Embed Size (px)
Citation preview


Ab initio Molekulardynamik-Simulationen von
Nanopartikeln
Diplomarbeit von
Georg Rollmann
vorgelegt dem Fachbereich Physik-Technologie der
Gerhard-Mercator-Universitat Duisburg
Duisburg, den 14. Marz 2001

Zusammenfassung
In dieser Arbeit werden chemische Reaktionen zwischen Precursor-Molekulen fur
die Bildung von Nanopartikeln im Rahmen der Dichtefunktionaltheorie unter-
sucht. Auf den Potentialflachen der reagierenden Systeme H2 + H ⇀↽ H + H2,
SiH4 + H ⇀↽ SiH3 + H2, SiCl4 + H ⇀↽ SiCl3 + HCl und TiCl4 + H ⇀↽ TiCl3 + HCl
werden Extremalpunkte identifiziert, mit deren Hilfe Aussagen uber energetische
und strukturelle Eigenschaften der Systeme gemacht werden konnen. Die fur die-
se Untersuchungen verwendete ab initio Methode ist die Hybrid-DFT-Methode
B3LYP. Diese Wahl war das Ergebnis von umfangreichen Testrechnungen in Be-
zug auf energetische und strukturelle Eigenschaften von Molekulen. Im Rahmen
dieser Rechnungen stellte sich heraus, daß die Ergebnisse stark von der verwen-
deten Methode sowie dem Basissatz fur die Entwicklung der Einteilchenwellen-
funktionen abhangig sind.
Die Energiebarriere der Reaktion SiH4 + H ⇀↽ SiH3 + H2 wurde ermittelt.
Der berechnete Wert stimmt mit dem experimentellen Wert aber nur qualitativ
uberein. Fur die Reaktion TiCl4 + H ⇀↽ TiCl3 + HCl wurde ein tiefliegendes
Minimum auf der PES lokalisiert. Dies kann als Grund dafur angesehen werden,
daß die entsprechende Reaktion ohne Aktivierungsenergie ablauft.
Ein Vergleich der Ergebnisse mit Resultaten, die unter Verwendung von ul-
traweichen Pseudopotentialen fur die Valenzelektronen erzielt wurden, laßt den
Schluß zu, daß letztere Methode fur die Beschreibung von Potentialflachen rea-
gierender Systeme geeignet sind.
Molekulardynamik-Simulationen von Stoßprozessen erweisen sich als geeigne-
te Methode, mit der das zeitliche Verhalten reagierender molekularer Systeme
analysiert werden kann. Fur Systeme mit freien Elektronen mussen die Simula-
tionen aber spinpolarisiert durchgefuhrt werden.
3

Abstract
In this work chemical reactions of precursor molecules for the creation of nanopar-
ticles are examined within the framework of density functional theory. Extremal
points on the potential energy surfaces of the reacting systems H2 +H ⇀↽ H+H2,
SiH4 + H ⇀↽ SiH3 + H2, SiCl4 + H ⇀↽ SiCl3 + HCl and TiCl4 + H ⇀↽ TiCl3 + HCl
are identified. This information can be used to learn about the structural and
energetical properties of the systems. The Hybrid-DFT method B3LYP was used
for the investigation of the potential energy surfaces, as this method has proved
to give the best results with respect to structural and energetical properties of
molecules. The calculations show that the results are strongly dependent on the
employed ab initio method and also on the basis set for the expansion of the
wavefunctions.
The energy barrier of the reaction SiH4 + H ⇀↽ SiH3 + H2 was calculated, but
the agreement with the experimental value is only qualitative. For the reaction
TiCl4 + H ⇀↽ TiCl3 + HCl a deep minimum on the potential energy surface was
localised, which is responsible for the reaction not showing an activation energy.
A comparison of the results with those obtained from calculations using ultra-
soft pseudopotentials for the valence electrons and a plane wave basis set shows,
that the latter method is also suited for the description of potential energy sur-
faces of reacting systems.
Scattering processes between precursor molecules are simulated using ab initio
molecular-dynamics simulations. The method proves to be able to analyse the
dynamics of reacting systems in a detailed way. The results of the simulations
show, that in order to obtain results of good quality the spin of the electrons has
to be included in the calculations if an open-shell system is considered.
4

Inhaltsverzeichnis
Zusammenfassung 3
Abstract 4
Inhaltsverzeichnis 4
Abbildungsverzeichnis 9
1 Einleitung 10
1.1 Nanopartikel aus der Gasphase . . . . . . . . . . . . . . . . . . . 10
1.2 Experimentelle Situation . . . . . . . . . . . . . . . . . . . . . . . 11
1.3 Motivation fur die durchgefuhrten Rechnungen . . . . . . . . . . . 12
2 Wechselwirkende Elektronen 13
2.1 Effektive Schrodingergleichung . . . . . . . . . . . . . . . . . . . . 13
2.2 Der Hartree-Fock Ansatz . . . . . . . . . . . . . . . . . . . . . . . 17
2.3 Post-Hartree-Fock-Methoden . . . . . . . . . . . . . . . . . . . . . 24
2.3.1 Configuration Interaction . . . . . . . . . . . . . . . . . . . 25
2.3.2 Coupled-Cluster-Methode . . . . . . . . . . . . . . . . . . 29
2.3.3 Andere Erweiterungen des Hartree-Fock-Ansatzes . . . . . 31
2.4 Dichtefunktionaltheorie . . . . . . . . . . . . . . . . . . . . . . . . 33
2.4.1 Die Elektronendichte als Variable . . . . . . . . . . . . . . 33
2.4.2 Funktionale fur Austausch und Korrelation . . . . . . . . . 36
2.5 Die Bedeutung der Randbedingungen . . . . . . . . . . . . . . . . 39
2.5.1 Freie Randbedingungen: Die Wahl der Basis . . . . . . . . 40
2.5.2 Periodische Randbedingungen: Ebene Wellen und Pseudo-
potentiale . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
5

6 INHALTSVERZEICHNIS
3 Energieflachen 48
3.1 Strukturoptimierung . . . . . . . . . . . . . . . . . . . . . . . . . 50
3.2 Molekulardynamik-Simulation . . . . . . . . . . . . . . . . . . . . 51
4 Ergebnisse 52
4.1 Vergleich der verwendeten Methoden . . . . . . . . . . . . . . . . 53
4.1.1 Wasserstoff . . . . . . . . . . . . . . . . . . . . . . . . . . 54
4.1.2 Bindungsenergien . . . . . . . . . . . . . . . . . . . . . . . 58
4.1.3 Rechenaufwand . . . . . . . . . . . . . . . . . . . . . . . . 68
4.1.4 Bindungslangen . . . . . . . . . . . . . . . . . . . . . . . . 70
4.1.5 Auswahl der Methode . . . . . . . . . . . . . . . . . . . . 73
4.2 Die Pseudopotentialmethode . . . . . . . . . . . . . . . . . . . . . 73
4.3 Potentialflachen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
4.3.1 Die Reaktion H2 + H ↔ H + H2 . . . . . . . . . . . . . . . 78
4.3.2 Die Reaktion SiH4 + H ↔ SiH3 + H2 . . . . . . . . . . . . 84
4.3.3 Die Reaktion SiCl4 + H ↔ SiCl3 + HCl . . . . . . . . . . . 89
4.3.4 Die Reaktion TiCl4 + H ↔ TiCl3 + HCl . . . . . . . . . . 90
4.3.5 Molekulardynamik-Simulationen . . . . . . . . . . . . . . . 95
5 Zusammenfassung und Ausblick 105
A Funktionale fur Austausch und Korrelation 111
A.1 Lokale Dichtenaherung . . . . . . . . . . . . . . . . . . . . . . . . 111
A.2 Gradientenmethoden . . . . . . . . . . . . . . . . . . . . . . . . . 111
A.3 Hybridmethoden . . . . . . . . . . . . . . . . . . . . . . . . . . . 112
B Die verwendeten Basissatze 113
B.1 Split-Valence Basissatze von Pople . . . . . . . . . . . . . . . . . 113
B.2 Correlation-Consistent Basissatze . . . . . . . . . . . . . . . . . . 116
C Der Algorithmus von Verlet 118

Abbildungsverzeichnis
2.1 Der Unterschied zwischen UHF und RHF . . . . . . . . . . . . . . 19
2.2 Die Dissoziation des H2-Molekuls . . . . . . . . . . . . . . . . . . 20
2.3 Besetzte und virtuelle Orbitale . . . . . . . . . . . . . . . . . . . . 22
2.4 Die Orbitale des Kohlenstoffatoms in einer minimalen STO-Basis 43
2.5 Die Orbitale des Kohlenstoffatoms in einer minimalen GTO-Basis 44
2.6 Periodische Randbedingungen in zwei Dimensionen . . . . . . . . 45
3.1 Schematische Darstellung einer Energieflache . . . . . . . . . . . . 49
4.1 Gesamtenergie des Wasserstoffatoms . . . . . . . . . . . . . . . . 54
4.2 Bindungsenergie des H2-Molekuls . . . . . . . . . . . . . . . . . . 56
4.3 Bindungsenergien von N2, O2 und CO . . . . . . . . . . . . . . . 60
4.4 Bindungsenergien von N2, NH3 und HCl . . . . . . . . . . . . . . 64
4.5 Bindungsenergien von SiH2, SiH3 und SiH4 . . . . . . . . . . . . . 67
4.6 Vergleich der benotigten Rechenleistung verschiedener Methoden . 69
4.7 Die Bindungslangen in N2, HCl und SiH4 Molekul . . . . . . . . . 71
4.8 Die Bindungslange im TiCl4-Molekul . . . . . . . . . . . . . . . . 72
4.9 Bindungsenergie von NH3 und SiH4 in Abhangigkeit von der Zel-
lengroße und der Cutoff-Energie . . . . . . . . . . . . . . . . . . . 74
4.10 Die Bindungslange im SiH4-Molekul in Abhangigkeit von der
Große der Simulationsbox . . . . . . . . . . . . . . . . . . . . . . 75
4.11 Graphische Darstellung der Atome . . . . . . . . . . . . . . . . . 77
4.12 H3-Molekul in Cs Symmetrie . . . . . . . . . . . . . . . . . . . . . 78
4.13 Hohenliniendiagramm der Potentialflache der Reaktion H2 + H ↔H + H2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
4.14 Energie des H3-Molekuls in Abhangigkeit von der raumlichen An-
ordnung der Atome . . . . . . . . . . . . . . . . . . . . . . . . . . 80
7

8 ABBILDUNGSVERZEICHNIS
4.15 Ladungsverteilung des H3-Molekuls in Abhangigkeit von der raum-
lichen Anordnung der Atome . . . . . . . . . . . . . . . . . . . . . 81
4.16 Der Ubergangszustand der Reaktion H2+H ↔ H+H2 in Abhangig-
keit vom Basissatz . . . . . . . . . . . . . . . . . . . . . . . . . . 82
4.17 LDA-Potentialflache der Reaktion H2 + H ↔ H + H2 . . . . . . . 83
4.18 Reaktionsenthalpie der Reaktion SiH4 + H → SiH3 + H2 . . . . . . 84
4.19 B3LYP/6-311G**-Potentialflache der Reaktion SiH4 + HSiH3 + H2 85
4.20 Der Ubergangszustand der Reaktion SiH44 + H → SiH3 + H2 in
Abhangigkeit vom verwendeten Basissatz . . . . . . . . . . . . . . 87
4.21 Energiebarriere der Reaktion SiH4 +H ↔ SiH3 +H2 unter Verwen-
dung der GGA- und der LDA-Methode . . . . . . . . . . . . . . . 88
4.22 Reaktionsenthalpie in Abhangigkeit vom verwendeten Basissatz . 89
4.23 Anordnung der Atome des Systems SiCl4 + H im Ubergangszustand 90
4.24 Der Ubergangszustand der Reaktion SiCl4 + H → SiCl3 + HCl in
Abhangigkeit vom verwendeten Basissatz . . . . . . . . . . . . . . 91
4.25 Reaktionsenthalpie der Reaktion TiCl4 + H → TiCl3 + HCl . . . . 92
4.26 Anordnung der Atome des Systems TiCl4 + H im energetischen
Minimum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
4.27 Das energetische Minimum des Systems TiCl4 + H . . . . . . . . . 94
4.28 Die Anordnung der Atome in einem weiteren Minimalpunkt auf
der Energieflache . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4.29 Momentaufnahmen der zeitlichen Entwicklung des Stoßprozesses
zwischen H und SiH4 . . . . . . . . . . . . . . . . . . . . . . . . . 96
4.30 Veranderung des Abstandes zwischen den Atomen . . . . . . . . . 97
4.31 Zeitlicher Verlauf der Geschwindigkeit des stoßenden H-Atoms . . 97
4.32 Momentaufnahmen der zeitlichen Entwicklung des Streuprozesses
zwischen Ar und SiCl4 . . . . . . . . . . . . . . . . . . . . . . . . 99
4.33 Abstande und Geschwindigkeiten von ausgewahlten Atomen des
reagierenden Systems SiCl4 + Ar . . . . . . . . . . . . . . . . . . . 100
4.34 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
4.35 Zeitliche Entwicklung der Anordnung der Atome im Laufe der Re-
aktion TiCl4 + H → TiCl3 + HCl . . . . . . . . . . . . . . . . . . 103
4.36 Entwicklung der Abstande im System TiCl4 + H . . . . . . . . . . 104
5.1 Molekulardynamiksimulation eines Ti2C7H20-Molekuls . . . . . . . 109

ABBILDUNGSVERZEICHNIS 9
5.2 Zeitliche Entwicklung des Diederwinkels im Ti2C7H20-Molekul . . 110
5.3 Verdrillung des Ti2C7H20-Molekuls . . . . . . . . . . . . . . . . . 110

Kapitel 1
Einleitung
1.1 Nanopartikel aus der Gasphase
Aufgrund von großen technischen Fortschritten, welche die Herstellung von Ma-
terialien auf der Nanometerskala ermoglichten, ist in den letzten Jahren die Na-
notechnologie als vielversprechendes Forschungsgebiet mit einem breiten Anwen-
dungsfeld entstanden. Nanostrukturierte Materialien haben aufgrund ihrer redu-
zierten Dimensionen oft Eigenschaften, die sie vom entsprechenden kompakten
Material unterscheiden und sind somit technologisch von großem Interesse.
Nanopartikel bilden die Brucke zwischen isolierten Atomen und dem
Festkorper. Ihre Charakterisierung stellt hohe Anforderungen an Theorie und
Experiment [1]. Die Eigenschaften von Nanopartikeln unterscheiden sich in vie-
lerlei Hinsicht von denen des entsprechenden Festkorpers und sind stark von ihrer
Große und Morphologie abhangig. Diese Tatsache eroffnet die Moglichkeit fur das
gezielte Design von Strukturen [2] mit einzigartigen Eigenschaften, die von der
Große und Zusammensetzung der Nanopartikel abhangig sind.
Eine Moglichkeit der Herstellung von Nanopartikeln ist ihre Erzeugung aus der
Gasphase. Als Ausgangsstoffe dienen dabei Precursor-Molekule. Infolge von re-
aktiven Prozessen zwischen diesen Molekulen und anschließender Nukleation und
Partikelbildung entstehen dann Cluster. Die kontollierte Herstellung großenselek-
tierter Cluster einer bestimmten Zusammensetzung in makroskopischen Mengen
stellt immer noch eine große Hurde dar, zu deren Bewaltigung die genaue Kennt-
nis der mikroskopischen Vorgange unumganglich ist.
Obwohl es inzwischen moglich ist, chemische Reaktionen mit Hilfe von ul-
10

1.2. EXPERIMENTELLE SITUATION 11
traschnellen Laserpulsen auf der Femtosekundenskala zu beoachten [3], kann man
die zeitliche Entwicklung eines isolierten atomaren Systems noch nicht direkt ver-
folgen. Aus diesem Grund ist fur die meisten reaktiven Prozesse nur wenig be-
kannt uber den genauen Reaktionsmechanismus und die Zwischenprodukte, die
wahrend einer Kollision entstehen.
Computersimulationen auf ab initio Basis stellen eine Moglichkeit dar, atoma-
re Vorgange zu analysieren. Dabei hat sich die Methode der Molekulardynamik-
Simulation in Verbindung mit der Dichtefunktionaltheorie als geeignetes Mittel
erwiesen, um die zeitliche Entwicklung eines reagierenden Systems mit vertret-
barem numerischem Aufwand zu untersuchen.
1.2 Experimentelle Situation
Eine interessante Gruppe von Nanopartikeln bilden die nichtoxidischen Nano-
Keramiken, zu deren Vertretern TiC, TiN und SiC zahlen. Aus solchen Partikeln
gesinterte Materialien weisen eine besonders große Harte und Bruchfestigkeit so-
wie hohe Schmelzpunkte auf, was sie pradestiniert fur die Verwendung als Werk-
stoffe in Bauteilen, die einer hohen mechanischen oder thermischen Belastung
ausgesetzt sind.
Die Erzeugung von TiN-Nanopartikeln aus der Gasphase unter Verwendung
der Precursor-Molekule TiCl4 und NH3 ist kurzlich von Herzler et al. nachgewie-
sen worden [4]. Als erster Schritt im Prozess ist hier die Dissoziation von TiCl4
wichtig, welche in Form der Reaktion TiCl4 + H ⇀↽ TiCl3 + HCl von Herzler et
al. untersucht worden ist und gemaß [5] keiner Aktivierungsenergie bedarf.
Als Precursor-Molekule fur die Erzeugung von SiC-Nanopartikeln kommen
beispielsweise SiCl4 sowie SiH4 in Betracht, wobei SiH4 außerdem bei der Her-
stellung von mikroelektronischen Bauteilen mittels CVD eine entscheidende Rolle
spielt. In diesem Zusammenhang wurde die Reaktion SiH4 + H ⇀↽ SiH3 + H2 von
Kunz et al. [6] untersucht, sowie die Dissoziation von SiCl4 durch Stoße mit Argo-
natomen [7]. Die ermittelten Aktivierungsenergien belaufen sich auf 2260 K bzw.
37760 K.

12 KAPITEL 1. EINLEITUNG
1.3 Motivation fur die durchgefuhrten Rech-
nungen
In dieser Arbeit sollen die Reaktionsmechanismen der fur die Bildung von TiN-,
TiC- und SiC-Nanopartikeln wichtigen Dissoziationen von TiCl4, SiCl4 und SiH4
untersucht werden. Fur den Vergleich mit experimentellen Energiebarrieren ist
die Kenntnis von Ubergangszustanden auf der hochdimensionalen Potentialflache
notwendig.
Es gibt eine Reihe von ab initio Methoden, die sich mit der Berechnung der
Energie eines atomaren Systems befassen. Die Ergebnisse, die man mit verschie-
denen Methoden erhalt, unterscheiden sich oftmals nicht nur quantitativ, sondern
auch qualitativ. Beispielsweise sagt die Hartree-Fock-Methode sowohl das Fluor-
Molekul F2 als auch das Ozonmolekul O3 als nicht stabil voraus. In der loka-
len Dichtenaherung im Rahmen der Dichtefunktionaltheorie ist das H3-Molekul
falschlicherweise stabil.
Deswegen wird in dieser Arbeit die Leistungsfahigkeit verschiedener ab in-
itio Methoden in Bezug auf energetische sowie geometrische Verhaltnisse bei
Precursor-Molekulen genauer untersucht. Fur eine aussagekraftige Interpretation
muß daruber hinaus untersucht werden, in welchem Maße der fur die Entwicklung
der Einteilchenwellenfunktionen verwendete Basissatz die Ergebnisse beeinflußt.
Im folgenden Kapitel wird auf die verwendeten ab initio Methoden ausfuhrlich
eingegangen, wobei ein Schwerpunkt auf der Herausstellung der ihnen anhaften-
den Naherungen sowie den daraus resultierenden Beschrankungen liegt. Danach
werden die unterschiedlichen Ansatze fur die Wahl des Basisfunktionen erlautert.
Die im Rahmen dieser Arbeit erzielten Ergebnisse finden sich im vierten Kapitel
im Anschluß an einen Uberblick uber die verwendeten Methoden zur Untersu-
chung von Energieflachen.

Kapitel 2
Wechselwirkende Elektronen
2.1 Effektive Schrodingergleichung
Die zeitliche Entwicklung des quantenmechanischen Zustands eines Systems wird
durch die Schrodingergleichung
H |Ψ〉 = ih∂
∂t|Ψ〉 (2.1)
beschrieben [8]. Der Hamiltonoperator H fur ein atomares System ohne außeres
Feld setzt sich aus den Operatoren fur die kinetische und die potentielle Energie
zusammen und hat in der nichtrelativistischen Naherung die folgende Gestalt:
Hat = Ti + Te + Vii + Vei + Vee. (2.2)
Hierbei stehen auf der rechten Seite die kinetische Energie der Atomkerne und
Elektronen sowie die Terme fur die Wechselwirkung der Teilchen untereinander.
Diese entspricht, sofern man die Teilchen wie geladene Massenpunkte behandelt,
dem nur vom Teilchenabstand abhangigen Coulombpotential.
Fur ein System aus K Atomkernen und N Elektronen erhalt man in der
Ortsdarstellung fur den Operator der kinetischen Energie den Ausdruck
Tat = Ti + Te = −K∑α=1
h2
2Mα∇2α −
N∑i=1
h2
2me∇2i , (2.3)
mit dem Planckschen Wirkungsquantum h = h/2π, den Kernmassen Mα und der
Elektronenmasse me. Die Wechselwirkungsterme lauten
13

14 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Vat = Vii + Vei + Vee (2.4)
=
K−1∑α=1
K∑β=α+1
ZαZβ|Rα − Rβ| −
K∑α=1
N∑i=1
Zα e
|Rα − ri| +
N−1∑i=1
N∑j=i+1
e2
|ri − rj| .
Hierbei bezeichnen {Rα} und {ri} die Positionen der Atomkerne bzw. Elektronen,
Zα die Kernladungszahl des Atoms α und e die Elementarladung.
In der Born-Oppenheimer-Naherung [9] geht man davon aus, daß die Elektro-
nen auf Grund ihrer im Vergleich zu den Kernen viel geringeren Masse den Bewe-
gungen der Atomkerne praktisch instantan folgen. Aus diesem Grund kann man
die Koordinaten der Atomkerne im Term Vei als Parameter auffassen und gelangt
so zum Hamiltonoperator des elektronischen Systems, welcher fur N Elektronen
die folgende Form hat:
Hel =
N∑i=1
− h2
2me∇2i︸ ︷︷ ︸
T
+
N∑i=1
vext(ri)︸ ︷︷ ︸V
+
N−1∑i=1
N∑j=i+1
e2
|ri − rj|︸ ︷︷ ︸W
. (2.5)
Die Atomkerne, die an den Orten {Rα} fixiert sind, ubernehmen jetzt die Rolle
eines außeren Potentials. Es gilt:
vext(ri) = −K∑α=1
Zα e
|Rα − ri| . (2.6)
In der Ortsdarstellung wird der N -Elektronen-Zustand durch die Wellenfunk-
tion Ψ(x1, . . . , xN , t) ∈ HN beschrieben, wobei HN der N -Teilchen-Hilbertraum
ist. Die Variable xi ist der kombinierte Index fur Ortskoordinate ri und Spin-
quantenzahl σi des Elektrons i.
Da der Hamiltonoperator Hel in (2.5) nicht explizit zeitabhangig ist, ist die
Gesamtenergie des elektronischen Systems eine Erhaltungsgroße. Die Zustande,
in denen sich das System befinden kann, sind Linearkombinationen der Eigenvek-
toren von Hel; diese genugen der Eigenwertgleichung
HelΨ(x1, . . . , xN , t) = EΨ(x1, . . . , xN , t). (2.7)
Die Wellenfunktion Ψ(x1, . . . , xN , t) laßt sich als Produkt aus einem Ortsanteil
ψ (x1, . . . , xN) und einem Zeitanteil f(t) schreiben. Letzterer stellt nur einen Pha-
senfaktor dar, so daß Erwartungswerte von zeitunabhangigen Operatoren zeitlich
konstant sind. Deshalb bezeichnet man die Losungen von (2.7) als stationar.

2.1. EFFEKTIVE SCHRODINGERGLEICHUNG 15
Druckt man die Koordinaten der Elektronen als Vielfache des Bohrschen
Radius aB = h2/2mee2 und die Energie als Vielfache der Hartree-Energie
Eh = mee4/h2 aus, so ergibt sich1 die dimensionslose, nichtrelativistische, zei-
tunabhangige Schrodingergleichung:(−1
2
N∑i=1
∇2i +
N∑i=1
vext(ri) +N−1∑i=1
N∑j=i+1
1
|ri − rj |
)ψ (x1, . . . , xN ) =
= Eψ (x1, . . . , xN ). (2.8)
Da die potentielle Energie des Systems durch ein 1/r-Potential bestimmt wird,
ist das Spektrum der Energieeigenwerte nach unten beschrankt [10]. Es ist zu
beachten, daß nicht jedem der Eigenfunktionen auch eine physikalische Bedeu-
tung zukommt. Da Elektronen ununterscheidbare Teilchen mit halbzahligem Spin
(Fermionen) sind, muß die Wellenfunktion antisymmetrisch in Bezug auf die Ver-
tauschung zweier Teilchen sein:
ψ (. . . , xi, . . . , xj, . . . ) = −ψ (. . . , xj , . . . , xi, . . . ). (2.9)
Diese Bedingung beschrankt die Eigenfunktionen von Hel auf den Unterraum
HN ⊂ HN der antisymmetrischen Wellenfunktionen. Den kleinsten, zu einer mit
(2.9) vertraglichen Wellenfunktion gehorenden Energieeigenwert bezeichnet man
als die Grundzustandsenergie E0 des Systems.
Der Grundzustand kann im Prinzip auch entartet sein. Die Grundzustandswel-
lenfunktion ψ0 ist dann, im Gegensatz zur Grundzustandsenergie, nicht eindeutig
bestimmt. Entsprechendes gilt fur die Grundzustandselektronendichte
ρ0(r) = N∑σi
∫dr2 . . .drN|ψ0 (x, x2, . . . , xN )|2. (2.10)
Aufgrund der Tatsache, daß der Hamiltonoperator (2.5) nicht vom Spin der
Teilchen abhangt, ist es moglich, Losungen von (2.8) so zu wahlen, daß sie zusatz-
lich Eigenfunktionen der Operatoren fur den projizierten und den totalen Spin
sind
Szψ0 = Msψ0 ,
S 2ψ0 = S(S + 1)ψ0 .(2.11)
1Eigentlich mußte in dieser Gleichung uberall ri = ri/aB anstelle von ri, E = E/Eh stattE und ψ (x1, . . . , xN ) anstelle von ψ (x1, . . . , xN ) stehen. Zugunsten der besseren Lesbarkeitwurden die alten Bezeichnungen aber weiter verwendet.

16 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Die exakte, analytische Losung von (2.8) ist fur Systeme mit mehr als ei-
nem Elektron prinzipiell unmoglich. Da der Raum aller antisymmetrischen N -
Teilchen-Wellenfunktionen die Dimension unendlich hat, ist aber auch eine be-
liebig genaue algebraische Losung ausgeschlossen, da man theoretisch unendlich
viele Basisfunktionen in die Rechnung miteinbeziehen mußte, um die Eigenfunk-
tionen von Hel und somit die wahre Grundzustandsenergie E0 zu finden.
Da E0 aber der kleinste, mit (2.9) vertragliche Eigenwert von Hel ist, gilt das
Variationsprinzip
E0 ≤ 〈ψ |Hel |ψ〉, ∀ψ ∈ HN , 〈ψ |ψ〉 = 1, (2.12)
wobei das Gleichheitszeichen fur ψ = ψ0 gilt. Es besagt, daß die Grundzustands-
energie eine untere Schranke der Erwartungswerte von Hel mit jeder beliebigen
antisymmetrischen, aber normierten Wellenfunktion ψ darstellt. Anders ausge-
druckt, ist
E0 = minψ∈HN
{〈ψ |Hel |ψ〉
∣∣∣ 〈ψ |ψ〉 = 1}. (2.13)
Somit gibt es ein Kriterium, mit dessen Hilfe man sich auf die Suche nach
dem Grundzustand eines Systems aus N Elektronen machen kann: man be-
rechne die Erwartungswerte von Hel mit allen nur erdenklichen N-Elektronen-
Wellenfunktionen und wahle den kleinsten Wert aus.
Naturlich ist diese Methode praktisch nicht durchfuhrbar. Es gibt aber eine
Reihe von Verfahren, die sich mit der Suche nach der besten Losung der effek-
tiven Schrodingergleichung im Sinne der Minimierung von 〈ψ | Hel |ψ〉 in einem
bestimmten Teilraum des HN befassen. In der Literatur werden diese oft als ab
initio-Methoden bezeichnet.
Einen ganz anderen Ansatz verfolgt die Dichtefunktionaltheorie. Die zu va-
riierende Große ist hier die Elektronendichte ρ(r), wie sie in (2.10) definiert ist.
Aber auch die Dichtefunktionaltheorie stutzt sich auf das Variationsprinzip, wenn
auch nicht mit dem exakten Hamiltonoperator Hel.
Im folgenden wird anstelle von ψ (x1, . . . , xN) wieder die Bezeichnung
Ψ(x1, . . . , xN) verwendet. Außerdem wird der elektronische Hamiltonoperator
Hel in atomaren Einheiten von nun an nur noch mit H bezeichnet.

2.2. DER HARTREE-FOCK ANSATZ 17
2.2 Der Hartree-Fock Ansatz
Eine Einteilchenwellenfunktion ϕ(x) = ϕ(r)χ(σ) mit∫d3r ϕ∗(r)ϕ(r) = 1 und 〈χ(σ)|χ(σ)〉 = 1 (2.14)
wird als Spinorbital bezeichnet, ihr Ortsanteil ϕ(r) als Orbital und χ(σ) ∈ {α, β}als Spinfunktion. Ein reines Produkt von solchen Spinorbitalen (ein sogenanntes
Hartree-Produkt [11])
ϕ1(x1)ϕ2(x2) · . . . · ϕN (xN)
erfullt offensichtlich nicht die Forderung nach Antisymmetrie. Laßt man jedoch
alle moglichen Permutationen der Elektronen zu, summiert dann uber alle die-
se Zustande und normiert die so entstandene Wellenfunktion, erhalt man eine
sogenannte Slaterdeterminante, die sich in der folgenden Form darstellen laßt:
Φ(x1, . . . , xN) =1√N !
∣∣∣∣∣∣∣∣ϕ1(x1) . . . ϕN (x1)
......
...
ϕ1(xN) . . . ϕN(xN)
∣∣∣∣∣∣∣∣ . (2.15)
Das Vertauschen zweier Teilchen ist in dieser Darstellung aquivalent mit dem
Vertauschen zweier Zeilen der Matrix, was mit einem Vorzeichenwechsel der De-
terminante verbunden ist. Somit ist die Wellenfunktion Φ(x1, . . . , xN ) antisym-
metrisch, sie ist außerdem eine Eigenfunktion des Sz-Operators (2.11) mit dem
Eigenwert Ms = 12(Nα − Nβ), wobei Nα bzw. Nβ die Zahl der Elektronen mit
χ(ms) = α bzw. χ(ms) = β bezeichnet. Man kann jedoch weder davon ausgehen,
daß sie eine Eigenfunktion von S 2 noch eine des (elektronischen) Hamiltonope-
rators H ist.
Falls die Menge {ϕi(x)} der Einteilchenwellenfunktionen vollstandig ist, bil-
det die Menge {Φ(x1, . . . , xN)} aller Slaterdeterminanten, die aus diesen Spinor-
bitalen gebildet werden konnen, eine Basis des HN . Somit kann die Grundzu-
standsenergie berechnet werden, indem man den Grundzustand in der Basis der
Slaterdeterminanten entwickelt
Ψ(x1, . . . , xN ) =∑n
αn Φn(x1, . . . , xN) (2.16)

18 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
und den Erwartungswert 〈Ψ| H |Ψ 〉 durch Variation der Koeffizienten αi mi-
nimiert, was bei vollstandiger Einteilchenbasis aufgrund der unendlichen Zahl
der zu variierenden Koeffizienten unmoglich ist.
Eine drastische Vereinfachung besteht nun darin, in der Summe (2.16) nur
einen einzigen Term zu berucksichtigen und durch geschickte Variation der
{ϕi(x)} diejenige Slaterdeterminante zu finden, welche den Erwartungswert
〈Ψ| H |Ψ 〉 minimiert. Dieser Ansatz wird als Hartree-Fock-Methode (HF) [12]
bezeichnet. Unterliegen die Spinorbitale {ϕi(x)} keinerlei Einschrankungen, ins-
besondere in Bezug auf ihre Spinfunktion, so spricht man von der Unrestricted
Hartree-Fock Methode (UHF). Allerdings ist eine einzelne Slaterdeterminante nur
in einigen Spezialfallen, wie dem, daß alle Elektronen gepaart sind, daß sich also
jeweils zwei der Spinorbitale in (2.15) nur in ihrer Spinfunktion unterscheiden,
eine Eigenfunktion des S2-Operators, so daß der mit dieser Methode gefunde-
ne Grundzustand im allgemeinen nicht den korrekten Gesamtspin aufweist. Man
spricht dann auch von”spin contamination“.
Diesen Mangel gleicht die Restricted Open-Shell Hartree-Fock-Methode
(ROHF) dadurch aus, daß - je nach Gesamtspin - eine gewisse Anzahl von Paaren
von Spinorbitalen in (2.15) in ihrem Orbital ubereinstimmt, vgl. Abbildung 2.1.
Zwar weist die so gebildete Slaterdeterminante nicht notwendigerweise die gesuch-
te Spinsymmetrie auf, es laßt sich aber immer eine endliche, fur jeden Wert des
Gesamtspins feste Linearkombination von Slaterdeterminanten (eine sogenann-
te Configuration-State Function (CSF)) konstruieren, die eine Eigenfunktion des
S 2-Operators ist. Dies ist in der UHF-Methode nicht moglich. Im Fall von ver-
schwindendem Gesamtspin spricht man von der Restricted Hartree-Fock-Methode
(RHF). Da die Wahl der Spinorbitale durch die erwahnte Nebenbedingung einge-
schrankt ist, ist die durch ROHF gefundene Grundzustandsenergie immer großer
oder gleich dem Wert in der UHF-Methode, die Wellenfunktion hat aber immer
die richtige Symmetrie in Bezug auf den Spinzustand.
Ein gutes Beispiel, um diesen Sachverhalt zu demonstrieren, bietet die Dis-
soziation des Wasserstoffmolekuls. Befinden sich die beiden Atome im Gleichge-
wichtsabstand, besetzen die beiden Elektronen dasselbe bindende Molekulorbital
und bilden ein Spinsingulett (S = 0). Im dissoziierten Zustand befindet sich je-
weils ein Elektron in der Nahe jedes Protons, die Elektronen besetzen jeweils
ein Atomorbital und bilden ein Spintriplett (S = 1). Die RHF-Rechnung behalt

2.2. DER HARTREE-FOCK ANSATZ 19
6
6
6
6
6
ϕ1,↑
ϕ2,↑
ϕ3,↑
ϕ4,↑
ϕ5,↑
?
?
?
?
ϕ1,↓
ϕ2,↓
ϕ3,↓
ϕ4,↓
ϕ5,↓
6
6
6
6
6
?
?
?
?
ϕ1
ϕ2
ϕ3
ϕ4
ϕ5
Abbildung 2.1: Der Unterschied zwischen der UHF-Methode (linke Seite) und der
RHF-Methode (rechte Seite).
nun den Singulett-Zustand fur alle Abstande der beiden Atome bei, was zu ei-
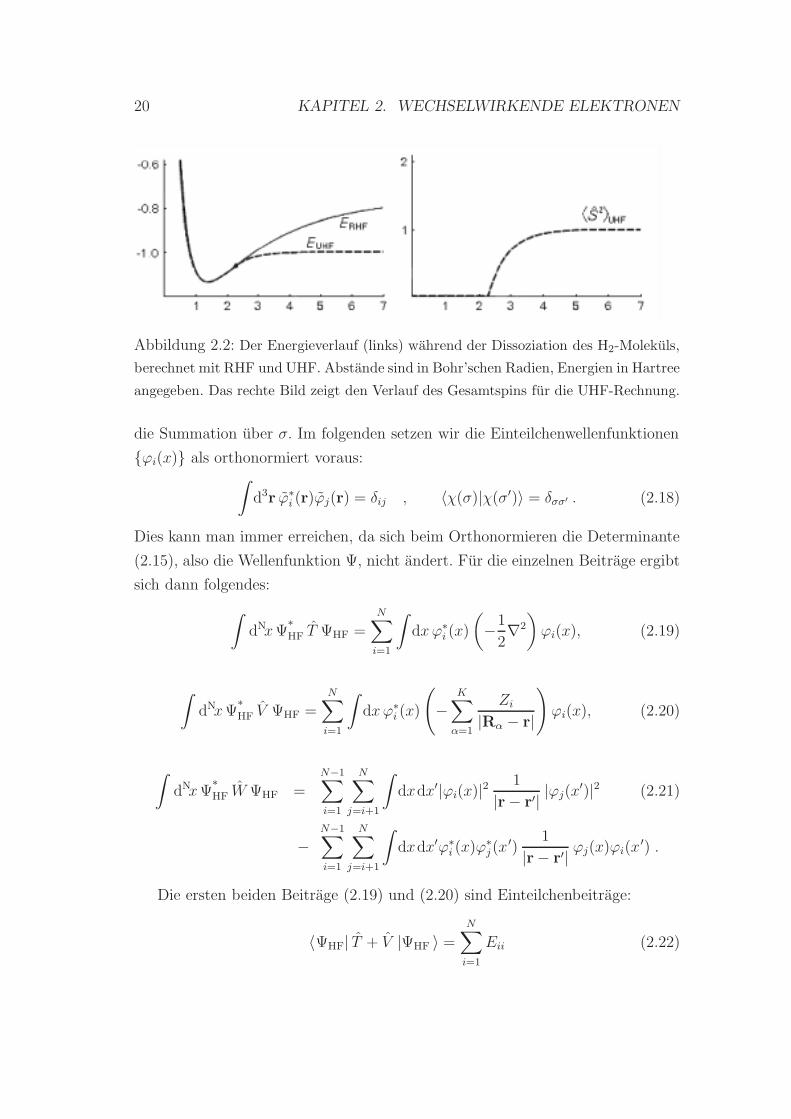
nem falschen Energieverlauf fuhrt (Abbildung 2.2 ). Bis zu einem Abstand von
r = 2.3 aB stimmt der Verlauf der UHF-Energie mit dem der RHF-Energie ube-
rein, die Wellenfunktionen sind vollig identisch. Bei großeren Abstanden jedoch
macht sich bemerkbar, daß die Spinorbitale der UHF-Methode nicht im Ortsan-
teil ubereinstimmen mussen. Ab diesem Punkt verlaufen die Energiekurven nicht
mehr parallel, die UHF-Energie konvergiert in den korrekten Wert bei vollstandi-
ger Dissoziation.
Ab r = 2.3 aB weist die UHF-Kurve fur den Gesamtspin eine Diskontinuitat
auf, der Wert des Spins steigt rasch von 0 auf 1 an. Die UHF-Wellenfunktion des
dissoziierten Wasserstoffmolekuls ist dann eine unphysikalische Uberlagerung aus
Singulett- und Triplett-Zustand.
Im Spezialfall von verschwindendem Gesamtspin stimmen RHF und UHF
uberein, wie man an vorigem Beispiel sehen konnte. Man spricht dann von der
Closed-Shell HF-Methode.
Der Erwartungswert des Hamiltonoperators berechnet sich mit (2.5) und (2.8)
zu
EHF = 〈ΨHF| T + V + W |ΨHF 〉 (2.17)
=
∫dNxΨ
∗HF
(−1
2
N∑i=1
∇2i +
N∑i=1
vext(ri) +
N−1∑i=1
N∑j=i+1
1
|ri − rj |
)ΨHF
Hierbei steht die Integration uber x symbolisch fur die Integration uber r und
20 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Abbildung 2.2: Der Energieverlauf (links) wahrend der Dissoziation des H2-Molekuls,
berechnet mit RHF und UHF. Abstande sind in Bohr’schen Radien, Energien in Hartree
angegeben. Das rechte Bild zeigt den Verlauf des Gesamtspins fur die UHF-Rechnung.
die Summation uber σ. Im folgenden setzen wir die Einteilchenwellenfunktionen
{ϕi(x)} als orthonormiert voraus:∫d3r ϕ∗
i (r)ϕj(r) = δij , 〈χ(σ)|χ(σ′)〉 = δσσ′ . (2.18)
Dies kann man immer erreichen, da sich beim Orthonormieren die Determinante
(2.15), also die Wellenfunktion Ψ, nicht andert. Fur die einzelnen Beitrage ergibt
sich dann folgendes:∫dNxΨ
∗HF T ΨHF =
N∑i=1
∫dxϕ∗
i (x)
(−1
2∇2
)ϕi(x), (2.19)
∫dNxΨ
∗HF V ΨHF =
N∑i=1
∫dxϕ∗
i (x)
(−
K∑α=1
Zi|Rα − r|
)ϕi(x), (2.20)
∫dNxΨ
∗HF W ΨHF =
N−1∑i=1
N∑j=i+1
∫dxdx′|ϕi(x)|2 1
|r − r′| |ϕj(x′)|2 (2.21)
−N−1∑i=1
N∑j=i+1
∫dxdx′ϕ∗
i (x)ϕ∗j (x
′)1
|r − r′| ϕj(x)ϕi(x′) .
Die ersten beiden Beitrage (2.19) und (2.20) sind Einteilchenbeitrage:
〈ΨHF| T + V |ΨHF 〉 =
N∑i=1
Eii (2.22)

2.2. DER HARTREE-FOCK ANSATZ 21
Das Ergebnis von (2.21) teilt man auf in den Coulomb- und den Austauschterm
〈ΨHF| W |ΨHF 〉 =
N−1∑i=1
N∑j=i+1
Cij −N−1∑i=1
N∑j=i+1
Jij. (2.23)
Fur die HF-Energie erhalt man schließlich, wenn man beachtet, daß Cii = Jii
ist, den etwas einfacheren Ausdruck
EHF =N∑i=1
Eii +1
2
N∑i=1
N∑j=1
(Cij − Jij). (2.24)
Der Coulombterm beschreibt die elektrostatische Wechselwirkung der Ladungs-
verteilungen der Elektronen i und j. Fur i = j beinhaltet Cii die sogenannte
Selbstenergie, die Wechselwirkung des Elektrons i mit seiner eigenen Ladungs-
verteilung. Dieser Term wird aber gerade durch den Austauschterm Jii aufgeho-
ben. Die Austauschenergie ist rein quantenmechanischer Natur und ist auf die
Antisymmetrie der Wellenfunktion zuruckzufuhren. Sie senkt die Energie stets
ab, verschwindet aber fur Elektronen mit verschiedener Spinquantenzahl, da das
Skalarprodukt 〈α | β 〉 nach (2.18) verschwindet.
Die Grundzustandsenergie EHF0 in der HF-Naherung wird nun dadurch be-
stimmt, daß die Variation von EHF nach den Spinorbitalen {ϕi(x)} verschwindet:
δEHF
δφi(x)= 0, ∀ i = 1 . . .N. ⇒ EHF = EHF
0 . (2.25)
Unter der Nebenbedingung, daß die Spinorbitale orthonormiert bleiben, ergeben
sich die Hartree-Fock-Gleichungen:
f ϕi(x) = εiϕi(x) (2.26)
mit dem Fock-Operator
f = −1
2∇2 + vext(r) + VHF(x). (2.27)
Das HF-Potential VHF setzt sich aus dem (lokalen) Coulomb- und dem (nicht-
lokalen) Austauschoperator zusammen:
VHF(x) =
N∑j=1
(Cj(x) − Jj(x)), (2.28)

22 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
...
6
6
6
ε1
ε3
...
εN
εN+2
...
?
?
?
ε2
ε4
...
εN−1
εN+1
...
6
E
E = 0
Abbildung 2.3: Die Orbitale mit den Energien ε1 bis εN sind besetzt. Die energetisch
hoherliegenden Orbitale werden als virtuelle Orbitale bezeichnet.
mit
Cj(x) =
∫dx′|ϕj(x′)|2 1
|r− r ′| (2.29)
und
Jj(x)ϕi(x) =
∫dx′ϕ∗
j (x′)
1
|r − r′| ϕi(x′) · ϕj(x). (2.30)
Da der Fock-Operator hermitesch ist, erhalt man als Losungen der HF-
Gleichungen eine unendliche Zahl von unterschiedlichen Eigenwerten mit den
dazugehorigen Eigenfunktionen. Die N energetisch niedrigsten Spinorbitale defi-
nieren den HF-Grundzustand ΨHF, alle anderen Eigenfunktionen sind zu ihnen
orthogonal und werden als virtuelle Orbitale bezeichnet (vgl. Abbildung 2.3). Das
Vielteilchenproblem (2.8) mit dem vollen Wechselwirkungsterm W und der Viel-
teilchenwellenfunktion wird im Hartree-Fock-Formalismus auf ein Einteilchenpro-
blem zuruckgefuhrt. Jedes Elektron bewegt sich unabhangig von den anderen in
einem Einteilchenpotential VHF(x), welches den Einfluß der anderen Elektronen
im Mittel wiederspiegelt. In diesem Sinne [13] kann man den Eigenwert |εi| als
die Energie auffassen, die man aufwenden mußte, um das Elektron im Spinor-
bital i aus dem System zu entfernen (fur i ≤ N), bzw. als die Energie, die frei

2.2. DER HARTREE-FOCK ANSATZ 23
wurde, wenn man dem System ein Elektron in diesem Orbital hinzufugte (fur
i > N). Diese Tatsache ist als Koopmans’sches Theorem bekannt. Da der Fock-
Operator die Spinorbitale selbst in Gestalt des HF-Potentials enthalt, mussen die
Gleichungen (2.26) selbstkonsistent gelost werden.
Eine analytische Losung der HF-Gleichungen ist ebenso unmoglich wie die
direkte Losung der Schrodingergleichung (2.8). Deshalb beschrankt man sich zu-
meist darauf, die Spinorbitale nach gewissen Basisfunktionen zu entwickeln
ϕi(x) =M∑µ=1
ciµχµ (2.31)
und die Koeffizienten {ciµ} so zu bestimmen, daß EHF minimal wird:
∂EHF
∂ ciµ= 0, ∀ i = 1 . . .N, µ = 1 . . .M. (2.32)
Dies fuhrt auf eine Eigenwertgleichung in Matrixform, deren Losung im Fall von
RHF nach dem Verfahren von Roothaan und Hall [14, 15] und im Fall von UHF
nach der Methode von Pople und Nesbet [16] erfolgen kann. Als Resultat ei-
ner solchen Rechnung erhalt man einen Satz von M Spinorbitalen, wovon die N
energetisch niedrigsten den HF-Grundzustand bilden. Die Energie, die man so
erhalt, hangt naturlich von dem Basissatz {χµ} ab, und wird, grob gesprochen,
umso niedriger, je umfangreicher der Basissatz ist. Im Grenzfall einer vollstandi-
gen Basis spricht man vom HF-Limit der Grundzustandsenergie, denn dann ist
auch die Menge der Spinorbitale, die aus den Basisfunktionen linear kombiniert
werden konnen, vollstandig.
Es gibt noch eine weitere Moglichkeit, den HF-Grundzustand zu ermitteln,
und zwar die numerische Losung der HF-Gleichungen. Da diese Gleichungen aber
komplizierte Integro-Differentialgleichungen sind, ist ihre Losung mit sehr hohem
Aufwand verbunden und im Moment nur fur Systeme moglich, die aus einem
einzigen Atom bestehen oder einen hohen Grad an Symmetrie aufweisen, wie
dies z.B. bei einem linearen Molekul der Fall ist. Ergebnisse von solchen Rech-
nungen sind oftmals von großer Genauigkeit und konnen dazu benutzt werden,
die Qualitat einer Einelektronenbasis {χµ} in Bezug auf die Reproduktion der so
gewonnenen (exakten) HF-Spinorbitale zu beurteilen. Durch den Vergleich mit
numerischen Ergebnissen hat sich z.B. herausgestellt, daß der Vorschlag von Sla-
ter [17], die Orbitale von isolierten Atomen als sogenannte Slater-Type Orbitals

24 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
(STO’s) darzustellen, die (im HF-Bild) exakten Orbitale ausgesprochen gut wie-
dergibt. Dieses Resultat (vgl. Kap. 2.5.1) laßt sich durch Linearkombinationen
weniger STO’s noch verbessern, was wiederum fur die Wahl solcher Funktionen
als Einelektronenbasis spricht.
Ergebnisse von HF-Rechnungen fur Atome zeigen, daß die Orbitale isolierter
Atome wasserstoffahnlichen Charakter haben. Dies ist der Grund, warum man
bei Elektronenkonfigurationen einzelner Atome von s-, p-, d-Orbitalen spricht.
Wahlt man fur die Basisfunktionen in (2.31) Atomorbitale, spricht man von der
LCAO-Methode, welche ursprunglich auf Bloch [18] zuruckgeht.
Die aus den Einteilchenwellenfunktionen {ϕi(x)} aufgebaute Slaterdetermi-
nante ΨHF lost die Schrodingergleichung
N∑i=1
fiΨHF =N∑i=1
εiΨHF, (2.33)
wobei die fi Einteilchenoperatoren sind. Damit wird noch einmal klar, daß dem
HF-Ansatz die Annahme zugrunde liegt, daß sich jedes Elektron unabhangig
von den anderen Elektronen in einem Einteilchenpotential bewegt. Korreliert ist
die Bewegung der Elektronen einzig und allein dadurch, daß die Wellenfunktion
ΨHF antisymmetrisch in Bezug auf die Vertauschung der Koordinaten zweier
Elektronen sein muß. Die Wahrscheinlichkeit dafur, daß sich zwei Elektronen
im selben Raumelement dr aufhalten, ist gegeben durch
ρ2(r, r) =N(N − 1)
2
∑σi
∫dr3 . . .drN|ψ0 (x, x2, . . . , xN)|2
∣∣∣∣∣r2=r
. (2.34)
Im HF-Bild verschwindet diese Wahrscheinlichkeit fur Elektronen mit gleichem
Spin:
ρHF2 (r, r) = 0 fur σ1 = σ2.
Diese Tatsache wird als Fermi- oder Austausch-Korrelation bezeichnet und die
dazugehorige Verminderung der Elektronendichte der ubrigen Elektronen um den
Ort eines jeden Elektrons herum als Fermi- oder Austausch-Loch.
2.3 Post-Hartree-Fock-Methoden
Die Wahrscheinlichkeit dafur, daß sich zwei Elektronen mit verschiedenem Spin in
den Raumelementen dr1 und dr2 aufhalten, entspricht im HF-Bild dem Produkt

2.3. POST-HARTREE-FOCK-METHODEN 25
der Wahrscheinlichkeitsdichten der einzelnen Elektronen:
ρHF2 (r1, r2) = ρ(r1)ρ(r2) fur σ1 6= σ2.
Fur r2 = r1 resultiert daraus eine endliche Wahrscheinlichkeit dafur, zwei
Elektronen mit unterschiedlichem Spin am selben Ort zu finden. In Wirklichkeit
sind aber auch die Bewegungen solcher Elektronen korreliert, und zwar durch die
abstoßende Coulombwechselwirkung, aufgrund derer sich die Elektronen gegen-
seitig ausweichen. Diese als Coulombkorrelation bezeichnete Tatsache wird durch
den Eindeterminantenansatz fur die HF-Wellenfunktion nicht beschrieben. Die
tatsachliche Coulombabstoßung wird im HF-Bild uberschatzt, was zu einer zu
großen Gesamtenergie fuhrt.
Legt man einen bestimmten Basissatz fur die Entwicklung der Einteilchen-
wellenfunktionen zugrunde, so bezeichnet man den Unterschied zwischen der in
diesem Basissatz exakten Grundzustandsenergie (vgl. 2.3.1) und der HF-Energie
in diesem Basissatz als Korrelationsenergie:
Ec = E0 − EHF0 . (2.35)
Es gibt viele Methoden, die sich, auf dem HF-Verfahren aufbauend, mit
der Suche nach der Korrelationsenergie befassen. Sie werden zumeist als Post-
Hartree-Fock Methoden bezeichnet, und ihnen ist gemeinsam, daß sie versuchen,
durch Erweiterungen des HF-Ansatzes eine Verbesserung (im Sinne von Verklei-
nerung) der Grundzustandsenergie eines elektronischen Systems zu erzielen.
2.3.1 Configuration Interaction
Sei M := {ϕi(x)} eine Menge von orthonormierten, quadratintegrablen Einteil-
chenwellenfunktionen und
S := { ‖ϕα1 , . . . , ϕαN‖ mit α1 < α2 < · · · < αN}
die Menge aller antisymmetrisierten Produkte, die man aus N solchen Funktio-
nen bilden kann (dann muß offensichtlich |M |> N sein). Wenn M vollstandig
ist, so ist S eine Basis des HN , d.h. man kann jede beliebige N -Elektronen-
Wellenfunktion durch Linearkombination von Slaterdeterminanten darstellen.
Wenn M jedoch endlich ist, gilt dieser Sachverhalt nicht mehr; S spannt dann

26 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
einen Unterraum U von HN auf. Die Suche nach der besten Losung der elektro-
nischen Schrodingergleichung im Sinne von (2.13) in diesem Unterraum wird als
Full Configuration Interaction (FCI) bezeichnet.
Jedes Element von S ist aufgrund der Definition eindeutig durch die Wahl
der ϕα1 , . . . , ϕαNbestimmt. Es lassen sich also alle Elemente von S dadurch
erzeugen, daß man einmal N beliebige Funktionen aus M auswahlt, und dann
bei diesem Referenzzustand ein oder mehrere Orbitale gegen andere austauscht.
Dieser Referenzzustand sei mit Φ0 := ‖ ϕ1, . . . , ϕN ‖ bezeichnet. Definiert man
folgende Operatoren uber die Art und Weise, wie sie auf den Referenzzustand
wirken
T ai ‖ϕ1 . . . ϕi . . . ϕN ‖ = ‖ϕ1 . . . ϕN ϕa‖ =: Φai
T abij ‖ϕ1 . . . ϕi . . . ϕj . . . ϕN ‖ = ‖ϕ1 . . . ϕN ϕa ϕb‖ =: Φabij
......
so lassen sich die Elemente von S nach einfach, zweifach oder mehrfach angeregten
Zustanden ordnen:
S = {Φ0,Φai ,Φ
abij ,Φ
abcijk , . . . } , i < j < · · · < N , N < a < b < . . .
Damit laßt sich jede N -Elektronen-Wellenfunktion aus U als Linearkombination
der folgenden Form schreiben:
Ψ = Φ0 +∑i,a
cai Φai +
∑i<ja<b
cabij Φabij +
∑i<j<ka<b<c
cabcijk Φabcijk + ... (2.36)
oder in Operatorschreibweise:
Ψ =(1 +
∑i,a
cai Tai︸ ︷︷ ︸
T1
+∑i<ja<b
cabij Tabij
︸ ︷︷ ︸T2
+∑
i<j<ka<b<c
cabcijk Tabcijk
︸ ︷︷ ︸T3
+ . . .)Φ0. (2.37)
Die FCI-Wellenfunktion hat dann fur ein System aus N Elektronen die Gestalt:
ΨFCI =(1 + T1 + T2 + · · ·+ TN
)Φ0. (2.38)
Der Erwartungswert des Hamiltonoperators bezuglich dieser Wellenfunktion de-
finiert die FCI-Energie:
EFCI = 〈ΨFCI| H |ΨFCI 〉. (2.39)

2.3. POST-HARTREE-FOCK-METHODEN 27
Gegenstand der Variation sind nun die Koeffizienten cai , cabij , . . . , die exakte Grund-
zustandsenergie bezuglich der gewahlten Einelektronenbasis ist gegeben durch die
Bedingungen
∂EFCI
∂ cabc...ijk...
= 0, ∀ i < j < . . .N , N < a < b < . . . ⇒ EFCI = E0. (2.40)
Somit ist die FCI-Wellenfunktion die beste Losung der elektronischen Schro-
dingergleichung in einem bestimmten Unterraum des HN ; im Grenzfall einer
vollstandigen Einelektronenbasis entspricht die FCI-Energie der exakten Grund-
zustandsenergie.
Oft wahlt man fur die Einteilchenfunktionen die M Spinorbitale, die sich aus
einer Hartree-Fock-Rechnung ergeben haben und als Referenzzustand den HF-
Grundzzustand ΨHF. Die Zahl der Slaterdeterminanten ist dann gegeben durch
die Zahl der Moglichkeiten, N Elektronen auf M Spinorbitale zu verteilen, und
zwar explizit durch (M
N
)=
M !
N ! (M −N)!. (2.41)
Diese Funktion wachst sehr schnell und nimmt schon bei moderatem N und M
sehr große Werte an. Zwar schrankt die Bedingung, daß die Wellenfunktion einen
gewissen Gesamtspin aufweisen soll, die
Zahl der in Frage kommenden Determi-
nanten noch erheblich ein, aber dennoch
ist eine FCI-Rechnung fur fast alle elek-
tronischen Systeme vollig unmoglich.
Zur Illustration dieses Sachverhalts
moge folgendes Beispiel dienen. Ein Sy-
stem aus 2N Elektronen befinde sich im
Spinsingulettzustand. Bezieht man fur
jedes Elektron ein Orbital in die Rech-
nung mit ein, so erhalt man insgesamt
M = 2N Spinorbitale, von denen N
besetzt sind. Die nebenstehende Tabel-
le verdeutlicht das rapide Wachstum der
Zahl der Slaterdeterminanten einer FCI
fur dieses Beispiel.
N Ndet
2 4
4 36
6 400
8 4900
10 63504
12 853776
14 11778624
16 165636900
18 2363904400
20 34134779536
Tabelle 2.1: Zahl der Determi-
nanten in einer FCI-Rechnung fur
M=2N .

28 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
In der Praxis ist man somit oft gezwungen, in die FCI-Entwicklung nur einen
Teil der Determinanten miteinzubeziehen. Dies fuhrt zu der Frage, nach wel-
chen Kriterien man bei der Auswahl der Determinanten vorgehen soll. Ein na-
heliegender Ansatz ware, die Entwicklung (2.36) nach einem bestimmten Term
abzubrechen. Das wurde bedeuten, daß man nur Anregungen bis zu einer ge-
wissen Ordnung berucksichtigt. Diese Methode wird als Configuration Interac-
tion (CI) bezeichnet. Da Einfach-Anregungen alleine die Grundzustandsenergie
in Bezug auf die HF-Energie nicht erniedrigen (Brillouin-Theorem), muß man
wenigstens Doppelt-Anregungen berucksichtigen (CID) oder aber Einfach- und
Doppelt-Anregungen (CISD). In letzterem Fall [19] ist die Wellenfunktion gege-
ben durch
ΨCISD =(1 + T1 + T2
)Φ0. (2.42)
Unterscheiden sich zwei Determinanten um mehr als zwei Spinorbitale, ver-
schwindet das entsprechende Matrixelement in (2.39), da der Hamiltonopera-
tor nur Ein- und Zwei-Teilchenterme enthalt, so daß z.B. Dreifach-Anregungen
alleine auch nicht die HF-Energie erniedrigen. Aus diesem Grund erhalt man
den großten Teil der Korrelationsenergie (2.35) durch die Berucksichtigung von
Doppelt-Anregungen. Die Grundzustandsenergie in der CISD-Naherung ist durch
das Minimum des folgenden Ausdrucks gegeben:
ECISD0 = min
{cai ,cabij }
〈ΨCISD| H |ΨCISD 〉 (2.43)
und ist somit eine obere Grenze fur die wahre Grundzustandsenergie, da die
Koeffizienten nach dem Variationsprinzip bestimmt werden.
Im Grenzfall großer Systeme steigt der Rechenaufwand einer CISD-Rechnung
mit der funften Potenz der Systemgroße, was zwar nicht mit dem fakultativen
Wachstum einer FCI-Rechnung zu vergleichen ist, jedoch der Art der behan-
delbaren Systeme klare Grenzen setzt. Laßt sich beispielsweise eine bestimmte
Eigenschaft eines Systems an einem Tag berechnen, so mußte man auf das glei-
che Ergebnis bei einem viermal großeren System unter Umstanden drei Jahre
warten. Deshalb ist man bemuht, die Zahl der Determinanten einer CI-Rechnung
moglichst klein zu halten. Dies gelingt beispielsweise durch die Frozen-Core Nahe-
rung, bei der keine Anregungen der Rumpfelektronen, sondern nur solche von
Valenzelektronen berucksichtigt werden. Damit vernachlassigt man zwar einen

2.3. POST-HARTREE-FOCK-METHODEN 29
betrachtlichen Teil der Korrelationsenergie, hofft aber, daß sich dieser Fehler
beim Bilden von Energiedifferenzen verschiedener Zustande eines Systems wie-
der heraushebt. Dem liegt die Annahme zugrunde, daß die Valenzelektronen der
entscheidende Faktor sind, wenn sich die chemische Umgebung eines Atoms z.B.
wahrend einer Reaktion andert, ein Gedanke, der auch bei der Idee der Pseudo-
potentiale (Kap. 2.5.2) eine Rolle spielt.
Im Vergleich zu HF-Rechnungen liefert schon die CISD-Methode oftmals
um ein Vielfaches genauere Werte fur die Grundzustandsenergie eines elektro-
nischen Systems. Daraus resultierend stimmen meist auch andere Großen, wie
z.B. Bindungsenergien, besser mit der Wirklichkeit uberein. Es gibt allerdings
einen großen Nachteil der CI-Methode. Besteht ein System aus mehreren, aber
(unendlich) weit voneinander entfernten Teilen, so ist die Gesamtwellenfunktion
ein Produkt der Wellenfunktionen der einzelnen Teilsysteme, da die Wechsel-
wirkung zwischen den Teilsystemen verschwindet. Aus diesem Grund sollte es
keinen Unterschied machen, ob man die Grundzustandsenergie fur das ganze Sy-
stem oder als Summe der Grundzustandsenergien der Teilsysteme berechnet, die
gefundenen Werte sollten ubereinstimmen. Eine CI-Rechnung liefert jedoch fur
die beiden Falle im allgemeinen verschiedene Ergebnisse. Dies ist darauf zuruck-
zufuhren, daß man immer nur Anregungen bis zu einer gewissen Ordnung in die
Rechnung miteinbezieht. Doppelt-Anregungen in beiden Teilen eines aus zwei
Teilsystemen zusammengesetzten System entsprechen z.B. Vierfach-Anregungen
im Gesamtsystem. Die fehlende Size Consistence der CI-Methode macht sich um-
so starker bemerkbar, je großer das zu untersuchende System ist. Ein Verfahren,
welches diesen Nachteil behebt, ist die sogenannte Coupled-Cluster Methode (CC)
[20, 21].
2.3.2 Coupled-Cluster-Methode
Jede N -Elektronen-Wellenfunktion laßt sich, wie oben gezeigt, bei gegebener Ein-
elektronenbasis als Summe (2.38) von angeregten Zustanden bezogen auf einen
bestimmten Referenzzustand darstellen. Aber auch durch den folgenden Expo-
nentialansatz
ΨCC = exp(T ) Φ0 , T = T1 + T2 + · · · + TN , (2.44)

30 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
wobei die Anregungsoperatoren Ti wie in (2.37) definiert sind, ist es moglich, jede
beliebige N -Elektronen-Wellenfunktion aus U zu beschreiben. Dieser Ansatz tragt
den Namen Coupled-Cluster-Ansatz (CC). Die Exponentialfunktion ist dabei in
der ublichen Weise als Neumannsche Reihe definiert
exp(T ) = 1 + T + 12T T + 1
6T T T + . . . (2.45)
und beinhaltet somit formal Anregungen jeglicher Ordnung. Aufgrund der Tat-
sache, daß die Anregungsoperatoren vertauschbar sind
[Ti, Tj] = 0, ∀ i, j < N , (2.46)
laßt sich Gleichung (2.44) durch Einsetzen von (2.45) umschreiben
ΨCC =(1 + T1 + T2 + 1
2T 2
1 + T3 + T1T2 + 16T 3
1 + . . .)
Φ0. (2.47)
Es wird ersichtlich, daß Anregungen einer bestimmten Ordnung i nicht nur durch
den entsprechenden Operator Ti hervorgerufen werden, sondern auch durch Pro-
dukte von Anregungen niedrigerer Ordnung. Die CC-Wellenfunktion ist in dieser
Formulierung mit der FCI-Wellenfunktion identisch, die einzelnen Beitrage sind
nur noch einmal in verschiedene Anteile aufgespalten worden.
Die Operatoren Ti sind in Analogie zu Gleichung (2.37) definiert, die Koef-
fizienten cai , cabij , . . . werden in diesem Zusammenhang meist als Amplituden be-
zeichnet. Fur die Grundzustandenergie erhalt man den Ausdruck
ECC0 = EHF
0 + 〈Φ0 | H | ( T2 + 12T 2
1 ) Φ0〉. (2.48)
Die Korrelationsenergie ist im CC-Bild, genau wie bei der FCI-Methode, allein
durch die Amplituden der einfach- und doppelt angeregten Slaterdeterminanten
bestimmt, da der Hamiltonoperator nur Ein- und Zwei-Teilchenbeitrage enthalt.
Im Gegensatz zum CI-Verfahren werden diese Koeffizienten hier jedoch nicht
durch Variation bestimmt, man kann vielmehr eine Reihe von Gleichungen her-
leiten, uber welche die einzelnen Amplituden in nichtlinearer Weise voneinander
abhangen. Auf diesem Wege macht sich der Einfluß der Amplituden hoherer Anre-
gungen bemerkbar, die selbstkonsistente Losung dieses Gleichungssystems liefert
schließlich die gesuchten Koeffizienten.
In der Praxis ist man somit wiederum gezwungen, aufgrund der ungeheuer
großen Zahl von Determinanten eine Auswahl der in die Rechnung eingehenden

2.3. POST-HARTREE-FOCK-METHODEN 31
Terme zu treffen. Bricht man die Summe (2.44) nach dem zweiten Glied ab, so
erhalt man die als CCSD-Naherung [22] bekannte Methode:
ΨCCSD = exp(T1 + T2) Φ0. (2.49)
Im Gegensatz zum CI-Verfahren bedeutet dies aber nicht, daß nur einfach und
zweifach angeregte Zustande in die Rechnung eingehen, denn durch den Exponen-
tialansatz werden, zumindest im Prinzip, Anregungen jedweder Ordnung, wenn
auch als Produkte in T1 und T2, berucksichtigt. Die oben angesprochene Na-
tur des Hamiltonoperators fuhrt dazu, daß man nun einen endlichen Satz von
Gleichungen fur die Koeffizienten erhalt, in denen formal nur Anregungen bis zur
vierten Ordnung auftauchen. Aber gerade diese Terme, welche in der CI-Methode
fehlen, sind der Grund dafur, daß eine CCSD-Rechnung die Forderung nach Size
Consistence erfullt.
Die Gleichungen, die die Amplituden miteinander verknupfen, sind jetzt al-
lerdings nicht mehr exakt, so daß die gefundene Grundzustandsenergie nur eine
Approximation der wahren Grundzustandsenergie ist.
Eine Verbesserung dieser Naherung laßt sich im Prinzip dadurch erzielen, daß
man mehr Terme in (2.44) berucksichtigt. Man spricht dann von der CCSDT,
CCSDTQ, usw. Der numerische Aufwand ist aber schon bei einer CCSDT-
Rechnung so gewaltig (die benotigte Rechenzeit skaliert mit der achten Potenz der
Systemgroße, im Vergleich zur sechsten Potenz bei CCSD), daß diese Verfahren
nur bei den allerkleinsten Systemen Anwendung finden.
Stattdessen erfreut sich eine andere Methode, welche unter der Bezeichnung
CCSD(T) bekannt ist, großer Beliebtheit. Dabei werden die Koeffizienten der
Einfach- und Doppelt-Anregungen wie beim CCSD-Verfahren berechnet, der Ef-
fekt von Dreifach-Anregungen wird hinterher storungstheoretisch [23] erfaßt. Die-
se Berechnung beinhaltet den großten Teil des Rechenaufwands, was zur Folge
hat, daß CCSD(T) prinzipiell eine N7-Methode ist. Der genaue Formalismus von
CCSD(T) kann in [24] nachgelesen werden.
2.3.3 Andere Erweiterungen des Hartree-Fock-Ansatzes
Neben CI und CC weit verbreitet ist eine dritte Post-Hartree-Fock Methode, die
auf Møller und Plesset [23] zuruckgeht. Dabei wird der Hamiltonoperator auf-
gespalten in eine Summe von Fockoperatoren (vgl. 2.27) und einen Storterm,

32 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
welcher die Korrelation der Elektronen beschreibt. Das Ergebnis einer solchen
Rechnung ist eine Wellenfunktion, die wie die CI-Wellenfunktion aus einem Re-
ferenzzustand und daraus abgeleiteten Slaterdeterminanten besteht.
Die storungstheoretische Berechnung der Beitrage bis zu einer gewissen Ord-
nung wird als MPN -Verfahren bezeichnet, wobei N die storungstheoretische Ord-
nung angibt. So bezieht MP2 Beitrage zweiter Ordnung Storungstheorie mit ein,
MP4 beinhaltet die Korrekturen bis zur vierten Ordnung. Der Rechenaufwand
von MP4 steigt mit der siebten Potenz der Systemgroße, ist also vergleichbar
mit dem einer CCSD(T)-Rechnung. Ein Vorteil der MP-Methoden ist ihre Size
Consistence. Die berechnete Energie stellt allerdings keine obere Schranke fur die
tatsachliche Grundzustandsenergie dar, da sie auf storungstheoretischem Wege
und nicht durch Variation berechnet wird.
Allen diesen Methoden ist gemeinsam, daß die entsprechenden Wellenfunktio-
nen auf einem einzigen Referenzzustand aufgebaut sind. Ist dieser Zustand keine
gute Naherung an die exakte N-Elektronen-Wellenfunktion, so macht sich diese
Tatsache im Ergebnis der entsprechenden Post-Hartree-Fock Verfahren bemerk-
bar. Einen Ausweg bietet die Multiconfiguration Self-Consistent Field (MCSCF)
Methode. Die zugehorige Wellenfunktion ist wie die CI-Wellenfunktion eine Sum-
me von Determinanten, aber zusatzlich zu den Entwicklungskoeffizienten sind die
zugrundeliegenden Orbitale ebenfalls variabel. Im Vergleich zu den sogenannten
Single-Reference Methoden ist die großere Flexibilitat mit einem erheblichen re-
chentechnischen Mehraufwand verbunden. So ist die Anwendung dieses Verfah-
rens auf die kleinsten Molekule beschrankt.
Mit dem Ziel, diese Methode dennoch auch bei großeren Systemen benut-
zen zu konnen, entstand das von Roos [25] entwickelte Complete Active-Space
Self-Consistent Field (CASSCF) Verfahren. Die Orbitale werden hierbei in be-
setzte, unbesetzte und aktive Orbitale unterteilt. Die CASSCF-Wellenfunktion
beinhaltet dann alle moglichen Zustande, welche durch Anregungen innerhalb
der aktiven Orbitale entstehen.
Die effizientesten Methoden zur Energieberechnung beruhen auf der Hinter-
einanderausfuhrung von Rechnungen auf unterschiedlichen theoretischen Ebenen
und daran anschließender Abschatzung der Korrelationsenergie, gegebenenfalls
unter Berucksichtigung eines empirischen Korrekturfaktors. Beispiele hierfur sind
die G1-Methode [26] sowie die daraus hervorgegangene G2-Methode [27]. In letz-

2.4. DICHTEFUNKTIONALTHEORIE 33
terem Fall werden HF-, MP2-, MP4- und QCISD(T)-Rechnungen unter Verwen-
dung verschiedener Basisatze (vgl Kap. 2.5.1) durchgefuhrt. Aus den daraus re-
sultierenden Energiedifferenzen wird die Korrelationsenergie mit Hilfe eines vom
Gesamtspin abhangigen Korrekturterms bestimmt.
Der Wert dieses Korrekturterms berechnet sich aus der Anpassung der
Bindungs- und Ionisationsenergien, Elektron- und Protonaffinitaten eines be-
stimmten Satzes von kleinen Molekulen, die hauptsachlich aus Elementen der
ersten Reihe des Periodensystems aufgebaut sind, an experimentell sehr genau
ermittelte Werte. Diese Verfahren, genauso wie die auf einer Extrapolation von
Basissatzen beruhende CBS-Methode [28], liefern Bindungsenergien, die im Rah-
men von etwa 50-100 meV mit dem Experiment ubereinstimmen.
2.4 Dichtefunktionaltheorie
Methoden, die uber den Hartree-Fock-Ansatz hinausgehen, haben haufig eine
deutliche Verbesserung der Ergebnisse zur Folge. Um dies zu erreichen, muß aber
in vielen Fallen eine sehr große Zahl von Determinanten berucksichtigt werden.
Da die erforderliche Rechenleistung der gangigen Post-Hartree-Fock Methoden
mit der funften bis achten Potenz der Systemgroße wachst, ist oftmals ein unver-
tretbarer numerischer Aufwand schon bei kleineren Systemen die Folge.
Eine Methode, die dieses Problem zu umgehen versucht, ist die Dichtefunktio-
naltheorie. Sie hat eine vollig andere Zugangsweise als die Hartree-Fock-basierten
Methoden, denn sie fuhrt die Berechnung der Grundzustandsenergie eines Sy-
stems aus N Elektonen auf eine Funktion zuruck, die nur von der Elektronen-
dichte ρ(r), abhangig ist.
2.4.1 Die Elektronendichte als Variable
In einer fundamentalen Arbeit [29] konnten Hohenberg und Kohn zeigen, daß
die Grundzustandselektronendichte ρ0(r) eines elektronischen Systems eindeutig
den zugehorigen Hamiltonoperator (2.5) und damit das außere Potential V sowie
alle weiteren physikalischen Eigenschaften des Systems bestimmt. Die Grundzu-
standsenergie
E0 = minΨ∈HN
{〈Ψ| H |Ψ 〉 | 〈Ψ |Ψ〉 = 1
}. (2.50)

34 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
ist somit auch ein Funktional von ρ0. Geht man zuerst alle moglichen Dichten
durch und dann zu jeder Dichte alle (normierten, antisymmetrischen, N-Teilchen-)
Wellenfunktionen, die zu dieser Dichte fuhren, so laßt sich nach [30] die Grund-
zustandsenergie schreiben als:
E0 = minρ
minΨ→ρ
{〈Ψ| H |Ψ 〉
}= min
ρminΨ→ρ
{〈Ψ| V + T + W |Ψ 〉
}= min
ρ
{∫drρ(r)vext(r) + min
Ψ→ρ
{〈Ψ| T + W |Ψ 〉
}}
= minρ
{∫drρ(r)vext(r) + F [ρ(r)]
}(2.51)
mit einem universellen, vom außeren Potential unabhangigen Funktional F [ρ].
Die Grundzustandselektronendichte ρ0 minimiert diesen Ausdruck - was eine Fol-
ge des Variationsprinzips (2.12) ist - und liefert die Grundzustandsenergie E0.
Dies trifft auch zu, wenn der Grundzustand entartet ist. In dem Fall erfullt jede
einzelne Dichte die Minimalitatsbedingung fur E0.
Die genaue Form des Funktionals F [ρ] ist nicht bekannt. Fur jede Approxi-
mation an das exakte F [ρ] gilt aber nicht das Variationsprinzip, so daß die mit
Dichtefunktionalmethoden gefundene Grundzustandsenergie eines elektronischen
Systems nicht mehr notwendigerweise eine obere Schranke fur E0 darstellt.
Kohn und Sham [31] schlugen vor, das Funktional F [ρ] in mehrere Anteile
aufzuspalten:
F [ρ] = TS[ρ] + U [ρ] + Exc[ρ]. (2.52)
Dabei entspricht TS der kinetischen Energie eines nichtwechselwirkenden Refe-
renzsystems mit derselben Grundzustandsdichte wie das betrachtete, wechselwir-
kende System. U [ρ] ist der sogenannte Hartreeterm, der die klassische Coulomb-
wechselwirkung der Ladungsdichte mit sich selbst beschreibt. Das Austausch-
korrelationsfunktional Exc[ρ] beinhaltet die Unterschiede zwischen der tatsachli-
chen kinetischen Energie und TS sowie zwischen der vollen Elektron-Elektron-
Wechselwirkung und U .
Da sich die Wellenfunktionen eines nichtwechselwirkenden Systems als Sla-
terdeterminante bzw. CSF (vgl.2.33) schreiben laßt, ergeben sich fur die dazu-

2.4. DICHTEFUNKTIONALTHEORIE 35
gehorigen Spinorbitale die folgenden Einteilchengleichungen:(−1
2∇2 + Veff(r)
)ϕi(x) = εiϕi(x) (2.53)
Die Bedingung, daß das nichtwechselwirkende System dieselbe Grundzustands-
elektronendichte wie das zu betrachtende System mit Wechselwirkung haben soll,
legt das effektive Potential Veff(r) fest:
Veff(r) = vext(r) + u(r) + vxc(r) (2.54)
Das Austauschkorrelationspotential ist dabei durch die Variationsableitung von
Exc[ρ] nach der Elektronendichte gegeben:
vxc(r) =δ Exc[ρ]
δ ρ(2.55)
und folglich rein lokaler Natur, im Gegensatz zum Fockoperator, welcher den
nichtlokalen Austauschterm enthalt. Die Gleichungen (2.53) in Verbindung mit
(2.54) und (2.55) werden als Kohn-Sham Gleichungen bezeichnet und mussen,
wie die HF-Gleichungen, selbstkonsistent gelost werden.
Den Kohn-Sham Orbitalen ϕi(x) kommt keine direkte physikalische Bedeu-
tung zu, bis auf die Tatsache, daß die Grundzustanselektronendichte
ρ0(r) =
N∑i=1
|ϕi(x)|2 (2.56)
als Summe uber die N Eigenfunktionen mit den niedrigsten Energieeigenwerten
von ihnen bestimmt wird. Es gibt somit kein Analogon zum Koopmans’schen
Theorem (vgl. 2.2) in der Dichtefunktionaltheorie, der großte Energieeigenwert
stellt jedoch im Fall eines isolierten Systems mit im Unendlichen verschwinden-
dem Potential das Negative der Ionisationsenergie dar.
Die verschiedenen Dichtefunktionalmethoden unterscheiden sich in der Form
des Austauschkorrelationsfunktionals Exc[ρ]. Da der Hamiltonoperator in nicht-
relativistischer Naherung bei Abwesenheit eines außeren Magnetfeldes vom Spin
der Elektronen unabhangig ist, reicht die Information uber die Elektronendichte
theoretisch zur Bestimmung der Grundzustandsenergie aus.
Da das exakte Funktional jedoch nicht bekannt ist und Naherungen an Exc[ρ]
oft nicht die notige Flexibilitat aufweisen, sind die in der Praxis benutzten Funk-
tionale oft explizit vom Spin der Elektronen, d.h. von der Spindichte
ζ =ρ↑ − ρ↓ρ↑ + ρ↓
(2.57)

36 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
abhangig und liefern so die zusatzliche Information uber die Magnetisierung. Die-
ses als spin-polarisierte Methode bezeichnete Vorgehen bringt jedoch eine Kom-
plikation mit sich. Zwei Zustande eines Systems, die die gleiche Dichte haben und
sich nur in der Spindichte unterscheiden, haben bei Verwendung solcher Funk-
tionale haufig eine unterschiedliche Energie. Außerdem weist das System bei die-
ser Behandlung meistens nicht den korrekten Gesamtpin auf. Das Problem der
Spin-Contamination ist dadurch begrundet, daß die Kohn-Sham Orbitale fur die
beiden Spinrichtungen, wie die HF-Orbitale im Rahmen der UHF-Methode, nicht
in ihren Ortsanteilen ubereinstimmen mussen. Eine detailierte Behandlung dieser
Aspekte findet sich in [32].
Ein Ausweg ist eine analog der ROHF-Methode auf mehreren Determinanten
aufgebaute Wellenfunktion, die so konstruiert ist, daß sie den korrekten Erwar-
tungswert des S 2-Operators liefert. Dies wurde von Filatov [33] vorgeschlagen.
Allerdings bußen die so gewonnenen Orbitale einen Teil ihrer Flexibilitat ein, da
alle doppelt besetzten Orbitale in ihrem Ortsanteil ubereinstimmen.
Sind alle Orbitale doppelt besetzt, so spricht man von der sogenannten nicht-
spin-polarisierten Methode, welche dem RHF-Verfahren ahnlich ist. Prinzipi-
ell ware diese Methode auch bei Systemen mit ungepaarten Elektronen, wel-
che naturlich einen nichtverschwindenden Gesamtspin aufweisen, in der La-
ge, die exakte Grundzustandsenergie zu ermitteln, da der Hamiltonoperator
spinunabhangig ist. Aufgrund des Naherungsausdruckes fur das Austausch-
Korrelationsfunktional sind die mit diesem Verfahren erzielten Ergebnisse aber
oftmals nicht von guter Qualitat.
2.4.2 Funktionale fur Austausch und Korrelation
Ublicherweise teilt man das Funktional fur die Austausch-Korrelationsenergie auf
in einen Austausch- und einen Korrelationsanteil,
Exc[ρ] = Ex[ρ] + Ec[ρ], (2.58)
wobei die beiden Summanden haufig von der Spindichte ζ abhangig sind. Uber
die tatsachliche Gestalt dieser Ausdrucke ist nichts bekannt, es lassen sich aber
eine Reihe von Randbedingungen herleiten, denen das exakte Funktional genugt.
Unter den vorgeschlagenen Naherungen fur Ex und Ec gibt es keine, die alle Rand-
bedingungen erfullt. Die analytischen Ausdrucke der einzelnen Formulierungen

2.4. DICHTEFUNKTIONALTHEORIE 37
unterscheiden sich betrachtlich, je nachdem aus welcher Uberlegung heraus sie
entstanden sind und welche der Randbedingungen sie erfullen.
Sie lassen sich in zwei Gruppen unterteilen. Funktionale, welche nur von
den Werten der Elektronendichte selbst abhangig sind, werden der lokalen Dich-
tenaherung (LDA) zugeordnet. Verallgemeinerte Gradientenmethoden (GGA’s)
beziehen außerdem noch den Betrag des Gradienten von ρ(r) mit ein.
Die Austauschkorrelationsenergie wird haufig in der folgenden Form angege-
ben:
Exc[ρ] =
∫dr ρ(r)
(εx[ρ(r)] + εc[ρ(r])
)(2.59)
Die explizite Form der verschiedenen Funktionale bezieht sich auf die Großen
εx[ρ] bzw. εc[ρ].
Die lokale Dichtenaherung
Der am haufigsten verwendete Term fur die Austauschenergie in der lokalen Dich-
tenaherung geht auf die exakte Austauschenergie des homogenen Elektronenga-
ses zuruck und wird oftmals mit dem Namen Slater in Verbindung gebracht. Der
analytische Ausdruck fur ein System ohne Spinpolarisation ist
εLDAx = −3
4
(3 ρ
π
)1/3
(2.60)
Die Berucksichtigung des Spins der Elektronen fuhrt auf die Austauschenergie in
der lokalen Spin-Dichtenaherung (LSDA)
εLSDAx = −3
2ρ1/3
((1 + ζ)4/3 + (1 − ζ)4/3
)(2.61)
mit der in (2.57) definierten Spindichte.
Die Korrelationsenergie eines Elektrons im homogenen Elektronengas ist als
tabellierte Funktion der Dichte bekannt [34]. Es existieren analytische Ausdrucke
fur εLDAc , die an diese Daten angepaßt wurden. In dieser Arbeit wurden die Funk-
tionale von Vosko et al. [35] sowie Ceperley und Alder [34] verwendet, welche aber
aufgrund ihrer langlichen und umfangreichen Form hier nicht explizit aufgefuhrt
sind.
Die lokale (Spin-)Dichtenaherung ist frei von empirischen Parametern. Sie ist
exakt fur Systeme mit konstanter Elektronendichte und liefert auch fur Systeme

38 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
mit langsam variierender Dichte brauchbare Ergebnisse. Ihr Erfolg selbst in Bezug
auf Systeme mit stark fluktuierender Dichte ist darauf zuruckgefuhrt worden,
daß die L(S)DA sehr viele der Randbedingungen erfullt, denen auch das exakte
Funktional genugen muß.
Allerdings lassen sich die meisten Molekule im Rahmen der L(S)DA nur
schlecht beschreiben. Bindungsenergien fallen haufig zu groß aus, was auf eine
Uberschatzung der Korrelationsenergie zuruckzufuhren ist.
Verallgemeinerte Gradientenmethoden
Eine einfache Taylorentwicklung der Elektronendichte bringt gegenuber der LS-
DA keine Verbesserung der Ergebnisse. Dies ist erst durch eine sogenannte ver-
allgemeinerte Gradientenmethode (GGA) moglich, im Rahmen derer Gradienten
der Dichte in die Berechnung der Austausch-Korrelationsenergie miteinbezogen
werden.
Die verschiedenen gebrauchlichen analytischen Ausdrucke fur εGGAx und εGGA
c
beziehen sich zumeist auf die dimensionslose Variable
x =|∇ρ|ρ4/3
(2.62)
Als Beispiel sei das von Becke [36] vorgeschlagene Funktional fur die Austausch-
energie genannt:
εB88x = εLDA
x − βρ1/3 x2
1 + 6βx sinh−1(x). (2.63)
Weitere Funktionale wurden zum Beispiel von Perdew und Wang [37, 38] ent-
wickelt.
Ausdrucke fur die Korrelationsenergie haben oftmals eine recht komplizierte
analytische Form und sind schwieriger zu ermitteln [39]. Sie unterscheiden sich
in den Randbedingungen, die sie erfullen oder nicht erfullen. Das von Lee et
al. [40] vorgeschlagene Funktional ist selbstwechselwirkungsfrei, liefert also keine
Korrelationsenergie bei einem System mit nur einem Elektron. Allerdings wird
im Rahmen dieser Naherung der Grenzfall des homogenen Elektronengases nicht
korrekt beschrieben. Die Verhaltnisse sind genau umgekehrt im Falle des von
Perdew und Wang entwickelten Funktionals [38].
Damit wird deutlich, daß es bei der Wahl eines bestimmten Funktionals fur
die Austausch- bzw. die Korrelationsenergie auf das zu untersuchende System

2.5. DIE BEDEUTUNG DER RANDBEDINGUNGEN 39
ankommt. Ein bestimmtes Funktional mag bei der Untersuchung eines speziellen
Systems ausgesprochen gute Ergebnisse liefern, im Falle eines anderen Systems
aber nicht zu verwenden sein.
Die Rolle des exakten Austauschterms
Eine Kombination aus lokaler Dichtenaherung, Gradientenkorrekturen und einem
Anteil der exakten HF-Austauschenergie wurde von Becke [41] vorgeschlagen. Die
Austausch-Korrelationsenergie ist in dieser Formulierung durch den Ausdruck
EHybridxc = ELSDA
xc + a(EHFx −ELSDA
x ) + b∆EGGAx + c∆EGGA
c (2.64)
gegeben. Fur die Gradientenkorrekturen schlug Becke ursprunglich die in [36]
bzw. [38] gegebenen und haufig mit der Kurzform B88 bzw. PW91 bezeichneten
Funktionale vor. Die Parameter a, b und c wurden von Becke durch Anpassung
an experimentelle Daten bestimmt.
In dieser Arbeit wurde die sogenannte B3LYP-Methode verwendet. Dabei
stellt sich die Korrelationsenergie durch die Interpolationsformel von Vosko et al.
[35] zuzuglich der Gradientenkorrektur mit Hilfe des in [40] angegebenen Aus-
druck dar. Die anderen Terme stimmen mit der ursprunglichen Fassung uberein.
Die Motivation hinter der Hybrid-Formulierung liegt in der sogenannten adia-
batischen Verbindungsformel, die ein adiabatisches Anschalten der Elektron-
Elektron-Wechselwirkung beschreibt. Im Grenzfall verschwindender Wechselwir-
kung stellt die HF-Austauschenergie den exakten Wert dar. Eine nahere Beschrei-
bung der Verhaltnisse findet sich z.B. in [42].
2.5 Die Bedeutung der Randbedingungen
Die Anwendungsmoglichkeiten von ab-initio Methoden zur Berechnung der Elek-
tonenstruktur erstrecken sich von Systemen, die nur aus wenigen Atomen be-
stehen, uber großere Molekule, dunne Schichten und Oberflachen bis hin zum
Festkorper. Ausgehend vom zu behandelnden System fuhrt man gewisse Randbe-
dingungen ein, um die Rechnungen zu erleichtern oder uberhaupt erst zu ermogli-
chen.
Da der ideale Festkorper in allen drei Raumrichtungen unendlich ausgedehnt
ist, besteht er aus einer unendlichen Zahl von einzelnen Atomen und somit auch

40 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Elektronen. Alle diese Teilchen in einer Rechnung zu berucksichtigen ist naturlich
nicht moglich. Nur einen kleinen Ausschnitt des Systems fur sich alleine zu be-
trachten, ware eine Moglichkeit, dieses Problem zu umgehen, brachte aber un-
erwunschte Grenzflacheneffekte mit ins Spiel. Aus diesem Grund fuhrt man haufig
sogenannte periodische Randbedingungen ein. Dabei wird ein endliches System
von Teilchen, das sich in einer von drei Vektoren aufgespannten2 sogenannten
Superzelle befindet, in alle Raumrichtungen periodisch fortgesetzt.
Ist ein isoliertes Atom oder Molekul Gegenstand der Untersuchung, so be-
steht der naturliche Ansatz in der Wahl von freien Randbedingungen. Es ist
aber auch moglich, periodische Randbedingungen zu verwenden, sofern gewisse
Kriterien bezuglich der Große der Superzelle erfullt sind. Periodische Randbe-
dingungen bieten sich zum Beispiel an, wenn das zu untersuchende Molekul von
einer Flussigkeit umgeben ist. Auch Kombinationen von freien und periodischen
Randbedingungen kommen manchmal zum Einsatz, beispielsweise bei der Unter-
suchung von Oberflachen oder dunnen Schichten.
Die Randbedingungen haben großen Einfluß auf den Verlauf einer Rechnung.
Sie sind außerdem ein entscheidender Aspekt bei der Wahl der Basisfunktionen,
in denen die Wellenfunktionen bzw. die Elektronendichte entwickelt werden.
2.5.1 Freie Randbedingungen: Die Wahl der Basis
Bei der Wahl der Einelektronenbasis fur die Entwicklung der Molekulorbitale
bzw. der Kohn-Sham-Wellenfunktionen sind einige Kriterien zu beachten. Zum
einen sollte der Basissatz Teilmenge eines im Raum der quadratintegrablen Funk-
tionen vollstandigen Funktionensystems sein, damit jede Wellenfunktion zumin-
dest im Prinzip durch systematische Erweiterung dieser Basis beliebig genau ap-
proximiert werden kann. Zum anderen sollte die Konvergenz in den gesuchten
elektronischen Zustand genugend schnell von statten gehen, denn fur die mei-
sten ab-initio-Methoden ist die maximale Systemgroße begrenzt durch die starke
Abhangigkeit der benotigten Rechenleistung von der Zahl der Basisfunktionen.
Auf der Suche nach einem Basissatz, der diesen beiden Kriterien Rechnung tragt,
ist ein moglicher Ansatz, sich zunachst auf einzelne Atome zu beschranken, da
2Die Superzelle muß nicht zwangslaufig solch ein Parallelepiped sein. Es gibt allerdingsnur funf verschiedene Typen von moglichen Polyedern, die bei wiederholtem Ausfuhren vonTranslationsoperationen den ganzen Raum ausfullen [43].

2.5. DIE BEDEUTUNG DER RANDBEDINGUNGEN 41
man sich in diesem Fall von physikalischen Einsichten in die elektronische Struk-
tur leiten lassen kann. Das sollte dabei helfen, eine moglichst geeignete Basis
fur die Atomorbitale zu finden, die Molekulorbitale werden dann in einem zwei-
ten Schritt durch Linearkombination solcher Atomorbitale (LCAO-Methode, [18])
konstruiert.
Bewegt sich ein einzelnes Elektron in einem kugelsymmetrischen Potential, so
konnen die Eigenfunktionen des dazugehorigen Hamiltonoperators als Produkt
einer Radialfunktion Rnl(r) und einer winkelabhangigen Kugelflachenfunktion
Ylm(θ, ϕ) mit den Haupt-, Drehimpuls- und Magnetquantenzahlen n, l undm dar-
gestellt werden. Die spezielle Form des Potentials bestimmt dabei einzig und allein
die Radialfunktionen. In Anbetracht der Tatsache, daß ein Elektron im Atom bei
Vernachlassigung der ubrigen Elektronen dem kugelsymmetrischen Kernpotential
ausgesetzt ist, scheint ein sinnvoller Ansatz fur die Wahl der Einelektronenbasis
der obige Produktansatz zu sein. Somit reduziert sich das Problem auf die Suche
nach einem geeigneten Satz von Radialfunktionen.
Im Falle des Coulombpotentials ergeben sich als exakte Losung der entspre-
chenden Schrodingergleichung fur Rnl(r) wasserstoffahnliche Funktionen, die sich
durch die Anwesenheit eines Exponentialterms exp(−Zr/n), eines assoziierten
Laguerrepolynoms in r vom Grade n − l − 1 sowie einer Potenz rl auszeichnen.
Ersetzt man Z/n durch ζ , so ergeben sich die sogenannten Laguerre-Funktionen:
RLFnl (r) = N rlL2l+2
n−l−1(2ζr) e−ζr (2.65)
mit dem Normierungsfaktor N . Fur festen Exponenten ζ und feste Drehimpuls-
quantenzahl l bilden diese Funktionen ein vollstandiges, othonormiertes Funktio-
nensystem. Doch es stellt sich heraus, daß fur die Beschreibung eines Atoms, bei
dem die Annahme eines Zentralpotentials nur eine Naherung ist, sehr viele Funk-
tionen der Form (2.65) linearkombiniert werden mussen, um eine hinreichende
Genauigkeit zu erzielen.
Eine bessere Konvergenz ließe sich erreichen, wenn man auch die Exponen-
ten ζ variieren wurde. Allerdings sind die Funktionen (2.65) dann nicht mehr
orthogonal, so daß die durch die Polynome L2l+2n−l−1 hervorgerufene komplizierte
Knotenstruktur der Laguerre-Funktionen, die ursprunglich allein der Wahrung
der Orthogonalitat diente, dann nicht mehr gerechtfertigt scheint.
Slater [17] hat als erster vorgeschlagen, nur den fuhrenden Term in den
Laguerre-Polynomen zu berucksichtigen, was auf Basisfunktionen der folgenden

42 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Form fuhrt:
RSTOn (r) = (2ζ)n+0.5(2n!)−0.5rn−1e−ζr. (2.66)
Diese Funktionen bilden ein vollstandiges System, sie haben aber keine Knoten
und sind deshalb nicht mehr orthogonal. Desweiteren sind sie von der Drehimpuls-
quantenzahll l unabhangig, so daß sich die Radialanteile s- und p-artiger Funk-
tionen nur durch ihren Exponenten unterscheiden. Die komplette Basisfunktion
der Gestalt
χSTOnlm (r) = RSTO
n (r)Ylm(θ, ϕ). (2.67)
wird als Slater-Type Orbital (STO) bezeichnet. Bei der Entwicklung einer Funk-
tion nach diesen Basisfunktionen
φ(r) =∑nlm
cnlmχSTOnlm (2.68)
sind die Parameter die Exponenten ζ und die Koeffizienten cnlm. Wie gut diese
Funktionen geeignet sind, um reale Atomorbitale zu beschreiben, sei am Beispiel
des Kohlenstoffatoms verdeutlicht. In der Abbildung 2.4 sind die aus einer nume-
rischen HF-Rechnung stammenden Radialverteilungsfunktionen des 2s-Orbitals
und des 2p-Orbitals eines Kohlenstoffatoms im 3P Grundzustand aufgetragen.
Zum Vergleich finden sich in dieser Abbildung die besten Naherungen (nach der
Methode der kleinsten Fehlerquadrate) an diese Orbitale in einer minimalen STO-
Basis, d.h. einer Basis, in der die Zahl der Basisfunktionen gerade so groß ist, wie
die der besetzten Orbitale.
Die Ubereinstimung mit den exakten Orbitalen ist erstaunlich gut, wenn man
bedenkt, daß das 2p-Orbital einer einzigen STO und das 2s-Orbital einer Line-
arkombination aus nur zwei STO’s entspricht. Durch Hinzufugen von wenigen
Basisfunktionen laßt sich dieses Ergebnis noch wesentlich verbessern, allerdings
sind die so gewonnenen Atomorbitale dann ausschließlich zur Beschreibung des
isolierten Kohlenstoffatoms im Grundzustand geeignet.
So gut die STO’s als Basisfunktionen fur die Atomorbitale geeignet zu sein
scheinen, so sind sie doch von Nachteil, was ihre Verwendung in Rechnungen
fur Molekulen oder Festkorper betrifft. Es existiert namlich kein effizienter Algo-
rithmus zur Auswertung von Integralen, deren Integrand aus dem Produkt von
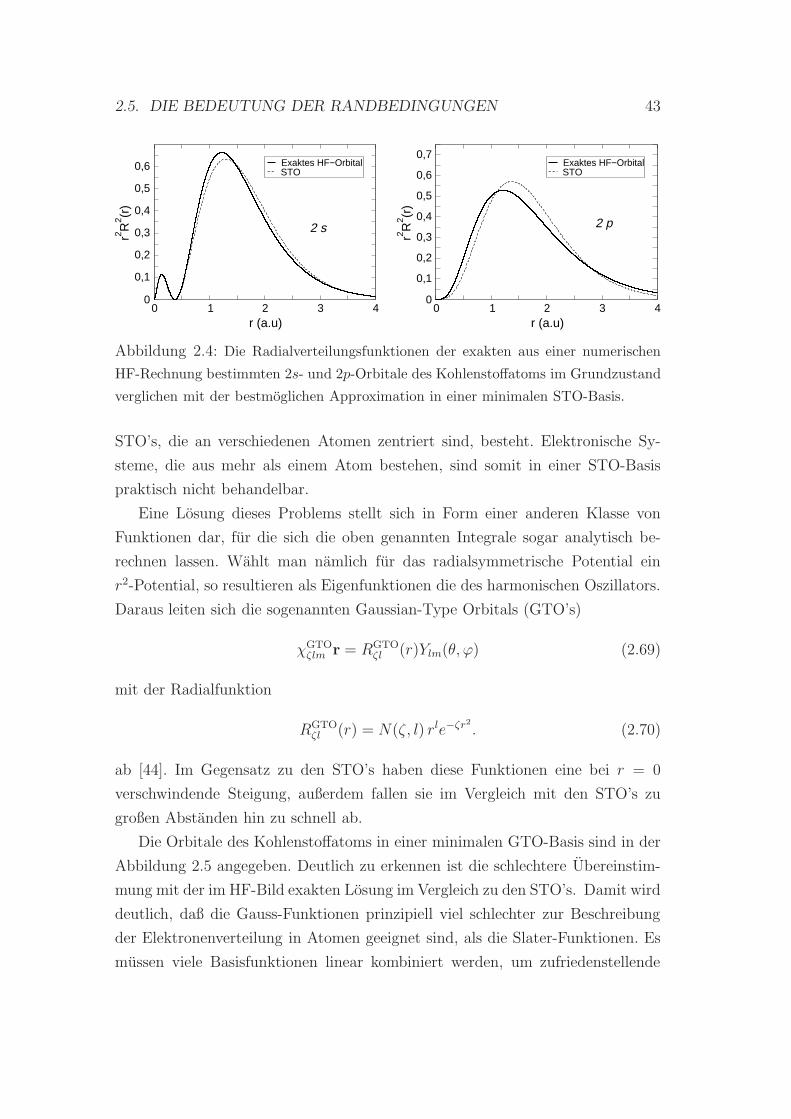
2.5. DIE BEDEUTUNG DER RANDBEDINGUNGEN 43
0 1 2 3 4r (a.u)
0
0,1
0,2
0,3
0,4
0,5
0,6
r2 R2 (r
)
Exaktes HF−OrbitalSTO
2 s
0 1 2 3 4r (a.u)
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
r2 R2 (r
)
Exaktes HF−OrbitalSTO
2 p
Abbildung 2.4: Die Radialverteilungsfunktionen der exakten aus einer numerischen
HF-Rechnung bestimmten 2s- und 2p-Orbitale des Kohlenstoffatoms im Grundzustand
verglichen mit der bestmoglichen Approximation in einer minimalen STO-Basis.
STO’s, die an verschiedenen Atomen zentriert sind, besteht. Elektronische Sy-
steme, die aus mehr als einem Atom bestehen, sind somit in einer STO-Basis
praktisch nicht behandelbar.
Eine Losung dieses Problems stellt sich in Form einer anderen Klasse von
Funktionen dar, fur die sich die oben genannten Integrale sogar analytisch be-
rechnen lassen. Wahlt man namlich fur das radialsymmetrische Potential ein
r2-Potential, so resultieren als Eigenfunktionen die des harmonischen Oszillators.
Daraus leiten sich die sogenannten Gaussian-Type Orbitals (GTO’s)
χGTOζlm r = RGTO
ζl (r)Ylm(θ, ϕ) (2.69)
mit der Radialfunktion
RGTOζl (r) = N(ζ, l) rle−ζr
2
. (2.70)
ab [44]. Im Gegensatz zu den STO’s haben diese Funktionen eine bei r = 0
verschwindende Steigung, außerdem fallen sie im Vergleich mit den STO’s zu
großen Abstanden hin zu schnell ab.
Die Orbitale des Kohlenstoffatoms in einer minimalen GTO-Basis sind in der
Abbildung 2.5 angegeben. Deutlich zu erkennen ist die schlechtere Ubereinstim-
mung mit der im HF-Bild exakten Losung im Vergleich zu den STO’s. Damit wird
deutlich, daß die Gauss-Funktionen prinzipiell viel schlechter zur Beschreibung
der Elektronenverteilung in Atomen geeignet sind, als die Slater-Funktionen. Es
mussen viele Basisfunktionen linear kombiniert werden, um zufriedenstellende
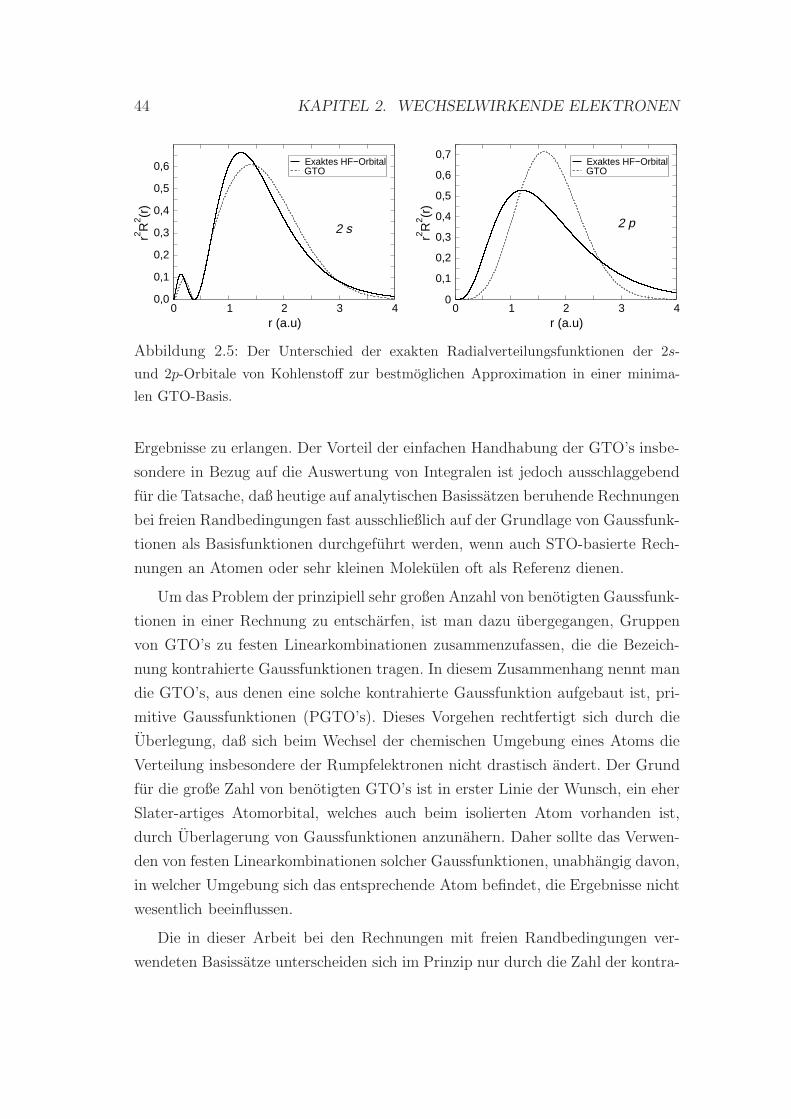
44 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
0 1 2 3 4r (a.u)
0,0
0,1
0,2
0,3
0,4
0,5
0,6
r2 R2 (r
)Exaktes HF−OrbitalGTO
2 s
0 1 2 3 4r (a.u)
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
r2 R2 (r
)
Exaktes HF−OrbitalGTO
2 p
Abbildung 2.5: Der Unterschied der exakten Radialverteilungsfunktionen der 2s-
und 2p-Orbitale von Kohlenstoff zur bestmoglichen Approximation in einer minima-
len GTO-Basis.
Ergebnisse zu erlangen. Der Vorteil der einfachen Handhabung der GTO’s insbe-
sondere in Bezug auf die Auswertung von Integralen ist jedoch ausschlaggebend
fur die Tatsache, daß heutige auf analytischen Basissatzen beruhende Rechnungen
bei freien Randbedingungen fast ausschließlich auf der Grundlage von Gaussfunk-
tionen als Basisfunktionen durchgefuhrt werden, wenn auch STO-basierte Rech-
nungen an Atomen oder sehr kleinen Molekulen oft als Referenz dienen.
Um das Problem der prinzipiell sehr großen Anzahl von benotigten Gaussfunk-
tionen in einer Rechnung zu entscharfen, ist man dazu ubergegangen, Gruppen
von GTO’s zu festen Linearkombinationen zusammenzufassen, die die Bezeich-
nung kontrahierte Gaussfunktionen tragen. In diesem Zusammenhang nennt man
die GTO’s, aus denen eine solche kontrahierte Gaussfunktion aufgebaut ist, pri-
mitive Gaussfunktionen (PGTO’s). Dieses Vorgehen rechtfertigt sich durch die
Uberlegung, daß sich beim Wechsel der chemischen Umgebung eines Atoms die
Verteilung insbesondere der Rumpfelektronen nicht drastisch andert. Der Grund
fur die große Zahl von benotigten GTO’s ist in erster Linie der Wunsch, ein eher
Slater-artiges Atomorbital, welches auch beim isolierten Atom vorhanden ist,
durch Uberlagerung von Gaussfunktionen anzunahern. Daher sollte das Verwen-
den von festen Linearkombinationen solcher Gaussfunktionen, unabhangig davon,
in welcher Umgebung sich das entsprechende Atom befindet, die Ergebnisse nicht
wesentlich beeinflussen.
Die in dieser Arbeit bei den Rechnungen mit freien Randbedingungen ver-
wendeten Basissatze unterscheiden sich im Prinzip nur durch die Zahl der kontra-

2.5. DIE BEDEUTUNG DER RANDBEDINGUNGEN 45
hierten Gaussfunktionen, aus denen sie aufgebaut sind. Naheres uber die genaue
Zusammensetzung der einzelnen Basissatze ist im Anhang B zu erfahren.
2.5.2 Periodische Randbedingungen: Ebene Wellen und
Pseudopotentiale
Ein anderes Bild ergibt sich bei der Verwendung von periodischen Randbedin-
gungen (Abbildung 2.6). In diesem Fall lassen sich die Wellenfunktionen der
Abbildung 2.6: Schematische Darstellung periodischer Randbedingunngen in zwei Di-
mensionen.
Elektronen darstellen als Produkt einer Funktion uk(r), welche die vorgegebene
Periodizitat aufweist und einer ebenen Welle :
ψk(r) = uk(r) eik·r. (2.71)

46 KAPITEL 2. WECHSELWIRKENDE ELEKTRONEN
Aufgrund der Periodizitat der Funktion uk(r) bietet sich eine Entwicklung nach
ebenen Wellen
χPWG (r) = ei (k+G)·r (2.72)
an, so daß man schließlich fur die Wellenfunktion des Elektrons den Ausdruck
ψk(r) =∑G
ck+G ei (k+G)·r (2.73)
erhalt. Die Große des Basissatzes wird durch die Angabe der Cutoff-Energie
Ecut =h2
2me|k + G|2 (2.74)
festgelegt, indem nur ebene Wellen mit einer Energie, die kleiner als Ecut ist, in
die Entwicklung (2.73) miteinbezogen werden.
Ein Vorteil der Verwendung von ebenen Wellen als Basisfunktionen liegt in
der einfachen Form, die die zu losenden Gleichungen fur die Koeffizienten ck+G
in diesem Fall annehmen. Außerdem laßt sich der Fehler, der aufgrund der endli-
chen Große des Basissatzes entsteht, kontinuierlich durch Vergroßerung der Cut-
toff Energie verringern. Allerdings sind fur eine ausreichende Beschreibung der
Valenzelektronen aufgrund der starken Oszillationen ihrer Wellenfunktionen in
Kernnahe sehr viele ebene Wellen notwendig, was eine Rechnung fur großere Sy-
steme erschwert.
Dieses Problem konnte durch die Entwicklung von Pseudopotentialmethoden
gelost werden. Dabei ersetzt man das Kernpotential sowie den Einfluß der Rumpf-
elektronen durch ein Pseudopotential, welches innerhalb einer Kugel mit dem Ra-
dius r um den betreffenden Atomkern hinreichend flach verlauft, außerhalb dieses
Gebietes aber mit dem exakten Potential identisch ist. Anstelle der Wellenfunk-
tionen der Valenzelektronen lassen sich Pseudowellenfunktionen konstruieren, die
außerhalb von r mit den tatsachlichen Wellenfunktionen ubereinstimmen, inner-
halb jedoch wesentlich flacher verlaufen.
Die Zahl der fur eine hinreichend gute Beschreibung des elektronischen Sy-
stems notwendigen ebenen Wellen wird somit auf zweierlei Weise reduziert. Zum
einen werden nur die Valenzelektronen in die Rechnung mit einbezogen. Dieses
Vorgehen ist gerechtfertigt, da das chemische Verhalten eines Atoms in unter-
schiedichen Umgebungen zum großten Teil durch die Valenzelektronen bestimmt

2.5. DIE BEDEUTUNG DER RANDBEDINGUNGEN 47
wird. Zum anderen ist aufgrund des flacheren Verlaufs der Pseudowellenfunktio-
nenen eine wesentlich geringere Zahl an ebenen Wellen fur deren Beschreibung
ausreichend.

Kapitel 3
Energieflachen
Der Born-Oppenheimer-Naherung (vgl. Kap. 2.1) liegt die Annahme zugrunde,
daß die Elektronen den Bewegungen der Atomkerne aufgrund ihrer viel geringeren
Masse nahezu instantan folgen. Daher ist die potentielle Energie eines atomaren
Systems bei vorgegebener Symmetrie allein eine Funktion der Kernkoordinaten
und stellt somit eine Hyperflache im 3N -dimensionalen Raum dar, wobei N die
Zahl der Atome im System bezeichnet.
Diese Potentialflache (Potential Energy Surface, PES) ist stetig. Das gilt
jedochnicht fur die Energieflache des Grundzustands, denn sie beschreibt die
kleinstmogliche Energie des Gesamtsystems in Abhangigkeit der Kernkoordina-
ten. Diese mag fur ein atomares System mit einer bestimmten Struktur z.B. einem
Triplett-Zustand des Elektronensystems entsprechen, fur eine andere Struktur
dem Singulett-Zustand. An den Punkten, wo sich die entsprechenden Potential-
flachen schneiden, weist die Flache der Grundzustandsenergie haufig Unstetig-
keitsstellen auf.
Nutzt man die Translations- und Rotationsinvarianz des Raumes aus, so laßt
sich die Zahl der Freiheitsgrade eines atomaren Systems auf 3N − 6 reduzieren.
Die Positionen der Atome werden dann durch sogenannte innere Koordinaten be-
schrieben. Dabei ist der Koordinatenursprung fest mit einem bestimmten Atom
verbunden, die Orientierung des Koordinatensystems ist durch zwei weitere Ato-
me vorgegeben. Die Positionen aller anderen Atome sind durch die Angabe eines
Abstands und zweier Winkel, von denen der eine ein Diederwinkel ist, definiert.
Durch Zwangsbedingungen ist eine weitere Reduktion der Zahl der Freiheitsgrade
moglich. Dabei spielt das Konzept der Symmetrie eine große Rolle, mit dessen
48
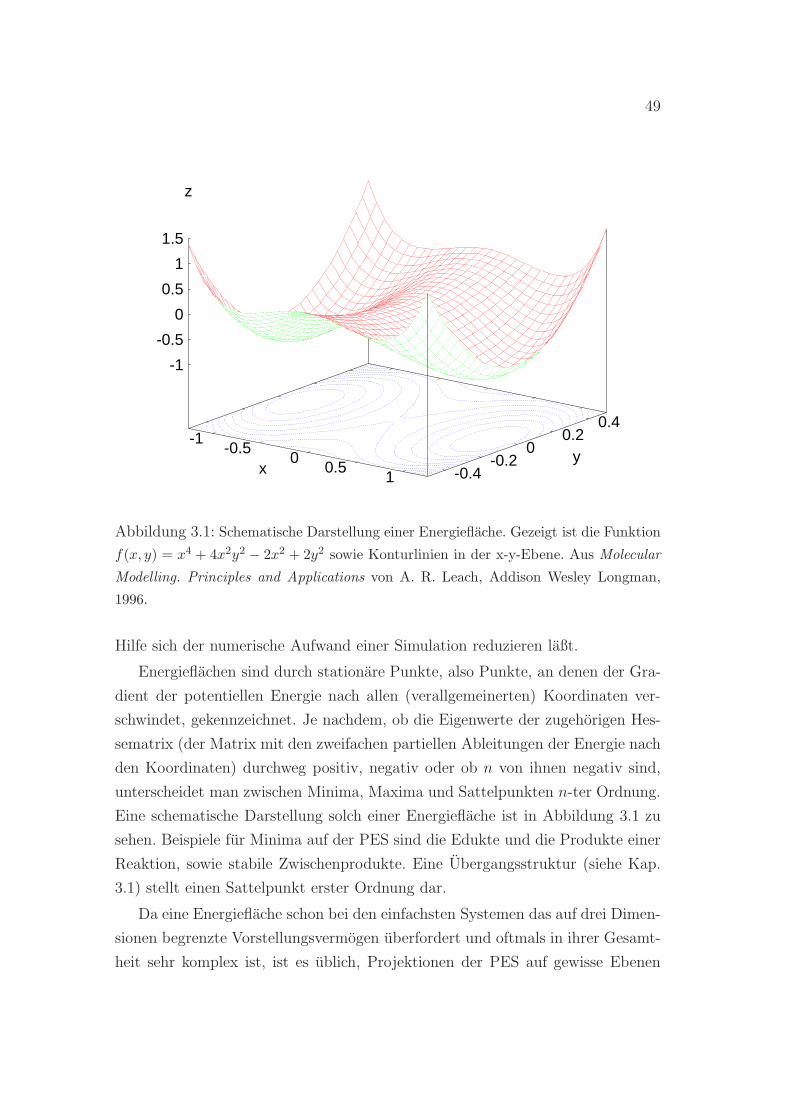
49
-1-0.5
00.5
1x -0.4-0.2
00.2
0.4
y
-1
-0.5
0
0.5
1
1.5
z
Abbildung 3.1: Schematische Darstellung einer Energieflache. Gezeigt ist die Funktion
f(x, y) = x4 + 4x2y2 − 2x2 + 2y2 sowie Konturlinien in der x-y-Ebene. Aus Molecular
Modelling. Principles and Applications von A. R. Leach, Addison Wesley Longman,
1996.
Hilfe sich der numerische Aufwand einer Simulation reduzieren laßt.
Energieflachen sind durch stationare Punkte, also Punkte, an denen der Gra-
dient der potentiellen Energie nach allen (verallgemeinerten) Koordinaten ver-
schwindet, gekennzeichnet. Je nachdem, ob die Eigenwerte der zugehorigen Hes-
sematrix (der Matrix mit den zweifachen partiellen Ableitungen der Energie nach
den Koordinaten) durchweg positiv, negativ oder ob n von ihnen negativ sind,
unterscheidet man zwischen Minima, Maxima und Sattelpunkten n-ter Ordnung.
Eine schematische Darstellung solch einer Energieflache ist in Abbildung 3.1 zu
sehen. Beispiele fur Minima auf der PES sind die Edukte und die Produkte einer
Reaktion, sowie stabile Zwischenprodukte. Eine Ubergangsstruktur (siehe Kap.
3.1) stellt einen Sattelpunkt erster Ordnung dar.
Da eine Energieflache schon bei den einfachsten Systemen das auf drei Dimen-
sionen begrenzte Vorstellungsvermogen uberfordert und oftmals in ihrer Gesamt-
heit sehr komplex ist, ist es ublich, Projektionen der PES auf gewisse Ebenen

50 KAPITEL 3. ENERGIEFLACHEN
zu betrachten. Haufig konnen aber einfache chemische Reaktionen durch einige
wenige starker variable Koordinaten charakterisiert werden, wahrend alle ande-
ren nahezu konstant bleiben. Mit dem Begriff Potentialflache einer Reaktion ist
dann eine Projektion der vollen Potentialflache auf eine von diesen Parametern
aufgespannten Ebene gemeint, und zwar derjenigen, die durch die Konstanz der
anderen Koordinaten gegeben ist.
Jeder (differenzierbare) Weg auf einer Potentialflache laßt sich parametrisie-
ren, beispielsweise nach der Bogenlange. Mit dem Begriff Reaktionskoordinate
ist ein Parameter gemeint, der den Weg zwischen zwei Minima auf der PES be-
schreibt. Eine Reaktion laßt sich somit in eindeutiger Weise durch die Energie
in Abhangigkeit der Reaktionskoordinate beschreiben und als zweidimensionaler
Graph darstellen. Dabei sind alle Koordinaten Funktionen dieser Reaktionskoor-
dinate; in der Praxis ist der genaue Zusammenhang meist nicht von Interesse, so
daß der Begriff oftmals eine abstrakte Bedeutung hat.
3.1 Strukturoptimierung
Minima auf der Potentialflache entsprechen stabilen Strukturen des atomaren
Systems. Wird die PES durch eine Funktion E(q1, . . . , qf ) beschrieben, wobei f
der Zahl der Freiheitsgrade des Systems angibt, so sind Minima {q01, . . . , q
0f} auf
dieser Flache durch die Bedingung
∂E
∂ qi
∣∣∣∣∣qi=q0i
= 0 ,∂ 2E
∂ q2i
∣∣∣∣∣qi=q0i
> 0 (3.1)
gekennzeichnet. Die Suche nach solchen Punkten wird als Strukturoptimierung
bezeichnet.
Da im allgemeinen kein analytischer Ausdruck fur die Form der PES zur
Verfugung steht, erfolgt diese Suche zumeist auf numerischem Wege, von einem
Punkt ausgehend, fur den die Energie bekannt ist. Hierfur stehen eine Reihe von
Algorithmen zur Verfugung, die sich in ihrer Effizienz unterscheiden.
Effiziente Algorithmen beziehen neben den Energiewerten an bestimmten
Punkten auch die entsprechenden Gradienten oder sogar die Matrix der zweiten
Ableitungen mit ein. Ein Beispiel fur einen solchen Algorithmus ist die Methode
der konjugierten Gradienten.

3.2. MOLEKULARDYNAMIK-SIMULATION 51
3.2 Molekulardynamik-Simulation
Im Gegensatz zu den beschriebenen Methode gibt die Molekulardynamiksimula-
tion Aufschluß uber die dynamische Entwicklung eines Systems. Dazu werden die
Newton’schen Bewegungsgleichungen fur die M Atomkerne
m ri = Fi , i = 1, . . . ,M (3.2)
numerisch gelost. Ein Algorithmus, der sich aufgrund seiner numerischen Stabi-
litat und der vergleichsweise einfachen Implementierung durchgestzt hat, ist der
Verlet-Algorithmus [45]. Eine nahere Beschreibung dieser Methode findet sich im
Anhang C.
Im Rahmen der Born-Oppenheimer-Naherung werden die Atomkerne als klas-
sische Teilchen behandelt. Die auf sie wirkende Kraft berechnet sich aus der
Ableitung der Gesamtenergie nach den jeweiligen Atomkoordinaten. Fur ein N -
Elektronen-System ergibt sich, sofern die Gesamtwellenfunktion eine Slaterdeter-
minante ist, der folgende Ausdruck:
Fi = −∂Eges
∂Ri−
N∑j=1
∂Eges
∂Ψj
∂Ψj
∂Ri−
N∑j=1
∂Eges
∂Ψ∗j
∂Ψ∗j
∂Ri(3.3)
Ist die Gesamtwellenfunktion eine Eigenfunktion des Hamiltonoperators, so he-
ben sich die die beiden letzten Terme in 3.3 gegeneinander auf. Dies wird als
Hellmann-Feynman Theorem bezeichnet.
Ist der Basissatz, nach dem die Wellenfunktionen entwickelt werden, nicht
vollstandig, so treten zusatzlich sogenannte Pulay-Krafte auf. Sind die Basisfunk-
tionen jedoch ebenen Wellen, verschwinden diese Krafte, da die Basisfunktionen
dann nicht von den Kernkoordinaten abhangig sind.
Aus den zu jedem Zeitpunkt vorliegenden Daten fur Orte und Geschwindig-
keiten der Atomkerne lassen sich nun thermodynamische Großen ermitteln. Als
Beispiel sei hier nur die instantane Temperatur aufgefuhrt. Fur sie besteht nach
dem Gleichverteilungssatz im Falle eines isolierten Systems aus M Atomen der
folgende Zusammenhang:
12(3M − 6) kB T =
M∑i=1
12mi v
2i (3.4)
Fur einen detaillierten Uberblick uber diese Thematik sei auf die Literatur [46, 47]
verwiesen.

Kapitel 4
Ergebnisse
Samtliche Rechnungen, deren Ergebnisse in dieser Arbeit dargestellt werden, wur-
den mit Hilfe der beiden Programmpakete Gaussian98 [48] und Vasp [49] durch-
gefuhrt, die sich in ihrer Arbeitsweise grundlegend unterscheiden. Bei Gaussi-
an98 handelt es sich um ein Programmpaket, das die Berechnung sowohl der
Gesamtenergie eines atomaren Systems als auch einzelner Anteile derselben
(z.B. Schwingungs- und Rotationsenergie) mit den unterschiedlichsten Metho-
den ermoglicht. Zur Verfugung stehen dabei neben den gebrauchlichen Post-HF-
Methoden eine große Auswahl an Dichtefunktionalmethoden. In der Rechnung
werden sowohl Valenz- als auch Rumpfelektronen explizit berucksichtigt, deren
Gesamtspin im Vorfeld deklariert werden muß und im Verlauf der Rechnung eine
Konstante darstellt. Die Einelektronenwellenfunktionen konnen in verschiedenen
Basissatzen entwickelt werden, die alle auf den an den Kernorten lokalisierten
Gaussfunktionen aufgebaut sind. Da Gaussian98 das Vorhandensein von freien
Randbedingungen voraussetzt, ist allerdings nur die Untersuchung molekularer
Systeme moglich.
Im Gegensatz dazu verwendet das Programmpaket Vasp periodische Rand-
bedingungen. Auch bei diesem Programm ist die Berechnung der Gesamtenergie
eines atomaren Systems ein zentraler Punkt. Als Methode steht hier jedoch aus-
schließlich die Dichtefunktionaltheorie zur Verfugung, wobei man zwischen der
lokalen Dichtenaherung (LDA) mit dem Austausch-Korrelationsfunktional von
Ceperley und Alder [34] und der verallgemeinerten Gradientenmethode (GGA)
von Perdew und Wang [38] wahlen kann. Wahlweise kann die Rechnung spinpo-
larisiert durchgefuhrt werden.
52

4.1. VERGLEICH DER VERWENDETEN METHODEN 53
Als Basisfunktionen fur die Kohn-Sham-Orbitale sowie die Ladungsdichte
kommen ebene Wellen zum Einsatz. Um die Zahl der benotigten Basisfunktio-
nen fur ein bestimmtes gefordertes Maß an Genauigkeit gering zu halten, werden
in der Rechnung nur die Valenzelektronen berucksichtigt, das effektive Kernpo-
tential wird durch ein Pseudopotential dargestellt. Die verwendeten ultraweichen
Pseudopotentiale sind stark an die von Vanderbilt [50] vorgeschlagenen angeleht
und wurden von Kresse [51] entwickelt.
Zusatzlich zur Berechnung der elektronischen Energie fur eine bestimmte
raumliche Anordnung von Atomen bietet Vasp die Moglichkeit der Strukturopti-
mierung und der Molekulardynamik-Simulation. Bei der Suche nach Energiemi-
nima mit Vasp wurde in dieser Arbeit ausschließlich die konjugierte Gradienten-
methode verwendet. Die Molekulardynamik-Simulationen wurden mit Hilfe des
Verlet-Algorithmus [45] durchgefuhrt.
Auch das Programm Gaussian98 bietet unter anderem die Moglichkeit der
Strukturoptimierung, wobei auf der Energieflache sowohl nach lokalen Minima als
auch nach Sattelpunkten beliebiger Ordnung gesucht werden kann. Außerdem ist
mit diesem Programm eine Frequenzberechnung mit anschließender thermodyna-
mischer Analyse moglich. Dabei wird die volle Matrix mit den zweiten Ableitun-
gen der Energie nach den Koordinaten berechnet, aus deren Diagonalisierung man
die Normalschwingungen mit den zugehorigen Frequenzen erhalt. Deren Kenntnis
ist unerlaßlich, wenn man eine Aussage uber die Natur eines Extremalpunktes
auf der Energieflache machen mochte. Ferner werden mit diesen Informationen
die Translations-, Rotations- und Schwingungsanteile der Zustandssumme ermit-
telt, wodurch sich die thermodynamischen Zustandsgroßen wie Entropie, Freie
Energie, Gibbs’sche Freie Energie, usw. berechnen lassen.
4.1 Vergleich der verwendeten Methoden
Eine Beschreibung der benutzten Methoden findet man in Kap. 2.2 und 2.3.2 so-
wie im Anhang A. Im einzelnen waren dies die auf der Wellenfunktion basierenden
Methoden HF und CCSD(T) sowie die Dichtefunktionalmethoden SVWN und
CA (beide lokale Spin-Dichtenaherung, LSDA), BP86, BPW91 und PW91PW91
(Gradientenmethoden, GGA) sowie B3LYP (Hybridmethode).
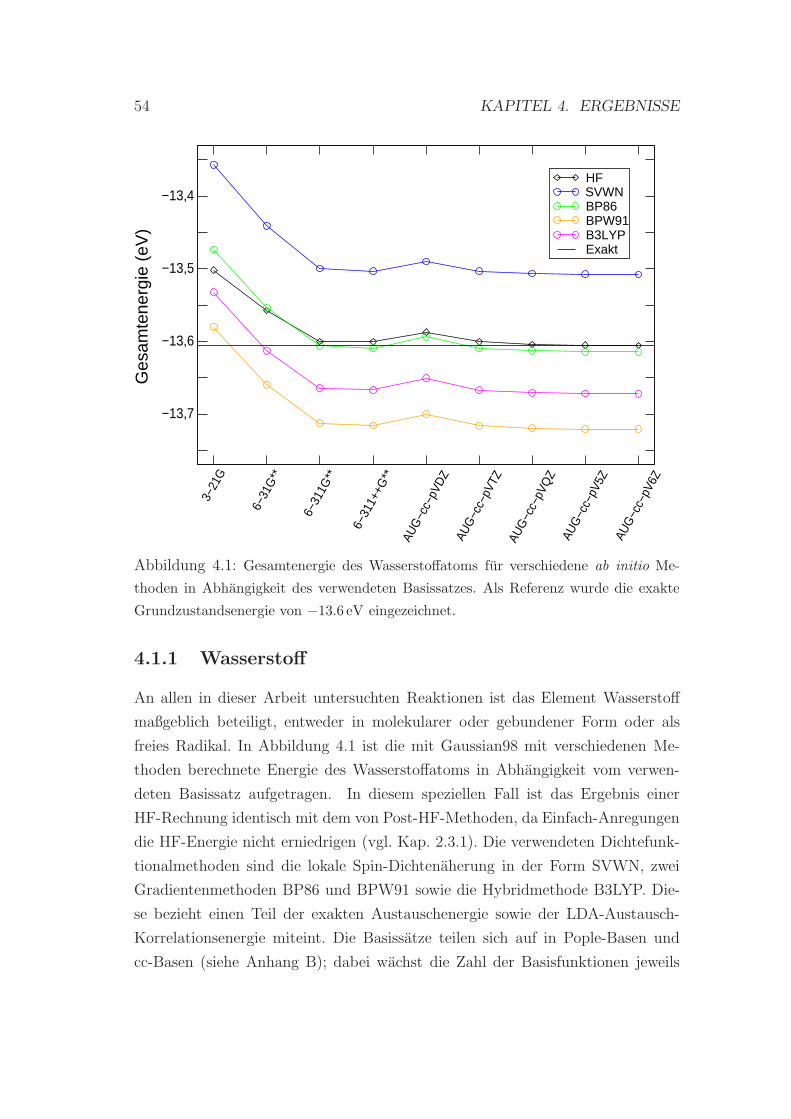
54 KAPITEL 4. ERGEBNISSE
3−21
G
6−31
G**
6−31
1G**
6−31
1++G
**
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
AUG
−cc−
pV5Z
AUG
−cc−
pV6Z
−13,7
−13,6
−13,5
−13,4
Ges
amte
nerg
ie (
eV)
HFSVWNBP86BPW91B3LYPExakt
Abbildung 4.1: Gesamtenergie des Wasserstoffatoms fur verschiedene ab initio Me-
thoden in Abhangigkeit des verwendeten Basissatzes. Als Referenz wurde die exakte
Grundzustandsenergie von −13.6 eV eingezeichnet.
4.1.1 Wasserstoff
An allen in dieser Arbeit untersuchten Reaktionen ist das Element Wasserstoff
maßgeblich beteiligt, entweder in molekularer oder gebundener Form oder als
freies Radikal. In Abbildung 4.1 ist die mit Gaussian98 mit verschiedenen Me-
thoden berechnete Energie des Wasserstoffatoms in Abhangigkeit vom verwen-
deten Basissatz aufgetragen. In diesem speziellen Fall ist das Ergebnis einer
HF-Rechnung identisch mit dem von Post-HF-Methoden, da Einfach-Anregungen
die HF-Energie nicht erniedrigen (vgl. Kap. 2.3.1). Die verwendeten Dichtefunk-
tionalmethoden sind die lokale Spin-Dichtenaherung in der Form SVWN, zwei
Gradientenmethoden BP86 und BPW91 sowie die Hybridmethode B3LYP. Die-
se bezieht einen Teil der exakten Austauschenergie sowie der LDA-Austausch-
Korrelationsenergie miteint. Die Basissatze teilen sich auf in Pople-Basen und
cc-Basen (siehe Anhang B); dabei wachst die Zahl der Basisfunktionen jeweils

4.1. VERGLEICH DER VERWENDETEN METHODEN 55
von links nach rechts.
Deutlich zu erkennen ist die Konvergenz der Energie mit großer werdendem
Basissatz zum jeweiligen Limit der Methode, welches bei HF naturlich dem ex-
akten Wert von −13.606 eV entspricht. Diesem Wert am nachsten kommt unter
den DFT-Methoden BP86 mit einer Energie von −13.614 eV, ansonsten schwan-
ken die Ergebnisse fur den großten Basissatz zwischen einem Fehler von 98 meV
bei SVWN und −115 meV bei BPW91. Die Hybridmethode B3LYP liefert einen
Wert von −13.672 eV, was einer betragsmaßigen Uberschatzung der Energie von
−66 meV entspricht.
Im Fall von SVWN zeigt sich die typische Unterschatzung der Austauschener-
gie in der lokalen Dichtenaherung. Die recht große Abweichung bei BPW91 laßt
sich darauf zuruckfuhren, daß das zugehorige Austauschkorrelationsfunktional
nicht frei von Selbstwechselwirkung ist. Eine Rechnung mit der BLYP-Methode,
deren Resultat hier nicht mit aufgefuhrt ist, liefert unter Verwendung der AUG-
cc-pV6Z-Basis eine Energie von −13.549 eV. Da das LYP-Funktional fur die Kor-
relation frei von Selbstwechselwirkung ist, berechnet sich daraus ein Fehler von
57 meV fur das entsprechende Austauschfunktional in diesem Basissatz. Die Aus-
tauschenergie im Fall von BP86 und BPW91 ist somit auch um diesen Betrag zu
gering, es ergeben sich also Werte fur die Korrelationsenergie von −65 meV bzw.
−172 meV. Dies ist in guter Ubereinstimmung mit den in [39] angegebenen Wer-
ten von −2 mH bzw. −6 mH. Der durch die Korrelationsenergie verursachte Fehler
hebt bei der BP86-Rechnung das Defizit in der Austauschenergie in etwa auf, was
zu der geringen Abweichung von nur −8 meV fuhrt. Bei der BPW91-Rechnung
uberwiegt der Fehler fur die Korrelation jedoch die zu kleine Austauschenergie,
das Resultat ist die recht hohe Abweichung in der Gesamtenergie.
Die Abweichung der mit der B3LYP-Methode erzielten Ergebnisse vom exak-
ten Wert ist auch auf die Selbstwechselwirkung zuruckzufuhren, jedoch liegt ihr
Ursprung in diesem Fall in der zu großen Korrelationsenergie der LSDA. Die be-
ste Ubereinstimmung liefert B3LYP mit dem relativ kleinen Basissatz 6-31G**,
der nur aus zwei kontrahierten Gaussfunktionen besteht. Der Grund dafur ist
naturlich eine gunstig ausfallende Fehlerkompensation, die Basis ist einfach zu
klein, um einen Großteil der - naturlich falschlicherweise vorhandenen - Korrela-
tionsenergie zu erfassen.
Selbst die großten hier diskutierten Abweichungen scheinen klein bezogen auf
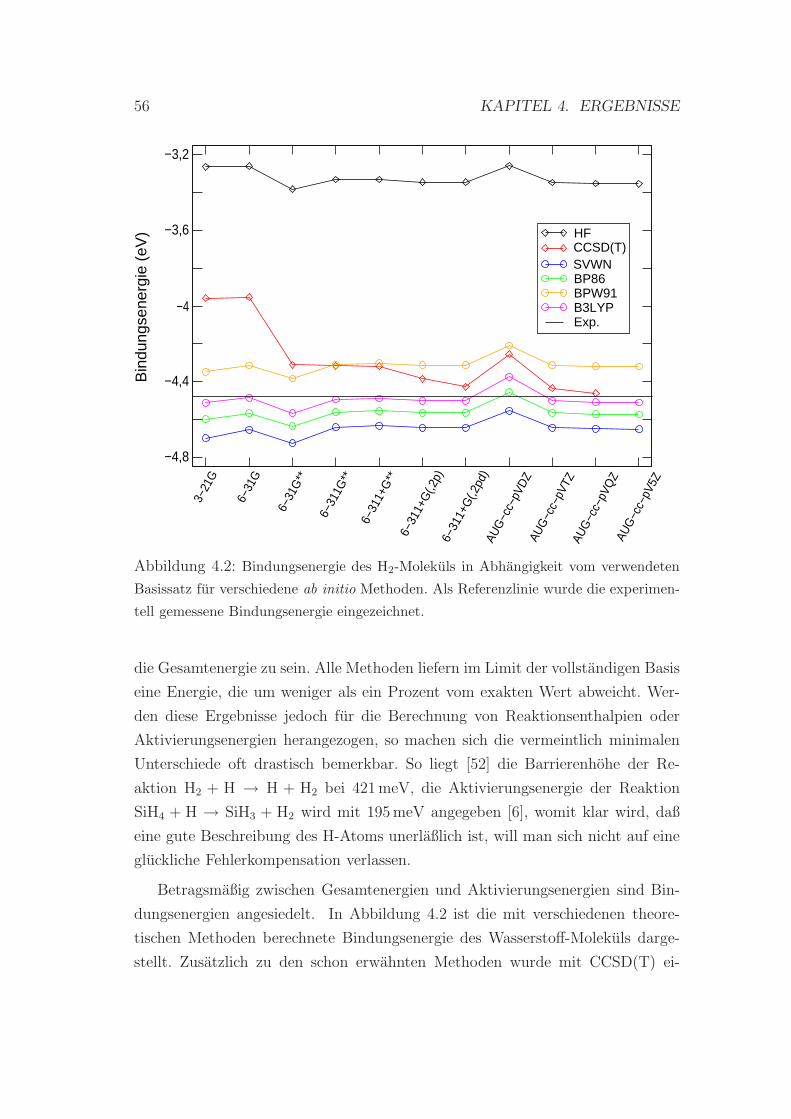
56 KAPITEL 4. ERGEBNISSE
3−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1+G
**6−
311+
G(,2
p)6−
311+
G(,2
pd)
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
AUG
−cc−
pV5Z
−3,2
−3,6
−4
−4,4
−4,8
Bin
dung
sene
rgie
(eV
) HFCCSD(T)SVWNBP86BPW91B3LYPExp.
Abbildung 4.2: Bindungsenergie des H2-Molekuls in Abhangigkeit vom verwendeten
Basissatz fur verschiedene ab initio Methoden. Als Referenzlinie wurde die experimen-
tell gemessene Bindungsenergie eingezeichnet.
die Gesamtenergie zu sein. Alle Methoden liefern im Limit der vollstandigen Basis
eine Energie, die um weniger als ein Prozent vom exakten Wert abweicht. Wer-
den diese Ergebnisse jedoch fur die Berechnung von Reaktionsenthalpien oder
Aktivierungsenergien herangezogen, so machen sich die vermeintlich minimalen
Unterschiede oft drastisch bemerkbar. So liegt [52] die Barrierenhohe der Re-
aktion H2 + H → H + H2 bei 421 meV, die Aktivierungsenergie der Reaktion
SiH4 + H → SiH3 + H2 wird mit 195 meV angegeben [6], womit klar wird, daß
eine gute Beschreibung des H-Atoms unerlaßlich ist, will man sich nicht auf eine
gluckliche Fehlerkompensation verlassen.
Betragsmaßig zwischen Gesamtenergien und Aktivierungsenergien sind Bin-
dungsenergien angesiedelt. In Abbildung 4.2 ist die mit verschiedenen theore-
tischen Methoden berechnete Bindungsenergie des Wasserstoff-Molekuls darge-
stellt. Zusatzlich zu den schon erwahnten Methoden wurde mit CCSD(T) ei-

4.1. VERGLEICH DER VERWENDETEN METHODEN 57
ne besonders aufwendige Post-HF Methode angewendet, deren Ergebnisse oft
als Referenz dienen, wenn keine experimentellen Daten vorhanden sind. In die-
ser Abbildung wie auch in allen folgenden Diagrammen sind die Ergebnisse
von HF-basierten Methoden mit eckigen, die von DFT-basierten Methoden mit
kreisformigen Symbolen versehen. Die Zahl der verwendeten Basissatze ist großer
als in Abbildung 4.1, da zusatzlich einige Basissatze ausgewahlt wurden, wel-
che beim einzelnen H-Atom aquivalente Ergebnisse liefern. Allerdings konnten
keine Rechnungen mit der AUG-cc-pV6Z-Basis durchgefuhrt werden, da sie die
verfugbaren Rechnerresourcen gesprengt hatten; das gleiche gilt fur die CCSD(T)-
Rechnung in der AUG-cc-pV5Z-Basis.
Die Bindungsenergien fur das H2-Molekul wie auch die fur die noch folgenden
Molekule wurden nach folgendem Verfahren berechnet. Zuerst wurde die Struktur
des Molekuls unter Verwendung des kleinsten Basissatzes optimiert. Das Ergebnis
diente dann als Startstruktur fur die Optimierung unter Verwendung der nachst-
großeren Basis. Diese Prozedur wurde bis zum großten Basissatz fortgefuhrt. An-
schließend wurde fur jeden Basissatz eine Frequenzberechnung fur den mit dieser
Basis gefundenen Gleichgewichtszustand durchgefuhrt, um die Nullpunktsschwin-
gungsenergie zu erhalten. Die Bindungsenergie ergibt sich dann als Differenz aus
der Gesamtenergie des Molekuls und - in diesem Fall - dem Zweifachen der Ener-
gie eines einzelnen H-Atoms. Dazu addiert wurden die entsprechenden Werte fur
die Nullpunktsenergie.
Verglichen mit der Gesamtenergie des H-Atoms sind die Verhaltnisse beim
H2-Molekul genau umgekehrt. Das Ergebnis der HF-Methode ist unter Verwen-
dung der großten Basis vom experimentellen Wert [53] von −4.48 eV mit einer
Diskrepanz von uber 1.12 eV am weitesten entfert. Bei kleinen Basissatzen findet
man mit der CCSD(T) Methode das zweitschlechteste Ergebnis. Dies andert sich
aber zu großeren Basissatzen hin, fur AUG-cc-pVQZ liegt die Post-Hartree-Fock
Methode mit 19 meV Unterschied schon am nachsten am experimentellen Wert.
Es ist davon auszugehen, daß sich dieses Ergebnis bei Vergroßern der Einteil-
chenbasis noch verbessert, da die CCSD(T)-Methode auf dem Variationsprinzip
beruht.
Den betragsmaßig hochsten Wert fur die Bindungsenergie liefert die SVWN-
Methode. Darin zeigt sich die typische Uberschatzung von Bindungsstarken in
der lokalen Dichtenaherung. Die Fehler der Gradientenmethoden belaufen sich

58 KAPITEL 4. ERGEBNISSE
auf −93 meV bei BP86 und 160 meV bei BPW91, was hauptsachlich auf das
unterschiedliche Ergebnis beim H-Atom zuruckzufuhren ist, da beide Methoden
in etwa gleiche Werte fur die Gesamtenergie des H2-Molekuls liefern.
Die B3LYP-Methode schneidet unter den DFT-Verfahren am besten ab. Gera-
de bei mittleren Basissatzen liefert sie eindeutig die besten Ergebnisse und kommt
unter Verwendung der 6-311+G** Basis bis auf 10 meV an den experimentellen
Wert heran. Das Limit der Methode liegt allerdings um etwa 20 meV darunter bei
−4.51 eV. Unter Berucksichtigung der Selbstwechselwirkungskorrektur laßt sich
dieses Resultat nicht weiter verbesseren [54], da die gunstig ausfallende Fehler-
kompensation zwischen Austausch- und Korrelationsanteil der jeweiligen Gesam-
tenergien in diesem Fall gestort wird.
Auffallend ist die geringe Abhangigkeit der DFT-Methoden vom verwendeten
Basissatz, es lassen sich schon bei relativ geringem numerischen Aufwand gute Er-
gebnisse erzielen. Allerdings ist mit dieser Tatsache auch ein Nachteil verbunden.
Im Gegensatz zur CCSD(T)-Methode - und im Prinzip zu allen Post-Hartree-
Fock Methoden - ist bei den DFT-Methoden keine systematische Verbesserung
der Ergebnisse durch die Verwendung von großeren Basissatzen moglich. Dies ist
naturlich nur von Bedeutung, solange die Ergebnisse außerhalb der chemischen
Genauigkeit von momentan etwa 1 kcalmol−1 bzw. 50 meV liegen, so daß im Fall
des H2-Molekuls dieser Nachteil praktisch nicht ins Gewicht fallt. Allerdings ist
dieses Molekul das kleinste (ungeladene) Molekul uberhaupt, wenn auch vielleicht
nicht das am einfachsten zu beschreibende. Wie sich die einzelnen Methoden bei
großeren Molekulen behaupten, wird im folgenden Abschnitt erlautert.
4.1.2 Bindungsenergien
Es gibt eine ganze Reihe von Veroffentlichungen, die sich mit der Frage beschafti-
gen, wie gut eine bestimmte theoretische Methode in Verbindung mit einer be-
stimmten Einteilchenbasis gewisse Eigenschaften wie Atomisierungs- und Ioni-
sierungsenergien, Standardbildungsenthalpien, Proton- und Elektronaffinitaten,
Bindungslangen und -winkel oder Schwingungsfrequenzen von Molekulen zu re-
produzieren in der Lage ist [27, 55, 56, 57, 58]. Als zugrundeliegender Satz von
Testmolekulen wird haufig eine als G2-Menge bezeichnete Ansammlung von Mo-
lekulen gewahlt, fur die sehr exakte experimentelle Daten vorliegen.
Zur Untersuchung von Bindungsenergien hat sich dabei die sogenannte G2-1

4.1. VERGLEICH DER VERWENDETEN METHODEN 59
Menge [27] durchgesetzt, welche aus 55 neutralen Molekulen, die aus Elemen-
ten der ersten und zweiten Reihe des Periodensystems zusammengesetzt sind,
besteht. Die genauesten chemischen Rechenmethoden liefern als Standardabwei-
chung in Bezug auf experimentelle Daten Werte um 50 meV, was im Rahmen der
chemischen Meßgenauigkeit liegt. Das Abschneiden der Dichtefunktionalmetho-
den ist nicht so gut, mit GGA-Methoden lassen sich, abhangig vom Basissatz,
Werte von etwa 150 meV erreichen. Hybridmethoden sind allerdings in der Lage,
eine Genauigkeit von unter 100 meV zu erzielen.
Das Problem bei solchen Vergleichsrechnungen ist zum einen, daß fur ein-
zelne Molekule die Abweichung sehr groß sein kann, wie es beispielsweise fur
SO2 oft der Fall ist. Vom guten Abschneiden einer bestimmten Methode laßt
sich also nicht unbedingt auf die Qualitat der Ergebnisse in Bezug auf das in
Betracht stehende System schließen. Zum anderen enthalten die zugrundeliegen-
den Molekule ausschließlich Elemente der ersten und zweiten Periode, außerdem
der dritten Periode im Fall der G2-2 Menge [58]. A priori Aussagen uber Sy-
steme mit Ubergangsmetallen oder Elementen aus hoheren Perioden sind somit
nicht moglich. Somit hangt die Wahl der theoretischen Methode meist vom in-
dividuellen Problem ab und muß im Vorfeld der Untersuchung im Rahmen von
Vergleichsrechnungen getroffen werden.
Zweiatomige Molekule
Die Bindungsenergien fur die auf den folgenden Seiten abgebildeten Molekule
wurden auf die gleiche Weise ermittelt, wie in Kapitel 4.1.1 beschrieben; sie sind
also schon mit der Nullpunktsschwingungsenergie korrigiert. Abbildung 4.3 zeigt
die Bindungsenergie einiger zweiatomiger Molekule, die aus Elementen der ersten
Periode zusammengesetzt sind, in Abhangigkeit vom verwendeten Basissatz. Da-
bei gelten die im untersten Diagramm angegebenen Bezeichnungen auch fur die
daruberliegenden.
Die Elektronen des Stickstoffmolekuls bilden im Grundzustand ein Spin-
Singulett. Aufgrund der im Vergleich zum Wasserstoff viel großeren Zahl von
Elektronen konnten keine Rechnungen in einer großeren als der AUG-cc-pVQZ-
Basis durchgefuhrt werden. Bei der CCSD(T)-Methode war sogar schon bei der
AUG-cc-pVTZ-Basis Schluß.
Auffallend ist die Tatsache, daß die Bindungsenergie auf der einen Seite durch
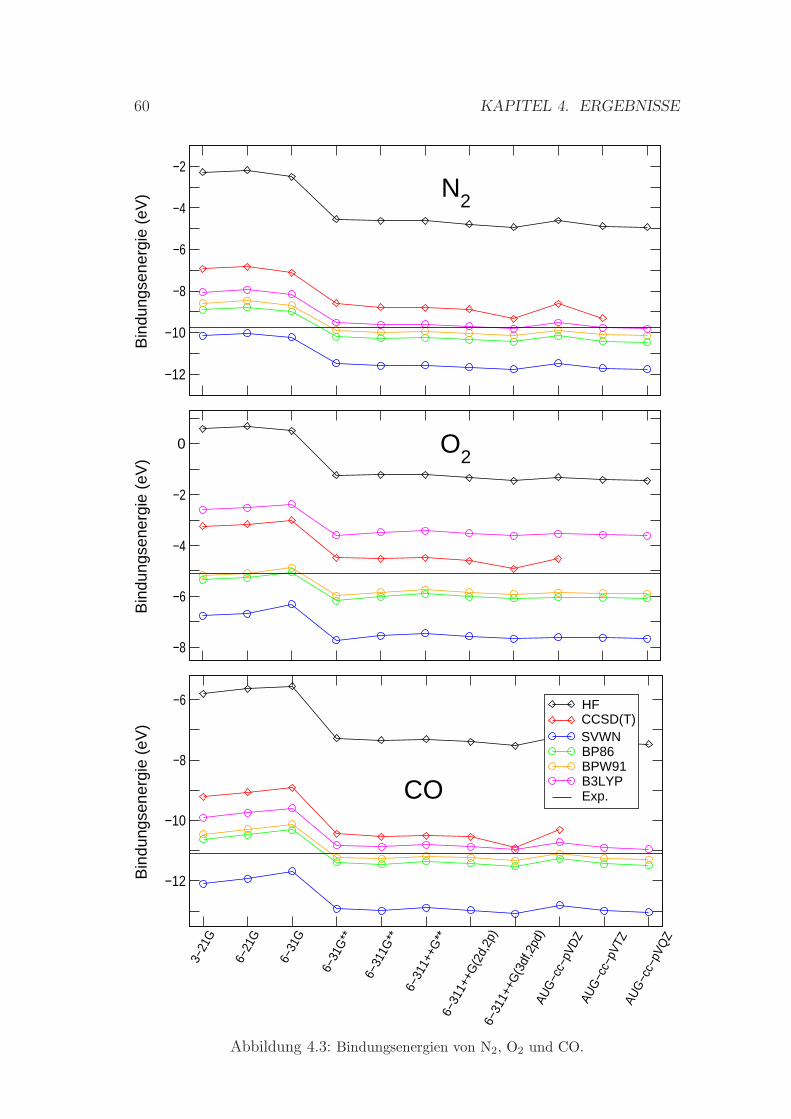
60 KAPITEL 4. ERGEBNISSE
−2
−4
−6
−8
−10
−12
Bin
dung
sene
rgie
(eV
) N2
0
−2
−4
−6
−8
Bin
dung
sene
rgie
(eV
)
O2
3−21
G
6−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**6−
311+
+G(2
d,2p
)6−
311+
+G(3
df,2
pd)
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
−6
−8
−10
−12Bin
dung
sene
rgie
(eV
)
HFCCSD(T)SVWNBP86BPW91B3LYPExp.CO
Abbildung 4.3: Bindungsenergien von N2, O2 und CO.

4.1. VERGLEICH DER VERWENDETEN METHODEN 61
die HF-Methode extrem unterschatzt (sie ergibt gerade mal 50 % des experi-
mentellen Wertes von −9.761 eV) und zum anderen durch SVWN um fast 2 eV
uberschatzt wird. Letzteres ist allerdings erst bei mittelgroßen Basissatzen der
Fall, die LDA liefert bei den kleinsten Basissatzen tatsachlich die besten Ergeb-
nisse unter allen betrachteten Methoden. Allerdings ist dieses Resultat auf eine
gunstige Fehlerkompensation zuruckzufuhren. Die kleinen Basissatze ergeben im
Vergleich zum Limit der Methode viel geringere Werte fur die Gesamtenergie des
N2-Molekuls, wahrend die Energie des einzelnen Stickstoffatoms vergleichsweise
konstant bleibt.
Die Gradientenmethoden sind der LDA gerade bei großeren Basissatzen weit
uberlegen, die Hybrid-Methode erreicht im Basissatzlimit als auch insgesamt (fur
die 6-311++G(3df,2pd)-Basis) das beste Resultat, mit Abweichungen von nur
62 meV bzw. 45 meV zum exakten Wert. Dagegen liefert das Coupled-Cluster-
Verfahren eher maßige Ergebnisse, die Berucksichtigung von d-und f -Orbitalen
als Polarisationsfunktionen ist fur eine hinreichend gute Bescheibung der π-
Bindungen im N2-Molekul offensichtlich notwendig, ergibt aber immer noch eine
Diskrepanz von 434 meV. Dagegen laßt sich festellen, daß die GGA-Methoden in
Bezug auf eine kleine Einteilchenbasis weit weniger anfallig sind.
Viel schwieriger zu behandeln ist das Sauerstoff-Molekul, dessen elektroni-
scher Grundzustand der Triplett-Zustand ist. Die CCSD(T)-Methode ist im Prin-
zip als einzige in der Lage, das O2-Molekul mit hinreichender Genauigkeit zu
beschreiben. Die Abweichung von der relativ kleinen Bindungsenergie von nur
5.117 eV betragt unter Verwendung der 6-311++G(3df,2pd)-Basis aber immer
noch 200 meV. Das Ansteigen der Energie zum nachsten Basissatz hin ist damit
zu begrunden, daß die Double-Zeta Basis weniger Flexibilitat aufweist als die in
den Abbildungen vor ihr stehende Valence Triple-Zeta Basis. Eine Rechnung in
der AUG-cc-pVTZ-Basis war fur das Sauerstoffmolekul mit der Coupled Cluster
Methode nicht durchfuhrbar, da der zur Verfugung stehende Hauptspeicher zu
gering war. Aufgrund der monotonen Anderung der Bindungsenergie mit großer
werdendem Basissatz ist aber anzunehmen, daß sich die Bindungsenergie im Ver-
gleich zur 6-311++G(3df,2pd)-Basis noch weiter verringern wurde.
Bei allen anderen Methoden kommt es augenscheinlich nur auf die Frage an,
ob im Basissatz Polarisationsfunktionen enthalten sind oder nicht. Denn HF sowie
alle DFT-Methoden liefern im wesentlichen nur zwei verschiedene Werte fur die

62 KAPITEL 4. ERGEBNISSE
Bindungsenergie, einen unter Verwendung der Basissatze ohne und einen fur die
mit Polarisationsfunktionen.
Die LDA in der Form SVWN weicht um etwa −1.5 eV bzw. −2.5 eV vom ex-
perimentellen Wert ab, die beiden GGA-Methoden uberschatzen die Starke der
O2-Bindung um etwa −0.9 eV unter Verwendung einer großen Basis, treffen bei
kleiner Basis allerdings nahezu den experimentellen Wert (vgl. Abbildung 4.3).
Die HF-Methode kann die Stabilitat des O2-Molekuls nicht vorhersagen, sofern
man in die Einteilchenbasis keine Polarisationsfunktionen miteinbezieht. Die Ge-
samtenergie zweier isolierter Sauerstoffatome ist niedriger als die des Sauerstoff-
molekuls, selbst ohne die Berucksichtigung der Nullpunktsschwingungsenergie.
Diese berechnet sich in der HF-Naherung, abhangig vom Basissatz, zu 108 meV
bis 126 meV. Der Einfluß der anteilig vorhandenen exakten Austauschenergie im
B3LYP-Funktional ist hier sehr gut zu erkennen und resultiert in einer effektiven
Verschlechterung in Bezug auf GGA. Mit einer Bindungsenergie von 3.609 eV
betragt der Unterschied zum Experiment 29.5 %.
Die hier dargestellte Schwierigkeit, das Element Sauerstoff gut zu beschrei-
ben, ist wohlbekannt und außert sich beispielsweise in dem Problem, die Ener-
gieflache des Ozon-Molekuls wiederzugeben. Der Grund fur diese Schwierigkeit
ist die Tatsache, daß die Bindung im Ozonmolekul einzig und allein durch Kor-
relationseffekte zustande kommt. Der Eindeterminantenansatz der HF-Methode
sagt fur dieses Molekul, wie auch fur F2, eine positive Bindungsenergie voraus
und ist somit nicht in der Lage, diese Molekule korrekt zu beschreiben.
In Verbindung mit anderen Elementen gelingt die Beschreibung des Sauer-
stoffs oft besser. Im untersten Graphen der Abbildung 4.3 ist die Situation fur das
zu N2 isoelektronische CO-Molekul wiedergegeben. Mit betragsmaßigen Abwei-
chungen von etwa 0.16 eV (BPW91 und B3LYP) bzw. 0.37 eV (BP86) bei großen
Basissatzen kommen die DFT-Methoden mit Gradientenkorrektur dem experi-
mentellen Wert von −11.11 eV am nachsten. Die LDA uberschatzt den Betrag der
Bindungsenergie um ca. 2 eV, HF liegt um etwa 4 eV zu tief. Einen ahnlichen Ver-
lauf wie beim O2-Molekul hat die Kurve der CCSD(T)-Methode, fur eine gute Be-
schreibung der Bindungsverhaltnisse sind Polarisationsfunktionen hoherer Ord-
nung obligatorisch. Dies bedeutet aber auch einen großen numerischen Aufwand.
Die Berechnung der Nullpunktsschwingungsenergie auf einem dual-Pentium 300
System nahm beispielsweise fur CCSD(T) in der 6-311++G(3df,2pd)-Basis etwa

4.1. VERGLEICH DER VERWENDETEN METHODEN 63
neun Stunden in Anspruch und dauerte somit etwa 100 mal langer als mit B3LYP
bzw. 150 mal langer als mit SVWN.
Eine analoge Rechnung fur das CO2-Molekul, deren Ergebnisse hier nicht wie-
dergegeben sind, lieferte prinzipiell das gleiche Bild. Jedoch war in diesem Fall
die CCSD(T)-Rechnung noch rechenintensiver, der umfangreichste verwendbare
Basissatz war die 6-311++G(2d,2p)-Basis. Rechnungen mit großerer Basis schei-
terten an dem enormen Speicherplatzbedarf der Post-HF Methode.
Wasserstoffverbindungen
In Abbildung 4.4 sind die Bindungsenergien einiger Molekule dargestellt, die das
Element Wasserstoff enthalten.
Das H2O-Molekul ist ein Objekt zahlreicher Untersuchungen, an dem jede
Methode getestet worden ist. Ein Beispiel fur einen Methodenvergleich fur die-
ses Molekul findet sich im Vorwort zu [59]. Die dort abgebildete Tabelle enthalt
die Abweichungen der mit verschiedenen Kombinationen von Methoden und Ba-
sissatzen berechneten Werte fur die Bindungsenergie des Wassermolekuls im Ver-
gleich zum experimentellen Wert von −9.51 eV und zum CCSD(T)/6-311G**
Ergebnis. In letzterem Fall belauft sich die Bindungsenergie auf −8.764 eV,
was in guter Ubereinstimmung mit dem in dieser Arbeit berechneten Wert von
−8.731 eV steht und einem Unterschied von 746 meV im Vergleich zum Expe-
riment entspricht. Die Ergebnisse der in [59] zitierten DFT-Methoden wurden
unter Verwendung der verschiedensten Basissatze erzielt, wobei z.B. das mit
BP86/6-31G** ermittele Ergebnis exakt mit dem hier berechneten Wert uber-
einstimmt; dasselbe gilt fur Resultate erhalten mit den vergleichbaren Hybrid-
DFT-Methoden B3PW91 und B3LYP. Grundsatzlich ist festzustellen, daß die in
der erwahnten Tabelle enthaltenen Ergebnisse der DFT-Methoden sehr gut (im
Bereich von 100 meV) mit dem experimentellen Wert ubereinstimmen, aus diesem
Grund aber auch eine betrachtliche Diskrepanz zum CCSD(T)-Wert aufweisen.
Mit dieser Beobachtung soll die Aussage wiederlegt werden, daß die Dichtefunk-
tionaltheorie generell fur die Berechnung von Bindungsenergien nicht geeignet
sei. Diese Aussage konnte sich ergeben, nahme man irrtumlicherweise oder in
Ermangelung des experimentellen Ergebnisses das CCSD(T)-Ergebnis als Refe-
renzwert. In der weiteren Argumentation wird das sogenannte Versagen der DFT
unter anderem auf die Verwendung der lokalen Dichtenaherung geschoben.
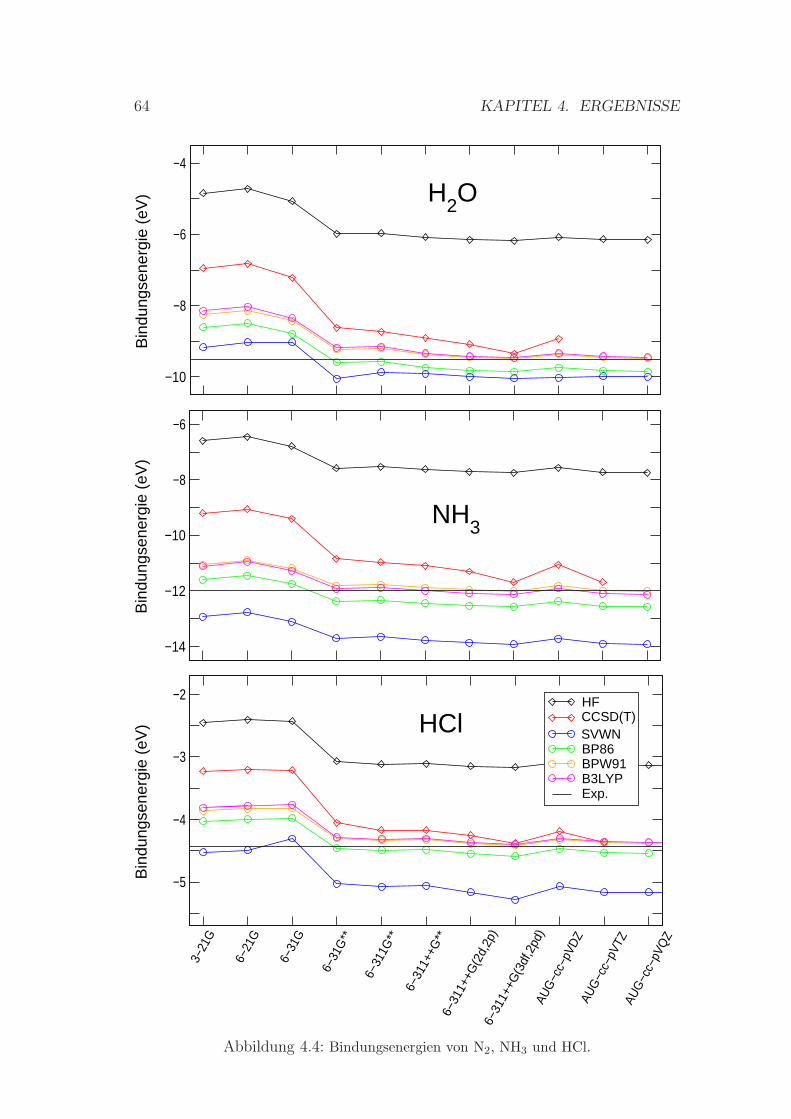
64 KAPITEL 4. ERGEBNISSE
−4
−6
−8
−10
Bin
dung
sene
rgie
(eV
) H2O
−6
−8
−10
−12
−14
Bin
dung
sene
rgie
(eV
)
NH3
3−21
G
6−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**6−
311+
+G(2
d,2p
)6−
311+
+G(3
df,2
pd)
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
−2
−3
−4
−5Bin
dung
sene
rgie
(eV
)
HFCCSD(T)SVWNBP86BPW91B3LYPExp.
HCl
Abbildung 4.4: Bindungsenergien von N2, NH3 und HCl.

4.1. VERGLEICH DER VERWENDETEN METHODEN 65
Beim Betrachten des obersten Diagramms in Abbildung 4.4 fallt auf, warum
die oben genannte Gegenuberstellung inklusive der darauf aufbauenden Argumen-
tation keinen Sinn macht und eher zum Nachteil der DFT als zu ihrem Vorteil
Nutzen ausgelegt werden kann. Klar zu erkennen ist die stetige Verbesserung des
CCSD(T)-Ergebnisses mit großer werdender Einteilchenbasis, so daß ein Gegner
der DFT behaupten konnte, die Deklarierung des CCSD(T)/6-311G** Resul-
tats als Referenzwert sei unsinnig. Unerlaßlich ist namlich, wie im Falle von O2
und CO schon angesprochen, die Einbeziehung von Polarisationsfunktionen hoher
Ordnung in den Basissatz, um mit der Post-Hartree-Fock Methode gute Resultate
in Bezug auf die durch den großen Elektronegativitatsunterschied von Wasserstoff
und Sauerstoff stark polarisierte Bindung zu erzielen.
Es fallt außerdem auf, daß die LDA nicht viel schlechtere Ergebnisse liefert
als die Gradientenmethode BP86. Das hervorragende Abschneiden der BP86/6-
31G** Kombination mit einem Unterschied von nur 78 meV zum experimentellen
Wert ist, wie ebenfalls aus Abbildung 4.4 ersichtlich, auf die kleine Einteilchenba-
sis zuruckzufuhren. Im Grenzfall der vollstangigen Basis betragt die Diskrepanz
der Gradientenmethode schon mehr als 350 meV und liegt damit recht nah am
LDA-Wert. Gute Ergebnisse unter Verwendung einer umfangreichen Einteilchen-
basis liefern BPW91 und die Hybrid-Methode B3LYP, sie konstatieren fur die
Bindungsenergie des H2O-Molekuls einen Wert von −9.446 eV in der AUG-cc-
pVQZ-Basis.
Damit wird deutlich, daß bei der Interpretation von Ergebnissen einzelner
Kombinationen von Methode und Basissatz Vorsicht geboten ist, ein entscheiden-
der Faktor ist das Konvergenzverhalten der Resultate in Richtung der vollstandi-
gen Basis. Die Ausfuhrungen im Vorwort von [59] haben u.a. diese Aussage zum
Inhalt, was durch die eigentlich nicht vergleichbaren Ergebnisse der Tabelle nicht
direkt unterstutzt wird.
Die Bindungsenergie des NH3-Molekuls ist in Abbildung 4.4 (Mitte) darge-
stellt. Der Verlauf der einzelnen Kurven ahnelt zum einen dem entsprechenden
Verlauf beim N2-Molekul. Gerade bei mittleren Basissatzen sind die Gradienten-
methoden, insbesondere das B3LYP-Hybrid, nicht zu schlagen. Die beste Uber-
einstimmung uberhaupt ergibt sich bei Wahl der 6-311++G**-Basis mit einem
Fehler von nur 0.6 meV. Den besten Wert unter Verwendung der großten Basis lie-
fert BPW91 mit −12.012 eV, was im Vergleich zum Experiment mit −11.999 eV

66 KAPITEL 4. ERGEBNISSE
einer Abweichung von −13 meV entspricht. Zum anderen weist der Kurvenverlauf
eine gewisse Ahnlichkeit zu dem zu NH3 isoelektronischen H2O-Molekul auf. Die
starke Basissatzabhangigkeit der CCSD(T)-Methode ist gut zu erkennen, doch
selbst die besten im Rahmen dieser Arbeit mit ihr erzielten Ergebnisse kommen
nicht an die Qualitat der GGA heran. LDA berechnet die Bindungsenergie auf
etwa −14 eV, HF schlagt mit einem Fehler von uber 30 % zu Buche.
Ein zentrales Molekul in dieser Arbeit ist das HCl-Molekul. Beachtet man die
gestreckte Darstellung in y-Richtung im Vergleich zu den voherigen Diagrammen,
so ergibt sich fur dieses Molekul eine gute Ubereinstimmung aller Methoden mit
dem experimentellen Wert von −4.43 eV. Die durch den großen Unterschied der
Elektronegativitaten von Wasserstoff und Chlor stark polarisierte Bindung laßt
sich im Falle der Pople-Basen am besten mit 6-311++G(3df,2pd) beschreiben.
In diesem Fall liefern BPW91, B3LYP und CCSD(T) in etwa gleiche Ergebnisse
von −4.41 eV, −4.39 eV bzw. −4.38 eV. Die Abweichung der LDA-Methode ist
mit 0.74 eV ahnlich gering wie beim Wassermolekul, der Unterschied des HF-
Ergebnisses zum experimentellen Ergebnis ist mit 1.27 eV nicht viel großer als
die Abweichung beim H2-Molekul und somit der zweitkleinste unter allen bisher
betrachteten Molekulen.
Silylen, Silen und Silan
Ein ahnliches Bild ergibt sich fur die drei in Abbildung 4.5 behandelten Molekule.
Im Gegensatz zu dem verwandten CH2, welches den Triplettzustand bevorzugt,
bilden die Elektronen im SiH2-Molekul (Silylen) im Grundzustand ein Spinsin-
gulett. SiH3 wird als Silyl bezeichnet und ist ein Radikal mit der Multiplizitat 2.
Das tetraedische Monosilan (SiH4) ist sehr stabil und bildet die Ausgangsbasis
fur die Siliziumherstellung [60].
Alle drei Molekule lassen sich durch Dichtefunktionalmethoden gut beschrei-
ben, wobei die Gradientenmethoden der LDA uberlegen sind. Allerdings liegen
die Abweichungen von SVWN nur zwischen 9.5 % (SiH4) und 11.8 % (SiH2) im
Grenzfall großer Einteilchenbasis.
Bemerkenswert ist das hervorragende Abschneiden der B3LYP-Methode
besonders im Bereich mittlerer Basissatze. Die Bindungsenergie des Silylen-
Molekuls von −6.262 eV wird mit einer Abweichung von nur 2 meV unter Ver-
wendung der 6-311G**-Basis nahezu genau getroffen, der Wert fur die großte
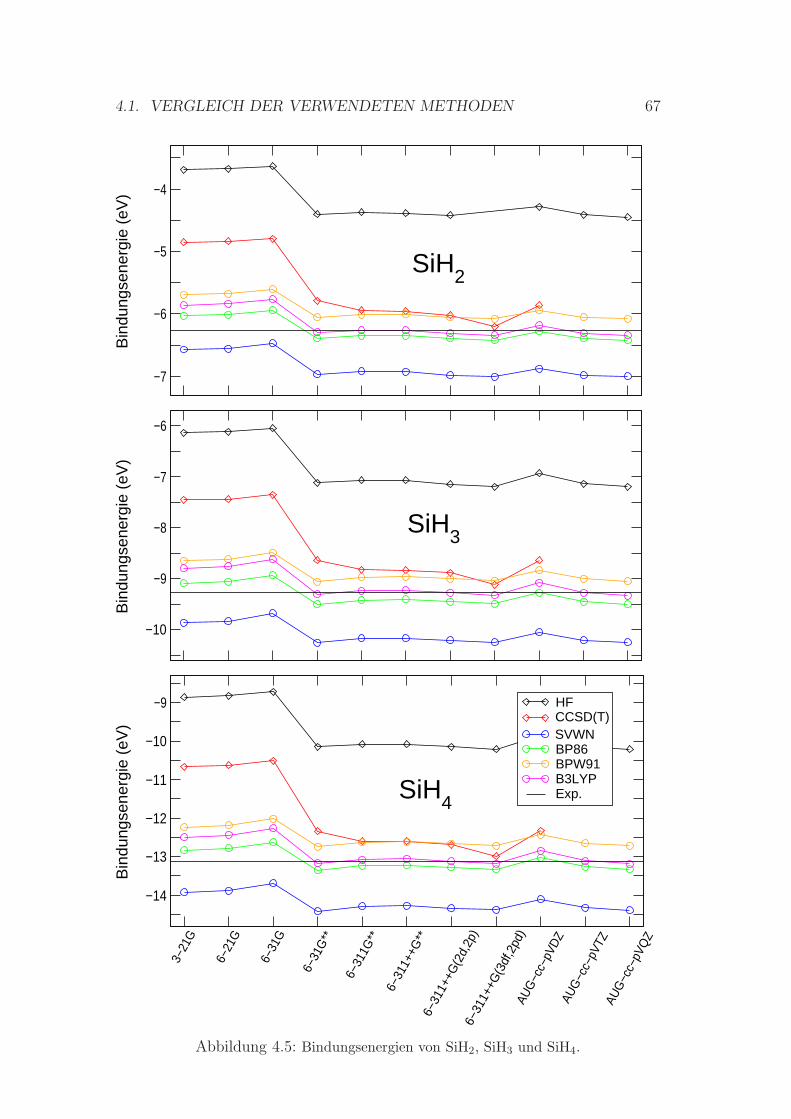
4.1. VERGLEICH DER VERWENDETEN METHODEN 67
−4
−5
−6
−7
Bin
dung
sene
rgie
(eV
)
SiH2
−6
−7
−8
−9
−10
Bin
dung
sene
rgie
(eV
)
SiH3
3−21
G
6−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**6−
311+
+G(2
d,2p
)6−
311+
+G(3
df,2
pd)
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
−9
−10
−11
−12
−13
−14
Bin
dung
sene
rgie
(eV
)
HFCCSD(T)SVWNBP86BPW91B3LYPExp.SiH4
Abbildung 4.5: Bindungsenergien von SiH2, SiH3 und SiH4.

68 KAPITEL 4. ERGEBNISSE
verwendete Basis liegt um 82 meV darunter.
Fur SiH3 und SiH4 ergeben sich die kleinsten Abweichungen von den experi-
mentellen Werten in Hohe von −9.28 eV bzw. −13.131 eV unter Verwendung der
6-311++G(2d,2p)-Basis. Sie betragen 1 meV bzw. 10 meV und liegen weit unter-
halb der chemischen Genauigkeitsgrenze. Fur die umfangreiche AUG-cc-pVQZ-
Basis belaufen sich die Unterschiede zum experimentellen Wert auf 54 meV bzw.
58 meV und sind damit immer noch die kleinsten fur diesen Basissatz unter allen
verwendeten Methoden. Durch die Berucksichtigung der Selbstwechselwirkung
wurde dieses Ergebnis wahrscheinlich eher verschlechtert, da sich dann die ver-
ringerte Gesamtenergie des H-Atoms verstarkend auf die Bindungsenergien der
Molekule auswirken wurde. Fur die BPW91-Methode ist das Gegenteil der Fall.
Die Abweichungen im Vergleich zu den experimentellen Werten liegen im Bereich
des durch die Selbstwechselwirkung bedingten Fehler beim H-Atom (vgl. 4.1.1)
multipliziert mit der Zahl der H-Atome im jeweiligen Molekul, so daß durch die
Berucksichtigung der Selbstwechselwirkungskorektur eine gute Ubereinstimmung
mit dem experimentellen Wert erreicht wurde.
Im Gegensatz zu den GGA-Methoden liefert CCSD(T) erst bei hinreichen-
der Große des Basissatzes gute Ergebnisse, wie dies auch schon bei den anderen
Molekulen der Fall war. Der systematischen Verbesserung steht allerdings der
enorme numerische Aufwand entgegen.
4.1.3 Rechenaufwand
In Abbildung 4.6 ist die fur eine Berechnung der Nullpunktsschwingungsenergie1
benotigte Rechenzeit auf einem dual Pentium 300 System uber der Große des ver-
wendeten Basissatzes aufgetragen. Die Abbildung zeigt den rasanten Anstieg der
Rechenzeit mit der Zahl der Basisfunktionen im Fall der CCSD(T)-Methode zu
erkennen, der nach einem N7-Gesetz (vgl. 2.3.2) verlauft. Gerade noch durchfuhr-
bar war eine Rechnung in der AUG-cc-pVTZ-Basis (107 Basisfunktionen), ei-
ne Rechnung unter Verwendung der nachstgroßeren Basis (142 Basisfunktionen)
hatte einen Anstieg des Rechenaufwandes um einen Faktor 7.25 bedeutet. Die
daraus resultierende Rechenzeit von etwa zehn Tagen ware zu vertreten gewesen.
1Die Berechnung der Bindungsenergie wurde nicht als Kriterium ausgewahlt, da die Zeit furdie in ihr enthaltene Strukturoptimierung von der ausgewahlten Startgeometrie abhangt undsomit nicht charakteristisch fur die verwendete Methode ist.
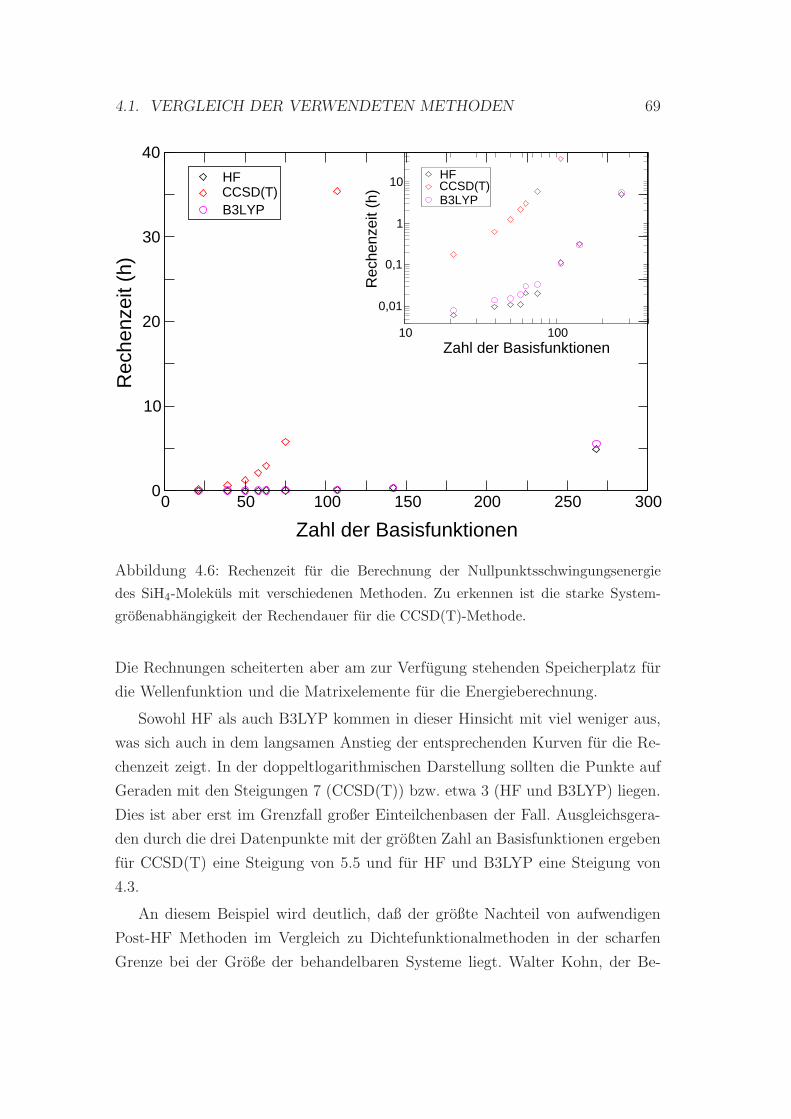
4.1. VERGLEICH DER VERWENDETEN METHODEN 69
0 50 100 150 200 250 300
Zahl der Basisfunktionen
0
10
20
30
40
Rec
henz
eit (
h)
HFCCSD(T)B3LYP
10 100Zahl der Basisfunktionen
0,01
0,1
1
10
Rec
henz
eit (
h)
HFCCSD(T)B3LYP
Abbildung 4.6: Rechenzeit fur die Berechnung der Nullpunktsschwingungsenergie
des SiH4-Molekuls mit verschiedenen Methoden. Zu erkennen ist die starke System-
großenabhangigkeit der Rechendauer fur die CCSD(T)-Methode.
Die Rechnungen scheiterten aber am zur Verfugung stehenden Speicherplatz fur
die Wellenfunktion und die Matrixelemente fur die Energieberechnung.
Sowohl HF als auch B3LYP kommen in dieser Hinsicht mit viel weniger aus,
was sich auch in dem langsamen Anstieg der entsprechenden Kurven fur die Re-
chenzeit zeigt. In der doppeltlogarithmischen Darstellung sollten die Punkte auf
Geraden mit den Steigungen 7 (CCSD(T)) bzw. etwa 3 (HF und B3LYP) liegen.
Dies ist aber erst im Grenzfall großer Einteilchenbasen der Fall. Ausgleichsgera-
den durch die drei Datenpunkte mit der großten Zahl an Basisfunktionen ergeben
fur CCSD(T) eine Steigung von 5.5 und fur HF und B3LYP eine Steigung von
4.3.
An diesem Beispiel wird deutlich, daß der großte Nachteil von aufwendigen
Post-HF Methoden im Vergleich zu Dichtefunktionalmethoden in der scharfen
Grenze bei der Große der behandelbaren Systeme liegt. Walter Kohn, der Be-

70 KAPITEL 4. ERGEBNISSE
grunder der Dichtefunktionaltheorie und Nobelpreistrager von 1998, beschreibt
diesen Sachverhalt in [61] mit den Worten”...systems of very many electrons whe-
re wave function methods encounter and are stopped by the exponential wall.“
4.1.4 Bindungslangen
Nachdem im vorangegangenen Abschnitt energetische Verhaltnisse diskutiert
wurden, geht es jetzt um Frage, wie gut die einzelnen Methoden in der Lage
sind, Molekulgeometrien zu beschreiben. Als Beispiele dienen dazu die Molekule
N2, HCl und SiH4. In Abbildung 4.7 ist die mit verschiedenen Methoden ermittel-
te Lange der N-N Bindung bzw. der X-H Bindung, X ∈ {Cl, Si} in der jeweiligen
optimierten Struktur in Abhangigkeit vom verwendeten Basissatz dargestellt.
Auffallend ist in allen drei Diagrammen die Unterschatzung der Bindungslangen
durch die HF-Methode. Diese Tatsache deckt sich mit Ergebnissen in der Litera-
tur.
Die beste Ubereinstimmung mit der experimentellen Bindungslange im Stick-
stoffmolekul von 1.098 A liefert im Bereich mittlerer Basissatze die LDA-Methode,
fur eine große Einteilchenbasis ist das Resultat von BPW91 am besten. Gut zu er-
kennen ist der Einfluß des exakten Austauschs in der B3LYP-Methode auf die be-
rechnete Bindungslange. So liegen die entsprechenden Werte zwischen dem GGA-
Ergebnis und der HF-Kurve. Die zu große Bindungslange der GGA-Methoden
wird nach unten korrigiert, was sich in einer ausgesprochen guten Ubereinstim-
mung der B3LYP-Kurve mit dem Experiment außert.
Beim HCl-Molekul sind die Verhaltnisse etwas anders. Im Bereich mittlerer
Basissatze kommt die CCSD(T)-Methode dem experimentellen Wert von 1.274 A
mit Abweichungen von weniger als 0.004 A am nachsten. Im Gegensatz zum
N2-Molekul uberschatzt die LDA hier die Lange der Bindung und liefert in
etwa die gleichen Ergebnisse wie die Gradientenmethoden. Sowohl bei kleinen
Basisatzen als auch im Limit der vollstandigen Einteilchenbasis schneidet die
B3LYP-Methode mit Fehlern im Bereich von 0.005 A unter allen DFT-Methoden
am besten ab, was wiederum durch den Einfluß des HF-ahnlichen Austauschterms
der Hybridmethode zu erklaren ist.
Die Situation beim tetraedischen Monosilan ist annahernd die gleiche. Aller-
dings kommt hier die B3LYP-Methode in allen Basissatzen mit Polarisations-
funktionen dem experimentellen Wert von 1.481 A im Vergleich zu allen anderen
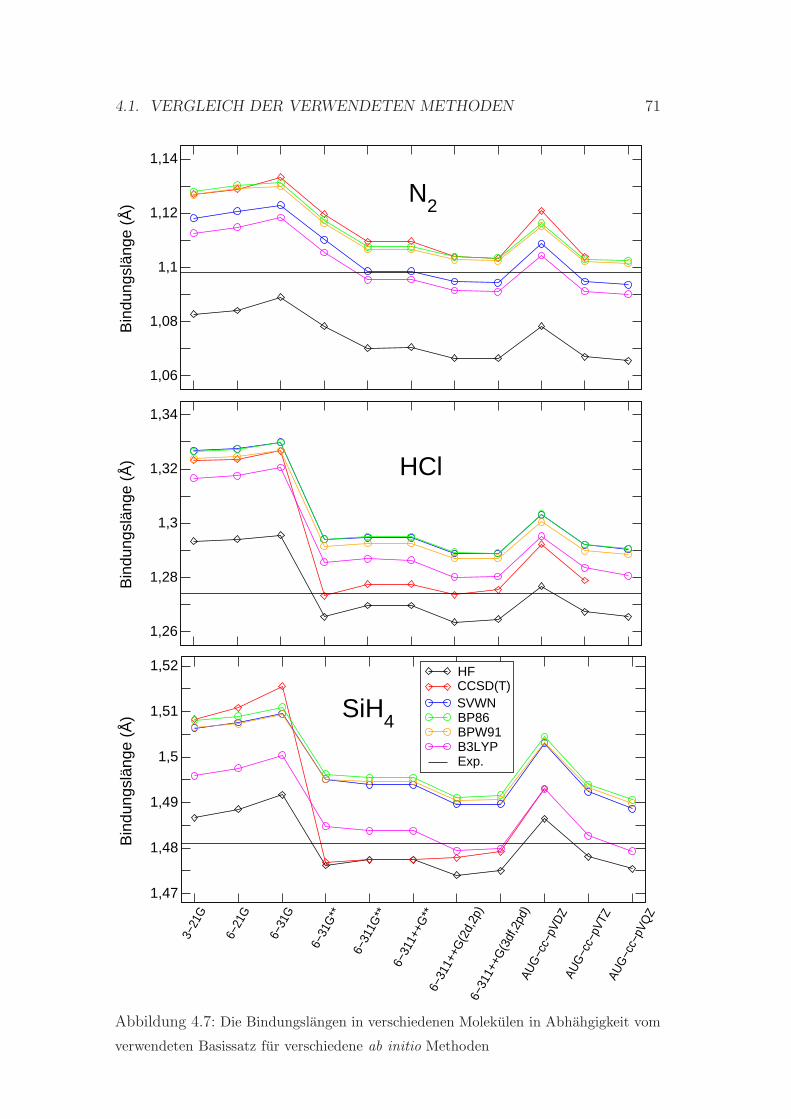
4.1. VERGLEICH DER VERWENDETEN METHODEN 71
1,06
1,08
1,1
1,12
1,14
Bin
dung
slän
ge (
Å) N2
1,26
1,28
1,3
1,32
1,34
Bin
dung
slän
ge (
Å) HCl
3−21
G
6−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**6−
311+
+G(2
d,2p
)6−
311+
+G(3
df,2
pd)
AUG
−cc−
pVD
ZAU
G−c
c−pV
TZAU
G−c
c−pV
QZ
1,47
1,48
1,49
1,5
1,51
1,52
Bin
dung
slän
ge (
Å)
HFCCSD(T)SVWNBP86BPW91B3LYPExp.
SiH4
Abbildung 4.7: Die Bindungslangen in verschiedenen Molekulen in Abhahgigkeit vom
verwendeten Basissatz fur verschiedene ab initio Methoden
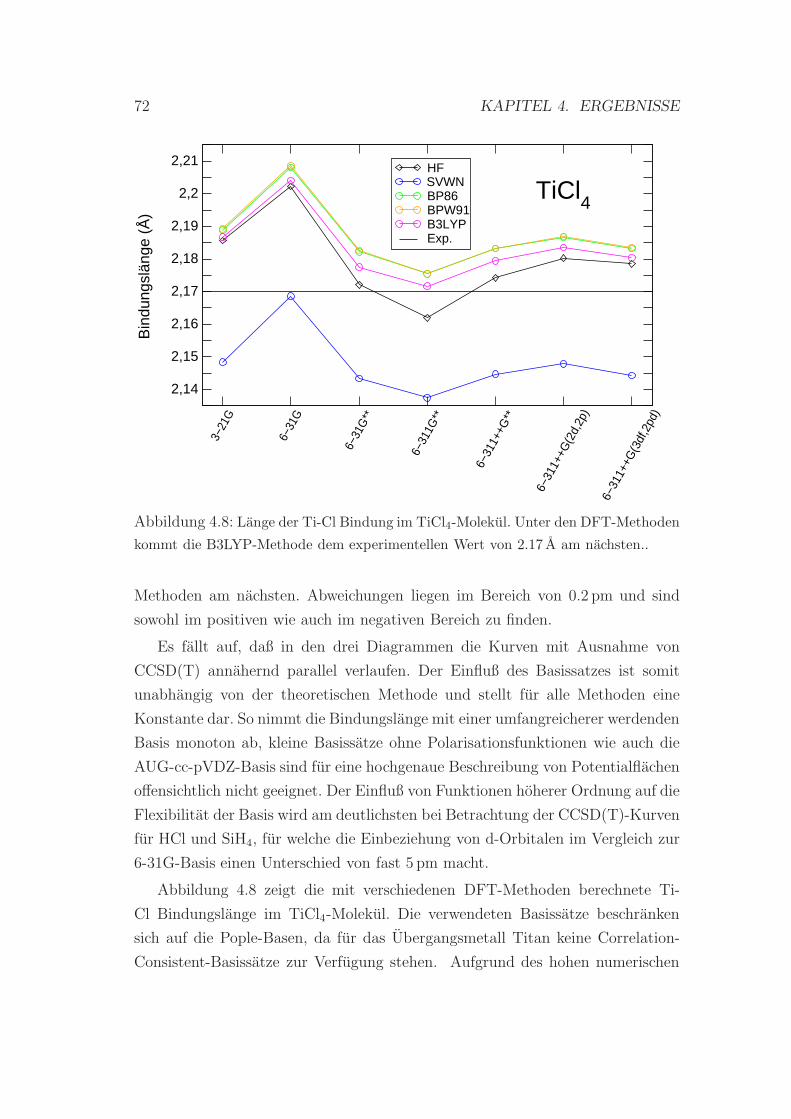
72 KAPITEL 4. ERGEBNISSE
3−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**
6−31
1++G
(2d,
2p)
6−31
1++G
(3df
,2pd
)
2,14
2,15
2,16
2,17
2,18
2,19
2,2
2,21B
indu
ngsl
änge
(Å
)
HFSVWNBP86BPW91B3LYPExp.
TiCl4
Abbildung 4.8: Lange der Ti-Cl Bindung im TiCl4-Molekul. Unter den DFT-Methoden
kommt die B3LYP-Methode dem experimentellen Wert von 2.17 A am nachsten..
Methoden am nachsten. Abweichungen liegen im Bereich von 0.2 pm und sind
sowohl im positiven wie auch im negativen Bereich zu finden.
Es fallt auf, daß in den drei Diagrammen die Kurven mit Ausnahme von
CCSD(T) annahernd parallel verlaufen. Der Einfluß des Basissatzes ist somit
unabhangig von der theoretischen Methode und stellt fur alle Methoden eine
Konstante dar. So nimmt die Bindungslange mit einer umfangreicherer werdenden
Basis monoton ab, kleine Basissatze ohne Polarisationsfunktionen wie auch die
AUG-cc-pVDZ-Basis sind fur eine hochgenaue Beschreibung von Potentialflachen
offensichtlich nicht geeignet. Der Einfluß von Funktionen hoherer Ordnung auf die
Flexibilitat der Basis wird am deutlichsten bei Betrachtung der CCSD(T)-Kurven
fur HCl und SiH4, fur welche die Einbeziehung von d-Orbitalen im Vergleich zur
6-31G-Basis einen Unterschied von fast 5 pm macht.
Abbildung 4.8 zeigt die mit verschiedenen DFT-Methoden berechnete Ti-
Cl Bindungslange im TiCl4-Molekul. Die verwendeten Basissatze beschranken
sich auf die Pople-Basen, da fur das Ubergangsmetall Titan keine Correlation-
Consistent-Basissatze zur Verfugung stehen. Aufgrund des hohen numerischen

4.2. DIE PSEUDOPOTENTIALMETHODE 73
Aufwands konnte keine entsprechende Rechnung mit dem CCSD(T)-Verfahren
durchgefuhrt werden. Aufffallig ist die gute Ubereinstimmung der B3LYP-
Methode mit dem experimentellen Wert von 2.17 A bei mittleren Basissatzen, was
wiederum auf die Einbeziehung der exakten Austauschenergie zuruckzufuhren ist.
Dies macht gegenuber den reinen Gradientenmethoden eine Verbesserung der Er-
gebnisse um etwa 0.5 A aus.
4.1.5 Auswahl der Methode
Die B3LYP-Methode bietet sich aufgrund ihrer durchweg guten Ergebnisse sowohl
in Bezug auf die energetischen Verhaltnisse als auch im Hinblick auf die Geometrie
als Methode fur die Untersuchung von Potentialflachen chemischer Reaktionen
an, welche die Elemente Wasserstoff, Silizium und Chlor sowie das Ubergangsme-
tall Titan beinhalten. Im Vergleich zu CCSD(T) ist sie um ein Vielfaches weniger
rechenintensiv, was gerade bei der Untersuchung einer PES aufgrund der fur eine
gute Beschreibung notwendigen großen Zahl von Energieberechnungen ein wich-
tiges Kriterium ist. Aus diesem Grund wurde die Hybridmethode B3LYP fur die
Behandlung der Potentialflachen verschiedener chemischer Reaktionen in Kap.
4.3 ausgewahlt.
4.2 Die Pseudopotentialmethode
Aus den vorangegangenen Betrachtungen geht hervor, daß die lokale Spin-
Dichtenaherung in der Form SVWN keine guten Ergebnisse bei der Beschreibung
der diskutierten Molekule liefert. Dies gilt auch fur die Resultate, die mit Hil-
fe des Programmpaketes Vasp unter Verwendung der LSDA in Form CA erzielt
wurden. Dies ist verstandlich, da sich die beiden Methoden nur in der expliziten
Form der Interpolationsfunktion unterscheiden, mit deren Hilfe die tabellierten
Werte fur die Korrelationsenergie eines Elektrons im homogenen Elektronengas
angenahert werden. Aus diesem Grund sind die mit Vasp im Rahmen der LSDA
erzielten Ergebnisse hier nicht naher aufgefuhrt.
Die zur Verfugung stehende GGA-Methode ist der in den vorangegangenen
Abschnitten diskutierten BPW91-Methode sehr ahnlich, sie unterscheidet sich
lediglich in der Form des Funktionals fur die Austauschenergie. Zusatzlich zum
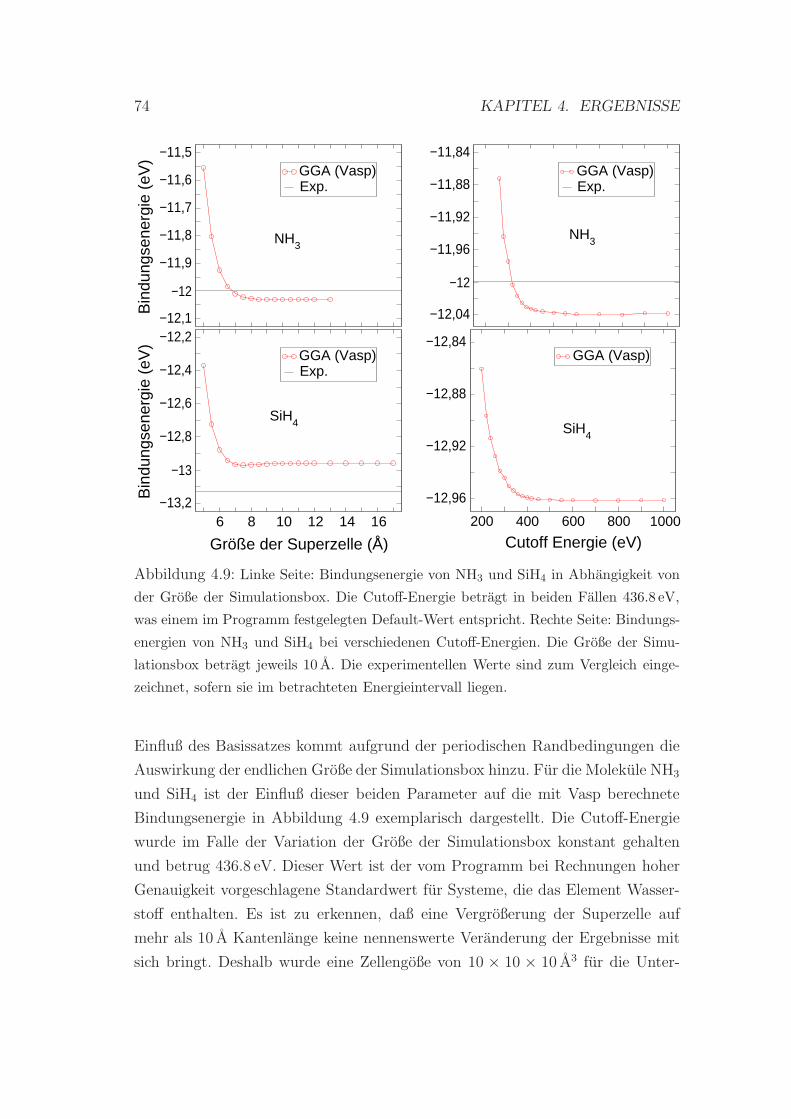
74 KAPITEL 4. ERGEBNISSE
−12,1
−12
−11,9
−11,8
−11,7
−11,6
−11,5B
indu
ngse
nerg
ie (
eV)
GGA (Vasp)Exp.
NH3
−12,04
−12
−11,96
−11,92
−11,88
−11,84GGA (Vasp)Exp.
NH3
6 8 10 12 14 16
Größe der Superzelle (Å)
−13,2
−13
−12,8
−12,6
−12,4
−12,2
Bin
dung
sene
rgie
(eV
)
GGA (Vasp)Exp.
SiH4
200 400 600 800 1000
Cutoff Energie (eV)
−12,96
−12,92
−12,88
−12,84GGA (Vasp)
SiH4
Abbildung 4.9: Linke Seite: Bindungsenergie von NH3 und SiH4 in Abhangigkeit von
der Große der Simulationsbox. Die Cutoff-Energie betragt in beiden Fallen 436.8 eV,
was einem im Programm festgelegten Default-Wert entspricht. Rechte Seite: Bindungs-
energien von NH3 und SiH4 bei verschiedenen Cutoff-Energien. Die Große der Simu-
lationsbox betragt jeweils 10 A. Die experimentellen Werte sind zum Vergleich einge-
zeichnet, sofern sie im betrachteten Energieintervall liegen.
Einfluß des Basissatzes kommt aufgrund der periodischen Randbedingungen die
Auswirkung der endlichen Große der Simulationsbox hinzu. Fur die Molekule NH3
und SiH4 ist der Einfluß dieser beiden Parameter auf die mit Vasp berechnete
Bindungsenergie in Abbildung 4.9 exemplarisch dargestellt. Die Cutoff-Energie
wurde im Falle der Variation der Große der Simulationsbox konstant gehalten
und betrug 436.8 eV. Dieser Wert ist der vom Programm bei Rechnungen hoher
Genauigkeit vorgeschlagene Standardwert fur Systeme, die das Element Wasser-
stoff enthalten. Es ist zu erkennen, daß eine Vergroßerung der Superzelle auf
mehr als 10 A Kantenlange keine nennenswerte Veranderung der Ergebnisse mit
sich bringt. Deshalb wurde eine Zellengoße von 10 × 10 × 10 A3 fur die Unter-
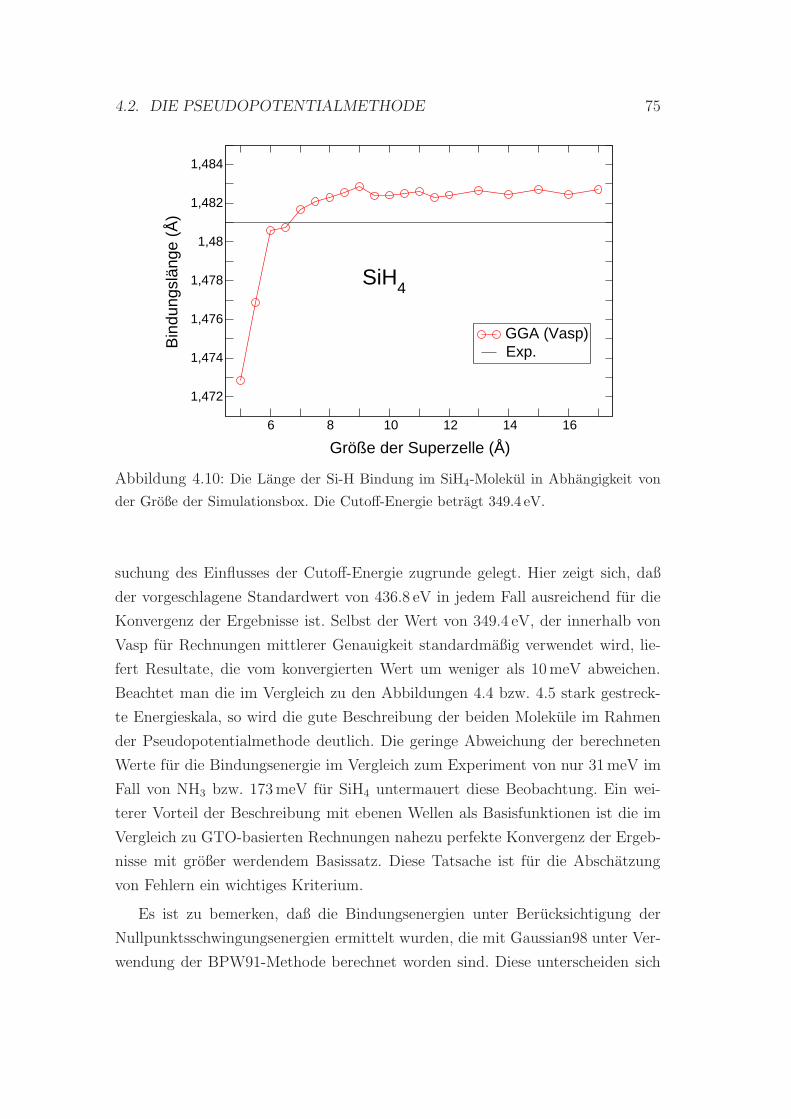
4.2. DIE PSEUDOPOTENTIALMETHODE 75
6 8 10 12 14 16
Größe der Superzelle (Å)
1,472
1,474
1,476
1,478
1,48
1,482
1,484B
indu
ngsl
änge
(Å
)
GGA (Vasp)Exp.
SiH4
Abbildung 4.10: Die Lange der Si-H Bindung im SiH4-Molekul in Abhangigkeit von
der Große der Simulationsbox. Die Cutoff-Energie betragt 349.4 eV.
suchung des Einflusses der Cutoff-Energie zugrunde gelegt. Hier zeigt sich, daß
der vorgeschlagene Standardwert von 436.8 eV in jedem Fall ausreichend fur die
Konvergenz der Ergebnisse ist. Selbst der Wert von 349.4 eV, der innerhalb von
Vasp fur Rechnungen mittlerer Genauigkeit standardmaßig verwendet wird, lie-
fert Resultate, die vom konvergierten Wert um weniger als 10 meV abweichen.
Beachtet man die im Vergleich zu den Abbildungen 4.4 bzw. 4.5 stark gestreck-
te Energieskala, so wird die gute Beschreibung der beiden Molekule im Rahmen
der Pseudopotentialmethode deutlich. Die geringe Abweichung der berechneten
Werte fur die Bindungsenergie im Vergleich zum Experiment von nur 31 meV im
Fall von NH3 bzw. 173 meV fur SiH4 untermauert diese Beobachtung. Ein wei-
terer Vorteil der Beschreibung mit ebenen Wellen als Basisfunktionen ist die im
Vergleich zu GTO-basierten Rechnungen nahezu perfekte Konvergenz der Ergeb-
nisse mit großer werdendem Basissatz. Diese Tatsache ist fur die Abschatzung
von Fehlern ein wichtiges Kriterium.
Es ist zu bemerken, daß die Bindungsenergien unter Berucksichtigung der
Nullpunktsschwingungsenergien ermittelt wurden, die mit Gaussian98 unter Ver-
wendung der BPW91-Methode berechnet worden sind. Diese unterscheiden sich

76 KAPITEL 4. ERGEBNISSE
von Basissatz zu Basissatz nur sehr wenig, so daß ein Einfluß durch diesen Faktor
ausgeschlossen werden kann.
Die endliche Große der Simulationsbox hat aufgrund der zuvor getroffe-
nen Feststellungen den starksten Einfluß auf die Resultate. Aus diesem Grund
wird bei der Untersuchung der Molekulgeometrie im Folgenden allein dieser
Faktor berucksichtigt. In Abbildung 4.10 ist die Bindungslange im Silanmo-
lekul in Abhangigkeit von der Große der Superzelle bei konstanter Cutoff-
Energie von 436.8 eVdargestellt. Wiederum wird deutlich, daß eine Zellengroße
von 10 × 10 × 10 A3 ausreichend fur die Beschreibung des Molekuls ist. Die Bin-
dungslange weicht vom experimentellen Wert um weniger als 0.003 A ab, die
Pseudopotentialmethode liefert somit ein Ergebnis, das im Vergleich zur entspre-
chenden Full-Potential-Rechnung mit Gaussian98 (vgl. Abbildung 4.7) wesentlich
besser und mit dem Resultat der B3LYP-Methode vergleichbar ist.
Als Fazit laßt sich festhalten, daß die Pseudopotentialmethode unter Verwen-
dung der Gradientenmethode hervorragende Ergebnisse in Bezug auf Bindungs-
energien und Molekulgeometrien liefert, sofern die Simulationsbox genugend groß
ist. Der Einfluß des Energie-Cutoffs ist in dieser Hinsicht vergleichsweise gering,
die vorgegebenen Standardwerte reichen fur eine gute Beschreibung der Molekule
aus.

4.3. POTENTIALFLACHEN 77
4.3 Potentialflachen
Die in dieser Arbeit behandelten Systeme beinhalten die Elemente Wasserstoff,
Kohlenstoff, Silizium, Chlor, Argon und Titan. Samtliche Bilder der raumlichen
Anordnung von Atomen, seien es Energieminima, Ubergangszustande oder Mo-
C: Z=61s
H: Z=1 Si: Z=14
Ti: Z=2222
Ar: Z=183p
Cl: Z=17
3p2p22s
3p
3s 2
23s23s
1
5 6 3d4s
42
Abbildung 4.11: Graphische Darstellung der beteiligten Elemente
mentaufnahmen aus Molekulardynamik-Simulationen wurden mit Hilfe des Pro-
gramms Molden2 erstellt. Dabei wurden die in Abbildung 4.11 dargestellten Sym-
bole fur die einzelnen Atomsorten verwendet.
Im folgenden werden vier Reaktionen untersucht, deren Gemeinsamkeit in
der Tatsache liegt, daß ein Reaktionspartner das Wasserstoffradikal ist. Sie un-
terscheiden sich jedoch in der Art des zweiten Reaktanden, was sich in der un-
terschiedlichen Komplexitat der entsprechenden Potentialflachen außert. Die Be-
rechnungen der Energieflachen wurden sowohl mit Hilfe von Gaussian98, als auch
mit Vasp durchgefuhrt.
2Naheres auf http://www.molden.org

78 KAPITEL 4. ERGEBNISSE
4.3.1 Die Reaktion H2 + H ↔ H + H2
Die einfachste Reaktion, an der mehr als zwei Atome beteiligt sind, ist die Reakti-
on H2+H → H+H2. Sie lauft unter zwischenzeitlicher Bildung eines H3-Molekuls
ab, welches aber nicht stabil ist. Die experimentelle Energiebarriere [54] belauft
sich auf 421 meV.
Im allgemeinsten Fall hat die raumliche Anordnung der drei Wasserstoffatome
Cs Symmetrie Die drei Atome liegen in einer Ebene, die Zahl der Freiheitsgrade
belauft sich auf drei.
r1
r2α
Abbildung 4.12: Anordnung der Atome im H3-Molekul in Cs Symmetrie.
Aufgrund der Symmetrie der Reaktion in Bezug auf die Edukte und Produkte
liegt die Annahme nahe, daß der Ubergangszustand einer symmetrischen Anord-
nung der Atome entspricht. Im Falle von α = 180 ◦ ist das System linear; die
entsprechende, unter Verwendung von B3LYP/6-31G** erstellte Potentialflache
ist in Abbildung 4.13 dargestellt. 4.14 in Abhangigkeit vom Winkel α dargestellt.
Dazu wurden die Abstande r1 und r2 auf einem Raster zwischen 0.6 A und 2.0 A
variiert und die entsprechende Gesamtenergie berechnet. Die Daten wurden dann
mit Hilfe von Tschebyschew-Polynomen 12. Grades approximiert und auf einem
feineren Raster ausgewertet, woraus dann das Hohenliniendiagramm entstanden
ist. In die Berechnung der PES wurden nur Anordnungen miteinbezogen, bei
denen kein Abstand großer als 2 A war. Denn bei großeren Abstanden traten
Diskontinuitaten in der Energiekurve auf, die auf das Umklappen einzelner Spins
zuruckgefuhrt werden konnten.
Die Existenz des Ubergangszustandes ist in Abbildung 4.13 gut zu erkennen.
Der Abstand zweier Linien entspricht einer Energiedifferenz von 50 meV. Die
einseitig geoffneten Aquipotentiallinien deuten auf einen Sattelpunkt hin. Die
Anwendung eines im Programm implementierten Algorithmus zur Lokalisierung
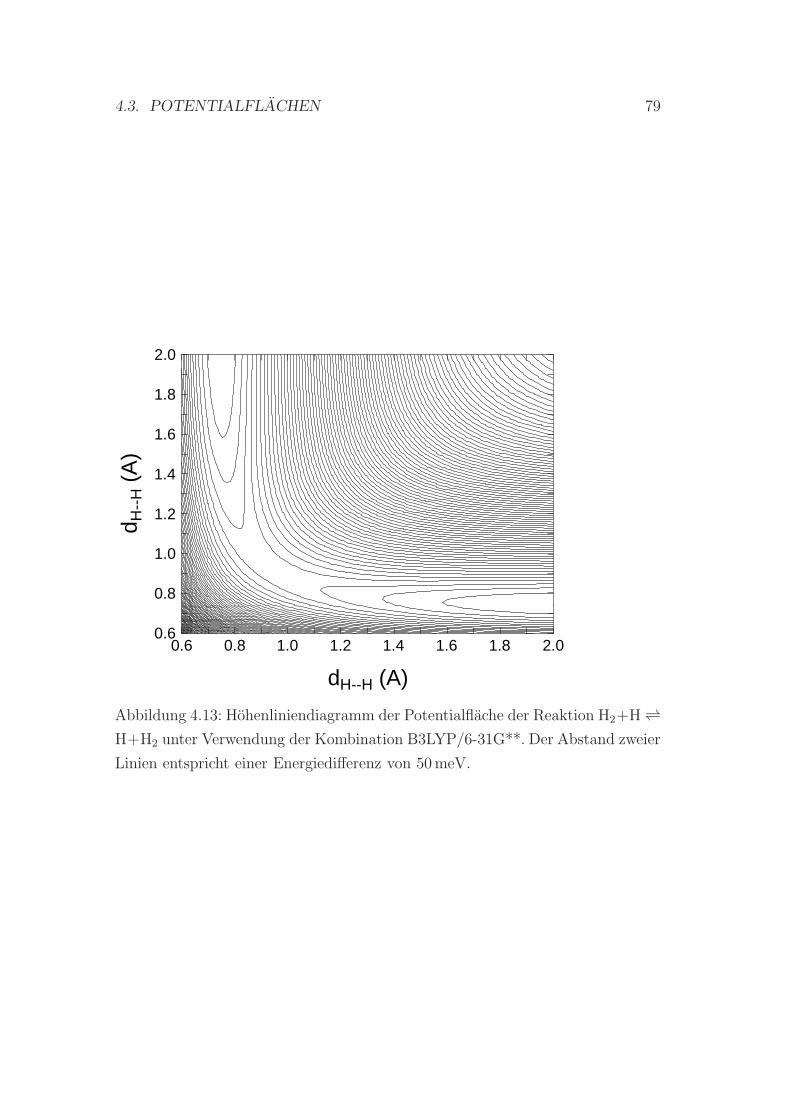
4.3. POTENTIALFLACHEN 79
0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0
dH--H (A)
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
d H--
H(A
)
Abbildung 4.13: Hohenliniendiagramm der Potentialflache der Reaktion H2+H ⇀↽
H+H2 unter Verwendung der Kombination B3LYP/6-31G**. Der Abstand zweier
Linien entspricht einer Energiedifferenz von 50 meV.
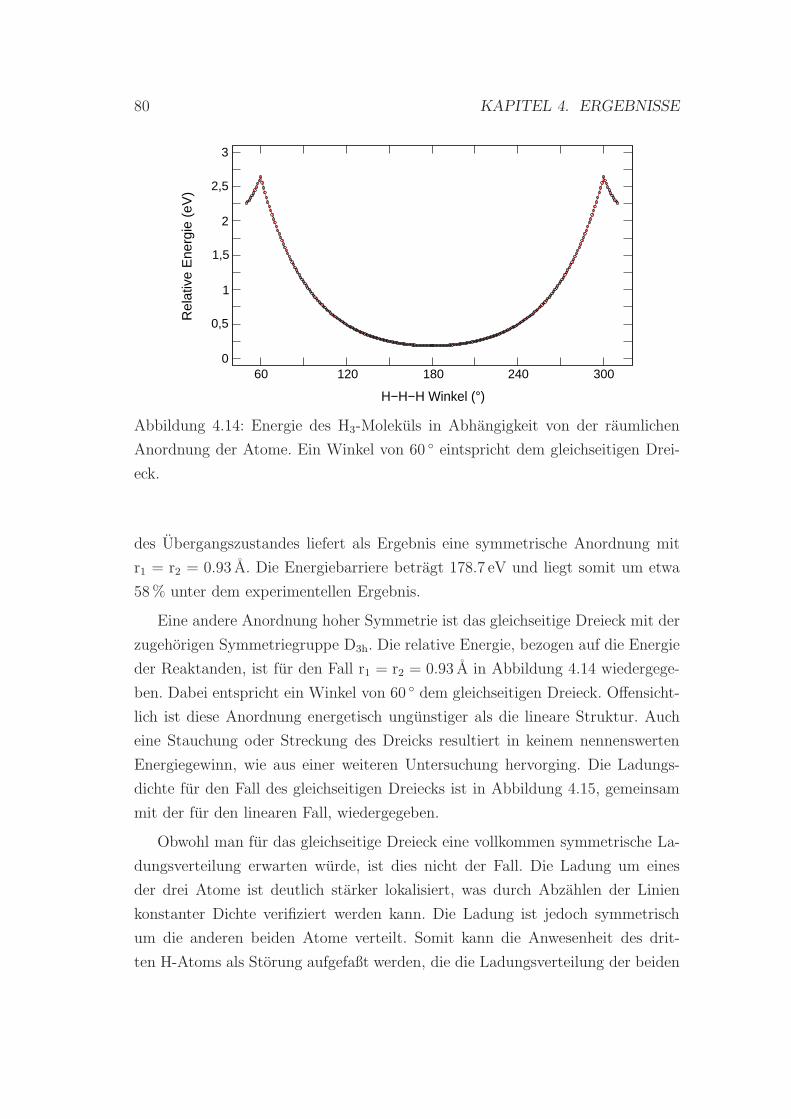
80 KAPITEL 4. ERGEBNISSE
60 120 180 240 300
H−H−H Winkel (°)
0
0,5
1
1,5
2
2,5
3
Rel
ativ
e E
nerg
ie (
eV)
Abbildung 4.14: Energie des H3-Molekuls in Abhangigkeit von der raumlichen
Anordnung der Atome. Ein Winkel von 60 ◦ eintspricht dem gleichseitigen Drei-
eck.
des Ubergangszustandes liefert als Ergebnis eine symmetrische Anordnung mit
r1 = r2 = 0.93 A. Die Energiebarriere betragt 178.7 eV und liegt somit um etwa
58 % unter dem experimentellen Ergebnis.
Eine andere Anordnung hoher Symmetrie ist das gleichseitige Dreieck mit der
zugehorigen Symmetriegruppe D3h. Die relative Energie, bezogen auf die Energie
der Reaktanden, ist fur den Fall r1 = r2 = 0.93 A in Abbildung 4.14 wiedergege-
ben. Dabei entspricht ein Winkel von 60 ◦ dem gleichseitigen Dreieck. Offensicht-
lich ist diese Anordnung energetisch ungunstiger als die lineare Struktur. Auch
eine Stauchung oder Streckung des Dreicks resultiert in keinem nennenswerten
Energiegewinn, wie aus einer weiteren Untersuchung hervorging. Die Ladungs-
dichte fur den Fall des gleichseitigen Dreiecks ist in Abbildung 4.15, gemeinsam
mit der fur den linearen Fall, wiedergegeben.
Obwohl man fur das gleichseitige Dreieck eine vollkommen symmetrische La-
dungsverteilung erwarten wurde, ist dies nicht der Fall. Die Ladung um eines
der drei Atome ist deutlich starker lokalisiert, was durch Abzahlen der Linien
konstanter Dichte verifiziert werden kann. Die Ladung ist jedoch symmetrisch
um die anderen beiden Atome verteilt. Somit kann die Anwesenheit des drit-
ten H-Atoms als Storung aufgefaßt werden, die die Ladungsverteilung der beiden

4.3. POTENTIALFLACHEN 81
Abbildung 4.15: Ladungsverteilung des H3-Molekuls in Abhangigkeit von der
raumlichen Anordnung der Atome.
anderen Atome beeinflußt und die gesamte potentielle Energie erhoht.
Im Gegensatz dazu weist die Ladungsverteilung im Falle des linearen H3-
Molekuls die gleiche Symmetrie wie die Anordnung der Atome im Molekul auf.
Die Ladung ist deutlich dichter verteilt als im Fall der dreizahligen Symmetrie.
Dies resultiert in der vergleichsweise niedrigeren Gesamtenergie dieser Konfigu-
ration.
Der Ubergangszustand laßt sich somit als eine bestimmte lineare Anordnung
der Atome mit gleichen interatomaren Abstanden identifizieren.
r r
Um den Einfluß des verwendeten Basissatzes auf diesen Abstand zu untersuchen,
wurde die Geometrie des H3-Molekuls unter der Nebenbedingungung r1 = r2 =
r in verschiedenen Basissatzen optimiert. Das Ergebnis ist in Abbildung 4.16
wiedergegeben.
Eine Vergroßerung der Einteilchenbasis bringt somit nur eine unwesentliche
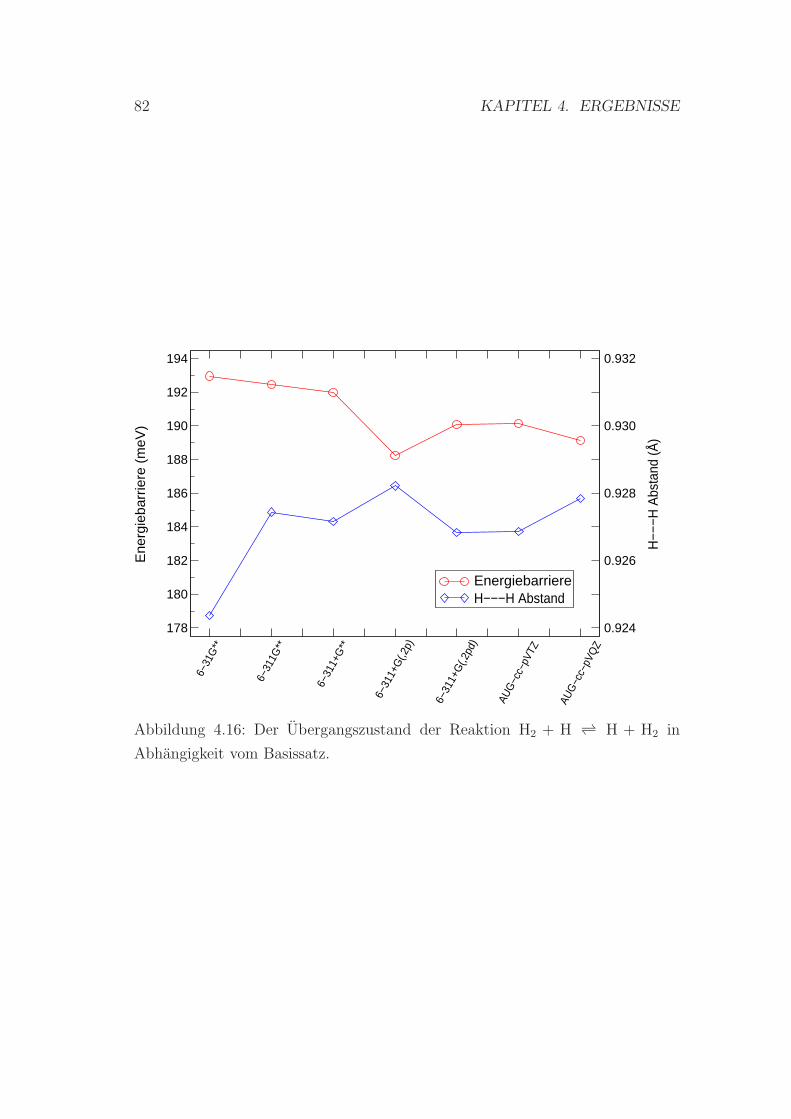
82 KAPITEL 4. ERGEBNISSE
6−31
G**
6−31
1G**
6−31
1+G
**
6−31
1+G
(,2p)
6−31
1+G
(,2pd
)
AUG
−cc−
pVTZ
AUG
−cc−
pVQ
Z
178
180
182
184
186
188
190
192
194
Ene
rgie
barr
iere
(m
eV)
0.924
0.926
0.928
0.930
0.932
H−−
−H A
bsta
nd (Å
)
EnergiebarriereH−−−H Abstand
Abbildung 4.16: Der Ubergangszustand der Reaktion H2 + H ⇀↽ H + H2 in
Abhangigkeit vom Basissatz.
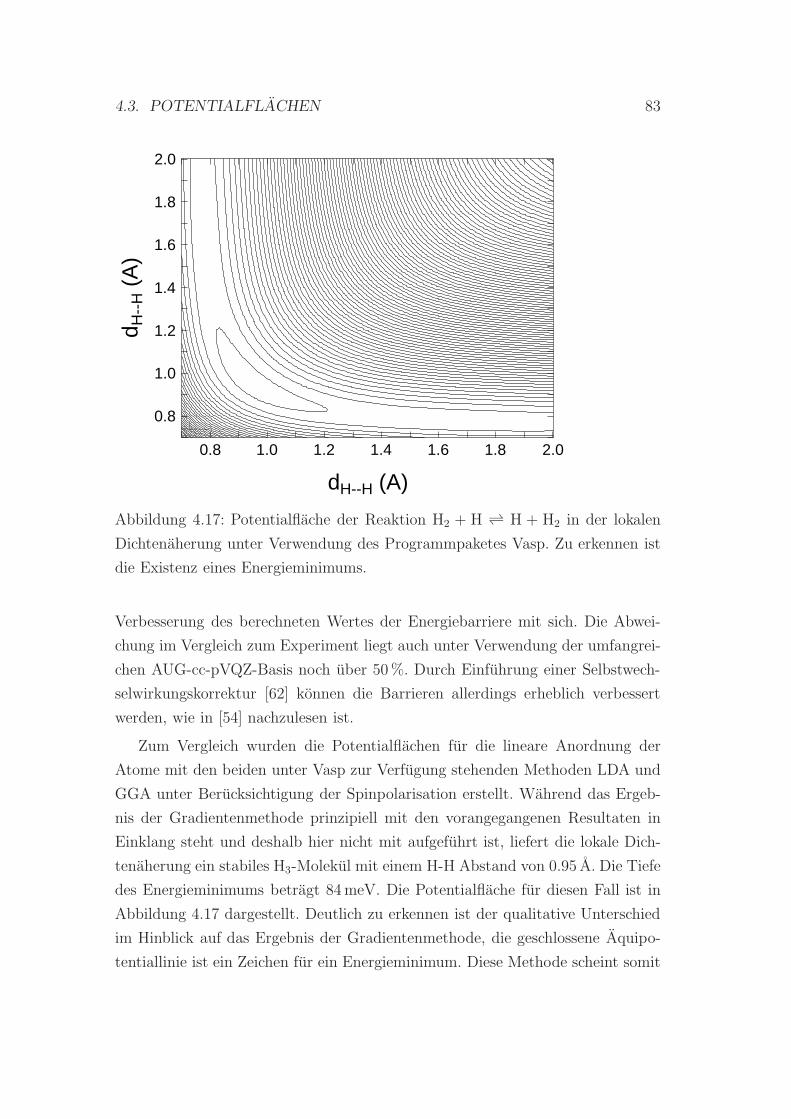
4.3. POTENTIALFLACHEN 83
0.8 1.0 1.2 1.4 1.6 1.8 2.0
dH--H (A)
0.8
1.0
1.2
1.4
1.6
1.8
2.0
d H--
H(A
)
Abbildung 4.17: Potentialflache der Reaktion H2 + H ⇀↽ H + H2 in der lokalen
Dichtenaherung unter Verwendung des Programmpaketes Vasp. Zu erkennen ist
die Existenz eines Energieminimums.
Verbesserung des berechneten Wertes der Energiebarriere mit sich. Die Abwei-
chung im Vergleich zum Experiment liegt auch unter Verwendung der umfangrei-
chen AUG-cc-pVQZ-Basis noch uber 50 %. Durch Einfuhrung einer Selbstwech-
selwirkungskorrektur [62] konnen die Barrieren allerdings erheblich verbessert
werden, wie in [54] nachzulesen ist.
Zum Vergleich wurden die Potentialflachen fur die lineare Anordnung der
Atome mit den beiden unter Vasp zur Verfugung stehenden Methoden LDA und
GGA unter Berucksichtigung der Spinpolarisation erstellt. Wahrend das Ergeb-
nis der Gradientenmethode prinzipiell mit den vorangegangenen Resultaten in
Einklang steht und deshalb hier nicht mit aufgefuhrt ist, liefert die lokale Dich-
tenaherung ein stabiles H3-Molekul mit einem H-H Abstand von 0.95 A. Die Tiefe
des Energieminimums betragt 84 meV. Die Potentialflache fur diesen Fall ist in
Abbildung 4.17 dargestellt. Deutlich zu erkennen ist der qualitative Unterschied
im Hinblick auf das Ergebnis der Gradientenmethode, die geschlossene Aquipo-
tentiallinie ist ein Zeichen fur ein Energieminimum. Diese Methode scheint somit
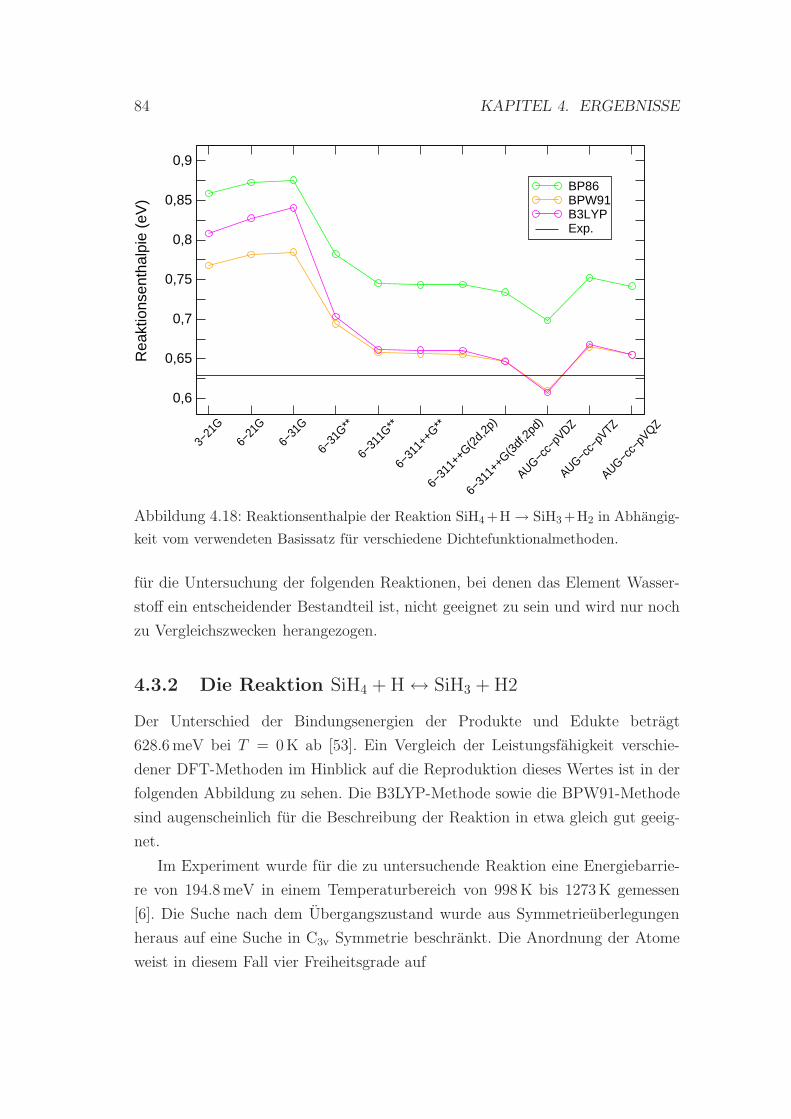
84 KAPITEL 4. ERGEBNISSE
3−21
G
6−21
G
6−31
G
6−31
G**
6−31
1G**
6−31
1++G
**
6−31
1++G
(2d,2
p)
6−31
1++G
(3df,
2pd)
AUG−cc−
pVDZ
AUG−cc−
pVTZ
AUG−cc−
pVQZ
0,6
0,65
0,7
0,75
0,8
0,85
0,9R
eakt
ions
enth
alpi
e (e
V)
BP86BPW91B3LYPExp.
Abbildung 4.18: Reaktionsenthalpie der Reaktion SiH4 +H → SiH3 +H2 in Abhangig-
keit vom verwendeten Basissatz fur verschiedene Dichtefunktionalmethoden.
fur die Untersuchung der folgenden Reaktionen, bei denen das Element Wasser-
stoff ein entscheidender Bestandteil ist, nicht geeignet zu sein und wird nur noch
zu Vergleichszwecken herangezogen.
4.3.2 Die Reaktion SiH4 + H ↔ SiH3 + H2
Der Unterschied der Bindungsenergien der Produkte und Edukte betragt
628.6 meV bei T = 0K ab [53]. Ein Vergleich der Leistungsfahigkeit verschie-
dener DFT-Methoden im Hinblick auf die Reproduktion dieses Wertes ist in der
folgenden Abbildung zu sehen. Die B3LYP-Methode sowie die BPW91-Methode
sind augenscheinlich fur die Beschreibung der Reaktion in etwa gleich gut geeig-
net.
Im Experiment wurde fur die zu untersuchende Reaktion eine Energiebarrie-
re von 194.8 meV in einem Temperaturbereich von 998 K bis 1273 K gemessen
[6]. Die Suche nach dem Ubergangszustand wurde aus Symmetrieuberlegungen
heraus auf eine Suche in C3v Symmetrie beschrankt. Die Anordnung der Atome
weist in diesem Fall vier Freiheitsgrade auf
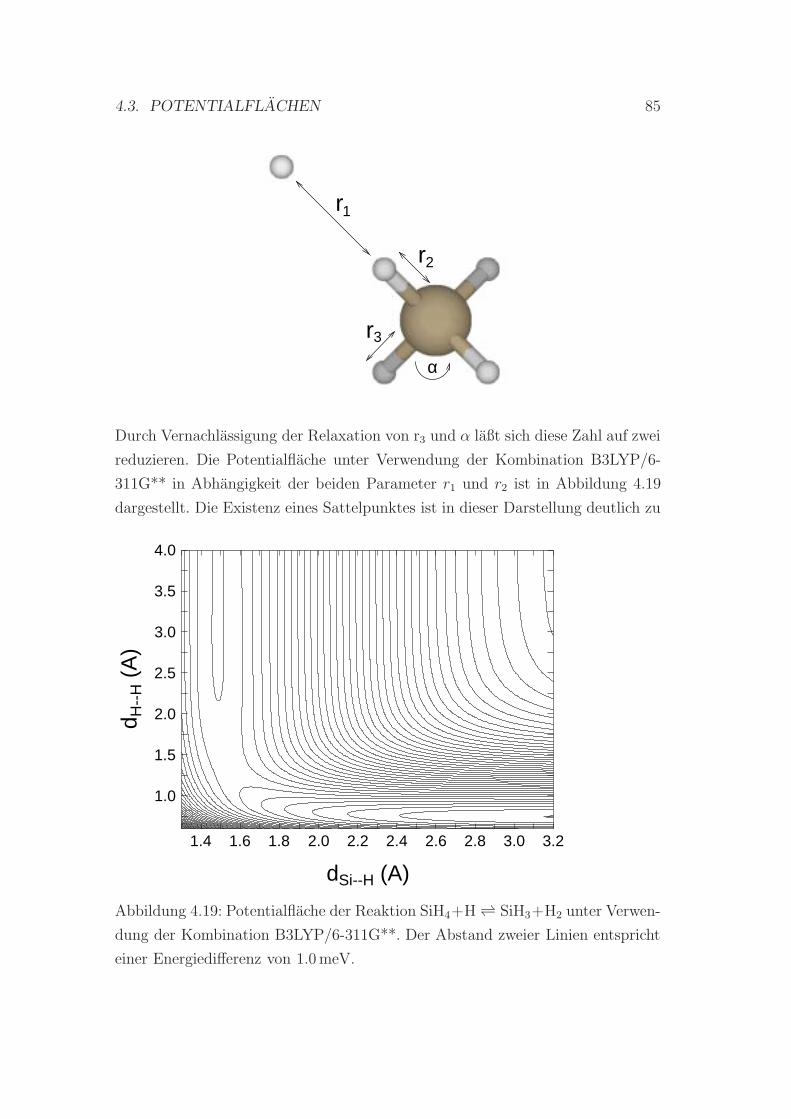
4.3. POTENTIALFLACHEN 85
r
3
1
r2
r
α
Durch Vernachlassigung der Relaxation von r3 und α laßt sich diese Zahl auf zwei
reduzieren. Die Potentialflache unter Verwendung der Kombination B3LYP/6-
311G** in Abhangigkeit der beiden Parameter r1 und r2 ist in Abbildung 4.19
dargestellt. Die Existenz eines Sattelpunktes ist in dieser Darstellung deutlich zu
1.4 1.6 1.8 2.0 2.2 2.4 2.6 2.8 3.0 3.2
dSi--H (A)
1.0
1.5
2.0
2.5
3.0
3.5
4.0
d H--
H(A
)
Abbildung 4.19: Potentialflache der Reaktion SiH4+H ⇀↽ SiH3+H2 unter Verwen-
dung der Kombination B3LYP/6-311G**. Der Abstand zweier Linien entspricht
einer Energiedifferenz von 1.0 meV.

86 KAPITEL 4. ERGEBNISSE
erkennen. Die entsprechende Si-H Bindung ist im Vergleich zum Gleichgewichts-
abstand von 1.48 A auf 1.54 A verlangert, der Abstand der beiden Wasserstoffa-
tome betragt 1.38 A.
Die Anwendung eines im Programm Gaussian98 implementierten Algorithmus
zur Suche von Sattelpunkten fuhrte zu dem Ergebnis, daß der Effekt der Rela-
xation der beiden Freiheitsgrade r3 und α auf die Lage des Ubergangszustandes
vernachlassigt werden kann. Die Abhangigkeit der Energiebarriere sowie der Geo-
metrie des Ubergangszustandes vom verwendeten Basissatz ist in Abbildung 4.20
wiedergegeben. Es zeigt sich ein ahnliches Bild, wie bei der zuvor behandelten
Reaktion H2 + H → H + H2. Die Abweichung der Energiebarriere vom experi-
mentellen Wert von 194.8 meV kann durch Vergroßern der Einteilchenbasis nicht
wesentlich verbessert werden.
Der Verlauf der Reaktion in Abhangigkeit der Parameter r1 und r2 wurde
mit den unter Vasp zur Verfugung stehenden Methoden LDA und GGA simu-
liert. Das Ergebnis ist in Abbildung 4.21 dargestellt. Fur verschiedene Werte des
Si-H Abstandes wurde der H-H Abstand variiert und die entsprechende Gesam-
tenergie berechnet. Man erkennt beim Betrachten des oberen Teils Abbildung
4.21, in dem die Ergebnisse der spinpolarisierten GGA-Rechnung gezeigt sind,
die Existenz einer Energiebarriere. Sie stellt sich als Ubergang von der schwarzen
Kurve, die einem Si-H Abstand von 1.48 A entspricht, zur roten, grunen, blauen
und schließlich violetten Kurve dar, der jeweils bei verschiedenen Werten fur den
H-H Abstand von statten geht. Der Grund fur die Energiebarriere liegt in der
Tatsache, daß die ersten Ubergange (schwarz-rot, rot-grun, usw.) bei positiver
Steigung der Kurven stattfinden, fur großere Si-H Abstande schneiden sich die
Kurven mit negativer Steigung.
Im Gegensatz dazu haben die Kurven im unteren Teil der Abbildung 4.21,
welche die Ergebnisse der LDA-Methode zeigt, durchweg negative Steigungen in
ihren gegenseitigen Schnittpunkten. Es liegt keine Energiebarriere vor. Das glei-
che Ergebnis liefert einer nicht-spinpolarisierten GGA-Rechnung, das hier nicht
weiter aufgefuhrt ist. Die Berucksichtigung des Spins ist somit von Bedeutung
fur eine angemessene Beschreibung der Potentialflache.
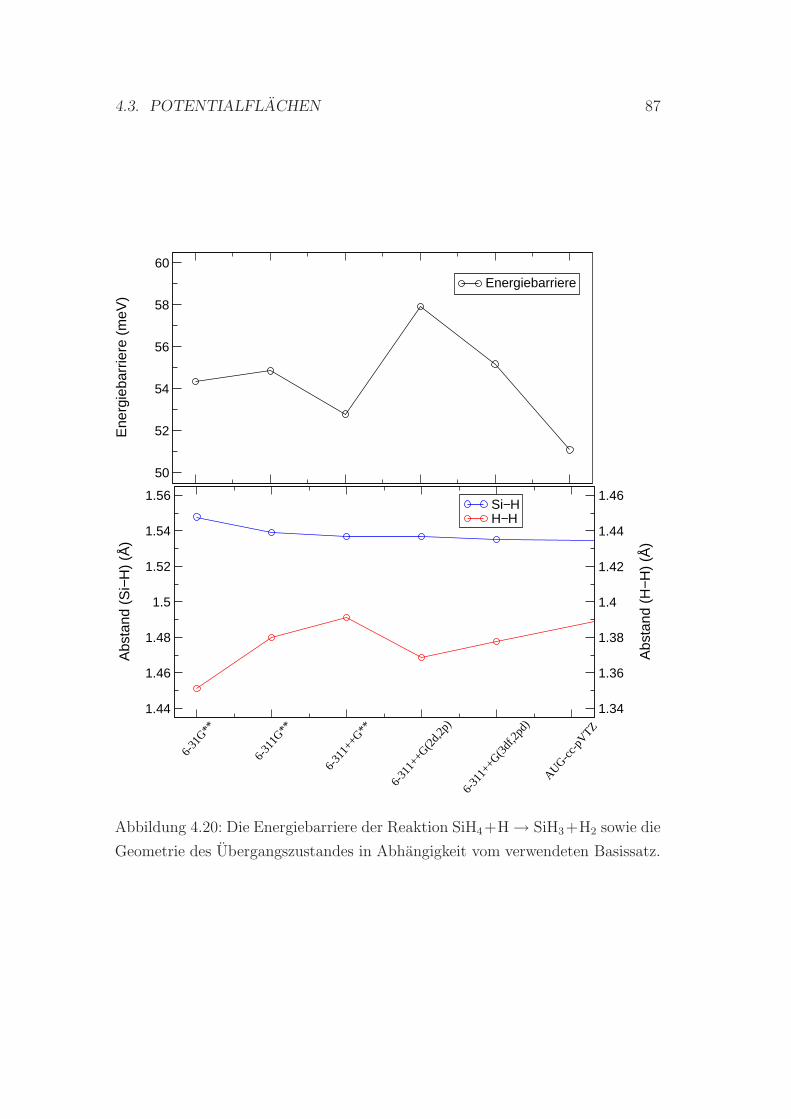
4.3. POTENTIALFLACHEN 87
50
52
54
56
58
60
Ene
rgie
barr
iere
(m
eV)
Energiebarriere
6-31
G**
6-31
1G**
6-31
1++G
**
6-31
1++G
(2d,
2p)
6-31
1++G
(3df
,2pd
)
AUG-c
c-pV
TZ
1.44
1.46
1.48
1.5
1.52
1.54
1.56
Abs
tand
(S
i−H
) (Å
)
1.34
1.36
1.38
1.4
1.42
1.44
1.46
Abs
tand
(H
−H)
(Å)
Si−HH−H
Abbildung 4.20: Die Energiebarriere der Reaktion SiH4+H → SiH3+H2 sowie die
Geometrie des Ubergangszustandes in Abhangigkeit vom verwendeten Basissatz.
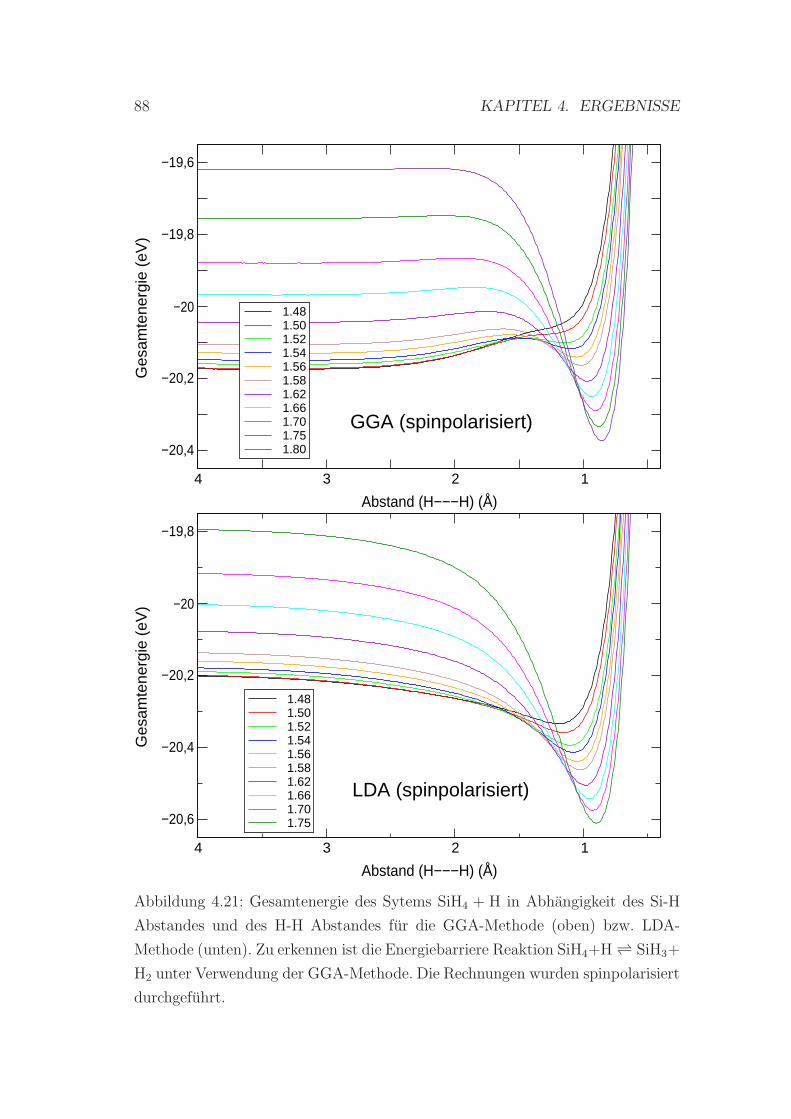
88 KAPITEL 4. ERGEBNISSE
1234
Abstand (H−−−H) (Å)
−20,4
−20,2
−20
−19,8
−19,6
Ges
amte
nerg
ie (
eV)
1.481.501.521.541.561.581.621.661.701.751.80
GGA (spinpolarisiert)
1234
Abstand (H−−−H) (Å)
−20,6
−20,4
−20,2
−20
−19,8
Ges
amte
nerg
ie (
eV)
1.481.501.521.541.561.581.621.661.701.75
LDA (spinpolarisiert)
Abbildung 4.21: Gesamtenergie des Sytems SiH4 + H in Abhangigkeit des Si-H
Abstandes und des H-H Abstandes fur die GGA-Methode (oben) bzw. LDA-
Methode (unten). Zu erkennen ist die Energiebarriere Reaktion SiH4+H ⇀↽ SiH3+
H2 unter Verwendung der GGA-Methode. Die Rechnungen wurden spinpolarisiert
durchgefuhrt.
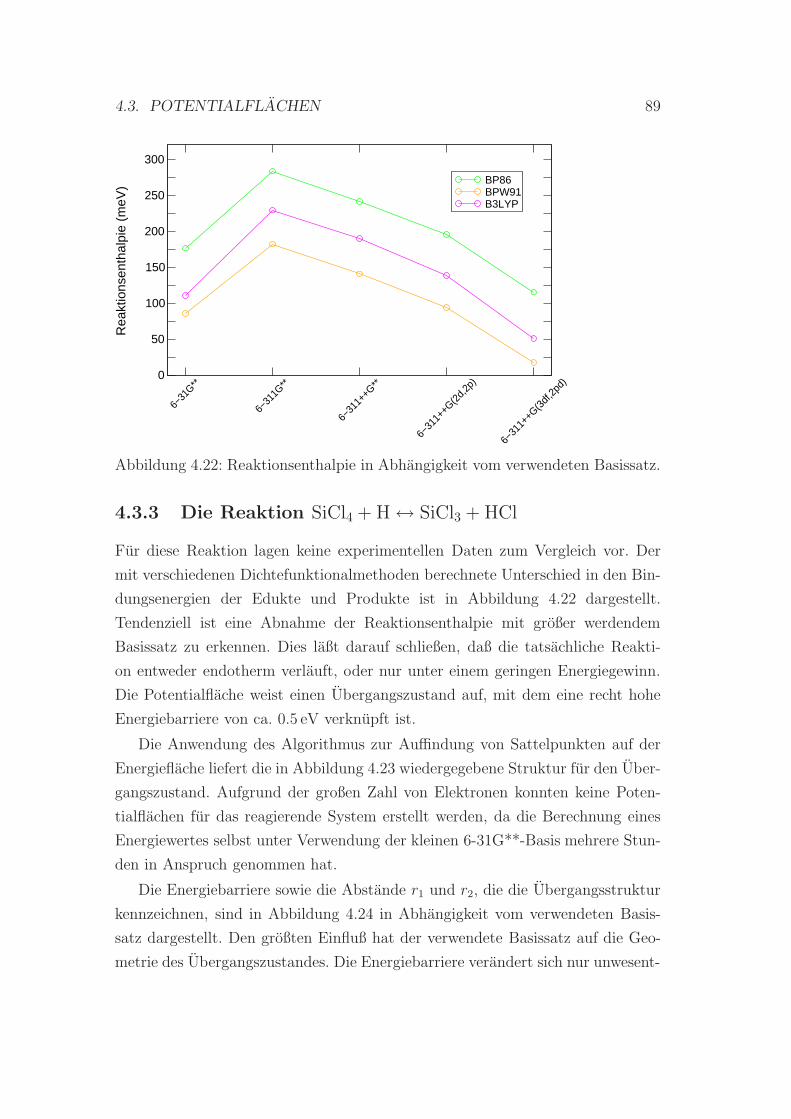
4.3. POTENTIALFLACHEN 89
6−31
G**
6−31
1G**
6−31
1++G
**
6−31
1++G
(2d,2
p)
6−31
1++G
(3df,
2pd)
0
50
100
150
200
250
300
Rea
ktio
nsen
thal
pie
(meV
) BP86BPW91B3LYP
Abbildung 4.22: Reaktionsenthalpie in Abhangigkeit vom verwendeten Basissatz.
4.3.3 Die Reaktion SiCl4 + H ↔ SiCl3 + HCl
Fur diese Reaktion lagen keine experimentellen Daten zum Vergleich vor. Der
mit verschiedenen Dichtefunktionalmethoden berechnete Unterschied in den Bin-
dungsenergien der Edukte und Produkte ist in Abbildung 4.22 dargestellt.
Tendenziell ist eine Abnahme der Reaktionsenthalpie mit großer werdendem
Basissatz zu erkennen. Dies laßt darauf schließen, daß die tatsachliche Reakti-
on entweder endotherm verlauft, oder nur unter einem geringen Energiegewinn.
Die Potentialflache weist einen Ubergangszustand auf, mit dem eine recht hohe
Energiebarriere von ca. 0.5 eV verknupft ist.
Die Anwendung des Algorithmus zur Auffindung von Sattelpunkten auf der
Energieflache liefert die in Abbildung 4.23 wiedergegebene Struktur fur den Uber-
gangszustand. Aufgrund der großen Zahl von Elektronen konnten keine Poten-
tialflachen fur das reagierende System erstellt werden, da die Berechnung eines
Energiewertes selbst unter Verwendung der kleinen 6-31G**-Basis mehrere Stun-
den in Anspruch genommen hat.
Die Energiebarriere sowie die Abstande r1 und r2, die die Ubergangsstruktur
kennzeichnen, sind in Abbildung 4.24 in Abhangigkeit vom verwendeten Basis-
satz dargestellt. Den großten Einfluß hat der verwendete Basissatz auf die Geo-
metrie des Ubergangszustandes. Die Energiebarriere verandert sich nur unwesent-

90 KAPITEL 4. ERGEBNISSE
r2
3r
1r
Abbildung 4.23: Anordnung der Atome des Systems SiCl4 + H im Ubergangszu-
stand.
lich mit großer werdeneder Einteilchenbasis. Legt man eine ahnliche Abweichung
der Ergebnisse von experimentellen Werten zugrunde, wie sie fur die Reaktion
SiH4 +H ⇀↽ SiH3 +HCl der Fall war, so laßt sich die tatsachliche Energiebarriere
zu mehr als 1 eV abschatzen. Dies bestatigt die Beobachtung einer starken Si-Cl-
Bindung im SiCl4-Molekul durch Kunz et al. [7]. In einem Stoßexperiment ermit-
telten sie die Aktivierungsenergie der Reaktion SiCl4 + Ar → SiCl3 + Cl + Ar zu
37760 K bzw. 3.25 eV. Die Dissoziationsenergie der Reaktion SiCl4 → SiCl3 + Cl
berechnet sich unter Verwendung der Kombination B3LYP/6-31G** zu −4.17 eV,
wobei die Nullpunktsschwingungsenergien berucksichtigt wurden. Eine Vergroße-
rung der Basis auf 6-311++G(3df,2pd) resultiert in einem Wert von −4.34 eV. In
diesem Zusammenhang wurden Molekulardynamik-Simulationen durchgefuhrt,
deren Ergebnisse in Kapitel 4.3.5 dargestellt sind.
4.3.4 Die Reaktion TiCl4 + H ↔ TiCl3 + HCl
Eine Berechnung der Bindungsenergien der beteiligten Molekule unter Beruck-
sichtigung der Nullpunktsschwingungsenergien zeigt, daß die Ergebnisse wie-
derum in hohem Maße von der gewahlten Methode abhangig sind (vgl. Abb.
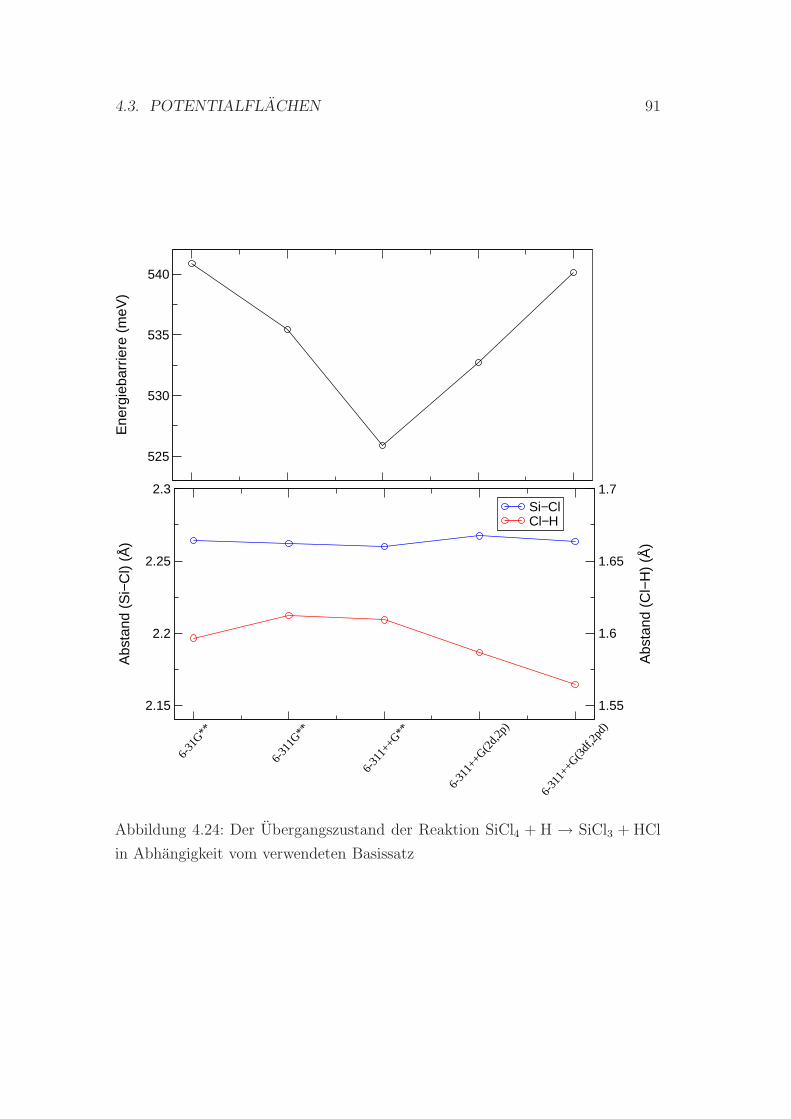
4.3. POTENTIALFLACHEN 91
525
530
535
540
Ene
rgie
barr
iere
(m
eV)
6-31
G**
6-31
1G**
6-31
1++G
**
6-31
1++G
(2d,
2p)
6-31
1++G
(3df
,2pd
)
2.15
2.2
2.25
2.3
Abs
tand
(S
i−C
l) (Å
)
1.55
1.6
1.65
1.7
Abs
tand
(C
l−H
) (Å
)
Si−ClCl−H
Abbildung 4.24: Der Ubergangszustand der Reaktion SiCl4 + H → SiCl3 + HCl
in Abhangigkeit vom verwendeten Basissatz
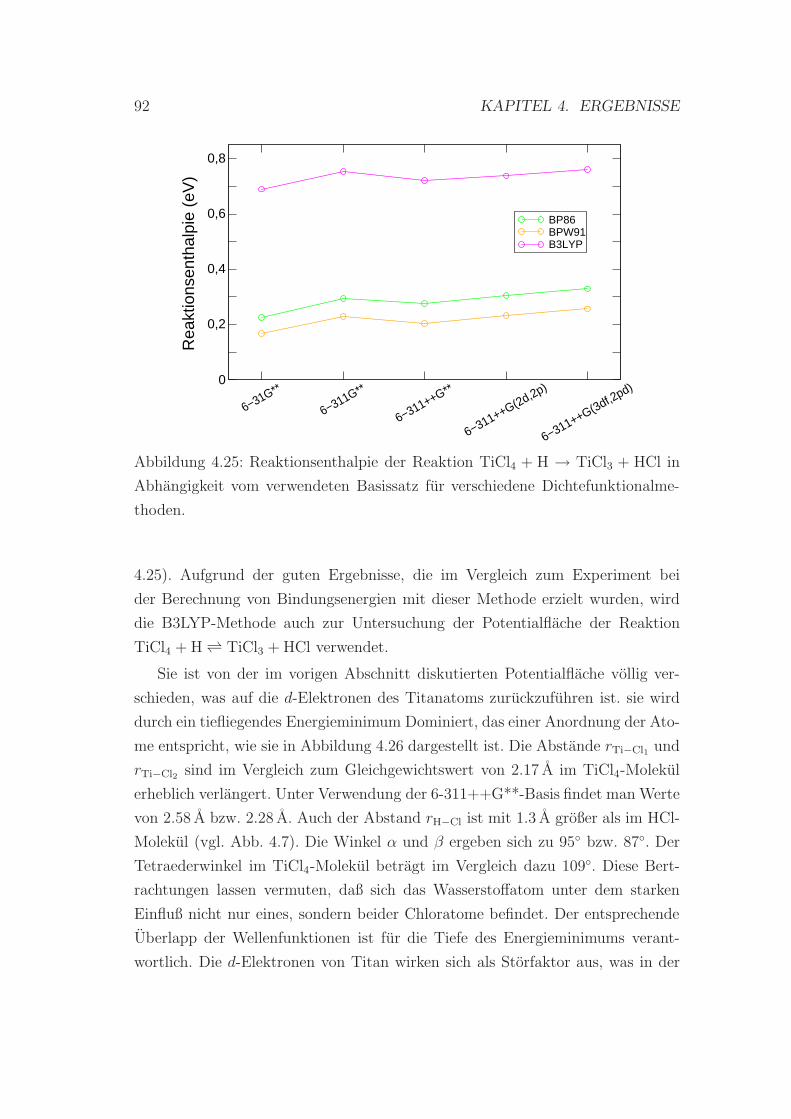
92 KAPITEL 4. ERGEBNISSE
6−31G**
6−311G**
6−311++G**
6−311++G(2d,2p)
6−311++G(3df,2pd)0
0,2
0,4
0,6
0,8
Rea
ktio
nsen
thal
pie
(eV
)
BP86BPW91B3LYP
Abbildung 4.25: Reaktionsenthalpie der Reaktion TiCl4 + H → TiCl3 + HCl in
Abhangigkeit vom verwendeten Basissatz fur verschiedene Dichtefunktionalme-
thoden.
4.25). Aufgrund der guten Ergebnisse, die im Vergleich zum Experiment bei
der Berechnung von Bindungsenergien mit dieser Methode erzielt wurden, wird
die B3LYP-Methode auch zur Untersuchung der Potentialflache der Reaktion
TiCl4 + H ⇀↽ TiCl3 + HCl verwendet.
Sie ist von der im vorigen Abschnitt diskutierten Potentialflache vollig ver-
schieden, was auf die d-Elektronen des Titanatoms zuruckzufuhren ist. sie wird
durch ein tiefliegendes Energieminimum Dominiert, das einer Anordnung der Ato-
me entspricht, wie sie in Abbildung 4.26 dargestellt ist. Die Abstande rTi−Cl1 und
rTi−Cl2 sind im Vergleich zum Gleichgewichtswert von 2.17 A im TiCl4-Molekul
erheblich verlangert. Unter Verwendung der 6-311++G**-Basis findet man Werte
von 2.58 A bzw. 2.28 A. Auch der Abstand rH−Cl ist mit 1.3 A großer als im HCl-
Molekul (vgl. Abb. 4.7). Die Winkel α und β ergeben sich zu 95◦ bzw. 87◦. Der
Tetraederwinkel im TiCl4-Molekul betragt im Vergleich dazu 109◦. Diese Bert-
rachtungen lassen vermuten, daß sich das Wasserstoffatom unter dem starken
Einfluß nicht nur eines, sondern beider Chloratome befindet. Der entsprechende
Uberlapp der Wellenfunktionen ist fur die Tiefe des Energieminimums verant-
wortlich. Die d-Elektronen von Titan wirken sich als Storfaktor aus, was in der

4.3. POTENTIALFLACHEN 93
rTi−Cl
rH−Cl
2
3
4
1
rTi−Cl
1
2
α
β
Abbildung 4.26: Raumliche Anordnung der Atome des Systems TiCl4 + H im
globalen Minimum der potentiellen Energie.
Verlangerung der Ti-Cl Bindungen resultiert.
Die Abhangigkeit der Tiefe dieses Minimums vom verwendeten Basissatz ist in
Abbildung 4.27 dargestellt. Die Einbeziehung von Polarisationsfunktionen hoher-
er Ordnung sowie diffusen Funktionen macht fur die energetischen Verhaltnisse
im Vergleich zu kleinen Basissatzen einen betrachtlichen Unterschied. Aufgrund
der großen Rechenintensitat wurden die folgenden Rechnungen aber nur unter
Verwendung der 6-311G**-Basis durchgefuhrt.
Außerhalb des Energieminimums hat die Potentialflache eine komplizierte
Struktur. Es konnte jedoch weder ein Ubergangszustand zwischen dem Eintritts-
kanal und dem Minimum, noch zwischen dem Minimum und dem Austrittskanal
lokalisiert werden. Eine Untersuchung des Eintrittskanals ließ den Schluß zu, daß
ein eventuell vorhandener Sattelpunkt mit einer Energiebarriere unterhalb von
1 meV verbunden ware.
Im Rahmen der Untersuchung des Austrittskanals wurde ein weiteres Energie-
minimum ausgemacht. Die Anordnung der Atome fur diesen Fall ist in Abbildung
4.28 wiedergegeben. Allerdings ist das Minimum nicht sehr tief (die Gesamtener-
gie liegt nur etwa 15 meV unterhalb der Energie der Produkte) und mag ein
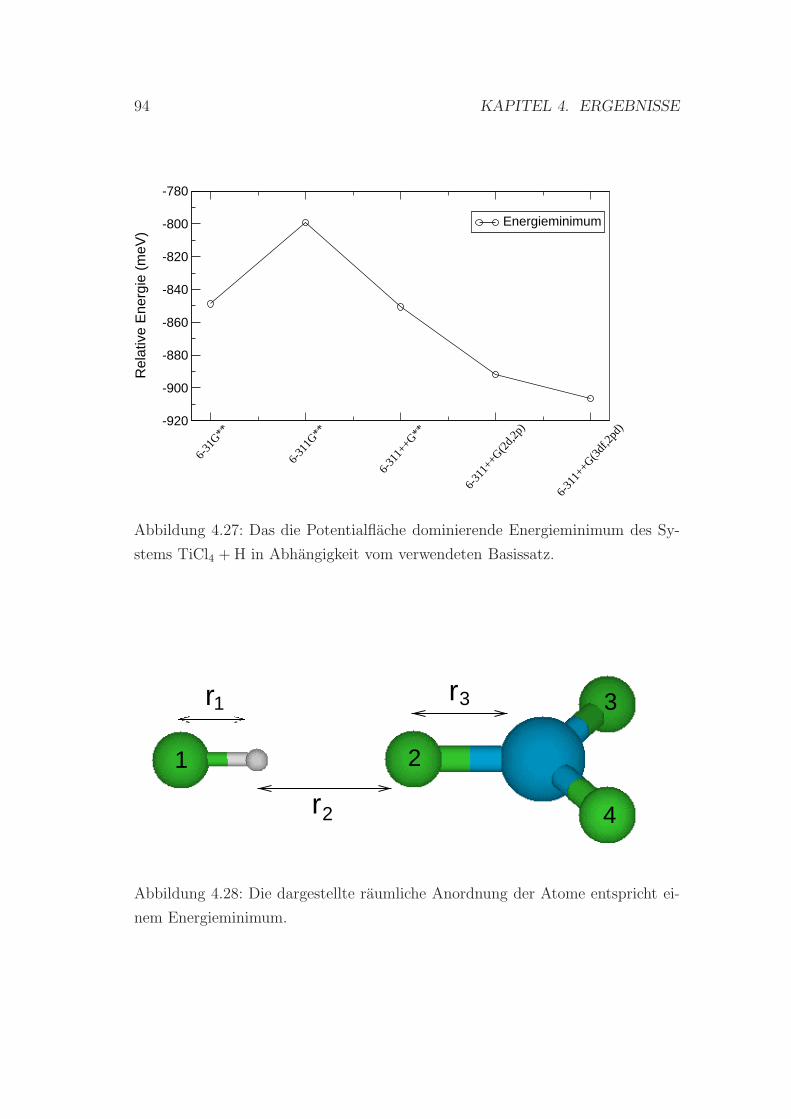
94 KAPITEL 4. ERGEBNISSE
6-31
G**
6-31
1G**
6-31
1++G
**
6-31
1++G
(2d,
2p)
6-31
1++G
(3df
,2pd
)-920
-900
-880
-860
-840
-820
-800
-780
Rel
ativ
e E
nerg
ie (
meV
)
Energieminimum
Abbildung 4.27: Das die Potentialflache dominierende Energieminimum des Sy-
stems TiCl4 + H in Abhangigkeit vom verwendeten Basissatz.
1r
r2
r3
2
3
4
1
Abbildung 4.28: Die dargestellte raumliche Anordnung der Atome entspricht ei-
nem Energieminimum.

4.3. POTENTIALFLACHEN 95
Artefakt der verwendeten Einteilchenbasis sein. Die Anordnung der Atome ist
der Atomkonfiguration der Produkte sehr ahnlich. Die Abstande r3 im TiCl3-
Molekul sind weitestgehend relaxiert, einzig der Abstand r1 ist noch etwas großer
als im Gleichgewichtszustand, was auf die Wechselwirkung des Wasserstoffatoms
mit dem Chloratom uber r2 zuruckgefuhrt werden kann.
Als eine Moglichkeit des Reaktionsverlaufes wird die Rotation des HCl-
Molekuls um das Chloratom mit der Nummer 2 mit gleichzeitiger interner Rota-
tion um den Mittelpunkt der H-Cl-Bindung angesehen. Um dies nachzuprufen,
wurden alle Freiheitsgrade des Systems gleichzeitig zwischen der Position des glo-
balen Minimums sowie dem Minimum im Austrittskanal auf lineare Weise variiert.
Die auf diese Weise gefundene Energiekurve wies ein Maximum etwa auf halbem
Weg zwischen Minimumszustand un Austrittskanal auf. Es konte jedoch kein ent-
sprechender Ubergangszustand gefunden werden. Erst eine Molekulardynamik-
Simulation gab einen Hinweis auf den Reaktionsverlauf (vgl. 4.3.5).
4.3.5 Molekulardynamik-Simulationen
Alle Simulationen wurden bei konstanter Gesamtenergie durchgefuhrt. Die hier
dargestellten Ergebnisse beruhen auf Verwendung der GGA-Methode. Es konn-
ten aber aus Zeitgrunden keine spinpolarisierten Rechnungen gemacht werden,
da dies einen Anstieg der Rechenzeit um den Faktor zehn bedeutet hatte. Der
Zeitschritt zur Integration der Bewegungsgleichungen betragt 0.5 fs. Dieser Wert
hatte sich als ausreichend fur die Simulationen erwiesen, was aus eine Analyse
von Fluktuationen in der Gesamtenergie hervorging.
Die Reaktion SiH4 + H → SiH3 + H2
Es wurden Molekulardynamik-Simulationen der Kollision des Wasserstoff-
Radikals mit einem SiH4-Molekul durchgefuhrt. Die dynamische Entwicklung des
Systems ist in Form von Momentaufnahmen in Abbildung 4.29 wiedergegeben.
Die anfangliche Temperatur betrug 1000 K. Drei Atome sind besonders gekenn-
zeichnet, um sie in der weiteren Diskussion von den anderen unterscheiden zu
konnen. Die Veranderung der Abstande zwischen diesen Atomen ist in Abbil-
dung 4.30 dargestellt.
Deutlich zu erkennen ist das nach etwa 75 fs einsetzende Aufbrechen der Si-
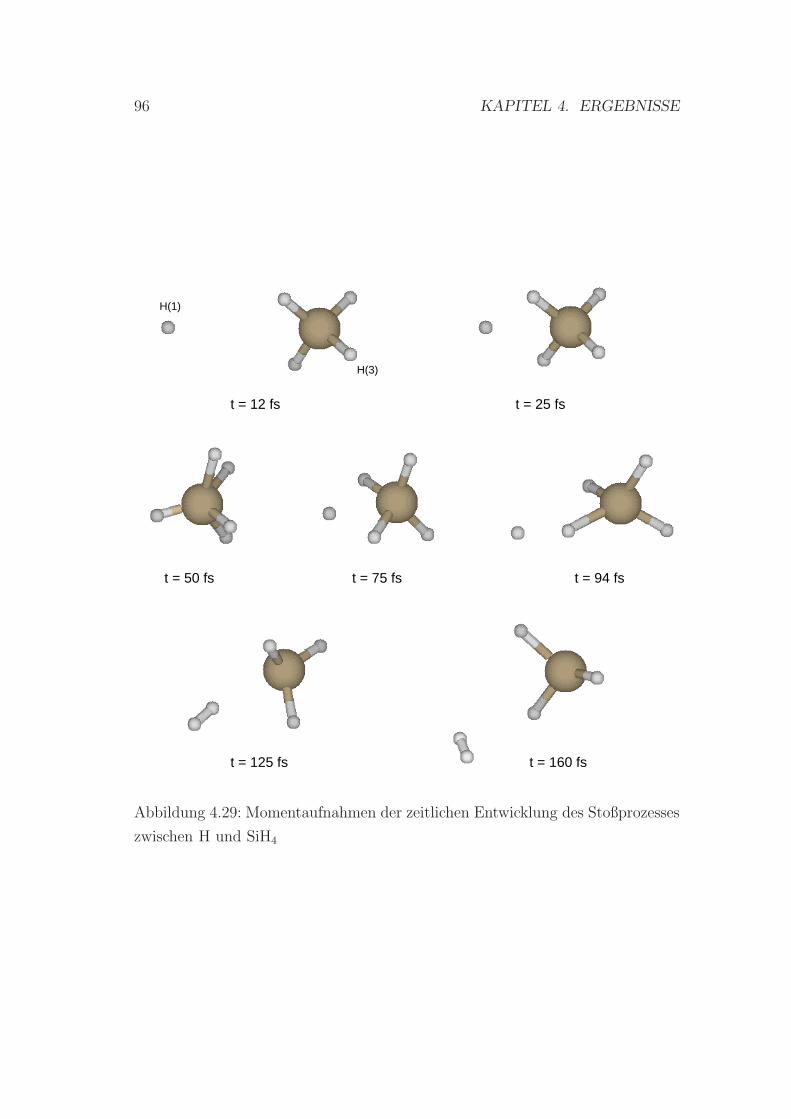
96 KAPITEL 4. ERGEBNISSE
H(2)
t = 125 fs t = 160 fs
t = 50 fs t = 75 fs t = 94 fs
t = 12 fs t = 25 fs
H(1)
H(3)
Abbildung 4.29: Momentaufnahmen der zeitlichen Entwicklung des Stoßprozesses
zwischen H und SiH4
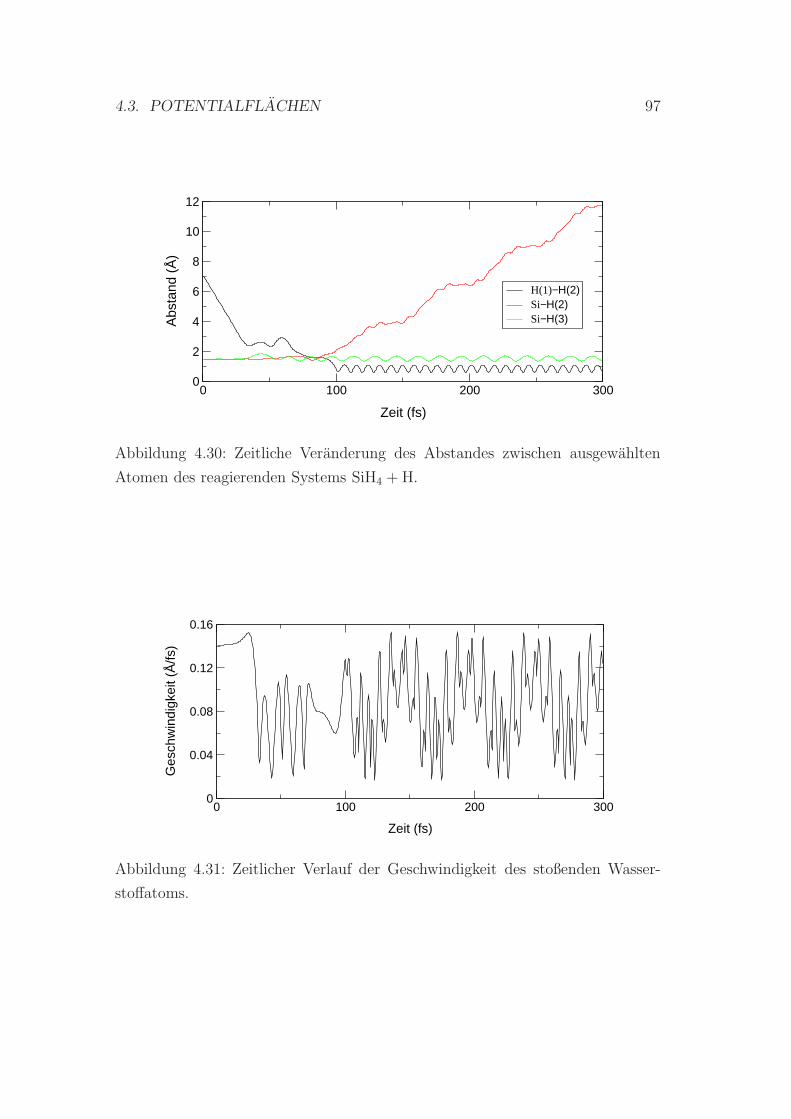
4.3. POTENTIALFLACHEN 97
0 100 200 300
Zeit (fs)
0
2
4
6
8
10
12
Abs
tand
(Å
)
H(1)−H(2) Si−H(2)Si−H(3)
Abbildung 4.30: Zeitliche Veranderung des Abstandes zwischen ausgewahlten
Atomen des reagierenden Systems SiH4 + H.
0 100 200 300
Zeit (fs)
0
0.04
0.08
0.12
0.16
Ges
chw
indi
gkei
t (Å
/fs)
Abbildung 4.31: Zeitlicher Verlauf der Geschwindigkeit des stoßenden Wasser-
stoffatoms.

98 KAPITEL 4. ERGEBNISSE
H(2) Bindung. Dadurch wird die Reaktion eingeleitet. Der Abstand zwischen den
Atomen H(1) und H(2) bleibt ab einem Zeitpunkt von etwa 100 fs im zeitlichen
Mittel konstant. Es ist zu sehen, daß ein Teil der anfanglichen kinetischen Energie
in innere Energie umgewandelt worden ist. Dies geht aus der vergroßerten Am-
plitude der Schwingungen des SiH3-Molekuls hervor und ist in Abbildung 4.30
anhand der grunen Kurve zu erkennen.
Aber auch in den Momentaufnahmen wird diese Tatsache in einer Verzerrung
des Molekuls deutlich. Der Reaktionsweg verlauft in der Art, wie man ihn auf-
grund der statischen Rechnungen zu dieser Potentialflache (vgl. 4.3.2) vermuten
wurde.
Die Tatsache, daß die Rechnungen nicht spinpolarisiert durchgefuhrt wurden,
zeigt sich in einem anderen Detail der Simulation. In Abbildung 4.31 ist die
Geschwindigkeit des Wasserstoffradikals in Abhangigkeit von der Simulationszeit
aufgetragen. Man kann daraus eine Beschleunigung innerhalb der ersten 25 fs
erkennen, was im Einklang mit dem Fehlen einer Energiebarriere bei Rechnungen
ohne Spinpolarisation (vgl. Kap. 4.3.2) steht. Die Beschleunigung ist jedoch nur
sehr gering, was den Schluß zulaßt, daß die nicht spinpolarisierte Rechnung auch
zur Beschreibung der Reaktion geeignet ist.
Streuung von Ar an SiCl4
Die Streuung von Argon-Atomen an SiCl4-Molekulen ist von Kunz et al. [7] ex-
perimentell durchgefuhrt worden. Eine Molekulardynamik-Simulation bei einer
anfanglichen Temperatur von 2000 K wurde als erster Schritt zur Simulation ei-
nes Dissoziationsprozesses durchgefuhrt. Dabei wurde das Edelgas durch ein von
Kresse entwickeltes [63] PAW-Potential beschrieben. Die dynamische Entwicklung
des Systems ist in Form von Momentaufnahmen in Abbildung 4.32 wiedergege-
ben. Die zeitliche Entwicklung einiger relevanter Abstande sowie der Geschwin-
digkeit des gestreuten Argon-Atoms ist in Abbildung 4.33 dargestellt.
Es ist zu erkennen, daß der Streuprozess im wesentlichen aus zwei nachein-
ander erfolgenden Stoßprozessen zusammengesetzt ist. Es wird aber insgesamt
nur ein relativ geringer Anteil an kinetischer Energie in Schwingungsenergie des
SiCl4-Molekuls umgewandelt.
Im Zusammenhang mit dem experimentellen Ergebnis bedeutet dies, daß die
Dissoziation des SiCl4-Molekuls wahrscheinlich nur infolge von etlichen aufein-
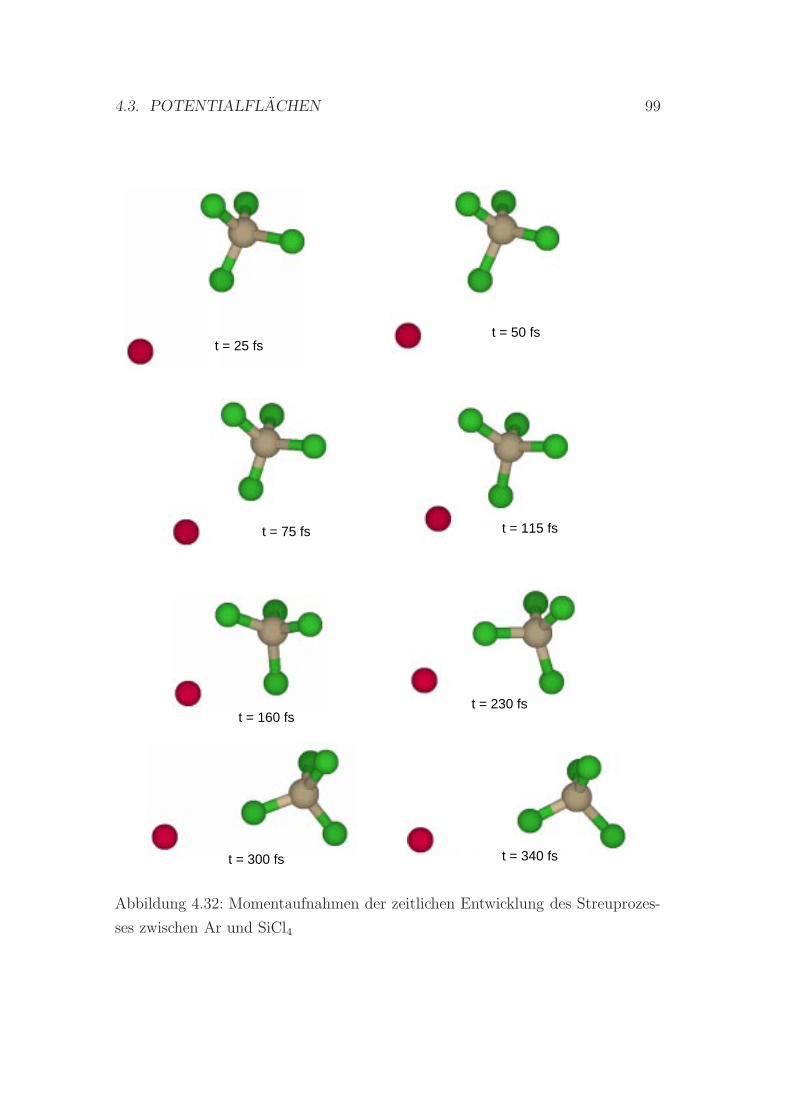
4.3. POTENTIALFLACHEN 99
t = 300 fs t = 340 fs
t = 25 fst = 50 fs
t = 75 fs t = 115 fs
t = 160 fst = 230 fs
Abbildung 4.32: Momentaufnahmen der zeitlichen Entwicklung des Streuprozes-
ses zwischen Ar und SiCl4
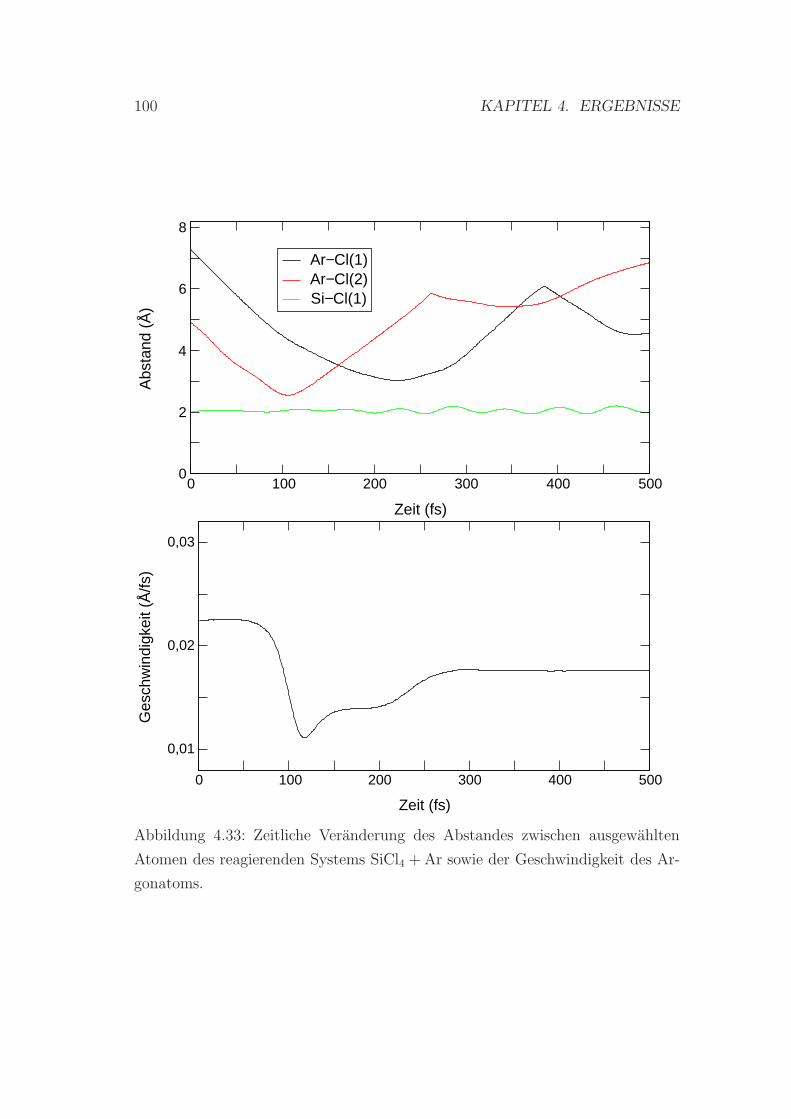
100 KAPITEL 4. ERGEBNISSE
0 100 200 300 400 500
Zeit (fs)
0
2
4
6
8
Abs
tand
(Å
)
Ar−Cl(1)Ar−Cl(2)Si−Cl(1)
0 100 200 300 400 500
Zeit (fs)
0,01
0,02
0,03
Ges
chw
indi
gkei
t (Å
/fs)
Abbildung 4.33: Zeitliche Veranderung des Abstandes zwischen ausgewahlten
Atomen des reagierenden Systems SiCl4 + Ar sowie der Geschwindigkeit des Ar-
gonatoms.
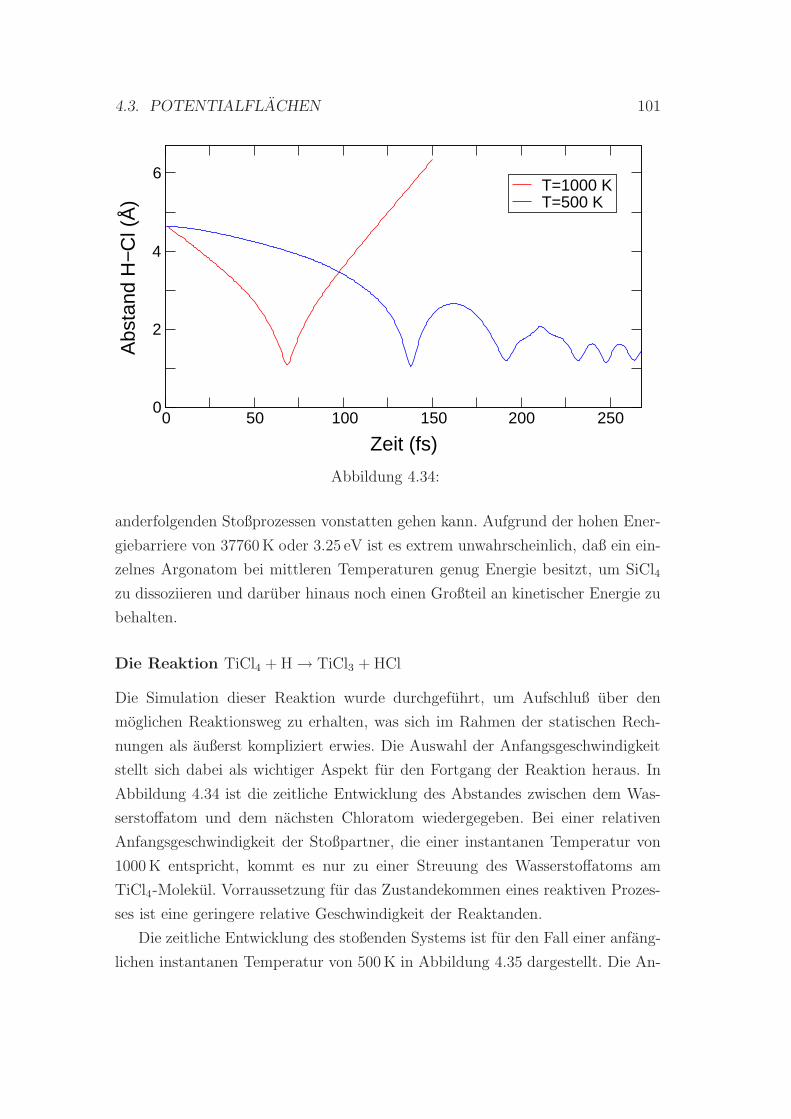
4.3. POTENTIALFLACHEN 101
0 50 100 150 200 250
Zeit (fs)
0
2
4
6
Abs
tand
H−C
l (Å
)
T=1000 KT=500 K
Abbildung 4.34:
anderfolgenden Stoßprozessen vonstatten gehen kann. Aufgrund der hohen Ener-
giebarriere von 37760 K oder 3.25 eV ist es extrem unwahrscheinlich, daß ein ein-
zelnes Argonatom bei mittleren Temperaturen genug Energie besitzt, um SiCl4
zu dissoziieren und daruber hinaus noch einen Großteil an kinetischer Energie zu
behalten.
Die Reaktion TiCl4 + H → TiCl3 + HCl
Die Simulation dieser Reaktion wurde durchgefuhrt, um Aufschluß uber den
moglichen Reaktionsweg zu erhalten, was sich im Rahmen der statischen Rech-
nungen als außerst kompliziert erwies. Die Auswahl der Anfangsgeschwindigkeit
stellt sich dabei als wichtiger Aspekt fur den Fortgang der Reaktion heraus. In
Abbildung 4.34 ist die zeitliche Entwicklung des Abstandes zwischen dem Was-
serstoffatom und dem nachsten Chloratom wiedergegeben. Bei einer relativen
Anfangsgeschwindigkeit der Stoßpartner, die einer instantanen Temperatur von
1000 K entspricht, kommt es nur zu einer Streuung des Wasserstoffatoms am
TiCl4-Molekul. Vorraussetzung fur das Zustandekommen eines reaktiven Prozes-
ses ist eine geringere relative Geschwindigkeit der Reaktanden.
Die zeitliche Entwicklung des stoßenden Systems ist fur den Fall einer anfang-
lichen instantanen Temperatur von 500 K in Abbildung 4.35 dargestellt. Die An-

102 KAPITEL 4. ERGEBNISSE
ordnung der Atome fur t = 140 fs entspricht dem Zustand mit der kleinsten
potentiellen Energie im Verlauf der ganzen Reaktion. Die Ahnlichkeit zu der
Konfiguration der Teilchen im Energieminimum, das mit Hilfe der Full-Potential
Methode ermittelt wurde (vgl. Abb. 4.26), fallt sofort ins Auge. Im Verlauf der
Reaktion halt sich das System eine Zeitlang in diesem Bereich der Potentialflache
auf. Es wurde somit ein moglicher Weg fur die Reaktion TiCl4+H → TiCl3+HCl
gefunden. Eine wiederholte Untersuchung der Reaktion mit Hilfe von Algorith-
men zur Strukturoptimierung und zur Suche nach Sattelpunkten auf der Potenti-
alflache wird auf diesen Ergebnissen aufbauen kunnen und somit die Frage nach
dem Reaktionsmechnismus beantworten kunnen.
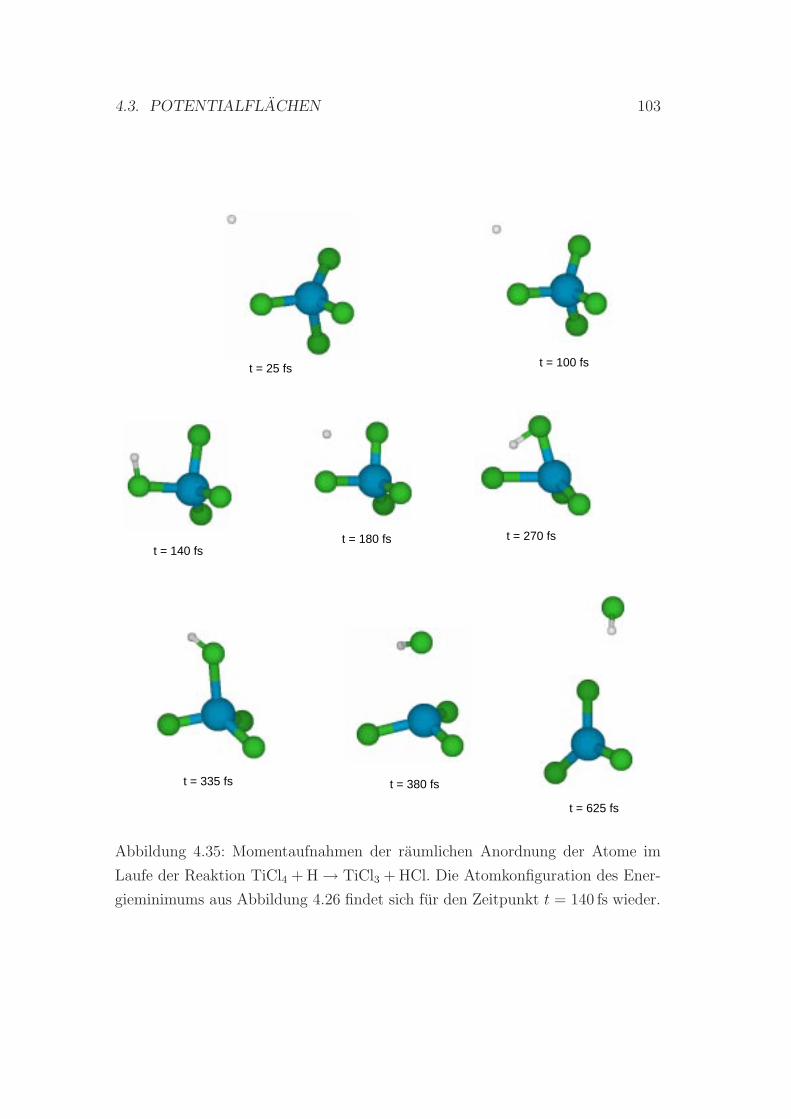
4.3. POTENTIALFLACHEN 103
t = 100 fst = 25 fs
t = 270 fst = 180 fst = 140 fs
t = 335 fs t = 380 fs
t = 625 fs
Abbildung 4.35: Momentaufnahmen der raumlichen Anordnung der Atome im
Laufe der Reaktion TiCl4 + H → TiCl3 + HCl. Die Atomkonfiguration des Ener-
gieminimums aus Abbildung 4.26 findet sich fur den Zeitpunkt t = 140 fs wieder.
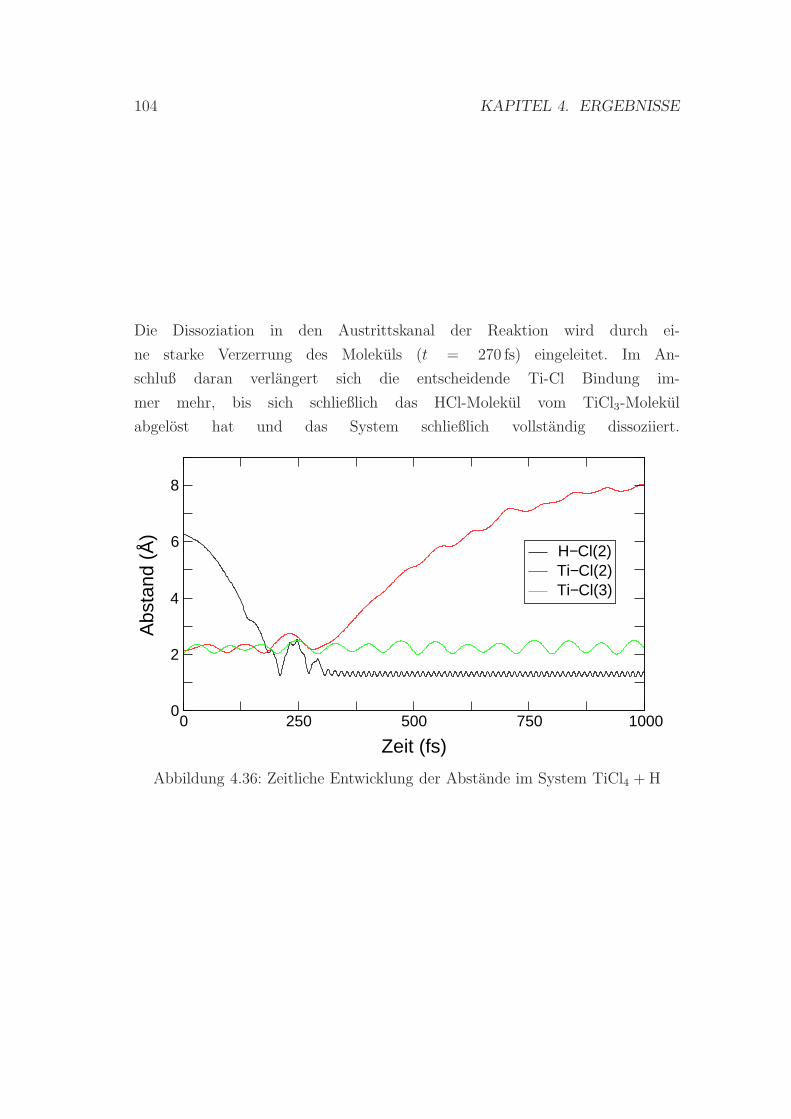
104 KAPITEL 4. ERGEBNISSE
Die Dissoziation in den Austrittskanal der Reaktion wird durch ei-
ne starke Verzerrung des Molekuls (t = 270 fs) eingeleitet. Im An-
schluß daran verlangert sich die entscheidende Ti-Cl Bindung im-
mer mehr, bis sich schließlich das HCl-Molekul vom TiCl3-Molekul
abgelost hat und das System schließlich vollstandig dissoziiert.
0 250 500 750 1000
Zeit (fs)
0
2
4
6
8
Abs
tand
(Å
)
H−Cl(2)Ti−Cl(2) Ti−Cl(3)
Abbildung 4.36: Zeitliche Entwicklung der Abstande im System TiCl4 + H

Kapitel 5
Zusammenfassung und Ausblick
Die Bindungsenergie von Molekulen wurde mit verschiedenen ab initio Metho-
den unter Berucksichtigung der Nullpunktsschwingungsenergie untersucht. Dabei
wurde gleichzeitig die Große des Basissatzes fur die Entwicklung der Einteilchen-
wellenfunktionen variiert. Es stellt sich heraus, daß die Ergebnisse sowohl von der
gewahlten Methode als auch vom Basissatz abhangig sind. Diese Abhangigkeit
wurde detailliert untersucht.
Es zeigt sich, daß beispielsweise das O2-Molekul im Rahmen der HF-Nahe-
rung unter Verwendung kleiner Basissatze nicht stabil ist (vgl. Abb. 4.3). Im
Gegensatz dazu wird die Bindungsenergie dieses Molekuls durch die LSDA-
Methode uberschatzt. Die besten Ergebnisse fur O2 lassen sich mit der CCSD(T)-
Methode (vgl. Kap. 2.3.2) erzielen. Der Nachteil dieser Methode liegt in der
großen Abhangigkeit der benotigten Rechenleistung von der Systemgroße. Die
prinzipiell mogliche systematische Verbesserung der Ergebnisse wird durch den
begrenzt zur Verfugung stehenden Festplattenspeicherplatz fur die Speicherung
von Matrixelementen eingeschrankt.
Bei viel geringerem Rechenbedarf liefern DFT-Methoden vergleichbare und
oftmals bessere Ergebnisse (vgl. Abb. 4.4). Dies ist insbesondere der Fall fur klei-
ne Basissatze, die keine hinreichende Erfassung der Korrelationsenergie durch
Post-HF-Methoden erlauben. Sehr deutlich macht sich diese Tatsache bei den
hier untersuchten Si-basierten Molekulen (vgl. Abb. 4.5) bemerkbar. Die Hybrid-
methode B3LYP liefert bei den untersuchten Molekulen die beste Beschreibung
der energetischen Verhaltnisse. Der benotigte Rechenaufwand ist mit einer HF-
Rechnung vergleichbar (Abb. 4.6).
105

106 KAPITEL 5. ZUSAMMENFASSUNG UND AUSBLICK
Die B3LYP-Methode ist auch fur die Beschreibung der geometrischen Eigen-
schaften sehr gut geeignet. Dies beruht darauf, daß B3LYP einen Teil der exakten
Austauschenergie miteinbezieht und so die Unterschatzung der Bindungslangen
im Rahmen der GGA ausgleicht. Dieses Verhalten zeigt sich bei den Molekulen
N2, HCl, SiH4 und TiCl4 (Abb. 4.7 und 4.8). Daher wurde die B3LYP-Methode
fur die weitere Untersuchung von Potentialflachen benutzt.
Des weiteren wurden die Reaktionen H2+H ⇀↽ H+H2, SiH4+H ⇀↽ SiH3+H2,
SiCl4 + H ⇀↽ SiCl3 + HCl und TiCl4 + H ⇀↽ TiCl3 + HCl untersucht. Diesen Re-
aktionen ist gemeinsam, daß einer der Reaktanden das Wasserstoff-Radikal ist.
Abweichungen der mit Dichtefunktionalmethoden berechneten Gesamtenergie des
H-Atoms sowie der Bindungsenergie des H2-Molekuls von den exakten bzw. expe-
rimentell ermittelten Werten konnten auf die Selbstwechselwirkung zuruckgefuhrt
werden.
Die einfachste Reaktion H2 + H ⇀↽ H + H2 laßt sich mit der B3LYP-Methode
qualitativ beschreiben. Die Hohe der Energiebarriere wird allerdings mit 179 meV
unter Verwendung der 6-31G** Basis um mehr als 50 % unterschatzt. Dabei er-
weist sich der Einfluß des Basissatzes als vergleichsweise gering. Fur den Fall der
AUG-cc-pVQZ-Basis berechnet sich die Energiebarriere zu 186 meV. Auch die
im Programmpaket Vasp implementierte GGA-Methode ergibt eine qualitativ
richtige Potentialflache, die mit der B3LYP/6-31G**-PES (Abb. 4.13) uberein-
stimmt. Im Gegensatz hierzu steht die mit der LDA-Methode berechnete PES
nicht im Einklang mit dem Experiment. Anstelle eines Sattelpunktes enthalt sie
ein Minimum und sagt somit das H3-Molekul als stabil voraus.
Fur die PES der Reaktion SiH4 + H ⇀↽ SiH3 + H2 ergibt sich ein vergleich-
bares Bild. Die mit der Kombination B3LYP/6-31G** berechnete Barrierenhohe
liegt bei 54 meV im Vergleich zum experimentellen Wert [6] von 195 meV. Dieser
Wert wurde allerdings in einem Temperaturbereich zwischen 998 K und 1273 K
gemessen. Fur einen genauen Vergleich mußte in der Rechnung die Temperatu-
rabhangigkeit des Ubergangszustandes berucksichtigt werden. Die lokale Dich-
tenaherung versagt bei der Beschreibung dieser Reaktion, indem sie keine Ener-
giebarriere findet. Auch mit Hilfe von nicht-spinpolarisierten Rechnungen laßt
sich der Ubergangszustand nicht lokalisieren. Im Rahmen des Programpakets
Vasp ist allein die spinpolarisierte GGA-Methode in der Lage, die Barriere der
Reaktion zu ermitteln.

107
Die Potentialflache der Reaktion SiCl4 +H ⇀↽ SiCl3 +HCl zeichnet sich durch
das Vorhandensein eines Begegnungsminimums sowie eines Ubergangszustands
aus. Die energetischen und strukturellen Eigenschaften erweisen sich wiederum
als nicht stark abhangig vom verwendeten Basissatz. Da die Energie des Uber-
gangszustandes um 0.5 eV uber der Energie der Ausgangsstoffe liegt, laßt sich
darauf schließen, daß die Reaktion nur unter Aufwendung einer Aktivierungs-
energie ablauft.
Im Gegensatz dazu zeichnet sich die PES der Reaktion SiCl4+H ⇀↽ SiCl3+HCl
durch ein energetisch sehr tief liegendes Energieminimum aus. Dies kann als
Erklarung dafur angesehen werden, daß die Reaktion ohne Aktivierungsenergie
ablauft [5]. Die genaue Form der PES stellt sich aufgrund der d-Elektronen von
Titan als kompliziert heraus. Fur die Untersuchung dieser PES erweist sich die
Methode der Molekulardynamik-Simulation als geeignet. Aufgrund von Ergeb-
nissen in Bezug auf die PES von SiH4 +H ⇀↽ SiH3 +H2 wurde die GGA-Methode
ausgewahlt. Die Rechnungen konnten nur nicht-spinpolarisiert durchgefuhrt wer-
den, da eine spinpolarisierte Rechnung eine Verlangerung der Rechenzeit um den
Faktor zehn bedeutet hatte. Das Ergebnis der Simulation ist, daß ein mogli-
cher Reaktionsweg in der Ablosung des HCl-Molekuls aus der Minimumstruktur
entlang der TiCl-Bindung verlaufen konnte. Die Untersuchung der PES mit Hilfe
der B3LYP-Methode laßt tatsachlich auf einen energetisch sehr flach verlaufenden
Austrittskanal in dieser Richtung schließen. Die Methode der Molekulardynamik-
Simulation erweist sich damit als wertvolles Mittel zur qualitativen Untersuchung
von Potentialflachen chemischer Reaktionen. Fur quantitative Aussagen mussen
die Simulationen spinpolarisiert durchgefuhrt werden, ungepaarte Elektronen im
System vorhanden sind.
Fur die Clusterbildung aus Precursormolekulen sind weitere Simulationen not-
wendig. Auch hier erweisen sich First-Principles-MD-Simulationen als sehr hilf-
reich, da die Simulationen insbesondere bei endlichen Temperaturen durchgefuhrt
werden konnen und es somit erlauben, Clusterbildung in der Gasphase zumindest
im Ansatz zu diskutieren.
Im Hinblick auf Clusterwachstum wurde ein aus zwei Titan- und sieben Koh-
lenstoffatomen aufgebautes Molekul simuliert. Die Bindungen der Kohlenstof-
fatome wurden durch zusatzliche Wasserstoffatome abgesattigt, um eine nicht-
spinpolarisierte Rechnung durchfuhren zu konnen.

108 KAPITEL 5. ZUSAMMENFASSUNG UND AUSBLICK
Dieser kleine Cluster wurde zuerst im Rahmen einer Strukturoptimierung un-
ter Verwendung der B3LYP-Methode optimiert. Die zeitliche Entwicklung des
Systems im Laufe einer Simulation bei T = 800K ist in Form von drei Moment-
aufnahmen in Abbildung 5.1 dargestellt. Am Anfang befand sich das Molekul in
einem symmetrischen Zustand, die mit 1, 2, 3 und 4 bezeichneten Atome bildeten
eine Ebene. Die zeitliche Entwicklung des von diesen Atomen gebildeten Dieder-
winkels ist in Abbildung 5.2 dargestellt. Das Molekul verdreht sich offenbar im
Laufe der Zeit aufgrund der Abstoßung der sterischen Methylgruppen (Abbildung
5.3).
Abschließend laßt sich festhalten, daß die Methode der Molekulardynamik-
Simulation in Verbindung mit der Dichtefunktionaltheorie ein geeignetes Werk-
zeug zur Beschreibung von Potentialflachen chemischer Reaktionen darstellt. Ins-
besondere dann, wenn die Potentialflache beispielsweise durch die Anwesenheit
von Ubergangsmetallelementen eine komplexe Struktur aufweist, oder wenn die
Zahl der Freiheitsgrade eines Molekuls fur eine systematische Untersuchung mit
statischen Methoden zu groß wird, lassen sich Aussagen uber mogliche Verlaufe
von Reaktionen treffen. Die genaue Struktur der PES kann dann entlang eines Re-
aktionsweges mit Hilfe von Optimierungsverfahren detailiert untersucht werden,
was die Berechnung eventuell vorhandener Energiebarrieren miteinschließt.
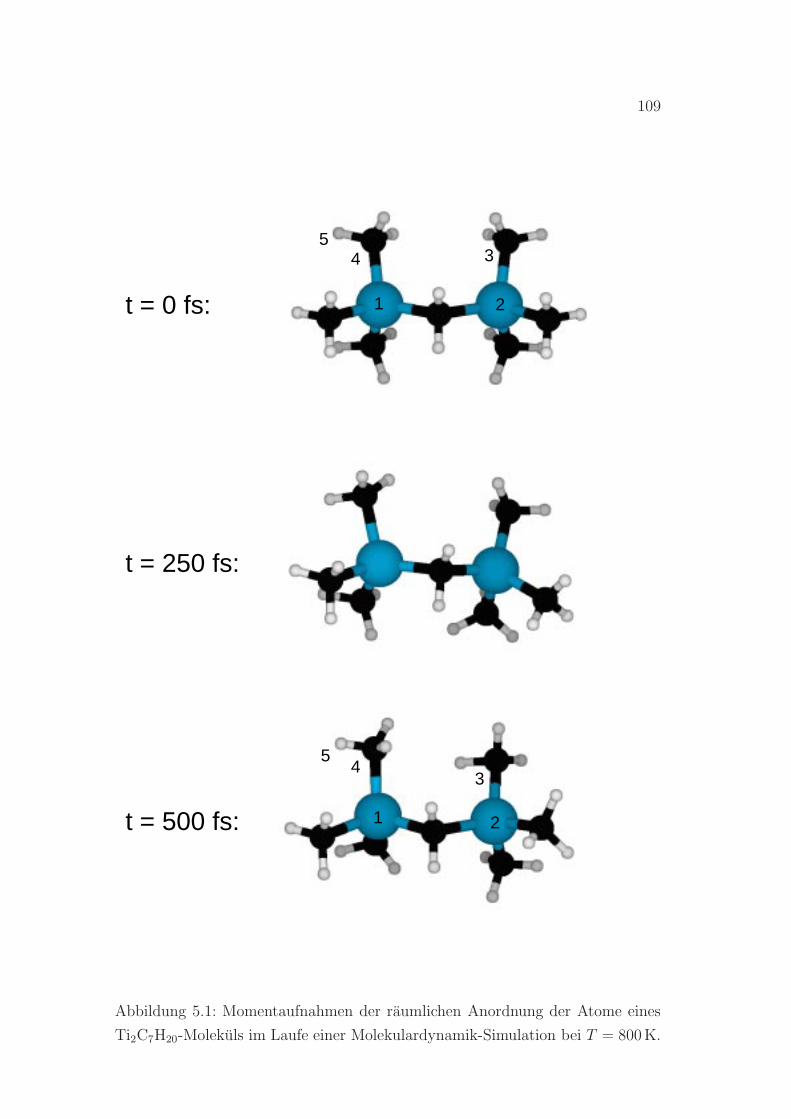
109
t = 500 fs:
t = 250 fs:
t = 0 fs:
5
21
34
54
1 2
3
Abbildung 5.1: Momentaufnahmen der raumlichen Anordnung der Atome eines
Ti2C7H20-Molekuls im Laufe einer Molekulardynamik-Simulation bei T = 800K.
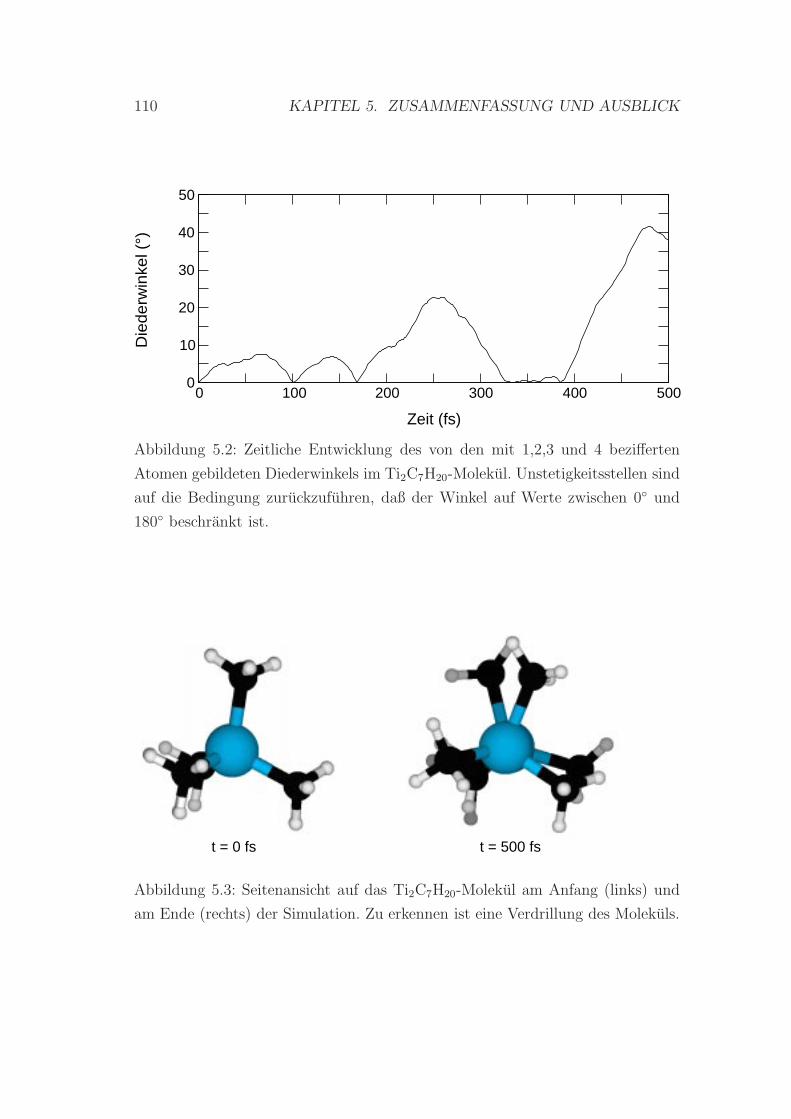
110 KAPITEL 5. ZUSAMMENFASSUNG UND AUSBLICK
0 100 200 300 400 500
Zeit (fs)
0
10
20
30
40
50
Die
derw
inke
l (°)
Abbildung 5.2: Zeitliche Entwicklung des von den mit 1,2,3 und 4 bezifferten
Atomen gebildeten Diederwinkels im Ti2C7H20-Molekul. Unstetigkeitsstellen sind
auf die Bedingung zuruckzufuhren, daß der Winkel auf Werte zwischen 0◦ und
180◦ beschrankt ist.
t = 0 fs t = 500 fs
Abbildung 5.3: Seitenansicht auf das Ti2C7H20-Molekul am Anfang (links) und
am Ende (rechts) der Simulation. Zu erkennen ist eine Verdrillung des Molekuls.

Anhang A
Funktionale fur Austausch und
Korrelation
Im folgenden sind die in dieser Arbeit verwendeten Kombinationen von Funktio-
nalen fur die Austausch- und die Korrelationsenergie wiedergegeben.
A.1 Lokale Dichtenaherung
SVWN: Die Austauschenergie ist in diesem Fall durch Gleichung (2.60) gegeben.
Die Korrelationsenergie beruht auf einer Interpotlationsformel von Vosko et al.
[35] fur die Energie eines Elektrons im homogenen Elektronengas.
CA: Das Funktional fur die Austauschenergie stimmt mit dem in SVWN ver-
wendeten uberein. Die Interpolationsformel fur die Korrelationsenergie stammt
von Ceperley und Alder und findet sich in [34].
A.2 Gradientenmethoden
BLYP: Die Austauschenergie ist durch den von Becke [36] vorgeschlagenen Aus-
druck gegeben. Die Korrelationsenergie berechnet sich nach der von Lee et al [40]
angegebenen Formel.
BP86: Dieses Funktional unterscheidet sich von BLYP nur im Ausdruck fur die
Korrelationsenergie, welcher von Perdew [37] gegeben wurde.
BPW91: Im Unterschied zu BP86 und BLYP beruht dieses Funktional auf der
Korrelationsenergie von Perdew und Wang [38].
111

112ANHANG A. FUNKTIONALE FUR AUSTAUSCH UND KORRELATION
PW91PW91: Die im Programmpaket Vasp enthaltene GGA-Methode beruht
auf den von Perdew und Wang in [38] vorgeschlagenen Ausdrucken fur die
Austausch- und Korrelationsenergie.
A.3 Hybridmethoden
B3LYP: Die Austauschenergie wird im Rahmen der B3LYP-Methode aus einer
Kombination der exakten Austauschenergie, der LDA-Austauschsenergie sowie
der Gradientenkorrektur von Becke [36] berechnet. Die Korrelationsenergie be-
rechnet sich aus der Korrelationsenergie der lokalen Dichtenaherung sowie einer
Gradientenkorrektur nach Lee et al.[40]. Die entsprechenden Anteile werden durch
Optimierung der drei Parameter im Rahmen einer Anpassung an experimentelle
Daten ermittelt.

Anhang B
Die verwendeten Basissatze
B.1 Split-Valence Basissatze von Pople
In der LCAO-Methode (Linear Combination of Atomic Orbitals, ursprunglich
von Bloch [18] vorgeschlagen) wird ein Molekulorbital als Linearkombination von
atomzentrierten Basisfunktionen dargestellt:
φi =M∑µ=1
ciµχµ. (B.1)
Die von Pople und Mitarbeitern vorgeschlagenen Basissatze [64, 65, 66, 67, 68]
zeichnen sich dadurch aus, daß die Basisfunktionen feste Linearkombinationen
von endlich vielen sogenannten primitiven Gaussfunktionen (PGTO’s) sind:
χµ =
K(µ)∑k=1
aµkχPGTOk , (B.2)
wobei die PGTO’s die Form (2.69) haben. Die Basisfunktionen werden deshalb
als kontrahierte Gaussfunktionen bezeichnet. Die Koeffizienten aµk sowie die Ex-
ponenten der primitiven Gaussfunktionen mussen im Vorfeld festgelegt werden
und bleiben im Verlauf einer Selbstkonsistenzrechnung konstant. Optimale Wer-
te fur diese Großen wurden in der Vergangenheit durch Anpassen von (B.2) an
numerisch ermittelte Atomorbitale bestimmt.
Die in dieser Arbeit verwendeten Basissatze unterscheiden sich zum einen
in der Zahl M der in die Rechnung eingehenden kontrahierten Gaussfunktionen
und zum anderen in den WertenK(µ), die die Anzahl zugrundeliegenden PGTO’s
bestimmen.
113

114 ANHANG B. DIE VERWENDETEN BASISSATZE
3-21G: Bei diesem Basisatz werden die Orbitale der Rumpfelektronen jeweils
durch eine einzige kontrahierte Gaussfunktion beschrieben, die sich aus drei pri-
mitiven Gaussfunktionen zusammensetzt. Die Valenzorbitale sind in diesem Fall
eine Linearkombination von zwei Basisfunktionen, von denen die eine eine Sum-
me von zwei PGTO’s ist und die andere eine einzelne primitive Gaussfunktion.
Fur das Element Stickstoff beispielsweise ergeben sich somit 9 Basisfunktionen
(1 × 1s und jeweils 2 × 2s, 2px, 2py, 2pz) oder 15 primitive Gaussfunktionen.
6-21G: Dieser Basissatz stimmt mit der 3-21G-Basis uberein, mit dem Un-
terschied, daß sich die Rumpforbitale jetzt als Summe von sechs PGTO’s darstel-
len. Somit ist die Zahl der Basisfunktionen fur jedes Element bei diesen beiden
Basissatzen identisch, sie unterscheiden sich lediglich in der Form der Basisfunk-
tionen der Rumpfelektronen.
6-31G: Eine bessere Beschreibung der Valenzelektronen im Vergleich zu den
beiden vorhergehenden Basisatzen wird durch diese Basis angestrebt. Sie bietet
wiederum keine Vergroßerung der Einelektronenbasis, jedoch werden jetzt drei
primitive Gaussfunktionen zur Beschreibung des inneren Teils der Außenelek-
tronen herangezogen. Fur das Element Stickstoff betragt die Zahl der PGTO’s
nunmehr schon 22.
6-31G** = 6-31G(d,p): Dieser sehr gebrauchliche Basissatz ist mit der
6-31G-Basis identisch, er besteht allerdings zusatzlich aus einem Satz von p-
Orbitalen im Falle von Wasserstoff bzw. (kartesischen) d-Orbitalen fur alle ande-
ren Elemente als Polarisationsfunktionen. Damit ist oftmals eine erheblich bessere
Beschreibung von Bindungen in Molekulen moglich. Jedes Stickstoffatom bringt
in dieser Basis 15 Basisfunktionen (zusatzlich dxy, dxz, dyz, dx2, dy2 und dz2) bzw.
28 PGTO’s in die Rechnung mit ein. Fur diesen Basissatz hat sich die Schreib-
weise mit den Sternchen anstelle von d und p eingeburgert.
6-311G** = 6-311G(d,p): Im Gegensatz zu den vorhergehenden Ba-
sissatzen, bei denen die Valenzorbitale durchweg durch zwei Basisfunktionen be-
schrieben wurden, ist diese Basis eine sogenannte Triple-Split Valence Basis. Denn
die Außenelektronen werden in diesem Fall durch eine Linearkombination von drei
kontrahierten Gaussfunktionen beschrieben, von denen zwei einer einzigen PGTO
entsprechen. Die Basisfunktion, die den inneren Teil der Valenzorbitale beschrei-
ben soll, ist wie in den vorhergehenden Fallen eine Summe von drei einfachen

B.1. SPLIT-VALENCE BASISSATZE VON POPLE 115
GTO’s. Das Resultat ist eine Zahl von 18 Basis- bzw. 32 primitiven Funktionen1
fur Stickstoff.
Fur Elemente aus der zweiten Reihe des Periodensystems steht das Acronym
6-311G** fur die von McLean und Chandler [69] aufgestellte Basis. Ubergangsme-
talle werden in diesem Fall durch die von Wachters und Hay [70, 71] entwickelten
Basisfunktionen beschrieben.
6-311++G** = 6-311++G(d,p): Zusatzlich zu den Polarisationsfunktio-
nen sind in diesem Fall noch sogenannte diffuse Funktionen involviert, welche
sich durch einen sehr kleinen Exponenten auszeichnen und somit die Beschrei-
bung von ausgedehnten Molekulorbitalen wesentlich verbessern. Besonders wich-
tig ist die Anwesenheit diffuser Funktionen bei der Beschreibung von Anionen
sowie schwach gebundenen Systemen, z.B. Wasserstoffbruckenbindungen. Das er-
ste +-Zeichen symbolisiert einen Satz p-artiger Funktionen fur alle Elemente mit
Ausnahme von Wasserstoff. Fur dieses Element gilt das zweite +-Zeichen, welches
fur ein diffuses s-Orbital steht.
6-311++G(2d,2p): Das Hinzufugen von weiteren Polarisationsfunktionen,
in diesem Fall zwei Satzen von p-Orbitalen fur Wasserstoff bzw. d-Orbitalen fur
alle anderen Elemente erhoht die Flexibilitat des Basissatzes in Bezug auf die
Reproduktion der exakten Wellenfunktion. Die Zahl der Spinorbitale zur Be-
schreibung der Elektronen eines Stickstoffatoms betragt hier 27. Sie sind aus 42
PGTO’s zusammengesetzt.
6-311++G(3df,2pd): Dieser Basissatz stellt eine der umfangreichsten bis
heute entwickelten Pople-Basen dar. Durch die Hinzunahme von f-artigen Ba-
sisfunktionen kann oftmals ein großer Teil der Korrelationsenergie eines elek-
tonischen Systems erhalten werden. Fur jedes im Molekul enthaltene N-Atom
beinhaltet dieser Basisatz 39 kontrahierte Gaussfunktionen, und zwar 5 s-artige,
12 p-artige, 15 d-artige und 7 f-artige. Ein großer Teil von ihnen besteht aber aus
einer einzigen primitiven Funktion, insgesamt benotigt man davon 58.
Charakteristisch fur diese nach Pople benannten Basissatze ist die Tatsache,
daß die Exponenten der fur die s- und p-Valenzorbitale verwendeten primitiven
Gaussfunktionen ubereinstimmen. Daruber hinaus gehort zu jeder Basisfunktion
1Die drei kartesischen d-Orbitale dx2 , dy2 und dz2 werden ublicherweise in zwei echte d-Orbitale dx2−y2 und d3z2−r2transformiert, so daß die Zahl der kontrahierten Gaussfunktionenum eins niedriger ist, als man bei sechs d-Orbitalen erwarten wurde.

116 ANHANG B. DIE VERWENDETEN BASISSATZE
aber ein ganz bestimmter Satz von PGTO’s. Außerdem sind die Basissatze nicht
immer sehr ausgeglichen, was sich in einer oft schlechten Konvergenz bestimmter
Eigenschaften eines behandelten Systems mit großer werdender Einteilchenbasis
außert. Es ist somit bisweilen schwierig bis unmoglich, den Wert einer Große im
Limit der vollstandigen Basis, also den fur die Methode charakteristischen Wert,
zu ermitteln.
Die folgenden Basissatze sind im Hinblick auf dieses Problem konzipiert wor-
den.
B.2 Correlation-Consistent Basissatze
Die von Dunning [72] vorgeschlagenen Basissatze, fur die haufig die Abkurzung cc-
pVXZ verwendet wird, gibt es im Moment nur fur die Hauptgruppenelemente von
Wasserstoff bis Argon mit Ausnahme der Alkali- und Erdalkalimetalle. Sie tragen
den Beinamen Correlation-Consistent, weil jede Basisfunktion einer Basis, zumin-
dest der Idee nach, eine vergleichbare Menge an Korrelationsenergie liefert. Das
Symbol X∈ {D,T,Q, 5, 6} steht fur die Zahl der Basisfunktionen, durch welche
die Valenzelektronen beschrieben werden. Diese Basissatze sind somit vom Typ
Split-Valence, Triple-Split Valence, usw., was durch das Kurzel VXZ fur Valence-
Double-Zeta, Valence-Triple-Zeta, usw. verdeutlicht werden soll. Der Buchstabe p
verdeutlicht die Anwesenheit von Polarisationsfunktionen, durch Voranstellen des
Symbols AUG- konnen den Basissatzen diffuse Funktionen hinzugefugt werde.
AUG-cc-pVDZ: Bei diesem Basissatz wird jedes Valenzorbital durch zwei
Basisfunktionen beschrieben. Daruber hinaus ist fur jedes Wasserstoffatom ein
Satz p-artiger Polarisationsfunktionen, fur jedes andere Element ein zusatzlicher
Satz d-Orbitale enthalten. Außerdem gibt es noch eine Reihe von diffusen Funk-
tionen, und zwar je eine s- und drei p-artige Funktionen fur Wasserstoff und
jeweils eine s-, drei p- und funf d-artige Funktionen fur jedes andere Element.
Um beim Beispiel Stickstoff zu bleiben: Jedes N-Atom ist in diesem Basissatz
mit 23 Basisfunktionen vertreten.
AUG-cc-pVTZ: Die nachstgroßere Basis ist eine Valence-Triple-Zeta Basis.
Um ihre Ausgeglichenheit zu bewahren, enthalt diese Basis mehr Polarisations-
funktionen als die Valence-Double-Zeta Basis, und zwar zwei Satze p- und einen
Satz d-Funktionen fur Wasserstoff bzw. zwei Satze d- und einen Satz f-Funktionen

B.2. CORRELATION-CONSISTENT BASISSATZE 117
fur jedes andere Element. Zusatzlich gibt es jetzt auch noch diffuse d- bzw. f-
Funktionen fur Wasserstoff bzw. Nicht-Wasserstoff.
AUG-cc-pVQZ: Fur großere Systeme schon nicht mehr verwendbar ist diese
Basis, bei der die Valenzorbitale durch vier Funktionen dargestellt werden. Sie
enthalt fur Elemente aus der zweiten und dritten Reihe des Periodensystems
schon zwei Satze g-Funktionen. Die Zahl der kontrahierten Gaussfunktionen fur
Stickstoff betragt 80. Sie sind aus 127 PGTO’s linear kombiniert.
AUG-cc-pV5Z: Die Zusammensetzung dieser Basis verlauft analog dem
oben angegebenen Schema. Fur Wasserstoff enthalt diese Basis die gleiche Zahl an
Basisfunktionen wie die vorhergehende Basis fur Elemente der zweiten Periode.
Die nachste Erweiterung ware eine AUG-cc-pV6Z Basis. Aufgrund der ungeheuer
großen Zahl an 189 Basisfunktionen fur Elemente der zweiten Periode bzw. 127
fur Wasserstoff ist die Verwendung dieser Basis bei der Behandlung der meisten
Molekule jedoch vollig ausgeschlossen.
Die Basissatze von Dunning sind darauf ausgelegt, bei Erhohen der Kardi-
nalnummer X eine systematische Verbesserung der Ergebnisse zu erzielen. Dies
zeichnet sie vor den Pople-Basen aus, deren Entwicklung eher historisch bedingt
ist und die sich auf eine recht vielfaltige Art und Weise mit dem Problem der
Korrelationsenergie befassen. Der Nachteil der Correlation-Consistent Basissatze
ist das rasche Ansteigen der Zahl der Basisfunktionen ∼ X3, was oftmals die
Verwendung einer anderen als der Valence-Double-Zeta Basis unmoglich macht.

Anhang C
Der Algorithmus von Verlet
Entwickelt1 man die Funktion r(t) vorwarts und ruckwarts in der Zeit um t0, so
ergeben sich folgende Gleichungen:
r(t+ h) = r(t0) + hdr
dt
∣∣∣∣t0
+1
2h2 d
2r
dt2
∣∣∣∣t0
+ · · ·
r(t− h) = r(t0) − hdr
dt
∣∣∣∣t0
+1
2h2 d
2r
dt2
∣∣∣∣t0
− · · ·
Addition dieser beiden Taylorreihen liefert2
r(t+ h) = 2r(t) − r(t− h) + h2a(t) + O(h4) . (C.1)
Die Position eines Teilchens zum Zeitpunkt t + h laßt sich also bei bekannter
Beschleunigung naherungsweise aus den Orten zum Zeitpunkt t und t − h be-
stimmen. Fur die Geschwindigkeit - allerdings zum Zeitpunkt t - ergibt sich aus
der Differenz der Taylorentwicklungen:
v(t) =r(t+ h) − r(t− h)
2h+ O(h2) . (C.2)
Gleichungen (C.1) und (C.2) stellen den Verlet-Algorithmus dar. Im Verleich zu
anderen Algorithmen braucht dieser recht wenig Speicherplatz; außerdem erhalt
1In der klassischen Mechanik sind Orte und Geschwindigkeiten von Teilchen stetige Funk-tionen der Zeit, d.h. die Taylorentwicklung konvergiert zu jedem Teitpunkt t.
2Der Ubersichtlichkeit halber ersetzen wir t0 durch t und schreiben fortan fur die zeitlicheAbleitung dr
dt
∣∣t0
v(t0), analog a(t0) fur d2rdt2
∣∣∣t0
118

119
er sehr gut die Geamtenergie, was ein Zeichen fur die gute Reproduktion der
Bahnkurve des Systems im Phasenraum ist. Formt man (C.2) nach r(t− h) um,
und setzt das Ergebnis in (C.1) ein, so folgt fur die Geschwindigkeit zum Zeit-
punkt t:
v(t) =2r(t+ h) − 2r(t) − h2a(t)
2h.
Somit erhalt man fur den Ort zur Zeit t+ h:
r(t+ h) = r(t) + hv(t) +1
2h2a(t) . (C.3)
Fur die Ableitungen der Orte nach der Zeit gilt nach (C.2) folgendes:
v(t+ h) − v(t) =r(t+ 2h) − r(t+ h) + r(t− h) − r(t)
2h.
Einsetzen der Terme fur r(t+2h) und r(t−h) in (C.1) und Auflosen nach v(t+h)
ergibt folgende Gleichung:
v(t+ h) = v(t) +h
2
[a(t) + a(t+ h)
](C.4)
Gleichungen (C.3) und (C.4) stellen die sogenannte Geschwindigkeits-
Formulierung des Verlet-Algorithmus dar und sind dem ursprungichen Algorith-
mus algebraisch vollig aquivalent. Koordinaten und Geschwindigkeiten werden in
naturlicher Art und Weise behandelt, gespeichert werden immer r,v und a zum
Zeitpunkt t.

Literaturverzeichnis
[1] Siehe z.B. Programm des SFB 445 uber”Nanopartikel aus der Gasphase:
Entstehung, Charakterisierung, Eigenschaften.
[2] P. Jena, S. N. Khanna und B. K. Rao, Cluster assembled materials, hrsg.
von K. Sattler (Materials Science Forum, Zuerich, 1996), Bd. 232, S. 1.
[3] A. H. Zewail, Femtochemistry: Atomic-scale dynamics of the chemical bond,
J. Phys. Chem. B 104, 5660 (2000).
[4] J. Herzler, L. R, H.-J. Mick und P. Roth, Shock tube study of the formation
of TiN molecules and particles, Nanostructured Mater. 10, 1161 (1998).
[5] J. Herzler und P. Roth, Shock tube study of the reaction of H atoms with
TiCl4, J. Phys. Chem. A 101, 9341 (1997).
[6] A. Kunz und P. Roth, A high temperature study of the reaction SiH4 + H ⇀↽
SiH3 + H2, Ber. Bunsenges. Phys. Chem. 102, 73 (1998).
[7] A. Kunz und P. Roth, Dissociation of SiCl4 based on Cl- and Si-concentration
measurements, 27th Symposium (Int.) on Combustion/The Combustion In-
stitute 261 (1998).
[8] E. Schrodinger, Quantisierung als Eigenwertproblem, Ann. Phys. 81, 109
(1926).
[9] M. Born und J. R. Oppenheimer, Zur Quantentheorie der Molekeln,
Ann. Phys. 84, 457 (1927).
120

LITERATURVERZEICHNIS 121
[10] L. D. Landau und E. M. Lifschitz, Lehrbuch der Theoretischen Physik
(Akadenie-Verlag Berlin, Berlin, 1986), Bd. III.
[11] D. R. Hartree, The wave mechanics of an atom with a non-Coulomb central
field, Proc. Cambr. Phil. Soc. 24, 89, 111 (1928).
[12] V. Fock, Naherungsmethode zur Losung des quantenmechanischen Mehrkor-
perproblems, Z. Phys. 61, 126 (1930).
[13] T. A. Koopmans, Uber die Zuordnung von Wellenfunktionen und Eigenwer-
ten zu den einzelnen Elektronen eines Atoms, Physica 1, 104 (1933).
[14] C. C. J. Roothaan, New developments in Molecular Orbital Theory,
Rev. Mod. Phys. 23, 69 (1951).
[15] G. G. Hall, The Molecular Orbital theory of chemical valency VIII: A method
for calculating ionisation potentials, Proc. Royal Soc. (London) A 205, 541
(1951).
[16] J. A. Pople und R. K. Nesbet, Self-consistent orbitals for radicals,
J. Chem. Phys. 22, 571 (1954).
[17] J. C. Slater, Atomic shielding constants, Phys. Rev. 36, 57 (1930).
[18] F. Bloch, Uber die Quantenmechanik der Elektronen in Kristallgittern, 52,
555 (1928).
[19] W. Meyer, Theory of self-consistent electron pairs. An iterative method for
correlated many-electron wave functions, J. Chem. Phys. 64, 2901 (1976).
[20] J. Cızek, On the correlation problem in atomic and molecular systems, calcu-
lation of wavefunction components in Ursell-type expansion using quantum-
field theoretical methods, J. Chem. Phys. 45, 4256 (1966).
[21] J. Cızek und J. Paldus, Correlation problems in atomic and molecular sy-
stems. III. Rederivation of the coupled-pair many-electron theory using the
traditional quantum chemical methods, Int. J. Quantum Chem. 5, 359 (1971).

122 LITERATURVERZEICHNIS
[22] G. D. Purvis und R. J. Bartlett, A full coupled cluster singles and doubles
model: The inclusion of disconnected triples, J. Chem. Phys. 76, 1910 (1982).
[23] C. Møller und M. S. Plesset, Note on an approximate treatment for many-
electron systems, Phys. Rev. 46, 618 (1934).
[24] K. Raghavachari, G. W. Trucks, J. A. Pople und M. Head-Gordon,
A fifth-order perturbation comparison of electron correlation energies,
Chem. Phys. Lett 157, 479 (1989).
[25] B. O. Roos, P. R. Taylor und E. M. Siegbahm, A complete active space SCF
method (CASSCF) using a density matrix formulated super-CI approach,
Chem. Phys. 48, 157 (1980).
[26] J. A. Pople et al., Gaussian-1 theory: A general procedure for prediction of
molecular energies, J. Chem. Phys. 90, 5622 (1989).
[27] L. A. Curtiss, K. Raghavachari, G. W. Trucks und J. A. Pople, Gaussian-
2 theory for molecular energies of first- and second-row compounds,
J. Chem. Phys 94, 7221 (1991).
[28] D. Feller und K. A. Peterson, An examination of intrinsic errors in electronic
structure methods using the Environmental Molecular Sciences Laboratory
computational results database and the Gaussian-2 set, J. Chem. Phys. 108,
154 (1998).
[29] P. Hohenberg und W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136,
864 (1964).
[30] M. Levy, Universal variational functionals of electron densities, first or-
der density matrices, and natural spin orbitals and solution of the v-
representability problem, Proc. Natl. Acad. Sci. (USA) 76, 6062 (1979).
[31] W. Kohn und L. J. Sham, Self-consistent equations including exchange and
correlation effects, Phys. Rev. A 140, 1133 (1965).

LITERATURVERZEICHNIS 123
[32] A. Savin, Recent advances in density functional methods, hrsg. von D. P.
Chong (World Scientific, Singapore, 1995), S. 129.
[33] M. Filatov und S. Shaik, Spin-restricted density functional approach to the
open-shell problem, Chem. Phys. Lett. 288, 689 (1998).
[34] D. M. Ceperley und B. J. Alder, Ground state of the electron gas by a sto-
chastic method, Phys. Rev. Lett. 45, 566 (1980).
[35] S. H. Vosko, L. Wilk und M. Nusair, Accurate spin-dependent electron liquid
correlation energies for local spin density calculations: a critical analysis,
Can. J. Phys. 58, 1200 (1980).
[36] A. D. Becke, Density-functional exchange-energy approximation with correct
asymptotic behavior, Phys. Rev. A 38, 3098 (1988).
[37] J. P. Perdew und Y. Wang, Accurate and simple density functional
for the electronic exchange energy: Generalized gradient approximation,
Phys. Rev. B 33, 8800 (1986).
[38] J. P. Perdew und Y. Wang, Accurate and simple analytic representation of
the electron-gas correlation energy, Phys. Rev. B 45, 13244 (1992).
[39] A. D. Becke, Density-functional thermochemistry. IV. A new dynami-
cal correlation functional and implications for exact-exchange mixing,
J. Chem. Phys. 104, 1040 (1996).
[40] C. Lee, W. Yang und R. G. Parr, Development of the Colle-
Salvetti correlation-energy formula int a functional of th electron density,
Phys, Rev. B 37, 785 (1988).
[41] A. D. Becke, Density-functional thermochemistry. III. The role of exact ex-
change, J. Chem. Phys. 98, 5648 (1993).
[42] W. Koch und M. Holthausen, A chemist’s guide to density functional theory
(Wiley-VCH, Weinheim, 2000).

124 LITERATURVERZEICHNIS
[43] D. J. Adams, Alternatives to the Periodic Cube in Computer Sinulation,
CCP5 Quaterly 10, 30 (1983).
[44] S. F. Boys, Electronic wave functions. I. A general method of calculation for
the stationary states of any molecular system, Proc. Royal Soc. (London) A
200, 542 (1950).
[45] L. Verlet, Computer ’experiments’ on classical fluids. I. Thermodynamical
properties of Lennard-Jones molecules, Phys. Rev. 159, 98 (1967).
[46] M. P. Allen und D. J. Tildesley, Computer simulation of liquids (Clarendon
Press, Oxford, 1993).
[47] R. Haberlandt, Molekulardynamik : Grundlagen und Anwendungen (Vieweg,
Braunschweig, 1995).
[48] Gaussian 98, Revision A.7, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G.
E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Mont-
gomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A.
D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M.
Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Och-
terski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A.
D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A.
G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.
Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng,
A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson,
W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S.
Replogle, and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
[49] G. Kresse und J. Furthmuller, Efficient iterative schemes for ab initio total-
energy calculations using a plane wave basis set, Phys. Rev. B 54, 11169
(1996).

LITERATURVERZEICHNIS 125
[50] D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenva-
lue formalism, Phys. Rev. B 41, 7892 (1990).
[51] G. Kresse und J. Hafner, Norm-conserving and ultrasoft pseudopotentials for
first-row and transition elements, J. Phys.: Condens. Matter 6, 8245 (1994).
[52] W. R. Schulz und D. J. Leroy, Kinetics of the reaction H+p−H2 ⇀↽ o−H2+H,
J. Chem. Phys. 42, 3869 (1965).
[53] J. M. W. Chase et al., JANAF Thermochemical Tables, 3rd edn,
J. Phys. Chem. Ref. Data 4, Monograph 9 (1998).
[54] G. I. Czonka und B. G. Johnson, Inclusion of exact eschange for self-
interaction corrected H3 density functional potential energy surface, The-
or. Chem. Acc. 99, 158 (1998).
[55] P. M. W. Gill, B. G. Johnson, J. A. Pople und M. J. Frisch, The performance
of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various
basis sets, Chem. Phys. Lett. 197, 499 (1992).
[56] J. C. W. Bauschlicher und H. Partridge, A modification of the Gaussian-2
approach using density functional theory, J. Chem. Phys 103, 1788 (1995).
[57] L. A. Curtiss et al., Extension of Gaussian-2 theory to molecules containing
third-row atoms Ga-Kr, J. Chem. Phys 103, 6104 (1995).
[58] L. A. Curtiss, K. Raghavachari, P. C. Redfern und J. A. Pople, Assessment of
Gaussian-2 and density functional theories for the computation of enthalpies
of formation, J. Chem. Phys 106, 1063 (1997).
[59] Recent developments and applications of modern density functional theory,
hrsg. von J. M. Seminario (Elsevier, Amsterdam, 1996).
[60] M. T. Swihart und S. L. Girshick, Thermochemistry and kinetics of si-
licon hydride cluster formation during thermal decomposition of silane,
J. Phys. Chem. B 103, 64 (1999).

126 LITERATURVERZEICHNIS
[61] W. Kohn, Nobel lecture: electronic structure of matter - wave functions and
density functionals, Rev. Mod. Phys. 71, 1253 (1999).
[62] J. P. Perdew und A. Zunger, Self-interaction correction to density-functional
approximations for many-electron systems, Phys. Rev. B 23, 5048 (1981).
[63] G. Kresse und D. Joubert, From ultrasoft pseudopotentials to the projector
augmented-wave method, Phys. Rev. B 59, 1758 (1999).
[64] W. J. Hehre, R. F. Stewart und J. A. Pople, Self-consistent molecular or-
bital methods. I. Use of gaussian expansions of slater-type atomic orbitals,
J. Chem. Phys 51, 2657 (1969).
[65] R. Ditchfield, W. J. Hehre und J. A. Pople, Self-consistent molecular orbital
methods. IX. An extended Gaussian-type basis for molecular orbital studies
of organic molecules, J. Chem. Phys 54, 724 (1971).
[66] W. J. Hehre, R. Ditchfield und J. A. Pople, Self-consistent molecular or-
bital methods. XII. Further Extensions of gaussian-type basis sets for use
in molecular orbital studies of organic molecules, J. Chem. Phys 56, 2257
(1972).
[67] J. S. Binkley, J. A. Pople und W. J. Hehre, Self-consistent molecular or-
bital methods. XXI. Small split-valence basis sets for first row elements,
J. Am. Chem. Soc. 102, 939 (1980).
[68] R. Krishnan, J. S. Binkley, R. Seeger und J. A. Pople, Self-consistent mo-
lecular orbital methods. XX. A basis sets for correlated wave functions,
J. Chem. Phys 72, 650 (1980).
[69] A. D. McLean und G. S. Chandler, Contracted Gaussian basis sets for mole-
cular calculations. I. Second row atoms, Z=11-18, J. Chem. Phys. 72, 5639
(1980).

LITERATURVERZEICHNIS 127
[70] A. J. H. Wachters, Gaussian basis set for molecular wavefunctions containing
third.row atoms, J. Chem. Phys. 52, 1033 (1970).
[71] P. J. Hay, Gaussian basis sets for molecular calculations. The representation
of 3d orbitals in transition-metal atoms, J. Chem. Phys. 66, 4377 (1977).
[72] J. T. H. Dunning, Gaussian basis sets for use in correlated molecular calcu-
lations. I. The atoms boron through neon and hydrogen, J. Chem. Phys. 90,
1007 (1989).

128 LITERATURVERZEICHNIS
An dieser Stelle mochte ich mich bei allen Personen bedanken, die zum Ge-
lingen dieser Arbeit beigetragen haben.
Zuerst gebuhrt mein Dank Herrn Prof. Entel fur die interessante Aufgaben-
stellung und dafur, daß er mir die Moglichkeit verschaffte, im Rahmen eines
Aufenthaltes an einem anderen Institut Erfahrungen zu sammeln.
Herrn Prof. Sauer mochte ich fur die freundliche Einladung nach Berlin und
die Aufnahme an seinem Institut danken. In diesem Rahmen mochte ich mich
bei Philipp und Max fur eine tolle Betreuung und viele interessante Gesprache
bedanken, die mir den Zugang zur Quantenchemie eroffnet haben.
Stefan, Lars, Minoru und Kai danke ich fur ihre standige Hilfsbereitschaft und
die ausgezeichnete Arbeitsatmosphare.
Ferner gilt mein Dank Egbert Hoffmann und Heike Herper fur aufschlußreiche
Diskussionen und die Beantwortung vieler Fragen. Ohne die Bereitstellung vieler
Tools ware die Bearbeitung der Ergebnisse nicht in dieser Form moglich gewesen.
Allen Leuten, die das Manuskript dieser Arbeit gelesen und mir durch ihre
Hilfe und Kritik vieles erleichtert haben, gilt mein spezieller Dank. Außerordent-
lich dankbar bin ich Egbert und Markus fur ihre großartige Hilfe bis zur letzten
Minute.
Besonders bedanke ich mich bei Sandra fur ihre Unterstutzung gerade in
schwierigen Phasen.
Bei meinen Eltern und meinem Bruder Stefan mochte ich mich fur die mo-
ralische Unterstutzung bedanken, die sie mir sowohl wahrend meines gesamten
Studiums als auch im Hinblick auf meine Diplomarbeit zukommen ließen. Ohne
ihre Hilfe hatte ich mein Studium nicht in dieser Weise absolvieren konnen.
Hiermit erklare ich, die vorliegende Diplomarbeit eigenstandig und nur mit den
angegebenen Hilfsmitteln erstellt zu haben.
Duisburg, den 14. Marz 2001





























