Embed Size (px)
Citation preview

Michael Krawczak
Genetische Epidemiologie
Genetische Epidemiologie ist eine multidisziplinäre Wissenschaft, die Erkennt-
nisse und Methoden der Genetik, der Statistik, der Epidemiologie und der Po-
pulationsgenetik verbindet. Sie beschäftigt sich mit der Untersuchung von gene-
tischen Faktoren, die im Zusammenspiel mit passenden Umwelteinflüssen bei
der Entstehung von Krankheiten des Menschen eine entscheidende Rolle spie-
len. Dabei unterscheidet sich die genetische dadurch von der klassischen Epi-
demiologie, dass genetische Ähnlichkeiten sowohl in Familien als auch auf Po-
pulationsebene bei der statistischen Modellbildung, der Studienplanung und der
Datenanalyse explizit berücksichtigt werden.
Genetische Epidemiologie – eine multidisziplinäre Wissenschaft
Die genetische Epidemiologie ist die Wissenschaft von den formalen Grund-lagen und den statistischen Eigenschaften solcher Erkrankungen des Menschen,bei deren Entstehung genetische Faktoren eine maßgebliche Rolle spielen1.Primärer Untersuchungsgegenstand der genetischen Epidemiologie sind daherFamilien von Patienten, vorzugsweise mehrere Generationen umfassend, an-hand derer den Ursachen und Weitergabemechanismen phänotypischerMerkmale nachgegangen werden kann. Daneben finden aber auch Kollektive(offensichtlich) unverwandter Merkmalsträger sowie deren Vergleich mit ent-sprechenden Kontrollgruppen zunehmendes Interesse bei der Erforschungerblicher Erkrankungen und Krankheitsprädispositionen2.
In vielerlei Hinsicht ist die genetische Epidemiologie eine interdisziplinä-re Wissenschaft, die eine größere thematische Nähe zur Populationsgenetik undzur Evolutionsgenetik aufweist als zur klassischen Epidemiologie3. Erst in letz-ter Zeit hat sich die Zielrichtung des Faches etwas geändert und schließt nunauch solche Erkrankungen ein, die durch niedrige familiäre Wiederholungsrisikengekennzeichnet sind, wie sie in der konventionellen epidemiologischen For-schung üblich sind. Dessen ungeachtet hat die genetische Epidemiologie ihregrößten Erfolge bislang bei der Aufklärung der genetischen Ursachen soge-nannter »monogenetischer« Störungen erzielen können. Dies sind Erkrankungen,die zumeist sehr selten sind, familiär stark gehäuft auftreten, und die aufVeränderungen in jeweils einem einzelnen Gen zurückzuführen sind. Zu denmonogenetischen Krankheiten zählen unter anderem die zystische Fibrose, dieHuntingtonsche Erkrankung und die Muskeldystrophie Typ Duchenne (Ta-belle 1). Für mehrere hundert dieser Erkrankungen konnten bereits jene Genelokalisiert und identifiziert werden, die bei Patienten kausal verändert sind4.Der dabei verfolgte methodische Ansatz wird als »positionale Klonierung«bezeichnet5. Statt auf entsprechendes Vorwissen bezüglich der ursächlichen bio-chemischen Defekte zurückzugreifen, stützt sich die positionale Klonierung von
35 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 35

Michael Krawczak
Krankheitsgenen allein auf die Vererbungsmuster geeigneter genetischer»Markergene« in betroffenen Familien, um daraus indirekt auf die Lokalisationder kausalen Genveränderungen im menschlichen Genom zu schließen. Je öftereine Krankheit und eine bestimmte Ausprägung des Markergens (die man als»Allel« des Gens bezeichnet) von Familienmitgliedern gemeinsam ererbt wer-den, um so stärker ist der Hinweis darauf, dass mindestens ein Gen in der phy-sikalischen Nähe des Markers die Krankheit ursächlich beeinflusst. Man sagtin diesem Fall, dass Markergen und Krankheitsgen chromosomal »gekoppelt«sind, weshalb die skizzierte Methode auch als »Kopplungsanalyse« bezeichnetwird.
36 Christiana Albertina 56 2003
Gen-Symbol Krankheit
ABCA1 Tangier Krankheit
APC familiäre adenomatöse Polypose
ATP2A2 Darier Krankheit
BRCA1, BRCA2 erblicher Brust- und Ovarialkrebs
CFTR zystische Fibrose
CYBB chronische Granulomatose
DMD Muskeldystrophie Type Duchenne
EFEMP1 Malattia leventinese
ELAC2 familiärer Prostatakrebs
FGFR3 Achondroplasie
FMR1 fragiles X Syndrom
FRDA Friedreichsche Ataxie
HD Huntingtonsche Krankheit
HFE Hämochromatose
JAG1 Alagille Syndrom
MEN1 multiple endokrine Neoplasie
MKKS McKusick-Kaufman Syndrom
MTMR2 Charcot-Marie-Tooth Krankheit
NF1, NF2 Neurofibromatose
PARK2 juveniler Morbus Parkinson
PKD1, PKD2 polyzystische Nierenerkrankung
PSEN1, PSEN2 früh manifestierender familiärer Morbus Alzheimer
RB1 Retinoblastom
RPGR geschlechtsgekoppelte Retinitis pigmentosa
SMN1 spinale Muskelatrophie
SRY „sex reversal“
TCF1 Diabetes mellitus Typ 3
TSC1, TSC2 tuberöse Sklerose
UBE3A Angelman syndrome
WT1 Wilms Tumor
Tabelle 1
Eine Auswahl mensch-
licher Gene, die ursäch-
lich an der Entstehung
monogenetischer Er-
krankungen beteiligt
sind und die durch
positionale Klonierung
identifiziert wurden
Genkartierung und meiotische Rekombination
Kopplungsanalysen dienen der Schätzung der Rate meiotischer (von»Meiose« = Reifeteilung) Rekombination zwischen je zwei Genloci, d.h. derHäufigkeit θ, mit der ein Kind von einem seiner Eltern das großmütterliche Allel
CA 56 Heft 31.07.2007 7:43 Uhr Seite 36
Genetische Epidemiologie
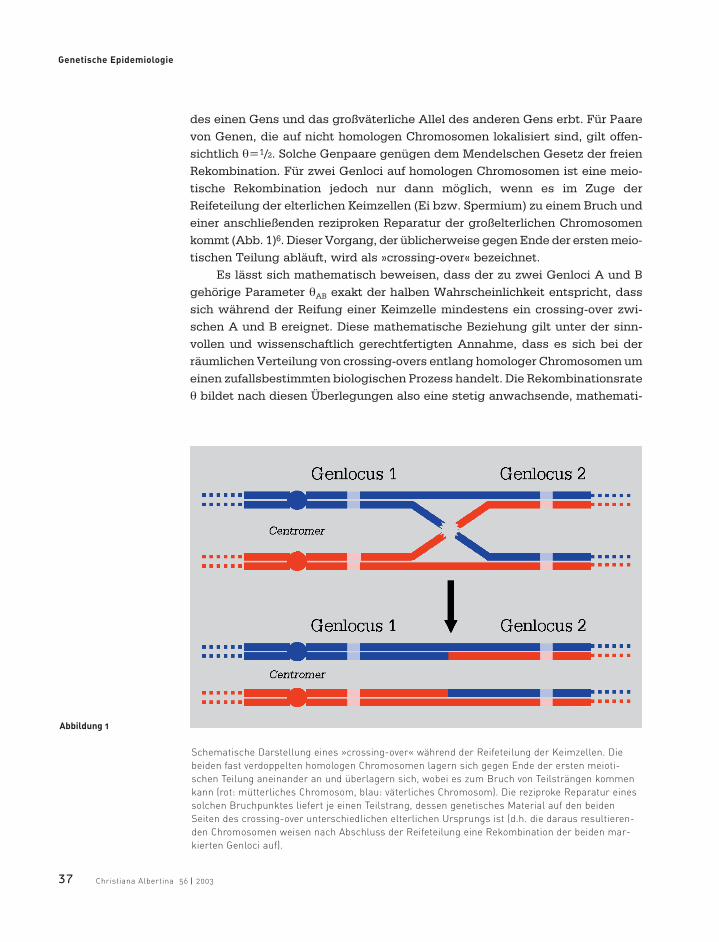
des einen Gens und das großväterliche Allel des anderen Gens erbt. Für Paarevon Genen, die auf nicht homologen Chromosomen lokalisiert sind, gilt offen-sichtlich θ=1/2. Solche Genpaare genügen dem Mendelschen Gesetz der freienRekombination. Für zwei Genloci auf homologen Chromosomen ist eine meio-tische Rekombination jedoch nur dann möglich, wenn es im Zuge derReifeteilung der elterlichen Keimzellen (Ei bzw. Spermium) zu einem Bruch undeiner anschließenden reziproken Reparatur der großelterlichen Chromosomenkommt (Abb. 1)6. Dieser Vorgang, der üblicherweise gegen Ende der ersten meio-tischen Teilung abläuft, wird als »crossing-over« bezeichnet.
Es lässt sich mathematisch beweisen, dass der zu zwei Genloci A und Bgehörige Parameter θAB exakt der halben Wahrscheinlichkeit entspricht, dasssich während der Reifung einer Keimzelle mindestens ein crossing-over zwi-schen A und B ereignet. Diese mathematische Beziehung gilt unter der sinn-vollen und wissenschaftlich gerechtfertigten Annahme, dass es sich bei derräumlichen Verteilung von crossing-overs entlang homologer Chromosomen umeinen zufallsbestimmten biologischen Prozess handelt. Die Rekombinationsrateθ bildet nach diesen Überlegungen also eine stetig anwachsende, mathemati-
37 Christiana Albertina 56 2003
Abbildung 1
Schematische Darstellung eines »crossing-over« während der Reifeteilung der Keimzellen. Die
beiden fast verdoppelten homologen Chromosomen lagern sich gegen Ende der ersten meioti-
schen Teilung aneinander an und überlagern sich, wobei es zum Bruch von Teilsträngen kommen
kann (rot: mütterliches Chromosom, blau: väterliches Chromosom). Die reziproke Reparatur eines
solchen Bruchpunktes liefert je einen Teilstrang, dessen genetisches Material auf den beiden
Seiten des crossing-over unterschiedlichen elterlichen Ursprungs ist (d.h. die daraus resultieren-
den Chromosomen weisen nach Abschluss der Reifeteilung eine Rekombination der beiden mar-
kierten Genloci auf).
CA 56 Heft 31.07.2007 7:43 Uhr Seite 37

Michael Krawczak
sche Funktion des physikalischen Abstands zweier Genloci, was sie zu einemSchlüsselparameter für die Kartierung von Genen macht. Unglücklicher Weiseist der Parameter θ jedoch kein additives Abstandsmaß, da er den Wert 1/2 for-mal nicht überschreiten kann. Um dennoch auf der Grundlage von Kopplungs-analysen eine Genkartierung zu ermöglichen, muss θ mit Hilfe einer sogenann-ten Kartierungsfunktion zunächst in einen additiven »genetischen Abstand«,d(θ), transformiert werden. Die dafür gebräuchlichste mathematische Funktionlautet d(θ)=1/2 ln(1-2θ); sie wurde für diesen Zweck erstmals 1919 von dem bri-tischen Genetiker J.B.S. Haldane vorgeschlagen7. Die genetische Entfernungzwischen zwei Genloci wird in der Einheit »Morgan« (M) gemessen. Durch dieseBezeichnungsweise wird der amerikanische Nobelpreisträger für Biologie T.H.Morgan geehrt, dem als Ersten der Nachweis gelang, dass die Chromosomeneine entscheidende Rolle bei der biologischen Vererbung spielen. Ein centi-Morgan (cM) entspricht der Erwartung, dass zwischen zwei Genloci unter 100Meiosen durchschnittlich eine Rekombination auftritt.
Parametrische Kopplungsanalyse
Die Kopplungsanalyse hat zwei wesentliche Aufgaben, nämlich (1) mit hin-reichender statistischer Sicherheit zu klären, ob θ=1/2 oder θ<1/2 gilt, und (2) imletztgenannten Fall den Parameter θ so präzise wie möglich zu schätzen8. BeideZiele lassen sich in Studien an Labortieren einfach erreichen, da man dort durchkontrollierte Züchtung schon nach wenigen Generationen die Träger von rekom-binanten und nicht-rekombinanten Chromosomen einfach abzählen kann. BeimMenschen (und bei Tieren mit langer Generationszeit) muss die Kopplungs-analyse jedoch auf vorhandene Familiendaten zurückgreifen. Ob und wie diesegenutzt werden können, um statistische Rückschlüsse auf die Kopplung vonGenen zu ziehen, hängt von der Komplexität der Daten ab und vom allgemei-nen Kenntnisstand hinsichtlich der genetischen, umweltbedingten und sto-chastischen Gegebenheiten des in Frage stehenden Merkmals.
Für die relativ einfachen Genotyp-Phänotyp-Beziehungen, die den meistenmonogenetisch bedingten Erkrankungen zu eigen sind, lässt sich die gemein-same Vererbung von Krankheits- und Markergenen in betroffenen Familien expli-zit mathematisch modellieren (»parametrische Kopplungsanalyse«). Diese Mo-dellbildung basiert auf den geschätzten Penetranzen, d.h. den Wahrscheinlich-keiten für die Manifestation der fraglichen Krankheitsgene bei einem potentiellenPatienten, sowie den populationsspezifischen Häufigkeiten aller in der Familienachgewiesenen Genotypen. Die Likelihood L= (θ = θ0 | D) einer Rekombina-tionsrate θ0 kann dann mit Hilfe geeigneter Computerprogramme aus den phä-notypischen Daten, D, der Familie errechnet werden. Dies führt zur Definitiondes sogenannten »lod score«
38 Christiana Albertina 56 2003
L(θ = θ0 | D),L(θ = 1/2| D)
z(θ0) = log10
CA 56 Heft 31.07.2007 7:43 Uhr Seite 38
Genetische Epidemiologie
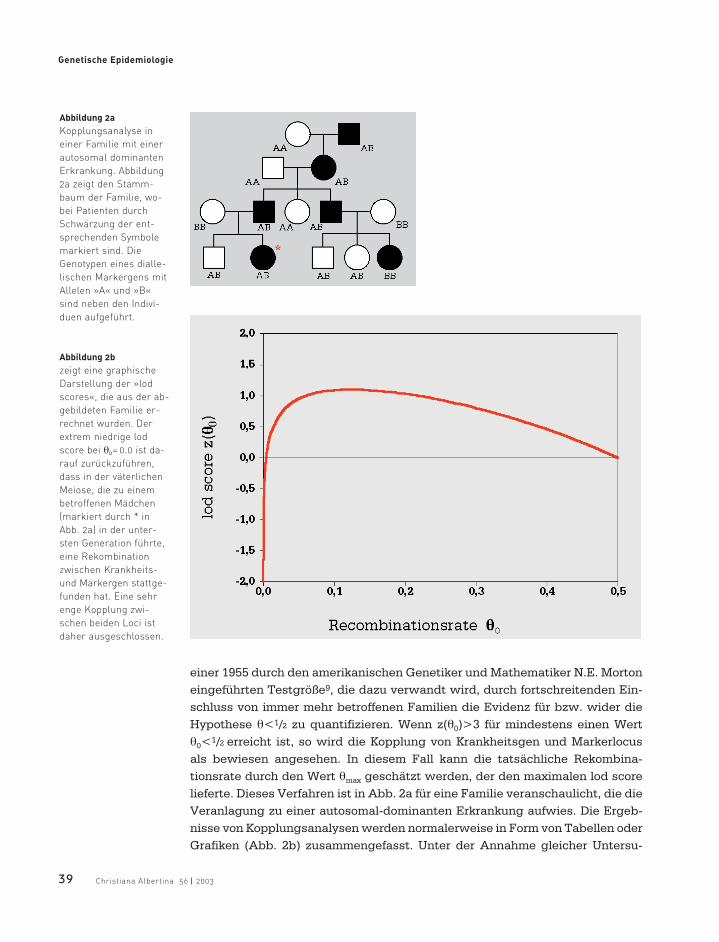
einer 1955 durch den amerikanischen Genetiker und Mathematiker N.E. Mortoneingeführten Testgröße9, die dazu verwandt wird, durch fortschreitenden Ein-schluss von immer mehr betroffenen Familien die Evidenz für bzw. wider dieHypothese θ<1/2 zu quantifizieren. Wenn z(θ0)>3 für mindestens einen Wertθ0<1/2 erreicht ist, so wird die Kopplung von Krankheitsgen und Markerlocus als bewiesen angesehen. In diesem Fall kann die tatsächliche Rekombina-tionsrate durch den Wert θmax geschätzt werden, der den maximalen lod scorelieferte. Dieses Verfahren ist in Abb. 2a für eine Familie veranschaulicht, die dieVeranlagung zu einer autosomal-dominanten Erkrankung aufwies. Die Ergeb-nisse von Kopplungsanalysen werden normalerweise in Form von Tabellen oderGrafiken (Abb. 2b) zusammengefasst. Unter der Annahme gleicher Untersu-
39 Christiana Albertina 56 2003
Abbildung 2a
Kopplungsanalyse in
einer Familie mit einer
autosomal dominanten
Erkrankung. Abbildung
2a zeigt den Stamm-
baum der Familie, wo-
bei Patienten durch
Schwärzung der ent-
sprechenden Symbole
markiert sind. Die
Genotypen eines dialle-
lischen Markergens mit
Allelen »A« und »B«
sind neben den Indivi-
duen aufgeführt.
Abbildung 2b
zeigt eine graphische
Darstellung der »lod
scores«, die aus der ab-
gebildeten Familie er-
rechnet wurden. Der
extrem niedrige lod
score bei θ0= 0.0 ist da-
rauf zurückzuführen,
dass in der väterlichen
Meiose, die zu einem
betroffenen Mädchen
(markiert durch * in
Abb. 2a) in der unter-
sten Generation führte,
eine Rekombination
zwischen Krankheits-
und Markergen stattge-
funden hat. Eine sehr
enge Kopplung zwi-
schen beiden Loci ist
daher ausgeschlossen.
CA 56 Heft 31.07.2007 7:43 Uhr Seite 39

Michael Krawczak
chungsbedingungen können diese Resultate aus unabhängigen Studien an un-verwandten Familien durch Summierung der lod scores zusammengeführt wer-den. Wenn die Lokalisation eines Krankheitsgens in Relation zu einer Vielzahlvon miteinander gekoppelten Markergenen ermittelt werden soll, so kann diesdurch die gleichzeitige Betrachtung aller Loci in einer multilokalen Kopplungs-analyse erfolgen. Multilokale Verfahren haben sich als doppelt so präzise undeffizient erwiesen wie paarweise Analysen (wie Anm. 8, S. 49).
Nicht-parametrische Kopplungsanalyse bei komplexen Erkrankungen
Es zeichnet sich ab, dass die wesentliche Herausforderung an die geneti-sche Epidemiologie zukünftig in der Analyse sogenannter »komplexer« Erkran-kungen bestehen wird, zu denen u.a. der Diabetes mellitus, die koronaren Herz-erkrankungen, Krebs und ein Vielzahl psychiatrischer Störungen gehören10. ImVergleich zu monogenetischen Erkrankungen zeichnen sich komplexe Krank-heiten durch folgende Charakteristika aus: (1) sie treten in Industriegesellschaften substantiell häufiger auf als monoge-netische Erkrankungen,(2) an ihrer Entstehung ist eine Vielzahl von Genen beteiligt, deren Variantenzudem miteinander interagieren, (3) einzelne Varianten haben nur eine relativ geringe, prädisponierende Wir-kung, und(4) Umweltfaktoren spielen für die tatsächliche Manifestation einer genetischenPrädisposition eine entscheidende Rolle.
Da es normalerweise keine Vorabinformation darüber gibt, wie die Va-rianten eines bestimmten Gens das Risiko für eine komplexe Erkrankung beein-flussen (d.h. das »genetische Modell« der Krankheit ist unbekannt), erfordertdie Genkartierung bei komplexen Erkrankungen den Einsatz statistischerVerfahren, die robust gegen eine fehlerhafte Modellbildung sind. Man sprichtin diesem Zusammenhang von »nicht-parametrischer« oder »modellfreier« Kopp-lungsanalyse11. Allerdings haben modellfreie Methoden den Nachteil, dass sieüber eine geringere statistische Macht zum Nachweis tatsächlich bestehender,kausaler Zusammenhänge verfügen.
Das gängigste Verfahren zur nicht-parametrischen Kopplungsanalyse be-steht in dem einfachen, voraussetzungsfreien Vergleich der genetischen Aus-stattung von Paaren eng verwandter Patienten. Die Idee, die dieser einfachenund unmittelbar einleuchtenden Methode zu Grunde liegt, geht auf einen 1935erschienenen Artikel des britischen Humangenetikers L.S. Penrose zurück12.Danach erben Vollgeschwister an einem autosomalen Genlocus entweder zwei,ein oder kein Allel in identischer Linie von ihren Eltern (Abb. 3). Im englischenSprachgebrauch wird diese Art der Übereinstimmung geschwisterlicher Alleleals »identity-by-descent« (»ibd«) bezeichnet, und der Begriff hat sich auch inder deutschen Fachwelt eingebürgert. Jeder der drei möglichen ibd-Zustände,
40 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 40

Genetische Epidemiologie
abgekürzt durch k=0, k=1 oder k=2, hat eine bestimmte Häufigkeit zk, und unterder Hypothese, dass zwischen Markergen und Erkrankung kein ätiologischerZusammenhang besteht, folgt die Vererbung der Markerallele allein den Men-delschen Regeln. Dies würde bedeuten, dass z0=1/4, z1=1/2 und z2=1/4 gilt, undzwar unabhängig davon, ob tatsächlich beide Geschwister erkrankt sind odernur eines von beiden. Jeder statistische Test, der auf eine Abweichung der beob-achteten ibd-Häufigkeiten von diesem Verhältnis gerichtet ist, repräsentierteinen Test bezüglich Kopplung des Markergens mit mutmaßlichen Krankheits-genen.
In seiner ursprünglichen Form verlangte der ibd-Test, dass der ibd-Statusam Markergen für alle Geschwisterpaare eindeutig bekannt ist. Dies setzt imAllgemeinen die genetische Untersuchung der Eltern voraus. Bei später Mani-festation, wie sie gerade für viele komplexe Erkrankungen typisch ist, lässt sichdiese Voraussetzung aber oft nur schwer erfüllen. Aus diesem Grund sind inletzter Zeit eine Vielzahl von Weiterentwicklungen des ibd-Tests vorgenommenworden, die den ibd-Status des erkrankten Geschwisterpaares indirekt aus gene-tischer Information über deren Verwandte rekonstruieren, die weitläufigereVerwandtschaften als nur Vollgeschwisterschaft zulassen, oder die allein aufdem identity-by-state (»ibs«) Status der geschwisterlichen Allele basieren. ImAllgemeinen sind diese Methoden weniger aussagekräftig als der Originaltest,doch lässt sich durch sie wertvolle Information in die Kopplungsanalyse einbe-ziehen, die anderweitig nicht genutzt werden könnte.
41 Christiana Albertina 56 2003
Abbildung 3
Mögliche Anzahl k der
Allele eines autosoma-
len Genlocus, die zwei
Geschwister „identical-
by-descent“ (ibd) geerbt
haben können. Für
jeden Wert von k (k=0,
k=1, oder k=2) sind die
ibd Allele beim zweiten
Geschwister rot mar-
kiert.
Quantitative Phänotypen
Nicht-parametrische Verfahren wurden auch für die Kopplungsanalysequantitativer Phänotypen entwickelt. Sie sind im Allgemeinen effizienter alsMethoden, die aus den ursprünglich quantitativen Merkmalen diskreteZustände ableiten (z.B. »normal« versus »erhöht«), da durch solcheTransformationen im Allgemeinen wichtige Information verloren geht. Diegrundsätzliche Idee der Kartierung ursächlicher Genveränderungen ist jedoch
CA 56 Heft 31.07.2007 7:43 Uhr Seite 41

Michael Krawczak
für quantitative und qualitative Merkmale die gleiche: Der am Markergen beob-achtete ibd-Status von Verwandten wird mit ihrer Ähnlichkeit bezüglich desPhänotyps in Beziehung gesetzt. Wenn das Markergen mit dem Merkmalsgengekoppelt ist, dann sollten Geschwister mit ähnlicher Merkmalsausprägung zueinem höheren ibd-Status am Markergen neigen, wohingegen für Geschwistermit deutlich unterschiedlichem Phänotyp das Gegenteil der Fall sein müsste.Ein formaler statistischer Test zum Nachweis dieses Effekts wurde 1972 vonden amerikanischen Statistikern J.K. Haseman und R.C. Elston entwickelt13. Fürjedes Geschwisterpaar wird neben ihrem ibd-Status k am Markergen auch dasQuadrat Y der Differenz ihrer phänotypischen Werte ermittelt. Eine einfache line-are Regressionsanalyse von Y und k vermag dann zu klären, ob die beiden Werteüberhaupt miteinander korreliert sind und, falls dies der Fall ist, ob die Art derKorrelation einen biologisch plausiblen Hinweis auf chromosomale Kopplungvon Merkmalsgen und Markergen liefert. Auch diese »Haseman-Elston Methode«wurde zwischenzeitlich mehrfach weiterentwickelt, um ihre Aussagekraft hin-sichtlich des Vorliegens von Kopplung zu erhöhen.
Eine alternative, wenngleich konzeptionell ähnliche Methode zur Kartierungquantitativer Merkmalsgene ist die »Varianzkomponentenanalyse«, die auf dieZerlegung der Variation eines quantitativen Merkmals in jene Anteile abzielt,die auf der Wirkung von gekoppelten bzw. nicht gekoppelten Genen beruhen.Auch dieses Verfahren geht davon aus, dass bei enger Kopplung zwischenMerkmalsgen und Markergen die phänotypische Ähnlichkeit zwischen Ver-wandten positiv mit deren ibd-Status am Markergen zusammenhängen sollte.Ein solcher Zusammenhang drückt sich dann unmittelbar in höherenSchätzwerten für die entsprechenden Varianzkomponenten aus.
Kopplungsungleichgewicht
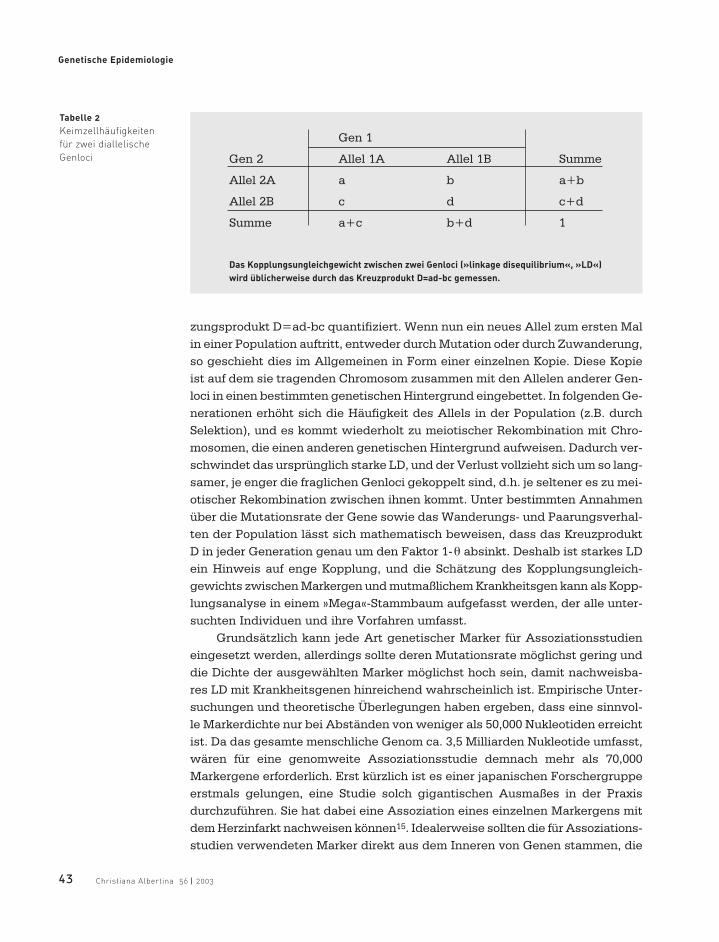
Auf biochemischer Ebene bemisst sich der Abstand zwischen zwei Genendurch die Anzahl der zwischen ihnen liegenden Nukleotide, d.h. der elementa-ren informationstragenden Bausteine A, T, G und C eines Chromosoms.Abstände von 1 Million Nukleotide gelten bei der Genkartierung als Untergrenzedes Auflösungsvermögens, das mit familienbasierten Kopplungsanalysenerreichbar ist. Eine präzisere Lokalisation von Krankheitsgenen lässt sich nurdurch populationsbasierte Assoziationsstudien erreichen, bei denen dasVorliegen eines sogenannten »Kopplungsungleichgewichts« (englisch »linkagedisequilibrium«, »LD«) zwischen Genen ausgenutzt wird14. Die gebräuchlichsteDefinition von LD besagt, dass sich zwei gekoppelte Genloci dann im Kopp-lungsungleichgewicht befinden, wenn einige ihrer Allele in den Keimzellen, diedie Mitglieder einer bestimmten Population produzieren, überzufällig häufiggemeinsam auftreten. Im einfachsten Fall zweier Loci mit je zwei Allelen las-sen sich die Keimzellhäufigkeiten a, b, c und d in einer 2x2 Tabelle zusammen-stellen (Tabelle 2). Anhand dieser Tabelle wird das LD dann durch das Kreu-
42 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 42
Genetische Epidemiologie
zungsprodukt D=ad-bc quantifiziert. Wenn nun ein neues Allel zum ersten Malin einer Population auftritt, entweder durch Mutation oder durch Zuwanderung,so geschieht dies im Allgemeinen in Form einer einzelnen Kopie. Diese Kopieist auf dem sie tragenden Chromosom zusammen mit den Allelen anderer Gen-loci in einen bestimmten genetischen Hintergrund eingebettet. In folgenden Ge-nerationen erhöht sich die Häufigkeit des Allels in der Population (z.B. durchSelektion), und es kommt wiederholt zu meiotischer Rekombination mit Chro-mosomen, die einen anderen genetischen Hintergrund aufweisen. Dadurch ver-schwindet das ursprünglich starke LD, und der Verlust vollzieht sich um so lang-samer, je enger die fraglichen Genloci gekoppelt sind, d.h. je seltener es zu mei-otischer Rekombination zwischen ihnen kommt. Unter bestimmten Annahmenüber die Mutationsrate der Gene sowie das Wanderungs- und Paarungsverhal-ten der Population lässt sich mathematisch beweisen, dass das KreuzproduktD in jeder Generation genau um den Faktor 1-θ absinkt. Deshalb ist starkes LDein Hinweis auf enge Kopplung, und die Schätzung des Kopplungsungleich-gewichts zwischen Markergen und mutmaßlichem Krankheitsgen kann als Kopp-lungsanalyse in einem »Mega«-Stammbaum aufgefasst werden, der alle unter-suchten Individuen und ihre Vorfahren umfasst.
Grundsätzlich kann jede Art genetischer Marker für Assoziationsstudieneingesetzt werden, allerdings sollte deren Mutationsrate möglichst gering unddie Dichte der ausgewählten Marker möglichst hoch sein, damit nachweisba-res LD mit Krankheitsgenen hinreichend wahrscheinlich ist. Empirische Unter-suchungen und theoretische Überlegungen haben ergeben, dass eine sinnvol-le Markerdichte nur bei Abständen von weniger als 50,000 Nukleotiden erreichtist. Da das gesamte menschliche Genom ca. 3,5 Milliarden Nukleotide umfasst,wären für eine genomweite Assoziationsstudie demnach mehr als 70,000Markergene erforderlich. Erst kürzlich ist es einer japanischen Forschergruppeerstmals gelungen, eine Studie solch gigantischen Ausmaßes in der Praxisdurchzuführen. Sie hat dabei eine Assoziation eines einzelnen Markergens mitdem Herzinfarkt nachweisen können15. Idealerweise sollten die für Assoziations-studien verwendeten Marker direkt aus dem Inneren von Genen stammen, die
43 Christiana Albertina 56 2003
Tabelle 2
Keimzellhäufigkeiten
für zwei diallelische
Genloci
Gen 1
Gen 2 Allel 1A Allel 1B Summe
Allel 2A a b a+b
Allel 2B c d c+d
Summe a+c b+d 1
Das Kopplungsungleichgewicht zwischen zwei Genloci (»linkage disequilibrium«, »LD«)
wird üblicherweise durch das Kreuzprodukt D=ad-bc gemessen.
CA 56 Heft 31.07.2007 7:43 Uhr Seite 43

Michael Krawczak
biologisch plausible Kandidaten für einen ätiologischen Zusammenhang mit derin Frage stehenden Erkrankung darstellen. Die Wahrscheinlichkeit für denNachweis einer Assoziation ließe sich dann noch weiter erhöhen, wenn die zuuntersuchenden Markerallele selbst funktionell signifikant wären, indem sie z.B.das Proteinprodukt oder regulatorische Elemente eines Gens verändern.
Familien-basierte Assoziationsstudien
Die einfachste Form der populationsbasierten Assoziationsstudie ist dasFall-Kontroll-Design, wie es auch in der klassischen Epidemiologie weit ver-breitete Anwendung findet. Durch den Vergleich der Häufigkeit bestimmterMarkerallele bei Patienten und bei gesunden Kontrollpersonen lassen sich dierelativen Erkrankungsrisiken abschätzen, die von diesen genetischen Einfluss-faktoren ausgehen. Dessen ungeachtet wurden aber immer wieder Bedenkengeäußert, dass genetische Unterschiede zwischen ethnischen, sozialen oder geo-graphischen Schichten in der zu untersuchenden Population zu einer Verfäl-schung der Risikoschätzungen führen könnten. Wenn Patienten und Kontroll-personen systematisch aus unterschiedlichen Schichten rekrutiert würden, könn-te dies in der Tat zu genetischen Unterschieden zwischen den Stichproben füh-ren, die dann aber nicht mit der Krankheit im Zusammenhang stünden. Um die-ses Problem zu lösen, haben genetische Epidemiologen geeignete Designs für
44 Christiana Albertina 56 2003
Abbildung 4
Test auf Vererbungsun-
gleichgewicht (englisch
„transmission disequili-
brium test“, „TDT“) für
diallelische Marker-
gene. Der TDT wird auf
Kernfamilien ange-
wandt, die die beiden
Eltern und ein betroffe-
nes Kind umfassen. Bei
der Bewertung der Ver-
erbung werden nur he-
terozygote Eltern be-
rücksichtigt (Zellen x
und y in der dargestell-
ten Tabelle). Die im TDT
errechnete Testgröße
(x-y)2/(x+y) wird mit ent-
sprechenden Statistik-
tabellen verglichen, um
zu beurteilen, ob in den
untersuchten Familien
eine signifikante Krank-
heitsassoziation vor-
liegt.
CA 56 Heft 31.07.2007 7:43 Uhr Seite 44

Genetische Epidemiologie
familienbasierte Assoziationsstudien entwickelt, von denen der Test auf Ver-erbungsungleichgewicht (englisch »transmission disequilibrium test«, »TDT«)das am weitesten verbreitete ist16. Ein TDT dient, wie der Name bereits sagt,dem Nachweis der bevorzugten Vererbung spezieller Markerallele von Elternauf ihre erkrankten Kinder (Abb. 4). Hierbei können allerdings nur Eltern berück-sichtigt werden, die heterozygot für das Markergen sind, d.h. auf den beidenhomologen Chromosomen unterschiedliche Allele aufweisen. Jede Abweichungdes Verhältnisses zwischen Vererbung und Nicht-Vererbung eines Allels vondem erwarteten Quotienten 1:1 deutet sowohl auf Kopplung als auch auf LDzwischen Marker- und Krankheitsgen hin.
Da beim TDT und bei ähnlichen Tests die Chromosomen enger Verwandtervon Patienten als interne Kontrollen fungieren, wurde es fast schon zum Para-digma der genetischen Epidemiologie komplexer Erkrankungen, dass familien-basierte Assoziationsstudien dem Fall-Kontroll-Design überlegen seien. Fami-lienbasierte Verfahren haben jedoch auch Nachteile, die nicht immer durch ihreoffensichtliche Robustheit gegenüber Populationsschichtung ausgeglichenwerden. So können z.B. Gen-Umwelt-Interaktionen in familienbasierten Studienüberhaupt nicht analysiert werden, da keine authentischen Kontrollen für einenVergleich mit Patienten zur Verfügung stehen (d.h. unverwandte Personen, dieden gleichen Umwelteinflüssen ausgesetzt sind wie die Patienten). Darüber hin-aus ist für den TDT die Kenntnis der elterlichen Genotypen erforderlich, was fürspät sich manifestierende Krankheiten in der Regel nicht oder nur schwer zuerreichen ist. Es wurden zwar statistische Verfahren entwickelt, die in solchenSituationen die genetische Information von anderen Familienmitgliedern zurRekonstruktion der elterlichen Genotypen heranziehen; diese Methoden sindjedoch kostspielig, ungenau und ineffizient. Auf der anderen Seite kann einermöglichen Verfälschung von Schätzungen relativer Risiken in genetischen Fall-Kontroll-Studien dadurch vorgebeugt werden, dass bei der Auswahl der Kon-trollpersonen sorgfältig auf eine Vergleichbarkeit hinsichtlich der ethnischen,sozialen und geographischen Herkunft der Patienten geachtet wird. Zudem kanndas Ausmaß an verbleibender Schichtung in einer konkreten Studie dadurchermittelt werden, dass eine hinreichend große Anzahl zusätzlicher Markergeneanalysiert wird, die nicht selbst Teil der eigentlichen Assoziationsstudie sind.Anzumerken bleibt schließlich auch noch, dass Populationsschichtung zwar einetheoretische Möglichkeit darstellt, dass aber eindeutige empirische Belege fürihre praktische Bedeutsamkeit bislang fehlen.
Klinische Relevanz
Leider wird die genetische Epidemiologie komplexer Erkrankungen nochimmer von einem offenkundigen Mangel an durchschlagenden Erfolgen geplagt,und die Gründe hierfür sind vielfältig. Ein besonders kritischer Faktor für dieAussagekraft genetisch-epidemiologischer Studien ist die genetische Hetero-
45 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 45

Michael Krawczak
genität der jeweils zu untersuchenden Studienkollektive. Genetische Hetero-genität kann auf zwei Ebenen auftreten, entweder in Form »allelischer He-terogenität« zwischen den Varianten eines bestimmten Gens oder als »Locus-Heterogenität« hinsichtlich der Art und Lokalisation verschiedener ursächlichbeteiligter Gene. Um die Aussagekraft genetischer Studien komplexer Erkran-kungen zu erhöhen, besteht daher ein vorrangiges Ziel der Planung und kom-petenten Durchführung solcher Studien in der Minimierung der genetischenHeterogenität auf allen Ebenen17. Zuvorderst verlangt dies die sinnvolle undwohl überlegte Auswahl der zu analysierenden Population. Idealerweise soll-ten für den Nachweis der Assoziation zwischen einem Krankheits- und einemMarkergen alle prädisponierenden Allele des Krankheitsgens direkte Nachfahreneiner einzelnen Vorläufermutation sein. Ansonsten ist es sehr unwahrschein-lich, dass alle prädisponierenden Allele den gleichen genetischen Hintergrundaufweisen, was jedoch eine Voraussetzung für das Vorliegen hinreichend star-ken LDs zum Markergen ist. Die am besten für die Analyse von Krankheitsasso-ziation geeigneten Populationen sind deshalb jene, die über einen langen Zeit-raum ihrer Geschichte klein und isoliert gewesen sind, oder die erst vor kurzerZeit durch rasche Ausbreitung aus einer kleinen Anzahl von Gründern hervor-gegangen sind. Es überrascht deshalb nicht, dass viele der bisherigen Erfolgeder genetischen Epidemiologie in Finnland und in einigen endogamen Ge-meinschaften in den USA (z.B. Amish, Hutterer oder Ashkenazim) erzielt wur-den. Neben der Berücksichtigung populationsgenetischer Gegebenheiten kanndie genetische Heterogenität eines Studienkollektivs auch dadurch reduziertwerden, dass das zu untersuchende Merkmal geeignet definiert wird. So kannes unter Umständen sinnvoll sein, die genetische Analyse getrennt nach Sub-typen einer Erkrankung durchzuführen oder Kovariate zu benutzen, durch diesich ätiologisch homogene Untergruppen des Kollektivs eingrenzen lassen. Indiesem Zusammenhang erweist sich die genetische Epidemiologie als eine sichpermanent weiterentwickelnde wissenschaftliche Disziplin, so dass auch vonder Entwicklung neuer analytischer Werkzeuge (z.B. multilokale Statistik, Zeit-reihenanalyse) eine Erhöhung der Aussagekraft genetischer Studien erwartetwerden kann (wie Anm. 7, Seite 49).
Komplexe Erkrankungen sind typischer Weise häufig und stellen eine nichtunerhebliche wirtschaftliche Belastung der nationalen Gesundheitssysteme dar.Das daraus resultierende öffentliche Interesse an einer verbesserten Diagnose,Therapie und Prävention macht Krankheiten wie z.B. Krebs, Herzinfarkt undDiabetes besonders attraktiv für die genetische Forschung. Dies führt dazu, dasssowohl auf nationaler wie auf internationaler Ebene oft eine große Anzahl ent-sprechender Studien parallel durchgeführt werden. Auf der anderen Seite istaus den oben genannten Gründen die Wahrscheinlichkeit für eine erfolgreicheKartierung und Charakterisierung kausaler Genveränderungen bei komplexenErkrankungen relativ gering. Auch bei höchsten Anforderungen an die statisti-sche Signifikanz werden daher die meisten positiven Resultate von Genkar-
46 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 46

Genetische Epidemiologie
tierungsstudien falsch sein. Dies hat zur Folge, dass die genetisch-epidemiolo-gische Forschung auch in Zukunft fast zwangsläufig zur Erzeugung falsch posi-tiver Ergebnisse getrieben wird. Meldungen über die erfolgreiche Aufklärungder genetischen Ursachen einer komplexen Erkrankung sollten deshalb immermit einer gewissen Vorsicht beurteilt werden und solange als vorläufig gelten,bis sie anhand unabhängiger Wiederholung der entsprechenden Studien, Meta-Analysen oder Laborversuche bestätigt wurden.
Genetische Epidemiologie in Deutschland
Letztlich kann die Aufdeckung der genetischen Ursachen von Erkrankungendes Menschen nur durch Untersuchungen am Menschen selbst gelingen. Es istsehr unwahrscheinlich, dass funktionelle Genomforschung an Modellorganismenallein die Vielzahl komplizierter Verknüpfungen aufdecken wird, durch die huma-ne genetische Variation in phänotypische Variation übersetzt wird. Zudem wer-den die allermeisten Geneffekte durch Umweltfaktoren initiiert, modifiziert oderinhibiert, was sich in Laborexperimenten nur unzureichend oder gar nicht nach-stellen lässt. Die enorme Bedeutung eines detaillierten Verständnisses der Ätio-logie komplexer Erkrankungen für die Medizin gebietet daher, dass die gene-tische Epidemiologie – in Verbindung mit humaner Populationsgenetik – inDeutschland als integraler Teil der nationalen Humangenom-Forschung fest eta-bliert wird. »Postgenom«-Forschung am Menschen ist national und internationalhoch kompetitiv, und die genetische Epidemiologie gilt zunehmend als eine deressentiellen Voraussetzungen dafür, dass langfristige Forschungsanstrengungenauf diesem Gebiet erfolgreich sein werden18.
Die für populations- und patientenbasierende Genomforschung notwendi-gen Qualifikationen sind vielfältig. Ihre Aneignung gestaltet sich jedoch fürStudierende und Nachwuchswissenschaftler in Deutschland immer noch alsschwierig. Eine Ausbildung in genetischer Epidemiologie ist derzeit nur punk-tuell und im wesentlichen erst auf Postgraduierten-Niveau möglich, währenddie Populationsgenetik fast ausschließlich eine Domäne der Forensik und dernichthumanen bzw. landwirtschaftlichen Biowissenschaften ist. Diese Um-stände haben u.a. dazu geführt, dass in Deutschland derzeit nur eine geringeAnzahl hinreichend qualifizierter Wissenschaftler zur Verfügung steht, um Pro-jekte der populations- und patientenbasierenden Genomforschung problemge-recht zu planen und kompetent analytisch und logistisch zu begleiten. Vergleichtman dies mit dem erstklassigen labortechnischen Entwicklungsstand der deut-schen Human-Genomforschung, so wird ein krasses Missverhältnis hinsichtlichpersoneller und finanzieller Ausstattung auf Seiten der theoretischen Wissen-schaften augenfällig. Angesichts der relativ bescheidenen Präsenz und dergleichzeitig dramatisch wachsenden Anforderungen, die z.B. die vor zwei Jahrenerfolgte Gründung des Nationalen Genomforschungsnetzes (NGFN)19 an diegenetische Epidemiologie in Deutschland stellen wird, sollte so schnell wie mög-
47 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 47

Michael Krawczak geb. 1959 in Göttingen; 1977
bis 1984 Mathematik-Studium in Göttingen;
dort bis 1991 wissenschaftlicher Mitarbeiter im
Institut für Humangenetik; 1987 Promotion zum
Doctor rerum naturalium; 1991-1994 wissen-
schaftlicher Mitarbeiter im Institut für
Humangenetik der Medizinischen Hochschule
Hannover; 1994 Habilitation zum Thema
»Molekulare Grundlagen und medizinische
Relevanz genetischer Variabilität«; 1996
Wechsel an das University of Wales College of
Medicine, Cardiff, zur Wahrnehmung eines
Heisenberg-Stipendiums; 1998 dort Ernennung
zum »Visiting Professor of Mathematical
Genetics«; seit April 2002 C4-Professur für
Medizinische Informatik und Statistik an der
CAU; Arbeitsschwerpunkte auf dem Gebiet der
Genetischen Epidemiologie und der
Populationsgenetik; Leiter eines Teilprojektes
des Nationalen Genomforschungsnetzes
Prof. Dr. Michael Krawczak
Institut für Medizinische Informatik und
Statistik
Universitätsklinikum Schleswig-Holstein
Brunswiker Str. 10, 24105 Kiel
Michael Krawczak
lich eine Möglichkeit geschaffen werden, Nachwuchs auf diesem Fachgebietsystematisch und kompetent auszubilden.
Auf Initiative entsprechend interessierter Epidemiologen und Biometrikerwurde im Herbst 2001 in Deutschland die Einrichtung sogenannter Genetisch-Epidemiologischer Methodenzentren (GEM) angeregt. Für deren materielle Aus-stattung konnten erfolgreich Finanzmittel im Rahmen des NGFN eingeworbenwerden. Den GEM, die an den Standorten München, Marburg, Göttingen, Bonn,Lübeck und Kiel eingerichtet wurden, obliegt zukünftig die zentrale Koordinationvon Planung, Dokumentation und Auswertung genetischer Studien im Rahmendes Genomforschungsnetzes. Der Verfasser des vorliegenden Artikels hat sichan dieser Initiative beteiligt, so dass auch die Medizinische Fakultät der Chris-tian-Albrecht-Universität als Standort für die Einrichtung eines GEM ausgewähltwurde. Neben seiner wissenschaftlichen Funktion wird diese Einrichtung mittel-fristig auch als Basis dafür dienen, eine systematische studentische Ausbildungin Genetischer Epidemiologie und Populationsgenetik in Form eines interna-tionalen Masterstudiengangs an der CAU Kiel zu etablieren.
Danksagung
Der Autor dankt Frau Petra Neumann, Institut für Medizinische Informatik und Statistik, CAU Kiel,
für ihre geduldige und bereitwillige Hilfe bei der Anfertigung des vorliegenden Artikels.
48 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 48

Genetische Epidemiologie
Anmerkungen
1 Morton NE (1982) Outline of Genetic Epide-
miology. Karger, Basel.
2 Collins FS, Patrinos A, Jordan E, Chakravarti
A, Gesteland R, Walters L (1998) New goals for
the U.S. Human Genome Project: 1998–2003.
Science 282: 682–689.
3 Schork NJ, Cardon LR, Xu X (1998) The future
of genetic epidemiology. Trends in Genetics 14:
266-272.
4 Online Mendelian Inheritance in Man
(http://www3.ncbi.nlm.nih.gov/omim/) und The
Human Gene Mutation Database
(http://www.hgmd.org/).
5 Siehe z.B. Collins FS (1995) Positional cloning
moves from perditional to traditional. Nature
Genetics 9: 347–350.
6 Siehe z.B. Vogel F, Motulsky AG (1996) Human
Genetics. Problems and approaches (3rd Edi-
tion). Springer Verlag, Berlin.
7 Haldane JBS (1919) The combination of linka-
ge values and the calculation of distance be-
tween the loci of linkage factors. Journal of
Genetics 8: 299-309.
8 Ott J (1999) Analysis of Human Genetic Link-
age. Johns Hopkins University Press, Baltimore.
9 Morton NE (1953) Sequential tests for the
detection of linkage. American Journal of
Human Genetics 7: 277-318
10 Risch NJ (2000) Searching for genetic deter-
minants in the new millenium. Nature 405:
847-856.
11 Siehe z.B. Sham P (2001) Shifting paradigms
in gene-mapping methodology for complex
traits. Pharmacogenomics 2: 195-202.
12 Penrose LS (1935) The detection of autoso-
mal linkage in data which consist of pairs of
brothers and sisters of unspecified parentage.
Annals of Eugenics 6: 133-138.
13 Haseman JK, Elston RC (1972) The investiga-
tion of linkage between a quantitative trait and
a marker locus. Behavioral Genetics 2: 3-19.
14 Siehe z.B. Elston RC (1998) Linkage and
association. Genetic Epidemiol 15: 565-576.
15 Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada
R, Tsunoda T, Sato H, Sato H, Hori M, Naka-
mura Y, Tanaka T (2002) Functional SNPs in the
lymphotoxin-a gene that are associated with
susceptibility to myocardial infarction. Nature
Genetics 32: 650-654.
16 Spielman R, McGinnis R, Ewens W (1993)
Transmission test for linkage disequilibrium:
The insulin gene region and insulin-dependant
diabetes mellitus (IDDM). American Journal of
Human Genetics 52: 506-516.
17 Siehe z.B. Terwilliger JD, Göring HHH (2000)
Gene mapping in the 20th and 21st centuries:
statistical methods, data analysis, and experi-
mental design. Human Biology 72: 63-132.
18 Vogel W (2000) Genetische Epidemiologie
oder zur Spezifität von Subdisziplinen der
Humangenetik. Medizinische Genetik 12: 395–
399.
19 Nationales Genomforschungsnetz
(http://www.rzpd.de/ngfn/)
49 Christiana Albertina 56 2003
Glossar
Allel Alternative Erscheinungsform eines Gens.
Allele besetzen denselben Genort, unterschei-
den sich aber hinsichtlich ihrer DNA-Sequenz.
Centromer Für die Zellteilung wichtiger Struk-
turbereich eines Chromosom.
Crossing-over Vorgang des Bruches und der re-
ziproken Reparatur eines homologen Chromo-
somenpaars, der zum Austausch von geneti-
schem Material zwischen den beteiligten Chro-
mosomen führt (Rekombination).
DNA Desoxyribonukleinsäure (englisch »acid«);
doppelsträngiges Molekül, das in der Abfolge
seiner Primärbausteine (Nukleotide) die Erbin-
formation eines Organismus trägt.
Gen Funktioneller Abschnitt einer DNA-Se-
quenz wie z.B. der Bauplan für ein Protein.
Genotyp Genetische Information, die in einem
einzelnen Locus, in mehreren Loci oder im Ge-
samtgenom enthalten ist.
Genetische Distanz Linearer Abstand zwischen
Genen auf einer Genkarte, gemessen in
Morgan (=100 centi-Morgan); eine genetische
Distanz von 1 centi-Morgan zwischen zwei
Genloci entspricht der Erwartung, dass die Loci
durchschnittlich einmal unter 100 Meiosen eine
Rekombination erfahren.
Heterozygotie Anwesenheit zweier verschiede-
ner Allele eines bestimmten Gens in einer
diploiden Zelle.
CA 56 Heft 31.07.2007 7:43 Uhr Seite 49

Michael Krawczak
Homozygotie Anwesenheit zweier identischer
Allele eines bestimmten Gens in einer diploiden
Zelle.
Locus Physikalische Position eines Gens auf
einem Chromosom oder einer Genkarte.
Kartierungsfunktion Mathematische Funktion,
die die Rekombinationsrate zwischen zwei Loci
in ein additives Abstandsmaß transformiert.
Kopplung Tendenz zur gemeinsamen Vererbung
von Allelen von Genen, die auf dem gleichen
Chromosom lokalisiert sind; Genloci mit einer
paarweisen Rekombinationsrate von weniger
als 50% werden als »gekoppelt« bezeichnet.
Kopplungsungleichgewicht (»linkage disequili-
brium«, »LD«) Überzufälliges gemeinsames
Auftreten von Allelen verschiedener Gene in
den Keimzellen, die von Mitgliedern einer
Population produziert werden.
Likelihood Maß für die Neigung, aufgrund vor-
liegender Beobachtungen eine bestimmte Hy-
pothese H hinsichtlich des Zustandekommens
der beobachteten Daten D als wahr zu akzep-
tieren. In der Statistik wird die Likelihood
L(H|D) mit der bedingten Wahrscheinlichkeit
P(D|H) der Daten gleichgesetzt.
lod score Allgemein der dekadische Logarith-
mus des Verhältnisses zweier Wahrscheinlich-
keiten oder Chancen; in der genetischen Kopp-
lungsanalyse quantifiziert der »lod score« den
empirischen Hinweis auf die Kopplung zweier
Genloci.
Marker DNA-Sequenz, deren Lage im Genom
bekannt ist.
Meiose Die beiden abschließenden Zellteilungen
der Keimzellentwicklung.
Nukleotid Baustein der Nukleinsäuren, beste-
hend aus einer Base (Adenin, Thymin bzw.
Uracil, Guanin, oder Cytosin), einem Zucker-
molekül und einer Phosphatgruppe.
Penetranz Anteil von Individuen mit einem prä-
disponierenden Genotyp, bei denen sich dieser
Genotyp durch das in Frage stehende Merkmal
(z.B. eine Erkrankung) manifestiert.
Phänotyp Die beobachtbaren (»äußeren«)
Merkmale eines Organismus, welche aus dem
Zusammenspiel von Umwelt und genetischen
Faktoren resultieren.
Rekombination Im Zuge der Vererbung von
einem Elternteil auf ein Kind stattfindender
Austausch von genetischem Material zwischen
den beiden großelterlichen Chromosomen.
50 Christiana Albertina 56 2003
CA 56 Heft 31.07.2007 7:43 Uhr Seite 50























![Perkutane Koronarintervention (PCI) und ... · Patientenbefragung 01 : Häufigkeit von Symptomen der Koronaren Herzkrankheit (KHK) vor und nach einer ... (Levitt et al. 2013 [III])](https://img.pdfslide.org/doc/110x75/5fecf32a4ac29f6d8530ae34/perkutane-koronarintervention-pci-und-patientenbefragung-01-hufigkeit.jpg)





