Embed Size (px)
Citation preview

1964 103
Aus der Universit/its-Kinderklinik Heidelberg (Direktor: Prof. Dr. Pm ~BANBERGEI~)
Angeborene Anomalien des Eiweiflstoffwechsels und deren analytische Probleme*
Von KUItT S CHREIER,
Mit 3 Textabbildungen
(Eingegangen am 14. September 1963)
Fiir einen Mediziner ist es eine Ehrenpflicht, zu Beginn seiner Betraehtungen tiber angeborene Stoffweehselanomalien in dankbarer Verehrung jener grogen Bio- ehemiker und Physiologen zu gedertken, deren bahnbreehende Leistungen nieht nur die Grundsteine zu dem Gebiude unserer Kenntnisse veto Metabolismus legten, sondern welehe geniale Helfer waren fiir die Formung eines vOllig neuen Lebens- und damit Weltbildes.
Mit der fiir das Jahr 1780 mutigen Feststellung yon LAVOISIER, dag ,,Leben eine ehemisehe Funktion" sei, beginnt die Biochemie aus dem HMbdunkel des l~{ystizismus in die niiehterne Sphere der exakten Wissensehaft zu treten. Eirt halbes Menschenalter spater erkannte FRiNcOIS MAGENDIE MS erster, dab Tier und l~'Ienseh ohne Eiweil3 nieht leben k6rmen. Wiederum 20 Jahre danaeh priigte der Hollgnder iKULDE~ das Epitheton ,,Protein" fiir jene ubiqnitgre Substanz im leben- den Kosmos. BE~ZELIVS zog erstmalig eine Parallele zwisehen KontaktkatMyse und Enzymwirkung.
Steigen wir die Stufenleiter der Erkenntnisgewinnung weiter hinaut; so leuehtet uns vor allem der Name yon J. vo• LIEBIG entgegen, weleher es vermoehte, in Deutschland einen Begeisterungssturm ftir die Biochemie zu entfaehen, wie er seitber nie mehr erweekt werden konnte. PFLffGEt~ formulierte: ,,Nur das Eiweig ist lebendig und vollzieht die eigentliehe Arbeit des Lebens." Sir ARCHIBALD GAB- ROD hat es im Jahre 1902 als erster gewagt, die Mendelsehen Gesetze aueh auf den lgensehen anzuwenden. Er pr~gte den Begriff der ,,inborn errors of meta- boIism". Aus der groBen Zahl bedeute~der Forseher seien nur noeh die Leistungen yon VoI% I%UBNE~, HO~'KINS und E~IL FISCHEg besonders hervorgehoben.
Seit etwa dem Jahre 1950 hat die gesamte Bioehemie und besonders die EiweilL ehemie einen grandiosen HShenflug begonnen, dessen vorlgufige Kulminations- punkte die Aufdeekung des Synthesemeehanismus der Proteine und die Auf- flndung des genetisehen Codes bilden. Damit fielen die ersten termini teehniei, welehe uns mitten hineinfiihren in die zu umreigenden Problemenkreise.
Als Diskussionsgrundlage dar f ieh kurz Vorgang und Steuerungs-
meehan i smen der Pro te insynthese er6rtern.
Das Wunderba re in der N a t u r ist wohl weniger die Vielfalt der Lebens-
iul3erungen und Ersehe inungsformen als v ie lmehr das VermSgen, jede
Ar t yon Lebewesen so kons t an t zu erhal ten, dag nu t eine re la t iv geringe Individuali t /~t des Erseheinungsbi ldes result ier t .
* Vorgetragen bei der Tagung der Faehgruppe ,,Analytisehe Chemie" in tIeidelberg im September t963.

104 KVRT SCIIREIER Bd. 20i
Dem Bemfihen zahlreicher Forscherkreise scheint es in den letzten Jahren gelungen zu sein, das Wesentliche dieser Meehanismen anfzu- hellen. Nach den derzeitigen Vorstellungen werden bei der Zellteilung Zahl und Sequenz der Purin- und Pyrimidinbasen im genetisehen Material, welches als Desoxyribonueleins/~ure (DNS) in Form der Gene auf den Chromosomen verteilt ist, im Rahmen einer identisehen oder besser ,,semikonservierenden" Reduplikation konstant erhalten. Die Geno wiederum garantieren die GleiehfSrmigkeit aller Proteine, Struktur- lipoide und Polysaccharide des gesamten Organismus, wobei die Eiweil~- k6rper nicht nur in ihrem Aminos/~uregehalt (AS), sondern auch in den spezifischen Eigenschaften und funktionellen Aufgaben festgelegt werden.
Wir glauben zu wissen, dab zwischen den Purin- und Pyrimidinbasen einer DNS und den einzelnen AS eines Proteins eine Beziehung besteht, wie etwa zwischen den Morsezeichen und dem Klartext. Vor fast 2 Jahren ist es NIR~NB~G u. MATTHA~I sowie OCHOA geg]fickt, durch Zugabe bestimmter Polynuc]eotide zu Bakterienkulturen Teile des Nuclein- s/~urecodes zu entziffern. Sehr wahrscheinlieh wird die Position jeder AS in der Polypeptidkette mittels sines nichtfiberlappenden Basentripletts (z.B. 3 • ffir Phenylalanin) festgelegt. Nach Uberlegungen und vorsichtigen Berechnungen vor ahem von BE~z~R, stehen dem Menschen bei Beginn seiner somatischen Entwicklung in der Keimzelle etwa 2 . 10 s Dreiercodes zur Verfiigung, oder anders ausgedrfiekt, etwa 80 Millionen Informationen je Chromosom.
Fehler im Code-System ffihren nat/irlieh zu einem unphysiologiseh zusammengesetzten Protein.
Die Purin- und Pyrimidinbasen-Einheiten vermitteln naturgem&B nicht nut Informationen fiber Art und Form neu zu synthetisierender Proteine und anderer Ze]lelemente, sondern in Form yon sogenannten Operatorgenen (MoxoD u. Mitarb.) steuern sie aueh das Zusammen- wirken der Strukturgene und lenken die Funktion der bereits in den ersten Synthesesehritten wirksamen Enzyme.
Ffir das Verstgndnis der Genese angeborener Stoffweehselanomalien ist es wohl erforderlich, sich den Syntheseweg eines Proteinmo]ekfils und damit auch den eines Enzympartikels mSglichst plastiseh vorzustellen, wei] mit Sieherheit anzunehmen ist, dal~ im I~ahmen eines erbliehen Defekts ]ede einzelne Stu]e gestSrt sein kann. Soll ein Protein neu ge- bildet werden, bedarf es folgender Voraussetzung:
a) Bauplan, b) alle erforderlichen Bausteine, c) Energiequelle, d) Mechanismen ffir die Verbindung der Bausteine entspreehend dem
Bauplan. Uber den Bauplan wurde bereits einiges gesagt.

1964 Anomalien des Eiweil3stoffwechsels 105
Zu Punk t b ist festzuhalten, dab unter physiologisehen Bedingnngen Eiweig lediglieh intracellulgr synthetisiert wird. Damit dies geschehen kann, miissen AS u. g. Bausteine zum Teil dureh die Zellmembranen transport ier t werden. Der genaue Mechanismus dieses Transportvor- ganges ist noch nieht bekannt. Mit einem hohen Grad yon Wahrschein- liehkeit handelt es sich dabei wohl um sogenannte Carrier-Meehanismen, bei denen eine AS oder ein Zucker locker an einen spezifisehen Tr/~ger gebunden wird, welcher die jeweilige Snbstanz, primitiv versinnbildlieht, wie ein aehsenmobiles Sehaufelrad in das Zellinnere befSrdert. Der Carrier muB (wenn die Vorstellung zutrifft) groB genug sein, um mit der AS o. ~. einen stereospezifischen Komplex bilden zu kSnnen und anderer- seits klein genug sein, um in der Zelle eine gewisse Wanderungsgesehwin- digkeit zu besitzen. Natiirlieh ist Art und Wirkungsgrad des Trgger- meehanismus genetisch fixiert und es gibt zweifelsohne Stoffwechsel- anomalien (z. B. aus der Gruppe der Nieren-Tubulopathien, das Har tnup- Syndrom; vielleieht aueh das Glucose-Galaktose-Malabsorptions-Syn- drom), welehe auf einer St6rung des Transportvorganges in best immte K6rperzellen beruhen.
Es sei hervorgehoben, dal3 bei den Vorg/Lngen an Membranen die ,,osmotisehe Barriere" eine wesentliche Rolle spielt und dab besondere , ,Permeasen" die extra- und intraeellulgren Grenzflgchen far verschiedene Stoffe durehggngiger machen. Dabei muB vor allem die Vorstellung ausger~Lumt werden, dab die Sgugetierzelle einem wassergefiillten S/Lck- ehen gleiche. Vielmehr wird das Cytoplasma dureh eine kaum vorstellbar vielf6rmig gefaRete Membran des sogenannten ,,Terminalretieulums" in zahllose Kam m ern (, ,Compartments") geteilt, in denen die metaboli- schen Prozesse in sehr untersehiedlieher Aktivit~t ablaufen. Die Trfim- mer dieses Terminalretieulums werden yon den Bioehemikern als Mikrosomen, welehe jetzt besser als Ribosomen bezeiehnet werden, isoliert und studiert.
Zum Punk t c. Ob es aueh angeborene Anomalien gibt, welehe anf einer t0ehlfunktion der energieproduzierenden bzw. transportierenden Cyelen in der Zelle beruhen, wissen wir noeh nicht. Sehr wahrscheinlich sind ernstere StSrungen nicht mit dem Leben vereinbar. Wir kennen allerdings ein Leiden - - die sogenannte Progerie - - zu deutsch das vor- zeitige Altern - - bei dem die Anomalietr/~ger bereits wghrend oder nach der Pubert~t vergreisen, was vielleieht auf einer Fehlsteuerung des gesamten Turnover beruht. Ob ein eehter ,,inborn error" vorliegt, bleibt noeh klarzustellen.
Zu Punk t d. Es wtirde zu weir fiihren, wenn ieh versuehen wollte, die Prozesse, welche in der Zelle znr Verknfipfung der AS zum spezifi- schen Polypeptid entwickelt sind, eingehender zu bespreehen. Gltiek- lieherweise haben in der Angewandten Chemie die wirklichen Exper ten

106 KIJt~T SCI-IREIER Bd. 201
zu diesen nicht einfachen Problemen Stellung genommen (WATsOr CRICK). Ira Stenogrammstil darf ich deshalb nur feststellen: Vom Struk- turgen wird die sogenannte Messenger-Ribonucleinsgure (RNS) als Bore mit der Information tiber das zu synthetisierende Protein ausgesandt. Die for die Peptidbildnng erforderliehen einzelnen AS werden durch Verbindung mit spezifischen relativ niedermolekularen t~NS-Molektilen ,,aktivier~". An der Ribosomenmatrize, welehe ebenfalls aus RNS be- steht, erfolgt die Verkniipfung der Peptidkette. In Abb. 1 habe ieh ver-
sucht, alle Vorggnge bild- Ze//membsa, lieh darzustellen*.
/ A o Nattir]ieh gibt es noch �9 vide Ltieken in diesem ex-
perimentell gut gesttitzten AY Vorstellungskomplex. Es
bleibt vor allem noch die g" &ez/f/'sche~ A,Z-akt/y/ependes Formung der Proteinmole-
~-nzrm ktile ans der soznsagen Ak//~'/ep/e ,45' eindimensional-orientierten
/
d~. L Ypez/f/sc/)e n/edepmolekt/Izpe Peptidkette, welehe yon der 2/72 (s.Z/Y2) Doppelspirale der riboso-
r AS malen RNS abgel6st wird, zum fertigen EiweiBmole-
Zp~,zsfepsysfem kill zu klgren. Da dieser
Vorgang offenbar nieht ge- ?o/en-~ netiseh fixiert ist, sondern
Z/~osom ~ yon intramolekulgren Rest- valenzen sowie Wasserstoff-
172. A/;/d'sua~'sme~aa/~mus und SS-Brtieken best immt wird, ist er hier nur yon
e/dmale/<o"/ geringem Interesse.
Das Auftreten abnor- Abb. 1. Vorggnge bei der Neubildung eines Proteins mal zusammengesetzter
Proteine mug nicht un- bedingt auf einem Fehler in der Matrize beruhen. Die s-RNS hat offenbar eine doppelte Ro]le, ngmlieh die Realisierung des eigentlichen Codifizierungsmeehanismus and die Erkennung des spezifisehen Akti- vierungsenzyms. Man daf t wohl annehmen, dab Code und Erkennung des Aktivierungsfermentes an zwei versehiedenen Stellen der Doppel- helix dieser s-RNS lokalisiert sind. So ergeben sieh zwei weitere M6gliehkeiten ftir die Entstehung yon angeborenen Stoffweehselano- malien:
* Unter Mitwirkung yon Herrn Dr. E. ZOLLEtL

1964 Anom~lien des Eiweigstoffweehsels 107
a) durch die irrttimliehe Einfiihrung yon AS in die Codifizierungs- region (so dal~ ein Aminoaeyl mit ,,falseher" AS entsteht),
b) durch cinen I r r tum bei der Erkennung des ,,riehtigen" Aktivie- rungsenzyms.
F/Jr das Uberleben mancher Mutationen ist es siehcr yon wesentlieher Bedeutung, dab wohl die meisten Proteine eine Art Wirkungskern besitzen, d .h . man kann unter Umst~nden ganze Polypeptidketten abspalten, ohne dab gr613ere FunktionseinbuBen auftreten miissen.
Die Kontrolle der Neubildung und der Aktivit/~t der Enzyme in einer Zelle geschieht naturgemgB nieht nur dutch Gene bzw. deren DNS, ~sondern aueh dureh spezifisehe Induktions- und Depressionsvorggnge. Jugendliehe und besonders embryonale Zellen bcsitzen eine augerordent- liehe F&higkeit, neue Fermente (sogenannte Adaptivfermente) zu synthe- tisieren, wenn ein bestimmtes Substrat angeboten wird. ~Jber diesen Vorgang wird derzeit nieht nur in der entwieklungsphysiologisehen Lite- ra tur viel diskutiert. Faszinierend und sehr einleuehtend seheint mir die Konzeption yon MoNoD, PARD~S U. a., dab dcr ,,inducer" keine neue Synthese katalysiert, sondern die Blockade eines an sieh bereits vor- handenen l~eaktionsablaufs aufhebt. Die Autoren nennen dies eine t~epression einer Depression.
Aus dem Gesagten ergibt sieh, dab die vielfaeh vorgetragenc Konzep- tion, alle angeborenen StoffweehselanomMien liegen sieh auf dem Boden des Ein-Gen-ein-Enzym-Mangels erkl/~ren, nicht ganz riehtig ist. Bei den meisten Anomalien steht his heute ja nur fest, dab irgendwie eine enzymatiseh katalysierte I{eaktion bloekiert ist. Zur leiehteren IJbersicht soll im folgenden die jetzt noeh/ibliche Einteilung in
A. St6rungen der molekularen Funktion (Enzymdefekt),
B. St6rnngen dcr molekularen Struktur ( = fehlgebildete Proteine),
C. St6rungen der molekularen Synthese ( = fehlende Proteine) bei- behalten werden.
Es ist keineswegs beabsiehtigt und zeitlieh aueh gar nieht mSglieh, such nur andeutungsweise zu jeder der etwa 100 bis jetzt beschriebenen StoffweehselanomMien etwas zu sagen. An einigen typisehen Beispielen will ieh versuehen, grunds&tzliehe geaktionsweisen und Seh&digungs- mechanismen hcrauszuarbeiten. Ieh darf vielleieht noch voraussehieken, dab im Grunde so gut wie alle bekannten St6rungen Anomalien im EiweiBstoffwechsel sind, denn eine Lipoidspeieherung oder eine Ver- /~nderung im Glykogenmolek/il hat ja wohl stets eine St6rnng in den zu- gehSrigen Enzymen zur Voraussetzung.
Die Abartigkeiten des Phenylalaninstoffweehsels sollen zuerst be- sproehen werden.

108 KUI~T SCH~EIER: AnomMien des Eiweigstoffwechsels Bd. 201
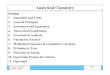
In Formelbild 1 ist der normale Abbauweg dieser AS mit den bekann- ten Bloekadestellen zusammengestellt. Die resultierenden Anomalien sind aueh dem Nieht-Mediziner gr6Btenteils bekannt.
Es handelt sieh:
a) Um den Albinismus, weleher bei Menseh und Tier in zwei Formen, sis
1. totaler Pigmentmangel und 2. als partieller Defekt (Weigseheekigkeit) vorkommt.
Vor allem dureh L E R ~ und seinen Arbeitskreis wurde naehgewiesen, dab bei den Anomalientr~gern in den Melanoeyten das Ferment Tyro- sinase fehlt.
Ieh bin sieher, dab Ihnen allen aueh
b) die Alkaptonurie gel~ufig ist -- jenes Leiden, welches mit einer Dunkelfgrbung des Urins einhergeht. Vor einigen Jahren ist es LADy gelungen, die alte Vermutung zu verifizieren, dab bei dieser reeessiv vererbten St6rung die ttomogentisins/tureoxydase in der Leber fehlt. Im Alter kann es zu einer Ablagerung eines Polymerisationsproduktes dieser S~ure im Knorpel, den Sehnen, Knoehen usw. kommen. Es resultiert die ,,ochronotisehe Osteopathie". Das Leiden verkiirzt das Leben nieht wesentlieh. Demnaeh ist eine Therapie nieht dringend er- forderlieh. Die Itomogentisinase ist strukturgebunden.
e) Die n/tehste Anomalie, welche auf dem Fehlen der Phenylalanin- oxydase beruht and deshalb mit l~eeht den Namen Phenylketonurie (F611ingsehe Krankheit) trs geh6rt in die gr6gere Gruppe der St6- rungen, welehe zu einer geistigen Retardation ffihren. Die friihest m6g- liehe Diagnose ist yon aussehlaggebender Bedeutung fiir das Sehicksal der Anomalientr/~ger. Nur wenn sie vor dem 4. Lebensmonat gelingt, kann eine normale geistige Entwieklung garantiert werden. Dutch das Fehlen des oxydierenden Fermentes h~uft sieh Phenylalanin in den K6rpersgften an nnd erreicht dort Werte, welehe das 20fache der Norm iiberschreiten k6nnen. Dureh Desaminasen und versehiedene Oxydasen reiehern sieh aueh Phenylbrenztraubens/~ure and versehiedene Ortho- oxyverbindungen usw. an. Daraus resultieren StSrungen des Stoff- weehsels (siehe nebenstehende Seite).
1. Kommt es zu kompetitiven Hemmungen des Membrantransports anderer AS. Dies macht sieh besonders in der Phase der st/~rksten Ge- hirnentwieklung (etwa ab dem 4.--12. Lebensmonat) negativ bemerkbar.
2. Ein oder das andere Abbauprodukt bzw. Phenylalanin selbst sind offenbar im eehten Sinne toxiseh; und zwar wieder besonders auf die Hirnzelle.
3. Seheint es zu Vergndernngen im 5-tIydroxytryptamin (Serotonin)- Stoffweehsel der Nervenzellen zu kommen.
Phenylalanin A
, / \ - - t ~ CN 2 I
CKNK 2 I COOK *$+0
Phenyl- brenz-
trauben- g~l l re
/Q
CK2 I
CO [ COOK r
Phenyl- essigsaure
I CK~ I COOK
* Trunsaminierung
Tyrosin OK / \
I ' \ / I CH~ !
C I t N H 2 I
COOK
* t + O
p-Kydroxyphenyl- brenztraubens~ure
OI{ I H. \ p-Kydroxy-
~ " - - ~ phenyl- milchs~ure
! [
CK~ I
CO I
COOK
.C
r 2,5-Dihydro- xyphenyl-
brenztrauben- S~tll'6
,,/~O K
I CK2 I
CO I
COOK Komogentisinsgure
/ZoK ! CKo
Cook ~ D
F u m a r y l a e e t e s s i g s ~ u r e l t0OC--CK CH--CO--CK2--CO CK2--COOK
F u m a r s ~ i u r e KOOC--CK= CIt--C00K
+
AeetessigsSoure CI~2CO--CK~--COOH
Tyralnin
/ ~ -~ AdrenMin \ /
[ CK2 I
CI-I~NK2 Dijodf, yrosin
OK J ~ J -~ Thyroxin
! CI-[s
I CtINH, I
COOK 3,4-Dioxy-
phenylalanin OK i Tyrosinase
I{O --> Me]anin
1 CK 2 E
I CKNH2
I COOK
A Oligophreni~ phenylpyruviea
B Tyrosinurie ** C Tyrosinose D Alkaptonbrie E Albinismus ** bei schwerem Lebersehaden
Formclbild J_ Atl$ : g . SCHKEIER, Die angebore- hen Btoffwechselanomalicn. Georg
Thieme Verl~g, S tu t tgar t 1963

110 KV~T SCmCEIER: Anomalien des EiweiBstoffwechsels Bd. 201
Klinisch resultiert jedenfalls ein gutmiitiger Vollidiot, welcher einen ziemlich penetranten Geruch nach Wolf- oder M~useurin ausstrSmt (hervorgerufen wohl durch Pheny]essigsi~ure). Ffir die Bundesrepublik ist mit einer js Geburt yon etwa 500 Phenylketonurikern zu rechnen.
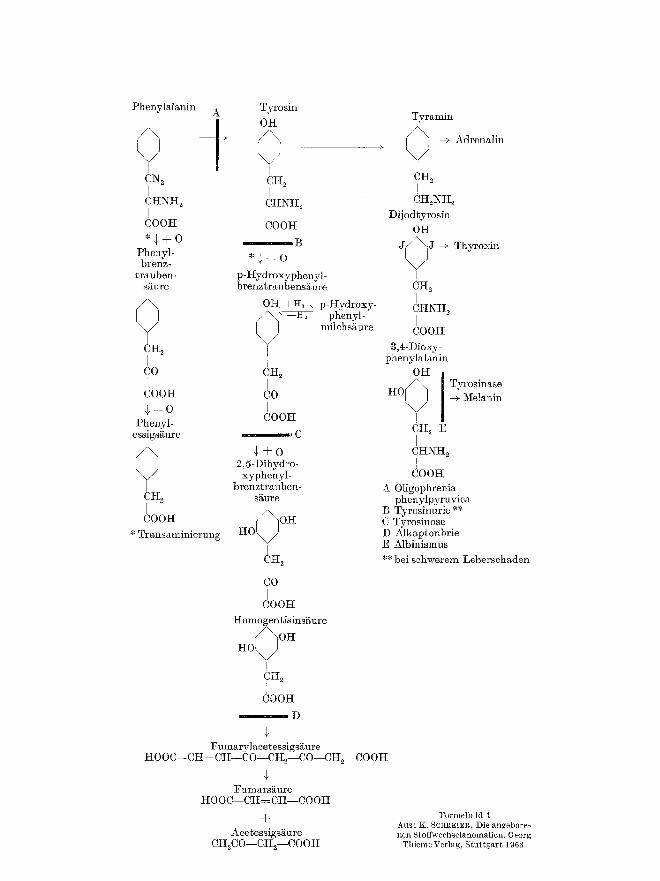
In allen Beziehungen ~hnlich wirkt sich der Histidinase-Mangel aus. Die homozygoten Anomalietri~ger sind allerdings nicht so stark ge- schi~digt wie die Phenylketonuriker.
In Formelbild 2 (siehe nebenstehende Seite) sind die bekannten Intermedi~rprodukte des }tistidinstoffwechsels (nach AUER]~ACK u. Mit- arb.) zusammengestellt.
Beide StSrungen sind auBerordentlich leicht durch einige dem Urin zugesetzten Tropfen einer 10~ EisenchloridlSsung zu diagnosti- zieren. Phenylbrenztraubensi~ure liefert einen tiefblau-grfinen Farbstoff; Imidazolbrcnztraubensi~ure gibt einen mehr braun-gr/inen Farbton.
Abb. 2. t ) ap i e r ch roma tog ramm des Ur ins eines an Ahorn-S i rup-Xrankhe i t leidenden 72atienten
Die schwerste bis jetzt bekannte, d. h. unmittelbar lebensbedrohende Enzymopathie ist die Ahorn-Sirup-Krankhei t (maple-syrup-urine- disease), welche treffender a]s Verzweigtketten-Aminosguren-Decarb- oxylasemangel zu definierea ist. Wit hatten selbst Gelegcnheit, eine Familie -- dieses bis jetzt noch selten diagnostizierten Leidens -- zu finden.
Abb. 2 zeigt das Papierchromatogramm des Urins dieses Patienten, welcher trotz unserer Bemfihungen am 9. Tag ad exitum kam. Wie
I H
H
H
H
I
Prot
ein-
Synt
hese
. ~
I ]
[ [
KC
----
C--
C--
C--
CO
OK
H
C--
C
--C
=C
--C
OO
K
1 un
d 3
Met
hyl-
His
tidi
n -
[ I
- "
I I
1['[
NH
2 It
istid
asc
N
NH
N
N
H
Car
n~
und
An
seri
~m
y
~ C
" ~
C /
H
H
H
/ H
isti
din
(Im
idaz
olal
anin
) U
roca
nins
Eur
e ~l
mst
id:n
- (I
mid
azol
acry
lsii
ure)
H
C--
C--
C~
-C--
NK
2
/[Tra
nsam
i ....
.
N NK
I
~c/
H
C=.
. C--C
--C--C
OO
H
jo J
H
ista
rnin
N
N
H
k~
~c/
H
Im
idaz
olac
etal
dehy
d Im
idaz
olbr
enzt
raub
ens~
ure
I I
] H
C~
C--
C--
CO
OH
H
C
C--
C--
C--
CO
OH
H
H
O
N
NH
N
H
H
~c/
~
c/
H
It
Imid
azol
essi
gs~i
ure
Imid
azoh
nilc
hs~u
re
Imid
azol
essi
gs~i
ure-
Rib
osid
e
H
H
H
I I
I 0
=C
C
--C
--C
--C
OO
K
' -
I I
I H
~H
N
NH
\~
O
=C -
-C--
C--
C--
CO
O~
p r
o 2:
~ Im
idac
olon
prop
ion-
\
~C
/
I I
I I
b H
C~
C--
C--
C--
CO
OH
H
OO
C--
C--
C--
CO
OH
[
t r
J H
H
H
N
NH
N
H
O=
C--
--C
-- C
--C
--C
OO
H
II I
Imid
azol
prop
ions
/iur
e N
I~
CH
For
mim
inog
luta
min
saur
e O
Glu
t!m
lnsa
ure
For
myl
isog
luta
min
Der
Ver
tika
lstr
ich
tren
nt
die
Met
abol
ite,
wel
che
hist
idas
eabh
~ngi
g si
nd (
rech
ts),
von
den
un
abh~
ngig
en.
Form
clbi
ld 2
,&us
: K. S
OH
I~,E
IER,
Die
ange
Dor
enen
Stof
fwec
hsel
anom
alic
n. G
eorg
Thie
me V
erla
g, S
tuttg
art 1
1963

112 Ku~T SC~]~[E~ Bd. 201
sehon die Bezeiehnung verrgt, liegt die Blockade im Abbau yon Leucin, Isoleuein und Valin.
Der Abbauweg yon Leuein verl~tuft folgendermagen:
(Die Block~destelle liegt offensichtlich auf der Stufe, wo die e-Ketosiure ~uf das Coenzym A-Molekiil iibertragen wird):
CI{ CH C~ L r I CH~ CttNH 2 CI-INH 2 I I I CttNI-I~ COOK COOK [ Isoleucin Valin COOI-I
Leucin
CK~\/CK~ CK~\/CI-&
CI-I~ Byridoxal- CI-12 Thiaminpyro- CK I phospha~ I phosphat i CKNtt~ ~- c~-Ketoglutar- CO +Lipons~ure CK2 I si~ure o. 5. I I COOI{ COOH COOK
Leuein ~-Ketoisoc~prons~ure Isovalerians~ure
Coenzym A verdriingt die intermedigr entstehende ])ihydro- licIons~ure, wobei Isovalerianyl-CoA gebildet wird.
Formelbild 3 Aus: K. SCHI~I~IEIr Die angeborenen Stoffwechselanomalien. Georg Thieme Yerlag, Stuttgart 1963
Den eigenartigen Namen hat das Leiden yon dem wirklich auf- fallenden Gerueh des Urins, weleher die Nase des Europgers am ehesten an Suppenwiirze erinnert.
Bei allen drei StSrungen kommt es darauf an, die AS, deren Stoff- weehsel bloekiert ist, nur in jener Menge zuzufiihren, welehe ffir ein normales Waehstum ausreiehend ist. Ffir Phenylketonuriker gibt es bereits entspreehende Prgparate im Handel. Gesehieht dies mit der nStigen Sorgfalt und Konsequenz, erreiehen die Anomalietri~ger einen normalen Intelligenzquotienten. Bei der Ahorn-Sirup-Krankheit mug die Diagnose vor dem 6. Lebenstag gestellt werden, sonst sterben die Neugeborenen oder werden trotz digtetiseher Einstellung nut tief- stehende Idioten.
In gleiehsinniger Weise ist therapeutiseh mit den Anomalien des Kohlenhydratstoffweehsels zu verfahren, bei denen ein Ferment, wel- ches den Zucker in den norm~len Glykolyseeyelus einsehleust, fehlt. Dies gilt z.B. fiir :

i964 Anomalien des Eiweif3s~offwechsels 113
1. Die Galaktoseintoleranz (es fehlt das Ferment Galaktose-1- Phosphat-Uridy]transferase),
2. Die Fructoseintoleranz (bei welcher die Phospho-Fructaldolase fehlt).
Beide StSrungen gehen mit sehweren tIypoglyk&mien (starke Er- niedrigung des Blutzuckers) einher und sind ]ebensbedrohend, wenn die jeweiligen Zueker nicht aus der Nahrung weggelassen werden.
]3el einer weiteren Anomalie ist eine geistige Retardation mit dcr vermehrten Ausscheidung yon Cystathionin vergesellschaftet. Die Ursache ist wohl in einem Defekt des Fermentsystems zu suchen, welches diese AS in Cystein und Itomoserin spaltet :
Sto#wechselweg des Methionins
Methionin
~--CH 3 Homocystein und Serin
I ~H~O
Cystathionin
~ - - wahrscheinliche Blockadeste]le
Homoserin und Cys~ein
Formelbild 4
Beim Arginin-Bernsteins~ure-Schwachsinn fehlt zumindest im Gehirn die Argininsuccinase, so da[3 sich diese Verbindung im ZNS stark an- reichert. Bei der eng verwanclten Citru]]inurie Hegt der ]Defekt eine Stufe hSher :
Citru]]in -~ Asparagins~ure
~ ~ N H s ~ CO x Argininbernsteins~ure l
O![flhin -~ Itarnstoff-~----Argi!in -V B~~
Formelbild 5
Hier einzuordnen w/ire sehlieBlich aueh die relativ h/~ufige Wilson- sche Krankheit . Da das kupfertragende Plasmaprotein Caeruloplasmin, welches mit seinen vier Kupferatomen als Oxydase wirkt, in der Leber nicht gebildet wird, resultiert eine Ablagerung des Metal]s in der Leber und in manchen Kernen des Zentralnervensystems, was zu einer Gelb- sucht und schliel3lich Lebercirrhose sowie einer schweren St6rung der Motorik f/ihrt.
Z. analyt. Chem., Bd. 201 8

114 KURT SCtIREIER Bd. 201
Als AbschluB dieser Reihe m6ehte ich fiber eine eigene, noeh unver- 5ffentl iehte Neuen tdeekung ber ichten . Es ha nde l t sieh u m die Glyeinose :
Wahrseheinliche Blockade
/ / / b e i der Oxalose
Kreatin Formiat Oxalat
Guanidoessigs~ure
Serin ~, G1 in P Proteine
!on n ure/ Cyclus
Gicht Formelbild 6
Welches F e r m e n t des Glycinstoffwechsels bei dem Anomal ie t r / iger fehlt , ve rm6gen wir n ich t zu sagen, weft das K i n d bere i ts am 9. Tage un te r dem Bride einer sehweren Vergi f tung - - also ga nz / i hn l i e h wie die P a t i e n t i n mR der A h o r n - S i r u p - K r a n k h e i t - - ve rs ta rb . I m Blur und Ur in war der Glye ingeha l t au f das 20faehe und mehr erh6ht . Kol lege
Tabello 1. Enzymmangelzust~inde (StSrungen der molekularen Funktion)
Syndrom Fehlendes Enzym
i. Albinismus Tyrosinase 2. Alkap~onurie Homogentisins~ureoxydase 3. Phenylketonurie Phenylalaninhydroxylase 4. Tyrosinose ? 5. Ahorn-Sirup-Krankheit Aminos/iuredecarboxylase ? 6. Akatalasie Katalase 7. Argininosuccinurio ? 8. Cystinose ? 9. Cystathinionurie ?
10. Citrullinurie ? 11. Glycinose ? 12. Har~nup-Syndrom ? 13. Hyperhistidin~mie ttistidase 14. Imidazol-Aminoazidurie ? 15. I-Iomocys~inurie ? 16. Hypophosphatasie Alkalische Phosphatase

1964 Anomalien des Eiweigstoffwechsels 115
Tabelle 2
Typ :Bezeichnung Eigenname kannte Enzymdefekl~ Fallzahl
1 v . GI~a~:~ 100 2 Po~eE 25 3 F o R ~ s 1~ 4 AND:EI~SEN
5 Muskelphosphorylasetyl o McARDLE 3 6 Leberphosphorylasetyp HEms 12
fl~lls: K. SCttREIER, Die angeborenen Stoffwechselanomalien. Georg Thieme Verlag, S tu t tgar t 1963
Hepatorenale Glykogenose Generalisierte Glykogenose Grenz-Dextrinose Amyloloektinose
Glucose-6-phosphatase ? Amylo-l,6-glucosidase Amylo-(1,4-- 1,6)-
transglykosidase Muskelphosphorylase Leberphosphorylase
Tabelle 3. Angeborene Stdrungen der tubuliiren Funktionen
I. Wasserriickresorption: JRcnalerDiabetes insipidus
II . Glucose: Renalc Glucosurie
III. Aminosauren: a) alycinurie b) Glycinurie ~ Glucosurie c) Arginin-Cystin-Lysinurie d) .Renale generalisierte Hyperaminoacidurie e) a~enale Hyperaminoacidurie mit Glucosurie
IV. Phosphat: Phosphatdiabetes (idiopathische Vitamin D-resistente Rachitis)
V. Sauren-Basen-tIomoiostase : Hyperchloriimische Acidose
VI. Komplexe St6rungen :
A. Prim/i, re Anomalien 1. De Toni Fanconi-Syndrom 2. Lowe-Syndrom 3. Familiiire renale Hyperprolinurie
]3. Sekund/~re Formen 1. Cystinose 2. Heioatorenale Glylcogenose 3. Galaktoseintoleranz 4. Fructoseintoleranz 5. Wilsonsche KranIcheit
DIEZEL vom Pa tho log i schen I n s t i t u t h a t schwerste Ver i inderungen i~t de r Zell- und Marks t ruk~ur des Gehirns gefunden.
I n Tab. 1 s ind die bis j e t z t b e k a n n t e n S t6rungen des AS-Stoff- weehsels zusammengestel l~. N ieh t n u t die excessive Vermehrung bzw.
8*

116 K v ~ SCgREIE~ Bd. 201
Verminderung yon Aminoss und Monosacchariden bzw. ihrer Metabolite, sondern aueh jene yon Porphyrinen und Metallen usw. alteriert das Zentralnervensystem.
Es ist vielleicht interessant zu erfahren, dab verschiedenartige Enzym- mangelzustgnde klinisch und biologisch das gMche Resultat haben k6nnen (z. B. die Speicherung yon Glykogen). Wie Tab. 2 zeigt, kennen wir nicht weniger als sechs Typen einer Glykogenose. Auch eine Meths
Abb. 3. Papierchromatogramm des Urins eines an Fanconi-Syndrom leidenden Patiengen
moglobingmie kann sowohl aus einem Fehler im Fermentsys tem des roten Btutk6rperchens als auch aus einer Fehlzusammensetzung des I-Igmoglobinmolekfils rcsultieren.
Einer gesonderten ganz kurzen Besprechung bedtirfen noch die an- geborenen St6rungen des Tubulusapparates der Niere. Wie aus Tab. 3 hervorgeht, kann im Rahmen einer angeborenen Stoffwechselanomalie entweder
a) die Wasserriickresorption, b) die Wiederaufnahme yon Glucose bzw. yon einzelnen oder zahl-
reichen AS gestSrt sein oder c) in Form einer komplexen Anomalie kann es zum Ausfall zahl-
reicher oder sogar fast aller Tubulusleistungen kommen. Aus der Fiille dcr Krankheitsbilder m6chte ich jenes Syndrom heraus-
greifen, welches eigentlich den Namen yon E~aIL ABD~HALD~ tragen

1964 Anomalien des Eiweiflstoffwechsels 117
miiBte, es wh'd gewShnlich als de Toni-Fanconi-Debr&sche Krankheit , im amerikanischen Schrffttum kurz als Faneoni-Syndrom, bezeichnet. Der Grund, warum ich sie gerade damit konfrontiere, ist folgender : Das Herz ]edes mit der AS-Chromatographie des Urins Vertrau~en schl~gt h6her, wenn er ein Chromatogramm sieht, wie es in Abb.3 dar- geste]l~ ist.
Ein sehr s Leiden geht zus~tzlich mit einer Speicherung yon Cystin in fast allen Organen einher. Diese sogenannte Cystinose f6hrt stets noch vor dem 10. Lebensjahr zum Tode, Im Examen und auch sp~ter verweehseln die Mediziner dieses Syndrom meist mit der Cystin- urie, deren Namen eigentlieh ,,renale Arginin-Cystin-Lysin-Ornithin- urie" ]auten mfiBte, denn von den Anoma]ietr~gern werden alle AS in mehr oder weniger grofier Menge ausgeschieden, bei denen der Abstand der Carboxylgruppe yon der endst~ndigen Aminogruppe etwa 7 ~ be- tr/~gt. Im Urin f~llt von den vier AS naturgem~B nur Cystin aus. Das Gen ffir diese Anomalie ist in Europa ungew6hn]ieh h~ufig. Mindestens ein Individuum unter 250 scheidet Cystin in abnorm hoher Menge aus. Nierensteine bilden sieh allerdings auBerordentlich viel seltener. Die Tatsache, dab sieh diese Aminos~ure in alkalisehem Milieu besser 16st, macht sich der Kliniker zunutze, indem er den Patienten mit Cystin- steinen in der Nacht noch einmal eine gr6Bere 1V[enge d/innen Tee trinken l~Bt und auBerdem groBe Mengen yon Natriumhydrogencarbonat ver- ordnet.
Bei einem anderen Nierensteinleiden, der Oxalose, fehlt ein Enzym des Glycinstoffwechsels, wahrscheinlich die Glyoxyls~turedecarboxylase, so dab zuviel Oxals~ture gebflde~ wird. Es treten nieht nur in der Niere Ablagernngen yon Caleiumoxalat auf.
Auch die Bildung der Enzyme des Magendarmtrakts kann als erb- licher Defekt ausbleiben. Bekannt und als Krankheitsbild besehrieben ist unter anderem das Fehlen der Laetase, Saeeharase und Isomaltase. Um das Bild abzurunden, sei betont, dab aueh zahlreiehe hormonelle St6rungen auf Enzymdefekten beruhen. Dies gilt ffir die Nebenniere, we fehlende Hydroxylasen, z.B. fiir C17 bzw. C m des Steroidringes zu dem Krankheitsbild des ,,adrenogenitalen Syndroms" ffihren (dabei handelt es sieh um genitale Pseudofriihreife mit oder ohne Salzverlnstsyndrom). Dies trifft aber aueh fiir die Sehilddrfise zu. Es konnten bereits 8 StS- rungen herausgefunden werden, bei denen der Kliniker die Diagnose einer Unterfunktion der Sehflddrfise stellt, die aber nieht auf einer Fehl- bfldung des Driisenepithels beruhen, sondern auf einem Mangel an Enzymen, welehe an der Einffihrung des Jods in die Wirkstoffe der Drfise beteiligt sind. Sehr wahrseheinlieh kann aueh die Syn~hese yon Proteohormonen der t typophyse ausbleiben. Aueh St6rungen im Purin- und Pyrimidinumsatz kommen vor, wie Tab. 4 zeigt.

118 KURT SCI{REIER Bd. 201
TabeHe 4
Syndrom Fehlendes Enzym
1. Xanthinurie Xanthinoxydase 2. T-AnomaHe ? 3. Gicht ? 4. Hyper-fi-Aminobutyricurie ? 5. Orotacidurie ?
Die Enzymgenetik 6finer uns auch ein Fenster zum Versts der individuell so verschiedenen Toleranz yon Heflmitteln, Giften und careinogenen Substanzen. Die Entgfftungsmeehanismen, ob sic dureh Glucuronidierung, Aeetylierung, dutch Koppelung mit Glyein, Sehwe- fels~ure oder dutch Hydroxylierungen erfolgen, stehen alle unter gene- tischer Kontrolle.
Bis jetzt sind unter anderen erbliche Unvertrs yon Primaquine sowie Suxamethonium besehrieben worden. Ferner ist auf- gefa]len, dab das soviel verwendete Isonicotinss yon etwa 500/0 der Mitteleurops raseh und yon der anderen Hs langsam inaktiviert wird.
Darfiber hinaus ls die Synthese immer neuer fakultativ mutations- auslSsender oder mutationsfSrdernder Medikamente und ihre Erprobung am Menschen der forschenden Medizin und der Chemic eine auBerordent- liche Verantwortung auf.
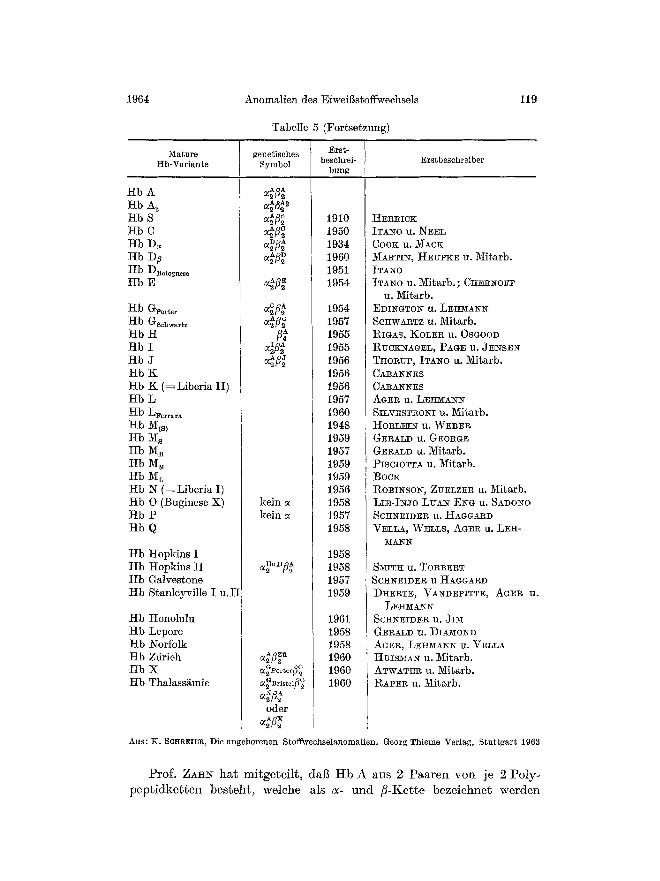
Musterbeispiele fiir angeborene St6rungen der mole]~ularen Strul~tur sind die Anomalien des menseh]iehen Blutfarbstoffs. Die Aufkls der Ursache f/Jr zahlreiche H~moglobinopathien ist eine Meisterleistung ehemischer Analy~ik. Das Globinmolek/il des menschlichen Blutfarb- stoffs hat vor der Geburt eine andere Struktur (Hb F = fetales Hb) als im spi~teren Leben (Hb A -~ adultes Hb). Neben dem normalen Er- waehsenen-Hb sind inzwisehen eine Vielzahl yon Varianten entdeckt worden. Die meisten sind in Tab. 5 aufgeffihrt.
Tabe~e 5
Fetale genetisches Erst- ~b-Variante Symbol beschrei- Erstbeschreiber
b u n g
Hb F lib Barfs Hb Fessas und
P~p~spyrou lib Alexandra
Hb Augusta I Hb Augusta II ttb Cyprus I
A F
7~ 1866 1958 1957
1959 1959 1960 1960 1959
E. v. K6~BER AGER U. LEH~ANN
FESSAS U. PAPASPYROU
FESSAS u. Mitarb. VELLA, AGE~ u. LEHI~ANN HUIS~AN I-IUISMAN GILLESPIE U. l~itarb.
1964 Anomalien des EiweiBs~offweehsels 119
Tabelle 5 (Fortsetzung)
Mature t tb-Variante
genetisches Symbol
Erst- besehrei-
bung
Hb A Hb Az Hb S Hb C l ib D~ Hb D# Hb DBolognese Hb E
Hb Gporter Hb Gs ,h ,~ Hb H Hb I Hb J Hb K Hb K (= Liberia II) Hb L Hb LF~r~ Hb M(s~ Hb M s Hb MB Hb MM Hb M~ Hb N (= Liberia I) Hb O (Buginese X) Hb P Hb Q
Hb Hopkins I Hb Hopkins n Hb Galvestone Hb Stanleyville I u. II
Hb Honolulu Hb Lepore Hb Norfolk Hb Ziirich Hb X Hb Thalassgmie
A A (X2~ 2
A S ~f12 1910 A C %fi2 1950 D A %fl~ 1934
e~fl~ 1960 1951
A E %fi~ 1954
G A %fl~ 1954 1957 ~r 1955
I A %f12 1955 A J %fl~ 1956
1956
kein kein
ttoIi A ~2 ~2
A Zfi ~2 ~2 ~gGPor t e r ~
~ B r l s t e l ~ X A
0~2/~ 2 oder A X
~2 ~2
Erstbeschreiber
I-IERRICK ITANO U. NEEL
COOK U. MACK MARTIN, HEUPKE U. M i t a r b . ITANO ITANO U. M i t a r b . ; C]~ERNOFF
u. Mitarb. EDINGTON U. LEIL~ANN
SCHWARTZ U. Mitarb. RIGAS, :[~OLER U. OSGOOD
RUCKNAGEL~ PAGE U. JENSEN
TEOR~P, ITANO U. Mitarb. CABANNES
CABANNES AGER U. LEIIMANN
SILVESTRONI U. Mitarb. HORLEIN U. WEBER
GERALD U. GEORGE
GERALD U. M i t a r b . PISCIOTTA u . M i t a r b .
BocK ROBINSON, ZUELZER U. Mitarb. LIE-INJ0 LUAN ENG u. SADONO SCHNEIDER U. HAGGARD
VELLA, WELLS, AGER U. LEg- I~IANN
SMITH u . TORBERT
NCHNEIDER U HAGGARD Dt tERTE, VANDEPITTE, AGER u .
LEHMANN SC]~NEIDER U. JIM GERALD U. DIAIVIOND
AGER, LEHIVlANN U. VELLA
HUIS~AN u. Mitarb. ATWATER U. Mitarb. RAPER U. Mitarb.
Aus: K. SCHREIER, Die angeborenen Stoffweehselanomalien. Georg Thieme Verlag, S tu t tgar t 1963
Prof. ZAHN hat mitgetei l t , dag H b A aus 2 Paaren yon je 2 Poly- pep t idke t t en besteht , welehe als a- u n d fi-Kette bezeiehnet werden

120 I~C1r SCHREIER Bd. 201
(Hb F besteht aus 2~- und 2~-Ketten). Obwohl das Molekfil aus 574 AS zusammengesetzt ist, konnte deren geihenfolge bei den normMen und einer ganzen Reihe pathologischer H/imoglobine bereits aufgekli~rt werden. Wie Tab. 6 demonstriert, sind bei den abnormalen Blutfarb- stoffen eine oder mehrere AS in einer Ket te dureh andere ersetzt. Da- durch/~ndert sich die elektrisehe Ladung des Molekiils und es resultiert eine erh6hte t~ragiliti~t der roten BlutkSrperehen, welehe bei manchen St6rungen wie der Mittelmeeran/imie das Leben der homozygoten Kranken stark verkfirzt. Die Zahl der Anomalietriiger mit abartig ge- stalteten Hgmoglobinen ist ungeheuer grog. Es gibt Landstriche, z .B. in Ghana, we 300/0 aller Menschen Sichelzellenhgmoglobin aufweisen und Ti ler in Nordburma, we Hb E/~hnlich frequent ist. Dies ist natfir- lich nicht nur ffir Erbbiologen, sondern genauso ffir V61kerkundler, tI istoriker usw. interessant.
Tabelle 6 Peptid T 4 Hb A val his leu thr pro glu glu lys Hb C val his leu thr pro lys glu lys Hb S val his leu thr pro val glu lys Hb GNeel val his leu thr pro glu gly lys Peptid 26 H b A val asp val asp glu val gly gly glu ala H b E vM asp val asp glu val gly gly lys ala
leu gly arg leu gly arg
Es gibt auch eine ganze Gruppe klinisch sehr eindrueksvoller Bei- spiele ffir das Fehlen einer Matrize zur Synthese eines wiehtigen Plasma- proteins (Siehe Tab. 7). Bei weitem die gef/~hrlichste Anomalie ist das Antik6rpermangel-Syndrom. Der Organismus dieser Kranken verffigt fiber keine Plasmazellen, in denen haupts/ichlieh die Bfldung yon Anti- kSrpern stattfindet, deshalb kommt es zu dauernd rezidivierenden bakteriellen Infektionen mit schwerstem Verlauf. Da der menschliche Neugeborene seine AntikSrper yon der Mutter transplacentar erhi~lt, kann die A-Gammag]obulingmie - - so nannte man das Leiden bis vor kurzer Zeit - - im allgemeinen nicht vor dem 6. Monat sieher diagnosti- ziert werden. Seit der Einffihrung der Breitspektrumantibiotica fiber- leben heute fast a]le Kranken die Infektionen, vor allem wenn sie gelegentlich einige Milliliter einer Gammaglobulinl6sung injiziert er- halten. Ffir den Arzt ist es wenig erfreulieh, dab es mehrere Typen yon Antik6rpermangelsyndromen gibt und dab dieses Leiden aueh als er worbene St6rung vorkommt.
Aus der groBen Gruppe der familii~ren Hypo- und Dysproteini~mien (das sind St6rungen mit abnormalem Plasmaproteinbestand bzw. fehlenden Fraktionen) sei nut erws dM~ das vSllige Fehlen der Albu- mine bekannt ist; dab etwa 1~ aller Individuen in Zentraleuropa kein

1964 AnomMien des Eiweigstoffwechsels
Tabelle 7. Plasmaproteine a) Fehlende Matrize
121
Syndrom Fehlende Proteine Klinische Symptome
1. Analbumin~imie 2. Antik6rpermangeL
syndrom 3. A-fl-Lipoprotein/~mie 4. Ahapgoglobulin-
Haptoglobin~mie 5. Wilsonsehe
Krankhei~
Albumin meist Gammaglobulin
fl-Lipoproteine
Coeruloplasmin
eventuell 0deme rezidivierende bakteNello
Infektionen Steatorrhoe
Lebercirrhose; cerebrale Symptomatik
b) Fehler in der Matrize
Syndrom
1. Doppelalbumini~mie 2. y-Globulinatypien 3. Haptoglobulinatypien 4. Transferrinatypien 5. Cholinesteraseatypien 6. verschiedene angeborene Dysprotein//mien 7. ,,neues" Plasmaprotein Frazer
Syndrom
Tabelle 8. Blutgerinnungs/aktoren
Fehlender Faktor
1. Afibrinogeniimie 2. Hypoprothrombin/~mie 3. It/~mophilie A 4. I-Igmophilie B 5. It/imophilie C 6. Parah/imophilie
(Itypoproaceeleringmie) 7. I-Iypoconvertin/~mie 8. Stuart-Faktor-Mangel 9. Itagemann-Faktor-Mangel
10. P1/~ttchenfermentmangelzustgnde 11. Multiple Mangelzustgnde
Fibrinogen (Faktor I) Prothrombin (Faktor II) AHG (Faktor VIII) PTC (Faktor IX) PTA Proaccelerin (Faktor V)
Proconvertin (Fak~or VII) Stuart-Faktor (Faktor X) Hagemarm-Faktor meist P1/it~chen-Faktor III meist AHG mit P1/ittehenfaktoren
tIaptoglobulin (= ein Mmoglobinbindendes Globulin) besitzen und dab es Individuen gibt, welehe keine/~-Lipoproteine haben. Letztere St6rung ist dadureh bemerkenswert, dag der Fett transport schwer gest6rt ist, was zu Fetts~/ihlen, Gedeihsg6rung und bemerkenswerterweise auch zu Formver/~nderungen der roten Blutk6rperchen ffihrt. Aueh die ]4tiologie zahlreieher Blutungsfibel (sie sind in Tab.8 zusammengestellt) kann dutch das Feh!en der Matrize ffir den jeweiligen Faktor erkl/~rt werden.

122 Kv~T Scm~E~s~ Bd. 201
Es bedarf wohl kaum einer Betonung, dal~ die Verschicdenartigkeit und Vielfalt der angeborenen Stoffwechselanomalien fiir den Arzt eine Unzahl analytischer Probleme mit sich bringt, denen auch der Er- fahrene grSl~tentefls hilflos gegenfibersteht. Dies gilt ffir die Diagnose- stellung der Leiden, aber ganz besonders f~r den Versuch, die bioehemi- sehe Xtiologie bis dahin unbekannter Syndrome aufzuklaren. Der Mediziner ist auf die Mithilfe yon an~lytisch gesehulten Chemikern an- gewiesen.
Die Wfinsche, welehe der klinisehe Chemiker, der reine Kliniker bzw. der praktisehe Arzt an die yon den analytischen Chemikern zu erarbei- tenden Methoden hat, sind natfirlich sehr different.
Fiir den Arzt in der Praxis ist das Idealziel ein Spot-Test f/ir alle ihn interessierenden StoffweehselgrSl~en. Etwa in der Art, wie wir ihn bereits in Form des Glueotests, des Clinistix, Phenistix usw. oder aueh in Form des Aeetests fiir Aceton besitzen. In Anbetraeht der mangel- haften chemischen Vorkenntnisse und der Unerfahrenheit mit chemisehen Reagentien kommen fiir die t~outinediagnostik des praktischen Arz~es Proben, die fiber das ~iveau eines Trommers oder einer Nylander-Probe hinausgehen, kaum in Frage. Es ist demnaeh eine in jeder Itinsieht lohnende Aufgabe, mSglichst simple, aber empfindliche und spezifische Verfahren in diesem gesteekten Rahmen zu entwickeln. Ganz besonders nStig ist z.B. eine Methode fiir ]tarns/~ure zur Gichtdiagnostik.
Der in einem kleineren Krankenhaus t/~tige Arzt, welehcr gezwungen ist, die Diagnose der Stoffwechselanomalien mit Hi]re einer medizini- schen technischen Assistentin zu machen, befindet sich in einer nicht wesentlieh besseren Situation als sein Kollege in der Allgemeinpraxis. Ohne eigene Erfahrungen mul~ er sieh meist auf die Befunde der medizi- nisch-technischen Assistentin verlassen, yon der man im al]gemeinen hSchstens die Beherrschung colorimetriseher Methoden erwarten kann. Die selbst/~ndige Auswertung yon Papierehromatogrammen o./~. geht aber fiber ihren Ausbildungsstand hinaus.
Die Diagnose der meisten Enzymopathien u./i. wird heute fast aus- schliel~lieh (vielleieht mit Ausnahme der Phenylketonurie) in den grol3en Krankenh~usern gestellt. Die Laboratorien der grol~en Kliniken werden vielfaeh yon Herren mit l/~ngerer chemischer Vorbildung geleitet. Dort werden demnaeh auch schwierigere analytische Verfahren gemeistert. Ffir die Ausarbeitung neuer Methoden fehlt aber meist die Zeit und auch die Erfahrung.
Das Hauptanliegen, welches vor allem die in den Labor~torien p~dia- trischer Klinik T/~tigen haben, ist die Entwieklung yon Mikromethoden oder noch besser yon Ultramikroverfahren, welche es gestatten, die cin- zelnen Metaboliten in einer kleinstmSglichen Menge Blur, Liquor o. ~. mSglichst exakt zu bestimmen. Die Fort~chritte, welehe gerade auf diesem

1964 Anomalien des EiweiBstoffwechsels 123
Gebiete in den letzten Jahren gemacht wurden, sind zwar betr~chtlich; sie betreffen aber im allgemeinen nur die fiblichen l~outinebestimmungen und nicht die Spezialmethoden. Ffir die Forschung auf dem Gebiete der angeborenen Stoffwechselanomalien sind besonders dringend Mikro- verfahren fiir Aminoss Purine, Pyrimidine, Glueurons~ure, Amino- zucker und phosphorylierte Intermedigrprodukte. GrS•te Sehwierigkeiten bereitet aueh der Isolierungsversuch yon Einzelproteinen. Die S~ulen- chromatographie ist auf vielen Gebieten natiirlich eine grol3e ttilfe. Leider ist sie aber zu aufwendig und kann fiir Serienuntersuchungen nicht eingesetzt werden. Die mikrobiologischen Verfahren zur quantitati- yen Bestimmung yon Aminosguren, Co-Fermenten usw. kranken an der sts MSglichkeit, dal3 das Bakterienwachstum dureh Metabolite, Pharmaka o. a. gehemmt wird.
Gerade ffir die Auffindung yon heterozygoten Anomalietr~gern, welehe ja im allgemeinen etwa 50~ des bei den homozygoten Individuen vSllig fehlenden jeweiligen Enzyms besitzen, ist die Ausarbeitung yon hoehempfindlichen Reaktionen bzw. die Verbesserung bestehender Ver- fahren ffir mSglichst alle Aminos~uren, deren Desaminierungs-, Oxy- dierungs- u. ~. Produkte ein dringendes Anliegen.
Die his vor kurzem mit so groBer Begeisterung in der genetischen Forschung verwendeten Nachweisreaktionen einze]ner Metabolite (z. B. der Phenylbrenztraubens~ure) ira Urin genfigt heute den Anforderungen nieht mehr, die wir an den Zeitpunkt der Diagnosestellung bzw. ftir die Auffindung yon heterozygoten Individuen stellen miissen.
Ftir die klinisch-chemisehe Routine und Forschung ist die Aus- arbeitung yon exakten Bestimmungsmethoden aller Enzyme des Inter- medi~rstoffweehsels ein erstrebenswertes Ziel. Das Problem seheint fiir die DPN-abhs l~eaktionen in etwa gelSst zu sein.
Ftir die Aufkl~rung der molekularen Struktur der Proteine, welehe aber wohl sowieso einigen Speziallaboratorien vorbehalten bleiben wird, ist die Vereinfachung und vor allem Standardisierung der in der Itgmo- globinforschung so erfolgreich verwendeten Methoden, wie des ,,Finger- Print"- Verfahrens usw. notwendig.
Durch die Auffindung yon ,,chelating agents", welehe nur ein Metall binden, ws nicht nur ftir die Analytik, sondern aueh ftir die Therapie yon Sehwermetallspeieherkrankheiten viel gewonnen.
Es gibt wohl kaum ein Gebiet der Medizin, in dem die Zusammen- arbeit zwisehen Bioehemiker und Kliniker so erfolgreieh war und ist, wie das der angeborenen Stoffweehselanomalien. Die Medizin hat ffir die Aufklgrung des St6rungsmechanismus zahlreicher Anomalien zu d~nken. Die Bioehemie erhielt wesentliehe neue Impulse und Kenntnis von bis dahin unbekannten Stoffwechselwegen. Es bleibt noch immer viel zu tun. Beide Disziplinen aber diirfen sieh vet den Leistungen des

124 KURT SCHlCEIER : Anomalien des EiweiBstoffwechsels Bd. 201
menschlichen Gehirns verneigen. Selbst aus EiweiB und Lipiden auf- gebaut, hat es dieses Organ vermocht, nicht nur die Gesetze der eigenen Struktur und Funktionen aufzudecken, sondern sogar die meist seltenen metabo]ischen Anomalien aufzufinden.
Zusammenfassung Nach einigen geschichtlichen Bemerkungen wird ganz kurz zu den
Problemen des Eiweil~synthesemechanismus und der genetischen Fixie- rung, der strukturellen und funktionellen Eigenschaften der Proteine Stellung genommen. Dann wird fiber die AnomaHen des Phenylalanin- stoffwechsels, die sogenannte Ahorn-Sh'up-Krankheit und einige andere zu geistiger Retardation ffihrende Anomalien des Aminos~ureumsatzes einsehliel~lieh der Glycinose berichtet. Kurz werden auch die angeborenen StSrungen der Tubulusfunktion, welche zu Nierensteinbildung u. ffihren, er6rtert. Es wird darauf hingewiesen, dab auch zahlreiche hor- monelle StSrungen und manche Unvertr~glichkeitserscheinung yon Medikamenten auf Enzymdeviationen beruhen. Als Musterbeispiel einer StSrung in der molekularen Struktur werden die H~moglobinopathien und fiir das Fehlen einer Matrize der Proteinsynthese die Dysprotein- ~mie des Blutes erw~hnt. Abschliel~end werden die Schwierigkeiten diskutiert, welche der praktische Arzt, der in einem kleinen Krankenhaus t~tige Mediziner und der Experte bei der Diagnosestellung und der Pro- sowie Metaphylaxe der angeborenen Anomalien hat.
Summary After a few introductory remarks about the history of biochemistry,
the problems of the protein synthesis mechanism and the genetic fixa- tion of the structural and functional characteristics of proteins are discussed. The anomalies of phenylalanine metabolism and also the so called "maple-syrup-urine disease" and some other errors, which lead to mental retardation--including glycinosis--are summarized. The inborn disturbances of the tubulus function, wlHch result in nephrotithiasis etc. are discussed shortly. I t is pointed out, that also several anomalies of the endocrine glands and some drug-intolerances have their basis in enzymatic deviations. As a model of the disturbances of the template of protein synthesis, the dysproteinaemias of the blood are mentioned. Finally the difficulties are discussed, which the practisizing physician, the doctor in small hospital and the expert has to meet, with the diagnosis and the pro- and metaphylaxis of inborn anomalies.
Literatur Zusammen/assende Darstellungen
SCnREIE~, K., unter Mitarbeit yon H. MATTERS, U. PORATH, J. SPRA~GER U. H. G. LAser: Die angeborenen Stoffwechselanomalien. Thieme-Verlag Stuttgart

1964 Bn~sro HESS und s BRAND: Bestimmung yon Enzymaktivitgten 125
1963. - - LINNEWEH, Y. (Hrsg.): Erbliche Stoffwechselkrankheiten/Genetic Defects of Biologically Active Proteins. Urban & Schwarzenberg, Mtinehen und Berlin 1962. - - STA~BVRY, J. B., J. B. Wu and D. S. F~ED~IeKSO~: The metabolic :Basis of Inherited Disease. McGraw Hill Book Company, Inc. London 1960. --
Einzeldar.stellung
BE~ZE~, S. : The Elementary Units of Heredity. ~cElroy and Glass Eds., The ]3iolochemical Basis of Heredity. Johns Hopkins Press, Baltimore 1957. -- C~IeK, F. H.: Angew. Chemie 75, 425 (1963). -- JACOB, F., and J. MO~OD: J. 1Viol. Biol. 3, 318 (1961). - - LADU, ]3. ~ . , E. G. Z~NNONI, L . LASTEt~, and J. E. SEEG~/)LLE~: J . biol. Chemistry 230, 251 (1958). - - LEI~NEE, A. B. , and T. ]3. FITZPATmeK: Physiol. l%ev. 80, 91 (1950). - - 1V[ATTKAEI, J . I ' i . , and W. M:. ~II~ENBEP~G: Prec. nat. Sci. USA 47, 1580 (1961). -- 1VI/)LLEI~, W., u. K. SCIIREIEtr Dtsch. Med.Wschr. 87, 2479 (1962). -- SP~'ZE~, J. F., P. LENGYEL, C. BASILIO, and S. 0c~o~: Prec. nat. Aead. Sci. USA 48, 441 (1961). -- WaTSOn, J. D.: Angew. Chem. 75, 439 (1963).
Prof. Dr. K. Se~Rnr~R, Universitgts-Kinderklinik, 69 Heidelberg
Aus dem Chemisehen Laboratorium der Medizinischen Universitgtsklinik (Ludolf Krehl-Klinik) Heidelberg
Grundlagen der Bestimmung yon Enzymaktivitiiten* Vor~
BENNO I~ESS und KARL BRAND
~ i t 15 Textabbildungen
(Eingegangen am 31. Oktober 1963)
I n e inem Sympos ion fiber ana ly t i sche Methoden der EiweiBchemie t r i t t neben die B e t r a c h t u n g der S t r u k t n r von E iwe igk6rpe rn wohl- begr f inde t die F r a g e nach ihrer F u n k t i o n , wenn m a n bedenkt , dab ein sehr groBer Teil der b e k a n n t e n Pro te ine Ms E n z y m e ka ta ly t i sohe Fghig- ke i t en en twieke ln : eine al lgemeine Er fahrung , die m i t der E n t d e e k u n g t ie r Urease dnreh den Amer ikane r S U M ~ R 2~ im gahre 1926 beginnt nnd zu r heu te a l lgemein gfi l t igen Pro te in -Theor ie der F e r m e n t w i r k u n g ge- ff ihrt ha t . Die ka t a ly t i sohe F u n k t i o n der E n z y m e be ruh t auf einer Fo lge e l emen ta r e r ehemiseher Reak t i onen zwisehen den ak t iven Zen t ren eines E n z y m s und seinen R e a k t i o n s p a r t n e r n , die du t ch Messung der Bi lanz sowie der in t e rme4 ig ren Tei l sehr i t te e r fag t werden kann . I m folgenden wird naeh e in le i tenden Defini t ionen ein l~berbl iek fiber d i rek te Methoden zu r Bes t immung yon E n z y m a k t i v i t g t e n sowie ihrer fo rmalk ine t i sohen Grund l agen gegeben (teilweise aus 17).
* Vorgetragen auf der Tagung der l%ehgruppe ,,Analytisehe Chemic" in J-Ieidelberg im September 1963.


















![[Chem] Schoops - Analytische Chemie](https://img.pdfslide.org/doc/110x75/5571f82249795991698cb7d7/chem-schoops-analytische-chemie.jpg)