Embed Size (px)
Citation preview

W. Klemm, W. Tilk nnd S. v. Miillenheim. Dilatometrische Messung usw. 1
Dilatometrische Messung der Warmeausdehnung zersetz- licher kristallisierter Sake.')
Von WILHELM KLEMM, WOLDEMAR TILK u. SIOELIN v. MULL EN HE IN.^) Mit 8 Figuren im Text.
Inhal teubersicht . I. Das MeBverfahren: GefaBmaterial. S. 3. - Mehapillaren. S. 3. - Fiillung. S. 3. - Quecksilberfullung. S. 4. - Vor- behandlung. S. 5. - Durchfuhrung der Messung. S. 6. - MeBgenauigkeit. S. 7. - 11. D a r s t e l l u n g d e r P r i i p a r a t e u n d D u r c h f i i h r u n g d e r Messungen: 1. Organische Substanzen. S. 9. - 2. KCl, BaCI,, LaCI,. S. 9. - 3. CuCl, CuBr, CuJ, TlCI, TIBr, PbCI,, PbJ,, CdJ,, HgCl. S. 12. - 4. ZnF,, CdF,, MgF,. S. 13. - 5. AlCl,, AIBr,, AlJ,, BiCl,, SbCI,. S. 14. - 6. TiBr,, TiJ,. S. 15. - 7. BeCI,, ZnCl,, CdC1,. S. 16 - 8. MgCI,, CaCI,, CaJ,. S. 16. - 9. ThCI,. 8. 17. - 10. ZrC1,. S. 17. - 11. LiOH, NaOH, KOH. S. 17. - 111. D i c h t e b e s t i m m u n g e n S. 19.
Die Bestimmung des Warmeausdehnungskoeffizienten kann ent- weder durch Messung der linearen Ausdehnung in den verschie- denen Kristallrichtungen erfolgen - direkte Messung; Methode von FIZEAU; rijntgenographisch 7 - oder durch Ermittlung der kubischen Ausdehnung, sei es durch Bestimmung der Dichte bei verschiedenen Temperaturen oder durch dilatometrische Nethoden. Die Messung der l i n e a r en Ausdehnung wird stets anzustreben sein; mit Ausnahrne des rijntgenographischen Verfahrens, das bei weiterer Ausgestaltung besonders aussichtsreich erscheint, verlangen jedoch die iibrigen bisher ausgearbeiteten Methoden zur Bestimmung des
~
l) Der vorliegende Bericht enthiilt nur die Beschreibung der Versuche iiber die Auswertung vgl. Z. Elektrochem. 34 (1928), 523; vgl. fernei das Referat Z. angew. Chem. 41 (1928), 32; eine erste Anwendung der Methode (Aluminiumchlorid) ist in einer zum 50. Geburtstage von W. BILTZ, heraus- gegebenen Festschrift (Hannover 1927) von W. KLEYM gegeben.
*) Uber den Anteil der Verfasser an dieser Untersuchung sei bemerkt, daE die Messungen an den Halogeniden der Hauptgruppen und an organischen Stoffen von W. TILK (Diplomarbeit, Hannover 1928), die an den Halogeniden der Nebengruppen und Alkalibydroxyden von S. v. M~LLENREIY (Diplomarbeit, Hannover 1928) ausgefiihrt sind. KC1, BaCI,, TlC1, MgF, sind von dem Unter- eeichneten gemessen. W. KLEHX.
3 K.BECKEB, Z. Physik 40 (1927), 37. Z. anorg. u. allg. Chem. Bd. 176. 1

2
linearen Koeffizienten groBe, wohlausgebildete Kristalle l), deren Dar- stellung in vielen Fallen noch nicht gegluckt ist. Aus Dichte- bestimmungen la& sich der kub i sche Ausdehnungskoeffizient u nur dann einigermaBen genau gewinnen, wenn man die Genauigkeit der Dichtemessung sehr hoch treibt ; HENGLEIN 2) konnte so die u-Werte der Alkalimetallhalogenide zwischen 0 und - 180 O auf etwa 4O/, bestimmen. 3, Eine dilatometrische Methode hat gegeniiber der Be- stimmung aus Dichtemessungen den Vorteil, daS sie auch bei weniger anspruchsvollen Anordnungen hinreichend genaue Werte liefert und da8 der Gang der thermischen Ausdehnung wiedergegeben wird und sich so eventuelle Modifikationsanderungen bei der Messung bemerkbar machen.
W. Klemm, W. Tilk und S. v. Miillenheim.
1. Das MeBverfahren.
Eine dilatometrische Methode erschien daher besonders ge- eignet. Als Fullflussigkeit wurde aus naheliegenden Qrunden Queck- silber gewahlt. Es gestattet, Messungen im Bereich von -38 bie 200° - vermutlich auch bei noch hoheren Temperaturen - aus- zufuhren.
Zur Messung verfuhr man so, da8 die Substanz (4-12 cm3) als aus dem SchmelzfluS erstarrte Brocken') in ein zylindrivches Glas- kBlbchen von 10-20 cm3 Inhalt gebracht wurde, an dessen oberen Teil ein MeBrEjhrchen angeschmolzen war. Dieses MeBgefaB wurde auf etwa 0,001 mm Hg evakuiert und im Vakuum fett- und luft- freies frisch destilliertes Quecksilber eingefullt. Das so vorbereitete Gerat wurde in einem Flussigkeitsbade erwarmt und bei ver- schiedenen Temperaturen der Stand des Quecksilbermeniskus in der Capillare notiert. Die Ausdehnung der Substanz selbst ergab sich dann als Diff erenz zwischen der beobachteten Volumanderung und der von Quecksilber und Glas. Nach grundsatzlich gleichen
I) Das Anschleifen eines aus der Schmelze erstarrten polykristallinen Priiparates geniigt nicht; man kann dabei, wie das Beispiel des metallischen Zinks zeigt, ganz fehlerhafte Werte erhalten (vgl. W. GTJERTLER, Handb. d. Metallogr., Bd. 11, 2. Teil, 5. Abschn. (1926), 151). Bei hygroskopischen Salzen kommt ein Anschleifen so wie so kaum in Frage.
9 F. A. HENGLEIN? Z. phys. Chem. 115 (1926); 117 (1925), 285. 3, Die Bestimmungen von G. P. BAXTEE u. C. C. WALLACE, Journ. h e r .
Ghem. SOC. 38 (1916), 259 und von G. P. BAXTEB u. C. F. HAWKINS, Journ. Amer. Chem. SOC. 38 (1916), 266 enthalten dagegen Fehler bis zu 40°/,!
*) Nnr bei HgCl benutzte man Pastillen, die mit einer Handpresse her- gestellt wurden.

Dilatometrische Messung der Wiirmeausdehnung zersetzlicher krist. Salze. 3
Methoden - aber rnit anderen Fiillfliissigkeiten, Wasser , Toluol, ErdGl usw. - haben bereits friiher guarbeitet: KoPP~), HAG EN^), HACESPIL 3; WURSCHMIDT 3 benutzte ebenfalls Hg.
Uber Einzelheiten der Durchfiihrung sei folgendes bemerkt.
V orbe r ei tun g zur Me s sung. GtefaPmaterial.
Anzustreben war ein Gefiihaterial, dessen Ausdehnungskoeffizient klein und sicher bekannt war und das keine thermischen Nachwirkungen zeigte - also etwa Quarzglas oder Therlnometerglas 59'*l oder 1611'. Da aber in vielen Fallen die umfangreiche Darstellungs- und Reinigungsapparatur mit dem DilatometergefiB verschmolzen werden multe, verwendete man Quarzglas nur d a m , wenn es filr die Darstellung der Halogenide notwendig war (BeCI,, ZnCl,, CdCl,, MgC12, CaCi,, CaJ,, LaCl,, ThCI,); fur ZrC1, benutzte man Kaliglas. In allen anderen Fallen verwendete man Dilatometer aus gewohn- lichem Thuringer Glas , so daB Gasreinigungsapparatur usw. bequem ange- schmolzen werden konnte. Irgendwelche Nachteile durch thermische Nach- wirkungen us w. zeigten sich innerhalb der angestrebten Genauigkeitsgrenze nicht.
Der A u s d e h n u n g k o e f f i z i e n t von Q u a r z g l a s ist bekannt und audem so klein, da6 seine Berucksichtigung nur eine uneshebliche Eorrektur aus- machte. Der Ausdehnungskoeffizient der benutzten RBhren aus Eali- und Thiiringer Glas wurde dadurch bestimmt, daB man Dilatometer aus den be- treffenden Glasarten nur mit Quecksilber fiillte und den Meniskusstand bei versehiedenen Temperaturen bestimmte. Als mi!tleren Ausdehnungskoeffizienten zwischen 20 und 180° ergaben verachiedene Bestimmungen fur K a l i g l a s 12.10-*, fur T h i i r i n g e r G l a s 28,5 bis 29-10-6.
NeBcapiUaren. Die benutzten Capillaren waren mit Skalenteilung versehen. Sie wurden
mit Quecksilber ausgewogen uud die erhaltenen Werte in Eiehkurven zu- aammengestellt. Die Glascapillaren waren in 0,Ol ema geteilt , Entfernung der Teilstriche voneinander ungefahr 0,2 em, so daS 0,001 ems mit einiger Sicher- heit geschatzt werden konnten. Die Quarzcapillaren waren etwas enger, auch lagen die einzelnen Teilstriche naher beieinander (- 0,l cm); ein Teilstrich entsprach N 0,004 emS, so daB die Ablesung etwas genauer war. Die nur einmal benutzte Ealiglascapillare war noch etwas enger.
Fullung. Bei Substanzen, die an der Luft eingefullt werden konnten, blieb das
GefilB (vgl. Fig. 1) zunilchat offen und wurde erst nach Einbringen der vorher gewogeneu Substanz bei a, also am Boden, zugeschmolzen. Die Wagung der Substanz, wie spater die des eingefullten Hg, erfolgte auf 0,05 g.
') KOPP, Ann. 93 (1855), 129. a) HA~EN, Wiedemanns Ann. 19 (1883), 436. 3, HACPSPIL, Compt. rend. 162 (1911), 261. ') WiiRscaxrDT, z. Metallkunde 13 (1921), 1.
1*

4 W. Klemm, W. Tilk und 6. v. Miillenheim.
Handelte es sich um h y g r o s k o p i a c h e Subatanzen, deren Einfiillen an der Luft untunlich war, so wurde das Dilatometer an die Darstcllungsapparatur angeschmolzen, das Priipmat in indifferenter Atmophiire oder in Vakuum
iibergeschmolzen bzw. eindestilliert und dann bei a und b ab- geschmolzen (vgl. Fig. 2.4). Hier wurde die Substanzmeoge so be- stimmt, da6 zuniichst das mit Schliffstopfen versehene Dilato- meter + Substanz gewogen wurde ; nach bcendcter bfessung sprengtc man die untere Spitze ab, loste das Priiparat heraus und wog das leere Glasgeriit zuriick.
Fig. 1. Fig. 2.
Gelegentlich ergab sich hei dieser Anordnung heim E i n d e s t i 1 lie ran ein Obeletand: Bei vielen Prliparaten kondensierte sich etwas Substanz in den MeSrijbrcben ; beim Versuch , dieses Kondensat durch Erhitzen zu entfernen, blieben manchmal geringe Riickstiinde, so dab nicht sicher war, oh die Eichung der Capillare noch giiltig war. Man zog daher hei spiiteren Versuchen vor, nur den unteren Teil des Dilatometergefabes in die Darstellungsapparatar ein- zuschmelzen (F ig 2 4 ; nach dem Abschmelzen bei a und b wurde dann c ab- geaprengt, vermittels eines kurzen Stiickv Vakuumschlauch mit der Queck- silbercinfillapparatur verbnnden, evakuiert und eine vorliiufige Quecksilber- fiillung (etwa bis d) vorgenommen. Jetzt konnte, da die Substanz geschiitzt war, die MeEcapillare hei c angeschmolzen werden. Darauf wurde ernent evakuiert und das fehlende Quecksilber nachgefiillt.
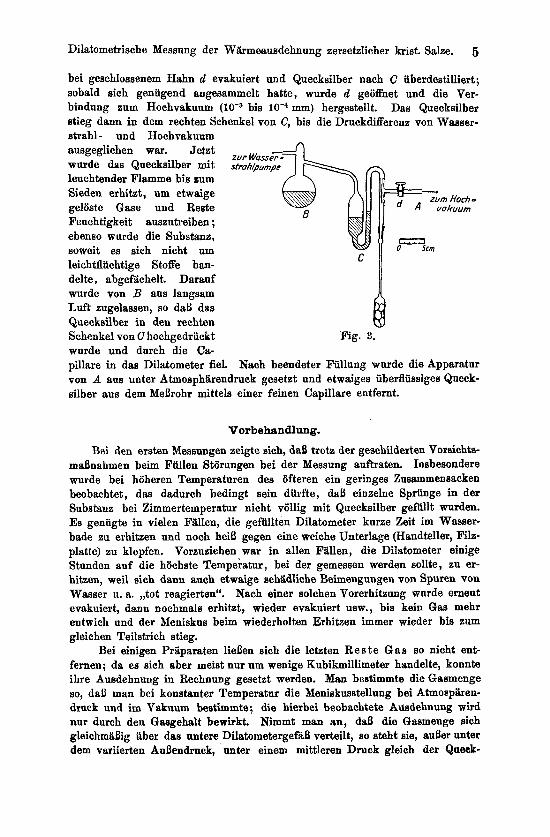
Qnecksilberfullnng. Urn fettfreies, reines und trockenes Quecksilber einzufullen, bediente man
sich folgender Anordnung: Das Dilatometer wurde mit einem, nur im oberen Teil gefetteten Schliff mit der Einfiillapparatur verbnnden (Fig. 3). Diese he- stand aus einer Queckailberdampfstrahlpumpe mit Vorpumpe und Manometer (A), einer Queckeilberdestillationseinrichtung (B), die im Waseeretrahlvakuum arbeitete, und der eigentlichen Fiilleinrichtung (C). Zungchst wurde von B aus
Dilatometrische Messung der Warmmusdehnung zersetzlicher krist. Salze. 5
bei geschlossenem Hahn d evakuiert und Quecksilber nach C iiberdestilliert ; sobald aich genugend angesammelt hatte, wurde d geoffnet und die Ver- bindung zum Hochvakuum bis lod4 mm) hergestellt. Das Queckailber stieg dann in dem rechten Schenkel von C, his die Druckdifferenz von Wasser- strahl- und Hochvakuum ausgeglichen war. Jetzt wurde das Quecksilber mit leuchtender Flamme bis zum Sieden erhibt, um etwaige gelSste Gase und Beste Feuch tigkeit auszutreiben ; ebenso wurde die Substanz, soweit es sich nicht um leichtflichtige Stoffe han- delte, abgefiichelt. Darauf wurde von B aus langsam Luft zugelassen, so daB das Quecksilber in den rechten Schenkel yon C hochgedriickt wurde und durch die Ca-
-
Fig. 3.
pillare in das Dilatometer fiel. Nach beendeter Fullung wurde die Apparatur von A aus unter Atmosphiirendruck gesetzt und etwaiges iibediissiges Queck- silber aus dem MeErohr mittels einer feinen Capillare entfernt.
Vorbehendlung.
Bei den ersten Measungen zeigte sich, da8 trotz der geschilderten Vorsichts- maJ3nahmen beim Fiillen Stiirungen bei der Messung auftraten. Insbesondere wurde bei hiiheren Temperaturen des iifteren ein geringes Zusammensacken beobachtet, das dadurch bedingt sein durfte, dab einzelne Sprunge in der Substanz bei Zimmertemperatur nicht vSllig mit Quecksilber gefullt wurden. Es geniigte in vielen Fallen, die gefullten Dilatometer kurze Zeit im Wasser- bade zu erhitzen und nocb hei6 gegen eine weiche Unterlage (Handteller, Filz- platte) zu klopfen. Vorzuziehen war in allen Fallen, die Dilatometer einige Stunden auf die hachste Temperatur, bei der gemessen werden sollte, zu er- hitsen, weil aich dann auch etwaige schiidliche Beimengungen von Spuren von Wasser u. a. ,,tot reagierten". Nach einer solchen Vorerhitzung wurde erneut evakuiert, dann nochmals erhitzt, wieder evakuiert usw., bie kein Gas mehr entwich und der Meniskus beim wiederholten Erhitzen immer wieder bis zum gleichen Teilstrich stieg.
Bei einigen Praparaten lieBen sich die letzten Res t e Gas so nicht ent- fernen; da es sich aber meist nur um wenige Rubikmillimeter handelte, konnte ihre Ausdehnung in Rechnung gesetzt werden. Man beetimmte die Gasmenge so, daU man bei konstanter Temperatur die Meniskusstellung bei Atmospiiren- druck und im Vakuum bestimmte; die hierbei beobachtete Ausdehnung wird nur durch den Gaegehalt bewirkt. Nimmt man an, daB die Gasmenge sich gleichm8;Big iiber das nntere DilatometergefaB verteilt, so steht sie, auJ3er unter dem variierten AuSendmck, unter einem mittleren Druck gleich der Queelr-

6 W. Klemm, W. Tilk und S. v. Mullenheim.
silberhshe von der Dilatometermitte bis zum Meniskus, so daB also aus der Volumauderung bei Variation des AuBendrucks die Gasmenge berechnet werden konnte. Die Korrektur war sehr gering.
Durchfiihrung d e r Messung. H e i z bad. Zur Messung wurde das Dilatometer in ein zylindrisches
GlasgefaS von etwa 11/, Liter Inhalt gehlingt, das rnit Paraffinijll) gefullt war uod durch zwei elektrische Heizbacken erhitzt wurdee), wobei an der Vorder- und Ruckseite des Heizbades je ein Spalt von etwa 3 cm Breite frei blieb, so da6 im durchfallenden Licht beobachtet werden konnte. Die Temperatur- beobachtung erfolgte mit einem Quecksilberthermometer, das von - 10 bis ZOO0 reichte und rnit geeichten Thermometern verglichen war; bei der Temperatur- messung konnten Zehntelgrade geschlitzt werden. Die Temperaturkonstanz des Ofens wurde dadurch gesichert , da5 ein rnit einem HeiBIuftmotor betriebenes Riihrwerk fur kraftige Durchmischung der Badfliissigkeit sorgte. Temperatur- differenzen > 0,3O traten nicht auf; nur in dem alleruntersten Teil des Bades war auf 2-3 cm LZinge eine etwas tiefere Temperatur als in dem ubrigen Teil; die DilatometergefaBe ragten niemals in diesen kuhleren Bereich hinein. Besondere Sorgfalt wurde darauf verwendet , daB die Thermometerkugel das weite DilatometergefaS genau in der Mitte beruhrte.
Bei t i e f e r e n T e m p e r a t u r e n zwischen -10 und -30° erfolgte die Temperaturmessung rnit einem SO, - Dampfdrucktherrnometer ; benutzt wurde ein Alkoholbad, das von Hand kraftig geruhrt wurde; durch Zugabe von fester Kohlensaure oder zimmerwarmem Alkohol wurden die gewunschten Tempe- raturen eingestellt.
G e s c h w i n d i g k e i t d e s T e m p e r a t u r a n s t i e g e s . Da Wert darauf gelegt wurde, den Verlauf der Meniskusanderung in mijglichst kleinen Tempe- raturintervallen festzulegen , wurde bei langsam steigender bzw. fallender Temperatur beobachtet. Die Erhitzungsgeschwindigkeit, die man einhalten konnte, ohne daB sich ein ,,Nachhinken" zeigte, war uberraschend grol : im allgemeinen lo pro 1-2 Minuten. Es riihrt dies offenbar daher, daB das sehr gut leitende Metal1 such bei schlecht wiirmeleitenden Stoffen vermijge der guten Durchdringung den Warmeaustausch schnell vermittelt. Nur bei sehr groSen und kompakten Schmelzkuchen war es notwendig, etwas langsamer zu crhitzen. DaB die verweudete Erhitzungs- bzw. Abkiihlungsgeschwindigkeit das zulassige MaB nicht uberschritten hatte, erkannte man einmal a n dem Zusamrnenfallen der Kurven bei steigender und fallender Temperatur und ferner daran, daB man gelegentlich bei einer Temperatur Konstanz fur 15 bis 20 Minuten abwartete: die d a m erhaltenen Werte deckten sich stets mit den bei den vorhergehenden Erhitzungsversuchen beobachteten.
Die M e s s u n g verlief nun so, daB in gewissen Zeitabstanden Temperatur und Quecksilbermeniskus im Dilatometer bestimmt wurde. Die graphisch auf-
l) Konzentrierte Schwefelsaure, die zuerst benutzt wurde, kann zu un-
*) Dieser Ofen ist bei W. BILTZ und 3'. MEYEB, Z. anorg. u. allg. Chem. angenehmen heftigen Reaktionen fuhren, falls ein SubstanzgefiiS platzt.
176 (1925), 26 beschrieben.

Dilatometrische Messung der Warmeausdehnung zersetzlicher krist. Salze. 7
getragenen Werte wurden durch eine Kurve verbunden, so da6 die Meniskus- stellung fiir jede beliebige Temperatur abgeleaen werden konnte.
A u s w e r t u n g . Die SO gefundene gesamte thermische Ausdehnung setzt sich zusammen aus der Volumanderung 1. der zu messenden Substanz, 2. des Quecksilbers, 3. des GefiiBes und zuweilen 4. des Gases. Die Ausdehnung des Quecksilbers 15Bt sich aus den bekannten Dicbton ohne weiteres angeben, da die Menge bekannt ist. Fur die Ausdehnung des GefaBes ist das Gesamt- volumen (Summe der Volumina von Snbstanz + Quecksilber zusatzlich der gefundenen Gesamtvolumeniinderung) mit dem k u b i s c h e n Ausdehnungs- koeffizienten des GefiBmaterials zu multiplizieren. Uber die Gaskorrektur
Die Ausdehnung des zu messenden Stoffes ergibt sich durch Addition der genannten GrGBen, wobei positiv eiogesetzt werden: gemessene Volumanderung und Ausdehnung des GefaEes, negativ die Ausdehnung von Quecksilber und Gas. Der mittlere kubische Ausdehnungskoeffizient n berechnet sich dann gem& a = -.- v2 - durch Division mit dem Anfangsvolumen (gegeben durch Einwage und Dichte) und Temperaturbereich der Messung. In den meisten Fallen war die Volumiinderung im untersuchten Temperaturgebiet eine nahezu lineare Funktion der Temperatur, so daB hier der mittlere Ausdehnungs- koeffizient mit dem wahren nahezu identisch ist. In einigen Fiillen zeigte der a-Wert einen leichten Anstieg mit der Temperatur, so da6 eine quadratische GIeichung benutzt werden mudte.
vgl. s. 5.
VI ta - 4
MeBgenauigkeit. Die A b l e s u n g von T e m p e r a t u r und Q u e c k s i l b e r m e n i s k u s ge-
stattete, die Einzelbeobachtungen rnit einer Genauigkeit von 0,001 bis 0,002 cma durchzufiihren; dadurch, daB alle Ablesungen zu einer Kurve vereinigt wurden, werden die nach beiden Richtungen liegenden Ablesefehler noch ausgeglichen, so daB die Bestimmung der Volumanderung auf 0,001 bis 0,002 zuverlassig ist. D a die mit lo8 multiplizierten a-Werte ungefiihr in derselben GriiBen- ordnung liegen, wie die Ausdehnung der Substanz in Kubikmillimeter, so diirften rein meltechnisch die gefundenen cc-Werte auf 1-2 Einheiten der letzten Stelle genau sein; das entspricht bei mittleren Werten (100-10*) einer Genauigkeit von 2O/,, bei kleineren (50-10-6) von 4°/v
Durch nicht ausgefiillte H o h l r a n m e in der Substanz kiinnen einmal Fehler dadurch hervorgerufen werden, dab ein griiberes Volumen vorgetiiuscht ist, J s sich aus Einwage und Dichte berechnet; dieser Fehler durfte nach Erfahrungen bei Dichtebestimmungen mit Quecksilber *) bei Salzen nicht groB sein.3 Sehr wesentlich k6nnte a l l m a h l i c h e s Eindringen von Quecksilber in solche Ritze bei Temperaturerhiihung stiiren3); wenn man aber die Beobachtung so lange
I) Vgl. W. KLEMY, Z. anorg. u. allg. Chem. 163 (1921), 239. a) GrbBer kann er dagegen nach unveriiffentlichten Messungen im hiesigen
Institut bei schlecht benetzenden, feinkristallinen Stoffen werden, bei denen roan bei Dichtebestimmungeu rnit Hg oft um loo/, zu niedrige Werte findet.
s, Meist beobachtet man dann allerdings einen spruoghaften Abfall, der SO auffallig ist, daB er nicht iibersehen werden kann.

8 W. Klemm, W. Tilk und S. v. Miillenheim.
0,423 0,125 0,134 0,058 0,015
wiederholt , bis man bei steigender und fallender Temperatur gleiche Werte erhiilt, so diirfte dieser Fehler ausgeschaltet sein.
Enthielt das Dilatometer Gas, so wurde dessen Ausdehnung beriicksichtigt (vgl. S. 5); die Korrektur betrug im ungunstigsten Falle 0,004 cm3. Sehr stiirend ist selbatversttindlich, wenn wiihrend dsr Messung Gasentwicklung auf- tritt; dies lieB sich durch entsprechende Vorbehandlung jedoch durchgehend vermeiden.
Zu beriicksichtigen ist schlieBlich, daB die Genauigkeit der Bestimmung von a nicht nur von der absoluten Menge des Praparates abhangig ist, sondern auch von dem Verha l tn i s d e r Subs tanemenge zu d e r Quecks i lber - menge. 1st dieses besonders ungunstig, dann sind die Korrekturen im Ver- haltnis zur Substanesusdehnung so grog, daB die Genauigkeit der Messung herabgemindert wird. Tabelle 1 gibt eine obersicht iiber Beispiele mit gunstigen nnd ungunstigen Verhiiltnissen.
0,1633 0,121 0,210 0,165 0,135**)
Tabelle 1.
Campher . . . . 10,92 Naphthalin . . . 7,76 AlJ,. . . . . . . 6,60
5,12 ZrC1, . . . . . . I 2,12 CaCl, . . . . . .
15,30 15,48 15,02 5,39 9,95 1
Ein Kriterium dsfur , da0 die rechnerisch ermittelte Genauigkeit auch wirklich erreicht wurde, ist in einigen Fiilleu durch Wiederholungs- messungen an unabhangigen PriLparaten gegeben. Dieses Ergebnis ist um so wichtiger, als es zeigt, d a 8 in den gepriiften Fallen auch die Priiparate hinreichend zuverliissig waren, urn die angegebene Genauigkeit zu rechtfertigen. Ein Vergleich mit Literaturwerten wurde beim KC1 durchgefiihrt; der ge- fundene Wert im Bereiche von 20-100° (113.10-6) stimmt mit dem von FIZEAU') bestimmten (114 susgezeichnet iiberein. $)
II. Darstellung der Praparate und Durchfuhrung der Messungen.
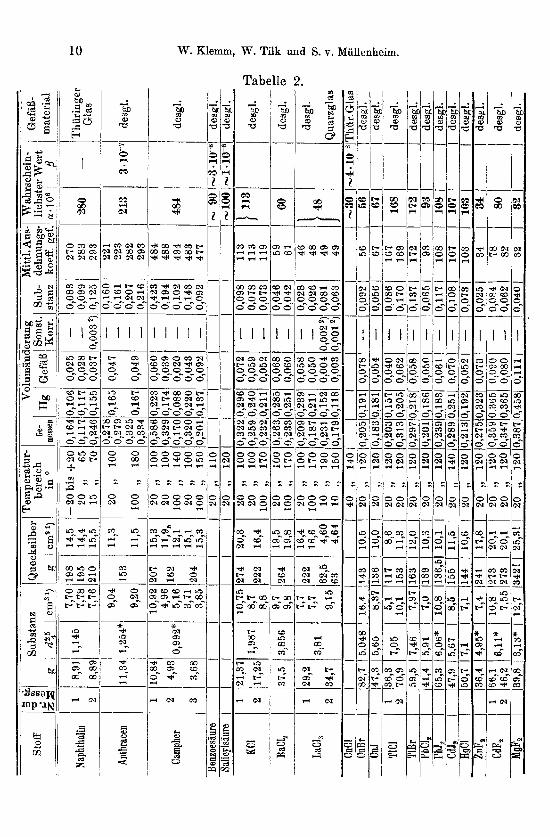
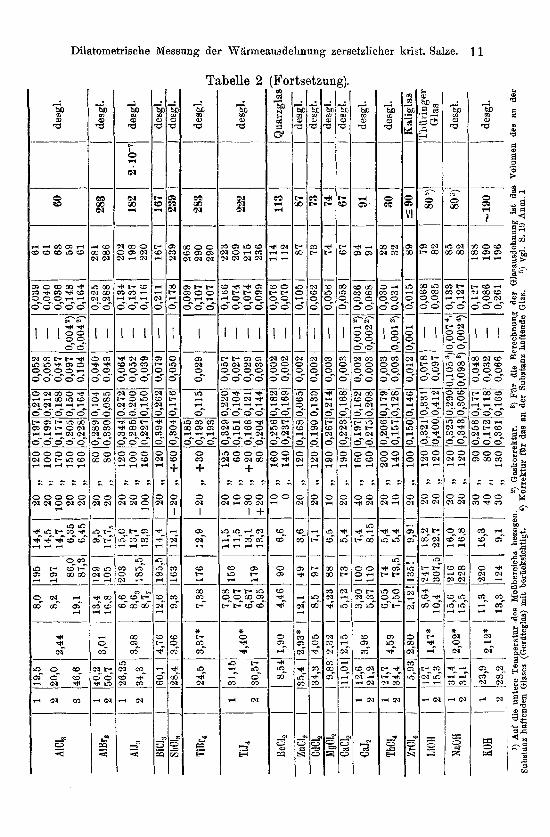
Tabelle 2, die iiber die Messungsergebnisee Auskunft gibt, dUrfte ohne besondere Erlauterung verstindlich sein. An Einzelheiten sei bemerkt, daB die Dichten der untersuchten Substanzen bei Zimmer- temperatur den Tabellen von LANDOLT-BORNSTEIN-ROTH bz w. dem
9 FIZEAU, Pogg. Ann. 332 (1867), 292. 3 Man kijnnte versuchen, die Qenauigkeit der Messungen daduroh zu
steigern, daB man engere Capillaren wahlt. Dies wurde dann auch den Vor- teil haben, da6 man mit geringeren Substanzmengen auskbe. Einige Ver- suche, die in dieser Richtung angestellt wurden, ermutigten jedoch nicht 5ur Fortsetzung.

Dilatometrische Messung der Warmeausdehnung zersetzlicher krist. Salze. 9
Handbuch von BEILSTEIN entnommen sind. Die mit * versehenen Werte sind neu bestimmt (Teil 111, S. 19). Als ,,wahrscheinlichster Wert" ist der Mittelwert aller Messungen angegeben, soweit nicht eine MeBreihe bevorzugt zu berucksichtigen war. Die Bezeichnungen a!
und ,!3 beziehen sich auf eine Darstellung gemaB der Formel
vt2 = fJtl [1 + a! ( tz - tl) + p (tz - 417 Die Reihenfolge ist so gewahlt, daB Substanzen von ahnlicher
Darstellungsart und ahnlichen Eigenschaften zusammengefaSt sind, so daB die Beschreibung der praparativen Bereitung und des Ein- bringens in die MeBgefaBe gruppenweise erfolgen kann.
1. Organische Snbstanzen: Die untersuchten Praparate stammten von KAHLBAUM; Naphthalin, Campher (Messung 2 und 3), Benzoe- saure und Salizylsaure waren ,,fur calorimetrische Bestimmungen". Mit Ausnahme von Campher lagen Kristallpulver vor. Da diese zur Messung weniger geeignet sind, wurden sie in einem Reagenzglase im Paraffinbade niedergeschmolzen und die erhaltenen kompakten Stucke in Dilatometer aus Thiiringer Glas gefllllt.
Man erwartete, daB die Untersuchung dieser Substanzen, die zu Vergleichs- zwecken erwunscht schien, zu besonders glatten Ergebnissen fuhren wurde; in der Tat ergaben sich auch bei N a p h t h a l i n und A n t h r a c e n keine be- sonderen Schwierigkeiten, wenn man erst einmal dafiir gesorgt hatte, da5 alle Hohlraume ausgefullt waren (vgl. S. 5 u. 7). Auch bei C a m p h e r verliefen die Messungen glatt; nur war es nicht moglich, Messungen oberhalb 140° durch- zufuhren, weil dann eine geringe Gasentwicklung stSrte.
Sehr groSe Schwierigkeiten ergaben sich dagegen bei Benzoe- und Sa l icy ls i iure ; es war hier, trotz einer groBen Reihe von Versuchen, nur innerhalb recht weiter Grenzen (f 5 mSglich, Ubereinstimmuug zu erzielen; manchmal trat nach zwei ganz normal verlaufenen Messungsreihen bei dem- selben Praparat bei der Wiederholung wieder eine $tarke Abweichung ein. Bei der Salicylsaure storte zudem eine bei 130° auftretende geringe Gasent- wicklung. Wieweit die Schwierigkeit, gut ubereinstimmende Resultate zu er- halten, dsmit zusammenhiingt , daS die Ausdehnungskoeffizienten sehr stark temperaturabhiingig sind, mu8 dahingestellt bleiben; auffdlig ist, da8 sich auch bei CuCl, bei dem ebenfalls eine starke Temperaturabhiingigkeit des a-Wertes gefunden wurde, sehr schwer iibereinstimmende Werte erzielen lieBen.
2. KC1, BaCI,, LaC1,: Bei diesen Priiparaten war die Vor- bereitung besonders einfach.
KCl (Karrisauar ,,zur Analyse") wurde im Platintiegel geschmolzen. Die Messung sollte die Brauchbarkeit des Verfabrens zeigen (vgl. d a m S. 8). Ob wirklich der Ausdehnungskoeffizient so stark temperaturabhangig ist , wie es nach Messung 2 scheint, sei dahingestellt.
Camp
her
I 1 I'::::',,,,,' 1 1
I 3,
85
Benz
oesa
ure
I I
I I
I Sa
1icyls
iui-e
i j
i I
I I
1 i
21.3
71
I 10
.75
I274
CnCI
I
I I
I I
14,5
1-
20
bis
+20
(0,
164/
0,10
6! 0.
025
1 - 1
0,08
3 1
270
65 I
U;ll
710;
117
0;02
8 -
I0;0
99 I
70 /
0,24
610,
1551
0,0
37 1
0,00
3211
0,
125
1 10
0 :$
;: 0,165
0,04
7
11,5
1 10
0 ,,
180
1::$
40,1
61
0,04
9 1 1
0 o:21
6 20
7 I
11.9
, 20
,,
100
/0~3
291O
i174
1 0.0
39 I -
10
1194
1
-
0 16
0 - I o
:161
15,3
I 2
0 ,,
100
05
86
02
23
0,
060
-
0 42
3
283
293
221
225
282
293
484
488
12;l
" I 1
00 ;;
140
0;17
0 0;
088l
0;
020
' -
494
15,l
20
,,
100
0,32
0 0,
220
0,04
3 -
01
43
48
3 O
il02
1 -_
__
15
,s
I 100
,, 15
0 10
,201
~0,1
37 0
,099
1 -
i0:0
92 1
477
I 20
,9
11
01 _
__
_
I I
I I
1
I1
I
20 ,,
120
I I
I I
1674
I 1:;
I: 17
0 /0
232/
0.21
1/
0.05
2 1 1 10
.073
I ::9"
0,09
8 11
3 20
,3
20 ,,
100
0,32
2 0,
296
0,07
2 -
100
0,25
9 0,
240
0,05
9 0,
078
16
4 I
20
,,
100
10.2
0910
,239
1 0,
058
1 - 1
0,02
8 I
46
,, 17
0 0;
1871
0;21
11 0
;050
1 - 10
;026
48
4:
60 1 'yo"
190
0,23
1 0,
152
0,00
4 0,
002'
) 0,
081
49
4,64
10
,,
150
10,1
79 0
,118
1 0,
003
O,0
Ol2
) 0,0
63
49
10,5
I
20
120
/0,2
0510
,191
1 0,
078
1 -
10,0
92 I
56
10,O
I
20 ,,
120
[0,1
8310
,181
1 0,0
54 I -
10,0
56 I
67 I
8,6
I 20
..
120
10.2
0310
.157
1 0,
040
I -
10.0
86 I
167
I I
t
'!
16
'6 I
40 ,,
1401
1
I -A.
11;3
I
20 i:
120
10;3
1310
;205
/ 0;0
62 I - I
Oil7
0 I
169
12
,02
0 ,,
120
(0,2
9710
,218
1 0,0
58 1 -
10,1
37 1
172
10,3
!
20 ,,
120
(0,2
0110
,186
1 0,
050
1 -
10,0
65 1
93
10,l
I
20 ,,
120
10,2
39(0
,183
( 0,0
61 I -
10,1
17 1
108
11,5
1
20 ,,
140
/0,2
8910
,251
1 0,
070
1 - I0,108 I
107
10,6
I
20 ..
120
10,2
1310
,192
1 0.
052
1 -
10,0
73 1
103
I.
,
17,8
i 2
0 ;;
120i
o,27
510,
323i
0,0
73 i -
10,0
25 I
34
2O,1
1
20 ,,
I20
10,3
5910
,365
1 0,
090
I -
10,0
84 I
78
20
,l I
20 ,;
120
10,3
4710
,365
) Ol0
80 I - 1
0;06
2 I
82
Z5.3!
I
20 ..
120
10.3
8710
.458
1 0.
111
I - 1
0.04
0 I
32
Wah
rsch
ein-
G
ef5S
- ' U'g
ert m
ater
ial
lichs
t cr.106
280
__
_
_
__
213
GO
-
1 Thurin
ger
Gla
v
desg
l.
desg
l. d
p.3
i desg
l. *
I I Quar
zgla
s
24. 1
0-81
Thu
r.Gla
s 1
desg
l. I
desg
l.
I desg
l.
__
__
__
_
__
- I I
desg
l. I
desg
l. 1
desg
l. I
dese
l. I
dese
l.
1
19,5
8,O
19
5 14
,4
20 ,,
120
0 19
7
I 146,6
I 1 19y1
1 2:;
I i: :: 1
60 1
O:22
8
20 ,,
100
01
99
02
12
150
0,21
0 0,
052
-
0,03
9 61
0
05
3
0,04
0
02
05
01
50
009
7 00
04*)
014
8 0:
164(
0k
04 1
0:00
4',1
O:16
* 1
deag
l.
U
r:
i; 1

12 W. Klemm, W. Tilk und S. v. Miillenheim.
BaCI, . 2H20 (KAELBAIJM ,,zur Analyse") wurde durch vorsichtiges Erhitzen entwgssert und das wasserfreie Salz im elektrischen Tiegelofen geschmolzen. Eine wiiSrige Lijsung der Schmelze reagierte gegen Phenolphthalein eben alkalisch.
LaCl, : Aus Lanthanammoniumnitrat, das das Institut der Liebenswiirdig- keit der D e u t s c h e n G a s g l i i h l i c h t - A u e r - G e s e l l s c h a f t verdankt, wurde La,08 dargestellt und daraus durch Chlorieren im Cl,/S,CI,-Strome LaCl, als Schmelzkuchen erhalten, der sich gut von der Wand des zur Darstellung be- nutzten Quarzrohres ablijsen lie& Praparat 1, das klar in Wasser lijslich war, wurde als Schmelzkuchen in ein Dilatometer aus Thiiringer Glas gefiillt. Prriparat 2 wurde, wie es spgter u. a. beim MgCl, (vgl. S. 16) beschrieben ist, mit der Gebllseflamme in ein Quarzdilatometer iibergeschmolzen, das an das Darstellungsrohr angeschmolzen war. Bei dem starken Erhitzen der Quarz- wand liek? sich natiirlich eine geringe Oxydbildung l) nicht ganz vermeiden; immerhin gibt der etwas zu niedrige C1-Gehalt (gef. 43,0°/,, ber. 43,40/,) zu keinen Bedenken Veranlassung , zumal da der Ausdehnungskoeffizient sehr klein ist.
3. CuC1, CuBr, CnJ; TlC1, TlBr; PbCI,, PbJ,; CdJ,; HgCl: Diese Substanzen sind durchweg leicht auf nassem Wege, zum groBten Teil durch Fallung, zu gewinnen, muBten jedoch im Vakuum nieder- geschmolzen werden, urn Zersetzung zu vermeiden (mit Ausnahme von HgC1, vgl. weiter unten).
Im einzelnen ist zu bemerken, daE die Farbe der geschmolzenen Cu(1)- H a l o g e n i d e denen der geschmolzenen Silberhalogenide Shnlich war - a h o etwa honiggelb. ') Auffiillig ist beim CuCl die starke Temperaturabhiingigkeit von a; ebenso wie bei den untersuchten organischen Sauren war die ffberein- stimmung der Messungen unter sich vie1 schlechter als sonst, so daS auf eine Wiedergabe im einzelnen verzichtet werden kann. Eine Reaktion mit dem Quecksilbermetall schien hier wie auch bei den T l ( 1 ) - H a l o g e n i d e n zu be- fiirchten, wurde jedoch nicht beobachtet.
Pb(I1)-Halogenide. PbCl,: gef. 74,3°/0, ber. 74,5°/0 Pb; PbJ,: gef. 45,0°/0, ber. 44,S0/, Pb. Der fur PbJ, gefundene a-Wert ist dem fur CdJ, ge- fundenen gleich; bei dem analogen Gitteraufbau der beiden Salze war darc zu erwarten. Fur diese Jodide sind von FIZEAIJ~) an angeschliffenen Schmele- kuchen lineare Ausdehuungskoeffizienten gemessen worden, die zu etwas niedrigeren a-Werten fiihren, als die von uns gefundenen. Mit Rucksicht auf die S. 2, Anm. 1 geguflerten Bedenken miichten wir den dilatometrisch be- stimmten Werten den Vorzug geben. Zudem ist fraglich, ob FIZEAU wirklich stabiles CdJ, untersucht hat; unsere Praparate zeigten die von COHEN und
l) Vgl. W. KLEXM u. J. ROCKSTIIOE, Z. snorg. u. allg. Chem. 152 (1926), 239. a) Das friiher zur Leitfihigkeitsmessung benutzte Priiparat von CuCl
[W. BILTZ u. W. KLEMM, Z. physik. Chemie 110 (1924), 3291 war im Schmelzflusse etwas dunkler gefiirbt, enthielt demnach wohl etwas CuCI,, obwohl die er- starrte Schmelze nahezu rein weiS aussah.
a) FIZEAU, Pogg. Ann. 132 (1867), 292; vermutlich ist auch der a-Wert fiir HgJ, etwas zu niedrig.

Dilatometrische Messung der Warmeausdehnung zersetzlicher krist. Salze. 13
MOESVELD I) als fur metastabile Gemische typisch beschriebenen Eigenschaften: trotz mehrtagigem Erwarmens auf looo trat bei 130-140° eine starke Kon- traktion ein, die der Umwandlung in die stabile Form entspricht; bei weiteren Messungen zeigten sich keine Sttirungen mehr.
Bei HgCl verbot sich ein Niederschmelzen mit Rucksicht auf die dabei stattfindende Zersetzung. 7 Man pre6te daher mit einer Handpresse Pastillen von ungefiihr 1,5 cm Durchmesser und 1 cm Hohe; es war hier fraglich, ob hinreichende Benetzung stattfinden wurde. Man konnte jedoch, abgesehen von einem geringen Zusammensacken wghrend der ersten Messung, nichts Auf- fiilliges beobachten. Der gefundene a-Wert nberrascht durch seine Kleinheit, da ja HgCl Molekuigitter besitzt.
4. ZnF,, Cdl?,, MgF,: Bei einem ersten Versuch wurden die Produkte, die man aus ZnC0,- und CdC08-L6sungen durch Behandeln mit HF erhielt, bei 150° im Vakuum entwassert und getrocknet und aus den so erhaltenen Praparaten Pastillen gepre6t; man erhielt vollig unbrauchbare Werte fiir a; auch zeigten die Priiparate eine etwas zu niedrige Dicbte (vgl. S. 21). Daher brachte man die gut vorgetrockneten Fluoride in einen Platinfingertiegel %) und bedeckte sie mit reichlich Ammoniumfluorid, um so beim Erhiteen fur eine Fluorwasserstoffxtmosphare zu sorgen. Der Ticgel wurde in einem beiderseitig mit ausgegliihter Asbestpappe verschlossenem Rohr aus Quarzgut in einen KohlerobrkurzschluSofen gebracht und etwa 10 Minuten nuf 1200 O (CdF,) bzw. 1000° (ZnF,) erhitzt. Nach dem Erkalten lieBen sich die erstarrten Schmelzkuchen durch geringes Klopfen gut aus dem Tiegel entfernen. Die Praparate waren in nicht zu dicker Schicht durchsichtig, CdF, schwach rosa, ZnF, leicht gelblich gefarbt. (Aufgenommenes Platin?) Der Tiegeldeckel zeigte einen ganz diinnen Beschlag, offenbar von Oxyd. Die Analyse ergab 74,3O/, Cd (ber. 74,7O/,,) und 63,2°/0 Zn (ber. 63,2O/,).
In einem paraffinierten Glase fie1 NgF2 aus MgSO,-Losung nach Zugabe von HF als gelatiniiser Niederschlag. Nach eintagigem Stehen wurde durch ein grol3es Faltenfilter filtriert und etwa eine Woche lang ausgewaschcn. Vorsichtiges Entwiissern im Trocken- schrank, beginnend bei l l O o , fuhrte zu einer kiirnigen Masse, die bei etwa l l O O o in der beschriebenen Anordnung unter Ammonium- fluorid stark sinterte. Das so erhaltene Produkt (gef. 38,S0/, Mg, her. 39,0°/,) war unter dem Nikroskop in diinner Schicht durch- sichtig; an vielen Stellen waren flache, glasklare Prismen aua der kompakten Masse herauskristallisiert. 3 Der gefundene ac-Wert ist
l) COHEN u. MOESVELD, Z. phys. Chem. 94 (1920), 471. 2) W. KLEMH u. W. BILTZ, Z. anorg. u. allg. Chem. 162 (1926), 226; 0. RUFF
u. R. SCHNEIDEB, Z. anorg. u. allg. Chem. 170 (1928), 42. %) Ein Eisentiegel reagierte mit CdF,; man fand nach dem Erhitzen zahl-
reiche Kiigelchen von Cadmiurnmetall und merkliche Mengen Eisenfluorid. 4, Man versuchte auch, MgF, aus einer NaC1-Schmelze umzukristallisieren;
dies Verfahren lieferte jedoch in einer Operation nur geringe Ausbeuten; zu- dem lie8 sich das erhaltene Produkt, das aus zerbrechlichen Niidelchen bestand, nicht zu haltbaren Pastillen pressen.
14 W. Klemm, W. Tilk und S. v. Mullenheim.
in Anbetracht des ungiinstigen Verhaltniases yon Hg zu MgF, nicht ganz so sicher wie die iibrigen Werte.
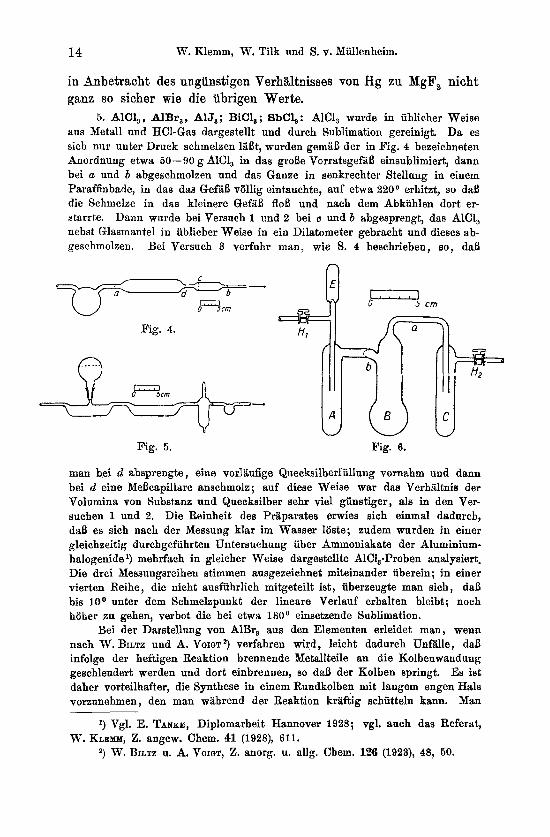
5. AlCl,, AlBr,, AlJ,; BiC1,; 8bCh: AIC1, wurde in iiblicher Weise aus Metal1 und HC1-Gas dargestellt und durch Sublimation gereinigt. D a es sich nur unter Druck schmelzen lilBt, wurden gemaB der in Fig. 4 bezeichneten Anordnung etwa 50-90 g AlC1, in das groBe Vorratsgef&B einsublimiert, d a m bei a uud b abgeschmolzen und das Game in senkrechter Stellung in einem Paraffinbade, in dae das GefiiB vollig eintauchte, auf etma 220° erhitzt, so daB die Schmelze in das kleinere GefaB floB und nach dem Abkiihlen dort er- starrte. D a m wurde bei Versuch 1 und 2 bei c und b abgesprengt, das AlC1, nebst Glasmantel in iiblicher Weise in ein Dilatometer gebracht und dieses ab- geschmolzen. Bei Versuch 3 verfuhr man, wie S. 4 heschrieben, so, daB
n
Fig. 4.
Fig. 5. Fig. 6.
man bei d absprengte , eine vorliiufige Quecksilberfiillnng vornahm und dann bei d eine Mehapillare anschmolz; auf diese Weise war das Verhaltnis der Volumina von Substanz und Quecksilber sehr vie1 giinstiger, als in den Ver- suchen 1 und 2. Die Reinheit des Praparates srwies sich einmal dadurch, daB es sich nach der Messung klar im Wasser loste; zudem warden in einer gleichzeitig durchgefiihrteu Untersuchung iiber Ammoniakate der Aluminium- halogenide l) mehrfach in gleicher Weise dargestellte AlCi,-Proben analysiert. Die drei Messungsreiheu stimmen ausgezeichnet miteinander iiberein; in einer vierten Reihe, die nicht ausfuhrlich mitgeteilt ist, iiberzeugte man sich, daB bis loo unter dem Schmelzpunkt der lineare Verlauf erhalten bleibt; noch hoher zu gehen, verbot die bei etwa 180° einsetzende Sublimation.
Bei der Darstellung von AlBr,, aus den Elementen erleidet man, wenn nach W. BILTZ und A. VOIQT~) verfahren wird, leicht dadurch Unf&lle, daB infolge der heftigen Reaktion brennende Metallteile an die Kolbenwandung geschleudert werden uud dort einbrennen, so da5 der Kolben springt. Es ist daher vorteilhafter, die Synthese in einem Rundkolben mit langem engen Hale vorzunehmen, den man wiihrend der Reaktion kraftig schiitteln kann. Man
1) Vgl. E. TANKE, Diplomarbeit Hannover 1928; vgl. auch das Referat, W. KLEMM, Z. angew. Chem. 41 (1928), 611.
W. BILTZ u. A. VOIGT, Z. anorg. u. allg. Chem. 126 (1923), 48, 50.

Dilatometrische Messung der Warmeausdehnung 5ersetzlicher krist. Salze. 15
gibt bei der Darstellung von AlBr, das Metal1 in sehr kleinen Anteilen zu fliissigem Brom (Eiskiihlung, die Reaktion kommt meist erst nach etwa einer Minute p l o t z l i c h in Gang!); bei AlJ, gibt man das Metal1 im UberschuB in den Kolben, f6gt etwas Jod zu und erwfirmt bis die Reaktion in Gang kommt; bei weiterer Jodzugabe geht die Einwirkung von selbst weiter. Nach be- endeter Umsetzung wird noch einige Zeit iiber den Schmelzpunkt erhitzt und dann in der in Fig. 5 ersichtlichen Weise in die Destillationsapparatur uber- geschmolzen, durch welche ein C0,-Strom geleitet wird. Die Destillation von AlBr, erfolgte im C0,-Strom, die von AlJ, im Vakuum der Olpumpe. Die Praparate sahen rein weiB aus, nur beobachtete man bei AlBr,, daS die ersten Tropfen des Destillats eine violettstichige Farbung besaBen, die offenbar durch eine geringf~gige Verunreinigung verursacht war. Gef. 89,6 O l 0 Br, ber. 89,9 o/o;
gef. 92,9% J, ber. 93,4O/,.
BiC1, wurde aus Netall, das nach MYLIUS und GROSCEUFF~) gereinigt war, durch Verbrennen im Cl,-Strome dargestellt ; gef. 33,6 O/,, C1, ber. 33,7 Ole. SbCI, lag als Sammlungsprkpiparat vor und wurde durch Destillation gereinigt.
6. Tar,, TiJ,: Zur Darstellung van TiBr, und TiJ, haben W. BILTZ und E. KEUNECKE~) die Einwirkung von fiiissigem HBr bzw. HJ atrf TiCI, empfohlm; bei der Darstellung gro5erer Mengen empfiehlt es sich, mehrfach mit frisch kondensiertem HBr bzw. HJ zu behandeln, da sonst die Umsetzung nicht mit Sicherheit voll- stindig vor sich geht.
Die benutzte Anordnung ist aus Fig. 6 ersichtlich. Das reine trockene HBr- bzw. HJ-Gas wurde im GedB B, das mit flussiger Luft bzw. CO, gekiihlt war, kondensiert; sobald geniigend Flussigkeit vorhanden war, wurde bei E abgesprengt, TiCl, in A gefiillt und wieder bei E abgeschmolzen. Nun wurde bei geschlossenem Hahn El und getiffnetem a. das TiC1, Iangsam in das mit COB gekiihlte GefiiB B hiniiberdestiliert, wobei anfangs eine ziemlich heftige Reaktion eintrat; durch zeitweiliges Wegnehmen der Kiihlfliissigkeit sorgte man dafiir, daB die Umsetzung moglichst vollstandig erfolgte. Das Gasgemisch (im wesentlichen HCl) entwich durch X., HBr bzw. HJ wurden in C durch geeignete Kiihlung kondensiert. Dann lieB man auf Zimmertemperatur er- warmen, damit die Umsetzung weiter fortschritt und wiederholte darxuf das Kondensieren von frischem HBr bzw. HJ einige Male. SchlieBlich wurde das Gef& B mit dem Bohprodukt bei 6 abgeschmolzen, bei a an eine Destilla- tionsapparatur angeschmolzen und i m Vakuum der olpumpe destilliert. Gef. 86,8°/0 Br, ber. 86,9O/,; gef. bei 3 Priiparaten 91,6°/0, 91,9O/,, 91,3O/, J, ber. 91,4°/0. Die Messung verlief bei TiBr, ohne Schwierigkeiten; hei TiJ, konnte sie trotz des hohen Schmelzpnnktes von TiJ, (150') nur in einem Falle bis 125 durehgefiihrt werden, in allen anderen Versuchen traten bei hoheren Temperaturen UnregelmaiBigkeiten auf (Zusammensinken, geringe Gasentwick- lung). Die Volumkurve scheint, wie bei AlJ,, schwach gekriimmt zu sein, da
'I MYLIUS u. GROSCBUFF, Z. anorg. u. allg. Chem. 96 (1916), 237. *) W. BILTZ und E. KEUXECKE, Z. anorg. u. allg. Chem. 147 (1925), 179.

16 W. Klemm, W. Tilk und S. v. Mullenheim.
jedoch die Unterschiede zwischen Versuch 1 und 2 ziemlich wesentlicb sind, wird nur ein Mittelwert fur a uber das ganze Temperaturgebiet gegeben.
7. BeCI,, ZnCI,, CdC1,: Die Darstellung erfolgt am zweck- ma6igsten durch Verbrennen der Metalle im Chlorstrome; wegen der hohen Siedepunkte wurden Quarzgerate benutzt, etwa in der Anordnung, die in Fig. 2 gegeben ist.
Be-Metal1 verdankt das Institut der Liebenswurdigkeit des W e r n e r - w e r k s d e r S i e m e n s & H a l s k e A,-G., dem auch an dieser Stelle herzlicher Dank ausgesprochen sei. Das Erhitzen erfolgte bei dcr Darstellung des BeCI, mit freier Flamme; das Metall reagierte unter Aufleuchten, das Reaktions- produkt sah rein weiB aus. Qef. 88,6, ber. 88,7u/0 c1.
Bei ZnC1, hatte man mit der Schwierigkeit zu klmpfen, daB das Destillat im MeSgefiiB leicht amorph erstarrte') und beim Anlassen infolge der Volum- anderung beim Kristallisieren das GefilB zerspredgte. Man destillierte daher mit mehrfachen Unterbrechungen und sorgte durch vorsichtiges Abfiicheln dafiir, daB die jeweils iibergegangenen Anteile kristallin wurden; gef. 52,2 Ole, ber. 52,0°/,, C1.
Bei der entsprechenden Darstellung von CdCI, waren friihere), als in einem sehr langen Quarzrohr im elektrischen Ofen erhitzt wurde, Schwierig- keiten nicht aufgetreten. Als man dagegen in der in Fig. 2 (S. 4) geneichneten Apparatur die Reaktion durchfuhrte, ergaben aich dadurch erhebliche Storungen, daB der Dampfdruck des Metalles groBer ist, als der dea Chlorids. Zwar ver- lief die Reaktion zunachat ganz ruhig; bald bildete aich jedoch iiber dem flussigen Metall eiue Schicht von geschmolzenem Chlorid, die das Metall vor weiterem Augriff schutete. Nur die geringe Metallmengc, die sich unter Braunfirbung in der Schmelze loate, vermittelte die weitere Reaktion. Ver- suchte man, den Fortgang der Chlorierung durch stlrkeres Erhitzen mit dem Bunsenbrenner zu beschleunigen, so traten an einzelnen Stellen explosions- artige Teilreaktionen auf, so daB Kliimpchen von Metall iiber die ganze Appa- retur verspritnten; dieae lieBen sich dann allerdings verhiiltnismiiBig leicht zu Chlorid umsetzen. Nach beendigter Chlorierung wurde im Chlorstrom destilliert und etwaiges gelastes C1, durch Erhitzen im Kohlensaurestrom vertrieben. Gef. 38,6, ber. 38,7O/, C1.
8. XgCI,, CaCl, , CaJ,: Die Darstellung erfolgte durch all- rnlihliches Erhitzen von Gemischen der Halogenidhydrate und der en tsprechenden Ammoniumsalze im Balogenwasserstoffstrom im Quarzgerat. Das Dilatometergefa6 befand sich dabei vor dem Ent- waisserungsgefa6, urn Verunreinigung durch abdestillierende Fremd- stoffe zu vermeiden.
Bei MgCI, und CaCl, wurde nach dem Vertreiben der NH4-Salze im Geblase in das Dilatometer ubergeschmolzen. MgCI,: gef. 74,3°/0, ber. 74,5 o/o C1; CaCI,: gef. 63,7%, ber. 63,9O/,, C1.
l) W. BILTZ und W. KLEMM, Z. phys. Chem. 110 (1924), 335. W. BILTZ und W. KLEMK, Z. phys. Chem. 110 (1924), 336.

Dilatometrische Messung der Wiirmeausdehnung zersetzlicher krist. Salze. 17
Das zur Entwlissernng von CaJ, benutzte Quarzgerat entsprach dem in Fig. 4 gezeichneten. Nach beendeter Entwasserung wurde wie bei AlCI, (Ver- such 3) im Vakuum bei a und b abgeschmolzen, das Ganze senkrecht in einen elektrischen Ofeu gestellt und bis 750° erhitzt, so daB das Priiparat in das MeE- gefaE schmolz, wo es nach dem Erkalten eine schiin kristallisierte, z. T. schwach rosa gefiirbte Masse bildete. Die Praparate lijsten sich nach der Messung klar in Wasser; Ca: J: Prgparat 1 : 1 : 1,94; Priiparat 2: 1 : 1,99.
9. ThCI, wurde, wie bei A. VOIQT und W. BILTZ~) beschrieben, dargestellt und in das MeBgefaB destilliert. Praparat 1: Es loste sich alles klar in Wasser bis auf einen Riickstand von 0,2%; Praparat 2: gef. 37,8O/, C1, ber. 37,Q0/,.
10. ZrC1,: I n einer Kaliglasapparatur, die der in Fig. 4 (S. 14) an- gegebenen entsprach, erhielt man durch Chlorieren von ZrOaa) im Cl,/CCi,-Strome in 24 Stunden etwa 6 g ZrC1,. Urn dieses sehr lockere Sublimat fur die Messung brauchbar zu machen ”), wurde bei a und b im Vakuum abgeschmolzen und soweit in einen elek- trischen Ofen eingefiihrt, daB nur ein ungefahr 3 cm langer Teil bei b herausragte. Bei 350-400° sublimierte das gesamte ZrC1, in die kalt gehaltene Spitze bei b, wo es sich in einigermaBen fester Kruste absetzte. Danach wurde bei d abgeschnitten und in der mehrfach beschriebenen Weise weiter behandelt.
Die trotz der genannten Behandlung noch recht lockere Beschaffenheit des Praparates brachte zwei ubelstilnde mit sich: einmal war es aulerordent- Iich schwer, das Prliparat auch nur einigermaBen gasfrei zu bekommen; an- dererseits war das Verhiiltnis der Volumina von Substanz und Quecksilber ein sehr ungunstiges, so daE man sich damit begnugen muBte, die GriiBenordnung des wWertes festzulegen, urn so zu entscheiden, ob festes ZrCl, den Molekiil- oder Ionengittern zuzuordnen ist. Der gefundene a-Wert spricht in Uberein- stimmung mit den Ergebnisseu von w. BILTZ und E. MEINECKE‘) fur das Letatere. Man verzichtete darauf, mit groEeren Mengeu den tr-Wert genauer zu bestimmen, da die Eigenart des Praparates dies als wenig aussichtsreich erscheinen lieb.
11. LiOH, NaOH, KOH: Auch bei den Alkalihydroxyden be- gniigte man sich damit, angenaherte Werte zu erhalten; denn bei
I) A. VOIGT und W. BILTZ, Z. anorg. u. allg. Chem. 133 (1924), 283. %) Auf eine Abtrennung eines etwaigen Hafniumgehaltes verzichtete man. 3, Ein Schmelzen des ZrC1, kam nach A. VOIGT und W. BILTZ, Z. anorg.
u. allg. Chem. 133 (1924), 303 nicht in Frage. 4, W. BILTZ und E. MEINECKE, Z. anorg. u. allg. Chem. 131 (1923), 4 ; es ist
natiirlich nicht erwiesen, dafl auch geschmolzenes ZrC1, ein guter Leiter ist; der Fliichtigkeit nach steht ZrC1, zwischen AICl, und ScC18 ; es wiire demnach auch miiglich, daB es sich iibnlich verhiilt wie AlCI,, d. h. beim Schmelzen aus der lonen- in die Molekulform ubergeht.
2. anorg. u. allg. Chem. Bd. 1%. 2

18 W. Klemm, W. Tilk und S. v. Mtillenheim.
der groJ3en Ltislichkeit dieser Stoffe im Wasser mu8 sich schon ein sehr geringer Wassergehalt, der recht schwer zu entfernen ist, in einer groBen Erhiihung der a-Werte ausdriicken. So zeigte z. B. ein Praparat NaOH von MERCK ,,in Platzchenform", das noch etwa 2 Wasser enthielt, einen um 100 O l 0 grSEeren Ausdehnungskoeffi- zienten als ein wasserfreies Praparat. Infolgedessen kam es nicht darauf an, den allen kauflichen Praparaten anhaftenden Karbonat- gehalt (bei NaOH und KOH von MERCK etwa 2 Ole, bei LiOH von KAHLBAUM 5-7 O/,,) zu entfernen, denn die a-Werte der Karbonate und Hydroxyde diirften sich nicht sehr stark unterscheiden l); viel- mehr muate nur ohne Vermehrung des Karbonatsgehalts das Wasser mliglichst weitgehend entfernt werden.
Fig. 7.
Hahn und kippte den Apparat
Geeignet erwies sich folgendes Ver- fahren: In ein Gerat aus Jenaer Glas, wie es Fig. 7 zeigt, war ein Silbertiegel ein- geschmolzen. Die Hydroxyde wurden von a aus eingefiullt und dann bei a abge- schmolzen. In einem geraumigen elek- trischen Ofen wurde im Wasserstrahlvakuum im Luftbade auf etwa looo iiber den Schmelz- purikt 2, erhitzt ; die Temperatursteigerung erfolgte sebr langsam , da die Substanzen zum StoBen neigten. Nachdem etwa eine Stunde lang die Temperatur auf etwa 100 O
iiber dem Schmelzpunkt e , gehalten war, ohne daB wahrend dieser Zeit weiteres Wasser sbgegeben wurde, schloB man den
s o , daB der Tiegelinhalt in den oberen zylindrischen Ansatz flieBen konnte, wo er erstarrte. Auf diese Weise wurden Stangen der Hydroxydc erhalten, die sich zur Messung ah sehr brauchbar erwiesen. Bei NaOR lieS sich das GIas schlecht von dem Schmelzkuchen W e n ; man entfernte es daher nur soweit, wie es sich rasch durchfuhren lie5. Die Analyse erfolgte durch Titration mit Schwefelsaure bis zum Umschlag mit Phenolphtalein bzw. Methylorange s, und ergab, daB die Wassermengen 0,1 bis 0,3 o/o nicht iiberstiegen und daB der Harbonatgehalt bei der Behandlung sich nicht vermehrt hatte.
l) Auf eine Korrektur wurde verzichtet, da die a-Werte an sich schon unsicherer sind als sonst.
2, Bei LiOH wurde nur bis dicht unter den Schmelzpunkt erhitzt, um die Bildung von L&O zu vermeiden; die Praparate waren stark gesintert.
3, Vgl. dazu MERCK'S, ,,Prufung der chemischen Reagenzien auf Reinheit", Darmstadt 1922, S. 175.
Dilatometrische Hessung der Warmeausdehnung zersetzlicher krist. Salze. 19
In den gefundenen ct-Werten druckt sieh ein auffalliger Unter- schied zwischen LiOH und NaOH l) einerseits , KOH andererseits aus ; diese groBe Differenz durfte weit auBerhalb der Versuchsfehler liegen.
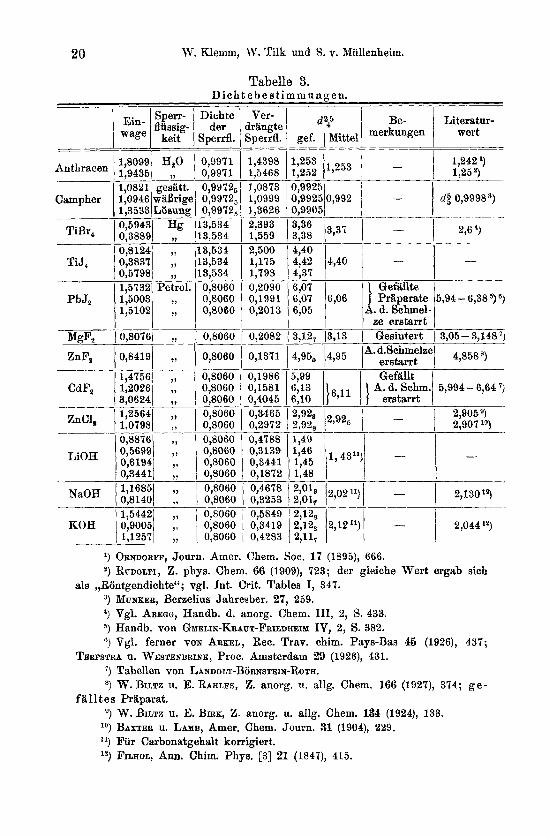
111. Dichtebestimmungen. Fur eine Reihe von Substanzen schien eine Nachprufung der
Literaturwerte fur die Dichte erwiinscht; die Dichten von LiOH und TiJ, wurden erstmalig bestimmt.
Benutzt wurde als Sperrflussigkeit fur Anthracen und Campher W'asser, bzw. eine gesattigte waBrige CampherlSsung, fur TiBr, und TiJ, Quecksilber, fur die iibrigen Substanzen Petroleum.
Die Ergebnisse zeigt Tabelle 3. Im einzelnen ist zu bemerken: Bei den Messungen von TiBr, und TiJ,
mufiten die Proben vor der Fullung liingere Zeit im Hochvakuum belassen werden, da schon die geringen Mengen Feuchtigkeit, die die Priiparate beim Einfiillen aufnahmen, geniigten , urn eine Zersetznng eintreten zu Iassen. Namentlich beim Jodid war der Halogenwasserstoff schwer zu entfernen, einige Proben musten verworfen werden, weil sie wahrend des Temperierens noch nachreagierten und Quecksilber herausdruckten.
TiBr, und TiJ, ergeben mit dem ccm' I I I
Die Mo 1 e ku l a r v o lumin a von MOLVO~.
von W. BILTZ und E. KEUNECKE~) bestimmten Molvolumen von TiC1, keine Linearbeziehung; eine Nach- priifung der Dichte von TiCl, ist in Aussicht genommen, da bei der friiheren Bestimmung mGg- licherweise durch LGslicheit in der Sperrflussigkeit ein Fehler ent- standen sein ksnn.
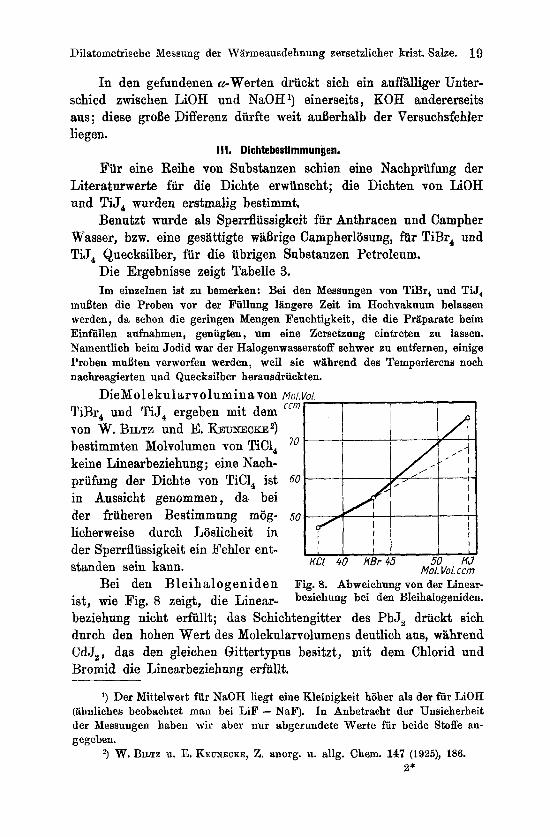
Bei den Bleihalogeniden ist, wie Fig. 8 zeigt, die Linear-
Fig. 8. Abweichung von der Linear- beziehung bei den Bleihalogeniden.
beziehung nicht erfullt; das Schichtengitter des PbJ, driickt sich durch den hohen Wert des Molekularvolumens deutlich aus, wahrend CdJ,, das den gleichen Qittertypus besitxt, mit dem Chlorid und Bromid die Linearbeziehung erfiillt.
I) Der Mittelwert fur NaOH liegt eine Kleinigkeit hoher als der fur LiOH (ahnliches beobachtet man bei LiF - NaF). In Anbetracht der Unsicherheit der Messungen haben wir aber nur abgerundete Werte fir beide Stoffe an- gegeben.
9 W. BILTZ u. E. EEUNECEE, Z. anorg. u. allg. Chem. 147 (1925), 186. 2*
20
Anthracen
Campher
TiBr,
Ti J,
1,8099 1,9435 1,0821 1,0946 1,3533
Cd Fa
H,O ,,
gesatt. wafirig Liiaung
~ _ _ _
_____
ZnC1, ____
LiOH
0,8124 0,3837 0,5798 1,5732 1,5003 1,5102
NaOH
KOH
____
,, ,, ,,
Petrol ,, ,,
W. Klemm, W. Tilk und S. v. Miiilenheim.
1,5442 0,9005 1,1257
Tabelle 3. D i c h t e b e s t i m m u n a e n .
,, ,, ,,
- Dichte
der Sperrfl.
0,9971 0,9971 0,9972, 0,99725 0,9972, 13,534 13,534 13,534 13,534 13,534 0,8060 0,8060 0,8060
0,8060
0,8060
0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060 0,8060
- _- Ver-
irgngte S p e d .
1,4398 1,5468 1,0873
1,3626 2,393 1,559 2,500 1,175 1,798 0,2090
0,2013
0,2082
0,1371
0,1986 0,1581 0,4045 0,3465 0,2972 0,4788 0,3139 0,3441 0,1872 0,4678 0,3253 0,5849 0,3419 0,4283
___ ___
1,0999 ____
-_
0,1991
__
__
___
__
--
0;9925/0,992 ! - 0.99051 1
4,40 4.42 14.40 1 -- 4j37 1 ' 1
G e f d r :$; 16,06 1 } Praparate 6,05 A. d. Schmel
I I ze erstarrt - 3,12" i3,13 i Gesintert
.- 4,950 14795 1 erstarrt 5.99 I I Geftillt
A. d.Schme1m
1,49 1 I
Literatur- wert
1,242 l) 1,25 8,
dg 0,9998s)
_____ ______
- 296 '1
-
-
5994 - 6,38 5, ')
3,05 - 3,148 ")
4,85S8)
5,994 - 6,64 '3 ____.
2,9OFi9) 2,907 lo)
-
2,130'9
2,044 12)
l) ORNDORFF, Journ. Amer. Chem. SOC. 1 7 (1895), 666. 2, RUDOLFI, Z. phys. Chem. 66 (1909), 723; der gleiche Wert ergab sich
als ,,Riintgendichte"; vgl. Int. Crit. Tables I, 347. 3, MUNKER, Berzelius Jahresber. 27, 259. 4, Vgl. ABEQG, Handb. d. anorg. Chem. 111, 2, S. 433. 5, Handb. von GMELIN-KRAUT-FRIEDHEIY IV, 2, S. 382. 6, Vgl. ferner VON ARKEL, Rec. Trav. chim. Pays-Bas 45 (1926), 437;
TEUPSTRA u. WESTENBRINE, Proc. Amsterdam 29 (1926), 431. ?) Tabellen von LAXDOLT-B~RNSTEIN-ROTH. 8, W. BILTZ u. E. RAHLFS, Z. anorg. 11. allg. Chem. 166 (1927), 374; g e -
8, W. BILTZ u. E. BIRK, 2. anorg. u. allg. Chem. 134 (1924), 133. f a l l t e s Praparat.
lo) BAXTER u. LAMB, Amer. Chem. Journ. 31 (1904), 229. 11) Fur Carbonatgehalt korrigiert. '3 FILHOL, Ann. Chim. Phys. [3] 21 (1847), 415.

Dilatometrische Messung der Wiirmeausdehnung zersetzlicher krist. Salze. 21
Bei den F luo r iden ergaben die Praparate von ZnF, und CdF, vor dem Schmelzen etwas niedrigere Werte als nachher; offenbar sind gefallte Praparate nicht einheitlich kristallin.
Bei ZnC1, handelte es sich um Kontrollmessungen, die den friiher gefundenen Wert bis auf eine unwesentliche Abweichung be- sfatigten; verfahren wurde so, wie es fur zersetzliche Stoffe friiher l)
beschrieben ist. Die Werte fur die Alkal ihydroxyde muBten fur den Carbonat-
gehalt korrigiert werden; die Korrektur ist nur bei LiOH von Be- lang; vielleicht hangt auch die starke Streuung der Werte fur LiOH mit einem wechselnden Carbonatgehalt zusammen.
Tabelle 4.
Anionenvolumen ber. 1 ,,Normalwert" 2,
TiBr, . . . . ' I 19.8 I 20 24;2! 1 26-28 -_ SJ,: . . . . ~
PbJ,. . . . . I 28 I 26-28
L__
@dF*, . . . . ___-
KOH . . . . ,
In Tabelle 4 sind schlieBlich nach dem Vorschlage von W. B ~ T Z die Anionenvolumina aus den - auf -273O extrapolierten - Molekularraumen dadurch berechnet , daB das Atomvolumen des Metalls - bei Mg und den Alkalimetallen das halbe Atomvolumen - vom Mol-Volumen abgezogen ist. Man erkennt, das die Normalwerte im allgemeinen erfiillt sind. Auffillig ist einmal der geringe Wert fur Jod in TiJ,, iiber den spater im Zusammenhang mit Dichte- bestimmungen an Molekiilgittern zu berichten sein wird, und ferner der auffallig niedrige Wert fiir die Hydroxylgruppe im KOE, der
l) W. KLEYH u. J. ROCKSTEOH, Z. anorg. u. allg. Chem. 162 (1926), 247. 4 Unter ,,Normalwert" ist das Volumen verstanden, das Rich in Ver-
bindungen besonders haufig findet; die Nullpunktsvolumina sind bei Br und namentlich J etwas kleiner, bei F erheblich griiber.
3, Vgl. W. BILTZ) Ann. 463 (1927), 273.

22 W. Klemm, W. Tilk und S. v. Miillenheim. Dilatometrische Messung usw.
dafiir spricht, daS beim Ubergang von NaOH zu KOH ein iihnlicher Wechsel im Qittertypus stattfindet, wie beim obergang von RbCl zu CsCI; es ware dies im Einklang mit der sprunghaften Knderung der Ausdehnungskoeffizienten (vgl. S. 19).
Herrn Professor W. BILTZ mochten wir auch an dieser Stelle fiir die Fiirderung dieser Untersuchung durch die Mittel des Instituts und fur wertvolle Ratschlage herzlichen Dank aussprechen.
Hammover, Twhnische Hochschzlle, Institzlt fuv unoTgunische Chemae.
Bei der Redaktion eingegangen am 10. August 1928.












![Methyl(trifluormethyl)haIogensulfonium-Salze [ 1 ] Halogen = …zfn.mpdl.mpg.de/data/Reihe_B/43/ZNB-1988-43b-0403.pdfMethyl(trifluormethyl)haIogensulfonium-Salze [ 1 ] Halogen = F,](https://img.pdfslide.org/doc/110x75/60b8a3e47a6cd577112f3fd0/methyltrifluormethylhaiogensulfonium-salze-1-halogen-zfnmpdlmpgdedatareiheb43znb-1988-43b-0403pdf.jpg)
















