Embed Size (px)
Citation preview

Molekulardynamik-Simulationenvon strukturellen Phasenumwandlungen
in Festkorpern, Nanopartikeln undultrad unnen Filmen
Vom Fachbereich Physik – Technologie derGerhard-Mercator-Universit¨at – Gesamthochschule Duisburg
zur Erlangung des akademischen Grades einesDoktors der Naturwissenschaften genehmigte Dissertation
vonKai Kadau
ausVoerde
Referent: Prof. Dr. P. EntelKorreferent: Prof. Dr. K. D. Usadel
Tag der mundlichen Pr¨ufung: 9. Marz 2001


Fur Anna und Antje, sowie meinenEltern und meinem Bruder Dirk.


5
Zusammenfassung
Im Rahmen dieser Arbeit wurden verschiedene strukturelle Eigenschaften von Festk¨orpern,Nanopartikeln und d¨unnen Filmen mit Hilfe von Molekulardynamik-Simulationen untersucht.Der Schwerpunkt lag dabei in der Untersuchung von martensitischen und austenitischen Trans-formationen in diesen Systemen, die in der Regel aus Fe und Ni bestanden. Dabei wurden zurBeschreibung der interatomaren WechselwirkungEmbedded-Atom Method-Potentiale verwen-det.
Die Berechnung der freien Energie in Abh¨angigkeit der Temperatur erlaubte die Ana-lyse der auftretenden strukturellen Phasen¨ubergange zwischen einer dicht gepackten Struk-tur (kubisch-flachenzentriert, hexagonal-dichteste Packung) und der kubisch-raumzentriertenStruktur. Es konnte gezeigt werden, daß Gitterdefekte ein entscheidender Faktor f¨ur die mar-tensitischenUbergange bei tiefen Temperaturen sind, wohingegen die austenitische Transfor-mation bei hohen Temperaturen kaum durch Gitterdefekte beeinflußt wird. Die Erkl¨arung desunterschiedlichen Verhaltens der martensitischen und der austenitischen Transformation ergabsich aus der Analyse der freien Energie entlang Bain-Wegs.
Das Studium der kristallographischen Orientierungsbeziehungen zwischen den Phasen vorund nach der Umwandlung gab Aufschluß ¨uber den Transformationsprozeß auf der atoma-ren Skala. Der gesamte in den Simulationen auftretende Transformationsprozeß ließ sich ent-sprechend der Wechsler-Liebermann-Read-Theorie mathematisch durch eine Kombination ausBain-Transformation, Rotation und einer gitterinvarianten Scherung bedingt durch Stapelfehlerund Zwillingsbildung darstellen.
Die Simulation von sehr großen Systemen, die bis zu acht millionen Atome enthielten,ermoglichte eine eingehende Untersuchung der homogenen und heterogenen Nukleationspro-zesse, die mit dem Auftreten der strukturellen Phasen¨ubergange verbunden sind. Bei Mo-lekulardynamik-Simulationen von schockinduzierten austenitischen Umwandlungen konntedie Dynamik der Korngrenzen zwischen den sich bildenden, verschieden orientierten Varian-ten der Austenitphase beobachtet werden. Temperaturgetriebene martensitische Umwandlun-gen in kleinen Partikeln mit einem Durchmesser von einigen Nanometern zeigten eine aus-gepragte heterogene Nukleation an vorhandenen Defekten und ein anschließendes Wachstumder verzwillten martensitischen Phase in der Austenitmatrix. Es zeigte sich wie auch im Ex-periment eine deutliche Abnahme derUbergangstemperaturen mit abnehmender Partikelgr¨oße.Dies konnte im Rahmen der verwendetenEmbedded-Atom Method-Potentiale durch eine un-terschiedliche Modifikation der freien Energie eines Partikels bedingt durch unterschiedlicheOberflachenenergien der austenitischen und der martensitischen Phase erkl¨art werden.
Das Zusammenspiel der Strukturen von Substrat- und Filmoberfl¨ache wurde intensivam experimentell h¨aufig untersuchten Modellsystem Fe auf Cu untersucht. Dabei wurdendie experimentellen Sachverhalte wie die Umwandlung der kubisch-fl¨achenzentrierten in diekubisch-raumzentrierte Struktur mit zunehmender Filmdicke und abnehmender Temperaturbestatigt. Simulationen von Aufwachsprozessen konnten den experimentell beobachteten Un-terschied bzgl. der strukturellen Stabilit¨at von kubisch-fl¨achenzentrierten d¨unnen Fe-Schichtenauf Cu(111)-Substratoberfl¨achen in Abh¨angigkeit des Filmherstellungsverfahrens beschreiben.


7
Abstract
In this work structural properties of bulk, nano-particles, and thin films were investigated byusing molecular-dynamics simulations. The focus was on the investigation of martensitic trans-formations in those systems, mainly consisted of Fe and Ni. For the describtion of the inter-atomic forces theEmbedded-Atom Methodwas used.
The calculation of the free energy as a function of temperature gave insight into the ther-modynamics of the system, and led to a correct interpretation of the structural transformationfrom a closed packed structure (face-centered-cubic, hexagonal-closed-packed) to the body-centered-cubic structure and vice versa. Pre-existing lattice defects turned out to be the domi-nant factor for the martensitic transformation at low temperatures, whereby the austenitic trans-formation at high temperatures is less affected by defects. The explanation of the different be-havior of the martensitc transformation process and the austenitic transformation process couldbe given by a detailed examination of the free energy along the Bain-path.
The study of the crystallographic orientational relationships of the austenitic and marten-sitic phases gave insight into the transformation process on the atomic-scale. The transforma-tion process observed in the molecular-dynamics simulations can be described in terms of theWechsler-Lieberman-Read theory as a combination of Bain-transformation, rotation, and lat-tice invariant shear due to stacking faults and twinning.
Simulations of very large supercells containing up to eight million atoms facilitated thestudy of the homogeneous and heterogeneus nucleation process of structural phase transitionswithin the solid state. Molecular-dynamics simulations of shock-induced austenitic transfor-mations gave valueable insight into the grain-boundary dynamics of the developing austeniticgrains. The heterogeneous nucleation process at different types of defects in nano-particles wasstudied. The burst-type growth of the martensitc phase starts at pre-existing defects with fur-ther growth of the twinned martensitic phase into the austenite-matrix. With decreasing sizeof the nano-particle, transition temperatures decreased as revealed in the few experiments thatexist. In the framework of the usedEmbedded-Atom Method-potential, this effect is due to thedifferent surface energies of the austenitic and the martensitic phases.
The interplay between the structure of films and an underlying substrate was intensivelystudied for the well known Fe on Cu-system. Experimental observations, like the increasingtendency for a structural transformation from the face-centered-cubic structure to the body-centered-cubic structure with increasing film thickness and decreasing temperature, were con-firmed. Simulations of the growth process gave insight into recently performed experiments ofthe dependence of the structural stability of face-centered-cubic Fe-films on Cu(111)-substratesas a function of the deposition process like thermal deposition and pulsed laser deposition.


9
Inhaltsverzeichnis
Zusammenfassung 5
Abstract 7
Inhaltsverzeichnis 9
Abbildungsverzeichnis 12
Tabellenverzeichnis 14
1 Einleitung 15
2 Molekulardynamik-Simulationen 182.1 Grundlagen .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.1.1 Verlet-Algorithmen . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.1.2 Potentiale endlicher Reichweite . . . . . . . . . . . . . . . . . . . . . 202.1.3 Randbedingungen. . . . . . . . . . . . . . . . . . . . . . . . . . . . 212.1.4 Temperatur . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.2 Auswertung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242.2.1 Druck . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242.2.2 Radiale Verteilungsfunktion . .. . . . . . . . . . . . . . . . . . . . . 252.2.3 Lokale Gitterstruktur der Atome . . . . . . . . . . . . . . . . . . . . . 262.2.4 Freie Energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.2.5 Quadratische Abweichung . . .. . . . . . . . . . . . . . . . . . . . . 28
2.3 Embedded-Atom Method (EAM)-Potentiale . . . . . . . . . . . . . . . . . . . 292.3.1 Idee und Formulierung nach Daw und Baskes . . . . . . . . . . . . . . 292.3.2 Anpassung der EAM-Potentiale. . . . . . . . . . . . . . . . . . . . . 30
2.4 Spezielle Ensemble und Simulationsmethoden . . . .. . . . . . . . . . . . . . 322.4.1 Nose-Hoover-Thermostat (NVT-Ensemble) . . . . . . . . . . . . . . . 322.4.2 Anderson-Methode (NPE-Ensemble) . . . .. . . . . . . . . . . . . . 342.4.3 Depositionsverfahren (Wachstum d¨unner Filme) . . . . .. . . . . . . 362.4.4 Induzierung von Schockwellen .. . . . . . . . . . . . . . . . . . . . . 37
2.5 Programmcode und Simulationsparameter . . . . . .. . . . . . . . . . . . . . 38
3 Struktur von Metallen 413.1 Allgemeines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 413.2 Martensitische Transformationen . . . .. . . . . . . . . . . . . . . . . . . . . 45
3.2.1 Einfuhrung und Ph¨anomenologie . . . . . . . . . . . . . . . . . . . . 453.2.2 Thermodynamik der Umwandlung und Nukleation . . . .. . . . . . . 48
3.3 StrukturelleAnderungen durch Schockeinwirkung . .. . . . . . . . . . . . . . 503.4 Festk¨orper, Nanopartikel und d¨unne Schichten . . .. . . . . . . . . . . . . . 51

10 Inhaltsverzeichnis
4 EAM-Potentiale fur Eisen und Nickel 534.1 Festk¨orper . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 534.2 Oberflachen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5 Simulationen metallischer Festkorper 575.1 Das Fe-Ni-System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
5.1.1 Martensitische und austenitische Transformationen. . . . . . . . . . . 575.1.2 Thermodynamik des Systems. . . . . . . . . . . . . . . . . . . . . . 605.1.3 (Freie) Energie entlang des Transformationswegs (Bain-Weg) .. . . . 62
5.2 Schockinduzierte strukturelleAnderungen in krz Fe . . . .. . . . . . . . . . . 655.2.1 Analyse des Schockeinflusses. . . . . . . . . . . . . . . . . . . . . . 655.2.2 Strukturelle Analyse der schockinduzierten Transformation . .. . . . 69
6 Untersuchungen an Fe80Ni20-Nanopartikeln 736.1 Einfluß der Oberfl¨ache auf die thermodynamische Gleichgewichtstemperatur . 736.2 Heterogene Keimbildung von Martensit . . .. . . . . . . . . . . . . . . . . . 75
6.2.1 Nukleation an Ecken . . . . . . . . . . . . . . . . . . . . . . . . . . . 766.2.2 Nukleation durch fl¨achenhafte Defekte . . . . . .. . . . . . . . . . . 80
7 Simulationen metallischer Filme auf Substraten 857.1 Voruberlegungen . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 857.2 Fe auf Cu(001) . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
7.2.1 Geschlossene Fe-Filme . . . .. . . . . . . . . . . . . . . . . . . . . . 877.2.2 Einzelne Fe-Insel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
7.3 Fe auf Cu(111) . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 927.3.1 Geschlossene Fe-Filme . . . .. . . . . . . . . . . . . . . . . . . . . . 927.3.2 Aufwachsprozesse . . . . . .. . . . . . . . . . . . . . . . . . . . . . 93
8 Zusammenfassung der Ergebnisse 988.1 Festk¨orper . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 988.2 Nanopartikel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 998.3 Dunne Filme . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1008.4 Ausblick . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
9 Anhang 1039.1 Atomare Einheiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1039.2 Parameter der EAM-Potentiale . . . .. . . . . . . . . . . . . . . . . . . . . . 1049.3 Ausgaben des MD-Programms . . . .. . . . . . . . . . . . . . . . . . . . . . 1069.4 Einfaches Oszillatormodell . . . . . .. . . . . . . . . . . . . . . . . . . . . . 109
Literaturverzeichnis 111
Stichwortverzeichnis 118

Inhaltsverzeichnis 11
Danksagung 123

12
Abbildungsverzeichnis
1.1 AustenitischeUbergangstemperaturen in Abh¨angigkeit vom Partikeldurchmesser . 17
2.1 Verlet-Listen und Zellenmethode . . . . .. . . . . . . . . . . . . . . . . . . . . . 212.2 Periodische Randbedingungen . . . . . .. . . . . . . . . . . . . . . . . . . . . . 222.3 Maxwell-Boltzmann-Verteilung . . . . . . . . . . . . . . . . . . . . . . . . . . . 232.4 Radiale Verteilungsfunktionen bei unterschiedlichen Temperaturen . . . . .. . . . 252.5 Standardabweichung des Mittelwerts des Abstands der 10 n¨achstgelegenen Atome 262.6 Einbettung eines Metallatoms in die Elektronendichte der umgebenden Atome . . . 292.7 Bindungsenergien f¨ur EAM-Potentiale von Al . . . . . . . . . . . . . . . . . . . . 312.8 Effektive Paarpotentiale f¨ur Al . . . . . . . . . . . . . . . . . . . . . . . . . . . . 322.9 Instantane Temperatur . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 342.10 Instantaner Druck . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 352.11 Volumenfluktuationen einer Superzelle . .. . . . . . . . . . . . . . . . . . . . . . 362.12 Epitaxieverfahren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 372.13 Momentum-Mirror-Verfahren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 382.14 Ablaufplan einer Molekulardynamik-Simulation. . . . . . . . . . . . . . . . . . 392.15 Verhalten der Erhaltungsgr¨oße in Abhangigkeit des Zeitschritts. . . . . . . . . . . 40
3.1 kfz, hdp und krz Gitter . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 413.2 (001)kfz- und(001)krz-Ebene . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 423.3 (111)kfz-, (0001)hdp- und(110)krz-Ebene . . . . . . . . . . . . . . . . . . . . . . 423.4 (110)kfz- und(1210)hdp-Ebene . . . . . . . . . . . . . . . . . . . . . . . . . . . . 433.5 Radiale Verteilungsfunktionen von krz, kfz und hdp Gittern . .. . . . . . . . . . . 443.6 Bain-Transformation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 463.7 Schematische Darstellung der Abl¨aufe beim Martensit¨ubergang . . . . . . . . . . . 473.8 Transmissionselektronenmikroskopische Aufnahme einer Martensitnadel .. . . . 473.9 Freie Enthalpie beim strukturellenUbergang . . . . . . . . . . . . . . . . . . . . . 493.10 Freie Keimbildungsenthalpie∆G eines Martensitkeims . . . . . . . . . . . . . . . 503.11 Phasendiagramm von Fe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
4.1 Energie in Abh¨angigkeit vom Volumen f¨ur Fe und Fe80Ni20 . . . . . . . . . . . . . 534.2 Druckkomponenten an Fe(001)krz-Oberflachen . . . . . . . . . . . . . . . . . . . 55
5.1 Hystereseschleifen f¨ur Fe80Ni20 bei der Simulation . . . . . . . . . . . . . . . . . 575.2 Phasendiagramm des Fe-Ni-Systems . . . . . . . . . . . . . . . . . . . . . . . . . 585.3 Verzwillte und verstapelte Austenitstruktur. . . . . . . . . . . . . . . . . . . . . . 595.4 Freie Energie einer Fe80Ni20-Legierung . . . . . . . . . . . . . . . . . . . . . . . 615.5 Vergleich von entropischem Beitrag und Energiebeitrag zur freien Energie .. . . . 615.6 Energie entlang des Bain-Wegs . . . . . . . . . . . . . . . . . . . . . . . . . . . . 635.7 Freie Energie entlang des Bain-Wegs . . . . . . . . . . . . . . . . . . . . . . . . . 635.8 Variation des Entropieanteils der freien Energie .. . . . . . . . . . . . . . . . . . 645.9 Geschockte Fe-Proben . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66

Abbildungsverzeichnis 13
5.10 Homogene Nukleation vonε-Fe induziert durch eine Schockwelle . .. . . . . . . 675.11 SchockgeschwindigkeitenUs als Funktion der KolbengeschwindigkeitUp fur Fe . . 685.12 Radiale Verteilungsfunktionen geschockter krz Fe-Kristalle . . . . . .. . . . . . . 695.13 Querschnitt des transformierten Kristalls senkrecht zur Schockrichtung. . . . . . . 705.14 Korngrenzendynamik eines geschockten Kristalls . . . .. . . . . . . . . . . . . . 71
6.1 TA in Abhangigkeit vom reziproken Partikeldurchmesser. . . . . . . . . . . . . . 756.2 Zeitliche Entwicklung der radialen Verteilungsfunktion eines kubischen Partikels . 766.3 Nukleation von Martensit an den Ecken eines kubischen Nanopartikels. . . . . . . 776.4 Verzwillte martensitische eines Nanopartikels . . . . . .. . . . . . . . . . . . . . 786.5 Zeitliche Entwicklung der radialen Verteilungsfunktion bei der Nukleation durch
einen flachenhaften Defekt . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . 806.6 Ausheilung eines fl¨achenhaften Defekts . .. . . . . . . . . . . . . . . . . . . . . 816.7 Nukleation durch fl¨achenhafte Defekte . . .. . . . . . . . . . . . . . . . . . . . . 826.8 Martensitbildung eines fehlerbehafteten sph¨arischen Nanopartikels . . . . . . . . . 836.9 Martensitanteil in Abh¨angigkeit von der Zeit . . . . . . . . . . . . . . . . . . . . . 84
7.1 Vergleich von kristallographisch verschieden orientierten Substraten und Filmen . . 867.2 Strukturelles Phasendiagramm von Fe auf Cu(001) . . .. . . . . . . . . . . . . . 867.3 Simulationszelle f¨ur einen geschlossenen Fe-Film auf einer Cu(001)-Oberfl¨ache . . 877.4 Struktur des Fe-Films auf einem Cu(001)-Substrat bei 150 K und 1 K. . . . . . . 887.5 Pitsch-Orientierung eines krz Fe-Films auf einem Cu(001)-Substrat .. . . . . . . 897.6 Simulationszelle einer Fe-Insel auf einer Cu(001)-Oberfl¨ache . . . . . . . . . . . . 917.7 Schichtweise Darstellung der Struktur einer Fe-Insel auf einem Cu(001)-Substrat. . 917.8 Schematische Darstellung der Kurdjumov-Sachs-Orientierung eines Fe-Films auf
einer Cu(111)-Oberfl¨ache . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 937.9 Lagenfullung eines aufwachsenden Fe-Films . . . . . . . . . . . . . . . . . . . . 947.10 Vier Monolagen Fe auf Cu(111) . . . . . .. . . . . . . . . . . . . . . . . . . . . 957.11 Acht Monolagen Fe auf Cu(111) . . . . . .. . . . . . . . . . . . . . . . . . . . . 957.12 Paarkorrelationsfunktionen von Fe-Filmen Cu(111) . . .. . . . . . . . . . . . . . 96
9.1 Freie Energien zweier einfacher Modellstrukturen . . . . . . . . . . . . . . . . . . 109

14
Tabellenverzeichnis
2.1 Vergleich von experimentellen Daten und EAM-Potential . . . . . . . . . . . . . . 31
3.1 Anzahl der Atome in den Nachbarschalen verschiedener Gitterstrukturen . . . . . . 433.2 Zwillingselemente verschiedener Strukturen . . .. . . . . . . . . . . . . . . . . . 443.3 Beispiele f¨ur Systeme, die martensitischeUbergange zeigen . . . . . . . . . . . . 45
4.1 Mittlere potentielle Energie von Fe f¨ur verschiedene Temperaturen . . . . .. . . . 544.2 Gitterkonstanten von Fe und Ni f¨ur verschiedene Temperaturen. . . . . . . . . . . 544.3 Oberflachenenergien von Fe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
6.1 Austenitische Umwandlungstemperaturen in Abh¨angigkeit der Partikelgr¨oße . . . . 74
7.1 krz Anteil eines Fe-Films auf Cu(001) mit Leerstellen . . . . .. . . . . . . . . . . 877.2 krz Anteil eines Fe-Films auf Cu(001) ohne Leerstellen . . . .. . . . . . . . . . . 907.3 krz Anteil eines Fe-Films auf Cu(111) mit Leerstellen . . . . .. . . . . . . . . . . 92
9.1 Atomare Einheiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1039.2 EAM-Potential fur Al . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1049.3 EAM-Potential fur Ni . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1049.4 EAM-Potential fur Fe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1059.5 EAM-Potential fur Cu . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1059.6 EAM-Potential fur Cu von Guiet al. . . . . . . . . . . . . . . . . . . . . . . . . . 105

15
1 Einleitung
Strukturelle Eigenschaften von Metallen und ihren Legierungen besch¨aftigen die Menschenschon seit der Antike. Bereits 800 Jahre vor unserer Zeitrechnung berichtete der Dichter Ho-mer [1] uber die Nutzung des strukturellen Phasen¨ubergangs in Eisen zur H¨artung von Schwer-tern der antiken Griechen. Die ersten wissenschaftlichen Untersuchungen struktureller Eigen-schaften von verschiedenen St¨ahlen wurden gegen Ende des 19. Jahrhunderts zun¨achst vonHenry Clifton Sorby und sp¨ater von Floris Osmond und Adolf Martens durchgef¨uhrt. Weiterein diesem Zusammenhang zu erw¨ahnende historische Namen sind Henry Marion Howe, Louis-Joseph Troost und William Roberts-Austen [2]. Zu Ehren von Roberts-Austen und Martens [3]wurde die strukturelle Hochtemperaturphase von Eisen-Kohlenstoff-Legierungen (kubisch-flachenzentrierte Struktur) Austenit, sowie die Tieftemperaturphase (kubisch-raumzentrierteStruktur) als Martensit benannt. Die Namensgebung hat sich bis heute gehalten und bezeichnetjetzt materialunabh¨angig spezielle strukturelle Phasen¨ubergange, die sogennannten martensi-tischen Umwandlungen [4, 5]. Bis zum heutigen Tag haben Ingenieure, Mediziner, Biologen,Metallurgen und Physiker zum Verst¨andnis struktureller Eigenschaften von Metallen und derenLegierungen beigetragen. Im einzelnen hier nicht aufz¨ahlbare Anwendungen und Forschungs-gebiete von der Schwerindustrie [6] bis hin zur Nanotechnologie [7] wurden geschaffen. Ge-nannt seien nur die Datenspeicherung [8], thermische Schalter [5] und medizinische Anwen-dungen wie Implantate, Endoskopsteuerung und Zahnspangen [9]. Schiffstanks f¨ur den Trans-port von verflussigtem Erdgas werden aus Fe-Ni Invar-Legierungen [10] hergestellt, die einenextrem niedrigen thermischen Ausdehnungskoeffizienten aufweisen. Trotz aller Erkenntnisseund der daraus resultierenden Entwicklung bleiben immer noch ungekl¨arte Fragen insbesonde-re im Hinblick auf die involvierten atomaren Prozesse bei strukturellen Phasenumwandlungen.Die verwendeten Materialien werden immer komplizierter und raffinierter, was auch durch dieWortschopfungSmart Materialszum Ausdruck gebracht wird.
Ein atomares Verst¨andnis des Festk¨orpers, insbesondere der Wechselwirkungen der Ato-me untereinander, wurde erst Anfang des 20. Jahrhunderts mit der Entwicklung der Quanten-mechanik (z. B. [11, 12]) m¨oglich. Seit der Postulierung der Bewegungsgesetze der klassi-schen Mechanik durch Newton [13] ist das L¨osen von Bewegungsproblemen vieler Teilchenim Prinzip moglich, aber erst durch den Einsatz von modernen Computersystemen f¨ur eine ho-he Anzahl von Teilchen durchf¨uhrbar. Molekulardynamik-Simulationen, d. h. das L¨osen klas-sischer Bewegungsgleichungen f¨ur eine große Anzahl von Atomen k¨onnen ein tiefgehendesVerstandnis des Festk¨orpers liefern [14–18].
Im Rahmen dieser Arbeit werden strukturelle Eigenschaften von Festk¨orpern, Nanopar-tikeln und dunnen Schichten unter Verwendung von Molekulardynamik-Simulationen unter-sucht. Der Schwerpunkt liegt in der Untersuchung struktureller Umwandlungen von Eisensund Eisen-Nickel-Legierungen unter dem Einfluß von Oberfl¨achen und anderen Defekten. Zieldieser Arbeit ist es, Vorstellungen atomarer Abl¨aufe zu entwickeln und darauf basierend An-regungen f¨ur die Interpretation von Experimenten und Inspirationen f¨ur neue Experimente zugeben. Außerdem werden M¨oglichkeiten und Grenzen des theoretischen Konzepts der Mo-lekulardynamik-Simulationen unter Verwendung der benutzten Kraftans¨atze ausgelotet.

16 1 Einleitung
Aussagekr¨aftige Ergebnisse insbesondere im Hinblick auf Nukleationsprozesse erfordernSimulationen mit einer großen Anzahl von Atomen. Dazu wurden in dieser Arbeit Zellen mitbis zu acht millionen Teilchen verwendet. Zum Vergleich sei angef¨uhrt, daß der Molekulardy-namik -Weltrekord (28.10.1999) auf einem massiv parallelen Rechnersystem des Typs CRAYT3E-1200 mit 512 Prozessoren (600 MHZ) und einem Hauptspeicher von 512MB/Prozessordes Forschungszentrum J¨ulich von Dr. J. Roth vom Institut f¨ur Theoretische und AngewandtePhysik der Universit¨at Stuttgart mit einer Simulation von ¨uber funf Milliarden Atomen fur sechsIntegrationschritte gehalten wird [19], wobei allerdings zur Beschreibung der interatomarenWechselwirkung ein einfaches Lennard-Jones-Potential Verwendung fand.
Im Anschluß an diese Einleitung wird die Methode der Molekulardynamik-Simulation vor-gestellt. Dies umfaßt auch die wichtige und mit zunehmender Anzahl von Atomen immerschwieriger werdende Auswertung der gewonnenen Daten und deren physikalische Interpre-tation. Es wird dieEmbedded-Atom Method(EAM), welche die interatomaren Kr¨afte von Me-tallen beschreibt, sowie verschiedene Algorithmen und Methoden der Molekulardynamik (MD)vorgestellt. Im 3. Kapitel werden die strukturellen Eigenschaften von Metallen und die damitverbundenen physikalischen Vorg¨ange kurz dargestellt, wobei speziell auf martensitische Pha-senubergange eingegangen wird.
In den vier daran anschließenden Kapiteln werden Simulationsergebnisse von Festk¨orpern,Partikeln und d¨unnen Filmen mit einer Ausdehnung bis zu einigen zehn Nanometern vorge-stellt. Strukturelle Eigenschaften werden anhand atomarer Prozesse und Eigenschaften deruntersuchten EAM-Kristalle erl¨autert. Die Ergebnisse werden als Diskussionsgrundlage f¨urdie Interpretation von experimentellen Beobachtungen verwendet. Die Kenntnis thermody-namischer Gr¨oßen wie der freien Energie und der Entropie bilden dabei die Grundlage derDiskussion der hier untersuchten strukturellenUbergange. Einen zentralen Punkt stellt da-bei der Verlauf der freien EnergieF(T,V) entlang des Transformationspfads dar, mit dessenHilfe die auftretenden Nukleationsprozesse analysiert und interpretiert werden. Am Beispielvon schockinduzierten strukturellen Transformationen wird der Mechanismus der homoge-nen Nukleation im Rahmen von Molekulardynamik-Simulationen diskutiert. Heterogene Nu-kleationsprozesse martensitischer Transformationen werden unter dem Einfluß verschiedenerGitterfehler untersucht und das Wachstum der martensitischen Keime verfolgt. Die systema-tische Untersuchung der Phasenstabilit¨at von Nanopartikeln unterschiedlichen Durchmessersgibt Aufschlußuber den unterschiedlichen Beitrag der Oberfl¨ache zur freien Energie der mar-tensitischen und der austenitischen Phase. In diesem Zusammenhang sei auf das Verhaltender Austenitubergangstemperaturen in Fe80Ni20-Partikeln hingewiesen. Im Unterschied zumMartensitubergang mit abnehmender Temperatur ist der Einfluß von Defekten auf den Auste-nitubergang als gering einzusehen. Ein wichtiges Ergebnis in Kapitel 6 ist die in Simulationengefundene Gr¨oßenabh¨angigkeit der Austenit¨ubergangstemperatur, die mit abnehmender Parti-kelgroße abnimmt (Abb. 1.1).
Das Zusammenspiel struktureller Eigenschaften von d¨unnen, aus wenigen Monolagenbestehenden Filmen und dem Substrat wird eingehend anhand von Molekulardynamik-Si-mulationen diskutiert. In Abh¨angigkeit der Filmdicke wird der Einfluß der Oberfl¨ache undder Grenzflache zwischen Substrat und Film auf die strukturellen Eigenschaften des Filmsuntersucht. Verschiedene kristallographische Orientierungen des Substrats ergeben dabei un-
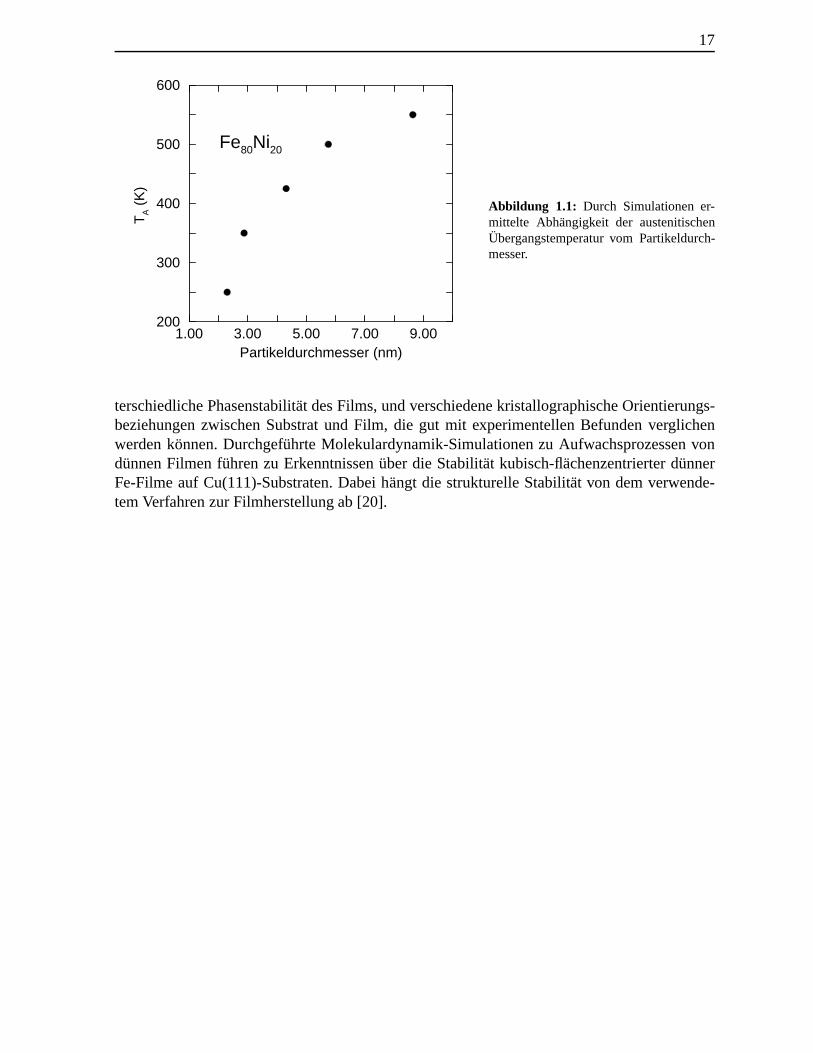
17
1.00 3.00 5.00 7.00 9.00Partikeldurchmesser (nm)
200
300
400
500
600
TA (
K)
Fe80Ni20
Abbildung 1.1: Durch Simulationen er-mittelte Abhangigkeit der austenitischenUbergangstemperatur vom Partikeldurch-messer.
terschiedliche Phasenstabilit¨at des Films, und verschiedene kristallographische Orientierungs-beziehungen zwischen Substrat und Film, die gut mit experimentellen Befunden verglichenwerden konnen. Durchgef¨uhrte Molekulardynamik-Simulationen zu Aufwachsprozessen vondunnen Filmen f¨uhren zu Erkenntnissen ¨uber die Stabilit¨at kubisch-flachenzentrierter d¨unnerFe-Filme auf Cu(111)-Substraten. Dabei h¨angt die strukturelle Stabilit¨at von dem verwende-tem Verfahren zur Filmherstellung ab [20].

18
2 Molekulardynamik-Simulationen
In Molekulardynamik-Simulationen werden die Newtonschen Bewegungsgleichungen f¨ur ei-ne gegebene Anzahl von Atomen oder Molek¨ulen numerisch gel¨ost. Erstmalig wurde eine Mo-lekulardynamik-Simulation von Alder und Wainwright 1957 [21,22] f¨ur harte Kugeln und 1964von Rahman f¨ur Teilchen, die der Lennard-Jones Wechselwirkung unterliegen, durchgef¨uhrt[15]. In dieser Arbeit sind die Simulationen auf Atome beschr¨ankt, die keiner zus¨atzlichenNebenbedingungen wie z. B. festen Abst¨anden unterliegen, man k¨onnte daher auch von einerAtomdynamik-Simulation sprechen. Zu l¨osen ist ein gekoppeltes System von gew¨ohnlichenDifferentialgleichungen zweiter Ordnung von entsprechend hoher Dimension. Ausgehend vonden Anfangsorten und Anfangsgeschwindigkeiten der Atome wird die Zeitentwicklung desatomaren Systems bestimmt. Im Limes unendlicher Beobachtungszeit k¨onnen somit Zeitmittel-werte eines Systems mit 3N Freiheitsgraden mit den zugeh¨origen generalisierten Koordinatenq = q1,q2, ...,q3N und Impulsenp = p1, p2, ..., p3N definiert werden [16,23,24]:
O = limt0→∞
1t0
∫ t0
0O(q(t),p(t))dt. (2.1)
Da der Grenzwert aus Gleichung (2.1) numerisch nicht gebildet werden kann und die Zeit ins aquidistante Zeitpunkte mit dem Abstand dt zerlegt wird, werden bei Molekulardynamik-Si-mulationen stattdessen Meßmittelwerte berechnet:
O =1l
s
∑l=1
O(q(ldt),p(ldt)). (2.2)
Bei entsprechend langer Simulationszeitsdt nahern sich die Meßmittelwerte dem Ausdruckaus Gleichung (2.1) an. Damit nun die Mittelwerte unabh¨angig von den speziellen Anfangsbe-dingungen sind, m¨ussen die Trajektorien chaotisches Verhalten zeigen. D. C. Rapaport [17] hatgezeigt, daß f¨ur ein System von Teilchen, die der Lennard-Jones Wechselwirkung unterliegen,ein exponentielles Auseinanderdriften von Trajektorien bei kleinsten Abweichungen der An-fangsbedingungen auftritt. Typisch ist, daß sogenannteinfrequent eventsin ihrer Erscheinungchaotisch in den Anfangsbedingungen sind. Als Beispiel kann hier angef¨uhrt werden, daß sicheinzelne lediglich bei exakt gleicher Anfangsbedingung und bei gleicher Rechengenauigkeitreproduzieren lassen.
Aus der Ergodenhypothese1 oder der Quasiergodenhypothese2 baut sich die Gibb-sche Formulierung der statistischen Mechanik auf, die die zu berechnenden Zeitmittelwertedurch Ensemblemittelwerte ersetzt. Ein Vorteil von Molekulardynamik-Simulationen ist diedirektere Bestimmung der Mittelwerte, und die damit verbundene Einfachheit mit der sichNichtgleichgewichtsph¨anomene beschreiben lassen, da Meßmittelwerte sich einfach auch imNichtgleichgewicht nach Gleichung (2.2) bestimmen lassen. Interessante Beispiele von Nicht-Gleichgewichts-Molekulardynamik-Simulationen sind Simulationen zu Rißausbreitungen in
1Die Phasenraumtrajektorie trifft im Laufe der Zeit jeden Punkt gleich oft, somit sind Zeitmittel gleich denEnsemblemittel.
2Eine Formulierung der Quasiergodenhypothese lautet (P. und T. Ehrenfest) [23]: Die im Phasenraum an dieEnergiehyperfl¨ache gebundene Trajektorie kommt im Laufe der Zeit jedem Punkt dieser Fl¨ache beliebig nahe.

2.1 Grundlagen 19
Festkorpern [25], Schockwellenstrukturen [26,27], Reibungsph¨anomenen [18,28] oder die da-mit im Zusammenhang stehenden Versetzungen in Metallen [29].
Im folgenden Kapitel werden einige technische Aspekte von Molekulardynamik-Si-mulationen dargestellt und erl¨autert. Dabei wird insbesondere auf die in dieser Arbeit verwen-deten und auf die nicht in der Literatur beschriebenen Verfahren eingegangen. F¨ur eine umfas-sendere Beschreibung wird auf die B¨ucher von D. C. Rapaport [17] und von M. P. Allen undD. J. Tildesley [15] verwiesen. Eine einfache Einf¨uhrung findet man in dem deutschsprachigenBuch von R. Haberland [16]. Interessante Aspekte von Computersimulationen, insbesondereim Zusammenhang mit makroskopischer Irreversibilit¨at sowie mikroskopischer Reversibilit¨at,sind in dem k¨urzlich erschienenen Buch von W. G. Hoover [14] zu finden.
2.1 Grundlagen
2.1.1 Verlet-Algorithmen
In einer Molekulardynamik-Simulation vonN Atomen wird ein gekoppeltes System vonN Dif-ferntialgleichungen zweiter Ordnung in Abh¨angigkeit von der Zeit numerisch gel¨ost. In dieserArbeit wurde zu diesem Zweck der Verlet-Algorithmus verwendet. Der Verlet-Algorithmus isteine einfache aber effektive Methode, die gestattet mit einer einzigen Kraftberechnung pro Zeit-schritt auszukommen und ¨uber eine gute numerische Stabilit¨at verfugt. Das Verlet-Verfahrenerhalt man, indem die beiden Taylor-Entwicklungen eines Ortsr i(t) nach der Zeit
r i(t +dt) = r i(t)+dt r i(t)+(dt)2
2r i +O[(dt)3]+O[(dt)4] + ..., (2.3)
r i(t−dt) = r i(t)−dt r i(t)+(dt)2
2r i −O[(dt)3]+O[(dt)4] + ..., (2.4)
addiert werden:
r i(t +dt) = 2r i(t)− r i(t−dt)+(dt)2r i +O[(dt)4] + ... (2.5)
Die Geschwindigkeiten werden bei Bedarf aus der Differenz
r i(t) =r i(t +dt)− r i(t −dt)
2dt+O[(dt)3] + ... (2.6)
bestimmt.Zur Bestimmung der Teilchenorte und Geschwindigkeiten habe sich verschiedene Metho-
den etabliert. Die Leapfrog-Version dieses Verfahrens bestimmt Orte und Geschwindigkeitenzu unterschiedlichen Zeiten [15]:
r i(t +dt) = r i(t)+dt r i(t +dt/2), (2.7)
r i(t +dt/2) = r i(t−dt/2)+dt r i(t). (2.8)

20 2 Molekulardynamik-Simulationen
Der aquivalente Velocity-Verlet-Algorithmus berechnet Orte und Geschwindigkeiten f¨ur glei-che Zeiten [15,30]:
r i(t +dt) = r i(t)+dt r i(t)+12(dt)2r i(t), (2.9)
r i(t +dt) = r i(t)+12
dt[r i(t)+ r i(t +dt)]. (2.10)
Die Krafte lassen sich aus dem Potentialφ = φ(r1, r2, ..., rN) bestimmen und sind gem¨aß dem2. Newtonschen Axiom mit den Beschleunigungen zu verkn¨upfen:
Fi(t) = mi r i(t) = −∂Φ∂r i
. (2.11)
Das Velocity-Verlet-Verfahren ist sehr effizient, da nur ein Satz von Variablen f¨ur die Orte, Ge-schwindigkeiten und Beschleunigungen der Atome ben¨otigt wird, die nach einem Zeitschrittalle zum gleichen Zeitpunkt vorliegen. In der Praxis werden w¨ahrend des ersten Velocity-Verlet-Schritts die neuen Orte sowie die ersten beiden Terme aus Gleichung (2.10) der neu-en Geschwindigkeiten berechnet. Anschließend werden die neuen Kr¨afte bestimmt, um im 2.Velocity-Verlet-Schritt die Verarbeitung des 3. Terms in Gleichung (2.10) zu vollziehen. Einweiterer Vorteil dieser Algorithmen liegt in deren Stabilit¨at bezuglich der Erhaltung der Ge-samtenergie des Systems. Das Verlet-Verfahren weist zwar Fluktuationen der Gesamtenergieauf aber keine Drift der Gesamtenergie im Laufe der Zeit. Verfahren h¨oherer Ordnung wiez. B. von Gear [16, 30] zeigen zwar weniger Fluktuationen, d. h. eine genauere Beschreibungvon einzelnen Trajektorien, aber eine Drift der Gesamtenergie.
2.1.2 Potentiale endlicher Reichweite
In Molekulardynamik-Simulationen wird ein Großteil der Rechenzeit f¨ur die Kraftberechnungverbraucht. Wird die Kraft zwischen allen vorhandenen Atompaaren berechnet, so steigt dernumerische Aufwand mitN2. Diese Methode wird also mit zunehmender Teichenzahl immerweniger praktikabel. Die in dieser Arbeit verwendeten Modellpotentiale haben eine endlicheReichweite, daher bietet es sich an, bei einer Kraftberechnung nur die Paare in Betracht zu zie-hen, die eine m¨ogliche Wechselwirkung haben. Dabei werden hier zwei Methoden verwendet(Abb. 2.1):
• Verlet-Listen
• Zellenmethode
Die Verlet-Listen nutzen die Tatsache aus, daß sich die Umgebung eines Atoms oder Mo-lekuls in einem Festk¨orper oder in einer Fl¨ussigkeit, nur langsam ver¨andert. Zu jedem Atomexistiert eine Liste von Atomen, die sich zu einer bestimmten Zeit innerhalb einescutt-offRadius befunden haben. Dieser Radius ist gr¨oßer gew¨ahlt als die Reichweite des benutzten Po-tentials. Bei einer Kraftberechnung wird nur noch die Kraft zwischen dem betrachteten Atomund Atomen aus dieser sogenannten Nachbarliste berechnet. W¨urde die anf¨angliche Nachbar-liste wahrend des Programmlaufs beibehalten werden, so w¨urde der Rechenzeitaufwand mit

2.1 Grundlagen 21
Abbildung 2.1: Die einfachste Methode die Kraftberechnung durchzuf¨uhren besteht darin, alle Paare zuberucksichtigen. Hierbei steigt der Rechenaufzeitaufwand wieN2 (links). Bei der Verlet-Listen-Methode (Mit-te) werden nur Wechselwirkungen von Atomen berechnet, die sich innerhalb eines Radius befinden, der etwasgroßer gew¨ahlt wird als die Wechselwirkungsreichweite. Die Zellenmethode (rechts) teilt das System in Zellenein, deren Kantenl¨ange mindestens gleich der Wechselwirkungsreichweite ist. Wechselwirkungen werden dabeinur zwischen benachbarten Zellen berechnet. Diese Methode eignet sich besonders gut f¨ur sehr große Teilchen-zahlen.
der TeilchenzahlN linear ansteigen. Allerdings muß die Liste in regelm¨aßigen Abst¨anden aufGrund moglicherAnderungen der Nachbarschaftsverh¨altnisse erneut aufgebaut werden. Dabeiwerden die Abst¨ande zwischen allen Paaren verglichen. Daher verzeichnet diese Methode beisehr großen Teilchenzahlen ein ¨uberproportionales Ansteigen der Rechenzeit mit der Teilchen-zahl.Bei extrem großen Teilchenzahlen von beispielsweise einigen Millionen empfiehlt sich daherdie Zellenmethode, bei der die gesamte Superzelle in kleine kubische Zellen mit einer Ausdeh-nung von mindestens der Wechselwirkungsreichweite aufgeteilt wird. Diese Aufteilung kannfur jeden Zeitschritt vorgenommen werden. Bei der Kraftberechnung werden dann nur Kr¨aftezwischen Atomen in benachbarten Zellen berechnet. Dieses Verfahren skaliert, da es nie allePaare mit einander verbindet linear mit der TeilchenzahlN.
2.1.3 Randbedingungen
Die Simulation von Clustern mit einem Durchmesser von einigen Atomabst¨anden erfordertkeine besonderen Randbedingungen, da in nat¨urlicher Weise der Cluster freie Oberfl¨achen hat.Sind Festk¨orpereigenschaften von Interesse, so sind periodische Randbedingungen sinnvoll,da diese bei endlich großen Systemen Oberfl¨acheneffekte unterdr¨ucken. Der gesamte Raumwird mit der simulierten Zelle periodisch fortgesetzt. Kr¨afte werden nun nach der sogenanntenminimum-image-conventionberechnet, wobei jedes Atom mit seinem n¨achst gelegenen peri-odischem Bild wechselwirkt (Abb. 2.2).
Der Vorteil von periodischen Randbedingungen ist einfach einzusehen: W¨ahrend ein Expe-rimentalphysiker eine Probe von vielleicht einem Zentimeter Kantenl¨ange vermißt und vonFestkorpereigenschaften spricht, ist dies insofern gerechtfertigt, als daß das Verh¨altnis vonOberflachenatomen zur Gesamtatomzahl verschwindend gering ist. In einer Molekulardy-namik-Simulation kann allerdings nicht auf so große Simulationszellen zur¨uckgegriffen wer-den. Hier muß auf Zellengr¨oßen zur¨uckgegriffen werden bei denen das Verh¨altnis von Ober-

22 2 Molekulardynamik-Simulationen
Abbildung 2.2: Periodische Randbedingungender Simulationsbox bedeuten eine periodischeFortsetzung der Box in alle Raumrichtungen.Die Gestalt der Zelle muß die Bedingungder periodischen Fortsetzbarkeit aufweisen. Dieminumum-image-conventionbewirkt, daß jedesAtom mit dem zu Ihm n¨achst gelegenen Bilddes Atoms wechselwirkt. Dies setzt voraus, daßdie Ausmaße der periodischen Superzelle gr¨oßersind als die Wechselwirkungsreichweite.
flachenatomen zur Gesamtatomzahl um sehr viel gr¨oßer ist. Somit bieten periodische Randbe-dingungen eine L¨osung des Problems. Allerdings ist Vorsicht geboten, da periodische Randbe-dingungen auch zu Artefakten und somit zu Mißinterpretationen der Ergebnisse f¨uhren konnen.Eine klarende Antwort auf die Frage, wie groß die Kantenl¨ange der Zelle sein muß, damit pe-riodische Randbedingungen keinen Einfluß auf die Ergebnisse haben, gibt es nicht. Klar ist,daß Korrelationsl¨angen oder L¨angen auf denen typische physikalische Ph¨anomene in Erschei-nung treten nicht unterschritten werden d¨urfen. Beispielsweise k¨onnen langwellige Phononenbei kleinen Zelldimensionen von vorneherein nicht in den Simulationen erfaßt werden.
Bei der Untersuchung von Oberfl¨achen werden periodische Randbedingungen in der Film-ebene angewendet und nicht senkrecht zur Oberfl¨ache. Hierbei entstehen zwei Oberfl¨achen,wobei es sinnvoll sein kann, eine von beiden einzufrieren, d. h. die ersten zwei bis sechs Lagenbei den Positionen des Festk¨orpers zu fixieren und von der Dynamik auszuschließen (Kap.7). Durch die Verwendung von periodischen Randbedingungen verringert sich die Anzahl derFreiheitsgrade um drei, d. h. in den Gleichungen (2.12), (2.30), (2.32) und (2.34) ist 3N durch3N−3 zu ersetzten.
2.1.4 Temperatur
Temperatur kann als ungeordnete Bewegung von Molek¨ulen oder Atomen aufgefaßt werden,wobei die Bewegung um so st¨arker ist, je h¨oher die Temperatur und je kleiner die Masse desentsprechenden Atoms oder Molek¨uls ist. Der Botaniker Brown entdeckte dies 1827 indirektunter dem Mikroskop an der unregelm¨aßigen Bewegung von Farbstoffen in Wasser [31, 32].Bei einer Molekulardynamik-Simulation kann diese ungeordnete Bewegung direkt beobachtetwerden. Um quantitative Aussagen ¨uber die Temperatur machen zu k¨onnen, kann der Gleich-verteilungssatz herangezogen werden:Jede kanonische Variable, die in die Hamiltonfunktionquadratisch eingeht, liefert im thermodynamischen Gleichgewicht einen BeitragkB
2 T zur mitt-leren Energie[33, 34]. Aus der kinetischen Energie eines Systems ausN-Atomen mit den Ge-schwindigkeitenr i ergibt sich somit die Temperatur bei Molekulardynamik-Simulationen in
2.1 Grundlagen 23
drei Raumdimensionen aus dem Mittelwert der kinetischen Energie der Atome:
T =2
3NkB〈
N
∑i=1
mi
2r2
i 〉, (2.12)
wobei etwaige Schwerpunktsbewegungen abgezogen werden m¨ussen. Entsprechend obigerGleichung kann auch die instantane Temperatur bestimmt werden, bei der die Zeitmittelungin Gleichung (2.12) entf¨allt. Sie fluktuiert in einer Simulation um den Mittelwert der Tempera-tur (Abschnitt 2.4.1).
Um wahrend der Simulation Einfluß auf die Temperatur nehmen zu k¨onnen, konnenbeispielsweise w¨ahrend der Anfangsphase der Simulation (Equilibrirungsphase) in kurzenAbstanden (etwa alle 10 Integrationsschritte) die Geschwindigkeiten mit dem Faktor√
T0
T(2.13)
skaliert werden.T0 ist die gewunschte Temperatur undT die instantane Temperatur entspre-chend Gleichung (2.12).
Die statistische Mechanik liefert die Verteilung der Geschwindigkeiten zu einer bestimmtenTemperatur. F¨ur kanonische und mikrokanonische Ensemble mit großen Teilchenzahlen ergibtsich die Maxwell-Boltzmann-Geschwindigkeitsverteilung :
w(v) ∼ v2e− miv
2
2kBT , (2.14)
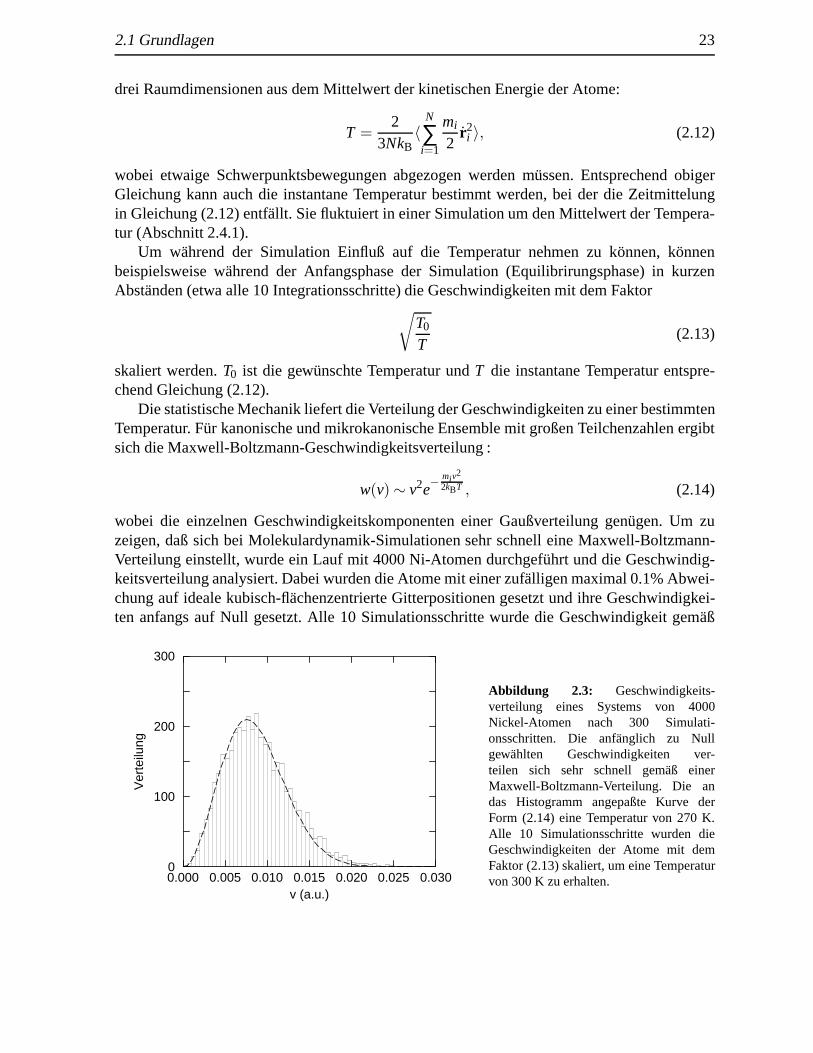
wobei die einzelnen Geschwindigkeitskomponenten einer Gaußverteilung gen¨ugen. Um zuzeigen, daß sich bei Molekulardynamik-Simulationen sehr schnell eine Maxwell-Boltzmann-Verteilung einstellt, wurde ein Lauf mit 4000 Ni-Atomen durchgef¨uhrt und die Geschwindig-keitsverteilung analysiert. Dabei wurden die Atome mit einer zuf¨alligen maximal 0.1% Abwei-chung auf ideale kubisch-fl¨achenzentrierte Gitterpositionen gesetzt und ihre Geschwindigkei-ten anfangs auf Null gesetzt. Alle 10 Simulationsschritte wurde die Geschwindigkeit gem¨aß
0.000 0.005 0.010 0.015 0.020 0.025 0.030v (a.u.)
0
100
200
300
Ver
teilu
ng
Abbildung 2.3: Geschwindigkeits-verteilung eines Systems von 4000Nickel-Atomen nach 300 Simulati-onsschritten. Die anf¨anglich zu Nullgewahlten Geschwindigkeiten ver-teilen sich sehr schnell gem¨aß einerMaxwell-Boltzmann-Verteilung. Die andas Histogramm angepaßte Kurve derForm (2.14) eine Temperatur von 270 K.Alle 10 Simulationsschritte wurden dieGeschwindigkeiten der Atome mit demFaktor (2.13) skaliert, um eine Temperaturvon 300 K zu erhalten.

24 2 Molekulardynamik-Simulationen
Gleichung (2.13) skaliert, um eine Temperatur von 300 K zu erhalten. Es stellte sich bereitsnach einigen Schritten eine Ann¨aherung an die Maxwell-Boltzmann-Verteilung ein. Abbildung2.3 (atomare Einheiten (a.u.) sind in Abschnitt 9.1 definiert) zeigt die Geschwindigkeitsvertei-lung nach 300 Schritten.
2.2 Auswertung
Neben den einfachen Gr¨oßen wie potentielle Energie, Temperatur, Teilchenpositionen, die an-hand von Gleichung (2.2) bestimmt werden k¨onnen, lassen sich auch subtilere Auswertungenwahrend oder nach einer Molekulardynamik-Simulation machen. Es ist sinnvoll, Auswertun-gen moglichst wahrend der laufenden Simulation durchzuf¨uhren. Anderenfalls m¨ußte im Prin-zip die gesamte Entwicklung des 6N dimensionalen Phasenraums abgespeichert werden, umhinterher entsprechende Mittelungen oder Systementwicklungen durchf¨uhren bzw. darstellenzu konnen. Die Mittelung w¨ahrend des Simulationslaufs setzt allerdings voraus, daß bereitsvor Simulationsbeginn eine genaue Kenntnis der interessanten Ph¨anomene vorhanden ist unddas Programm Mittelungen von wichtigen Gr¨oßen durchf¨uhrt und ausgibt. Schwieriger gestal-tet es sich bei der visuellen Darstellung von Systementwicklungen, wie etwa einem Keim-bildungsprozeß an dem unter Umst¨anden mehrere Millionen Atome beteiligt sind. Das Pro-grammpaketSPaSM(ScalableParallel Short-rangeMolecular dynamics) (Abschnitt 2.5) bietetdie Moglichkeit Auswertungsroutinen zu programmieren und zu implementieren, die w¨ahrendder Simulation Bildsequenzen erstellen, welche die Auswertung von Simulationen von mehre-ren Millionen Atomen unterst¨utzt.
2.2.1 Druck
Mit Hilfe des Virialsatzes3 [34] kann der Druck in einer Simulationszelle ermittelt werden [16]:
P =1
3V〈
N
∑i=1
mi r2i 〉︸ ︷︷ ︸
kinetischer Anteil
+1
3V〈
N
∑j=1
N
∑i< j
Fi j r i j 〉︸ ︷︷ ︸Kra f tanteil
, (2.15)
dabei istV das Systemvolumen,Fi j die Kraft die Atom j auf Atomi ausubt undr i j = r i −r j derAbstandsvektor zweier Atome. Der Druck setzt sich aus einem kinetischen Anteil (vgl. idea-les Gas) und einem Kraftanteil zusammen. Bei dieser Schreibweise wurde angenommen, daßsich die Krafte in entsprechende Paaranteile aufteilen lassen (Abschnitt 2.3). Ist dies nicht derFall, muß der allgemeinere Ausdruck f¨ur den Kraftanteil des Drucks13V 〈∑N
i Fi=1r i〉 verwendetwerden.
Durch Aufhebung der Mittelung kann entsprechend der instantanen Temperatur (Abschnitt2.1.4), der instantane Druck definiert werden. Unter Benutzung des dyadischen Produkts kann
3Eine mogliche Formulierung des Virialsatzes von Rudolf Clausius 1870: F¨ur mechanisch stabile Systeme istdie gemittelte kinetische Energie gleich dem gemittelten Virial der wirksamen Kr¨afte: 3
2NkBT = − 12〈∑N
i Fi=1r i〉
2.2 Auswertung 25
der sogenannte Drucktensor definiert werden [15]:
Pαβ =1V〈
N
∑i=1
mi r iαr iβ〉+1V〈
N
∑j=1
N
∑i< j
Fi j αr i j β〉. (2.16)
Die drei DiagonalelementePxx,Pyy,Pzz konnen als Druck in die entsprechenden Richtungen,die Nicht-DiagonalelementePxy,Pyz,Pzx als Scherdr¨ucke interpretiert werden. Die invarianteSpur entspricht dem Druck:P = 1
3SpPαβ. Der instantane Druck kann zur Regelung des Vo-lumens beim Anderson-Verfahren benutzt werden (Abschnitt 2.4). Beim Parinello-Rahmann-Verfahren dienen die einzelnen Komponenten des instantanen Drucktensors zur Regelung derKantenlangen der Simulationsbox und zur Regelung der Winkel zwischen den Kanten der Box.Interessante Untersuchungen lassen sich mit Hilfe des lokalen Drucktensors durchf¨uhren. Hier-bei wird die Summation in Gleichung (2.16) nur ¨uber Atome innerhalb eines Teilvolumensvdurchgefuhrt [35].
2.2.2 Radiale Verteilungsfunktion
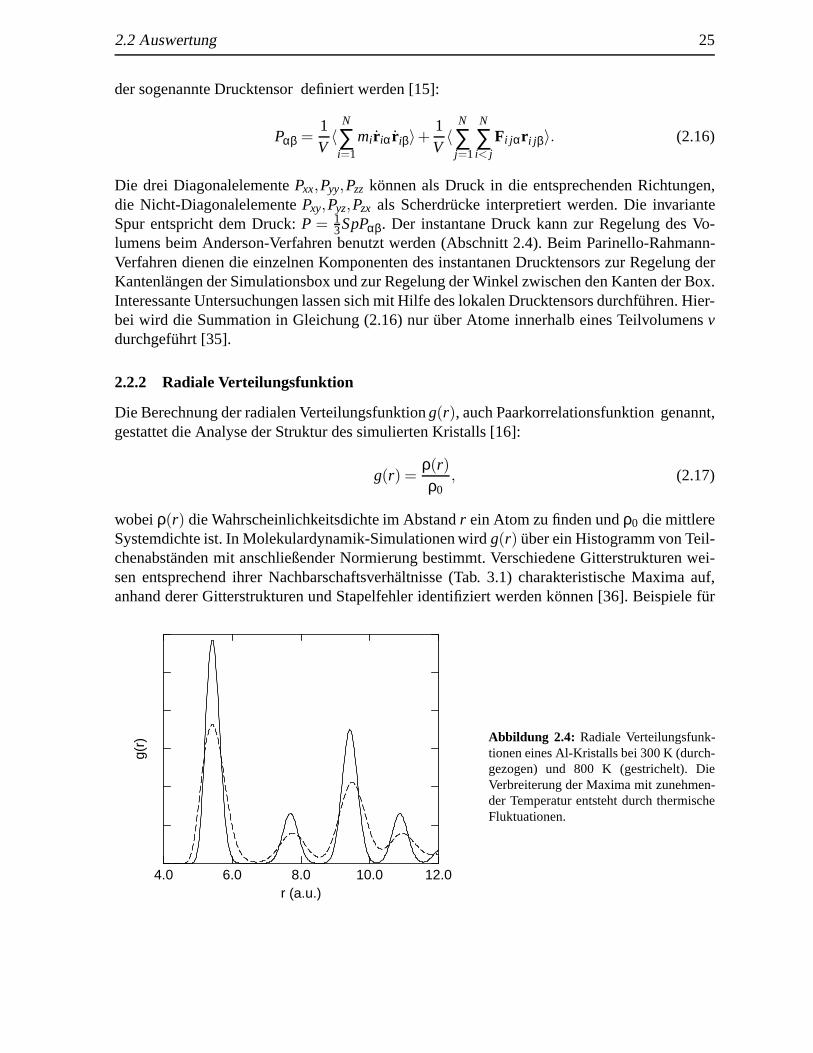
Die Berechnung der radialen Verteilungsfunktiong(r), auch Paarkorrelationsfunktion genannt,gestattet die Analyse der Struktur des simulierten Kristalls [16]:
g(r) =ρ(r)ρ0
, (2.17)
wobeiρ(r) die Wahrscheinlichkeitsdichte im Abstandr ein Atom zu finden undρ0 die mittlereSystemdichte ist. In Molekulardynamik-Simulationen wirdg(r) uber ein Histogramm von Teil-chenabst¨anden mit anschließender Normierung bestimmt. Verschiedene Gitterstrukturen wei-sen entsprechend ihrer Nachbarschaftsverh¨altnisse (Tab. 3.1) charakteristische Maxima auf,anhand derer Gitterstrukturen und Stapelfehler identifiziert werden k¨onnen [36]. Beispiele f¨ur
4.0 6.0 8.0 10.0 12.0r (a.u.)
g(r) Abbildung 2.4: Radiale Verteilungsfunk-
tionen eines Al-Kristalls bei 300 K (durch-gezogen) und 800 K (gestrichelt). DieVerbreiterung der Maxima mit zunehmen-der Temperatur entsteht durch thermischeFluktuationen.
26 2 Molekulardynamik-Simulationen
radiale Verteilungsfunktionen von idealen Gittern findet man in Abschnitt 3.1. Mit zunehmen-der Temperatur tritt eine Verbreiterung der Maxima durch thermische Fluktuationen auf (Abb.2.4), was zu einem Verschmelzen von benachbarten Maxima f¨uhren kann. Um ortsaufgel¨osteInformationen ¨uber die Gitterstruktur zu erhalten, ist es sinnvoll den simulierten Kristall inGebiete aufzuteilen und jeweils ¨ortlich getrennte Paarkorrelationsfunktionen zu bestimmen.
2.2.3 Lokale Gitterstruktur der Atome
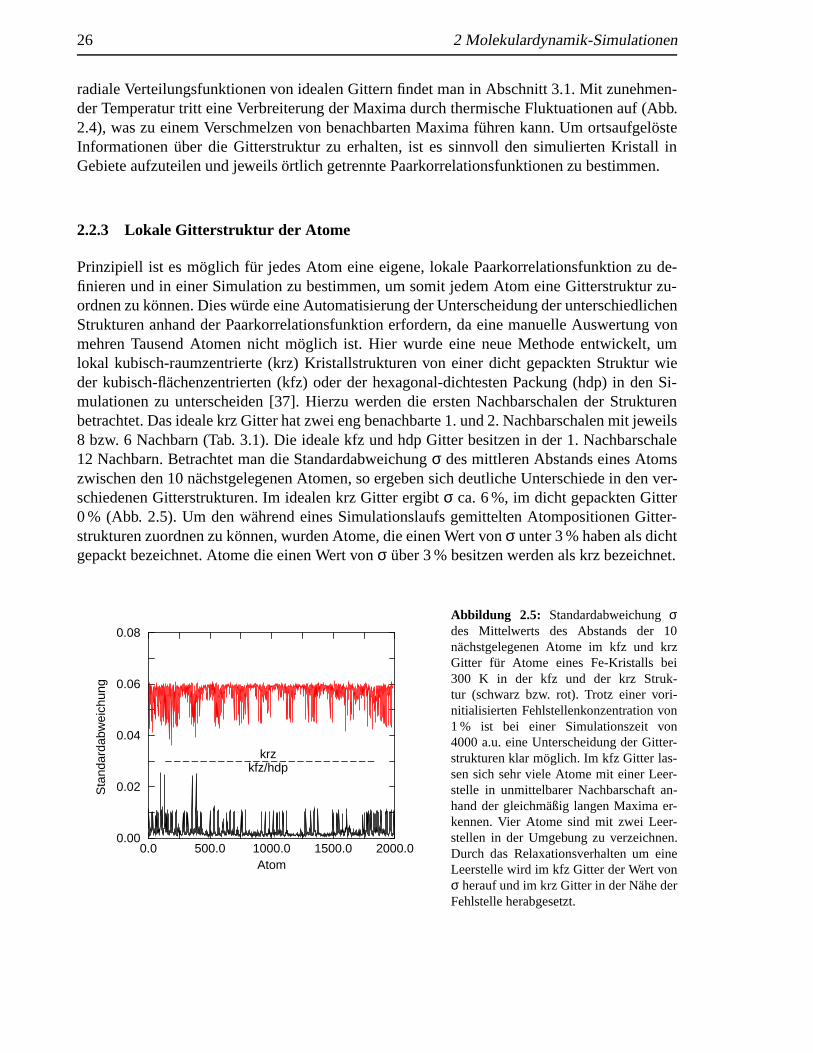
Prinzipiell ist es m¨oglich fur jedes Atom eine eigene, lokale Paarkorrelationsfunktion zu de-finieren und in einer Simulation zu bestimmen, um somit jedem Atom eine Gitterstruktur zu-ordnen zu k¨onnen. Dies w¨urde eine Automatisierung der Unterscheidung der unterschiedlichenStrukturen anhand der Paarkorrelationsfunktion erfordern, da eine manuelle Auswertung vonmehren Tausend Atomen nicht m¨oglich ist. Hier wurde eine neue Methode entwickelt, umlokal kubisch-raumzentrierte (krz) Kristallstrukturen von einer dicht gepackten Struktur wieder kubisch-flachenzentrierten (kfz) oder der hexagonal-dichtesten Packung (hdp) in den Si-mulationen zu unterscheiden [37]. Hierzu werden die ersten Nachbarschalen der Strukturenbetrachtet. Das ideale krz Gitter hat zwei eng benachbarte 1. und 2. Nachbarschalen mit jeweils8 bzw. 6 Nachbarn (Tab. 3.1). Die ideale kfz und hdp Gitter besitzen in der 1. Nachbarschale12 Nachbarn. Betrachtet man die Standardabweichungσ des mittleren Abstands eines Atomszwischen den 10 n¨achstgelegenen Atomen, so ergeben sich deutliche Unterschiede in den ver-schiedenen Gitterstrukturen. Im idealen krz Gitter ergibtσ ca. 6 %, im dicht gepackten Gitter0 % (Abb. 2.5). Um den w¨ahrend eines Simulationslaufs gemittelten Atompositionen Gitter-strukturen zuordnen zu k¨onnen, wurden Atome, die einen Wert vonσ unter 3 % haben als dichtgepackt bezeichnet. Atome die einen Wert vonσ uber 3 % besitzen werden als krz bezeichnet.
0.0 500.0 1000.0 1500.0 2000.0Atom
0.00
0.02
0.04
0.06
0.08
Sta
ndar
dabw
eich
ung
krzkfz/hdp
Abbildung 2.5: Standardabweichungσdes Mittelwerts des Abstands der 10nachstgelegenen Atome im kfz und krzGitter fur Atome eines Fe-Kristalls bei300 K in der kfz und der krz Struk-tur (schwarz bzw. rot). Trotz einer vori-nitialisierten Fehlstellenkonzentration von1 % ist bei einer Simulationszeit von4000 a.u. eine Unterscheidung der Gitter-strukturen klar m¨oglich. Im kfz Gitter las-sen sich sehr viele Atome mit einer Leer-stelle in unmittelbarer Nachbarschaft an-hand der gleichm¨aßig langen Maxima er-kennen. Vier Atome sind mit zwei Leer-stellen in der Umgebung zu verzeichnen.Durch das Relaxationsverhalten um eineLeerstelle wird im kfz Gitter der Wert vonσ herauf und im krz Gitter in der N¨ahe derFehlstelle herabgesetzt.

2.2 Auswertung 27
Wurden zur Berechnung der Standardabweichungσ die 12 nachstgelegenen Atome zu ei-nem Atom ber¨ucksichtigt werden, so w¨urden Defekte wie Fehlstellen den Wert vonσ von dichtgepackten Strukturen erh¨ohen, da fehlende Atome in der 1. Nachbarschale durch Atome ausder 2. Nachbarschale bei der Berechnung vonσ ersetzt werden. Dadurch, daß nur 10 Nachbarnberucksichtigt werden, ist das Verfahren gegen lokale Defekte wie Leerstellen resistent (Abb.2.5). Das Verfahren ist unabh¨angig von der Gitterkonstante und l¨aßt sich auch unter Kompres-sion anwenden. Allerdings ist das Verfahren auf eine Mittelung der Atompositionen ¨uber einigeSimulationschritte angewiesen, da ansonsten thermische Fluktuationen der Atompositionen dieStandardabweichungσ vergroßern.
Eine andere M¨oglichkeit in den Simulation den Atomen lokal eine Gitterstruktur zuzuordenbesteht darin, die Anzahl der Atome innerhalb einer Kugel mit dem Radiusr zu zahlen. Wirdder Radius so gew¨ahlt, daß er zwischen dem Abstand der 1. und 2. Nachbarschale der krzStruktur liegt, und gr¨oßer als der Abstand der 1. Nachbarschale der dicht gepackten Strukturist, betragt die Anzahl der Atome innerhalb der Kugel mit Radiusr im krz Gitter 8 und im dichtgepackten Gitter 12. Dieses Verfahren ist nicht so stark auf eine Mittelung der Atompositionenangewiesen, allerdings werden die Gitterkonstanten der Strukturen ben¨otigt. Beide Verfahrensind nur fur Festkorper geeignet. Zur Bestimmung der Struktur von Oberfl¨achen m¨ussen andereMethoden verwendet werden.
2.2.4 Freie Energie
Wahrend sich der Erwartungswert des HamiltonoperatorsE(S,V,N) (aufgrund der hier vorlie-genden Konstanz der Teilchenzahl wird im FolgendenE = E(S,V) geschrieben) als Summeaus potentieller und kinetischer Energie in einer Molekulardynamik-Simulation einfach be-rechnen laßt, ist die Berechnung anderer thermodynamischer Potentiale nicht trivial. Um diethermodynamische Stabilit¨at eines Systems bei vorgegebener Temperatur und unter vorgege-benem Volumen oder Druck zu analysieren, muß die freie EnergieF(T,V) = E − TS bzw.die Freie EnthalpieG(T,P) = E−TS+PV berechnet werden. Schwierigkeiten macht hierbeidie Berechnung der EntropieS. Als einfaches Beispiel sei die Schwingungsentropie [38] einesklassischen harmonischen Oszillators mit der Kreisfrequenzω angefuhrt. Sie ergibt sich imkanonischen Ensemble zu [34] (vgl. auch Abschnitt 9.4):
S= kB
[ln
kBThω
+1
]. (2.18)
Beschreibt man den Festk¨orper mit einem einzigen harmonischen Oszillator, so bedeutet Glei-chung (2.18), daß Festk¨orper die durch eine niedrige Frequenz (weiches Gitter) beschriebenwerden eine hohe Schwingungsentropie haben. Festk¨orper, die durch eine hohe Frequenz (har-tes Gitter) beschrieben werden, haben eine niedrige Schwingungsentropie.
Eine attraktive Moglichkeit, den Gitteranteil der Entropie durch Vergleich mit harmoni-schen Oszillatoren direkt aus einer Molekulardynamik-Simulation abzusch¨atzen wurde 1995von J. R. Morris und K. M. Ho vorgestellt [39]. Hierbei werden KorrelationsmatrizenCi j furdie Komponenten der Ortsvariablenxi , i = 1...3N bestimmt:
Ci j = 〈xixj〉−〈xi〉〈xj〉. (2.19)

28 2 Molekulardynamik-Simulationen
Eine Diagonalisierung der Korrelationsmatrizen f¨uhrt auf die inversen quadratischen Eigenfre-quenzen des Systems und kann zur Berechnung der Entropie benutzt werden:
S≤ 12
kB ln
[(2πλ2
)3N
detC
]+kB3N, λ = h/
√2πmkBT. (2.20)
Der Vorteil dieser Methode besteht darin, die Berechnung der Entropie und somit der freienEnergie durch einen einzigen Simulationslauf zu ermitteln. Allerdings ist diese Methode wegender notwendigen Berechnung der entsprechend hochdimensionalen KorrelationsdeterminanteC numerisch aufwendig. So m¨ussen z. B. bei einem System von 432 Atomen 1296× 1296Determinanten berechnet werden. Dies f¨uhrte im Rahmen dieser Arbeit zum Einsatz von ma-thematischen Routinen der Firma IMSL [40] zur Berechnung der Determinanten.
Eine praktischere Methode, um Differenzen der freien Energie zwischen der kfz und krzStruktur eines Festk¨orpers zu bestimmen, kann durch die thermodynamische Relation [41]
dF = −PdV −SdT (2.21)
gewonnen werden. Werden bei konstanter Temperatur die Komponenten des Drucktensorsgemaß Gleichung (2.16) entlang der Bain-Transformation (Abschnitt 3.2.1), die die kfz Struk-tur in die krz Struktur ¨uberfuhrt, berechnet, so ergibt sich aus Gleichung (2.21) (sofern dieTatsache ber¨ucksichtigt wird, daß der Druck nicht mehr isotrop ist [42]):
dFT=konst.= −PxxAyzdx−PyyAzxdy−PzzAxydz. (2.22)
Wobei Pii die Druckkomponenten entlang der drei kartesischen Axen, undAi j die Flachesenkrecht dazu bezeichnet. Mit dieser Methode lassen sich nur Differenzen zwischen freienEnergien berechnen. Allerdings erlaubt die Methode neben einer thermodynamischen Stabi-lit atsanalyse die Interpretation der entlang des Reaktionswegs m¨oglicherweise auftretendenEnergiebarriere.
2.2.5 Quadratische Abweichung
Aussagen ¨uber die Mobilitat der Atome k¨onnen mit Hilfe der mittleren quadratischen Abwei-chung von der Ausgangsposition getroffen werden [17]:
A =1N
N
∑i=1
[r i(t)− r i(0)]2. (2.23)
Da in dieser Arbeit auch das Filmwachstum auf Substratoberfl¨achen betrachtet wird, bestehteine interessante Anwendung darin, von Atomen die auf Oberfl¨achen deponiert wurden, diequadratische Abweichung in der Oberfl¨achenebene zu studieren. Somit kann die Mobilit¨at derdeponierten Atome charakterisiert werden (Abschnitt 2.4.3 und 7.3.2).

2.3 Embedded-Atom Method (EAM)-Potentiale 29
2.3 Embedded-Atom Method (EAM)-Potentiale
Die moglichst korrekte Beschreibung der interatomaren Wechselwirkung ist entscheidend f¨urdie physikalische Bedeutung der gewonnen Resultate. Paarpotentiale, wie das Lennard-Jones-Potential erfassen die Eigenschaften von Metallen nicht hinreichend. Das elastische Verhal-ten wird unzureichend beschrieben, da f¨ur PaarpotentialeC12 = C44 gilt, was fur Metalle imallgemeinen nicht der Fall ist. Paarpotentiale besitzen Leerstellenbildungsenergien, die denKohasionsenergien entsprechen, wohingegen Metalle meist deutlich niedrigere Leerstellenbil-dungsenergien aufweisen. Bei Metallen wird eine Verkleinerung des Ebenenabstands an Ober-flachen beobachtet, eine Beschreibung durch reine Paarpotentiale f¨uhrt zu einer Vergr¨oßerungdes Ebenabstands an Oberfl¨achen [30]. Diese Unzul¨anglichkeiten treten bei derEmbedded-Atom Method(EAM) nicht auf, sie ber¨ucksichtigt den metallischen Charakter durch einen Ein-bettungsanteil der Energie.
2.3.1 Idee und Formulierung nach Daw und Baskes
Daw and Baskes [43, 44] beschreiben die Energie eines Atoms als Summe von einer Ein-bettungsfunktionF der Elektronendichte am Kernort und einem abgeschirmten Coulomb-Potential mit den abstandsabh¨angigen effektiven LadungenZi(ri j ):
E =N
∑i=1
F(ρi)︸ ︷︷ ︸Einbettungsenergie
+12
N
∑i=1
N
∑j 6=i
φi j (ri j )︷ ︸︸ ︷2
Zi(ri j )Zj(ri j )ri j︸ ︷︷ ︸
abgeschirmtes Coulomb-Potential
. (2.24)
Die Elektronendichten ergeben sich aus der Superposition der Elektronendichten der umgeben-den Atome:
ρi =N
∑i 6= j
ρatj (ri j ). (2.25)
w w w w
w w w
w w w w
w
............................................................
w
ρi
w
w w w w
ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp
pppppppppppppppppppppppppp
pppppppppppppp
pppppppppppppppppppppppppppppppppppppppppppppp
pppppppppppppppppppppppppppppppppppppppppppppp
ppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp
pppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppppp
pppppppppppppppppppppppppppppppppppppppppppppp
pp
Abbildung 2.6: Einbettung eines Metallatoms in die Elek-tronendichte der umgebenden Atome. Die Einbettungsenergiehangt von der Elektronendichte am Kernort ab, welche sich ausder Superposition der Elektronendichten der umgebenden Ato-me ergibt.

30 2 Molekulardynamik-Simulationen
Die Krafte in EAM-Systemen ergeben sich gem¨aß Gl. (2.11) zu:
Fi = ∑j 6=i
[F ′
i (ρi)ρatj′(ri j )+F ′
j (ρ j)ρati′(ri j )+φ′i j (ri j )
] r j − r i
ri j. (2.26)
Fur die numerische Berechnung m¨ussen geeignete Ans¨atze fur den EinbettungsanteilF(ρ),die effektive LadungZ(r) und die Elektronendichteρat
j (r) gefunden werden. In dieser Ar-beit werden die FunktionenF und Z in Form von kubischen Splines [45] dargestellt, d. h.als stuckweise kubische Funktionen, die zweimal stetig differenzierbar aneinander gesetzt wer-den. Die Ladungsdichteρat(r) wurde aus densingle-determinantHartree-Fock Rechnungenfreier Atome von Clementiet al. [46] bestimmt, wobei sich die Ladungsdichte aus dens- undp- bzw.d-Anteilen zusammensetzt:
ρat(r) ={
Nsρats (r)+(N−Ns)ρat
p/d(r)−ρc : r < rc
0 : r ≥ rc,(2.27)
dabei istN die Anzahl der gesamten Valenzelektronen undNs die Anzahl ders-Elektronen. Umdie Reichweite der Potentiale endlich zu gestalten, begrenzt dercut-off Radiusrc die Reich-weite der Elektronendichte. Die Konstanteρc wird so gewahlt, daßρat amcut-off Radius stetigbleibt.
2.3.2 Anpassung der EAM-Potentiale
Die EinbettungsenergieF und die effektive LadungZ werden an St¨utzstellen vorgegeben, umdie strukturellen Eigenschaften der Elemente Fe, Ni, Cu und Al mit der EAM beschreiben zukonnen. Die Werte an diesen St¨utzstellen wurden so bestimmt, daß die Eigenschaften des ent-sprechenden Kristalls des chemischen Elements m¨oglichst gut wiedergegeben werden. Hier-zu zahlen die Bindungsenergie, Gitterkonstante, Leerstellenbildungsenergie, elastische Kon-stanten sowie einzelne Phononenfrequenzen am Zonenrand [47]. Neben den experimentellzuganglichen Daten k¨onnen auch aus Elektronenstrukturrechnungen [48] hervorgegangeneDaten mit in die Anpassung einbezogen werden. Die Summe der quadratischen Abweichun-gen zwischen den experimentellen Meßwerten und den Elektronenstrukturdaten, die den Re-alkristall beschreibenMExp.
i und den entsprechenden Daten des EAM-KristallsMEAMi , muß
minimiert werden:
min∑i
ci
[MExp.
i −MEAMi
]2. (2.28)
Die Gewichtskoeffizientenci werden je nach Wichtigkeit der entsprechenden Gr¨oße per Handgewahlt. Die Anpassung geschieht in Abh¨angigkeit von der Anzahl der St¨utzstellen fur dieFunktionenF undZ in 5 bis 9 Dimensionen und wurde mit Hilfe einerleast squares fitRoutinedurchgefuhrt [40].
Im folgenden werden zwei verschieden angepaßte EAM-Modelle f¨ur das Element Al vor-gestellt, die den Einfluß der in die Anpassungsprozedur aufgenommenen Daten eines Kristallsveranschaulichen. Neben der Anpassung der zuvor genannten experimentellen Daten (Tab. 2.1)wurde bei einem EAM-Potential f¨ur Al noch die Volumenabh¨angigkeit der EnergieE(V), die
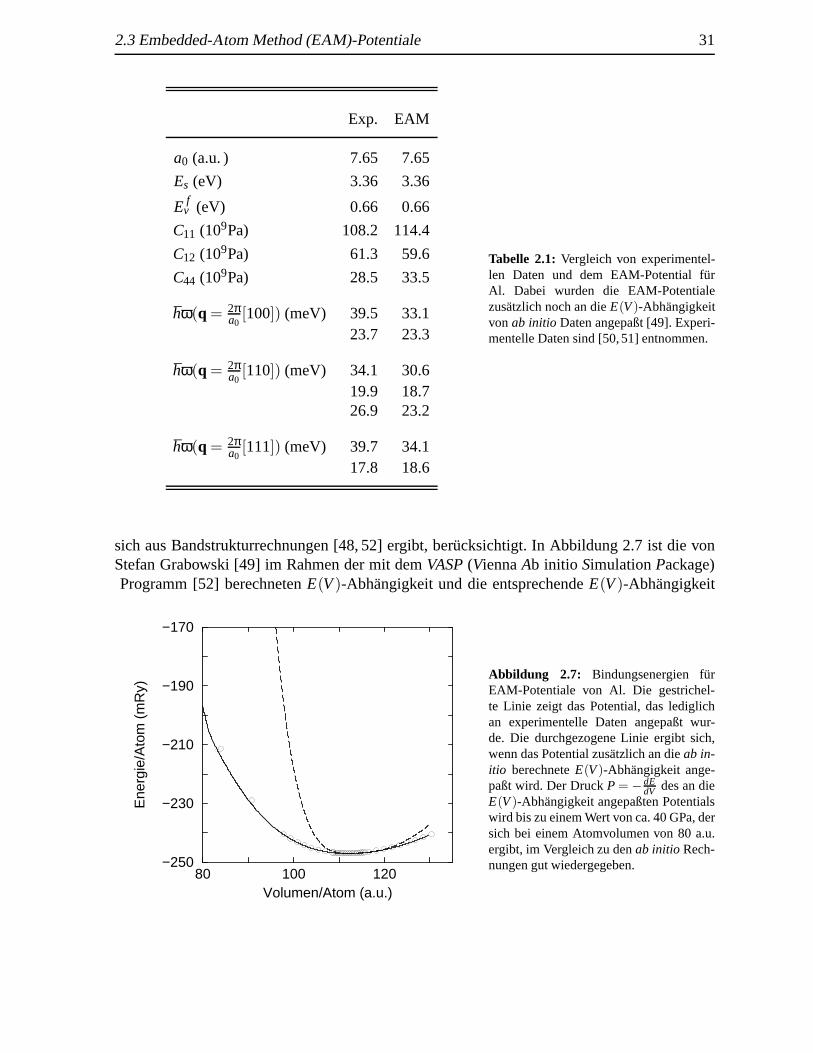
2.3 Embedded-Atom Method (EAM)-Potentiale 31
Exp. EAM
a0 (a.u. ) 7.65 7.65
Es (eV) 3.36 3.36
E fv (eV) 0.66 0.66
C11 (109Pa) 108.2 114.4
C12 (109Pa) 61.3 59.6
C44 (109Pa) 28.5 33.5
hω(q = 2πa0
[100]) (meV) 39.5 33.123.7 23.3
hω(q = 2πa0
[110]) (meV) 34.1 30.619.9 18.726.9 23.2
hω(q = 2πa0
[111]) (meV) 39.7 34.117.8 18.6
Tabelle 2.1: Vergleich von experimentel-len Daten und dem EAM-Potential f¨urAl. Dabei wurden die EAM-Potentialezusatzlich noch an dieE(V)-Abhangigkeitvon ab initio Daten angepaßt [49]. Experi-mentelle Daten sind [50,51] entnommen.
sich aus Bandstrukturrechnungen [48, 52] ergibt, ber¨ucksichtigt. In Abbildung 2.7 ist die vonStefan Grabowski [49] im Rahmen der mit demVASP(ViennaAb initio SimulationPackage)Programm [52] berechnetenE(V)-Abhangigkeit und die entsprechendeE(V)-Abhangigkeit
80 100 120Volumen/Atom (a.u.)
−250
−230
−210
−190
−170
Ene
rgie
/Ato
m (
mR
y)
Abbildung 2.7: Bindungsenergien f¨urEAM-Potentiale von Al. Die gestrichel-te Linie zeigt das Potential, das lediglichan experimentelle Daten angepaßt wur-de. Die durchgezogene Linie ergibt sich,wenn das Potential zus¨atzlich an dieab in-itio berechneteE(V)-Abhangigkeit ange-paßt wird. Der DruckP = − dE
dV des an dieE(V)-Abhangigkeit angepaßten Potentialswird bis zu einem Wert von ca. 40 GPa, dersich bei einem Atomvolumen von 80 a.u.ergibt, im Vergleich zu denab initio Rech-nungen gut wiedergegeben.
32 2 Molekulardynamik-Simulationen
4 6 8 10r(a.u.)
−8
−6
−4
−2
0
2E
ffekt
ives
Paa
rpot
entia
l (m
Ry)
Abbildung 2.8: Effektive Paarpotentia-le fur EAM-Potentiale von Al. Die ge-strichelte Linie zeigt das Potential, dassich ohne Verwendung vonab initioDaten ergibt. Die durchgezogene Kurvezeigt das Potential, das zus¨atzlich an dieE(V)-Abhangigkeit angepaßt wurd. F¨urAbstande kleiner als der n¨achste Nach-barabstand besitzt letzteres einen gerin-geren Anstieg. Die Kreise markieren dieAbstande der drei n¨achsten Nachbarn inder kfz Struktur.
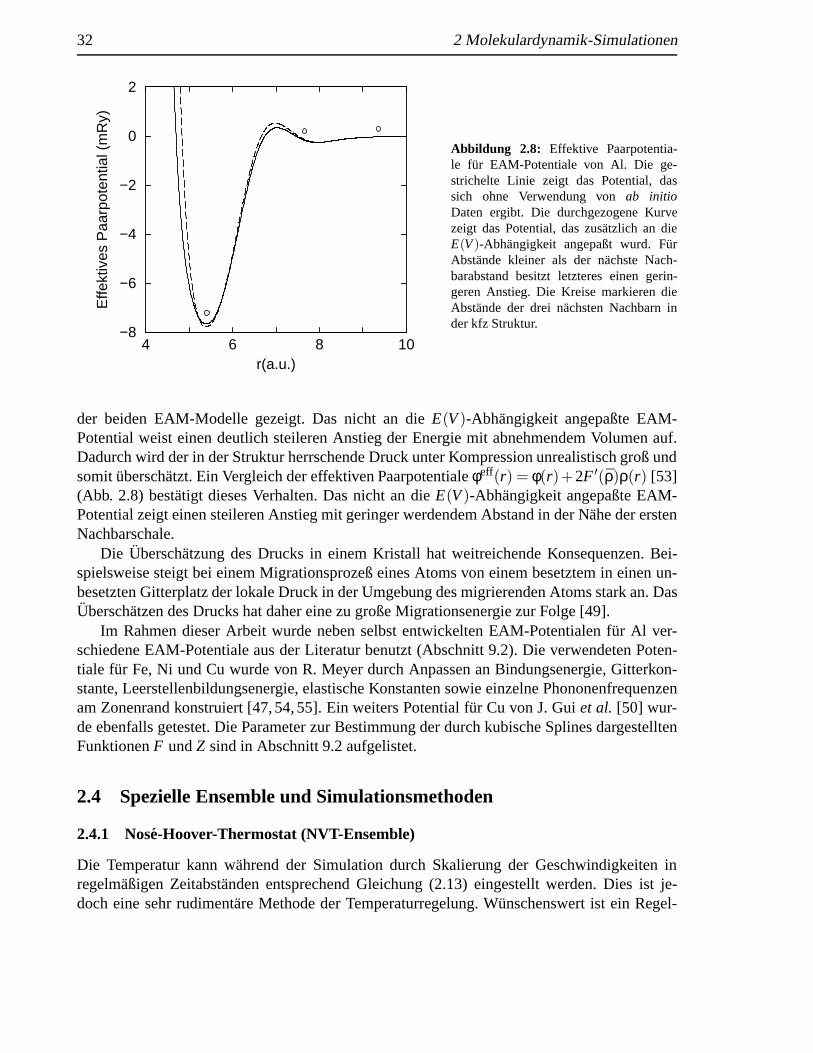
der beiden EAM-Modelle gezeigt. Das nicht an dieE(V)-Abhangigkeit angepaßte EAM-Potential weist einen deutlich steileren Anstieg der Energie mit abnehmendem Volumen auf.Dadurch wird der in der Struktur herrschende Druck unter Kompression unrealistisch groß undsomitubersch¨atzt. Ein Vergleich der effektiven Paarpotentialeφeff(r) = φ(r)+2F ′(ρ)ρ(r) [53](Abb. 2.8) best¨atigt dieses Verhalten. Das nicht an dieE(V)-Abhangigkeit angepaßte EAM-Potential zeigt einen steileren Anstieg mit geringer werdendem Abstand in der N¨ahe der erstenNachbarschale.
Die Uberschatzung des Drucks in einem Kristall hat weitreichende Konsequenzen. Bei-spielsweise steigt bei einem Migrationsprozeß eines Atoms von einem besetztem in einen un-besetzten Gitterplatz der lokale Druck in der Umgebung des migrierenden Atoms stark an. DasUberschatzen des Drucks hat daher eine zu große Migrationsenergie zur Folge [49].
Im Rahmen dieser Arbeit wurde neben selbst entwickelten EAM-Potentialen f¨ur Al ver-schiedene EAM-Potentiale aus der Literatur benutzt (Abschnitt 9.2). Die verwendeten Poten-tiale fur Fe, Ni und Cu wurde von R. Meyer durch Anpassen an Bindungsenergie, Gitterkon-stante, Leerstellenbildungsenergie, elastische Konstanten sowie einzelne Phononenfrequenzenam Zonenrand konstruiert [47, 54, 55]. Ein weiters Potential f¨ur Cu von J. Guiet al. [50] wur-de ebenfalls getestet. Die Parameter zur Bestimmung der durch kubische Splines dargestelltenFunktionenF undZ sind in Abschnitt 9.2 aufgelistet.
2.4 Spezielle Ensemble und Simulationsmethoden
2.4.1 Nose-Hoover-Thermostat (NVT-Ensemble)
Die Temperatur kann w¨ahrend der Simulation durch Skalierung der Geschwindigkeiten inregelmaßigen Zeitabst¨anden entsprechend Gleichung (2.13) eingestellt werden. Dies ist je-doch eine sehr rudiment¨are Methode der Temperaturregelung. W¨unschenswert ist ein Regel-

2.4 Spezielle Ensemble und Simulationsmethoden 33
mechanismus der kontinuierlich arbeitet. Nos´e schlug vor, die physikalische Zeitt ′ zu skalie-ren und somit die kinetischen Energien entsprechend zu verkleinern (s> 1) oder zu erh¨ohen(s< 1) [56,57]:
dt = s(t ′)dt ′. (2.29)
Hierzu fuhrte Nose folgende Lagrange-Funktion mit der virtuellen Zeitt fur N Atome ein:
L =12
N
∑i=1
mis2r2
i −Φ(r1, r2, ..., rN)+Q2
s2− (3N+1)kBT0 lns, (2.30)
wobeiΦ(r1, r2, ..., rN) die potentielle Energie,sder neue Freiheitsgrad des W¨armebads ist demeine effektive MasseQ zugeordnet wird.
Hieraus ergeben sich nach Anwendung des Lagrange II Verfahrens [58] die Bewegungs-gleichungen f¨ur die dynamischen Variablen in Abh¨angigkeit von der virtuellen Zeitt:
r i =1
s2miFi − 2s
sr i (2.31)
Qs =N
∑i=1
misr2i −
(3N+1)kBT0
s. (2.32)
Da es praktischer ist, die Simulation in der physikalischen Zeit durchzuf¨uhren, werden Glei-chung (2.31) und Gleichung (2.32) in Abh¨angigkeit von der physikalischen Zeit ausgedr¨uckt.Wird nun t ′ mit t bezeichnet, und die Abk¨urzungζ = s/s eingefuhrt, ergibt sich die Nos´e-Hoover-Formulierung [59]:
r i =1mi
Fi − ζr i︸ ︷︷ ︸Zusatzkra f t/mi
, (2.33)
12
Qζ =12
N
∑i=1
mi r2i︸ ︷︷ ︸
Ist−kinetische Energie
− (3N+1)kBT0
2︸ ︷︷ ︸Soll−kinetische Energie
. (2.34)
Fur die Bestimmung der Erhaltungsgr¨oße des Gesamtsystems, bestehend aus Atomen undThermostat (vgl. Gl. 2.36), wird lns benotigt:
d lnsdt
= ζ. (2.35)
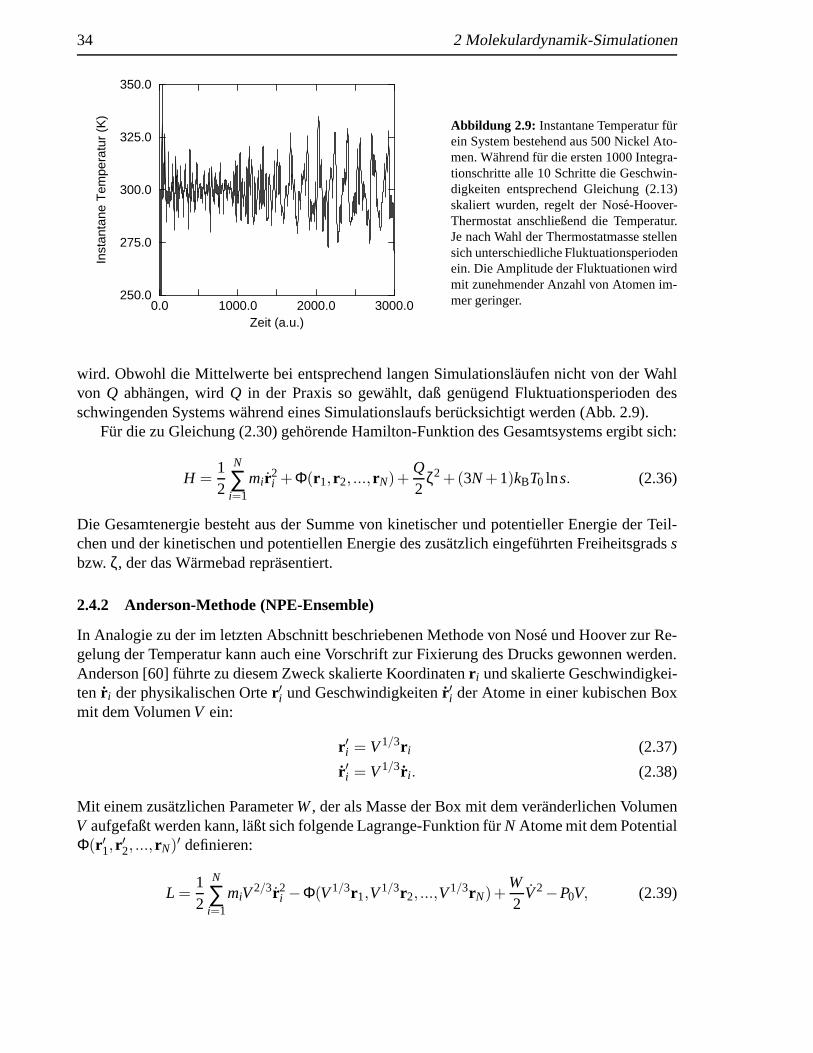
In dieser Formulierung wird der Regelmechanismus sofort deutlich. Die Bewegungsglei-chungen erhalten eine zus¨atzliche Reibungskraftζ, die je nach dem, ob die Ist-kinetische Ener-gie großer oder kleiner als die Soll-kinetische Energie des Systems ist, gr¨oßer bzw. kleinerwird und auch negative Werte annehmen kann. Der Nos´e-Hoover-Thermostat stellt einefeed-backMethode zur Temperaturregelung dar. Es kann gezeigt werde, daß die so erzeugten Zeit-mittelwerte denen des kanonischen Ensembles entsprechen [56]. Der als Thermostatmassebezeichnete ParameterQ bestimmt die Tr¨agheit, mit der die instantane Temperatur geregelt
34 2 Molekulardynamik-Simulationen
0.0 1000.0 2000.0 3000.0Zeit (a.u.)
250.0
275.0
300.0
325.0
350.0In
stan
tane
Tem
pera
tur
(K)
Abbildung 2.9: Instantane Temperatur f¨urein System bestehend aus 500 Nickel Ato-men. Wahrend fur die ersten 1000 Integra-tionschritte alle 10 Schritte die Geschwin-digkeiten entsprechend Gleichung (2.13)skaliert wurden, regelt der Nos´e-Hoover-Thermostat anschließend die Temperatur.Je nach Wahl der Thermostatmasse stellensich unterschiedliche Fluktuationsperiodenein. Die Amplitude der Fluktuationen wirdmit zunehmender Anzahl von Atomen im-mer geringer.
wird. Obwohl die Mittelwerte bei entsprechend langen Simulationsl¨aufen nicht von der Wahlvon Q abhangen, wirdQ in der Praxis so gew¨ahlt, daß gen¨ugend Fluktuationsperioden desschwingenden Systems w¨ahrend eines Simulationslaufs ber¨ucksichtigt werden (Abb. 2.9).
Fur die zu Gleichung (2.30) geh¨orende Hamilton-Funktion des Gesamtsystems ergibt sich:
H =12
N
∑i=1
mi r2i +Φ(r1, r2, ..., rN)+
Q2
ζ2 +(3N+1)kBT0 lns. (2.36)
Die Gesamtenergie besteht aus der Summe von kinetischer und potentieller Energie der Teil-chen und der kinetischen und potentiellen Energie des zus¨atzlich eingef¨uhrten Freiheitsgradssbzw.ζ, der das W¨armebad repr¨asentiert.
2.4.2 Anderson-Methode (NPE-Ensemble)
In Analogie zu der im letzten Abschnitt beschriebenen Methode von Nos´e und Hoover zur Re-gelung der Temperatur kann auch eine Vorschrift zur Fixierung des Drucks gewonnen werden.Anderson [60] fuhrte zu diesem Zweck skalierte Koordinatenr i und skalierte Geschwindigkei-ten r i der physikalischen Orter ′i und Geschwindigkeitenr ′i der Atome in einer kubischen Boxmit dem VolumenV ein:
r ′i = V1/3r i (2.37)
r ′i = V1/3r i . (2.38)
Mit einem zusatzlichen ParameterW, der als Masse der Box mit dem ver¨anderlichen VolumenV aufgefaßt werden kann, l¨aßt sich folgende Lagrange-Funktion f¨ur N Atome mit dem PotentialΦ(r ′1, r
′2, ..., rN)′ definieren:
L =12
N
∑i=1
miV2/3r2
i −Φ(V1/3r1,V1/3r2, ...,V
1/3rN)+W2
V2−P0V, (2.39)
2.4 Spezielle Ensemble und Simulationsmethoden 35
wobeiP0 der vorgegebene Druck des Systems ist. F¨ur die dynamischen Variablen folgen dannunter Anwendung des Lagrange II Verfahrens [58] die Bewegungsgleichungen f¨ur die skalier-ten Koordinaten und Geschwindigkeiten in Gleichung (2.37) und (2.38):
r i =1
V1/3miFi − 2V
3Vr i (2.40)
WV =V2/3
3V
N
∑i=1
mi r2i +
V−2/3
3
N
∑i=1
Fir i −P0. (2.41)
Die Rucktransformation auf die physikalischen Koordinaten und Geschwindigkeiten, die jetztmit r i und r i bezeichnet werden, liefern die folgenden Bewegungsgleichungen:
r i =1mi
Fi − V3V
r i︸ ︷︷ ︸Zusatzkra f t/mi
, (2.42)
WV =1
3V
N
∑i=1
mi r2i +
13V
N
∑i=1
Fir i︸ ︷︷ ︸instantaner Druck
−P0. (2.43)
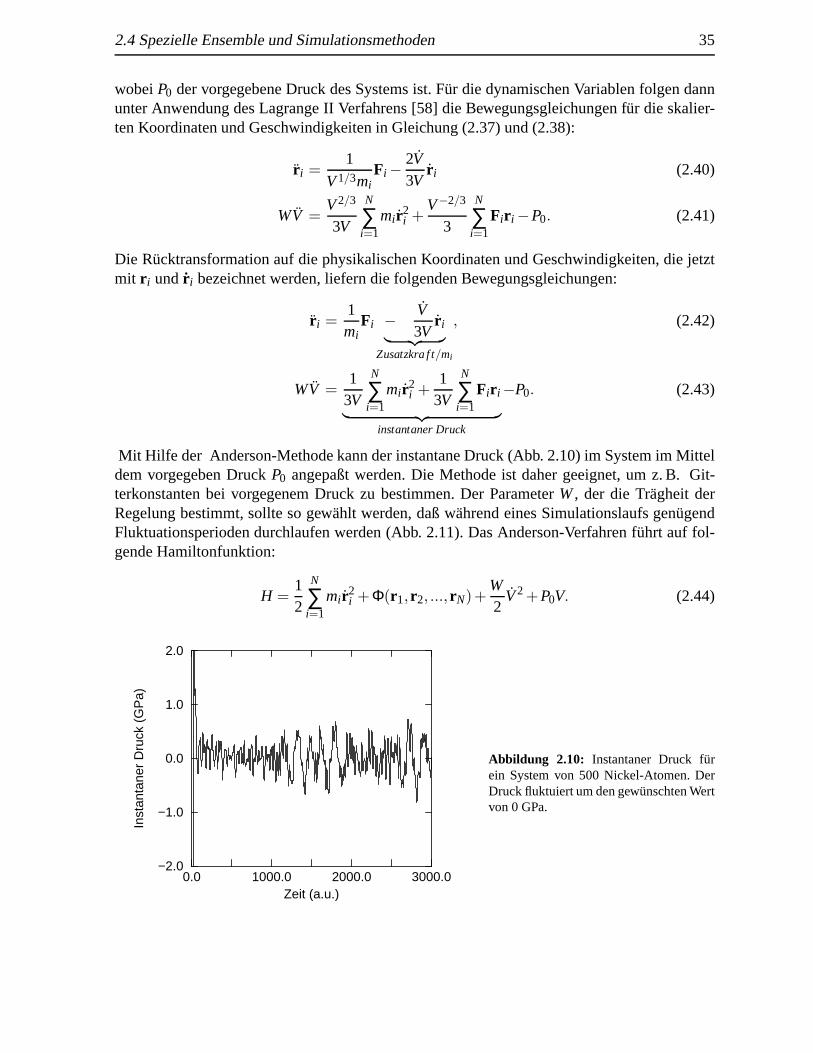
Mit Hilfe der Anderson-Methode kann der instantane Druck (Abb. 2.10) im System im Mitteldem vorgegeben DruckP0 angepaßt werden. Die Methode ist daher geeignet, um z. B. Git-terkonstanten bei vorgegenem Druck zu bestimmen. Der ParameterW, der die Tragheit derRegelung bestimmt, sollte so gew¨ahlt werden, daß w¨ahrend eines Simulationslaufs gen¨ugendFluktuationsperioden durchlaufen werden (Abb. 2.11). Das Anderson-Verfahren f¨uhrt auf fol-gende Hamiltonfunktion:
H =12
N
∑i=1
mi r2i +Φ(r1, r2, ..., rN)+
W2
V2 +P0V. (2.44)
0.0 1000.0 2000.0 3000.0Zeit (a.u.)
−2.0
−1.0
0.0
1.0
2.0
Inst
anta
ner
Dru
ck (
GP
a)
Abbildung 2.10: Instantaner Druck f¨urein System von 500 Nickel-Atomen. DerDruck fluktuiert um den gew¨unschten Wertvon 0 GPa.
36 2 Molekulardynamik-Simulationen
0.0 1000.0 2000.0 3000.0Zeit (a.u.)
76.00
76.10
76.20
76.30V
olum
en/A
tom
(a.
u.)
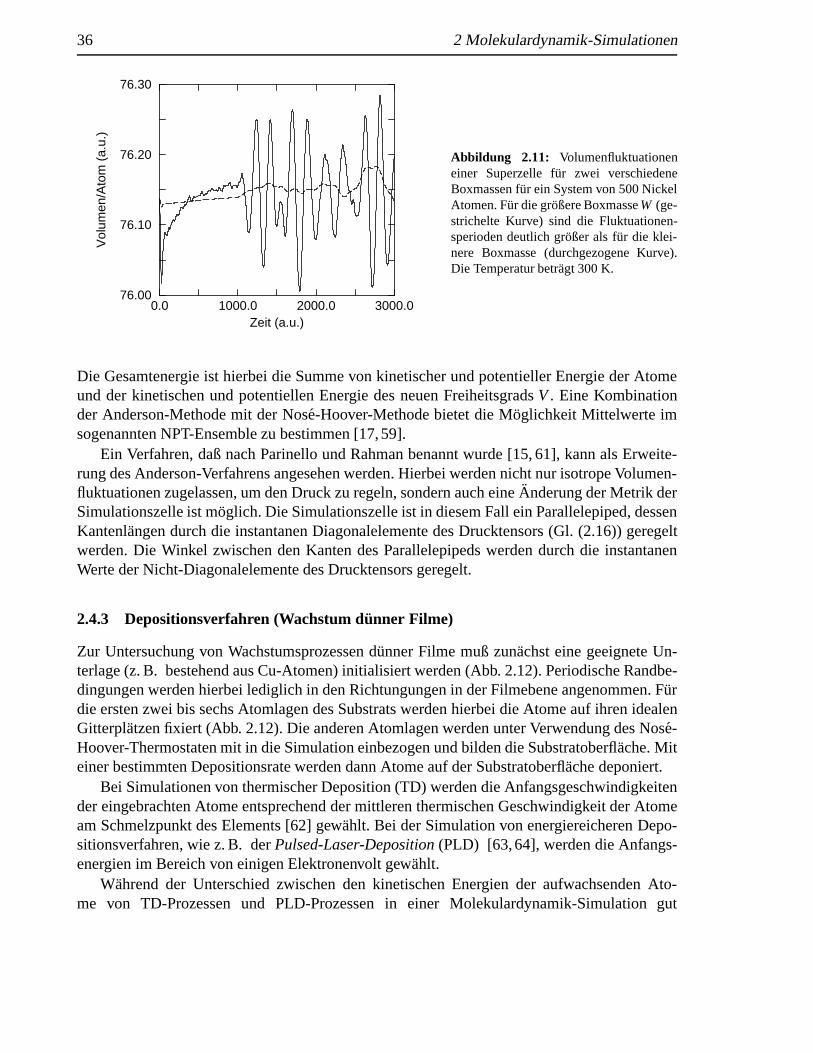
Abbildung 2.11: Volumenfluktuationeneiner Superzelle f¨ur zwei verschiedeneBoxmassen f¨ur ein System von 500 NickelAtomen. Fur die großere BoxmasseW (ge-strichelte Kurve) sind die Fluktuationen-sperioden deutlich gr¨oßer als f¨ur die klei-nere Boxmasse (durchgezogene Kurve).Die Temperatur betr¨agt 300 K.
Die Gesamtenergie ist hierbei die Summe von kinetischer und potentieller Energie der Atomeund der kinetischen und potentiellen Energie des neuen FreiheitsgradsV. Eine Kombinationder Anderson-Methode mit der Nos´e-Hoover-Methode bietet die M¨oglichkeit Mittelwerte imsogenannten NPT-Ensemble zu bestimmen [17,59].
Ein Verfahren, daß nach Parinello und Rahman benannt wurde [15, 61], kann als Erweite-rung des Anderson-Verfahrens angesehen werden. Hierbei werden nicht nur isotrope Volumen-fluktuationen zugelassen, um den Druck zu regeln, sondern auch eineAnderung der Metrik derSimulationszelle ist m¨oglich. Die Simulationszelle ist in diesem Fall ein Parallelepiped, dessenKantenlangen durch die instantanen Diagonalelemente des Drucktensors (Gl. (2.16)) geregeltwerden. Die Winkel zwischen den Kanten des Parallelepipeds werden durch die instantanenWerte der Nicht-Diagonalelemente des Drucktensors geregelt.
2.4.3 Depositionsverfahren (Wachstum d¨unner Filme)
Zur Untersuchung von Wachstumsprozessen d¨unner Filme muß zun¨achst eine geeignete Un-terlage (z. B. bestehend aus Cu-Atomen) initialisiert werden (Abb. 2.12). Periodische Randbe-dingungen werden hierbei lediglich in den Richtungungen in der Filmebene angenommen. F¨urdie ersten zwei bis sechs Atomlagen des Substrats werden hierbei die Atome auf ihren idealenGitterplatzen fixiert (Abb. 2.12). Die anderen Atomlagen werden unter Verwendung des Nos´e-Hoover-Thermostaten mit in die Simulation einbezogen und bilden die Substratoberfl¨ache. Miteiner bestimmten Depositionsrate werden dann Atome auf der Substratoberfl¨ache deponiert.
Bei Simulationen von thermischer Deposition (TD) werden die Anfangsgeschwindigkeitender eingebrachten Atome entsprechend der mittleren thermischen Geschwindigkeit der Atomeam Schmelzpunkt des Elements [62] gew¨ahlt. Bei der Simulation von energiereicheren Depo-sitionsverfahren, wie z. B. derPulsed-Laser-Deposition(PLD) [63,64], werden die Anfangs-energien im Bereich von einigen Elektronenvolt gew¨ahlt.
Wahrend der Unterschied zwischen den kinetischen Energien der aufwachsenden Ato-me von TD-Prozessen und PLD-Prozessen in einer Molekulardynamik-Simulation gut

2.4 Spezielle Ensemble und Simulationsmethoden 37
v
Filmatome
thermalisierteSubstratatome
fixierteSubstratatome
Abbildung 2.12: Bei Simulationen von Film-wachstum auf Substratoberfl¨achen werden dieuntersten Atomlagen in der Simulationszellefixiert. Die restlichen Substratatome werdenmit Hilfe des Nose-Hoover-Verfahrens thermali-siert. Die aufwachsenden Atome werden mit ei-ner bestimmten Geschwindigkeit auf die Ober-flache geschossen. Die Geschwindigkeit der auf-wachsenden Atome wird bei der Analyse vonthermischen Depositionsverfahren entsprechendder thermischen Geschwindigkeit der Atomeam Schmelzpunkt des Materials gew¨ahlt. Beider Simulation des Pulsed-Laser-Depositions-Verfahrens werden die Anfangsenergien im Be-reich von einigen Elektronenvolt gew¨ahlt. DasSystem weist nur in Richtungen in der Film-ebene periodische Randbedingungen auf.
berucksichtigt werden kann, ist der Unterschied zwischen den instantanen Depositionsratenbeider Verfahren in der Molekulardynamik-Simulation aus Rechenzeitgr¨unden nur schwer zuberucksichtigen. Sowohl TD- als auch PLD-Prozesse haben mittlere Aufwachsraten, die in derGroßenordnung von einigen Monolagen/Sekunde (ML/s) liegen. Das gepulste PLD-Verfahrenhat allerdings instantane Depositionsraten, die zwischen 104 und 106 ML/s liegen [63,64]. DieAnalyse des Einflusses der Depositionsrate ist aber m¨oglich im Rahmen von Wachstumsmo-dellen unter Verwendung kinetischer Monte-Carlo-Simulationen [65], die als ideale Erg¨anzungzu den Molekulardynamik-Simulationen angesehen werden k¨onnen.
2.4.4 Induzierung von Schockwellen
Eine ganz andere Methode strukturelle Phasen¨anderungen in Festk¨orpern zu generieren, bestehtdarin, Schockwellen [66] durch das System laufen zu lassen. Zur Erzeugung von Schockwel-len in Molekulardynamik-Simulationen gibt es verschiedene Methoden, die aber alle ¨ahnlicheErgebnisse liefern [26]. Bei der hier verwendetenMomentum-Mirror-Methode wird allen Ato-men anfanglich (zus¨atzlich zu den thermischen Geschwindigkeiten) eine GeschwindigkeitUP
in Richtung eines virtuellen Momentenspiegels, der sich bei z=0 senkrecht zur z-Achse be-findet, aufgepr¨agt (Abb. 2.13). Alle Atome, die den Spiegel treffen werden entsprechend ih-rer Impulsanderungpz → −pz reflektiert. Im Koordinatensystem in dem Spiegel ruht bewegtsich eine Schockfront mit der GeschwindigkeitUS−UP, wobeiUS die Geschwindigkeit derSchockfront im Festk¨orper ist. Die Situation entspricht einem Kolben unendlicher Masse, dermit der GeschwindigkeitUP von links kommend auf das System auftrifft und eine Schockwellemit der GeschwindigkeitUS nach rechts erzeugt (Abb. 2.13). Bei diesem Verfahren werden inaller Regel periodische Randbedingungen in transversaler Richtung angenommen, um Rand-effekte zu vermeiden. Da es sich um Nichtgleichgewichtsprozesse handelt, wird bei dieser Art

38 2 Molekulardynamik-Simulationen
UP
UP
PU
t=0
Spiegel t>0US-
Schockfront
SpiegelAbbildung 2.13: In Molekulardynamik-Si-mulationen k¨onnen Schockwellen mit Hilfedes Momentum-Mirror-Verfahrens induziertwerden. Alle Atome erhalten anf¨anglich Ge-schwindigkeitenUP in Richtung des sich beiz=0 befindlichen Spiegels. Atome, die denSpiegel treffen, ver¨andern die z-Komponenteihres Impulses entsprechendpz → −pz. EineSchockwelle mit der Ausbreitungsgeschwin-digkeit US, die sich im Koordinatensystem indem der Spiegel ruht mit der GeschwindigkeitUS −UP bewegt, wird erzeugt. Die Situationentspricht einem Kolben mit unendlicher Masse,der mit der GeschwindigkeitUP auf das Systemaufprallt.
von Simulationen kein weiterer Eingriff in die Bewegungsgleichungen, wie z. B. in Form desNose-Hoover-Thermostaten, vorgenommen.
2.5 Programmcode und Simulationsparameter
Die in dieser Arbeit vorgestellten Simulationen wurden unter Verwendung von drei verschiede-nen Programmen durchgef¨uhrt. Zum einen wurde mit dem Programm MDNTP (Molekulardy-namik des NTP-Ensemble) von Ralf Meyer [54] und dem von Peter Lomdahl und Dave Beazleyentwickelten CodeSPaSM(ScalableParallel Short-rangeMolecular dynamics) [67, 68] gear-beitet und entsprechend notwendige Modifikationen und Erweiterungen vorgenommen. Zumanderen wurde der eigene Code MD entwickelt, der die Simulation verschiedener Ensemble,verschiedener Potentiale und das Anpassen der EAM-Potentiale an Realkristalle gestattet (Ab-schnitt 2.3.2). Jeder Code hat spezifische Vor- und Nachteile, so erlaubt derSPaSMCode dieSimulation und Auswertung von sehr großen Atomzahlen (mehrere Millionen) auf parallelenRechnersystemen. Das Paket ist modular aufgebaut und die Durchf¨uhrung der Simulation ge-schieht unter Benutzung der ScriptsprachePython, die es dem Benutzer erlaubt, individuellgewunschte Routinen aufzurufen. Die Erweiterung des Pakets durch den Benutzer ist einfacherals fur ein geschlossenes C-Programm.
Der praktische Ablauf einer Molekulardynamik-Simulation wird im folgenden anhand desselbstentwickelten Programms MD beschrieben. Abbildung 2.14 zeigt dazu einen Ablaufplan.Zunachst wird durch das Initialisierungsprogramm eine Startkonfiguration der Atome erstellt.Dabei kann die Initialisierung der Atome auf einem Kristallgitter stattfinden. Das eigentlicheMolekulardynamik-Simulationsprogramm liest mehrere Dateien ein:
• Initialisierungsdatei, mit der die Simulation gesteuert wird. In ihr stehen Parameter wieLange des Simulationslaufs, Gr¨oße des Zeitinkrements, Temperatur, Druck, Randbe-dingungen, Aufdampfrate, Art der interatomaren Wechselwirkung, im Falle von EAM-

2.5 Programmcode und Simulationsparameter 39
Wechselwirkungen der Name der Datei, in der sich die Wechselwirkungsparameter befin-den, einige Parameter mit deren Hilfe Anzahl und Umfang der Ausgabedateien gesteuertwerden.
• Datei mit den Parametern der benutzten EAM-Wechselwirkung.
• Mit dem Initialisierungsprogramm erzeugte Atomkonfigurationsdatei.
Da der reine C-Quellcode ca. 10 000 Programmzeilen umfaßt, was einen Umfang von etwa250 DIN A4-Seiten entspricht, wurde auf den Abdruck des Programms verzichtet. Um dieArbeitsweise des Programms zu dokumentieren, befindet sich in Abschnitt 9.3 ein Abdruckder sich bei der Standardausgabe ergebenden Arbeitsschritte und Ausgaben. In Abschnitt 9.1befindet sich eine Tabelle der im Programm benutzten atomaren Einheiten.
Die Simulationsparameter des Nos´e-Hoover-Thermostaten und der Druckregelungsverfah-ren wurden so gew¨ahlt, daß die Fluktuationen der regulierten Gr¨oßen wie Temperatur undDruck genugend Perioden w¨ahrend eines Simulationslaufs durchlaufen um eine korrekte Mit-telung der Systemgr¨oßen zu gew¨ahrleisten. In meiner Diplomarbeit habe ich gezeigt, daß sichdie Wahl dieser Parameter in großen Bereichen nicht kritisch auf die Untersuchungen aus-wirkt [69]. Vor der eigentlichen Mittelung wird in der Regel unter wiederholter Skalierung derGeschwindigkeiten nach Gleichung (2.13) eine Equilibrirungsphase durchgef¨uhrt.
1. Velocity-Verlet Schritt
Neue Kräfte berechnen
Ja
2. Velocity-Verlet Schritt
Auswertung (instantane Größen)
Laufende?
Ergebnisse ausgeben
Ende der Simulation
Auswertung (gemittelte Größen)
Ergebnisse ausgeben
Simulationsparameter Startkonfiguration
Nein
Abbildung 2.14: Ablaufplan einer Molekular-dynamik-Simulation unter Verwendung desVelocity-Verlet-Algorithmus. Eine gezielte undeffiziente Auswertung und Ausgabe von instan-tanen Daten spart Festplattenplatz. Am Ende derSimulation werden die aktuellen Daten der Ato-me sowie sonstige Simulationsvariablen in eineDatei geschrieben, so daß die Simulation an die-ser Stelle fortgesetzt werden kann.

40 2 Molekulardynamik-Simulationen
0.0 1000.0 2000.0Zeit (a.u.)
−115.825
−115.775
−115.725
H (
Ry)
∆t=1 a.u.∆t=2 a.u.∆t=4 a.u.∆t=8 a.u.
Abbildung 2.15: Verhalten der Erhal-tungsgroße in Abhangigkeit des Zeit-schritts fur eine Simulationstemperaturvon 800 K. Ahnliches Verhalten ist beiZeitschritten von 1 a.u. und 2 a.u. zu ver-zeichnen. Bei Zeitinkrementen von 4 a.u.sind bereits deutlich st¨arkere Fluktuationenals bei kleineren Zeitschritten zu beobach-ten. Bei Integrationschritten von 8 a.u. isteine starke Drift der Erhaltungsgr¨oße zuerkennen, die schließlich zu einer numeri-schen Instabilit¨at fuhrt.
Der Zeitschritt in den Simulationen sollte so klein gew¨ahlt werden, daß die erzeugten Re-sultate nicht von ihm abh¨angen und die Simulation numerisch stabil verl¨auft. Aus Rechenzeit-grunden sollte der Zeitschritt auch nicht beliebig klein gew¨ahlt werden. Um einen Kompromißzu finden, wurden zwei Gesichtspunkte betrachtet. Physikalisch vern¨unftige Ergebnisse erfor-dern einen so kleinen Zeitschritt, daß die Periode der h¨ochsten im Festk¨orper auftretendenPhononenfrequenzen deutlich gr¨oßer ist als das Zeitinkrement. Die hier untersuchten Metallehaben longitudinale Zonenrandfrequenzen von bis zu etwa 40 meV. Hieraus ergibt sich eineminimale Schwingungsdauer von ca. 70 a.u. (102 fs). Somit ist die Wahl des Zeitinkrementsvon 1 a.u. (1.5 fs) sinnvoll. Als zweiter Gesichtspunkt wurde das Verhalten der Erhaltungsgr¨oßein Abhangigkeit des Zeitschritts betrachtet (Abb. 2.15). Die Fluktuationen der Erhaltungsgr¨oßefur eine Simulation eines Aluminium Kristalls bei 800 K sind bei einem Zeitschritt von 1 a.u.und 2 a.u. kaum zu unterscheiden. Bei einem Zeitschritt von 4 a.u. treten bereits deutliche Fluk-tuationen der Erhaltungsgr¨oße auf, w¨ahrend die Simulation aber noch w¨ahrend einer Simulati-onszeit von 3000 a.u. numerisch stabil ist. Bei einem Zeitschritt von 8 a.u. kommt es zu einerdeutlichen Drift der Erhaltungsgr¨oße, die in einer numerischen Instabilit¨at endet und schließ-lich zum Absturz des Programms f¨uhrt. Bei niedrigeren Temperaturen als den hier untersuchten800 K fallen die Fluktuationen und die Drift geringer aus. Dies rechtfertigt in den Simulatio-nen die Wahl des Zeitschritts von ca. 1 a.u., der auch bereits von S. Rubini und P. Ballone [51]gewahlt wurde, um strukturelle Phasen¨ubergange in Ni-Al-Legierungen mittels Molekulardy-namik-Simulationen zu untersuchen.

41
3 Struktur von Metallen
3.1 Allgemeines
Da in dieser Arbeit strukturelle Umwandlungen in metallischen Legierungen behandelt wer-den und es bei der Klassifikation von Phasen auf atomare Nachbarschaftsverh¨altnisse an-kommt, werden im folgenden der Vollst¨andigkeit halber einige elementare strukturelle Eigen-schaften aufgef¨uhrt. Metallische Bindungen zeichnen sich durch Delokalsierung der Valen-zelektronen der beteiligten Atome aus. Durch die Delokalisierung senkt sich die kinetischeEnergie der Valenzelektronen stark ab und liefert somit einen Beitrag zur Bindung. Bei denUbergangsmetallen, wie z.B. Fe, Co, Ni, Pd oder Pt, spielen zus¨atzlich kovalente Wechselwir-kungen der nicht aufgef¨ullten d- oder f-Orbitale eine Rolle. Die nicht oder nur schwach rich-tungsabh¨angig wirkende Wechselwirkung der Atome untereinander f¨uhrt zu einer m¨oglichstdicht gepackten Struktur bei Metallen. Das sind in der Regel die kubisch-fl¨achenzentrierte, diehexagonal-dichteste Packung oder das kubisch-raumzentrierte Gitter (Abb. 3.1). Die krz Struk-tur zeichnet sich durch eine geringere Raumausf¨ullung (68%) gegen¨uber den beiden anderenGittertypen (Raumausf¨ullung jeweils 74%) aus. Desweiteren treten besonders in Metallegie-rungen tetragonal verzerrte Typen dieser drei Gitter auf. Die verschiedenen Strukturen sind inden Abbildungen 3.2 bis 3.4 aus verschiedenen Blickrichtungen dargestellt.
Die hdp Struktur ist der kfz Struktur sehr ¨ahnlich, sie unterscheidet sich nur durch die un-terschiedliche Stapelfolge der dicht gepackten Ebenen. Anhand der Nachbarschftsverh¨altnisseder drei Gittertypen wird dieAhnlichkeit der beiden Strukturen nochmals verdeutlicht (Tab.
[001]
[100]
[010] [1120]
[1210]
[0001][2110][010]
[100]
[001]
Abbildung 3.1: Die drei wichtigsten Grundstrukturen von Metallen: kfz, hdp und krz Gitter. Der Kugelradiuswurde so gew¨ahlt, daß gleiche Raumausf¨ullung in den verschiedenen Gittern herrscht (z. B. akfz = 3
√2akrz). Zu
beachten ist, daß bei hdp Strukturen anstatt der drei Millerschen-Indizes(hkl) haufig vier Miller-Bravais-Indizes(hkil), mit i =−(h+k), verwendet werden. Das hat den Vorteil, daß kristallopgraphisch gleichwertige Richtungengleichartig indiziert werden, welches bei der Indizierung nach den drei Miller-Indizes nicht der Fall ist. Die imhdp Gitter eingezeichneten Richtungen ([2110], [1210], [1120] und [0001]) stellen somit eine erweiterte Basisdar [70,71].

42 3 Struktur von Metallen
[010]
[110]
[100]
kfz(001)
[100]
krz(001)
[010]
[110]
Abbildung 3.2: Das kfz und das krz Gitter aus der [001]-Richtung betrachtet . Zu erkennen sind zwei (001)-Ebenen (grau bzw. schwarz ) in der Stapelfolge ABAB. Der Atomabstand in[110]kfz-Richtung betr¨agt 1/
√2, in
[110]krz -Richtung√
2 kubische Gitterkonstanten.
[110]
[112]
[011]
kfz(111)
[1210]
hdp(0001)
[1120]
[1100] [110]
krz(110)
[001]
[111]
Abbildung 3.3: Sicht auf die dicht gepackten Ebenen der drei wichtigen Metallstrukturen kfz, hdp und krz. Diekfz und hdp Gitter unterscheiden sich lediglich durch die Stapelfolge. Das kfz Gitters ist ABCABC gestapelt, daßhdp Gitter hat die Stapelfolge ABAB der dicht gepackten Ebenen. Die ABAB gestapelten dicht gepackte Ebenedes krz Gitters ist im Vergleich zu den beiden anderen Gittertypen leicht tetragonal verzerrt, somit sehr ¨ahnlich.
3.1). Das hdp Gitter weist lediglich zus¨atzliche Nachbarschalen im Vergleich zum kfz Gitterauf. Der Vergleich der dicht gepackten Ebenen des kfz oder des hdp Gitters, d. h. der (111)-bzw. (0001)-Ebenen, mit der des krz Gitters zeigt, daß sich die dicht gepackten Ebenen deskfz- bzw. des hdp Gitters durch leichte Verzerrung in die(110)krz-Ebene transformieren lassen(Abb. 3.3). Das krz Gitter hat in der ersten Nachbarschale vier Nachbarn weniger als das kfz-oder hdp Gitter, daf¨ur hat es sechs nahe gelegene ¨ubernachste Nachbarn (Tab. 3.1).
Die mit Hilfe von Molekulardynamik-Simulationen erzeugten radialen Verteilungsfunktio-nen (Abschnitt 2.2.2) geben Aufschluß ¨uber die Struktur von Kristallen. In Abbildung 3.5 sindfur ideale krz, kfz und hdp Strukturen die radialen Verteilungsfunktionen bei einer Tempera-tur von 300 K dargestellt. Dabei wurden die Simulationszellen so pr¨apariert, daß sich jeweilseine gleiche Raumausf¨ullung einstellte. Die einzelnen Maxima spiegeln die Nachbarschafts-verhaltnisse der Kristallstrukturen wider. Die Breite der Maxima h¨angt von der Temperatur ab,

3.1 Allgemeines 43
[111]
[110]
[112]
kfz(110)
[0001]
[1010]
[1011]
hdp(1210)
Abbildung 3.4: Blick auf die ABAB gestapelten(110)kfz- und (1210)hdp-Ebenen. Sowohl die ABCABC Sta-pelfolge fur das kfz Gitter als auch die ABAB f¨ur das hdp Gitter der dicht gepackten Ebenen in[111]- bzw.[0001]-Richtung (senkrecht im Bild) sind deutlich zu erkennen. Der Abstand der Atome in[110]-Richtung betr¨agt1/
√2, in [112]
√3/2 kubische Gitterkonstanten.
was zu einem Verwaschen von zwei eng benachbarten Maxima durch thermische Fluktuationenfuhren kann (vgl. Abschnitt 2.2.2).
Da es in der Natur keine defektfreien Kristalle gibt, muß noch der Einfluß von Kristallbau-fehlern diskutiert werden. Kristallbaufehler bestimmen maßgeblich mit das makroskopischephysikalische Verhalten von Metallen. Typische Kristallbaufehler sind Leerstellen, Zwischen-gitteratome, Versetzungen, Korngrenzen oder Oberfl¨achen. Durch die Dynamik von Verset-zungen wird das plastische Verhalten bestimmt. Versetzungsdynamik wird wiederum durchKorngrenzen begrenzt. Auch Nukleationprozesse werden stark von bereits vorhanden Fehlernder Struktur beeinflußt. Eine Kristallprobe ist in der Regel aus verschieden orientierten Kri-stallbereichen, den sogenannten K¨ornern, aufgebaut. Die Grenzfl¨achen der K¨orner nennt man
Tabelle 3.1: Anzahl der AtomeN in den Nachbarschalen verschiedener Gitterstrukturen. Der AbstandA derNachbarschalen ist in Einheiten der kubischen Gitterkonstantea an gegeben. Um das kfz und das hdp Gittervergleichen zu k¨onnen, wurden die Abst¨ande des hdp Gitters in der kubischen Gitterkonstante des kfz Gitters mitgleicher Raumausf¨ullung angegeben.
Nachbarschale: 1. 2. 3. 4. 5.Struktur N A N A N A N A N A
kfz 12 0.7071 6 1.0 24 1.2247 12 1.4142 24 1.5811hdp 12 0.7071 6 1.0 2 1.1547 18 1.2247 12 1.3540krz 8 0.8660 6 1.0 8 1.4142 24 1.6583 8 1.7321
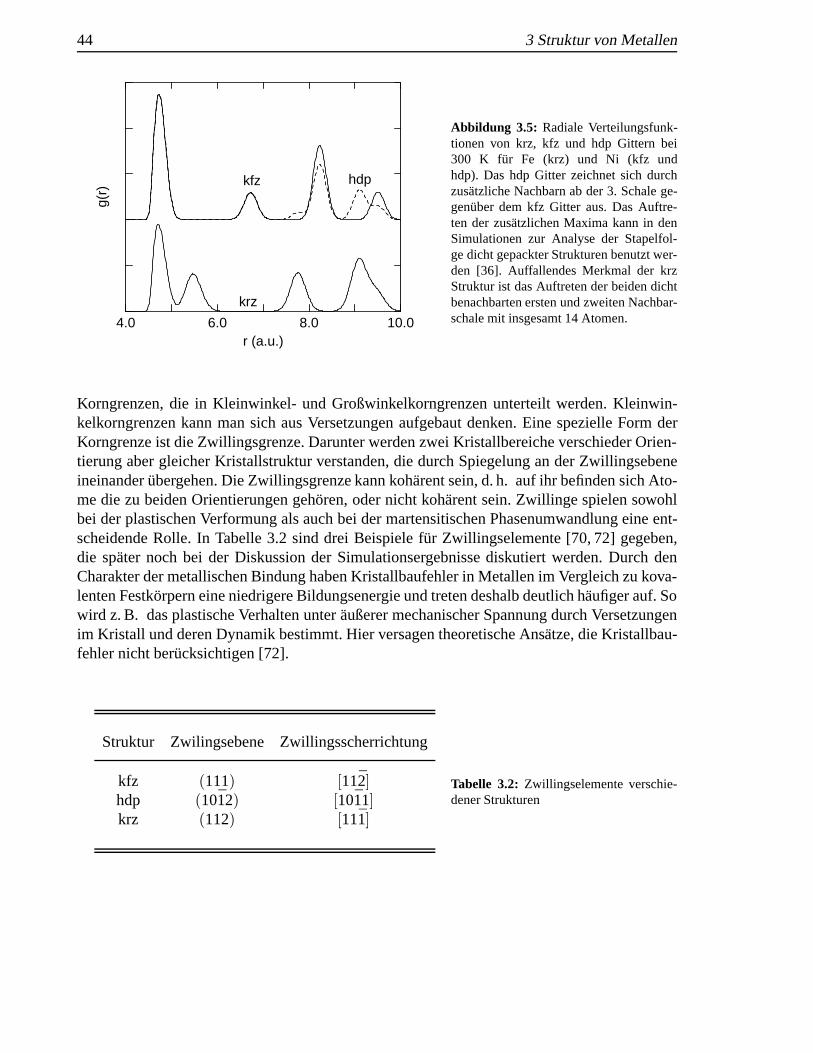
44 3 Struktur von Metallen
4.0 6.0 8.0 10.0r (a.u.)
g(r)
krz
kfz hdp
Abbildung 3.5: Radiale Verteilungsfunk-tionen von krz, kfz und hdp Gittern bei300 K fur Fe (krz) und Ni (kfz undhdp). Das hdp Gitter zeichnet sich durchzusatzliche Nachbarn ab der 3. Schale ge-genuber dem kfz Gitter aus. Das Auftre-ten der zus¨atzlichen Maxima kann in denSimulationen zur Analyse der Stapelfol-ge dicht gepackter Strukturen benutzt wer-den [36]. Auffallendes Merkmal der krzStruktur ist das Auftreten der beiden dichtbenachbarten ersten und zweiten Nachbar-schale mit insgesamt 14 Atomen.
Korngrenzen, die in Kleinwinkel- und Großwinkelkorngrenzen unterteilt werden. Kleinwin-kelkorngrenzen kann man sich aus Versetzungen aufgebaut denken. Eine spezielle Form derKorngrenze ist die Zwillingsgrenze. Darunter werden zwei Kristallbereiche verschieder Orien-tierung aber gleicher Kristallstruktur verstanden, die durch Spiegelung an der Zwillingsebeneineinander ¨ubergehen. Die Zwillingsgrenze kann koh¨arent sein, d. h. auf ihr befinden sich Ato-me die zu beiden Orientierungen geh¨oren, oder nicht koh¨arent sein. Zwillinge spielen sowohlbei der plastischen Verformung als auch bei der martensitischen Phasenumwandlung eine ent-scheidende Rolle. In Tabelle 3.2 sind drei Beispiele f¨ur Zwillingselemente [70, 72] gegeben,die spater noch bei der Diskussion der Simulationsergebnisse diskutiert werden. Durch denCharakter der metallischen Bindung haben Kristallbaufehler in Metallen im Vergleich zu kova-lenten Festk¨orpern eine niedrigere Bildungsenergie und treten deshalb deutlich h¨aufiger auf. Sowird z. B. das plastische Verhalten unter ¨außerer mechanischer Spannung durch Versetzungenim Kristall und deren Dynamik bestimmt. Hier versagen theoretische Ans¨atze, die Kristallbau-fehler nicht ber¨ucksichtigen [72].
Struktur Zwilingsebene Zwillingsscherrichtung
kfz (111) [112]hdp (1012) [1011]krz (112) [111]
Tabelle 3.2: Zwillingselemente verschie-dener Strukturen

3.2 Martensitische Transformationen 45
3.2 Martensitische Transformationen
3.2.1 Einfuhrung und Phanomenologie
Strukturelle Ubergange, d. h. Phasenumwandlungen innerhalb der festen Phase von einerGitterstruktur in eine andere Gitterstruktur, k¨onnen sowohl Phasen¨ubergange erster als auchzweiter Ordnung sein. Phasen¨ubergange zweiter Ordnung sind h¨aufig diffusionsgetrieben undzeichnen sich durch einen kontinuierlichen Verlauf materialspezifischer Gr¨oßen, wie z. B. derDichte, aus [73]. Eine spezielle Klasse struktureller Phasen¨ubergange erster Ordnung stel-len die martensitischen Transformationen dar, wobei die Hochtemperaturphase als Austenit,die Tieftemperaturphase als Martensit bezeichnet wird. Martensitische Transformationen oderdie Rucktransformationen (austenitische Transformationen) k¨onnen sowohl durch Tempera-turanderungen als auch durchAnderungen des Drucks oder auch nur einzelner Druckkompo-nenten [5, 69] induziert werden. Tabelle 3.3 zeigt Beispiele von Systemen, die martensitischeTransformationen aufweisen.
System Austenitstruktur Martensitstruktur
Fe-C kfz krz (tetragonal verzerrt)Fe-Ni kfz krz
Zr krz hdpNi-Al krz (B2) kfz (tetragonal verzerrt,7R,14M)
Tabelle 3.3: Beispiele furSysteme, die martensitischeUbergange zeigen.
Dieser Abschnitt stellt die f¨ur diese Arbeit wichtigen Aspekte martensitischerUbergangedar. Ausfuhrliche Arbeiten zu diesem Thema sind in den Artikeln von Wayman [4] und De-laey [5] und den Sonderb¨anden anl¨aßlich der regelm¨aßig stattfindenden Konferenzen ¨uber mar-tensitische Materialien (z. B.Journal de Physique 2000, ESOMAT 2000) enthalten.
Als allgemein akzeptierte Definition f¨ur martensitischeUbergange gilt [5]:
• Displazive, gitterverzerrende Umwandlung mit scherungsartiger Gestalts¨anderung,
• Strukturelle Phasenumwandlung, die ohne Diffusion ablaufen kann,
• Kinetik und Morphologie der Umwandlung werden durch Verzerrungsenergie bestimmt.
Dabei zeigt der sogenannte thermoelastische Martensit¨ubergang, wie er z. B. im Ni-Al-Systemvorkommt, eine kleine Hystereseschleife. Hierbei wachsen die Martensitplatten mit zunehmen-der Unterkuhlung weiter und verkleinern sich beim anschließendem Erhitzen wieder, da dasSystem sich in einer Art thermoelastischen Gleichgewicht befindet. Der Grenzfl¨achenanteilder Keimbildungsenergie (s. Gl. (3.2)) ist sehr klein und die Energie zur Bildung von Keimenbleibt elastisch in den Martensitplatten gespeichert. Der sogenannte ausbruchsartige Martensit

46 3 Struktur von Metallen
[001]
[010]
[010]
[001]
[100]
12.2
5% E
xpan
sion
[100
]
12.25% Expansion
20.63% Kontraktion
Abbildung 3.6: Die Bain-Transformationuberfuhrt die kfz Struktur (Einheitszelledurch schwarze Linien dargestellt) durchKontraktion in [001]-Richtung und Deh-nung in [010]- und [100]-Richtung in diekrz Struktur (Einheitszelle durch rote Lini-en dargestellt). Die hellblau eingezeichne-te dicht gepackte(111)kfz-Ebene transfor-miert in die dicht gepackte(011)krz-Ebene.Durch Rotation um die [100]-Achsen ent-stehen die f¨ur martensitische Transforma-tionen typische kristallographischen Ori-entierungsbeziehungen zwischen den be-teiligten Strukturen.
(burst-type) tritt z. B. in Fe-Ni- oder Fe-Pd-Legierungen auf. Er besitzt eine große Hystere-se, die einige 100 K umfassen kann [4, 72, 74] und zeichnet sich durch Bildung irreversiblerMartensitplatten mit typischer durch innere Verspannungen bestimmte Morphologie der kri-stallographisch verschieden orientierten Martensitplatten in der Austenitmatrix aus. Die Mar-tensitplatten wiederum weisen Zwillinge und Stapelfehler auf.
Gemaß der Wechsler-Lieberman-Read-Theorie kann die gesamte mikroskopische Verzer-rungPm durch Kombination von homogener GittertransformationB, inhomogener gitterinva-rianter ScherungP und RotationR dargestellt werden:
Pm = RPB. (3.1)
Als mikroskopische Verzerrung wird dabei die Transformationsmatrix verstanden, die sich ausder Veranderung verschiedener gerader Kratzer auf der urspr¨unglich glatten Austenitprobenach einer martensitischen Umwandlung ergibt. Die homogene GittertransformationB kannz. B. die Bain-Tranformation sein, die das kfz Gitter durch Stauchung und Dehnung in daskrz Gitter uberfuhrt (Abb. 3.6). Die inhomogene gitterinvariante Scherung wird durch Zwil-lingsbildung oder Stapelfehler realisiert. Die Grenzfl¨ache zwischen Martensit und Austenitwird hierbei als Habitusebene bezeichnet. Der Mechanismus der Wechsler-Lieberman-Read-Theorie beim Martensit¨ubergang ist schematisch in Abbildung 3.7 dargestellt. Klar zu erken-nen ist die mikroskopische Verkippung zwischen der urspr¨unglichen Austenit- und der neugebildeten Martensitphase. Anhand mikroskopischer Aufnahmen kann, bedingt durch die Ver-kippung der Martensit sowie verschiedene Martensitvarianten innerhalb einer Martensitnadelbeobachtet werden (Abb. 3.8).

3.2 Martensitische Transformationen 47
R
Austenit Martensit
B P
Abbildung 3.7: Schematische Darstellung der Abl¨aufe beim Martensit¨ubergang nach der Wechsler-Liebermann-Read-Theorie. Durch die homogene GittertransformationB wird die Martensitstruktur erzeugt. Die inhomogenegitterinvariante ScherungP wird hier schematisch durch eine Zwillingsbildung dargestellt. Die abschließendeRotationR fuhrt die Austenit- und Martensitphase an ihrer Grenzfl¨ache, der sogenannten Habitusebene zusammen.
Abbildung 3.8: Transmissionselektronen-mikroskopische Aufnahme einer Marten-sitnadel (gestreiftes Gebiet) eingebettet inder Austenitmatrix (weiß). Die bei tie-fen Temperaturen gebildete Martensitnadelweist feine Zwillinge (Streifen) auf. DieFe75Ni25 Legierung (Ms= 243 K) wurdebei Raumtemperatur mikroskopiert. (Quel-le: E. Hornbogen, Institut f¨ur Werstof-fe, Lehrstuhl Werstoffwissenschaft, Ruhr-Universitat Bochum)
Ein weiteres typisches Merkmal martensitischerUbergange sind feste kristallographischeOrientierungsbeziehungen zwischen der Austenit- und der Martensitstruktur, die eine Folge desgleichzeitigen Auftretens von Austenit und Martensit sind (z. B. [4,5,75]):
• Kurdjumov-Sachs:(111)kfz||(110)krz und[110]kfz||[111]krz.
Es transformieren dicht gepackte Ebenen in dicht gepackte Ebenen, sowie dicht gepackteRichtungen in dicht gepackte Richtungen.
• Nishiyama-Wassermann:(111)kfz||(101)krz und[121]kfz||[101]krz oderaquivalent dazu:(111)kfz||(110)krz und[110]kfz||[001]krz.
• Pitsch:(001)kfz||(110)krz und[110]kfz||[111]krz.
Die Kurdjumov-Sachs- und die Nishiyama-Wassermann-Orientierungsbeziehungen werdenhaufig in Fe-Legierungen in r¨ontgenographischen Untersuchungen festgestellt. Beispielswei-se findet man in Fe-1.4%C die Kurdjumov-Sachs-, in Fe-30%Ni die Nishiyama-Wassermann-Beziehung. Bei einer Fe-22%Ni-0.8%C Legierung liegt die Orientierungsbeziehung zwischen

48 3 Struktur von Metallen
den beiden oben genannten Extrema. In diesen F¨allen ist die Habitusebene jeweils eine hochindizierte Ebene. Zwischen d¨unnen krz Fe-Filmen und einem kfz Cu(001)-Substrat wird diePitsch-Orientierung [75] gefunden [76]. D¨unne krz Fe-Filme auf kfz Cu(111)-Oberfl¨achenzeigen die Kurdjumov-Sachs-Orientierungsbeziehung [77–79]. Die entsprechende Nishiyama-Wassermann-Beziehung f¨ur die Transformation einer hdp- in eine krz Struktur lautet:
• (0001)hdp||(110)krz und[1010]hdp||[110]krz oderaquivalent dazu:(0001)hdp||(110)krz und[1210]hdp||[001]krz.
Der von Burgers vorgeschlagene Mechanismus f¨ur die Transformation von der krz in die hdpStruktur, wie er z. B. in Ti bei 1156 K oder in Zr bei 1165 K vorkommt, f¨uhrt zu folgendenBeziehungen [80]:
• (110)krz||(0001)hdp und[111]krz||[2110]hdp.
Die Parallelitat von dicht gepackten Richtungen l¨aßt diese Beziehung damit als hdp-Analogonder Kurdjumov-Sachs Beziehung erscheinen.
Die Bain-Transformation (Abb. 3.6) ergibt folgende Orientierungsbeziehung:
• (001)kfz||(001)krz und [100]kfz||[110]krz
Die experimentell beobachteten Orientierungsbeziehungen k¨onnen durch die beschriebeneBain-Verzerrung sowie eine Rotation erf¨ullt werden. Dieser Sachverhalt demonstriert die Wich-tigkeit der Bain-Verzerrung als m¨oglichen Reaktionsweg einer martensitischen Umwandlung:Da die Energie entlang des Reaktionswegs sich durch Rotation nicht ¨andert, gibt die Bain-Transformation eine geeignete Grundlage um beispielsweise Energiebarrieren entlang des Re-aktionswegs zu analysieren.
Allen Orientierungsbeziehungen ist derUbergang von einer dicht gepackten Ebene in ei-ne andere dicht gepackte Ebene gemein. Eine Ausnahme bildet die Pitsch-Orientierung, diehauptsachlich in dunnen Filmen auftritt (Kap. 7). Selbstverst¨andlich konnen auch entsprechen-de kristallographisch ¨aquivalente Ebenen und Richtungen ineinander transformieren, um dieOrientierungsbeziehungen zu erf¨ullen. Beispielsweise kann eine der vier ¨aquivalenten(111)kfz-Ebenen,{111} = (111),(111),(111),(111), in eine(011)krz-Ebene transformieren.
3.2.2 Thermodynamik der Umwandlung und Nukleation
Die Ursache einer strukturellen Umwandlung ist dieAnderung der freien EnthalphienG derunterschiedlichen Strukturen in Abh¨angigkeit der intensiven Variablen wie Temperatur, Druckoder Magnetfeld, die denUbergang induzieren. Das System strebt in den Zustand niedrigsterfreier Enthalpie bzw. freier EnergieF, falls keinaußerer Druck vorliegt (Abschnitt 2.2.4).
Im Zusammenhang mit martensitischen Umwandlungen gibt es f¨unf verschiedene relevan-te Temperaturen (Abb. 3.9). Die thermodynamische GleichgewichtstemperaturM0, die experi-mentell nicht direkt meßbar ist, so daß man auf Absch¨atzungen angewiesen ist. Die TemperaturMs bei der beim Abk¨uhlen die martensitische Umwandlung einsetzt, sowie die TemperaturMf ,bei der der Prozeß der Martensitbildung abgeschlossen ist. Beim anschließenden Erhitzen be-zeichnetAs die Temperatur, bei der die austenitische Phase anf¨angt sich wieder auszubilden,
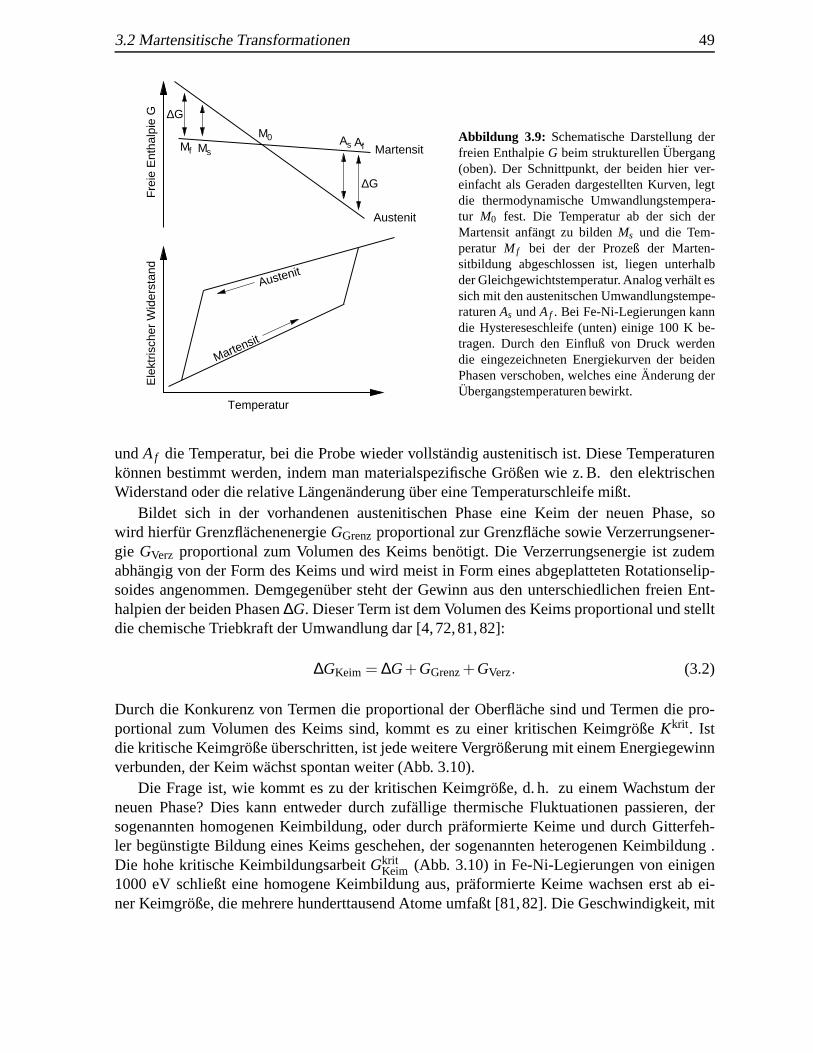
3.2 Martensitische Transformationen 49
0
∆G
∆G
M
Temperatur
Martensit
Austenit
AAs fMf MsF
reie
Ent
halp
ie G
Martensit
Austenit
Ele
ktris
cher
Wid
erst
and
Abbildung 3.9: Schematische Darstellung derfreien EnthalpieG beim strukturellenUbergang(oben). Der Schnittpunkt, der beiden hier ver-einfacht als Geraden dargestellten Kurven, legtdie thermodynamische Umwandlungstempera-tur M0 fest. Die Temperatur ab der sich derMartensit anfangt zu bildenMs und die Tem-peratur Mf bei der der Prozeß der Marten-sitbildung abgeschlossen ist, liegen unterhalbder Gleichgewichtstemperatur. Analog verh¨alt essich mit den austenitschen Umwandlungstempe-raturenAs undAf . Bei Fe-Ni-Legierungen kanndie Hystereseschleife (unten) einige 100 K be-tragen. Durch den Einfluß von Druck werdendie eingezeichneten Energiekurven der beidenPhasen verschoben, welches eineAnderung derUbergangstemperaturen bewirkt.
undAf die Temperatur, bei die Probe wieder vollst¨andig austenitisch ist. Diese Temperaturenkonnen bestimmt werden, indem man materialspezifische Gr¨oßen wie z. B. den elektrischenWiderstand oder die relative L¨angenanderung ¨uber eine Temperaturschleife mißt.
Bildet sich in der vorhandenen austenitischen Phase eine Keim der neuen Phase, sowird hierfur GrenzflachenenergieGGrenzproportional zur Grenzfl¨ache sowie Verzerrungsener-gie GVerz proportional zum Volumen des Keims ben¨otigt. Die Verzerrungsenergie ist zudemabhangig von der Form des Keims und wird meist in Form eines abgeplatteten Rotationselip-soides angenommen. Demgegen¨uber steht der Gewinn aus den unterschiedlichen freien Ent-halpien der beiden Phasen∆G. Dieser Term ist dem Volumen des Keims proportional und stelltdie chemische Triebkraft der Umwandlung dar [4,72,81,82]:
∆GKeim = ∆G+GGrenz+GVerz. (3.2)
Durch die Konkurenz von Termen die proportional der Oberfl¨ache sind und Termen die pro-portional zum Volumen des Keims sind, kommt es zu einer kritischen Keimgr¨oßeKkrit . Istdie kritische Keimgr¨oßeuberschritten, ist jede weitere Vergr¨oßerung mit einem Energiegewinnverbunden, der Keim w¨achst spontan weiter (Abb. 3.10).
Die Frage ist, wie kommt es zu der kritischen Keimgr¨oße, d. h. zu einem Wachstum derneuen Phase? Dies kann entweder durch zuf¨allige thermische Fluktuationen passieren, dersogenannten homogenen Keimbildung, oder durch pr¨aformierte Keime und durch Gitterfeh-ler begunstigte Bildung eines Keims geschehen, der sogenannten heterogenen Keimbildung .Die hohe kritische KeimbildungsarbeitGkrit
Keim (Abb. 3.10) in Fe-Ni-Legierungen von einigen1000 eV schließt eine homogene Keimbildung aus, pr¨aformierte Keime wachsen erst ab ei-ner Keimgroße, die mehrere hunderttausend Atome umfaßt [81,82]. Die Geschwindigkeit, mit
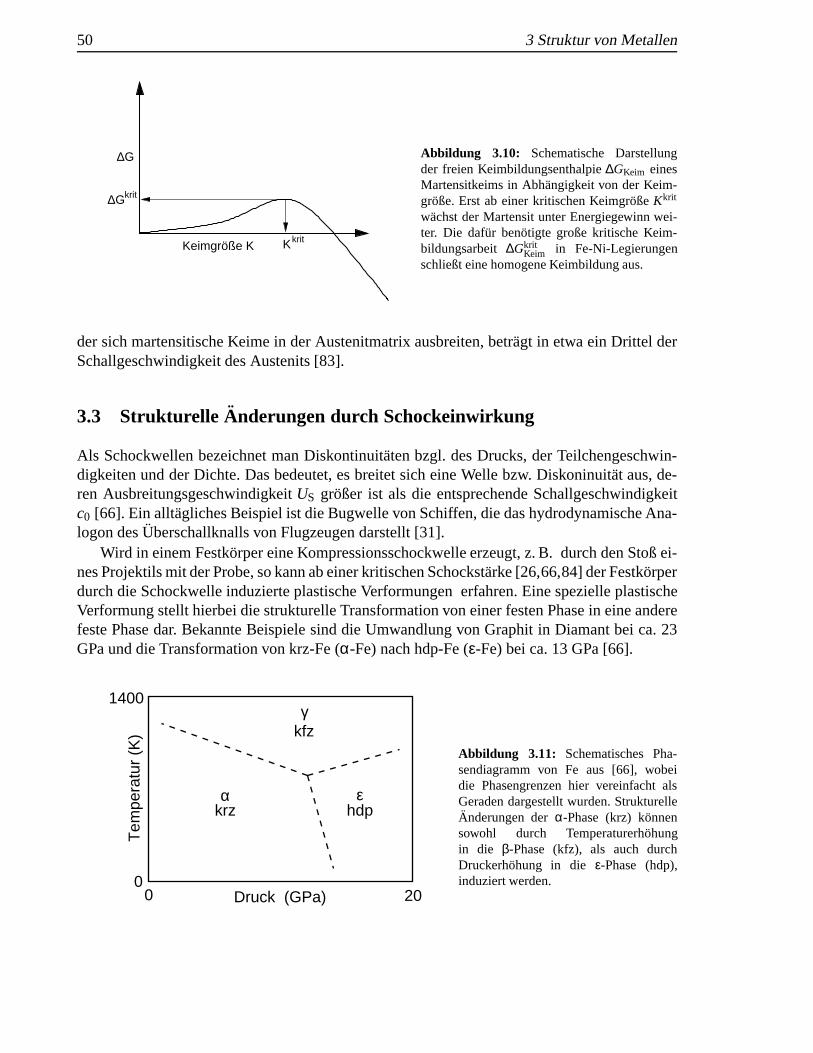
50 3 Struktur von Metallen
G
∆G
∆
kritKeimgröße K
krit
K
Abbildung 3.10: Schematische Darstellungder freien Keimbildungsenthalpie∆GKeim einesMartensitkeims in Abh¨angigkeit von der Keim-große. Erst ab einer kritischen Keimgr¨oßeKkrit
wachst der Martensit unter Energiegewinn wei-ter. Die dafur benotigte große kritische Keim-bildungsarbeit ∆Gkrit
Keim in Fe-Ni-Legierungenschließt eine homogene Keimbildung aus.
der sich martensitische Keime in der Austenitmatrix ausbreiten, betr¨agt in etwa ein Drittel derSchallgeschwindigkeit des Austenits [83].
3.3 Strukturelle Anderungen durch Schockeinwirkung
Als Schockwellen bezeichnet man Diskontinuit¨aten bzgl. des Drucks, der Teilchengeschwin-digkeiten und der Dichte. Das bedeutet, es breitet sich eine Welle bzw. Diskoninuit¨at aus, de-ren AusbreitungsgeschwindigkeitUS großer ist als die entsprechende Schallgeschwindigkeitc0 [66]. Ein alltagliches Beispiel ist die Bugwelle von Schiffen, die das hydrodynamische Ana-logon desUberschallknalls von Flugzeugen darstellt [31].
Wird in einem Festk¨orper eine Kompressionsschockwelle erzeugt, z. B. durch den Stoß ei-nes Projektils mit der Probe, so kann ab einer kritischen Schockst¨arke [26,66,84] der Festk¨orperdurch die Schockwelle induzierte plastische Verformungen erfahren. Eine spezielle plastischeVerformung stellt hierbei die strukturelle Transformation von einer festen Phase in eine anderefeste Phase dar. Bekannte Beispiele sind die Umwandlung von Graphit in Diamant bei ca. 23GPa und die Transformation von krz-Fe (α-Fe) nach hdp-Fe (ε-Fe) bei ca. 13 GPa [66].
krz
kfz
hdpα
γ
ε
Druck (GPa)
Tem
pera
tur
(K)
0 20
1400
0
Abbildung 3.11: Schematisches Pha-sendiagramm von Fe aus [66], wobeidie Phasengrenzen hier vereinfacht alsGeraden dargestellt wurden. StrukturelleAnderungen derα-Phase (krz) k¨onnensowohl durch Temperaturerh¨ohungin die β-Phase (kfz), als auch durchDruckerhohung in die ε-Phase (hdp),induziert werden.

3.4 Festk¨orper, Nanopartikel und d¨unne Schichten 51
In Abbildung 3.11 ist das Phasendiagramm von Fe als Funktion der Temperatur und desDrucks schematisch gezeigt. Zu erkennen ist der durch Druck induzierte strukturelleUbergangderα-Phase in dieε-Phase. Man kann somit durch Kompressionswellen in Fe eine Transforma-tion der krz Struktur in eine dicht gepackte Struktur induzieren. Umgekehrt kann eine Trans-formation der dicht gepackten Phase in die krz Phase durch Dekommpressionswellen induziertwerden, die bei der Reflexion von Kompressionswellen entstehen [66].
3.4 Festkorper, Nanopartikel und dunne Schichten
Nimmt die Oberflache im Verh¨altnis zum Volumen einer Probe einen signifikanten Anteil ein,so werden physikalische Eigenschaften maßgeblich durch die Oberfl¨ache mitbestimmt. Da-mit kann im Vergleich zum Festk¨orper eine dramatischeAnderung des physikalischen Ver-haltens eintreten. An der Oberfl¨ache kommt es zu einer Ver¨anderung des Atomlagenabstandssenkrecht zur Oberfl¨ache. Es kann zur sogenannten Oberfl¨achenrekonstruktion kommen, eineVeranderung der Symmetrie der Oberfl¨ache im Vergleich zum Festk¨orper. Dies tritt im allge-meinen bei nicht dicht gepackten Oberfl¨achen auf, wie z. B. der Au(110) (1x2) Rekonstruktion,bei der jeweils ganze Reihen von Atomen fehlen. Bei Metallen wird meist eine Verringerungdes Ebenenabstands der Oberfl¨achenebene und der darunter liegenden Ebene um einige Pro-zent festgestellt. Die Abst¨ande der darunter liegenden Ebenen verhalten sich in der Regel alter-nierend bzgl. des Vorzeichens der Relaxation [85]. An Oberfl¨achen bildet sich eine sogenannteOberflachenspannung, so daß an der Oberfl¨ache die Komponenten in der Oberfl¨achenebene deslokalen Drucktensors (vgl. Abschnitt 2.2.1) negativ sind, und daher die Oberfl¨ache verkleinernmochten, wahrend die senkrechte Komponente auf Grund der m¨oglichen Relaxation senkrechtzur Oberflache Null ist (Abschnitt 4.2) [86].
Experimentelle Werte der Relaxationen an Oberfl¨achen und Oberfl¨achenenergien streuenzum Teil stark, so daß der Vergleich mit Modellen erschwert wird. F¨ur die Relaxation desersten Lagenabstands f¨ur Cu(100)-Oberfl¨achen werden Werte von -2.1% bis -1.0% angege-benen. Werte f¨ur die Relaxation der Cu(111)-Oberfl¨ache streuen von -0.7% bis -0.3% [87].Fur Ni werdenahnliche Werte angegeben. Die aus zwei experimentellen Werten gemittelteOberflachenenergie f¨ur die krz Fe(100)- und Fe(110)-Oberfl¨ache ergibt 2.1 10−3 a.u. bzw.1.88 10−3 a.u. Der Vergleich mit dem hier benutzten EAM-Modell zeigt eine zufriedenstel-lendeUbereinstimmung (Tab. 4.3).
Fur die Diskussion von strukturellen Phasen¨ubergangen in Nanopartikeln und d¨unnen Fil-men muß neben den Betrachtungen aus Abschnitt 3.2.2 auch ber¨ucksichtigt werden, daß derGewinn an freier Energie von der Oberfl¨ache abh¨angen kann, und zwar um so st¨arker je großerdas Verhaltnis von Oberfl¨ache zu Volumen ist [88]. Beispielsweise fanden 1996 T. Tadakiet al.eine Verminderung der martensitischen StarttemperaturMs an Fe-Ni-Nanopartikeln [89]. DerEinfluß der Partikelgr¨oße auf martensitische Umwandlungstemperaturen wird in Kapitel 6 mitHilfe von Molekulardynamik-Simulationen untersucht und mit entsprechenden Experimentenverglichen.
Dunne Filme von einigen Atomlagen lassen sich in nicht-Gleichgewichtsstrukturen aufSubstratoberfl¨achen aufwachsen. So ist es m¨oglich, bei Raumtemperatur und auch bei nied-rigeren Temperaturen, d¨unne Fe-Schichten von einigen Atomlagen auf Substraten wie Cu, Pd,

52 3 Struktur von Metallen
Ag oder Ni in der kfz Struktur stabilisieren, obwohl Fe in diesem Temperaturbereich eigentlichin der krz Struktur vorliegt. Die kritische Fe-Schichtdicke bis zu der sich die Schicht in derkfz Phase stabilisieren l¨aßt, hangt dabei empfindlich vom gew¨ahlten Substrat sowie von derkristallographischen Orientierung des Substrats ab [76,77,79,79,90–99].Uber den Einfluß desVerfahrens zur Filmpr¨aparation gibt es intensive Studien bez¨uglich dunner Fe-Filme auf Cu-Oberflachen [20, 64, 100]. Diese zeigen, daß bei thermisch deponiertem Fe auf Cu-SubstratenInselwachstum oder Multilagenwachstum vorliegt. Im Gegensatz dazu zeigen mittels gepul-ster Laser-Deposition aufgewachsene Fe-Filme ein Lage-f¨ur-Lage Wachstum. Der strukturelleUbergang, der mit gepulster Laser-Deposition erzeugten Fe-Filme findet erst bei h¨oheren Film-dicken als bei den thermisch deposition Fe-Filmen statt. In Kapitel 7 wird die strukturelle Sta-bilit at dunner Filme im Hinblick auf kristallographisch verschieden orientierte Substrate undunterschiedlicher Depositionsverfahren untersucht.
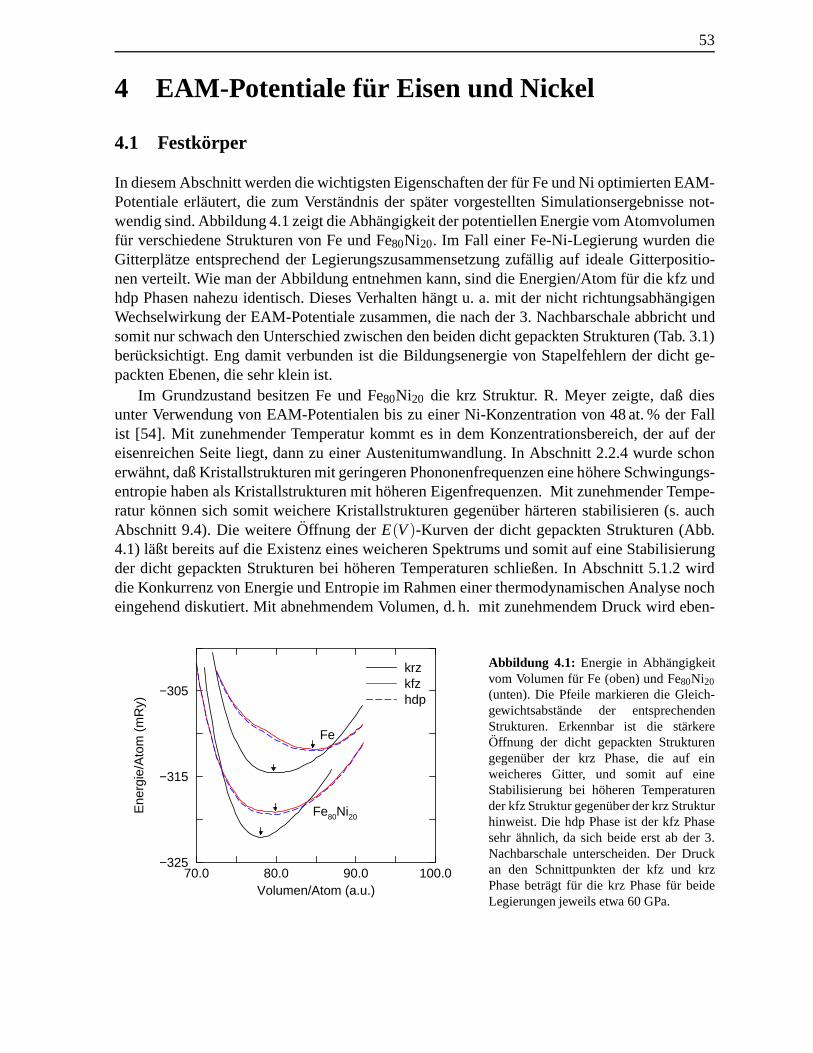
53
4 EAM-Potentiale fur Eisen und Nickel
4.1 Festkorper
In diesem Abschnitt werden die wichtigsten Eigenschaften der f¨ur Fe und Ni optimierten EAM-Potentiale erl¨autert, die zum Verst¨andnis der sp¨ater vorgestellten Simulationsergebnisse not-wendig sind. Abbildung 4.1 zeigt die Abh¨angigkeit der potentiellen Energie vom Atomvolumenfur verschiedene Strukturen von Fe und Fe80Ni20. Im Fall einer Fe-Ni-Legierung wurden dieGitterplatze entsprechend der Legierungszusammensetzung zuf¨allig auf ideale Gitterpositio-nen verteilt. Wie man der Abbildung entnehmen kann, sind die Energien/Atom f¨ur die kfz undhdp Phasen nahezu identisch. Dieses Verhalten h¨angt u. a. mit der nicht richtungsabh¨angigenWechselwirkung der EAM-Potentiale zusammen, die nach der 3. Nachbarschale abbricht undsomit nur schwach den Unterschied zwischen den beiden dicht gepackten Strukturen (Tab. 3.1)berucksichtigt. Eng damit verbunden ist die Bildungsenergie von Stapelfehlern der dicht ge-packten Ebenen, die sehr klein ist.
Im Grundzustand besitzen Fe und Fe80Ni20 die krz Struktur. R. Meyer zeigte, daß diesunter Verwendung von EAM-Potentialen bis zu einer Ni-Konzentration von 48 at. % der Fallist [54]. Mit zunehmender Temperatur kommt es in dem Konzentrationsbereich, der auf dereisenreichen Seite liegt, dann zu einer Austenitumwandlung. In Abschnitt 2.2.4 wurde schonerwahnt, daß Kristallstrukturen mit geringeren Phononenfrequenzen eine h¨ohere Schwingungs-entropie haben als Kristallstrukturen mit h¨oheren Eigenfrequenzen. Mit zunehmender Tempe-ratur konnen sich somit weichere Kristallstrukturen gegen¨uber harteren stabilisieren (s. auchAbschnitt 9.4). Die weitereOffnung derE(V)-Kurven der dicht gepackten Strukturen (Abb.4.1) laßt bereits auf die Existenz eines weicheren Spektrums und somit auf eine Stabilisierungder dicht gepackten Strukturen bei h¨oheren Temperaturen schließen. In Abschnitt 5.1.2 wirddie Konkurrenz von Energie und Entropie im Rahmen einer thermodynamischen Analyse nocheingehend diskutiert. Mit abnehmendem Volumen, d. h. mit zunehmendem Druck wird eben-
70.0 80.0 90.0 100.0Volumen/Atom (a.u.)
−325
−315
−305
Ene
rgie
/Ato
m (
mR
y)
krzkfzhdp
Fe80Ni20
Fe
Abbildung 4.1: Energie in Abhangigkeitvom Volumen fur Fe (oben) und Fe80Ni20
(unten). Die Pfeile markieren die Gleich-gewichtsabst¨ande der entsprechendenStrukturen. Erkennbar ist die st¨arkereOffnung der dicht gepackten Strukturengegen¨uber der krz Phase, die auf einweicheres Gitter, und somit auf eineStabilisierung bei h¨oheren Temperaturender kfz Struktur gegen¨uber der krz Strukturhinweist. Die hdp Phase ist der kfz Phasesehr ahnlich, da sich beide erst ab der 3.Nachbarschale unterscheiden. Der Druckan den Schnittpunkten der kfz und krzPhase betr¨agt fur die krz Phase f¨ur beideLegierungen jeweils etwa 60 GPa.

54 4 EAM-Potentiale fur Eisen und Nickel
Struktur Legierung 100 K 300 K
krz Fe 0.313634 0.311729kfz Fe 0.310846 0.309449
Unterschied 0.002788 0.002280
Tabelle 4.1:Mittlere potentielle Energie(Ry) von Fe fur verschiedene Temperatu-ren
falls eine dicht gepackte Struktur gegen¨uber der krz Struktur stabilisiert. Der Schnittpunkt derjeweiligenE(V)-Kurven unterschiedlicher Strukturen liegt bei einem DruckP = −dE
dV in derkrz Phase von ca. 60 GPa sowohl f¨ur Fe als auch f¨ur das Fe80Ni20-System. Dies bestimmt denkritischen Druck fur eine strukturelle Umwandlung unter Verwendung von EAM-Potentialen.
Tabelle 4.1 zeigt die gemittelten potentiellen Energien von Fe in verschiedenen Gitterstruk-turen bei 100 K und 300 K sowie den Unterschied der gemittelten potentiellen Energien der krzund kfz Struktur. Die gemittelte potentielle Energie und damit auch die innere Energie nimmtin der krz Phase st¨arker mit der Temperatur zu als in der kfz Phase. Somit kann auf eine h¨oherespezifische W¨armecp = T (∂S/∂T)p = (∂E/∂T)p=0 [34,101] der krz Struktur im Unterschiedzur kfz Struktur geschlossen werden. Aufgrund des st¨arkeren Ansteigens der inneren Energiein der krz Phase als in der kfz Struktur, steigt auch die Entropie in der krz Phase st¨arker als inder kfz Phase. R. Meyer berechnete die spezifische W¨armecp aus derAnderung der innerenEnergie fur das EAM-Modell von Fe zu 25.7 J/mol K und 21.4 J/mol K f¨ur die krz- bzw. kfzStruktur [54]. Die entsprechenden experimentellen Werte sind 38.6 J/mol K und 33.1 J/mol Kfur die krz bzw. kfz Struktur [102].
Die Abstande zwischen Ni-Atomen sind kleiner als die Abst¨ande zwischen Fe-Atomen(Tab. 4.2). In einer Simulation bei konstantem Druck von Fe-Ni-Legierungen stellt sich entspre-chend der Konzentration der Ni-Atome ein Kompromiß bez¨uglich des Atomvolumens von Niund Fe ein. Eine Analyse des lokalen Drucks der Atome (Abschnitt 2.2.1) in einer krz Fe80Ni20-Legierung bei 300 K ergibt einen mittleren lokalen Druck der Ni-Atome von ca.−11.06 GPa.Fur die Fe-Atome erh¨alt man einen mittleren Druck von ca. 2.85 GPa. Insgesamt addierensich die Drucke entsprechend der Legierungszusammensetzung zu Null. Der lokale positiveDruck der Fe-Atome spiegelt die Tatsache wider, daß Fe ein h¨oheres Gleichgewichtsvolumen
System 0 K 300 K
krz Fe 5.424 5.492kfz Fe 6.969 6.976kfz Ni 6.652 6.728
Tabelle 4.2: Gitterkonstanten von Fe und Ni (a.u.) f¨ur verschiedeneTemperaturen
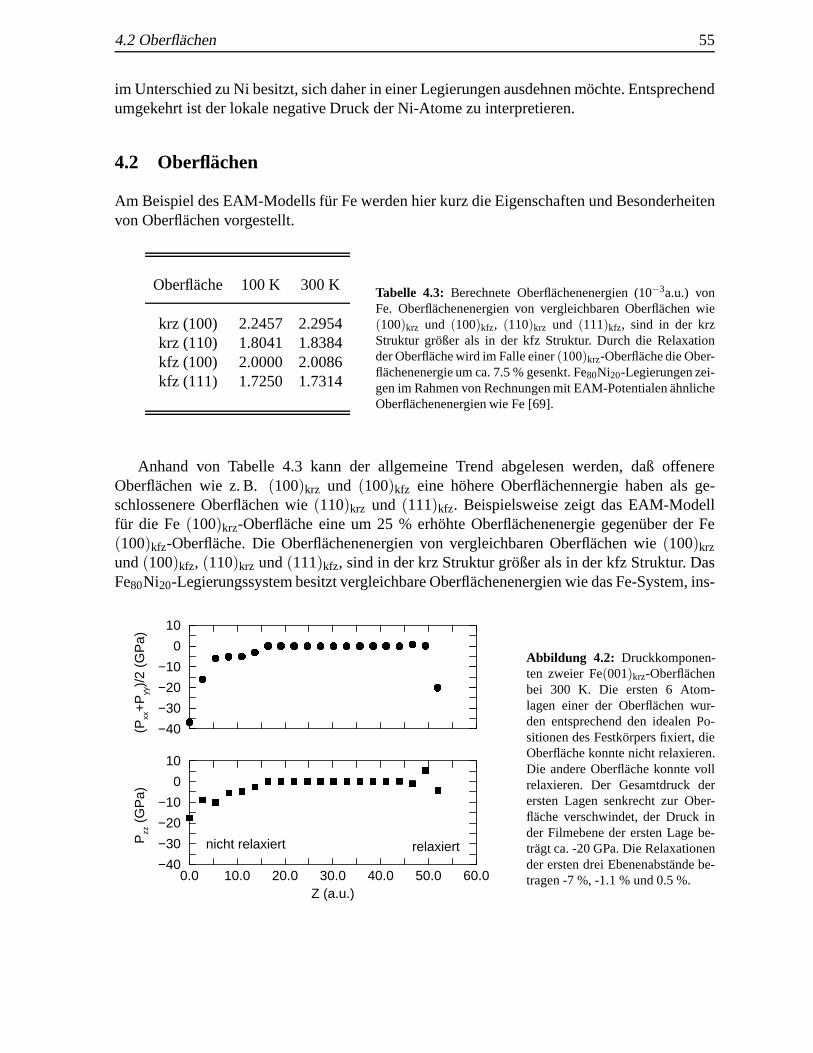
4.2 Oberflachen 55
im Unterschied zu Ni besitzt, sich daher in einer Legierungen ausdehnen m¨ochte. Entsprechendumgekehrt ist der lokale negative Druck der Ni-Atome zu interpretieren.
4.2 Oberflachen
Am Beispiel des EAM-Modells f¨ur Fe werden hier kurz die Eigenschaften und Besonderheitenvon Oberflachen vorgestellt.
Oberflache 100 K 300 K
krz (100) 2.2457 2.2954krz (110) 1.8041 1.8384kfz (100) 2.0000 2.0086kfz (111) 1.7250 1.7314
Tabelle 4.3: Berechnete Oberfl¨achenenergien (10−3a.u.) vonFe. Oberflachenenergien von vergleichbaren Oberfl¨achen wie(100)krz und (100)kfz, (110)krz und (111)kfz, sind in der krzStruktur großer als in der kfz Struktur. Durch die Relaxationder Oberflache wird im Falle einer(100)krz-Oberflache die Ober-flachenenergie um ca. 7.5 % gesenkt. Fe80Ni20-Legierungen zei-gen im Rahmen von Rechnungen mit EAM-Potentialen ¨ahnlicheOberflachenenergien wie Fe [69].
Anhand von Tabelle 4.3 kann der allgemeine Trend abgelesen werden, daß offenereOberflachen wie z. B. (100)krz und (100)kfz eine hohere Oberfl¨achennergie haben als ge-schlossenere Oberfl¨achen wie(110)krz und (111)kfz. Beispielsweise zeigt das EAM-Modellfur die Fe(100)krz-Oberflache eine um 25 % erh¨ohte Oberflachenenergie gegen¨uber der Fe(100)kfz-Oberflache. Die Oberfl¨achenenergien von vergleichbaren Oberfl¨achen wie(100)krzund(100)kfz, (110)krz und(111)kfz, sind in der krz Struktur gr¨oßer als in der kfz Struktur. DasFe80Ni20-Legierungssystem besitzt vergleichbare Oberfl¨achenenergien wie das Fe-System, ins-
0.0 10.0 20.0 30.0 40.0 50.0 60.0Z (a.u.)
−40
−30
−20
−10
0
10
Pzz
(G
Pa)
−40
−30
−20
−10
0
10
(Pxx
+P
yy)/
2 (G
Pa)
nicht relaxiert relaxiert
Abbildung 4.2: Druckkomponen-ten zweier Fe(001)krz-Oberflachenbei 300 K. Die ersten 6 Atom-lagen einer der Oberfl¨achen wur-den entsprechend den idealen Po-sitionen des Festk¨orpers fixiert, dieOberflache konnte nicht relaxieren.Die andere Oberfl¨ache konnte vollrelaxieren. Der Gesamtdruck derersten Lagen senkrecht zur Ober-flache verschwindet, der Druck inder Filmebene der ersten Lage be-tragt ca. -20 GPa. Die Relaxationender ersten drei Ebenenabst¨ande be-tragen -7 %, -1.1 % und 0.5 %.

56 4 EAM-Potentiale fur Eisen und Nickel
besondere sind die Verh¨altnisse der Oberfl¨achenenergien von krz und kfz Struktur denen desFe Systems sehr ¨ahnlich [69].
In Abbildung 4.2 sind die lokalen Druckkomponenten eines d¨unnen krz Fe-Films im Rah-men einer Simulation von 4000 Fe-Atomen dargestellt. Periodische Randbedingungen wurdenur in Richtungen in der Filmebene(x,y) angenommen. Die Druckkomponenten wurden beieiner Temperatur von 300 K ¨uber ein Zeitintervall von 100 000 a.u. gemittelt. Eine der Ober-flachen wurde fixiert, so daß dort keine Relaxationen stattfinden konnten. Die Mittellung derlokalen Druckkomponenten der Atome an der relaxierten Oberfl¨achen ergibt f¨ur die Kompo-nente senkrecht zur Oberfl¨ache (Pzz) den erwarteten Druck von null GPa. Der Druck in derFilmebene ((Pxx+Pyy)/2) der ersten Lage betr¨agt ca. -20 GPa. Dies spiegelt die Tatsache wi-der, daß die Oberfl¨ache in senkrechter Richtung relaxieren kann, welches zu einer f¨ur Metalletypischen Absenkung der Abst¨ande zwischen der ersten und zweiten Lage f¨uhrt. Eine Relaxati-on des Drucks in der Oberfl¨achenebene, ist nicht vollst¨andig moglich. Es existiert ein negativerlokaler Druck in der Oberfl¨achenebene, der zur Oberfl¨achenspannung f¨uhrt [86]. Wird kei-ne Relaxation der Oberfl¨ache zugelassen, so erh¨ohen sich die lokalen Dr¨ucke. Dies fuhrt beiMetalloberflachen meist zu einem negativen Druck senkrecht zur Oberfl¨ache (Pzz), d. h. eineVerringerung des Lagenabstands der ersten beiden Lagen an der Oberfl¨ache wird favorisiert.

57
5 Simulationen metallischer Festkorper
5.1 Das Fe-Ni-System
5.1.1 Martensitische und austenitische Transformationen
Dieser Abschnitt stellt Simulationen zur strukturellen Untersuchung von Umwandlungen imFe-Ni-System vor (siehe auch [54,69]), die z. T. aus meiner Diplomarbeit entnommen sind undhier weitergehend diskutiert werden. Es wurden unterschiedliche Legierungszusammensetzun-gen mit einer zuf¨alligen Verteilung der Ni- und Fe-Atome auf einem krz Gitter untersucht. Beikonstanter Temperatur unter Verwendung des Nos´e-Hoover-Thermostaten wurde f¨ur P= 0 GPa(Parinello-Rahman-Verfahren) 6000 a.u. lang verschiedene Mittelwerte wie Teilchenpositio-nen, Atomvolumen und Paarkorrelationsfunktionen berechnet. Dies wurde f¨ur jede der Tempe-raturen ausgehend von 25 K in Schritten von 25 K bis 900 K und anschließender Abk¨uhlung inSchritten von 25 K durchgef¨uhrt.
Eine Auftragung der Temperaturabh¨angigkeit des atomaren Volumens zeigt, daß eine ande-re Steigung beim Aufheizen vorliegt als beim Abk¨uhlen. Dies weist auf strukturelleUbergangehin. Abbildung 5.1 zeigt dies exemplarisch f¨ur Fe80Ni20 (686 Atome in der Simulationszelle).Ist der Ausgangspunkt der Simulationen eine perfekte (v¨ollig defektfreie) krz Struktur, so findetman beim Aufheizen zwar den austenitischenUbergang, aber der Martensit¨ubergang bei tiefenTemperaturen stellt sich beim Abk¨uhlen nicht ein. Dies ist bei Simulationen anderer Legie-rungskonzentrationen und verschieden großer Simulationzellen ebenfalls beobachtet worden.Offensichtlich erfordert die Martensitumwandlung preexistierende Martensitkeime oder hinrei-chend hohe Konzentrationen von Defekten, die als Nukleationskeime dienen. Werden Fehlerz. B. in Form von zuf¨allig verteilten Leerstellen in das System eingebaut, kommt es bei tiefenTemperaturen zu einer Martensitumwandlung, wobei dieUbergangstemperatur von der Leer-stellenkonzentration abh¨angt (Abb. 5.1). Der austenitischeUbergang bei hohen Temperaturenwird durch den Einbau von Leerstellen kaum beeinflußt. Dies ist ein Hinweis, daß Defekte den
0 200 400 600 800 1000Temperatur (K)
2.65
2.70
2.75
2.80
Wig
ner−
Sei
tz R
adiu
s (a
.u.)
0% Leerstellen2% Leerstellen3% Leerstellen
Fe80Ni20 A
MAbbildung 5.1: Hystereseschleifen f¨urFe80Ni20 bei der Simulation f¨ur ver-schiedene Leerstellenkonzetrationen [69]).Wahrend die austenitische Umwandlungs-temperatur A kaum von der Leerstel-lenkonzentration abh¨angt, zeigt sich, daßdie martensitischeUbergangstemperaturM als stark von der Leerstellenkonzentra-tion abhangt. Im fehlerfreien Kristall trittder martensitischeUbergang auch bei tief-sten Temperaturen nicht auf.
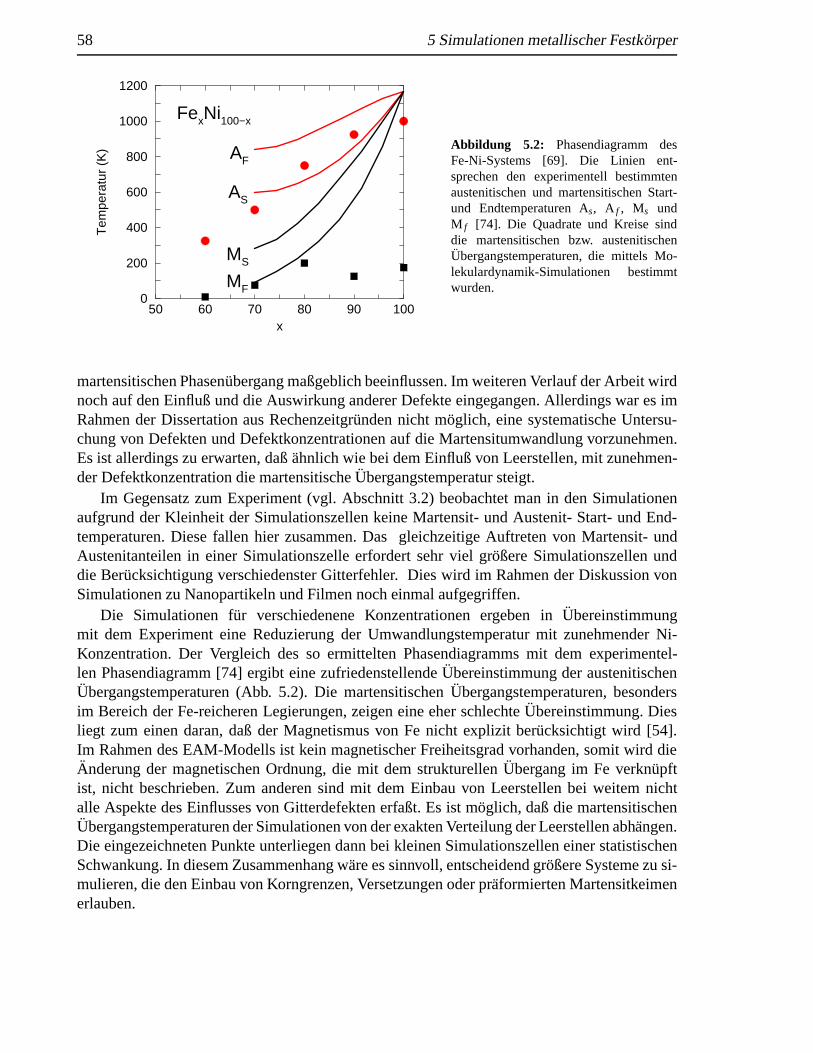
58 5 Simulationen metallischer Festk¨orper
50 60 70 80 90 100x
0
200
400
600
800
1000
1200T
empe
ratu
r (K
)FexNi100−x
MF
MS
AS
AFAbbildung 5.2: Phasendiagramm desFe-Ni-Systems [69]. Die Linien ent-sprechen den experimentell bestimmtenaustenitischen und martensitischen Start-und Endtemperaturen As, A f , Ms undM f [74]. Die Quadrate und Kreise sinddie martensitischen bzw. austenitischenUbergangstemperaturen, die mittels Mo-lekulardynamik-Simulationen bestimmtwurden.
martensitischen Phasen¨ubergang maßgeblich beeinflussen. Im weiteren Verlauf der Arbeit wirdnoch auf den Einfluß und die Auswirkung anderer Defekte eingegangen. Allerdings war es imRahmen der Dissertation aus Rechenzeitgr¨unden nicht m¨oglich, eine systematische Untersu-chung von Defekten und Defektkonzentrationen auf die Martensitumwandlung vorzunehmen.Es ist allerdings zu erwarten, daß ¨ahnlich wie bei dem Einfluß von Leerstellen, mit zunehmen-der Defektkonzentration die martensitischeUbergangstemperatur steigt.
Im Gegensatz zum Experiment (vgl. Abschnitt 3.2) beobachtet man in den Simulationenaufgrund der Kleinheit der Simulationszellen keine Martensit- und Austenit- Start- und End-temperaturen. Diese fallen hier zusammen. Das gleichzeitige Auftreten von Martensit- undAustenitanteilen in einer Simulationszelle erfordert sehr viel gr¨oßere Simulationszellen unddie Berucksichtigung verschiedenster Gitterfehler. Dies wird im Rahmen der Diskussion vonSimulationen zu Nanopartikeln und Filmen noch einmal aufgegriffen.
Die Simulationen f¨ur verschiedenene Konzentrationen ergeben inUbereinstimmungmit dem Experiment eine Reduzierung der Umwandlungstemperatur mit zunehmender Ni-Konzentration. Der Vergleich des so ermittelten Phasendiagramms mit dem experimentel-len Phasendiagramm [74] ergibt eine zufriedenstellendeUbereinstimmung der austenitischenUbergangstemperaturen (Abb. 5.2). Die martensitischenUbergangstemperaturen, besondersim Bereich der Fe-reicheren Legierungen, zeigen eine eher schlechteUbereinstimmung. Diesliegt zum einen daran, daß der Magnetismus von Fe nicht explizit ber¨ucksichtigt wird [54].Im Rahmen des EAM-Modells ist kein magnetischer Freiheitsgrad vorhanden, somit wird dieAnderung der magnetischen Ordnung, die mit dem strukturellenUbergang im Fe verkn¨upftist, nicht beschrieben. Zum anderen sind mit dem Einbau von Leerstellen bei weitem nichtalle Aspekte des Einflusses von Gitterdefekten erfaßt. Es ist m¨oglich, daß die martensitischenUbergangstemperaturen der Simulationen von der exakten Verteilung der Leerstellen abh¨angen.Die eingezeichneten Punkte unterliegen dann bei kleinen Simulationszellen einer statistischenSchwankung. In diesem Zusammenhang w¨are es sinnvoll, entscheidend gr¨oßere Systeme zu si-mulieren, die den Einbau von Korngrenzen, Versetzungen oder pr¨aformierten Martensitkeimenerlauben.

5.1 Das Fe-Ni-System 59
krz (T= 100 K) [001] 45.0° (011)
(111) Zwillings- ebene
A
ABC
C B A C A C A B C A B C A
BACAC
BC
A
kfz (T= 600 K) [001] 35.3°
Abbildung 5.3: Ausgehend von einer idea-len krz Struktur (oben) bildet sich un-ter erhitzen einer Fe70Ni30-Legierung ei-ne verzwillte und verstapelte dicht ge-packte Austenitstruktur (unten) [69]. Dieschwarzen und grauen Kreise markierenunterschiedliche Ebenen. Dicht gepackte(011)-Ebenen des krz Gitters transformie-ren in die dicht gepackten (111)-Ebenendes kfz Gitters, wobei eine leichte Rota-tion der Ebenen um die[100]krz zu ver-zeichnen ist. Die Richtung senkrecht zurPapierebene ist die[100]krz- bzw. [110]kfz-Richtung. Abgesehen von der leichten Ro-tation um Richtung senkrecht zur Pa-piereben entspricht dies der Nishiyama-Wassermann-Orientierungsbeziehung. DieZwillingsebene ist des hier auftretendenZwillings ist die (111)-Ebene, die Scher-richtung die [112]-Richtung. Eingezeich-net sind jeweils zwei kubische Elementar-zellen sowie Bereiche, die nach der Trans-formation zwillig zueinander liegen. DieStapelfolgen der dicht gepackten Ebenenzeigen Bereiche mit ideale kfz Struktur(ABCABC) und Stapelfehler bzw. Berei-che die hdp (ABAB) geordnet sind.
Die Analyse der radialen Verteilungsfunktionen erm¨oglicht leicht die Identifikation der sichin den Simulationen gebildeten Phasen und erlaubt dar¨uber hinaus auch eine Unterscheidungder auftretenden Stapelfehler in einer dicht gepackten Struktur [36]. Dar¨uber hinaus erm¨oglichteine Auftragung der atomaren Positionen vor und nach der Umwandlung eine optische Ana-lyse der durch denUbergang induzierten Gitterfehler wie Zwillingselemente oder Stapel-fehler. Außerdem kann man durch Vergleich der Strukturen vor und nach der Transformati-on, Ruckschlusse auf den Tranformationmechanismus gewinnen. Am Beispiel einer Fe70Ni30-Legierung mit 4394 Atomen in der Simulationsbox ist dies in Abbildung 5.3 dargestellt. Diedicht gepackten (011)-Ebenen der krz Struktur transformieren unter leichter Rotation um die[100]krz-Richtung in die dicht gepackten (111)-Ebenen der kfz Struktur, wobei die[100]krz-Richtung des krz Gitters in die[110]kfz-Richtungubergeht. Die Struktur weist im wesentlichenzwei verschieden orientierte zwillig zueinander liegende Bereiche der Austenitstruktur auf.

60 5 Simulationen metallischer Festk¨orper
Die hier auftretende (111)-Zwillingsebene sowie die[112]-Scherrichtung ist typisch f¨ur kfzStrukturen (Abschnitt 3.1). Im System sind einige Stapelfehler enthalten. Das Auftreten vonStapelfehlern w¨ahrend der Simulationen ist eng verkn¨upft mit der in Abschnitt 4.1 diskutier-ten niedrigen Stapelfehlerenergie. Mathematisch kann der in den Simulationen beschriebeneUbergang aus einer Kombination von Bain-Verzerrung, Rotation um die[100]krz-Richtung undBildung von Zwillingen und Stapelfehlern beschrieben werden. Die Elemente der in Abschnitt3.2.1 eingef¨uhrten phanomenologischen Martensittheorie finden sich somit in den Simulatio-nen wieder.
Die hier auftretende Orientierungsbeziehung zwischen der urspr¨unglich initialisierten Mar-tensitstruktur und der neu gebildeten Austenitstruktur entspricht bis auf eine kleine Rotation derin Fe-Ni-Legierungen h¨aufig gefundenen Nishiyama-Wassermann-Orientierungsbeziehung.Die im Experiment beobachteten festen Orientierungsbeziehungen der beiden kristallographi-schen Phasen h¨angen mit dem gemeinsamen Auftreten von Austenit und Martensit in der Probezusammen. Die hier gefundenen kristallographischen Beziehungen sind somit nicht direkt mitden experimentell beobachteten zu vergleichen. Im Rahmen der Diskussion struktureller Um-wandlungen in d¨unnen Filmen auf Substraten werden Beispiele gezeigt, bei denen der direkteVergleich der kristallographischen Orientierungsbeziehungen mit dem Experiment m¨oglich ist(Abschnitt 7).
Die Analyse von austenitisch pr¨aparierten Simulationszellen zeigt, daß dabei ebenfallsZwillinge in der entstandenen Martensitstruktur auftreten, die mit experimentell beobachte-ten Zwillingssystemen des krz Gitters ¨ubereinstimmen (Tab. 3.2). Auch in diesem Fall l¨aßt sichder gesamte Umwandlungsmechanismus im Rahmen der ph¨anomenologischen Martensittheo-rie beschreiben.
5.1.2 Thermodynamik des Systems
Im Rahmen der Simulationen k¨onnen auch Aussagen ¨uber die thermodynamische Stabilit¨at desFe-Ni-Systems mit Hilfe der freien EnergienF = E−TSgewonnen werden. Am Beispiel einerFe80Ni20-Legierung sind entsprechende Ergebnisse in den Abbildungen 5.4 und 5.5 gezeigt.Die Simulation wurde ausgef¨uhrt fur ein System von 432 Atomen mit zuf¨allig verteilten Fe-und Ni-Atomen auf einem krz bzw. kfz Gitter f¨ur verschiedene Temperaturen und f¨ur P= 0 GPauber ein Zeitintervall von 200 000 a.u. Der Entropieanteil wurde mit Hilfe der in Abschnitt 2.2.4vorgestellten Methode der Korrelationsdeterminanten bestimmt.
Abbildung 5.4 zeigt die freie Energie in Abh¨angigkeit von der Temperatur f¨ur diekrz und die kfz Struktur. Die ermittelte thermodynamische Gleichgewichtstemperatur liegtbei ca. 300 K. Damit ist die Gleichgewichtstemperatur um etwa 200 K zu niedrig vergli-chen mit dem Mittelwert aus experimentell bestimmter austenitscher und martensitischerUbergangstemperatur (Abb. 5.2). F¨ur das in dieser Arbeit benutzte EAM-Potential f¨ur Fe hatR. Meyer einen ¨ahnlichen Verlauf der freien Energien f¨ur krz Fe und kfz Fe mit einer thermo-dynamischen Gleichgewichtstemperatur von 500 K [54] berechnet.
Die Abbildung 5.5 erlaubt einen direkten Vergleich der AnteileE und −TS der freienEnergien. W¨ahrend bei tiefen Temperaturen die chemische Triebkraft aus dem EnergietermE = Upot+Ukin uberwiegt, verh¨alt es sich bei h¨oheren Temperaturen in der N¨ahe des austeniti-
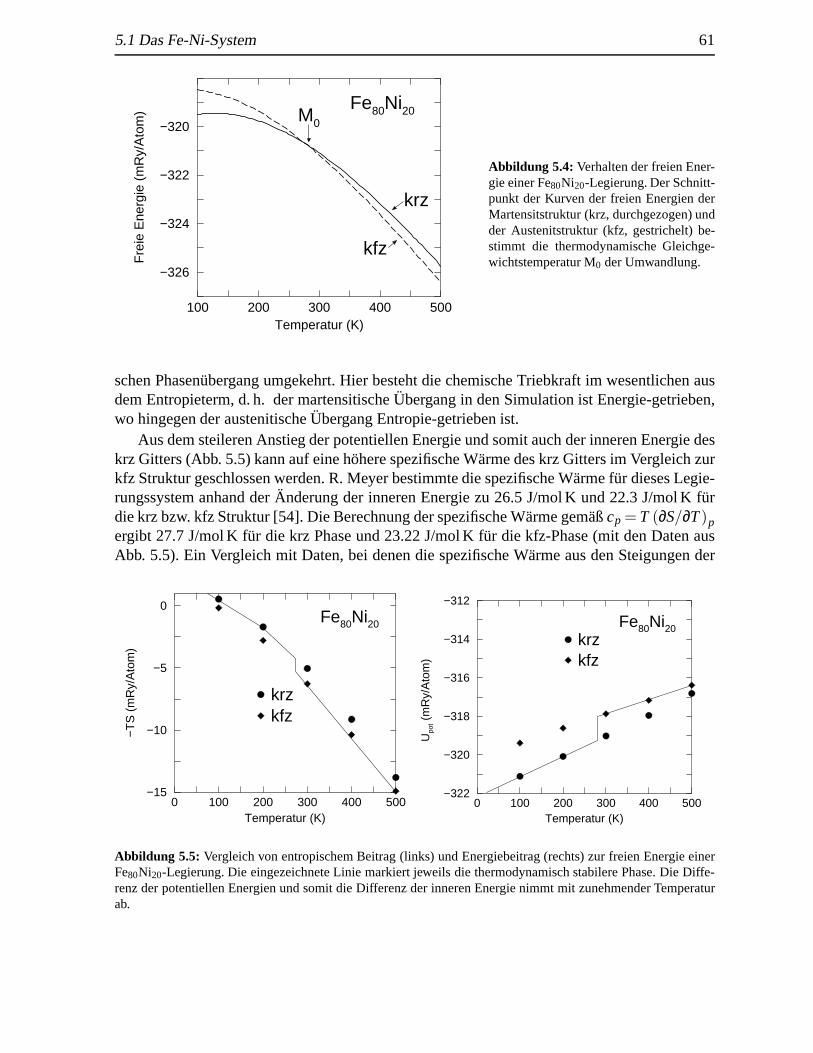
5.1 Das Fe-Ni-System 61
100 200 300 400 500Temperatur (K)
−326
−324
−322
−320F
reie
Ene
rgie
(m
Ry/
Ato
m) Fe80Ni20
kfz
krz
M0
Abbildung 5.4: Verhalten der freien Ener-gie einer Fe80Ni20-Legierung. Der Schnitt-punkt der Kurven der freien Energien derMartensitstruktur (krz, durchgezogen) undder Austenitstruktur (kfz, gestrichelt) be-stimmt die thermodynamische Gleichge-wichtstemperatur M0 der Umwandlung.
schen Phasen¨ubergang umgekehrt. Hier besteht die chemische Triebkraft im wesentlichen ausdem Entropieterm, d. h. der martensitischeUbergang in den Simulation ist Energie-getrieben,wo hingegen der austenitischeUbergang Entropie-getrieben ist.
Aus dem steileren Anstieg der potentiellen Energie und somit auch der inneren Energie deskrz Gitters (Abb. 5.5) kann auf eine h¨ohere spezifische W¨arme des krz Gitters im Vergleich zurkfz Struktur geschlossen werden. R. Meyer bestimmte die spezifische W¨arme fur dieses Legie-rungssystem anhand derAnderung der inneren Energie zu 26.5 J/mol K und 22.3 J/mol K f¨urdie krz bzw. kfz Struktur [54]. Die Berechnung der spezifische W¨arme gem¨aßcp = T (∂S/∂T)pergibt 27.7 J/mol K f¨ur die krz Phase und 23.22 J/mol K f¨ur die kfz-Phase (mit den Daten ausAbb. 5.5). Ein Vergleich mit Daten, bei denen die spezifische W¨arme aus den Steigungen der
0 100 200 300 400 500Temperatur (K)
−15
−10
−5
0
−T
S (
mR
y/A
tom
)
krzkfz
Fe80Ni20
0 100 200 300 400 500Temperatur (K)
−322
−320
−318
−316
−314
−312
Upo
t (m
Ry/
Ato
m)
krzkfz
Fe80Ni20
Abbildung 5.5: Vergleich von entropischem Beitrag (links) und Energiebeitrag (rechts) zur freien Energie einerFe80Ni20-Legierung. Die eingezeichnete Linie markiert jeweils die thermodynamisch stabilere Phase. Die Diffe-renz der potentiellen Energien und somit die Differenz der inneren Energie nimmt mit zunehmender Temperaturab.

62 5 Simulationen metallischer Festk¨orper
Energie gewonnen wurde, zeigt guteUbereinstimmung.Die gesamte Entropie, die in den Simulationen auftritt und f¨ur den Phasen¨ubergang ver-
antwortlich ist, besteht aus dem Gitteranteil. Entropiebeitr¨age, die z. B. durch magnetischeOrdnung entstehen, werden von diesem Modell nicht erfaßt. Der strukturelleUbergang von derkrz Phase in die kfz Phase kommt somit allein dadurch zustande, daß das kfz Gitter verglichenmit dem krz Gitter niedrigere Schwingungsfrequenzen hat. Die Konkurrenz zwischen Energieund Entropie kann bereits anhand eines einfachen harmonischen Oszillator-Modells verdeut-licht werden (Abschnitt 9.4). Der hier auftretende Unterschied der spezifischen W¨armencp
ergibt sich aus den unterschiedlichen Anharmonizit¨aten der verschiedenen Gitterstrukturen.Die in diesem System berechneteAnderung der Entropie bei der Gleichgewichtstemperatur
M0 liegt bei T∆S≈ 1 mRy/Atom. Aus den experimentell bestimmten Phononenspektren f¨urα-Fe undγ-Fe bestimmte J. Neuhaus die Differenz der Schwingungsentropien in Fe bei derUbergangstemperatur zuT∆S≈ 1 mRy/Atom [103] in ausgezeichneterUbereinstimmung mitdem theoretischen Wert. Dieser gutenUbereinstimmung steht der Tatsache gegen¨uber, daß dieim Rahmen dieses Modells errechneten Gleichgewichtstemperaturen zu klein sind.
5.1.3 (Freie) Energie entlang des Transformationswegs (Bain-Weg)
Nach der thermodynamischen Stabilit¨atsanalyse f¨ur das Fe80Ni20-System wird im folgendeneine thermodynamische Analyse der Dynamik der strukturellenUbergange und damit ver-bundener Nukleationsprozesse vorgestellt. Um geeignete Aussagen machen zu k¨onnen, wur-de als Transformationspfad die Bain-Transformation (Abschnitt 3.2.1) genommen. Dasc/a-Verhaltnis parametrisiert dabei den Transformationsweg von der krz Struktur (c/a = 1/
√2) in
die kfz Struktur (c/a = 1). Der in den Simulationen auftretendeUbergang laßt sich als eineKombination von Bain-Verzerrung, Rotation sowie der Bildung von Stapelfehlern und Zwil-lingen darstellen (Abschnitt 5.1.1). Dies rechtfertigt es bei der Diskussion der Dynamik denBain-Weg als Reaktionsweg anzunehmen.
Eine Simulationszelle mit 432 Atomen wurde bei verschiedenen Temperaturen und fest-gehaltenem Volumen an 15 Punkten entlang des Transformationswegs simuliert. Es wurdenjeweils uber eine Zeit von 6000 a.u. die Druckkomponenten und die potentielle Energie desSystems gemittelt. Dabei wurde das Systemvolumen linear von der krz Struktur in die kfzStruktur entlang des Bain-Wegs so ge¨andert, daß kein Druck in der krz sowie in der kfz Struk-tur herrscht. Anschließend wurde aus Gleichung (2.21) mit Hilfe der ermittelten Druckkom-ponenten der Unterschied der freien Energie entlang des Transformationspfads bestimmt. DieBetrage einzelner Druckkomponenten sind entlang des Transformationswegs nicht ¨uber 2 GPaangestiegen. DiePxx- undPyy-Anteile uberschreiten im allgemeinen 1 GPa nicht.
Die potentielle Energie des System entlang des Bain-Wegs (Abb. 5.6) hat bei tiefen Tem-peraturen ein Minimum, das der krz Struktur entspricht und das mit zunehmender Tempera-tur langsam verschwindet. Analog verh¨alt es sich mit dem Minimun f¨ur die kfz-Struktur, dasbei hohen Temperaturen ausgepr¨agt ist und bei tiefen Temperaturen ebenfalls verschwindet.Bei dieser Betrachtungsweise wirkt der Prozeß der Umwandlung von krz in kfz symmetrischzu der Umwandlung der kfz in die krz Phase. Das heißt, daß der martensitischeUbergang in
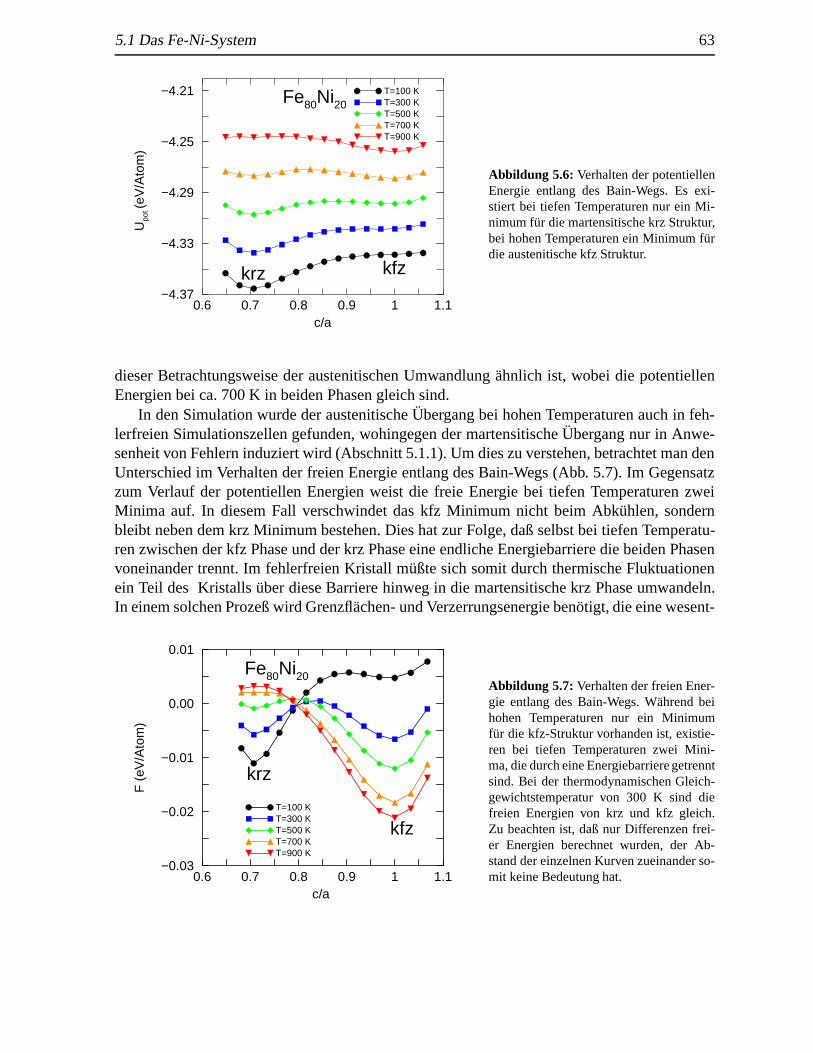
5.1 Das Fe-Ni-System 63
0.6 0.7 0.8 0.9 1 1.1c/a
−4.37
−4.33
−4.29
−4.25
−4.21
Upo
t (eV
/Ato
m)
T=100 KT=300 KT=500 KT=700 KT=900 K
Fe80Ni20
krz kfz
Abbildung 5.6: Verhalten der potentiellenEnergie entlang des Bain-Wegs. Es exi-stiert bei tiefen Temperaturen nur ein Mi-nimum fur die martensitische krz Struktur,bei hohen Temperaturen ein Minimum f¨urdie austenitische kfz Struktur.
dieser Betrachtungsweise der austenitischen Umwandlung ¨ahnlich ist, wobei die potentiellenEnergien bei ca. 700 K in beiden Phasen gleich sind.
In den Simulation wurde der austenitischeUbergang bei hohen Temperaturen auch in feh-lerfreien Simulationszellen gefunden, wohingegen der martensitischeUbergang nur in Anwe-senheit von Fehlern induziert wird (Abschnitt 5.1.1). Um dies zu verstehen, betrachtet man denUnterschied im Verhalten der freien Energie entlang des Bain-Wegs (Abb. 5.7). Im Gegensatzzum Verlauf der potentiellen Energien weist die freie Energie bei tiefen Temperaturen zweiMinima auf. In diesem Fall verschwindet das kfz Minimum nicht beim Abk¨uhlen, sondernbleibt neben dem krz Minimum bestehen. Dies hat zur Folge, daß selbst bei tiefen Temperatu-ren zwischen der kfz Phase und der krz Phase eine endliche Energiebarriere die beiden Phasenvoneinander trennt. Im fehlerfreien Kristall m¨ußte sich somit durch thermische Fluktuationenein Teil des Kristalls ¨uber diese Barriere hinweg in die martensitische krz Phase umwandeln.In einem solchen Prozeß wird Grenzfl¨achen- und Verzerrungsenergie ben¨otigt, die eine wesent-
0.6 0.7 0.8 0.9 1 1.1c/a
−0.03
−0.02
−0.01
0.00
0.01
F (
eV/A
tom
)
T=100 KT=300 KT=500 KT=700 KT=900 K
krz
kfz
Fe80Ni20Abbildung 5.7: Verhalten der freien Ener-gie entlang des Bain-Wegs. W¨ahrend beihohen Temperaturen nur ein Minimumfur die kfz-Struktur vorhanden ist, existie-ren bei tiefen Temperaturen zwei Mini-ma, die durch eine Energiebarriere getrenntsind. Bei der thermodynamischen Gleich-gewichtstemperatur von 300 K sind diefreien Energien von krz und kfz gleich.Zu beachten ist, daß nur Differenzen frei-er Energien berechnet wurden, der Ab-stand der einzelnen Kurven zueinander so-mit keine Bedeutung hat.
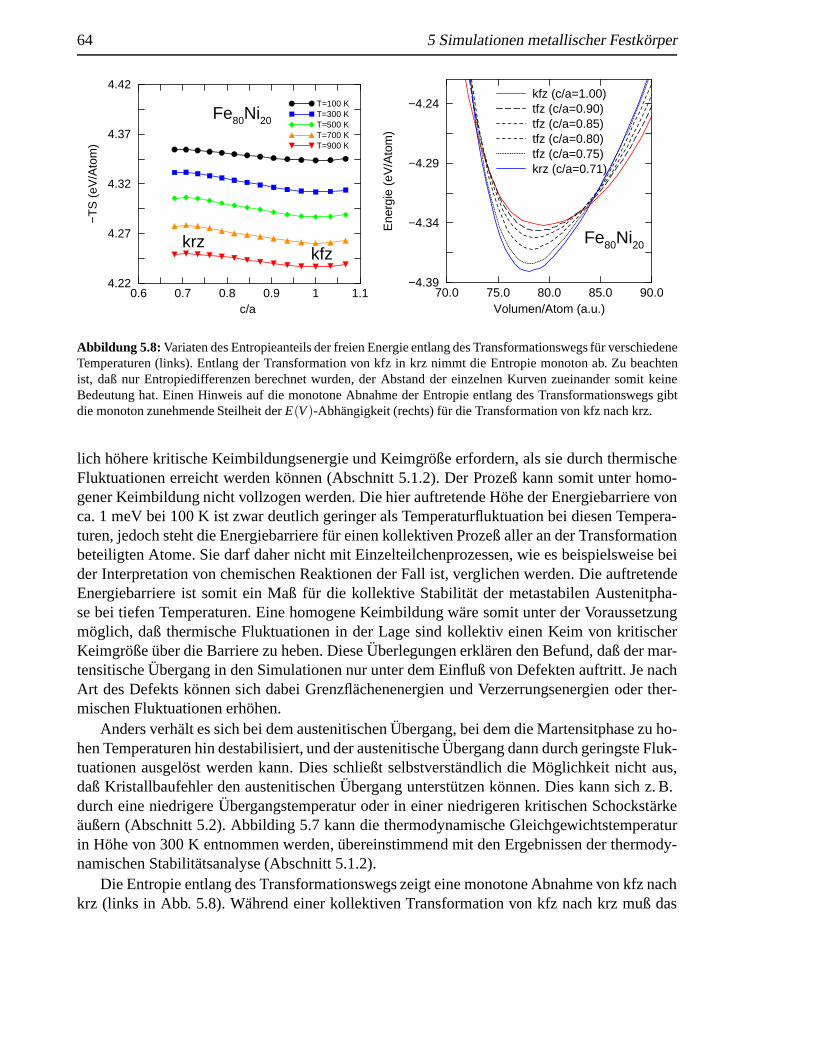
64 5 Simulationen metallischer Festk¨orper
0.6 0.7 0.8 0.9 1 1.1c/a
4.22
4.27
4.32
4.37
4.42−
TS
(eV
/Ato
m)
T=100 KT=300 KT=500 KT=700 KT=900 K
krzkfz
Fe80Ni20
70.0 75.0 80.0 85.0 90.0Volumen/Atom (a.u.)
−4.39
−4.34
−4.29
−4.24
Ene
rgie
(eV
/Ato
m)
kfz (c/a=1.00)tfz (c/a=0.90)tfz (c/a=0.85)tfz (c/a=0.80)tfz (c/a=0.75)krz (c/a=0.71)
Fe80Ni20
Abbildung 5.8: Variaten des Entropieanteils der freien Energie entlang des Transformationswegs f¨ur verschiedeneTemperaturen (links). Entlang der Transformation von kfz in krz nimmt die Entropie monoton ab. Zu beachtenist, daß nur Entropiedifferenzen berechnet wurden, der Abstand der einzelnen Kurven zueinander somit keineBedeutung hat. Einen Hinweis auf die monotone Abnahme der Entropie entlang des Transformationswegs gibtdie monoton zunehmende Steilheit derE(V)-Abhangigkeit (rechts) f¨ur die Transformation von kfz nach krz.
lich hohere kritische Keimbildungsenergie und Keimgr¨oße erfordern, als sie durch thermischeFluktuationen erreicht werden k¨onnen (Abschnitt 5.1.2). Der Prozeß kann somit unter homo-gener Keimbildung nicht vollzogen werden. Die hier auftretende H¨ohe der Energiebarriere vonca. 1 meV bei 100 K ist zwar deutlich geringer als Temperaturfluktuation bei diesen Tempera-turen, jedoch steht die Energiebarriere f¨ur einen kollektiven Prozeß aller an der Transformationbeteiligten Atome. Sie darf daher nicht mit Einzelteilchenprozessen, wie es beispielsweise beider Interpretation von chemischen Reaktionen der Fall ist, verglichen werden. Die auftretendeEnergiebarriere ist somit ein Maß f¨ur die kollektive Stabilitat der metastabilen Austenitpha-se bei tiefen Temperaturen. Eine homogene Keimbildung w¨are somit unter der Voraussetzungmoglich, daß thermische Fluktuationen in der Lage sind kollektiv einen Keim von kritischerKeimgroßeuber die Barriere zu heben. DieseUberlegungen erkl¨aren den Befund, daß der mar-tensitischeUbergang in den Simulationen nur unter dem Einfluß von Defekten auftritt. Je nachArt des Defekts k¨onnen sich dabei Grenzfl¨achenenergien und Verzerrungsenergien oder ther-mischen Fluktuationen erh¨ohen.
Anders verh¨alt es sich bei dem austenitischenUbergang, bei dem die Martensitphase zu ho-hen Temperaturen hin destabilisiert, und der austenitischeUbergang dann durch geringste Fluk-tuationen ausgel¨ost werden kann. Dies schließt selbstverst¨andlich die Moglichkeit nicht aus,daß Kristallbaufehler den austenitischenUbergang unterst¨utzen konnen. Dies kann sich z. B.durch eine niedrigereUbergangstemperatur oder in einer niedrigeren kritischen Schockst¨arkeaußern (Abschnitt 5.2). Abbilding 5.7 kann die thermodynamische Gleichgewichtstemperaturin Hohe von 300 K entnommen werden, ¨ubereinstimmend mit den Ergebnissen der thermody-namischen Stabilit¨atsanalyse (Abschnitt 5.1.2).
Die Entropie entlang des Transformationswegs zeigt eine monotone Abnahme von kfz nachkrz (links in Abb. 5.8). Wahrend einer kollektiven Transformation von kfz nach krz muß das

5.2 Schockinduzierte strukturelleAnderungen in krz Fe 65
System sich in Richtung abnehmender Entropie bewegen. Einen Hinweis auf dieses Verhal-ten kann bereits aus der Volumenabh¨angigkeit der Energie entlang des Transformationswegs(rechts in Abb. 5.8)) entnommen werden. Die kontinuierlich entlang des Transformationswegsvon kfz nach krz steiler (h¨arter) werdendenE(V)-Abhangigkeiten deuten auf eine abnehmendeEntropie hin (Abschnitt 4.1 und 9.4). Das System muß eine Entropieschneise durchlaufen.
5.2 Schockinduzierte strukturelleAnderungen in krz Fe
Strukturelle Umwandlungen zwischen zwei festen Phasen werden nicht nur in Abh¨angigkeitder Temperatur beobachtet. Auch durch Schockeinwirkung kann krz Fe in eine dicht ge-packte Struktur, wie hdp Fe, umgewandelt werden (Abschnitt 3.3). Um die atomaren Prozes-se, die dabei auftreten zu analysieren, wurden Molekulardynamik-Simulationen von krz Si-mulationszellen durchgef¨uhrt. Unter Benutzung derMomentum-Mirror-Methode (Abschnitt2.4.4) wurden Schockwellen unterschiedlicher St¨arke, d. h. unterschiedlicher Kolbenge-schwindigkeitenUp, in [001]krz-Richtung induziert. Die Simulationszellen hatten die Dimensi-on 40.2 nm x 40.2 nm x 57.4 nm und beinhalten ca. 8 Millionen Atome, die anfangs auf dieidealen Positionen des krz Gitters gesetzt wurden. Die Anfangstemperatur betrug 50 K. Die Si-mulationen wurden in Los Alamos auf einer 12-Prozessorshared-memorySun Enterprise 4000vorgenommen, wobei die CPU-Zeit pro Simulation (eine Schockst¨arke) ca. 50 h betrug .
Die Analyse der Simulationen wurde unter zwei verschiedenen Gesichtspunkten vorge-nommen. Im folgendem Abschnitt wird der Schockeinfluß untersucht. Dazu geh¨ort das Aus-breitungsverhalten der sich bildenenden Schockwellen und die Untersuchung von Nukleations-prozessen. In Abschnitt 5.2.2 wird dann eine Analyse der strukturellen Umwandlung, die sichdurch den Schockeinfluß ergibt, durchgef¨uhrt. Dazu z¨ahlt neben der Analyse der kristallogra-phischen Strukturen auch die Betrachtung der auftretenden Korngrenzendynamik w¨ahrend derNukleation der hdp Phase.
5.2.1 Analyse des Schockeinflusses
Unter dem Einfluß von geringen Kolbengeschwindigkeiten von maximalUP = 850 m/s brei-tet sich im Kristall eine Kompressionsschockwelle aus, die das krz Gitter in Schockrich-tung staucht, somit tetragonal verzerrt (links oben in Abb. 5.9). Kurz ¨uber der kritischenSchockst¨arkeUP = 850 m/s wird neben der nun alselastic precursorbezeichneten elastischenKompressionswelle eine langsamere der elastischen Kompressionswelle hinterher laufende pla-stische Verformungsschockwelle beobachtet (rechts oben in Abb. 5.9). Die plastische Verfor-mung stellt die auch im Experiment beobachtete strukturelle Transformation vonα-Fe inε-Fedar. Kristallographisch verschieden orientierte Bereiche werden durch Korngrenzen voneinan-der getrennt. Mit zunehmender St¨arke der Schockwelle nimmt die Geschwindigkeit der pla-stischen Verformungsschockwelle zu (links unten in Abb. 5.9). Bei hohen Schockst¨arken exi-stiert schließlich nur noch die plastische Welle, die denelastic precursoreingeholt hat (rechtsunten in Abb. 5.9). Molekulardynamik-Simulation f¨ur kfz Strukturen ergaben ¨ahnlicheela-stic precursorErscheinungen bei Schockwellen in [011]-Richtung und Schockwellen in [111]-Richtung [104], nicht aber bei Schockwellen in [001]-Richtung [26].
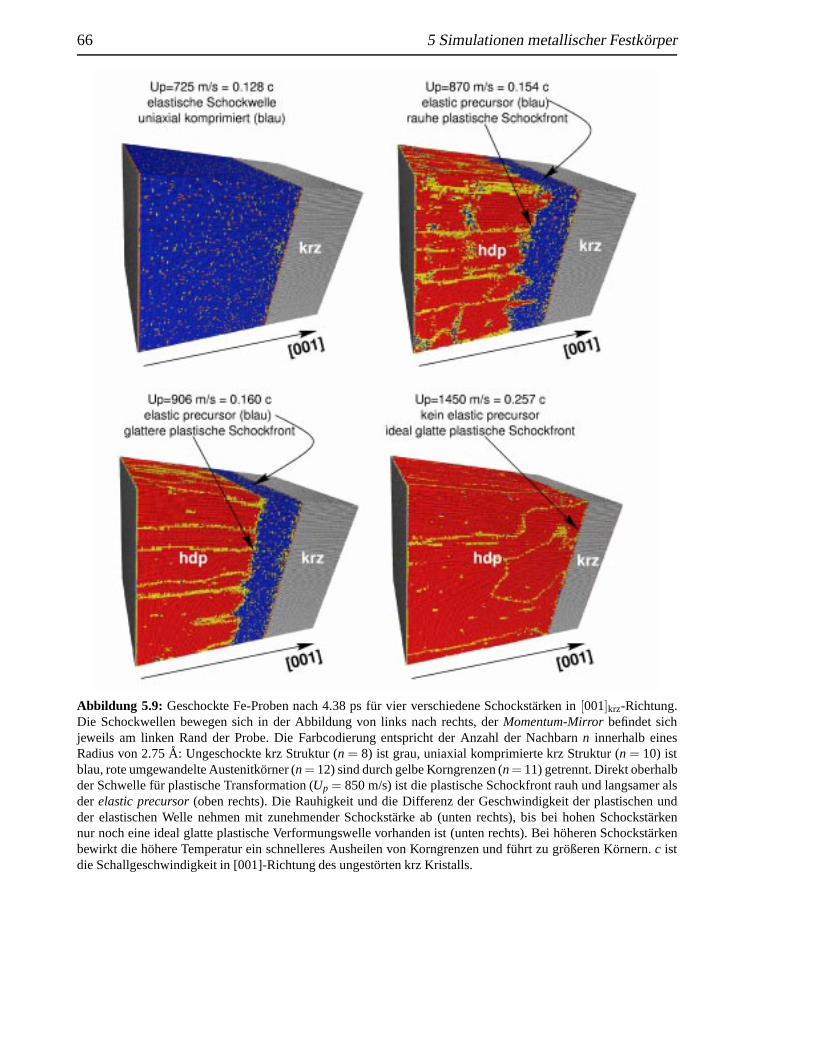
66 5 Simulationen metallischer Festk¨orper
Abbildung 5.9: Geschockte Fe-Proben nach 4.38 ps f¨ur vier verschiedene Schockst¨arken in[001]krz-Richtung.Die Schockwellen bewegen sich in der Abbildung von links nach rechts, derMomentum-Mirrorbefindet sichjeweils am linken Rand der Probe. Die Farbcodierung entspricht der Anzahl der Nachbarnn innerhalb einesRadius von 2.75A: Ungeschockte krz Struktur (n = 8) ist grau, uniaxial komprimierte krz Struktur (n = 10) istblau, rote umgewandelte Austenitk¨orner (n= 12) sind durch gelbe Korngrenzen (n= 11) getrennt. Direkt oberhalbder Schwelle f¨ur plastische Transformation (Up = 850 m/s) ist die plastische Schockfront rauh und langsamer alsder elastic precursor(oben rechts). Die Rauhigkeit und die Differenz der Geschwindigkeit der plastischen undder elastischen Welle nehmen mit zunehmender Schockst¨arke ab (unten rechts), bis bei hohen Schockst¨arkennur noch eine ideal glatte plastische Verformungswelle vorhanden ist (unten rechts). Bei h¨oheren Schockst¨arkenbewirkt die hohere Temperatur ein schnelleres Ausheilen von Korngrenzen und f¨uhrt zu großeren K¨ornern.c istdie Schallgeschwindigkeit in [001]-Richtung des ungest¨orten krz Kristalls.
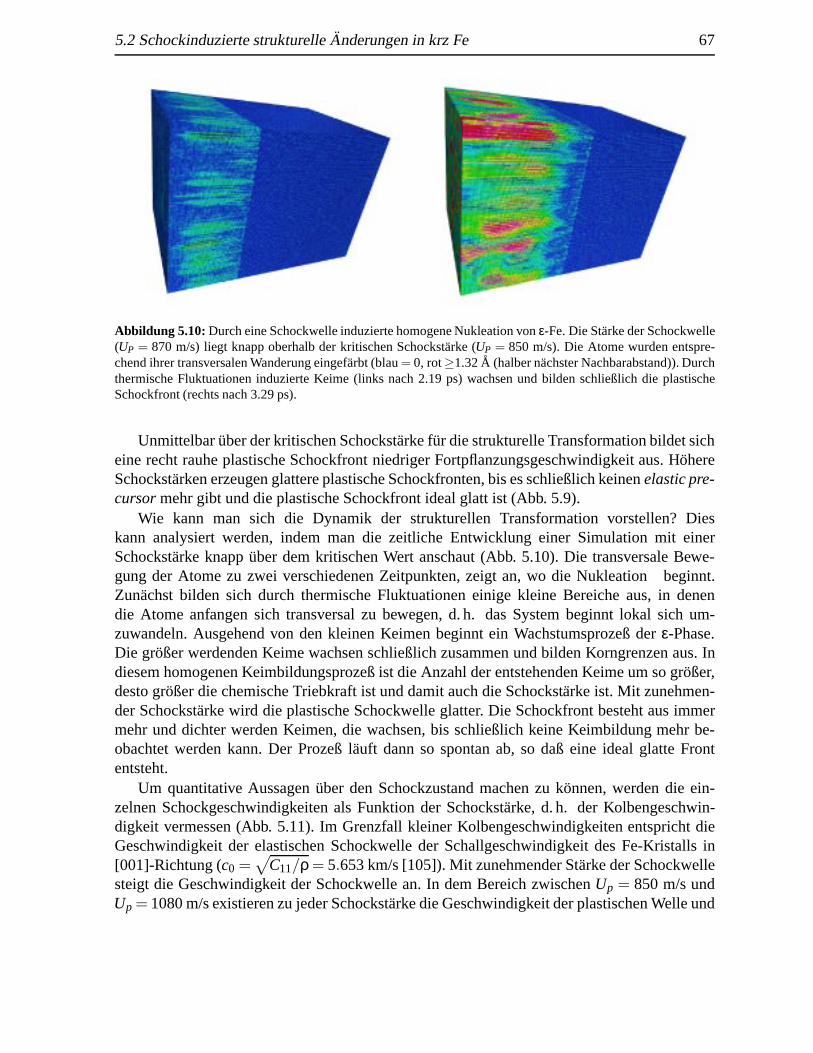
5.2 Schockinduzierte strukturelleAnderungen in krz Fe 67
Abbildung 5.10: Durch eine Schockwelle induzierte homogene Nukleation vonε-Fe. Die Starke der Schockwelle(UP = 870 m/s) liegt knapp oberhalb der kritischen Schockst¨arke (UP = 850 m/s). Die Atome wurden entspre-chend ihrer transversalen Wanderung eingef¨arbt (blau= 0, rot≥1.32A (halber nachster Nachbarabstand)). Durchthermische Fluktuationen induzierte Keime (links nach 2.19 ps) wachsen und bilden schließlich die plastischeSchockfront (rechts nach 3.29 ps).
Unmittelbaruber der kritischen Schockst¨arke fur die strukturelle Transformation bildet sicheine recht rauhe plastische Schockfront niedriger Fortpflanzungsgeschwindigkeit aus. H¨ohereSchockst¨arken erzeugen glattere plastische Schockfronten, bis es schließlich keinenelastic pre-cursormehr gibt und die plastische Schockfront ideal glatt ist (Abb. 5.9).
Wie kann man sich die Dynamik der strukturellen Transformation vorstellen? Dieskann analysiert werden, indem man die zeitliche Entwicklung einer Simulation mit einerSchockst¨arke knapp ¨uber dem kritischen Wert anschaut (Abb. 5.10). Die transversale Bewe-gung der Atome zu zwei verschiedenen Zeitpunkten, zeigt an, wo die Nukleation beginnt.Zunachst bilden sich durch thermische Fluktuationen einige kleine Bereiche aus, in denendie Atome anfangen sich transversal zu bewegen, d. h. das System beginnt lokal sich um-zuwandeln. Ausgehend von den kleinen Keimen beginnt ein Wachstumsprozeß derε-Phase.Die großer werdenden Keime wachsen schließlich zusammen und bilden Korngrenzen aus. Indiesem homogenen Keimbildungsprozeß ist die Anzahl der entstehenden Keime um so gr¨oßer,desto großer die chemische Triebkraft ist und damit auch die Schockst¨arke ist. Mit zunehmen-der Schockst¨arke wird die plastische Schockwelle glatter. Die Schockfront besteht aus immermehr und dichter werden Keimen, die wachsen, bis schließlich keine Keimbildung mehr be-obachtet werden kann. Der Prozeß l¨auft dann so spontan ab, so daß eine ideal glatte Frontentsteht.
Um quantitative Aussagen ¨uber den Schockzustand machen zu k¨onnen, werden die ein-zelnen Schockgeschwindigkeiten als Funktion der Schockst¨arke, d. h. der Kolbengeschwin-digkeit vermessen (Abb. 5.11). Im Grenzfall kleiner Kolbengeschwindigkeiten entspricht dieGeschwindigkeit der elastischen Schockwelle der Schallgeschwindigkeit des Fe-Kristalls in[001]-Richtung (c0 =
√C11/ρ = 5.653 km/s [105]). Mit zunehmender St¨arke der Schockwelle
steigt die Geschwindigkeit der Schockwelle an. In dem Bereich zwischenUp = 850 m/s undUp = 1080 m/s existieren zu jeder Schockst¨arke die Geschwindigkeit der plastischen Welle und
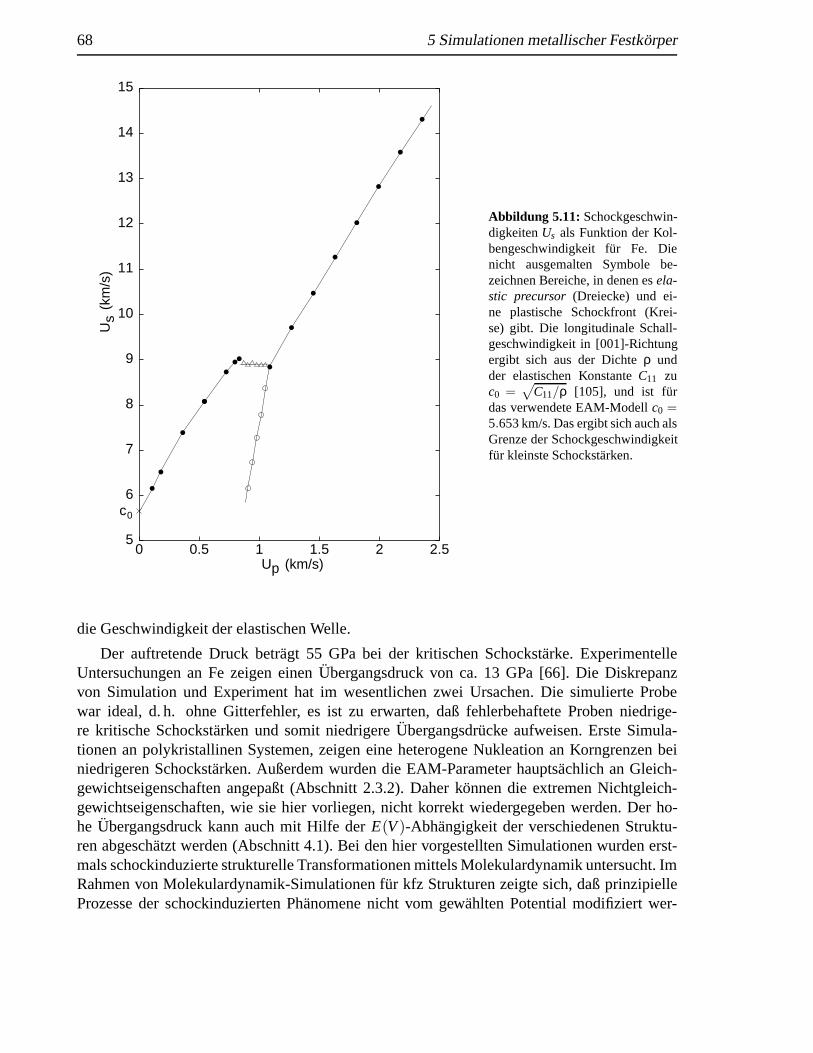
68 5 Simulationen metallischer Festk¨orper
5
6
7
8
9
10
11
12
13
14
15
0 0.5 1 1.5 2 2.5
Us
(km
/s)
Up (km/s)
c0
Abbildung 5.11: Schockgeschwin-digkeitenUs als Funktion der Kol-bengeschwindigkeit f¨ur Fe. Dienicht ausgemalten Symbole be-zeichnen Bereiche, in denen esela-stic precursor (Dreiecke) und ei-ne plastische Schockfront (Krei-se) gibt. Die longitudinale Schall-geschwindigkeit in [001]-Richtungergibt sich aus der Dichteρ undder elastischen KonstanteC11 zuc0 =
√C11/ρ [105], und ist fur
das verwendete EAM-Modellc0 =5.653 km/s. Das ergibt sich auch alsGrenze der Schockgeschwindigkeitfur kleinste Schockst¨arken.
die Geschwindigkeit der elastischen Welle.
Der auftretende Druck betr¨agt 55 GPa bei der kritischen Schockst¨arke. ExperimentelleUntersuchungen an Fe zeigen einenUbergangsdruck von ca. 13 GPa [66]. Die Diskrepanzvon Simulation und Experiment hat im wesentlichen zwei Ursachen. Die simulierte Probewar ideal, d. h. ohne Gitterfehler, es ist zu erwarten, daß fehlerbehaftete Proben niedrige-re kritische Schockst¨arken und somit niedrigereUbergangsdr¨ucke aufweisen. Erste Simula-tionen an polykristallinen Systemen, zeigen eine heterogene Nukleation an Korngrenzen beiniedrigeren Schockst¨arken. Außerdem wurden die EAM-Parameter haupts¨achlich an Gleich-gewichtseigenschaften angepaßt (Abschnitt 2.3.2). Daher k¨onnen die extremen Nichtgleich-gewichtseigenschaften, wie sie hier vorliegen, nicht korrekt wiedergegeben werden. Der ho-he Ubergangsdruck kann auch mit Hilfe derE(V)-Abhangigkeit der verschiedenen Struktu-ren abgesch¨atzt werden (Abschnitt 4.1). Bei den hier vorgestellten Simulationen wurden erst-mals schockinduzierte strukturelle Transformationen mittels Molekulardynamik untersucht. ImRahmen von Molekulardynamik-Simulationen f¨ur kfz Strukturen zeigte sich, daß prinzipielleProzesse der schockinduzierten Ph¨anomene nicht vom gew¨ahlten Potential modifiziert wer-

5.2 Schockinduzierte strukturelleAnderungen in krz Fe 69
den [26]. Somit ist zu erwarten, daß sich die hier beobachteten Prozesse sich unter Modifika-tion des Potentials qualitativ nicht ¨andern. Um einen quantitativ besseren Vergleich mit demExperiment zu erhalten, m¨ussen dieE(V)-Abhangigkeiten im Rahmen von EAM-Potentialenstarker ber¨ucksichtigt werden.
5.2.2 Strukturelle Analyse der schockinduzierten Transformation
Eine genaue Strukturbestimmung der neu gebildeten Phase kann durch die Analyse der radia-len Verteilungsfunktion sowie durch ein entsprechend aufwendigeres Studium der Gitterstruk-tur im Ortsraum erfolgen (Abschnitt 2.2). In Abbildung 5.12 sind die radialen Verteilungs-funktionen jeweils f¨ur verschiedene Bereiche der Simulationszelle aufgetragen. Die Bereicheentsprechen dem nicht geschockten Material, dem uniaxial komprimierten und dem strukturellumgewandelten Material. Die radiale Verteilungsfunktion eines unkomprimierten krz Gittersunterscheidet sich von der Verteilungsfunktion eines uniaxial komprimierten krz Gitters da-durch, daß die 8 Nachbarn aus der ersten Nachbarschale alle n¨aherrucken. Die 4 Nachbarn der2. Schale, die senkrecht zur Ausbreitungsrichtung der Schockwelle liegen (transversale Nach-barn) verandern ihren Abstand nicht, wohingegen die 2 Nachbarn in Ausbreitungsrichtung derSchockwelle (longitudinale Nachbarn) bedingt durch die Kompression n¨aherrucken. Dies f¨uhrtzu einer Aufspaltung der Beitr¨age der 2. Nachbarschale in Form von zwei Maxima der radialenVerteilungsfunktion.Ahnliche Aufspaltungen beobachtet man f¨ur die weiter entfernten Nach-barschalen. DieAhnlichkeit der radialen Verteilungsfunktionen f¨ur das uniaxial komprimiertekrz Gitter und das ideale krz Gitter ist dennoch deutlich zu erkennen, w¨ahrend die Verteilungs-funktion fur das umgewandelte Material sich deutlich von den anderen beiden radialen Vertei-lungsfunktionen unterscheidet. Der Vergleich der Verteilungsfunktionen f¨ur das umgewandelteMaterial mit den radialen Verteilungsfunktionen f¨ur das kfz und hdp Gitter bei vergleichbarenDruck- und Temperaturverh¨altnissen, zeigt eine nahezu perfekteUbereinstimmung mit der hdpStruktur.
4.0 6.0 8.0 10.0r (a.u.)
−7e−14
1
2
3
4
g(r)
hdp
uniaxial komprimiertes krz
krz
Abbildung 5.12: Radiale Verteilungsfunk-tionen eines krz Kristalls nach dem Durch-laufen unterschiedlich starker Schockwel-len. Das ungeschockte krz Gitter zeigt dieersten 8 und ¨ubernachsten 6 Nachbarndicht beieinander. Der elastisch kompri-mierte Kristall zeigt eine Aufteilung derNachbarschalen ab der 2. Nachbarschale.Bei hohen Schockst¨arken (oben) zeigt derVergleich der radialen Verteilungsfunktionund idealen Gitterpositionen von kfz (ge-strichelte Linie, Kreise) und hdp Gittern(gepunktete Linie, Dreiecke) deutlich, daßdie gebildete Phase eine nahezu ideale hdpStruktur aufweist.

70 5 Simulationen metallischer Festk¨orper
B
A
B
A
B
AA
B
A
B
C
BC
Abbildung 5.13: Querschnitt des transformierten Kristalls senkrecht zur Schockrichtung f¨ur eine Kolbenge-schwindigkeit vonUP = 1450 m/s. Zwillig zueinander liegende dicht gepackte Bereiche (n = 12, offene Krei-se) sind durch ein Muster von nicht koh¨arenten Korngrenzen (n ≤ 11) von einander getrennt.n ist die Anzahlder Nachbarn innerhalb eines Radius von 2.75A. Die dicht gepackten Ebenen der urspr¨unglichen krz Strukturtransformieren in die dicht gepackten Ebenen der nahezu perfekt geordneten hdp Struktur (Vergr¨oßerung). Zweikristallographisch verschieden orientierte austenitische Bereiche zeigen eine fast v¨ollig fehlerfreie ABAB Stapel-folge. Ein Stapelfehler befindet sich auf der rechten Seite.
Die Analyse der Gitterstruktur im Ortsraum ergibt tats¨achlich eine dicht gepackte Strukturmit nahezu perfekter ABAB Stapelfolge (hdp Gitter) (Abb. 5.13). Es bilden sich zwei kri-stallographisch verschieden orientierte Bereiche aus, die zwillig zueinander sind und durchnicht koharente Korngrenzen voneinander getrennt sind. Die ABAB gepackten(110)krz-oder (110)krz-Ebenen transformieren ohne Rotation in die(0001)hdp-Ebenen (entsprichtder (111)kfz-Richtung). Weiterhin ist die[110]krz- oder [110]krz-Richtung parallel zu der
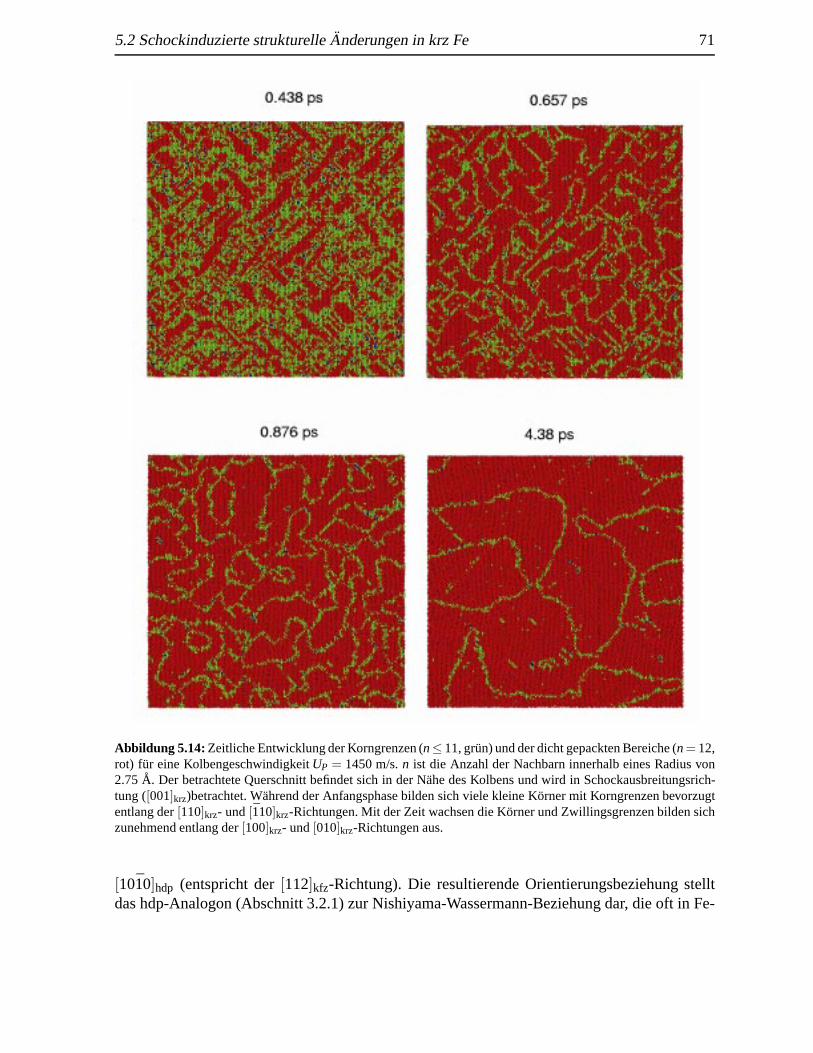
5.2 Schockinduzierte strukturelleAnderungen in krz Fe 71
Abbildung 5.14:Zeitliche Entwicklung der Korngrenzen (n≤ 11, grun) und der dicht gepackten Bereiche (n= 12,rot) fur eine KolbengeschwindigkeitUP = 1450 m/s.n ist die Anzahl der Nachbarn innerhalb eines Radius von2.75A. Der betrachtete Querschnitt befindet sich in der N¨ahe des Kolbens und wird in Schockausbreitungsrich-tung ([001]krz)betrachtet. W¨ahrend der Anfangsphase bilden sich viele kleine K¨orner mit Korngrenzen bevorzugtentlang der[110]krz- und[110]krz-Richtungen. Mit der Zeit wachsen die K¨orner und Zwillingsgrenzen bilden sichzunehmend entlang der[100]krz- und[010]krz-Richtungen aus.
[1010]hdp (entspricht der[112]kfz-Richtung). Die resultierende Orientierungsbeziehung stelltdas hdp-Analogon (Abschnitt 3.2.1) zur Nishiyama-Wassermann-Beziehung dar, die oft in Fe-

72 5 Simulationen metallischer Festk¨orper
Legierungen und deren strukturellen Transformationen experimentell gefunden wird [4,5].Im Rahmen des EAM-Modells kann man die hdp Phase kaum von der kfz Phase unter-
scheiden, da die Stapelfehlerenergien sehr gering sind (vgl. Abschnitt 4.1). Das Auftreten derhdp Phase und nicht etwa der kfz Phase ist somit im wesentlichen auf den Mechanismus desUbergangs zur¨uckzufuhren (ABAB gepackte(110)krz-Ebenen transformieren in ABAB ge-packte(0001)hdp-Ebenen). Erste Simulationen zu schockwelleninduzierter struktureller Trans-formationen in(011)krz-Richtung best¨atigen dies. DerUbergangsmechanismus ist anders be-dingt durch die andere kristallographische Orientierung des Gitters im Vergleich zur Ausbrei-tungsrichtung der Schockwelle. Es bildet sich verst¨arkt eine kfz Struktur aus.
Betrachtet man die Dynamik der K¨orner, so bilden sich anf¨anglich viele kleine K¨orneraus, wobei die Korngrenzen bevorzugt entlang der[110]krz- und[110]krz-Richtung liegen (Abb.5.14). Im weiteren Verlauf wachsen die K¨orner und die Korngrenzen bilden sich nun bevorzugtentlang der urspr¨unglichen[100]krz- und[010]krz-Richtungen aus. Dabei wachsen die gr¨oßerenKorner, indem kleinere K¨orner absorbiert werden. Durch den geringeren Innendruck oder dergeringeren Oberfl¨achenenergie pro Volumen absorbieren die gr¨oßeren Korner die kleineren.
Die hier und in Abschnitt 5.2.1 vorgestellten Simulationen sind in Zusammenarbeit mitT. C. Germann, P. S. Lomdahl und B. L. Holian (Los Alamos) entstanden. Die Daten aus Ab-bildung 5.11 sind zum Teil aus von T. C. Germann durchgef¨uhrten Simulationen entnommen.Das Layout von Abbildung 5.11 wurde maßgeblich von T. C. Germann gestaltet. Das Lay-out von Abbildung 5.13 wurde gleichermaßen von T. C. Germann und K. Kadau erstellt. InLos Alamos sollen demn¨achst ultra-schnelle zeitaufgel¨oste Rontgenbeugungsexperimente anFe-Einkristallen erfolgen, die dann einen direkten Vergleich mit den hier gewonnenen struktu-rellen Informationen erlauben w¨urden.

73
6 Untersuchungen an Fe80Ni20-Nanopartikeln
Dieses Kapitel besch¨aftigt sich mit einem zus¨atzlichen Aspekt martensitischer Umwandlungen.Dies betrifft die Abhangigkeit derUbergangstemperatur von der Gr¨oße des Partikels, wennman von Festk¨orpern zu Partikeln unterschiedlicher Gr¨oßeubergeht. Außerdem ist dies engmit der Frage verkn¨upft, was die minimale Gr¨oße eines Partikels f¨ur die martensitische Nu-kleation und das Wachstum der martensitischen Phase ist. R. E. Cech und D. Turnbull haben1956 Experimente an Fe-Ni-Legierungspulver durchgef¨uhrt, die zeigten, daß mit abnehmenderPartikelgroße die martensitische Umwandlungstemperaturen sinken. Die Martensitbildung inkleinen Partikeln wird dabei unterdr¨uckt [106]. Partikeldurchmesser von einigen 10µm zeigteneine Verschiebung der martensitischen Umwandlungstemperatur um bis zu ca. 200 K. Die Au-toren diskutierten ihr Ergebnis dahingehend, daß mit einer Reduktion der Partikelgr¨oße auchdie Wahrscheinlichkeit reduziert wird, eine Inhomogenit¨at im Partikel anzutreffen, an dem eineheterogene Nukleation stattfinden kann. Sie gingen nicht davon aus, daß sich die thermodyna-mische GleichgewichtstemperaturM0 (Abschnitt 3.2) von Austenit und Martensit ¨andert.
T. Tadaki et al. untersuchten 1996 Nanopartikel von Fe-Ni-Legierungen mit einer Fe-Konzentration von ca. 80% [89]. Die Gr¨oße der Partikel lag zwischen 3 und 16 nm mit einemHaufungspunkt bei 6 nm (ca. 10 000 Atome). Die Autoren fanden ebenfalls eine Reduktion derMartensittemperatur. In diesem Fall wurde dies auf eine Verschiebung der thermodynamischenGleichgewichtstemperatur von Martensit und Austenit zur¨uckgefuhrt. Dies wurde 1999 durchUntersuchungen von K. Asakaet al. [107,108] best¨atigt.
In den nachsten beiden Abschnitten wird anhand von Absch¨atzungen der energetischen Ver-anderungen in Abh¨angigkeit von der Partikelgr¨oße und mit Hilfe von Simulationen verschiedengroßer Partikel gezeigt, daß der Unterschied der Oberfl¨achenenergien von Austenit und Mar-tensit bei Partikelgr¨oßen im Nanometerbereich einen entscheidenden Einfluß auf die Gleich-gewichtstemperaturM0 hat. Anhand von Simulationszellen mit bis zu einer Millionen Atomenwird der heterogene Nukleationscharakter bei martensitischen Umwandlungen untersucht. ImRahmen des hier benutzten EAM-Potentials wird deutlich gemacht, daß zwei verschiedene Ur-sachen f¨ur die Unterdr¨uckung des Martensits in kleinen Partikeln existieren. Bei Partikeln voneinigen 10 nm Durchmesser und dar¨uber hinaus ist das Fehlen von Keimen entscheidend f¨urdie Untersruckung der Martensitbildung, w¨ahrend die thermodynamische Gleichgewichtstem-peratur sich wenig ¨andert. Bei Partikeln von nur wenigen Nanometern nimmt die thermodyna-mische GleichgewichtstemperaturM0 ab, wodurch die martensitische Umwandlung auch beivorhandenen Defekten unterdr¨uckt werden kann.
6.1 Einfluß der Oberflache auf die thermodynamische Gleichgewicht-stemperatur
Die Oberflachenenergien der hier verwendeten krz Kristalle liegen im Mittel knapp 10 % ¨uberdenjenigen der kfz Phase (Tab. 4.3). Durch die Anwesenheit von Oberfl¨achen wird die kfz Pha-se, d. h. der Austenit, energetisch gegen¨uber dem Martensit favorisiert. Quantitative Aussagenkonnen aus dem Verh¨altnis der Energiedifferenz der Oberfl¨achenenergien von kfz und krz Pha-

74 6. Untersuchungen an Fe80Ni20-Nanopartikeln
se ∆Ekfz−krzO , und der Energiedifferenz der entsprechenden Festk¨orperenergien∆Ekfz−krz
Fk , inAbhangigkeit des Partikelradius gewonnen werden. Im Fall von Fe ergibt sich f¨ur T = 100 K(Tab. 4.3 und Tab. 4.1)
∆Ekfz−krzO
∆Ekfz−krzFk
≈ 5d, (6.1)
d ist der Partikeldurchmesser in Gitterkonstanten des krz Gitters. Hierbei wurde∆Ekfz−krzO als
Mittelwert der beiden Differenzen der Oberfl¨achenenergien ((100)kfz, (100)krz) und ((111)kfz,(110)krz) bestimmt. Die Oberfl¨achen des Nanopartikels wurden somit f¨ur diese Absch¨atzungals je zur Halfte aus(100)kfz- und(111)kfz- bzw.(100)krz- und(110)krz-Oberflachen aufgebautangenommen. Vergleichbare Werte ergeben sich f¨ur Fe80Ni20 Legierungen [69].
Kleine Partikel mit Durchmessern bis zu f¨unf Gitterkonstanten weisen einen Beitrag∆Ekfz−krz
O zur Energiedifferenz auf, der im Vergleich zur Energiedifferenz des Festk¨orpers∆Ekfz−krz
Fk dominierend ist. Bei Partikeldurchmessern von 50 Gitterkonstanten betr¨agt der Ober-flachenanteil nur noch 10%. Es ist zu erwarten, daß bei entsprechend kleinen Partikeldurch-messern die austenitische kfz Struktur gegen¨uber der martensitischen krz Struktur thermody-namisch stabilisiert wird. Die thermodynamische GleichgewichtstemperaturM0 sollte mit ab-nehmendem Partikeldurchmesser geringer werden.
Zur Untersuchung der Umwandlungstemperatur wurden Simulationen von krz initialisier-ten Fe80Ni20 Nanopartikeln unterschiedlichen Durchmessers durchgef¨uhrt. Ausgehend von25 K wurden die Partikel in 25 K Schritten auf 700 K erhitzt und ¨uber ein Zeitintervall von12 000 Integrationschritte simuliert. Um sicherzustellen, daß die Partikel nicht f¨ur langere Si-mulationszeiten bereits bei tieferen Temperaturen in die austenitische Struktur umwandeln,wurden weitere 24 000 Integrationschritte bei einer um 25 K niedrigeren Temperatur als derzuvor gefundenenUbergangstemperatur durchgef¨uhrt.
Partikel mit einem Durchmesser bis zu 6 Gitterkonstanten (228 Atome) weisen beitiefsten Temperaturen keine stabile krz Struktur auf. Tabelle 6.1 zeigt die austenitischeUbergangstemperatur in Abh¨angigkeit des Partikeldurchmessers. Die kleinsten Partikel zei-gen die niedrigstenUbergangstemperaturen. Mit zunehmender Partikelgr¨oße stabilisiert sichdie krz Struktur. Partikel von 30 Gitterkonstanten Durchmesser (28 276 Atome) zeigen bereitsUmwandlungstemperaturen von 550 K. Sie liegen damit schon relativ nahe an der austeni-
D N TA
8 544 25010 1056 35015 3582 42520 8388 50030 28276 550
Tabelle 6.1: Austenitische Umwandlungstemperaturen (TA (K)) inAbhangigkeit der Partikelgr¨oße (D (akrz), N (Anzahl der Atome)). Mitabnehmendem Partikeldurchmesser wird die martensitische Phase desta-bilisiert. Im Rahmen des benutzten EAM-Modells kann dieses Verhaltenauf die unterschiedlichen Oberfl¨achenenergien von Martensit und Austenitzuruckgefuhrt werden.
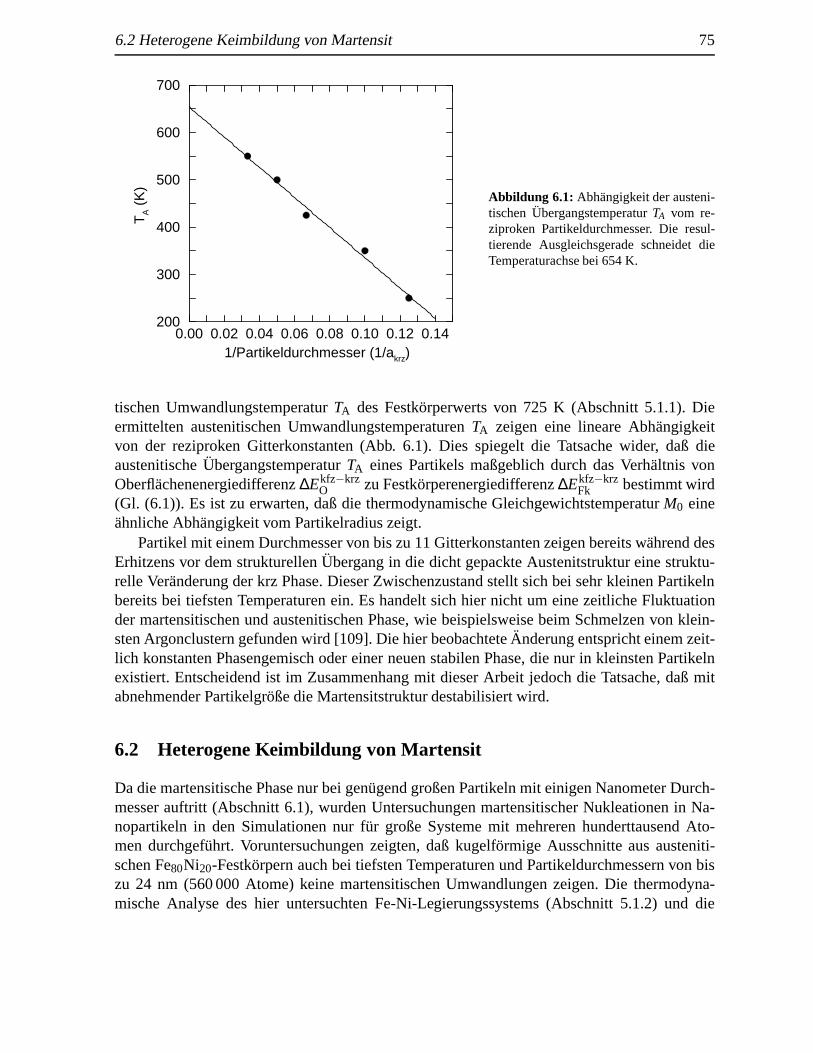
6.2 Heterogene Keimbildung von Martensit 75
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.141/Partikeldurchmesser (1/akrz)
200
300
400
500
600
700
TA (
K)
Abbildung 6.1: Abhangigkeit der austeni-tischenUbergangstemperaturTA vom re-ziproken Partikeldurchmesser. Die resul-tierende Ausgleichsgerade schneidet dieTemperaturachse bei 654 K.
tischen UmwandlungstemperaturTA des Festk¨orperwerts von 725 K (Abschnitt 5.1.1). Dieermittelten austenitischen UmwandlungstemperaturenTA zeigen eine lineare Abh¨angigkeitvon der reziproken Gitterkonstanten (Abb. 6.1). Dies spiegelt die Tatsache wider, daß dieaustenitischeUbergangstemperaturTA eines Partikels maßgeblich durch das Verh¨altnis vonOberflachenenergiedifferenz∆Ekfz−krz
O zu Festkorperenergiedifferenz∆Ekfz−krzFk bestimmt wird
(Gl. (6.1)). Es ist zu erwarten, daß die thermodynamische GleichgewichtstemperaturM0 eineahnliche Abhangigkeit vom Partikelradius zeigt.
Partikel mit einem Durchmesser von bis zu 11 Gitterkonstanten zeigen bereits w¨ahrend desErhitzens vor dem strukturellenUbergang in die dicht gepackte Austenitstruktur eine struktu-relle Veranderung der krz Phase. Dieser Zwischenzustand stellt sich bei sehr kleinen Partikelnbereits bei tiefsten Temperaturen ein. Es handelt sich hier nicht um eine zeitliche Fluktuationder martensitischen und austenitischen Phase, wie beispielsweise beim Schmelzen von klein-sten Argonclustern gefunden wird [109]. Die hier beobachteteAnderung entspricht einem zeit-lich konstanten Phasengemisch oder einer neuen stabilen Phase, die nur in kleinsten Partikelnexistiert. Entscheidend ist im Zusammenhang mit dieser Arbeit jedoch die Tatsache, daß mitabnehmender Partikelgr¨oße die Martensitstruktur destabilisiert wird.
6.2 Heterogene Keimbildung von Martensit
Da die martensitische Phase nur bei gen¨ugend großen Partikeln mit einigen Nanometer Durch-messer auftritt (Abschnitt 6.1), wurden Untersuchungen martensitischer Nukleationen in Na-nopartikeln in den Simulationen nur f¨ur große Systeme mit mehreren hunderttausend Ato-men durchgef¨uhrt. Voruntersuchungen zeigten, daß kugelf¨ormige Ausschnitte aus austeniti-schen Fe80Ni20-Festkorpern auch bei tiefsten Temperaturen und Partikeldurchmessern von biszu 24 nm (560 000 Atome) keine martensitischen Umwandlungen zeigen. Die thermodyna-mische Analyse des hier untersuchten Fe-Ni-Legierungssystems (Abschnitt 5.1.2) und die
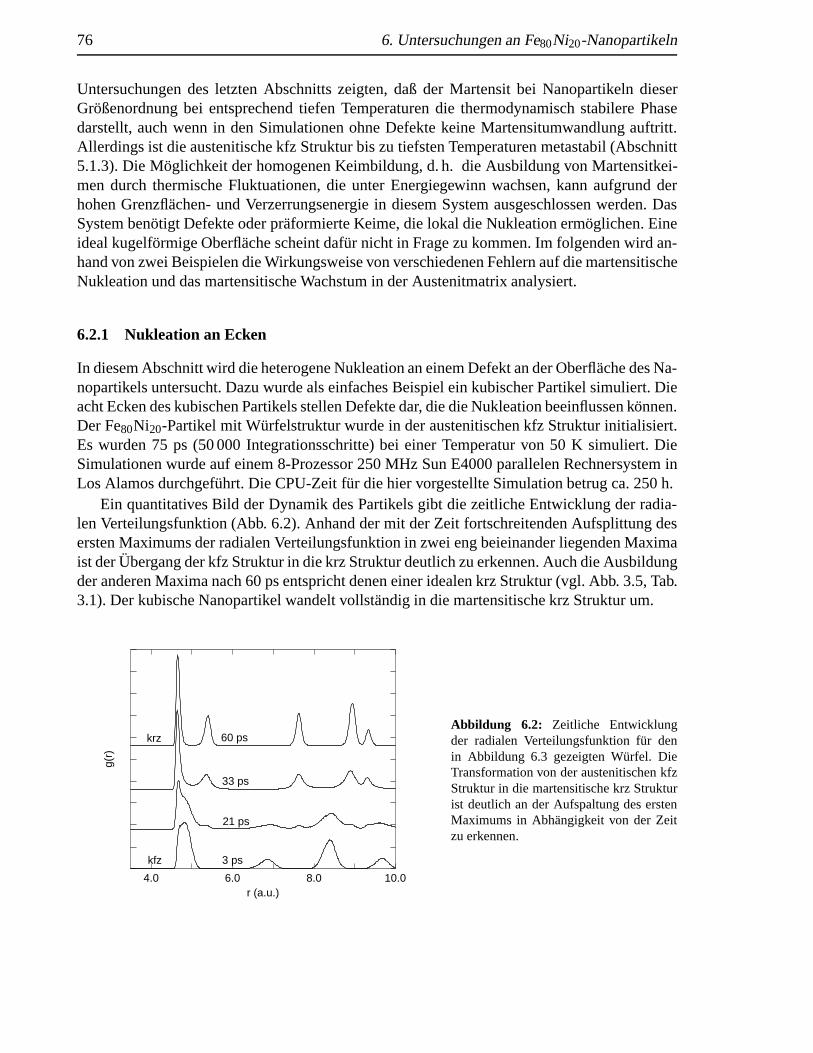
76 6. Untersuchungen an Fe80Ni20-Nanopartikeln
Untersuchungen des letzten Abschnitts zeigten, daß der Martensit bei Nanopartikeln dieserGroßenordnung bei entsprechend tiefen Temperaturen die thermodynamisch stabilere Phasedarstellt, auch wenn in den Simulationen ohne Defekte keine Martensitumwandlung auftritt.Allerdings ist die austenitische kfz Struktur bis zu tiefsten Temperaturen metastabil (Abschnitt5.1.3). Die Moglichkeit der homogenen Keimbildung, d. h. die Ausbildung von Martensitkei-men durch thermische Fluktuationen, die unter Energiegewinn wachsen, kann aufgrund derhohen Grenzfl¨achen- und Verzerrungsenergie in diesem System ausgeschlossen werden. DasSystem ben¨otigt Defekte oder pr¨aformierte Keime, die lokal die Nukleation erm¨oglichen. Eineideal kugelformige Oberflache scheint daf¨ur nicht in Frage zu kommen. Im folgenden wird an-hand von zwei Beispielen die Wirkungsweise von verschiedenen Fehlern auf die martensitischeNukleation und das martensitische Wachstum in der Austenitmatrix analysiert.
6.2.1 Nukleation an Ecken
In diesem Abschnitt wird die heterogene Nukleation an einem Defekt an der Oberfl¨ache des Na-nopartikels untersucht. Dazu wurde als einfaches Beispiel ein kubischer Partikel simuliert. Dieacht Ecken des kubischen Partikels stellen Defekte dar, die die Nukleation beeinflussen k¨onnen.Der Fe80Ni20-Partikel mit Wurfelstruktur wurde in der austenitischen kfz Struktur initialisiert.Es wurden 75 ps (50 000 Integrationsschritte) bei einer Temperatur von 50 K simuliert. DieSimulationen wurde auf einem 8-Prozessor 250 MHz Sun E4000 parallelen Rechnersystem inLos Alamos durchgef¨uhrt. Die CPU-Zeit fur die hier vorgestellte Simulation betrug ca. 250 h.
Ein quantitatives Bild der Dynamik des Partikels gibt die zeitliche Entwicklung der radia-len Verteilungsfunktion (Abb. 6.2). Anhand der mit der Zeit fortschreitenden Aufsplittung desersten Maximums der radialen Verteilungsfunktion in zwei eng beieinander liegenden Maximaist derUbergang der kfz Struktur in die krz Struktur deutlich zu erkennen. Auch die Ausbildungder anderen Maxima nach 60 ps entspricht denen einer idealen krz Struktur (vgl. Abb. 3.5, Tab.3.1). Der kubische Nanopartikel wandelt vollst¨andig in die martensitische krz Struktur um.
4.0 6.0 8.0 10.0r (a.u.)
g(r)
kfz
krz
3 ps
21 ps
33 ps
60 psAbbildung 6.2: Zeitliche Entwicklungder radialen Verteilungsfunktion f¨ur denin Abbildung 6.3 gezeigten W¨urfel. DieTransformation von der austenitischen kfzStruktur in die martensitische krz Strukturist deutlich an der Aufspaltung des erstenMaximums in Abhangigkeit von der Zeitzu erkennen.
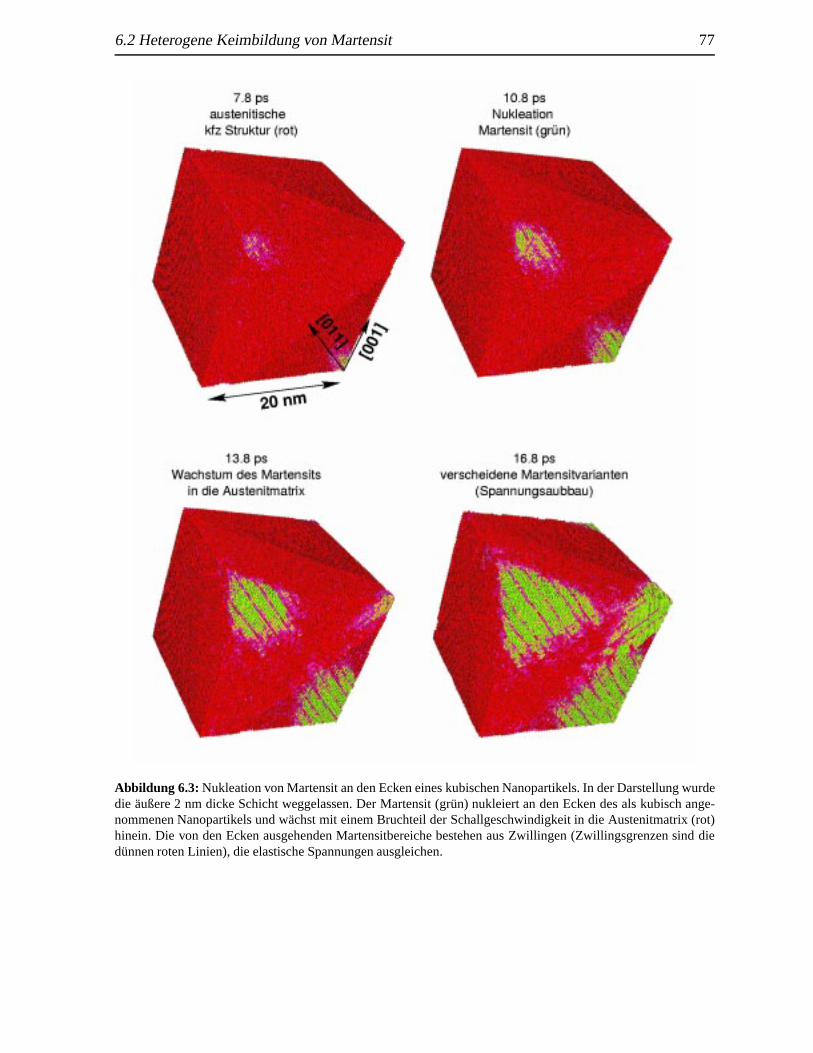
6.2 Heterogene Keimbildung von Martensit 77
Abbildung 6.3: Nukleation von Martensit an den Ecken eines kubischen Nanopartikels. In der Darstellung wurdedie außere 2 nm dicke Schicht weggelassen. Der Martensit (gr¨un) nukleiert an den Ecken des als kubisch ange-nommenen Nanopartikels und w¨achst mit einem Bruchteil der Schallgeschwindigkeit in die Austenitmatrix (rot)hinein. Die von den Ecken ausgehenden Martensitbereiche bestehen aus Zwillingen (Zwillingsgrenzen sind diedunnen roten Linien), die elastische Spannungen ausgleichen.

78 6. Untersuchungen an Fe80Ni20-Nanopartikeln
In Abbildung 6.3 ist die Dynamik der Umwandlung anhand der Farbcodierung des Mar-tensits (gr¨un) dargestellt. Die Martensitumwandlung beginnt an den verschiedenen Ecken desPartikels, die als Nukleationszentren dienen (Abb. 6.3). Bedingt durch thermische Fluktua-tionen beginnt die Bildung der martensitischen Phase zu unterschiedlichen Zeiten auf derPicosekunden-Skala, so startet die Nukleation an der vorderen oberen linken Ecke und an dervorderen unteren rechten Ecke bei ca. 7.8 ps, w¨ahrend nach 16.8 ps die vordere untere linkeEcke noch keinen Beginn der Transformation erkennen l¨aßt (Abb. 6.3). Ausgehend von denEcken beginnt der Martensit in das Innere des Partikels zu wachsen. Die Wachstumsgeschwin-digkeit hangt dabei von der kristallographischen Richtung ab. Die Wachstumsgeschwindig-keit in [001]-Richtung betr¨agt ca. 1200 m/s, die in [011]-Richtung ca. 1500 m/s. Die Schall-geschwindigkeit f¨ur das verwendete EAM-Potential in der krz Struktur betr¨agt 5353 m/s in[001]-Richtung und 6277 m/s in [011]-Richtung (Abschnitt 5.2, [105]). Die Ausbreitungsge-schwindigkeit der martensitischen Phase ist somit gr¨oßer in kristallographischen Richtungen,
Abbildung 6.4: Verzwillte martensitische Struktur des umgewandelten Nanopartikels. Die Farbkodierung gibt dieAnzahl (4-12) der Nachbaratome innerhalb eines Abstands von 2.75A. Die Verkippung der (001)-Ebene (dunkel)und der (011)-Ebene (hell) kommt durch das Zusammentreffen von zwei verschieden orientierten Martensitvari-anten zustande (oben im Bild). Die vordere Oberfl¨ache zeigt entlang der Diagonalen eine Zwillingsgrenze (s. Ver-großerung): Der lange Pfeil markiert die[111]krz-Zwillingsscherrichtung, die beiden kurzen Pfeile markieren die[001]krz-Richtung der unterschiedlichen Varianten. Die verzerrten Hexagone sind typisch f¨ur die (011)-Oberfl¨acheeines krz Gitters.

6.2 Heterogene Keimbildung von Martensit 79
in denen die Schallgeschwindigkeit gr¨oßer, das Material also h¨arter ist.Die einzelnen von den Ecken des Partikels ausgehenden martensitischen Bereiche, einge-
bettet in der Austenitstruktur, weisen kristallographisch verschieden orientierte Martensitvari-anten auf (Zwillinge). Dieses Verhalten ¨ahnelt dem experimentellen Verhalten einzelner ver-zwillter Martensitnadeln in der Austenitmatrix (vgl. Abb. 3.8). Nach diesem ausbruchsartigenProzeßes stoßen die an den Ecken gebildeten kristallographisch unterschiedlichen Martensit-bereiche zusammen und treten in Konkurrenz zueinander. In dieser Phase ist der Anteil desAustenits bereits gering, die feinen Zwillinge (einige Monolagen dick) wachsen teilweise voninnen aus dem Kristall heraus und vergr¨oßern die kristallographisch gleich orientierten marten-sitischen Bereiche.
Nach Abschluß dieses dynamischen Prozesses der einige 10 ps lang dauert, bildet sichein aus mehreren großen Martensitvarianten bestehender martensitischer Nanopartikel, dervollstandig in die martensitische krz Phase umgewandelt ist (Abb. 6.4). Die verschiedenen Va-rianten liegen teilweise zwillig zueinander unter Ausbildung des f¨ur krz Strukturen typischenZwillingssystems (Abschnitt 3.1, Tab. 3.2). Die verschiedenen, auf der hier beobachteten Zeits-kala, stabilen Martensitvarianten sind das Resultat der in Konkurrenz zueinander stehenden Nu-kleationsprozesse, die an den Ecken des W¨urfels beginnen. Durch die unterschiedlichen Varian-ten entsteht auf der Oberfl¨ache des Nanopartikels eine Verkippung zwischen einer (001)-Ebeneund einer (011)-Ebene. In mikroskopischen Untersuchungen werden Oberfl¨achenverkippungenausgenutzt, um verschieden orientierte martensitische Bereiche innerhalb der Austenitmatrixsichtbar zu machen (Abschnitt 3.2.1, Abb. 3.8).
Ein entscheidender Unterschied zwischen der Bildung von Martensit in Nanopartikeln undFestkorpern besteht darin, daß es in Nanopartikeln keine in der Austenitmatrix eingebettetenstabilen Martensitbereiche gibt, da keine Austenitmatrix ¨ubrigbleibt. Wahrend der Bildung unddes Wachstums von Martensit bilden sich jedoch im Nanopartikel verzwillte martensitischeVarianten, um die elastischen Spannungen in der Austenitmatrix abzubauen, somit tritt das imFestkorper statische Verhalten in den Nanopartikeln nur als dynamischer Zwischenzustand auf.
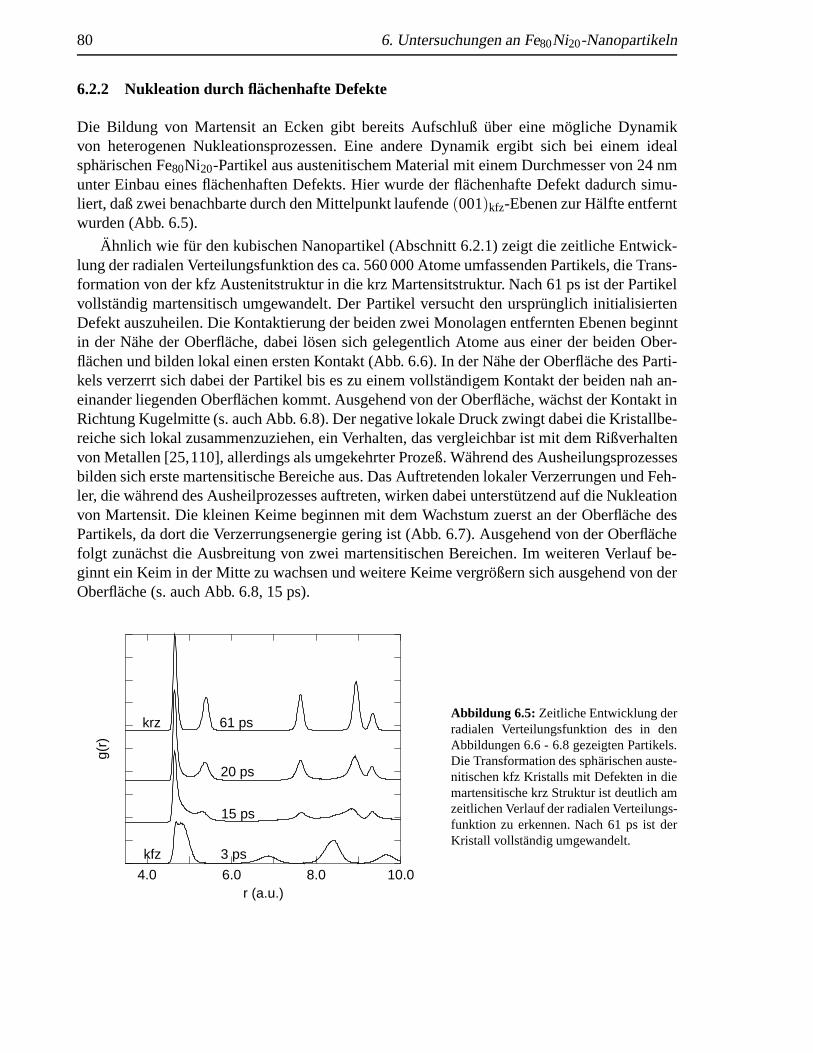
80 6. Untersuchungen an Fe80Ni20-Nanopartikeln
6.2.2 Nukleation durch flachenhafte Defekte
Die Bildung von Martensit an Ecken gibt bereits Aufschluß ¨uber eine m¨ogliche Dynamikvon heterogenen Nukleationsprozessen. Eine andere Dynamik ergibt sich bei einem idealspharischen Fe80Ni20-Partikel aus austenitischem Material mit einem Durchmesser von 24 nmunter Einbau eines fl¨achenhaften Defekts. Hier wurde der fl¨achenhafte Defekt dadurch simu-liert, daß zwei benachbarte durch den Mittelpunkt laufende(001)kfz-Ebenen zur H¨alfte entferntwurden (Abb. 6.5).
Ahnlich wie fur den kubischen Nanopartikel (Abschnitt 6.2.1) zeigt die zeitliche Entwick-lung der radialen Verteilungsfunktion des ca. 560 000 Atome umfassenden Partikels, die Trans-formation von der kfz Austenitstruktur in die krz Martensitstruktur. Nach 61 ps ist der Partikelvollstandig martensitisch umgewandelt. Der Partikel versucht den urspr¨unglich initialisiertenDefekt auszuheilen. Die Kontaktierung der beiden zwei Monolagen entfernten Ebenen beginntin der Nahe der Oberfl¨ache, dabei l¨osen sich gelegentlich Atome aus einer der beiden Ober-flachen und bilden lokal einen ersten Kontakt (Abb. 6.6). In der N¨ahe der Oberfl¨ache des Parti-kels verzerrt sich dabei der Partikel bis es zu einem vollst¨andigem Kontakt der beiden nah an-einander liegenden Oberfl¨achen kommt. Ausgehend von der Oberfl¨ache, wachst der Kontakt inRichtung Kugelmitte (s. auch Abb. 6.8). Der negative lokale Druck zwingt dabei die Kristallbe-reiche sich lokal zusammenzuziehen, ein Verhalten, das vergleichbar ist mit dem Rißverhaltenvon Metallen [25,110], allerdings als umgekehrter Prozeß. W¨ahrend des Ausheilungsprozessesbilden sich erste martensitische Bereiche aus. Das Auftretenden lokaler Verzerrungen und Feh-ler, die wahrend des Ausheilprozesses auftreten, wirken dabei unterst¨utzend auf die Nukleationvon Martensit. Die kleinen Keime beginnen mit dem Wachstum zuerst an der Oberfl¨ache desPartikels, da dort die Verzerrungsenergie gering ist (Abb. 6.7). Ausgehend von der Oberfl¨achefolgt zunachst die Ausbreitung von zwei martensitischen Bereichen. Im weiteren Verlauf be-ginnt ein Keim in der Mitte zu wachsen und weitere Keime vergr¨oßern sich ausgehend von derOberflache (s. auch Abb. 6.8, 15 ps).
4.0 6.0 8.0 10.0r (a.u.)
g(r)
kfz
krz
3 ps
15 ps
20 ps
61 psAbbildung 6.5: Zeitliche Entwicklung derradialen Verteilungsfunktion des in denAbbildungen 6.6 - 6.8 gezeigten Partikels.Die Transformation des sph¨arischen auste-nitischen kfz Kristalls mit Defekten in diemartensitische krz Struktur ist deutlich amzeitlichen Verlauf der radialen Verteilungs-funktion zu erkennen. Nach 61 ps ist derKristall vollstandig umgewandelt.
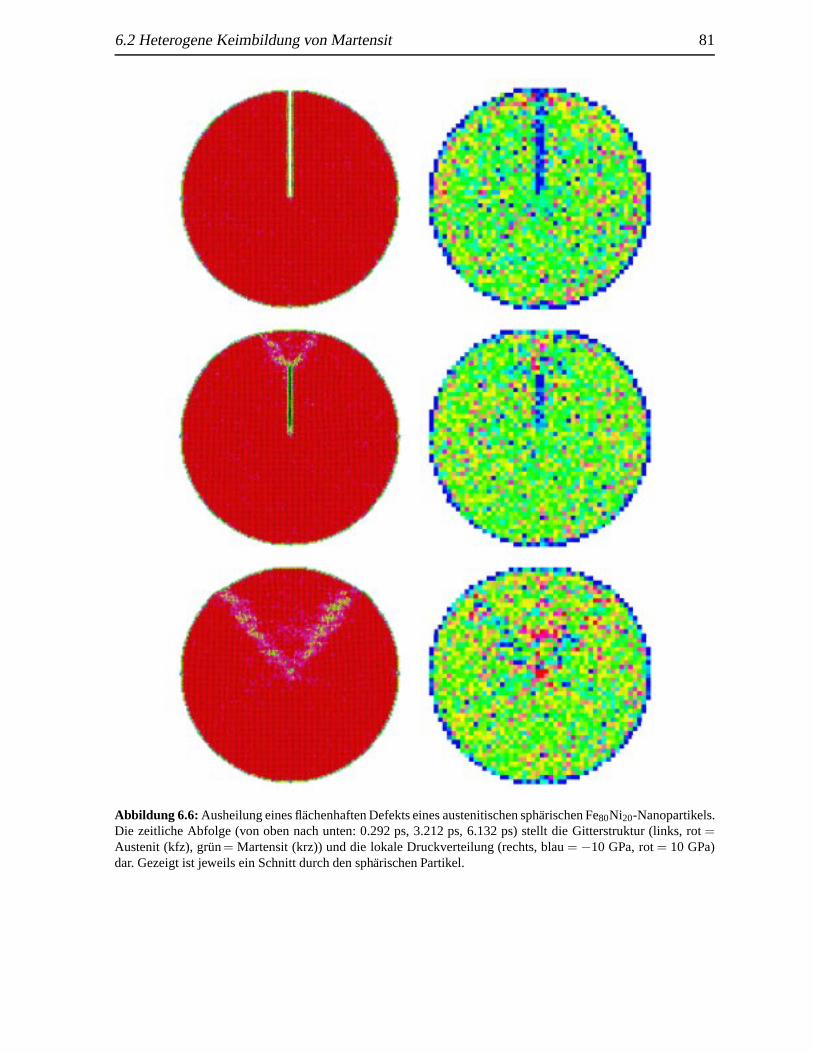
6.2 Heterogene Keimbildung von Martensit 81
Abbildung 6.6: Ausheilung eines fl¨achenhaften Defekts eines austenitischen sph¨arischen Fe80Ni20-Nanopartikels.Die zeitliche Abfolge (von oben nach unten: 0.292 ps, 3.212 ps, 6.132 ps) stellt die Gitterstruktur (links, rot=Austenit (kfz), grun= Martensit (krz)) und die lokale Druckverteilung (rechts, blau= −10 GPa, rot= 10 GPa)dar. Gezeigt ist jeweils ein Schnitt durch den sph¨arischen Partikel.
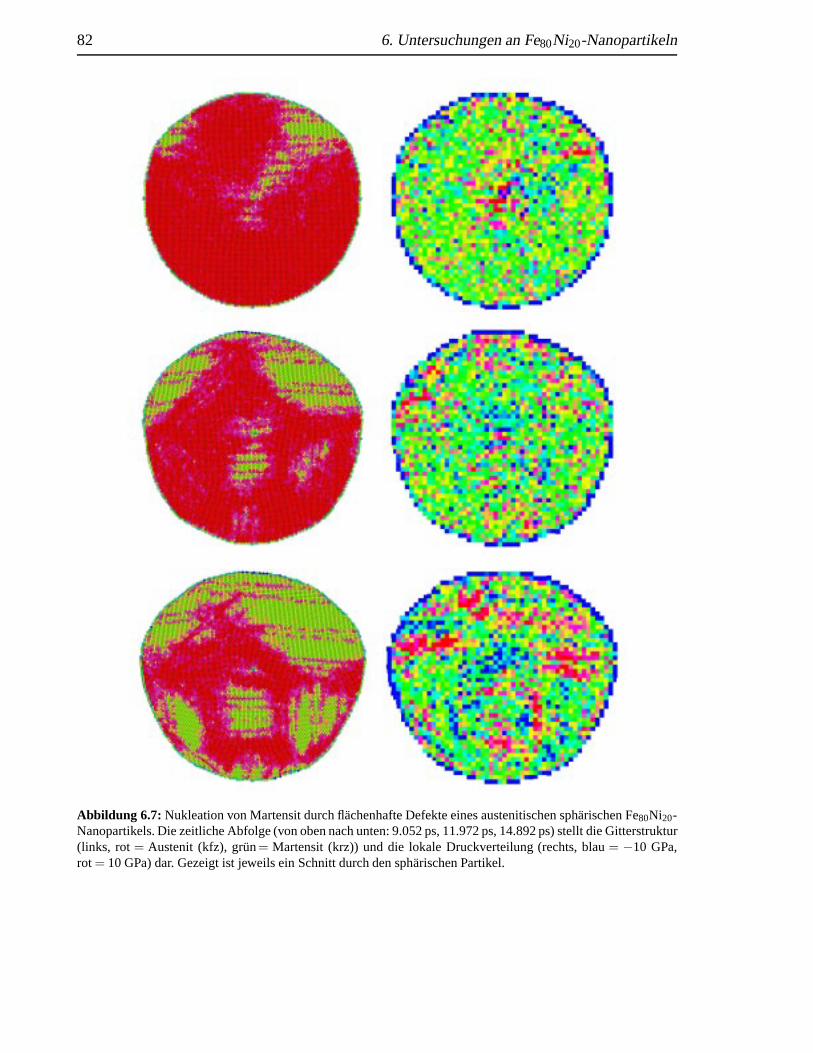
82 6. Untersuchungen an Fe80Ni20-Nanopartikeln
Abbildung 6.7: Nukleation von Martensit durch fl¨achenhafte Defekte eines austenitischen sph¨arischen Fe80Ni20-Nanopartikels. Die zeitliche Abfolge (von oben nach unten: 9.052 ps, 11.972 ps, 14.892 ps) stellt die Gitterstruktur(links, rot= Austenit (kfz), grun= Martensit (krz)) und die lokale Druckverteilung (rechts, blau= −10 GPa,rot = 10 GPa) dar. Gezeigt ist jeweils ein Schnitt durch den sph¨arischen Partikel.

6.2 Heterogene Keimbildung von Martensit 83
Abbildung 6.8: Martensitbildung w¨ahrend der Ausheilung eines fl¨achenhaften Defekts. Gezeigt ist ein Schnittdurch den sph¨arischen Fe80Ni20-Nanopartikel . Die kleinen Martensitkeime wachsen in der Austenitmatrix unterAusbildung von verschieden orientierten Varianten. Nach 60 ps ist der Partikel vollst¨andig martensitisch (gr¨un)umgewandelt und besteht aus vier verschiedenen Bereichen (getrennt durch durch d¨unne rote Linien).
Die Martensitbereiche bestehen wie im Fall des kubischen Nanopartikels (Abschnitt 6.2.1)aus verschiedenen Varianten. Beim Zusammentreffen der verschiedenen Bereiche treten dieVarianten zueinander in Konkurrenz. Durch die verschieden orientierten Martensitbereiche, diein die Austenitmatrix eingebettet sind, entstehen hohe lokale Dr¨ucke (Abb. 6.7).
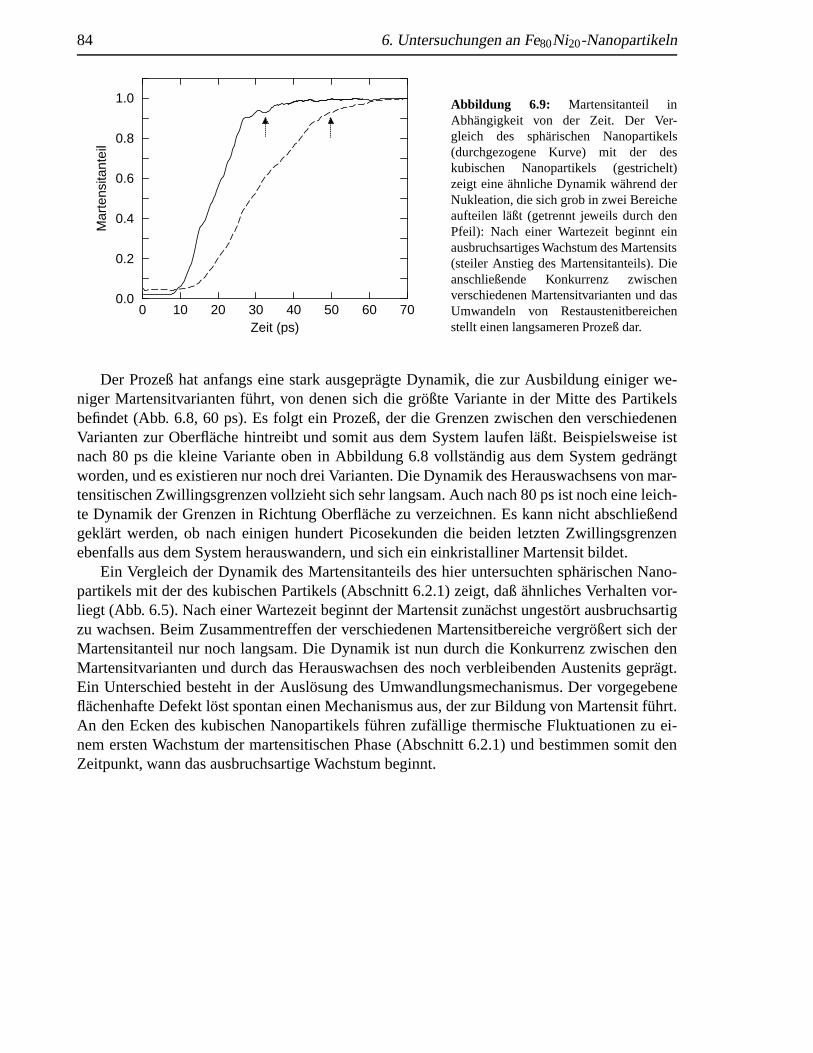
84 6. Untersuchungen an Fe80Ni20-Nanopartikeln
0 10 20 30 40 50 60 70Zeit (ps)
0.0
0.2
0.4
0.6
0.8
1.0M
arte
nsita
ntei
lAbbildung 6.9: Martensitanteil inAbhangigkeit von der Zeit. Der Ver-gleich des sph¨arischen Nanopartikels(durchgezogene Kurve) mit der deskubischen Nanopartikels (gestrichelt)zeigt eineahnliche Dynamik w¨ahrend derNukleation, die sich grob in zwei Bereicheaufteilen laßt (getrennt jeweils durch denPfeil): Nach einer Wartezeit beginnt einausbruchsartiges Wachstum des Martensits(steiler Anstieg des Martensitanteils). Dieanschließende Konkurrenz zwischenverschiedenen Martensitvarianten und dasUmwandeln von Restaustenitbereichenstellt einen langsameren Prozeß dar.
Der Prozeß hat anfangs eine stark ausgepr¨agte Dynamik, die zur Ausbildung einiger we-niger Martensitvarianten f¨uhrt, von denen sich die gr¨oßte Variante in der Mitte des Partikelsbefindet (Abb. 6.8, 60 ps). Es folgt ein Prozeß, der die Grenzen zwischen den verschiedenenVarianten zur Oberfl¨ache hintreibt und somit aus dem System laufen l¨aßt. Beispielsweise istnach 80 ps die kleine Variante oben in Abbildung 6.8 vollst¨andig aus dem System gedr¨angtworden, und es existieren nur noch drei Varianten. Die Dynamik des Herauswachsens von mar-tensitischen Zwillingsgrenzen vollzieht sich sehr langsam. Auch nach 80 ps ist noch eine leich-te Dynamik der Grenzen in Richtung Oberfl¨ache zu verzeichnen. Es kann nicht abschließendgeklart werden, ob nach einigen hundert Picosekunden die beiden letzten Zwillingsgrenzenebenfalls aus dem System herauswandern, und sich ein einkristalliner Martensit bildet.
Ein Vergleich der Dynamik des Martensitanteils des hier untersuchten sph¨arischen Nano-partikels mit der des kubischen Partikels (Abschnitt 6.2.1) zeigt, daß ¨ahnliches Verhalten vor-liegt (Abb. 6.5). Nach einer Wartezeit beginnt der Martensit zun¨achst ungest¨ort ausbruchsartigzu wachsen. Beim Zusammentreffen der verschiedenen Martensitbereiche vergr¨oßert sich derMartensitanteil nur noch langsam. Die Dynamik ist nun durch die Konkurrenz zwischen denMartensitvarianten und durch das Herauswachsen des noch verbleibenden Austenits gepr¨agt.Ein Unterschied besteht in der Ausl¨osung des Umwandlungsmechanismus. Der vorgegebeneflachenhafte Defekt l¨ost spontan einen Mechanismus aus, der zur Bildung von Martensit f¨uhrt.An den Ecken des kubischen Nanopartikels f¨uhren zufallige thermische Fluktuationen zu ei-nem ersten Wachstum der martensitischen Phase (Abschnitt 6.2.1) und bestimmen somit denZeitpunkt, wann das ausbruchsartige Wachstum beginnt.

85
7 Simulationen metallischer Filme auf Substraten
Bei Festkorpern wurde gezeigt, daß die Austenittransformation mit zunehmender Temperaturauch in defektfreien Systemen auftritt, w¨ahrend die Martensittransformation mit abnehmenderTemperatur preexistierende Nukleationszentren erfordert. Nanopartikel zeichnen sich dadurchaus, daß dieUbergangstemperatur mit abnehmender Partikelgr¨oße abnimmt, wobei Partikel miteinigen Nanometern Durchmesser bereits nahe der Transformationstemperatur des Festk¨orpersliegen. Außerdem haben sich Nanopartikel als geeignet erwiesen, den Einfluß von Defektenverstandlich diskutieren zu k¨onnen. Bei d¨unnen Filmen auf Substraten kommt ein weitererAspekt hinzu, der auf dem Einfluß des Substrats beruht. Es wird in diesem Kapitel mit Hilfevon Molekulardynamik-Simulationen die Abh¨angigkeit der Stabilit¨at von Nichtgleichgewichts-strukturen auf kristallographisch verschieden orientierten Substraten in Form von d¨unnen nurwenige Monolagen umfassenden Schichten untersucht. Ein Schwerpunkt stellt dabei die Stabi-lisierung von kfz Fe-Filmen dar. Die Simulationen wurden in einem Temperaturbereich durch-gefuhrt, in dem Fe normalerweise krz ist (Abschnitt 5.1.1 und 5.1.2). Es werden sowohl Si-mulationen von d¨unnen (geschlossenen) Filmen auf Substratoberfl¨achen vorgestellt, als auchSimulationen von Inseln auf Substraten. Erg¨anzend werden Simulationen vorgestellt, die deninteressanten Unterschied zwischen einem thermischen Depositionsverfahren und einem ener-giereicheren Depositionsverfahren wie demPulsed-Laser-DepositionVerfahren [63] beschrei-ben (Abschnitt 2.4.3).
7.1 Voruberlegungen
Bevor die Ergebnisse f¨ur die unterschiedlichen Filme diskutiert werden, ist es sinnvoll, sich dieAhnlichkeit verschiedener Orientierungen zwischen Substrat und Film anzuschauen (Abb. 7.1).Dies ermoglicht es den Einfluß des Substrats und der kristallographischen Orientierung besserzu verstehen. Bedingt durch die unterschiedlichen Gitterkonstanten von Cu, Pd und Ag sinddie Atomabst¨ande auf der Substratoberfl¨ache unterschiedlich. Je nach kristallographischer Ori-entierung zwischen Substrat und krz Fe-Film sind die entsprechenden Atomabst¨ande des Sub-stratsahnlich den Atomabst¨anden des krz Films oder nicht. Die Ag(001)kfz-Substratoberfl¨achehat einen n¨achsten Nachbarabstand von 0.289 nm, welcher nur um 0.7 % gr¨oßer ist als dernachste Nachbarabstand einer Fe(001)krz-Ebene. Der n¨achste Nachbarabstand der Cu(111)kfz-Substratoberfl¨ache ist um 2.4 % gr¨oßer als der n¨achste Nachbarabstand einer Fe(110)krz-Ebene.Es ist daher nicht zu erwarten, daß Ag(001)kfz und Cu(111)kfz geeignet sind, kfz Fe zu stabi-lisieren, da das Substrat den Atomabstand von krz Fe fast ideal vorgibt und die krz Strukturrelativ unverspannt aufw¨achst. Dies steht im Einklang mit experimentellen Befunden. Auf derAg(001)-Oberflache wachst krz Fe in einer Bain-Orientierung auf, kfz Fe l¨aßt sich nicht stabi-lisieren [111], kfz Fe-Filme lassen sich nur wenige Monolagen auf einer Cu(111)-Oberfl¨achestabilisieren [77, 78]. Die Ag(111)-, Cu(001)-, Pd(001)- und Pd(111)-Oberfl¨ache gestatten eskfz Fe von einigen Monolagen zu stabilisieren [76,91,97–99].
Ein weiterer interessanter Aspekt h¨angt mit der Frage nach der Stabilit¨at der kfz Phasein Abhangigkeit von Temperatur und Schichtdicke zusammen. Abbildung 7.2 zeigt das struk-
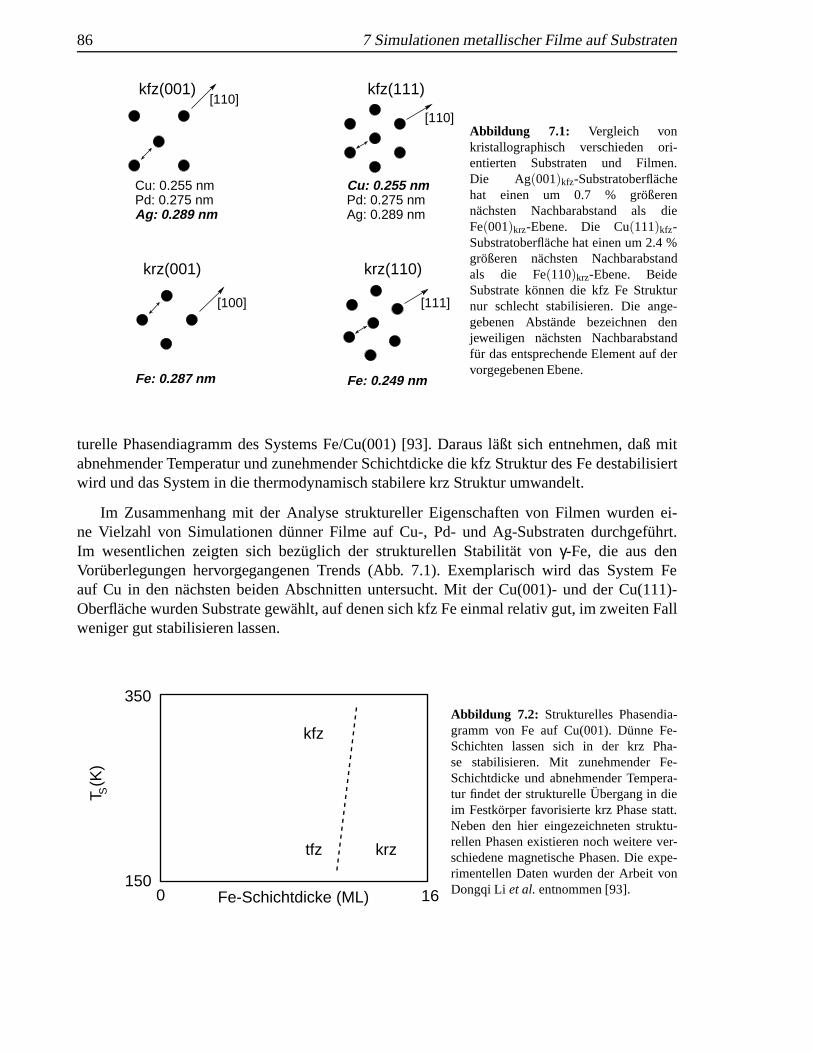
86 7 Simulationen metallischer Filme auf Substraten
[111]
kfz(001) kfz(111)
Cu: 0.255 nm Pd: 0.275 nm Ag: 0.289 nm
Fe: 0.249 nm Fe: 0.287 nm
krz(001) krz(110)
Cu: 0.255 nm Pd: 0.275 nm Ag: 0.289 nm
[100]
[110][110]
Abbildung 7.1: Vergleich vonkristallographisch verschieden ori-entierten Substraten und Filmen.Die Ag(001)kfz-Substratoberfl¨achehat einen um 0.7 % gr¨oßerennachsten Nachbarabstand als dieFe(001)krz-Ebene. Die Cu(111)kfz-Substratoberfl¨ache hat einen um 2.4 %großeren n¨achsten Nachbarabstandals die Fe(110)krz-Ebene. BeideSubstrate k¨onnen die kfz Fe Strukturnur schlecht stabilisieren. Die ange-gebenen Abst¨ande bezeichnen denjeweiligen nachsten Nachbarabstandfur das entsprechende Element auf dervorgegebenen Ebene.
turelle Phasendiagramm des Systems Fe/Cu(001) [93]. Daraus l¨aßt sich entnehmen, daß mitabnehmender Temperatur und zunehmender Schichtdicke die kfz Struktur des Fe destabilisiertwird und das System in die thermodynamisch stabilere krz Struktur umwandelt.
Im Zusammenhang mit der Analyse struktureller Eigenschaften von Filmen wurden ei-ne Vielzahl von Simulationen d¨unner Filme auf Cu-, Pd- und Ag-Substraten durchgef¨uhrt.Im wesentlichen zeigten sich bez¨uglich der strukturellen Stabilit¨at von γ-Fe, die aus denVoruberlegungen hervorgegangenen Trends (Abb. 7.1). Exemplarisch wird das System Feauf Cu in den n¨achsten beiden Abschnitten untersucht. Mit der Cu(001)- und der Cu(111)-Oberflache wurden Substrate gew¨ahlt, auf denen sich kfz Fe einmal relativ gut, im zweiten Fallweniger gut stabilisieren lassen.
S
350
T (
K)
0 Fe-Schichtdicke (ML) 16
krz
kfz
tfz
150
Abbildung 7.2: Strukturelles Phasendia-gramm von Fe auf Cu(001). D¨unne Fe-Schichten lassen sich in der krz Pha-se stabilisieren. Mit zunehmender Fe-Schichtdicke und abnehmender Tempera-tur findet der strukturelleUbergang in dieim Festkorper favorisierte krz Phase statt.Neben den hier eingezeichneten struktu-rellen Phasen existieren noch weitere ver-schiedene magnetische Phasen. Die expe-rimentellen Daten wurden der Arbeit vonDongqi Li et al.entnommen [93].

7.2 Fe auf Cu(001) 87
7.2 Fe auf Cu(001)
7.2.1 Geschlossene Fe-Filme
Die Abhangigkeit der Struktur von der Schichtdicke und der Temperatur wurde mittels Mo-lekulardynamik-Simulationen untersucht. Simulationszellen bestehend aus einer Cu-Schichtvon ca. 5 nm (30 Monolagen) und einer Fe-Schicht von bis zu 2.5 nm (15 Monolagen)(Abb. 7.3) wurden in der kfz Struktur initialisiert. Senkrecht zur Grenzfl¨ache Film/Substratwurde zus¨atzlich noch ein Vakuum erzeugt, so daß nur in Richtung in der Oberfl¨ache pe-riodische Randbedingungen wirksam waren. Es entstehen somit zwei freie Oberfl¨achen in(001)-Richtung, eine Cu-Oberfl¨ache und die in diesem Zusammenhang interessierende Fe-Oberflache. Unter Variation der Fe-Schichtdicke wurde bei verschiedenen Temperaturen un-
Fe 2.5 nm
5 nm
7.5 nm
Cu(001)
Abbildung 7.3: Schematische Darstellung der Simulations-zelle fur einen geschlossenen Fe-Film auf einer Cu(001)-Oberflache. Die Fe-Schichtdicke von 2.5 nm entspricht 15 Mo-nolagen. Periodische Randbedingungen wurden in den beidenin der Filmebene liegenden Richtungen verwendet. Die Anzahlder simulierten Atome betr¨agt ca. 40 000.
ter Verwendung des Nos´e-Hoover-Thermostaten und des Parinello-Rahman Verfahrens ¨ubereinen Zeitraum von 9 ps bis 40.5 ps simuliert. Anschließend wurde die Struktur des Fe-Filmsanalysiert (Abschnitt 2.2.3). Um den Einfluß von Defekten mit einbeziehen zu k¨onnen, wer-den zuerst Simulationen mit zuf¨allig verteilten Leerstellen in der Simulationszelle mit einerKonzentration von 2 % vorgestellt. Anschließend folgt eine Diskussion von Simulationen ohnevorinitialisierte Leerstellen in den Zellen.
Die Simulationen zeigen, daß in Abh¨angigkeit von der Temperatur die kfz Struktur desFe-Films instabil wird, und der Fe-Film teilweise in die krz Struktur umwandelt. Bereits nach9 psandert sich der Anteil an umgewandeltem Fe nicht mehr, eine weitere Relaxation ist bis zu40.5 ps nicht zu verzeichnen. Der Anteil des in die krz Struktur transformierten Fe-Films h¨angt
ML Fe 50 K 300 K
7 13.68 % 0.49 %15 43.28 % 0.03 %
Tabelle 7.1:Prozentanteil des in die krz Struktur umgewandeltenFe-Films auf Cu(001) mit einer Leerstellenkonzentration von 2 %nach einer Simulationszeit von 9 ps.

88 7 Simulationen metallischer Filme auf Substraten
stark von der Temperatur und von der Fe-Schichtdicke ab (Tab. 7.1). Mit zunehmender Schicht-dicke und abnehmender Temperatur w¨achst wie im Experiment der Anteil der krz Struktur an(vgl. Abb. 7.2). Die Zunahme des krz Anteils mit sinkender Temperatur ist durch die Zunah-me der chemischen Triebkraft zu erkl¨aren (Abschnitt 5.1.2). Die krz Struktur wird mit abneh-mender Temperatur gegen¨uber der kfz Struktur stabilisiert. Die Zunahme des krz Anteils mitzunehmender Schichtdicke hat im wesentlichen zwei Ursachen. Zum einen hat die Oberfl¨acheEinfluß auf die freie Energie des Systems. Die Oberfl¨achenenergien, die sich aus dem EAM-Potential fur Fe ergeben, favorisieren die kfz Struktur und verschieben die thermodynamischenGleichgewichtstemperaturen mit abnehmender Schichtdicke zugunsten der kfz Struktur (vgl.Abschnitt 6.1). Zum anderen spielt die Grenzfl¨ache zwischen Cu-Substrat und Fe-Film eineentscheidende Rolle. Die Vorgabe der kfz Struktur durch das Cu-Substrat bindet die ersten La-gen des Fe-Films an die kfz Struktur, bei einer Umwandlung in die krz Struktur entsteht einverzerrter Grenzbereich innerhalb der Fe-Schicht (Abb. 7.4). Beides sind Effekte deren Gr¨oßeim wesentlichen von der Fl¨ache abh¨angen und somit nicht von der Dicke des Films beein-flußt werden. Die chemische Triebkraft w¨achst mit dem Volumen und somit mit zunehmenderFilmdicke. Hierbei sei erw¨ahnt, daß zus¨atzlich durchgef¨uhrte Molekulardynamik-Simulationen
Abbildung 7.4: Struktur des Fe-Films auf einem Cu(001)-Substrat bei 150 K (links) und 1 K (rechts). Die Nuklea-tion von krz Bereichen (blau) innerhalb der kfz Schicht (orange) ist deutlich sichtbar. Der Anteil des umgewandel-ten Fe nimmt in Richtung des Cu-Substrats (unten im Bild) deutlich ab. Der Fe-Film ist schichtweise dargestellt,das Cu-Substrat ist nicht miteingezeichnet.

7.2 Fe auf Cu(001) 89
Abbildung 7.5: Pitsch-Orientierung eines krz Fe-Films auf einem Cu(001)-Substrat in der Simulation. Linksim Bild ist die ursprunglich initialisierte(001)kfz-Oberflache des Fe-Films mit einer eingezeichneten[010]kfz-Richtung (schwarzer Pfeil) und einer[110]kfz-Richtung (heller Pfeil) dargestellt. Nach der partiellen Transforma-tion des Fe-Films in die krz Struktur bilden sich krz Zwillinge, die bez¨uglich des Substrats Pisch-orientiert sind(Mitte), die schwarzen Pfeile markieren die[001]krz-Richtungen, der helle Pfeil markiert die[111]krz-Richtung.Die eingezeichneten verzerrten Hexagone sind typisch f¨ur die(110)krz-Ebenen. Rechts im Bild ist schematisch dieresultierende Pitsch-Orientierung zwischen Substrat (nicht gef¨ullte Kreise) und aufgewachsenem Film (gef¨ullteKreise) dargestellt.
freier Fe-Filme zeigen, daß austenitische Fe-Filme ohne das Cu-Substrat bereits bei 300 K abeiner Schichtdicke von 7 Monolagen vollst¨andig martensitisch umwandeln. Dies unterstreichtden großen Einfluß des Substrats auf die Struktur des aufgewachsenen Films.
Um einen dynamischen Eindruck von dem Umwandlungsprozeß von d¨unnen Filmen aufSubstraten zu erhalten, wurden die pr¨aparierten Simulationszellen (Abb. 7.3) ausgehend von300 K in Schritten von 5 K abgek¨uhlt und bei jeder Temperatur 0.375 ps lang simuliert. Beihoheren Temperaturen ist eine thermische Fluktuation von krz Keimen zu verzeichnen. DieVerteilung der fluktuierenden krz Keime ist noch unabh¨angig von der Entfernung zum Cu-Substrat. Zudem finden sich kleinere, einige Atome umfassende, stabile Keime, die an dievorgegebene Defektstruktur gebunden sind. Mit abnehmender Temperatur bilden sich stabile-re krz Bereiche, die im weiteren Verlauf wachsen. Der Anteil des krz Fe nimmt dabei in derNahe des Cu-Substrats ab. Auch bei tiefsten Temperaturen ist der Fe-Film nicht vollst¨andigin die krz Struktur umgewandelt, die dadurch entstehende Grenzfl¨ache zwischen krz und kfzStruktur in dem Fe-Film stellt sich als sehr rauh und eher fraktal dar (rechts in Abbildung7.4). Eine Analyse der fraktalen Eigenschaften von kfz-krz-Grenzfl¨achen wurde aber bishernicht durchgef¨uhrt. Die unvollstandige Transformation stimmt mit experimentellen Befundenuberein. W. Keuneet al.[90] beobachteten in Konversionselektronen-M¨oßbauer-Spektroskopie(CEMS) Experimenten an 42 Monolagen dicken Fe-Filmen auf Cu(001) ebenfalls eine unvoll-standige Transformation der tiefsten Fe-Lagen.
Untersuchungen der relaxierten Struktur ergaben in den Simulationen die auch im Experi-ment [76] gemessene Pitsch-Orientierungsbeziehung (Abschnitt 3.2.1) zwischen kfz Substratund gebildeter krz Phase (Abb. 7.5). In den Simulationen wird beobachtet, daß die(001)kfz-Ebene in die(011)krz-Ebene transformiert und die[110]kfz-Richtung in die[111]krz-Richtungubergeht. Es bilden sich zwei kristallographisch verschieden orientierte Bereiche der krz Struk-

90 7 Simulationen metallischer Filme auf Substraten
tur aus, die zwillig zueinander liegen. Das in den Simulationen auftretende Zwillingssystembestehend aus einer(112)-Zwillingsebene und einer[111]-Scherrichtung ist typisch f¨ur die krzStruktur (Abschnitt 3.1). Eine Variante hat jeweils eine typische Breite von 5-10(112)-Ebenen.Es bildet sich somit ein Streifenmuster auf der Oberfl¨ache aus. Die typische Breite von 5-10(112)krz-Ebenen einzelner Varianten blieb unter Reduzierung der Ausdehnung der Oberfl¨acheder Simulationszelle erhalten. Das deutet darauf hin, daß die Breite der Varianten kein Arte-fakt der periodischen Randbedingungen ist. Bildete sich in dem System beispielsweise nur eineVariante aus, so w¨urde durch die scherungsartige martensitische Transformation eine verzerr-te makroskopische Form des Films gegen¨uber dem Cu-Substrat entstehen. Der Mechanismusder Zwillingsbildung mit einer typischen Gr¨oße der auftretenden Varianten gleicht diese ma-kroskopische Verzerrung wieder aus (s. auch Abb. 3.7). Das Auftreten der streifigen Dom¨anenwird experimentell u. a. von Wuttiget al. [76] beobachtet.
Experimentelle Ergebnisse aus niederenergetischen Elektronenbeugungsexperimenten(LEED) zeigen vier verschiedene Varianten der Pitsch-orientierten krz Bereiche [76,94,112]. Inden Simulationen werden nur zwei verschiedene Varianten der krz Phase w¨ahrend eines Laufsgefunden. Es treten in der Simulation, bedingt durch Scherung entweder in[110]kfz oder in[110]kfz-Richtung, vier verschiedene Varianten in den Simulationen auf, aber immer nur zweigleichzeitig innerhalb einer Zelle (Abb. 7.5). Eine m¨ogliche Ursache hierf¨ur ist, daß die Simu-lationszellen zu klein sind, um weitere Varianten zuzulassen. Der Fe-Film in den Simulationenwurde als ideal glatt angenommen, experimentell w¨achst der Film nicht ideal glatt auf undverschiedene Varianten k¨onnen sich leichter unabh¨angig voneinnander ausbilden.
Simulationen f¨ur entsprechende Fe-Filme ohne Leerstellen zeigen Unterschiede zu den Si-mulationen mit Leerstellen. Der Anteil des umgewandelten Fe h¨angt stark von der Leerstellen-konzentration ab (vgl. Tab. 7.1 und 7.2). Filme, in denen keine Leerstellen initialisiert wurden,zeigen deutlich weniger Anteil des umgewandelten Fe. Die Struktur und die kristallographi-sche Orientierung von Substrat und Film ¨andern sich ebenfalls. Anstatt der experimentell be-obachteten Streifenanordnung martensitischer Varianten, bildet sich nun ein einfaches Quadrat-gitter, bestehend aus zwei verschiedenen Martensitvarianten. Die Umwandlung vollzieht sichnicht mehr scherungsartig, sondern ergibt die Orientierungsbeziehung der Bain-Transformation(Abb. 3.6). Die krz Bereiche bilden nun regelm¨aßig angeordnete Kan¨ale, die zum Cu-Substrathin dunner werden. Dies unterstreicht den Einfluß von Fehlern auf strukturelle Transformatio-nen in dunnen Filmen und gibt einen Hinweis auf die Abh¨angigkeit der strukturellen Stabilit¨atin Abhangigkeit der G¨ute des Filmherstellungsverfahrens.
ML Fe 50 K 300 K
7 1.32 % 0.00 %15 17.93 % 0.00 %
Tabelle 7.2:Prozentanteil des in die krz Struktur umgewandeltenFe-Films auf einer Cu(001)-Oberfl¨ache ohne initialisierte Leer-stellen nach einer Simulationszeit von 9 ps.
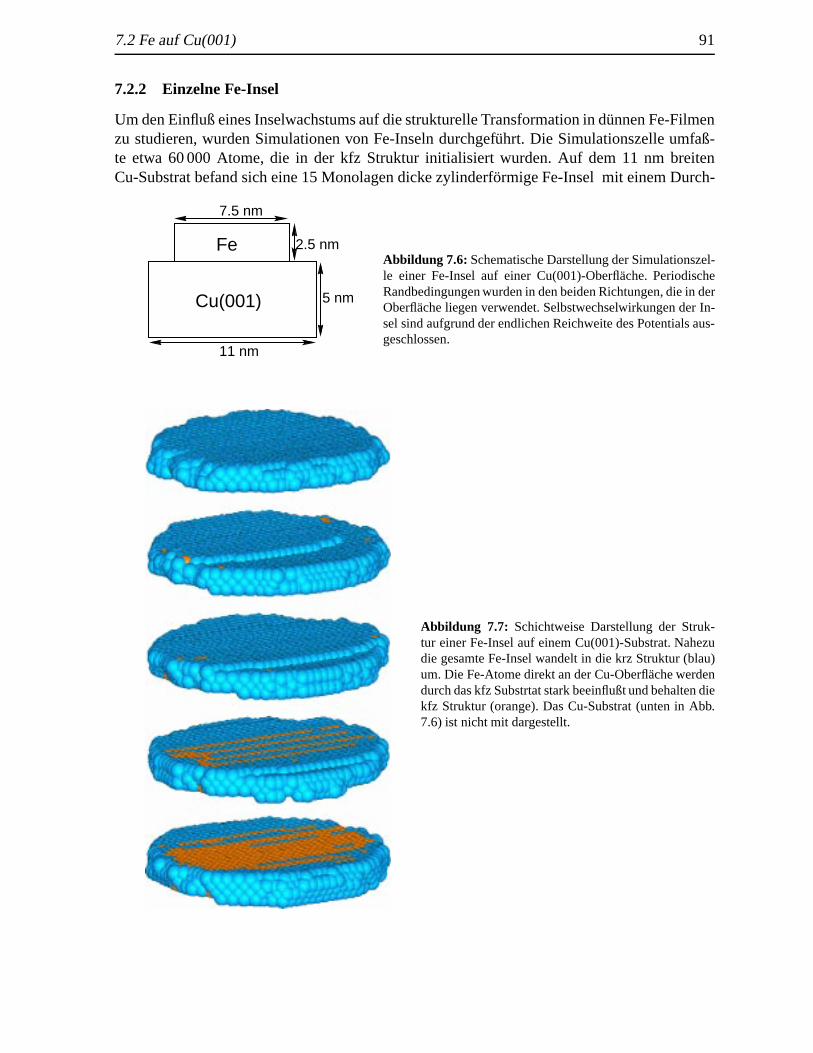
7.2 Fe auf Cu(001) 91
7.2.2 Einzelne Fe-Insel
Um den Einfluß eines Inselwachstums auf die strukturelle Transformation in d¨unnen Fe-Filmenzu studieren, wurden Simulationen von Fe-Inseln durchgef¨uhrt. Die Simulationszelle umfaß-te etwa 60 000 Atome, die in der kfz Struktur initialisiert wurden. Auf dem 11 nm breitenCu-Substrat befand sich eine 15 Monolagen dicke zylinderf¨ormige Fe-Insel mit einem Durch-
Cu(001)
Fe 2.5 nm
7.5 nm
11 nm
5 nm
Abbildung 7.6: Schematische Darstellung der Simulationszel-le einer Fe-Insel auf einer Cu(001)-Oberfl¨ache. PeriodischeRandbedingungen wurden in den beiden Richtungen, die in derOberflache liegen verwendet. Selbstwechselwirkungen der In-sel sind aufgrund der endlichen Reichweite des Potentials aus-geschlossen.
Abbildung 7.7: Schichtweise Darstellung der Struk-tur einer Fe-Insel auf einem Cu(001)-Substrat. Nahezudie gesamte Fe-Insel wandelt in die krz Struktur (blau)um. Die Fe-Atome direkt an der Cu-Oberfl¨ache werdendurch das kfz Substrtat stark beeinflußt und behalten diekfz Struktur (orange). Das Cu-Substrat (unten in Abb.7.6) ist nicht mit dargestellt.

92 7 Simulationen metallischer Filme auf Substraten
messer von 7.5 nm (Abb. 7.6). Unter Verwendung des Nos´e-Hoover-Thermostaten wurden 9 psbei 50 K simuliert. Im Gegensatz zu dem in Abschnitt 7.2.1 gefundenen Verhalten eines ge-schlossenen Fe-Films, transformiert die Fe-Insel fast vollst¨andig in die krz Struktur (Abb. 7.7).Lediglich die Lagen, die direkt an das Cu-Substrat angrenzen, behalten die kfz Struktur. DieInsel weist im Gegensatz zum Film ein h¨oheres Relaxationsverm¨ogen parallel zur Oberfl¨acheauf, wodurch die Transformation beg¨unstigt wird.
Die umgewandelte Insel besitzt wie der geschlossene Fe-Film zwei zwillig zueinander ori-entierte krz-Bereiche, die ein streifen¨ahnliches Muster auf der Oberfl¨ache bilden. Eine Variantehat dabei ¨ahnliche Ausmaße (5-10(112)krz-Ebenen) wie im Fall des geschlossenen Fe-Films.Die Insel weist eine leichte nach oben zunehmende Verj¨ungung auf. Die Abst¨ande der n¨achstenNachbarn der krz Fe sind etwa um 2.5 % geringer als die des kfz Cu-Substrats, welches dieVerjungung als elastische Relaxation erkl¨art.
7.3 Fe auf Cu(111)
7.3.1 Geschlossene Fe-Filme
Der Einfluß der kristallographischen Orientierung des Substrats wird anhand des Beispiels Feauf Cu(111) dargestellt. Hierzu wurden analog zu Abschnitt 7.2.1 Simulationszellen pr¨apariert,die einen Fe-Film einer Dicke von bis zu 2.5 nm (14 Monolagen) auf einem Cu(111)-Substratbeinhalteten (vgl. Abb. 7.3). Um Fehler beim Filmwachstum zu ber¨ucksichtigen, wurden Leer-stellen mit einer Konzentration von 2 % zuf¨allig im System verteilt. Unter Verwendung desNose-Hoover-Thermostaten und des Parinello-Rahman-Verfahrens wurden Simulationen beiverschiedenen Temperaturen f¨ur verschiedene Fe-Schichtdicken durchgef¨uhrt. Senkrecht zurGrenzflache von Substrat und Film wurde ein Vakuum eingebaut, so daß periodische Randbe-dingungen nur in den beiden parallel zur Oberfl¨ache liegenden Richtungen wirksam waren.
Ahnlich wie bei Simulationen von geschlossenen Fe-Filmen auf Cu(001)-Substraten (Ab-schnitt 7.2.1) zeigt sich eine partielle Transformation der Fe-Schicht in die krz-Struktur. DerAnteil des umgewandelten Fe h¨angt dabei von der Temperatur und der Fe-Schichtdicke ab (Tab.7.3). Wiederum ist zu erkennen, daß mit zunehmender Fe-Schichtdicke und abnehmender Tem-peratur der Anteil des umgewandelten Fe steigt (vgl. Aschnitt 7.2.1).
Eine Analyse der umgewandelten Struktur des Films ergibt zwei zwillig zueinander lie-gende krz Varianten, die in der Kurdjumov-Sachs-Orientierung zum Cu(111)-Substrat liegen(Abb. 7.8). Die(111)kfz-Ebene transformiert dabei in eine(110)krz-Ebene und eine dicht ge-
ML Fe 50 K 300 K
6 8.43 % 1.10 %14 91.67 % 1.46 %
Tabelle 7.3:Prozentanteil des in die krz Struktur umgewandeltenFe-Films auf Cu(111) mit einer Leerstellenkonzentration von 2 %nach einer Simulationszeit von 9 ps.

7.3 Fe auf Cu(111) 93
Abbildung 7.8: Die Bildung von zwilligen krz Berei-chen mit Kurdjumov-Sachs-Orientierung in einem Fe-Films auf einer Cu(111)-Oberfl¨ache kann in den Simu-lationen beobachtet werden. Gef¨ullte Kreise markierendie verzerrten krz(110)-Hexagone, die nicht gef¨ulltenKreise die kfz(111)-Hexagone des Substrats.
packte[110]kfz-Richtung transformiert ebenfalls in eine dicht gepackte[111]krz-Richtung. Diebeiden krz Varianten bilden wie im Fall des Cu(001)-Substrats (Abschnitt 7.2) Streifen aus,die entlang der dicht gepackten Richtung des Substrats liegen. Experimentelle Untersuchun-gen [77, 78, 113] ergeben ebenfalls die hier beobachtete Kurdjumov-Sachs-Orientierung vonSubstrat und krz transformiertem Fe-Film unter der Ausbildung von drei mal zwei zwillig zu-einander liegender Varianten entlang der drei gleichberechtigten dicht gepackten Richtungenin der Cu(111)-Oberfl¨ache. Experimentelle Analysen in [78] und [64] zeigen diese Zwillingejeweils als streifige Dom¨anen, die als Inseln voneinander separiert sind. In diesem Zusammen-hang ist einzusehen, daß in den Simulationen von geschlossenen Filmen nur zwei verschiedeneVarianten auftreten, und nicht die experimentell gefundenen sechs oder vier f¨ur Fe-Filme aufCu(111)- bzw. Cu(001)-Substraten.
Der Vergleich des umgewandelten Fe-Anteils auf einem Cu(111)-Substrat und einemCu(001)-Substrat (vgl. Tab. 7.3 und 7.1) zeigt bei Schichtdicken von 14-15 Monolagen,einen wesentlich h¨oheren Anteil an umgewandeltem Fe auf dem Cu(111)-Substrat als aufdem Cu(001)-Substrat. Experimentelle Untersuchungen best¨atigen, daß d¨unne Fe-Filme aufCu(111)-Substraten bereits nach wenigen Monolagen in die krz Struktur umwandeln, wohin-gegen Fe-Filme auf Cu(001)-Substratoberfl¨achen erst bei deutlich h¨oheren Bedeckungen diestrukturelle Transformation von kfz in die krz Struktur vollziehen [76–78,94,112,113].
7.3.2 Aufwachsprozesse
Neuere experimentelle Untersuchungen von d¨unnen Fe-Schichten auf Cu(111)-Substratenergeben deutliche Unterschiede bez¨uglich der Stabilitat der aufwachsenden kfz Phase inAbhangigkeit des Aufwachsverfahrens [64]. Thermisch deponierte Fe-Filme auf Cu(111) zei-gen ein inselartiges Wachstum und zeigen bereits ab einer nominalen Bedeckung von dreibis vier Monolagen eine strukturelle Transformation von der kfz Struktur in die krz Struk-tur [64, 114]. Mit Hilfe des energiereichenPulsed-Laser-DepositionVerfahrens aufgewachse-ne Filme zeigen ein gutes Lagenwachstum. Die Stabilit¨at der kfz Struktur auf dem Cu(111)-Substrat nimmt deutlich zu im Vergleich zu thermisch deponierten Filmen.
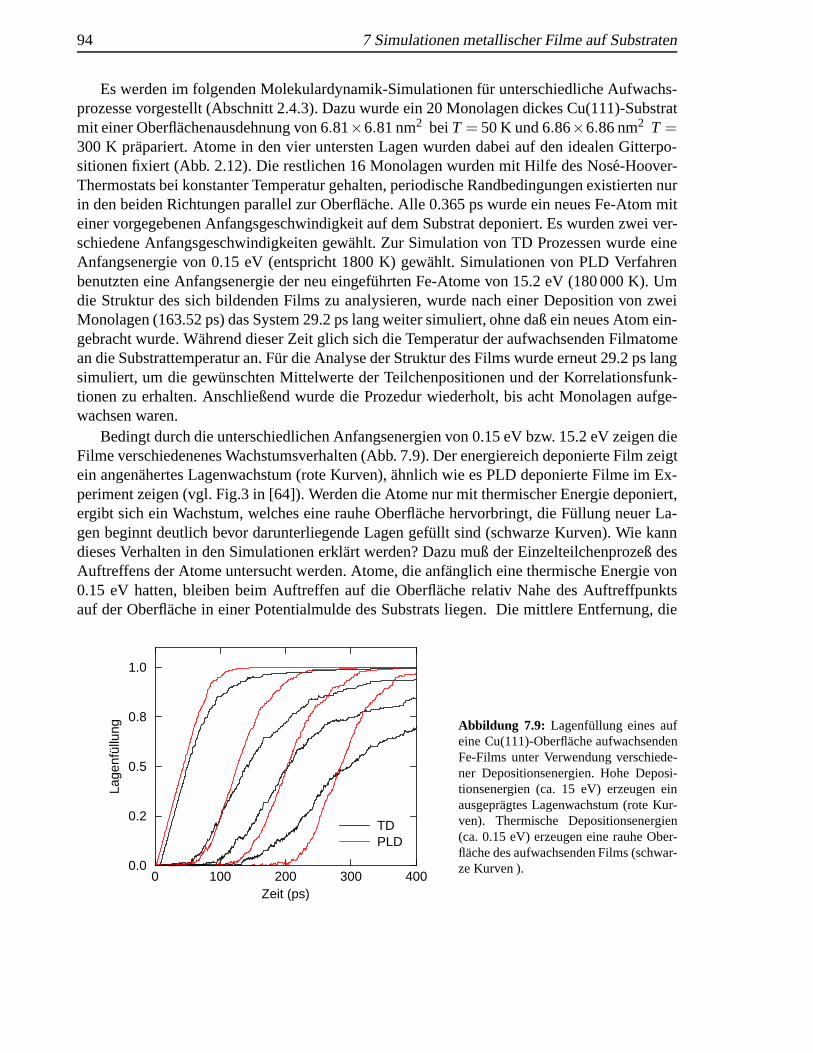
94 7 Simulationen metallischer Filme auf Substraten
Es werden im folgenden Molekulardynamik-Simulationen f¨ur unterschiedliche Aufwachs-prozesse vorgestellt (Abschnitt 2.4.3). Dazu wurde ein 20 Monolagen dickes Cu(111)-Substratmit einer Oberflachenausdehnung von 6.81×6.81 nm2 beiT = 50 K und 6.86×6.86 nm2 T =300 K prapariert. Atome in den vier untersten Lagen wurden dabei auf den idealen Gitterpo-sitionen fixiert (Abb. 2.12). Die restlichen 16 Monolagen wurden mit Hilfe des Nos´e-Hoover-Thermostats bei konstanter Temperatur gehalten, periodische Randbedingungen existierten nurin den beiden Richtungen parallel zur Oberfl¨ache. Alle 0.365 ps wurde ein neues Fe-Atom miteiner vorgegebenen Anfangsgeschwindigkeit auf dem Substrat deponiert. Es wurden zwei ver-schiedene Anfangsgeschwindigkeiten gew¨ahlt. Zur Simulation von TD Prozessen wurde eineAnfangsenergie von 0.15 eV (entspricht 1800 K) gew¨ahlt. Simulationen von PLD Verfahrenbenutzten eine Anfangsenergie der neu eingef¨uhrten Fe-Atome von 15.2 eV (180 000 K). Umdie Struktur des sich bildenden Films zu analysieren, wurde nach einer Deposition von zweiMonolagen (163.52 ps) das System 29.2 ps lang weiter simuliert, ohne daß ein neues Atom ein-gebracht wurde. W¨ahrend dieser Zeit glich sich die Temperatur der aufwachsenden Filmatomean die Substrattemperatur an. F¨ur die Analyse der Struktur des Films wurde erneut 29.2 ps langsimuliert, um die gew¨unschten Mittelwerte der Teilchenpositionen und der Korrelationsfunk-tionen zu erhalten. Anschließend wurde die Prozedur wiederholt, bis acht Monolagen aufge-wachsen waren.
Bedingt durch die unterschiedlichen Anfangsenergien von 0.15 eV bzw. 15.2 eV zeigen dieFilme verschiedenenes Wachstumsverhalten (Abb. 7.9). Der energiereich deponierte Film zeigtein angen¨ahertes Lagenwachstum (rote Kurven), ¨ahnlich wie es PLD deponierte Filme im Ex-periment zeigen (vgl. Fig.3 in [64]). Werden die Atome nur mit thermischer Energie deponiert,ergibt sich ein Wachstum, welches eine rauhe Oberfl¨ache hervorbringt, die F¨ullung neuer La-gen beginnt deutlich bevor darunterliegende Lagen gef¨ullt sind (schwarze Kurven). Wie kanndieses Verhalten in den Simulationen erkl¨art werden? Dazu muß der Einzelteilchenprozeß desAuftreffens der Atome untersucht werden. Atome, die anf¨anglich eine thermische Energie von0.15 eV hatten, bleiben beim Auftreffen auf die Oberfl¨ache relativ Nahe des Auftreffpunktsauf der Oberfl¨ache in einer Potentialmulde des Substrats liegen. Die mittlere Entfernung, die
0 100 200 300 400Zeit (ps)
0.0
0.2
0.5
0.8
1.0
Lage
nfül
lung
TDPLD
Abbildung 7.9: Lagenfullung eines aufeine Cu(111)-Oberfl¨ache aufwachsendenFe-Films unter Verwendung verschiede-ner Depositionsenergien. Hohe Deposi-tionsenergien (ca. 15 eV) erzeugen einausgepr¨agtes Lagenwachstum (rote Kur-ven). Thermische Depositionsenergien(ca. 0.15 eV) erzeugen eine rauhe Ober-flache des aufwachsenden Films (schwar-ze Kurven ).

7.3 Fe auf Cu(111) 95
Abbildung 7.10: Vergleich von thermisch deponierten (links) und energiereich deponierten (rechts) Fe-Filmen aufeinem Cu(111)-Substrat nach einer nominalen Deposition von vier Monolagen. Die Substratatome sind dunkel, dieaufgewachsenen Fe-Atome hell gezeichnet. Der thermisch deponierte Film zeigt im Gegensatz zum energiereichdeponierten Film bereits Verzerrungen der hexagonalen Struktur auf, welche auf die krz Struktur hinweisen.
Abbildung 7.11: Vergleich von thermisch deponierten (links) und energiereich deponierten (rechts) Fe-Filmen aufeinem Cu(111)-Substrat nach einer nominalen Deposition von acht Monolagen. Die Substratatome sind dunkel,die aufgewachsenen Fe-Atome hell gezeichnet. Der thermisch deponierte Film zeigt im Gegensatz zum energie-reich deponierten Fe-Film die krz Struktur, wobei zwischen Substrat und Film die Kurdjumov-Sachs-Beziehungbesteht.
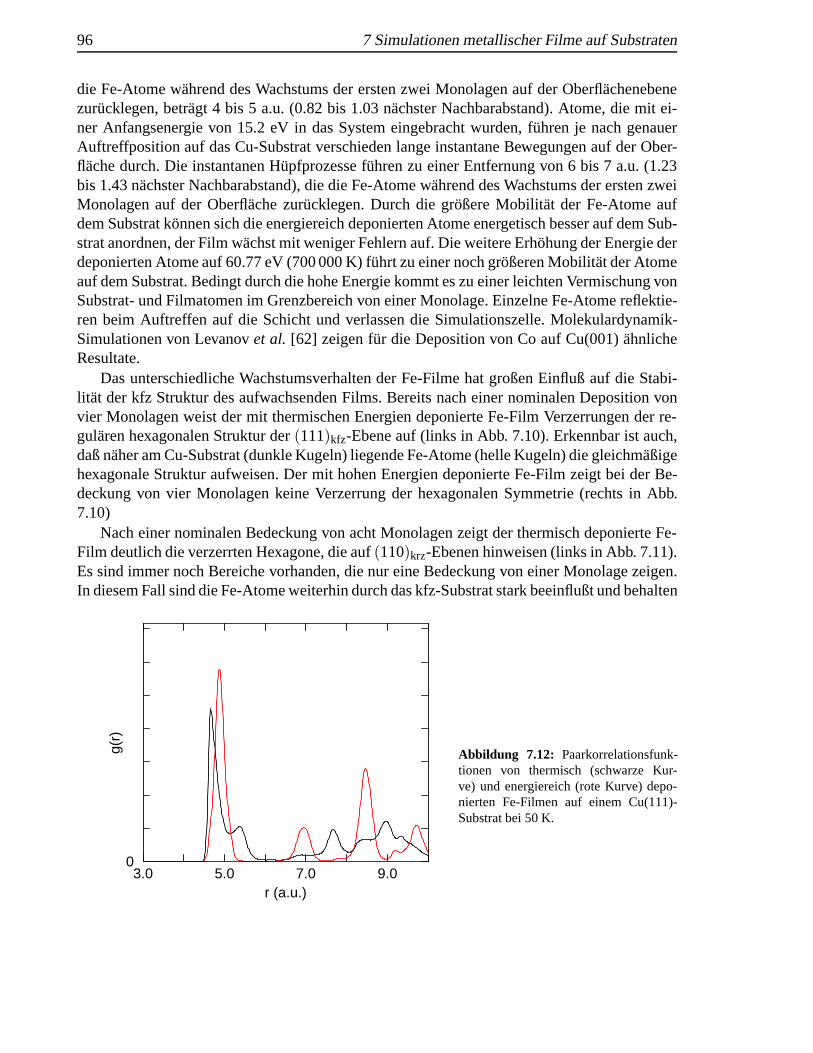
96 7 Simulationen metallischer Filme auf Substraten
die Fe-Atome w¨ahrend des Wachstums der ersten zwei Monolagen auf der Oberfl¨achenebenezurucklegen, betr¨agt 4 bis 5 a.u. (0.82 bis 1.03 n¨achster Nachbarabstand). Atome, die mit ei-ner Anfangsenergie von 15.2 eV in das System eingebracht wurden, f¨uhren je nach genauerAuftreffposition auf das Cu-Substrat verschieden lange instantane Bewegungen auf der Ober-flache durch. Die instantanen H¨upfprozesse f¨uhren zu einer Entfernung von 6 bis 7 a.u. (1.23bis 1.43 nachster Nachbarabstand), die die Fe-Atome w¨ahrend des Wachstums der ersten zweiMonolagen auf der Oberfl¨ache zur¨ucklegen. Durch die gr¨oßere Mobilitat der Fe-Atome aufdem Substrat k¨onnen sich die energiereich deponierten Atome energetisch besser auf dem Sub-strat anordnen, der Film w¨achst mit weniger Fehlern auf. Die weitere Erh¨ohung der Energie derdeponierten Atome auf 60.77 eV (700 000 K) f¨uhrt zu einer noch gr¨oßeren Mobilitat der Atomeauf dem Substrat. Bedingt durch die hohe Energie kommt es zu einer leichten Vermischung vonSubstrat- und Filmatomen im Grenzbereich von einer Monolage. Einzelne Fe-Atome reflektie-ren beim Auftreffen auf die Schicht und verlassen die Simulationszelle. Molekulardynamik-Simulationen von Levanovet al. [62] zeigen fur die Deposition von Co auf Cu(001) ¨ahnlicheResultate.
Das unterschiedliche Wachstumsverhalten der Fe-Filme hat großen Einfluß auf die Stabi-lit at der kfz Struktur des aufwachsenden Films. Bereits nach einer nominalen Deposition vonvier Monolagen weist der mit thermischen Energien deponierte Fe-Film Verzerrungen der re-gularen hexagonalen Struktur der(111)kfz-Ebene auf (links in Abb. 7.10). Erkennbar ist auch,daß naher am Cu-Substrat (dunkle Kugeln) liegende Fe-Atome (helle Kugeln) die gleichm¨aßigehexagonale Struktur aufweisen. Der mit hohen Energien deponierte Fe-Film zeigt bei der Be-deckung von vier Monolagen keine Verzerrung der hexagonalen Symmetrie (rechts in Abb.7.10)
Nach einer nominalen Bedeckung von acht Monolagen zeigt der thermisch deponierte Fe-Film deutlich die verzerrten Hexagone, die auf(110)krz-Ebenen hinweisen (links in Abb. 7.11).Es sind immer noch Bereiche vorhanden, die nur eine Bedeckung von einer Monolage zeigen.In diesem Fall sind die Fe-Atome weiterhin durch das kfz-Substrat stark beeinflußt und behalten
3.0 5.0 7.0 9.0r (a.u.)
0
g(r)
Abbildung 7.12: Paarkorrelationsfunk-tionen von thermisch (schwarze Kur-ve) und energiereich (rote Kurve) depo-nierten Fe-Filmen auf einem Cu(111)-Substrat bei 50 K.

7.3 Fe auf Cu(111) 97
ihre hexagonale Symmetrie. Auch bei einer Bedeckung von acht Monolagen zeigt der energie-reich aufgebrachte Fe-Film eine hexagonale Symmetrie. Eine Umwandlung in die krz Strukturfindet aber nicht statt.Ahnlich wie im Fall der Fe-Insel (Abschnitt 7.2.2) ist die Umwandlungbei den thermisch aufgebrachten Filmen durch die gr¨oßere Freiheit parallel zur Oberfl¨ache desSystems m¨oglich.
Die quantitative Analyse der Struktur der aufgewachsenen Fe-Filme nach einer nomina-len Bedeckung von 8 Monolagen mit Hilfe der Paarkorrelationsfunktion zeigt den thermischaufgewachsenen Film als nahezu ideale krz Struktur (schwarze Kurve in Abb. 7.12). Es sindnur sehr schwache Maxima der kfz Struktur erhalten geblieben, die durch die untersten an dasCu-Substrat angrenzenden Fe-Lagen erzeugt werden (vgl. links in Abb. 7.11). Der energie-reich aufgewachsenen Fe-Film zeigt die f¨ur die kfz Struktur typischen Maxima mit lediglichkleinsten Abweichungen von der kfz Struktur (rote Kurve in Abb. 7.12).

98
8 Zusammenfassung der Ergebnisse
In der vorliegenden Arbeit wurden Molekulardynamik-Simulationen von Festk¨orpern, Nano-partikeln und Filmen vorgestellt. Unter Verwendung derEmbedded-Atom Methodwurden dieSysteme Fe-Ni und Fe-Cu untersucht. Mittels der Simulationen wurden strukturelle Eigen-schaften in Festk¨orpern, Partikeln und d¨unnen Filmen betrachtet, wobei der Schwerpunkt in derUntersuchung von strukturellen Transformationen wie der martensitischen und austenitischenTransformation lag. Die Bestimmung der freien Energie in Abh¨angigkeit von der Tempera-tur erlaubte die Analyse der auftretenden strukturellen Phasen¨ubergange zwischen einer dichtgepackten Struktur (kfz, hdp) und der krz Struktur. Dabei stellten sich Gitterdefekte als be-stimmender Faktor f¨ur martensitische Transformationen bei tiefen Temperaturen in allen dreiSystemklassen heraus. Die Durchf¨uhrung von Simulationen mit bis zu acht millionen Ato-men ergab einen Einblick in homogene und heterogene Nukleationsprozesse struktureller Pha-senubergange. Die Untersuchung der kristallographischen Orientierungsbeziehungen zwischenden Phasen vor und nach der Umwandlung gab Aufschluß ¨uber den Transformationsprozeßauf atomare Skala. Der gesamte in den Simulationen auftretende Transformationsprozeß l¨aßtsich analog der Wechsler-Liebermann-Read-Theorie mathematisch durch eine Kombinationaus Bain-Transformation, Rotation und gitterinvarianter Scherung bedingt durch Stapelfehlerund Zwillingsbildung darstellen.
8.1 Festkorper
Die Simulationen haben gezeigt, daß der austenitische Phasen¨ubergang bei hohen Temperatu-ren auch ohne das Einbeziehen von Gitterdefekten auftritt. Martensitische Transformationenbei tiefen Temperaturen werden in den Simulationen nur bei der Ber¨ucksichtigung von Git-terdefekten beobachtet. Die Berechnung der freien Energien der krz und kfz Strukturen inAbhangigkeit der Temperatur bot eine Grundlage f¨ur die Diskussion der im Rahmen dieserArbeit behandelten strukturellen Phasen¨ubergange. Der Verlauf der freien Energie entlang desBain-Wegs gibt Aufschluß ¨uber den Unterschied von austenitischer und martensitischer Trans-formation. Wahrend bei hohen Temperaturen entlang des Transformationswegs von der mar-tensitischen krz Struktur in die austenitische kfz Struktur keine Energiebarriere vorhanden ist,zeigt sich bei tiefen Temperaturen von der austenitischen Struktur in die martensitische Struktureine Energiebarriere entlang des Transformationswegs. Bei tiefen Temperaturen existiert nebender thermodynamisch stabilen martensitischen Phase eine metastabile austenitische Phase, wasden großen Einfluß von Defekten und preexistierenden martensitischen Keimen auf den mar-tensitischenUbergang bei tiefen Temperaturen unterstreicht.
Simulationen von schockinduzierten austenitschen Tranformationen von perfekt martensi-tischen krz Fe-Einkristallen gaben Aufschluß ¨uber den Mechanismus der homogenen Keimbil-dung innerhalb der festen Phase. Mit zunehmender Schockst¨arke, d. h. mit zunehmender che-mischer Triebkraft geht das Wachstum der neuen Phase von einer wachsenden Anzahl von klei-nen thermisch aktivierten Keimen aus. Beim Zusammenwachsen der Keime bilden sich nichtkoharente Korngrenzen aus. Das Ausheilen von Korngrenzen vollzieht sich bei den untersuch-

8.2 Nanopartikel 99
ten Nichtgleichgewichtsverh¨altnissen auf der Picosekundenskala und geschieht mit zunehmen-der Schockst¨arke schneller. Der Druck, bei dem sich schockbedingt in den Simulationen einestrukturelle Transformation einstellt, liegt deutlich ¨uber dem experimentell ermitteltem Druck.Dies hat im wesentlichen zwei Ursachen. Erstens sind die Simulationen an idealen Kristall-strukturen ohne Fehler durchgef¨uhrt worden, so daß heterogene Keimbildung von vornehereinausgeschlossen ist. Zweitens zeigte sich, daß das verwendete EAM-Potential den Druck unterKompression ¨ubersch¨atzt. Am Beispiel eines EAM-Potentials f¨ur Aluminium wurde deutlichgemacht, daß eine explizite zus¨atzliche Anpassung der EAM-Potentiale an die unter Verwen-dung vonab initio Methoden bestimmteE(V)-Abhangigkeit in diesem Zusammenhang sinn-voll ist. Erste Versuche, ein Potential f¨ur Eisen unter zus¨atzlicher Ber¨ucksichtigung der Volu-menabh¨angigkeit der Energie, die anhand vonab initio Methoden bestimmt wurde [115, 116],anzupassen, waren zwar erfolgreich in Bezug auf die Anpassung der Daten, zeigten aber in denSimulationen keinen temperaturgetriebenen strukturellenUbergang der krz Struktur in die kfzStruktur. Das heißt die Optimierung der EAM-Potentiale ist nach wie vor ein subtiler Punkt, dadie Berucksichtigung (neben experimentellen Daten) vonab initio Rechnungen nicht immer zueinem besseren Potential f¨uhrt.
8.2 Nanopartikel
Am Beispiel von Fe80-Ni20-Nanopartikeln wurde die Abh¨angigkeit der strukturellenUbergangstemperaturen von der Partikelgr¨oße untersucht. Es zeigte sich eine deutliche Abnah-me derUbergangstemperaturen mit abnehmender Partikelgr¨oße. Im Rahmen der verwendetenEAM-Potentiale konnte dieser Sachverhalt durch eine Modifikation der freien Energie einesPartikels durch den unterschiedlichen Oberfl¨achenbeitrag der austenitischen und der marten-sitischen Phase erkl¨art werden. Martensitische(100)krz- und(110)krz-Oberflachen haben einehohere Oberfl¨achenenergie als austenitische(100)kfz- bzw. (111)kfz-Oberflachen. In kleinenPartikeln mit einer Ausdehnung von einigen Nanometern wird dadurch der Austenit favorisiert.Ob die experimentell beobachtete Abnahme der martensitischen StarttemperaturMS auf denUnterschied zwischen Oberfl¨achenenergien von vergleichbaren Oberfl¨achen wie der(100)krz-und der(100)kfz-Oberflache bzw. der(110)krz- und der(111)kfz-Oberflache zur¨uckzufuhrenist, oder in der Ausbildung von energetisch ung¨unstigen Oberfl¨achen des Martensits [117] zubegrunden ist, kann nicht abschließend gekl¨art werden.
Aus diesem Grund konnte die Untersuchung von martensitischen Phasen¨ubergangen erstbei großeren Partikeln von einigen 10 Nanometern untersucht werden. Hier zeigten Simula-tionen fur große Systeme (million Atome) ¨uber einen Zeitraum von 70 ps erneut den Einflußvon Defekten als bestimmenden Faktor f¨ur die Nukleation von Martensit. Die heterogene Nu-kleation an unterschiedlichen Defekten wie Ecken und fl¨achenhaften Defekten zeigen jeweilsahnliches Verhalten. Ausgehend von der Oberfl¨ache findet ein ausbruchsartiges Wachstum vonkleinen Keimen ins Innere des Partikels statt. Es bilden sich verschieden orientierte Variantendes Martensits, die ein streifenartiges Muster in der Austenitmatrix ergeben. Das Anwachsendes Martensits f¨uhrt zu konkurrierendem Verhalten der verschiedenen Martensitvarianten un-tereinander. Die Umwandlung des noch verbleibenden Rests an Austenit geschieht im Vergleichzu dem ausbruchsartigen Anfang der Umwandlung viel langsamer. Als Resultat ergibt sich ein

100 8 Zusammenfassung der Ergebnisse
aus wenigen großen Varianten aufgebauter martensitischer Kristall. Ob noch vorhandene Zwil-lingsgrenzen bei l¨angeren Simulationeszeiten aus dem System gedr¨angt werden, und somit einEinkristall entsteht, ist offen.
8.3 Dunne Filme
Das Zusammenspiel zwischen der Struktur der Substratoberfl¨ache und der Struktur der Fil-moberflache wurde am experimentell h¨aufig verwendeten Modellsystem Fe auf Cu untersucht.Dabei wurden die experimentellen Sachverhalte wie die Umwandlung der kfz Struktur in diekrz Struktur mit zunehmender Filmdicke und abnehmender Temperatur best¨atigt, und im Rah-men des benutzten Modells verstanden.
Je nachdem, ob ein geschlossener Fe-Film oder eine Fe-Insel auf einer Cu(001)-Substratoberfl¨ache simuliert wurde, ergab sich eine unterschiedliche Grenzfl¨achentopologie derkrz und kfz Struktur in der N¨ahe des Substrats. Der geschlossene Film zeigte eine sehr rauheGrenzflache zwischen der krz Struktur und der kfz Struktur, die Grenzfl¨ache erstreckte sichuber mehrere Monolagen. Die Insel zeigte nur eine Grenzfl¨ache von sehr wenigen Monolagennahe der Substratoberfl¨ache. Beiden F¨allen ist gemeinsam, daß nicht der gesamte Fe-Film indie krz-Struktur umwandelt, sondern daß nahe des Cu-Substrats der Film nicht vollst¨andig indie krz Struktur umwandelt. Dies best¨atigen experimentelle Konversionselektronen-M¨oßbauer-Spektroskopie (CEMS) Untersuchungen, die eine nicht vollst¨andige Umwandlung des Fe na-he des Cu-Substrats zeigen [90]. Sowohl im Experiment als auch in der Simulation findetman das streifige Ausbilden von zwilligen krz Varianten in Pitsch-Orientierung zum Substrat.Dies laßt sich auf die guteUbereinstimmung des Atomabstands von Cu[110]kfz und Fe[111]krzund die Fehlanpassung der Atomreihen senkrecht zu diesen Richtungen befindlichen Ebenenzuruckfuhren.
Der Vergleich von Simulationen von Fe/Cu(001) mit Simulationen von Fe/Cu(111) ergabeinen deutlichen Anstieg des Anteils von in die krz Struktur umgewandeltem Fe. Die rela-tiv gute Ubereinstimmung der Cu(111)kfz-Ebene und der Fe(110)krz-Ebene besonders ent-lang der dicht gepackten Richtungen, kann als Argument f¨ur die verstarkte Umwandlung indie krz Struktur angef¨uhrt werden. Die auch im Experiment festgestellte Kurdjumov-Sachs-Orientierungsbeziehung ist das Resultat der sehr gutenUbereinstimmung des n¨achsten Nach-barabstands von kfz Cu und krz Fe.
Die Simulation des Aufwachsprozesses der Fe-Atome auf das Cu(111)-Substrat zeigte deut-lich die Abhangigkeit des Wachstumsprozesses des Films von der kinetischen Energie, mit derdie Atome auf das Substrat aufgebracht wurden. Thermische Energien der eingebrachten Ato-me fuhrten dazu, daß der Film rauh aufw¨achst und die Filmqualit¨at somit schlecht ist. Da-durch wurde der strukturelleUbergang der Fe-Filme von der kfz Struktur in die krz Strukturbereits bei einigen Monolagen vollzogen. Fe-Atome, die mit Energien von einigen Elektro-nenvolt in das System eingebracht wurden, bildeten relativ glatte, fehlerfreie Filme, die inder kfz Phase wesentlich stabiler sind als die Filme, die aus thermisch deponierten Atomenentstanden sind. Der Vergleich mit neuesten Experimenten mit thermisch deponierten Filmenund mittelsPulsed-Laser-Depositionerzeugter Filme [64] zeigt eine guteUbereinstimmungbzgl. des Wachstumsverhaltens und der strukturellen Stabilit¨at der kfz Phase der aufgewach-

8.4 Ausblick 101
senen Fe-Filme. In den Molekulardynamik-Simulationen kann aufgrund der extrem hohenDepositionsraten nicht zwischen dem Unterschied der instantanen Depositionsraten von ther-mischen Depositionsverfahren desPulsed-Laser-DepositionVerfahrens unterschieden werden.Die unterschiedliche Mobilit¨at der Atome auf der Substratoberfl¨ache kommt im wesentlichennur durch die von der Energie abh¨angigen instantanen H¨upfprozesse zustande. Das unter-schiedliche Diffusionsverhalten in Abh¨angigkeit von instantanen Depositionsraten kann durchdie Untersuchung von Wachstumsmodellen, unter Verwendung von kinetischen Monte-Carlo-Simulation [65], in Erg¨anzung zu den Molekulardynamik-Simulationen studiert werden.
8.4 Ausblick
Viele strukturelle Eigenschaften von Metallen konnten unter Benutzung derEmbedded-AtomMethodgut wiedergegeben werden. W¨ahrend die unter Verwendung von Molekulardynamik-Simulationen erfassten Zeitbereiche ausreichen, um beispielsweise Schockwellen zu simulie-ren, konnten Diffusionsvorg¨ange auf Oberfl¨achen nicht ausreichend beschrieben werden, dadie von den Simulationen erfaßte Zeit nur eine knappe Nanosekunde umfaßt. Ans¨atze, die-ses Problem zu l¨osen, gibt es in der Literatur in Form derHyperdynamics, die das Auftretenseltener Prozesse wie Diffusionspr¨unge durch eine Modifikation des Potentials erh¨oht [118].Dies konnte aber im Rahmen dieser Arbeit nicht mehr implementiert werden und sollte aber inzukunftigen Arbeiten ber¨ucksichtigt werden.
Die Große der untersuchten Simulationszellen, die bis zu acht millionen Atome umfassten,konnte bereits gut die Bildung von kristallographisch verschieden orientierten Kristallbereichenbeschreiben. Noch gr¨oßere Zellen sind m¨oglicherweise n¨otig, um den Einfluß von Gitterfehlernwie Korngrenzen auf die Nukleationsprozesse analysieren zu k¨onnen. Erste von T. C. Germanndurchgefuhrte Simulationen mit ca. 25 Millionen Atomen, die bereits vorinitialisierte Korn-grenzen enthalten, zeigen eine hetrogene Nukleation der austenitischen Phase an den Korn-grenzen unter Schockeinfluß. Eine andere M¨oglichkeit Molekulardynamik-Simulationen imHinblick auf die Große der Simulationszellen zu unterst¨utzen, konnte in der Kombination vonMolekulardynamik-Simulationen mit mesoskopischen Methoden wie z. B. Ginzburg-Landau-Methoden [119] liegen.
Einen weiteren zukunftsweisenden Aspekt stellt die Untersuchung von Nanophasen-Material dar [7, 120–122]. Beispielsweise steigt zun¨achst die Festigkeit von Nanophasen-Metallen, wenn der Festk¨orper aus K¨ornern mit nur einigen Nanometer Durchmesser aufge-baut ist. Urs¨achlich hierfur scheint die Unterdr¨uckung von Versetzungen in kleinen Nanopar-tikeln zu sein. Mit zunehmender Verkleinerung des Korndurchmessers erniedrigt sich die Fe-stigkeit aufgrund von Gleitprozessen zwischen den K¨ornern wieder. Erste Molekulardynamik-Simulationen von Kompaktierungsprozessen von Fe-Ni-Nanopartikeln, zeigen plastische Ver-formungen durch das Abgleiten von dicht gepackten Ebenen auch ohne das Auftreten von Ver-setzungen in dem System. In Zukunft sollten diesbez¨uglich systematische Simulationen durch-gefuhrt werden, die die Analyse mechanischer Eigenschaften sowie das Studium der lokalenDruckverteilung in Nanophasen-Metallen beeinhalten.
Das hier benutzte EAM-Modell f¨ur Fe-Ni-Legierungen erlaubte bereits eine realistischeBeschreibung der strukturellen Transformationen in diesem Legierungssystem [47, 54, 69]. Es

102 8 Zusammenfassung der Ergebnisse
reflektiert effektiv Eigenschaften, die aus einem quantenmechanischem Verst¨andnis von Me-tallen gewonnen wurden, und ist somit geeignet zum Verst¨andnis der Dynamik der Atomeeines metallischen Festk¨orpers beizutragen. Es hat sich aber gezeigt, daß die den Simulatio-nen entnommene Gleichgewichtstemperatur der krz und der kfz Phase zu tief liegt. Verbesse-rungsmoglichkeiten konnten sich ergeben, wenn sowohl die krz als auch die kfz Phase anabinitio Daten angepaßt w¨urden. Erste Versuche (Abschnitt 8.1) zeigten, daß dies im Rahmender hier verwendeten Potentiale nicht ohne weiteres m¨oglich ist. Eine Erweiterung des EAM-Modells aufmodifiedEAM-Modelle [123], die eine Winkelabh¨angigkeit der Kraft beinhalten,konnte eine Verbesserung darstellen. Eine weitere interessante Verbesserung der Beschreibungließe sich durch die Einf¨uhrung eines magnetischen Freiheitsgrads in Form eines Ising- oderHeisenberg-Modells erzielen. Die Analyse eines solchen Modells ist dann durch eine Kombi-nation von Molekulardynamik- und Monte-Carlo-Simulation denkbar.
Im Prinzip ist unter Verwendung vonab initio Methoden die L¨osung von vielen physika-lischen Problemen der uns umgebenden Welt, eingeschlossen der in dieser Arbeit behandeltenProbleme, m¨oglich [124]. Allerdings lassen sich damit dynamische Prozesse f¨ur eine großeAnzahl von Atomen ¨uber einen l¨angeren Zeitraum mit heutigen Rechnersystemen nicht l¨osen.Vielleicht wird in Zukunft mit zunehmender Rechenleistung von Computern das L¨osen vonMolekulardynamik-Simulationen unter Verwendung vonab initio Methoden fur eine hohe An-zahl von Atomen m¨oglich sein.

103
9 Anhang
9.1 Atomare Einheiten
Die folgende Tabelle gibt die im Rahmen dieser Arbeit verwendeten atomaren Einheiten (a.u.)wieder.
Tabelle 9.1:Atomare Einheiten
atomare Einheit sonstige Einheit SI Einheit
Energie in Ry (Rydberg) ≡ 13.6054 eV ≡ 2.17985 10−18 JLange in a0 (Bohrscher Radius) ≡ 0.529177 A ≡ 5.29177 10−11 mMasse in u (atomare Masseneinheit) ≡ 1.66044 10−27 kg
Zusammmengesetzte Einheiten:Zeit in a0
√u/Ry ≡ 1.46005 10−15 s
Geschwindigkeit in√
Ry/u ≡ 3.62328 104 m/s
Volumen in a30 ≡ 0.148185 A3 ≡ 1.48185 10−31 m3
Kraft in Ry/a0 ≡ 4.11932 10−8 J/mDruck in Ry/a30 ≡ 1.47104 104 GPa ≡ 1.47104 1013 Pa
Fundamentalkonstanten:h = 0.0331346 Rya0
√u/Ry ≡ 1.05457 10−34 Js
kB = 6.33369 10−6 Ry/K ≡ 1.38065 10−23 J/K
Eine weitere n¨utzliche Umrechnung ist:
1 J/(mol K) = 1.0364 10−5eV/(Atom K)= 0.23892 cal/(Atom K)= 7.617810−7 Ry/(Atom K)= 0.12027 kB/Atom.

104 9 Anhang
9.2 Parameter der EAM-Potentiale
Nachfolgende Tabellen bestimmen die durch kubische Splines dargestellten Einbettungsfunk-tionenF(ρ) und die effektiven LadungsfunktionenZ(r) der in dieser Arbeit verwendeten EAM-Potentiale f¨ur Al, Fe, Ni und Cu. Die Energien sind in Ry, die Ladungen ine− angegeben.
ρ/ρ0 F F ′′ r/a0 Z Z′
0.0 0.0 0.0 0.0 13.0 0.00.25 -0.1730 0.43 2.25430.50 -0.2273 0.65 0.19191.50 -0.2972 0.71 0.08612.0 -0.3308 0.85 -0.07542.5 -0.2623 1.1 0.0 0.02.8 0.0 0.0
Tabelle 9.2: Parameter des EAM-Potentials fur Al unter BerucksichtigungdesE(V)-Verlaufs (Abschnitt 2.3.2). DieGleichgewichtsladungsdichte betr¨agtρ0 = 4.3345 10−3 a.u., die Gleichge-wichtsgitterkonstantea0 = 7.6534 a.u.,der cut-off Radius rc = 10.1 a.u., dieSumme der ValenzelektronenN = 3und die Anzahl der s-ValenzelektronenbetragtNS = 2.
ρ/ρ0 F F ′′ r/a0 Z Z′
0.0 0.0 0.0 0.0 28.0 0.00.50 -0.2695 0.6 0.98741.0 -0.3961 0.71 0.15962.0 -0.2654 0.85 0.0 0.02.3 0.0 0.0
Tabelle 9.3: Parameter des EAM-Potentials fur Ni [47, 54]. DieGleichgewichtsladungsdichte betr¨agtρ0 = 4.1867 10−3 a.u., die Gleichge-wichtsgitterkonstantea0 = 6.6518 a.u.,der cut-off Radiusrc = 8.7769 a.u., dieSumme der ValenzelektronenN = 10und die Anzahl der s-ValenzelektronenbetragtNS = 0.85.

9.2 Parameter der EAM-Potentiale 105
ρ/ρ0 F F ′′ r/a0 Z Z′
0.0 0.0 0.0 0.0 26.0 0.00.50 -0.2823 0.7 1.44031.0 -0.4276 0.87 0.24522.0 -0.3030 0.94 0.14912.3 0.0 0.0 1.0 0.0734
1.2 0.0 0.0
Tabelle 9.4: Parameter des EAM-Potentials fur Fe [47, 54]. DieGleichgewichtsladungsdichte betr¨agtρ0 = 2.7762 10−3 a.u., die Gleichge-wichtsgitterkonstantea0 = 5.4235 a.u.,der cut-off Radiusrc = 8.3319 a.u., dieSumme der ValenzelektronenN = 8und die Anzahl der s-ValenzelektronenbetragtNS = 0.57.
ρ/ρ0 F F ′′ r/a0 Z Z′
0.0 0.0 0.0 0.0 29.0 0.00.50 -0.2068 0.6 0.95661.0 -0.3082 0.71 0.13902.0 -0.2047 0.85 0.0 0.02.3 0.0 0.0
Tabelle 9.5: Parameter des EAM-Potentials fur Cu [55]. Die Gleich-gewichtsladungsdichte betr¨agtρ0 = 6.0417 10−3 a.u., die Gleich-gewichtsgitterkonstantea0 = 6.8308 a.u.,der cut-off Radiusrc = 9.0131 a.u., dieSumme der ValenzelektronenN = 11und die Anzahl der s-ValenzelektronenbetragtNS = 1.0.
ρ/ρ0 F F ′′ r/a0 Z Z′
0.0 0.0 0.0 0.0 29.0 0.00.50 -0.2111 0.43 6.87051.0 -0.3138 0.65 0.33932.0 -0.2143 0.71 0.1431 0.02.3 0.0 0.0 0.85 0.0 0.0
Tabelle 9.6: Parameter des EAM-Potentials fur Cu [50]. Die Gleich-gewichtsladungsdichte betr¨agtρ0 = 6.180 10−3 a.u., die Gleich-gewichtsgitterkonstantea0 = 6.831 a.u.,der cut-off Radiusrc = 13.663 a.u., dieSumme der ValenzelektronenN = 11und die Anzahl der s-ValenzelektronenbetragtNS = 1.0.

106 9 Anhang
9.3 Ausgaben des MD-Programms
Durch den AufrufINIT des interaktiven Initialisierungs-Programm wird die bin¨are Konfigura-tionsdatei300k.cfgerzeugt:
********************* INIT MD CONFIGURATION *******************Informationen gew unscht (0=Nein, Zahl=Ja) ?0Gittertyp 1=FCC[100],2=BCC[100],3=FCC[111],4=BCC[110],5=HCP[0001]?1Anzahl der Gitterkonstanten in x-Richtung ?5Anzahl der Gitterkonstanten in y-Richtung ?5Anzahl der Gitterkonstanten in z-Richtung ?5Wieviele GK sollen unten festgehalten werden ?0Kubische Gitterkonst. in der benutzen Basiseinheit(z.B.in Sigma) ?6.652GK-Prozentzahl der zuf alligen Auslenkungen ?0.1Maximale Geschwindigkeit der gaußverteilten Komponenten ?0Faktor des Elementes 1 an der Legierung (1=nur El.1,0=nur El. 2))?0Leerstellenanteil im Gitter ?0Kugeldurchmesser in GK (0=keine Kugel) ?0Zufallszahlenstart (seed) ?47110815Dateiname ?300k.cfgTats achlicher Legierungsanteil :0.000000Tats achlicher Leerstellenanteil :0.000000Anzahl der erzeugten Atome :500
Durch den Aufruf des eigentlichen MD Programms mit dem Namen der Initialisierungs-datei MD 300k.ini wird neben den gew¨unschten Ausgabedateien folgende Standardausgabeerzeugt:
******************* MOLECULAR DYNAMICS SIMULATION ******************************** Version 1.2 (9/25/2000) ************************************* Copyright Kai Kadau ***********************
INFORMATION: Falls Informationen zum Programm oder zu Ein- undAusgabedateien des Programms gewuenscht sind, einfach das Programmohne Eingabedatei *.ini starten: MD <enter>
Neustart (0=Nein,1=Ja)_________________________________________: 1Soll-Temperatur________________________________________________: 300.000000Thermalisierungsschritte_______________________________________: 1000Laufschritte___________________________________________________: 50000Zeitschritt____________________________________________________: 1.000000Periodische Randbedingungen in x- y- z-Richtung (1=an, 0=aus)__: 1 1 1Vorgegebener Systemdruck_______________________________________: 0.000000’Masse’ der Simulationsbox (u/abohrˆ4)_________________________: 0.000500Relaxationszeit des Nose-Hoover-Thermostaten (Q=g*K_B*T*tauˆ2) : 30.000000Cut_off Radius f ur die Nachbarlisten___________________________: 12.000000Geschwindigkeitsskalierungsintervall der Thermalisierung : 10Geschwindigkeitsskalierungsintervall des Laufs : 0Nachbarlisten Update-Intervall_________________________________: 100Log-Buch Ausgabeintervall______________________________________: 10Phasenraum der Substratatome Ausgabeintervall__________________: 0Phasenraum der Film-(aufgewachsenen) Atome Ausgabeintervall____: 0Atompositionsausgabeintervall__________________________________: 0

9.3 Ausgaben des MD-Programms 107
Euler-Flag (0=aus, sonst Euler-Schritt voran)__________________: 0Lokales Drucktensor-Flag_______________________________________: 1Lokale Gr oßen-Flag_____________________________________________: 1Just-calc-Flag_________________________________________________: 0Lokales Gitterstruktur-Flag____________________________________: 1
Initialisierungsdatei__________________________________________: 300k.iniKonfigurationssdatei___________________________________________: 300k.cfgLogbuchdatei___________________________________________________: 300k.logGemittelte Atompositionsdatei (Typ,x,y,z)______________________: 300k.x0Lokale Drucktensorsdatei (Typ,x,y,z,px,py,pz,pxy,pxz,pz)_______: 300k.pressLokale Gr oßendatei (Typ,x,y,z,epot,T,rho)______________________: 300k.epot_T_rhoGemittelte Gitterstrukturdatei (Gst.,x,y,z)__________________: 300k.idPaarkorreltionsfunktion des Substrats__________________________: 300k_sub.corPaarkorreltionsfunktion des Films______________________________: 300k_film.cor
Epitaxieintervall______________________________________________: 0Anfangs z-Position des neue eingef uhrten Atoms_________________: 135.000000Maximale Anfangs x-Geschwindigkeit des neu... _________________: 0.000000Maximale Anfangs y-Geschwindigkeit des neu...__________________: 0.000000Anfangs z-Geschwindigkeit des neu ... _________________________: 0.000000Zufallszahlenanfangswert_______________________________________: 1234Startwert der Lagenz ahlung (in z-Richtung)_____________________: 22.360000Dicke einer Lage (in z-Richtung)_______________________________: 3.440000Anzahl der zu z ahlenden Lagen__________________________________: 0
Art der Wechselwirkung_________________________________________: EAM
SPEICHERBEDARF fur die Atomkoordinaten ... Kr afte in KB________: 113SPEICHERBEDARF fur Nachbarschaftslisten in KB__________________: 1171SPEICHERBEDARF Tabellen der e-Dichte und deren Abl. (KB/El.)___: 937SPEICHERBEDARF Tabellen der Splines und deren Ableitung(KB/El.): 625
Arbeite mit tabellierten e-Dichtefunktionen. Intervalle________: 60000Arbeite mit tabellierten Splinefunktionen. Intervalle__________: 20000EAM-Datei Atomsorte 1, prozentualer Anteil an den Filmatomen___:/users/kai/y_molekulardynamics_sources/y_potentials/iron.eam, 0.000000EAM-Datei Atomsorte 2, prozentualer Anteil an den Filmatomen___:/users/kai/y_molekulardynamics_sources/y_potentials/nickel.eam,100.000000Anzahl der eingelesenen Atome__________________________________: 500Anzahl der Atome nach Deposition_______________________________: 500Anzahl der zu thermalisierenden Atom___________________________: 500Anzahl der festgehaltenen Atome________________________________: 0Eingelesenen Kantenl angen (x,y,z)______________________________: 33.662042,33.662042, 33.662042Anf angliches Eingelesenes Volumen/Atom_________________________: 76.287146Anzahl der f ur die thermalisierung relevanten Freiheitsgrade___: 1498Benutzte Thermostat’masse’ in Masse*L angeˆ2(u*abohr*abohr)_____: 2.56175e+03Benutzte Wand’masse’ in Masse/L angeˆ4 (EAM=u/abohrˆ4)__________: 5.00000e-04
NEUSTART der Simulation, alte bereits gemachte Simulationsschrit-

108 9 Anhang
te werden bei der Mittelwertbildung nicht ber ucksichtigt, das Log-buch wird neu begonnen !
BEMERKUNG: Bei der Eingabe von Nullen bei Intervallen oder derThermostat- oder Wandmasse werden die Werte INT_MAX bzw. FLT_MAXzugeordnet. Damit ergibt sich die M oglichkeit den Thermostaten sowiedie fluktuierende Box abzuschalten. (Nicht numerisch, son-dern durch Fallunterscheidung).
Molekulardynamik Startzeit : Thu Sep 28 16:49:19 2000...ENDE DER SIMULATION:
ANZAHL der Atome mit lokaler fcc-Struktur :500ANZAHL der Atome mit lokaler bcc-Struktur :0Unterscheidungskriterum fcc-bcc versagt an Oberfl achen, zu kurzenMittelungszeiten (starke Fluktuationen) oder aber auch beitetragonalen Verzerrungen. In solchen F allen wird zugunsten der bcc Struk-tur entschieden.
Molekulardynamik Startzeit : Thu Sep 28 16:49:19 2000Molekulardynamik Endzeit : Thu Sep 28 17:51:20 2000Laufzeit : 0 Tage, 1 Stunden, 2 Minuten,
1 Sekunden
Laufzeitmass(Laufz./100Atome*100Schritte) : 1.459187 Sekunden*********************** ENDE DER SIMULATION *****************
Anzahl der vollzogenen Schritte : 51000Gemitteltes Volumen der Box : 3.807287e+04Gemittelte Kantenl ange in x-Richtung : 3.364122e+01Gemittelte Kantenl ange in y-Richtung : 3.364122e+01Gemittelte Kantenl ange in z-Richtung : 3.364122e+01Gemittelter Druck in x-Richtung : 2.559028e-07Gemittelter Druck in y-Richtung : -3.859544e-08Gemittelter Druck in z-Richtung : -4.241634e-07Gemittelter Gesamtdruck=1/3(PX+PY+PZ) : -6.895201e-08Gemittelter Scherdruck in xy-Richtung : -2.662773e-07Gemittelter Scherdruck in xz-Richtung : -4.757843e-08Gemittelter Scherdruck in yz-Richtung : -2.170535e-07Gemittelte pot. Energie des Sys. pro Atom : -3.247005e-01Gemittelte kin. Energie des Sys. pro Atom : 2.844729e-03Zahl der von der Quelle emittierten Atome : 0Gesamtzahl der Atome : 500
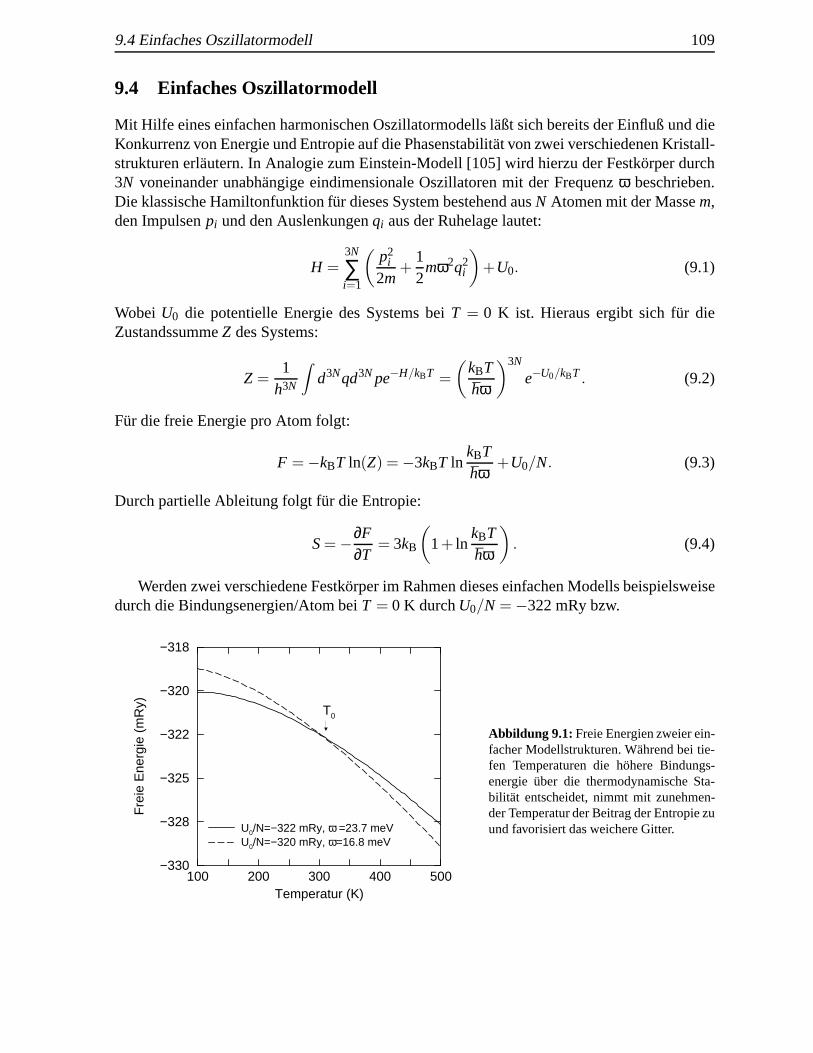
9.4 Einfaches Oszillatormodell 109
9.4 Einfaches Oszillatormodell
Mit Hilfe eines einfachen harmonischen Oszillatormodells l¨aßt sich bereits der Einfluß und dieKonkurrenz von Energie und Entropie auf die Phasenstabilit¨at von zwei verschiedenen Kristall-strukturen erl¨autern. In Analogie zum Einstein-Modell [105] wird hierzu der Festk¨orper durch3N voneinander unabh¨angige eindimensionale Oszillatoren mit der Frequenzω beschrieben.Die klassische Hamiltonfunktion f¨ur dieses System bestehend ausN Atomen mit der Massem,den Impulsenpi und den Auslenkungenqi aus der Ruhelage lautet:
H =3N
∑i=1
(p2
i
2m+
12
mω2q2i
)+U0. (9.1)
Wobei U0 die potentielle Energie des Systems beiT = 0 K ist. Hieraus ergibt sich f¨ur dieZustandssummeZ des Systems:
Z =1
h3N
∫d3Nqd3Npe−H/kBT =
(kBThω
)3N
e−U0/kBT . (9.2)
Fur die freie Energie pro Atom folgt:
F = −kBT ln(Z) = −3kBT lnkBThω
+U0/N. (9.3)
Durch partielle Ableitung folgt f¨ur die Entropie:
S= −∂F∂T
= 3kB
(1+ ln
kBThω
). (9.4)
Werden zwei verschiedene Festk¨orper im Rahmen dieses einfachen Modells beispielsweisedurch die Bindungsenergien/Atom beiT = 0 K durchU0/N = −322 mRy bzw.
100 200 300 400 500Temperatur (K)
−330
−328
−325
−322
−320
−318
Fre
ie E
nerg
ie (
mR
y)
U0/N=−322 mRy, ω =23.7 meVU0/N=−320 mRy, ω=16.8 meV
T0
Abbildung 9.1: Freie Energien zweier ein-facher Modellstrukturen. W¨ahrend bei tie-fen Temperaturen die h¨ohere Bindungs-energie ¨uber die thermodynamische Sta-bilit at entscheidet, nimmt mit zunehmen-der Temperatur der Beitrag der Entropie zuund favorisiert das weichere Gitter.

110 9 Anhang
U0/N = −320 mRy und ¯hω = 23.7 meV bzw.hω = 16.8 meV beschrieben, liegt der Schnitt-punkt der freien Energien bei ca. 300 K (Abb. 9.1). Bei tiefen Temperaturen ist die Strukturmit der niedrigeren Bindungsenergie thermodynamisch stabiler. Umgekehrt wird bei hohenTemperaturen die weichere Struktur (beschrieben durch die niedrigere Frequenz) stabilisiert.Die Konkurrenz der AnteileE und−TSvon der freien EnergieF = E−TS in Abhangigkeitder Temperatur kann bereits anhand des einfachen harmonischen Oszillatormodells veran-schaulicht werden. Es ist allerdings nicht m¨oglich den Kurvenverlauf der freien Energiendieses einfachen Modells an die freien Energien der untersuchten EAM-Kristalle anzupas-sen, da die durch EAM-Potentiale beschriebenen Strukturen nicht harmonisch sind und sichnicht nur durch eine Frequenz darstellen lassen. Im Unterschied zum EAM-Modell besitzt dasharmonische Modell keine thermische Ausdehnung und die W¨aremkapazit¨aten sind konstant
Cp = Cv = T(
∂S∂T
)P=0
= 3kB.

111
Literaturverzeichnis
[1] Homer,Homer principales, Abendland 700 BC.
[2] E. R. Petty,Introductory, in Martensite: Fundamentals and Technology, herausgegebenvon E. R. Petty, Kap. 1, UMI Books on Demand, Michigan 1970.
[3] H. Czichos,Adolf Martens and the Research on Martensite, Kap. 1, S. 3, E. Hornbogenund N. Jost, Oberursel (1989).
[4] C. M. Wayman und H. K. D. H. Bhadeshia,Phase transformations, nondiffusive, Bd. 2,Kap. 16, S. 1507, Herausg. von R. W. Cahn and P. Haasen, North-Holland, Amsterdam(1996).
[5] L. Delaey,Diffusionless transformations, in Phase transformations in materials, heraus-gegeben von R. W. Cahn, P. Haasen und E. J. Kramer, Bd. 5 vonMaterials Science andTechnology, S. 339, VCH, Weinheim 1991.
[6] P. Neumann,Stahl - ein traditioneller Werkstoff mit hohem Innovationspotential, Spek-trum der Wissenschaft, DIGTEST: Moderne Werkstoffe3, 116 (1996).
[7] R. W. Siegel,Nanophasen-Materialien und ihre paradoxen Eigenschaften, Spektrum derWissenschaft3, 62 (1997).
[8] M. Alexe, C. Harnagea und D. Hesse,Nano-Engineering fur nichtfluchtige ferroelek-trische Speicher, Phys. Blatt.10, 47 (2000).
[9] E. Hornbogen,Werkstoffe mit Formgedachtnis: neue Legierungen und Anwendungen,Spektrum der Wissenschaft, DIGTEST: Moderne Werkstoffe3, 111 (1996).
[10] E. F. Wassermann, P. Entel und M. Acet,Vom Urmeter zur Schattenmaske: 100 JahreGrundlagenforschung an Eisenlegierungen, Phys. Blatt.56, 27 (2000).
[11] E. Schrodinger, Quantisierung als Eigenwertproblem, Annalen der Physik81, 109(1926).
[12] F. Schwabl,Quantenmechanik, Springer, Heidelberg 1990.
[13] I. Newton,Philosophiae naturalis principia mathematica, Cambridge 1687.
[14] W. G. Hoover,Time Reversibility, Computer Simulation, and Chaos, World Scientific,Singapore 1999.
[15] M. P. Allen und D. J. Tildesley,Computer Simulation of Liquids, Clarendon Press, Ox-ford 1987.
[16] R. Haberland, S. Fritzsche, G. Peinel und K. Heinzinger,Molekulardynamik, Vieweg,Braunschweig 1995.

112 Literaturverzeichnis
[17] D. C. Rapaport,The Art of Molecular Dynamics Simulation, Cambridge UniversityPress, Cambridge 1995.
[18] B. N. J. Persson,Sliding Friction, Springer, Heidelberg 1998.
[19] S. Hofler-Thierfeldt, Neuer Molekulardynamik-Weltrekord mituber 5 MilliardenAtomen, ZAM Aktuell 82, 1 (2000).
[20] J. Shen, P. Ohresser, C. V. Mohan, M. Klaua, J. Barthel und J. Kirschner,Magnetic Mo-ment of fcc Fe(111) Ultrathin Films by Ultrafast Deposition on Cu(111), Phys. Rev. Lett.80, 1980 (1998).
[21] B. J. Alder und T. E. Wainwright,Phase Transition for a Hard Sphere System,J. Chem. Phys.27, 1208 (1957).
[22] B. J. Alder und T. E. Wainwright,Studies in Molecular Dynamics. I. General Method,J. Chem. Phys.31, 459 (1959).
[23] W. Nolting,Statistische Physik, Zimmermann-Neufang, Ulmen 1994.
[24] H. Haug,Statistische Physik, Vieweg, Braunschweig 1997.
[25] B. L. Holian und R. Ravelo,Fracture simulations using large-scale molecular dynamics,Phys. Rev. B51, 11275 (1995).
[26] B. L. Holian und P. S. Lomdahl,Plasicity Induced by Shock Waves in NonequilibriumMolecular-Dynamics Simulations, Science280, 2085 (1998).
[27] V. V. Zhakhovskii, S. V. Zybin, N. Nishihara und S. I. Anisimov,Shock Wave Structurein Lennard-Jones Crystal via Molecular Dynamics, Phys. Rev. Lett.83, 1175 (1999).
[28] B. N. J. Persson,Theory of friction: Dynamical phase transitions in adsorbed layers,J. Chem. Phys.103, 3849 (1995).
[29] S. J. Zhou, D. L. Preston, P. S. Lomdahl und D. M. Beazley,Large-Scale MolecularDynamics Simulations of Dislocation Intersection in Copper, Science279, 1525 (1998).
[30] Y. K. K. Ohno, K. Esfarjana,Computational Materials Science, Springer, Heidelberg1999.
[31] Bergmann und Sch¨afer,Mechanik Akustik Warme, Walter de Gruyter, Berlin 1990.
[32] C. Gerthsen, H. O. Kneser und H. Vogel,Physik, Springer, Heidelberg 1989.
[33] W. Brenig,Statistische Theorie der Warme, Springer, Heidelberg 1975.
[34] W. Greiner, L. Neise und H. St¨ocker,Thermodynamik und Statistische Physik, HarriDeutsch, Frankfurt am Main 1987.

Literaturverzeichnis 113
[35] U. Landman, U. Luedtke und W. D. Ringer,Atomistic mechanisms of adhesive contactformation and interfacial processes, Wear153, 3 (1992).
[36] P. Entel, H. C. Herper, M. Schr¨oter, E. Hoffmann, K. Kadau und R. Meyer,Ab InitioDescription of Displacive Phase Transformations, in Displacive Phase Transformationsand Their Applications in Materials Engineering, herausgegeben von K. Inoue, S. 319,The Minerals, Metals & Materials Society, Palo Alto 1998.
[37] K. Kadau, R. Meyer und P. Entel,Molecular-Dynamics Study of Thin Iron Films onCopper, Surf. Rev. Lett.6, 35 (1999).
[38] J. R. Morris und R. J. Gooding,Vibrational entropy effects at a diffusionless first ordersolid-to-solid transition, Phys. Rev. B43, 6057 (1991).
[39] J. R. Morris und K. M. Ho,Calculating Accurate Free Energies of Solids directly fromSimulations, Phys. Rev. Lett.74, 940 (1995).
[40] IMSL MATH/LIBRARY, IMSL, Inc., Houston, Texas, U.S.A. (1989).
[41] L. D. Landau und E. M. Lifshitz,Statistical Physics, Pergamon Press, Oxford 1993.
[42] K. Kadau, R. Meyer und P. Entel,A molecular-dynamics study: Nucleation processesand structural transformation of thin iron films on Cu(001), in Structure and Dynamicsof Heterogeneous Systems, herausgegeben von P. Entel und D. E. Wolf, S. 210, WorldScientific, Singapore 2000.
[43] M. S. Daw und M. I. Baskes,Semiempirical, Quantum Mechanical Calculation of Hy-drogen Embrittlement in Metals, Phys. Rev. Lett.50, 1285 (1983).
[44] M. S. Daw und M. I. Baskes,Embedded-atom method: Derivation and application toimpurities, surfaces, and other defects in metals, Phys. Rev. B29, 6443 (1984).
[45] W. H. Press, B. P. Flannery, S. A. Teukolsky und W. T. Vetterling,Numerical Recipes inC, Cambridge University Press, Cambridge 1988.
[46] E. Clementi und C. Roetti,Roothaan-Hartree-Fock Atomic Wavefunctions, At. DataNucl. Data Tables14, 177 (1974).
[47] R. Meyer und P. Entel,Martensite-austenite transition and phonon dispersion curves ofFe1−xNix studied by molecular-dynamics simulations, Phys. Rev. B57, 5140 (1998).
[48] J. Kubler und V. Eyert,Electronic Structure Calculations, in Electronic structure andmagnetic properties of metals and ceramics, Part I, herausgegeben von K. H. J.Buschow, Bd. 3A vonMaterials Science and Technology, Kap. 1, S. 1, VCH, Weinheim1994.
[49] S. Grabowski, K. Kadau und P. Entel,Atomistic modelling of Diffusion in Al, erscheintin Phase Transitions (2001).

114 Literaturverzeichnis
[50] J. Gui, Y. Cui, S. Xu, Q. Wang, Y. Ye, M. Xiang und R. Wang,Embedded-atom methodstudy of the effect of order degree on the lattice parameters of Cu-based shape memoryalloys, J. Phys.: Condens. Matter6, 4601 (1994).
[51] S. Rubini und P. Ballone,Quasiharmonic and molecular-dynamics study of the marten-sitic transformation in Ni-Al alloys, Phys. Rev. B48, 99 (1993).
[52] G. Kresse und J. Furthm¨uller, Efficient iterative schemes for ab initio total-energy cal-culations using a plane wave basis set, Phys. Rev. B54, 11169 (1996).
[53] R. A. Johnson,Implications of the Embedded-Atom Method Format, in Many-Atom In-teractions in Solids, herausgegeben von R. M. Nieminen, M. J. Puska und M. J. Manni-nen, S. 85, Springer, Berlin 1990.
[54] R. Meyer, Computersimulationen martensitischer Phasenubergange in Eisen-Nickel-und Nickel-Aluminium-Legierungen, Dissertation, Gerhard-Mercator-Universit¨at Duis-burg 1998.
[55] R. Meyer, private Mitteilung.
[56] S. Nose, A molecular dynamics method for simulations in the canonical ensemble,Mol. Phys.52, 255 (1984).
[57] S. Nose,A unified formulation of the constant temperature molecular dynamics methods,J. Phys. Chem.81, 511 (1984).
[58] W. Greiner,Mechanik Bd. 2, Harri Deutsch, Frankfurt am Main 1980.
[59] W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions,Phys. Rev. A31, 1695 (1985).
[60] H. C. Anderson,Molecular dynamics simulations at constant pressure and/or tempera-ture, J. Phys. Chem.72, 2384 (1980).
[61] M. Parinello und A. Rahman,Crystal Structure and Pair Potentials: A Molecular-Dynamics Study, Phys. Rev. Lett.45, 1196 (1980).
[62] N. Levanov, V. S. Stepanyuk, W. Herget, O. S. Trushin und K. Kokko,Molecular dy-namics simulation of Co thin films growth on Cu(001), Surf. Sci.400, 54 (1998).
[63] H. U. Krebs,Characteristic Properties of Laser-Deposited Metallic Systems, Int. J. Non-Equilibrium Processing10, 3 (1997).
[64] P. Ohresser, J. Shen, J. Barthel, M. Z. C. V. Mohan, M. Klaua und J. Kirschner,Growthstructure and magnetism of fcc ultrathin films on Cu(111) by pulsed laser deposition,Phys. Rev. B59, 3696 (1999).

Literaturverzeichnis 115
[65] F. Westerhoff, L. Brendel und D. E. Wolf,Layer-by-Layer Pattern Propagation andPulsed Laser Deposition, in Structure and Dynamics of Heterogeneous Systems, her-ausgegeben von P. Entel und D. E. Wolf, S. 140, World Scientific, Singapore 2000.
[66] M. A. Meyers,Dynamic Behavior of Materials, Wiley-Interscience, New York 1994.
[67] P. S. Lomdahl, P. Tamayo, N. G. Jensen und D. M. Baezley,50 GFlops Molecular Dy-namics on the Connection Machine 5, in Proceedings of Supercomputing ’93, heraus-gegeben von G. S. Ansell, S. 520, IEEE Computer Society Press 1993.
[68] D. M. Beazley und P. S. Lomdahl,Controlling the Data Glut in Large-Scale Molecular-Dynamics Simulations, Computers in Physics11, 230 (1997).
[69] K. Kadau,Molekulardynamik Simulationen der martensitischen Phasenumwandlung imEisen-Nickel-Legierungssystem, Diplomarbeit, Gerhard-Mercator-Universit¨at Duisburg1996.
[70] H. Bohm,Einfuhrung in die Metallkunde, B.I. -Wissenschaftsverlag, Mannheim 1968.
[71] G. Gottstein,Physikalische Grundlagen der Materialkunde, Springer, Heidelberg 1998.
[72] P. Haasen,Physikalische Metallkunde, Springer, Heidelberg 1974.
[73] R. D. Doherty,Diffusive phase transformation in the solid state, Bd. 2, Kap. 15, S. 1363,Herausg. von R. W. Cahn und P. Haasen, North-Holland, Amsterdam (1996).
[74] M. Acet, T. Schneider und E. F. Wassermann,Magnetic Aspects of Martensitic Trans-formations in FeNi Alloys, J. Phys. IV (Paris)5, C2–111 (1995).
[75] W. Pitsch,The Martensite Transformation in Thin Foils of Iron-Nitrogen alloys, Phi-los. Mag.4, 577 (1959).
[76] M. Wuttig, B. Feldmann, J. Thomassen, F. May, H. Zillgen, A. Brodde, H. Hannemannund H. Neddermeyer,Structural transformations of fcc iron films on Cu(100), Surf. Sci.291, 14 (1993).
[77] D. Tian, F. Jona und P. M. Marcus,Structure of ultrathin films of Fe on Cu{111} andCu{110}, Phys. Rev. B45, 11216 (1992).
[78] J. Shen, R. Skomski, M. Klaua, H. Jenniches, S. S. Manoharan und J. Kirschner,Mag-netism in one dimension: Fe on Cu(111), Phys. Rev. B56, 2340 (1997).
[79] J. Shen, C. Schmidthals, J. Woltersdorf und J. Kirschner,Structural phase transforma-tion under reversed strain: a comparative study of iron ultrathin film growth on nickeland copper (100), Surf. Sci.407, 90 (1998).
[80] A. Heiming, W. Petry und J. Trampenau,Are Martensitic Phase Transitions in PureGroup 3 and 4 Metalls Driven by Lattice Vibrations, J. Phys. IV (Paris)1, C4–83 (1991).

116 Literaturverzeichnis
[81] V. Raghavan und M. Cohen,A Nucleation Model for Martensitic Transformations inIron-Base Alloys, Acta Metall.20, 333 (1972).
[82] M. Cohen,Operatinal Nucleation in Martensitic Transformations, Metall. Trans.3, 1095(1972).
[83] R. E. Reed-Hill,Physical Metallurgy Principles, D. Van Nostrand Company, New York1973.
[84] B. L. Holian,Modelling shock-wave deformation via molecular dynamics, Phys. Rev. A37, 2562 (1988).
[85] M. A. van Hove,Crystal Surfaces, in Structure of Solids, herausgegeben von R. W. Cahn,P. Haasen und E. J. Kramer, Bd. 1 vonMaterials Science and Technology, S. 483, VCH,Weinheim 1992.
[86] J. Wan, Y. L. Fan, D. W. Gong, S. G. Shen und X. Q. Fan,Surface relaxation and stressof fcc metals: Cu, Ag, Au, Ni, Pd, Pt, Al, and Pb, Modelling Simul. Mater. Sci. Eng.7,189 (1999).
[87] A. M. Rodriguez, G. Bozzolo und J. Ferrante,Multilayer relaxation and surface energiesof fcc and bcc metals using equivalent crystal theory, Surf. Sci.289, 100 (1993).
[88] A. Kara und T. S. Rahman,Vibrational Properties of Metallic Nanocrystals,Phys. Rev. Lett.81, 1453 (1998).
[89] T. Tadaki, Y. Murai, A. Koreeda, Y. Nakata und Y. Hirotsu,Structure and phase transfor-mation of nano-scale particles of Fe-Ni alloys, Mater. Sci. Eng. A217/218, 235 (1996).
[90] W. Keune, A. Schatz, R. D. Ellerbrock, A. Fuest, K. Wilmers und R. A. Brand,Moss-bauer effect study of face-centered cubic-like Fe on Cu(001), J. Appl. Phys.79, 4265(1996).
[91] M. Wuttig und J. Thomassen,Structure determination Fe films on Cu(100), Surf. Sci.282, 237 (1993).
[92] K. Kalki, D. D. Chambliss, K. E. Johnson, R. J. Wilson und S. Chiang,Evidence ofmartensitic fcc-bcc transition of thin Fe films on Cu(100), Phys. Rev. B48, 18344 (1993).
[93] D. Li, M. Freitag, J. Pearson, Z. Q. Qui und S. D. Bader,Magnetic Phases of UltrathinFe Grown on Cu(100) as Epitaxial wedges, Phys. Rev. Lett.72, 3112 (1994).
[94] F. Scheurer, R. Allespach, P. Xhonneux und E. Courtens,Magnetic coupling of structuralmicrodomains in bcc Fe on Cu(001), Phys. Rev. B48, 9890 (1993).
[95] J. Shen, M. Klaua, H. Jenniches, J. Barthel, C. V. Mohan und J. Kirschner,Structural andmagnetic phase transitions of Fe on stepped Cu(111), Phys. Rev. B56, 11134 (1997).

Literaturverzeichnis 117
[96] S. Lu, Z. Q. Wang, D. Tian, Y. S. Li und F. Jona,Epitaxial Growth ofγ-Fe on Ni{001},Surf. Sci.221, 35 (1993).
[97] C. Muller, H. Muhlbauer und G. Dumpich,Epitaxial growth of Pd/Fe/Pd trilayers ontosapphire, Thin Solid Films310, 81 (1997).
[98] T. Steffl, H. Muhlbauer, B. Rellinghaus und G. Dumpich, in-situRHEED and STM in-vestigations on the epitaxial growth of Fe/Pd multilayers on Pd(001)-buffered sapphire,in Structure and Dynamics of Heterogeneous Systems, herausgegeben von P. Entel undD. E. Wolf, S. 226, World Scientific, Singapore 2000.
[99] S. Fahler, M. Weissheit und H. U. Krebs,In-situ Characterization of Laser depositedFe/Ag Multilayers by combination of time-of-flight, RHEED and Resistence Measure-ments, Mater. Res. Soc. Symp. Proc.502, 139 (1998).
[100] H. Jenniches, J. Shen, C. V. Mohan, S. S. Manoharan, J. Barthel, P. Ohresser, M. Klauaund J. Kirschner,Structure and magnetism of pulsed-laser-deposited ultrathin films ofFe on Cu(100), Phys. Rev. B59, 1196 (1999).
[101] T. Fließbach,Statistische Physik, Spektrum Akademischer Verlag, Heidelberg 1995.
[102] W. Bendick und W. Pepperhoff,On theα/γ Phase Stability of Iron, Acta Metall.30, 679(1982).
[103] J. Neuhaus, W. Petry und A. Krimmel,Phonon softening and martensitic transformationin α-Fe, Physica B234-236, 897 (1997).
[104] T. C. Germann, B. L. Holian, P. S. Lomdahl und R. Ravelo,Orientation Dependence inMolecular Dynamics Simulations of Shocked Single Crystals, Phys. Rev. Lett.84, 5351(2000).
[105] C. Kittel, Introduction to solid state physics, Wiley & Sons, New York 1971.
[106] R. E. Cech und D. Turnbull,Heterogeneous Nucleation of the Martensite Transforma-tion, Trans. Metall. Soc. AIME206, 124 (1957).
[107] K. Asaka, Y. Hirotsu und T. Tadaki,Martensitic transformation in nanometer-sized par-ticles of Fe-Ni alloys, Mater. Sci. Eng. AA273-275, 262 (1999).
[108] K. Asaka, Y. Hirotsu und T. Tadaki,Tansmission electron microscopy and electrondiffraction study on structure and phase transformation on nanometer-sized Fe-15-30at.% Ni alloy particles, J. Electron Microscopy48, 387 (1999).
[109] K. L. Nierholz, K. P. Nos und P. Entel,Freezing and melting of clusters and examina-tion of potential energy surfaces, in Structure and Dynamics of Heterogeneous Systems,herausgegeben von P. Entel und D. E. Wolf, S. 167, World Scientific, Singapore 2000.

118 Literaturverzeichnis
[110] M. Clossen,Molekulardynamik-Simulationen von Materialermudungseffekten, Diplo-marbeit, Gerhard-Mercator-Universit¨at Duisburg 1996.
[111] B.-U. Runge, M. Dippel, G. Filleb¨ock, K. Jacobs und G. Schatz,Induced Magnetic Hy-perfine Field at Ag Sites near an Fe(100)/Ag(100) Interface, Phys. Rev. Lett.79, 3054(1997).
[112] A. Schatz,Wachstum, Struktur und Magnetismus von epitaktischen kubisch-flachenzen-trierten (fcc) Fe(100)-Schichten, Dissertation, Gerhard-Mercator-Universit¨at Duisburg1996.
[113] G. Gubbiotti, L. Albini, G. Carlotti, S. Loreti, C. Minarini und M. D. Crescenzi,fcc-bccphase transition of epitaxial Fe/Cu(111) films: a structural and magnetic study, Surf. Sci.433-435, 680 (1999).
[114] H. Jenniches, M. Klaua, H. H¨oche und J. Kirschner,Comparison of pulsed laserdeposition and thermal deposition: Improved layer-by-layer growth of Fe/Cu(111),Appl. Phys. Lett.69, 3339 (1996).
[115] H. C. Herper, E. Hoffmann und P. Entel,Ab initio full-potential study of the structuraland magnetic phase stability of iron, Phys. Rev. B60, 3839 (1999).
[116] H. C. Herper, Ab-initio-Untersuchungen magnetischer und struktureller Eigenschaftenvon 3d-Ubergangsmetallen und ihren Legierungen, Dissertation, Gerhard-Mercator-Universitat Duisburg 2000.
[117] K. Asaka, Y. Hirotsu und T. Tadaki,Facets of nonometer-sized Fe-Ni alloy particlesand theitr change upon the martensitic transformation, Mater. Sci. Forum (Trans TechPublications)327-328, 409 (2000).
[118] A. F. Voter, Hyperdynamics: Accelerated Molecular Dynamics of infrequent events,Phys. Rev. Lett.78, 3908 (1997).
[119] S. R. Shenoy, T. Lookman, A. Saxena und A. R. Bishop,Martensitic textures: Multiscaleconsequences of elastic compatibility, Phys. Rev. B60, R12537 (1999).
[120] J. Schiotz, F. D. D. Tolla und K. W. Jacobsen,Softening of nanocrystalline metals at verysmall grains, Nature (London)391, 561 (1998).
[121] S. Yip,The strongest size, Nature (London)391, 532 (1998).
[122] J. Weissm¨uller und J. W. Cahn,Mean stresses in microstructures due to interfacestresses: A generalization of a capillary equation for solids, Acta mater.5, 1899 (1997).
[123] M. I. Baskes,Modified embedded-atom potentials for cubic materials and impurities,Phys. Rev. B46, 2727 (1992).
[124] R. B. Laughlin und D. Pines,The Theory of Everything, Proc. Nat. Acad. Sci. U.S.A.97,28 (2000).

Index
α-Fe, 50ε-Fe, 50
a.u. (atomare Einheiten), 103Ablaufplan, 38Anderson-Methode, 35atomare Einheiten (a.u.), 103Atomdynamik-Simulation, 18Aufwachsprozeß d¨unner Filme, 94Ausbreitungsgeschwindigkeit der martensi-
tischen Phase, 78ausbruchsartiger Martensit, 45ausbruchsartiges Wachstum, 79, 84Austenit, 45austenitischeUbergangstemperaturen, 58austenitische Transformation, 45
Bain-Tranformation, 46Bain-Transformation,c/a, 62Burgers-Mechanismus, 48burst-type, 46
chaotisches Verhalten, 18chemische Triebkraft, 49, 60, 88CPU-Zeit, 65, 76cutt-off, 20
dunner Film, 85Dekommpressionswellen, 51Depositionsrate, 36Domanen, streifige, 90Druck in einer Simulationszelle, 24Druckregelung, 35Drucktensor, 25
EAM-Potentiale, 29, 104Einbettungsanteil der Energie, 29Einheiten, 103Einstein-Modell, 109elastic precursor, 65elastische Relaxation, 92Elektronestrukturrechnungen, 30
Embedded-Atom Method, 29Energie-getrieben, 61Energiebarriere, 28, 63Entropie entlang des Bain-Wegs, 64Entropie-getrieben, 61Entropieanteil, 60Equilibrirungsphase, 23, 39Ergodenhypothese, 18
Fe-Ni-System, 57Filmdicke, 85, 92freie Energie, 27, 48, 60, 109freie Energie, Berechnung, 27, 28freie Enthalpie, 27, 48freier Fe-Film, 89
Gear, 20Geschwindigkeit einer Schockwelle, 67Gitterfehler, 58Gleichgewichtstemperatur, 60Gleichverteilungssatz, 22Grenzflache zwischen krz und kfz, 89Grenzflachenenergie, 49Grundzustand, 53
Habitusebene, 46heterogene Keimbildung, 49, 63, 76, 80hexagonal-dichteste Packung, 41Hochtemperaturphase, 45homogene Keimbildung, 49, 63, 67Hysterese, 58
infrequent events, 18instantane H¨upfprozesse, 96instantane Temperatur, 23instantaner Druck, 24interatomare Wechselwirkung, 29
Keimbildung, 49kinetische Monte-Carlo-Simulation, 37kinetische Monte-Carlo-Simulation, 101kinetischer Anteil des Drucks, 24
119

120 INDEX
Kompressionswelle, elastische, 65Kompressionswelle, plastische, 65Konkurrenz zwischen Energie und En-
tropie, 60Korngrenze, 70Korngrenzendynamik, 72Korrelationsdeterminante, 27, 60Kraftanteil des Drucks, 24Kristallbaufehler, 43kristallographische Orientierung des Sub-
strats, 85kristallographische Orientierungsbeziehun-
gen, 47Kristallstruktur, harte, 27, 53Kristallstruktur, weiche, 27, 53kritische Keimbildungsarbeit, 49kritische Keimgroße, 49kubisch-flachenzentriertes Gitter, 41kubisch-raumzentriertes Gitter, 41Kurdjumov-Sachs-Orientierung, 47, 92
Lagrange-Funktion, 33, 34Leapfrog-Version, 19least squares fit, 30Leerstellen, 57Leerstellenbildungsenergie, 29lokale Gitterstruktur, 26lokaler Drucktensor, 25
Martensit, 45Martensit in kleinen Partikeln, 73martensitischeUbergangstemperaturen, 58martensitische Transformation, 45Martensitkeime, 57Martensitvarianten, 79, 84Maxwell-Boltzmann-Verteilung, 23MD, 18Meßmittelwerte, 18metallischer Charakter, 29minimum-image-convention, 21Molekulardynamik-Simulationen, 18Molekulardynamikprogramm MD, 38Momentum-Mirror-Methode, 37, 65
Nachbarliste, 20Nanopartikel, 73, 76Nichtgleichgewichtsph¨anomene, 18, 37, 65Nishiyama-Wassermann-Orientierung, 47Nose-Hoover-Formulierung, 33Nose-Thermostat, 33Nukleation, 49, 62, 67, 75, 80Nukleationskeime, 57numerisch stabil, 40
Oberflachen, 55Oberflacheneffekte, 21Oberflachenenergie, 55Oberflachenenergie, Einfluß auf martensi-
tische Transformationen, 73Oberflachenrelaxation, 56Oszillatormodell, 109
Paarkorrelationsfunktion, 25Parinello-Rahman-Verfahren, 36periodische Randbedingungen, 21Phasendiagramm, 58Phasenumwandlungen, 45Pitsch-Orientierung, 47, 89plastische Verformung, 65plastische Verformungen, 50PLD (Pulsed-Laser-Deposition), 36, 93Python, 38
radiale Verteilungsfunktion, 25Rechenzeit, 65, 76Relaxation der Oberfl¨ache, 56
Schallgeschwindigkeit, 67Schockwelle, 50, 65Schwingungsentropie, 27, 53SPaSM, 24, 38spezifische W¨arme, 54, 61Stapelfehler, 53, 59statistische Mechanik, 23streifenahnliches Muster, 92
TD (thermal-deposition), 36, 93Temperatur, 22, 23, 33Thermodynamik des Fe-Ni-Systems, 60

INDEX 121
thermodynamische Gleichgewichtstemper-atur, 73
thermoelastischer Martensit, 45Thermostatmasse, 33Tieftemperaturphase, 45Transformationsmatrix, 46
Umrechnung von Einheiten, 103unvollstandige Transformation, 89
Velocity-Verlet-Algorithmus, 20Verlet-Algorithmus, 19Verlet-Listen, 20Verzerrungsenergie, 49Virialsatz, 24
Warmebad, 34Wechsler-Lieberman-Read-Theorie, 46
Zellenmethode, 20, 21Zwillinge, 44, 46, 59, 79, 90Zwillingsgrenze, 70


123
Danksagung
Mein besonderer Dank gilt Prof. Dr. P. Entel, der diese Arbeit erm¨oglicht hat. Besonders dankenmochte ich ihm fur die vielen kreativen Vorschl¨age und Anregungen sowie die vielen Freir¨aumezum Verwirklichen eigener Ideen. Ihm habe ich z. B. zu verdanken, daß ich an einer Reihe voninteressanten Konferenzen teilnehmen konnte, die sehr wertvoll f¨ur die Erstellung dieser Arbeitwaren.
Prof. Dr. D. Wolf mochte ich fur seine inspirierenden Vorlesungen und Diskussionen danken.
Weiterhin danke ich Ralf Meyer (Montreal), Christian Helm (Los Alamos) und Stefan Gra-bowski fur viele fruchtbare Diskussionen, wertvolle Kritik und Anregungen aller Art. RalfMeyer hat mir wahrend meiner Diplomzeit den Einstieg in die Methode der Molekulardynamik-Simulation erleichtert und stand mir auch sp¨ater mit Rat und Tat zur Seite.
Heike Herper danke ich f¨ur die standige Diskussionsbereitschaft ¨uber Themen aller Art undihre Elektronenstrukturdaten.
Vielen Dank an Stefan, Heike, meinen Vater, meinen Bruder Dirk und Antje f¨ur Anregungenund Kritik zur vorliegenden Arbeit und dar¨uber hinaus.
Den Mitarbeitern der Abteilung f¨ur Theoretische Tieftemperaturphysik danke ich f¨ur alltaglicheHilfestellungen sowie die gute Arbeitsatmosph¨are. Meinen B¨urokollegen Lars Roters, StefanGrabowski, Minoru Siguhara, Georg Rollmann sowie allen G¨asten m¨ochte ich fur die auf-lockernde Atmosph¨are im Buro danken. Den Systemverwaltern Fred Hucht, Michael Staats,Markus Gruner und Ralf Meyer danke ich f¨ur die Bereitstellung der exzellenten Arbeitsumge-bung, sowie f¨ur viele andere Tricks und Tips.
Ein spezieller Dank richtet sich an Timothy C. Germann, Peter S. Lomdahl und Brad LeeHolian (Los Alamos, U.S.A.) f¨ur die vielen Hilfestellungen und hervorragende Kollaborationwahrend meiner Zeit in der W¨uste, Los Alamos. Thank you guys. I survived theCerro GrandeFire.
Weiterhin mochte ich mich f¨ur die vielen Antworten auf Fragen zur Interpretation von experi-mentellen Befunden bei Professoren und Mitarbeitern aus der Experimentalphysik bedanken.
Prof. Dr. E. Hornbogen m¨ochte ich fur das zur Verf¨ugung stellen einer Mikroskopaufnahmedanken.
Diese Arbeit wurden aus Mitteln der Deutschen Forschungsgemeinschaft im Rahmen des Gra-duiertenkollegStruktur und Dynamik heterogener Systeme, sowie des SFB 445 gef¨ordert.
Vielen Dank an meine Familie. Ohne die vielen physikalischen- und jede Menge unphysikali-schen Spielchen (Vadders Rakete ¨ubers Haus!) und Diskussionen w¨are mein Interesse an derPhysik wohl nie entstanden. Antje, Anna und meiner Mutter danke ich f¨ur alles mogliche.






























