Embed Size (px)
Citation preview

KOLLOID-ZEITSCHRIFT & ZEITSCHRIFT FUR POLYMERE ZUR Z E I T VEREINIGT M I T DEN K O L L O I D . B E I H E F T E N �9 ORGAN DER KOLLOID-GESELLSCHAFT
Band 210 Mai 1966 Heft 1
Polymere A us dem Deutsche~ Kunststo]-Instltut, Darmstadt
Die Bestimmung der Mikrostruktur von Vinylpolymeren durch hochaufl6sende Protonenresonanz*)
Von U. J o h n s e n
Mit 16 Abbildungen in 20 Einzeldarstellungen (Eingegangen am 18. Februar 1966)
1. Einleitung Die heute benutzte Nomenklatur zur Be-
zeichnung der stereoregulEren Polymeren ist von Natta eingefiihrt worden.
Nach seiner Definition (1-3) hat ein stereo- regul/~res Polymeres eine vollst/~ndig regul/~re Struktur, es ist frei yon Verzweigungen, und alle seine Monomereinheiten folgen in den Molekelketten in Kopf-Schwanz-Anordnung aufeinander. RSntgenographische Untersu- chungen, insbesondere die Dimensionen der Einheitszellen der kristMlisierten Molekel- ketten ftihrten 2Vatta zu dem Schlug, dab Vinylpolymere zwei stereoregul/~re Formen annehmen kSnnen, in denen die aufeinander- folgenden pseudoasymmetrischen Kohlen- stoffatome entweder durchweg diese]be Kon- figuration oder die entgegengesetzte Kon- figuration besitzen. Diese beiden Formen nannte Natta isotaktisch und syndiotaktisch.
Naeh Definition sind diese Bezeiehnungen nur anwendbar, wenn die gesamte Molekelket- te die angegebene Struktur aufweist. Alle m5g- lichen anderen Strukturen mit einem geringe- ren Ordnungsgrad nannte ~Vatta ataktisch.
Weitergehende Untersuchungen zeigten jedoch sehr bald, dab die beiden Formen nach Natta nur Endpunkte einer langen SkMa der Taxie darstellen, die sich yon dem voll- st/~ndig syndiotaktischen his zu dem vol]- st//ndig isotaktischen Polymeren erstreckt. Als Folge wurde Nattas Definition erweitert und der :Begriff des Stereoblock-Polymeren (4) geschaffen, in dessen Molekelketten iso- taktische nnd syndiotaktische Bereiche ein- ander abwechseln. Damit taueht sofort die ];rage nach tier L/~nge der stereoregul//ren B15cke auf.
Diese Frage kann nur mit solchen experi- mentellen Methoden beantwortet werden,
*) Fachbe r i ch t d e r P h y s i k e r t a g u n g in F r a n k f u r t a. M.- HSchs t a m 8. Oktober 1965.
welche die Wirkung der n~chsten Nachbarn innerhalb der Molekelketten erfassen, die also Monomereinheiten innerhalb der beiden Arten von Sequenzen nach ihrer Konfigura- tion und diese yon Monomereinheiten am Rande der Sequenzen unterscheiden.
Fiir diese Untersuchungen der Mikrotaxie oder der Mikrostruktur haben sich die Kern- spin-Resonanz und in le~z~er Zeit aueh die Ultrarot-Spektroskopie (5-8) als geeignetes MJttel erwiesen.
Uber Untersuchungen mit Itilfe der Kern- spin-Resonanz wird hier berichtet. Am SchluB werden neuere Arbeiten dargestellt, durch die zum ersten Male aueh die Struktur eines ehemischen Copolymeren bestimmt wurde.
2. Die Methode der Kernspin-Resonanz Die Kernspin-Resonanz ist ein spezieller
Zweig der Absorptionsspektroskopie. Sic benutzt die Atomkerne der Molekeln als ma- gnetische Sonden. Dazu eignen sich in der Polymerchemie die Protonen und die Atom- kerne des Fluors. Beide haben die Spin- Quantenzahl 1/2. Info]ge ihres magnetischen Moments besitzen die Protonen in einem starken magnetischen GMchfeld H0 zwei diskrete Energieniveaus, zwischen denen bei der l~esonanz mit einem zweiten hochffe- quenten Wechselfeld l~berg/~nge induziert werden. Die dabei absorbierte Energie wird registriert.
Die Energiedifferenz zwischen den Termen oder die beobachtete Resonanzfrequenz wird allein durch die St//rke des am Oft der Kerne wirksamen lokMen Magnetfeldes bestimmt. Dieses unterseheidet sich infolge der Wechsel- wirkung der Kerne untereinander und der Wechselwirkung der Kerne mit den Elektro- hen ihrer Umgebung yon dem auBen angeleg- ten Felde H o. Da diese Weehselwirkungen yon der molekularen und der stereoehemisehen

2 Kolloid-Zeitschrift und Zeitschrift fiir Polymere~ Band 210 �9 Heft I
Struktur der untersuchten Substanzen ab- hangen, ist es mSglich, aus der Lage und aus der Intensifier der im Spektrum auftretenden Resonanzlinien auf die Struktur zurfickzu- schlieBen.
2.1. Magnetische Dipol- WechselwirIcung Den grSl~ten EinfluB auf das Magnetfeld
am Oft eines Kerns haben die magnetischen ~elder der Nachbarkerne. Der Beitrag eines Protons im Abstand yon 1 A liegt in der GrS~enordnung yon 10 0rsted. Er h~ngt ab yon dem Kernabstand und der Orientie- rung der Kernverbindungslinie zur Achse des ~uBeren Feldes. Da in amorphen FestkSrpern sowohl die Abst~nde als auch die gegenseitige Orientierung statistisch verteilt sind, i~ndert sich bei homogenem i~ul]eren Feld das wirk- same Feld yon Kern zu Kern. Bei fester Mel~- frequenz - fast alle Spektrometer arbeiten mit einer festen Frequenz und veri~nderlichem Feld - erstreckt sich daher die Resonanz fiber einen Feldstiirkebereieh von 10 0rsted. Die im Spektrum beobachtete Absorptions- linie ist die Umhfillende einer Vielzahl yon dicht nebeneinanderliegenden nicht getrenn- ten Einzellinien. Mit einem derartigen Spek- trum sind Strukturbestimmungen offensicht- lich nicht mSglich. In Flfissigkeiten und Gasen dagegen ist infolge der nngeordneten Wi~rmebewegung der Molekeln das Zusatz- feld am err jedes Kernes sehr stark ver~nder~ lich, und sein zeitlieher Mittelwert ist sogar Null. Wenn die Anderung des Zusatzfeldes rasch genug erfolgt, n~mlieh sehr oft w~h- rend der Lebensdauer eines Spinzustandes, dann kSnnen die Kerne die wechselnden Zu- satzfelder nicht unterseheiden und registrie- ten nur den Mittelwert Null.
Die Resonanz erfolgt so, als ob die magne- tische Dipolwechselwirkung nieht vorhanden w~re. Die Linien werden dabei gegenfiber den Linien in den FestkSrperspektren um den Faktor 10 ~ schm~ler. In Flfissigkeiten und LSsungen, und nur in diesen kSnnen Strukturuntersuchungen vorgenommen wer- den, wird dann die Struktur der Molekel durch zwei magnetische Effckte auf die beob- achteten Kerne abgebildet: die diamagne- tische Abschirmnng und die indirekte Spin- kopplung.
2.2. Der diamagnetische E~ekt (Chemische Verschiebung)
Das ~ul~ere 1Viagnetfeld induziert in den Elektronenhfillen der Molekel diamagnetische ~omente. Dadurch entsteht zus~tzlich ein
induziertes Feld, das dem ~uBeren Feld pro- portional ist, aber in entgegengesetzte Rich- tung zeigt. Am Ort der Kerne in der Molekel wirkt daher nieht das i~uBere Feld H0, sondern das schwi~chere Feld
H = H0(1 -- ~).
Die dimensionslose Abschirmkonstante a hat ffir Protonen die GrSBenordnung 10 -5. Da die Absehirmung durch das diamagnetisehe Verhalten der Elektronen hervorgerufen wird, ~ndert sich die Abschirmkonstante mit der chemischen Gruppierung, in welche die Kerne eingebaut sind. Dabei gehSrt zu jeder che- mischen Baugruppe eine charakteristische Abschirmkonstante. Infolgedessen sind die Resonanzlinien der Protonen im Spektrum geordnet naeh den chemischen Gruppen, in denen sic sich befinden. Die Erscheinung wird ,,Chemische Versehiebung" genannt.
Diejenigen Kerne einer Molekel, die sich dnreh Symmetrieoperationen ineinander fiberffihren ]assen, wie z. B. die sechs Pro- tonen eines Benzolringes, besitzen dieselbe Abschirmkonstante. Sic werden als i~qui- valente Protonen bezeichnet,ihre Linien fallen im Spektrum in eine Linie zusammen.
Das Resonanzspektrum einer Molekel be- steht also aus den getrennt liegenden Reso- nanzlinien der chemischen Gruppen wie z. B. -CH~, -COOCH 8, - O H oder andere,
deren jede nur einen Satz gquivalenter Pro- tonen enthiilt.
2.3. Die indirekte Spin]copplung Die l~esonanzlinie eines Satzes yon s
valenten Protonen kann aber wieder aufge- spalten werden durch die indirekte Spinkopp- tung mit den ~quivalenten Protonen eines anderen Satzes, im allgemeinen einer anderen chemischen Gruppe, die mit der beobaehteten fiber Hauptvalcnzen verbunden ist. Diese Wechselwirkung ffihrt zu der Multiplett- struktur des Spektrums:
Die indirekte Spinkopplung wirkt auf dem Wege fiber die Bindungselektronen. Sic beruht darauf, dal3 ein orientierter Kernspin die Spins der Elektronen in seiner Umgebung polarisiert. Diese polarisieren wiederum die Spins anderer Elektronen, so dal~ durch alter- nierende Polarisation fiber mehrere Bindun- gen hinweg am Oft eines Nachbarkerns ein Zusatzfeld entsteht.
Die GrS~e und Richtung des so entstehen- den Feldes wird durch die Anzahl und die Orientiernng der benaehbarten Kerne be- stimmt. Die Wechselwirkung nimmt iedoch mit der Anzahl der zwischen zwei Kernen

Johnsen, Die Bestimmung der Mikrostruldur von Vinylpolymeren 3
liegenden Bindungen stark ab u n d i s t bei Protonen, zwischen denen mehr als drei a- Bindungen liegen, nieht mehr naehweisbar.
:Die Spinkopplung zwisehen den Kernen eines/~quivalenten Satzes yon Kernen wirkt sieh im Spektrum nieht aus. Wenn weiterhin die ehemischen Versehiebungen zwisehen den einzelnen/~quivalenten S/~tzen einer Molekel, also die Frequenzabst/inde zwisehen den Resonanzlinien der S/itze im Spektrum groB sind gegenfiber der Linienaufspaltung infolge der Spinkopplung - und nur solehe F/~lle werden bier behandelt- , dann werden die Verhgltnisse leicht tibersehaubar.
Zum Beispiel haben dann zwei S//tze A und X yon /~quivalenten Protonen die Energie- niveaus
E(m A, mx) = hvo(1 -- ~A)mA -b by0(1 - - a x ) m x
-{- h J A x mA m x .
Dabei bedeuten: h Plancksche Konstante , v0 t lesonanzfrequenz eines isolierten I(ernes, a Abschirmkonstange,
JAxKopplungskonstange zwischen den S~gzen A und X,
m magnetische Quantenzahl.
Sie kann bei n Protonen (Spin-Quan~enzahl 1/2) in einem Satz die n + 1 verschiedenen Werte n /2 ; n/2 -- 1 ; . . . ; -- n/2 annehmen.
:Die ersten beiden Terme der obigen Glei- chung stellen die Wechselwirkungsenergie der t)rotonen in den S/itzen A und X mit dem wirksamen ~uBeren Feld H ( 1 - a ) dar, der dritte Term besehreibt die Wechselwirkungs- energie infolge der Spinkopplung.
Ffir die beobachteten ~berg/~nge gilt die Auswahlregel
A m = •
Bei einem lJbergang im Satz A treten daher folgende Frequenzen auf:
vA(mx) = % 0 -- aA) -k J A x m x .
Hat der Satz X z. B. zwei Protonen, kann mx die Werte 1, 0, - 1 besitzen. In der Resonanz des Satzes A treten daher drei oder allgemein nx + 1 Linien auf.
Durch die Spinkopplung werden also die Spektren kompliziert nnd ihre :Deutung und Auswertung oft sehr ersehwert. Ihre Wir- kung liiBt sich jedoeh durch die Methode der :Doppelresonanz verhindern.
2.4. Spinentkopplung dutch Doppelresonanz Um die Linienaufspaltung infolge der Spin-
kopplung zwischen den Kernen der S/~tze A und X einer Molekel auszusehalten, wird die t~esonanzfrequenz der Gruppe X sehr stark eingestrahlt, so dab die Kerne der Gruppe X
infolge yon Absorptions- und induzierten Emissionstiberg/~ngen sehr h~ufig ihre Orien- tierung zum ~uBeren Feld weehseln. Infolge- dessen/~ndert aueh das dureh die Elektronen- spins auf die Kerne der Gruppe A fiber- tragene 1%1d sehr h/iufig seine Stiirke und seine Polarit//t. In der l~esonanz der Kerne A wirkt daher, //hnlieh wie bei der direkten Dipolweehselwirkung, nur der verschwin- dende zeitliehe Mittelwert des Zusatzfeldes. Infolgedessen f//llt das Multiplett der Kerne A wieder in eine Linie zusammen, die leieht beobachtet und gedeutet werden kann.
3. Beispiele In diesem Absehnitt wird gezeigt, wie mit
der Methode der magnetisehen Kernspin- l%sonanz insbesondere bei Spinentkopplung die Mikrostruktur yon Vinylpolymeren mit einer Komponente nnd die Kettenstruktur yon chemischen Copolymeren bestimmt wer- den kann. Dazn wird zun~ehst das Verfahren am Beispiel eines solchen Vinylpolymeren er- 1/~utert, dessen Spektrum sehr einfaeh ist und ohne Spinentkopplung analysiert werden kann. Es stellt zugleich den ersten (1961) in der Literatur angegebenen Fall dar (9-15).
3.1. P olymethylmethacrylat
Das Polymere hat die Strukturformel
CI-I 8
Es besitzt drei S/~tze versehieden abgesehirm- ter Protonen in den CH2-, Ctt 3- und COOCH a- Gruppen, wobei zwisehen den Protonen in versehiedenen ehemisehen Baugruppen je- wells vier e-Bindungen liegen. Da die beob- aehtete Spinkopplung nur fiber drei a-Bin- dungen reieht, kSnnen die Linien im Spek- trum nut dutch Kopplung inn erhalb der Gruppen aufgespalten werden. Das abet ist nur mSglich in den Methylen(Ctt2)-Gruppen, wenn die beiden Protonen nieht iiquivalent sind, d. h. jedes ffir sich einen Satz bilden. In den CHa- und den COOI-I~-Gruppen sind die Protonen in ]eder festgehaltenen Orientie- rung der Gruppen zwar keineswegs /iqui- valent, infolge der Rotation dieser Gruppe in den untersuehten LSsungen werden sie aber effektiv //quivalent, so dab ihre gegen- seitige Kopplung in den Spektren nicht in Erseheinung tritt. Fiir diese Polymere ist daher ein einfaches nnd, infolge der grogen Anzahl/iquivalenter Protonen in mindestens
1"

4 Kolloid-Zeltschrifl und Zeitschrift fi~r Polymere, Band 210 �9 Heft I
zwei Baugruppen, auch ein intensives Spek- t rum zu erwarten.
In der Abb. 1 sind die Spektren yon L5sun- gen zweier, mit verschiedenen Polymerisa- tionsverfahren hergestellter Pr~parate ein- ander zum Vergleich gegeniibergestellt.
In diesen Spektren gehSrt die Referenzlinie ganz reehts zum Tetramethylsilan, das den LSsungen beigemischt wurde. Die iibrigen drei Liniengruppen mtissen, wie in der Ab-
die in den einzelnen Spektren verschiedene Intensiti~ten besitzen.
Diese erheblichen Unterschiede in den Spektren werden folgendermai~en mit der Konfiguration der Ket ten in Beziehung ge- setzt :
Das Singulett in der CH~-Resonanz zeigt an, da]~ die beiden Methylen-Protonen i~qui- valent sind. Das aber ist nur mSglich, wenn sieh die Methylen-Gruppe an einer syndiotak-
/ -COOCH3 I - CH3 a)
(c%),~si
) 11 I -COOCH 3.
-CH 2-
6iq'l ~83 8~18 8 , 4 6 . ,
b)
I -CH 3 (CH3)4S[
.~ 8(80 8,96 9p9 i t-
Abb. 1. Polymethylmethacrylat (PMMA). 200/0 in Chloroform; 120 ~ 56,4 MHz. a) PMMA mit 95% isotakti- schen Verkniipfungen; b) PMMA mit 85% syadiotaktischen Verkniipfungen
bildung angegeben, den Protonen in den drei ehemischen Baugruppen des Polymeren zu- geordnet werden. Die eine Linie der Meth- oxylgruppen (COOCH3) erseheint in beiden Spektren bei dem gleiehen Wert T = 6,41. In den Resonanzen der CH~- und der CH~- Gruppen treten charakteristische Unter- sehiede z~Sschen den beiden Spektren auf~ die auf die versehiedene sterische Struktur der Molekelketten hinweisen. In der CH 2- Resonanz erseheinen ein Linienquartett mit den Sehwerpunkten der i~ul~eren Linienpaare bei T = 7,83 und 8,46 und eine Einzellinie bei T = 8,18. Weiterhin zeigt die CHs-Re- sonanz drei Linien bei T = 8,80; 8,96; 9,09,
tischen Verkntipfung zweier Monomerein- heiten befindet. Hier sind die beiden Pro- tonen in der speziellen Konformation des gestreckten Kohlenstoff-Geriistes ~quivalent. Aber aueh zu ieder anderen Konformation, in der die Protonen eine versehiedene mole- kulare Umgebung besitzen, gibt es eine ener- getisch gleiehwertige Konformation, in der die molekulare Umgebung spiegelsymmetrisch ist. Wi~hrend der statistischen W~rmebewe- gung der Molekelke~ten in der LSsnng haben daher beide Protonen im zeitlichen )/Iittel dieselbe molekulare Umgebung und sind effek~iv ~quivalent. Im Spektrum zeigen sie daher nur eine Linie. An einer isotaktischen

Johnsen, Die Bestimmung der Mikrostruktur von Vinylpolymereu 5
Verkntipfung jedoch trifft diese 13berlegung nicht zu, so dab die Resonanz der beiden ver- schieden abgeschirmten und daher nicht /~quivatenten Protonen in ein Linienquartett aufgespalten ist. Das obere Spektrum der Abb. 1 gehSrt daher zu einem isotaktischen, d~s untere zu einem iiberwiegend syndiotak- tischen PMlVLA.
Die drei Linien der Methyl-Resonanz wer- den CHa-Gruppen in so]chen Monomerein- heiten zugeordnet (9), die
a) nach beiden Seiten hin isotaktisch ver- kniipft sind (v = 8,80);
b) an einem Ende isotaktisch, am anderen syndiotaktisch verkniipft sind (v = 8,96), und
c) zwischen zwei syndiotaktisch verkntipf- ten Grundbausteinen liegen (T = 9,09).
Die Reihenfolge in der Zuordnung der drei Linien folgt aus dem Intensit~svergleich mit den beiden Resonanzen der Methylen- Gruppen und ist eindeutig.
In allen drei aufgefiihrten ,,Dreiergruppen" yon Monomereinheiten wechselt die Kette wiihrend der statistischen W~rmebewegung aufgrund der verschiedenen Verkntipfungen (Konfigurationen) auch zwisehen verschie- denen stabilen Konformationen. Daher be- finder sich jeweils die Methyl-Gruppe in der zentralen Monomereinheit im zeitlichen Mit- tel in verschiedenen molekularen Umge- bungen und ist deshalb in den drei ,,Triaden" unterschiedlich stark magnetisch abge- schirmt, so dab ihre Resonanzlinien im Spek- t rum getrennt erscheinen.
Die Konzentrationen der isotaktischen und der syndiotaktischen Verkniipfungen sowie die Konzentrationen der Verkniipfungs- paare 1) folgen aus den Linienintensiti~ten im Spektrum.
In den Spektren yon Polymeren mit nicht extremer Stereoregulariti~t wie in der Abb. 1 sind in der Cttg-Resonanz das Linienquartett und die Einzellinie deutlich sichtbar (s. Abb. 2). In diesen F~llen ]iefern die normier- ten Intensit~ten des Quartetts und des Singuletts unmittelbar die Konzentrationen P(i) und P(s) der beiden Verkntipfungsarten (i = isotaktisch, s = syndiotaktisch).
Einen weitaus genaueren und tieferen Ein- blick in den Aufbau der Ket ten geben jedoch die drei Linien der ~r Ihre normierten Intensiti~ten entsprechen den
1) Zur Terminologie: Eine Verkniipfung besteht zwischen einem Paar (Diade) yon Monomereinheiten, ein Verkniipfungspaar in einer Dreiergruppe (Triade) yon Monomeren, usw.
Konzentrationen der Verkniipfungspaare P(ii); P(is) + P(sl); P(ss).
Die heterotaktischen Verkntipfungspaare (is) und (si) sind experimentell nicht unter- scheidbar, so dab ihre Konzentrationen P(is) + P(si) gemeinsam durch die Intensiti/t der mittleren Linie erfM~t werden.
Diese Paarkonzentrationen sind nun nicht unabh~ngig (14, 16) von den Konzentra- tionen P(i) und P(s) der Einzelschritte.
Addiert man n~mlich die isotaktisehen Verkntipfungen, die auf eine gleiche Ver- kntipfung folgen, und die isotaktischen Ver- knfipfungen, die auf eine syndiotaktische
I
I , ' ) I L 723 ~,18 8,46
a,80 a, ge 9,o9 ,-ff
Abb. 2. CH~- und CHs-Resonanz eines Polymethyl- methacrylaSs mi~ 81% isoSaktischen Verkniipfungen.
20% in Chloroform; 120 ~ 56,4 MFIz
Verkntipfung folgen, dann erh/~lt man die Anzahl aller isotaktischen Verkniipfungen, so dab
P(si) + P(ii) = P(i).
Andererseits kann eine isotaktische Ver- kniipfung nur vor einer gleichen oder vor einer syndiotaktischen Verkniipfung stehen, so dab auch
P(is) + P(ii) = P(i).
Daraus folgt, dab die Konzentrationen der gemeinsam gemessenen heterotaktischen Paare (is) und (si) ein~nder gleich sind. ])as heiB~, aus den Paarkonzentrationen kSnnen die Konzentrationen der einzelnen Ver- kntipfungen P(i) und P(s) abgelei~et wer- den nach der Gleichung
1/2 [P(is) + P(si)] + P(ii) = P(i).
Vorausgesetzt ist immer, dab jeder Mono- mereinheit eine Einheit folgt und eine Ein-
6 Kolloid-Zeitschr{ft und Zeitschrifl fiir Polymere, Band 210 �9 Heft I
heir vorausgeht, dab also der Polymerisa- tionsgrad groB ist und die Endeinheiten ver- nachl/~ssigt werden k6nnen. Das ist aber bei den teehnischen Polymeren immer der Fall.
Mit den Konzentrationen P(is) und P(si) lassen sich nun die mittleren Lgngen der Se- quenzen bestimmen. Denn eine isotaktisehe Sequenz endet mit einer syndiotaktischen Verkniipfung, der eine isotaktisehe Ver- kntipfung vorausgeht. Das heiBt, zu jeder iso- taktisehen Sequenz gehSrt solch ein isotak- tisch-syndiotaktisches Verkniipfungspaar. Seine Konzentration P(is) ist daher identiseh mit der Konzentration der isotaktisehen Se- quenzen. Dividiert man nun die Konzen- tration aller isotaktisehen Verkniipfungen durch die Konzentration ihrer Sequenzen, dann erh/ilt man die mittlere L/inge/~(i) der isotaktischen B15cke, angegeben in Anzahlen yon Monomereinheiten, also
P(i) P (is)" = # (i).
Die entspreehenden GrSgen fiir den syn- diotaktischen Fall erh/flt man durch Ver- tauschen yon i und s. Er wird daher im folgenden nieht mehr erw//hnt.
In sehr vielen F/fllen abet kann man nicht nur die mittleren Sequenzlgngen, sondern sogar die Sequenzl//ngenverteilung aus den Spektren ableiten.
Die gemessenen Konzentrationen der Ein- zelschritte k6nnen n/~mlich naeh ihrer Defi- nition als Wahrseheinliehkeiten, d. h. als An- zahl der gtinstigen F/file zur Anzahl der Ge- samtf/flle aufgefaBt werden. Danach ist P(i) die Wahrseheinliehkeit dafiir, dab eine an beliebiger Stelle der Kette herausgegriffene Bindung zwisehen zwei Monomereinheiten gerade eine isotaktische Verkniipfung ist.
Andererseits wird aber die Art der Ver- kniipfung w/~hrend der Polymerisation fast- gelegt. In dieser Betraehtungsweise ist daher P(i) die Wahrseheinliehkeit daftir, dab w//h- rend der Polymerisation eine Monomerein- heir isotaktiseh an die Endeinheit der wach- senden Kette angelagert wird, d. h. die Wahr- scheinlichkeiten Pi ftir den isotaktisehen Polymerisationssehritt.
Analog wird die Paarkonzentration P(ii) aufgefagt als die Wahrscheinliehkeit, dab wghrend der Polymerisation zwei isotak- tische Verknfipfungen aufeinander folgen. In dem einfaehsten denkbaren Fall ist w/ih- rend der Polymerisation die Art der Anlage- rung unabh/~ngig yon der Art des vorherge- gangenen Sehrittes. Dann ist aber die Wahr- seheinliehkeit ftir ein Paar yon aufeinander
folgenden Polymerisationsschritten gleich dam Produkt der Wahrseheinlichkeiten fiir die Einzelschritte. Das heiBt, as gilt:
P (ii) = Pi ~
P(is) + P(si) = 2PiPs
P(ss) = Ps ~.
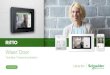
Ob dieser Fall vorliegt, kann sehr leicht naeh- geprtift werden. Dazu warden die gemessenen Wahrscheinlichkeiten der Paare gegen die gemessenen Wahrseheinlichkeit eines Einzel- sehrittes aufgetragen (9).
Das ist in Abb. 3 ftir eine groBe Anzahl von Methylmethacrylaten getan, die unter ver-
1,0
0,9
0,8.
~[ 0,7
0,6 P
0,5
0,4.
\ /
2 ps pi
O. 0 o,l 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0
k
Pi Abb. 3. Die Pa~rkonzen~rationen P (ii), P (is + si) und P(ss) einiger Polymethylmethaerylate aufgetmgen gegen die Wahrscheinliehkeit einer isotaktischen
Wachstumsreaktion
versehiedenen Bedingungen und Verfahren hergestellt wurden. Drei tibereinanderliegende MeBpunkte gehSren zu einer Substanz. Dieaus- gezogenen Parabeln geben die theoretischen Werte fiir die gesehilderte einfaehe Statistik wieder. Diese liegt bei einer groBen Anzahl yon Polymerisaten vor, insbesondere auf der linken Seite, die zu tiberwiegend syndiotak- tischen Prgparaten gehSrt. Fiir diese Poly- mere sind daher das Gesetz, nach dem ihre Ketten wachsen, und die bestimmenden Parameter bekannt. Damit kann die Sequenz- I/ingenverteilung berechnet werden.
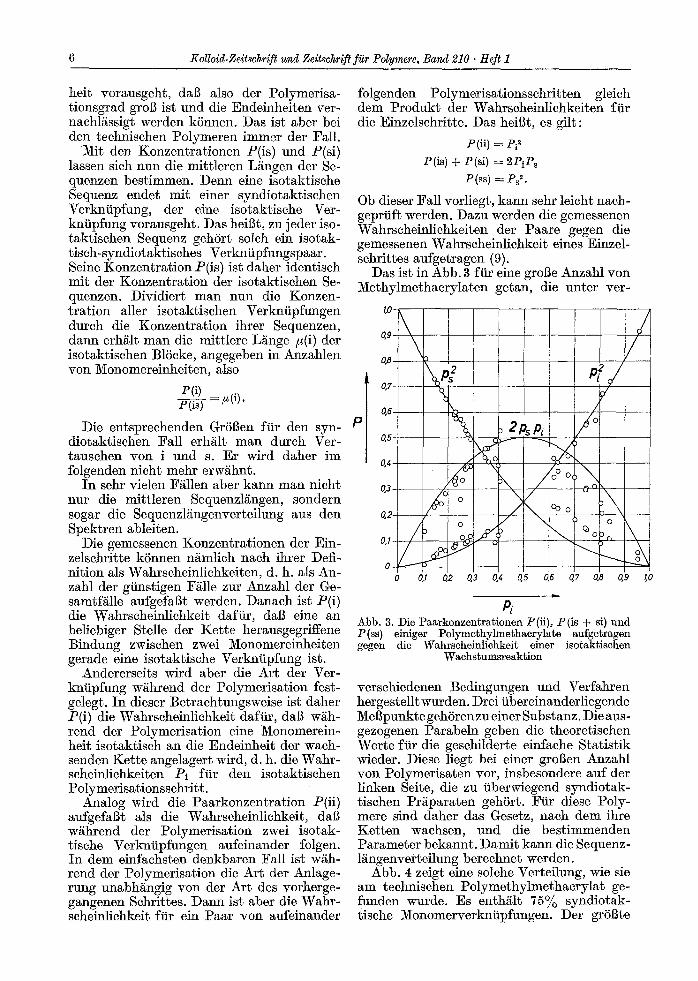
Abb. 4 zeigt eine solehe Verteilung, wie sie am technisehen Polymethylmethacrylat ge- funden wurde. Es enth/flt 75% syndiotak- tische Monomerverkntipfungen. Der grSBte
Johnsen, Die Bestimmung der Mikrostruktur yon ginylpolymerer~ 7
Teil der 3lonomereinheiten befindet sich in syndiotaktischen Sequenzen mit etwa 6 Mo- nomereinheiten, syndiotaktische Sequenzen mit mehr als 20 Einheiten kommen praktisch nicht vor. Die isotaktischen Sequenzen sind augerordentlich kurz, sie enthalten praktisch h6chsten vier Monomereinheiten.
F
"14" %
I0
6-
2
n 0 --- I I I I I
I 4 8 12 16 2 o
Abb. 4. Gewichtsanteil der abgeschlossenen isotak~i- schen (i, senkrechte Kurve) und syndiotaktischen (s) Sequenzen eines technischen Polymethylmethaerylats als ~unktion ihrer LEngen n in Anzahlen yon Monomer-
einheiten
Fiir andere Polymere, deren Megwerte nicht auf den Parabeln liegen, deren Molekelketten also nach einem komplizierteren Gesetz wachsen, kann man ohne spezielle Annahmen nut die mittlere Sequenzl/inge angeben.
3.2. Poly-~-methylstyrol Wie am Polymethylmethacrylat kann auch
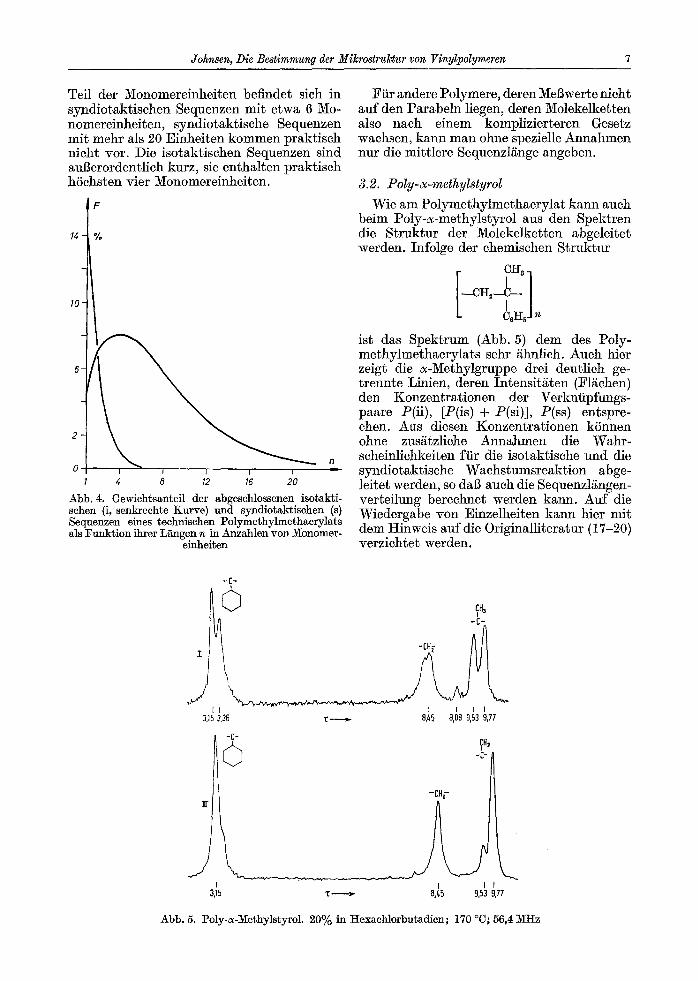
beim Poly-c~-methylstyrol aus den Spektren die Struktur der Molekelketten abgeleitct werden. Infolge der chemischen Struktur
ist das Spektrum (Abb. 5) dem des Poly- methylmethacrylats sehr ~hnlieh. Aueh hier zeigt die ~-3~ethylgruppe drei deutlich ge- trennte Linien, deren Intensitgten (Flgehen) den Konzentrationen der Verkniipfungs- paare P(ii), [P(is) + P(si)], P(ss) entspre- chen. Aus diesen Konzentrationen k6nnen ohne zusgtzliche Annahmen die Wahr- scheinlichkeiten ftir die isotaktische und die syndiotaktische Wachstumsreaktion abge- leitet werden, so da6 auch die Sequenzl/ingen- verteilung berechnet werden kann. Auf die Wiedergabe yon Einzelhciten kann hier mit dem Hinweis auf die 0riginalli~eratur (17-20) verzichtet werden.
lr
/ 3.15 3.36
-C-
1[ I I I I x ~ 8,45 9,08 9,53 9/77
-c~ c,,
I 3,15 T
-CHs
8,45
L 9,53 9,77
Abb. 5. Poly-c~-Methylstyrol. 20% in I-Iexachlorbu~adien; 170 ~ 56,4 MHz
8 Kolloid.Zeitschrift und Zeitschrift fi~r Polymere, Band 210 �9 Heft I
4. Beispiele fiir Spinentkopplung Sehr viel komplizierter sind die S3?ektren
derjenigen Vinylpolymeren
die start einer ~-~ethylgruploe nur ein ein- faehes H-Atom tragen; denn dieses a-Proton ist mit den ~ethylen-Protonen der eigenen ~[onomereinheit und mit den beiden Pro- tonen in der folgenden Naehbareinheit ge- koppelt. I)aher sind die I~esonanzlinien des o~-Protons und auch die Linien der Methylen- Protonen in Multipletts aufgespalten, welche die geringen Linienversehiebungen infolge der sterisehen Effekte iiberdeeken. Bei die- sen Substanzen handelt es sieh also um geeig- nete Objekte ftir die Anwendung der Doppel- resonanz-Methode. Im folgenden werden das Polyvinylehlorid (21-33), der Polyvinyl- alkohol (37-40) und die VC-VDC-Copoly- meren behandelt.
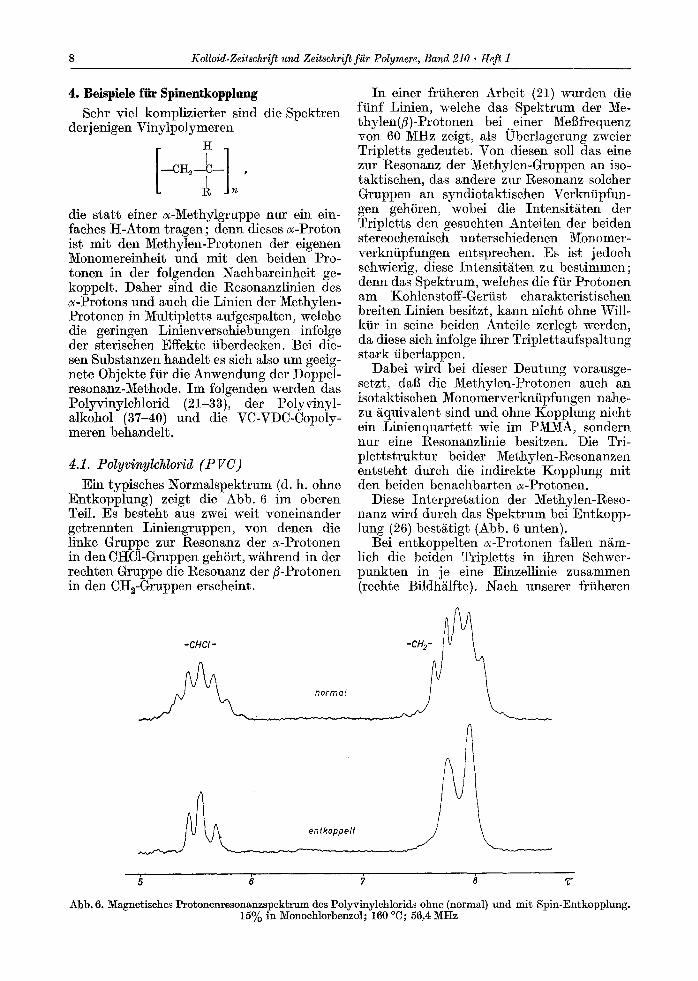
4.1. Polyvinylchlorid (P VC) Ein typisehes Normalspektrum (d. h. ohne
Entkopplung) zeigt die Abb. 6 im oberen Tell. Es besteht aus zwei welt voneinander getrennten Liniengruppen, yon denen die linke Gruppe zur Resonanz der ~-~rotonen in den CI-IC1-Gruppen gehSrt, w/~hrend in der reehten Gruppe die I~esonanz der fl-Protonen in den CH2-Gruppen erseheint.
In einer frtiheren Arbeit (21) wurden die fiinf Linien, welehe das Spektrum der Me- thylen(~)-Protonen bei einer Meftfrequenz yon 60 MHz zeigt, als Uberlagerung zweier Tripletts gedeutet. Von diesen soil das eine zur l~esonanz der Methylen-Gruppen an iso- taktischen, das andere zur Resonanz solcher Gruppen an syndiotaktischen Verkniipfun- gen gehSren, wobei die Intensit/~ten der Tripletts den gesuehten Anteilen der beiden stereochemisch unterschiedenen Monomer- verkniipfungen entsprechen. Es ist jedoch schwierig, diese Intensit/iten zu bestimmen; denn das Spektrum, welches die ffir Protonen am Kohlenstoff-Geriist charakteristisehen breiten Linien besitzt, kann nieht ohne Will- kiir in seine beiden Anteile zerlegt werden, da diese sich infolge ihrer Triplettaufspaltung stark fiberlappen.
Dabei wird bei dieser Deutung vorausge- setzt, daft die ~ethylen-Protonen aueh an isotaktisehen Monomerverkniipfungen nahe- zu/~quivalent sind und ohne Kopplung nieht ein Linienquartett wie im PMMA, sondern nut eine Resonanzlinie besitzen. Die Tri- plettstruktur beider Methylen-I~esonanzen entsteht durch die indirekte Kopplung mit den beiden benachbarten ~-Protonen.
Diese Interpretation der Methylen-Reso- nanz wird dutch das Spektrum bei Entkopp- lung (26) best/~tigt (Abb. 6 unten).
Bei entkoppelten cr fallen n/s ]ieh die beiden Tripletts in ihren Schwer- punkten in je eine Einzellinie zusammen (reehte Bildh/~lfte). Nach unserer friiheren
~ t
-CHC( - -CH2- A
Abb. 6. Magnetisehes Protonenresonanzspektrum des Polyvinylchlorids ohne (normal) und mit Spin-Entkopplung. 15% in Monochlorbenzol; 160 ~ 56,4 MHz

Johnsen, Die Bestlmmung der Mikrostruktur von Vinylpolymeren 9
Interpretation wird die Linie bei v = 7,78 den Protonen in solchen 3/Iethylen-Gruppen zugeordnet, die isotaktiseh mit der n/iehsten Monomereinheit verkntipft sind, die zweite Linie bei ~ = 7,96 gehSrt folglich zu Pro- tonen in Methylen-Gruppen an syndiotak- tisehen Verkniipfungen. Die Intensit/~t dieser zweiten Linie w//chst in den Spektren auf Kosten der anderen, wenn die Po]ymerisa- t ionstemperatur der untersuchten Substan- zen f/~llt. Auf diesem Befund und auf der Be- hauptung, dab mit fallender Polymeri- sationstemperatur die Anzahl der syndiotak- tischen Verkniipfungen ansteigt (34-36), be- ruht die angegebene Zuordnung. Sie wird weiterhin durch die Spektren niedermoleku- larer Nodellsubstanzen gesttitzt und kann damit als sicher angesehen werden.
Die normierten Intensit/iten der beiden Linien der Methylen-Resonanz bei Entkopp- lung entspreehen den relativen Anzahlen der beiden stereochemiseh verschiedenen Methy- len-Gruppen und damit den Konzentratio- hen P(i) und P(s) der isotaktisehen und der syndiotaktisehen 31onomerverkntipfungen in den Molekelketten. Da sieh bei Entkopplung nur noeh die Auslgufer der beiden Linien fiberlappen, kSnnen hier P(i) nnd P(s) er- heblieh genauer ermittelt werden als durch Zerlegung des Normalspektrums.
Werden dutch ein stark eingestrahltes I-Iochfrequenzfeld die 3/Iethylen-Protonen ent- koppelt, dann erscheinen im Spektrum der entkoppelten ~-Protonen (Abb. 6 links) start der komplizierten Multiplettstruktur nun drei Linien bei v = 5,48; 5,59 und 5,71. Diese drei Linien der entkoppelten ~-Protonen im PVC-Spektrum entsprechen den drei Linien der ~-Methylgruppen in den Normalspektren des Polymethylmethacrylats und des Poly-c~- methylstyrols.
Die Zuordnung der drei Linien zu den drei versehiedenen Verkniipfungspaaren (ii), (is + si) und (ss) im PVC kann mit Hilfe der Spin-Entkopplung in folgender Weise (26) an die gesicherte Zuordnung der beiden Linien in der Methylen-Resonanz angeschlossen werden :
Wghlt man die Entkopplungsfrequenz so, dab nur die Protonen der Methylen-Gruppen in syndiotaktisehen Sequenzen (3 = 7,96) zu hgufigen l~)berg/~ngen angeregt werden, dann sind aueh nur ~-Protonen in syndiotaktiseh verkniipften Triaden yon Monomereinheiten yon den fl-Protonen entkoppelt; aus der breiten ~-Resonanz wi/ehst dann nur an der linken Seite bei ~ :~ 5,48 eine scharfe Linie heraus. Sie gehSrt folglieh zu c~-Protonen in
solchen Monomereinheiten, die mit beiden Naehbareinheiten syndiotaktisch verkniipft sind.
Entkoppelt man andererseits nnr die Methylen-Protonen in isotaktisehen Se- quenzen (v = 7,78), dann wird nur die Linie der ~-Protonen in solchen Monomereinheiten scharf, die mit beiden Naehbareinheiten iso- taktisch verkniipft sind. Die dritte Linie der r in der zentralen Einheit yon heterotaktisch (is) und (si) verkntipften Dreiergruppen liegt in der Mitte bei v = 5,59. Die normierten Intensit/iten dieser Linien sind proportional den Konzentrationen der zugehSrigen Triaden, d. h. der Verkntipfungs- paare.
Aus diesen Paarkonzentrationen kSnnen wiederum die Konzentrationen der einzelnen Verkniipfunge.n P(i) und P(s) abgeleitet werden. Die Ubereinstimmung dieser Werte mit den Konzentrationen P(i) und P(s), die direkt in der Methylen-Resonanz beob- aehtet werden, stfitzt wiederum die ange- gebene Deutung des Spektrums.
Teehnische Polyvinylchloride besitzen einen Anteil yon etwa 55~o syndiotaktisehen Verkniipfungen in ihren Nolekelketten. Die mit I-Iilfe der Paarkonzentrationen nnmittel- bar gemessenen mittleren Sequenzl/ingen sind daher sehr kurz und liegen fiir isotaktisehe und fiir syndiotaktische Sequenzen bei unge- fghr zwei Nonomereinheiten. Wie bei radi- kaliseh polymerisiertem Polymethylmeth- aerylat wurde auch hier keine Abh/~ngigkeit der Anlagerungswahrseheinlichkeiten yon der Art der vorhergehenden Wachstums- reaktion beobachtet, so dab auch die Sequenz- lgngenverteilung ohne zusgtzliehe Voraus- setzung bereehnet werden kann.
4.2. Polyvinylalkohol ( P VA )
Der Polyvinylakohol wird aus Polyvinyl- /~thern nnd Polyvinylestern der allgemeinen Formel
I-I
; Jn hergestellt. Seine stereoehemisehe Mikro- struktur wird dabei yon den Strukturen der Ausgangspolymeren bestimmt. Auf dem Um- weg fiber den PVA kann man deshalb auf die Strukturen der Potyvinyl/~ther und Polyvinyl- ester zuriieksehlieBen, wenn diese sieh nieht direkt ermitteln lassen.
Die Abb. 7 zeigt die 3lethylen-Resonanz (Normalspektrnm) eines PVA bei Verwen-
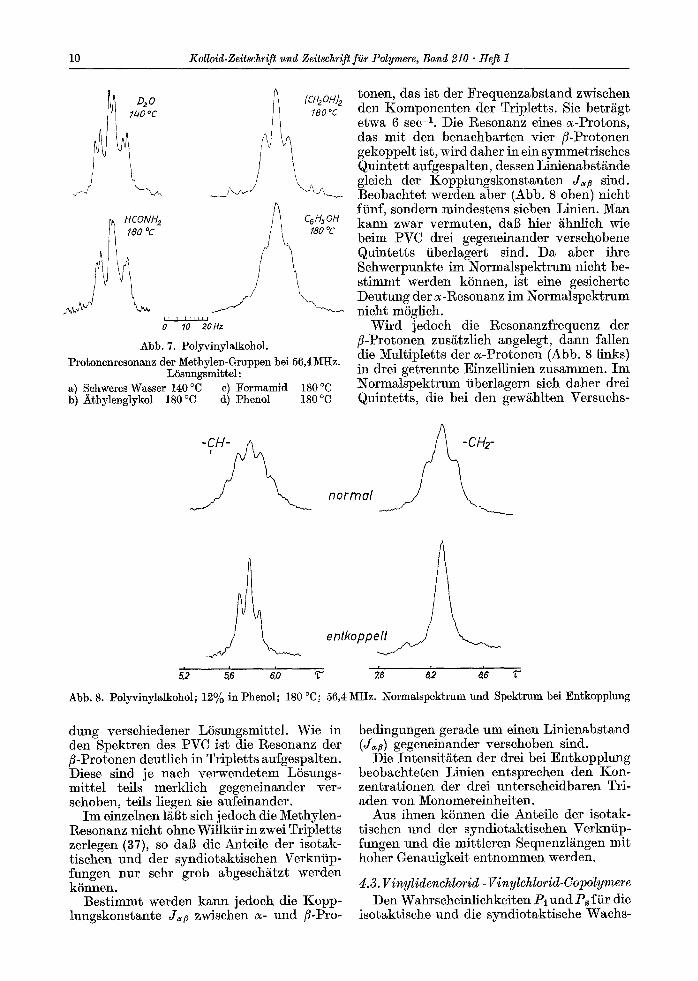
10 Kolloig-zeitschrift und Zeltschrift fi~r Polymere, Band 210 �9 Heft I
~o 1#0 ~
HCONHz
tt 180 % ~ ~ C6HSOH
10 20 Hz
Abb. 7. Polyvinylalkohol. Pro~onenresonanz der ~ethylen-Gruppen bei 56,4MHz.
LSsungsmit~el: a) Sehweres Wasser 140 ~ c) Formamid 180 ~ b) Athylenglykol 180 ~ d) Phenol 180 ~
normal
tonen, das ist der Frequenzabstand zwischen den Komponenten der Tripletts. Sie betr~gt etwa 6 sec -1. Die Resonanz eines cr das mit den benachbarten vier /%Protonen gekoppelt ist, wird daher in ein symmetrisches Quintett aufgespalten, dessen Linienabst~nde gleich der Kopplungskonstanten J~a sind. Beobachtet werden aber (Abb. 8 oben) nicht fiinf, sondern mindestens sieben Linien. Man kann zwar vermuten, dal~ hier ~hnlich wie beim PVC drei gegeneinander verschobene Quintetts tiberlagert sind. Da abet ihre Schwerpunkte im Normalspektrum nicht be- stimmt werden kSnnen, ist eine gesicherte Deutung der ~-Resonanz im Normalspektrum nicht mSglich.
Wird jedoch die Resonanzfrequenz der /%Protonen zusgtzlich angelegt, dann fallen die Multipletts der c~-Protonen (Abb. 8 links) in drei getrennte Einzellinien zusammen. Im Normalspektrum iiberlagern sich daher drei Quintetts, die bei den gewghlten Versuchs-
entkoppe~~.~.~ Abb. 8. Polyvinylalkohol; 12% in Phenol; 180 ~ 56,4 MtIz. Normalspektrum und Spektrum bei En~kopplung
dung verschiedener LSsungsmittel. Wie in den Spektren des PVC ist die Resonanz der fl-Protonen deutlich in Tripletts aufgespalten. Diese sind ]e nach verwendetem LSsungs- mittel teils merklich gegeneinander ver- schoben, teils liegen sie aufeinander.
Im einzelnen l~6t sich jedoch die Methylen- Resonanz nicht ohne Willkiir in zwei Tripletts zerlegen (37), so daf~ die AnLeile der isotak- tischen und der syndiotaktischen Verkniip- fungen nur sehr grob abgeschgtzt werden kSnnen.
Bestimmt werden kann jedoch die Kopp- lungskonstante J ~ zwischen c~- und fl-Pro-
bedingungen gerade um einen Linienabst~nd (J~) gegeneinander verschoben sind.
Die Intensit~ten der drei bei Entkopplung beobachteten Linien entsprechen den Kon- zentrationen der drei unterscheidbaren Tri- aden yon Monomereinheiten.
Aus ihnen kSnnen die Anteile der isotak- tischen und der syndiotaktischen Verknfip- fungen und die mittleren Sequenzl~ngen mit hoher Genauigkeit entnommen werden.
6.3. V iny l idenchlor id- V inylchlorid-Copolymere Den Wahrscheinlichkeiten Pi und Ps ftir die
isotaktische und die syndiotaktische Wachs-
Johnsen, Die Bestimmung der Mikrostruktur yon Vinylpolymeren 11
tumsreaktion wi~hrend der Polymerisation der sterischen Copolymeren entsprechen bei chemischen Copolymeren die Wahrschein- ]ichkeiten P n und P2s fiir die Anlagerung der Komponenten M 1 und M s an die gleiche Ein- heir und die Wahrscheinlichkeiten Pls und Psi fiir die Anlagerung an das Comonomere.
Nach der kinetischen Theorie der Copoly- merisation besteht folgender Zusammenhang zwischen diesen Wahrscheinlichkeiten und der Zusammensetzung des Polymerisations- ans&tzes:
r~X~ Pn = rlX1 + X2 P,~ = 1 - Pn
r~X2 P21 -= 1 - - P2~. P~2 ~ X1 + r2X~
Dabei bedcuten X 1 und X s die Molenbrtiche der Monomeren M~ und M~ im Ansatz und r 1 und r~ die Copolymerisationsparameter.
Zusiitzlich werden hier die GrSBen P1 und P2 eingeffihrt. Sie bezeichnen die Wahrschein- ]ichkeiten, mit denen die Monomereinheiten M 1 und M s bei einer beliebigen Wachstums- reaktion an die Kct ten eingelagert werden. Diese GrSBen kSnnen aus den vorher ange- gebenen Wahrscheinlichkeiten Pn, P~, P~s und P21 abgeleitet werden nach folgenden Beziehungen:
P1 = -PIPn + P~P~I
Bei bekannt m Polymerisationsansatz und bekannten Polymerisationsparametern r 1 und r~ l~Bt sich mit diesen Wahrscheinlichkeiten die molekulare Struktur in den Ket ten des Copolymeren berechnen.
Im folgenden werden die berechneten mit den spektroskopisch bestimmten Konzen- trationen der Diaden und Tetraden ver- glichen.
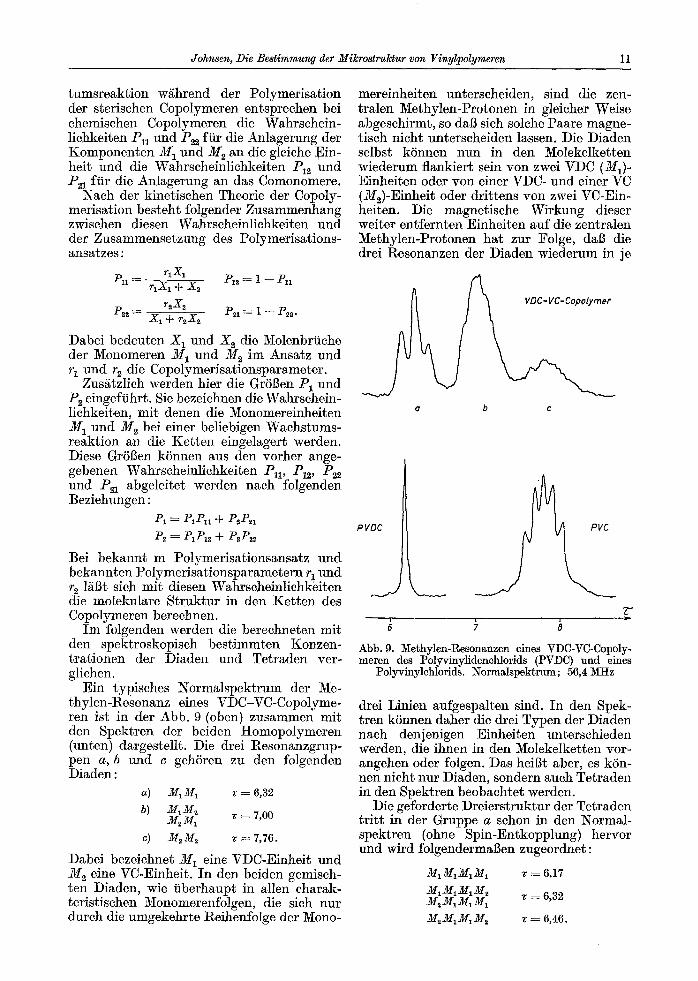
Ein typisches Normalspektrum der Me- thylen-Resonanz eines VDC-VC-Copolyme- ten ist in der Abb. 9 (oben) zusammen mit den Spektren der beiden Homopolymeren (unten) dargestellt. Die drei Resonanzgrup- pen a, b u n d c gehSren zu den folgenden Diaden:
a) M1M1 T ----- 6 ,32
b) MIM~ M~M~ ~ = 7,00
c) M2M2 ~ = 7,76.
Dabei bezeichnet M 1 ein6 VDC-Einheit und M seine VC-Einheit. In den beiden gemisch- ten Diaden, wie tiberhaupt in allen charak- teristischen Monomerenfolgen, die sich nur durch die umgekehrte Reihenfolge der Mono-
mereinheiten unterscheiden, sind die zen- tralen Methylen-Protonen in gleicher Weise abgeschirmt, so dab sich solche Paare magne- tisch nicht unterscheiden lassen. Die Diaden selbst kSnnen nun in den Molekelketten wiederum flankiert sein yon zwei VDC (M1)- Einheiten oder yon einer VDC- und einer VC (M~)-Einheit oder drittens von zwei VC-Ein- heiten. Die magnetische Wirkung dieser weiter entfernten Einheiten auf die zentralen Methylen-Protonen hat zur Folge, dab die drei Resonanzen der Diaden wiederum in je
Q b c
P VDC
Abb. 9. Methylen-Resonanzen eines VDC-VC-Copoly- meren des Polyvinylidenchlorids (PV])C) und eines
Polyvinylchlorids. Normalspektrum; 56,4 MHz
drei Linien aufgespalten sind. In den Spek- tren kSnnen daher die drei Typen der Diaden nach denjenigen Einheiten unterschieden werden, die ihnen in den Molekelketten vor- angehen oder folgen. Das heiBt aber, es kSn- nen nicht nur Diaden, sondern auch Tetraden in den Spektren beobachtet werden.
Die geforderte Dreierstruktur der Tetraden tr i t t in der Gruppe a schon in den Normal- spektren (ohne Spin-Entkopplung) hervor und wird folgendermaBen zugeordnet:
M1M1M1M1 ~ = 6,17 M1MIM1M~ M~M1M1M1 T = 6 ,32
M2M1MIM~ v-= 6,46.
12 Kollold-Zeitschrifi und Zeitschrifl f4ir PoIymere, Band 210 �9 Heft I
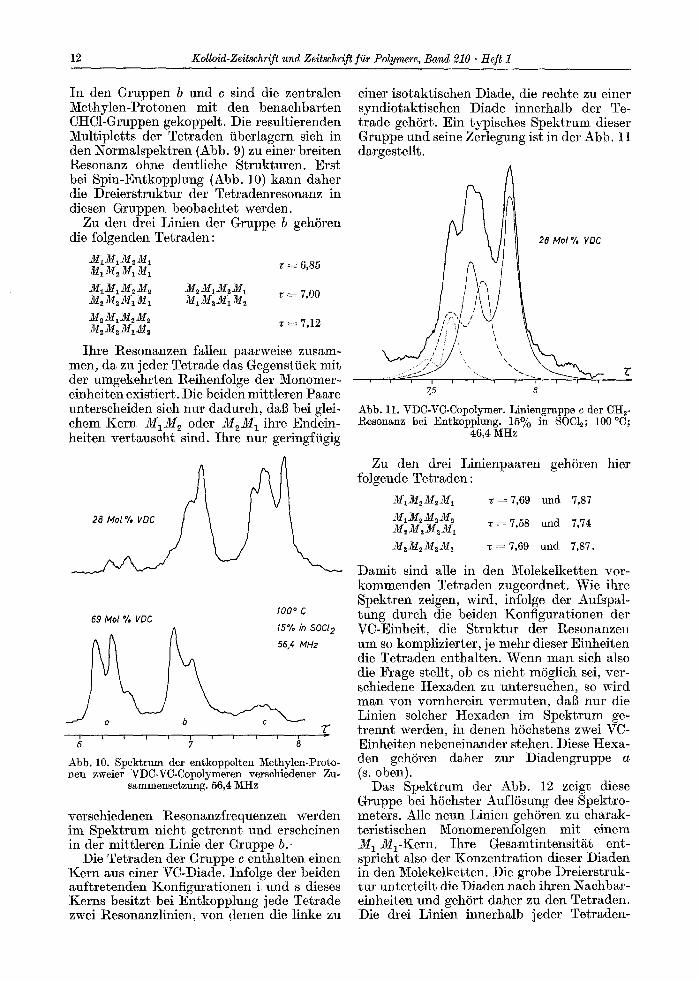
In den Gruppen b und c sind die zentralen Methylen-Protonen mit den benaehbarten CHC1-Gruppen gekoppelt. Die resultierenden Multipletts der Tetraden fiberlagern sieh in den Normalspektren (Abb. 9) zu einer breiten l~esonanz ohne deutliche Strukturen. Erst bei Spin-Entkopplung (Abb. 10) kann daher die Dreierstruktur der Tetradenresonanz in diesen Gruppen beobaehtet werden.
Zu den drei Linien der Gruppe b gehSren die folgenden Tetraden:
M1MIM~M1 M1M~ Mi M1 v -~- 6,85
M~M1M2M~ 2/I2M1M2MI M2M2M~M~ M~M2MIM~ v = 7,00
M,M~M, M2 M~M2M~M~ v = 7,12
Ihre Resonanzen fallen paarweise zusam- men, da zu j eder Tetrade das Gegenstfiek mit der umgekehrten Reihenfolge der Monomer- einheiten existiert. Die beiden mittleren Paare unterscheiden sieh nur dadureh, dal~ bei glei- chem Kern M 1 M 2 oder M 2 M 1 ihre Endein- heiten vertauseht sind. Ihre nur geringffigig
100 ~ C 69 b4ol % VDC
15% in 50Cl 2
Abb. 10. Spektrum der entkoppelten Methylen-Proto- hen zweier VDC-VC-Copolymeren verschiedener Zu-
sammensetzung. 56,4 3aHz
verschiedenen l~esonanzfrequenzen werden im Spektrum nieht getrennt und erseheinen in der mittleren Linie der Gruppe b.
Die Tetraden der Gruppe c enthalten einen Kern aus einer VC-Diade. Infolge der beiden auftretenden Konfigurationen i und s dieses Kerns besitzt bei Ent.kopplung jede Tetrade zwei Resonanzlinien, yon denen die linke zu
einer isotaktischen Diade, die reehte zu einer syndiotaktischen Diade innerhalb der Te- trade gehSrt. Ein typisches Spektrum dieser Gruppe und seine Zerlegung ist in der Abb. 11 dargestellt.
28 Mol % VDC
7,5 o
Abb. lI. VDC-VC-Copolymer. Liniengruppe e der CH~- P~esonanz bei Entkopplung. 15~ in SOCI~; 100 ~
46,4 MHz
Zu den drei Linienpaaren gehSren hier folgende Tetraden:
M1M~M~M1 ~ 7 , 6 9 und 7,87 M1M2 M~ M2 M~M2MaM1 v ~ 7,58 und 7,74
M2M~M2M~ ~=7,69 und 7,87.
Damit sind alle in den 3/[olekelketten vor- kommenden Tetraden zugeordnet. Wie ihre Spektren zeigen, wird, infolge der Aufspal- tung durch die beiden Konfigurationen der VC-Einheit, die Struktur der t~esonanzen um so komplizierter, je mehr dieser Einheiten die Tetraden enthalten. Wenn man sich also die Frage stellt, ob es nicht mSglich sei, ver- schiedene Hexaden zu untersuehen, so wird man yon vornherein vermuten, dal~ nur die Linien solcher t texaden im Spektrum ge- trennt werden, in denen hSchstens zwei VC- Einheiten nebeneinander stehen. Diese t texa- den gehSren daher zur Diadengruppe a (s. oben).
Das Spektrum der Abb. 12 zeigt diese Gruppe bei hSchster AuflSsung des Spektro- meters. Alle neun Linien gehSren zu eharak- teristischen Monomerenfolgen mit einem M1M1-Kern. Ihre Gesamtintensits ent- spricht also der Konzentration dieser Diaden in den Moleke]ketten. Die grebe Dreierstruk- fur unterteilt die Diaden nach ihren Nachbar- einheiten und gehSrt daher zu den Tetraden. Die drei Linien innerhalb ]eder Tetraden-
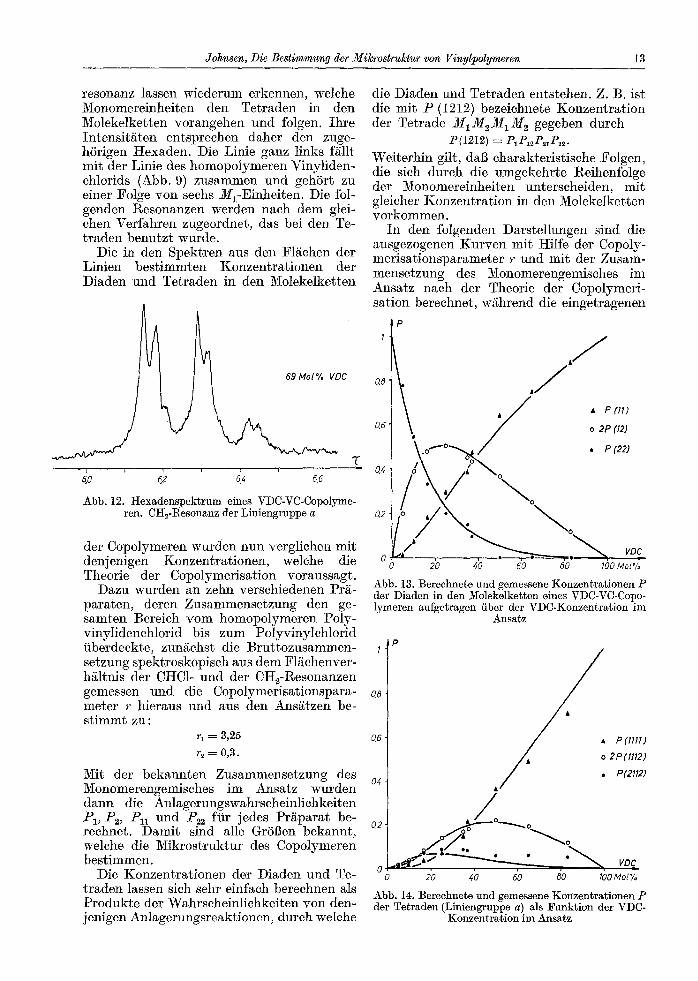
Johnsen, Die Bestimmung der Mikrostruktur yon Vinylpolymeren 13
resonanz lassen wiederum erkennen, welehe Monomereinheiten den Tetraden in den Molekelketten vorangehen und folgen. Ihre Intensit.~ten entsprechen daher den zuge- h6rigen Hexaden. Die Linie ganz links f/~llt mit der Linie des homopolymeren Vinyliden- chlorids (Abb. 9) zusammen und gehSrt zu einer Folge yon sechs M1-Einheiten. Die fol- genden Resonanzen werden nach dem glei- chen Verfahren zugeordnet, das bei den Te- traden benutzt wurde.
Die in den Spektren aus den F1/ichen der Linien bestimmten Konzentrationen der Diaden und Tetraden in den Molekelketten
S i L
s,0
C
6,2 6.4 ,6
T
Abb. 12. t t exadenspekt rum eines VDC-VC-Copolyme- ren. CH~-Resonanz der Liniengruppe a
der Copolymeren wurden nun vergliehen mit denjenigen Konzentrationen, welehe die Theorie der Copo]ymerisation voraussagt.
D~zu wurden an zehn verschiedenen Pr/~- paraten, deren Zusammensetzung den ge- samten ]3ereich vom homopo]ymeren Poly- vinylidenchlorid bis zum PolyvinyIehlorid tiberdeckte, zungehst die Bruttozusammen- I setzung spektroskopisch aus dem F1/~chenver- h~ltnis der CHC1- und der CH2-Resonanzen gemessen und die Copolymerisationspara- a~ meter r hieraus und aus den Ans/~tzen be- st immt zu :
r 1 = 3,25 0,6
r2 = 0,3.
Mit der bekannten Zusammensetzung des Monomerengemisches im Ansatz wurden a4 dann die Anlagerungswahrscheinlichkeiten P1, Pc, Pn und /'~2 fiir jedes Pr/~parat be- reehnet. Damit sind alle Gr56en bekannt, o,2 welche die Mikrostruktur des Copolymeren bestimmen.
Die Konzentrationen der Diaden und Te- ~ traden lassen sich sehr einfach bereehnen als Produkte der Wahrscheinlichkeiten yon den- jenigen Anlagerungsreaktionen, dureh welehe
die Diaden und Tetraden entstehen. Z. B. ist die mit P (1212) bezeiohnete Konzentration der Tetrade M1M2M1M 2 gegeben durch
P(1212) ---- P1Pl~P~,P12.
Weiterhin gilt, dab charakteristische Folgen, die sich durch die umgekehrte Reihenfolge der Monomereinheiten unterscheiden, mit gleicher Konzentration in den Molekelketten vorkommen.
In den folgenden Darstellungen sind die ausgezogenen Kurven mit Hilfe der Copoly- merisation@arameter r u n d mit der Zusam- mensetzung des Monomerengemisches im Ansatz nach der Theorie der Copolymeri- sation berechnet, w/~hrend die eingetrsgenen
P 1
o,6
. J . /
j / �9 P (11) o 2P (12)
o,
0 J �9 0 20 40 60 80 100 Mol%
Abb. 13. Bereehnete und gemessene Konzentra t ionen P dec Diaden in den Molekelketten eines VDC-VC-Copo- lymeren aufgetragen fiber der VDC-Konzentrat ion im
Ansatz
IP
A~ P(nll) A o 2P(II12)
/ . P(2112) t -
20 40 60 80 100 Mol%
Abb. 14. Berechnete und gemessene Konzentra t ionen P der Tetraden (Liniengruppe a) als Funkt ion der VDC-
Konzentra t ion im Ansatz
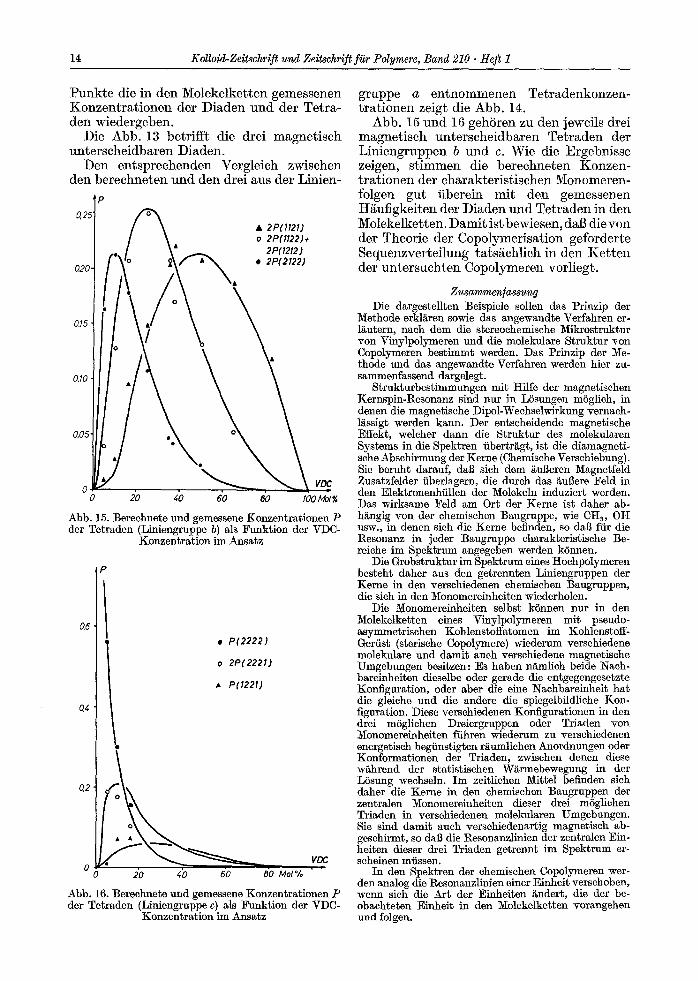
14 Kolloid-Zeitschrif t und Zeitschrift fi~r Polymere, Band 210 �9 Hef t 1
P u n k t e d ie in d e n M o l e k e l k e t t e n g e m e s s e n e n K o n z e n t r a t i o n e n d e r D i a d e n u n d d e r T e t r a - d e n w i e d e r g e b e n .
D ie A b b . 13 b e t r i f f t d ie d re i m a g n e t i s c h u n t e r s c h e i d b a r e n D i a d e m
D e n e n t s p r e c h e n d e n V e r g l e i c h z w i s c h e n d e n b e r e c h n e t e n u n d d e n d re i aus d e r L i n i e n -
O,2
0,2(
0.15
0,16
0,0'.
0 0 20 40 60 60 100 Mot%
Abb. 15. Berechnete und gemessene Konzentrationen P der Tetraden (Liniengruppe b) als Funktion der VDC-
Konzentration im Ansatz
O,6'
0,4
O,2
,P
�9 P(2222)
o 2P(2221)
�9 P(1221)
~00, , . 0 20 40 60
V~
#0 Mot% " '
Abb. 16. Bereehnete und gemessene Konzentrationen P der Tetraden (Liniengruppe c) als Funktion der VDC-
Konzentration im Ansatz
g r u p p e a e n t n o m m e n e n T e t r a d e n k o n z e n - t r a t i o n e n ze ig t d ie A b b . 14.
A b b . 15 u n d 16 g e h S r e n zu d e n j ewei l s dre i m a g n e t i s c h u n t e r s c h e i d b a r e n T e t r a d e n d e r L i n i e n g r u p p e n b u n d c . W i e d ie E r g e b n i s s e ze igen , s t i m m e n d ie b e r e c h n e t e n K o n z e n - t r a t i o n e n d e r c h a r a k t e r i s t i s c h e n M o n o m e r e n - fo lgen g u t i i be re in m i t d e n g e m e s s e n e n H i i u f i g k e i t e n d e r D i a d e n u n d T e t r a d e n in d e n M o l e k e l k e t t e n . D a m i t i s t b ewiesen , d a b d i e v o n d e r T h e o r i e d e r C o p o l y m e r i s a t i o n g e f o r d e r t e S e q u e n z v e r t e i l u n g t a t s ~ c h l i c h in d e n K e t t e n d e r u n t e r s u c h t e n C o p o l y m e r e n v o r l i e g t .
Zusammen/assung Die d~rgestellten Beispiele sollen d~s Prinzip der
Methode erkl/~ren sowie das angew~ndte Verfahren er- l~utern, nach dem die s~ereochemische Mikrostruktur yon Vinylpolymeren und die molekulare Struktur yon Copolymeren bestimmt werden. Das Prinzip der Me- rhode und das angewandte Verfahren werden hier zu- sammenfassend dargelegt.
Strukturbestimmungen mit Hilfe der magnetischen Kernspin-Resonanz sind nur in L6sungen m6glich, in denen die magnetische Dipol-Wechselwirkung vernach- 1/~ssigt werden kann. Der entscheidende magnetische Effekt, welcher dann die Struktur des molekularen Systems in die Spektren fibertr~gt, ist die diamagneti- sche Abschirmung der Kerne (Chemische Verschiebung). Sie beruht darauf, dab sich dem ~uBeren Magnetfeld Zusatzfelder fiberlagern, die dutch das ~ul~ere Feld in den Elektronenhfillen der ~[olekeln induziert werden. ]:)as wirksame Feld am Ort der Kerne ist daher ab- h~ngig yon der chemischen Baugruppe, wie CtI a, OH usw., in denen sich die Kerne befinden, so dab fiir die i~esonanz in jeder ~augruppe charakteristische Be- reiehe im Spektrum angegeben werden k6nnen.
Die Grobstruktur im Spektrum eines Hochpolymeren besteht daher aus den getrennten Liniengruppen tier Kerne in den verschiedenen chemischen Baugruppen, die sich in den Monomereinheiten wiederholen.
Die Monomereinheiten selbst kSnnen nur in den Molekelketten eines Vinylpolymeren mR pseudo- asymmetrischen Kohlenstoffatomen im Kohlenstoff- Gerfist (sterische Copolymere) wiederum versehiedene molekulare und damit auch versehiedene magnetische Umgebungen besitzen: Es haben n/~mlich beide Nach- bareiuheiten dieselbe oder gerade die entgegengesetzte Konfiguration, oder aber die eine Nachbareinheit hat die gleiche und die andere die spiegelbildliche Kon- figuration. Diese verschiedenen Konflgurationen in den drei mSglichen Dreiergruppen oder Triaden yon ~onomereinheiten ffihren wiederum zu verschiedenen energetisch begiinstigten raumlichen Anordnungen oder Konformationen der Triaden, zwischen deuen diese w/~hrend der s~a~istischen W/~rmebewegung in der L6sung wechseln. Im zeitlichen Mittel befinden sich daher die Kerne in den chemischen Baugruppen der zentralen Monomereinheiten dieser drei m6glichen Triaden in verschiedenen molekularen Umgebungen. Sie sind damit auch verschiedenarbig magnetisch ab- geschirmt, so dab die Resonanzlinien der zentralen Ein- heiten dieser drei Triaden getrennt im Spektrum er- scheinen miissen.
In den Spektren der chemischen Copolymeren wer- den analog die I~esonanzlinien einer Einheit verschoben, wenn sich die Art der Einheiten ~ndert, die der be- obachteten Einheit in den Molekelketten vorangehen und folgen.

Johnsen, Die JBestimmung der Mikrostru~ur yon Vinylpolymeren 15
Mit der Mebhode der Kernspin-Resonanz kSnnen so charakteristische Monomerenfolgen, wie Diaden, die oben als Beispiel geschilderten Triaden, Tetraden usw., als Strukturelemente der Ketten bes%immt werden. Die normierten Intensit~ten der beobaehteten Resonanz- linien in den Spektren entspreehen den Konzentratio- nen der zugehSrigen Monomerenfolgen in den Molekel- ketten. Dabei hangt es vom chemischen Aufbau der Monomereinheiten und besonders yon der Magnetfeld- st/~rke des benutzten Spektrometers ab, wie lang die Folgen sind, die tats/~chlich noch getrennt erfa$t wer- den k6nnen.
Bei sterischen Copolymeren gelingt es bisher meist, nur die Konzentrationen yon Diaden und Triaden zu bestimmen. Da hier die Konfiguration einer Monomer- einheit nur in bezug auf die Konfiguration der vorher- gehenden Einheit defmiert ist, wird die Mikrostruktur solcher Ketten durch die Hs der isotaktischen und der syndiotaktischen Verknfipfungen, sowie dureh die L/~ngen der stereochemiseh einheitlichen Sequenzen charakterisiert. Dabei entspreehen die experimentell be- stimmten Konzentrationen der Monomerentriaden den Konzentrationen der in den Molekelketten vorhandenen verschiedenen Verkniipfungspaare. Wie am Beispiel des Polymethylmethacrylats und des Poly-~-methyl- styrols gezeigt wurde, folgen aus ihnen unmittelbar die Konzentrationen der isotaktischen nnd der syndio- taktischen Verkniipfungen nnd die mittleren Sequenz- 1/ingen. In vielen Fallen kann dariiber hinaus auch die Sequenzl/~ngenverteilung abgeleitet werden.
Erheblich komplizierter sind jedoch die Spektren derjenigen Polymeren, die start einer cr nur ein einfaches H-Atom in den Monomereinheiten besitzen. Durch die hier vorhandene indirekte Spin- Kopplung zwischen den Kernen in den versehiedenen ehemischen Baugruppen der Monomereinheiten werden die Linien in Multipletts aufgesp~lten. Dabei iiber- decken die breiten Multipletts die geringen Linien- versehiebungen infolge der sterischen Effekte. Wie an den Beispielen des Polyvinylchlorids und des Polyvinylalkohols gezeigt wurde, ist aber oft auch in diesen F/~llen eine gesicherte Strukturbestimmung mSg- lich, wenn die Kerne durch Doppelresonanz voneinander entkoppelt werden. Die durch Spinkopplung in Multi- pletts aufgespaltenen Linien fallen dann in einfache Linien zusammen, so da]~ die Triadenstruktur in den Spektren deutlieh hervortritt.
Am Vinylidenchlorid-Vinylchlorid-Copolymeren, in dessen Vinylchlorid-B15cken wiederum eine sterisehe Mikrostruktur vorhanden ist, gelingt es mit Hilfe der Doppelresonanztechnik, die Konzentrationen aller Diaden und Tetraden und sogar einiger Hexaden zu bestimmen. Die experimentell ermittelten Konzentra- tionen stimmen quantitativ iiberein mit denjenigen Konzentrationen, welehe nach der Theorie der Copoly- merisation berechnet wurden. Diese Messungen be- st/~tigen daher die yon der Theorie vorausgesagte mole- kulare Struktur der Molekelketten eines Copolymeren.
Die dargestellten Beispiele zeigen, daI3 die Methode der Kernspin-Resonanz wesentliche Einblicke in die Mikrostruktur hochpolymerer Molekelketten erlaubt. Es ist zu erwar~en, dal~ sie bei zukfinftigen Verbesse- rungen der experimentellen Technik sicher mit welter waehsendem Erfolg eingesetzt werden wird.
Ziteratur
1) iVatta, G., Atti. Accad. Naz. Lineei, Mem 8, Ser. II , 4, 61 (1955).
2) iYatta, G., J. Polymer. Sci. 16, 143 (1955). 3) ~atta, G., P. Pino, P. Corradini, F. Danusso,
E. Mantica, G. Mazzanti und G. Moraglio, J. Amer. Chem. Soe. 77, 1708 (1955).
4) Fox, T. G., S. Garret, W. E. Goode, S. Gratch, J. F. Kincaid, A. Spell und J. D. Stroupe, J. Amer. Chem. Soe. 80, 1768 (1958).
5) Germar, H., K. H. HeUwege und U. Johusen, Makromolekulare Chem. 69, 106 (1963).
6) Germar, H., Kolloid-Z. u. Z. Polymere 193, 25 (1963).
7 ) Germar, H., Makromolekulare Chem. 84, 36 ( 1965). 8) Germar, H., Makromolekulare Chem. 86, 89 (1965). 9) Bovey, F. A. und G. V. D. Tiers, J. Polymer
Sci. 44, 173 (1960). 10) Johnsen, U. und K. Tessmar, Kolloid-Z. u. Z.
Polymere 168, 160 (1960). 11) Nishioka, A., H. Watanabe, I. Yamaguchi und
H. Shimizu, J. Polymer Sci. 45, 232 (1960). 12) Nishioka, A., H. Watauabe, K. Abe und Y. Sono,
J. Polymer Sci. 48, 241 (1960). 13) Bovey, F. A., J. Polymer Sci. 46, 59 (1960). 14) Johnsen, U., Kolloid-Z. u. Z. Polymere 178, 16l
(1961), 15) Braun, D., M. Herner, U. Johnsen und W. Kern,
Makromolekulare Chem. 51, 15 (1962). 16) Coleman, B. D. und T. G. Fox, J. Polymer Sci.
A, 1, 3183 (1963). 17) Heu]er, H., Diplomarbeit, TIt (Darmstadt
1961/62). 18) Brownstein, S., S. Bywater lmd D. J. Wor]old,
Makromolekulare Chem. 48, 127 (1961). 19) Sakurada, Y., M. Matsumoto, K. Imai, A.
Nishioka und Y. Kato, J. Polymer Sei. B, 1, 633 (1963). 20) Braun, D., G. Heu/er, U. Johnsen und K. Kolbe,
Ber, Bunsenges. phys. Chem. 68, 959 (1964). 21) Johnsen, U., J. Polymer Sci. 54, 56 (1961). 22) Chu]o, R., S. Sato, T. Ozeki und E. Nagai, J.
Polymer Sci. 61, 12 (1962). 23) Bovey, F. A. und G. V. D. Tiers, A. C. S. Division
of Polymer ChemistryAtlantie City Meeting 3,115 (1962). 24) Bovey, F. A. und G. V. D. Tiers, Chem. and
Ind. 1962, 1826. 25) Tincher, W. C., J. Polymer Sei. 62, 148 (1962). 26) Bovey, F. A., E. W. Anderson, P. U. Douglas und
J. A. Manson, J. Chem. Phys. 39, 1199 (1963). 27) Sato, S., .R. Chu]o und E. IVagai, Rep. Progr.
Polymer Physics, Japan 7, 301 (1964). 28 Doskocilova, D., J. Polymer Sci. B 2, 421 (1964). 29 Doskocilova, D., Coll. Czech. Chem. Commun. 29,
2290 (1964). 30 Doskocilova, D., Faserforschg. u. Textiltechn. 12,
610 (1964). 31 Yoshino, T. und J. Komiyama, J. Polymer SoL
B 3, 311 (1965). 32 Tincher, W. C., Makromolekulare Chem. 85, 20
(1965). 33 Bargon, J., K. H. Hellwege und U. Johnson,
Makromolekulare Chem. (ira Druck). 34 Fordham, J. W. L., P. H. Burleigh und C. L.
Strum. J. Polymer Sei. 41, 73 (1959). 35 Krimm, S., A. R. Bevens, V. L. Folt und J. J.
Shipman, Chem. and Ind. 1958, 1512; 1959, 433. 36) Natta, G. und P. Corradini, J. Polymer Sci. 20,
251 (1956). 37) Danno, A. und ~ . Hayakawa, Bull. chem. Soc.
Japan 35, 1748 (1962). 38) Bargon, J., K. H. ttellwege und U. Johnson,
Makromolekulare Chem. 85, 291 (1965). 39) Ramay, J~. U. und/IT. D..Field, J. Polymer Sei.
B 3, 63 (1965). 40) Tichner, W. U., Makromolekulare Chem. 85, 46
(1965).
Anschrlft des Verfassers : Dr. U. Johnsen, I)eutschesKunststoff-Institut,
6100 Darmstadt, Schlo~gartenstr. 6 1~