
E'. Foemtei; F. Lange, 0. DroPbach u. W. Seidel. Scltweflige Saure uszo. 245
Beitrage zur Kenntnis der schwefligen Saure und ihrer Salze.
1. Uber die Zersetzung der schwefligen Saure und ihrer Sake in wii8riger Liisung.
Von F. FOEBSTER, F. LANGE, 0. DBOSSBACH und W. SELDEL. Mit 9 Figoren im Text.
1. Einleitung. \Vie die salpetrige Saure, die phosphorige SBure, die Chlorsaure
und andere Sauren mittlerer Oydntionsstnfen das Bestreben zeigen. in wallriger Liisung von selbst in die hochste Oxydationsstufe deb sburebildenden Elementes und in eine tiefere von ihm uberzugelien, so neigt auch die schweflige Saure dam, von selbst in die bestiindigere Kchwefelsaure sich au verwandeln. Schon PRIESTLEY hat festgestellt , dall dabei aber als niedere Oxydationsstufe elementarer Schwefel Pntsteht . Die Umsetzung kann durch die Eruttogleichnng
3H,SO,-t 2H2S04 + 8 + H,O
wiedergegeben werclen. In gleicher Richt,ung verliinft, der Zerfall des Schwefeldioxyds bei hoher Ternperatur, d h r e n d die festen schwefligsanren Sillze der Allinlien bei Rotglut in Sulfat und Sulfid sich umsetzen, ahnlich wie festes Chlorat in Perchlorat und Chloricl in der Hitze sich vermnndelt. Die Neigung des vierwertigen Schwefels zum Ubergange in den sechswertigen ist also allgemein. Es fragt sich aber, in welcher vlieisc der Ubergang der dabei ihre Wertig- lieit vermindernden Schwefela tome in elementaren Schwefel TW
sich geht. Die Antwort hierauf darf man am ehesten erwarten aus cler Untersuchung der in wiifiriger Losung vor sich gehenden Umsetzung der schwefligen Siiure bzw. ihrer sauren Salze. Fur diese ist zu prufen, ob Cfleichuiig (1) '(Vlrldich dem Meohanismus des Umslztzes ent- spricht, oder ob nicht zunschst niedere Oxydationsstufen des Schwefels dabei sich bilden, die - unbestandiger als die schweflige Sgure -
2. anorg. U. allg. Chem Bd. 138. 17

246 El Foerster, li: Langc, 0. Dropbaeh wad W. Seidel.
ihrerseits wtiiter zerfdlen und erst ddurch zur Abscheidung des freien Schwefels fiihren. Dieser Fmge gelten die im folgenden mit- zuteilenden Untersuchungen.
Der Zerfall der schwefligen Saure in wSBriger Losung verlauft sehr trage, so daB sein von BERTHOLLEI niid BERGMAN in Bestatigung von PRIESTLEY s Beobachtungen nufs Neue festgestellter Eintritt von FOURCROY und VAGQUELIW wieder best,ritten wurde. Erst, C. BEITNER~) hat den Sachverhslt sichergestellt , indem er ermittelte, (la13 in der Tat waBrige Losu-ngen von schwefliger Saure sich in $chxefelsaure und Schwefel zersetzten, wenn er sie im geschlossenen Rohr anf mindestens 170--180° erhitzte. Dabei bemerkte er die iiberrascliende Tatsache, daB in verdiinnterer Losung dieser Umsatz weit schneller verliiuft als in konzentriertrr. Wahrend in sehr ver- dunnter Losung 2 Stunden geniigten, um bei 170-1800 den Umsa,tz vollstandig zu machen, war in konzeiitrierter Losung auch nach 8 tiigigem Erhitzen der Geruch nach Schwefeldioxyd noch nicht verschwunden. Das wurde aber erreicht, als die konzentrierte Losung von schwefliger Siiure 14 Ta,ge auf 2000 erhitzt wurde. ,4uch BERIHELOT~) stellte et8vrra gleichzeitig fest, dnB bei 150-1800 die schweflige Siiure sich im Sinne der Gleichung (1) zu zersetzea vermag. Bei 110-115° aber lionnte er Gleiches nicht errnitteln.
Etwas eingehendere Mitteilungen iiim den Vorga,ng bracht,e eine 1913 von E. JUNGFLEISCII und L. BRUNEL~) veroffentlichte Arbeit. Sie fanilen, daIj er, wenn mch CuBerst langsam, schon bei gewohnlicher Temperatur sich abspielt : im zugeschmobenen Rohr im, Dunkeln aufbewahrt, zeigte eine waRrige Losung von schwefliger Saure nach 10 Jahren einen deutlichen Gehalt an Schwefelsaurc. Auch bei loo0 ist der Zerfall noch sehr langsam. Er mwrde aber - im Gegensatz zu GEIINERS Beobachtungen - in konzentrierterer Losung als lebhafter gefunden. In solcher trat nnch 72 stiindigem Erhitzen auf loOD eine schwache Cklbfiirbung auf, wie sie auch in der bei gewohnlicher Temperatur lange aufbemhrten Probe sich gezeigt hatte; bei 96stiindigem Erhitzen hatte sich diese Gelb- farbung erheblich verstiirkt. Erst jetzt begann die Ausscheidung von Schwefel, wahrend gleichzeitig die Gelbfiirbung zuriickging. Da diese gelbe Losung Indigo entfarbt, nalimen JUNGFLEISCH und
Lieb. A m . 129 (1864), 350.
Compt. r e d . 356 (1913,). 1719. 2, Ann de Chim. Phys. 171, 14 (1898), 289.

Zersetmng der schwefligen Saurs und ,ihw Sake ,in wlipriger Losung. 24 7
BRUNEL an, da13 die Umsetzung primar zu hyposchwefliger Saurel) und Schwefelsaure fuhrt :
3S0, + 2H20 -+ H2S204 + H2S04
H2S204 -+ S + H2S04.
(2)
(3)
und jene erst zur Bildung des Schwefels AnlaB gabe:
Die Xiiglichkeit eines solchen Vorganges schliel3en sie aus der Beob- nchtung, daB eine Losung von hyposchwefliger Saure beim Erwarmen Schwefel und Schwefelsaure liefert. Den genaueren Vergleich im quantitativen Verlauf der Vorgange haben sie aber ebenso unter- lassen, wie die nahere Verfolgung ihres Befundes, ,,daS auch noch andere Realrtionsprodukte bei der Umsetzung auftreten".
Wie durch gesteigerte Temperatur, wird der Zerfall der schwef- ligen Saure auch durch Belichtung sehr beschleunigt. Dies wurde zuerst von LOEW~) beobachtet ; er fand, daB im zugeschmolzenen Glasrohr der Sornmersonne ausgesetzte T,osungen von schwefliger Saure, nachdem sie zwei Monate lang klar geblieben waren, Schwefel i\ bschieden und nun auch Schwefelsaure enthielten.
Der schwefligen Saure analog verhalten sich ihre sauren Salze. BARBAGLJA und G U C C I ~ ) fanden, daB gesattigte Lbsungen der Bi- sulfite von Xatrium, Ammonium und Magnesium sich bei 1500 ver- IiiiltnismaBig schnell, in 7 Stunden, vollstandig im Sinne der Brutto- gleichung :
3KaHSO,+ Nii2S04 + NaHSO, + S + H20 (4)
umzusetzen vermogen. Bei 1400 aber fanden sie fur eine Natrium- liisulfitlosung in 8 Stunden noch keine Umsetzung.
In der Folgezeit ist letztere Umsetzung mehrfach mit Ruck- sicht auf ihre technische Bedeutung untersucht worden. Dcnn unterliegen ihr Cnloinmbisulfitlosungen wahrend ihrer gewohnlich bei 120-1300 im hutoklaven vor sich gehenden Einwirkung auf Holzstoff, so verlieren sip dadurch einerseits mehr oder weniger weit ihren Wirliungsgrad, bevor sie die Extralition des Holzstoffes durchfiihren, andererseits kann, da diese Bisulfitlosungen auch schwefligc! Sauure enthalten, die entstehende Schwefelsaure zu starker
1) Durch diese Bezeichnung wird die alte unzweckmiiBige Bezeichnung ,,hydroschweflige" Saure ersetzt; vgl. K. A. HOFMAXN, d~wrganische Chemie, 4. Aufl. S. 166.
2, Jahreaber. f. Chenz. (1873), 164. 3) Ber. d. D. Chem. Cea. 13 (1886), 2325.
17*

248 F. Foeruter, l? Lange, 0. Dropbach und W. Sei&l.
Zerstorung der Ligninstoffe, zur Entstehung geschwiirzter Produkte fuhren. Die Untersuchungenl) iher die naheren Umstiinde, unter denen diese Storungen eintraten, lehrten, da13 dies einerseits geschah, xt-enn die Bisulfitlosungen freien Schwefel, andererseits, wenn sie Selen enthielten, wenn also in dem zu ihrer Herstellung benntzten Schwefeldioxyd freier Schwefel oder Selen bzw. Selendioxyd ent - halten war. Damit wLtren Schwefel nnd Selen als Katalysatoren fur den Zerfall der Bisalfite erkannt, von denen namentlich das letztere sich XIS ungemcin wirksam erwiesen hat, da schon sehr geringe Mengen von ihm, 0,3--0,4 mg Se nuf 1 Liter der Sulfitlosnng. die schadliche Xersetzung dieser Losungeu herbeizufuhren vermochten.
In diesem immerhin noch wenig geklarten Zustandez) befand sich die Angelegenheit, als der eine von uns im Sommer 1914 Herrn F. LANGE veranIaGte, sie einer genaueren systematischen Unter- suchung zu unterziehen. Der Kriegsausbruch unterbrach aber die Versuche.
Inzwischen erfuhren unsere Frslgen nach zwei Seiten eine weitere Forderung. Da Ammoniumbisulfit bei seiner Zersetzung Ammoniumsulfat gibt, limn dieses auf solchem Wege hergestellt werden, ohne da13 es erforderlich ist, Schwefeldioxyd durch einen besonderen Oxydationsvorgang in Schwefelsaure uberzufdxen; man gewinnt vielmehr neben dem Ammoniumsulht aus einem Teil des vom Ammoniali gehundenen Schwefeldioxyds noch wertvollen elementaren Sehwefel in sehr reiner Gestalt. Von diesem Gesichtspunkte aus wurde bei der Badischen Anilin- und Sodafabrik die Umsetzung eingehender untersucht. Dariiber hat Herr BOSCH niiher berichtet '3) Es ergab sich, daB die Umsetzung konzentrierter Ammoniumbisulfit- losungen bei looo im Autoklaven unter starker Temperaturerhohung explosionsartig vor sicli geht, da der Vorgang autokatalytisch ver- lauft, fndem er durch den von ihm erzeugten Schwefel sich selbst aufierordentlich stark beschleunigt . Nach dieser Erkenntnis war es moglich, durch Zugabe von, Schwefel oder anderen Katalysatoren, unter denen besonders das Selen hervorgehoben wird, den Vorgang bei etwas niedrigerer, uud dann durch Kiddung gut aufrecht zu erhaltender, Temperatur und doch in bequemer Zeit durchzufiihren.
l) P. KLASON, Papierzeitzlng 85 (1910), 374, 415; TH. KIAER, Dip1.-Arb.,
2, Vgl. G. N. LEWIS, M. RANDALL und F. R. V. RICHOWSKI, Jount. Americ.
3, 2. f. Elektrdma. 34 (1918), 306f.
Dresden 1911.
SOC. 40 (1918), 359.

Zersetxung der sohwefligera Saure uttd ihrer Sake in wupriger Lomng. 249
Er gibt, wie Pormel (4) l e h t , BUS Ammoniumbisnlfit neben Ammon- sulfat auch Bisulfat ; ciemzufolge entweicht , wenn man Bisulfit sich zersetzen l&Bt, &was Schwefeldioxyd. Doch genugt es, statt von reinem Bisulfi t von einer Losung von 2 Mol Ammonbisulfit und 1 1101 Aminoiisulfit auszugehen, welche sowohl fur Ammoiliak wie fur Schwefeldioxyd so kleinen Psrtialdruck besitzt, da13 sie ohne Verlust auch zur Absorption von an Schwefeldioxyd armen Gasen clienen bnn. Eine solche Ausgangslosung lcann glatt in Ammonium- sulfat nnd Schwefel verwandelt werden. Bezuglich des Reaktions- inechanismus gibt BOSCH an, daJ3 der Vorgang hochst wahrscheinlich Yiber Thiosulfat und Polythionnt fuhrt , eine Auffassung, zu der schon KLASON gelangt war .I) Eine weitere technische Verwertung der Bidfitxersetzung strebte STREHLENERT anz), indem er die Ablauge der Sulfitcellulose im verbleiten Autoklaveii auf 2000 erhitzte, um niit Hilfe des hierbei entstehenden Bisulfats die in der Losung enthaltenen Ligninstoffe in unlosliche kohlige Substanzen zu ver- wandeln und so den Heizwert der in der Sulfitablauge vorhantlenen organischen Verbindungen verwerten zu konnen.
So mertvoll fur die Theorie der Eisulfitzersetzung die von BOSCH gernachten Angaben waren uber die Rolle, die dabei der Schwefel a,lq Katalysator spielt, sowie uber das Auftreten von Thiosulfat nnd Polythionat als Zwischenprodukte, so gaben sie doch nooh kein vollstandiges Bild uber den Reaktionsmeohanismus ; aueh blieb zu enlscheiden, wie jene Erfahrungen uber die Zwischenprodukte sich mit der Vermutung von JUNGFLEISCH und BRUNEI,, daS die hypo- schweflige Same die Zersetzung vermittele, sich vereinigen k B t .
Deshalb erschien es nicht uberflussig, nach Ihiegsende die im Sommer 1914 hegonnenen Untersuchungen wieder aufznnehmen.
2. Die bei den Untereuchungen benutzten analytiechen Verfahren. Fur die Verfolgung der Selbstzersetzung der schwefligen Saure
und ihrer sauren Salze bedurfte es der analytischen Bestimmung unxersetzten Ausgangsmaterials sowie von Thiosulfat, Polythionat , Hyposulfit , Sulfat und Schwefel.
Fur die Bestimrnung des freien, des Polythionat- und des Sulfat- schwefels wurde in der von dem einen von tins und A. H O R N I G ~ ) kurz- lich beschriebenen Weise verfahren.
1) 1. c. z, Pa/perfabrikant (1917), 143, 167, 257, 376, 383, 395, 409, 421. 3, 2. anmg. u. allg. Chem. 1% (1922), 106.

F. Foerster, F. La?&$e, 0. Dropbach und W. Seidel.
Die in der LDsung verbliebene schweflige SBure bzw. das Bi- sulfit mufiten, um Verfliichtigung der ersteren und Oxydat,ion durch Luftsauerstoff auszuschliefien, moglichst rasch und ohne Filtration der Losung bestimmt merden. Das war stet< mbglich, dit der ab- geschiedene Schwefel sicli leicht Mar absetzt. Daher wurde ein IJestimmter Teil der Lijsung sbpipettiert - bei Untersuchung der Losungen der schwefligen Skure nach deren Einstellung in Ei:, und mit eisgeliiihlter Pipette - und in iiberschussige Jodlosung einfliefien gelassen und deren UlwrschnB mit Thiosulfat zuriick- gemessen.
bus einem anderen gemessenen Tcil der Liisung wurcle - Lei Hisnlfitlosungen nach genugendem Ansaucrn mit SalzsBure - durch einen Strom reinen Stickstoffs das Schwefelclioxyd griincllich sb- geblasen. Trat dabei eine Abscheidung von Schwefel auf, so zeigte sie das Vorhandensein von Thioschwefelsiiure an. Der hierbei ab- gescliiedene, gewohlich schon durch die voni Stickstoffstroni wr- snlal3te Flussigkeitsbeweguiig, im lkdarfsfalle durch Zusatz von ein wenig Lanthanchlorid l), znsflminenjie1J:illte Schwefel wurde ahfiltriert und nach der Gleichung
ditr:ius die rorhandene Thiosulfatmenge sowie der ihr eiitsprechendr Jodverbrauch berechne t und dieser von dem gesamten Jodverbraucli in Abzug gebracht. Dieses Verfalirrn ist nicht genaii; clemi, da die freie Thioschwefelsaure sicli immer zu eineni Teil in Pentathionsaure T-erwandelt, bleibt die am dem abgeschiedenen Schwefel ermittelte Menge Thioschwefelsaure etwas hinter der vorhandenen zuriicli. Durch Versuche mit Isekannten Mengen Thiosulfat murde festgestellt, daB bei unserer Arbeitsweise dieser Fehler ziemlicli konstant TO/,,
des Thiosulfatschwefels betrug. Da es sich sehr oft nur um ganz lileine Mengen dieser Saure hsndelte, lionnte er oft 1-ernachlilssigt werden; bei grijfieren Rlengen wurde cine Korrelitur von + 70/0 ilires Betrages angebracht.
Indigo entfgrbte, und gegebenenfdls die Rleiige einer auf Na,S,O, eingestellten, verdunnten ammoniahlischen Losung von indigosulfo- wurem Natrium ermittelt, 'die ein gegebenes Volumen der Losung mi ent f arbeii vermeg .
Zur Untersuchung auf Hyposulfit wurde gepriift, ob die Lijsung .
l) E. HEINZE, ,702r.m. prakt. Clmn,. 99 (1919), 138.

Zersetxung der schwefligen Sawe und ihrer Salxe in wabnger L6sung. 25 1
3. Die Selbstzersetznng der fieien sohwefligen Saure. (nach Versuchen von F. LANGE).')
Die Durchfiihrung und Verfolgung der Zersetzung der schwef- ligen Saure wird, da sic erhebliche Zeit und gesteigerte Temperatur erfordert , zweckrniil3ig im geschlossenen Rohre vorgenommPli.
Urn dabei von Losungen behnnten Gehalts an schwefliger Saure auszugehen , wurde jedes Urnfullen vermieden , die Lo- sungen unmittelbar in den fur die Zersetzung dienenden Rohren hergestellt. Zu diesem Zwecke muden diese Rohre an einer Stelle ilires oberen Teils soweit ausgezogen, dal3 sie spiiter hier leicht abgeschmolzen tverden konnten. Dann wurden sie mit der erforder- lichen, gemessenen Menge ausgeliochten Wassers lwschiclit nnd in Eiswasser gestellt, und nun wurde auf ihre obrre Mundung ein doppelt. durchbohrter Stopfen auf gesetzt . Durcli dessen eine Bohrung ging ein enges, durch die verjiingte Stelle noch leicht liindurclixnfiihrende~ Glasrohr auf den Boden des Rohres, miihrrnd in iler andcren Bohrung ein kurzes Rohr sich befand, das zu einrr mit Natronlauge beschickten Flasche fuhrte. Auch diese trug in einem doppelt durchbohrten Stopfen ein Einleitnngsrohr nnd andererseits ein liapillar ausgezogenes -4bleitungsrohr, das mit der Luft in Verbindung stand. In das so vorbereitete Hohr wurde nun ein langsanier Strom voii Schwefel- dioxyd von genau gemessener Geschwindigkei t eine ebenfalls genau gemessene Zeit hindurch eingeleitet . Der dabei nicht vom Wasser anfgenommene Teil des Schwefelclioxyds wurde von der vorgelegten Xatronlauge gelost . Sobald die gewiinschte Menge Schmefeldioxyd zugef&rt war, wurden Stopfen und Einleitungsrohr cntfernt und das noch im Eismsser stehende Rohr sofort abgeschmolzen. Darauf wurde die von der Na t ronlauge aufgenommene hfenge cles Schwefel- dioxyds jodometrisch bestimmt ; durch ihren Abzug von der ins- gesamt eingeleiteten ergab sich die im Rohr geloste Xenge an schwefliger Siiure. Der freie Gasraum uber der Losung murde stets so klein wie moglich gehalten ; das durch die Losung hindurchgeleitete Schwefeldioxyd verdringte aus ihm die Luft.
Die Ermittelung der insgesamt zugefiihrten Menge von Schwefel- dioxyd erforderte eine Vorrichtung zur genauen Ermittelung nnd Einstellung der Stromungsgeschwindigkeit dieses Gases, das dam aus flussigem, in einer Eisenbombc enthaltenem Schwefeldioxyd ent- wiclielt tvurde. Dezu erivies sich ein Striimungsmesser. an welchem
Dissertation. Dresden 1919.

252 $! Ebersler, F. Lange, 0. Dropbmh ujtd W. Seidel.
cler h i m Dnrchtritt des Gases clurch eiii kapillsres Rohr cintretende Druckabfall an &em einpfinclliclieii Flussigkeitsmanometer gemessen wnrdel), als sehr zwecliiniiBig. Er wnrde zuniichst geeicht, indeni m m bri bestimnitcw husschl8gen des Nanometers wv8hrcnd einrr qt.iian gemessenen Zeit einen Sch~~~efeldioxydstroni dnrch ihii hin- ,lurch uiid d a m in Satronlauge treteii lieB, und die T ’ O ~ dieser auf- genonimeiien Mengen Schwefeldioxyd jodomrtrisch bestinimte. Auf .;olehe TVeise war es nioglich, wenti der Stromungsmesser imrnrr h i e t w cIer glricheii Teinperatur beiiutzt wurde, an ihm ohne weiteres die in bestimmter Zeit, 1iiiidnrch~egangL.iicri Gewichtsmengen ,211
Schwfeldioxyd niit, hiiueichencl rr Gonauigkeit abzulrsen. Die so vorbereiteten Rolire ~ ~ u r d r n , iind zwar ineist mehrere
zu gleicher Zeit, in Pinem Iio1iei1, \ on untrn durch einen Breiiner zu heizeiidcn Eisenzylindrr ini Kasser- oder im Olbade bestimmte Zeiten liinclurcli auf der Versuchstemprrat~ir gehalten. Nach deren ljrrricliguiig w-ur&> iler Breniier gelijwht und die Rohre ini Bade c~lralten gelassea, x-obei auch cler zeitliche ‘I’rmperaturruckgaiig beobachtet wurde. Nacli d.em ABkilhlen wirden die Rohre, iii denen clrr sbgeschiedene Sclin rfel nieist wllstiiiidig lilar sich abgesetzt hatte, geoffnet nod ihr Inhalt in clrr oben beschriebenen Weise i i m lysiert .
Die Ergelmishe dcr Ailill y e n bind, uin sit: in gut vergleichbarc. Form z u bringen, immer so berechnet, daB sie die in den verschieden& Verbinclnngen bzw. im freien Znstande jeweils gefundenen Jiengeii Schwefel sngeben. Ein T’ergleich der so wiedergefundenc-n mil der ids SO, angervandten Sch~wfelmenge rrlaubt eiiie Iiontrolle rler tnrlialtenen Ergehnisse.
Vorversuche lehrten, da13 bei 1000 rs tagelsngen Erhitzeiis i l e ~ Lostingen der sclnvefligen Siiure Bedurfte, nin auch nur die ron JUXGFLEISCH irnd BRUNEL beschriebene GelbfCirbung w-ahrnehrnbar imd kleine Sch\~,rfelsCiiireniPng~ii nachrveisbar zu niachen. Zweck- m&Biger erschien es tLiher, zunachst zur Erforschung des allgemeinen Verlaufes des Vorgnnges bei hiiherer Temperstur zu arbeiten ; einc solchc voii 1500 twiip< sicli hicrfur 81s besonders geeignet.
:L) Versuche be i 1500.
Dic Erldznng cler init cleii Losungeii cler schwefligeii Stiiire gofullten Rohrc \wrde ini @bade vorgenommen, die Rohre anf einern Drahtgcstelle in d a s I d t e 01 eingesetzt. Bis die Ternperatur von
1) Vgl. Z. nnyew. C‘bnz. %3 (1920), 116.

hselaung der schwefligen Suure und iher Sake i 7 t tuu,6riger Losulzy. 253
looo erreicht war, dauerle es etwa Stunik, bis 1500 eine weitere Stunde ; die Temperatur 77011 1500 lionnte niit Schwankungen von i 23" aufrecM erhalten nwden. Die Versuchszeit wurde von der Zeit, in der die Temperntur ~01-1 1500 erreicht war, bis zu der, bei welcher der Brenner gelosclit wurde, gezahlt. Das Bbkuhlen von 150" auf looo beanspruchte pine halbe Stunde, und von loo0 auf 5O0 eine weitere halbe Stunde. Dann konnten die Rohre ohnc Gefahr t w s dem Olbade herausgehoben werden. Gegenuber kurzeren Versuchszeiteiz \-on z. B. zwei Stunden sind also die Zriten des Snheizcns und Abkiihlens verhaltnismaBig betrzchtlich. D;t aber die Reaktion iiberhaupt nur trage verliiuft und in ihrer Geschmindig- lieit mit sinkender Temperatur in bekanntem MaBe stark abfallt , durften auch dann die vor und nacli der eigentlichrn Reaktionszeit T ' O ~ sich gehencleii Umsetzungen iiicht allzu schwr ins Gewicht fallen; sie sind bei der Bewertung der Ergehnisse auBer Betracht, geblieben.
Die erhaltenen Ergebnisse sincl in der folgenden Ubersicht 1 i s . 254) zusammengestcllt.
13ei allen diescn Vorsuchen, nuch denen von liiirzc hter Dauer, freier Schn rfel abgeschietlen, die Lijsnng blieb nbcr immer
farblos, ohne einc sichtbare Gelbfiirbung. Neben der Schwefelsiiure uiid dem freien Schwefel traten 31s Reaktionsprodukte Tor allem Polythionsiiureii auf, und zwar im mesentlichen TetmthionsBurr , rind in nzanchen Fallen geringe, rben nachweisbare 3lengen voii 'L'hioschwefelsaure. Letztere zeigen sich abrr nur dort, 1% o nocli verhiiltnismiiBig grol3e Anteile der schwefligen Siiuiure ubrig sinrl, sri es, daB die Versuche nur kdrzerc Zeit gedauert haben, sei es, da13 lioiuentriertere Ausgangslosnngen bcnutxt wurden. 8hnliches Vrgdb sich, nur w-eit cleutlicher, auch liinsichtlich der ain Ende der Ver- suche gefundencn 31engen der PolythionsBnren ; diese treten gegeii- iiber den ancleren Reaktionsproduktrii, znmal dem freien Schwefel, uin so starker hervor, je kiirxer die Versuchszeit ist, bzw. bei gleicher Versuchsdauer, je grol3er die Konzentralion der ubriggebliehenen SO, ist. Da die Entstehung der Polythionsiiuren vom angewandten SO,-Schwefel eine gewisse Menge in Ansprnch nimmt , bleibt das Verhiilhis des entstanderien SO,-Schwefels zum elementtaren Schwef4 um so weiter hinter dem nach Gleichung (1) zu erwartenden Werte 2 : 1 zuruck, je mehr der Polythionsiiureschmefel vom verbmuchtm Ausgangsmaterial ausmacht. Das tritt besonders bei Versuch Nr. 1 hervor, RO in verdiinter SO,-Losung bei kurzer Versuc21sdaner

-
-
33
2
ti
$$
*
a
-
~
1 2 3 4 5 6 7 8 9 10
11
12
13
14
15
16
17
18
19
20
*Is
so,
Tin-
:e
rset
zt
-
-
Ver
- uc
hs-
kyer
in
st
un-
den 2 2 3l
/,
3l/:
7 7 7 7 16
16
16
32
32
32
50
50
50
96
96
90
-
~
6' so
, in
lo
o cc
m
kung
s.
rolu
men
in
ccn
i
20
20
20
20
40
40
12
40
40
20
20
40
20
20
40
20
20
15
10
I5
0,03
0 Sp
ur
0,03
4 Sp
ur
0,02
7 ni
chts
0,
029
Spur
0,04
0 ni
chts
0,
022
nich
ts
0,01
4 Sp
ur
0,04
8 Sp
ur
0,02
3 ni
chts
?
,, ?
,,
Ang
ewan
d t
0,02
0 0,
100
0,03
8 0,
113
0,09
1 0,
150
0,09
1 0,
471
0,19
2 0,
232
0,27
0
gs
tbl
s so
,
0,42
5 1,
589
0,42
8 1,
555
0,49
1 0,
791
0,51
0 3,
770
0,79
6 1,
199
1,60
3
0,79
4 0,
803
1,67
3
0,79
4 1,
650
1,94
0
0,32
0 0,
585
1,36
1
0,42
1 1,
586
0,42
1 1,
550
0,48
5 0,
798
0,50
7 3:
753
0,79
0
1,50
5
0,78
2 0,
822
1,66
4
0,78
4 1,
644
1,92
5
0.31
1
1,19
8
g so
, in
10
0 cc
m
4,25
15
,97
4,27
15
,55
2,45
3,
94
8,50
X6
,4 3,97
12
,o
16,0
3,97
8,
02
3,97
16,7
16,5
19
,4
11,7
18
,3
4,27
25,l
21,9
35,3
24
,l
63,l
63,O
56
,s
38,9
79,2
57
,5
50,2
88,2
74
,9
63,2
95,6
73
,2
74,l
96.7
Ubt
rsic
h t 1
. V
e r s u
c hs t
e m pe
r at u
r 15
00.
0,09
2 0,
200
0,57
3
0,03
4 0,
424
0,50
0
0,01
0 0,
044
0,31
3
Gcf
und.
cn n
ech
Bee
ndig
ung
des
Erh
itzen
s
0,46
2,
oo
5,72
0,17
4,
23
5,OO
0,13
0,
87
4,lB
0,31
5 3,
15
1,23
8 12
,4
gs
51
s so
,
0,05
6 0,
215
0,08
4 0,
232
0,18
1 0,
310
0,18
4 0,
941
0,40
3 0,
475
0,55
2
0,45
1 0,
419
0,70
7
0,49
8 0,
810
0,93
1
0,20
0 0,
356
0,69
8
g so
, in
10
0 cc
m
0,69
2,
69
1,04
2,
90
1,12
1,
93
3,80
5,
87
2,51
5,
52
6,89
2,82
5,
02
8,81
3,ll
10
J 11
,6
3,32
8,
90
11,6
0,02
2 ,,
0,21
7 0,
203
0,03
4 ?
1 :: ~
0,34
9
0,01
5 ,,
0,23
8 0,
010
1 :: I 0,401
0,
033
0,46
1
0,00
2
0,00
3 0,
244
Ver
hkilt
- iis
SO
,- ic
hwef
el:
elem
en-
tare
m S
2 : 0
,715
2 : 0
,93
2 : 0
,95
2 : 0
,97
2: 1
,oo
2 : 0
,97
2 : 0
,98
2: 1,
oo
2 : 0
,95
2 : 0
,98
2 : 0
,98
2 : 0
,96
2 : 0
,97
2 :.0
,99
2 : 0
,96
2 : 0
,97
2 : 0
,99
2 : 0
,99
2 : 0
,99
2 : 0
,99

Zersetxung der schwefligen Saure und ilrer Sake i~ w?i,&>ar Liisung. 255
etvi-a halb soviel Polythionsiiure-Schwefel als SO,-Schmefel sich vorfand und das gedachte Verhaltnis 2 : 0,715 betragt.
Aus diesen Beobachtungen ergibt sich, daS - in Bestiitigung der Erfahrungen voii BOSCH uber den Zerfall der Bisulfite - die Selbstzersetxung der schwefligeii SBure wahrscheinlich uber l'hio- sehwfelsaure, jedenfalls iiber Polythionsliuren als Zwischenprodukte fuhrt . Als Produkte eines Nebenvorganges konnen sie nicht angeselien merden, da. sie sich d a m urn so reichlicher ansammeln miifiten, je weiter der Zerfall der scliwefligen SBure fortschreitet. Ferner wird es aus Versuch Nr. 1 wa,hrscheinlich, daS der primare Vorgang zwar bereitls Schwefelsiiure, aber nocli nicht elementureii Schwefel gibt, da13 also der Zerfdl der schwefligen SBure in der Tat zundchst neben c1c.m hiiheren Osyde des Schwefels SO, zu einem niederen Oxyde fuhrt,, welches zur Entstehung von ThioschwefelsBure und Polythion- saureir AiilaB gibt. Als solches kiiine SO in Betritcht, von dem sicli sowohl die Thioschwefelsaure (H,S,O, - H,O + .,SO), nls such die Pentathioiniiure (H,SSO, - H,O + 5SO) xbleitet. Erst dcr nachtjrBgliche Zerfall dieser Sauren gilit offmbar neben weiterer Sch-\i-efels$ure freien Schx-efc-1.
Vergleicht man den zeitlichen Verlauf unseres Vorganges init den beksnnten Gesetzen der Reakt,ionsgeschwindiglieit, nach denen die Zeiten gleichen Umsatzes je na.ch der Reaktionsordnung von der Xnfaiigskonzent.ration unabhiingig sind, oder sich umgekehrt wie die erste bzw. eine hohere Potenz von ihr verhalten, so zeigen die Ver- Ruche der Ubersicht 1 ein vollig abweichendes Verhslten. Denn sie lehren, daS vielfach die Zeiteii gleichen Umsatzes uin so kurzer sind, je kleiiie 1: die anftinglichrn Konzentrationen der schwefligen Sauren sind, so daB der Umsatz einer verdiinntwen Ldsung dieser Siiure schneller fortschreitet und vollstlindiger wird als der einer konzentrierteren. Im Anfaiige cles Vorgsnges rrscheint der beob- achtete Umsatz dlerdings als von der Snfangskonzentration ziemlich unabhangig. und erst im weiteren Verluufe der Umsetzung tritt dns angegebene Verhalten deutlicher hervor. Es wird durch die Kurven- zeichung Fig. 1 (S. 256) fur LBsungen der Anfangskonxentrationen von 4°/0 und 160/, SO, veranscha.ulicht. Diese beweist die Itichtigkeit der von GEITNER gemachten, von JUXGFLEISCH und BRUNEI, a,ber iiicht bestiitigten Beobachtungen. Seine Erkltirung h n n es niir dadurch finden, da.B der Vorgnng selbst sich eineii negativen Iiata- Igdator schafft, der mit steigender Konzentration irnmer wirlisa,mer wircl, a,lso bei hoherer A iifangskonzentrat,ion schon nach Irlcinerem
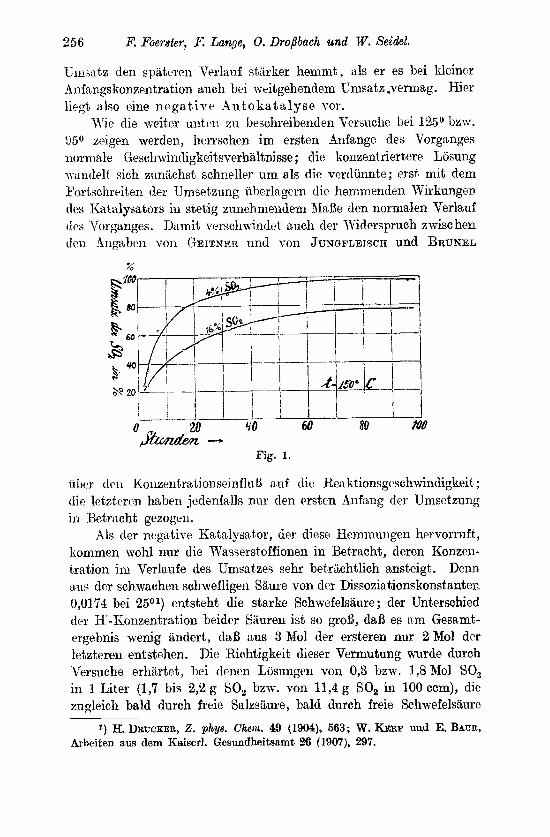
256 B! Fowster, I{; Lange, 0. DroPbach und W. See'deE.
Umsatz den spatercn Veylanf stiirlier hemrnt, als er es bei lileiner -4nfangskonzentration auch bei meitgehendem Umsntz .vermag. Hicr lipgt also eine nega t ive Au to l i a t a lyse vor.
W e die weiter unteii zn beschreibeiiden Versuche bei 125O Lzw. !W aeigen werden, lierrschen im ersten dnfange des Vorgnnges normale Gescli.\?iiiidigkeitligkeitsverh~ltnisse; die konzentriertere Liisung xanclelt sich znn&chst schneller urn als die vertliinntre ; erst mit dem Portschreiten drr Umsetzung iiherlagern die hemmenden Wirkungen des Katalysators in stetig zunehmendem AIaBe den normalen Verlauf ( 1 ~ s Tiorganges. Damit verschuindet auch der Tliiderspruch swisc hen dm rlngnhrn 17011 GEITNER und 5-011 JUNGFLEIWH und BRUNEL
., 4.
I
40 60 80 190
Fig. 1.
u h r t1t.11 2(oiizentratiolzseinflul3 nnf die Heahtioiibgeschwint~~eit; c I k leteteren haben jedenfalls nur den ersten -4nfang der Umsetzung in Betracht gezogen.
hl.; cler negative Katalysator, der diese Hemmungen hervorrnft, hommen wohl nur die Wasserstoffionen in Betracht, deren Konsen- tration im Verlaufe des Umsatzes sehr betrkhtlich ansteigt . Denn a u s (lor schwachen sohwefligen SBure von der Dissoziationskonstanteii 0,0174 bei 2501) entsteht die starke Schwefelsaure ; der Unterschied tier W -I<onzentration beider S8uren ist so grol3, daB es ain Gesamt- ergebnis wenig bndert, claB aus 3 Mol der ersteren nur 2 Mol der letzteren entstehen. Die Richtigkeit dieser Vermutung wtirde durch Versnche erhbrtet, bei denen Losungen voii 0,3 bzw. 1,s Mol SO2 in 1 Liter (1,7 bis 2,2 g SO, bem. von 11,4 g SO, in 100 ccm), die zngleich bald durch freie Salzsaure, bald dnroh freie Schwefelsaure
1 ) H. DRUCEER, 2. phga. Chem 49 (1904), 563; W. KERP uqd E. BAUR, -4rheiten aus dam Kdserl. Gesundheibmt 06 (19M), 297.

Zersetxung dw sehwofligen Saum und i b w Salze iu wuj’riger Li;szcng. 257
2 fach oder 4 fach normal maren, jedesmal 7 Stunden lang auf 1500 erhitzt wurden. NUP in der durch Schwefelsaure 2 fach normalen Losung von 1,7 g SO, in 100 ccm konnte dabei eine Schwefel- abscheidung beobachtet werden, die einem Umsatz von etwa 4OI0 entsprach; in allen anderen Fallen war ein Unisatz entwecler uber- haupt nicht, oder nur in Spnren nacliweisbar. Die H henimen also in der Tat sehr stark die Selbstzersetzung w5Briger Losungen voii Schwefeldioxyd. Eei den Versuchen der Ubersicht 1 stieg die Iion- zentration der durch den Unisatz entstandenen Schwefelsanre ill den nrspriinglich etwa lSO/O SO, enthaltenden Losungen snf et w i
3 fach normal, in den 4°/0 SO, enthaltenden auf 0,s fach norma,l, und gerade in jenen Fallen t,rsteii die Verziigerungserscheinungen besonders hervor .
Man konntc nuch an die Moglichkeit denken, da13 die Hem- mungen durch einen Gegenvorgang veranla5t seien. Als soleher kame nur die Einwirkung cier Schwefelsaure auf den Schwefel: 2H28O4 + S --t 3 SO, + 2H,O in Frage, die unter dem EinfluB konzentrierter Schwefelsfjnre, wie man weiS, in cler Tat cintritt. Da G. N. LEWIS~) angesichts d i e m Tatsache die Moglichkeit eriirtert hat, daB durch solche Gegenwirkung der Zerfall der wBBrigen Losungell der schwefligen Saure zu dem Gleichgewicht SH.$03 + 2&SO, -+ S + H,O fuhren konnte, wurde auch nicht unterlassen, durch den Versuch zu prufen, ob jener Yorgang auch unter den beim Zerfall walSriger Losungen von sohwefliger Sllure herrschenden Bedingungen wirklich vor sich geht. Schmefebiiurelosungen von 11 und 2W/, SO,, d. h. 1,37 und 2,87 Mol H,SO,Lit,er, cleren erstere also den Endkonzen- trationen der Versuohe 17 und 20 entspricht, m r d e n dazu mit freiem Sohwefel 24 Stunden h n g auf 1800 erhitzt. Beim Offnen der Rohre zeigte sich in ihnen lieine Spur von Schmefeldioxyd; iiur ill
der 11% SO, enthaltenden Losung waren Spuren von Schwefel- wasserstoff und Polythionsaure entstanden, \vie sie sich stets scli011 unter der Einwirkung heiljen Wassers auf Schwefel bilden,). Dadurcli ist erwiesen, da5 die Hemmungen beim Zerfall von waBrigen Ldsungen von schwefliger Saure, wie sie mit SO, von Atmospharendruck zii
erhalten sind, nicht durch das Auftreten eines Gleichgewichts bedingt sind. Ein solches komte sich vielleicht mit weit konzentrierteren unter hoheren SO,-Drucken ZU erhaltenden Ldsungen einstellen,
I) Journ. Americ. C h . Soc. 40 (1918), 359. e) C. GEITNEB,~. c.

258 F. E71ercrter, E Laiigc, 0. Drofibacib und W. Seidel.
P-000.1 a000 0- 0- 0" .. .. .. m0.1-
-, a GO o bzw. aus konzentrierterer 0- a? o? 0- 7 0 0 y SchwefelsPure und Schwefel 0.1mmC.I . . im geschlossenen GefaiB zn G O - ~ T ~ SO,-Drucken hoherer Betrage 1-a a fuhren.
2%%% b) Versuche be i ;ddr; 120-1250 u n d be i 1800.
t--t-o
t-10-I..
t- o.l 3 $ u s a m 0-0 0 0
Uni dieErscheinungen auch --qq. bei anderen Temperaturen als
bei 150° zu untersuohen, wur-
peratur je eine Versuchsreihe nuch bei 120-1250 und bei
gebnisse sind in Cbersicht 2
m den ganz wie bei dieser Tem- +a + : : :
w 180° durchgefdxt; ihre Er- g $ : ? ? z
.- d
3
I - O 0- zusammengest ellt . 3 2 2 ?om% -aH- Diese Versuche bestiitigen &( (0 P) 0 s ;m---n- 7 2 - - inalleiidiejenigenderubers. 1.
Insbesondere eeigt sich wieder "- k- 2 d i a l
6. *- m- 0- 000 2 s6.0-w- ringen Umsatz, der bei 120
3 1% 125O erzielt wurde, dns 2 wlamb" di o Hervortreten der Polythion-
i dd" m ZLO0.1
- a m cD schwefelsgure) und das Zu- Z%:d GOaocQ 0.1r-n 2882 ruckbleiben cler Menge des
elementaren Schwefels gegen- uber dem demVerhaltnis SO,- =- 10. m*
a a m - 4 t- '0 - 2 Schwefel : element. Schwefel = 2 : 1 entsprechenden. Bri
Z g S 2 q 2 den beiden konzentrierteren 2 dd 2 SO,-Ldsungen der Versuche
-~ Nr. 21 und 22 trat auch o o o o o o o eine deutliche Gelbfarbung
cler Losung ein und wurde im Verlaufe der Versuche
2 t- t- 3 schwlcher, wenn die anfangs - noch geringfugige Schwefelab-
0.1 CJ 0.1 m 0.1 CJ c.1 scheidung reichlicher wurde.
p l r D e - a b g g g g bei dein verhaltnism%iBig ge-
ci ; sauren (neben der Thio-
e s- 0 -
> --
- 0 0
0.1- -. *- **
y - 2 t-10-t-
C J m n 0.1m0.10.1
-- ~
-01(m dimcot-

Zersetxung der schwefligen Saure und ihrw S a k e it t wapriger Losictag. 259
Besonders die verdunnte SO,-Liisung, wie sie bei Versnch 23 benutzt wurde, gab eine reichliche , ini Verhaltnis zur entstandenen Schwefel- saure sogar uberwiegende Menge von Polythionsauren, und die Schwefel- sbscheidung hatte bei Beendigung des Versuchs gerade erst begonnen ; eine Gelbfarbung trat aber hier nicht hervor. Im Gegensatz hierzu waren bei 1800, wo infolge der bei dieser Temperatur bereits sehr gesteigerten Reaktionsgeschwindigkeit meist schon nach 7 Stunden sehr weitgehender Umsatz eintrat, die Mengen der iibriggebliebenen Zwischenprodukte sehr gering.
Der Umsatz zeigte sich bei 1800 Tvieder in gleiclien Zeiten [urn so grofler, je kleinere Anfangskonzentrationen benntzt waren ; in der 17°/oigen SO,-Losung ging der Umsatz, der nach 7 Stunden 77,S0/, betrug, in miteren 41 Stunden nur auf 90,40/, herauf, nachdem in den ersten 7 Stunden die Konzentration der entstandenen Schwefel- saure fast auf 1,s MoljLiter gestiegen war.
Bei den geringeren Umsatzen bei 1200 aber zeigt sich derjenige in der konzentrierteren Losung in der gleichen Zeit noch erheblich groBer als in de; verdiinnteren Losung, immerhin verhielten sicb beide etwa = 2 : 1, wahrend das Verhaltnis der Anfangskonzen- trationen S : 1 betrug ; die Konzentration der freien Schwefelsaure aber belief sich dabei nuf 0,37 bzw. 0,05 Mol/Liter.
c) Versnche bei 1000 bzw. 90-95O. Wenn schon die Umsetzung bei 1200 sehr langsam verlauft,
so ist dies, wie oben schon bemerkt, bei 1000 in solchem MaBe der Fall, daB hier ein viele Tage langes Hrhitzen der Losung erforderlich ist, wenn man nennenswerte Mengen der Reaktionsprodukte erhalten will. Man kann die Zeit aber sehr abkiirzen, Venn man der Losung von vornherein etwas freien Schwefel zusetzt. Um diesen der Losung leicht zuganglich zu machen, wmrde eine lrolloide Schwefelmilch benutzt, doch iuBten , UM diese am Ausflocken zu verhindern, dic angewandten Schwefelmengen auf -kleine Betrage beschriinkt werden. Das mgestrebte Ziel wurde auch mit diesen erreicht.
Zur Bereitung der Schwefelmilch wurde Schwefelwasserstoff in ausgekochtes Wasser geleitet und c l a m nur soviel Schwefeldioxyd vorsichtig eingeleitet, da13 die Losung noch stark nach dem ersteren Gase roch. Sie wurde, um den Schwefelwasserstoff Gelegenheit zu gebes, etwa entstandene kleine Mengen von Polythionsauren zu Schwefel zu reduzieren, 24 Stunden stehen gelassen, und nun der uberflussige Schwefelwasserstoff durch einen Wasserstoffst'rom ab-

260 K Fotrster, l? Lawe, 0. Drobbach und W. Seidel.
geblasen. Zur Ermittelung des Gehalts der Losung an suspendierterr] Schwefel wurde er mitt Lanthanchloricl ausgeflockt, auf getrocknetem Filter gesammelt und bei 100-1050 zur Qewichtskonstanz getrocknet. Etwas langer haltbare Suspensionen lionnten bis zu 0,03 g S in 100 ccni rrhalten werden. Von diesen wnrden gewissc Anteile den in rler oben beschriebenen Weise hergestellten SO,-Losungen zugefugt.
Die Wirlisamkeit dieser Zusiitze zeigtc. sich daran, dsfi Liisungen, die ohnr sie erst nach mehreren Tagen bei looo die den begonnenen Umsatz ankiindigende Qelbfarbung iinntihmen, diese schon nach einem Tege erlieniien liefien uiid m c h zwei Tagen Schwefel abschieden. Dies geschah um so eher, je grofier der Schwefelzusatz war: 1,s mg S in 50 ccm einer SO,-Liisung veranlaSten das Erscheinen der Gelb- fiirbnng nach 24 Stunden, 0,2 mg S nach 34 Stunden.
Als Heizfliissigkeit diente bei diesen Versuchen Wasser ; a d dds Heizbad \vim- dabei eiii BuckfluSkuhler aufgesetzt. Wurdr das Wasser in liraftigem Sieden gehalten, so sprangen die Rohre haufig, rermutlich durch die in ihrem oberen Teile durch das lebhaft zuriick- fliefiencle Iiondenswasser veranlal3ten Temperatursch.wankungerl. Ti'urdt: aber das Heizwasser auf etwa 95O gehalten. so lieJ3en sicli tliese Storungen fast beseitigen. Daher wurden nur wenigr Versuche iiii siedenden Wasser, die Mehrzahl bei 950 ausgefiihrt. lhre Ergeb- iiisse sind in der Ubersicht 3 zusammengestellt. Da sich zeigte, dafi der zngesetzte Sohwefel zunichst in Lbsung geht,, kann him iiber das Verhaltnis des bri der Reaktion ebgeschiedenen elemen- 1 .Lren Schmefels zuin ciitstandenen Sulfatschwfel niohts sicheres :iusgesagt werden; die Angebe dieses Trerh<nisses ivurde da1it.r liier unterlassen.
Von diesen Versuchen zeigen die, 1x4 denen der Umsatz nur gering ist, wie bei Nr. 28, 32, 33 und 35, die eben erwahnte Er- scheinung, dal3 der als Schwefelmilch zugesetzte Schwefel zunachst in die Losung ubergeht; denn bei ihnen wird am SchluB weniger freier Schwefel gefunden als nrsprimglich zugesetzt war. Erst im weiteren Verlaufe der Umsetzung tritt fortschreitencle Abscheidung voii Schwefel ein , der sich dann in faxnkranhrtig angeordneten Kristallgebildeii am Boden der Rohre abscheidet . Das Inlosunggehen des Schwefels kann kaum anders vor sich gehen, als daS er darch seine Wechselwirkung mit der schwefligen Saure zuniichat Thio- schwefelsaure bildet ; diese findet sich auch in allen Liisungen iii kleiner Menge vor. Uberwiegend traten aber wieder Polythionsiiureii nuf; ihre Konzentration wachst iii der bei 950 untersnchten ver-

diinnten Losung von 2,60/, SO, stetig mit fortschrei- tender Reaktion an, an- fangs etwa im gleichen Xa13e wie die Schwefel- siiure, spiiter weit lang- samer als diese; in der lionzentxierteren steigt sic dagegen nur anfangs, um d a m wieder zu sinken. Bei den Versuchen mit lileinem Umsatz betriigt tler Polytliionsiiureschwe- fel ein Mehrfaches des ent- standenen Sulfatschwefels ; dies rnacht cs wnhrschein- lich, daB drr in LO- s ring gegangc-ne elementare Schwfel, ebenso wie er :in der Bildung der Thio- +
schwefelsiiure beteiligt ist, -2 os auch fur die Poly- 2 tliionsiiuren ist. Erst wenn '2 diese sich hinreichend in der Losung angesammelt haben, tritt der Zerfall der schwefligen Saure leb- hafter ein. So lcommt es, dalj bei 950im Anfange die Xeaktion cine starke Be- schleunigung zeigt , der Umsatz z. B. bei der ver- diinnteren Lbsung nach 48 Stunden 7,4, nach der doppelten Zeit aber schon 35,50/0 betragt .
Der Verlauf des Um- satzes ist also, wenn von vornherein freier Sohwefel zugegen ist, im Anfange
-2
2. anoig. u. all& Chem. Bd. 1%.
-/I -
magcD 0.1 mocoooco 0 T: 0- 0- 0- m- d
0 0 0 0 m m m m

2 62 F. Fowatw, F. L a y e , 0, DroPbaoh und W. Scidel.
der einer pos i t i ven A u t o k a t a l y s e ; die katslytische Wirkung des zugefugten Schwefels besteht, wie die Versuche lehren, darin, da8 er die Entstehung von Thioschwefelsaure bzw. Polythion- sauren veranla8t und durch deren Vermittelung die schnellere Erzeugung von Schwefel und Schwefelsiiure bewirkt . Da sich Thioschwefelsaure und Polythionsauren auch ohne besonderen Schwefelzusatz am Anfsnge der Zersetzung der schwefligen Saure shets neben Schwefelsaure bilden, so mul3 dieser Vorgang auch ohne Schwefelzusatz in seinem Anfange ein autokatalytisch beschleunigter sein. Wenn dies in den ohne Zugabe von freiem Schwefel durch- gefiihrten Versuchen der Ubersichten 1 und 2 nicht hervortritt, so liegt das daran, da13 bei 150° schon in der kurzesten Versuchsdauer von 2 Stunden die Umsetzung so weit fortgcschritten ist, daB die Anfangswirkungen nicht mehr erkennbar sind, und bei 120° Versuche von kiirzerer Dauer nicht angestellt wurden. Solche sind aber, wie weiter unten geschildert werden wird, bei den Bisulfitlosungen nach- geholt worden und lassen dort keinen Zweifel daruber, daS auch die Umsetzung der freien schwefligen Saure anfangs autokatalytisch beschleunigt ablauft .
Bei diespm Vorgange lirgt also ein recht eigenartiger Fall von Autokatalyse vor : eines der Zerfallsprodukte, der Schwefel, wirkt als positiver Katalysator, ein anderes, die Wasserstoffionen, als negativer Katalysator. Wie die Versuche der Ubersicht 3 lehren, g+niigm schon sehr kleine Mengen von Schwefel, urn die erstere Wirkung betrachtlich werden zu lassen, wahrend die Wasserstoff- ionen erst allmahlich mit ihrer starker anwachsenden Konzentration zu immer groljerer Wirkung gelangen. Dadurch wird veranlal3t , da8 im Anfange der Umsetzung die positive Autokatalyse, im weiteren Verlaufe die negative uberwiegt.
Dies tritt in den Versuchen der Ubersicht 3 deutlich hervor. Denn hier zeigt sich bei Versuch 34 und 35 der durch die positive Katslyse des Schwefels im Verlauf von 48 Stunden lebhsfter gewordene Umsatz in der konzentrierteren Losung noch wesentlich groljer als in der verdiinnteren, wenngleich der Unterschied gegen- uber dem Konzentrationsabstand verhaltnismaflig gering ist . Aber nach 96 Stunden hat bci Versuch 36 und 37 die negative Katalyw der in der konzentrierteren Losung auf etwa 0,4 Mollliter an- gewachsenen Schwefelsaure den Umsatz so weit verzogert , daS er von dem in der verdiinnteren nur auf 0,08 Mol H,SO,/Liter an- gereichprten LOsung gleichzeitig erreichten annahernd Pingeholt ist.

d) Die Ze r se t zung de r wii13rigen Losung de r schwef- l igen S a u r e i m Sonnenl ich t .
Wie in der Einleitung bemerkt, wird der bei gewohnlicher Temperatur sehr trage Zerfall der wal3rigen Losung der schwefligen Same durch das Sonnenlicht erheblich beschleunigt . Um den Verlauf des Vorganges anch unter dieser Bedingung zu untersuchen, wurden von drei mit lronzentrierter Losung von Schwefeldioxyd beschickten Rohren das eine im Dunkeln aufbewahrt, die beiden anderen vom Januar ab an einen fur die Resonnung gunstigen Ort ins Freie gestellt. Das erstere war auch nach 6 Monaten noch vollig farblos; die Losung enthielt aber doch schon eben nachweisbsre Mengen von Schwefelsiiure und von Polythionsauren. Die belichteten Rohre hegannen im Juni eine schwache gelbliche Farhe x u zeigen, die unter der starken Sonnenbestrahlung dieses Monats im Verlaufe von drei Wochen sich sehr erheblich verstarkte, um dann wahrend des Juli keine sichtbare weitere Vertiefung der Farbe anzunehmen. Jetzt, also nach 7 Monaten, zeigte die Untersuchung der Losung, daB sie kleine Mengen von Thioschwefelsaure, und etwm grijfiere, sehr deutlich nachweisbare Mengen von Schwefelsaure und von Polythionsiiuren enthielt . Freier Schwefel war aber noch nicht ausgeschieden. Der Verlauf der durch das Sonnenlicht beschleunigten Zersetzung ist also im ersten Stadium ganz der gleiche, wie er bei den bei 950 ausgefiihrten Versuchen in deren Anfangen hervortrat.
e) Theore t i s che E r o r t e r u n g des Vorganges. Der Zustand in einer wiiflrigen Losung des Schwefeldioxylis
Man nimmt darin gewohnlich die ist ein ziemlich verwickelter. Gleichgewiohte an:
80% + H20 + H,SO,, (5 )
(6)
(7)
H,SO, + HSO,' 4- H', HSO,I == SO," + H'.
Angesichts des Bestehens der kristallisierten, sehr bestandigen sogenannten Metabisulfite M2S20, mu13 auch das Anion S20;' in Retracht gezogen werden ; es durfte mit den Bestandteilen der ersten drei Gleichgewichte wohl durch das Gleichgewicht :
S,O,l' c+ $0;' + so, (8)
in Beziehung stehen. Hierzu kommen noch die Gleichgewichte der Tautomerie :
18*

384 P. Pberster, F. Lamp, 0. Dropbach und W. Soidel.
Die Dissoziationskonst,ante fk das erste 13’ der schwefligen Sgurc? ist bei 250 z t ~ 1,74 * 10-2 festgestellt worcleiil); sie ist, ciansch nillie gleich der Konstanten der zweiten Dissoziationsstufe: der Schwefc!l- saure, die von K. JELLINEK~) in nnna’hornder Ubereinstimniung mit R. LUTHER^) ebenfalls zu 3,7-10-2 gefnnden ~ u r d e . ~ ) Die Kon- stante der zweiten Dissoziationsstufe der schwefligen SBure, also fur Gleichgewicht (7), liegt nnch KOLTHOFF~) fur 15O bei 1,0 * 10-7. Dsnnch wird, wenn ma.n von den verdunntesten LBsiingen absieht , Gleichgcwicht (7) sehr stark liiiksseitig verschoben. sein. I11 einer wal3rigen Losung von SO, werden also als Anionen praktisch nur HSO,’ und S,O,” in Betrrxht kommen, wie gleiches auch fur Bi- sulfitlosungen gilt. Da,mit im Einklsnge steht, da.13 die wffl3rige Losung des Schwefeldioxyds im Ultraviolett einc IJichtabsorpt.;ou zeigtc), die einerseits der des SO,-Gases, andwerseits cler der Eisulfif - bzw. der Met,abisul~itldsungen sehr iihnlich, von der der Sulfitlosungen aber wesentlich verschieden ist7) ; in den Losungen der schwefligrn
1) K. DRUCKER, 2. phys. Chem. 49 (1904), 580; W. KEEP und E. B.4na.
z, 2. phys. C h m . 76 (1911), 331. 3, 2. Elektrochent. 18 (1907), 294. 4) A. NOYES und M. A. STEWART, Journ. Smwic. C h m . Soc. 3% (lolo),
1133, fanden den Wert 3,2-10-2. 5) 2 umrg. 21.. allg. Chem. 109 (1Q20), 69; vgl. such K. JZLLINEK, a. a. 0.
S. 352. e, J. LINDNER, Monmtsh. f . Ch.em. 38 (1912), 613; IZ. SCHAETER, Z. EZeXt,,o-
chem. 21 (1915), 181. 7) Die schweflige S ~ ~ L X C ist als mittelstarkc &re anzusehen; es eatspriclit,
nur der Analogie mit anderen Reihen von Sauersbffsauren, deren Stiirke auch mit dem Sauerstoffgehalt abnimmt, wenn sic wefientlich schwiicher ist als die ,%hwefelsaure. Wenn man aber ihre Stiirke zahJenmaBig awdruckt, indeln man die in ihren Losungen gemessene H-Konzentration mit der gesamten darin titrierbaren Konzentration an Sulfitschwefel in Beziehung setzt, so hat man swar einen iibersichtlichcu Ausdruck des Messungsergebnisses, gelsngt aher ZII
einer zu niedrigen Bewertung der StSirke der Saure, weil fur diese nur die als H2S0, vorhandenen Molekeln, nicht aber die gesamte Konzentration dea Sulfit- schwefels der Liisung bestimmend ist, und ein erheblicher Teil von diesrm als SO, vorhanden ist. Da Gleichgewicht (5) streng genommen nrtch
( 5 )
- _ _ _
Arbeiten &us dem Reichrrgesundheitsamt 26 (1006), 299.
SO, -f H,O -i4 SO,. * . IT,O =+ H,SO,

Zersp,tx%?ig der schw@igm S E u m urd i h m Sulxo in wtifiriger Liisung. 265
Stiure werden also die dnionen denen in Bisulfitlosungen vergleich- bar seira.
Die vorbeschriebenen Versuche zeigten, da% der Zerfall der schwefligen Satire durch Zunahme der H’-Konzentration gehemmt wird. Da dadurch die Gleichgewichte (6) und (7) zuungunsten der Ionen der schwefligen Saure verschoben werden, so folgt daraus, daB es diese und nicht das freie Schwefeldioxyd sind, an denen sich der Zerfall vollzieht . Da andererseits selbst eine konzentrlerte Natriumsulfitlosung auch bei 16 stiindigem Erhitzen auf 1500 keine merkliche hderung erfuhr, Bisulfite aber sich ganz iihnlioh wie die schweflige Saure verhalten, so bilden offenbar die Ionen HSO,’ bzw. S20i ’ das Material fur die beobachteten Zerfallsvorgiinge,
Dieser SchluB steht scheinbar im Widerspruch zu einer eigen- artigen Beobachtung von J. VOLHARD.~) Er fand, daB bei Gegen- wart starker Jodwssserstoffsawe schweflige Saure in kurzer Zeit in Schwefelsaure und Schwefel zerfallt. Er erkliirt die Erscheinung dahin, daB die Jodwasserstoffsaure den an sich sehr 6ragen Zerfall dcr schwefligen Saure katalytisch beschleunigt . Da wir jetzt wissen, da% die Wasserstoffionen auf diesen Vorgang nur hemmend wirlien, konnen nur die Jodionen die positive katalytische Wirliung hervor- bringen, und diese muB die entgegengesetzte der Wasserstoffionen wcit iibertrelen. Immerhin aber sind in stsrker Jodwasserstoffsaure PO wenig Ionen der schwefligen Saure anzunehmen, daB in diesem Fdle die Zersetznng sich h u m anders als an SO,-Molekeln voll- xiehen Eann.
Es fragt sich nun, wie diese verschiedenen Tatsachen sich mit- uinander vereinigen lassen. VOLHARD beobachtete, daB beim Ein- leiten von SO2 in die Jodwasserstoffsaure vom Siedepunkt 126O diese zu freiem Jod oxydiert, jene zu freiem Schwefel reduziert wird, da13 aber beim Verdiinnen der Losung der umgekehrte Vor- gang eintritt. Daraus schlieBt VOLHARD, daB die Einwirkung der
aufzufassen ist, kann auch unter Beriicksichtigung der etwa aus Verteilungs- messungen zu ermittelnden Konzentration des freien SO, eine sichere Zahlen- angabe uber die wahre Disaodationskonstante cler H,SO, nicht gegeben werden. Noch weniger kann zur Zeit iiber die fiir das chemische Verhrblten der schwefligen SSure maogebenden Aktivitatsgrade ihrer Ionen angegeben werden. Da es sich deshalb im folgenden nur um die Erorterung der Richtungen handeln kann, in denen durch die sich abspielenden Vorgange die obwaltenden Gleichgewichte verschoben werden, wird es geviigen, statt der Aktivitiiten der beteiligten Ionen :bra vermutlich vorhandenen Konzentrationen zur Erorterung heranzuziehen.
1) Lieb. A m . 242 (1887), 93.

266 El Foerster, F. Larqe, 0. Dq6bach und W. Soidel.
Jodwasserstoffsiiure auf schweflige Saure auf einer abwechselnclen Reduktion von dieser zu Schwefel und einer Oxydation durch das gleichzeitig entstehende Jod zu Schwefelsaure beruhe, also in den Vorg angen bestehe :
SO2 + 4 H J -> S + 2 J, + 2H,O, 250, + 2 J z 4- 4H2O -P 2HzSO4 + 4 H J ,
3 so2 + 2Hz0 3 2€I,SO,j -k 8.
Wenn auch die Moglichkeit solcher Vorgangsfolge nicht zu leugnen ist, so zeigen doch schon die VoLHARDschen Versuche, daS die zur oxydierenden Wirkung der sohwefligen Siiure auf Jodion erforder- liche gegenseitige Potentialla ge der Vergange :
SO, + 4H‘ 3 S + 2H20 + 4 0 ,
4 J‘ + 4 0 +- 2 J,
nur in hochlionzentrierter Jodwasserstoffsaure gegeben zu sein scheint, wahrend VOLHAHD schon fand, daW auch in ganz verdunntcr saurer Losung von J’ ihre katalytische Wirkung auf den Zerfall der xchwefligen Saure nicht ansbleibt, und sich bei unseren Ver- suchen herausstellte, daB rin kleiner J’-Zusate auch den Zerfall des Bisulfits sehr erheblich xu beschlennigen vermag.
Die Deutnng dieser Wirkung durfte in anderrr, allgemeinerer Ilichtnng x u snchen sein, und zwar unter Benutsung einer ebenfalls von VOLHARD gemachten Brobachtung. Er fand namlich, daB schweflige S h r e in verdunnterer Jodwasserstoffsiiure stets eine gelbe Farbe hervorruft, die nicht, \ on frciem Jocl herruhrt. Dic gleiche Farbe trit8t beim Einleiten w n Schwefeldioxyd in Jodkalium hervor. Die spiiteren Untersuchungen von E. PACHARD’) und \-on P. WALDEN und M. CENTNERSZW’ER~) haben nun gelehrt, daB dies(. gelbe Farbe durch komplcxr Toncii zwischen J’ imd SO,. von ciciien 11) der Losung das Ion [J(SO,),]’ auftritt, also durch koordinntivc hlagernng von SO2, bedingt ist. Es: befindet sich also in einer Jodwasserstofflosung mindestens rin ‘l‘eil der SO, nicht mehr irn Pi c+n Zustande.
Schon A. BERG^) sprach nun die Vermutung aus, da13 es die fit 1 1 1 ~ Verbindung von Jodion mit Schwefeldioxyd ist, welche sich
I ) C‘ompt. rend. 180 (IQOO), 1188. ?) %. phys. Chem. 43 (1903), 432. ’) BUZZ. SOC. Chim. [3] 28 (lQOO), 409.

Ze:el.sclnung der sclawefligm Saure und ihrer Sdxe in wzpriger Losung. 26 7
in Schwefel, Schwefelsiiure und freies Jodion zersetzt. Geht ma,n im Einklang hiermit von der Annahme aus, daB es die nicht im freien Zustande, sondern in irgendeiner Form der Bindung befind- lichen SO,-Molekeln sind, welche die Umsetzung in Schwefelsanre nnd Schwefel erfahren, so verschwindet der Widerspruch zwischen den VoLHARDschen Beobachtungea und dem aus unserem Befunde gezogenen Schlusse, und erhalt dieser eine nllgemeinere Form. Denn auch in den Ionen HS0,l und S,Oi’ befindet sich SO, in Bindung an andere Ionen, nur daB die anfangs auch hier wahrscheinlich auftretenden koordinativen Bindungen nach
SO,. * * OH’ += HSO,’, so,. - * SO,”+ szoa”
mehr oder weniger weit in konstitutive ubergegangen sind. W e weit darauf, daB die koordinntive Bindung hier weit geringer ist 81s im Ion [J(SO,),J der Geschwindigkeitsunterschiecl bei der Um- setzung in beiden Fallen beruht , mu13 dahingestellt bleiben. Jedrn- falls sind es gebundene SO,-Molekeln, die zur Umsetzung von Sohwefelsiiure und Schwefel neigen, und als solche kommen in einer Losung von schwefliger Saure, wenn keine anderen SO, binden- den Ionen da sind, nur HSO,’ oder S205)‘ in Betracht.
Wgren HSO,’ die wirksamen Ionen, so miiBte etwa eines von ihnen als Oxydationsmittel, das andere nls Reduktionsmittel wirken, muaten also wohl zwei verschiedene, der von der SO, in wiiBriger Losung gebildeten Molekelarten niiteinander in Wechselwirkung treten. Als solche kamen vielleicht die beiden Tautomeren der HSO,’ in Betracht. Wenn andererseits S205)’ der Trager des Umwandlungs- vorganges ware, so bestande dieser darin, daB zwischen zwei koordinativ benachbarten SO,-Molekeln xunachst eine Verschiebung der mittleren Vierwertigkeit des Schwefelatoms in eine hohere und eine niedere Wertigkeit vor sich ginge. Daruber, ob die eine oder die snclere Vorstellungsweise der Natur des Vorganges naher lirgt , kann man znrzeit nichts aussagen. Sehr wahrscheinlich aber ist es, daB zunachst z wei Molekeln der schwefligen Saure in Wechselwirkung treten. Diese kann dann nur im Sinne der Gleichungen:
2HS03’ 3 SOL’ + H2S02 (lOa> baw . S,OQ‘ 3 so:’ + so (lob) vor sich gehen. Es ist also die Annahme wahrscheinlich, daB beim Zerfall dcr Anioncn der schwefligen Siiure primar das stabile Anion

268 F. Focrster, F. Lange, 0. Drobbaoh u& W. ~SeideeE.
der Schwefelsaure und die ganz unbcstandige Sulfoxylsaurc haw. das Sulfoxyd SO sich bilden.
Letztere mussen dann alsbadd stabilere Gestalt annehmen. Das konnen sie, indem sie in das wesentlich bestandigere Hydrst des Sulfoxyds, in die Thioschwefelsaure, iibergehen, die Verbindung, die ja haufig dort suftlritt, wo man das Entstehen dcr Sulfoxylsiiurc~ vermuten dhrf:
2SO + 2&0 -+ 2HZSO2-+ H,Sz03 + H20. (11)
Damit geht atioh eines der zunachst zweiwertig aufgetretcnen Schwefclatome in den bei Sauerstoffbindung stabilen Zustand der Sechswertigkeit uber :
so + S/OH -f 0s: s <OH-rO,S(oH OH SH - \OH
Die Thioschwefelsiiure ist bei den Versnohen, zumal bei (35-1000, regelmiiBig in den noch in der Umsetznng befindlichen Losungen der schwefligen Saure gefunden worden. In solchen nehmen Thio- sulfatlosungen, wie lange bekannt ist, eine gelbe Farbe an; eine solche trat auch, wenigstens da, wo Thioschwefelsaure deutlich nachweisbar ist, stets auf und verblaBte, wenn die Umsetzuiig ihrem Ende zuging. Man darf ohne weiteres diese gelbe Farbe als &as Zeichen der Gegenwart von Thioschwefelsaure deuten.
Diese gelbe Farbe stimmt in ihrem Ton vollkommen uberein mit der, welche Jodion in Losungen von schwefliger Satire erzeugen. Sehr wahrscheinlich ruhrt sie auch in Thiosulfatlosungen von dcr Bildung eines komplexen Ions [S,O,( SO2),]” hor, also dem Eestehen eines Gleichgewiohtes
S,O,” + xso, += [s203(so,)x]”. (13)
\f7enn nun im Ion [J(SO,),]’, wie wir sahen, SO, als zu besohleu- nigtem Umsatz befahigt anzunehmen ist, so ist gleiches auoh fur SO, in [S,03(S02),]” nicht von der Hand ou weisen. Die Entstehung der Thiosohwefelsaure wird also die Umsetzung der schwefligen Siiure katalytisch beschleunigen, also fur den anfangs positiv auto- kstalytisch vor sich gehenden Verlauf dieses Vorganges jedenfalls mitbestimmennd sein. Im gleichen Sinne wirkt der Zusatz vGn Schwefel, duroh den nach
IiBO,’ + 8 -> 8,0,” f- €1 (14)

Thioschwefelsaure erzeugt wild ; deren St8rke ist von etwa gleicheiu Betrage wie die der Sohwefelsiiurel), also die Formulierung von C+leichnng (14) fur ihre Entstehung ails HSO,' geboten.
Diese Wirkung der Thioschwefelsaure ist aber nicht die einzige, mit der dieses ZwischeIiprodukt an den weiteren Umsetzungen in den Losungen der schwefljgen Saure teilnehmen kann.
])em die Anionen S,O," sind in Gegenwart von Waseerstoff- ionen nicht besttindig, sondern unterli~gen, Tvie kurzlich dargetamn2), den Umsetzungen:
nnd
Vorgang (15) ist die Umkehrung von (14) und fuhrt bei Gegenwart grol3erer HS0,'-Konzentretionen zu dem Gleichgewicht :
S,O," + H' 3 1380,' +- S (1 5)
0 6 ) 5 9 ' 2 0 3 " -t 6H'+ SS,O," -t 3H;').
8,0," + H' =+ HSO,' + S, P5e)
we1chf.s zusamnieii mit Gleichgewicht (13) eine groBere S20,''. Konzentration fur Vorgang (16) zur Verfugung halt. Dieser eigen- itrtige Vorgang, auf dessen Natur am Ende dieser Untcrsnchungen noch einmal zuruckzukommen ist, geht, zumd bei gesteigerter Tempesatur, ziernlich rasch vonstatten und fuhrt daher die in der Liisung verbliebene Thioschwefelsanrc sehr bald groBenteils in Penta- t hionsaure uber.
Bei Gegenwart von schwefliger Saure ist auoh diesc nichl bestandig , sondern unterliegt den1 verhaltnismiifiig scbnell ver- laufenden Vorgange2) :
SSO," + HSO,' =+ 840," + S203" -t H', (17)
dem sich der, freilioh wesentlich langsarnerez), aber bei hohwer Temperatur immer noch recht rasche Vorgang
S40;' + HSO,' += S,O," -t- S20i ' + H' (1 8)
aiischliefit. wiederum reclit schnell verLuEcnde Verseifung :
Die von ihm erzeugte Trithionsgure aber crf5lirt die
SSO," + H2O -> SO," + 820," -t 911' . (1 9)
Dieae liefert nlso von den best'hndigen Endprodukten aller dieser
l) K. JELLINEP, 2. p1zy8. Chena. 76 (1911), 333. *) F. FOERSTEB u m d A. HORNIG, Dieso Zei tsdrif t 126 (l922), I lOf f . , 138ff.

2 70 111 3’oerstei; 1$ L U I I ~ P , 0. Iho/Uiach iind W. Seidel.
Unlsetungen die SO:’, vahrmd Vorgang (1 5) den elementaren Schwefel erzeugt.l)
Von diesen Vorgangen verbrauchen (1 0) , (1 7) und (1 8) schweflige Saure. Von ihnen verlauft (10) au13erordentlich trage, (18) und mmentlich (17) aber nngleich schneller. Sie sind also in erstcr Linie fur das Verschwinden der schwefligen SBure maBgebend ; daneben kame hierfur nur in Betracht, daS durch dieiEntstehung der Ionen [S,O,(SO,),]” auch Vorgang (10) unmittelbar beschlcunigt werden kann, eine Wirkung, die aber, wie wir noch 4! sehen werden, jedenfalls gegenuber der ersteren stark zuriicktritt. Da nun Vor- gang (17) und (18) den Vorgang (16), die Umwandlung von Thio- d f a t in Pentathionat, voraussetzen nnd allem Anscheine nach nicht langsamer verlaufen als diese , ist Vorgang (16) mohl der in erster Linie geschwindigkeitsbestimmende fur die Umsetzung der schwefligen Saure. MaSgebend fur ihn ist die Konzen- tration der Thioschwefelsaure. Diese wird durch die Vorgiinge (17), (18) und (19) immer wieder neu gebildet und ist somit der eigentliche Katttlysator fur den Zerfall der schwefligen Saure, und dessen Beschleunigung durch Schwefel ist nur eine mittelbare, indem dieser mit HSO,’ nach (14) Thiosulfation liefert.
Im Verlaufe der geschilderten Umsetzungen erzeugt Vorgang (1‘3) iieben SO,“ eine sich immer sleigernde Konzentration von H . Dadurch mu13 sich Gleichgewicht (15a): S,O,” + H‘ e HSO,’ + S rechtsseitig verschieben, d. h. von den beiden auf S,O,” wirkenden Vorgangen (15) und (16) mu13 crsterer immer rnehr hervor-, letzterer immer mehr zuriicktreten. Da nur Vorgang (16) den wesentlicheri Teil der positiv htalytischen Wirkung des Thiosulfats bxw. des Schwefels vermittelt, wird diese durch die Zunahme cler H’ immer mehr unterdruckt und schliefilich verhindert . Dadurch, da13 in s tLker saurer Losung S,O,“ in merklicher Konzentration nicht mehr bestehen kormen, wird also ihr katalytischer Einflu13 durch gesteigerte H-Konzentration aufgehoben. Der negative katalytische EinfluS der H’ auf den Zerfall der schwefligen Saure beruht also
1) Auf die Bedeutung des Zeifdls der Trithionsiiure fiir den Zerfall der achwefligen Sure und der Bisulfite hat schon KLASON (1. c.) hingewiesen; die von ihm hierfiir angenommene Umsetzungsgleichung H2&Oe -+ H2S0, + S + SO, wird den Tatsachen nicht ganz gerecht. Auch sein Befund, daB Dithionsiiure bei der Zersetzung von sauren Calciumbisulfitlijsugen auftritt, konnte nicht bestiitigt werden. Da er nicht angibt, wie er diese schwer bestimmbare Siiure festgestellt hben will, konnte auch dem Irrtum nioht naohgegengen werden.

eintxseits darauf, daD er die am Vorgange beteiligten Ionen HSO,' bzw. S20," in ihrer Konzentration vermindert, andererseits darauf, da8B er die Konzentration des positiven Katalysators S203" dem Nullwert nahert. Ware letzteres nicht der Fall, so muBte durch Zusatz von Schwefel auch in stilrker saurer Losung dcr Zerfall der achwefligen Siiure mit merklicher Geschwindigkeit herbeigefuhrt werden konnen. Das wird aber, wie oben erwahnt, miiglioh, wenn Jodionen den positiven Katalysa,tor bilden, da deren Konzentration durch gesteigerte H'-Konzentration ebensowenig vermindcrt wie die Entstehung der hier allein fur die Bcschleunigung des Vorganges in Betracht kommenden Ionen [ J(S02),]' aufgehoben wird.
Anch mit den beim Zerfall der schwefligen S h r e beobachteten Einzelheiten stelicn die Folgerungeii aus der hier entwickelten Theorie uber den Mechanismus dieses Vorganges im Einklange :
Gibt man, urn das s toch iomet r i s che Verha l tn i s von Ausgangs- u n d E n d p r o d u k t e n nach dieser Theorie zu ermitteln, die VorgBnge dnrch folgenda Formulierung der oben gegebenen Gleichungen wieder, die als Benk t ions fo lge I bezeichnet sei:
5H&O, -+ !?IH~S,O, 4- 3H20, (1 6) 2H2S50, + 2&S03 -+ 2H,S40, + 2H2S203, (17) 2H28,0, + 2H2S03 -* 2H2S30, + 2H2S203, (1 8) 2H2S30, + 2H20 -+ 2H,SO, + 2H2S203, (19)
80 c rgibt dereii Addition :
4H2SO3 -+ 2H,S04 + H2S20, -t- H20. EY bliebe also 1 Mol H2S203 ubrig, das sich in der zugleich an J l ' ungereicherten Losung aber nicht halten kann, sondern sich nach
H2S203 -+ H2S03 + S (1 5 )
(1)
tveitgehend zersctzen muD. Das Endergebnis ist also 3H2803-+ 2HzS0.3 + 8 + KO
in 0 bereinstimmung mit den Beobachtungen.
der Reakt ionsfo lge 11: Zum gleichen Ergebnis gelangt man aus folgenden Gleichungen,
3 2H2S04 f 2HzS02, (10) 2H2S02 -> H,S,O, + H,O, (11) H,S203+ H2S03 + S (1 5 )
3H2S03 -+ 2H2S0, + S + H20. (1 ) -_______-

Diese Reaktionsfolge kann frcilich riur soweit in Betracht kommen, als Vorgang (1 0) bzw. seine Beschleunigung durch komplexe Ionen wie [S,O,(SO,)J’ oder [ J(S0,)J am Verbranch der schwefligen Saure beteiligt sind, also xohl im wesentlichen bei der katalytischen Betatigung der Jodionen.
Fur d e n ze i t l i chen Verlauf de r K o n z e n t r a t i o n s a n d c - r u n g e n des S u l f a t - u n d des Po ly th iona t schwefe l s lehren die den Anfang der Umsetzung am besten beleuchtenden Versucha der Ubersicht 3 das Folgende : Schwefelsaure und Thioschwefelsiiurc bzw. die an deren Stelle trotlenden Polythionsauren treten zunachst in etwa gleichzeitig ansteigendeii nlengen nuf ; auch wenn Schwefel zugcgen ist, geschieht dies bei etwa 100° mit immerhin geringer Geschwindigkeit, da die Losung des Schwefels nur langsnm von- statten geht. Dabei bleibt der Thioschwefelsavre trotz ihrer kleinen Konzentration Zeit, in Pentsthionsiiure iibermgehen, und dieser, die weiteren Umwandlungen nach Vorgang (17), (18) und (19) en vollziehen. Aber das geschieht zunachst nicht so schnell, ilaB nicht cine langsame Snreicherung cler Polythionsauren eintrate. Je mehr aber ihre Konzentrmtion anw&clist, um so schneller liefern sie Schwcfcl- saure, urn so langsemer reichern sie sich”a1so weitcr an, wie es die Versuche lehren. Zu einem stationBen Zustande zwischen Ent- stehung nnd Verbrauch dieser Saurcn kann es aber nicht kommen; denn die clurch die Verseifung der Trithionsaure frei merdenden €I bewirken es, dal3 mit der Zeit daq Gleichgewicht S,O,” + H’ e= HSO,’ + S zur Sattigung der Losung an Schwefel fuhrt, und nun- mehr neu entstehende S,O,” in immer stkkerem UniPrnge freieii Schwefel und immer weniger Pentathionsaure liefern, die Konzen- tration der ubrigbleibenden Polythionsauren also wieder abnimmf . Dies alles zeigen die Versnche im Einklange mit der Theorie. So lange in den Losungen noch die Konzentration der S,O,” ansteigt, tut sich dies durch das gleichzeitige Anwachsen der Konzentration rles gelbgefarbten Anions [S,O,(SO,),]” auch dem Auge kund. In dem MaBe wie dnrch Steigerung der H -Konzentration .die Bestandig- keit der S,O,” in der Losung sich vermindert, wird auch die Gelb- farbung schwacher und verschwindet schliefilich.
Von der K o n z e n t r a t i o n de r a n g e w a n d t e n schwef l igen Siiure erscheint die der Polythionsgnren als recht unrogelm8;fiig beeinflufit. Vergleicht man aber solche Versuche, bei denen der Umsatz auf mittlere Werte gelangt ist, bei denen wenigstens annihernd wohl ein stationarer Zuatand anzunehmen ist, so zeigt

Zersetxung der schwe@qe% Stiuw und ilirer Sake iw wa,5riger Lb’wng. 273
sich (Vers. 22 und 23, 30 und 31, 36 und 37), daf3 mit ninehmender SO,-Konzentration die der Polyt,hionsBuren sinlit; dies ist zu erwarten, da bei hoherer Konzentration der schwefligen Siiure diese auch schneller mit den Polythionsauren sich umsetzt..
Je hoher die T e m p e r a t u r ist, je schneller also alle VorgBnge verlaufen, um so schneller mcrden auch der primare Vorgang (10) ixnd seine Folgevorgange (II), (15) und (16) sich abspielen, urn 80
kleinere Koiizentrationen von den als rasch reagierende Zwischen- produkte auftretenden PolythionsBuren und von Thioschwefelsiiure wcrden jeweils in der Losung sich halten. So weit die Versuche gut vergleichbar sind, wird diese Folgerung bis 150° erfiillt. Bei dieser Temperatur zeigt sich auch nicht mehr die sonst beobachtete Gelbfarbung. Bei den, freilich meist his zu weitgehentlem Umsetx fortgeftihrten, Versucheii bei 180° waren nur noch Spurta der Yoly- thionsauren nachweisbar. Es ware denkbar, daB jetzt die Vor- gange (lo), (11) und (15) so schnell verlaufen, dalj hir die Poly- merisation der Thioschwefelsaure zu Pentathionsgure nur sehr kleiiic, Ptlcngen cler ersteren iibrigblieben, der Gesamtvorgang jetzt also irn wesentlichen ohne Vermittelung der Pentathionsiinre sich a b- spielte, nnd d d 8 uberhaupt mit steigender Temperatur die Heakiionsl- folge I immer stiirker von der Reaktionsfolge I1 uberlagert und schliel3lich ganz von ihr ersetzt wurde. Sicheres Beobnchtungs- material liegt freilich in dieser Richtung noch nicht vor.
f) Der Zer fa l l de r schwef l igen S a u r e bei 1000 i n Gegenwaxt; v a n Thiosul fa t .
Fur die im vorangehenden dargelegte Theorie uber den Mechanis- mus des Zerfalls der schwefligen Saure ist es wesentlich, daB dieser Vorgang durch Vermittelung der Thioschwefelsaure bzw. der Poly- thionsauren weit schneller vor sich geht als ohne deren Mitwirkung. Dal3 dies der Fall sein mulj, ergab sich, soweit die Polythionsauren in Frage kommen, aus den Erfahrungen iiber die Einwirkung der schwefligen Siiure auf Pentathionsaure und auf Tetrathionsaure.1) Die hieruber vorlifgenden Versuchc wurden bei gewijhnlicher Tem- peratur ausgefuhrt. Urn aber die Erscheinungen auch uiiter den Bedingungen, unter denen die schweflige Saure zersetzt wurde, ctwas ngher kennen zu lernen, und auch zu prufen, in welchem MaBe etwa die Umsetzung der gelben Komplexverbindung von S,03”
1) F. FCJEHSTER und A. HORNIQ, 1. c.
214 K J'oerster, lil Lange, 0. LkoPbach und W. Seidel.
~~
3,6 12,l 22,l 8,6
53,6 4,O
iind SO, iieben der Bildung der Polythionshren fur den Verlauf der Zersetzungen mitbeslimmend ist, ist es erwiinscht, da+s Ver- halten der schwefligen Saure in der Hitze naher kennen zu lemen, wenn von vornherein etwas groI3ere Konzentrationen des Thio- sulfatanions zugegen sind.
Die Versuche wurdeii wie die fruheren ausgefuhrt, nur wnrdr das Schwefeldioxyci jetzt in ein bestimmtes Volumen einer wieder gut gekuhlten Losung von Natrinmthiosulfat eingeleitet. Die Losung farbte sich dabei je nach der Thiosulfatkonzentration mehr oder weniger intensiv gelb ; sie wurde d a m im zugeschmolzenen Rohw auf looo erhitzt und einige Zeit anf dieser Tewperatur gehalten. Die Versuche sind also mit denen der Ubl rsicht 3 vergleichbar; ihre Ergebnisse sind in Ub micht 4 zusamniengestcllt. Da, bpi ihnen nicht immer das gleich- Volunien angewandt wurde, sind die Ergebnisse der Vergleichbarkeit halber auf 100 ccm umgerechnet . Als ,,Umsatz" konnte hier nicht wie fruher der verschwundene Anteil der angewandten schwefligen Siiure in Rechnung gesetzl werden, da bei der Abscheidung von freiem Schwefel aus Thio- schwefelsaure bzw. Trithionsiiure schweflige Siiure zuruckgebildet. andererseits solche von den Pol ythionsa~iren verbrancht und in Thioschwefelsiiure verwandelt wird. Deshalb wurde hier der ,,Urn- satz" :LUS dem Verhgltnis des entstandenen SO,-SchwefelR zum angewanclten SO,-Schwefel berechnet .
- 0,449 - 0,062 + 0,325 - 0,680 + 1,671 + 0,569
Ubersicht 4. Zerfall von SO,-Losungen bei 1000 in Gegenwart von Thiosulfat.
42 j 2 41a 1 2
5,549 4,148
) ccm R 8 818 'hlusulfa
0,685 0,685 0,685 1,125 4,503 1,229
0,5&
-
___ ~
~
g 8 PI8 80,
5,29 1 4,175 3,394 3,986 1,593 4,182
4,564
.~ --__
Gefunden in 100 ccm nach beendig __
!J 8 818 SO,
0,194 0,637 1,094 0,440 2,975 0,168
Spur
~ -_
- g 8 als
Polytl~inns
0,981 0,681 0,326 1,470 2,027 0,536
0,038
- ~- --
~ . .
:m Erhitzen 9 8 als
lhlo811lfa
0,153 0,066 0,034 0,235 0,205 0,124
nichtg
~- .
9 s 8lemeular -_ ~ - _ _ -
0,366 0,738 0,111 2,638 0,357
0,232
O 1 -
Die Konzentration der schwefligcn Saure bei cliesen Versuchen Vergleioht man das loetyug etwas mehr els 0,15 Mol in 100 ccm.

Zersetxmg der schwefligen fi'uicre und ihrer Satxe in wa/higev L6sung. 215
Ergebnis von Versuch 38 rnit dem von Versnch 28 in Ubrrsicht, 3, SO zeigt siuh, da13 rnit Hilfe von 0,Ol Mol Thiosulfat in 2 Stunden etwa der 7 fache Betrag des Umsatzes erreicht wird als dort in 22 Stunden rnit 0,0015 g-Atomen Schwefel und in der 11/, fachen Anfangskonzentration. Die beschleunigende Wirkung des Thio- sulfats auf den Umsatz kann nicht deutlicher hervortreten.
TJm auch die Wirkuiig noch viel kleinerer Thiosulfatkonzen- trationen kennen zu lernen,lwurde ein Parallelversuch zu Versuch 32 angestellt. Bei letzterem war eine Losung von 0,16 Mol S0,in 100 corn mit 0,0009 g-Atom Schwefel 24 Stunden auf 950 erhitzt worden. Nun wurde eine etwa gleich verdiinnte SO,-Losung in der gloichen Weise, jedoch rnit 0,001 Mol Thiosulfat, erhitzt, d. h. derjeiiigon Menge von S203", die bei jenem Versuohe aus dem gesnmten freien Schwefel etwa hatte entstehen konnen. Dabei bildet,rn sich auf 100 ccm:
rnit 0,001 Mol rnit 0,0009 g- s80;' Atom S
S 0,- Schwefel 52 mg 14 mg Polythionatschwefel 126 mg 60 mg
Man erkennt, v ie viel starker beschleunigend das Thiosulfnt wirkt, wenn es von Beginn der Umsetzung an in der Liisung ist, als wenn die gleiche Nenge sich erst durch die langsame Auflosung des Schwefels bilden muB, daB aber beide Wirkungen parallel laufen, die des Schwefels als Katalysator also, wie oben schon ausgesprochea, nur darin besteht, daB er HSO,' in S,O," verwandelt.
Steigert man die Thiosulfatkonzentration, wie es in Versuch 41 und besonders in Versuch 42 geschah, so vermehrt sich etwa ent- sprechend die Menge der entstandenen Schwefelsaure. Dabei aber tritt hervor, daB schon im Anfange des Umsatzes (Versuch 38 und 41) das Thiosulfatanion schnell zum groBen Teil verschwindet und an seine Stelle Polythionatanionen treten, wahrend die Sulfatbildung dagegen noch stark zuriickbleibt. Dies entspricht dem Reaktions- schema I, nach dem das Sulfat aus dem Thiosulfat mindestens in der Hauptsache uber das Polythionat hinweg sich bildet. Die Poly- merisation von S,O," zu S,O," geht also weit schneller vor sich als etwa eine unmittelbare dem Reaktionsschema I1 entsprechende Sulfatbildung aus den koordinativ an S,O," gebundenen SO,, fur welche, nach der intensiv gelben Farbe der Losung zu schlieBen, hei diesen Versuchen die Bedingungen besonders giinstige waren. Mnn ist also berechtigt, wenigstens bei looo, das Renktionsschemo I

216 F. FoersteT, I? Lange, 0. DroPbach und W. Seidel.
als dem Hauptteil des Umsetzungsmechanismus der schrefligen Baure entsprechend anzusehen.
Auoh die verh8ltnismaIjig groIje Geschwindigkeit, mit der die Pentathionsaure schweflige Saure verbrauoht, und dabei neue Thio- schwefelsiiure und damit auch neue Polythionsauren erzeugt, tritt in den Versuchen hervor. Denn bei kurzer Versuchsdauer (Versuch 38 und 41) ist, wie die let'zte Spalte der Ubersicht 4 zeigt, die Menge iles als Polythionat und als Thiosulfat in der Losung vorhandenen Schwefels weit grol3er als die des angewandten ThiosulfatschwefelP war : durch die Vorgange des Abbaus der hoheren Polythionsauren durch schweflige Satire und der immer wieder vor sioh gehenden Polymerisation des dabpi entstehenden Thiosulfats wird also SO,- Fchwefel in erlieblichem MaBe in Polythionat- bzw. Thiosulfat- schwefel verwandelt, wahrend die Uniwandlung des erstereii in Sulfat- schwefel noch stark zuruckbleibt . Erst bei fortgesetzter Steigerung iler Polythionatkonzentration, sei es infolge langerer Versuchsdauer (Versuch 40), sei es infolge hoher Anfangskonzentration des Thio- sulfats (Versuch 42), andert sich die Sachlage ; wahrend sich immer mehr Sulfat in der Losung anreichert, treten Polythionat und nament- lich Thiosulfat immer mehr zm3pii&, und immer grol3er werden dafur die Mengen an elementarem Schwefel, indem die mit der Sulfat- bildung steigende H-Konzentration ihre oben beschriebene Wirk- s~ mkeit ausubt .
Diese Wirkung der H wird durch die Versuohe 41a und 41b noch besonders erlautert ; sie sind mit Versuch 41 vergleiohbar . Die LGsung enthielt auf 100 ccm 0,019 Mol Na,S,O,. Wird sie mit 0,019 Mol HC1 versetzt, also der Halfte der dem Thiosulfat aquivalenten Menge, so trubt sie sich alsbald unter Schwefel- aussoheidung , und naoh zweistiindigem Erhitzen auf 1000 ist die Menge entst andenen Sulfats und Polythionats, sowie die des nooh ubrigen Thiosulfats weit kleiner, die des frei gewordenen Schwefels dagegen dreimal so groB als ohne Salzsaurezusatz, wahrend die Konzentration der schwefligen Saure nicht nur nictht abgenommen, sondern sich ein wenig gesteigert hat. Wird dann bei Versuoh 41 b ein UbersohuIj an Salzsaure, mehr als das 10fache der dem Thiosulfat iiquivalenten Menge, angewandt, so tritt fast keine Sulfatbildung mehr ein, und es maoht sich lediglioh die bekannte Zersetzung des Thiosulfats durch H bemerkbar: 9SO/o von ihm zerfallen unter Abgabe von freiem Schwefel und Vermehung der SO,-Konzentration und 7% gehen in Polythionsliuren uber.

Zeraetzung der schwetlige% Sawre und ihrer Sdxe in wabriger Losung. 27 7
Durch diese Versuchsreihe wird also die oben entwiclielte Theorie in allen Einzelheiten bestatigt .
g) Die Rolle der hyposchwefligen S&ure bei der Zersetzung der schwefligen Saure.
Eine wesentlich andere Vorstellung uber den niiheren Verlauf der Selbstzersetzung der schwefligen Saure, als im vorstehenden begriindet wurde, haben JUNGFLEISCH und BRUNEL~) entwickelt. Da eine Hyposulfitlosung beim Ansauern sich braun bis gelbbraun farbt, glaubten sie, die Gelbfarbung, die auch sie stets im Anfange znmal bei etwas niedrigerer Temperatur beim Zerfall von Schweflig- saurelosungen beobachteten, auf Gegenwart von hyposchwefliger Saure zuruckfuhren zu durfen. Sie wurden in ihrer Ansicht besttkkt durch die Beobachtung, da13 die gelben Losungen, die im Beginn des Zerfalls der schwefligen Saure auftreten, Indigo- oder Lackmus- losung entfarben und aus Silberlosung sofort metallisches Silber abscheiden, sich auch gegen die Ionen anderer edlerer Metalle ganz analog der hyposchwefligen Saure verhalten. DemgemaB nehmen sie an, wie in der Einleitung schon erwahnt, daB der Zerfall dm schwefligen Saure primar im Sinne der Gleichung:
3 SO, + 2H, 0 -+ H2S,0, + H,SO,
H2S204 + HzS04 + S
(2)
(3)
vor sich geht, und die hyposchweflige Saure dann weiter nach
zerfallt, eine Vorgangsfolge, die auch zu dem durch die Brutto- gleichung :
3 SO, + 2 H20 3 2 &SO4 + S
dargestellten Ergebnis fuhren wiirde, und von JUNGFLEISCH und BRUNEL dadurch gestiitzt wird, daB sie in einer verdiinnten Losung von hyposchwofliger Saure unter iihnlichen Umstanden, wie denen, unter denen die gelbgewordenen Losungen der schwefligen Saure Schwefel abscheiden, die Entstehung von Schwefelsaure und von Schwefel beobachteten.
Diesen Befund von JUNGFLEISOH und BRUNEL konnen wir nur teilweise bestatigen. Zutreffend ist, daB die gelben Losungen, die aus Thiosulfat durch schweflige VOLHARD 2, beobacht et e, Indigo- und
1) 1. c. ') 1. c.
Z. nnorg. U. allg. Chem. Bd. 128.
Saure entstehen, wie schon Lackmuslosung alsbald ent-
19

278
farben. An umeren Ltisungen haben wir jedoch rnit Silberlaung niemals eine Metallabscheidung, sondern sowohl rnit sokhen Losungen wie mit denen des Quecksilbers nichts als die bekannten Thiosulfat- bzw. Polythionatreaktionen beobnchten konnen.
Vor allem aber fanden wir den Verlauf des Zerfalls der Losungen von hyposchwefliger Saure ganz anders als ihn JUNGFLEISCR und BRUNEL angeben. Wir haben diesen gerfall in Gegenwart von uber- schiissiger schwefliger Saure untersucht . Setzt man hyposchweflig- saures Natrium zu einer Losung von dieser Saure, so tritt alsbald die fur sie kennzeichnende Braunfarbung auf. Diese aber verblafit schon bei gewohnlicher Temperatur ziemlich schnell, wahrend sich etwas Schwefel abscheidet, der sich aber bei kurzem Erwarmen anf looo wieder vollstandig auflost, indem die die Thioschwefelsaure itnzeigende Gelbfarbung auftritt, aber kcine, oder fast keine Sehwefelsaure sich bildet. Quantitativ wurden die Erscheinungen mieder im geschlossonen Rohr in folgenden Versuchen verfolgt.
Versuch 43. 40 ccm mit 2,304 g S als SO, wurden mit 10 ocm einer Losung von 0,87 g Na,S,O, N 0,805 g Hyposulfitsehwefel bei 00 vermischt und das Rohr zugeschmolzen.
K Foerster, liI Lunge, 0. Ih.oj3baola md W. Seidel.
Hach zweistiindigem Ekhitzen auf looo enthielt es :
2,403 g S als SO,, 0,086 g S als SO,, 0,296 g S als Polythionsiiuren
und eine Spur von Thiosulfatschwefel ; elementarer Schwefel war nicht abgeschieden.
Versuch44. 40ccm mit 2,205 g S als SO, wurden in der gleichen Weise wie beim vorigen Versuch mit 10 ccm einer Losung von 0,5398 g Hyposulfitschwefel versetzt und das R o b 14 Stunden auf 1000 erhitzt. Es enthielt jetat:
2,060 g 6 als SO,, 0,154 g €3 als SO,, 0,282 g S als Polythionsiiure, 0,008 g S a16 Thiosah~refelsPure, 0,0925 g S elementar.
Hieraus ergibt sich, wie zumal die Zunahme des SO,-Schwefels uncl die gleichzeitig entstandene kleine Menge SO,-Schwefel in Versuch 43 lehren, daB die hyposchweflige Saure in schweflige Saure und Thioschwefelsaure ubergeht , welch’ letztere in der Losung der

Zewetxung d&r schwefligen S&hye und ihrer Sake in w&ijlriger Losung. 279
schwefligen Siiure wie immer alsbald sich im wesentlichen in Poly- thionsauren verwandelt, ubd daB eTie bei langerer Erhitzhng in TTersuch 44 entstmdenen Mengen VOB Schwefelsaure und Schwefel lediglich in def stets bebbachteteb Weise von der Wechselwirkung der Polythionsiiuren mit der schwefligen Saure herriihren.
DaB die hyposchweflige Sfiure, die man' ja dls Kondensations- produkt yon schwefliger Saure und von Sulfoxylsanre aufzufassen berechtigt ist, im Sinne der Gleichung:
ZH,S,O, + E&O 3 HzS2O3 + 2H2S03 (20)
zerfhllt, hat J. M E Y E R ~ ) nachgewiesen. Nimmt man an, daS im kleinen MaSe auch der umgekehrfe Vorgang eintreten bnn, also clas Gleichgewicht :
sich einstellen kann, so versteht man, daB in einer Losung von Thio- schwefelsaure und schwefliger Siiure auch kleine Mengen der Indigo entfarbenden hyposchwefligen Siiure auftreten konnen. Diese erscheint dann nur als Nebenprodukt, nicht aber als Zwischen- yrodukt; ihr Zerfall in dem von JUNGFLEISCH und BRUNEL an- genommenen Sinne konnte ja auch die positive Autokatalyse, welche die Polythionate veranlassen, nicht hervorbringen.
Die Auffassung von JUNGFEEISCH und BRUNEL, daS der Zerfall cler sckwefligen Saure im wesentlichen uber die hyposchweflige Saure fuhrt, durfte dadurch als den Tatsachen nicht genugend Rechnung tragend erwiesen sein. Allerdings steht diese Auffassung insofern mit der hier entwickelten Ansicht uber den Verlauf des Vorganges in Beziehung, als auch naoh dieser beim primaren Zerfall der schwefligen Saure neben der Schwefelsiiure die Fhtstehung von Sulfoxylsaure angenommen wird. Wahrend nach unserer Auffassung diese fur den weiteren Verlauf der Umsetmngen dadurch wirksam wird, daB sie sich nach
&S203 + 2H,S03 + 2H,S20, + H,O P a )
HZSO, + HZSO2 + H,S,O, + q O (11)
in Thioschwefelsaure verwandelt, muBte sie, wenn die Ansicht von JUNGFLEISCH und BRUNEL zu Recht bestande, nach
H,SO, + H,S03 3 H,S,O, + H,O (21) sich mit der schwefligen Sliure zu hyposchwefliger Same ver'einigen. Vermutlich ist die erstere Umsetmng in saurer Losung die schnellere,
l ) 2. anorg. u. allg. Chein. 34 (1903), 43. 19*

280 F. Foerster, l? Lave, 0. Dropbach und W. Seidel.
und bleibt daher die letztere auf die Rolle einer Nebenreaktion beschrankt. Aber selbst wenn sie in groBerem Umfange eintrate, wiirde sie, wie die Versuche 43 und 44 lchren, doch alsbald in der Losung der schwefligen Saure in das gleiche Produkt wie die Thio- schwefelsaure, namlich die Polythionsauren ubergehen, in denen wir ja die fi ir den Verbrauch der schwefligen Saure wesentlichen Zwischenprodukte zu erblioken haben.
4. Die Selbstzersetznng der Bisnlflte. (Nach Versuchen von 0. DROSSBACH.)~)
LI) Versuohe i n zugeschmo1zene.n R o h r e n bei 100-1500.
Erhitzt man Alkalibisulfitlosungen im zugeschmolzenen Rohre in derselben Weise, wie es fur die Losungen der sohwefligen SSiure geschah, so beobachtet man ein ganz ahnliches Verhalten wie bei diescr. Um einen sicheren Vergleich zu erhalten, wurde zuniichst eine Natriumbisulfitlosung angewandt , welche der mit 160/, SO, hergestellten Losung der Saure aquivalent war. Sie gab bei 1000 auch bei 22 stiindigem Erhitzen noch keine Ausscheidung von freiem Schwefel. Diese blieb auch noch aus, als solche Losung 7 Stunden auf 1500 erhitzt wurde. Bei Zugabe von freiem Schwefel, dessen Menge Sol0 des in Gestalt von Bisulfit angewandten betrug, wurde aber in der gleichen Zeit ein Umsatz von 99,50/, des an- gewandten Bisulfits erreicht. Eine Losung der h a l b e n Bisulfit- konzentration konnte ohne Zusatz von freiem Schwefel 19 Stunden lang auf 1500 erhitzt werden, ohne mehr als eine schwache Gelb- farbung zu zeigen; im Verlauf weiterer 7 Stunden trat aber die Umsetzung ein und zwar alsbald fast vollstandig, zu 97O/, des an- gewandten Bisulfits. Wurde aber eine Natriumbisulfitlosung von etwa der doppelten Konzentration der zuerst angewandten, welche einer mit SO, von Atmospharendruck nicht herstellbaren Losung von 32,80/, SO, entsprache, ohne Zusatz suf 150° erhitzt, so wurde in 7 Stunden ein Umsatz von 99,70/,, erreicht. Fast gleich stark war in so konzentrierter Losung der Umsatz bei 120-1250 inner- halb 16 Stunden, und auch bei 1000 konnte ein Umsatz von etwa 980/, in 62 Stunden erzielt werden mit Hilfe eines Zusatzes von 501,
des Bisulfitschwefels in elementarer Form. Bei diesem Versuche zeigte sich, daB der zugegebene Schwefel wieder unter starker Gelb-
1) Dissertation. Dresden 1921. Die ersten orientierenden Versuohe d e n von F. llluva~ ausgefiihrt.

Zerselxung der schw6fl~en Saure und ihrer Sake in wapriger Losung. 28 1
fkbung der Losung zunachst groaenteils verschwand, und daB erst nach der 40. Stunde die Zersetzung unter Abscheidung von Schwefel eintrat, um d a m in wenigen weiteren Stunden gr6Btenteils voll- endet zu sein. Bei allen diesen Versuchen war am Ende der Um- setzung Polythionatschwefel in der Losung zugegen, jedoch nur in ganz geringer Menge.
Was die Erscheinungen bei der Bisulfitzersetzung besondes kennzeichnet, ist hiernach die Tatsache, da13 sie in aquivalenter Konzentration wesentlich rascher und weit vollstandiger verlauft als die der freien schwefligen,Saure, und daB auch bei Konzentrationen, die weit hoher sind als die hochsten, die bei der freien Siiure angewandt wurden, der Umsatz in verhiiltnismaBig kurzer Zeit ein sehr weit- gehender ist. Das ist nach den Erfahrungen uber den EinfluB der H auf die Umsetzung des HSO,’ aucb nicht anders zu erwarten, da der Umsatz:
3Na’ + SHSO,’ 2 3Na’ + SO,” + HS04’ + S + H,O (4)
nach den Gleichgewichtskonstanten von HSO,’ und H$O, nur hoohstens I/, der H’-Konzentration erzeugt, als in aquimolekularer Losung fur den Zerfall
3 H + 3HS0,’ -+ 2HS0,’ + 2H‘ + S + H,O (1)
von Hause aus zugebote steht. Dadurch wird die nachtragliche ,
negative Autokatalyse weitgehend verhindert , und zugleich bewirkt , daB die anfangliche positive Autokatalyse durch den freien Schwefel bzw. das von ihm erzeugte Thiosulfat uneingeschrankt zur Geltung kommt. Das zeigen auch die beschriebenen Versuche. Denn bei Zusatz von Schwefel tritt bei 1000 zunachst ein Verbrauch von Schwefel ein, und erst hiernach wird verhaltnismB13ig spat der Vor- gang lebhaft. ilhnlich war auoh ohne Schwefelzusatz bei 1500 zu beobachten, daB die Zersetzung zunachst einer gewissen Inkubations- zeit bedarf. Diese dient dazu, die den raschen Umsatz erst ver- mittelnde Konzentration an Thiosulfat bzw. Polythionat hervor- zubringen.
Um diese fiir die Theorie unseres Vorganges wichtige Seite der Zersetzung etwas genauer zu untersuchen, wurde fiir zwei etwas konzentriertere Losungen von Natriumbisulfit bei 150° und ohne jeden Zusatz der zeitliche Verlauf der Umsetzung verfolgt, wie sie sioh aus der Menge des jeweils entstandenen Sulfatschwefels ergibt .

282 P. Foerster, E: Lamge, -0. B@bach und W. S e a l .
Es geniigt, die Ergebnisse durc-h die Kurvenzeiohnung 2 zu ver- unschauji&en. Sie zeigt das a o f h g W e langsame Einsetzen der Reaktba und ihre immer starkq und schlieIjlicb sprunghaft sich steigernde Geschwindjgkeit, ,&is tyBisc4e Bild einq positiven Auto- katalyse. Dab& waren bei den snfPnglicb noch geringeren Um- satzen immer hochstens Spuren von freiem Schwefel abgeschieden und in der Losung reiohlich Polythionat entblte,n, wahrend bei annahernd vollstiindigem Umsatz die hfenge des elementaren
Schwefels zum Sulfatschwefel wieder sehr nahe sich wie 1 : 2 verhielk, und &a Polythionat in seiner Menge stark zuriickgegangen war.
Aus diesen Erfahrungen ergab sich, daB die Bisulfitlijsungen weih geeigneter ads diejenigen der froien schwefhgen Saure sind urn die von der oben entwickelten Theorie gefarderten beiden U m t z - stadien, anfkngliche EntshAung von Sulfat und Thiosulfat bzw. Polythionat und darauffolgende Umsetzung von Thiosulfat bzw. Polykhionat in Sohwefel und Sukfat, jedes f l i r sioh und beide in ihrer zeitlichen Aufeinaaderfoolge niiher zu untersuohen. Denn hier kann, so lange das Bisulfit nooh nioht weifgehend verschwunden ist, die zur Absoheidung des freien Schwefels erforderliche geslteigerte H'- Konzentration uberhaupt nicht eintreten. Bei 1500 freilich ist die naoh dem slnflwlglichen geringereg Uaaatz eintretende Reaktions- bewhleunfgqg so groB, da13 RS niobt gelang, die fiir Rtwa mifitlerep Umsatz auwekhenden Reaktionseeiten zu treffan ; entweder war der Umsat7; qur in seinen Anfiingen eingetl;eltev, Qder er war f a t vdstandig verlaufen ; jedenfalls konnte so der Zustad der Losung,

Zersekung der schwefligm Saure und ihrer Salxs i,n wa/hGer Losung. 283
der fiir das Einsetzen der Ausscheidung des elementaren Schwefels bestimmend war, nicht gefunden werden.
Bei looo erforderte auch fur die starks$e der ohen benutzten Natriurnbisulfitlijsungen der Umsatz ohne Schwefebusatz eine un- erwiinscht lange Versuchsdauer. Durch Benutzung noch konaen- trierterer Losungen ia8t siob diese abkkzen. Deren Bereitung ist fur Ammoniumbisulfit besonders bequem. Eei iiquivalenter Kon- zentration besitzen dessen Lijsungen in ihrer Umsrttzgefichwindigkeit vor den Natriumbisulfitlthungen allerdings keiaen Vorzug. Zn eimr Ammoniumbisulitlosung yon etwa 24 g Bisulfitmbwefel ia 100 ccm, also etwa der doppelten Kowntretion der stiirksten, oben benutzten Losung des Natriumsalzes, Fvurde in 76 Stuaden ein Umsatz von 870/, erreicht , wabrend er in einer Ammoniumbisulfitlosung mit 16 g Bisulfitscrhwefel in 1IK) com in der gleichen Zeit die Inkubations- zeit nmh kaum iibersahritten hatte und aJlr etwa jenes Um- satzes erreichte. Geht man ao noch stkkeren -Losungen, z. B. von 33 g Bisulfitschwefel in 100 corn uber, so treten stets aufierordentlich heftige Explosionen ein. Da dies fast imrper schon wahrend des Anheizens geschah, durften sie wohl hier wesentlioh darauf mriick- zufiihren sein, da13 die Sbsrohre dem Druck des bei 1000 durch die Hydrolyse frei werdenden Schwefeldioxyds nioht mehr stand hielten. Sie sind also in ihrer Ursache vermutlich verschieden von den von Boscs nach den Erfahrungen beim Arbeiten im grolleren Mas- stabe erwiihnten Explosionen, die, wie BOSCH wohl mit Recht awimmt, darwf beruhen, daB yon groBeren Losungsmengen die durah das plijtzliche autobtalytische Eintreten der erheblich exothermep Um- setzung: 3H,SL),, aq = 9H2SQ,, aq + S + 48400 cal. entwiokelten Wlirmemengen nkht rasch genug abgeleitet werden konnen, und dem- entsprechend plofaJichs und sebr bsdeutende Steigerungen des Wasser- dampfdruokes der Lijsung veranlassen. Angesichts der verhaltnismiiBig kuraen Zeiten, weldw die Uf~roaeteung konzentrierter Arnmoniumbisui- fitlasungen bei 1 0 0 0 beanaprucht, erschien es moglich, dime in oftenen GefgRen und d a m auob mit griihren Mengen der koohenden Losung durchzufiihren uQd dab4 den gevunschten tbferen Einblick in den zeit- licherr Kerbuf des Vorganges und seiner eimelnen Phasen zu g e b n .
b) Versuche mit kgnzenkrierter, koehender Ammonium- bisulf i t losung i m offenen GefiiBe.
Vorversuohe migten, drq@ eine in offewm Gefiil3 hei 1080 kochende starke Apmoniumbisulfitlosung sioh weit langsamer urnsetst, ak

284 EI Fowsfer, l% Larye, 0. Dropbach wnd W. Seidel.
im geschlossenen Rohr bei 1000; erst nach etwa drei Wochen tritt unter solchen Umstiinden die Schwefelabsoheidung ein. Der Grund clieser Verzogerung liegt darin, daB in der Siedehitze die Aziditat der Losung, also die Konzentration der HSO,', stark zuruckgeht, indem die durcb die Hydrolyse frei werdende schweflige Saure reichliche Mengen von SO, abgibt, bis ihre Konzentration auf den bei ihrem bei 1080 liegenden Siedepunkte mit SO, von 1 Atm. im Gleich- gewicht stehenden sehr verminderten Betrag herabgegangen ist. Sollte die mit diesen Versuchen beabsichtigte nahere Verfolgung des Vorganges durchgefuhrt werden, so muljte also die Apparatur
der durch wochenlanges ununter- brochenes Sieden einer konzen- trierten Losung bedingten hohen Beanspruchung gewachsen sein. Erst nach sehr zeitraubenden Ver- suchen wurde dies erreicht.
Der Apparat durfte keinen Kork- oder GummiverschluB haben. Die Ldsung wird wahrend des Vor- ganges immer viskoser und liiljt noch vor der Abscheidung des Schwefels Ammoniumsulfat kri- stallisieren. Dadurch gerat sie leicht in ein auBerordentlich hef- tiges, schlieBlich zur Zertrumme- rung des Apparats fiihrendes StoBen. Es zeigte sich, daB dies nur mit Hilfe einer die Losung dauernd aufrihrenden Vorrichtung m ver- huten war. Diesen Anforderungen
entsprach die durch Fig. 3 veranschaulichte Apparatur. Kolben A aus Jenaer Glas fal3t etwn 1200 ecm; er ist durch eine eingeschliffene Kappe verschlossen, die durch einen WasserverschluB abgedichtet ist. Sie trsgt einerseits den durch Quecksilberversohlufi J ab- gedichteten Riihrer R, andererseits das zur Probenahme bestimmte Rohr 11, das durch einen Glasstopfen verschlossen wird. Mittels des seitlichen Rohres C ist der RuckfluBkuhler K an den Kolben angeschlossen und zwar derart , da13 der verbindende Gummischlauch dhrch eine mehrfache Umwickelung mit Isolierband gesichert ist. Mit ihm wurden zwei Waschflaschen verbunden, deren meite, mit
Fig. 3.

Zersetzung dw schwefligen Saure und ihrer Sake in wapriger Losung. 285
Natronlauge beschickt, zur Zuriickbaltung des austretenden Schwefel- dioxyds dient, wahrend die erste leer und so angeordnet war, daB auch bei Unterdruck im Kolben ein Zuriicksteigen der Lauge in diesen verhindert war.
Fiir die Analyse der von Zeit zu Zeit entnommenen Losungs- proben wurden sie mit ausgekochtem Wasser auf ein bestimmtes Volumen gebracht und gemessene Anteile von diesem, die je nach Bedarf 0,1--0,5 ccm der Hauptlosung entsprachen, in der oben angegebenen Weise zur Bestimmung von Sulfit-, Sulfat-, Thiosulfat-, Polythionat- und Hgposulfitschwefel untersucht. SchlieBlich wurde auch die Aaiditat der Losung gegenuber Phenolphthalein verfolgt .
Die Abscheidung des elementaren Schwefels tritt hier erst in h e m spateren St'adium der Umsetzung ein. Um auch sie mogliohst eingehend zu verfolgen, wurde von der noch zu erorternden Tatsache Gebrauch gemaaht, daB die Umsetzung bis zum Eintritt der Schwefel- fBllung durch einen kleinen Selenzusatz auf wenige Stunden verkiirzt werden kann, und dsB das Selen sich quantitativ vor dem Schwefel wieder abscheidet . In einer besonderen Versuchsreihe wurden je 100 ccm der Ammoniumbisulfitlosung in einem kleinen Rundkolben gekocht, der jetzt kein Riihrwerk brauchte, sondern nur einen RiickfluBkuhler trug und eine besondere, mit Glasstopfen verschliea- bare AusguBoffnung besaB. In diesem wurde die Losung zunachst linter Zugabe von 0,2 g Selen soweit umgesetzt, daB das Selen wieder abgeschieden war und eben das Auftreten freien Schwefels sichtbar wurde. Jetzt wurde das Erhitzen unterbrochen, die ganze Losung rasch filtriert und das Filtrat nach Entnahme einer P r h e in den Kolben zuriickgebracht und weiter gekocht, und nun von Zeit zu Zeit der inzwischen abgeschiedene Schwefel abfiltriert und die Losung immer wieder in der gleichen Weise weiter behandelt, bis die Schwefelabscheidung nur noch geringfugig war. Die jeweilig abfiltrierten Schwefelmengen wurden gewaschen und gewogen, naoh- dem sie bei 1080 zur Gewichtskonstanz gelangt waren. Der erste Niederschlag enthielt neben etwas Schwefel die gesamte Menge des als Katalysator angewandten Selens. Die aus der von ihm ab- filtrierten Losung entnommene Probe zeigte, daB die Veriinderung der Losung bis zum Beginn der Schwefelsbscheidung im wesent- lichen die gleiche war wie ohne Benutzung des Katslysators.
Von den bei diesen Versuchen erhaltenen Ergebnissen sind in Uber- Rich t 5 (S.286) die Mittelwerte von 4Versuchsreihen zusammengestellt, welche 170 einwandfreie Einzelbestimmungen umfassen. Die Kurven
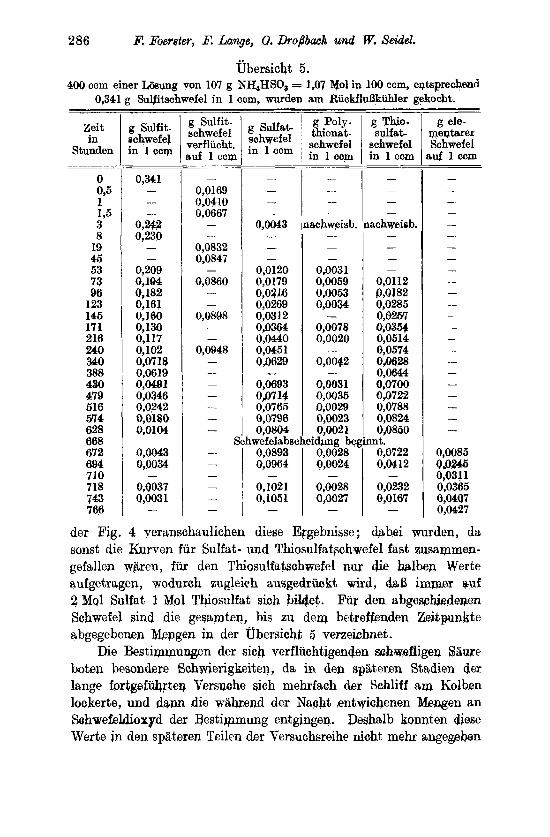
206 El Fi8r6ta; F. Lange, 0. Drobbwh urad W. SeideE.
_ ~ .
g TI&- sulfat-
schwefel in 1 ccm
Ubersicbt 5. 400 ccm einer Lijsung von 107 g NH,HSQ, = 1,07 &fo1 in 100 ccm, eqtaprechend
Sulfitschwefel in 1 ccm, wurden am RiickfluBkiihler gekocbt.
g ele- meQtarer Schwefel
suf 1 ccm
0,34 - . ~ -
Zeit in
Stunden
0.0179
~~
0
1
3 8
19 45 53 73 96
123 146 17 1 216 240 340 388 430 479 516 574 628 668 672 694 710 718 743 766
0,5
1,5
0.0059
-- g Sulfit- schwefer in 1 ccm
0,341 - - -
0,242 0,230
0,209 0,104 0,182 0,161 0,160 0,130 0,117 0,102 0,0718 0,0619 0,0491 0,0346 0,0242 0,OlrbO 0,0104
0,0043 0,0034
0,0037 0,003 1
- -
-
-
~ _ _ ~ __ g Sulfit- schwefel rerflucht. r u f 1 ccm
0,0169 0,0410 0,0667
-
- -
0,0832 0,0847
0,0860
0,0898
-
- - - -
0,0948 - _- - - - - -
L -_ - - - - -
g Sulfat- schyef el in 1 ccm
g Poly- thionat- schwefel in 1 corn
-~
- I -
- I - - 1 1 -
0,0043 inacbweisb. ,nachweisb. ' - - 1 - _ I - ' - - / - , -
0;021f3 0,0269 0,03 1 2
0,0440 0,0451 0,0629
0,0693 04719 0,0765 0,0796 0,0804
iwefelabsc 0,0893 0,0964
0,1021 0,1051
-
-
-
0;0053 0,0034
0,0074 0,0020
0,0042
0,003 1 0,0035 0,0029 0,0023
!idpng be( 0,0028 0,0024
0,0028 0,0027
-
- -
0,0021
-
-
0,0112 0,0182 0,0285 0,0267
0,0514 0,0674 0,0628 0,0644 0,0700 0,0722 0,0788 0,0824 O,Q860
0,0354
mt. 0,0722 0,0412
- 1 00:::; i -
0.0085 Oh246 0.031 1 0;0365 0,0407 0,0427
der Fig. 4 yeranschaulichen diese Ergebnisse; &bei wurden, da sonst die Kurven fur Sulfat- und Thiosulfatachwefel fast zusarpmen- gefallen whren, fur den Thiosulfatschwefel nuy die ha1b.e~ Werte aufgetragen, woduroh zugleich ausgedriickt wkd, deIj imwr @uf 2 Mol Sulfat 1 Idol Thiosulfat skh biwet. Fiir den abgeaobbdeaen Schwefel sind die gesamten, bis zu dew betreffenden Zeikpunkte abgegebenep Nepgen in der Ubersicbt 5 verzeichnet.
Die Bestirprnungen der sick verfluchtigemjen d m d i g e n Saure botep besondere Schwierigkeiteq, da in den spaterm Stadien der lange fortgefarteq Versuche sich mehrfach der Schliff am Kolben lockerte, und &an die wahrend der Nacht e n t ~ c b e n e n Merqpn an Srrhwekldioxyd der Bestirpmung entgjngen. Deshalb konnten dime Werte in den spateren Teilen der Yersuchsreihe nioht mehr ongegebn
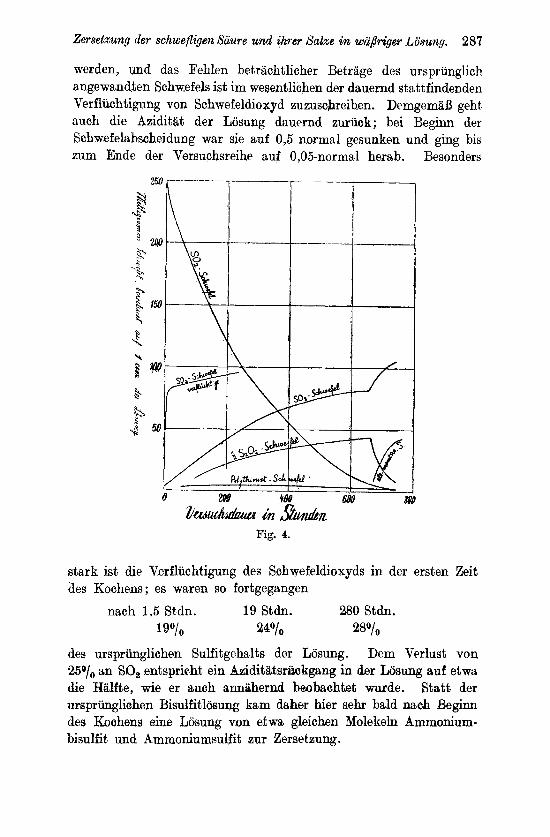
Zersetxung dey schwefligen Saure und ihrer Salxc in wa,higw Losung. 257
werden, und das Fehlen betriichtlicher Betrage des urspriinglich angewandhen SchFefels ist im wesentlichen der dauernd stattfindenden Verfliichtigung von Schwefeldioxyd zuzuscbreihen. DemgemaB geht auch die Azidit&t der Losuug dsuernd zuriick; bei Beginn der Scbwefelabsoheidung war sie auf 0,5 normal gesunken und ging bis zum Besonders
Fig. 4.
stark ist die Verfliichtigung des Schwefeldioxyds in der ersten Zeit des Kochens ; es waren so fortgegangen
nach 1,5 Stdn. 19 Stdn. 280 Stdn. 19O/0 24°/o 28Yo
des urspriinglichen Sulfitgehalts der Losung. Dem Verlust von 250/, an SO, sntspricht ein Aziditatsriiokgang in der Losung auf etwa die Halfte, wie er auch annahernd beobachtet wurde. Statt der urspriinglichen Bisulfitlijsulrg kam d e b r hier sehr bald nach Beginn des Koohens eine Losung von etwa gleicbn Molekeln Ammonium- bisulfit und Ammoniumsulfit m r Zersetzung.

288 li! Foerster, F. Langa, 0. Dropbach und TV. Seidel.
Die Kurven lehren, daB der Zerfall dieser Bisulfit-Sulfit- losung mit der Abnahme der Konzentration des Sulfitschwefels zeitlich ganz stetig fortschreitet, also bei ihm kein autokatalytischer Verlauf hervortritt. Ferner zeigen sie, daB dieser Zerfall in zwei aufeinanderfolgenden Vorgangen sich abspielt : Im ersten entsteht Sulfat- und Thiosulfatschwefel in etwa gleichen Gewichtsmengen neben untergeordneten, freilich etwas schwankend gefundenen Mengen an Polythionatschwefel. Dieser Vorgang schreitet ohne Abscheidung von Schwefel so lange weiter, bis fast aller Sulfitschwefel verschwunden ist, nur noch 1,3O/, von ihm vorhanden und 98,’2O/, in Sulfot und Thiosulfat, 0,5O/, in Polythionet ubergegangen sind.
Erst jetzt beginnt der zweite, weit schneller verlaufende Vorgang, durch welchen das Thiosulfat verschwindet und aus ihm Sulfat und freier Schwefel ent stehen.
Das Sulfat bildet sich also durch zwei aufeinanderfolgende, in ihrem Wesen verschiedene Vorgange, der freie Schwefel nur durch den letzten von diesen. Dieser scheidet sich dabei nicht nur in der Losung ab, sondern auch im Kiihler und zwar in so reichlichem MaBe, da13 dies nicht allein von der bekannten Verfluchtigung von Schwefel mit TVasserdampfen herruhren kann. Der Grund fur diese Er- scheinung liegt darin, daB wahrend der Zersetzung des Thiosulfats auch Schwefelwasserstoff auftritt, der mit gleichzeitig entweichendem Schwefeldioxyd und dem im Kuhler kondensierten Wasser sich zu freiem Schwefel umsetzt.
Neben den bisher erwahnten Reaktionsprodukten tritt bei dieser Zersetzung der Bisulfit-Sulfitlosung in kleiner Menge such Hyposulfit auf ; seine Konzentration steigt anfangs langsam auf 0,004 Mol Na,S,O, in 1 ccm, und sinkt dann, wenn die Sulfit- konzentration unter die Halfte ihres Anfangswertes herabgegangen ist, allmiihlich wieder ; beim Auftreten des freien Schwefels ist das Hyposulfit verschwunden.
c) Versuohe m i t k o n z e n t r i e r t e r Ammoniumbisu l f i t l o sung b e i gewohnl icher T e m p e r a t u r i m of fenen GefiiB.
Die im vorstehenden Abschnitte beschriebenen Versuche geben insofern kein eindeutiges Bild uber das Verhalten der Bisulfite, ah ihre Anordnung den teilweisen Ubergang des aauren in das gesiittigte Sulfit bedingte. Das lie13 sich vermeiden, wenn der Vor- gang bei gewohnlicher Temperatur verfolgt wurde.

Zersetzung der schwefligen $awe and ihrer SalM in wapriger Loswlzg. 209
Das ist aber in bequemer Zeit nur moglich, wenn man diese durch geeignete K a t a l y s a t o r e n hinreichend verkiirzt. Wie die Zersetzung der schwefligen Saure, kann auch die des Bisulfites durch Zusatz von Polythionaten oder von freiem Schwefel erheblich beschleunigt werden. So vermindert in einer 5,5-molaren Natrium- bisulfitlosung ein Zusatz von 4% ihres Salzgehaltes an Kalium- pentathionat die Zeit, welche bis zum Eintritt der Schwefel- abscheidung vergeht, wenn die Bisulfitzersetzung bei 700 im geschlossenen GefaBe, einer Anordnung, von der spiiter noch ein- gehender die Rede sein wird, vorgenommen wird, auf die Halfte, von 12 auf 6 Tage.
AuBerordentlich vie1 starker aber wirkt das schon bei einem Teil der vorbeschriebenen Versuche als Katalysator benutzte Selen ; es ist dabei gleichgultig, ob man es in elementarer Gestalt oder als selenige Saure der Losung zusetzt. Im ersteren Falle lost es sich in ihr in kurzer Zeit auf, und zwar sehr visl schneller a19 die gleiche Menge von Schwefel; 0,5 g fein gepulvertes Se losten sich z. B. in 8,5 n-Ammoniumbisulfitlosung in 10 Minuten, wahrend 0,5 g S unter den gleichen Umstanden dazu 55 Minuten brauchten. Benutzt man selenige Saure, so scheidet die Bisulfitlosung daraus freies Selen ab, das aber schon in der Kalte in wenigeri Augenblicken sich wieder lost. Wie stark die besohleunigende Wirkung von SelenzusBtzen ist, lehrt die Beobachtung, dal3 auf Zugabe von 0,65O/, vom Bisulfit- gehalt die eben erwahnte 5,5-molare Natriumbisulfitlosung schon nach 30 Stunden bei 700 im geschlossenen Gefal3 sich so weit zersetzt hat, dal3 die Schwefelabscheidung beginnt. Im offenen Gef&B und bei Kochhitze tritt in 5-molarer Ammoniumbisulfitlosung bei Gegen- wart von 0,lo/, des Salzes an seleniger Saure, also von 0,77 Millimol H,SeO, auf 1 Mol NH,HSO, nach 10 Stunden, bei der 15fachen Menge des Katalysators nach 2l/, Stunden die Sohwefelabscheidung ein, die ohne Katalysator sich erst nach 600 Stunden einstellt.
DieseWirkung des Selens wurde schon 1865 VonRATFiKE1) gefunden. Spater hat KLASON und dann KIAER2)sie bei der Zersetzung vonCalcium- bisulfitlosungen naher verfolgt und festgestellt, daB schon 2,7 - 10-60/o Selen in einer solchen Losung genugen, um ihre Neigung zur Um- setzung in Sulfat, Schwefel und Wasserstoffion merklich beschleunigt hervortreten zu lassen. Auch stellte er fest, daB Tellur eine ungleich schwAchere Wirkung als Selen hervorruft ; mit sorgfaltig gereinigtem,
1) Joum. prakt. C h . 96 (1865), 10. 2) 1. c.

290 El Foersler, I? Lange, 0. Dro,8baoh und W. Seidcl.
selenfreiem Tellur konnten wir in kochender Ammoniumbisulfit- losung iiberhaupt keine beschleunigende Wirkung erzielen.
Bei gewohnlicher Temperatur genugt ein Selenzusatz von 0,5 bis 0,70/, der umzusetzenden Salzmenge, um den Eintritt der Schwefelabscheidung bei der Zepsetzung veil Bisulfitlbsungen nach etlichen Tagen, z. B. in 7-molaref Ammoniumbisulfitlosung nach 9-10 Tagen, herbeizufuhren. Die katalytische Wirkung kleiner Selenzusatze ermoglicht also auch bei gewohnlicher Tempera tur die Verfolgung des Verlaufs der Bisulfitzersetzung, die ohne solchen Kakalysator nur aul3erst trage vor sich geht.
Es zeigte sich, dal3 auch unter diesen Umstiinden der Bisulfit- zerfall in zwei aufeinnnder folgenden Abschnitten vor sich geht : im ersten tritt wieder kein freier Schwefel auf, in der Losung entsteht Sulfat, daneben aber in hochstens ganz untergeordnetem MaSe Thiosulfat und dafiir fast ansschliel3lich Polythionat . Erst wenn der Sulfitschwefel nahezu verschwunden ist, beginnt mit der Aus- scheidung des Schwefels das zweite Stadium der Zersetzung: in ihm vrrschwindet das Polythioiiat und liefert den freien Schwefel und weiteres Sulfat. Am Ende des ersten Stadiums scheidet sich alles Selen wieder ab, und zwar so vollstiindig, dal3 es auch mit der sehr empfindlichen Probe mit Codeinphosphat 1) in der Losung nicht mehr nachgewiesen werden kann ; seinem katalytischen Einflusse nnterliegt also wieder nur das erste Stadium der Zersetzung, wie es schon bei den Versucheii in der Kochhitze hervortrat.
Zur naheren Verfolgung der Umsetzung wurden die Versuche in Erlenmeyerkolben von der durch Fig. 5 gekennzeichneten Ein- richtung ausgefiihrt. Rohr C cliente dazu, die Losung unter schwachem Wasserstoffdruck zu halten, so dal3 auch bei den Probenahmen durch B die Luft nicht in storendem MaBe eindringen konnte. Die Ergebnisse einer in solcher Art durchgefiihrten Versuchsreihe sind in Ubersicht 6 (S. 292) zusammengestellt und durch die Kurven der Fig. 6 veranschaulicht . Ausgegangen wurde von 200 ccrn einer 70 g NH,HSO, in 100 ccm enthaltenden, d. h. etwa 7-molaren Losung. Die An- gaben der Ubersicht 6 beziehen sich auf je 1 ccm von ihr. Beim An- sauern einer Probe schied sich im ersten Teile des Versuches Selen ab, dem etwas BUS Thiosulfat stammender Schwefel beigemischt sein konnte. E r wurde nicht besonders bestimmt, da das Gewicht der so gefallten Selenniederschlage niemals merklich hoher, sondern immer
I) ERNST SCIWIDT, Arch. d. Pharm. 2663 (1914), 161.
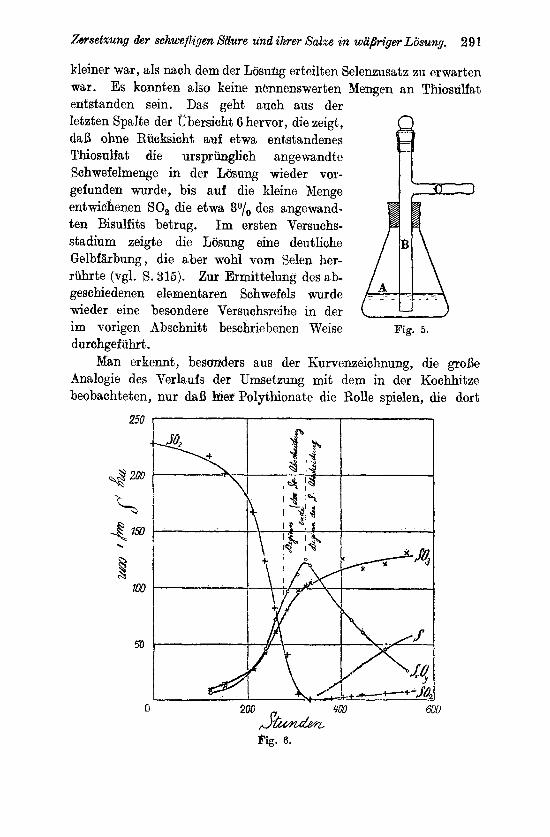
Zersetxung der schwefligen S&re und ihrer Sake in w&?riyw Liisung. 29 1
kleiner war, als nach dem der Lbsung erteilten Selenzusatz zu erwarten war. Es konnten also keine nknnenswerten Mengen an Thiosulfat entstanden sein. Das geht auch aus der Ietzten Spalte der Ubersicht 6 hervor, die zeigt, da13 o h 0 Riicksicht euf etwa entstandenes Thiosulfat die urspriingfioh angewandte Sohwefelmenge in der Msung wieder vor- gefunden wurde, bis auf die kleine Menge entwichenen SO, die etwa Solo des angewand- ten Bisulfits betrug. Im ersten Versuchs- stadium zeigte die Losung eine deutIiche Gelbfiirbung, die aber wohl vom Selen her- riihrte (vgl. S. 315). Zur Ermittelung des ab- geschiedenen elementaren Schwefels wurde wieder eine besondere Versuchsreihe in der
durohgefiihrt . Man erkennt, besmders aus der Kurvenzeichnung, die grol3e
Analogie des Verlaufs der Umsetzung mit dem in der Kochhitze beobachteten, nur dafi bet Polythionate die
im vorigen Abschnitt beschriebenen Weise Fig. 5.
r I
Rolle spielen, die dort
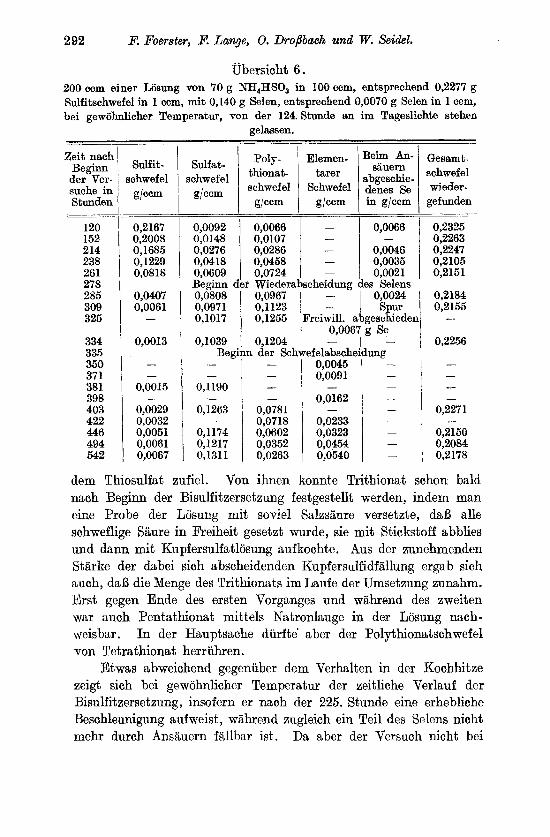
292 P. Foerster, l? Lange, 0. Dropbach ulad W. Seidel.
Ubersioht 6. 200ccm einer Lijsung von 70g NH,HSO, in 100ccm, entsprechend 0,2277 g Sulfitschwefel in 1 ccm, mit 0,140 g Selen, entsprechend 0,0070 g Selen in 1 ccm, bei gewohnlicher Temperatur, von der 124.Stunde an im Tageslichte stehen
gelassen.
Sulfat- schwefel
deem
____-
Zeit nach Beginn der Ver- suche in Stunden
120 152 214 238 261 278 285 309 325
334 335 350 371 381 398 403 422 446 494 542
____- ~-
P O l Y - thionat- schwef el
Sulfit- 3chwefel deem
0,2167 0,2008 0,1685 0,1229 0,0818
0,0407 0,0061 -
0,0013
- -
0,0015
0,0029 0,0032 0,0051 0,0061 0,0067
-
- -
- -
-
-
0,0066
0,0046 0,0035 0,0021
- 0,0276 0,0418 0,0609
0,0286 0,0458 0,0724
0;0971 0,1017
Oil123 0,1255
Gesamt- schwefel wieder-
gefunden
0,2325 0,2263 0,2247 0,2105 0,2151
0,2184 0,2155 -
0,2256
- - - -
0,227 1
0,2150 0,2084 0,2178
-
- -
0,1190
0,1263 -
0,1174 0,1217 0,1311
-
dem Thiosulfat z-Sel. Von ihnen -3nnte Trithionat :-__on bald nach Beginn der Bisulfitzersetzung festgestellt werden, indem man eine Probe der Losung mit soviel Salzsaure versetzte, daB alle schweflige Siiure in Freiheit gesetzt wurde, sie mit Stiokstoff abblies und d a m mit Kupfersulfatlosung aufkochte. Aus der zunehmenden Starke der dabei sich abscheidenden Kupfersulfidfallung ergab sioh auch, da13 die Menge des Trithionats im Laufe der Umsetzung zunahm. Erst gegen Ende des ersten Vorganges und wahrend des zweiten war auoh Pentathionat mittels Natronlauge in der Losung nach- weisbar. In der Hauptsache durfte' aber der Polythionatschwefel von Tetrathionat herruhren.
Etwas abweichend gegeniiber dem Verhalten in der Kochhitze zeigt sich bei gewohnlicher Temperatur der zeitliohe Verlauf der Bisulfitzersetzung, insofern er naoh der 225. Stunde eine erhebliche Besohleunigung aufweist, wahrend zugleich ein Teil des Selens nicht mehr durch Ansiiuern fallbar ist. Da aber der Versuch nicht bei
- - - -
0,0781 0,0718 0,0602 0,0352 0,0263
0,0233 0,0323 0,0454 0,0540
- - - -

Zerdxung dsr uchwefiigen Sidure uizd ihrer Salze in wapriger Losung. 293
genau innegehaltener Temperatur durchgefuhrt wurde, muS es z-unachst dahingestellt bleiben, ob die hier beobachtete Beschleunigung wirklich der Natur des Vorganges entspricht . Die Sulfitkonzentration sinkt wahrend des ersten Vorganges bis xu Beginn der Schwefel- abscheidung auf O,6O/, ihres Anfangswertes, um d a m - im Gegen- satz zum Verhalten in der Kochhitze - whhrend des zweiten Vor- ganges, so weit er beobachtet wurde, bis auf etwa Sol0 desdnfangs- wertes wieder zu steigen. Ware der Versuch bis zum Ende des zweiten Vorganges genauer verfolgt worden, so ware, wie ein spiiterer Versuch zeigen wird, auch der im zweiten Vorgange nachgebildete Sulfit-S wieder verschwunden. Nach vollstandiger Umsetzung, die etwa 3 Monate nach Beginn der Schwefelabscheidung eingetreten war, wurde nur die Aziditat der Losung bestimmt. Diese war im Anfange des Versuches 5,7-normal und wurde nech vollendeter Um- setzung zu 1,g-normal gefunden, d. h. der anfanglichen, wie es Gleichung (4) verlangt. Auch die in der Ubersicht 6 fur den SchluS des Versuchs angegebenen Werte des Sulfatschwefels und des elementaren Schwefels entsprachen noch nioht dem Endzustande, da noch betrachtliche Mengen Polythionatschwefel vorhanden waren; demgemaI3 ist ihr Verhaltnis nur 2 : 0,82.
Berucksichtigt man bei dem in der Kochhitze ausgefuhrten Versuchen die verfluchtigte Menge des Schwefeldioxyds, so sind die dort und bei der Versuchsreihe der Ubersicht 6 untersuchten Konzentrationen an Sulfitschwefel einander annahernd gleich, beide Versuchsreihen lassen sich also in bezug auf den fur die ein- zelnen Stadien erforderlichen Zeitaufwmd vergleichen. Dieser ist fur das erste Versuchsstadium in der Kochhitze etwa der doppelte dessen, der bei gewohnlicher Temperatur unter dem Einflusse des Selemusatzes beobachtet wurde ; im zweiten Stadium aber fuhrt dort die Umsetzung im dritten Teile der Zeit etwa so weit wie hier.
Als ein weiterer Unterschied beider Arbeitsweisen ergab sich schlieBlich, dal3, wenn bei gewohnlicher Temperatur die Hydrolyse des Bisulfits vermieden wurde, im ersten Stadium der Bisulfit- zersetzung kein Hyposulfit nachweisbar war.
d) Versuche bei 70-800 in1 geschlossenen GefaBe. Die beiden in den vorangehenden Abschnitten beschriebenen
Versuchsreihen uber die Zersehng der Bisulfitlosungen unterscheiden sich einerseits hinsichtlich der Reaktionstemperatur, andererseits hinsichtlich der Wasserstoffionenkonzentration, die bei ihnen herrschte,
Z. anorg. U. allg. Cham. Bd. 1%. 20

294 F. l%er&r, 2% Lange, 0. Dropbach und W. Seidel.
insofern diese in der Kochhitze sehr vermindert, bci gewohnlicher Temperatur aber die dem Bisulfit entsprechende war. Die Frage, welohes von beiden Momenten in erster Linie dafur bestimmend war, da5 im ersten Zersetzungsstadium dort Thiosulfat, hier Poly- thionat neben Sulfat mtsteht, lie13 sich entscheiden, wenn man die Zersetzung ohne wesentliche Aziditatsverminderung, also ohne Ent- weichen von Schwefeldioxyd, auch bei hoherer Temperatur ver- folgen konnte. Es zeigte sich, daJ3 dies bei 70-800 im geschlossenen
A
d U
Fig. 7.
GefaBe moglich ist, da unter diesen Umstanden der Schwefeldioxyd- druck starkerer Bisulfitlosungen 3Iq bis 1 Atmosphare nicht iiber- steigt. Solche Drucke helten gute Rundkolben nus, so daI3 man die Vorgiinge mit dem Auge und durch Probenahme verfolgen kann.
Fur die Versuche wurde fol- gende durch Fig. 7 erlauterte Apparatur benutzt. A ist ein stark- wandiger Rundkolben von 300 ccm Fassungsvermogen, der durch einen Gummistopfen fest verschlossen werden kann. Dieser tragt die Rohre B und C. Ersteres dient zur Probenahme und besitzt dazu den sehr gut eingeschliffenen Hahn D, duroh welchen der im Kolben herrschende Druck bei vorsichtiger a fnung etwas vom Kolbeninhslt heraustreibt. Rohr C fuhrt zu
dem Quecksilbermanometer H ; das mit Druckschlauch angeschlossene Verbindungsstuck zwisohen beiden tragt den Hahn E, der den Druck- ausgleich erforderlichenfalls herzustellen erlaubt. Das Manometer dient zugleich ads Sicherheitsventil, indem seine Liinge und Queck- silberfullung so bemessen sind, da13, wenn der aberdruck im Kolben 1 Atmosphare ubersteigt , das Quecksilber herausgedruckt wird ; zu seiner Aufnahme dient die Schale P. Der Kolben hangt in einem als Wasserbad dienenden groBen Becherglase.
Mit solcher Anordnung erwies sich der durch Selen katalysierte Verlauf der Bisulfitzersetzung bei 70-800 ganz iibereinst8immend
Zersetzung der schwefligen $ a w e und ihyer 8ahe in wapriger Losung. 2 96
mit dem bei gewohnlicher Temperaitur : im ersten Stadium fanden sich in der Losung Sulfat und Polythionat neben sehr w-enig Thio- sulfat ; unter Ausscheidung von Schwefel verschwanden die letzteren im zweiten Stadium. Darnit wird bewiesen, daB der eigenartige Verlauf der Umsetzung einer im offenen GefB13 kochenden B i d f i t - losung durch die Verminderung der Wasserstoffionenkonzentration iii ihr hervorgerufen ist. Von ihr hBngt es also ab, ob als Zwischen- produkte in einer sich zersetzenden Bisulfitlosung wesentlich Thio-
t
0
Fig. 8.
sulfat oder Polythionate auftreten; geht man von Gemischen von Sulfit und Bisulfit Bus, so wird das erstere, zersetzt ma.n Bisulfit und sorgt dafur, daB kein SO, entweicht, so werden die letzteren die Entstehung des elementaren Schwefels vermitteln.
Von Interesse war es bei diesen Versuchen, den zeitlichen Gang des SO,-Druckes whhrend der Zersetzung zu verfolgen, da, wie oben schon erwahnt, wenn Polythionate im ersten Stadium als Zwischen- produkte auftreten, im zweiten Stadium anfs neue Sulfitschwefel sich zeigt. Die beiden folgenden Versuche und die sie veranschau- lichende Kurvenzeichnung, Fig. 8, geben hieruber duslrunft ; sie wurden mit Natriumbisulfit, dnrchgefuhrt.
20*

296 F. Roerstw, R Lange, 0. Dro/3bach zcnd W. Seidel.
26 30 40 90
68 ' + 503 74 + 523 74,5 1 +519 75,3 + 519
Versuch 46.
Losung wie bei Versuch 45.
15 20 30 45 60 110 130 160
~ ~~
70 80,2 79,O 79,O 79,l 79,O 80.2 79.6
240 250 325 400 450 480 555 635 710 1305
Se - Ahchei 200 I 80.7
80,O 80,3 81,O 81,5 79,2 82,O 80,O 82,3 80,2 70
Druck in mm Hg
+ 328 + 493 + 588 + 586 + 576 + 566 + 574 + 582 + 335 + 210 + 60 + 33 + 170 + 318 + 417 + 439 + 433 + 410 + 368
u g
ginnt
+ 50 Abgeschiedene Menge S 8,4 g,
d. h. 92O/, Ausbeute.
Man erkennt, wie mit der beginnenden Se-Abscheidung, also gegeh Ende des ersten Stadiums der Umsetzung, der vorher wenig veranderte Druck schnell herabsinkt und im Anfange der Schwefel- abscheidung noch weiter fallt, um nun aufs neue iiberraschend stark anzusteigen und dann ganz allmahlich wieder sich dem NulIwert zu nahern.1) Bei 75O liegt das offenbar mit dem Minimum der Sulfit- konzentration zusammenfallende Druckminimum sogar bei negativen Werten; die Losung nimmt jetzt das vorher in den Raum iiber ihr abgegebene Schwefeldioxyd wieder auf, und da jedenfalls ein Teil der im Kolben urspriinglioh vorhandenen Luft zur Oxydation von schwefliger Saure gedient hat, so wird der Druck negativ, bis das im zweiten Stadium sich entwickelnde SO, dies wieder ausgleioht. Bei 800 ist wohl der SO,-Druck auch sehr verdiinnter Bisulfitlosungen schon so grol3, daB ein Unterdruck nicht mehr zustande kommt.
1) Die Kurven von Fig. 8 wwden der Raumersprnia wegen beim &it- punkt 800 Minuten abgebrochen.

Zersetxung dev schwefligelz Saure ulzd ihrer Sa2xe in w@rigw Losung. 297
Vergleicht man die Tatmehe, da13 im zweiten Stadium nach diesen Versuchen der Drnck auf eine ahnliche Hohe wie im ersten zuruokkehrt, mit der aus Obersioht 7 sioh ergebenden, da13 die Menge des in jenem neu entstehenden Sulfitschwefels nur ein geringer Bruchteil von der ist, die im ersten Stadium anfangs vorlag, so folgt, daB der Sulfitschwefel in beiden Fiillen in versohiedener Form vor- liegen muB, im zweiten Stadium offenbar als freie schweflige SBure bzw. ihr Anhydrid. Es war daher von Interesse, auoh den zeitlichen Gang der Aziditat der Losung etwas niiher zu verfolgen. Dazu wurden Proben der Losung mit Natronlauge unter Anwendung von Phenolphthalein als Indikator titriert. Wenn hierbei auoh die alkalisahe Reaktion des Sulfits gegen diesen Indikator einen Febler bedingt , so war doch der Zweck des Versuchs zu erreiohen; er wurde zugleich benutzt, um die Art der in der Losung auftretenden Poly- thionate zu verfolgen. Die erhaltenen Ergebnisse sind die folgenden :
Versuch 47. 100 ccm NaRSO, mit 0,178 g Sulfit-Schwefel in 1 ccm,
Temperatur 800. 0,55 Mol in 100ccm. 1 g H,SeO, als Katsllysator.
Zeit in Min.
0 40 90
135 155 160 210 225 250 270 390 465 670
_ - _ __ ___.
1 ccm ver- braucht ccm
n-NaOH
I 4,30 ' 4,lO
1 237 I 1,78 ' 1,38 j 0,33
0,30 I 0,40 1 0,50
1 1J4
i 3,69
1 0,98
I 1945
Bemerkungen
Polythionat nachweisbar. Trithiomt nachweisbar. Pentathionat nicht nachweisbar. Trithionat reichlich vorhanden. Selenabscheidung beginnt.
Pentathionat nachweisbar. Schwefelabscheidung beginnt.
Pentathionat nachweisbar.
Die Beobachtungen uber die Polythionate bestiitigen die Uber- einstimmung im Verlaufe der Umsetzung bei $00 mit der bei Zimmer- temperatur.
Den Gang der Aziditat veranschaulicht die Kurve in Fig. 9; sie zeigt, wie im ersten Stadium bis zum Beginn der Schwefel- abscheidung der Verlauf der Aziditat mit dem des S0,Druckee iibereinstimmt ; bier bestimmt das Bisulfit die Aziditat. Zu Beginn
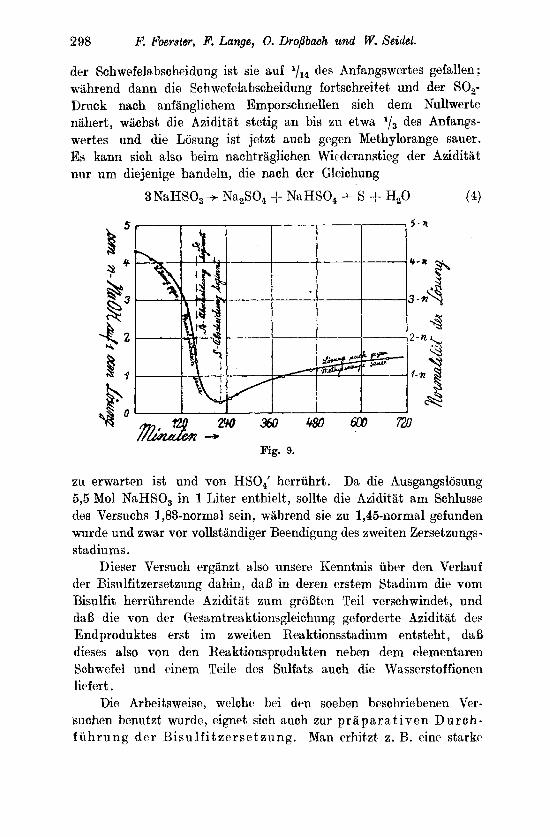
298
der Schwefelabscheidung ist sie auf des Anfangswertes gefallen : wzhrend dann die Schwe felabscheidung fortschreitet und der SO,- Druck nach anfanglichem Emporschnellen sich dem Nulkwerte niihert, wachst die Aziditat stetig an bis zu etwa I/, des Anfangs- wertes und die Losung ist jetzt auch gegen Methylorange sauer. EH kann sich also beim nachtraglichen Wideranstieg der Aziditiit nnr urn diejenige handeln, die nach der Gleichung
R li'oerstw, F. Lange, 0. DroPbaoh und W. Seidel.
3 NaHSO, -+ Na,S04 4- NaHSO, + S + &O (4)
$0
zti erwarten ist und von HSO,' herruhrt. Da die Ausgangslosung 5,5 Mol NaHSO, in 1 Liter enthielt, sollte die Aziditat am Schlusse des Versuchs 1,83-normal sein, wahrend sie zu 1,45-normal gefunden wurde und zwar vor vollstandiger Beendigung des zweiten Zersetzungs- stadiums.
Dieser Versuch erganzt also unsere Kenntnis uber den Verlauf der Bisulfitzersetzung dahin, daB in deren erstem Stadium die vom Bisulfit herruhrende Aziditat zum groBten Teil verschwindet, und daB die von der Gesamtreaktionsgleichung geforderte Aziditat des Endproduktes erst im zweiten Reaktionsstadium entsteht, daB dieses also von den Reaktionsprodukten neben dem elementaren Pchwefel und einem Teile des Sulfats auch die Wasserstoffionen liefert .
Die Arbeitsweise, welohe bei den soeben beschriebenen Ver- snchen benatzt wurde, eignet sich auch zur p r a p a r a t i v e n Durch- f u h r u n g de r Bisu l f i tzerse tzung. Man erhitzt a. B. eine starke

Zersetwng der schwefligen Sawe und ihrer Salxe in wapriger LSsung. 299
Ammoniumbisulfitlosung nach Zugabe voA etwa 0,20/, Se in einem mit Quecksilbermanometer versehenen Rundkolben so lange auf 70-80°, bis der Druck am Apparat ein Minimum erreicht und alles Selen neben etwas Schwefel sich abgeschieden hat. Man filtriert die heil3e Losung rasch von dem aufs Neue zu benutzenden Selen ab, bringt sie in den Kolben zuruck und erhitzt weiter, bis wieder der Uberdruck fast verschwunden ist . Der ausgeschiedene Schwefel wird von der heiBen Losung getrennt und aus dieser das Sulfat kristallisieren gelassen.
e) T heore t i s che E r o r t e r u n g der Versuchsergebnisse .
Die vorstehend mitgeteilten Versuchsergebnisse stehen durchaus im Einklange mit der fur den Mechanismus des Zerfalls der schwef- ligen Saure gegebenen Theorie. Der Unterschied im Verhalten der freien Saure gegeniiber dem ihrer Sake besteht, wie oben sohon erwahnt , wesentlich darin, da13 die H'-Konzentration von vornherein von wesentlich niederer Gr.bBenordnung ist und auch durch die allmahliche Anreicherung der SO," hieran sich kaum etwss Hndern kann, so lange noch gewisse Konzentrationen von Sulfit- bzw. Bisulfit vorhenden sind. Sie sorgen dafiir, daB die H'-Konzentration zunachst nicht uber die in einer Losung von Bisulfit und schwefliger Saure mogliche GroSenordnung hinauswachst . Dadurch wird die nur bei hoherer H'-Konzentration erhebliche Rechtsverschiebung des Gleichgewichts
S,O," + H' ~t HSO,' + S (15a)
so lange hintenan gehalten, bis das Bisulfit, beinahe verschwunden ist. Bis dahin kann je nach dem Verhaltnis der Konzentrationen von HSO,' und SO,'' und der dadurch bedingten H'-Konzentration, entweder das primar entstehende Thiosulfatanion bestehen bleiben oder sich in das Anion der Pentathionsaure umwandeln und d a m die an ihm sich durch die HSO,' vollziehenden Folgereaktionen erfahren. Erst wenn das Bisulfit weitgehend verbraucht ist, kann die H'-Konzmtration steigen. Dann erst kann aus Thiosulfat bzw. Polythionat die eine gewisse Aziditat voraussetzende Abscheidung von freiem Schwefel vor sich gehen. Dadurch gelangen die bei der Zersetzung cler freien Saure sich uberlagernden Umsetzungsvorgange beim Zerfall des Bisulfits in eine zeitliche Aufeinanderfolge, durch die ein noch sicherer Einblick in den Mechanismus des Vorganges gewghrt wird, als es beim Zerfall der freien Saure moglich war.

300 F! liberster, F. Lange, 0. Bopbach und W. Sdd.
Fur die nahere Erorterung der unter solchen Umstanden ab- laufenden Vorgange sol1 zunachst die Mitwirkung des Selens a u k Betracht bIeiben; am ganzen Geprage der Vorgange wird ja, wie oben bemerkt, nichts geandert, wenn sie ohne diesen Katalysator sich abspielen. Die Art seiner Wirkung wird in einem spaterm Abschnitt zu besprechen sein.
Die beiden Stadien im Bisulfitzerfall sollen gesondert betrechtet werden. Der Kurze wegen sind dabei die bis fast zum Verschwinden des Bisulfits sblaufendrn Vorgange als erster Hauptvorgang, die d a m an den hierbei entst andenen Zwischenprodukten unter Schwefelabscheidung sich vollziehenden Vorgange als zweiter Haupt- vorgang bezeichnet.
Dem ersten bis zum Ver- schwinden der Hauptmenge des Bisulfits fiihrenden Vorgange diirfte als primare Umsetzung des Bisulfits wieder diejenige in Sulfat und Sulfoxylsaure zugrunde liegen :
I. Der e r s t e Haup tvorgang .
2H80,’ 3 SO,” +. H,SO,. v o a >
Als wichtige Stiitze dieser Auffa ssung kann die Tatsache angesehen werden, daB da, wo be im Kochen e iner Bisu l f i t losung i m of fenen GefaBe SO, entweichen kann, also eine genugendniedrige H’-Konzentration das Bestehenbleiben kleiner Mengen von Hypo- sulfit, als dem Kondensationsprodukt von Sulfit- und Sulfoxylsaure, gestattet, solches auch nachgewiesen werden konnte ; das war in der kochenden Ammoniumbisulfitlosung der Fall, in der durch das Entweichen von SO, in dem Gleichgewicht
HSO,’ == H + SO,” ‘ (7) die SO,”-Konzentration sehr gesteigert, die H -Konzentration ent- sprechend vermindert war.
Zum weitaua groBten Teil geht aber auch unter diesen Urn- standen die Sulfoxylsaure in Thioschwefelsaure iiber, deren Anion sich nun neben dem der Schwefelsaure stetig in der Losung snreichert und zwar, wie Ubcrsicht 5 und die Kurvenzeichnung Fig. 4 lehren, derart, daB praktisch auf 2S0, immer 1 S,O, entsteht. Das ist nach Gleichung (10) und (11) eu erwarten, deren Gesamtergebnis durch die Gleichung
msammenzufassen wlre. Einen merklichen katalytischen EinfluB hnn bier das Thiosulfat nicht ausiiben, da ihm fur den dazu
4HSOi -+ 250,” + S,O,” + Q O + 2H‘ (22)

Zersetxzing der schwefligen Sawe und ilwey Sake in wapmger Lomfig. 301
erforderlichen Ubergang in Pentathionat die H-Konzentration fehlt, und andererseits angesichts der kleinen SO,-Konzentration auch das gelbe komplexe Anion [ S,O,( SO2),]" nur in sehr kleiner Konzentration auftritt und auch, wie oben gezeigt , zu schnellem Umsatze nicht besonders befahigt ist .
Dieser Befund ist sehr wichtig, denn er zeigt, daB de r k a t a - l y t i s che E in f luB des Schwefels bzw. des Th iosu l f a t s a u f d e n B i su l f i t ze r f a l l n u r auf e in b e s t i m m t e s K o n z e n t r a t i o n s - i n t e rva l1 des H' besch rank t i s t . Wie friher gezeigt, ist dieses nach oben begrenzt, da bei Gegenwart groBerer Konzentrationen starker Sauren jener EinfluB nicht mehr besteht; jetzt zeigt sich, daB auch nech unten, bei der H-Konzentration einer Sulfit-Bisulfit- losung, eine Grenze fiir ihn vorhanden ist. Wahrend im ersteren Falle die vollige Unbestandigkeit des S203" bei Gegenwart grol3erer H-Konzentrationen die Ursache der Erscheinung war, ist jetzt die groSe Bestandigkeit des S20it bei sehr kleiner H-Konzentration der Grund fiir das Ausbleiben ihrer katalytischen Wirksamkeit. Nur in dem Konzentrationsintervall der H , in welchem S,O," in genugender Konzentration bestehen und doch auch wieder mit groBerer Geschwindigkeit sich umsetzen kann, vermag es seine katalytische Tatigkeit zu entfalten, bzw. die des Schwefels zu vei- mitteln. Es ist besonders interessant, daB auch bei der kleinen H-Konzentration, bei der die katalytische Wirkung des Schwefels aufhort, die des Selens noch fortbesteht. Darauf wird spater noch zuriickzukommen sein.
Da sowohl die Schwefelsaure wie die Thioschwefelshure starke Sauren sind, so werden die bei den durch Gleichung (22) ausgedriickten Vorgiingen freiwerdenden H' Gleichgewicht (7) mgunsten der HSO,' verschieben und dadurch einerseits weiteres Entweiohen von SO, a m der im offenen GefaBe kochenden Losung veranlassen, wie es den Reobachtungen entspricht . Andererseits aber erzeugen sie aus SO," das HSO,', an dem die weiteren Urnwandlungen sich voll- ziehen konnen, so daB, wie BOSCH mitteiltel), auch eine von vorn- herein Sulfit und Bisulfit enthaltende Losung sich glattauf in Sulfat und Schwefel zersetzen kann, obwohl das gesattigte Sulfit allein hierzu nicht imstande ware. Durch die in Gleichung (22) zusammen- gefaBten Vorgange verschwindet also langsam fortschreitend das Bisulfit aus der Losung, geschwindigkeitsbestimmend diirfte hierfiis
1) 1. c.

302 F. Fowster, F. Lunge, 0. DroPbach und W. Seidel.
wesentlich Vorgang (10a) sein. Am Ende dieser ersten Vorgangs- reihe finden sich Sulfat uiid Thiosulfat in schwach saurer Losung neben Resten von Sulfit in der Losung.
Etwas anders gestalten sich die Erscheinungen, w e n n e in E n t w e i o h e n von SO, v e r h i n d e r t wird, sei es, dal3 bei gewohn- lieher Temperatur im offenen oder bei hoherer im geschlossenen [fef&B gearbeitet wird. Unter dem Einflul3 der H’ der Bisulfit.18sung I)olymerisiert sich jetzt die Sulfoxylsanre, sei es unmittelbar, sei es mittelbar uber die Thioschwefelsaure zu Pentathionsaure (im Sinne von Gleichung (16). Diese unterliegt dann alsbald der weiteren Ein- wirkting des Bisulfits [Gleichnng (17) und (IS)] und uber Tetra- thiouat- und Trithionatanion entsteht [Gleichung (19)] das Sulfat und daneben das Thiosulfatanion, ans dem aufs neue Pentathion- saure sich bildet. So lange noch reichlich Bisulfit zugegen ist, ver- schwindet die Pentathionsiiure sehr schnell und der Polythionat- schwefel ruhrt wesentlich von Tet,rathionat her. Mit fortschreitender Umsetzung gesellt sich ihm in st,eigendem Ma& Trithionat bei, <lessen Anreicherung aber durch seine verhaltnismaBig schnelle Zcrsetzung eingeschrankt ist . Erst wenn die Bisulfitkonzentration stark abgenommen hat, kann die a m der Thioschwefelsiiure zuriick- gebildete Pentathionsaure sich so weit in der Losung halten, daS ihr Nachweis mit Hilfe von Natronlange gelingt. Das haben die Versuche, zumal Versuch 47, gelehrt.
Fur diesen Verlauf der Umsetzung ist es yon Bedeutung, dal3 diese selbst auf die H-Konzentration regelnd einwirkt . Man erkennt dies am besten, wenn man die Gleiehungen fiir die hier zussmmen- Miirkenden Vorgange folgendermafien formuliert :
20HSO3’ --t 10S0,” + 5S20,” + 5€&0 + lOH’, (‘22)
5 S,03” + 10 H’ 2S,O,“ + 2HS03’-+ 2S4O6” + 2S,03” + 2H’, 2S,O,” 3 2HSO,’ -F 2S,O,” + 2S,O,” + 2H’, 2S,O,” + 2H20 --t %SO,” + 2S20,” + 4H’.
-> 2 S50,” + 3 H,O + 4H’, (16)
(18)
(1 9)
(1 7)
Von den 20H’ des Bisulfits, die unter Anwendung von Phenol- phthalein als Indikator titrierbar sind, wurde Vorgang (22) die Hiilfte ubrig lassen. Dsran Bndert sich nichts, wenn die HSO,’ lediglich nach Vorgang (17) bis (19) unter Snlfatbildung verschwinden, da die Summe der Gleiohungen (16) bis (19) wieder 4HS0,‘ -+ 2S0,” + S,O,” $- H,O + 2H’ ergibt. Durch die, Polymerisation des Thio-

Zersekzcng der schwefligen S u m uizd ihrer Sake in wa/j’riger Losung. 303
sulfats aber werden von deli bei dessen Entstehung auftretenden 10H nur 4 ubrigbleiben. Freilich kommen durch die folgenden Vor. gange noch 8H hinzu, aber c‘s entstehen zugleich GS,O,”, also ins- gesamt wieder auf 1 S,O,“ 2€T, und bei neuer Pentnthionatbildung bleiben davon wieder nur 4/10 ubrig, und so geht es weiter. Die Abnahme der Aziditat wurde ihr HochstmaB erreichen, wenn das Bisulfit ganz in Sulfat und Pentathionat, also im Sinne der Gleichung
20 H803‘-+ 10 SO,” + 2 S506” f H H20 -t 4 H (23) sich umset2;t.l) Imrnerhin kann hiernach mit dem volligen Ver- schwinden der HSO,’ die Aziditat nicht atif Null herabgehen. So lange noch Bisulfit vorhanden ist, werden die entstehenden H‘ die G leichgewichte
zugunsten von SO, verschieben. Da aber die Gesamt konzentration des Bisulfitschwefels herabgeht. wird mit ihr nuch der SO,-Druck dauernd sinken, wie es auch beobachtet ist (Versuch 45 und 46). Bis das Bisulfit weitgehend verschwunden ist, behalten im Gleioh- gewicht
HSO; + H’ + &SO, + H,O + SO, (5) u. (6)
S,O,” + IT p. HSO,’ + S (1 5a)
die Konzentrationen von S,O,” iind H‘ diejenigen Werte, die er- forderlich sind, damit das Thiosulfat ausschliekilich die Umwandlung in Pentathionat erfahrt .
Geschwindigkeitsbestimmend fur denverbmuch von HSO,’ und fur die Entstehung von Sulfat und Polythionat sind bei diesen Reaktionen wieder (vergl. S. 270) die Vorgange (16) bis (19). 1st von Anfang an kein Thiosulfat oder Polythionat zugegen, so regeln zuniichst die sehr langsamen, in Gleichung (22) zusamknengefafiten Vorgange den zeitlichen Verlauf der Umsetzung. In dem Mafie aber, wie hierbei Thiosulfat entsteht, treten immtlr mehr die bei der H’-Konzentration von Bisulfitlosungen an diesem sich vollziehenden, weit schnelleren Vorgange (16) bis (19) in den Vordergrund. Darauf beruht es, daB, wie Fig. 2 zeigte, bei der Zersetzung im geschlossenen Rohr eine
1) Der bei Versuch 47 beobachtete Riickgang der Gesamtaziditiit ist noch groBer, ah der iiach dieser Gleichung dafur zu erwartende Hochstwert. Hierbei wurde freilicli mit Selen als Kablysator gearbeitet, wobei, wie weiter mten auszufuhren ist, ein noch etwas starkerer Aziditiitsruckgang als ihn Gleichung (23) fordert, zu erwarten ware. Die absoluten Fehlbetrage sind zwar nicht bedeutend; es bedarf weiterer Versuche, um ihrer Ursache nachzugehen; das Entweichen einer geringen Menge SO, genugte, urn sie zu erklaren.
--

304 3. Foerster, l% Lange, 0. Dropbach und W. Seidel.
gewisse Irikubationszeit dem spiiteren, weit schnelleren Verlauf der Bisulfitzersetzung voransgeht, und das positiv autokatalytische Ge- prage dieses Vorganges hervortritt. Sobald aber genugende Konzen- trationen von Thiosulfat oder Polythionat der Losung von vorn- herein erteilt oder durch Schwefelzusatz in ihr erzeugt werden, mill3 die Inkubationszeit mehr odpr weniger wegfallen, der Bisulfitzerfall also durch cliese von ihm immer neugebildeten Verbindungen kata- lytisch sehr beschleunigt werden. Bedingung hierfur ist die in Bisulfit- bzv. Bisulfit-Schwefligsaurelosungen herrschende H'-Konzentration.
11. Der z w e i t e H a u p t v o r g a n g . Die Natur des zweiten, nach Vmchwinden der Hauptmenge des Bisulfits sich abspielenden Vorganges ist nach den bisher erorterten Erfahrungen am leich- testen dort zu ubersehen, wo die Ver suchsbed ingungen d a s E n t w e i c h e n v o n Schwefe ld ioxyd ve rh inde rn .
Die wahrend des ersten Hauptvorganges auftretenden H' werden, wie wir sahen, in ihrer Konzentration durch das noch vorhandene Bisnlfit auf den kleinen Betragen einer Bisulfit-Schwefligsaure- losung gehalten. Mit dem Ruckgange der HS0,'-Konzentretion muI3 dann drr Punlit kommen, an dem das Gleichgewicht
S,O," + H == HSO,' + S W e )
sich soweit rechtsseitig verschiebt, daB die Abscheidung von elemen- tarem Schwefel eintritt. Diese kennzeichnet den zweiten Hsupt- vorgang und hat weitere wichtige Erscheinungen zur Folge ;
1. In dem MaBe, wie der Vorgang
S203" + H' -+ HSO,' + S (15) infolge der jetzt dauernden Steigerung der H'-Konzentration a,n Umfang zunimmt, muB der bisher allein das S,O," verbrauchende Vorgang (1 S), die Polymerisation zu S506", zuriicktreten. Anderer- seits wird auch die Geschwindigbeit des Abbaus der hoheren Poly- thionate durch HSO,', also mittelbar die Neuentstehung von Poly- thionat aus HSO; durch das Anwachsen der H'-Konzentration vermindert , es wird also die Nachlieferung von Polythionat verlang- samt, und in der Losung bleibt Pentathionat nachweisbar. D s andererseits die Verseifung des Trithionats in ihrer Geschwindigkeit von der H'-Konzentration nicht merklich beeinflufit istl), schreitet der Verbrauch von Polythionat und seine Umwandlung in Sulfat verhaltnismalBig schnell voran : die Polythionatkonzentration gehf
F. FOERSTIOR und A. HORNIQ, 2. anorg. u. allg. Chem. 126 (1922), 134.

Zersetrung der schwefligen. Saura und ihrer Sake in waphgev Losung. 305
zuruck, wahrend die Sulfatkonzentration zunachst etwa im gleichen MaBe wie vorher weiter ansteigt. Das lehrt Ubersicht 6 und Fig. 6.
2. Wahrend des ersten Hauptvorganges wurde HSO,’ lediglich verbraucht ; Vorgang (15) aber erzeugt HSO,’. Zuntichst wird E s, nachdem der zweite Hauptvorgang eingesetzt hat, jedenfalls groljenteils noch beim Abbau der hoheren Polythionate verbraucht . Allmahlich aber wird durch deren Konzentrationsruckgang und die zunehmende E-Konzmtration dieser Verbrauch immer langsamer , HSO,’ mu13 sieh also in der Losung allmahlich wieder anreichern. Anch dies lehrte Ubersicht 6 und Fig. 6 .
3. Wenn alles Bisulfit verschwunden ist, muB nach GI. (23) (S.303) eine gewisse Menge von H noch in der Losung vorhanden sein. Diese stehen jetzt nur den Anionen starker Sauren gegenuber, diirften also in der Hauptsache frei vorhanden sein. Dadurch werden, sobald durch Ruckgang des Polythionats und immer starkeres Hervortreten von Vorgang (15) die HSO; sich etwas anzureichern beginnen, die Gleichgewichte (5) und (6) (S. 263) stark zugunsten des freien Schwefeldioxyds verschoben, und trotz verhaltnismiiBig kleiner Konzentration an Sulfitschwefel der SO,-Druck der Losung erheblich gesteigert. So kommt es, dalj sehr bald nach Beginn der Schwefel- sbscheidung der SO,-Druck nach Durchlaufen eines Mindestwertes bald einen neuen, ziemlich lioch liegenden Hochst’wert erreicht, wie es Fig. 8 nach den Versuchen 45 und 46 zeigt.
4. 1st dieser Zustand erreicht, so liegt eine Losung von freiem Schwefeldioxyd neben Sulfat, dem Rest des Polythionats und einer ge- wissen Menge der freien Sauren vor. In dieser ist es nun das Schwefel- dioxyd, welches unter dem katalytischen Einflusse des Polythionats und unter steter Nachbildung aus diesem, also verhaltnismaBig langsam, sich weiter zu Schwefelsaure uncl Schwefel zersetzt, bis alles Schwefel- dioxyd und alles Polythionat schliefilich in diese Stoffe ubergegangen sind. Die Vorgange sind dabei die oben (S. 269ff.) fur den Zerfall der schwefligen Saure erorterten, und fiihren, wie es auch Versuch 45 und 46 lehren, nur triige zum vollstandigen Umsatz in die Endprodukte.
Das Wesen und Ineinandergreifen dieser Vorgknge lkBt sich durch ohemische Gleichungen am einfachsten ausdriicken, wenn man dem Umstande, dal3 die beim Abbau der hoheren Polythionate entstehenden S,O,” immer weniger der Polymerisation unterliegen, dadurch Rechnung tragt, da8 man das Ergebnis des ersten Haupt- vorganges so darstellt, als hiitte er zu Trithionat gefiihrt, und wiire alles debei entstehende Thiosulfat zunachst erhalten geblieben.

306 F. Foevstev, 8’. Lanye, 0. Dropbach und W. Seidel.
Dann ware die nach Gleichung (22) und (16) bis (18) im ersten Hauptvorgange vollzogene Urnwandlung des Bisulfits die folgende :
12Na‘ + 12HS0,’ +- SSO,” + S,O,” + 2S,O,” + 4 H + 12%’ +
und der zweite Hauptvorgang bestiindr in den S‘orgiiigen: 4 q 0 , (24)
(19) (1 5) (1 1
S,O,” + H,O -+ SO,” + S,O,” + 2H’ 3S,O,” + 6 H 3 3HS0,’ + 3H’ + 35 SHSO,’ + 3H‘+ 280,’’ + S + 4 H + H,O
Die Summe von allen ergibt:
d. h.
Msn sieht, da13 die Wiederverniehrung der Aziditat auf l/,des Anfangs- wertes nur im zweiten Hauptvorgange und irn wesentlichen durch den Zerfall der freien schwefligen Saure eintreten kann und daher zu einer von Methylorange anzuzeigenden H’-Konzentration fuhren mu13, wie es Versuch 47 und Fig. 9 lehren.
Erheblich anders gestaltet sich der zweite Hauptvorgang, wenn d u r c h E n t w e i c h e n von SO, i m e r s t e n H a u p t v o r g a n g e die Aziditat erheblich niedriger gehalten, ist als im vorbesprochenen Falle. Unter solchen Umstanden enthielt die Losung nach Ubcr- sicht 5 bei Beginn der Schwefelabscheidung
12Na + 12HS0,’ +8SOLf + 4 8 -+ 4H‘ + 12Na’ + 4H,O, 3Na’ + 3HS0,’ -+ .ZSO,lf + 3Ka’ + H’ $- S + H,O. (4)
in 1 ccin in 1 1
0,005 g Sulfitschwefel 0,080 g Sulfatschwefel 0,085 g Thiosulfatschwefel 0,002 g Polythionatschwefel
0,15 Mol Sulfit 2,50 1101 Sulfat 1,32 Mol Thiosulfat 0,015 Mol Tetrathionat .
Bei dieser Versuchsreihe wurde keine Bestimmung des Sauregehalts vorgenommen; in einer anderen aber wurde die AziditLt kurz nach Beginn der Sch wefelabscheidung, bei Benutzung von Phenolphthalein als Indikator, 0,48-normal gefunden. Fur den zweiten Hauptvorgang liegt also hier eine schffach saure, ziemlich konzentrierte Thiosulfat- losung vor, die neben uberwiegenden Mengen Sulfat ein wenig Polythionat , das oben als Tetrathionat angenommen mrde , und kleine Reste von Sulfitschmefel enthalt, der nnch der Aziditat kaum anders denn als freie schweflige Saure zogegen sein durfte.
Uber das Verhalten einer solcheii Thiosnlfatlbsung in der Koch- hitze kann nnch den oben dnrgelegtpn Erfahruiigen nicht:; bicheres

Zersetrulzg der schzuefligen Saure und ihwr Salxe in wap-igep Losung. 307
von x-ornherein ausgt’yagt werden. Pie Versuche der Ubwsicht 5 ergaben, da13 in einer solchen Losung beim Kochen das Thiosulfat sich vcrhaltnisma5ig schnell in Sulfat und Schwefel zersetzt und zwar so, da5 auf 1 Atom neu entstehenden Sulfatschwefels 2 Atome freien Schwefels sich abscheiden. Dabei trat aber auch Schwefel- wasserstoff auf und schied sich auffallend vie1 Sohwefel im unteren Teile des RuckfluBkuhlers ab. Letzteres mu5 als Zeichen dafur angesehen werden, daS auch Schwefeldioxyd bei diesen Umsetzungen frei wird, von dem in der Kochhitze ein Teil mit dem Schwefel- wasserstoff sich verfluchtigt, und im Kuhler in Beriihrung mit dem dort niedergeschlagenen Wasser sich in bekannter Weise zu freiem Schvefel umsetzt.
Die Bildung von Sulfat und Schwefelwasserstoff kann nur durch einen in der kochenden, schwach sauren Thiosulfatlosung sich abspielenden Verseifungsvorgang eintreten:
8,O;’ + H,O 3 SO,” + H,S. (25) In diesem Sinne verlauft, wie man weiB, die Zersetzung des Thio- sulfats in Gegenwart von Srhwermetallkationen, wie Ag’ oder CU”~) ; aber auch ohne solche kann das Thiosulfat sich in dieser Richtung zersetzen. (!OLSON*) beobachtete, da5, wenn eine 0,50/, ige Natrium- thiosulfatlosung in kochende verdunnte Salzsiiure langsam eintropft, sie sich quanlitativ in Sulfat und Schwefelwasserstoff zersetzt. Er bermerkt dabei, daS dazu die starke Verdunnung des Thiosulfats wesentlich ist, da bei hoherer Konzentration der Umsatz stets von einer Schwefelabscheidung begleitet ist. Das trifft nicht ganz zu, da auch eine konzentrierte kochende Thiosulfatlosung beim Zutropfen von 0,Ol-normaler Salzsaure langsam Schwefelwasserstoff entwickelt, und dabei in ihr Sulfat entsteht, wahrend sie vollig klar bleibt.
Das Thiosulfat kann sich also in saurer Losung nach drei Richtungen umwandeln : ____
1) dhnlich verhitlt sich Amen. 13ei Gegenwart von etwas arseniger S u r e geht nach einem Versuch von SALZER, auf den RASCHIG aufmerksam machte, eine mit Salzsiiure angesauerte Thiosulfatlosung in bedeutendem MeBe in Penta- thionsawe iiber. F. FOERSTER und A. HORNIG (1. c., S. 93), bemerkten nun, daB auch in diesem Falle die Thiosulfatlosung nach einiger Zeit sicb trubte, und sprachen den Niederscblag als Schwefel an. Herr R. VOGEL fand aber inzwischen im hiesigen Laboratorium, daB es Arsensulfid ist, was sich hierbei abscheidet. Dies sei hier zur Richtigstellung unseres Irrtums bemerkt, auf den auch Herr ltASCHIG uns aufmerksam zu machen die Giite hatte.
2, Bull. Xoc. Clhim. 34 (1880), 66.

308 F. Foemter, li: Lange, 0. DroljDach und W. Seihl.
A. in Schwefeldioxyd und Schwefel, B. in Pentathionsiiure, C. in Sulfat und Schwefelwasserstoff.
Die Bedingungen, unter clenen jeder dieser einzelnen VorgBnge sich glatt abspielt, sind noch nicht systematisch untersucht . I ) Meist, iiberlagern sie sich, indem einer dieser Vorgange sls Hauptvorgang von dem einen oder von beiden der anderen Vorgiinge begleitet wird.
Auch bei den im geschlossenen GefiiB durchgefuhrten Zer- setzungen des Bisulfits lie6 sich wahrend des zweiten Haupl- vorganges gelegentlich ein schwacher Schwefelwasserstoffgeruch bemerken. Also auch hier scheint Vorgang C an der Umsetzung des &us dem Polythionat vorubergehend entstehenden Thiosulfats teil- zunehmen ; wahrscheinlich ist dies aber nur in ganz untergeordnetem MaSe der Fall. Deshdb wurde dieser Umstand oben anch unerwahnt gelassen.
Ebenso wirken bei der Zersetzung der im offenen GefiiBe kochen- den schwach angeshuerten Thiosulfatlosung offenbar Vorgang C und A zusammen. Dies kann durch die Gleichungen:
2S,03” + 2H20 -* 2S0,“ + 2H,S, S,O,” + H’ =+ HSO; + S, HSO,’ + H’ + SO, + H,O, 2H,S + SO, -P 3 S + 2H,O,
(25) (15a)
(6) (26)
8S,O,” + 2 H -+ 2S0,” + 4s + H,O (27)
zum Ausdruck gebracht werclen, deren Endergebnis mit dem Befunde des Versuchs ubereinstilnmt. Das Gleichgewicht (15a) wiirde hierbei dadurch dauernd rechtsseitig verschoben, daB der Schwefelwasser- stoff nach (26) das Schwefeldioxyd verbraucht.
Diese Wirkung des Schwefelwasserstoffs wird dadurch bestiitigt, da13 schon bei dessen Einleiten in heiBe, nicht zu verdiinnte Thio- sulfatlosung sich nach einiger Zeit Schwefel abscheidet : selbst die sehr kleine H’-Konzentration des Schwefelwasserstoffs kann also bei hinreichender S,O,”-Konzentration Gleichgewicht (1 5a) so weit verschieben, daB es zusammen mit Vorgang (26) zur Schwefel- abscheidung fuhrt.
Wie die oben angegebene Analyse zeigt, enthalt die Losung, in der der zweite Hauptvorgang im offenen GefaB eintritt, auch
1) Die Untersuchung ist in Angriff genommen.

Zersetxung der sehwefligen Saure wad ihrer Salm in w$riger Liisung. 309
kleine Mengen Polythionatschwefel. Diese scheinen wenigstens fur den zeitlichen Verlauf der VorgSinge hcht ganz bedeutungslos zu sein. Als eine Losung, welche der Ausgangslosung fur den zweiten Hauptvorgang entsprechend durch Losen von 33 g (NH4),S04, 20 g (NH4),S203 und 0,5 g &S40, in 100 ccm hergestellt und durch NH4HS03 oder H,SO, O,%normal sauer gemacht und zum Kochen erhitzt wurde, begann alsbald die Schwefelabscheidung. Als aber eine von Polythionat freie, im ubrigen gleich zusammengesetzte Losung ebenso behandelt mrde , tmt die Schwefelabsoheidung erst nach 3/4 Stunden ein. Das Polythionat wirkt hierbei also als Kata- lysator, vermutlich dadurch, dafi es das durch die H sich einstellende , nur schwach rechtsseitig liegende Gleichgewicht (15a) durch den Vorgang :
S40," + RSO,' .i= S 3 0 i ' + S,O," + H' zugunsten der Schwefelabscheidung verschiebt . Wie man sieht, ist auch die Zersetzung des Thiosulfats im zweiten Hauptvorgange ziemlich verwickelt. Wie schon die letzterwahnten Beobachtungen zeigen , liegt die Moglichkeit vor, daf3 auch Vorgang B neben A und C hier eine gewisse Rolle spielt, wie es eigentlich h u m anders zu erwarten ist, da er ja auch einer kleineren H-Konzentration bedarf als A. Eine grundlichere Untersuchung dieser Erscheinungen is t noch erwiinscht.
5. Die Poglichkeit technisoher Anwendung der vorher beschriebenen Erscheinnngen.
DaB man die freiwillige Zersetzung von Ammoniumbisulfit - losungen, oder besser von Gemisohen von Ammoniumbisulfit- und Ammoniumsulfitlosungen, wie man sie beim Auffangen von Schwefel- dioxyd in Ammoniaklosungen geeigneter Konzentration erhalt , zur Gewinnung von Ammoniumsulfat und Schwefel nutzlich verwerten kann, haben die von BOSCH mit'geteilten, in der Einleitung schon erwahnten Arbeiten der Badischen Anilin- und Sodafabrik gelehrt .
Hier sol1 nur auf wenige Punkte hingewiesen werden, in denen die Zersetzung, zumal der Bisulfite in der Technik unwillkommene Folgen hat. DaB auch reine Bisulfitlosungen bei gewohnlicher Temperatur nicht beliebig lange haltbar sind, sandern nach langerer Zeit sich immer schneller zersetzen werden, leluen die oben mit- geteilten Erfahrungen. Besonders leicht mu6 dies eintreten, wenn die Losungen auch nur sehr kleine Mengen der ihre Zersetzung katalytisch beschleunigenden Stoffe enthalten. Dazu wird beim
1;. anorg. u. allg. Chem. Rd. 1'28. 21

310 I? Foerstar, I% Lunge, 0. Dropbach w a d W. SeideE.
technischen Arbeiten leicht Gelegenheit geboten. Das zur Herstellung der Bisulfite benotigte Schwefeldioxyd wird entweder durch Ver- brennen von Schwefel oder durch Abrosten von Pyrit gewonnen. Im ersteren Falle konnen sich, bei ungenugender Wirksamkeit des Schwefelbrenners, Dampfe von unverbmnntem Schwefel dem Schwefeldioxyd beiniischen ; sie werden dann beim Eintritt dieser Gase in die Losungen von Alkalihydrat oder Alkalicarbonat die entstehende Bisulfitlosung mit Thiosulfat oder Polythionat ver- unreinigen. Im eweiten Falle kam, wenn im Pyrit Selen vorkommt, die Bisulfitlosung selenhaltig werden. KLASON~) fand in einer Probe Pyrit von Falun 0,035c/, Se und bemerkt, da 0,4 mg Se in 1 Liter auf Calciumbisulfitlosung schon sehr storend wirken, daB dazu nur 2,40/, des Selens des Pyrits in die Bisnlfitlosnng zu gelangen brauchen. Da das Selen in den Rostgasen des Pyrits als feinster Staub von SeO, vorhanden seiii diirfte, ist also bei irgend merklich selenhaltigeii Kiesen die Gefahr der storenden Einwirkung eine sehr groBe.
Der Art der Storungen, die hierdurch bei der Herstellung der Sulfitzellulose sich ergeben konnen, wurde schon in der Einleitung gedacht,. Weniger bekannt scheint es zu sein, daB diese Wirkungeii sogar noch in den kristallisierten, sauren Alkalisulfiteii, also besonders dem Kaliummetasulfit Ii2Sz05, sich bemerkbar machen konnen. In einer Fabrik, die d i e m Salz herstellte, war es vorgekommen, daB lagernde Best,ande eine mehr oder weniger gelbliche Farbe an- genommen hatten, und nnn erhebliche Mengen Sulfat und Schwefel enthielten ; gelegentlich war diese Gelbfarbung sogar unter starker Selbsterhitlzung des Salzes eingetreten. Es zeigte sich, daB die gelblich bis gelb gewordenen Salze nachweisba.re Mengen Selen ent hielt en.
6. Die katalytische Wirkung des Belens a d den ereten Eaup tvorgang .
(Nach Versuchen voii W. SEIDEL.)
Die Beobachtungen, welche oben uber die katalytische Wirkung des Selens auf die Umwandlung des Bisulfits in Sulfat und Thio- sulfat bzw. Polythionate niitgeteilt wurden, haben gelehrt, dalS man es bier mit einem t ypischen Fall eines iiber Zwischenreaktionen wrlaufenden katalytisch beschleunigten Vorganges zu tun hat ; denn sie haben geeeigt, da,B clas als Xatalysator einer Bisulfitlosung

Zerstxkiny der sclmxfligen Suure und ihrer S a k e in wa,briqqer Liisu~~g. 3 1 1
zugesetzte Seleii zuniichst rasch als solches verschwindet und ern Ende des ersten IJauptvorgangrs, nachdem es den Ubergang des Bisulfits in Sulfat uiid Thiosulfat bzw. Polythionat sehr stark hescbleunigt 'hat, quantitativ in elementarer Form wieder vorliegt . Seine Wirkuiig kann also Lhnlich der des Schwefels nur darin bestehen, ctnB das Selen in der Bisulfitlosiing Verbindungen billet, unter deren EinjluB das Bisulfit weit schiieller nls durch die vom Schwefel erzeugten Verbindungen in Snlfat und Tliiosulfat sich umwnndelt . Es koinmt also fur die Dentune, seiner Wirksamkeit darauf an, zu untersucheii, welche Verbindungen des Selens und Schwefels nnd welche Vorgange an ihnen niit groBer Rea,lrtionsgeschwindig~~rit, Sulfnt und Thiosu1fa.t bzw. Polythionaf liefern kiinnen.
a ) Sc lenosa l fa t u n d Se lenodi th iona t .
Dnr3 Verbindungen bestehen, in denen das fielen je ein Atom Schwefel im Thiosulfat wir im Tritliionat ersetzt, ist schon 1-01' Janger Zeit \'on B. RATHKE~) iestgestellt worden. Er stellte dic IXiunisalze der Selenoschw?-efelsaure X,SeSO, nnd cier Seleno- tlithioiisiiure K,SeS,O, in kristdlisierber Form her, ersteres allcr- dings noch nicht in gang reinem Zustandr. K- und Na-Salz der cier Trithioiisaurr entsprechenden Selenodithionaaure wurdeii neuerdings ,iuch voii G. T. MORGAN vnd H. D. K. DREW~) aus der Acetyl- acetonverbindung des Selens durch Eeliandeln mit Bisnlfiten dar- gestellt :
Se : C6H6O2 + BKHSO, -+ Se(SO,K), + CH3COCH,COCH3;
also durch eiiieii Vorgang, welcher das Anion dieser Salze als 8e/ SO,' \so;
aufzu fassen erlaubt . Der Tiorgang, durch den RA'CIIKE die Alkaliselenosulfate in
lidsung erhielt, war der der Aufl6sung von Selen in warmer Alkali- sulfitlosung. Benutzte er dabei das sehr leicht losliche Kalinx- sulfit in lronzeiitrierter Losung, so konnte nach reichlicher Anf- uahmp voii Seleii beim Eindunsten der Losung, mchdem suerst in geriiigem MaBe I<risballe voii I<,SeS,O, sich abgeschieden hatten, ilas I<% li u ins e le no s u l f a t I<,SeSO, in sechseitigen Tafeln kristal- lisierf erlialteii werden, die : i b a noch von etwas Kaliumthio.sulfat veruiireinigt warm.
l) Jourct. pralct. Chem. 95 (1865), I. 2) Journ. Chem.. SOC. 117 (1920), 1456.
21*

312 l? Fomter, El Latage, 0. Iho@bach und W. Seidal.
Wir fanden es ZweckmaBiger, folgenden Weg einzuschlagen , der an den der Darstellung von Thiosulfat durch Einwirkung von schwefliger Saure auf Schwefelnatrium erinnert: In 10 n-KOH wird in der Warme Selen bis zur SBttigung gelost ; das hikrbei benutzte geschmolzene rind fein gepulverte Selen lost sich groBenteils sehr schnell auf, doch blieben stets kleine Mengen einer nur langsam loslichen Form des Selens zuruck, deren Auftreten uber den erreichten Sattigungsgrad nicht tauschen darf. In die entstandene dunkelrote Polyselenidlosung wird nun eine mit freier schwefliger Saure gesattigte etwa 5-molare Kaliumbisulfitlosung in der Warme vorsichtig ein- getragen, bis die rote Farbe der Losung verschwunden ist. Sie kann nun iiber freier Flamme bis zur beginnenden Kristallisation ein- gedampft werden, beim Erkalten scheiden sich grol3e Mengen des Selenosulfats ab. Sie sind unter diesen Umstanden meist durch etwas freies Selen gefarbt. Versetzt man aber eine bei Zimmer- temperatur etwa gesattigte Losung bei dieser Temperatur schwach mit Salzsaure, so daB eine geringe Selenabscheidung eintritt, so scheidet die filtrierte Losung beim Eindunsten uber Schwefelsaure ganz farblose Kristalle des Salzes ab.
Um diese umzukristallisieren, verfahrt man so, daB man zu- iiachst einen kleinen Teil der Kriztalle unter allmahlicher Zugabe von warmer 10 n-KOH bis zur Sattigung in dieser lost, und d a m iibwecliselnd immer etwas Wasser und weitere Kristalle bei hochstens 400 zufugt, bis alles gelost ist, und nun die Losung im Exsikkator uber Schwefelsaure eindunstet, eine freilich recht zeitraubende Operation. Das Saki, welches in den von RATHKE beschriebenen, groI3eri sechsseitigen Tafeln kristallisiert, gab, bei 1000 getrocknet, liei der nach den Angaben des genannten Forschers ausgefuhrten Analyse folgende Werte :
Gefunden Berechnet fur K,SeSO, Se 33,Q o/o 33,36 O/,,
S 13,O 13,48
1)as Selenosulfat lost sich nur in starker Alkalilauge oder in Sulfit- bzw. Bisulfitlosung ohne bleibende Selenabscheidung auf. Durch Wasser wird es ebenso wie seine alkalische Losung teilweise zersetzt unter Bildung von freiem Selen ; durch verdunnte Salzsaure wird dieser Zerfall vollstandig. Zum Unterschiede von dem weit bestandigeren Thiosulfnt geniigt also hier in SO,”-freier Losung

Zerselzung der schwefliyen Saure u n d ihrw Salne in wapriger Losung. 3 13
schon die H -Konzentration des Wassers, ja einer schwachen Alkali- lOsung, um das Gleichgewicht :
SeSO,” + H’ e HSO,’ + Se,
HSO,’ + OH’ + SO,” + H,O, SO,’‘ + Se + SeSO,”
(28)
(7)
(29 1
dern die weiteren Gleichgewichte
sich beigesellen, soweit rechtsseitig zu verschieben, daB eine reich- liche Menge freien Selens sich abscheidet. Wird dabei das Verhaltnis SeSO,” zu HSO,’ so bemessen, dal3 bei gewohnlicher Temperatur eben kleine Mengen Selen sich abscheiden, so verschwinden sie beim Ermarmen wieder, nm beim Erkalten aufs neue aufzutreten. Das Gleichgewicht (28) liegt also bei hoherer Temperatur etwas starker linlisseitig als bei niederer. Bemerkenswert und fur das Folgende wichtig, ist es, daB mahrend in starker Bisulfitlosung die Selen- fibscheidung durch deren H‘-Konzentration unter dem Einflusse der HSO,’ vollstiindig verhindert wird, und der Zerfall des Thio- sulfclts auch gegenuber freier schwefliger Saure ausbleibt, das Seleno- snlfat durch diese Saure sowie auch durch die selenige Sanre voll- ctandig zersetzt wird.
Durch Erwarmen der Losung des Selenosnlfats in Bisulfitlosung erhielt RATHKE das dem Trithionat analoge Kal iumselenodi - t h i ona t K,SeS,O,. Die Konzentrationsverhaltnisse, unter denen dieses Salz unter solchen Bedingungen reichlich entsteht, gibt RATHKE n i c k a n ; wir konnten sie noch nicht ermitteln. Dagegen entsteht es mit grooer Leichtigkeit in der auch schon von diesem Forscher angegebenen Weise, dal3 man die Losung des Selenosulfats in Bisulfit mit Kaliumselenit versetzt. Man verfahrt dabei folgender- mal3en: In 4 ccm 10 n-KOH wird von 4,76 g K,SeSO, (0,02 Mol) c twNn die Halfte langsam eingetragen ; nach eingetretener Losung wird etwas Wasser zutropfen gelassen und der Rest des Salzes gelost. Dam gibt man 0,l Mol KHSO, in 5fach molarer Losung und alsdann eine Losung von 2,22 g SeO, in soviel 10 n-KOH, daS sie gegen Lackmus neutral ist. Unter starker Selbsterwarmung tritt sofort,igeReaktion ein, und groBe, beimErkalten sich nochvermehrende Mengen schoner, glanzender N adelchen des neuen Salzes scheideii sich ab. Nach dem Absaugen und Waschen mit 5O0/,igem Alkohol erhalt man 10 g fast reinen Selenodithionats, d. h. SOO/,, des an- gewandten Selens in Gestalt dieses Salzes, wahrend aus der Mutter-

314 B! Fgeyster, F. Lange, 0. Dropbach und W. Seidel.
hnge noch kleine Rlengen weniger reinen Salzes unter der Einwirkung des Alkohols sicli ahscheiden. Durcli einmaliges Umkristallisiereiz x i s warmem Wasser erhalt man das Salz analysenrein; beine A])- scheidung wird dabei zweckmhl3ig dnrch Alkoholxusatz hefordert . Xnch dtlm Trocknen bci 1000 ergab die Analyse:
Gefiinden Rerechnet fur I<,SrP,O, Se 24,7 O/,, 24995 O/,)
Das durch verhaltnism8Big groBe Bestandigkeit ausgezeichnete Sulz gibt mit Quecksilberchlorid eine orangegelbe, beim Erhitzen weir, werilende Fdlnng. Im tibrigen wird sein chemisches Verhalten w i t er unten besprochen werden.
Die hier benutzte Art der Darstellung des Selenodithionats c~innert, worauf schon RATHKE hinwies, sehr an die des Kalium- trithionats durch Einleiten yon schwefliger SBure in eine lionzen- trierte Losung von Kaliumthiosulfat . Wir werden aber, wenn weiter uiiten der Mechanismus des Vorganges erortert werden wird, dartun, daB die Ahnlichlreit h ide r Vorgange keine ganz volls tiindip ist .
h) Se leno th iona te vom T e t r a - uncl P e n t a t y p u s ; d i e o s y d i e r e n d e Wirl iung de r se len igcn Saure auf s chwef l ig r
S a u r e , T1i i o s c h we f e 1 s a u r e u n d S e len o s c hme f e 1 s 5iu r e.
Wenn somit dem Thiosulfat und dem Trithionat analoge Selen- T-erbindungen leicht erhdtlich sind, in denen ein Teil des Schwefel:; jwer durch Selen ersetzt ist, so liegt der Gedanke nahe, daB auch den T&-a- und den Pentathionaten entsprechende Selenverbindungen hestehen. Das laBt Rich in der Tat sehr wahrscheinlich machen. Es ist eine bekannte Tatsache, daB beim Einleiten von Schwefel- clioxyd in eine Losung von scleniger Saure die Selensbscheidung uicht sofort beginnt, sondern erst nach einiger Zeit und d a m als- bald in reichlichem Ma Be eintritt . Entsprechendes geschieht , wenn zit einem UberschuB von schwefliger Saure selenige Saure hinzugefugt wird. In beiden Fallen tritt aber die Selenabscheidung auch bei starkclm Vorwalten eines der Reaktionsteilnehmer sofort ein, wenn die Losung Salzsaure oder eine andere stlirkere Saure enthalt. Diese Erscheinungen sind von HANS SCHULZE~) naher untersucht worden. Er fand, daB die Fallung des Selens beim Zusammenwirken von SO, uiid SeO, in waBriger Losung um so schneller uncl um so roll-
l ) Journ. prakt. G'hem. N. F. 32 (1885), 390.

Zersetxung der schweflipn Saure zcnd ihrsr Sake in wapriger Liisung. 315
standiger in dem nach den angewandten Mengen im Sinne der Gleichung 2H,S03 + H,Se03 -+ 2H,SO, + Se + H,O zu erwarten- (!en Umfange vor sich geht, je mehr das Mengenverhaltnis, in dem die Reaktionsteilnehmer angewandt werden, dieser Gleichung genugt. hber auch dann hiingt es von der Geschwindigkeit ab, mit dem der pine von beiden Stoffen Zuni anderen gefugt wird, ob die Um- setaung eine vollstandige ist ; je langsamer der Zusatz geschieht, urn so unvollstandiger erweist sich auch beim stoohiometrischen Verhaltnis 2 SO, : 1 SeO, die Selenabscheidung. Das in der Losung verbleibende Selen ist darin in verhaltnismaBig bestandiger Bindung enthalten; aus der beim UberschuB von SO, erhaltenen Losung ist r s auch beim Kochen nicht abauscheiden, wahrend die mit SeO, im UberschuB erhaltene Losung in der Siedehitze Selen abscheidet , wobei es freilich dahingestellt bleiben muB, ob dieser Unterschied Lzllein auf der verschiedenen Bestiindigkeit der in beiden Fallen zu erwartenden verschiedenen Verbindungen oder auch darauf beruht , daB beim Kochen der UberschuB von SO, entfernt wird, der von SeO, aber nicht.
An einer durch Wechselwirlsung von SeO, mit einem UberschuB von SO, hergestellten Losung beobachtete H. SCHULZE das Folgende : Wird nach vollzogener Umsetzung der SO,-UberschuB be1 gewohn- licher Temperatur abgeblasen und die naturlich erhebliche Mengen von Schwefelsaure enthaltende Losung eingedunstet, so findet dabei dauernd zunehmende Abscheidung von Selen und Entwicklung von Schwefeldioxyd statt. Wird die frisch abgeblasene Losung in uber- nchussige Alkalilauge gegossen, so scheidet sich sofart reichlkh freies Selen ab ; auch bei ihrer Neutralisation mit Bariumcarbonat fallt neben Bariumsulfat Selen aus, wahrend in der Losung ein Barium- salz verbleibt, das nach der Analyse wahrscheinlich der Seleno- dithionsaure angehort.
Diese Beobachtungen konnen wir nur bestiitigen und in folgender Hinsicht erganZen: Wird die Losung, in der selenige Saure mit iiberxchussiger schwefliger Sliure zusammengebracht witr, einige Zeit stehen gelassen und dann der UberschuB des SO, durch einen Stickstoffstrom abgeblasen, so erhiilt man eine hellgelbe Losung. Diese scheidet beim Stehen in langsam fortschreitender Reaktion Selen ab ; auf SaJzsaurezusatz tritt keine sofortige Selenabscheidung ein, doch wird diese dadurch beschleunigt: Durch uberschiissige Alkali- huge wird die Losung nicht immer gefallt; es hangt von der Gro5e des Uberschusses an schwefliger Saure und von der Zeit ab, uber die

316 A Foersts; F. Laage, 0. Dropbaoh wad W. Seidel.
cliese wirkte, ob die Alkalilauge Selen abscheidet. Bleibt die Fiillung <:us, so tritt sie aber sofort in reichlichem MaBe ein, wenn man die alkalisohe Losung mit Salzsaure wieder ansauert.
Die Tatsache, daB die Losung einer Selenoschwefelverbindung riuf Alkalizusatz Selen abscheidet , erinnert sehr eindringlich an die die Pentathionate kennzeichnende Reaktion, daB Alkali aus ihnen Schwefel in Freiheit setzt; man wird aus dieser Beobachtung also den SchluB ziehen durfen, daB auch Selenoschwefelverbindungen xon diesem Typus, also Anionen SsSe(5-,)Ogl’ zu bestehen vermogen. Andererseits entspricht die Erscheinung, daB eine Selenoschwefel- T erbindung auf Alkalizusatz kein Selen abscheidet, wohl aber beim Wiederansauern der alkalischen Losung, durchaus dem Verhalten der Tetrathionate ; denn durch Alkalilauge wird aus diesen Thio- sulfat gebildet , das beim Ansbuern unter Schwefelabscheidung zer- fallt. Da, wie wir wissen, Selenosulfat auf Saurezusatz sofort Selen abscheidet, darf man also schlieBen, daB auch Selenoschwefel- verbindungen vom Typus der Tetrathionate, also Anionen S,Se(,- 9l 06” cxistenzfahig sind. Wie die Pentathionate durch schweflige Siiure in Tetrathionat, diese aber durch Schwefel wieder in Pentathionat verwandelt werden, so treten nach diesen Beobachtungen bei den Selenverbiiidungen solehe vom Pentat ypus urn so mehr hervor, je geringer der UberschuB a n SO, und je naher damit die Moglichkeit der Abscheidung des Selens geruckt ist. Mail wird also nicht zweifeln d*fen, daB die ganze Reihe der Polythiona,te auch bei den Seleno- thionaten besteht, auch wenn die Isolierung der einzelnen, durch stufenweiseb Ersatz des Schwefels durch Selen hier denkbaren, sehr mannigfachen Einzelglieder dieser Typen bisher noch nicht gelungen ist .
Fur die Entstehung dieser Verbindungen bei der Wechsel- n-irkuag von schwefliger und seleniger Saure konnte man zu der Annahme geneigt sein, da13 das bei der oxydierenden Betatigung der selenigen Saure aus dieser entstehende Selen in die Gleichgewichte :
Se + HSO,’ + SeSO,” + H (28) bzw. Se .f HSeO,’ e Se20;’ + H (30)
triite, und in drr sauren Losung, analog wie ex bei S203“ der Fall ist, mit betrachtlicher Geschwindigkeit die Polymerisierung der Anionen SeSO,” bezw. SeZO3” zu solchen vom Pentatypus vor sich ginge. Wie a ber oben gezeigt, bleiben bei der E-Konzentration der schwefligen Same keine merklichen Konzentrationen von SeS0,” im Gleich-

Zersetxmg der sclzwefligen Saure ufid ihrer Sulze in wa$'riger Lomng. 31 7
gewicht (28) bestehen; noch weniger kann das der Fall sein, wenn durch die oxydierende Wirkung der selenigen Saure auf schweflige Saure freie Schwefelsaure in der Losung auftritt. Wir haben uns daher auch vergeblioh bemiiht , die Einwirkung einer Selenosulfat- losung auf einen groBen UberschuB von schwefliger Saure so zu leiten, daD dabei eine Losung von der gleichen Art wie bei der Ein- wirkung der selenigen Saure auf schweflige Saure entstand. Die oben ausgesprochene Vermutung muD man also wohl fallen lassen.
Es ist aber noch ein anderer Weg denkbar, auf dem die gedachten Verbindungen ents tehen konnm. Bei der Einwirkung der selenigen Saure auf die Schwefelsaure ist es das Wahrscheinlichste, da13 je 1 Mol von beiden zunachst in Wechselwirkung treten; der Vorgang sri dann folgendermaBen formiiliert :
SeO, + HSO,' --t SeO + SOLf + H'. (31) Das SeO konnte dann auf andere HSO,' nach zwei Richtungen wirken :
SeO + HSO,' -+ Se + SO," + H' SeO + 2HS0,' -+ Se(SO,)," + H,O.
(31)
(33) Es entstande also Selenodithionat, das nun, je groBer der Umfang yon Vorgang (32) ware, also ja mehr SeO, zur Verfiigung stande, die Yorgange :
SeS,O," + Se -+ Se,S,O,"
SezSz06,' + Se -+ Se3Sz0," (34)
(35) geben, also Verbindungen vom Tetra- und vom Pentatypus erzeugen miiBte, wie solche nach den Beobachtungen entstehen. Da hiernach die Selenothioverbindungen der oxydierenden Wirkung der selenigen Saure ihre Entstehung verdanken, so muBte aus iiberschiissiger schwefliger Saure auf 1 Mol SeO, weniger als 2 Mol &SO, ent- stehen, wahrend, wenn jene lediglich aus freiwerdendem Selen und ill Folge einer Polymerisierung der dabei entstehenden Verbindung sich bildeten, das Molverhaltnis 1 : 2 gefunden werden muBte. Der Versuch ergab, als 20 ccm 0,l-molarer Se0,-Losung, also 2 Millimol SeO, in einen groBen UberschuB einer S0,Losung gebracht, der SO,-UberschuB alsbald mit Stickstoff abgeblasen und die Losung kalt unter Zugabe von Filterschleim mit Chlorbarium gefallt wurde, 0,784 g BaSO, cu 3,36 Millimol SO,, d. h. die verbrauchte SeO, und die entstandene Schwefelsaure standen im Molverhaltnis 1 : 1,68. LaBt man andererseits 0,02 Mol SO, auf 0,Ol Mol mit KOH neu-

318 2;". Foerster, El Lange, 0. Dropbach und W. Seidel.
tralisiertes SeO, (znsamnien etwa 25 ccm Losung) in 50 ccm gesiittigter Natriumacetatlosung wirken, so ist sofort alles SO, verschwunden, lind die Losung cnthalt neben vie1 Sulfat Selenodithionat und ein Selenosulfat vom Tetratypus, wiihrend kein freies Selen abgeschieden ist. Hier ist also die Umsetzung etwa so verlaufen, wie es oben angrnommen ist .
Eine weitere wichtige Bestkitigung dieser Auffassung ergibt sicli aus dem Verha l t en de r se len igen S a u r e gegen Thioschwefe l - sBnre. Hieriiber liegen schon altere Beobachtnngen von J. F. NORRIS und H. FAY^) vor. Sie fanden, da13 man selenige Sanre glatt titri- mctrisch bestimmen lrann, wenn man ihre mit Eiswasser verdunntt. Losung rnit Salzsaure und alsdann mit einem UberschuB einer 0,l n - 'Chiosulfatlosung versetzt und nun den von dieser un- verbraucht gebliebenen Anteil mit Jodlosung zuruclrmi5t. Hierbei werden auf 1 Mol SeO, 4 Mol Na2S,03 verbraucht, und in der saureii Liisung findet, zunachst wenigstens, lieine Selenabscheidung statt ; sie tritt aber sofort ein, wenn die Losung allialisch gemacht mird. Demnach formulieren die genannten Forscher den Vorgang folgender- ma13en :
ScO, +4Na,S,03 + 4HCJ -+ Na,S,O,+Na,SeS,O, + 4 Na Cl-1- 2 H,O. (36'
Das Anion SeS,O," ist, vom Pentatypus und mu13 daher leicht dem Angriff der Natronlauge unterliegen. Diese Beoba,chtungen lionnten wir nur bestatigen; in den nach dieser Gleichung sich ergebenden Mengenverhaltnissen zusammengebracht (Versuch 48), rcagieren schon in verdunnterer Losung diese Korper momentan unter fuhlbarer Warmeentwicklung glatt aufeinander. Aber die erhaltjene Losung ist nicht bestandig ; kuhlt man sie auf Zimmer- temperatur ab, und uberlaBt eino solche von 0,Ol Mol SeO,, 0,04 Mol Na,S,03 und 0,04 Mol HC1 in 100 ccm sich selbst, so beginnt manch- ma1 nach wenigen Minuten, manchmal erst nach 11, Stunde Selen- abscheidung, die, such wenn sie spat eingesetat hat, nach 15 Stunden nahezu, nach mehreren Tagen ganz vollstandig ist, so da13 jetzt liein Selen in der Losung niehr nachweisbar ist, und diese jetzt neben Chlornatrium nur noch Tetrathionat enthalt. Es kann nach dvr Entstehung uncl dem Verhalten dieser Losung im Vergleich mit dem des Pentathionats lrein Zweifel bcstehen, da13 in ihr eine dem -
l ) Amer. Chem. Journ. 18 (1895), 705; 23 (1900), 119.

Zersetzung der schaoefligen Saure undihrer Sake in wti/kiyev Ltisung. 31 9
Pentathionat analoge Selenverbindung n i t dem Anion SeS,O," .;orhanden ist . Wie S,O," durch Alkali zur sofortigen Abscheidung 1-on 1 S veranlaDt wird, und eine neutrale oder saure Losung dieses Sdzes langsam nach S,O," + S,O," + S zerfallt , so gibt das in momentaner Reaktion entstandene SeS,O," anf Alkalizusatz sofort , heim Stehen in schwach saurer Losung langsam nach
SeS,O," -+ S,O;' + Se sein Selenatom ab.
folgenderma8en denken: Den Mechanismus dieser Umsetzungeii darf man sich wohl
SeO, + 2H' + 2S20c 3 SeO + S,O," + H,O, SeO + 2 H' + 2 S,O," -F Se(S,O,)," + H20, (37)
(38) (39) Se(S,O&" 3 S,O," + Se.
Ihre Summe entspricht Gleichung (36). Nach diesen Gleichungen iiehrnen H' a n den Umsetzungen teil; ihre Koneentration ist fur die Geschwindigkeit der Vorgange sehr bestimmend. Wahrencl diese unter der Mitwirkung der Salzsanre rnomentan zu Ende gehen, dauert es bis .zum vollstandigen Verbrauch des . Thio- d f a t s Stunden, wenn man statt der Salzsaure die aquivalente Menge Essigsaure bei der Umsetzung anwendet. Noch langsanirr verliiuft die Umsetzung bei Oegenwart von Natriumacetat . Sie lileibt aber auch zwischen den neutralen Losungen von Thiosulfat m d Selenit nicht aus, doch verlauft sie dann anscheinend ver- mickelter als in saurer Losung; die Flussigkeit wird schwach alkalisch, bcheidet sofort viel Selen ab und enthalt noch viel Thiosulfat un- verandert. Die selenige SBnre vermag also jedenfalls in saurer Losung mit gro13ter Geschwincligkeit auf Thiosulfat in ahnlicher Richtung oxydierend zu wirken wie Jod, nur werden von dem dabei freiwerdenden Selenatom voriibergehend zwei S,O," zu einem dem Pent at hiona t ent s prec henden .4nion ge bunden. Diese Tat sache erinnert daran, daB auch Pentathionat unter Umstanden durch &en sohnell verlaufenden Vorgang in hoherer Konzentration auf- treten als es im Gleichgewicht mit anderen anwesenden Anionen bestehen bleiben kann, wenn diese Gleichgewichte sich nur tragc einstellen. 1)
Aus diesen Vorgangen ergibt sich nun auch die Deutung fur die Umse tzung v o n Se len i t mi t Se lenosul fa t , die oben fur die Darstellung des Selenodithionats benutzt wurde. Die groBe
l) 2. a w g . u. allg. Ckm. 126 (1921), 125.

320 l? Foerster, El Lange, 0. Dropbaeh und W. Seidel.
Empfindlichkeit des Selenosulfats gegen H’ verbietet hierbei die Benutzung einer salzsauren Losung oder auch nur die Anwendung freier seleniger Saure und erlaubt nur eine Ansauerung durch Bi- sulfit; dessen Anionen aber sind gegenuber denen der hoheren Poly- thionate nicht bestandig und das gleiche gilt von denen der hoheren Selenothionate, wie schon daraus folgt, daB bei Gegenwart schon yon 1 Mol KHSO, auf 4 Mol Na,S,O, bei deren Umsetzung mit SeO, nach Gleichung (36) die durch Alkali abscheidbare und spater yon selbst ausfallende Selenmenge nur noch eine ganz genngfiigige ist . Durch die Folgereaktion mit HSO,’ ist das Endergebnis der Oxydation des Selenosulfats durch selenige Same etwas verschieden von dem tles Thiosulfats, jedoch in klar vorauszusehender Weise. Die Vor- gknge sind hier die folgendcn :
(40) (41 1 (42)
2Se,S,06 + 2HS0,’ =+ 2SeS,O,” + 2SeS0,’’ + 2 H . (49)
SeO, + 2H’ + ”ieSO,” -F SeO -+ Re2S206” + H,O,
Sc(SeSO,),” + HSO,’ + Se2S206)f + SeSO,” + H , SeO + 2H’ + 2SeS0,” -> Se(SeSO,),” + H,O,
Bei OberschuS von HSO,’ ist das Endergebnis:
SeO, + SeSO,” + H’ + 3HS0,’ -+ 2SeS,O,” + 2H,O. (44) Wahrend vom Thiosulfat 4 Anionen von 1 SeO, oxydiert werden. ist es also beim Selenosulfat nur 1 Bnion; dafur aber werden 3HS0,’ rnit herangezogen, und statt der PenCa- und Tetrastufe tritt hier die Tristufe im Endergebnis hervor. XMan erkennt auch, dal3 im Vergleich mit der Entstehung des Trithionats a m Thiosulfat und Schwefeldioxyd hier die oxydierende Betatigung der selenigen Saure gegen Se SO,” wider an die Stelle des Polymerisierungsvorganges der S,O,” tritt.
Da13 in der Tat die Umsetzung im Sinne von Gleichung (44) stattfindet, ergibt sich in1 Hinblick auf die am Umsatz teilnehmeiiden Selenverbindungen aus der oben angegebenen Tatsache, daB beim Zusammrnwirken von je 1 1101 Selenosulfat und Selenit SOO/O des gesamten angewandten Selens in Gestalt des auskristallisierten Selenodithionats gewonnen werden lionnen. Da13 auch der Verbrauch an Bisulfit der Gleichung (44) entspricht, lionnte erhartet werdeii (Versuch 49) als K,SeO,, K,SeSO, und KHSO, im Molverhaltnis 1 : 1 : 3,7 so zusammengebracht wurden, da13 keine Kristallisation eintrat und alsdann die Losung rnit Salzsaure angesauert und der QberschuB an SO, mit Stickstoff abgeblasen, in Alkali anfgefangen

Zersetcnt1n.q der schzcefiigen SGure und dwer Sake in tvufi'riger Losung. 32 1
und hier mit Jod titriert wurde. Es ergab sich auf 2Atome Selen r4n Verbrauch von 2,85 Mol KHSO,. Der geringe Unterschied gegenuber der Erwartung von 3,O Mol KHSO, ruhrt wohl vorwiegend daher, da13 K,SeS,O, in angesauerter Losung sich verhaltnismaBig leicht, wie wir noch sehen werden, unter SO,-Entwicklung zersetzt. Bemerkenswert bei dieser Reaktion ist, daB die Sulfatbildung, die man zwischen KHSO, und K,SeO, erwarten sollte, und die auch bei Abwesenheit von K2SeS0, reichlich und rasch eintritt , bei seiner Gegenwart nur geringfugig ist, die Oxydation des SeSO," durch SeO, d s o weit schneller vonstatten geht als die von SO," oder HSO,'. Immerhin muB bei der Darstellung des Selenodithionats dieser Umstand im Auge behalten werden, also die Reihenfolge, in der die Reagentien dabei zusammengc3bracht werden, sachgemafi gewahlt werden.
Oben wurde schon erwahnt, daB Selenodithionat auch ans 3eleniger Saure und schwefliger Siiure bei Gegenwart von Natrium- acetat entsteht. DerngemaiB ist es auch moglich, dieses Salz durch Einwirkung von Kaliumselenit auf Bisulfit allein zu erhalten. Dabei wird zunachst letzteres momentan zu Sulfat oxydiert, und das dabei freiwerdende Selen tritt mit HSO,' in das Gleichgewicht :
Se + HSO; + SeSO," -+ H' (28)
nnd nunmehr lenkt das SeSO," die weitere oxydierende Wirkung der selenigen Saure auf sich; die in diesem Falle das Selenodithionat begleitenden betrachtlichen Sulfat mengen erschweren aber seine Reindars t ellung.
c) Die Ze r se t zung des Se lenod i th iona t s . Wie im Eingange dieses Abschnittes bemerkt, kommt es fiir
die Deutung der katalytischen Wirkung des Selens auf die Zersetzung des Bisulfits darauf an, den Vorgang zu ermitteln, durch den die Selenverbindungen mit sehr viel groBerer Geschwindigkeit als die entsprechenden Schwefelverbindungen den Ubergang des Bisulfits in Sulfat und Thiosulfat herbeifiihren. Da, wie fruher gezeigt, bei letzterem Vorgange die katalytische Wirkung des Schwefels im Endergebnis darin zu erblicken ist, daB er Trithionat entstehen lBBt, und dieses verhaltnismaBig schnell zu Sulfat und Thiosulfat sufgespalten wird, so wurde vermutet, daB die analoge Zersetzung des Selenodithionats vielleicht mit so viel groBerer Geschwindigkeit vonstatten ging, daB sie fur die rasche Sulfatbildung bei Gegenwart

322 El Fowster, I?. Lunge, 0. Dro/3ba& und W. Seidel.
des Selens als der in erster Link geschwindigkeitsbestimmende T'organg aiizusehen wiire.
Den gleichen Gedanken hat in1 Grunde schon RATHKE~) aw- gesprochen; er sagt : ,,Kocht man eine Losung von zweifach-schweflig- hamem Kali mit Seleii, so wird das fortwahrend sich bildende denodithionsaure Kali immer sogleich wieder zersetzt, und wem man nach Iangerer Zeit zu kochen aufhort, so enthalt die LOsung sehr wenig Selen, hingegen vie1 schwefelsaures und unterschweflig- saures Kali. Es tritt also die eigentumliche Erscheinung ein, daB. infolge des temporaren Eingeliens des Selens in eine Verbindung. das schwefligsaure Kali gespalten und in sehr konzentrierter Losung durch Bildung und Zerpetzung von trithionsaurem Kali sogar Schwefel ausgeschieden wird."l) Auch KLASON~) hat die Vermutnng Ausgesprochen, daB, Venn nach seinem Befuiide Selen etwa 500 ma1 so stark als Schwefel katalytisch auf die Bisulfitzersetzung einwirkt . dies daher ruhre, da13 ,,TrithionsBure in einer 500 ma1 gro13eren Konzentration als Selenodithionsiiure bestandig ist". Die JTer- mutung, da13 das Selenodithionat der Trager dieser katalytisclieiz M7irkung des Selens sei, hat sich bei genauer Prufung aber nicht iiestatigt.
Fur den Zerfall des Seleiiodithionats in wahiger Losung, den- 4e ts mit der Zeit eintritt, stellte RATHKE fest, da13 er zu Sulfa[. Schwefeldioxyd und Selen fuhrt, also in den Endprodukten analog (?em des Trithionats ist. Vergleicht man aber die beiden Salzc liierin genauer, so fallt sofort ein wesentlicher Unterschied in die Augen: Uiitersucht man ihre Losungeii bei hoherer Temperatur. etwa bei 80°, also unter den fur den glatten Zerfall des Trithionats iiach K,S30,+ K,SO,+ SO,+ S giinstigsten Beding~ngen~), so tritt- in einer 0,l-molaren Trithionatlosung sehr bald Schwefelabscheidung ( in, wiihrend SO, entweicht ; aus der aquivalenten Selenodithionat - Iosung aber findet zwar auch das letztere statt, aber Selen scheidet sich zunachst nicht ab, soiidern die Losung farbt sich gelb und enthalt nun das Selenothionat des Tetratypus, d. h. sie scheidet iiach Alkalizusatz und folgendem Ansauern Selen ab. Erst iiach langerer Zeit tritt freies Seleii auf, wahrend dauernd Schwefeldioxyd entweicht und in der Losung Sulfat entsteht.
1) 1. c., s. 10-11. 2, 1. c., S. 415. 3, 2. unorg. u. allg. Chew 125 (1922), 121.

Zersetzuq der schwefligea Sawe m d ihrer Sake i m wapriger Liisuag. 323
Die Zersetzung der waBrigen Losung des Selenodithionats verlauft also bei 80° folgendermaBen :
SeS,O," + H,O -> SO," + SeSO," + 2H, (45) SeSO," + 2H* -+ SO, + Se + H20, (28)
SeS,O," + Se =+ Se2S206/', (34)
(46) ___
2 SeS,O," -f SO," + Se2S20i' + SO,. Verfolgt man (Versuch 50) die Entwicklung des SO, durch
Ubertreiben mittels Stickstoff in Alkali und jodometrische Unter- suchung der vorgelegten Losung, so findet man z. B. fur eine Losung von 0,Ol 1401 K,SeS,O, in 50 ccm bei SOo, da8 auf 1 Mol K,SeS,O, his zur beginnenden Selenabscheidung 0,36 Mol SO, abgegeben sind, wahrend alsdann unter dauernder Selenabscheidung noch weitere 0,60 Mol SO, aufgefangen wurden, ohne da8 die Zersetzung auch jetzt eine ganz vollstandige war. Wahrend im ersten Stadium 0,36 Mol SO, abgeblasen wurden, wsren 0,40 Mol SO," in der Losung entstanden. Aus diesen Beobachtungen folgt, daB Gleichgewicht (34) sich bei den Selenoverbindungen auch bei SOo in erheblichem Mal3e einstellt, und zwar in 0,Bmolarer Lijsung so, daB darin 1 SeS,O," auf 2Se,S,O," kommen; die Umsetzungen im Sinne der Gesamt- gleichung (46) finden also bei den Selenverbindungen glatter statt als die analogeii des Trithionats, die bei diesem Salze bei gewohnlicher Temperatur sich vollziehen.1) Wird schliel3lich durch die fort- schreitende Zersetzung des Selenodithionats seine Konzentration kleiner, als sie im Gleichgewicht mit dem vorher entstandenen Se2S20i' besteht, so wird sie aus diesem zuriickgebildet, wahrend das gleichzeitig entstehende und das weiter aus SeS,O," sich bildende Selen frei bleibt. Da somit die f i i r den Vorgang verfugbare Konzen- tration des SeS20," durch das Gleichgewicht (34) weit schneller vermindert wird, als es fur S,Oi' durch das analoge Gleichgewicht S 3 0 i ' + S += S,O," geschieht, lassen sich die Geschwindigkeiten der Verseifungsvorgange von SeS,O," und S,O," nicht streng ver- gleichend be trachten.
Fur unsere Frage kommt es aber vor allem darauf an, zu er- mitteln, ob SO," aus SeS,O," wesentlich schneller entsteht als aus S30i'. Wie der vorangehende Versuch (50) lehrt, geht die SO4- Bildung in der Losung mit der Abgabe von SO, parallel; es genugte also, den Vergleich durch aeitliche Verfolgung der letzteren durch-
l) 2. anorg. u. allg. Chem. 125 (1922), 127.

324 F. Foerster, F. h n p , 0. Dropbach und W. Seidel.
Zeit nach Beginn des Versuchs
in Minuten
18 31 54 77 97
~. - - ~ _ _ _ _ _ _ ~
zufuhren. Dazu wurde (Versuch 51) je 0,Ol Mol K,S30, und K,SeS20, in 50 ccm Wasser gelost und die die Losung enthaltenden Kolbchen I und I1 mit Ein- und Ableitungsrohr fur Stickstoff versehen und an zwei parallel geschaltete Vorlagen A und B mit Natronlauge an- geschlossen. Wahrend die Kolbchen im Wasserbade von 800 standen, wurde ein Stickstoffstrom von genau geregelter, konstanter Geschwindigkeit durch Kolbchen I, Vorlage I A , Kolbchen 11, Vor- lage I1 A hintereinander eine bestimmte Zeit geleitet. Nach deren Ablauf wurde der Stickstoffstrom auf die Vorlagen I B und I1 B umgestellt, der Inhalt von I A und I1 A titriert, diese Vorlagen neu gefiillt, wieder eingeschaltet, und nun nach bestimmter Zeit der Stickstoffstrom wieder auf diese geleitet, I B und I1 B untersucht usf. Die erhaltenen Ergebnisse sind die folgenden, wobei immer die gesamten, bis zu dem betreffenden Zeitpunkte abgegebenen SO,- Nengen, in ccm 0,l n- Jodlosung ausgedriickt, verzeichnet sind; bei vollstandiger Zersetzung, also Abgabe von 0,Ol Mol SO,, wiirde dies 200 ccm der Jodlosung entsprochen haben.
__ I - ~~ - _-
5,20 15,O 30,3 43,9 56,4 90,s
158,8
________ - - -__-_______ --
1,4 8 3
23,5 35,51) 44,l 80,5
178,O l) Se-Abscheidung beginnt.
Aus diesen Werten erkennt man, daB die Zersetzung des Seleno- dithionats durch eine kurze (noch nicht zu deutende) Inkubations- zeit eingeleitet wird, die beim Trithionat fehlt, im ubrigen aber die SO,-Abgabe in beiden Fallen etwa im gleichen MaBe ansteigt, also auch die Sulfatbildung aus dem Selenodithionat keineswegs schneller verlauft als aus dem Trithionat.
Wahrend nun aber das Trithionat sich in saurer Losung nicht, wesentlich schneller zersetzt als in anfangs neutraler,l) wird der Zerfall des Selenodithionats duroh Saurezusatz sehr wesentlich beschleunigt. Lost man z. B. 0,Ol Mol K,SeS20, in 50 ccm n-HCI und erwarmt auf 80°, so tritt alsbald starke Selenabscheidung ein, .- ____
I) 2. anorg. u. allg. Chem. 145 (1922), 134.

Zsrsctxung a h schwefligcn Saurc wad ihrer Salxe in wapriflr Losung. 326
und SO, entweicht in Stromen, so daB nach kurzer Zeit die Zer- setzung beendet ist. Nimmt man zur Losung 0,l n- bzw. 0,Ol n-HCl, so dauert es entsprechend liinger, aber auch im letzteren Falle noch erheblich khrzere Zeit aJs in neutraler Losung, bis die Selenabscheidung einsetzt ; dabei ksnn man sich davon uberzeugen, daB der Charakter der Umsetzung auch in saurer Losung der oben geschilderte ist. Diese Wirkung der Saure tritt auch bei gewohnlicher Temperatur hervor: vermischt man e k e Losung des Selenodithionats mit dem gleichen bis dem doppelten Volumen an konzentrierter Salzsaure, so tritt auch bei groBer Verdunnung des Salzes sofort Selenabscheidung ein. Da weder die Selenothionate vom Pentatypus noch die vom Tetratypus diese Reaktion zeigen, so eignet sie sich gut zur Erkennung des Selenodithionats. Sie ist auch zu beriicksiehtigen, wenn man in angesiiuerter Ldung Sulfat neben Selenodithionat durch Chlor- barium bestimmen will; man muB dabei stark verdunnen, den Saure- zusatz auf das MindestmaB beschriinken und die FPllung nur tun- lichst kurze Zeit stehen lassen ; andernfalls verliert der Niederschlag seine rein weil3e Farbe und nimmt an Gewicht zu.
Ein weiterer wichtiga Unterschied zwischen Selenodithionat und Trithionat zeigt sich im Verhalten gegen Alhlikuge ; wiihrend das letztere dabei kein Sulfat bildet, seine Verseifung hier also in anderer Richtung els in saurer Losung verlauft, wird das erstere auch durch Alkali unter Bulfatbildnng langsam zerlegt. Eine genauere Untersuohung hieriiber steht noch aus.
Angesichts des Umstandes, da13 H’ die Verseifung des Seleno- dithionats beschleunigt , erschien es schlieBlioh noch moglioh , daR es bei Gegenwart eines Uberschusses von Bisulfit, d. h. unter den Bedingungen, unter denen das Selen seine katalytisehe Wirkung ausubt, erheblich schneller sich zersetzt als Trithionat. Urn dies zu priifen, wurden (Versuch 52) zwei Losungen, deren eine 0,Ol Mol K,SeS,O, und 0,08 Mol KHSO,, deren andere 0,Ol Mol K,S30, und 0,03 Mol KHS03 in je 50 ccm enthielt, nebeneinander 20 Minuten lsng auf 800 im Wasserbade erhitzt und zwar so, daB die Reaktions- gefiil3e durch einen mit einem Manometer versehenen Gummistopfen fest verschlossen waren, ako kein Schwefeldioxyd entweichen lassen konnten, und dann beide Losungen rssch abgekiihlt, mit Salzsiiure angesauert und durch einen Stickstoffstrom vom Schwefeldioxyd befreit. Darauf wurde in beiden das entstsndene Sulfat bestimmt. Die Losung des Selenodithionats ergab 0,787 g BaSO,, die Lo- sung des Trithionats 0,933 g BaSO,. Danach bildet auch bei
/, nnorg. II nllg. Chem. Bd. 128. 22

32 6 E Foersfer, E: Lanp, 0. Dropbbach wnd W. Seidsl.
Gegenwart der kleinen H-Konzentration des Bisuhits das Seleno- dithionat nicht sohneller, sondern auch hier etwas langsamer Sulfat als das Trithionat. Die katalytische Wirkung des Selens auf den Zerfall des Bisulfits kann also nicht darauf beruhen, daB Seleno- dithionat wesentlieh sohneller unter Sulfatbildung der Verseifung unterliegt als Trithionat.
d) Versuch, denMechanismus der ka ta ly t i schen Wirkung des Selens zu deuten.
Gibt man zu einer Losung von Bisulfit elementares Selen, so geht es, wie oben gesagt, so lange jenes im Uberschusse ist, unter Bildung von Selenosulfat in Losung. Diese Verbindung mu6 also die kata- lytische Wirkung des Selens auf die Bisulfitzersetzung vermitteln. Betreffs der Art dieser Wirksamkeit war zunachst die Annahme naheliegend, daB sie wie die des Thiosulfats darin bestiinde, daB das Selenosulfat uber Selenothionate hinweg zum Sulfat fuhrt. Die vorangehenden Beobachtungen muBten nun aber Zweifel erwecken, ob das Selenosulfat wirklich auf ganz gleiohartigem Wege wie das Thiosulfat zu wirken vermag; denn sie zeigten, dad der fur das Verhal ten des Thiosulfats so wiohtige Polymerisierungsvorgang zu Pentathionat sich beim Selenosulfat nicht nachweisen lie& und daB die Sulfatbildung aus dem Selenodithionat gegenuber der aus Tri- tbiolzat keineswegs den groBen Geschwindigkeitsvorsprung besitzt , den sie nach der starken katalytischen Wirkung des Selens haben muate, wenn das Selenosulfat auf Bisulfit in ganz gleicher Weise wie das Thiosulfat einwirkte.
DaB dies in der Tat nicht der Fall ist, lehrte folgender Versuoh 53: 1/30 Mol K,SeSO, und Ys Mol KHS03 wurden zu 100 ocm gelost und die Losung, mit einem ein Manometer tragenden Gummistopfen ver- sehlossen, bei SOo im Wasserbade langere Zeit erhitzt. Von Zeit zu Zeit wurden Proben genommen und auf die in ihnen enthaltenen Selenverbindungen qualitativ untersucht. Es zeigte sioh, daB die weit iiberwiegende Menge des Selens als Selenosulfat , also durch verdiinnte Salzsiiure sofort flillbar, in der Losung blieb; nach dem hbblasen des SO, aus der angesiiuerten und filtrierten Probe lieB sich in ihr schon bald nach Beginn des Versuohes durch starke Salzsaure eine kleine Menge von Selenodithionat nachweisen, aber durch Natronlauge keine Selenverbindung vom Penta- oder Tetrat ypus. Die Menge der ersteren Verbindung aber nahm im Verlaufe des Versuches nicht sichtbar zu. SchlieBlioh, nach 9 Stunden, war das

Zersetxung d8r schwef2~gm Saurc und ihrer Salxe in wapriger Losung. 327
Bisulfit, das immer reichlicher in Sulfat uberging, soweit verschwunden, da5 die Losung beim Erkalten Selen abzuscheiden begann. Jetzt war das Selenodithionat verschwunden, statt seiner aber enthielt die Losung in kleiner Menge eine auf Zusatz von Natronlange Selen abscheidende Verbindung, also eine solche vom Pentatypus, wahreiid die weit iiberwiegende Menge des Selens in ihr immer noch als Selenosulfat enthalten war. Eine Thiosulfatlosung ware unter gleichen Umstanden dagegen weitgehend in Polythionate iiber- gegangen.
Hieraus ergibt sich, da13 dss Selenosulfat nicht durch einen Ubergang in Selenothionate den Zerfall des Bisulfits beschleunigen liann, ja es scheint angesichts des langsamen Zerfalls des Bisulfits bei obigem Versuche, als sei das Selenosulfat selbst uberhaupt nicht der unmittelbar wirkende Katalysator, sondern nnr die Quelle fiir diesen. Die oben beschriebenen Beobachtungen an den Seleno- schwefelverbindungen hatten nun aber gelehrt, da13 bei ihnen stets iind nur dort, wo selenige Siiure oder Selenite oxydierend wirken, grofie Reaktionsgeschwindigkeiten auftreten. Das fuhrt zu der Ver- mutung, daB die Anionen der selenigen SBure durch ihre oxydierenden Wirkungen die eigentlichen Trager der katalytischen Wirkung des Selens auf die Bisulfitzersetzung sind.
Da nun aber, solange Selenverbindungcn diese Wirkuiig aus- iibeii, nachweislich immer Selenosulfat zugegen ist , konnen die genannten Anionen liaum a nclers als dnrch clas Anftreten der Gleich- gewichte :
HSO,’ + H + SOa”, (7)
(47)
(48)
(49)
SeSO,” + SO,” + SeO,” + S20i’, SeO,” + H‘ + HSeO,’,
also auch
in der Losung sich bi1den.l)
SeSO,” + HSO,’ + HSe0; -+ SIOi’
-____
1) Es wiiren auch folgende Gleichgewichte denkbar: SeSO,” + 2 H =r SeO + SO + H,O,
2 SO + H,O + S,O,” + 2H. Des Selenoxyd SeO konnte dam die rasch oxydierend wirkende Substanz sein. Bei solcher Annahme befiinde man sich aber ganz auf dem Gebiete der Hypothese, wkhrend die oben angegebene sich a d die Tatsache stutzt, daB selenige %we bzw. HSe03‘ ihre oxydierende Wirkung sehr rasch ausiibt. Deshalb soll nur die oben erorterte Annahme naher verfolgt werden.
22*

328 E: Fowstw, B! Lapage, 0. Drobbaeh und W. Seidd.
Diese Annahme wird gestutzt durch die Beobachtung von RATHKE, daB, wenn aus Losungen von Selenosulfat sich Seleno- dithionat abgeschieden hat, also Selenit in ihnen entstanden sein muBte, die Mutterlauge Thiosulfat enthielt. Ferner mu13 mch den GIeichgewichten (47) und (49) die Konzentration der Anionen der selenigen Saure einen um so grofieren Bruchteil des ursprunglichen Selenosulfets ausmachen, je grol3er ihm gegenuber der UberschuB der Anionen der schwefligen Saure ist ; kleine Mengen Selenosulfat rnuBten daher in groBer Bisulfitkonzentration verhaltnismiiljig wirk- samer sein, als groBere Mengen zumal gegenuber einer kleineren Bisulfitkonaentrstion. Damit steht das Ergebnis von Versuch 53 im Einklsng, sowie die oben (S. 289) mitgeteilte Beobachtung, da13 die beschleunigende Wirkung des Selens langsamer ansteigt als seine, Piner gegebenen Bisulfitlosung zugefugte Menge. Endlich spricht dafur auch die Tatsache, daI3 Trithionat, melches ja, wie bekannt, im Gleichgewichte
S,O," + S + S,O,"
stark entschwefelnd wirken kann, also vermutlich auch in einem Gleichgewicht
S,O," + SeSO," ~t S,O," + St.0,"
die Entstehung der Anionen der selenigen Saure begiinstigen konnte, die Bisulfitzersetzung bei Gegenwart von Selenosulfat sehr beschleunigt. Beim Erwarmen einer Lijsung von 1/30 Mol K,SO, und ll6 Mol KHSO, auf 95-980 (Versuch 54) trat bei Gegenwart von 1/30 Mol K,S,O, die, stets einen bestimmten Verbrauch des Bisulfits anzeigende Selenabscheidung schon nach 15 Minuten, in der gleichen, aber von Trithionat freien Losung jedoch erst nach 95 Minuten ein. Auch verschwindet in KHSO, gelostes K,SeSO,, welches einer mit KHSO, versetzten Trithionatlosung zugesetzt wird, schon bei gewohnlicher Temperatur so weit, daB es durch verdiinnte Sttlmaure nicht mehr fallbar ist, wenn die Menge des K,SeSO, nicht mehr als 1 Mol auf 2 Mol Trithionat betragt. D a m geht wahrscheinlich das Seleno- sulfat mit dem entstehenden Selenit in Selenodithionat uber.
Hiernach erscheint die Annahme der Gleichgewichte (47) und (49) sls nicht unberechtigt. Sie sol1 i m folgenden a l s Arbe i t shypo- t h e s e dienen.
Dann kann der Mechanismus, in welchem das Selen katalytisoh

Zersetxung dsr scla~efligem Sawe unddhrer Salze in wapriger Losung. 329
auf die Bisulfitzersetzung einwirkt, in folgenden Vorgangen erblickt werden :
(28)
(49)
(31)
(32)
HSO,' + Se =+ SeSO," + H', SeSO," + HSO,' + HSeO,' + S203", HSeO,' + HSO,' -+ SeO + SO," + H,O,
SeO + HSO,' -+ Se + SO," + H , ___-___
4HS0,' -+ 2S0," + S203" + 2H' + H,O. (22)
Alle 4 TeilvorgBnge kiinnen sehr schnell verlaufen und mussen daher auch bei kleinen Selenmengen, also kleiner Konzentration von Se SO," bzw. HSeO,', eine schnelle Umwandlung des Bisulfits in Sulfat und Thiosulfat und eine gleich schnelle Riickbildung von SeSO," be- wirken, so lange fur das Gleichgewicht (28) die Konzentration des HSO,' noch geniigend hoch, und die des H' no& genugend klein ist, also in der Zeit, in der der erste Hauptvorgang der Bisulfit- zersetzung sich abspielt. Sobald die eine oder andere von diesen Voraussetzungen nicht mehr zutrifft, kann das bei Vorgang (32) freiwerdende Selen nicht mehr in die Losung zuruckkehren; dam hat seine katalytische Wirkung ihr Ende erreicht. Day wie oben bemerkt , die H-Konzentration, bei der Gleichgewicht (28) ganz linksseitig verschoben ist, weit tiefer liegt als die, bei der Gleiches fur das entsprechende Thiosulfatgleichgewicht (15a) eintritt, mu13, wenn die Bisulfitbonzentration sinkt, und die H'-Honzentrstion dabei ansteigt, das Selen vor dem Schwefel sich abscheidon und barn fur den unter Schwefelabscheidung vor sich gehenden zweiten Hauptvorgang als Katalysator nicht mehr wirksam sein, wie es die Beobachtungen auch gelehrt bben .
Die Gleichungen (B), (49), (31) und (32) erlautern nun freilich nur die gleichzeitige Entstehung vou Sulfat und Thiosulfat unter dem Einflusse des &lens, wie sie in Sulfit-Bisulfitlosungen beob- achtet ist. Fur die Deutung der Tatsache aber, daB in reiner Bi- suUit1osung neben Sulfat und rnit ihm schritthaltend Polytbionate entstehen, bedarf es eines weiteren rasch verlaufenden Vorganges, da der bei Abwesenheit von Selen das Thiosulfat in Polythionate uberfuhrende Polymerisationsvorgang nach allem, was wir von ihm wissen, obne Katalysator nioht schnell genug ablauft, um sein volliges Schrittllalten mit der vom Selen beschleunigten Bildung des Sulfats und Thiosulfats zu ermoglichen. Auch einen solchen Vor- gang kann die selenige Stiure veranlassen. Denn wir sahen oben,

330 F! Foerstel; F. Lunge, 0. Droj?bach und If’. Soidel.
tlaB sie in angesauerter Losung Thiosulfat mit sehr groBer Geschwindiglieit in Tetrathionat zu verwandeln vermag :
HSeO,’ + 2S,O,” + 3H’ -+ SeO + S40;’ + 2H20, (37) SeO + 2S,O,” + 2 H -+ H,O + SeS,OL’, (38)
SeS,O,” + HSO,’ 3 S,O,” + SeSO,” + H’. (50)
Die Reaktionsgeschwindigkeit dieser Vorgange erwies sich bpi groBerer H’-Xonzentration als auBerordentlich grofi, zugleich aber als abhangig von dieser. Aber auch in schwacher saurer Losung ist sie allem Anschein nach groB genug, dafi vermutlich auch bei der H’-Konzentration einer starkeren Eisulfitlosung die oxydierende Wirkung der selenigen Saure sich gleichzeitig auf das Bisulfit und tlas Thiosulfat erstrecken kann. Das Ergebnis des Zusaminenwirkens clieser Vorgange lraiin folgendermafien dargestellt wcrden :
4HS0,’ + 4Se =+ 4H’ + 4SeSO,”,
4SeS0,“+4HS03‘ + 4HSe0,’ + 4S,O,”, (28)
(49) HSeO,’ + 4S,O,’ + 5H’ --t 2S,O,” + Se + 3H,O, (37), (38), (39)
3HSc0,’ + 6HS0,’ --f 6S0,” + 3Se + 3 H + 3H20, (Sl), (32)
(51)
Die Endgleichung lehrt, daB, wenn alles S,O,” der oxydierenden Wirkung von HSeO,’ unterliegt, und alles dabei nicht verbrauchte HSe0; zur Sulfatbildung aus HSO,‘ dient, in der Losung Sulfat- nnd Polythionatschwefel im Verhaltnis 6 : 8 = 1 : 1,33 snftreten und die mit Phenolphthalein als Indikator titrierbare Aziditat auf I/,
ihres Anfangswertes zuruckgegangen sein muBte, wenn alles Bi- hulfit umgewandelt wLre. Der Axiditatsruckgang miirjte hier also iiooh etwas groBer sein, als wenn unter der katalytischen Wirkung des Schwefels das Bisulfit nach Gleichung (23) glatt in Sulfat unci Pentathionat sich verwandeln konnte, wobei er auf der Anfangs- aziditiit fuhren miil3te (S.303). Gber den AziditatPruckgang der Losung liegt freilich bisher nur Versuch 47 vor, bei dem dieser sogar bis auf
des Anfangswertes gefuhrt hat. Uber die Ursa8che dieser, absolit genommen, nicht erheblichen Abweichung kann zunachst nichts gesagt werden. Das Verhaltnis von Sulfat- zu Polythionatschwefel wurde bei der Versuchsreihe der Ubersicht 6 stets kleiner als 1, und gegen Ende des ersten Hauptvorganges = 1 : 1,2 gefunden. DaS dieses gegenuber dem erwarteten etwas zuruckbleibt, hat ohne
__ _ _ _ _ _ ~ ~ 14HS0,’ --f 6S0,” + 9S,Oi’ + 6H20 + 2H’.

Zersetmng der schweflQen Saure und ihrer Sake in wapriqer Losung. 331
Zweifel seinen Grund darin, daB die Geschwindigkeit, mit der S20,” bei Gegenwart von H’ in S,O,” von selbst iibergeht, durchaus nicht als verschwindend anzusehen ist, und dalj andererseits auch Tetra- thionat neben jenen Vorgangen in recht merklichem MaBe rm Tri- thionat abgebaut und dieses durch Verseifung und unter teilweiser Ruckbildung von Polythionat, wie oben dargetan, an der Sulfat- bildung teilnehmen muB.
Ebenso wie auf das Thiosulfat kann sich die oxydierende Betlitigung der selenigen Skire auch auf dap neben ihr im Gleich- gewicht (49) stehende Selenosulfat erstrecken; wir sahen, daIj dabei mit groBer Geschwindigkeit Selenodithionat entsteht :
(40) HSeO,’ + 2SeS0,“ + 3H’ -+ SeO + 2H20 + Se2S206)’, SeO + 2SeS0,” + 2H‘ -+ Se3S20i’ + &O, (41)
Se,S,O,“ + HSO,’ + SeS20g‘ + SeSO,” + H’ , Se3S20i’ + 2HS0, + SeS,O,” + SeSO,” + 2 H .
(43)
(42) Denkbar ware noch, daS neben Vorgang (41) und (38) auch der Vorgang
stattfande. Durch diese Vorgange mu6 stets auch in gewissem Grade das Selenosulfat in Selenodithionat, bzw. bei Ruckgang des Bi- sulfits und Gegenwart hinreichender Selenosulfatkonzentration, im Sinne von Gleichgewioht (42) und (43) in ein Selenothionat vom Pentatypus verwandelt werden. DaB letzteres eintreten kann, lehrt Versuch 53 sowie der in Ubersicht 6 hervortretende Umstand, claB wahrend der katalytischen Bettitigung des Selens ein gewisser Teil von ihm duroh verdunnte Sliuren nicht mehr fiillbar ist. Da aber das Selenodithionat immerhin ziemlich schnell zu Sulfat und Selenosulfat verseift wird, mu13 0.4, in dem MaSe als seine Entstehung sich verlangsamt, d. h. die HS0,’-Konzentration abnimmt, auch wieder verschwinden, so daB, wenn das Bisulfit verbraucht ist, auch das in das Selenodithionat ubergegangene Selen wieder ab- geschieden sein muB. DaIj dieses aber nicht als Zwischenprodukt fur den Hauptteil des Vorganges, sondern nur als das Ergebnis eines Nebenvorganges aufgefaBt werden darf, lehren die oben uber die hschwindigkeit seiner Verseifung angegebenen Beobachtungen.
Aus unserer Arbeitshypothese uber den Mechanismus der katalflischen Wirksamkeit des Selens bei der Bisulfitzersetzung und BUS den uber die oxydierende Wirkung der selenigen S h r e
SeO + 2HS0,’ -+ SeS,Oi’ + H20 (52)

332 E Foemter, El Lange, 0. &@bad und W. &idat.
gemachten Ekobachtungen laBen sich ako die allgemeinen Richt- linien, nach denen dieser Vorgang in seinen Einzelteilen stattfindet, vollstiindig ableiten. Ob die Vorstellungen, zu denen wir uber das Zusltrnmenwirken der verschiedenen Oxydationsvorgange gelangten, in jeder Hinsicht deren Eigenart ontsprechen, bedarf noch genauerer Untersuchungen; aber es ist kein Grund zu sehen, der sie unwahr- xheinlich machte. Der Umfang, in dem die einzelnen, von HSeO,‘ bzw. von SeO ausgeubten Oxydationswirkungen nebeneinander ver- laufen, ist durch ihre Reaktionsgeschwindigkeiten bestimmt . Diese hangen neben der Art und der Konzentration der der Oxydation unterliegenden Molekeln auch vorn Oxydations potential der HSeO,’ ab, welches mit steigender H’-Konzentration steigt. Aus den Unter- suehungen’) iiber die elektrolytische Oxydation von SO,” weifi man, wie stark das Anodenpotential auch die Richtung des Oxy- dationsvorganges beeinflufit, so da8 z. B. das Sulfit bald ausschliefi- lich nach
also analog Vorgang (31) nnd (32) oxydiert wird, bald daneben in erheblichem Mafie nach
so,” + 0 --f so;/, (53)
2S0,” + 2H‘ + 0 3 S,O,“ + H,O, (54) (1. h. unter Zusammentritt von zwei einfacheren zu einem kompli- zierteren Anion, analog den Vorgangen (37) und (38). Auch letztere Art von Vorgiingen, die f~ ihren Verlauf die Mitwirkung von H’ verlangen, miiDten, da sie nach der oben entwickelten Vorstellung nur in Bisulfit-, nicht in Sulfit-Bisulfitlosungen sich abspielen sollen, erst durch ein gesteigertes Oxydationspotential von HSeO,’ ihre Wettbewerbfghigkeit mit der Sulfatbildung erlangen, wiihrend die Oxydation zu Sulfat auch in Sulfit-Bisulfitlosung, also bei niedrigerem Oxydationspotential die durchaus uberwiegende Geschwindigkeit besitzen miil3te. In der Tat hat sich, wie oben gesagt m d e , fur die Einwirknng der selenigen Saure auf Thiosulfatanion die hiernach zu erwartende Abhiingigkeit ihrer Geschwindigkeit von der H - Konzentration nachweisen lassen.
Die Art, in der nach dieser Auffassung das Selen seine katalytische Wirkung bei der Bisulfitzersetzung ausiibt, scheint auf den ersten Blick eine ganz andere zu sein, als sie oben fur die katalytische
1) F. FOERSTER u. A. FRIESSNER, Ber. d . D . Chem. Gm. 35 (1902). 2515; A. FRIESSNER, 2. Elektrochem. 10 (1904), 731; vergl. auch J. 0. THATCHEB, z. phys. chem. 47 (1904), 641.

Zerselxzlng dcr schwefligen Sawe und ihrer Salze in wapriger Losung. 333
Betatigung des Scliwefels dargelegt wurde. Ifan konnte d a w h an der Wahrscheinlichkeit der fur das Selen angenommenen Wirkungs- weise zu zweifeln sich fur berechtigt halten. Ein solcher tiefgehender Unterschied aber besteht bei naherem Zusehen nicht.
Als den primaren Vorgang der Bisulfitzersetzung nahmen wir oben (S . 267) die Selbstoxydation des HSO,’ an:
(l0a) Das dabei auftretende SO kann nun allem Anschein nach bei niederem Oxydationspotentiale, also bei der kleinen H-Konzentration einer Sulfit-Bisulfitlosung keine oxydierende Wirkung ausuben, sondern sich nur alsbald nach
zu Thiosulfat polymerisieren , das unter diesen Umstanden weit- gehend stabil ist, daher wie 8.301 gezeigt, keine erhebliche kata- lytische Wirkung in einer Sulfit-Bisulfitlosung auszuuben vermag.
(31) hat nun Zuni Unterschiede von (loa) auch bei ganz kleiner H - Konzentration eine grol3e Geschwindigkeit, und eine ahnliche besitzt die Wirkung von SeO auf HSO,’. Das dabei freiwerdende Selen erzeugt sofort SoSO,”, und dieses bildet wiederum sehr rasch nnch
das Oxydationsmittel zuruck, wiihrend nach (31) SO;’, nach (49) S,O,” sehr vie1 schneller entstehen als nach (10a) und (11). Darum vermag Selen bzw. SeSO,” in Sulfit-Bisulfitlosung, im Gegensatz zu Schwefel bzw. SZO,’‘, stark katalytisch zu wirken. Der Unterschied in beiden Fallen liegt nur in der verschiedenen Geschwindigkeit, mit der Selenoxycle oder Schwefeloxyde oxydierend zu wirken ver- magen.
Die Bestandigkeit des S20Ql’ nimmt ab, wenn die H‘-Konzen- tration steigt. 1st diese noch so klein wie in einer Bisulfitlosnng, so tritt die Unbestandigkeit von S,O,” in dern Vorgange
in die Erecheinung. Diese Qleichung driickt nur die stochiometrischen Verh%ltnisse von Anfangs- und Endzustand aus, sagt aber uber den Mechanismus des Vorganges nichts Bus. Nimmt man an, dal3 mit et was gesteigert er H‘-Konzent ra tion irn Gleic hge wic ht,
HBO,’ + HSO,’ -> SO,” + SO + &O.
2 6 0 + &O e S,O,” + 2H’ (11)
Der zu (loa) analoge Vorgang HSeOi + €€SOL -+ SO,” + SeO + H20
SeSO,” + HS03‘ + HSeO,’ $. S20,” (49)
5S,Q,” + 6 H -+ 2S,O,” + 3H,O (1 6)
S,03” + 2H’ Z= 250 + H,O (11)

334 F. Fooerster, F. Lange, 0. Dro/7baeh uad W. Seidel.
auch das Sulfoxyd eine genugende Komentrati onssteigerung erfahrt, um bei dem zugleich gesteigerten Oxydationspotential nach
SO + 2 S,O," + 2H' -+ S50i' + H20 (55) oxydierend zu wirken, so gewinnt man, da Gleichung (11) und (55) Gleichung (16) ergeben, fur die sonst so ratselhafte Pentathionat- bildung eine einfaache Deutung. Diese tritt damit in Analogie zu
SeO + 2S,O," + 2 H 3 SeS,O," + H,O (38) Vorgang (55) vermittelt in einer Bisulfitlosung die katalytische Wirksamkeit des Schwefels bzw. des Thiosulfats, Vorgang (38) die des Selens bzw. des Selenosulfats. DaB Ietztere hier eine weit stgrkere ist als erstere, beruht wieder nur auf dcr verschiedenen Oxydations- geschwindigkeit von Selenoxyd und Sulfoxyd.
Unter diesen Gesichtspunkten betrachtet, erscheint die kata- lytische Wirkung des Selens und des Schwefels als durchaus wesens- gleich, nur dem Grade nach verschieden.
Es ergibt sich weiter daraus die wiohtige, im voraufgehenden srhon mehrfach betonte Eigenart dieser katalytischen Wirkung, daB sie durch die Ha-Konzentration der Losung sowohl nach unten wie nach oben eine Begrenzung erfahrt, und daB diese bei den beiden Katalysatoren, Schwefel und Selen, eine verschiedene ist. Wird die H'-Konzentration so klein, daB Vorgang (55) unmerklich langsam wird, so ist fur die katalytische Wirkung des Schwefels bzw. des Thiosulfats die uiitere Grenze erreicht, wiihrend man an diese fur die Wirkung des Selens bew. Selenosulfats erst zu gelangen ver- mochte, wenn Vorgang (31) unmerklich wiirde. Ersteres liegt, wie oben betont, in einer Sulfit-Bisulfitlosung vor, letzteres ist hier aber noch lange nicht erreicht, und es liegen vorlaufig auch keine Er- fahrungen vor, ob man bei weiterer Abnahme der H an diese Grenze uberhaupt gelangen kann. So versteht man die oben erorterte Er- scheinung, da8 die katalytische Wirkung des Schwefels in einer Sulfit-Bisulfitlosung aufhiirt, wzhrend die des Selens in einer solchen sehr kraftig ist.
Die Begrenzuiig nach oben ist bestimmt durch die verschiedene H-Konzentration, bei der die die katalytischen Wirkungen ver- mittelnden Ionen S203" und SeSO," noch in merklicher Konzen- tration bestehen bleiben, also wiederum durch deren Bestandigkeit . Diese erwies sich aIs sehr verschieden: wahrend im Gleichgewicht
S,O," + H' f: HSO,' + S (15a)

Zersetxzcng der schwefligen Sazcre und ihrer Sake in wapriger Loswng. 335
selbst neben der der Sattigung der Losung rnit Schwefeldioxyd entsprechenden H’-Konzentration beliebige Konzentrationen von S,O,” uber geraume Zeit bestehen bleiben konnen, und erst die H-Konzentrationen starker Sauren diese zum Verschwinden bringt , wird das Gleichgewicht
schon durch einen geringen UberschuB \’on freier schwefliger Saure vollig rechtsseitig verschoben. Daraus folgt, daB die katalytische Wirksamkeit des Schwefels auf die Umsetzung des Sulfitschwefels nach obenhin erst ihre Grenze findet, wenn in der Losung starke Sauren von etwas grohrer Konzentration auftreten, die Grenze fiir die Wirksamkeit des Selens aber schon erreicht ist, wenn durch die Steigerung der H’-Komentration freie schweflige Saure in etwas starkerem MaBe in der Losung aufzutreten vermag. Das Bereioh der katalytischen Wirksamkeit des Schwefels ist also gegen das des Selens insofern erheblich verschoben, als jenes in das Gebiet hoherer H-Konzentrationen weiter hinauf-, diews in das Gebiet kleinerer H’-Konzentrationen weiter hinabreicht.
Diese SchluBfolgerungen sind durch die oben mitgeteihen Tatsachen vollig bestatigt. Sie fiihren zu dem Ergebnis, daB die Zersetzung der freien schwefligen Skure, die durch Schwefel, wie wir sahen, sehr stark katalytisch gefordert wird, durch Selen keine katalytische Beeinflussung erfahren sollte. Wie weit dies der Fall ist, ergibt sich aus folgendem Versuch (55): Eine Losung von 1,0 Mol SO,/Liter wurde auf 4 Rohre, die rnit Stickstoff gefullt waren, zu je 20 ccm verteilt; zwei der Rohre erhielten einen Zusatz von je 0,Ol g-Atom Selen, das dritte von 0,01 g-Atom Schwefel, und das vierte blieb ohne Zusatz. Nach dem Zuschmelzen wurden die Rohre auf einer Scheibe befestigt, die in einem Wasserbade in langsamer Umdrehung um eine wagerechte Achse gehalten werden konnte, und wurden so zweimal 12 Stunden auf 850 erhitzt. Nach dem Abkuhlen und dem Abblasen des Schwefeldioxyds fanden sich folgende Menge Sulfatschwefel in den Rohren vor:
ohne Zusatz . . . . . 3,7 mg Sulfat-S Rohr 1 rnit Se . . . . 9,9 ,, Y Y
Rohr 2 rnit Se . . . . 9,5 7 , 1 )
Rohr rnit S . . . . . 38,l ., 1 )
SeSO,“ + 13’ += HSO,’ + Se (28)
I n letzterem Rohre war die Losung gelb gefarbt und enthielt auch betrachtliche Mengen von Thioschwefelsaure. Wenn auch eine die

336 F. Ebar&r, F. Lanq8, 0. UroPbaok wnal W. Seidel.
Reaktion fbrdernde Wirkung des Selens nicht ganz ausgeblieben ist, so erwies sie sich doch als ungleich schwiicher gegeniiber der des Schwefels ; das aufierordentlich verschiedene Verhalten beider Ihtalysatoren gegen Bisulfitlijsungen einerseits, schweflige Saure andererseits, wie es die Theorie verlangt, wird aber durch cliesen Versuch lilar beleuchtet. Der Rest von Wirksamkeit, den das Selen auch bei diesem Versuche noch zeigt, hiingt damit zusammen, da,S es mit der schwefligen Saure Selenothiosauren bildet , von denen sich die des Pentatypus wie die des Tetratypus spurenweise in d m mit Selen behandelten Losungen nachweisen lieBen.
Die oben entwickelte Auffassung uber die Art der ka talytischen Wirkung des Selens auf die Umsetzung von Sulfitschwefel erlaubt also, diese Wirkung einheitlich mit der des Schwefels zu uberblicken und dabei die Eigenarten beider Katalysatoren gegeniiber den frag- lichen Vorgangen in ihrer GesetzmaSigkeit vorauszusagen.
Die grof3en Geschwindigkeits- und Bestandigkeitsunterschiede, welche die niederen Osyde des Schwefels gegenuber denen des Selens zeigen, erinnern lebhaft an ahnliche Unterschiede zwischen den niederen Oxydationsstufen der Halogene. Auch hier nimmt mit, steigendem Atorngewichte des Halogens die Geschwindigkeit der oxydierenden Wirkung der niederen Oxydationsstufen stark zu, deren Bestandigkeit stark ab.
So wirkt HOBr auf Hypobromih mit mehr als lOOmal so groBer Geschwindigkeit als HOG1 auf Hypochlorit 1); jene konnte bisher nur in hiichstens 0,l-rnolarer Konzentration, diese bis zu 2-molar und mehr erhalten werden. Eine der sehr unbestiindigen chlorigen S i h e und ihren Salzen entsprechende Bromverbindung konnte uberhaupt noch nicht aufgefunden werden. I n noch gesteigertem MaBe tritt gleiches bei der niedrigsten Oxydationsstufe des Jodsz), der unterjodigen Saure und ihren Salzen, bervor. Entsprechend scheinen auch die niedrigen Oxyde des Tellurs und die zugehorigen Salze von sehr geringer Bestandigkeit zu sein, SD daa dem Thio- sulfat analoge Tellurverbindungen, etwa K,TeSO,, ebenso wie deren als bestandiger zu erwartende Umsetzungsprodukte wie K,TeS,O, auc h nnter Bedingungen, unter denen die analogen Selenverbindungen glatt entstehen, bisher nioht gewonnen werden k ~ n n t e n . ~ ) Es ist
l) H. KRETZSCIIMAR, 2. Elektrochem. 10 (1904), 798. ?) A. SKRABAL, Se'tzunpber. (1. Wiener A k d . d . Wissensch. 120 (1911). 53. 3, G. T. MORGAN ti. H. D. K. DREW, 1. c.

daher nicht uberraschend, daB Tellur suf den Bisulfitzerfall keine katalytische Wirkung auszuiiben vermag.
Die hier mitgeteilten Untersuohungen bediirfen noah vielfach der genaueren und vertiefteren Durcharbeitung. Sie werden fort- gesetzt; es ware nur zu begruBen, wenn Fachgenossen geneigt waren, auf diesen groBen und noch so wenig gepflegten Gebieten mit- zuarbeiten.
7. Zusammenfaernng.
1. Die seit lange bekannte Selbstzerset~ung der schwefligen Saure :
3H2s0, -+ 2S0," + 4 H + S + H,O (a>
besitzt eine sehr geringe Gesohwindigkeit, Auch bei 1000 braucht sie viele Tage, bei 1500 etwa zwei Tage, urn ihr Ende zu erreichcn. Sie wird durch die von ihr erzeugten Stoffe, von Schwefel und Wasser- stoffion, in ihrer Geschwindigkeit beeinflufit, verliiuft also auto- ka talytisch.
2. Die Wirkung der beiden Katalysatoren ist dabei eine ent- gegengesetzte : Schwefel beschleunigt, Wasserstoffion verzogert den Vorgang. Die positiv katalytische Wirkung des Schwefels, auch eines von vornherein der Losung erteilten Schwefelzusatzes, tritt nur im Anfange der Umsetzung deutlich hervor; mit deren Fortgang wird sie immer starker von der negativ katalytischen Wirkung des Wasserstoffions uberlagert .
3. Diese bewirkt, da13 verdiinnte Losungen der schwefligen Sgure sich sohneller umsetzen als konzentrierte, und daB dort der Umsatz anniihernd vollstandig ist, hier aber selbst in langerer Versuchsdauer und bei 1800 unvollstandig bleibt.
4. Schon in 2 n-Salzsaure als Losungsmittel tritt die Zersetzung der schwefligen Saure nioht mehr ein.
5. Der primare Vorgang besteht sehr wahrscheidich in den Umsetzungen :
2HS0,' -+ SO," + SO +KO, (b)
SSO + H20 S2031' + 2H', (c)
deren erste sehr langsam verlauft.
daB er weit schneller als die Vorgiinge (b) und (c) nach 6. Die positiv katalytische Wirkung des Schwefels besteht darin,
HS0; + S += S,O," + H'

338 B! Foerste?’, El Lange, 0. Dropbach zcnd W. Seidel.
Thioschwefelsaure erzeugt , diese nach
5S,O,” + 6H’ -+ 2S,O,” + 3H,O (el Pentathionsaure gibt, letztere unter Verbrauch von HSO,’ zu Tetra- und Trithionsaure abgebaut wird :
S,O,” + HSO,’ 3 S,Oi‘ + Sz03” + H S,O,” + HSO,’ -+ S,O,“ + S203” + H
M,O,” + H,O -t SO;‘ + S2031’ -I- 2 H
(fi (€9
(11)
und schlieBlich die Trithionsaure zu Schwefelsaure verseift wird :
8
uiid daB alle diese Vorgiinge S,O,” zuriickliefern und im Vergleich mit (b) sehr schnell verlaufen. Das Thiosulfatanion ist also der eigent- liche positive Katalysator, wenn cine fur Vorgang (e) ausreichende H‘-Konzentration vorhanden ist.
7. Dieser Theorie entsprechend treten walxend des Zerfall:, der schwefligen Saure stets S,Oi’ und daneben, und zwar stark vorwiegend, die Anionen der Polythionsiiuren als Zwischenprodukte auf; ihre Konzentration steigt snfangs an und geht spiiter wieder zuruck; je mehr von diesen Zwischenprodukten noch vorhanden ist , um so mehr bleibl das VerhBltnis des freien Schwefels zum SO,,“ hinter dem von Gleichung (a) geforderten Verhaltnis 1 : 2 zuriick.
8. Die negativ katalytische Wirkung des Wasserstoffions beruht darauf, da13 es einerseits unter Versehiebung des Gleichgewichts (d) den Vorgang
S,O,” + H’ -+ HSO,’ + S immer mehr an Stelle von Vorgang (e) treten laat und daniit den positiven Katalysator zerstort, und daB es andererseits in den Gleichgewichten der sohwefligen Saure
HSO,’ + H + SO, + H,O S205)’ + 2H’ =+ 2S0, + &O
(9 (4)
die all& fur den Umsatz geeigneten Ionen HSO,’ bzw. iS20,” in ihrer Konzentration vermindert.
9. I m Gegensstz eur Salzsaure wirkt nach VOLHARD Jodwasser- stoffsaure, selbst in hoher Konzentration, sehr beschleunigend auf den Zerfall der schwefligen Saure. Es miissen also ihre Anionen durch positiv katalytische Wirliung die negative des H weit uber- treffen.
10. Da J’ sich mit SO, zu gelben, komplexen Anionen, wie [J(SO,)J’ verbindet, ergibt sich hiernach als Bedingung fur die

Zersetxung der schwefligen Saure und ihrer Salxe in wa/+iger Losung. 339
Zersetzung der schwefligen Siiure in Losung, daS iu ihr SO, in koordinativer oder konstitutiver Bindung enthalten sein mu13. Durch die Bindungen
SO,. * - H,O + H’ + HSO,‘ (U [SO,. . SO,]” =+ S,O,” w
bieten die Anionen HSO,’ bzw. S,O,” nur e inen , allerdings besonders wichtigen Fall der Verwirklichung dieser Bedingung.
11. Auch S,O,” gibt in SO,-haltiger Losung gelbgefarbte, komplexe Ionen [S,O,(SO,),]”. S203” unterliegt aber in solchen Lmosungen weit schneller der Pentathionsaurebildung nach (e) als einer unmittelbaren Sulfatbildung aus jenen komplexen IoEeo. Dadurch verlauft seine katalytische Wirksarnkeit wesentlkh so, wie in (6) angegeben.
12. Die Selbstzersetzung der Bisulfite unterscheidet sich von der der schwefligen Siiure dadurch, dal3 bis zum weitgehenden Ver- brauch des Bisulfits die H-Konzentration auf den niedrigen Werten einer Bisulfit-Schwefligsaurelosung verharren mu13.
13. Dadurch tritt die positiv lrat’alytische Wirkung des S,O,” stark hervor ; die Zersetzung einer anfangs reinen Bisulfitlosung verlauft stark positiv autokatalytisch, also vie1 schneller als die der schwefligen Saure von gleicber Mollionzentration und fuhrt auch in konzentrierter Losung zum vollstandigen Umsatz :
3HS0,’ 3 2S0,” + H + S + H,O. (4 Von vornherein der Losung gegebene ZusBtze von Schwefel oder Polythionat verkiirzen die Umsetzungszeit erheblich.
14. Eine Bisulfitlosung setzt sich bei gewohnlicher Temperiltur wie bei 800 oder hoher bis nahe zum Verschwinden des Bisulfits nach Vorgang (b) bis (h) unter Bildung von Sulfat und Polythionat urn, ohne daS freier Schwefel auftritt (Hauptvorgang I). Dabei geht, da jene Vorggnge auch K’ liefern, die h i d i t a t nicht ganz so stark zuruck wie das Bisulfit, und es wird, wenn dieses weitgehend ver- braucht ist, Vorgang (d,) in Gleichgewicht (d) begiinstigt, und wahrend die von (h) gelieferten H’ sich unter fortschreitender Sulfat- bildung immer weiter anreichern, freier Schwefel erzeugt, das Poly- thionat verbraucht und die h i d i t s t der Losung gesteigert (Ehupt- vorgang 11). Dabei steigt z u n ~ h s t durch die von Vorgang (d,) gelieferten HSO,’ der Sulfitschwefel wieder an; er ist jetzt als freie &SO3 vorhanden, und diese zersetzt sich langsam nach (a), unter- stutzt durch die katalytische Wirkung der Polythionate.

340 3’. Fuemter, El Lange, 0. Drupbach und W. Seidel.
15. I m einzelnen entsprechen die bei diesen Umsetznngen zu beobachtenden Erscheinungen denen, die nzbch den Erfahrnngen iiber Entstehung und Zersetzung der Polythionate zu erwarten sind.
16. Bedingung fur die in Satz (14) beschriebene Art des Reaktionsverlaufs ist es, da13 durch die Versuchsanordnung das Entweichen von Schwefeldioxyd verhindertl wird.
17. Wird eine starke Bisulfitlosung im offenen GefaBe gekocht, so daB sie durch Entweichen des durch Hydrolyse in ihr entstandenen freien SO, in eine Bisulfit-Sulfitlosung iibergeht, RO bildet der erste Hauptvorgang nur Sulfat und Thiosulfat zu etwa gleichen Molekeln neben ganz wenig Polythionat . Bei der sehr geringen H-Konzentration der Losung bewirkt das Thiosulfat keine merkliche Autokatalyse, und die Umsetzung, wesentlich nach @) und (c), ist sehr langwierig. Da sie aber wieder H‘ liefert, ist beim Verschwinden des Sulfitsohwefels die Lijsung schwach sauer. Der jetzt eintretende zweite Haupt- vorgang liefert unter Zersetzung des Thiosulfats Sulfat und Schwefel durch folgende Vorgange :
S,03” + &O -+ SO,” + H28, S203” + 2 H -+ SO, + S + H,O, 2H,S + SO, -+ 3s + 2H,O.
(1) ( 4 ) (m)
Ein Wiederanstieg der Aziditat oder eine merkliche voriibergehende Wiederanreicherung von Sulfitschwefel findet hier nicht statt.
18. Reine Bisulfit- odm Bisulfit-Sulfitlosungen werden in ihrer Zersetzung durch kleine Mengen 8elen sehr stark katalytisch be- schleunigt ; aber immer nur der erste Hauptvorgang nnterliegt dieser Beschleunigung. Das in elementarer Form oder als selenige Saure zugesetzte Selen geht dabei zunachst als Selenosulfat SeSO,” in Losung und scheidet sich vor Beginn der Schwefelabscheidung quantitativ wieder aus.
19. Die von B. RATHKE schon vor langer Zeit hergestellten Salze Selenosulfat K,SeSO, und Selenodithionat K,SeS,O, wurden, jenes zum ersten Male ganz rein, hergestellt. Auch wird gezeigt, daB neben dem Selenodithionat, das dem Trithionat entspricht , Selenothionate vom Tetra- und Pentatypus existenzfahig sind.
20. Das Selenosulfat bildet analog zu (d) das Gleichgewicht
SeSO,“ + H’ + HSO,’ + Se . (n) Seine Verschiebung unter Selenabscheidung gesohieht schon bei weit kleinerer H’-Konzentration als unter sonst gleiohen Umstiinden

die entsprechende Verschiebung von (d) unter Schwefelabscheidung. Da (n) durch freie schweflige Saure weitgehend rechtsseitig verschoben wird, wirkt elementares Selen auf den Zerfall der schwefligen S h r e nur schwach - weit geringer als Schwefel - katalytisch ein, und muB sich bei der Bisulfitzersetzung gegen Ende des ersten Hauptvor- ganges abscheiden, sobald die H - Konzentration etwa auf die der H2S0, entsprechenden Betriige gelangt i t .
21. Tn einer Bisulfitlosuig verlhuft wcder die> Umwandlung von SeSO,“ in SrS,O,” noch dessm Verseifung nach
SeS20,” + &O -+ SO;’ + SeSO,” + 2 H (0)
zu Sulfat rnit groberer Geschwindigkeit als die analogen Um- setzungen der Schwefelverbinduiigen.
22. Dngegen sind die oxydierenden Wirkungen der selenigen Saure :
SeO, + HSO,‘ -+ SeO + SO,” + H‘, SeO + HSOQl + Se + SOL‘ + H ,
SeO, + 2S,O,” + 2 H -+ SeO + S,O,“ + H,O, SeO + 2S,O,” + 2 H -+ SeS40i‘ + &O,
(P,
(Pl)
(9)
(91)
(42) SeS,O,“ -+ S40i ’ + Se
als Vorgiinge von sehr hoher, bei (9) und (ql) mit steigender H‘- Konzentration wachsender Reaktionsgeschwindigkeit nachgewiesen, die auch durch das bei ihnen freiwerdende Selen in einer Bisulfit- losung inimer neues Se SO,” erzeugen.
23. Macht man die durch mancherlei Tatsachen gestutzte An- mhme, daB Selenosulfat in einer Bisulfitlosung das Gleichgewicht
HSO,‘ + SeSO,” =: HSeO,’ + S,O,” (r)
bildet, also die fi ir die Vorgiinge (p) und (9) erforderliche selenige Saure zu liefern vermag, so gelangt man zu einer den Tatsachen nllenthalben genugenden Deutung der starken katalytischen Wirkung des Selens auf den Bisulfitzerfall.
24. Bei der H-Konzentration einer Sulfit -Bisulfitlosung wiirde dann die selenige Siiure bzw. ihr Anion HSeO,’ nach (p) und (pl) HSO,’ zu SO,” oxydieren, und das neben HSeO,’ nach (r) entstehende S,O,“ sich in der Losung anreichern; bei der H-Konzentration einer Bisulfitlosung wiirde dagegen auch S,O,” nach (q), (ql), (4,) zu Tetrathiomt oxydiert werden.
2, pnorg. P. ail#. Chem. BIL 128 23

26. In Amlogie zu einem solchen Reaktionsmecthanismus tritt auch der der htalytischen Wirkung des Schwefeb, wenn man an- nimmt, daI3 der hierfur ausschlaggebende Vorgang (e) der Penta- thionatbildung naoh
s,o," + 2 H * 2so + q o , (0)
63) so + 2s,o," + 2 H --t s(s,o,);J + H20 verlhuft .
26. Da das Tellur nach den bisherigen Erfahrungen kein eigenes bestlndiges Anion analog SeSO," liefern kann, hat selenfreies Tellur aui den Bisulfitzerfa,ll keinen katdytischen EinfluB.
Dresdem, Anorganisch- Chernischcs Laboratoriuvr der Tehniseherc Hochschule.
Bei der Redaktion eingegangen am 5. bllire 1923.
Recommended

















