Embed Size (px)
Citation preview

9 NierenzellkarzinomeJochen Decker und Hiltrud Brauch
Nicht-hereditäre TumorerkrankungenD. Ganten / K. Ruckpaul (Hrsg.)© Springer-Verlag Berlin Heidelberg 2002
Inhaltsverzeichnis
9.1 Einführung, geschichtlicher Abriss . . . . . . 257
9.2 Krankheitsbild . . . . . . . . . . . . . . . . . . 2589.2.1 Epidemiologie . . . . . . . . . . . . . . . . . . . 2589.2.2 Klinische Symptome . . . . . . . . . . . . . . . 2589.2.3 Histopathologische Klassifikation
und Staging . . . . . . . . . . . . . . . . . . . . 2599.2.3.1 Klarzelliges Nierenzellkarzinom . . . . . . . . 2609.2.3.2 Papilläres (chromophiles) Nierenzellkarzinom 2609.2.3.3 Chromophobes Nierenzellkarzinom . . . . . . 2619.2.3.4 Sammelgangkarzinome (Ductus-Bellini-
Karzinome) . . . . . . . . . . . . . . . . . . . . . 2619.2.3.5 Nichtklassifizierbare Nierenzellkarzinome . . 2619.2.4 Ätiologie und Risikofaktoren . . . . . . . . . . 2619.2.4.1 Umweltbedingte Faktoren . . . . . . . . . . . . 2619.2.4.2 Erbliche Faktoren – genetische
Prädispositionen . . . . . . . . . . . . . . . . . 262
9.3 Molekularbiologische Grundlagen . . . . . . 2669.3.1 Genetik der klarzelligen Nierenzell-
karzinome . . . . . . . . . . . . . . . . . . . . . 2669.3.1.1 Balancierte chromosomale Translokationen
bei Familien mit HCCRCC . . . . . . . . . . . 2669.3.1.2 Verlust der Heterozygotie (loss of
heterozygosity, LOH) bei sporadischenNierenzellkarzinomen als Hinweis auf einTumorsuppressorgen . . . . . . . . . . . . . . . 267
9.3.1.3 vhl-Tumorsuppressorgen . . . . . . . . . . . . 267
9.3.1.4 Konstitutionelle Risikofaktorenfür das sporadische Nierenzellkarzinom . . . 277
9.3.2 Genetik des papillären Nierenzellkarzinoms . 2789.3.2.1 Rolle des met-Protoonkogens . . . . . . . . . . 2799.3.3 Andere Nierenzellkarzinomgene . . . . . . . . 2799.3.3.1 tsc1- und tsc2-Gene . . . . . . . . . . . . . . . . 2799.3.4 Sonstige molekulare Faktoren
und Mechanismen . . . . . . . . . . . . . . . . . 280
9.4 Diagnostik des Nierenzellkarzinoms . . . . . 2809.4.1 Klinische Präsentation . . . . . . . . . . . . . . 2809.4.2 Labordiagnostik und molekularpathologische
Untersuchungen . . . . . . . . . . . . . . . . . . 2809.4.3 Bildgebende Verfahren . . . . . . . . . . . . . . 2809.4.4 Invasive diagnostische Verfahren . . . . . . . . 281
9.5 Therapie des Nierenzellkarzinoms . . . . . . . 2819.5.1 Tumornephrektomie . . . . . . . . . . . . . . . . 2819.5.2 Strahlentherapie . . . . . . . . . . . . . . . . . . 2819.5.3 Systemtherapie beim metastasierten
Nierenzellkarzinom . . . . . . . . . . . . . . . . 2829.5.3.1 Immunmodulation . . . . . . . . . . . . . . . . . 2829.5.3.2 Chemotherapie . . . . . . . . . . . . . . . . . . . 2829.5.3.3 Sonstige Substanzen . . . . . . . . . . . . . . . . 2829.5.3.4 Experimentelle Ansätze . . . . . . . . . . . . . . 282
9.6 Hilfreiche Internetadressen . . . . . . . . . . . 283
9.7 Literatur . . . . . . . . . . . . . . . . . . . . . . . 283
9.1 Einführung, geschichtlicher Abriss
Das Nierenzellkarzinom wurde bereits im frühen19. Jahrhundert beschrieben und sein Ursprungdem tubulären Epithel zugeordnet. Zunächst setztesich, aufbauend auf den Arbeiten von Grawitz(1883), die Lehrmeinung durch, dass das Nieren-zellkarzinom aus ektopen adrenalen Zellen im Nie-rengewebe abzuleiten sei, da sich die Karzinomzel-len in ihrem Fettgehalt nicht von Zellen der Ne-benniere unterscheiden (Grawitz 1883). In der älte-ren Literatur wird daher das Nierenzellkarzinomauch als Grawitz-Tumor oder hypernephroidesKarzinom bezeichnet (Grawitz 1883). Auch der Be-griff des Hypernephroms wurde in diesem Zusam-
menhang geprägt und findet sich fortan zwar kon-zeptionell falsch, aber dennoch nachhaltig auchheute noch in Lehrbüchern.
Bei den Nierenzellkarzinomen handelt es sichum eine heterogene Gruppe von Tumoren, derenBeschreibung anhand morphologischer und zyto-logischer Kriterien vorgenommen wurde (Mostofi1981). Über viele Jahre hinweg wurde diese Eintei-lung recht uneinheitlich angewendet. In den 80erJahren wurde ein Klassifikationssystem erarbeitet,das unter Berücksichtigung elektronenmikroskopi-scher und immunhistologischer Befunde eine Ein-teilung in Subtypen und die histogenetische Zu-ordnung ermöglicht (Thoenes et al. 1986). Jetztkonnte bestätigt werden, dass die Haupttypen,

• das klarzellige Nierenzellkarzinom und• das papilläre (chromophile) Nierenzellkarzinom,
aus dem proximalen Tubulusepithel hervorgehen(Thoenes et al. 1986). Ein heute international emp-fohlenes und anerkanntes Klassifikationssystembasiert auf dieser histogenetischen Einteilung undwird durch aktuelle genetische und molekularge-netische Befunde ergänzt und gestützt (Störkel etal. 1997).
Zum Verständnis der Entstehung von Nierenzell-karzinomen sind detaillierte molekulare und patho-logische Grundlagen erforderlich. In den folgendenKapiteln werden daher die Zusammenhänge zwi-schen den klinischen Erscheinungsbildern des Nie-renzellkarzinoms und den zugrunde liegendenmolekularen Pathomechanismen dargestellt. DieKenntnis dieser Zusammenhänge lässt im Rahmendes Krankheitsmanagements bereits Anwendungenin der molekularen Diagnostik erkennen, aber auchim Bereich der Therapie sind erste Möglichkeitenhin zu innovativen Ansätzen möglich.
9.2 Krankheitsbild
9.2.1 Epidemiologie
Das Nierenzellkarzinom ist das häufigste Malig-nom der Niere. Von allen durch Krebs verursach-ten Todesfällen steht es in Deutschland im Westenan 8. (Anteil 3,3%) und im Osten an 6. Stelle (An-teil 4,3%) (Becker u. Wahrendorf 1998). Die Zahlder jährlichen Neuerkrankungen wird für Gesamt-deutschland auf >10000 Fälle geschätzt (5700Männer und 4500 Frauen) (Arbeitsgemeinschaftbevölkerungsbezogener Krebsregister in Deutsch-land 1997). Im internationalen Vergleich liegt dieInzidenzrate des Nierenzellkarzinoms in Deutsch-land (Männer 11:100 000; Frauen 8:100 000) höherals in den USA (Männer 10:100 000; Frauen5:100 000), sie ist jedoch niedriger als in der be-nachbarten Niederrheinregion Frankreichs (Män-ner 15:100000; Frauen 6:100 000) (Boring et al.1994, McLaughlin et al. 1995, Parkin u. Muir1992). In industrialisierten Ländern einschließlichDeutschland wird eine 2- bis 3%ige jährliche Zu-nahme der Nierenzellkarzinominzidenz vermutet(Motzer et al. 1996).
Nierenzellkarzinome treten in der Regel solitärim Alter zwischen 50 und 70 Jahren auf, der Mor-biditätsgipfel liegt um das 65. Lebensjahr. In selte-nen Fällen wurde ein Nierenzellkarzinom aber
auch schon im Kleinkindalter beschrieben (Line-han et al. 1996).
9.2.2 Klinische Symptome
Die klinischen Symptome des Nierenzellkarzinomssind initial nicht spezifisch und häufig erst imfortgeschrittenen Stadium richtungsweisend. Ins-besondere wird die rechtzeitige Diagnose dadurcherschwert, dass sehr häufig nichturologischeSymptome andere Leiden wie z.B. Systemerkran-kungen vortäuschen. In diesem Zusammenhangwurde der Begriff des „urologischen Chamäleons“geprägt. Dieses facettenreiche Bild ist die Folge pa-raneoplastischer Syndrome, die anhand serologi-scher Marker (Tabelle 9.1) erfasst werden können.Spezifische Hinweise sind die folgenden 3, als„klassische Trias“ bekannten Symptome:1. schmerzlose, plötzlich einsetzende Hämaturie,2. ein tastbarer Tumor im Bereich der Nieren und3. ein Flankenschmerz.
Darüber hinaus können auch subfebrile Temperatu-ren, Gewichtsabnahme, erhöhte Senkungsgeschwin-digkeit der Blutkörperchen, Nachtschweiß und Anä-mie sowie in 10% der Fälle auch Polyglobulie auftre-ten. Fakultativ und meistens als Spätsymptom fin-den sich der Einbruch in die V. renalis und in dasLymphsystem mit metastatischer Streuung bevor-zugt in Lunge, Leber, Gehirn und Knochen. DieHäufigkeit der Metastasierung zum Zeitpunkt derDiagnose liegt bei 25–35%. Die vermehrte Anwen-
258 J. Decker und H. Brauch
Tabelle 9.1. Paraneoplastische Symptome beim Nierenzell-karzinom durch ektope Bildung von Hormonen, Hormonde-rivaten oder ähnlichen endogenen Faktoren
Ektope Hormoneoder ähnliche Faktoren
Systemische Symptome
Erythropoetin Sekundäre Polyzythämiea
Parathormonund ähnliche Substanzen
Hyperkalzämie
Prostaglandin ARenin HypertonieProstaglandine(vasodilatierend)
Hypotonie
Prolaktin GalaktorrhöGonadotropin (HCG) Gynäkomastie, LibidoverlustAdrenokortikotropesHormon (ACTH)
Cushing-Syndrom
Eine sekundäre Polyzythämie kann auch bei Hirnmetastasenbedingt durch ein Nierenzellkarzinom und beim Hämangio-blastom des Kleinhirns auftreten. Beim Von-Hippel-Lindau-Syndrom kann dies ein differenzialdiagnostisches Problemdarstellen.

dung technisch kontinuierlich verbesserter, moder-ner bildgebender Verfahren hat dazu geführt, dassNierenzellkarzinome heute oft als Zufallsbefund be-reits präsymptomatisch diagnostiziert werden.
9.2.3 Histopathologische Klassifikationund Staging
Das Nierenzellkarzinom stellt keine einheitliche Tu-morentität dar. Vielmehr verbirgt sich unter diesemBegriff eine heterogene Gruppe von Karzinomenmit charakteristischen histopathologischen und ge-netischen Eigenschaften. Die aktuelle Klassifikationwurde durch die Kenntnis und Verknüpfung mor-phologischer und genetischer Merkmale wesentlichbeeinflusst und stetig geändert. Granuläre oder sar-komatoide Karzinome stellen keine spezifischenEntitäten mehr dar, da Karzinome mit granulärenoder Spindelzellmerkmalen verschiedentlich in Tu-mortypen vorkommen können. Die Einteilung inunterschiedliche Entitäten basiert auf den Ergeb-nissen klassischer pathologischer Untersuchungs-techniken wie Hämatoxilin-Eosin(HE)-Färbung,
Histo- und Immunhistochemie sowie Elektronen-mikroskopie. In Verbindung mit zytogenetischenund molekulargenetischen Merkmalen setzt sichgemäß den Empfehlungen der Union InternationaleContre le Cancer (UICC) und des American JointCommittee on Cancer (AJCC) international immermehr die Einteilung in die Grundtypen• klarzellige Nierenzellkarzinome,• papilläre (chromophile) Nierenzellkarzinome,• chromophobe Nierenzellkarzinome,• Sammelgangkarzinome (Ductus-Bellini-Karzino-
me) und• nichtklassifizierbare Nierenzellkarzinome
durch (Störkel et al. 1997). Als Unterscheidungs-kriterien werden zytologische Merkmale, Wachs-tumsmuster, Grading sowie histopathogenetischeund molekulargenetische Parameter herangezogen(Störkel et al. 1997, Thoenes et al. 1986, 1990).Histogenetisch werden die klarzelligen und papil-lären (chromophilen) Nierenzellkarzinome demproximalen Tubulus, die chromophoben und Sam-melgangkarzinome dem Sammelrohrsystem zuge-ordnet (Abb. 9.1).
a 9 Nierenzellkarzinome 259
Abb. 9.1. Schematische Darstellung der Grundformen epithe-lialer Nierentumoren (Thoenes et al. 1986, 1990). Klarzelligeund papilläre (chromophile) Nierenzellkarzinome entstehenim proximalen, chromophobe Nierenzellkarzinome und On-kozytome im distalen Tubulus. Als erbliche Erkrankung tre-ten klarzellige Nierenzellkarzinome im Rahmen des Von-Hippel-Lindau-Syndroms (VHL), papilläre Nierenzellkarzi-nome beim hereditären papillären Nierenzellkarzinom(HPRCC) und chromophobe Nierenzellkarzinome sowie On-kozytome im Rahmen des Birt-Hogg-Dubé-Syndroms (BHD)auf. Typische genetische Veränderungen klarzelliger Nieren-zellkarzinome sind der Verlust eines Chromosom-3p-Allels
und Alterationen des vhl-Gens. vhl-Keimbahnmutationenprädisponieren für die VHL-Erkrankung, somatische vhl-Al-terationen treten bei etwa 45% der sporadischen Nierenzell-karzinome auf. Papilläre Nierenzellkarzinome sind durchTrisomien 7 und 17 sowie den Verlust eines Y- oder X-Chro-mosoms gekennzeichnet. Keimbahnmutationen im met-Pro-toonkogen kommen bei 86% der HPRCC vor, somatischemet-Mutationen treten aber nur bei etwa 3% der sporadi-schen Fälle auf. Chromophobe Nierenzellkarzinome sinddurch Monosomien der Chromosomen 1, 2, 6, 10, 13, 17und 21 gekennzeichnet
Das Staging kann nach dem Robson-System(Robson et al. 1969) und dem von der UICC emp-fohlenen, kürzlich überarbeiteten TNM-System(Fleming 1998) erfolgen. Entsprechend der TNM-Klassifikation wurde die Grenze der Größe vonT1-Tumoren von <2,5 cm auf ≥7 cm angehoben(Russo 2000).
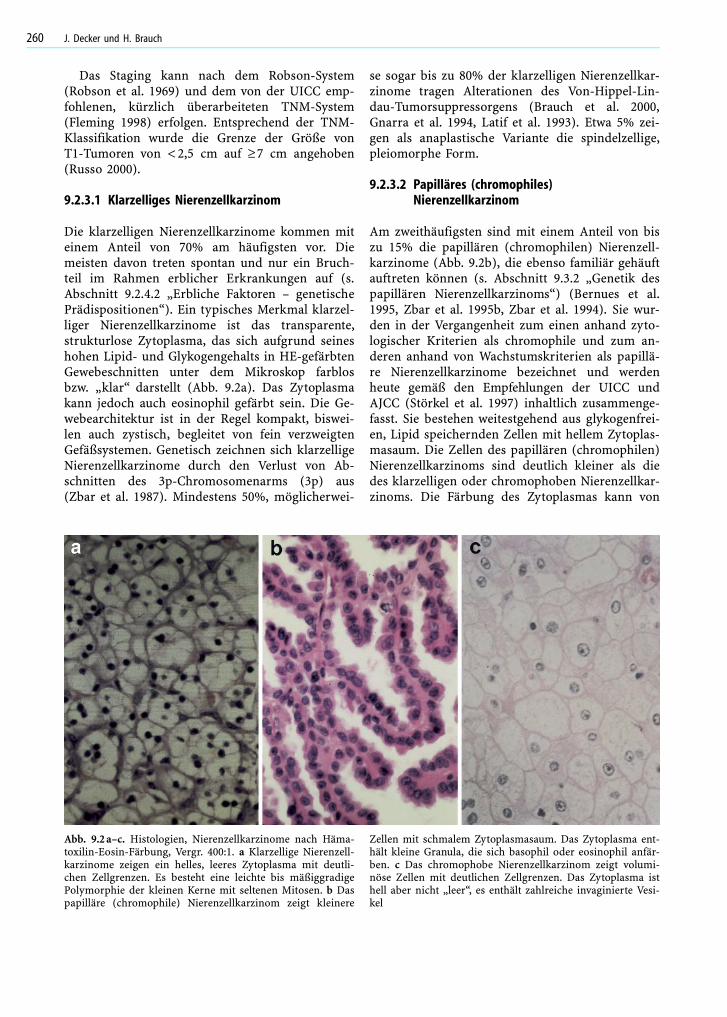
9.2.3.1 Klarzelliges Nierenzellkarzinom
Die klarzelligen Nierenzellkarzinome kommen miteinem Anteil von 70% am häufigsten vor. Diemeisten davon treten spontan und nur ein Bruch-teil im Rahmen erblicher Erkrankungen auf (s.Abschnitt 9.2.4.2 „Erbliche Faktoren – genetischePrädispositionen“). Ein typisches Merkmal klarzel-liger Nierenzellkarzinome ist das transparente,strukturlose Zytoplasma, das sich aufgrund seineshohen Lipid- und Glykogengehalts in HE-gefärbtenGewebeschnitten unter dem Mikroskop farblosbzw. „klar“ darstellt (Abb. 9.2a). Das Zytoplasmakann jedoch auch eosinophil gefärbt sein. Die Ge-webearchitektur ist in der Regel kompakt, biswei-len auch zystisch, begleitet von fein verzweigtenGefäßsystemen. Genetisch zeichnen sich klarzelligeNierenzellkarzinome durch den Verlust von Ab-schnitten des 3p-Chromosomenarms (3p) aus(Zbar et al. 1987). Mindestens 50%, möglicherwei-
se sogar bis zu 80% der klarzelligen Nierenzellkar-zinome tragen Alterationen des Von-Hippel-Lin-dau-Tumorsuppressorgens (Brauch et al. 2000,Gnarra et al. 1994, Latif et al. 1993). Etwa 5% zei-gen als anaplastische Variante die spindelzellige,pleiomorphe Form.
9.2.3.2 Papilläres (chromophiles)Nierenzellkarzinom
Am zweithäufigsten sind mit einem Anteil von biszu 15% die papillären (chromophilen) Nierenzell-karzinome (Abb. 9.2b), die ebenso familiär gehäuftauftreten können (s. Abschnitt 9.3.2 „Genetik despapillären Nierenzellkarzinoms“) (Bernues et al.1995, Zbar et al. 1995b, Zbar et al. 1994). Sie wur-den in der Vergangenheit zum einen anhand zyto-logischer Kriterien als chromophile und zum an-deren anhand von Wachstumskriterien als papillä-re Nierenzellkarzinome bezeichnet und werdenheute gemäß den Empfehlungen der UICC undAJCC (Störkel et al. 1997) inhaltlich zusammenge-fasst. Sie bestehen weitestgehend aus glykogenfrei-en, Lipid speichernden Zellen mit hellem Zytoplas-masaum. Die Zellen des papillären (chromophilen)Nierenzellkarzinoms sind deutlich kleiner als diedes klarzelligen oder chromophoben Nierenzellkar-zinoms. Die Färbung des Zytoplasmas kann von
260 J. Decker und H. Brauch
Abb. 9.2a–c. Histologien, Nierenzellkarzinome nach Häma-toxilin-Eosin-Färbung, Vergr. 400:1. a Klarzellige Nierenzell-karzinome zeigen ein helles, leeres Zytoplasma mit deutli-chen Zellgrenzen. Es besteht eine leichte bis mäßiggradigePolymorphie der kleinen Kerne mit seltenen Mitosen. b Daspapilläre (chromophile) Nierenzellkarzinom zeigt kleinere
Zellen mit schmalem Zytoplasmasaum. Das Zytoplasma ent-hält kleine Granula, die sich basophil oder eosinophil anfär-ben. c Das chromophobe Nierenzellkarzinom zeigt volumi-nöse Zellen mit deutlichen Zellgrenzen. Das Zytoplasma isthell aber nicht „leer“, es enthält zahlreiche invaginierte Vesi-kel
a

eosinophil bis basophil variieren. Die Gewebear-chitektur zeichnet sich durch ein tubulopapilläresWachstum aus und ist bei den meisten Karzino-men mindestens fokal ausgeprägt. Auf der Basismorphologischer und immunhistochemischer Cha-rakteristika werden papilläre Nierenzellkarzinomein 2 Typen unterteilt (Delahunt u. Eble 1997):• Typ 1 zeichnet sich durch Papillen mit 1–2 La-
gen epithelialer Zellen aus, deren Zytoplasmaklar bzw. hell ist;
• Typ 2 ist durch Papillen mit irregulären Schich-ten epithelialer Zellen gekennzeichnet, die reichan eosinophilem Zytoplasma sind.
Typ-1-Tumoren treten häufiger als Typ-2-Tumorenassoziiert mit multiplen, mikroskopisch kleinenpapillären Tumoren auf (Zbar u. Lerman 1998).
9.2.3.3 Chromophobes Nierenzellkarzinom
Die chromophoben Nierenzellkarzinome stehen an3. Stelle der Häufigkeiten mit einem Anteil von biszu 5%. Sie haben wie die klarzelligen Nierenzell-karzinome ein blasses bzw. eosinophiles Zytoplas-ma von variabler Größe (Abb. 9.2c), das sich mitHales kolloidaler Eisenfärbung blau anfärben lässtund viele Mikrovesikel enthält. Die Gewebearchi-tektur zeigt in der Regel kompaktes Wachstum mitZellen variabler Größe, wovon die größten entlangkleiner Blutgefäße liegen. Genetisch zeichnen sichdie chromophoben Nierenzellkarzinome durch Hy-podiploidie, insbesondere durch die Monosomiender Chromosomen 1, 2, 6, 10, 13, 17 und 21 aus(Bugert et al. 1997).
9.2.3.4 Sammelgangkarzinome(Ductus-Bellini-Karzinome)
Ductus-Bellini-Karzinome sind mit einem Anteil<1% sehr selten. Sie zeichnen sich durch verschie-dene Erscheinungsformen aus. In der Regel ist dieMorphologie von irregulären Kanälen mit unregel-mäßiger Epithelauskleidung mit „Hufnagel“(hobnail)-Erscheinung gekennzeichnet, die vonentzündetem desmoplastischem Stroma umgebensind (Abb. 9.1d). Ductus-Bellini-Karzinome sindso selten, dass es bis heute keine einheitlichen Be-funde über genetische Aberrationen gibt.
9.2.3.5 Nichtklassifizierbare Nierenzellkarzinome
Für den Fall, dass Nierenzellkarzinome keinem dergenannten Subtypen zugeordnet werden können,gelten sie als nichtklassifizierbar. Merkmale sind
• gemischte Zelltypen,• sarkomatoide Zellen ohne erkennbare epithelia-
len Anteile,• Muzinproduktion,• Mischungen aus Epithel- und Stromazellen so-
wie• nicht zuzuordnende Zelltypen.
Obwohl sich die sarkomatoiden Veränderungen inallen Grundtypen der Nierenzellkarzinome finden,gibt es keine Hinweise darauf, dass es sich hierbeium eine eigenständige Klasse handelt. Es wirdeher davon ausgegangen, dass sie hochgradigeKarzinome der jeweiligen Grundtypen darstellen.Für den Fall, dass ein Grundzelltyp nicht mehr er-kennbar ist, werden sarkomatoide Nierenzellkarzi-nome als nichtklassifizierbar bezeichnet.
9.2.4 Ätiologie und Risikofaktoren
9.2.4.1 Umweltbedingte Faktoren
Als ätiologische Faktoren des Nierenzellkarzinomswerden umweltbedingte Faktoren einschließlichder Wirkung hormoneller, zellulärer und geneti-scher Ursachen diskutiert und untersucht. Obwohldie Ursachen noch nicht genau verstanden werden,gibt es immer mehr Hinweise dafür, dass wahr-scheinlich berufliche Expositionen Risikofaktorendarstellen. Die International Agency for Researchon Cancer (IARC) zieht ein Risiko für Beschäftigtein der Koks-, Eisen- und Stahlindustrie in Betracht(IARC 1987). Eine internationale, multizentrischeFall-Kontroll-Studie ermittelte ein Nierenzellkarzi-nomrisiko für Personen mit beruflicher Expositiongegenüber Asbest, Schwermetallverbindungen wieBlei und Kadmium, Lösungsmitteln, Treibstoffenund anderen Petroleumprodukten (Mandel et al.1995). Speziell in Deutschland wurde von1991–1995 in einer bevölkerungsbezogenen Fall-Kontroll-Studie ein Risiko für Personen mit star-ken Expositionen gegenüber organischen Lösungs-mitteln, Kadmium- und Bleiverbindungen sowieLötdämpfen ermittelt (Pesch et al. 2000). Dabeiwurden als Risikofaktoren berufliche Expositionengegenüber Farben, Mineralölen, Kühlschmierstof-fen, Benzin, polyzyklischen aromatischen Kohlen-wasserstoffen und Asbest bestätigt. Zu den poten-tiell gefährdeten Personen gehören Beschäftigte inden Bereichen Groß- und Einzelhandel, Reinigungund Textilreinigung, Elektronikgerätemontage,Gummi- und Plastikherstellung sowie Gleisarbei-ter, Maler, Färber, Gerber und Personen mit ähnli-
a 9 Nierenzellkarzinome 261

chen Aufgaben. Auch Lebensstilfaktoren wie Rau-chen und Übergewicht, Letzteres besonders beiFrauen, gelten als Risikofaktoren (McLaughlin etal. 1995, Mellemgaard et al. 1995). Vorerkrankun-gen der Niere werden mit unterschiedlichem Risi-ko bewertet. Während Patienten mit Niereninfek-tionen und Diabetes ein nur leicht erhöhtes Risikofür ein Nierenzellkarzinom haben (Schlehofer etal. 1996), ist das Risiko bei Patienten mit termina-ler Nierenerkrankung höher. So haben z.B. Patien-ten mit ACKD (acquired cystic kidney disease)gegenüber der Normalbevölkerung ein 30facherhöhtes Risiko, besonders dann, wenn eine chro-nische Dialyse notwendig geworden war (Chung-Park et al. 1989, Matson u. Cohen 1990). Ein ver-mutetes Risiko bei regelmäßiger Einnahme phena-zetinhaltiger und anderer Analgetika (Parazetamol,Salizylate und Pyrazolonderivate) konnte entwedernicht bestätigt oder nur bei sehr hohen Dosen auf-gezeigt werden (McCredie et al. 1995).
9.2.4.2 Erbliche Faktoren –genetische Prädispositionen
Ungefähr 4% aller Nierenzellkarzinome treten in-folge einer genetischen Prädisposition auf (Line-han et al. 1995). Dazu gehören Nierenzellkarzino-me, die im Rahmen des hereditären Von-Hippel-Lindau(VHL)-Syndroms (Linehan et al. 1995, Mel-mon u. Rosen 1964, Neumann 1987b), dem heredi-tären klarzelligen Nierenzellkarzinom (Cohen et al.1979, Li et al. 1993) und dem hereditären papillä-ren Nierenzellkarzinom HPRCC (Zbar et al. 1994,1995b) auftreten (Tabelle 9.2).
Erbliche Nierenzellkarzinome treten charakte-ristischerweise multifokal und bilateral auf. Dabeikönnen in einer Niere Läsionen verschiedener Sta-dien auftreten, die von der mikroskopischen dys-plastischen Läsion bis hin zu Karzinomen mit ei-
nem Durchmesser von 15 cm reichen können. DieAnzahl der pro Niere vorkommenden Tumorenliegt in der Regel bei 1–2, kann aber auch bis >50Tumoren betragen. Einer der stärksten Risikofak-toren für das erbliche Nierenzellkarzinom ist einepositive Familiengeschichte für die in Tabelle 9.2aufgeführten Erkrankungen. Die damit assoziier-ten Nierenzellkarzinome werden jeweils auto-somal-dominant vererbt. Wie viele und welcheGenträger letztendlich erkranken, hängt vom je-weiligen Gen und der spezifischen, der Krankheitzugrunde liegenden Mutation ab. Die erblichenNierenzellkarzinome können anhand ihrer Histolo-gie und dem Fehlen bzw. Auftreten begleitender,unabhängiger Tumoren unterschieden werden. Diemit dem HCCRCC (hereditären klarzelligen Nie-renzellkarzinom) und dem VHL-Syndrom assozi-ierten Nierenzellkarzinome gehören dem klarzelli-gen, die Nierenzellkarzinome des HPRCC dem pa-pillären Grundtyp an. Die VHL-Erkrankung wirdauf der Basis der Ausprägung unabhängiger Tumo-ren in anderen Organsystemen zu den Syndromengerechnet.
Das Auftreten eines einzelnen Nierenzellkarzi-noms bei Verwandten 1. und 2. Grads scheint keinausreichendes Indiz für ein erhöhtes Risiko zu sein(Schlehofer et al. 1996), vielmehr weist ein jungesErkrankungsalter auf eine mögliche genetischePrädisposition hin.
Trotz des niedrigen prozentualen Anteils neh-men die erblich bedingten Nierenzellkarzinome ei-ne übergeordnete Bedeutung für das Verständnisder Genese des Nierenzellkarzinoms ein. DieGründe dafür liegen in der erfolgreichen Identifi-kation der ursächlichen Gene und Gendefekte, dieheute für alle 3 erblich bedingten Formen desNierenzellkarzinoms bekannt sind. Sie werden imAbschnitt 9.3 „Molekularbiologische Grundlagen“entsprechend ihrer Entdeckungsgeschichte chrono-logisch dargestellt. Dabei kommt dem VHL-Syn-drom und dem nach ihm benannten vhl-Tumor-suppressorgen eine besondere Rolle zu, da das vhl-Gen auch bei der Entstehung sporadischer Nieren-zellkarzinome eine entscheidende Rolle spielt.
In selteneren Fällen kommen Nierenzellkarzino-me auch im Rahmen der tuberösen Sklerose vor(Jones et al. 1997).
Von-Hippel-Lindau-Syndrom. Das VHL-Syndrom isteine autosomal-dominant vererbbare Erkrankung,die mit einer Häufigkeit von etwa 1:45000 vor-kommt. Es handelt sich dabei um ein Syndrom, imRahmen dessen Genträger Tumoren in verschiede-nen Organsystemen entwickeln können. Die be-
262 J. Decker und H. Brauch
Tabelle 9.2. Bekannte hereditäre Formen des Nierenzellkarzi-noms
Erkrankungen Mutationen
Hereditäres klarzelligesNierenzellkarzinom (HCCRCC)
Translokation 3;8
Von-Hippel-Lindau-Syndrom(VHL)
vhl-Mutationen
Hereditäres papilläresNierenzellkarzinom (HPRCC)
met-Mutationen
Tuberöse Sklerose Möglicherweisetsc1/tsc2
Birt-Hogg-Dubé-Syndrom(BHD)
Unbekannt

troffenen Organe sind außer den Nieren auch Au-gen, ZNS, Nebennieren, Pankreas, Innenohr undNebenhoden. Das VHL-Syndrom wurde zuerst alsAngiomatosis retinae bei Zwillingen entdeckt (Col-lins 1894) und später als familiäre Erkrankung er-kannt (von Hippel 1904). Nachdem auch die his-tomorphologische Beziehung zu Hämangioblasto-men des ZNS (Lindau-Tumoren) und die gemein-same Vererbung der Augen- und ZNS-Läsionen er-kannt waren (Lindau 1926), wurden in der Folge-zeit immer mehr Tumoren diesem Formenkreiszugeschrieben. Die Benennung als VHL-Syndromnach den Entdeckern von Hippel und Lindau so-wie eine Evaluation der Literatur und Beschrei-bung einer großen Familie wurden jedoch erst1964 von Melmon u. Rosen vorgenommen. Die Pe-netranz des VHL-Syndroms liegt im Alter von 65Jahren bei 90%.
Die mit dem VHL assoziierten Tumoren (Tabel-le 9.3) treten mit unterschiedlichen Häufigkeitenin scheinbar beliebigen Kombinationen und vari-ablem Erkrankungsalter auf. Oft beginnt die Er-krankung mit einem retinalen Angiom, wobei dasmittlere Erkrankungsalter bei 25 Jahren liegt. ZNS-Tumoren treten im Mittel im Alter von 30 Jahrenund Nierenzellkarzinome in der Regel erst am En-de der 3. Lebensdekade auf. Die belasteten Famili-en unterscheiden sich durch die in ihren Familienauftretenden Tumoren (Chen et al. 1995b, Glenn etal. 1991, Neumann u. Wiestler 1991). Zur besserensystematischen Beschreibung wird die VHL-Er-krankung in Typen eingeteilt, wobei das Unter-scheidungskriterium das Auftreten oder Fehlenvon Phäochromozytomen ist:
1. VHL Typ 1Zum VHL Typ 1 gehören alle VHL-Familien, diekeine Phäochromozytome, aber alle anderen Tu-moren einschließlich Nierenzellkarzinome aus-prägen können (Glenn et al. 1991, Neumann u.Wiestler 1991);
2. VHL Typ 2Bei VHL-Typ-2-Familien treten Phäochromozy-tome und potentiell auch alle anderen Läsionenauf. Es werden unterschieden:– VHL Typ 2A (Brauch et al. 1995),– VHL Typ 2B (Brauch et al. 1995, Chen et al.
1995b) und– VHL Typ 2C.
Phäochromozytome treten bei den VHL-Typ-2-Fa-milien häufig als erste Manifestation auf.
Manifestationen in der Niere. Genträger für dasVHL-Syndrom können in den Nieren sowohlbenigne Zysten als auch Nierenzellkarzinome ent-wickeln. Etwa 76% der Patienten entwickeln Zystenund etwa 24–28% Karzinome. Bei Zysten kann dieZystenwand mit Karzinomzellen ausgekleidet sein.Obwohl diese Tumoren oft schon klein und imStadium der Begrenzung auf die Niere entdecktwerden, handelt es sich um potentiell maligne undmöglicherweise schon um metastasierende Tumo-ren. Eine Untersuchung des National Cancer Insti-tute zeigt, dass die malignen und potentiell mali-gnen Läsionen primär dem klarzelligen Grundzell-typ der Nierenzellkarzinome zuzuordnen sind(Poston et al. 1995). Wichtige und charakteristi-sche Merkmale der renalen VHL-Läsionen sind ih-re Multiplizität (Abb. 9.3) und das bilaterale Auf-treten. Die histologische Aufarbeitung und Extra-polation der Anzahl identifizierter Läsionen erga-ben bei Patienten im mittleren Alter von 37 Jahreneinen Schätzwert von ungefähr 1100 nicht malig-nen Zysten mit klarzelliger Auskleidung und 600Nierenzellkarzinomen (Walther et al. 1995). Dieszeigt deutlich, dass alle Nierenläsionen bei VHL-Patienten als potentiell maligne eingestuft werdenmüssen. Für die Praxis gilt gemäß des ConsensusReports (4th International Symposium on von Hip-pel-Lindau Disease 2000, Mayo Clinic, Rochester,MN) die 3-cm-Regel als Grenzwert der Einstufungder Malignität. Für Tumoren mit kleinerem Durch-messer beträgt das Metastasierungsrisiko <1%. Dadieses Risiko von Familie zu Familie variiert, soll-ten die familienspezifischen Nierenzellkarzinom-charakteristika mit in die Bewertung eingehen.
a 9 Nierenzellkarzinome 263
Tabelle 9.3. Manifestationen beim VHL-Syndrom, Zusam-menstellung aus der Literatur (Neumann 1987, Maher 1990u.a.)
Organ Tumoren %
Auge Angiomatosis retinae 50–57Zentrales Nerven-system
Zerebelläre Hämangio-blastome
55–59
Spinale Hämangio-blastome
13–14
Nebenniere Phäochromozytome 7–19Niere Zysten 76
Klarzelliges Nieren-zellkarzinom
24–28
Bauchspeicheldrüse ZystenInselzellkarzinome
22Selten
Innenohr EndolymphatischeZystadenome
<10
Nebenhoden Zystadenome 10–15Leber (und andere pa-renchymatöse Organe)
Zysten Gelegent-lich

Andere häufige LäsionenAngiomatosis retinae. Sie gehört zu den häufigstenTumoren des VHL-Syndroms und tritt bei 50–57%der Patienten oft als erster Tumor auf (Glenn et al.1990, Lamiell et al. 1989, Maher et al. 1990, Neu-mann 1987a). Obwohl die retinalen Angiome keinemalignen Tumoren darstellen, besteht für den Pa-tienten die Gefahr der Netzhautablösung und derErblindung, was sich heute aber durch die recht-zeitige Erkennung und eine Laserbehandlung ver-meiden lässt.
Hämangioblastome des zentralen Nervensystems. Sietreten in bis zu 70% der von VHL betroffenen Pa-tienten auf (Glenn et al. 1990, Lamiell et al. 1989,Maher et al. 1990, Neumann 1987a). Es handeltsich ebenfalls um benigne Tumoren, die jedoch er-hebliche Komplikationen auslösen und zum Todführen können.
Phäochromozytome. Sie kommen bei etwa 7–19%der VHL-Patienten vor (Glenn et al. 1990, Lamiellet al. 1989, Neumann 1987a) und treten „nur“ inbestimmten Familien auf. Sie werden daher alsUnterscheidungskriterium für die Zuordnung zuden VHL-Phänotypen herangezogen.
Pankreasläsionen. Sie kommen als Zysten in etwa22% der Fälle vor, seltener sind seröse Zystadeno-me und Inselzelltumoren (Green et al. 1986, Neu-mann et al. 1991).
Papilläre Zystadenome der Epididymis und des gro-ßen Ligaments. Papilläre Zystadenome der Neben-hoden treten bei 10–15% der VHL-erkranktenMänner auf. Es handelt sich dabei ausschließlichum gutartige Tumoren. Die histologisch ähnliche,analoge Läsion bei Frauen kann im embryologischverwandten Gewebe des großen Ligaments auftre-ten (Manski et al. 1997).
Tumoren des Innenohrs (endolymphatic sac tumors –ELST). Der endolymphatische Sack liegt am Endedes endolymphatischen Sackkanals zwischen Duraund Posterior fossa im Innenohr. ELST können lo-kal invasiv wachsen und zu Hörverlust und Fa-zialisparese führen (Manski et al. 1997).
Hereditäres papilläres Nierenzellkarzinom. Das here-ditäre papilläre Nierenzellkarzinom (HPRCC) warlange als eigenständige Nierenzellkarzinomerkran-kung unerkannt geblieben. Zwar wurden im Rah-men der Beschreibungen familiärer Nierenzellkar-zinome auch immer wieder Tumoren mit papillä-
264 J. Decker und H. Brauch
Abb. 9.3a, b. Hereditäres und sporadisches Nierenzellkarzi-nom. a Multiple und klarzellige Nierenzellkarzinome undZysten in einer Niere eines Patienten mit dem erblichen
Von-Hippel-Lindau-Syndrom; b solitäres, sporadisches klar-zelliges Nierenzellkarzinom (aus Murphy et al. 1994)
� �

rem Wachstum beschrieben, jedoch gehörten diePatienten innerhalb dieser Familien jeweils dergleichen Generation an, sodass unklar war, ob essich dabei um eine eigenständige erbliche Formdes Nierenzellkarzinoms handelte (Bernues et al.1995, Franksson et al. 1972, Pearson 1969). Erstnachdem Zbar et al. (1994) papilläre Nierenzellkar-zinome bei Familienangehörigen in jeweils 2 oder3 Generationen erkannten, war klar, dass dasHPRCC eine eigenständige Entität unter den here-ditären Nierenzellkarzinomen darstellt (Zbar et al.1994). HPRCC ist mit multiplen, bilateralen Tumo-ren mit histologisch papillärem Aussehen assozi-iert. Patienten belasteter Familien entwickeln zwarähnlich wie Patienten mit der VHL-Erkrankung inbeiden Nieren multiple Tumoren unterschiedlicherGrößen, jedoch gehören diese ausschließlich dempapillären Typ an und sind in der Regel gut diffe-renziert (Zbar et al. 1994). Das mittlere Diagnose-alter des hereditären, papillären Nierenzellkarzi-noms liegt bei 40 Jahren, der jüngste Patient war24 Jahre alt. Die Zeitspanne zwischen Diagnoseund Metastasierung ist unklar. Im Gegensatz zumVHL-Syndrom scheinen die Manifestationen desHPRCC auf die Niere begrenzt zu sein. Obwohl beiPatienten oder ihren Verwandten gelegentlich auchandere Tumoren wie z.B. ein Pankreas-, Mamma-,Magen-, Rektum-, Gallengang- oder Lungenkarzi-nom sowie ein Fibrosarkom oder ein malignes Me-lanom beobachtet wurden, ist die Anzahl der bis-her erfassten Familien zu klein, um eine abschlie-ßende Bewertung mit Hinblick auf die Ausprägunganderer Tumoren vorzunehmen (Schmidt et al.1997, Zbar 1995, Zbar et al. 1994). Jedoch scheintes mit Hinblick auf die Anzahl, die Uni- bzw. Bilate-ralität sowie den Differenzierungsgrad und die Zell-morphologie phänotypische Unterschiede bei denpapillären Nierenzellkarzinomen zu geben. Aus Un-tersuchungen des National Cancer Institute gehthervor, dass zwischen den Familien phänotypischeUnterschiede bei der Ausprägung der papillärenNierenzellkarzinome bestehen. Zum Beispiel zeigtendie erkrankten Mitglieder einer Familie im Gegen-satz zum typischen Bild der multiplen und bilatera-len Tumoren jeweils nur singuläre papilläre Nieren-zellkarzinome in einer Niere. In einer anderen Fa-milie waren die Tumoren schlecht differenziertund zeigten innerhalb des Tumors unterschiedlicheZellmorphologien (Zbar u. Lerman 1998).
Erfahrungsgemäß können HPRCC im Rahmender radiologischen Vorsorge bei asymptomatischenFamilienmitgliedern aufgrund ihrer hypovaskulä-ren Eigenschaften mit Zysten verwechselt werden(Press et al. 1984).
Nierenzellkarzinome bei der tuberösen Sklerose. Dietuberöse Sklerose (TSC) ist eine erbliche System-erkrankung mit stark variablem klinischem Bild,deren Erstbeschreibung schon vor 100 Jahren er-folgte (Bourneville 1880). Sie zeichnet sich durchdas gehäufte Auftreten von Hamartomen in ver-schiedenen Organen, vornehmlich Herz, Haut, Ge-hirn und Lunge, aus. Oft stehen die zerebralenManifestationen mit mentaler Retardierung undEpilepsieneigung im Vordergrund. Etwa 40–80%der Erkrankten entwickeln renale Läsionen, diesich überwiegend als Angiomyolipome (Jones etal. 1997), in seltenen Fällen (2–2,5%) aber auch alsNierenzellkarzinome manifestieren. Die Angabenzur Häufigkeit der Nierenzellkarzinome schwankenerheblich (Cook u. Lawless 1996, van Slegtenhorstet al. 1997, Washecka u. Hanna 1991), sie dürfte je-doch deutlich niedriger als beim VHL-Syndromsein (Sampson 1996). Dabei handelt es sich vor-wiegend um klarzellige Nierenzellkarzinome, diehäufig der spindelförmigen oder anaplastischenVariante angehören (Bjornsson et al. 1996). Mögli-cherweise unterscheiden sich die TSC-assoziiertenklarzelligen Nierenzellkarzinome in ihrer Genesevon anderen erblichen klarzelligen Nierenzellkarzi-nomen. TSC-assoziierte Nierenzellkarzinome ex-primieren nämlich das HMB-45-Antigen, das cha-rakteristischerweise von Melanozyten exprimiertwird und ein Marker für Melanome und Tumorender Neuralleiste ist. Da dieses Antigen auch beisporadischen Angiomyolipomen exprimiert wird,wird bei der TSC ein Zusammenhang zwischenAngiomyolipomen und Nierenzellkarzinomen ver-mutet (Bjornsson et al. 1996). Auch die im Rah-men der TSC vorkommenden erblichen Nierenzell-karzinome treten häufig bilateral und früher aufals die sporadischen Tumoren.
Andere erbliche NierenzellkarzinomeBirt-Hogg-Dubé-Syndrom. Das Birt-Hogg-Dubé-Syn-drom (BHD) ist eine autosomal-dominant vererb-bare Erkrankung mit Prädisposition für kleinerundliche, multiple Papeln im Bereich des Ge-sichts, des Halses und des Oberkörpers. Histolo-gisch sind diese Hautläsionen Fibrofollikulomen,Trichoadenomen und Akrochordonen zuzuordnen.Zusammen mit BHD können in belasteten Famili-en auch Kolonpolypen und renale Tumoren seg-regieren (Toro et al. 1999). Bei den renalen Tumo-ren handelt es sich überwiegend um Onkozytome,jedoch wurden bei einem Patienten auch bilateralechromophobe Adenokarzinome mit klaren und eo-sinophilen Zellen beobachtet (Roth et al. 1993). Ineiner anderen Familie trat eine Variante des papil-
a 9 Nierenzellkarzinome 265

lären Nierenzellkarzinoms auf (Toro et al. 1999).Somit besteht für BHD-Patienten und ihre An-gehörigen ein Risiko, an einem Nierenzellkarzinomzu erkranken.
Nicht-VHL-, Nicht-HPRCC- und Nicht-TSC-assoziierteNierenzellkarzinome. Erbliche Nierenzellkarzinomesind nicht immer den assoziierten bekannten fami-liären Erkrankungen zuzuordnen. Gelegentlich lie-gen nur schwache Hinweise auf eine erbliche Kom-ponente vor, und eine molekulargenetische Zuord-nung ist nicht möglich. Klarzellige Nierenzellkarzi-nome können z.B. singulär bei einem Elternteilund einem Kind auftreten. Gelegentlich könnenein singulärer Tumor bei einem Elternteil, aber bi-laterale Tumoren bei einem Kind auftreten (Zbaru. Lerman 1998). Stärkere Hinweise auf eine erb-liche Komponente auch bei kleinen Familien erga-ben sich aus der Beobachtung singulärer Nieren-zellkarzinome bei einem Vater und seinen 3 Kin-dern, die von 2 verschiedenen Müttern stammten(Zbar u. Lerman 1998). Schließlich wurden singu-läre klarzellige Nierenzellkarzinome auch in größe-ren Familien beobachtet: In einer Familie tratensie über 3 Generationen hinweg auf und in eineranderen Familie waren 5 Mitglieder in 2 Genera-tionen betroffen (Teh et al. 1997). Diese klarzelli-gen Nierenzellkarzinome haben wahrscheinlich ei-ne andere Genese als die VHL-, HPRCC- und TSC-assoziierten Nierenzellkarzinome. Dennoch sollhier auf eine epidemiologische Studie hingewiesenwerden, die zeigte, dass die meisten Nierenzellkar-zinome, die in kleinen Familien bei 2 Erstgradver-wandten auftreten, nicht erblich bedingt sind.Wahrscheinlich entstehen sie „zufällig“ und sindmöglicherweise durch gemeinsame Lebensgewohn-heiten zu erklären (Schlehofer et al. 1996).
9.3 Molekularbiologische Grundlagen
9.3.1 Genetik der klarzelligenNierenzellkarzinome
9.3.1.1 Balancierte chromosomale Translokationenbei Familien mit HCCRCC
Die ersten Hinweise auf die Existenz und Lokalisa-tion eines Nierenzellkarzinomgens stammen ausBeobachtungen am genetischen Material von Pa-tienten, die der gleichen Familie angehörten. Über3 Generationen hinweg wurden bei 10 Mitgliedern
klarzellige Nierenzellkarzinome (HCCRCC) beob-achtet, wobei das mittlere Alter bei der Diagnosebei 46 Jahren und damit etwa 10 Jahre früher alsbeim sporadischen Nierenzellkarzinom lag. Die zy-togenetische Analyse der konstitutionellen Chro-mosomen ergab, dass alle Patienten den gleichenchromosomalen Defekt in der Keimbahn aufwie-sen. Dabei handelte es sich um eine balancierteTranslokation zwischen den Chromosomen 3 und8 [t(3;8) (p14;q24)] (Cohen et al. 1979, Li et al.1982) (Abb. 9.4). Nachdem weitere Familien mitHCCRCC und balancierten Translokationen unterBeteiligung der Chromosomen 3 und 11 [(t(3;11)(p13;p15)] (Pathak et al. 1982) sowie 3 und 6[t(3;6) (p13;q25.1)] (Kovacs et al. 1989a) identifi-ziert waren, war klar, dass der relevante Genortfür HCCRCC auf dem kurzen Arm des Chromo-soms 3 liegen musste.
266 J. Decker und H. Brauch
Abb. 9.4. 3;8-Translokation. Darstellung einer konstitutionel-len Translokation t (3;8) (p14.2; q24) eines Patienten mit ei-nem familiären klarzelligen Nierenzellkarzinom. Es handeltsich um eine balancierte reziproke Translokation mit denBruchpunkten 3(p14.2) und 8 (q24). Dabei wird ein Chro-mosomensegment (3p14.2–3ptel) mit einem anderen(8q24–8qtel) ausgetauscht. Oben partielle Karyogrammenach G-Bänderungsfärbung, links jeweils ein normalesChromosom 3 bzw. normales Chromosom 8, rechts patholo-gisch veränderte Derivate beider Chromosomen, Pfeil Bruch-punkt, unten schematische Darstellung der partiellen Karyo-gramme in Form von Ideogrammen, modifiziert nach Wangu. Perkins (1984)

9.3.1.2 Verlust der Heterozygotie(loss of heterozygosity, LOH)bei sporadischen Nierenzellkarzinomenals Hinweis auf ein Tumorsuppressorgen
Auch bei nicht-erblich bedingten (sporadischen)Nierenzellkarzinomen führten zytogenetische undmolekulargenetische Studien auf die Spur eines re-levanten Gens auf Chromosom 3. Dabei erwiesensich Chromosom-3p-Defekte (Abb. 9.5) bei klarzel-ligen Nierenzellkarzinomen als gängige Aberratio-nen (Decker et al. 1988, Kovacs et al. 1987).
Auf der Basis von Restriktionsfragmentlängen-polymorphismus(RFLP)-Analysen wurde erstmaligklar, dass es sich bei diesen häufigen 3p-Aberratio-nen um den Verlust der Heterozygotie (LOH) unddamit um den Verlust von 3p-Material handelt. Die-ses im Tumor fehlende genetische Material mussteein für die Tumorgenese kritisches, noch unbekann-tes Gen enthalten (Abb. 9.6) (Anglard et al. 1991,Brauch et al. 1994, Kovacs et al. 1989b, Morita etal. 1991, Ogawa et al. 1991, van der Hout et al. 1991,Zbar et al. 1987). Über 90% der Nierenzellkarzino-me zeigten dieses genetische Merkmal, jedoch wardas Ausmaß der verloren gegangenen 3p-Bereicheunterschiedlich groß (Anglard et al. 1991).
9.3.1.3 vhl-Tumorsuppressorgen
In der Regel weist der Verlust genetischen Materi-als auf den Verlust eines für die Tumorgenese kri-tischen Gens hin. In diesem Fall kann auch dashomologe Allel, z.B. durch eine Punktmutation,mutiert sein. Gene, die durch homologe Mutatio-nen (two hit mechanism) die Tumorgenese auslö-sen können, heißen Tumorsuppressorgene (Abb.9.7). In normalen Zellen trägt das von ihnen ko-dierte Protein mit seiner biologischen Funktionzur Aufrechterhaltung der Homöostase bei und„unterdrückt“ die Tumorentstehung. Die Existenzvon Tumorsuppressorgenen wurde in den 70er Jah-ren auf der Grundlage klinischer, genetischer undepidemiologischer Beobachtungen sowie mathema-tisch-statistischer Berechnungen postuliert (Co-mings 1973, Hethcote u. Knudson 1978, Knudson1971).
Die Identifikation eines fehlenden Gens in Tu-moren gestaltete sich jedoch generell schwierigund gelang erst mit Hilfe moderner molekularbio-logischer „Werkzeuge“, die es ermöglichten, homo-loge Allele zu unterscheiden (Kan u. Dozy 1978,Solomon u. Bodmer 1979). Ein Nachteil dabei war,dass sich sporadische Tumoren dafür nicht eig-neten. Bei Familien mit erblichen Tumorerkran-
a 9 Nierenzellkarzinome 267
Abb. 9.5. Zytogenetische Untersuchungen beim sporadischenNierenzellkarzinom: 3p-Deletionen. Verlust genetischen Ma-terials beim Nierenzellkarzinom als Hinweis für Tumorsup-pressorloci auf Chromosom 3. Zusammenfassung veröffent-lichter zytogenetischer Ergebnisse, die Veränderungen amChromosom 3 beschreiben [Zentrale Datenbank des Natio-nalen Krebsforschungsinstituts (NCI), Stand September2000]: Großes Bildsegment Deletionen chromosomaler Ab-schnitte mit Angaben des Deletionsbruchpunkts, kleinerBildausschnitt Unbalancierte Translokationen, die durch denVerlust eines der Translokationsderivate zum Verlust vonChromosom-3-Material führen. Dargestellt sind die Bruch-punkte des Chromosoms 3 und des Translokationspartners:z.B. bedeutet 3(p13)
´51, 5(q22)´32 bzw. 5(q11)
´9, dass die Chro-mosomenbande 3(p13) 51-mal betroffen war, 5(q22) 32-malund 5(q11) 9-mal. Zusammen mit LOH-Daten ergeben sichHinweise auf mehrere Tumorsuppressorgene auf Chromo-som 3, insbesondere 3p. Diese könnten bei der Kanzeroge-nese von Nierenzellkarzinomen eine Rolle spielen. Ein häufi-ger Bruchpunkt kann Hinweise auf die direkte Lokalisationeines relevanten Gens sein
268 J. Decker und H. Brauch
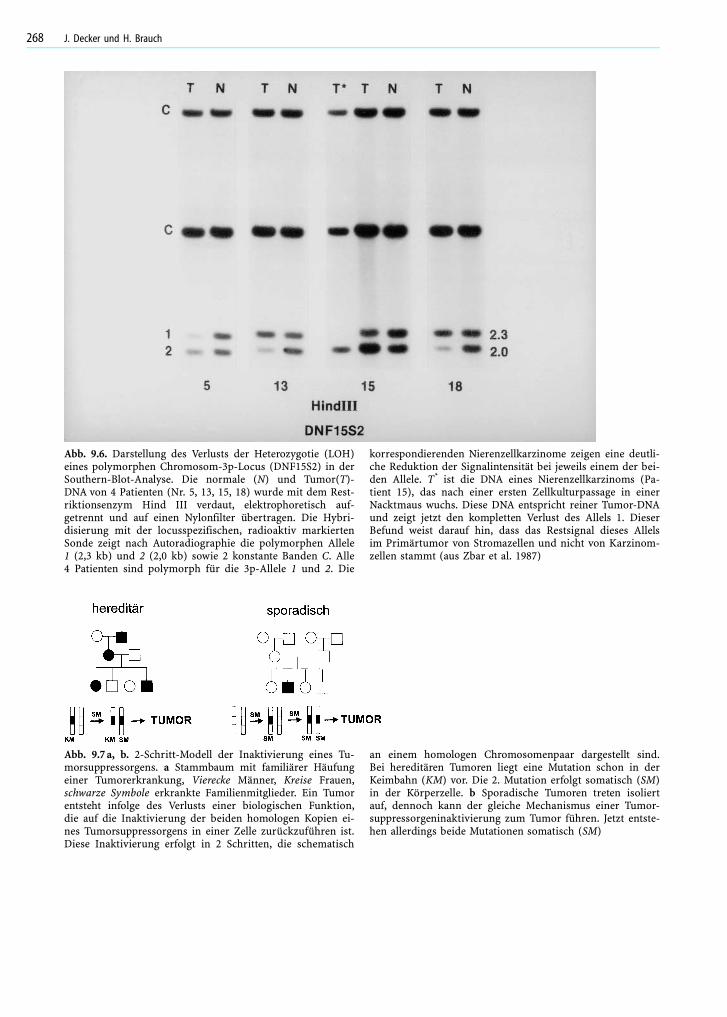
Abb. 9.6. Darstellung des Verlusts der Heterozygotie (LOH)eines polymorphen Chromosom-3p-Locus (DNF15S2) in derSouthern-Blot-Analyse. Die normale (N) und Tumor(T)-DNA von 4 Patienten (Nr. 5, 13, 15, 18) wurde mit dem Rest-riktionsenzym Hind III verdaut, elektrophoretisch auf-getrennt und auf einen Nylonfilter übertragen. Die Hybri-disierung mit der locusspezifischen, radioaktiv markiertenSonde zeigt nach Autoradiographie die polymorphen Allele1 (2,3 kb) und 2 (2,0 kb) sowie 2 konstante Banden C. Alle4 Patienten sind polymorph für die 3p-Allele 1 und 2. Die
korrespondierenden Nierenzellkarzinome zeigen eine deutli-che Reduktion der Signalintensität bei jeweils einem der bei-den Allele. T* ist die DNA eines Nierenzellkarzinoms (Pa-tient 15), das nach einer ersten Zellkulturpassage in einerNacktmaus wuchs. Diese DNA entspricht reiner Tumor-DNAund zeigt jetzt den kompletten Verlust des Allels 1. DieserBefund weist darauf hin, dass das Restsignal dieses Allelsim Primärtumor von Stromazellen und nicht von Karzinom-zellen stammt (aus Zbar et al. 1987)
Abb. 9.7a, b. 2-Schritt-Modell der Inaktivierung eines Tu-morsuppressorgens. a Stammbaum mit familiärer Häufungeiner Tumorerkrankung, Vierecke Männer, Kreise Frauen,schwarze Symbole erkrankte Familienmitglieder. Ein Tumorentsteht infolge des Verlusts einer biologischen Funktion,die auf die Inaktivierung der beiden homologen Kopien ei-nes Tumorsuppressorgens in einer Zelle zurückzuführen ist.Diese Inaktivierung erfolgt in 2 Schritten, die schematisch
an einem homologen Chromosomenpaar dargestellt sind.Bei hereditären Tumoren liegt eine Mutation schon in derKeimbahn (KM) vor. Die 2. Mutation erfolgt somatisch (SM)in der Körperzelle. b Sporadische Tumoren treten isoliertauf, dennoch kann der gleiche Mechanismus einer Tumor-suppressorgeninaktivierung zum Tumor führen. Jetzt entste-hen allerdings beide Mutationen somatisch (SM)

kungen gelang jedoch die Identifikation tumoras-soziierter Allele durch die Bestimmung des krank-heitsassoziierten Allels anhand der gemeinsamenVererbung der Erkrankung und eines Markerallels(Cavenee et al. 1983). Im Fall des Nierenzellkarzi-noms gelang dies über die inhaltliche Querverbin-dung zum VHL-assoziierten Nierenzellkarzinom,das die gleichen histologischen und molekularenCharakteristika wie das sporadische klarzelligeNierenzellkarzinom aufweist (Abb. 9.8) (Decker etal. 1988, Tory et al. 1989).
Darauf aufbauend wurden im Folgenden posi-tionelle Klonierungsverfahren zur weiteren Ein-grenzung eines Nierenzellkarzinomtumorsuppres-sorgens angewendet. Ausschlaggebend dafür wardie Generierung von über 2000 Einzelkopie-DNA-Fragmenten aus dem kritischen 3p-Chromosomen-abschnitt (Lerman et al. 1991), die zur Unterschei-dung eines Nierenzellkarzinom prädisponierendenAllels vom Wildtypallel in genetischen Kopplungs-analysen bei VHL-Familien herangezogen wurden(Hosoe et al. 1990, Seizinger et al. 1988). Die
Kopplungsanalysen in über 100 VHL-Familien eng-ten die kritische 3p25-Region auf 2,5 Mbp ein (La-tif et al. 1993). Schließlich gelang die Isolierung ei-ner kleinsten gemeinsam überlappenden Regiondreier Keimbahndeletionen (Cosmid 11) (Yao et al.1993) und die Identifikation der Kandidatengeneg7 und g6 (Latif et al. 1993). Expressionsstudienzeigten, dass g7-Transkripte in allen humanen Ge-weben einschließlich Gehirn und Niere exprimiertwaren. In Mutationsanalysen gelang mit g7 derNachweis von Keimbahndeletionen bei VHL-Pa-tienten sowie der Nachweis von Punktmutationenbei B-Zell-Linien von VHL-Patienten und Zelllini-en sporadischer Nierenzellkarzinome (Latif et al.1993). Somit war gezeigt, dass g7 das relevanteNierenzellkarzinomgen ist. Es wurde mit dem Gen-symbol vhl belegt, da es bei Familien mit demVHL-Syndrom identifiziert wurde. Der Tumorsup-pressorcharakter des vhl-Gens wurde funktionellin In-vitro-Experimenten bestätigt, bei denen dieTransfektion einer intakten vhl-Kopie in vhl-defek-te Nierenzellkarzinomzelllinien A498 und UMRC6das Wachstum der Tumorzellen unterdrückte(Chen et al. 1995a). In ähnlicher Weise unter-drückte die Rekonstitution vhl-defekter 786-O-Nie-renzellkarzinomzellen mit intaktem vhl die Tumor-bildung in Nacktmäusen (Iliopoulos et al. 1995).
Genstruktur, Expression und biologische Funktion.Die genomische Sequenz des vhl-Gens nimmt12–15 kb in der 3p25-Region ein (Lerman 2001).Die zunächst identifizierte Nukleotidsequenz um-fasst 852 Nukleotide, die auf 3 Exons verteilt sind(Abb. 9.9) (Latif et al. 1993). Die Identifikation derTranskriptionsstartkodons und der Promotorregi-on ergab jedoch eine kürzere Gensequenz von 639Nukleotiden, die für ein Protein von 213 Amino-säuren mit einem errechneten Molekulargewichtvon 24000 kodiert (Gen-Bank accession no.AF010238) (Kuzmin et al. 1995). Die Transkripti-onsinitiationsstellen liegen in der Nähe einerSp1/AP2-Stelle. Strukturelle Charakteristika desvhl-Gens sind das Fehlen von TATA- und CCAAT-Strukturelementen im Promotorbereich sowie dasVorliegen GC-reicher Sequenzen, die den Pro-motorbereich und Exon 1 als CpG-Insel ausweisen(Herman et al. 1994, Kuzmin et al. 1995). Die Pro-motorregion enthält Bindungsstellen für die Tran-skriptionsfaktoren NRF1 und PAX, von denenNRF1 die Expression nukleärer Gene in Mito-chondrien und PAX2 die Konversion mesenchyma-ler in epitheliale Zellen während der Entwicklungder Niere kontrolliert (Kuzmin et al. 1995, Lerman2001).
a 9 Nierenzellkarzinome 269
Abb. 9.8. Darstellung des Verlusts des Wildtyp-3p-Allels inden Nierenzellkarzinomen (RCC) eines VHL-Patienten unterBerücksichtigung der elterlichen Allele. Die Genotypisierungder Blut-DNA des gesunden Vaters (leeres quadratischesSymbol), der erkrankten Mutter (schwarzes Kreissymbol) unddes erkrankten Sohnes (Patient) (schwarzes quadratischesSymbol) am 3p-DNF15S2-Locus zeigt, dass der Vater homo-zygot für das Allel 1 (1,1) und Mutter und Sohn jeweils he-terozygot für Allel 1 und 2 (1,2) sind. Somit stammt Allel 1des erkrankten Sohns vom Vater und entspricht dem Wild-typallel. Allel 2 stammt von der Mutter und entspricht demKrankheitsallel. In den Nierenzellkarzinomen der linken undrechten Niere ist jeweils das vom Vater stammende Allel 1verlorengegangen. Das von der Mutter stammende Allel 2,das mit der prädisponierenden vhl-Mutation assoziiert ist,ist in beiden Tumoren vorhanden. Dieser Befund bestätigtindirekt die homologe Inaktivierung des relevanten Tumor-suppressorgens unter Berücksichtigung der Bedeutung vonFamilienanalysen bei der Zuordnung von Allelen (Zbar, pers.Mitteilung)

Das vhl-Gen wird universal sowohl in embryo-nalen als auch adulten Geweben und in Zelllinienexprimiert. Während der Entwicklung wird esbeim Menschen und bei der Maus sowohl zeitlichals auch räumlich kontrolliert in den Organen desUrogenitaltrakts, der Retina, dem Gehirn und demRückenmark, der Milz und dem Bronchialepitheli-um exprimiert (Kessler et al. 1995, Latif et al.1993, Richards et al. 1996). Dieses Expressions-muster korreliert z.T. mit den Organen, in denenbei vhl-Genträgern Tumoren entstehen können.Die Eliminierung des vhl-Gens durch Knockoutführt bei Mäusen zur gestörten Plazentavaskulari-sierung und ist in utero letal (Gnarra et al. 1997).Im Gegensatz dazu wurde bei Drosophila währendder Embryogenese bei Stämmen mit reduzierterVHL-Aktivität ein Abbruch der Tracheaentwick-lung beobachtet (Adryan et al. 2000). Der Haupt-stamm der Trachea war abgebrochen und von ei-ner exzessiven ektopen Ausknospung kleiner Kol-lateraläste begleitet. Dagegen führte die Über-expression zur Unterdrückung der Vaskularisation,einem Gain-of-function-Phänotyp, der bei Droso-phila auch durch Überexpression des humanen vhlinduziert werden konnte. Damit ist es wahrschein-lich, dass VHL während der Entwicklung die Zell-migration am Ende des vaskulären Mikrosystemskontrolliert.
Das vhl-Gen kann sowohl in der Keimbahn alsauch in der Körperzelle durch Mutationen gestörtwerden. Keimbahnmutationen verursachen beiGenträgern die VHL-Erkrankung (Brauch et al.1995, Chen et al. 1995a, Crossey et al. 1994, Deckeret al. 1996, Glavac et al. 1996, Klein et al. 2001, La-tif et al. 1993, Zbar et al. 1996). Somatische Muta-tionen treten im Nierenepithel auf und führen bei
betroffenen Patienten zum sporadischen klarzelli-gen Nierenzellkarzinom (Brauch et al. 1999, 2000,Foster et al. 1994, Gnarra et al. 1994, Kenck et al.1996, Shuin et al. 1994, Whaley et al. 1994).
Die Aminosäuresequenz und die Anzahl derTranskripte (mRNA) lässt 4 potentielle VHL-Pro-teine und Funktionen zu. Rein rechnerisch bestehtdas VHL-Protein aus 213 Aminosäuren (pVHL24),die mit Ausnahme einer 8fachen sauren GXEEX-Sequenz (Position 14–53) keine Homologien zuanderen Proteinen aufweisen (Latif et al. 1993, Ler-man 2001). Beginnt die Transkription erst amMethionin in Position 54, dem 2. internen Tran-skriptionsstartkodon, resultiert das 160 Aminosäu-ren umfassende basische pVHL18. Beide Proteinekommen jeweils in 2 weiteren Formen vor, die sichdurch das Vorhandensein oder Fehlen der Exon-2-kodierten Aminosäuren unterscheiden.
Beide Isoformen, pVHL24 und pVHL18, könnenaufgrund zweier konservierter Strukturmotive(Aminosäuren 65–127 und Aminosäuren 155–188)mit anderen Proteinen in Wechselwirkung treten.Daraus leitet sich eine Funktion als Adaptormo-lekül in regulatorischen Prozessen ab (Lerman2001). Da funktionelle Unterschiede zwischenpVHL24 und pVHL18 bislang nicht bekannt sind,werden im weiteren Verlauf die BezeichnungenpVHL für das Protein und VHL für die Funkti-on(en) verwendet.
Biochemische Analysen zeigten zunächst, dasspVHL einen ternären Komplex VCB mit den Pro-teinen Elongin C und Elongin B ausbildet (Abb.9.10) (Duan et al. 1995a, Kishida et al. 1994, 1995).Dieser Komplex kann durch vhl-Mutationen, wiesie in Tumoren vorkommen, destabilisiert werden,insbesondere dann, wenn Mutationen im 2. kon-
270 J. Decker und H. Brauch
Abb. 9.9. Schematische Darstellung der vhl-Genstruktur unddes kodierten Tumosuppressorproteins (aa). Die kodierendeSequenz wurde zunächst mit 852 Nukleotiden angegeben(Latif et al. 1993), die auf 3 Exons verteilt sind. Die kor-rigierte kodierende Sequenz von 639 Nukleotiden beginntmit der Nukleotidposition 214 (Kuzmin et al. 1995). Charak-teristisch sind 2 Transkriptionsstartkodons (ATG), eine Not-I-Restriktionsschnittstelle und die 8fach repetitive Sequenz
für 5 Aminosäuren [Gly-X-Glu-Glu-X]8 im Exon 1. Das kor-respondierende Protein von 213 Aminosäuren enthält Pro-teinbindungsdomänen. Die von Exon-1-Sequenzen kodiertep18-p34-Domäne (grau) ist wahrscheinlich für die Erken-nung von Zielmolekülen erforderlich, während die vonExon-3-Sequenzen kodierte Elongin-C-B-Bindungsdomäne(schwarz) (Kodon 155–167) die Verbindung zum Ubiquitin-ligasesystem herstellt

servierten Strukturmotiv auftreten (Duan et al.1995b, Kibel et al. 1995, Kishida et al. 1995, Loner-gan et al. 1998). Zunächst wurde in vitro gezeigt,dass VHL die Transkriptionselongation von Zielge-nen durch Bindung an Elongin B und Inhibitionder Elonginfunktion reguliert (Duan et al. 1995b,Kishida et al. 1995). Diese Funktion konnte jedochin vivo nicht bestätigt werden.
Der VCB-Komplex kann jedoch um das CUL-2-Protein zum VCB-CUL-2-Komplex erweitert wer-den (Pause et al. 1997), der ähnlich den Hefe-SCF-Komplexen E3-Ubiquitin-Ligase-Aktivität aufweist(Krek 1998, Mukhopadhyay et al. 1997). AußerCullin (C) enthalten diese Ligasen noch SKP1 (S)und F-Box-Proteine (F). Je nach Substratspezifitätder F-Box-Proteine markieren SCF im Rahmen derUbiquitinierung Zielmoleküle zur Degradierungim 26S-Proteasom. In Analogie nimmt das VHL-Protein die Funktion eines F-Box-Proteins wahr,das zum einen spezifische Substratmoleküle er-kennt und zum anderen die Verbindung zum Ubi-quitinligasesystem herstellt (Cockman et al. 2000)(Abb. 9.11). Die dafür erforderlichen Eigenschaftendes VHL-Proteins wurden durch Röntgenstruktur-analysen des VCB-Komplexes bei einer Auflösungvon 2,7 Å bestätigt (Stebbins et al. 1999), wonaches sich aus einer größeren NH2-terminalen �-Do-mäne mit �-Faltblattstruktur (Aminosäurereste63–154) und einer kleineren �-Domäne (Amino-säurereste 155–204) mit �-helikaler Struktur zu-sammensetzt. Die Interaktion mit Elongin C er-folgt weitestgehend über die �-Domäne (Amino-säurereste 155–192). Ungefähr die Hälfte aller tu-morigenen vhl-Mutationen betreffen die �-Domäneund die für die Elongin-C-Bindungen kritischenAminosäuren der �-Domäne (Stebbins et al. 1999).
Als physiologische, zelluläre Zielmoleküle einerVHL-assoziierten Ligaseaktivität kommen dieTranskriptionsfaktoren HIF-1� und HIF-2� (Krieget al. 2000, Maxwell et al. 1999) sowie Sp1 (Cohenet al. 1999, Mukhopadhyay et al. 1997), Fibronek-tin (Bonicalzi et al. 2001, Ohh et al. 1998) und 2
a 9 Nierenzellkarzinome 271
Abb. 9.10. Sekundärstruktur des ternären VCB-Komplexesgemäß Röntgenstrukturanalyse. Das VHL-Protein (rot) inter-agiert hauptsächlich über eine �-helikale Struktur mit Elon-gin C (blau). Während Elongin C ebenfalls mit Elongin B(grün) in Wechselwirkung tritt, interagiert das VHL-Proteinnicht mit Elongin B (aus Stebbins et al. 1999)
Abb. 9.11. Funktion des VHL-Proteins (pVHL). Das VHL-Protein nimmt im Rahmen des kontrollierten Proteinabbausdie Funktion eines Adaptormoleküls wahr. Dazu muss eszum einen Zielmoleküle, z. B. HIF erkennen, und zum ande-ren die Verbindung zu Faktoren herstellen, die den Abbauim Proteasom vermitteln. Die Erkennung der Zielmoleküle
erfolgt über die �-Domäne, während die �-Domäne für dieInteraktion mit Elongin C erforderlich ist. Die weitere Inter-aktion der Elongine mit hCUL-2 sowie dessen Interaktionmit weiteren Faktoren des Ubiquitinkomplexes (UBC)ermöglichen den Abbau des Zielmoleküls (HIF) im Protea-som (nach Kaelin 1999)

Isoformen der Proteinkinase C (Pal et al. 1997) inFrage. HIF-1� reguliert die normalerweise durchSauerstoffentzug induzierbare Expression von Ge-nen der Angiogenese und des Glukosemetabolis-mus. Beispiele sind der vaskuläre endothelialeWachstumsfaktor (VEGF), der Glukosetransporter1 (GLUT1), glykolytische Enzyme und die NO-Synthetase (Semenza 1998, Wenger u. Gassmann1997). HIF-1� wird normalerweise schnell durchdas Ubiquitin-Proteasom-System abgebaut (Kamu-ra et al. 2000), bei Hypoxie kommt es jedoch zurStabilisierung (Huang et al. 1998, Pugh et al. 1997,Salceda u. Caro 1997). HIF-2� ist ein weiterer, inStruktur und Funktion ähnlicher Faktor (Wengeru. Gassmann 1997). Beide Transkriptionsfaktorenwerden auch bei normalem Sauerstoffpartialdruckin VHL-funktionsdefekten Zellen stabilisiert, wiees für VHL–/VHL–-Nierenzellkarzinomzellen ge-zeigt wurde (Krieg et al. 2000, Maxwell et al.1999). Somit übt pVHL eine weiter reichende ne-gative regulatorische Funktion auf die ProteineVEGF, GLUT1 und Platelet derived growth factor �aus (Gnarra et al. 1996, Iliopoulos et al. 1995, Sie-meister et al. 1996). Bestätigt wurde dies in zell-biologischen Experimenten, die bei VHL–/VHL–-Nierenzellkarzinomzellen eine verstärkte vegf- undglut1-Expression im Vergleich zu VHL+/VHL+-Nie-renzellkarzinomzellen zeigte, die durch Rekonsti-tution des vhl-Wildtyps unterdrückt werden konn-te (Gnarra et al. 1997, Krieg et al. 2000). DieHochregulation HIF-regulierter Angiogenesefak-toren erklärt den hohen Vaskularisierungsgrad derVHL-Syndrom-assoziierten Tumoren und von Nie-renzellkarzinomen.
Im Rahmen des kontrollierten Proteinabbaus zel-lulärer Proteine könnte das VHL-Protein darüberhinaus eine übergeordnete Schutzfunktion wahr-nehmen, indem es falsch oder inkomplett prozes-sierte Proteine der ubiquitinvermittelten Proteolysezuführt. Zellbiologische Experimente zeigten, dassNierenzellkarzinomzellen mit positiver VHL-Ex-pression weniger toxisch auf Glukoseentzug reagier-ten als VHL-defiziente Zellen (Gorospe et al. 1999).Da VHL-positive Zellen unter diesen Stressbedin-gungen nachweislich mehr ubiquitinierte Proteineaufweisen als VHL-negative Zellen, könnten Letzteredurch Anreicherung und Überladung mit falschprozessierten Proteinen in der Aufrechterhaltung vi-taler zellulärer Funktionen kompromittiert sein.
Ähnlich einer VEGF-Überexpression zeigen Nie-renzellkarzinome im Vergleich zum normalen Nie-renepithel eine verstärkte Expression der karbo-nischen Anhydrasen CA IX (Liao et al. 1997, Oos-terwijk et al. 1986) und CA XII (Sahin et al. 1995).
In VHL-defekten Nierenzellkarzinomzelllinien wirddie CA-IX- und CA-XII-Expression nach Transfek-tion von intaktem vhl herunterreguliert (Ivanov etal. 1998). Dabei werden für die Regulation von CAXII beide VHL-Domänen benötigt, jedoch reichtdie Elonginbindungsdomäne für die Regulationvon CA IX aus. Physiologisch katalysieren karbo-nische Anhydrasen die reversible CO2-Hydrierungzu Karbonsäure, die durch Dissoziation in HCO3
–
und H3O+ den pH absenkt. Eine Überexpression
karbonischer Anhydrasen im Tumorgewebe könntesomit zur Azidifizierung des extrazellulären Mi-lieus führen und so Tumorwachstum und -ausbrei-tung begünstigen. Der molekulare Zusammenhangzwischen vhl-Defekt und CA-Überexpressionkönnte einen Beitrag zum Verständnis des bereitsseit Otto Warburg (Warburg 1926) diskutiertensauren Milieus von Tumorgeweben im Vergleichzu Normalgeweben liefern (Griffiths 1991).
pVHL ist auch für die Ausbildung einer intaktenextrazellulären Fibronektinmatrix erforderlich. Esbindet in Assoziation mit dem endoplasmatischenRetikulum und Golgi-Apparat Fibronektin, eine In-teraktion, die durch vhl-Mutationen aufgehobenwerden kann (Ohh et al. 1998). Im Gegensatz zuvhl-intakten Nierenzellkarzinomzellen, die in vitrolose Aggregate mit Anzeichen epithelialer Differen-zierung einschließlich trabekulärer und tubulärerStrukturen bilden, haben vhl-defekte Nierenkarzi-nomzellen diese Fähigkeit verloren (Lieubeau-Teil-let et al. 1998). Dies korreliert mit Unterschiedenin der Fibronektinablagerung. Auch embryonaleFibroblasten von vhl-Knockout-Mäusen sowie vhl-Knockout-Mausembryonen können in vitro bzw. invivo keine intakte Fibronektinmatrix mehr aufbau-en (Ohh et al. 1998). Eine Erklärung dafür könntesein, dass VCB auch für die Eliminierung von Fib-ronektin verantwortlich ist, das im endoplasmati-schen Retikulum falsch gefaltet oder falsch prozes-siert und zur zytoplasmatischen Oberfläche zu-rücktransportiert wird.
Schließlich wird VHL auch für den Abbruch desZellzyklus benötigt (Pause et al. 1998) und schütztZellen vor UV-induzierter Apoptose (Schoenfeld etal. 2000).
Das VHL-Protein kann sowohl im Zellkern alsauch im Zytoplasma lokalisieren und sich abhängigvom Zellzyklus zwischen den beiden Kompartmen-ten hin und her bewegen. Während der G1-G0-Phaseist pVHL ausschließlich im Nukleus und währendder S-Phase im Zytoplasma lokalisiert (Ye et al.1998). Der nukleäre Export benötigt in vitro die Hy-drolyse von ATP, die Gegenwart von GTP, eine RNA-Polymerase-II-Aktivität und die Gegenwart der
272 J. Decker und H. Brauch

GTPase Ran (Groulx et al. 2000). Dabei benutztpVHL wahrscheinlich ein eigenes nukleäres Export-signal, das der von Exon 2 kodierten �-Domäne zu-geordnet wurde (Bonicalzi et al. 2001).
Keimbahnmutationen. Die vhl-Mutationen verschie-dener Patienten unterscheiden sich zwar, jedochsegregiert innerhalb einer betroffenen Familie einedefinierte Mutation vom erkrankten Elternteil aufdie zu erkrankenden Nachkommen und weist diesesomit als vhl-Genträger aus. Etwa 160 verschiedeneKeimbahnmutationen wurden bislang in über 450VHL-Familien aus Europa, USA und Japan identi-fiziert und erfasst (Beroud et al. 1998, 2000, Zbaret al. 1996). Somit tragen die meisten VHL-Famili-en ihre eigene familienspezifische Keimbahnmuta-tion. Nur wenige Mutationen treten häufiger, d.h.in mehreren Familien auf. Dies lässt darauf schlie-ßen, dass die meisten Mutationen entwicklungs-geschichtlich vor nicht allzu langer Zeit entstandensind und sich noch nicht durch Migration geogra-phisch verteilt haben. Eine Ausnahme ist dieC:T-Mutation des Nukleotids 505, die in Europaund Nordamerika speziell bei 20 Familien inDeutschland und Pennsylvania nachgewiesen wur-de und die auf einen gemeinsamen Vorfahren imSchwarzwald zurückgeht (Brauch et al. 1995). An-
dere häufige Mutationen wie die 712C:T- und713G:A-Mutationen treten bei nichtverwandten Fa-milien aus Nordamerika und Europa auf und wei-sen diese Nukleotidpositionen als Hot spots fürMutationen aus (Chen et al. 1995b, 1996, Crosseyet al. 1994, Klein et al. 2001).
Etwa 80% aller Keimbahnmutationen entfallenauf kleine, intragenische Mutationen bestehendaus Nukleotidsubstitutionen, die zum Austauscheiner Aminosäure (Missense), zum vorzeitigen Ab-bruch der Proteinsynthese durch die Entstehungeines neuen Stoppkodons (Nonsense) oder zurLängenalteration des Proteins durch Veränderungeiner Spleißkonsensusregion führen. Darüber hi-naus treten Deletionen und Insertionen einzelneroder mehrerer Nukleotide auf, die zum Frameshiftführen und somit ein in der Länge modifiziertesProtein mit veränderter Funktion hervorbringen.Die Methode der Wahl zum Nachweis dieserPunktmutationen ist die Sequenzanalyse mittelsgeeigneter Oligonukleotide zur Amplifikation derkodierenden Abschnitte des vhl-Gens (Chen et al.1995b, Glavac et al. 1996, Klein et al. 2001). Unge-fähr 20% aller Keimbahnmutationen bestehen ineiner partiellen oder kompletten Deletion des vhl-Gens, die mit Hilfe der einfachen und quantitati-ven Southern-Blot- (Abb. 9.12) sowie der FISH-
a 9 Nierenzellkarzinome 273
Abb. 9.12. 3-Generationen-Stammbaum einer VHL-Familiemit häufigem Nierenzellkarzinom und Nachweis der Genträ-gerschaft für einen nierenzellkarzinomassoziierten Keimbahn-defekt, Kreise weibliche, Quadrate männliche Familienmitglie-der, schwarze Symbole VHL-Patienten, Diagonale Verstorbene.Alle nummerierten Familienmitglieder wurden der vhl-Keim-bahnanalyse unterzogen. Der familienspezifische vhl-Keim-bahndefekt wurde mittels Southern-Blot-Analyse (EcoRI-Ver-dauung) mit der radioaktiv markierten vhl-cDNA-Sonde g7identifiziert. Alle Personen tragen 2 gleich lange 3p-Allele,
die sich als konstantes DNA-Fragment darstellen. Die Patien-ten 3, 8, 10, 12, 13 und 17 zeigen ein konstantes (c) und einemutiertes Allel (m), das um etwa 10 kb verkürzt ist. Alle aufder Basis dieser Prädisposition erkrankten Personen ent-wickelten ein Nierenzellkarzinom. Die Personen 10 und 12wurden präsymptomatisch (schwarzes Oval) als Genträgeridentifiziert. Bei einer mit x gekennzeichneten Person hatsich ein Verdacht auf die VHL-Erkrankung molekulargene-tisch nicht bestätigt (aus Glavac et al. 1996)

Analyse detektiert werden können. Der Umfangder Deletionen kann zwischen 1 kb und 380 kbbetragen (Stolle et al. 1998, Yao et al. 1993).
Genotyp-Phänotyp-Korrelationen. Die hohe Variabili-tät des klinischen Bilds der VHL-Erkrankung, dieVielzahl und die Streuung der verschiedenenKeimbahnmutationen innerhalb des Gens und ihreverschiedenen biologischen Konsequenzen auf Pro-teinebene erschweren eine aussagefähige Genotyp-Phänotyp-Korrelation. Zur Beurteilung eines Nie-renzellkarzinomrisikos und anderer Risiken wurdedie VHL-Erkrankung in Typen eingeteilt. Nieren-zellkarzinome treten überwiegend mit dem VHLTyp 1 und den damit assoziierten Keimbahnmuta-tionen auf (Chen et al. 1995b, Crossey et al. 1994,Glavac et al. 1996, Zbar et al. 1996). Eine 1996veröffentlichte Zusammenfassung der internationa-len Ergebnisse zu vhl-Keimbahnmutationen undihren assoziierten Phänotypen zeigte, dass voninsgesamt 414 Familien mit identifizierten vhl-Mu-tationen 81% (n=336) dem VHL Typ 1 angehörten(Zbar et al. 1996). Diese Mutationen setzten sichzum einen aus Deletionen oder Punktmutationenzusammen, die grobe, Längen alterierende Schä-den des Proteins bewirken können, oder bestandenzum anderen aus Missense-Mutationen, die imProtein nur kleine strukturelle Veränderungen inForm eines Aminosäureaustausches bewirkenkönnen. Nierenzellkarzinome sind sowohl mit den„groben“ als auch den „feineren“ Mutationen asso-ziiert. Die Häufigkeit des Nierenzellkarzinomsbeim VHL-Typ-1-Phänotyp kann bis zu 70% be-tragen (Glavac et al. 1996). Keimbahnmutationen,deren phänotypische Ausprägung in belasteten Fa-milien besonders häufig zu Nierenzellkarzinomenführen, sind in Tabelle 9.4 aufgelistet. Zu diesen
gehören große Deletionen, die das gesamte Genoder große Teile davon eliminieren. Zum Beispielhatten alle 10 erkrankten Mitglieder einer slowaki-schen VHL-Familie vom VHL Typ 1 Nierenzystenund 6 Patienten (60%) ein Nierenzellkarzinom.Abbildung 9.12 zeigt einen Teil dieser Familie unddie molekulare Analyse (Glavac et al. 1996).
Aber auch einzelne Missense-Mutationen kön-nen, wie die Beispiele in Tabelle 9.4 zeigen, ein ho-hes Risiko für Nierenzellkarzinome übertragen. Indiesem Fall sind sie jedoch häufig mit dem VHLTyp 2 assoziiert. In dieser Gruppe lässt sich das Ri-siko für Nierenzellkarzinome je nach Mutation gra-duell bewerten, was sich in der Zuordnung zu denTypen VHL-2A, VHL-2B und VHL-2C widerspiegelt(Brauch et al. 1995, Glavac et al. 1996, Zbar et al.1996). Da Nierenzellkarzinome im Gegensatz zuden anderen VHL-Läsionen ein Potential zur Metas-tasierung besitzen, ist die Abschätzung des Risikosfür vhl-Genträger von großer Bedeutung.
Beispiele für VHL-Typ-2-Mutationen mit häufigem Nie-renzellkarzinom (VHL Typ 2B). Ein hohes Nierenzell-karzinomrisiko wurde bei einer großen hawaiani-schen Familie beobachtet. Die Patienten in dieserFamilie sind Genträger einer Missense-Mutation(T:C) des Nukleotids 686, die im Protein den Ami-nosäureaustausch eines Leucins gegen ein Prolin(Kodon 158) verursacht. Von 51 Patienten hatten29 (57%) ein Nierenzellkarzinom. Nur 1 Patient(2%) entwickelte ein Phäochromozytom, das Risi-ko für Retina- und ZNS-Läsionen lag bei 49% bzw.63% (Chen et al. 1995b).
Auch Genträgern der 712- (C:T) oder 713-Muta-tionen (G:A), die den Austausch eines Arginins ge-gen ein Tryptophan bzw. gegen ein Glutamin (Ko-don 167) bewirken, haben ein hohes Risiko fürNierenzellkarzinome. Diese Mutation wurde seitihrer Entdeckung bei etwa 30 Familien aus denUSA, Japan und Europa (Deutschland, Frankreich,Großbritannien, Finnland, Schweiz und Türkei)identifiziert und gilt als Hot-spot-Mutation, d.h.sie findet sich bei nichtverwandten Familien undauch de novo (Chen et al. 1996, Crossey et al.1994, Klein et al. 2001, Zbar et al. 1996).
Dieser Phänotyp wurde zunächst bei einer gro-ßen Familie aus Neufundland beobachtet (Green etal. 1986). Bei 41 Patienten traten in 15 Fällen(37%) Nierenzellkarzinome und in 28 Fällen (68%)Phäochromozytome auf, 29 Patienten (73%) hattenretinale Angiome und 10 Patienten (24%) ZNS-Lä-sionen (Chen et al. 1995b). Im erweiterten Patien-tengut von 73 Patienten mit einer 712/713-Hot-spot-Mutation wurde das Risiko für Nierenzellkar-
274 J. Decker und H. Brauch
Tabelle 9.4. Beispiele nierenzellkarzinomassoziierter vhl-Keimbahnmutationen bei Familien mit mindestens 3 Fällenvon Nierenzellkarzinomen
Größe, Lokalisation Mutationen Referenz
4–380 kb Deletion Chen et al. (1994)10 kb Deletion Glavac et al. (1996)393delG Frameshift Chen et al. (1994)439delTTC delPhe Chen et al. (1994)617T:A Stopp Chen et al. (1994)715insTTGTCCGT Frameshift Chen et al. (1994)769G:T Stopp Chen et al. (1994)452G:A Ser80Asn Glavac et al. (1996)479T:C Leu89Pro Glavac et al. (1996)564G:T Trp117Cys Chen et al. (1994)686T:C Leu158Pro Chen et al. (1994)712C:T Arg167Trp Chen et al. (1994)713G:A Arg167Gln Chen et al. (1994)

zinome mit 33%, das Risiko für Phäochromozyto-me mit 64% und das Risiko für Augen- und ZNS-Läsionen mit 64% bzw. 38% errechnet (Chen et al.1996). Allgemein ist auf der Basis des molekularenBefunds einer VHL-Typ-2B-Mutation die klinischeVorsorgeuntersuchung zur Früherkennung vonNierenzellkarzinomen zu empfehlen.
Beispiele für VHL-Typ-2-Mutationen mit seltenem Nie-renzellkarzinom (VHL Typ 2A). Die vhl-Mutation C:Tim Nukleotid 505, die im Protein einen Aminosäu-reaustausch eines Tyrosins gegen ein Histidin (Ko-don 98) bewirkt, kommt aufgrund eines Founder-Effekts in deutschen und deutschstämmigen VHL-Familien zwar öfter vor, spielt aber mit Hinblickauf ein Nierenzellkarzinomrisiko keine große Rolle(Brauch et al. 1995). Von 116 Patienten hatten nur4 (3%) ein Nierenzellkarzinom, aber 73 (63%) einPhäochromozytom, 57 (49%) ein retinales Angiomund 17 (15%) ZNS-Läsionen. Somit ist das Risikofür Träger dieser Founder-Mutation zwar gering,aber nicht ganz auszuschließen.
Beispiele für VHL-Typ-2-Mutationen ohne Nierenzellkar-zinome (VHL Typ 2C). Nierenzellkarzinome könnenbei einigen VHL-Familien ganz fehlen. Bei kleinenFamilien mit nur wenigen erkrankten Personen isteine solche Beurteilung zwar kaum möglich, je-doch erlauben größere Familien mit dem gleichen„minimalen“ Phänotyp eine Aussage. Ein Beispielist die Missense-Mutation des Nukleotids 775 (C:G,Kodon 188), die zu einem Austausch von Leucingegen Valin führt und bei 9 Patienten einer Fami-lie „nur“ Phäochromozytome verursachte (Glavacet al. 1996, Neumann et al. 1995). Die anderenüblichen VHL-assoziierten Läsionen einschließlichder Nierenzellkarzinome fehlten völlig. WeitereBeispiele sind die Missense-Mutationen des Nu-kleotids 547 T:C im Kodon 112, die ein Tyrosingegen ein Histidin austauscht (Tisherman et al.1993, Zbar et al. 1996), und eine Mutation des Nu-kleotids 709 im Kodon 166 (G:T), die den Einbaueines Valins statt eines Phenylalanins bewirkt(Gross et al. 1996). Die differenzialdiagnostischeProblematik dieses minimalen VHL-Phänotyps be-steht darin, dass die betroffenen Patienten auf-grund des Krankheitsbilds irrtümlicherweise dermultiplen endokrinen Neoplasie Typ II oder derNeurofibromatose Typ I zugeordnet werdenkönnten (Neumann et al. 1995). Hier muss daraufgeachtet werden, dass auch beim Fehlen von Nie-renzellkarzinomen die Zuordnung zu einem nie-renzellkarzinomassoziierten Syndrom wie demVHL-Syndrom wahrscheinlich sein kann.
Somatische vhl-Mutationen beim sporadischen Nieren-zellkarzinom. Das vhl-Gen ist ein klassisches Tu-morsuppressorgen, das gemäß eines postulierten2-Schritt-Inaktivierungsmechanismus die Tumor-genese anstößt (Abb. 9.7). Das Tumorsuppressor-modell stellt vererbbare und sporadische Tumorenauf der Ebene der erforderlichen grundlegendenGendefekte gleich, indem es die vererbbaren undsporadischen Tumoren gleicher Organspezifitätdurch Inaktivierung des gleichen Tumorsuppres-sorgens erklärt. Auf das Nierenzellkarzinom unddas vhl-Gen übertragen entstehen demnach ver-erbbare, VHL-assoziierte Nierenzellkarzinome in-folge einer genetischen Prädisposition, d.h. aufder Grundlage einer vorliegenden vhl-Keimbahn-mutation, in Kombination mit einer somatischenvhl-Mutation der bis dahin intakten Genkopie ineiner Zielzelle des Nierenepithels. Nichterblicheklarzellige Nierenzellkarzinome sollten demnachebenfalls durch 2, beide Allele betreffenden vhl-Mutationen hervorgerufen werden, die in diesemFall jedoch beide in der Körperzelle, d.h. einerepithelialen Nierenzelle, entstehen. Eine dieser Mu-tationen ist in der Regel ein Verlust des vhl-Gens,wie er Mitte der 80er Jahre anhand von 3p-LOH-Analysen als häufigstes genetisches Merkmal vonNierenzellkarzinomen beobachtet wurde. Nach derIdentifikation des vhl-Gens wurden diese Karzino-me systematisch auf Alterationen der verbleiben-den vhl-Genkopie hin untersucht. Analysen zurIdentifikation von Mutationen innerhalb der ko-dierenden vhl-Sequenz brachten eine Vielzahl so-matischer Mutationen zutage, deren Verteilungund Vielfalt der der Keimbahnmutationen ähneln(Beroud et al. 1998, Brauch et al. 2000, Foster etal. 1994, Gnarra et al. 1994, Herman et al. 1994,Kenck et al. 1996, Shuin et al. 1994, Whaley et al.1994). Die Häufigkeit intragenischer somatischervhl-Mutationen variierte in den Studien zwischen33 und 57%. Sie betreffen in der Regel klarzelligeNierenzellkarzinome und gelegentlich papilläre,chromophile Nierenzellkarzinome, sie treten je-doch nicht in chromophoben Nierenzellkarzino-men und auch nicht in gutartigen renalen Onkozy-tomen auf (Brauch et al. 2000). Möglicherweisesind vhl-Mutationen mit der Fähigkeit zum schnel-len Wachstum klarzelliger Nierenzellkarzinome as-soziiert. Obwohl sie in allen pT-Stadien nachweis-bar sind, treten sie signifikant häufig in Karzino-men des pT3-Stadiums auf (Brauch et al. 2000).Ob es sich dabei um einen Prognosefaktor für dasklarzellige Nierenzellkarzinom handelt, muss nochüberprüft werden.
a 9 Nierenzellkarzinome 275

Auch bei den sporadischen klarzelligen Nieren-zellkarzinomen zeigte sich, dass vhl-Mutationenprinzipiell über die gesamte vhl-Sequenz auftretenkönnen (Abb. 9.13). Im Unterschied zur Keimbahntreten vermehrt Mutationen und Alterationen auf,die den Verlust der Funktion nach sich ziehen, wiez.B. Frameshift-Mutationen und Hypermethylie-rung des Promotor- und Exon-1-Bereichs (Brauchet al. 2000, Foster et al. 1994, Gnarra et al. 1994,Herman et al. 1994, Kenck et al. 1996, Shuin et al.1994, Whaley et al. 1994). Im Unterschied zuKeimbahndefekten treten somatische Defekte häu-fig im Exon 2 auf. Dies ist bemerkenswert, da nurknapp 20% der kodierenden vhl-Sequenz auf die-sen Genabschnitt entfallen, aber fast die Hälfte dersomatischen Alterationen in diesem Abschnittidentifiziert wurden (Brauch et al. 2000, Foster etal. 1994, Gnarra et al. 1994). Die Verteilung soma-tischer Mutationen, insbesondere häufige Mutatio-nen im Exon 2 (ATT-TTT-Cluster) könnte mögli-cherweise auf Umwelteinflüsse bei der Entstehungder Mutationen hinweisen (Brauch et al. 2000,Gnarra et al. 1994). Während Studien in den USA,England und Japan keine Hinweise auf eine häufi-ge vhl-Mutation beim sporadischen Nierenzellkar-zinom zeigten, hatten in der deutschen Studie 9Patienten mit einem klarzelligen Nierenzellkarzi-nom eine Mutation dieses T-Pentamers im Exon 2.
Dies betrifft insgesamt 12% der Patienten mit vhl-Defekten aus dem Süddeutschen Raum (Brauch etal. 2000). Obwohl keinerlei Angaben zu voran-gegangenen Fremdstoffbelastungen vorliegen,könnten die Herkunft der Patienten und somit diegeographische Nähe zur französischen Nieder-rheinregion mit der weltweit höchsten Nierenzell-karzinominzidenz (Parkin u. Muir 1992) auf diemögliche Beteiligung von Umwelteinflüssen hin-weisen (Brauch et al. 2000). Ein Zusammenhangzwischen einem Nierenzellkarzinomrisiko undKanzerogenen wurde am Beispiel des in den 50er–70er Jahren häufig verwendeten industriellenLösungsmittels Trichlorethylen (TRI) bereits kon-trovers diskutiert (Vamvakas 1998, McLaughlin1997, Henschler 1995, Bruning 2000), auf moleku-larer Ebene jedoch bestätigt (Bruning 1997, Brauch1999). Die vhl-Mutationsanalyse der Nierenzellkar-zinome ehemals beruflich TRI-exponierter Patien-ten zeigte ein spezifisches Mutationsmuster (Abb.9.14), das den Zusammenhang zwischen der Kan-zerogenexposition und den nierenzellkarzinomspe-zifischen DNA-Schäden unterstreicht (Brauch et al.1999).
Darüber hinaus ist aus toxikologischen Unter-suchungen bekannt, dass ein TRI-Stoffwechselwegüber die Niere führt (Abb. 9.15), wo lokal reaktiveMetaboliten entstehen und mit DNA interagieren
276 J. Decker und H. Brauch
Abb. 9.13. Häufigkeit und Typen somatischer vhl-Alteratio-nen beim sporadischen klarzelligen Nierenzellkarzinom(CCRCC). Jedes Symbol (Pfeile, Balken, Sterne) charakteri-siert eine somatische Mutation oder Hypermethylierung (X)eines CCRCC. Die Mutationen treten stromabwärts der Not-I-Restriktionsschnittstelle auf. Von >150 klarzelligen Nieren-zellkarzinomen hatten etwa 45% somatische Aberrationen.Die meisten Mutationen (Frameshift, Spleißmutation, Non-sense) führen zu groben Längenalterationen des Proteinsund wahrscheinlich zum Verlust der Funktion. Im Fall einer
Hypermethylierung des Gens wird dieses nicht mehr tran-skribiert, in diesem Fall fehlt die VHL-Funktion ebenfalls.Missense-Mutationen führen zum Austausch einer Amino-säure und kompromittieren ebenso die normale Protein-funktion. Exon 2 trägt zwar nur etwa 20% zur kodierendenvhl-Sequenz bei, jedoch entfallen etwa die Hälfte aller soma-tischen Alterationen in diesen Sequenzabschnitt. Die Anhäu-fung der Abwärtspfeile im Exon 2 weist auf einen somati-schen Mutations-Hot-spot einer ATT-TTT-Sequenz der Ko-dons 147 und 148 hin (Brauch et al. 2000)
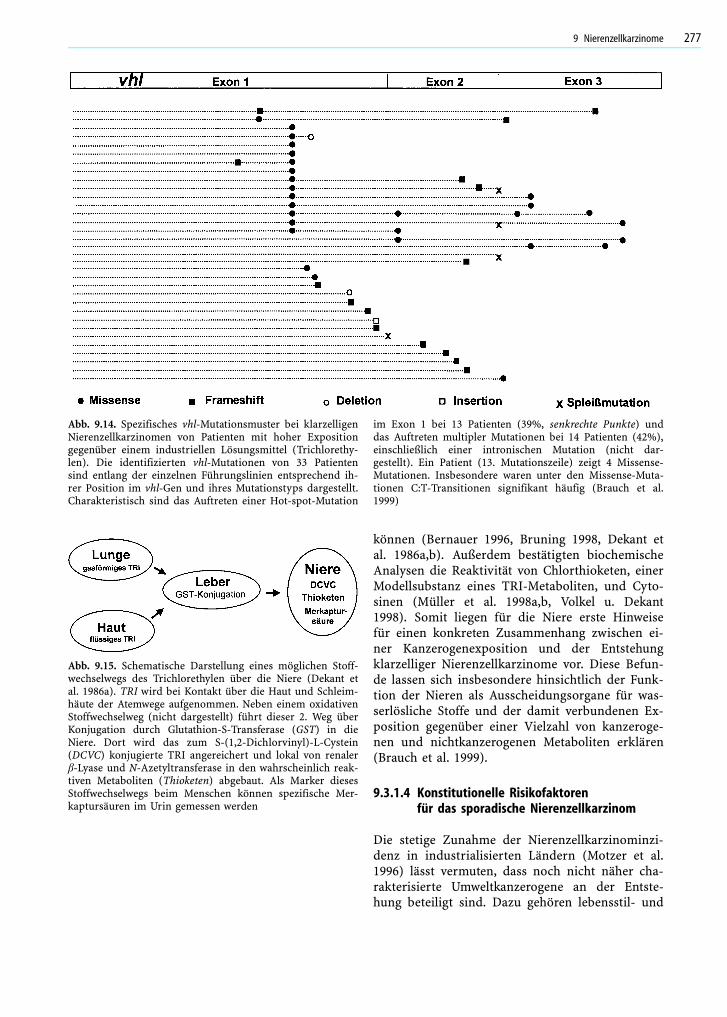
können (Bernauer 1996, Bruning 1998, Dekant etal. 1986a,b). Außerdem bestätigten biochemischeAnalysen die Reaktivität von Chlorthioketen, einerModellsubstanz eines TRI-Metaboliten, und Cyto-sinen (Müller et al. 1998a,b, Volkel u. Dekant1998). Somit liegen für die Niere erste Hinweisefür einen konkreten Zusammenhang zwischen ei-ner Kanzerogenexposition und der Entstehungklarzelliger Nierenzellkarzinome vor. Diese Befun-de lassen sich insbesondere hinsichtlich der Funk-tion der Nieren als Ausscheidungsorgane für was-serlösliche Stoffe und der damit verbundenen Ex-position gegenüber einer Vielzahl von kanzeroge-nen und nichtkanzerogenen Metaboliten erklären(Brauch et al. 1999).
9.3.1.4 Konstitutionelle Risikofaktorenfür das sporadische Nierenzellkarzinom
Die stetige Zunahme der Nierenzellkarzinominzi-denz in industrialisierten Ländern (Motzer et al.1996) lässt vermuten, dass noch nicht näher cha-rakterisierte Umweltkanzerogene an der Entste-hung beteiligt sind. Dazu gehören lebensstil- und
a 9 Nierenzellkarzinome 277
Abb. 9.14. Spezifisches vhl-Mutationsmuster bei klarzelligenNierenzellkarzinomen von Patienten mit hoher Expositiongegenüber einem industriellen Lösungsmittel (Trichlorethy-len). Die identifizierten vhl-Mutationen von 33 Patientensind entlang der einzelnen Führungslinien entsprechend ih-rer Position im vhl-Gen und ihres Mutationstyps dargestellt.Charakteristisch sind das Auftreten einer Hot-spot-Mutation
im Exon 1 bei 13 Patienten (39%, senkrechte Punkte) unddas Auftreten multipler Mutationen bei 14 Patienten (42%),einschließlich einer intronischen Mutation (nicht dar-gestellt). Ein Patient (13. Mutationszeile) zeigt 4 Missense-Mutationen. Insbesondere waren unter den Missense-Muta-tionen C:T-Transitionen signifikant häufig (Brauch et al.1999)
Abb. 9.15. Schematische Darstellung eines möglichen Stoff-wechselwegs des Trichlorethylen über die Niere (Dekant etal. 1986a). TRI wird bei Kontakt über die Haut und Schleim-häute der Atemwege aufgenommen. Neben einem oxidativenStoffwechselweg (nicht dargestellt) führt dieser 2. Weg überKonjugation durch Glutathion-S-Transferase (GST) in dieNiere. Dort wird das zum S-(1,2-Dichlorvinyl)-L-Cystein(DCVC) konjugierte TRI angereichert und lokal von renaler�-Lyase und N-Azetyltransferase in den wahrscheinlich reak-tiven Metaboliten (Thioketen) abgebaut. Als Marker diesesStoffwechselwegs beim Menschen können spezifische Mer-kaptursäuren im Urin gemessen werden

ernährungsassoziierte Faktoren, Umweltverschmut-zungen und Chemikalien sowie Medikamente. Um-weltfaktoren werden häufig durch Phase-I-Enzyme,z.B. den Isoenzymen der Cytochrom-P450-Familie(CYP) zu potentiellen Kanzerogenen verstoffwech-selt. Umgekehrt erfolgt die Detoxifizierung vonKanzerogenen durch Konjugation an Phase-II-En-zyme wie z.B. Glutathiontransferasen (GST) undN-Azetyltransferasen (NAT). Auch Transportermo-leküle können dabei eine Rolle spielen. SowohlFremdstoff metabolisierende Enzyme als auchTransportermoleküle sind häufig genetisch undfunktionell polymorph und können für die interin-dividuellen Unterschiede hinsichtlich der Suszepti-bilität gegenüber umweltassoziierten kanzerogenenEffekten verantwortlich gemacht werden (Idle et al.1992, Miller et al. 1997, Nebert et al. 1996, Puga etal. 1997).
Ein Zusammenhang zwischen einem erhöhtenNierenzellkarzinomrisiko und polymorphen,Fremdstoff metabolisierenden Enzymen wurde be-reits gezeigt. So hatten z.B. Personen mit einemmutanten cyp1a1-Genotyp ein erhöhtes Nierenzell-karzinomrisiko (Longuemaux et al. 1999). DiesesRisiko war noch verstärkt, wenn der cyp1a1-Geno-typ in Kombination mit einem mutanten gstp1-Ge-notyp, einem positiven gstt1-Genotyp oder einemnegativen nat2-Genotyp vorlag. Ein erhöhtes Risi-ko wurde auch für Personen mit einem negativengstm1-Genotyp und einem mutanten gstp1-Geno-typ beobachtet, wenn eine Assoziation entwedermit einem negativen nat2-Genotyp oder einemmutanten cyp1a1-Genotyp vorlag (Longuemaux etal. 1999). Das Nierenzellkarzinomrisiko war ins-besondere bei Patienten mit beruflich hoher Tri-chlorethylenexposition erhöht, wenn Patienten ei-nen positiven gstt1- oder positiven gstm1-Genotypaufwiesen (Bruning 1997). Diese Befunde erschei-nen umso wichtiger, wenn zusätzlich die mit demNierenzellkarzinom assoziierten vhl-Mutationen indie Betrachtung mit einbezogen werden. Hier wur-de bei Patienten mit positivem gstt1-Genotyp unddefektem nat1-Genotyp eine Assoziation mit demAuftreten eines bestimmten vhl-Mutationstyps ge-zeigt (Gallou et al. im Druck).
In einer anderen unabhängigen Studie wurdegezeigt, dass Patienten mit einem Nierenzellkarzi-nom signifikant häufig einen mutanten Genotypdes mdr1 (multi drug resistance)-Gens hatten(Siegsmund et al. eingereicht). Personen mit einemmutanten Genotyp zeigen im Vergleich zu Per-sonen mit einem Wildtyp eine reduzierte Expressi-on des vom mdr1-Gen kodierten PGP-Glykopro-teins in der Niere. Bei Patienten mit einem Nieren-
zellkarzinom und vhl-Mutationen lag die Häufig-keit des mutanten Genotyps um 40% höher als inder normalen Bevölkerung (Siegsmund pers. Mit-teilung). Zusammengenommen weisen diese Be-funde auf ein Zusammenspiel zwischen geneti-schen Suszeptibilitätsfaktoren und (unbekannten)Umweltfaktoren hin, die ein Risiko für die Entste-hung somatischer vhl-Genschäden und somit fürdie Entstehung von Nierenzellkarzinomen darstel-len.
9.3.2 Genetik des papillären Nierenzellkarzinoms
Eine familiäre Häufung papillärer Nierenzellkarzi-nome wurde gelegentlich beobachtet. Da jedochalle Betroffenen derselben Generation angehörten,war unklar, ob dieser familiären Häufung einegenetische Prädisposition zugrunde lag (Bernueset al. 1995, Franksson et al. 1972, Zbar u. Lerman1998). Erst die Beobachtung multipler und bilate-raler Nierenzellkarzinome mit papillärem Wachs-tumsmuster in 2 und mehr Generationen verstärk-te den Verdacht auf ein hereditäres, papilläres Nie-renzellkarzinom (HPRCC) (Schmidt et al. 1997,Zbar et al. 1994, 1995a). Die autosomal-dominanterbliche HPRCC ist nicht mit 3p-Defekten odervhl-Mutationen assoziiert, was auf eine vhl-unab-hängige Tumorgenese hinweist. Mit Hilfe zytoge-netischer und molekularer Kopplungsanalysenkonnte das relevante Gen bei belasteten Familienauf dem langen Arm des Chromosoms 7(7q31.1–34) lokalisiert werden (Bernues et al.1995, Schmidt et al. 1997). Diese Arbeiten stütztendie ursprüngliche Beobachtung einer Trisomie 7bei erblichen und nichterblichen papillären Nie-renzellkarzinomen (Kovacs 1993a). Allerdingsweist die Trisomie nicht auf einen tumorsuppres-sorassoziierten Mechanismus der Tumorgenese,sondern eher auf den Mechanismus einer Protoon-kogenaktivierung hin. Schließlich konnten beiHPRCC-Patienten Keimbahnmutationen im met-Protoonkogen identifiziert werden, das in der kri-tischen 7q-Region lokalisiert ist. Zur Klärung desZusammenhangs zwischen einer Trisomie 7 undmet-Mutationen wurde mit Hilfe der Fluoreszenz-in-situ-Hybridisierung (FISH) und Bestimmungdes Krankheitsallels in Familien gezeigt, dass imTumor immer das mutierte met-Allel dupliziertwar (Fischer et al. 1998, Zhuang et al. 1998). Wäh-rend etwa 86% der HPRCC-Familien Keimbahn-mutationen im met-Protoonkogen haben, tretensporadische Mutationen nur in etwa 3% der Fälleauf (Schmidt et al. 1997, 1999). Charakteristisch
278 J. Decker und H. Brauch

sind Polysomien der Chromosomen 7 und 17 (Ko-vacs 1989), wobei sich insbesondere maligne Tu-moren durch Polysomien von 3q, 7, 12, 16, 17 und20 sowie den Verlust des Y-Chromosoms auszeich-nen (Kovacs 1989, 1993a,b, Kovacs et al. 1989b,van den Berg et al. 1994). Diagnoseweisend sinddie Trisomien 7 und 17 sowie der Verlust des X-oder Y-Chromosoms, während die anderen geneti-schen Veränderungen eher auf eine Tumorprogres-sion hinweisen.
9.3.2.1 Rolle des met-Protoonkogens
Das met-Protoonkogen kodiert für eine Rezeptor-tyrosinkinase, die bei der Transduktion von Sig-nalen für Motilität, Proliferation und Morphogene-se eine Rolle spielt (Park et al. 1987, Weidner et al.1993). Wie andere Rezeptortyrosinkinasen bestehtmet ebenfalls aus• einer extrazellulären Ligandenbindungsdomäne,• einer Transmembrandomäne und• einer intrazellulären katalytischen Domäne
und wird in epithelialen Geweben exprimiert (Abb.9.16) (Ullrich u. Schlessinger 1990). Die Bindungdes Liganden Hepatozytenwachstumsfaktor/Scatter-Faktor (HGF/SF) an der Ligandenbindungsdomäneder Zelloberfläche führt zur Rezeptordimerisie-rung und Aktivierung der intrinsischen Tyrosinki-nase und somit zur Autophosphorylierung beiderRezeptoren (Schlessinger u. Ullrich 1992), wo-durch die Signalkaskade in Gang gesetzt wird. DerHGF/SF-MET-Signalweg spielt während der Em-bryogenese bei der normalen Entwicklung von Le-ber und Muskeln eine Rolle (Bladt et al. 1995). Au-ßer der Kontrolle zellulärer Prozesse ist MET indie Metastasierung und Invasion von Tumoren in-volviert (Jeffers et al. 1996). Die in HPRCC identi-fizierten met-Mutationen betreffen in der Regel dieKinasedomäne (Exon 16–19) (Schmidt et al. 1997,1999, Zbar u. Lerman 1998), ein Mutationsvertei-lungsmuster, das auch schon bei anderen Re-zeptortyrosinkinasen identifiziert wurde. Beispielesind c-KIT, das in Knochenmarkstammzellen ex-primiert wird und bei der Mastozytose hämatolo-gischer Erkrankungen mutiert ist (Nagata et al.1995), oder RET, das im Rahmen der multiplenendokrinen Neoplasie medulläre Schilddrüsenkar-zinome, Phäochromozytome, Neurome und Ne-benschilddrüsenhyperplasie verursacht (Hofstra etal. 1994). Wahrscheinlich interferieren diese Muta-tionen mit der Selbstinhibition der Tyrosinkinaseund bewirken so eine Aktivierung des Signalwegs.Funktionell führen diese Mutationen nach Trans-
fektion in NIH3T3-Zellen zur konstitutiven Phos-phorylierung von MET (Schmidt et al. 1999). DieDiskrepanz zwischen der Häufigkeit von met-Keimbahnmutationen und somatischen met-Muta-tionen lässt vermuten, dass met-Mutationen dieEntstehung von HPRCC verursachen, die Entste-hung sporadischer papillärer Nierenzellkarzinomejedoch überwiegend nach einem anderen Mecha-nismus verläuft.
9.3.3 Andere Nierenzellkarzinomgene
9.3.3.1 tsc1- und tsc2-Gene
Sowohl für tsc1 als auch für tsc2 besteht ein Zu-sammenhang mit Nierenzellkarzinomen. Bei 14von 42 sporadischen Nierenzellkarzinomen wurdeein LOH in der tsc1-Region gefunden (Cairns et al.1995), nach der Klonierung des Gens konnte sogarder homozygote Verlust von tsc1 in einem heredi-
a 9 Nierenzellkarzinome 279
Abb. 9.16. Darstellung der MET-Transmembranrezeptortyro-sinkinase mit �- und �-Kette sowie Lokalisation der bekann-ten Keimbahn- und somatischen Mutationen im met-Pro-toonkogen. Alle Mutationen bei Patienten mit HPRCC undsporadischem papillärem Nierenzellkarzinom sind vom Mis-sense-Typ und auf die Kinasedomäne (K) beschränkt

tären Nierenzellkarzinom gezeigt werden (vanSlegtenhorst et al. 1997). Für das tsc2-Gen konntenKeimbahnmutationen im Eker-Ratten-Modell derautosomal-dominant hereditären Nierenzellkarzi-nome beschrieben werden (Kobayashi et al. 1995,Yeung et al. 1994). Auch für die tsc2-Region wurdeein LOH in einem TSC-assoziierten Nierenzellkar-zinom nachgewiesen (Bjornsson 1996). Zur ge-nauen Beurteilung der molekularpathologischenBedeutung der Aberrationen der beiden tsc-Genefür die renale Kanzerogenese liegen keine ausrei-chenden Daten vor.
Ein Gen für das Birt-Hogg-Dubé-Syndrom istbisher nicht bekannt.
9.3.4 Sonstige molekulare Faktorenund Mechanismen
Verschiedene angiogenetische Wachstumsfaktorenwurden im Nierenzellkarzinom als erhöht expri-miert beschrieben (Wechsel et al. 1999), insbeson-dere VEGF (vascular endothelial growth factor)(Brieger et al. 1999, Tomisawa et al. 1999). Diese Ex-pressionserhöhung fand sich v. a. beim klarzelligenNierenzellkarzinom, nicht jedoch bei anderen his-tologischen Nierenzellkarzinomtypen (Brieger etal. 1999). Eine erhöhte VEGF-Expression scheintmöglicherweise mit einem fortgeschrittenen Tumor-stadium einherzugehen. VEGF stellt einen signifi-kanten Prognosefaktor dar, wobei umstritten ist,ob es sich dabei um einen unabhängigen oder einenabhängigen Prognosefaktor handelt (Jacobsen et al.2000, Paradis et al. 2000). VEGF kann auch im Se-rum von Tumorpatienten gemessen werden (Satoet al. 1999). Zwischen der VEGF-Expression, der Tu-morvaskularisation und der Expression von VHLbesteht im Nierenzellkarzinom eine inverse Bezie-hung (Brieger et al. 1999). Je weniger VHL expri-miert wird, umso höher ist die Expression desVEGF. Die Blockade der VEGF-Funktion könnte da-her ein Therapieansatz im Rahmen antiangiogeneti-scher Strategien darstellen (Harris 2000).
Auch andere Wachstumsfaktoren scheinen beimNierenzellkarzinom erhöht exprimiert zu sein (At-las et al. 1992, Freeman et al. 1989, Weidner et al.1990). Hier ist v. a. EGFR (epidermal growth factorreceptor) zu nennen (Moch et al. 1998, Yoshida u.Tosaka 1994), dessen Amplifikation mit einemschnelleren Tumorwachstum assoziiert sein soll(Moch et al. 1997). Die direkte klinische und zell-biologische Relevanz dieser und anderer Beobach-tungen sind Gegenstand aktueller Untersuchungen(Ramp et al. 2000, Yoshida et al. 1997):
Störungen auf der Ebene der Telomeraseaktivi-tät (Sugimura 1999) und der Methylierung (Her-man et al. 1994, Schulz 1998) von Genen und Me-chanismen des Imprintings (Mai et al. 1998, Odaet al. 1998) werden beim Nierenzellkarzinom dis-kutiert. Abschließende Ergebnisse liegen nicht aus-reichend vor und sind daher nicht Gegenstanddieses Kapitels.
9.4 Diagnostik des Nierenzellkarzinoms
9.4.1 Klinische Präsentation
Die im Abschnitt 9.2.2 „Klinische Symptome“ be-schriebenen klinischen Symptome sind entwedersehr unspezifisch oder aber sie entsprechen, imFall der klassischen Trias, einem fortgeschrittenenStadium.
9.4.2 Labordiagnostik und molekular-pathologische Untersuchungen
Einen labormedizinischen Blutparameter im Sinneines etablierten spezifischen Tumormarkers gibtes bisher nicht. Die Laborbefunde sind insgesamteher unspezifisch, eine erhöhte Blutsenkungs-geschwindigkeit spricht für das Vorliegen vonFernmetastasen, ohne dass diese beweisend dafürwäre. Bei Knochenmetastasen ist oft die alkalischePhosphatase erhöht, ebenso kann eine Hyperkalzä-mie vorliegen, die aber meist Ausdruck eines para-neoplastischen Syndroms ist (s. Abschnitt 9.2.2„Klinische Symptome“ und Tabelle 9.1).
Erste molekulare Studien sollen Hinweise dafürliefern, ob spezifische DNA-Veränderungen (z.B.LOH) auch im Blut nachweisbar und diagnose-oder prognoseweisend sind. Tumoren werden aufspezifische genetische Veränderungen, wie bei-spielsweise chromosomale 9p-Verluste hin unter-sucht, die möglicherweise prognostische Relevanzgewinnen könnten (Moch 1996).
9.4.3 Bildgebende Verfahren
Die beiden Verfahren, die heute zur Stadieneintei-lung und auch zur Planung des operativen Vor-gehens als unbedingt notwendig angesehen wer-den, sind Ultraschalluntersuchung und Computer-tomographie. In der Tat werden heute die meisten
280 J. Decker und H. Brauch

Nierenzellkarzinome durch die Sonographie ent-deckt. Hierbei ist es besonders wichtig, atypischeZysten bei Wandunregelmäßigkeiten genauer abzu-klären, da sich dort häufiger inzidentale Nieren-zellkarzinome verstecken können. Ausscheidungs-urogramm und Magnetresonanztomographiekönnen im Einzelfall, insbesondere zur Operati-onsplanung, sinnvoll sein.
9.4.4 Invasive diagnostische Verfahren
Angiographie und Feinnadelbiopsie werden heutenicht als Routinediagnoseverfahren beim Nieren-zellkarzinom angesehen. Die Indikationen zu die-sen Untersuchungen müssen sehr streng gestelltwerden, insbesondere da das letztere Verfahren so-gar die Gefahr der Tumorzellverschleppung in sichbergen mag. In Ausnahmefällen kann auch – z.B.bei komplizierten Nierenzysten mit Verdacht aufein Nierenzellkarzinom – eine operative Freilegungmit offener Biopsie und Schnellschnittunter-suchung indiziert sein.
9.5 Therapie des Nierenzellkarzinoms
Es ist wichtig, zwischen lokal begrenztem und me-tastasierendem Tumor zu unterscheiden. Danachbestimmen sich im Wesentlichen die Prognoseund die Möglichkeit der Therapie. Liegt ein fort-geschrittenes Stadium mit Fernmetastasierung vor,beträgt die mittlere Überlebenszeit <12 Monate.Ein großer Anteil von Patienten mit Nierenzellkar-zinom kommt erst bei bereits fortgeschrittenemTumorstadium mit Fernmetastasen zur Erstdiagno-se. Werden die Patienten mit einbezogen, die au-ßerdem im weiteren Verlauf der Erkrankung Me-tastasen entwickeln, haben heute nahezu noch im-mer die Hälfte der Patienten ein nicht mehr im lo-kalen Stadium befindliches Nierenzellkarzinom. Dadiese Karzinome gegenüber einer systemischenTherapie resistent sind, verstirbt die Mehrzahl die-ser Patienten. Die Zahl der im frühen Stadium zu-fällig entdeckten Tumoren nimmt durch den ver-mehrten Einsatz von Vorsorgeuntersuchungen, ins-besondere der Ultraschalluntersuchung, deutlichzu. Dies erhöht kontinuierlich die Zahl der Patien-ten mit Nierenzellkarzinomen im lokalisierten Sta-dium. In solchen Fällen ist der Tumor tatsächlichnur auf die Niere begrenzt. Die operative Tumor-entfernung bietet diesen Patienten eine echte Hei-
lungschance (Russo 2000, Vogelzang u. Stadler1998).
9.5.1 Tumornephrektomie
Beim nichtmetastasierten Nierenzellkarzinom istdie radikale Tumornephrektomie die Therapie derWahl. Neben der gesamten Niere werden meistgleichzeitig und gleichseitig die Nebenniere unddie regionalen Lymphknoten entfernt (Blom et al.1999, Naitoh et al. 1999, Steinbach et al. 1994).Zwar wird die Entfernung der Nebenniere kontro-vers diskutiert, die Überlebensvorteile bei Durch-führung der regionalen Lymphadenektomie wer-den jedoch allgemein anerkannt (Phillips u. Mes-sing 1993). Bei besonderen, elektiven Situationenwird die Organ erhaltende Nierenteilresektion dis-kutiert, hierzu gehören besonders kleine, vermut-lich Low-grade-Tumoren, insbesondere die desVon-Hippel-Lindau-Syndroms, wobei ein hohes Ri-siko besteht, dass die kontralaterale Niere durcheinen unabhängigen, 2. Primärtumor betroffenwird (Steinbach et al. 1991, 1992). Eine absoluteIndikation für die Organ erhaltende Chirurgie sindPatienten mit Einzelniere, mit eingeschränkter Nie-renfunktion und mit bilateralen Nierentumoren.Bei lokalen Rezidiven verschlechtert sich die Prog-nose entscheidend, hier besteht die Therapie häu-fig in der operativen Entfernung und einer System-therapie (Campbell u. Novick 1994). Bei einemmetastasierten Nierenzellkarzinom wird aus pallia-tiven Gründen eine Tumorresektion durchgeführt,u. a. weil dadurch Komplikationen vorgebeugt wer-den kann. Eine Metastasenchirurgie ist entwedernur bei solitären Metastasen oder zur Vorbeugungvon Komplikationen durch die Metastase indiziert.Ein Überlebensvorteil ist damit sehr wahrschein-lich nicht verbunden.
9.5.2 Strahlentherapie
Das Nierenzellkarzinom ist strahlenunempfindlich.Nur in ausgewählten, palliativen Situationen ist ei-ne Radiatio sinnvoll, etwa bei starken, durch Me-tastasen bedingten Schmerzen. Zeigen Knochen-metastasen die Gefahr, zu frakturieren, sollten sieebenfalls prophylaktisch bestrahlt werden. Beimultiplen oder bei symptomatischen Hirnmetasta-sen kann die Schädelbestrahlung indiziert sein.
a 9 Nierenzellkarzinome 281

9.5.3 Systemtherapie beim metastasiertenNierenzellkarzinom
9.5.3.1 Immunmodulation
Nierenzellkarzinome zeigen in seltenen Fällenspontane Remissionen. Es wird vermutet, dass diesim Zusammenhang mit einer Reaktion des Im-munsystems steht. Zum Beispiel wurde die Infiltra-tion des Tumorgewebes durch T-Lymphozyten be-obachtet (Storkel et al. 1992).
Am intensivsten sind die immunmodulatorischwirkenden Substanzen Interferon � und Interleu-kin-2 untersucht. Nach anfänglichen, viel verspre-chenden Ergebnissen, die z.T. nur an hoch selek-tionierten Patienten erhoben wurden, zeigten dieMehrzahl der Folgestudien ernüchternde Ergebnis-se. In einer großen Metaanalyse kamen Motzer u.Russo (2000) zu dem Schluss, dass nur sehr weni-ge Patienten eine komplette oder partielle Remis-sion auf die Gabe dieser beiden Substanzen zeigen.Noch weniger Patienten hatten einen echten Lang-zeitüberlebensvorteil. In der Tat wurde bereits1993 gezeigt, dass bei >1000 mit Interferon � be-handelten Patienten mit metastasierendem Nieren-zellkarzinom die Gesamtansprechrate nur bei 12%lag. Im Anschluss an diese ernüchternden Ergeb-nisse wurden verschiedene Kombinationen undDosierungen untersucht. Interferon � ist als Be-standteil von Kombinationstherapien unverändertein Standard beim Nierenzellkarzinom (Fossa2000). Interferone sind natürlich vorkommendeGlykoproteine, die als Antwort auf virale Infektio-nen, Antigen- oder Mitogenstimulation hauptsäch-lich von Leukozyten produziert werden.
9.5.3.2 Chemotherapie
Nierenzellkarzinome gehören zu den therapieresis-tenten Tumoren. Es gibt keine Chemotherapeutika,die in den bisher ausgetesteten Dosierungen undApplikationsformen für sich allein eine ausreichen-de Wirksamkeit gezeigt hätten (Motzer u. Russo2000). Ein Grund dafür könnte die hohe Expressi-on des mdr1-Gens sein (Gamelin et al. 1999, Mi-ckisch 1994, Yu et al. 1998). mdr1 kodiert für eintransmembranöses Transporterprotein (P-Glyko-protein), das Fremdstoffe vom Innern der Zellenach außen transportieren kann. Zu diesen Fremd-stoffen gehören eine Vielzahl von Chemotherapeu-tika, die aufgrund der P-Glykoprotein-Transporter-funktion nicht wirksam werden können. Die Über-windung dieses Multi-drug-resistance-Mechanis-
mus wird als therapeutischer Ansatz gewertet undkomplementär zur Gabe von Chemotherapeutikaerprobt (Braybrooke et al. 2000, Volm et al. 1991,Yu et al. 1999). Vinblastin und andere Vincaalka-loide werden heute in verschiedenen Kombinatio-nen mit den genannten Immunmodulatoren einge-setzt (Motzer u. Russo 2000).
9.5.3.3 Sonstige Substanzen
Viele Substanzen wurden aufgrund von Erfolg ver-sprechenden, experimentellen Anfangsergebnissenauch in die ersten klinischen Therapiestudien ein-gebracht, bisher gab es aber den gewünschten gro-ßen Durchbruch nicht (Pagliaro et al. 2000, Vogel-zang u. Stadler 1998).
9.5.3.4 Experimentelle Ansätze
Die Tumorvakzination mag in einigen Punktenviel versprechend sein, ist aber noch in den expe-rimentellen Anfängen und wird zurzeit nur an we-nigen Zentren angeboten (Berg et al. 2000).
Der monoklonale Antikörper G250 gegen nie-renzellkarzinomspezifische Epitope brachte in denersten Studien ermutigende Ergebnisse, die zurFortführung der Untersuchungen berechtigen(Divgi et al. 1998, Grabmaier et al. 2000, Luiten etal. 1996).
In einem selektierten Patientengut mit metasta-sierenden Nierenzellkarzinomen führte eine alloge-ne, nichtmyeloablative Transplantation von peri-pheren hämatopoetischen Stammzellen bei 10 von19 Patienten zu einem deutlichen Therapieerfolg(Childs et al. 2000). Dabei spielt die Graft-versus-host-Reaktion der allogenen Zellen eine entschei-dende Rolle. Ob sich diese viel versprechendenAnfangserfolge fortsetzen lassen, müssen Folgestu-dien zeigen.
Der Tyrosinkinaserezeptorhemmer SU 5416 be-wirkt eine Inhibition des VEGF-vermittelten Sig-nalweges (Harris 2000). Normalerweise führt dieBindung seines Liganden an den VEGF-Rezeptorzur Autophosphorylierung und damit zur Aktivie-rung. SU 5416 blockiert diesen Aktivierungsweg.Erste Untersuchungen mit dieser Substanz werdenbei VHL-Patienten mit fehlenden therapeutischenAlternativen durchgeführt (Harris 2000).
282 J. Decker und H. Brauch

9.6 Hilfreiche Internetadressen
Hilfreiche Internetadressen sind:http://www.meb.uni-bonn.de/cancernet/101070.html#1_GENERALINFORMATIONhttp://cancernet.nci.nih.gov/wyntk_pubs/kid-ney.htm
9.7 Literatur
Adryan B, Decker HJ, Papas TS, Hsu T (2000) Tracheal de-velopment and the von Hippel-Lindau tumor suppressorhomolog in Drosophila. Oncogene 19:2803–2811
Anglard P, Tory K, Brauch H et al. (1991) Molecular analysisof genetic changes in the origin and development ofrenal cell carcinoma. Cancer Res 51:1071–1077
Atlas I, Mendelsohn J, Baselga J, Fair WR, Masui H, KumarR (1992) Growth regulation of human renal carcinomacells: role of transforming growth factor alpha. CancerRes 52:3335–3339
Becker N, Wahrendorf J (1998) Atlas of cancer mortality inthe Federal Republic of Germany 1981–1990, 3rd edn.Springer, Berlin Heidelberg New York
Berg WJ, Divgi CR, Nanus DM, Motzer RJ (2000) Novel in-vestigative approaches for advanced renal cell carcinoma.Semin Oncol 27:234–239
Bernauer U, Birner G, Dekant W, Henschler D (1996) Bio-transformation of trichloroethene: dose-dependent excre-tion of 2,2,2-trichloro-metabolites and mercapturic acidsin rats and humans after inhalation. Arch Toxicol70:338–346
Bernues M, Casadevall C, Miro R et al. (1995) Cytogeneticcharacterization of a familial papillary renal cell carcino-ma. Cancer Genet Cytogenet 84:123–127
Beroud C, Joly D, Gallou C, Staroz F, Orfanelli MT, Junien C(1998) Software and database for the analysis of muta-tions in the VHL gene. Nucleic Acids Res 26:256–258
Beroud C, Collod-Beroud G, Boileau C, Soussi T, Junien C(2000) UMD (Universal mutation database): a genericsoftware to build and analyze locus-specific databases.Hum Mutat 15:86–94
Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP (1996)Tuberous sclerosis-associated renal cell carcinoma. Clini-cal, pathological, and genetic features. Am J Pathol149:1201–1208
Bladt F, Riethmacher D, Isenmann S, Aguzzi A, BirchmeierC (1995) Essential role for the c-met receptor in the mi-gration of myogenic precursor cells into the limb bud.Nature 376:768–771
Blom JH, Poppel H van, Marechal JM et al. (1999) Radicalnephrectomy with and without lymph node dissection:preliminary results of the EORTC randomized phase IIIprotocol 30881. EORTC Genitourinary Group. Eur Urol36:570–575
Bonicalzi ME, Groulx I, Paulsen N de, Lee S (2001) Role ofexon 2-encoded beta-domain of the von Hippel-Lindautumor suppressor protein. J Biol Chem 276:1407–1416
Boring CC, Squires TS, Tong T, Montgomery S (1994) Can-cer statistics, 1994. CA Cancer J Clin 44:7–26
Bourneville D (1880) Sclereuse tubereuse des circonvolu-tions cerebrales. Idiote et epilepsie hemiplegique. ArchNeurol (Paris) 1:81
Brauch H, Pomer S, Hieronymus T, Schadt T, Lohrke H, Ko-mitowski D (1994) Genetic alterations in sporadic renal-cell carcinoma: molecular analyses of tumor suppressorgene harboring chromosomal regions 3p, 5q, and 17p.World J Urol 12:162–168
Brauch H, Kishida T, Glavac D et al. (1995) Von Hippel-Lin-dau (VHL) disease with pheochromocytoma in the BlackForest region of Germany: evidence for a founder effect.Hum Genet 95:551–556
Brauch H, Weirich G, Hornauer MA, Storkel S, Wohl T,Bruning T (1999) Trichloroethylene exposure and specificsomatic mutations in patients with renal cell carcinoma.J Natl Cancer Inst 91:854–861
Brauch H, Weirich G, Brieger J et al. (2000) VHL alterationsin human clear cell renal cell carcinoma: association withadvanced tumor stage and a novel hot spot mutation.Cancer Res 60:1942–1948
Braybrooke JP, Vallis KA, Houlbrook S et al. (2000) Evalua-tion of toremifene for reversal of multidrug resistance inrenal cell cancer patients treated with vinblastine. CancerChemother Pharmacol 46:27–34
Brieger J, Weidt EJ, Schirmacher P, Storkel S, Huber C,Decker HJ (1999) Inverse regulation of vascular endothe-lial growth factor and VHL tumor suppressor gene insporadic renal cell carcinomas is correlated with vasculargrowth: an in vivo study on 29 tumors. J Mol Med77:505–510
Bruning T, Bolt HM (2000) Renal toxicity and carcinogeni-city of trichloroethylene: key results, mechanisms, andcontroversies. Crit Rev Toxicol 30:253–285
Bruning T, Lammert M, Kempkes M, Thier R, Golka K, BoltHM (1997) Influence of polymorphisms of GSTM1 andGSTT1 for risk of renal cell cancer in workers with long-term high occupational exposure to trichloroethene. ArchToxicol 71:596–599
Bruning T, Vamvakas S, Makropoulos V, Birner G (1998)Acute intoxication with trichloroethene: clinical symp-toms, toxicokinetics, metabolism, and development ofbiochemical parameters for renal damage. Toxicol Sci41:157–165
Bugert P, Gaul C, Weber K et al. (1997) Specific geneticchanges of diagnostic importance in chromophobe renalcell carcinomas. Lab Invest 76:203–208
Cairns P, Tokino K, Eby Y, Sidransky D (1995) Localizationof tumor suppressor loci on chromosome 9 in primaryhuman renal cell carcinomas. Cancer Res 55:224–227
Campbell SC, Novick AC (1994) Management of local recur-rence following radical nephrectomy or partial nephrec-tomy. Urol Clin North Am 21:593–599
Cavenee WK, Dryja TP, Phillips RA et al. (1983) Expressionof recessive alleles by chromosomal mechanisms in reti-noblastoma. Nature 305:779–784
Chen F, Kishida T, Duh FM et al. (1995a) Suppression ofgrowth of renal carcinoma cells by the von Hippel-Lin-dau tumor suppressor gene. Cancer Res 55:4804–4807
Chen F, Kishida T, Yao M et al. (1995b) Germline mutationsin the von Hippel-Lindau disease tumor suppressor gene:correlations with phenotype. Hum Mutat 5:66–75
Chen F, Slife L, Kishida T, Mulvihill J, Tisherman SE, Zbar B(1996) Genotype-phenotype correlation in von Hippel-Lindau disease: identification of a mutation associatedwith VHL type 2A. J Med Genet 33:716–717
a 9 Nierenzellkarzinome 283

Childs R, Chernoff A, Contentin N et al. (2000) Regressionof metastatic renal-cell carcinoma after nonmyeloablativeallogeneic peripheral-blood stem-cell transplantation. NEngl J Med 343:750–758
Chung-Park M, Parveen T, Lam M (1989) Acquired cysticdisease of the kidneys and renal cell carcinoma inchronic renal insufficiency without dialysis treatment.Nephron 53:157–161
Cockman ME, Masson N, Mole DR et al. (2000) Hypoxia in-ducible factor-alpha binding and ubiquitylation by thevon Hippel-Lindau tumor suppressor protein. J BiolChem 275:25 733–25741
Cohen AJ, Li FP, Berg S et al. (1979) Hereditary renal-cellcarcinoma associated with a chromosomal translocation.N Engl J Med 301:592–596
Cohen HT, Zhou M, Welsh AM et al. (1999) An importantvon Hippel-Lindau tumor suppressor domain mediatesSp1-binding and self-association. Biochem Biophys ResCommun 266:43–50
Collins ET (1894) Intra-ocular growths. Two cases, brotherand sister, with pecular vascular new growth, probablyprimarily retinal, affecting both eyes. Trans OphthalmolSoc UK 14:141
Comings DE (1973) A general theory of carcinogenesis. ProcNatl Acad Sci USA 70:3324–3328
Cook RJ, Lawless JF (1996) Interim monitoring of longitudi-nal comparative studies with recurrent event responses.Biometrics 52:1311–1323
Crossey PA, Richards FM, Foster K et al. (1994) Identifica-tion of intragenic mutations in the von Hippel-Lindaudisease tumour suppressor gene and correlation with dis-ease phenotype. Hum Mol Genet 3:1303–1308
Decker HJ, Neumann HP, Walter TA, Sandberg AA (1988) 3pinvolvement in a renal cell carcinoma in von Hippel-Lin-dau syndrome. Region of tumor breakpoint clustering on3p? Cancer Genet Cytogenet 33:59–65
Decker HJ, Neuhaus C, Jauch A et al. (1996) Detection of agermline mutation and somatic homozygous loss of thevon Hippel-Lindau tumor-suppressor gene in a familywith a de novo mutation. A combined genetic study, in-cluding cytogenetics, PCR/SSCP, FISH, and CGH. HumGenet 97:770–776
Dekant W, Metzler M, Henschler D (1986a) Identification ofS-1,2-dichlorovinyl-N-acetyl-cysteine as a urinary meta-bolite of trichloroethylene: a possible explanation for itsnephrocarcinogenicity in male rats. Biochem Pharmacol35:2455–2458
Dekant W, Schulz A, Metzler M, Henschler D (1986b) Ab-sorption, elimination and metabolism of trichloroethy-lene: a quantitative comparison between rats and mice.Xenobiotica 16:143–152
Delahunt B, Eble JN (1997) Papillary renal cell carcinoma: aclinicopathologic and immunohistochemical study of 105tumors. Mod Pathol 10:537–544
Divgi CR, Bander NH, Scott AM et al. (1998) Phase I/IIradioimmunotherapy trial with iodine-131-labeled mono-clonal antibody G250 in metastatic renal cell carcinoma.Clin Cancer Res 4:2729–2739
Duan DR, Humphrey JS, Chen DY et al. (1995a) Characteri-zation of the VHL tumor suppressor gene product: locali-zation, complex formation, and the effect of natural inac-tivating mutations. Proc Natl Acad Sci USA 92:6459–6463
Duan DR, Pause A, Burgess WH et al. (1995b) Inhibition oftranscription elongation by the VHL tumor suppressorprotein. Science 269:1402–1406
Fischer J, Palmedo G, Knobloch R von et al. (1998) Duplica-tion and overexpression of the mutant allele of the METproto-oncogene in multiple hereditary papillary renal celltumours. Oncogene 17:733–739
Fleming ID (1998) Revision of TNM classification for oro-pharyngeal carcinoma. Cancer 82:1611–1612
Fossa SD (2000) Interferon in metastatic renal cell carcino-ma. Semin Oncol 27:187–193
Foster K, Prowse A, Berg A van den et al. (1994) Somaticmutations of the von Hippel-Lindau disease tumour sup-pressor gene in non-familial clear cell renal carcinoma.Hum Mol Genet 3:2169–2173
Franksson C, Bergstrand A, Ljungdahl I, Magnusson G, Nor-denstam H (1972) Renal carcinoma (hypernephroma) oc-curring in 5 siblings. J Urol 108:58–61
Freeman MR, Washecka R, Chung LW (1989) Aberrant ex-pression of epidermal growth factor receptor and HER-2(erbB-2) messenger RNAs in human renal cancers. Can-cer Res 49:6221–6225
Gamelin E, Mertins SD, Regis JT et al. (1999) Intrinsic drugresistance in primary and metastatic renal cell carcino-ma. J Urol 162:217–224
Gallou C, Longemeaux S, Deloménie C et al. (in press) Asso-ciations of GSTT1 non-null and NAT1 slow/rapid geno-types with VHL transversions in sporadic renal cell carci-noma. Pharmacogenetics in press
Glavac D, Neumann HP, Wittke C et al. (1996) Mutations inthe VHL tumor suppressor gene and associated lesionsin families with von Hippel-Lindau disease from centralEurope. Hum Genet 98:271–280
Glenn GM, Coyke PL, Zbar B, Linehan WM (1990) Von Hip-pel-Lindau disease. Clinical review and molecular genet-ics. Probl Urol 4:312–330
Glenn GM, Daniel LN, Choyke P et al. (1991) Von Hippel-Lindau (VHL) disease: distinct phenotypes suggest morethan one mutant allele at the VHL locus. Hum Genet87:207–210
Gnarra JR, Tory K, Weng Y et al. (1994) Mutations of theVHL tumour suppressor gene in renal carcinoma. NatGenet 7:85–90
Gnarra JR, Zhou S, Merrill MJ et al. (1996) Post-transcrip-tional regulation of vascular endothelial growth factormRNA by the product of the VHL tumor suppressorgene. Proc Natl Acad Sci USA 93:10 589–10 594
Gnarra JR, Ward JM, Porter FD et al. (1997) Defective pla-cental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA 94:9102–9107
Gorospe M, Egan JM, Zbar B et al. (1999) Protective func-tion of von Hippel-Lindau protein against impaired pro-tein processing in renal carcinoma cells. Mol Cell Biol19:1289–1300
Grabmaier K, Vissers JL, De Weijert MC et al. (2000) Molec-ular cloning and immunogenicity of renal cell carcino-ma-associated antigen G250. Int J Cancer 85:865–870
Grawitz PA (1883) Die sogenannten Lipoma der Niere.Virchows Arch 93:39–63
Green JS, Bowmer MI, Johnson GJ (1986) Von Hippel-Lin-dau disease in a Newfoundland kindred. CMAJ 134:133–146
Griffiths JR (1991) Are cancer cells acidic? Br J Cancer64:425–427
Gross DJ, Avishai N, Meiner V, Filon D, Zbar B, AbeliovichD (1996) Familial pheochromocytoma associated with anovel mutation in the von Hippel-Lindau gene. J Clin En-docrinol Metab 81:147–149
284 J. Decker und H. Brauch

Groulx I, Bonicalzi ME, Lee S (2000) Ran-mediated nuclearexport of the von Hippel-Lindau tumor suppressor pro-tein occurs independently of its assembly with cullin-2. JBiol Chem 275:8991–9000
Harris AL (2000) Von Hippel-Lindau syndrome: target foranti-vascular endothelial growth factor (VEGF) receptortherapy. Oncologist 5:32–36
Henschler D, Vamvakas S, Lammert M et al. (1995) In-creased incidence of renal cell tumors in a cohort ofcardboard workers exposed to trichloroethene. Arch Tox-icol 69:291–299
Herman JG, Latif F, Weng Y et al. (1994) Silencing of theVHL tumor-suppressor gene by DNA methylation in re-nal carcinoma. Proc Natl Acad Sci USA 91:9700–9704
Hethcote HW, Knudson AG Jr (1978) Model for the inci-dence of embryonal cancers: application to retinoblasto-ma. Proc Natl Acad Sci USA 75:2453–2457
Hofstra RM, Landsvater RM, Ceccherini I et al. (1994) Amutation in the RET proto-oncogene associated withmultiple endocrine neoplasia type 2B and sporadic med-ullary thyroid carcinoma. Nature 367:375–376
Hosoe S, Brauch H, Latif F et al. (1990) Localization of thevon Hippel-Lindau disease gene to a small region ofchromosome 3. Genomics 8:634–640
Huang LE, Gu J, Schau M, Bunn HF (1998) Regulation ofhypoxia-inducible factor 1alpha is mediated by an O2-de-pendent degradation domain via the ubiquitin-protea-some pathway. Proc Natl Acad Sci USA 95:7987–7992
IARC IAfRoC (1987) IARC Monographs on the evaluation ofcarcinogenic risks to humans. Overall evaluations of car-cinogenicity: an updating of IARC monographs Vols 1–42. IARC, Lyon, Suppl 7
Idle JR, Armstrong M, Boddy AV et al. (1992) The pharma-cogenetics of chemical carcinogenesis. Pharmacogenetics2:246–258
Iliopoulos O, Kibel A, Gray S, Kaelin WG (1995) Tumoursuppression by the human von Hippel-Lindau gene prod-uct. Nat Med 1:822–826
Ivanov SV, Kuzmin I, Wei MH et al. (1998) Down-regulationof transmembrane carbonic anhydrases in renal cell car-cinoma cell lines by wild-type von Hippel-Lindau trans-genes. Proc Natl Acad Sci USA 95:12 596–12601
Jacobsen J, Rasmuson T, Grankvist K, Ljungberg B (2000)Vascular endothelial growth factor as prognostic factor inrenal cell carcinoma. J Urol 163:343–347
Jeffers M, Rao MS, Rulong S et al. (1996) Hepatocyte growthfactor/scatter factor-Met signaling induces proliferation,migration, and morphogenesis of pancreatic oval cells.Cell Growth Differ 7:1805–1813
Jones AC, Daniells CE, Snell RG et al. (1997) Molecular ge-netic and phenotypic analysis reveals differences betweenTSC1 and TSC2 associated familial and sporadic tuberoussclerosis. Hum Mol Genet 6:2155–2161
Kaelin G Jr (1999) Many vessels, faulty gene. Nature 399:203–204
Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC,Conaway JW (2000) Activation of HIF1alpha ubiquitina-tion by a reconstituted von Hippel-Lindau (VHL) tumorsuppressor complex. Proc Natl Acad Sci USA 97:10 430–10450
Kan YW, Dozy AM (1978) Polymorphism of DNA sequenceadjacent to human beta-globin structural gene: relation-ship to sickle mutation. Proc Natl Acad Sci USA 75:5631–5635
Kenck C, Wilhelm M, Bugert P, Staehler G, Kovacs G (1996)Mutation of the VHL gene is associated exclusively withthe development of non-papillary renal cell carcinomas. JPathol 179:157–161
Kessler PM, Vasavada SP, Rackley RR et al. (1995) Expres-sion of the Von Hippel-Lindau tumor suppressor gene,VHL, in human fetal kidney and during mouse embryo-genesis. Mol Med 1:457–466
Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG Jr (1995)Binding of the von Hippel-Lindau tumor suppressor pro-tein to Elongin B and C. Science 269:1444–1446
Kishida T, Yao M, Chen F, Orcutt ML, Lerman MI, Zbar B(1994) A novel donor splice site mutation associated withtwo mRNAs in von Hippel-Lindau disease. Hum MolGenet 3:1191–1192
Kishida T, Stackhouse TM, Chen F, Lerman MI, Zbar B(1995) Cellular proteins that bind the von Hippel-Lindaudisease gene product: mapping of binding domains andthe effect of missense mutations. Cancer Res 55:4544–4548
Klein B, Weirich G, Brauch H (2001) DHPLC for germlinemutation screening in the analysis of the VHL tumorsuppressor gene: usefulness and limitations. Hum Genet108:376–384
Knudson AG Jr (1971) Mutation and cancer: statistical studyof retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Kobayashi T, Hirayama Y, Kobayashi E, Kubo Y, Hino O(1995) A germline insertion in the tuberous sclerosis(Tsc2) gene gives rise to the Eker rat model of domi-nantly inherited cancer. Nat Genet 9:70–74
Kovacs G (1989) Papillary renal cell carcinoma. A morpho-logic and cytogenetic study of 11 cases. Am J Pathol134:27–34
Kovacs G (1993a) Molecular cytogenetics of renal cell tu-mors. Adv Cancer Res 62:89–124
Kovacs G (1993b) Molecular differential pathology of renalcell tumours. Histopathology 22:1–8
Kovacs G, Szucs S, de Riese W, Baumgartel H (1987) Specificchromosome aberration in human renal cell carcinoma.Int J Cancer 40:171–178
Kovacs G, Brusa P, de Riese W (1989a) Tissue-specific ex-pression of a constitutional 3;6 translocation: develop-ment of multiple bilateral renal-cell carcinomas. Int JCancer 43:422–427
Kovacs G, Wilkens L, Papp T, de Riese W (1989b) Differen-tiation between papillary and nonpapillary renal cell car-cinomas by DNA analysis. J Natl Cancer Inst 81:527–530
Krek W (1998) Proteolysis and the G1-S transition: the SCFconnection. Curr Opin Genet Dev 8:36–42
Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH(2000) Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in re-nal carcinoma cells by von Hippel-Lindau tumor suppres-sor gene loss of function. Oncogene 19:5435–5443
Kuzmin I, Duh FM, Latif F, Geil L, Zbar B, Lerman MI(1995) Identification of the promoter of the human vonHippel-Lindau disease tumor suppressor gene. Oncogene10:2185–2194
Lamiell JM, Salazar FG, Hsia YE (1989) Von Hippel-Lindaudisease affecting 43 members of a single kindred. Medi-cine (Baltimore) 68:1–29
Latif F, Tory K, Gnarra J et al. (1993) Identification of thevon Hippel-Lindau disease tumor suppressor gene.Science 260:1317–1320
a 9 Nierenzellkarzinome 285

Lerman M (2001) VHL protein function in health and dis-ease. Wiley, New York
Lerman MI, Latif F, Glenn GM et al. (1991) Isolation and re-gional localization of a large collection (2,000) of single-copy DNA fragments on human chromosome 3 for map-ping and cloning tumor suppressor genes. Hum Genet86:567–577
Li FP, Marchetto DJ, Brown RS (1982) Familial renal carci-noma. Cancer Genet Cytogenet 7:271–275
Li E, Beard C, Jaenisch R (1993) Role of DNA methylationin genomic imprinting. Nature 366:362–365
Liao SY, Aurelio ON, Jan K, Zavada J, Stanbridge EJ (1997)Identification of the MN/CA9 protein as a reliable diag-nostic biomarker of clear cell carcinoma of the kidney.Cancer Res 57:2827–2831
Lieubeau-Teillet B, Rak J, Jothy S, Iliopoulos O, Kaelin W,Kerbel RS (1998) Von Hippel-Lindau gene-mediatedgrowth suppression and induction of differentiation inrenal cell carcinoma cells grown as multicellular tumorspheroids. Cancer Res 58:4957–4962
Lindau A (1926) Studien über Kleinhirnzysten. Bau, Patho-genese und Beziehungen zur Angiomatosis retinae. ActaPathol Microbiol Scand [Suppl I] p1
Linehan WM, Lerman MI, Zbar B (1995) Identification ofthe von Hippel-Lindau (VHL) gene. Its role in renal can-cer. JAMA 273:564–570
Linehan WM, Shipley W, Parkinson D (1996) Cancer of thekidney and ureter. In: DeVita VT, Hellman S, RosenbergSA (eds) Cancer: principles and practice of oncology.Lippincott, Philadelphia, p 19
Lonergan KM, Iliopoulos O, Ohh M et al. (1998) Regulationof hypoxia-inducible mRNAs by the von Hippel-Lindautumor suppressor protein requires binding to complexescontaining elongins B/C and Cul2. Mol Cell Biol 18:732–741
Longuemaux S, Delomenie C, Gallou C et al. (1999) Candi-date genetic modifiers of individual susceptibility to re-nal cell carcinoma: a study of polymorphic human xeno-biotic-metabolizing enzymes. Cancer Res 59:2903–2908
Luiten RM, Coney LR, Fleuren GJ, Warnaar SO, Litvinov SV(1996) Generation of chimeric bispecific G250/anti-CD3monoclonal antibody, a tool to combat renal cell carcino-ma. Br J Cancer 74:735–744
Maher ER, Yates JR, Harries R et al. (1990) Clinical featuresand natural history of von Hippel-Lindau disease. QJM77:1151–1163
Mai N, Qian C, Yokomizu A et al. (1998) Loss of imprintingand allele switch of p53 in renal cell carcinoma. Onco-gene 17:1739–1741
Mandel JS, McLaughlin JK, Schlehofer B et al. (1995) Inter-national renal-cell cancer study. IV. Occupation. Int JCancer 61:601–605
Manski TJ, Heffner DK, Glenn GM et al. (1997) Endolym-phatic sac tumors. A source of morbid hearing loss invon Hippel-Lindau disease. JAMA 277:1461–1466
Matson MA, Cohen EP (1990) Acquired cystic kidney dis-ease: occurrence, prevalence, and renal cancers. Medicine(Baltimore) 69:217–226
Maxwell PH, Wiesener MS, Chang GW et al. (1999) The tu-mour suppressor protein VHL targets hypoxia-induciblefactors for oxygen-dependent proteolysis. Nature399:271–275
McCredie M, Pommer W, McLaughlin JK et al. (1995) Inter-national renal-cell cancer study. II. Analgesics. Int J Can-cer 60:345–349
McLaughlin JK, Blot WJ (1997) A critical review of epide-miology studies of trichloroethylene and perchloroethy-lene and risk of renal-cell cancer. Int Arch Occup Envi-ron Health 70:222–231
McLaughlin JK, Lindblad P, Mellemgaard A et al. (1995) In-ternational renal-cell cancer study. I. Tobacco use. Int JCancer 60:194–198
Mellemgaard A, Lindblad P, Schlehofer B et al. (1995) Inter-national renal-cell cancer study. III. Role of weight,height, physical activity, and use of amphetamines. Int JCancer 60:350–354
Melmon KL, Rosen SW (1964) Lindau’s disease: review ofthe literature and study of a large kindred. Am J Med36:595–617
Mickisch GH (1994) Chemoresistance of renal cell carcino-ma: 1986–1994. World J Urol 12:214–223
Miller MS, McCarver DG, Bell DA, Eaton DL, Goldstein JA(1997) Genetic polymorphisms in human drug metabolicenzymes. Fundam Appl Toxicol 40:1–14
Moch HP, Sauter G, Buchholz N et al. (1996) Genetic aberra-tions detected by comparative genomic hybridization areassociated with clinical outcome in renal cell carcinoma.Cancer Res 56:27–30
Moch H, Sauter G, Buchholz N et al. (1997) Epidermalgrowth factor receptor expression is associated with rap-id tumor cell proliferation in renal cell carcinoma. HumPathol 28:1255–1259
Moch H, Sauter G, Gasser TC et al. (1998) EGF-r gene copynumber changes in renal cell carcinoma detected byfluorescence in situ hybridization. J Pathol 184:424–429
Morita R, Saito S, Ishikawa et al. (1991) Common regions ofdeletion on chromosomes 5q, 6q, and 10q in renal cellcarcinoma. Cancer Res 51:5817–5820
Mostofi FK (1981) Histological typing of kidney tumors. In:Mostofi FK (ed) International histological classificationof tumours, vol 25. WHO, Geneva
Motzer RJ, Russo P (2000) Systemic therapy for renal cellcarcinoma. J Urol 163:408–417
Motzer RJ, Bander NH, Nanus DM (1996) Renal-cell carcino-ma. N Engl J Med 335:865–875
Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S,Sukhatme VP (1997) The von Hippel-Lindau tumor sup-pressor gene product interacts with Sp1 to repress vascu-lar endothelial growth factor promoter activity. Mol CellBiol 17:5629–5639
Müller M, Birner G, Dekant W (1998a) Reactivity of haloke-tenes and halothioketenes with nucleobases: chemicalcharacterization of reaction products. Chem Res Toxicol11:454–463
Müller M, Birner G, Sander M, Dekant W (1998b) Reactivityof haloketenes and halothioketenes with nucleobases: re-actions in vitro with DNA. Chem Res Toxicol 11:464–470
Murphy WM, Beckwith JB, Farrow GM (1994) Atlas of tu-mor pathology, tumors of the kidney, bladder, and re-lated urinary structures. Armed Forces Institute ofPathology, Washington DC, p 102
Nagata H, Worobec AS, Oh CK et al. (1995) Identification ofa point mutation in the catalytic domain of the protoon-cogene c-kit in peripheral blood mononuclear cells of pa-tients who have mastocytosis with an associated hemato-logic disorder. Proc Natl Acad Sci USA 92:10 560–10 564
Naitoh J, Kaplan A, Dorey F, Figlin R, Belldegrun A (1999)Metastatic renal cell carcinoma with concurrent inferiorvena caval invasion: long-term survival after combination
286 J. Decker und H. Brauch

therapy with radical nephrectomy, vena caval thrombecto-my and postoperative immunotherapy. J Urol 162:46–50
Nebert DW, McKinnon RA, Puga A (1996) Human drug-me-tabolizing enzyme polymorphisms: effects on risk oftoxicity and cancer. DNA Cell Biol 15:273–280
Neumann HP (1987a) Basic criteria for clinical diagnosisand genetic counselling in von Hippel-Lindau syndrome.Vasa 16:220–226
Neumann HP (1987b) Prognosis of von Hippel-Lindau syn-drome. Vasa 16:309–311
Neumann HP, Wiestler OD (1991) Clustering of features ofvon Hippel-Lindau syndrome: evidence for a complex ge-netic locus. Lancet 337:1052–1054
Neumann HP, Dinkel E, Brambs HJ et al. (1991) Pancreaticlesions in the von Hippel-Lindau syndrome. Gastroenter-ology 101:465–471
Neumann HP, Eng C, Mulligan LM et al. (1995) Conse-quences of direct genetic testing for germline mutationsin the clinical management of families with multiple en-docrine neoplasia, type II. JAMA 274:1149–1151
Oda H, Kume H, Shimizu Y, Inoue T, Ishikawa T (1998) Lossof imprinting of igf 2 in renal-cell carcinomas. Int J Can-cer 75:343–346
Ogawa O, Kakehi Y, Ogawa K, Koshiba M, Sugiyama T,Yoshida O (1991) Allelic loss at chromosome 3p charac-terizes clear cell phenotype of renal cell carcinoma. Can-cer Res 51:949–953
Ohh M, Yauch RL, Lonergan KM et al. (1998) The von Hip-pel-Lindau tumor suppressor protein is required forproper assembly of an extracellular fibronectin matrix.Mol Cell 1:959–968
Oosterwijk E, Ruiter DJ, Hoedemaeker PJ et al. (1986)Monoclonal antibody G 250 recognizes a determinantpresent in renal-cell carcinoma and absent from normalkidney. Int J Cancer 38:489–494
Pagliaro L, Daliani D, Amato R et al. (2000) A phase II trialof bryostatin-1 for patients with metastatic renal cell car-cinoma. Cancer 89:615–618
Pal S, Claffey KP, Dvorak HF, Mukhopadhyay D (1997) Thevon Hippel-Lindau gene product inhibits vascular perme-ability factor/vascular endothelial growth factor expres-sion in renal cell carcinoma by blocking protein kinase Cpathways. J Biol Chem 272:27 509–27 512
Paradis V, Lagha NB, Zeimoura L et al. (2000) Expression ofvascular endothelial growth factor in renal cell carcino-mas. Virchows Arch 436:351–356
Park M, Dean M, Kaul K, Braun MJ, Gonda MA, VandeWoude G (1987) Sequence of MET protooncogene cDNAhas features characteristic of the tyrosine kinase familyof growth-factor receptors. Proc Natl Acad Sci USA84:6379–6383
Parkin DM, Muir CS (1992) Cancer incidence in five conti-nents. Comparability and quality of data. IARC Sci Publ120:45–173
Pathak S, Strong LC, Ferrell RE, Trindade A (1982) Familialrenal cell carcinoma with a 3;11 chromosome transloca-tion limited to tumor cells. Science 217:939–941
Pause A, Lee S, Worrell RA et al. (1997) The von Hippel-Lin-dau tumor-suppressor gene product forms a stable com-plex with human CUL-2, a member of the Cdc53 familyof proteins. Proc Natl Acad Sci USA 94:2156–2161
Pause A, Lee S, Lonergan KM, Klausner RD (1998) The vonHippel-Lindau tumor suppressor gene is required for cellcycle exit upon serum withdrawal. Proc Natl Acad SciUSA 95:993–998
Pearson HH (1969) Familial renal tumours. Aust N Z J Surg38:333–338
Pesch B, Haerting J, Ranft U, Klimpel A, Oelschlagel B,Schill W (2000) Occupational risk factors for renal cellcarcinoma: agent-specific results from a case-controlstudy in Germany. Int J Epidemiol 29:1014–1024
Phillips E, Messing EM (1993) Role of lymphadenectomy inthe treatment of renal cell carcinoma. Urology 41:9–15
Poston CD, Jaffe GS, Lubensky IA et al. (1995) Characteriza-tion of the renal pathology of a familial form of renal cellcarcinoma associated with von Hippel-Lindau disease:clinical and molecular genetic implications. J Urol153:22–26
Press GA, McClennan BL, Melson GL, Weyman PJ, MauroMA, Lee JK (1984) Papillary renal cell carcinoma: CT andsonographic evaluation. AJR Am J Roentgenol 143:1005–1009
Puga A, Nebert DW, McKinnon RA, Menon AG (1997) Ge-netic polymorphisms in human drug-metabolizing en-zymes: potential uses of reverse genetics to identify genesof toxicological relevance. Crit Rev Toxicol 27:199–222
Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ(1997) Activation of hypoxia-inducible factor-1; defini-tion of regulatory domains within the alpha subunit. JBiol Chem 272:11 205–11 214
Ramp U, Reinecke P, Gabbert HE, Gerharz CD (2000) Differ-ential response to transforming growth factor (TGF)-al-pha and fibroblast growth factor (FGF) in human renalcell carcinomas of the clear cell and papillary types. EurJ Cancer 36:932–941
Richards FM, Schofield PN, Fleming S, Maher ER (1996) Ex-pression of the von Hippel-Lindau disease tumour sup-pressor gene during human embryogenesis. Hum MolGenet 5:639–644
Robson CJ, Churchill BM, Anderson W (1969) The results ofradical nephrectomy for renal cell carcinoma. J Urol101:297–301
Roth JS, Rabinowitz AD, Benson M, Grossman ME (1993)Bilateral renal cell carcinoma in the Birt-Hogg-Dube syn-drome. J Am Acad Dermatol 29:1055–1056
Russo P (2000) Renal cell carcinoma: presentation, staging,and surgical treatment. Semin Oncol 27:160–176
Sahin U, Tureci O, Schmitt H et al. (1995) Human neoplasmselicit multiple specific immune responses in the auto-logous host. Proc Natl Acad Sci USA 92:11 810–11 813
Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha(HIF-1alpha) protein is rapidly degraded by the ubiqui-tin-proteasome system under normoxic conditions. Itsstabilization by hypoxia depends on redox-inducedchanges. J Biol Chem 272:22 642–22 647
Sampson JR (1996) The kidney in tuberous sclerosis: mani-festations and molecular genetic mechanisms. NephrolDial Transplant 11:34–37
Sato K, Tsuchiya N, Sasaki R et al. (1999) Increased serumlevels of vascular endothelial growth factor in patientswith renal cell carcinoma. Jpn J Cancer Res 90:874–879
Schlehofer B, Pommer W, Mellemgaard A et al. (1996) Inter-national renal-cell-cancer study. VI. The role of medicaland family history. Int J Cancer 66:723–726
Schlessinger J, Ullrich A (1992) Growth factor signaling byreceptor tyrosine kinases. Neuron 9:383–391
Schmidt L, Duh FM, Chen F et al. (1997) Germline and so-matic mutations in the tyrosine kinase domain of theMET proto-oncogene in papillary renal carcinomas. NatGenet 16:68–73
a 9 Nierenzellkarzinome 287

Schmidt L, Junker K, Nakaigawa N et al. (1999) Novel muta-tions of the MET proto-oncogene in papillary renal carci-nomas. Oncogene 18:2343–2350
Schoenfeld AR, Parris T, Eisenberger A et al. (2000) The vonHippel-Lindau tumor suppressor gene protects cells fromUV-mediated apoptosis. Oncogene 19:5851–5857
Schulz WA (1998) DNA methylation in urological malignan-cies. Int J Oncol 13:151–167
Seizinger BR, Rouleau GA, Ozelius LJ et al. (1988) Von Hip-pel-Lindau disease maps to the region of chromosome 3associated with renal cell carcinoma. Nature 332:268–269
Semenza GL (1998) Hypoxia-inducible factor 1 and the mo-lecular physiology of oxygen homeostasis. J Lab ClinMed 131:207–214
Shuin T, Kondo K, Torigoe S et al. (1994) Frequent somaticmutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor gene in primary human renalcell carcinomas. Cancer Res 54:2852–2855
Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Ba-ron G, Marme D (1996) Reversion of deregulated expres-sion of vascular endothelial growth factor in human renalcarcinoma cells by von Hippel-Lindau tumor suppressorprotein. Cancer Res 56:2299–2301
Solomon E, Bodmer WF (1979) Evolution of sickle variantgene. Lancet 1:923
Stebbins CE, Kaelin WG Jr, Pavletich NP (1999) Structure ofthe VHL-ElonginC-ElonginB complex: implications forVHL tumor suppressor function. Science 284:455–461
Steinbach F, Stöckle M, Riedmiller H, Weingartner K, Ho-henfellner R (1991) Tumor enucleation of renal cell carci-noma: operative technique, DNA cytometry results, com-plications. Prog Clin Biol Res 370:1–7
Steinbach F, Stöckle M, Müller SC et al. (1992) Conservativesurgery of renal cell tumors in 140 patients: 21 years ofexperience. J Urol 148:24–30
Steinbach F, Stöckle M, Griesinger A et al. (1994) Multifocalrenal cell tumors: a retrospective analysis of 56 patientstreated with radical nephrectomy. J Urol 152:1393–1396
Stolle C, Glenn G, Zbar B et al. (1998) Improved detectionof germline mutations in the von Hippel-Lindau diseasetumor suppressor gene. Hum Mutat 12:417–423
Störkel S, Keymer R, Steinbach F, Thoenes W (1992) Reac-tion patterns of tumor infiltrating lymphocytes in differ-ent renal cell carcinomas and oncocytomas. Prog ClinBiol Res 378:217–223
Störkel S, Eble JN, Adlakha K et al. (1997) Classification ofrenal cell carcinoma: Workgroup No. 1. Union Internatio-nale Contre le Cancer (UICC) and the American JointCommittee on Cancer (AJCC). Cancer 80:987–989
Sugimura K, Yoshida N, Hisatomi H, Nakatani T, Ikemoto S(1999) Telomerase in human renal cell carcinoma. B JUrol Int 83:693–697
Teh BT, Giraud S, Sari NF et al. (1997) Familial non-VHLnon-papillary clear-cell renal cancer. Lancet 349:848–849
Thoenes W, Störkel S, Rumpelt HJ (1986) Histopathologyand classification of renal cell tumors (adenomas, onco-cytomas and carcinomas). The basic cytological and his-topathological elements and their use for diagnostics.Pathol Res Pract 181:125–143
Thoenes W, Störkel S, Rumpelt HJ, Moll R (1990) Cytomor-phological typing of renal cell carcinoma – a new approach.Eur Urol 18:6–9
Tisherman SE, Tisherman BG, Tisherman SA, Dunmire S,Levey GS, Mulvihill JJ (1993) Three-decade investigation
of familial pheochromocytoma. An allele of von Hippel-Lindau disease? Arch Intern Med 153:2550–2556
Tomisawa M, Tokunaga T, Oshika Y et al. (1999) Expressionpattern of vascular endothelial growth factor isoform isclosely correlated with tumour stage and vascularisationin renal cell carcinoma. Eur J Cancer 35:133–137
Toro JR, Glenn G, Duray P et al. (1999) Birt-Hogg-Dubesyndrome: a novel marker of kidney neoplasia. Arch Der-matol 135:1195–1202
Tory K, Brauch H, Linehan M et al. (1989) Specific geneticchange in tumors associated with von Hippel-Lindau dis-ease. J Natl Cancer Inst 81:1097–1101
Ullrich A, Schlessinger J (1990) Signal transduction by re-ceptors with tyrosine kinase activity. Cell 61:203–212
Vamvakas S, Bruning T, Thomasson B et al. (1998) Renalcell cancer correlated with occupational exposure to tri-chloroethene. J Cancer Res Clin Oncol 124:374–382
Van den Berg E, Oosterhuis JW, Störkel S et al. (1994)Pathogenesis of renal cell tumors: a cytogenetic model.44th European Workshop on Cytogenetics and MolecularGenetics of Human Solid Tumors, Noordwijkerhout, TheNetherlands, A:45
Van der Hout AH, Brown RS, Li FP, Buys CH (1991) Locali-zation by in situ hybridization of three 3p probes withrespect to the breakpoint in a t(3;8) in hereditary renalcell carcinoma. Cancer Genet Cytogenet 51:121–124
Van Slegtenhorst M, Hoogt de R, Hermans C et al. (1997)Identification of the tuberous sclerosis gene TSC1 onchromosome 9q34. Science 277:805–808
Vogelzang NJ, Stadler WM (1998) Kidney cancer. Lancet352:1691–1696
Volkel W, Dekant W (1998) Chlorothioketene, the ultimatereactive intermediate formed by cysteine conjugate beta-lyase-mediated cleavage of the trichloroethene metaboliteS-(1,2-Dichlorovinyl)-L-cysteine, forms cytosine adductsin organic solvents, but not in aqueous solution. ChemRes Toxicol 11:1082–1088
Volm M, Pommerenke EW, Efferth T, Lohrke H, Mattern J(1991) Circumvention of multi-drug resistance in humankidney and kidney carcinoma in vitro. Cancer 67:2484–2489
Von Hippel E (1904) Über eine seltene Erkrankung derNetzhaut. Graefes Arch Ophthalmol 59:83–106
Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM(1995) Prevalence of microscopic lesions in grossly nor-mal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no re-nal disease: clinical implications. J Urol 154:2010–2015
Wang N, Perkins KL (1984) Involvement of band 3p14 in t(3;8)hereditary renal carcinoma. Cancer Genet Cytogenet11:479–481
Warburg O (1926) Über den Stoffwechsel von Tumoren.Cambridge University Press, Cambridge
Washecka R, Hanna M (1991) Malignant renal tumors in tu-berous sclerosis. Urology 37:340–343
Wechsel HW, Bichler KH, Feil G, Loeser W, Lahme S, PetriE (1999) Renal cell carcinoma: relevance of angiogeneticfactors. Anticancer Res 19:1537–1540
Weidner U, Peter S, Strohmeyer T, Hussnatter R, AckermannR, Sies H (1990) Inverse relationship of epidermal growthfactor receptor and HER2/neu gene expression in humanrenal cell carcinoma. Cancer Res 50:4504–4509
Weidner KM, Sachs M, Birchmeier W (1993) The Met recep-tor tyrosine kinase transduces motility, proliferation, and
288 J. Decker und H. Brauch

morphogenic signals of scatter factor/hepatocyte growthfactor in epithelial cells. J Cell Biol 121:145–154
Wenger RH, Gassmann M (1997) Oxygen(es) and the hypox-ia-inducible factor-1. Biol Chem 378:609–616
Whaley JM, Naglich J, Gelbert L et al. (1994) Germ-line mu-tations in the von Hippel-Lindau tumor-suppressor geneare similar to somatic von Hippel-Lindau aberrations insporadic renal cell carcinoma [published erratum appearsin Am J Hum Genet 56:356]. Am J Hum Genet 55:1092–1102
Yao M, Latif F, Orcutt ML et al. (1993) Von Hippel-Lindaudisease: identification of deletion mutations by pulsed-field gel electrophoresis. Hum Genet 92:605–614
Ye Y, Vasavada S, Kuzmin I, Stackhouse T, Zbar B, WilliamsBR (1998) Subcellular localization of the von Hippel-Lin-dau disease gene product is cell cycle-dependent. Int JCancer 78:62–69
Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG(1994) Predisposition to renal carcinoma in the Eker ratis determined by germ-line mutation of the tuberoussclerosis 2 (TSC2) gene. Proc Natl Acad Sci USA 91:11413–11 416
Yoshida K, Tosaka A (1994) Epidermal growth factor bind-ing by membranes of human renal cell carcinomas: es-tablishment of an epidermal growth factor receptor assayfor clinical use. Int J Urol 1:319–323
Yoshida K, Hosoya Y, Sumi S et al. (1997) Studies of the ex-pression of epidermal growth factor receptor in humanrenal cell carcinoma: a comparison of immunohisto-chemical method versus ligand binding assay. Oncology54:220–225
Yu DS, Chang SY, Ma CP (1998) The expression of mdr-1-related gp-170 and its correlation with anthracycline re-sistance in renal cell carcinoma cell lines and multidrug-resistant sublines. Br J Urol 82:544–547
Yu DS, Sun GH, Ma CP, Chang SY (1999) Cocktail modula-tor mixtures for overcoming multidrug resistance in re-nal cell carcinoma. Urology 54:377–381
Zbar B (1995) Von Hippel-Lindau disease and sporadic renalcell carcinoma. Cancer Surv 25:219–232
Zbar B, Lerman M (1998) Inherited carcinomas of the kid-ney. Adv Cancer Res 75:163–201
Zbar B, Brauch H, Talmadge C, Linehan M (1987) Loss of al-leles of loci on the short arm of chromosome 3 in renalcell carcinoma. Nature 327:721–724
Zbar B, Tory K, Merino M et al. (1994) Hereditary papillaryrenal cell carcinoma. J Urol 151:561–566
Zbar B, Glenn G, Lubensky I et al. (1995a) Hereditary papil-lary renal cell carcinoma: clinical studies in 10 families. JUrol 153:907–912
Zbar B, Glenn G, Lubensky I et al. (1995b) Hereditary papil-lary renal cell carcinoma: clinical studies in 10 families. JUrol 153:907–912
Zbar B, Kishida T, Chen F et al. (1996) Germline mutationsin the Von Hippel-Lindau disease (VHL) gene in familiesfrom North America, Europe, and Japan. Hum Mutat8:348–357
Zhuang Z, Park WS, Pack S et al. (1998) Trisomy 7-harbouringnon-random duplication of the mutant MET allele in her-editary papillary renal carcinomas. Nat Genet 20:66–69
a 9 Nierenzellkarzinome 289





























