Embed Size (px)
Citation preview

Klinische Bewertungg2016.07.07
Dr. Bassil AkraGlobal DirectorClinical Centre of Excellence
TÜV SÜD Product Service

Worauf steuern wir zu?
Stand Heute • MEDDEV 2 7 1 rev 3
Übergang • MEDDEV 2 7 1 rev
In naher Zukunft• MEDDEV 2 7 1 rev MEDDEV 2.7.1 rev. 3
and DirectivesMEDDEV 2.7.1 rev. 4, Directives and MDR/IVDR
MEDDEV 2.7.1 rev. 5? and MDR/IVDR
TÜV SÜD Product Service Slide 2
http://ec.europa.eu/growth/sectors/medical-devices/guidance/index_en.htm
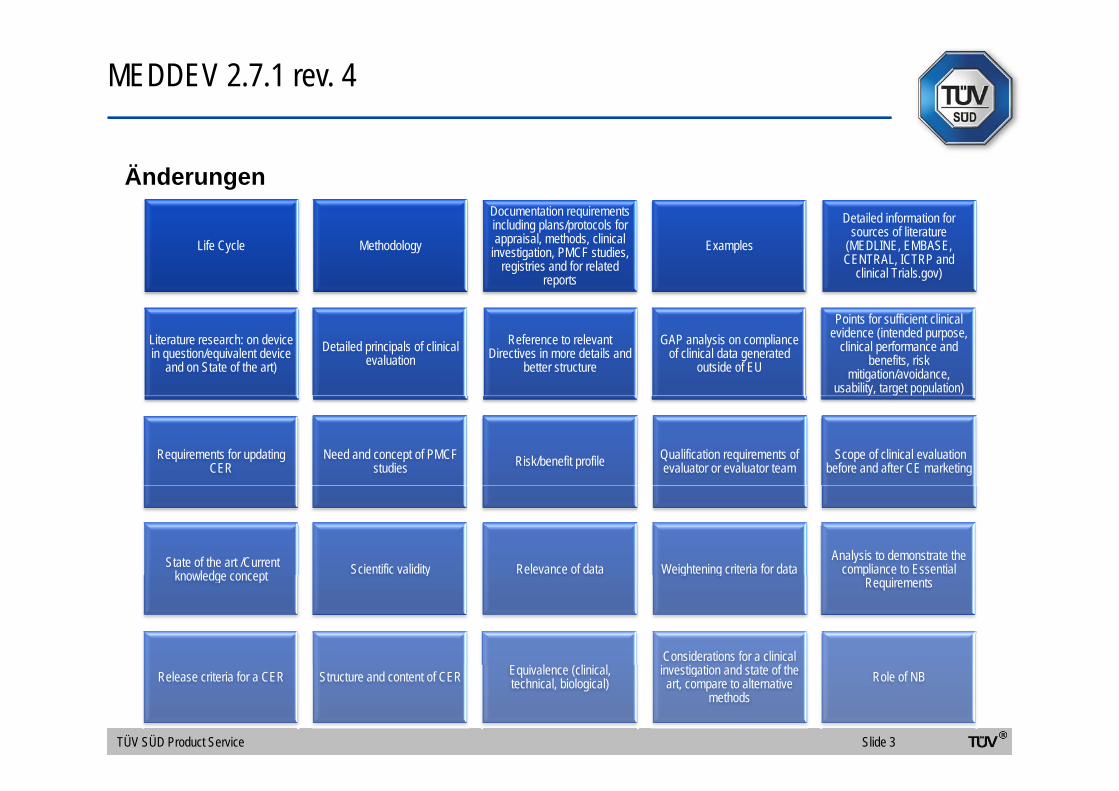
MEDDEV 2.7.1 rev. 4
ÄnderungenDocumentation requirements Detailed information for
Life Cycle Methodology
qincluding plans/protocols for appraisal, methods, clinical
investigation, PMCF studies, registries and for related
reports
Examples
Detailed information for sources of literature
(MEDLINE, EMBASE, CENTRAL, ICTRP and
clinical Trials.gov)
Literature research: on device in question/equivalent device
and on State of the art)Detailed principals of clinical
evaluationReference to relevant
Directives in more details and better structure
GAP analysis on compliance of clinical data generated
outside of EU
Points for sufficient clinical evidence (intended purpose,
clinical performance and benefits, risk
mitigation/avoidance, usability, target population)
Requirements for updating CER
Need and concept of PMCF studies Risk/benefit profile Qualification requirements of
evaluator or evaluator team Scope of clinical evaluation
before and after CE marketing
State of the art /Current knowledge concept Scientific validity Relevance of data Weightening criteria for data
Analysis to demonstrate the compliance to Essential knowledge concept y g g p
Requirements
(Considerations for a clinical
f
TÜV SÜD Product Service Slide 3
Release criteria for a CER Structure and content of CER Equivalence (clinical, technical, biological)
investigation and state of the art, compare to alternative
methodsRole of NB

Äquivalenz Verfahren in der MEDDEV 2.7.1 rev. 4
Der Nachweis der Äquivalenz wird sowohl für klinische, technische als auch biologische Eigenschaften verlangt
Für die vermutete Äquivalenz gilt: • Vergleich gilt für ein einziges weiteres Produkt• Erfüllung aller drei Charakteristika (klinisch, technisch, biologisch)• Kein klinisch signifikanter Unterschied bei der Sicherheit undg
Leistungsfähigkeit des Medizinproduktes• Die Unterschiede zwischen dem zu begutachtetem Medizinprodukt
und dem vermuteten Äquivalenzprodukt müssen identifiziert,vollständig offengelegt, und ausgewertet werden
• Spezielle Fertigungsverfahren (z.B. Oberflächenbehandlung,Herstellungs-prozesse zur Änderung der Materialeigenschaften)
TÜV SÜD Product Service Slide 4
• Wenn möglich, klinisch relevante Spezifikationen und Eigenschaftendes Medizinprodukts und des Äquivalenzproduktes messen.

MEDDEV 2.7.1 rev. 4
Demonstration der Äquivalenz
biologischbiologischVerwendung der selben Materialien oder Substanzen welche mit Verwendung der selben Materialien oder Substanzen welche mit dem selben Gewebe oder Körperflüssigkeiten in Kontakt stehen
A h kö fü G ät äh t d l h it i t kt Ausnahmen können für Geräte gewährt werden, welche mit intakter Haut in Kontakt stehen und nur für Nebenkomponenten.
In diesen Fällen kann basierend auf Risikoanalysen die Verwendung ähnlicher Materialien unter Berücksichtigung ihrer Funktion und Beschaffenheit erlaubt sein
TÜV SÜD Product Service Slide 5

MEDDEV 2.7.1 rev. 4
Beispiele für Studien mit fehlender wissenschaftlicher Validität zurDemonstration der adäquaten klinischen Leistung und / oder klinischenSicherheit
Fehlende Information zu elementaren Aspekten
Zu geringe Fallzahlen für den Nachweis statistischer Signifikanz
Verwendung ungeeigneter statistischer Methoden
Fehlen adequater Kontrollgruppen
Unsachgemäße Sammlung von Fallzahlen zur Mortalität und schwerwiegend unerwünschten Ereignissen
Fehlinterpretation durch die Autoren
Illegale Aktivitäten
TÜV SÜD Product Service Slide 6

MEDDEV 2.7.1 rev. 4
Wer sollte eine klinische Bewertung durchführen?
• Gerätetechnologie und ihrer Anwendung;• Forschungsmethodik (inkl Planung klinischer Bewertungen und Biostatistik)
Der Gutachter sollte Kenntnisse besitzen zu
• Forschungsmethodik (inkl. Planung klinischer Bewertungen und Biostatistik)• Diagnose und Behandlung der Erkrankung mittels des bestimmungsmäßigen Gebrauch des Gerätes,
Kenntnisse zu alternativen Behandlungsoptionen, Behandlungsstandards und Technologien (z.B. klinischeFachexperte in dem jeweiligen medizinischen Fachgebiet);
• Informationsmanagement (z.B. wissenschaftlicher Hintergrund oder Qualifikation in Bibliothekswesen;Erfahrung in Umgang mit relevanten Datenbanken wie Embase und Medline)
• Regulatorischen Anforderungen; und• Medical Writing (z.B. Dissertation in einem relevanten wissenschaftlichen Gebiet oder Medizin; Training und
Erfahrung im Schreiben medizinsicher Publikationen, systematische Review und Bewertung klinischerDaten);
• Hochschulabschluss & 5 Jahre dokumentierte Berufserfahrung• 10 Jahre dokumentierte Berufserfahrung (sollte ein Hochschulabschluss nicht gefordert sein)
Mindestanforderung an die Kenntnisse des Gutachters
TÜV SÜD Product Service Slide 7
10 Jahre dokumentierte Berufserfahrung (sollte ein Hochschulabschluss nicht gefordert sein)
Bemerkung: Unter Umständen kann der Kenntnissgrad des Gutachter geringer sein. Dies sollte dokumentiert und hinreichend begründet werden.

Aktualisierung des Clinical Evaluation Report MEDDEV 2.7.1 rev. 4
• wenn dem Hersteller neue Informationenaus den Post Market Aktivität bekanntwerden welche eine Auswirkung auf diewerden, welche eine Auswirkung auf dieaktuelle Bewertung haben;
• Wenn solche Art von Infomationen nichtbekannt ist, spätestents
Im Normalfall wird die • jährlich bei Geräten mit erheblichem
Risiko und / oder welche noch nicht aufdem Markt etabliert sind
• Alle 2 bis 5 Jahre bei Geräten mit
wird die klinische Bewertung aktualisiert: • Alle 2 bis 5 Jahre, bei Geräten mit
geringen Risikopotential und gut imMarkt etablierten
• In begründeten Fällen
aktualisiert:
g
TÜV SÜD Product Service Slide 8

Vorschlag der EU Verordnung
Der Hersteller muss den Grad des klinischen Nutzens angebenund rechtfertigen sowie den Nachweis zur Erfüllunggrundlegender Anforderungen im Hinblick auf die Sicherheit undgrundlegender Anforderungen im Hinblick auf die Sicherheit undLeistungsfähigkeit erbringen, welche für die Eigenschaften desGerätes und den bestimmungsgemäßen Gebrauch des Gerätesgeeignet sind.g g
TÜV SÜD Product Service Slide 9

GAP Analyse – MDD vs. MDR
A X 1 1
MDD• Annex X, 1.1a• Bei implantierbaren Medizinprodukten und Produkten der Klasse III sind klinische
Prüfungen durchzuführen, außer bei den Produkten für welche bereits klinischeDaten vorliegen .MDD
• (46) Um ein hohes Maß an Sicherheit und Leistungsfähigkeit zu gewährleisten mussder Nachweis der Einhaltung der allgemeinen Sicherheits- und Leistungs-anforderungen durch klinische Daten erbracht werden, für Produkte der Klasse III und
MDRanforderungen durch klinische Daten erbracht werden, für Produkte der Klasse III undimplantierbare Medizinprodukte muss der Nachweis grundsätzlich durch klinischeUntersuchungen erbracht werden, die unter der Verantwortung eines Sponsorsdurchgeführt werden, welcher sowohl der Hersteller als auch eine andere natürlicheoder juristische Person darstellen kann und die Verantwortung für die Erfüllung derMDR klinischen Bewertung trägt
TÜV SÜD Product Service Slide 10

Vorschlag der EU Verordnung
Für implantierbare Medizinprodukte und Produkte der Klasse IIIü kli i h U t h d h füh t d ß imüssen klinische Untersuchungen durchgeführt werden, außer in
begründeten Ausnahmefällen, wenn• das Medizinprodukt auf der Modifikation eines bereits durch den gleichendas Medizinprodukt auf der Modifikation eines bereits durch den gleichen
Hersteller vermarkteten Gerätes basiert.• eine Äquivalenz des modifizierte Medizinprodukt zu dem bereits von dem
Hersteller vermarkteten Gerät nachgewiesen wurde.• die klinische Bewertung des vermarkteten Medizinproduktes ausreicht um die
Konformität des modifizierten Medizinproduktes im Hinblick auf die Sicherheit undLeistungsfähigkeit zu demonstrieren.
TÜV SÜD Product Service Slide 11
*Clear contract between different device manufacturer

Vorschlag der EU Verordnung - Artikel (49) 2.aa
Der Hersteller eines Medizinproduktes bei welchem dieDer Hersteller eines Medizinproduktes, bei welchem dieÄquivalenz mit dem Produkt eines anderen Herstellersnachgewiesen wurde, kann sich auch auf Absatz 2a berufeng ,auf die Durchführung von klinischen Untersuchungenverzichten, sofern Anforderungen aus Absatz 2a und die imfolgenden genannten Bedingungen erfüllt werden:folgenden genannten Bedingungen erfüllt werden:• Beide Hersteller regeln vertraglich fest, dass der Hersteller des neuen
M di i d kt k ti i li h di t h i h D k t tiMedizinproduktes kontinuierlich die technische Dokumentation zumÄquivalenzprodukt einsehen darf.
• die ursprüngliche klinische Bewertung wurde in Übereinstimmung mit denA f d di V d d h füh tAnforderungen dieser Verordnung durchgeführt,
• Der Hersteller des neuen Medizinproduktes bringt den Nachweis.
TÜV SÜD Product Service Slide 12

Vorschlag der EU Verordnung - Artikel (49) 2.ab
• welche rechtmäßig in Verkehr gebrachtoder in Betriebe genommen wurde gemäßder Richtlinie 90/385/EEC oder der
Die Anforderung zur Durchführung der Richtlinie 90/385/EEC oder der
Richtlinie 93/42/EEC und für welche dieklinische Bewertung:
• auf ausreichenden klinischen Daten
Durchführung der klinischen Untersuchungen gemäß Absatz
basiert• in Übereinstimmung mit den jeweiligen
produktspezifischen geltenden Spezifi-k ti (C S ifi ti ) di fü
g2a gilt nicht für solche implantierbare Medizinprodukte kationen (Common Specification), die für
die klinische Bewertung des besagtenProduktes verfügbar sind
Medizinprodukte und Geräte der Klasse III:
TÜV SÜD Product Service Slide 13

Vorschlag der EU Verordnung - Artikel (49) 2.ab
Die Anforderung zur Durchführung der klinischen Untersuchungen gemäß Absatz 2a gilt nicht für solche g g gimplantierbare Medizinprodukte und Geräte der Klasse III wie: • Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen , Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen ,
Schrauben, Keile, Platten, Drähte, Stifte, Klammern oder Anschlüsse, und für solche bei denen die klinische Bewertung auf ausreichenden klinischen Daten basiert und in Übereinstimmung ist mit den jeweiligen produktspezifischen geltenden Spezifikationen die für die klinisch produktspezifischen geltenden Spezifikationen, die für die klinisch Bewertung des besagten Produktes verfügbar sind
TÜV SÜD Product Service Slide 14

Vorschlag der EU Verordnung – Scrutiny Procedure
Für Klasse III Produkte kann ein Hersteller durch eine Expertenguppe im Hinblick auf die gewählte Für Klasse III Produkte kann ein Hersteller durch eine Expertenguppe im Hinblick auf die gewählte Strategie zur klinischen Entwicklung und zu Vorschlägen für klinische Prüfungen konsultiert werden .
*
TÜV SÜD Product Service Slide 15

Vorschlag der EU Verordnung – Scrutiny Procedure
Der Hersteller muss die geäußerten Ansichten des Expertengremiums
berücksichtigen
ÜDiese Überlegungen sind der klinischen Bewertung zu dokumentieren.
Der Hersteller kann sich rechtlich nicht auf die Ansichten des Expertengremiums bei
kü fti K f ität b t f h künftigen Konformitätsbewertungsverfahren berufen.
TÜV SÜD Product Service Slide 16

Vorschlag der EU Verordnung – Aktualisierung des Sicherheitsberichts
Pro Gerät und gegeben falls pro Gerätekategorie oder Gruppe muss der Hersteller Pro Gerät und gegeben falls pro Gerätekategorie oder Gruppe, muss der Hersteller regelmäßig einen aktualisierten Sicherheitsbericht erstellen, in dem die Ergebnisse und
Schlussfolgerungen zu den gesammelten Daten aus der Post Market Überwachung gemäß Anhang IIa zusammengefasst werden und daraus resultierend eine begründete g g g g
Beschreibung aller präventiven und korrektiven Maßnahmen dargelegt wird.
Über die gesamte Lebensdauer des Medizinproduktes muss der Bericht folgende Über die gesamte Lebensdauer des Medizinproduktes muss der Bericht folgende Angaben enthalten
Absatzmenge und eine
Schlussfolgerungen zur Nutzen-Risiko-Bewertung
Die wichtigsten Ergebnisse des kliniscehn Post-Market
Follow-up Berichts
Absatzmenge und eine Schätzung zur Zahl der
Population, welche das Gerät verwendet, und soweit möglich
di N t hä fi k it d
TÜV SÜD Product Service Slide 17
Follow up Berichts die Nutzungshäufigkeit des Gerätes

Vorschlag der EU Verordnung – Aktualisierung des Sicherheitsberichts
CE k i h t CE-gekennzeichneten Medizinprodukte
• Der Bericht muss mindestens
Custom-made Devices
• Für Sonderanfertigung ist der Der Bericht muss mindestens jährlich aktualisiert werden und außer für Sonderanfertigung (Custom-
Für Sonderanfertigung ist der Bericht Teil der Dokumentation gemäß Abschnitt 2 des Annex XI.Sonderanfertigung (Custom
made), Teil der technischen Dokumentation darstellen, gemäß Annex II und IIa.
Abschnitt 2 des Annex XI.
TÜV SÜD Product Service Slide 18
g

Vorschlag der EU Verordnung – Herstellerpflichten
Hersteller von Geräten der Klasse III oder implantierbaren Hersteller von Geräten der Klasse III oder implantierbaren Medizinprodukten sollen gemäß Artikel 66a Berichte durch
Nutzung von elektronischen Systemen bei der in das Bewertungsverfahren involvierten Benannten Stelle, gemäß
Artikel 42, einreichen.
TÜV SÜD Product Service Slide 19

Vorschlag der EU Verordnung – Pflichten der Benannten Stellen
Die Benannte Stelle prüft den Bericht und fügt ihre Bewertung zu der Datenbank inklusiver der einzelnen Beschreibung aller getroffenen Maßnahmen hinzu.
Alle Berichte und die Bewertung durch die Benannten Stelle werden den zuständigen Behörden über das elektronische System zur Verfügung gestellt.
TÜV SÜD Product Service Slide 20

Vorschlag der EU Verordnung – Zusammenfassung zur Sicherhietund der klinischen Leistungsfähigkeit
Für implantierbare Medizinprodukte und Geräte der Klasse III, all andere
als custom-made oder Prüfpräparate, muss der Hersteller p p ,
eine Zusammenfassung zur Sicherheit und der klinischen Leistungsfähigkeit verfassen Leistungsfähigkeit verfassen
TÜV SÜD Product Service Slide 21
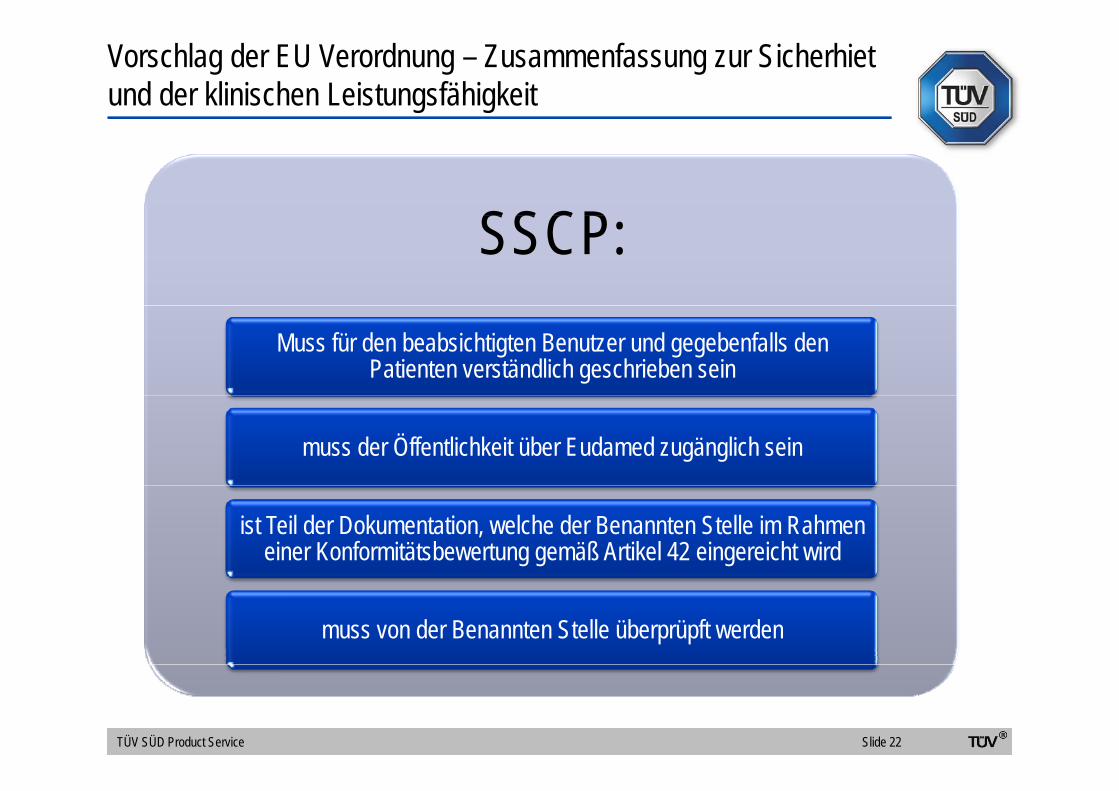
Vorschlag der EU Verordnung – Zusammenfassung zur Sicherhietund der klinischen Leistungsfähigkeit
SSCPSSCP:Muss für den beabsichtigten Benutzer und gegebenfalls den
Patienten verständlich geschrieben sein
muss der Öffentlichkeit über Eudamed zugänglich sein
ist Teil der Dokumentation, welche der Benannten Stelle im Rahmen einer Konformitätsbewertung gemäß Artikel 42 eingereicht wird
muss von der Benannten Stelle überprüpft werden
TÜV SÜD Product Service Slide 22

Vorschlag der EU Verordnung – Zusammenfassung zur Sicherhietund der klinischen Leistungsfähigkeit
Die Zusammenfassung zur Sicherheit und der klinischen Leistungsfähigkeit sollteDie Zusammenfassung zur Sicherheit und der klinischen Leistungsfähigkeit solltemindestens die folgenden Aspekte adressieren:
• Benennung des Medizinproduktes und des Hersteller, einschließlich der UDI-DI und derRegistrierungsnummerRegistrierungsnummer
• Zweckbestimmung, Einschließlich der therapeutischen Indikation und , Kontraindikation sowieZielgruppe
• Beschreibung des Gerätes, einschließlich des Verweises auf Vorgängermodell(e) oder VariationenBeschreibung des Gerätes, einschließlich des Verweises auf Vorgängermodell(e) oder Variationenwenn vorhanden, sowie eine Beschreibung der Unterschiede, des Zubehörs, und andereMedizinprodukte und nicht medizinische Geräte, die dazu bestimmt sind mit dem beschriebenenMedizinprodukt in Kombination verwendet zu werden.Mögliche diagnostische oder therapeutische Alternativen;• Mögliche diagnostische oder therapeutische Alternativen;
• Verweis auf harmonisierte Normen und gemeinsame Spezifikationen• Zusammenfassung der klinischen Bewertung gemäß Annex XIII, sowie relevante Information zu
klinischen Nachverfolgung nach Markteinführung;klinischen Nachverfolgung nach Markteinführung;• Vorschläge zu den Schulungen der Anwender und zu dem Profil;• Informationen über eventuelle Restrisiken und unerwünschte Wirkung, Warnhinweise und
Vorsichtsmaßnahmen.
TÜV SÜD Product Service Slide 23

Vorschlag der EU Verordnung – Zusammenfassung zur Sicherhietund der klinischen Leistungsfähigkeit
Für Medizinprodukte der Klasse III und implantierbare Medizinprodukte muss die
f SZusammenfassung zur Sicherheit und der klinischen Leistungsfähigkeit gemäß
A tik l 26(1) jäh li h kt li i t d Artikel 26(1) jährlich aktualisiert werden.
TÜV SÜD Product Service Slide 24

Stay informed about the expected changes and prepare for transition
Prepare for the transitionPrepare for the transition We are closely following the regulation
developments and will provide updates to developments, and will provide updates to medical devices manufacturers to help them stay informed about the transition.
For the latest information, visit the following webpages:
Medical Device Regulation: www.tuv-sud.com/mdr
Contact us today for one-stop In Vitro Diagnostic Device
Regulation:www.tuv-sud.com/ivdr
Co tact us today o o e stopquality, safety and sustainability solutions.
• www.tuv-sud.com/medicaldevice
di ld i @t d
TÜV SÜD Product Service Slide 25











![· 2015-11-23 · li2zahl Define LibPub li2zahl (liste) Func © (Ziffernliste)—> Zahl Local z,li,i li:=liste: z:=0 For i, l,dim(liste 10+1i[ 1] li:=mid(li,2) EndFor Return z EndFunc](https://img.pdfslide.org/doc/110x75/5f9948de1503ab6c6c39cfbf/2015-11-23-li2zahl-define-libpub-li2zahl-liste-func-ziffernlistea.jpg)
















