Embed Size (px)
Citation preview

Z. anorg. allg. Chem. 607 (3992) 101-108
Zeitschrift fur anorganische und allgemeine Chemie 0 Johann Ambrosius Barth 1992
Acyl- und Alkylidenarsane. VII [ 11
Tetrakis(2,2-dimethylpropionyl)diarsan - Darstellung und Struktur
G. Becker*, M. Schmidt und M. Westerhausen
Stuttgart, Institut fur Anorganische Chemie der Universitat Stuttgart
Bei der Redaktion eingegangen am 26. April 1991.
Herrn Professor Peter Sartori zum 60. Geburtstage am 19. Dezember 1991 gewidmet
Inhaltsiibersicht. Wird das jeweilige, aus Lithium- dihydrogenarsenid und 2,2-Dimethylpropionylchlorid im Molverhaltnis 3 : 2 bei -40 bis -50°C in Tetrahydro- furan*) oder 1 ,2-Dimethoxyethani) gebildete, aber nicht isolierte Etherat des Lithium-bis(2,2-dimethylpropionyl)ar- senids (2 a) mit 85proz. Tetrafluoroborsaure Diethyl- ether-Addukt weiter umgesetzt, so erhalt man nach der ublichen Aufarbeitung des Ansatzes nicht Bis(2,2-di- methylpropiony1)arsan (44 , sondern mit 64- bzw. 62proz.
I ) Tetrahydrofuran (THF); 1,2-Dimethoxyethan (DME); Bis( 1,2-dimethylamino)ethan (TMEDA); Tetramethylsilan (TMS).
Ausbeute Tetrakis(2,2-dirnethylpropionyl)diarsan (5). Die auBerst oxydationsempfindliche gelborange Verbindung kristallisiert monoklin in der Raumgruppe P2,/n I - 100 f 3 OC; a = 1224,6(3); b = 1419,7(3); c = 1333,1(3) pm; B = 96,22(2)"; Z = 41. Nach den Ergebnis- sen der Rontgenstrukturanalyse (R, = 0,036) weist das Molekul mit zwei urn 86" gegeneinander verdrehten Half- ten die synclinale Konformation auf; die As-C-Abstande liegen mit 203 bis 205 pm wie in einem anderen Acylarsan [l] deutlich uber dem Standard von 196 pm. Weitere cha- rakteristische Bindungslangen und -winkel sind: As-As 242; C-0 120 bis 121 pm; As-As-C 88 bis 107"; As-C-0 118 bis 122".
Acyl- and Alkylidenearsines VII
Synthesis and Structure of Tetrakis(2,2-dimethylpropionyl)diarsine
Abstract. Lithium dihydrogenarsenide and 2,2-di- methylpropionyl chloride in a molar ratio of 3 : 2 react at - 40 to - 50 "C in tetrahydrofuran') or 1,2-dimethoxy- ethane') to give the corresponding etherate of lithium bis(2,2-dimethyIpropionyl)arsenide (2a). Treatment of these solutions with stoichiometric amounts of 85% te- trafluoroboric acid a diethylether adduct yields yellow- orange tetrakis(2,2-dimethylpropionyl)diarsine (5) in 64 or 62% yield resp., but not the expected bis(2,2-dimethylpro- piony1)arsine (4 a). The very air-sensitive compound crystallizes in the monoclinic space group P2,/n [ - 100 k 3 "C; a = 2 224.6(3); b = 1419.7(3); c = 1333.1(3)pm; f l = 96.22(2)"; Z = 4). According to
the X-ray structure analysis (R, = 0.036) the molecule shows synclinal conformation; the two diacylarsyl- subunits are twisted against one another by an angle of 86". As in another acylarsine [I] the As-C distances (203 to 205 pm) were found to be significantly longer than the standard value of 196 pm. Further characteristic bond lengths and angles are: As-As 242; C-0 120 to 121 pm; AS-AS-C 88 to 107"; AS-C-0 118 to 122".
Key words: Lithium bis(2,2-dimethylpropionyl)arse- nide DME; tetrakis(2,2-dimethylpropionyl)diarsine; X- ray structure determination

102 Z . anorg. allg. Chem. 607 (1992)
Einleitung Nach neueren Untersuchungen unserer Arbeitsgruppe [2 - 41 reagieren Lithium-dihydrogenphosphid - 2 THF sowie das homologe Arsenid H,As-Li - 2 THF [3] und Acylchloride im Molverhaltnis 3:2 zum ent- sprechenden Lithium-diacylphosphid * 2 THF (1) bzw. -arsenid - 2 THF (2) (Gl. (1)). An die wahrend der Um- setzung insgesamt zweimal ablaufende Substitution eines
ZTHF ji,
3 H2E-Li. ZTHF
1: E=P; 2: E=As
+ZR-CO-CL; cTHF. DME) -2LiCL; -2EHx
d II c
'0 I r
/\ &\ R E R
R = Organyl; a: (H$J3C- I, 2
Lithiumatoms durch den Acylrest schliel3t sich unter Ent- wicklung von Phosphor- oder Arsenwasserstoff jeweils ein Lithium-Wasserstoff-Austausch an. Die Verbindun- gen 1 und 2 gewinnen durch Ausbildung eines Chelates rnit dem an die beiden Sauerstoffatome des Diacylphos- phids bzw. -arsenids gebundenen Lithium zusatzliche Stabilitat. Die anschliel3ende Reaktion rnit 85proz. Tetrafluoroborsaure * Diethylether-Addukt fuhrt zu den Diacylphosphanen (3) und -arsanen (4) (Gl. (2)) [3]. Das fur diese beiden Verbindungsklassen charakteristische Gleichgewicht zwischen Keto- und Enol-Tautomerem ist wie bei den lY3-Diketonen (R-CO--),CH, abhangig von der Dielektrizitatskonstanten des Losungsmittels und der Temperatur ([3, 5-91; [lo], s. auch [ill).
ZTHF
1,2 1, 3: E=P; 2, 4: E=As
Darstellung und Charakterisierung
Lithium-dihydrogenarsenid . 2 OR, (OR, = 1 /2 DME, THF) reagiert in der zuvor beschriebenen Weise auch rnit 2,2-Dimethylpropionylchlorid. Da aber das in 1,2-Di- methoxyethan gebildete DME-Addukt des Lithium- bis(2,2-dimethylpropionyl)arsenids nach dem iiblichen Aufarbeiten nur als sehr unbestandiges, orangefarbenes 0 1 erhalten werden konnte, haben wir bei nachfolgenden Umsetzungen in Tetrahydrofuran oder 1,2-Dimethoxy- ethan auf eine Isolierung der Verbindung verzichtet und die jeweilige Lijsung bei -40 "C rnit 85proz. Tetrafluoroborsaure - Diethylether-Addukt versetzt. Der nach Entfernen aller bei Zimmertemperatur im Vakuum fliichtigen Anteile verbleibende Ruckstand wird rnit n- Pentan extrahiert. Aus der etwas eingeengten LiSsung scheiden sich bei +4 "C gelborange, sehr oxydations- empfindliche Kristalle ab; die Ausbeute betragt 64 bzw. 62%.
Die zunachst als Bis(2,2-dimethylpropionyl)arsan angesproche- ne Verbindung ist thermisch so labil, dal3 sie auch bei +4 "C im Kuhlschrank nicht iiber mehrere Wochen hinweg ohne betracht- liche Zersetzung aufbewahrt werden kann. Aufgrund dieser In- stabilitat lie13 sich die Molmasse kryoskopisch in Benzol nicht ermitteln; im Laserlicht des Raman-Spektrometers trat soforti- ge Zersetzung ein. Die fur eine As-H-Gruppe typische IR-Ab- sorption um 2100 t 40cm-' [I21 wird nicht beobachtet. Das 'H-NMR-Spektrum der in d6-Benzol gelosten Verbindung zeigt neben dem intensiven Singulett der tert-Butyl-Gruppen (6 = 1,11 ppm) ein schwacheres Signal bei einer chemischen Verschiebung von 1,34 ppm, das zunachst einer As-H-Gruppe zugeordnet wurde, sich aber wie eine Reihe weiterer Signale im Laufe der Zeit verstarkte und somit auch von Zersetzungspro- dukten herriihren kann. Fur uns unerwartet lie13 sich bei sehr tiefem Feld die fur 0 . - H . * 0-Brucken in enol-tautomeren Diacylphosphanen und -arsanen charakteristische Resonanz um +I8 ppm [3, 6, 71 nicht nachweisen. Diese zum Teil wider- spruchlichen Befunde waren zwar rnit dem alleinigen Auftreten des Keto-Tautomeren annahernd zu erklaren; die Beobachtung aber, da13 das homologe Bis(2,2-dimethylpropionyl)phosphan in Lijsungsmitteln mit geringer Dielektrizitatskonstanten wie etwa Benzol (DKZg8 = 2,274 [13]) iiberwiegend als Enol-Tauto- mer vorliegt [7], ist mit dieser Annahme nicht vereinbar.
Die zur endgultigen Klarung durchgefuhrte Rontgen- strukturanalyse fiihrt zu dem iiberraschenden Ergebnis, dal3 Tetrakis(2,2-dimethylpropionyl)diarsan (5) entstan- den ist. Offenbar tritt bereits wahrend der Reaktion bzw. beim vorsichtigen Aufarbeiten des Ansatzes eine rasche, entweder thermisch oder durch diffuses Tageslicht photo- chemisch induzierte Spaltung der vermutlich durch die beiden Acylgruppen zusatzlich geschwachten As-H-Bin- dung und die sich anschliel3ende Kombination zweier Bis(2,2-dimethylpropionyl)arsyl-Fragmente zum Diarsan 5 ein. Allerdings konnte die Entwicklung von Wasserstoff in Form aufsteigender Blaschen nicht beobachtet werden. Das Vorliegen eines Tetraacyldiarsans erklart nicht nur die bereits geschilderte Befunde, sondern steht auch im Einklang rnit weiteren spektroskopischen Ergebnissen. Das j3C-NMR-Signal der Carbonylgruppen tritt rnit ei- nem 8-Wert von 198,Oppm bei sehr tiefem Feld auf; im Vergleich hierzu weisen aber Verbindungen wie Tris(2,2-dimethylpropionyl)- (6 = 224,5 ppm) und Bis(2,2-dimethylpropionyl)phenylarsan (6 = 225,O ppm) sowie einige (2,2-Dimethylpropionyl)organyl-trimethylsi- lylarsane (8 = 228 bis 231 ppm) noch starkere Tieffeld- verschiebungen auf [ 141. Die grofite, im Massenspek- trum zu beobachtende Masse ist der um eine 2,2-DimethylpropionyI-Einheit reduzierten Molekul- masse zuzuordnen.
Das Tetrakis(2,2-dimethylpropionyl)-Derivat 5 ist der erste Vertreter der bislang unbekannten Verbindungsklas- se der Tetraacyldiarsane. Auch die homologen Tetraacyl- diphosphane wurden noch nicht in praparativen Mengen dargestellt. Lediglich iiber die Bildung des NMR-spek- troskopisch nachgewiesenen Tetraacetyl-Derivates als Ne- benprodukt bei der Reaktion von Tris(trimethy1- sily1)phosphan mit Acetylchlorid haben wir vor einigen

G. Becker u. a., Tetrakis(2,2-dimethylpropionyl)diarsan 103
Jahren berichtet [I 51. In diesem Zusammenhang sei auch auf die Synthese und die Struktur des 1,2-Bis(2',2'-di- methylpropiony1)-i ,2-diphenyldiphosphans [ 161 hinge- wiesen.
Kristalldaten, MeStechnik und Strukturbestimmung
Beim Abkiihlen einer bei Zimmertemperatur gesattigten Ld- sung des Tetrakis(2,2-dimethylpropionyl)diarsans (5) in n-Pen- tan auf +4 "C erhalt man unregelmaoig geformte, aber fur eine Rontgenstrukturanalyse geeignete Einkristalle. Wegen der Emp- findlichkeit der Verbindung gegeniiber Rontgenlicht bei + 20 "C konnten keine Buerger-Pr3zessionsaufnahmen angefer- tigt werden, die Aufnahme eines Datensatzes bei -100 f 3 "C am Vierkreisdiffraktometer war aber problemlos moglich. Die iiber diese Messung ermittelten Ausloschungen (h01: h + 1 = 2n + 1 ; OkO : k = 2n + 1) fiihrten zur zentrosym- metrischen Raumgruppe P2Jn [17 a]; die Berechnung der Raumerfullung iiber Volumeninkremente (Tab. 1) ergab vier Formeleinheiten in der Elementarzelle. Die rnit den 28-Werten von 15 Reflexen im Bereich (20" 5 28 5 25") bei -100 k 3 "C am Vierkreisdiffraktometer ermittelten und verfeinerten Gitter- konstanten sind in Tab. 1 zusammengestellt. Angaben zur Mes-
sung der Reflexintensitaten und zur Strukturbestimmung kon- nen Tab. 2 entnommen werden.
Das Programmsystem SHELXS-86 [I7 el fiihrte mit den Posi- tionen von zwei Arsen- und vier Sauerstoffatomen zu einem brauchbaren Strukturvorschlag; die Lagen der noch fehlenden Kohlenstoffatome lieOen sich den im Laufe der weiteren Struk- turbestimmung jeweils gerechneten Differenz-Fouriersynthesen entnehmen. Beim Ubergang von isotropen zu anisotropen Aus- lenkungsparametern fur alle schwereren Atome sank der Giite- faktor R von 0,069 auf 0,057. Die noch fehlenden Ortskoordi- naten aller Wasserstoffatome ergaben sich aus der bei einem R- Wert von 0,053 gerechneten Differenz-Fouriersynthese; rnit we- nigen Ausnahmen lieBen sie sich ebenso wie ihre isotropen Aus- lenkungsparameter zu sinnvollen Werten verfeinern. Die Ergeb- nisse der Strukturbestimmung sind in Tab. 3 zusammengestellt; Bindungslangen und -winkel sowie einige charakteristische Tor- sionswinkel finden sich in Tab. 4.
Molekiil- und Kristallstruktur
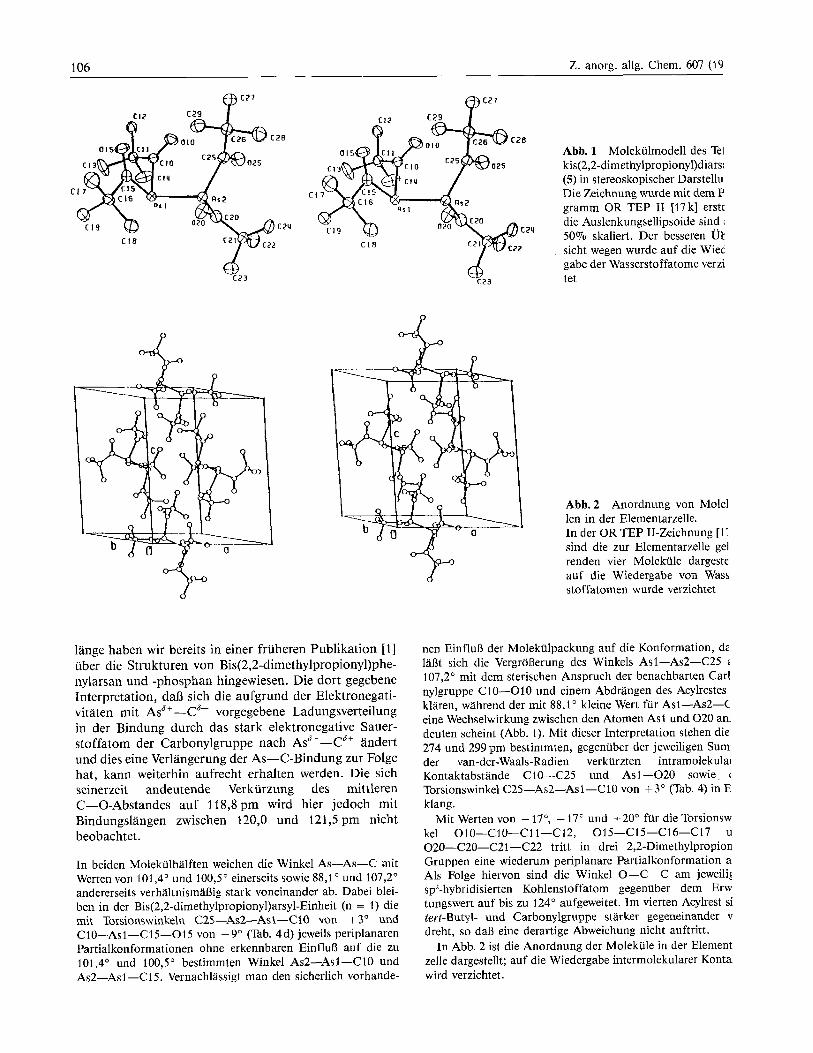
Abb. 1 zeigt ein Molekiilmodell des Tetrakis(2,2-di- methylpropiony1)diarsans (5) in stereoskopischer Darstel- lung.
Tabelle 1 CZOH360&Z; monoklin; Raumgruppe P2Jn (Nr. 14 [17a], Z = 4; MeBtemp. - I00 k 3 "C; Zersp.: 118 * 121 "C (im abge- schmolzenen Rohrchen unter Argon); Raumerfiillung nach Kituigorodskii [ 17 b]: 8O%*.
Kristalldaten des %trakis(2,2-dimethylpropionyl)diarsans (5).
a = 1224,6(3) pm b = 1419,7(3) pm c = 1333,1(3) pm f i = 96,22(2)" V = 2 304,O . lo-'' m3 d, = 1,414 g * ~ m - ~ F(000) = 1016 M = 490,4
* Der Berechnung liegen die in Tab. 4 angegebenen mittleren Bindungslangen, ein C-H Abstand von 108 pm und folgende Werte fur die intermolekularen Radien zugrunde: As 200; C 170; 0 136; H 117 pm [17b, c]
Tabelle 2 Angaben zur Messung der Reflexintensitaten, zur Strukturbestimmung und zur Verfeinerung. Vierkreisdiffraktometer P2, der Firma Syntex, Cupertino (USA); MoKa-Strahlung; Graphitmonochromator; Kontrolle von Inten- sitat und Orientierung durch je zwei Messungen in einem Interval1 von 98 Reflexen; Aufbereitung der Daten und Ermittlung der Struktur mit den Programmsystemen X-RAY 76 [ 17 d] und SHELXS 86 [ 17 el; keine Absorptionskorrektur; Atomformfaktoren fur die neutralen Atome As, C und 0 nach Cromer und Mann [ 17 fJ, fur H nach Stewart, Duvidson und Simpson [ 17 g]; jeweils mehrere Verfeinerungszyklen rnit vollstandiger Matrix und anschliefiender Differenz-Fouriersynthese; Minimalisierung der Funktion Zw(F,, - IF, I)*; Gewichtung nach dem statistischen Fehler der Messung.
Kristallabmessungen (mm) und -gestalt MeDbereich und -temperatur gemessener Bereich des reziproken Raumes Scanmodus, -breite") und -geschwindigkeit symmetrieunabhangige Reflexe MeOwerte rnit [F, > 4a(F,)jb) hearer Absorptionskoeffizient p [I7 h] Konvergenz bei einem R,-Wertc) von maximale Restelektronendichte
0,3 x0,3 x0,6; quaderf6rmig 2" I 2 0 5 50"; -100 * 3 "C 0 5 h 5 14; 0 5 k I 16; - 15 5 1 5 15 w-/20-Scan; 2"; 2"/min 4 535 3 250 33,Ol . 1O2mm-' 0,036 0,92 * m - 3
") Messung des Untergrundes rnit einer der MeBzeit des Reflexes entsprechenden Dauer b, Die unbeobachteten Reflexe blieben bei den Verfeinerungen unberiicksichtigt ') R,=Z.(& lF,-IFF,ll)/Z.(&~F,)
104 Z. anorg. allg. Chem. 607 (1992)
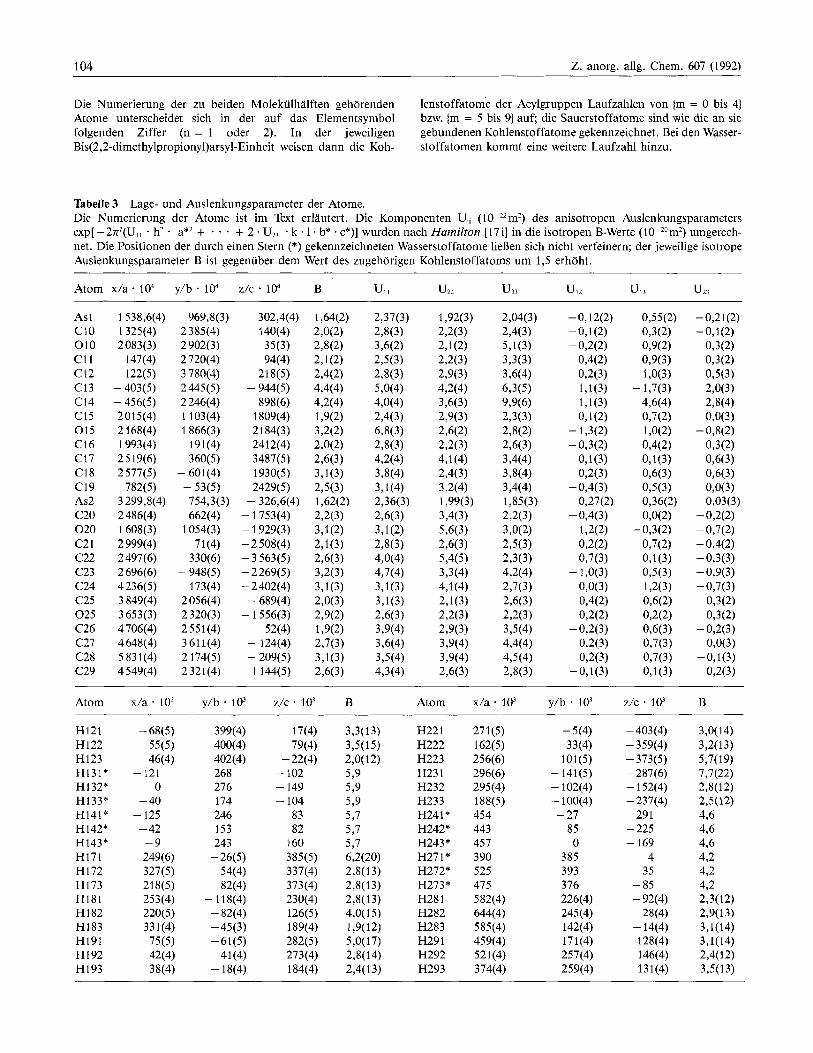
Die Numerierung der zu beiden Molekiilhalften gehorenden lenstoffatorne der Acylgruppen Laufzahlen von (m = 0 bis 4) Atome unterscheidet sich in der auf das Elementsymbol bzw. (m = 5 bis 9) auf; die Sauerstoffatome sind wie die an sie folgenden Ziffer (n = 1 oder 2). In der jeweiligen gebundenen Kohlenstoffatome gekennzeichnet. Bei den Wasser- Bis(2,2-dimethylpropionyl)arsyl-Einheit weisen dann die Koh- stoffatomen kommt eine weitere Laufzahl hinzu.
Tabelle 3 Lage- und Auslenkungsparameter der Atome. Die Numerierung der Atome ist im Text erlautert. Die Komponenten U, ( 10-22m2) des anisotropen Auslenkungsparameters exp[ -27r2(U,, . h2 . a*2 + * * . + 2 * U23 . k . l . b* * c*)] wurden nach Hamilton [17i] in die isotropen B-Werte (10-Zom2) umgerech- net. Die Positionen der durch einen Stern (*) gekennzeichneten Wasserstoffatome lieBen sich nicht verfeinern; der jeweilige isotrope Auslenkungsparameter B ist gegenuber dem Wert des zugehorigen Kohlenstoffatoms um 1,5 erhoht.
Atom x/a . la? y / b . lo4 z/c * la? B Ul, uzz U,, UlZ UP uz3
As 1 c10 010 c11 c12 C13 C14 C15 015 C16 C17 C18 C19 As2 c20 020 c 2 1 c22 C23 C24 C25 025 C26 C27 C28 C29
1538,6(4) 1325(4) 2 083(3)
147(4) 122( 5 )
- 403(5) - 456(5) 2015(4) 2 168(4)
2 5 19(6)
782(5) 3 299,8(4) 2486(4) 1608(3)
2497(6) 2 696(6) 4 236(5) 3 849(4) 3 653(3) 4 706(4) 4648(4) 5831(4) 4 549(4)
1993(4)
2 577(5)
2 999(4)
969,8(3) 2 385(4) 2 902(3) 2 720(4) 3 780(4) 24435) 2 246(4) 1103(4) 1866(3)
191(4) 360(5)
- 53(5)
662(4) 1054(3)
7 1 (4) 330(6)
- 948(5) 173(4)
2 056(4) 2 320(3) 2 5 5 l(4) 3611(4) 2 174(5) 2321(4)
- 601 (4)
754,3(3)
302,4(4) 140(4) 35(3) 94(4)
- 944(5) 218(5)
898(6) 1809(4) 2184(3) 24 1 2( 4) 3487(5) 1930(5) 2429(5)
- 1753(4) - 1929(3) - 2 508(4) - 3 563(5) - 2269(5) -2402(4) - 689(4) - 1556(3)
52(4)
- 326,6(4)
- 124(4) - 209(5)
1 144(5)
Atom x / a . lo3 y /b . lo3 z/c . 10’ B Atom x/a . lo3 y/b . lo3 z/c * lo3 B
Hi21 H122 HI23 H131* H132* H133* H141* H142* H143* HI71 HI72 HI73 HI81 HI82 HI83 HI91 HI92 HI93
-68(5) 55(5 ) 46(4)
- 121 0
- 40 - 125 - 42
-9 249(6) 327(5) 218(5) 253(4) 220(5) 33 l(4) 75(5) 42(4) 38(4)
399(4) 400(4) 402(4) 268 276 174 246 153 243 - 26(5)
54(4) 82(4)
- 1 l8(4) - 82(4) -45(3) -61(5)
41(4) - 18(4)
17(4) 79(4)
- 22(4) - 102 - 149 - 104
83 82
160 385(5) 337(4) 373(4) 230(4) 126(5) 189(4) 282(5) 273(4) 184(4)
H221 H222 H223 H23 I H232 H233 H241* H242* H243* H271* H272* H273* H28 1 H282 H283 H29 1 H292 H293
27 1 ( 5 ) 162(5) 256(6) 296(6) 295(4) 188(5) 454 443 457 390 525 475 582(4) 644(4) 585(4)
521(4) 459(4)
374(4)
- 5(4) 33(4)
lOl(5) - 141(5) - 102(4) - 1 OO(4) - 27
85 0
385 393 376 226(4) 245(4) 142(4) 171(4) 257(4) 259(4)
- 403(4) - 359(4) -373(5) -287(6) - 152(4) -237(4)
29 1 - 225 -169
4 35
- 85 - 92(4)
28(4) - 14(4) 128(4) 146(4) 131(4)
G. Becker u. a., Tetrakis(2,2-dimethylpropionyl)diarsan 105
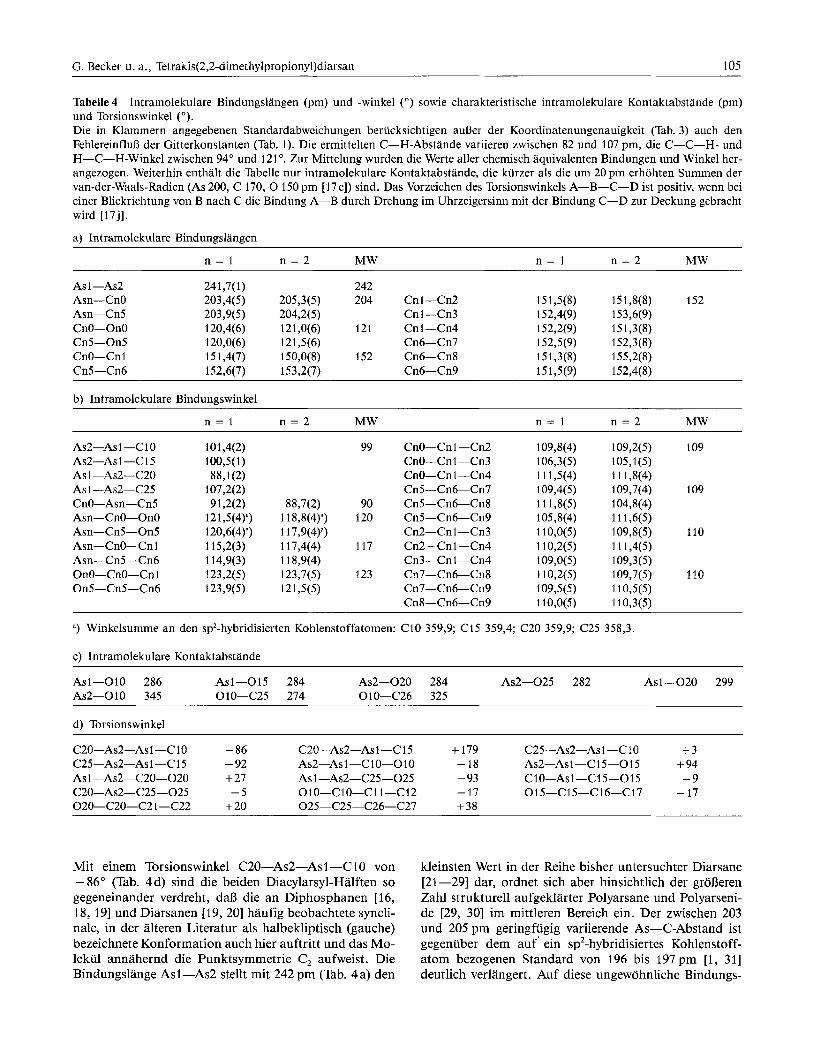
Tabelle 4 Intramolekulare Bindungslangen (pm) und -winkel (") sowie charakteristische intramolekulare Kontaktabstande (pm) und Torsionswinkel ("). Die in Klammern angegebenen Standardabweichungen beriicksichtigen aul3er der Koordinatenungenauigkeit (Tab. 3) auch den Fehlereinflulj der Gitterkonstanten (Tab. 1). Die ermittelten C-H-Abstande variieren zwischen 82 und 107 pm, die C-C-H- und H-C-H-Winkel zwischen 94" und 121 O. Zur Mittelung wurden die Werte aller chemisch aquivalenten Bindungen und Winkel her- angezogen. Weiterhin enthalt die %belle nur intramolekulare Kontaktabstande, die kiirzer als die um 20 pm erhohten Summen der van-der-Waals-Radien (As 200, C 170, 0 150 pm [ I ~ c ] ) sind. Das Vorzeichen des Torsionswinkels A-B-C-D ist positiv, wenn bei einer Blickrichtung von B nach C die Bindung A-B durch Drehung im Uhrzeigersinn mit der Bindung C-D zur Deckung gebracht wird [17j].
a) Intramolekulare Bindungslangen
n = l n = 2 MW n = l n = 2 MW
AS 1 -As2 241,7(1) 242 Am-CnO 203,4(5) 205,3(5) 204 Cnl-Cn2 151,5(8) 151,8(8) 152 Asn-Cn5 203,9(5) 204,2(5) Cnl-Cn3 152,4(9) 153,6(9) Cn0-On0 120,4(6) 121,0(6) 121 Cnl-Cn4 152,2(9) 15 1,3(8) Cn5-On5 120,0(6) 121,5(6) Cn6-Cn7 152,5(9) 1 52,3(8) CnO-Cn1 15 1,4(7) 150,0(8) 152 Cn6-Cn8 151,3(8) 155,2(8) Cn5-Cn6 152,6(7) 153,2(7) Cn6-Cn9 1 5 1,5 (9) 152,4(8)
b) lntramolekulare Bindungswinkel
n = l n = 2 MW n = l n = 2 MW
As~-AsI-C~O As~-AsI-CI~ Asl-As2-C20 AS 1 -As2-C25 CnO-Asn-Cn5 Asn-Cn0-On0 Asn-Cn5-On5 Asn-CnO-Cnl Asn-Cn5-Cn6 On0-Cn0-Cnl 0115-Cn5-Cn6
101,4(2) 100,5( 1)
107,2(2)
12 1,5 (4)") 120,6(4)") 115,2(3) 114,9(3) 123,2(5) 123,9(5)
88,1(2)
91,2(2)
99
88,7(2) 90 118,8(4)") 120
117,4(4) 117 1 18,9(4) 123,7(5) 123 121,5(5)
117,9(4)")
CnO-Cnl-Cn2 CnO-Cnl-Cn3 CnO-Cn1 -Cn4 Cn5-Cn6-Cn7 Cn5-Cn6-Cn8 Cn5-Cn6-Cn9 Cn2-Cnl-Cn3 Cn2-Cnl-Cn4 Cn3-Cnl-Cn4 Cn7-Cn6-Cn8 Cn7-Cn6-Cn9 Cn8-Cn6-Cn9
109,8(4) 106,3(5) 11 1,5(4) 109,4(5) 11 1,8(5) 105,8(4) 1 10,0(5) 110,2(5) 109,0(5) 110,2(5) 109,5(5) 1 10,0(5)
109,2(5) 109 105,1(5) 1 1 1,8(4) 109,7(4) 109 104,8(4) 11 1,6(5) 109,8(5) 110 1 1 1,4(5)
109,7(5) 110 109,3( 5 )
110,5(5) 1 10,3(5)
") Winkelsumme an den sp2-hybridisierten Kohlenstoffatomen: C10 359,9; C15 359,4; C20 359,9; C25 358,3
c) Intramolekulare Kontaktabstande
Asl-010 286 Asl-015 284 As2-020 284 As2-025 282 Asl-020 299 As2-010 345 010-C25 274 010-C26 325
d) Torsionswinkel
C~O-AS~-ASI-CIO - 86 C20-As~-As I-C 15 + 179 C~~-AS~-ASI-CIO +3 C25-A~2-A~l-C15 - 92 AD-As 1 -C 10-010 - 18 AS~-ASI--CI 5-015 + 94 AS 1 -A~2-C20-020 + 27 A~I-A~2-C25-025 - 93 C10-A~l-C15-015 -9 C2O-A~2-C25-025 - 5 010-c10-c11-c12 - 17 015-C15-C16-C17 - 17 020-c20-c21 -C22 + 20 025-C25-C26-C27 + 38
Mit einem Torsionswinkel C2O-As2-Asl -C10 von -86" (Tab. 4d) sind die beiden Diacylarsyl-Halften so gegeneinander verdreht, dan die an Diphosphanen [ 16, 18, 191 und Diarsanen 119, 201 haufig beobachtete syncli- nale, in der alteren Literatur als halbekliptisch (gauche) bezeichnete Konformation auch hier auftritt und das Mo- lekiil annahernd die Punktsymmetrie C, aufweist. Die Bindungslange Asl-As2 stellt mit 242 pm (Tab. 4a) den
kleinsten Wert in der Reihe bisher untersuchter Diarsane [21-291 dar, ordnet sich aber hinsichtlich der groneren Zahl strukturell aufgeklarter Polyarsane und Polyarseni- de [29, 301 im mittleren Bereich ein. Der zwischen 203 und 205 pm geringfugig variierende As-C-Abstand ist gegeniiber dem auf' ein sp2-hybridisiertes Kohlenstoff- atom bezogenen Standard von 196 his 197 pm [I, 311 deutlich verlangert. Auf diese ungewohnliche Bindungs-
106
@ c 2 7
a10 c% c 2 0
t l 2
c2u
d C Z 3
-3-
Z. anorg. allg. Chem. 607 (19
C29
Abb. 1 Molekulmodell des Tei kis(2,2-dimethylpropionyl)diarsi (5) in stereoskopischer Darstellu Die Zeichnung wurde rnit dem P gramm OR TEP I1 117 k] erste die Auslenkungsellipsoide sind 50% skaliert. Der besseren Ut sicht wegen wurde auf die Wiec - gabe der Wasserstoffatome verzi
C P 3 tet
lange haben wir bereits in einer friiheren Publikation [ I ] iiber die Strukturen von Bis(2,2-dimethylpropionyl)phe- nylarsan und -phosphan hingewiesen. Die dort gegebene Interpretation, dafi sich die aufgrund der Elektronegati- vitaten rnit As'+-C'- vorgegebene Ladungsverteilung in der Bindung durch das stark elektronegative Sauer- stoffatom der Carbonylgruppe nach Asst -Cs+ andert und dies eine Verlangerung der As-C-Bindung zur Folge hat, kann weiterhin aufrecht erhalten werden. Die sich seinerzeit andeutende Verkiirzung des mittleren C-0-Abstandes auf 118,8 pm wird hier jedoch rnit Bindungslangen zwischen 120,O und 1213 pm nicht beobachtet.
In beiden Molekiilhalften weichen die Winkel As-As-C rnit Werten von 101,4" und 100,5" einerseits sowie 88,l" und 107,2" andererseits verhaltnismaRig stark voneinander ab. Dabei blei- ben in der Bis(2,2-dimethylpropionyl)arsyl-Einheit (n = 1) die mit Torsionswinkeln C25-As2-Asl-C10 von + 3" und ClO-Asl-C15-015 von - 9" (Tab. 4d) jeweils periplanaren Partialkonformationen ohne erkennbaren EinfluD auf die zu 101,4" und 100,5" bestimmten Winkel As2-Asl-C10 und As2-Asl-CI5. Vernachlassigt man den sicherlich vorhande-
Abb.2 Anordnung von Mole1 len in der Elementarzelle. In der OR TEP 11-Zeichnung [I: sind die zur Elementarzelle gel renden vier Molekiile dargeste auf die Wiedergabe von Wass stoffatomen wurde verzichtet
nen EinfluR der Molekiilpackung auf die Konformation, da laRt sich die VergroRerung des Winkels AsI-As2-C25 i
107,2" mit dem sterischen Anspruch der benachbarten Carl nylgruppe C10-010 und einem Abdrangen des Acylrestes klaren, wahrend der mit 88, l" kleine Wert fur Asl-As2--C eine Wechselwirkung zwischen den Atomen As1 und 020 an. deuten scheint (Abb. 1). Mit dieser Interpretation stehen die 274 und 299 pm bestimmten, gegeniiber der jeweiligen Sum der van-der-Waals-Radien verkurzten intramolekulai Kontaktabstande CZO-C25 und Asl-020 sowie (
Torsionswinkel C25-As2-Asl-CIO von +3" (Tab. 4) in E klang.
Mit Werten von - 17", - 17" und +20" fur die Torsionsw kel 010-C1O-C11-C12, 015-C15-C16-C17 u 020-C2O-C21-C22 tritt in drei 2,2-Dimethylpropion Gruppen eine wiederum periplanare Partialkonformation a Als Folge hiervon sind die Winkel 0-C-C am jeweilig sp'-hybridisierten Kohlenstoffatom gegeniiber dem Erw tungswert auf bis zu 124" aufgeweitet. Im vierten Acylrest si tert-Butyl- und Carbonylgruppe starker gegeneinander v dreht, so daR eine derartige Abweichung nicht auftritt.
In Abb. 2 ist die Anordnung der Molekule in der Element zelle dargestellt; auf die Wiedergabe intermolekularer Kontal wird verzichtet.

G. Becker u. a., Tetrakis(2,2-dimethylpropionyl)diarsan 107
Priiparativer Teil
Samtliche Arbeiten wurden unter nachgereinigtem (BTS-Kataly- sator [32], Phosphor(V)-oxid) Argon als Schutzgas durchge- fiihrt. Die verwendeten Losungsmittel haben wir zunachst ent- weder (Ether) rnit Natrium in Gegenwart von Benzophenon oder (Kohlenwasserstoffe) mit Natriumdraht vorgetrocknet, dann uber Lithiumalanat destilliert und anschliefiend mit Ar- gon gesattigt. Bei den NMR-Spektren stehen positive 6-Werte fur Tieffeldverschiebungen.
Lithium-dihydrogenarsenid - 2THF. Die Darstellung des THF- Adduktes wird in Veroffentlichung [3] beschrieben.
Lithium-dihydrogenarsenid . DME. Darstellung: Zu einer auf - 78 "C gekuhlten Losung von 15,59 g (200 mmol) Arsan [33] in etwa 300 ml 1,2-Dimethoxyethan, dargestellt durch Konden- sieren des Gases auf das rnit fliissigem Stickstoff eingefrorene Solvens und anschlierjendes, vorsichtiges Erwarmen, tropft man - analog zur Darstellung des entsprechenden Phosphids [34] - langsam und unter Ruhren bei einem Gesamtdruck von 400 rnbar in der Apparatur 108 ml einer 1,664 M (1 80 mmol) Gsung von Lithium-n-butanid in n-Hexan. Nach einer weiteren Stunde werden die im Vakuum bei -78 "C fluchtigen Reak- tionspartner und ein Teil des Losungsmittels vorsichtig abdestil- liert; dabei fallt ein weifier, pulvriger Niederschlag aus. Bewahrt man derartige Ansatze bei -10 "C im Kuhlschrank auf, so bil- den sich im Laufe von Tagen farblose, durchsichtige Kristalle. Sie sind nur im Kontakt mit der Mutterlauge bestandig und wer- den nach weitgehendem Abpipettieren der uberstehenden Lii- sung ohne weitere Reinigung fur anschlierjende Umsetzungen verwendet. Ausbeute 11,4 g (65,5 mmol); 36%. Auf die Charak- terisierung des Lithium-dihydrogenarsenids . DME werden wir spater im Zusammenhang rnit dem entsprechenden TMEDA- Adduktj) naher eingehen [35].
Lithium-bis(2,2-dimethylpropionyl)arsenid . DME. Darstel- lung: Zu einer auf -40 "C gekiihlten Suspension von 7,86g (45,2 mmol) Lithium-dihydrogenarsenid - DME in 150 ml 1,2-Dimethoxyethan tropft man langsam und unter Ruhren 3,5 g (29,O mmol; 3,6 ml) 2,2-Dimethylpropionylchlorid in 50 ml des gleichen Solvens. Die sofort einsetzende Reaktion gibt sich durch ausfallendes Lithiumchlorid und eine Farbanderung des Ansatzes nach schlierjlich dunkelgelb zu erkennen. Man 1aBt langsam auf Zimmertemperatur erwarmen, destilliert das Lo- sungsmittel einschliefilich des gebildeten Arsenwasserstoffs bei +20 "C im Vakuum weitgehend ab und filtriert. Nach vollstan- diger Entfernung des restlichen Solvens bleibt ein orangefarbe- nes, auRerst protolyse- und oxydationsempfindliches 0 1 zuruck, das sich aufgrund seiner thermischen Instabilitat kaum charak- terisieren 1Mt. Ausbeute 3,4 g (9,9 mmol); 68%. Charakterisierung. NMR-Spektren. 'H (L.M. d,-BenzoVDME; int. Stand. TMSI)): (H3C)3C 6 = 1,38ppm. Ein I3C- NMR-Spektrum konnte nicht aufgenommen werden.
IR-Spektrum (cm-' ; kapillar zwischen KBr-Scheiben, Dich- tungsmittel Lithelen; sehr stark (vs), stark (s), mittel (m), schwach (w), sehr schwach (vw), Schulter (sh)): 2960 vs, 2930 sh, 2900 sh, 2865 m, 1714 m, 1672 m, 1574 vs, 1 560 sh, 1473sh, 1459vs, 1386s, 1361 s, 1297m, 126Ow, 1246w, 1225 m, i 195 ms, 1 158 sh, 1 128 m, 1 108 w, 1089 ms, 1074 ms, 1031 ms, 979 w, 954 w, 902 vs, 870 w, 852 w, 824 w, 800 ms, 790 sh, 642 w, 630 ms, 512 m, br, 430 m.
Tetrakis(2,2-dimethyIpropionyl)diarsan (5). Darstellung: Zu ei- ner Suspension von 18,5 g (81 mmol) Lithium-dihydrogen- arsenid . 2THF in 200 ml Tetrahydrofuran tropft man langsam unter Ruhren bei -40 bis -50 "C und einem Druck von etwa 60mbar eine Losung von 6,5 g (54mmol; 6,6ml) 2,2-Dimethylpropionylchlorid in 25 ml des gleichen Solvens. Die Bildung des fluchtigen Arsans fuhrt zu einem Druckan- stieg; die Farbe des Ansatzes andert sich uber gelb nach oliv- grun. Nach vollstandiger Zugabe wird auf - 10 "C erwarmt, entstandenes Arsan weitgehend im Vakuum entfernt und ohne weitere Aufarbeitung umgehend bei -40 "C rnit 5,l g (27 mmol) 85proz. Tetrafluoroborsaure . Diethylether-Addukt versetzt. Wahrend der Zugabe farbt sich der Ansatz gelborange. Man larjt auf Zimmertemperatur erwarmen, destilliert alle bei +20 "C fluchtigen Anteile im Vakuum ab und extrahiert den Ruckstand sorgfaltig rnit n-Pentan. Beim langsamen Abkiihlen der so erhaltenen, gegebenenfalls vorsichtig eingeengten Lljsung auf + 4 "C kristallisiert Tetrakis(2,2-dimethylpropionyl)diarsan (5) aus. Zur Kontrolle wurde die Reaktion unter sonst gleichen Bedingungen auch in 1 ,ZDimethoxyethan durchgefuhrt; die Ausbeute variierte rnit 62% nur geringfugig. Ausbeute 4,2 g (8,6 mmol); 64%. Charakterisierung. Zersp. 118 . . 121 "C (im abgeschmolzenen Rohrchen unter Argon); aufierst oxydationsempfindliche, gelb- orange Kristalle.
Elementaranalyse: CZOH360A~Z; C 48,7 (ber. 49,O); H 7,2 (7,4)%.
Verbindung 5 zersetzt sich sehr leicht, so da13 die Molmasse kryoskopisch in Benzol nicht bestimmt werden konnte.
NMR-Spektren. 'H (L.M. d,-Benzol, int. Stand. TMS): (H,C)$2 6 = 1,li ppm. I3C ('HI (L.M. und int. Stand. d,-Ben-
27,O ppm. IR-Spektrum (cm-'; Verreibung in Nujol, kapillar zwischen
KBr-Scheiben): 1815 s, 1750 s, 1710 s, 1 665 m, 1265 w, 1220 w, 1195 w, 1040 m, 1030 m, 1005 s, 905 m, 890 s, 805 s, 790 sh, 750 w, 720 w, 600 m, 590 m, 475 w, 440 w.
Charakteristische Massen aus dem Massenspektrum (Mas- senspektrometer MAT 7 11 der Firma Varian; Ionisierungsener- gie 20 eV; Quellentemperatur 330 K): M+-[CO-C(CH3)?] 405,O
85,l (7,O); (H3C)$+ 57,l (16,lVo). Das Auftreten der Molekul- masse M+ konnte nicht beobachtet werden.
~01): CO 6 = 198,O; C(CH3)3 6 = 48,O; C(CH3), 6 =
(59,2); M/2+ 245,l (100); AS^' 194,9 (7,6); (H3C)jC--CO+
Die fur die Rontgenstrukturanalyse erforderlichen Rechnun- gen fiihrten wir an den Anlagen CYBER 174 und CRAY 2 des Rechenzentrums der Universitat Stuttgart durch. Der Deut- schen Forschungsgemeinschaft, 5300 Bonn-Bad Godesberg und dem Fonds der Chemischen Industrie, 6000 Frankfurt a. M. so- wie der Firma Hoechst AG, 6230 Frankfurt a. M.-Hbchst sei fur grorjziigige Unterstutzung gedankt.
Literatur
[I] Vorausgehende Mitteilung: G. Becker, B. Becker, M. Birk- hahn, 0. Mundt, R. E. Schmidt: Z. anorg. allg. Chem. 529 (1 985) 97
[2] G. Becker, M. Birkhahn, U.: Massa, FX Uhl: Angew. Chem. 92 (1980) 756; Int. Ed. Engl. 19 (1980) 741
[3] (3. Becker, JK Becker, M. Schmidt, FK Schwarz, M. We- sterhausen: Z. anorg. allg. Chem. 605 (1991) 7

108 Z. anorg. allg. Chem. 607 (1992)
[4] G. Becker, H.-D. Hausen, M. Schmidt, W Schwarz, W Uhl, M. Westerhausen, M. Birkhahn, W Massa: in Vorbereitung
[ 5 ] G. Becker, H. l? Beck: Z . anorg. allg. Chem. 430 (1977) 77 [6] G. Becker: Z. anorg. allg. Chem. 480 (1981) 38 [7] G. Becker, M. Rossler; G. Uhl: Z . anorg. allg. Chem. 495
(1982) 73 [8] C. L. Liotta, M. L. McLaughlin, B. A. O’Brien: Tetrahe-
dron Lett. 25 (1984) 1249 [9] G. Markl, H. Sejpka: Tetrahedron Lett. 27 (1986) 1771
[lo] L. Weber, K. Reizig, M. Frebel: Chem. Ber. 119 (1986) 1857
[ i l l L. Weber, D. Bungardt: J. Organomet. Chem. 311 (1986) 269
[ 121 J. Weidlein, U Miiller, K. Dehnicke: Schwingungsfrequen- Zen I; Stuttgart: Thieme Verlag, 1981, S. 257
[I31 R. C Weast (Hrsg.): CRC Handbook of Chemistry and Physics, 66. Aufl., Boca Raton (Florida): CRC Press,
[I41 G. Becker, G. Gutekunst: Z. anorg. allg. Chem. 470 (1980)
[I51 G. Becker: Z. anorg. allg. Chem. 480 (1981) 21 [16] G. Becker, 0. Mundf, M. Rossler: Z . anorg. allg. Chem.
468 (1 980) 55 [I71 a) Th. Hahn (Hrsg.): International Tables for Crystallo-
graphy, Bd. A Space-Croup Symmetry, 2. Aufl., Dor- drecht (NL): Reidel Publ. Comp. 1984; b) A. I. Kitaigo- rodskii: Organic Chemical Crystallography, New York: Consultants Bureau 1961; c) J. E. Huheey: Anorganische Chemie, Berlin: de Gruyter 1988, S. 278; E A . Cotton, G. Wilkinson: Anorganische Chemie, Weinheim: Verlag Chemie 1970, S. 107; d) J. M. Stewart (Hrsg.): The XRAY System - Version of 1976, Technical Report TR-446; Col- lege Park (Maryland, USA): Computer Science Center, University of Maryland 1976; e) G. M. Sheldrick, in G. M. Sheldrick, C. Kruger, R. Goddard (Hrsgg.): SHELXS-86, Crystallographic Computing 3; Oxford: Uni- versity Press 1985, S. 175; f ) D. 7: Cromer; L B. Mann: Acta Crystallogr. A 2 4 (1968) 321; g) R. E Stewart, E. R. Davidson, K 7: Simpson: J. Chem. Phys. 42 (2965) 3175; h) J. A. Zbers, W C. Hamilton (Hrsgg.): Interna- tional Tables for X-ray Crystallography, Bd. IV, Birming- ham (CB): Kynoch Press 1974, S. 47; i) W C Humilton: Acta Crystallogr. 12 (1959) 609; j ) R. S. Cahn, Sir C. In- gold, K Prelog: Angew. Chem. 78 (1966) 413; k) C. K. Johnson: OR TEP 11: A FORTRAN Thermal El- lipsoid Plot Program for Crystal Structures Illustrations
1985 - 1986, S. E-49
131
(ORNL-5 138); Oak Ridge (Tennessee, USA): Oak Ridge National Laboratory, 1976
[I 81 L. Maier, in G. M. Kosolapofj L. Maier (Hrsgg.): Organic Phosphorus Compounds, Vol. 1, New York: Wiley-hter- science, 1972, S. 314
[I93 0. Mundf, H. Riffee[, G. Becker; A. Simon: Z . Natur- forsch. 43b (1988) 952 u. dort zit. Lit
[20] E Kober: Chem. Ztg. 105 (1981) 199 [21] A. J. Ashe IZZ, W M. Butler, 7: R. Diephouse: Organome-
tallics 2 (1983) 1005 [22] G. Becker, G. Gutekunst, C. Witfhauer: Z . anorg. allg.
Chem. 486 (1982) 90 [23] C. Ni, Z. Zhang, 2. Xie, C. Qian, Y I Huang: Jiegou Hua-
xue 5 (1986) 181; C.A. 107 (1987) 145210f [24] A 4 Baudler, K AktaZay, 7: Heinlein, K.-F: Tebbe: Z . Na-
turforsch. 37b (1982) 299 [25] A. L. Rheingold, D. L. Staley, M. E. Fountain: J. Organo-
met. Chem. 365 (1989) 123 [26] R. Borowski, D. Rehder, K. von Deuten: J. Organomet.
Chem. 220 (1981) 45 [27] A. Boardman, R. u! ZZ. Small, I. J. Worrall: Inorg. Chim.
Acta 121 (1986) L35 [28] G. Baum, A. Greiling, W Massa, B. C. Hui, J. Lorberth:
Z. Naturforsch. 44b (1989) 560 [29] JK Honle, J . Wolf; H. G. von Schnering: Z. Naturforsch.
43b (1988) 219; K Honle, H. G. von Schnering, M. So- mer: Z. Kristallogr. 174 (1986) 82
[30] B. K Eichhorn, R. C. Haushalter, J. C. Huffman: Angew. Chem. 101 (1989) 1081; R. C. Haushalter: J. Chem. Soc., Chem. Commun. 1987, 196; R. C. Haushalter, B. W Eich- horn, A. L. Rheingold, S. J. Geib: J. Chem. SOC., Chem. Commun. 1988, 1027
[31] A. Haaland: J. Mol. Struct. 97 (1983) 115; G. Becker, G. Gutekunst: Z . anorg. allg. Chem. 470 (1980) 157
[32] M. Schiitze: Angew. Chem. 70 (1958) 697 [33] E. Wiberg, K. Modritzer: Z. Naturforsch. 12b (1957) 123 [34] H. Schbyer, G. Fritz, JK Holderich: Z . anorg. allg. Chem.
[35] G. Becker, D. Kashummer, M. Schmidt, lK Schwarz: 428 (1977) 222
unveroffentlicht
Anschr. d. Verf.:
Prof. Dr. C. Becker, Dr. Martina Schmidt, Dr. M. Westerhausen Institut fur Anorganische Chemie der Universitat Stuttgart Pfaffenwaldring 55 W-7000 Stuttgart 80 (Vaihingen), Bundesrepublik Deutschland


![Polysulfonylamine, XII [1] N-Acyl-dimesylamine (N.N ...zfn.mpdl.mpg.de/data/Reihe_B/43/ZNB-1988-43b-1495.pdf · 1497 A. Blaschette et al. N-Acyl-dimesylamine Tab. II. Spektroskopische](https://img.pdfslide.org/doc/110x75/5eccdbe82a00c011850e8902/polysulfonylamine-xii-1-n-acyl-dimesylamine-nn-zfnmpdlmpgdedatareiheb43znb-1988-43b-1495pdf.jpg)



![Acyl- und Alkylidenphosphane, XV [1] 2.2 ...zfn.mpdl.mpg.de/data/Reihe_B/36/ZNB-1981-36b-0016.pdf · G. Becker et al. Acyl- und Alkylidenphosphane 17 [11, s. auch 12] ergibt für](https://img.pdfslide.org/doc/110x75/5e117fb1d56d93523e1f3b17/acyl-und-alkylidenphosphane-xv-1-22-zfnmpdlmpgdedatareiheb36znb-1981-36b-0016pdf.jpg)








![En route to multi-bridged metallocenes · PDF filePG Protecting Group . VII Ph Phenyl ... 5.4.7 2,2-Dimethyl-1,1,1-tris[1’-(tributylstannyl)ferrocenyl]propane (175 ... (1-methoxy-1-methylethyl)oxazoline](https://img.pdfslide.org/doc/110x75/5a972f777f8b9ad96f8d0ded/en-route-to-multi-bridged-metallocenes-protecting-group-vii-ph-phenyl-547.jpg)













