Embed Size (px)
Citation preview
MPG
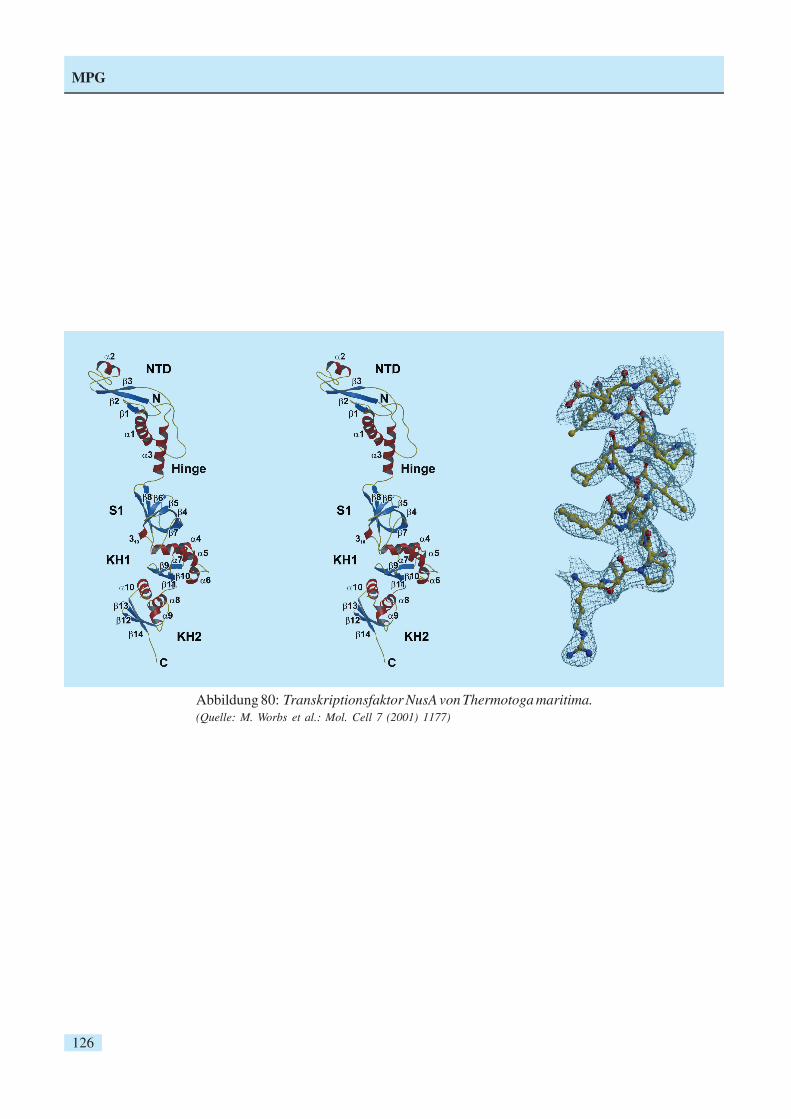
Abbildung 80: Transkriptionsfaktor NusA von Thermotoga maritima.(Quelle: M. Worbs et al.: Mol. Cell 7 (2001) 1177)
126

MPG
Max-Planck-GesellschaftArbeitsgruppen für StrukturelleMolekularbiologieLeiter: H.-D. Bartunik, E. Mandelkow (Sprecher), A. Yonath
Die Max-Planck-Arbeitsgruppen beschäftigen sichmit den Beziehungen zwischen der Struktur undder Funktion von biologischen Makromolekülen.Thematische Schwerpunkte sind
– die Enzyme und ihr katalytischer Mechanismus,
– das Zytoskelett und seine Rolle in Zellbewegungund Alzheimer-Krankheit,
– das Ribosom und seine Funktion in der Protein-biosynthese.
Die Proben werden mit biochemischen Methodenisoliert oder mit molekularbiologischen Methodensynthetisiert. Die wesentliche Methode der Struk-turuntersuchung ist die Röntgenbeugung von Pro-teinkristallen, Fasern oder Lösungen; daneben wer-den weitere biophysikalische Analyseverfahren wieSpektroskopie, Elektronenmikroskopie, Bildverar-beitung und andere eingesetzt. Schwerpunkte me-thodischer und instrumenteller Entwicklungen sindneue Kristallisationsverfahren, Einsatz von elektro-nischen Detektoren, Laue-Methoden und eine Mess-strecke für die Proteinkristallographie.
Forschungsschwerpunkte
Proteindynamik
Die MPG-Arbeitsgruppe für Proteindynamik unter-sucht Struktur-Funktionsbeziehungen von Proteinen.Sie setzt dabei Methoden der Proteinkristallographiebei ultra-hoher Auflösung, der Kryokristallographiesowie der Nanosekunden-zeitaufgelösten Röntgenbeu-gung ein. Ein weiterer Schwerpunkt ist die Entwick-lung von Methoden anomaler Phasenlösung und ihreAnwendung auf de-novo-Bestimmungen von Protein-strukturen. Die Gruppe betreibt eine Messstation an derWiggler-Beamline BW6 an DORIS.
Ein Schwerpunkt der wissenschaftlichen Arbeiten lagbei der weiteren Entwicklung von Verfahren experi-menteller Phasierung. Mit Hilfe anomaler Streuungbei einer Röntgenwellenlänge (SAD-Methode) bzw.bei mehreren Wellenlängen (MAD) wurde eine Reiheneuer Proteinstrukturen aufgeklärt. Ein wichtiges Bei-spiel stellt die Struktur des Transkriptionsfaktors NusAvon Thermotoga maritima dar (Abb. 80), die auf derGrundlage anomaler Streuung an der Se-K-Kante ge-löst wurde. Durch die Anordnung multipler Domänen(S1, KH) entsteht eine ausgedehnte Oberfläche für dieBindung von RNA. Durch kooperative Effekte könnendie Stärke und Spezifizität der RNA-Bindung moduliertwerden.
Ein weiterer Schwerpunkt lag bei der Entwicklung vonVerfahren und Techniken zur Automatisierung der Beu-gungsmessungenund ihrer Auswertung. Damit wurdenwesentliche Voraussetzungen für die Lösung von Pro-teinstrukturen in hohem Durchsatz und damit für An-wendungen in der Strukturgenomik an der BeamlineBW6 geschaffen. In einem ersten Schritt sollen vorge-frorene Proteinkristalle automatisch auf ein Diffrakto-meter transferiert werden. Dazu wurde ein Robotersys-tem konzipiert, das in Kürze fertig gestellt und installiertwerden soll. Ein zweiter Schritt, bei dem der montierteKristall automatisch identifiziert und in den Röntgen-strahl positioniert wird, wurde bereits abgeschlossen;dieses System wurde schon in Routinemessungen ein-gesetzt. Ein dritter Schritt, der die Möglichkeit zu au-tomatischer Beugungsdatensammlung sowie zur sofor-tigen Reduktion der Daten schafft, wurde ebenfallsbereits realisiert. Weitere Schritte, die sich in der Ent-wicklung befinden, zielen auf eine automatische Be-urteilung der Kristallqualität, Wahl der Messstrategiesowie Lösung der Patterson als Basis für automatisierteSAD/MAD-Phasierung; diese Schritte bedürfen bis-her der Expertise erfahrener Kristallographen. Es wirderwartet, dass sich die Auswahl geeigneter Kristalle(Screening) sowie die eigentliche Kristallstrukturana-
127

MPG
lyse für einen erheblichen Teil aller Proteine, die alsTargets für Projekte der Strukturgenomik von Interessesind, automatisieren lässt.
Alle Röntgenbeugungsmessungen wurden an derBeamline BW6 an DORIS durchgeführt, die von MPGund GBF gemeinsam betrieben wird.
Zytoskelett
Die MPG-Gruppe ,,Zytoskelett“ befasst sich mitder Stukturbestimmung von Proteinen des Zytoske-letts mit Hilfe der Synchrotronstrahlung, insbeson-dere mit der Untersuchung des Struktur-Funktions-Zusammenhangs von Mikrotubuli und Mikrotubuli-assoziierten Proteinen. Mikrotubuli sind hohlzylindri-sche Proteinfasern, die durch spontane Polymerisationvon Tubulin entstehen. Sie spielen bei der Organisationzellulärer Strukturen eine wichtige Rolle und sind anvielen dynamischen Prozessen in der Zelle beteiligt.
Es gibt eine große Zahl von Proteinen und Protein-komplexen, die mit Mikrotubuli assoziiert sind. Vondiesen stehen zwei Gruppen im Vordergrund des Inter-esses: Die erste ist die Gruppe der Motorproteine, diefür den Transport zellulärer Bestandteile entlang derMikrotubuli verantwortlich sind. Zu dieser Gruppe ge-hören Dynein und die Familie der Kinesine. Die zweiteGruppe sind die so genannten ,,MAPs“ (Mikrotubuli-assoziierte Proteine); sie haben hauptsächlich stabili-sierende bzw. regulative Wirkung auf das Mikrotubuli-Gerüst der Zelle. Hierzu gehört das Tau-Protein, daseine besondere Rolle bei der Entstehung verschiedenerFormen neuronaler Erkrankungen, wie FTDP-17 undAlzheimer-Krankheit, spielt. Neuere Untersuchungenbelegen, dass es einen direkten, funktionellen Zusam-menhang zwischen den beiden Gruppen, den Motorpro-teinen und den MAPs, gibt. Dies eröffnet neue Perspek-tiven für die Erforschung der molekularen Ursachen dergenannten Krankheiten.
Konventionelles Kinesin, der Hauptvertreter derKinesin-Familie, besteht in seiner nativen Form auszwei schweren und zwei leichten Peptidketten. Jede derbeiden schweren Ketten hat an ihrem N-Terminus eineetwa 350 Aminosäuren umfassende, globuläre ,,Mo-tordomäne“. Diese Motordomänen sind für die Inter-aktion mit den Mikrotubuli und für die Umwandlung
chemischer Energie in gerichtete Bewegung (mecha-nische Arbeit) verantwortlich. Dabei wird die ener-giereiche Verbindung ATP (Adenosin-tri-phosphat) zuADP (Adenosin-di-phosphat) und Phosphat hydro-lysiert.
Mittlerweile sind Röntgenstrukturen von Motordomä-nen verschiedener Kinesine bekannt, unter anderem auskonventionellem Kinesin von Mensch und Ratte. Vorkurzem ist es gelungen, die Motordomäne des Pilzki-nesins NcKin (Neurospora crassa Kinesin) zu kristalli-sieren und die Struktur mit Röntgendiffraktion zu be-stimmen. NcKin weist gegenüber anderen Kinesineneinige Besonderheiten auf. NcKin hat keine leichtenKetten und bewegt sich drei- bis fünfmal schneller alsdas konventionelle Kinesin aus tierischen Zellen. DerVergleich der Struktur des Pilzkinesins mit den bereitsbekannten Kinesinstrukturen liefert Hinweise auf denMechanismus der Motoraktivität von Kinesinen im all-gemeinen und auf die Ursachen für die hohe Geschwin-digkeit des Pilzkinesins im besonderen: Kleine struk-turelle Änderungen in der ATP-Bindungstasche (her-vorgerufen durch die Spaltung von ATP in ADP undPhosphat)werdendurcheindynamischesNetzwerkvonSalzbrücken auf benachbarte Regionen in der Motor-domäne übertragen und verstärkt. Diese Regionen sindeinerseits für die Wechselwirkung mit Mikrotubuli undandererseits für die Kommunikation mit der zweitenMotordomäne verantwortlich. Der geschwindigkeits-bestimmende Schritt ist der Ersatz der SpaltprodukteADP und Phosphat durch frisches ATP. Im Vergleichmit den anderen Kinesinen hat das schnelle Pilzkinesineine viel weitere ATP-Bindungstasche. Dadurch kannder Austausch von ADP und ATP schneller erfolgen.Weiterhin trägt vermutlich auch die höhere Beweglich-keit der Region, über die die beiden Motordomänenmiteinander kommunizieren, zur Geschwindigkeit desPilzkinesins bei.
Das Mikrotubuli-assoziierte Protein Tau hat in Lö-sung keine bestimmte Struktur, die Tau-Molekülenehmen vielmehr weitgehend zufällige Konforma-tionen an. Das Fehlen einer definierten globulärenStruktur führt bei Proteinen häufig zu unerwünsch-ter Aggregation und zur Bildung großer Polymerkom-plexe (Fasern), die die Zelle schädigen. Dieser Pro-zess ist die Grundlage verschiedener Demenzen desMenschen (Beispiel: Alzheimer-Krankheit, Parkinson-Krankheit, Creutzfeld-Jacob-Krankheit) und verwand-
128

MPG
0.47 nm
1.07 nm
filam
ent a
xis
0.47
3.0 1.07
a b
Abbildung 81: Röntgendiffraktion von teilweise orientierten Fasern der Mikrotubuli-Bindungsdomäne des Tau-Proteins (a) und daraus abgeleitete Organisation der für dieAggregation der Tau-Moleküle wesentlichen β-Stränge im Kernbereich der Filamente (b).
ter Erkrankungen bei Tieren (Beispiel: BSE = Rin-derwahn). Das hier untersuchte Tau-Protein aggre-giert zu paarigen helikalen Filamenten (PHFs) inder Alzheimer-Krankheit, und Mutationen des Tau-Proteins lösen so genannte ,,frontotemporale“ Demen-zen aus (FTDP-17). Der Aggregationsprozess konntein vitro nachvollzogen werden. Dadurch war es mög-lich, die Struktur der Filamente mit Hilfe der Röntgen-faserdiffraktion zu untersuchen (Abb. 81). BestimmteTau-Mutanten der frontotemporalen Demenz neigenbesonders stark zur Fibrillenbildung, verglichen mitder langsameren Aggregation des Tau-Proteins bei derAlzheimer-Krankheit. In beiden Fällen liegt aber einähnliches Strukturprinzip zugrunde: Das Tau-Proteinenthält ein Hexapeptid-Motiv, welches von einer unge-ordneten Struktur in die Konformation eines β-Strangesübergehen kann. Die Umwandlung geschieht zunächstspontan in einem ersten, langsamen Prozess (Nuklea-tion), später wird sie beschleunigt durch den Kontaktmit bereits umgewandelten Tau-Molekülen (Polyme-risationsphase). Dabei kommt es zur Ausbildung ei-ner β-Faltblatt-ähnlichen, filamentösen Struktur. Einesystematische Untersuchung der Faktoren, die die Ag-gregation zu Tau-Filamenten begünstigen, könnte dazubeitragen, die Entstehungsursachen der Alzheimer-Krankheit besser zu verstehen und neue Therapiean-sätze zu entwickeln.
Struktur der Ribosomen
Der genetische Code ist eine Bibliothek von Bauplänen,die die Sequenz von Proteinen beschreibt. Die Über-setzung des genetischen Codes und die Produktion derProteine wird durch ein universelles Zellorganell, dasRibosom, geleistet.
Das Ribosom besteht aus zwei Untereinheiten, dieverschiedene Funktionen im Rahmen der Protein-Biosynthese erfüllen. Die kleine Untereinheit (30S inProkaryonten, bestehend aus der 16S rRNA und 20 ri-bosomalen Proteinen) ist für die Übersetzung des gene-tischen Codes, dessen Blaupause auf der mRNA abge-legt ist, verantwortlich. Die große Untereinheit (50S inProkaryonten, bestehend aus 5S rRNA, 23S rRNA und33 ribosomalen Proteinen) fügt die einzelnen Amino-säuren zu einer langen Peptid-Kette entsprechend demBauplan zusammen. Aufgrund der zentralen Rolle desRibosoms in der Protein-Biosynthese ist das Ribosomzugleich das primäre Target der meisten Antibiotika.
Im Jahr 2000 konnte die Struktur der 30S ribosoma-len Untereinheit von Thermus thermophilus bei einerAuflösung von 3.3 Å röntgen-kristallographisch aufge-klärt werden und im Folgenden bis zu einer Auflösungvon 3.1Å verfeinert werden. Darauf basierend konnte
129

MPG
die Arbeitsgruppe Komplexe der 30S Untereinheit mitdem Initiationsfaktor IF3 sowie zwei Antibiotika, Te-tracyclin und Edeine, aufklären.
Der Initiationsfaktor IF3 erfüllt wesentliche Funktio-nen in der Initiationsphase der Protein-Biosynthese. Zudiesem Zeitpunkt sind die 30S und 50S Untereinhei-ten getrennt, die mRNA wird in die 30S Untereinheiteingefädelt und die Initiationsfaktoren IF1-3 sowie dieInitiator-tRNA werden angelagert. IF3 stellt sicher, dasskeine vorzeitige Bindung der ribosomalen Unterein-heiten stattfindet und die mRNA mit dem korrektenStart-Codon eingebaut wird. Die Kristallstruktur desKomplexes der 30S Untereinheit mit IF3 konnte ei-nige der Funktionen von IF3 auf struktureller Ebeneverständlich machen.
Tetracyclin war das erste Breitband-Antibiotikum, dasentdeckt wurde, und ist noch immer eines der am weites-ten verbreiteten Antibiotika, obwohl Resistenzen gegenTetracyclin den Anwendungsbereich in den vergange-nenJahrendramatischeingeschränkthaben.Tetracyclinwar bekannt dafür, ein so genannter ,,A-site-inhibitor“zu sein, also die Bindung von tRNA an der ,,A-site“ zuunterbinden und dadurch die Protein-Biosynthese zumErliegen zu bringen. Die Struktur des 30S-TetracyclinKomplexes zeigt, dass für die Wirkung von Tetracy-clin eine Bindungsstelle verantwortlich ist, die mit derBindungsstelle der tRNA übereinstimmt und so dieBlockade des Ribosoms verursacht. Daneben wurdensechs weitere Bindungsstellen von Tetracyclin gefun-den, die aber in erster Linie die Konstituierung desRibosoms beeinflussen und keine direkte bakterizideWirkung entfalten.
Edeine ist ein universelles Antibiotikum. Es unterbindetalso im Gegensatz zu Tetracyclin, das nur an bakteri-ellen Ribosomen seine Wirkung entfaltet, die Protein-Biosynthese in jeder Zelle unabhängig von ihrem phy-logenetischen Ursprung. Aus diesem Grunde ist Edeinekein Antibiotikum pharmazeutischer Relevanz, aberwegen seiner Universalität ein ausgezeichnetes Modell,um allgemeine Aspekte der Protein-Biosynthese zu un-tersuchen. Die Untersuchung des 30S-Edeine Komple-xes hat gezeigt, dass Edeine – wenig überraschend– in einer universell konservierten Region des Ribo-soms bindet, die durch dynamische Konformations-Änderungen direkt an der Initiation beteiligt ist. Edeineunterbindet diese Konformations-Änderungen und be-wirkt damit einen Stillstand des Prozesses. Darüber
hinaus blockiert Edeine den Pfad der mRNA und ver-hindert die korrekte Positionierung der Initiator-tRNAauf der kleinen ribosomalen Untereinheit.
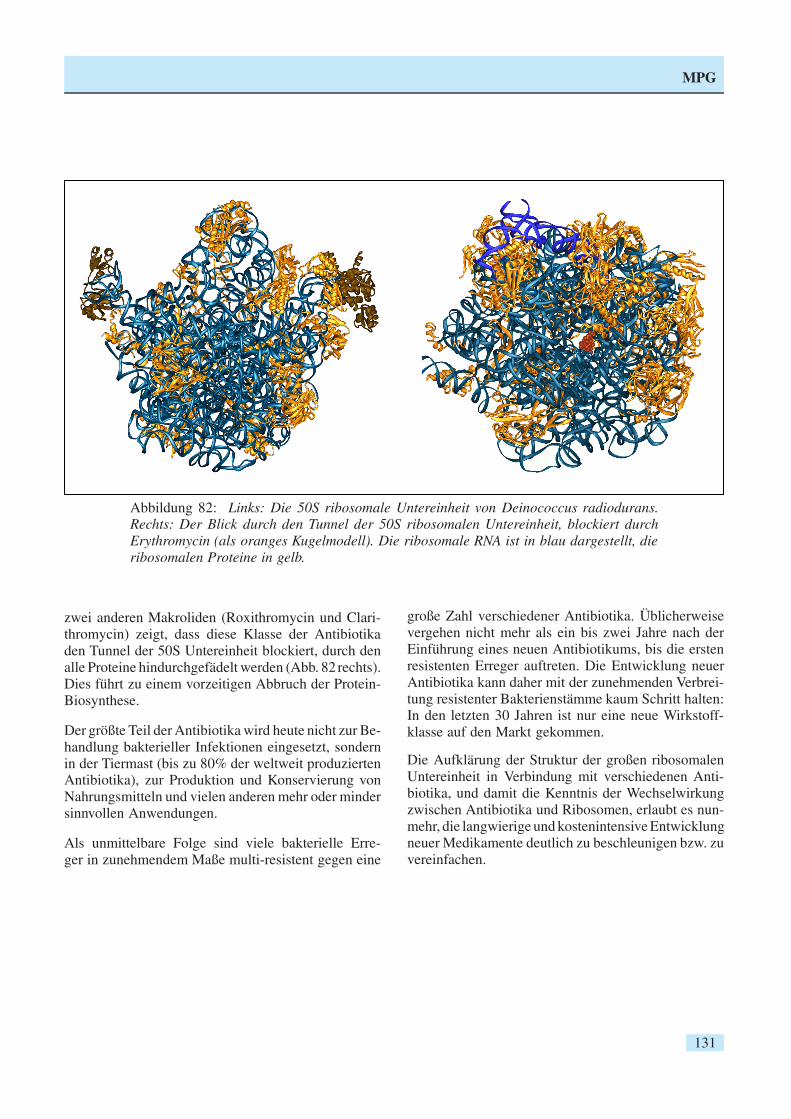
Ebenfalls im Jahr 2000 wurde die Struktur der 50S ri-bosomalen Untereinheit von Haloarcula marismortuipubliziert. Dieser Organismus ist zum einen wenig ge-eignet, um die Wechselwirkung des Ribosoms mit An-tibiotika zu untersuchen, zum anderen führen die Kris-tallisationsbedingungen zu einem inaktiven Ribosom.Das hat unter anderem zu einem inkorrekten Modell fürdie katalytische Aktivität der 50S Untereinheit geführt.In 2001 konnte die Arbeitsgruppe aber die Struktur der50S ribosomalen Untereinheit von Deinococcus radi-odurans (Abb. 82 links) aufklären. Dieses Bakterium,das seinen Namen einer extremen Stabilität gegenüberradioaktiver Strahlung verdankt, bietet ein nahezu per-fektes Modell-System, um die Ribosomen-Antibiotika-Wechselwirkungen zu untersuchen. Als Folge konntendie Strukturen von therapeutisch wichtigen Antibiotikawie Clindamycin, Chloramphenicol und Erythromy-cin im Komplex mit der 50S ribosomalen Untereinheitermittelt werden.
Chloramphenicol ist sehr wirkungsvoll in der Behand-lung eines breiten Spektrums bakterieller Infektionen,einschließlich schwerer anaerober Infektionen. Clinda-mycin wird unter anderem zur Bekämpfung anaeroberInfektionen und Cocci-Bakterien sowie zur Behandlungvon Pneumocystis induzierter Lungenentzündung vonAIDS-Patienten verwendet. Beide Antibiotika bindendirekt im Peptidyltransferase-Zentrum des Ribosoms.Die Strukturen der ribosomalen Untereinheit mit Clin-damycin und Chloramphenicol bestätigen die Vermu-tung, dass diese Antibiotika die Protein-Biosynthesedurch ,,molekulare Mimikry“ unterbrechen: Sie be-setzen nämlich Bereiche, die denen von zelleige-nen Aminosäuren ähnlich sind, und werden deshalbdurch das Ribosom gebunden. Da sie aber naturgemäßkeine Peptidbindung eingehen können, bringen sie diePeptidyltransferase-Reaktion zum Stillstand.
Die so genannten Makrolide-Antibiotika, deren be-kanntester Vertreter Erythromycin ist, werden zur Be-kämpfung einer Vielzahl bakterieller Infektionen ver-wendet. Anhand biochemischer Daten war bereitsbekannt, dass Erythromycin die Peptidyltransferase-Reaktion erst nach der Bildung einer kurzen Amino-säurekette unterbindet. Die Struktur der 50S Unter-einheit im Komplex mit Erythromycin sowie mit den
130
MPG
Abbildung 82: Links: Die 50S ribosomale Untereinheit von Deinococcus radiodurans.Rechts: Der Blick durch den Tunnel der 50S ribosomalen Untereinheit, blockiert durchErythromycin (als oranges Kugelmodell). Die ribosomale RNA ist in blau dargestellt, dieribosomalen Proteine in gelb.
zwei anderen Makroliden (Roxithromycin und Clari-thromycin) zeigt, dass diese Klasse der Antibiotikaden Tunnel der 50S Untereinheit blockiert, durch denalle Proteine hindurchgefädelt werden (Abb. 82 rechts).Dies führt zu einem vorzeitigen Abbruch der Protein-Biosynthese.
Der größte Teil der Antibiotika wird heute nicht zur Be-handlung bakterieller Infektionen eingesetzt, sondernin der Tiermast (bis zu 80% der weltweit produziertenAntibiotika), zur Produktion und Konservierung vonNahrungsmitteln und vielen anderen mehr oder mindersinnvollen Anwendungen.
Als unmittelbare Folge sind viele bakterielle Erre-ger in zunehmendem Maße multi-resistent gegen eine
große Zahl verschiedener Antibiotika. Üblicherweisevergehen nicht mehr als ein bis zwei Jahre nach derEinführung eines neuen Antibiotikums, bis die erstenresistenten Erreger auftreten. Die Entwicklung neuerAntibiotika kann daher mit der zunehmenden Verbrei-tung resistenter Bakterienstämme kaum Schritt halten:In den letzten 30 Jahren ist nur eine neue Wirkstoff-klasse auf den Markt gekommen.
Die Aufklärung der Struktur der großen ribosomalenUntereinheit in Verbindung mit verschiedenen Anti-biotika, und damit die Kenntnis der Wechselwirkungzwischen Antibiotika und Ribosomen, erlaubt es nun-mehr, die langwierige und kostenintensive Entwicklungneuer Medikamente deutlich zu beschleunigen bzw. zuvereinfachen.
131