Embed Size (px)
Citation preview

974 H. Pauisen und W. Roben Liebigs Ann. Chem. 1985, 974-994
Cyclit-Reaktionen, XI1
Enantioselektive Synthese der zentralen Pseudodisaccharid- Einheit von a-Glycosidase-Inhibitoren des Oligostatin-Typs
Hans Paulsen * und Wolfgang Roben
Institut fur Organische Chemie der Universitat Hamburg, Martin-Luther-King-Platz 6, D-2000 Hamburg 13
Eingegangen am 27. August 1984
Eine Pseudodisaccharid-Einheit der a-Glycosidase-Inhibitoren des Oligostatin-Typs wurde synthetisiert. Hierzu wird zunachst eine Synthese des enantiomerenreinen Derivates des verzweig- ten Aminoinosits 26 entwickelt. Eine Verknupfung von 26 mit der reaktiven Epoxidgruppe des 1,6 : 3,4-Dianhydrozuckers 36 fuhrt zum Pseudodisaccharid 45, unter Herstellung der gewiinsch- ten Verkniipfung iiber eine Iminogruppe. Die Offnung des 1,6-Anhydroringes im Pseudo- disaccharid 46 gelingt mit Acetanhydrid/Bortrifluorid. Vom Pseudodisaccharid sind die Octa- acetate 51 und 52 der a- und p-Anomeren in reiner Form zu gewinnen. Bei Entblockierung von 51 und 52 lagert sich das freie Pseudodisaccharid spontan in den Tricyclus 53 urn, der eine Pyrroli- dinzucker-Einheit enthalt. Das Pseudodisaccharid ist in der Pyranoseform nur als blockierte Ver- bindung oder als Glycosid bestandig.
Cyclitol Reactions, XII1). - Enantioselective Synthesis of the Central Pseudodisaccharide Unit of a-Glycosidase Inhibitors of the Oligostatine Type A pseudodisaccharide unit of the a-glycosidase inhibitors of the oligostatine type has been synthe- sized. At first a synthesis of the enantiomeric pure derivative of the branched amino inositol26 has been developed. A linkage of 26 with the reactive epoxide group of the 1,6: 3,4-dianhydro sugar 36 yields the pseudodisaccharide 45, producing the desired linkage via an imino group. The opening of the 1,6-anhydro ring in the pseudodisaccharide 46 succeeds with acetanhydridel boron trifluoride. From the pseudodisaccharide the pure octaacetates 51 and 52 of the a- and b-anomers can be obtained. Deblocking of 51 and 52 to the free pseudodisaccharide causes a spontaneous rearrangement to the tricyclus 53 which contains a pyrrolidine sugar unit. In the pyranose form, the pseudodisaccharide can only exist as blocked compound or as glycoside.
Substanzen mit a-Glycosidase-Hemmung gewinnen in jungster Zeit immer starker an Bedeu- rung. Insbesondere ist durch ihre Hemmwirkung auf intestinale Glycosidhydrolasen eine Steuerung der Glucoseaufnahme und damit des Blutzuckerspiegels moglich, woraus sich ein neu- artiges Therapieprinzip bei Diabetes mellitus eroffnet.
Schmidt und Mitarbeiterz) berichteten erstmalig iiber die Isolierung von Pseudooligosacchariden aus Kulturbriihen von Stammen der Ordnung Actinomycelaks, die eine aunerordentlich starke a-Glycosidase-Hemmwirkung aufwiesen3). Die wirksamste Substanz mit vier Kohlenhydrat- Einheiten wurde als Acarbose bezeichnetz). Spater isolierten Omoto und Mitarbeiter4) aus Strep- tomyces myxogenes eine ahnliche Klasse von Oligosacchariden, die gleichfalls eine starke a-Glycosidase-Hemmung zeigte und die sie als Oligostatine bezeichneten. Weitere Pseudo-
0 V C H Verlagsgesellschaft mbH, D-6940 Weinheim, 1985 0170- 2041/85/0505 -0974 $02.50/0

Cyclit-Reaktionen, XI1 975
oligosaccharide mit vergleichbarer Struktur und einer Wirksamkeit der a-Glycosidase-Hemmung wurden anschlieBend isoliert 5 ) .
,OH
0- I L
Oligostatin C : OLigostatin D: Oligostatin E:
HO HO
Valienamin
2
m n 0 3 0 4 1 4
1 L Jn
Allen a-Glycosidase-Inhibitoren gemeinsam ist als Strukturelement eine Pseudodisaccharid- Struktur, bestehend aus einem verzweigten Cyclit-Teil rnit o-Konfiguration, der pseudo-a-N- glycosidisch an die 4-NH2-Gruppe einer 4-Amino-4,6-didesoxy-o-glucose-Einheit gebunden ist. Die Struktur der Oligostatine ist in 1 wiedergegeben. Die Acarbose unterscheidet sich von den Oligostatinen dadurch, da8 der zentrale Cyclit-Teil in 1 durch das sogenannte Valienamin6) (2) ausgetauscht wird. Dabei hat sich das Pseudotetrasaccharid mit m = 0 und n = 2 als bisher stark- ster a-Glycosidase-Inhibitor erwiesen2). Das Valienamin (2), uber dessen enantiomerenreine Synthese wir kiirzlich berichteten’), zeigt eine wenn auch erheblich schwachere a-Glycosidase- Hemmung 8). Die weiteren Kohlenhydrat-Komponenten sind somit neben dem Pseudodisaccharid fur die starke Wirkung unbedingt notwendig.
In der vorliegenden Verdffentlichung wird die Synthese einer Pseudodisaccharid- Einheit beschrieben, die der zentralen Einheit der Oligostatine entspricht. Die Reaktio- nen und die Eigenschaften dieser Pseudodisaccharid-Einheit sind von erheblichem Interesse. Das Hauptproblern der Synthese ist hierbei die Herstellung einer Verkniip- fung iiber eine Irninogruppe zwischen dern verzweigten Cyclit und einer 4-Amino- glucose-Einheit. Die Herstellung derartiger Verknupfungstypen bereitete bisher erheb- liche Schwierigkeiten. Insbesondere sei in diesern Zusarnmenhang auf die Arbeiten von Suarnig) hingewiesen, wobei von racernischen Cyclitstrukturen ausgegangen und dann im Verlauf der Synthese eine Diastereornerentrennung durchgefuhrt wird.
Am aussichtsreichsten erschien es uns, die gewunschte Verknupfungsart durch Offnung eines Epoxides rnit einern Arnin herzustellen. Voraussetzung sollte allerdings sein, da13 es sich urn ein reaktives, leicht zu offnendes Epoxid handelte. Darnit gliederte sich die Synthese in die folgenden Aufgaben: Zunachst muBte ein Amino-Derivat des verzweigten Cyclits rnit richtiger Stereochernie enantiornerenrein synthetisiert werden. Als Kupplungskornponente karn ein leicht zu offnendes Epoxid-Saccharid in Frage, das bei der 0 ffnung die D-ghco-Konfiguration regioselektiv ergeben sollte. Die Kupplungs- reaktion selbst war moglicherweise rnit geeigneten Modellsubstanzen vorher zu iiber- priifen.
Liebigs Ann. Chem. 1985
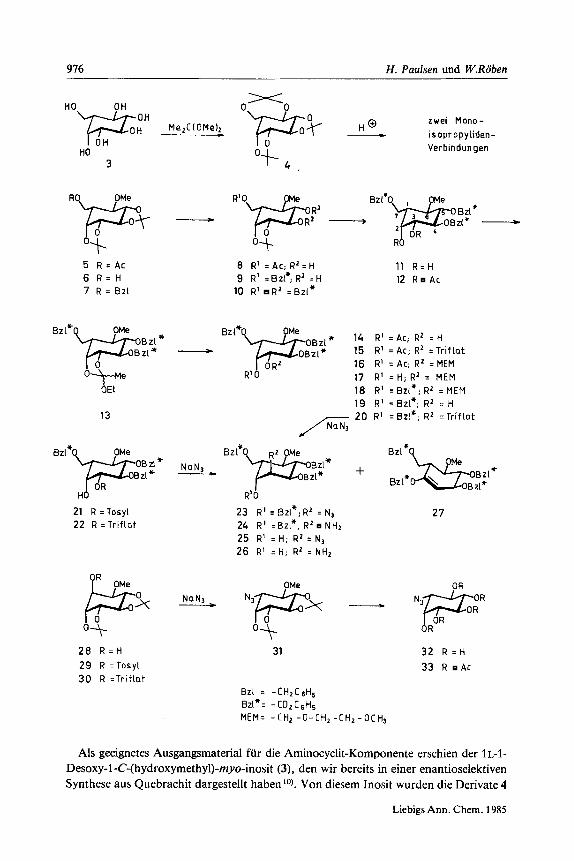
976 H. Paulsen und W.Roben
zwei Mono- HO 0
*HOH MezC(OMe)L isopropyl iden-
4 Verbindungen I OH
3 HO
O-t- 5 R = A t 6 R = H 7 R = 821
13
"l*pBZl* 00 zl * OR
2 1 R = T o s y l 22 R = T r i f l a t
O-f- 2 8 R = H 29 R = T o s y l 30 R = T r i f l a t
'"+,.' - B z " p y * - O3-
8 R' : A t ; R'=H 11 R = H 9 R' =Bzl'; R' = H 10 R' R' = Bzl*
12 R A C
14 R' = Ac; R z = H - 15 R' = A t ; R' = T r i f l a t 16 R' A t ; I?' = MEM 17 R ' H; R' = MEM 18 R ' =BZl*; R' =MEM 1 9 R ' = Bz~*; R' = H
N a N 2
R' 0
23 24 25 26
R ' = Bzl*; R' = N3 R ' =BZl*, R' NHz R' = H; R z 5 N3 R' = H; Rz = NHz
27
31 3 2 R = H 33 R A C
Als geeignetes Ausgangsmaterial fur die Aminocyclit-Komponente erschien der 1 L-I- Desoxy-1-C-(hydroxymethy1)-myo-inosit (3), den wir bereits in einer enantioselektiven Synthese aus Quebrachit dargestellt haben lo). Von diesem Inosit wurden die Derivate 4
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 971
und 5 untersucht. Die Triisopropyliden-Verbindung 4 ist leicht durch Umsetzung des freien Inosits 3 rnit 2,2-Dimethoxypropan herzustellen. Das Derivat 5 wurde bereits fruher von uns synthetisiert lo). Eine partielle Hydrolyse von 4 zum Aufbau eines geeig- neten Schutzgruppenmusters erwies sich jedoch als wenig geeignet. Bei der partiellen Hydrolyse von 4 erhalt man als Hauptprodukte ein Gemisch zweier Monoisopropyli- denverbindungen. Da dieses Gemisch sich als nicht trennbar herausstellte, wurde dieser Weg nicht weiter verfolgt, sondern trotz Anwesenheit einer 0-Methylgruppe wurden die Reaktionen des Produktes 5 weiter untersucht. Dies ist keine Beeintrachtigung, da die 0-Methylgruppe im Endprodukt der Synthese gerade an der Stelle des Pseudo- disaccharides gebunden ist, die im Oligostatin mit einem weiteren Glucoserest ver- knupft ist.
Die selektive saure Hydrolyse von 5 liefert in der Tat in hoher Ausbeute das Mono- isopropyliden-Produkt 8, bei dem weitere unerwunschte Umlagerungen der Isopropy- liden-Gruppen nicht auftreten konnen. Zur Uberfiihrung in ein stabil rnit Benzylether blockiertes Produkt wurde in 8 die 7-0-Acetylgruppe alkalisch abgespalten und das erhaltene Produkt anschlieaend zu 10 benzyliert. Fur die Benzylierung wurde ([D,]Brommethyl)benzol als Benzylierungsreagenz verwendet. Dadurch lassen sich die 'H-NMR-Spektren erheblich vereinfachen. Die Methylenprotonensignale der Benzylether liegen normalerweise im gleichen Bereich wie die Ringprotonen des Cyclits und uberlappen sich gegenseitig. Durch den Austausch der Methylenprotonen durch zwei Deuteriumatome fallen im 'H-NMR-Spektrum die Methylensignale weg, was die Interpretation der dann ungestort sichtbaren Cyclit-Signale erheblich erleichtert. Die Anzahl der im Molekiil vorhandenen Benzylether ist ohne weiteres dann durch Integra- tion der aromatischen Protonen zu ermitteln.
Der hierbei erhaltene Tribenzylether 10 ist auRerdem noch auf einem anderen Wege zuganglich. Die Entacetylierung von 5 ergibt 6 , dessen Benzylierung rnit ([D,]Brom- methyl)benzol das Produkt 7 liefert. Mit 7 ist ebenfalls eine partielle Hydrolyse der 4,5-O-Isopropylidengruppe moglich zu 9, dessen Nachbenzylierung ebenfalls zu 10 fuhrt. Die saure Hydrolyse von 10 ergibt in quantitativer Ausbeute das 2,3-Diol11, das als entsprechendes Diacetat 12 lH-NMR-spektroskopisch charakterisiert wurde. Im 2,3-Diol sol1 jetzt selektiv unter Inversion am C-3 eine Azidogruppe eingefiihrt werden, um zum gewunschten verzweigten Aminocyclit zu gelangen.
Die Einfuhrung einer Azidogruppe in das Cyclit-System ist eine oft diffizile Reaktion, die von der jeweiligen Konfiguration des Cyclits erheblich beeinflufit wird. Einige Testreaktionen wurden daher zunachst an dem uns zur Verfiigung stehenden Cyclit 2 8 ' ~ ' ~ ) durchgefiihrt. Bei Umsetzung des aus 28 erhaltlichen Tosylats 29 rnit Natriumazid erhalt man neben dem gewunschten Produkt 31 in erheblichen Mengen ein Eliminierungsprodukt, bei dem die Abspaltung der 0-Tosyl-Gruppe in der Weise erfolgt, da8 man zum Methyl-Enolether des entsprechenden Kondurits gelangt. Diese unerwiinschte Eliminierungsreaktion tritt wesentlich schwacher in Erscheinung, wenn man das aus 28 ebenfalls erhaltliche Triflat 30 rnit Natriumazid umsetzt. In diesem Falle ist die unter Inversion gebildete Azidoverbindung 31 das Hauptprodukt, wahrend das Eliminierungsprodukt nur in kleiner Menge auftritt. Es sei erwahnt, daR 31 auch bei Gegenwart einer Azidogruppe mit Bortribromid in Dichlormethan bei - 20°C ent- methyliert werden kann. Man gelangt nach Methylether-Spaltung zu 32, das als Acetat
Liebigs Ann. Chem. 1985

978 H . Paulsen und W. Roben
33 charakterisiert wurde. Das Azid 33 stellt eine Vorstufe zum 1 L-1-Amino-1-desoxy- myo-inosit dar, der auf diesem Wege ebenfalls zuganglich ist.
Um in 11 eine nucleophile Substitution durchfiihren zu konnen, ist eine selektive Blockierung der OH-Gruppe am C-2 erforderlich. Aus diesem Grunde wurde 11 in den Orthoester 13 iibergefiihrt, der unmittelbar einer saurekatalysierten Offnungsreaktion unterworfen wurde. Nach der Regel von King und Allbutt'3) erfolgt die Offnung des Orthoesters 13 in der Weise, daR die axiale Hydroxygruppe am C-2 acetyliert ist, und man gelangt zu dern Monoacetyl-Derivat 14. Das aus 14 erhaltliche Triflat 15 lafit sich jedoch nicht analog zu 30 rnit Natriurnazid substituieren. Es findet praktisch keine Reaktion statt.
Da verrnutet wurde, dafi die 2-0-Acetylgruppe die nucelophile Reaktion verhindern wurde, sollte diese Gruppe durch einen Benzylether ausgetauscht werden. Die Umset- zung von 14 rnit [(ZMethoxyethoxy)methyl-(MEM-)chlorid fuhrte zu 16, aus dem die Acetylgruppe alkalisch zu 17 abgespalten wurde. Die Benzylierung von 17 ergibt das Produkt 18, aus dem wiederurn durch saure Hydrolyse die MEM-Gruppe abgespalten werden kann zu 19. Die Urnsetzung von 19 rnit Trifluormethansulfonsaureanhydrid fuhrt dann zurn recht labilen Triflat 20. Die Umsetzung von 20 rnit Natriumazid in Dirnethylforrnarnid lieferte bereits bei Raurnternperatur unter Inversion das gesuchte Azid 23. Allerdings wurden nicht unerhebliche Anteile von Eliminierungsprodukt 27 gebildet, was sich auch durch Variation der Reaktionsbedingungen nicht unterdriicken lien. Die Reduktion der Azidofunktion in 23 mit Natriurnborhydrid/Nickel(II)- chlorid j4) fuhrte dann in guter Ausbeute zum blockierten Hydroxyvalidamin 24.
Der so aufgezeigte Reaktionsweg war jedoch hinsichtlich der Stufenzahl recht auf- wendig. Es wurde daher gepruft, ob rnit 11 direkt eine selektive Sulfonylierung unter Verzicht auf einen selektiven Schutz der 2-OH-Gruppe rndglich war. Die 3-OH-Gruppe in 11 erwies sich in der Tat als erheblich reaktiver. Eine selektive Tosylierung von 11 fuhrt in guten Ausbeuten zurn Monotosylat 21. Bei -40°C ist 11 rnit Trifluormethan- sulfonsaureanhydrid ebenfalls in ein Monotriflat 22 zu iiberfiihren.
Die nucleophile Substitution der Tosylatgruppe in 21 gelingt erst in Hexamethyl- phosphorsauretriamid bei 135 "C. Neben der Azido-Verbindung 25 wird unter den drastischen Bedingungen auch Elirninierungsprodukt gebildet. Mit dem Triflat 22 ge- lingt die nucleophile Substitution rnit Natriumazid zu 25 bereits bei 30°C. Ein uner- wiinschter Anteil an Eliminierungsprodukt tritt jedoch auch bei dieser Reaktion in etwa gleicher Menge auf. Aus Griinden der besseren Handhabbarkeit wurde daher im wesentlichen 21 zu 25 umgesetzt. Die Reduktion der Azidogruppe in 25 gelingt rnit Natriurnborhydrid/Nickel(II)-chlorid in gleicher Weise und ergibt das Arnin 26. Mit 26 steht der entsprechend verzweigte Aminocyclit zur Verfiigung, der fur eine Kupplung rnit einer Epoxid-Saccharid-Kornponente eingesetzt werden kann.
Als Kupplungskomponente fur den Arninocyclit 26 kommt das Derivat der 1,6: 3,4- Dianhydro-P-D-galactopyranose 34j5) in Frage. Es ist bekannt, dafi der Epoxidring in 34 relativ leicht durch nucleophile Reagenzien geoffnet wird. Ferner verlauft nach den bisherigen Erfahrungen diese Offnung regioselektiv und liefert, gemaR der Regel von Fiirst-Plattner 16), ein Offnungsprodukt der D-gluco-Konfiguration, wie es bei der Kupplungsreaktion erwiinscht wird. Nachteilig in 34 ist die 2-O-Tosylgruppe, die unbe- dingt entfernt werden mul3. Bei einer Enttosylierung von 34 mit Natriurn in fliissigern
Liebigs Ann. Chem. 1985
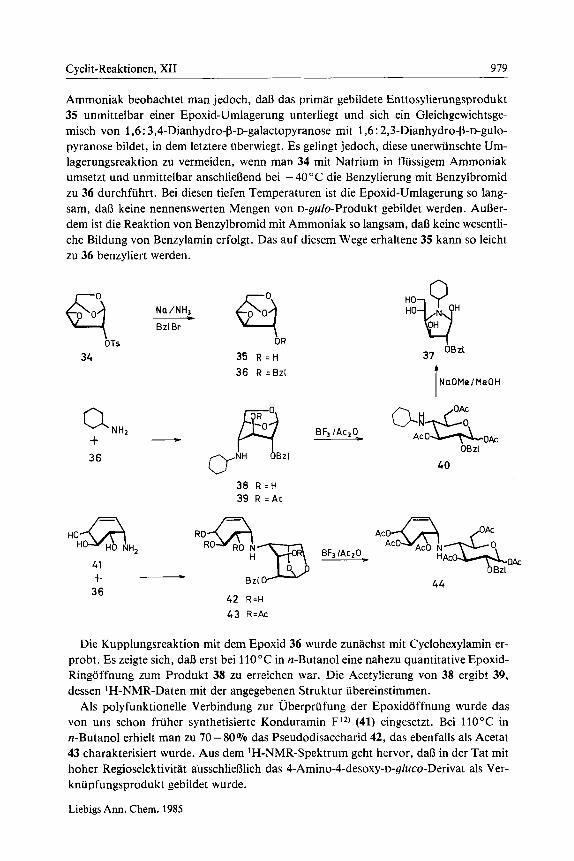
Cyclit-Reaktionen, XI1 979
Ammoniak beobachtet man jedoch, da13 das primar gebildete Enttosylierungsprodukt 35 unrnittelbar einer Epoxid-Umlagerung unterliegt und sich ein Gleichgewichtsge- misch von 1,6 : 3,4-Dianhydro-b-~-galactopyranose rnit 1,6 : 2,3-Dianhydro-b-~-gulo- pyranose bildet, in dem letztere uberwiegt. Es gelingt jedoch, diese unerwunschte Um- lagerungsreaktion zu vermeiden, wenn man 34 mit Natrium in flussigem Ammoniak umsetzt und unmittelbar anschlieflend bei - 40 "C die Benzylierung rnit Benzylbromid zu 36 durchfuhrt. Bei diesen tiefen Ternperaturen ist die Epoxid-Umlagerung so lang- sam, da13 keine nennenswerten Mengen von D-gulo-Produkt gebildet werden. AuBer- dem ist die Reaktion von Benzylbromid rnit Ammoniak so langsam, da13 keine wesentli- che Bildung von Benzylamin erfolgt. Das auf diesem Wege erhaltene 35 kann so leicht zu 36 benzyliert werden.
OTs 34
+ 36
H O G
41 Ho HO NH,
Na/NHJ
Bzl Br -
OR
35 R = H 36 R =Bzl
- OBzl oNH
38 R = H 39 R = A c
6Bzl 37
NaOMelMeOH T OAc
BF / A t 0 3 0621
40
Ro Ro Em BF3/Ac20, Ac:GN/&+ HAC OAK
R O G
Bzl ___1
44 + Bzl 36 42 R=H
4 3 R=Ac
Die Kupplungsreaktion rnit dem Epoxid 36 wurde zunachst mit Cyclohexylamin er- probt. Es zeigte sich, da8 erst bei 110°C in n-Butanol eine nahezu quantitative Epoxid- Ringoffnung zurn Produkt 38 zu erreichen war. Die Acetylierung von 38 ergibt 39, dessen 'H-NMR-Daten rnit der angegebenen Struktur ubereinstimmen.
Als polyfunktionelle Verbindung zur Uberprufung der Epoxiddffnung wurde das von uns schon fruher synthetisierte Konduramin F1*) (41) eingesetzt. Bei 110°C in n-Butanol erhielt man zu 70- 80% das Pseudodisaccharid 42, das ebenfalls als Acetat 43 charakterisiert wurde. Aus dem 'H-NMR-Spektrum geht hervor, da13 in der Tat rnit hoher Regioselektivitat ausschliefllich das 4-Amino-4-desoxy-~-gluco-Derivat als Ver- knupfungsprodukt gebildet wurde.
Liebigs Ann. Chem. 1985
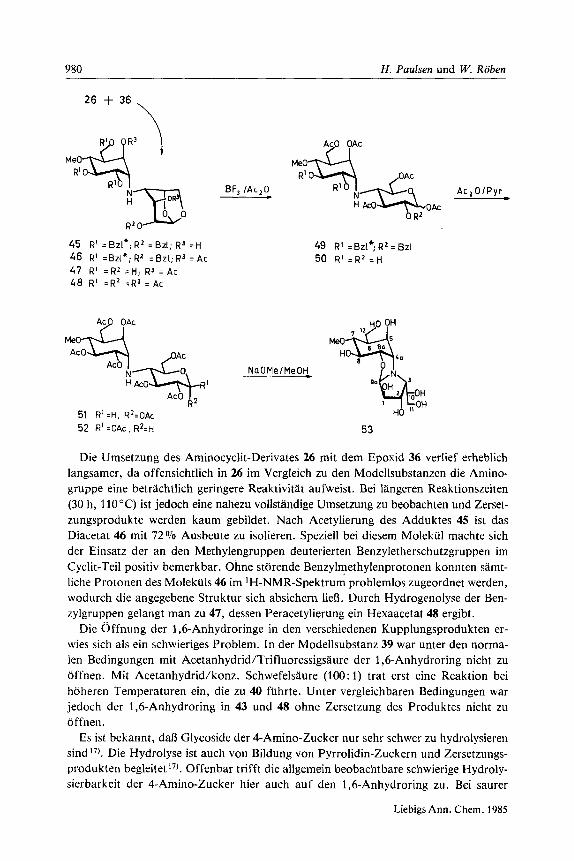
980 H . Paulsen und W. Roben
R‘ 0
AcO
51 R’=H. R2=OAc
BF, /Ac,O c
A c , O / P y r I
OAC
NaOMe/MeOH
HO
52 R‘ zOAC. Rz=H 53
Die Urnsetzung des Arninocyclit-Derivates 26 rnit dem Epoxid 36 verlief erheblich langsarner, da offensichtlich in 26 irn Vergleich zu den Modellsubstanzen die Amino- gruppe eine betrachtlich geringere Reaktivitat aufweist. Bei langeren Reaktionszeiten (30 h, 110°C) ist jedoch eine nahezu vollstandige Urnsetzung zu beobachten und Zerset- zungsprodukte werden kaum gebildet. Nach Acetylierung des Adduktes 45 ist das Diacetat 46 mit 72% Ausbeute zu isolieren. Speziell bei diesem Molekul rnachte sich der Einsatz der an den Methylengruppen deuterierten Benzyletherschutzgruppen irn Cyclit-Teil positiv bernerkbar. Ohne storende Benzylmethylenprotonen konriten sarnt- liche Protonen des Molekiils 46 im ‘H-NMR-Spektrurn problernlos zugeordnet werden, wodurch die angegebene Struktur sich absichern lieR. Durch Hydrogenolyse der Ben- zylgruppen gelangt man zu 47, dessen Peracetylierung ein Hexaacetat 48 ergibt.
Die 0 ffnung der 1 ,bAnhydroringe in den verschiedenen Kupplungsprodukten er- wies sich als ein schwieriges Problem. In der Modellsubstanz 39 war unter den norrna- len Bedingungen rnit Acetanhydrid/Trifluoressigsaure der 1,6-Anhydroring nicht zu offnen. Mit Acetanhydrid/konz. Schwefelsaure (100: 1) trat erst eine Reaktion bei hoheren Temperaturen ein, die zu 40 fuhrte. Unter vergleichbaren Bedingungen war jedoch der 1,6-Anhydroring in 43 und 48 ohne Zersetzung des Produktes nicht zu offnen.
Es ist bekannt, da13 Glycoside der 4-Amino-Zucker nur sehr schwer zu hydrolysieren sind 17). Die Hydrolyse ist auch von Bildung von Pyrrolidin-Zuckern und Zersetzungs- produkten begleitet 17). Offenbar trifft die allgemein beobachtbare schwierige Hydroly- sierbarkeit der 4-Amino-Zucker hier auch auf den 1,6-Anhydroring zu. Bei saurer
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 981
Hydrolyse wird vermutlich der Aminostickstoff protoniert, wodurch die Protonierung und Hydrolyse am anomeren Zentrum erschwert wird. Bei Anwendung von Lewis- Sauren als Katalysatoren sollte zwar auch eine Reaktion rnit dem Stickstoff auftreten, jedoch ware die Positivierung des Stickstoffes abgeschwacht, so da13 rnit diesem Reagenz eine leichtere Spaltung der glycosidischen Bindung am anomeren Zentrum zu erwarten war.
In der Tat lie13 sich mit Acetanhydrid/Ether ~ Bortrifluorid (1 To) der 1,6-Anhydro- ring in 39 bei Raumtemperatur in 3 h ohne Bildung von Nebenprodukten glatt offnen zu 40. Man erhalt ein Anomerengemisch von c( : P wie 1 : 1. Mit 43 gelingt die gleich- artige Spaltung in Acetanhydrid mit erhohter Ether - Bortrifluorid-Konzentration von 8 YO. Auch hier wird in guter Ausbeute das Anomerengemisch 44 in einem CI : P-Verhalt- nis von 1.9: 1 isoliert.
Mit 48 war jedoch selbst bei einer Erhohung der Ether - Bortrifluorid-Konzentration auf 15 Vo in Acetanhydrid keine Offnung des 1,6-Anhydroringes zu erzielen. Mbgli- cherweise waren die Acetatgruppen verantwortlich fur diese hohe Stabilitat. Die benzy- lierte Verbindung 46 erwies sich als gunstiger und reaktiver. Bei Behandlung von 46 rnit Acetanhydrid/Ether - Bortrifluorid (10: 1) bildet sich zunachst ein Primarprodukt, bei dem der 7’-O-Benzylether in 46 abgespalten und die dabei freigesetzte primare 7’-Hydroxygruppe acetyliert wird. Anschlieljend erfolgt dann die Aufspaltung des 1,6-Anhydroringes, und man gelangt zum geoffneten Produkt 49, das wiederum als Anomerengemisch, a : j3 wie 2.7: 1, vorliegt. Es gelingt jedoch, beide Anomeren rnit Hilfe eines 2-D-COSY-NMR-Spektrums vollstandig zuzuordnen. Damit ist ein neuarti- ger Weg fur schwierig zu offnende 1,6-Anhydroringe gefunden.
Durch Hydrogenolyse der Benzylethergruppen in 49 erhalt man das Derivat 50. Die Nachacetylierung ergibt ein Anomerengemisch der Octaacetate 51 und 52. Beide Ano- meren lassen sich chromatographisch trennen, und von beiden Anomeren sind die ‘H-NMR-Spektren vollstandig zu analysieren. Die Befunde stehen rnit den angegebe- nen Strukturen in guter Ubereinstimmung.
Mit der Abspaltung der Acetylgruppen in 51 und 52 stellte sich die Frage, ob das ent- stehende 4-Amino-4-desoxy-~-gluco-Derivat mit einer freien Hydroxygruppe am anomeren Zentrum uberhaupt in der pyranoiden Form stabil ist. An einfachen 4-Amino- Zuckern wurde ein leichter Ubergang in Pyrrolidin-Zucker beobachtet 17). Behandelt man die Modellsubstanz 40 rnit katalytischen Mengen Natriummethylat in Methanol, so entsteht ein einheitliches, auljerst saurelabiles Produkt. Das ‘H-NMR-Spektrum zeigt nicht mehr die grorjen Vicinalkopplungen der Pyranose-Form. Statt dessen sind die beobachteten Kopplungen rnit einem furanoiden System in Ubereinstimmung zu bringen. Dies zeigt, dalj nach Abspaltung der Acetylgruppen spontan eine Umlagerung zum Pyrrolidin-Zucker 37 erfolgt. Bei der Hydrolyse von 40 war von einem Anomeren- gemisch ausgegangen worden. Das entstandene Produkt 37 ist einheitlich und ergibt fur das anomere Proton eine Kopplung von J l ,2 = 1.9 Hz. Diese Kopplung durfte darauf hinweisen, dal3 in der furanoiden Form mit Stickstoff im Ring die 0-Konfiguration vor- liegt, wie in Formel 37 angegeben.
Die Entacetylierung von 51 oder 52 mit Natriummethylat liefert in beiden Fallen ein gleiches einheitliches Produkt, das sich als recht stabil herausstellt. Das ‘H-NMR- Spektrum zeigt durch Vergleich des Kopplungsmusters rnit den Protonen-Signalen des
Liebigs Ann. Chem. 1985

982 H . Paulsen und W. R6ben
Pyrrolidin-Zuckers 37, da8 auch hier im Aminozucker-Teil eine Ringverengung zum Pyrrolidin-Zucker stattgefunden haben mu8. Das Spektrum ist bestens vereinbar rnit dem Tricyclus 53. Insbesondere deutet auch die Kopplung von J4a,8a = 6.3 Hz in 53 gegeniiber den Kopplungen der blockierten Derivate von Jl , ,2 , = 4.6 Hz in 51 und J,,,,, = 4.4 Hz in 52 auf eine Verzerrung des Cyclitringes durch die tricyclische Struktur hin. Vergleichbare tricyclische Strukturen werden auch bei dem sauren hydrolytischen Abbau von Acarbose2) und von Oligostatin4) erhalten.
Die Befunde zeigen, daR das Pseudodisaccharid, das das invariante Mittelstuck des Oligostatins und anderer Glycosidase-Hemmer darstellt, in freier, nicht glycosidierter Form nicht stabil ist. Selbst unter neutralen, milden Bedingungen erfolgt spontan eine Cyclisierung zum Pyrrolidin-Zucker, wobei sich ein besonders stabiler Tricyclus der Form 53 bildet. Nur die Glycoside des Pseudodisaccharides sind, wie Formel 1 zeigt, stabil und weisen eine entsprechende a-Glycosidase-Inhibitorwirkung auf. Der Tricyclus 53 ist in dieser Beziehung nicht mehr aktiv.
Der Fa. BayerAG, Wuppertal, danken wir sehr fur die Unterstiitzung bei den Untersuchungen. Der Deuischen Forschungsgemeinschaft und dem Fonds der Chemischen Indusirie sind wir gleichfalls fur die Bereitstellung der Mittel zu Dank verpflichtet.
Experimenteller Teil Alle Reaktionen wurden dunnschichtchromatographisch auf Kieselgel-Fertigfolie (Merck,
GF254) verfolgt. Anfarbung: Ethanol/Schwefelsaure (10: l ) , Naphthoresorcin/Schwefelsaure/ Ethanol (1 : 50: 500) bei Aziden und Olefinen, NinhydridEthanol (1 : 500) bei freien Aminen. Saulenchromatographische Trennungen: Kieselgel 60, 230 - 400 mesh (Merck). - Optische Drehungen: Perkin-Elmer-Polarimeter 241 in 10-cm-Kiivetten bei 589 nm. - NMR-Spektren: Bruker WM 270, Bruker WM 400, innerer Standard TMS.
I ~-l-Desoxy-l-C-(hydroxymelhyl)-2,3 : 4,s : 6,7-tri-O-isopropyliden-myo-inosii (4): Zu einer Sus- pension aus 300 mg 1 L-l-Desoxy-l-C-(hydroxymethyI)-myo-inosit10) (3) (1.5 mmol) in 5 rnl absol. Dimethylformamid werden 2 rnl2,2-Dimethoxypropan und 0.1 m13 N methanolische HCI gege- ben, und der Ansatz wird bei 50°C geruhrt. Nach Beendigung der Umsetzung wird HCI rnit Triethylamin neutralisiert. Der Ansatz wird zurn Sirup eingeengt, in 10 ml CHCI, aufgenommen und zweirnal rnit Wasser gewaschen. Die organische Phase wird getrocknet und eingeengt. Zur Abtrennung von Nebenprodukten wird in ToluoVEthylacetat (5 : 1, v/v) chrornatographiert. Ausb. 430 rng (89%), [a]? = -46.5 (c = 1.2 in CHCI,). - 'H-NMR (400 MHz, C,D,): 6 =
1.14, 1.29, 1.39, 1.42, 1.51 ( 5 ~ , 18H, 3(CH3),C), 1.65 (dddd, J1,, = 3.8 Hz, J1,, = 10.0 Hz, J1,Ta = 11.2 Hz, Jl,7e = 4.2 Hz, 1 H , 1-H), 3.41 (t. J4,5 = 9.3 Hz, J5,, = 9.6 Hz, 1 H, 5-H), 3.61 (dd,J2,, = 4 . 9 H ~ , l H , 2 - H ) , 3 . 7 4 ( d d , J ? ~ , ? ~ = 1 1 . 2 H z , 1 H , 7 e - H ) , 3 . 8 1 ( t , J 3 , 4 = 8 . 8 H z , 1 H , 4-H), 3.96 (dd, 1 H , 3-H), 4.13 (t, 1 H , 7a-H), 4.28 (t, 1 H , 6-H).
C,,H,,O, (314.4) Ber. C61.13 H8.34 Gef. C 61.10 H8.35
I L- 7-0-A cetyl-I -desoxy-I-C-(hydroxymeihyl)-2,3 : 4,5-di-O-isopropyliden-6-O-meihyl-myo-inosii (5): 2.0 g 610) (6.9 mmol) werden in 10 ml absol. Pyridin gelost und rnit ca. 5 Aquiv. Acetanhydrid versetzt. Es wird auf 40°C erwarmt, bis die Urnsetzung beendet ist, und anschlieRend auf 0 ° C ge- kuhlt. Uberschussiges Acetanhydrid wird mit wenig Wasser hydrolysiert. Nach 10 min wird zu- nachst CHCI, und dann 10proz. KHS04-Losung zugegeben. Die organische Phase wird bis zur Entfernung des Pyridins rnit 10proz. KHS04-Losung geschiittelt, mit gesattigter NaHC0,- Losung und anschlieRend mit Wasser gewaschen. Die organische Phase wird rnit MgSO, getrock-
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 983
net und i. Vak. eingeengt. Ausb. quantitativ, [a]$ = +31.4 (c = 0.6 in CHCI,). - 'H-NMR (400 MHz, C6D6): S = 1.20, 1.36, 1.37, 1.41 (4s, 12H, 2(CH3),C), 1.72 (s, 3H, CH,CO), 1.99 ( d d d d , J 1 , 2 = 4 . 8 H z , J 1 , 6 = 9 . 1 H z , J 1 , 7 a = 9 . 6 H ~ , J ~ , ~ ~ = 4 . 4 H z , l H , l - H ) , 3 . 3 1 - 3 . 2 3 ( m ,
J2,3 = 5.0 Hz, 1 H, 3-H), 4.08 (t, 1 H, 2-H), 4.47 (dd, J7a,7e = 10.5 Hz, 1 H, 7a-H), 4.71 (dd, 1 H , 7e-H).
J3.4 = 9.0 Hz, J5,6 = 9.3 Hz, 2H, 4-H, 5-H), 3.40 (s, 3H, OCH,), 3.68 (t, 1 H, 6-H), 3.91 (dd,
C,,H,,O, (330.4) Ber. C 58.17 H 7.93 Gef. C 57.96 H 7.99
I L- fO-Benzyl-I-desoxy-I-C-(hydroxymethyl)-2,3 : 4, S-di-0-isopropyliden-6-0-methyl-myc-inosit (7): Zur Benzylierung werden 2.5 g 6'0) (8.7 rnmol) in 20 ml absol. Dimethylformamid geldst und mit 1.2 Aquiv. ([D2]Brommethyl)benzol versetzt. Der Ansatz wird auf 0°C gekuhlt und 1.5 Aquiv. olfreies Natriumhydrid in mehreren kleinen Portionen unter jeweiliger dunnschicht- chromatographischer Kontrolle wahrend ca. 2 h hinzugefugt. Es wird weitere 5 h bei Raumtemp. geruhrt. Zur Aufarbeitung vernichtet man uberschussiges Natriumhydrid mit wenig Methanol und engt i. Vak. ein. Es wird in CHCI, aufgenommen, zweimal mit verd. NaC1-Losung und ein- ma1 mit Wasser gewaschen. Die organische Phase wird mit MgSO, getrocknet und i. Hochvak. von Benzylmethylether, Benzylalkohol etc. befreit. Chromatographische Reinigung mit Toluol/ Ethylacetat (12: 1). Ausb. 3.09 g (94%), [a]? = +32.0 (c = 2.6 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.22, 1.38, 1.39, 1.41 [ 4 ~ , 12H, 2(CH3)2C], 1.97 (dddd, J1,2 = 4.7 Hz, J 1 , 6 = ~O.OHZ,J~,~,=~.~HZ,J,,~~=~.~HZ,~H,~-H),~.~~(~~,J~,~ =9 .8Hz , J5 , ,=9 .6Hz , 1 H, 5-H), 3.48 (s, 3H, OCH,), 3.50 (m, 2H, 6-H, 7a-H), 3.56 (t, J3,4 = 9.7 Hz, l H , 4-H), 3.68 (dd,J,,,,,= 10.3H~,lH,7e-H),3.90(dd.5~, , =4.8Hz,lH,3-H),4.10(t,lH,2-H),7.26(m,
C,,H,,D,O, (308.5) Ber. C 66.29 H 8.48 Gef. C 66.22 H 8.50 5 H, C6H5).
1 L- 7-0-Acetyl- I-desoxy-I -C-(hydroxymeIhyl)-2,3-O-isopropyliden-6-O-methyl-myo-inosit (8): 1.2 g 5 (3.6 mmol) werden in 20 ml98proz. Essigsaure gelost und bei 30°C stehengelassen. Nach Beendigung der Umsetzung wird die Essigsaure mit Toluol azeotrop abdestilliert. Der sirupose Ruckstand wird in ToluoVEthanol (5 : 1) gelost und zur Trockne eingeengt, urn restliche Essig- saure zu entfernen. Chromatographie mit Toluol/Ethanol (4: 1). Ausb. 790 mg (75 %), [a]$ =
+ 18.3 (c = 2.2 in CHCI,). - 'H-NMR (400 MHz, CDCI, + 5 % CD,OD): 6 = 1.39, 1.42 [2s, 6H, (CH3)2C], 2.12 (dddd, J1.2 = 3.7 Hz, 51.6 = 10.8 Hz, J1,7a = 9.6 Hz, J1,7e = 4.4 Hz, 1 H, l-H),3.30(dd9J5,6 = 9.1 Hz,~H,~-H),~.~O(S,~H,OCH~),~.~~(~~,J~,~ = 9.0Hz, lH,5-H) , 3.73 (t, 53,4 = 8.6 HZ, 1 H, 4-H), 4.08 (dd, Jz,3 = 5.0 Hz, 1 H, 3-H), 4.42 (dd, J7a,7e = 10.3 Hz, 1 H, 7a-H), 4.46 (dd, 1 H, 2-H), 4.68 (dd, 1 H, 7e-H), 7.26-7.38 (m, 5H, C6HS).
C,,H,,O, (290.3) Ber. C 53.78 H 7.64 Gef. C 53.82 H 7.68
I ~-7-O-Benzyl-l-desoxy-l-C-(hydroxyme~hy~-2,3-O-iso~ropyliden-6-O-~et~yl-myo-inosit (9): 2.50 g 7 (6.6 mmol) werden in 50 ml98proz. Essigsaure gelost und bei 30°C stehengelassen. Nach vollstandiger Abspaltung der 4,5-Isopropylidengruppe wird die Essigsaure azeotrop mit Toluol abdestilliert. Kristallisation aus n-Hexan/Ethanol (40: 1). Ausb. 1.82 g (81 %), Schmp. 89.3"C, [a]: = + 14.0 (c = 4.8 in CHCI,). - 'H-NMR (400 MHz, CDCI,): 6 = 1.36, 1.49 [2s, 6H, (CH&C], 2.14 (dddd, .It,, = 3.8 Hz, J1.6 = 11.0 Hz, J1,7a = 9.8 Hz, = 4.3 Hz, l H , 1-H), 3 .13(dd,55,6=9.1H~,lH,6-H),3.36(dd,J~,~ = lO.OHz,lH,5-H),3.46(~,3H,OCH3),3.57 (dd, (dd, J,,, = 4.9 Hz, 1 H, 3-H), 4.46 (dd, 1 H, 2-H), 7.31 (m, 5H, C,H,).
= 7.6 Hz, 1 H, 4-H), 3.72 (dd, J7a,7e = 8.7 Hz, 1 H, 7a-H), 3.81 (dd, 1 H, 7e-H), 3.90
C,,H,D,O, (340.4) Ber. C63.51 H8.29 Gef. C63.55 H8.18
I L-4, 5,7- Tri-O-ben~l-l-desoxy-I-C-(hydroxymethyl)-2,3-O-isopropyliden-6-O-methyl-myo-inosil (10): a) 1.80 g 9 (5.3 mmol) werden in 20 ml Dimethylformamid gelost und mit ([D,]Brommethyl)- benzol wie bei 7 benzyliert. Ausb. 2.53 g (91 %).
Liebigs Ann. Chem. 1985

984 H. Paulsen und W. Roben
b) 650 mg 8 (2.2 mmol) werden in 10 ml absol. Methanol gelCist und mit 0.1 ml 1 N methanoli- schem NaOCH, versetzt. Nach 1 h ist die Umsetzung beendet. Die Na+-Ionen werden mit wenig Kationenaustauscher IR 120 entfernt. Es wird abfiltriert und zur Trockene eingeengt. Der Roh- sirup wird in 20 ml Dimethylformamid aufgenommen und mit 3.5 Aquiv. ([D,]Brommethyl)- benzol wie bei 7 benzyliert. Ausb. 109 mg (93 %), [a]kO = + 10.4 (c = 1.9 in CHCI,). - 'H-NMR (400 MHz, CDCI,): 6 = 1.37, 1.43 [ 2 ~ , 6H, (CH,),C], 2.14 (dddd, J1,, = 3.7 Hz, J , , , = 11.1 Hz,J1,Ta = 9.9Hz, J1,Te = 4.4Hz, l H , l-H),3.26(dd, J5,6 = 9.1 Hz, 1H,6-H), 3.38(t, 14.5 = 9.4 Hz, l H , 5-H), 3.50 ( s , 3H, OCH,), 3.63 (dd, J3.4 = 7.0 Hz, I H , 4-H), 3.71 (dd, J7a,7e = 8.6 Hz, 1 H, 7a-H), 3.83 (dd, 1 H, 7e-H), 4.10 (dd, J2,, = 5.1 Hz, 1 H, 3-H), 4.47 (dd, l H , 2-H), 7.19-7.44 (m, 15H, 3C,H,).
C,,H,,D,O, (524.7) Ber. C73.25 H8.45 Gef. C73.20 H8.39
I ~ - 4 , 5 , 7 - Tri-O-benzyl-I-desoxy-I-C-(hydroxymethyl)-6-O-methyl-myo-inosit (11): In einer Mischung aus Essigsaure, Tetrahydrofuran und Wasser (2: 1 : 1, v/v/v) werden 4.0 g 10 (7.6 mmol) gelost und bei 60 "C geriihrt. Nach vollstandiger Abspaltung der Isopropylidengruppe wird zum Sirup eingeengt und noch jeweils zweimal mit Toluol zur azeotropen Entfernung der Essigsaure abdestilliert. Zur Kristallisation wird in wenig Ether aufgenommen und bis zum Ein- treten einer Opaleszenz mit n-Hexan versetzt. Ausb. 3.36 g (91 Vo), Schmp. 97.7% [a]? = + 15.2 (c = 1.0 in CHCI,).
C,,H,,D,O, (484.6) Ber. C 71.87 H 8.32 Gef. C 71.80 H 8.29
I L-2, 3-Di-O-acetyl-4,5,7-tri-O-benzyl-l -desoxy-1 -C-(hydroxymethyl)-6-O-methyl-myo-inosit (12): 100 mg 11 (0.21 mmol) werden in 2 ml absol. Pyridin gelost und analog der Darstellung von 5 acetyliert. Kristallisation aus n-Hexan. Ausb. 114 mg (97 Vo), Schmp. 89.8 "C, [a]? = + 26.2 (c = 5.2 in CHCI,). - 'H-NMR (400 MHz, CDCI,): 6 = 1.88, 1.91 (2s, 6H, ZCH,CO), 1.93 (m,
9.0 Hz, l H , 6-H), 3.42 (dd, J,a,7e = 9.0 Hz, l H , 7a-H), 3.51 (s, 3H, OCH,), 3.54 (t, J4,5 = 9.2 Hz, l H , 5-H), 3.63 (dd, l H , 7e-H), 3.82 (dd, J3,4 = 10.0 Hz, I H , 4-H), 4.88 (dd, J2,, = 3.0 Hz, I H , 3-H), 5.62 (t, l H , 2-H), 7.12-7.37 (m, 15H, 3C6H5).
C,,H,,D,O, (568.7) Ber. C 69.70 H 7.80 Gef. C 69.63 H 7.72
JI.2 = 2.6 Hz, JI.6 = 11.2 Hz, J1,7a = 8.4 Hz, J1,7e = 3.4 Hz, 1 H, 1-H), 3.41 (dd, J5,6 =
I ~-2-0-Acetyl-4,5,7-tri-O-ben~yl-l-desoxy-I-C-(hydroxymethyl)-6-O-methyl-myo-inosit (14): 300 mg 11 (0.62 mmol) werden in 6 ml absol. Toluol gelost, mit 0.6 ml Orthoessigsaure-triethyl- ester sowie 15 mg p-Toluolsulfonsaure versetzt und der Ansatz bei Raumtemp. belassen. Nach 10 min zeigt die diinnschichtchromatographische Kontrolle vollstandigen Umsatz. Es werden 4 ml 90proz. Essigsaure zugefugt, dann wird geriihrt. Nach 5 min ist die Offnung des Orthoesters beendet. Es wird mit 15 ml Toluol verdiinnt und zum Sirup eingeengt. Die chromatographische Reinigung erfolgt mit Toluol/Ethylacetat (4: 1). Kristallisation aus n-Hexan. Ausb. 278 mg (85%), Schmp. 118.9'C, [a]? = 43.3 (c = 2.7 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.69 (m, J , , 2 = 2.5 Hz, J1,6 = 11.0 Hz, J1,7e = 3.0 Hz, 1 H, 1-H), 1.72 (s, 3H, CH3CO), 3.39 (s, 3H, OCH,), 3.40-3.45 (m, 3H, 3-H, 5-H, 7a-H)*), 3.48 (dd, J5,6 = 9.2 Hz, 1 H, 6-H), 3.62 (dd, J7a,7e = 8.8 Hz, l H , 7e-H), 3.76 (t, 53,4 = J4,5 = 9.3 Hz, 1 H, 4-H)*), 5.65 (t, J2,3 = 3.0 Hz, I H , 2-H), 7.08-7.39(m, 15H, 3C6H,).
C,,H,,D607 (526.7) Ber. C 70.70 H 8.04 Gef. C 70.69 H 8.12
I L-2-0-A cetyl-4, 5,7-tri-O-benzyl-l -d~oxy-l-C-(hydroxymethyl)-~O-methyl-3-O-[(tr1~uormethy~- sulfonyll-myo-inosit (15): 60 rng 14 (0.1 1 mmol) werden in 5 ml absol. Dichlormethan geldst und unter Stickstoff auf - 4OoC gekuhlt. Es werden 0.5 ml absol. Pyridin und 27 PI Trifluormethan-
*) 4-H und 5-H konnen vertauscht sein.
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 985
sulfonsaureanhydrid (0.16 mmol) zugegeben. Nach beendeter Umsetzung wird rnit 10 ml Dichlorme- than verdiinnt und zweimal rnit eiskaltem Wasser gewaschen. Die organische Phase wird rnit MgSO, getrocknet, zum Sirup eingeengt und rnit Toluol/Ethylacetat (1 2: 1) chromatographiert. Ausb. 52 mg (69%), [ u ]g = +43.4 ( c = 0.9 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.21 (dddd, J1,, = 2.3 Hz, J1,6 = 11.4 Hz, J1,7a = 6.4 Hz, = 2.9 Hz, l H , LH), 1.60 (s, 3H, COCH,), 3.23 (t, J4.5 = 9.6 Hz, J5,6 = 9.2 Hz, l H , 5-H), 3.31 (s, 3H, OCH,), 3.25 (dd, J7a,7e = 9.2 Hz, 1 H, 7a-H), 3.40 (dd, 1 H, 7e-H), 3.46 (dd, 1 H, 6-H), 3.93 (t, J3,4 = 10.0 Hz, 1H,4-H),4.71 (dd,J2,, = 3.1 Hz, l H , 3-H),5.87(dd, lH,2-H),7.00-7.40(m, 15H,3C6H5).
C32H,,D,F,0,S (658.7) Ber. C 58.35 H 6.27 S 4.87 Gef. C 58.29 H 6.31 S 4.80
1 L-2-0-A cetyl-4.5, 7-tri-0- benzyl-I-desoxy-1 -C-(hydroxymethyl)-3-0-[(2-methoxyethoxy)methy~- 6-0-methyl-myo-inosit (16): Zu einer Losung von 600 mg 14 (1.1 mrnol) in 10 ml absol. Dichlor- methan werden 0.3 rnl Ethyldiisopropylamin (1.8 mmol) und 0.18 ml[(2-Methoxyethoxy)methyl]- chlorid (1.4 mrnol) gegeben. Nach Beendigung der Reaktion (DC: Toluol/Ethanol30: 1) wird mit 0.5 ml Triethylamin versetzt und mehrmals rnit Wasser gewaschen. Die organische Phase wird mit MgSO, getrocknet und zum Sirup eingeengt. Ausb. 687 mg (89%), [ u ]g = +38.5 (c = 4.7 in CHCI,). - 'H-NMR (400 MHz, C,D,): 6 = 1.66 (dddd, J1,, = 2.0 Hz, Jl,6 = 11.2 Hz, Jl,7a =
7.8 Hz, J,,,,=3.0 Hz, l H , 1-H), 1.73 (s, 3H, CH,CO), 3.11 (s, 3H, OCH, [MEMI), 3.37 (m,
(dd, J,a,7e = 8.8 Hz, l H , 7a-H), 3.57 (dd, l H , 6-H), 3.62 (dd, l H , 7e-H), 3.75 (dd, J2,3 = 3.0 Hz,J,,, = 9.8 Hz, 1 H, 3-H), 3.69-3.80(m, 2H, OCH,C [MEMI), 3.93(dd,lH,4-H),4.86 (d, J = 7.0 Hz, 1 H, OCH20 [MEMI), 4.91 (d, 1 H, OCH,O [MEMI), 5.89 (t. 1 H, 2-H), 7.03-7.40(m, 15H, 3C6H,).
2H, OCH2C [MEMI), 3.39(~, 3H, OCH,), 3.44(t, J4,5 = 9.1 Hz, J5.6 = 9.0 Hz, l H , 5-H), 3.45
C,,H,,D,O, (624.8) Ber. C 68.38 H 8.20 Gef. C 68.22 H 8.26
1 ~-4,5,7-Tri-O-benzyI-l-desoxy-l -C-(hydroxymethyc)-3-O-[(2-methoxyetho~)methy~-~O-methyl- myo-inosii (17): 650 mg 16 (1.06 mmol) werden in 10 ml absol. Methanol geldst und mit 0.1 mi 1 N methanolischem NaOCH, versetzt. Nach Beendigung der Reaktion werden die Na+-Ionen rnit wenig Kationenaustauscher IR 120 entfernt. Es wird filtriert und zur Trockene eingeengt. Ausb. quantitativ, [a]$ = +39.8 (c = 1.4 in CHCI,).
C,,H,,D,O, (572.8) Ber. C 69.20 H 8.45 Gef. C 69.11 H 8.52 ,
I L-2, 4,5,7- Tetra-0- benzyl- I-desoxy-1 -C-(hydroxymethyl)-3-0-[(2-melhoxyethoxy)methy~-6-0- methyl-myo-inosit (18): 600 mg 17 (1.05 rnmol) werden in 10 ml absol. Dimethylformamid gelast und rnit ([D,]Brornrnethyl)benzol analog der Darstellung von 7 benzyliert. Saulenchrornatogra- phische Reinigung rnit Toluol/Ethylacetat (8: 1). Ausb. 635 mg (91 %), [u ]g = + 33.3 (c = 3.8 in CHCI,). - 'H-NMR (400 MHz, C,D,): 6 = 1.95 (m, J!,, = 2.0 Hz, J,,7e = 2.0 Hz, 1 H, 1-H), 3.09 ( s , 3H, OCH, [MEMI), 3.25 (s, 3H, OCH,), 3.31 (m, Jdq5 = 9.1 Hz, 3H, 5-H, OCH,C [MEMI), 3.45 (m, J7a,7e = 8.5 Hz, 2H, 6-H, 7a-H), 3.57 (ddd, 1 H, OCH,C [MEMI), 3.69(ddd, 1 H, OCH2C [MEMI), 3.73 (dd, J2.3 = 2.4 Hz, J,,, = 9.9 Hz, l H , 3-H), 3.77 (dd, l H , 7e-H),
OCH,O [MEMI), 7.06-7.42 (rn, 20H, 4C6H5). 4.16 (t, 1 H, 4-H), 4.36 (t, 1 H, 2-H), 4.71 (d, J = 6.6 Hz, 1 H, OCHZO [MEMI), 4.89 (d, 1 H,
C,H,D,O, (664.9) Ber. C 72.26 H 8.49 Gef. C 72.12 H 8.55
I ~-2,4,5,7-Tetra-O-ben~l-l-desoxy-l-C-(hydroxyme1hyc)-6-0-me1hy1-myo-in~it (19): 500 mg 18 (0.75 rnmol) werden in 10 rnl einer Mischung aus 3 N HCVTetrahydrofuran (1 : 1, v/v) gelbst und auf 60°C erwarmt. Nach ca. 10 h ist die MEM-Schutzgruppe abgespalten (DC: Toluol/Ethanol 30: 1, zweimal entwickeln, denn 19 ist geringfugig unpolarer als 18). Zur Aufarbeitung wird der Ansatz auf die Hllfte eingeengt, rnit 15 ml Dichlormethan versetzt und rnit NaHC03-Losung, dann rnit Wasser gewaschen. Die organische Phase wird mit MgSO, getrocknet und eingeengt. Ausb. 410 mg (95%). [a]? = + 14.6 (c = 2.6 in CHCI,). - 'H-NMR (400 MHz, C,D6): 6 =
Liebigs Ann. Chern. 1985

986 H . Paulsen und W. Roben
1.94 (dddd, Jl,2 = 2.3 Hz, Ji,6 = 11.0 Hz, J1,7a = 8.8 Hz, J1,7e = 5.4 Hz, l H , 1-H), 3.27 (s, 3H, OCH,), 3.42 (dd, J5,, = 9.0 Hz, l H , 6-H), 3.49 (t, = 9.2 Hz, l H , 5-H), 3.63 (dd, J2,, = 2.7 Hz, J,,, = 9.8 Hz, l H , 3-H), 3.72 (m, 2H, 7a-H, 7e-H), 3.98 (dd, l H , 4-H), 4.22(t, 1 H, 2-H), 7.09-7.40 (rn, 20H, 4C,H5).
C,,H,,D,O, (576.8) Ber. C 74.97 H 8.39 Gef. C 74.82 H 8.46
1 L-2.4, 5,7- Tetra-0- benzyl-l-d~oxy-l-C-(hydroxymethyl)-6-O-melhyl-3-O-~ftr~iuormethyl)su~o- nylj-myo-inosil (20): 200 mg 19 (0.35 mmol) werden in 10 ml absol. Dichlormethan gelost und unter Stickstoff auf -40°C gekiihlt. AnschlieRend werden 0.1 ml Pyridin und 85 p1 Trifluorme- thansulfonsaureanhydrid (0.5 mmol) zugefugt. Nach Beendigung der Umsetzung wird mit 15 ml Dichlorrnethan verdunnt und zweimal rnit eiskaltem Wasser gewaschen. Die organische Phase wird mit MgSO, getrocknet und zum Sirup eingeengt. Evtl. vorhandene Pyridinreste konnen durch azeotrope Destillation mit Toluol entfernt werden. Die Substanz sollte nicht uber 4OoC erwarmt werden. Ausb. 219 rng (89%), [ c Y ] ~ = +24.0 (c = 2.2 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1 . 6 6 ( d d d d , J 1 , 2 = 2 . 1 H ~ , J ~ , ~ = ~ ~ . ~ H Z , J ~ , ~ , = ~ O . O H Z , J ~ , ~ ~ = ~ . ~ H Z , ~ H , ~ - H ) , ~ . ~ ~ ( s , 3H, OCH,), 3.22 (t, J4,5 = 9.0 Hz, Js,6 = 9.2 Hz, 1 H, 5-H), 3.26 (dd, 1 H, 6-H), 3.58 (dd, J7a,7e = 8.8 Hz, 1 H, 7a-H), 3.63 (dd, l H , 7e-H), 4.10 (dd, J,,, = 10.1 Hz, 1 H, 4-H), 4.44 (t, J2,, = 2.7 Hz, l H , 2-H), 4.78 (dd, l H , 3-H), 7.00-7.37 (m, 20H, 4C,H5).
C,,H,,D,F,O,S (708.8) Ber. C 62.70 H 6.68 S4.52 Gef. C 62.62 H 6.75 S 4.48
I ~ - 4 J . 7 - Tri-0-benzyl-I-desoxy-1 -C-(hydroxymethyl)-6-O-methyl-3-O-(p-to~~u~ony~-myo-inosit (21): 2.0 g 11 (4.1 mrnol) werden in 30 ml absol. Pyridin gelost, mit 800 mg p-Toluolsulfonyl- chlorid (4.2 rnmol) versetzt und auf 40°C erwarmt. Nach Beendigung der Reaktion (DC: Toluol/ Ethylacetat 6: 1) wird uberschiissiges p-Toluolsulfonylchlorid rnit 2 ml Wasser zersetzt. Nach 10 min wird auf 10 rnl eingeengt, in 60 ml Dichlormethan aufgenommen und mit 1 N HCI pyridin- frei gewaschen. Es wird mit NaHC0,-Losung und mit Wasser gewaschen. Die organische Phase wird rnit MgSO, getrocknet und zurn Sirup eingeengt. Kristallisation aus n-Hexan. Ausb. 2.58 g (98%), Schrnp. 102.8"C, [a]? = +9.9(c = 14.1 inCHC1,). - 'H-NMR(270MHz, C6D,): 6 = 1.43 (dddd, Jl,2 = 1.8 Hz, J1,6 = 11.3 Hz, J1,7a = 6.0 Hz, J,,7e = 3.0 Hz, 1 H, 1-H), 1.78 (s, 3H, C,H,CH,), 3.27 (s, 3H, OCH,), 3.31 (t, J4,5 = 9.2 Hz, J5,6 = 9.0 Hz, 1 H, 5-H), 3.36 (dd, J,a,7e = 8.9 Hz, 1 H, 7e-H), 3.61 (dd, 1 H, 6-H), 3.67 (dd, 1 H, 7a-H), 4.54 (t, J3,, = 9.6 Hz, 1 H, 4-H), 4.63 (dd, J2, , = 2.6 Hz, l H , 3-H), 4.68 (m, l H , 2-H), 6.57 (m, 2H, AA'-Tosyl), 7.04-7.27 (rn, 15H, 3C,H5), 7.81 (m, 2H, Tosyl).
Ber. C 67.69 H 7.26 S 5.02 C,,H,,D,O,S (638.8) Gef. C 67.75 H 7.33 S4.95
I L-4, 5,7- Tri-0- benzyl-1 -desoxy-l-C-(hydroxymethyl)-6-O-methyl-3-O-~ftri~luormethyl)su~ony~- myo-inosit (22): Zu einer Losung aus 300 mg 11 (0.6 mmol) in 10 ml absol. Dichlormethan werden 0.3 ml Pyridin gegeben und unter Stickstoff auf - 40°C gekuhlt. Es werden 7 ml einer 0.1 M Ld- sung von Trifluorrnethansulfonsaureanhydrid in absol. Dichlormethan langsam zugetropft. Nach Beendigung der Umsetzung (DC: Toluol/Ethylacetat 6 : 1) wird uberschussiges Anhydrid rnit 0.5 rnl Methanol zersetzt und zweimal rnit eiskaltem Wasser gewaschen. Die organische Phase wird mit MgSO, getrocknet und eingeengt. Zur Entfernung evtl. vorhandener Pyridinreste wird mit 10 ml absol. Toluol erneut zur Trockene eingeengt. Wahrend der Aufarbeitung sollte das Produkt nicht dber 40°C erwarmt werden. Ausb. 340 rng (89%), [a]? = +18.1 (c = 2.9 in CHCI,). -
'H-NMR (400 MHz, C6D6): 6 = 0.93 (dddd, Jl,2 = 2.2 Hz, J l , 6 = 11.4 Hz, Ji,7a = 4.4 Hz, J,,7e = 2.6 Hz, l H , 1-H), 3.12 (dd, J,a,7e = 9.1 Hz, l H , 7e-H), 3.21 (t, J4,5 = 9.5 Hz, J5,, =
9.2 Hz, 1 H, 5-H), 3.23 (s, 3H, OCH,), 3.55 (dd, 1 H, 7a-H), 3.58 (dd, 1 H, 6-H), 4.20 (t, J3,4 =
9.9Hz, 1H,4-H),4.31 (t, J2,3 = 2.7Hz, lH,2-H) ,4 .63(dd , l H , 3-H),7.04-7.40(m, 15H, 3C,H5).
C,,H,,D,F,O,S (616.7) Ber. C 58.43 H 6.37 S 5.20 Gef. C 58.28 H 6.45 S 5.13
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 987
I ~-(IN,2,4/3,5,6)-I-Azido-2.3,6,7-tetra-O-ben~yl-5-C-(hydroxymethy[)-4-O-methy1-2,3,4,6-cyclo- hexantetrol (23) und I~-(1,3/2,4)-1,2,5,7-Tetra-O-benzyl-4-C-(hydrox~methy/)-S-O-methyl-5- cyclohexen-1,2,3,5-tetrol(27): Zu einer Losung aus 160 mg 20 (0.23 mmol) in 10 ml absol. Dimethyl- formamid werden 250 mg Natriumazid gegeben, und die Suspension wird bei Raumtemp. ge- riihrt. Nach Beendigung der Reaktion wird i. Hochvak. auf 2 ml eingeengt, in 20 ml Dichlorme- than aufgenommen und zweimal mit 5prOZ. NaC1-Losung gewaschen. Die organische Phase wird mit MgSO, getrocknet und zum Sirup eingeengt. Die Produkte werden saulenchromatographisch mit ToluoVEthylacetat (25: 1) getrennt.
23: Ausb. 71 mg (52%), [ a ] f = +28.6 (c = 0.8 in CHCI,). - 'H-NMR (400 MHz, C,D,): 6 =
J3,4 = 8.0 Hz, l H , 4-H), 3.29 (s, 3H, OCH,), 3.70 (dd, J7a,7e = 8.6 Hz, 7a-H), 3.71 (t, J , , , =
2.8 Hz, J1,, = 3.7 Hz, l H , 1-H), 3.79 (dd, l H , 7e-H), 3.87 (dd, l H , 6-H), 3.94 (dd, J2?, = 9.5 Hz, 1 H, 2-H), 3.99 (dd, 1 H, 3-H), 7.05 -7.37 (m, 20H, 4C,H,).
C,,H,,D,N,O, (601.8) Ber. C71.85 H7.87 N6.98 Gef. C71.89 H7.91 N6.91
27: Ausb. 46 mg (36%), [ a ] g = +47.5 (c = 0.6 in CHCI,). - 'H-NMR (400 MHz, C6D,): S = 2.61 (m, J 1 , , = 2.4 Hz, J,, , = 9.0 Hz, J4,, = 1.9 Hz, J4,7 = 2.5 Hz, J4,,, = 2.7 Hz, 1H,4-H),
2.47 (dddd, J4,5 = 11.4 Hz, Js,6 = 3.1 Hz, Js,Ta = 10.7 Hz, J5,7e = 4.3 Hz, 1 H, 5-H), 3.28(dd,
3.52(s,3H,OCH3),3.70(dd9J7,7s =8.9Hz, lH,7-H) ,3 .84(dd ,J1 , , = 7.1 Hz,J2,, = 10.0Hz, 1 H, 2-H), 3.93 (dd, 1 H, 3-H), 3.96 (dd, 1 H, 7'-H), 4.38 (dt, J1,6 = 2.4 Hz, 1 H, 1-H), 4.76 (t, l H , 6-H), 7.07-7.45 (m, 20H, 4C,H,).
C,,H,,D,O, (558.8) Ber. C77.39 H 8.30 Gef. C77.31 H 8.22
I o-(IN,2,4/3,5,6)-1 -Arnino-2,3,6,7-tetra-O-ben~l-5-C-(hydroxymethyl)-4-O-methyl-2,3,4,6-cyclo- hexantetrol(24): Zu einer Lbsung von 50 mg 23 (83 pmol) in 5 ml Ethanol werden 2 ml einer 4proz. NiC12-Losung in Ethanol gegeben. Unter diinnschichtchromatographischer Kontrolle (Laufmittel: Toluol/Ethanol 6: 1) wird eine gesattigte Losung von NaBH, in Ethanol unter Riihren zugetropft. Nachdem die Reduktion beendet ist, wird mit 40 ml Dichlormethan verdiinnt und mit 50 ml konz. NH3-Wasser geschuttelt, bis die organische Phase farblos ist. Die Nickelsalze werden dabei als Hexammin-Nickel-Komplex in der waorigen Phase gelost. Die organische Phase wird mit MgSO, getrocknet und zum Sirup eingeengt. Fur die NMR-spektroskopische Probe empfiehlt sich eine saulenchromatographische Reinigung mit Toluol/Ethanol (8 : l) , um vorhan- dene Spuren von Nickelsalzen sicher abzutrennen. Ausb. 38 mg (79%), [a]? = + 22.5 (c = 0.4 in CHCI,). - 'H-NMR (400 MHz, CDCI,): 6 = 1.25 (s, 2H, NH,), 2.42 (dddd, J4,5 = 11.2 Hz, Js,6 = 3.3 Hz, J5,Ta = 9.8 Hz, J5,7e = 4.6 Hz, l H , 5-H), 3.23 (dd, J3,, .= 8.8 Hz, l H , 4-H), 3.42 (t, 51,2 = 4.0 Hz, J1,6 = 3.5 Hz, 1 H, 1-H), 3.44 (s, 3H, OCH,), 3.68 (dd, J7a,7e = 8.8 Hz, 1 H, 7a-H), 3.72 (dd, 1 H, 7e-H), 3.74 (dd, J2,3 = 9.6 Hz, 1 H, 2-H), 3.78 (t. 1 H, 6-H), 3.81 (dd, l H , 3-H), 7.15-7.39(m,20H,4C6H,).
C,,H,,D,NO, (575.8) Ber. C75.10 H8.58 N2.43 Gef. C75.22 H8.61 N2.38
I D-(I N,2,4/3,5,6)-1 -Azido-2,3,7-tri-O-benzyl-5-C-(hydroxymethyl)-4-O-methyl-2,3,4,6-cyclohewan- tetrol (25): a) 2.0 g 21 (3.1 mmol) werden in 50 ml absol. Hexamethylphosphorsauretriamid (HMPT) gelost und mit 4.0 g Natriumazid versetzt. Der Ansatz wird bei 135 "C geriihrt, bis das Edukt vollstandig reagiert hat (das DC wird vor dem Entwickeln 10 min i. Hochvak. vom HMPT befreit; Toluol/Ethylacetat 6 : 1). Es wird i. Hochvak. (lo-, mbar) bei 80°C direkt in eine an den Reaktionskolben angesetzte, mit fliissigem Stickstoff gekiihlte Falle destilliert. Nach ca. 15 min ist fast das gesamte HMPT aus dem Ansatz entfernt. Es wird in 50 ml Dichlormethan auf- genommen und zweimal mit Wasser gewaschen. Die organische Phase wird mit MgSO, getrock- net und eingeengt. Das gebildete Eliminierungsprodukt wird saulenchromatographisch mit Toluol/Ethylacetat (12: 1) abgetrennt. Ausb. 1.02 g (64 Yo).
Liebigs Ann. Chem. 1985
64

988 H . Paulsen und W. Roben
b) 200 mg 22 (0.3 rnrnol) werden in 5 ml absol. HMPT gelost und rnit 400 rng Natriurnazid ver- setzt. Der Ansatz wird bei 30°C geruhrt, bis das Edukt vollstandig umgesetzt ist. Die Aufarbei- tung erfolgt wie oben beschrieben. Ausb. 114 rng (69%), [a]$ = +20.8 (c = 2.5 in CHCI,). - 'H-NMR (270 MHz, C6D6, 6-OH ausgetauscht mit CD,OD): 6 = 1.86 (dddd, J4,5 = 11.3 Hz, J5,6 = 2.4 Hz, J5,7a = 4.0 Hz, J5,,e = 2.4 Hz, 1 H, 5-H), 3.32 (dd, J7a,7e = 9.2 Hz, 1 H, 7e-H), 3.38 (s, 3H, OCH,), 3.64 (dd, J,,, = 9.1 Hz, I H , 4-H), 3.78 (dd, l H , 7a-H), 3.83 (t, J1,, =
l H , 2-H),6.99-7.38(rn, 15H, 3C6H5). 3.5 Hz, J l , 6 = 3.8 Hz, l H , 1-H), 3.91 (t,J2,3 = 9.5 Hz, lH,3-H),3,92(dd, lH,6-H),4.14(dd,
C2,H2,D6N,05 (509.7) Ber. C68.35 H 7.71 N 8.24 Gef. C 68.39 H 7.78 N 8.19
1 ~-(lN,2,4/3,5,6)-I-Amino-2,3,7-tri-O-benzyl-5-C-(hydroxyme~hyl)-4-O-mefhyl-2,3,4,6-cyclo- hexantefrol (26): 1 .O g 25 (2.0 mrnol) werden analog der Umsetzung von 23 zu 24 rnit NiCI,/ NaBH, reduziert und aufgearbeitet. Ausb. 815 mg (86%), [a]E = +42.5 (c = 2.4 in CHCI,). - 'H-NMR (270 MHz, CDCI,): 6 = 1.50 (br. s, 2H, NH,), 2.13 (dddd, J4,5 = 11.0 Hz, J5,6 =
2.3 Hz, JS, , = 2.4 Hz, J5.7' = 3.1 Hz, 1 H, 5-H), 3.44 (t, J l , 2 = 4.0 Hz, J1,6 = 3.3 Hz, I H , 1-H), 3.50 (s, 3H, OCH,), 3.68 (dd, J3,4 = 9.0 Hz, l H , 4-H), 3.71 (dd, J7,,, = 9.1 Hz, 1 H, 7-H),3.79(t9J2,3 =9.3H~,lH,3-H),3.89(dd,1H,2-H),4.00(dd,lH,6-H),4.06(dd,lH,7'-H).
C2,H,,D6N05 (483.7) Ber. C 72.02 H 8.54 N 2.90 Gef. C 71.95 H 8.50 N 2.86
I ~-3,4: 5,6-Di-O-isopropyliden-2-O-methyl-l-O-[(tr1~uormefhyl)su~ony~-chiro-inosif (30) : Zu einer Losung von 200 mg 28 (0.7 mrnol) in 5 rnl absol. Dichlorrnethan werden bei -40°C unter Stickstoff 0.3 rnl absol. Pyridin und 0.17 ml Trifluormethansulfonsaureanhydrid gegeben. Nach 10 rnin ist die Umsetzung beendet. Es wird mit 10 ml Dichlorrnethan verdunnt, in 20 rnl Eis/Wasser gegeben und zweirnal rnit Wasser gewaschen. Die organische Phase wird rnit MgSO, getrocknet und eingeengt. Ausb. 279 rng (94%), [a]$ = + 23.1 (c = 6.3 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.07, 1.26, 1.27, 1.38 [4s, 12H. 2 (CH3)2C], 3.30 (s, 3H, OCH,), 3.47 (dd, J2,, =
7.3 Hz, 53.4 = 10.8 Hz, 1 H, 3-H), 3.55 (dd, J4,5 = 7.5 Hz, 1 H, 4-H), 3.68 (dd, J1,2 = 6.3 Hz, 1 H, 2-H), 4.05 (t, J5.6 = 7.4 Hz, 1 H, 5-H), 4.22 (t, Jl,6 = 7.5 Hz, 1 H, 6-H), 4.96 (dd, l H , I-H).
C,,H,,F,O,S (406.4) Ber. C41.38 H5.21 S7.89 Gef. C41.30 H5.16 S7.80
I ~-l-Azido-l-desoxy-2,3: 4,5-di-O-isopropyliden-6-O-rnethyl-myo-inosif (31) und I ~ - ( I , 2 , 4 / 3 - 1,2:3,4-Di-O-isopropyliden-5-O-methyl-cyclohexen-1,2,3,4,5-pentol: Zu einer Losung aus 100 mg 30 (0.25 rnrnol) in 5 rnl absol. Dirnethylformarnid werden 300 rng Natriumazid gegeben, und die Suspension wird bei Raurnternp. geriihrt. Nachdern das Edukt urngesetzt ist, wird rnit 20 ml Toluol verdiinnt und zum Sirup eingeengt. Es wird in 20 rnl Dichlorrnethan suspendiert und rnit Wasser gewaschen. Die organische Phase wird rnit MgSO, getrocknet und eingeengt. Die chromatogra- phische Trennung von entstandenern 31 und dern durch Elirninierung gebildeten Cyclohexen- Derivat erfolgt mit Toluol/Ethylacetat (10 : 1).
31: Ausb. 62 mg (84%), [a]$ = +41.3 (c = 2.2 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 =
1.20, 1.34, 1.37, 1.50 [ 4 ~ , 12H, 2 (CH3)2C], 3.25 (dd, Ji,6 = 8.4 Hz, J5,6 = 10.4 Hz, 1 H, 6-H), 3.32 (s, 3H, OCH,), 3.44 (t, J1,2 = 4.9 Hz, J2,, = 4.4 Hz, l H , 2-H), 3.59 (dd, l H , 1-H), 3.98-4.05 (m, 2H, 4-H, 5-H), 4.09 (dd, J,,, = 5.8 Hz, 1 H, 3-H).
C13H21N305 (299.3) Ber. C 52.16 H 7.07 N 14.04 Gef. C 52.25 H 7.19 N 13.93
Cyclohexen-Nebenprodukf: Ausb. 5.2 rng (8.3%). [a]$ = -32.4 (c = 0.5 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.28, 1.36, 1.38, 1.50 [4s, 12H, 2 (CH,),C], 3.17 (s, 3H, OCH,), 3.94 (ddd, J1,4 = 1.2 Hz, J3,4 = 8.8 Hz, 54.6 = 2.0 Hz, 1 H, 4-H), 4.03 (dd, J2,3 =
9.4 Hz, 1 H, 3-H), 4.13 (dd, Jj,, = 7.0 Hz, 1 H, 2-H), 4.44 (dd, Jl,6 = 3.5 Hz, 1 H, 6-H), 4.64 (ddd, 1 H, I-H).
C,,H2005 (256.3) Ber. C 60.92 H 7.87 Gef. C 60.80 H 7.95
Liebigs Ann. Chern. 1985

Cyclit-Reaktionen, XI1 989
I ~-2,3,4,5,6-Penta-O-acetyl-I-azido-l-desoxy-myo-inosit (33): 200 mg 31 (0.67 mmol) werden in 10 ml absol. Dichlormethan gelost und unter Stickstoff auf - 50°C gekiihlt. Es werden 3.5 ml einer 1 M Losung von Bortribromid in Dichlormethan unter Riihren langsam zugetropft, dann wird auf - 10°C erwarmt. Nach Beendigung der Umsetzung (DC: CH,Cl,/MeOH 3: 1) wird auf -30°C gekiihlt, langsam 1 ml Methanol zugegeben und 10 min geriihrt. Es wird mit 10 ml Dichlormethan verdiinnt, auf Raumtemp. gebracht und rnit 10proz. NaHCO,-Losung neutralisiert. Die waBrige Phase enthalt neben Salzen reines 32. Zur Abtrennung der Salze wird die wanrige Phase zur Trockene eingeengt, in 10 rnl Pyridin suspendiert (Ultraschall) und analog der Darstellung von 5 acetyliert. Ausb. 118 mg (43 To), Schmp. 153.0°C, [a]E = - 3.5 (c = 2.9 in CHCI,). - 'H-NMR (400MHz. C6D6): 6 = 1.47, 1.67, 1.68, 1.69, 1.71 ( 5 ~ , 15H, 5CH3CO), 2.49(dd,J1,, = 2.7 Hz, J1.6 = 10.7 Hz, l H , I-H), 4.95 (dd, J2.3 = 2.8 Hz, J,,, 7 10.5 Hz, I H , 3-H), 5.21 (t, J4,5 = 9.9 Hz, J5,b = 9.7 Hz, 1 H, 5-H), 5.54 (t. 1 H, 2-H), 5.70 (dd, 1 H, 6-H), 5.75 (t, 1 H, 4-H).
C,,H,,N,O,, (415.4) Ber. C46.27 H5.10 N10.12 Gef. C46.32 H5.19 N10.18 1,6: 3,4-Dianhydro-2-O-benzyl-/3-~-galactopyranose (36): Zu einer Suspension von 5.0 g 34
(16.8 mmol) in 25 ml absol. Tetrahydrofuran werden 60 ml fliiss. Ammoniak gegeben. Eine Kiih- lung des Ansatzes ist nicht notig, da die Eigenkiihlung durch Verdampfen von Ammoniak aus- reicht. Evtl. mu0 etwas Ammoniak erganzt werden, um das Gesamtvolumen auf ca. 70 ml zu hal- ten. Unter kraftigem Riihren wird Natrium in kleinen Stiicken zugesetzt, bis der Ansatz nach ei- ner Zugabe einige Minuten blaugrau bleibt (DC: Toluol/Ethanol 6 : l).'Es wird langsam 1.0 g NH,CI zugegeben und unter Riihren durch Anlegen von Vak. auf ca. 30 ml eingeengt, wobei die Temp. auf ca. - 60°C sinkt. Es werden 4.0 ml Benzylbromid zugegeben, und der Ansatz wird auf 20 ml eingeengt. Dabei muR die Temp. unter -20°C bleiben. Der Riickstand wird in 50 ml auf -20°C vorgekiihltem Dimethylformamid aufgenommen. Diinnschichtchromatographische Kontrolle zeigt fast vollstandige Umsetzung zu 36, die durch Zugabe von wenig Natriumhydrid vervollstandigt wird. Wahrend der Benzylierungsreaktion bleibt das Vak. angelegt, um das Am- moniak weitgehend zu entfernen. Zur Aufarbeitung 1aBt man aufwarmen, versetzt mit 100 ml Toluol und zentrifugiert vom NH,Br und NaBr ab. Der klare Riickstand wird dekantiert und eingeengt. Es wird in Dichlormethan aufgenommen und zweimal mit Wasser gewaschen. Die organische Phase wird getrocknet, eingeengt und i. Hochvak. von hochsiedenden Resten befreit. Es wird rnit ToluoVEthylacetat (8: 1) chromatographiert. Ausb. 3.86 g (98%), [a]E = -44.9 (c = 0.7 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 2.89 (dd, J I s 3 = 1.4 Hz, J,,, = 4.0 Hz, 1 H, 3-H), 2.92 (t, J4.5 = 4.8 Hz, 1 H, 4-H), 3.15 (dd, J5,6 = 4.8 Hz, J6.6, = 6.4 Hz, 1 H, 6-H), 3.43 ( s , I H , 2-H), 3.76 (d, l H , 6'-H), 4.01 (t, l H , 5-H), 4.17 ( s , 2H, PhCHZ), 5.37 (s, 1 H, 1-H), 7.00-7.20 (m, 5H, C,H,).
C,,H1404 (234.3) Ber. C 66.66 H 6.02 Gef. C 66.53 H 6.15 2-O-Benzy~-4-(cyc~ohexylam~no)-4-desoxy-~-~-g~ucofuranose (37): 150 mg 40 (0.31 mmol) wer-
den in 10 ml absol. Methanol gelost und rnit 4 Tropfen 1 N methanolischem NaOCH, versetzt. Die Umsetzung ist nach 10 min beendet. Zur diinnschichtchromatographischen Kontrolle mu13 die DC-Folie vorher ca. 10 s einer NH,-Atmosphare ausgesetzt werden, da sonst auf der Folie Zerset- zung eintritt, die einen uneinheitlichen Reaktionsverlauf vortauscht (Toluol/Ethanol 6 : 1). Es wird rnit wenig Kationenaustauscher IR 120 unter standiger pH-Kontrolle geriihrt, um die Na+ -1onen zu entfernen. Es darf keinesfalls ein AustauscheriiberschuR eingesetzt werden, da dann unter den sauren Bedingungen Zersetzung zu Pyrrolen eintreten kann. Ausb. 93 mg (84vo), Icc]~,~ = + 10.1 (c = 1.7 in CH,OH). - 'H-NMR (400 MHz, CD30D): S = 1.25, 1.80 (2m, 10H, Cyclohexyl), 2.66 (mc, 1 H, Cyclohexyl-1'-H), 3.68 (dd, J5,6a = 6.0 Hz, J6a,6b = 11.2 Hz, I H , 6a-H), 3.76 (dd, J5,6b = 4.5 Hz, l H , 6b-H), 3.90 (ddd, J4,j = 6.0 Hz, l H , 5-H), 3.92 (dd,
4.66 (2d, 2H, PhCH,), 7.25-7.40 (m, 5H, C6H5). J1,2 = 1.9 Hz, J2,3 = 4.5 Hz, 1 H, 2-H), 4.24 (dd, J3,4 = 6.2 Hz, l H , 3-H), 4.37 (d, 1 H, 1-H),
C,,H,,NO, (351.4) Ber. C 64.93 H 8.32 N 3.99 Gef. C65.08 H 8.45 N 3.85
Liebigs Ann. Chem. 1985
64'

990 H. Puulsen und W. Roben
1,6-Anhydro-2-O-benzyl-4-(cyclohexylumino)-4-desoxy-/3-~-g1ucopyranose (38): 800 mg 36 (3.4 mmol) werden in 10 ml n-Butanol gelost und rnit 2 ml Cyclohexylamin (17.0 mmol) versetzt. Es wird auf 110°C erwarmt, bis die Umsetzung beendet ist (DC: Toluol/Ethanol6: 1). Es wird rnit 20 ml Toluol verdiinnt und zur Trockene eingeengt. Kristallisation aus n-Hexan. Ausb. 935 mg (82%). Schmp. 127.3"C, [a]: = -48.8 (c = 1.4 in CHCI,).
C,,H,,NO, (333.4) Ber. C 68.44 H 8.16 N 4.20 Gef. C 68.58 H 8.22 N 4.28
3-0-Acetyl-I, 6-anhydro-2-O-ben~l~-(cyclohexylamino)-4-d~oxy-/3-~-glucopyranose (39): 500 mg 38 (1.5 mmol) werden analog zu 5 mit 1.2 Aquiv. Acetanhydrid bei - 10°C acetyliert. Saulen- chromatographische Reinigung rnit ToluoVEthanol (10: 1). Ausb. 513 mg (91 Vo), [a]: = - 48.2 (c = 3.4 in CHCI,). - 'H-NMR (270 MHz, CDCI,): 6 = 1.18, 1.73 (2m, 10H, Cyclohexyl), 1.93
3.6 Hz, I H , 1'-H), 2.73 (br. s, J2,4 = 1.5 Hz, I H , 4-H), 3.26(m, J1,2 = 1.5 Hz, J2,, = 1.5 Hz, l H , 2-H), 3.75 (dd, J5,6e = 5.6 Hz, J6a,6e = 7.1 Hz, l H , 6e-H), 4.09 (dd, .I5,,= = 1.0 Hz, l H , 6a-H), 4.55 (d, I H , 5-H), 4.63 (d, J = 12.0 Hz, l H , PhCH), 4.79 (d, I H , PhCH), 5.00 (ddd, J1,, = 1.5 Hz, l H , 3-H), 5.35 (t, l H , 1-H), 7.22-7.35 (m, 5H, C,H&
(s, 1 H, NH), 2.08 (s, 3H, CH,CO), 2.67 (dddd, J1r,2,a = J1a,6,a = 10.2 Hz, J1t,2,e = J1,,6re =
C,,H,,NO, (375.5) Ber. C 67.18 H 7.79 N 3.73 Gef. C 67.03 H 7.85 N 3.58
I,3,6-Tri-O-acetyl-2-O-ben~l-4-(cycloh~y1amino)-4-desoxy-~-g1ucopyranose (40): 100 mg 39 (0.27 rnmol) werden in 5 ml Acetanhydrid gelost und mit 0.5 ml Et,O- BF, versetzt. Nach 20 min ist die Bildung einer unpolareren Komponente auf dem DC zu beobachten. Nach 3 h ist die Um- setzung beendet. Es wird rnit 20 ml Toluol verdunnt und auf 2 ml eingeengt. Die Kodestillation rnit Toluol zur azeotropen Entfernung des Acetanhydrids wird zweimal wiederholt. Es wird in 10 rnl Dichlormethan aufgenornrnen, mit gesattigter NaHC0,-Losung und mit Wasser gewa- schen, rnit MgSO, getrocknet und eingeengt. Ausb. 119 mg (94%), a: p = 1 : 1 (aus 'H-NMR), [a]: = +22.6 (c = 3.1 in CHCI,). - 'H-NMR (400 MHz, C,D,) fur a-o-Anomeres: 6 = 0.85 - 1.13 (m, 10H, Cyclohexyl), 1.44 (br. s, 1 H, NH), 1.58, 1.69, 1.75 (3s, 9H, 3 CH,CO), 2.34 (m, 1 H, 1'-H), 2.64 (t, J,,, = 10.2 Hz, = 10.4 Hz, 1 H, 4-H), 3.45 (dd, J,,2 = 3.6 Hz,
12.1 Hz, I H , PhCH), 4.41 (dd, J6a,6e = 11.6 Hz, 1 H, 6a-H), 4.52 (d, l H , PhCH), 4.54 (dd, 1 H, 6e-H), 5.46 (t. 1 H, 3-H), 6.67 (d, l H , 1-H), 7.09-7.23 (m, 5H, C,H,); fur B-D-Anomeres: 6 = 0.85-1.13(m,10H,Cyclohexyl),1.56(br.s, lH ,NH) , 1 .58 ,1 .69 (2~ ,9H,3 CH,CO),2.34 (m, I H , l'-H), 2.63 (t, = 10.1 Hz, J4,5 = 10.2 Hz, l H , 4-H), 2.99 (ddd, J5,6a = 5.1 Hz,
11.7 Hz, I H , 6a-H), 4.44 (dd, I H , 6e-H), 4.54 (d, J = 11.6 Hz, l H , PhCH), 4.66 (d, l H , PhCH), 5.07 (t, l H , 3-H), 5.79 (d, l H , 1-H), 7.09-7.23 (m, 5H, C,H,).
J2,3 = 9.8 Hz, l H , 2-H), 3.83 (ddd, J5,6a = 5.3 Hz, J5,se = 2.2 Hz, I H , 5-H), 4.26 (d, J =
J5,6e = 2.4 Hz, 1 H, 5-H), 3.49 (dd, Jl,2 = 8.6 Hz, Jz, , = 9.0 Hz, 1 H, 2-H), 4.31 (dd, J6a,6e =
CZ5H3,NO8 (477.6) Ber. C62.88 H 7.39 N2.93 Gef. C62.76 H7.46 N2.81
3-0-A celyl-l, 6-unhydro-2-O-ben~I-4-desoxy-4-~l~-(lN,2,4/3)-(2,3,4-tri-O-acelyl-2,3,4-trihydroxy- 5-cyc~ohexen-~-yl)-amino]-/3-~-glucopyranose (43): 100 rng 41 (0.69 mmol) werden in 4 ml n-Buta- no1 gelbst, mit 400 mg 36 (1.71 mmol) versetzt und auf 110°C erwarmt. Nach ca. 20 h ist die Um- setzung beendet (DC: CHCI,/CH,OH 3 : 1). Es wird mit 20 ml Toluol verdunnt und zum Sirup von 42 eingeengt. Es wird in 5 ml absol. Pyridin aufgenommen und analog zu 5 acetyliert. Zur Ab- trennung des im UberschuB eingesetzten 36 wird mit Toluol/Ethylacetat (10: 1) chromatogra- phiert. Ausb. 288 mg (76 To), [a]g = - 37.8 (c = 2.8 in CHCI,). - 'H-NMR (270 MHz, C,D,):
1 H, NH), 3.23 (s, 1 H, 2-H), 3.41 (dd, J5,6a = 5.6 Hz, J6a,6b = 7.2 Hz, 1 H, 6a-H), 3.66 (d, 1 H, 6b-H), 3.90 (m, JI9*? = 4.2 Hz, J1,,4' = 0.8 Hz, .I1,,,, = 5.3 Hz, l H , 1'-H), 4.01 (d, I H , 5-H),
6 = 1.56, 1.61, 1.73, 1.76 ( 4 ~ , 12H, 4 CH,CO), 2.44 (d, J 4 , N H = 12.0 Hz, 1 H, 4-H), 2.66 (d,
4.30 (d, J = 12.0 Hz, 1 H, PhCH), 4.49 (d, 1 H, PhCH), 5.20 (s, 1 H, 3-H), 5.27 (dd, Jz,3, =
10.7 Hz, 1 H, 2'-H), 5.45 (s, 1 H, 1-H), 5.53 (dd, J4,,5t = 2.8 Hz, J5,,,, = 10.0 Hz, 1 H, 5'-H), 5.70
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 991
(dddd, J3,,4t = 7.2 Hz, J4t,6f = 1.9 Hz, 1 H, 4'-H), 5.91 (ddd, 1 H, 6'-H), 6.18 (dd, 1 H, 3'-H), 7.00-7.25 (m, 5H, C6H,).
C27H,,N0,, (547.6) Ber. C 59.23 H 6.07 N 2.56 Gef. C 59.12 H 6.20 N2.42
1,3,6- Tr~-O-acetyl-2-O-benzy1-4-desoxy-4-[1~-(1 N,2,4/3)-(2,3,4-tri-O-acety1-2,3,4-trihydroxy-5- cyc/ohexen-~-yl)arnino]-~-g~ucopyranose (44): 100 mg 43 (0.18 mmol) werden in 5 ml Acetan- hydrid gelost und mit 0.4 ml Et,O ~ BF, versetzt. Nach 8 h ist die Umsetzung zum Gemisch der anomeren Acetate beendet. Die Aufarbeitung erfolgt analog der Umsetzung von 39 zu 40. Ausb. 104 mg (88%), a : p = 1.9: 1 (aus 'H-NMR), [a];' = +69.7 (c = 0.8 in CHCI,). - 'H-NMR (400 MHz, C6D6) fur ap-Anomeres: 6 = 1.61,1.68, 1.73, 1.81, 1.88,1.98 (6s, 18H, 6 CH,CO), 2.68 (ddd, J4,NH = 8.5 Hz, J3,4 = 10.0 Hz, J4,5 = 10.5 Hz, l H , 4-H), 3,39(dd, J1,* = 3.8 Hz,
Jlr,2, = 4.3 Hz, J1t,6, = 5.3 Hz, l H , 1'-H),4.21 (d, J = 12.1 Hz, l H , PhCH),4.28(dd, J6a,6b = J2.3 = 9.8 Hz, l H , 2-H), 3.53 (ddd, J5,ba = 4.0 Hz, J5,bb = 2.5 Hz, I H , 5-H), 3.74 (ddd,
12.0 Hz, 1 H, 6b-H), 4.36 (dd, 1 H, 6a-H), 4.48 (d, 1 H, PhCH), 5.05 (dd, J2',,, = 10.0 Hz, 1 H,
2.8 Hz, 1 H, 5'-H), 5.58 (m, J3,,4, = 6.9 Hz, 1 H, 4'-H), 6.05 (dd, 1 H, 3'-H), 6.61 (d, I H , 1-H), 6.99-7.21 (m, 5H, C,H,); fur p-D-Anomeres: 6 = 1.58, 1.65, 1.71, 1.81, 1.83, 2.04 (6s, 18H,
2'-H), 5.42 (t, 1 H, 3-H), 5.48 (ddd, Je,6, = 1.4 Hz, J5,,6* = 10.0 Hz, lH, 6'-H), 5.55 (dd, J4',5z =
6 CHSCO), 2.40 (ddd, J4,j = 10.3 Hz, J5,sa = 3.5 Hz, J5,6b = 2.6 Hz, l H , 5-H), 2.62 (ddd, J3.4 = 9.8 Hz, J4,NH = 8.8 Hz, IH, 4-H), 3.42 (dd, J1,2 = 8.1 Hz, J2,3 = 9.4 Hz, 1 H, 2-H), 3.65(1n,J~.,~. = ~ . ~ H Z , J , , , ~ =5.3Hz,lH,l'-H),4.21(d,J=12.0Hz,lH,PhCH),4.21(dd, Jba,6b = 12.0 Hz, 1 H, 6b-H), 4.48 (dd, l H , 6a-H), 4.62 (d, l H , PhCH), 5.01 (dd, l H , 3-H),
5.50 (dd, J4f,5r = 2.8 Hz, 1 H, 5'-H), 5.58 (m, = 7.0 Hz, 1 H, 4'-H), 5.66 (d, 1 H, 1-H), 6.03 (dd, l H , 3'-H), 6.99-7.21 (m, 5H, C6H,).
5.01 (dd, J2',3- = 11.0 Hz, l H , 2'-H), 5.42 (ddd, J4t,V = 1.5 Hz, Jsr,6s = 10.0 Hz, l H , 6'-H),
C31H39N0,4 (649.7) Ber. C 57.31 H 6.05 N 2.16 Gef. C 57.22 H 6.18 N 2.09 3-0-A cetyl-4- / 1~-(1N,2,4/3,5,6)-[6-0-acetyl-2,3,7-tri-O-benzyl-2,3,4,6-tetrahydroxy-5-C-(hydro~-
methyl)-4-O-methylcyclohexyl]amino~-I,6-anhydro-2-O-benzyl-4-desoxy-~-~-glucopyranose (46): 400 mg 26 (0.83 mmol) werden in 5 ml n-Butanol gelost, mit 600 mg 36 (2.6 rnmol) versetzt und bei 110°C geruhrt. Nach 30 h ist die Umsetzung beendet (DC: Toluol/Ethanol 3: 1). Zur Auf- arbeitung wird mit 10 ml Toluol verdunnt und zum Sirup eingeengt. Der Ruckstand wird in 10 ml absol. Pyridin aufgenommen und entsprechend der Darstellung von 5 acetyliert. Die saulen- chromatograpische Abtrennung des im Uberschufi eingesetzten 36 erfolgt an Kieselgel mit ToluoVEthylacetat (4: 1). Ausb. 479 mg (72%), [a]: = +8.5 (c = 1.2 in CHCI,). - 'H-NMR (400 MHz, C6D6): S = 1.58, 1.67 ( 2 ~ , 6H, 2 CH,CO), 2.73 (d, J4,NH = 12.4 Hz, 1 H, 4-H), 2.98 (d, l H , NH), 2.98 (dddd, J4,,,, = 11.3 Hz, Js,,6, = 3.0 Hz, Js,,7,a = 9.6 Hz, Jst,7,e = 3.4 Hz, l H ,
8.8 Hz, 1 H, 4'-H), 3.40 ( s , 3H, OCH,), 3.55 (dd, J7ta,7te = 8.8 Hz, 1 H, 7'-H), 3.56 (dd, Jl,,2, =
4.2 Hz, J1,,6t = 3.0 Hz, 1 H, 1'-H), 3.60(d, 1 H, 6b-H), 3.83 (dd, J2,,,, = 9.9 Hz, I H , 2'-H), 3.83
5'-H), 3.33 (s, 1 H, 2-H), 3.36 (dd, J5,6a = 5.5 Hz, J6a,6b = 7.4 Hz, 1 H, 6a-H), 3.37 (dd, J3,,4t =
(dd, 1 H, 7'e-H), 4.09 (dd, 1 H, 3'-H), 4.32 (d, l H , 5-H), 4.44 (d, J = 12.4 Hz, l H , PhCH), 4.55 (d, l H , PhCH), 5.37 (s, 1 H, 3-H), 5.49 (s, 1 H, 1-H), 5.69 (t, 1 H, 6'-H), 6.99-7.43 (m, 20H,
Ber. C 68.89 H 7.42 N 1.75 C6H5).
C46H47D6N011 (802.0) Gef. C 68.96 H 7.55 N 1.66 3-0-Acetyl-4-j 1~-(1N,2,4/3,5,6)-[6-0-acetyl-2,3,4,6-tetrahydroxy-5-C-(hydroxyrnethyl)-4-0-
rnethylcyclohexyl]amino /-1,6-anhydro4-daody-p-D-glucopyranme (47): Eine Losung von 100 mg 46 (0.12 mmol) in 5 ml absol. Methanol wird mit 20 mg Palladiumkohle (10% Pd) in einem 20-ml- Autoklaven mit Wasserstoff bei 8 bar unter Ruhren hydriert. Nach 2 h ist die Hydrogenolyse der Benzylether beendet. Es wird von der Pd-Kohle abfiltriert und eingeengt. Ausb. 51 mg (94%), [a]: = + 17.6 (c = 2.2 in CH,OH).
C,,H2,NOI, (435.4) Ber. C49.65 H 6.71 N 3.22 Gef. C 49.60 H.6.62 N 3.30
Liebigs Ann. Chem. 1985

992 H. Paulsen und W. Roben
2,3-Di-O-acetyl-l, 6-anhydro-4-desoxy-4-/ 1~-(1N,2,4/3,5,6)-[2,3,6,7-tetra-O-acetyl-2,3,4,6-tetra- hydroxy-5-~C-hydroxyniethyl)-4-O-methylcyclohexy~aminol-~-o-glucoyranose (48): 40 mg 47 (92 Fmol) werden in 5 ml absol. Pyridin gelost und analog zu 5 acetyliert. Ausb. 55 mg (99%), [a];' = + 13.2 ( c = 4.0 in CHC1,). - 'H-NMR (400 MHz, C,D,): S = 1.56, 1.63, 1.75, 1.76, 1.83, 1.85 ( 6 ~ , 18H, 6 CH,CO), 2.36(d, J 4 , N H = 11.1 Hz, l H , 4-H), 2.47 (d, l H , NH), 2.90 (dddd, J4,,5, = 11.3 Hz, J5,,6r = 2.8 Hz, J5,.7ta = 9.2 Hz, J5,,7se = 4.6Hz, 1 H, 5'-H), 3 .20 (~ , 3H, OCH,), 3.36 (dd, J5,6a = 5.6 Hz, J6a,6b = 7.5 Hz, 1 H, 6a-H), 3.47 (dd, J38,4, = 9.2 Hz, 1 H,
4.35 (dd, J7a,7e = 10.9 Hz, l H , 7'a-H), 4.40 (dd, l H , 7'e-H), 4.83 (m, J1,, = 1.6 Hz, J2,, = 1.5 Hz, J2,4 = 1.5 Hz, l H , 2-H), 4.97(m, J1,, = 1.5 Hz, J3,4 = 1.5 Hz, 1 H, 3-H), 5.43 (t, l H ,
4'-H), 3.51 (d, lH,6b-H) , 3.60(dt,Jl. ,z = 4.3 H z , J ~ . , ~ = 3.2 Hz, l H , lf-H),4.17(d, lH,S-H),
6'-H), 5.53 (t, 1 H, 1-H), 5.56 (dd, JZ.3, = 10.8 Hz, 1 H, 2'-H), 5.74 (dd, 1 H, 3'-H).
C2,H,,N0,, (603.6) Ber. C 51.74 H 6.18 N 2.32 Gef. C 51.88 H 6.32 N 2.26
3-0-A cetyl-I, 6-anhydro-2-0-benzyl-4-desoxy-4-j 1~-(1N,2,4/3,5,6)-[6,7-di-O-ace~yl-2,3-di-O-benzyl- 2,3,4,6-lelrahydroxy-5-C-(hydroxymethy/)-4-O-meIhylcyclohexy~amino~-~-o-glucopyranose: 100 mg 46 (0.12 rnmol) werden in 5 ml Acetanhydrid gelost und mit 50 1.11 Et,O - BF, versetzt. Nach 4 h ist die Umsetzung beendet (DC: Toluol/Ethylacetat 2: 1). Es wird das Acetanhydrid zweimal azeo- trop rnit Toluol i. Vak. abdestilliert. Dabei sollte nur bis auf ein Restvolumen von ca. 1 ml ein- geengt werden, um keine zu hohen BF3-Konzentrationen zu erhalten. Es wird in 20 rnl Dichlor- methan aufgenommen, mit gesattigter NaHC03-Losung und mit Wasser gewaschen. Die organi- sche Phase wird rnit MgS04 getrocknet und eingeengt. Ausb. 83 mg (89%), [ a ] g = + 10.1 (c =
1.0 in CHCI,). - 'H-NMR (400 MHz, C,D,): 6 = 1.64, 1.65, 1.71 (3s, 9H, 3 CH,CO), 2.69 (d, = 3.3 Hz, l H ,
J5,6a = 5 . 5 Hz, J6a.6b = 7.4 Hz, 1 H, 6a-H), 3.50 (t, Jla,2, = 4.2 Hz, J1,,,, = 3.3 Hz, 1 H, 1'-H),
4.42 (d, J = 12.4 Hz, 1 H, PhCH), 4.46 (m, 2H, 7'a-H, 7'e-H), 4.55 (d, 1 H, PhCH), 5.35 (s, 1 H, 3-H), 5.46 (t, 1 H, 6'-H), 5.50 (s, 1 H, 1-H), 6.99-7.40 (m, 15H, 3 C,H,).
= 12.0 Hz, 1 H, 4-H), 2.96 (d, 1 H, NH), 2.96 (m, J4',5p = 11.3 Hz, 5'-H), 3.30 (dd, J3t,4, = 8.9 Hz, l H , 4'-H), 3.33 (s, I H , 2-H), 3.39 (s, 3H, OCH,), 3.43 (dd,
3.66 (d, 1 H, 6b-H), 3.79 (dd, J7.3' = 9.9 Hz, 1 H, 2'-H), 4.04 (dd, 1 H, 3'-H), 3.43 (d, 1 H, 5-H),
C4,H4SD4N0,2 (751.9) Ber. C65.50 H7.11 N1.86 Gef. C65.39 H7.20 N1.88
1.3.6- Tri-0-acetyl-2-0-benzyl-4-desoxy-4-j 1~-(1N,2,4/3,5,6)-[6,7-di-O-acetyl-2,3-di-O-ben~l- 2,3,4,6-tetrahydroxy-5-C-(methy~-4-O-methy1cycloh~y~amino -0-glucose (49): 300 mg 46 (0.37 mmol) in 20 rnl Acetanhydrid werden rnit 2 ml Et,O - BF, versetzt. Nach 15 h ist die Urnset- zung zum Anomerengemisch 49 beendet. Die Aufarbeitung des Ansatzes erfolgt analog zu 40. Ausb. 230 mg (72%), u : p = 2.7: 1 ('H-NMR), [a]? = +67.1 ( c = 0.8 in CHCI,). - 'H-NMR (400 MHz, C6D6) fur u-o-Anomeres: S = 1.59, 1.66, 1.80, 1.81, 1.85 (5 s, 15 H, 5 CH,CO), 1.97 (m, 1 H, NH), 2.66 (m, J4,,5, = 11.2 Hz, J5,,6. = 3.2 Hz, J5,,7,a = 9.6 Hz, J5,,7,e = 4.2 Hz, 1 H, 5'-H), 3.20 (m, J,,,, = J4,5 = 9.0 Hz, 1 H, 4-H), 3.35 (dd, J3,,4, = 9.0 Hz, 1 H, 4'-H), 3.42 (dd,
(dd, J28,3s = 10.0 Hz, 1 H, 2'-H), 3.78 (m, Js,6a = 4.8 Hz, = 2.2 Hz, 1 H, 5-H), 3.95 (dd, 1 H, 3'-H), 4.14 (d, J = 11.6 Hz, 1 H, PhCH), 4.39 (dd, J6a,6b = 12.0 Hz, 1 H, 6a-H), 4.40 (dd, J7,a,7ee = 10.8 Hz, l H , 7'e-H), 4.42 (d, I H , PhCH), 4.47 (dd, l H , 7'a-H), 4.72 (dd, l H , 6b-H),5.56(t,lH,3-H),5.56(t,lH,6'-H),6.55(d,lH,l-H),7.00-7.53(m,15H,3 C6H5);fUr 0-D-Anorneres: 6 = 1.55, 1.65, 1.76, 1.80, 1.83 (5s, 15H. 5 CH,CO), 2.58 (m, J4,s = 9.8 Hz, J5,6a = 4.2 Hz, J5,6b = 2.4 Hz, l H , 5-H), 2.58 (m, J4,,5, = 11.8 Hz, J5,,6, = 3.5 Hz, J5,,7,a = 10.0 Hz, J5,,,le = 4.2 Hz, lH,S'-H), 3.20(m, J3,4 = 8.8 Hz, 1H,4-H), 3.34(dd,J3.,,. = 9.0 Hz,
JI.1 = 3.7 Hz, 52.3 = 9.1 Hz, 1 H, 2-H), 3.62 ( t , J1s,2, = 4.2 Hz, JIt,,, = 3.8 Hz, 1 H, 1'-H), 3.78
1 H, 4'-H), 3.44 (dd, J i , , = 7.7 Hz, J2,3 = 8.7 Hz, l H , 2-H), 3.56 (t, J,, , , = 4.0 Hz, Jl,,6, = 3.8 Hz, 1 H, l'-H), 3.75 (dd, J2,,3' = 9.8 Hz, 1 H, 2'-H), 3.97 (dd, 1 H, 3'-H), 4.23 (dd, J6a,6b = 12.0 Hz, 1 H, 6a-H), 4.33 (dd, J7ja,7se = 10.9 Hz, 1 H, 7'e-H), 4.42 (d, J = 12.0 Hz, 1 H, PhCH),
Liebigs Ann. Chem. 1985

Cyclit-Reaktionen, XI1 993
4.48 (dd, 1 H, 7'a-H), 4.57 (d, 1 H, PhCH), 4.61 (dd, 1 H, 6b-H), 5.19 (t, 1 H, 3-H), 5.56 (t, 1 H, 6'-H), 5.73 (d, l H , 1-H), 7.00-7.53 (m, 15H, 3 C6H5).
C4,H5,D4N0,, (854.0) Ber. C63.29 H6.96 N1.64 Gef. C63.33 H7.08 N1.69
I ,2,3,6- Tetra-0-acetyl-4-desoxy-4- I~-(1N,2,4/3,5,6)-[2,3,6,7-tetra-O-acetyl-2,3,4,6-tetrahydroxy- 5-(C-hydroxyrnethyl)4-O-methylcyclohexylloj-a- und -b-D-glucose (51 und 52): 200 mg 49 (0.23 mmol) werden in 10 ml absol. Methanol gelost, mit 100 mg Palladiumkohle (10% Pd) versetzt und im Autoklaven bei 12 bar H2-Druck geriihrt. Nach 2 h ist die Umsetzung beendet. Es wird ab- filtriert, eingeengt und analog zu 5 acetyliert. Die Octaacetate 51 und 52 werden saulenchromato- graphisch mit Toluol/Aceton (4: 1) getrennt.
a-o-Anomeres 51: Ausb. 101 mg (61 %), [ a ] g = +51.2 (c = 1.6 in CHCI,). - 'H-NMR (400 MHz, C,D6): 6 = 1.53 (d, J4,NH = 10.2 Hz, l H , NH), 1.60, 1.61, 1.63, 1.73, 1.78, 1.81, 1.84, 1.99 (SS, 24H, 8 CH,CO), 2.68 (dddd, J4',5t = 11.4 Hz, J5s,6f = 3.1 Hz, J5t,7,a = 8.1 Hz,
OCH,), 3.46 (ddd, J5,6a = 4.4 Hz, J5,6b = 2.3 Hz, l H , 5-H), 3.50 (dd, = 9.2 Hz, l H ,
6a-H), 4.38 (dd, J7a,7e = 11.3 Hz, l H , 7'a-H), 4.45 (dd, l H , 7'e-H), 4.52 (dd, l H , 6b-H), 5.06
J5*,7Je = 3.8 Hz, 1H, 5'-H), 3.16 (ddd, J3,4 = 9.9 Hz, J4,5 = 10.5 Hz, l H , 4-H), 3.20 (s, 3H,
4'-H), 3.74 (dd, Jj'f,zt = 4.6 Hz, J1,,6t = 3.2 Hz, l H , 1'-H), 4.11 (dd, J6a,6b = 12.0 Hz, l H ,
(dd, Jl,2 = 3.7 Hz, J2.3 = 10.0 Hz, 1 H, 2-H), 5.45 (dd, J Z , , , = 10.6 Hz, 1 H, 2'-H), 5.47 (t, 1 H, 3-H), 5.50 (t, 1 H, 6'-H), 5.72 (dd, 1 H, 3'-H), 6.55 (d, 1 H, 1-H).
B-D-Anorneres 52: Ausb. 46 mg (28%), [a]? = +23.5 (c = 1.0 in CHCI,). - 'H-NMR (400 MHz, C6D6): 6 = 1.18 (d, J4,NH = 10.4 Hz, 1 H, NH), 1.50, 1.59, 1.66, 1.73, 1.75, 1.80, 1.81, 2.03 ( 8 ~ , 24H, 8 CH,CO), 2.36 (ddd, J4,5 = 10.4 Hz, J5,6a = 4.1 Hz, J5,6b = 2.2 Hz, 1 H, 5-H), 2.67 (dddd, J4#,5s = 11.3 Hz, J5s.C = 3.2 Hz, JSt,78a = 7.6 Hz, J5,,78e = 4.4 Hz, 1 H, 5'-H), 3.09 (ddd, J3,4 = 9.6 Hz, l H , 4-H), 3.19 ( s , 3H, OCH,), 3.47 (dd, J3,,4, = 9.4 Hz, l H , 4'-H), 3.70 (t, Jls,z, = 4.4 Hz, Jlt,6' = 3.5 Hz, 1 H, 1'-H), 4.05 (dd, J6a,6b = 12.0 Hz, 1 H, 6a-H), 4.37 (dd, J7,a,7.e = 11.2 Hz, l H , 7'a-H), 4.41 (dd, l H , 7'e-H), 4.49 (dd, I H , 6b-H), 5.02 (dd, J2,, =
9.2 Hz, 1 H, 3-H), 5.18 (dd, J1.2 = 8.5 Hz, l H , 2-H), 5.41 (t, 1 H, 6'-H), 5.42 (dd, J2,,3t = 10.6 Hz, 1 H, 2'-H), 5.71 (dd, 1 H, 3'-H), 5.79 (d, 1 H, 1-H).
C30H4,N0,, (705.7) Ber. C 51.06 H 6.14 N 1.98 51: Gef. C51.13 H6.19 N1.90 52: Gef. C 51.20 H 6.06 N 1.88
( I R,2S,3 R, 4aS,SR, 6S, 7R, 8s. 8aS,9aR)-3-[(I S)- 1,2-DihydroxyethylJ-I,2,3,4a,5,6,7,8,8a,9a-decahy- dro-6-~hydroxyrnethyl)-7-rnethoxypyrrolo[2, I-b]benzoxazol-I,2,5,8-tetrol(53): 60 mg 51 (85 pmol) in 10 ml absol. Methanol werden mit 3 Tropfen 1 N methanolischem NaOCH, versetzt. Die Umset- zung ist nach 1 h beendet. Die Aufarbeitung erfolgt analog zu 37. Ausb. 26 mg (83 %), [a]g = + 58.6 (c = 0.8 in CH,OH). - 'H-NMR (400 MHz, CD,OD): 6 = 2.00 (dddd, J5,6 = 3.2 Hz,
4.8 Hz, 1 H, 3-H), 3.18 (dd, J4a,5 = 4.4 Hz, J4a,8a = 6.3 Hz, 1 H, 4a-H), 3.32 (dd, J7,8 =
lla-H), 3.74(dd,J,0,11b = 4.6Hz, l H , llb-H), 3.78(dd,J8,8a = 7.5 Hz, l H , 8-H), 3.84(dd, J12a,12b = 10.6 Hz, 1 H, 12a-H), 3.85 (dd, 1 H, 12b-H), 3.87 (ddd, 1 H, 10-H), 3.93 (t. J,,9a =
= 3.5 Hz, 1 H, 1-H), 4.20 (dd, 1 H, 5-H), 4.35 (dd, 1 H, 2-H), 4.36 (dd, 1 H, 8a-H), 5.09 (d, 1 H, 9a-H).
J6,7 = 9.1 Hz, J6,11a = 6.0 Hz, J6,11b = 5.0 Hz, l H , 6-H), 3.14 (dd, J2,, = 5.2 Hz, J3,10 =
8.6 Hz, l H , 7-H), 3.50 ( s , 3H, OCH,), 3.66 (dd, JIOslla = 5.8 Hz, Jlla,llb = 11.0 Hz, 1 H,
C,,H,,NO, (366.4) Ber. C 45.90 H 6.88 N 3.82 Gef. C 45.86 H 6.95 N 3.89
Liebigs Ann. Chem. 1985

994 H. Paulsen und W. Roben
I ) X I . Mitteilung: H. Paulsen und H. Schmidt-Lewerkiihne, Liebigs Ann. Chem. 1985, 959, voranstehend.
2, D. D. Schmidt, W. Frommer, B. Junge, L. Miiller, W. Wingender und E. Tnrscheit, Natur- wissenschaften 64,535 (1977); E. Truscheit, W. Frommer, B. Junge, L. MiiIler> D. D. Schmidt und W. Wingender, Angew. Chem. 93, 738 (1981); Angew. Chem., Int. Ed. Engl. 20, 744 (1 981).
3) W. Puls, U. Keup, H. P. Krause, G. Thomas und F. Hoffmeister, Naturwissenschaften 64, 536 (1977).
4) S. Omoto, J . Itoh, H. Ogino, K. Iwamatsu, N. Nishizawa und S. Inouye, J. Antibiot. 34, 1429 (1 981 ).
5 ) S. Namaiki, K. Kangouri, T. Nakate, H. Hara, K. Sugita und S. Omura, J. Antibiot. 35, 1234 (1982); S. Murao, K. Ohyama und S. Ogura, Agric. Biol. Chem. 41,919 (1977); K. Fukuhara, H. Murai und S. Murao, ebenda 46, 1941 (1982).
6 ) Y. Kameda und S. Horii, J. Chem. SOC., Chem. Commun. 1912,146. 7) H. Paulsen und F. R. Heiker, Liebigs Ann. Chem. 1981, 2180. 8) Y. Kameda, N. Asano, M. Yoshikawa und K. Matsui, J . Antibiot. 33, 1575 (1980). 9) S. Ogawa, Y. Iwasawa, T. Toyokuni und T. Suami, Chem. Lett. 1982, 1729; S. Ogawa, N.
Chida und T. Suami, ebenda 1980,1559; S. Ogawa, Y. Iwasawa, T. Toyokuni und T. Suami, ebenda 1983, 331.
10) H. Paulsen und W. Roben, Liebigs Ann. Chem. 1983, 1073. I I ) H. Paulsen, W. Roben und F. R. Heiker, Tetrahedron Lett. 21, 3687 (1980). 12) H. Paulsen, W. Roben und F. R. Heiker, Chem. Ber. 114, 3243 (1981). '3) J. F. King und A . D. Allbutt, Can. J. Chem. 48, 1754 (1970). 14) H . Paulsen und V. Sinnwell, Chem. Ber. 111, 879 (1978). 15) M. Cernj, L. Kalooda und J . Pacuk, Collect. Czech. Chem. Commun. 33, 1143 (1968). 16) A. Fiirst und P. A . Plattner, Helv. Chim. Acta 32, 275 (1949). 17) H. Paulsen und K. Todt, Adv. Carbohydr. Chem. 23, 115 (1968).
[169/84]
Liebigs Ann. Chern. 1985



![Enantioselektive Katalyse, IX. Neue optisch reine 3,4-Bis ...anorganik.uni-tuebingen.de/aknagel/VEROEFFEN/19931260505_ftp.pdf · U. Nagel, T. Krink Enantioselektive Katalyse, IX"]](https://img.pdfslide.org/doc/110x75/5e1385baf543157800699fef/enantioselektive-katalyse-ix-neue-optisch-reine-34-bis-u-nagel-t-krink.jpg)

























