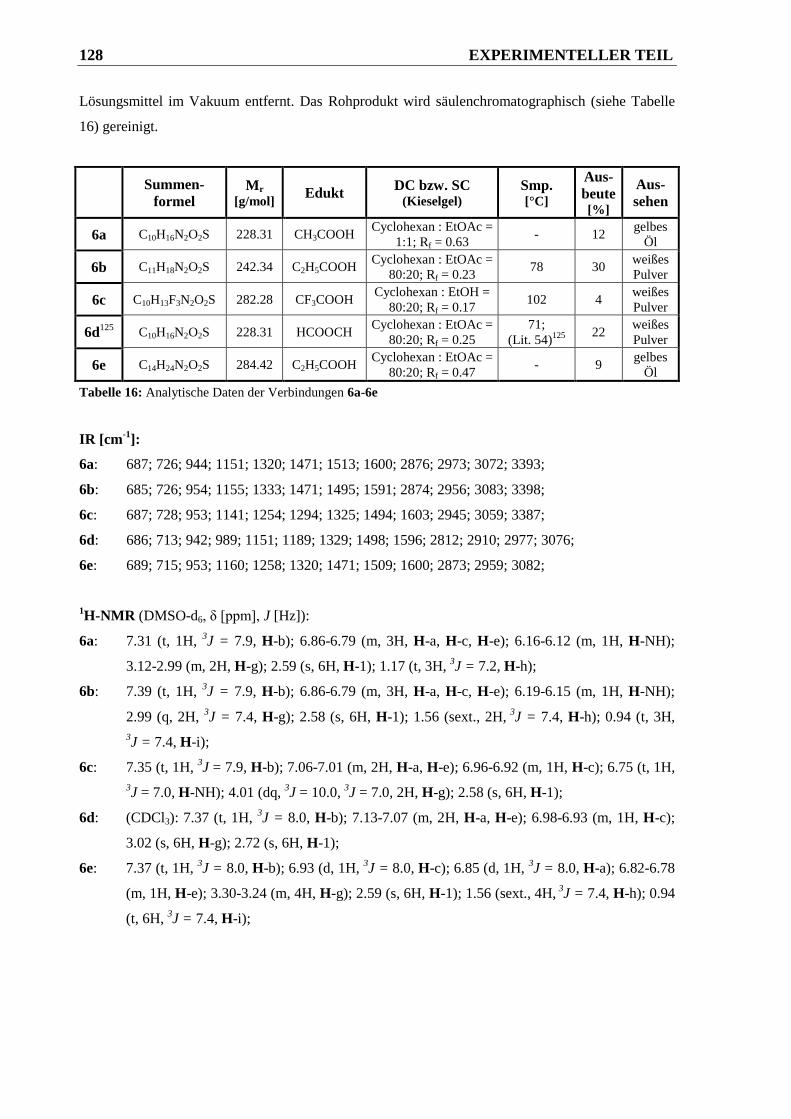
128 EXPERIMENTELLER TEIL
Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird säulenchromatographisch (siehe Tabelle
16) gereinigt.
Summen-
formel Mr
[g/mol] Edukt
DC bzw. SC (Kieselgel)
Smp. [°C]
Aus-
beute [%]
Aus-
sehen
6a C10H16N2O2S 228.31 CH3COOH Cyclohexan : EtOAc =
1:1; Rf = 0.63 - 12
gelbes
Öl
6b C11H18N2O2S 242.34 C2H5COOH Cyclohexan : EtOAc =
80:20; Rf = 0.23 78 30
weißes
Pulver
6c C10H13F3N2O2S 282.28 CF3COOH Cyclohexan : EtOH =
80:20; Rf = 0.17 102 4
weißes
Pulver
6d125
C10H16N2O2S 228.31 HCOOCH Cyclohexan : EtOAc =
80:20; Rf = 0.25
71;
(Lit. 54)125
22
weißes
Pulver
6e C14H24N2O2S 284.42 C2H5COOH Cyclohexan : EtOAc =
80:20; Rf = 0.47 - 9
gelbes
Öl
Tabelle 16: Analytische Daten der Verbindungen 6a-6e
IR [cm-1
]:
6a: 687; 726; 944; 1151; 1320; 1471; 1513; 1600; 2876; 2973; 3072; 3393;
6b: 685; 726; 954; 1155; 1333; 1471; 1495; 1591; 2874; 2956; 3083; 3398;
6c: 687; 728; 953; 1141; 1254; 1294; 1325; 1494; 1603; 2945; 3059; 3387;
6d: 686; 713; 942; 989; 1151; 1189; 1329; 1498; 1596; 2812; 2910; 2977; 3076;
6e: 689; 715; 953; 1160; 1258; 1320; 1471; 1509; 1600; 2873; 2959; 3082;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
6a: 7.31 (t, 1H, 3J = 7.9, H-b); 6.86-6.79 (m, 3H, H-a, H-c, H-e); 6.16-6.12 (m, 1H, H-NH);
3.12-2.99 (m, 2H, H-g); 2.59 (s, 6H, H-1); 1.17 (t, 3H, 3J = 7.2, H-h);
6b: 7.39 (t, 1H, 3J = 7.9, H-b); 6.86-6.79 (m, 3H, H-a, H-c, H-e); 6.19-6.15 (m, 1H, H-NH);
2.99 (q, 2H, 3J = 7.4, H-g); 2.58 (s, 6H, H-1); 1.56 (sext., 2H,
3J = 7.4, H-h); 0.94 (t, 3H,
3J = 7.4, H-i);
6c: 7.35 (t, 1H, 3J = 7.9, H-b); 7.06-7.01 (m, 2H, H-a, H-e); 6.96-6.92 (m, 1H, H-c); 6.75 (t, 1H,
3J = 7.0, H-NH); 4.01 (dq,
3J = 10.0,
3J = 7.0, 2H, H-g); 2.58 (s, 6H, H-1);
6d: (CDCl3): 7.37 (t, 1H, 3J = 8.0, H-b); 7.13-7.07 (m, 2H, H-a, H-e); 6.98-6.93 (m, 1H, H-c);
3.02 (s, 6H, H-g); 2.72 (s, 6H, H-1);
6e: 7.37 (t, 1H, 3J = 8.0, H-b); 6.93 (d, 1H,
3J = 8.0, H-c); 6.85 (d, 1H,
3J = 8.0, H-a); 6.82-6.78
(m, 1H, H-e); 3.30-3.24 (m, 4H, H-g); 2.59 (s, 6H, H-1); 1.56 (sext., 4H, 3J = 7.4, H-h); 0.94
(t, 6H, 3J = 7.4, H-i);

EXPERIMENTELLER TEIL 129
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
6a: 149.2 (C-d); 134.9 (C-f); 129.5 (C-b); 115.4 (C-c); 113.6 (C-a); 109.8 (C-e); 37.4 (C-1);
37.0 (C-g); 13.9 (C-h);
6b: 149.4 (C-d); 135.1 (C-f); 129.6 (C-b); 115.5 (C-c); 113.6 (C-a); 109.9 (C-e); 44.4 (C-g);
37.6 (C-1); 21.6 (C-h); 11.5 (C-i);
6c: 147.7 (C-d); 134.8 (C-f); 129.3 (C-b); 128.1 (q, 1J = 239.4, C-h); 115.8 (C-a); 115.2 (C-c);
110.2 (C-e); 43.2 (q, 2J = 37.8, C-g); 37.1 (C-1);
6d: (CDCl3): 150.3 (C-d); 135.2 (C-f); 129.6 (C-b); 116.0 (C-c); 114.1 (C-a); 109.6 (C-e); 37.6
(C-1); 26.3 (C-g);
6e: 147.5 (C-d); 134.9 (C-f); 129.4 (C-b); 115.0 (C-c); 112.7 (C-a); 108.6 (C-e); 51.4 (C-g);
37.1 (C-1); 19.3 (C-h); 10.6 (C-i);
6.2.2 Synthese der Piperidin-2-carbonsäureethylester 7a und 7b
Allgemeine Vorschrift zur Synthese der Verbindungen 7a und 7b:
1 Äq (6.4 mmol) der Piperidin-2-carbonsäure bzw. der S-Piperidin-2-carbonsäure wird unter
Argonatmosphäre in 40 ml abs. EtOH gelöst. Unter Rühren werden langsam 3 Äq (19.2 mmol)
SOCl2 zugetropft und die Reaktionsmischung 4 h unter Rückfluss erhitzt. Nach vollständiger
Umsetzung (DC-Kontrolle, siehe Tabelle 17) wird das Lösungsmittel im Vakuum entfernt, der ölige
Rückstand in 40 ml ges. wässriger NaHCO3-Lösung suspendiert und viermal mit je 30 ml CHCl3
extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet und das
Lösungsmittel im Vakuum entfernt.
Summenformel Mr
[g/mol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
7a126
C8H15NO2 157.21 Cyclohexan : EtOAc = 1:1;
Rf = 0.1
90;
(Lit. 60)126
gelbes Öl;
(Lit. Öl)126
7b127
C8H15NO2 157.21 Cyclohexan : EtOAc = 1:1;
Rf = 0.1
91;
(Lit. 88)127
gelbes Öl
Tabelle 17: Analytische Daten der Verbindungen 7a-7b

130 EXPERIMENTELLER TEIL
In der Literaturstelle128
sind nur 1H-NMR-, und
13C-NMR-Daten von 7a und 7b als Hydrochlorid
angegeben.
IR [cm-1
]:
7a: 1046; 1205; 1370; 1456; 1740; 2695; 2906; 3256;
7b: 1045; 1186; 1371; 1444; 1732; 2854; 2933; 3289;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7a: 4.05 (q, 2H, 3J = 7.1, H-b); 3.22 (dd, 1H,
3J = 9.8,
3J = 3.0, H-d); 3.01-2.92 (m, 1H, H-h);
2.58-2.49 (m, 1H, H-h); 2.07 (s, 1H, H-i); 1.88-1.80 (m, 1H, H-e); 1.70-1.63 (m, 1H, H-e);
1.49-1.28 (m, 4H, H-f, H-g); 1.15 (t, 3H, 3J = 7.1, H-a);
7b: 4.11 (q, 2H, 3J = 7.1, H-b); 3.28 (dd, 1H,
3J = 9.8,
3J = 3.1, H-d); 3.08-2.98 (m, 1H, H-h);
2.64-2.55 (m, 1H, H-h); 2.02 (s, 1H, H-i); 1.95-1.85 (m, 1H, H-e); 1.78-1.66 (m, 1H, H-e);
1.56-1.30 (m, 4H, H-f, H-g); 1.20 (t, 3H, 3J = 7.1, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
7a: 173.2 (C-c); 60.3 (C-b); 58.4 (C-d); 45.5 (C-h); 28.9 (C-e); 25.6 (C-g); 23.8 (C-f); 13.9
(C-a);
7b: 173.4 (C-c); 60.6 (C-b); 58.6 (C-d); 45.7 (C-h); 29.1 (C-e); 25.8 (C-g); 24.0 (C-f); 14.1
(C-a);
6.2.3 Synthese von 1-(2-Ethoxy-2-oxoacetyl)piperidin-2-carbonsäureethylester 8
0.96 g (7.0 mmol) Monoethyloxalylchlorid und 1.42 g (14.0 mmol) Et3N werden unter
Argonatmosphäre in 30 ml abs. Dioxan gelöst. 1.1 g (7.0 mmol) der Verbindung 7a werden unter
Argonatmosphäre in 30 ml abs. Dioxan gelöst, unter Rühren zum ersten Reaktionsansatz getropft und
6 h bei RT gerührt. Nach vollständiger Umsetzung (DC-Kontrolle: Kieselgel; Cyclohexan : EtOAc =

EXPERIMENTELLER TEIL 131
1 : 1; Rf = 0.64) wird das Lösungsmittel im Vakuum entfernt. Der ölige Rückstand wird in 30 ml
CHCl3 suspendiert, viermal mit je 30 ml wässriger 2 M HCl-Lösung und danach viermal mit je 30 ml
dem. H2O gewaschen. Die organische Phase wird über Na2SO4 getrocknet und das Lösungsmittel im
Vakuum entfernt. Man erhält 1.7 g (6.6 mmol) von Verbindung 8 in Form eines braunen Öls.
Summenformel: C12H19NO5
Mr = 257.28 g/mol
Ausbeute: 94 % (Lit. 86 %)129
Aussehen: braunes Öl
Die IR-, und 1H-NMR-Daten zu 8 entsprechen der Literaturstelle
129.
IR [cm-1
]: 1020; 1083; 1258; 1445; 1661; 1738; 2961;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
5.20 (d, 1H, 3J = 5.7, H-d); 4.33 (q, 2H,
3J = 7.1, H-b); 4.26-4.16 (m, 2H, H-k); 3.63-3.56 (m, 1H,
H-h); 3.41-3.31 (m, 1H, H-h); 2.34-2.20 (m, 1H, H-e); 1.81-1.62 (m, 3H, H-e, H-f, H-g); 1.59-1.24
(m, 8H, H-a, H-f, H-g, H-l);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
170.0 (C-c); 162.8 (C-j); 161.7 (C-i); 62.1 (C-b); 61.5 (C-k); 51.6 (C-d); 44.2 (C-h); 26.4 (C-e); 25.0
(C-g); 21.0 (C-f); 14.2 (C-a); 14.0 (C-l);
6.2.4 Synthese von 3-Methyl-2-oxopentansäure 9
2.0 g (13.0 mmol) 3-Methyl-2-oxopentansäure-Natriumsalz werden in 100 ml dem. H2O gelöst und
konz. HCl bis pH 1 zugegeben. Nach mehrfacher Extraktion mit je 50 ml CHCl3 (DC-Kontrolle:
Kieselgel; PE : EtOAc = 1: 1; Rf = 0.26 (Ehrlich)) werden die vereinigten organischen Phasen über
Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Der organische Rückstand wird auf
2 °C abgekühlt und man erhält 1.5 g (11.3 mmol) von Verbindung 9 in Form weißer Nadeln.

132 EXPERIMENTELLER TEIL
Summenformel: C6H10O3
Mr = 130.14 g/mol
Smp. = 46 °C
Ausbeute: 86 % (Lit. 48 %)130
Aussehen: weiße Nadeln (Lit. Öl)130
Die IR-, und 1H-NMR-Daten zu 9 entsprechen der Literaturstelle
130.
IR [cm-1
]: 1120; 1185; 1291; 1326; 1460; 1747; 2968; 3041-3588 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
4.32 (br, 1H, H-g); 3.35-3.24 (m, 1H, H-c); 1.82-1.69 (m, 1H, H-b); 1.52-1.37 (m, 1H, H-b); 1.13 (d,
3H, 3J = 7.0, H-d); 0.89 (t, 3H,
3J = 7.5, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
199.4 (C-e); 159.6 (C-f); 41.7 (C-c); 25.2 (C-b); 14.8 (C-d); 11.3 (C-a);
6.2.5 Synthese von 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäureethylester 1063
1.47 g (11.0 mmol) der Verbindung 9 werden unter Argonatmosphäre in 30 ml abs. DMF gelöst, 1.4
g (11.0 mmol) Oxalylchlorid langsam zugetropft und 12 h bei RT gerührt. Im Anschluss werden 1.71
g (13.2 mmol) N,N- Diisopropylethylamin und nach 5 min 1.73 g (11.0 mmol) der Verbindung 7a
zugegeben. Diese Mischung wird 1-2 d bei RT gerührt. Nach vollständiger Umsetzung (DC-
Kontrolle: Kieselgel; EtOAc : EtOH = 80 : 20; Rf = 0.93) wird das Lösungsmittel im Vakuum
entfernt. Der ölige Rückstand wird in 50 ml CHCl3 suspendiert und zunächst viermal mit je 40 ml
wässriger 2 M HCl-Lösung und danach viermal mit je 40 ml dem. H2O gewaschen. Die organische
Phase wird über Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Anschließend wird

EXPERIMENTELLER TEIL 133
das Rohprodukt säulenchromatographisch (Kieselgel; PE : EtOAc = 70 : 30; Rf = 0.82) gereinigt.
Man erhält 0.6 g (2.2 mmol) von Verbindung 10 in Form eines weißen Öls.
Summenformel: C14H23NO4
Mr = 269.34 g/mol
Ausbeute: 20 %
Aussehen: weißes Öl
IR [cm-1
]: 1024; 1196; 1235; 1422; 1642; 1734; 2872; 2936;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
5.41 (dd, 1H, 3J = 11.8,
3J = 5.3, H-d); 4.23-4.11 (m, 2H, H-b); 3.86 (d, 1H,
2J = 13.5, H-h); 3.26-
3.17 (m, 1H, H-h); 2.73-2.59 (m, 1H, H-k); 2.30-2.20 (m, 1H, H-e); 1.84-1.52 (m, 4H, H-e, H-f,
H-g, H-m); 1.50-1.29 (m, 3H, H-f, H-g, H-m); 1.24 (t, 3H, 3J = 7.1, H-a); 1.13-1.05 (m, 3H, H-l);
0.95-0.80 (t, 3J = 7.1, 3H, H-n);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
216.4 (C-j); 176.4 (C-c); 171.5 (C-i); 60.9 (C-b); 51.8 (C-d); 43.3 (C-h); 36.8 (C-k); 26.6 (C-m);
25.5 (C-e); 21.1 (C-g); 17.2 (C-f); 16.7 (C-l); 14.2 (C-a); 11.9 (C-n);
6.2.6 Darstellung der Pipecolinsäureethylester-Amide 11 und 12
6.2.6.1 Synthese der Carbonsäureamide 11a-11c aus den entsprechenden Säurechloriden
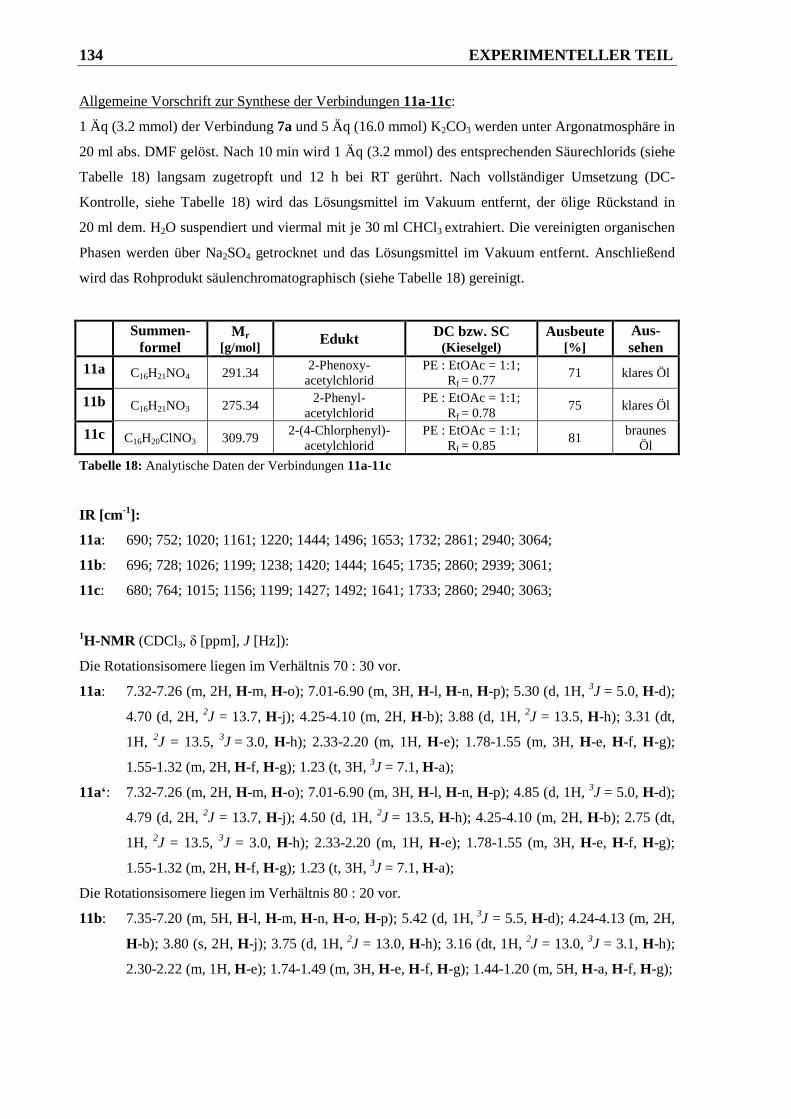
134 EXPERIMENTELLER TEIL
Allgemeine Vorschrift zur Synthese der Verbindungen 11a-11c:
1 Äq (3.2 mmol) der Verbindung 7a und 5 Äq (16.0 mmol) K2CO3 werden unter Argonatmosphäre in
20 ml abs. DMF gelöst. Nach 10 min wird 1 Äq (3.2 mmol) des entsprechenden Säurechlorids (siehe
Tabelle 18) langsam zugetropft und 12 h bei RT gerührt. Nach vollständiger Umsetzung (DC-
Kontrolle, siehe Tabelle 18) wird das Lösungsmittel im Vakuum entfernt, der ölige Rückstand in
20 ml dem. H2O suspendiert und viermal mit je 30 ml CHCl3 extrahiert. Die vereinigten organischen
Phasen werden über Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Anschließend
wird das Rohprodukt säulenchromatographisch (siehe Tabelle 18) gereinigt.
Summen-
formel Mr
[g/mol] Edukt
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
11a C16H21NO4 291.34 2-Phenoxy-
acetylchlorid
PE : EtOAc = 1:1;
Rf = 0.77 71 klares Öl
11b C16H21NO3 275.34 2-Phenyl-
acetylchlorid
PE : EtOAc = 1:1;
Rf = 0.78 75 klares Öl
11c C16H20ClNO3 309.79 2-(4-Chlorphenyl)-
acetylchlorid
PE : EtOAc = 1:1;
Rf = 0.85 81
braunes
Öl
Tabelle 18: Analytische Daten der Verbindungen 11a-11c
IR [cm-1
]:
11a: 690; 752; 1020; 1161; 1220; 1444; 1496; 1653; 1732; 2861; 2940; 3064;
11b: 696; 728; 1026; 1199; 1238; 1420; 1444; 1645; 1735; 2860; 2939; 3061;
11c: 680; 764; 1015; 1156; 1199; 1427; 1492; 1641; 1733; 2860; 2940; 3063;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
11a: 7.32-7.26 (m, 2H, H-m, H-o); 7.01-6.90 (m, 3H, H-l, H-n, H-p); 5.30 (d, 1H, 3J = 5.0, H-d);
4.70 (d, 2H, 2J = 13.7, H-j); 4.25-4.10 (m, 2H, H-b); 3.88 (d, 1H,
2J = 13.5, H-h); 3.31 (dt,
1H, 2J = 13.5,
3J = 3.0, H-h); 2.33-2.20 (m, 1H, H-e); 1.78-1.55 (m, 3H, H-e, H-f, H-g);
1.55-1.32 (m, 2H, H-f, H-g); 1.23 (t, 3H, 3J = 7.1, H-a);
11a‘: 7.32-7.26 (m, 2H, H-m, H-o); 7.01-6.90 (m, 3H, H-l, H-n, H-p); 4.85 (d, 1H, 3J = 5.0, H-d);
4.79 (d, 2H, 2J = 13.7, H-j); 4.50 (d, 1H,
2J = 13.5, H-h); 4.25-4.10 (m, 2H, H-b); 2.75 (dt,
1H, 2J = 13.5,
3J = 3.0, H-h); 2.33-2.20 (m, 1H, H-e); 1.78-1.55 (m, 3H, H-e, H-f, H-g);
1.55-1.32 (m, 2H, H-f, H-g); 1.23 (t, 3H, 3J = 7.1, H-a);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
11b: 7.35-7.20 (m, 5H, H-l, H-m, H-n, H-o, H-p); 5.42 (d, 1H, 3
J = 5.5, H-d); 4.24-4.13 (m, 2H,
H-b); 3.80 (s, 2H, H-j); 3.75 (d, 1H, 2J = 13.0, H-h); 3.16 (dt, 1H,
2J = 13.0,
3J = 3.1, H-h);
2.30-2.22 (m, 1H, H-e); 1.74-1.49 (m, 3H, H-e, H-f, H-g); 1.44-1.20 (m, 5H, H-a, H-f, H-g);

EXPERIMENTELLER TEIL 135
11b‘: 7.35-7.20 (m, 5H, H-l, H-m, H-n, H-o, H-p); 4.64-4.54 (m, 2H, H-d, H-h); 4.13-4.03 (m,
2H, H-b); 3.71 (s, 2H, H-j); 2.73-2.65 (m, 1H, H-h); 2.19-2.11 (m, 1H, H-e); 1.74-1.49 (m,
3H, H-e, H-f, H-g); 1.44-1.20 (m, 5H, H-a, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
11c: 7.35-7.24 (m, 2H, H-l, H-p); 7.23-7.15 (m, 2H, H-m, H-o); 5.39 (d, 1H, 3J = 5.5, H-d); 4.24-
4.14 (m, 2H, H-b); 3.75 (s, 2H, H-j); 3.74-3.68 (m, 1H, H-h); 3.18 (dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.32-2.23 (m, 1H, H-e); 1.75-1.54 (m, 3H, H-e, H-f, H-g); 1.40-1.20 (m, 5H,
H-a, H-f, H-g);
11c‘: 7.35-7.24 (m, 2H, H-l, H-p); 7.23-7.15 (m, 2H, H-m, H-o); 4.58 (d, 1H, 2J = 13.0, H-h); 4.52
(d, 1H, 3J = 5.5, H-d); 4.14-4.06 (m, 2H, H-b); 3.66 (s, 2H, H-j); 2.74-2.67 (m, 1H, H-h);
2.22-2.15 (m, 1H, H-e); 1.75-1.54 (m, 3H, H-e, H-f, H-g); 1.40-1.20 (m, 5H, H-a, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
11a: 170.9 (C-c); 167.9 (C-i); 158.0 (C-k); 129.5 (C-m, C-o); 121.5 (C-n); 114.7 (C-l, C-p); 67.4
(C-j); 61.2 (C-b); 52.3 (C-d); 43.1 (C-h); 26.6 (C-e); 25.2 (C-g); 20.9 (C-f); 14.2 (C-a);
11a‘: 170.9 (C-c); 167.9 (C-i); 158.0 (C-k); 129.5 (C-m, C-o); 121.5 (C-n); 114.7 (C-l, C-p); 67.4
(C-j); 61.2 (C-b); 55.9 (C-d); 40.0 (C-h); 26.6 (C-e); 25.2 (C-g); 20.9 (C-f); 14.2 (C-a);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
11b: 171.3 (C-c); 170.8 (C-i); 134.9 (C-k); 128.6 (C-l, C-m, C-o, C-p); 126.7 (C-n); 61.1 (C-b);
52.0 (C-d); 44.1 (C-h); 41.1 (C-j); 26.6 (C-e); 25.2 (C-g); 20.8 (C-f); 14.2 (C-a);
11b‘: 171.3 (C-c); 170.8 (C-i); 134.9 (C-k); 128.6 (C-l, C-m, C-o, C-p); 126.7 (C-n); 61.1 (C-b);
56.6 (C-d); 41.3 (C-j); 39.5 (C-h); 27.2 (C-e); 24.5 (C-g); 20.7 (C-f); 14.2 (C-a);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
11c: 171.1 (C-c); 170.4 (C-i); 133.4 (C-k); 132.6 (C-n); 130.1 (C-m, C-o) 128.7 (C-l, C-p); 61.2
(C-b); 52.1 (C-d); 44.0 (C-h); 40.3 (C-j); 26.6 (C-e); 25.2 (C-g); 20.8 (C-f); 14.2 (C-a);
11c‘: 171.1 (C-c); 170.4 (C-i); 133.4 (C-k); 132.6 (C-n); 130.1 (C-m, C-o) 128.7 (C-l, C-p); 61.5
(C-b); 56.6 (C-d); 40.4 (C-j); 39.6 (C-h); 27.3 (C-e); 24.5 (C-g); 20.8 (C-f); 14.2 (C-a);
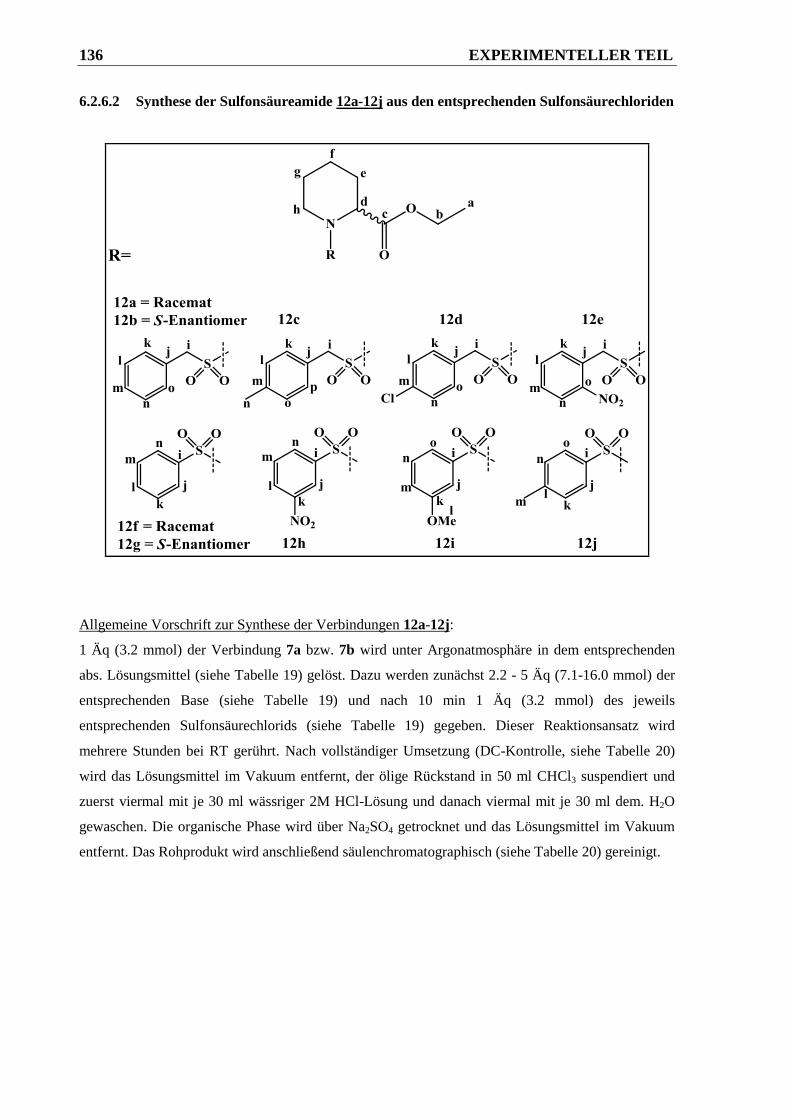
136 EXPERIMENTELLER TEIL
6.2.6.2 Synthese der Sulfonsäureamide 12a-12j aus den entsprechenden Sulfonsäurechloriden
Allgemeine Vorschrift zur Synthese der Verbindungen 12a-12j:
1 Äq (3.2 mmol) der Verbindung 7a bzw. 7b wird unter Argonatmosphäre in dem entsprechenden
abs. Lösungsmittel (siehe Tabelle 19) gelöst. Dazu werden zunächst 2.2 - 5 Äq (7.1-16.0 mmol) der
entsprechenden Base (siehe Tabelle 19) und nach 10 min 1 Äq (3.2 mmol) des jeweils
entsprechenden Sulfonsäurechlorids (siehe Tabelle 19) gegeben. Dieser Reaktionsansatz wird
mehrere Stunden bei RT gerührt. Nach vollständiger Umsetzung (DC-Kontrolle, siehe Tabelle 20)
wird das Lösungsmittel im Vakuum entfernt, der ölige Rückstand in 50 ml CHCl3 suspendiert und
zuerst viermal mit je 30 ml wässriger 2M HCl-Lösung und danach viermal mit je 30 ml dem. H2O
gewaschen. Die organische Phase wird über Na2SO4 getrocknet und das Lösungsmittel im Vakuum
entfernt. Das Rohprodukt wird anschließend säulenchromatographisch (siehe Tabelle 20) gereinigt.
EXPERIMENTELLER TEIL 137
Summen-
formel Mr
[g/mol] Edukt Reaktionsbedingungen
LM Base Temperatur /
Zeit
12a C15H21NO4S 311.40 Benzylsulfonsäure-
chlorid Dioxan
2.2 Äq (7.1 mmol)
Diisopropylethylamin RT / 4.5 h
12b C15H21NO4S 311.40 Benzylsulfonsäure-
chlorid DMF
5 Äq (16.0 mmol)
K2CO3 RT / 24 h
12c C16H23NO4S 325.42 4-Methylbenzyl-
sulfonsäurechlorid DMF
5 Äq (16.0 mmol)
K2CO3 RT / 12 h
12d C15H20ClNO4S 345.84 4-Chlorbenzyl-
sulfonsäurechlorid DMF
5 Äq (16.0 mmol)
K2CO3 RT / 12 h
12e C15H20N2O6S 356.39 2-Nitrobenzyl-
sulfonsäurechlorid DMF
5 Äq (16.0 mmol)
K2CO3 RT / 12 h
12f131
C14H19NO4S 297.37 Phenylsulfonsäure-
chlorid CH2Cl2
3 Äq (9.6 mmol)
Et3N
0 °C / 1 h
RT / 16 h
12g C14H19NO4S 297.37 Phenylsulfonsäure-
chlorid THF
3 Äq (9.6 mmol)
Et3N RT / 12 h
12h C14H18N2O6S 342.37 3-Nitrophenyl-
sulfonsäurechlorid CH2Cl2
3 Äq (9.6 mmol)
Et3N
0 °C / 1 h
RT / 16 h
12i C15H21NO5S 327.4 3-Methoxyphenyl-
sulfonsäurechlorid CH2Cl2
3 Äq (9.6 mmol)
Et3N
0 °C / 1 h
RT / 14 h
12j132
C15H21NO5S 311.40 4-Methylphenyl-
sulfonsäurechlorid DMF
5 Äq (16.0 mmol)
K2CO3 RT / 12 h
Tabelle 19: Synthetische Daten der Verbindungen 12a-12j
Aussehen DC bzw. SC
(Kieselgel)
Ausbeute [%]
Smp. [°C]
12a gelbes Öl Cyclohexan : EtOAc = 1:1;
Rf = 0.73 77 -
12b gelbes Öl Cyclohexan : EtOAc = 80:20;
Rf = 0.46 63 -
12c weißes Öl PE : EtOAc = 1:1;
Rf = 0.85 50 -
12d klares Öl PE : EtOAc = 1:1;
Rf = 0.85 46 -
12e gelbes Öl PE : EtOAc = 1:1;
Rf = 0.74 17 -
12f131
gelbes Öl Cyclohexan : EtOAc = 80:20;
Rf = 0.41
92;
(Lit. 37)131
-
12g braunes Öl Cyclohexan : EtOAc = 1:1;
Rf = 0.87 79 -
12h brauner
Feststoff
Cyclohexan : EtOAc = 80:20;
Rf = 0.23 97 94
12i gelbes Öl Cyclohexan : EtOAc = 80:20;
Rf = 0.29 76 -
12j132
klares Öl;
(Lit. FS*)132
PE : EtOAc = 1:1;
Rf = 0.73
71;
(Lit. 82)132
(Lit. 167)
132
Tabelle 20: Analytische Daten der Verbindungen 12a-12j
* Angaben beziehen sich auf das Dicyclohexylamin-Salz
Die IR-, und 1H-NMR-Daten zu 12f und 12j entsprechen den Literaturstellen
131,132.

138 EXPERIMENTELLER TEIL
IR [cm-1
]:
12a: 697; 739; 962; 1060; 1148; 1200; 1335; 1455; 1732; 2860; 2940; 3062;
12b: 688; 739; 963; 1075; 1145; 1222; 1335; 1455; 1738; 2860; 2942; 3065;
12c: 697; 767; 813; 1024; 1159; 1280; 1444; 1514; 1730; 2857; 2934; 3060;
12d: 717; 730; 961; 1059; 1128; 1199; 1335; 1492; 1732; 2861; 2942; 3062;
12e: 714; 737; 962; 1060; 1147; 1199; 1337; 1528; 1732; 2863; 2943; 3060;
12f: 689; 737; 960; 1093; 1155; 1181; 1337; 1446; 1733; 2862; 2943; 3062;
12g: 689; 737; 959; 1093; 1155; 1182; 1337; 1446; 1736; 2862; 2943; 3064;
12h: 671; 714; 955; 1067; 1125; 1156; 1209; 1350; 1527; 1731; 2863; 2958; 3092;
12i: 685; 727; 959; 1036; 1152; 1240; 1338; 1478; 1597; 1737; 2862; 2942; 3067;
12j: 654; 727; 959; 1093; 1154; 1179; 1337; 1446; 1738; 2862; 2942; 3065;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
12a: 7.40-7.35 (m, 2H, H-k, H-o); 7.31-7.24 (m, 3H, H-l, H-m, H-n); 4.57 (d, 1H, 3
J = 4.0, H-d);
4.29-4.20 (m, 4H, H-b, H-i); 3.50-3.41 (m, 1H, H-h); 3.15 (dt, 1H, 2J = 12.8,
3J = 3.1, H-h);
2.22-2.11 (m, 1H, H-e); 1.72-1.54 (m, 3H, H-e, H-f, H-g); 1.51-1.38 (m, 1H, H-g); 1.33-1.20
(m, 4H, H-a, H-f);
12b: 7.57-7.40 (m, 2H, H-k, H-o); 7.39-7.33 (m, 3H, H-l, H-m, H-n); 4.60-4.55 (m, 1H, H-d);
4.30-4.17 (m, 4H, H-b, H-i); 3.49-3.41 (m, 1H, H-h); 3.15 (dt, 1H, 2J = 12.7,
3J = 3.1, H-h);
2.20-2.12 (m, 1H, H-e); 1.73-1.35 (m, 3H, H-e, H-f, H-g); 1.32-1.18 (m, 5H, H-a, H-f, H-g);
12c: 7.20 (d, 2H, 3J = 8.0, H-k, H-p); 7.11 (d, 2H,
3J = 8.0, H-l, H-o); 4.21 (q, 2H,
3J = 7.0, H-b);
3.76 (d, 1H, 2J = 13.2, H-i); 3.37 (d, 1H,
2J = 13.2, H-i); 3.10 (dd, 1H,
3J = 8.0,
3J = 4.3,
H-d); 2.96-2.89 (m, 1H, H-h); 2.33 (s, 3H, H-n); 2.16-2.08 (m, 1H, H-h); 1.89-1.74 (m, 2H,
H-e, H-g); 1.67-1.50 (m, 3H, H-e, H-f, H-g); 1.39-1.24 (m, 4H, H-a, H-f);
12d: 7.42 (d, 2H, 3J = 8.5, H-l, H-n); 7.34 (d, 2H,
3J = 8.5, H-k, H-o); 4.62 (d, 1H,
3J = 4.7, H-d);
4.31-4.20 (m, 4H, H-b, H-i); 3.46 (d, 1H, 2J = 12.7, H-h); 3.12 (dt, 1H,
2J = 12.7,
3J = 3.1,
H-h); 2.24-2.17 (m, 1H, H-e); 1.75-1.56 (m, 3H, H-e, H-f, H-g); 1.53-1.40 (m, 1H, H-g);
1.33-1.21 (m, 4H, H-a, H-f);
12e: 8.01 (d, 1H, 3J = 8.0, H-n); 7.59-7.56 (m, 2H, H-k, H-l); 7.52-7.45 (m, 1H, H-m); 4.91 (d,
1H, 2J = 13.7, H-i); 4.81 (d, 1H,
2J = 13.7, H-i); 4.61 (d, 1H,
3J = 4.7, H-d); 4.26-4.18 (m,
2H, H-b); 3.64-3.56 (m, 1H, H-h); 3.20 (dt, 1H, 2J = 12.6,
3J = 3.0, H-h); 2.23-2.16 (m, 1H,
H-e); 1.76-1.59 (m, 3H, H-e, H-f, H-g); 1.59-1.41 (m, 1H, H-g); 1.31-1.18 (m, 4H, H-a,
H-f);
12f: 7.82-7.77 (m, 2H, H-j; H-n); 7.58-7.43 (m, 3H, H-k, H-l, H-m); 4.73 (d, 1H, 3J = 5.3, H-d);
4.09-3.87 (m, 2H, H-b); 3.83-3.76 (m, 1H, H-h); 3.23 (dt, 1H, 2J = 12.4,
3J = 3.0, H-h); 2.17-
2.08 (m, 1H, H-e); 1.82-1.57 (m, 3H, H-e, H-f, H-g); 1.56-1.41 (m, 1H, H-g); 1.37-1.20 (m,
1H, H-f); 1.13 (t, 3H, 3J = 7.1, H-a);

EXPERIMENTELLER TEIL 139
12g: 7.79-7.75 (m, 2H, H-j, H-n); 7.69-7.56 (m, 3H, H-k, H-l, H-m); 4.65-4.60 (m, 1H, H-d);
4.05-3.82 (m, 2H, H-b); 3.70-3.63 (m, 1H, H-h); 3.18-3.06 (m, 1H, H-h); 2.03-1.93 (m, 1H,
H-e); 1.63-1.52 (m, 3H, H-e, H-f, H-g); 1.32-1.10 (m, 2H, H-f, H-g); 1.06 (t, 3H, 3J = 7.1,
H-a);
12h: 8.63-8.59 (m, 1H, H-j); 8.43-8.38 (m, 1H, H-l); 8.12-8.09 (m, 1H, H-n); 7.70 (t, 1H, 3J = 8.1,
H-m); 4.80 (d, 1H, 3J = 5.4, H-d); 4.09-3.92 (m, 2H, H-b); 3.88-3.80 (m, 1H, H-h); 3.18 (dt,
1H, 2J = 12.6,
3J = 3.0, H-h); 2.25-2.17 (m, 1H, H-e); 1.88-1.66 (m, 3H, H-e, H-f, H-g);
1.64-1.51 (m, 1H, H-g); 1.34-1.19 (m, 1H, H-f); 1.16 (t, 3H, 3J = 7.1, H-a);
12i: 7.40-7.35 (m, 2H, H-j, H-o); 7.32-7.30 (m, 1H, H-m); 7.10-7.04 (m, 1H, H-n); 4.73 (d, 1H,
3J = 5.2, H-d); 4.10-3.90 (m, 2H, H-b); 3.84 (s, 3H, H-l); 3.81-3.73 (m, 1H, H-h); 3.25 (dt,
1H, 2J = 12.7,
3J = 3.0, H-h); 2.17-2.08 (m, 1H, H-e); 1.84-1.58 (m, 3H, H-e, H-f, H-g);
1.57-1.46 (m, 1H, H-g); 1.36-1.23 (m, 1H, H-f); 1.15 (t, 3H, 3J = 7.1, H-a);
12j: 7.67 (d, 2H, 3J = 8.0, H-j; H-o); 7.27 (d, 2H,
3J = 8.0, H-k, H-n); 4.73 (d, 1H,
3J = 5.0, H-d);
4.09-3.91 (m, 2H, H-b); 3.79-3.72 (m, 1H, H-h); 3.22 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.41
(s, 3H, H-m); 2.16-2.08 (m, 1H, H-e); 1.80-1.39 (m, 4H, H-e, H-f, H-g); 1.35-1.22 (m, 1H,
H-f); 1.15 (t, 3H, 3J = 7.1, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
12a: 171.6 (C-c); 131.1 (C-k, C-o); 129.5 (C-j); 128.7 (C-l, C-n); 128.6 (C-m); 61.5 (C-b); 58.9
(C-i); 56.1 (C-d); 43.6 (C-h); 28.0 (C-e); 25.2 (C-g); 20.5 (C-f); 14.4 (C-a);
12b: 171.4 (C-c); 131.0 (C-k, C-o); 129.4 (C-j); 128.5 (C-l, C-n); 128.4 (C-m); 61.4 (C-b); 58.7
(C-i); 56.0 (C-d); 43.4 (C-h); 27.8 (C-e); 25.1 (C-g); 20.3 (C-f); 14.2 (C-a);
12c: 174.0 (C-c); 136.6 (C-m); 130.7 (C-j); 129.3 (C-k, C-p); 128.8 (C-l, C-o); 64.6 (C-d); 60.3
(C-i); 60.2 (C-b); 50.2 (C-h); 29.6 (C-e); 25.3 (C-g); 22.6 (C-f); 21.1 (C-n); 14.3 (C-a);
12d: 171.4 (C-c); 134.6 (C-m); 132.3 (C-l, C-n); 128.8 (C-k, C-o); 127.9 (C-j); 61.5 (C-b); 57.9
(C-i); 55.0 (C-d); 43.5 (C-h); 27.9 (C-e); 25.1 (C-g); 20.4 (C-f); 14.2 (C-a);
12e: 171.4 (C-c); 149.7 (C-o); 134.1 (C-l); 133.1 (C-k); 129.5 (C-m); 125.6 (C-n); 124.4 (C-j);
61.6 (C-b); 56.1 (C-d); 54.1 (C-i); 43.1 (C-h); 27.9 (C-e); 25.0 (C-g); 20.3 (C-f); 14.2 (C-a);
12f: 170.6 (C-c); 140.1 (C-i); 132.3 (C-l); 128.8 (C-k, C-m); 127.1 (C-j, C-n); 61.0 (C-b); 55.0
(C-d); 42.6 (C-h); 27.9 (C-e); 24.7 (C-g); 20.0 (C-f); 14.0 (C-a);
12g: 170.8 (C-c); 140.5 (C-i); 132.4 (C-l); 128.9 (C-k, C-m); 126.3 (C-j, C-n); 60.4 (C-b); 54.3
(C-d); 42.0 (C-h); 26.8 (C-e); 23.6 (C-g); 19.1 (C-f); 13.5 (C-a);
12h: 170.2 (C-c); 148.1 (C-k); 142.1 (C-i); 132.8 (C-n); 130.0 (C-m); 126.8 (C-j); 122.5 (C-l);
61.4 (C-b); 55.5 (C-d); 42.9 (C-h); 28.0 (C-e); 24.9 (C-g); 20.0 (C-f); 14.1 (C-a);
12i: 170.7 (C-c); 159.8 (C-k); 141.2 (C-i); 129.8 (C-o); 119.3 (C-j); 118.7 (C-n); 112.0 (C-m);
61.1 (C-b); 55.6 (C-l); 55.1 (C-d); 42.7 (C-h); 27.9 (C-e); 24.7 (C-g); 20.0 (C-f); 14.0 (C-a);
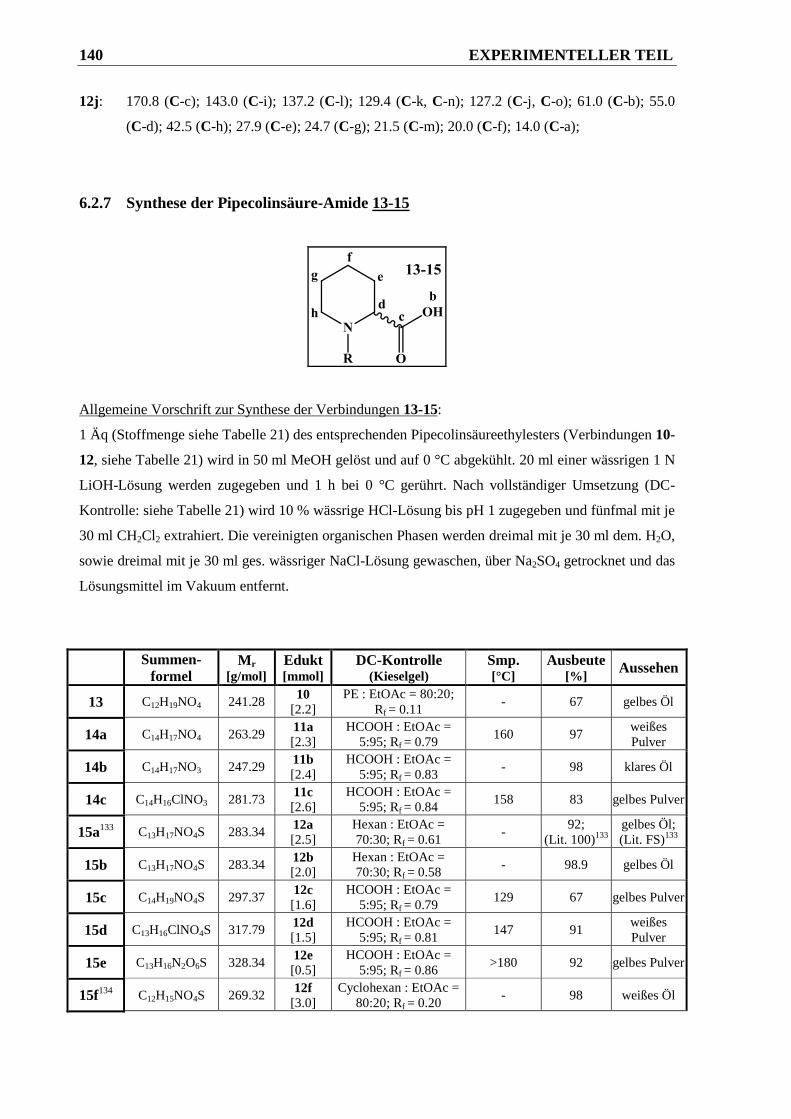
140 EXPERIMENTELLER TEIL
12j: 170.8 (C-c); 143.0 (C-i); 137.2 (C-l); 129.4 (C-k, C-n); 127.2 (C-j, C-o); 61.0 (C-b); 55.0
(C-d); 42.5 (C-h); 27.9 (C-e); 24.7 (C-g); 21.5 (C-m); 20.0 (C-f); 14.0 (C-a);
6.2.7 Synthese der Pipecolinsäure-Amide 13-15
Allgemeine Vorschrift zur Synthese der Verbindungen 13-15:
1 Äq (Stoffmenge siehe Tabelle 21) des entsprechenden Pipecolinsäureethylesters (Verbindungen 10-
12, siehe Tabelle 21) wird in 50 ml MeOH gelöst und auf 0 °C abgekühlt. 20 ml einer wässrigen 1 N
LiOH-Lösung werden zugegeben und 1 h bei 0 °C gerührt. Nach vollständiger Umsetzung (DC-
Kontrolle: siehe Tabelle 21) wird 10 % wässrige HCl-Lösung bis pH 1 zugegeben und fünfmal mit je
30 ml CH2Cl2 extrahiert. Die vereinigten organischen Phasen werden dreimal mit je 30 ml dem. H2O,
sowie dreimal mit je 30 ml ges. wässriger NaCl-Lösung gewaschen, über Na2SO4 getrocknet und das
Lösungsmittel im Vakuum entfernt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Smp. [°C]
Ausbeute [%]
Aussehen
13 C12H19NO4 241.28 10
[2.2]
PE : EtOAc = 80:20;
Rf = 0.11 - 67 gelbes Öl
14a C14H17NO4 263.29 11a
[2.3]
HCOOH : EtOAc =
5:95; Rf = 0.79 160 97
weißes
Pulver
14b C14H17NO3 247.29 11b
[2.4]
HCOOH : EtOAc =
5:95; Rf = 0.83 - 98 klares Öl
14c C14H16ClNO3 281.73 11c
[2.6]
HCOOH : EtOAc =
5:95; Rf = 0.84 158 83 gelbes Pulver
15a133
C13H17NO4S 283.34 12a
[2.5]
Hexan : EtOAc =
70:30; Rf = 0.61 -
92;
(Lit. 100)133
gelbes Öl;
(Lit. FS)133
15b C13H17NO4S 283.34 12b
[2.0]
Hexan : EtOAc =
70:30; Rf = 0.58 - 98.9 gelbes Öl
15c C14H19NO4S 297.37 12c
[1.6]
HCOOH : EtOAc =
5:95; Rf = 0.79 129 67 gelbes Pulver
15d C13H16ClNO4S 317.79 12d
[1.5]
HCOOH : EtOAc =
5:95; Rf = 0.81 147 91
weißes
Pulver
15e C13H16N2O6S 328.34 12e
[0.5]
HCOOH : EtOAc =
5:95; Rf = 0.86 >180 92 gelbes Pulver
15f134
C12H15NO4S 269.32 12f
[3.0]
Cyclohexan : EtOAc =
80:20; Rf = 0.20 - 98 weißes Öl
EXPERIMENTELLER TEIL 141
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Smp. [°C]
Ausbeute [%]
Aussehen
15h C12H14N2O6S 314.31 12h
[3.1]
Cyclohexan : EtOAc =
80:20 (+ 5 % HCOOH);
Rf = 0.63
143 92 weißes
Pulver
15i135
C13H17NO5S 299.34 12i
[2.4]
Cyclohexan : EtOAc =
1:1 (+ 5 % HCOOH);
Rf = 0.52
133 87;
(Lit. 31*)135
weißes
Pulver
15j136
C13H17NO4S 283.34 12j
[2.3]
HCOOH : EtOAc =
5:95; Rf = 0.88
96;
(Lit. 100)136
99;
(Lit. 76)136
weißes
Pulver;
(Lit. FS)136
Tabelle 21: Analytische Daten der Verbindungen 13-15 * Angabe gilt für S-Enantiomer
Die IR-, und 1H-NMR-Daten zu 15j entsprechen der Literaturstelle
136.
IR [cm-1
]:
13: 992; 1091; 1154; 1415; 1654; 1718; 2885; 2941; 2759-3390 (b);
14a: 694; 753; 1019; 1082; 1201; 1239; 1432; 1618; 1737; 2858; 2952; 3052; 2642-3429 (b);
14b: 696; 723; 865; 1025; 1140; 1204: 1446; 1592; 1725; 2861; 2939; 3028; 2752-3348 (b);
14c: 689; 746; 1013; 1089; 1160; 1221; 1453; 1600; 1716; 2865; 2933; 3014; 2684-3422 (b);
15a: 699; 732; 959; 1060; 1118; 1317; 1455; 1728; 2854; 2952; 3062; 3012-3393 (b);
15b: 699; 732; 959; 1060; 1118; 1317; 1455; 1728; 2854; 2952; 3065; 3012-3393 (b);
15c: 681; 758; 820; 1050; 1185; 1283; 1454; 1732; 2185; 2868; 2954; 3052; 2785-3405 (b);
15d: 661; 730; 928; 1146; 1214; 1308; 1335; 1495; 1711; 2859; 2933; 3091; 2737-3414 (b);
15e: 715; 733; 963; 1060; 1149; 1259; 1347; 1525; 1704; 2863; 2951; 3016; 2716-3474 (b);
15f: 688; 730; 958; 1075; 1100; 1325; 1445; 1724; 2878; 2960; 3065; 3115-3205 (b);
15h: 710; 736; 878; 1044; 1085; 1353; 1536; 1714; 2894; 2974; 3033; 2785-3428 (b);
15i: 679; 724; 1027; 1099; 1251; 1314; 1487; 1599; 1703; 2874; 2943; 3012; 2703-3327 (b);
15j: 667; 728; 955; 1114; 1157; 1214; 1334; 1445; 1711; 2860; 2937; 3034; 2991-3351 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
13: 5.45-5.36 (m, 1H, H-d); 3.86-3.78 (m, 1H, H-h); 3.22-3.12 (m, 1H, H-h); 2.72-2.60 (m, 1H,
H-k); 2.27 (d, 1H, 2J = 13.4, H-e); 1.86-1.54 (m, 4H, H-e, H-f, H-g, H-m); 1.53-1.31 (m, 3H,
H-f, H-g, H-m); 1.15-1.04 (m, 3H, H-l); 0.88 (t, 3H, 3J = 7.4, H-n);
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
14a: (CD3CN): 9.46 (br, 1H, H-b); 7.32-7.25 (m, 2H, H-m; H-o); 6.99-6.87 (m, 3H, H-l, H-n, H-
p); 5.15 (d, 1H, 3J = 5.7, H-d); 4.73 (d, 2H,
2J = 14.8, H-j); 3.75 (d, 1H,
2J = 13.0, H-h); 3.23
(dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.22-2.15 (m, 1H, H-e); 1.77-1.27 (m, 5H, H-e, H-f, H-g);
14a‘: (CD3CN): 9.46 (br, 1H, H-b); 7.32-7.25 (m, 2H, H-m; H-o); 6.99-6.87 (m, 3H, H-l, H-n,
H-p); 4.85 (d, 2H, 2
J = 14.8, H-j); 4.78-4.64 (m, 1H, H-d); 4.36 (d, 1H, 2J = 13.0, H-h); 2.66
(dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.22-2.15 (m, 1H, H-e); 1.77-1.27 (m, 5H, H-e, H-f, H-g);

142 EXPERIMENTELLER TEIL
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
14b: (CD3CN): 9.51 (br, 1H, H-b); 7.35-7.18 (m, 5H, H-l, H-m; H-n, H-o, H-p); 5.23 (d, 1H,
3J = 5.5, H-d); 3.83 (d, 1H,
2J = 13.5, H-h); 3.73 (d, 2H,
2J = 15.5, H-j); 3.13 (dt, 1H,
2J = 13.5,
3J = 3.1, H-h); 2.20-2.09 (m, 1H, H-e); 1.72-1.44 (m, 3H, H-e, H-f, H-g); 1.39-
1.25 (m, 2H, H-f, H-g);
14b‘: (CD3CN): 9.51 (br, 1H, H-b); 7.35-7.18 (m, 5H, H-l, H-m; H-n, H-o, H-p); 4.73 (d, 1H,
3J = 5.5, H-d); 4.43 (d, 1H,
2J = 13.5, H-h); 3.60 (d, 2H,
2J = 15.5, H-j); 2.61 (dt, 1H,
2J = 13.5,
3J = 3.1, H-h); 2.20-2.09 (m, 1H, H-e); 1.72-1.44 (m, 3H, H-e, H-f, H-g); 1.39-
1.25 (m, 2H, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
14c: (CD3CN): 9.46 (br, 1H, H-b); 7.36-7.28 (m, 2H, H-l, H-p); 7.24-7.16 (m, 2H, H-m, H-o);
5.22 (d, 1H, 3J = 5.3, H-d); 3.81 (d, 1H,
2J = 13.0, H-h); 3.71 (d, 2H,
2J = 17.0, H-j); 3.14 (dt,
1H, 2J = 13.0,
3J = 3.0, H-h); 2.20-2.13 (m, 1H, H-e); 1.71-1.52 (m, 3H, H-e, H-f, H-g);
1.42-1.28 (m, 2H, H-f, H-g);
14c‘: (CD3CN): 9.46 (br, 1H, H-b); 7.36-7.28 (m, 2H, H-l, H-p); 7.24-7.16 (m, 2H, H-m, H-o);
4.71 (d, 1H, 3J = 5.3, H-d); 4.41 (d, 1H,
2J = 13.0, H-h); 3.58 (d, 2H,
2J = 17.0, H-j); 2.60 (dt,
1H, 2J = 13.0,
3J = 3.0, H-h); 2.20-2.13 (m, 1H, H-e); 1.71-1.52 (m, 3H, H-e, H-f, H-g);
1.42-1.28 (m, 2H, H-f, H-g);
15a: 7.49-7.33 (m, 5H, H-k, H-l, H-m, H-n, H-o); 4.60-4.55 (m, 1H, H-d); 4.27 (s, 2H, H-i);
3.49-3.41 (m, 1H, H-h); 3.15 (dt, 1H, 2J = 12.4,
3J = 3.1, H-h); 2.20-2.12 (m, 1H, H-e); 1.73-
1.52 (m, 3H, H-e, H-f, H-g); 1.51-1.18 (m, 2H, H-f, H-g);
15b: 7.48-7.33 (m, 5H, H-k, H-l, H-m, H-n, H-o); 4.58-4.54 (m, 1H, H-d); 4.26 (s, 2H, H-i);
3.48-3.40 (m, 1H, H-h); 3.13 (dt, 1H, 2J = 12.8,
3J = 2.9, H-h); 2.20-2.11 (m, 1H, H-e); 1.75-
1.51 (m, 3H, H-e, H-f, H-g); 1.49-1.21 (m, 2H, H-f, H-g);
15c: 7.32 (d, 2H, 3J = 8.0, H-k, H-p); 7.17 (d, 2H,
3J = 8.0, H-l, H-o); 4.58 (d, 1H,
3J = 5.0, H-d);
4.23 (s, 2H, H-i); 3.51-3.44 (m, 1H, H-h); 3.14 (dt, 1H, 2J = 12.9,
3J = 3.0, H-h); 2.35 (s, 3H,
H-n); 2.21-2.14 (m, 1H, H-e); 1.77-1.55 (m, 3H, H-e, H-f, H-g); 1.50-1.25 (m, 2H, H-f,
H-g);
15d: (CD3CN): 9.43 (br, 1H, H-b); 7.45-7.38 (m, 4H, H-k, H-l, H-n, H-o); 4.42 (d, 1H, 3J = 5.1,
H-d); 4.28 (d, 2H, 2J = 4.1, H-i); 3.44 (d, 1H,
2J = 13.0, H-h); 3.18 (dt, 1H,
2J = 13.0,
3J = 3.0, H-h); 2.13-2.05 (m, 1H, H-e); 1.70-1.57 (m, 3H, H-e, H-f, H-g); 1.45-1.22 (m, 2H,
H-f, H-g);
15e: (CD3CN): 9.53 (br, 1H, H-b); 8.03-7.99 (m, 1H, H-n); 7.74-7.68 (m, 1H, H-l); 7.64-7.58 (m,
2H, H-k, H-m); 4.90-4.79 (m, 2H, H-i); 4.47-4.44 (m, 1H, H-d); 3.50-3.43 (m, 1H, H-h);
3.21 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.19-2.08 (m, 1H, H-e); 1.73-1.62 (m, 3H, H-e, H-f,
H-g); 1.49-1.21 (m, 2H, H-f, H-g);

EXPERIMENTELLER TEIL 143
15f: 7.83-7.78 (m, 2H, H-j; H-n); 7.59-7.45 (m, 3H, H-k, H-l, H-m); 4.78 (d, 1H, 3
J = 4.8, H-d);
3.80-3.72 (m, 1H, H-h); 3.23 (dt, 1H, 2J = 12.4,
3J = 3.0, H-h); 2.20-2.12 (m, 1H, H-e); 1.80-
1.60 (m, 3H, H-e, H-f, H-g); 1.51-1.23 (m, 2H, H-g, H-f);
15h: (DMSO-d6): 12.97 (br, 1H, H-b); 8.53-8.41 (m, 2H, H-j, H-l); 8.25-8.21 (m, 1H, H-n); 7.89
(t, 1H, 3J = 8.0, H-m); 4.65 (d, 1H,
3J = 4.2, H-d); 3.76-3.68 (m, 1H, H-h); 3.10 (dt, 1H,
2J = 12.8,
3J = 2.7, H-h); 2.10-2.02 (m, 1H, H-e); 1.68-1.53 (m, 3H, H-e, H-f, H-g); 1.38-
1.09 (m, 2H, H-f, H-g);
15i: 7.52-7.36 (m, 2H, H-j, H-o); 7.34-7.31 (m, 1H, H-m); 7.12-7.05 (m, 1H, H-n); 4.79 (d, 1H,
3J = 4.9, H-d); 3.84 (s, 3H, H-l); 3.81-3.73 (m, 1H, H-h); 3.22 (dt, 1H,
2J = 12.8,
3J = 3.0,
H-h); 2.20-2.12 (m, 1H, H-e); 1.81-1.59 (m, 3H, H-e, H-f, H-g); 1.54-1.21 (m, 2H, H-f,
H-g);
15j: (CD3CN): 9.34 (br, 1H, H-b); 7.68 (d, 2H, 3
J = 8.0, H-j; H-o); 7.35 (d, 2H, 3J = 8.0, H-k,
H-n); 4.66 (d, 1H, 3J = 5.3, H-d); 3.70-3.64 (m, 1H, H-h); 3.18 (dt, 1H,
2J = 12.9,
3J = 3.0, H-
h); 2.41 (s, 3H, H-m); 2.09-2.01 (m, 1H, H-e); 1.65-1.52 (m, 3H, H-e, H-f, H-g); 1.31-1.23
(m, 2H, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
13: 216.4 (C-j); 177.6 (C-c); 175.5 (C-i); 51.9 (C-d); 43.5 (C-h); 37.0 (C-k); 26.7 (C-m); 25.4
(C-e); 20.9 (C-g); 17.2 (C-f); 16.6 (C-l); 11.9 (C-n);
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
14a: (CD3CN): 172.6 (C-c); 168.8 (C-i); 159.3 (C-k); 130.3 (C-m, C-o); 122.0 (C-n); 115.5 (C-l,
C-p); 67.0 (C-j); 52.6 (C-d); 43.4 (C-h); 27.2 (C-e); 25.7 (C-g); 21.5 (C-f);
14a‘: (CD3CN): 172.6 (C-c); 168.8 (C-i); 159.3 (C-k); 130.3 (C-m, C-o); 122.0 (C-n); 115.5 (C-l,
C-p); 67.0 (C-j); 56.0 (C-d); 40.3 (C-h); 27.7 (C-e); 25.3 (C-g); 21.5 (C-f);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
14b: (CD3CN): 172.8 (C-c); 171.7 (C-i); 136.6 (C-k); 129.8 (C-l, C-p); 129.2 (C-m, C-o); 127.2
(C-n); 52.4 (C-d); 44.5 (C-h); 40.8 (C-j); 27.0 (C-e); 25.6 (C-g); 21.3 (C-f);
14b‘: (CD3CN): 172.8 (C-c); 171.7 (C-i); 136.6 (C-k); 129.8 (C-l, C-p); 129.2 (C-m, C-o); 127.2
(C-n); 55.1 (C-d); 40.6 (C-j); 40.0 (C-h); 27.6 (C-e); 25.2 (C-g); 21.3 (C-f);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
14c: (CD3CN): 172.9 (C-c); 171.4 (C-i); 135.7 (C-k); 132.7 (C-n); 131.8 (C-m, C-o) 129.2 (C-l,
C-p); 52.6 (C-d); 44.6 (C-h); 40.1 (C-j); 27.2 (C-e); 25.8 (C-g); 21.5 (C-f);
14c‘: (CD3CN): 172.9 (C-c); 171.4 (C-i); 135.7 (C-k); 132.7 (C-n); 131.8 (C-m, C-o) 129.2 (C-l,
C-p); 56.9 (C-d); 40.2 (C-j); 39.9 (C-h); 27.8 (C-e); 25.3 (C-g); 21.7 (C-f);
15a: 176.6 (C-c); 130.9 (C-k, C-o); 129.1 (C-m); 128.6 (C-l, C-n); 126.0 (C-j); 58.9 (C-i); 55.7
(C-d); 43.5 (C-h); 27.5 (C-e); 24.9 (C-g); 20.3 (C-f);

144 EXPERIMENTELLER TEIL
15b: 175.8 (C-c); 130.9 (C-k, C-o); 129.2 (C-m); 128.6 (C-l, C-n); 126.0 (C-j); 58.9 (C-i); 55.7
(C-d); 43.5 (C-h); 27.6 (C-e); 24.9 (C-g); 20.3 (C-f);
15c: 176.1 (C-c); 138.5 (C-m); 130.8 (C-k, C-p); 129.3 (C-l, C-o); 126.0 (C-j); 58.6 (C-i); 55.6
(C-d); 43.5 (C-h); 27.5 (C-e); 24.9 (C-g); 21.2 (C-n); 20.3 (C-f);
15d: (CD3CN): 172.9 (C-c); 134.8 (C-m); 133.5 (C-l, C-n); 130.0 (C-j); 129.4 (C-k, C-o); 58.1
(C-i); 56.3 (C-d); 44.0 (C-h); 28.4 (C-e); 25.6 (C-g); 20.9 (C-f);
15e: (CD3CN): 172.7 (C-c); 146.2 (C-o); 135.3 (C-k); 134.2 (C-l); 130.8 (C-m); 126.3 (C-n);
125.1 (C-j); 56.4 (C-d); 54.9 (C-i); 43.9 (C-h); 28.5 (C-e); 25.6 (C-g); 20.9 (C-f);
15f: 176.6 (C-c); 139.9 (C-i); 132.6 (C-l); 128.9 (C-k, C-m); 127.1 (C-j, C-n); 54.8 (C-d); 42.6
(C-h); 27.5 (C-e); 24.5 (C-g); 20.0 (C-f);
15h: (DMSO-d6): 171.3 (C-c); 147.5 (C-k); 141.3 (C-i); 132.7 (C-n); 131.0 (C-m); 127.0 (C-l);
121.3 (C-j); 54.8 (C-d); 42.3 (C-h); 27.1 (C-e); 24.0 (C-g); 19.4 (C-f);
15i: 175.5 (C-c); 159.8 (C-k); 141.0 (C-i); 130.0 (C-o); 119.3 (C-j); 118.9 (C-n); 111.9 (C-m);
55.6 (C-l); 54.7 (C-d); 42.7 (C-h); 27.5 (C-e); 24.5 (C-g); 20.0 (C-f);
15j: (CD3CN): 172.3 (C-c); 144.4 (C-i); 138.7 (C-l); 130.5 (C-k, C-n); 127.9 (C-j, C-o); 55.7
(C-d); 43.4 (C-h); 28.1 (C-e); 25.1 (C-g); 21.4 (C-m); 20.8 (C-f);
6.2.8 Synthese von 3-(3,4,5-Trimethoxyphenyl)propan-1-ol 16
2.4 g (60.0 mmol) LiAlH4 werden langsam unter Argonatmosphäre in 60 ml abs. Et2O suspendiert
und eine Suspension von 1.0 g (4.2 mmol) 3-(3,4,5-Trimethoxyphenyl)propionsäure in 20 ml abs.
Et2O zugetropft. Dieser Ansatz wird 6 h unter Rückfluss erhitzt und danach auf RT abgekühlt. Nach
vollständiger Umsetzung (DC-Kontrolle: Kieselgel; Cyclohexan : EtOAc = 1 : 1; Rf = 0.25) wird
tropfenweise zuerst EtOH, danach dem. H2O zugegeben. Sobald keine Gasentwicklung mehr
stattfindet, wird die Mischung mit wässriger 2 M NaOH-Lösung überschichtet und 12 h bei RT
stehen gelassen. Anschließend wird der Niederschlag abfiltriert, der wässrige Rückstand viermal mit
je 30 ml CHCl3 extrahiert, die vereinigten organischen Phasen über Na2SO4 getrocknet und das
Lösungsmittel im Vakuum entfernt. Man erhält 0.6 g (2.7 mmol) von Verbindung 16 in Form einer
klaren Flüssigkeit.

EXPERIMENTELLER TEIL 145
Summenformel: C12H18O4
Mr = 226.27 g/mol
Ausbeute: 64 % (Lit. 88 %)137
Aussehen: klare Flüssigkeit (Lit. Flüssigkeit)137
Die 1H-NMR-Daten zu 16 entsprechen der Literaturstelle
138.
IR [cm-1
]: 686; 701; 1082; 1258; 1418; 1590; 2360; 2961; 3059; 3164-3598 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
6.42 (s, 2H, H-f, H-m); 3.85 (s, 6H, H-h, H-l); 3.82 (s, 3H, H-j); 3.70 (t, 2H, 3J = 6.4, H-b); 2.69-
2.62 (m, 2H, H-d); 1.94-1.85 (m, 2H, H-c); 1.50 (br, 1H, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
153.0 (C-g, C-k); 137.6 (C-e); 136.0 (C-i); 105.2 (C-f, C-m); 62.0 (C-d); 60.7 (C-j); 55.9 (C-h, C-l);
34.2 (C-b); 32.4 (C-c);
6.2.9 Synthese von 1,3-Diphenylpropan-1-ol 17139
0.26 g (11.0 mmol) Mg-Späne werden bei RT unter Argonatmosphäre in 20 ml abs. Et2O suspendiert
und eine Lösung von 2.0 g (11.0 mmol) 2-Phenylethylbromid in 30 ml abs. Et2O zugetropft. Danach
wird die Reaktionsmischung solange unter Rückfluss erhitzt, bis die Mg-Späne komplett abreagiert
sind. Nach dem Erkalten wird eine Lösung von 1.17 g (11.0 mmol) Benzaldehyd in 30 ml abs. Et2O
zugetropft und 2 h auf dem Wasserbad erhitzt. Nach dem Abkühlen wird Eis zugegeben und der
dadurch entstandene Niederschlag mit halbkonz. wässriger HCl-Lösung gelöst. Die organische Phase
wird abgetrennt und die wässrige Phase zweimal mit je 30 ml Et2O extrahiert. Die vereinigten
organischen Phasen werden dreimal mit je 40 ml ges. wässriger NaHCO3-Lösung, dreimal mit je
40 ml ges. wässriger NaHSO3-Lösung, sowie dreimal mit je 40 ml dem. H2O gewaschen, über
Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird anschließend

146 EXPERIMENTELLER TEIL
säulenchromatographisch gereinigt (Kieselgel; Cyclohexan : EtOAc = 1 : 1; Rf = 0.69). Man erhält
1.4 g (6.8 mmol) von Verbindung 17 in Form eines klaren Öls.
Summenformel: C15H16O
Mr = 212.29 g/mol
Ausbeute: 61 % (Lit. 54 %)140
Aussehen: klares Öl
Die IR-, 1H-NMR-, und
13C-NMR-Daten zu 17 entsprechen der Literaturstelle
140.
IR [cm-1
]: 697; 746; 1015; 1259; 1453; 1494; 3062; 3115-3520 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.30-7.07 (m, 10H, H-aromat.); 4.60 (dd, 1H, 3J = 7.8,
3J = 5.4, H-i); 2.72-2.53 (m, 2H, H-g); 2.10-
1.89 (m, 2H, H-h); 1.59 (br, 1H, H-p);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
144.6 (C-j); 141.8 (C-f); 128.5 (C-aromat.); 128.4 (4C, C-aromat.); 127.7 (C-aromat.); 125.9 (4C,
C-aromat.); 73.9 (C-i); 40.5 (C-h); 32.1 (C-g);
6.2.10 Darstellung der Piperidinamid-2-carbonsäureester-Derivate 18-29
Allgemeine Vorschrift zur Synthese der Verbindungen 18-29:
1 Äq (Stoffmenge siehe Tabelle 22 - Tabelle 33) des entsprechenden Pipecolinsäureamids
(Verbindungen 13-15, siehe Tabelle 22 - Tabelle 33), 1.1 Äq des entsprechenden Alkohols (siehe
Tabelle 22 - Tabelle 33), 1.6 Äq DCC, 0.35 Äq DMAP und 0.35 Äq p-Toluolsulfonsäure werden
unter Argonatmosphäre in 40 ml abs. CH2Cl2 gelöst. Diese Reaktionsmischung wird 72 h bei RT
gerührt und nach vollständiger Umsetzung (DC-Kontrolle, siehe Tabelle 22 - Tabelle 33) der
Niederschlag abfiltriert. Das Lösungsmittel des Filtrats wird im Vakuum entfernt, der ölige
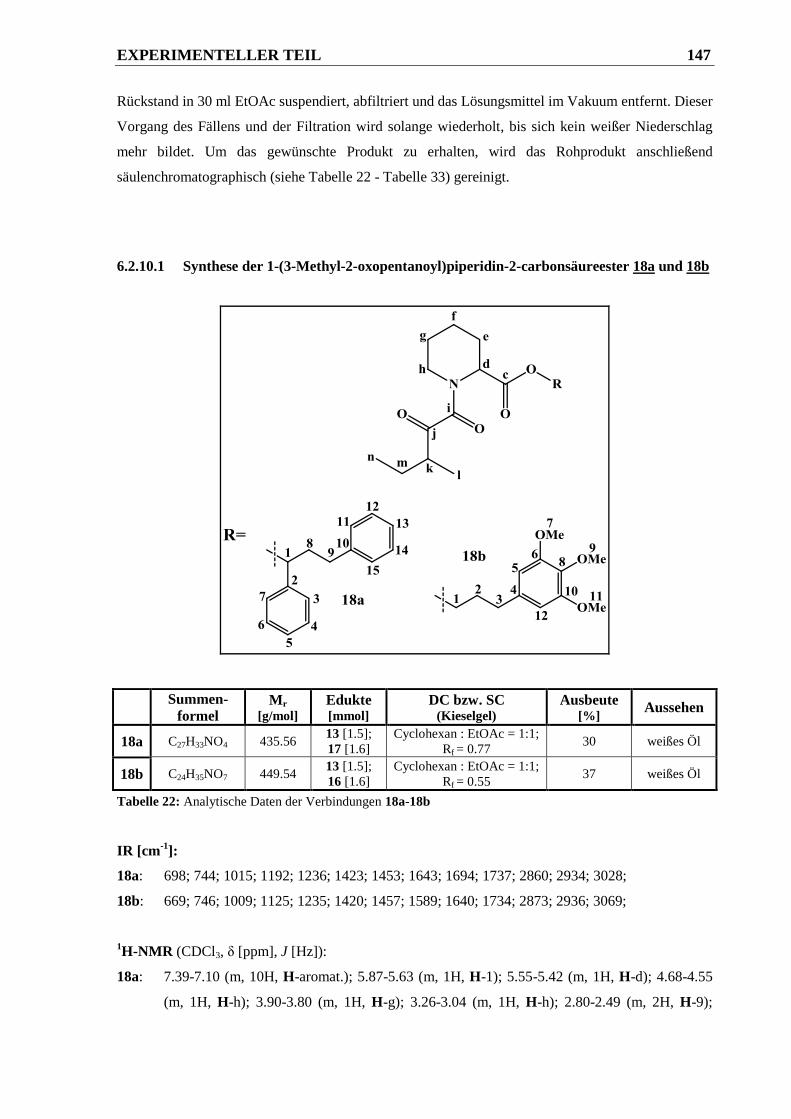
EXPERIMENTELLER TEIL 147
Rückstand in 30 ml EtOAc suspendiert, abfiltriert und das Lösungsmittel im Vakuum entfernt. Dieser
Vorgang des Fällens und der Filtration wird solange wiederholt, bis sich kein weißer Niederschlag
mehr bildet. Um das gewünschte Produkt zu erhalten, wird das Rohprodukt anschließend
säulenchromatographisch (siehe Tabelle 22 - Tabelle 33) gereinigt.
6.2.10.1 Synthese der 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäureester 18a und 18b
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aussehen
18a C27H33NO4 435.56 13 [1.5];
17 [1.6]
Cyclohexan : EtOAc = 1:1;
Rf = 0.77 30 weißes Öl
18b C24H35NO7 449.54 13 [1.5];
16 [1.6]
Cyclohexan : EtOAc = 1:1;
Rf = 0.55 37 weißes Öl
Tabelle 22: Analytische Daten der Verbindungen 18a-18b
IR [cm-1
]:
18a: 698; 744; 1015; 1192; 1236; 1423; 1453; 1643; 1694; 1737; 2860; 2934; 3028;
18b: 669; 746; 1009; 1125; 1235; 1420; 1457; 1589; 1640; 1734; 2873; 2936; 3069;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
18a: 7.39-7.10 (m, 10H, H-aromat.); 5.87-5.63 (m, 1H, H-1); 5.55-5.42 (m, 1H, H-d); 4.68-4.55
(m, 1H, H-h); 3.90-3.80 (m, 1H, H-g); 3.26-3.04 (m, 1H, H-h); 2.80-2.49 (m, 2H, H-9);
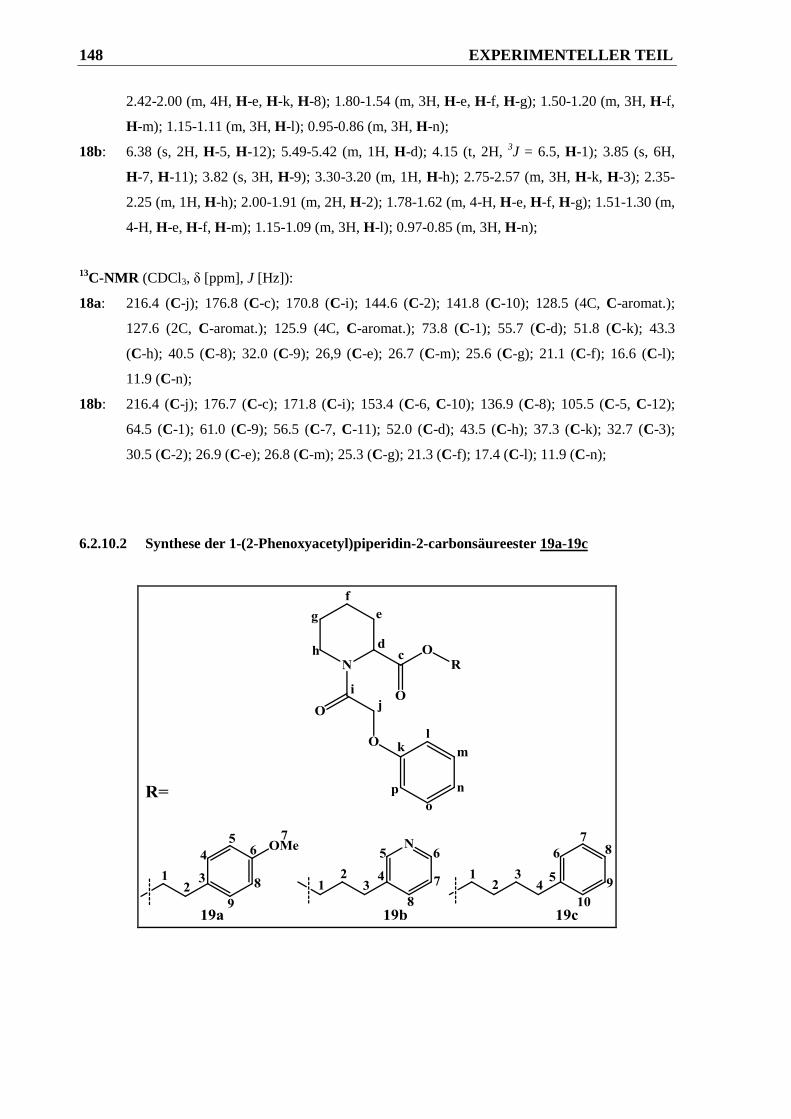
148 EXPERIMENTELLER TEIL
2.42-2.00 (m, 4H, H-e, H-k, H-8); 1.80-1.54 (m, 3H, H-e, H-f, H-g); 1.50-1.20 (m, 3H, H-f,
H-m); 1.15-1.11 (m, 3H, H-l); 0.95-0.86 (m, 3H, H-n);
18b: 6.38 (s, 2H, H-5, H-12); 5.49-5.42 (m, 1H, H-d); 4.15 (t, 2H, 3J = 6.5, H-1); 3.85 (s, 6H,
H-7, H-11); 3.82 (s, 3H, H-9); 3.30-3.20 (m, 1H, H-h); 2.75-2.57 (m, 3H, H-k, H-3); 2.35-
2.25 (m, 1H, H-h); 2.00-1.91 (m, 2H, H-2); 1.78-1.62 (m, 4-H, H-e, H-f, H-g); 1.51-1.30 (m,
4-H, H-e, H-f, H-m); 1.15-1.09 (m, 3H, H-l); 0.97-0.85 (m, 3H, H-n);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
18a: 216.4 (C-j); 176.8 (C-c); 170.8 (C-i); 144.6 (C-2); 141.8 (C-10); 128.5 (4C, C-aromat.);
127.6 (2C, C-aromat.); 125.9 (4C, C-aromat.); 73.8 (C-1); 55.7 (C-d); 51.8 (C-k); 43.3
(C-h); 40.5 (C-8); 32.0 (C-9); 26,9 (C-e); 26.7 (C-m); 25.6 (C-g); 21.1 (C-f); 16.6 (C-l);
11.9 (C-n);
18b: 216.4 (C-j); 176.7 (C-c); 171.8 (C-i); 153.4 (C-6, C-10); 136.9 (C-8); 105.5 (C-5, C-12);
64.5 (C-1); 61.0 (C-9); 56.5 (C-7, C-11); 52.0 (C-d); 43.5 (C-h); 37.3 (C-k); 32.7 (C-3);
30.5 (C-2); 26.9 (C-e); 26.8 (C-m); 25.3 (C-g); 21.3 (C-f); 17.4 (C-l); 11.9 (C-n);
6.2.10.2 Synthese der 1-(2-Phenoxyacetyl)piperidin-2-carbonsäureester 19a-19c
EXPERIMENTELLER TEIL 149
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
19a C23H27NO5 397.46
14a [2.0];
2-(4-Methoxyphenyl)-
ethanol [2.2]
PE : EtOAc = 1:1;
Rf = 0.69 60 klares Öl
19b C22H26N2O4 382.45
14a [2.0];
3-(Pyridin-3-yl)-
propan-1-ol [2.2]
EtOAc;
Rf = 0.35 55 klares Öl
19c C24H29NO4 395.49 14a [2.0];
4-Phenylbutan-1-ol [2.2]
PE : EtOAc = 1:1;
Rf = 0.72 82 gelbes Öl
Tabelle 23: Analytische Daten der Verbindungen 19a-19c
IR [cm-1
]:
19a: 691; 753; 1016; 1161; 1244; 1444; 1512; 1599; 1651; 1733; 2860; 2938; 3065;
19b: 691; 754; 1016; 1162; 1220; 1422; 1496; 1599; 1652; 1733; 2859; 2941; 3029;
19c: 691; 750; 1017; 1162; 1220; 1451; 1496; 1599; 1653; 1734; 2859; 2937; 3026;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19a: 7.32-7.26 (m, 2H, H-m; H-o); 7.13-7.06 (m, 2H, H-4, H-9); 7.01-6.88 (m, 3H, H-l, H-n,
H-p); 6.85-6.81 (m, 2H, H-5, H-8); 5.30 (d, 1H, 3J = 5.0, H-d); 4.75 (d, 1H,
2J = 13.7, H-j);
4.67 (d, 1H, 2J = 13.7, H-j); 4.34-4.22 (m, 2H, H-1); 3.84-3.75 (m, 4H, H-h, H-7); 3.15 (dt,
1H, 2J = 13.4,
3J = 3.0, H-h); 2.89-2.78 (m, 2H, H-2); 2.27-2.17 (m, 1H, H-e); 1.69-1.20 (m,
5H, H-e, H-f, H-g);
19a‘: 7.32-7.26 (m, 2H, H-m; H-o); 7.13-7.06 (m, 2H, H-4, H-9); 7.01-6.88 (m, 3H, H-l, H-n,
H-p); 6.85-6.81 (m, 2H, H-5, H-8); 4.82 (d, 1H, 3J = 5.0, H-d); 4.60-4.57 (m, 2H, H-j); 4.45
(d, 1H, 2J = 13.4, H-h); 4.34-4.22 (m, 2H, H-1); 3.84-3.75 (s, 3H, H-7); 2.89-2.78 (m, 2H,
H-2); 2.59 (dt, 1H, 2J = 13.4,
3J = 3.0, H-h); 2.27-2.17 (m, 1H, H-e); 1.69-1.20 (m, 5H, H-e,
H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19b: 8.50-8.40 (m, 2H, H-5, H-6); 7.50-7.43 (m, 1H, H-8); 7.30-7.24 (m, 2H, H-m; H-o); 7.23-
7.19 (m, 1H, H-7); 6.99-6.90 (m, 3H, H-l, H-n, H-p); 5.32 (d, 1H, 3
J = 5.5, H-d); 4.81-4.63
(m, 2H, H-j); 4.19-4.04 (m, 2H, H-1); 3.90 (d, 1H, 2J = 13.5, H-h); 3.31 (dt, 1H,
2J = 13.5,
3J = 3.0, H-h); 2.69-2.59 (m, 2H, H-3); 2.32-2.20 (m, 1H, H-e); 1.98-1.85 (m, 2H, H-2);
1.79-1.60 (m, 3H, H-e, H-f, H-g); 1.55-1.29 (m, 2H, H-f, H-g);
19b‘: 8.50-8.40 (m, 2H, H-5, H-6); 7.50-7.43 (m, 1H, H-8); 7.30-7.24 (m, 2H, H-m; H-o); 7.23-
7.19 (m, 1H, H-7); 6.99-6.90 (m, 3H, H-l, H-n, H-p); 4.89 (d, 1H, 3J = 5.5, H-d); 4.81-4.63
(m, 2H, H-j); 4.55-4.49 (m, 1H, H-h); 4.19-4.04 (m, 2H, H-1); 2.74 (dt, 1H, 2J = 13.5,
3J = 3.0, H-h); 2.69-2.59 (m, 2H, H-3); 2.32-2.20 (m, 1H, H-e); 1.98-1.85 (m, 2H, H-2);
1.79-1.60 (m, 3H, H-e, H-f, H-g); 1.55-1.29 (m, 2H, H-f, H-g);

150 EXPERIMENTELLER TEIL
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19c: 7.29-7.22 (m, 4H, H-m, H-o, H-7, H-9) 7.19-7.11 (m, 3H, H-6, H-8, H-10); 6.99-6.87 (m,
3H, H-l, H-n, H-p); 5.29 (d, 1H, 3
J = 5.5, H-d); 4.78-4.60 (m, 2H, H-j); 4.16-4.01 (m, 2H,
H-1); 3.85 (d, 1H, 2J = 13.5, H-h); 3.27 (dt, 1H,
2J = 13.5,
3J = 3.0, H-h); 2.75-2.55 (m, 2H,
H-4); 2.29-2.20 (m, 1H, H-e); 1.74-1.55 (m, 7H, H-e, H-f, H-g, H-2, H-3); 1.50-1.27 (m,
2H, H-f, H-g);
19c‘: 7.29-7.22 (m, 4H, H-m, H-o, H-7, H-9) 7.19-7.11 (m, 3H, H-6, H-8, H-10); 6.99-6.87 (m,
3H, H-l, H-n, H-p); 4.84 (d, 1H, 3
J = 5.5, H-d); 4.78-4.60 (m, 2H, H-j); 4.48 (d, 1H,
2J = 13.5, H-h); 4.16-4.01 (m, 2H, H-1); 2.75-2.55 (m, 3H, H-h, H-4); 2.29-2.20 (m, 1H,
H-e); 1.74-1.55 (m, 7H, H-e, H-f, H-g, H-2, H-3); 1.50-1.27 (m, 2H, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19a: 170.9 (C-c); 167.9 (C-i); 158.4 (C-k); 158.0 (C-6); 129.8 (C-4, C-9); 129.6 (C-3); 129.5
(C-m, C-o); 121.5 (C-n); 114.7 (C-l, C-p); 113.9 (C-5, C-8); 67.3 (C-j); 65.7 (C-1); 55.3 (C-
7); 52.3 (C-d); 43.0 (C-h); 34.1 (C-2); 26.5 (C-e); 25.2 (C-g); 20.8 (C-f);
19a‘: 170.9 (C-c); 167.9 (C-i); 158.4 (C-k); 158.0 (C-6); 129.8 (C-4, C-9); 129.6 (C-3); 129.5
(C-m, C-o); 121.5 (C-n); 114.7 (C-l, C-p); 113.9 (C-5, C-8); 68.1 (C-j); 65.7 (C-1); 55.9 (C-
d); 55.3 (C-7); 39.9 (C-h); 34.1 (C-2); 27.2 (C-e); 24.5 (C-g); 20.8 (C-f);
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19b: 170.9 (C-c); 168.0 (C-i); 158.0 (C-k); 149.9 (C-5); 147.7 (C-6); 136.2 (C-4); 135.8 (C-8);
129.5 (C-m, C-o); 123.4 (C-7); 121.6 (C-n); 114.7 (C-l, C-p); 67.4 (C-j); 64.1 (C-1); 52.3
(C-d); 43.2 (C-h); 29.9 (C-2); 29.2 (C-3); 26.5 (C-e); 25.2 (C-g); 20.9 (C-f);
19b‘: 170.9 (C-c); 168.0 (C-i); 158.0 (C-k); 149.9 (C-5); 147.7 (C-6); 136.2 (C-4); 135.8 (C-8);
129.5 (C-m, C-o); 123.4 (C-7); 121.6 (C-n); 114.7 (C-l, C-p); 68.3 (C-j); 64.1 (C-1); 56.0
(C-d); 40.0 (C-h); 29.9 (C-2); 29.2 (C-3); 27.3 (C-e); 24.5 (C-g); 20.9 (C-f);
Die Rotationsisomere liegen im Verhältnis 70 : 30 vor.
19c: 171.0 (C-c); 167.9 (C-i); 158.0 (C-k); 141.9 (C-5); 129.5 (C-m, C-o); 128.4 (C-7, C-9);
128.3 (C-6, C-10); 125.9 (C-8); 121.5 (C-n); 114.6 (C-l, C-p); 67.4 (C-j); 65.1 (C-1); 52.3
(C-d); 43.1 (C-h); 35.3 (C-4); 28.1 (C-3); 27.7 (C-2); 26.6 (C-e); 25.2 (C-g); 21.0 (C-f);
19c‘: 171.0 (C-c); 167.9 (C-i); 158.0 (C-k); 141.9 (C-5); 129.5 (C-m, C-o); 128.4 (C-7, C-9);
128.3 (C-6, C-10); 125.9 (C-8); 121.5 (C-n); 114.6 (C-l, C-p); 67.8 (C-j); 65.1 (C-1); 56.0
(C-d); 40.0 (C-h); 35.3 (C-4); 28.1 (C-3); 27.7 (C-2); 27.2 (C-e); 24.1 (C-g); 21.0 (C-f);
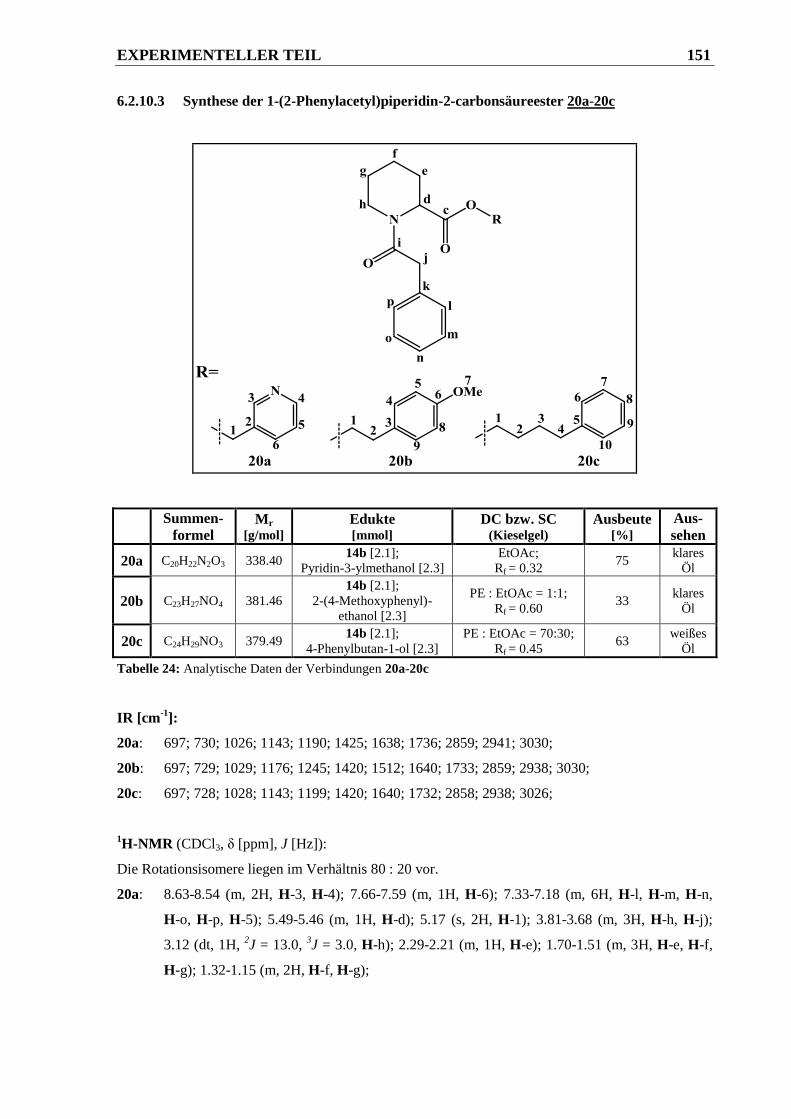
EXPERIMENTELLER TEIL 151
6.2.10.3 Synthese der 1-(2-Phenylacetyl)piperidin-2-carbonsäureester 20a-20c
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
20a C20H22N2O3 338.40 14b [2.1];
Pyridin-3-ylmethanol [2.3]
EtOAc;
Rf = 0.32 75
klares
Öl
20b C23H27NO4 381.46
14b [2.1];
2-(4-Methoxyphenyl)-
ethanol [2.3]
PE : EtOAc = 1:1;
Rf = 0.60 33
klares
Öl
20c C24H29NO3 379.49 14b [2.1];
4-Phenylbutan-1-ol [2.3]
PE : EtOAc = 70:30;
Rf = 0.45 63
weißes
Öl
Tabelle 24: Analytische Daten der Verbindungen 20a-20c
IR [cm-1
]:
20a: 697; 730; 1026; 1143; 1190; 1425; 1638; 1736; 2859; 2941; 3030;
20b: 697; 729; 1029; 1176; 1245; 1420; 1512; 1640; 1733; 2859; 2938; 3030;
20c: 697; 728; 1028; 1143; 1199; 1420; 1640; 1732; 2858; 2938; 3026;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20a: 8.63-8.54 (m, 2H, H-3, H-4); 7.66-7.59 (m, 1H, H-6); 7.33-7.18 (m, 6H, H-l, H-m, H-n,
H-o, H-p, H-5); 5.49-5.46 (m, 1H, H-d); 5.17 (s, 2H, H-1); 3.81-3.68 (m, 3H, H-h, H-j);
3.12 (dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.29-2.21 (m, 1H, H-e); 1.70-1.51 (m, 3H, H-e, H-f,
H-g); 1.32-1.15 (m, 2H, H-f, H-g);

152 EXPERIMENTELLER TEIL
20a‘: 8.63-8.54 (m, 2H, H-3, H-4); 7.66-7.59 (m, 1H, H-6); 7.33-7.18 (m, 6H, H-l, H-m, H-n,
H-o, H-p, H-5); 5.04 (s, 2H, H-1); 4.66-4.56 (m, 2H, H-d, H-h); 3.81-3.68 (m, 2H, H-j);
2.67 (dt, 1H, 2J = 13.0,
3J = 3.0, H-h) 2.18-2.10 (m, 1H, H-e); 1.70-1.51 (m, 3H, H-e, H-f,
H-g); 1.32-1.15 (m, 2H, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20b: 7.35-7.17 (m, 5H, H-l, H-m, H-n, H-o, H-p); 7.12-7.07 (m, 2H, H-4, H-9); 6.87-6.80 (m,
2H, H-5, H-8); 5.43-5.39 (m, 1H, H-d); 4.34-4.18 (m, 2H, H-1); 3.80-3.77 (m, 5H, H-j,
H-7); 3.73-3.65 (m, 1H, H-h); 3.08-2.98 (m, 1H, H-h); 2.91-2.79 (m, 2H, H-2); 2.24-2.15
(m, 1H, H-e); 1.65-1.47 (m, 3H, H-e, H-f, H-g); 1.39-1.13 (m, 2H, H-f, H-g);
20b‘: 7.35-7.17 (m, 5H, H-l, H-m, H-n, H-o, H-p); 7.12-7.07 (m, 2H, H-4, H-9); 6.87-6.80 (m,
2H, H-5, H-8); 4.59-4.49 (m, 2H, H-d, H-h); 4.34-4.18 (m, 2H, H-1); 3.80-3.77 (m, 5H, H-j,
H-7); 2.91-2.79 (m, 2H, H-2); 2.58 (dt, 1H, 2J = 13.3,
3J = 3.0, H-h) 2.12-2.05 (m, 1H, H-e);
1.65-1.47 (m, 3H, H-e, H-f, H-g); 1.39-1.13 (m, 2H, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20c: 7.35-7.11 (m, 10H, H-aromat.); 5.45 (d, 1H, 3J = 5.0, H-d); 4.19-4.02 (m, 2H, H-1); 3.84-
3.70 (m, 3H, H-h, H-j); 3.18 (dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.75-2.61 (m, 2H, H-4); 2.31-
2.23 (m, 1H, H-e); 1.74-1.54 (m, 7H, H-e, H-f, H-g, H-2, H-3); 1.46-1.23 (m, 2H, H-f, H-g);
20c‘: 7.35-7.11 (m, 10H, H-aromat.); 4.66-4.56 (m, 2H, H-d, H-h); 4.19-4.02 (m, 2H, H-1); 3.84-
3.70 (m, 2H, H-j); 2.75-2.61 (m, 3H, H-h, H-4); 2.19-2.12 (m, 1H, H-e); 1.74-1.54 (m, 7H,
H-e, H-f, H-g, H-2, H-3); 1.46-1.23 (m, 2H, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20a: 171.1 (C-c); 171.0 (C-i); 149.8 (C-3); 149.5 (C-4); 135.9 (C-6); 134.8 (C-k); 131.3 (C-2);
128.6 (C-l, C-m, C-o, C-p); 126.7 (C-n); 123.5 (C-5); 64.3 (C-1); 52.0 (C-d); 44.1 (C-h);
41.1 (C-j); 26.5 (C-e); 25.0 (C-g); 20.8 (C-f);
20a‘: 171.1 (C-c); 171.0 (C-i); 149.8 (C-3); 149.5 (C-4); 135.9 (C-6); 134.8 (C-k); 131.3 (C-2);
128.6 (C-l, C-m, C-o, C-p); 126.7 (C-n); 123.5 (C-5); 64.3 (C-1); 56.6 (C-d); 41.3 (C-j);
39.6 (C-h); 27.3 (C-e); 24.5 (C-g); 20.8 (C-f);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20b: 171.2 (C-c); 170.8 (C-i); 158.4 (C-6); 134.9 (C-k); 129.8 (C-4, C-9); 129.6 (C-3); 128.6
(C-l, C-m, C-o, C-p); 126.7 (C-n); 113.9 (C-5, C-8); 65.6 (C-1); 55.3 (C-7); 52.0 (C-d);
44.0 (C-h); 41.1 (C-j); 34.1 (C-2); 26.5 (C-e); 25.1 (C-g); 20.7 (C-f);
20b‘: 171.2 (C-c); 170.8 (C-i); 158.4 (C-6); 134.9 (C-k); 129.8 (C-4, C-9); 129.6 (C-3); 128.6
(C-l, C-m, C-o, C-p); 126.7 (C-n); 113.9 (C-5, C-8); 65.6 (C-1); 56.6 (C-d); 55.3 (C-7);
41.3 (C-j); 39.5 (C-h); 34.1 (C-2); 27.1 (C-e); 24.5 (C-g); 20.7 (C-f);
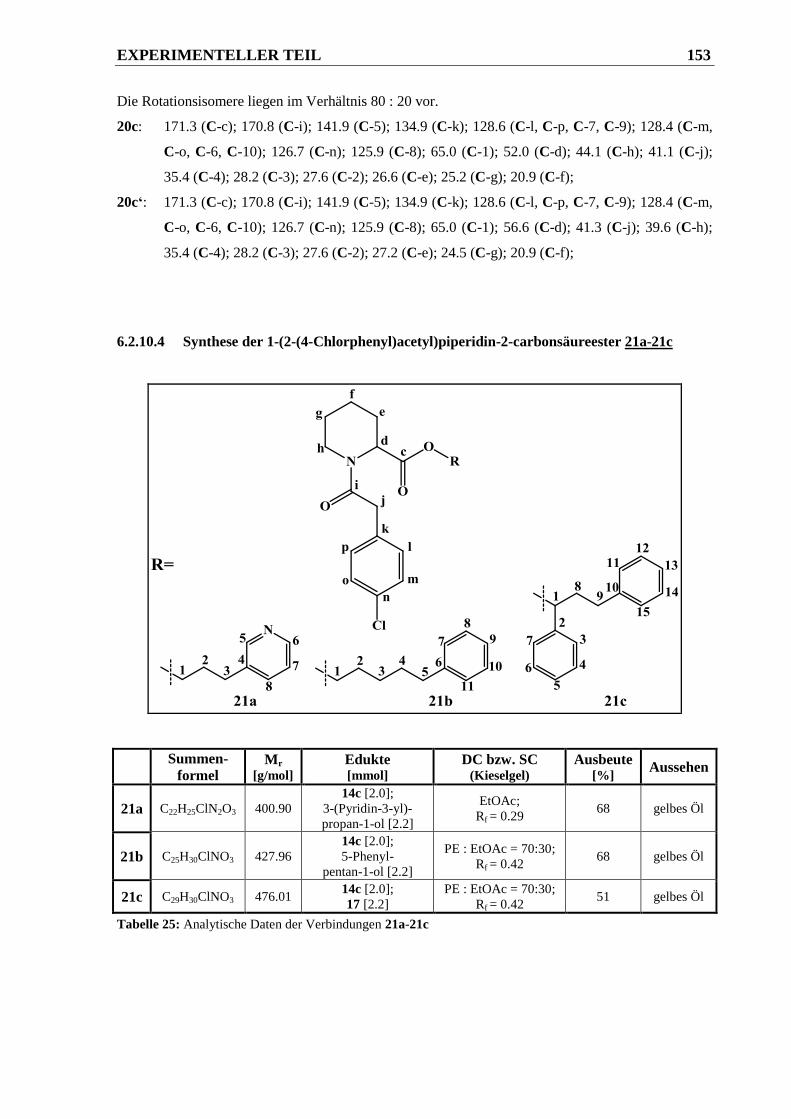
EXPERIMENTELLER TEIL 153
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
20c: 171.3 (C-c); 170.8 (C-i); 141.9 (C-5); 134.9 (C-k); 128.6 (C-l, C-p, C-7, C-9); 128.4 (C-m,
C-o, C-6, C-10); 126.7 (C-n); 125.9 (C-8); 65.0 (C-1); 52.0 (C-d); 44.1 (C-h); 41.1 (C-j);
35.4 (C-4); 28.2 (C-3); 27.6 (C-2); 26.6 (C-e); 25.2 (C-g); 20.9 (C-f);
20c‘: 171.3 (C-c); 170.8 (C-i); 141.9 (C-5); 134.9 (C-k); 128.6 (C-l, C-p, C-7, C-9); 128.4 (C-m,
C-o, C-6, C-10); 126.7 (C-n); 125.9 (C-8); 65.0 (C-1); 56.6 (C-d); 41.3 (C-j); 39.6 (C-h);
35.4 (C-4); 28.2 (C-3); 27.6 (C-2); 27.2 (C-e); 24.5 (C-g); 20.9 (C-f);
6.2.10.4 Synthese der 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäureester 21a-21c
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aussehen
21a C22H25ClN2O3 400.90
14c [2.0];
3-(Pyridin-3-yl)-
propan-1-ol [2.2]
EtOAc;
Rf = 0.29 68 gelbes Öl
21b C25H30ClNO3 427.96
14c [2.0];
5-Phenyl-
pentan-1-ol [2.2]
PE : EtOAc = 70:30;
Rf = 0.42 68 gelbes Öl
21c C29H30ClNO3 476.01 14c [2.0];
17 [2.2]
PE : EtOAc = 70:30;
Rf = 0.42 51 gelbes Öl
Tabelle 25: Analytische Daten der Verbindungen 21a-21c

154 EXPERIMENTELLER TEIL
IR [cm-1
]:
21a: 714; 768; 1015; 1196; 1422; 1492; 1640; 1733; 2859; 2941; 3031;
21b: 698; 747; 1015; 1200; 1428; 1492; 1644; 1734; 2857; 2933; 3026;
21c: 698; 750; 1015; 1195; 1426; 1493; 1643; 1735; 2857; 2932; 3029;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21a: 8.49-8.41 (m, 2H, H-5, H-6); 7.50-7.46 (m, 1H, H-8); 7.30-7.15 (m, 5H, H-l; H-m, H-o,
H-p, H-7); 5.40 (d, 1H, 3
J = 5.5, H-d); 4.15 (t, 2H, 3J = 6.5, H-1); 3.78-3.65 (m, 3H, H-j, H-
h); 3.20 (dt, 1H, 2J = 13.0,
3J = 3.0, H-h); 2.68-2.63 (m, 2H, H-3); 2.28-2.21 (m, 1H, H-e);
2.00-1.89 (m, 2H, H-2); 1.75-1.55 (m, 3H, H-e, H-f, H-g); 1.35-1.23 (m, 2H, H-f, H-g);
21a‘: 8.49-8.41 (m, 2H, H-5, H-6); 7.50-7.46 (m, 1H, H-8); 7.30-7.15 (m, 5H, H-l; H-m, H-o,
H-p, H-7); 4.63-4.51 (m, 2H, H-d, H-h); 4.15 (t, 2H, 3J = 6.5, H-1); 3.78-3.65 (m, 2H, H-j);
2.68-2.63 (m, 3H, H-h, H-3); 2.28-2.21 (m, 1H, H-e); 2.00-1.89 (m, 2H, H-2); 1.75-1.55 (m,
3H, H-e, H-f, H-g); 1.35-1.23 (m, 2H, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21b: 7.34-7.26 (m, 4H, H-l, H-p, H-8, H-10); 7.24-7.15 (m, 5H, H-m, H-o, H-7, H-9, H-11); 5.41
(d, 1H, 3
J = 5.5, H-d); 4.14 (t, 2H, 3J = 6.7, H-1); 3.78-3.63 (m, 3H, H-j, H-h); 3.17 (dt, 1H,
2J = 13.0,
3J = 3.0, H-h); 2.74-2.57 (m, 2H, H-5); 2.30-2.15 (m, 1H, H-e); 1.73-1.54 (m, 9H,
H-e, H-f, H-g, H-2, H-3, H-4); 1.46-1.25 (m, 2H, H-f, H-g);
21b‘: 7.34-7.26 (m, 4H, H-l, H-p, H-8, H-10); 7.24-7.15 (m, 5H, H-m, H-o, H-7, H-9, H-11);
4.62-4.51 (m, 2H, H-d, H-h); 4.14 (t, 2H, 3J = 6.7, H-1); 3.78-3.63 (m, 2H, H-j); 2.74-2.57
(m, 3H, H-h, H-5); 2.30-2.15 (m, 1H, H-e); 1.73-1.54 (m, 9H, H-e, H-f, H-g, H-2, H-3,
H-4); 1.46-1.25 (m, 2H, H-f, H-g);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21c: 7.36-7.03 (m, 14H, H-aromat.); 5.80-5.71 (m, 1H, H-1); 5.48-5.42 (m, 1H, H-d); 3.75-3.51
(m, 3H, H-h, H-j); 3.16 (dt, 1H, 2J = 13.1,
3J = 3.0, H-h); 2.74-2.48 (m, 2H, H-9); 2.35-2.16
(m, 2H, H-8); 2.14-2.03 (m, 1H, H-e); 1.76-1.15 (m, 5H, H-e, H-f, H-g);
21c‘: 7.36-7.03 (m, 14H, H-aromat.); 5.80-5.71 (m, 1H, H-1); 5.13-5.08 (m, 1H, H-d); 4.62-4.47
(m, 1H, H-h); 3.75-3.51 (m, 2H, H-j); 2.74-2.48 (m, 3H, H-h, H-9); 2.35-2.16 (m, 2H, H-8);
2.14-2.03 (m, 1H, H-e); 1.76-1.15 (m, 5H, H-e, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21a: 171.2 (C-c); 170.5 (C-i); 149.9 (C-5); 147.7 (C-6); 135.8 (C-8); 133.3 (C-4); 130.7 (C-k);
130.1 (C-l, C-p); 128.8 (C-m, C-o); 127.0 (C-n); 123.4 (C-7); 64.1 (C-1); 52.1 (C-d); 44.0
(C-h); 40.2 (C-j); 29.9 (C-2); 29.3 (C-3); 26.6 (C-e); 25.2 (C-g); 20.8 (C-f);

EXPERIMENTELLER TEIL 155
21a‘: 171.2 (C-c); 170.5 (C-i); 149.9 (C-5); 147.7 (C-6); 135.8 (C-8); 133.3 (C-4); 130.7 (C-k);
130.1 (C-l, C-p); 128.8 (C-m, C-o); 127.0 (C-n); 123.4 (C-7); 64.1 (C-1); 56.6 (C-d); 40.4
(C-j); 39.7 (C-h); 29.9 (C-2); 29.3 (C-3); 27.3 (C-e); 24.5 (C-g); 20.8 (C-f);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21b: 171.2 (C-c); 170.4 (C-i); 142.3 (C-6); 133.4 (C-k); 132.6 (C-n); 130.1 (C-l, C-p); 128.7
(C-m, C-o); 128.4 (C-8, C-10); 128.3 (C-7, C-11); 125.7 (C-9); 65.1 (C-1); 52.1 (C-d); 44.0
(C-h); 40.2 (C-j); 35.7 (C-5); 30.9 (C-4); 28.5 (C-3); 26.6 (C-e); 25.5 (C-2); 25.2 (C-g); 20.8
(C-f);
21b‘: 171.2 (C-c); 170.4 (C-i); 142.3 (C-6); 133.4 (C-k); 132.6 (C-n); 130.1 (C-l, C-p); 128.7
(C-m, C-o); 128.4 (C-8, C-10); 128.3 (C-7, C-11); 125.7 (C-9); 65.1 (C-1); 56.6 (C-d); 40.4
(C-j); 39.6 (C-h); 35.7 (C-5); 30.9 (C-4); 28.5 (C-3); 27.3 (C-e); 25.5 (C-2); 24.5 (C-g); 20.8
(C-f);
Die Rotationsisomere liegen im Verhältnis 80 : 20 vor.
21c: 178.7 (C-c); 170.5 (C-i); 140.7 (C-10); 137.7 (C-2); 133.4 (C-k, C-n); 130.1 (C-l, C-p);
128.8 (C-m, C-o); 128.5 (4C, C-aromat.); 128.3 (4C, C-aromat.); 126.3 (2C, C-aromat.);
76.5 (C-1); 52.1 (C-d); 44.1 (C-h); 40.3 (C-j); 38.0 (C-8); 31.7 (C-9); 26.6 (C-e); 25.2 (C-g);
20.6 (C-f);
21c‘: 178.7 (C-c); 170.5 (C-i); 140.7 (C-10); 137.7 (C-2); 133.4 (C-k, C-n); 130.1 (C-l, C-p);
128.8 (C-m, C-o); 128.5 (4C, C-aromat.); 128.3 (4C, C-aromat.); 126.3 (2C, C-aromat.);
76.5 (C-1); 56.6 (C-d); 40.3 (C-j); 39.7 (C-h); 38.0 (C-8); 31.7 (C-9); 27.5 (C-e); 24.8 (C-g);
20.6 (C-f);
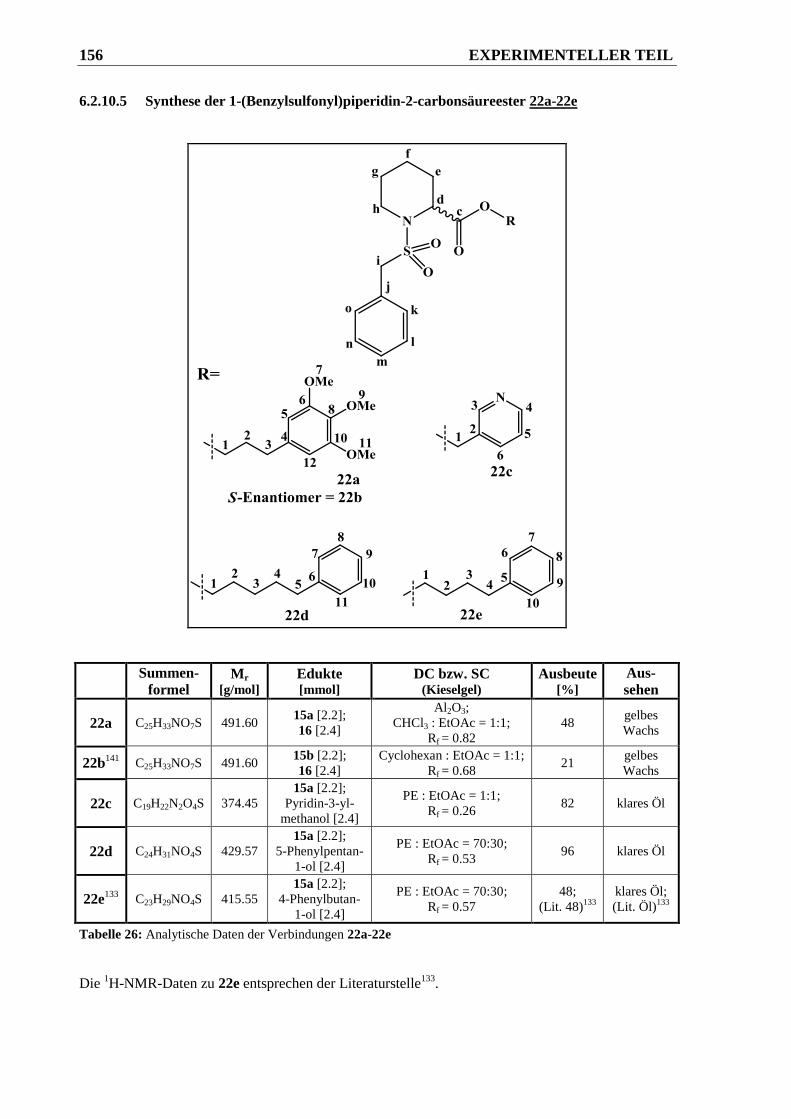
156 EXPERIMENTELLER TEIL
6.2.10.5 Synthese der 1-(Benzylsulfonyl)piperidin-2-carbonsäureester 22a-22e
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
22a C25H33NO7S 491.60 15a [2.2];
16 [2.4]
Al2O3;
CHCl3 : EtOAc = 1:1;
Rf = 0.82
48 gelbes
Wachs
22b141
C25H33NO7S 491.60 15b [2.2];
16 [2.4]
Cyclohexan : EtOAc = 1:1;
Rf = 0.68 21
gelbes
Wachs
22c C19H22N2O4S 374.45
15a [2.2];
Pyridin-3-yl-
methanol [2.4]
PE : EtOAc = 1:1;
Rf = 0.26 82 klares Öl
22d C24H31NO4S 429.57
15a [2.2];
5-Phenylpentan-
1-ol [2.4]
PE : EtOAc = 70:30;
Rf = 0.53 96 klares Öl
22e133
C23H29NO4S 415.55
15a [2.2];
4-Phenylbutan-
1-ol [2.4]
PE : EtOAc = 70:30;
Rf = 0.57
48;
(Lit. 48)133
klares Öl;
(Lit. Öl)133
Tabelle 26: Analytische Daten der Verbindungen 22a-22e
Die 1H-NMR-Daten zu 22e entsprechen der Literaturstelle
133.

EXPERIMENTELLER TEIL 157
IR [cm-1
]:
22a: 698; 739; 1010; 1124; 1238; 1336; 1455; 1589; 1733; 2857; 2938; 3063;
22b: 698; 739; 1059; 1124; 1238; 1336; 1455; 1589; 1732; 2858; 2938; 3033;
22c: 698; 739; 1059; 1127; 1148; 1335; 1455; 1579; 1738; 2859; 2944; 3061;
22d: 697; 739; 1059; 1128; 1199; 1336; 1454; 1496; 1732; 2857; 2934; 3061;
22e: 697; 738; 1059; 1128; 1199; 1336; 1454; 1603; 1732; 2859; 2939; 3061;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
22a: 7.49-7.33 (m, 5H, H-k, H-l, H-m, H-n, H-o); 6.41 (s, 2H, H-5, H-12); 4.55-4.50 (m, 1H,
H-d); 4.27 (s, 2H, H-i); 4.24-4.15 (m, 2H, H-1); 3.84 (s, 6H, H-7, H-11); 3.82 (s, 3H, H-9);
3.46-3.40 (m, 1H, H-h); 3.17 (dt, 1H, 2J = 12.4,
3J = 3.1, H-h); 2.66 (t, 2H,
3J = 7.5, H-3);
2.20-2.12 (m, 1H, H-e); 2.03-1.95 (m, 2H, H-2); 1.75-1.50 (m, 3H, H-e, H-f, H-g); 1.49-1.18
(m, 2H, H-f, H-g);
22b: 7.47-7.32 (m, 5H, H-k, H-l, H-m, H-n, H-o); 6.41 (s, 2H, H-5, H-12); 4.54-4.50 (m, 1H,
H-d); 4.27 (s, 2H, H-i); 4.25-4.15 (m, 2H, H-1); 3.84 (s, 6H, H-7, H-11); 3.82 (s, 3H, H-9);
3.48-3.40 (m, 1H, H-h); 3.17 (dt, 1H, 2J = 12.4,
3J = 3.1, H-h); 2.67 (t, 2H,
3J = 7.5, H-3);
2.19-2.12 (m, 1H, H-e); 2.03-1.95 (m, 2H, H-2); 1.75-1.50 (m, 3H, H-e, H-f, H-g); 1.49-1.15
(m, 2H, H-f, H-g);
22c: 8.63 (d, 1H, 4J = 2.0, H-3); 8.60 (dd, 1H,
3J = 5.0,
4J = 2.0, H-4); 7.74-7.70 (m, 1H, H-6);
7.45-7.41 (m, 2H, H-k; H-o); 7.39-7.30 (m, 4H, H-l, H-m, H-n, H-5); 5.26 (d, 1H, 2J = 12.6,
H-1); 5.18 (d, 1H, 2J = 12.6, H-1); 4.56 (d, 1H,
3J = 4.0, H-d); 4.25 (d, 2H,
4J = 1.3, H-i);
3.46-3.40 (m, 1H, H-h); 3.14 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.16-2.10 (m, 1H, H-e); 1.70-
1.51 (m, 3H, H-e, H-f, H-g); 1.48-1.34 (m, 1H, H-g); 1.20-1.08 (m, 1H, H-f);
22d: 7.50-7.43 (m, 2H, H-k, H-o); 7.39-7.33 (m, 3H, H-l, H-m, H-n); 7.30-7.25 (m, 2H, H-8,
H-10); 7.20-7.15 (m, 3H, H-7; H-9, H-11); 4.58-4.54 (m, 1H, H-d); 4.27 (s, 2H, H-i); 4.24-
4.10 (m, 2H, H-1); 3.48-3.41 (m, 1H, H-h); 3.13 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.65-2.60
(m, 2H, H-5); 2.16-2.10 (m, 1H, H-e); 1.74-1.57 (m, 7H, H-e, H-2, H-3, H-4); 1.47-1.36 (m,
3H, H-f, H-g); 1.28-1.15 (m, 1H, H-f);
22e: 7.48-7.42 (m, 2H, H-k, H-o); 7.39-7.33 (m, 3H, H-l, H-m, H-n); 7.31-7.26 (m, 2H, H-7,
H-9); 7.21-7.15 (m, 3H, H-6; H-8, H-10); 4.56 (d, 1H, 3J = 4.0, H-d); 4.27 (s, 2H, H-i); 4.25-
4.11 (m, 2H, H-1); 3.48-3.42 (m, 1H, H-h); 3.14 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.69-2.62
(m, 2H, H-4); 2.18-2.11 (m, 1H, H-e); 1.76-1.57 (m, 7H, H-e, H-2, H-3, H-f, H-g); 1.50-
1.37 (m, 1H, H-g); 1.31-1.15 (m, 1H, H-f);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
22a: 171.5 (C-c); 153.2 (C-6, C-10); 137.9 (C-4); 136.7 (C-8); 131.0 (C-k, C-o); 129.3 (C-j);
128.6 (C-l, C-n); 128.5 (C-m); 105.4 (C-5, C-12); 64.6 (C-1); 60.9 (C-9); 58.9 (C-i); 56.1
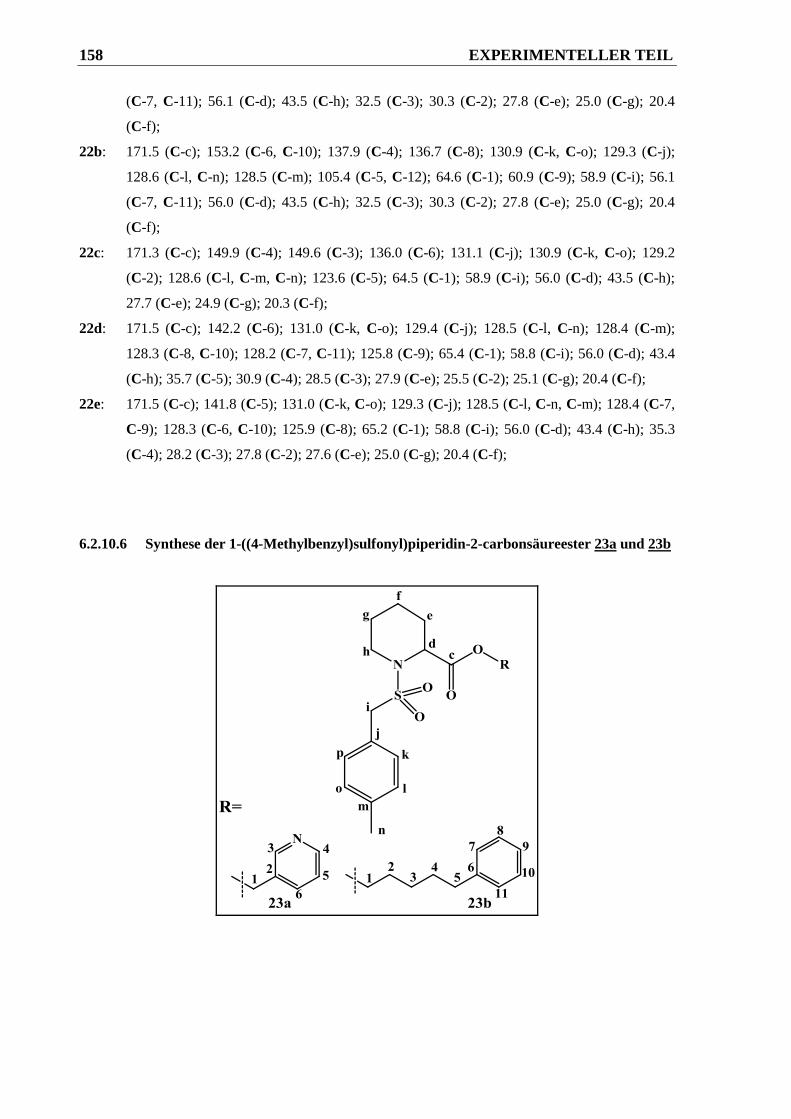
158 EXPERIMENTELLER TEIL
(C-7, C-11); 56.1 (C-d); 43.5 (C-h); 32.5 (C-3); 30.3 (C-2); 27.8 (C-e); 25.0 (C-g); 20.4
(C-f);
22b: 171.5 (C-c); 153.2 (C-6, C-10); 137.9 (C-4); 136.7 (C-8); 130.9 (C-k, C-o); 129.3 (C-j);
128.6 (C-l, C-n); 128.5 (C-m); 105.4 (C-5, C-12); 64.6 (C-1); 60.9 (C-9); 58.9 (C-i); 56.1
(C-7, C-11); 56.0 (C-d); 43.5 (C-h); 32.5 (C-3); 30.3 (C-2); 27.8 (C-e); 25.0 (C-g); 20.4
(C-f);
22c: 171.3 (C-c); 149.9 (C-4); 149.6 (C-3); 136.0 (C-6); 131.1 (C-j); 130.9 (C-k, C-o); 129.2
(C-2); 128.6 (C-l, C-m, C-n); 123.6 (C-5); 64.5 (C-1); 58.9 (C-i); 56.0 (C-d); 43.5 (C-h);
27.7 (C-e); 24.9 (C-g); 20.3 (C-f);
22d: 171.5 (C-c); 142.2 (C-6); 131.0 (C-k, C-o); 129.4 (C-j); 128.5 (C-l, C-n); 128.4 (C-m);
128.3 (C-8, C-10); 128.2 (C-7, C-11); 125.8 (C-9); 65.4 (C-1); 58.8 (C-i); 56.0 (C-d); 43.4
(C-h); 35.7 (C-5); 30.9 (C-4); 28.5 (C-3); 27.9 (C-e); 25.5 (C-2); 25.1 (C-g); 20.4 (C-f);
22e: 171.5 (C-c); 141.8 (C-5); 131.0 (C-k, C-o); 129.3 (C-j); 128.5 (C-l, C-n, C-m); 128.4 (C-7,
C-9); 128.3 (C-6, C-10); 125.9 (C-8); 65.2 (C-1); 58.8 (C-i); 56.0 (C-d); 43.4 (C-h); 35.3
(C-4); 28.2 (C-3); 27.8 (C-2); 27.6 (C-e); 25.0 (C-g); 20.4 (C-f);
6.2.10.6 Synthese der 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäureester 23a und 23b

EXPERIMENTELLER TEIL 159
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
23a C20H24N2O4S 388.48 15c [1.1];
Pyridin-3-ylmethanol [1.2]
EtOAc;
Rf = 0.49 55
braunes
Öl
23b C25H33NO4S 443.60 15c [1.1];
5-Phenylpentan-1-ol [1.2]
PE : EtOAc = 1:1;
Rf = 0.68 58
klares
Öl
Tabelle 27: Analytische Daten der Verbindungen 23a-23b
IR [cm-1
]:
23a: 711; 766; 969; 1059; 1128; 1148; 1335; 1448; 1514; 1738; 2856; 2929; 3031;
23b: 699; 748; 966; 1059; 1129; 1149; 1336; 1453; 1514; 1733; 2858; 2934; 3026;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
23a: 8.63 (d, 1H, 4J = 1.7, H-3); 8.60 (dd, 1H,
3J = 4.8,
4J = 1.7, H-4); 7.74-7.69 (m, 1H, H-6);
7.34-7.28 (m, 3H, H-k, H-p, H-5); 7.16 (d, 2H, 3J = 7.8, H-l, H-o); 5.26 (d, 1H,
2J = 12.6,
H-1); 5.18 (d, 1H, 2J = 12.6, H-1); 4.56 (d, 1H,
3J = 3.7, H-d); 4.20 (d, 2H,
4J = 1.1, H-i);
3.48-3.40 (m, 1H, H-h); 3.14 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.35 (s, 3H, H-n); 2.16-2.09
(m, 1H, H-e); 1.71-1.09 (m, 5H, H-e, H-f, H-g);
23b: 7.34 (d, 2H, 3J = 8.0, H-k, H-p); 7.30-7.24 (m, 2H, H-8, H-10); 7.21-7.14 (m, 5H, H-l, H-o,
H-7, H-9, H-11); 4.56 (d, 1H, 3J = 4.8, H-d); 4.24-4.09 (m, 4H, H-i, H-1); 3.48-3.42 (m, 1H,
H-h); 3.13 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.63 (t, 2H,
3J = 8.0, H-5); 2.35 (s, 3H, H-n);
2.16-2.09 (m, 1H, H-e); 1.74-1.56 (m, 7H, H-e, H-f, H-g, H-2, H-4); 1.50-1.16 (m, 4H, H-f,
H-g, H-3);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
23a: 171.3 (C-c); 149.9 (C-4); 149.6 (C-3); 138.4 (C-m); 136.0 (C-6); 131.1 (C-2); 130.8 (C-k,
C-p); 129.3 (C-l, C-o); 126.1 (C-j); 123.5 (C-5); 64.5 (C-1); 58.6 (C-i); 56.0 (C-d); 43.4
(C-h); 27.7 (C-e); 24.9 (C-g); 21.2 (C-n); 20.3 (C-f);
23b: 171.6 (C-c); 142.2 (C-6); 138.3 (C-m); 130.8 (C-k, C-p); 129.2 (C-l, C-o); 128.4 (C-8,
C-10); 128.3 (C-7, C-11); 126.2 (C-j); 125.7 (C-9); 65.3 (C-1); 58.4 (C-i); 56.0 (C-d); 43.4
(C-h); 35.7 (C-5); 30.9 (C-2); 28.5 (C-3); 27.9 (C-e); 25.5 (C-4); 25.1 (C-g); 21.2 (C-n);
20.4 (C-f);
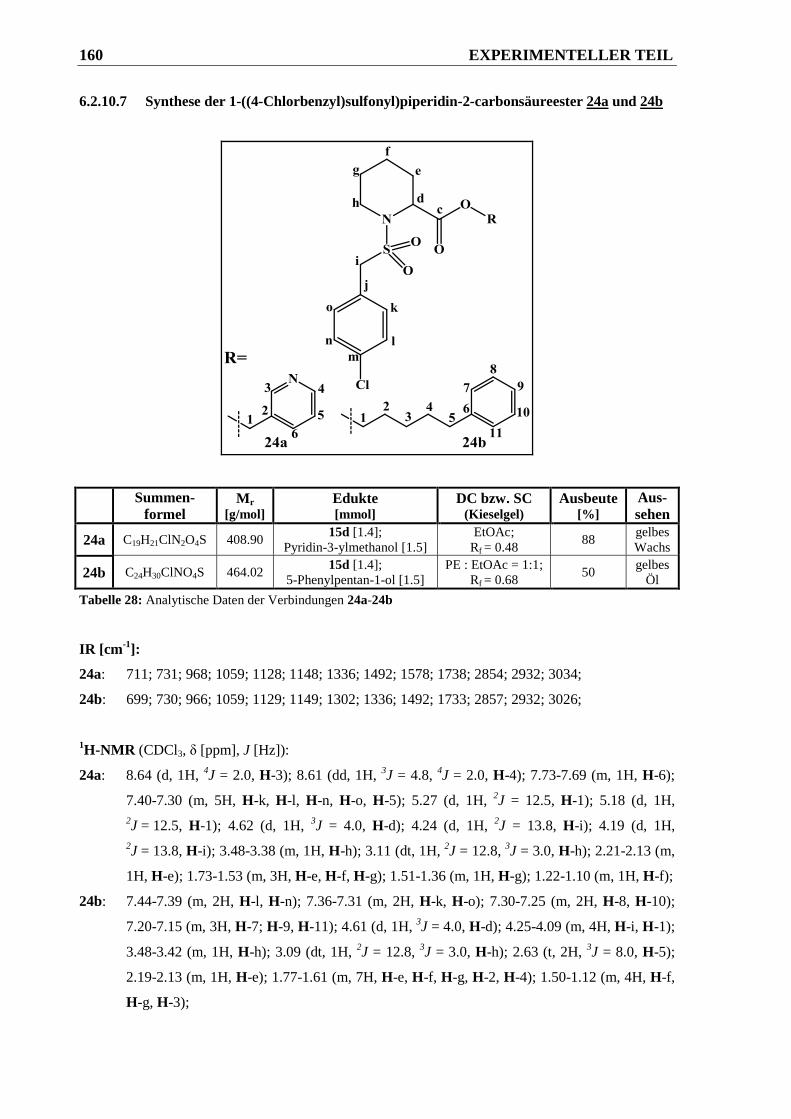
160 EXPERIMENTELLER TEIL
6.2.10.7 Synthese der 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäureester 24a und 24b
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
24a C19H21ClN2O4S 408.90 15d [1.4];
Pyridin-3-ylmethanol [1.5]
EtOAc;
Rf = 0.48 88
gelbes
Wachs
24b C24H30ClNO4S 464.02 15d [1.4];
5-Phenylpentan-1-ol [1.5]
PE : EtOAc = 1:1;
Rf = 0.68 50
gelbes
Öl
Tabelle 28: Analytische Daten der Verbindungen 24a-24b
IR [cm-1
]:
24a: 711; 731; 968; 1059; 1128; 1148; 1336; 1492; 1578; 1738; 2854; 2932; 3034;
24b: 699; 730; 966; 1059; 1129; 1149; 1302; 1336; 1492; 1733; 2857; 2932; 3026;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
24a: 8.64 (d, 1H, 4J = 2.0, H-3); 8.61 (dd, 1H,
3J = 4.8,
4J = 2.0, H-4); 7.73-7.69 (m, 1H, H-6);
7.40-7.30 (m, 5H, H-k, H-l, H-n, H-o, H-5); 5.27 (d, 1H, 2J = 12.5, H-1); 5.18 (d, 1H,
2J = 12.5, H-1); 4.62 (d, 1H,
3J = 4.0, H-d); 4.24 (d, 1H,
2J = 13.8, H-i); 4.19 (d, 1H,
2J = 13.8, H-i); 3.48-3.38 (m, 1H, H-h); 3.11 (dt, 1H,
2J = 12.8,
3J = 3.0, H-h); 2.21-2.13 (m,
1H, H-e); 1.73-1.53 (m, 3H, H-e, H-f, H-g); 1.51-1.36 (m, 1H, H-g); 1.22-1.10 (m, 1H, H-f);
24b: 7.44-7.39 (m, 2H, H-l, H-n); 7.36-7.31 (m, 2H, H-k, H-o); 7.30-7.25 (m, 2H, H-8, H-10);
7.20-7.15 (m, 3H, H-7; H-9, H-11); 4.61 (d, 1H, 3J = 4.0, H-d); 4.25-4.09 (m, 4H, H-i, H-1);
3.48-3.42 (m, 1H, H-h); 3.09 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.63 (t, 2H,
3J = 8.0, H-5);
2.19-2.13 (m, 1H, H-e); 1.77-1.61 (m, 7H, H-e, H-f, H-g, H-2, H-4); 1.50-1.12 (m, 4H, H-f,
H-g, H-3);
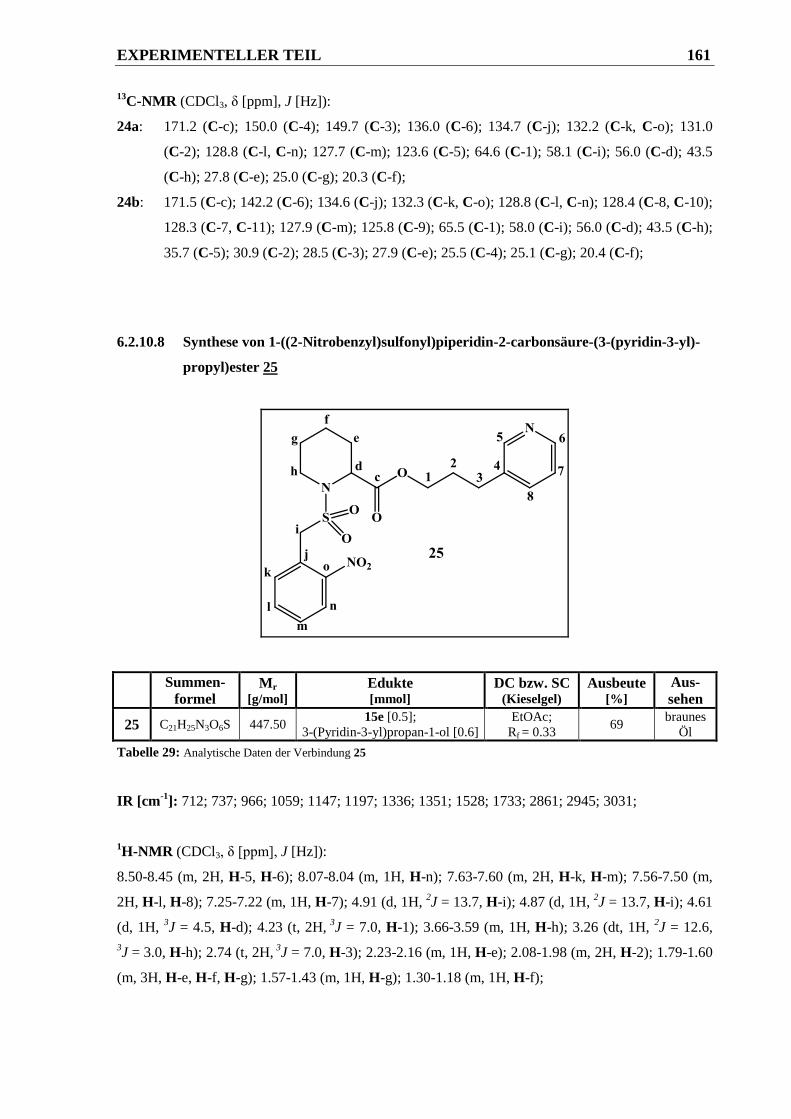
EXPERIMENTELLER TEIL 161
13C-NMR (CDCl3, δ [ppm], J [Hz]):
24a: 171.2 (C-c); 150.0 (C-4); 149.7 (C-3); 136.0 (C-6); 134.7 (C-j); 132.2 (C-k, C-o); 131.0
(C-2); 128.8 (C-l, C-n); 127.7 (C-m); 123.6 (C-5); 64.6 (C-1); 58.1 (C-i); 56.0 (C-d); 43.5
(C-h); 27.8 (C-e); 25.0 (C-g); 20.3 (C-f);
24b: 171.5 (C-c); 142.2 (C-6); 134.6 (C-j); 132.3 (C-k, C-o); 128.8 (C-l, C-n); 128.4 (C-8, C-10);
128.3 (C-7, C-11); 127.9 (C-m); 125.8 (C-9); 65.5 (C-1); 58.0 (C-i); 56.0 (C-d); 43.5 (C-h);
35.7 (C-5); 30.9 (C-2); 28.5 (C-3); 27.9 (C-e); 25.5 (C-4); 25.1 (C-g); 20.4 (C-f);
6.2.10.8 Synthese von 1-((2-Nitrobenzyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)-
propyl)ester 25
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
25 C21H25N3O6S 447.50 15e [0.5];
3-(Pyridin-3-yl)propan-1-ol [0.6]
EtOAc;
Rf = 0.33 69
braunes
Öl
Tabelle 29: Analytische Daten der Verbindung 25
IR [cm-1
]: 712; 737; 966; 1059; 1147; 1197; 1336; 1351; 1528; 1733; 2861; 2945; 3031;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
8.50-8.45 (m, 2H, H-5, H-6); 8.07-8.04 (m, 1H, H-n); 7.63-7.60 (m, 2H, H-k, H-m); 7.56-7.50 (m,
2H, H-l, H-8); 7.25-7.22 (m, 1H, H-7); 4.91 (d, 1H, 2J = 13.7, H-i); 4.87 (d, 1H,
2J = 13.7, H-i); 4.61
(d, 1H, 3J = 4.5, H-d); 4.23 (t, 2H,
3J = 7.0, H-1); 3.66-3.59 (m, 1H, H-h); 3.26 (dt, 1H,
2J = 12.6,
3J = 3.0, H-h); 2.74 (t, 2H,
3J = 7.0, H-3); 2.23-2.16 (m, 1H, H-e); 2.08-1.98 (m, 2H, H-2); 1.79-1.60
(m, 3H, H-e, H-f, H-g); 1.57-1.43 (m, 1H, H-g); 1.30-1.18 (m, 1H, H-f);
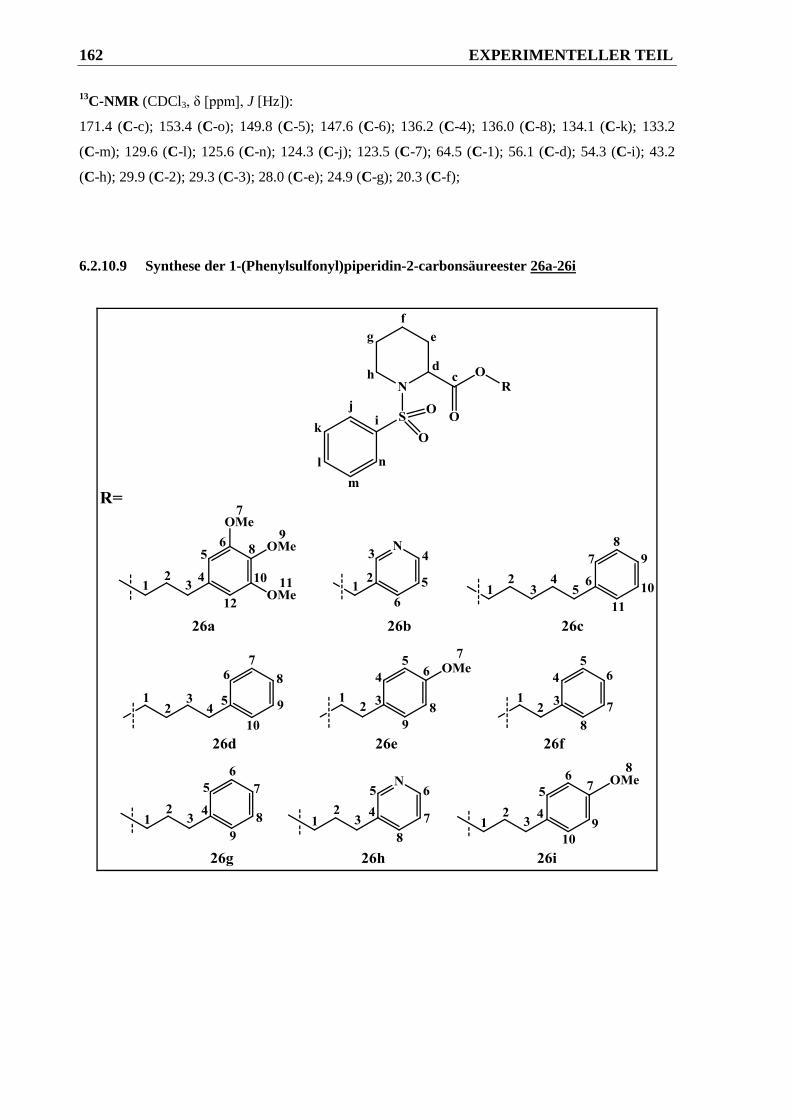
162 EXPERIMENTELLER TEIL
13C-NMR (CDCl3, δ [ppm], J [Hz]):
171.4 (C-c); 153.4 (C-o); 149.8 (C-5); 147.6 (C-6); 136.2 (C-4); 136.0 (C-8); 134.1 (C-k); 133.2
(C-m); 129.6 (C-l); 125.6 (C-n); 124.3 (C-j); 123.5 (C-7); 64.5 (C-1); 56.1 (C-d); 54.3 (C-i); 43.2
(C-h); 29.9 (C-2); 29.3 (C-3); 28.0 (C-e); 24.9 (C-g); 20.3 (C-f);
6.2.10.9 Synthese der 1-(Phenylsulfonyl)piperidin-2-carbonsäureester 26a-26i
EXPERIMENTELLER TEIL 163
Summen-
formel Mr
[g/mol]
Edukte
[mmol] DC bzw. SC
(Kieselgel) Ausbeute
[%]
Aus-
sehen
26a C24H31NO7S 457.57 15f [2.5];
16 [2.8]
Al2O3;
Cyclohexan : EtOAc =
80:20; Rf = 0.33
28 gelbes
Wachs
26b C18H20N2O4S 360.43
15f [2.5];
Pyridin-3-yl-
methanol [2.8]
PE : EtOAc = 1:1
(+5 % Et3N);
Rf = 0.35
8 gelbes Öl
26c C23H29NO4S 415.55
15f [2.5];
5-Phenyl-
pentan-1-ol [2.8]
PE : EtOAc = 1:1
(+5 % Et3N);
Rf = 0.82
23 klares Öl
26d133
C22H27NO4S 401.52
15f [2.5];
4-Phenyl-
butan-1-ol [2.8]
PE : EtOAc = 1:1;
Rf = 0.87 16 klares Öl
26e C21H25NO5S 403.49
Verb. 15f [2.5];
2-(4-Methoxy-
phenyl)ethanol [2.8]
PE : EtOAc = 1:1
(+5 % Et3N);
Rf = 0.74
22 klares Öl
26f C20H23NO4S 373.47 Verb. 15f [2.5];
2-Phenylethanol [2.8]
PE : EtOAc = 1:1
(+5 % Et3N);
Rf = 0.82
9 weißes
Öl
26g C21H25NO4S 387.49
Verb. 15f [2.5];
3-Phenyl-
propan-1-ol [2.8]
Cyclohexan : EtOAc =
1:1; Rf = 0.77 76 gelbes Öl
26h C20H24N2O4S 388.48
Verb. 15f [2.5];
3-(Pyridin-3-yl)-
propan-1-ol [2.8]
Cyclohexan : EtOAc =
1:1 (+5 % Et3N);
Rf = 0.42
83 gelbes Öl
26i142
C22H27NO5S 417.52
Verb. 15f [2.5];
3-(4-Methoxy-phenyl)-
propan-1-ol [2.8]
PE : EtOAc = 1:1
(+5 % Et3N);
Rf = 0.79
19 klares Öl
Tabelle 30: Analytische Daten der Verbindungen 26a-26i
IR [cm-1
]:
26a: 690; 738; 1056; 1115; 1160; 1334; 1449; 1591; 1739; 2860; 2947; 3059;
26b: 689; 738; 970; 1056; 1111; 1154; 1336; 1446; 1578; 1741; 2858; 2943; 3061;
26c: 690; 738; 963; 1093; 1157; 1338; 1446; 1603; 1735; 2857; 2933; 3061;
26d: 689; 737; 964; 1093; 1156; 1338; 1446; 1604; 1736; 2859; 2941; 3061;
26e: 689; 738; 1034; 1111; 1156; 1245; 1337; 1445; 1513; 1612; 1736; 2852; 2932; 3063;
26f: 690; 736; 1056; 1093; 1156; 1240; 1338; 1446; 1735; 2859; 2945; 3062;
26g: 689; 737; 1093; 1112; 1157; 1339; 1446; 1734; 2115; 2855; 2929; 3059;
26h: 690; 738; 1093; 1111; 1160; 1336; 1446; 1576; 1737; 2862; 2944; 3062;
26i: 689; 737; 1056; 1111; 1156; 1244; 1337; 1445; 1512; 1612; 1736; 2859; 2943; 3062;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
26a: 7.82-7.78 (m, 2H, H-j, H-n); 7.58-7.41 (m, 3H, H-k, H-l, H-m); 6.38 (s, 2H, H-5, H-12);
4.80-4.75 (m, 1H, H-d); 4.08-3.93 (m, 2H, H-1); 3.84 (s, 6H, H-7, H-11); 3.82 (s, 3H, H-9);
3.80-3.73 (m, 1H, H-h); 3.25 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.62-2.56 (m, 2H, H-3); 2.19-

164 EXPERIMENTELLER TEIL
2.10 (m, 1H, H-e); 1.93-1.82 (m, 2H, H-2); 1.80-1.60 (m, 3H, H-e, H-f, H-g); 1.53-1.39 (m,
1H, H-g); 1.37-1.20 (m, 1H, H-f);
26b: 8.59 (d, 1H, 3J = 3.6, H-4); 8.52 (s, 1H, H-3); 7.78-7.73 (m, 2H, H-j; H-n); 7.63-7.58 (m, 1H,
H-6); 7.56-7.50 (m, 1H, H-l); 7.47-7.41 (m, 2H, H-k, H-m); 7.32-7.27 (m, 1H, H-5); 5.07 (d,
1H, 2
J = 12.5, H-1); 4.95 (d, 1H, 2
J = 12.5, H-1); 4.80 (d, 1H, 3
J = 5.0, H-d); 3.80-3.73 (m,
1H, H-h); 3.19 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.16-2.09 (m, 1H, H-e); 1.82-1.38 (m, 3H,
H-e, H-f, H-g); 1.28-1.15 (m, 2H, H-f, H-g);
26c: 7.84-7.73 (m, 2H, H-j, H-n); 7.54-7.41 (m, 3H, H-k, H-l, H-m); 7.29-7.24 (m, 2H, H-8,
H-10); 7.20-7.12 (m, 3H, H-7, H-9, H-11); 4.72 (d, 1H, 3J = 4.7, H-d); 3.99-3.80 (m, 2H, H-
1); 3.78-3.71 (m, 1H, H-h); 3.17 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.61-2.55 (m, 2H, H-5);
2.12-2.04 (m, 1H, H-e); 1.79-1.20 (m, 11H, H-2, H-3, H-4, H-e, H-f, H-g);
26d: 7.82-7.73 (m, 2H, H-j, H-n); 7.54-7.48 (m, 1H, H-l); 7.46-7.41 (m, 2H, H-k, H-m); 7.32-
7.27 (m, 2H, H-7, H-9); 7.22-7.14 (m, 3H, H-6, H-8, H-10); 4.75 (d, 1H, 3
J = 5.0, H-d);
4.04-3.85 (m, 2H, H-1); 3.81-3.75 (m, 1H, H-h); 3.21 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.61
(t, 2H, 3J = 7.4, H-4); 2.15-2.08 (m, 1H, H-e); 1.80-1.42 (m, 7H, H-e, H-f, H-g, H-2, H-3);
1.33-1.23 (m, 2H, H-f, H-g);
26e: 7.84-7.72 (m, 2H, H-j, H-n); 7.59-7.43 (m, 3H, H-k, H-l, H-m); 7.06 (d, 2H, 3J = 8.7, H-4,
H-9); 6.83 (d, 2H, 3J = 8.7, H-5, H-8); 4.73 (d, 1H,
3J = 5.0, H-d); 4.19-4.02 (m, 2H, H-1);
3.80-3.71 (m, 4H, H-h, H-7); 3.11 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.75 (dt, 2H,
3J = 7.0,
4J = 3.0, H-2); 2.08-2.01 (m, 1H, H-e); 1.80-1.10 (m, 5H, H-e, H-f, H-g);
26f: 7.81-7.71 (m, 2H, H-j, H-n); 7.57-7.42 (m, 3H, H-k, H-l, H-m) 7.32-7.17 (m, 3H, H-5, H-6,
H-7); 7.15-7.11 (m, 2H, H-4, H-8); 4.71 (d, 1H, 3J = 5.3, H-d); 4.24-4.04 (m, 2H, H-1);
3.75-3.67 (m, 1H, H-h); 3.07 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.82-2.76 (m, 2H, H-2); 2.06-
1.97 (m, 1H, H-e); 1.79-1.20 (m, 5H, H-e, H-f, H-g);
26g: 7.84-7.70 (m, 2H, H-j, H-n); 7.53-7.48 (m, 1H, H-l); 7.47-7.40 (m, 2H, H-k, H-m); 7.31-
7.25 (m, 2H, H-6, H-8); 7.22-7.16 (m, 1H, H-7); 7.15-7.11 (m, 2H, H-5, H-9); 4.75 (d, 1H,
3J = 5.0, H-d); 4.04-3.85 (m, 2H, H-1); 3.81-3.73 (m, 1H, H-h); 3.23 (dt, 1H,
2J = 12.7,
3J = 3.0, H-h); 2.63-2.57 (m, 2H, H-3); 2.16-2.08 (m, 1H, H-e); 1.94-1.59 (m, 5H, H-2, H-e,
H-f, H-g); 1.38-1.18 (m, 2H, H-f, H-g);
26h: 8.51-8.43 (m, 2H, H-5, H-6); 7.82-7.78 (m, 2H, H-j, H-n); 7.63-7.57 (m, 1H, H-8); 7.56-7.44
(m, 3H, H-k, H-l, H-m); 7.33-7.27 (m, 1H, H-7); 4.77-4.74 (m, 1H, H-d); 4.06-3.95 (m, 2H,
H-1); 3.79-3.72 (m, 1H, H-h); 3.23 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.71-2.66 (m, 2H, H-3);
2.18-2.07 (m, 1H, H-e); 1.93-1.84 (m, 2H, H-2); 1.81-1.58 (m, 3H, H-e, H-f, H-g); 1.52-1.37
(m, 1H, H-g); 1.35-1.20 (m, 1H, H-f);
26i: 7.82-7.76 (m, 2H, H-j, H-n); 7.55-7.42 (m, 3H, H-k, H-l, H-m); 7.11-7.03 (m, 2H, H-5,
H-10); 6.86-6.81 (m, 2H, H-6, H-9); 4.76 (d, 1H, 3
J = 4.8, H-d); 4.04-3.86 (m, 2H, H-1);
3.82-3.75 (m, 4H, H-h, H-8); 3.24 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 2.59-2.53 (m, 2H, H-3);

EXPERIMENTELLER TEIL 165
2.16-2.10 (m, 1H, H-e); 1.86-1.61 (m, 5H, H-e, H-f, H-g, H-2); 1.54-1.42 (m, 1H, H-g);
1.35-1.25 (m, 1H, H-f);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
26a: 170.8 (C-c); 153.2 (C-6, C-10); 140.2 (C-i); 136.6 (C-4); 136.4 (C-8); 132.4 (C-l); 128.8
(C-k, C-m); 127.1 (C-j, C-n); 105.4 (C-5, C-12); 64.4 (C-1); 60.8 (C-9); 56.1 (C-7, C-11);
55.1 (C-d); 42.7 (C-h); 32.4 (C-3); 30.2 (C-2); 27.8 (C-e); 24.6 (C-g); 20.1 (C-f);
26b: 170.5 (C-c); 149.9 (C-4); 149.6 (C-3); 140.0 (C-i); 136.6 (C-2); 136.0 (C-6); 132.5 (C-l);
128.8 (C-k, C-m); 127.1 (C-j, C-n); 123.5 (C-5); 64.2 (C-1); 55.1 (C-d); 42.7 (C-h); 27.8
(C-e); 24.6 (C-g); 20.0 (C-f);
26c: 170.7 (C-c); 142.2 (C-6); 140.1 (C-i); 132.3 (C-l); 128.8 (C-k, C-m); 128.4 (C-8, C-10);
128.3 (C-7, C-11); 127.1 (C-j, C-n); 125.8 (C-9); 65.0 (C-1); 55.1 (C-d); 42.6 (C-h); 35.7
(C-5); 30.9 (C-4); 28.3 (C-3); 27.9 (C-e); 25.4 (C-2); 24.7 (C-g); 20.0 (C-f);
26d: 170.7 (C-c); 141.8 (C-5); 140.1 (C-i); 132.3 (C-l); 128.8 (C-k, C-m); 128.4 (C-7, C-9);
128.3 (C-6, C-10); 127.1 (C-j, C-n); 125.9 (C-8); 64.9 (C-1); 55.1 (C-d); 42.6 (C-h); 35.3
(C-4); 28.0 (C-3); 27.9 (C-2); 27.6 (C-e); 24.7 (C-g); 20.1 (C-f);
26e: 170.6 (C-c); 158.4 (C-6); 140.1 (C-i); 132.3 (C-l); 129.8 (C-4, C-9); 129.3 (C-3); 128.8
(C-k, C-m); 127.2 (C-j, C-n); 114.0 (C-5, C-8); 65.6 (C-1); 55.3 (C-d); 55.1 (C-7); 42.5
(C-h); 34.0 (C-2); 27.8 (C-e); 24.7 (C-g); 19.9 (C-f);
26f: 170.6 (C-c); 140.1 (C-i); 137.4 (C-3); 132.3 (C-l); 128.8 (C-k, C-m, C-5, C-7); 128.5 (C-4,
C-8); 127.2 (C-j, C-n); 126.7 (C-6); 65.3 (C-1); 55.0 (C-d); 42.5 (C-h); 34.8 (C-2); 27.8
(C-e); 24.6 (C-g); 19.9 (C-f);
26g: 170.4 (C-c); 140.5 (C-i); 139.8 (C-4); 132.0 (C-l); 128.5 (C-k, C-m); 128.2 (C-6, C-8);
128.0 (C-5, C-9); 126.8 (C-j, C-n); 125.8 (C-7); 64.1 (C-1); 54.7 (C-d); 42.3 (C-h); 31.7
(C-3); 29.7 (C-2); 27.6 (C-e); 24.4 (C-g); 19.7 (C-f);
26h: 170.7 (C-c); 148.7 (C-5); 146.6 (C-6); 140.2 (C-i); 137.1 (C-8); 136.8 (C-4); 132.4 (C-l);
128.9 (C-k, C-m); 127.1 (C-j, C-n); 123.8 (C-7); 63.9 (C-1); 55.1 (C-d); 42.7 (C-h); 29.7
(C-2); 29.2 (C-3); 27.7 (C-e); 24.6 (C-g); 20.1 (C-f);
26i: 170.7 (C-c); 158.0 (C-7); 140.1 (C-i); 132.9 (C-4); 132.4 (C-l); 129.3 (C-5, C-10); 128.8
(C-k, C-m); 127.1 (C-j, C-n); 113.9 (C-6, C-9); 64.4 (C-1); 55.3 (C-8); 55.1 (C-d); 42.7 (C-
h); 31.1 (C-3); 30.3 (C-2); 27.9 (C-e); 24.7 (C-g); 20.1 (C-f);
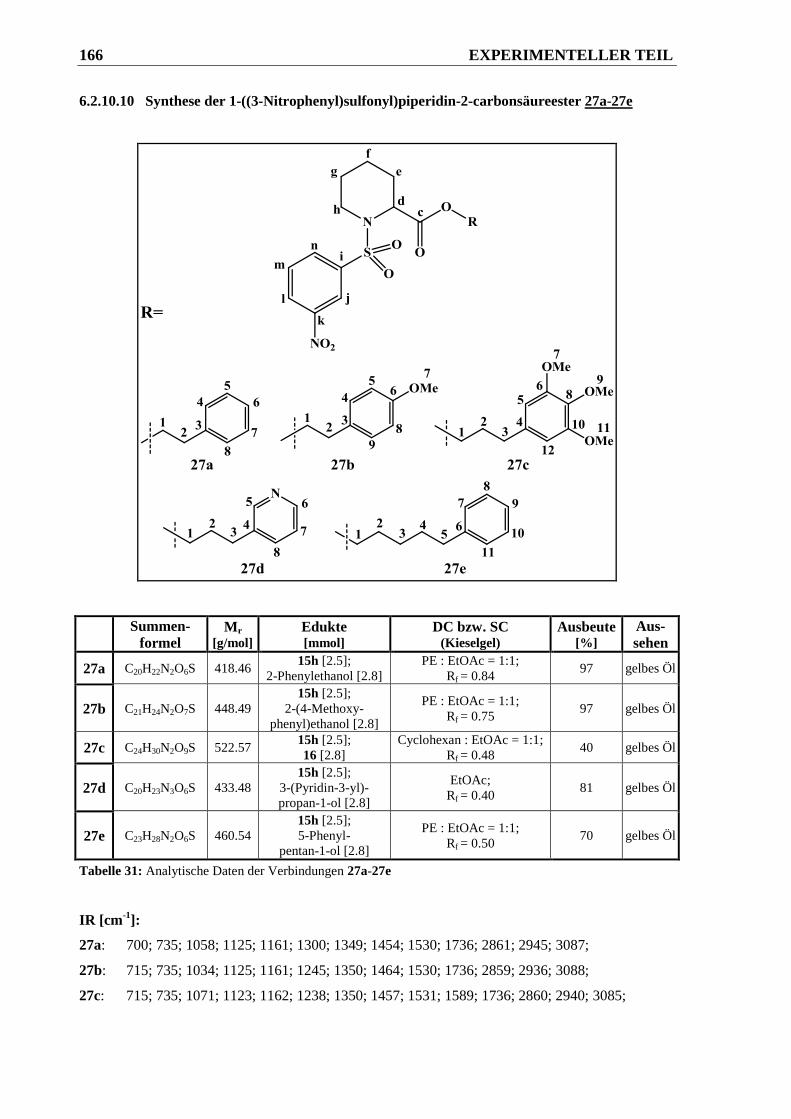
166 EXPERIMENTELLER TEIL
6.2.10.10 Synthese der 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäureester 27a-27e
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
27a C20H22N2O6S 418.46 15h [2.5];
2-Phenylethanol [2.8]
PE : EtOAc = 1:1;
Rf = 0.84 97 gelbes Öl
27b C21H24N2O7S 448.49
15h [2.5];
2-(4-Methoxy-
phenyl)ethanol [2.8]
PE : EtOAc = 1:1;
Rf = 0.75 97 gelbes Öl
27c C24H30N2O9S 522.57 15h [2.5];
16 [2.8]
Cyclohexan : EtOAc = 1:1;
Rf = 0.48 40 gelbes Öl
27d C20H23N3O6S 433.48
15h [2.5];
3-(Pyridin-3-yl)-
propan-1-ol [2.8]
EtOAc;
Rf = 0.40 81 gelbes Öl
27e C23H28N2O6S 460.54
15h [2.5];
5-Phenyl-
pentan-1-ol [2.8]
PE : EtOAc = 1:1;
Rf = 0.50 70 gelbes Öl
Tabelle 31: Analytische Daten der Verbindungen 27a-27e
IR [cm-1
]:
27a: 700; 735; 1058; 1125; 1161; 1300; 1349; 1454; 1530; 1736; 2861; 2945; 3087;
27b: 715; 735; 1034; 1125; 1161; 1245; 1350; 1464; 1530; 1736; 2859; 2936; 3088;
27c: 715; 735; 1071; 1123; 1162; 1238; 1350; 1457; 1531; 1589; 1736; 2860; 2940; 3085;

EXPERIMENTELLER TEIL 167
27d: 714; 735; 1058; 1125; 1161; 1300; 1350; 1423; 1530; 1736; 2861; 2947; 3086;
27e: 715; 734; 1058; 1125; 1161; 1300; 1349; 1453; 1531; 1736; 2857; 2932; 3085;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
27a: 8.59 (t, 1H, 4J = 2.0, H-j); 8.39 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-l); 8.04 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-n); 7.65 (t, 1H,
3J = 8.0, H-m); 7.32-7.19 (m, 3H, H-4, H-6,
H-8); 7.16-7.12 (m, 2H, H-5, H-7); 4.77 (d, 1H, 3J = 5.0, H-d); 4.26-4.14 (m, 2H, H-1);
3.80-3.73 (m, 1H, H-h); 3.05 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.85 (dt, 2H,
3J = 7.0,
4J = 3.5,
H-2); 2.14-2.07 (m, 1H, H-e); 1.82-1.43 (m, 4H, H-e, H-f, H-g); 1.12-1.05 (m, 1H, H-f);
27b: 8.60 (t, 1H, 4J = 2.0, H-j); 8.39 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-l); 8.04 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-n); 7.66 (t, 1H,
3J = 8.0, H-m); 7.08-7.03 (m, 2H, H-4, H-9);
6.86-6.81 (m, 2H, H-5, H-8); 4.78 (d, 1H, 3J = 5.0, H-d); 4.21-4.10 (m, 2H, H-1); 3.81-3.74
(m, 4H, H-h, H-7); 3.07 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.79 (dt, 2H,
3J = 7.0,
4J = 3.0,
H-2); 2.16-2.08 (m, 1H, H-e); 1.83-1.44 (m, 3H, H-e, H-f, H-g); 1.32-1.10 (m, 2H, H-f,
H-g);
27c: 8.62-8.60 (m, 1H, H-j); 8.41-8.37 (m, 1H, H-l); 8.12-8.08 (m, 1H, H-n); 7.68 (t, 1H, 3J = 8.0,
H-m); 6.37 (s, 2H, H-5, H-12); 4.82 (d, 1H, 3J = 4.9, H-d); 4.07-3.96 (m, 2H, H-1); 3.85 (s,
6H, H-7, H-11); 3.83 (s, 3H, H-9); 3.75-3.69 (m, 1H, H-h); 3.20 (dt, 1H, 2J = 12.7,
3J = 3.0,
H-h); 2.60-2.54 (m, 2H, H-3); 2.24-2.17 (m, 1H, H-e); 1.94-1.83 (m, 2H, H-2); 1.94-1.66
(m, 3H, H-e, H-f, H-g); 1.62-1.47 (m, 1H, H-g); 1.35-1.12 (m, 1H, H-f);
27d: 8.62-8.59 (m, 1H, H-j); 8.50-8.44 (m, 2H, H-5, H-6); 8.41-8.37 (m, 1H, H-l); 8.13-8.09 (m,
1H, H-n); 7.69 (t, 1H, 3J = 8.0, H-m); 7.59-7.55 (m, 1H, H-8); 7.32-7.27 (m,1H, H-7); 4.81
(d, 1H, 3J = 4.9, H-d); 4.08-3.99 (m, 2H, H-1); 3.83-3.76 (m, 1H, H-h); 3.18 (dt, 1H,
2J = 12.7,
3J = 2.9, H-h); 2.71-2.66 (m, 2H, H-3); 2.24-2.16 (m, 1H, H-e); 1.96-1.87 (m, 2H,
H-2); 1.87-1.67 (m, 3H, H-e, H-f, H-g); 1.61-1.47 (m, 1H, H-g); 1.32-1.10 (m, 1H, H-f);
27e: 8.60 (t, 1H, 4J = 2.0, H-j); 8.39 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-l); 8.09 (ddd, 1H,
3J = 8.0,
4J = 2.0,
4J = 1.0, H-n); 7.67 (t, 1H,
3J = 8.0, H-m); 7.30-7.25 (m, 2H, H-8, H-10);
7.21-7.14 (m, 3H, H-7, H-9, H-11); 4.79 (d, 1H, 3J = 5.0, H-d); 4.00-3.88 (m, 2H, H-1); 3.81
(d, 1H, 2J = 12.7, H-h); 3.14 (dt, 1H,
2J = 12.7,
3J = 2.9, H-h); 2.60 (t, 2H,
3J = 7.6, H-5);
2.21-2.14 (m, 1H, H-e); 1.85-1.49 (m, 9H, H-e, H-f, H-g, H-2, H-3, H-4); 1.35-1.15 (m, 2H,
H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
27a: 170.2 (C-c); 142.1 (C-k); 137.1 (C-3); 135.3 (C-i); 132.8 (C-n); 130.0 (C-m); 128.8 (C-5,
C-7); 128.6 (C-4, C-8); 126.8 (C-l, C-6); 122.5 (C-j); 65.6 (C-1); 55.5 (C-d); 42.8 (C-h);
34.9 (C-2); 27.9 (C-e); 24.8 (C-g); 19.9 (C-f);
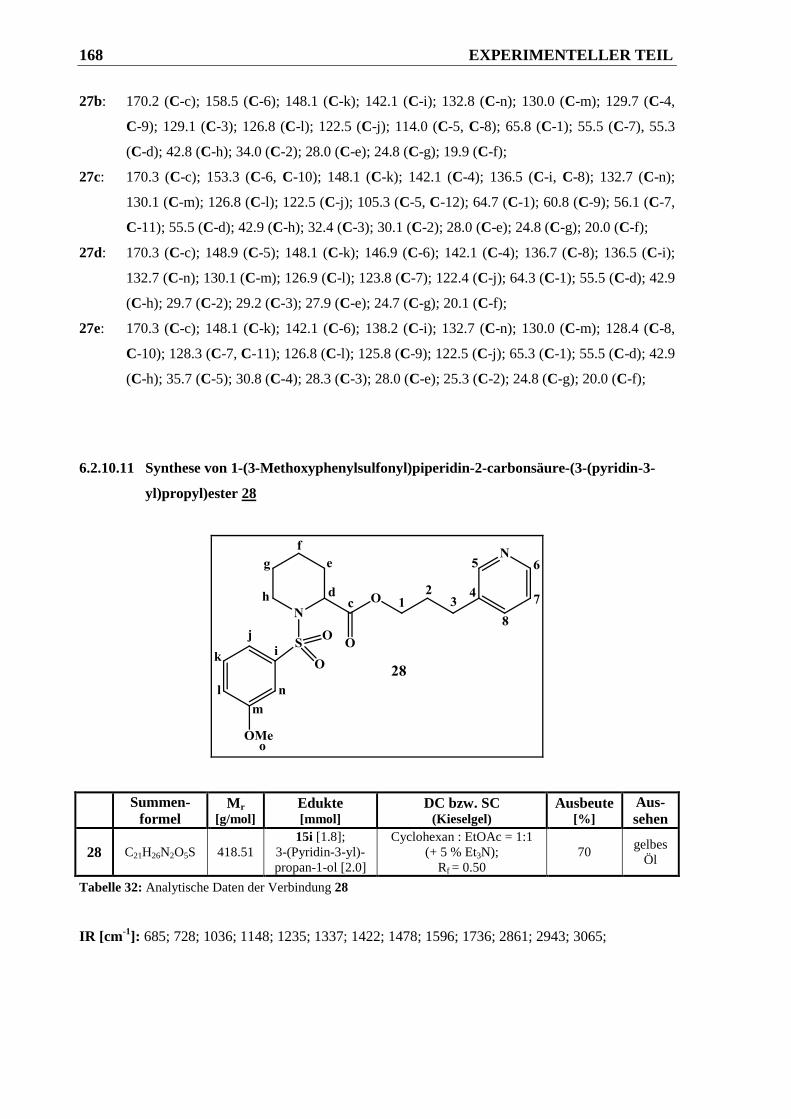
168 EXPERIMENTELLER TEIL
27b: 170.2 (C-c); 158.5 (C-6); 148.1 (C-k); 142.1 (C-i); 132.8 (C-n); 130.0 (C-m); 129.7 (C-4,
C-9); 129.1 (C-3); 126.8 (C-l); 122.5 (C-j); 114.0 (C-5, C-8); 65.8 (C-1); 55.5 (C-7), 55.3
(C-d); 42.8 (C-h); 34.0 (C-2); 28.0 (C-e); 24.8 (C-g); 19.9 (C-f);
27c: 170.3 (C-c); 153.3 (C-6, C-10); 148.1 (C-k); 142.1 (C-4); 136.5 (C-i, C-8); 132.7 (C-n);
130.1 (C-m); 126.8 (C-l); 122.5 (C-j); 105.3 (C-5, C-12); 64.7 (C-1); 60.8 (C-9); 56.1 (C-7,
C-11); 55.5 (C-d); 42.9 (C-h); 32.4 (C-3); 30.1 (C-2); 28.0 (C-e); 24.8 (C-g); 20.0 (C-f);
27d: 170.3 (C-c); 148.9 (C-5); 148.1 (C-k); 146.9 (C-6); 142.1 (C-4); 136.7 (C-8); 136.5 (C-i);
132.7 (C-n); 130.1 (C-m); 126.9 (C-l); 123.8 (C-7); 122.4 (C-j); 64.3 (C-1); 55.5 (C-d); 42.9
(C-h); 29.7 (C-2); 29.2 (C-3); 27.9 (C-e); 24.7 (C-g); 20.1 (C-f);
27e: 170.3 (C-c); 148.1 (C-k); 142.1 (C-6); 138.2 (C-i); 132.7 (C-n); 130.0 (C-m); 128.4 (C-8,
C-10); 128.3 (C-7, C-11); 126.8 (C-l); 125.8 (C-9); 122.5 (C-j); 65.3 (C-1); 55.5 (C-d); 42.9
(C-h); 35.7 (C-5); 30.8 (C-4); 28.3 (C-3); 28.0 (C-e); 25.3 (C-2); 24.8 (C-g); 20.0 (C-f);
6.2.10.11 Synthese von 1-(3-Methoxyphenylsulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-
yl)propyl)ester 28
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
28 C21H26N2O5S 418.51
15i [1.8];
3-(Pyridin-3-yl)-
propan-1-ol [2.0]
Cyclohexan : EtOAc = 1:1
(+ 5 % Et3N);
Rf = 0.50
70 gelbes
Öl
Tabelle 32: Analytische Daten der Verbindung 28
IR [cm-1
]: 685; 728; 1036; 1148; 1235; 1337; 1422; 1478; 1596; 1736; 2861; 2943; 3065;
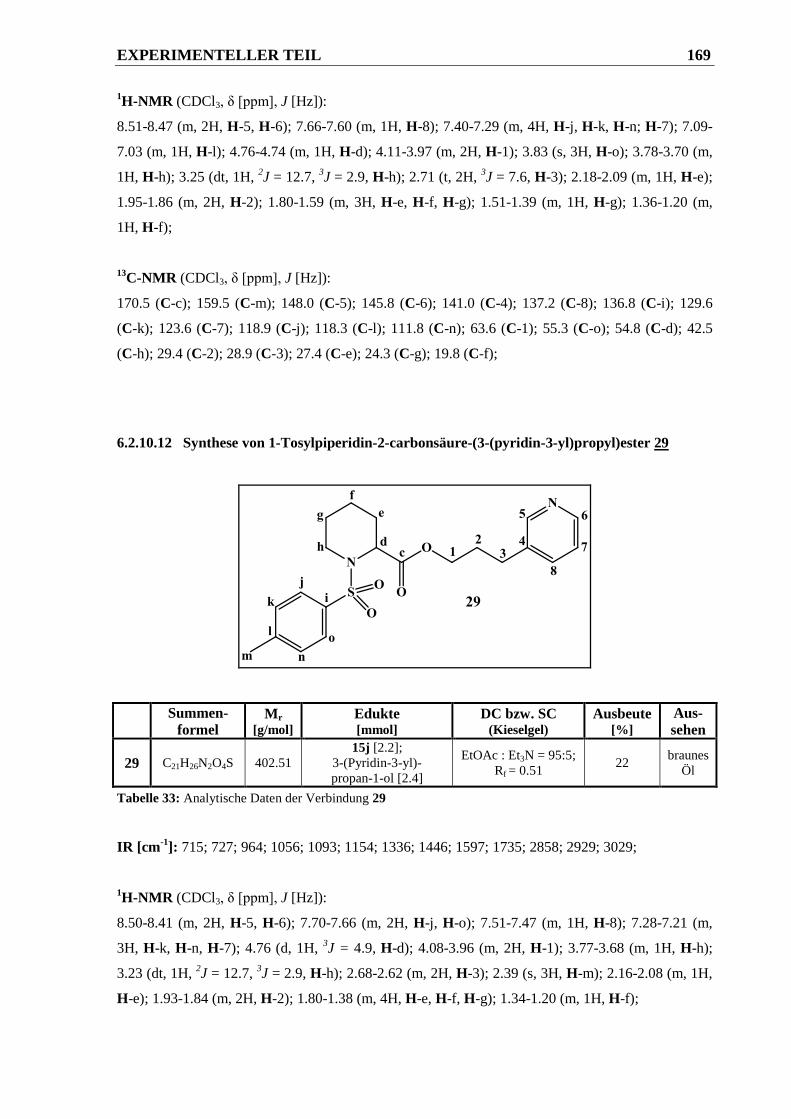
EXPERIMENTELLER TEIL 169
1H-NMR (CDCl3, δ [ppm], J [Hz]):
8.51-8.47 (m, 2H, H-5, H-6); 7.66-7.60 (m, 1H, H-8); 7.40-7.29 (m, 4H, H-j, H-k, H-n; H-7); 7.09-
7.03 (m, 1H, H-l); 4.76-4.74 (m, 1H, H-d); 4.11-3.97 (m, 2H, H-1); 3.83 (s, 3H, H-o); 3.78-3.70 (m,
1H, H-h); 3.25 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.71 (t, 2H,
3J = 7.6, H-3); 2.18-2.09 (m, 1H, H-e);
1.95-1.86 (m, 2H, H-2); 1.80-1.59 (m, 3H, H-e, H-f, H-g); 1.51-1.39 (m, 1H, H-g); 1.36-1.20 (m,
1H, H-f);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
170.5 (C-c); 159.5 (C-m); 148.0 (C-5); 145.8 (C-6); 141.0 (C-4); 137.2 (C-8); 136.8 (C-i); 129.6
(C-k); 123.6 (C-7); 118.9 (C-j); 118.3 (C-l); 111.8 (C-n); 63.6 (C-1); 55.3 (C-o); 54.8 (C-d); 42.5
(C-h); 29.4 (C-2); 28.9 (C-3); 27.4 (C-e); 24.3 (C-g); 19.8 (C-f);
6.2.10.12 Synthese von 1-Tosylpiperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester 29
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
29 C21H26N2O4S 402.51
15j [2.2];
3-(Pyridin-3-yl)-
propan-1-ol [2.4]
EtOAc : Et3N = 95:5;
Rf = 0.51 22
braunes
Öl
Tabelle 33: Analytische Daten der Verbindung 29
IR [cm-1
]: 715; 727; 964; 1056; 1093; 1154; 1336; 1446; 1597; 1735; 2858; 2929; 3029;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
8.50-8.41 (m, 2H, H-5, H-6); 7.70-7.66 (m, 2H, H-j, H-o); 7.51-7.47 (m, 1H, H-8); 7.28-7.21 (m,
3H, H-k, H-n, H-7); 4.76 (d, 1H, 3J = 4.9, H-d); 4.08-3.96 (m, 2H, H-1); 3.77-3.68 (m, 1H, H-h);
3.23 (dt, 1H, 2J = 12.7,
3J = 2.9, H-h); 2.68-2.62 (m, 2H, H-3); 2.39 (s, 3H, H-m); 2.16-2.08 (m, 1H,
H-e); 1.93-1.84 (m, 2H, H-2); 1.80-1.38 (m, 4H, H-e, H-f, H-g); 1.34-1.20 (m, 1H, H-f);
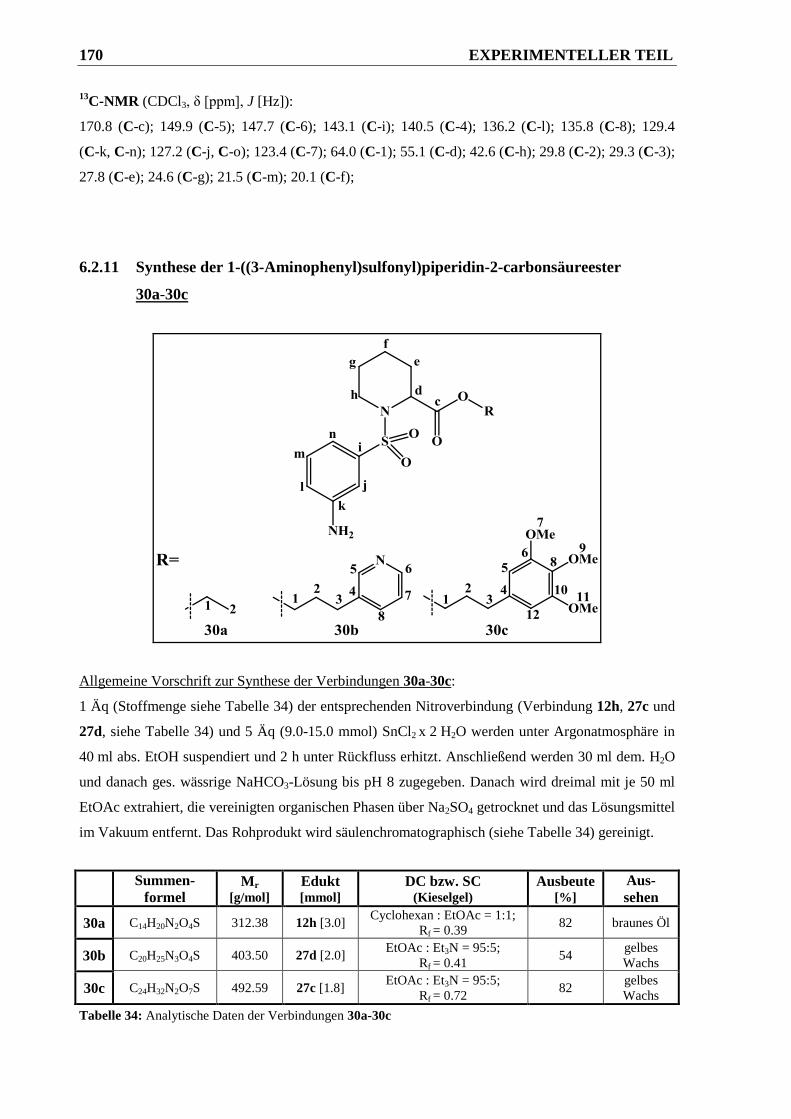
170 EXPERIMENTELLER TEIL
13C-NMR (CDCl3, δ [ppm], J [Hz]):
170.8 (C-c); 149.9 (C-5); 147.7 (C-6); 143.1 (C-i); 140.5 (C-4); 136.2 (C-l); 135.8 (C-8); 129.4
(C-k, C-n); 127.2 (C-j, C-o); 123.4 (C-7); 64.0 (C-1); 55.1 (C-d); 42.6 (C-h); 29.8 (C-2); 29.3 (C-3);
27.8 (C-e); 24.6 (C-g); 21.5 (C-m); 20.1 (C-f);
6.2.11 Synthese der 1-((3-Aminophenyl)sulfonyl)piperidin-2-carbonsäureester
30a-30c
Allgemeine Vorschrift zur Synthese der Verbindungen 30a-30c:
1 Äq (Stoffmenge siehe Tabelle 34) der entsprechenden Nitroverbindung (Verbindung 12h, 27c und
27d, siehe Tabelle 34) und 5 Äq (9.0-15.0 mmol) SnCl2 x 2 H2O werden unter Argonatmosphäre in
40 ml abs. EtOH suspendiert und 2 h unter Rückfluss erhitzt. Anschließend werden 30 ml dem. H2O
und danach ges. wässrige NaHCO3-Lösung bis pH 8 zugegeben. Danach wird dreimal mit je 50 ml
EtOAc extrahiert, die vereinigten organischen Phasen über Na2SO4 getrocknet und das Lösungsmittel
im Vakuum entfernt. Das Rohprodukt wird säulenchromatographisch (siehe Tabelle 34) gereinigt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
30a C14H20N2O4S 312.38 12h [3.0] Cyclohexan : EtOAc = 1:1;
Rf = 0.39 82 braunes Öl
30b C20H25N3O4S 403.50 27d [2.0] EtOAc : Et3N = 95:5;
Rf = 0.41 54
gelbes
Wachs
30c C24H32N2O7S 492.59 27c [1.8] EtOAc : Et3N = 95:5;
Rf = 0.72 82
gelbes
Wachs
Tabelle 34: Analytische Daten der Verbindungen 30a-30c

EXPERIMENTELLER TEIL 171
IR [cm-1
]:
30a: 686; 730; 1152; 1292; 1451; 1598; 1633; 1732; 2869; 2942; 3065; 3375; 3469;
30b: 686; 731; 1150; 1176; 1313; 1481; 1598; 1731; 2860; 2945; 3057; 3376; 3453;
30c: 686; 731; 1122; 1238; 1315; 1453; 1589; 1732; 2859; 2940; 3062; 3371; 3464;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
30a: 7.17 (t, 1H, 3J = 7.8, H-m); 6.94-6.92 (m, 1H, H-j); 6.84-6.80 (m, 1H, H-n); 6.77-6.74 (m,
1H, H-l); 5.55 (s, 2H, H-NH2); 4.51 (d, 1H, 3J = 4.0, H-d); 4.04-3.87 (m, 2H, H-b); 3.62-
3.56 (m, 1H, H-h); 3.15 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h); 1.97-1.90 (m, 1H, H-e); 1.63-1.50
(m, 3H, H-e, H-f, H-g); 1.31-1.13 (m, 2H, H-f, H-g); 1.10 (t, 3H, 3J = 7.1, H-a);
30b: 8.43-8.39 (m, 2H, H-5, H-6); 7.64-7.60 (m, 1H, H-8); 7.34-7.29 (m, 1H, H-7); 7.16 (t, 1H,
3J = 7.9, H-m); 6.97-6.94 (m, 1H, H-j); 6.86-6.82 (m, 1H, H-n); 6.77-6.73 (m, 1H, H-l); 5.57
(s, 2H, H-NH2); 4.56 (d, 1H, 3J = 5.1, H-d); 4.05-3.85 (m, 2H, H-1); 3.63-3.55 (m, 1H, H-h);
3.21-3.11 (m, 1H, H-h); 2.64-2.58 (m, 2H, H-3); 2.00-1.92 (m, 1H, H-e); 1.86-1.77 (m, 2H,
H-2); 1.65-1.53 (m, 3H, H-e, H-f, H-g); 1.31-1.10 (m, 2H, H-f, H-g);
30c: 7.13 (t, 1H, 3J = 7.9, H-m); 6.93-6.90 (m, 1H, H-j); 6.82-6.79 (m, 1H, H-n); 6.75-6.71 (m,
1H, H-l); 6.45 (s, 2H, H-5, H-12); 5.54 (s, 2H, H-NH2); 4.55-4.50 (m, 1H, H-d); 4.03-3.84
(m, 2H, H-1); 3.72 (s, 6H, H-7, H-11); 3.60-3.52 (m, 4H, H-h, H-9); 3.19-3.09 (m, 1H, H-h);
2.53-2.45 (m, 2H, H-3); 1.96-1.91 (m, 1H, H-e); 1.82-1.74 (m, 2H, H-2); 1.61-1.50 (m, 3H,
H-e, H-f, H-g); 1.29-1.11 (m, 2H, H-f, H-g);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
30a: 169.9 (C-c); 149.2 (C-k); 139.8 (C-i); 129.2 (C-m); 117.2 (C-l); 113.0 (C-n); 110.8 (C-j);
60.4 (C-b); 54.2 (C-d); 41.9 (C-h); 26.7 (C-e); 23.5 (C-g); 19.0 (C-f); 13.5 (C-a);
30b: 169.9 (C-c); 149.1 (C-5); 149.0 (C-k); 146.8 (C-6); 139.7 (C-4); 135.9 (C-8); 135.3 (C-i);
129.1 (C-n); 123.0 (C-m); 117.0 (C-l); 112.7 (C-7); 110.5 (C-j); 63.4 (C-1); 54.1 (C-d); 41.8
(C-h); 28.7 (C-2); 27.9 (C-3); 26.5 (C-e); 23.3 (C-g); 18.9 (C-f);
30c: 170.1 (C-c); 152.4 (C-6, C-10); 149.2 (C-k); 139.9 (C-4); 136.4 (C-8); 135.3 (C-i); 129.3
(C-n); 117.2 (C-m); 113.0 (C-l); 110.7 (C-j); 105.2 (C-5, C-12); 63.7 (C-1); 59.6 (C-9); 55.5
(C-7, C-11); 54.3 (C-d); 41.9 (C-h); 31.4 (C-3); 29.3 (C-2); 26.7 (C-e); 23.5 (C-g); 19.2
(C-f);

172 EXPERIMENTELLER TEIL
6.2.12 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbon-
säureethylester 31
0.17 g (1.6 mmol) Dimethylcarbaminsäurechlorid werden unter Argonatmosphäre in 20 ml abs.
CH2Cl2 gelöst und auf 0 °C abgekühlt. Danach werden 0.5 g (4.8 mmol) Et3N und nach 10 min 0.5 g
(1.6 mmol) der Verbindung 30a zugetropft und 1 h bei 0 °C sowie 14 h bei RT gerührt. Anschließend
wird der Niederschlag abfiltriert und das Lösungsmittel des Filtrats im Vakuum entfernt. Der ölige
Rückstand wird in 50 ml CHCl3 suspendiert, dreimal mit je 50 ml wässriger 2 M HCl-Lösung und
danach dreimal mit je 50 ml dem. H2O gewaschen. Die organische Phase wird über Na2SO4
getrocknet und das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird
säulenchromatographisch (Kieselgel; PE : EtOAc = 1 : 1; Rf = 0.11) gereinigt und man erhält 0.14 g
(0.4 mmol) von Verbindung 31 in Form eines klaren Öls.
Summenformel: C17H25N3O5S
Mr = 383.46 g/mol
Ausbeute: 23 %
Aussehen: klares Öl
IR [cm-1
]: 687; 736; 959; 1150; 1180; 1241; 1303; 1530; 1651; 1735; 2863; 2940; 3064; 3387;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.79 (td, 1H, 3J = 7.5,
4J = 2.0, H-j); 7.62 (br, 1H, H-n); 7.42-7.32 (m, 2H, H-k, H-l); 6.45 (br, 1H,
H-o), 4.67 (d, 1H, 3J = 4.5, H-d); 4.05-3.86 (m, 2H, H-b); 3.78-3.70 (m, 1H, H-h); 3.23 (dt, 1H,
2J = 12.8,
3J = 3.0, H-h); 3.01 (s, 6H, H-q); 2.11-2.05 (m, 1H, H-e); 1.79-1.55 (m, 3H, H-e, H-f,
H-g); 1.49-1.37 (m, 1H, H-g); 1.31-1.18 (m, 4H, H-a, H-f);

EXPERIMENTELLER TEIL 173
13C-NMR (CDCl3, δ [ppm], J [Hz]):
170.7 (C-c); 155.1 (C-p); 140.5 (C-i); 140.0 (C-m); 129.5 (C-k); 123.4 (C-j); 121.1 (C-l); 117.7
(C-n); 61.1 (C-b); 55.2 (C-d); 42.7 (C-h); 36.5 (C-q); 27.8 (C-e); 24.7 (C-g); 20.0 (C-f); 14.1 (C-a);
6.2.13 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbon-
säure 32
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.7 durchgeführt.
Summenformel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
32 C15H21N3O5S 355.41 31 [0.4]
EtOH : EtOAc = 1:1
(+ 5 % HCOOH);
Rf = 0.76
97 klares Öl
Tabelle 35: Analytische Daten der Verbindung 32
IR [cm-1
]: 688; 723; 963; 1150; 1376; 1464; 1533; 1649; 1735; 2851; 2953; 3069-3478 (b);
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
12.86 (br, 1H, H-b); 8.60 (s, 1H, H-o); 8.06-7.98 (m, 1H, H-n); 7.79-7.74 (m, 1H, H-j); 7.41 (t, 1H,
3J = 8.0, H-k); 7.33-7.29 (m, 1H, H-l), 4.51 (d, 1H,
3J = 4.9, H-d); 3.65-3.58 (m, 1H, H-h); 3.26-3.18
(m, 1H, H-h); 2.94 (s, 6H, H-q); 2.01-1.94 (m, 1H, H-e); 1.60-1.41 (m, 3H, H-e, H-f, H-g); 1.25-
1.13 (m, 2H, H-f, H-g);
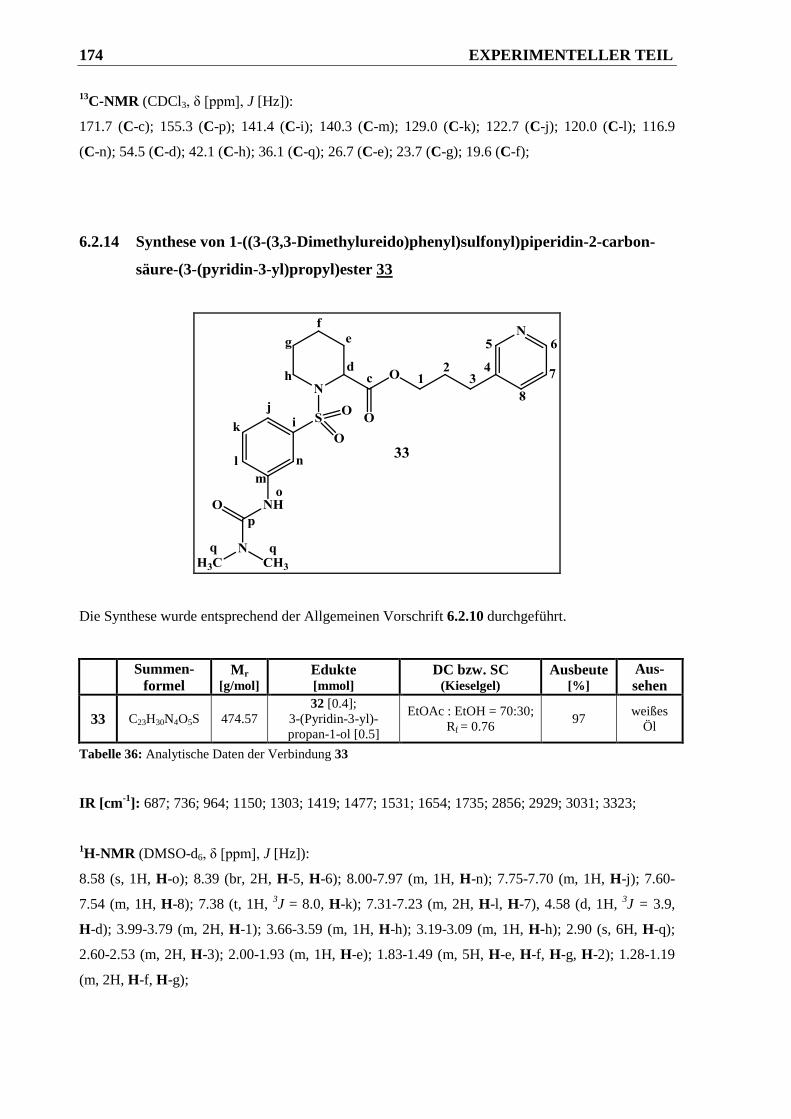
174 EXPERIMENTELLER TEIL
13C-NMR (CDCl3, δ [ppm], J [Hz]):
171.7 (C-c); 155.3 (C-p); 141.4 (C-i); 140.3 (C-m); 129.0 (C-k); 122.7 (C-j); 120.0 (C-l); 116.9
(C-n); 54.5 (C-d); 42.1 (C-h); 36.1 (C-q); 26.7 (C-e); 23.7 (C-g); 19.6 (C-f);
6.2.14 Synthese von 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbon-
säure-(3-(pyridin-3-yl)propyl)ester 33
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.10 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
33 C23H30N4O5S 474.57
32 [0.4];
3-(Pyridin-3-yl)-
propan-1-ol [0.5]
EtOAc : EtOH = 70:30;
Rf = 0.76 97
weißes
Öl
Tabelle 36: Analytische Daten der Verbindung 33
IR [cm-1
]: 687; 736; 964; 1150; 1303; 1419; 1477; 1531; 1654; 1735; 2856; 2929; 3031; 3323;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
8.58 (s, 1H, H-o); 8.39 (br, 2H, H-5, H-6); 8.00-7.97 (m, 1H, H-n); 7.75-7.70 (m, 1H, H-j); 7.60-
7.54 (m, 1H, H-8); 7.38 (t, 1H, 3J = 8.0, H-k); 7.31-7.23 (m, 2H, H-l, H-7), 4.58 (d, 1H,
3J = 3.9,
H-d); 3.99-3.79 (m, 2H, H-1); 3.66-3.59 (m, 1H, H-h); 3.19-3.09 (m, 1H, H-h); 2.90 (s, 6H, H-q);
2.60-2.53 (m, 2H, H-3); 2.00-1.93 (m, 1H, H-e); 1.83-1.49 (m, 5H, H-e, H-f, H-g, H-2); 1.28-1.19
(m, 2H, H-f, H-g);

EXPERIMENTELLER TEIL 175
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
169.9 (C-c); 155.0 (C-p); 149.2 (C-5); 146.9 (C-6); 141.3 (C-k); 139.4 (C-4); 136.1 (C-i); 135.4
(C-8); 128.7 (C-m); 123.1 (C-7); 122.6 (C-j); 118.9 (C-l); 116.6 (C-n); 63.6 (C-1); 54.3 (C-d); 42.0
(C-h); 35.8 (C-q); 28.8 (C-2); 28.1 (C-3); 26.7 (C-e); 23.5 (C-g); 19.1 (C-f);
6.2.15 Darstellung von BOC-geschützten Aminosäuren 34
6.2.15.1 Synthese von 4-((tert.-Butoxycarbonyl)amino)buttersäure 34a und 3-((tert.-Butoxy-
carbonyl)amino)propionsäure 34b
Allgemeine Vorschrift zur Synthese der Verbindungen 34a und 34b:
1 Äq der entsprechenden Aminosäure (Stoffmenge siehe Tabelle 37) wird unter Argonatmosphäre in
100 ml abs. MeOH gelöst, 50 ml einer 10 % methanolischen Et3N-Lösung langsam zugetropft und
10 min unter Rückfluss erhitzt. Danach werden 2 Äq (49.2-52.8 mmol) (BOC)2O zugegeben und
solange unter Rückfluss erhitzt, bis sich alle Bestandteile gelöst haben. Nach vollständiger
Umsetzung (DC-Kontrolle, siehe Tabelle 37) wird das Lösungsmittel im Vakuum entfernt, der
Rückstand in 10 ml eiskalter HCl (pH 2) gelöst und 10 min gerührt. Anschließend wird die wässrige
Phase viermal mit je 40 ml EtOAc extrahiert, die vereinigten organischen Phasen über Na2SO4
getrocknet und das Lösungsmittel im Vakuum entfernt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
34a143
C9H17NO4 203.24 4-Aminobutter-
säure [24.6]
Cyclohexan : EtOAc =
70:30; Rf = 0.38
98;
(Lit. 95)143
klares Öl;
(Lit. FS)143
34b144
C8H15NO4 189.21 3-Aminopropion-
säure [26.4]
PE : EtOAc = 70:30
(+ 5 % HCOOH);
Rf = 0.75
54;
(Lit. 89)144
klares Öl;
(Lit. FS)144
Tabelle 37: Analytische Daten der Verbindungen 34a-34b
Die 1H-NMR-, und
13C-NMR-Daten zu 34a und 34b entsprechen den Literaturstellen
145,146.

176 EXPERIMENTELLER TEIL
IR [cm-1
]:
34a: 1045; 1163; 1247; 1366; 1522; 1707; 2978; 2837-3492 (b);
34b: 1068; 1163; 1249; 1365; 1509; 1690; 2978; 3199-3580 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
34a: 4.74 (br, 1H, H-d); 3.21-3.10 (m, 2H, H-e); 2.36 (t, 2H, 3J = 7.2, H-g); 1.80 (p, 2H,
3J = 7.2,
H-f); 1.42 (s, 9H, H-a);
34b: 5.22 (br, 1H, H-d); 3.41-3.31 (m, 2H, H-e); 2.48 (t, 2H, 3J = 6.0, H-f); 1.43 (s, 9H, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
34a: 177.4 (C-h); 162.9 (C-c); 79.3 (C-b); 39.8 (C-e); 31.2 (C-g); 28.3 (C-a); 25.2 (C-f);
34b: 176.8 (C-g); 156.0 (C-c); 79.2 (C-b); 36.5 (C-e); 35.3 (C-f); 28.4 (C-a);
6.2.15.2 Synthese von 2-((tert.-Butoxycarbonyl)amino)essigsäure 34c
0.75 g (10.0 mmol) Glycin sowie 0.44 g (11.0 mmol) NaOH-Plätzchen werden in 100 ml dem. H2O
gelöst und eine Lösung von 2.6 g (12.0 mmol) (BOC)2O in 30 ml THF zugetropft. Der Ansatz wird
anschließend in der Mikrowelle in 6 min auf 70 °C erwärmt und 80 min bei dieser Temperatur
gerührt. Nach vollständiger Umsetzung (DC-Kontrolle: Kieselgel; PE : EtOAc = 1: 1; Rf = 0.23
(KMnO4)) wird wässrige 6 M HCl-Lösung bis pH 2.5 zugegeben und 30 min bei RT gerührt. Die
wässrige Phase wird dreimal mit je 50 ml CHCl3 extrahiert, die vereinigten organischen Phasen über
Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Das entstandene Rohprodukt wird
aus CH umkristallisiert und man erhält 1.3 g (7.4 mmol) von Verbindung 34c in Form eines weißen
Feststoffs.
Summenformel: C7H13NO4
Mr = 175.18 g/mol
Smp. = 88 °C (Lit. 89 °C)147
Ausbeute: 74 % (Lit. 77 %)147
Aussehen: weißer Feststoff (Lit. FS)147

EXPERIMENTELLER TEIL 177
Die IR-, 1H-NMR-, und
13C-NMR-Daten zu 34c entsprechen der Literaturstelle
148.
IR [cm-1
]: 1055; 1157; 1196; 1409; 1529; 1666; 1738; 2978; 2773-3446 (b);
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
7.03 (br, 1H, H-d); 3.57 (d, 2H, 3J = 6.0, H-e); 1.38 (s, 9H, H-g);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
171.5 (C-f); 155.5 (C-c); 77.7 (C-b); 41.5 (C-e); 27.9 (C-a);
6.2.16 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)-
sulfonyl)piperidin-2-carbonsäureethylester 35a und 1-((3-(2-((tert.-Butoxy-
carbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäureethyl-
ester 35b
Allgemeine Vorschrift zur Synthese der Verbindungen 35a und 35b:
1 Äq (2.0 mmol) der Verbindung 34b bzw. 34c (siehe Tabelle 38), 1 Äq (2.0 mmol) N-
Hydroxysuccinimid und 1 Äq (2.0 mmol) DCC werden unter Argonatmosphäre in 50 ml abs. CH2Cl2
gelöst und 2 h bei RT gerührt. Anschließend wird eine Lösung von 1 Äq (2.0 mmol) von Verbindung
30a in 20 ml abs. CH2Cl2 zugetropft und 8 h bei RT gerührt. Nach vollständiger Umsetzung (DC-
178 EXPERIMENTELLER TEIL
Kontrolle, siehe Tabelle 38) wird das Lösungsmittel im Vakuum entfernt und der ölige Rückstand in
50 ml EtOAc suspendiert. Der dadurch entstandene Niederschlag wird abfiltriert und das
Lösungsmittel des Filtrats im Vakuum entfernt. Der Vorgang des Fällens und der Filtration wird
solange wiederholt, bis sich kein Niederschlag mehr bildet. Das entstandene Rohprodukt wird
säulenchromatographisch (siehe Tabelle 38) gereinigt.
Summen-
formel Mr
[g/mol] Edukt
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aussehen
35a C22H33N3O7S 483.58 34b Cyclohexan : EtOAc = 1:1;
Rf = 0.43 78 braunes Öl
35b C21H31N3O7S 469.55 34c PE : EtOAc = 60:40;
Rf = 0.31 21 braunes Öl
Tabelle 38: Analytische Daten der Verbindungen 35a-35b
IR [cm-1
]:
35a: 687; 733; 960; 1058; 1150; 1250; 1301; 1478; 1541; 1595; 1685; 2937; 3085; 3347;
35b: 686; 733; 960; 1056; 1149; 1246; 1300; 1479; 1525; 1596; 1683; 2936; 3082; 3348;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
35a: 8.39 (s, 1H, H-o); 7.94 (s, 1H, H-j); 7.88 (d, 1H, 3J = 8.0, H-n); 7.51 (d, 1H,
3J = 8.0, H-l);
7.42 (t, 1H, 3J = 8.0, H-m); 5.51 (br, 1H, H-s); 4.71 (d, 1H,
3J = 4.8, H-d); 4.08-3.89 (m, 2H,
H-b); 3.81-3.73 (m, 1H, H-h); 3.50-3.43 (m, 2H, H-r); 3.26 (dt, 1H, 2J = 12.7,
3J = 3.0, H-h);
2.66 (t, 2H, 3J = 6.1, H-q); 2.18-2.10 (m, 1H, H-e); 1.82-1.55 (m, 3H, H-e, H-f, H-g); 1.43
(s, 9H, H-v); 1.20-1.12 (m, 5H, H-a, H-f, H-g);
35b: 8.38 (s, 1H, H-o); 7.85 (s, 1H, H-j); 7.78 (d, 1H, 3J = 8.0, H-n); 7.51-7.47 (m, 1H, H-l); 7.40
(t, 1H, 3J = 8.0, H-m); 5.17 (br, 1H, H-r); 4.68 (d, 1H,
3J = 4.8, H-d); 4.05-3.87 (m, 4H, H-b,
H-q); 3.77-3.71 (m, 1H, H-h); 3.22 (dt, 1H, 2J = 12.8,
3J = 3.0, H-h); 2.14-2.06 (m, 1H,
H-e); 1.77-1.55 (m, 3H, H-e, H-f, H-g); 1.45 (s, 9H, H-u); 1.30-1.19 (m, 2H, H-f, H-g); 1.11
(t, 3H, 3J = 7.1, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
35a: 172.6 (C-c); 170.6 (C-p); 169.9 (C-t); 140.8 (C-k); 138.7 (C-i); 129.5 (C-m); 123.4 (C-n);
122.4 (C-l); 118.0 (C-j); 79.9 (C-u); 61.1 (C-b); 55.2 (C-d); 42.8 (C-h); 37.8 (C-q); 34.9 (C-
r); 28.4 (C-v); 27.8 (C-e); 24.7 (C-g); 20.0 (C-f); 14.1 (C-a);
35b: 179.3 (C-c); 170.6 (C-p); 168.0 (C-s); 141.0 (C-k); 138.1 (C-i); 133.9 (C-m); 133.4 (C-n);
129.6 (C-l); 123.5 (C-j); 80.3 (C-t); 61.2 (C-b); 55.2 (C-d); 44.6 (C-q); 42.8 (C-h); 28.3
(C-u); 27.8 (C-e); 24.7 (C-g); 20.0 (C-f); 14.1 (C-a);

EXPERIMENTELLER TEIL 179
6.2.17 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)-
sulfonyl)piperidin-2-carbonsäure 36a und 1-((3-(2-((tert.-Butoxycarbonyl)-
amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäure 36b
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.7 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
36a C20H29N3O7S 455.53 35a [1.6] EtOAc;
Rf = 0.16 89 weißes Wachs
36b C19H27N3O7S 441.50 35b [0.4] EtOAc : Et3N = 95:5;
Rf = 0.79 80 braunes Wachs
Tabelle 39: Analytische Daten der Verbindungen 36a-36b
IR [cm-1
]:
36a: 685; 728; 960; 1058; 1149; 1257; 1302; 1478; 1525; 1595; 1679; 2960; 3084; 2811-3562 (b);
36b: 685; 777; 960; 1056; 1150; 1249; 1299; 1478; 1537; 1596; 1680; 2935; 3090; 2857-3478 (b);
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
36a: 12.84 (br, 1H, H-b); 10.21 (s, 1H, H-o); 8.09 (s, 1H, H-j); 7.77 (d, 1H, 3J = 8.0, H-n); 7.47
(t, 1H, 3J = 8.0, H-m); 7.42-7.38 (m, 1H, H-l); 6.85 (br, 1H, H-s); 4.48 (d, 1H,
3J = 4.3,
H-d); 3.62-3.55 (m, 1H, H-h); 3.24-3.15 (m, 3H, H-h, H-r); 2.51-2.44 (m, 2H, H-q); 2.03-
1.91 (m, 1H, H-e); 1.60-1.44 (m, 3H, H-e, H-f, H-g); 1.35 (s, 9H, H-v); 1.22-1.11 (m, 2H,
H-f, H-g);
36b: 12.84 (br, 1H, H-b); 10.22 (s, 1H, H-o); 8.06 (s, 1H, H-j); 7.82-7.75 (m, 1H, H-n); 7.49 (t,
1H, 3J = 7.9, H-m); 7.43-7.41 (m, 1H, H-l); 7.06 (t, 1H,
3J = 6.0, H-r); 4.49 (d, 1H,
3J = 4.3,
H-d); 3.72 (d, 2H, 3J = 6.0, H-q); 3.63-3.56 (m, 1H, H-h); 3.24-3.10 (m, 1H, H-h); 2.00-1.93

180 EXPERIMENTELLER TEIL
(m, 1H, H-e); 1.57-1.45 (m, 3H, H-e, H-f, H-g); 1.38 (s, 9H, H-u); 1.24-1.10 (m, 2H, H-f,
H-g);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
36a: 174.6 (C-c); 171.4 (C-p); 169.6 (C-t); 140.4 (C-k); 139.5 (C-i); 129.3 (C-m); 122.1 (C-n);
120.6 (C-l); 116.4 (C-j); 77.3 (C-u); 54.3 (C-d); 41.9 (C-h); 36.5 (C-q); 36.0 (C-r); 27.9
(C-v); 26.6 (C-e); 23.5 (C-g); 19.3 (C-f);
36b: 171.3 (C-c); 168.5 (C-p); 155.6 (C-s); 140.4 (C-k); 139.2 (C-i); 129.5 (C-m); 122.2 (C-n);
120.8 (C-l); 116.4 (C-j); 77.8 (C-t); 54.4 (C-d); 43.4 (C-q); 41.9 (C-h); 27.9 (C-u); 26.6
(C-e); 23.5 (C-g); 19.3 (C-f);
6.2.18 Synthese von 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)-
sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester 37a und 1-((3-
(2-((tert.-Butoxycarbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-
carbonsäure-(3-(pyridin-3-yl)propyl)ester 37b
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.10 durchgeführt.
EXPERIMENTELLER TEIL 181
Summen-
formel Mr
[g/mol]
Edukte
[mmol] DC bzw. SC
(Kieselgel) Ausbeute
[%] Aussehen
37a C28H38N4O7S 574.69
36a [1.4];
3-(Pyridin-3-yl)-
propan-1-ol [1.6]
Al2O3;
EtOAc : EtOH = 90:10
(+ 5 % Et3N); Rf = 0.74
71 weißes Öl
37b C27H36N4O7S 560.66
36b [0.3];
3-(Pyridin-3-yl)-
propan-1-ol [0.4]
EtOAc;
Rf = 0.18 40
weißes
Wachs
Tabelle 40: Analytische Daten der Verbindungen 37a-37b
IR [cm-1
]:
37a: 687; 733; 966; 1057; 1151; 1245; 1301; 1423; 1478; 1541; 1595; 1691; 2941; 3044; 3329;
37b: 686; 734; 964; 1054; 1151; 1250; 1300; 1424; 1478; 1526; 1595; 1700; 2932; 3065; 3334;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
37a: 10.25 (s, 1H, H-o); 8.40-8.37 (m, 2H, H-5, H-6); 8.12 (s, 1H, H-j); 7.74 (d, 1H, 3J = 8.0,
H-n); 7.60-7.55 (m, 1H, H-8); 7.46 (t, 1H, 3J = 8.0, H-m); 7.42-7.38 (m, 1H, H-l); 7.31-7.27
(m, 1H, H-7); 6.85 (s, 1H, H-s); 4.59 (d, 1H, 3J = 3.9, H-d); 3.97-3.82 (m, 2H, H-1); 3.61 (d,
1H, 2J = 13.3, H-h); 3.23-3.08 (m, 3H, H-h, H-r); 2.56 (t, 2H,
3J = 7.7, H-3); 2.48-2.43 (m,
2H, H-q); 2.00-1.93 (m, 1H, H-e); 1.81-1.71 (m, 2H, H-2); 1.65-1.51 (m, 3H, H-e, H-f,
H-g); 1.34 (s, 9H, H-v); 1.28-1.18 (m, 2H, H-f, H-g);
37b: (CDCl3): 8.61 (s, 1H, H-o); 8.48 (dd, 1H, 3J = 4.8,
4J = 1.5, H-6); 8.41 (d, 1H,
4J = 1.5,
H-5); 7.91 (s, 1H, H-j); 7.77 (d, 1H, 3J = 8.0, H-n); 7.56-7.47 (m, 2H, H-l, H-8); 7.42 (t, 1H,
3J = 8.0, H-m); 7.25-7.22 (m, 1H, H-7); 5.44 (s, 1H, H-r); 4.73 (d, 1H,
3J = 4.2, H-d); 4.10-
4.02 (m, 1H, H-1); 3.96-3.89 (m, 3H, H-q, H-1); 3.79 (d, 1H, 2J = 12.8, H-h); 3.24 (dt, 1H,
2J = 12.8,
3J = 3.0, H-h); 2.64 (t, 2H,
3J = 7.6, H-3); 2.18-2.10 (m, 1H, H-e); 1.90-1.60 (m,
5H, H-2, H-e, H-f, H-g); 1.49 (s, 9H, H-u); 1.35-1.28 (m, 2H, H-f, H-g);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
37a: 169.8 (C-c); 169.6 (C-p); 161.0 (C-t); 149.2 (C-5); 147.0 (C-6); 139.9 (C-k); 139.6 (C-4);
136.1 (C-i); 135.5 (C-8); 129.3 (C-m); 123.1 (C-7); 122.4 (C-n); 120.7 (C-l); 116.5 (C-j);
71.3 (C-u); 63.6 (C-1); 54.4 (C-d); 42.1 (C-h); 38.4 (C-q); 38.1 (C-r); 28.9 (C-2); 28.1
(C-3); 27.9 (C-v); 26.8 (C-e); 23.6 (C-g); 22.8 (C-f);
37b: (CDCl3): 175.2 (C-c); 170.7 (C-p); 168.1 (C-s); 149.7 (C-5); 147.7 (C-6); 141.0 (C-k); 138.4
(C-4); 136.3 (C-i); 136.1 (C-8); 129.6 (C-m); 124.4 (C-7); 123.6 (C-n); 122.7 (C-l); 118.2
(C-j); 83.1 (C-t); 64.0 (C-1); 55.2 (C-d); 44.9 (C-q); 42.8 (C-h); 29.7 (C-2); 29.2 (C-3); 28.3
(C-u); 27.8 (C-e); 24.7 (C-g); 20.1 (C-f);
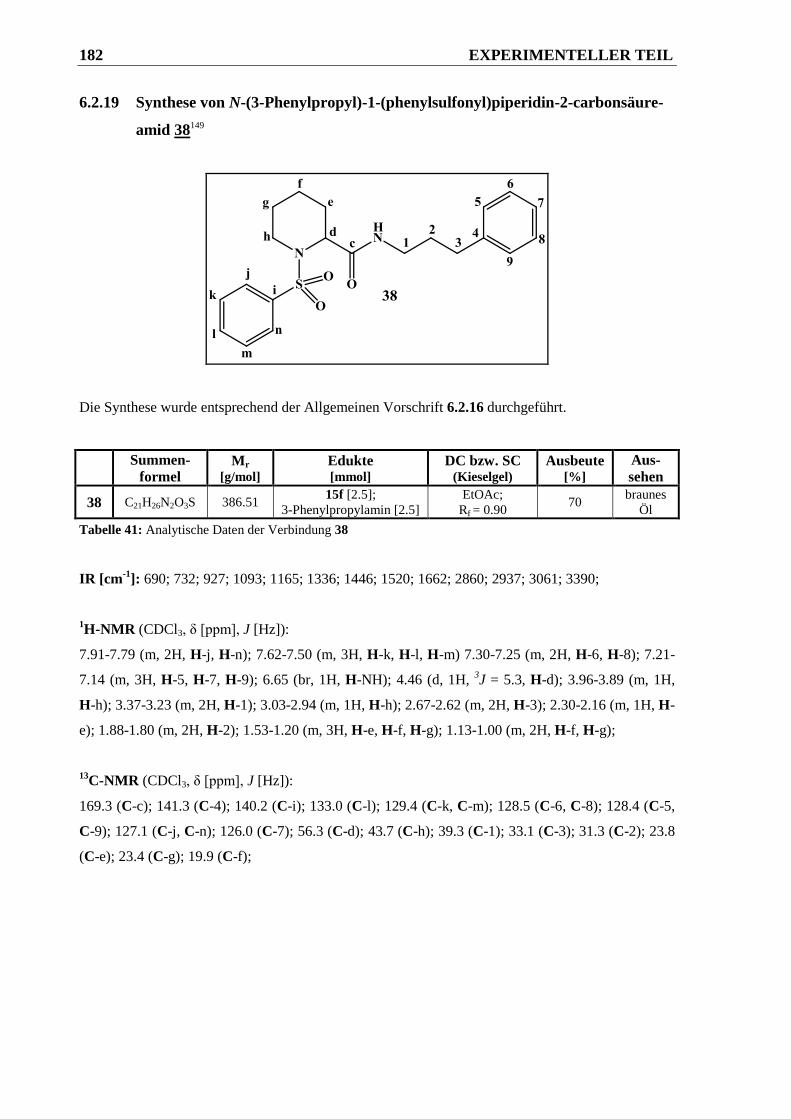
182 EXPERIMENTELLER TEIL
6.2.19 Synthese von N-(3-Phenylpropyl)-1-(phenylsulfonyl)piperidin-2-carbonsäure-
amid 38149
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.16 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
38 C21H26N2O3S 386.51 15f [2.5];
3-Phenylpropylamin [2.5]
EtOAc;
Rf = 0.90 70
braunes
Öl
Tabelle 41: Analytische Daten der Verbindung 38
IR [cm-1
]: 690; 732; 927; 1093; 1165; 1336; 1446; 1520; 1662; 2860; 2937; 3061; 3390;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.91-7.79 (m, 2H, H-j, H-n); 7.62-7.50 (m, 3H, H-k, H-l, H-m) 7.30-7.25 (m, 2H, H-6, H-8); 7.21-
7.14 (m, 3H, H-5, H-7, H-9); 6.65 (br, 1H, H-NH); 4.46 (d, 1H, 3J = 5.3, H-d); 3.96-3.89 (m, 1H,
H-h); 3.37-3.23 (m, 2H, H-1); 3.03-2.94 (m, 1H, H-h); 2.67-2.62 (m, 2H, H-3); 2.30-2.16 (m, 1H, H-
e); 1.88-1.80 (m, 2H, H-2); 1.53-1.20 (m, 3H, H-e, H-f, H-g); 1.13-1.00 (m, 2H, H-f, H-g);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
169.3 (C-c); 141.3 (C-4); 140.2 (C-i); 133.0 (C-l); 129.4 (C-k, C-m); 128.5 (C-6, C-8); 128.4 (C-5,
C-9); 127.1 (C-j, C-n); 126.0 (C-7); 56.3 (C-d); 43.7 (C-h); 39.3 (C-1); 33.1 (C-3); 31.3 (C-2); 23.8
(C-e); 23.4 (C-g); 19.9 (C-f);
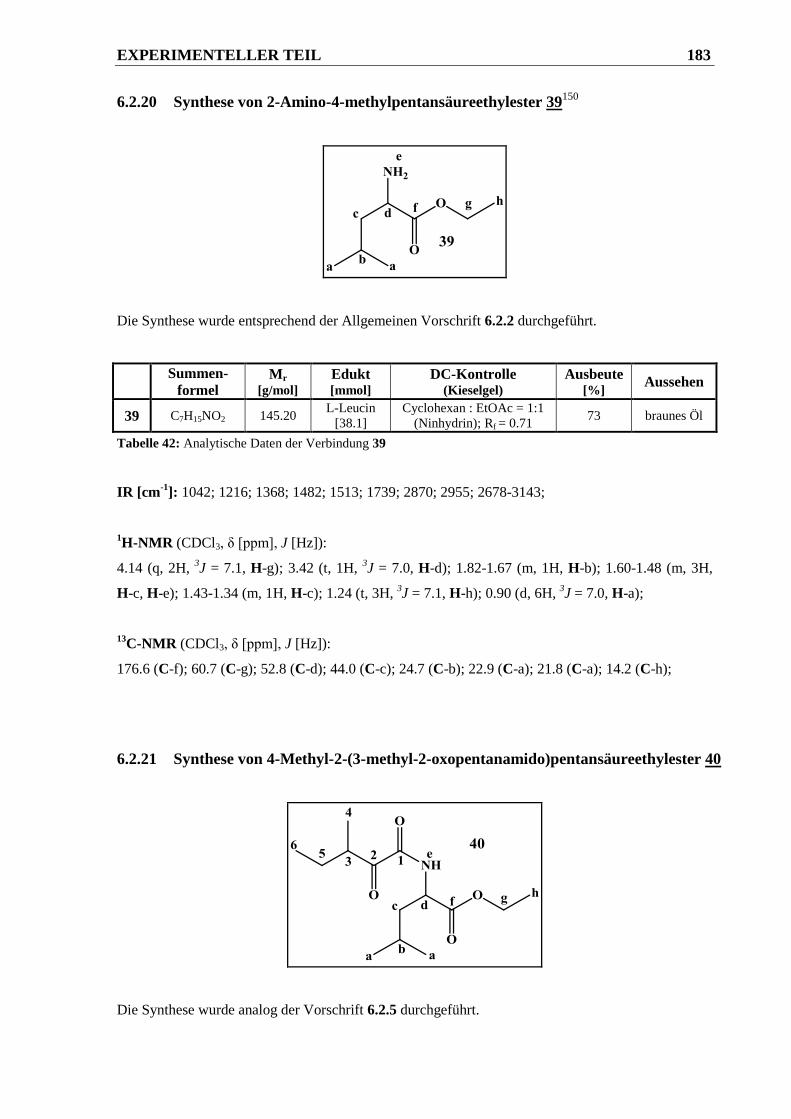
EXPERIMENTELLER TEIL 183
6.2.20 Synthese von 2-Amino-4-methylpentansäureethylester 39150
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.2 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
39 C7H15NO2 145.20 L-Leucin
[38.1]
Cyclohexan : EtOAc = 1:1
(Ninhydrin); Rf = 0.71 73 braunes Öl
Tabelle 42: Analytische Daten der Verbindung 39
IR [cm-1
]: 1042; 1216; 1368; 1482; 1513; 1739; 2870; 2955; 2678-3143;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
4.14 (q, 2H, 3J = 7.1, H-g); 3.42 (t, 1H,
3J = 7.0, H-d); 1.82-1.67 (m, 1H, H-b); 1.60-1.48 (m, 3H,
H-c, H-e); 1.43-1.34 (m, 1H, H-c); 1.24 (t, 3H, 3J = 7.1, H-h); 0.90 (d, 6H,
3J = 7.0, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
176.6 (C-f); 60.7 (C-g); 52.8 (C-d); 44.0 (C-c); 24.7 (C-b); 22.9 (C-a); 21.8 (C-a); 14.2 (C-h);
6.2.21 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäureethylester 40
Die Synthese wurde analog der Vorschrift 6.2.5 durchgeführt.

184 EXPERIMENTELLER TEIL
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
40 C13H23NO4 257.33 9 [10.0];
39 [10.0]
Cyclohexan : EtOAc = 70:30;
Rf = 0.78 77
braunes
Öl
Tabelle 43: Analytische Daten der Verbindung 40
IR [cm-1
]: 1031; 1150; 1197; 1463; 1527; 1650; 1740; 2873; 2961; 3315;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.95 (br, 1H, H-e); 4.54-4.45 (m, 1H, H-d); 4.13 (q, 2H, 3J = 7.3, H-g); 3.45-3.34 (m, 1H, H-3);
1.78-1.48 (m, 4H, H-b, H-c, H-5); 1.42-1.26 (m, 1H, H-5); 1.21 (t, 3H, 3J = 7.3, H-h); 1.08-1.02 (m,
3H, H-4); 0.90-0.80 (m, 9H, H-a, H-6);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
201.5 (C-2); 171.9 (C-f); 159.7 (C-1); 61.5 (C-g); 50.8 (C-d); 41.4 (C-3); 40.4 (C-c); 25.4 (C-5);
24.8 (C-b); 22.7 (C-a); 21.8 (C-a); 15.0 (C-4); 14.1 (C-h); 11.4 (C-6);
6.2.22 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure 41151
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.7 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aussehen
41 C12H21NO4 243.30 40 [6.0] Cyclohexan : EtOAc = 1:1;
Rf = 0.53 87 gelbes Öl
Tabelle 44: Analytische Daten der Verbindung 41
IR [cm-1
]: 1150; 1237; 1462; 1532; 1634; 1719; 2874; 2961; 2858-3323 (b);
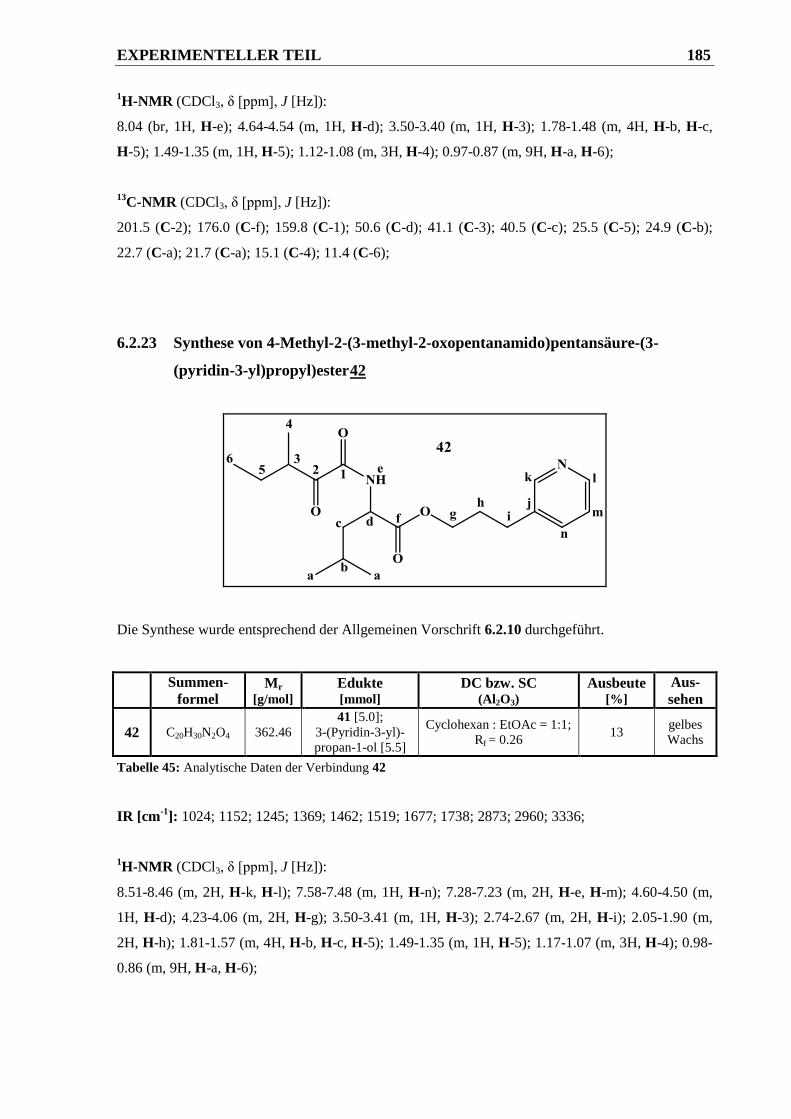
EXPERIMENTELLER TEIL 185
1H-NMR (CDCl3, δ [ppm], J [Hz]):
8.04 (br, 1H, H-e); 4.64-4.54 (m, 1H, H-d); 3.50-3.40 (m, 1H, H-3); 1.78-1.48 (m, 4H, H-b, H-c,
H-5); 1.49-1.35 (m, 1H, H-5); 1.12-1.08 (m, 3H, H-4); 0.97-0.87 (m, 9H, H-a, H-6);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
201.5 (C-2); 176.0 (C-f); 159.8 (C-1); 50.6 (C-d); 41.1 (C-3); 40.5 (C-c); 25.5 (C-5); 24.9 (C-b);
22.7 (C-a); 21.7 (C-a); 15.1 (C-4); 11.4 (C-6);
6.2.23 Synthese von 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure-(3-
(pyridin-3-yl)propyl)ester 42
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.10 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Al2O3)
Ausbeute [%]
Aus-
sehen
42 C20H30N2O4 362.46
41 [5.0];
3-(Pyridin-3-yl)-
propan-1-ol [5.5]
Cyclohexan : EtOAc = 1:1;
Rf = 0.26 13
gelbes
Wachs
Tabelle 45: Analytische Daten der Verbindung 42
IR [cm-1
]: 1024; 1152; 1245; 1369; 1462; 1519; 1677; 1738; 2873; 2960; 3336;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
8.51-8.46 (m, 2H, H-k, H-l); 7.58-7.48 (m, 1H, H-n); 7.28-7.23 (m, 2H, H-e, H-m); 4.60-4.50 (m,
1H, H-d); 4.23-4.06 (m, 2H, H-g); 3.50-3.41 (m, 1H, H-3); 2.74-2.67 (m, 2H, H-i); 2.05-1.90 (m,
2H, H-h); 1.81-1.57 (m, 4H, H-b, H-c, H-5); 1.49-1.35 (m, 1H, H-5); 1.17-1.07 (m, 3H, H-4); 0.98-
0.86 (m, 9H, H-a, H-6);

186 EXPERIMENTELLER TEIL
13C-NMR (CDCl3, δ [ppm], J [Hz]):
201.5 (C-2); 171.9 (C-f); 159.7 (C-1); 149.4 (C-k); 147.3 (C-l); 136.4 (C-j); 136.2 (C-n); 123.6
(C-m); 64.4 (C-g); 50.9 (C-d); 41.4 (C-3); 40.5 (C-c); 29.8 (C-h); 29.3 (C-i); 25.4 (C-5); 24.9 (C-b);
22.7 (C-a); 21.8 (C-a); 15.1 (C-4); 11.5 (C-6);
6.2.24 Synthese von Pyrrolidin-2-carbonsäureethylester 43152
Die Synthese wurde entsprechend der Allgemeinen Vorschrift 6.2.2 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukt [mmol]
DC-Kontrolle (Kieselgel)
Ausbeute [%]
Aus-
sehen
43 C7H13NO2 143.18 Pyrrolidin-2-
carbonsäure [8.7]
CHCl3 : MeOH = 1:1
(Ninhydrin); Rf = 0.56
73;
(Lit. 71)153
klares Öl;
(Lit. Öl)153
Tabelle 46: Analytische Daten der Verbindung 43
Die 1H-NMR-Daten zu 43 entsprechen der Literaturstelle
153.
IR [cm-1
]: 1025; 1093; 1258; 1430; 1663; 1730; 2873; 2963; 3312;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
3.90 (q, 2H, 3J = 7.1, H-g); 3.49-3.41 (m, 1H, H-e); 2.83-2.75 (m, 1H, H-b); 2.66-2.58 (m, 1H, H-b);
2.52 (br, 1H, H-a); 1.89-1.79 (m, 1H, H-d); 1.64-1.41 (m, 3H, H-c, H-d); 1.00 (t, 3H, 3J = 7.1, H-h);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
174.7 (C-f); 60.2 (C-g); 59.1 (C-e); 46.4 (C-b); 29.6 (C-d); 24.9 (C-c); 13.6 (C-h);
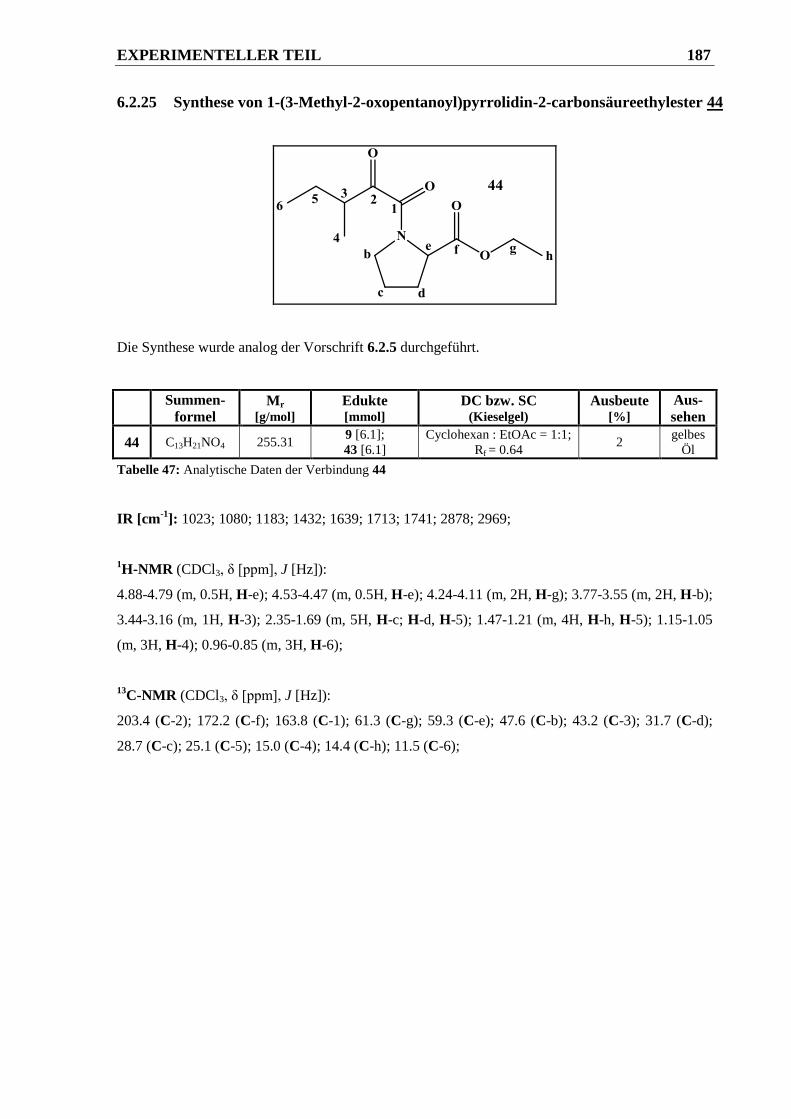
EXPERIMENTELLER TEIL 187
6.2.25 Synthese von 1-(3-Methyl-2-oxopentanoyl)pyrrolidin-2-carbonsäureethylester 44
Die Synthese wurde analog der Vorschrift 6.2.5 durchgeführt.
Summen-
formel Mr
[g/mol]
Edukte [mmol]
DC bzw. SC (Kieselgel)
Ausbeute [%]
Aus-
sehen
44 C13H21NO4 255.31 9 [6.1];
43 [6.1]
Cyclohexan : EtOAc = 1:1;
Rf = 0.64 2
gelbes
Öl
Tabelle 47: Analytische Daten der Verbindung 44
IR [cm-1
]: 1023; 1080; 1183; 1432; 1639; 1713; 1741; 2878; 2969;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
4.88-4.79 (m, 0.5H, H-e); 4.53-4.47 (m, 0.5H, H-e); 4.24-4.11 (m, 2H, H-g); 3.77-3.55 (m, 2H, H-b);
3.44-3.16 (m, 1H, H-3); 2.35-1.69 (m, 5H, H-c; H-d, H-5); 1.47-1.21 (m, 4H, H-h, H-5); 1.15-1.05
(m, 3H, H-4); 0.96-0.85 (m, 3H, H-6);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
203.4 (C-2); 172.2 (C-f); 163.8 (C-1); 61.3 (C-g); 59.3 (C-e); 47.6 (C-b); 43.2 (C-3); 31.7 (C-d);
28.7 (C-c); 25.1 (C-5); 15.0 (C-4); 14.4 (C-h); 11.5 (C-6);

188 EXPERIMENTELLER TEIL
6.2.26 Synthese von 2-(4-Isopropylphenoxy)essigsäure 45154
1.0 g (7.3 mmol) p-Isopropylphenol werden in 30 ml 10.5 M methanolischer NaOH gelöst, auf 80 °C
erhitzt und 0.7 g (7.3 mmol) Chloressigsäure zugetropft. Anschließend wird die Reaktionslösung 2 h
bei 80 °C gerührt und 12 h bei RT stehen gelassen. Nach vollständiger Umsetzung (DC-Kontrolle:
Kieselgel; PE : EtOAc = 1 : 1 (+ 5 % HCOOH); Rf = 0.25) werden 30 ml dem. H2O und danach
konz. HCl bis pH 2 zugegeben. Die entstandene freie Säure wird viermal mit je 30 ml EtOAc
extrahiert, die vereinigten organischen Phasen viermal mit je 30 ml 10 % wässriger Na2CO3-Lösung
gewaschen, über Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält 1.4 g
(7.2 mmol) von Verbindung 45 in Form eines braunen Öls.
Summenformel: C11H14O3
Mr = 194.23 g/mol
Ausbeute: 98 %
Aussehen: braunes Öl
IR [cm-1
]: 689; 786; 1120; 1197; 1454; 1513; 1738; 2854; 2924; 2867-3425 (b);
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.11-7.07 (m, 2H, H-d, H-f); 6.79-6.74 (m, 2H, H-e, H-g); 5.88 (br, 1H, H-k); 3.49 (s, 2H, H-i); 2.85
(sept., 1H, 3J= 6.9, H-b); 1.22 (d, 6H,
3J= 6.9, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
174.1 (C-j); 153.4 (C-h); 141.2 (C-c); 127.4 (C-d, C-f); 115.1 (C-e, C-g); 69.2 (C-i); 33.2 (C-b);
24.2 (C-a);

EXPERIMENTELLER TEIL 189
6.2.27 Synthese von 2-(4-Isopropylphenoxy)-1-morpholinessigsäureamid 46155
0.86 g (4.4 mmol) der Verbindung 45 werden in 30 ml DMF gelöst, nacheinander langsam 0.4 g
(4.4 mmol) Morpholin, 1.34 g (13.3 mmol) NMM und 2.8 g (8.8 mmol) PPA zugetropft und
2 Wochen bei RT gerührt. Nach vollständiger Umsetzung (DC-Kontrolle: Kieselgel; Cyclohexan :
EtOAc = 1:1; Rf = 0.40) wird das Lösungsmittel im Vakuum entfernt, der ölige Rückstand in 50 ml
EtOAc suspendiert und dreimal mit je 30 ml wässriger 2 M HCl-Lösung, dreimal mit je 30 ml dem.
H2O und dreimal mit je 30ml ges. wässriger NaHCO3-Lösung gewaschen. Die organische Phase wird
über Na2SO4 getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält 0.4 g (1.6 mmol)
von Verbindung 46 in Form eines braunen Öls.
Summenformel: C15H21NO3
Mr = 263.33 g/mol
Ausbeute: 36 %
Aussehen: braunes Öl
IR [cm-1
]: 689; 785; 1026; 1112; 1222; 1433; 1509; 1657; 2895; 2959;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
7.15-7.12 (m, 2H, H-d, H-f); 6.87-6.84 (m, 2H, H-e, H-g); 4.65 (s, 2H, H-i); 3.67-3.56 (m, 8H, H-k,
H-l, H-m, H-n); 2.85 (sept., 1H, 3J= 6.9, H-b); 1.21 (d, 6H,
3J= 6.9, H-a);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
166.8 (C-j); 155.7 (C-h); 142.2 (C-c); 127.4 (C-d, C-f); 114.3 (C-e, C-g); 67.8 (C-i); 66.7 (C-l,
C-m); 45.9 (C-k, C-n); 33.2 (C-b); 24.1 (C-a);

190 EXPERIMENTELLER TEIL
6.2.28 Synthese von 5-Methyl-2-phenyl-1H-pyrazol-3(2H)-on 47156
2.0 g (15.4 mmol) Acetessigsäureethylester werden unter Argonatmosphäre in 50 ml abs. EtOH
gelöst, 1.66 g (15.0 mmol) Phenylhydrazin zugegeben, 3 h unter Rückfluss erhitzt und anschließend
12 h bei RT stehen gelassen. Nach vollständiger Umsetzung (DC-Kontrolle: Kieselgel; Cyclohexan :
EtOAc = 1 : 1; Rf = 0.37) wird das Lösungsmittel im Vakuum entfernt und das Rohprodukt aus n-
Pentan umkristallisiert. Man erhält 1.7 g (9.8 mmol) von Verbindung 47 in Form eines gelben
Pulvers.
Summenformel: C10H10N2O
Mr = 174.2 g/mol
Smp. = 130 °C (Lit. 126 °C)157
Ausbeute: 65 % (Lit. 88 %)157
Aussehen: gelbes Pulver (Lit. FS)157
Die IR-, 1H-NMR-, und
13C-NMR-Daten zu 47 entsprechen der Literaturstelle
157.
IR [cm-1
]: 692; 754; 1012; 1092; 1157; 1321; 1446; 2924; 3077; 3261;
1H-NMR (DMSO-d6, δ [ppm], J [Hz]):
11.42 (s, 1H, H-e); 7.70 (d, 2H, 3J = 7.9, H-g, H-k); 7.41 (t, 2H,
3J = 7.9, H-h, H-j); 7.20 (t, 1H,
3J = 7.9, H-i); 5.36 (s, 1H, H-c); 2.11 (s, 3H, H-a);
13C-NMR (DMSO-d6, δ [ppm], J [Hz]):
166.1 (C-d); 157.4 (C-b); 128.7 (C-h, C-j); 124.9 (C-i); 120.5 (C-g, C-k); 117.9 (C-f); 87.5 (C-c);
14.2 (C-a);

EXPERIMENTELLER TEIL 191
6.2.29 Synthese von 2-Amino-4-(methylsulfinyl)buttersäureethylester 48
0.5 g (3.0 mmol) 2-Amino-4-(methylsulfinyl)buttersäure werden unter Argonatmosphäre in 20 ml
abs. EtOH suspendiert, 1.08 g (9.09 mmol) SOCl2 langsam zugetropft, 3 h unter Rückfluss erhitzt
und anschließend 12 h bei RT gerührt. Nach vollständiger Umsetzung (DC-Kontrolle: Kieselgel;
EtOAc : Et3N = 95 : 5; Rf-Wert = 0.35) wird das Lösungsmittel im Vakuum entfernt, der ölige
Rückstand in 30 ml ges. wässriger NaHCO3-Lösung gelöst und viermal mit je 20 ml CHCl3
extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet und das
Lösungsmittel anschließend im Vakuum entfernt. Man erhält 0.2 g (1.0 mmol) von Verbindung 48 in
Form eines gelben Öls.
Summenformel: C7H15NO3S
Mr = 193.26 g/mol
Ausbeute: 35 %
Aussehen: gelbes Öl
IR [cm-1
]: 1057; 1335; 1441; 1674; 2917; 2974; 2805-3023;
1H-NMR (CDCl3, δ [ppm], J [Hz]):
4.17 (q, 2H, 3J = 7.0, H-g); 3.60-3.52 (m, 1H, H-d); 2.61 (t, 2H,
3J = 7.3, H-b); 2.11-1.99 (m, 4H,
H-a, H-c); 1.88-1.71 (m, 1H, H-c); 1.59 (s, 2H, H-e); 1.27 (t, 3H, 3J = 7.0, H-h);
13C-NMR (CDCl3, δ [ppm], J [Hz]):
175.7 (C-f); 61.0 (C-g); 53.4 (C-d); 34.0 (C-c); 30.5 (C-b); 15.4 (C-a); 14.2 (C-h);

192 EXPERIMENTELLER TEIL
6.3 Expression von Mip
6.3.1 Verwendete Geräte
Rotationsschüttler: Innova 4330 Incubator Shaker
New Brunswick Scientific Co., Edison, USA
Zentrifuge: Sorvall RC5B Plus Zentrifuge
Thermo Fisher Scientific, Schwerte, Deutschland
French-Press: Hochdruckzellaufschlussapparatur Modell TS 0.75 KW
Constan Systems Ltd., Daventry, UK
Ultrazentrifuge: Ultrazentrifuge Rotor 45Ti
Beckman Coulter GmbH, Krefeld, Deutschland
6.3.2 Klonierung und Transformation des Mip-Gens
Die Isolation und die Klonierung der Mip-DNA-Sequenz aus L. pneumophila-Stamm Philadelphia I,
sowie seine Transformation in E. coli-K12-Stamm HB101 wurden am Max-Planck-Institut für
Enzymologie der Proteinfaltung in Halle durchgeführt und die für die nachfolgende Expression
benötigten E. coli-Zellen zur Verfügung gestellt.
6.3.3 Anzucht von E. coli-Bakterien
Herstellung des LB-Mediums:
16.0 g Pepton, 10.0 g Hefeextrakt und 5.0 g NaCl werden unter Rühren in 1.0 L dem. H2O gelöst, mit
1 M HCl bzw. 1 M NaOH auf pH 7 eingestellt und 20 min in heißem gesättigtem Wasserdampf bei
121 °C autoklaviert. Insgesamt werden 6 x 1.0 L LB-Medium hergestellt, so dass schließlich 6
einzelne Behältnisse für die anschließende Kultivierung zur Verfügung stehen.
Herstellung der Vorkulturen:
Aus allen 6 LB-Medien werden jeweils 5.0 ml entnommen und mit 5.0 µl Ampicillin (100 µg/ml)
sowie einer E. coli-K12-HB101-Kultur versehen. Diese Vorkulturen werden 12 h im Rotations-
schüttler bei 150 rpm und 37 °C inkubiert.

EXPERIMENTELLER TEIL 193
Kultivierung der E. coli-K12-HB101-Bakterien:
Zeigen alle Proben ein Bakterienwachstum, wird jeweils eine dieser Vorkulturen (5.0 ml) sowie
1.0 ml Ampicillin (100 µg/ml) in 1.0 L LB-Medium gegeben und anschließend 24 h im
Rotationsschüttler bei 150 rpm und 37 °C inkubiert. Danach werden die gewachsenen Zellen 10 min
bei 5.000 rpm in Zentrifugengefäßen pelletiert und das Filtrat verworfen.
6.3.4 Aufschluss der E. coli-K12-HB101-Zellkulturen
Herstellung des 2 mM Trizin-Puffers pH 8.5:
1.08 g N-Tris(hydroxymethyl)methylglycin (Trizin) werden unter Rühren in 3.0 L dem. H2O gelöst
und mit 1 M HCl bzw. 1 M NaOH auf pH 8.5 eingestellt.
Mechanische Lyse:
Die Zellpellets werden auf Eiskühlung in 2 mM Trizin-Puffer pH 8.5 suspendiert und 50.0 µl
Phenylmethylsulfonylfluorid sowie 100.0 µl Proteaseinhibitor (Cocktail von Roche) zugegeben.
Diese Suspension wird in den vorgekühlten Stahlzylinder der French-press-Anlage gefüllt und ein
Druck von 1200 bar angelegt. Unter diesem Druck werden die Bakterienzellen aufgeschlossen und
die flüssige Phase anschließend 45 min bei 100.000 rpm und 4 °C ultrazentrifugiert. Der pelletierte
Rückstand wird verworfen und die flüssige Phase einer anschließenden Reinigung zugeführt.
6.3.5 Säulenchromatographische Reinigung von Mip
Herstellung des 2mM Trizin-Puffers pH 8.5:
Siehe Kapitel 6.3.4
Herstellung der 35 %igen Ammoniumsulfat-Lösung:
350.0 g (NH4)2SO4 werden unter Rühren in 1.0 L dem. Wasser gelöst.
Säulenchromatographische Reinigung:
Die gesamte Reinigung wird bei einer Temperatur von 4 °C durchgeführt.

194 EXPERIMENTELLER TEIL
1. Säule: Fractogel-DEAE (Diethylaminoethyl):
Der flüssige Rückstand der Ultrazentrifugation wird mit 1 M HCl bzw. 1 M NaOH auf pH 8.5 ein-
gestellt, auf eine mit 2 mM Trizin-Puffer pH 8.5 equilibrierte DEAE-Säule (Fractogel®) gegeben und
anschließend mit 2 mM Trizin-Puffer pH 8.5 eluiert. Das gewünschte Mip befindet sich im Eluat.
2. Säule: Fractogel-TMAE (Trimethylammoniumethyl):
Der flüssige Rückstand der ersten Säule wird auf eine mit 2 mM Trizin-Puffer pH 8.5 equilibrierte
TMAE-Säule (Fractogel®) gegeben und anschließend mit 2 mM Trizin-Puffer pH 8.5 eluiert. Das
gewünschte Mip befindet sich im Eluat.
3. Säule: Affigel-Blue:
Der flüssige Rückstand der zweiten Säule wird auf eine mit 2 mM Trizin-Puffer pH 8.5 equilibrierte
Affigel-Blue-Säule gegeben und anschließend mit 2 mM Trizin-Puffer pH 8.5 gewaschen. Das Eluat
wird gesammelt und das an die Säule gebundene Mip anschließend zweimal mit 100.0 ml eines KCl-
Gradienten (0-2 M) in Fraktionen eluiert. Die einzelnen Fraktionen dieses zweiten Durchlaufs
werden zusammen mit dem ersten Eluat der dritten Säule mittels SDS-Page-Gelelektrophorese auf
anwesendes Mip untersucht.
4. Säule: Fractogel-Propyl:
Die Protein-enthaltenen Fraktionen werden vereinigt, in 35 %iger Ammoniumsulfat-Lösung
suspendiert, auf eine mit 2 mM Trizin-Puffer pH 8.5 und mit 35 %iger Ammoniumsulfat-Lösung
equilibrierte Fractogel Propyl-Säule gegeben und anschließend mit 35 %iger Ammoniumsulfat-
Lösung gewaschen. Das Eluat wird verworfen und das gebundene Mip anschließend zweimal mit
40.0 ml eines Ammoniumsulfat-Gradienten (0-35 %) in Fraktionen von der Säule eluiert. Die
einzelnen Fraktionen des zweiten Durchlaufs werden mittels SDS-Page-Gelelektrophorese auf
anwesendes Mip untersucht.
Natriumdodecylsulfat-Polyacrylamidgelelektrophorese (SDS-PAGE) (nach Lämmli 1970)158
:
Das Elektrophoresegel auf Polyacrylamidbasis (bestehend aus Trenn- und Sammelgel) wurde nach
einer allgemeinen Vorschrift am Max-Planck-Institut hergestellt und für diese Arbeit zur Verfügung
gestellt.159
Die zu untersuchenden Fraktionen, der Molekularmarker (M) und ggf. der Durchlauf (D)
werden in SDS-haltigem Probenpuffer† gelöst und mit dem Farbstoff Bromphenolblau versetzt. Das
Polyacrylamidgel wird in die Elektrophoresekammer eingesetzt und mit den gefärbten
Untersuchungslösungen beladen. Als Elektrolysepuffer wird ein SDS-haltiges Tris-Glycin-
Puffersystem‡ verwendet. Der Elektrophoreselauf erfolgt bei einer Stromstärke von 25 mA. Das
† SDS-Probenpuffer (pH 6.8): 78.0 mM Tris; 11.5 g/L SDS; 0.25 % (v/v) ß-Mercaptoethanol; 2.0 % (v/v)
Glycin; ‡ SDS-Laufpuffer (pH 8.8): 25.0 mM Tris; 192.0 mM Glycin; 3.5 mM SDS;

EXPERIMENTELLER TEIL 195
entnommene Elektrophoresegel wird mit Coomassie-Brilliant-Blau-Färbelösung§ für 12 h inkubiert.
Nach vollständiger Färbung wird das Gel anschließend durch Einlegen in eine Entfärbelösung**
entfärbt. Dieser Vorgang wird so oft wiederholt, bis der Gelhintergrund klar ist und alle
Proteinbanden eindeutig sichtbar sind. Der Vergleich der Banden der einzelnen Fraktionen mit den
Banden des Proteinmarkers identifiziert die Proben, die Mip enthalten.
6.3.6 Dialyse von gereinigtem Mip
Herstellung des 35 mM HEPES-Puffers:
25.0 g 2-(4-(2-Hydroxyethyl)-1-piperazinyl)ethansulfonsäure (HEPES) werden unter Rühren in 3.0 L
dem. H2O gelöst und mit 1 M HCl bzw. 1 M NaOH auf pH 7.8 eingestellt.
Dialyse:
Die Protein-enthaltenen Fraktionen werden vereinigt und in einen dünnwandigen Dialyseschlauch
gefüllt. Anschließend wird der Schlauch in 3.0 L 35 mM HEPES-Puffer für 12 h eingelegt und das
Protein aufkonzentriert.
6.4 Enzymatischer Assay
6.4.1 Verwendete Geräte
Temperaturschrank: Serie MKKL (+ 2 - + 45 °C)
Flohr Instruments, Utrecht, Holland
Spektrometer: Appliscan, 96-Well-Plate-Reader
Thermo Fisher Scientific, Schwerte, Deutschland
Mikrotiter-Platten: MicroWellTM
96-Well-Platten
Thermo Fisher Scientific, Schwerte, Deutschland
§ Coomassie-Färbelösung: 0.2 % (w/v) Coomassie R250; 0.05 % (w/v) Coomassie G250; 10.0 % (v/v)
Essigsäure; 30.0 % (v/v) Methanol; 17.5 % (v/v) Ethanol; **
Entfärbelösung: 45.0 % (v/v) Methanol; 10.0 % (v/v) Essigsäure;

196 EXPERIMENTELLER TEIL
6.4.2 Herstellung der Puffer- bzw. Stammlösungen
Herstellung des 10 mM HEPES-Puffers:
2.38 g HEPES und 5.84 g NaCl werden unter Rühren in 1.0 L dem. H2O gelöst und mit 1 M HCl
bzw. 1 M NaOH auf pH 7.8 eingestellt.
Herstellung der Substrat-Stammlösung:
0.01 g synthetisches Peptid werden in 0.1 ml DMSO gelöst und mit 10 mM HEPES-Puffer 1 : 600
verdünnt, so dass eine Substrat-Stammlösung mit einer Endkonzentration von 0.24 mM entsteht.
Herstellung der α-Chymotrypsin-Stammlösung:
0.01 g α-Chymotrypsin werden in 1.0 ml 10 mM HEPES-Puffer gelöst und mit demselben Puffer
1 : 50 verdünnt.
Herstellung der Mip-Stammlösung:
Die Konzentration des gereinigten Proteins wurde UV-spektrometrisch über eine Differenzmessung
der Absorptionen bei 280 nm und 310 nm bestimmt und beträgt 40.6 µM. Davon wird ein Aliquot
von 10.0 µl entnommen und mit 10 mM HEPES-Puffer so verdünnt, dass eine Mip-Stammlösung mit
einer Endkonzentration von 600 nM entsteht.
Herstellung der Inhibitor-Stammlösung:
Von jeder zu untersuchenden Substanz werden 500.0 µl einer 10 mM Stammlösung in EtOH p.a.
hergestellt. Davon werden vier Aliquoten von 10.0 µl entnommen und diese jeweils so verdünnt, dass
Lösungen mit den Endkonzentrationen 30 µM, 150 µM, 300 µM und 600 µM entstehen.
6.4.3 Protease-gekoppelter PPIase-Assay
Der PPIase-Assay wird in einem Temperaturbereich von 9-11 °C durchgeführt. Die Substanzen
werden nach folgendem Pipettierschema in die 96-Well-Platte vorgelegt:
Substrat [240 µM]
Puffer [10 mM]
Mip [0.6 µM]
Inhibitor [30, 150, 300, 600 µM]
Nullwert 100 µl 50 µl - -
Kontrolle 100 µl 30 µl 20 µl -
Probe 100 µl 20 µl 20 µl 10 µl
Tabelle 48: Pipettierschema für die Durchführung des PPIase-Assays

EXPERIMENTELLER TEIL 197
Von jedem Inhibitor, dem Nullwert sowie der Kontrolle wird eine Dreifachbestimmung
durchgeführt. Unmittelbar vor der Messung werden je 150.0 µl der α-Chymotrypsinlösung durch das
Spektrometer in jede Kavität zugegeben, so dass jeweils 300.0 µl Untersuchungslösung mit den
folgenden Endkonzentrationen der einzelnen Komponenten entsteht: Substrat: 80 µM, Mip: 40 nM,
Inhibitor: 1 µM, 5µM, 10 µM, 20 µM. Die Reaktion startet mit der α-Chymotrypsin-Zugabe und es
wird über einen Zeitraum von 200 s alle drei Sekunden die Absorption bei 390 nm bestimmt.

198 EXPERIMENTELLER TEIL

ANHANG 199
7 ANHANG
7.1 Überblick synthetisierter Verbindungen
1a N,N-Dimethylphenylsulfonsäureamid
1b 1-(Phenylsulfonyl)piperidin
1c 1-(Phenylsulfonyl)pyrrolidin
1d N-(Phenethyl)phenylsulfonsäureamid
1e (S)-N-(1-Phenylethyl)phenylsulfonsäureamid
1f N-(Prop-2-yn-1-yl)phenylsulfonsäureamid
1g N-Propylphenylsulfonsäureamid
1h N-Ethylphenylsulfonsäureamid
1i N-Isopropylphenylsulfonsäureamid
1j N-Cyclopentylphenylsulfonsäureamid
1k N-(4-Methoxybenzyl)phenylsulfonsäureamid
1l N-(4-Methylbenzyl)phenylsulfonsäureamid
1m N-Cyclopropylphenylsulfonsäureamid
1n 1-(Phenylsulfonyl)piperazin
1o N-Benzyl-N-methylphenylsulfonsäureamid
1p N-(2-(1H-Indol-3-yl)ethyl)phenylsulfonsäureamid
2 N-Benzylphenylsulfonsäureamid
3a N-Ethyl-N-methylphenylsulfonsäureamid
3b N-Methyl-N-propylphenylsulfonsäureamid
3c N-Isopropyl-N-methylphenylsulfonsäureamid
4a N,N-Dimethyl-3-(trifluormethyl)phenylsulfonsäureamid
4b N,N-Dimethylcyclohexylsulfonsäureamid
4c N,N-Dimethylbenzylsulfonsäureamid
4d 4-Acetyl-N,N-dimethylphenylsulfonsäureamid
4e N,N-Dimethyl-3-nitrophenylsulfonsäureamid
5 3-Amino-N,N-dimethylphenylsulfonsäureamid
6a 3-(Ethylamino)-N,N-dimethylphenylsulfonsäureamid
6b N,N-Dimethyl-3-(propylamino)phenylsulfonsäureamid
6c N,N-Dimethyl-3-((2,2,2-trifluorethyl)amino)phenylsulfonsäureamid
6d 3-(Dimethylamino)-N,N-dimethylphenylsulfonsäureamid
6e 3-(Dipropylamino)-N,N-dimethylphenylsulfonsäureamid
7a Piperidin-2-carbonsäureethylester

200 ANHANG
7b (S)-Piperidin-2-carbonsäureethylester
8 1-(2-Ethoxy-2-oxoacetyl)piperidin-2-carbonsäureethylester
9 3-Methyl-2-oxopentansäure
10 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäureethylester
11a 1-(2-Phenoxyacetyl)piperidin-2-carbonsäureethylester
11b 1-(2-Phenylacetyl)piperidin-2-carbonsäureethylester
11c 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäureethylester
12a 1-(Benzylsulfonyl)piperidin-2-carbonsäureethylester
12b (S)-1-(Benzylsulfonyl)piperidin-2-carbonsäureethylester
12c 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäureethylester
12d 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäureethylester
12e 1-((2-Nitrobenzyl)sulfonyl)piperidin-2-carbonsäureethylester
12f 1-(Phenylsulfonyl)piperidin-2-carbonsäureethylester
12g (S)-1-(Phenylsulfonyl)piperidin-2-carbonsäureethylester
12h 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäureethylester
12i 1-((3-Methoxyphenyl)sulfonyl)piperidin-2-carbonsäureethylester
12j 1-Tosylpiperidin-2-carbonsäureethylester
13 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäure
14a 1-(2-Phenoxyacetyl)piperidin-2-carbonsäure
14b 1-(2-Phenylacetyl)piperidin-2-carbonsäure
14c 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäure
15a 1-(Benzylsulfonyl)piperidin-2-carbonsäure
15b (S)-1-(Benzylsulfonyl)piperidin-2-carbonsäure
15c 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäure
15d 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäure
15e 1-((2-Nitrobenzyl)sulfonyl)piperidin-2-carbonsäure
15f 1-(Phenylsulfonyl)piperidin-2-carbonsäure
15g 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure
15h 1-((3-Methoxyphenyl)sulfonyl)piperidin-2-carbonsäure
15i 1-Tosylpiperidin-2-carbonsäure
16 3-(3,4,5-Trimethoxyphenyl)propan-1-ol
17 1,3-Diphenylpropan-1-ol
18a 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
18b 1-(3-Methyl-2-oxopentanoyl)piperidin-2-carbonsäure-(1,3-diphenylpropyl)ester
19a 1-(2-Phenoxyacetyl)piperidin-2-carbonsäure-(4-methoxyphenethyl)ester
19b 1-(2-Phenoxyacetyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester
19c 1-(2-Phenoxyacetyl)piperidin-2-carbonsäure-(4-phenylbutyl)ester

ANHANG 201
20a 1-(2-Phenylacetyl)piperidin-2-carbonsäure-(pyridin-3-ylmethyl)ester
20b 1-(2-Phenylacetyl)piperidin-2-carbonsäure-(4-methoxyphenethyl)ester
20c 1-(2-Phenylacetyl)piperidin-2-carbonsäure-(4-phenylbutyl)ester
21a 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester
21b 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
21c 1-(2-(4-Chlorphenyl)acetyl)piperidin-2-carbonsäure-(1,3-diphenylpropyl)ester
22a 1-(Benzylsulfonyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
22b (S)-1-(Benzylsulfonyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
22c 1-(Benzylsulfonyl)piperidin-2-carbonsäure-(pyridin-3-ylmethyl)ester
22d 1-(Benzylsulfonyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
22e 1-(Benzylsulfonyl)piperidin-2-carbonsäure-(4-phenylbutyl)ester
23a 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäure-(pyridin-3-ylmethyl)ester
23b 1-((4-Methylbenzyl)sulfonyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
24a 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäure-(pyridin-3-ylmethyl)ester
24b 1-((4-Chlorbenzyl)sulfonyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
25 1-((2-Nitrobenzyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester
26a 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
26b 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(pyridin-3-ylmethyl)ester
26c 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
26d 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(4-phenylbutyl)ester
26e 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(4-methoxyphenethyl)ester
26f 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(phenethyl)ester
26g 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(3-phenylpropyl)ester
26h 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)ester
26i 1-(Phenylsulfonyl)piperidin-2-carbonsäure-(3-(4-methoxyphenyl)propyl)ester
27a 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure-(phenethyl)ester
27b 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure-(4-methoxyphenethyl)ester
27c 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
27d 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl))propyl)ester
27e 1-((3-Nitrophenyl)sulfonyl)piperidin-2-carbonsäure-(5-phenylpentyl)ester
28 1-((3-Methoxyphenyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl))propyl)ester
29 1-Tosylpiperidin-2-carbonsäure-(3-(pyridin-3-yl))propyl)ester
30a 1-((3-Aminophenyl)sulfonyl)piperidin-2-carbonsäureethylester
30b 1-((3-Aminophenyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl))propyl)ester
30c 1-((3-Aminophenyl)sulfonyl)piperidin-2-carbonsäure-(3-(3,4,5-trimethoxyphenyl)propyl)ester
31 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäureethylester
32 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäure

202 ANHANG
33 1-((3-(3,3-Dimethylureido)phenyl)sulfonyl)piperidin-2-carbonsäure-(3-(pyridin-3-yl)propyl)-
ester
34a 4-((tert.-Butoxycarbonyl)amino)buttersäure
34b 3-((tert.-Butoxycarbonyl)amino)propionsäure
34c 2-((tert.-Butoxycarbonyl)amino)essigsäure
35a 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)piperidin-2-carbonsäure-
ethylester
35b 1-((3-(2-((tert.-Butoxycarbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäure-
ethylester
36a 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)piperidin-2-carbonsäure
36b 1-((3-(2-((tert.-Butoxycarbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäure
37a 1-((3-(3-((tert.-Butoxycarbonyl)amino)propanamido)phenyl)sulfonyl)piperidin-2-carbonsäure-
(3-(pyridin-3-yl)propyl)ester
37b 1-((3-(2-((tert.-Butoxycarbonyl)amino)acetamido)phenyl)sulfonyl)piperidin-2-carbonsäure-(3-
(pyridin-3-yl)propyl)ester
38 N-(3-Phenylpropyl)-1-(phenylsulfonyl)piperidin-2-carbonsäureamid
39 2-Amino-4-methylpentansäureethylester
40 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäureethylester
41 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure
42 4-Methyl-2-(3-methyl-2-oxopentanamido)pentansäure-(3-(pyridin-3-yl)propyl)ester
43 Pyrrolidin-2-carbonsäureethylester
44 1-(3-Methyl-2-oxopentanoyl)pyrrolidin-2-carbonsäureethylester
45 2-(4-Isopropylphenoxy)essigsäure
46 2-(4-Isopropylphenoxy)-1-morpholinessigsäureamid
47 5-Methyl-2-phenyl-1H-pyrazol-3(2H)-on
48 2-Amino-4-(methylsulfinyl)buttersäureethylester
7.2 logP-Werte synthetisierter Verbindungen
Die logP-Werte wurden mit Hilfe des Computerprogramms MOE berechnet und sind in Tabelle 49
dargestellt.
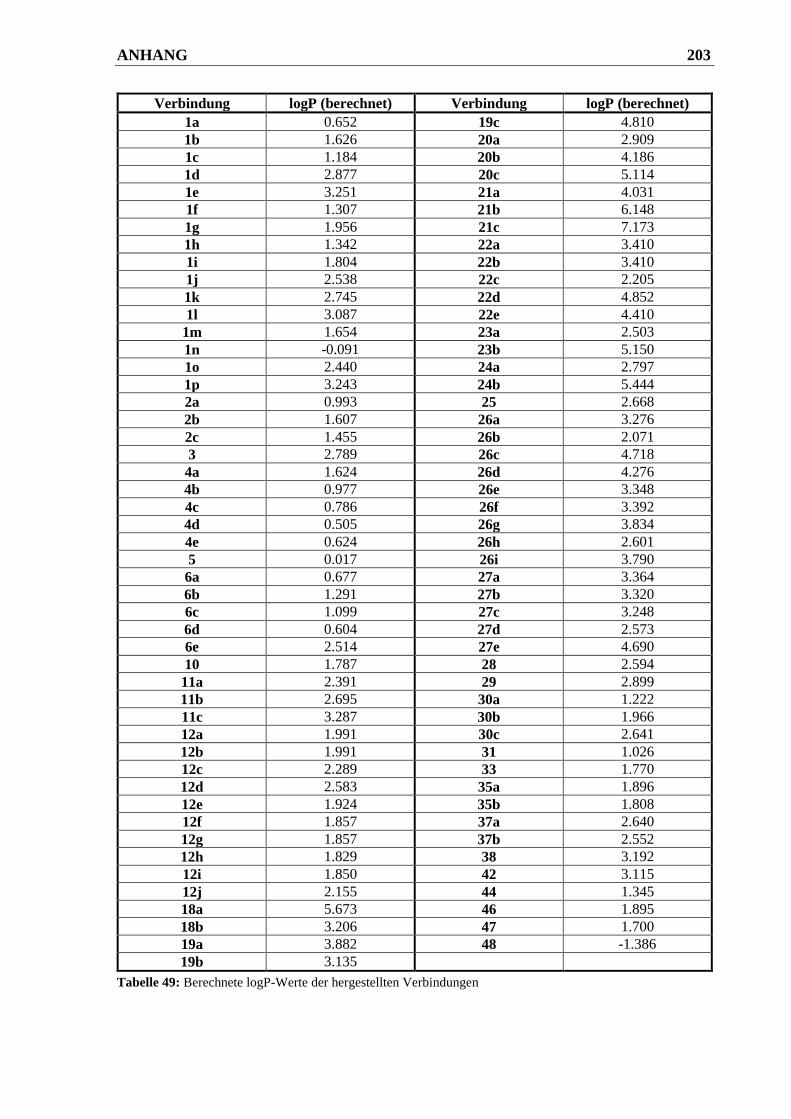
ANHANG 203
Verbindung logP (berechnet) Verbindung logP (berechnet)
1a 0.652 19c 4.810
1b 1.626 20a 2.909
1c 1.184 20b 4.186
1d 2.877 20c 5.114
1e 3.251 21a 4.031
1f 1.307 21b 6.148
1g 1.956 21c 7.173
1h 1.342 22a 3.410
1i 1.804 22b 3.410
1j 2.538 22c 2.205
1k 2.745 22d 4.852
1l 3.087 22e 4.410
1m 1.654 23a 2.503
1n -0.091 23b 5.150
1o 2.440 24a 2.797
1p 3.243 24b 5.444
2a 0.993 25 2.668
2b 1.607 26a 3.276
2c 1.455 26b 2.071
3 2.789 26c 4.718
4a 1.624 26d 4.276
4b 0.977 26e 3.348
4c 0.786 26f 3.392
4d 0.505 26g 3.834
4e 0.624 26h 2.601
5 0.017 26i 3.790
6a 0.677 27a 3.364
6b 1.291 27b 3.320
6c 1.099 27c 3.248
6d 0.604 27d 2.573
6e 2.514 27e 4.690
10 1.787 28 2.594
11a 2.391 29 2.899
11b 2.695 30a 1.222
11c 3.287 30b 1.966
12a 1.991 30c 2.641
12b 1.991 31 1.026
12c 2.289 33 1.770
12d 2.583 35a 1.896
12e 1.924 35b 1.808
12f 1.857 37a 2.640
12g 1.857 37b 2.552
12h 1.829 38 3.192
12i 1.850 42 3.115
12j 2.155 44 1.345
18a 5.673 46 1.895
18b 3.206 47 1.700
19a 3.882 48 -1.386
19b 3.135
Tabelle 49: Berechnete logP-Werte der hergestellten Verbindungen

204 ANHANG

LITERATURVERZEICHNIS 205
8 LITERATURVERZEICHNIS
1. Gesetz zur Verhütung und Bekämpfung von Infektionskrankheiten beim Menschen
(Infektionsschutzgesetz - IfSG). http://www.gesetze-im-internet.de/bundesrecht/ifsg/gesamt.pdf
(accessed 25.01.2012).
2. Fraser, D. W.; Tsai, T. R.; Orenstein, W.; Parkin, W. E.; Beecham, H. J.; Sharrar, R. G.; Harris, J.;
Mallison, G. F.; Martin, S. M.; McDade, J. E.; Shepard, C. C.; Brachman, P. S., Legionnaires'
disease: description of an epidemic of pneumonia. N. Engl. J. Med. 1977, 297 (22), 1189-1197.
3. McDade, J. E.; Shepard, C. C.; Fraser, D. W.; Tsai, T. R.; Redus, M. A.; Dowdle, W. R.,
Legionnaires' disease: isolation of a bacterium and demonstration of its role in other respiratory
disease. N. Engl. J. Med. 1977, 297 (22), 1197-1203.
4. Osterholm, M. T.; Chin, T. D.; Osborne, D. O.; Dull, H. B.; Dean, A. G.; Fraser, D. W.; Hayes, P.
S.; Hall, W. N., A 1957 outbreak of Legionnaires' disease associated with a meat packing plant. Am.
J. Epidemiol. 1983, 117 (1), 60-67.
5. Thacker, S. B.; Bennett, J. V.; Tsai, T. F.; Fraser, D. W.; McDade, J. E.; Shepard, C. C.; Williams,
K. H., Jr.; Stuart, W. H.; Dull, H. B.; Eickhoff, T. C., An outbreak in 1965 of severe respiratory
illness caused by the Legionnaires' disease bacterium. J. Infect. Dis. 1978, 138 (4), 512-519.
6. Glick, T. H.; Gregg, M. B.; Berman, B.; Mallison, G.; Rhodes, W. W., Jr.; Kassanoff, I., Pontiac
fever. An epidemic of unknown etiology in a health department: I. Clinical and epidemiologic
aspects. Am. J. Epidemiol. 1978, 107 (2), 149-160.
7. Newton, H. J.; Ang, D. K. Y.; van Driel, I. R.; Hartland, E. L., Molecular pathogenesis of
infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 2010, 23 (2), 274-298.
8. Rogers, J.; Dowsett, A. B.; Dennis, P. J.; Lee, J. V.; Keevil, C. W., Influence of temperature and
plumbing material selection on biofilm formation and growth of Legionella pneumophila in a model
potable water system containing complex microbial flora. Appl. Environ. Microbiol. 1994, 60 (5),
1585-1592.
9. Kwaik, Y. A., The phagosome containing Legionella pneumophila within the Protozoan
Hartmannella vermiformis is surrounded by the rough endoplasmic reticulum. Appl. Environ.
Microbiol. 1996, 62 (6), 2022-2028.
10. Bozue, J. A.; Johnson, W., Interaction of Legionella pneumophila with Acanthamoeba
castellanii: uptake by coiling phagocytosis and inhibition of phagosome-lysosome fusion. Infect.
Immun. 1996, 64 (2), 668-673.
11. Garduno, R. A: Life cycle, growth cycles and developmental cycle of Legionella pneumophila.
http://microbiology.medicine.dal.ca/people/garduno/Garduno%20chapter.pdf (accessed 10.08.2011).
12. Steinert, M.; Hentschel, U.; Hacker, J., Legionella pneumophila: an aquatic microbe goes astray.
FEMS Microbiol. Rev. 2002, 26 (2), 149-162.
13. Machner: Modulation of Human Macrophages by the Intracellular Pathogen Legionella
pneumophila. http://2009annualreport.nichd.nih.gov/ump.html (accessed 05.08.2011).
14. Hoge, C. W.; Breiman, R. F., Advances in the epidemiology and control of Legionella infections.
Epidemiol. Rev. 1991, 13, 329-340.
15. Centers-for-Disease-and-Control-prevention Patient Facts: Learn More about Legionnaires'
disease. http://www.cdc.gov/legionella/patient_facts.htm (accessed 20.08.2011).
16. Fraser, D. W.; Wachsmuth, I.; Bopp, C.; Feeley, J. C.; Tsai, T. F., Antibiotic treatment of guinea-
pigs infected with agent of Legionnaires' disease. Lancet 1978, 1 (8057), 175-178.

206 LITERATURVERZEICHNIS
17. Robert-Koch-Institut: Ratgeber für Ärzte - Legionellose.
http://www.rki.de/cln_162/nn_468478/DE/Content/Infekt/EpidBull/Merkblaetter/Ratgeber_
Legionellose.html (accessed 10.08.2011).
18. Pedro-Botet, M. L.; Yu, V. L., Treatment strategies for Legionella infection. Expert Opin.
Pharmacother. 2009, 10 (7), 1109-1121.
19. Miyamoto, M.; Yamaguchi, Y.; Sasatsu, M., Disinfectant effects of hot water, ultraviolet light,
silver ions and chlorine on strains of Legionella and nontuberculous mycobacteria. Microbios. 2000,
101 (398), 7-13.
20. Bundesministerium-für-Gesundheit: Trinkwasserverordnung und Legionellen (Stand
19.10.2011).
http://www.bundesgesundheitsministerium.de/ministerium/presse/pressemitteilungen/2011-
02/aenderung-der-trinkwasserverordnung/trinkwasserverordnung-und-legionellen.html (accessed
10.11.2011).
21. Augsburger-Allgemeine: Legionellen: Augsburger Experte: "beunruhigende Häufung".
www.augsburger-allgemeine.de/bayern/Augsburger-Experte-beunruhigende-Haeufung-
id7153461.html (accessed 25.08.2011).
22. Augsburger-Allgemeine: Legionellen: Firma unter Verdacht. www.augsburger-
allgemeine.de/neu-ulm/Legionellen-Firma-unter-Verdacht-id15758466.html (accessed 25.08.2011).
23. Robert-Koch-Institut: SurvStat. http://www3.rki.de/SurvStat/ (accessed 10.08.2011).
24. von Baum, H.; Ewig, S.; Marre, R.; Suttorp, N.; Gonschior, S.; Welte, T.; Luck, C., Community-
acquired Legionella pneumonia: new insights from the German competence network for community
acquired pneumonia. Clin. Infect. Dis. 2008, 46 (9), 1356-1364.
25. Luck, P. C.; Steinert, M., Pathogenesis, diagnosis and therapy of Legionella infections.
Bundesgesundhbl. Gesundheitsforsch. Gesundheitsschutz 2006, 49 (5), 439-449.
26. Shevchuk, O.; Jäger, J.; Steinert, M., Virulence properties of the Legionella pneumophila cell
envelope. Front. Microbiol. 2011, 2 (74), 1-12.
27. Riboldi-Tunnicliffe, A.; Konig, B.; Jessen, S.; Weiss, M. S.; Rahfeld, J.; Hacker, J.; Fischer, G.;
Hilgenfeld, R., Crystal structure of Mip, a prolylisomerase from Legionella pneumophila. Nat. Struct.
Biol. 2001, 8 (9), 779-783.
28. Fischer, G.; Bang, H.; Ludwig, B.; Mann, K.; Hacker, J., Mip protein of Legionella pneumophila
exhibits peptidyl-prolyl-cis/trans isomerase (PPIase) activity. Mol. Microbiol. 1992, 6 (10), 1375-
1383.
29. Schmid, F. X., Prolyl isomerase: Enzymic catalysis of slow protein-folding reactions. Annu. Rev.
Biophys. Biomol. Struct. 1993, 22, 123-143.
30. Cianciotto, N. P.; Eisenstein, B. I.; Mody, C. H.; Toews, G. B.; Engleberg, N. C., A Legionella
pneumophila gene encoding a species-specific surface protein potentiates initiation of intracellular
infection. Infect. Immun. 1989, 57 (4), 1255-1262.
31. Wintermeyer, E.; Ludwig, B.; Steinert, M.; Schmidt, B.; Fischer, G.; Hacker, J., Influence of site
specifically altered Mip proteins on intracellular survival of Legionella pneumophila in eukaryotic
cells. Infect. Immun. 1995, 63 (12), 4576-4583.
32. Koehler, R.; Fanghaenel, J.; Koenig, B.; Lueneberg, E.; Frosch, M.; Rahfeld, J.-U.; Hilgenfeld,
R.; Fischer, G.; Hacker, J.; Steinert, M., Biochemical and functional analyses of the Mip protein:
Influence of the N-terminal half and of peptidylprolyl isomerase activity on the virulence of
Legionella pneumophila. Infect. Immun. 2003, 71 (8), 4389-4397.
33. Wagner, C.; Khan, A. S.; Kamphausen, T.; Schmausser, B.; Uenal, C.; Lorenz, U.; Fischer, G.;
Hacker, J.; Steinert, M., Collagen binding protein Mip enables Legionella pneumophila to

LITERATURVERZEICHNIS 207
transmigrate through a barrier of NCI-H292 lung epithelial cells and extracellular matrix. Cell.
Microbiol. 2007, 9 (2), 450-462.
34. Wikipedia: Edward Jenner. http://de.wikipedia.org/wiki/Edward_Jenner (accessed 25.08.2011).
35. Dawson, T. M.; Steiner, J. P.; Lyons, W. E.; Fotuhi, M.; Blue, M.; Snyder, S. H., The
immunophilins, FK 506 binding protein and cyclophilin, are discretely localized in the brain:
relationship to calcineurin. Neuroscience 1994, 62 (2), 569-580.
36. Szep, S.; Park, S.; Boder, E. T.; Van Duyne, G. D.; Saven, J. G., Structural coupling between
FKBP12 and buried water. Proteins Struct., Funct., Bioinf. 2009, 74 (3), 603-611.
37. Brillantes, A. M. B.; Ondrias, K.; Scott, A.; Kobrinsky, E.; Ondriasova, E.; Moschella, M. C.;
Jayaraman, T.; Landers, M.; Ehrlich, B. E.; Mars, A. R., Stabilization of calcium release channel
(Ryanodine receptor) function by FK506-binding protein. Cell 1994, 77 (4), 513-523.
38. Jayaraman, T.; Brillantes, A. M.; Timerman, A. P.; Fleischer, S.; Erdjument-Bromage, H.;
Tempst, P.; Marks, A. R., FK506 binding protein associated with the calcium release channel
(ryanodine receptor). J. Biol. Chem. 1992, 267 (14), 9474-9477.
39. Breiman, A.; Camus, I., The Involvement of Mammalian and Plant FK506-Binding Proteins
(FKBPs) in Development. Transgenic Res. 2002, 11 (4), 321-335.
40. Cameron, A. M.; Nucifora, F. C., Jr.; Fung, E. T.; Livingston, D. J.; Aldape, R. A.; Ross, C. A.;
Snyder, S. H., FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400-1401)
and anchors calcineurin to this FK506-like domain. J. Biol. Chem. 1997, 272 (44), 27582-27588.
41. Huse, M.; Chen, Y.-G.; Massague, J.; Kuriyan, J., Crystal structure of the cytoplasmic domain of
the type I TGFβ receptor in complex with FKBP12. Cell 1999, 96 (3), 425-436.
42. Wang, T.; Donahoe, P. K.; Zervos, A. S., Specific interaction of type I receptors of the TGF-β
family with the immunophilin FKBP-12. Science 1994, 265 (5172), 674-676.
43. Ruegger, A.; Kuhn, M.; Lichti, H.; Loosli, H. R.; Huguenin, R.; Quiquerez, C.; Von Wartburg,
A., Cyclosporin A, a peptide metabolite from Trichoderma polysporum (Link ex Pers.) Rifai, with
immunosuppressive activity. Helv. Chim. Acta 1976, 59 (4), 1075-1092.
44. Mutschler, E.; Geisslinger, G.; Kroemer, H. K.; Ruth, P.; Schaefer-Korting, M., Mutschler
Arzneimittelwirkungen. Wissenschaftliche Verlagsgesellschaft mbH Stuttgart: 2008; Vol. 9.
45. Sawada, S.; Suzuki, G.; Kawase, Y.; Takaku, F., Novel immunosuppressive agent, FK506. In
vitro effects on the cloned T cell activation. J. Immunol. 1987, 139 (6), 1797-1803.
46. Tocci, M. J.; Matkovich, D. A.; Collier, K. A.; Kwok, P.; Dumont, F.; Lin, S.; Degudicibus, S.;
Siekierka, J. J.; Chin, J.; Hutchinson, N. I., The immunosuppressant FK506 selectively inhibits
expression of early T cell activation genes. J. Immunol. 1989, 143 (2), 718-726.
47. Liu, J.; Farmer, J. D., Jr.; Lane, W. S.; Friedman, J.; Weissman, I.; Schreiber, S. L., Calcineurin
is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66 (4),
807-815.
48. Schreiber, S. L., Chemistry and biology of the immunophilins and their immunosuppressive
ligands. Science 1991, 251 (4991), 283-287.
49. Sabers, C. J.; Martin, M. M.; Brunn, G. J.; Williams, J. M.; Dumont, F. J.; Wiederrecht, G.;
Abraham, R. T., Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells.
J. Biol. Chem. 1995, 270 (2), 815-822.
50. Dumont, F. J.; Su, Q., Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1995,
58 (5), 373-395.

208 LITERATURVERZEICHNIS
51. Bierer, B. E.; Somers, P. K.; Wandless, T. J.; Burakoff, S. J.; Schreiber, S. L., Probing
immunosuppressant action with a nonnatural immunophilin ligand. Science 1990, 250 (4980), 556-
559.
52. Bierer, B. E.; Mattila, P. S.; Standaert, R. F.; Herzenberg, L. A.; Burakoff, S. J.; Crabtree, G.;
Schreiber, S. L., Two distinct signal transmission pathways in T lymphocytes are inhibited by
complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci.
USA 1990, 87 (23), 9231-9235.
53. Fischer, G.; Bang, H.; Mech, C., Determination of enzymatic catalysis for the cis-trans-
isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 1984, 43
(10), 1101-1111.
54. Kofron, J. L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D. H., Determination of kinetic
constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay.
Biochemistry 1991, 30 (25), 6127-6134.
55. Holt, D. A.; Luengo, J. I.; Yamashita, D. S.; Oh, H. J.; Konialian, A. L.; Yen, H. K.; Rozamus, L.
W.; Brandt, M.; Bossard, M. J.; Levy, M. A.; Eggleston, D. S.; Liang, J.; Schultz, L. W.; Stout, T. J.;
Clardyt, J., Design, synthesis, and kinetic evaluation of high-affinity FKBP ligands and the X-ray
crystal structures of their complexes with FKBP12. J. Am. Chem. Soc. 1993, 115 (22), 9925-9938.
56. Steiner, J. P.; Hamilton, G. S.; Ross, D. T.; Valentine, H. L.; Guo, H.; Connolly, M. A.; Liang,
S.; Ramsey, C.; Li, J.-H. J.; Huang, W.; Howorth, P.; Soni, R.; Fuller, M.; Sauer, H.; Nowotnik, A.
C.; Suzdak, P. D., Neurotrophic immunophilin ligands stimulate structural and functional recovery in
neurodegenerative animal models. Proc. Natl. Acad. Sci. USA 1997, 94 (5), 2019-2024.
57. Engleberg, N. C.; Carter, C.; Weber, D. R.; Cianciotto, N. P.; Eisenstein, B. I., DNA sequence of
mip, a Legionella pneumophila gene associated with macrophage infectivity. Infect. Immun. 1989, 57
(4), 1263-1270.
58. Sippel, M. Computational Structure-based Design Approaches: Targeting HIV-1 Integrase and
the Macrophage Infectivity Potentiator of Legionella pneumophila. Dissertation, Würzburg, 2010.
59. Ceymann, A.; Horstmann, M.; Ehses, P.; Schweimer, K.; Paschke, A.-K.; Steinert, M.; Faber, C.,
Solution structure of the Legionella pneumophila Mip-rapamycin complex. BMC Struct. Biol. 2008,
8, 17.
60. Kang, C. B.; Hong, Y.; Dhe-Paganon, S.; Yoon, H. S., FKBP Family Proteins: Immunophilins
with Versatile Biological Functions. Neurosignals 2008, 16 (4), 318-325.
61. Van Duyne, G. D.; Standaert, R. F.; Schreiber, S. L.; Clardy, J., Atomic structure of the
rapamycin human immunophilin FKBP-12 complex. J. Am. Chem. Soc. 1991, 113 (19), 7433-7434.
62. Michnick, S. W.; Rosen, M. K.; Wandless, T. J.; Karplus, M.; Schreiber, S. L., Solution structure
of FKBP, a rotamase enzyme and receptor for FK506 and rapamycin. Science 1991, 252 (5007), 836-
839.
63. Juli, C.; Sippel, M.; Jaeger, J.; Thiele, A.; Weiwad, M.; Schweimer, K.; Roesch, P.; Steinert, M.;
Sotriffer, C. A.; Holzgrabe, U., Pipecolic Acid Derivatives As Small-Molecule Inhibitors of the
Legionella MIP Protein. J. Med. Chem. 2011, 54 (1), 277-283.
64. Robert-Koch-Institut: RKI-Ratgeber für Infektionskrankheiten - Merkblätter für Ärzte:
Infektionen durch Chlamydien (Teil 1): Erkrankungen durch Chlamydia trachomatis.
Epidemiologisches Bulletin 2009, 37, 369-376.
65. Lundemose, A. G.; Birkelund, S.; Fey, S. J.; Mose Larsen, P.; Christiansen, G., Chlamydia
trachomatis contains a protein similar to the Legionella pneumophila mip gene products. Mol.
Microbiol. 1991, 5 (1), 109-115.

LITERATURVERZEICHNIS 209
66. Lundemose, A. G.; Kay, J. E.; Pearce, J. H., Chlamydia trachomatis Mip-like protein has
peptidyl-prolyl cis/trans isomerase activity that is inhibited by FK506 and rapamycin and is
implicated in initiation of chlamydial infection. Mol. Microbiol. 1993, 7 (5), 777-783.
67. Centers-for-Disease-and-Control-prevention: Trypanosomiasis, American.
www.dpd.cdc.gov/dpdx/html/trypanosomiasisamerican.htm (accessed 14.11.2011).
68. World-Health-Organization: Chagas disease (American trypanosomiasis).
http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed 14.11.2011).
69. Moro, A.; Ruiz-Cabello, F.; Fernandez-Cano, A.; Stock, R. P.; Gonzalez, A., Secretion by
Trypanosoma cruzi of a peptidyl-prolyl cis-trans isomerase involved in cell infection. EMBO J. 1995,
14 (11), 2483-2490.
70. Pereira, P. J. B.; Vega, M. C.; Gonzalez-Rey, E.; Fernandez-Carazo, R.; Macedo-Ribeiro, S.;
Gomis-Ruth, F. X.; Gonzalez, A.; Coll, M., Trypanosoma cruzi macrophage infectivity potentiator
has a rotamase core and a highly exposed α-helix. EMBO Rep. 2002, 3 (1), 88-94.
71. McGee, Z. A.; Stephens, D. S.; Hoffman, L. H.; Schlech, W. F., 3rd; Horn, R. G., Mechanisms of
mucosal invasion by pathogenic Neisseria. Rev. Infect. Dis. 1983, 5 (4), 708-714.
72. Leuzzi, R.; Serino, L.; Scarselli, M.; Savino, S.; Fontana, M. R.; Monaci, E.; Taddei, A.; Fischer,
G.; Rappuoli, R.; Pizza, M., Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a
peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages.
Mol. Microbiol. 2005, 58 (3), 669-681.
73. Robert-Koch-Instiut: RKI-Ratgeber für Infektionskrankheiten - Merkblätter für Ärzte:
Salmonellose (Salmonellen-Gastroenteritis). Epidemiologisches Bulletin 2009, 13, 117-126.
74. Norville, I. H.; Harmer, N. J.; Harding, S. V.; Fischer, G.; Keith, K. E.; Brown, K. A.; Sarkar-
Tyson, M.; Titball, R. W., A Burkholderia pseudomallei macrophage infectivity potentiator-like
protein has rapamycin-inhibitable peptidylprolyl isomerase activity and pleiotropic effects on
virulence. Infect. Immun. 2011, 79 (11), 4299-4307.
75. Jones, A. L.; Beveridge, T. J.; Woods, D. E., Intracellular survival of Burkholderia pseudomallei.
Infect. Immun. 1996, 64 (3), 782-790.
76. Becker, H. G. O.; Berger, W.; Domschke, G.; Fanghänel, E.; Faust, J.; Fischer, M.; Gentz, F.;
Gewald, K.; Gluch, R.; Mayer, R.; Müller, K.; Pavel, D.; Schmidt, H.; Schollberg, K.; Schwetlick,
K.; Seiler, E., Organikum: Organisch-chemisches Grundpraktikum. Barth Verlagsgesellschaft mbH:
Edition Deutscher Verlag der Wissenschaften: 1993; Vol. 19.
77. Bellamy, F. D.; Ou, K., Selective reduction of aromatic nitro compounds with stannous chloride
in nonacidic and nonaqueous medium. Tetrahedron Lett. 1984, 25 (8), 839-842.
78. Gribble, G. W.; Lord, P. D.; Skotnicki, J.; Dietz, S. E.; Eaton, J. T.; Johnson, J., Reactions of
sodium borohydride in acidic media. I. Reduction of indoles and alkylation of aromatic amines with
carboxylic acids. J. Am. Chem. Soc. 1974, 96 (25), 7812-7813.
79. Mutule, I.; Suna, E., Arylzinc species by microwave assisted Grignard formation-transmetalation
sequence: application in the Negishi coupling. Tetrahedron 2005, 61 (47), 11168-11176.
80. Neises, B.; Steglich, W., 4-Dialkylaminopyridines as acylation catalysts. 5. Simple method for
the esterification of carboxylic acids. Angew. Chem. 1978, 90 (7), 556-557.
81. Pracejus, G., Optical activation of N-phthaloyl-α-amino acid derivatives by catalysis with tertiary
bases. Liebigs Ann. Chem. 1959, 622, 10-22.
82. Ponnusamy, E.; Fotadar, U.; Spisni, A.; Fiat, D., A novel method for the rapid, non-aqueous tert-
butoxycarbonylation of some 17O-labeled amino acids and oxygen-17 NMR parameters of the
products. Synthesis 1986, (1), 48-49.

210 LITERATURVERZEICHNIS
83. Schiller, M. Neue Derivate der Etacrynsäure: Synthese und biologische Aktivität. Dissertation,
Würzburg, 2009.
84. Holzgrabe, U.; Heller, E., A new synthetic route to compounds of the AFDX-type with affinity to
muscarinic M2-receptor. Tetrahedron 2003, 59 (6), 781-787.
85. Burkhart, F.; Hoffmann, M.; Kessler, H., Stereoselective synthesis of a C-glycosidic analog of N-
glucoasparagine. Angew. Chem., Int. Ed. Engl. 1997, 36 (11), 1191-1192.
86. Knorr, L., Synthetical experiments by means of ethyl acetoacetate. Liebigs Ann. Chem. 1886,
238, 137-219.
87. Lehmann, F.; Holm, M.; Laufer, S., Three-Component Combinatorial Synthesis of Novel
Dihydropyrano[2,3-c]pyrazoles. J. Comb. Chem. 2008, 10 (3), 364-367.
88. Hacker, J.; Ott, M.; Ludwig, B.; Rdest, U., Intracellular survival and expression of virulence
determinants of Legionella pneumophila. Infection 1991, 19 (4), 198-201.
89. Ludwig, B.; Schmid, A.; Marre, R.; Hacker, J., Cloning, genetic analysis, and nucleotide
sequence of a determinant coding for a 129-kilodalton peptidoglycan-associated protein (Ppl) of
Legionella pneumophila. Infect. Immun. 1991, 59 (8), 2515-2521.
90. Bio-Rad: Affi-Gel® Blue Affinity Chromatography Gel for Enzyme and Blood Protein
Purification. http://www3.bio-rad.com/cmc_upload/Literature/13362/Bulletin_1107.pdf (accessed
24.02.2012).
91. Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F. X., Cyclophilin and
peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337 (6206), 476-
478.
92. Stryer, L., Biochemie. Spektrum Akademischer Verlag GmbH Heidelberg - Berlin: 1999; Vol. 4.
93. Chaplin, H. O.; Hunter, L., Associating effect of the hydrogen atom. I. Amides and sulfonamides.
J. Chem. Soc. 1937, 1114-1118.
94. Levchenko, E. S.; Bal'on, Y. G., Synthesis of arylsulfonyl derivatives of pyrrolidine, pyrroline,
and pyrrole. Zh. Org. Khim. 1967, 3 (8), 1509-1512.
95. Snyder, H. R.; Heckert, R. E., A method for the rapid cleavage of sulfonamides. J. Am. Chem.
Soc. 1952, 74, 2006-2009.
96. Malyshev, O. R.; Vinogradov, M. G., Convenient synthesis of π-acceptor chiral stationary phases
for high-performance liquid chromatography from halogen-substituted 3,5-dinitrobenzoylamides. J.
Chromatogr. A 1999, 859 (2), 143-151.
97. Block, D. G.; Bailey, R. E.; Towle, J. L. N-Alkynylarenesulfonamides and -sulfonimides.
FR1438772, 1966.
98. Goldstein, M.; Russell, M. A.; Willis, H. A., Infrared spectra of N-substituted sulfonamides.
Spectrochim. Acta, Part A 1969, 25 (7), 1275-1285.
99. Watt, G. W.; Otto, J. B., Ammonolysis of ethyl iodide by liquid ammonia. J. Am. Chem. Soc.
1947, 69, 836-838.
100. v. Braun, J.; Jostes, F.; Wagner, H., Decomposition of the six-carbon chain of adipic acid. II.
Ber. Dtsch. Chem. Ges. B 1928, 61B, 1423-1431.
101. Bollinger, F. W.; Hayes, F. N.; Siegel, S., Base-catalyzed decomposition of N-nitroso-N-
cyclopentylurethan. J. Am. Chem. Soc. 1953, 75, 1729-1730.
102. Park, K. H.; Lee, J. B., Indirect oxidative deamination of benzylamines to benzaldehydes. Synth.
Commun. 1992, 22 (7), 1061-1065.

LITERATURVERZEICHNIS 211
103. Chung, K. H.; Kim, J. N.; Ryu, E. K., Friedel-Crafts alkylation reactions of benzene with amide
bond containing compounds. Tetrahedron Lett. 1994, 35 (18), 2913-2914.
104. Jacob, R. M. 1-Alkylsulfonyl-4-alkylpiperazines. US2507408, 1950.
105. Katritzky, A. R.; Zhang, G.; Wu, J., 1-Phenylsulfonylbenzotriazole: A novel and convenient
reagent for the preparation of benzenesulfonamides and aryl benzenesulfonates. Synth. Commun.
1994, 24 (2), 205-216.
106. Ho, B.-T.; McIsaac, W. M.; Tansey, L. W.; Kralik, P. M., Hydroxyindole-O-methyltransferase.
II. Inhibitory activities of some N-acyltryptamines. J. Pharm. Sci. 1968, 57 (11), 1998-2000.
107. Chapman, C. J.; Frost, C. G.; Mahon, M. F., Structure and reactivity of new phosphine ligands
containing the hemi-labile sulfone moiety. J. Chem. Soc., Dalton Trans. 2006, (18), 2251-2262.
108. De Luca, L.; Giacomelli, G., An Easy Microwave-Assisted Synthesis of Sulfonamides Directly
from Sulfonic Acids. J. Org. Chem. 2008, 73 (10), 3967-3969.
109. Garcia Ruano, J. L.; Parra, A.; Marzo, L.; Yuste, F.; Mastranzo, V. M., One-pot synthesis of
sulfonamides from methyl sulfinates using ultrasound. Tetrahedron 2011, 67 (16), 2905-2910.
110. Burns, B.; King, N. P.; Tye, H.; Studley, J. R.; Gamble, M.; Wills, M., Chiral phosphinamides:
new catalysts for the asymmetric reduction of ketones by borane. J. Chem. Soc., Perkin Trans. 1
1998, (6), 1027-1038.
111. Harmata, M.; Zheng, P.; Huang, C.; Gomes, M. G.; Ying, W.; Ranyanil, K.-O.; Balan, G.;
Calkins, N. L., Expedient Synthesis of Sulfinamides from Sulfonyl Chlorides. J. Org. Chem. 2007,
72 (2), 683-685.
112. Molander, G. A.; Fleury-Bregeot, N.; Hiebel, M.-A., Synthesis and Cross-Coupling of
Sulfonamidomethyltrifluoroborates. Org. Lett. 2011, 13 (7), 1694-1697.
113. Benard, S.; Neuville, L.; Zhu, J., Copper-Mediated N-Cyclopropylation of Azoles, Amides, and
Sulfonamides by Cyclopropylboronic Acid. J. Org. Chem. 2008, 73 (16), 6441-6444.
114. Naito, H.; Hata, T.; Urabe, H., Selective Deprotection of Methanesulfonamides to Amines. Org.
Lett. 2010, 12 (6), 1228-1230.
115. Schuster, I. I.; Doss, S. H.; Roberts, J. D., Natural-abundance nitrogen-15 nuclear magnetic
resonance spectroscopy. Electronic effects in benzenesulfonamides. J. Org. Chem. 1978, 43 (25),
4693-4696.
116. Choi, J. H.; Lee, B. C.; Lee, H. W.; Lee, I., Competitive Reaction Pathways in the Nucleophilic
Substitution Reactions of Aryl Benzenesulfonates with Benzylamines in Acetonitrile. J. Org. Chem.
2002, 67 (4), 1277-1281.
117. Short, W. F.; Oxley, P. Substituted carboxamides. GB587810, 1947.
118. McKay, A. F., The reaction of primary amines with 1-nitroso-2-nitramino-2-imidazoline. J.
Org. Chem. 1951, 16, 1395-1404.
119. Agbalyan, S. G.; Esayan, G. T.; Magakyan, P. O.; Nshanyan, A. O., Esters of sulfonic acids.
XIII. Synthesis of some cyclohexanesulfonic acid derivatives. Izv. Akad. Nauk Arm. SSR, Khim.
Nauki 1964, 17 (1), 69-74.
120. Johnson, T. B.; Ambler, J. A., Amines. IV. Alkylation and hydrolysis of aliphatic sulfonamides,
New synthesis of sarcosine. J. Am. Chem. Soc. 1914, 36, 372-385.
121. Gregory, W. A. 1-(p-Sulfamoylphenyl)-2-(2,2-dichloroacetamido)-1,3-propanediols.
US2680135, 1954.
122. Waletzky, E. Anticoccidal agent. US2531755, 1950.

212 LITERATURVERZEICHNIS
123. Hill, B.; Liu, Y.; Taylor, S. D., Synthesis of α-Fluorosulfonamides by Electrophilic
Fluorination. Org. Lett. 2004, 6 (23), 4285-4288.
124. Shavyrina, V. V.; Ermakova, Z. I., Synthesis and study of some N-aminopropyl and N-
aminopropionyl derivatives of 3-substituted diphenylamines. Khim. Farm. Zh. 1969, 3 (11), 30-34.
125. Schmitz-Josten, R.; Sueling, C.; Boemer, B.; Brinkmeyer, H. Photopolymerization methods and
suitable photoactivators. DE3135115, 1983.
126. Reckhow, W. A.; Tarbell, D. S., Hexahydroindolo[2,3-a]quinolizine. J. Am. Chem. Soc. 1952,
74, 4960-4962.
127. Armistead, D. M.; Saunders, J. O. Preparation of N-(aryloxalyl)piperidine-2-carboxylates and
analogs as multidrug resistance inhibitors. WO9736869, 1997.
128. Titanyuk, I. D.; Beletskaya, I. P., Synthesis of non-natural cyclic amino acids from available
unsaturated tertiary amines. Russ. J. Org. Chem. 2010, 46 (9), 1277-1281.
129. Bender, D. R.; Brennan, J.; Rapoport, H., Periodate oxidation of α-keto γ-lactams. Enol
oxidation and β-lactam formation. Mechanism of periodate hydroxylation reactions. J. Org. Chem.
1978, 43 (17), 3354-3362.
130. Niznik, G. E.; Morrison, W. H., III; Walborsky, H. M., Metallo aldimines. Masked acyl
carbanion. J. Org. Chem. 1974, 39 (5), 600-604.
131. McFarlane, A. K.; Thomas, G.; Whiting, A., Diastereoselectivity and assignment of absolute
stereochemistry in the aza-Diels-Alder reaction of a sulfonylimino acetate with 1-methoxy-3-
trimethylsilyloxybuta-1,3-diene. J. Chem. Soc., Perkin Trans. 1 1995, (21), 2803-2808.
132. Fujii, T.; Miyoshi, M., Novel synthesis of L-pipecolic acid. Bull. Chem. Soc. Jpn. 1975, 48 (4),
1341-1342.
133. Hamilton, G. S.; Li, J.-h.; Steiner, J. P. Method of using neurotrophic sulfonamide compounds.
US5721256, 1998.
134. Familoni, O. B., Synthesis of tricyclic tetrahydro 1,2-benzothiazinones via Friedel-Craft anionic
equivalents. J. Pharm. Res. Dev. 1998, 3 (1), 21-29.
135. Kosley, R. W., Jr.; Baron, B.; Jimonet, P.; Jurcak, J. G.; Shimshock, S. J.; Zhao, X.-Y.; Sher, R.;
Mueller, P. J.; Beall, J.; Barrague, M.; Guiles, J. W. 1-Arylsulfonylpiperidine derivatives and their
preparation, pharmaceutical compositions, FKBP binding compositions, and pharmaceutical use for
treatment of conditions associated with neuronal degeneration. WO2006012256, 2006.
136. Moss, W. O.; Wakefield, E.; Mahon, M. F.; Molloy, K. C.; Bradbury, R. H.; Hales, N. J.;
Gallagher, T., Ketene S,S-acetals as 1,3-dipolarophiles towards azides. A new synthetic entry into
cyclic amino acids. Tetrahedron 1992, 48 (36), 7551-7569.
137. Rapoport, H.; Campion, J. E., Synthesis of 2,3,4-trimethoxybenzocyclohepten-6-one. J. Am.
Chem. Soc. 1951, 73, 2239-2241.
138. Rosowsky, A.; Chen, H.; Fu, H.; Queener, S. F., Synthesis of new 2,4-Diaminopyrido[2,3-
d]pyrimidine and 2,4-Diaminopyrrolo[2,3-d]pyrimidine inhibitors of Pneumocystis carinii,
Toxoplasma gondii, and Mycobacterium avium dihydrofolate reductase. Bioorg. Med. Chem. 2003,
11 (1), 59-67.
139. Pfeiffer, P.; Kalckbrenner, E.; Kunze, W.; Levin, K., Hydrochalcones and hydrochalcols. J.
Prakt. Chem. 1928, 119, 109-130.
140. Martinez, R.; Ramon, D. J.; Yus, M., Easy α-alkylation of ketones with alcohols through a
hydrogen autotransfer process catalyzed by RuCl2(DMSO)4. Tetrahedron 2006, 62 (38), 8988-9001.
141. Holt, D. A.; Konialian-Beck, A. L.; Oh, H.-J.; Yen, H.-K.; Rozamus, L. W.; Krog, A. J.; Erhard,
K. F.; Ortiz, E.; Levy, M. A.; Brandt, M.; Bossard, M. J.; Luengo, J. I., Structure-activity studies of

LITERATURVERZEICHNIS 213
synthetic FKBP ligands as peptidyl-prolyl isomerase inhibitors. Bioorg. Med. Chem. Lett. 1994, 4
(2), 315-320.
142. Choi, C.; Li, J.-H.; Vaal, M.; Thomas, C.; Limburg, D.; Wu, Y.-Q.; Chen, Y.; Soni, R.; Scott,
C.; Ross, D. T.; Guo, H.; Howorth, P.; Valentine, H.; Liang, S.; Spicer, D.; Fuller, M.; Steiner, J.;
Hamilton, G. S., Use of parallel-synthesis combinatorial libraries for rapid identification of potent
FKBP12 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12 (10), 1421-1428.
143. Schnabel, E., Improved synthesis of tert-butoxycarbonyl amino acids by a constant pH reaction.
Liebigs Ann. Chem. 1967, 702, 188-196.
144. Bentley, P. H.; Gregory, H.; Laird, A. H.; Morley, J. S., Polypeptides. I. The synthesis of
peptides related to eledoisin. J. Chem. Soc., Suppl. 1964, 6130.
145. Ejim, L.; Mirza, I. A.; Capone, C.; Nazi, I.; Jenkins, S.; Chee, G.-L.; Berghuis, A. M.; Wright,
G. D., New phenolic inhibitors of yeast homoserine dehydrogenase. Bioorg. Med. Chem. 2004, 12
(14), 3825-3830.
146. Weitman, M.; Lerman, K.; Nudelman, A.; Major, D. T.; Hizi, A.; Herschhorn, A., Structure-
activity relationship studies of 1-(4-chloro-2,5-dimethoxyphenyl)-3-(3-propoxypropyl)thiourea, a
non-nucleoside reverse transcriptase inhibitor of human immunodeficiency virus type-1. Eur. J. Med.
Chem. 2011, 46 (2), 447-467.
147. Anderson, G. W.; McGregor, A. C., tert-Butoxycarbonylamino acids and their use in peptide
synthesis. J. Am. Chem. Soc. 1957, 79, 6180-6183.
148. Houen, G.; Struve, C.; Sondergaard, R.; Friis, T.; Anthoni, U.; Nielsen, P. H.; Christophersen,
C.; Petersen, B. O.; Duus, J. O., Substrate specificity of the bovine serum amine oxidase and in situ
characterization of aminoaldehydes by NMR spectroscopy. Bioorg. Med. Chem. 2005, 13 (11), 3783-
3796.
149. Wei, L.; Wu Yon, Q.; Wilkinson, D. E.; Chen, Y.; Soni, R.; Scott, C.; Ross, D. T.; Guo, H.;
Howorth, P.; Valentine, H.; Liang, S.; Spicer, D.; Fuller, M.; Steiner, J.; Hamilton, G. S., Solid-phase
synthesis of FKBP12 inhibitors: N-sulfonyl and N-carbamoylprolyl/pipecolyl amides. Bioorg. Med.
Chem. Lett. 2002, 12 (10), 1429-1433.
150. Losse, G.; Jeschkeit, H., Racemate resolution of amino acid esters. IV. Preparation of optically
active esters of simple amino acids from their DL-compounds by resolution with di-O-benzoyl-D-
tartaric acid. Chem. Ber. 1957, 90, 1275-1278.
151. Gerling, K.; Heinemann, H.; Meier, A.; Langer, K. Preparation of acylated amino acid
derivatives as pharmaceuticals and dietetics. EP400499, 1990.
152. Karrer, P.; Portmann, P.; Suter, M., The reduction of α-amino carboxylic acid esters with
lithium aluminum hydride. Helv. Chim. Acta 1948, 31, 1617-1623.
153. Lochead, A. W.; Proctor, G. R.; Caton, M. P. L., Use of chloroalkenylamines for the synthesis
of 1-azabicyclo[3.3.0]octane and 1-azabicyclo[4.3.0]nonane derivatives. J. Chem. Soc., Perkin Trans.
1 1984, (11), 2477-2489.
154. Aberg, B., Plant-growth regulators. IX. p-Alkylphenoxyacetic and -propionic acids, and some
related derivatives of naturally occurring phenols. Physiol. Plant. 1954, 7, 241-252.
155. Stebbins, J. L.; Zhang, Z.; Chen, J.; Wu, B.; Emdadi, A.; Williams, M. E.; Cashman, J.;
Pellecchia, M., Nuclear Magnetic Resonance Fragment-Based Identification of Novel FKBP12
Inhibitors. J. Med. Chem. 2007, 50 (26), 6607-6617.
156. Kitamura, R., Constitution of antipyrine and related compounds. V. Experimental evidence of
the third form (the third oscillation form) of antipyrine. Yakugaku Zasshi 1940, 60, 3-9.
157. Pal, S.; Mareddy, J.; Devi, N. S., High speed synthesis of pyrazolones using microwave-assisted
neat reaction technology. J. Braz. Chem. Soc. 2008, 19 (6), 1207-1214.

214 LITERATURVERZEICHNIS
158. Laemmli, U. K., Cleavage of structural proteins during the assembly of the head of
bacteriophage T4. Nature 1970, 227 (5259), 680-685.
159. Jansohn, M.; Rothhämel, S. Gentechnische Methoden: Eine Sammlung von Arbeitsanleitungen
für das molekularbiologische Labor. http://www.springer.com/978-3-8274-2429-7 (accessed
05.03.2012).