Embed Size (px)
Citation preview

157
9. Zusammenfassung und Ausblick
Die Aldolreaktion spielt in der organischen Synthesechemie eine wichtige Rolle für
den Zugang zu industriellen Produkten im Großmaßstab aber auch zu
feinchemischen Vorstufen für die Pharmaindustrie. Insbesondere für letztere ist es
im Sinne einer ökonomischen und Ressourcen schonenden Synthese notwendig,
die Reaktionsprodukte reproduzierbar in enantiomerenangereicherter oder gar
enantiomerenreiner Form zu erhalten. Die Menge an einzusetzendem Katalysator
zum Erreichen möglichst schneller Umsetzungen spielt ebenfalls eine erhebliche
Rolle. Die Aldolreaktion ist in der klassischen organischen Synthese als entweder
säure- oder basenkatalysiert bekannt, ohne Gegenwart entweder der einen oder
der anderen Spezies verläuft die Reaktion unmerklich langsam oder sie läuft gar
nicht ab.
Es sind unterschiedliche Ansätze verfolgt worden, um die Aldolreaktion katalytisch
zu beschleunigen und stereoselektiv zu steuern. Sehr häufig werden beispielsweise
vorsynthetisierte Silylenolate in Gegenwart von Lewissäuren wie Titanchlorid in
wasserfreiem Dichlormethan oder THF bei -78°C umgesetzt (Mukaiyama, 1973) [12].
Eine Abwandlung ist die Enolisierung mit LDA (Lithiumdiisopropylamid) bei tiefen
Temperaturen. Verwendet werden außerdem Metalle wie Bor, Magnesium,
Aluminium, Zirconium, Rhodium, Cer, Molybdän, Rhenium, Cobalt, Eisen und Zink.
Andere Methoden bedienen sich nach der Reaktion abzuspaltenden Auxiliaren, wie
z.B. dem Evans-Oxazolidinon [15], des weiteren sind Shibasakis BINOL-Barium-
komplexe [16] oder dinucleare organische Zinkkomplexe nach Trost [17] als Methode
zur direkten Aldolreaktion verbreitet. Die dabei erforderlichen Bedingungen, wie
Abwesenheit von Wasser (auch aufwändig getrocknete Edukte) und tiefe
Temperaturen stellen hohe apparative Ansprüche bei der Durchführung der
Reaktion.
Die Verwendung von teilweise toxischen Metallen verursacht umwelttechnische
Probleme, des weiteren müssen die Metalle später aufwendig recyclisiert werden.
Die dabei üblichen Metallkatalysatoren werden daher zunehmend in neuerer Zeit
durch rein organische, also metallfreie Katalysatoren ersetzt. Ziel dabei ist es unter
anderem auch umweltpolitische Aspekte zu berücksichtigen, was nicht nur die

158
Katalysatoren angeht, sondern auch die verwendeten Reaktionsmedien,
Temperaturen und die Möglichkeit der Gegenwart von Wasser beinhaltet, des
weiteren die einfachere Abtrennbarkeit der Katalysatoren von Reaktionsgemischen
und der damit verbundenen Schonung der Umwelt aufgrund ihrer relativen
toxikologischen Unbedenklichkeit.
Zur Evaluierung der Eigenschaften neuer Katalysatoren für die Aldolkatalyse
wurden in dieser Arbeit eine Reihe nicht natürlicher Aminosäuren synthetisiert, die
als Monomere in Verbindung mit anderen natürlichen Aminosäuren wie L- und D-
Prolin, L-Dopamin, L-Tyrosin, L-Phenylalanin, 4-Trans-L-Hydroxyprolin und β-Alanin
zu Peptidkatalysatoren verknüpft wurden. Hierbei dienten verschiedene
Festphasen-Syntheseharze als Trägermedium während der Synthese nach
bewährten, gängigen und speziell angepassten Festphasen-Peptidsynthese-
protokollen. Eine Übersicht der verwendeten und nicht natürlichen Monomere gibt
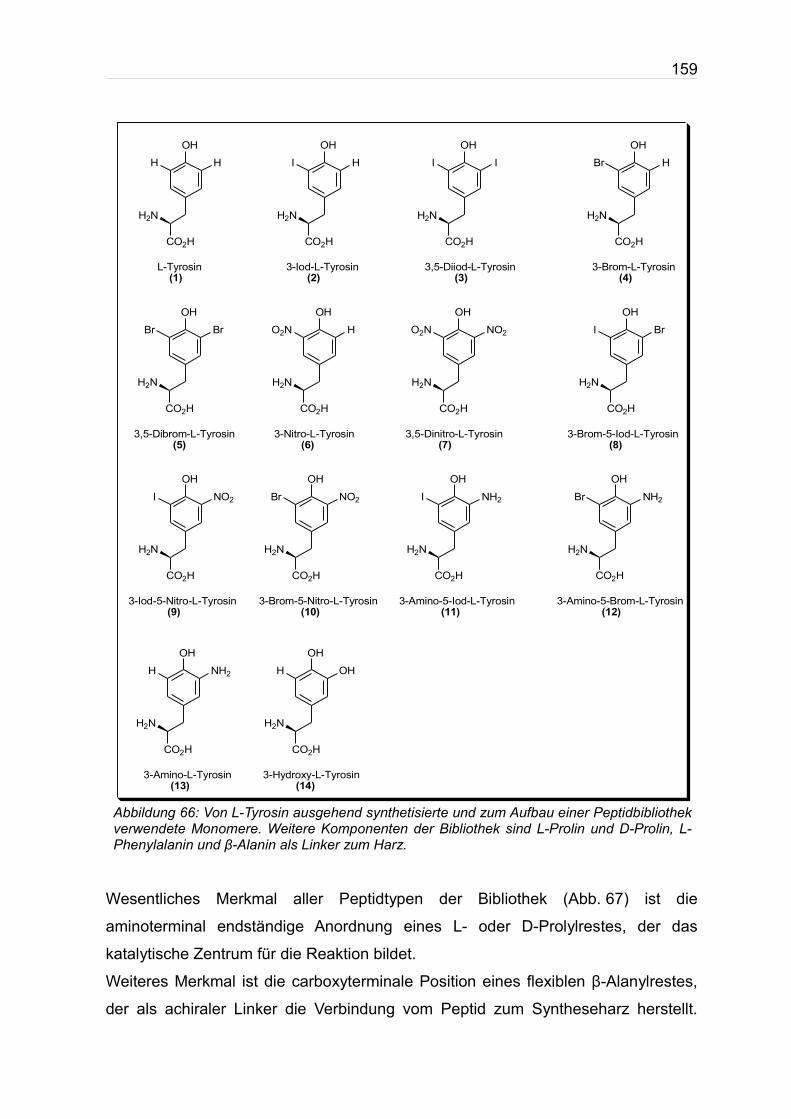
Abb. 66 auf der nächsten Seite. Alle Verbindungen sind Abwandlungen von Tyrosin,
da erwartet wird, das die OH-Funktion und das aromatische π-System eine Rolle
bei der Aldol-Katalyse spielen. Diese wurden zunächst in Vortests an
verschiedenen Harzen zu Peptiden synthetisiert und in ihrer Eignung zur
enantioselektiven Aldolkatalyse untersucht, ebenso erfolgten Katalysetests der
freien Monomere (ohne Prolin) in Lösung, um die Konstellation der
Reaktionsvolumina, der erforderlichen zeitlichen Dimension solcher Katalysetests,
Methoden zur Ausbeutebestimmung und einzusetzende Substratmengen zu
ermitteln. Gleichzeitig erfolgten Katalysetests in Lösung und unter Einsatz
festphasengebundener Peptide mit Verwendung von L- und D-Prolin. Die
literaturbekannte Katalysereaktion des Prolins oder festphasengebundener
struktureller Analoga des Prolins dienten als Standard bezüglich Lösungsmittel,
Reaktionsdauer und dabei erzieltem Grad an Umsetzung und später nach
Evaluierung zuverlässiger NMR-Methoden unter Zuhilfenahme chiraler NMR-
Shiftreagenzien auch zur Ermittlung und Vergleich erzielter Enantiomeren-
überschüsse bei Katalyse durch eigene Peptide. Als Ausgangspunkt wurde
zunächst eine Peptidbibliothek mit insgesamt 110 verschiedenen Tri- und
Tetrapeptiden hergestellt, um dann nach geeigneten Kandidaten im Hinblick auf
den notwendigen Kompromiss zwischen Reaktionsdauer und Umsetzung an
Edukten zum erwünschten Produkt verbunden mit möglichst geringem Anteil an
Reaktionsnebenprodukten zu suchen.
159
Wesentliches Merkmal aller Peptidtypen der Bibliothek (Abb. 67) ist die
aminoterminal endständige Anordnung eines L- oder D-Prolylrestes, der das
katalytische Zentrum für die Reaktion bildet.
Weiteres Merkmal ist die carboxyterminale Position eines flexiblen β-Alanylrestes,
der als achiraler Linker die Verbindung vom Peptid zum Syntheseharz herstellt.
Abbildung 66: Von L-Tyrosin ausgehend synthetisierte und zum Aufbau einer Peptidbibliothek verwendete Monomere. Weitere Komponenten der Bibliothek sind L-Prolin und D-Prolin, L-Phenylalanin und β-Alanin als Linker zum Harz.
OH
CO2H
H2N
HH
L-Tyrosin (1)
OH
CO2H
H2N
HI
3-Iod-L-Tyrosin (2)
OH
CO2H
H2N
II
3,5-Diiod-L-Tyrosin (3)
OH
CO2H
H2N
HBr
3-Brom-L-Tyrosin (4)
OH
CO2H
H2N
BrBr
3,5-Dibrom-L-Tyrosin (5)
OH
CO2H
H2N
HO2N
3-Nitro-L-Tyrosin (6)
OH
CO2H
H2N
NO2O2N
3,5-Dinitro-L-Tyrosin (7)
OH
CO2H
H2N
BrI
3-Brom-5-Iod-L-Tyrosin (8)
OH
CO2H
H2N
NO2I
3-Iod-5-Nitro-L-Tyrosin (9)
OH
CO2H
H2N
NO2Br
3-Brom-5-Nitro-L-Tyrosin (10)
OH
CO2H
H2N
NH2I
3-Amino-5-Iod-L-Tyrosin (11)
OH
CO2H
H2N
NH2Br
3-Amino-5-Brom-L-Tyrosin (12)
OH
CO2H
H2N
NH2H
3-Amino-L-Tyrosin (13)
OH
CO2H
H2N
OHH
3-Hydroxy-L-Tyrosin (14)

160
Ergänzt wurde die Bibliothek durch den Einsatz von trans-Hydroxyprolin in
gewöhnlicher Peptidbindung und einige über eine Esterbrücke zum Hydroxyprolin
verknüpfte Depsipeptide in somit für Peptide ungewöhnlicher Bindungsform. Eine
solche Bibliothek kann enormen Umfang annehmen, erwähnt werden sollte hierbei,
dass die Bibliothek keinen Anspruch auf Vollständigkeit erhebt und mit Rücksicht
auf den zu erwartenden Syntheseaufwand auch gar nicht will, da die Peptide ohne
Zuhilfenahme automatisierbarer Hilfsmittel wie Peptidsynthesizer komplett von
Hand hergestellt wurden.
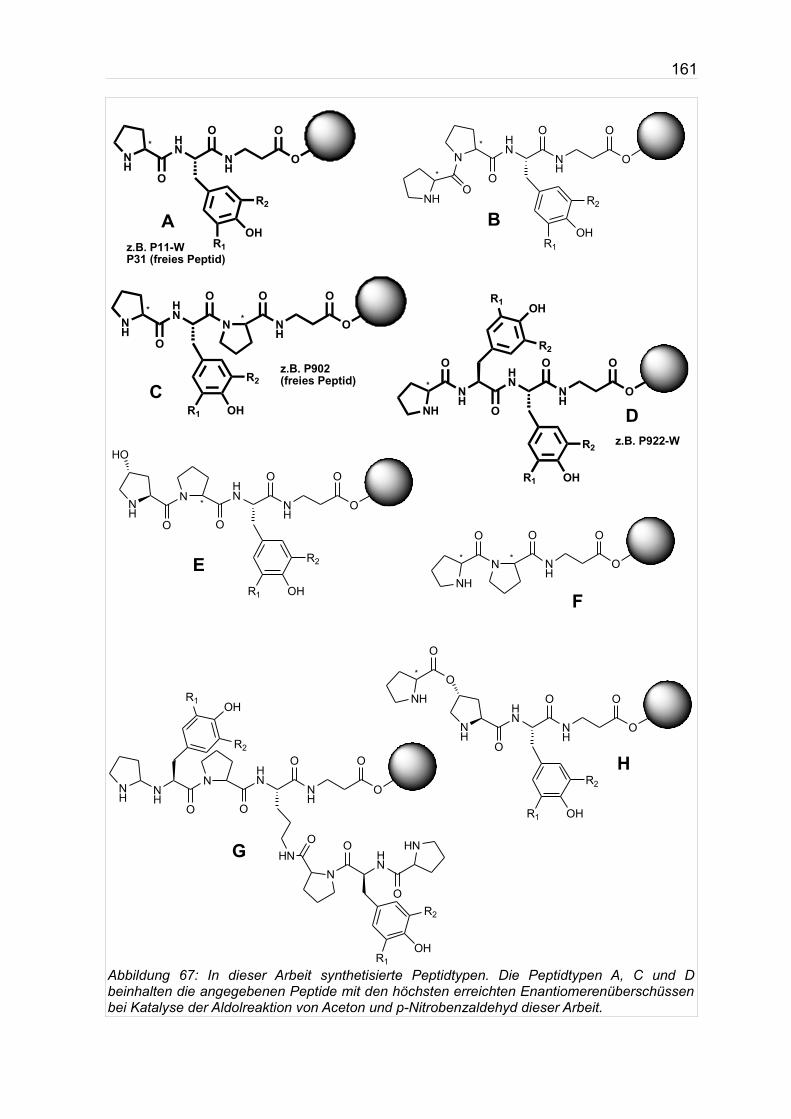
Eine schematische Übersicht der synthetisierten Peptidvarianten gibt Abb. 67 auf
der nächsten Seite.
In der folgenden Aufzählung sind die in Abb. 67 aufgeführten und
gekennzeichneten Peptide in der für Peptidsequenzen üblichen Text-Schreibweise
angegeben. (modTyr steht hier für modifiziertes Tyrosin)
A. H-Pro-modTyr-β-Ala-Harz
B. H-Pro-Pro-modTyr-β-Ala-Harz
C. H-Pro-modTyr-Pro-β-Ala-Harz
D. H-Pro-modTyr-modTyr-β-Ala-Harz
E. H-L-Hyp-Pro-modTyr-β-Ala-Harz
F. H-Pro-Pro-β-Ala-Harz
G. H-Pro-(O)-Hyp-modTyr-β-Ala-Harz (Depsipeptid)
H. (H-Pro-modTyr-Pro)2-α,ε-Lys-β-Ala-Harz (verzweigtes Peptid)
Die Peptidtypen A-D (siehe dazu auch nächste Seite) stellen hierbei den Hauptteil
der Bibliothek dar. Peptidtyp E ist eine Erweiterung des Typs B und enthält nur
wenige Mitglieder.
Peptidtyp F dient zum Vergleich längerer und Tyrosin enthaltender Peptide mit rein
aliphatischen Peptiden.
Die Peptidtypen A, C und D beinhalten hierbei die Peptide mit den höchsten in
dieser Arbeit erzielten Enantiomerenüberschüssen.
161
Abbildung 67: In dieser Arbeit synthetisierte Peptidtypen. Die Peptidtypen A, C und D beinhalten die angegebenen Peptide mit den höchsten erreichten Enantiomerenüberschüssen bei Katalyse der Aldolreaktion von Aceton und p-Nitrobenzaldehyd dieser Arbeit.
O
HN*
NH
OH
O
NH
O
O
R2
R1
O
HN*
N
OH
O
NH
O
O
R2
R1
*
ONH
O
NH
O
ON *
O
OH
R2
R1
HN*
O
NH
O
NH
O
O
R2
R1 OH
HN
O
OHR1
R2
NH
*
O
NH
O
NH
O
O
R2
R1 OH
HN
O
N*
O
NH
HO
O
NH
O
O
R2
R1 OH
HN
O
NH
O*
O
NH
O
NH
O
O
HN
HN
O
N
N
O
O
NH
R2
OHR1
NH
HN
O
OHR1
R2
O
HN
O
NH
O
ON **
O
NH
A B
CD
E
F
G
H
z.B. P922-W
z.B. P11-WP31 (freies Peptid)
z.B. P902(freies Peptid)

162
Die festphasengebundenen Peptide wurden nach der Synthese für die
Katalysetests der Reaktion aus p-Nitrobenzaldehyd und Aceton als
Reaktionspartner und Lösungsmittel zugleich verwendet. Die Peptidharze wurden
mit Stammlösungen des Aldehyds und einer genau bemessenen Menge Aceton
versetzt, um einheitliche Bedingungen für die Reaktion zu gewährleisten. Nach
einer festgelegten Zeitspanne zur Reaktion konnte der Reaktionsfortschritt mittels
NMR durch Vergleich der Eduktsignale mit den Produktsignalen erfasst werden.
Die Auswertung zeigte sehr unterschiedliche Ausbeuten von Aldolprodukt
(zwischen 3 % und über 86 %) und ermöglichte eine Unterteilung in schnell
umsetzende und weniger schnell oder langsam umsetzende Peptid-Harze. Bei der
Katalyse spielten die beteiligten Monomere und die Sequenzen der Peptide eine
Rolle. Beispielsweise erwiesen sich Sequenzen mit Diiodtyrosin, Dinitrotyrosin und
L-Dopa als für schnelle Katalyse ungeeignet, wohingegen 3-Nitrotyrosin, 3-Brom-5-
Iodtyrosin und insbesondere 3-Iod-5-Nitrotyrosin in der Sequenz gute bis
hervorragende Umsetzungsraten ermöglichten. Neben 3-Iod-5-Nitrotyrosin und 3-
Nitrotyrosin in der Peptidsequenz erzielen insbesondere Peptide der Anordnung A,
C und D (siehe Abb. 67) hohe Umsetzungsraten, auch innerhalb eines
Sequenztyps lassen sich Tendenzen erkennen, je nach eingesetztem Monomer.
Für die Bestimmung von Enantiomerenüberschüssen wurden chirale NMR-
Verschiebungsreagenzien verwendet, da keine funktionsfähige chirale HPLC zur
Verfügung stand. Dies erforderte eine chromatographische Trennung reiner
Produkte von Hand bei etwa der Hälfte der synthetisierten Mitglieder der Bibliothek
und anschließenden stufenweisen Zusatz der chiralen Lanthanoiden-Shift-
reagenzien.
Die erreichten Enantiomerenüberschüsse reichen von 0 % Überschuss des einen
gegenüber dem anderen Enantiomer (Racemat) bis zu 56 % Überschuss für das R-
oder S-Aldolisomer. Bei festphasengebundenen Peptiden bewirkt aminoterminal
endständiges L-Prolin in der Regel die Entstehung des R-Aldols, bei D-Prolin ist es
genau umgekehrt. Bei racemischen Produktgemischen der Peptidkatalyse besteht
kein erkennbarer Zusammenhang mit der aminoterminal-endständigen Chiralität
der L-Prolyl- oder D-Prolylreste. Es gibt also sowohl endständig L-Prolin tragende,
das Racemat katalysierende Peptide, als auch endständig D-Prolin tragende

163
Peptide, die ein racemisches Produktgemisch katalysieren. Die Entstehung eines
bestimmten Enantiomerenverhältnisses wird demzufolge auch durch die Sequenz
und die verwendeten Tyrosinderivate stark mitbeeinflusst. Dies bestätigt sich auch
insbesondere durch Ausnahmen, bei denen Peptide mit endständigem L- und D-
Prolin bei ansonsten identischer Restsequenz dasselbe Aldolenantiomer erzeugen
können. Zu Ausnahmen siehe auch Abb. 70.
Sowohl in den Peptiden des Typs A, als auch in den Peptidtypen C und D finden
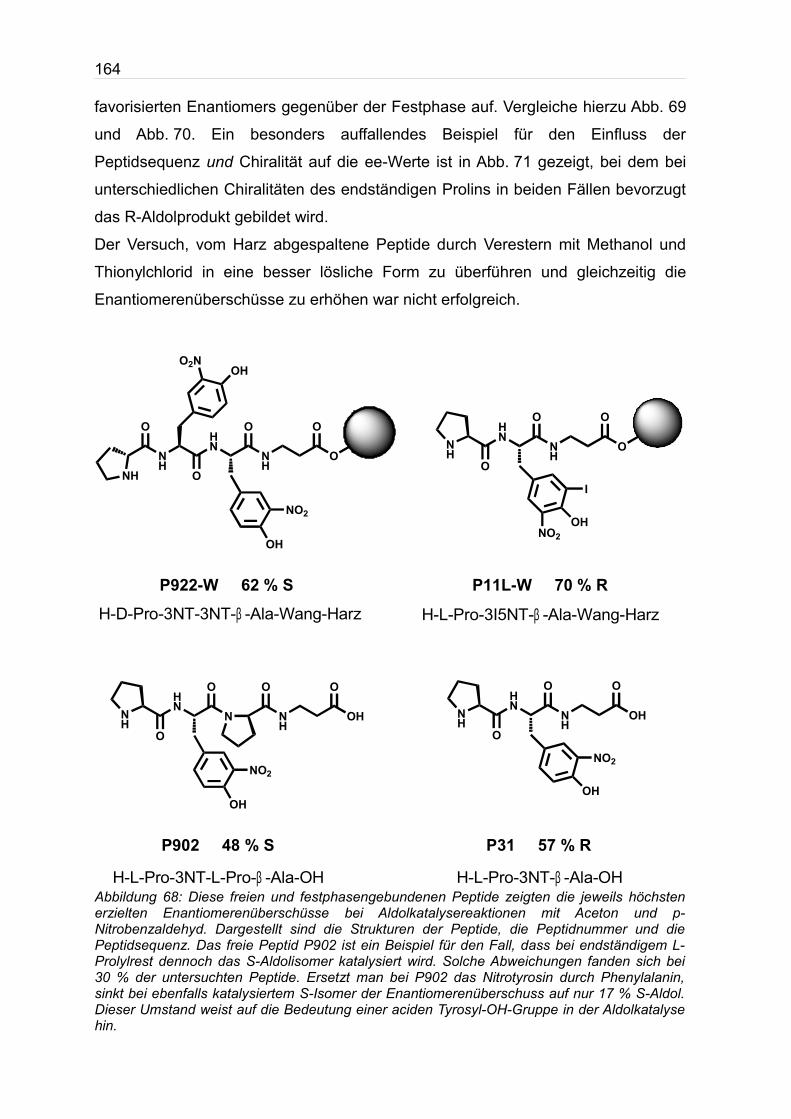
sich Sequenzen mit hohen Enantiomerenüberschüssen. Einige Beispiele sind in
Abb. 68 gezeigt. Die Länge der Peptide zeigt starke Variationen in der
Reaktionsgeschwindigkeit gegenüber den erzielten Enantiomerenüberschüssen.
Ein allgemeiner Trend scheint zu sein, dass kürzere Peptide tendenziell höhere
Umsatzraten und etwas höhere Enantiomerenverhältnisse versprechen.
Eine Aldolkatalyse in Lösung sollten 28 zusätzlich an Polystyrol-Wang-Harz in
größerer Menge synthetisierte Peptide ermöglichen. Einige dieser Peptide
beinhalten neue Peptidequenzen, die eine Erweiterung der zuvor erzeugten
Bibliothek darstellen. Diese wurden nach der Synthese bis auf eine kleine
zurückbehaltene Menge für Festphasenkatalysetests vom Trägerharz abgespalten
und in Aldolkatalysetests in Lösung mit freien Peptiden eingesetzt. Bei
Katalysetests in Lösung gelangen Umsetzungen von 50 % bis über 96 % in 24 h
mit Enantiomerenüberschüssen von bis zu 57 % R-Aldol und bis zu 48 % S-Aldol.
Zwei Beispiele sind in Abb. 68 aufgeführt. Festphasengebundene Peptide
katalysieren die Reaktion vermutlich diffusionsbedingt erheblich langsamer,
erreichten jedoch bis zu 70 % R-Aldol und bis zu 62 % S-Aldolprodukt.
Somit lagen die erzielten Enantiomerenüberschüsse bei einigen Peptiden teilweise
erheblich über denen mit L-Prolin oder D-Prolin allein erreichbaren Werten.
Die Suche nach Peptidkatalysatoren für die Aldolreaktion, die das Prolin im Hinblick
auf Enantiomerenüberschüsse übertreffen, kann so als erfolgreich angesehen
werden.
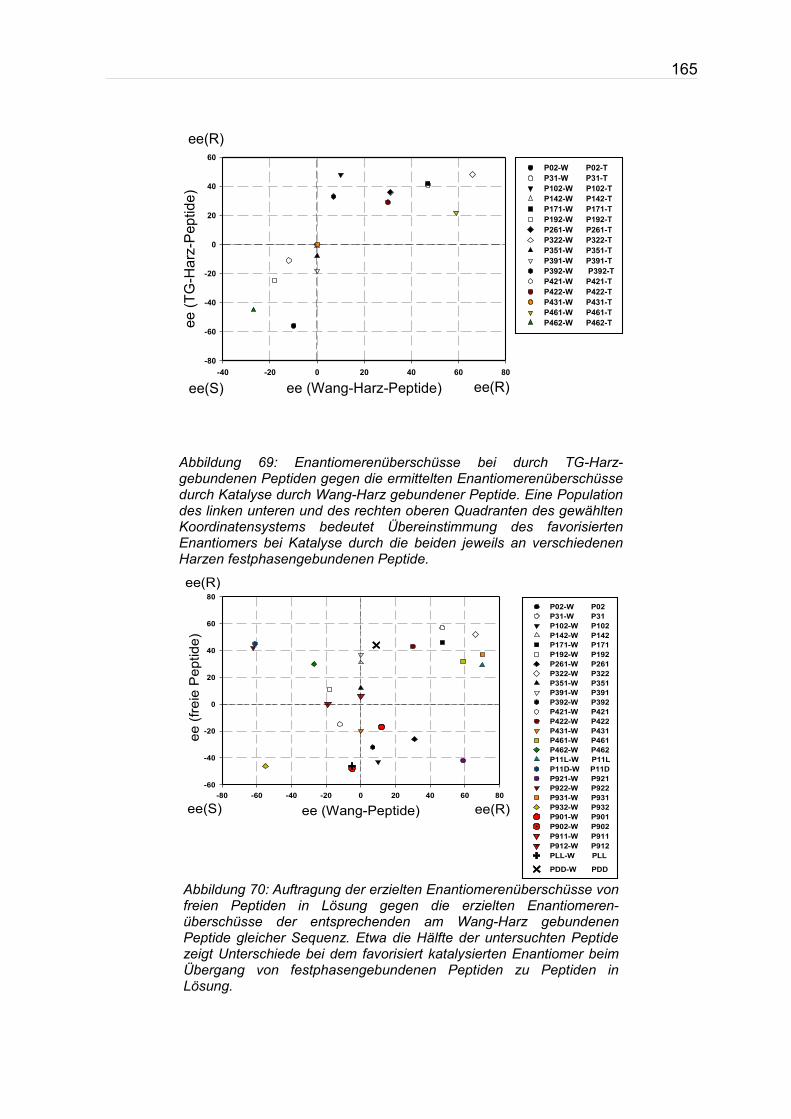
Bei Vergleich der Enantiomerenüberschüsse und insbesondere des favorisiert
katalysierten Enantiomers festphasengebundener Peptide untereinander und im
Vergleich mit Katalysetests in Lösung fallen die Ergebnisse bei festphasen-
mediierter Aldolkatalyse übereinstimmend zusammen und unterscheiden sich
lediglich im Grad des erreichten Enantiomerenüberschusses. Katalysetests in
Lösung weisen dagegen bei 13 von 28 der getesteten Peptide eine Inversion des
164
favorisierten Enantiomers gegenüber der Festphase auf. Vergleiche hierzu Abb. 69
und Abb. 70. Ein besonders auffallendes Beispiel für den Einfluss der
Peptidsequenz und Chiralität auf die ee-Werte ist in Abb. 71 gezeigt, bei dem bei
unterschiedlichen Chiralitäten des endständigen Prolins in beiden Fällen bevorzugt
das R-Aldolprodukt gebildet wird.
Der Versuch, vom Harz abgespaltene Peptide durch Verestern mit Methanol und
Thionylchlorid in eine besser lösliche Form zu überführen und gleichzeitig die
Enantiomerenüberschüsse zu erhöhen war nicht erfolgreich.
Abbildung 68: Diese freien und festphasengebundenen Peptide zeigten die jeweils höchsten erzielten Enantiomerenüberschüsse bei Aldolkatalysereaktionen mit Aceton und p-Nitrobenzaldehyd. Dargestellt sind die Strukturen der Peptide, die Peptidnummer und die Peptidsequenz. Das freie Peptid P902 ist ein Beispiel für den Fall, dass bei endständigem L-Prolylrest dennoch das S-Aldolisomer katalysiert wird. Solche Abweichungen fanden sich bei 30 % der untersuchten Peptide. Ersetzt man bei P902 das Nitrotyrosin durch Phenylalanin, sinkt bei ebenfalls katalysiertem S-Isomer der Enantiomerenüberschuss auf nur 17 % S-Aldol. Dieser Umstand weist auf die Bedeutung einer aciden Tyrosyl-OH-Gruppe in der Aldolkatalyse hin.
O
HNN
H
OH
O
NH
O
O
I
NO2
O
NH
O
O
NO2
OH
HN
O
OHO2N
NH
O
NH
O
HNN
H
OH
O
NH
O
OH
NO2
O
NH
O
OHN
O
OH
NO2
HN
O
NH
P11L-W 70 % R
P31 57 % RP902 48 % S
P922-W 62 % S
H-D-Pro-3NT-3NT-β -Ala-Wang-Harz H-L-Pro-3I5NT-β -Ala-Wang-Harz
H-L-Pro-3NT-L-Pro-β -Ala-OH H-L-Pro-3NT-β -Ala-OH
165
Abbildung 69: Enantiomerenüberschüsse bei durch TG-Harz-gebundenen Peptiden gegen die ermittelten Enantiomerenüberschüsse durch Katalyse durch Wang-Harz gebundener Peptide. Eine Population des linken unteren und des rechten oberen Quadranten des gewählten Koordinatensystems bedeutet Übereinstimmung des favorisierten Enantiomers bei Katalyse durch die beiden jeweils an verschiedenen Harzen festphasengebundenen Peptide.
ee (Wang-Harz-Peptide)-40 -20 0 20 40 60 80
ee (T
G-H
arz-
Pep
tide)
-80
-60
-40
-20
0
20
40
60P02-W P02-T P31-W P31-T P102-W P102-T P142-W P142-T P171-W P171-T P192-W P192-T P261-W P261-T P322-W P322-T P351-W P351-T P391-W P391-T P392-W P392-T P421-W P421-T P422-W P422-T P431-W P431-T P461-W P461-T P462-W P462-T
ee(R)
ee(S) ee(R)
Abbildung 70: Auftragung der erzielten Enantiomerenüberschüsse von freien Peptiden in Lösung gegen die erzielten Enantiomeren-überschüsse der entsprechenden am Wang-Harz gebundenen Peptide gleicher Sequenz. Etwa die Hälfte der untersuchten Peptide zeigt Unterschiede bei dem favorisiert katalysierten Enantiomer beim Übergang von festphasengebundenen Peptiden zu Peptiden in Lösung.
ee (Wang-Peptide)-80 -60 -40 -20 0 20 40 60 80
ee (f
reie
Pep
tide)
-60
-40
-20
0
20
40
60
80P02-W P02 P31-W P31 P102-W P102 P142-W P142 P171-W P171 P192-W P192 P261-W P261 P322-W P322 P351-W P351 P391-W P391 P392-W P392 P421-W P421 P422-W P422 P431-W P431 P461-W P461 P462-W P462 P11L-W P11L P11D-W P11D P921-W P921 P922-W P922 P931-W P931 P932-W P932 P901-W P901 P902-W P902 P911-W P911 P912-W P912 PLL-W PLL
PDD-W PDD
ee(R)
ee(R)
ee(S)

166
Ein weiterer Schwerpunkt der Arbeit liegt in der Untersuchung der Kinetik von
peptidkatalysierten Aldolreaktionen. Hier wurde erstmals gezeigt, dass sich der
Reaktionsverlauf festphasengebundener und in Lösung befindlicher Peptide
während der Katalysereaktion bei Verwendung von deuteriertem Aceton als
Lösungsmittel (oder anderer deuterierter Lösungsmittel) mittels online-NMR
problemlos verfolgen lässt. Aufgrund von Löslichkeitsproblemen der Peptide bei
Katalysen in Lösung und der Schwierigkeit bei der Realisierung identischer
Reaktionsbedingungen aufgrund stark unterschiedlicher Löslichkeiten wurde mit
festphasengebundenen Peptiden gearbeitet. Obwohl die NMR-Messungen so
praktisch mit Peptidharz in Suspension erfolgten, konnten sehr gute NMR-Spektren
erhalten werden, die eine präzise Angabe über die Menge entstandenen
Aldolproduktes machen.
Die Katalysereaktion ist eine zweistufige Reaktion, die im ersten Schritt die Bildung
eines Enamins aus Aceton und der sekundären Aminogruppe des aminoterminalen
Prolylrestes beinhaltet. Der zweite Schritt der Reaktion ist dann der Angriff des p-
Nitrobenzaldehyds auf das Enamin. Da Aceton im riesigen Überschuss vorliegt, ist
anzunehmen, dass der Prolylrest in Enaminform vorliegt. Die Erfassung des
Umsatzes nach der Zeit eröffnete einen Zugang zu wichtigen kinetischen
Konstanten und eine Zuordnung eines kinetischen Modells aus der Enzymkinetik,
der Michaelis-Menten-Kinetik, bei der der eigentlichen Reaktion ein vorgelagertes
Abbildung 71: Zwei Peptidsequenzen,die sich nur in der Chiralität des endständigen Prolins unterscheiden. Dennoch katalysieren sie das gleiche Enantiomer des Aldolproduktes. Dies legt die Vermutung nahe, dass nicht nur Sequenz, sondern die individuell resultierende Sekundärstruktur erheblich mitbestimmend für das favorisierte Aldolenantiomer ist. Hier liegen wahrscheinlich starke strukturelle Ähnlichkeiten beider betrachteter Peptide vor.
O
HNN
H
OH
O
NH
O
OH
I
NO2
P11L 29 % R
H-L-Pro-3I5NT-β -Ala-OH
O
HNN
H
OH
O
NH
O
OH
I
NO2
P11D 45 % R
H-D-Pro-3I5NT-β -Ala-OH

167
Gleichgewicht im Sinne einer Vorkomplexierung voraus läuft. Es wurde für 6
Peptide, deren Katalyseleistungen herausragend waren, die Michaelis-Konstante
bestimmt, die eine Aussage über die Stärke der Assoziation oder Dissoziation
macht, indem die Entstehung der Aldolprodukte nach der Zeit bei 3 verschiedenen
Konzentrationen an Substrat ermittelt wurden. Zu den kinetisch relevanten
Auswertungen siehe Kap. 7 „Auswertung der Kinetikdaten“.
Aus den Kurvenverläufen wurde durch globale nichtlinare Regression für jedes
Peptid bei 3 Konzentrationen der Wert der Anfangsgeschwindigkeit ermittelt und in
verschiedenen Diagrammtypen in nichtlinearisierter und linearisierter Form
aufgetragen. Aus den linearisierten Auftragungen wurde jeweils die Michelis-
Konstante und die sich bei maximaler Sättigung an verfügbarem Substrat
einstellende Maximalgeschwindigkeit erhalten. Aus Km für verschiedene unter-
suchte Peptide folgt, dass die Reaktion über eine für jedes Peptid individuelle,
jedoch relativ geringe Assoziation ablaufen sollte, daher liegen die Konzentrationen
bei Substratsättigung mit 100-400 mM erheblich höher, als für die meisten Enzyme,
bei denen hier bestimmte Konzentrationswerte bereits weit über den physiologisch
anzutreffenden Werten liegen. Es konnten ebenfalls Werte für kcat, der Wechselzahl
oder turnover number, der katalytischen Effizienz kcat / Km und der spezifischen
Aktivität berechnet werden. Die gewonnen Werte wurden bezüglich der erhaltenen
Daten mit Literaturdaten aus Enzymdatenbanken (BRENDA) verglichen und
innerhalb dieser eingeordnet und diskutiert.
Die hier entwickelte und erstmals in der Organokatalyse eingesetzte online-NMR-
Methode zur nichtinvasiven Untersuchung reaktionskinetisch relevanter Vorgänge
bei laufenden Katalysereaktionen ermöglicht so relativ schnell eine Evaluierung der
katalytischen Eigenschaften neuer festphasengebundener Peptidkatalysatoren.
Vorteilhaft dabei ist bei einmal vorbereiteten Proben die Möglichkeit eines relativ
schnell möglichen Austausches der Substrate gegen davon abweichende
Mengenverhältnisse oder andere Substrate. Die einzige Einschränkung ist die
Notwendigkeit der Verwendung deuterierter Lösungsmittel. In verschiedenen
Lösungsmitteln können jedoch so auch Polaritätseinflüsse des Lösungsmittels auf
einfache Art und Weise mit erfasst und studiert werden.
Es wurde auch der Versuch unternommen, mittels einer Modellingstudie mögliche
geometrische Anordnungen verschiedener Peptide in bereits vorgenerierter

168
Enaminform zum Substrat p-Nitrobenzaldehyd zu ermitteln und mit ihrer Hilfe
Vorhersagen zur Enantioselektivität bei der Reaktion zu treffen, die hierbei
auftretenden Schwierigkeiten bei der Auswahl einer günstigen Ausgangsstruktur
wurde durch Vergleich mit bereits literaturbekannten Daten zum Teil verringert. So
konnten Houk et al. durch quantenchemische Berechnungen günstige Kon-
formationen der möglichen Übergangszustände von ungünstigen unterscheiden.
Diese Berechnungen wurden im wesentlichen jedoch an starren Systemen, wie
Prolin oder ähnlich unflexiblen Molekülen durchgeführt, was die Betrachtung
erheblich vereinfacht, da nicht aus verschiedenen möglichen Konformationen
selektiert werden muss. Rechnungen an Systemen ähnlicher Komplexität wie an in
dieser Arbeit vorliegenden Peptidkatalysatoren sind nach dem gegenwärtigen
Kenntnisstand bisher nicht durchgeführt worden. In Analogie zu den Arbeiten von
Houk et al. [77] wurde von einem präformierten Enamin ausgegangen, sowie die für
die Katalyse notwendige Gegenwart einer aciden Funktionalität in unmittelbarer
räumlicher Nähe zum Enamin. Diese unerlässliche Rolle, die bei Prolin von der
Carboxyfunktion übernommen wird, sollte hier vom modifizierten Tyrosin ausgeübt
werden, wobei zusätzliche sterische und elektrostatische Einflüsse der
aromatischen Substituenten eingebracht werden. Eine räumliche Nähe kann hier in
dieser Arbeit nicht nur durch bloße Nachbarschaft des Tyrosin-aryl-OH realisiert
werden, sondern auch konformationsbedingt durch in der Sequenz weiter entfernte
Tyrosyl-Reste. Die bei Peptiden auftretende Problematik erheblicher Unsicherheit
bei der Strukturvorhersage günstiger und ungünstiger Konformationen bei teilweise
nahezu identischen Energieunterschieden, insbesondere bei
festphasengebundenen und weniger bei freien Peptiden macht es schwierig,
analoge Ergebnisse zu erhalten. Die bei (relativ großen) Enzymen mit bekannten
(Kristall)-Strukturen übliche Methode des systematischen Dockings ist hier
aufgrund der unbekannten Bindungsstelle nicht möglich. Es wurden daher durch
systematische Konformationssuchen bei Freilassen und Variation aller relevanten
Torsionswinkel im Peptid eine Reihe durch das OPLS-Kraftfeld energieminimierter
Strukturen erhalten, unter denen diejenigen mit in Einklang mit der Hypothese der
räumlichen Nähe des Aryl-OH zum Enamin stehenden Strukturen selektiert wurden.
In diesen Strukturen wurden Abstände zwischen Enamin und Aryl-OH bestimmt und
mit Literaturwerten aus quantenchemischen Rechnungen verglichen. Ein Beispiel
ist in der Abb. 72 gezeigt. Die in Abb. 72 gezeigt Modellingstruktur kurz vor
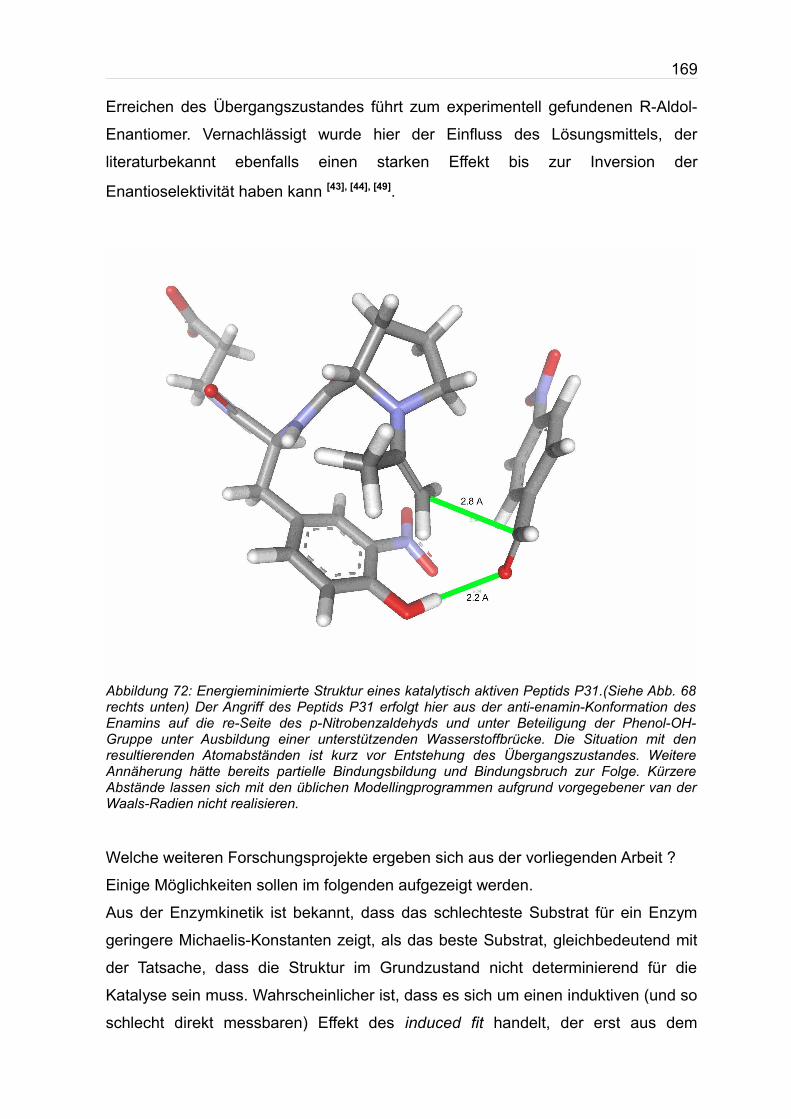
169
Erreichen des Übergangszustandes führt zum experimentell gefundenen R-Aldol-
Enantiomer. Vernachlässigt wurde hier der Einfluss des Lösungsmittels, der
literaturbekannt ebenfalls einen starken Effekt bis zur Inversion der
Enantioselektivität haben kann [43], [44], [49].
Welche weiteren Forschungsprojekte ergeben sich aus der vorliegenden Arbeit ?
Einige Möglichkeiten sollen im folgenden aufgezeigt werden.
Aus der Enzymkinetik ist bekannt, dass das schlechteste Substrat für ein Enzym
geringere Michaelis-Konstanten zeigt, als das beste Substrat, gleichbedeutend mit
der Tatsache, dass die Struktur im Grundzustand nicht determinierend für die
Katalyse sein muss. Wahrscheinlicher ist, dass es sich um einen induktiven (und so
schlecht direkt messbaren) Effekt des induced fit handelt, der erst aus dem
Abbildung 72: Energieminimierte Struktur eines katalytisch aktiven Peptids P31.(Siehe Abb. 68 rechts unten) Der Angriff des Peptids P31 erfolgt hier aus der anti-enamin-Konformation des Enamins auf die re-Seite des p-Nitrobenzaldehyds und unter Beteiligung der Phenol-OH-Gruppe unter Ausbildung einer unterstützenden Wasserstoffbrücke. Die Situation mit den resultierenden Atomabständen ist kurz vor Entstehung des Übergangszustandes. Weitere Annäherung hätte bereits partielle Bindungsbildung und Bindungsbruch zur Folge. Kürzere Abstände lassen sich mit den üblichen Modellingprogrammen aufgrund vorgegebener van der Waals-Radien nicht realisieren.

170
entstehenden Enzym-Substrat-Komplex im Moment der Entstehung des
Übergangszustandes die eigentliche katalytisch aktive Spezies erzeugt. Dieser
Vorgang ist mit einer Konformationsänderung verbunden. Da solche Vorgänge stark
temperaturabhängig sein sollten, könnte eine Variation der Reaktionstemperatur in
mehreren Stufen und die damit verbundene Änderung von Reaktions-
geschwindigkeit und erzieltem Enantiomerenverhältnis das Verständnis solcher
Vorgänge verbessern. Eine Erweiterung dieses Themenbereiches wäre eine
Verwendung verschiedener, anderer Substrate für enaminkatalysierte Reaktionen,
sowie die Ausdehnung der Katalysetests auch auf andere Reaktionstypen, z.B.
Mannich-Reaktion.
Im Falle des Tyrosins können zusätzliche Untersuchungen, die eine Modifikation
der Tyrosyl-OH-Gruppe zum Ziel haben den Einfluss dieser Funktionalität zu tunen
und die Auswirkungen daraus zu ermitteln.
Zusätzlich wäre eine Modifikation an anderer Position des Tyrosins, zum Beispiel
durch Einbau größerer Gruppen an Position der Tyrosyl-β-Protonen mit der Folge
einer geringeren sterischen Freiheit des aromatischen Tyrosyl-Ringes denkbar.
Zukünftige Forschungen auf diesem Gebiet sollten neben dem Einsatz einer
schnellen chiralen HPLC, die den Experimentator von aufwändigen, sich ständig
wiederholenden Tätigkeiten entlastet, auch den Zusatz verschiedener löslicher
Additive zu den Reaktionsansätzen beinhalten. Der Zusatz komplexierender
Metallkationen, wie zum Beispiel löslicher Zinksalze sollte eine erhöhte strukturelle
Komplexität ermöglichen, die eine Steigerung der Enantiomerenausbeute und
vielleicht der Umsatzgeschwindigkeiten zur Folge haben kann. Auch der Einfluss
von unterschiedlichen Mengen Wasser auf die Katalysereaktion kann zu einer
Verbesserung der katalytischen Fähigkeiten führen, wie kürzlich von einigen
Arbeitsgruppen bei Aldolreaktion und anderen organokatalytisch durchgeführten
Reaktionen gezeigt wurde [84], [85], [86].
Eine weitere Möglichkeit schließt die Verwendung auf Naturstoffen basierender
möglichst preiswerter chiraler Additive ein, beispielsweise aus natürlichen
Aminosäuren gewonnene peraminomethylierte quartäre Ammoniumverbindungen,
deren besondere Eigenschaft zur Ausbildung starker π-aryl-Kation-
Wechselwirkungen als lösliches chirales Auxiliar für ansonsten festphasen-
gebundene katalytisch aktive Peptide dienen kann. Solche Wechselwirkungen sind
bei Acetylcholin mit den aromatischen Strukturbereichen von Tryptophan und

171
Phenylalanin an der Bindungsstelle bekannt. Der Vorteil einer quartären
Ammoniumverbindung wäre eine relativ einfache Rückgewinnung des extrem
polaren Moleküls durch Ionenaustauschchromatographie. Dies wiegt die bekannte
Toxizität quartärer Ammoniumverbindungen durch Verwendung geschlossener
Kreisläufe vollständig auf. Die Edukte und Produkte der Aldolkatalyse sollten durch
ihre doch erheblich apolarere Natur einen solchen Reinigungsvorgang unverändert
überstehen und die Säule unbeeinflusst passieren können.
Ziel zukünftiger Untersuchungen können Systeme sein, die als primar katalytisch
aktives Zentrum kein endständiges Prolin tragen, sondern achirale lineare oder
cyclische sekundäre Amine beinhalten, so wäre auch der Einsatz von Foldamer-
teilen und Rotameren in der Struktur zum Erreichen einer sterischen Komplexität
denkbar.
















![Synonymaverzeichnis 6. Synonymaverzeichnis Acetaldehyd [Acetaldehyd] Acetan [Ethylen] Aceton [Aceton] Acetoxylsäure [Essigsäure] Acetylentrichlorid [Trichlorethylen] Acetylhydrür](https://img.pdfslide.org/doc/110x75/5e1d817c0b1a5723a07c9249/synonymaverzeichnis-6-synonymaverzeichnis-acetaldehyd-acetaldehyd-acetan-ethylen.jpg)












