Embed Size (px)
Citation preview

Ab–initio Statistikzur Modellierungdes Systems NiAl
Diplomarbeit
von
Frank Lechermann
Hauptberichter : Prof. Dr. H.-E. Schaefer
Mitberichter : Prof. Dr. D. Schweitzer
Institut fur Theoretische und Angewandte Physik
Universitat Stuttgart
Januar 2000


Inhaltsverzeichnis
1 Einleitung 1
2 Cluster-Variationsmethode (CVM) 4
2.1 Einfuhrung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2 Cluster-Entwicklung (CE) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2.1 Orthonormale Clusterfunktionen . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.2.2 Cluster-Entwicklung der Energie . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2.3 Besetzungswahrscheinlichkeit der Cluster . . . . . . . . . . . . . . . . . . . . 12
2.2.4 Berucksichtigung von Symmetrien . . . . . . . . . . . . . . . . . . . . . . . . 13
2.2.5 Nichtorthogonale Basissatze . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.2.6 V-Matrix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.3 Das Variationsprinzip . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
2.3.1 Zur Statistik im kanonischen Ensemble . . . . . . . . . . . . . . . . . . . . . . 19
2.3.2 Variation der freien Energie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.3.3 Bragg-Williams-Naherung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3.4 Bethe-Naherung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.3.5 Entropie in der CVM . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
2.3.6 Das Funktional der freien Energie . . . . . . . . . . . . . . . . . . . . . . . . 27
2.3.7 CVM-Gleichungen in orthogonaler Basis . . . . . . . . . . . . . . . . . . . . . 28
2.4 Diskussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.4.1 Mean-Field-Charakter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.4.2 Problem der CVM . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.4.3 Vergleich mit Reihenentwicklung . . . . . . . . . . . . . . . . . . . . . . . . . 31
3 Thermodynamik von Phasendiagrammen binarer Systeme 32
3.1 Beschreibungsgroßen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
i

ii INHALTSVERZEICHNIS
3.1.1 Energien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
3.1.2 Potentiale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
3.2 Phasenstabilitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.2.1 Stabilitatsbedingungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.2.2 Phasenkoexistenz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
3.2.3 Mischungslucke (miscibility gap) . . . . . . . . . . . . . . . . . . . . . . . . . 40
3.2.4 Systeme am Phasenubergang . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
4 Elektronentheorie 43
4.1 Eigenwertproblem . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
4.2 Dichtefunktionaltheorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
4.2.1 Theoreme von Hohenberg und Kohn . . . . . . . . . . . . . . . . . . . . . . . 44
4.2.2 Kohn-Sham-Formalismus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
4.2.3 Lokale Dichtenaherung (LDA) . . . . . . . . . . . . . . . . . . . . . . . . . . 45
4.3 Mixed-basis-Pseudopotentialmethode . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
4.3.1 Periodisches Potential und Wellenfunktionen . . . . . . . . . . . . . . . . . . 46
4.3.2 Berechnung von Kraften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
5 Ab-initio Statistik 48
5.1 Energieterme in realen Systemen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
5.2 Bestimmung der ECIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
5.2.1 Direct Configurational Averaging (DCA) . . . . . . . . . . . . . . . . . . . . 50
5.2.2 Structure Inversion Method (SIM) . . . . . . . . . . . . . . . . . . . . . . . . 51
5.2.3 Berucksichtigung von Relaxationsphanomenen . . . . . . . . . . . . . . . . . 52
5.3 Natural Iteration Methode (NIM) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
5.3.1 Tetraedernaherung auf fcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.3.2 Tetraedernaherung auf bcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . 57
5.3.3 Zur Numerik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
5.3.4 Ablaufplan der ab-initio Modellierung . . . . . . . . . . . . . . . . . . . . . . 60
6 System NiAl 63
6.1 Grundzustandsbetrachtungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
6.1.1 Auswahl der Strukturen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
6.1.2 Abschneideenergie und Anzahl der k-Punkte . . . . . . . . . . . . . . . . . . 64

INHALTSVERZEICHNIS iii
6.1.3 Energien der geordneten Strukturen . . . . . . . . . . . . . . . . . . . . . . . 66
6.1.4 ECIs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72
6.1.5 Relaxation der L10-Struktur . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
6.1.6 Die Struktur Ni5Al3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
6.2 Thermodynamik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
6.2.1 CVM-Programm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
6.2.2 System auf fcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
6.2.3 System auf bcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
6.2.4 Inkoharentes System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
6.2.5 Berucksichtigung von Leerstellen . . . . . . . . . . . . . . . . . . . . . . . . . 105
7 Zusammenfassung 108
A Zur orthogonalen Entwicklung 111
A.1 Orthonormalitat und Vollstandigkeit der Tschebyscheff Polynome . . . . . . . . . . . 111
A.2 Orthonormalitat und Vollstandigkeit der Clusterfunktionen . . . . . . . . . . . . . . 112
B CVM-Naherungen 114
C Strukturen 115
C.1 fcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
C.2 bcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
C.3 Ni5Al3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
D Prototyp-Rechnungen 121
D.1 fcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
D.1.1 Ising-Modell . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
D.1.2 Binare Legierung mit antiferromagnetischer reiner NN-WW . . . . . . . . . . 123
D.2 bcc-Gitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 124
D.2.1 Binare Legierung mit antiferromagnetischer NN- und NNN-WW . . . . . . . 124
E Zur Phasenstabilitat im (µ∗A,Ω)−Formalismus 125
F T − µ∗A−Diagramme fur NiAl 127

Kapitel 1
Einleitung
Mit der Formulierung der fundamentalen Gesetze der Thermodynamik und Statistik Ende des19. Jahrhunderts wurde der Grundstein fur das theoretische Verstandnis temperaturabhangigerPhanomene gelegt. Die Erkenntnis, daß das thermodynamische Verhalten eines Systems aus demkomplizierten Wechselspiel von Energie und Entropie resultiert, kann als eine der großten physika-lischen Errungenschaften gewertet werden. Dabei war der erste Gedanke, daß die Physik bei derBeschreibung von Vielteilchensystemen aufgrund der gigantischen Anzahl von Freiheitsgraden anihre Grenzen gestoßen ware. Die Uberwindung dieser Problematik lag in dem Verstandnis, daß allerelevanten thermodynamischen Eigenschaften als Ergebnis eines Mittelungsprozesses uber die ein-zelnen moglichen Zustande eines solchen Systems gewonnen werden konnen. So elegant und befrie-digend sich dies auch auf dem Papier darstellte, so schwierig gestaltete sich die explizite Berechnungsolcher gemittelten Großen in der Realitat. Bereits die Analyse der Wechselwirkung mikroskopi-scher Teilchen mit klassischen Methoden erwies sich als unzureichend. Uber lange Zeit hinweg wardie Statistik daher auf qualitative Aussagen bezuglich klassischer Phanomene beschrankt.Mit der Entwicklung der Quantenmechanik in den 20er Jahren dieses Jahrhunderts wurde dannerstmals eine korrekte Beschreibung der physikalischen Welt auf mikroskopischer Ebene erzielt.Aber schon die analytische Behandlung von kleinsten realen Mehrteilchensystemen ist nicht mehrhandhabbar, von der Ausfuhrung des statistischen Mittelungsprozesses ganz zu schweigen.Im Zuge der fortschreitenden Entwicklung der Rechnertechnologie entwickelte man nun neuarti-ge physikalische Ansatze, die es moglich machen sollten, sowohl die Berechnung der energetischenWechselwirkungen in Mehrteilchensystemen als auch das sampling-Problem in den Griff zu bekom-men. Beide Problemkreise wurden zuerst getrennt angegangen.Schon in den 30er Jahren machten Bragg und Williams [1] sowie Bethe [2] erste einfache Ansatze, umdie wegen der hohen Teilchenzahl in realen thermodynamischen Systemen unmogliche Durchfuhrungdes kompletten statistischen Mittelungsprozesses geeignet zu nahern. Dabei wurden sie von demGrundgedanken geleitet, daß sich die Wahrscheinlichkeit fur das Auftauchen eines bestimmtenSystemzustands genahert aus den Wahrscheinlichkeiten damit kompatibler Subsystemzustande ge-nerieren lassen sollte. Diese Subsysteme bestanden in den besagten Naherungen fur den Fall vonGittersystemen lediglich aus einzelnen Gitterpunkten bzw. Paaren von Gitterpunkten. Dadurchließen sich die kombinatorischen Schwierigkeiten auf ein Mindestmaß reduzieren. Die ersten Schrit-te hinsichtlich einer Verallgemeinerung dieses Gedankens auf großere Subsysteme und einer damitverbundenen Verbesserung in der Naherung wurden erstmals Ende der 40er Jahre von Yang [3, 4]und Li [5] getatigt. In ihrer quasichemischen Methode waren die Uberlappungen dieser Subsystemejedoch nicht korrekt berucksichtigt. Eine dahingehend konsistente Methode stellte Kikuchi [6] imJahre 1951 mit der ersten Formulierung einer Cluster-Variationsmethode (CVM) vor. Parallel zu
1

2 KAPITEL 1. EINLEITUNG
diesem Ansatz wurden etwa zur gleichen Zeit die erste Zuge eine andere Moglichkeit zur Handha-bung des Mittelungsprozesses entwickelt, die Monte-Carlo-Simulation (MC).Diese und andere Methoden waren jedoch lange Zeit auf die Beschreibung von Prototypsystemenbeschrankt, da erst mit der Geburt der Dichtefunktionaltheorie Mitte der 60er Jahre und ihrerrechenbaren Version, der lokalen Dichtenaherung (LDA), eine Moglichkeit entdeckt wurde, dieGrundzustandsenergie eines realen Festkorpers ab-initio, also ohne Verwendung von empirischenAnpaßparametern, nahezu exakt zu bestimmen. Es dauerte dann noch bis Mitte der 80er Jahre, bisdie elektronentheoretischen Computer-Codes derart weit entwickelt und die Rechner ausreichendschnell waren, um den alten Traum Wirklichkeit werden zu lassen und erstmals Phasendiagrammevon mehrkomponentigen Systemen ”from first principles” rechenbar waren. Neben der CVM undder MC als statistische Methode ist in diesem Zusammenhang auch die Molekulardynamik in ihrerab-initio Version nach Car und Parrinello [7] als herausragendes Beispiel zu nennen.
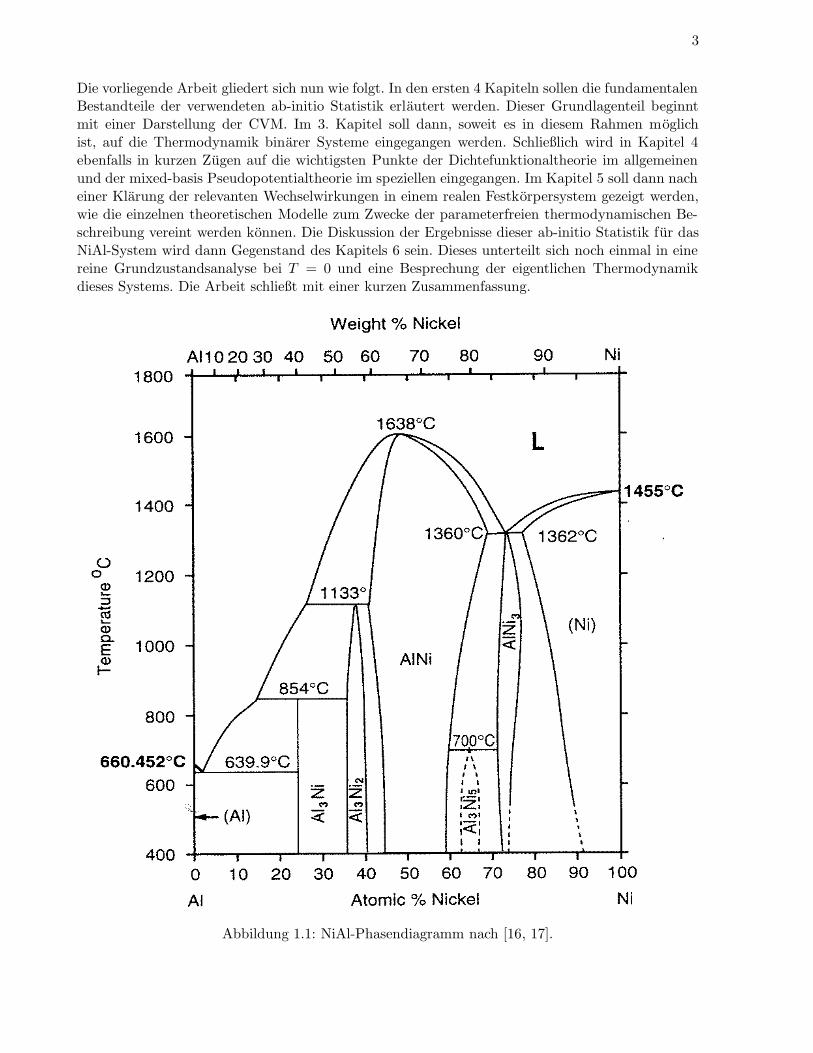
Ziel dieser Arbeit war es nun, eine ab-initio Statistik aufbauend auf der CVM zur theoretischen Mo-dellierung des binaren Systems NiAl zu formulieren. Hierzu sollten nach einem 1983 von Connollyund Williams [8] vorgestellten Verfahren ab-initio gerechnete Grundzustandsenergien auf geeigne-te Wechselwirkungsparameter abgebildet werden. Die auf der Dichtefunktionaltheorie basierendenab-initio Rechnungen wurden mit einer mixed-basis Pseudopotentialmethode durchgefuhrt. DerComputer-Code hierzu wurde in der Elektronentheoriegruppe von Prof. Fahnle maßgeblich vonBernd Meyer entwickelt [9]. Zentrales Anliegen war nun eine parameterfreie Bestimmung des NiAl-Phasendiagrammes. Diese Aufgabe wurde in der Vergangenheit, basierend auf anderen elektronen-theoretischen Methoden, bereits von mehreren Gruppen angegangen [10, 11, 12, 13, 14].Das NiAl-System zeichnet sich durch seine hohe Schmelztemperaturen aus. Diese Eigenschaft so-wie die geringe Dichte, verbunden mit einer hohen Zugfestigkeit, machen die NiAl-Verbindungen zuhochinteressanten Werkstoffen. Das experimentelle NiAl-Phasendiagramm ist in Abbildung (1.1)dargestellt. Die stabilen festen Phasen der reinen Elemente grunden auf einem fcc-Gitter. Im Ni-armen Gebiet bilden die orthorhombische D011-NiAl3- und die trigonale D519-Ni2Al3-Struktur diereinen Phasen. Nahe der aquiatomaren Zusammensetzung befindet sich eine NiAl-Phase auf einembcc-Grundgitter mit Fernordnung B2, die ohne vollig zu entordnen bei 1638 C in der Stochiometriein die flussige Phase ubergeht. In der Ni-reichen Region ist in der Umgebung der Stochiometrie 0.75wiederum eine fcc-Struktur, Ni3Al-L12, bis 1360 C stabil. Im Ubergangsbereich zwischen diesenbeiden Grundgittern befindet sich eine Tieftemperaturphase mit der Zusammensetzung Ni5Al3, dieeine Ga3Pt5-Struktur mit orthorhombischer Symmetrie besitzt. Die Regionen ohne Kennzeichnungin (1.1) entsprechen Mischphasen der horizontal angrenzenden reinen Phasen.Die Berechnung eines solchen Phasendiagrammes mittels der CVM kann nun in verschiedenenNaherungen durchgefuhrt werden. Diese zeichnen sich durch die Wahl der maximalen Cluster, al-so der Große der generierenden Subsysteme, aus. In dieser Arbeit wurden die Moglichkeiten undGrenzen der sogenannten Tetraedernaherung untersucht.Da sich Ni und Al um einen Faktor von etwa 1.5 im atomaren Volumen unterscheiden, ist zu er-warten, daß Volumeneffekte im Phasendiagramm eine wichtige Rolle spielen. Die Berucksichtigungsolcher Effekte stellte ebenfalls ein Anliegen dieser Arbeit dar. Wie aus experimentellen Unter-suchungen weiter bekannt ist, gibt im NiAl-System nahe der Stochiometrie 0.5 die vergleichsweisegroße Anzahl von Antistrukturatomen und Leerstellen Anlaß zu einer ausgebildeten Defektstruktur.Da eine solche Defektstruktur in einer realistischen thermodynamischen Behandlung nicht ausge-schlossen werden darf, war die Erweiterung der CVM zur ab-initio Berucksichtigung von Leerstellenbei beliebiger Zusammensetzung und Temperatur ein weiteres Ziel. Die einzige bekannte CVM-Rechnung zur Defektkonzentration in B2-NiAl unter Berucksichtigung von Leerstellen arbeitet mitempirischen Daten und geeigneten Fitparametern [15].
3
Die vorliegende Arbeit gliedert sich nun wie folgt. In den ersten 4 Kapiteln sollen die fundamentalenBestandteile der verwendeten ab-initio Statistik erlautert werden. Dieser Grundlagenteil beginntmit einer Darstellung der CVM. Im 3. Kapitel soll dann, soweit es in diesem Rahmen moglichist, auf die Thermodynamik binarer Systeme eingegangen werden. Schließlich wird in Kapitel 4ebenfalls in kurzen Zugen auf die wichtigsten Punkte der Dichtefunktionaltheorie im allgemeinenund der mixed-basis Pseudopotentialtheorie im speziellen eingegangen. Im Kapitel 5 soll dann nacheiner Klarung der relevanten Wechselwirkungen in einem realen Festkorpersystem gezeigt werden,wie die einzelnen theoretischen Modelle zum Zwecke der parameterfreien thermodynamischen Be-schreibung vereint werden konnen. Die Diskussion der Ergebnisse dieser ab-initio Statistik fur dasNiAl-System wird dann Gegenstand des Kapitels 6 sein. Dieses unterteilt sich noch einmal in einereine Grundzustandsanalyse bei T = 0 und eine Besprechung der eigentlichen Thermodynamikdieses Systems. Die Arbeit schließt mit einer kurzen Zusammenfassung.
Abbildung 1.1: NiAl-Phasendiagramm nach [16, 17].

Kapitel 2
Cluster-Variationsmethode (CVM)
2.1 Einfuhrung
Wie in der Einleitung bereits erwahnt, wurde der Grundstein der Cluster-Variationsmethode imJahre 1951 von Ryo Kikuchi gelegt [6]. Dessen verallgemeinerte Mean-Field-Theorie stellt eine Nahe-rungsmethode zur Behandlung der thermodynamischen Eigenschaften von periodischen Systemendar. Fernab von kritischen Bereichen liefert die Methode prinzipiell beliebig genaue Resultate undist aufgrund ihres analytischen Charakters in diesen Bereichen als eine wahre Alternative zu auf-wendigen MC- oder MD-Simulationen zu werten. Zwar kann sich die Algebra zur Formulierung derrelevanten Gleichungssysteme mitunter aufwendig gestalten, sind diese jedoch einmal aufgestellt,so ist der zeitliche Aufwand zur numerischen Losung im Vergleich zu einer Monte-Carlo-Simulationum einen Faktor 100 geringer.Auf die ersten Arbeiten von Kikuchi folgten Umformulierungen und Vereinfachungen in der verall-gemeinerten Darstellung der Theorie [18, 19, 20]. Da die CVM ursprunglich als verbesserter Ansatzfur die Beschreibung von kritischen Phanomenen konzipiert wurde, geriet sie mit Aufkommen derRenormierungsgruppentheorie fast in Vergessenheit. Erst die Anwendbarkeit der Methode auf prak-tische Fragestellungen zur Phasenstabilitat, motiviert durch Prototyp-Rechnungen von van Baal[21], verhalf der CVM Ende der 70er Jahre zu neuem Aufschwung.Nach ersten Berechnungen von Phasendiagrammen mittels empirischen Wechselwirkungsparame-tern wurden 1984 die uber die Jahre erzielten Verallgemeinerungen der CVM von Sanchez et al.[22] in einen rigorosen mathematischen Formalismus eingebettet, der gerade die Verknupfung derTheorie mit elektronentheoretischen ab-initio Methoden nahelegt. Die hier gewahlte Darstellungder Theorie orientiert sich besonders in der Formulierung der reinen Cluster-Entwicklung stark andiesem Artikel.
2.2 Cluster-Entwicklung (CE)
2.2.1 Orthonormale Clusterfunktionen
Die Grundaussage der Theorie lautet, daß bei Vorgabe eines Grundgitters bekannter Symmetriejede konfigurationsabhangige Große des Systems als exakte Funktion der Besetzungszahlen dar-stellbar ist. Um dies einzusehen, betrachtet man ein Gitter bestehend aus N Gitterpunkten. Einebestimmte Konfiguration des Gitters wird nun durch die Besetzung der Gitterpunkte mit Teilchen
4

2.2. CLUSTER-ENTWICKLUNG (CE) 5
der am System beteiligten M Komponenten bestimmt. Die Besetzung eines Gitterpunkts p wirdmathematisch durch eine Besetzungszahl σp ausgedruckt. Der Wertebereich dieser Besetzungszahlist prinzipiell willkurlich wahlbar, durchgesetzt hat sich jedoch in Anlehnung an das Ising-Modelldie Verwendung von ”Spinvariablen”, die eine symmetrische Behandlung der auftretenden Kompo-nenten ermoglicht
M gerade : σp = ±m,±(m− 1), . . . ,±1 mit M = 2m
M ungerade : σp = ±m,±(m− 1), . . . ,±1, 0 mit M = 2m+ 1 .
(2.1)
Eine bestimmte Konfiguration des Gitters wird somit eindeutig durch einen N-dimensionalen Vektorfolgender Gestalt beschrieben
σ = σ1, σ2, . . . , σN . (2.2)
Durch Definition eines inneren Produkts ist es nun moglich, einen dem Raum der Konfigurationenzugeordneten euklidischen Vektorraum V von Funktionen auf dem betrachteten Gitter zu bilden.Seien f(σ) und g(σ) zwei Elemente des Vektorraums V, so soll folgende Zuordnung gelten :
V × V → R
f, g 7→ 〈f, g〉 = ρ0NTr(N)f · g , (2.3)
wobei unter Tr(N) der Spuroperator uber die MN Konfigurationen zu verstehen und ρ0N eine No-
mierungskonstante ist
Tr(N) =∑
σ1
∑
σ2
· · ·∑
σN
∧ ρ0N = M−N . (2.4)
Es laßt sich leicht verifizieren, daß obige Zuordnung den Anforderungen an ein inneres Produktgenugt. Die weitere Vorgehensweise besteht nun darin, eine vollstandige orthonormale Basis in die-sem Vektorraum zu konstruieren. Wahrend die Orthonormalitat keine notwendige Bedingung furdie Anwendung der Clustertheorie bedeutet, ist die Forderung nach Vollstandigkeit zwingend furdie Exaktheit der Methode im thermodynamischen Limes.1
Die Konstruktion einer vollstandigen orthonormalen Basis fur einen Gitterpunkt p, d.h. zur Dar-stellung einer beliebigen, von σp abhangigen Funktion, ist denkbar einfach, wenn man sich klarmacht, daß bei Vorgabe von n Stutzstellen ein Polynom vom Grad (n − 1) in eindeutiger Weiseeinen geforderten Funktionsverlauf beschreibt. In unserem Fall ist die Zahl der Stutzstellen durchdie Zahl der Komponenten M bestimmt. Als Ausgangspunkt fur die Konstruktion einer Familievon orthonormalen Polynomen dient allgemein der Satz linear unabhangiger Polynome
1, σp, σ2p, . . . , σ
M−1p . (2.5)
Die lineare Unabhangigkeit dieser Polynome ist eine Konsequenz der Tatsache, daß
a0 + a1σp + · · · + aM−1σM−1p ≡ 0 , σp ∈ ±m,±(m− 1), . . . ,±1, (0) (2.6)
gilt, wenn und nur wenn a0 = a1 = · · · = aM−1 = 0.Mittels des Gram-Schmidtschen Orthogonalisierungsverfahren laßt sich daraus ein Satz orthonor-maler Polynome θn(σp) erzeugen. Da es sich bei σp um eine diskrete Variable handelt, hat die Wahl
1Hiervon abweichend existiert auch eine andere Definition des inneren Produkts, bei der die Summation uberdie Konfigurationen auf solche mit konstanter Konzentration der Komponenten beschrankt ist [23]. Die Vor- undNachteile dieses alternativen Aufbaus der Theorie sollen hier jedoch nicht weiter diskutiert werden (s. hierzu [23],[24]und [25]).

6 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
des inneren Produkts zur Folge, daß die so konstruierten orthonormalen Polynome gleichbedeutendmit den M ersten diskreten Tschebyscheff Polynomen sind2
θ2s(σp) =s∑
k=0
c(s)k σ2k
p mit s = 0, 1, . . . ,m− 1, (m) (2.7)
θ2s+1(σp) =s∑
k=0
d(s)k σ2k+1
p mit s = 0, 1, . . . ,m− 1 . (2.8)
Diese Polynome genugen damit folgender Orthonormalitatsrelation
〈θn(σp), θn′(σp)〉 = δnn′ . (2.9)
Außerdem bilden die so erzeugten θn(σp) eine vollstandige Basis in dem Funktionenraum, der demRaum der Konfigurationen des Gitterpunkts p zugeordnet ist (s. (A.1))
1
M
M−1∑
n=0
θn(σp)θn(σ′p) = δσpσ′p . (2.10)
Betrachtet man Funktionen, die von den Spinvariablen σp an vielen Gitterpunkten p abhangen, dannerhoht sich entsprechend die Dimension des zugehorigen Funktionenraums. Vermoge des direktenProdukts kann nun jedoch in einfacher Weise eine Basis in diesem erweiterten Funktionenraum kon-struiert werden, welche wiederum die Eigenschaft der Vollstandigkeit und Orthonormalitat besitzt.So gilt fur die Basis B in einem MN -dimensionalen Funktionenraum folgende Konstruktionsvor-schrift
B =
1θ1(σ1)
...θM−1(σ1)
⊗
1θ1(σ2)
...θM−1(σ2)
⊗ · · · ⊗
1θ1(σN )
...θM−1(σN )
. (2.11)
Die einzelnen Basisfunktionen lassen sich nun nach sogenannter Clusterzugehorigkeit und weiternach dem Grad sortieren.Ein Cluster in einem Kristall mit bekanntem Grundgitter (z.B. sc, fcc, ...) ist eindeutig durchdie in ihm enthaltenen Gitterpunkte definiert. Dabei ist wichtig, daß sich diese Cluster sehr wohldurchdringen konnen (s. Abb. (2.1)). Auf einem Gitter, bestehend aus N Gitterplatzen, existierengenau 2N Cluster.3
Eine charakteristische Clusterfunktion fur einen bestimmten Cluster α = p, p′, . . . , p′′ mit einemspeziellen Satz von Indizes s = n, n′, . . . , n′′ schreibt sich jetzt folgendermaßen
φαs(σp, σp′ , . . . , σp′′) = θn(σp)θn′(σp′) · · · θn′′(σp′′) . (2.12)
Die Menge aller Clusterfunktionen eines bestimmten Clusters α bilden nun wiederum eine vollstandi-ge, orthogonale Basis in dem Unterfunktionenraum, der dem Raum der Clusterkonfigurationenzugeordnet ist. Die verschiedenen Unterraume sind naturlich ebenso orthogonal zueinander
〈φαs, φβs′〉 = δαβδss′ , (Orthonormalitat) (2.13)
ρ0α
∑
s
φαs(σ)φαs(σ′) = δσσ′ . (Vollstandigkeit) (2.14)
2Der Term (m) in (2.7) kommt im Fall einer ungeraden Zahl M von Komponenten zum Tragen.3Dies laßt sich leicht dadurch nachvollziehen, daß man jedem Gitterpunkt gedanklich eine 1 fur im Cluster enthalten
und eine 0 fur nicht enthalten zuordnen kann. Es gibt nun eben genau 2N Moglichkeiten, die Einsen und Nullen aufdie Gitterpunkte zu verteilen.

2.2. CLUSTER-ENTWICKLUNG (CE) 7
mit ρ0α = M−|α|, wobei hier und im weiteren |α| die Zahl der Gitterpunkte im Cluster α kennzeich-
net. Die Vollstandigkeit wird in Anhang (A.2) gezeigt.Aus dem bisherigen folgt nun, daß jede beliebige Große, welche eine Funktion der Konfigurationist, eindeutig und formal exakt nach Clusterfunktionen entwickelt werden kann :
f(σ) = f0 +∑
α
∑
s\0fαsφαs(σ) mit fαs = 〈φαs, f〉 . (2.15)
Der Ausdruck f0 bezeichnet den Entwicklungskoeffizient, der zum sogenannten leeren Clustergehort, welcher bei der Bildung des direkten Produkts (1.9) entsteht und als konfigurationsun-abhangige Große die Rolle des Aufpunkts ubernimmt. Daher muß bei der Summation uber dieGrade der Clusterfunktionen dieser ”nullte” Grad ausgeschlossen werden, d.h. n, n ′, . . . , n′′ 6= 0.Aufgrund der orthonormalen Clusterfunktionen ist es also moglich, die Entwicklungskoeffizientenfαs durch Projektion zu erhalten. Dies ist eine direkte Folge der unterliegenden Linearen Algebra,die ja beispielsweise auch den Formalismus der Quantenmechanik mitbestimmt.Im nachsten Abschnitt soll am Beispiel der Cluster-Entwicklung der Konfigurationsenergie dieAnwendung der dargestellten Grundgedanken der Theorie demonstriert sowie die physikalische Be-deutung der Entwicklungskoeffizienten fαs aufgezeigt werden.
Abbildung 2.1: Ausgewahlte Cluster auf dem fcc-Gitter (aus [23]).

8 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
2.2.2 Cluster-Entwicklung der Energie
Bei den meisten Anwendungen der Theorie kommt der Entwicklung der Konfigurationsenergie einezentrale Bedeutung zu. Es soll hier aber nochmal darauf hingewiesen werden, daß jede Funktion derKonfiguration (z.B. Volumen, phononische freie Energie, elektr. Widerstand, ...) prinzipiell Cluster-entwickelbar ist. Im Rahmen der CVM mit dem Ziel der Beschreibung der Thermodynamik einesKristallsystems wird sich jedoch die Entwicklung der Energie als eine fundamentale Problemstellungerweisen, weshalb im weiteren einige grundsatzliche Bemerkungen hierzu gemacht werden sollen.Fur das folgende wollen wir uns auf den Spezialfall eines binaren Systems beschranken, da diebesprochenen Prinzipien allgemein gelten und die vereinfachte Bezeichnungsweise das Verstandniserleichtert. Da namlich im binaren Fall der Grad der Basisfunktionen fur einen Gitterpunkt maximaln = 1 ist, kann man auf die Summation uber s verzichten und infolgedessen die Energie schreibenals
E = K0 +∑
α
Kαφα mit s = 1 . (2.16)
Die Clusterfunktionen φα reduzieren sich nun auf einfache Produkte, gebildet aus den Spinvariabenσp der im Cluster α enthaltenen Gitterpunkte p
φα =∏
p∈ασp . (2.17)
Die Entwicklungskoeffizienten, in diesem Fall auch effective cluster interactions (ECI) genannt,ergeben sich wiederum zu
Kα = 〈φα, E〉 ⇔ Kα = 2−N2N∑
σφα(σ)E(σ) . (2.18)
Um die Bedeutung der ECIs zu verstehen, eignet sich eine etwas andere Schreibweise von (2.18)
Kα = 2−|α|∑
σα
(σp1σp2 · · · σp|α|)︸ ︷︷ ︸
=φα
2|α|−N∑
σN−α
E(σα;σN−α)
︸ ︷︷ ︸
=Wσp1σp2 ···σp|α|
. (2.19)
Zwei Merkmale der ECIs sind zu vermerken.
1. Da in (2.18) uber samtliche Konfigurationen von Cluster und Restkristall summiert wird,sind die ECIs konfigurationsunabhangig.
2. Die ECIs entsprechen keineswegs Wechselwirkungspotentialen mit kurzer Reichweite, wie dieAnalogie zum Ising-Modell nahelegt, sondern sie enthalten formal samtliche Wechselwirkun-gen, vorausgesetzt die Konfigurationsenergie E(σ) ist hinreichend genau bekannt. So ist derAusdruck WIJ...K gleichzusetzen mit der mittleren Energie aller Konfigurationen mit einemI-Atom an p1, J an p2,..., K an pn. Speziell fur die effective pair interactions (EPI) ergibt sich
Kpp′ =1
4(WAA +WBB −WAB −WBA) . (2.20)
Die ECIs sind also als Differenzen totaler Konfigurationsenergien zu verstehen, wobei dieGewichtung derselben, im obigen Fall uber das Vorzeichen, durch die Clusterfunktionen fest-gelegt ist.

2.2. CLUSTER-ENTWICKLUNG (CE) 9
Bei der bisherigen Betrachtung wurde immer stillschweigend angenommen, daß bei der Summeuber die Konfigurationen tatsachlich jede Konfiguration berucksichtigt wird. Im Realfall ist dasbei einer Zahl von 2N Konfigurationen und einem N in der Großenordnung der Loschmidt’schenZahl jedoch utopisch. Das entsprechende gilt fur die Zahl der betrachteten Cluster, die aquivalentskaliert.In diesem Zusammenhang muß weiter auch noch geklart werden, wie die Systeminformation in denFormalismus zu integrieren ist. Die Berechnungsvorschrift (2.18) geht davon aus, daß die EnergienE(σ) bereits vorliegen. Dies ist der Ansatzpunkt fur eine Anwendung der Theorie auf reale Sy-steme. Durch Vorgabe bekannter Konfigurationsenergien des betrachteten Systems lassen sich diegesuchten ECIs genahert ermitteln. Die genaue Diskussion, welche Konfigurationen dafur geeignetund wieviele erforderlich sind, soll auf das Kapitel 5 verschoben werden.Im folgenden sollen die fur eine Begrenzung hinsichtlich der Clusterzahl wichtigen Eigenschaftender ECIs betrachtet werden. Um bei einer bestimmten Clustergroße in der Entwicklung (2.16) ab-brechen zu konnen, sollte gewahrleistet sein, daß der Betrag der ECIs mit wachsender Clustergroßehinreichend schnell abfallt, damit der Fehler in der Energieberechnung vertretbar bleibt. Ein allge-meines Argument hierfur oder gar ein analytischer Ausdruck fur das Konvergenzverhalten der ECIsist jedoch nicht bekannt, da die Problematik stark systemabhangig ist. Prinzipiell sollten jedochfur die Anwendbarkeit der Cluster-Entwicklung zwei grundsatzliche Aussagen fur das betrachte-te System gelten. Zum einen mussen die ECIs dem Betrag nach abnehmen, wenn die Abstandeder Gitterpunkte bei großeren Clustern anwachsen. Zum anderen haben die Clusterwechselwirkun-gen mit steigender Zahl von im Cluster enthaltenen Gitterpunkten ebenfalls abzunehmen. BeideKriterien sollen kurz materialunabhangig andiskutiert werden
• Fur den Fall von effektiven Paarwechselwirkungen ist zu erwarten, daß wenn der Abstandzwischen den Gitterpunkten anwachst, die GroßenWIJ in die korrespondierenden PunkttermeWI und WJ aufspalten sollten. Also zum Beispiel
WAB ≈ 1
2(WA +WB) , (2.21)
da bei zunehmender Entfernung die energetische Korrelation verschwinden sollte. Verwendetman diese Naherung fur samtliche Terme in (2.20), so verschwindet Kpp′ identisch. DieseBetrachtungen zu reinen Paarclustern lassen sich naturlich in gleicher Weise auf Mehrkorper-cluster anwenden. Woraus sich allgemein folgern laßt, daß die ECIs fur Cluster, die weitvoneinander getrennte Gitterpunkte beinhalten, betragsmaßig klein sein sollten.
• Das zweite Kriterium soll wiederum ohne Beschrankung der Allgemeinheit anhand des Triplettcluster-ECI Kpp′p′′ untersucht werden. Dieser Clusterterm schreibt sich mit (2.19) als
Kpp′p′′ =1
8(WAAA+WABB−WAAB−WABA−WBAA−WBBB +WBBA +WBAB) . (2.22)
Die ersten 4 Terme in (2.22) entsprechen der Paarwechselwirkung mit einem A-Atom an p.Die zweiten 4 Terme sind zusammengefaßt wieder die Paarwechselwirkung nun mit einemB-Atom an p. Dies legt folgende Schreibweise fur Kpp′p′′ nahe:
Kpp′p′′ =1
2
([Kp′p′′
]
p=A − [Kp′p′′]
p=B
)
. (2.23)
Wenn also[Kp′p′′
]
p=I von der selben Großenordnung wie Kpp′ ist, dann sollte der Triplett-
ECI wegen (2.23) im Allgemeinfall betragsmaßig kleiner sein als der Paar-ECI. Da fur denPaar-ECI, wie sich leicht nachprufen laßt, die Beziehung
Kp′p′′ =1
2
([
Kp′p′′]
p=A +[
Kp′p′′]
p=B
)
(2.24)

10 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
gilt, sollten die ECIs mit steigender Zahl der im Cluster enthaltenen Gitterpunkte i. a. ver-schwinden.
In den bekannten Rechnungen fur reale Systeme wurde die Entwicklung in binaren Verbindungenmeist bis zum Tetraeder als maximalen Korpercluster gefuhrt, in den schwerer zu behandelndenternaren Systemen werden oftmals gar keine 3-dimensionalen Cluster berucksichtigt. Es wurde aberbeispielsweise fur das binare PdV-System auch schon bis zum 13- und 14-Punkt-Cluster auf demfcc-Gitter (s. Abb. (2.2)), welche 4. NN-Wechselwirkungen enthalten, entwickelt [26]. Ein Abbruchnach der NNN-Wechselwirkung fuhrte in diesem System zu falschen Grundzustanden.In der bisherigen Darstellung der Clusterentwicklung wurden Cluster, die sich durch eine Symme-trieoperation des Grundgitters ineinander uberfuhren lassen, einzeln in der Energie-Entwicklungaufgefuhrt. Es ist aber augenscheinlich klar, daß es moglich sein muß, den Beitrag solcher Clu-ster in der Entwicklung zusammenzufassen, wenn man makroskopische Systeme behandeln will.Die genaue Berucksichtigung der Symmetrie in der CE wird im Abschnitt 2.2.4 diskutiert werden.Demnach wird sich im Realfall die Summation in (2.16) allein uber die symmetrieinaquivalentenCluster erstrecken, wobei dann die reinen Clusterfunktionen durch die uber das gesamte Gittergemittelten Clusterfunktionen zu ersetzen sind. Deren Symmetrie bezuglich der Zusammensetzungan A- und B-Atomen spielt nun fur die Konvergenz der CE eine nicht unerhebliche Rolle.So besitzen in binaren Systemen die Cluster mit gerader (ungerader) Anzahl an Gitterpunkten auchgerade (ungerade) gemittelte Clusterfunktionen. Demzufolge tragen in der aquiatomaren Zusam-mensetzung die ungeraden Clusterwechselwirkungen nicht zur Energie bei, da die entsprechendengemittelten Clusterfunktionen an dieser Stelle einen Knoten aufweisen. Reine Paarwechselwirkun-gen wurden daher auf Energien symmetrisch zu c = 1
2 fuhren. Die asymmetrischen Anteile werdendemnach allein von den ungeraden Clustertermen getragen. Dieser Sachverhalt muß in der Energie-Entwicklung fur Systeme mit eben solchen starken Asymmetrien berucksichtigt werden.
Abbildung 2.2: Maximale Cluster der 13+14-Punkt-Naherung.

2.2. CLUSTER-ENTWICKLUNG (CE) 11
Zum Schluß dieses Abschnitts soll noch einmal anhand eines kleines Beispiels die Handhabungder Methode sowie die formale Exaktheit bei Mitnahme aller Konfigurationen und aller Clusterverdeutlicht werden.
Beispiel : Der binare 2-Punkt-Cluster
p’p
E(σ) = K0 +∑
α
Kαφα(σ) = K0 +Kpφp +Kp′φp′ +Kpp′φpp′
mit φp = σp ∧ φp′ = σp′ ∧ φpp′ = σpσp′
Berechnung der Kα mit (2.18) und σp(A) = +1 und σp(B) = −1 :
Kα = 2−|α|∑
σα
(σp1σp2 · · · σp|α|)
2|α|−2∑
σ2−α
E(σα;σ2−α)
⇒ K0 =1
4(EAA +EAB +EBA +EBB) =
1
4(EAA + 2EAB +EBB)
Kp =1
4(EAA +EAB −EBA −EBB) =
1
4(EAA −EBB)
Kp′ =1
4(EAA −EAB +EBA −EBB) =
1
4(EAA −EBB)
Kpp′ =1
4(EAA −EAB −EBA +EBB) =
1
4(EAA − 2EAB +EBB)
⇒ E(σ) = (1 + φp + φp′ + φpp′)EAA
4+ (1 − φpp′)
EAB2
+ (1 − φp − φp′ + φpp′)EBB
4
⇒ σp = σp′ = +1: E(σ) = EAA
σp = −σp′: E(σ) = EAB
σp = σp′ = −1: E(σ) = EBB
Bei der Rechnung wurde die Relation EAB = EBA benutzt, da die Punkte p und p′ hier symme-trieaquivalent sind. Dies erkennt man auch am identischen Auftreten von φp und φp′ . Offensichtlichlaßt sich die Rechnung unter Berucksichtigung der Symmetrie stark vereinfachen. Dies wird imubernachsten Abschnitt naher untersucht werden.

12 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
2.2.3 Besetzungswahrscheinlichkeit der Cluster
Um den Anschluß an die Gleichgewichtsthermodynamik zu erhalten, muß die Theorie eine Aussageuber die Wahrscheinlichkeit machen, mit der eine bestimmte Clusterkonfiguration im thermodyna-mischen Gleichgewicht auftaucht. Da die Wahrscheinlichkeit ρ(σ) fur eine bestimmte Kristallkon-figuration σ naturlich eine eindeutige Funktion eben dieser Konfiguration sein muß, laßt sich diesefolgendermaßen schreiben
ρ(σ) = ρ0N
1 +∑
α
∑
s\0ξαsφαs(σ)
. (2.25)
Die ξαs werden als multisite correlation functions bezeichnet
ξαs =1
ρ0N
〈φαs, ρ〉 = Tr(N)ρφαs , (2.26)
und sind gleichbedeutend mit den thermodynamischen Erwartungswerten der Clusterfunktionen.Die Wahrscheinlichkeit fur das Auftreten einer bestimmten Clusterkonfiguration τ des Cluster βerhalt man durch Ausspuren
ρβ(τ ) = Tr(N−β)ρ . (2.27)
Der Operator Tr(N−β) wirkt dabei allein auf die Clusterfunktionen φαs in (2.25)
Tr(N−β)φαs =∑
σ\σp∈βφαs . (2.28)
Es sind nun zwei Falle zu unterscheiden :
• Cluster α ist nicht in Cluster β enthalten (α 6⊆ β):Dann wird uber alle Konfigurationen von α summiert, und es gilt
Tr(N−β)φαs = M |N−β|〈φαs, 1〉(2.13)=
0 : α 6= 1
M |N−β| : α = 1.
Liegt eine Uberlappung der Cluster vor, so lassen sich die in β enthaltenen Gitterpunkte ausder Summe ziehen, und fur den verbleibenden Cluster gilt wieder obige Beziehung.
• Cluster α ist in Cluster β enthalten (α ⊆ β):Dann wird die entsprechende Clusterfunktion φαs einfach M |N−β|-fach aufsummiert,
Tr(N−β)φαs = M |N−β|φαs .
Nach Verrechnung der Normierungskonstanten erhalt man also schließlich
ρβ(τ ) = ρ0β
1 +∑
α⊆β
∑
s\0φαs(τ )ξαs
mit ρ0β = M |β| . (2.29)
Die Cluster-Theorie ist also insofern konsequent, daß die Cluster-Entwicklung auch fur Subsysteme,in diesem Fall durch den Cluster β beschrieben, anwendbar ist. Desweiteren garantieren Gleichung(2.25) und (2.29) auch die wichtigen thermodynamischen Relationen
Tr(N)ρ = 1 . (2.30)
Tr(α−β)ρα = ρβ . (2.31)
Dies ist hier eine direkte Folge der Entwicklung nach orthogonalen Basisfunktionen.

2.2. CLUSTER-ENTWICKLUNG (CE) 13
2.2.4 Berucksichtigung von Symmetrien
Im Beispiel von Abschnitt 2.2.2 wurden bereits Symmetriebetrachtungen zur Vereinfachung derRechnung verwendet. Da mit der Cluster-Theorie im Allgemeinfall Kristallsysteme behandelt wer-den, die zu einer bestimmten Raumgruppe gehoren, ist der Gedanke naheliegend, symmetrieaqui-valente Terme in der Entwicklung nach Clustern zusammenzufassen.Sei R ein Element der Raumgruppe G des Grundgitters (z.B. sc, fcc, ...) . Dann gilt fur jedekonfigurationsabhangige Funktion, die auf diesem Gitter definiert ist,
R−1 : f = f(Rσ) = f(σ) , (2.32)
weil durch die Wirkung von R nurmehr die Konfiguration als Ganzes umorientiert wird, die in-ternen Relationen zwischen den Gitterpunkten aber erhalten bleiben. Entsprechendes gilt fur dieClusterfunktionen φαs, wenn man bedenkt, daß durch Wirkung des Gruppenelements auch derCluster selbst transformiert wird,
R−1 : φαs(σ) = φR(αs)(Rσ) = φαs(σ) . (2.33)
Nun existieren MN Cluster und ebenso viele Konfigurationen σ. Da nun durch eine Gruppenope-ration sowohl die Clusterfunktionen φαs als auch die Funktion f(σ) selbst unverandert bleiben,folgt aus dem eindeutigen Zusammenhang zwischen diesen Großen, daß auch die Entwicklungsko-effizienten fαs invariant unter der Raumgruppe des Grundgitters sein mussen,
R−1 : fαs = fR(αs) = fαs . (2.34)
Es sei vermerkt, daß bei der Argumentation die auf (2.34) fuhrte die Orthogonalitat der Cluster-funktionen keine Bedingung war. Demzufolge gelten obige Symmetriebeziehungen auch fur nichtor-thogonale Entwicklungen, welche im nachsten Abschnitt betrachtet werden.Die Invarianz der Entwicklungskoeffizienten unter der Raumgruppe des Grundgitters fuhrt jedochscheinbar auf ein Problem. In der Begrundung dieser Invarianz wurde an keiner Stelle verwen-det, daß es sich hierbei um Entwicklungskoeffizienten einer ausgewahlten konfigurationsabhangigenFunktion handelt. Folgerichtig mußten also auch die Koeffizienten der Entwicklung der Clusterent-wicklung der Besetzungswahrscheinlichkeit diese Symmetrieeigenschaft besitzen. Bei Verwendungorthonormaler Basissatze sind aber diese Koeffizienten ξαs proportional zu den inneren Produkten〈φαs, ρ〉. Dies wurde jedoch auf den ersten Blick bedeuten, daß keine geordneten Strukturen mitdefinierten Untergittern mit der Cluster-Theorie behandelt werden konnten, da die Korrelations-funktionen der Symmetrie des ungeordneten Systems gehorchen mußten.Die Losung dieser Problematik liegt darin begrundet, daß der Formalismus der Statistik allge-mein von sich aus nicht in der Lage ist, ferngeordnete Strukuturen ”hervorzubringen”. Betrachtetman beispielsweise eine geordnete Struktur mit zwei Untergittern, so ist a priori nicht klar, welcheAtomsorte welches Untergitter einzunehmen hat. Beide Moglichkeiten haben gleiches statistischesGewicht. Der spontane Symmetriebruch muß vielmehr von Hand durchgefuhrt werden, indem manwahrend der Rechnung kleine externe Felder an die betreffenden Gitterpunkte anlegt, welche wiederausgeschaltet werden, nachdem der thermodynamische Limes vollzogen wurde [27].Man kann nun unter Vorgabe der Raumgruppe H einer ferngeordneten Struktur auf dem Grund-gitter die aquivalenten Cluster hinsichtlich dieser Raumgruppe ausfindig machen. Eine Menge sol-cher symmetrieaquivalenter Cluster bildet einen sogenannten Orbit. Die Cluster innerhalb einesOrbits besitzen nun die identischen Korrelationsfunktionen ξis, wobei i eben den Orbit der aqui-valenten Cluster αi indiziert. Dies erlaubt es, die grundlegenden Relationen der orthonormalenCluster-Entwicklung symmetrie-adaptiert niederzuschreiben, indem man zusatzlich zu den reinen

14 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
Clusterfunktionen nun neue ”Clusterorbitfunktionen” φlis definiert,
φlis =∑
αi⊆βl
φαis . (2.35)
Die Summe lauft dabei uber alle aquivalenten Cluster αi, die in einem Cluster des Typs l enthaltensind. Seien also i < l und k < l drei Orbits von symmetrieaquivalenten Clustern unter der speziellenRaumgruppe H, dann gilt
• Orthogonalitatsrelation
〈φlis, φlks′〉 = δikδss′ . (2.36)
• Reduzierte Besetzungswahrscheinlichkeit
ρl(τ ) = ρ0l
1 +∑
i<l
∑
s\0ξisφ
lis(τ )
. (2.37)
• Konfigurationsenergie
E(σ) = K0 +N∑
i∗
∑
s\0νi∗Ki∗sηi∗s(σ) . (2.38)
Dabei bezeichnet νi∗ die Zahl symmetrieaquivalenter Cluster pro Gitterpunkt, und ηi∗s istder Mittelwert der betreffenden Clusterfunktion auf dem betrachteten Orbit
ηi∗s(σ) =1
|G|
|G|∑
R′
φR′αi∗s(σ) =
1
Nνi∗
∑
αi∗
φαi∗s(σ) . (2.39)
Der Index ”i*” symbolisiert, daß es sich hierbei um einen Orbit zur Raumgruppe des Grund-gitters handelt. Die Darstellung der Konfigurationsenergie genugt bekanntlich der Symmetriedes Grundgitters.
Zum Abschluß dieses Abschnitts soll noch einmal deutlich auf den Unterschied zwischen den Sym-metrieoperationen des unterliegenden Grundgitters und denen einer bestimmten Kristallkonfigura-tion hingewiesen werden. Die Gleichung (2.34) gilt speziell fur die ECIs unabhangig von der Kri-stallkonfiguration, d.h. obwohl zum Beispiel in der B2-Struktur von NiAl das Ni- und Al-Untergitterbezuglich der Symmetriegruppe, die zu dieser Struktur gehort, nicht aquivalent sind, besitzen dieGitterpunkte beider Untergitter den gleichen ECI K1, da sie bezuglich der Symmetriegruppe derunterliegenden bcc-Struktur aquivalent sind. Auf der anderen Seite bedeutet dies nicht, daß somitauch die Korrelationsfunktionen auf beiden Untergittern identisch sein mussen.

2.2. CLUSTER-ENTWICKLUNG (CE) 15
2.2.5 Nichtorthogonale Basissatze
Der bisherige Aufbau der CE beruhte auf der Verwendung eines orthogonalen Basissatzes. Dieserwurde fur eine beliebige Zahl von Komponenten aus den M ersten Tschebyscheff Polynomen (2.7)und (2.8) gebildet. Im Fall eines binaren Systems sind diese Funktionen identisch mit den einfachenSpinfunktionen, d.h.
θ0 = 1 ∧ θ1 = σ ,
mit σ ∈ −1, 1 .
Bei Beibehaltung der Spinvariablen und Hinzunahme weiterer Komponenten werden in diesem For-malismus die Basisfunktionen zu Polynomen mit allgemein irrationalen Entwicklungskoeffizienten.Dies kann sich bei der meist numerisch aufwendigen Anwendung der Theorie im Rahmen der CVMals unvorteilhaft erweisen. Desweiteren sind dann die Korrelationsfunktionen ξαs nicht mehr aufdas Intervall [−1, 1] beschrankt.Es besteht nun jedoch die Moglichkeit, unter Verzicht auf die Orthogonalitat zu einer einfacherenund numerisch gunstigeren Basis uberzugehen. Prinzipiell laßt sich die Theorie der CE in jedervollstandigen Basis formulieren. Im folgenden sollen nun die Konsequenzen einer nichtorthogonalenBasis am Beispiel der 1, σ, σ2-Basis fur ein ternares System diskutiert werden.Die 1, σ, σ2-Basis ist weder orthogonal noch normiert. Sie hat jedoch den Vorzug, daß wie imbinaren Fall die Korrelationsfunktionen ξαs auf das Intervall [−1, 1] beschrankt sind und die darausgebildeten Clusterfunktionen immer ganzzahlige Werte annehmen. Die Vollstandigkeit dieser Basisdruckt sich in der exakten Transformation auf die orthonormale Tschebyscheff-Basis aus, die javollstandig ist.
θ0(σp)θ1(σp)θ2(σp)
=
1 0 0
0√
32 0√
2 0 − 3√2
1σpσ2p
.
⇔ θ = A¯
P (2.40)
Die Clusterfunktionen in dieser Basis gewinnt man erneut aus dem direkten Produkt der Basis-funktionen zu den Gitterpunkten p. Auch die Darstellung der Energie und die Betrachtungen zurSymmetrie bleiben erhalten. Aufgrund der Nichtorthogonalitat konnen jedoch die ECIs in dieserBasis nicht mehr uber das innere Produkt bestimmt werden. Die ECIs sind auch nicht unbedingtidentisch mit denen der Tschebyscheff-Basis. Durch den Vergleich der CE fur die Energie und mit-tels (2.40) laßt sich folgender Zusammenhang zwischen den ECIs in den beiden Basen ableiten[28],
K(NO)αs = c(s)K(O)
αs +∑
β⊆α
mβ
mαµβα
∑
(s′)
d(s, s′)K(O)βs′ , (2.41)
wobei m die Multiplizitat der Cluster und µβα die Zahl der Subcluster β in α wiedergibt. DieKonstanten c(s) und d(s, s′) erhalt man aus den Tensorprodukten von A
¯mit sich selbst. Der
analytische Ausdruck fur c(s) lautet
c(s) =
(√
3
2
)n1 (
− 3√2
)n2
, (2.42)
mit ni als der Zahl der i-Potenzen in s. Da nun die Großen c(s) und d(s, s′) fur wachsende Cluster
ebenfalls betragsmaßig zunehmen, kann es unter Umstanden sein, daß die Konvergenz der K(NO)αs
langsamer verlauft als die der K(O)αs in der Tschebyscheff-Basis.

16 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
Die Darstellung der Besetzungswahrscheinlichkeiten in der Form (2.25) und (2.74) ist zwar wei-terhin moglich, da die ξαs jedoch nicht mehr uber das innere Produkt gewonnen werden konnen,haben diese dann nicht mehr die Bedeutung einer thermodynamischen Korrelationsfunktion. Willman die Besetzungswahrscheinlichkeiten auch in einer nichtorthogonalen Basis als lineare Funktio-nen der thermodynamischen Korrelationsfunktionen ξαs darstellen, so kann dies durch einen etwasallgemeineren Zugang uber die sogenannte V-Matrix erreicht werden. Diese rechentechnisch außerstwichtige Große soll im letzten Abschnitt dieses Unterkapitels vorgestellt werden.
2.2.6 V-Matrix
Sowohl die Korrelationsfunktionen ξαs als auch die Besetzungswahrscheinlichkeiten ρα beschreibendie Clusterbesetzung im thermodynamischen Gleichgewicht. Beide sind also im mathematischenSinn wieder als verschiedene Basen im Raum der Gitterkonfigurationen aufzufassen. Eine Trans-formation zwischen diesen beiden Basen muß daher auch bei Verwendung eines nichtorthogonalenBasissatzes bezuglich der CE gegeben sein. Diese allgemeine Transformation, die naturlich denSpezialfall der orthogonalen Entwicklung enthalt, wird vermoge der sogenannten V-Matrix durch-gefuhrt [29, 30].Der denkbar einfachste Satz von Basisfunktionen besteht aus den Potenzen der σp
B = 1, σp, σ2p, . . . , σ
M−1p . (2.43)
Es erweist sich fur das weitere als gunstig, die Potenzen von σp durch den Besetzungsoperator
Γ(r)p (σ) darzustellen. Dieser hat den Wert 1, wenn der Gitterpunkt p in der Kristallkonfiguration
σ mit einem Atom der Sorte r besetzt ist, und den Wert 0 wenn nicht. Fur die Potenz von σp giltdann
σkp =M∑
m=1
τkr Γ(r)p . (2.44)
Fur jede Konfiguration ist σp identisch mit τr. Die Großen τr sind jedoch auf die Atomsorte bezogeneParameter, wohingegen die σp mit einem Gitterpunkt verknupfte Variablen symbolisieren.Die Gleichung (2.44) laßt sich nun auch in Matrixform schreiben
1σpσ2p...
σM−1p
=
1 1 1 · · · 1τ1 τ2 τ3 · · · τMτ21 τ2
2 τ23 · · · τ2
M...
......
. . ....
τM−11 τM−1
2 τM−13 · · · τM−1
M
Γ(1)p
Γ(2)p
Γ(3)p...
Γ(M)p
.
⇔ Σp = T¯
Γp (2.45)
Die Eintrage der M ×M -Matrix T¯
lauten
Tij = τ i−1j . (2.46)
Dies ist wiederum die allgemeine Form einer van der Monde Matrix, fur deren Determinante allge-mein gilt
|T¯| =
∏
i≥j(τi − τj) . (2.47)

2.2. CLUSTER-ENTWICKLUNG (CE) 17
Somit ist die Gleichung (2.45) immer invertierbar, sofern die τr verschieden gewahlt werden. DieVerwendung von Spinvariablen als Konfigurationsvariablen ist daher nur eine Moglichkeit von vie-len. Die lineare Unabhangigkeit der sukzessiven Potenzen von σp wurde bereits in Abschnitt 2.2.1diskutiert. Die Vollstandigkeit dieser Basis ist im Fall diskreter Variablen augenscheinlich klar.Der Besetzungsoperator Γp kann also seinerseits durch eine Linearkombination der σkp ausgedrucktwerden
Γp = T¯−1Σp . (2.48)
Der nachste Schritt besteht nun darin, vom Besetzungsoperator eines Gitterpunkts auf einen Clu-sterbesetzungsoperator Γα(τ ;σ) zu verallgemeinern,
Γα(χ;σ) =
|α|∏
p∈αΓ(r(χ))p (σ) . (2.49)
Der Operator Γα(χ;σ) hat wiederum den Wert 1, wenn Cluster α in Kristallkonfiguration σ dieKonfiguration χ aufweist, und 0 wenn nicht. Dies erreicht man eben dadurch, daß im Produkt von
(2.49) mittels Γ(r(χ))p (σ) jeder Gitterpunkt im Cluster α ”abgefragt” wird, ob er im Einklang mit
der Konfiguration χ besetzt ist. Nach den Regeln der Statistik ist die Wahrscheinlichkeit fur dasAuftreten einer bestimmten Clusterkonfiguration im statistischen Mittel durch folgenden Ausdruckgegeben
ρα(χ) =∑
σρ(σ)Γα(χ;σ) = 〈Γα(χ;σ)〉 . (2.50)
Mit den Gleichungen (2.48) und (2.49) laßt sich dies auch schreiben als
ρα(χ) =M−1∑
r1=0
M−1∑
r2=0
· · ·M−1∑
r|α|=0
(T−1i1r1
T−1i2r2
· · · T−1i|α|r|α|
)〈σr1p1σr2p2 · · · σr|α|p|α|〉 , (2.51)
wobei i die durch χ festgelegten Komponenten indiziert. Die Erwartungswerte 〈. . .〉 sind wiederidentisch mit den Korrelationsfunktionen ξαs, die zur Basis B gehoren. Die Summationen uber dieKomponenten an den in α enthaltenen Gitterpunkten p entspricht aufgrund der Vollstandigkeiteiner Summation uber β ⊆ α und s. Damit folgt
ρα(χ) =∑
β⊆α
∑
s\0
∏
p∈αT−1ipnp
︸ ︷︷ ︸
≡Vα(χ),βs
ξβs , (2.52)
wobei darauf zu achten ist, daß uber den ”leeren” Cluster mitzusummieren ist.Die Matrix T
¯−1 ist zeilenweise nach den Spinvariablen und spaltenweise nach den Potenzen von
σp indiziert. Der Index ip wahlt die Zeile nach der Besetzung von p in Clusterkonfiguration χ. DerIndex np hat den Wert der Potenz von σp an p hinsichtlich s wenn i ∈ β, und den Wert 0 wenni 6∈ β.Die Großen Vα(χ),βs sind die Elemente der sogenannten V-Matrix. Diese hat die Dimension M |α|×M |α|. Die V-Matrix erlaubt es also, in allgemeiner Weise die Korrelationsfunktionen und Cluster-wahrscheinlichkeiten aufeinander abzubilden. Bei Berucksichtigung der unterliegenden Raumgrup-pensymmetrie des Systems laßt sich die Relation (2.52) weiter vereinfachen, was notwendig ist,da bei Behandlung von mehrkomponentigen Systemen und großeren Clustern die Dimension derV-Matrix schnell anwachst.Ebenso wie bei der Darstellung der Clusterwahrscheinlichkeiten in orthogonaler Basis werden auchmit (2.52) die Nebenbedingungen (2.30) und (2.31) aufgrund von (2.48) automatisch erfullt. Dies

18 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
bedeutet, daß bei der Erweiterung der Cluster-Entwicklung auf thermodynamische Systeme keineLagrange-Parameter erforderlich sind, sofern man die Clusterwahrscheinlichkeiten als Funktionender unabhangigen Korrelationsfunktionen ξαs darstellt.

2.3. DAS VARIATIONSPRINZIP 19
2.3 Das Variationsprinzip
Es existieren in der Literatur mehrere verschiedene Zugange zur Cluster-Variationsmethode. Prin-zipiell laßt sich die CVM, wie auch ein Großteil der allgemeinen Thermodynamik, axiomatischaufbauen. Die Darstellung in dieser Arbeit orientiert sich an dem Ubersichtsartikel von Finel [31].Diesem ist es gelungen, die physikalische Bedeutung der verschiedenen Cluster-Naherungen an-schaulich herauszuarbeiten.
2.3.1 Zur Statistik im kanonischen Ensemble
Bevor direkt auf die Herleitung der CVM-Gleichungen eingegangen wird, sollen einige Grundrela-tionen der statistischen Physik diskutiert werden. Dies geschieht zum einen, um die Terminologiefestzulegen, und zum anderen, um die Problematik, die in der Berechnung der relevanten Großenliegt, herauszuarbeiten. Die Beschrankung auf das kanonische Ensemble bedeutet keine wirklicheEinschrankung, da sich samtliche Gedanken leicht auf beliebige Kontakte erweitern lassen.Die zentrale Große in der Statistik des kanonischen Ensembles ist die Zustandssumme Z
Z =∑
σe−βE(σ) , (2.53)
wobei β = 1kBT
und E(σ) die bereits bekannte Energie in Konfiguration σ darstellt. Die Wichtigkeitvon Z liegt darin begrundet, daß in ihr die gesamte thermodynamische Information enthalten ist,was sich darin ausdruckt, daß sich aus ihr jede thermodynamische Zustandsgroße berechnen laßt.So gilt beispielsweise
innere Energie : U = −∂(lnZ)
∂β. (2.54)
Entropie : S = −kβ2 ∂
∂β
(lnZ
β
)
. (2.55)
freie Energie : F = −kBT lnZ = U − TS . (2.56)
Anhand eines Modellsystems soll nun einmal die Berechnung der Zustandssumme nachvollzogenwerden. Das System der Wahl ist das 2-dimensionale Ising-Modell eines Ferromagneten im Nullfeldauf einem Quadrat-Gitter. Dabei kann ein Spin am Ort p entweder im Zustand ”up”, indiziert mit1, oder im Zustand ”down”, indiziert mit 2, sein. Die Energie einer speziellen Konfiguration σ wirdallein uber die Wechselwirkung nachster Nachbarpaare bestimmt,
E(σ) = −J∑
<p,p′>
σpσp′ mit J > 0 und σ ∈ −1, 1 (2.57)
Dies laßt sich auch schreiben als
E(σ) = −1
2(qN1 −N12)J +N12J − 1
2(qN2 −N12)J
=
(
2N12 −1
2qN
)
J . (2.58)
Dabei sind N1,N2 und N12 die Anzahl der Spins ”up”, Spins ”down” und Spin-Paare ”up-down”sowie q die Koordinationszahl des Gitters. Dies Zustandssumme errechnet sich daraus mittels (2.53)zu
Z = e−12βqNJ
∑
N,N1,N12g(N,N1, N12)e
−2βN12J , mit N = N1 +N2 = const. . (2.59)

20 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
Die Schwierigkeit besteht nun darin, den Entartungsfaktor g(N,N1, N12), der die Konfigurationengleicher Energie zahlt, zu berechnen. Ist dies fur kleines N noch handhabbar, so wird es im ther-modynamischen Limes (N → ∞) meist unmoglich.4
Fur thermodynamische Systeme wachst der Entartungsfaktor g(N,N1, N12) mit zunehmender Sy-stemenergie stark an. Auf der anderen Seite geht der Exponentialterm in der Summe von (2.59)bei steigender Wechselwirkungsenergie schnell gegen null. Demzufolge tragt zur Zustandssumme Zmaßgeblich ein gewisser Satz N,N1, N12 bei. Im thermodynamischen Limes ist es nun erlaubt,allein den zu diesem Satz gehorenden Term in der Summe (2.59) mitzunehmen und die freie Energiemittels (2.56) direkt in folgender Form niederzuschreiben
F = U − TS = Emax − kBT ln gmax(N,N1, N12) . (2.60)
Ist die Energie Emax zu diesem relevanten Satz bekannt, so kann auf diese Weise mittels einergeeigneten Darstellung von gmax die freie Energie direkt berechnet werden. In realen Systemenbesteht die zusatzliche Schwierigkeit darin, daß nun auch E(σ) genahert werden muß.Eine verallgemeinerte Naherungsmethode, aufbauend auf einer Variation der freien Energie, soll imnachsten Abschnitt vorgestellt werden.
2.3.2 Variation der freien Energie
Im kanonischen Ensemble, also bei konstanter Temperatur, Volumen und Teilchenzahl, errechnetsich die exakte Wahrscheinlichkeit ρeq(σ) fur eine bestimmte Kristallkonfiguration im thermody-namischen Gleichgewicht folgendermaßen
ρeq(σ) =e−βE(σ)
Z. (2.61)
Diese Relation gewinnt man durch Maximierung der Entropie bezuglich ρ,
S = maxρ
−kTr(ρ ln ρ);U = Tr(ρH); Trρ = 1 , (2.62)
wobei H die Hamiltonfunktion des Systems beschreibt. Der Maximierung der Entropie unter denangegebenen Randbedingungen entspricht eine Minimalisierung der freien Energie F . Es soll nuneine Funktional der freien Energie in folgender Form definiert werden
F [ρ] = −kBTTrρ lne−βH
ρ= U [ρ] − TS[ρ] . (2.63)
Da nun der Logarithmus eine konkave Funktion ist, d.h.
1
2(lnx+ ln y) ≤ ln
(x+ y
2
)
(2.64)
gilt, genugt der Erwartungswert einer Große X der Ungleichung
Trρ lnX ≤ lnTrρX , (2.65)
sofern die Normierungsbedingung Trρ = 1 erfullt ist. Daraus folgt nun, daß fur jede Verteilung ρ,fur die die Normierungsbedingung gilt, folgende Relation erfullt ist :
F [ρ] ≥ −kBT lnTrρe−βH
ρ= −kBT lnZ . (2.66)
4Fur den Fall des hier behandelten Isingmodells in zwei Dimensionen wurde zwar von Onsager [32] eine exakteLosung des Problems gefunden, der hierzu erforderliche theoretische Aufwand war jedoch immens.

2.3. DAS VARIATIONSPRINZIP 21
Demnach liegt das definierte Funktional der freien Energie fur jede normerhaltende Verteilung ρoberhalb der wahren freien Energie und stimmt mit dieser allein dann uberein, wenn die exakteGleichgewichtsverteilung ρeq in (2.63) eingesetzt wird,
F [ρ] ≥ F ∧ F [ρeq] = F . (2.67)
Somit lautet dann die zu (2.62) korrespondierende Variationsgleichung
F = minρ
TrρH + kBTTrρ ln ρ; Trρ = 1 . (2.68)
Minimiert man nun also (2.63) hinsichtlich aller erlaubten Verteilungen ρ, so erhalt man die exaktefreie Energie.Damit ist aber unser ursprungliches Problem, die Berechnung des Entartungsfaktors g im Spezial-fall des Ising-Modells, nurmehr auf das hierzu aquivalente Problem, namlich der Konstruktion allermoglicher Verteilungen ρ, umgeschrieben worden. Der Ansatz zur Losung des Problems ist jetztjedoch naheliegend. Es muß eine Methode entwickelt werden, die es erlaubt aus einem vergleichs-weise geringen Satz von Variationsparametern einen ausreichend großen Satz von realistischen,normerhaltenden Verteilungen ρ zu erzeugen, bezuglich derer dann die Minimierung durchzufuhrenist. Die Hoffnung ist, daß mit diesem Verfahren eine gute Annaherung an die wahre freie Energiegewonnen wird. Die beiden einfachsten auf diesem Prinzip beruhenden Methoden sollen in dennachsten beiden Abschnitten, wiederum bezugnehmend auf das Ising-Modell, diskutiert werden.
2.3.3 Bragg-Williams-Naherung
Viele Problemstellungen der Statistik und Quantenmechanik behandeln die Beschreibung von Sub-systemen. Dabei wird oftmals die Wahrscheinlichkeitsverteilung des gesamten Systems durch Aus-spuren exakt auf die entsprechende Verteilung im Subsystem reduziert. Nach dem Ausspurungspro-zess ist ein Teil der ursprunglichen Information unwiederbringlich verlorengegangen. Man muß sichalso daruber im klaren sein, daß man beim Beschreiten des umgekehrten Weges, also der Konstruk-tion einer gesamten Wahrscheinlichkeitsverteilung aus der eines Subsystems, immer eine Naherungin Kauf nehmen muß.Die Bragg-Williams-Methode [1] betrachtet die kleinste Einheit eines Kristalls, einen einzelnenGitterpunkt p, als Subsystem und entspricht folgender Konstruktionsvorschrift fur die Besetzungs-wahrscheinlichkeit ρ des Gesamtsystems
ρBW =∏
p
ρp . (2.69)
Wie sich leicht nachprufen laßt, erfullen die damit erzeugten Verteilungen die Normierungsbedin-gung Trρ = 1. Im folgenden einige Anmerkungen zu dieser Naherung :
• Wie sich an (2.69) ablesen laßt, werden die Spinausrichtungen an den einzelnen Gitterpunktenals statistisch unabhangig betrachtet. Dies hat zur Konsequenz, daß jeder Verteilung, die zurselben vorgegebenen Anzahl fur die beiden Spinsorten gehort, die gleiche Wahrscheinlichkeitzugeordnet wird. Dies steht naturlich im krassen Widerspruch zu jeder Art von Nahwechsel-wirkung im Kristall. Zwar lassen sich auch innerhalb der BW-Naherung Untergitter definie-ren, was jedoch aufgrund der Tatsache, daß solche Untergitter gerade wegen weitreichendenBesetzungskorrelationen existent sind, als zweifelhafte Beschreibung gewertet werden muß.

22 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
• Die BW-Naherung ist die alteste Mean-field-Theorie. Diese Namensgebung wird durch fol-gende Naherung von (2.58) einsichtig
E(σ) =
(
2N12 −1
2qN
)
J ≈ mH ′(N1 −N2)
mit mH ′ = −qJ (N1 −N2)
2N⇒ N12 = q
N1N2
N(2.70)
Hierbei ist m das magnetische Moment eines Spins und H ′ das sogenannte mean-field. DieEinfuhrung von H ′ diente der Eliminierung von N12, dessen Existenz die freie Permutati-on der Ni verhinderte. Physikalisch hat H ′ eben die Bedeutung eines Feldes, das an einembestimmten Spin infolge der vorhandenen Nachbarspins anliegt. Die Ortsabhangigkeit diesesFeldes wird jedoch vernachlassigt, sodaß H’ allein den Mittelwert des tatsachlichen Feldesreprasentiert.
• Es existieren zwei Grenzfalle, in denen diese Naherung exakt ist. Der erste Grenzfall folgtdirekt aus der statistischen Unabhangigkeit der Einzelspins bei beliebiger Wechselwirkung imLimes T → ∞.Desweiteren wird die Zuteilung gleicher Wahrscheinlichkeit fur Konfigurationen mit festerAnzahl der Spinsorten dann richtig, wenn man sich ein Modellsystem uberlegt, bei dem eineabstandsunabhangige Wechselwirkung vorliegt (s. z.B. [33]). Dies laßt den Schluß zu, daß dieBW-Naherung umso besser wird, je hoher die Dichte des betrachteten Systems ist.
2.3.4 Bethe-Naherung
E
A C
B D
Abbildung 2.3: Aufbau eines Quadratgitters mit der Bethe-Methode
Eine Verbesserung der BW-Naherung ließe sich durch die Berucksichtigung der Besetzungskor-relationen zwischen Paaren von Gitterpunkten erreichen. In Analogie zu (2.69) sollte sich eineGesamtverteilung konstruieren lassen, die beispielsweise uber die nachsten Nachbarpaare faktori-siert
ρ =∏
<p,p′>
ρpp′ . (2.71)
Bei genauer Betrachtung ist jedoch diese Konstruktion unphysikalisch, da sie im Grenzfall T → ∞nicht mit der dort exakten BW-Naherung ubereinstimmt. Dies folgt aus der Tatsache, daß in diesemLimes die Paarwahrscheinlichkeit ρpp′ notgedrungen uber die einzelnen Gitterpunkte faktorisierenmuß, also ρpp′ → ρpρp′ , was aufgrund der Uberlappung der NN-Paare zu Mehrfachzahlungen derGitterpunkte in (2.71) fuhrt. Schließlich wurde sich in diesem Grenzfall
ρ∞ = limT→∞
∏
<p,p′>
ρpp′ =∏
p
ρqp (2.72)

2.3. DAS VARIATIONSPRINZIP 23
ergeben, wenn q wiederum die Koordinationszahl bezeichnet. Fordert man die Aquivalenz beiderNaherungen in diesem Limes, so ist (2.71) folgendermaßen zu korrigieren,
ρ =
∏
<p,p′> ρpp′∏
p ρq−1p
. (2.73)
Dies wird als Bethe-Naherung bezeichnet [2].Die Problematik dieser Methode soll an einem Beispiel verdeutlicht werden [34]. Gegeben sei dasbereits beschriebene Ising-Modell auf einem Quadratgitter (s. Abb. (2.3)). Es seien die Punkt- undPaarwahrscheinlichkeiten ρp und ρpp′ vorgegeben. Dann laßt sich daraus die zugehorige Verteilungρ fur das gesamte Quadratgitter anschaulich dadurch konstruieren, daß man gedanklich ein Ensem-ble von N Quadratgittern aufbaut. Hierzu startet man bei entsprechend vielen NN-Paaren, derenBesetzung derart gewahlt wird, daß fur das Ensemble ρp und ρpp′ den gegebenen Werten entspre-chen. Durch sukzessives Hinzufugen von Gitterpunkten soll nun das Ensemble von Quadratgitternunter Wahrung der Punkt- und Paarwahrscheinlichkeiten aufgebaut werden.Wird nun beim Gitteraufbau ein Punkt C hinzugefugt, so muß dessen Besetzung im Ensemblediesen Zwangsbedingungen genugen. Da nun jedoch eben nur Punkt- und Paarwahrscheinlichkei-ten vorgegeben sind, kann diese Besetzung lediglich hinsichtlich einem der beiden Punkte A oderD in exakter Weise durchgefuhrt werden. Die Konstruktionsvorschrift (2.73) beschreibt in dieserHinsicht einen Mittelweg [34], welcher zur Folge hat, daß bei energetischer Wechselwirkung dierelevanten geordneten Spinstrukturen nicht korrekt reproduziert werden, da die Korrelation der imNN-Paar nicht enthaltenen Spins unberucksichtigt bleibt. Somit liefert die Bethe-Methode geradebei antiferromagnetischer WW auf frustrierten Gittern wie dem fcc-Gitter qualitativ falsche Resul-tate.Die Bethe-Methode ist wiederum auf Gittern exakt, die keine geschlossenen Cluster enthalten (s.Abb. (2.4) u. (2.5)). Diese Modellgitter werden dementsprechend Bethe-Gitter genannt.
Abbildung 2.4: Bethe-Gitter mit Koordi-nationszahl 3.
Abbildung 2.5: Bethe-Gitter mit Koordi-nationszahl 4.

24 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
2.3.5 Entropie in der CVM
Um realistischere Wahrscheinlichkeitsverteilungen zu erzeugen, wird nun in der CVM ein Schemaentwickelt, das es erlaubt, prinzipiell beliebige große Subsysteme, d.h. Cluster, auszuwahlen, umdann mit deren Verteilung die Besetzungswahrscheinlichkeit fur das Gesamtsystem zu erstellen.In Analogie zur Darstellung von ρ durch Korrelationsfunktionen soll sich die Wahrscheinlichkeitfur einen bestimmten Cluster durch zugeordnete Großen der darin enthaltenen Cluster darstellenlassen. Genauer gesagt soll gelten
ρα =∏
β⊆αρβ ⇔ ln ρα =
∑
β⊆αln ρβ . (2.74)
Es sei angemerkt, daß dieser Ansatz im Gegensatz zu den vorherigen Faktorisierungen prinzipiellexakt sein kann, da der Cluster α selbst im Produkt enthalten ist. Diese Tatsache ermoglicht esnun, die noch unbestimmten ρβ durch eine Umkehrung von (2.74) rekursiv zu berechnen. So folgtsofort fur die Punkte und Paare :
ln ρp = ln ρp .
ln ρpp′ = ln ρpp′ − ln ρp − ln ρp′ .
Fur beliebige Cluster entspricht die Umkehrung einer sogenannten Mobius-Transformation, welchefolgende Gestalt hat :
ln ρα =∑
β⊆α(−1)|α|−|β| ln ρβ . (2.75)
Diese basiert auf folgender Relation∑
β;α⊆β⊆γ(−1)(|β|−|α|) = δαγ , (2.76)
welche mittels Induktion anhand der Tatsache verifiziert werden kann, daß die Zahl der Cluster βmit r Gitterpunkten und α ⊆ β ⊆ γ gleich
(|γ|−|α||α|−r
)
ist. Fur eine detaillierte Diskussion der Mobius-
Transformation sei auf [35, 36] verwiesen.Der Sinn dieser Transformation der Wahrscheinlichkeiten ρα auf die ρα wird ersichtlich, wenn mansich das Verhalten dieser Großen im Limes T → ∞ betrachtet. Dort gilt, wie bereits bemerkt,
limT→∞
ρα =∏
p∈αρp . (2.77)
Es laßt sich nun mithilfe der Cluster-Algebra zeigen, daß sich fur die hoheren Cluster in diesemGrenzfall die ρα reduzieren auf
limT→∞
ρα = 1 wenn |α| > 1 , (2.78)
was auch nachvollziehbar ist, da dann in (2.74) lediglich die in α enthaltenen Punktcluster dieClusterwahrscheinlichkeit ρα bestimmen. Diese Eigenschaft der ρα erlaubt es nun, das bei der Bethe-Methode angewandte Naherungsprinzip auf weitergehende Ansatze mit großeren Maximalclusternauszudehnen. Zu diesem Zweck wahlt man zuerst einen bestimmten Satz von MaximalclusternαM aus. Im nachsten Schritt setzt man dann fur beliebige Temperaturen die ρα aller der Cluster,die nicht in einem der Maximalcluster enthalten sind, gleich 1. Demzufolge laßt sich die vollstandigeBesetzungswahrscheinlichkeit im Rahmen der CVM schreiben als
ρCVM =∏
β⊆αMρβ ≡
′∏
β
ρβ (2.79)

2.3. DAS VARIATIONSPRINZIP 25
Mit der weiterhin exakten Relation (2.75) kann man nun wiederum auf die eigentlichen ρβ zuruck-transformieren :
ρCVM =′∏
α
ρα
(2.75)=
′∏
α
′∏
β⊆αρ(−1)|α|−|β|
β . (2.80)
Die beiden Produkte in (2.80) kann man vertauschen, wenn man sich klar macht, daß man ebensogutzuerst alle α, die einen gewissen Cluster β enthalten, betrachten kann und erst anschließend dieProduktbildung uber die verschiedenen β vollzieht :
ρCVM =′∏
β
′∏
α⊇βρ(−1)|α|−|β|
β
=′∏
β
ρ
∑′
α⊇β(−1)|α|−|β|
β
=′∏
β
ρaβ
β .
Also :
ρCVM =′∏
β
ρaβ
β mit aβ =′∑
α⊇β(−1)|α|−|β| . (2.81)
Die Koeffizienten aβ berucksichtigen dabei die Uberlappungen der Untercluster und verhinderneventuelle Mehrfachzahlungen. Alternativ lassen sich diese Koeffizienten auch mittels folgenderRekursionsformel berechnen
∀α :′∑
β⊇αaβ = 1 . (2.82)
Diese Gleichung wird in manchen Arbeiten [22] als direkte Definition der aβ verwendet. Die an-schauliche Bedeutung dieser Relation ist, daß die aβ gerade so bestimmt werden mussen, daß jederCluster β exakt einmal gezahlt wird. Es sei vermerkt, daß diese Koeffizienten allein von der unter-liegenden Gittergeometrie abhangen und somit konfigurationsunabhangig sind.Die aufgestellten Relationen machen es nun moglich, einen analytischen Ausdruck fur die Entro-pie anzugeben. Dies ist ein großer Vorteil der CVM gegenuber dem Monte-Carlo-Verfahren, dasauf die direkte Berechnung von Erwartungswerten abzielt. Ersetzt man namlich in dem bekanntenAusdruck fur die Entropie die exakte Verteilung ρ durch die genaherte Große ρCVM so erhalt man
S = −kTr(N)ρ ln ρ(2.81)≈ −kTr(N)ρ
′∑
β
aβ ln ρβ = −k′∑
β
aβTr(N)ρ ln ρβ . (2.83)
Ein fester Cluster β kann nun in m|β| verschiedenen Konfigurationen τ auftreten. Damit laßt sichdie Spurbildung in (2.83) aufspalten in eine Summe uber diese Konfigurationen des einzelnen Clu-sters sowie in eine verbleibende Summe uber die zu jeder einzelnen Clusterkonfiguration erlaubtenKonfigurationen χ des umschließenden Gitters,
Tr(N)ρ ln ρβ =m|β|∑
τ
mN−|β|∑
χρ(χ)
︸ ︷︷ ︸
=ρβ(τ )
ln ρβ(τ )
= Tr(β)ρβ ln ρβ . (2.84)

26 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
Demzufolge wird in der CVM die Entropie des kompletten Systems durch eine Linearkombinationvon Clusterentropien angenahert5 :
SCVM =′∑
β
aβSβ mit Sβ = −kBTr(β)ρβ ln ρβ . (2.85)
Fur eine gegeben Raumgruppensymmetrie schreibt sich die Entropie dann als
SCVM = −NkB
∑
i≤nbiTr(i)ρi lnρi mit bi = νiai . (2.86)
In Gleichung (2.86) beschreibt n den Orbit eines Maximalclusters. Es sei auf den Unterschiedzwischen ai und bi hingewiesen. Der Koeffizient ai ist allein von der Topologie des unterliegendeGrundgitters abhangig, wohingegen bi die Symmetrie einer auf diesem Grundgitter realisierten Kri-stallstruktur widerspiegelt.Zum Abschluß dieses Abschnitts noch ein kleines Beispiel zur Berechnung der aβ-Koeffizienten.
Beispiel: CVM auf Dreiecksgitter
Die Menge der maximalen Cluster soll sich auf einen einzelnen solchen Cluster reduzieren. Als αMsoll das Dreieck dienen (s. Abb (2.6)). Die einzelnen aβ-Koeffizienten sollen zum Test sowohl mittels(2.81) als auch rekursiv uber (2.82) bestimmt werden.
• aβ =∑′α⊇β(−1)|α|−|β|
a∆ = (−1)0 = 1
a− = (−1)0 + 2(−1)1 = −1
a· = (−1)0 + 6(−1)2 + 6(−1)1 = 1
• ∑′β⊇α aβ = 1
a∆ = 1
2a∆ + a− = 1 ⇔ a− = 1 − 2a∆ = 1 − 2 = −1
6a∆ + 6a− + a· = 1 ⇔ a· = 1 − 6a∆ − 6a− = 1 − 6 + 6 = 1
. . .
. . .
. . .
.
. . .
.
.
.
.
.
Abbildung 2.6: Ausschnitt aus dem Dreiecksgitter.
Jeder Gitterpunkt gehort zu 6 Dreiecken und 6 NN-Paaren. Ein NN-Paar gehort zu 2 Dreiecken. Dermaximale Cluster, in diesem Fall das Dreieck, besitzt definitionsgemaß den Koeffizienten aαM
= 1.
5Es existiert noch eine andere, vereinfachte Cluster-Methode, die allein die Uberlappung in den Punkten dermaximalen Cluster berucksichtigt. Diese quasichemische Naherung [37] liefert jedoch im direkten Vergleich sowohlqualitativ als auch quantitativ schlechtere Resultate als die CVM. Fur den Fall der Bethe-Naherung stimmen beideMethoden uberein.

2.3. DAS VARIATIONSPRINZIP 27
2.3.6 Das Funktional der freien Energie
Nachdem nun ein analytischer Ausdruck fur die Entropie im Rahmen der CVM gefunden wurde,fehlt zur Berechnung des Funktionals F [ρ] jetzt noch der entsprechende Ausdruck fur die innereEnergie
U = TrρH . (2.87)
Da aber bekanntlich die Konfigurationsenergie eindeutig nach Clustern entwickelbar ist, erscheintes konsistent, dieselbe nun ebenfalls bis zum gleichen Satz maximaler Cluster αM zu entwickeln.Dann laßt sich (2.87) schreiben als
U ≈ Trρ′∑
αs
Kαsφαs =′∑
αs
KαsTrρφαs =′∑
αs
Kαsξαs . (2.88)
Somit ist man jetzt in der Lage, die komplette freie Energie in Abhangigkeit von Clustertermenauszudrucken
F =′∑
α
(∑
s
Kαsξαs + kBTaαTr(α)ρα ln ρα
)
. (2.89)
Zur Bestimmung der freien Energie in dieser Naherung mussen jetzt noch die Variationsparameterermittelt werden. In (2.89) stehen mit den Korrelationsfunktionen ξαs und den Besetzungswahr-scheinlichkeiten ρα offensichtlich zwei verschiedene Kandidaten hierfur zur Verfugung. Wie aus denvorherigen Abschnitten jedoch bereits bekannt ist, lassen sich diese beiden verschiedenen Beschrei-bungsgroßen eindeutig aufeinander abbilden. Entsprechend existieren zwei verschiedene numerischeLosungsverfahren zur Minimierung der freien Energie, je nachdem ob man die ξαs durch die ρα aus-druckt, oder die umgekehrte Beschreibung wahlt.Im ersten Fall erweist es sich weiter als sinnvoll, direkt auf Wahrscheinlichkeiten ραM
der maxi-malen Cluster zu transformieren. Dann sind die Besetzungswahrscheinlichkeiten ρα der Subclusternicht mehr frei variierbar, da sie uber die Nebenbedingung (2.31) mit den Wahrscheinlichkeitender Maximalcluster verknupft sind. Desweiteren muß fur jeden Cluster α die Relation Tr(α)ρα = 1erfullt sein. Diese beiden Zwangsbedingungen mussen also im Minimierungsprozess geeignet, zu-meist mittels Lagrange-Parameter, berucksichtigt werden. Die in dieser Arbeit verwendete Natural-Iteration-Methode (NIM) zur Minimierung des relevanten thermodynamischen Potentials beruhteben auf diesem Prinzip und wird im Kapitel 5.3 detailliert beschrieben.Fur den Fall der komplette Darstellung des relevanten thermodynamischen Potentials in Abhangig-keit von den Korrelationsfunktionen ξαs soll im folgenden Abschnitt das zu losende Gleichungssy-stem hergeleitet werden.

28 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
2.3.7 CVM-Gleichungen in orthogonaler Basis
Die Beschreibung mittels Korrelationsfunktionen hat den Vorteil, daß hierbei keine Lagrange-Parameter erforderlich sind. Im folgenden sollen die CVM-Gleichungen fur den Spezialfall der Ver-wendung eines orthonormalen Basissatzes bestimmt werden. Mittels den Korrelationsfunktionenξαs lassen sich die reduzierten Dichtematrizen in diesem Fall mit (2.29) schreiben als
ρβ(τ ) = ρ0β
1 +∑
α⊆β
∑
s\0ξαsφαs(τ )
. (2.90)
Wie bereits angemerkt, werden in dieser Darstellung die angesprochenen Nebenbedingungen auto-matisch erfullt. Die durch (2.89) beschriebene Große F wird somit zum Funktional der Funktionenξαs,
F =′∑
α
∑
s\0Kαsξαs + kBT
′∑
α
aαTr(α)ρα ln ρα , (2.91)
und die Minimierungsbedingung lautet dann
∂F∂ξαs
= 0 ∀αs . (2.92)
Unter Benutzung von
∂ρβ∂ξαs
=
ρ0βφαs : β ⊇ α
0 : β 6⊇ α(2.93)
erhalt man daraus folgenden Satz von Gleichungen
∂F∂ξβs
= Kβs + kBT′∑
α⊇β
[
aαρ0αTr(α)φβs ln ρα + aαρ
0αTr(α)φβs
]
. (2.94)
Der letzte Term in der Klammer verschwindet aufgrund von (2.13), womit die relevanten CVM-Gleichungen dann folgende Gestalt annehmen :
Kβs
kBT+
′∑
α⊇βaαρ
0αTr(α)φβs ln ρα = 0 . (2.95)
Die zu aquivalenten Clustern gehorenden Gleichungen lassen sich wiederum zusammenfassen, sodaßfur jeden Orbit j gilt
νjKjs
kBT+∑
i≥jbiρ
0iTr(i)φijs ln ρi = 0 (2.96)
Die Losung der Gleichungen (2.95) bzw. (2.96) liefert die Korrelationsfunktionen ξβs im thermody-namischen Gleichgewicht. Damit lautet die freie Energie in der CVM-Naherung,
Feq = NkBT∑
i≤nbiρ
0iTr(i) ln ρi , (2.97)
wobei n den Orbit der maximalen Cluster kennzeichnet.

2.4. DISKUSSION 29
2.4 Diskussion
Nachdem in den vorausgegangenen Abschnitten die Grundlagen der CVM formuliert wurden, solldas letzte Unterkapitel fur eine Diskussion hinsichtlich der Gute dieser statistischen Methode ge-nutzt werden.
2.4.1 Mean-Field-Charakter
Im Rahmen der Behandlung der Bragg-Williams-Naherung in Abschnitt 2.3.3 wurde diese als klassi-sche Mean-field-Theorie identifiziert. Da der CVM das gleiche Prinzip, d.h. der Darstellung von Ge-samtwahrscheinlichkeitsverteilungen aus denen von Subsystemen, zugrundeliegt und diese somit alskonsequente Weiterentwicklung der BW-Naherung anzusehen ist, sollte der Mean-Field-Charakterauf einer hoheren Stufe erhalten geblieben sein. Um dies nachzuvollziehen, soll in diesem Abschnittgezeigt werden, daß die systematische Konstruktion der gesamten Besetzungswahrscheinlichkeit ρaus Clusterwahrscheinlichkeiten ρα gleichbedeutend mit einer genaherten Zerlegung der tatsachli-chen Hamiltonfunktion in effektive Hamiltonfunktionen Heff
α ist.Fur den Fall des Ising-Modells schrieb sich die Hamiltonfunktion H folgendermaßen :
H = −J∑
<p,p′>
σpσp′ = −J∑
p
σp∑
p′
[〈σp′〉 + (σp′ − 〈σp′〉)] . (2.98)
In der Bragg-Williams-Naherung wird die Summe uber den letzten Term in der eckigen Klammerin (2.98) vernachlassigt und H approximiert als
HBW =∑
p
−∑
p′
J〈σp′〉
︸ ︷︷ ︸
=heff
σp =∑
p
Heffp . (2.99)
Die Große Heffp stellt dabei die effektive Hamiltonfunktion des Punktes im mittleren Feld der
Umgebung dar. Die Ersetzung σpσp′ → σp〈σp′〉 entspricht im Mittel der Vernachlassigung derBesetzungskorrelation zwischen den NN-Punkten 〈σpσp′〉 → 〈σp〉〈σp′〉. In diesem Fall faktorisiertdie Zustandssumme uber die einzelnen Gitterpunkte, sodaß sich ρp direkt berechnen laßt zu
ρp(σp) =e−βh
effσp
∑
σ′pe−βh
effσ′p. (2.100)
Diese Beschreibung wird nun in der CVM dergestalt verallgemeinert, daß hohere effektive Hamil-tonfunktionen in die Naherung von H aufgenommen werden
HCVM =′∑
α
aαHeffα , (2.101)
wobei nun eben Heffα dem effektiven Hamiltonoperator des Clusters α im mittleren Feld der Um-
gebung entspricht und aα wiederum die Clusteruberlappungen berucksichtigt.Ist also uber die Cluster-Entwicklung (2.16) eine eindeutige Hamiltonfunktion definiert, so gehtdiese in den thermodynamischen Formalismus im Rahmen der CVM in der Form von (2.101) ein.

30 KAPITEL 2. CLUSTER-VARIATIONSMETHODE (CVM)
Der dadurch deutlich gewordene Mean-field-Charakter der CVM manifestiert sich nunmehr in fol-genden Relationen :
ρα =exp (−Heff
α /kBT )
Zα. (2.102)
Feq =′∑
α
aαFα . (2.103)
Fα = −kBT lnZα . (2.104)
Die in den vorherigen Abschnitten begrifflich gemachte Naherung der CVM uber die Einschrankungdes Raums der Wahrscheinlichkeitsverteilungen laßt sich also als Begrenzung der Wechselwirkungs-reichweite der effektiven Hamiltonfunktion HCVM umdeuten, die mit der der ursprunglichen Ha-miltonfunktion H nicht identisch sein muß.Die Identifizierung der CVM als verallgemeinerte Mean-Field-Theorie wird auch wichtig, wenn zuuntersuchen ist, inwiefern die Methode die thermodynamischen Relationen, die sich aus den Ablei-tungen der freien Energie als beschreibendes thermodynamisches Potential ergeben, erhalt. So laßtsich beispielsweise fur die klassische BW-Naherung zeigen, daß in dieser Approximation die Rela-tionen, die sich aus den ersten Ableitungen der freien Energie ergeben, fur diese Naherung exakterhalten bleiben. Dies kann als direkte Folge des Variationsprinzip unter Beachtung der Nebenbe-dingung Trρ = 1 gedeutet werden. Im nachsten Abschnitt wird ersichtlich, daß es bei Ausdehnungdieses Prinzips auf die CVM Probleme auftreten konnen.
2.4.2 Problem der CVM
Bei der Formulierung des Variationsprinzips wurde auf die Beachtung der Nebenbedingung Trρ = 1bestanden. Diese wurde jedoch bei der Herleitung der Methode nicht benutzt, da das einzige physi-kalische Kriterium bei der Konstruktion der Theorie die Exaktheit der Methode im Grenzfall hoherTemperaturen war. Daher ist nicht garantiert, daß fur Cluster α, die nicht in einem maximalenCluster enthalten sind, die Relation Tr(α)ρα = 1 ebenfalls erfullt ist.Diese Problematik kann bei einer bestimmten Wahl von maximalen Clustern zu unphysikalischenErgebnissen fuhren, da beispielsweise die Grundrelation des Variationsprinzips, F ≥ F , nicht ge-sichert ist. Desweiteren laßt sich keine eindeutige Hierarchie der Clusternaherungen festlegen, d.h.Entwicklungen mit großeren maximalen Clustern konnen sich als schlechter erweisen als solche mitkleineren (s. Tab. (D.1)).Aus diesem Grund ist eine gewisse Unsicherheit der Methode inharent. In diesem Zusammenhangist auch die oftmals erwunschte Berechnung der Nahordnung als kritisch anzusehen, da sich diesein den meisten Fallen auf thermodynamische Relationen stutzt, die sogar die zweite Ableitung derfreien Energie beinhalten, wie zum Beispiel das Fluktuations-Dissipationstheorem. Im Zweifelsfallbleibt daher meist nur der Vergleich mit anderen Methoden der Statistischen Mechanik. Eine be-liebte Vergleichsmethode ist beispielsweise die Monte-Carlo-Simulation.Diese Problematik der CVM soll jedoch nicht den falschen Eindruck erwecken, daß es sich hierbeium eine unzureichende Modellierung handelt. So sind heute die Kriterien fur die Auswahl geeig-neter Clusternaherungen nahezu geklart [31, 38], und auch bei der Berechnung der Nahordnungliefert die CVM weitaus bessere Resultate [39, 31] als beispielsweise die auf der BW-Naherung sichgrundende Krivoglaz-Clapp-Moss-Formel [40, 41].

2.4. DISKUSSION 31
2.4.3 Vergleich mit Reihenentwicklung
Zum Abschluß dieses Unterkapitels soll noch einmal zu dem ursprunglichen Beispiel, dem 2d-Ising-Modell eines Ferromagneten auf einem Quadratgitter, zuruckgekommen werden. Der Unterschiedder Bethe-Methode und der CVM mit einem einfachen Quadrat als maximalen Cluster soll anhanddes Vergleichs mit einer Tieftemperatur-Reihenentwicklung der Zustandssumme Λ pro Gitterpunktnoch einmal verdeutlicht werden [34] :
exakt Λ = z−1(1 + z4 + 2z6 + 5z8 + 7z10 + 44z12 + . . .) ,
CVM Λ = z−1(1 + z4 + 2z6 + 5z8 + 7z10 + 43z12 + . . .) ,
Bethe Λ = z−1(1 + z4 + 2z6 + 4z8 + 8z10 + 16z12 + . . .)
mit z = e−2J/kBT . In der Bethe-Naherung konnen bekanntlich keine geschlossenen Konfigurationenkorrekt beschrieben werden, weswegen das Quadrat, reprasentiert durch den z8-Term, falsch gezahltwird. Der erste falsche Term der CVM-Darstellung ist von Ordnung z12 und entspricht einemAchteck.

Kapitel 3
Thermodynamik vonPhasendiagrammen binarer Systeme
In diesem Kapitel sollen einige Grundtatsachen der Thermodynamik und Statistik, die fur dasVerstandnis und die Berechnung von Phasendiagrammen binarer Systeme erforderlich sind, dis-kutiert werden. Aus verstandlichen Grunden kann im Rahmen dieser Abhandlung nur auf diewichtigsten Aspekte dieser bucherfullenden Thematik eingegangen werden. Fur eine tiefergehen-de Einarbeitung in dieses Teilgebiet der Statistischen Physik sei auf die umfassende Fachliteraturhierzu verwiesen (z.B. [42]).Im wesentlichen ist das Gebiet der Phasendiagrammberechnung ein Musterbeispiel fur die Anwen-dung der wohlbekannnten Relationen der Thermodynamik und Statistik. Eingeschlossen darin sindsowohl die klassische Thermodynamik in der Gibbsschen Formulierung als auch eine so aktuelleEntwicklung wie die Theorie der kritischen Phanomene. Im Vordergrund dieser Arbeit steht jedochweniger die Untersuchung des kritische Verhalten eines Systems am Phasenubergang als vielmehrdie Berechnung der globalen Topologie eines Phasendiagrammes. Aus diesem Grund wird im fol-genden auch nur rudimentar auf die Physik der Phasenubergange eingegangen und mehr Gewichtauf das Verstandnis der relevanten physikalischen Großen, die eine Phase gegenuber der anderenauszeichnen, gelegt.
3.1 Beschreibungsgroßen
Ziel dieses Abschnitts ist es, die der Problemstellung angepassten Kenngroßen zu identifizieren undihre physikalische Bedeutung zu klaren.
3.1.1 Energien
Die Berechnung des Phasendiagrammes eines intermetallischen Systems muß die stabilen Phasenbei vorgegebener Temperatur und Konzentration der beiden Komponenten liefern. Eine wahre ab-initio Berechnung eines solchen Diagrammes ware somit zum Beispiel nach geeigneter Definitioneiner Hamiltonfunktion durch eine Car-Parrinello Molekulardynamik [7] realisiert. In diesem Fallwurden sich die stabilen Phasen nach Konvergenz des Verfahrens von selbst einstellen.Die andere Vorgehensweise beruht darauf, Grundzustandsenergien bekannter geordneter Struktu-
32

3.1. BESCHREIBUNGSGROSSEN 33
ren zu bestimmen und uber ein Auswahlverfahren die fur das interessierende System gunstigstenStrukturen zu ermitteln. Im Rahmen dieses Ensembles muß dann eine Statistik entwickelt werden,die es erlaubt, aus diesen vorgegebenen moglichen Phasen die bei gegebenen Parametern jeweilsstabile zu identifizieren. Wie eine solche ab-initio Statistik zu formulieren ist, soll im Kapitel 5dargelegt werden.Mit dieser Vorgehensweise im Hinterkopf erscheint es jedoch wichtig, geeignete energetische Para-meter einzufuhren, die es ermoglichen, sowohl die Energieunterschiede der moglichen Strukturenals auch deren Auswirkung auf das Phasendiagramm zu quantifizieren. Die totale Energie Etot ei-ner Struktur ist zwar fur die eigentliche Rechnung gunstig, enthalt jedoch einen großen Anteil anfur die Phasenkonkurrenz irrelevanter ”Aufpunktsenergie”. Es existieren daher daraus abgeleiteteEnergiegroßen, deren Werte oftmals bereits qualitative Aussagen zur Phasenstabilitat erlauben.
• Die Kohasionsenergie Ecoh ist fur eine Gitterstruktur ψ folgendermaßen definiert
Eψcoh = Eψtot −E∞ , (3.1)
wobei E∞ die Summe der totalen Energien der unendlich weit voneinander entfernten Atomeder Komponenten ist. Mittels der Vorschrift (3.1) wird die fur das Bindungsverhalten unerheb-liche Rumpfenergie der Atome eliminiert. Betragsmaßig bewegt sich diese Kohasionsenergieim Bereich einiger eV pro Atom.
• Eine fur die Berechnung der Phasendiagramme essentielle Große ist die BildungsenergieEψform,
Eψform = Eψcoh −∑
i
ciEψ0
coh(i) , (3.2)
mit den Konzentrationen ci der Komponenten in der Struktur ψ und den KohasionsenergienEψ0
coh(i) derselben auf dem Grundgitter ψ0 von ψ. In koharenten Phasendiagrammen, alsosolchen mit voraussichtlich lediglich einem Grundgitter (z.B. fcc), ist in binaren Systemen einpositives (negatives) Vorzeichen der Bildungsenergie oftmals ein qualitatives Indiz dafur, daßdas Gemisch bei tiefen Temperaturen dazu neigt zu entordnen (ordnen). Schon in ternarenSystemen ist diese einfache Argumentation nicht mehr aufrechtzuerhalten, da zwar eine nega-tive Bildungsenergie die Struktur ψ gegenuber einem Gemisch aus (A)+(B)+(C) auszeichnet,jedoch beispielsweise eine Mischung (A,B)+(B,C) wiederum gunstiger sein kann. Fur eine ge-nauere Stabilitatsbetrachtung in ternaren Systemen sei auf [43, 44] verwiesen.Wichtig erscheint, daß bei inkoharenten Systemen die Definition von Eform nicht mehr voll-kommen eindeutig ist, da sich dort die Frage nach der jeweils geeigneten Wahl des Grundgit-ters ψ0 in (3.2) stellt.Im Vergleich zur Kohasionsenergie nimmt die Bildungsenergie nurmehr das Intervall von 0.1-1eV pro Atom ein.
• Die Unordnungsenergie Edis entspricht der Bildungsenergie eines vollig ungeordneten Gemi-sches aus den Komponenten.
• Als Ordnungsenergie Eord wird die Differenz zwischen der Bildungsenergie einer geordne-ten Struktur und der stochiometrisch aquivalenten Unordnungsenergie bezeichnet. Von derGroßenordnung liegt diese Energie etwa um einen Faktor 10 unterhalb einer typischen Bil-dungsenergie.
• Desweiteren laßt sich auf folgende Weise eine Strukturenergiedifferenz Estruct definieren :
Estruct = Eψcoh −Eψ′
coh , (3.3)
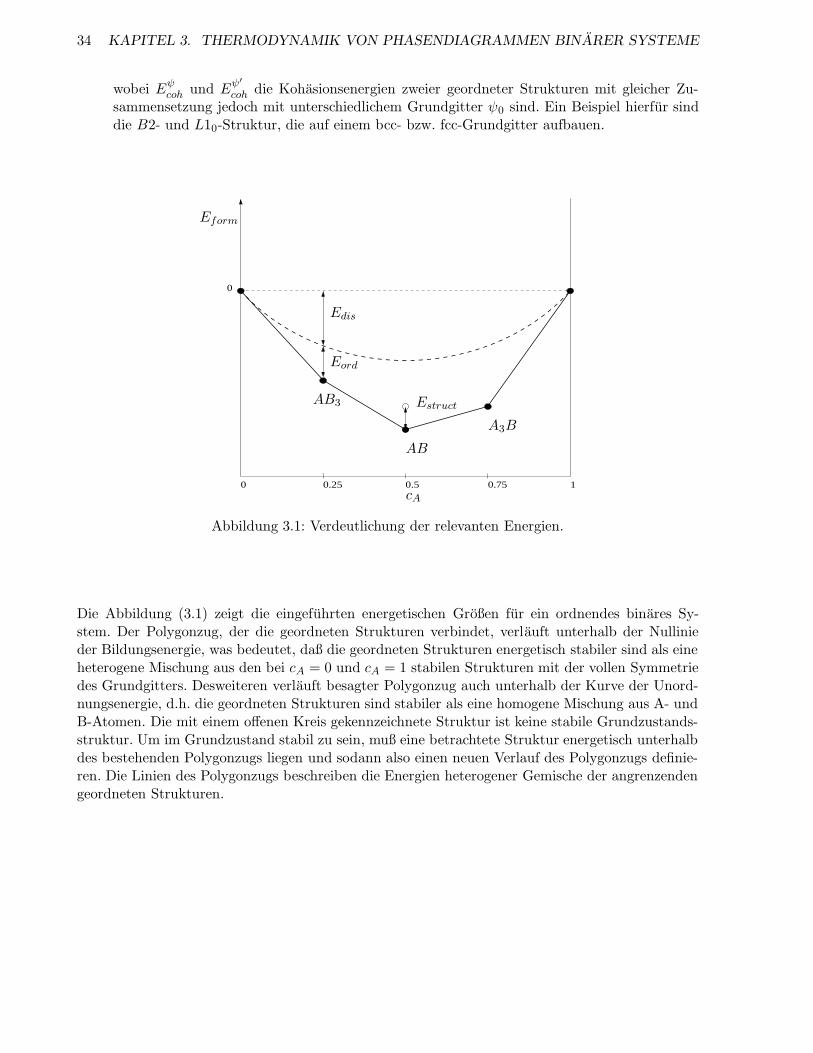
34 KAPITEL 3. THERMODYNAMIK VON PHASENDIAGRAMMEN BINARER SYSTEME
wobei Eψcoh und Eψ′
coh die Kohasionsenergien zweier geordneter Strukturen mit gleicher Zu-sammensetzung jedoch mit unterschiedlichem Grundgitter ψ0 sind. Ein Beispiel hierfur sinddie B2- und L10-Struktur, die auf einem bcc- bzw. fcc-Grundgitter aufbauen.
0 0.25 0.5
0
0.75 1
PSfrag replacements
Eform
Edis
Eord
EstructAB3
AB
A3B
cA
Abbildung 3.1: Verdeutlichung der relevanten Energien.
Die Abbildung (3.1) zeigt die eingefuhrten energetischen Großen fur ein ordnendes binares Sy-stem. Der Polygonzug, der die geordneten Strukturen verbindet, verlauft unterhalb der Nullinieder Bildungsenergie, was bedeutet, daß die geordneten Strukturen energetisch stabiler sind als eineheterogene Mischung aus den bei cA = 0 und cA = 1 stabilen Strukturen mit der vollen Symmetriedes Grundgitters. Desweiteren verlauft besagter Polygonzug auch unterhalb der Kurve der Unord-nungsenergie, d.h. die geordneten Strukturen sind stabiler als eine homogene Mischung aus A- undB-Atomen. Die mit einem offenen Kreis gekennzeichnete Struktur ist keine stabile Grundzustands-struktur. Um im Grundzustand stabil zu sein, muß eine betrachtete Struktur energetisch unterhalbdes bestehenden Polygonzugs liegen und sodann also einen neuen Verlauf des Polygonzugs definie-ren. Die Linien des Polygonzugs beschreiben die Energien heterogener Gemische der angrenzendengeordneten Strukturen.

3.1. BESCHREIBUNGSGROSSEN 35
3.1.2 Potentiale
Nachdem im vorherigen Abschnitt einige grundlegende energetische Großen eingefuhrt wurden, sollnun auf die fur die thermodynamische Beschreibung geeigneten Potentiale eingegangen werden. Eskann gezeigt werden, daß je nach Problemstellung gewisse Freiheiten hinsichtlich der Wahl der un-abhangigen und abhangigen Großen existieren. Dabei soll die detaillierte Diskussion der damit engverknupften Phasenstabilitatsbedingungen auf einen spateren Abschnitt verschoben werden.Gibt man bei der Berechnung der Phasendiagramme die Konzentrationen ci der Komponenten isowie die Temperatur T und den Druck p vor, so ist das entsprechende Potential zur thermodyna-mischen Beschreibung die freie Enthalpie G :
G = U − TS + PV . (3.4)
Aufgrund des extensiven Charakters von G laßt sich zeigen, daß ebenso gilt :
G =∑
i
µiNi . (3.5)
Der durch (3.5) beschriebene Zusammenhang zwischen freier Enthalpie G und chemischen Poten-tialen µi der Komponenten i wird auch als Gibbs-Duhem-Relation bezeichnet und laßt sich mittelsder Grundrelation dU = TdS − PdV +
∑
i µidNi auch differentiell schreiben,
SdT − V dP +∑
i
Nidµi = 0 . (3.6)
Wiederum wegen des extensiven Charakters der relevanten thermodynamischen Großen ist esmoglich, zu sogenannten molaren Großen uberzugehen. Dabei werden die extensiven Funktionenauf eine fur die Problemstellung geeignete Teilchenzahl normiert. Fur die Behandlung eines reinbinaren Systems erweist sich aufgrund der Nebenbedingung cB = 1 − cA die Normierung auf dieGesamtteilchenzahl N = N1 +N2 als gunstig. Es sei vermerkt, daß dies nicht unbedingt zwingendist und durchaus andere Normierungen denkbar sind. Mit diesen molaren Großen schreiben sichdie vorherigen Gleichungen nun als
g = u− Ts+ Pv =∑
i
µici , (3.7)
0 = sdT − vdP +∑
i
cidµi , (3.8)
wobei die molaren Großen hier und im folgenden mit Kleinbuchstaben geschrieben werden.Bisher wurden die ci und µi als Paare konjugierter Variabler angesehen. Die NebenbedingungcB = 1− cA legt es jedoch nahe, ein anderes Paar als solches anzusehen. Dies wird ersichtlich, wennman (3.8) unter Verwendung dieser Nebenbedingung nach dµi umformt :
dµA = −sdT + vdP − cBd(µB − µA) , (3.9)
dµB = −sdT + vdP − cAd(µA − µB) . (3.10)
Die Interpretation der Gleichungen (3.9) und (3.10) ist folgende. Zum einen erscheint es nicht mehrzweckmaßig, die µi als unabhangige Potentiale anzusehen. Dies impliziert jedoch, jetzt cA und(µA − µB) sowie cB und (µB − µA) als Paare konjugierter Variablen zu identifizieren. Diese neuenproblemangepaßten effektiven Potentiale werden im weiteren mit einem ”*” gekennzeichnet :
µ∗A =1
2(µA − µB) , (3.11)
µ∗B =1
2(µB − µA) = −µ∗A . (3.12)

36 KAPITEL 3. THERMODYNAMIK VON PHASENDIAGRAMMEN BINARER SYSTEME
Der Faktor 12 erweist sich bei weitergehenden Rechnungen als gunstig, hat aber keine tiefere Be-
deutung. Um ein besseres ”Gefuhl” fur diese Effektivgroßen zu bekommen, ist es sinnvoll, eineanschauliche Bedeutung herauszuarbeiten. Zu diesem Zweck sei daran erinnert, daß das chemischePotential als 1. Ableitung der freien Enthalpie nach der Konzentration dargestellt werden kann :
µA =∂G
∂NA
∣∣∣∣NB ,P,T
= G(NA + 1, NB , P, T ) −G(NA, NB , P, T ) . (3.13)
Somit hat das chemische Potential der Komponente A die Bedeutung des Gewinns an freier Ent-halpie bei Hinzufugen eines Teilchens der Sorte A. Die Nebenbedingung cB = 1 − cA mit derNormierung auf N geht jedoch von einer festen Gesamtteilchenzahl N aus. Unter diesem Gesichts-punkt kann man beispielsweise 2µ∗
A als eine Art Operator verstehen, der ein Teilchen der Sorte A”erzeugt” und gleichzeitig eines der Sorte B ”vernichtet”, um eben N konstant zu halten. Mathe-matisch ist µ∗A daher proportional zur ersten Ableitung der freien Enthalpie nach der Konzentrationbei konstanter Gesamtteilchenzahl,
µ∗A =1
2
∂g
∂cA
∣∣∣∣N,T,P
. (3.14)
Mit µ∗A und µ∗B hat man also zwei der Problemstellung angepasste Potentiale gefunden. Man kannnun ein weiteres solches neues Potential konstruieren, wenn man die Gleichungen (3.9) und (3.10)addiert, mit 1
2 multipliziert und zu den integralen Großen ubergeht
1
2(µA + µB) = g − µ∗AcA − µ∗BcB ≡ Ω . (3.15)
Mathematisch entspricht der Ubergang von g zu Ω einer allgemeinen Legendre-Transformation.Demnach steckt in Ω diesselbe thermodynamische Information wie in g.Dieses Ω wird in der Literatur oft etwas irrefuhrend als ”großes Potential” bezeichnet. Es ist jedochvon dem in der Thermodynamik allgemein als großen Potential bekannten J zu unterscheiden :
J = F −∑
i
µiNi = −PV . (3.16)
j = f − µAcA − µBcB = −Pv . (3.17)
Zur Unterscheidung wird im weiteren Ω als effektives großes Potential und j weiterhin als großesPotential bezeichnet. Im folgenden seien noch einige wichtige Relationen angeschrieben, denen diebesprochenen Potentiale genugen :
0 = µ∗A + µ∗B . (3.18)
µA = Ω + µ∗A . (3.19)
µB = Ω + µ∗B . (3.20)
g = Ω + µ∗AcA + µ∗BcB . (3.21)
Im nachsten Unterkapitel soll auf die Kriterien fur die Stabilitat einer Phase eingegangen werden.Dabei werden sich die hier eingefuhrten Effektivgroßen als außerst nutzlich erweisen.

3.2. PHASENSTABILITAT 37
3.2 Phasenstabilitat
3.2.1 Stabilitatsbedingungen
Aus der Konkurrenz von Energie und Entropie folgt, daß bei gegebenem Druck, Temperatur undKonzentration diejenige Phase stabil ist, welche sich im Minimum der freien Enthalpie G einstellt.In der Praxis ist es nun oftmals einfacher, wie eingangs schon erwahnt, eine Menge von l moglichenPhasen ψi zu betrachten und fur diese dann jeweils eine Große G zu berechnen. Dieses G hat dieBedeutung der freien Enthalpie bei vorgegebnen T,P und cA unter der Zwangsbedingnung, daß diePhase ψi vorliege. Das Minimum dieses Satzes von Gψi wird sodann als thermodynamische freieEnthalpie G identifiziert
G ≡ min
Gψ1 , Gψ2 , . . . , Gψl
. (3.22)
Fur die Bestimmung dieses minimalen Gψi wird sich im folgenden eine Eigenschaft der freienEnthalpie in Abhangigkeit von der Konzentration cA als wichtig erweisen, die sich aus der hinrei-chenden Bedingung fur das Minimum der freien Enthalpie im thermodynamischen Gleichgewichtfolgern laßt.Die notwendige Bedingung fur die Minimaleigenschaft der freien Enthalpie bei vorgegebenem Tund P lautet
dg = µAdcA + µBdcB = 0 . (3.23)
Mit der Nebenbedingung cA + cB = 1 gilt nun ebenso
dg = (µA − µB)dcA , (3.24)
⇔ dg = 2µ∗AdcA ,
⇔ dg =∂g
∂cA
∣∣∣∣N,T,P
dcA .
Ein stabiles Gleichgewicht liegt nur dann vor, wenn die freie Enthalpie minimal bezuglich derKonzentration ist, d.h. folgendes gilt
∂
∂cA
(∂g
∂cA
)∣∣∣∣N,T,P
> 0 . (3.25)
Diese Stabilitatsbedingung bedeutet nichts anderes, als daß die Krummung der freien Enthalpie-kurve im g − cA−Diagramm immer positiv zu sein hat.Warum dies so sein muß, ist auch anschaulich klar. Ware an einer Stelle die Krummung negativ, soware das System instabil gegenuber Konzentrationsfluktuationen. Durch Aufspalten in zwei Sub-systeme mit verschiedenen Konzentrationen cA konnte das Gesamtsystem aufgrund des extensivenCharakters der freien Enthalpie diese absenken und somit einen gunstigeren Zustand einnehmen.Es stellt sich an dieser Stelle naturlich die Frage, ob diese Gesetzmaßigkeit auch fur eine Mean-Field-Behandlung des betreffenden Systems zutreffend ist, da in (3.25) bereits eine 2. Ableitung vong steht. Die Gultigkeit muß im Fall verschiedener Clusternaherungen durch die explizite Rechnungverifiziert werden.

38 KAPITEL 3. THERMODYNAMIK VON PHASENDIAGRAMMEN BINARER SYSTEME
3.2.2 Phasenkoexistenz
Hat man bei einem bestimmten Satz T, P, cA eine geordnete Struktur als stabile Phase iden-tifiziert, so stellt sich naturlich sofort die Frage, wie ausgedehnt diese Phase im Diagramm ist.Fur die Berechnung der Phasengrenzen benotigt man wiederum thermodynamische Relationen,die die Bedingungen der Phasenkoexistenz regeln. Betrachtet man der Einfachheit halber nur dasGleichgewicht zweier geordneter Strukturen ψ1 und ψ2, so lauten diese Relationen
Tψ1 = Tψ2 , (3.26)
Pψ1 = Pψ2 , (3.27)
µψ1
A = µψ2
A , (3.28)
µψ1
B = µψ2
B , (3.29)
Es stellt sich also das Problem, diese Vielzahl an Parametern, die in den Gleichungen (3.26-3.29)stehen, fur zwei thermodynamisch stabile Strukturen rechnerisch aufeinander abzustimmen. ZurLosung dieser Aufgabe erweist es sich als zweckmaßig, nicht mit der freien Enthalpie als beschrei-bendem Potential, sondern mit dem effektivem großen Potential Ω als solchem zu hantieren. Dieswird ersichtlich, wenn man die Gleichungen (3.28) und (3.29) einmal addiert und ein andermalvoneinander subtrahiert
µψ1A + µψ1
B = µψ2A + µψ2
B ⇒ Ωψ1 = Ωψ2 , (3.30)
µψ1A − µψ1
B = µψ2A − µψ2
B ⇒ µ∗ψ1A = µ∗ψ2
A . (3.31)
Stimmen also unter Vorgabe von T, P und µ∗A fur zwei reine Phasen ψ1 und ψ2 die zugehorigen
Potentiale Ωψ1 und Ωψ2 uberein, so hat man den Phasenkoexistenzbereich ermittelt.Um eindeutig klaren zu konnen, ob die gefundenen koexistierenden Phasen auch tatsachlich die indem zugehorigen Konzentrationsbereich thermodynamischen stabilen Phasen sind, mußte eigentlichwiederum auf die freie Enthalpie zuruckgegriffen werden, da diese ja aufgrund der Minimaleigen-schaft die stabilen Phasen kennzeichnet. Es zeigt sich jedoch, daß das effektive große PotentialΩ allein ausreicht, um sowohl die Phasengrenzen zu bestimmen als auch die stabilen Phasen zuidentifizieren. Um dies einzusehen, sind noch einige weitergehende erklarende Bemerkungen zurBedeutung der eingefuhrten Effektivgroßen vonnoten.Wie aus Gleichung (3.14) hervorgeht, entspricht 2µ∗
A gerade der Steigung der freien Enthalpiekurveim g−cA−Diagramm. Setzt man in (3.19) die Relationen (3.14) und (3.18) ein, so erhalt man unterVerwendung der Nebenbedingung cA + cB = 1 folgende Gleichung fur das chemische Potential µAbei fester Konzentration c′A :
µA = g(T, p, c′A) + 2µ∗A(1 − c′A) (3.32)
Dies bedeutet anschaulich, daß sich µA als Schnitt der an g in c′A anliegenden Tangente mit derAchse cA = 1 ergibt (s. Abb. (3.2)). Fur den Spezialfall c′A = 1 erhalt man naturlich µA = g.Entsprechend ergibt sich µB als Schnitt der gleichen Tangente mit der Achse cA = 0.Damit hat man nun auch die Moglichkeit, eine graphische Deutung des Potentials Ω zu geben. Alsarithmetischen Mittelwert der chemischen Potentiale µA und µB gewinnt man Ω mittels Schnittder besagten Tangente mit der Geraden cA = 1
2 . Die Transformation von g auf Ω stimmt also auchgraphisch mit einer allgemeinen Legendre-Transformation uberein.Durch Vorgabe eines Wertes fur µ∗A, womit wegen (3.18) auch µ∗
B festgelegt ist, wahlt man also einebestimmte Steigung im g− cA-Diagramm aus. Hat man fur ein solches µ∗A das zugehorige minimaleΩ gefunden, so ist die entsprechende Phase an der Stelle T, P, c′A als die stabile Phase unter demgewahlten Ensemble an moglichen Strukturen zu werten (s. Abb. (3.2)). Dies folgt bereits alleinanhand von graphischen Uberlegungen aus der Tatsache, daß jegliche freie Enthalpiekurve immer
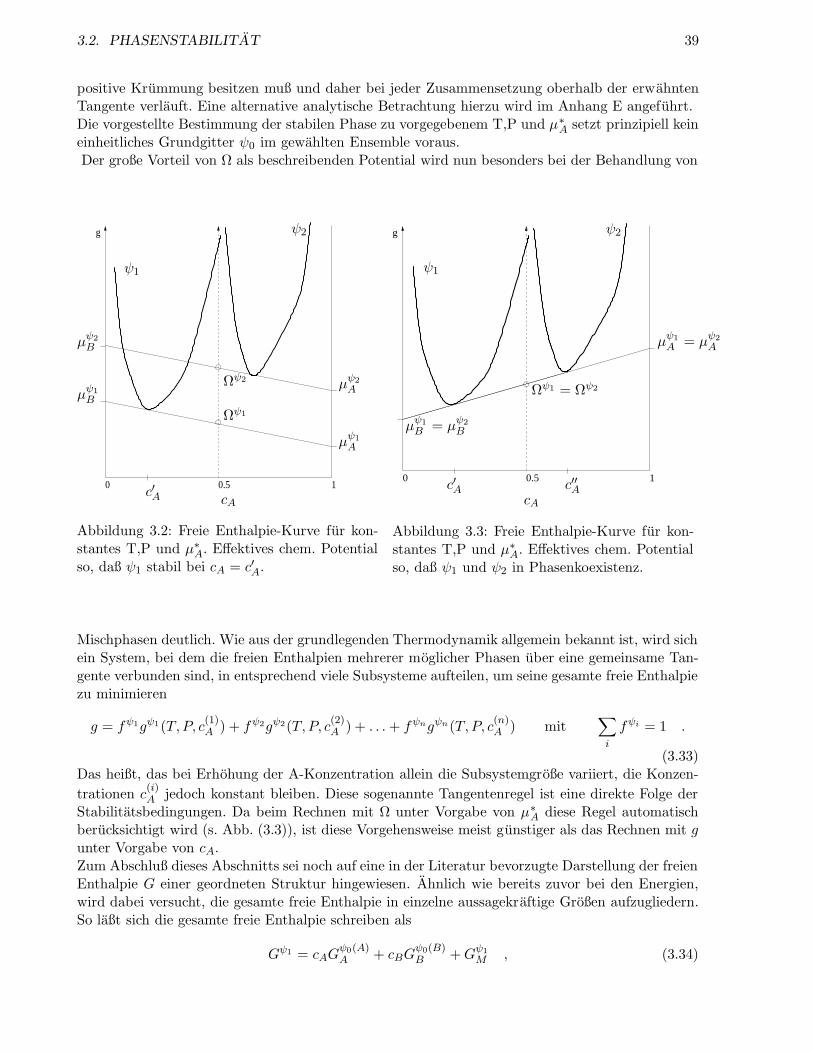
3.2. PHASENSTABILITAT 39
positive Krummung besitzen muß und daher bei jeder Zusammensetzung oberhalb der erwahntenTangente verlauft. Eine alternative analytische Betrachtung hierzu wird im Anhang E angefuhrt.Die vorgestellte Bestimmung der stabilen Phase zu vorgegebenem T,P und µ∗
A setzt prinzipiell keineinheitliches Grundgitter ψ0 im gewahlten Ensemble voraus.Der große Vorteil von Ω als beschreibenden Potential wird nun besonders bei der Behandlung von
g
0 0.5 1
PSfrag replacements
µψ1A
µψ2Aµψ1
B
µψ2B
µψ1A = µψ2
A
µψ1B = µψ2
B
cAc′A
c′′A
ψ1
ψ2
Ωψ1
Ωψ2
Ωψ1 = Ωψ2
ψ0(A)ψ0(B)
Gψ0(A)A
Gψ0(B)B
Gψ1M
Abbildung 3.2: Freie Enthalpie-Kurve fur kon-stantes T,P und µ∗A. Effektives chem. Potentialso, daß ψ1 stabil bei cA = c′A.
g
0 0.5 1
PSfrag replacements
µψ1A
µψ2A
µψ1B
µψ2B
µψ1A = µψ2
A
µψ1B = µψ2
B
cAc′A c′′A
ψ1
ψ2
Ωψ1
Ωψ2
Ωψ1 = Ωψ2
ψ0(A)ψ0(B)
Gψ0(A)A
Gψ0(B)B
Gψ1M
Abbildung 3.3: Freie Enthalpie-Kurve fur kon-stantes T,P und µ∗A. Effektives chem. Potentialso, daß ψ1 und ψ2 in Phasenkoexistenz.
Mischphasen deutlich. Wie aus der grundlegenden Thermodynamik allgemein bekannt ist, wird sichein System, bei dem die freien Enthalpien mehrerer moglicher Phasen uber eine gemeinsame Tan-gente verbunden sind, in entsprechend viele Subsysteme aufteilen, um seine gesamte freie Enthalpiezu minimieren
g = fψ1gψ1(T, P, c(1)A ) + fψ2gψ2(T, P, c
(2)A ) + . . .+ fψngψn(T, P, c
(n)A ) mit
∑
i
fψi = 1 .
(3.33)Das heißt, das bei Erhohung der A-Konzentration allein die Subsystemgroße variiert, die Konzen-
trationen c(i)A jedoch konstant bleiben. Diese sogenannte Tangentenregel ist eine direkte Folge der
Stabilitatsbedingungen. Da beim Rechnen mit Ω unter Vorgabe von µ∗A diese Regel automatisch
berucksichtigt wird (s. Abb. (3.3)), ist diese Vorgehensweise meist gunstiger als das Rechnen mit gunter Vorgabe von cA.Zum Abschluß dieses Abschnitts sei noch auf eine in der Literatur bevorzugte Darstellung der freienEnthalpie G einer geordneten Struktur hingewiesen. Ahnlich wie bereits zuvor bei den Energien,wird dabei versucht, die gesamte freie Enthalpie in einzelne aussagekraftige Großen aufzugliedern.So laßt sich die gesamte freie Enthalpie schreiben als
Gψ1 = cAGψ0(A)A + cBG
ψ0(B)B +Gψ1
M , (3.34)

40 KAPITEL 3. THERMODYNAMIK VON PHASENDIAGRAMMEN BINARER SYSTEME
wobei Gψ0(i) die freie Enthalpie der bei ci = 1 vorliegenden stabilen Kristallstruktur und Gψ1
M diesogenannte freie Mischungsenthalpie ist. Der energetische Anteil dieser freien Mischungsenthalpielaßt sich entsprechend mit der bereits eingefuhrten Bildungsenergie Eform berechnen :
Gψ1 = Eψ1tot + pV − TS ,
Gψ1 = Eψ1
coh + cAEat(A) + cBEat(B) + pV − TS ,
Gψ1 = Eψ1
form + cA[
Eat(A) +Eψ0
coh(A)]
+ cB[
Eat(B) +Eψ0
coh(B)]
+ pV − TS ,
Gψ1 = Eψ1
form + cAEψ0tot(A) + cBE
ψ0tot(B) + p(cAVA + cBVB + Vform)
−T[
cASψ0(A) + cBS
ψ0(B) + Sψ1
form
]
,
Gψ1 = cA[
Eψ0tot(A) + pVA − TSψ0(A)
]
+ cB[
Eψ0tot(B) + pVB − TSψ0(B)
]
+Eψ1
form + pVform − TSψ1
form , (3.35)
Gψ1 = cAGψ0(A)A + cBG
ψ0(B)B +Gψ1
M ,
wobei Eat die atomaren Energien der Komponenten kennzeichnet.Der Vorteil dieser Darstellung liegt in der besseren Interpretierbarkeit der errechneten Großen, wasjedoch, wie bereits erwahnt, mit der Wahl eines geeigneten Referenzzustands erkauft wird.
3.2.3 Mischungslucke (miscibility gap)
Zur Vertiefung des in den letzten Abschnitten eingefuhrten wesentlichen Handwerkzeugs zur Be-rechnung eines binaren Phasendiagrammes soll nun hier die sogenannte Mischungslucke oder auchmiscibility gap besprochen werden. Eine solche Mischungslucke beschreibt einen Bereich im Phasen-diagramm, in dem eine heterogene Mischung aus zwei homogenen Phasen mit gleicher Symmetrieaber unterschiedlicher Atomsortenkonzentration existent ist. Verantwortlich hierfur ist eine negativeKrummung in der freien Enthalpie-Kurve der entsprechenden Phase, welche zu thermodynamischinstabilen Zustanden fuhrt.Die wesentlichen physikalischen Zusammenhange sind in Abb. (3.4) verdeutlicht. Im Bereich I
ist die Krummung der freien Enthalpiekurve positiv, und somit befindet man sich im thermodyna-misch stabilen Bereich. Die Krummung ist zwar im Bereich II ebenfalls noch positiv, jedoch erlaubtdie Tangentenregel ein Absenken der freien Enthalpie, da die Punkte A und D eine gemeinsameTangente verbindet. Bei vorgegebener Temperatur T1 ist fur alle Konzentrationen cA im BereichII eine heterogene Mischung zweier homogener Phasen mit Konzentrationen c ′A und c′′A gunstigerals die homogene Phase mit Konzentration cA. Durch geeignete Zwangsbedingungen ist es jedochmoglich, auch in diesem Konzentrationsbereich eine homogene, wenn auch metastabile Phase zurealisieren. Zu thermodynamisch instabilen Einzelphasen kommt es aber im Bereich III, da dorteine negative Krummung der freien Enthalpiekurve vorliegt.Die Grenze zwischen II und III wird allgemein als Spinodal-Kurve bezeichnet, da die darauf liegen-den Punkte in einem Ω-µ∗
A-Diagramm durch Punkte an den Enden scharfer Spitzen ausgewiesensind (Abb. (3.5)). Im kritischen Punkt E mussen folgende Bedingungen erfullt sein :
∂2g
∂c2A= 0 ∧ ∂3g
∂c3A= 0 . (3.36)
Der kritische Punkt ist daher der einzige Punkt, bei dem die Stabilitatsgrenze von einem stabilenhomogenen System erreicht werden kann. Bei der Mischungslucke ist es also moglich, von einerhomogenen Phase in die andere zu gelangen, ohne daß eine Phasengrenzlinie gekreuzt werden muß.

3.2. PHASENSTABILITAT 41
G
T
I
I
II IIIII
a
b
0 1
0 1
miscibility gap
spinodal
A
B C
D
E
I II III II
PSfrag replacements
cA
cA
ψ1 ψ′1
T = T1
T1
µ∗AΩ
Abbildung 3.4: (a) Kurve freier Enthal-pie bei T = T1.(b) Miscibility gap undSpinodal-Kurve.
CSpinodal-Punkte
AD
B
PSfrag replacements
cAψ1
ψ′1
T = T1
T1
µ∗A
Ω
Abbildung 3.5: Potentialdiagramm eines Sy-stems mit miscibility gap fur T und P konstant.
Eine Analogie hierzu existiert in einem einkomponentigen flussig-gasformig System, wo dies durchUmlaufen des kritischen Punkts ebenfalls moglich ist. In beiden Fallen andert sich wohlgemerktdie Symmetrie des Systems nicht.Eine Mischungslucke laßt sich ebenfalls im (µ∗
A,Ω)-Formalismus berechnen. Das Verfahren hierzuwird am Beispiel des NiAl-Systems auf dem reinen fcc-Gitter in Abschnitt 6.2.2 detailliert beschrie-ben.
3.2.4 Systeme am Phasenubergang
Ein Phasenubergang in einem System macht sich durch eine Unstetigkeit in mindestens einer derAbleitungen des beschreibenden thermodynamischen Potentials bemerkbar. Der Grad der Ablei-tung, in der eine solche Unstetigkeit auftritt, charakterisiert die Ordnung des betreffenden Pha-senubergangs.Die Beschreibung eines Systems in der Umgebung eines Phasenubergangs gehort zu den anspruchs-vollsten Problemstellungen der statistischen Physik. Ein Grund hierfur ist die Tatsache, daß indiesem Bereich die Fluktuationen in den physikalischen Observablen zur dominierenden Große wer-den, so daß einfache Mean-Field-Theorien fur eine adaquate Beschreibung nicht ausreichend sind.Desweiteren erfordert die theoretische Behandlung dieser Thematik die Rechnung im thermodyna-mischen Limes, da nur in diesem Grenzfall ein Phasenubergang auftreten kann. Von besonderemtheoretischen Interesse sind die Phasenubergange 2. Ordnung, da es dort zu kritischen Phanomenenkommt. Die Bewaltigung speziell dieser Problematik ist Ziel einer Vielzahl von Theorien, wobei diesogenannte Renormierungstheorie in diesem Zusammenhang eine herausragende Stellung einnimmt.

42 KAPITEL 3. THERMODYNAMIK VON PHASENDIAGRAMMEN BINARER SYSTEME
In unserem Fall laßt sich die Konzentration cA als erste Ableitung von Ω nach dem effektiven chem.Potential schreiben
cA =∂Ω
∂µ∗A
∣∣∣∣∣N,T,P
(3.37)
Betrachtet man nun den Ubergang zwischen zwei geordneten Strukturen ψ1 und ψ2 im Phasen-diagramm, so handelt es sich fur den Fall, daß die Konzentrationen cψ1
A und cψ2A im Phasenkoexi-
stenzbereich ubereinstimmen, d.h. keine Mischphasen auftreten, um einen Ubergang 2. Ordnung.Demzufolge weist sich ein Phasenubergang 1. Ordnung durch das Auftreten einer Mischphase aus(s. Abb. (3.6)). Die Charakterisierung der Ordnung des Phasenubergang ist jedoch trotzdem nichttrivial, da eine Mischphase beliebig dunn sein kann und beim expliziten Rechnen daher oftmalskeine eindeutige Entscheidung gefallt werden kann.In diesem Zusammenhang sind einige Bemerkungen zum Begriff des Ordnungsparameters am Bei-
b
a
PSfrag replacements
G
T
T
cA
cA
ψ1
ψ1
ψ2
ψ2
T1
T2
T3
T4
ζζe
Abbildung 3.6: Phasenubergangezwischen Kristallstrukturen.(a) 2.Ordnung, (b) 1. Ordnung.
metastabil
a
b
instabil
instabil
PSfrag replacements
G
GT
TcAψ1
ψ2
T1
T1
T1
T1
T2
T2
T2
T2
T3
T3
T3
T3
T4
T4
ζ
ζ ζe
ζe
Abbildung 3.7: Phasenubergange.(a) 2. Ordnung, (b)1. Ordnung. Die Lage der Extrema in G wird durch ζebeschrieben
spiel eines Ordnungs-Unordnungsubergangs bei Temperaturerhohung hilfreich. Diese von Landaueingefuhrte systemspezifische Große ist, vereinfacht gesagt, die relevante Beschreibungsgroße in derunmittelbaren Umgebung des Phasenubergangs. Dies druckt sich dadurch aus, daß der Ordnungs-parameter ζ fur die stabile Phase ψ1 unterhalb des Phasenubergangs ungleich null und oberhalbdes Phasenubergangs fur die nunmehr stabile Phase ψ2 gleich null sein soll (Abb. (3.7)).In der unmittelbaren Umgebung des Phasenubergangs soll sich nun das beschreibende Potentialnach Potenzen von ζ entwickeln lassen. Dies setzt jedoch voraus, daß ζ in dieser Region klein ist.Eine einfache Rechnung zeigt, daß fur einen Phasenubergang 2. Ordnung der Ordnungsparameterbei Annaherung aus der Phase ψ1 kontinuierlich gegen null geht, weswegen diese Art des Ubergangsauch kontinuierlicher Phasenubergang genannt wird. Beim Ubergang 1. Ordnung dagegen springt ζ

3.2. PHASENSTABILITAT 43
von einem endlichen Wert bei einer gewissen Temperatur auf null. Im ”ubersprungenen” Wertebe-reich von ζ konnen metastabile Zustande des Systems liegen. Der Sprung des Ordnungsparametersist auf die Ausbildung eines gunstigeren Minimums im beschreibenden thermodynamischen Poten-tial zuruckzufuhren (Abb. (3.7)).Bei Phasenubergangen zwischen Kristallstrukturen ist der Ordnungsparameter eine symmetrie-abhangige Funktion. Dies hat zur Folge, daß im Fall eines kontinuierlichen Ubergangs aufgrundder Identitat beider Systeme im Ubergangspunkt gewisse Relationen zwischen den beiden Symme-triegruppen der relevanten Strukturen existieren mussen. Hingegen sind beim Phasenubergang 1.Ordnung keinerlei Beschrankungen hinsichtlich der Symmetrieanderungen existent. Dies ermoglichtes, anhand von Symmetriebetrachtungen Regeln dafur abzuleiten, ob ein Phasenubergang 2. Ord-nung uberhaupt auftreten kann. Das Ergebnis solcher Uberlegungen sind die notwendigen aberallgemein nicht hinreichenden Landau-Lifschitz-Regeln. Sie lauten folgendermaßen :
1. Die Gruppe G der geordneten Phase muß eine Untergruppe der Gruppe G0 der ungeordnetenPhase sein.
2. Der mit dem Ubergang verbundene Stern muß der Stern eines speziellen Punkts in derBrillouin-Zone der ungeordneten Phase sein.
3. Es muß unmoglich sein, drei Vektoren des betrachteten Sterns zu einem Vektor des reziprokenGitters der ungeordneten Phase aufsummieren zu konnen.
Zwei Begriffe mussen in diesem Zusammenhang geklart werden. Der Stern eines Vektors ist dieMenge aller Vektoren, die durch Anwendung aller Symmetrieoperationen des Gitters auf eineneinzelnen Vektor entsteht. Ein spezieller Punkt ist ein Punkt im reziproken Gitter, an dem dieFouriertransformierte K(k) der reinen Paarwechselwirkungen extremal wird.Eine genaue Diskussion der Regeln sowie deren Ausnahmen wurde den Rahmen dieser Abhandlungsprengen. Eine ausfuhrliche Diskussion der Landau-Lifschitz-Theorie findet sich in dem allgemeinempfehlenswerten Buch von Ducastelle [33] und den dortigen Verweisen.Zur Verdeutlichung soll jedoch der B2-A2-Ubergang auf dem bcc-Gitter als Beispiel angefuhrt wer-den. Die 1. Regel ist fur diesen Fall, wie man sofort einsieht, erfullt. Weiter ist das reziproke Gitterder bcc-Struktur ein fcc-Gitter. Die speziellen Punkte des fcc-Gitters in der 1. Brillouin-Zone sind〈000〉, 〈100〉, 〈 1
212
12〉 und 〈1
2120〉. Der ordnende Wellenvektor der B2-Struktur lautet 〈100〉. Damit ist
auch Regel 2 erfullt. Außerdem ist es nicht moglich, drei 〈100〉-Vektoren zu einem fcc-Gittervektoraufzusummieren (Regel 3). Es laßt sich daher sagen, daß der B2-A2-Ubergang sowohl 1. als auch2. Ordnung sein kann.Die Landau-Lifschitz-Theorie der Phasenubergange ist von ihrer Natur her eine Mean-Field-Theorie,was bedeutet, daß die Korrelationen der Fluktuationen vernachlassigt werden. Allgemein liegen spe-ziell die im Rahmen dieser Theorie errechneten kritischen Temperaturen zu hoch. Dies laßt sichanschaulich dadurch verstehen, daß die unzureichende Berucksichtigung der weitreichenden Fluk-tuationen in der Mean-Field-Theorie dazu fuhrt, daß das System erst dann entordnet, wenn dieFluktuationen mit kurzer Reichweite dominant werden. Demzufolge ist das wahre System bei gege-bener Temperatur in Wirklichkeit viel starker ungeordnet als durch die Landau-Lifschitz-Theoriebeschrieben.Die Theorie wird jedoch weiterhin als Ausgangspunkt fur die Beschreibung von kontinuierlichenPhasenubergangen verwendet. Fur Phasenubergange 1. Ordnung kann die Entwicklung nach ζ aberaufgrund des Sprungs des Ordnungsparameters problematisch sein.

Kapitel 4
Elektronentheorie
Die Zielsetzung der Berechnung des Phasendiagrammes eines vorgegebenen intermetallischen Sy-stems erfordert es naturlich, spezifische Wechselwirkungsparameter zur Berechnung der innerenEnergie vorzugeben. Neben der einen Moglichkeit, experimentelle Daten hierzu heranzuziehen, exi-stieren seit einigen Jahren auch Verfahren, diese Wechselwirkungsparameter ”ab initio”, d.h. alleindurch Vorgabe der Kernladungszahl der Komponenten und der vorliegenden Kristallstruktur, zuberechnen.Die Bestimmung der relevanten Parameter auf diesem Weg erfordert die Vorgabe von ausgewahl-ten Grundzustandsenergien (s. Kapitel 5). Zu diesem Zweck soll hier als weiterer Baustein einer”ab-initio Statistik” in kurzen Zugen die Dichtefunktionaltheorie (DFT) vorgestellt werden. Dieseist elektronentheoretische ab-initio Methode zur Berechnung von Grundzustandsobservablen.
4.1 Eigenwertproblem
Prinzipiell besteht die Aufgabe bei der Bestimmung der Kristallenergie einer bestimmten Strukturdarin, die zeitunabhangige Vielteilchen-Schrodinger-Gleichung zu losen :
H|Ψ(R, r)〉 = E|Ψ(R, r)〉 , (4.1)
mit R : Orts- und Spinkoordinaten der Kerne ,
r : Orts- und Spinkoordinaten der Elektronen .
Die exakte Losung dieses Eigenwertproblems ist aufgrund der Tatsache, daß sich die Vielteilchen-wellenfunktion wegen der komplizierten Wechselwirkung nicht als Produkt von Einteilchenwellen-funktionen darstellen laßt, schlichtweg unmoglich.Eine erste Vereinfachung von (4.1) erhalt man durch die sogenannte Born-Oppenheimer-Naherung[45]. Dieser liegt zugrunde, daß wegen der vergleichsweise viel hoheren Geschwindigkeit der Elektro-nen gegenuber der der Kerne in erster Naherung angenommen werden kann, daß sich die Elektronenzu jedem Zeitpunkt im Grundzustand bezuglich den Kernkoordinaten befinden. Diese Annahmeschlagt sich mathematisch in einer Entkoppelung der Wellenfunktion nieder :
|Ψ(R, r)〉 = |φ(R)〉|ψ(r)〉 (4.2)
Dabei gehen in |ψ(r)〉 die Kernkoordinaten lediglich als Parameter ein. Desweiteren kann man beider Behandlung von unmagnetischen Systemen auf die explizite Abhangigkeit der Wellenfunktion
44

4.2. DICHTEFUNKTIONALTHEORIE 45
von den Spinkoordinaten verzichten. Trotz dieser Vereinfachungen liegt eine Losung des Problemsnoch in weiter Ferne, da bei bekanntem Hamiltonoperator weiterhin eine Vielteilchenwellenfunktionmit 3Ne-Elektronenkoordinaten als Argument zu ermitteln ist.
4.2 Dichtefunktionaltheorie
4.2.1 Theoreme von Hohenberg und Kohn
Der wichtige Gedanke zur grundlegenden Vereinfachung des Problems liegt in der Erkenntnis, daßes zur Berechnung von Grundzustandsobservablen ausreichend ist, allein die Elektronendichte n(r)bei T = 0 zu kennen. Aus der Kenntnis der nurmehr von drei Koordinaten abhangigen Funktionn(r) laßt sich sodann die Grundzustandsenergie uber ein Variationsverfahren berechnen. DieseAussagen stutzen sich auf die beiden Theoreme von Hohenberg und Kohn [46] :
1. Seien n(r) und n′(r) zwei Grundzustandsdichten eines wechselwirkenden Elektronensystemsin den außeren Potentialen V (r) und V ′(r), dann gilt
n(r) = n′(r) ⇒ V ′(r) = V (r) + const. (4.3)
Dies erlaubt es, samtliche Grundzustandsgroßen als Funktionale der Dichte n(r) zu schreiben.So ergibt sich fur die Energie
E[n] = F [n] +
∫
V (r)n(r)d3r , (4.4)
wobei das Funktional F [n] die vom außeren Potential unabhangigen Energieterme beinhaltet.
2. Das Energiefunktional E[n] ist minimal fur die richtige Grundzustandsdichte n(r).
4.2.2 Kohn-Sham-Formalismus
Durch die Identifizierung der Elektronendichte als relevante Beschreibungsgroße zur Berechnungder Grundzustandsenergie hat sich die Zahl der Variablen von 3Ne-Elektronenkoordinaten auf die3 Ortskoordinaten von n(r) reduziert.Es verbleibt das Problem, eine geeignete Darstellung der Energieterme in diesem gekoppelten Sy-stem zu finden. Die weitere Vorgehensweise besteht darin, das reale wechselwirkende Elektronensy-stem im außeren Potential V (r) formal exakt auf ein virtuelles nichtwechselwirkendes Elektronen-system im außeren Potential Veff (r) abzubilden. Mit der Forderung, daß dieses virtuelle Systemdieselbe Grundzustandsdichte wie das reale System besitzen soll, erhalt man durch Minimalisierendes Energiefunktionals bezuglich n(r) fur Veff (r) folgende Form
Veff (r) = V (r) + e2∫
d3r′n(r′)|r − r′| +
δExc[n(r)]
δn(r), (4.5)
mit Exc[n] = T − TS +Ex +Ec (4.6)
T : kinetische Energie des realen wechselwirkenden Elektronensystems
TS : kinetische Energie des virtuellen nichtwechselwirkenden Elektronensystems
Ex : Austauschenergie
Ec : Korrelationsenergie .

46 KAPITEL 4. ELEKTRONENTHEORIE
Im virtuellen System laßt sich die Vielteilchenwellenfunktion als Produkt von Einteilchenwellen-funktionen darstellen. Diese genugen der entsprechend modifizierten Schrodingergleichung, auchKohn-Sham-Gleichung genannt :
[
− h2
2m∆i + Veff (ri)
]
ϕi(ri) = εiϕi(ri) . (4.7)
Die Elektronendichte erhalt man bei Kenntnis der ϕi(ri) aus
n(r) =Ne∑
i
|ϕi(r)|2 . (4.8)
Die korrekte Grundzustandsdichte gewinnt man durch die Vorgabe einer Startladungsdichte n0(r)und anschließender Iteration der Gleichungen (4.5), (4.7) und (4.8), bis sich die Ladungsdichteannahernd selbst reproduziert und somit Selbstkonsistenz sichergestellt ist. Die Grundzustands-energie des realen Elektronensystems ergibt sich dann zu
E[n] =Ne∑
i
εi −e2
2
∫
d3rd3r′n(r)n(r′)|r− r′| −
∫
d3rδExcδn
n(r) +Exc[n] . (4.9)
Dieses Variationsverfahren wird auch Kohn-Sham-Formalismus genannt [47].
4.2.3 Lokale Dichtenaherung (LDA)
Bisher wurde bis auf die Born-Oppenheimer-Naherung keinerlei weitere Naherung vorgenommen.Die Abbildung auf ein virtuelles nichtwechselwirkendes Elektronensystem ist exakt. Allein es fehltnoch eine Darstellung fur die Austausch-Korrelationsenergie Exc und deren FunktionalableitungδExc
δn . Besteht keine allzu starke Korrelation, so laßt sich Exc durch folgenden Ansatz nahern :
Exc[n(r)] ≈∫
d3rεxc(n(r))n(r) ≡ ELDAxc [n(r)] , (4.10)
wobei εxc die Austausch-Korrelationsenergie pro Elektron eines homogenen Elektronengases der
Dichte n(r) ist. Die Funktionalableitung δELDAxc
δn kann daraufhin durch geeignete Interpolationsfor-meln ermittelt werden. Diese Naherung wird als lokale Dichtenaherung (LDA) bezeichnet.
4.3 Mixed-basis-Pseudopotentialmethode
Ihrem Charakter nach ist die Dichtefunktionaltheorie eine Mean-Field-Theorie, da auch hier einVielteilchen-Hamiltonoperator auf einen effektiven Einteilchen-Hamiltonoperator abgebildet wird.Aufbauend auf der DFT existiert nun ein ganzer ”Zoo” von elektronentheoretischen Methodenzur Behandlung von Vielteilchensystemen. Diese unterscheiden sich gerade in der Festkorperphysikhauptsachlich durch die Konstruktion des periodischen Potentials V (r) und die Wahl des Basissat-zes, nach dem die Einteilchenwellenfunktionen ϕi(ri) entwickelt werden. Im folgenden soll lediglichdie in dieser Arbeit verwendete mixed-basis-Pseudopotentialmethode kurz vorgestellt werden. Fureine eingehende Diskussion dieser Methode sei auf [48] verwiesen.

4.3. MIXED-BASIS-PSEUDOPOTENTIALMETHODE 47
4.3.1 Periodisches Potential und Wellenfunktionen
Ausgangspunkt der Pseudopotentialtheorie ist die Feststellung, daß fur die relevanten Festkorperei-genschaften nahezu ausschließlich die Valenzelektronen verantwortlich sind, weil die Rumpfelektro-nen nicht zu der chemischen Bindung zwischen den Atomen beitragen. Demzufolge reicht es aus,allein die Valenzelektronen im Kohn-Sham-Formalismus zu behandeln und die verbleibenden Rump-felektronen in geeigneter Weise in das außere Potential V (r) ”einzuarbeiten”.Desweiteren laßt sich die unmittelbare Umgebung eines Kristallatoms in einen Rumpf- und einenZwischenbereich einteilen. Fur die Festkorperphysik ist nun lediglich die Streueigenschaft des imRumpfbereich wirkenden Potentials VR(r) von Bedeutung. Zur richtigen Beschreibung der Valenz-wellenfunktion ϕiZ(ri) im Zwischenbereich sind daher eine Vielzahl von Potentialen VR(r) denkbar,die alle diesselbe Streueigenschaft haben, von denen das wahre rechnerisch jedoch schwer handhab-bar ist.Der Grund hierfur ist, daß fur das Coulombpotential am Kernort die Valenzwellenfunktionen imRumpfbereich ϕiR(ri) stark oszillieren und dementsprechend viele Knoten aufweisen. Gunstiger furdie Entwicklung nach ebenen Wellen als Basisfunktionen ware es jedoch, wenn ϕiR(ri) keinerlei Kno-ten aufweisen wurde. Dies kann durch die Einfuhrung eines passenden Pseudopotentials V PS(r),daß das wahre Potential ersetzt, erreicht werden. Neben der Beibehaltung der ursprunglichen Streu-eigenschaften muß naturlich gewahrleistet sein, daß das Pseudopotential im Zwischenbereich in dasauslaufende Coulombpotential ubergeht.Bei der Konstruktion eines geeigneten Pseudopotentials V PS(r) bestehen trotzallem gewisse Frei-heiten. Grundsatzlich lassen sich Pseudopotentiale ebenfalls ab-initio berechnen. Hierzu ermitteltman zuerst ein atomares V PS
at (r) und bildet dann die translationsinvariante Fortsetzung auf demGitter.Mit dem so konstruierten Pseudopotential berechnet man daraufhin die Valenzwellenfunktionen,Grundzustandselektronendichte und Grundzustandsenergie im Kohn-Sham-Formalismus. Dabei wer-den, wie schon erwahnt, die Valenzwellenfunktionen nach einer geeigneten Basis entwickelt. Diedenkbar einfachste Basis ist diejenige, die aus ebenen Wellen aufgebaut ist. Fur die Behandlungvon Nebengruppenmetallen mit lokalisierten d-Zustanden ist eine reine ebene Wellen-Basis jedochunzureichend, da zur korrekten Beschreibung sehr viele ebene Wellen benotigt werden, was sich ineiner außerst hohen Rechenzeit niederschlagt.Es ist daher von Vorteil, eine gemischte Basis aus ebenen Wellen und zusatzlichen lokalisiertenFunktionen zu verwenden. Aufgrund der Gultigkeit des Bloch-Theorems ist es weiter sinnvoll, dengesamten Kohn-Sham-Formalismus im k-Raum durchzufuhren. Dazu wahlt man in der 1. Brillouin-Zone einen Satz irreduzibler k-Punkte aus und verwendet diese als Stutzstellen. Die Valenzwellen-funktionen lassen sich somit folgendermaßen darstellen :
ϕn,k(r) =1√Ω
∑
G
αn,kG
ei(k+G)r +∑
γlm
βn,kγlmφk
γlm(r) . (4.11)
n : Bandindex
Ω : Kristallvolumen
G : primitiver Gittervektor des reziproken Gitters
αn,kG
: Entwicklungskoeffizient
γ : γ-tes Atom in der Einheitszelle
lm : Bahndrehimpuls- und magnetische Quantenzahl
βn,kγlm : Entwicklungskoeffizient
φk
γlm(r) : Blochfunktion aus lokalisierten Funktionen

48 KAPITEL 4. ELEKTRONENTHEORIE
Das Eigenwertproblem schreibt sich in Matrixdarstellung als
Hkϕn,k = εn,kSkϕn,k , (4.12)
wobei Sk eine Uberlappmatrix ist, die der Tatsache Rechnung tragt, daß die ebenen Wellen undlokalisierten Funktionen nicht unbedingt orthogonal zueinander sind.
4.3.2 Berechnung von Kraften
Gerade bei Strukturen mit geringer Symmetrie treten bei unterschiedlichem Atomvolumen derKomponenten oftmals Relaxationen, d.h. Verschiebungen der Teilchen aus hochsymmetrischen Git-terpositionen, auf. Die Einstellung einer neuen abstandsveranderten Konfiguration ermoglicht esdem System, seine Gesamtenergie weiter abzusenken.Zur Berucksichtigung dieser wichtigen Energiebeitrage muß eine Moglichkeit gefunden werden, diees erlaubt Krafte auf die Atome zu berechnen, entlang derer diese dann verschoben werden konnen.Zur Losung dieses Problems macht man sich klar, daß die Grundzustandsenergie Ee eine Hyper-flache im Konfigurationsraum der Atomkernpositionen Rα bildet. Demnach ist die Kraft auf dasAtom i gegeben durch
Fi = −δEeδRi
. (4.13)
Dabei gilt fur δEe in erster Naherung :
δEe = − ∂Ee∂Ri
∣∣∣∣δRi=0
δRi +
∫
d3rδEeδn
∣∣∣∣n0
δn . (4.14)
Die Verschiebung der Grundzustandsenergie δEe setzt sich also aus einer expliziten Abhangigkeitvon der Atomposition und einer impliziten Abhangigkeit, die dadurch entsteht, daß eine Verschie-bung δRi eine infinitesimale Anderung in der Grundzustandsdichte bewirkt, zusammen. Es laßtsich nun zeigen, daß der implizite Term fur den Fall, daß n0 die exakte Grundzustandsdichte ist,verschwindet. Somit gilt
Fi = −δEeδRi
=∂Ee∂Ri
∣∣∣∣δRi=0
. (4.15)
Dies wird auch als Hellmann-Feynman-Theorem [49, 50] bezeichnet. Kennt man also die exakteGrundzustandsdichte, so hat sich die Berechnung der Krafte enorm vereinfacht, da nurmehr die ex-plizit von den Kernkoordinaten abhangigen Terme im Energiefunktional betrachtet werden mussen.Da man in der Praxis niemals mit einer vollstandigen Basis rechnen kann und so die exakte Grund-zustandsdichte nicht wirklich hat, muß man bei der Verwendung lokalisierter Basisfunktionen aberi. a. zusatzliche Beitrage zur Gesamtkraft, sogenannte incomplete-basis-set corrections, verwenden[48].

Kapitel 5
Ab-initio Statistik
Nachdem in den vorherigen Kapiteln die Grundlagen der CVM, Thermodynamik von Phasendia-grammen und Elektronentheorie behandelt wurden, soll nun in diesem Kapitel gezeigt werden,wie diese Themenkreise in einer allgemeinen ab-initio Statistik mit dem Ziel der parameterfreienBerechnung von Phasendiagrammen miteinander verbunden werden konnen.
5.1 Energieterme in realen Systemen
Bevor detailliert auf den Formalismus eingegangen wird, muß zuerst noch geklart werden, welcheWechselwirkungen und Anregungen in einem realen System relevant sind und demnach in derRechnung berucksichtigt werden mussen.Im folgenden sollen kurz die wichtigsten Energieterme in einem unmangnetischen System aufgezahltwerden, deren eindeutige Trennung jedoch nicht immer einfach ist.
• chemische Energie EchemDieser Term liefert den Hauptbeitrag zur Kristallenergie bei einer festen Konfiguration σ.Er ist als reine Wechselwirkungsenergie infolge der Kristallbindung zu verstehen. Wird zumBeispiel bei T = 0 und definierten Atompositionen in einem binaren Kristall ein Atom Adurch ein Atom der Sorte B ersetzt, so andert sich, wenn A und B gleiches Volumen besitzen,bis auf den energetischen Beitrag durch die Nullpunktsschwingungen allein Echem.Beim Rechnen mit der totalen Energie sind in diesem Term zusatzlich die atomaren Energiender unendlich weit voneinander getrennten Atome ”verarbeitet”.
• Relaxationsenergie ErelIn dieser Energie sollen alle Modifikationen der totalen Energie zusammengefaßt werden, dieals Folge von Volumenanderungen oder veranderten Atompositionen aufgrund von Relaxatio-nen in Erscheinung treten. Die Beschreibung dieses Terms gestaltet sich beispielsweise geradefur ungeordnete Systeme schwierig. In der Literatur wird Erel haufig noch in weitere Termezerlegt, worauf in Abschnitt 5.2.3 eingegangen werden soll.
• Schwingungsenergie EphonBekanntermaßen ist das Kristallgitter nicht starr, sondern es kann zu Schwingungen ange-regt werden. Dieser phononische Energiebeitrag ist i. a. nicht zu vernachlassigen und kannzusammen mit der in einer thermodynamischen Behandlung naturlich ebenfalls zu berucksich-
49

50 KAPITEL 5. AB-INITIO STATISTIK
tigenden Schwingungsentropie Sphon durchaus relevante Auswirkungen auf das zu erstellendePhasendiagramm haben.
• elektronische Anregungsenergie EelBei T 6= 0 wird es im System elektronische Anregungen geben. Die Thermodynamik dermeisten bisher betrachteten realen Systeme bleibt von dem Beitrag der freien elektronischenAnregungsenergie jedoch in den meisten Fallen unbeeinflußt, da dieser gegenuber den anderenTermen vergleichsweise klein ist. Es existieren aber Systeme, bei denen die Berucksichtigungdieses Beitrag durchaus relevant wird [51].
Fur die thermodynamische Beschreibung muß eigentlich, wie in Kapitel 3 gesehen, die freie Ent-halpie G herangezogen werden. Im Realfall ist jedoch der Druck P derart klein, daß der Term PVin G keinen nennenswerten Beitrag macht. Aus diesem Grund wird hier und im folgenden alleinmit der freien Energie F gearbeitet. Dies andert naturlich nichts an den in Kapitel 3 gemachtenAussagen.Zusammenfassend laßt sich also fur die Zustandssumme Z eines realen Systems bei minimalerKopplung der einzelnen Beitrage folgender Ausdruck angeben
Z =∑
σe−β[Echem+Erel] ×
∑
phon
e−βEphon∑
el
e−βEel , (5.1)
wobei ”∑
phon” und ”∑
el” die Summationen uber die phononischen und elektronischen Freiheits-grade symbolisieren soll.Es ist nun in der Regel gerechtfertigt anzunehmen, daß bei Ankopplung an ein Warmebad wahrendder Lebensdauer einer bestimmten Konfiguration das thermodynamische Gleichgewicht bezuglichden phononischen und elektronischen Anregungen vergleichsweise schnell erreicht wird. Demzufol-ge sollte es erlaubt sein, die Summation uber die phononischen und elektronischen Freiheitsgradejeweils fur eine feste Konfiguration σ separat durchzufuhren. Somit schreibt sich (5.1) als
Z =∑
σZphon(σ)Zel(σ)e−β[Echem+Erel] . (5.2)
Die freie Energie ergibt sich daraus in altbekannter Weise
F = −kBT lnZ = U − TSconf , (5.3)
mit
U =∑
σρ(σ)[Echem(σ) +Erel(σ) +Ephon(σ) − TSphon(σ) +Eel(σ) − TSel(σ)] , (5.4)
wobei Sconf die Konfigurationsentropie sowie Sphon und Sel die Schwingungs- respektive elektr.Anregungsentropien sind.Da in dieser Arbeit keine Schwingungs- oder elektr. Anregungsbeitrage zur freien Energie beruck-sichtigt werden, soll die Behandlung dieser Terme nicht genauer diskutiert werden. Da diese Beitrageaber ebenfalls konfigurationsabhangig sind, steht einer Behandlung im Rahmen der CVM prinzipi-ell nichts im Wege. So muß bei Miteinbeziehung dieser Terme einfach eine durch (5.4) nahegelegteCluster-Entwicklung der konfigurationsabhangigen freien Energie erfolgen.Fur einen Uberblick uber die bereits existierenden Verfahren zur genaherten Berucksichtigung die-ser Effekte sei der Ubersichtsartikel von de Fontaine [52] mit den dortigen Verweisen angefuhrt.

5.2. BESTIMMUNG DER ECIS 51
5.2 Bestimmung der ECIs
Es stellt sich nun die Frage, in welcher Weise ab-initio gerechnete Großen als Parameter in ei-ne verallgemeinerte statistische Methode eingehen konnen. Die thermodynamische Beschreibungeines System ist uber (5.1) prinzipiell eindeutig durch die zugrundeliegende Hamiltofunktion Hbestimmt. Die erste Aufgabe besteht also darin, diese fur ein reales Vielteilchensystem zu for-mulieren. Unter der Annahme, daß zwischen den Energien zu verscheidenen Konfigurationen σ
lineare Abhangigkeiten bestehen, kann zur Losung dieser Aufgabe die in Abschnitt (2.2) vorgestell-te Cluster-Entwicklung herangezogen werden. Im diesem Formalismus hat die Hamiltonfunktion Hdes betrachteten Systems folgende Gestalt :
H(σ) = K0 +N∑
i∗
∑
s\0νi∗Ki∗sηi∗s(σ) , (5.5)
wobei i∗ wiederum den Orbit eines Clusters unter der Symmetrie des Grundgitters indiziert. Zurab-inito Bestimmung der Ki∗s sollen in diesem Unterkapitel zwei Methoden vorgestellt werden.
5.2.1 Direct Configurational Averaging (DCA)
Da die ηi∗s normierten Clusterorbitfunktionen nach (2.35) entsprechen, wobei das gesamte Gitterdie Rolle von l ubernimmt, gehorchen diese bei Verwendung eines orthogonalen Basissatzes ebenfallseiner Orthogonalitatsrelation. In diesem Fall lassen sich die ECIs bekanntlich durch eine Umkehrungvon (5.5) gewinnen :
Ki∗s =1
NmN
mN∑
σηi∗sEdirekt(σ) . (5.6)
Die Gleichung (5.6) sagt also aus, daß die ECIs Ki∗s eindeutig aus direkt ab initio gerechnetenEnergien Edirekt(σ) zu allen denkbaren Konfigurationen σ gewonnen werden konnen.Die Berechnung der ECIs mittels (5.6) birgt jedoch einige Probleme in sich, die im folgenden kurzandiskutiert werden sollen.
• Die Formel (5.5) ist exakt, wenn uber alle Konfigurationen σ summiert wird, was aber imthermodynamischen Limes fur reale Anwendungen nicht durchfuhrbar ist. Es besteht jedochdie Hoffnung, daß die Ki∗s hinreichend schnell konvergieren und daher ein beschrankter Satzvon Konfigurationen fur die explizite Rechnung ausreicht.Trotzallem mussen ausreichend viele zufallige Konfigurationen mitgenommen werden, umdie Orthogonalitat naherungsweise sicherzustellen. Allgemein besitzen jedoch die wenigstenKonfigurationen Translationssymmetrie, sodaß die Rechnungen hierzu meist im Ortsraumdurchgefuhrt werden. Mit anderen Worten, die sehr genauen Dichtefunktionalmethoden sindfur diese Belange inpraktikabel, da schlichtweg zu zeitaufwendig.Implementiert werden kann die Berechnung mittels (5.6) daher bis heute lediglich fur tight-binding- oder embedded-atom-Methoden. Dabei werden Systemgroßen von N ≥ 1000 Git-terpunkten gewahlt, um die Randeffekte gering zu halten. Die Anzahl der Konfigurationen,uber die summiert wird, belauft sich auf ca. 50-100.
• Da die ECIs kleinen Differenzen von vergleichsweise großen Energiewerten entsprechen, sinddie Anforderungen an die Gute der ab-initio Methode allgemein sehr hoch.
• Notwendige Bedingung fur den Gebrauch von (5.6) ist die Entwicklung nach orthogonalenBasisfunktionen. Die von der Interpretation und Numerik her oftmals gunstigeren nichtor-thogonalen Clusterfunktionen konnen hier nicht verwendet werden.

52 KAPITEL 5. AB-INITIO STATISTIK
Trotz dieser Einschrankungen hat sich die Implementation von (5.6) in der Form des Direct Con-figurational Averaging (DCA) ([53, 54]) bewahrt.Der Vorteil dieser Methode ist, daß die einzelnen ECIs unabhangig voneinander bestimmt werdenkonnen.
5.2.2 Structure Inversion Method (SIM)
Neben der Ermittlung der ECIs uber das innere Produkt existiert noch eine weitere Methode,die im Gegensatz zum DCA vornehmlich auf der Berechnung von geordneten Strukturen σ mitausgewiesener Translationssymmetrie basiert. Diese 1983 von Connolly und Williams eingefuhrteMethode [8] wird auch als structure inversion method (SIM) bezeichnet.Hierbei lost man sich ein wenig vom strengen Formalismus der CE. Man geht zwar wiederum von(5.5) aus, nimmt jedoch unter der Voraussetzung, daß sowieso lediglich X mN Wechselwirkungenwichtig sein werden, von vornerein nur ebensoviele Clusterterme in der Entwicklung mit. DurchY ab initio gerechnete totale Energien ausgewahlter Strukturen lassen sich diese X ECIs dannentweder durch least-mean-square Verfahren
Y∑
σ
∣∣∣∣∣
X∑
i∗νi∗Ki∗sηi∗s(σ) − Edirekt(σ)
N
∣∣∣∣∣
2
= min. , (5.7)
oder im Fall von Y = X durch direkte Matrixinversion
Ki∗s =1
Nνi∗
X∑
ση−1σ,i∗sEdirekt(σ) (5.8)
gewinnen. Die Große η−1σ,i∗s entspricht dabei dem (σ, i ∗ s)-Element der Inversen der quadratischen
Y × X-Matrix η. Die SIM ist exakt wenn Y = X = mN gilt, wohingegen das DCA im Fall vonY = mN , unabhangig von X, exakt ist.Es stellt sich nun naturlich die Frage, welche Konfigurationen bei dieser Methode verwendet werdensollen. Da meist nur 10-20 Clusterterme in der Entwicklung (5.5) mitgenommen werden, ist es auf-grund der geringen Anzahl naheliegend, die genauen Dichtefunktionalverfahren zuganglichen, alsoperiodische geordnete Strukturen, hierfur heranzuziehen. Prinzipiell ist die einzige Einschrankungin der Wahl der Konfigurationen im Fall der Matrixinversion gegeben. Dort muß gewahrleistet sein,daß die Konfigurationen so gewahlt werden, daß die Matrix η nicht singular wird. Kriterien fur dieAuswahl der Konfigurationen hinsichtlich einer verbesserten Konvergenz der CE existieren sowohlfur den Fall Y ≥ X [55] als auch fur Y = X [56].Der Vorteil der SIM besteht zum einen in der vergleichsweise geringen Anzahl ab initio zu rech-nender Strukturen und zum anderen darin, daß es sich hierbei um rein geordnete Strukturen mitvergleichsweise kleiner Einheitszelle handelt. Damit bleibt, jedenfalls fur binare Systeme, der Re-chenaufwand auch fur anspruchsvollere elektronentheoretische Methoden vertretbar. Die physika-lische Bedeutung der ECIs ist jedoch nicht mehr eindeutig, da die eigentliche Deutung als totaleEnergiedifferenzen nur im Zusammenhang mit der Abbildung (5.6) Sinn macht. Desweiteren konnenmit dieser Methode die einzelnen ECIs nicht unabhangig voneinander bestimmt werden.Sowohl das DCA als auch die SIM finden Anwendung in der aktuellen Forschung. Vom physikali-schen Standpunkt ist keine der beiden Methoden der anderen vorzuziehen. Fur einen systemspezi-fischen Vergleich beider Verfahren sei auf [57] verwiesen.Um die Konvergenz der ECIs zu testen, wird bei beiden Methoden versucht, die Energien vonStrukturen, welche nicht in die Bestimmung der ECIs eingegangen sind, mit den erhaltenen Ki∗szu reproduzieren. Stimmen die so errechneten Energien mit den tatsachlich ab initio bestimmten

5.2. BESTIMMUNG DER ECIS 53
Werten gut uberein, so sollte Konvergenz erreicht sein. Ist dies nicht der Fall, so muß gegebenen-falls die Entwicklung weitergetrieben werden. Dieses trial and error-Verfahren fuhrt in der Praxiszu guten Ergebnissen.
5.2.3 Berucksichtigung von Relaxationsphanomenen
Bevor auf die Problematik der Berucksichtigung der Relaxationen eingegangen wird, soll zuerstgeklart werden, welche Relaxationsarten prinzipiell in einem realen System auftreten konnen. Diessoll anhand der Klassifizierung von Zunger et al. [10, 58] geschehen.
1. VolumendeformationDieser erste Relaxationsmechanismus ist auch der einfachste und beruht auf einer homogenenDeformation der Einheitszelle des Kristalls. Gerade bei geordneten kubischen Strukturen, beidenen lediglich symmetrieerhaltende Relaxationen auftreten konnen, ist dies aber zugleichauch der wichtigste Mechanismus.
2. Externe ZellrelaxationGeht man uber zu Strukturen geringerer Symmetrie, beispielsweise von einer kubischen zueiner tetragonalen Einheitszelle, so entstehen weitere neue Freiheitsgrade der Relaxation. Sokann gerade bei tetragonaler Symmetrie das Verhaltnis von c- zu a-Achse variieren und wirddaher auf den energetisch gunstigsten Wert relaxieren.
3. Interne ZellrelaxationReduziert man nun die Symmetrie des System noch weiter, indem man entweder Defekte wieLeerstellen oder Antistrukturatome berucksichtigt oder gar zum Extremfall einer ungeordne-ten Struktur ohne Fernordnung ubergeht, so konnen weitere lokale Relaxationen auftreten.Diese konnen mit Atomverlagerungen ohne Berucksichtigung der Symmetrie des Grundgittersverbunden sein.
Bei der ab-initio Behandlung der direkt berechneten Strukturen und der anschließenden Abbildungauf die ECIs mittels (5.7) oder (5.8)1 stellt sich schon allein die profane Frage, bei welchem Kri-stallvolumen die einzelnen Energien zu berechnen sind. Daruberhinaus andert sich meist mit derBesetzung des Grundgitters im kraftefreien Gleichgewicht auch die Symmetrie desselben aufgrundvon externen Zellrelaxationen. Problematisch hierbei ist, daß bei der bekannten Clusterentwicklungdie Symmetrie des Grundgitters eindeutig vorgegeben ist. So werden demnach ehemals entarteteECIs symmetrieinaquivalent. Gerade bei dem vorzeitigen Abbruch der CE kann diese Tatsache zufalschen Beschreibungen der reinen Energie als auch der Thermodynamik des Systems fuhren. Sobesteht bei letzterem die Problematik darin, die im Phasendiagramm auftretenden verschiedenenRelaxationsarten durch die SIM mit vergleichsweise wenig Clustertermen korrekt zu beschreiben.Gerade die am Ordnungs-Unordnungs Ubergang außerst wichtige interne Zellenrelaxation bereitetdabei aufgrund der großen Anzahl an Freiheitsgraden enorme Schwierigkeiten.Im folgenden sollen die existierenden Verfahren zur Behandlung der Relaxation bei Beibehaltungeines festen Grundgitters mit vergleichsweise hoher Symmetrie in der Klassifizierung nach Carlsson[59] vorgestellt werden.
1In der weiteren Diskussion soll allein die in dieser Arbeit verwendete SIM betrachtet werden.

54 KAPITEL 5. AB-INITIO STATISTIK
I. Vernachlaßigung der RelaxationDie Energien samtlicher fur die Abbildung verwendeter Strukturen werden bei einer festvorgegegebenen Form und Große der Einheitszelle berechnet. Diese Methode ist denkbarschlecht und fuhrt meist zu einer falschen Topologie des Phasendiagrammes.
II. Globale RelaxationFur jede geordnete Struktur ψ wird die Eψ(V )-Kurve im relevanten Volumenintervall be-stimmt. Dies erreicht man durch die Berechnung der Energie an mehreren Stutzstellen umdas Gleichgewichtsvolumen und anschließende Interpolation mit einer geeigneten Fitformel.Mittels der Abbildung (5.7) bzw. (5.8) wird die Volumenabhangigkeit der Eψ auf die Ki∗subertragen, so daß man schließlich volumenabhangige ECIs erhalt. Im thermodynamischenFormalismus wird dann bei einer bestimmten Zusammensetzung das zugehorige Volumendurch Minimalisieren der entsprechenden Gleichungen bezuglich V gewonnen. Aufgrund die-ser Tatsache wird diese Art, Relaxationsphanomene zu berucksichtigen, auch als Postinver-sions-Methode bezeichnet.Die physikalische Naherung dieses Verfahrens liegt in der Annahme begrundet, daß jeder Clu-sterkonfiguration ein gleiches mittleres Volumen zugeordnet werden kann. Diese zugegebenunrealistische Idealisierung liefert bereits annehmbare Resultate. Doch gerade bei Systemen,in denen sich die die Atomvolumina der Komponenten merklich unterscheiden, entstehendeutliche Diskrepanzen zwischen den experimentellen Daten und theoretischen Ergebnissen.Allgemein werden zum Beispiel bei dieser Methode die kritischen Temperaturen zumeistuberschatzt.Neben dem Volumen V ist es auch moglich die Abhangigkeit der ECIs nach dem gleichenVerfahren auf weitere Kristallparameter, wie etwa das c/a-Verhaltnis, zu erweitern.
III. Lokale RelaxationBei diesem Verfahren gehoren die Energien Eψ der fur die SIM verwendeten Strukturen zu denjeweiligen vollstandig ausrelaxierten Zustanden dieser Verbindungen. Die berechneten ECIssind dann wiederum volumenunabhangig. Diese Vorgehensweise entspricht der Annahme, daßjede Clusterkonfiguration im Kristall ideal relaxieren kann, d.h. ohne Berucksichtigung vonInkompatibilitaten. Diese Modellierung ist naturlich ebenso unrealistisch, da das Relaxati-onsverhalten sicherlich stark von der chemischen Umgebung abhangen wird.Da die Relaxationsenergie prinzipiell auch als direkte Funktion der Konfiguration des unrela-xierten Gitters verstanden werden kann, entspricht diese Art, die Relaxationen zu behandeln,am ehesten dem mathematischen Formalismus der CE. Weil jedoch in diesem Fall die CEallgemein sehr langsam konvergiert, kann der vorzeitige Abbruch der Entwicklung zu unrea-listischen Ergebnissen fuhren. Trotzallem ist der Stellenwert dieser Preinversions-Methodedem der globalen Relaxation gleichzusetzen.
Die Methode I wird aufgrund der unzureichenden Beschreibung so gut wie nicht verwendet. Aberauch die verwendete Methoden II und III liefern nur bedingt gute Ergebnisse. Es existieren ebensoMischformen und alternative Verfahren [11, 60], die aber ebenfalls noch nicht zufriedenstellend sind,da der lokale Charakter der Relaxationen bei beliebiger Zusammensetzung und Temperatur bishermathematisch noch nicht einwandfrei in den Cluster-Formalismus eingebaut werden konnte.Derzeit wird bei uns in der Gruppe versucht, dies durch eine Kombination der CVM mit derInkompatibilitatsmethode der stochastischen Elastizitatstheorie [61] zu erreichen.

5.3. NATURAL ITERATION METHODE (NIM) 55
5.3 Natural Iteration Methode (NIM)
Nachdem die ECIs Ki∗s mit der SIM bestimmt worden sind, konnen die thermodynamischen Ei-genschaften des System durch Minimierung des beschreibenden Potentials im Rahmen der CVMgewonnen werden. In Kapitel 2.3.6 wurde das zu variierende Funktional der freien Energie sym-metrieunabhangig formuliert. Fur das weitere ist es gunstig, dieses Funktional o.B.d.A. unter derAnnahme des Vorliegens minimaler Symmetrie innerhalb der verwendeten Clusternaherung darzu-stellen
F = N′∑
i
(∑
s
νiKisξis + kBTbiTr(i)ρi ln ρi
)
. (5.9)
Im Gegensatz zu Kapitel 2.3.7 sollen nun die ρi die Variationsparamter sein. Die einfachste Moglich-keit, dabei die Nebenbedingung (2.31) zu berucksichtigen, besteht darin, die ρi schon im Funktionaldurch die Wahrscheinlichkeiten ρn der Maximalcluster auszudrucken. Dies ist der grundlegende An-satz der von Kikuchi entwickelten Natural Iteration Methode zur Minimierung des nach Clusterentwickelten relevanten Potentials [62].Die ξis hangen allgemein uber die V-Matrix mit den ρn zusammen
ρn(τ ) =∑
i≤n
∑
s
Vn(τ ),isξis . (5.10)
Bei Verwendung einer orthogonalen Basis sind die Matrixelemente Vn(τ ),is identisch mit den Clu-sterorbitfunktionen φnis. Die Umkehrung von (5.10) lautet
ξis = Tr(n)ρn(τ )V −1n(τ ),is . (5.11)
Somit erhalt man folgenden Ausdruck fur die inneren Energie :
U = NTr(n)ρn(τ )∑
is
νiKisV−1n(τ ),is ≡ NTr(n)ρn(τ )Kn(τ ) . (5.12)
Dabei sind die Kn jetzt konfigurationsabhangige effektive Clusterenergien der Orbits der maximalenCluster n.Entsprechend laßt sich die Entropie mittels (2.31) allein durch die Clusterorbitwahrscheinlichkeitenρn darstellen,
S = −NkB
∑
i≤nbiTr(i)
(
Tr(n−i)ρn lnTr(n−i)ρn)
. (5.13)
Damit schreibt sich das Funktional der freien Energie als
F = N
Tr(n)ρnKn + kBT∑
i≤nbiTr(i)
(
Tr(n−i)ρn lnTr(n−i)ρn)
. (5.14)
Bei der Berechnung von Phasendiagrammen und den damit verbundenen physikalischen Großen istes oft gunstiger, nicht mit der freien Energie bzw. der freien Enthalpie zu arbeiten, sondern mitdem großen Potential J bzw. dem effektiven großen Potential Ω. In binaren Systemen sind diesePotentiale folgendermaßen definiert :
J = F −∑
l
µlNl = −PV bzw. j = f − µAcA − µBcB = −Pv , (5.15)
Ω = g − µ∗AcA − µ∗BcB . (5.16)

56 KAPITEL 5. AB-INITIO STATISTIK
Da der Term −Pv nur bei hohen Drucken relevant wird, gilt f ≈ g und somit genahert
J = F −∑
i
µiNi = 0 bzw. j = f − µAcA − µBcB = 0 , (5.17)
Ω = f − µ∗AcA − µ∗BcB . (5.18)
Im weiteren wird die Minimierung anhand des effektiven großen Potentials Ω behandelt. Der For-malismus ist jedoch der gleiche fur j, und auch die Erweiterung auf mehrkomponentige Systemeist einfach durchzufuhren.Fur eine bestimmte Kristallsymmetrie lautet Ω demnach
Ω = Tr(n)ρnKn + kBT∑
i≤nbiTr(i)
(
Tr(n−i)ρn lnTr(n−i)ρn)
− µ∗AcA(ρn) − µ∗BcB(ρn) . (5.19)
Bisher wurde lediglich die Nebenbedingung (2.31) durch Umschreiben des Funktionals auf dieWahrscheinlichkeiten der maximalen Cluster als alleinige Variationsparameter berucksichtigt. Esmuß jedoch bei der Minimierung sichergestellt sein, daß zudem Tr(n)ρn = 1 gilt. Dies kann mandurch Einfugen eines Lagrange-Parameter λ in (5.19) gewahrleisten
Ω = Tr(n)ρnKn+kBT∑
i≤nbiTr(i)
(
Tr(n−i)ρn lnTr(n−i)ρn)
−µ∗AcA(ρn)−µ∗BcB(ρn)+λ(1−Tr(n)ρn) .
(5.20)Nachdem das effektive große Potential in diese Form gebracht wurde, kann die Minimierung von Ωerfolgen, d.h. es mussen die CVM-Gleichungen
∂Ω
∂ρn= 0 (5.21)
aufgestellt werden. Zum besseren Verstandnis soll dies nun gleich an zwei konkreten Beispielenerfolgen. Es sind dies die auch in dieser Arbeit verwendeten Tetraedernaherungen auf dem fcc- undbcc-Gitter.
5.3.1 Tetraedernaherung auf fcc-Gitter
In dieser Naherung ist der maximale Cluster der regulare Tetraeder, bei dem die Kantenlangen denNN-Abstanden auf dem fcc-Gitter entsprechen.Zur Behandlung von Superstrukturen muß das fcc-Grundgitter in Untergitter aufgeteilt werden,die das Einstellen einer Fernordnung erlauben. Mit dem Tetraeder als maximalem Cluster ist eineUnterteilung in 4 sc-Gitter derart moglich, daß die 4 Ecken des Tetraeders je zu einem dieser Unter-gitter gehoren (Abb. (5.1)). Eine Unterteilung in mehr Untergitter ist zum einen nicht mehr ganz soeinfach handhabbar, zum anderen in dieser Naherung aber auch nicht unbedingt sinnvoll, da einesolche Untergitterstruktur uber NN-Abstande hinausgehen wurde und die Besetzungskorrelationenvon dem einfachen Tetraeder dann nicht sauber erfaßt werden konnten.Zur Charakterisierung der Strukturen, die auf besagten Untergittern dargestellt werden konnen,werden folgende Fernordungsparameter definiert :
ζ1 = 〈σ1〉 + 〈σ2〉 − 〈σ3〉 − 〈σ4〉 , (5.22)
ζ2 = 〈σ1〉 − 〈σ2〉 , (5.23)
ζ3 = 〈σ3〉 − 〈σ4〉 , (5.24)
wobei 〈σl〉 den Erwartungswert der Spinvariablen auf dem Untergitter l wiedergibt. Damit konnendie Strukturen A1, L10, L12 und P4/mmm beschrieben werden (Tab. (5.2)).

5.3. NATURAL ITERATION METHODE (NIM) 57
Neben der Bestimmung der Tetraederwechselwirkungen Kn besteht die zentrale Aufgabe darin, denEntropieterm fur diese Clusternaherung aufzustellen. Hierzu mussen zuerst die aβ-Koeffizientenfur die im Tetraeder enthaltenen Subcluster bestimmt werden (s. Tab. (5.1)). Wie man leichtnachrechnet, verschwindet der aβ-Koeffizient des Dreiecks. Dies liegt daran, daß das Dreieck keineFigur ist, die durch Uberlappung der Tetraeder entsteht. Da es durch die Tetraeder schon mitgezahltwird, taucht dieser Cluster nicht mehr eigenstandig in der Darstellung der Entropie auf.Die Mulitplizitat wird mit νi∗ allein fur das Grundgitter angegeben. Die Symmetrieeinschrankungen,die sich durch die Beschreibung verschiedener Kristallstrukturen ergeben, werden, wie im folgendengezeigt wird, beim Losen der CVM-Gleichungen der NIM implizit berucksichtigt.Die fur die NIM relevante Clusterwahrscheinlichkeit ρn hat in der Tetraedernaherung folgendeGestalt :
ρn = ρ(1234)abcd , (5.25)
wobei die oberen Indizes die Untergitter beschreiben und die unteren Indizes die Besetzung aufdiesen angeben. Die Entropie ergibt sich nach (2.86) zu
S
NkB
= −∑
i≤nbiTr(i)ρi ln ρi . (5.26)
Fur einen allgemeinen geordneten Zustand laßt sich dies auch schreiben als
S
NkB
= −∑
i∗
aβi∗νi∗r
r∑
i∈i∗Tr(i)ρi ln ρi
= −5
4
∑
a
[
L(ρ(1)a ) + L(ρ(2)
a ) + L(ρ(3)a ) + L(ρ(4)
a )]
+∑
ab
[
L(ρ(12)ab ) + L(ρ
(13)ab ) + L(ρ
(14)ab ) + L(ρ
(23)ab ) + L(ρ
(24)ab ) + L(ρ
(34)ab )
]
−2∑
abcd
L(ρ(1234)abcd ) , (5.27)
mit L(x) = x lnx. Diese Entropiedarstellung fur das fcc-Gitter in Tetraedernaherung wurde erst-mals von Golsov et al. [63] und van Baal [21] aufgestellt.Druckt man nun die Subclusterwahrscheinlichkeiten in (5.27) wieder durch (5.25) aus, setzt dieseEntropiedarstellung in (5.20) ein und fuhrt (5.21) durch, so ergeben sich folgende Bestimmungs-
gleichungen fur die ρ(1234)abcd [62]
ρ(1234)abcd = Y
12abcdX
− 58
abcd exp
(
−4K(fcc)abcd + µ∗a + µ∗b + µ∗c + µ∗d
8kBT
)
exp
(λ
2kBT
)
, (5.28)
mit
Yabcd = ρ(12)ab ρ(13)
ac ρ(14)ad ρ
(23)bc ρ
(24)bd ρ
(34)cd , (5.29)
Xabcd = ρ(1)a ρ
(2)b ρ(3)
c ρ(4)d . (5.30)
Bildet man nun auf beiden Seiten von (5.28) die Spur uber die Tetraederwahrscheinlichkeiten, soergibt sich
exp
(
− λ
2kBT
)
=∑
abcd
Y12abcdX
− 58
abcd exp
(
−4K(fcc)abcd + µ∗a + µ∗b + µ∗c + µ∗d
8kBT
)
. (5.31)

58 KAPITEL 5. AB-INITIO STATISTIK
Die Gleichung (5.31) hat die Form der Berechnung einer Zustandssumme. Auf der rechten Seitewird uber alle Tetraederkonfigurationen summiert. Dies ist konsistent mit der Annahme, alle Kri-stallkonfigurationen mittels Tetraederkonfigurationen darstellen zu konnen. Die Faktoren X undY tragen der Tatsache Rechnung, daß hierzu bereits ein Mittelungsprozeß stattgefunden habenmuß. Die fur die Rechnung benotigten Paar- und Punktwahrscheinlichkeiten gewinnt man durchAusspuren der Tetraederwahrscheinlichkeiten :
ρ(12)ab =
∑
cd
ρ(1234)abcd ρ(13)
ac =∑
bd
ρ(1234)abcd . . . , (5.32)
ρ(1)a =
∑
bcd
ρ(1234)abcd ρ
(2)b =
∑
acd
ρ(1234)abcd . . . . (5.33)
Die Gleichungen (5.28),(5.31),(5.32) und (5.33) bilden nun einen Iterationszyklus.Im ersten Schritt des Verfahrens werden Punktwahrscheinlichkeiten vorgegeben. Aus diesen werdendann Paarwahrscheinlichkeiten anhand der Bragg-Williams-Methode erzeugt, d.h.
ρ(12)ab = ρ(1)
a ρ(2)b . . . (5.34)
Danach wird der Exponentialterm exp(
− λ2kBT
)
mittels (5.31) berechnet. Daraufhin konnen die
Tetraederwahscheinlichkeiten ρ(1234)abcd nach (5.28) bestimmt werden. Die neuen Paar- und Punkt-
wahrscheinlichkeiten ergeben sich dann aus (5.32) und (5.33). Diese gehen im nachsten Zykluswieder in (5.31) ein usw. .Der Iterationszyklus wird als konvergiert angesehen, wenn die Tetraederwahrscheinlichkeiten sichselbst reproduzieren, also folgende Bedingung erfullt ist :
∆p =∑
abcd
[
ρ(1234)abcd (p) − ρ
(1234)abcd (p− 1)
]
< ε , (5.35)
wobei p die Zahl der Iterationen indiziert.Ist Selbstkonsistenz erreicht, so ergibt sich, wie anhand (5.31) leicht zu verifizieren ist, daß daseffektive große Potential Ω gleich dem Lagrange-Parameter λ ist
Ω = λ . (5.36)
Unter Verwendung der Relationen (3.19),(3.20) und (3.21) konnen aus Ω dann die chemischen Po-tentiale sowie die freie Enthalpie bzw. freie Energie gewonnen werden. Neben diesen thermodyna-mischen Großen liefert die Minimierung naturlich auch die Clusterbesetzungswahrscheinlichkeitenim thermodynamischen Gleichgewicht. Aus diesen konnen dann bei Bedarf Nah- oder Fernord-nungsparameter ermittelt werden.
5.3.2 Tetraedernaherung auf bcc-Gitter
Auf dem bcc-Gitter ist der irregulare Tetraeder mit zwei NN-Abstanden und zwei NNN-Abstandenals Kantenlangen die einfachste Moglichkeit, einen dreidimensionalen Cluster zu bilden. Auch dasbcc-Gitter laßt sich in 4 Untergitter aufteilen, wobei wiederum jede Ecke des Tetraders zu einemanderen Untergitter gehort. Diese Untergitter sind jedoch im Gegensatz zu den sc-Gittern im fcc-Kristall fcc-artig (Abb. (5.2)). In dieser Aufteilung bilden die Gitterpunkte der Untergitter 1 und2 sowie die von 3 und 4 NNN-Paare.Die Definition der Fernordnungsparameter ζi entspricht der des fcc-Gitters. Mit dieser Untergitter-struktur lassen sich die Strukturen A2, B32, B2, D03 und F 43m beschreiben (Tab. (5.2)).Die Bezeichnung der Tetraderwahrscheinlichkeiten ist identisch mit der auf dem fcc-Gitter. Auf-grund der veranderten Topologie des Gitters ergeben sich nun jedoch andere Uberlappungskoeffi-zienten aβ und Multiplizitaten νi∗ (Tab. (5.1)).

5.3. NATURAL ITERATION METHODE (NIM) 59
Die Entropie in dieser CVM-Naherung stellt sich nach [64, 65] in Analogie zu (5.27) folgendermaßendar :
S
NkB
=1
4
∑
a
[
L(ρ(1)a ) + L(ρ(2)
a ) + L(ρ(3)a ) + L(ρ(4)
a )]
−∑
ac
[
L(ρ(13)ac ) + L(ρ(14)
ac ) + L(ρ(23)ab ) + L(ρ
(24)ab )
]
−3
2
∑
ab
[
L(ρ(12)ab ) + L(ρ
(34)ab )
]
+3∑
abc
[
L(ρ(123)abc ) + L(ρ
(124)abc ) + L(ρ
(134)abc ) + L(ρ
(234)abc )
]
−6∑
abcd
L(ρ(1234)abcd ) . (5.37)
Die Minimierung fuhrt wiederum zu folgenden Gleichungen fur die ρ(1234)abcd (z. B. [66]) :
ρ(1234)abcd = U
12abcdV
− 14
abcdY− 1
6abcdX
124abcd exp
−4K
(bcc)abcd + µ∗i + µ∗j + µ∗k + µ∗l
24kBT
exp
(λ
6kBT
)
, (5.38)
mit
Uabcd = ρ(123)abc ρ
(124)abd ρ
(134)acd ρ
(234)bcd (5.39)
Vabcd = ρ(12)ab ρ
(34)cd (5.40)
Yabcd = ρ(13)ac ρ
(14)ad ρ
(23)bc ρ
(24)bd (5.41)
Xabcd = ρ(1)a ρ
(2)b ρ(3)
c ρ(4)d (5.42)
Die Losung von (5.38) erfolgt nach dem gleichen Schema wie im Fall des fcc-Gitters.
5.3.3 Zur Numerik
Die in den vorausgegangenen Abschnitten beschriebene NIM ist ein numerisches Verfahren zurMinimierung des relevanten thermodynamischen Potentials. Im Gegensatz zur Newton-RaphsonMethode (NRM) (z.B [67]), die ein allgemeines numerisches Verfahren zur Losung nichtlinearerGleichungssysteme darstellt, ist es eine speziell fur die Clusterentwicklung konzipierte Methode, diesich direkt die bestehenden Relationen zwischen den Clusterwahrscheinlichkeiten zu Nutze macht.Im folgenden sollen einige Eigenschaften dieser Methode dargelegt werden :
• Ein Vorteil der NIM gegenuber der NRM ist die mathematische Einfachheit, da weder Ablei-tungen noch Matrix-Inversionen durchzufuhren sind und bei festem Grundgitter diesselbenIterationsgleichungen fur samtliche Strukturen auf diesem Gitter verwendet werden konnen.
• Es kann gezeigt werden, daß wahrend der Iteration das beschriebene Potential monoton fallt[62]. Dies fuhrt dazu, das der Iterationszyklus immer in eine stabile oder metastabile Losungkonvergiert.
• Fern von kontinuierlichen Phasenubergangen ist die Zahl der erforderlichen Iterationsschrit-te p bei einem ε von 10−9 meist kleiner als 103. An einem solchen Ubergang divergiert dasVerfahren [62]. Aufgrund der Einfachheit des Formalismus ist die erforderliche CPU-Zeit in

60 KAPITEL 5. AB-INITIO STATISTIK
Anbetracht der heutigen Rechnerleistungen aber auch in der Nahe von kritischen Punkten,gerade im Vergleich zu aufwendigen MC- oder MD-Simulationen, gering. Jedoch konvergiertdie NIM i.a. langsamer als die NRM.Bei obigem ε ist das relevante Potential in der Tetraedernaherung auf ca. 10−14 Ry auskon-vergiert.
• Der Iterationszyklus der NIM ist einfacher zu handhaben als der der NRM, da aufgrund derNahe des Verfahren zur konkreten Problemstellung keine total unphysikalischen Losungen wieetwa negative Wahrscheinlichkeiten auftreten konnen. Dies kann jedoch in der NRM durchauspassieren, was eine sensiblere Wahl der Anfangswerte als in der NIM erfordert.
• Durch die Vorgabe der Punktwahrscheinlichkeiten ρ(i)a im ersten Iterationsschritt (5.34) wird
zugleich eine bestimmte Kristallstruktur vorgegeben. Diese ”aufgepragte” Symmetrie kannim Verlauf der Iteration auch dann erhalten bleiben, wenn die Temperatur und die effektivenchemischen Potentiale derart gewahlt sind, daß besagte Kristallstruktur lediglich metasta-bil ist. Dies bedeutet, daß die NIM in eine metastabile Losung lauft. Metastabile Losungenkonnen auch bei eigentlich stabilen Strukturen durch eine ungunstige Wahl der Anfangswerteauftreten. Desweiteren konnen auch Symmetrieanderungen im Iterationszyklus entstehen.Diese Tatsachen machen die Identifzierung der zu den vorgegebenen Parametern stabilenStruktur nicht immer einfach. Die allgemeine Vorgehensweise besteht darin, bei festem Pa-rametersatz samtliche dafur in Frage kommenden Strukturen durch die Vorgabe eines Satzesvon Anfangswerten mit der NIM zu behandeln. Entscheidendes Kriterium bei der Auswahl derstabilen Struktur ist anschließend naturlich das effektive große Potential Ω. Oftmals mussenaber auch die konvergierten Clusterwahrscheinlichkeiten fur die endgultige Entscheidung zu-gunsten einer Struktur herangezogen werden.Das Durchlaufen metastabiler Bereiche klingt zuerst wie ein Nachteil der CVM. Diese Losun-gen konnen jedoch auch ein Gewinn bedeuten, da zwar die Identifizierung des wahren Mini-mums etwas Erfahrung erfordert, man aber zusatzliche thermodynamische Information erhalt.So lassen sich dadurch zum Beispiel Spinodal-Kurven oder Phasenubergange 1. Ordnung in-nerhalb der Clusternaherung beliebig genau behandeln.
• Die globale Volumenrelaxation laßt sich in der NIM dadurch realisieren, daß nach jedemIterationsschritt das Potential hinsichtlich des Volumens minimiert wird. Dabei erfahren alleindie K
(fcc)abcd bzw. K
(bcc)abcd eine Veranderung. Deren neue Werte werden dann einfach im nachsten
Iterationsschritt verwendet. Es zeigt sich, daß das Volumen meist binnen weniger Iterationen(p < 100) konvergiert ist.
Zum Abschluß dieses Kapitels sollen im letzten Abschnitt noch einmal die wichtigsten Eckpunkteder hier vorgestellten ab-initio Statistik in Form eines allgemeinen Ablaufplans zusammengefaßtwerden.

5.3. NATURAL ITERATION METHODE (NIM) 61
5.3.4 Ablaufplan der ab-initio Modellierung
1. Zu Beginn muß das Grundgitter, oder im Fall von inkoharenten Systemen der Satz von Grund-gittern, festgelegt werden. Desweiteren muß man klaren, welcher Basisatz fur die Clusterent-wicklung gewahlt werden soll. Bei Verwendung des DCA ist man auf eine orthogonale Basisbeschrankt.
2. Im nachsten Schritt muß man sich fur den Grad der Clusternaherung entscheiden, d.h. denSatz maximaler Cluster αM bestimmen. Prinzipiell konnen fur die CE der Energie und dieClusterdarstellung der Entropie auch verschiedene Satze von maximalen Clustern gewahltwerden. Die CE der Hamiltonfunktion nimmt dann folgende Gestalt an :
H(σ) = K0 +N∑
i∗≤n∗
∑
s\0νi∗Ki∗sηi∗s(σ) .
Der Indexzusatz ”*” bedeutet dabei, daß es sich bei dem betreffenden Orbit um einen solchenunter der Raumgruppe des gewahlten Grundgitters handelt. Fur die explizite Rechnung istweiter die Bestimmung der Multiplizitaten νi∗ wichtig, d.h. die Zahl der symmetrieaquivalen-ten Cluster pro Gitterpunkt.
3. Bei Verwendung der SIM sind dann die Y Referenzkonfigurationen fur die Connolly-Williams-Abbildung auszuwahlen. Dies muß so geschehen, daß im Fall Y = X keine lineare Abhangig-keiten zwischen diesen einzelnen Konfigurationen bestehen.Anschließend muß man sich daruber klar werden, wie die Relaxationen zu behandeln sind.Dies hangt naturlich vom gewahlten System ab. Bei einer reinen Volumenrelaxation mussendann die totalen Energien Edirekt besagter Konfigurationen ab initio volumenabhangig be-rechnet werden. Im lokalen Relaxationsschema sind fur das weitere allein die Energien zu denGleichgewichtsvolumina relevant, im globalen Schema ist die gesamte Etot(V )-Kurve jederStruktur wichtig.
4. Sodann hat man die gittergemittelten Korrelationsfunktionen ηi∗s fur die Referenzkonfigu-rationen in der gewahlten Basis zu bestimmen. Ist dies geschehen, konnen die X Ki∗s imuberbestimmten Fall durch
Y∑
σ
∣∣∣∣∣
X∑
i∗νi∗Ki∗sηi∗s(σ) − Edirekt(σ)
N
∣∣∣∣∣
2
= min. ,
oder im Fall von Y = X durch direkte Matrixinversion
Ki∗s =1
Nνi∗
X∑
ση−1σ,i∗sEdirekt(σ)
ermittelt werden.
5. Allgemein ist es fur die thermodynamische Behandlung gunstig, die V-Matrix fur die jeweiligeClusternaherung aufzustellen. So ist dies bei der Verwendung eines nichtorthogonalen Basis-satzes eine Bedingung, um die Transformation zwischen den Clusterwahrscheinlichkeiten undden Korrelationsfunktionen zu ermoglichen
ρn(τ ) =∑
i≤n
∑
s
Vn(τ ),isξis .

62 KAPITEL 5. AB-INITIO STATISTIK
Da diese Transformation auch bei orthogonaler Basis wichtig ist und dort die Eintrage derV-Matrix gerade den Clusterfunktionen entsprechen, ist die Konstruktion dieser Matrix auchin diesem Fall angebracht.
6. Fur die explizite Rechnung muß nun noch entschieden werden, ob die ξis oder die ρi alsVariationsparameter fur die Minimierung von
F = N′∑
i
(∑
s
νiKisξis + kBTbiTr(i)ρi ln ρi
)
.
dienen sollen. Im ersten Fall hat man die Newton-Raphson-Methode zur Losung der CVM-Gleichungen
νjKjs
kBT+∑
i≥jbiρ
0iTr(i)φijs ln ρi = 0
zu implementieren. Bei Wahl der ρi wird die vorgestellte NIM verwendet. Dabei mussen dieKi∗s mittels der V-Matrix zuerst noch auf die Kn transformiert werden
Kn =∑
is
νiKisV−1n(τ ),is .
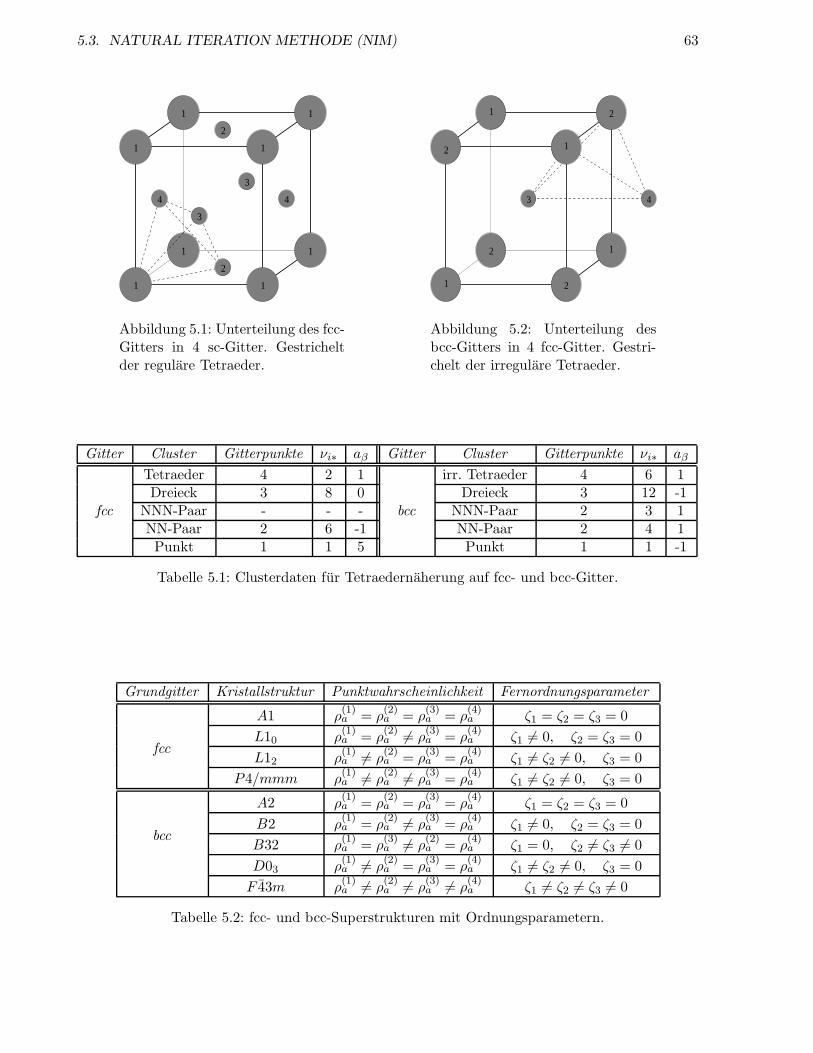
5.3. NATURAL ITERATION METHODE (NIM) 63
3
2
1
1 1
1
1
11
2
3
44
1
Abbildung 5.1: Unterteilung des fcc-Gitters in 4 sc-Gitter. Gestricheltder regulare Tetraeder.
1
4
1
1
1
2
2
2
2
3
Abbildung 5.2: Unterteilung desbcc-Gitters in 4 fcc-Gitter. Gestri-chelt der irregulare Tetraeder.
Gitter Cluster Gitterpunkte νi∗ aβ Gitter Cluster Gitterpunkte νi∗ aβ
Tetraeder 4 2 1 irr. Tetraeder 4 6 1Dreieck 3 8 0 Dreieck 3 12 -1
fcc NNN-Paar - - - bcc NNN-Paar 2 3 1NN-Paar 2 6 -1 NN-Paar 2 4 1Punkt 1 1 5 Punkt 1 1 -1
Tabelle 5.1: Clusterdaten fur Tetraedernaherung auf fcc- und bcc-Gitter.
Grundgitter Kristallstruktur Punktwahrscheinlichkeit Fernordnungsparameter
A1 ρ(1)a = ρ
(2)a = ρ
(3)a = ρ
(4)a ζ1 = ζ2 = ζ3 = 0
L10 ρ(1)a = ρ
(2)a 6= ρ
(3)a = ρ
(4)a ζ1 6= 0, ζ2 = ζ3 = 0
fccL12 ρ
(1)a 6= ρ
(2)a = ρ
(3)a = ρ
(4)a ζ1 6= ζ2 6= 0, ζ3 = 0
P4/mmm ρ(1)a 6= ρ
(2)a 6= ρ
(3)a = ρ
(4)a ζ1 6= ζ2 6= 0, ζ3 = 0
A2 ρ(1)a = ρ
(2)a = ρ
(3)a = ρ
(4)a ζ1 = ζ2 = ζ3 = 0
B2 ρ(1)a = ρ
(2)a 6= ρ
(3)a = ρ
(4)a ζ1 6= 0, ζ2 = ζ3 = 0
bccB32 ρ
(1)a = ρ
(3)a 6= ρ
(2)a = ρ
(4)a ζ1 = 0, ζ2 6= ζ3 6= 0
D03 ρ(1)a 6= ρ
(2)a = ρ
(3)a = ρ
(4)a ζ1 6= ζ2 6= 0, ζ3 = 0
F 43m ρ(1)a 6= ρ
(2)a 6= ρ
(3)a 6= ρ
(4)a ζ1 6= ζ2 6= ζ3 6= 0
Tabelle 5.2: fcc- und bcc-Superstrukturen mit Ordnungsparametern.

Kapitel 6
System NiAl
6.1 Grundzustandsbetrachtungen
Bevor die Thermodynamik der NiAl-Systems angegangen werden kann, muß zuerst das Verhaltendieses Systems bei T = 0 untersucht werden. Hierzu gehort die Klarung der Grundzustande inAbhangigkeit von der Ni-Konzentration. Wie bereits erwahnt, wird in dieser Arbeit keine reine ab-initio Betrachtung in der Weise durchgefuhrt, daß keinerlei Strukturinformation in die Rechnungeingeht, wie es etwa die Car-Parrinello Methode erlaubt.In diesem Sinn sind die hier angefuhrten Betrachtungen als Diskussionsgrundlage fur das spaterzu besprechende thermodynamische Verhalten des Systems NiAl zu verstehen, da eine detaillierteGrundzustandssuche ein Thema fur sich darstellt und den Rahmen dieser Abhandlung sprengenwurde.Die in diesem Kapitel besprochenen Rechnungen wurden mit dem in unserer Gruppe entwickeltenmixed-basis Pseudopotentialprogramm [9] durchgefuhrt.
6.1.1 Auswahl der Strukturen
Die Basis der in dieser Arbeit getatigten Betrachtungen zum Grundzustand und thermodynami-schen Verhalten des NiAl-Systems bildete ein ausgewahlter Satz von geordneten Kristallstrukturen.Zusammengestellt wurde dieses Ensemble zum einen hinsichtlich den experimentellen Gegebenhei-ten und zum anderen bezuglich einer sinnvollen statistischen Modellierung im Rahmen der CVM inTetraedernaherung. Da im experimentellen Phasendiagrammm sowohl fcc- als auch bcc-Strukturenrealisiert sind, mußte eine gekoppelte CVM auf diesen beiden Grundgittern entwickelt werden. Diekomplexe orthorhombische D011-NiAl3- und die trigonale D519-Ni2Al3-Struktur im Ni-armen Ge-biet wurden in dieser Arbeit nicht berucksichtigt.Die experimentell stabilen reinen Phasen im NiAl-System sind Ni-fcc, L12-Ni3Al und Al-fcc ebenauf einem fcc-Grundgitter sowie B2-NiAl auf einem bcc-Grundgitter. Zur Durchfuhrung der SIM inTetraedernaherung mit Matrixinversion waren demnach noch 2 fcc-Strukturen und 5 bcc-Strukturenerforderlich.Die besagten stabilen Strukturen lassen sich jeweils durch eine bestimmte Tetraederkonfigurationim fcc- bzw. bcc-Gitter charakterisieren. Da nun die Zahl der unter der Symmetrie des Grundgittersinaquivalenten Tetraederkonfigurationen genau der Zahl der ECIs entspricht, erschien es sinnvoll, zuden verbleibenden Tetraederkonfigurationen korrespondierende Kristallstrukturen in das Ensemblemitaufzunehmen. Weiteres Kriterium war die Nahe der zusatzlichen Strukturen zum Experiment
64

6.1. GRUNDZUSTANDSBETRACHTUNGEN 65
sowie die Darstellbarkeit auf den Grundgittern im Rahmen der veranschlagten Untergitterstruktur(Abb. (5.1) und (5.2)). Auf dem fcc-Gitter kamen so noch die L10-NiAl- und L12-NiAl3-Strukturhinzu. Die reinen bcc- und die beiden D03-Strukturen sowie die B32-Struktur vervollstandigten denzum bcc-Gitter gehorenden Teil des Ensembles. Aufgrund der Tatsache, daß jede der Struktureneiner bestimmten Tetraederkonfiguration entspricht, kann man diese auch als kanonisch bezeichnen.Die notwendige lineare Unabhangigkeit dieser Strukturen ist augenscheinlich klar.Samtliche in dieser Arbeit behandelten Kristallstrukturen sind in Anhang C dargestellt.
6.1.2 Abschneideenergie und Anzahl der k-Punkte
Bei der ab-initio Berechnung der Grundzustandsenergie einer gegebenen Struktur werden die Va-lenzwellenfunktionen des Kristalls nach ebenen Wellen und lokalisierten Funktionen entwickelt. Dievon den Elektronen zu besetzenden Bander werden uber ein geeignet gewahltes Stutzstellennetz inder 1. Brillouin-Zone bestimmt.Wollte man die Energie innerhalb der Pseudopotentialtheorie in LDA exakt berechnen, so mußtenunendlich viele ebene Wellen und ein unendlich dichtes k-Punkt-Netz verwendet werden. In derRealitat wird man versuchen, durch Naherungen im Entwicklungssatz und Stutzstellennetz einmoglichst kleines Energieintervall anzustreben, innerhalb dessen die exakte Energie liegt. Die Be-schrankung im Entwicklungssatz manifestiert sich in einer gewissen Abschneideenergie Ecut :
h2
2m|k + G|2 ≤ Ecut , (6.1)
wobei G ein reziproker Gittervektor ist. Das k-Punkt-Netz im reziproken Raum kann auf verschie-dene Weise erzeugt werden. Die einfachste Methode nach Chadi-Cohen (CC) [68] geht von einemsc-Netz aus. Dabei werden als Parameter die Zahl der Unterteilungen der Basisvektoren im rezi-proken Raum angegeben.Die Abschneideenergie und die Zahl der k-Punkte sind Konvergenzparameter, hinsichtlich dererman die Ergebnisse der Rechnung auskonvergieren mußte. In der praktischen Rechnung wird manalso die Konvergenzparameter so lange vergroßern, bis man den Eindruck gewonnen hat, daß sichdie Ergebnisse bei einer weiteren Vergroßerung nur noch innerhalb einer vorgegebenen Schrankeverandern. Diese Konvergenztests erfolgten in der vorliegenden Arbeit anhand der bcc-StrukturB2-NiAl, da diese im Phasendiagramm von NiAl die dominierende Struktur darstellt. Es wurdedann angenommen, daß die Resultate fur alle anderen Strukturen ganz ahnliches Konvergenzver-halten aufweisen.In den Abbildungen (6.1) und (6.2) ist die Abhangigkeit der Gesamtenergie von besagten Großenaufgetragen. Zu erkennen ist, daß Etot offensichtlich weniger stark von der Anzahl der k-Punkte alsvon der Abschneideenergie abhangt. Bei der Wahl der beiden Großen muß zwischen der Genauig-keit und dem Rechenaufwand abgewogen werden.Die fur die spatere Behandlung der Thermodynamik des binaren Systems benotigten ab-initio Rech-nungen erlauben geordnete Strukturen mit vergleichsweise kleiner Einheitszelle, so daß in diesemFall der Rechenaufwand auch bei relativ hoher Genauigkeit in der Gesamtenergie vertretbar ist.Will man jedoch auch Defekte im System beschreiben, so waren bei den hierzu bisher angestelltenRechnungen meist Superzellenrechnungen notig (z.B. [69]) . Bei diesen wird aufgrund der in derRealitat meist geringen Wechselwirkungen der Defekte untereinander ein solcher in eine entspre-chend große Superzelle der geordneten Struktur eingebaut und diese dann periodisch fortgesetzt.Die Energieberechnung solch großer Zellen ist bei hohen Anforderungen an die absolute Genauig-keit sehr zeitaufwendig. In diesen Fallen mussen die Konvergenzparameter kleiner gewahlt werden,um zu vertretbaren CPU-Zeiten zu gelangen.
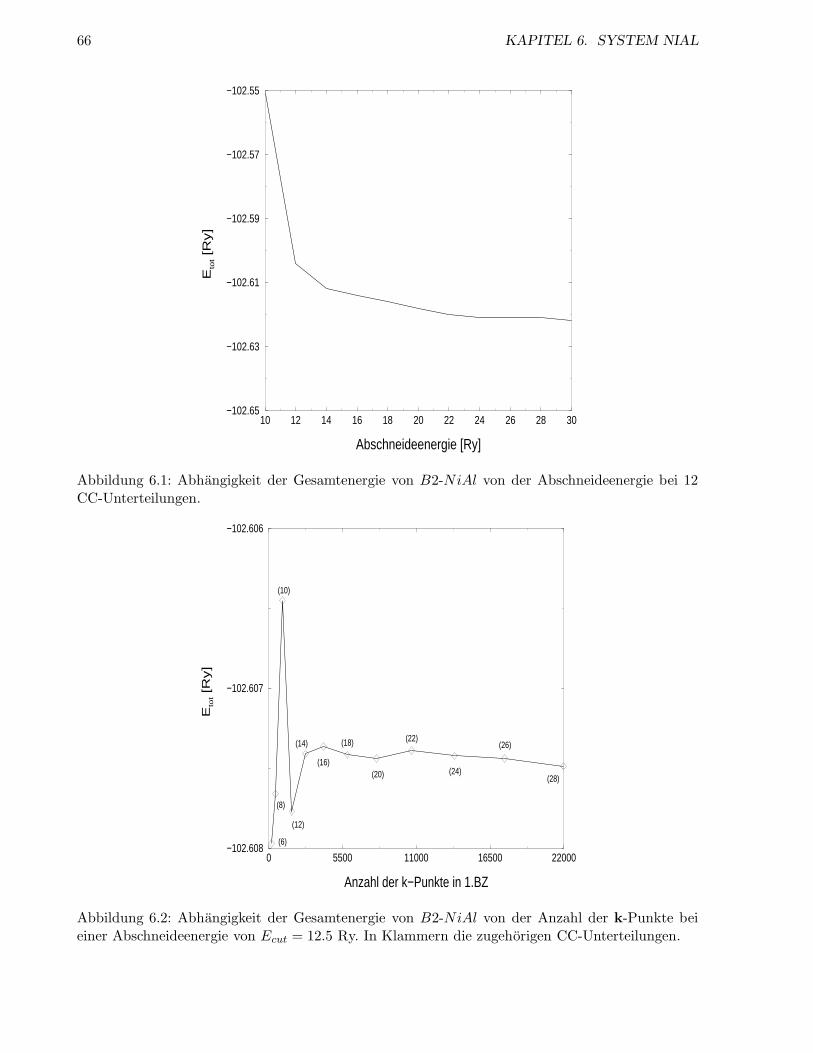
66 KAPITEL 6. SYSTEM NIAL
10 12 14 16 18 20 22 24 26 28 30
Abschneideenergie [Ry]
−102.65
−102.63
−102.61
−102.59
−102.57
−102.55
Eto
t [R
y]
Abbildung 6.1: Abhangigkeit der Gesamtenergie von B2-NiAl von der Abschneideenergie bei 12CC-Unterteilungen.
0 5500 11000 16500 22000
Anzahl der k−Punkte in 1.BZ
−102.608
−102.607
−102.606
Eto
t [R
y]
(6)
(8)
(10)
(12)
(14)
(16)
(18)
(20)
(22)
(24)
(26)
(28)
Abbildung 6.2: Abhangigkeit der Gesamtenergie von B2-NiAl von der Anzahl der k-Punkte beieiner Abschneideenergie von Ecut = 12.5 Ry. In Klammern die zugehorigen CC-Unterteilungen.

6.1. GRUNDZUSTANDSBETRACHTUNGEN 67
Um nun die Anforderungen an die Genauigkeit gerade in Hinblick auf die thermodynamischenEigenschaften des System untersuchen zu konnen, wurden zwei Parametersatze von Abschneide-energie und k-Punkt-Zahl gewahlt. Diese werden im weiteren mit I und II bezeichnet.
I. Ecut=12.5 Ry und 12 CC-UnterteilungenDiese Parameter wurden bei den erwahnten, bereits durchgefuhrten Defektrechnungen ver-wendet. Somit ist eine Vergleich der in dieser Arbeit gewonnen Ergebnisse mit denen derSuperzellenrechnungen moglich.
II. Ecut=25 Ry und 22 CC-UnterteilungenDie Gesamtenergie ist bei diesen Parametern im Rahmen der Methode auf ∆Etot ≤ 1mRyauskonvergiert.
In beiden Satzen wurde das Austauschkorrelationsfunktional Exc[n] in der Parametrisierung vonCeperley und Alder [70] verwendet.
6.1.3 Energien der geordneten Strukturen
Zur Berechnung
Im letzten Abschnitt wurden die Abschneideenergie und Zahl der k-Punkte hinsichtlich dem Kon-vergenzverhalten der Gesamtenergie der B2-Struktur gewahlt. Bei Behandlung verschiedener Struk-turen variiert jedoch die Große der Brillouin-Zone, und somit ware bei konstanter CC-Unterteilungauch die Dichte des k-Punkt-Netzes verschieden. Eine solche strukturabhangige k-Punkt-Dichtewurde jedoch bedeuten daß die Genauigkeit der Energieberechnung ebenfalls strukturabhangigware, da eine erhohte k-Punkt-Dichte eine gesteigerte Genauigkeit nach sich zieht. Demzufolgemuß, um die Energien der Strukturen vergleichen zu konnen, darauf geachtet werden, daß die CC-Unterteilungen dahingehend abgestimmt werden, daß bei allen betrachteten Strukturen annahernddie gleiche Dichte des k-Punkt-Netzes vorliegt.Desweiteren macht die Rechnung mit geringen Konvergenzparametern nur dann Sinn, wenn die be-rechtigte Hoffnung besteht, daß die Energiedifferenzen nahezu auskonvergiert sind, wenn dies auchnoch nicht fur die absolute Energie gilt. Fur die in dieser Arbeit angestellten Stabilitatsbetrach-tungen sind gerade die strukturellen Energie- bzw. freien Energiedifferenzen die entscheidendenGroßen und weniger die absoluten Werte. Die tatsachliche Konvergenz der Differenzen kann bei-spielsweise an den Werten der ECIs getestet werden, da diese definitionsgemaß Differenzen vonabsoluten Energien entsprechen. Allgemein gilt in diesem Zusammenhang jedoch die Aussage, daßdie Differenzen weitaus schneller konvergieren sollten als die absoluten Energien.Samtliche geordneten Strukturen wurden mit kubischer Symmetrie behandelt. Die Gesamtenergienwurden dabei volumenabhangig gerechnet. Bei jeder Struktur σ wurde dabei aus den totalen Ener-gien zu 9 verschiedenen Volumina mittels Interpolation eine Etot(σ, V )-Kurve erstellt. Die hierbeiverwendete Interpolationsformel lautet1
Etot(σ, V ) = c0(σ) + c1(σ)V +c2(σ)
V 3. (6.2)
Diese wurde auch in der Arbeit von Pasturel et al. [13] benutzt.
1Im folgenden sollen alle Energien, wenn nicht explizit anders angegeben, als Energien pro Atom verstandenwerden. Ebenso wird das Systemvolumen auf die Zahl der Atome normiert.

68 KAPITEL 6. SYSTEM NIAL
Eine Fitkurve in Potenzen von V ist deshalb gunstig, da dies zu einer vergleichsweise einfachenMinimierungsprozedur beim globalen Volumenrelaxationsverfahren fuhrt. Desweiteren scheinen imHinblick auf die Naherung in der statistischen Methode, keine allzu hohen Anspruche bezuglich derGute der Interpolationsformel erforderlich.Die Bildungsenergie ist im NiAl-System folgendermaßen definiert :
Eform(σ, V ) = Ecoh(σ, V ) − cNiEcoh(Ni− fcc, Veq) − cAlEcoh(Al − fcc, Veq) . (6.3)
Es wird also auf die reinen fcc-Phasen bei deren Gleichgewichtsvolumina normiert. Dies ist sinn-voll, da die Kohasionsenergien der A1-Phasen (ungeordnet fcc) tiefer liegen als die der A2-Phasen(ungeordnet bcc) und somit fur die reine Ni- und Al-Phase das fcc-Grundgitter das energetischgunstigere darstellt.
Diskussion
In den Tabellen (6.1) und (6.2) sind die Ergebnisse der ab-initio Rechnungen fur die beiden gewahl-ten Parametersatze zusammengestellt. Vergleicht man die errechneten Energien, so ist zu erkennen,daß sich beim Parametersatz II gegenuber I noch Anderungen von typischerweise 10 mRy in derGesamtenergie vollziehen. Hingegen ist die fur Stabilitatsbetrachtungen relevante Bildungsenergiebis auf 10 meV unverandert. Eine Energiedifferenz von ∆E = 10 meV entspricht jedoch nach Boltz-mann immer noch einer Temperatur von etwa 100 Grad Kelvin.Fur die im Phasendiagramm stabilen Strukturen kann bei einigen Strukturdaten der Vergleich mitexperimentellen Daten gemacht werden (s. Tab.(6.4). Fur die anderen Strukturen wurden keine ex-perimentellen Werte gefunden, da diese, wenn uberhaupt, dann nur als metastabile Phasen existentsind. Die berechneten Kohasionsenergien fur die Elemente liegen deutlich hoher als die experimen-tellen Werte [71], und der Vergleich mit den ab initio bestimmten Gitterkonstanten zeigt, daß dieserund 3 Prozent unterhalb den gemessenen Werte liegen. Dieses ”overbinding” ist eine wohlbekannteSchwache der LDA. Der berechnete Wert fur die Bildungsenergie von B2-NiAl liegt innerhalb dervon Desai [72] angegebenen Fehlergrenzen.In der Tabelle (6.3) wurden außerdem zum Vergleich die von Pasturel et al. gewonnenen Strukturda-ten aufgefuhrt. Diese wurden ebenfalls ab initio, jedoch mit einer anderen elektronentheoretischenMethode, der sogenannten LMTO-ASA mit combined correction [73, 74], berechnet. Ohne auf dieDetails dieser Methode einzugehen, ist zu sagen, daß von der Theorie her dieses Verfahren schlechterist als die mixed-basis-Pseudopotentialmethode. Dies liegt hauptsachlich daran, daß in der LMTO-ASA Methode nicht mit dem wahren, sondern einem genaherten Kristallpotential gearbeitet wird.Der direkte Vergleich der beiden Methoden zeigt folgendes auf. Zum einen liegen die Gitterkonstan-ten von Pasturel et al. naher an den experimentell bestimmten. Zum anderen ist in dieser Arbeitgerade die Differenz der Bildungsenergien zwischen den experimentell stabilen Strukturen B2-NiAlund L12-Ni3Al zu Gunsten der B2-Struktur erhoht. Der Wert der Bildungsenergie von B2-NiAlliegt dabei weiter vom experimentellen Wert entfernt als der mixed-basis-Wert.Es soll noch ein weiterer illustrativer Vergleich angefuhrt werden. Die Bildungsenergien der reinenbcc-Strukturen lassen sich auch als direkte energetische Kristallstrukturdifferenzen verstehen. Furreines Al-bcc betragt der Unterschied dieser Große durch die veranderte ab-initio-Methode knapp50 meV, was einer Temperatur von ca. 600 K entspricht. Es existieren nun auch Rechnungen mitder FLAPW-Methode [75], einem weiteren ab-inito-Verfahren, fur diese Große. So wird von wirdSluiter et al. [76] mit FLAPW eine Bildungsenergie fur Al-bcc von 0.0626 eV angegeben. DieserWert der als von der Gute als zur mixed-basis Pseudopotentialmethode aquivalent einzustufendenFLAPW-Methode liegt naher am LMTO-Wert. Einer CALPHAD-Untersuchung zufolge lautet dieBildungsenergie fur Al-bcc wiederum 0.1044 eV [77].
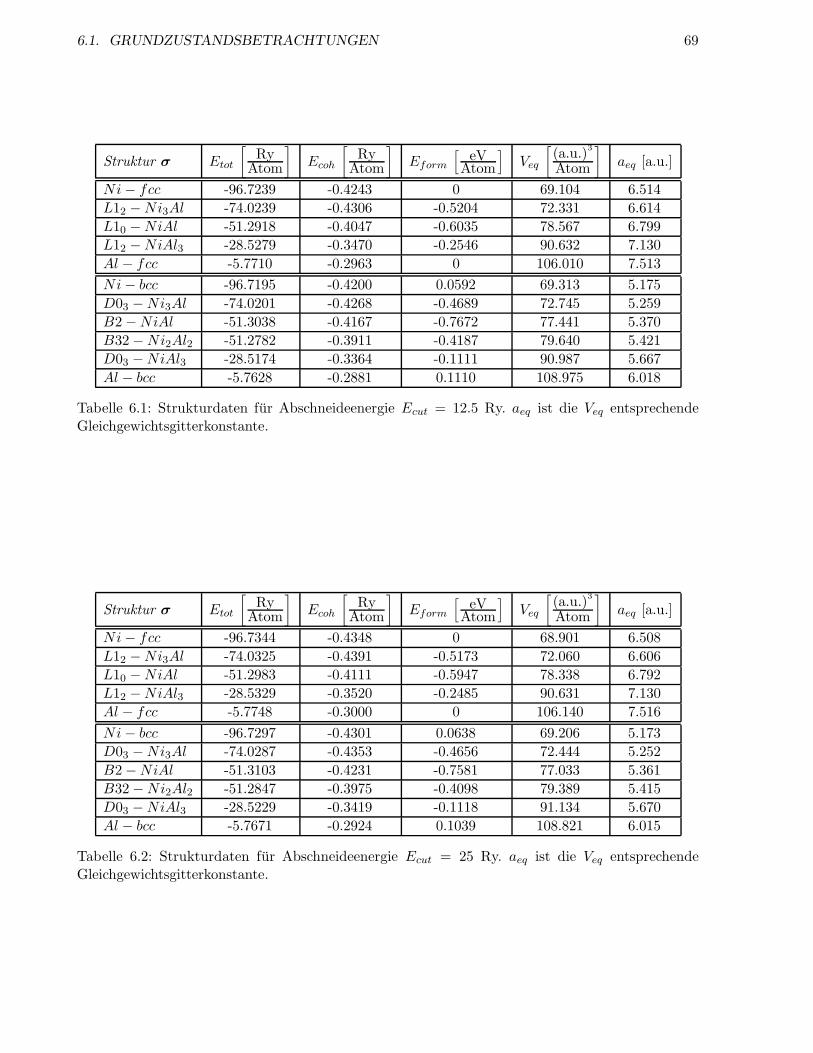
6.1. GRUNDZUSTANDSBETRACHTUNGEN 69
Struktur σ Etot
[
RyAtom
]
Ecoh
[
RyAtom
]
Eform[
eVAtom
]
Veq
[(a.u.)
3
Atom
]
aeq [a.u.]
Ni− fcc -96.7239 -0.4243 0 69.104 6.514
L12 −Ni3Al -74.0239 -0.4306 -0.5204 72.331 6.614
L10 −NiAl -51.2918 -0.4047 -0.6035 78.567 6.799
L12 −NiAl3 -28.5279 -0.3470 -0.2546 90.632 7.130
Al − fcc -5.7710 -0.2963 0 106.010 7.513
Ni− bcc -96.7195 -0.4200 0.0592 69.313 5.175
D03 −Ni3Al -74.0201 -0.4268 -0.4689 72.745 5.259
B2 −NiAl -51.3038 -0.4167 -0.7672 77.441 5.370
B32 −Ni2Al2 -51.2782 -0.3911 -0.4187 79.640 5.421
D03 −NiAl3 -28.5174 -0.3364 -0.1111 90.987 5.667
Al − bcc -5.7628 -0.2881 0.1110 108.975 6.018
Tabelle 6.1: Strukturdaten fur Abschneideenergie Ecut = 12.5 Ry. aeq ist die Veq entsprechendeGleichgewichtsgitterkonstante.
Struktur σ Etot
[
RyAtom
]
Ecoh
[
RyAtom
]
Eform[
eVAtom
]
Veq
[(a.u.)
3
Atom
]
aeq [a.u.]
Ni− fcc -96.7344 -0.4348 0 68.901 6.508
L12 −Ni3Al -74.0325 -0.4391 -0.5173 72.060 6.606
L10 −NiAl -51.2983 -0.4111 -0.5947 78.338 6.792
L12 −NiAl3 -28.5329 -0.3520 -0.2485 90.631 7.130
Al − fcc -5.7748 -0.3000 0 106.140 7.516
Ni− bcc -96.7297 -0.4301 0.0638 69.206 5.173
D03 −Ni3Al -74.0287 -0.4353 -0.4656 72.444 5.252
B2 −NiAl -51.3103 -0.4231 -0.7581 77.033 5.361
B32 −Ni2Al2 -51.2847 -0.3975 -0.4098 79.389 5.415
D03 −NiAl3 -28.5229 -0.3419 -0.1118 91.134 5.670
Al − bcc -5.7671 -0.2924 0.1039 108.821 6.015
Tabelle 6.2: Strukturdaten fur Abschneideenergie Ecut = 25 Ry. aeq ist die Veq entsprechendeGleichgewichtsgitterkonstante.
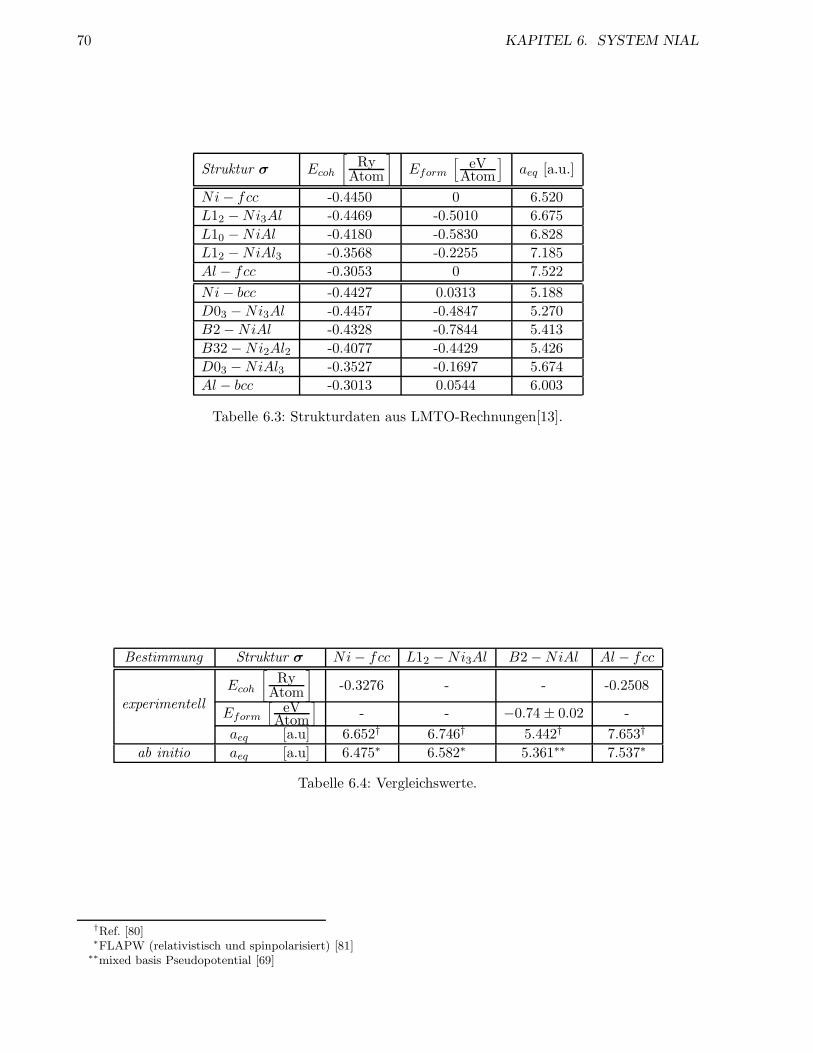
70 KAPITEL 6. SYSTEM NIAL
Struktur σ Ecoh
[
RyAtom
]
Eform[
eVAtom
]
aeq [a.u.]
Ni− fcc -0.4450 0 6.520
L12 −Ni3Al -0.4469 -0.5010 6.675
L10 −NiAl -0.4180 -0.5830 6.828
L12 −NiAl3 -0.3568 -0.2255 7.185
Al − fcc -0.3053 0 7.522
Ni− bcc -0.4427 0.0313 5.188
D03 −Ni3Al -0.4457 -0.4847 5.270
B2 −NiAl -0.4328 -0.7844 5.413
B32 −Ni2Al2 -0.4077 -0.4429 5.426
D03 −NiAl3 -0.3527 -0.1697 5.674
Al − bcc -0.3013 0.0544 6.003
Tabelle 6.3: Strukturdaten aus LMTO-Rechnungen[13].
Bestimmung Struktur σ Ni− fcc L12 −Ni3Al B2 −NiAl Al − fcc
Ecoh
[
RyAtom
]
-0.3276 - - -0.2508
experimentellEform
[eV
Atom
]
- - −0.74 ± 0.02 -
aeq [a.u] 6.652† 6.746† 5.442† 7.653†
ab initio aeq [a.u] 6.475∗ 6.582∗ 5.361∗∗ 7.537∗
Tabelle 6.4: Vergleichswerte.
†Ref. [80]∗FLAPW (relativistisch und spinpolarisiert) [81]
∗∗mixed basis Pseudopotential [69]

6.1. GRUNDZUSTANDSBETRACHTUNGEN 71
Dieser Wert fur Eform(Al-bcc) ist mit dem mixed-basis-Wert nahezu identisch.Der uneingeschrankte Vergleich der Ergebnisse, gewonnen mit verschiedenen DFT-Methoden inLDA, ist aber allgemein mit Vorsicht zu genießen. Neben der oftmals grundverschiedenen Darstel-lung von Kristallpotential und Wellenfunktionen innerhalb der einzelnen Verfahren spielt auch dieWahl des Austauschkorrelationsfunktionals Exc[n] eine wichtige Rolle. In der Arbeit von Pasturelet al. wurde die Parametrisierung von Barth-Hedin nach Moruzzi et al. [78] verwendet. Bei denRechnungen von Sluiter et al. kam die Hedin-Lundquist Parametrisierung [79] zur Anwendung.Sowohl Pasturel et al. als auch Sluiter et al. geben an, mit den erwahnten Parametrisierungen dieGesamtenergie auf 0.1 mRy auskonvergiert zu haben.Auch beim Vergleich mit experimentellen Daten ist Vorsicht geboten, da in den gemessenen ener-getischen Großen meist auch Anregungen, die in den Rechnungen gar nicht berucksichtigt wurden,wie zum Beispiel phononische Effekte, enthalten sind. Dies liegt daran, daß die Experimente hierzumeist bei Raumtemperatur durchgefuhrt wurden.
Grundzustande
Mit den mittels des mixed-basis-Pseudopotentialprogrammes berechneten Energien laßt sich nuneine Grundzustandsanalyse durchfuhren. Die hohen negativen Bildungsenergien zeigen, daß imNiAl-System bei beliebiger Zusammensetzung die geordneten Verbindungen energetisch gunstigersind als eine heterogene Mischung aus Ni-fcc und Al-fcc. Wie aus den Abbildungen (6.3) bzw. (6.4)hervorgeht, ist in der Stochiometrie 0.5 die Bildungsenergie der B2-Struktur minimal, und daherist diese im Gegensatz zur L10- und B32-Struktur eine stabile Grundzustandstruktur.Im Ni-armen Gebiet ist den Rechnungen zufolge eine heterogene Mischung der B2- und der A1-Struktur im Grundzustand stabil. Die in diesem Bereich in der Stochiometrie 0.25 sich befindendeD03- und L12-Struktur liegen oberhalb des Polygonzugs der stabilen Strukturen und konnen daherbei T = 0 nicht existieren. Allein fur den Fall T 6= 0 besteht die Moglichkeit, daß diese Verbindun-gen entropisch stabilisiert werden. Dies ist jedoch aufgrund des energetisch weiten Abstands vonder Stabilitatslinie nicht zu erwarten. Es sei bereits an dieser Stelle daran erinnert, daß nach demexperimentellen Phasendiagramm im Ni-armen Bereich die nahezu lediglich in der Stochiometrieexistenten Strukturen D011-NiAl3 mit orthorhombischer und D519-Ni2Al3 mit trigonaler Symme-trie stabil sind. Diese wurden in dieser Arbeit jedoch nicht berucksichtigt.Im Ni-reichen Gebiet setzt sich in der Stochiometrie 0.75 die auf einem fcc-Gitter basierende L12-Struktur gegenuber der bcc-artigen D03-Struktur durch und bildet an dieser Stelle eine weiterestabile Grundzustandsstruktur. Bei Erhohung der Ni-Konzentration von cNi = 0.5 zu cNi = 0.75andert sich also das Grundgitter von bcc nach fcc. In dieser Region sind daher starke Relaxationser-scheinungen zu erwarten, die bekanntlich gerade die thermodynamische Beschreibung im Rahmender CVM erschweren.Fur die Grundzustandsbetrachtungen ergeben sich zwischen den beiden Konvergenzparametersatzenkeine qualitativen Unterschiede.Um genauere Aussagen sowohl bezuglich der ordnenden Eigenschaften des NiAl-Systems als auchuber den quantitativen Unterschied der beiden Parametersatze machen zu konnen, ist es sinnvoll,die ECIs fur dieses System zu betrachten, die ja ohnehin fur die thermodynamische Beschreibungvonnoten sind.
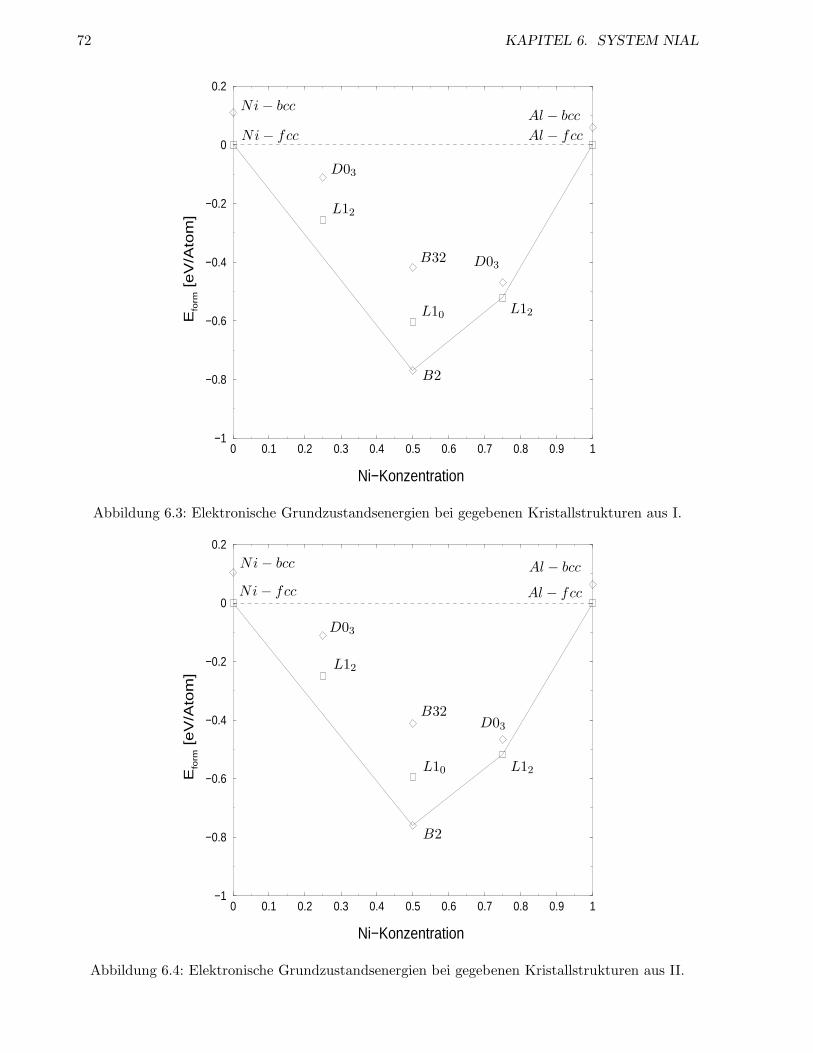
72 KAPITEL 6. SYSTEM NIAL
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
−1
−0.8
−0.6
−0.4
−0.2
0
0.2
Efo
rm [
eV
/Ato
m]
PSfrag
replacem
ents
Eform
Ni− fcc
Ni− bcc
Al − fcc
Al − bcc
B2
B32 D03
D03
L12
L12
L10
Abbildung 6.3: Elektronische Grundzustandsenergien bei gegebenen Kristallstrukturen aus I.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
−1
−0.8
−0.6
−0.4
−0.2
0
0.2
Efo
rm [eV
/Ato
m]
PSfrag
replacem
ents
Eform
Ni− fcc
Ni− bcc
Al − fcc
Al − bcc
B2
B32D03
D03
L12
L12
L10
Abbildung 6.4: Elektronische Grundzustandsenergien bei gegebenen Kristallstrukturen aus II.

6.1. GRUNDZUSTANDSBETRACHTUNGEN 73
6.1.4 ECIs
Fur die Klarung von Stabilitatsfragen ist allein die Bildungsenergie Eform einer bestimmten Konfi-guration von Relevanz. Sowohl die Gesamt- als auch die Kohasionsenergie enthalten fur die Phasen-konkurrenz irrelevante Energieanteile. Da die Bildungsenergie naturlich ebenfalls eine eindeutigeFunktion der Konfiguration σ ist, ist es also in diesem Zusammenhang ausreichend und vor allemim Hinblick auf die Interpretierbarkeit sinnvoll, im folgenden Eform nach Clustern zu entwickeln.2
Die Cluster-Entwicklung wurde in der naheliegenden orthogonalen 1, σ-Basis durchgefuhrt. Da-bei wurde einem Ni-Atom die Spinvariable +1 und einem Al-Atom die Spinvariable -1 zugeordnet.Auf dem fcc-Gitter hat diese Entwicklung in Tetraedernaherung folgende Form :
E(fcc)
form(σ) = K0 +K1η1 + 6K2η2 + 8K3η3 + 2K4η4 . (6.4)
Die Faktoren vor den ECIs geben die Multiplizitaten νi∗, also die Zahl der Cluster pro Gitterpunkt,wieder. Entsprechend lautet die Cluster-Entwicklung fur das bcc-Gitter mit dem irregularen Tetra-eder als maximalen Cluster :
E(bcc)
form(σ) = K0 +K1η1 + 4K2,1η2,1 + 3K2,2η2,2 + 12K3η3 + 6K4η4 . (6.5)
Die ECIs fur beide Gitter wurden mittels der SIM durch Matrixinversion gewonnen. In den Tabellen(6.5) und (6.6) sind die hierfur erforderlichen gemittelten Clusterfunktionen ηi der geordnetenStrukturen aufgefuhrt.Setzt man diese gemittelten Clusterfunktionen in die Gleichungen (6.4) und (6.5) ein, so erhaltman zwei Matrix-Gleichungen der Form
Eform = η¯K . (6.6)
Die Inversion dieser Gleichungen liefert
K = η¯
−1Eform . (6.7)
Dabei ist K der Vektor der ECIs, η¯−1 die Inverse der Matrix der gemittelten Clusterfunktionen
und Multiplizitaten sowie Eform der Vektor der Bildungsenergien der geordneten Strukturen. DieseEnergien wurden, wie bereits erwahnt, volumenabhangig gerechnet. Demzufolge laßt sich Eform
auch schreiben alsEform = C
¯V , (6.8)
wobei C¯
die Matrix der Entwicklungskoeffizienten der Interpolationsformel (6.2) darstellt und Vder Vektor (1, V, 1
V 3 ) ist. Setzt man nun (6.8) in (6.7) ein, so erhalt man
K = η¯
−1C¯︸ ︷︷ ︸
=Ξ¯
V . (6.9)
Die Matrix Ξ¯
ermoglicht es, bei vorgegebenem Volumen direkt die volumenabhangigen ECIs, diefur das globale Volumenrelaxationsschema benotigt werden, zu berechnen :
K(V ) = Ξ¯V . (6.10)
Entsprechend gewinnt man die volumenunabhangigen ECIs fur das lokale Volumenrelaxationssche-ma durch Verwendung der Gleichung (6.7), wobei jetzt die Energien beim jeweiligen Gleichgewichts-volumen einzusetzen sind
K = η¯
−1Eform(Veq) . (6.11)
2Im Rahmen der thermodynamischen Betrachtungen erfolgte aufgrund der Normierung der chemischen Potentialeauf das Vakuum wieder eine CE der Gesamtenergie.
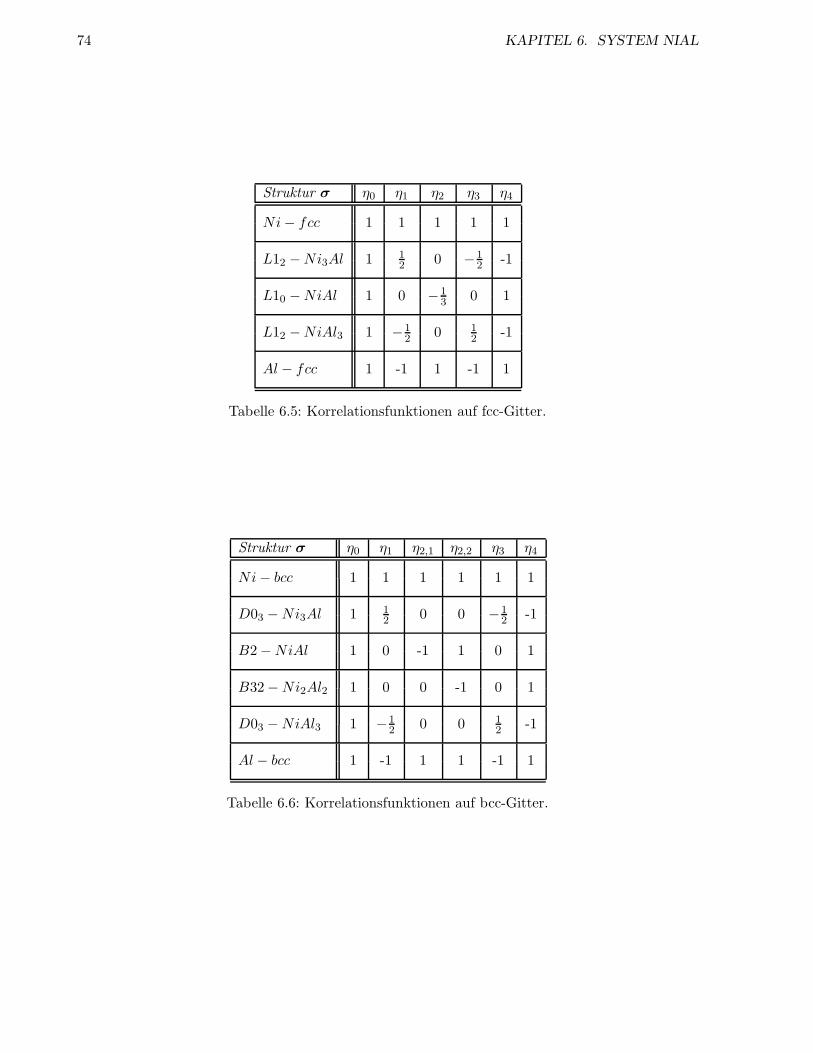
74 KAPITEL 6. SYSTEM NIAL
Struktur σ η0 η1 η2 η3 η4
Ni− fcc 1 1 1 1 1
L12 −Ni3Al 1 12 0 −1
2 -1
L10 −NiAl 1 0 − 13 0 1
L12 −NiAl3 1 −12 0 1
2 -1
Al − fcc 1 -1 1 -1 1
Tabelle 6.5: Korrelationsfunktionen auf fcc-Gitter.
Struktur σ η0 η1 η2,1 η2,2 η3 η4
Ni− bcc 1 1 1 1 1 1
D03 −Ni3Al 1 12 0 0 −1
2 -1
B2 −NiAl 1 0 -1 1 0 1
B32 −Ni2Al2 1 0 0 -1 0 1
D03 −NiAl3 1 −12 0 0 1
2 -1
Al − bcc 1 -1 1 1 -1 1
Tabelle 6.6: Korrelationsfunktionen auf bcc-Gitter.

6.1. GRUNDZUSTANDSBETRACHTUNGEN 75
Volumenabhangige ECIs
Die volumenabhangigen ECIs fur die beiden Kristallstrukturen, gewonnen mit Parametersatz I,sind in den Abbildungen (6.5) und (6.6) wiedergegeben. Die Auftragung des Volumens wurde der-art gewahlt, daß man im Diagramm von links nach rechts vom Al- zum Ni-Gleichgewichtsvolumenwandert. Somit hat man uber die volumenabhangigen ECIs quasi durch die ”Hintertur” eine Art vonAbhangigkeit der ECIs von der Konzentration erreicht. Obwohl die Separierung der energetischenBedeutung der einzelnen ECIs mit Vorsicht zu genießen ist, da es sich ja nicht um Mehrkorper-potentiale (s. 2.2.2) handelt, soll dennoch versucht werden, den volumenabhangigen Verlauf dieserGroßen verstandlich zu machen. Dabei ist es aufgrund der komplizierten Verknupfung von chemi-scher Energie und Relaxationsenergie im globalen Volumenrelaxationsschema weiter nicht eindeutigmoglich, diese Energieterme in der vorliegenden Auftragung sauber zu trennen.
• K0
Dieser ECI gehort zum ”leeren” Cluster und tragt nurmehr die allen Strukturen gemeinsameAufpunktsenergie. Daher entspricht der volumenabhangige Verlauf dieser Große grob einergemittelten volumenabhangigen Energiekurve.
• K1
Der ECI zum Punkt-Cluster wertet den Energiebeitrag, der allein durch den Unterschiedan Ni- und Al-Atomen im System entsteht. In diesem Sinne ist K1 als eine Art chemischesPotential bei T = 0 zu verstehen. Da nun ein Al-Atom die zugeordnete Spinvariable -1 besitzt,hatK1, angefangen beim Al-Gleichgewichtsvolumen, einen positiven Wert. Bei einem gewissenVolumen wechselt dann das Vorzeichen von K1. Dies muß auch so sein, da bei erhohterVolumendeformation und gestiegener Ni-Konzentration irgendwann die Ni- gegenuber denAl-Atomen energetisch dominieren.
• K2 bzw. K2,1
Der ECI des NN-Paares hat sowohl auf dem fcc- als auch auf dem bcc-Gitter einen positivenWert. Dies laßt sich in Analogie zum Ising-Modell als eine antiferromagnetische Wechselwir-kung deuten. Beide Gitter sind also in der ersten Schale stark ordnend. Der leichte Anstiegdieses ECI bei Verringerung des Volumens macht noch einmal deutlich, daß es sich bei die-ser Große nicht um ein reines Zweikorperpotential handelt. So ist im Ni-reichen Gebiet dieordnende Wirkung erhoht, was ein Indiz fur die Stabilitat der L12-Struktur in dieser Regionist.
• K2,2
Dieser Clusterterm tritt aufgrund der Wahl der maximalen Cluster nur auf dem bcc-Gitterauf. Der NNN-Paar ECI hat immer noch antiferromagnetischen Charakter, jedoch ist derBetrag dieser Große schon deutlich gegenuber dem des K2,1-Werts verringert. Dies konnteein Indiz fur die kurze Reichweite der relevanten Wechselwirkung sein.
• K3 und K4
Diese ECIs sind ihrem Betrag nach deutlich kleiner als die der anderen Clusterterme. In-teressant ist, daß der ungewichtete ECI K3 betragsmaßig kleiner ist als K4. Berucksichtigtman aber zusatzlich die Multiplizitaten der Cluster, so kehrt sich die Hierarchie zwischen denbeiden wieder um.
Gerade die letzten Bemerkungen zu den hoheren Clustertermen machen deutlich, wie schwierig eineeinwandfreie physikalische Interpretation der einzelnen ECIs ist.
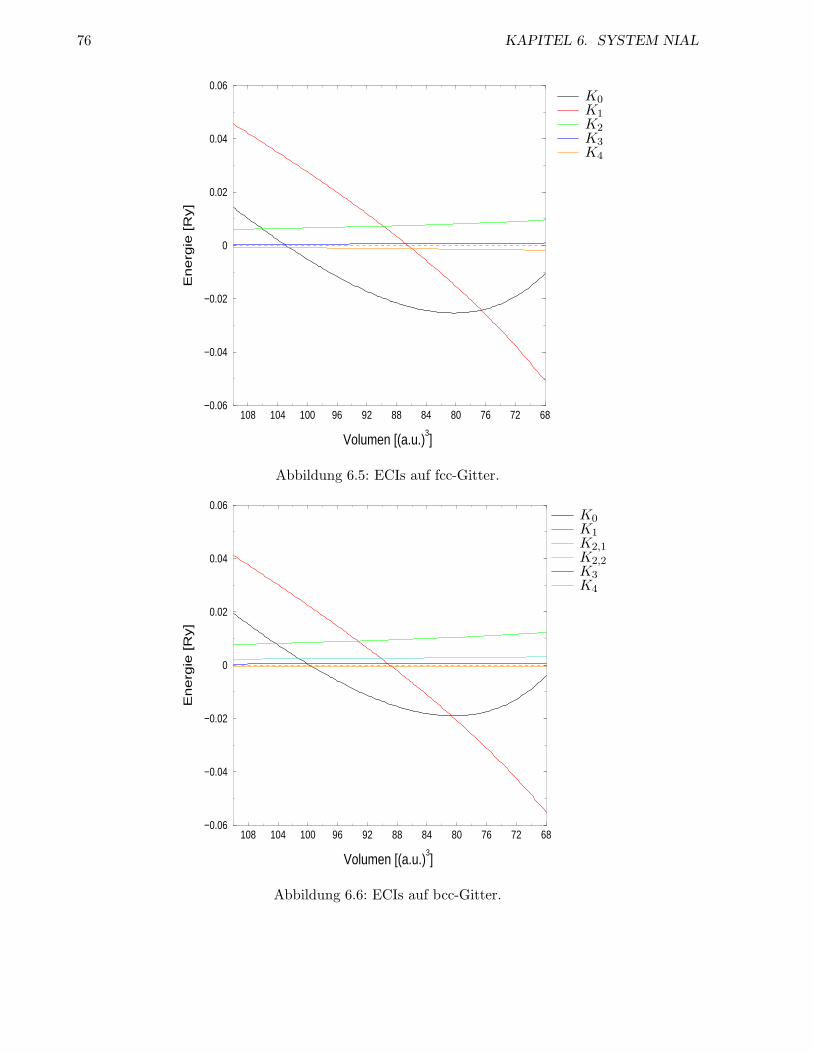
76 KAPITEL 6. SYSTEM NIAL
6872768084889296100104108
Volumen [(a.u.)3]
−0.06
−0.04
−0.02
0
0.02
0.04
0.06
Energ
ie [R
y]
PSfrag
replacem
ents
K0K1K2K3K4
K2,1
K2,2
Abbildung 6.5: ECIs auf fcc-Gitter.
6872768084889296100104108
Volumen [(a.u.)3]
−0.06
−0.04
−0.02
0
0.02
0.04
0.06
Energ
ie [R
y]
PSfrag
replacem
ents
K0K1
K2
K3K4
K2,1K2,2
Abbildung 6.6: ECIs auf bcc-Gitter.
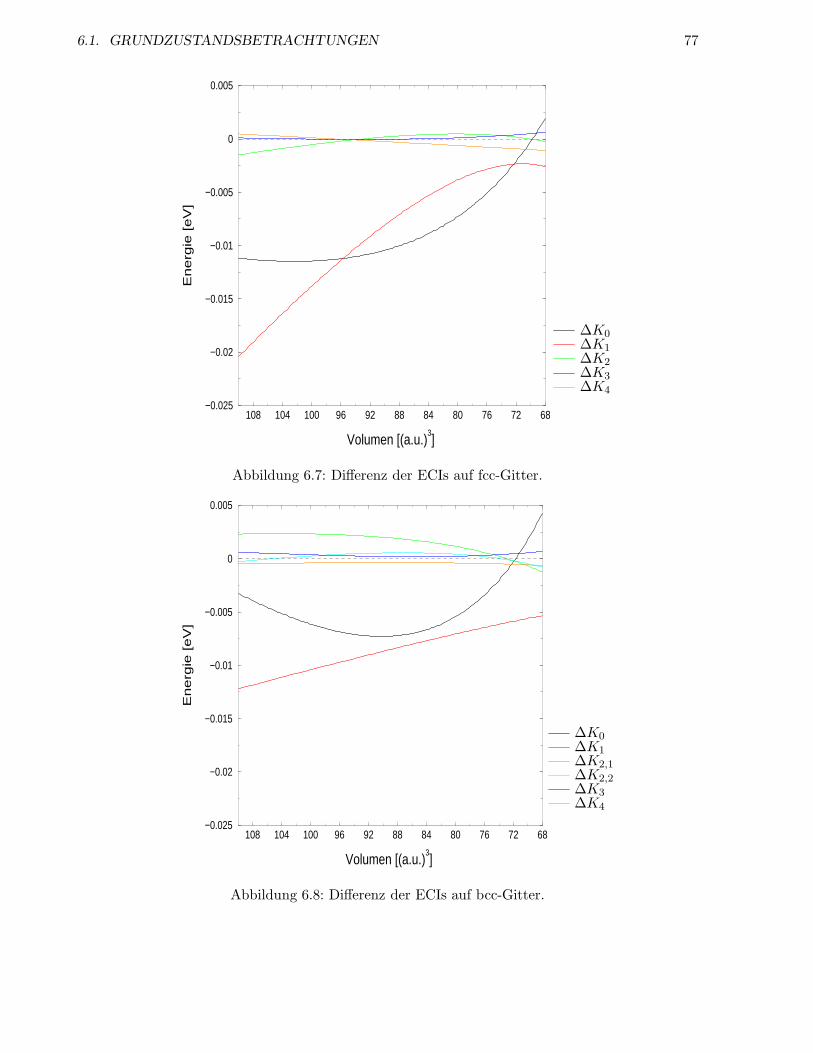
6.1. GRUNDZUSTANDSBETRACHTUNGEN 77
6872768084889296100104108
Volumen [(a.u.)3]
−0.025
−0.02
−0.015
−0.01
−0.005
0
0.005
Energ
ie [eV
]
PSfrag
replacem
ents
∆K0∆K1∆K2∆K3∆K4
∆K
2,1
∆K
2,2
Abbildung 6.7: Differenz der ECIs auf fcc-Gitter.
6872768084889296100104108
Volumen [(a.u.)3]
−0.025
−0.02
−0.015
−0.01
−0.005
0
0.005
Energ
ie [eV
]
PSfrag
replacem
ents
∆K0∆K1
∆K
2
∆K3∆K4
∆K2,1∆K2,2
Abbildung 6.8: Differenz der ECIs auf bcc-Gitter.

78 KAPITEL 6. SYSTEM NIAL
Die dargestellten volumenabhangigen ECIs wurden mit den Energien zum Parametersatz I be-rechnet. Die entsprechenden ECIs zum Parametersatz II haben naturlich den gleichen qualitativenVerlauf, weichen jedoch quantitativ geringfugig von ersteren ab (Abb. (6.7) und Abb. (6.8)). In dengezeigten Abbildungen ist nur die Differenz
∆Ki = K(I)i −K
(II)i .
der ungewichteten ECIs aufgetragen. In die Connolly-Williams-Abbildung gehen jedoch zusatz-lich die Multiplizitaten ein, so daß diese bei der Diskussion der Abweichung berucksichtigt werdenmussen.Die Abweichung in K1 sollte bei der spateren Statistik unter Verwendung von chemischen Potentia-len nicht allzu stark ins Gewicht fallen, weil auch diese linear an η1 ankoppeln und so ein Fehler inK1 kompensiert werden kann. Die Unterschiede der anderen ECIs sind im Bereich 1-10 meV, wasfur thermodynamische Belange immer noch energetisch bedeutsam ist. Aufgrund der Variation imVorzeichen uber den relevanten Volumenbereich sind jedoch auch Kompensationseffekte zu erwar-ten. Eine letzte Entscheidung hinsichtlich der Gute der ECIs bezuglich den Konvergenzparameternder DFT-Methode kann jedoch erst nach Vergleich der Ergebnisse der CVM-Rechnungen getroffenwerden.
Konvergenztest mit volumenunabhangigen ECIs
Um nun die Konvergenz der Cluster-Entwicklung im Rahmen der Tetraedernaherung zu testen,wurde versucht, mit den berechneten ECIs die Bildungsenergien anderer, nicht in der SIM ver-wendeter Strukturen zu reproduzieren. Aufgrund des fehlenden zusatzlichen Variationsparameterssollte die Konvergenz der CE im lokalen Volumenrelaxationsschema langsamer verlaufen als imGlobalen. Um quasi eine Obergrenze fur die Konvergenz festlegen zu konnen, wurden daher fur denKonvergenztest allein die ECIs des lokalen Volumenrelaxationsschemas (Tab. (6.7)) herangezogen.Als Teststrukturen ausgewahlt wurden fur das bcc-Gitter die beiden C11b-Strukturen NiAl2 undNi2Al sowie die B11-Struktur Ni2Al2. Im Fall des fcc-Gitters fiel die Wahl auf die ”40”-StrukturNi2Al2 und die Z2-Struktur Ni2Al2. Mit den erhaltenen ECIs und den Korrelationsfunktionen furbesagte Strukturen (Tab. (6.8)) wurde uber die Gleichungen (6.4) und (6.5) sodann der Test durch-gefuhrt. Obwohl samtliche Teststrukturen tetragonale Symmetrie aufweisen, wurden alle ab-initio-Rechnungen bei idealem fcc- bzw. bcc-Grundgitter durchgefuhrt. Die Ergebnisse sind in Tabelle(6.9) zusammengefaßt.
• ′′40′′ −Ni2Al2Diese Struktur ist bezuglich der Korrelationsfunktionen in Tetraedernaherung mit der L10-Struktur entartet. Die Abweichung zwischen der direkten Rechnung und der indirekten Be-stimmung der Bildungsenergie uber die ECIs ist mit 4.8 meV im Bereich des Konvergenzfeh-lers der Pseudopotentialmethode. Die Struktur ordnet in der NN-Schale und wird offenbardeshalb gut durch die Tetraedernaherung beschrieben.
• Z2 −Ni2Al2Diese Verbindung entspricht einer Schichtstruktur Ni2Al2 in [001]-Richtung. Offensichtlich istdie CE in dieser Form nicht in der Lage, ein solches Schichtsystem energetisch zu beschreiben.Trotz der Tatsache, daß auch in vergleichsweise kleinen Clustern prinzipiell noch weitreichendeWechselwirkungen enthalten sind, wird die Wechselwirkung zwischen den einzelnen Schichtenvon den ECIs, gewonnen mit der SIM durch Matrixinversion und dem Tetraeder als alleinigemmaximalen Cluster, offensichtlich nicht korrekt wiedergegeben.
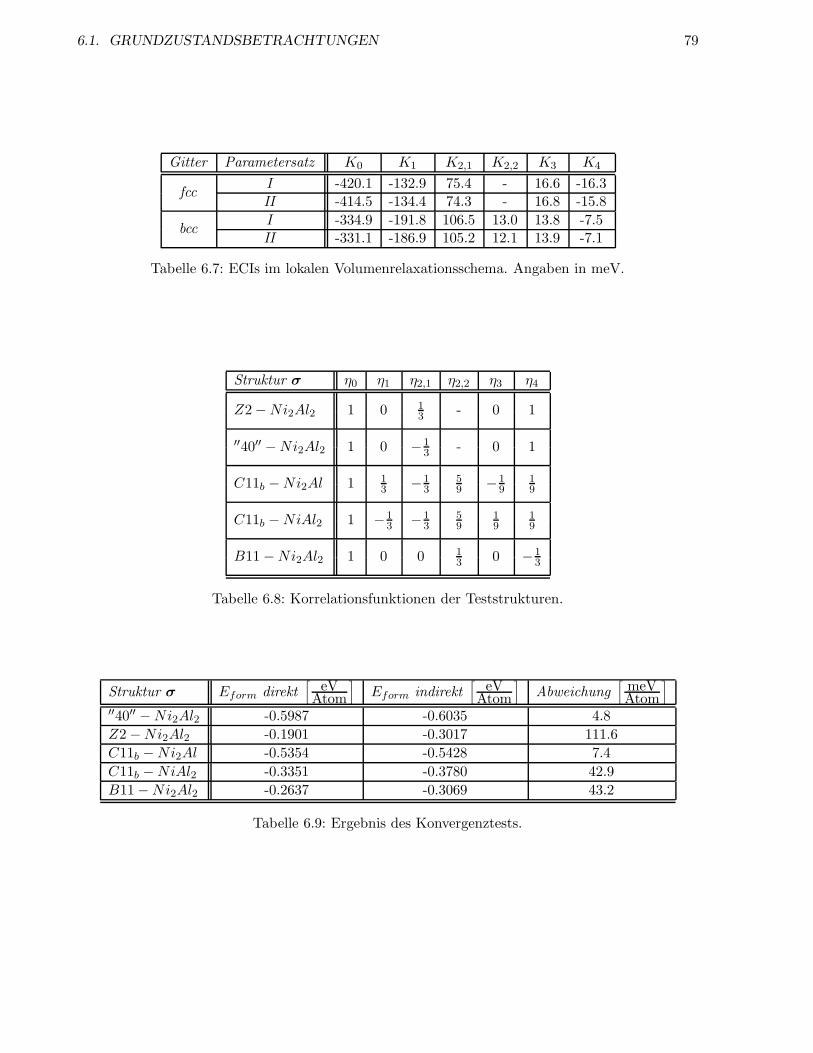
6.1. GRUNDZUSTANDSBETRACHTUNGEN 79
Gitter Parametersatz K0 K1 K2,1 K2,2 K3 K4
I -420.1 -132.9 75.4 - 16.6 -16.3fcc
II -414.5 -134.4 74.3 - 16.8 -15.8
I -334.9 -191.8 106.5 13.0 13.8 -7.5bcc
II -331.1 -186.9 105.2 12.1 13.9 -7.1
Tabelle 6.7: ECIs im lokalen Volumenrelaxationsschema. Angaben in meV.
Struktur σ η0 η1 η2,1 η2,2 η3 η4
Z2 −Ni2Al2 1 0 13 - 0 1
′′40′′ −Ni2Al2 1 0 −13 - 0 1
C11b −Ni2Al 1 13 −1
359 −1
919
C11b −NiAl2 1 −13 −1
359
19
19
B11 −Ni2Al2 1 0 0 13 0 −1
3
Tabelle 6.8: Korrelationsfunktionen der Teststrukturen.
Struktur σ Eform direkt[
eVAtom
]
Eform indirekt[
eVAtom
]
Abweichung[
meVAtom
]
′′40′′ −Ni2Al2 -0.5987 -0.6035 4.8
Z2 −Ni2Al2 -0.1901 -0.3017 111.6
C11b −Ni2Al -0.5354 -0.5428 7.4
C11b −NiAl2 -0.3351 -0.3780 42.9
B11 −Ni2Al2 -0.2637 -0.3069 43.2
Tabelle 6.9: Ergebnis des Konvergenztests.

80 KAPITEL 6. SYSTEM NIAL
• C11b1 −Ni2Al und C11b1 −NiAl2Zwar handelt es sich auch hier um ein Schichtsystem (Ni2Al bzw. NiAl2 entlang [001]),jedoch ist im irregularen Tetraeder das NNN-Paar enthalten. Somit sollte die Darstellungdieser Ubergitterstrukturen mit den vorhandenen ECIs besser moglich sein. Der gravierendeUnterschied in den Abweichungen bei den beiden Strukturen ist jedenfalls nicht ganz klar.
• B11Wiederum eine tetragonale Struktur mit Schichten Ni2Al2 in [001]-Richtung, welche aberwiederum kubisch behandelt wird. Hier ist eher verstandlich, daß diese Doppelschichten ausNi bzw. Al nicht mehr korrekt durch den Tetraeder erfaßt werden. Jedoch zeigt der direkteVergleich mit der Z2-Struktur, daß die Berucksichtigung der NNN-Wechselwirkung auf dembcc-Gitter den Fehler in Grenzen halt.
Es zeigt sich, daß die Beschreibung von Strukturen mit großerer Einheitszelle von der Tetra-edernaherung in Kombination mit der SIM unter Matrixinversion erwartungsgemaß nicht mehrzuverlassig gemeistert wird. Eine verbesserte Beschreibung der Teststrukturen unter Beibehaltungder reinen Tetraedernaherung ließe sich vermutlich durch Vorgabe mehrere geordneter Strukturenin einer SIM mit least-mean-square-Verfahren erzielen. Dies wurde aber in dieser Arbeit nicht ver-sucht.Die gewonnenen Ergebnisse lassen jedoch die Vermutung zu, daß die starke Ordnungstendenz desNiAl-Systems in der NN-Schale im Rahmen der reinen Tetraedernaherung augenscheinlich dazufuhrt, daß die Bildungsenergie der Schichtstrukturen uberschatzt wird. Demzufolge reicht der Te-traeder als maximaler Cluster allein offensichtlich nicht aus. Durch Hinzunahme einiger weitererentfernter Nachbarpaare waren verbesserte Resultate mit der CE zu erwarten. SIM mit least-mean-square-Verfahren von Lu et al. [10] unter Mitnahme hoherer Clusterpaare verstarken dieseAnnahme.Fur eine realistische Beschreibung des NiAl-Systems mussen jedoch auch aufgrund des asymmetri-schen Charakters der Bildungsenergien bezuglich der aquiatomaren Zusammensetzung wahrschein-lich weitere (ungerade) Mehrkorpercluster in der CE mitgefuhrt werden.

6.1. GRUNDZUSTANDSBETRACHTUNGEN 81
6.1.5 Relaxation der L10-Struktur
In den bisherigen Rechnungen wurde die L10-Struktur wie eine fcc-Struktur, also mit kubischerSymmetrie behandelt. In Wirklichkeit besitzt diese Struktur jedoch tetragonale Symmetrie, da sieaus abwechselnden Schichten von Ni- und Al-Atomen aufgebaut ist. Demzufolge ist eine Relaxationder c-Achse des Systems mit dem Ziel, die Gesamtenergie abzusenken, moglich.Diese zellexterne Relaxation wurde mit dem mixed-basis-Pseudopotentialprogramm durchgefuhrt.Hierzu wurden bei konstantem Volumen 9 verschiedene c/a-Verhaltnisse vorgegeben und jeweilsdie Gesamtenergie berechnet. Die Interpolation erfolgte mittels folgender Formel :
Etot(c/a, V ) = b0 + b1(c/a) +b2
(c/a)2+
b3(c/a)3
+b4
(c/a)4. (6.12)
Diese Interpolation wurde fur jedes der 9 Volumina durchgefuhrt. Dadurch konnte bei jedem vor-gegebenen Volumen das energetisch gunstigste c/a-Verhaltnis bestimmt werden. Wie aus Tabelle(6.10) ersichtlich, ist dieses Verhaltnis im betrachteten Bereich nahezu volumenunabhangig.Es wurde nun die Gesamtenergie der ideal relaxierten Struktur dem jeweils entsprechenden Volu-men zugeordnet und somit eine neue Energiekurve Etot(V ) fur die tetragonale L10-Struktur erstellt.Im Vergleich mit der rein kubischen fcc Struktur (Abb. (6.10)) ist eine deutliche Absenkung derEnergie um knapp 10 mRy zu erkennen. Desweiteren hat sich das Gleichgewichtsvolumen von demder fcc-Struktur mit 78.56 (a.u.)3 pro Atom auf 77.40 (a.u.)3 pro Atom reduziert.Dieses neue Gleichgewichtsvolumen ist nahezu identisch mit dem der B2-Struktur. Diese Uberein-stimmung kommt nicht von ungefahr, da bei Betrachtung der L10-Struktur schnell klar wird, daßwenn das c/a-Verhaltnis den Wert 1√
2annimmt, diese Struktur identisch mit der B2-Struktur ist
(Abb. (6.9)). Die Tabelle (6.10) zeigt, daß das c/a-Verhaltnis innerhalb der Rechengenauigkeit demder Relaxation auf die B2-Struktur entspricht. Dieses Ergebnis wird von Zou et al. bestatigt [82].In der Stochiometrie 0.5 ist also das bcc-Gitter als Grundgitter offensichtlich energetisch begunstigt.Eine einwandfreie Begrundung hierfur erfordert zusatzlich eine detaillierte Untersuchung der Bin-dungsverhaltnisse im NiAl-System, was z.T. von Zou et al. durchgefuhrt wurde. Allgemeinere Be-trachtungen liefern jedoch bereits erste Indizien fur die Stabilitat der B2-Struktur.So wird grundsatzlich in einem System mit starker Ordnungstendez in der NN-Schale aufgrund vonFrustrationseffekten auf dem fcc-Gitter das bcc-Gitter in der Stochiometrie 0.5 begunstigt sein.Weiter spielt sicherlich auch der Unterschied in der Atomgroße der beiden Komponenten eine Rol-le, da sich beispielsweise aufgrund dieses deutlichen Unterschieds im NiAl-System Verspannungenin der L10-Struktur ergeben, welche sich durch den Ubergang zur B2-Struktur absenken lassen.Dies liegt daran, daß in der bcc-Struktur die großen Al-Atome vom ehemals NN-Abstand in denNNN-Abstand ”ubersiedeln” konnen. Diese beiden, zugegeben einfachen, Argumente geben z.B.bereits eine erste Erklarung fur die Stabilitat der L10-Struktur im TiAl- im Gegensatz zum NiAl-System [55], da in ersterem sowohl die Ordnungstendenz weitaus schwacher ausgebildet ist als auchTi und Al in etwa dieselben Atomradien aufweisen.Fur die SIM ist die Verwendung der ”kunstlichen” kubischen L10-Struktur nicht unproblematisch,da im lokalen Volumenrelaxationsschema eigentlich nur vollstandig ausrelaxierte Strukturen furdie Abbildung herangezogen werden durfen. Naturlich ergibt sich demzufolge auch ein Problem imglobalen Schema, da dort ebenso die c/a-Relaxation komplett vernachlassigt wird und allein dieVolumenrelaxation uber die Variation desselben explizit in die Energetik eingeht.
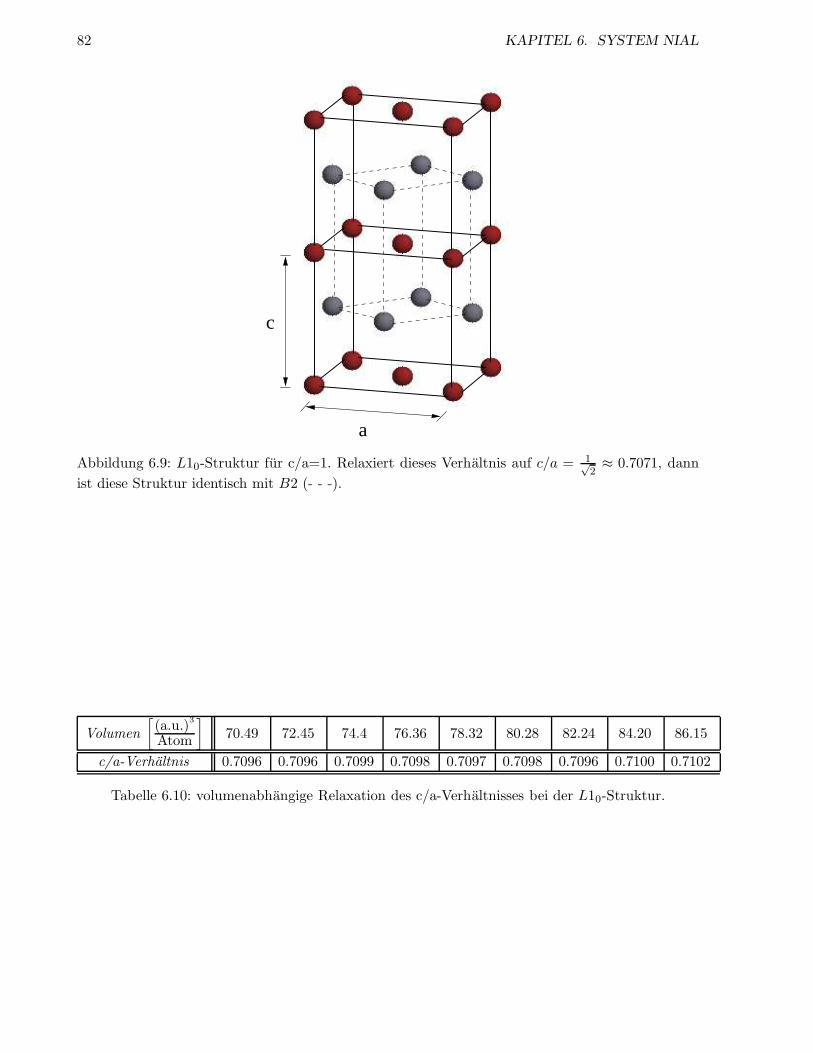
82 KAPITEL 6. SYSTEM NIAL
a
c
Abbildung 6.9: L10-Struktur fur c/a=1. Relaxiert dieses Verhaltnis auf c/a = 1√2≈ 0.7071, dann
ist diese Struktur identisch mit B2 (- - -).
Volumen
[(a.u.)
3
Atom
]
70.49 72.45 74.4 76.36 78.32 80.28 82.24 84.20 86.15
c/a-Verhaltnis 0.7096 0.7096 0.7099 0.7098 0.7097 0.7098 0.7096 0.7100 0.7102
Tabelle 6.10: volumenabhangige Relaxation des c/a-Verhaltnisses bei der L10-Struktur.
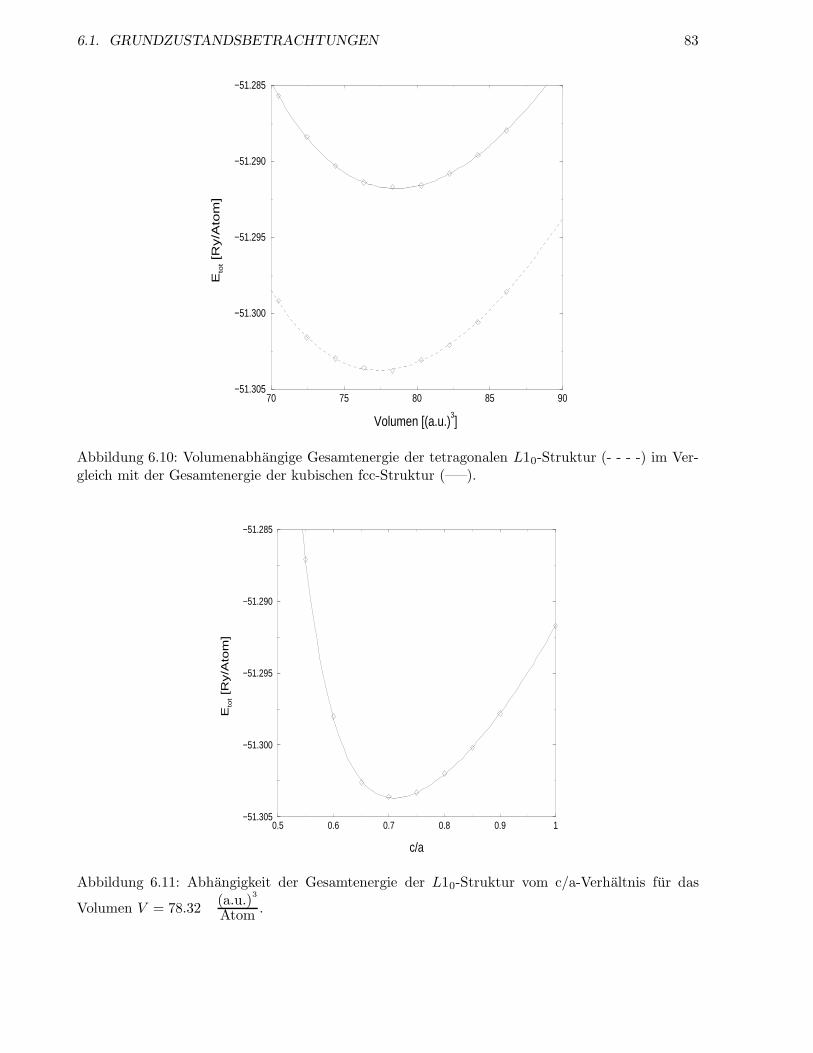
6.1. GRUNDZUSTANDSBETRACHTUNGEN 83
70 75 80 85 90
Volumen [(a.u.)3]
−51.305
−51.300
−51.295
−51.290
−51.285
Eto
t [R
y/A
tom
]
Abbildung 6.10: Volumenabhangige Gesamtenergie der tetragonalen L10-Struktur (- - - -) im Ver-gleich mit der Gesamtenergie der kubischen fcc-Struktur (—–).
0.5 0.6 0.7 0.8 0.9 1
c/a
−51.305
−51.300
−51.295
−51.290
−51.285
Eto
t [R
y/A
tom
]
Abbildung 6.11: Abhangigkeit der Gesamtenergie der L10-Struktur vom c/a-Verhaltnis fur das
Volumen V = 78.32(a.u.)
3
Atom .

84 KAPITEL 6. SYSTEM NIAL
6.1.6 Die Struktur Ni5Al3
Dem experimentellen Phasendiagramm zufolge wechselt das Grundgitter des NiAl-Systems von bccnahe der Stochiometrie 0.5 zu fcc im Ni-reichen Gebiet. Im Ubergangsbereich wurde im Experimenteine ferngeordnete Tieftemperaturphase mit der Zusammensetzung Ni5Al3 als stabil bis ca. 700C identifiziert [83] (Abb (1.1)). In der Literatur wird die entsprechende Kristallstruktur oftmalsals ”Ga3Pt5-Struktur” bezeichnet.Im Fall von Ni5Al3 basiert diese zwar auf einem fcc-Gitter, besitzt jedoch eine starke orthor-hombische Verzerrung. Es gilt als gesichert, daß, ausgehend von der B2-Struktur, außerhalb derStochiometrie martensitische Transformationen fur die Umwandlung von bcc nach fcc eine maß-gebliche Rolle spielen. So soll es dadurch zur Umwandlung der (110)bcc-Ebenen in (111)fcc-Ebenenkommen [84]. Die experimentellen Gitterkonstanten fur die Ni5Al3-Struktur lauten a=14.060 a.u.,b=12.623 a.u. und c=7.030 a.u. [80].Die starke Konkurrenz von bcc und fcc laßt sich, ohne auf die Dynamik der Strukturumwandlungeinzugehen, theoretisch dadurch untersuchen, daß man diese Ni5Al3-Struktur sowohl auf einembcc-Gitter als auch einem Gitter, das von dem fcc-Gitter abgeleitet ist, darstellt (s. Anh. C.3).Wahrend die bcc-Struktur kubische Symmetrie besitzt, hat die fcc-artige Struktur eben orthor-hombische Symmetrie.Es wurden nun mit dem mixed-basis-Pseudopotentialprogramm die Energien der beiden Struktu-ren im Grundzustand berechnet. Desweiteren erfolgte eine strukturelle Relaxation der orthorhom-bischen Struktur, bis die Krafte auf die Atome unterhalb 1 mRy/a.u. lagen. Samtliche Rechnungenwurden wiederum volumenabhangig durchgefuhrt.Der Vergleich der Energien zeigt, daß die bcc-Struktur offenbar energetisch gunstiger ist als dieunrelaxierte fcc-Struktur. Erst die orthorhombische Verzerrung fuhrt zu einer Begunstigung der fcc-artigen Struktur. Die Rechnungen erlauben es jedoch nicht, die orthorhombische Ni5Al3-Strukturals stabile Grundzustandsstruktur zu verifizieren (Abb. (6.12)). Zwar kommt die Bildungsenergierecht nah an die die B2- und L12-Struktur verbindende Stabilitatslinie heran, verbleibt aber trotz-allem oberhalb davon.Die experimentellen Arbeiten beziehen sich jedoch ausschließlich auf Temperaturen uber 400 C.Von daher ist es auch nicht moglich, Ni5Al3 als eindeutige Grundzustandsstruktur zu identifizieren.Es waren auch Entropieeffekte als Ursache fur die Stabilitat dieser Phase gegenuber einer Mischungaus B2 und L12 denkbar.Von Lu et. al [10] wurde die Ni5Al3-Struktur ebenfalls auf ihre Stabilitat im Grundzustand mittelsder LMTO-ASA-Methode getestet. Diese Rechnungen wurden allein fur die kubische bcc-Strukturdurchgefuhrt. In dieser Arbeit von Lu et. al liegt diese geringfugig unterhalb der die B2- und L12-Struktur verbindenden Stabilitatslinie und wird somit dort als Grundzustandstruktur identifiziert.Es sei an dieser Stelle jedoch noch einmal deutlich darauf hingewiesen, daß der Erfinder der LMTO-Methode selbst (Andersen) die ASA-Naherung, also die Ersetzung des wahren Kristallpotentialsdurch ein Atomkugelpotential, als unzureichend fur die Bestimmung von strukturellen Energieun-terschieden beurteilt.Bei der Diskussion der Stabilitat der Ni5Al3-Phase wird deutlich, wie wichtig Relaxationseffekteim System NiAl sind. Ursache hierfur durfte die vergleichsweise große Differenz der Atomvoluminavon Ni und Al, in Verbindung mit dem unterschiedlichen chemischen Verhalten der beiden Elemen-te, sein. Demzufolge muß eine unzureichende Berucksichtigung oder gar Vernachlassigung dieserEffekte zu falschen Ergebnissen fuhren.
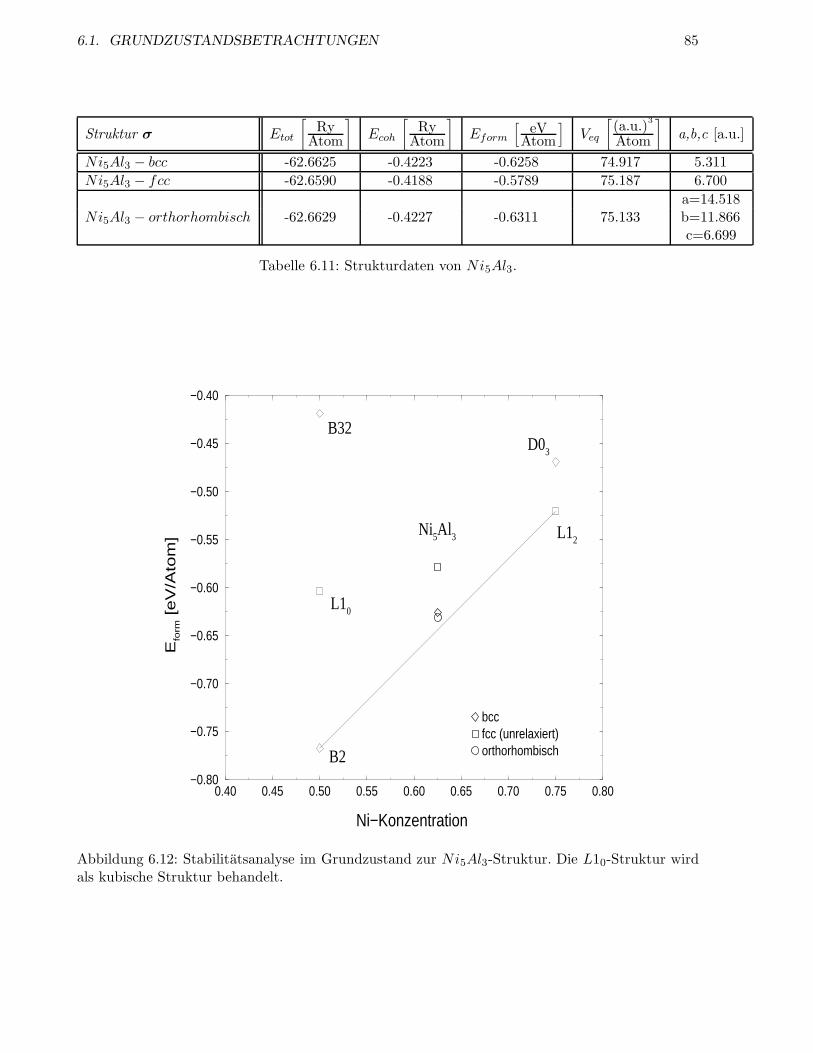
6.1. GRUNDZUSTANDSBETRACHTUNGEN 85
Struktur σ Etot
[
RyAtom
]
Ecoh
[
RyAtom
]
Eform[
eVAtom
]
Veq
[(a.u.)
3
Atom
]
a,b,c [a.u.]
Ni5Al3 − bcc -62.6625 -0.4223 -0.6258 74.917 5.311
Ni5Al3 − fcc -62.6590 -0.4188 -0.5789 75.187 6.700
a=14.518Ni5Al3 − orthorhombisch -62.6629 -0.4227 -0.6311 75.133 b=11.866
c=6.699
Tabelle 6.11: Strukturdaten von Ni5Al3.
0.40 0.45 0.50 0.55 0.60 0.65 0.70 0.75 0.80
Ni−Konzentration
−0.80
−0.75
−0.70
−0.65
−0.60
−0.55
−0.50
−0.45
−0.40
Efo
rm [
eV
/Ato
m]
bccfcc (unrelaxiert)orthorhombisch
B32
L10
B2
D03
L12Ni5Al3
Abbildung 6.12: Stabilitatsanalyse im Grundzustand zur Ni5Al3-Struktur. Die L10-Struktur wirdals kubische Struktur behandelt.

86 KAPITEL 6. SYSTEM NIAL
6.2 Thermodynamik
Nachdem im letzten Teilkapitel die Eigenschaften des NiAl-Systems im Grundzustand in Abhangig-keit von der Konzentration untersucht wurden, kann nun mit dem in Kapitel 5 entwickelten For-malismus das thermodynamische Verhalten dieser intermetallischen Verbindung analysiert werden.Die fur die Rechnungen erforderlichen Wechselwirkungskoeffizienten, d.h. die ECIs, wurden bereitsbestimmt. Dabei hat sich gezeigt, daß die Tetraedernaherung mit der SIM durch Matrixinversionerwartungsgemaß nicht in der Lage ist, weitreichende Besetzungskorrelationen in der Elementar-zelle, wie sie beispielsweise bei Schichtstrukturen eine wichtige Rolle spielen, zu beschreiben.Obwohl zwar im NiAl-System nach dem experimentellen Phasendiagramm, abgesehen vom Ni-armen Gebiet, lediglich Strukturen mit vergleichsweise kleiner Einheitszelle dominant sind, ist klar,daß gerade im Ubergangsbereich zweier Phasen weitreichende Korrelationen auch die Thermody-namik solcher Strukturen bestimmen. Nichtsdestotrotz soll auch fur endliche Temperaturen an derTetraedernaherung, nun auch fur die Entropie, festgehalten werden, um deren Moglichkeiten undGrenzen auch fur diese Belange zu testen.
6.2.1 CVM-Programm
Bevor nun detailliert auf die Resultate der angestellten Rechnungen eingegangen wird, sollen ineinem ersten Abschnitt zuerst einige Bemerkungen zum Rechenverfahren selbst gemacht werden.Fur die statistischen Berechnungen wurde ein Programm entwickelt, das es erlaubt, die CVM inTetraedernaherung sowohl auf dem fcc- als auch auf dem bcc-Gitter durchzufuhren. Die Systemin-formation geht uber die volumenabhangigen totalen Energien der geordneten, fur die SIM benotig-ten Strukturen als Eingabeparameter ein. Die SIM selbst ist ebenfalls direkt in das Programmimplementiert.Es sei daran erinnert, daß fur die energetischen Diskussionen immer auf die Bildungsenergien ab-gehoben wurde. Fur die explizite thermodynamische Rechnung wurde nun jedoch die totale freieEnergie nach Clustern entwickelt. Dies erweist sich rechnerisch als gunstiger, da in diesem Falltatsachlich das Vakuum den Energienullpunkt reprasentiert. Die Wahl dieses Nullpunktes ist je-doch, solange fur bcc- und fcc-Strukturen einheitlich gemacht, prinzipiell willkurlich (s. Gl (3.34)).Aus Grunden der Anschaulichkeit wird aber auch bei Diskussion der thermodynamischen Stabi-litaten vereinzelt auf die entsprechenden Bildungsgroßen eingegangen.In den thermodynamischen Betrachtungen wurden keinerlei phononischen oder elektronischen An-regungen berucksichtigt. Somit wurde die Entropie auch wie eine reine Konfigurationsentropiebetrachtet und die Großen Sψ0(A) und Sψ0(B) in Gl. (3.35) gleich null gesetzt.Die CVM kann wahlweise mit dem lokalen oder globalen Volumenrelaxationsschema durchgefuhrtwerden. Unter Vorgabe der effektiven chemischen Potentiale µ∗
Ni bzw. µ∗Al wird unter Verwendungder NIM das effektive große Potential Ω fur jede hinsichtlich den Startwerten vorgegebene Strukturberechnet. Aufgrund der positiven Krummung der freien Energie-Kurven kann wahlweise durchgeeignete Variation der effektiven chemischen Potentiale auch bei fester Konzentration gerechnetwerden, was zum Beispiel die Bestimmung temperaturabhangiger Ordnungsparameter ermoglicht.Die Funktionsfahigkeit des Programmes, insbesondere der NIM, wurde bei der Berechnung vonPrototyp-Phasendiagrammen getestet. Die Resultate hierzu sind im Anhang D aufgefuhrt.In den Rechnungen fur das NiAl-System wurden nur die Strukturen berucksichtigt, die auch in derSIM verwendet wurden. Aufgrund der veranschlagten Zerlegung des fcc- und bcc-Grundgitters in 4sc- bzw. 4 fcc-Untergitter waren prinzipiell auch die Strukturen F 43m und P4/mmm (Tab. (5.2))darstellbar. Angesichts der geringen Zahl der abgebildeten Strukturen und der daraus folgendeneingeschrankten Beschreibungsmoglichkeit des Systems erscheint es aber nicht vertretbar, Aussagen

6.2. THERMODYNAMIK 87
uber das thermodynamische Verhalten nicht in der Abbildung verwendeter Strukturen zu machen.Die reinen fcc- und bcc- Phasendiagramme wurden samtlich mit den mittels Parametersatz I ge-wonnenen ECIs berechnet. Das komplette Phasendiagramm wurde sowohl mit den ECIs aus I alsauch aus II erstellt.Die explizite Berechnung eines solchen Phasendiagrammes gestaltet sich derart, daß bei Vorgabeeiner bestimmten Temperatur das effektive chemische Potential µ∗
Ni fur alle darstellbaren reinenPhasen durchgefahren wird. Bei einem bestimmten Wert von µ∗
Ni ist die Phase mit minimalem Ωstabil. Aus numerischen Grunden ist es gunstig, fur den Start der Rechnung eine Zusammensetzungnahe der Stochiometrie einer aus den Grundzustandbetrachtungen bekannten stabilen Struktur zuwahlen. Wechselt im Lauf der Rechnung dann bei einem gewissen µ∗
Ni die stabile Phase, so sindfur die Berechnung der Phasengrenze zwei Falle zu unterscheiden :
• Handelt es sich um einen Phasenubergang 1. Ordnung, so lassen sich die Ω-Kurven der betref-fenden reinen Phasen in die jeweiligen metastabilen Regionen fortsetzen. Die Phasengrenzeentspricht bei fester Temperatur dann dem Schnittpunkt mit gleichem Ω und gleichem µ∗
Ni
dieser beiden Kurven. Dieser Punkt laßt sich uber ein geeignetes Iterationsverfahren beliebiggenau berechnen. Im Programm wurde eine Ubereinstimmung von 10−12 Ry in Ω gefordert.Bei einem durch Variation des effektiven chemischen Potential induzierten Ubergangs 1. Ord-nung entsteht ein Mischphasenbereich (Abb. (3.6). Dessen Breite ergibt sich als Differenz derzugehorigen Konzentrationen im Schnittpunkt der Ω-Kurven.
• Hat man es mit einem kontinuierlichen Phasenubergang zu tun, so gestaltet sich die Berech-nung der Phasengrenze etwas schwieriger. Dies liegt daran, daß es nun nicht mehr moglich ist,die Ω-Kurven der reinen Phasen in den Stabilitatsbereich der jeweils anderen Phase fortzu-setzen. Es ist eine allgemeine Aussage der Theorie der Phasenubergange (Abb. 3.7), daß beieinem kontinuierlichen Phasenubergang die ursprunglich stabile Phase im Bereich oberhalbdes Ubergangspunktes instabil ist. Daher muß man bei der Berechnung der Phasengrenzedurch Wahl kleiner Schrittweiten in µ∗
Ni versuchen, moglichst nahe an den Ubergangspunktheranzukommen. Da die NIM an einem solchen Ubergangspunkt jedoch divergiert, ist dieRechenzeit in diesem Fall ungleich langer.
Speziellere Angaben zur jeweiligen Rechentechnik werden im Rahmen der Diskussion der erhaltenenResultate dargelegt.
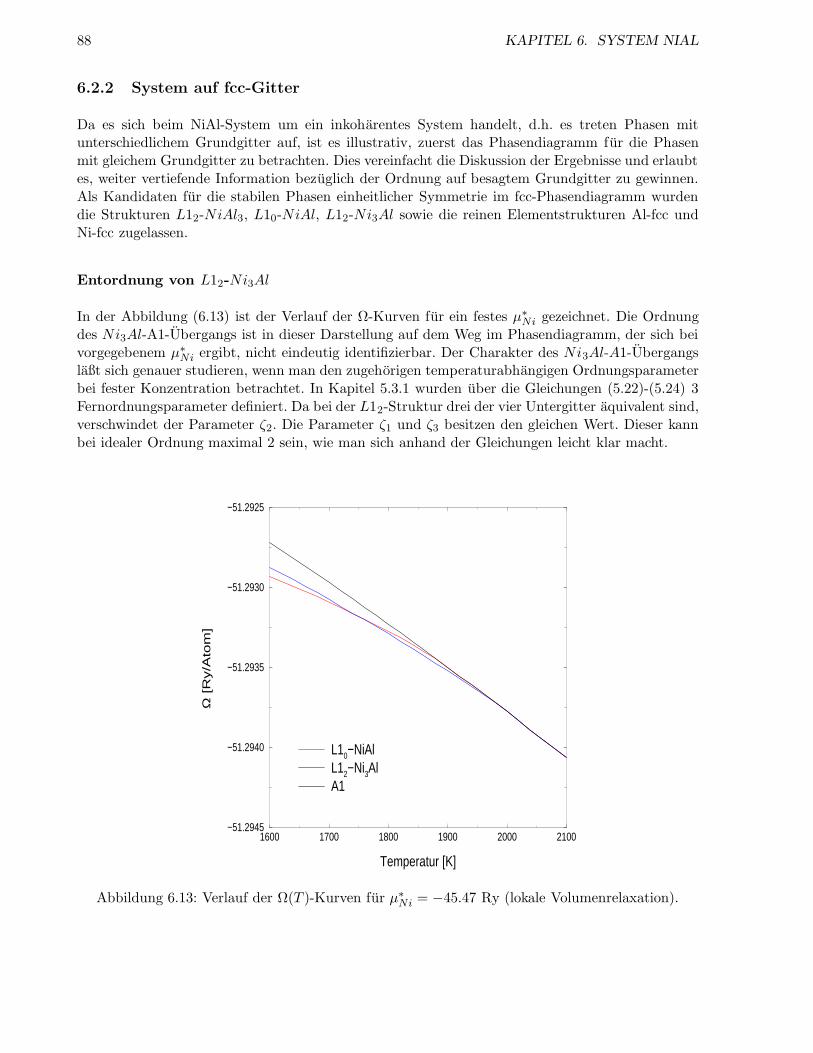
88 KAPITEL 6. SYSTEM NIAL
6.2.2 System auf fcc-Gitter
Da es sich beim NiAl-System um ein inkoharentes System handelt, d.h. es treten Phasen mitunterschiedlichem Grundgitter auf, ist es illustrativ, zuerst das Phasendiagramm fur die Phasenmit gleichem Grundgitter zu betrachten. Dies vereinfacht die Diskussion der Ergebnisse und erlaubtes, weiter vertiefende Information bezuglich der Ordnung auf besagtem Grundgitter zu gewinnen.Als Kandidaten fur die stabilen Phasen einheitlicher Symmetrie im fcc-Phasendiagramm wurdendie Strukturen L12-NiAl3, L10-NiAl, L12-Ni3Al sowie die reinen Elementstrukturen Al-fcc undNi-fcc zugelassen.
Entordnung von L12-Ni3Al
In der Abbildung (6.13) ist der Verlauf der Ω-Kurven fur ein festes µ∗Ni gezeichnet. Die Ordnungdes Ni3Al-A1-Ubergangs ist in dieser Darstellung auf dem Weg im Phasendiagramm, der sich beivorgegebenem µ∗Ni ergibt, nicht eindeutig identifizierbar. Der Charakter des Ni3Al-A1-Ubergangslaßt sich genauer studieren, wenn man den zugehorigen temperaturabhangigen Ordnungsparameterbei fester Konzentration betrachtet. In Kapitel 5.3.1 wurden uber die Gleichungen (5.22)-(5.24) 3Fernordnungsparameter definiert. Da bei der L12-Struktur drei der vier Untergitter aquivalent sind,verschwindet der Parameter ζ2. Die Parameter ζ1 und ζ3 besitzen den gleichen Wert. Dieser kannbei idealer Ordnung maximal 2 sein, wie man sich anhand der Gleichungen leicht klar macht.
1600 1700 1800 1900 2000 2100
Temperatur [K]
−51.2945
−51.2940
−51.2935
−51.2930
−51.2925
Ω [R
y/A
tom
]
L10−NiAlL12−Ni3AlA1
Abbildung 6.13: Verlauf der Ω(T )-Kurven fur µ∗Ni = −45.47 Ry (lokale Volumenrelaxation).
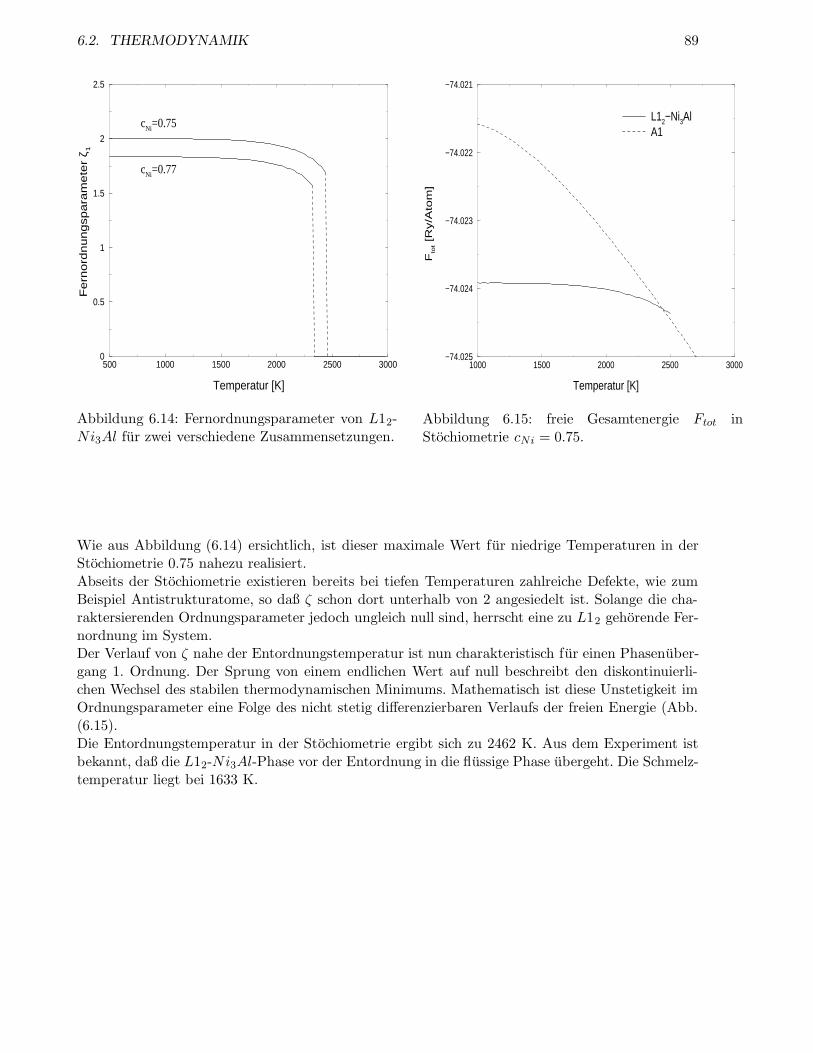
6.2. THERMODYNAMIK 89
500 1000 1500 2000 2500 3000
Temperatur [K]
0
0.5
1
1.5
2
2.5F
ern
ord
nungspara
mete
r ζ 1
cNi=0.77
cNi=0.75
Abbildung 6.14: Fernordnungsparameter von L12-Ni3Al fur zwei verschiedene Zusammensetzungen.
1000 1500 2000 2500 3000
Temperatur [K]
−74.025
−74.024
−74.023
−74.022
−74.021
Fto
t [R
y/A
tom
]
L12−Ni3AlA1
Abbildung 6.15: freie Gesamtenergie Ftot inStochiometrie cNi = 0.75.
Wie aus Abbildung (6.14) ersichtlich, ist dieser maximale Wert fur niedrige Temperaturen in derStochiometrie 0.75 nahezu realisiert.Abseits der Stochiometrie existieren bereits bei tiefen Temperaturen zahlreiche Defekte, wie zumBeispiel Antistrukturatome, so daß ζ schon dort unterhalb von 2 angesiedelt ist. Solange die cha-raktersierenden Ordnungsparameter jedoch ungleich null sind, herrscht eine zu L12 gehorende Fer-nordnung im System.Der Verlauf von ζ nahe der Entordnungstemperatur ist nun charakteristisch fur einen Phasenuber-gang 1. Ordnung. Der Sprung von einem endlichen Wert auf null beschreibt den diskontinuierli-chen Wechsel des stabilen thermodynamischen Minimums. Mathematisch ist diese Unstetigkeit imOrdnungsparameter eine Folge des nicht stetig differenzierbaren Verlaufs der freien Energie (Abb.(6.15).Die Entordnungstemperatur in der Stochiometrie ergibt sich zu 2462 K. Aus dem Experiment istbekannt, daß die L12-Ni3Al-Phase vor der Entordnung in die flussige Phase ubergeht. Die Schmelz-temperatur liegt bei 1633 K.

90 KAPITEL 6. SYSTEM NIAL
Phasendiagramme in beiden Volumerelaxationsschemata
Das fcc-Phasendiagramm (s. Abb. (6.16) und (6.17)) weist zwei reine Phasen mit FernordnungL10-NiAl und L12-Ni3Al auf. Die nicht gekennzeichneten Regionen stellen Mischphasen der an-grenzenden reinen Phasen dar. Im Ni-armen Gebiet kann sich die L12-NiAl3-Phase nicht durchset-zen, ein Indiz hierfur war bereits die ungunstige Bildungsenergie dieser Struktur. In diesem Bereichbildet unter den moglichen Phasen die Mischung aus A1 und L10 im thermodynamischen Gleich-gewicht das Minimum. Wie bereits mehrfach angemerkt, ist von experimenteller Seite bekannt,daß im Ni-armen Bereich die D519-Ni2Al3-Phase mit trigonaler Symmetrie, sowie die orthorhom-bische D011-NiAl3-Phase stabil sind. Diese vergleichsweise komplexen Strukturen wurden jedochin diesen Rechnungen nicht berucksichtigt. Die beiden verschiedenen Volumenrelaxationsschematafuhren erwartungsgemaß zu unterschiedlichen Ergebnissen. Zwar entstehen keine Anderungen in derAnzahl der verschiedenen stabilen Phasen, jedoch differieren die Verlaufe der Phasengrenzen dochstark. Auffallend ist, daß bei der globalen Relaxation im Grenzfall tiefer Temperaturen die reinenPhasen nahezu ausschließlich in der Stochiometrie stabil sind. Dies laßt sich dadurch verstehen,daß die freie Energie bei diesen geringen Temperaturen praktisch vollstandig durch den energeti-schen Term bestimmt wird. Allein dieser Anteil wird im globalen Schema hinsichtlich des mittlerenVolumens minimiert. Entfernt man sich in diesem Temperaturbereich von der stochiometrischenZusammensetzung, so bewirkt das veranschlagte effektive chemische Potential eine ”Auslenkung”des Volumens, weg vom in der Stochiometrie vorliegenden Gleichgewichtsvolumen. Aufgrund derDominanz des energetischen Terms enspricht dies nahezu komplett dem Entfernen aus dem Mini-mum der volumenabhangigen Energiekurve der stabilen geordneten Struktur in der Stochiometrie.Die Volumenminimierung hat dann in diesem Bereich zur Folge, daß das System alsbald in besagtesMinimum zurucklauft und die stochiometrische Zusammensetzung auch bei veranderten chemischenPotentialen nahezu erhalten bleibt. Erst wenn die Volumenauslenkung derart groß wird, daß einUberwechseln in die Energiekurve einer anderen stabilen Verbindung mit veranderter Stochiometriemoglich wird, wechselt eben auch die stabile Phase. Dieses Artefakt der globalen Volumenminimie-rung wird erst mit Anwachsen des entropischen Terms in der freien Energie ausgeglichen.Desweiteren unterscheiden sich die Entordnungstemperaturen der L10- und L12-Phase in beidenFallen deutlich voneinander. Bei der globalen Relaxation liegen sie um mehrere 100 K hoher als beider lokalen Relaxation. Wiederum kann man die volumenabhangige Clusterenergie verantwortlichmachen. Bei steigender Temperatur entordnet zwar aufgrund des Anwachsens des Entropietermsder Kristall, jedoch variiert bei fester Zusammensetzung das mittlere Volumen im globalen Relaxa-tionsschema nur geringfugig. Daraus folgt nun, daß zwar mehr Tetraeder im System auftreten, derenKonfiguration nicht im Einklang mit der ferngeordneten Phase ist, diesen jedoch die jetzt fur sieungunstige Clusterenergie, welche zu einem gunstigen Volumen der geordneten Verbindung gehort,zugeordnet wird, so daß sie mit geringerem statistischen Gewicht erscheinen. Diese energetischeUnterschatzung der Defekttetraeder bei hohen Temperaturen hat eine erhohte Entordnungstempe-ratur zur Folge.Im Gegensatz dazu wird im lokalen Volumenrelaxationsschema jeder Tetraederkonfiguration diehinsichtlich der Besetzung gunstigste Energie zugeordnet. Dieser andere Grenzfall uberschatzt dem-zufolge energetisch die Defektbildung und fuhrt zu einer verfruhten Entordnung. Diese Ausfuhrun-gen erlauben es jedoch nicht, einer der beiden Relaxationsschemata grundsatzlich den Vorzug zugeben. Die realistischeren Ubergangstemperaturen im lokalen Schema lassen sich teilweise auchdurch Kompensationseffekte deuten. Die verfruhte Entordnung aufgrund der unzureichenden ener-getischen Beschreibung kompensiert sicherlich zum Teil die verspatete Entordnung infolge der Tat-sache, daß es sich bei der CVM um eine Mean-Field-Theorie handelt.
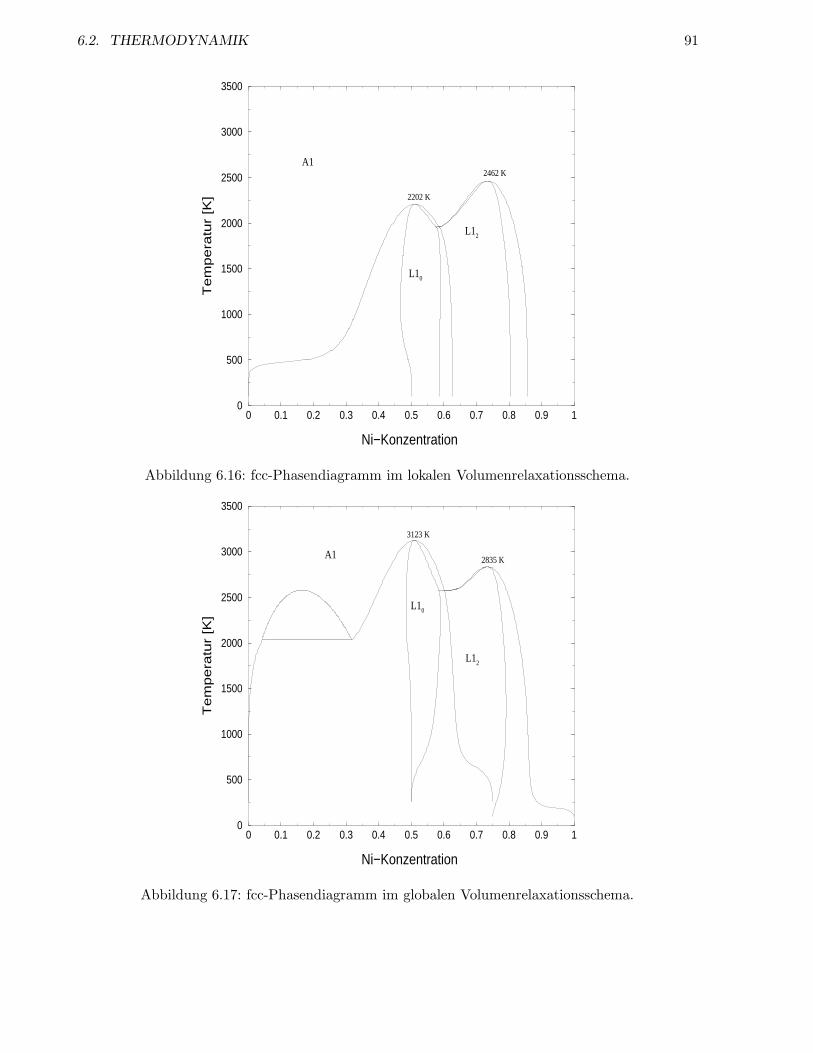
6.2. THERMODYNAMIK 91
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
3500
Tem
pera
tur
[K]
A1
L10
L12
2462 K
2202 K
Abbildung 6.16: fcc-Phasendiagramm im lokalen Volumenrelaxationsschema.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
3500
Tem
pera
tur
[K]
A1
L10
L12
2835 K
3123 K
Abbildung 6.17: fcc-Phasendiagramm im globalen Volumenrelaxationsschema.

92 KAPITEL 6. SYSTEM NIAL
Mischungslucke in der A1-Phase
Ein weiterer offensichtlicher Volumeneffekt ist die Mischungslucke im Ni-armen Gebiet, die bei derglobalen Relaxation auftritt. Die Berechnung dieses Gaps ist etwas mit Schwierigkeit verbunden, daes sich hierbei nicht um eine Phasengrenze zwischen zwei Phasen mit unterschiedlicher Symmetriehandelt. Vielmehr hat man es mit einer heterogenen Mischphase zweier ungeordneter Phasen der-selben Symmetrie aber unterschiedlicher Zusammensetzung zu tun. Daher konnen die erwahntenBerechnungsmethoden hier nicht angewandt werden.Eine mogliche Berechnung gestaltet sich so, daß man außerhalb des Gaps wiederum bei fester Tem-peratur startet und dann das effektive chemische Potential solange modifiziert, bis sich ein Sprungin der Konzentration vollzieht (Abb. (6.18a)). Dadurch ist man von der stabilen Einzelphase bisan die Spinodalgrenze (Punkt C) gewandert. Dadurch, daß auf diesem Weg bei jeder Anderungvon µ∗Ni die vorherigen Besetzungswahrscheinlichkeiten als Startwerte fur den neuen Iterationszy-klus der NIM verwendet werden, ist eine metastabile Einzelphase zwischen den Punkten B und Crealisierbar (Abb. (6.18b)). Nach dem Sprung von C nach D fahrt man dann µ∗Ni wieder soweitzuruck, bis sich ein erneuter Sprung von F nach A vollzieht und man also die gegenuberliegendeSpinodalgrenze erreicht hat.Mit dieser Prozedur hat man durch den Weg A-B-C-D-E-F nun eine Art Hystereschleife im Ω-µ∗
Ni-Diagramm definiert, die den Schnittpunkt der beiden Phasen unterschiedlicher Zusammensetzungenthalt (Abb. (6.18c). Durch Speichern des jeweiligen Ω-Werts beim Durchlaufen der Schleife kannman dann am Ende den besagten Schnittpunkt (B u. E) durch Vergleich der gespeicherten Datenermitteln. Im Schnittpunkt sind wiederum µ∗
Ni und Ω identisch, d.h. nur an diesem Punkt konnendie beiden Phasen unterschiedlicher Zusammensetzung koexistieren. Somit charakterisiert dieserPunkt dann die beiden Punkte der Phasengrenze des Entmischungsgaps im T -cNi-Diagramm zurgegebenen Temperatur.Die Existenz dieses Entmischungsgaps im Ni-armen Gebiet des fcc-Diagrammes wird auch vonCarlsson und Sanchez [85] sowie Pasturel et al. [13] bestatigt. Die ersten beiden machen hierfur ei-ne negative Krummung in der Mischungsenthalpie ∆Hform der ungeordeten A1-Struktur in diesemBereich verantwortlich. Diese wiederum soll eine Folge der Drei- und Vierkorperwechselwirkungsowie der Volumenabhangigkeit sein.Es existiert von Colinet et al. auch eine Bestimmung des fcc-Phasendiagrammes, bei dem NN-Paar-Wechselwirkungsparameter mittels semiempirischer tight-binding-Rechnungen ermittelt wer-den [12]. In diesem Fall taucht die besagte Mischungslucke nicht auf. Außerdem existiert bei diesenRechnungen auch eine stabile NiAl3-L12-Phase.
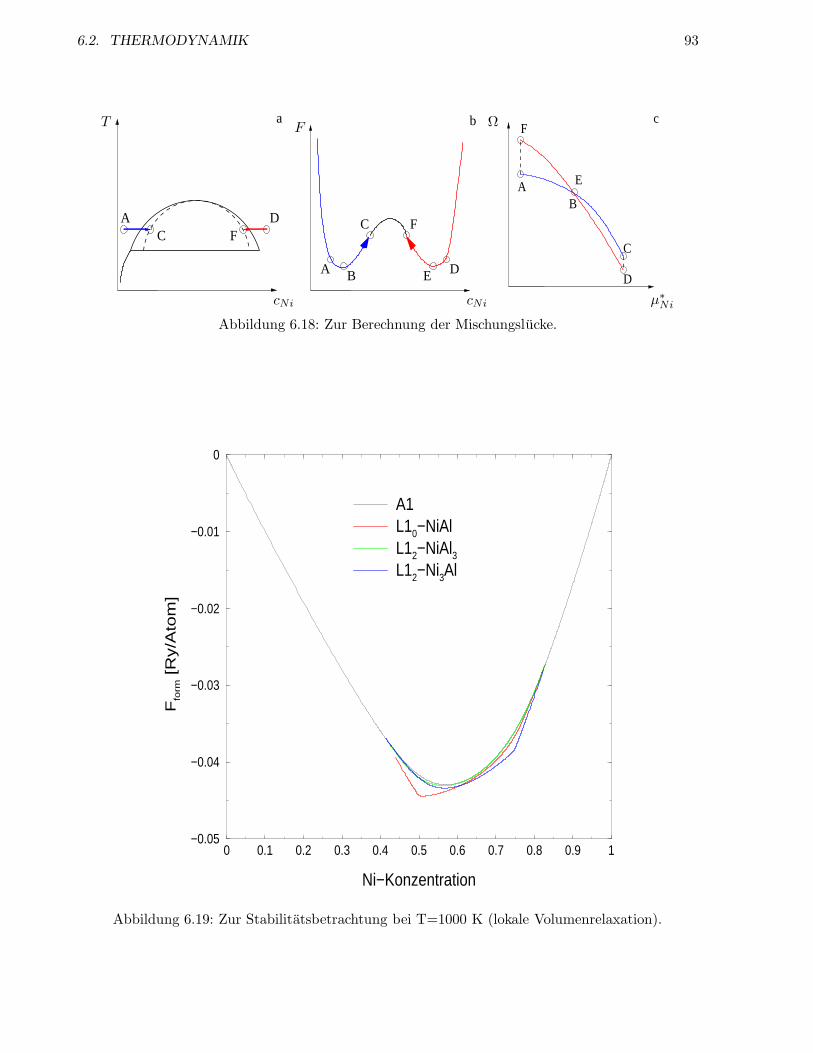
6.2. THERMODYNAMIK 93
DA
a
C F
PSfrag replacements
Ωµ∗Ni
T
FcNi
C
A
b
B E D
F
PSfrag replacements
Ωµ∗NiT
F
cNi
F
A
c
E
D
C
BPSfrag replacements
Ω
µ∗Ni
TF
cNi
Abbildung 6.18: Zur Berechnung der Mischungslucke.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
−0.05
−0.04
−0.03
−0.02
−0.01
0
Ffo
rm [
Ry/A
tom
]
A1L10−NiAlL12−NiAl3L12−Ni3Al
Abbildung 6.19: Zur Stabilitatsbetrachtung bei T=1000 K (lokale Volumenrelaxation).

94 KAPITEL 6. SYSTEM NIAL
Freie Bildungsenergien
Bei den Grundzustandsbetrachtungen wurden die Bildungsenergien der geordneten Strukturen kon-zentrationsabhangig aufgetragen (vgl. Abb. (6.3) u. (6.4)). Zur Veranschaulichung der Rechenme-thode ist es nun hilfreich, in Analogie dazu bei Betrachtung des Systems bei endlichen Temperaturendie freien Mischungssenergien nach (3.34) entsprechend darzustellen (Abb. (6.19)).Die durch A1 beschriebene Kurve entspricht dabei der Energie Edis im Fall T = 0. Die Auftragungzeigt, daß die Krummungen der Kurven immerzu positiv sind. Obwohl also die CVM eine Naherungder wahren freien Energie beschreibt, bleiben die thermodynamischen Stabilitatsrelationen erwar-tungsgemaß erhalten. Es ist zu erkennen, daß die freie Mischungsenergie der L12-NiAl3-Phase uberden kompletten Konzentrationsbereich thermodynamisch ungunstig ist. Desweiteren fallt auf, daßdie metastabilen Phasenbereiche weiter ins Ni-reiche Gebiet vordringen als ins Ni-arme. Offensicht-lich ist die Ordnung auf dem fcc-Gitter in dieser Region kaum ausgepragt. Zur Erinnerung seian dieser Stelle noch einmal erwahnt, daß das effektive chemische Potential µ∗
Ni proportional zurSteigung der dargestellten Kurven ist (Gl. (3.14)). Es ist nachzuvollziehen, wie sich durch Anlegeneiner gemeinsamen Tangente beispielsweise an die NiAl- und Ni3Al-Kurve der Mischphasenbereichabgrenzen laßt.
6.2.3 System auf bcc-Gitter
In gleicher Weise wie fur das fcc-Gitter wurden auch fur das bcc-Gitter Rechnungen angestellt. Inder Statistik wurden dabei die Strukturen D03-NiAl3, B2-NiAl, B32-Ni2Al2, D03-Ni3Al sowiedie beiden reinen Element-Strukturen Ni-bcc und Al-bcc berucksichtigt.In Abbildung (6.20) ist wiederum exemplarisch der Verlauf der relevanten Ω-Kurven bei einemfesten µ∗Ni wiedergegeben. Die Rechnungen waren auf diesem Grundgitter aufwendiger, da im NiAl-System auf dem bcc-Gitter zum Teil kontinuierliche Phasenubergange stattfinden.
Entordnung von B2-NiAl
Von besonderem Interesse ist der B2-A2-Ubergang. Im experimentellen Phasendiagramm ist eineausgedehnte B2-Phase existent, welche jedoch ebenso wie bereits die L12-Ni3Al-Phase vor derEntordnung in die flussige Phase ubergeht. Da in der B2-Struktur die Untergitter 1 und 2 sowie3 und 4 aquivalent sind, verschwinden die Fernordnungsparameter ζ2 und ζ3. Es verbleibt ζ1 alsrelevanter Ordnungsparameter fur diese Struktur. Dieser nimmt bei idealer Ordnung den Wert 4an (s. Abb.(6.21)).Bei der Berechnung der temperaturabhangigen Ordnungsparameter wurde allgemein die Tempera-tur in 2 K Schritten erhoht. Die Entordnungstemperaturen liegen bei 7770 K in der Stochiometriesowie 7814 K bei der Ni-Konzentration 0.55. Abseits der Stochiometrie ist die B2-A2-Ubergang-stemperatur also geringfugig hoher.Naturlich kann auf diese Weise nicht eindeutig geklart werden, ob es sich um einen kontinuierlichenPhasenubergang handelt, da nicht beliebig genau an den Ubergangspunkt herangegangen werdenkann. Daher entsteht aufgrund der diskreten Schrittweite in der Temperatur letztendlich immerein, wenn auch kleiner, Sprung auf ζ1 = 0. Alle Indizien weisen jedoch auf einen kontinuierlichenPhasenubergang hin.

6.2. THERMODYNAMIK 95
Phasendiagramme in beiden Volumerelaxationsschemata
In den Abbildungen (6.22) und (6.23) sind nun wiederum die Phasendiagramme auf dem bcc-Gitterin den beiden Volumenrelaxationsschemata dargestellt. Deutlich ist die Dominanz der B2-Phasezu erkennen. Im Ni-reichen Gebiet ist die D03-Ni3Al-Struktur stabil, wohingegen in der Ni-armenRegion die entsprechende D03-Struktur nicht stabilisiert werden kann. Das gleiche gilt fur die B32-Struktur.In diesem Zusammenhang ist der Vergleich mit den Prototyp-Phasendiagrammen mit reiner NN-und NNN-Wechselwirkung und der Entropie in Tetraedernaherung interessant [86]. Allgemein sindPrototyp-Systeme durch die Art und Große der Wechselwirkung auf einem festem Grundgitterdefiniert3. Entscheidend bei den angesprochenen bcc-Prototypdiagrammen ist nun das Verhaltnisε = K2,2/K2,1. Die moglichen Phasendiagramme lassen sich bei rein antiferromagnetischen Wech-selwirkungen in 3 Kategorien einteilen
• ε < 23
Bei diesen ε-Werten tauchen nur die Phasen A2, B2 und D03 im Phasendiagramm auf.
• ε > 23
Diagramme mit A2, B32 und D03.
• ε = 23
Sowohl B2 als auch B32 treten neben A2 und D03 im Phasendiagramm auf.
Die Wahrscheinlichkeit fur das Auftauchen der B32-Struktur steigt also mit wachsendem K2,2.Dies ist auch anschaulich klar, da eine Ordnung im NNN-Abstand in dieser Clusternaherung undverwendeter Aufteilung des bcc-Gitters in 4 fcc-Gitter jedenfalls nahe der Stochiometrie auf B32fuhren muß, da unter gleichen Voraussetzungen die B2-Phase einer Ordnung im NN-Abstand ent-spricht.Obwohl in den Rechnungen zum NiAl-System nun weitere Wechselwirkungen als die reinen Paar-wechselwirkungen mitaufgenommen wurden, zeigt ein Blick auf die Tabelle (6.7), daß besagtesSystem fur diesen Vergleich zur ersten Kategorie gehoren muß. Dies wird durch die berechnetenPhasendiagramme bestatigt.Weiter kann mit der Landau-Theorie, d.h. speziell mit den Landau-Lifschitz-Regeln, die Moglich-keit des Auftretens eines kontinuierlichen Phasenubergangs zwischen den betreffenden Strukturenvorhergesagt werden. Demnach konnen alle bis auf den Ubergang D03-A2 sowohl 1. als auch 2.Ordnung sein. Besagter Ubergang sollte nach den Landau-Lischitz-Regeln 1. Ordnung sein.Im besonderen zeigen die Prototyp-Rechnungen [86], daß der B2-D03-Ubergang 2. Ordnung ist,wenn die B2-Phase naher an der aquiatomaren Zusammensetzung liegt. Umgekehrt ist der Uber-gang 1. Ordnung, wenn die Verhaltnisse vertauscht sind. Die gleiche Gesetzmaßigkeit gilt augen-scheinlich auch bei den bcc-Diagrammen des NiAl-Systems.Der Einfluß der verschiedenen Relaxationsarten auf das Phasendiagramm ist qualitativ derselbe wieim Fall des fcc-Gitters. Die verstarkte Tendenz zur Mischphasenbildung im Tieftemperaturbereichbei der globalen Volumenrelaxation hat einen Ubergang 1. Ordnung zwischen B2 und D03 in die-sem Bereich zur Folge. Aufgrund der stark ordnenden B2-Struktur ist der fur das Phasendiagrammrelevante Temperaturbereich um ein vielfaches großer als auf dem fcc-Gitter. Eine Mischungsluckewie auf dem fcc-Gitter bleibt aus.
3Fur das fcc-Gitter mit reiner NN-WW existiert beispielsweise nur ein Phasendiagramm, da die Topologie desDiagrammes vollstandig durch die reduzierte Temperatur kBT/K2,1 festgelegt ist (s.a. Anhang D.1).
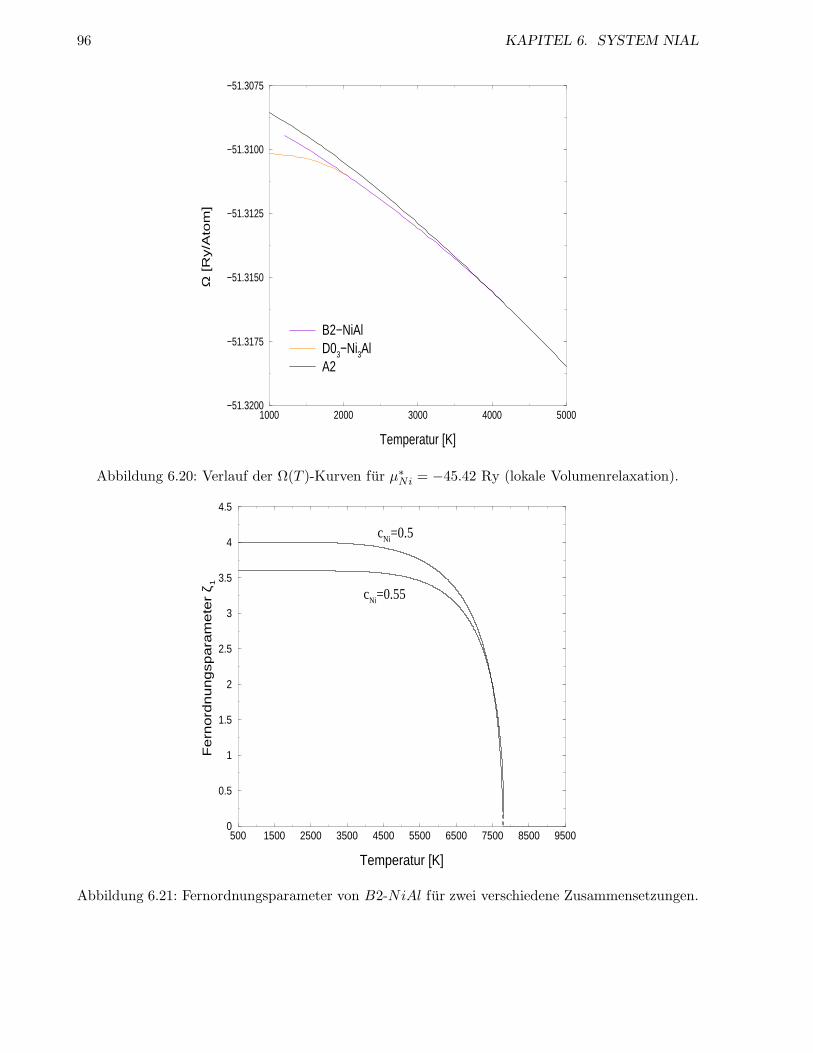
96 KAPITEL 6. SYSTEM NIAL
1000 2000 3000 4000 5000
Temperatur [K]
−51.3200
−51.3175
−51.3150
−51.3125
−51.3100
−51.3075
Ω [R
y/A
tom
]
B2−NiAlD03−Ni3AlA2
Abbildung 6.20: Verlauf der Ω(T )-Kurven fur µ∗Ni = −45.42 Ry (lokale Volumenrelaxation).
500 1500 2500 3500 4500 5500 6500 7500 8500 9500
Temperatur [K]
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
Fern
ord
nungspara
mete
r ζ 1
cNi=0.55
cNi=0.5
Abbildung 6.21: Fernordnungsparameter von B2-NiAl fur zwei verschiedene Zusammensetzungen.
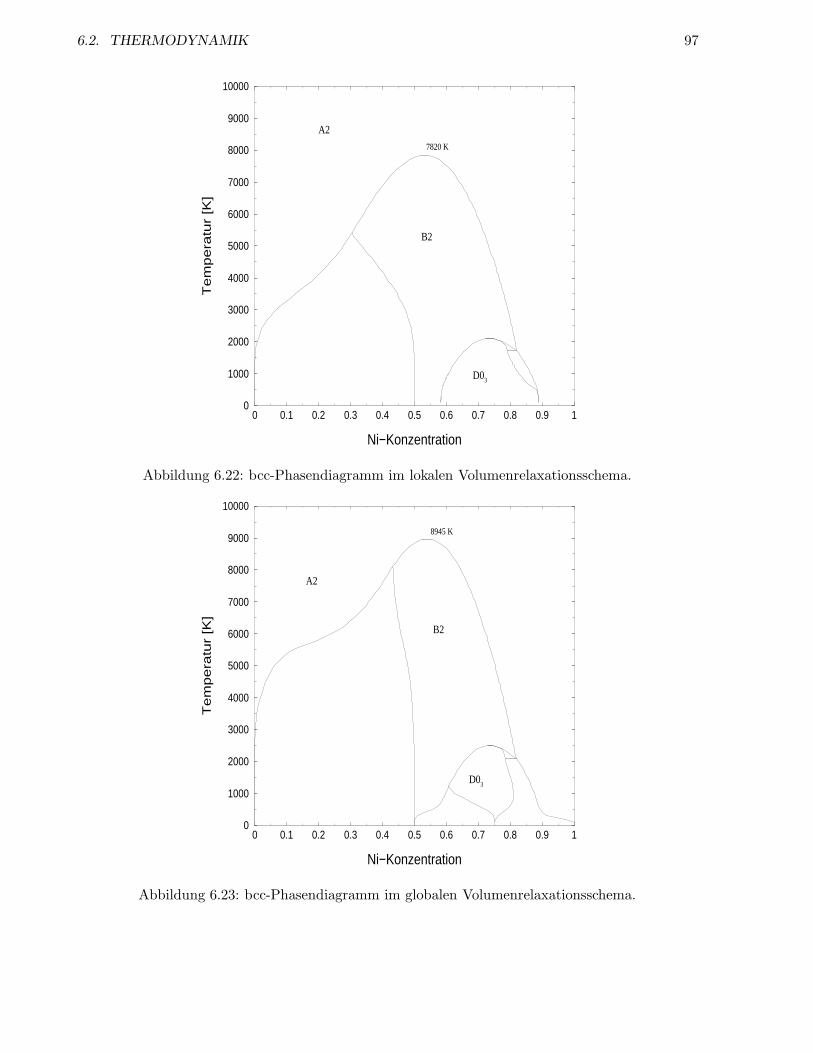
6.2. THERMODYNAMIK 97
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
Tem
pera
tur
[K]
B2
A2
D03
7820 K
Abbildung 6.22: bcc-Phasendiagramm im lokalen Volumenrelaxationsschema.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
Tem
pera
tur
[K]
B2
D03
A2
8945 K
Abbildung 6.23: bcc-Phasendiagramm im globalen Volumenrelaxationsschema.
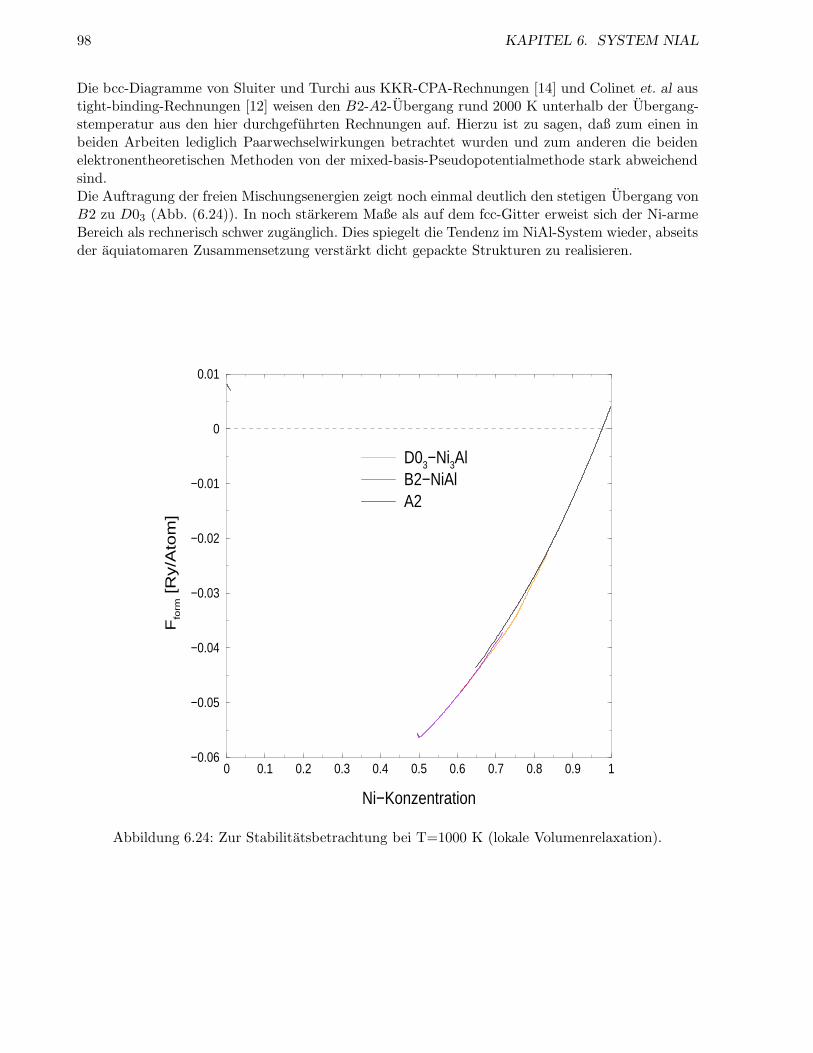
98 KAPITEL 6. SYSTEM NIAL
Die bcc-Diagramme von Sluiter und Turchi aus KKR-CPA-Rechnungen [14] und Colinet et. al austight-binding-Rechnungen [12] weisen den B2-A2-Ubergang rund 2000 K unterhalb der Ubergang-stemperatur aus den hier durchgefuhrten Rechnungen auf. Hierzu ist zu sagen, daß zum einen inbeiden Arbeiten lediglich Paarwechselwirkungen betrachtet wurden und zum anderen die beidenelektronentheoretischen Methoden von der mixed-basis-Pseudopotentialmethode stark abweichendsind.Die Auftragung der freien Mischungsenergien zeigt noch einmal deutlich den stetigen Ubergang vonB2 zu D03 (Abb. (6.24)). In noch starkerem Maße als auf dem fcc-Gitter erweist sich der Ni-armeBereich als rechnerisch schwer zuganglich. Dies spiegelt die Tendenz im NiAl-System wieder, abseitsder aquiatomaren Zusammensetzung verstarkt dicht gepackte Strukturen zu realisieren.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
−0.06
−0.05
−0.04
−0.03
−0.02
−0.01
0
0.01
Ffo
rm [R
y/A
tom
]
D03−Ni3AlB2−NiAlA2
Abbildung 6.24: Zur Stabilitatsbetrachtung bei T=1000 K (lokale Volumenrelaxation).

6.2. THERMODYNAMIK 99
6.2.4 Inkoharentes System
Nachdem in den beiden letzten Abschnitten die Phasen auf dem fcc- und bcc-Gitter getrennt unter-sucht wurden, soll nun das komplette Phasendiagramm diskutiert werden. Die Rechnungen hierfurlaufen wiederum analog zu den koharenten Diagrammen. Unter Vorgabe von µ∗
Ni und T wird daseffektive große Potential Ω fur die moglichen reinen fcc- und bcc-Phasen berechnet. Anschließendwerden die gewonnenen Werte verglichen und diejenige Phase mit dem minimalen Ω(µ∗
Ni, T ) alsstabil gewertet.
Modellierung der flussigen Phase
Wie schon des ofteren bemerkt und auch bei Ansicht des experimentellen Phasendiagrammes er-sichtlich, gehen die B2-NiAl- sowie die L12-Ni3Al-Phase vor der Entordnung in die flussige Phaseuber. Eine Modellierung des kompletten Phasendiagrammes muß daher auch die flussige Phaseberucksichtigen. Obwohl schon Fortschritte in der ab-initio Beschreibung von Flussigkeiten erzieltworden sind [87], ist eine konsistente Eingliederung solcher Rechenmethoden in den Formalismus derCluster-Entwicklung derzeit noch nicht realisiert. Um trotzdem eine wenn auch grobe Beschreibungder Thermodynamik einer flussigen NiAl-Phase zu erhalten, kann man folgenderweise verfahren.Da aus Experimenten [88, 89] bekannt ist, daß flussiges NiAl eine Nahordnung aufweist und dielokale Koordination ungefahr 11 betragt, kann versucht werden, die flussige Phase als ungeordnetefcc-Phase zu beschreiben. Damit kann man naturlich keinerlei strukturelle Information uber dieFlussigkeit gewinnen, jedoch ist bei der Berechnung des Phasendiagrammes der Beitrag der che-mischen Ordnung zu den thermodynamischen Potentialen von zentralem Interesse. Dieser Beitragist aber allein von den Konzentrationsfluktuationen abhangig. Um diese zu beschreiben, sollte dieProjektion des kontinuierlichen Phasenraums der Atombewegung in der Flussigkeit auf ein diskre-tes Gitter annehmbar sein. Die Volumenabhangigkeit der freien Energie der flussigen Phase wurdein gleicher Weise wie fur das ungeordnete fcc-Gitter gehandhabt, da die Variation dieser Große inbeiden Phasen ebenfalls ahnlich verlauft [90].Zur Illustration der Nahordnung ist in Abbildung (6.25) der NN-Nahordnungsparameter αNN indieser Naherung fur verschiedene Zusammensetzungen aufgetragen. Die funktionale Gestalt die-ser auch als isotroper Cowley-Warren-Nahordnungsparameter bezeichneten Große schreibt sichbezuglich zweier beliebiger Gitterpunkte p und q wie folgt :
αpq =〈σpσq〉 − 〈σp〉〈σq〉
1 − 〈σp〉〈σq〉. (6.13)
Der Parameter impliziert fur den NN-Abstand im NiAl-System erwartungsgemaß eine antiferro-magnetische Ordnung. Dabei ist erneut zu erkennen, daß das Ni-arme Gebiet weitaus schwacherordnet als das Ni-reiche.Um nun die so modellierte flussige Phase von einer ”herkommlichen” A1-Phase unterscheiden zukonnen, muß man naturlich noch eine Angabe bezuglich der Differenz der freien Energien dieserbeiden Phasen machen. In den Rechnungen wurde zu diesem Zweck die freie Energie der A1-Phaseentsprechend modifiziert. Hierzu startet man bei der allgemeinen Darstellung der freien Energie furein binares System auf dem Gitter ψ0 (vgl. (3.34))
Fψ0 = cNiFψ0
Ni + cAlFψ0
Al + Fψ0
M . (6.14)
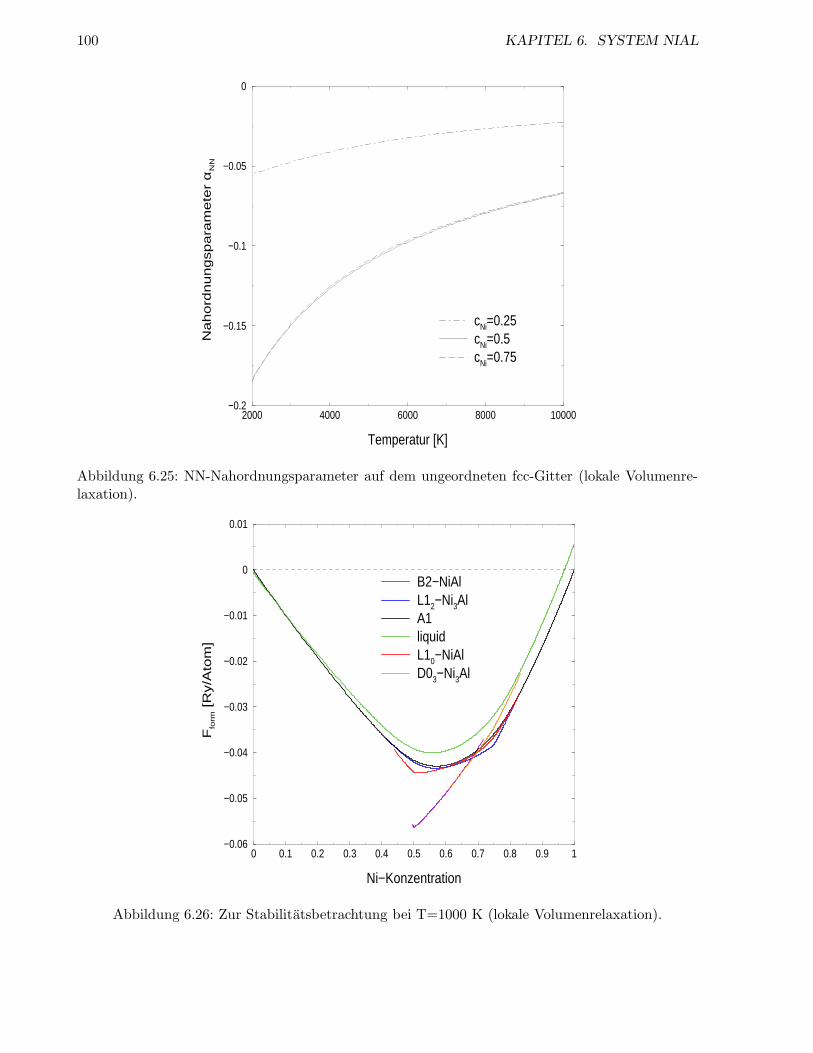
100 KAPITEL 6. SYSTEM NIAL
2000 4000 6000 8000 10000
Temperatur [K]
−0.2
−0.15
−0.1
−0.05
0
Nahord
nungspara
mete
r α
NN
cNi=0.25cNi=0.5cNi=0.75
Abbildung 6.25: NN-Nahordnungsparameter auf dem ungeordneten fcc-Gitter (lokale Volumenre-laxation).
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
−0.06
−0.05
−0.04
−0.03
−0.02
−0.01
0
0.01
Ffo
rm [R
y/A
tom
]
B2−NiAlL12−Ni3AlA1liquidL10−NiAlD03−Ni3Al
Abbildung 6.26: Zur Stabilitatsbetrachtung bei T=1000 K (lokale Volumenrelaxation).

6.2. THERMODYNAMIK 101
In der Schmelze stellen sich nun die freien Energien F ψ0i der Elemente folgendermaßen dar [13] :
F liqi (T ) = Efcci + ∆Em
i − T
(
∆EmiTmi
)
, (6.15)
wobei ∆Emi−fcc die Schmelzwarmen und Tmi−fcc die Schmelztemperaturen der Elemente beschreiben.
Diese Werte wurden thermodynamischen Tabellen [91] entnommen und stellen also die einzigenempirischen Parameter in den vorliegenden Rechnungen dar.Die totale freie Energie der flussigen Phase schreibt sich in besagter Naherung dann als
F liq(T ) = FA1 + cNi
(
∆EmNi−fcc − T∆EmNi−fccTmNi−fcc
)
+ cAl
(
∆EmAl−fcc − T∆EmAl−fccTmAl−fcc
)
. (6.16)
In den hier getatigten Rechnungen wurde diese Formel so implementiert, daß erst nach der Kon-vergenz der NIM hinsichtlich der A1-Phase die gewichteten empirischen Daten zu Ω hinzudaddiertwurden. Ein anderer semiempirischer Zugang zur Modellierung der fussigen NiAl-Phase findet sichin [92].
Phasendiagramme
Die Auftragung der Mischungsenergien bei T=1000 K zeigt deutlich, daß sowohl die L10- als auchdie D03-Phase im gemeinsamen Phasendiagramm von fcc- und bcc-Strukturen keine stabilen Pha-sen mehr darstellen (Abb.(6.26)). Neben der ungeordneten A1-Struktur bilden die im experimentel-len Phasendiagramm auf diesen Gittern ebenfalls existente B2- und L12-Struktur im thermodyna-mischen Gleichgewicht die alleinigen ferngeordneten homogenen stabile Phasen. Daß dies nicht nurfur eine bestimmte Temperatur, sondern generell gilt, zeigen die vollstandigen Phasendiagramme(Abb. (6.27)-(6.30). Diese wurden zur Klarung des Einflusses der Konvergenzparameter mit beidenECI-Satzen berechnet. In Anhang F sind zusatzlich die Phasendiagramme im T − µ∗
Ni-Raum wie-dergegeben.Die Topologie des experimentellen Phasendiagrammes ist in groben Zugen korrekt wiedergegeben.Naturlich trifft dies nicht auf den Ni-armen Bereich zu, da die dort stabile D519-Ni2Al3- und D011-NiAl3-Phase in den Rechnungen nicht berucksichtigt wurden.Problematisch ist außerdem der Bereich des Ubergangs der Ni3Al-L12-Phase in die flussige Phase.In einem engen Konzentrations- und Temperaturbereich treffen dort die L12-, A1- und flussigePhase aufeinander. Demzufolge sind die Phasengrenzen dort besonders empfindlich gegenuber Un-genauigkeiten in den beschreibenden thermodynamischen Potentialen. Die rechnerisch existenteMischphase L12-flussig (im experimentellen Phasendiagramm existiert diese Phase nicht) ruhrt ne-ben solchen Ungenauigkeiten sicherlich auch von der unzureichenden Beschreibung der flussigenPhase her. Allgemein sind in den errechneten Diagrammen die Mischphasenbereiche zu stark aus-gepragt. Dies trifft besonders auf die Ubergangsregionen zwischen den beiden Kristallstrukturenfcc und bcc zu. Obwohl zwar die flussige Phase auch als ”fcc-Struktur” beschrieben wird, dient ihreBerucksichtigung aufgrund der angesprochenen Schwierigkeiten vornehmlich der Terminierung der”festen” Phasen. Wichtiger erscheint daher das offensichtliche Unvermogen der Tetraedernaherungmit Volumenrelaxation, den B2-L12-Ubergang zu beschreiben. Wie bei den Grundzustandsbetrach-tungen angefuhrt, induzieren weitreichende martensitische Transformationen die Umwandlung vonbcc zu fcc. Dies wird eben durch die Stabilitat der strukturell vergleichsweise komplizierten Ni5Al3-Phase impliziert. Obwohl nun zwar die energetischen Differenzen der bcc- und fcc-Strukturen abinito sehr genau berechenbar sind, laßt sich diese Genauigkeit nicht in einfacher Weise auf die sta-tistische Modellierung ubertragen. Wahrend die aquiatomaren Verbindungen L10 und B2 in den

102 KAPITEL 6. SYSTEM NIAL
koharenten Diagrammen einen weiten Konzentrationsbereich belegten, ist die energetisch eigentlichdominate B2-Phase im inkoharenten Diagramm eindeutig zu schwach ausgepragt.Das Problem in der Modellierung inkoharenter Systeme mit der CVM liegt in der inaquivalentenCluster-Entwicklung. Gerade bei kleinen maximalen Clustern kommt es dadurch zu inkonsisten-ten Beschreibungen. So wurden in diesen Rechnungen auf dem bcc-Gitter auch Wechselwirkungenim NNN-Abstand berucksichtigt, wohingegen auf dem fcc-Gitter lediglich WW im NN-Abstandin die Energetik eingingen. Da die Ordnungstendenz im NiAl-System mit dem Abstand deutlichabnimmt, wird die Ordnung auf dem fcc-Gitter daher gegenuber dem auf dem bcc-Gitter scheinbaruberschatzt. Dies ist ein grundsatzliches Problem der CE, da die Gittertopologien nunmal verschie-den sind. Da jedoch die energetischen Beitrage mit wachsender Clustergroße abnehmen sollten,ware zu erwarten, daß durch Berucksichtigung hoherer Cluster auch der Einfluß dieser Inkonsistenzverringert werden wurde.In diesem Zusammenhang darf nicht vergessen werden, daß diese Inkonsistenz auch fur die Entropi-en gilt, die ja ebenfalls in Tetraedernaherung behandelt werden. So ergeben beispielsweise Monte-Carlo-Simulationen von Prototyp-Systemen im Fall des bcc-Gitters kaum nennenswerte Unterschie-de im Vergleich zur CVM-Tetraedernaherung [66], wohingegen auf dem fcc-Gitter dieselbe Naherungaufgrund der Frustrationseffekte noch nicht ausreichend ist.Als weiteres Kriterium fur die Qualitat der Naherung ist die Beschreibung der charakteristischenUbergangstemperaturen zu werten (Tab. (6.12)). Dazu ist zu sagen, daß in den gerechneten Dia-grammen eine Temperaturschrittweite von 10 K gewahlt wurde. Eine bessere Auflosung ist zwarrechnerisch durchaus moglich, jedoch aufgrund der begrenzten Aussagekraft der Naherung wenigsinnvoll. Allein beim Ubergang B2-flussig wurde eine kleinere Schrittweite von 2 K gewahlt, dadie mit den Temperaturschritten entstehenden Konzentrationssprunge dort vergleichsweise hochwaren.
Diagramm Ni-fcc-liq B2-liq L12-liq Al-fcc-liq
lokale Relaxation (I) 930 2584 1690 1720
lokale Relaxation (II) 930 2570 1690 1720
globale Relaxation (I) 930 2830 1750 1720
globale Relaxation (II) 930 2824 1760 1720
experimentell 933 1911 1633 1728
Tabelle 6.12: Ubergangstemperaturen. Angaben in K.
Der Vergleich mit den experimentellen Daten zeigt, daß die fcc-flussig Ubergangstemperaturendurch die Rechnung erstaunlich gut wiedergegeben werden. Ursache hierfur ist der verschwindendeBeitrag der freien Mischungsenergie am Rand des Phasendiagrammes. Fur diese Ubergangstempe-raturen zeichnen daher hauptsachlich die empirischen Schmelztemperaturen und -warmen verant-wortlich. Diese sind naturlich denkbar gut. Mit einer Differenz von 60 K bei der lokalen und 120K bei der globalen Volumenrelaxation ist der L12-flussig-Ubergang in Anbetracht der zugehorigenEnergieunterschiede auch noch relativ gut beschrieben. Der Schmelzpunkt der B2-Phase ist jedochdenkbar schlecht bestimmt.

6.2. THERMODYNAMIK 103
Die Uberschatzung der Ordnungstendenz bei der Bestimmung der Energieparameter sowie dieteilweise unzureichende Entropieberucksichtigung in der Tetraedernaherung sind die maßgeblichenGrunde fur die allgemein zu hohen Ubergangstemperaturen, auch in den reinen fcc- und bcc-Phasendiagrammen. Speziell fur die B2-Phase konnte hierfur jedoch auch die Vernachlassigungder Leerstellenbildung mitverantwortlich sein, da in dieser Phase die Konzentration von Leerstellenvergleichsweise hoch ist. Weiter erscheint es auch moglich, die ausbleibende Stabilitat der B2-Phaseim Ni-armen Gebiet nahe der Stochiometrie wenigstens zum Teil ebenfalls als eine Folge der Ver-nachlassigung der Leerstellen zu deuten. So kann ohne diese Defekte die Entordnung in diesemBereich lediglich uber Antistrukturatome erfolgen, welche aber energetisch ungunstig sind und esinfolge dessen zum schnellen Zusammenbruch der B2-Phase kommt.Außerdem konnte auch allgemein die Vernachlassigung des phononischen Beitrags zur freien Ener-gie eine Mitursache fur die zu hohen Ubergangstemperaturen sowie den teilweise unrealistischenVerlauf der Phasengrenzen sein.Der Einfluß der verschiedenen Konvergenzparamter auf die Topologie und die charakteristischenTemperaturen ist marginal. Die Ungenauigkeiten in diesem Bereich werden von der unzureichendenTetraedernaherung uberschattet.Desweiteren liefert keines der beiden Volumenrelaxationsschemata zufriedenstellende Ergebnisse.Zwar liegen die Schmelztemperaturen bei der lokalen Relaxation etwas besser, jedoch sind diePhasengrenzen im Ni-reichen Gebiet fernab des Schmelzbereichs schlechter wiedergegeben als beider globalen Relaxation. So ist die Einschnurung der L12-Phase zu tiefen Temperaturen hin imglobalen Volumenrelaxationsschema ansatzweise verwirklicht. Bei der globalen Relaxation kommtes im Ni-armen Gebiet erneut zu einem der CVM unzuganglichen heterogenen Bereich. Aufgrundder beschriebenen Modellierung der flussigen Phase war es jedoch nicht moglich, diesen Bereichthermodynamisch eindeutig zu charakterisieren. Demnach mussen die Unzulanglichkeiten in dieserRegion als Artefakt der Rechnung gewertet werden.Das Phasendiagramm von Pasturel et. al [13] mit den LMTO-Werten aus Tabelle (6.3) unterschei-det sich von den hier gerechneten hauptsachlich dadurch, daß die B2-Phase geringfugig starkerausgepragt ist und die L12-Phase etwas schmaler ausfallt. Dies kann durch die bereits angespro-chene großere Differenz der zugehorigen Bildungsenergien in dieser Arbeit erklart werden. Weitererfolgte dort die Behandlung der Schmelze mit geringfugig anderen Energieparametern.Trotz allen Problemen und fehlerhaften Beschreibungen sollte nicht vergessen werden, daß das Pha-sendiagramm eines realen Systems das Ergebnis beliebig komplizierter Vielteilchenwechselwirkun-gen ist. In Anbetracht dieser Tatsache ist aufgrund der Einfachheit des zugrundeliegenden Modellesund des vergleichsweise geringenen Rechenaufwands das Resultat vielversprechend.
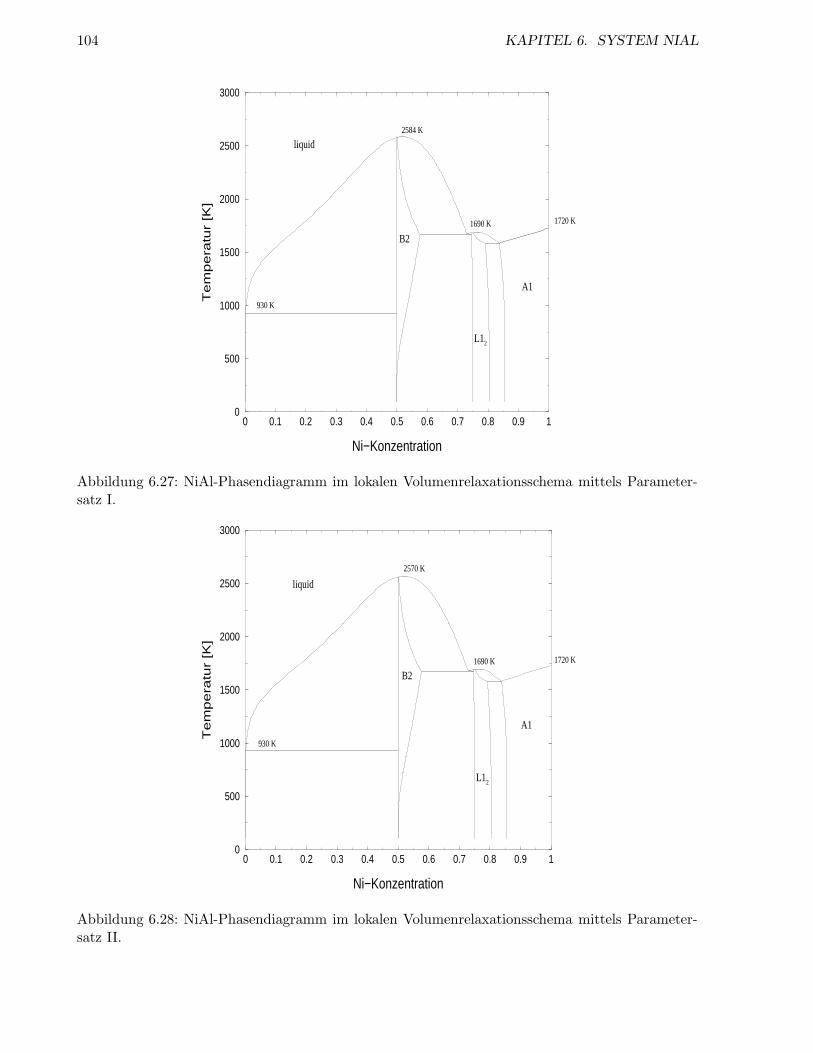
104 KAPITEL 6. SYSTEM NIAL
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
B2
liquid
L12
A1
930 K
2584 K
1690 K 1720 K
Abbildung 6.27: NiAl-Phasendiagramm im lokalen Volumenrelaxationsschema mittels Parameter-satz I.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
B2
L12
liquid
A1
2570 K
930 K
1690 K 1720 K
Abbildung 6.28: NiAl-Phasendiagramm im lokalen Volumenrelaxationsschema mittels Parameter-satz II.
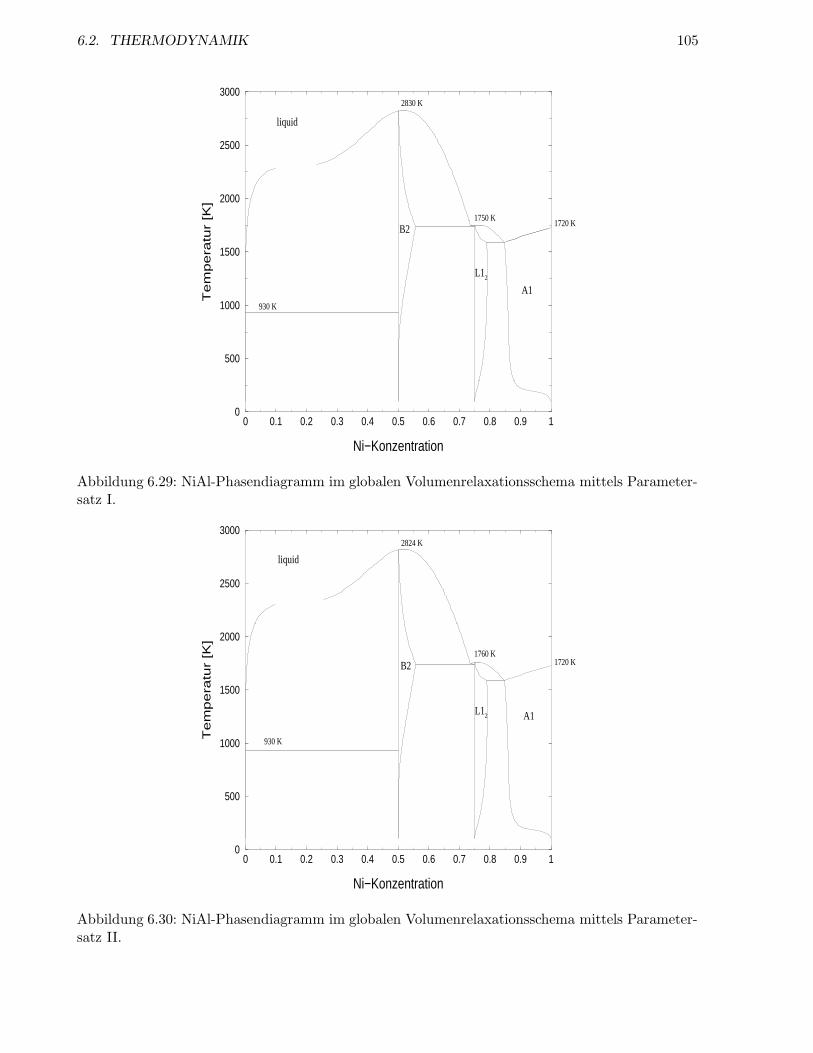
6.2. THERMODYNAMIK 105
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
B2
liquid
A1
L12
930 K
2830 K
1750 K1720 K
Abbildung 6.29: NiAl-Phasendiagramm im globalen Volumenrelaxationsschema mittels Parameter-satz I.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Ni−Konzentration
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
liquid
B2
L12 A1
930 K
1760 K1720 K
2824 K
Abbildung 6.30: NiAl-Phasendiagramm im globalen Volumenrelaxationsschema mittels Parameter-satz II.

106 KAPITEL 6. SYSTEM NIAL
6.2.5 Berucksichtigung von Leerstellen
In der bisherigen Behandlung des NiAl-Systems wurde davon ausgegangen, daß jeder Gitterplatzmit einem Ni- oder Al-Atom besetzt ist. Im Realfall bleiben jedoch im thermodynamischen Gleich-gewicht einige Gitterplatze unbesetzt. Durch den Einbau solcher Leerstellen kann das System seinefreie Enthalpie namlich weiter absenken. In einem einkomponentigen System gilt allgemein fur dieAnderung der freien Enthalpie durch die Berucksichtigung von Leerstellen :
∆G = Nvg + kBNT
(
lnN
N +Nv+Nv
Nln
Nv
N +Nv
)
. (6.17)
Hierbei ist g als Bildungsenergie einer einzelnen Leerstelle anzusehen. Die Zahl der Leerstellen wirddurchNv, die der Atome durchN beschrieben. Bei der Herleitung von (6.17) wird eine vergleichswei-se geringe Leerstellenkonzentration cv angenommen, so daß keine Wechselwirkung zwischen diesenvorliegt. In dieser Naherung ergibt sich fur cv folgender Ausdruck :
cv = exp
(
− g
kBT
)
. (6.18)
Je hoher also die Temperatur ist, umso großer ist auch die Leerstellenkonzentration. Daher sprichtman in diesem Zusammenhang auch von thermischen Leerstellen, deren Bildung als reiner Entro-pieeffekt zu werten ist.In mehrkomponentigen Systemen konnen zusatzlich sogenannte strukturelle Leerstellen abseits derStochiometrie auftreten, welche auch im Grenzfall T = 0 im Gitter verbleiben.Allgemein ist die Leerstellenbetrachtung in mehrkomponentigen Systemen komplizierter, da dortnicht nur das Grundgitter variieren kann, sondern je nach Zusammensetzung auch verschiedenegeordnete Strukturen realisiert sind. Die Berucksichtigung von Leerstellen in einem solchen Sy-stem unterhalb der Entordnungstemperaturen, muß daher in eine allgemeine Untersuchung dermoglichen Defekte in den geordneten Verbindungen eingegliedert werden. Neben den Leerstellensind auch Antistrukturatome als Defekte zu werten. So tritt in der Realitat meist eine Korrelationzwischen den Antistrukturdefekten und den Leerstellen auf, was auf mitunter komplexe Defekt-mechanismen fuhrt. Es scheint nun klar, daß gerade im Hinblick auf Entordnungsphanomene dieBerucksichtigung von Leerstellen bei einer thermodynamischen Behandlung eines realen Systemsin vielen Fallen unerlaßlich ist.Fur das NiAl-System wurden solche Betrachtungen von theoretischer Seite mit ab-initio Metho-den bereits nahe der Stochiometrie 0.5 und fernab der Entordnungstemperatur in der B2-Strukturgetatigt [69]. Demnach sind bei cNi < 0.5 Ni-Leerstellen auf dem Al-Untergitter die maßgebli-chen strukturellen Defekte. Oberhalb der aquiatomaren Zusammensetzung treten dann vermehrtNi-Antistrukturatome auf dem Al-Untergitter auf. Wegen des großen Volumenunterschieds von Niund Al sind Al-Antistrukturatome auf dem Ni-Untergitter energetisch ungunstiger. In der Stochio-metrie kommt es mit steigender Temperatur zur Ausbildung eines sogenannten ”Triple-Defekts”.Dabei werden jeweils zwei Ni-Leerstellen und ein Ni-Antistrukturatom unter Ausbildung einer neu-en B2-Einheitszelle an der Oberflache des Systems erzeugt.Mittels der Cluster-Entwicklung sollte es nun moglich sein, die Leerstellenkonzentrationen sowie dieDefektstruktur fur nahezu beliebige Zusammensetzungen und Temperaturen berechnen zu konnen.Prinzipiell konnten die Antistrukturdefekte auch bereits mit der bisher verwendeten Methodik ana-lysiert werden, da im CVM-Formalismus die Besetzungen der verschiedenen Untergitter ausgege-ben werden. Jedoch erscheint eine solche Untersuchung ohne Berucksichtigung der hierfur wichtigenLeerstellen nicht serios. Diese lassen sich nun dadurch in den Formalismus integrieren, daß man vomStandpunkt der Cluster-Entwicklung nicht ein rein binares, sondern ein ternares System betrachtet.Als dritte Komponente neben Ni und Al wird nun eben die Leerstelle angesehen. Die Tatsache, daß

6.2. THERMODYNAMIK 107
es sich dabei nicht um eine reale chemische Komponente handelt, ist fur die reine CE unerheblich.Eine Cluster-Entwicklung fur ternare Systeme ist aber aufwendiger als der binare Fall, da jetztdie Clusterfunktionen nicht mehr als reine Spinprodukte darstellbar sind. Die gewachsene Zahl vonmoglichen Kristallkonfigurationen (3N statt 2N ) erfordert, daß zur Erhaltung der Vollstandigkeitder Entwicklung auch die Quadrate der Spinfunktionen als Basisfunktionen hinzukommen.Im Binaren bildete die 1, σ-Basis eine orthogonale Basis. Wollte man die Orthogonalitat erhal-ten, so mußte man im ternaren Fall zu den entsprechenden Tschebyscheff Polynomen ubergehen.Die Orthogonalitat ist jedoch beim Connolly-Williams-Verfahren keine notwendige Bedingung, sodaß die etwas einfachere, nichtorthogonale 1, σ, σ2-Basis ebenso verwendet werden kann. Die Ver-teilung der Spinvariablen ist beliebig. In Analogie zum Binaren ist 1 fur Ni, -1 fur Al und 0 furdie Leerstellen naheliegend. In besagter Basis hat die clusterentwickelte Konfigurationsenergie proGitterpunkt auf dem bcc-Gitter in Tetraedernaherung dann folgenden Gestalt :
E
N= K0/N +
K(1)1 σ1 +K
(2)1 σ2
1 +
4(
K(11)2,1 σ1σ3 +K
(12)2,1 σ1σ
23 +K
(22)2,1 σ2
1σ23
)
+
3(
K(11)2,2 σ1σ2 +K
(12)2,2 σ1σ
22 +K
(22)2,2 σ2
1σ22
)
+ (6.19)
12(
K(111)3 σ1σ2σ3 +K
(112)3 σ1σ2σ
23 +K
(121)3 σ1σ
22σ3 +K
(122)3 σ1σ
22σ
23
+K(212)3 σ2
1σ2σ23 +K
(222)3 σ2
1σ22σ
23
)
+
6(
K(1111)4 σ1σ2σ3σ4 +K
(1112)4 σ1σ2σ3σ
24 +K
(1122)4 σ1σ2σ
23σ
24 +K
(2121)4 σ2
1σ2σ23σ4
+K(1222)4 σ1σ
22σ
23σ
24 +K
(2222)4 σ2
1σ22σ
23σ
24
)
,
wobei die Indizes an den Spinvariablen die Platze auf dem Cluster bezeichnen (s. Abb. (5.2)). Essei nochmal darauf hingewiesen, daß die Entwicklung der Symmetrie des ungeordneten bcc-Gittergehorcht, somit also beispielsweise die Clusterfunktionen σ1σ
22 und σ2
1σ2 denselben ECI besitzen.Desweiteren wird nicht auf die Zahl der Atome normiert, sondern tatsachlich auf die Zahl N derGitterpunkte, d.h N enthalt bereits die Leerstellenzahl. Die Entwicklung (6.19) beinhaltet 21 ECIs.Demzufolge mussen bei Anwendung der SIM mindestens ebensoviele ab-initio Energien von gegeig-net gewahlten Referenzkonfigurationen vorgegeben werden.Es existieren nun wiederum zahlenmaßig genausoviele energetisch inaquivalente Tetraederkonfigu-rationen wie ECIs. Demzufolge ist die Transformation der ECIs auf Tetraederenergien vermoge derV-Matrix durchfuhrbar.Die CVM mit der NIM lauft im Ternaren prinzipiell genauso wie fur das binare System. In derthermodynamischen Behandlung dieses pseudo-ternaren Systems hat jedoch die Tatsache, daß essich bei der zusatzlichen dritten Komponente um eine Leerstelle handelt, Folgen fur das chemischePotential derselben. Das chemische Potential eines Teilchens ist als Ableitung der freien Enthalpienach der Zahl der entsprechenden Teilchen bei konstantem Druck und Temperatur definiert (Gl.(3.13)). Im thermodynamischen Gleichgewicht stellt sich die Zahl der Leerstellen nun selbststandigso ein, daß die freie Enthalpie minimal wird. Demzufolge ist das chemische Potential einer Leer-stelle immer null. Will man Aussagen uber die Leerstellenkonzentrationen in Abhangigkeit von denchemischen Potentialen von Ni und Al gewinnen, so bietet sich die Rechnung im großkanonischenEnsemble an. Als beschreibendes Potential ist also direkt das große Potential J anzusehen :
j =J
N= f − µNic
∗Ni − µAlc
∗Al . (6.20)

108 KAPITEL 6. SYSTEM NIAL
Die Konzentrationen in (6.20) sind mit einem * versehen, da es sich hierbei um Konzentrationenhandelt, die auf N , also die Zahl der Gitterpunkte, normiert sind. Dies ist erforderlich, um die furdie NIM notwendige Bedingung
c∗Ni + c∗Al + c∗v = 1 (6.21)
zu gewahrleisten. Die tatsachlichen, auf die Atomzahl im System normierten Konzentrationen erhaltman, wie sich leicht verifizieren laßt, mittels
cNi =c∗Ni
c∗Ni + c∗Al, cAl =
c∗Alc∗Ni + c∗Al
, cv =c∗v
c∗Ni + c∗Al. (6.22)
Da die Zahl der Leerstellen im Vergleich zur Zahl der Ni- und Al-Atome gering sein sollte, kann manals Startwerte fur die chemischen Potentiale bei einer bestimmten Zusammensetzung die bereitsermittelten entsprechenden Großen des rein binaren Systems verwenden.Die Bestimmung der fur die SIM notwendigen Strukturen ist fur dieses spezielle System nicht trivial.Fur die Abbildung mit Matrix-Inversion ist essentiell, daß alle 21 inaquivalenten Tetraederkonfi-gurationen im Satz der ausgewahlten Strukturen verwirklicht sind, da ansonsten die Matrix η
¯aus
(6.6) nicht invertierbar ist. Da vorerst die Leerstellen allein in der B2-Phase berucksichtigt werdensollten, wurden, um eine moglichst realistische Beschreibung der Verhaltnisse zu bekommen, dieseTetraederkonfigurationen jeweils in eine 32-atomige B2-Superzelle eingebaut. Genauer gesagt, mankonstruiert eine zuerst ideale B2-Superzelle und fahrt dann auf einem ausgewahlten irregularen Te-traeder die moglichen Leerstellenkonfigurationen durch. Dabei muß aufgrund der Forderung nachKonsistenz in der Durchfuhrung der Cluster-Entwicklung, beispielsweise auch ein, zugegeben un-realistischer, kompletter Leerstellentetraeder ab initio in exakter Weise behandelt werden.Mit dieser Wahl der Referenzkonfigurationen sollte es moglich sein, die lokalen Relaxationen inder Umgebung der Leerstellen auch in den thermodynamischen Betrachtungen im Rahmen derCVM ansatzweise berucksichtigen zu konnen, da die besagten Superzellenkonfigurationen mittelsdes mixed-basis Programmes strukturell relaxiert werden konnen. Unter den 21 Tetraederkonfigu-rationen befinden sich naturlich auch 6 rein binare Konfigurationen. Diese wurden wiederum durchdie im Binaren bereits verwendeten geordneten Strukturen reprasentiert.Diese Cluster-Entwicklung mit dem Ziel der Gewinnung von ECIs zur Beschreibung der B2-Phasemit Leerstellen erscheint aufgrund der typischen geringen Konzentration dieser Defekte in einemKristall (10−4−10−2) wenigstens fur die Tetraeder mit Einzelleerstellen eine annehmbare Naherungder Realitat.Fur die Ermittlung der Korrelationsfunktionen dieser Superzellenkonfigurationen zur Konstruktionder Matrix η
¯wurde ein Computer-Programm entwickelt, da dies von Hand gerade im Ternaren
sehr aufwendig werden kann. Auch die allgemeine 81×81-V-Matrix fur den ternaren Fall wurde miteinem Computer-Algorithmus generiert.Fur die Berechnung der ECIs waren ohne strukturelle Relaxation ingesamt 3 × 15 = 45 Superzel-lenrechnungen erforderlich, da zum Zweck der Ermittlung des Gleichgewichtsvolumens der Defekt-strukturen jeweils 3 Gesamtenergien zu Volumina um das B2-Gleichgewichtsvolumen berechnetwurden. Die Interpolation erfolgte wiederum anhand der Formel (6.2).Im zeitlichen Rahmen der Diplomarbeit konnten diese und die anschließenden CVM-Rechnungenmit Leerstellen jedoch nicht zu einem Abschluß gefuhrt werden. Erst im Laufe der Fertigstellung derZweitabgabe war es moglich, diese Rechnungen zu komplettieren. Dabei konnte sowohl der Triple-Defekt in der Stochiometrie 0.5 als auch eine Stabilisierung der B2-Phase im Ni-armen Bereichnahe dieser Stochiometrie aufgrund von strukturellen Ni-Leerstellen verifiziert werden.

Kapitel 7
Zusammenfassung
Die Berechnung des Phasendiagrammes des technologisch wichtigen intermetallischen Systems NiAlstellt aus folgenden Grunden eine große Herausforderung dar :
1. Da Ni- und Al-Atome stark unterschiedliche Atomradien haben, sollten strukturelle Relaxa-tionseffekte, die i.a. theoretisch sehr schwierig zu erfassen sind, eine wesentliche Rolle spielen.
2. Da im NiAl viele Leerstellen, u.a. auch strukturelle Leerstellen, vorhanden sind, sollte derenEinfluß auf das Phasendiagramm und auf den Ordnungs-Unordnungs-Ubergang wichtig sein.
In der vorliegenden Arbeit wurden diese Fragestellungen ab initio, d.h. (weitgehend) ohne Verwen-dung von Anpaßparametern oder experimentellen Daten, angegangen. Dazu wurde die ab-initioElektronentheorie mit der statistischen Mechanik kombiniert, wobei die Elektronentheorie die Ha-miltonfunktion des Systems liefert und die statistische Mechanik daraus naherungsweise das rele-vante thermodynamische Potential berechnet, das dann die gesamte thermodynamische Informationdes Systems enthalt.Die Hamiltonfunktion wurde in Form der Cluster-Entwicklung dargestellt, bei der die Konfigura-tionsenergie im Prinzip exakt durch Beitrage aller denkbarer Clusterfiguren (Punkt, Paare, Drei-ecke, Tetraeder, usw.) auf einem Grundgitter reprasentiert wird. Die Koeffizienten der Cluster-Entwicklung wurden dabei mit der sogenannten Struktur-Inversionsmethode (SIM) nach Connolly-Williams uber die Energien von speziell ausgewahlten Referenzkonfigurationen bestimmt, die mitder ab-initio Elektronentheorie (Dichtefunktionaltheorie in lokaler Dichtenaherung in Kombinationmit der ab-initio Pseudopotentialmethode) ermittelt wurden.Die Naherung besteht darin, daß man sich in der Cluster-Entwicklung auf relativ kleine Cluster (inder vorliegenden Arbeit maximal Tetraeder) beschrankt. Fur die so bestimmte Hamiltonfunktionwird dann das thermodynamische Potential mit der Cluster-Variationsmethode berechnet, welchedas Potential im Prinzip exakt aus den Besetzungswahrscheinlichkeiten aller denkbarer Clusterbestimmt und somit uber die einfache Bragg-Williams-Naherung hinausgeht. Die Naherung be-steht wieder in einer Beschrankung auf relativ kleine Cluster. In der vorliegenden Arbeit wurdewie fur die Cluster-Entwicklung die Tetraedernaherung verwendet. Die Tetraederbesetzungswahr-scheinlichkeiten wurden durch Minimalisieren des thermodynamischen Potentials bestimmt.Diese ab-initio statistische Mechanik wurde schon von anderen Autoren fur das System NiAl ver-wendet, ohne daß dabei die Relaxationseffekte in zufriedenstellenderweise berucksichtigt und ohnedaß Leerstellen zugelassen wurden. Ziel der Diplomarbeit war es, dieses Verfahren zu implementie-ren, zu testen und erste Schritte zur Beantwortung der beiden oben genannten Fragestellungen zuunternehmen.
109

110 KAPITEL 7. ZUSAMMENFASSUNG
Mit dem implementierten Verfahren wurden die Grundzustandsstrukturen untersucht und die Pha-sendiagramme unter der Annahme bestimmt, daß nur Phasen auf dem bcc- und dem fcc-Grundgittersowie auf beiden Grundgittern gemeinsam moglich sind. Da man aus Experimenten weiß, daß dasSystem NiAl vor der volligen Entordnung schmilzt, mußten die thermodynamischen Eigenschaf-ten der Schmelze durch die einer modifizierten ungeordneten fcc-Phase modelliert werden, wozuexperimentelle Daten verwendet werden mußten. Somit hatte man sich bei der Behandlung derTerminierung des Phasendiagrammes durch die flussige Phase vom reinen ab-initio Verfahren zulosen.Die Ergebnisse fur die Grundzustandsstrukturen erwiesen sich im Einklang mit den experimentel-len Daten. Allein die im experimentellen Phasendiagramm existente Ni3Al5-Struktur konnte nichtals stabile Grundzustandsstruktur verifiziert werden. Es konnte aber gezeigt werden, daß die L10-Struktur im NiAl-System im Grundzustand ideal auf die B2-Struktur relaxiert.Der Vergleich mit anderen elektronentheoretischen Verfahren zeigte, daß in den jeweils gewonne-nen Resultaten zwischen diesen verschiedenen Methoden noch signifikante Unterschiede bestehen.In diesem Zusammenhang erscheint es unerlaßlich, daß zur Beantwortung der hier gestellten Fra-gen im Rahmen der elektronentheoretischen ab-initio Methode mit dem wahren Kristallpotentialgearbeitet wird, wie in der vorliegenden Arbeit durchgefuhrt, da gerade fur die Bestimmung vonEnergieunterscheiden bezuglich zweier verschiedener Kristallstrukturen die Naherung durch einAtomkugelpotential unzureichend ist.
Die Phasendiagramme wurden in einem ersten Schritt ohne Berucksichtigung von Leerstellen be-stimmt. Dabei spiegeln die erhaltenen Diagramme qualitativ die realen Verhaltnisse wieder, wennauch die Topologie und die Ubergangstemperaturen noch nicht vollstandig reproduziert werdenkonnten. Aufgrund der Rechnungen kann im Ni-armen Gebiet eine weitaus schwachere Ordnungkonstatiert werden als in der Ni-reichen Region. Dies geht einher mit der experimentellen Feststel-lung, daß in diesem Gebiet die komplizierten Strukturen D519 und D011 stabil sind. So hat einSystem mit vergleichweise einfachem Grundgitter zwei Moglichkeiten, seine freie Energie abzusen-ken. Zum einen durch Ausbildung einer Untergitterstruktur, was gleichbedeutend mit dem Aufbaueiner Ordnung ist, und zum anderen durch symmetrieandernde Gittertransformationen. Die kom-plexen Strukturen im Ni-armen Gebiet konnen demnach als eine Folge der schwach ausgebildetenOrdnungstendenz in dieser Region gedeutet werden. In den expliziten Rechnungen blieben dieseStrukturen jedoch unberucksichtigt.Die experimentellen Phasen im Ni-reichen Gebiet konnten in den Rechnungen bestatigt werden. Indiesem Zusammenhang wurde klar gezeigt, daß die reinen Phasen B2 und L12 vor der Entordnungin die flussige Phase ubergehen.Die Abweichungen der berechneten von den realen Phasendiagrammen konnen folgende Grundehaben :
a. Die Beschrankung auf die Tetraedernaherung bei der Cluster-Entwicklung [10, 93] und derCluster-Variationsmethode.
b. Eine noch nicht adaquate Beschreibung der Relaxationseffekte. In der vorliegenden Arbeitwurden zwei konkurrierende Relaxationsschemata, lokal und global, verwendet, und es er-geben sich dabei deutliche quantitative Unterschiede. Da beide Schemata die kompliziertenInkompatibilitatseffekte der Mischung von kleinen (Ni) und großen (Al) Atomvolumina nurunzureichend berucksichtigen, ist geplant, diese Effekte im Rahmen der Inkompatibilitatsme-thoden der stochastischen Elastizitatstheorie besser zu erfassen.
c. Die Vernachlassigung der Leerstellen. Im Rahmen der vorliegenden Arbeit wurden ersteSchritte zur Berucksichtigung der Leerstellen unternommen, indem man diese wie eine dritte

111
Komponente mit chemischen Potential Null behandelt. Die Berechnung eines quasiternarenPhasendiagrammes ist erheblich aufwendiger als die eines binaren Phasendiagrammes undkonnte aus Zeitgrunden im Rahmen der Diplomarbeit nicht abgeschlossen werden.
d. Die Vernachlassigung des phononischen Beitrags zur freien Energie.

Anhang A
Zur orthogonalen Entwicklung
A.1 Orthonormalitat und Vollstandigkeit der Tschebyscheff Poly-
nome
Die Bestimmungsgleichung fur die Entwicklungskoeffizienten c(s)k und d
(s)k der Tschebyscheff Poly-
nome (2.7) und (2.8) ist durch die Forderung nach Orthogonalitat gegeben,
〈Θn(σp),Θn′(σp)〉 = ρ(0)N Tr(N)Θn(σp)Θn′(σp)
=1
M
m∑
σp=−mΘn(σp)Θn′(σp)
.= δnn′ . (A.1)
Sofort ersichtlich, gilt fur das Polynom Θ0 allgemein
Θ0 = 1 . (A.2)
In einem ternaren System haben die Polynome beispielsweise folgende Gestalt
Θ0(σp) = 1 , Θ1(σp) = d(0)0 σp , Θ2(σp) = c
(1)0 + c
(1)1 σ2
p ,
σp ∈ −1, 0, 1. Mit (A.1) ergeben sich die Entwicklungskoeffizienten in diesem Fall zu
d(0)0 =
√
3
2, c
(1)0 =
√2 , c
(1)1 = − 3√
2.
Die Vollstandigkeit dieser orthonormalen Basis laßt sich folgendermaßen zeigen. Fur beliebige Kom-binationen von n und n′ laßt sich (A.1) auch als Matrixrelation schreiben :
1
M
Θn(σp) Θn(σ′p) · · · Θn(σ
′′p)
Θn′(σp) · · · · · · Θn′(σ′′p)...
. . .. . .
...Θn′′(σp) · · · · · · Θn′′(σ′′p)
Θn(σp) Θn′(σp) · · · Θn′′(σp)Θn(σ
′p) · · · · · · Θn′′(σ′p)
.... . .
. . ....
Θn(σ′′p) · · · · · · Θn′′(σ′′p)
=
1 0 · · · 00 1 · · · 0...
. . .. . .
...0 · · · · · · 1
.
112

A.2. ORTHONORMALITAT UND VOLLSTANDIGKEIT DER CLUSTERFUNKTIONEN 113
Da die beiden Matrizen auf der linken Seite offensichtlich invers zueinander sind, darf die Reihen-folge der Multiplikation vertauscht werden, ohne daß sich das Ergebnis andert :
Θn(σp) Θn′(σp) · · · Θn′′(σp)Θn(σ
′p) · · · · · · Θn′′(σ′p)
.... . .
. . ....
Θn(σ′′p) · · · · · · Θn′′(σ′′p)
1
M
Θn(σp) Θn(σ′p) · · · Θn(σ
′′p)
Θn′(σp) · · · · · · Θn′(σ′′p)...
. . .. . .
...Θn′′(σp) · · · · · · Θn′′(σ′′p)
=
1 0 · · · 00 1 · · · 0...
. . .. . .
...0 · · · · · · 1
.
Komponentenweise ergibt sich dadurch folgende Relation
1
M
M−1∑
n=0
Θn(σp)Θn(σ′p) = δσpσ′p . (A.3)
Diese Gleichung ist identisch mit der Vollstandigkeitsrelation (2.10).
A.2 Orthonormalitat und Vollstandigkeit der Clusterfunktionen
Um die Relationen (2.13) und (2.14) beweisen zu konnen, ist es hilfreich, zuerst die Orthogonalitatder Tschebyscheff Polynome zu verschiedenen Gitterpunkten p und p′ nachzuvollziehen. Das innereProdukt zweier solcher Polynome schreibt sich als
〈Θn(σp),Θn′(σp′)〉 = ρ(0)N Tr(N)Θn(σp)Θn′(σp′)
=1
M2
m∑
σp=−m
m∑
σ′p=−mΘn(σp)Θn′(σp′) .
Fur den Fall p = p′ reduziert sich dieses Produkt auf (A.1). Ist p 6= p′, so kann man mit (A.2)umformen zu
1
M2
m∑
σp=−m
m∑
σ′p=−mΘn(σp)Θn′(σp′) =
1
M2
m∑
σp=−mΘn(σp)
m∑
σ′p=−mΘ0(σp′)Θn′(σp′)
A.1=
δ0n′
M
m∑
σp=−mΘn(σp)
A.2=
δ0n′
M
m∑
σp=−mΘn(σp)Θ0(σp)
A.1= δn0δn′0 .
Die beiden Falle lassen sich zu einer Gleichung zusammenfassen :
< Θn(σp)Θn′(σp′) >= δpp′δnn′ fur n ≥ 1, n′ ≥ 1 . (A.4)
Die Orthogonalitat beschrankt sich also auf die Polynome n ≥ 1, was jedoch, wie sich zeigen wird,ausreichend ist.Die Clusterfunktion φαs hat nun folgende Gestalt :
φαs(σp, σp′ , . . . , σp′′) = θn(σp)θn′(σp′) · · · θn′′(σp′′) . (A.5)

114 ANHANG A. ZUR ORTHOGONALEN ENTWICKLUNG
Das innere Produkt zweier Clusterfunktionen beliebiger Cluster α und β und beliebigen Grad slautet
〈φαs, φβs′〉 = ρ(0)N Tr(N)φαsφβs′
∼m∑
σp=−mp∈α
· · ·m∑
σq=−mq∈α
m∑
σp′
=−m
p′∈β
· · ·m∑
σq′
=−m
q′∈β
φnα(σp) · · · φn′′α(σq)φnβ
(σp′) · · · φn′′β(σq′) .
Es sind nun 3 Falle zu unterscheiden
1. α und β besitzen keine gemeinsamen Gitterpunkte:Dann verschwindet das innere Produkt aufgrund des Kroneckerdelta δpp′ in (A.4).
2. α und β besitzen einige gemeinsame Gitterpunkte:Wiederum verschwindet das Produkt, da ein δpp′ = 0 hierfur ausreicht.
3. α und β sind identisch:Das Produkt liefert die Zahl 1, wenn der Grad der Funktionen ebenfalls identisch ist (A.4).
Damit ist die Orthonormalitatsrelation (2.13) bewiesen.Die Vollstandigkeitsrelation (2.14) kann auf die gleiche Weise wie (A.3) bewiesen werden. Die Ma-trizen haben dann die Dimension M |α|, und die Komponenten werden durch die verschiedenenClusterfunktionen φαs reprasentiert.Daß sich die Orthogonalitat nurmehr uber die Polynome n ≥ 1 erstreckt, bedeutet keinen Nachteil,da das Polynom fur n = 0 per Definition gleich 1 ist (A.2). Demzufolge ”verschwindet” fur diesenWert von n einfach eine Gitterpunktabhangigkeit in (A.5). Die resultierende Clusterfunktion istsomit einfach die des Clusters, bestehend aus den |α|−1 verbleibenden Gitterpunkten. Mit anderenWorten, wahrend sich die Zahl der σp-abhangigen Clusterfunktionen bei erweiterter Gitterpunkt-zahl naturlich gemaß dem direkten Produkt-Formalismus erhohen muß, reicht auch weiterhin eineeinzelne σp-unabhangige Clusterfunktion aus.

Anhang B
CVM-Naherungen
Abbildung B.1: Eine Auswahl von maximalen Cluster fur das sc-, bcc- und fcc-Gitter hinsichtlicheiner im Rahmen der Naherung moglichst erfolgreichen physikalischen Beschreibung (nach [38]).Der Koeffizient p indiziert den maximalen Abstand von Gitterpunkten im maximalen Cluster.
115

Anhang C
Strukturen
An dieser Stelle sollen die im Rahmen dieser Arbeit betrachteten Strukturen dargestellt werden.Ein A-Atom hat die Farbe Rot, ein B-Atom ist grun gezeichnet. Die Einheitszellenvektoren sindin kartesischen Koordinaten, die Koordinaten der Basisatome bezuglich den Einheitszellenvektorenangegeben. Die Charakterisierung der Raumgruppe erfolgt durch die Schonflies-Notation sowie dasPearsonsymbol.
C.1 fcc-Gitter
fcc
Formel : A
Bravaisgitter : face-centered cubic
EZ-vektoren :(0, 1, 1)(1, 0, 1)(1, 1, 0)
Basisatome A : (0, 0, 0)
B : −Raumgruppe : O5
h , cF4
116
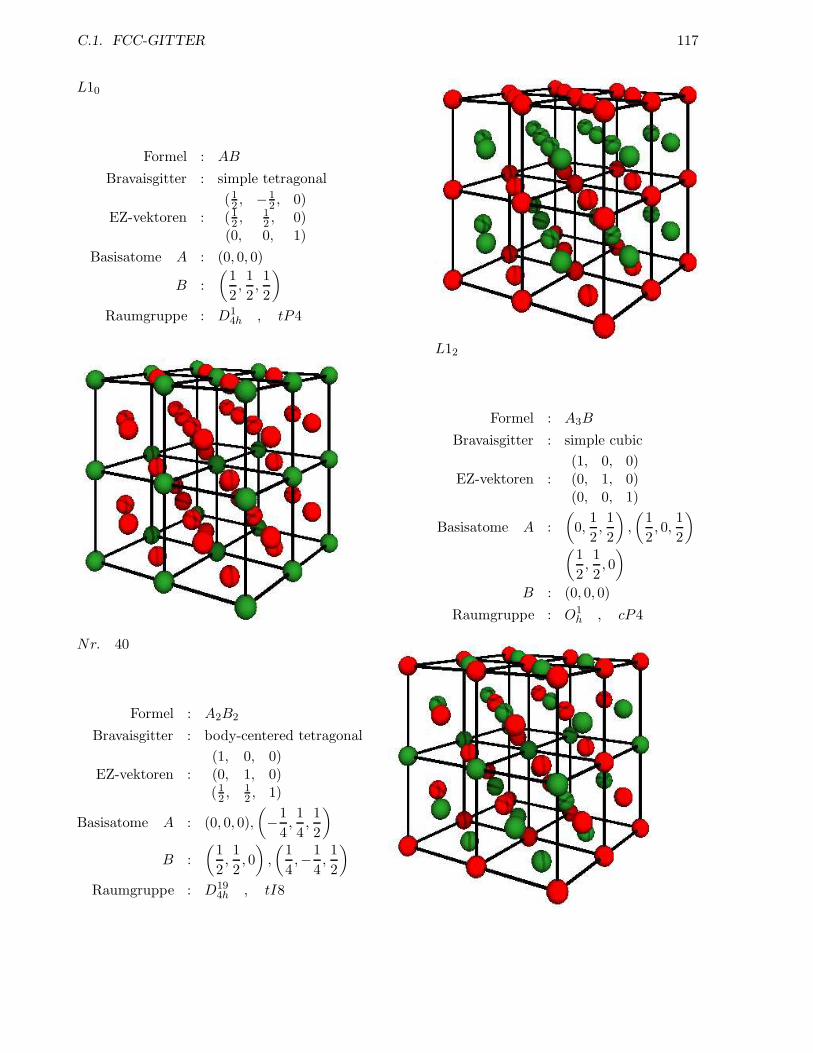
C.1. FCC-GITTER 117
L10
Formel : AB
Bravaisgitter : simple tetragonal
EZ-vektoren :(12 , −1
2 , 0)(12 ,
12 , 0)
(0, 0, 1)
Basisatome A : (0, 0, 0)
B :
(1
2,1
2,1
2
)
Raumgruppe : D14h , tP4
L12
Formel : A3B
Bravaisgitter : simple cubic
EZ-vektoren :(1, 0, 0)(0, 1, 0)(0, 0, 1)
Basisatome A :
(
0,1
2,1
2
)
,
(1
2, 0,
1
2
)
(1
2,1
2, 0
)
B : (0, 0, 0)
Raumgruppe : O1h , cP4
Nr. 40
Formel : A2B2
Bravaisgitter : body-centered tetragonal
EZ-vektoren :(1, 0, 0)(0, 1, 0)(12 ,
12 , 1)
Basisatome A : (0, 0, 0),
(
−1
4,1
4,1
2
)
B :
(1
2,1
2, 0
)
,
(1
4,−1
4,1
2
)
Raumgruppe : D194h , tI8
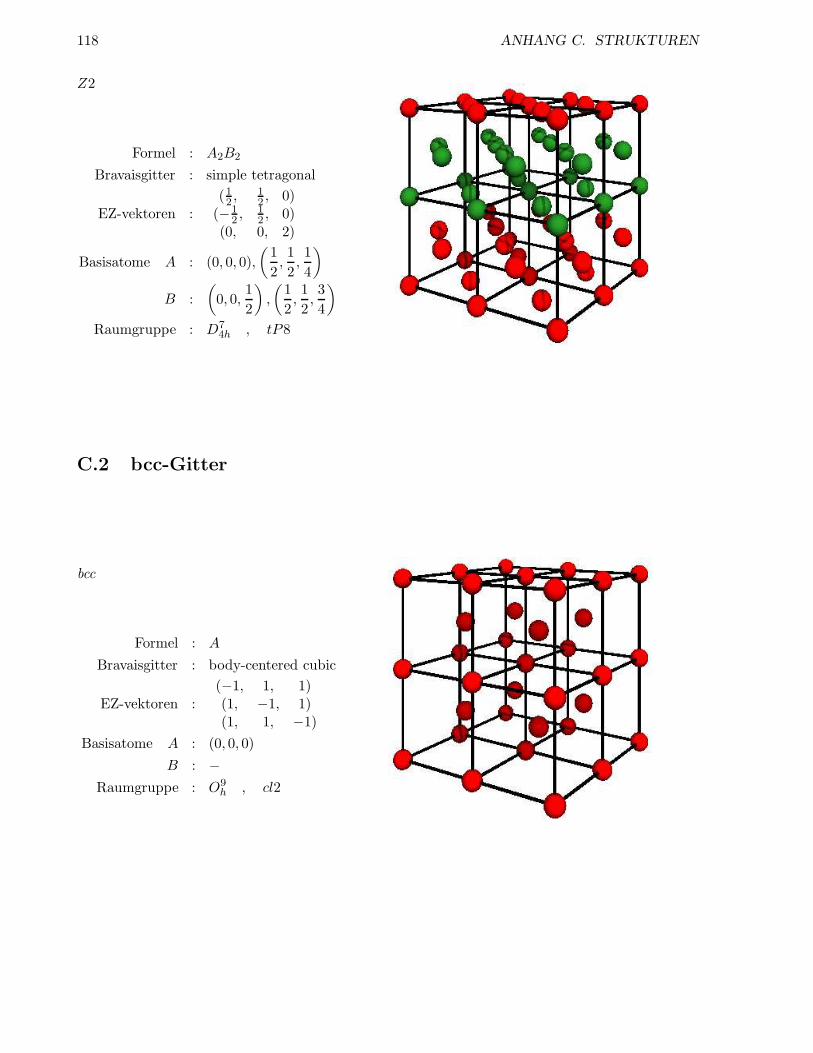
118 ANHANG C. STRUKTUREN
Z2
Formel : A2B2
Bravaisgitter : simple tetragonal
EZ-vektoren :(12 ,
12 , 0)
(−12 ,
12 , 0)
(0, 0, 2)
Basisatome A : (0, 0, 0),
(1
2,1
2,1
4
)
B :
(
0, 0,1
2
)
,
(1
2,1
2,3
4
)
Raumgruppe : D74h , tP8
C.2 bcc-Gitter
bcc
Formel : A
Bravaisgitter : body-centered cubic
EZ-vektoren :(−1, 1, 1)(1, −1, 1)(1, 1, −1)
Basisatome A : (0, 0, 0)
B : −Raumgruppe : O9
h , cl2
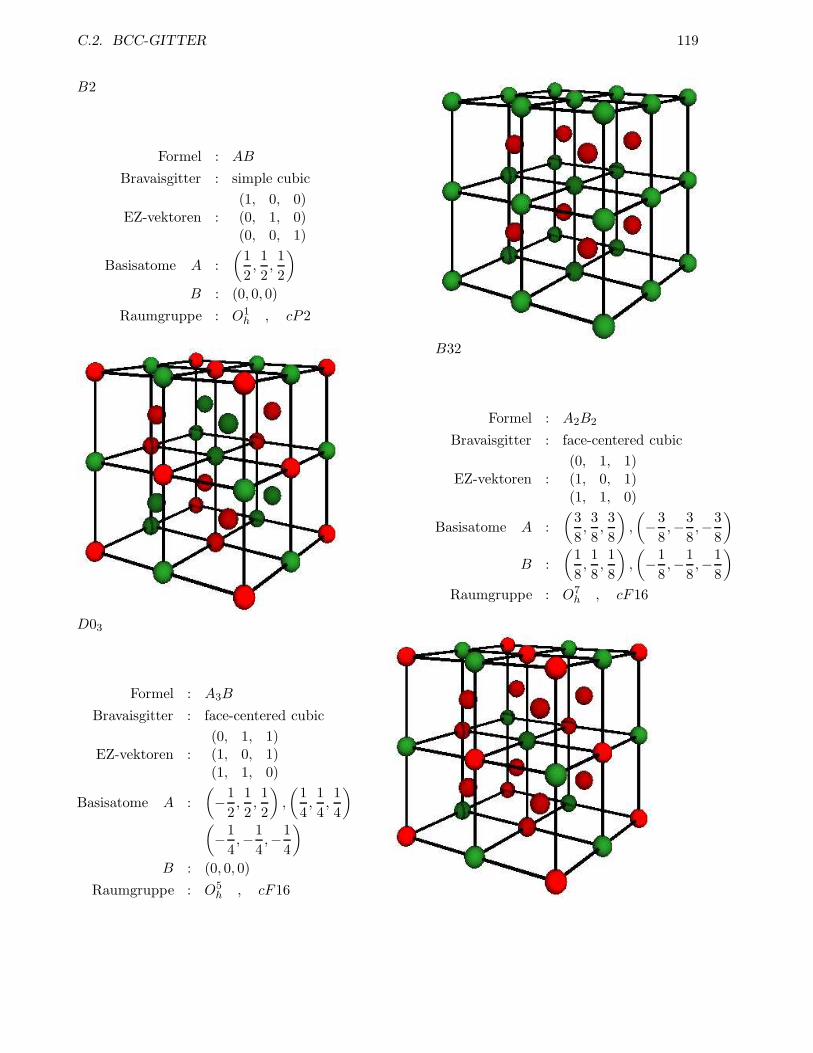
C.2. BCC-GITTER 119
B2
Formel : AB
Bravaisgitter : simple cubic
EZ-vektoren :(1, 0, 0)(0, 1, 0)(0, 0, 1)
Basisatome A :
(1
2,1
2,1
2
)
B : (0, 0, 0)
Raumgruppe : O1h , cP2
B32
Formel : A2B2
Bravaisgitter : face-centered cubic
EZ-vektoren :(0, 1, 1)(1, 0, 1)(1, 1, 0)
Basisatome A :
(3
8,3
8,3
8
)
,
(
−3
8,−3
8,−3
8
)
B :
(1
8,1
8,1
8
)
,
(
−1
8,−1
8,−1
8
)
Raumgruppe : O7h , cF16
D03
Formel : A3B
Bravaisgitter : face-centered cubic
EZ-vektoren :(0, 1, 1)(1, 0, 1)(1, 1, 0)
Basisatome A :
(
−1
2,1
2,1
2
)
,
(1
4,1
4,1
4
)
(
−1
4,−1
4,−1
4
)
B : (0, 0, 0)
Raumgruppe : O5h , cF16
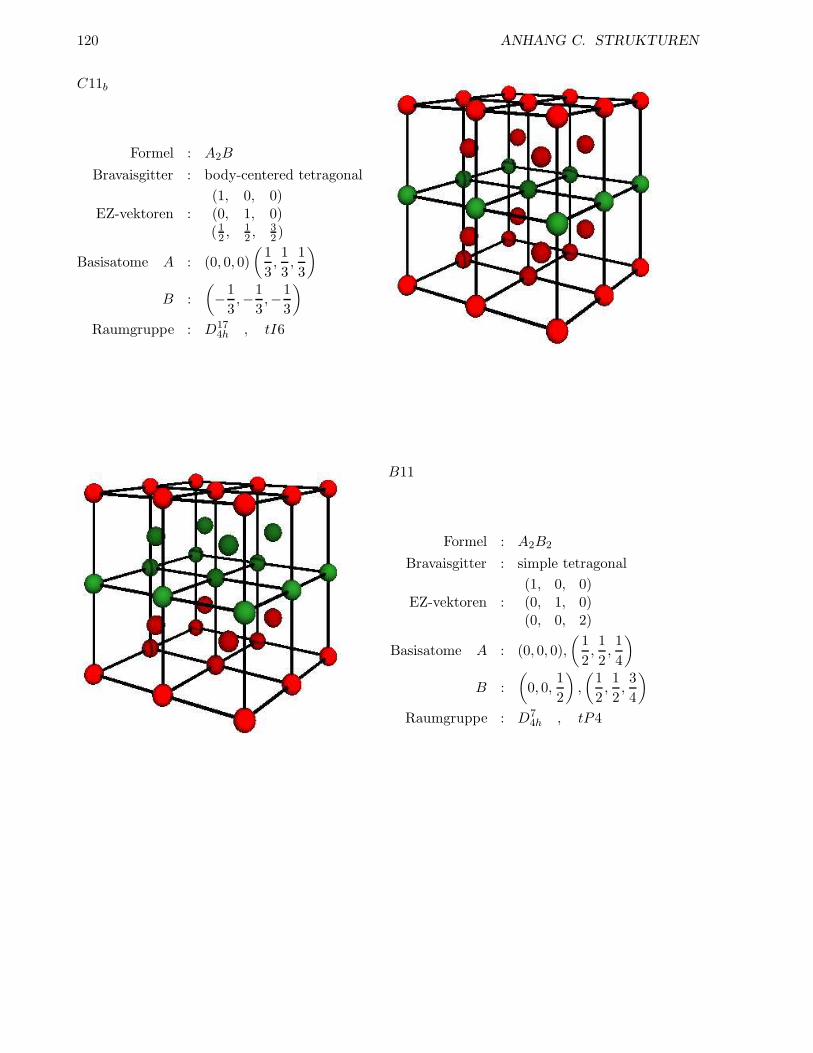
120 ANHANG C. STRUKTUREN
C11b
Formel : A2B
Bravaisgitter : body-centered tetragonal
EZ-vektoren :(1, 0, 0)(0, 1, 0)(12 ,
12 ,
32 )
Basisatome A : (0, 0, 0)
(1
3,1
3,1
3
)
B :
(
−1
3,−1
3,−1
3
)
Raumgruppe : D174h , tI6
B11
Formel : A2B2
Bravaisgitter : simple tetragonal
EZ-vektoren :(1, 0, 0)(0, 1, 0)(0, 0, 2)
Basisatome A : (0, 0, 0),
(1
2,1
2,1
4
)
B :
(
0, 0,1
2
)
,
(1
2,1
2,3
4
)
Raumgruppe : D74h , tP4
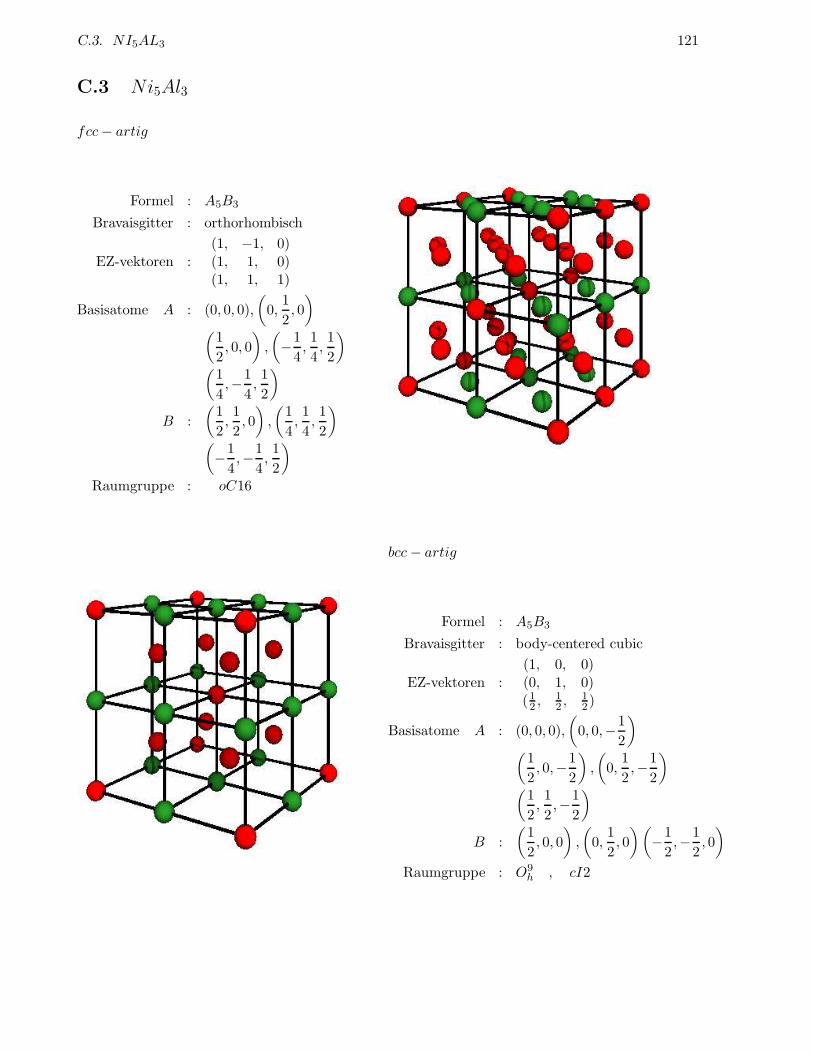
C.3. NI5AL3 121
C.3 Ni5Al3
fcc− artig
Formel : A5B3
Bravaisgitter : orthorhombisch
EZ-vektoren :(1, −1, 0)(1, 1, 0)(1, 1, 1)
Basisatome A : (0, 0, 0),
(
0,1
2, 0
)
(1
2, 0, 0
)
,
(
−1
4,1
4,1
2
)
(1
4,−1
4,1
2
)
B :
(1
2,1
2, 0
)
,
(1
4,1
4,1
2
)
(
−1
4,−1
4,1
2
)
Raumgruppe : oC16
bcc− artig
Formel : A5B3
Bravaisgitter : body-centered cubic
EZ-vektoren :(1, 0, 0)(0, 1, 0)(12 ,
12 ,
12)
Basisatome A : (0, 0, 0),
(
0, 0,−1
2
)
(1
2, 0,−1
2
)
,
(
0,1
2,−1
2
)
(1
2,1
2,−1
2
)
B :
(1
2, 0, 0
)
,
(
0,1
2, 0
)(
−1
2,−1
2, 0
)
Raumgruppe : O9h , cI2

Anhang D
Prototyp-Rechnungen
Die Funktionsfahigkeit des im Rahmen dieser Arbeit entwickelten CVM-Programms wurde anPrototyp-Systemen getestet. Ein solches ist durch das Grundgitter und die Art (z.B. Paar-), Reich-weite (z.B. NN oder NNN) sowie die Große der Wechselwirkung(en) definiert. Der Test war erfor-derlich, da die Numerik der NIM nicht unproblematisch ist und eine Vielzahl von rein numerischenFragestellungen zuerst geklart werden mußte. Auf Details dieser rein technischen Problematik sollan dieser Stelle nicht weiter eingegangen werden.Die Qualitat der in diesem Abschnitt vorgestellten Diagramme ist zum Teil nicht so hoch wie die furdie Diagramme des NiAl-System. Dies ist jedoch keine Schwache des Programms, sondern kommtdaher, daß bei ausreichender Wiedergabe der bereits bekannten Resultate nicht versucht wurde, dieGenauigkeit noch hoher und die Auflosung noch feiner zu wahlen. Eine vollstandige Diskussion dervielfaltigen Prototyp-Systeme hatte den Rahmen dieser Arbeit gesprengt. Die Physik der Prototyp-Systeme ist ein wichtiger Aspekt bei der Klarung grundlegender Fragestellungen und aufgrund desUmfangs ein Thema fur sich. Fur eine ausfuhrliche Diskussion dieses Gebiets sei auf das Buch vonDucastelle und den dortigen Zitaten verwiesen [33].In den folgenden beiden Abschnitten ist nun eine Auswahl der durchgefuhrten Testrechnungendargestellt.
D.1 fcc-Gitter
D.1.1 Ising-Modell
Die Abbildung (D.1) zeigt den Anteil an Spinpaaren mit antiparalleler Spinausrichtung auf einemfcc-Gitter mit ferromagnetischer NN-Wechselwirkung (K2 < 0) in Abhangigkeit von der Tem-peratur. Die Temperatur ist dabei, wie bei Prototyp-Systemen ublich, in reduzierten Einheitenangegeben. Die reduzierte Temperatur τ = kBT/K2 ist einheitenlos. Wahrend die energetischeWechselwirkung durch die Systemwahl vorgegeben ist, kann der Entropieterm bei Behandlung mitder CVM durch die Wahl des maximalen Clusters in verschiedener Weise genahert werden. In derAbbildung wurde als maximaler Cluster einmal das NN-Paar und ein andermal der regulare Tetra-eder bestehend aus NN-Paaren gewahlt.In der Literatur wird der Wert fur die Ubergangstemperatur als τ = kBTc/12K2 skaliert (Tab.D.1). In den hier durchgefuhrten Rechnungen wurde τ in Schritten von 0.001 durchgefahren. Dabeiergab sich fur die Paarnaherung ein τ von 0.91417. Die Tetraedernaherung lieferte τ = 0.8355. AmUbergangspunkt waren bis zur Konvergenz der NIM 3 · 105 Iterationen notig.
122
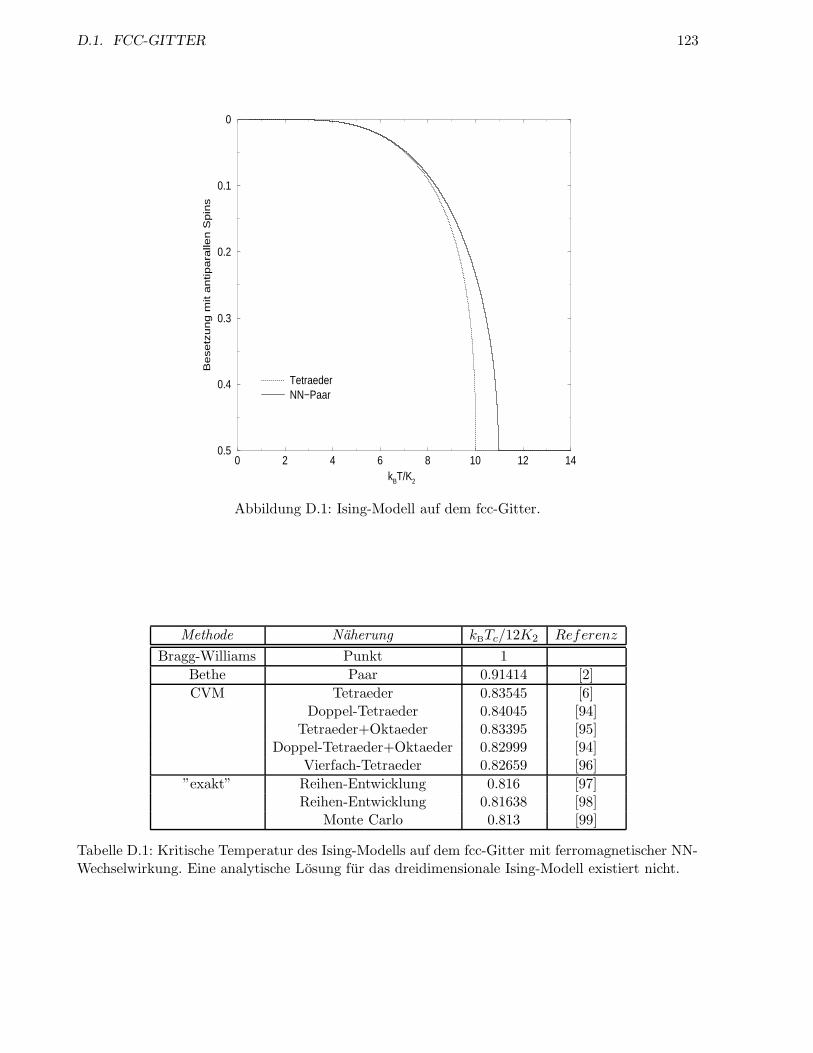
D.1. FCC-GITTER 123
0 2 4 6 8 10 12 14kBT/K2
0
0.1
0.2
0.3
0.4
0.5
Be
se
tzu
ng
mit a
ntip
ara
llen
Sp
ins
TetraederNN−Paar
Abbildung D.1: Ising-Modell auf dem fcc-Gitter.
Methode Naherung kBTc/12K2 Referenz
Bragg-Williams Punkt 1
Bethe Paar 0.91414 [2]
CVM Tetraeder 0.83545 [6]Doppel-Tetraeder 0.84045 [94]
Tetraeder+Oktaeder 0.83395 [95]Doppel-Tetraeder+Oktaeder 0.82999 [94]
Vierfach-Tetraeder 0.82659 [96]
”exakt” Reihen-Entwicklung 0.816 [97]Reihen-Entwicklung 0.81638 [98]
Monte Carlo 0.813 [99]
Tabelle D.1: Kritische Temperatur des Ising-Modells auf dem fcc-Gitter mit ferromagnetischer NN-Wechselwirkung. Eine analytische Losung fur das dreidimensionale Ising-Modell existiert nicht.
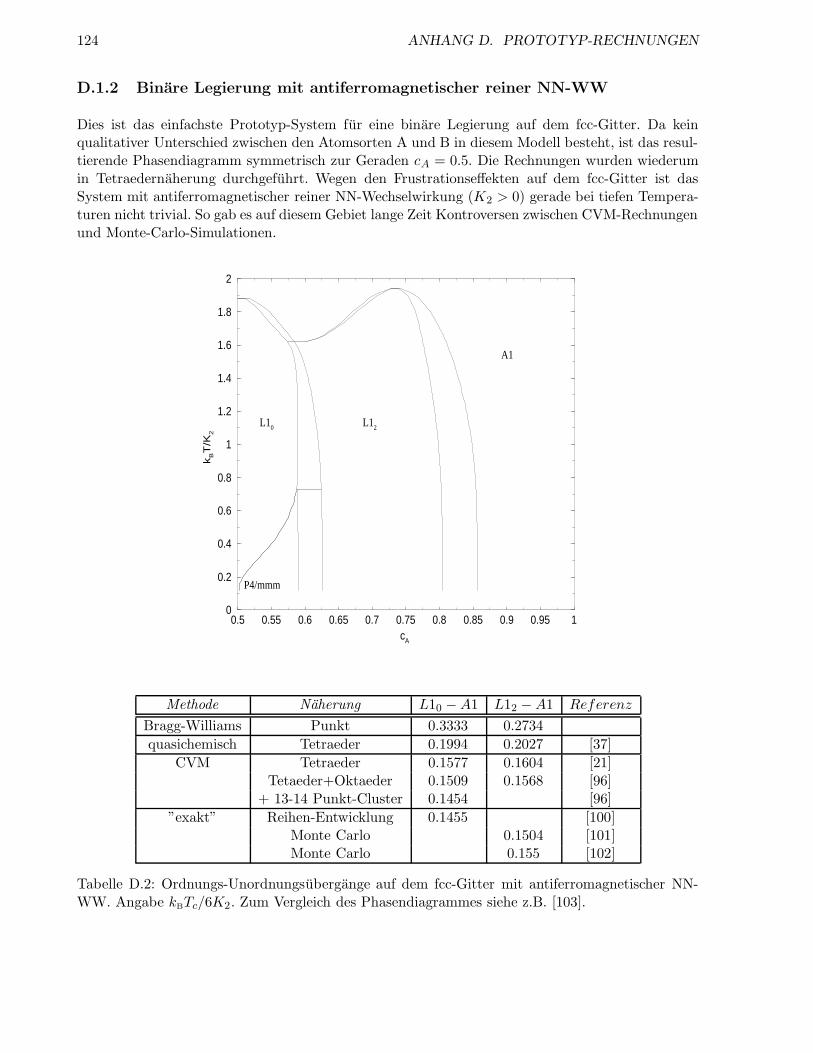
124 ANHANG D. PROTOTYP-RECHNUNGEN
D.1.2 Binare Legierung mit antiferromagnetischer reiner NN-WW
Dies ist das einfachste Prototyp-System fur eine binare Legierung auf dem fcc-Gitter. Da keinqualitativer Unterschied zwischen den Atomsorten A und B in diesem Modell besteht, ist das resul-tierende Phasendiagramm symmetrisch zur Geraden cA = 0.5. Die Rechnungen wurden wiederumin Tetraedernaherung durchgefuhrt. Wegen den Frustrationseffekten auf dem fcc-Gitter ist dasSystem mit antiferromagnetischer reiner NN-Wechselwirkung (K2 > 0) gerade bei tiefen Tempera-turen nicht trivial. So gab es auf diesem Gebiet lange Zeit Kontroversen zwischen CVM-Rechnungenund Monte-Carlo-Simulationen.
0.5 0.55 0.6 0.65 0.7 0.75 0.8 0.85 0.9 0.95 1cA
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
kBT
/K2
L10 L12
A1
P4/mmm
Methode Naherung L10 −A1 L12 −A1 Referenz
Bragg-Williams Punkt 0.3333 0.2734
quasichemisch Tetraeder 0.1994 0.2027 [37]
CVM Tetraeder 0.1577 0.1604 [21]Tetaeder+Oktaeder 0.1509 0.1568 [96]
+ 13-14 Punkt-Cluster 0.1454 [96]
”exakt” Reihen-Entwicklung 0.1455 [100]Monte Carlo 0.1504 [101]Monte Carlo 0.155 [102]
Tabelle D.2: Ordnungs-Unordnungsubergange auf dem fcc-Gitter mit antiferromagnetischer NN-WW. Angabe kBTc/6K2. Zum Vergleich des Phasendiagrammes siehe z.B. [103].

D.2. BCC-GITTER 125
D.2 bcc-Gitter
D.2.1 Binare Legierung mit antiferromagnetischer NN- und NNN-WW
Auf dem bcc-Gitter enthalt der kleinste dreidimensionalen Cluster, der irregularen Tetraeder, so-wohl Gitterpunkte mit NN-Abstand als auch solche mit NNN-Abstand. Die Prototyp-Systeme sinddaher meist solche mit NN- und NNN-Wechselwirkung [86, 66]. Die verschiedenen zugehorigenPhasendiagramme werden allgemein durch das Verhaltnis ε = K2,2/K2,1 von NNN- zu NN-WW(beide antiferromagnetisch) charakterisiert. Die Struktur F 43m wurde in der Arbeit von Sluiter etal. [86] nicht berucksichtigt, weshalb die Diagramme zu ε = 1.0 verschieden ausfallen. Rubin [104]hat diese Phase jedoch fur ε = 1.0 berucksichtigt und kommt zu dem Schluß, daß die F 43m-Phasezwischen der B32- und D03-Phase liegt. Weiter sollen beide Phasenubergange zu den angrenzendenPhasen 2. Ordnung sein. Diese Aussagen konnen im Rahmen der Genauigkeit der durchgefuhrtenRechnungen bestatigt werden.
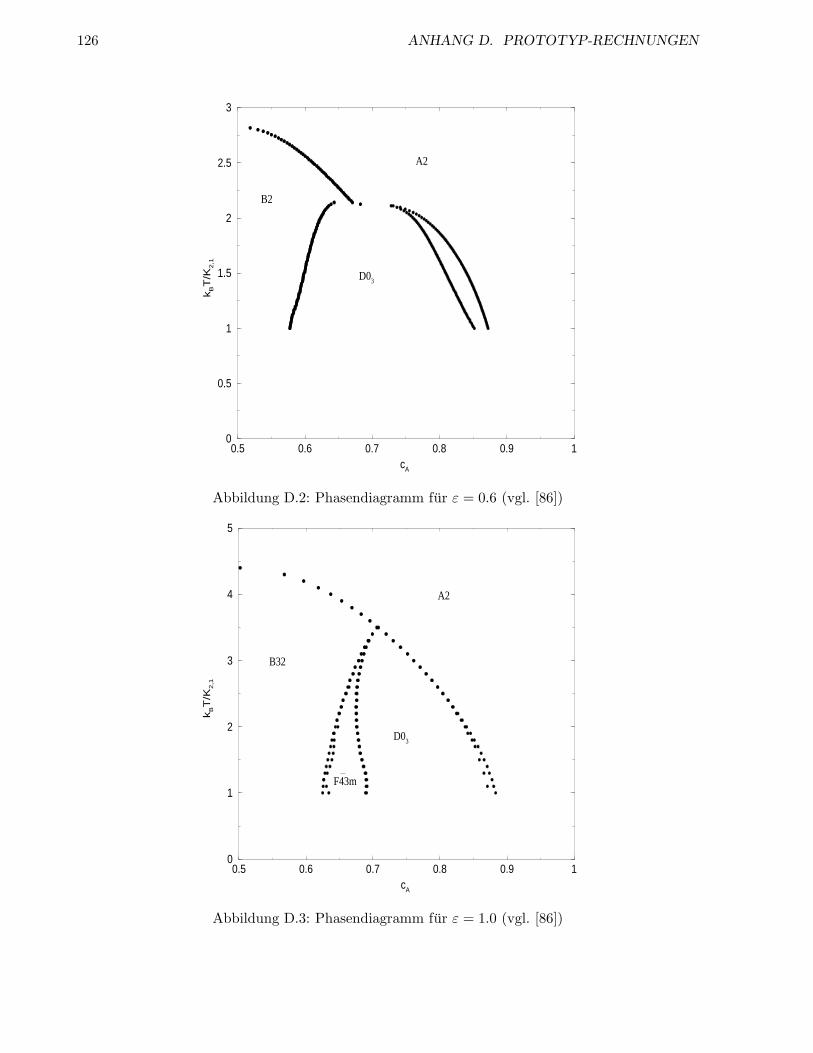
126 ANHANG D. PROTOTYP-RECHNUNGEN
0.5 0.6 0.7 0.8 0.9 1cA
0
0.5
1
1.5
2
2.5
3
kBT
/K2
,1
B2
A2
D03
Abbildung D.2: Phasendiagramm fur ε = 0.6 (vgl. [86])
0.5 0.6 0.7 0.8 0.9 1cA
0
1
2
3
4
5
kBT
/K2
,1
B32
A2
D03
F43m
Abbildung D.3: Phasendiagramm fur ε = 1.0 (vgl. [86])

Anhang E
Zur Phasenstabilitat im(µ∗A, Ω)−Formalismus
In Abschnitt 3.2.2 wurde anhand graphischer Betrachtungen gezeigt, daß bei Vorgabe des effektivenchemischen Potentials µ∗
A das mittels einer allgemeinen Legendre-Transformation aus der freienEnthalpie G hervorgehende effektive große Potential Ω fur die Diskussion der thermodynamischenStabilitat bezuglich konkurrierender Phasen ausreichend ist. An dieser Stelle soll hierfur nun einealternative analytische Begrundung, aufbauend auf einer Taylor-Entwicklung der freien Enthalpieg pro Gitterpunkt, gegeben werden.In Abschnitt 3.1.2 wurde fur Ω folgender Ausdruck hergeleitet :
Ω = g − µ∗AcA − µ∗BcB .
Mit cA + cB = 1 und (3.18) laßt sich dies auch schreiben als
Ω = g − 2µ∗AcA + µ∗A . (E.1)
Im folgenden sollen nun o.B.d.A zwei Phasen ψ1 und ψ2 betrachtet werden (Abb. (E.1)). UnterVorgabe von µ∗A, T und p stelle sich im thermodynamischen Gleichgewicht fur ψ1 die Konzentration
c(1)A und fur ψ2 die Konzentration c
(2)A ein. Wiederum o.B.d.A gelte gψ1
(
c(1)A
)
< gψ2
(
c(1)A
)
, d.h. bei
c(1)A soll die Phase ψ1 thermodynamisch stabil sein.
Die zugehorigen effektiven großen Potential schreiben sich mittels (E.1) dann als
Ωψ1(µ∗A) = gψ1
(
c(1)A
)
− 2µ∗Ac(1)A + µ∗A , (E.2)
Ωψ2(µ∗A) = gψ2
(
c(2)A
)
− 2µ∗Ac(2)A + µ∗A . (E.3)
Die Differenz dieser beiden Potentiale zu den verschiedenen Phasen ergibt sich zu
Ωψ1(µ∗A) − Ωψ2(µ∗A) = gψ1
(
c(1)A
)
− gψ2
(
c(2)A
)
− 2µ∗A(
c(1)A − c
(2)A
)
. (E.4)
Die entscheidende Frage ist nun, ob folgender Schluß gilt :
Ωψ1(µ∗A) < Ωψ2(µ∗A) ⇒ gψ1
(
c(1)A
)
< gψ2
(
c(1)A
)
?
Zur Beantwortung dieser Frage soll eine Taylor-Entwicklung der Große gψ2
(
c(1)A
)
um die Konzen-
tration c(2)A erfolgen :
gψ2
(
c(1)A
)
= gψ2
(
c(2)A
)
+∂gψ2
∂cA
∣∣∣∣∣c(2)A
(
c(1)A − c
(2)A
)
+1
2
∂2gψ2
∂c2A
∣∣∣∣∣c(2)A
(
c(1)A − c
(2)A
)2. (E.5)
127
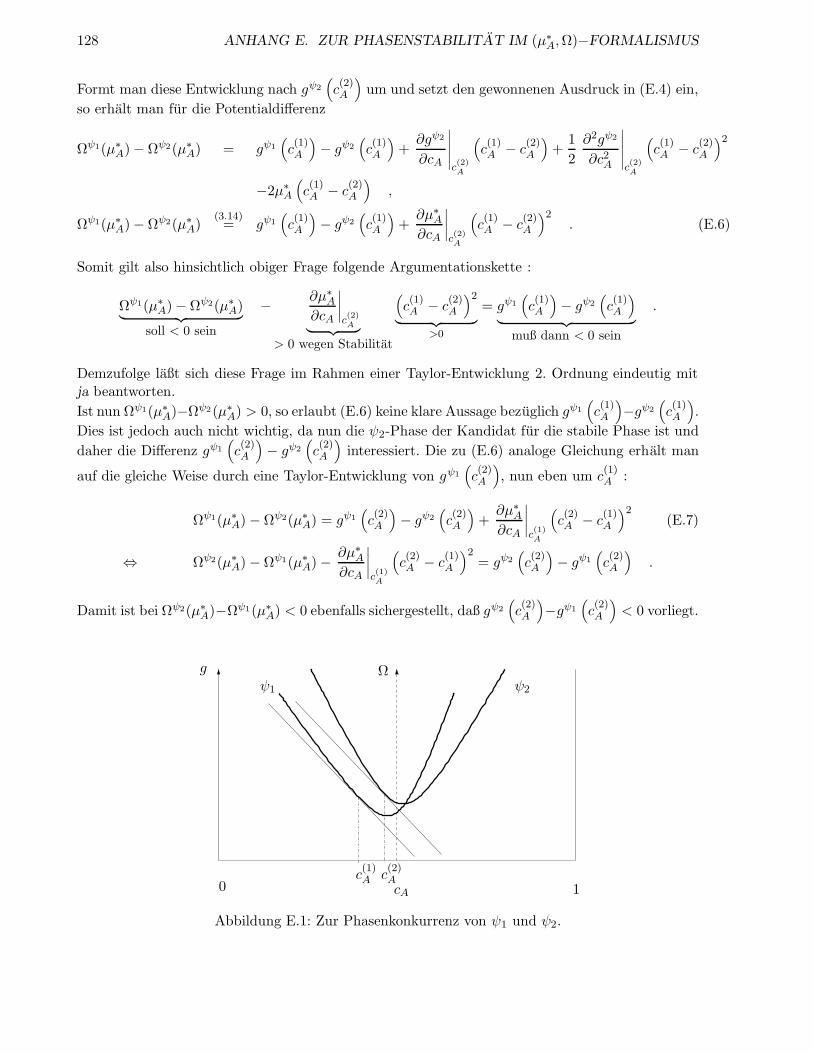
128 ANHANG E. ZUR PHASENSTABILITAT IM (µ∗A,Ω)−FORMALISMUS
Formt man diese Entwicklung nach gψ2
(
c(2)A
)
um und setzt den gewonnenen Ausdruck in (E.4) ein,
so erhalt man fur die Potentialdifferenz
Ωψ1(µ∗A) − Ωψ2(µ∗A) = gψ1
(
c(1)A
)
− gψ2
(
c(1)A
)
+∂gψ2
∂cA
∣∣∣∣∣c(2)A
(
c(1)A − c
(2)A
)
+1
2
∂2gψ2
∂c2A
∣∣∣∣∣c(2)A
(
c(1)A − c
(2)A
)2
−2µ∗A(
c(1)A − c
(2)A
)
,
Ωψ1(µ∗A) − Ωψ2(µ∗A)(3.14)= gψ1
(
c(1)A
)
− gψ2
(
c(1)A
)
+∂µ∗A∂cA
∣∣∣∣c(2)A
(
c(1)A − c
(2)A
)2. (E.6)
Somit gilt also hinsichtlich obiger Frage folgende Argumentationskette :
Ωψ1(µ∗A) − Ωψ2(µ∗A)︸ ︷︷ ︸
soll < 0 sein
− ∂µ∗A∂cA
∣∣∣∣c(2)A
︸ ︷︷ ︸
> 0 wegen Stabilitat
(
c(1)A − c
(2)A
)2
︸ ︷︷ ︸
>0
= gψ1
(
c(1)A
)
− gψ2
(
c(1)A
)
︸ ︷︷ ︸
muß dann < 0 sein
.
Demzufolge laßt sich diese Frage im Rahmen einer Taylor-Entwicklung 2. Ordnung eindeutig mitja beantworten.
Ist nun Ωψ1(µ∗A)−Ωψ2(µ∗A) > 0, so erlaubt (E.6) keine klare Aussage bezuglich gψ1
(
c(1)A
)
−gψ2
(
c(1)A
)
.
Dies ist jedoch auch nicht wichtig, da nun die ψ2-Phase der Kandidat fur die stabile Phase ist und
daher die Differenz gψ1
(
c(2)A
)
− gψ2
(
c(2)A
)
interessiert. Die zu (E.6) analoge Gleichung erhalt man
auf die gleiche Weise durch eine Taylor-Entwicklung von gψ1
(
c(2)A
)
, nun eben um c(1)A :
Ωψ1(µ∗A) − Ωψ2(µ∗A) = gψ1
(
c(2)A
)
− gψ2
(
c(2)A
)
+∂µ∗A∂cA
∣∣∣∣c(1)A
(
c(2)A − c
(1)A
)2(E.7)
⇔ Ωψ2(µ∗A) − Ωψ1(µ∗A) − ∂µ∗A∂cA
∣∣∣∣c(1)A
(
c(2)A − c
(1)A
)2= gψ2
(
c(2)A
)
− gψ1
(
c(2)A
)
.
Damit ist bei Ωψ2(µ∗A)−Ωψ1(µ∗A) < 0 ebenfalls sichergestellt, daß gψ2
(
c(2)A
)
−gψ1
(
c(2)A
)
< 0 vorliegt.
PSfrag replacements
cAc(1)A c
(2)A
ψ1 ψ2
Ωg
0 1
Abbildung E.1: Zur Phasenkonkurrenz von ψ1 und ψ2.

Anhang F
T − µ∗A−Diagramme fur NiAl
In Abschnitt 6.2.4 wurde das vollstandige NiAl-Phasendiagramm im Rahmen der Cluster-Entwicklungund CVM in Tetraedernaherung mit zwei verschiedenen Volumenrelaxationsschemata vorgestellt.Neben der dort gewahlten T − cNi−Auftragung lassen sich diese Diagramme auch mit dem effek-tiven chemischen Potential µ∗
Ni als Ordinate darstellen. In dieser Auftragung treten nun naturlichkeine Mischphasen mehr auf, da µ∗
Ni bekanntlich uber einen solchen Mischphasenbereich hinwegkonstant ist.Die beiden Diagramme (F.1) und (F.2) unterscheiden sich wiederum in dem in der zugehorigenRechnung gewahlten Volumenrelaxationsschemata.Die Dominanz der B2-Struktur ist in dieser Darstellung im Gegensatz zu der in Abschnitt 6.2.4gewahlten gewohnten T − cA−Auftragung in beiden Diagrammen deutlich zu erkennen.Die Unterschiede in den Parametersatzen haben dabei offensichtlich nahezu ausschließlich eine Ver-schiebung des effektiven chemischen Potentials zur Folge. Der Einfluß der gewahlten Differenz inden Konvergenzparametern auf die fur die Topologie der Diagramme verantwortlichen Kopplungenist anscheinend gering.
129
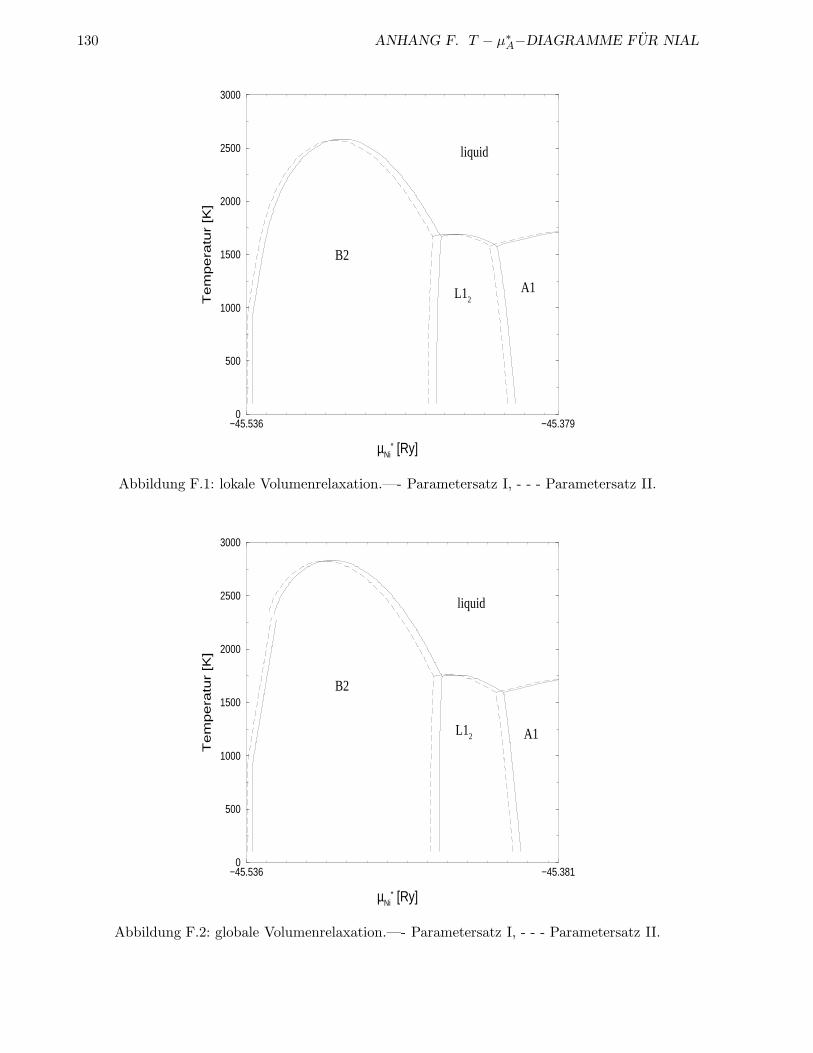
130 ANHANG F. T − µ∗A−DIAGRAMME FUR NIAL
−45.536 −45.379
µNi* [Ry]
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
B2
L12
liquid
A1
Abbildung F.1: lokale Volumenrelaxation.—- Parametersatz I, - - - Parametersatz II.
−45.536 −45.381
µNi* [Ry]
0
500
1000
1500
2000
2500
3000
Tem
pera
tur
[K]
liquid
A1
B2
L12
Abbildung F.2: globale Volumenrelaxation.—- Parametersatz I, - - - Parametersatz II.

Literaturverzeichnis
[1] W. L. Bragg und E. J. Williams, Proc. Roy. Soc. (London) A 145, 699 (1934).
[2] H. A. Bethe, Proc. Roy. Soc. (London) A 150, 552 (1935).
[3] C. N. Yang, J. Chem. Phys. 13(2), 66 (1945).
[4] C. N. Yang und Y. Y. Li, Chinese J. Phys. 7, 59 (1947).
[5] Y. Y. Li, J. Chem. Phys. 17(5), 447 (1949).
[6] R. Kikuchi, Phys. Rev. 81(6), 988 (1951).
[7] R. Car und M. Parrinello, Phys. Rev. Lett. 55, 2471 (1985).
[8] J. W. D. Connolly und A. R. Williams, Phys. Rev. B 27, 5169 (1983).
[9] B. Meyer, C. Elsasser und M. Fahnle, Fortran 90 Program for Mixed-Basis PseudopotentialCalculations for Crystals, Max-Planck-Institut fur Metallforschung, Stuttgart.
[10] Z. W. Lu, S. H. Wei, A. Zunger, S. Frota-Pressoa und L. G. Ferreira, Phys. Rev. B 44(2),512 (1991).
[11] J. M. Sanchez, In Structural and Phase Stability of Alloys (1992).
[12] C. Colinet, P. Hicter und A. Pasturel, Phys. Rev. B 45(4) (1992).
[13] A. Pasturel, C. Colinet, A. T. Paxton und M. van Schilfgaarde, J. Phys.: Condens. Matter4, 945 (1992).
[14] M. Sluiter und P. E. A. Turchi, In Statics and Dynamics of Alloy Phase Transformations(1993).
[15] T. Shinoda, H. Hosoda und Y. Mishima, Materials Science and Engineering 192/193, 930(1995).
[16] P. Nash, M. F. Singleton und J. L. Murray, In Phase Diagrams of Binary Nickel Alloys, ASMInt’l., Materials Park, OH (1991).
[17] H. Okamoto, J. Phase Equil. 14, 257 (1993).
[18] J. A. Barker, Proc. R. Soc. A 216, 45 (1953).
[19] J. Hijmans und J. D. Boer, Physica A 21, 471, 485, 499 (1955).
[20] T. Morita, J. Math. Phys. 13(1), 115 (1972).
131

132 LITERATURVERZEICHNIS
[21] C. M. van Baal, Physica A 64, 571 (1973).
[22] J. M. Sanchez, F. Ducastelle und D. Gratias, Physica A 128, 334 (1984).
[23] C. Wolverton, M. Asta, H. Dreysse und D. de Fontaine, Phys. Rev. B 44(10), 4907 (1991).
[24] D. de Fontaine, In Solid State Physics Vol. 47, Academic Press (1994).
[25] J. M. Sanchez, In Theory and Applications of the Cluster Variation and Path ProbabilityMethods (1996).
[26] C. Wolverton, G. Ceder, D. de Fontaine und H. Dreysse, Phys. Rev. B 45, 13105 (1992).
[27] R. B. Griffiths, In Phase transitions and critical phenomena Vol. 1, Academic Press (1972).
[28] C. Wolverton und D. de Fontaine, Phys. Rev. B 49(13), 8627 (1994).
[29] G. Inden und W. Pitsch, In Materials Science and Technology Vol. 5, VCH (1991).
[30] G. Ceder, G. Garbulsky, D. Avis und K. Fukuda, Phys. Rev. B 49(1), 1 (1994).
[31] A. Finel, In Statics and Dynamics of Alloy Phase Transformations (1993).
[32] L. Onsager, Phys. Rev. 65, 117 (1944).
[33] F. Ducastelle, Order and Phase Stability in Alloys. North-Holland (1991).
[34] D. M. Burley, In Phase transitions and critical phenomena Vol. 2, Academic Press (1972).
[35] G. C. Rota, Z. Wahrscheinlichkeitstheorie 2, 340 (1964).
[36] C. Domb, In Phase Transitions and Critical Phenomena Vol. 3, Academic Press (1974).
[37] J. M. Bell und J. Oitmaa, Physica A 129, 17 (1984).
[38] D. A. Vul und D. de Fontaine, Mat. Res. Soc. Symp. Proc. 291, 401 (1993).
[39] J. M. Sanchez, Physica A 111, 200 (1982).
[40] M. A. Krivoglaz und A. A. Smirnov, The Theory of Order-Disorder in Alloys. McDonald,London (1964).
[41] P. C. Clapp und S. C. Moss, Phys. Rev. 142, 418 (1966).
[42] M. Hillert, Phase Equilibria, Phase Diagrams and Phase Transformations. Cambridge (1998).
[43] L. Meijering, Philips Res. Rep. 5, 333 (1950).
[44] L. Meijering, Philips Res. Rep. 6, 183 (1950).
[45] M. Born und J. R. Oppenheimer, Ann. Phys. 84, 457 (1927).
[46] P. Hohenberg und W. Kohn, Phys. Rev. 136(B), 864 (1964).
[47] W. Kohn und L. J. Sham, Phys. Rev. 140(A), 1133 (1965).
[48] B. Meyer, Doktorarbeit, MPI fur Metallforschung (1998).
[49] H. Hellmann, Einfuhrung in die Quantenchemie. Deuticke, Leipzig (1937).

LITERATURVERZEICHNIS 133
[50] R. P. Feynman, Phys. Rev. 56, 340 (1939).
[51] C. Wolverton und A. Zunger, Phys. Rev. B 49, 16058 (1994).
[52] D. de Fontaine, In Theory and Applications of the Cluster Variation and Path ProbabilityMethods (1996).
[53] H. Dreysse, A. Berera, L. T. Wille und D. de Fontaine, Phys. Rev. B 39, 2442 (1989).
[54] C. Wolverton, G. Ceder, D. de Fontaine und H. Dreysse, Phys. Rev. B 48, 726 (1993).
[55] M. Asta, D. de Fontaine und M. van Schilfgaarde, J. Mater. Res. 8(2554), 4907 (1993).
[56] L. G. Ferreira, S. H. Wei und A. Zunger, Phys. Rev. B 40, 3197 (1989).
[57] C. Wolverton und A. Zunger, Phys. Rev. B 50(15), 10548 (1994).
[58] D. B. Laks, L. G. Ferreira, S. Froyen und A. Zunger, Phys. Rev. B 46(19), 12587 (1992).
[59] A. E. Carlsson, Phys. Rev. B 35, 4858 (1987).
[60] C. Amador, W. R. L. Lambrecht, M. van Schilfgaarde und B. Segall, Phys. Rev. B 47(22),15276 (1993).
[61] M. Fahnle, J. Furthmuller und R. Pawellek, In Continuum Models and Discrete Systems,Longman (1991).
[62] R. Kikuchi, J. Chem. Phys. 60(3), 1071 (1974).
[63] N. S. Golsov, L. Y. Pudan, G. S. Golosova und L. E. Popov, Soviet Phys. Solid State 14,1280 (1972).
[64] R. Kikuchi und H. Sato, Acta Metallurgica 22, 1099 (1974).
[65] R. Kikuchi und C. M. van Baal, Scripta Metall. 8, 425 (1974).
[66] H. Ackermann, G. Inden und R. Kikuchi, Acta Metallurgica 37(1), 1 (1989).
[67] R. A. Buckingham, Numerical Methods. Pitman and Sons, London (1957).
[68] D. J. Chadi und M. L. Cohen, Phys. Rev. B 8, 5747 (1973).
[69] B. Meyer und M. Fahnle, Phys. Rev. B 59(9), 6072 (1999).
[70] D. M. Ceperley und B. J. Alder, Phys. Rev. Lett. 45, 566 (1980).
[71] C. Kittel, Einfuhrung in die Festkorperphysik, 6. Auflage. John Wiley and Sons Inc, NewYork (1986).
[72] P. D. Desai, J. Phys Chem Ref. Data 16, 109 (1987).
[73] O. K. Andersen, Phys. Rev. B 12, 3060 (1975).
[74] H. Skriver, The LMTO-Method. Springer-Verlag, Berlin (1984).
[75] H. J. F. Jansen und A. J. Freeman, Phys. Rev. B 30, 561 (1984).
[76] M. Sluiter, D. de Fontaine, X. Guo, R. Podloucky und A. Freeman, Phys. Rev. B 42(16),10460 (1990).

134 LITERATURVERZEICHNIS
[77] N. Saunders, A. P. Miodownik und A. T. Dinsdale, CALPHAD 12, 351 (1988).
[78] V. L. Moruzzi, J. F. Janak und A. R. Williams, In Calculated Electronic Properties of Metals,New York Pergamon (1978).
[79] L. Hedin und B. I. Lundquist, J. Phys. C: Solid State Phys. 4, 2064 (1955).
[80] W. B. Pearson, A Handbook of Lattice Spacings and Structures of Metals and Alloys, Vol. 2.Pergamon Press (1967).
[81] S. Kohlhammer, personliche Mitteilung.
[82] J. Zou und C. Fu, Phys. Rev. B 51(4), 2115 (1995).
[83] K. Enami und S. Nenno, JIM 19, 571 (1978).
[84] S. M. Shapiro, B. X. Yang, G. Shirane, Y. Noda und L. E. Tanner, Phys. Rev. Lett. 62, 1298(1989).
[85] A. E. Carlsson und J. M. Sanchez, Solid State Communications 65(6), 527 (1988).
[86] M. Sluiter, P. Turchi, F. Zezhong und D. de Fontaine, Physica A 148A, 61 (1988).
[87] G. A. de Wijs, G. Krasse und M. J. Gillan, Phys. Rev. B 57, 8223 (1998).
[88] M. Maret, T. Pomme, A. Pasturel und P. Chieux, Phys. Rev. B 42, 1598 (1990).
[89] M. Maret, P. Chieux, J. M. Dubois und A. Pasturel, J. Phys.: Condens. Matter 3, 2801(1991).
[90] G. D. Ayuyshina, E. S. Levin und P. V. Geled, Russian J. Phys. Chem. 43, 11 (1969).
[91] G. V. Samsonov, Handbook of the Physicochemical Properties of the Elements. Plenum (1968).
[92] A. Pasturel, In Statics and Dynamics of Alloy Phase Transformations (1993).
[93] R. Drautz, personliche Mitteilung.
[94] J. M. Sanchez und D. de Fontaine, Phys. Rev. B 17(7), 2926 (1978).
[95] S. K. Aggarwal und T. Tanaka, Phys. Rev. B 16(9), 3963 (1977).
[96] A. Finel, Doktorarbeit, Universite Pierre et Marie Curie (1987).
[97] M. E. Fisher und R. J. Bruford, Phys. Rev. 156(2), 583 (1967).
[98] S. McKenzie, J. Phys. A 8, L102 (1975).
[99] O. G. Mouritsen, S. J. K. Jensen und B. Frank, Phys. Rev. B 24(1), 347 (1981).
[100] D. F. Styer, Phys. Rev. B 32(1), 393 (1985).
[101] H. T. Diep, A. Ghazali, B. Berge und L. Lallemand, Europhys. Lett. 2(8), 603 (1986).
[102] K. Binder, J. L. Lebowitz, M. K. Phani und M. H. Kalos, Acta Metallurgica 29, 1655 (1981).
[103] R. Tetot, A. Finel und F. Ducastelle, Journal of Statistical Physics 61(1/2), 121 (1990).
[104] G. Rubin, Doktorarbeit, Universitat Paris 6 (1994).

Danksagung
Die vorliegende Arbeit ist das Ergebnis einer Zusammenarbeit des Instituts fur Theoretische undAngewandte Physik der Universitat Stuttgart mit dem Max-Planck-Institut fur Metallforschung inStuttgart. Allen, die mich dabei unterstutzt haben danke ich sehr herzlich !Insbesondere bedanke ich mich bei :
• Herrn Prof. Dr. H.-R. Trebin und Herrn Prof. Dr. H. Kronmuller fur die Ermoglichung dieserDiplomarbeit.
• Herrn Prof. Dr. H.-E. Schaefer und Herrn Prof. Dr. M. Fahnle fur die interessante Themen-stellung.Weiter gebuhrt Herrn Prof. Dr. M. Fahnle Dank fur die ausgezeichnete Betreuung, seinestandige Gesprachsbereitschaft und das Vertrauen in meine Arbeit.
• Herrn Prof. Dr. D. Schweitzer fur die Ubernahme des Mitberichts.
• Herrn Dipl.-Phys. Gabriel Bester fur die Einarbeitung in das mixed-basis Programm und dievielen hilfreichen Diskussionen.
• Frau Prof. C. Colinet und Herrn Prof. A. Pasturel, die sich wahrend meines kurzen Aufenthaltsin Grenoble Zeit nahmen und mich bei der Implementation der CVM sowie dem Verstandnisder Thermodynamik von Phasendiagrammen einen großen Schritt weiterbrachten.
• allen Kollegen fur die außerst angenehme Arbeitsatmosphare.
• allen Freunden und speziell bei meiner Familie, ohne deren Unterstutzung all daß hier niemoglich gewesen ware.















