Embed Size (px)
Citation preview

Z. Anal. Chem. 279, 23-27 (1976) - �9 by Springer-Verlag 1976
Atomabsorptions-spektrometrische Bestimmung von Elementspuren in reinem Kupfer und Kupfersalzen Spurenanreicherung durch AusfS.11en der Matrix als CuS
E. Jackwerth und P. G. Willmer
Institut ftir Spektrochemie und angewandte Spektroskopie, Dortmund
Eingegangen am 4. Juli 1975
AAS Determination of Trace Elements in Pure Copper and Copper Salts. Preconcentration by Precipitation of the Matrix as CuS. For the determination of traces of elements such as Cd, Co, Fe, In, Mn, Ni, Pb, T1 and Zn in
pure copper and copper salts the matrix is precipitated as CuS using thioacetamide as precipitating reagent. In solutions containing 3 N hydrochloric acid, the recovery of the elements is > 90 % (T1 75 %). By addition of HNO3 and evaporation of the filtrate to dryness a trace concentrate is received which is practically free of a salt residue and in which the preconcentrated elements are determined by flame AAS. The relative standard deviation s/2 is generally about 0.03. Using samples of I g of copper the limit of detection for different elements was found to be between 0.2 and 3 ppm.
Zusammenfassung. Zur Bestimmung der Elementspuren Cd, Co, Fe, In, Mn, Ni, Pb, T1 und Zn in reinem Kupfer und Kupfersalzen wird die Matrix als CuS mit Thioacetamid als F/illungsreagens ausgeffillt. In 3 N salzsaurer L6sung werden die Elemente mit Ausbeuten >_ 90 %, T1 zu 75 %, im Filtrat wiedergefunden. Nach Zusatz yon HNO3 und Einengen des Filtrats, erhfilt man ein praktisch rfickstandsfreies Spurenkonzentrat, in dem die an- gereicherten Elemente durch Flammen-Atomabsorptions-Spektrometrie bestimmt werden. Die relative Stan- dardabweichung s/2 liegt im allgemeinen bei Werten um 0,03. Ffir Einwaagen von 1 g Kupfer werden Nachweis- grenzen erzielt, die, je nach Element, zwischen 0,2 und 3 ppm liegen.
Analyse von Kupfer, Kupfersalzen; Spektralphotometrie, Atomabsorption; Ausf/illen der Matrix als CuS.
1. Einleitung
Eine der Grundregeln der Spurenanalyse verbietet die Ausfallung gr6Berer Niederschlagsmengen im Ver- laufe chemischer Trenn- und Anreicherungsprozesse: Schwerl6sliche und mit einer groBen OberflS.che rein verteilt ausfallende Stoffe neigen dazu, zahlreiche Ionen zu meist unreproduzierbaren Anteilen mitzu- reiBen.
In dem im folgenden beschriebenen Verfahren zur Analyse von reinem Kupfer und Kupfersalzen wird das Matrixelement zur Spurenanreicherung, entgegen dieser Regel, als Kupfersulfid ausgeffillt und ab- getrennt. Die Tatsache, dab eine Reihe wichtiger Elementspuren in hoher Ausbeute im Filtrat wieder- gefunden wird, zeigt, dab es auch bei Sulfid-Nieder- schlfigen Ausnahmen vom allgemeinen Verbot der
Matrixf/illung gibt und daB es sich lohnt, nach solchen Ausnahmen zu suchen. Wie eine Durchsicht der Literatur ergab, gilt dies ffir die Reinheitskontrolle des Kupfers in besonderem MaBe.
Ftir Kupfermetall und Kupfersalze sind Verfahren zur Gruppen-Abtrennung bisher kaum beschrieben worden; eher findet man Trennverfahren fiir einzelne oder fiir nur wenige Elemente [4]. Die gleichzeitige Anreicherung mehrerer Spuren gelingt hier vor allem durch Sorption an einen Kollek- tor-Niederschlag, z.B. an Fe(OH)3 [15] oder an La(OH)3 [16]. Zur Abtrennung der Matrix Kupfer wird in einigen Arbeiten die Elektrolyse [1- 3, 6], die AusfSllung als CuSCN [7] oder die Sorption des Chlorokomplexes an stark basischen Ionenaustauschern [11] vorgeschlagen. Die in der Reinststoff- Analyse sonst weit verbreitete Anwendung chelatbildender Gruppenreagentien st6Bt beim Kupfer auf groBe Schwierig- keiten: Wegen der besonderen Neigung des Kupfers, selbst stabile Chelate zu bilden, werden gewfinschte Spurenreaktio-

24 Z. Anal. Chem., Band 279, Heft 1 (1976)
nen meist vollst~indig durch die Reaktion der Matrix iiber- deckt. Es fehlen auBerdem genfigend selektive Maskierungs- reagentien ffir Kupfer, so dab es ffir die gemeinsame Abtren, nung gr6Berer Gruppen von Schwermetallspuren bisher so gut wie keine M6glichkeiten gibt.
Nicht zuletzt waren es diese grunds/itzlichen Pro- bleme bei der Spurenanalyse des Kupfers, die den AnstoB dazu gaben, auch solche als ,,verboten" geltenden F/illungsreaktionen nfiher zu untersuchen.
2. Zur Ausfiillung der Matrix Kupfer als CuS
Matrixf/illungen sind zur Spurenanreicherung nur dann brauchbar, wenn die Ffillungsreaktion so gelenkt werden kann, dab keine Mitreil3vorgfinge auftreten. Einige M6glichkeiten hierzu haben wir im Zusammen- hang mit der Untersuchung von Silbersalz-Nieder- schlS.gen bereits frfiher diskutiert [8, 9]. In jedem Fall sind Arbeitsvorschriften, bei denen ein Hauptbestand- teil zur Spurenanreicherung ausgeffillt wird, nur fiir diejenigen Elementspuren und nur fiir solche Kon- zentrationsbereiche giiltig, bei denen die Brauchbar- keit des Verfahrens durch Eichzus/itze fiberprfift worden ist.
Jede Extrapolation auf geringere oder h6here Spu- renkonzentrationen oder Analogieschliisse auf das Sorptionsverhalten nicht im einzelnen untersuchter Elemente sind allein schon wegen der Kompliziertheit aller Sorptionsprozesse und wegen der noch geringen Kenntnisse fiber Sorptionsmechanismen unsicher und daher unzulfissig. In dieser eingeschr~inkten Form muB das F/illungsverbot ffir Hauptbestandteile in der Spurenanalyse verstanden und aufrechterhalten wer- den.
Sulfidffillungen sind im allgemeinen aus mehreren Grtinden zur Matrixabtrennung wenig geeignet.
Viele der zur Charakterisierung der Reinheit von Metallen wichtigen Spurenverunreinigungen bilden stabile Sulfide; H2S ist ein ausgesprochen unselektives F/illungsmittel. Sulfide geh6ren zu den am wenigsten 16slichen Metallverbindungen; auf Zusatz des Ffillungsmittels erh/ilt man vielfach Nieder- schlfige yon undefinierter Zusammensetzung und mit erheb- lichen kolloiden Anteilen oder groBer innerer Oberflfiche. Die Folge ist ein stark ausgepr~igtes Sorptionsverm6gen ffir Kat- ionen und Anionen. Die geringe L6slichkeit der Metall- sulfide macht es aul3erdem schwierig, die Selektivit/it der FS.11ungsreaktion durch Verwenden von Hilfskomplexbild- nern zu verbessern. Sulfide werden also nicht zuf'~illig besonders vielseitig in der Spurenanalyse als Kollektor-Niederschlfige eingesetzt [14].
Auch CuS wird in der Literatur als Spurenf'~inger vor- geschlagen [14]; es gibt Arbeitsvorschriften z.B. ffir Spuren Zn [13], Pb [12] oder Sb [17] bzw. ffir Au, Bi, Cd, Hg, Mo, Pd und Sb als Gruppe [5], die als Sulfide an CuS angereichert werden.
Kupfersulfid besitzt also offensichtlich gerade die- jenigen Eigenschaften, die bei einem zur Matrix-
abtrennung ausgef/illten Niederschlag unerwfinscht sind.
Ganz allgemein nimmt mit ansteigender Acidit/it einer Probenl6sung die Zahl der ausf/illbaren Elemente ab; dies gilt ebenso auch ftir die Zahl der von einem sulfidischen Spurenf/inger mitgerissenen Elementspu- ren. Im Fall von CuS als Kollektor arbeitet man in neutralen bis schwach sauren L6sungen, um beim Einleiten von H2S die L6slichkeitsprodukte m6g- lichst vieler Spurensulfide zu tiberschreiten. Ob dies ffir ein Spurenelement tatsfichlich gelingt, h/ingt je- doch neben dem pH-Wert auch von der Spuren- konzentration sowie von Nebenreaktionen ab, die das Element z.B. mit zugesetzten Komplexbildnern ein- geht.
Sandell weist nachdrficklich darauf hin, dab der Erfolg der Anreicherung an sulfidischen Spurenf'~ingern stark yon der L6slichkeit der Spurensulfide bestimmt wird. In besonderem AusmaB gilt dies, wenn keine Mischkristalle zwischen den Sulfiden yon Spur und Spurenf'~inger gebildet werden [17].
Um Kupfer als Hauptbestandteil ohne gr613eren Spurenverlust abtrennen zu k6nnen, mfissen die Ffillungsbedingungen also so gew/ihlt werden, dab sie den Erfordernissen einer Spurenffinger-Reaktion genau entgegengerichtet sind:
a) Die Acidit/it der Probenl6sung mul3 so hoch sein, dab das Kupfer zwar noch weitgehend quantitativ ausgef~llt wird, die L6slichkeitsprodukte m6glichst vieler Spurensulfide jedoch nicht mehr erreicht werden.
b) Das eigentliche F/illungsmittel H2S mul3 in ,,homogener L6sung" gebildet werden.
c) Die Matrix Kupfer muB in der Siedehitze aus- gef/illt werden; Niederschlag und fiberstehende L6- sung mtissen rasch durch Filtrieren voneinander getrennt werden.
3. Versuche zur Spurenanreicherung
Unter diesen allgemeinen Bedingungen wurde das Verhalten yon Mikrogramm-Mengen einiger Metall- ionen bei der Ausffillung von 1 g Kupfer als CuS untersucht. Als Ffillnngsreagens wurde das ffir die gravimetrische Metallsulfid-Ffillung aus homogener L6sung bevorzugte Thioacetamid ausgewfihlt, da es in verhfiltnismfiBig reiner Form im Handel ist. Aller- dings mfissen dabei bestimmte Arbeitsbedingungen eingehalten werden, urn, wie mit H2S als Ffillungs- reagens, salzfreie Spurenkonzentrate zu erhalten: Unabh/ingig vom Anion des eingesetzten Kupfer- salzes wird Thioacetamid hydrolytisch zu Schwefel- wasserstoff und Ammoniumacetat zersetzt; beim Ein- dampfen des Filtrats der Kupfersulfidf/illung verbleibt ein betrfichtlicher Salzr/ickstand. Allein nitrathaltige Filtrate, die gleichzeitig Salzsfiure enthalten, dampfen ohne gr6Beren Salzrfickstand ein; tiberschfissiges Ffillungsreagens sowie seine Folgeprodukte werden
E. Jackwerth und P. G. Willmer: Bestimmung yon Elementspuren in reinem Kupfer und Kupfersalzen 25
hier offensichtlich durch entstehendes ,,K6nigswas- ser" vollstfindig zu leicht flfichtigen Stoffen oxidiert. Lediglich Schwefel wird in geringer Menge ausgeschie- den, der aber rasch zu Sulfat oxidiert wird. Man erh/ilt ein Spurenkonzentrat, welches zuletzt aus einem Tropfen Schwefelsfiure mit den aus dem Kupfer angereicherten Elementen besteht. Wird das Kupfer aus salzsauren L6sungen des Kupferchlorids oder -sulfats ausgef~illt, so erh/ilt man ebenfalls salzfreie Spurenkonzentrate dann, wenn man das Filtrat der CuS-F~illung mit einigen Millilitern HNO3 versetzt und einengt. Durch die Oxidation des Ammonium- acetats kann das Mel3volumen ffir die Spuren, wenn erforderlich, sehr klein gehalten werden; Empfind- lichkeit und Nachweisverm6gen des Analysenverfah- tens lassen sich so fiber einen gr613eren Bereich den zu erwartenden Spurenkonzentrationen anpassen. Bei Abtrennung des Kupfers aus L6sungen des Metalls erhfilt man salzfreie Spurenkonzentrate, wenn zum L6sen des Probenmaterials H N Q verwendet wird. Allerdings ist die HNO3-Konzentrat ion der Proben- 16sung nicht ohne Einflul3 auf den Reaktionsablauf: [st sie sehr hoch, so wird bereits das F/illungsreagens in stfirmischer Reaktion oxidiert, ohne dab es zur Ausffillung des Kupfersulfids kommt. Die ftir die Selektivit/it der Ffillungsreaktion erforderliche hohe Acidit/it darf deshalb nicht mit Salpeters/iure, sondern mul3 mit Salzs/iure eingestellt werden. In H N O 3 gel6stes Kupfermetall wird daher zunfichst zur Trockne eingedampft. Nitratverluste, die durch teil- weise hydrolytische Zersetzung beim Erhitzen des verbleibenden Salzrfickstandes entstehen, werden durch Zusatz von 0,5 ml HNO3 zum Filtrat der CuS- F/illung wieder ausgeglichen, wenn das aus dem F/il- lungsreagens entstehende Ammoniumacetat beim Ein- dampfen des Filtrats vollstfindig oxidiert werden soll.
Zur AusFgllung des Kupfers wird Thioacetamid in geringem fJberschul3 der siedenden Probenl6sung zu- gesetzt. Es f/illt zunfichst ein volumin6ser gelblicher Niederschlag aus, der sich jedoch rasch unter H2S- Entwicklung zu Kupfersulfid umsetzt. Nach wenigen Minuten setzt sich der schwarze Niederschlag ab, und es entsteht eine praktisch klare fiberstehende L6sung, die ohne Schwierigkeit durch ein Filter von mittlerer H/irte abfiltriert werden kann.
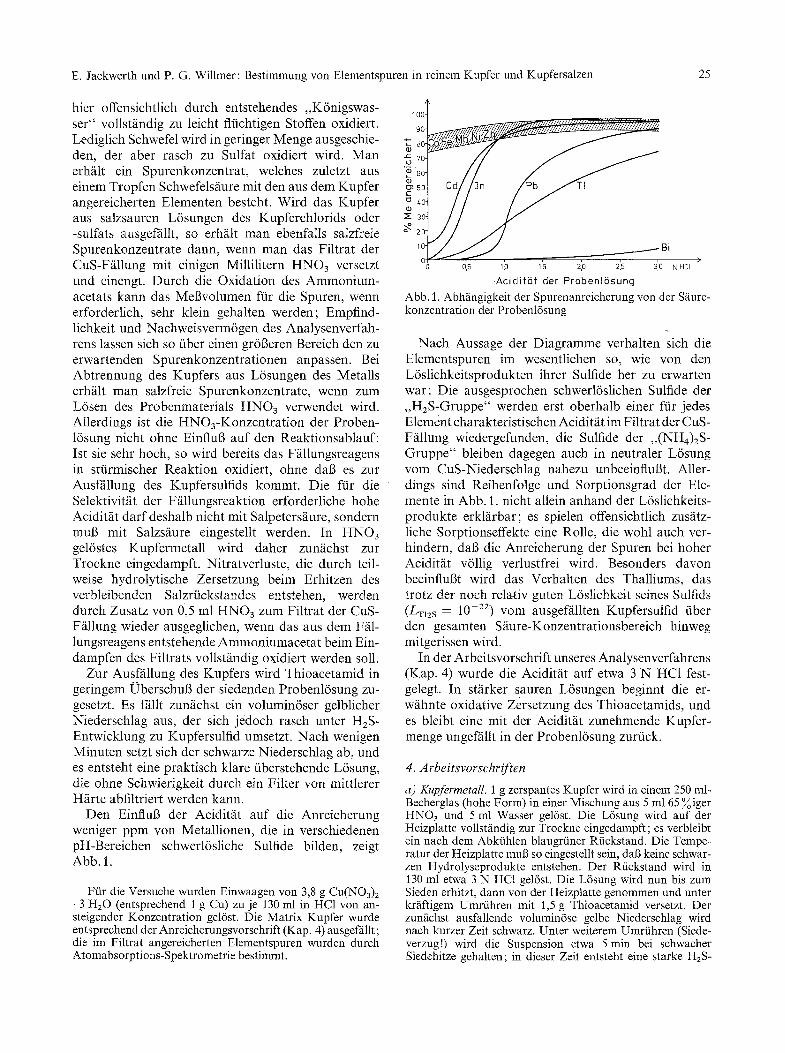
Den EinfluB der Acidit~it auf die Anreicherung weniger ppm yon Metallionen, die in verschiedenen pH-Bereichen schwerl6sliche Sulfide bilden, zeigt Abb. 1.
Ffir die Versuche wurden Einwaagen von 3,8 g Cu(NO3) 2 �9 3 HzO (entsprechend 1 g Cu) zu je 130 ml in HC1 von an- steigender Konzentration gel6st. Die Matrix Kupfer wurde entsprechend der Anreicherungsvorschrift (Kap. 4) ausgeffillt; die im Filtrat angereicherten Elementspuren wurden durch Atomabsorptions-Spektrometrie bestimmt.
/
100-
90-
~ a0- 0)
70-
60"
c ~ sc~ 40-
3O
2~ 10"
0 o5
rr ( ( H r / . . . . . . . . . . . . . . . irl, , ,
/
/ . ~ B i 1,0 1,'5 210 215 3'0 N HC[
Acidi t f i t der Probenl6sung
Abb. 1. Abhfingigkeit der Spurenanreicherung yon der S~ure- konzentration der Probenl6sung
Nach Aussage der Diagramme verhalten sich die Elementspuren im wesentlichen so, wie von den L6slichkeitsprodukten ihrer Sulfide her zu erwarten war: Die ausgesprochen schwerl6slichen Sulfide der , ,HzS-Gruppe" werden erst oberhalb einer ffir jedes Element charakteristischen Aciditfit im Filtrat der CuS- Ffillung wiedergefunden, die Sulfide der ,,(NHg)2S- Gruppe" bleiben dagegen auch in neutraler L6sung vom CuS-Niederschlag nahezu unbeeinflul3t. Aller- dings sind Reihenfolge und Sorptionsgrad der Ele- mente in Abb. 1. nicht allein anhand der L6slichkeits- produkte erkl/irbar; es spielen offensichtlich zusfitz- liche Sorptionseffekte eine Rolle, die wohl auch ver- hindern, dab die Anreicherung der Spuren bei hoher Aciditfit v611ig verlustfrei wird. Besonders davon beeinflul3t wird das Verhalten des Thalliums, das trotz der noch relativ guten L6slichkeit seines Sulfids (LT|zs = 10 -22) VOlTI ausgef/illten Kupfersulfid fiber den gesamten S/iure-Konzentrationsbereich hinweg mitgerissen wird.
In der Arbeitsvorschrift unseres Analysenverfahrens (Kap. 4) wurde die Aciditfit auf etwa 3 N HC1 fest- gelegt. In stfirker sauren L6sungen beginnt die er- w/ihnte oxidative Zersetzung des Thioacetamids, und es bleibt eine mit der Aciditfit zunehmende Kupfer- menge ungeffillt in der Probenl6sung zurfick.
4. Arbeitsvorschriften
a) Kupfermetall. I g zerspantes Kupfer wird in einem 250 ml- Becherglas (hohe Form) in einer Mischung aus 5 m! 65 ~iger H N Q und 5 ml Wasser gel6st. Die L6sung wird auf der Heizplatte vollst/indig zur Trockne eingedampft; es verbleibt ein nach dem Abkfihlen blaugrfiner Rfickstand. Die Tempe- ratur der Heizplatte mul3 so eingestellt sein, dab keine schwar- zen Hydrolyseprodukte entstehen. Der Rfickstand wird in 130 ml etwa 3 N HC1 gel6st. Die L6sung wird nun bis zum Sieden erhitzt, dann vonder Heizplatte genommen und unter krS.ftigem Umrtihren mit 1,5 g Thioacetamid versetzt. Der zunfichst ausfallende volumin6se gelbe Niederschlag wird nach kurzer Zeit schwarz. Unter weiterem Umrfihren (Siede- verzug!) wird die Suspension etwa 5 rain bei schwacher Siedehitze gehalten; in dieser Zeit entsteht eine starke HzS-

26 Z. Anal. Chem., Band 279, Heft 1 (1976)
Entwicklung und der CuS-Niederschlag setzt sich ab. Die Suspension wird filtriert, Glas und Filter werden mit wenigen Millilitern 3 N HC1 gewaschen. Das Filtrat wird nach Zugabe von 0,5ml 65~iger HNO3 bis beinahe zur Trockne ein- gedampft. Der Spurenriickstand wird unter leichtem ErwS, r- men in 5 ml 4 N HNO3 gel6st; die L6sung wird im MeB- kolben mit Wasser zu 10,0 ml erg/inzt.
Zur Bestimmung des Chemikalien-Blindwertes werden alle Reagentien entsprechend den Mengen aus der Arbeits- vorschrift eingedampft und analysiert. Wichtig ist, dab das Thioacetamid zun/ichst allein mit der zur Analyse notwendigen 3 N Salzs/iure erhitzt wird, bevor die Salpetersfiure zugesetzt wird. Andernfalls entstehen st6rende Mengen an Schwefel und andere Zersetzungsprodukte.
MeBgerfit. Varian-Techtron Atomabsorptions-Spektro- meter Modell 1000 mit zugeh6rigen Hohlkathodenlampen; Acetylen-Luft-Flamme.
b) Kupfernitrat. 3,80 g Cu(NO3)z �9 3 H20 (entsprechend 1 g Cu) werden in 130 ml etwa 3 N HC1 gel6st und entspre- chend der Vorschrift ftir Kupfermetall weiter behandelt.
c) Kupferchlorid, Kupfersulfat. 2,68 g CuC12 �9 2 H20 bzw. 3,93 g CuSO4 - 5 HeO (entsprechend I g Cu) werden in 130 ml etwa 3 N HC1 gel6st und entsprechend der Vorschrift fiir Kupfermetall weiter behandelt. Hier wird lediglich die nach dem Abfiltrieren des CuS-Niederschlages zuzusetzende HNO3- Menge auf 5 ml erh6ht (s. Kap. 3).
5. Analysenergebnisse
Nach Ffillung von 1 g Kupfer entsprechend unserer Arbeitsvorschrift gelangen etwa 1 - 2 mg Cu 2+ ins Spurenkonzentrat; eine St6rung bei der AAS-Bestim- mung der Elementspuren durch Kupfer in dieser
Tabelle 1
Ele- ment- spur
unter- suchter Konzen- trations- bereich [ppm]
Anrei- rel. Standard- Nach- cherungs- abweichung weis- ausbeute grenze [ %] s/2 bei (3 a-
(N= 12) ppm Grenze) [ppm]
Cd Co Fe In Mn Ni Pb T1 Zn
1 . . 10 1 .. 10 5 .. 50 5 .. 50 1 .. 10 1 .. 10 2 . . . 20 5 . . . 50 1 . . . 10
94 0,027 1,6 0,2 95 0,021 5,4 1 95 0,032 14,5 0,6 96 0,020 24,5 3 90 0,025 1,6 0,3 95 0,017 6,0 0,7 90 0,026 9,8 2 75 0,035 20,0 3 93 0,072 2,2 0,2
Menge wird nicht mehr beobachtet. Das gilt auch ftir die in konzentrierteren Kupferl6sungen gest6rte Ana- lysenlinie des Zinks (Zn: 213,856 nm; Cu: 213,851 nm). Die unter den Bedingungen der Arbeitsvorschrift erhaltenen Zahlenwerte f/Jr die Anreicherung von 9 Elementspuren sind in Tab. 1 zusammengestellt. Zugesetzte Spuren Bi wurden zu nur etwa 8 ~ , Au, Ag und Sn wurden gar nicht angereichert. Es soll auch hier noch einmal darauf hingewiesen werden, dab die tabellierten Zahlen ffir die Anreicherungs-Ausbeuten der Elemente zunfichst nur ftir den jeweils unter- suchten Konzentrationsbereich gelten. Vor allem bei geringeren Spurenkonzentrationen mul3 mit h6heren Sorptionsverlusten, d.h. kleineren Ausbeuten ge- rechnet werden. Deshalb k6nnen auch die aus den Untergrundschwankungen und mit Hilfe extrapotierter Eichgeraden ermittelten Werte ffir die Nachweis- grenzen (3 a-Grenzen) lediglich als Orientierungs- daten angesehen werden. Die relativen Standard- abweichungen s/2 des Analysenverfahrens wurden an 12 Analysen von Elektrolyt-Kupfer bestimmt, dem die nicht nachweisbaren Elemente in den in der Tabelle angegebenen Gehalten zugesetzt wurden.
Tab. 2 enthfilt Vergleichswerte ftir Fe, Ni und Pb, die an photometrisch analysiertem Elektr01yt-Kupfer gewonnen wurden. Die Werte unseres Analysen- verfahrens sind dabei Mittelwerte aus je 12"Einzel- analysen; die angegebenen Streubereiche sind die aus den Einzelwerten dieser Probenmaterialien errechneten 2 o--Werte. In Tab. 3 sind schliel31ich die von uns ermittelten Analysendaten f/Jr das bei den Unter- suchungen als Probenmaterial vorwiegend eingesetzte Kupfermetal l und ffir verschiedene analysenreine Kupfersalze zusammengestellt. Bei einem Teil der analysierten Elemente kann lediglich die Garantie- grenze ftir Reinheit [10] angegeben werden. Die f/Jr die einzelnen Prfiparate unterschiedlichen Garantie- grenzen ergeben sich aus den dem Kupfergehalt ent- sprechenden Probeneinwaagen (s. Arbeitsvorschrif- ten). Soweit Daten ftir die untersuchten Elemente in den Garantiescheinen bzw. Typanalysen der Materia- lien enthalten sind, wurden die vom Hersteller (Merck, Darmstadt) zugelassenen H6chstgehalte an Spuren- verunreinigungen - in Klammern gesetzt - in der Tabelle mit aufgenommen.
Tabelle 2
Fe ~ Ni ~ Pb
Probe 360 photometrische Werte 2 nach unserem Verfahren ermittelt (2,5 + 0,1)
Probe 361 photometrische Werte 1,9 nach unserem Verfahren ermittelt (1,7 + 0,3)
10-3 10-3
10-3 10 -3
2,3 10 -3 (2,3 + 0,1) 10 -3
8 10 - 3 (7,3 -t- 0,4). 10 -3
12- 10 -3 (13,4 + 0,4)- 10 -3
4,8 �9 10 -3 (5,4 + 0,2). 10 -3

E. Jackwerth und P. G. Willmer: Bestimmung von Elementspuren in reinem Kupfer und Kupfersalzen 27
Tabelle 3. Spurenverunreinigungen der benutzten Kupfer- prfiparate [ppm]
Element- Kupfer- Cu(NO3)2 CuCI2 C u S Q spur Blech �9 3 HzO �9 2 H20 �9 5 H20
Cd 0,4 a 0,09 6,0 0,6 Co 2" 0,5" 0,7 a 21,9 Fe 5,8 (50) 1,5(20) 2,1 (5) 18,0 (30) In 6 a 1,6 a 2,2" 1,5" Mn 0,3 0,2 a 0,9 7,5 Ni 5,5 0,4 (20) 2,3 (10) 10,0 (20) Pb 3,0 (500) 1,1" (20) 3,8 (40) 4,1 (50) T1 6 a 1,6 a 2,2" 1,5" Zn 0,2 0,2 (10) 0,4 48,9 (300)
Garantiegrenze ffir Reinheit [10]. ( ) zulfissige H6chstgehalte bzw. Werte der Typanalyse von
Cu-Blech.
Wir danken der Deutschen Forschungsgemeinschaft sowie dem Fonds der Chemischen Industrie ftir die finanzielle Unterst/Rzung dieser Arbeit.
Literatur
1. Babko, A. K., Marchenko, P. V., Nazarchuk, T. N.: Zavodsk. Lab. 21, 662 (1955); vgl. Anal. Abstr. 3, 932 (1956)
2. Barabas, S., Cooper, W. C.: Metallurgia 56, 101 (1957) 3. van Dijk, G., Verbeek, F. : Anal. Chim. Acta 54, 475
(1971) 4. Dozinel, O. M. : Chemiker-Ztg. 87, 698 (1963) 5. Duke, J. F. : A spectrochemical method for the determi-
nation of trace impurities in indium. In: M. S. Brooks and J. K. Kennedy: Ultrapurification of semiconductor mate- rials. New York: Macmillan 1962
6. Eve, A. J., Verdier, E. T. : Anal. Chem. 28, 537 (1956) 7. Hair, R. P., Newman, E. J. : Analyst 89, 42 (1964) 8. Jackwerth, E., Graffmann, G. : diese Z. 241, 96 (1968) 9. Jackwerth, E., Lohmar, J., Schwark, G. : diese Z. 260,
101 (1972) 10. Kaiser, H. : diese Z. 209, 1 (1965) 11. Ligka, K., Klir, L.: Chem. Listy 51, 1549 (1957); vgl.
Anal. Abstr. 5, 782 (1958) 12. Lucas, R., Grassner, F. : Mikrochemie (Emich-Fest-
schrift) 1930, 197 13. Lutz, R. E. : J. Ind. Hyg. Toxicol. 7, 273 (1925) 14. Mizuike, A.: Separation and preconcentration techniques.
In: R. F. Bunshah: Modern analytical techniques for metals and alloys. New York: Intersci. Publ. 1970
15. Neiman, E. Y., Trukhacheva, L. N. : Zavodsk. Lab. 38, 1058 (1972); vgl. Anal. Abstr. 24, 2096 (1973)
16. Reichel, W., Bleakley, B. G. : Anal. Chem. 46, 59 (1974) 17. Sandell, E. B.: Colorimetric determination of traces of
metals. New York: Intersci. Publ. 1959
Prof. Dr. E. Jackwerth, Institut ffir Spektrochemie und angewandte Spektroskopie, D-4600 Dortmund 1, Bunsen-Kirchhoff-Stral3e 11, Bundesrepublik Deutschland





























