Embed Size (px)
Citation preview

1
Analytik I
Vorlesungsskript

2
Einführung In der Vorlesung „Analytische Chemie I werden Sie mit den theoretischen Grundlagen und praktischen Anwendungen der quantitativen Analyse vertraut gemacht; genauer gesagt: mit den klassischen Methoden der quantitativen Analyse. Die physikal.-chemischen Methoden werden in Analytik II behandelt. Dabei sollen Sie nicht nur lernen, wie man nach Vorschrift eine Quantitative analytische Bestimmung durchführt, bzw. nachkocht (das ist die Ausbildung eines Chemielaboranten). Sie sollen vielmehr auch beurteilen lernen, warum eine Vorschrift gerade diese oder jene Form hat und warum diese Form sinnvoll ist. Auch ist es wichtig zu wissen, welche der für die Bestimmung etwa eines Elementes in einem chemischen System zahlreichen existierenden Methoden zur Lösung eines spezifischen Problems die geeignetste sein wird.
Die klassische quantitative Analyse basiert im wesentlichen auf einer quantitativen Verfolgung der Chemie von Ionen in wässriger Lösung. Die charakteristischen Reaktionen von Ionen in Lösung selbst können zu deren Identifizierung, d.h. zu ihrem qualitativen Nachweis herangezogen werden. Mit dieser Problematik befasst sich auch die einschlägige Vorlesung im Hauptseminar, die zugleich Grundlage für die Vorlesung Anal. I ist. Ein Problem hierbei ist, dass diese Veranstaltungen parallel laufen.
Was ist analytische Chemie? Αναλυειν = auflösen
Der Begriff Analyse entstammt der Philosophie und lässt sich etwa wie folgt charakterisieren: Erscheinungen der objektiven Realität werden in ihre Elemente und in Relationen zwischen diesen Elementen zergliedert. Durch eine gedankliche Synthese wird das primär vorhandene Ganze rekonstruiert. Die Übertragung auf die chemische Analyse führt etwa zu folgendem Schema:
Chemisches System
reiner Stoff, Gemisch u.a.
Analyse
"Elemente" = Atome, Ionen, Moleküle Gruppen, Gemischbestandteile
Reaktionen zwischen "Elementen"
Qualität Quantität Strukturen
gedankliche Synthese
Anders formuliert: Die analytischen Methoden dienen der Beantwortung folgender Fragen:
a) Was liegt vor? => qualitative Analyse
b) Wie viel liegt vor? => Quantitative Analyse
c) Wie liegt es vor? =>Strukturanalyse
Demonstration an einem einfachen Beispiel: Kochsalz
a) Na+ und Cl-
b) 1 mol Na+ und 1 mol Cl-

3
c) Jedes Na+ ist 6 Cl-, jedes Chlorid von 6 Natriumionen als nächsten Nachbarn in regelmäßig kubischer Anordnung umgeben: Steinsalzgitter
Häufig besteht die Problemstellung allerdings nicht in einer derart vollständigen Beschreibung des gegebenen Systems, man begnügt sich dann damit, einen bestimmten Anteil in der Probe gespeicherter Information über deren qualitative und/oder quantitative Zusammensetzung zu erschließen.
Literatur K. Doerffel, R. Geyer, H. Müller „Analytikum. Methoden der analytischen Chemie und ihre Grundlagen“ (auch instrumentell) Deutscher Verlag f. Grundstoffindustrie, 9. Auflage 1994 E. Fluck, M.Becke-Goehring “Einführung in die Theorie der quantitativen Analyse” Verlag Steinkopff, 7. Auflage 1990 J.S: Fritz, G.H. Schenk „quantitative Analytische Chemie“ (auch instrumentell) Vieweg 1989 U.R. Kunze, G. Schwedt „Grundlagen der qualitativen und quantitativen Analyse“ VCH Wiley Verlag, 5. Auflage 2001, 32,90 € D.C. Harris „Lehrbuch der quantitativen Analyse“ (auch instrumentell) Springer 1998 M. Otto „Analytische Chemie“(auch instrumentell) VCH 1995 62,90 € G. Jander, E. Blasius „Einführung in das anorganisch-chemische Praktikum“ Hirzel Verlag 14. Auflage 34.80 € G. Jander, K.F. Jahr „Maßanalyse“ W. de Gruyter 16. Auflage 2003 24.95 € H. Lux, W. Fichtner „Quantitative Anorganische Analyse“ Springer 9. Auflage 1992 Methodenübersicht Die Methoden der qual. und quant. Elementar- bzw. Strukturanalyse haben sich im Laufe der Zeit stark gewandelt. Am Anfang standen die 1. klassischen chemischen Methoden. Ihnen liegt jeweils eine stöchiometrisch vollständig ablaufende chemische Reaktion zugrunde.
m C + n R → CmRn
C: zu bestimmender Bestandteil R: Reagens CmRn Produkt
Hierzu zählen:

4
1.1. Gewichtsanalyse = Gravimetrie bzw. Elektrogravimetrie Bei der Gravimetrie wird CmRn als schwerlösliches Produkt erzeugt und ausgewogen. Bei der Elektrogravimetrie wird das schwerlösliche Produkt durch Elektrolyse erzeugt und ausgewogen. Die Menge des Reagenzes wird nicht bestimmt.
1.2. Maßanalyse = Titrimetrie Bestimmung der Menge an Reagenz (Volumen einer Lösung bekannten Gehalts) das bis zum Endpunkt (vollständige Umsetzung) verbraucht wird. Der Gehalt der Maßlösung wird zuvor durch eine Wägung eingestellt (indirekte Wägung). Die Produktmenge CmRn wird nicht bestimmt.
1.3. Gasanalyse auf der Basis von Gewichts – oder Volumenmessungen
Eine Komponente in einem Gasgemisch wird z.B. durch eine chemische Reaktion in einer flüssigen Phase gebunden, es erfolgt Messung der Volumenänderung der Gasphase.
Die moderne Entwicklung ist gekennzeichnet durch die zunehmende Einführung von physikalisch-chemischen Methoden
Hierzu lässt sich praktisch jede physikalische Eigenschaft von Stoffen heranziehen. Man unterteilt in vier wichtige große Gruppen:
2.1. Elektrische Methoden: Beispiel Coulometrie = Messung der Ladungsmenge, die für eine quantitative Umsetzung erforderlich ist.
2.2. optische Methoden (Spektroskopie) Messung der Lichtabsorption eines gefärbten Stoffes in Lösung
2.3. Thermische Methoden
2.4. Nukleare Methoden; Überführung eines Elementes in ein radioaktives Isotop (durch Neutronenbestrahlung) und Messung der Radioaktivität
Die physikalischen Methoden zeichnen sich durch folgende Vorteile aus:
• Geringer Zeitaufwand
• Erweiterung des Messbereiches in den Ultramikrobereich
• Simultane oder sequentielle Bestimmung mehrerer Bestandteile
• Ermöglichung vorher praktisch undurchführbarer Strukturanalysen.
In der quantitativen Analytik sind die klassischen Methoden jedoch nicht passee, denn es sind absolute Methoden, deren Genauigkeit innerhalb des Messbereiches die der instrumentellen Methoden häufig übertrifft. Die instrumentellen erfordern überdies Eichung.
Die mangelnde Spezifität von chemischen oder physikalisch-chemischen Verfahren macht häufig eine der eigentlichen Bestimmungen vorgelagerte Trennung erforderlich.
Übersicht der Trennverfahren:

5
Übersicht über analytische Bestimmungsmethoden
1. Klassische Methoden 2. Physikalisch-chemische Methoden
Bestimmung der gesuchten Substanz erfolgt über direkte oder indirekte Wägung oder Volumenmessung: 1.1. Gewichtsanalyse (Gravimetrie) einschließlich Elektrogravimetrie 1.2. Maßanalyse (Volumetrie, Titrimetrie) 1.3. Methoden der Gasanalyse auf der
Basis von Gewichts- und Volumen- messung
Bestimmung der gesuchten Substanz beruht auf Messung von physikalischen Eigenschaften mit Hilfe spezieller Geräte: 2.1. Elektrische Methoden 2.2. Optische (spektroskopische)
Methoden 2.3. Thermische Methoden 2.4. Nukleare Methoden
Übersicht über analytische Trennverfahren
Trennung durch 1. Fällung 2. Elektrolyse 3. Komplexbildung (Maskierung) 4. Destillation 5. Extraktion 6. Chromatographie 7. Ionenaustausch 8. Elektrophorese 9. Dialyse

6
Analysengang Die Lösung eines analytischen Problems gliedert sich in mehrere „strategische“ Abschnitte. Gesetzt den Fall, Sie sollen ein x-beliebiges System auf einen oder mehrere Bestandteile hin untersuchen: ein Werkstoff, ein Mineral, biologisches Material, Nahrungsmittel, u.s.w. Dann würde sich folgendes Schema anbieten: siehe Umdruck
Probennahme: Ist ein Problem für sich und kann sehr kompliziert sein, wird bei wertvollen Produkten oder etwa in der forensischen Chemie oder der Nahrungsmittelkontrolle häufig sogar von vereidigten Sachverständigen vorgenommen.
Auflösen bzw. Aufschluss: Auflösen in Wasser oder ggf. org. Solventien, dabei oft Zusätze von Nöten: Säuren, oxidierende Säuren, Komplexbildner, Basen.
Versagen derartige Lösungsmittel ist ein Aufschluss vorzuschalten, in der Regel eine chem. Reaktion bei höherer Temperatur (>200°C): konz. H2SO4, oxidierende reduzierende, sulfidierende, saure oder alkalische Salzschmelzen, Gase. Problem der Lösungs- oder Aufschlussmittel: Verunreinigungen, wichtig ist die richtige Dosierung.
Gravimetrie Gegenüberstellung Gravimetrie – Titrimetrie
Bei den gravimetrischen Verfahren werden die zu bestimmenden Ionen oder Moleküle unter genau festgelegten Arbeitbedingungen in Form einer Schwerlöslichen Verbindung als Niederschlag abgeschieden.
Beispiel: Fe-Bestimmung
Durch Vorbehandlung wird alles Fe in Fe3+ überführt = Oxidation in saurer Lösung mit H2O2 oder HNO3.
H2O2 + 2 Fe2+ + 2 H+ → 2 H2O + 2 Fe3+
Die Ausfällung von Fe3+ erfolgt als Hydroxid. Hierzu wird die H+-Konzentration herabgesetzt, der pH-Wert also erhöht. Dies kann durch Zusatz von NH3, Acetat oder Urotropin erfolgen:
1) NH3 + H+ → NH4+
2) NaOAc + H+ → Na+ + HOAc
3) (CH2)6N4 + 6 H2O → 4 NH3 + 6 HCHO
N
CH2 CH2CH2
NN N
CH2CH2
CH2
Durch Verringerung der Protonenkonz. hydrolysieren die Fe3+ Ionen in der Lösung, es scheidet sich ein rotbrauner wasserhaltiger Niederschlag ab: Fe(OH)3⋅ x H2O (Fällungsform) des zu bestimmenden Stoffes. X ist nicht definiert, die Zusammensetzung des Niederschlages schwankt also, die Auswaage ist zur Berechnung des Eisengehaltes ungeeignet. Daher muss der Nd. durch geeignete Operationen in eine Wägeform mit stöchiometrisch genau definierter Zusammensetzung überführt werden.
Dies geschieht im Beispiel durch Abfiltrieren, Auswaschen (Cl—frei) Veraschen des Filters und Glühen bei 600-700 °C. man erhält als Wägeform Fe2O3.
Zusammenstellung der Arbeitsschritte: 1) Vorbehandlung: Oxidieren/Reduzieren, Eindampfen/verdünnen
2) Fällung: Abscheidung des zu bestimmenden Stoffes unter festgelegten Bedingungen in Form einer schwerlöslichen Verbindung (Fällungsform)
3) Filtration: Trennung von Nd. und Mutterlauge
4) Waschen: Beseitigung von anhaftenden Fremdstoffen.

7
5) Prüfung auf Vollständigkeit der Fällung: Nochmalige Reagenzzugabe zur Mutterlauge.
6) Trocknung, ggf. Glühen: Fällungsform in Wägeform überführen.
7) Abkühlen: im Exsikkator (Zeitbedarf)
8) Wägen: ggf. Wiederholung von Operationen 6-8 bis zur Gewichtskonstanz.
9) Berechnung: des zu best. Stoffes aus der Auswaage.
Bei den maßanalytischen Verfahren bringt man ebenfalls die Stoffe in Form zweier Lösungen zur Reaktion, die Zugabe von Reagenzien erfolgt jedoch in Form einer Lösung genau bekannten Gehaltes: Titerlösung oder Maßlösung.
Die Zugabe der Lösung erfolgt bis zur quantitativen Umsetzung, d.h. bis zum Reaktionsendpunkt bzw. bis zum Äquivalenzpunkt.
Beispiel: HCl-Bestimmung
In einem Becherglas oder einem Weithals-Erlenmeyerkolben wird eine bestimmte Menge einer HCl-Lösung unbekannten Gehaltes vorgelegt. Nun wird aus einer Bürette (kalibriertes Glasrohr mit Hahn und Abtropfspitze am unteren Ende) die Reagenzlösung zugegeben. Man verwendet eine NaOH-Lsg. Genau bekannten Gehaltes. Zur Erkennung des Äquivalenzpunktes (hier Neutralpunkt) muss man einen sog. Indikator zugeben: ein Farbstoff, der bei einem bestimmten pH-Wert seine Farbe ändert. Man titriert genau bis zum Farbumschlag und liest an der Bürette das Volumen der zur Neutralisation verbrauchten Natronlauge ab. Bei Kenntnis der Reaktionsablaufes lässt sich aus dem Verbrauch an Reagenzlösung (aus Volumen und Konzentration) die gesuchte Stoffmenge berechnen. Dieser Vorgang heißt Titration.
Normalerweise beinhaltet also die Titration 3 Teilschritte:
1) Vorbehandlung
2) Titration
3) Berechnung
Die Waage lässt sich allerdings auch bei der Volumetrie nicht umgehen, man benötigt sie zur Einstellung des Gehaltes der Maßlösung (wird später eingehend behandelt). Dieser Vorgang erfolgt bei der Volumetrie jedoch nur einmal und muss erst wiederholt werden, wenn der Vorrat an Maßlösung verbraucht ist.
Vergleich: Gravimetrie – Titration Gravimetrische Bestimmungsverfahren sind langwieriger und zeitraubender als volumetrische, es entfallen: Filtrieren, Waschen Trocknen, Glühen und die Kontrolle der Gewichtskonstanz. Die Volumetrie ist schneller und von geringerem Schwierigkeitsgrad. Wir kommen auf den Vergleich später nochmals zurück. Zuvor seien einige andere Aspekte behandelt.

8
Fehler in der Analytik Normalerweise werden an einer Probe mehrere Bestimmungen durchgeführt und es wird der Mittelwert aus den Einzelmessungen gebildet.
∑=
=n
iixn
x1
1
x = Mittelwert
xi = Einzelwert
n = zahl der Messungen
Für die Einschätzung der Qualität analytischer Ergebnisse sind vorrangig 2 Begriffe wichtig:
• Richtigkeit = Genauigkeit = Maß für die Abweichung des Mittelwertes vom Sollwert
• Präzision = Wiederholbarkeit = Maß für die Streuung der Einzelwerte
Die Verhältnisse lassen sich gut anhand der Treffer auf einer Zielscheibe veranschaulichen, deren Mittelpunkt der Sollwert darstellt. S. Umdruck
Präzision schlecht
Richtigkeit schlecht
Präzision schlecht
Richtigkeit gut
Präzision gut
Richtigkeit schlecht
Präzision gut
Richtigkeit gut
••
•
•
• •••
•
•
••••
•
•
••
••••••••
••
•• •••••••
•
••
••
•
••
• •••
Gibt man als Ergebnis nur den Mittelwert an, kann man weder über die Richtigkeit noch über die Präzision aussagen machen. Das Aufspüren der Ursachen für mangelhafte Richtigkeit und/oder Präzision ist umso schwieriger, je mehr Arbeitsschritte ein Analysengang erfordert, da jeder einzelne Arbeitsschritt genau untersucht werden muss.

9
Mengen-, Gehalts- und Konzentrationsangaben Der allg. Begriff für eine begrenztes Materiesystem ist die Stoffportion, in der Analytik z.B. Probe, Einwaage, Auswaage. Die Quantität einer Stoffportion lässt sich durch Angabe ihrer Eigenschaften Masse, Volumen, Anzahl der enthaltenen Teilchen erfassen. Letzteres ist für die Chemie von maßgeblicher Bedeutung und hat zur Festlegung der Größe „Stoffmenge“ geführt.
Das Mol (Einheitenzeichen mol) wurde 1971 als Basiseinheit für die Stoffmenge in das SI-System eingeführt. Die Stoffmenge ist eine messbare Eigenschaft einer Stoffportion (eine Substanzprobe hat eine Stoffmenge).
Definition:
Das Mol ein die Stoffmenge eines Systems, das aus ebensoviel Einzelteilchen besteht, wie Atome in 12 g des Kohlenstoffnuklides 12C enthalten sind.
Das Mol ist damit eine reine Zählgröße mit dem Größenzeichen n.
Bei Berechnungen soll die Stoffmenge durch eine Größengleichung angegeben werden. Da sich die Einheit Mol nur auf die Teilchenzahl, nicht auf die Teilchenart X bezieht, muss letztere immer angegeben werden:
n (x) = a mol z.B. n(S8) = 3,5 mol Größe Teilchenart Maßzahl Einheit
Die experimentell messbare Teilchenzahl pro Mol wird als Avogadro-Konstante (molare Teilchenanzahl, Symbol NA) bezeichnet: NA = (6,022045 ± 0,000031) 1023 mol-1
Verknüpfung von Stoffmenge n (x) und Teilchenzahl N(x) einer Teilchenart x: ANxNxn )()( =
Molare Masse Beim praktischen Arbeiten erfolgt die Messung der Quantität der bei einer Reaktion umgesetzten Stoffe nicht aufgrund ihrer Teilchenzahl, sondern mittels der Waage über ihre Masse. Die Größe, mit der man Masse m und Stoffmenge n ineinander umrechnen kann, ist die molare Masse M:
m = M (x) ⋅ n(x)
Damit ist M(x) die Masse der Stoffmenge 1 mol. Die SI-Einheit ist kg/mol, aber üblich ist g/mol. Da man M auf verschiedene Teilchen beziehen kann, wird das Symbol für das Teilchen in Klammern hinter das Größenzeichen gesetzt, z.B.:
M(S) = 32,06 g /mol
M(SO42-) = 96,06 g /mol
M(H2SO4) = 98,07 g /mol
Durch die Definition der Basiseinheit Mol ist die molare Masse von 12C als Bezugswert vorgegeben: M(12C) = 12 g/mol
Zwischen der molaren Masse M und der Masse eines Einzelteilchens mT besteht folgender Zusammenhang:
M = mT ⋅ NA NA = 6,022 1023 mol-1
Die molare Masse ist also die Masse von NA Molekülen, Atomen oder Ionen.
Übungsaufgabe: Wie viele S-Atome befinden sich in 1 g Schwefelsäure?

10
m = 1 g M(H2SO4) = 98,07 g/mol m/M = n(H2SO4) = 1 / 98,07 = 0,010197 mol
N = n ⋅ NA = 0,010197 ⋅ 6,022 ⋅1023= 6,14 ⋅ 1021 H2SO4 – Moleküle = S-Atome
Gehalt von Lösungen - Konzentration Lösungen kommt in der Maßanalyse eine zentrale Bedeutung zu. Gehalt ist der allgemeine (qualitative) Oberbegriff (z.B. Bleigehalt eines Erzes); werden Zahlenwerte angegeben, so verwendet man die Begriffe Anteil und Konzentration. Die quantitative Zusammensetzung einer Lösung lässt sich mittels der Größen Masse m, Stoffmenge n oder Volumen V beschreiben.
Anteile Sind Verhältnisgrößen, (keine Einheit) in denen eine der genannten Größen eines Bestandteiles i (mi, ni Vi) auf dieselbe Größe aller Bestandteile einer Stoffportion (m, n, V0) bezogen ist. Man unterscheidet:
Massenanteil mm
mmw i
ii
ii ==∑
z.B. g / kg
Stoffmengenanteil nn
nnx i
ii
ii ==∑
z.B. mol/mol
Volumenanteil 0VV
VV i
ii
ii ==∑
ϕ z.B. ml/l
V0 ist das Volumen aller Bestandteile vor dem Vermischen; ist mit dem Gesamtvolumen V der Mischung nur identisch, wenn beim Mischen keine Volumenänderung eintritt (ideale Mischung).
Die Angabe erfolgt entweder durch Dezimalbruch (0,001), Verhältnisbezeichnungen (%, ‰) oder gleichartige Einheiten in Zähler und Nenner. Typische Angaben für Spurenbestandteile:
ppm (parts per million) 1 : 106 µg/g mg/kg
ppb (parts per billion) 1 : 109 ng/g µg/kg
ppt (parts per trillion) 1 : 1012 pg/g ng/kg
Konzentration Gehaltsgröße bei denen die Quantitätsgröße des Bestandteils i auf das Volumen der Lösungsportion bezogen wird. Man unterscheidet:
Massenkonzentration Vmi
i =β g/l z.B.: 1 g / 100 ml = 10 g /l H2SO4
Stoffmengenkonzentration Vnc i
i = mol/l z.B.: 0,0102 mol /100 ml = 0,102 mol/l H2SO4
Volumenkonzentration VVi
i =σ z.B.: 1 g H2SO4= 0,55 ml => 0,55 ml / 100 ml = 0,0055
Für ideale Mischungen ist σi = ϕi
Wenn V = f(T) sind alle 3 Konzentrationsgrößen temperaturabhängig!

11
Massenkonzentration βi und Massenanteil wi lassen sich über die Dichte ρ = m/V der Lösung ineinander umrechnen:
ρρβ ⋅=⋅== iii
i wmm
Vm
bi = 0,01097 mol / 99,45 g = 1,1⋅10-4 mol/g
Die wichtigste Gehaltsgröße in der Maßanalyse ist aber die Stoffmengenkonzentration c. Um den Teilchenbezug von n auszudrücken, setzt man wieder die Teilchenart in Klammern:
c(x) = n(x) / V => n(x) = c(x) ⋅ V
Früher wurde die Stoffmengenkonzentration auch als Molarität M bezeichnet, Beispiel:
Früher: 0,05 M H2SO4 heute: c(H2SO4) = 0,05 mol/l
Die Gegenseitige Umrechnung von Massen- und Stoffmengenkonzentration erfolgt mit Hilfe der molaren Masse des gelösten Stoffes:
βi= c(x) ⋅ M(x) bzw. c(x) = βi / M(x)
Eine T-unabhängige Größe ist die Molalität b; bi ist die auf die Masse einer Lösungsmittelportion m(Lm) bezogene Stoffmenge ni der darin gelösten Stoffportion:
bi = ni / mLösungsmittel

12
Äquivalentteilchen bzw. Äquivalent Unter einem Äquivalent versteht man rein formal den Bruchteil 1/z* eines Teilchens X (Atom, Molekül, Ion, Atomgruppe), der bei einer bestimmten Reaktion jeweils am Austausch von einer positiven oder negativen Ladung beteiligt ist. Die immer ganzzahlige Zahl z* wird als Äquivalentzahl bezeichnet.
Arten von Äquivalentteilchen Säure-Base-Äquivalent (Neutralisationsäquivalent)
Es charakterisiert ein gedachtes Teilchen, das bei einer Säure-Base-Reaktion ein Proton freisetzen oder binden kann, z.B. HCl, 1/2 H2SO4, 1/3 H3PO4, NaOH, 1/2 Ba(OH)2, 1/3 Al(OH)3. Die Äquivalentzahl z* ist gleich der Anzahl H+- oder OH−-Ionen, die das Teilchen bei vollständiger Umsetzung abgibt.
Redox-Äquivalent Kennzeichnet ein Teilchen, das bei einer Redoxreaktion ein Elektron aufnehmen oder abgeben kann, z.B. Fe2+, 1/5 KMnO4, 1/6 KBrO3. Die Äquivalenzzahl z* ist gleich dem Betrag der Differenz der Oxidationszahlen vor und nach der Reaktion desjenigen Atoms, das dabei seine Oxidationszahl ändert.
Ionen-Äquivalent Ist als Bruchteil eines Ions zu betrachten, der seiner positiven oder negativen Ladung entspricht, z.B. Na+, 1/2 Mg2+, 1/3 Al3+, Cl−, 1/2 SO4
2−. Die Äquivalentzahl z* ist gleich dem Betrag der Ladungszahl des an der Reaktion beteiligten Ions, z.B. beim Ionenaustausch oder bei der elektrolytischen Abscheidung.
Für einen bestimmten Stoff kann die Äquivalentzahl je nach betrachteter Reaktion variieren.
Beispiel Permanganat (MnO4−):
In stark saurer Lösung erfolgt Reduktion zu Mn2+ gemäß
Mn(+7)O4− + 8 H+ + 5 e− → Mn2+ + 4 H2O z* = 5
In schwach saurer oder neutraler Lösung erfolgt Reduktion zu MnO2 gemäß
Mn(+7)O4− + 2 H2O + 3 e− → Mn(+4)O2 + 4 OH− z* = 3
Die Stoffmenge von Äquivalenten, n(1/z* X), wird ebenfalls in Mol angegeben.
Beispiel H2SO4:
Für eine vorgegebene Schwefelsäureportion sei die Stoffmenge, bezogen auf H2SO4-Moleküle
n(H2SO4) = 0,1 mol
Bezieht man sie auf Äquivalente, ist die Stoffmenge
n(½H2SO4) = 2 · 0,1 mol
Daraus folgt
2n(H2SO4) = n(½H2SO4) (d.h. jedes Molekül H2SO4 entspricht 2 Äquivalenten ½H2SO4)
Allgemein besteht für eine Stoffportion zwischen der Stoffmenge der Teilchen X und der Stoffmenge ihrer Äquivalentteilchen die Beziehung
n(1/z* X) = z* · n(X)

13
Vergleich: Gravimetrie – Titration (2. Teil) Warum Gravimetrie, wenn Titration soviel einfacher ist?
Bei der Gravimetrie lässt sich eine höhere Genauigkeit erzielen. Wir wollen hierzu nun die Ablesefehler der Messgeräte betrachten:
Rechenbeispiel: Vorgelegte Fe-Lösung: V = 20 ml, c(Fe) = 0,1 mol/l
=> Stoffmenge (Fe): n = c ⋅ V = 0,1 mol/l ⋅ 2 ⋅10-2 l = 2 ⋅10-3 mol = 2 mmol
Masse m(Fe): m = n ⋅ M = 2 mmol ⋅ 55,847 g/mol = 111,694 mg
1) Bestimmung mittels Gravimetrie
Normale Analysenwaage ablesbar auf 0,1 mg => der max. Fehler sein 0,05 mg.
Fe → Fe3+ → Fe(OH)3⋅x H2O → Fe2O3
Fällungsform Wägeform
Theoretische Auswaage: 159,692 mg (2 Fe = 111,694 mg + 3 O =47,997 mg)
Praktische Auswaage: 159,7 mg, max. Fehler 0,05 mg
Rel. Fehler = 0,05 mg / 159,7 mg = 0,00031 = 0,031 %
2) Bestimmung durch Titration
Normale 50 ml-Bürette ablesbar auf 0,1 ml => max. Fehler sei 0,05 ml
Fe → Fe2+ Titration mit KMnO4 mit c = 0,02 mol/l
5 Fe2+ + MnO4- + 8 H+ → 5 Fe3+ + Mn2+ + 4H2O
5 mol (Fe) = 1 mol (KMnO4)
2 mmol (Fe) = 2/5 mmol (KMnO4) = 2/5 mmol / 0,02 mol/l = 20 ml
Es würden also 20 ml Maßlösung gebraucht. Max. Fehler 0,05 ml
Rel Fehler: 0,05 / 20 = 0,0025 = 0,25 %
Folgerung: Vorteil der Maßanalyse: Geringer Zeitbedarf und geringere Schwierigkeitsgrad; Nachteil gegenüber Gravimetrie: geringere Genauigkeit. Weiterhin ist die Maßanalyse oft unspezifisch. Liegen mehrere Bestandteile in einer Probe vor, können sie u.U. nebeneinander titriert werden. Dagegen existiert bei der Gravimetrie bessere Kontrollmöglichkeiten durch qualitative und quantitative Analyse des Niederschlages, man kann ihn weiter reinigen oder andere Korrekturen anbringen.
Bedeutsamer Nachteil der Gravimetrie: nicht automatisierbar.
Fazit: Gravimetrische Verfahren werden heute in der Praxis routinemäßig bis auf wenige Ausnahmen nicht mehr durchgeführt. Sie bilden aber die Grundlage für die Kalibrierung und Kontrolle der modernen instrumentellen Bestimmungsverfahren.

14
Einteilung der Gravimetrie Erfolgt nach der Art der Abscheidung
1) Abscheidung des gesuchten Elements, meist jedoch einer schwerlöslichen stabilen Verbindung des betreffenden Elements auf chemischem Wege (Zugabe von Fällungsreagenz)
2) Abscheidung des gesuchten Elementes oder Verbindung mit Hilfe des elektrischen Stromes = Elektrogravimetrie.
Einteilung der Volumetrie Es empfiehlt sich die Einteilung nach der Art der Endpunktsermittlung vorzunehmen:
1) Endpunktsanzeige durch einen chem. Indikator
2) Endpunktsanzeige durch physik.-chem. Methoden (Konduktometrie, Potentiometrie, HF-Titration, Amperometrie, Voltametrie, photometr. Titration. Term. Titration u.s.w. Anal II)
Eine weitere Unterteilung wird später behandelt.
Exkurs: Chemisches Gleichgewicht und Massenwirkungsgesetz Keine chemische Reaktion läuft vollständig ab. Lässt man beispielsweise Wasserstoff und Iod miteinander reagieren, bildet sich Iodwasserstoff.
H2 + I2 → 2 HI
Es reagieren aber nicht alle H2- und I2-Moleküle miteinander zu HI-Molekülen, sondern die Reaktion verläuft unvollständig. Bringt man in ein Reaktionsgefäß genau 1 mol H2 und 1 mol I2, so bilden sich z.B. bei 490 °C nur 1,544 mol HI im Gemisch mit je 0,228 mol H2 und I2. Auf der Teilchenebene sieht es so aus, dass ja ein H2 mit einem I2-Molekül zusammenstoßen muss, damit sich die beiden Produktmoleküle HI bilden können. Dabei ist natürlich nicht jeder Zusammenstoß erfolgreich. Gleichzeitig können aber auch zwei HI-Moleküle zusammenstoßen und wieder H2 und I2 bilden, dies bezeichnet man als die Rückreaktion.
2 HI → H2 + I2
Dass diese tatsächlich abläuft kann man sehen, wenn man reines HI (2 mol) in das Reaktionsgefäß einfüllt und ebenfalls auf 490 °C erhitzt. Wieder stellt sich das gleiche Verhältnis von H2, I2 und HI ein (0,228 mol : 0,228 mol : 1,544 mol).
Zwischen allen drei Molekülen bildet sich also ein Zustand, bei dem keine weitere Änderung der Zusammensetzung des Reaktionsgemisches erfolg. Diesen Zustand nennt man chemisches Gleichgewicht. Der Gleichgewichtszustand ist kein Ruhezustand. Nur makroskopisch sind im GGW-Zustand keine Veränderungen feststellbar. Tatsächlich erfolgt aber dauernd Zerfall und Bildung von HI-Teilchen. Pro Zeiteinheit werden genauso viele Teilchen gebildet wie zerfallen.
H2
I2
HI

15
Zeit
Konz
1
2
[H2] [I2]
[HI]
Zeit
Konz
1
2
[H2] [I2]
[HI]
Das Auftreten eines Gleichgewichts wird bei der Formulierung von Reaktionsgleichungen durch einen Doppelpfeil wiedergegeben.
Bei vielen chemischen Reaktionen sind allerdings im Gleichgewicht überwiegend die Komponenten einer Seite vorhanden. Man sagt dann, das GGW liegt auf einer Seite.
Die Lage eines chemischen GGWs wird durch das Massenwirkungsgesetz beschrieben. Es lautet für die Iodwasserstoffreaktion:
H2 + I2 2 HI H2 + I2 HI + HI
][][][][
][][][
2222
2
IHHIHI
IHHIK
⋅⋅=
⋅=
K ist die GGW-Konstante für die jeweilige Reaktion, sie ist von der Temperatur abhängig. Für unser obiges Beispiel ergibt sich 45,9 = K.
Liegt das GGW auf der Produktseite ist K sehr groß, ist K hingegen klein, liegen vorwiegend die Edukte vor.
Für eine allgemeine chem. Reaktion lautet das MWG:
aA + bB cC + dD ba
dc
BADCK][][][][
⋅⋅=
Das Produkt der Konzentrationen der Produkte erscheint im Zähler, das der Edukte im Nenner. Im MWG sind die Konzentrationen der Stoffe multiplikativ verknüpft, die stöchiometischen Zahlen a,b,c und d treten daher als Exponenten der Konzentrationen auf.
Alle im MWG auftauchende Stoffe müssen sich in der gleichen Phase (gasförmig, oder flüssig bzw gelöst) befinden. Feststoffe besitzen keine Konzentration und werden aus c = 1 mol/l gesetzt.
Beispiel 1: Boudouard-GGW CO2 + C 2 CO 1][
][
2
2
⋅=COCOK
Beispiel 2: Auflösung von AgCl AgCl Ag+ + Cl- 1
]][[ +−= AgClK
GGW, bei denen nicht alle Stoffe in der gleichen Phase sind, bezeichnet man als heterogene GGW.
GGW lassen sich verschieben durch Konzentrations-, Temperatur- oder Druckänderungen. (Prinzip von Le Chatelier)
•Eine Temperaturerhöhung begünstigt die endotherme Reaktion (Änderung der GGW-Konstanten) •Eine Druckerhöhung begünstigt die Reaktion, bei der die Teilchenzahl vermindert wird.
Beispiel: N2 + 3 H2 2 NH3 DH = -92 kJ (exotherm)
Temperaturerhöhung : GGW verschiebt sich nach links Druckerhöhung: GGW verschiebt sich nach rechts.

16
Theoretische Grundlagen der Fällung Grundsatz: keine Fällung ist vollständig! Dies folgt aus der Tatsache, dass jede Niederschlagsbildung eine Gleichgewichtsreaktion ist.
A + B AB↓
Somit tritt auch Rückreaktion ein, das GGW liegt jedoch stark auf der rechten Seite.
Kinetische Deutung des thermodynamischen GGW: ständig gehen Ionen von der Oberfläche des Nd. aus in Lösung, ständig kristallisieren Ionen aus der Lsg. An die Nd.-Oberfläche an. Im GGW-Zustand lösen sich in der Zeiteinheit ebenso viele Teilchen wie auskristallisieren, so dass ein scheinbarer Stillstand nach außen hin herrscht.
Folge: Jeder Stoff besitzt eine gewissen wenn vielleicht auch äußerst geringe Löslichkeit. Es ist also sachlich unsinnig, von unlöslichen Verbindungen zu sprechen. Besser ist der Terminus: Schwerlöslich!
Für eine quant. Analyse braucht eine Fällung auch gar nicht vollständig zu sein, Sie muss jedoch quantitativ sein. Was heißt das?
Quantitativ ist eine Fällung dann, wenn die in der Lösung verbliebene Restmenge auf der Waage nicht mehr erfasst werden kann, also unterhalb der Wägegenauigkeit liegt. Wie wir sahen liegt letzteres bei 0,1 mg = 10-4 g.
Von einer quantitativen Fällung spricht man also dann, wenn sich im Liter weniger als 10-4g des Niederschlages lösen.
Da man in der Praxis bei Fällung meist im Bereich von 100 – 300 ml Lösung arbeitet, unterschreitet man mit Sicherheit die Wägegenauigkeit.
Löslichkeit und Löslichkeitsprodukt Das Löslichkeitsprodukt leitet sich vom MWG her.
Wir wenden das MWG auf den Lösungsvorgang eines schwerlöslichen Salzes an, z.B. AgCl
AgCl Ag+ + Cl-
Im Wasser besteht ein GGW zwischen der festen Verbindung AgCl und den Ionen, die im Wasser nicht „nackt“, sondern hydratisiert d.h. von einer Wasserhülle umgeben sind.
Im Gegensatz zur homogenen Iodwasserstoff-GGW handelt es sich hier um ein heterogenes GGW mit der festen Phase AgCl. Ihre Gegenwart ist zwar für die GGW-Reaktion erforderlich, es ist aber gleichgültig, in welcher Menge sie vorliegt (ob 0,2 oder 20 g). Feste Phasen haben keine veränderliche Konzentrationen, es treten daher im MWG für feste Phasen keine Konzentrationsglieder auf. Wir müssen daher das MWG anders formulieren:
KL= [Ag+]⋅[Cl-] Löslichkeitsprodukt
In Worten: Unter definierten äußeren Bedingungen (z.B. feste Temperatur) ist das Produkt der Konzentrationen der Ionen in der Lösung über ihrem Niederschlag (also in ges. Lösung) konstant.
Beispiel nebst Dimension:
AgCl: KL= [Ag+]⋅ [Cl-] = 10-10 [mol²/l²]
Ag2CrO4: KL= [Ag+]²⋅ [CrO42-] = 10-12 [mol³/l³]
Die Löslichkeit L ist dagegen eine Konzentrationsangabe (mol/l). Sie gibt die Stoffmenge an, die sich in einem best. Volumen einer gesättigten Lösung gelöst vorfindet. Neben der üblichen Angabe in mol/l finden sich aber auch g/l, g/100g und g/100 ml.

17
Zusammenhang zwischen KL und L Beispiel: BaSO4 1:1-Elektrolyt
KL= [Ba2+] [SO42-] = 10-10 [mol²/l²]
In reinem Wasser gilt: [Ba2+] = [SO42-] einsetzen in KL: KL= [Ba2+]² = [SO4
2-]²
[Ba2+] = [SO42-] = KL
1/2 = 10-5 mol/l = L Löslichkeit.
M(BaSO4) = 233.43 g/mol
β = c(x) ⋅ M(x) = 10-5 ⋅ 233,43 g/l = 0,23 mg/l
Diese einfache Beziehung gilt nur für 1:1-Elektrolyte der Form AB. Wir betrachten den allgem. Fall einer Verbindung MaXb
Typ: AB Beispiel: BaSO4
A2B Ag2CrO4
A2B3 Fe2(SO4)3
A3B2 Zn3(PO4)2
AB3 CrCl3
AB4 ZrCl4
Zunächst sei der Typ A2B am Beispiel Ag2CrO4 behandelt: Ag2CrO4 2 Ag+ + CrO4
2-
m= 2 n = 1
KL = [Ag+]²⋅ [CrO42-] = 4⋅ 10-12 mol³/l³
L = [Ag+]/2 = [CrO42-] => [Ag+] = 2⋅ L , [CrO4
2-] = 1⋅ L
Einsetzen in das Löslichkeitsprodukt: KL = (2L)² ⋅ L
=> 34LKL = mol/l = 10-4 mol/l
M(Ag2CrO4) = 331,73 g/mol, es lösen sich also β = L M = 331,73 g/mol ⋅10-4 mol/l= 331,73⋅10-4 g/l = 33,2 mg/l
Allgemeiner Fall:
” “
festes Salz
Na+
Cl-
Na+
Cl-
Na+
Cl-
Na+
Cl- ” “ ” “
” “ ”“
” “ ” “
Dissoziation
NaCl Na+ + Cl-
Salz Kation Anion
Kristallgitter hydratisierte Ionen
Gitterenergie Hydratationsenergie
AmBn m An+ + n Bm-
[AmBn] = 1/m [An+] = 1/n [Bm-]
[An+] = m ⋅ [AmBn]
[Bm-] = n ⋅ [AmBn]

18
Löslichkeitsprodukt: KL= [An+]m ⋅⋅⋅⋅ [Bm-]n Einheit: molm+n/ lm+n
KL= mm ⋅ [AmBn]m ⋅ nn ⋅ [AmBn]n
[ ] nmnmnm
L BAnm
K +=⋅
Löslichkeit L (in mol/l): [ ] nm nmL
nm nmKBA +⋅
=
Diese Formel schließt natürlich auch den einfachen Fall der Verbindung AB mit m=1 und n=1 ein.
LL KKL =⋅
= +1111 11
Das folgende Beispiel möge zeigen, dass für die Löslichkeit einer Verbindung nicht so sehr der Zahlenwert, sondern vor allem die Dimension von KL maßgeblich ist. Hierzu vergleichen Wir die Löslichkeiten von CdCO3 und Ag2CO3:
KL(CdCO3) = 5,2 10-12 mol²/l²
KL(Ag2CO3) = 8,2 10-12 mol³/l³
Die Zahlenwerte sind also annähernd gleich.
L (CdCO3) = lmolKL /103,2 6−⋅=
L(Ag2CO3) = lmolKKL LL /103,11005,2412
43 1231212
−−+ ⋅=⋅==⋅
=
Ag2CO3 ist also ca. 100 x besser löslich als CdCO3!
Aufgabe: Wie viel mg Ba(IO3)2 sind in 150 ml Wasser bei 25°C löslich? KL = 1,57⋅10-9 mol³/l³
lmolKKL LL /1073,0103925,0412
33 931212
−−+ ⋅=⋅==⋅
=
M[Ba(IO3)2]= 137,33 + [126,90 + 3 ⋅16]⋅2 = 487,13 g/mol
β = M c = 0,73 ⋅10-3 mol/l ⋅ 487,13 g/mol = 355,6 mg/l
In 150 ml : 355,6 mg ⋅ 150 ml / 1000 ml = 53,3 mg
Beeinflussung einer Fällung aufgrund des Löslichkeitsproduktes Wir betrachten einen schwerlöslichen Stoff AB. Das Löslichkeitsprodukt lautet: [A+][B-] = KL
Ist nun die Aufgabe gestellt, die Ionen A+ durch Zugabe von B- möglichst vollständig aus einer Lösung auszufällen, so folgt aus dem Ausdruck für KL, dass die Restkonzentration an A+ an der Lösung umso kleiner ist, je größer die Konzentration an B- ist: Fällung mit einem Überschuss an Reagenz!
Beispiel AgCl: Wie groß ist die Löslichkeit von AgCl KL= 1,1⋅10-10 mol²/l²

19
a) in reinem Wasser
b) in einer KCl-Lösung (10-3 mol/l)
zu a) L = (KL)1/2 =10-5 mol/l
M(AgCl) = 143 g/mol => 1,43 mg/l AgCl lösen sich in 1 l Wasser.
zu b) [Ag+] = KL / [Cl-] = 10-10 / 10-3 = 10-7 mol/l
Die Löslichkeit beträt dann 1,43⋅10-5 g/l
Der Wert ist auf den hundertsten Teil des Wertes in reinem Wasser abgesunken.
Die Berechungen sind nur dann sinnvoll, wenn in der Lösung wenige Fremdionen vorhanden sind und wenn die Niederschläge keine hydrolytischen Eigenschaften besitzen. Außerdem gelten sie auch nur dann exakt, wenn z.B. der Überschuss an Fällungsmittel nicht zu groß wird.
Jahn hat die Konzentration an Chlorid und Silberionen in Abhängigkeit von der KCl-Konzentration gemessen und das jeweilige Löslichkeitsprodukt für AgCl betrachtet. Er fand: Mit zunehmender Konzentration an KCl wird der Wert des Löslichkeitsproduktes von AgCl größer (geringer aber deutlicher Effekt) KL ist also keine Konstante, mit anderen Worten: Die Löslichkeit von Silberchlorid nimmt nicht in dem Maße ab, wie es von einem konstanten Wert KL gefordert würde. Die Ursache hierfür ist die Abnahme der Aktivitätskoeffizienten. Die Ableitung des MWG (und KL) basiert auf der Annahme idealer Bedingungen, Erscheinen im MWG Konzentrationen, so behält es seine Gültigkeit nur in sehr verdünnter, sog. „idealen“ Lösungen. In konzentrierteren Lösungen treten zunehmend Wechselwirkungen (z.B. elektrostat. Anziehungskräfte) zwischen den Ionen auf. Diese Wechselwirkungen muss man mit Hilfe von Korrekturen berücksichtigen. Als Korrekturgröße wurde der Aktivitätskoeffizient f eingeführt: a = f ⋅ c 0 < f < 1
Erst wenn wir in den Ausdruck für das Löslichkeitsprodukt statt der Konzentrationen c die Aktivitäten a einsetzen, gilt das Gesetz exakt.
Für c → 0 wird f = 1, d.h. a = c
Die Abhängigkeit des Aktivitätskoeffizienten f von der Konzentration einer Lösung wird durch eine Debye und Hückel abgeleitete Beziehung wiedergegeben, die hier kurz erläutert sei.
Zunächst Begriff der Ionenstärke I: Wenn eine Lösung die Ionen A,B, C, … mit den Ladungen ZA, ZB, Zc,… und den Konzentrationen cA, cB, cC,… enthält, ist die Ionenstärke durch folgenden Ausdruck definiert:
I = ½ (cAZA² + cBZB² + cCZC² + …) = lmolZci
ii /21 2∑
Beispiele: Jeweils c = 0,01 mol /l
KCl ZK = ZCl = 1 I = ½ (0,01 ⋅1² + 0,01 ⋅1²) = 0,01 mol/l
CaCl2 ZCa = 2, ZCl = 1 I = ½ (0,01 ⋅2² + 0,02 ⋅1²) = 0,03 mol/l
MgSO4 ZMg = ZSO4 = 2 I = ½ (0,01 ⋅2² + 0,01 ⋅2²) = 0,04 mol/l
In einer Lösung, die KCl und MgSO4 (jeweils 0,01 mol/l) enthält, ist die Ionenstärke = 0,01 + 0,04 = 0,05 mol/l
Für niedrige Ionenstärken (ca. 0,01) gilt nun folgende Beziehung:
- lg fi = 0,5 ⋅ zi² ⋅ I0,5
fi Ionenaktivitätskoeffizient für ein Ion i der Ladung zi . Der Faktor 0,5 gilt für Wasser als Lösungsmittel bei 20°C.
Beispiel: 0,01 mol/l KCl-Lösung I = 0,01 mol/l (s. oben)

20
lg fK = lg fCl = - 0,5 1² (10-2)1/2 = - 0,05 => fK = fCl = 0,89
Mit anderen Worten: Die Kalium und Chlorid-Aktivität einer 0,01 mol/l KCl-Lösung beträgt nur 0,0089 mol/l.
Es ist unmöglich, eine Formel aufzustellen, die eine allgemeingültige Berechnung der Ionenaktivitätskoeffizienten bei höherer Ionenstärke zulässt. Für Ionenstärken I bis 0,1 gilt folgende Näherung:
- lg fi = IIzi
+⋅⋅
15,0 2
Für kleine Werte von I geht diese Gleichung in obige über (da √I im Nenner für kleine I vernachlässigbar ist.)
2. Beispiel: MgSO4 0,01 mol/l I = 0,04 (s.o.)
lg fMg = lg fSO4 = 33,02,14,0
04,0104,0²25,0
−=−=+
⋅⋅−
fMg = fSO4 = 10-0,33 = 0,46
Ein weiterer Effekt, der KL beeinflusst, kann in der Bildung von löslichen Komplexen bei größerem Überschuss an Fällungsmittel bestehen.
AgCl + Cl- [AgCl2]-
Verlauf der Löslichkeit von AgCl in KCl-Lösung
c(KCl)0 10-3
10-5
L(AgCl)
berechnet über KL
Konsequenz: geringer Überschuss des Fällungsmittels ist genug, denn er verringert die Löslichkeit. Ein großer Überschuss ist zu vermeiden, denn er hat meist nur störende Effekte (z.B. Verringerung der Aktivität, Komplexbildung).

21
Fällungsgrad Der Fällungsgrad ist eine Größe, die das Ausmaß einer Fällung abzuschätzen gestattet.
Definition: In einer Lösung seinen vor der Fällung nA mol einer Ionensorte enthalten. Nach Zugabe des Fällungsmittels sollen sich aufgrund der Löslichkeit des Niederschlages noch nE mol der Ionen in Lsg. befinden.
Ausgefällte Stoffmenge: nA - nE
Fällungsgrad: A
EAnnn −
=ϕ = Verhältnis ausgefällter Stoffmenge zu ursprünglicher Stoffmenge
Die Angabe erfolgt üblicher Weise in %.
Für eine (praktisch nicht mögliche) vollständige Fällung wäre ϕ = 1 bzw. 100%, da dann nE=0.
Werden von 1 mol 0,1 mol nicht gefällt, ist der Fällungsgrad 90%.
Üblicher weise arbeiten wir mit Konzentrationen: c = n/V mol/l
Am Anfang: cA = Anfangskonzentration VA = Anfangvolumen nA = Anfangsstoffmenge
Am Ende: cE = Endkonzentration VE = Endvolumen nE = Endstoffmenge (nicht ausgefällte Stoffmenge)
%100)1(1 ⋅⋅⋅−=−=−=AA
EE
A
E
A
EAVcVc
nn
nnnϕ
Beispiel: Bestimmung von Barium durch Fällung als BaSO4
Wie viel Sulfat ist der Probelösung zuzusetzen, damit die Fällung quantitativ ist?
Annahme: cA (Ba2+) = 0,01 mol/l = 10-2 mol/l VA = 100 ml c(Na2SO4) = 0,01 mol/l (Reagenz)
a) Wann beginnt die Fällung? Immer dann, wenn Löslichkeitsprodukt überschritten ist. KL= [Ba2+]⋅[SO4
2-] = 10-10 mol²/l² => [SO42-] = KL / [Ba2+] = 10-10 / 10-2 = 10-8 mol/l
Die Fällung beginnt also, wenn die Sulfatkonzentration in der Lösung den Wert 10-8 erreicht hat.
b) Wie viel Reagenzlösung muss man bis zum Fällungsbeginn zugeben?
In 100 ml Lösung soll [SO42-] = 10-8 mol/l sein. Die Volumenvermehrung kann – wie wir
sehen werden – vernachlässigt bleiben.
n(SO42-) = VA ⋅c(SO4
2-) = 0,1 ⋅10-8 = 10-9 mol
Wie viel ml Na2SO4-Lösung mit c= 0,01 mol/l ist das?
V(Na2SO4) = n/c = 10-9 / 10-2 = 10-7 l = 10-4 ml = 0,1 µl also sehr wenig!!! Zum Vergleich: Ein Tropfen = 30 µl
c) Wie groß ist der Fällungsgrad ϕ, wenn wir zur Bariumlösung die stöchiometrisch äquivalente Sulfatmenge zufügen?
D.h.: 100 ml Ba-Lösung c = 0,01 mol/l + 100 ml Sulfat-Lösung c = 0,01 mol/l

22
cA = 10-2mol/l cE = 10-5 mol/l (aus L = KL0,5 )
VA = 100 ml VE = 200 ml
%8,99%100)1021(%100)1,0102,0101(%100)1( 3
2
5=⋅⋅−=⋅
⋅⋅−=⋅
⋅⋅
−= −−
−
AA
EEVcVcϕ
D.h. in der Lösung sind noch 0,2 % des Ba2+ enthalten!
d) Ist die Fällung damit quantitativ?
M(BaSO4) = 233 g/mol nA = c⋅ V = 10-3 mol
Theoretische Auswaage bei vollständiger Fällung: 10-3 ⋅ 233 g/mol = 233 mg BaSO4
Bei einer Wägeungenauigkeit von 0,1 mg ist der max. Fehler also 0,1 mg / 233 mg = 0,043%
Dieser Wert liegt um den Faktor 5 unter den 0,2 % nicht erfassten Bariums => Fällung nicht quantitativ!
Oder: in Lösung befinden sich noch 0,2 % des BaSO4 = 0,2 % 233 mg = 0,466 mg. Das ist mehr als die Wägeungenauigkeit von 0,1 mg. => Fällung nicht quantitativ!
e) Wie viel Sulfat muss noch zugegeben werden, damit die Fällung quantitativ ist?
Hierzu: Wie verbessert sich der Fällungsgrad, wenn ein Überschuss von 1 ml NaSO4 zugegeben wird?
SO42—Menge setzt sich zusammen aus vorhandener Sulfat-Menge aus BaSO4 und der
Überschussmenge:
c(SO42-) = 10-5 + lmol
mllmolml
VSOcV
ges/10610510
201/101)( 555
224 −−−
−−⋅=⋅+=⋅=
⋅
Aus KL ergibt sich die Bariumkonzentration zu 10-10 /6 ⋅10-5 = 1/6 ⋅10-5 mol/l = cE
Der Fällungsgrad ist dann:
%97,99%100)10311(%100)
1,0102,0106/11(%100)1( 3
2
5=⋅⋅−=⋅
⋅⋅⋅−=⋅
⋅⋅
−= −−
−
AA
EEVcVcϕ
In der Lösung verbleiben 0,03 % des Bariums, dieser Wert liegt unterhalb der Wägeungenauigkeit von 0,04 %. Die Fällung ist also quantitativ!
0,03 % von den 233 mg BaSO4 = 0,07 mg nicht mehr auf der Waage erfassbar!
Folgerung: Ein kleiner Überschuss an Fällungsmittel genügt bereits, um die Fällung quantitativ zu machen!

23
Gleichzeitige Fällung zweier Niederschläge In einer Lösung sollen sich zwei verschiedene Ionensorten befinden, die beide mit dem zugesetzten Reagenz ausgefällt werden können. Beispiel: Fällung von Iodid und Chlorid mit Silbernitrat.
[Ag+] ⋅ [Cl-] = 10-10 mol²/l² [Ag+] ⋅ [I-] = 10-16 mol²/l²
In der Lösung über den Niederschlägen befinden sich : Ag+, Cl- und I-. Frage: In welchem Mengenverhältnis stehen Cl- und I- zueinander, wenn Gleichgewicht herrscht.
Im GGW: [Ag+] = [Ag+]
Somit ist : KL/[Hal]= 10-10 / [Cl-] = 10-16 / [I-]
[Cl-] / [I-] = 106 /1
Auf ein Iodid kommen 1 Mio Chloridionen. Das gilt natürlich nur dann, wenn ausreichend Silbernitrat zur Fällung zugefügt worden ist.
Betrachten wir die Fällung von Beginn her, so wird zunächst nur AgI ausfallen bis [Cl-] / [I-] = 106 /1 ist. Erst dann beginnt auch AgCl auszufallen und zwar so, dass das Verhältnis 1: 1 Mio. stets konstant bleibt. Frage: Ist auf diesem Wege eine quantitative Trennung der beiden Ionen möglich?
Hierzu ein Rechenbeispiel: VA = 100 ml mit cA(I-) = c(Cl-) = 0,1 mol/l Reagenz: c(Ag+) = 0,1 mol/l
Zur Ausfällung von Iodid sein ca. 100 ml Ag-Lösung erforderlich, eine genaue Berechnung ergibt 100 ml – 20 µl(1 Tropfen), damit AgCl gerade auszufallen beginnt, d.h. VE≈ 200 ml.
Für die Endkonzentration cE von Iodid muss gelten:
[I-] / [Cl-] = [I-] / [5 ⋅10-2] = 10-6 d.h. [I-] = 5 ⋅10-8 mol/l
ausgefällte Menge: n = nA – nE = cA ⋅VA – cE ⋅VE = 10-1 mol/l ⋅10-1 l – 2 ⋅10-1 l ⋅ 5 ⋅10-8 mol/l = 10-2 – 10-8 ≈ 10-2 mol
Berechnung des Fällungsgrades:
ϕ = (1 – 10-8 / 10-2) 100% = 100 – 10-4 % = 99,9999%
In der Lösung verbleiben 0,0001% AgI. = 0,000235 mg
Auswaage von 0,01 mol AgI = 2,35 g. (M[AgI] = 235 g/mol)
Wägeungenauigkeit = 0,1 mg 0,1 mg / 2,35 g = 0,043 %
Damit ist die Fällung von Iodid quantitativ bevor die Chloridfällung beginnt. Somit wäre theoretisch auch eine Quantitative Trennung von Iodid und Chlorid durch Fällung möglich.
Für AgBr gilt: KL= [Ag+]⋅ [Br-] = 5 ⋅ 10-13 mol²/l²
Führt man denselben Gedankengang für eine Trennung von Chlorid und Bromid durch, so ergibt sich ein Fällungsgrad von 99% für AgBr in dem Moment, wo die AgCl-Fällung einsetzt. Die Rechnung zeigt weiterhin, dass die AgBr-Fällung zu diesem Zeitpunkt noch nicht quantitativ ist.
Folgerung: Für eine quantitative Trennung zweier Ionensorten durch ein gemeinsames Fällungsreagenz ist Voraussetzung, dass sich die Löslichkeitsprodukte der auszufällenden Verbindungen genügend stark voneinander unterscheiden. (z.B. um 6 Zehnerpotenzen)

24
Gekoppelte Salzlösungen und Salzfällungen Das Verhältnis [Cl-] / [I-] = 106 /1 muss sich auch dann im GGW-Zustand einstellen, wenn wir AgCl und AgI nebeneinander in Wasser suspendieren. Für die Löslichkeit der Halogenide gilt zunächst:
L(AgCl) = (10-10)0,5 = 10-5 mol/l = [Ag+] = [Cl-]
L(AgI) = (10-16)0,5 = 10-8 mol/l = [Ag+] = [I-]
Frage: Wie groß ist die Konzentration der 3 Ionensorten im GGW in einer AgCl / AgI-Suspension?
[Ag+] ⋅ [Cl-] = 10-10 mol²/l² (1) [Ag+] ⋅ [I-] = 10-16 mol²/l² (2)
Es gibt also 2 Gleichungen mit 3 Unbekannten, als 3. Gleichung ziehen wir die Elektroneutralität heran: [Ag+] = [Cl-] + [I-] (3)
Wir setzen [Cl-] und [I-] aus Gl. (1) und (2) in Gl. (3) ein:
[Ag+] = 10-10 / [Ag+] + 10-16/ [Ag+]
[Ag+]² = 10-10 + 10-16 ≈ 10-10 => [Ag+] = 10-5 mol/l Für [Cl-] im Gleichgewicht ergibt sich dann: [Cl-] = KL / [Ag+] = 10-10 / 10-5 = 10-5 mol/l Die Chloridkonzentration ist also gegenüber einer reinen AgCl-Suspension praktisch unverändert.
Für [I-] ergibt sich aber: [I-] = KL / [Ag+] = 10-16 / 10-5 = 10-11 mol/l Sie ist also um 3 Zehnerpotenzen gegenüber dem Wert in reiner AgI-Suspension gesunken.
Wie eingangs postuliert, ist also wieder das Verhältnis Iodid : Chlorid = 1 : 106 erreicht.
Vorgänge dieser Art bei denen letztlich ein schwerlösliches Salz in ein noch schwerer lösliches übergeht, bezeichnet man als gekoppelte Salzlösungen und Salzfällungen.
Man spricht von einer gekoppelten Salzlösung, wenn zwei schwerlösliche Salze vorliegen, die ein gemeinsames Ion (Kation oder Anion) besitzen. Die Konzentrationen der beiden Gegenionen sind dann in einer Suspension vom Verhältnis der Löslichkeitsprodukte abhängig.
Weitere Beispiele:
• Umwandlung von weißem AgCl (KL= 10-10) in schwarzes Ag2S (KL=10-48,8) in Sulfidlösungen
• Umwandlung von weißem PbSO4 (KL= 10-8) in schwarzes PbS (KL=10-29) in Sulfidlösungen
• Umwandlung von weißem CaSO4 (KL= 10-4,3) in CaCO3 (KL=10-7,9) in Carbonatlösungen
Durch die gekoppelte Salzlösung und Salzfällung kann umgekehrt auch ein schwerer lösliches Salz in ein leichter lösliches umgewandelt werden, wenn
a) die Konzentration des Fällungsmittels sehr hoch gewählt wird,
b) der Unterschied der KL-Werte der Salze nicht allzu groß ist.

25
Beispiel:
Umwandlung von BaSO4 durch lösliche Carbonate (Soda) in BaCO3 = Sodaauszug oder Sodaschmelze
KL(BaSO4) = 10-10 mol²/l²
KL(BaCO3) = 10-8,16 mol²/l²
Aus dem Verhältnis: 015,0101010
][][
][][][][ 84,1
16,8
10
23
24
23
2
24
2====
⋅⋅ −
−
−
−
−
−+
−+
COSO
COBaSOBa
[SO42-] = 0,015 [CO3
2-]
Beim Sodaauszug wird Na2CO3 in großem Überschuss eingesetzt, z.B. sei [CO32-] = 1 mol/l .
Dann wird vorhandenes festes BaSO4 soweit umgewandelt, dass in der Lösung eine [SO42-]
= 0,015 mol/l herrscht. Nehmen wir an, wir haben 100 ml dieser Sodalösung. Dann löst sich darin eine Stoffmenge von n(BaSO4) = c ⋅V = 0,015 mol/l ⋅ 0,1 l = 1,5 ⋅ 10-3 mol
Das entspricht: n ⋅ M(BaSO4) = m(BaSO4) = 1,5 ⋅ 10-3 ⋅ 233 g/mol = 349,5 mg BaSO4
Fazit: Man kann ca. 350 mg BaSO4 mit 100 ml Soda-Lösung (c = 1 mol/l) vollständig in BaCO3 umwandeln unter Freisetzung der äquivalenten Sulfat-Menge.

26
Säuren und Basen Unter einer Säure versteht man nach Brönsted einen Protonenspender, eine Base ist das Gegenteil, ein Protonenempfänger.
Früher war die Definition von Arrenius an das Wasser als Lösungsmittel gebunden, bei Brönsted ist sie unabhängig vom Lösungsmittel.
HCl H+ + Cl-konjugierte Säure = Proton + konjugierte Base
Dissoziation
NH4+H+ + NH3
Proton + Base = konjugierte Säure Da keine freien Protonen existieren können, müssen sie von einem anderen Stoff aufgenommen werden, dieser reagiert dann als Base. Es müssen also immer zwei Säure-Base-Paare miteinander reagieren. Im Allgemeinen nimmt man dazu Wasser.
H2O + H+ H3O+
Base + Proton konj. Säure An einer Protolysereaktion sind immer zwei Säure-Base-Paare beteiligt, zwischen denen ein GGW existiert, d.h. wir fassen die beiden Säure-Base-Paare zusammen:
HA + H2O A- + H3O+
Man kann also ganz allgemein auch schreiben:
H2O + H2O H3O+ + OH-
Säure 1 + Base 2 Base 1 + Säure 2
Einige Teilchen – wie beispielsweise Wasser – können sowohl Säure als auch Base sein. Man nennt diese Teilchen amphother. Reines Wasser unterliegt einer Eigenprotolyse gemäß obiger Gleichung. Auf diese GGW-Reaktion wendet man das MWG an.
K = [H2O]2
[OH-] • [H3O+]
K [H2O]² = KW = [H3O+] [OH-] = 10-14 mol² / l²
Ionenprodukt
Da die Konzentration des Wassers konstant ist (55,34 mol/l) wird sie in die Konstante mit hineingezogen, man erhält eine neue konstante KW. Diese hat den Zahlenwert 10-14(bei 25°C), also sehr klein. Es liegen demnach nur wenige Hydroniumionen und Hydroxidionen im Wasser vor. Wenn die Lösung neutral ist, liegen genauso viele Hydronium- wie Hydroxidionen vor. Also 10-7 mol/l.
An dieser Stelle sollte dann auch der pH-Wert eingeführt werden.
pH = - lg [H3O+] bzw. pOH = - lg [OH-]
Eine neutrale Lösung hat demnach einen pH-Wert von 7. Ist der pH-Wert kleiner, ist die Lösung sauer, ist der Wert größer, ist die Lösung alkalisch.
Saure Lösung: [H+] > 10-7 pH < 7
Alkalische Lösung: [H+] < 10-7 pH > 7
Der Wert des Ionenproduktes KW kann gleichermaßen logarithmiert ausgedrückt werden:

27
pKW = - lg(KW) = + 14
[H+] [OH-] = KW = 10-14
pH + pOH = pKW = 14
Somit werden wechselseitige Umrechnungen von pH und pOH stark vereinfacht. Mit diesen Kenntnissen können wir nun solche Fällungen behandeln, bei denen pH-Werte eine Rolle spielen, nämlich die Fällung von Hydroxiden, Carbonaten und Sulfiden.
Fällung von Hydroxiden AgCl und BaSO4 sind einfache Fällungen. Zur Vermeidung von Störungen arbeitet man in saurer Lösung, doch besteht keine unmittelbare Abhängigkeit vom pH-Wert. Bei vielen Fällungen ist jedoch der Fällungsgrad vom pH-Wert der Lösung abhängig, wenn nämlich die Ionen des Fällungsmittels im GGW mit den H+-Ionen der Lösung stehen. Wie Beispielsweise bei der Fällung von Hydroxiden mit OH- als Fällungsmittel. Beide Ionen sind über das Ionenprodukt des Wassers verknüpft.
pH + pOH = pKW = 14
Wir betrachten die Beispiele Fe(OH)3 und Mn(OH)2 mit deren Löslichkeitsprodukten:
[Fe3+] ⋅ [OH-]³ = 10-37,4 mol4/l4 [Mn2+]⋅ [OH-]² = 10-14,2 mol³/l³
Bei welchem pH-Wert muss man die Fällung vornehmen, um eine Trennung zu erzielen, es soll also nur Fe(OH)3 ausfallen.
Wir substituieren die OH—Konzentration im Löslichkeitsprodukt durch den Ausdruck 10-14/[H+].
332,1428
2444,3742
3 /10]²[
10][/10]³[
10][ lmolH
MnlmolH
Fe −+
−+−
+
−+ =⋅=⋅
[Fe3+] = [H+]³ ⋅104,6 [Mn2+] = [H+]² ⋅ 1013,8 mol³/l³
lg [Fe3+] = 3 lg [H+] + 4,6 lg [Mn2+] = 2 lg [H+] + 13,8 logarithmiert
-lg [Fe3+] = - 3 lg [H+] - 4,6 - lg [Mn2+] = -2 lg [H+] - 13,8 x (-1)
pFe3+ = - 4,6 + 3 pH pMn2+ = - 13,8 + 2 pH Trägt man die pMen+ gegen pH auf, so müssen Geraden resultieren, dabei wird pMen+ in der Regel nach unten aufgetragen, um anzudeuten, dass die Me-Konzentration mit steigendem pMe-Wert sinkt. Um die Gerade einzeichnen zu können, berechnen wir jeweils 2 Punkte, in dem wir 2 beliebige pMe-Werte vorgeben.
pMe = 0 pMe = 5
Fe: 3 pH = 4,6 3 pH = 5 + 4,6 = 9,6 pH = 1,53 pH = 3,2
Mn: 2 pH = 13,8 2 pH = 5 + 13,8 = 18,8 pH = 3,2 pH = 9,4
Derartige Diagramme nennt man Fällungsdiagramme.
Jeweils links von der Geraden erfolgt Auflösung rechts von der Geraden Fällung des betreffenden Hydroxids.
Betrachten wir die Metallsalzlösungen mit der Konzentration 10-2 mol/l. Bei VA = 1 l liegt die Auswaage in der Größenordnung von 1 g [Fe(OH)3: 1,07 g; m = c· V· M = 10-2mol/l ·1 l ·107 g/mol]. Bei einem typischen Wägefehler von 0,1 mg ist der 10-4 –te Teil der Auswaage. Mit

28
Sicherheit liegen wir unterhalb der Wägeungenauigkeit, wenn der 10-5te Teil in Lösung verbleibt, d.h. die Endkonzentration betrage 10-7 mol/l. Die untere gestrichelte Linie bei pMe = 7 entspricht also der quantitativen Ausfällung.
Aus dem Diagramm lassen sich nun die pH-Werte entnehmen, bei denen die Fällung jeweils beginnen und bei denen sie beendet sind: Schnittpunkte der Fällungsgeraden mit den gestrichelten Linien bei pMe = 2 bzw. 7. Wir erhalten folgende Ergebnisse:
Fe3+ Mn2+
Beginn der Fällung pH 2,2 7,9
Quantitative Fällung pH 3,9 10,4
Durch Wahl eines geeigneten pH-Wertes zwischen 3,9 und 7,9 kann Fe3+ praktisch vollständig ausgefällt werden, ohne dass Mn2+ mitfällt. Eine Trennung ist ohne weiteres möglich.
In der Literatur finden sich Fällungsdiagramme für zahlreiche Hydroxide.
Amphotere Hydroxide Darunter versteht man solche, die sich sowohl in Säure unter Bildung der hydratisierten Metallionen, als auch in Basen unter Bildung von komplexer Hydroxo-Anionen lösen.
Allgemein: Men+ + n OH-
- n OH- Me(OH)n + m OH-
- m OH- [Me(OH)n+m]m-
Beispiel: Al3+ + 3 OH- Al(OH)3
KL = [Al3+] [OH-]³ = 10-34,3 mol4/l4
Berechnung der Fällungsgeraden: 37,73443,3442
3 ][10][/10]³[
10][ ++−+
−+ ⋅==⋅ HAllmol
HAl
pAl = - 7,7 + 3 pH
Wiederauflösung erfolgt, wenn man die Protonenkonzentration weiter verringert gemäß:
Al(OH)3 + OH- [Al(OH)4]-
MWG: 1][]])([[ 4 ≈=
−
−K
OHOHAl
Wie schon bei der Betrachtung zu KL wird die feste Phase Al(OH)3 nicht berücksichtigt.
Der Wert für K kann um etwa 2 Zehnerpotenzen schwanken und hängt stark vom Zustand (Alterungsgrad) des Al(OH)3 ab.
Berechnung der Fällungsgeraden: [(Al(OH)4)-] ≈ [OH-] = 10-14 / [H+]
pAl* ≈ 14 - pH
Der pAl*-Wert charakterisiert hier ebenfalls die Menge des Al in der Lösung, auch wenn es als [Al(OH)4]- vorliegt.
Insgesamt resultieren also zwei Fällungsgeraden. Berechnung der charakteristischen Punkte: cAnfang = 10-2 mol/l
Fällung von Al(OH)3 Auflösung von Al(OH)3
pAl3+ = 2 pAl* = 2
3 pH = 9,7 pH = 12 pH = 3,2
Dies sind die pH-Werte des Fällungsbeginns, einmal von der sauren einmal von der alkalischen Seite kommend.

29
Die Al(OH)3-Fällung ist in jedem Fall quantitativ, wenn die Al-Konzentration um 5 Zehnerpotenzen gesunken ist.
pAl3+ = 7 pAl* = 7
3 pH = 14,7 pH = 7 pH = 4,9
Wir erhalten also ein Diagramm mit 2 sich schneidenden Geraden.
pH
pMe2+
0 1 2 3 4 5 6 7 8 9 10 11 12 13 140
1
2
3
4
5
6
7
8
9
Vorgabe 10-2 mol/l
quant. Ausfällung
Fällungsdiagramm für Hydroxide
FeMnAl
Fe3+ Fe(OH)3
Al(OH)3 Al(OH)4-Al3+
Mn2+ Mn(OH)2
Fassen wir zusammen: In einer Al3+-Lösung mit [Al3+] = 10-2 mol/l beginnt die Fällung bei pH = 3,2 mit steigendem pH bewegen wir uns nun auf der Fällungsgeraden abwärts, bis bei pH = 4,9 die Fällung quantitativ ist. Die Wiederauflösung des Hydroxids wird merklich, wenn die [Al(OH)4
-] 10-7 zu überschreiten beginnt. (wir bewegen uns auf der gestrichelten Linie pMe = 7). Mit steigendem pH bewegen wir uns weiter entlang der 2. Geraden aufwärts, bis die [Al(OH)4
-] bei pH = 12 wieder bei 10-2 mol/l liegt. Jetzt ist das Al(OH)3 wieder quantitativ aufgelöst.
Fazit: Al(OH)3 lässt sich also nur zwischen pH 4,9 und 7 quantitativ fällen. Auf der Ausfällung als Hydroxid basieren einige sehr wichtige Methoden zur Trennung und Bestimmung von Metallionen. Z.B. Trennung von Me2+ von Me3+ und Me4+ , d.h.
Fe3+, Al3+, Cr3+, Ti4+, Zr4+ von Zn2+, Ni2+, Co2+, Mn2+ (Hydrolysentrennung)
Die Hydroxidfällung wird ausgeführt, obwohl die ausgefällten Verbindungen analytisch unerfreulich sind:
a) sie bilden voluminöse, gallertige, schwer filtrier- und auswaschbare Niederschläge
b) aufgrund ihrer großen Oberfläche neigen sie zur Adsorption von Fremdstoffen
c) es besteht Tendenz zu Mitfällung basischer Salze.
Wegen b und c kann die Zusammensetzung der Niederschläge stark von der erwarteten Stöchiometrie abweichen.
Die Fällungsmethoden können recht unterschiedlich sein, je nach Lage der Fällungsgeraden. Im allgemeinen geht man von kleinen zu großen pH-Werten über. Manchmal genügt bereits Wasser als Fällungsmittel, z.B. Hydrolyse von Zinn IV:
Sn4+ + 8 H2O Sn(OH)4 ↓+ 4 H3O+
Bei M3+-Ionen ist zur Fällung durch Hydrolyse Acetat, NH3 oder Urotropin erforderlich, um die entstehenden Protonen zu binden.
Al3+ + 3 H2O Al(OH)3 ↓+ 3 H+
3 H+ + 3 OAc- 3 HOAc

30
Fällung von Sulfiden Ähnlich wie bei den Hydroxiden liegen die Verhältnisse bei der Fällung von Sulfiden. Auch hier ist die Konzentration des Fällungsmittels Sulfid abhängig vom pH-Wert. H2S ist eine sehr schwache zweibasige Säure:
H2S + H2O → H3O+ + HS−
HS− + H2O → H3O+ + S2−
H2S + 2 H2O → 2 H3O+ + S2−
K
K
K
1
2
1
= =
= =
⋅ = =
+ −−
+ −
−−
+ −−
[ ][ ][ ]
/
[ ][ ][ ]
/
[ ] [ ][ ]
/
H O HSH S
mol l
H O SHS
mol l
KH O S
H Smol l
3
2
7
32
13
23
2 2
2
20 2 2
10
10
10
H2S ist ein Gas, das sich nicht besonders gut in Wasser löst, bei Atmosphärendruck lösen sich nur ca. 0,1 mol/l, d.h. [H2S] = 0,1 mol/l. Daraus ergibt sich:
[H2S] = 10-1 mol/l ⇒ [ ] /[ ]
S mol lH O
221 3 3
32
10−−
+=
Über die Protonenkonzentration, d.h. den pH-Wert kann also die Sulfidkonzentration eingestellt werden, Je größer [H+], desto kleiner ist [S2-] und umgekehrt.
Sulfide mit sehr kleinen KL-Werten lassen sich bereits in saurer Lösung quantitativ fällen, Bei pH = 1 gilt dies für die Sulfide der Kationen: Hg2+, Cu2+, Ag+, Pb2+, Sn2+/4+, Bi3+, As3+, Sb3+, Cd2+. Dagegen fallen die Sulfide der Kationen Zn2+, Co2+, Ni2+, Fe2+, Mn2+ nicht oder nicht quantitativ.
Darauf basiert das bedeutsame Verfahren der Sulfid-Trennung. Bei Fällung von Sulfiden innerhalb der genannten Gruppen werden die Verhältnisse komplizierter, da sich die Bereiche teilweiser Fällungen überschneiden können. Wir wollen die Verhältnisse am Beispiel der Fällung von Fe und Mn als Sulfid untersuchen.
[Fe2+] [S2-] = 10-18 mol²/l² [Mn2+] [S2-] = 10-15 mol²/l²
23221
182
182
212
][10][1010][
10][
10][
++−
−+
−+
−+
=⋅=
=⋅
HHFe
HFe
26221
152
152
212
][10][1010][
10][
10][
++−
−+
−+
−+
=⋅=
=⋅
HHMn
HMn
lg [Fe2+] = 3 + 2 lg [H+] lg [Mn2+] = 6 + 2 lg [H+]
pFe2+ = -3 + 2 pH pMn2+ = -6 + 2 pH
Ist die Anfangskonzentration cAnfang = 0,01 mol/l gilt für den Fällungsbeginn:
Fe: 2 = -3 + 2pH Mn: 2 = -6 + 2pH
2pH = 5 2 pH = 8
pH = 2,5 pH = 4
Für die quantitative Fällung mit [Me2+] = 10-7 mol/l gilt
Fe: 7 = -3 + 2pH Mn: 7 = -6 + 2pH
2pH = 10 2 pH = 13
pH = 5 pH = 6,5

31
2
7
4
6
1 2 3 4 5 6 7 8 pH
pMe2+
5
3
1Fe2+ FeS
MnSMn2+
Überschneidungsbereich
Zwischen pH 4 und 5 resultiert eine Überschneidung der Fällungsbereiche, d.h. FeS ist noch nicht quantitativ gefällt, wenn die Fällung von MnS bereits beginnt.
Wie lässt sich trotzdem eine quantitative Trennung bewerkstelligen?
Wir wählen zunächst den pH-Wert so, dass er in das Überschneidungsgebiet fällt, also z.B. pH = 4,1.
Berechnung des in Lösung verbleibenden Fe nach der Fällung:
pFe2+ = - 3 + 2 pH = -3 + 2⋅ 4,1 = 5,2 => [Fe2+] = 10-5,2 mol/l
Die Berechnung des Fällungsgrades ist einfach, da durch Einleitung von H2S praktisch keine Volumenvermehrung erfolgt, d.h. VE =VA
ϕ = (1 – cE / cA) = (1 - 10-5,2 / 10-2) 100 % = 99,94 % 0,6 % des Eisens sind nicht ausgefällt worden, wie sich leicht nachprüfen lässt, entspricht dies bereits einer nahezu quantitativen Fällung. Der Nd. ist jedoch stark durch MnS verunreinigt. Berechnung von [Mn2+] in Lösung:
pMn2+ = -6 + 2pH = -6 + 8,2 = 2,2 => [Mn2+] = 10-2.2 = 0,63 ⋅ 10-2 mol/l
Es waren ursprünglich 1⋅10-2 vorhanden, 0.63⋅10-2 mol/l sind nicht ausgefallen, d.h. 0,37⋅10-2 mol/l sind als MnS ausgefallen, Das sind 37% der gesamten Mn-Menge. Der FeS-Niederschlag enthält demnach 37 % des Mn2+ als MnS.
Eine Trennung lässt sich dennoch erzielen durch Umfällung: Der Niederschlag wird in Säure gelöst und erneut pH 4,1 eingestellt.
Gleiche Lösemittelmenge vorausgesetzt bleiben bei erneuter Fällung mit H2S wieder 0,06% der 99,94% Fe in Lösung zurück. Der Niederschlag enthält damit 99,88 % des gesamten Eisens, diese Fällung ist immer noch nahezu quantitativ.
MnS fällt aber nicht mehr mit, da bei pH = 4,1 0,63⋅10-2 mol/l in Lösung verbleiben können, aber nur 0,37 ⋅10-2 mol/l Mn2+ vorhanden sind. Die Filtrate der 1. und 2. Fällung werden nun vereinigt, dann fällt man MnS bei einem höheren pH quantitativ aus; Dieser Niederschlag ist nun durch sehr wenig (0,12 %) FeS verunreinigt.
Die Bildung eines Niederschlages Wir haben bislang Fällungen nur unter dem Gesichtspunkt von Löslichkeitsprodukt und Löslichkeit, d.h. unter dem Aspekt des termodynamischen Gleichgewichts zwischen Ionen in der Lösung und Niederschlag betrachtet. Für die Praxis der quantitativen Analyse ist aber auch der Zustand eines Niederschlages und seine Reinheit von großer Bedeutung.
Zustand: Der Nd. soll gut filtrierbar und auswaschbar sein, d.h. kristallin und nicht schleimig (Hydroxid, Sulfide); je grobkristalliner ein Nd., desto leichter und schneller seine Verarbeitung.

32
Reinheit: Ein großes Problem ist das Mitfällen von Verunreinigungen (Entstehung systematischer Fehler):
a) durch Adsorption an der Nd.-Oberfläche (besonders stark bei feinkristallinen Nd. mit entspr. Großer Oberfläche) b) durch Okklusion (Einschluss) von Fremdsubstanzen oder sogar Mutterlauge. Diese Gefahr ist besonders groß bei grobkristallinen Fällungen.
Zustand und Reinheit des Niederschlages werden durch diverse physikalisch-chemische Faktoren bei der Fällung beeinflusst. Wir wollen deshalb die Bildung eines Nd. genauer analysieren (wurden von vielen Autoren eingehend untersucht).
Zunächst ist folgendes festzustellen: Die Löslichkeit eines Nd. ist umso größer, je geringer die Teilchengröße ist; kleinere Teilchen stehen also mit einer konzentrierteren Lösung im Gleichgewicht als größere. Erreicht die Teilchengröße jedoch ein gewissen Wert (etwa 10-4 – 10-3 cm³), so wird bei weiterer Steigerung der Größe die Löslichkeitsänderung unmessbar klein.
Bei Lösungen kann Übersättigung eintreten, gibt man zu einer Ionenlösung Fällungsreagenzlösung zu, so tritt häufig der Fall ein, dass bei Erreichen von KL noch keine Fällung eintritt. Dieser Effekt lässt sich auf die zuvor beschriebene Abhängigkeit Teilchengröße – Löslichkeit zurückzuführen.
Eine übersättigte Lösung kann jedoch den gelösten Stoff spontan als neue Phase abscheiden, wenn nämlich genügend gelöste Teilchen durch Wärmebewegung zusammentreffen und einen – zunächst submikroskopischen – Keim bilden. Dieser wächst nun weiter, da sich die Teilchengröße vergrößert hat, die Löslichkeit entsprechend abgenommen hat.
Die Wahrscheinlichkeit für die Keimbildung steigt (bei sonst konst. Bedingungen) sehr stark mit der Konzentration der Lösung an, damit wächst auch die Zahl der gebildeten Keime, denn
a) wächst die Wahrscheinlichkeit von Zusammenstößen mit wachsender Zahl von Teilchen je Volumeneinheit
b) sinkt die erforderliche Mindestkeimgröße, je konzentrierter die Lösung ist.
Wenn jeder Keim die Bildung eines Kristalls veranlasst, muss nach Beendigung der Fällung gelten:
Durchschnittsmasse eines Kristalls = Gesamtkristallmasse / Keimzahl
Ist die Keimzahl infolge hoher Konzentration und entsprechend hoher Übersättigung der ursprünglichen Lösung sehr groß, so wird der Niederschlag sehr feinkristallin.
Eine quantitative Betrachtung führt zu folgenden Näherungen: rc
ccLL o σ~−=∆
∆L = Übersättigung c0 = Anfangskonzentration (vor Fällung) c = Endkonzentration (nach Fällung) σ = Oberflächenspannung des Kristalls bez. Keims r = Teilchenradius
Geschwindigkeit der Keimbildung : k ~ (∆L)n n = 4 bis 8
Wachstumsgeschwindigkeit: k’ ~ A (∆L) A: Oberfläche des Nd.
Für optimale Fällungsbedingungen soll k’ > k sein, d.h. ∆L (Übersättigung) soll klein sein, wird erreicht, wenn Anfangskonzentration co klein und c groß ist (erhöhte Temperatur).
Folge für die Praxis: Bei der Fällung soll Übersättigung vermieden werden durch

33
• Verwendung verdünnter Lösungen
• Langsames Zugeben und Umrühren (Vermeidung lokaler Übersättigung)
• Fällung in der Wärme (Erhöhung der Löslichkeit)
Nach der Fällung der Primärteilchen treten Sekundärprozesse auf, die den Niederschlag verändern: Alterung. Daran können mehrere Vorgänge beteiligt sein:
a) Teilchenwachstum der größeren auf Kosten der kleineren Kristalle, geht umso schneller, je höher die Temperatur und je größer die Löslichkeit des Stoffes selbst ist.
b) Rekristallisation: Je rascher die Bildung der Primärkristalle ist, desto eher weisen sie eine skelettartige Struktur auf (wie Schneekristalle). An einem skelettartigen Kristall, der das Lösungsvolumen möglichst vollständig durchsetzt, können sich weitere Ionen aus der Lösung viel schneller anlagern (kürzere Wege) als an einen kugelförmigen; ihre freie Oberfläche und damit das Adsorptionsvermögen sind zudem wesentlich größer. Im Alterungsprozess werden die Spitzen der Skelettkristalle aufgrund ihrer höheren Löslichkeit (Krümmungsradien) zugunsten der glatten Flächen abgebaut; begünstig durch Wärme.
c) Phasenumwandlung: Wenn verschiedene Modifikationen des Nd. möglich sind,
entsteht bei der Fällung häufig zunächst die bei den herrschenden Bedingungen thermodynamisch instabilere. Da diese aber grundsätzlich die höhere Löslichkeit hat, wandelt sie sich (wiederum begünstigt durch Wärme) allmählich in die stabilere, weniger lösliche um.
Folge für die Praxis: Den Nd. – wenn möglich – nicht gleich nach der Fällung filtrieren, sondern in Berührung mit der Mutterlauge einige Zeit warm stehen lassen.
Fällung aus homogener Lösung PFHS precipitation from homogenous solution (Lit: Cartwright, Newman, Wilson, Analyst 92, 663 (1967)
Üblicher Weise werden Niederschläge dadurch gebildet, dass man die Lösung mit dem Fällungsreagenz in die Probelösung eingießt. Das hat den Nachteil, dass selbst bei schnellem Rühren nicht zu vermeiden ist, dass an der Eintropfstelle hohe lokale Konzentrationen auftreten. Folge: hohe Übersättigungen => starke Keimbildung (Keimbildungsgeschw. ist groß gegenüber Kristallwachstumsgeschw.) => feinverteilte Niederschläge.
Abhilfe: Fällung aus homogener Lösung.
Man verwendet eine Reagenzlösung, die das Fällungsmittel nicht frei sondern in chemisch gebundener Form enthält. Nach gutem Vermischen von Proben- und Reagenzlösung entsteht eine homogene Lösung, die zunächst klar bleibt. Nun wird das Fällungsmittel langsam aus seinem „latenten“ Zustand freigesetzt, in den meisten Fällen durch Hydrolyse, die in der Kälte sehr langsam ist. Durch Erwärmen jedoch beschleunigt wird. Folge: Die Konzentration an Fällungsmittel ist in der gesamten Lösung nicht nur klein, sondern völlig gleichmäßig. Die Zahl der gebildeten Keime ist gering; das im Laufe der Zeit allmählich freigesetzte Fällungsmittel wird durch das Kristallwachstum aufgebraucht.: Es entstehen grobkörnige, leicht filtrier- und auswaschbare Niederschläge.

34
Homogen gefällte Nd. neigen wegen ihrer geringen Oberfläche kaum zur Adsorption von Verunreinigungen auch Okklusionen sind infolge des langsamen Kristallwachstums stark eingeschränkt.
Beispiele:
1) Grobkristallines BaSO4 bei Fällung mit Dimethyl- oder Diethylsulfat oder Amidosulfonsäure:
OS
OHO
HO OS
OCH3O
CH3O OS
OHO
H2N
(RO)2SO2 + 2 H2O 2 ROH + H2SO4
H2NSO3H + 2 H2O NH4+ + H3O+ + SO4
2-
2) Ca2+, Zn2+ Lanthanoide, Th4+ bilden schwerlösliche Oxalate. Man erhält sie in ausgezeichnet kristalliner Form, wenn man das Oxalat-Ion bei 100°C durch Hydrolyse (Verseifung) des Dimethyl- oder Diethylesters der Oxalsäure freisetzt. (RO)2C2O2 + 2 H2O 2 ROH + 2 H++ C2O4
2-
3) Langsamer Entzug von Protonen aus der Lösung durch Hydrolyse von Urotropin: (CH2)6N4 + 6 H2O CH2O + 4 NH3
4) Langsame Bildung von Sulfid-Ionen bei der Hydrolyse von Thioacetamid: CH3CSNH2 + 2 H2O CH3COO- + NH4
+ + H2S
Die homogene Fällung gewinnt steigende Bedeutung.
Adsorption Unter einer Phase versteht man die Zustandsform eines Stoffes, die in sich physikalisch gleichartig ist, sich aber durch Grenzflächen von einer anderen Phase abgrenzt. Im physikalisch homogenen Innern einer solchen Phase herrscht eine symmetrische Verteilung der von den Bausteinen des Stoffes ausgehenden Bindungskräfte, dagegen herrscht an der Grenzfläche zweier Phasen eine unsymmetrische Kräfteverteilung.
Phase II (Lösung)
Phase I (Kristall)
Grenzfläche
Die unsymmetrische Kräfteverteilung führt oft dazu, dass ein in einer der beiden Phasen gelöster Stoff unmittelbar an der Phasengrenze in ganz anderer Konzentration vorliegt als im Innern der Lösung: Adsorption.
Von Bedeutung für die analytische Chemie ist die Adsorption zuwischen einer flüssigen und festen Phase, also zwischen Lösung und Niederschlag. Es können also z.B. leichtlösliche Stoffe an der Oberfläche von schwerlöslichen Niederschlägen festgehalten werden.
Die Adsorptionskräfte sind von der gleichen Art wie die Kräfte einer chemischen Bindung; bei Ionen kommt die Bildung einer sogenannten Oberflächenverbindung also durch elektrostatische Kräfte zustande. Die adsorbierte Stoffmenge ist der Phasenoberfläche proportional; bei feinverteilten Niederschlägen mit sehr großer Oberfläche kann die Adsorption beträchtliche Werte annehmen.
Die Abhängigkeit der adsorbierten Stoffmenge von der Konzentration des Stoffes in der Hauptmenge der Lösung beschreibt die Freundlich’sche Adsorptions-Isotherme – eine

35
empirische Funktion: x = αααα⋅⋅⋅⋅ cββββ ββββ<1
c [mol/l]
x
Grenzwert
x: Im GGW von 1 g fester Phase adsorbierte Stoffmenge
α und β: charakteristische Konstanten des Systems, abhängig vom Dispersionsgrad (also nur konstant, wenn auch die Dispersität konstant ist)
Die Konstante β ist immer <1, daher muss die Kurve konvex sein (von oben her gesehen). Daraus folgt: Die adsorbierte Stoffmenge x ändert sich langsamer als die Konzentration.
Ist z. B. β = ½ und sinkt c auf ¼ seines ursprünglichen Wertes, so fällt x nur auf den halben Wert:
x = α c1/2 x = α (¼ c)1/2 = ½ α c1/2
Dies ist der Grund für die bekannte Tatsache, dass sich die letzten Spuren eines adsorbierten Stoffes durch Auswaschen nur sehr schwer entfernen lassen.
Linearisierung der Adsorptionsisotherme nach Freundlich durch logarithmieren: log x = log α + β log c
Damit ergibt sich β als Steigung der Geraden, α aus dem Ordinatenabschnitt.
Diese Beziehung beschreibt Adsorptionserscheinungen gut in kleinen und mittleren Konzentrationsbereichen.
Einen größeren Gültigkeitsbereich hat die Adsorptionsisotherme nach Langmuir:
cbcax⋅+⋅=
1
Diese geht für kleine Werte von c über in x = a ⋅⋅⋅⋅c hier ist also x direkt proportional zu c.
Für große Werte von c wird 1 vernachlässigbar klein (gegen bc) und wir erhalten die Beziehung: x = a/b d.h. die adsorbierte Menge wird konstant! Die Kurve erreicht einen Grenz- oder Sättigungswert. Das bedeutet: die gesamte Oberfläche ist mit dem adsorbierten Stoff bedeckt.
Für die Analytik von besonderer Bedeutung ist die Adsorption von Ionen. Wird eine Ionensorte stärker adsorbiert als die vorhandenen anderen Ionen, so wird die Adsorptionsschicht elektrisch aufgeladen. Dies wiederum bewirkt eine Anreicherung von „Gegenionen“ in der Adsorptionsschicht unmittelbar angrenzenden Lösungsschicht.
Kristall
“ “ “ “ “ “ “ “ ” ” ” ” ” ” ” ”
Adsorptionsschicht “ fest haftend
Gegenionen (” ) ggf. austauschbar
Lösung ” “ ” ” “ ” ” “
Beispiel: AgI-Fällung durch Versetzen einer KI-Lösung mit einer AgNO3-Überschuss.
Es besteht nun die Möglichkeit, dass sich in der Adsorptionsschicht entweder Ag+ oder K+ oder NO3
—Ionen anreichern. Die relative Adsorbierbarkeit hängt von der Stärke ab, mit der die Ionen im Gitter gebunden werden (Gitterenergie) und lässt sich anhand der relativen Löslichkeiten abschätzen. Von den möglichen Kombinationen Ag+ - I-, K+ - I-, Ag+ - NO3
- hat Ag+ - I- bei weitem die geringste Löslichkeit (höchste Gitterenergie). Folge: Adsorptionsschicht = Ag+ Gegenionen = NO3
-
Die relative Menge x der adsorbierten Ionen hängt nach Freundlich außer von den Werten α und β (die der relativen Adsorbierbarkeit Rechnung tragen) auch von der Konzentration ab. Da α und β für Ag+ ohne dies relativ groß sind, werden Ag+ bevorzugt adsorbiert, solange [Ag+] nicht zu kleine Werte annimmt.

36
AgI
K+ I- NO3-
AgI
K+ Ag+ NO3-
Schematisch lässt sich also die Zusammensetzung eines Bereiches der festen Phase und ihrer Oberfläche durch die Formel darstellen: [(AgI)m Ag+
n] (NO3-)n Gegenion wobei m >> n ist.
Bestimmt man Ag+, so versetzt man eine AgNO3-Lösung mit einem Überschuss an KI.
Von den Ionen in der Lösung wir aus analogen Gründen nunmehr I- am stärksten adsorbiert. Schematische Formel des Niederschlages: [(AgI)m I-n] (K+)n Gegenion
Folgerung: Ein Salzkristall adsorbiert bevorzugt seine eigenen Ionen! In einem Fall wir also AgNO3 als Oberflächenverbindung im anderen Fall KI „mitgefällt. Man bezeichnet diesen Vorgang auch als Mitziehen. Werden die mitgefällten Teilchen im Niederschlag eingebaut, so spricht man von Okklusionen. Hier muss dem Auswaschvorgang eine Zerkleinerung der Kristalle vorausgehen.
Noch gefährlicher, weil noch schwerer zu trennen sind die Mischkristalle, die sich dann bilden können, wenn die in Betracht kommenden Verbindungen den gleichen Gitterbau aufweisen, z.B. AgCl/AgBr BaSO4/KMnO4 KClO4/KMnO4
Vorbeugende Maßnahmen: (zur Vermeidung der durch Adsorption bedingten systematischen Analysenfehler).
• Erzeugung von grobkörnigen Niederschlägen mit kleiner Oberfläche durch Wahl geeigneter Fällungsbedingungen oder durch teilchenvergrößernde Sekundärprozesse (s.o.)
• Fällung mit einem Reagenz, das sich leicht entfernen lässt oder Umwandlung der Oberflächenverbindung in eine leicht entfernbare.
1. Beispiel: Bestimmung von Fe3+ durch Fällung mit OH- als Fe(OH)3 und verglühen zu Fe2O3
a) Fällt man aus salzsaurer Lsg. Mit NaOH, so werden überschüssige OH-Ionen adsorbiert – [(Fe(OH)3)m (OH-)n] (Na+) bildet sich, beim Glühen würde sich Na-Ferrat bilden und das Ergebnis verfälschen.
b) Fällt man aus salzsaurer Lösung mit NH3, so bildet sich dagegen [(Fe(OH)3)m (OH-)n] (NH4
+). Beim Glühen entweichen die Verunreinigungen vollständig als NH3 und H2O. selbst eingeschlossenes NH4Cl würde leicht entweichen, nicht dagegen eingeschlossenes NaCl.
2. Beispiel: Umwandlung eines adsorbierten Stoffes (AgI)
Wie wir sahen, wird bei der Ag+-Bestimmung KI vom AgI-Niederschlag zurückgehalten. Wäscht man mit reinem Wasser aus, so lässt sich KI nur sehr schwer entfernen, da I- sehr stark adsorbiert wird. Es besteht jedoch die Möglichkeit eines Ionenaustausches in der Gegenionenschicht, wenn andere Ionen in der Lösung im Überschuss vorliegen. Wäscht man mit NH4NO3-Lösung oder HNO3 aus, so sind folgende Austauschvorgänge möglich:
[(AgI)m I-n] (K+)n + n NH4+ [(AgI)m I-n] (NH4
+)n + n K+
[(AgI)m I-n] (K+)n + n H+ [(AgI)m I-n] (H+)n + n K+
Wegen der hohen NH4+ bzw. H+-Konzentration in der Waschflüssigkeit liegt das GGW nach
dem MWG weit rechts. NH4I bzw. HI lassen sich nun durch Erhitzen des Niederschlages ( im Gegensatz zu KI) leicht verflüchtigen.
Nachfällung

37
Wenn ein Niederschlag längere Zeit mit einer Lösung bestimmter Zusammensetzung in Berührung steht, so kann eine Nachfällung erfolgen.
Beispiel Cu2+ und Fe2+lassen sich aufgrund der Löslichkeitsunterschiede ihrer Sulfide trennen. CuS fällt aus saurer Lösung, FeS erst aus ammoniakalischer. Nun stellt man fest, dass CuS sein Gewicht vergrößert, wenn man aus Fe2+-haltiger Lösung fällt und stehen lässt.
Das gefällte CuS adsorbiert Sulfidionen (starke Bindung) als Gegenionen der Doppelschicht treten H+-Ionen auf. Die Sulfidkonzentration an der Oberfläche ist also wesentlich größer als im Innern der Lösung. Die H+-Gegenionen können durch Fe2+-Ionen ausgetauscht werden. Wegen der hohen Sulfidkonzentration kommt es an der CuS-Oberfläche zur Abscheidung von FeS, das wiederum Sulfidionen adsorbiert u.s.w.
Diese Überlegungen führten zu einer Methode, die Nachfällungen zu verhindern und z.B. Cu2+und Fe2+ (oder Cu2+ / Zn2+ oder Zn2+ / Co2+ )über Sulfidfällungen sauber zu trennen:
Setzt man ungesättigte Aldehyde (R-CH=CH-CHO) zu, so lagern sich diese aufgrund spezieller Oberflächenwechselwirkungen über der adsorbierten primären H2S-Schicht an und verhindern so die Nachfällung. So erhält man tadellos saubere Fällungen bei der Zn/Co-Trennung durch Zusatz von Acrolein (R = H) Cu/Zn-Trennung durch Zusatz von Crotonaldehyd (R = Me).
Filtrieren Nach Erzeugung einer quantitativen Fällung müssen Niederschlag und Lösung getrennt werden durch die Trennoperation Filtrieren. Die Art ihrer Durchführung hängt ab von:
- Zustand des Niederschlages (man unterscheidet zwei Gruppen: nichtkristalline gallertartige oder kristalline Fällungen)
- Der Art der Weiterverarbeitung (entweder nur Trocknen oder Trocknen + Glühen)
Filtriertechnik: Entweder unter Normaldruck (Flüssigkeit läuft aufgrund ihrer eigenen Schwere durch das Filter) oder unter vermindertem Druck (Absaugen). In Spezialfällen wendet man auch überhöhten Druck oberhalb des Filters an.
Es stehen prinzipiell 3 Filterarten zur Verfügung:
- Papierfilter
- Glasfiltertiegel
- Porzellanfiltertiegel
Die Filterarten unterscheiden sich vor allem im Material und in der Porenweite und sind in folgender Tabelle zusammengefasst: Siehe Umdruck Erläuterungen zur Bezeichnung von Glasfiltertiegeln:
Zahl Buchstabe Zahl 1 D 5 Größe Material Porendurchmesser
G = Geräteglas 20 D = Duranglas 50 N = Normalglas 16 III B = Quarzglas
Das geeignete Filter muss nach Art des ND. und nach dessen Korngröße ausgewählt werden.

38
Filterarten:
Filterart mittlerer Poren-durchmesser (mmmmm)
Verwendungs- beispiel
Papierfilter: (z.B. Schleicher & Schüll) Schwarzband 7 Fe(OH)3, Al(OH)3 Weißband 5 Sulfide Rotband 3
Blauband 2 BaSO4
Glasfiltertiegel: (z.B. Schott) D00 200-500
D0 150-200
D1 90-150
D2 40-90
D3 15-40
D4 3-15 AgCl D5 <1-3
Porzellanfiltertiegel: (z.B. Haldenwanger) A3 ≅8
A2 ≅7
A1 ≅6 BaSO4
Bezeichnung von Glasfiltertiegeln:
1 D 5
↑ ↑ ↑ Größe Material Poren-Æ
1 = 30 ml D = Duran s.o. 2 = 50 ml B = Quarzglas
10= 15 ml

39
Papierfilter: für gallertartige Nd. (z.B. Hydroxide) sind relativ engporig, Poren verstopfen nicht so leicht wie etwa bei Porzellantiegeln. Es handelt sich um sog. „Quantitative Papierfilter“; das Material ist mit HCl und HF vorbehandelt, um möglichst alle mineralischen Bestandteile herauszulösen. Folge: Sie lassen sich praktisch aschefrei verbrennen (während der nicht stöchiometrische Nd. durch Glühen von der Fällungs- in die Wägeform überführt wird.)
Bei der Verbrennung verkohlt das Filter zunächst, der entstehende Kohlenstoff wirkt reduzierend. Daher darf man bei leicht reduzierbaren Nd. (z.B. AgCl) keine Papierfilter verwenden.
Masse des Verbrennungsrückstandes auf Filterpackung vermerkt, z.B. Fa. Schleicher u. Schüll: bei 11 cm Durchmesser: „Pondus cineris unius filtri“: 0,07 mg Auf Analysenwaage nicht erfassbar.
Quant. Papierfilter werden i.a. durch Farbbezeichnungen unterschieden. Bezeichnung Porenweite [µm] Filtrationszeit [s] Verwendung
Schwarzband 7,4 – 3,7 40 – 60 SiO2, Sulfide, Hydroxide
Weißband 6,8 – 3,4 100 – 150
Blauband (Barytfilter) 2,2 1000 - 1500 ZnS, BaSO4
Praxis des Filtrierens Größe des Filters nach Menge des Nd. bemessen, Filter darf höchstens ½ mit Nd. gefüllt sein.
Verwendung von Analysentrichtern (Urbanti-Trichter). Diese bilden eine saugende Flüssigkeitssäule durch Aussparungen und entsprechend langes Rohr. Filter nur bis 1 cm unter Trichterrand; nie vollständig mit Flüssigkeit füllen. Filterrohr soll an Becherglaswand anliegen. Nach Möglichkeit heiß filtrieren, da Viskosität dann geringer ist (ggf. Dampftrichter verwenden).
Porzellan- und Glasfiltertiegel: Nd. kann nach Filtration unmittelbar getrocknet bzw. geglüht (Porzellan) werden. Verwendung von Saugflaschen, Gummiringen und Vorstoß. Ggf. mehrfach filtrieren, wenn anfangs Trübung auftritt. Nicht zu hohen Unterdruck verwenden und nicht Trockensaugen.

40
Auswaschen
Gehört zu den schwierigsten Operationen in der Gravimetrie. Durch das Auswaschen sollen anhaftende Verunreinigungen (Fremdelektrolyt, Fällungsmittel) vom Nd. entfernt werden ohne dass der Nd. selbst in Lösung geht.
Im folgenden seien mögliche Komplikationen und entsprechende Gegenmaßnahmen besprochen.
1) Löslichkeit des Nd. zu groß, durch die Waschflüssigkeit werden merkliche Mengen gelöst. Gegenmaßnahme:
a) Wechseln des Waschmittels, i.a. Wahl eines geeigneten organischen Lösungsmittels anstelle von Wasser.
b) Zusätze zum Waschmittel, i.a. Zusatz von Fällungsmittel in leicht flüchtiger Form: Für Chloride HCl, für Sulfate H2SO4, für Sulfide H2S, ansonsten möglichst Ammoniumsalze. Zurückdrängung der Löslichkeit aufgrund des MWG!
c) Kombination von a und b
Beispiele:
Ca-Oxalat: man wäscht mit (NH4)2C2O4-haltigerem Wasser. Beim Glühen entsteht CaCO3 und (NH4)2C2O4 zersetzt sich.

41
PbSO4: Auswaschen mit 50%ig. Alkohol, der etwas H2SO4 enthält.
KClO4: Auswaschen mit absol. Alkohol, der etwas HClO4 enthält.
2) Der Nd. geht beim Waschen in den kolloidalen Zustand über (Durchlaufen oder Verstopfung des Filters): Besonders bemerkbar bei Al(OH)3. Grund: Das Ausflocken von Nd. beruht oft auf der Aggregation von Teilchen kolloidaler Dimensionen unter der Einwirkung adsorbierter Elektrolyte. Werden diese entfernt, so erfolgt der umgekehrte Vorgang. Gegenmaßnahme: Zusatz kleiner Mengen Elektrolyt, z.B. NH4NO3 das sich beim Glühen zu N2O und H2O zersetzt. Gilt auch für Fe(OH)3
3) Schlechte Löslichkeit der Verunreinigung in der Waschflüssigkeit. Die Verunreinigungen müssen bekannt sein! Beispiel: Fällung von Ni2+ mit Diacetyldioxim, welches in Wasser relativ schwer löslich ist und beim Fällungsvorgang mit ausfallen kann. Gegenmaßnahme: Waschen mit Alkohol.
4) Auftreten schwerlöslicher Niederschläge durch Reaktion mit der Waschflüssigkeit. Tritt als Folge einer Hydrolyse auf, wenn man einen Nd., der z.B. dreiwertige Metallionen (Fe3+, Bi3+, Sb3+ ) mitgerissen hat, mit reinem Wasser wäscht. Abhilfe: Zusatz von etwas HCl zum Waschwasser. Wenn ND. in Säure löslich (Chromate, Phosphate, Arsenate) setzt man NH3 zu, z.B. beim Waschen von MgNH4PO4
Fazit: Nur in den seltensten Fällen lässt sich reines Wasser als Waschflüssigkeit verwenden. Oft müssen mehrere der genannten Methoden kombiniert werden.
Wie viel Waschflüssigkeit darf man nehmen, wie wäscht man am wirkungsvollsten aus?
Das maximal vertretbare Volumen an Waschflüssigkeit lässt sich aufgrund der Werte von KL und L berechnen.
Für die Art des Auswaschens gilt: Es ist wesentlich wirkungsvoller, öfter mit jeweils wenig Solvens auszuwaschen als einmal mit viel. Dies lässt sich durch einfache Verteilungsgesetze begründen. Die Regel ist jedoch nur dann sinnvoll anwendbar, wenn kristalline Niederschläge mit kleiner Oberfläche vorliegen. Bei gelartigen Nd. mit sehr großer Oberfläche macht sich die Adsorption stark bemerkbar. Hier ist es günstiger, eine Umfällung zur Reinigung des Nd. vorzunehmen.
Von der Vollständigkeit des Auswaschens überzeuge man sich durch quantitative Prüfung der Waschflüssigkeit (z.B. chloridfrei waschen).
__________________________________________________________________________
Beispielrechnung zur Begründung: VR = Restvolumen der dem Nd. anhaftenden Flüssigkeit co = Konzentration der Verunreinigung in VR xo = Menge der Verunreinigung (xo = co VR) V = Volumen der Waschflüssigkeit Allgemein gilt für Verdünnungen: c1 V1 = co Vo
c1 = coVo/V1 mit V1 = Vo+VR

42
1. Waschung: Nach Zugabe von V sei die Konzentration c1 = VV
VcVV
x
R
Ro
R
o+
⋅=+
Verbleibt nach Ablauf oder Abgießen dasselbe Restvolumen VR, so ist die darin
nochvorhandene Menge x1 = c1 VR = VV
VxVV
VcR
Ro
R
Ro +
⋅=+
⋅22
x1< xo
2. Waschung: c2 = )²(
²1VV
VcVV
x
R
Ro
R +⋅=
+ x2 = c2 VR =
)²(
3
VVVcR
Ro +⋅
n. Waschung: cn = nR
nR
oVV
Vc)( +
⋅ xn = cn VR = nR
nR
oVV
Vc)(
1
+⋅
+
Beispiel: VR= 1 ml a) V = 100 ml, n = 1 b) V = 20 ml, n = 5 (gleiches Gesamtvolumen!)
a) c1 1011
oR
Ro c
VVVc =+
⋅= b) c5 = 555
5
211
)201(1
)(⋅=
+⋅=
+⋅ oo
R
Ro cc
VVVc
c5 << c1
__________________________________________________________________________
Trocknen und Glühen Die gefällten und gewaschenen Niederschläge müssen getrocknet werden, ggf. auch geglüht, denn sie sollen in einer definierte, stöchiometrische Wägeform überführt werden. Auch feste Analysenproben werden in der Regel nicht luftgetrocknet eingewogen, sondern vorher zusätzlich getrocknet.
Die Art der Trocknung bzw. Glühens hängt von der Natur der Substanz ab. Das Trocknen lässt sich beschleunigen, indem man am Ende des Waschvorganges mit Alkohol und dann mit Ether nachwäscht. In der Regel geschieht das Trocknen im Trockenschrank bei 110°C etwa 3 h lang. Das Abkühlen erfolgt im Exsikkator über einem Trockenmittel (mind. 30 min). Substanzen, die sich bei 110°C zersetzen könnten oder ihre Kristallwasser verlieren, trocknet man direkt im Exsikkator bei Raumtemperatur.
Das Trocknen sollte generell bis zur Gewichtskonstanz durchgeführt werden, z.B. durch die Folge: Trocknen – Abkühlen – Wägen – Trocknen – Abkühlen – Wägen – ...
Wirkungsweise des Exsikkators Beruht auf einer isothermen Destillation des Lösungsmittels.
In einem geschlossenen System befinden sich 2 Behälter mit Lösungsmittel bzw. Lösung.
po p
po > p
Lsgm. Trockenmittel
po = Dampfdruck des Lsgm. p = Dampfdruck der Lösung bzw. Trockenmittels
Wie wir sehen werden, ist po>p, falls der Dampfdruck des gelösten Stoffes = 0 ist.
Die Beziehung zwischen po und p ergibt sich aus dem Raoultschen Gesetz:
nN
Npp o +⋅=
N = Stoffmenge des Lösungsmittels
n = Stoffmenge des gelösten Stoffes

43
Der Quotient N/(N+n) = Stoffmengenanteil des Lösungsmittels Für reines Lsgm. ist n = 0, dann ist p = po
po – p =∆p = Dampfdruckerniedrigung, die das Lsgm. durch den gelösten Stoff erfährt.
∆p / po = relative Dampfdruckerniedrigung
nN
nnNNnN
nNN
pp
ppp
pp
oo
o
o +=
+−+=
+−=−=−=∆ 11
Die relative Dampfdruckerniedrigung ist also gleich dem Stoffmengenanteil des gelösten Stoffes!
Im Falle der Trocknung im Exsikkator ist der Dampfdruck über dem Trocknungsgut praktisch gleich dem Dampfdruck po von reinem Wasser, da der Feststoff in Wasser nahezu unlöslich ist. Das Trockenmittel hat i.a. die Eigenschaft, sich in Wasser sehr gut zu lösen bzw. Wasser sogar chem. zu binden: P4O10 + 6 H2O → 4 H3PO4 oder als Kristallwasser aufzunehmen: CaCl2 2 H2O→ CaCl2 2 H2O 2 H2O→ CaCl2 4 H2O 2 H2O→ CaCl2 6 H2O
Wenn wir nun den Lösungsvorgang betrachten und praktisch wasserfreies Trocknungsmittel einsetzen, so ist:
≈+
(da N →0), folglich ist p≈ 0, denn 1≈−=∆
o
o
o ppp
pp
Die Wirkung eines Trockenmittels ist umso besser, je kleiner p ist (Vakuum).
Einige Trocknungsmittel für Exsikkatoren:
H2SO4 (konz.) Vorteil: Sehr rasche Trocknung, hohes H2O-Aufnahmevermögen
Nachteil: Erschöpfung nicht äußerlich erkennbar
Mg(ClO4)2 Sehr wirksam, bildet wie CaCl2 Hydrate mit 2, 4, und 6 H2O
P4O10 Wirksamstes Trockenmittel, allgemein anwendbar. Verbrauch an glasigem Aussehen erkennbar, kommt auch auf mineralischem Träger im Handel
CaCl2 Wird am häufigsten verwendet. Je poröser, desto schneller wirksam; kann bis 97,5 % der Trockenmasse an Wasser aufnehmen, dann zerfließt es.
Silicagel: Zerfließt nicht (0,02 mg H2O /l) Aufnahmevermögen für Wasser viel geringer als bei H2SO4 oder CaCl2. Dafür durch längeres Erhitzen auf 150°C regenerierbar ohne Einbuße an Trocknungsvermögen.
Einige Trocknungsmittel werden auch mit Indikator geliefert, z.B. H2SO4, P4O10, Silicagel.
„Blaugel“ enthält z.B.CoCl2 bei 3,5 mmHg (4,66 hPa) Wasserdruck umschlag nach violett, bei 5 mmHg nach rosa.
Blaugel ist krebserregend (K2, Symbol T) und soll durch andere SIlicagele ersetzt werden, die kein CoCl2 enthalten:
Orangegel Silicagel mit org. Farbstoff: orange = trocken, weiß = feucht 6 % Wasser
Sorbsil® C Chamäleon (Oker Chemie)
enthält (NH4)Fe(SO4) orange = trocken, weiß = feucht 19 % Wasser
regenerierbar bei 140°C

44
Glühen Elektrische Tiegelöfen (bis 1100°C und mehr). Üblicher Weise 800 –900°C im Porzellantiegel langsam aufheizen, vorher Trocknung! Verwendung von Deckel und Tiegelschuh. (Deckel vor allem, wenn Glühen mit Brenner oder Gebläse erfolgen soll.)
Das Glühen dient vor allem 2 Zwecken:
1) Entfernung von Verunreinigungen, falls diese hinreichend flüchtig sind, z.B. Reste von Wasser (etwa okkludiertes – oder Kristallwasser), adsorbierte Fremdstoffe. An sich wäre das Glühen aus diesem Grunde generell wünschenswert, doch sind hier durch die Zersetzlichkeit der Substanzen Grenzen gesetzt.
2) Überführung der nicht stöchiometrischen Fällungsform eines Nd. in eine stabile, wohldefinierte Wägeform.
Beispiele:
• BaSO4 Bei 800°C entweichen letzte Spuren von Wasser sowie adsorbierte H2SO4 oder (NH4)2SO4. Die Zersetzung zu BaO + SO3 erfolgt erst oberhalb 1400°C.
• MgNH4PO4 ⋅6 H2O = Fällungsform, beim Glühen auf 900°C tritt nach Wasserverlust folgender Prozess ein: 2 MgNH4PO4 → Mg2P2O7 + 2 NH3 + H2O Wägeform
• Fe(OH)3⋅x H2O = Fällungsform. Glühen bei 800-900°C → Fe2O3 (Wägeform) Bei 1100°C oder unter reduzierenden Bedingungen (Flammengase, Filterrückstände) erfolgt Bildung von Fe3O4, daher für genügend Luftzutritt sorgen und Temperatur kontrollieren.
• Al(OH)3⋅x H2O = Fällungsform Al(OH)3 >400°→ γ-Al2O3 >1100°C→ α-Al2O3
kubisch hexagonal
stark hygroskopisch nicht hygroskopisch
Je nach Problem können also die Bedingungen des Trocknen oder Glühens recht unterschiedlich sein.
Veraschen Filter mit Nd. möglichst feucht veraschen. Zuerst zusammenfalten und in Porzellantiegel einlegen. Erhitzen im Tiegelofen unter Luftzutritt. Veraschen sollte bei möglichst niederer Temperatur geschehen. Bei Veraschen mit Brenner darauf achten, dass Filter nicht in Brand gerät. Filter verkohlt zunächst, bevor es verglimmt.
Thermogravimetrie Einige thermische Umwandlungen beim Glühen verlaufen stufenweise. Die Methode der Thermogravimetrie gestattet nun ein genaueres Verfolgen derartiger Vorgänge. Dabei wird in einer speziellen Apparatur das Gewicht einer Probe über eine gewisse Zeit verfolgt, während zugleich die Temperatur stetig mit konst. Rate erhöht wird.

45
m
t (~T)
100°C
228°C
398°C
420°C
660°C
838°C
CaC2O4
CaCO3
CaO
- CO2
- CO
- H2O
CaC2O4 H2O
Am Beispiel des CaC2O4 H2O wollen wir sehen, dass die Zersetzung zu CaO ebenfalls stufenweise erfolgt.
Entsprechend den jeweils abgegebenen Gasmengen steht die Stufenhöhe im Verhältnis von 18:28:44.
Man kann also die T-Existenzbereiche der einzelnen Spezies aus einem derartigen Diagramm ablesen.
Ferner erkennt man natürlich, welche Substanzstufen überhaupt auftreten.
Bei MgC2O4 2 H2O treten z.B. nur 2 Stufen auf, die folgenden Vorgängen entsprechen:
MgC2O4 2 H2O → MgC2O4 + 2 H2O
MgC2O4 → MgO + CO + CO2
Die Stufe des MgCO3 wir also nicht durchlaufen.
Es gibt zahlreiche Anwendungen für die Thermogravimetrie, u.a. Bestimmung der möglichen Hydratformen eines Salzes.
Organische Fällungsmittel Anforderungen an die Wägeform:
- eindeutig stöchiometrische Zusammensetzung
- Stabilität, nicht hygroskopisch
- Wünschenswert ist eine möglichst günstiger analytischer Faktor
Der analytische Faktor F ist diejenige Zahlengröße, mit der man die Auswaage A zu multiplizieren hat, um die gesuchte Menge m des zu bestimmenden Elementes bzw. der Verbindung zu erhalten: gesuchte Menge m = A ⋅ F
F errechnet sich aus den bekannten stöchiometrischen Gesetzmäßigkeiten. Hierzu ein Beispiel: Mg – Bestimmung Fällungsform: MgNH4PO4 6 H2O Wägeform Mg2P2O7
Mg2P2O7 mit der Molmasse 222,55 g/mol enthält 2 Mg mit der Masse 2⋅24,305 = 48,61 g/mol
In 1g Mg2P2O7 sind demnach 48,61/222,55 = 0,2184 g Mg enthalten. => F = 0,2184
Wie sollte F generell beschaffen sein? theor. Auswaage = A Fehler der Auswaage = ∆A Praktische Auswaage = A + ∆A Daraus ergibt sich das Ergebnis m = F ⋅(A + ∆A) = F⋅A + F ⋅∆A mit dem Fehler ∆m = F ⋅∆A
Ist F groß, so ist auch der Fehler F∆A groß; ist F klein, so fällt der Fehler gegenüber der exakten Größe F⋅A weniger ins gewicht.
Ziel der Entwicklung gravimetrischer Verfahren ist also, Wägeformen mit möglichst kleinem analytischen Faktor zu entwickeln.

46
Fällt man Magnesium als Carbonat, so muss bei 900°-1000°C zu MgO als Wägeform verglüht werden. M(MgO)=40,304 g/mol
F = 24,305 / 40,304 = 0,603
F ist also größer und damit ungünstiger als bei Mg2P2O7!
Damit F klein wird, muss der Nenner groß sein, d.h. das Element muss in einer Verbindung mit möglichst hoher Molmasse eingebaut werden. Dieses Ziel wird z.B. durch relativ schwere organische Fällungsreagenzien erreicht.
N
OHOxin
Im Falle des Mg2+ bietet sich 8-Hydroxychinolin (Oxin) an. Es bildet einen 1:2 –Komplex, der mit 2 Molekülen Kristallwasser kristallisiert: Mg(Oxin)2⋅2 H2O M = 348,640 g/mol => F = 24,305/248,64 = 0,0971 Dieser Faktor ist sehr günstig.
Hat man bei den 3 Mg-Bestimmungen jeweils (durch Verspritzen) einen Fehler Da von 1,0 mg gemacht, so ist der Analysenfehler für den Mg-Wert: bei MgO = 0,6030 ⋅ 1 mg = 0,603 mg ≈ 0,60 mg bei Mg2P2O7 = 0,2184 mg ≈ 0,22 mg bei Mg(Ox)2 ⋅2 H2O = 0,0697 mg ≈ 0,07 mg
Anderes Beispiel: Bestimmung von Phosphor
a) als Mg2P2O7 F = 2⋅30,97 / 222,55 = 0,2783
b) Fällung als (NH4)3[P(Mo3O10)4] d.h. P in das sehr schwere Anion einer Heteropolysäure eingebaut. (M = 1876,13 g/mol) F = 30,97 / 1876,13 = 0,01651
c) Man kann noch einen Schritt weitergehen und das NH4-Kation durch das Chinoliniumkation ersetzen und (C9H8N)3[P(Mo3O10)4] ausfällen. (M = 2212,52 g/mol) => F = 0,013998 Dieser Faktor liegt bereits extrem niedrig, die Ergebnisse lassen sich mit wesentlich höherer Genauigkeit angeben.
N
H+
Die Zahl der verfügbaren organischen Fällungsreagenzien ist bereits recht groß. Sie gestatten nicht nur ein genaueres sondern meist auch ein schnelleres Arbeiten. Daneben werden auf diese Weise Trennungen möglich, die nach den klassischen Methoden nur schwierig und sehr zeitaufwendig auszuführen waren. Grund: hohe Selektivität und Spezifität. Auch ist die Empfindlichkeit oft größer. Meist handelt es sich hierbei um Chelatliganden.
Indirekte Analyse Das Verfahren der indirekten Analyse wendet man an, um mehrere Verbindungen nebeneinander nachzuweisen und zu bestimmen, die sich aufgrund ihrer chemischen Ähnlichkeit nur sehr schwer oder gar nicht voneinander trennen lassen.
Beispiele für derartige Gemische:
KCl + KBr K2CO3 + Na2CO3
NaCl + KBr MgCO3 + CaCO3 (Dolomit)
NaCl + KCl NaOH + KOH
Das Prinzip der indirekten Analyse besteht nun darin, dass man durch Anwendung geeigneter Operationen die in dem Gemisch vorhandenen Verbindungen in neue Verbindungen überführt, deren Gesamtmenge man gravimetrisch oder titrimetrisch bestimmt.
Voraussetzung: Das Gemisch muss rein sein, es darf also keine weiteren Stoffe enthalten.

47
1. Beispiel: Hartsalz-Probe (KCl + NaCl) vom Gewicht E mg (=Einwaage). Die Menge an NaCl und KCl ist zu bestimmen.
Probe lösen und mit AgNO3 das Chlorid fällen.
M(NaCl) = 58,443 g/mol M(KCl) = 74,551 g/mol M(AgCl) = 143,321 g/mol
stöchiometrischer Faktor: F1= )()(Pr
EduktModuktM
Wäre die Probe reines NaCl, so müsste für die Masse des ausgefällten AgCl gelten: m(AgCl) = F1E mit F1=M(AgCl)/M(NaCl) = stöchiom. Faktor F1 = 143,321 / 58,443 = 2,4523
Bei reinem KCl gilt entsprechend: m(AgCl) = F2E mit F2=M(AgCl)/M(KCl) = stöchiom. Faktor F2 = 143,321 / 74,551 = 1,9225
Die Tatsächliche Auswaage A müsste folglich zwischen diesen beiden Grenzwerten liegen, also 1,9225 E < A < 2,3425 E KCl NaCl
490,5
384,5
100% NaCl 0 % NaCl0 % KCl 100 % KCl
Nehmen wir an, unsere Einwaage E sei 200 mg, dann wären die Grenzen für die Auswaage A : 384,5 mg < A < 490,5 mg
Zur Auswertung könnte man eine graphische Methode heranziehen:
Rechnerische Auswertung X(NaCl) + Y(KCl) = E (1)
F1X(NaCl) + F2Y(KCl) = A (2)
Auflösen von Gl. (1) jeweils x oder Y einsetzen in Gl.(2) und umformen ergibt die Lösung. Eleganter ist der Weg über Determinanten:
12
1
21
1
12
2
21
2
11
1
11
1
FFFEA
FF
AFE
YFFAFE
FF
FAE
X−⋅−
==−−⋅
==
2. Beispiel: Gemisch von KCl und KBr
m(KCl) = X m(KBr) = Y X + Y = E (Einwaage) (1)
Nun behandelt man das Gemisch mit konz. H2SO4 und dampft zur Trockne ein (=Abrauchen). Dabei werden die Halogenide in K2SO4 überführt gemäß:
2 KCl + H2SO4 → K2SO4 + 2 HCl
2 KBr + H2SO4 → K2SO4 + 2 HBr
Die Stöchiometrischen Faktoren ergeben sich wie folgt:
7322,0001,238262,174
)(2)(
1687,1102,149262,174
)(2)(
422
421
==⋅
=
==⋅
=
KBrMSOKMF
KClMSOKMF

48
Als 2. Gleichung ergibt sich wieder: F1X + F2Y = A (2)
Gleiche Lösungen wir oben!
Zahlenbeispiel: E = 500 mg, A = 460 mg
KBrmgFFFEAY
KClmgFFAFEX
9,2844365,0
1687,1500460
1,2154365,046010,366
1687,17322,04607322,0500
12
1
12
2
=−
⋅−=−⋅−
=
=−
−=−−⋅=
−−⋅=
Kontrolle: X + Y =215,1 + 284,9 = 500 mg = E
3. Beispiel: Bestimmung von Cl- und Br- durch Ausfällung als AgCl + AgBr. Dann bei ca. 300°C Cl2überleiten, alles wandelt sich in AgCl um (Auswaage).
m(AgCl) = X m(AgBr) = Y X + Y = E (Einwaage) (1)
76327,0772,187321,143
)()( ===
AgBrMAgClMF X + FY =A (Auswaage) (2)
1111
11
1111
1
−−==
−−⋅==
FEA
F
AE
YF
AFE
F
FAE
X
Zahlenbeispiel: E = 500 mg, A = 452,6 mg
AgBrmgFEAY
AgClmgF
AFEX
23,20023673,04,47
4365,05006,452
1
77,29923673,0965,70
176327,06,45276327,0500
1
=−−=
−−=
−−=
=−−=
−−⋅=
−−⋅=
Kontrolle: X + Y =299,77 + 200,23 = 500 mg = E
4. Beispiel: Analyse von Dolomit = CaCO3 + MgCO3
Glühen bis zur Gewichtskonstanz: CaCO3 + MgCO3 → CaO + MgO + 2 CO2
Oder Abrauchen mit H2SO4: CaCO3 + MgCO3 → CaSO4 + MgSO4 + 2 CO2 + 2 H2O
X(CaCO3) + Y(MgCO3) = E XF1+ YF2 = A (m von CaO + MgO)
Sind in einem Gemisch 3 Bestandteile enthalten, so wird eine 3. Bestimmungsgleichung erforderlich, d.h. es ist eine weitere Stoffumwandlung nötig.
Zahlenbeispiel: E = 500 mg, A = 250 mg F1 = 0,5603; F2= 0,478
312
1
312
2
3,3660823,015,30
0823,05603,0500250
7,1330823,011
5603,0478,0250478,0500
MgCOmgFFFEAY
CaCOmgFFAFEX
==⋅−=−⋅−
=
==−
−⋅=−−⋅=
Kontrolle: X + Y =133,7 + 366,3 = 500 mg = E

49
Elektrogravimetrie Bisher wurden in der Gravimetrie nur Methoden behandelt, bei denen die Abscheidung der zu bestimmenden Ionensorte mit Hilfe eines geeigneten Fällungsmittels vorgenommen wurde. Bei der Ausfällung wird die in Lösung befindliche solvatisierte Ionenform eines Elementes (Ag+) oder einer Elementgruppe (SO4
2-) durch Reaktion mit einem Gegenion(Cl- oder Ba2+) in die elektroneutrale Form eines unlöslichen Feststoffes überführt,z.B. Ag+ + Cl- → AgCl
Die Neutralisation der Ladung kann aber auch durch ein Elektron direkt erfolgen, ohne dass ein Gegenion zu Hilfe genommen wird: Ag+ + e- → Ag
In diesem Fall ist der elektrische Strom bzw. die elektrische Ladung das „Fällungsmittel“: Elektrogravimetrische Methoden (ältere Bezeichnungen: Elektroanalyse, Elektrolyse)
Prinzip: Der zu bestimmende Stoff wird aus einer Lösung mit Hilfe des elektrischen Stromes an einer geeigneten Elektrode als fest haftender Überzug abgeschieden. Die Stoffmenge errechnet sich direkt aus der Gewichtszunahme der Elektrode, quantitative Abscheidung vorausgesetzt.
Versuchsanordnung Bestehende aus: Elektrolysezelle, Untersuchungslösung, 2 Elektroden, Gleichstromquelle mit variabler Spannung (Trafo mit Gleichrichter), Rührer.
UI
Potentiostat
”œ“
” “
AnodeKathode
Reduktion Oxidation
e-
e-
Spannungswahl z.B. durch in Serie geschaltete variable Widerstände.
In der Regel Pt- oder Pt-Ir-Elektroden. Die Elektrode, auf der die Abscheidung statt findet, wird als Schale, Scheibe, Zylinder, Konus oder als Drahtnetz ausgebildet. Die Oberfläche sollte möglichst groß sein, weshalb die Netzelektrode bevorzugt wird. Als Gegenelektrode wir ein starker, spiralig aufgewundener Pt-Draht verwendet.
Bei kleiner Spannung U fließt noch kein Strom. Erst wenn U einen bestimmten Wert (Zersetzungs-spannung) überschritten hat, beginnt der Stromfluss.
Die angelegte Spannung bedingt ein elektrisches Feld zwischen den Elektroden, unter dessen Einwirkung die Ionen wandern: die positiven Kationen zur negativen Kathode, die negativen Anionen zur positiven Anode.
An den Elektroden erfolgt Entladung der Ionen, d.h. es finden Redoxprozesse statt. Die Anionen geben ihre Ladung an der Anode ab, werden also oxidiert, die Kationen nehmen an der Kathode Elektronen auf, werden also reduziert. Den Elektronentransport übernimmt die Stromquelle („Elektronenpumpe“), die die erforderliche Redoxenergie als elektrische Energie liefert. Der Ladungstransport erfolgt in Lösung durch positiv und negativ geladene Ionen, Im äußeren Stromkreis durch Elektronen.
Anwendung In der Regel Bestimmung von Metallen. Unter geeigneten Bedingungen lassen sich Cu, Ag, Au, Hg, Ni, Co, Zn, Sb u.a.m. an der Kathode abscheiden.
In wenigen Fällen erfolgt die Abscheidung von Kationen an der Anode (Oxidation): Pb2+ als PbO, Mn2+ als MnO2
An einer Ag-Anode lassen sich ferner die Halogenide Cl-, Br- und I- als AgX abscheiden.

50
Gesetzmäßigkeiten
1791 - 1867
Wir suchen zunächst nach einer Beziehung zwischen abgeschiedener Masse und einer elektrischen Größe. Diese Wurde zuerst empirisch von Michael Faraday 1834 gefunden.
1. Faradaysches Gesetz Die bei der Elektrolyse abgeschiedene Stoffmenge der Masse m ist der durchgeflossenen Ladung Q direkt proportional.
m ~ Q
Untersuchen wir die Proportionalität, dazu stellen wir folgende Überlegungen an:
1 mol Elektronen sind NA ⋅e NA = 6,022⋅1023 mol-1 (Avogadro-Zahl) e = 1,6022⋅10-19 As (Elementarladung)
NA⋅e = 9,6485 ⋅104 As/mol ≈ 96 500 As/mol = 1 F = 1 Faraday
Die Ladungsmenge 1 F scheidet gerade ab: 1 mol Me1+-Ionen (Ag+, Na+) ½ mol Me2+-Ionen (Cu2+, Zn2+) oder 1/3 mol Me3+-Ionen (Au3+, Fe3+) allgemein 1/z mol Mez+
FzQMm⋅⋅= 1. Faraday’sches Gesetz
Ionenäquivalent zM
= Masse eines Stoffes, die man mit 1 F abscheiden kann
Beispiel: Ag+ M = 107,868 g/mol z = 1
Die Ladungsmenge 1 As scheidet ab: mgmolAsmolgAs
FzQMm 118,1
/485961/868,1071 =
⋅⋅=
⋅⋅=
Die Masse FzM⋅
wurde früher auch elektrochemisches Äquivalent genannt.
=
2
2
1
1
2
1
zMzM
mm
2. Faraday’sches Gesetz: Die von gleicher Ladung Q abgeschiedenen Massen zweier Stoffe verhalten sich wie die molaren Massen ihrer Ionenäquivalente (M/z).
Ist die Stromstärke I während der elektrolytischen Abscheidung konstant, so gilt:
Q = I⋅t
In der Praxis ist das jedoch nicht der Fall. Durch die Abscheidung verarmt die Lösung an leitenden Ionen, der Widerstand steigt ständig, die Stromstärke sinkt ständig ab, man erhält einen Exponentiellen Abfall. (gilt natürlich nur bei konst. Spannung). Die Ladung entspricht der Fläche unter der Kurve, d.h.
∫
∫
⋅⋅
=
==
t
t
t
dtIFzMm
dtIQ
0
0
I0
t
Q
U = const.

51
Wichtige Einflussfaktoren bei der elektrolytischen Abscheidung sind:
• die Spannung U zwischen den Elektroden
• die Stromdichte = Stromstärke I pro Flächeneinheit der Elektrode
• die Temperatur der Lösung
• das Elektrodenmaterial.
Zersetzungsspannung Welche Klemmspannung U muss man an die Elektrolysezelle anlegen, damit die elektrolytische Abscheidung stattfindet? Bei welcher Spannung fließt also ein Strom I bzw. die Ladung Q = I⋅t durch die Zelle?
Im Falle eines metallischen Leiters gilt das Ohmsche Gesetz: U = I⋅R bzw. I = U/R
I
UEZ
I = ———U-Ez
R
Graphisch resultiert im I/U-Diagramm eine Gerade der Steigung 1/R, die durch den Koordinatenursprung geht.
Bei Elektrolytlösungen dagegen fließt ein Strom erst dann, wenn U einen bestimmten Wert EZ erreicht hat, die sog. Zersetzungsspannung. Erst wenn U > EZ, erlangt das Ohmsche Gesetz wieder Gültigkeit (allerdings nur in einem begrenzten Bereich).
U = EZ + I⋅R oder I = (U – EZ)/R
Erst nach Erreichen von EZ beginnen auch die sichtbaren chemischen Abscheidungen an den Elektroden.
Betrachtet man Zahlenwerte für die Zersetzungsspannung verschiedener Elektrolytlösungen, in denen jeweils gleiche Konzentrationen vorliegen, so mach man folgende Feststellung:
Die EZ-Differenzen, die sich beim Übergang von einer zur anderen Ionensorte bei jeweils gleichem Gegenion ergeben, sind ziemlich konstante Werte. Die Differenzen lassen sich unter der Annahme interpretieren, dass sich EZ jeweils aus einem kationischen und einem anionischen Anteil zusammensetzt: EZ = EA - EK
Das Minuszeichen folgt daraus, dass beide Anteile umgekehrte Vorzeichen haben.
Die Formel für die Klemmspannung lautet dann: U = I⋅R + EA – EK
EA und EK werden auch als Abscheidungspotentiale bezeichnet. Sie lassen sich aus der Nernstschen Gleichung errechnen, die im folgenden diskutiert sei.
Elektrodenpotentiale, Nernstsche Gleichung Wir betrachten die Vorgänge an den beiden Elektroden getrennt am Beispiel von CuSO4.
Kathode: Abscheidung von Kupfer Cu2+ + 2e- Cu
Anode: Abscheidung von Sauerstoff 2 H2O O2 + 4 H+ + 4e- (oder 4 OH- O2 + 2 H2O + 4e- )

52
Wir betrachten eine Cu-Elektrode. An der Grenzfläche zwischen Kupfer-Metall und Cu2+-Ionen in Lösung bildet sich ein Potential aus. Anschauung von Nernst: Es besteht ein GGW zwischen Lösungstension des Metalls (ist für jedes Metall eine charakteristische Größe) und Abscheidungsbestreben des Metalls (ist eine Funktion des osmotischen Druckes und damit der Cu2+-Konzentration in Lösung). Im Falle Cu ist der osmotische Druck > Lösungstension, an der Cu-
Cu
+ ++ ++ +
--- -
--
Cu2+
Oberfläche scheiden sich einige Cu2+-Ionen ab, in Lsg. verbleiben einige überschüssige Anionen; es bildet sich eine „Helmholtzsche Doppelschicht“ aus. Bei unedlen Metallen ist diese Schicht umgekehrt polarisiert. Das entstehende Potential lässt sich jedoch nicht messen.
Messbar ist jedoch die Potentialdifferenz zwischen zwei derartigen Halbzellen. Man hat nun folgende Konventionen eingeführt:
a) als Bezugselektrode wählt man die Standard-Wasserstoffelektrode und setzt ihr Potential = 0 V.
b) Das Standardpotential eines Elementes, z.B. eines Metalls, die aus dem Metall und einer Metallionen-Lösung der Aktivität α = 1 mol/l bei 25°C besteht, gemessen gegen die Standard-Wasserstoffelektrode.
Redoxpaar: H2 + H2O → 2H3O
+ + 2 e-
V0][H][H
lg2
V0,059EE
2
230 =+=
+O
Auf diese Weise resultiert die Spannungs-reihe der Elemente.
• Unedle Metalle besitzen ein negatives Standardpotential Eo
• Edle Metalle besitzen ein positives Standardpotential Eo
Standardpotentiale, E0 [V]
Li+
K+
Na+
Al3+
Zn2+
Cr3+
Sn2+
Pb2+
2H+
Cu2+
Ag+
Hg2+
O2
Cl2Au3+
++++
+++++
+++++++++ e-
e-
3 e-
2 e-
3 e-
2 e-
2 e-
2 e-
2 e-
e-
2 e-
Ca2+
Mg2+
4 H+
2 e-
e-2 e-
2 e-
+ 4 e-
3 e-
LiKCaNaMgAlZnCrFe
SnPbH2
CuAgHg2 H2O2 Cl-
Au
- 3,05- 2,93- 2,87- 2,71- 2,36- 1,66- 0,76- 0,74- 0,442 e-Fe2+
- 0,14- 0,130,000
+ 0,34+ 0,80+ 0,86+ 1,23+ 1,36+ 1,50
Co2+
Ni2+++
2 e-
2 e-CoNi
- 0,28- 0,25
F2 + 2 H+ + 2 e- 2 HF + 3,06
Für die unedlen Metalle kann obige einfache Versuchsanordnung nicht Verwendung finden!
Die in der Tabelle angegebenen Paare stellen sog. Redoxsysteme dar.:
Oxz+ + z⋅e- Red
Beispiel: Ca2+ + 2e- Ca oxidierte reduzierte Form Cl2 + 2e- 2 Cl-
Die Konzentrationsabhängigkeit des Potentials einer Redoxsystems ist durch die Nernstgleichung gegeben, die sich aus thermodynamischen Beziehungen ableiten lässt.

53
)(Re)(lnda
OxaFz
RTEE o⋅
+=
Eo = Standardpotential R = Gaskonstante = 8,3144 J /K⋅mol T = Temperatur (K) 25°C = 298 K F = Faraday-Konst. = 96 485 C/mol z = Zahl der umgesetzten Elektronen
Einsetzen der Konstanten und Umrechnung von ln x = 2,3026 lg x
)(Re)(lg059,0
)(Re)(lg
485963026,22983144,8
daOxa
zEE
daOxa
zEE
o
o
+=
⋅⋅⋅+=
In sehr verdünnten Lösungen lassen sich anstelle der Aktivitäten ai die Konzentrationen ci einsetzen. Das Standardpotential Eo ergibt sich für den Fall, dass die Aktivitäten aller beteiligten Stoffe = 1 sind; lg 1 = 0 => E = Eo
Dabei sind folgende Konventionen zu beachten für a:
Gase: Standardzustand (Partialdruck =1,013 bar = po) ansonsten gilt a = p/po
Festkörper: a = 1 (in jedem Fall!)
Lösungsmittel: z.B. Wasser a = 1 (in jedem Fall!)
Gelöster Stoff: a = 1 = ao ansonsten gilt a = jeweilige Aktivität
Liegen alle beteiligten Stoffe in den Standardzuständen vor, so ist E = Eo.
Beispiel: Metallelektrode M Mz+ + z e- Cu: z = 2
VVE
CuEEMaz
EE CuCuzo
31,01,0lg0295,034,0
]lg[2059,0)(lg059,0 2
=+=
+°=+= ++
Wasserstoffelektrode: H2 2 H+ + 2 e-
Für p(H2)=1,013 bar gilt pHHaE ⋅−=+= + 059,0)(lg2059,00 2
bei pH = 7: E = 0,413 V
Weicht der Partialdruck p(H2) vom Standarddruck po ab, so lautet der Ausdruck:
)(/)(
)(lg2059,00
22
2
HpHpHaE o
++=
Sauerstoffelektrode: 2 H2O O2 + 4 H+ + 4e-
Für p(O2)=1,013 bar gilt pHpHEHaEE oo 059,023,1059,0)(lg4059,0 4 −=⋅−=+= +
Für reines Wasser (pH=7) ergibt sich E = 1,23 – 0,059 7 = 0,82 V
Da der O2-Partialdruck über einer gesättigter O2-Lösung an Luft ca. 0,2 po. Bei pH = 7 erhalten wir dann
V
pHEOpHaOpEE o
oo
81,08067,0413,00103,023,1
059,02,0lg4059,0
)()()(
lg4059,0
2
42
≈=−−=
⋅−+=⋅
+=+

54
Es resultiert also praktisch obiger Wert (Der Einfluss des O2-Partialdruckes ist sehr klein).
Galvanische Zellen Schalten wir zwei Elektroden (Halbzellen) zusammen, so erhalten wir ein galvanisches Element. Ein klassisches Beispiel ist das Daniel-Element: Cu-Elektrode + Zn-Elektrode. Die Potentialdifferenz wird als elektromotorische Kraft EMK bezeichnet.
Zn Cu
Cu2+Zn2+
e-
KathodeAnode
Salzbrücke
Potentialdifferenz = EMK = ∆E = EKathode - EAnode= ECu – EZn
Kupfer: ][Culg0,0295V0,34[Cu]
][Culg2
V0,059EE 22
0 ++
+=+=
Zink: ][Znlg0,0295V0,76[Zn]
][Znlg2
V0,059EE 22
0 ++
+−=+=
EMK = ECu - EZn = 0,34 – (- 0,76) + 0,0295 lg([Cu2+]/[Zn2+])
= 1,10 V + 0,0295 lg([Cu2+]/[Zn2+])
Wenn [Cu2+]=[Zn2+], dann ergibt sich eine EMK von 1,1 V. Dabei ist Zn die negative, Cu die positive Elektrode! Bei Herstellung einer leitenden Verbindung fließt ein Elektronenstrom vom Zink zum Kupfer und es findet die Redoxreaktion statt:
Zn + Cu2+ Zn2+ + Cu
Dabei nimmt das Potential des Cu allmählich ab (wird negativer), das der Zn-Elektrode nimmt zu (wird positiver) bis schließlich EMK = 0. Dann herrsch GGW.
29,371029,37 =⇒=⇒= +
+
+
+
+
+
][Cu][Zn
][Cu][Znlg
][Cu][Znlg
2V0,0591,10 2
2
2
2
2
2

55
E
EM
K =
1,1
V
0
0,34 V
- 0,76 V
Cu/Cu2+
Zn/Zn2+
Wir können den Vorgang, der reversibel ist, jedoch auch umkehren, indem wir von außen eine EMK entgegengesetzte und dem Betrag nach etwas größere Spannung (zur Überwindung des ohmschen Widerstandes der Lösung) anlegen, wenn wir also Elektronen von der Cu- zur Zn-Elektrode pumpen. Prinzipiell ist eine solche Umkehrung einer Elektrolyse gleichzusetzen. Während des Ablaufes der umgekehrten (Unfreiwillig verlaufenden) Reaktion steigt die Potentialdifferenz zwischen den Elektroden ständig an.
_________________________________________________________
Konzentrationskette (Einschub)
Es ist möglich, galvanische Elemente zu konstruieren, bei denen die EMK ausschließlich auf der Konzentrationsdifferenz beruht, d.h. die Elektroden besteen aus dem gleichen material und tauchen in Lösungen unterschiedlicher Konzentrationen des entsprechenden Ions.
Cu Cu
Cu2+Cu2+
e-
KathodeAnode
Salzbrücke
E1 = E0 + ½ 0,059 V lg [Cu2+]
Da Konzentration hier kleiner, löst sich Cu hier auf => Oxidation = Anode
E2 = E0 + ½ 0,059 V lg [Cu2+]
Da Konzentration hier größer, scheidet sich Cu2+ hier ab => Reduktion = Kathode
EMK = EKat – EAn = E2 – E1 = ½ 0,059 V lg [Cu2+]2 / [Cu2+]1
Für ein konkretes Beispiel sei [Cu2+]1 =10-5 mol/l und [Cu2+]2 = 10-1 mol/l
EMK = ½ 0,059 V lg 104 = 0,118 V
________________________________________________________________
Polarisation Zum besseren Verständnis der elektrolytischen Abscheidung müssen wir jedoch noch den Begriff der Polarisation behandeln.
Wir betrachten zunächst folgende Anordnung. Es tritt keine Potentialdifferenz auf.

56
Cu Cu
Cu2+
KathodeAnode
Pt Pt
Cu2+
Pt Pt
Cu2+
A
AnodeKathode
Cu O2
Wäre die Zelle durch ein Diaphragma getrennt und wären die Cu-Konzentrationen auf beiden Seiten ungleich, so würde bei einer solchen „Konzentrationskette“ eine Potentialdifferenz resultieren.
Legen wir eine Spannung an, so fließt sofort ein Strom der dem Ohmschen Gesetzt folgt. Cu steht mit Cu2+ im thermodynamischen GGW, der Vorgang ist völlig reversibel.
Nun ersetzen wir die Cu- durch Pt-Elektroden, diese stehen nicht im Thermodynamischen GGW mit Cu2+, sie sind indifferent („irreversibel“); auch hier tritt keine Potentialdifferenz auf.
Legen wir eine Spannung an, so fließt nun ein sehr geringer Strom (Polarisationsstrom), der aber eine gewisse Änderung der Elektrodenoberfläche bewirkt: An der Kathode scheidet sich etwas Cu ab, an der Anode etwas O2. Dadurch werden die Elektroden ungleichartig, es baut sich eine Polarisationsspannung P auf, die der angelegten Spannung entgegengesetzt und annähernd gleich ist. Steigern wir die äußere Spannung weiter, so steigt auch die Polarisationsgegenspannung, der Strom steigt wiederum nur sehr wenig an; er hält die Beladung (Polarisation) der Elektroden gerade aufrecht, bis diese soweit fortgeschritten ist, dass die Pt-Kathode gerade etwa mit einer einatomigen Cu-Schicht belegt ist und der O2-Druck an der Anode = 1 bar ist: Es ist eine Cu/Cu2+ und eine O2/H2O-Elektrode entstanden.
Potentialdifferenz = „maximale“ Polarisationsspannung für 1 mol/l neutrale Cu2+-Lsg.
ECu = 0,34 V EO2=0,82 V
EA-EK = EO2 – ECu = 0,48 V
Bei weiterer äußerer Spannungssteigerung wächst die Polarisation nur noch unwesentlich an, sie bleibt praktisch hinter der äußeren Spannung zurück, die Folge ist ein starker Stromanstieg, entsprechend einem vollen Einsatz der Abscheidung.
I
UEZ
P ≈ U
P<U
EZ entspricht also der Potentialdifferenz zw. Einer Cu und einer O2-Elektrode, vermehrt um einen geringen Spannungsanteil zur Überwindung des ohmschen Wiederstandes der Lösung. EZ = I⋅R + EA - EK
Im Laufe der elektrolytischen Abscheidung wird ECu kleiner und EO2 größer, d.h. EO2 – ECu wächst! Um eine quantitative Cu-Abscheidung zu erhalten, müssen wir also die Spannung allmählich steigern. In der Praxis arbeitet man allerdings von vornherein bei einer höheren Spannung.
Überspannung Berechnet man die Zersetzungsspannungen von Elektrolytlösungen nach der Nernst’schen Gleichung und vergleicht sie mit den experimentell bestimmten Werten, so stellt man mehr oder weniger große Diskrepanzen fest, die praktischen Werte liegen wesentlich höher als die theoretischen.
In einer 0,5 molaren H2SO4 würde sich errechnen: pH = 0!
H2-Elektrode: 0 V

57
O2-Elektrode: 1,23 V => EZ = 1,23 V
Praktisch findet man einen Wert von 1,67 V, d.h. einen um 0,44V zu hohen Wert (an Pt-Elektroden).
Weitere Beispiele: Lösungen von Sulfaten zweiwertiger Ionen (Berechnung von Ez mit folgenden Annahmen: a[Me2+] = 0,5, pO2=1,013 bar, pH = 5)
Me2+ EZ (Berechnet) [V] EZ (gemessen) [V] Differenz [V]
Zn2+ 1,70 2,35 0,65
Ni2+ 1,19 2,09 0,90
Co2+ 1,22 1,92 0,70
Cd2+ 1,34 2,03 0,69
Cu2+ 0,60 1,49 0,89
Man bezeichnet diese Differenz also Überspannung η. Sie zerfällt ebenso wie die Zersetzungsspannung in 2 Anteile, einen anionischen und einen kationischen:
η = ηA - ηK
Sie muss zusätzlich bei der elektrolytischen Abscheidung überwunden werden. ηK ist in der Regel bei Metall-Kationen klein: ηK (Cu/Cu2+) = 0,01 V, ηK (Fe2+Fe) = 0,1 V
ηA kann dagegen reicht hohe Werte annehmen, vor allem wenn Gase abgeschieden werden. So beruhen die obigen Werte vor allem auf der hohen Überspannung des O2 an Pt. Ursache: kinetische Hemmung eines der mit der Abscheidung verbundenen Einzelschritte.
Vorzeichen der Überspannung: negativ. Wenn ein kathodischer Reduktionsvorgang erfolgt positiv: wenn ein anodischer Oxidationsvorgang erfolg.
Auf diese Weise wirken η in jedem Falle erhöhend auf die Potentialdifferenz!
Außer von der Natur des abzuscheidenden Stoffes hängt die Überspannung von folgenden Faktoren ab:
1) Elektrodenmaterial: Hierzu als Beispiele die η-Werte von H2 an verschiedenen Metallen:
Pt (platiniert) 0,00 V Ag 0,15 V Ni 0,21 V Die hohe Überspannung von H2 an Metallen Cu 0,23 V ermöglicht die elektrogravimetrische Bestimmung Pb 0,64 V unedler Metalle. Zn 0,70 V Hg 0,78 V
Die Überspannung von O2 an blankem Pt beträgt in saurer Lösung 0,4 V
2) Oberflächenbeschaffenheit der Elektrode (bei gleichem Elektrodenmaterial) an platiniertem Pt ist die Überspannung kleiner an blankem Pt.
3) Stromdichte: Stromstärke pro Fläche I/F [A/cm²] Die Überspannung steigt bei Erhöhung der Stromdichte an. Beispiel H2-Abscheidung aus 1 mol/l H2SO4 an Pb:
Stromdichte [mA/cm²] 1 10 100 1000
Überspannung η [V] 0,52 1,09 1,18 1,26
Im stromlosen Zustand tritt keine Überspannung auf, hier entsprechen also die Potentiale den Gleichgewichtspotentialen nach Nernst.

58
4) Temperatur: Temperaturerhöhung bewirkt Abnahme der Überspannung, denn die kinetische Hemmung wird vermindert. (Erhöhung der Reaktionsgeschwindigkeit)
Klemmspannung: Aus den vorhergegangen Überlegungen ergibt sich zusammenfassend für die Klemmspannung, die wir zur Erzwingung einer elektrolytischen Abscheidung anlegen müssen:
U = I⋅R + EZ + η
Nach Aufteilung der Terme ergibt sich:
U = I⋅R + (EZA –EZK) + (ηA – ηK)
Beispiel: Elektrolyse einer CuSO4-Lösung an Pt-Elektroden im Sauren, [Cu2+] = 1, [H+] = 1 I = 0,1 A, R = 2 Ω
U = 0,1 A ⋅ 2Ω + 1,23 V – 0,34 V + 0,44 V + 0,01 V = 1,54 V
U = I⋅R + (EZO2 –EZCu) + (ηO2 – ηCu)
Sind in einer Elektrolytlösung mehrere entladbare Ionensorten enthalten, so erhebt sich die Frage, welche Vorgänge bei niedrigster Klemmspannung ablaufen werden.
Man errechnet nun zunächst unter Zuhilfenahme der Normalpotentiale und der Konzentrationen nach der Nernstgleichung die Redoxpotentiale aller in Frage kommenden Elektrodenreaktionen und bestimmt schließlich unter Berücksichtigung der Überspannungen die den einzelnen Redoxpaaren zuzuordnenden Abscheidungspotentiale. Dazu wendet man folgende allgemeine Regel an:
1
2
3
4
∆E
Anode
Kathode
Stets verläuft an der Kathode zunächst der Reduktionsvorgang, der das Redoxpaar mit dem höchsten (positivsten) Potential und an der Anode derjenige Oxidationsvorgang, der das Redoxpaar mit dem niedrigsten Potential erzeugt. Auf diese Weise resultiert die niedrigste Potentialdifferenz zwischen Anode und Kathode!
Elektrogravimetrische Trennung Aufgrund der Berechnung zur Abscheidung eines Elementes erforderlichen Klemmspannung lässt sich entscheiden, ob die elektrolytische Trennung zweier Ionensorten möglich ist. Wir betrachten als Beispiel die Ag-Cu-Trennung.
U = I⋅R + (EZO2 –EZMe) + (ηO2 – ηMe)
Unter den gleichen Bedingungen (I = 0,1 A, R = 2 Ω, EZO2=1,23 V, ηO2 = 0,44 V) ergibt sich: U = 0,2 + 1,23 + 0,44 - EZMe - ηMe = 1,87 - EZMe - ηMe
Und da ηMe ≈ 0,01 V => U = 1,88 V - EZMe
Es seien die Ausgangskonzentrationen [Cu2+] = [Ag+] =0,1 mol/l
EZcu = E0cu + ½ 0,059 V lg [Cu2+] = 0,34 – 0,03 = 0,31 V
EZAg = E0Ag + 1⋅ 0,059 V lg [Ag+] = 0,80 – 0,06 = 0,74 V
Dann gilt für die erforderliche Klemmspannung U:
UCu = 1,88 – 0,31 = 1,57 V UAg = 1,88 – 0,74 = 1,14 V
Wir können also durch geeignete Wahl von U erreichen, dass nur Ag abgeschieden wird. Die quantitative Abscheidung von Ag sei erreicht, wenn [Ag+] = 10-6 mol/l ist, dann gilt für EZAg
EZAg= 0,80 + 0,059 lg 10-6 = 0,80 – 0,35 = 0,45 V und somit

59
UAg(Ende) = 1,88 – 0,45 = 1,43 V
Daraus folgt, dass sich Ag elektrolytisch von Cu2+ trennen lässt, wenn man unter dem gegebenen Bedingungen bei einer Klemmspannung von 1,43 V arbeitet, bei der noch keine Kupferabscheidung erfolgt.
Betrachten wir die I/U-Diagramme:
U [V]
I
1,14 1,43 1,57
[Ag+]= 10-1 [Ag+]= 10-6 [Cu2+]= 10-1
0,29 V 0,14 V
Generell gilt, dass man Ag+ quantitativ von Cu2+ elektrogravimetrisch trennen kann, wenn man bei einer Klemmspannung arbeitet, die um 0,14 V unterhalb der für die Abscheidung von Cu2+ erforderlichen Klemmspannung arbeitet.
Hierzu müssen wir die Spannung genau kontrollieren. Vorteilhaft arbeitet man mit einem Potentiostaten als Spannungsquelle.
Elektrogravimetrische Trennungen sind elegant, sauber und relativ einfach durchzuführen.
Abscheidung unedler Metalle Aufgrund der Spannungsreihe sollte man schließen, dass sich aus wässriger Lösung nur Metalle abscheiden lassen, die edler sind als Wasserstoff, also ein positives Normalpotential besitzen. Bei unedlen Metallen sollte ausschließlich H2-Entwicklung stattfinden. Aus 2 Gründen lassen sich jedoch auch unedle Metalle abscheide:
1) Das Potential der H2/H+-Elektrode lässt sich durch Wahl des pH-Wertes variieren. E = - 0,059 pH pH = 0 => E = 0 V (in HCl 1,18 mol/l) pH = 7 => E = -0,41 V pH = 11 => E = - 0,65 V
Je höher der pH-Wert, desto mehr rutscht E in den Bereich unedler Metalle.
Beispiel: Ni-Abscheidung [Ni2+]Anfang= 0,1 mol/l [Ni2+]Ende= 10-6 mol/l EoNi= -0,25 V
Potential am Ende der Abscheidung: E = - 0,25 + ½ 0,059 lg10-6 = - 0,25 – 0,18 = -0,43 V
Dieser Wert entspricht der Zersetzungsspannung einer H2Elektrode bei pH = 7. Mit anderen Worten: Nickel lässt sich bereits aus neutraler Lösung (sicherheitshalber aus NH3-Lösung) quantitativ abscheiden.
2) Einfluss der Überspannung
Wir nehmen an, das Potential der Metallelektrode sei tatsächlich negativer als das der H-Elektrode. Dann scheidet sich jedenfalls zunächst Wasserstoff ab. Bei hinreichend hoher Stromdichte verarmt der Raum der Kathodennähe an Protonen, da der Verlust an H+ durch Entladung nicht durch die Diffusion aus dem Innern der Lösung wettgemacht werden kann. Folge, H-Potential an Kathodenoberfläche wird negativer, dadurch kommt das unedlere Metall zum Zuge und wird abgeschieden. Aus der Pt-Elektrode ist eine aus dem abgeschiedenen Metall bestehende Elektrode geworden. Wegen der nun existierenden höheren Überspannung dieser Elektrode für H2-Abscheidung würde selbst bei niederer Stromdichte keine H+-Entladung mehr erfolgen, nur noch Metallabscheidung. Durch die Überspannungseffekte wird der Anwendungsbereich der Elektroanalyse ganz erheblich erweitert.
Beispiel: Abscheidung von Zn [Zn2+]Ende= 10-6 mol/l EoZn=- 0,76 V

60
Potential am Ende der Abscheidung: E = - 0,76 + ½ 0,059 lg10-6 = - 0,76 – 0,18 = -0,94 V
Selbst aus stark alkalischer Lösung z.b. bei pH = 14 wäre Zn nicht abscheidbar, des es gilt:
EH2 = - 0,059 ⋅14 = -0,83 V
Nun ist jedoch die Überspannung von H2 an einer Zn-Elektrode = 0,70 V Damit wird
EH2 = EZ(H2) – η(H2) = EZ(H2) - 0,70 V
Mit anderen Worten: Wir können Zn bereits bei pH = 4,1 theoretisch quantitativ abscheiden, denn es gilt dann: EH2= - 0,059⋅ pH – 0,70 V = -0,059 ⋅4,1 – 0,70 = - 0,94 V
Der hohen Überspannung des Wasserstoffs an Pb verdanken auch der Bleiakku seine Existenz!
Alkali und Erdalkalimetalle sowie Aluminium lassen sich jedoch unter üblichen Bedingungen aus wässrigen Lösungen nicht mehr elektrolytisch abscheiden, die hierzu erforderlichen hohen pH-Werte sind nicht mehr realisierbar.
Beispiel: EoK = - 2,9 V EH2= - 0,059 pH
-2,9 V = -0,059 pH => pH = 2,9/0,059 = 49,1 ≈ 50
Demnach wäre eine OH—Konzentration von 1036 mol/l erforderlich.
Trotzdem gibt es einen Ausweg: Verwendung einer Hg-Kathode hat 2 wesentliche Effekte:
1) hohe Überspannung für H2 (0,78 V oder noch höher bei höheren Stromdichten)
2) Das abgeschiedene Metall reagiert unter Amalgambildung; es wird nicht kompakt sondern in stark „verdünnter“ Form abgeschieden, seine Lösungstension ist stark herabgesetzt: es wird edler.
Praktische Anwendung: Chloralkalielektrolyse!
Allgemein ergibt sich ansonsten folgende Bedingung für die quantitative Abscheidung eines Metalls [Men+]Ende= 10-6 mol/l
EoMe + 1/z ⋅ 0,059 lg 10-6 > η(H2) – 0,059 pH
=> EoMe - 0,35 / z > ηηηη(H2) – 0,059 pH
Nach dieser Beziehung erscheint ein möglichst hoher pH-Wert für die kathodische Fällung eines Metalls günstig zu sein. Einer beliebigen Erhöhung des pH-Wertes sind jedoch durch die Gefahr der Ausfällung des Metallhydroxids aus der Lösung Grenzen gesetzt. Dies lässt sich nun durch Zugabe von Komplexbildnern, die in alkal. Lsg. existenzfähige Komplexe liefern, verhindern.
Beispiele: [Ni(NH3)6]2+, [Zn(OH)4]2-,[Cd(CN)4]2-,
Die Komplexbildung hat einen weiteren Vorteil: sie hält die Konzentration an freien Metallionen in Lösung klein. Die Erfahrung hat gezeigt, dass die Abscheidung dann in Form eines glatten, feinkristallinen, an der Elektrode gut haftenden Nd. erfolgt.
Beispiel Ni-Abscheidung ist zwar aus saurer Lösung möglich, denn es gilt nach :
EoMe - 0,35 / z > η(H2) – 0,059 pH für pH = 4
- 0,25 – 0,35/2 > - 0,21 – 0,059 ⋅ 4
- 0,43 > - 0,45 Jedoch würde Ni als schwammiger Nd. abgeschieden.
Deshalb fällt man Ni aus NH3-Lsg. elektrolytisch. Ni2+ + 6 NH3 [Ni(NH3)6]2+,

61
MWG (Komplexzerfallskonstante): 7,8263
63
210
])([][][ −+
+==
⋅K
NHNiNHNi
63
7,82632
][10])([
][NH
NHNiNi
−++ ⋅=
Es sei [Ni2+]Anfang = 0,1 mol/l, [NH3] = 1 mol/l (großer Überschuss)
Durch Zusatz von (NH4)2SO4 wird der pH auf ca. 10 abgepuffert. Unter diesen Bedingungen liegt praktisch alles Nickel als Amminkomplex vor, d.h. [Ni(NH3)6
2+] = 0,1 mol/l
=> lmolNH
NHNiNi /10
1101,0
][10])([
][ 7,96
7,8
63
7,82632 −
−−++ =⋅=
⋅=
ENi= - 0,25 + ½ 0,059 lg 10-9,7 = -0,25 – 0,29 = – 0,54 V
Für quantitative Abscheidung muss [Ni2+]Ende = 10-6 mol/l werden, auch hier liegt Nickel überwiegend als Komplex vor, d.h. [Ni(NH3)6
2+] = 10-6 mol/l
lmolNH
NHNiNi /1010
11010
][10])([
][ 157,146
7,86
63
7,82632 −−
−−−++ ≈=⋅=
⋅=
ENi= - 0,25 + ½ 0,059 lg 10-15 = -0,25 – 0,44 = – 0,69 V
Bei pH = 10 ist die Bedingung für die quantitative Abscheidung erfüllt, denn es gilt:
- 0,69 V > η(H2) – 0,059 pH = 0,21 – 0,59 = - 0,80 V
Einfluss der Anionen Anionen können elektrolytische Abscheidungen auf verschiedene Weise hemmen. Am günstigsten ist, wenn sie bei der Elektrolyse nicht verändert werden. Wir untersuchen die wichtigsten Fälle.
Sulfat Verhält sich indifferent!
Die kathodische Reduktion nach SO42- + 3H+ + 2e- HSO3
- + H2O hat zwar ein Standardpotential von + 0,20 V ist jedoch sehr stark gehemmt. Die anodische Oxidation zu Peroxodisulfat nach 2 SO4
2- S2O82- + 2 e- (Eo=2,0 V) ist nur bei sehr hohen
Stromdichten möglich. Bei der Elektrolyse von Sulfatlösungen wird also an der Anode nur H2O zu O2 oxidiert! Nitrat Verhältnisse wenig günstig. Die elektrochemische Reduktion über HNO2 und NO bis zu NH3 ist möglich.
NO3- + 3 H+ + 2e- HNO2 + H2O Eo = 0,94 V
NO3- + 4 H+ + 3e- NO + 2 H2O Eo = 0,96 V
NO3- + 10 H+ + 8e- NH4
+ + 3 H2O Eo = 0,88 V
Diese Vorgänge sind zwar von Überspannungen begleitet, aber nicht völlig gehemmt.

62
Nur der Tatsache, dass Nitrat sehr langsam reduziert wird, ist zu verdanken, dass Metalle mit Eo<0,94 V (Ag und Cu) aus nitrathaltiger Lösung abgeschieden werden können. Blei lässt sich dagegen kathodisch nicht mehr abscheiden (geht auch nicht aus SO4
2-, da PbSO4 schwerlöslich ist). Bei der Elektrolyse von Cu und Ag aus NO3
-Lösung setzt man Harnstoff zu, um die entstehende, stark oxidierend wirkende HNO2 zu vernichten:
2 HNO2 + O=C(NH2)2 2 N2 + CO2 + 3 H2O
Chlorid
Denkbare Anodenvorgänge: 2 Cl- Cl2 + 2 e- Cl2 reagiert mit Pt!!! oder 2 H2O O2 + 4 H+ + 4 e-
22 36,1 Cl2-2
Cl][Cl][Cllg
2V0,059E η++=
EO2 = 1,23 – 0,059 pH + ηO2
Trotz des höheren Eo-Wertes wird Chlorid vor Wasser anodisch oxidiert, da ηCl2 << ηO2
Störend wirken sich nur die relativ gute Löslichkeit von Chlorgas in Wasser (0,1 mol/l) aus; Chlor gelangt in den Kathodenraum und wird dort wieder zu Chlorid reduziert; dadurch kann u.U. die Metallabscheidung völlig unterbunden werden. Man bezeichnet diesen Vorgang als „Depolarisation“, was nach folgender Beziehung durchaus sinnvoll ist. Wir berechnen die Zersetzungsspannung einer aus Chlor-Anode und Chlor-Kathode bestehenden Zelle.
EZ = EA – EK = 1,36 – 1,36 + ½ 0,059 lg 2-2
*][Cl]*[Cl
+ ½ 0,059 lg 2-2
][Cl][Cl
Anodenraum Kathodenraum
= ½ 0,059 lg ][Cl][Cl
][Cl]*[Cl
22*-
2-2
⋅⋅
Wenn sich die Chlor und die Chloridkonzentrationen im Anoden und Kathodenraum durch starkes Rühren praktisch ausgleichen, geht dieser Ausdruck und damit EZ gegen Null.
Eine besondere Komplikation tritt bei der Elektrolyse chloridhaltiger Kupfer-Lösungen auf, dadurch dass Cu2+ auch zu Cu+ reduziert werden kann, das mit Cl- schwerlösliches CuCl bildet. Wir müssen also folgende Prozesse an der Kathode nebeneinander betrachten.
a) Cu2+ + 2e- Cu Eo= 0,34 V
b) Cu2+ + e- Cu+ Eo= 0,159 V
Cu+ + Cl- CuCl KL= 10-6 mol²/l²
Elektrolysiert man eine CuCl2-Lösung mit c=0,1 mol/l so ergeben sich folgende Potentiale:
a) ECu/Cu2+= 0,34 + ½ 0,059 lg 0,1 = 0,31 V
b) ECu+/Cu2+= 0,159 + 0,059 lg [Cu2+]/[Cu+]
Und da [Cu+] = 10-6 / [Cl-], ergibt sich
ECu+/Cu2+= 0,159 + 0,059 lg6-
-2
10][Cl][Cu ⋅+
Da anfangs CuCl2 vorliegt, ist [Cu2+] = 0,1 mol/l und [Cl-] = 0,2 mol/l. Daraus ergibt sich:

63
ECu+/Cu2+= 0,159 + 0,059 lg =⋅⋅−
6-
-1
10]10 1021
0,159 + 0,059 lg 2 10-4 = 0,413 V
Da ECuCl > ECu muss an der Kathode Vorgang b ablaufen, es scheidet sich CuCl anstelle von Cu ab.
Anodische Fällung
Wie erwähnt, lässt sich Blei aus salpetersaurer Lösung nicht kathodisch als metallisches Blei abscheiden, da das Standardpotential von Blei mit – 0,13 V bereits zu niedrig liegt, vorher also Nitrat reduziert wird. Schwefelsaure und salzsaure Lösungen scheiden ebenfalls aus, da PbSO4 und PbCl2 schwerlöslich sind.
Man kann nun jedoch Blei anodisch oxidieren und als PbO2 zur Abscheidung bringen.
Bedingungen: c(Pb(NO3)2)=0,1 mol/l in HNO3 (pH = 0 => [HNO3] > 1 mol/l)
Mögliche Kathodenvorgänge:
1) Pb2+ + 2e- Pb EPb= - 0,13 + ½ 0,059 lg [Pb2+] = - 0,16 V
2) H3O+ + e- ½ H2 + H2O EH2= - 0,059 pH + η(H2) = η(H2)
3) NO3- + 3 H+ + 2e- HNO2 + H2O EHNO2 = 0,94 V + ½ 0,059 lg
][]³][[
2
3HNO
HNO +− + η(HNO2)
EHNO2≈ 0,94 + η(HNO2)
Da [NO3-] und [H+] = 1 und [HNO2] klein, wenn Harnstoff vorhanden ist. Die übrigen
Reduktionsprozesse von Nitrat haben ähnliche Potentiale.
Mögliche Anodenvorgänge:
4) 2 H2O O2 + 4 H+ + 4 e- EO2= 1,23 + η(O2) = 1,23 + 0,44 = 1,67 V
5) Pb2+ + 2 H2O PbO2 ↓+ 4 H+ + 2 e- EPbO2= 1,46 + ½ 0,059 lg ][][2
4
+
+
PbH = 1,49 V
Die Praxis zeigt, dass die Kombination der Reaktionen 3 und 5 die niedrigste Zersetzungsspannung liefert. An der Anode scheidet sich PbO2 ab, an der Kathode wird NO3
- zu HNO2 reduziert.
Gesamtgleichung: Pb2+ + NO3- + H2O PbO2 + HNO2 + H+
Man erkennt auch, dass die anodische PbO2-Bildung erst durch die hohe Überspannung des Sauerstoffs ermöglicht wird. Auch hier ist die Entfernung des HNO2 durch Zusatz von Harnstoff wichtig, da sich sonst PbO2 wieder auflöst.
Für die Praxis ist wichtig, dass sich an der Kathode trotz allem geringe Mengen Pb abscheiden können. Dies wird durch Zugabe von etwas Cu2+ verhindert, dann scheidet sich nämlich Cu ab (Eo= 0,35 V).
Anode = Pt-Schale
Kathode = rotierende Pt-Scheibe
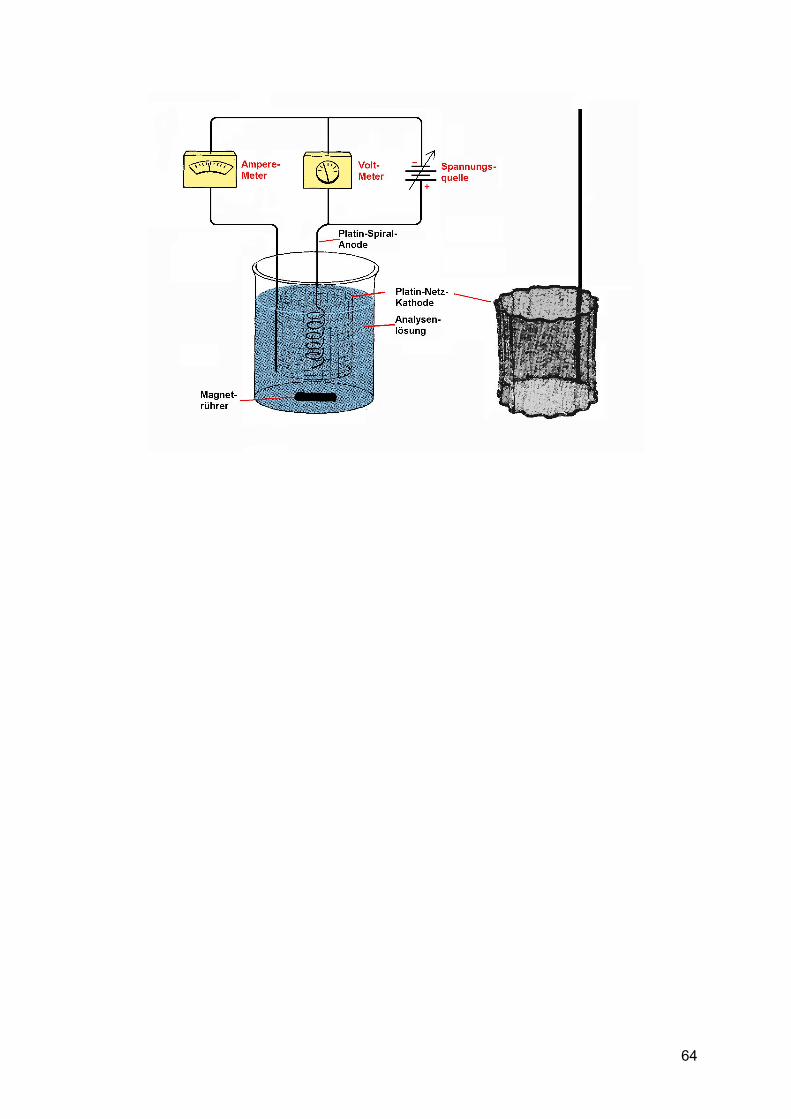
64

65
Durchführung von Elektrolysen Das Metall soll sich als glatter, fest haftender Nd. auf der Elektrode abscheiden und zwar quantitativ.
1) Standardelektrolyse
“”
Wird ohne Rühren durchgeführt, eventuell unter gelindem Erwärmen (wodurch eine gewisse Zirkulation bewirkt wird). Man arbeitet bei kleinen Stromdichten aus folgenden Gründen:
a) damit kein zu großes Ungleichgewicht zwischen (schneller) Entladung und (langsamer) Nachdiffusion von Ionen aus der Lösung zum Elektrodenoberfläche entsteht, die Verarmung an Ionen in der Diffusionsschicht in Elektrodennähe also nicht zu groß wird.
b) Damit sich nicht H2 abscheidet (als Folge von der unter a genannten neg. Erscheinungen)
c) Damit der Niederschlag nicht schwammig wird. Schwammige Niederschläge entstehen gerade dann, wenn die H2-Entwicklung zu stark wird.
Die geringe Stromdichte hat zur Folge, dass die Elektrolyse viel Zeit beansprucht. Deshalb lässt man die Standardelektrolyse häufig über Nacht laufen. Prüfung auf Vollständigkeit der Elektrolyse geschieht durch Senken der vorher nicht ganz eintauchenden Elektrode und Kontrolle, ob sich an der blanken Oberfläche noch etwas abscheidet.
Das Herausziehen der Elektroden muss unter Spannung geschehen, damit das Abgeschiedene Metall sich im sauren Medium nicht wieder auflöst. Die Elektrode wird abgespritzt, getrocknet und ausgewogen. Nachteil: sehr langsam!
2) Schnellelektrolyse Um die Elektrolysedauer auf ca. ½ h abzukürzen, muss man bei Stromdichten um ca. 5 – 10 A/dm² arbeiten. Um der starken Verarmung der Ionenkonzentration an der Abscheide-Elektrode entgegenzuwirken, muss kräftig gerührt werden. Elektromagnetische Rührer oder die Spiral-Anode wird direkt an Rührmotor angeschlossen. Rühen allein genügt noch nicht, daher arbeitet man zusätzlich bei 70-80°C , um Diffusion zu erhöhen.
Die Standardelektrolyse wird praktisch immer in H2SO4-Lösung durchgeführt. Da Metall und Legierungen meist in HNO3 aufgelöst werden bedeutet dies: vorher mit H2SO4 abrauchen (Zeitaufwand!)
Bei der Cu-Bestimmung verbietet sich HCl als Lösungsmittel: Es wird CuCl anstelle von Cu abgeschieden, überdies greift anodisch gebildetes Cl2 die Pt-Anode an.
Die Schnellelektrolyse kann auch aus HNO3 erfolgen, wenn Harnstoff zum Abfangen der HNO2 zugesetzt wird. Gerade hier ist gutes Rühren erforderlich, denn in stark NO3-haltigen Lösungen kann Reduktion von NO3
- bis zu NH3 erfolgen, wodurch an der Kathode Metallhydroxide ausgefällt werden können.
3) Innere Elektrolyse Bei der Besprechung des Daniel-Elementes hatten wir gesehen, dass bei Herstellung einer leitenden Verbindung zwischen Zn- und Cu-Elektrode folgender Vorgang abläuft:
Cu2+ + Zn Cu + Zn2+
Dieser Vorgang findet freiwillig auch dann statt, wenn wir einen Zinkstab in eine Cu2+-Lösung eintauchen. Der Zn-Stab überzieht sich mit einer Cu-Schicht, Zn2+geht in Lösung, es besteht eine direkte leitende Verbindung zwischen beiden Metallen: Innere Elektrolyse.
Allgemein besteht das Prinzip der inneren Elektrolyse darin, dass das Ion eines edleren Metalls durch ein unedleres Metall ausgefällt wird. Derartige Prozesse erfordern keine

66
äußere Energiezufuhr. Die praktische Durchführung in quant. Analytischer Hinsicht sei am Beispiel der Cu-Bestimmung durch innere Elektrolyse veranschaulicht:
“”
Zn
Cu2+
Diaphragma (Tonzelle)
Pt-Netzelektrode
Cu
Zn2+
Zn
Zn-Stab, Diaphragma und Pt-Netzelektrode sind in der Praxis konzentrisch angeordnet. An der Anode (Zn-Stab) geht Zink als Zn2+ in Lösung, die verbleibenden Elektronen fließen über den Schließungsdraht zur Pt-Elektrode, die dadurch zur Kathode wird, an der sich Cu abscheidet. Für diese Art der Elektrolyse benötigen wir keine äußere Spannungsquelle. Das Verfahren hat einige weitere Vorteile:
Bei der äußeren (üblichen) Elektrolyse ist die Cu-Bestimmung in Gegenwart von Fe nicht möglich, enthält die Lsg. Fe3+, so spielt sich an der Kathode der Vorgang ab: Fe3+ + e- Fe2+ Eo= 0,77 V
Und nicht Cu2+ + 2e- Cu Eo = 0,35 V
Das entstandene Fe2+ diffundiert zur Anode und wird hier wieder zu Fe3+ oxidiert (Depolarisation) es erfolgt also lediglich dauernde Umladung von Fe und keine Cu-Abscheidung. Bei der inneren Elektrolyse wird zwar auch Fe2+ kathodisch gebildet, das Diaphragma verhindert jedoch die Diffusion zur Anode. Wenn nur noch Fe2+ vorliegt, kann die Cu-Abscheidung beginnen.
Chlorid stört allerdings auch hier, da die Bildung von CuCl ein kathodischer Vorgang ist. Andere Metallbestimmungen können jedoch in Chloridhaltiger Lösung durchgeführt werden, denn durch das Diaphragma kommt es nicht zur Depolarisation (d.h. anodisch entwickeltes Chlorgas kann nicht kathodisch zu Chlorid reduziert werden).
Als Anoden können auch die Metalle Mg, Zn, Cd, Pb u.a. verwendet werden. Durch richtige Wahl des sich lösenden Anodenmaterials und der Konzentration der Gegenionen im Anodenraum ist es möglich, die Spannung zwischen Anode und Kathode auf ganz bestimmte Werte einzustellen (wichtig für Trennung).Innere Elektrolyse ist u.a. wichtig für selektive Abscheidung kleiner Metallmengen, u.a. für folgende Probleme:
a) Trennung geringer Mengen Bi, Cu, Ag von großen Pb-Mengen
b) Bestimmung von Cd und Cu in Zn
c) Bestimmung von Sn in Al
d) Trennung von Pb und Sb.



![Index [application.wiley-vch.de] · 312 Index – Zink-Elektrode 163, 171 –zweiterArt167 Elektrodenpolarisation 232 Elektrodenpotenzial 162 Elektrogravimetrie 211, 232, 235 Elektrogravimetrische](https://img.pdfslide.org/doc/110x75/5d56686e88c993e0768ba78b/index-312-index-zink-elektrode-163-171-zweiterart167-elektrodenpolarisation.jpg)

























