Embed Size (px)
Citation preview

Fresenius Z. Anal. Chem. 306, 136-143 (1981) Fresenius Zeitschrift fiir
,~) Springer-Verlag 1981
Zur Invers-Voltammetrie einiger Kupferchelate an der Kohlepaste-Elektrode. Bestimmung von Kupfer in Trinkwasser durch Oxidation von Kupferdithiooxamid*
H. Monien** und Ute Gerlach-~
Univ.-Gesamthochschule Siegen, Analyt. Chemie, Adolf-Reichwein-Str. 2, D-5900 Siegen 2l
P. Jacob
Univ. Dortmund, Abt. Chemie, Otto-Hahn-Stra/3e, D-4600 Dortmund 50
Inverse-Voltammetry of some Copper Chelates using a Carbon Paste Electrode. Determination of Copper in Drinking Water by Oxidation of Copper Dithio-oxamide
Summary. Dithio-oxamide, 2,2'-bichinoline, neocu- proine and bathocuproine can be oxidized at the carbon paste electrode as well as the corresponding copper chelates. Particularly, dithio-oxamide is a convenient reagent for the determination of copper in the ppb level in presence of a 105-fold excess of other metals. - For the d.c. inverse-voltammetric determination of copper in drinking water a sample volume between 1 and 10 ml is sufficient and 3 rain for pre-electrolysis. The oxida- tion of the copper dithio-oxamide chelate occurs near + 0.25 V (reference electrode: Ag/AgC1/KC1 sat.) in a base electrolyte (pH 4.1) containing acetate buffer. - The procedure described offers some advantages in comparison with the copper determination using mer- cury electrodes since interferences are less. A single determination requires about 15 rain applying the ad- dition technique.
Zusammenfassung. Dithiooxamid, 2,2'-Bichinolin, Neocuproin und Bathocuproin lassen sich ebenso wie die entsprechenden Kupferchelate an der Kohlepaste- Elektrode oxidieren. Besonders Dithiooxamid ist ffir die Bestimmung von Kupfer im ppb-Bereich auch in Gegenwart eines 105-fachen fJberschusses anderer Me- talle geeignet. - Zur invers-voltammetrischen Kupfer- bestimmung als Gleichstromverfahren in Trinkwasser gentigt bei 3 rain Vorelektrolysedauer ein Probenvolu- men zwischen 1 und 10 ml. Die Oxidation des Kupfer- dithiooxamid-chelats erfolgt in acetatgepuffertem Leit- elektrolyten (pH 4,1) bei +0,25 V (Bezugselektrode:
* Herrn Prof. Dr. E. Blasius zum 60. Geburtstag gewidmet ** Korrespondenzanschrift
Ag/AgC1/KC1 ges/itt.). - Das beschriebene Verfahren hat gegeniiber der Kupferbestimmung an einer Queck- silberelektrode den Vorteil, weniger st6ranf~illig zu sein. Die Durchf/ihrung einer Bestimmung nach der Addi- tionsmethode nimmt etwa 15 rain in Anspruch.
Key words: Best. von Kupfer in Wasser; Voltammetrie, inverse; Dithiooxamid, Trinkwasser
Die ~berffihrung eines Kations in ein Chelat ftihrt immer zu einer )~nderung des elektrochemischen Ver- haltens dieses Kations, die sich in einer oft mehrere 100 mV betragenden Verschiebung des oder der Peak- potentiale bemerkbar macht [2, 8]. Diese Erscheinung ist ein brauchbares Mittel, um St6rungen dutch andere L6sungspartner zu vermeiden und so die Selektivit/it der voltammetrischen Bestimmung eines Elements zu steigern [1, 5]. Die Selektivitfitssteigerung kann dabei gegentiber den als Matrix vorliegenden Elementen so grol3 sein, dab sich die Abtrennung der Elementspur erfibrigt und Direktbestimmungen ausgeffihrt werden k6nnen [5-7]. Eine solche Verfahrensweise, die auf Trennvorgfinge verzichten kann, wird grunds~itzlich anzustreben sein, denn jeder zus/itzliche Verfahrens- schritt vergr613ert die Zahl der systematischen Fehler und stellt so das Analysenergebnis hinsichtlich seiner Richtigkeit mehr und mehr in Frage [12].
In dieser Arbeit wird fiber die Invers-Voltammetrie von Kupfer vor und nach der Chelatisierung mit Dithiooxamid, 2,2'-Bichinolin, Neocuproin und Ba- thocuproin berichtet. Das Ziel ist die Entwicklung eines Verfahrens zur Bestimmung von Kupfer als Chelat, das selektiver aber doch ebenso nachweisstark ist wie die Kupferbestimmung an Quecksilber- und anderen Fest-
0016-1152/81/0306/0136/$01.60
H. Monien et al. : Bestimmung von Kupfer in Trinkwasser 137
11 nglm Cu /
I !
/ -0,2 0 + 0,5 + 1,0
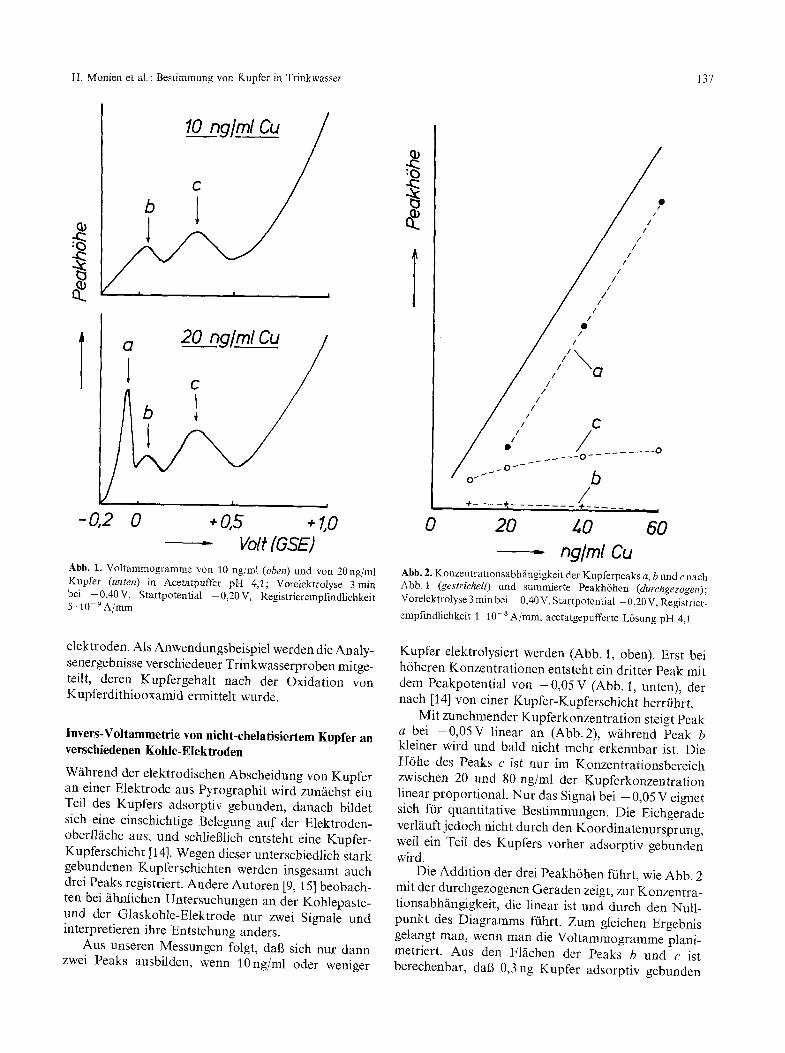
vo# (OSE) Abb. 1, Voltammogramme yon 10 ng/ml (oben) und yon 20ng/ml Kupfer (unten) in Acetatpuffer pH 4,1; Vorelektrolyse 3rain bei -0,40V, Startpotential -0,20V, Registrierempfindlichkeit 5.10 9A/ram
elektroden. Als Anwendungsbeispiel werden die Analy- senergebnisse verschiedener Trinkwasserproben mitge- teilt, deren Kupfergehalt nach der Oxidation von Kupferdithiooxamid ermittelt wurde.
Invers-Voitammetrie yon nicht-chelatisiertem Kupfer an verschiedenen Kohle-Elektroden
W~ihrend der elektrodischen Abscheidung yon Kupfer an einer Elektrode aus Pyrographit wird zun/ichst ein Teil des Kupfers adsorptiv gebunden, danach bildet sich eine einschichtige Belegung auf der Elektroden- oberflfiche aus, und schlieBlich entsteht eine Kupfer- Kupferschicht [14]. Wegen dieser unterschiedlich stark gebundenen Kupferschichten werden insgesamt auch drei Peaks registriert. Andere Autoren [9, 15] beobach- ten bei ~ihnlichen Untersuchungen an der Kohlepaste- und der Glaskohle-Elektrode nur zwei Signale und interpretieren ihre Entstehung anders.
Aus unseren Messungen folgt, dab sich nur dann zwei Peaks ausbilden, wenn 10ng/ml oder weniger
i i I
y / ' /
b / 0 20 40 60
ng/ml Cu Abb. 2. Konzentrationsabhfingigkeit der Kupferpeaks a, b undc nach Abb. 1 (gestricheh) und summierte Peakh6hen (durchgezogen); Vorelektrolyse 3 rain bei - 0,40 V, Startpotential - 0,20 V, Registrier- empfindlichkeit 1.10-8 A/ram, acetatgepufferte L6sung pH 4,1
Kupfer elektrolysiert werden (Abb. 1, oben). Erst bei h6heren Konzentrationen entsteht ein dritter Peak mit dem Peakpotential yon -0,05 V (Abb. 1, unten), der nach [14] yon einer Kupfer-Kupferschicht herrfihrt.
Mit zunehmender Kupferkonzentration steigt Peak a bei -0,05V linear an (Abb. 2), wfihrend Peak b kleiner wird und bald nicht mehr erkennbar ist. Die H6he des Peaks c ist nut im Konzentrationsbereich zwischen 20 und 80 ng/ml der Kupferkonzentration linear proportional. Nut das Signal bei - 0,05 V eignet sich ffir quantitative Bestimmungen. Die Eichgerade verl~iuft jedoch nicht durch den Koordinatenursprung, weil ein Teil des Kupfers vorher adsorptiv gebunden wird.
Die Addition der drei Peakh6hen fiihrt, wie Abb. 2 mit der durchgezogenen Geraden zeigt, zur Konzentra- tionsabhfingigkeit, die linear ist und durch den Null- punkt des Diagramms ffihrt. Zum gleichen Ergebnis gelangt man, wenn man die Voltammogramme plani- metriert. Aus den Flfichen der Peaks b und c ist berechenbar, dab 0,3ng Kupfer adsorptiv gebunden
138 Fresenius Z. Anal. Chem., Band 306 (1981)
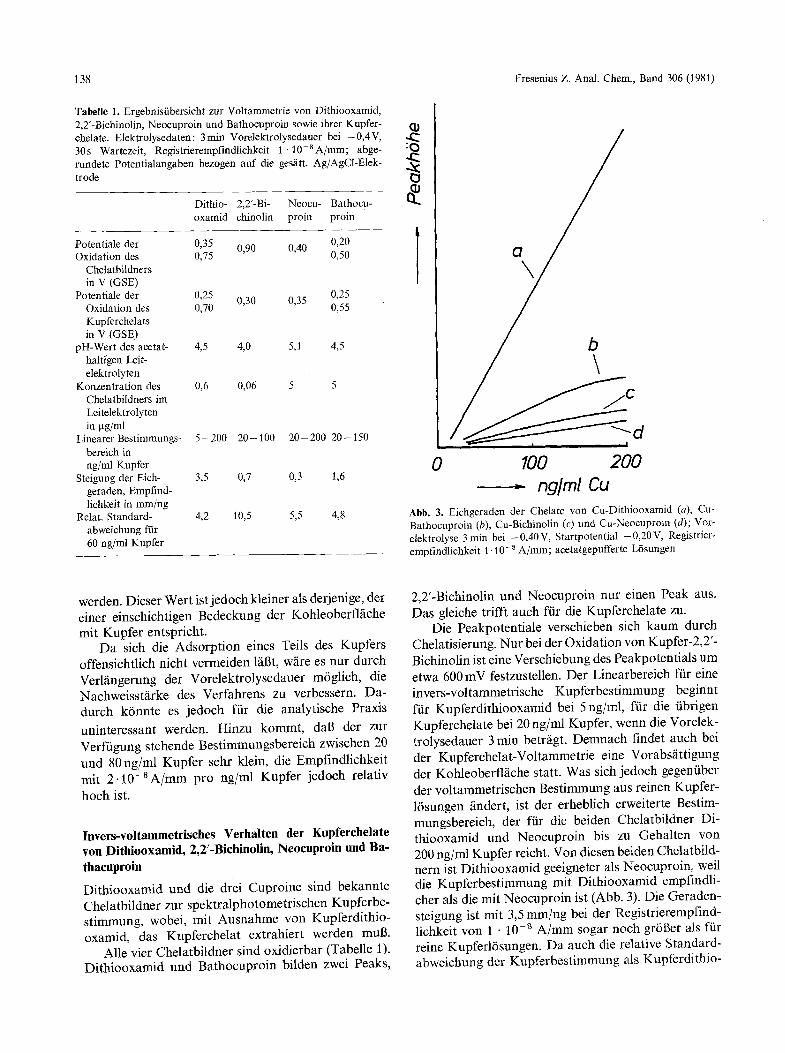
Tabelle 1. Ergebnistibersicht zur Voltammetrie von Dithiooxamid, 2,2'-Bichinolin, Neocuproin und Bathocuproi• sowie ihrer Kupfer- (11 chelate. Elektrolysedaten: 3 rain Vorelektrolysedauer bei - 0,4 V, 30 s Wartezeit, Registrierempfindlichkeit I �9 10-8 A/mm; abge- rundete Potentialangaben bezogen auf die ges~itt. Ag/AgC1-Elek- , ~ trode
Dithio- 2,2'-Bi- Neocu- Bathocu- oxamid chinolin proin proin
Potentiale der 0,35 0,90 0,40 0,20 Oxidation des 0,75 0,50
Chetatbildners in V (GSE)
Potentiale der 0,25 0,30 0,35 0,25 Oxidation des 0,70 0,55 Kupferchelats in V (GSE)
pH-Wert des acetat- 4,5 4,0 5,1 4,5 haltigen Leit- elektrolyten
Konzentration des 0,6 0,06 5 5 Chelatbildners im Leitelektrolyten in gg/ml
Linearer Bestimmungs- 5-200 20-100 2 0 - 200 20-150 bereich in ng/ml Kupfer
Steigung der Eich- 3,5 0,7 0,3 1,6 geraden, Empfind- lichkeit in mm/ng
Relat. Standard- 4,2 10,5 5,5 4,8 abweichung ffir 60 ng/ml Kupfer
#.
O \
b \ j c
0 700 200 --- ng/ml Cu
Abb. 3. Eichgeraden der Chelate von Cu-Dithiooxamid (a), Cu- Bathocuproin (b), Cu-Bichinolin (c) und Cu-Neocuprnin (d); Vor- elektrolyse 3 rain bei - 0,40 V, Startpotential - 0,20 V, Registrier- empfindlichkeit 1.10 -8 A/mm; acetatgepufferte L6sungen
werden. Dieser Wert ist jedoch kleiner als derjenige, der einer einschichtigen Bedeckung der Kohleoberfl/iche mit Kupfer entspricht.
Da sich die Adsorption eines Teils des Kupfers offensichtlich nicht vermeiden lfil3t, wfire es nur durch Verl/ingerung der Vorelektrolysedauer m6glich, die Nachweisstfirke des Verfahrens zu verbessern. Da- durch k6nnte es jedoch f/Jr die analytische Praxis uninteressant werden. Hinzu kommt, dab der zur Verfiigung stehende Bestimmungsbereich zwischen 20 und 80 ng/ml Kupfer sehr klein, die Empfindlichkeit mit 2-10-8A/mm pro ng/ml Kupfer jedoch relativ hoch ist.
Invers-voltammctrisches Verhalten der Kupferchelate yon Dithiooxamid, 2,2'-Bichinolin, Neocuproin und Ba- thacuproin
Dithiooxamid und die drei Cuproine sind bekannte Chelatbildner zur spektralphotometrischen Kupferbe- stimmung, wobei, mit Ausnahme yon Kupferdithio- oxamid, das Kupferchelat extrahiert werden mull
Alle vier Chelatbildner sind oxidierbar (Tabelle 1). Dithiooxamid und Bathocuproin bilden zwei Peaks,
2,2'-Bichinolin und Neocuproin nur einen Peak aus. Das gleiche trifft auch f/ir die Kupferchelate zu.
Die Peakpotentiale verschieben sich kaum durch Chelatisierung. Nur bei der Oxidation von Kupfer-2,2'- Bichinolin ist eine Verschiebung des Peakpotentials um etwa 600 mV festzustellen. Der Linearbereich ftir eine invers-voltammetrische Kupferbestimmung beginnt fiir Kupferdithiooxamid bei 5 ng/ml, fiir die iibrigen Kupferchelate bei 20 ng/ml Kupfer, wenn die Vorelek- trolysedauer 3 rain betr~igt. Demnach findet auch bei der Kupferchelat-Voltammetrie eine Vorabsfittigung der Kohleoberflfiche statt. Was sich jedoch gegentiber der voltammetrischen Bestimmung aus reinen Kupfer- 16sungen /indert, ist der erheblich erweiterte Bestim- mungsbereich, der ffir die beiden Chelatbildner Di- thiooxamid und Neocuproin bis zu Gehalten von 200 ng/ml Kupfer reicht. Von diesen beiden Chelatbild- nern ist Dithiooxamid geeigneter als Neocuproin, weil die Kupferbestimmung mit Dithiooxamid empfindli- cher als die mit Neocuproin ist (Abb. 3). Die Geraden- steigung ist mit 3,5 mm/ng bei der Registrierempfind- lichkeit yon 1 - 10 -s A/mm sogar noch gr6ger als fiir reine KupferlBsungen. Da auch die relative Standard- abweichung der Kupferbestimmung als Kupferdithio-
H. Monien et al. : Bestimmung von Kupfer in Trinkwasser | 39
I I J
-C 2 0 +0,5 + 1,0 = - Volt (GSE)
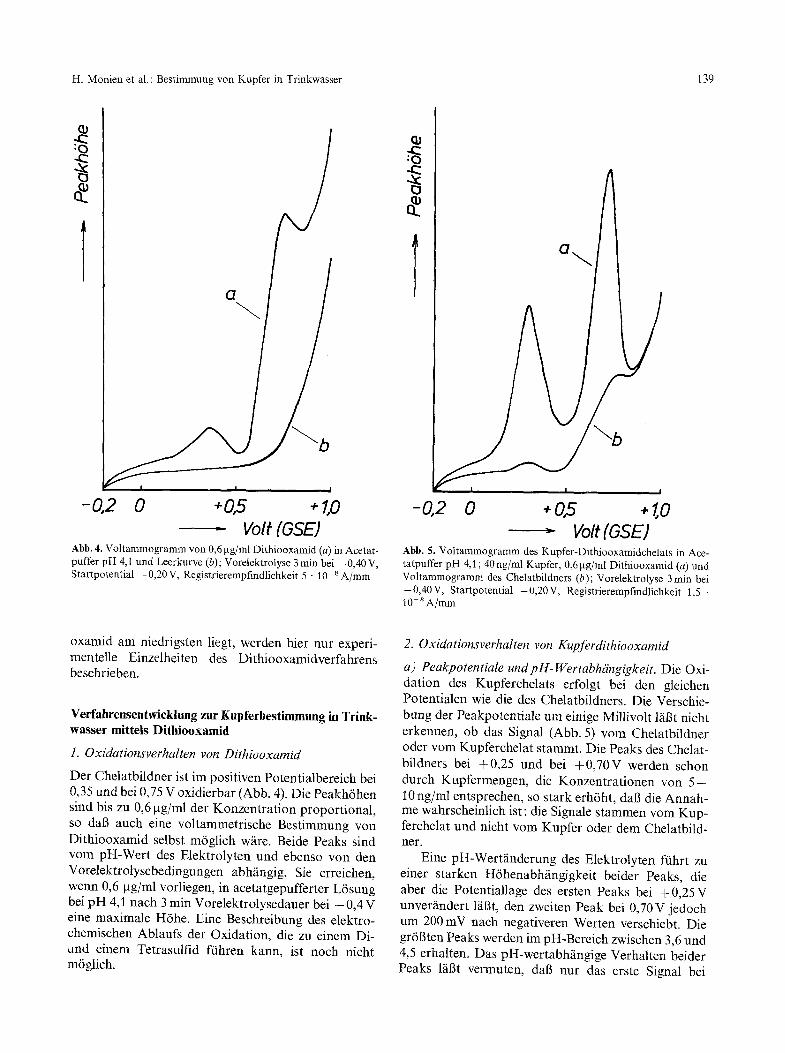
Abb. 4. Voltammogramm von 0,6 pg/ml Dithiooxamid (a) in Acetat- puffer pH 4,1 und Leerkurve (b); Vorelektrolyse 3 min bei - 0,40 V, Startpotential - 0,20 V, Registrierempfindlichkeit 5 - 10- 8 A/mm
I I
-0 ,2 0 + 0,5 + 1,0 = Volt (GSE)
Abb. 5. Voltammogramm des Kupfer-Dithiooxamidchelats in Ace- tatpuffer pH 4,1 ; 40 ng/ml Kupfer, 0,61xg/mI Dithiooxamid (a) und Voltammogramm des Chelatbildners (b); Vorelektrolyse 3 rain bei -0,40V, Startpotential -0,20V, Registrierempfindlichkeit 1,5 . 1 O- 8 A/ram
oxamid am niedrigsten liegt, werden hier nur experi- mentelle Einzelheiten des Dithiooxamidverfahrens beschrieben.
Verfahrensentwicklung zur Kupferbestimmung in Trink- wasser mittels Dithiooxamid
1. Oxidationsverhalten yon Dithiooxamid
Der Chelatbildner ist im positiven Potentialbereich bei 0,35 und bei 0,75 V oxidierbar (Abb. 4). Die Peakh6hen sind bis zu 0,6 gg/ml der Konzentration proportional, so dab auch eine voltammetrische Bestimmung yon Dithiooxamid selbst m6glich wfire. Beide Peaks sind vom pH-Wert des Elektrolyten und ebenso yon den Vorelektrolysebedingungen abh~ingig. Sie erreichen, wenn 0,6 gg/ml vorliegen, in acetatgepufferter L6sung bei pH 4,1 nach 3 rain Vorelektrolysedauer bei - 0,4 V eine maximale H6he. Eine Beschreibung des elektro- chemischen Ablaufs der Oxidation, die zu einem Di- und einem Tetrasulfid fiihren kann, ist noch nicht m6glich.
2. Oxidationsverhalten yon Kupferdithiooxamid
a) Peakpotentiale und pH-Wertabhiingigkeit. Die Oxi- dation des Kupferchelats erfolgt bei den gleichen Potentialen wie die des Chelatbildners. Die Verschie- bung der Peakpotentiale um einige Millivolt l~il3t nicht erkennen, ob das Signal (Abb. 5) vom Chelatbildner oder vom Kupferchelat stammt. Die Peaks des Chelat- bildners bei +0,25 und bei +0 ,70V werden schon durch Kupfermengen, die Konzentrationen yon 5 - 10 ng/ml entsprechen, so stark erh6ht, dab die Annah- me wahrscheinlich ist: die Signale stammen vom Kup- ferchelat und nicht yore Kupfer oder dem Chelatbild- ner.
Eine pH-Wertfinderung des Elektrolyten ftihrt zu einer starken H6henabh~ingigkeit beider Peaks, die aber die Potentiallage des ersten Peaks bei +0,25 V unver/indert lfiBt, den zweiten Peak bei 0,70 V jedoch um 200 mV nach negativeren Werten verschiebt. Die gr6Bten Peaks werden im pH-Bereich zwischen 3,6 und 4,5 erhalten. Das pH-wertabhfingige Verhalten beider Peaks laBt vermuten, dab nur das erste Signal bei
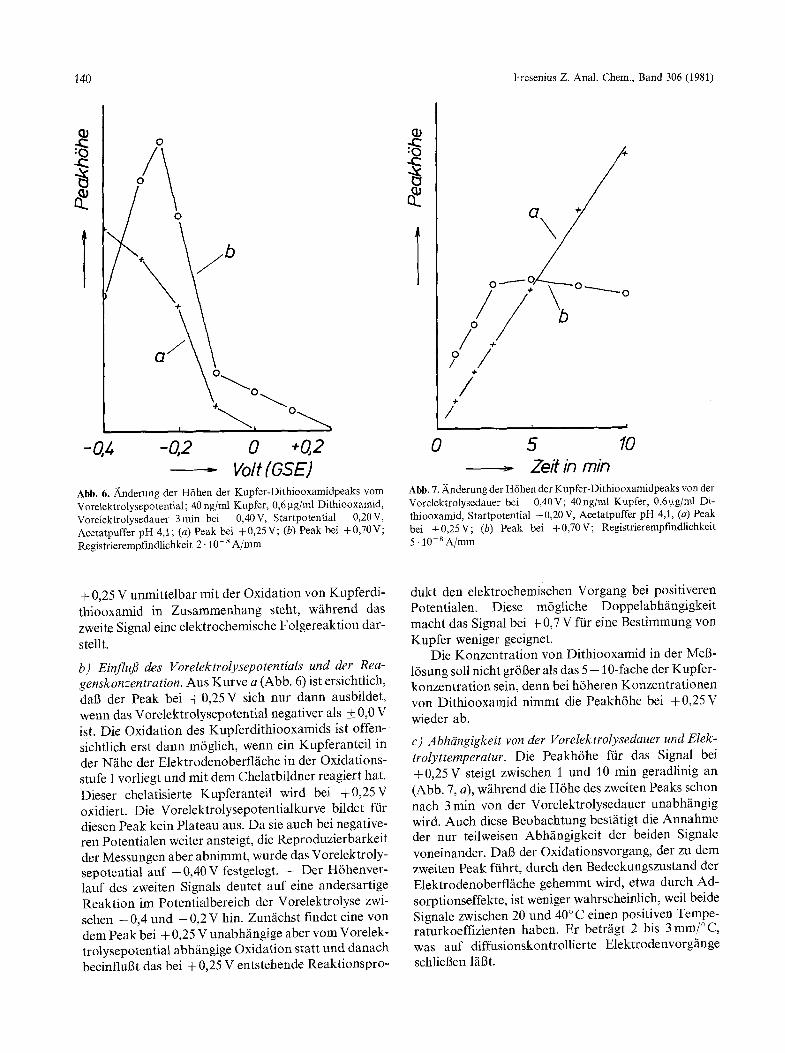
140 Fresenius Z. Anal. Chem., Band 306 (1981)
Abb. 6. Anderung der H6hen der Kupfer-Dithiooxamidpeaks vom Vorelektrolysepotential; 40 ng/ml Kupfer, 0,6 ~tg/ml Dithiooxamid, Vorelektrolysedauer 3rain bei -0,40V, Startpotential -0,20V, Acetatpuffer pH 4,1; (a) Peak bei +0,25V; (b) Peak bei +0,70V; Registrierempfindlichkeit 2.10- s A/ram
o
, . ,
-0,4 -0,2 0 +0,2 0 5 10 = Volt (GSE) :,. Zeit in min
AbI0.7. Anderung der H6hen der Kupfer-Dithiooxamidpeaks von der Vorelektrotysedauer bei - 0,40 V; 40 ng/ml Kupfer, 0,6 gg/ml Di- thiooxamid, Startpotential -0,20 V, Acetatpuffer pH 4,1, (a) Peak bei +0,25V; (b) Peak bei +0,70V; Registrierempfindlichkeit 5- 10- 8 A/mm
+ 0,25 V unmittelbar mit der Oxidation von Kupferdi- thiooxamid in Zusammenhang steht, w~ihrend das zweite Signal eine elektrochemische Folgereaktion dar-
stellt.
b) EinfluJ3 des Vorelektrolysepotentials und der Rea- genskonzentration. Aus Kurve a (Abb. 6) ist ersichtlich, daB der Peak bei +0,25 V sich nur dann ausbildet, wenn das Vorelektrolysepotential negativer als + 0,0 V ist. Die Oxidation des Kupferdithiooxamids ist of ten- sichtlich erst dann m6glich, wenn ein Kupferanteil in der N~he der Elektrodenoberfl/iche in der Oxidations- stufe I vorliegt und mit dem Chelatbildner reagiert hat. Dieser chelatisierte Kupferanteil wird bei +0 ,25V oxidiert. Die Vorelektrolysepotentialkurve bildet ffir diesen Peak kein Plateau aus. Da sie auch bei negative- ren Potentialen weiter ansteigt, die Reproduzierbarkeit der Messungen aber abnimmt, wurde das Vorelektroly- sepotential auf - 0 , 4 0 V festgelegt. - Der H6henver- lauf des zweiten Signals deutet auf eine andersartige Reaktion im Potentialbereich der Vorelektrolyse zwi- schen - 0,4 und - 0,2 V hin. Zunfichst findet eine von dem Peak bei + 0,25 V unabh/ingige aber vom Vorelek- trolysepotential abh~ingige Oxidation statt und danach beeinfluBt das bei + 0,25 V entstehende Reaktionspro-
dukt den elektrochemischen Vorgang bei positiveren Potentialen. Diese m6gliche DoppelabhS~ngigkeit macht das Signal bei + 0,7 V ftir eine Bestimmung von Kupfer weniger geeignet.
Die Konzentration von Dithiooxamid in der MeB- 16sung soll nicht gr613er als das 5 - 10-fache der Kupfer- konzentration sein, denn bei h6heren Konzentrationen von Dithiooxamid nimmt die Peakh6he bei +0,25 V wieder ab.
c) Abhdngigkeit yon der Vorelektrolysedauer und Elek- trolyttemperatur. Die Peakh6he ftir das Signal bei +0 ,25V steigt zwischen 1 und 10 min geradlinig an (Abb. 7, a), w/ihrend die H6he des zweiten Peaks schon nach 3 rain v o n d e r Vorelektrolysedauer unabh/ingig wird. Auch diese Beobachtung bestiitigt die Annahme d e r n u r teilweisen Abhgngigkeit der beiden Signale voneinander. DaB der Oxidationsvorgang, der zu dem zweiten Peak ftihrt, durch den Bedeckungszustand der Elektrodenoberfl/iche gehemmt wird, etwa durch Ad- sorptionseffekte, ist weniger wahrscheinlich, weil beide Signale zwischen 20 und 40~ einen positiven Tempe- raturkoeffizienten haben. Er betr/igt 2 bis 3 mm/~ was auf diffusionskontrollierte Elektrodenvorg/inge schlieBen 1N3t.

H. Monien et al. : Bestimmung yon Kupfer in Trinkwasser
Tabelle 2. Zulfissiger fJberschul3 eines Kations bei der invers- voltammetrischen Bestimmung van 50 ng/ml Kupfer als Kupferdi- thiooxamid
Zulftssiger Neben Kupfer vorliegendes Kation ~berschul3
> l0 s Alkali- und Erdalkalimetalle, Ce, Cd, Co, Cr, In, La, Mn, Mo, Nb, Ni, Ta, Th, U, W, Zn, Zr
104_ 105 AI, As, Ga, Sb, Te, Ti, V 103-104 Fe, Pb, Rh, Ru, Se, TI 102-103 Ir, Os, Pt I0 -- 102 Ag < 10 Au, Bi, Hg, Pd
3. Beeinflussung der Oxidation yon Kupferdithiooxamid durch Kationen und Anionen
Zur 15berprfifung des Einflusses von Kationen und Anionen wurden 50 ng/ml Kupfer mit steigenden Mengen eines Salzes unter sonst konstant gehaltenen Elektrolysebedingungen aber z.T. ge~inderter Elektro- lytzusammensetzung voltammetriert. Die noch zul/issi- gen Oberschfisse gibt Tabelle 2 wieder.
Die Beeinflussung des Kupfersignals bei +0 ,25V macht sich durch eine Abnahme der Peakh6he bemerk- bar. Sie beruht in den L6sungen mit 105fachen Ober- schfissen vor allem auf der Zunahme der Ionenkonzen- tration. Chemische Einfltisse sind von Blei, Kobalt, Eisen, Nickel, Quecksilber, Silber und einigen Platin- metallen zu erwarten, die ebenfalls mit dem Chelatbild- her reagieren. Die Chelatbildung dieser Metalle ist pH- wertabhfingig. Unter den experimentellen Bedingun- gen, wie sie hier vorliegen, ist eine Kupferbestimmung auch bei grol3en Oberschtissen von Kobalt und Nickel m6glich, da bei pH 4 die Bildung des Kupferchelats vorherrscht [4].
Neben dem EinfluB der Ionenst/irke und einer m6glichen chemischen Beeinflussung ffihren besonders elektrochernische Ursachen zu St6rungen. Kationen, die zwischen - 0 , 4 und + 0,3 V oxidiert oder reduziert werden, st6ren merklich. Das trifft fiir die Elemente Gold, Palladium, Quecksilber, Silber und Wismut zu. Am stfirksten ist die Beeinflussung dutch Wismut. So- lange Kupfer und Wismut in annfihernd gleicher Kon- zentration vorliegen, werden zwei nahe beieinander liegende Signale registriert. Aber schon bei etwa 10fachem Wismutfiberschul3 ist ein gesondertes Kupfersignal nicht mehr zu erkennen. Die St6rungen, die diese ftinf Metalle verursachen, lassen sich zwar verringern, wenn die Elektrolytzusammensetzung ge- findert wird, aber doch nur in verh/iltnism/il3ig gerin- gem MaBe.
Von den wichtigsten Anionen verursachen nut Chloridionen, wenn sie in ungeffihr 100fachcm Ober- schul3 vorhanden sind, eine Signald~impfung. Aus
141
Tabeile 3. Ergebnisse invers-voltammetrischer Bestimmung yon Kupfer nach dessen Chelatisierung mit Dithiooxamid in verschiede- nen Proben Trinkwasser
Probenherkunft Kupfer Relat. Ein- gtg/1 Standard- gesetztes
abweichung Proben- (N = 11) volumen
ml
Universitfit- 5 l 0 0,04 0,2 Gesamthochschule Siegen ~
Universitfit- 22,5 0,08 1,0 Gesamthochschule Siegen b
Siegen-Geisweid 0,5 0,09 9,0 Siegen-Weidenau 1,0 0,20 9,0 Bad Nauheim 1,5 0,15 9,0 Erlangen 2,0 0,10 5,0
a Nach kurzem Ablauf b Nach mehrst/indigem Ablauf des Leitungswassers
diesem Grunde ist das Agar-KNO3-Diaphragma not- wendig, das auch die Diffusion yon Silberionen aus der Bezugselektrode in den Elektrolyten verhindert.
Kupferbestimmung in Trinkwasser
Ein Beispiel zur Anwendung des Dithiooxamidverfah- rens ist die Bestimmung des Kupfergehalts yon Trink- wasser (Tabelle 3). Die in vorkonditionierten Polyethy- lenflaschen aufbewahrten Proben wurden unfiltriert nach der unten angegebenen Arbeitsweise voltamme- triert.
Nur die beiden ersten Proben stammen aus dersel- ben Wasserleitung, die bis zur Versorgungsstelle aus Kupferrohren besteht. Die Proben unterscheiden sich in bezug auf die Verweilzeit des Wassers in der Leitung. Wie das unterschiedliche Ergebnis dieser beiden Analy- sen zeigt, kann sich der Kupfergehalt des Wassers bei lfingerem Stehen um den Faktor 20 vergr6Bern.
Bei den iibrigen Proben handelt es sich um solche, die in verschiedenen Haushalten entnommen wurden. Die Probenahme erfolgte zu einer beliebigen Tageszeit, nachdem das Wasser 3rain abgelaufen war. Soweit bekannt, wird das Trinkwasser bei diesen Probenahme- stellen durch verzinkte Eisenrohre geleitet.
Die relat. Standardabweichung ist bei den Proben mit den kleinen Kupfergehalten verhfiltnismfiBig hoch. Dieser Nachteil mag jedoch dureh die Tatsache kom- pensiert werden, dab man mit geringen Probenvolu- mina auskommt und auf eine chemische Anreicherung des Kupfers verzichten kann. - Ffir die Durchffihrung einer Bestimmung mit zwei Eichzusfitzen werden unge- ffihr 15 rain ben6tigt.

142 Fresenius Z. Anal. Chem., Band 306 (1981)
D i s k u s s i o n der Ergebnisse Exper imente l le r Teil
Ein ffir die Vo l t ammet r i e von Kupfe r interessantes Ergebnis ist zun/ichst, dab die vier Chela te yon Kupfe r mit D i th iooxamid , 2 ,2 ' -Bichinol in , N e o c u p r o i n und Ba thocup ro in an der Koh lepas t e -E l ek t rode oxidier t werden k6nnen; wenn auch mi t sehr unterschiedl icher Empfindl ichkei t . Das Verfahren mit D i t h i o o x a m i d ist im Vergleich mit dem an einer Quecks i lbe re lek t rode gent igend empf ind l ich und auch nachweiss ta rk genug.
Vor al lem ist es aber bedeu tend selekt iver als eine
Bes t immung von n icht -chela t i s ie r tem Kupfer , weil das Ana lysens igna l um e twa 300 mV verschoben im posi t i - ven Poten t ia lbere ich auftr i t t . D a d u r c h erg ib t sich die M6gl ichkei t , K u p f e r als E lementspur in anorgan i schen Subs tanzen zu bes t immen, deren Mat r ixe lemen te nur im negat iven Poten t ia lbere ich e lek t rochemisch akt iv sind. Dies ist der wichtigste Vorte i l des vo l t ammet r i -
schen Kupfe rd i th iooxamid -Ver fah rens . Von den M6gl ichke i ten zur A n w e n d u n g dieses
Verfahrens ist die Bes t immung yon Kupfe r in Tr ink- wasser, wie sie hier beschr ieben wird, sicher das einfach- ste Anwendungsbe i sp ie l . Das Verfahren b e d a r f auger der H e r s t e l l u n g der Reagens l6sung ebensowenig einer P robenvo rbe re i t ung wie die bekann ten vo l t ammet r i - schen Bes t immungen von K u p f e r an Quecks i lbere lek-
t roden. Ein weiterer Vortei l des D i th iooxamid -Ver f a h re ns
ist j edoch , dab es zwei sys temat ische Fehle r vermeidet ,
die gerade bei der Vo l t ammet r i e von K u p f e r an Queck- s i lbere lek t roden auftreten. Besonders die geringe L6s- l ichkei t von Kupfe r in Quecks i lber [8, 13] k a n n zu un- kon t ro l l i e rba ren Kupfe rver lus ten und dami t zu e inem sys temat ischen Fehler ffihren. A u c h der an Queck- s i lbere lek t roden auf t re tende s ta rk anste igende G r u n d - s t rom im Bereich des Kupfers igna ls stellt eine Feh le r - quelle dar , die aber durch das , , subt rakt ive Ver fah ren" mi t e inem 4-E lek t rodensys tem (zwei Arbe i t se lek t ro -
den) besei t igt werden kann [101. Ein andere r sys temat ischer Fehler , der, wenn m a n
Quecks i lbe re lek t roden benutz t , n icht zu besei t igen ist u n d mi t dem st~indig gerechnet werden mug, ist die neben Kupfe r m6gl iche M i t a b s c h e i d u n g von A n t i m o n und Wismut , die zu S t6rungen verschiedener A r t A n l a g geben kann, sofern m a n n icht spezielle Lei te lekt ro ly te
zu ihrer Verme idung einsetzt. Die in neuester Zei t entwickel ten Gold-Zwi l l ings -
e l ek t roden zur Bes t immung kleiner Kupfe rgeha l t e z.B. in Meerwasse r [11] t ragen ebenfaUs zur Verr in- gerung sys temat ischer Fehle r bei. A u g e r d e m sind sie sehr nachweiss tark . - Insgesamt gesehen, stellt sich wohl heraus, dab Fes t e l ek t roden aus Koh le oder G o l d ftir K u p f e r b e s t i m m u n g e n im N a n o - und Pico- g rammbere i ch geeigneter als Quecks i lbe re lek t roden
sind.
Gerdte. Polarograph E 506 mit dazugeh6render Ausrfistung. Ar- beitselektrode: Kohlepaste-Elektrode EA 267; Bezugselektrode: Ag/AgCI-Elektrode EA420 in gesfitt. KC1-L6sung mit Agar- KNOa-Diaphragma; Hilfselektrode: Platinelektrode EA 202. AUe Gerfite geh6ren zum Lieferprogramm der Fa. Metrohm (Herisau, Schweiz).
Reagentien, L6sungen. Kupfer-Stamml6sung mit 1,0 mg/ml; Kupfer- Standardl6sung zur ~-Eichung mit 10,0 ~tg/ml. Acetatpufferl6sung pH 4,3 : 200 g Ammoniumacetat (krist.) p. a. werden mit 100 ml dest. Wasser und 200ml Eisessig 96 % versetzt. Zur Entfernung von Schwermetallen wird die Pufferl6sung mit 0,2 g Natriumdiethyldi- thiocarbamat versetzt und 15 min geschtittelt. Danach wird in der Pufferl6sung 0,5 g Aktivkohle aufgeschlhmmt, die Suspension aber- reals einige Minuten geschfittelt und fiber ein Aktivkohlescheibchen filtriert [3]. Reagensstamml6sung: 0,1% Dithiooxarnid in abs. Etha- nol; zur Herstellung der ftir die Bestimmung ben6tigten Reagensl6- sung werden 10ml der Stamml6sung mit einem Gemisch gleicher Volumina von abs. Ethanol und bidest. Wasser auf 100 ml aufgeftillt. - Kohlepaste: 5 g Spektralkohle RWB (Ringsdorff-Werke, Bonn- Bad Godesberg) werden mit 1 ml Paraffin (flfissig) zu einer Paste verrieben. - Agar-KNO3-Diaphragma: 1,5 g Agar-Agar werden in 50 ml bidest. Wasser dutch langsames Erwfirmen gel6st und danach 10 g Kaliumnitrat in die L6sung eingetragen. Die klare Agarl6sung wird in den unteren Teil des Schliffes der Bezugselektrode eingegos- sen, so dab die erstarrte Agarschicht nicht mehr als ungef'~ihr 5 mm betrggt. Das Frittenrohr unterhalb des Schliffes ist mit verdfinnter Acetatpufferl6sung (1:5) geffillt und durch ein Schlauchstfick mit dem Schliffteil verbunden [7]. - Es wurden ausschlieglich z.A.- Chemikalien der Fa. Merck, Darmstadt, verwandt,
Arbeitsweise. Man pipettiert in die Mel3zelle I ml Pufferl6snng, danach zwischen 1 und 8 ml Probenl6sung und ergfinzt das Volumen mit bidest. Wasser auf 10ml. Nun wird die MeBzelle am Stativ mit den bereits meBfertigen Elektroden befestigt und der Magnetrfihrer eingeschaltet. Die L6sung zu entlfiften, ist nicht erforderlich. Zu der L6sung in der Mel3zelle werden 60 gl Reagensl6sung zugesetzt und sofort mit der Vorelektrolyse bei der Einstellung yon -0,4V begonnen. Nach der Anreicherungsdauer von 3 rain schaltet man den Rtihrer ab, stellt das Startpotential auf 0,0V ein und 1/il3t nach insgesamt 30s Wartezeit die Spannung mit 10mV/s in positiver Richtung ablaufen (GerS.teeinstellung: U = 2,0V, tdrop = 0,4 und mm/tarop = 0,5). -- Zur Konzentrationsbestimmung nach der Zusatzmethode wird die Messung zweimal mit einem Zusatz yon 20 und 40 gl der Kupfer-Standardl6sung (10lxg/ml) wiederholt. Dazu wird sowohl die Megl6sung als aueh die Oberflhche der Kohlepaste- Elektrode jedes Mal erneuert.
Literatur
1. Brajnina, Ch. Z.: Talanta 18, 513 (1971) 2. Brajnina, Ch. Z.: Stripping Voltammetry in Chemical Analysis.
New York: John Wiley & Sons, 1972 3. Jackwerth, E., Lohmar, J., Wittler, G.: Fresenius Z. Anal.
Chem. 270, 6 (1974) 4. Jacobs, W. D., Yoe, J. H. : Anal. Chim. Acta 20, 332 (1959) 5. Monien, H.: Chem.-Ing.-Techn. 42, 857 (1970); 43, 666 (1971) 6. Monien, H., Zinke, K. : Fresenius Z. Anal. Chem. 250, 178 (1970) 7. Monien, H., Jacob, P., JS.hnisch, B. : Fresenius Z. Anal. Chem.
267, 108 (1973)

H. Monien et al. : Bestimmung von Kupfer in Trinkwasser 143
8. Neeb, R. : Inverse Polarographie und Voltammetrie. Weinheim: Verlag Chemie, 1969
9. Popov, G. N., Pnev, V. V., Zacharov, M. S. : Z. Anal. Chim. 27, 1760 (1972)
10. Sipos, L., Kozar, S,, Kontu~i6, I., Branica, M.:J . Electroanal. Chem, 87, 347 (1978)
11. Sipos, L., Golimowski, J., Valenta, P., Niirnberg, H. W.: Fresenius Z. Anal. Chem. 298, l (1979)
12. Tsch6pel, P., Kotz, L., Schulz, W., Veber, M., TNg, G.: Fresenius Z. Anal. Chem. 302, 1 (1980)
13. Valenta, P., Mart, L., Riitzel, H. :J. Electroanal. Chem. 82, 327 (1977)
14. Vassos, B. H., Mark, Jr. H. B. : J. Electroanal. Chem. 13, l (1967) 15. Vydra, F., Stullkovfi, M., Pet~.k, P. : J. Electroanal. Chem. 40, 99
(1972)
Eingegangen am 24. Oktober 1980





![DIE NEUE THEORETISCHE MOPED- PRÜFUNG · 1. Verkehrszeichen Anmerkung Basis Prüfungsmodul Moped Mopedauto Kapitel Zeit [min] 2. Verkehrszeichen invers 3. Rechtskunde Vorrang 4. Lückentexte](https://img.pdfslide.org/doc/110x75/5f0722387e708231d41b78f8/die-neue-theoretische-moped-proefung-1-verkehrszeichen-anmerkung-basis-prfungsmodul.jpg)