Embed Size (px)
Citation preview

J . .Jannek zc. J . Meyer. Eiize neue Bestimmzing des Atomngewichtes usw. 51
Eine neue Bestimmung des Atomgewichtes des Selens. Von
JOSEB JANNEK und JULIUS MEYER. Mit 4 Figuren im Text.
Geschichte und Beurteilnng der bisherigen Atomgewichtsbestimmungen des Selens.
Der Entdecker dieses Elementes, das in der neueren Zeit durch seine eigentiimlichen photochemischen und chemotherapeutischen Eigenschaften mehr und mebr an Bedeutung gewinnt,, bestimmte auch sein Aquivalent- und damit auch sein Atomgewicht. J. J. B E R Z E L I ~ fand, daB 100 Gewichtsteile Selen beim Sattigen mit Chlorgas ihr Gewicht um 179 Teile vermebren, wobei aus dem Selen das Selen- tetrachlorid SeC1, entsteht.z Auf 0 = 100.00 bezogen, berechnet BERZELIUS das Atomgewicht des Selens zu 494.59. Fur die jetzt dlgemein angenommene Basis 0 = 16.000 erhalt man aus den Messungen des BERZELIUS den Wert Se = 79.13. Wie ERDMANN~ bemerkt, ist in der WEBERSChen Atomgewichtstabelle als Ergebnis der Messungen des BERZELIUS der Wert 495.285 enthalten, aus dem sich Se = 79.25 ergibt. In einer wertvollen Zusammenstellung und Durchrechnung der Atomgewichte siimtlicher Elemente hat F. W. CLARKE,$ offenbar ebenfallv aus diesen Angaben, fir das Selen den Wert 79.25 berechnet. Uber den Wert der Bestimmungen des BERZELIUS laBt sich heute nur noch schwer ein sicheres Urteil ftillen. Aber es ist doch sehr fraglich, ob diese an sich so wertvollen und einst grundlegenden Untersuchungen noch den Anforderungen geniigen, die wir auf Grund der Fortscbritte der Naturwissenschaften heute
Pogg. Am%. 6 (1826), 1. Es sei hier auf eine irrtiimliche Augabe V. LEWIIERS, J. Arne?. Clterrb.
Sac. 20 (1898), 555 aufmerksam gemacht, die auch F. W. CLAEKE ubernommen zu haben scheint, wonach bei BERZELIUS ails 100 Teilen Se 179 Trile SeCI, ent- standen sein sollen, wiihrend in Wirklichkeit die Gewichtszunahrne 179 Teile betrug.
Jotma. prnkt. Chem. 55 (1852), 202. 4 The Constants of Nature, Part V, A Recalculation of the Atomic:
Weights, 1910, p. 381. 4"

52 J. Jannelz und J. 31eye.r.
an die Reinheit einer Substanz, an die Unangreifbarkeit der GefaB- materialien und an die Vollstandigkeit der Ilurchfuhrung der chemischen Reaktionen stellen miissen, die bei den Bestimmungen der dtomgewichte in Frage kommen. Es mag nur eine Frage hier aufgeworfen werden, die auch schon CLARKE stellt, ob es denn iiber- haupt miiglich ist, nach der von BERZELIUS angewendeten Methode reines, von jeder Spur Selenoxychlorid freies Selentetrachlorid zu gewinnen. Eine geringe Menge SeOCI, wurde aber das scheinbare Atomgewicht des Se merklicli erhohen.
Die chronologisch nun folgenden Arbeiten von E. MITSCEERLICH und NITZSCH,~ welche Alkaliselenate analysierten, brauchen nicht naher erortert zu werden, da sie nicht in erster Ilinie die Bestimmung des Atomgewichtes des Selens, sondern nur die Festlegung der Zu- sammensetzung der Selensaure bezweckten, und da E. MITSCHERLICH selbst zugibt, da8 bei der Analyse etwas Selen verloren ging.
Den Arbeiten von M. F. S A C C ~ liegt der an sich sehr gesunde Gedanke zugrunde, da8 man bei Atomgewichtsbestimmungen mog- lichst verschiedene Methoden anwenden miisse. Denn bei der Analyse ein und derselben Verbindung nach stets derselben Methode kann ein und derselbe Versuchsfehler in allen Bestimmungen ent- halten sein, so daB auch eine grof3ere Anzahl von Bestimmungen dann keine wahrheitsgetreueren Werte liefern kann. SACC hat zur Ver- meidung und Erkennung derartiger systematischer Versuchsfehler zuerst elementares Selen in ,,selenige Saure", gemeint ist aber Selendioxyd, verwandelt; d a m hat er Selendiuxyd und Selenite zu Selen reduziert, und schlieBlich hat e r Selenite vollstandig analysiert. Er reinigte das Selen, in dem sich auBer Tellur noch Schwefel und Calciumsulfat vorfand, durch Auflosen in Salpetersaure und einmaliges Sublimieren des Selendioxyds, aus dessen wasseriger Lijsung das elementare reine Selen dann mit Ammoniumbisulfit und Salzsaure gefallt wurde. Auf den erfolglosen Versuch einer trockenen Oxydation des Selens durch Sauerstoff und Stickoxyde merden wir spater zu- riicklrornmen. Bei der Oxydation des Se zu SeO, durch Salpetersaure arbeitete SACC mit ungewijhnlich groBen Mengen (42-60 g Se) und erhielt bei drei Versuchen wenig ubereinstimmende Ergebnisse: Se = 500.00, 506.30 und 489.52, oder Se = 80.00, 81.01 und 78.32. SACC hat zu dem letzten Werte das meiste Zutrauen und glnubt,
* Pogg. Ann. 9 (1827), 623. 'd Ann. Chim. Phys. [3] 21 (1847), 119.

Eine neue Bestimmung des Atomgewichtes des Selens. 53
daf3 das zu hohe Atomgewicht der beiden ersteri Versuche auf einen SalpetersLuregehalt des Selendioxyds zuriickzufihren sei. Indessen hatten geringe Mengen von HNO, in SeO, gerade umgekebrt den Wert herabgedriickt, nicht aber erhijht. Es ist hingegen nach unseren Versuchen unzweifelhaft, daB sich beim Eindampfen der salpeter- sauren Selendioxydlosung zur Trockne merkliche Mengen von SeO, verfliichtigt haben, wahrend andererseits noch Spuren von Wasser darin zuriickgeblieben waren. Denn wie wir weiter unten auf Grund der Dampfdruckkurven des Selendioxyds und der selenigen Siiure zeigen werden, ist es unmoglich, durch Eindampfen einer wiisserigen Losung von seleniger Saure absolut wasserfreies Selendioxyd ohne Verluste zu erhalten, das nicht eiumal beim Sublimieren des Ruck- standes vollig einwandfrei zu erzielen ist. SACC hat bei seinen Versuchen auf3erdem die Bildung der Selenstiure ganz auger acht gelassen. SACC hat dann ,,trockene selenige Saure((, gemeint ist wiederum Selendioxyd, durch Ammoninmbisulfit und Saizsaure zu Selen reduziert. Er verwendet 0.5, 3.5 und 5 g SeO,. Die Ergebnisse dieser Versuche waren: Se = 78.34, 78.05 und 78.96, bezogen auf 0 = 16.000. Auch bei diesem Verfahren liegen recht erhebliche Fehler vor, die auf die Verunreinigung des Selendioxyds durch Feuchtigkeit zuriickzufiihren sind. Ferner sind im nichtumsub- limierten Selendioxyd j e nach der Darstellungsweise mehr oder weniger betdchtliche Mengen von Stickosyden vorhanden.
Diese bisher recht diirftigen Ergebnisse sucht SAW nun noch durch Analysen von Bariumselenit BaSeO, zu stiitzen, die aber unter sich Abweichungen bis zu 2 "II, zeigen. Das Bariumseleiiit wurde hei diesen Versuchen durch Ammoniumbisulfit und Salzsaure in ein Gemisch von Selen und Bariumsulfat verwandelt, das zur Wagung gebracht wurde. SACC gibt zu, daB es ihm unmoglich gewesen sei, den Niederschlag von Se und BaSO, vollstandig &us den Oefafien zu entfernen, d a er sehr fest an den Wandungen sa8. AnBerdem hatte sich, angeblich infolge von Hyposulfitbildung, beim EingieBen von Salzsaure in die heifie Losung auch etwas Schwefel abgeschieden. Demnach sind auch die hier erhaltenen Zahlen un- zuverlassig.
SACC analysierte weiterhin auch noch mehrere andere Selenite. Aus neutralem Bariumnitrat und Natriumselenit stellte er Barium- selenit dar, das gewaschen und gegliiht wurde. Aus dem Riickstande wurde durch Behandeln mit iiberschiissiger Schwefelsaure Barium- sulfat gewonnen und zur Wagung gebracht. Aus vier Analysen,

54 J. Jartnek zcnd J. Meyei.
bei denen 0.2-1.0 g BaSeO, angewendet wurden, ergab sich so im Mittel Se = 78.63. Ferner fallt SACC aus AgN0,-Losung durch selenige Saure das Silberselenit Ag,SeO, nus, das aber durch Be- handeln mit Salzsiiure iiur in unvollkonimener Weise in AgCl ver- waiidelt werden konnte. Durch Hinwirkung von konzentrierter Schwefelsiiure auf Silberselenit erhielt er dann beim Eindampfen Silbersulfat, das zwar frei von Se, aber stets durch metallisches Ag verunreinigt war. Aus zwei Analysen berechnet er hierbei das Btorngewiclit Se = 79.06. In ghnlicher Weise zersetzte SACC J31ei- selenit mit konzentrierter Schwefelsaure und wog das gegliihte Blei- snlfat, das aber stets etwas Selen zuriickbehahen hatte. Aus drei unter sich sehr scblecht ubereinstimmenden Analysen, die bis 8 o/i , Unterschied aufweisen, berechnet er Se = 76.21. Auch bei dieser Zahl, die ihm selbst etwas zu lrlein erscheiiit, hiilt er den Fehler nicht fur so bedeutend, um sie von der endgultigen Bereclinung ausxuschlieflen. Einwandfrei halt er allerdings nur die Reduktioii des Selendioxyds zu Se (78.46) und die Umsetzung des Barium- selenits xu Bariumsulfat (78.63); das Mittel daraus ist Se = 78.55. Wenn nian aber, wie es SACC dann tut, auch die anderen Analysen, niimlich die Oxydation des Se zu SeO,, Umwandlung des ,Ag,YeO, in Ag,SO, und ties PbSeO, in PbSO, mit in Betra,cht zieht, so hat man angeblich eine grabere Wahrscheinlichlreit, dem wirklichen Atom- gewichte des Selens sich zu niihern, da man sich dann dem BER- zELIusschen Werte 79.13 nahert. Tatsachlich ist das aber gar nicht der Pall, d s das Mittel aus den samtlichen Werten 78.31 ist.
E’;tssen wir die Ergebnisse der Saccschen Arbeit zusammen, so darf sie keinesfalls a19 ein Fortschritt gegeniiber der Untersuchung des BERZELIUS betrachtet werden. Trotz der Mannigfaltigkeit der benutzten Methoden ist die Bedeutung der Arbeit fur die Atom- gewieh tsbestimmung des Selens nur gering.
011 den Untersuchungen von 0. L.ERDMANN und MARCH AND,^ denen CLARKE sorgfaltiger Arbeiten nachriihmt, groBeres Wert beizumessen ist, steht clahin. Mit der griiBtei Sorgfdt dargestelltes, wiederholt sublimiertes und schon kristallisiertes Selenquecksilber wurde mit reincm Kupfer gemischt, worauf das Quecksilber abdestilliert, kon- densiert iind gewogen wurdc. Als Mittelwert ergab sich Se = 78.80. Niihere hngaben felilen leider in der kurzen VeriifTentlichung, wahrhrend die in Aussicht gestellte ausfuhrliche Arbeit nicht erschienen zu sein scheint. -
Joz6t.n. pmk t . Chem. 66 (1552), 202.

Eine neue Beslimmuiag des Atomgewichtes des Xelencs. 55
Die nachsten Bestimmungen riihren von DUN AS^ her, der zur ursprunglichen Uethode von BERZELIUS zuruckkehrte, indem er reines Se durch Behandeln mit Chlor in SeCl, verwandelte, wobei das uberschiissige Chlor zur vollstindigen Kondensation des Tetra- chlorids auf -20° abgekuhlt wurde. Die Ergebnisse von 7 Analysen, bei denen immer ungefahr 2 g Se angewendet wurden, schwanken zwischen Se = 79.20 und 79.66. Der Mittelwert ist 79.38. Ab- geselien von der ziemlich schlechten Ubereinstimmung der W erte uiiter sich gilt hier derselbe Einwand, der oben gegen BERZELIIJS erhoben wurde, daf3 sich namlich merkliche Mengen von SeOCl, gebildet haben, worauf auch die hohen Zahlen fur Se hindeuten.
Nach neuen Bestimmungsmethoden haben dann mit grolSem Eifer ERMAN und PETTERSSON gesucht. Nach vergeblichen Versuchen mit mancherlei Salzen (CaSe0,. 2 H,O , MgSeO, . 6 H,O , (NH,),SeO,. AI,(SeO,), .24H,O, Ag,SeO,, Ag,SeO,) kommen sie zu der Ansicht, ,.daW nur wenige Selenverbindungen sich zu einer genauen Analyse eignen. Neben der eigentlichen Reaktion gehen niimlich andere Um- setzungen vor sich, die von Massenwirkung oder Dissoziation herriihren und einen allerdings sehr geringen, aber doch fur die' Genauigkeit der Resultate verhangnisvollen EinfluB ausiiben". Darum wollen sie nicht einmal ihre Analysen des Silberselenits, die nach ihrer eigenen Sngabe einen normalen, einwandfreien Verlauf nahmen, zur Berechnung des Atomgewichtes herangezogen wissen, zum Teil auch aus dem Grunde, weil ihnen die angewendeten Substanzmengen im Verhaltnis zu der GroBe des zu bestimmenden Atomgewichtes zu klein erscheinen. Bei 7 Analysen, die mit 5-7 g Ag,SeO, aus- gefuhrt wurden, erhalten sie Zahlen, die zwischen 78.90 und 79.18 schwanken, was allerdings keine gute Ubereinstimmung bedeutet. Nach den Erfahrungen, die die modernen Untersuchungen iiber Atomgewichtsbestimmungen geliefert haben, sind die angewendeten Substanzmengen aber keineswegs zu gering, und der Grund fur die mangelhafte Ubereinstimmung der Ergebnisse durfte in den Mangeln der benutzten Methode zu suchen sein. EKMANN und PETTERSSON erhitzten namlich reines Ag,SeO,, wobei das Selendioxyd von der Oberflache der geschmolzenen Substanz allmahlich ohne Spritzen verdampf'en und nach dem Zerfall des Silberoxyds reines Silber zu- ruckbleiben 5011. Dies ist aber, worauf auch schon CLARKE hinweist,
' Lieb. Awn. 113 (1860), 32. * Bed. Ber. 9 (1576), 1210.

56 J. Jannek und J. Meyer.
durchaus nicht der Fall, da das geschmolzene Silber immer Spuren von Selen zuriickbehalt, die das Atomgewicht zu klein erscheinen lassen. DaB es uberhaupt unmoglich ist, Silber durch Gluhen selen- frei zu erhnlten, hat auch schon JACKSON~ naahgewiesen. EKMAN uncl PETTERSSON fuhren dann als ,,tadellos und vollkommen zuver- lassig" funf Reduktionen des Selendioxyds zu Selen an , das aus einer erwarmten LGsung durch Zusatz von Salzsaure und Einleiten von SO, ausgefallt und auf einem Glasfilter gesammelt wurde. Es lramen 11-31 g SeO, zur hnwendung. Zweimal wurcle der Wert 79.06, dreimal 79.08 uiid einmal 79.10 erhalten, so daD man als mittleren Wert 79.08 erhalt. Gegen die Arbeitsweise von PBTTERSSON und EEMAN ist einzuwenden, daB sie kein absolut waseerfreies Selendioxyd in den Handen gehabt haben, und ein geringer Feuchtigkeitsgehalt druckt das Atomgewicht herab. Auger- dem scheint es nun nach unseren Erfahrungeii doch nicht ganz zweifellos zu sein, daB die Reduktion mit schwefliger Saure so uber- aus quantitativ verlauft, wie es hier bei derartigen Prazisionsver- suchen zu verlangen ist.
A h nachste Untersuchung kommt eine Arbeit von V. LEN HER^ in Betracht, der zunachst reines Silberselenit durch einen Chlor- wasserstoffstrom unter Erhitzen in AgCl uberfiihrte, wahrend das Selendioxyd in die fliichtige Verbindung SeO,. 2HC1 iiberging und entwich. Nachdem das Chlorsilber gewogen war, wurde es noch im Wasserstoffstrom zu metallischem Silber reduziert, das dann eben- falls gewogen wurde. Das verwendete Silber wm nach der Methode von STAS, das Selen durch wiederholtes Umsublimieren des SeO, gereinigt worden. Das Ag,SeO, ist ein sehr gut kristallisierendes Salz ohne Kristallwasser von groBer Bestandigkeit. Indessen be- zweifeln wir, daB es bei Temperaturen von 7 O - S O 0 , bei welchen Temperaturen es getrocknet wurde, alle Feuchtigkeit verliert. Mit diesem Versuchsfehler wurde aber ein zu hohes Atomgewicht her- auskommen. LENHER gibt an, daB seine Priifung des Chlorsilbers auf Se negativ ausfiel, ohne aber eine Methode zum Nachweise geringer Se-Mengen anzufuhren. Die bisher bekannten Methoden reichen aber nicht aus, um sehr geringe Selenmengen, 0.1 mg und weniger, mit Sicherheit nachzuweisen. Trifft der Einwand, da8 das AgCl selenhaltig war, aber zu, so mul3 der dadurch entstehende
Lieb. Ann. 179 (1875), S. Jourm. Amer. Chenz. Sac. 20 (1895), 555.

Eine neue Bestimmung des Alorngewicl~tes des Selens. 57
Fehler auf das Atomgewicht verkleinernd wirken. LENHER hat ferner sein Porzellanschiffchen im HC1-Strome erhitzt. Nach unseren Er- fahrungen ist es nicht moglich, hierbei das Gemicht eines Porzellan- gefaBes geniigend konstant zu halten. Die hier dargelegten Fehler- quellen zeigen sich auch deutlich in den Ergebnissen. Bei elf' Umsetzungen von Ag,SeO, zu AgCI liegt das Atomgewicht des Se zwischen 79.263 und 79.373, im Mittel bei 79.329. Bei 8 Reduk- tionen des AgCl zu Ag zwischen 79.250 und 79.369, im Mittel wiederum bei 79.329. Die bei demselben Versuche aus der Umsetzung des Ag,SeO, zu AgCl und Reduktion des AgCl zu Ag erhaltenen Werte miiBten bei sorgfaltigem Arbeiten iibereinstimmen. Auger in 2 Fallen ist dies aber keineswegs der Fall, ein weiterer Beweis, da8 die LENHEasche Methode, wenn nicht unbrauchbar, so doch fiir Atomgewichtsbestimmungen noch bedeutend besser aus- gearbeitet werden muB.
LENHER hat dann noch eine zweite Reihe von Bestimmungen ausgefiihrt, indem er Ammoniumbromoselenat (NH,),SeBr, zu Selen reduzierte. Ammoniumbromid wurde durch Umkristallisieren sorg- faltig gereinigt. Von dem reinen Produkte wurden 89 Teile in Wasser geliist, mit 4 Teilen Selen und mit Brom, das durch Be- handelnmit MnO, und konzentrierter H,SO, von Chlor und organischen Bestandteilen befreit war, in geringem UberschuB bis zur vollstandigen Losung versetzt. Beim Eindampfen schied sich das Bromoselenat ab. Es wurde mehrmals aus verdunnter Bromwasserstoffsaure um- kristallisiert und nach dem Trocknen im Natronkalkexsikkator unter dem Xikroskop auf Reinheit gepriift, wobei zwischen den roten Bromoselenatkristallen keine weiBen Ammoniumbromidkristalle ge- funden wurden. Diese Reinheitspriifung ist mehr a19 zweifelhaft, cia das NH,Br j a auch in Form von Mischkristallen in fester Losung mit dem Bromoselenat vorliegen kann. Aus der kalten Losung einer gewogenen Menge des Bromoselenats wurde das Selen nach Zusatz von etwas Ammoniak durch Hydroxylaminchlorhydrat ausgefiillt und durch Erwarmen in die schwarze Modifikation uber- gefiihrt. Das schwarze Selen wurde auf ein Asbestfilter gebracht, gewaschen, bei l ooo getrocknet und gewogen. Die Anwendung eines Asbestfilters ist indessen bei Prazisionsarbeiten nicht ange- bracht, und LENHER gibt auch nicht an, ob seine Asbestfilter beim Auswaschen mit Wasser geniigend gewichtskonstant geblieben sind. Wir bezweifelii es. Auch die Ergebnisse, die LERHER nach dieser Methode erhielt, beweisen die geringe Zuverlassigkeit des Verfahrens.

58 J. Jannek und J. Meyev.
Denn bei 8 Bestimmungen lagen die Werte fur Se zwischen 79.226 und 79.367. Der Mittelwert ist S e = 79.285. Mit den vorher- gehenden beiden Ergebnissen vereinigt ergibt sich Se = 79.314, bozogen auf Ag = 107.92, Br = 79.95, C1= 35.45, N = 14.04 und H = 1.005.
Ohne Bedeutung sind die beiden Atomgewichtsbestimmungen, die STEINER~ bei einer Untersuchung iiber clas Tellur nebenbei ausgefuhrt hat , indem er durch Verhrennen des Phenylselenids (C,HJ,Se den C-Gehalt bestimmte und daraus die Werte Se = 78.8 und 70.1 berechnete. Es brauclit wolil nicht weiter ausgefuhrt zu wertlen, dd3 eine organische Verbrennung kaum die richtige BIethode zur Bestinimung von Atomgewichten ist.
Kine neue Art der Analyse des Silberselenits ist dann von JULIU~ MEYER~ rersucht worden, cler das Silber aus eirier cyan- kalischen Ag,SeO,-Losung elektrolytisch auf einer mattierten Platin- schale niederschlug. Die Reinigung des Silbers und Selens geschah iiach den bekannten Methoden. Ein offenkundiger Mangel dieser Methode bestelit darin, daB das Silber aus der cyankalischen Losung auf elektrolytischem Wege nicht absolut quantitativ ausgeschiederi wird. 1)enn nach den Untersuchungen von H. NERNST und von FAR UP^ lost sich das ausgeschiedene Silber unter dem Einfiusse des Luft- sauerstoffs in der Kalinmcyanidliisung durch Autoxydation wieder auf. Dementsprechend gahen die entsilberten Lijsungen aus funf Restimmungen nach dern Eindampfen mit Salpetersaure und Weg- sublimieren des Selendioxyds bei erneuter Elelrtrolyse noch 0.2 mg Silber. Die so erhaltenen Zahlen schwanken zwischen 79.17 und 79.28. Das Mittel ist 79.23 und erniedrigt sich noch unter Berucksich- tigung des nachtraglich ausgeschiedenen Silhers auf 79.21. Das geringe Zutrauen, das ,JULIUS MEYER zu dieser Zahl hat, ist die Veranlassung gewesen, das Atomgewicht des Selens neu zu be- stimmen.
Zum SchluB mussen wir noch auf eine Untersuchung von Kuzwa und KREHLIK hinweisen, die von B. BRAUNER in den Ab- handlungen der K. I(. Franz- Josefs-Akademie in tschechischer Sprache mitgeteilt worden ist. Das von ihnen gefunclene Atomgewicht ist i 9 27, auf des Vakuum bezogen Se = 79.26. und ist in dem Be- richte der Internationalen Atomgewichtskommission fur 1913 ver-
Uerl. Ber. 36 (1901), 570. 2. a m r g . Chem. 31 (1902), 391: Berl. Ber. 35 (19021, 1591. 2. f. Elektroohenz. S (1902), 569.

Eine new Bestimmung des Atomgewichtes des Selens. 59
offentlicht worden. Sie ist das Mittel von zehn Bestimmungen, die auf der Reduktion von seleniger Saure durch Schwefeldioxyd be- ruhen. E s war uns leider nicht moglich, uber die Arbeitsweise etwas Genaueres zu erfahren, weder in der Literatur noch von Herrn Hofrat €3. BRAUNER selbst, der iibrigens nach einer schriftlichen AuBerung diese Untersuchung noch nicht als abgeschlossen be- trachtet. Aus diesem Grunde ist eine Beurteilung des KIJZMA- KREHLIKschen Wertes noch nicht moglich. Eine kiirzlich er- schienene Untersuchung der Gasdichte des Selenwasserstoffs durch P. BRUTLANTS und A. BYTXBIER~ f ihr t fur das Atomgewicht des Selens zu dem bemerkenswert niederen Werte Se = 79.18.
Vorarbeiten. Seit den Versuchen von E ~ M A ~ ; und PETTERSSON, die nach den
oben gegebenen Darlegungen nicht den Anspruch auf Genauigkeit erhehen konnen, wie sie bei den Atomgewichtsbestimmungen der neueren Zeit angestrebt werden mi& seit diesen Versuchen ist eine direkte Bestimmung des Verhaltnisses Se : 0 nicht wieder ausgefuhrt worden, wenn wir die noch nicht diskutierbare Untersuchung van KEEHLIK und KUZMA auBer acht lassen. Und dabei stellt gerade die Uberflihrung eines Elementes in sein Oxyd oder-der umge- kehrte Weg die ideale Methode einer Atomgewichtsbestimmung dar, weil das betreffende Element in direkte Beziehung zur experimen- tellen Basis der Atomgewiclite gesetzt wird und jede Umrechnung iiber das mehr oder weniger genau bestimmte Atomgewicht eines Hilfselementes und damit eine haufig erhebliche Zahl an Versuchs- fehlern fortfallt. Freilich bietet gerade die quantitative Ausfuhrung der Oxydation bei Beobacbtung der allergroBten Genauigkeit nicht geringe Schwierigkeiten, wie sich nuch im Verlaufe dieser Unter- suchung noch zeigen wird. Bei der Peststellung des quantitativen Verhsltnisses zwichen Se und SeO, haben ferner die Versuchsfehler, die auch bei den sorgfaltigsten Arbeiten niemals vollstandig zu ver- meiden sind, den geringsten EinfluB auf das Ergebnis. Daher haben wir es doch versucht, die experimentellen Schwierigkeiten einer quanti- tativen Oxydation von Se zu SeO, und einer zur Kontrolle sich an- schliegenden Reduktion des erhaltenen SeO, wiederum zu Se zu iiberwinden, was denn auch schlieBlich in verhaltnismaBig einfacher Weise gelungen ist. Ein Vergleich der angewendeten und wieder-
-
Bull. Bead. rroy. Belg. 1912, 856-70. 2 Vgl. W. OSTWALD, Lehrb. d. allgem. Chem. I, S. 21.

60 J. Jannek und J. Meyer.
erhaltenen Selenmengen lie6 dann auch sogleich erkennen, ob bei dem betreffenden Versuch etwa Verluste eingetreten waren. Damit dieses en tscheidende Kriterium aber auch reichlich einwandfrei he- nutzt werden konnte, wurde erst festgestellt, ob sich Selen mit den zur Verwendung gelengenden Losungsmitteln und Reagenzien etwa verfluchtigt oder darin loslich ist. An zweiter Stelle muBte uber das Verhalten von SeO, und H,SeO, bei hoheren Temperaturen Klarheit geschaffen werden. Vor allem war festzustellen, ob selenige Saure durch geriiigendes Erwgrmen samtliches Wasser abgibt nnd in Form von chemisch reinem Dioxyd zuriickbleibt. Bisher war ja keine andere Methode zur Darstellung von SeO, bekannt. Die groBere oder geringere Leichtigkeit der Wssserabgabe eines Hy- drats laWt sich nun aus dem Verlaufe der Tensionskurve dieses Hydrates entnehmen. Daher wurde die Tension der selenigen Saure bestimmt. Es war ferner zu untersuchen, ob sich bei der Entwas. serung der selenigen Saure nicht auch das entstehende Dioxyd ver- fliichtigt, weshalb auch die Dampfdruckkurve des Selendioxyds ge- messen wurde.
Die Tension der selenigen Saure.
Zur Darstellung der reinen selenigen Saure wurde Selen in Salpetersaure aufgelost , das beim Eindampfen zur Trockne ent- standene Dioxyd umsublimiert und in wenig Wasser aufgel6st. Die LGsung wurde im Porzellanschiilchen unter sorgfaltigem AusschluB von Staubteilchen, die schon in den geringsten Mengen reduzierend wirken, bis zur beginnenden Kristallisation auf dem Wasserbade ein- geengt. I m Schwefelsaureexsikkator erhalt man bald eine feste Kristallmasse, die man so lange im Exsikkator stehen laBt, bis sie trocken erscheint. Diese Bristallmasse wird zerrieben, wobei man ein Kristallpulver erhalt, das man erforderlichenfalls noch einmal bis zu r Trockne uber Schwefelsaure stehen 1aBt. Eine -4nalyse des trockenen Kristallpulvers lieferte 61.10°/, Se, wahrend sich 61.30°/, Se fur die Formel H,SeO, berechnen. Die so erhaltene farblose trockene selenige Saure wurde stets uber Schwefelsaure aufbewahrt. Eine hierbei eintretende Dehydratation des H,SeO, zu SeO, ist fur die Tensionsbestimmung ohne Belang. Die Dampfdruckmessungen wurden in dem iiblichen Differentialtensimeter ausgefuhrt, das auf der einen Seite mit konzentrierter Schwefelsaure, spater mit dem besser ge- eigneten Phosphorpentoxyd beschickt war. Als Manometerfliissigkeit erwies sich Paraffin61 als ungeeignet, da infolge einer Reaktion

Eine neue Bestimmung des Atonzgewichtes des Selens. 61
zwischen H,SeO, und Paraffinoldampfen geringe Gasbildung, ver- mutlich C0,-Bildung , auftrat , was sich aul3er der unregelmafiigen Tensioii auch an der Rotfarbung der selenigen Saure zu erkennen gab. Bei den niclit unbetrachtlichen Dampfdrucken erwies sich Quecksilber als Manometerfliissigkeit recht geeignet. Bei steigenden Temperaturen wurden in den verschiedenen Versuchsreihen befrie- digend ubereinstimmende Ergebnisse erhalten, bei fallenden Tempe- raturen aber nicht. Uer Augenschein lehrt, daf3 das frei gewordene Wasser beim Abkuhlen nicht wieder vollstandig von dem Dioxyd aufgenommen wird. Es wird vielmehr zuerst von der obersten Schicht absorbiert und die so wieder entstandene H,SeO, bildet dann eine feste, zusammenhangende Schicht, die die weitere Wasseraufnahme erschwert. Es bilden sich dann stellenweise mehr oder weniger gesattigte Losungen von seleniger SBure in Wasser , deren Dampf- druck je nach Iionzentration und TropfchengroBe verschieden sein kann. Die Dauer eines Versuches betrug 10-12 Stunden. Die in drei Versuchen beobachteten Werte sind in folgender Tabelle wieder- gegeben. Bei Zimmertemperatur trat selbst bei tagelangem Stehen keine Niveaudifferenz auf.
I1
19.0 25.0 :3 2 35 37 38.5 40 40.5 44 44.5 47 50 53 53.4 56 59 61 62 63 66 69 70 70.8 72.4 75 75.4
I11 Tension ( p ) in mm nach Versuch
3.5
8.7
15.0
39.0
46.4
5.3
8.3
18.2 24.2
25.4 33.4 38.8
40.0 42.6
1.2
25.2
35.6
36.6
49.6

62
Temperatoren _ _ ~ - I- __
LI
Y s a ir
a B 2
77.6 79.4 so.0 84.0 84.6 85.0 88.0 89.0 91.2 94.0 96.6
105.0 107.0 110.4 111.0
106.6 99.4 97.0 86.0 85.6 76.4 7 1.4 70.0 67.6 64.0 62.4 60.6 56.0 50.0 36.0 1 s.0
Tension ( p ) in mm naoh Versuch I
52.2 55.8
67.0
74.6 78.4
_ _ ~ _ _ ~ ~ ~ _ _ ~
75.4
41.8 35.6 34.1 32.0 29.6
15.8
2.9
I1
50.8 57.8 59.8
70.8 72.8
89.8 96.4
107.4 115.0 149.4 162.S 178.8 183.9
163.4 134.0 125.4
90.0
~ ~~
53.2
41.2 37.8
30.9
15.4 10.2
59.0 69.2 71.0
53.0
31.0
26.5
Die Tension des Selendioxyds. Zur Xessung des Dampfdruckes wurde ein aus HartgIas an-
gefertigtes Spiralmanometer verwendet, das nach dem Prinzip des LADEPI’BuKG-LEHMANNschen Manometers konstruiert worden war. Es war derselbe Apparst, den friiher JOHNSON zur Bestimmung der Dampfdichte des Salmiaks benutzt hatte. Fur die Abhangigkeit der Spiegellage von der Temperatur sind wir zu ganz anderen Er- gebnissen wie JOHNSON gekommeu. Bis 300° war keine Verande- rung der Spiegellage wahrzunehmen, yon da an war sie aber ziem- licli unregelmiI3ig. Auf die sehr sch!echt kontrollierbare Abhlngig- keit der Spiegeldrehung von der Temperatur bei Spiralmanometern haben neuerdings auch G. PREUNER und J. BROCKX~LLEI~ hingewiesen.
.Zreitsc/ir. plrysik. Chenz. 61 (1907), 457. &hclw. pliysik. Chela. S1 (1912), 129.
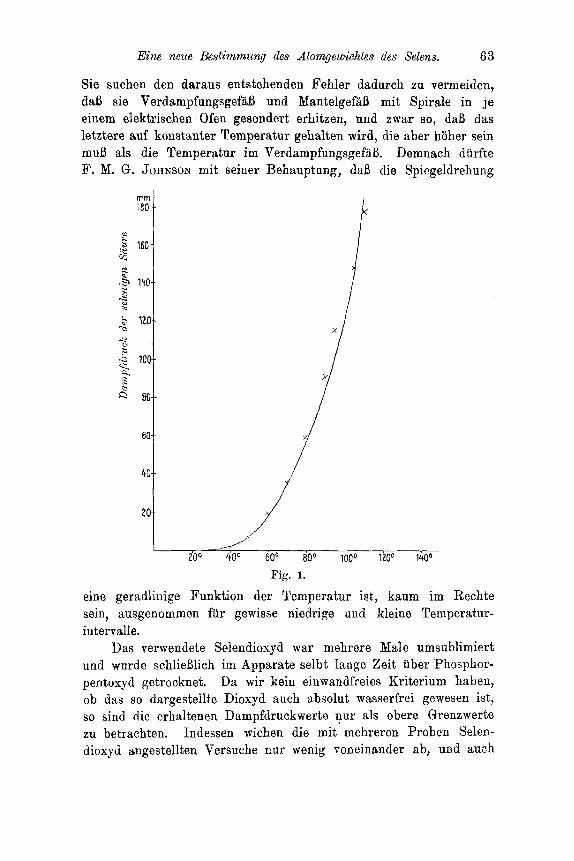
Eine neue Bestimmung des Atorngewichtes des Selens. 63
Sie suchen den daraus entstehenden Fehler dadurch zu vermeiden, daB sie VerdampfungsgefaB und MantelgefaB mit Spirale in j e einem elektrischen Ofen gesondert erhitzen, nnd zwar so, daB das letztere auf konstanter Temperatur gehalten wird, die aber htiher sein muB als die Temperatur im VerdampfungsgefaB. Demnach diirfte F. M. G. JOHNSON mit seiner Behauptung, daB die Spiegeldrehung
1 , /
200 400 60° 800 1000 1200 1400
Fig. 1.
eine geradlinige Funktion der Temperatur ist, kaum im Rechte sein, ausgenommen fur gewisse niedrige und kleine Temperatur- intervalle.
Das verwendete Selendioxyd war mehrere Male umsublimiert und wurde schlieBlich im Apparate selbt lange Zeit iiber Phosphor- pentoxyd getrocknet. Ya wir kein einwandfreies Kriterium haben, ob das so dargestellte Dioxyd auch absolut wasserfrei gewesen ist, so sincl die erhaltenen Dampfdruckwerte nur als obere Grenzwerte zu betrachten. Indessen wichen die mit mehreren Proben Selen- dioxyd angestellten Versuche RUP wenig voneinander ab, und auch
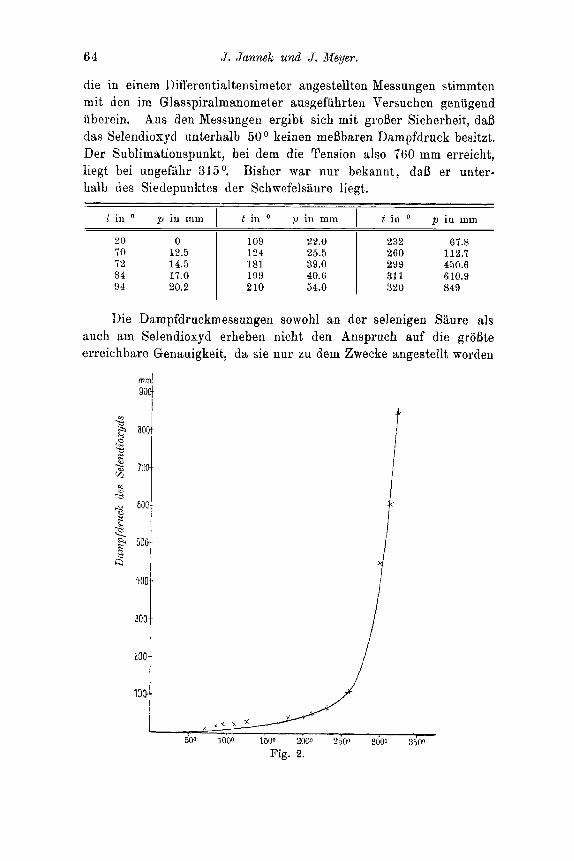
64 J. Jannek und J. Meyer.
die in einem Differentialtensimeter angestellten Messungen stimmten mit den im Glasspiralmanometer ausgefiihrten Versuchen geniigend iiberein. Aus den Messungen ergibt sich mit groBer Sicherheit, daB das Selendioxyd unterhalb 50 O keinen meBbaren Dampfdruck besitzt. Der Sublimationspunkt, bei dem die Tension also 760 mm erreicht, liegt bei ungef'iihr 315O. Bisher war nur hekannt, daB er unter- halb des Siedepunktes der Schwefelsaure liegt.
1 t in a p in mm , t in O p in mm 1 t in O p i n mm
20 0 109 22.0 232 67.8 i 0 12.5 1 124 25.5 260 112.7 72 14.5 181 39.0 1 299 450.6 84 17.0 199 40.6 311 610.9 94 20.2 210 54.0 1 320 849
- - ~ ______-________ - - . _ _ _____ - - - -
300.-
200--
100-
Die Dampfdruckmessungen sowobl an der selenigen Saure als auch am Selendioxyd erheben nicht den Anspruch auf die groBte erreichbare Genauigkeit, da sie nur zu dem Zwecke angestellt worden
I

Eine rLeue Uestimmurq des Atomgewicktes des Selens. G 5
waren, um iiber die Temperaturen zu unterrichten, bei der die Wasserabspaltung aus der selenigen SBure und die Verfliichtigung des Selendioxyds merklich zu werden beginnt. Die Tensionskurven zeigen mit aller wiinschenswerten Deutlichkeit, daB der Zerfall der selenigen Sauren in Wasser und Dioxyd schon weit unterhalb der Wasserbadtemperatur beginnt, dalj aber bei dieser Temperatur das Selendioxyd auch schon einen merklichen Dampfdruck besitzt und daher nicht unmerklich fliichtjg ist.
Wir haben versucht, die Abhangigkeit des Dampfdruckes der selenigen Saure sowohl als auch des Selendioxyds von der Tempe- ratur durch Formeln darzustellen und haben in erster Linie die NERNsTschen Formeln darauf anzuwenden gesucht. Indessen ist die Ubereinstimmung zwischen unseren beobachteten und den be- rechneten Werten eine recht mangelhafte. Wir beabsichtigen, dieses an sich recht interessante Problem spater, wenn wir im Besitze der erforderlichen Apparate sind, noch einmal eingehender zu behandeln.
Es sei nur hervorgehoben, da8 wir mit Hilfe der NEHNsTschen Dampfdruckformeln fur die molekulare Hydratationswarrne der sele- nigen Saure Werte erhielten, die zwischeii 3800 und 5000 cal lagen. Dies fiihrte iins dazu, die thermochemischen Verhaltnisse dieser Sauren etwas naher zu untersuchen.
Bestimmung der Kydratationswarme der selenigen Saure.
I n ein weithalsiges DEwARsches GefiD wurde eine bestimmte Menge destillierten Wasser von Zimmertemperatur gegeben, so daB das GefaB etwa zu gefiillt war. I n dieses Kalorimeter tauchte ein besonders konstruierter Intensivriihrer, der durch einen HeiBluft- motor angetrieben wurde , und ein BEcKMANNsches Thermometer, dessen Skala 0.01 O zeigte. Nachdem sich unter fortwahrendem Riihren eine konstante Temperatur eingestellt hatte, wurde aus einem schmalen Wageglaschen eine bestimmte Menge Selendioxyds oder seleniger Saure hinzugefiigt und die Temperaturanderung wahrend der Aufiosung bebbachtet. Die Auflosung war nach ungefihr 3 Mi- nuten beendet und die Temperatur wieder konstant. Aus der be- obachteteri Temperaturanderung, der Menge des angewendeten Wassers und der angewendeten Substanz berechuet sich d a m leicht die molelrulare Auflosungswarme des Selendioxyds und der selenigen -~
.Jahrb. f . h’/i~litrochenz. 13 (19Oti), 151. Vgl. die Dissert. von J. JANNEI, Breslsu 1913.
Z aiioiq. Chem Bd. 63. 5

66
Saure. Der Unterscliied zwischen beiden entspriclit der gesuchten Hydrntationswlrine des Selcndioxyds.
Eine ganz entsprechende kalorinietrische Versuchsaiiordnung ist jetzt, auch von E. ANDERSON uiid H. A. NOYES beschrieben uiid emp- foirlen worden. Unsere Spparatur liaben wir bereits im Sommer 1912 mit bestem Erfolge benutzt, wie folgende ZaEilen beweisen, die sich auf Zimmertemperiitur beziehen.
.I. .Jclntzel; u?bd J . &!eyer.
Wasserrnenge in g
150 160 160 lea 160 160 160
, Y 1 etr ipcr~t~~~BnrJer t ing in ('
-0.OS3 - 0.100 -0.115 -0.148 -0.48 )
-0.514 -0.50s
Mol. LGsungswiirmv in cal
- 944
- 940 - 936 -4116 -4123
- 5941
-4102
Als rnittlere Werte ergeben sich aus diesen Beobachtungen fur die molekulare LosungswBrme des Selendioxyds in Wasser - 928 ca1, fur die der selenigen SBure -4114 cal. Die Hydratationswiirme des Selendioxyds berechnet sic11 dnraus zu +3192 cal, so daB wir die thermochemische Gleichung erhalteu:
SeO, + H,O = H,SeO, + 3192 cal.
gibt fur die LGsungswiirme der selenigen Siiure den Wert -918 cal m. Es liegt hier zweifellos von seiten des Altmeisters der Thermochemie eine Verwechselung der Namen Seleiidioxyd und selenige Skure vor, die auch der Ubersetzer nicht, beseitigt hat.
Es 1st bemerkenswert, daJ3 die aus den Danipfdrucken der selenigeii SBure entnomnieiie HydratationsiTarme von der experi- mentell gefundenen nicht allzu erheblicli abn.eicht, ein Zeichen, daJ3 die gemessenen Dampfdruclre wolii im groBen und gaiizen richtig sind.
Der Dampfdruck des Selens. urn festzustelleu, his zu welchen Temperaturen Selen ohne Ver-
clampfungsverluste getrocknet werden kana, war es notwendig, auch den Dampfdruck des elementaren Selens zu beslimmen. Die be- nutzte Apparatur ist im Prinzip der von 0. RUFF und H. G R A P ~
JUL. THOATSEN
T T
' Jow-iz. PInys. Cl~em. 17, 249. ' Tliermochemisclie Untersuchnngen. Deutsch roil J. TI~AKJBG , Stuttgart
1906, s. 15s. J d t 6. f. Xlekfrochem. I4 (1907), 90.

Einc neue B c s f i ~ ~ r n u ~ ~ ~ des Atomgewichks des S’eleit Y. 6 7
angewendeten nachgeahmt worclen, die den Dampfdruck des Schwefels zwisclien 78 O und 210 O messen wollten. Zwei aneinandergeschmolzene U-Rohren enthielten ein Gemenge von gekorntem, metallischem Selen und Glasperlen, die nur zur VergroBerung der ObcrflHclie dienten. Uber dem Selen befand sich etwas Glaswolle, um das MitreiBen yon Selenstaub zu verhindern. An diese beiden U-Rohre schloB sich vermittels eines gasdichten Glasschliffes ein drittes, ebenfalls mit Selen gefulltes U-Rohr, das als Kontrollrohr diente und vor und nach jedem Versuche gewogen wurde. An dieses Kontrollrohr wurde vermittels eines Glasschliffes eine Kiihlschlange angesetzt, die in einem WEINHoLDSchen GefaBe durch Eiswasser auf 0’ gehalten wurde. Die drei U-Rohre mitsamt den beiden Schliffen - also auch das eine Ende der Kuhlschlange - standen vollstandig unter 6 1 in einem Paraffinolthermostaten. Die Glasschliffe wurden dmch je zwei Metallfedern zusammengehalten. Uber das Selen murde nun bei bestimmter konstanter Temperatur ein gewisses Volumen reinen Kohlendioxyds geleitet, das sich in den ersten beiden U-Rohren mit Selendampf stittigte, im dritten nichts mehr aufnahm und in der Kiihlschlange dann wieder abgab. Aus der Menge D des in der Kiihlschlange kondensierten Selendampfes und der Menge K des durch den Apparat hindurchgeleiteten Kohlendioxyds in Molen be-
rechnet sich dann der Dampfdrucli des Selens zu p = 13 E - j - D ’
wo B der Barometerstand wahrend des Versuches ist. Es wurde ein Versuch unterhalb des Schmelzpunktes 21 7 O des
Selens, bei 200 O, und ein zweiter dariiber, bei 230 O, ausgefuhrt. Das verfluchtigte Selen setzte sich schon oberhalb der eigentlichen Kiihlschlange als roter Beschlag fest, der beim nachherigen Trocknen der Kuhlschlange bei 100O von selbst in die schwarze Modifikation iiberging. Die Kontrollrijhre zeigte bei diesen Versuchen keine Gewichtshderung.
’
D
D i n 3101. ~ iIl:m _ _ ~ ~ -
~ .__ ___ - _____~ ~- 200 758 8 6 ~ 0.3572 1 0.0018 1 0.00001138 10.&415 230 ’ 757 1 5 6 0.3572 0.0023 000001453 0.03080
Die hier festgestellten Dampfdrucke sind so gering, daB sie bei den Versuchstemperaturen gar nichi in Frage konimen, wenn die Versuche nicht iiberaus lange Zeit erfordern. I n der Tat konnte pulverformiges Selen bei einer Temperatur von 1 70-200’ unter
5 *

Daruberleiten von trockener Luft stundenlang getrocknet werden, ohne daB ein Gewichtsverlust festzustellen war. F u r hohere Tem- peraturen ist der Dampfdruck des Selens kurzlich von G. PREUNER und J. BROCKM~LLER mit Hilfe eines Spiralmanometers aus Quarz- glas gernesseii worden. Uer niedrigste von ihnen beobachtete Wert war 3 inm bei 390°, was mit unseren Messungen gut harmoniert. Der Dampfdruck des Selens ist unter der Annahme berechnet worden, daB Clem Dampfmolekul die Forrnel Se, zukommt. Nun ist die MolekulargroBe des gasformigen Selens beim Siedepunkte unter Atmospharendruck selir nahe Se,, niihert sich mit wachsenden Temperaturen dem Werte Se,, um bei 2070°2 noch kleiner zu werden. Indessen ist es sehr unwahrscheinlich, daB die Molekel- groBe des Selendampfes unter den vorliegenden Verhaltnissen gleich Se, oder gar noch groBer ist. Denn bei den auI3erordentlich ge- ringen Dampfdrucken, die bei unseren Versuchsbedingungen fest- gestellt wurden, werden die Selenmolekiile weitgehend dissoziiert, wie die entsprechenden Versuche beim Schwefel erwiesen haben. Es diirfte daher angebrachter sein, der gasformigen Selenmolekel die Formel Se, als Se, zuzuerteilen.
Die Loslichkeit des Selens in Wasser. Urn sicher zu sein, daB beim Auswaschen des weiter unten
durch Reduktion gewonnenen Selens init heibem Wasser kein Ma- terial verloren geht, wurde das kristallinische Selen auf seine Los- lichkeit in Wasser untersucht. Dies geschah in der Weise, daI3 in einem Platin-Neubauertiegel, in dem sich etwas pulverformiges Selen befand, mehrere Male bestimmte Mengen heiljen Wassers hindurch- gegeben wurden. Ein sorgfkltig gereinigter Neubauertiegel wog nach dem Trocknen im Anilinbade bei ungefahr 170° 27.48060 g; nachdem 300 ccm heiDes Wasser langsam hindurchgegeben und der Tiegel unter denselben Bedingungen wieder getrocknet iiiid gewogen worden war, liatte sich sein Gewicht nicht im geringsten verandert, es war wiederum 27.480GO g. Nun wurde etwas pulverformiges Selen hineingegeben, das durch Krhitzen des roten amorphen Selens gewonnen wurclen war. Nachdem 600 ccni heiBes Wasser hindurch- gegeben worden waren, wog der Neubauertiegel mit den1 Seleii
* Zeiiachr. phys. Cheni. 51 (1912), 129. ’ v. WARTENI~E~ZO, Juhrb. f. ~ 1 e X t r o c A e ~ n . 14 (1907), 226. a Vgl. die 13einerkungen von JUL. MEIER zu den a~inlogen Dampfdruck-
bestimmungen im Julirb. f. I?kktroe/ienz. 14 (1907), 90.

Eine neue Beestinamuny des Atorngez&hles des Selens. 69
28.04071 g. Es wurden dann viermal je 300 ccm siedend hei8es Wasser langsam hindurchfiltriert, wie oben jedesmal getrocknet und gewaschen. Das Gewicht des Tiegels mit dem Selen anderte sich nicht im geringsten. Demnach ist das Selen in Wasser als voll- kommen unloslich zu betrachten.
Die Bestimmung des Atomgewichtes des Selena. Das Ziel der vorliegenden Arbeit war nun, unter Beachtung
der im vorliergehenden Teile gemachten Beobachtungen durch quan- titative Uberfuhrung reinen Selens in absolut wasserfreies Selen- dioxyd das Verhaltnis der Gewichtsmenge dieser beiden Stoffe zu bestimmen, und zur Kontrolle das erhaltene Dioxyd wiederum zu reinem Selen zu reduzieren. Hierbei bot zunachst namentlich der erste Teil, die quantitative Durchfiihrung der Oxydation des Selens zutn Selendioxyd, groBe Schwierigkeiten. Die Reindarstellung des Selens und das Trocknen einer gowogenen Menge unter Wahrung der Gewichtskonstanz war leicht auszufiihren, da der Dampfdruck des Selens bei der fur das Trocknen in Betracht kommenden Tem- peratur keine Rolle spielt. Dagegen erwies es sich als vollkommen unmoglich, das Selendioxyd, das zunachst nach der alten Methode durch Oxydation des Selens niit Salpetersaure und Dehydratation der so erhaltenen selenigen Saure durch mehrfaches Urnsublimieren gewonnen worden war, ohne Verlust vollstandig vom Wasser zu befreien. Bei Zimmertemperatur ist die Tension der selenigen Saure verschwindend klein und das Selendioxyd erscheint demnach sogar hygroskopisch. Bei hoheren Ternperaturen kann zwar, wie die Tensionskurven lehren , eine Wasserabspaltung aus der selenigen Saure bewirkt werden. Sol1 die Uberfuhrung der selenigen Saure in das Dioxyd aber in mehbarer Zeit erfolgen, so muB die Tem- peratur so hoch gesteigert werden, da8 sich auch das Selendioxyd zu verfluchtigen beginnt. Und eine genugende Gewichtskonstanz durch mehrmaliges Trocknen erreichen zu wollen, erwies sich a19 vollstandig vergeblich. Es erwies sich daher als notwendig, die bei dem Trocknen sich verfluchtigenden Selendioxydmengen aufzufangen und genau zu bestimmen. Es handelte sich meistens urn Mengen von 0.3-0.4 mg, die in einer Vorlage, die mit konzentrierter Schwefel- saure gefullt war, aufgefangen wurden. Da eine gravimetrische Be- stimmung dieser kleinen Mengen nicht zweckmaBig war, wurden andere Methoden ausgearbeitet, und es erwiesen sich schlieBlich zwei kolorimetrische Methoden als gut geeignet, um geringe und

70 J. . J m w k ?bud .I. L\fr!jer.
geringste Yengen von Selendioxyd oder von seleniger Siiure in Wasser uiid in konzentriertor Schwefelsiiure nachweisen zu ltonnen.
Indessen wurdc diese erstc BIethode schliel3lich doch aufgegebeii, da die technischen Srhwieriglieiten zu groB waien und da vor allem kein einwandfreies Kriterium gefnnden werden konnte, mit desben Hilfe erkannt werden konnte, dd.3 tlas getrocknete Selendioxyd wirk- lich rein und wasserfrei war. Weiin aber erst dieser Zweifel be- rechtigt war, so wurde auch die zweite Ergiinzungs- und Kontroll- Lestiminurig - n&mlich die Reduktion des Dioxyds zu 8elen - hinfiillig, selbst danu, wenu unter Nitberechnung des geschiitzteii Seleiitlioxyds die ursprunglich in Anwendung gebrachte Selenmenge resultierte. Nachdem Oxydationsrersuche mi l reinem, troclienen Sauerstofjt' urid niit Ozon nicht recht befriedigend ausgefallen waren, wurde sclilieBlich ein geradezu ideales Oxydationsrnittel im Stick- stoffdioxyd aufgefunden? niit dem unter Anwendung geeigneter Ap- partztureii ohne besondere Schwierigkeiten einwandfreie trockerie Oxydationen unter volligem AusschluB von Wasser durchgefuhrt werden ltonnten. Die Reduktion des so erhaltenen absolut wasser- freien Selendioxyds bot nun keine besoncleren Schwierigkeiten niehr.
Obwohl die erste Methode der Oxydation des Selens zu Selen- diosyd niit Salpetersaure nicht zu dem gewunschten Ziele fiihrte, sollen hier docli einige Punkte daraus dargelegt werden, weil bei ihrer Ausarbeitung eine Anzahl von bemorkenswerten Beobachtungen gemacht worden sind, die uns unter anderem auch bei dem end- gultigeii Verfahren I on groBem Nutzen geweseu sind.
Reindarstellung der Materialien. Selen. Eine griil3ere Menge liauflichen gefgllten Selens (zum Teil vori
C. A. b'. K a h l b a u m , zum Teil von Giu l in i ) wurde in eiiier Por- zellanschale auf dem U'asserbade mit reiiier konzentrierter Salpeter- skure osydiert, die Losung der elitstandenen seleiiigen S%ure auf dem Sandbade zur Trockne eingedampft, mit Wasser aufgenomnien uiid filtricrt, du in wieder zur Trockne eingedampft, worauf der feste farblose Ruclistaiid aus einem groBeri Porzellantiegel in ein daruber gestiilptes Hecherglas subliiniert wurde. I>as :tus losen, farhlosen Kri- stallen bestehende Selendioxyd, nicht aber die an der Glaswandurig sitzencle glasige Masse, wurde in Wasser gclost und iiach Reclarf noch einmal filtriert. Die so erhaltene selenige Saure wurde bei 7vl'asser\)'3.Ilteniperatur zu Selen reduziert, wobei darauf zu achten war, tlaB dasselbe in fein verteilter Forni, nicht in Klnnipen oder

Eke izeue Ilestimwauny des Atompwichtes des Selem. 7 1
Krusten, ausgefallt wurde. Als Reduktionsmittel dienten 1. schwef- lige Saure, 2. Hydrazinsulfat, 3. Hydroxylaminchlorhydrat. Das so erhaltene Selen wurde filtriert , mehrere Male mit heiBem Wasser dekantiert und so lange gewaschen, his bei 1. und 2. keine Sulfat- reaktion, bei 3. keine Chloridreaktion inehr eintrat. Der erhaltene Seleiistaub wurde dann in einer Porzellanschale auf dem Wasser- bade getrocknet. Von jedem dieser drei Praparate wurde dann die Hiilfte nochmals mit Salpetersaure oxydiert, eingedampft, sublimiert und mit deinselben Reduktionsmittel wie das erstemal reduziert. Auf diese Weise wurcien noch drei neue Selenpraparate erhalten, die moglicherweise einen noch hoheren Reinheitsgrad besagen. Ton einer Destillation des Selens im Vakuum, die zur Reinigung haufig empfohlen und ausgeubt wird, wurde abgesehen, da das Umsub- limieren des Dioxyds doch dieselbe, wenn nicht eine bessere Gewahr fur die Reinheit gibt. Das so erhaltene Selen ist ein graublaues bis schwarzes , kristallinisches, lockeres Pulver, das sich bei der Oxydation durch Salpetersaure klar lost und beim Eindampfen dann einen reinen weiBen Riickstand liefert.
Priifung der Salpetersaure auf Reinheit. Salpetersaure gehort zu den vielgebrauchten Reagenzien, deren
Reinheitsgrad im allgemeinen erheblich uberschatzt wird. Beim Eindampfen von 10 ccm reiner konzeritrierter Salpetersaure in einem Platintiegel im Nickelluftbade bei etwa 200 O wurde ein Riickstand von 0.00064 g festgestellt, der sich nach dem Gliihen des Tiegels auf 0.00013 verminderte. Diese Salpetersgure wurde daher aus einer Glasretorte unter AusschluB von Kork und Gummi direkt in einen langhalsigen Glaskolben iiberdestilliert, der durch flieBendes Wasser gekiihlt wurde. 10 ccm dieser Saure gaben beim Ein- dampfen wie oben einen Ruckstand von 0.00068 g, nach Clem Gluhen von 0.000 17 g. Salpetersaure, die aus PlatingefaBen destilliert war, zeigte bemerkenswerterweise einen erheblich groBeren Ruckstand, namlich 0.00228 g. In einer neueren Arbeit von B A X T E R ~ uber das Atomgewicht des Phosphoi s wird Salpetersaure dadurch gereinigt, daB sie durch ein Platinrohr destilliert wird. Indessen wird nicht weiter angegeben, ob die so gewonnene Salpetershure auch wirklich riickstandfrei ist. Nach unseren Erfahrungen miissen wir das be- zweifeln, da Salpetersaure bei Benutzung von PlatingefaBen in geringer Weise verunreinigt wird, indem das Platin wahrscheinlich etwas oxydiert wird und sich dann wohl in Spuren auflost.
~
%. nnorg. Chem. SO (1913), 186.

7 2 .I. ,Jannek wad J. Meyel-.
Es wurde weiterhin Saipeters%ure aus einem Quarzglaskolben destilliert. 10 ccm dieser Saure gaben 0.00026 g Ruckstand. Da es ausgeschlossen zu sein schien, daI3 Quarzglas von Salpetersaure in merklichen Mengen aufgelost wird, so lag der SchluB nahe, tiaB das Platin selbst von der Salpetersaure, die iibrigens keine Spur Salzsgure enthielt, nngegriffen wurde. Darauf deutet j a auch der bei cler aus Platin destillierten Salpetersgure erhaltene Ruck- stand hin. I n der Tat war beim Eindampfen von 10 ccm der aus Quarz destillierten Salpetersaure in einem Quarztiegel nicht der geringste Ruckstand nachzuweisen. Beim Eiiidampfen von 10 ccm in HartglasgefaBen betrug der Ruckstand 0.05 nig, was wohl auf einen geringen Angriff des Hartglases zuruckzufiihren ist.
Da die Destillation am QuarzgefaBen sich in so vorzuglicher Weise zur Reinigung der Salpetersaure geeignet gezeigt hatte, wurde auch die Sa lz sau re , die allerdings in nur geringer Menge gebraucht wurde, und das Wasse r auf diese Weise gereinigt. Es ergab sich, daI3 10 ccrn dieser Substanzen beim Eindampfen im Quarztiegel keinen Ruckstand hinterliellen.
Reinigung des Hydrazinhydrats. Das Hydrazinhydrat war als etwa 90°/,ige Losung bezogen
worden und stellte eine Elare, wasserhelle E’lussigkeit dar. 5 ccm einer etwa 50°/,igen Losung wurden bei 160 O im Platintiegel ein- gedampft und hinterlieI3en einen Ruckstand von 0.00031 g. Weitere 5 ccm, in demselben Tiegel eingedampft, erhohteii das Gewicht des Ruckstandes auf 0.00062 g. Nach dem Gliihen des Tiegels wurden nur noch 0.00031 g Ruckstand gewogen. Urn das Hydrazinhydrat, das sich zur Reduktion der seleiiigeii Skure vie1 besser eignet als das schwer losliche Sulfat, zu reinigen, wurde es aus einem Platinkolben destilliert. Das wasserklare Destillat, das eine Nenge kleiner Stick- stoifblaschen enthielt, die von einer geringen Zersetzung des Hydra- zins herriihren und sich bald an den Wanden absetzten, wurde in einem gr6Beren Silbertiegel aufbewahrt , der zur Vermeidung der Kohlrndioxydabsorption in einem Natronkalkexsikkator stand. Dieses Hydrazinhydrat war vollkommen riickstandfrei. Sowohl Platintiegel als auch HartglasgefaBe zeigten nach dem Abdampfen einiger Kubik- zentimeter nicht die geringste Gewichtszunahme.
Oxydationsversuche mit Salpetersaure. Bei einer jeden Atomgewichtsbestimmung ist es von groBem
Vortsil, wenn man Operationen wie Filtrieren, UmgieBen usw. ver-

Eine neue Beslitnmuiiy des Atonzgewichtes dps Selens. 73
meiden und die vorzunehmenden Operationen von der ersten bis zur letzten Wlgung in ein und demselben GefaBen ausfuhren kann. Um die Uberfuhrung des Selens in das Dioxyd mittels Salpetersiiure in einer einzigen Apparatur durchfuhren zu konnen, wurde ein flaschen- formiges GefaB aus Hartglas angewendet, das unterhalb des Halses noch eine kugelfijrmige Erweiterung besaB. In dem Hahe war mittels Schliffes ein rechtwinkliges Glasrohr angesetzt, das mit einem Dreiweghahn in Verbindung stand. Auf diese Weise war es mog- lich, das HartglasgefaB abwechselnd zu evakuieren und mit trockener Luft zu fiillen. Die eingefiihrte Luft wurde durch Calciumchlorid und konzentrierte Schwefelsaure getrocknet. Ton der Verwendung des Phosphorpentoxyds wurde zuerst Abstand genommen, da es sich nicht als genugend rein erwies und gelegentlich beim Feuchtwerden ein reduzierendes Gas entwickelte, welches mit der zu trocknenden Luft mitgefuhrt wurde und eine geringe Reduktion des Selendioxyds bewirkte, die durch schwache Rotfarbung kenntlich wurde.
In das HartglasgefaB wurde nun eine gewisse Menge Selen gebracht und langsam durch maiBig verdiinnte Salpetersaure gelost. Die Abgase passierten hinter dem Dreiweghahn eine mit Wasser gefullte Schlange, urn jede Spur entweichenden Selens aufzufangen. Wenn die Salpetersaure verdampft war, wurde eine Schwefelsgure- schlange eingeschaltet und das HartglasgefaB abwechselnd evakuiert und mit trockener Luft gefiillt. Auf diese Weise sollte eine voll- standige Dehydratation der selenigen Saure bewirkt und jede Spur entweichenden Selens in der konzentrierten Schwefelsaure aufgefangen werden. Dieses Ziel hat sich auf diese Weise nicht erreichen lassen, weshalb diese Methode auch bald verlassen wurde. Das Bestreben aber, die in den Vorlagen enthaltenen geringen Mengen Selendioxyds und seleniger Saure quantitativ zu bestimmen, hat zu zwei empfind- lichen Methoden zum quantitativen Nachweis geringer Selendioxyd- mengen auf kolorimetrischem Wege gefiihrt.
Uber den qnantitativen Nachweis geringer Mengen seleniger Saure in Wasser und von Selendioxyd in konzentrierter Schwefelsaure
mittels Natriumhydrosulfit.
Nach Beobachtungen van JUL. MEYER und spiiter von 0. BBUNCK~ wird durch Natriumhydrosulfit selbst aus sehr verdunnten Losungen
-~ ~
2. alzorg. Chem. 34 (1903), 51. Lid . Ann. 336 (1904), 281.

74 J. Jcmnel, urd cJ. &'e!je?-.
von seleniger Saure kolloidales Selen ausgeschieden, das infolge seiner iiitensiven Fc'iirbung noch in sehr geringen Mengen erkannt werden kann. Ks ist alter zu beachten, daW die freie hydroschweflige Saure i m Gegensatz zu ihren Salzen nicht farblos ist, sondern ihren LO- sungen eine gelbe bis orangegelbe Farbe verleiht, die leicht zu Ver- wechselungen mit der Farbe des kolloidalen Selens in geringen Mengen fiihren kann. Zur Empfindlichlieitspriifung wurden reine wasserige Liisungeri von seleniger SBure mit 0.1, 0.02, 0.01, 0.003 und 0.002 Proz. SeO, angewendet. J e 1 ccm dieser Losungen wurde nlit 0.1 g Na,S,O, versetzt und geschiittelt. Die freie hydroschweflige Saure, die teils durch Einwirkurig der freien selenigen Siure ent- steht, teilweise aber durch Ninwirkung des Sauerstoffw der Luft :
NazS,O, + 0, + H,O = NaHSO, + NaHSO, uncl
wird durch Zusatz von etwas fester Soda neutralisiert und entfarbt. I n den ersten drei Losungen bildete sich eine deutliche rote Losung von kolloidalem Selen, die also 0.001, 0.0002 und 0.0001 g SeO, entspracli. Die vierte Losung mit 0.00005 g SeO, zeigte noch eine schwach gel bliche bis orangegelbe F%rbung, w%hrend bei der letzten, vertlhntesten Losung eine solche kaum noch zu bemerken war. Die Empfindlichkeitsgrenze liegt unter diesen Urnstanden also bei 0.005 O l i o selcniger Skure. Starkere Mineralsauren storen die Reaktion dadurch, daB sie aus dem Hydrosulfit weiBen Schwefel abscheiden. Zwar erhalt man bei Gegenwart von seleniger Siiure stets eine gelbliche bis rotlichgelbe Mischung, doch ist es unbedingt vorteil- hafter, die Hauptmenge der freien Mineralsauren vorher durch Soda abzustumpfen. Sucht man das Selen in salpetersaurer Losung nach- zuweisen, so darf man die Saure nicht durch Abdampfen bis zur Trockne vertreiben, weil die geringen Mengen seleniger Saure da- bei verloren gehen. Ebenso ist beim Einengen verdiinnter Losungen von seleniger SBure auf dem Wasserbade die Bildung von Rand- krusten zu vermeiden. Denn wahrerid sich, wie einige Versuche zeigten, aus der Losung selbst kein SeO, oder H,SeO, verfluchtigt, ist der Dampfdruck des trockenen Dioxyds urid der trockenen selenigen Saure bei Wasserbtidtemperatur bereits SO groB, daB all- niahlich rnerkliche Verluste eintreten kijnnen.
?rTa,,S,O, 2 . . + XaHSO, + NaHSO, = H,S,O, + Na,SO, + Na,SO,,
A4usfuhrlichcr siehe Jnr.. MEYER 11. Jos. JANNICK, Zeilsckr-. ccnul?/t. Clmn.
JUL. MEYER, Z. aizorg. C/LrnL. 34 (1903), 52. 62 (1913), 534.

Eine m i e L'eslirri.mziii!j des dtotugeln;ichlcs des Selem. 75
Die Beobachtung, daB der Schwefel, welcher durch starke Sauren a u s dem Hydrosulfit ausgeschieden wird, in Gegenwart von seleniger Saure gelb bis rotlich-gelb gef&rbt erscheint, machte es wahrschein- lich, daB auf diese Weise auch in konzentrierter Schwefelsiiure ge- ringe Mengen im Selendioxyd nachzuweisen waren. Es ist ratsam, mit den1 Zusatz von Na,S,O, sparsam zu sein, damit die gelbe Farbe nicht durch allzugroBe Massen weiBen Schwefels verdeclrt wird. Es genugt meist weniger als 0.1 g Na,S,O, auf 1 ccm H,SO,. Die Intensitiit der Gelbfarbung ist der vorhandenen Selendioxyd- menge entsprechend abgestuft. Zur Priifung der Empfindlichkeit dieser Reaktion wurden Losungen von SeO, in konzentrierter Schwefelshre mit 0.1, 0.02, 0.01, 0.005 und 0.002 SeO, an- gewendet. In den ersten vier Losungen war das Dioxyd auf Grund der Gelbfarbung des durch Hydrosulfit ausgeschiedenen Schwefels scharf nachzuweisen. 0.002 SeO, wird man bei gutem Tages- lichte und beim Vergleich mit einer Probe reiner Schwefelsaure mit einiger Sicherheit nachweisen konnen. Der Versuch, das Selendioxyd durch Wegdampfen der Schwefelsaure anzureichern, fiihrt nicht Z U ~
Ziel, da das Schwefeldioxyd, dessen Sublimationspunkt nach unseren Tensionsmessungen in der Nahe des Siedepunktes der SchwefelsLure liegt, ebenfalls entweicht.
Uber die kolorimetrische Bestimmung sehr geringer Mengen seleniger Same mit Jodkalium.
Die quantitative Schatzung des Selendiosyds in salpetersaurer Losung sowie in konzentrierter Schwefelsaure nach der Hydrosulfit.8 methode besitzt einige Nachteile, durch welche ihre Empfindlichkeit und Genauigkeit hgufig recht erheblich herabgedriickt werden kann. Die reichliche Schwefelabscheidung einerseits und andererseits der Umstand, da8 sich das rote kolloidal geloste Selen in stark saurer Losung und auch bei Gegenwart groBerer Mengen von Neutralsalzen, wie sie durch die Neutralisation mit Soda entstehen, leicht zu- sammenballt, machen diese an sich sonst sehr empfindliche Methode haufig xecht undeutlich und zu einer genauen kolorimetrischen Messung wenig geeignet. SchlieBlich war uns die Empfindlichkeit fur unsere Zwecke nicht weitgehend genug. A11e diese Miingel treten in der zweiten Methode nicht auf. Sie beruht in der Reduktions- wirkung ron Jodkalium in saurer Losung auf selenige Saure:
SeG, + 4 H J = Se + SH,O + 4 J .

J. ,Januel; und ,J. N!?JcT.
Das ausgeschiedene Jod farbt bei Anwesenheit uberschiissigen Kaliumjodids die Losung noch in den geringsten Mengen deutlich gello, und nian kann selbst bei Losungen mit 0.0005 SeO, mit bloBem duge noch eine sehr schone Gelbfiirbung beobachten. Bei Anwendung eines KRussschen Kolorirneters mit LUMMER-BRODHUN- schem Prisma lLBt sich die Erkennung der Gelbfarbung noch vie1 weiter treiben und quantitativ gestalten. SchlieBlich kann man die Empfindlichkeit des Nachweises durch Starkezusatz noch um min- destens das Zehnfache steigern, so daB man qualitativ rnit dem Auge noch ungefahr 0.0000005 g SeO, in 1 ccm Losung nachweisen kann. Bei der Ausfiihrung der Versuchel ist z u beachten, daB sich Jod- wssserstoff unter dem EinfluD des Lichtes und des Luftsauerstoffes allmkhlich unter Jodabscheidung zersetzt. Ferner ist die Anwen- dung konzeritrierter Salpeter- und Schwefelsaure zu vermeiden. Zur Vermeidung der Ausfallung des kolloidal gelijsten Selens ist es empfehlenswert, der Losung von vornherein ein Schutzkolloid, etwas Gummiarabikum, zuzusetzen. Die fur das Kolorimeter notwendigen Vergleichsliisungen miissen frisch sein und unter denselben Ver- suchsbedingungen wie die zu priifenden Losungen angesetzt werden.
Auch in konzentrierter Schwefelsaure ist die kolorimetrische Be- stirnmung des darin gelosten Selendioxyds nach dieser Methode moglich, wenn man die Saure auf das Zwanzigfache verdunnt. Aller- dings leidet die Empfindlichkeit des Nachweises durch die Ver- dunnung. Verdunnt man weniger stark und stumpft man den grogten Teil der uberschussigen Saure durch Soda ab, so kann man immer- hin noch 0.000 25 SeO, in konzentrierter Schwefelsaure nach- weisen, d. h. 0.0000025 g Se0, im Kubikzentimeter Saure.
Uber die Bildung von Selensaure bei der Oxydation des Selens durch Salpetersaure und uber ihre Entfernung.
Wie schon lange bekannt ist, geht die Oxydation des Selens durch Salpetersaure zu einem sehr geringen Teile weiter bis zur Bildung von Selensaure. Uber das Verhalten der Selensgure beim Erhitzen liegen nur einige, sich teilweise widersprechende Angaben vor. Bei einem Versuche, bei dem Selen in einem HartglasgefaBe mit reinster Salpetersaure oxydiert worden war, blieb beim s tkkeren Erhitzen, nachdem die selenige Saure dehydratisiert und als Se0,
Die Darlegung der genauen Versuchsbedingungen, die Herr VON GARN ausgearbeitet hat, wird in der Zeitschr. analyt. Chem. erfolgen.

Eim neue Bcstiminum~ des Atoimpwichtes des Seleizs. 77
hochsublimiert war, eine diinne gelbe Kruste am Boden, die selbst beim Erhitzen auf 400° nicht verschwand. Um einen Einblick in das Verhalten der SelensBure heim Erhitzen zn erhalten, wurde etwas reine Selensaure ( K a h l b a u m ) in Wasser gelost und in ein SublimiergeWB aus Hartglas gebracht, das im LuftSade erhitzt wurde. Zuerst ging das Wasser weg, das abgesaugt und in einer mit Wasser beschickten Vorlage gesammelt wurde. Bei hoherer Temperatur (250-300 O) biideten sich hellgrune Uampfe, die sich im oberen Teile des Apparates zu einem weil3en Pulver verdichteten. Wahrenddem war am Boden noch etwas kochende Plussigkeit. Diese erstarrte allmahlich und nun sublimierte bei noch hoherer Tempe- ratur (500-600°) der grijBte Teil nach den oberen Partien des Apparates. Ein kleiner , gelblich aussehender Rest wollte selbst beim Erhitzen mit freier Flamme nicht vom Boden weichen. In der Wasservorlage lieB sich nur etwas selenige Saure nachweisen. Bariumhydrat gab in salpetersaurer Losung keine Fallung von Bariumselenat. Auch das sublimierte weiBe Pulver erwies sich als reines Selendioxyd. Der gelbliche Ruckstand, der als dunne Kruste am Boden festsa6, war in Wasser unliislich. Aber auch in konzen- trierter Salz- und Salpetersaure und selbst in Konigswasser Iijste er sich nur zum kleinen Teile auf, ohne sich jedoch zu zersetzen. Seine Natur lronnte bisher nicht festgestellt werden. Wahrschein- lich liegt ein Oxyd des Selens vor, wie sich aus der Unloslichkeit in konzentrierter Saure und vor allem daraus ergibt, daf3 dieser Ruckstand in konzentriertem Hydrazinhydrat sich mit roter Farbe auflost. Auf Zusatz von Saure fillt aus dieser Lijsung dann wieder rotes Selen aus.
Eine zweite Probe derselben Selensaure wurde nach den An- gaben von MITSCHERLICH und NITZSCH rnit etwas konzentrierter Salzsaure eingedampft und der Ruckstand wie oben erhitzt. Die ganze Masse sublimierte als farbloses Selendioxyd hinauf. Die Bariumselenatprobe fie1 negativ aus. Auch die anderen Halogen- wasserstoffsiiuren sind imstande, Selensaure zu reduzieren, und beim Jodwasserstoff geht die Reduktion sogar his zum elementaren Selen. Bromwasserstoff wirkt nicht vie1 starker als Chlorwasserstoff und fiihrt zur selenigen Saure.
Aus diesen Versuchen geht hervor, daB man, urn selensaure- freies Dioxyd zu erhalten, gleich bei der Oxydation durch Salpeter-
~
Pogg. A m . 9 (18'27), 630.

'is J. Janwrk und J . illeyer.
siiure otler wenigstens vor dem Sublimicren etwas Salzsgure hinzu- fiigen muW. Auch in anderer Beziehung erwies sich der Chlorwasser- stoffzusatz als vorteilhaft. Penn wiihrend man bei der Oxydation mit HSO, nieist ein etwas gelb bis rotlich gefirbtes Dioxyd erhklt, entsteht bei Gegenwart eiiiiger Tropfen Salzsiiure stets ein rein weiBer Riickstand, der sich nuch beiin Sublimieren nicht im gering- sten verfirbt. Der einzige, bei Atomgewichtsbestimmungen aller- dings selir bedeiililiche Nuchteil dieses Zusatzes konrite die Bildung von Spurell Chlorselen untl voii Selenoxychloriden sein, die leichtor als das OXJ d eiitmeichen und sich in der Wasservorlage hydrolytisch zersetzeii wurden.
Loslichkeit des Selens in Hydrazinhydrat und in Alkalien.
Bei der Reduktion des Selendioxyds durch Hydrazinhydrat wurde die iiberrsschende Beobnchtung gemacht, da8 sich die Losung nach vollstandigsr Reduktion der selanigen SBure zu Selen bei iiber- schiissigem Hydrazinhydrat intensiv rot f i rb t und bei Gegenwart von viel Selen und von lronzentriertem Hydrazinhydrat dickflussig wurde. Beim Eindampfen dieser Selenlosung zur Trockne ver- schwanil die rote E'arbe vollstkndig und es blieb nur schwarzes Selen zuriick. Die Auflijsnng des §elens in dem konzentrierten Hydrazinhydrat ist auf die Bildung von Hydrazinsalzen des Selen- wasserstoft'es urid der selenigen Saure zuruckzufuhren:
GH,N.NH,OH + 3Se = 2(H2N.NH,),Se + (H,N.NH,),SeO, + 3H,O.
An das Hydrazoniumselenid lagert sich d a m wahrscheinlich noch nielir Selen an unter Bildung von Polyseleniden. Der Vor- gang stcht der Auflosung von Selen, Schwefel und Halogenen in den Allialien in jeder Beziehung eng zur Seite, ist aber bemerkens- wert dadurch, daB die Reaktion beim Verdiinnen von rechts nach liiiks verliiuft, und zwar scheidet sich das Selen in kolloidaler Form aus. Damit ist aber ein selir einfaches und elegantes Verfahren zur Dustellung kolloidaler Selenlosungen gegeben. Die entstehenden Losungen sind intensiv gefdrbt und auBerordentlich haltbar.
Dieselben Vorgange findet man auch beim Schwefel, nicht aber beim Tellur wieder. Auch Scliwefel lost sich mit gro8er Leichtig- keit in liochprozentigem Hydrazinhydrat reichlicb zu einer dunkel- orangegelben, dickfiussigen Masse auf, die deutlich riach Schwefel- wasserstoff riecht und beim Eintropfen in sehr viel U'asser eine gelb- gefarhte Losung liefert, aus der sich allmkhlich Schwefel aussclieidet.

I h e v m e Bcstiitzmsng tips -Itomgewiehtr.s [lev Selcus. 79
Da die Losung von Schwefel in Hydrazinhydrnt deutlich n:ich Schwefelmasserstoff riecht, so war es nicht ausgeschlossen, datl auch geringe Mengen Selen bei der Reduktion des L)icJxyds durch Hydra- zinhydrat bis zum Selenwasserstoff reduziert wurtleiz. Zur I h t - scheidung dieser Frage wurde folgender Versucli angestellt. Auf einen Rundkolben war ein Kugelkuhler nufgesetzt , dessen oberes Elide durch Schlauch mit zwei Schwefels~ureschlangeu verbunden war, die eine Losung von Rleiacetat enthielten. Durch einen ‘I’ropf- trichter wurde allmahlich 10 Ofoige Hydrazinhydratlosung zu der im Kolben befindlichen, kochenden Losung von seleniger S h r e gegeben. Zugleich wurde durcfi ein drittes Rohr ein Luftstrom geleitet, der mit Pernimganat und Wasser gewaschen war uiid den etwa ent- standenen Selenwasserstoff nach dem Filtrieren durch Bauinwolle durch die Rleiacetatschlangen fuhrte. Selbst bei lange wahrenden Versucheri zeigte sich niemals die geringste Dunkelf&rbung in den Acetatlosungen, wie pie durch Rildung von noch so wenig Bleiselenid eingetreten ware. Dagegen fie1 ein geringer, rein weiBer Nieder- schlag aus, der sich als Bleiselenit erwies. Es ist zweifellos, daB dime selenige Stlure infolge des energischen Kochens und der stdr- mischen Stickstoffentwickelung bei der Eedulition arif mechauischem Wege als Wssserstaub mitgerissen wurde.
Das Ergebnis, daB sich bei der Reduktion der selenigen Saure durch wenig Hydrazinhydrat kein Selenwasserstoff bildet, steht mit dem Verhalten des Selenwasserstoffes im Einklang. Nach BODEX- STEIN verlauft die Reaktion zwischen Selen und Wasserstoff selbst bei proWer Oberflache des Selens sehr langsam. Bei 324O werden innerhttlb 24 Stunden 5 0 1 0 , innerhalb 48 Stunden 1 7 Oi lo H,Se ge- bildet. Zur Erreichung des Gleichgewichtes sind nach BODENSTEIN und nach PJ~LABON viele T’age notwendig. Da man pro l o n Tempe- raturerniedrigung die Reaktionsgeschwindigkeit auf die Hlilfte her- absetzen liann, so wiirde man fur die Bildungsgeschwindigkeit des Selenwasserstoffes bei 100 O einen auBerordentlich geringen Wert er- warten miissen, der noch urn so geringer ausfallen diirfte, als bei unseren Versucheri der zur Reduktion erforderliche Wassers toff nich t einmal in freier Form, sondern in gebundenem Zustande vorlag. Dadurch werden unsere Versuche auch theoretisch gestiitzt. so daB man einen Selenverlust durch Bildung von Selenwasserstoff bei der Reduktion der selenigen Saure durch HydrazinhJTtlrat nicht zu be- fiirchten hat.
Zeitschi-. phys. Chem. 89 (1899), 429.

80 J. Junnel; und J. Dfe.yrr.
Eine Reihe von Versuchen uberzeugten uns von der Uiim6glich- keit, durch Oxydation mit Salpetersiiure zu einem absolut wasser- freien Uioxyd zu gelangen. Dementsprechend schwanken die Atom- gewichtswerte, die sich am der Restimmung des Verhaltnisses Se : SeO, nach dieser nicht vollig einwandfreien Methode ergaben, innerhalb ziemlich weiter Grenzen; denn es wurclen die Zahlen 79.18, 79.23 und 79.20 erhalten. Bei der Reduktion des durch Salpeter- saureoxydation erhaltenen Dioxyds mittels Hyclrazinhydrat zu Selen ergaberi sich die Zahlen 79.11 und 79.24. Ks muBte demnach, da die technischen Schwierigkeiten riicht iiberwunden werden konnten, eine neue Methode z u r Oxydation des Selens bei vollstandigem Aus- schluB von Feuchtigkeit aufgesucht werden.
Versuche znr Oxydation des Selens auf trockenem Wege rnit Saner- stoff, Ozon und mit Stickstofftetroxyd.
Selen im Sauerstoffstrom quantitativ in Dioxyd iiberzufiihren, erwies Rich als nur sehr schwierig moglich. Denn im Gegensatz zum Schwefel verbrelint Selen nicht ohne weiteres in der Luft und auch nicht im Sauerstoffstrom, wenn es nicht genugend erhitzt wird. Hierbei werden immer unverbrannte Selendampfe mitgerissen, die das entstehende Dioxyd stark verfiirben. Pdverformiges Selen mittels Sauerstoffes zu oxydieren, erwies sich niclit als miiglich. Bei geniigen- dem Erhitzen schmilzt das Selen und oxydiert sich, wobei gelegent- lich eine blauliche Flamme auftritt. Aber dann beginnt auch schon die Verdarnpfung des Selens. Wir ko~inen demnach die Beobach- tungen, die M. F. S A C C ~ eicst bei dem Versuch, Selen direkt mit Sauerstoff zu oxydieren, geniaclit hat , vollauf bestiitigen. Das so erhaltene selenhaltige Dioxyd lieB sich auch durch lnehrmaliges Um- sublimieren im trockenen Sauerstoffstrom nicht nbsolut weiB erhalten.
Bedeutend erfolgreicher waren die Osydationsversuche mit Ozon. Schon bei schwach oznnhaltigeni Sauerstoff, wie ihri eine einfache Ozonrijhre lieferte, war bei gewohnlicher Temperatur eine deutlich energische Oxydation an der Bildung eines weiBeri Beschlages zu erkeiinen. Der Versuch wurde deshalb mit einem ozonreicheren Sauerstoff wiederholt. Eine Batterie von 5 Ozonrdiren nach dem System STOLZENBERG, die an ein Induktorium angeschlossen waren, das init 70 Volt und 8 Amp. betrieberi wurde, lieferte einen Sauer-
' Eingehenderes dnruber siche Jos. JANLEE, Dissert , Breslau 1913 ' dm. c h h . phys. 131 21 (184T), 121.

Ene i i e m Bestimmung des Alonzgewichtes des Selens. 81
stoffstrom mit ungefahr loo/, 0,. Der einer Bombe entnommene Sauerstoff wurde durch lronzentrierte Schwefelsaure getrocknet und nach dem Ozonieren auf den Boden eines Reagenzglases geleitet, in dem sich etwas pulverfiirmiges Selen befand. Schon bei Zimmer- temperatur war eine deutliche Oxydation des Selens wahrzunehmen, indem die oberen Schichten grauweiB wurden. Dann wurde die Temperatur langsam gesteigert, wobei schlieblich das gesamte Selen oxydiert wurde und als Dioxyd nach oben sublimierte. Durch zwei- bis dreimaliges Umsublimieren im Ozonstrome wurde das anfangs etwas schmutzige und rotliche Produkt schlieblich rein wei6.
Obwohl nach diesen Versuchen die Anwendung von Ozon zur trockenen Oxydation des Selens, trotz ihrer Kostspieligkeit , nicht aussichtslos erschien, so wurde sie doch nicht weiter ausgearbeitet, weil wir ein noch viel energischeres und dabei in seiner Anwendung fast als ideal zu bezeichnendes Oxydationsmittel im Stickstofftetroxyd auffanden.
Schon M. F. S A C C ~ hatte Selen durch Stickoxyde (oxyde nitreux) zu oxydieren versucht, allerdings ohne Erfolg, denn er schreibt, daf3 das Selen in diesem Gase ebenso wie in Kohlensaure sublimiert werden konne, weil dieses Metalloid angeblich zum Sauer- stoff weniger Affinitat besa6e als der Stickstoff.
Die Einwirkung des Stickstofftetroxyds auf Selen beginnt schon bei Zimmertemperatur, wird aber mit steigender Temperatur erheb- lich intensiver. Daher ist es vorteilhaft, in der Hitze zu arbeiten, die aber nicht iiber den Schmelzpunkt des Selens hinaus gesteigert werden darf. Denn das geschmolzene Selen hat gegenuber dem pulverformigen eine sehr geringe Oberflache, so daB die Oxydation dadurch wieder langsamer erfolgt. Bei der Oxydation unter diesen Versuchsbedingungen wird das Stickstofftetroxyd zu Stickoxyd redu- ziert, das sich aber durch Zufuhr von Sauerstoff immer wieder in das Di- bzw. Tetroxyd verwandeln IaiBt. Man ist so imstande, mit wenig N,O, und viel 0, beliebig gro6e Mengen Selen in das Dioxyd iiberzufuhren. Das Stickstofftetroxyd dient im Grunde also nur, ebenso wie beim BleikammerprozeB, als Sauerstoffiibertrager.
Diese Methode der Selenoxydation hat gegenuber der nassen mit Salpetersaure mehrere sehr groBe Vorziige. Erstens erhalt man auf diese Weise wirklich wasserfreies Selendioxyd, da sich Stickstoff- tetroxyd und Sauerstoff durch Phosphorpentoxyd gut und scharf
' Ann. C ~ ~ W L . p h p . [3] 21 (1847), 121. Z. anorg. Chem. Bd. 83. 6

82 ,I. Jannel; uud J. illeyer.
trocknen lsssen uiid da bei dcr Reaktion kein Wasser entsteht. Zweitens erzielt man beim Unisublimieren in der SauerstoE-Stick- stofftetroxydatrnosphiire stets ein absolut reinos, blendend weiBes, aus kleinen Kristallchen bestehendes, hervorragend schones Produkt. Dritteiis 1st die Bilclung von Seleiisiiure infolge der Abwesenheit von Feuclitigkeit ausgesclilossen , und schlietllich sind hier keine Verluste ail Selendioxyd zu befurchten, wie sie beim Osydieren des Selens rnit Salpetersiiure schon wiihrend des ]{indampfens der Lo- sung zur Trockne eiiitreten, gan:! besonders aber bei den Versuchen, das Selendiosyd durch Urnsublimieren absolut wasserfrei zu erhalten. Man hat deninach hier eine quantitative, viillig einwandfreie und selir saubere Methode der Oxydation des Selens zu Selendioxyd, die auch zur priiparativen Darstellung griitlerer Uengen reinen, absolut w'isserfreien Selendioxyds sehr geeignet ist.
Nachdem sich gezeigt hatte, dafl sich das Selen mittels N,O, leicht und quarititativ in SeO, verwandeln M t , wurde diese Methode soweit ausgearbeitet, daB sie den an eine Atomgewichtshestimmung zu stellenden E'orderungen in jeder Weise geniigt. An Materialien waren nur reines Selen notwendig, von dem die bei der ersteri Methode beschriebenen sechs Praparate noch vorhanden waren, ferner reines Stickstofftetroxyd und reiner Sauerstoff.
Reinigung des Stickstofftetroxyds.
E s stand uns neben einem Priiparate, das durch Rehandeln yon Arsentrioxyd mit Salpetersaure gewonnen war, noch eine griiBere Menge fabrikmiiBig gewonnenen Stickstofftetroxycls aus den Hochster Farbwerken zur Verfugung, das aber beim Umfullen mit Gummi in Beruhrung gekommen war und dalier organische Verunreinigungen enthielt. Diese organischen Produkte scheiden sich beim EingieBen des Tetroxyds in Wasser in Form fettiger Tropfchen ab. Das Tetroxyd wurde von diesen Verunreinigungen dadurch mit Erfolg befreit, daB es im schwachen Snuerstoff'strom uber gelinde gluhende Kupferoxydspane geleitet und d a m in zwei W aschflaschen auf- gefangen wurde, die in einer Kiiltemischung standen. Die einzelnen Teile : Destillierkolbeii mit GlasstGpsel , Verbrennungrrohr und Waschflasche waren zu einem Stiicke zusammerigeschmolzen worden. I n das schmutziggelbe Destillat wurde solange Sauerstoff' eingeleitet, bis die Firbung wieder in Gelbbraun iibergegangen war. Die Auf- liisuiig eiiies Tropfens dieses Sticlrstofftetroxyds erwies sich nun

als frei von organischen Substanzen, zeigte aber deutliche Chlor- reaktion. Deshalb wurde das Praparat mit Silbernitrat durch- geschiittelt und davon abdestilliert. Das Chlor war auf cliese Weise, wie mehrere Kontrollreaktionen zeigten, aus dem Tetroxyd voll- standig entfernt worden. Zuletzt wurde das Destillat mit Phosphor- pentoxyd geschuttelt und dariiber aufbewahrt. Auf diese Weise war es gelungen, ein absoIut wasser- und chlorfreies Proclukt zu erhalten, wie die zu verschiedenen Zeiten wieder vorgenommenen Priifungen zeigten.
Priifung des Sauerstoffs auf Reinheit.
Es war dann notwendig, daB auch der zur Verwendung ge- langende Sauerstoff genugentl rein, vor allem wasserstoff frei war, eine Forderung, die z. B. beim Elektrolytsauerstoff nur schwer zu erfullen ist. Der von uns benutzte Bombensauerstoff enthielt nach wiederholt angestellten Gasanalysen cat. 3 - - 4 O / , Stickstoff, aber keine gasanalytisch nachweisbaren Mengen Wasserstoff. Wenn geringe Wasserstoffmengen doch vorhanden gewesen sein sollten, so durften diese Spuren doch keine Wasserbildung verursachen und keine E’ehlerquelle sein. Denn da unser Sauerstoff erst durch konzen- trierte Schwefelsaure und dann durch P,05 absolut trocken gemacht worden waren, so diirfte, wie die BaEERschen Versuche beweisen,’ selbst bei den von uns angewendeten Temperaturen bis 400O eine Wasserbildung nicht eingetreten sein. AuBerdem ist nach den BoDENsTEINschen Versuchen die Geschwindigkeit der Wasserbildung unter normalen Verhaltnissen selbst bei 5000 noch aufierordentlich gering,3 so dab die Entstehung eines Versuchsfehlers wohl ausge- schlossen ist, selbst wenn der verwendete Sauerstoff minimale. gas- analytisch nicht mehr nachweisbare Mengen Wasserstoff enthalten haben sollte.
Die Apparatnr.
Die Konstruktion des zur quantitativen trnckenen Oxydation des Selens bestimmten Apparate ist ohne weitere Beschreibung aus Fig. 3 ersichtlich. Der Sauerstoff, der einem Gasometer entnommen wurde und durch eine Waschflasche mit ofters erneuerter konzen-
Journ. Cheni. SOC. 1894, 603. Zeitschr. phys. Chem. 29 (1S99), 665. Vgl. z. B. JUL. MEYER, Chern. Reaktionsgeschwindigkeit, S. 36 .
6*

84 J. ,Jannek uncl ,J. Dhyer.
trierter Schwefelsaure perlte, wurde durch den seitlichen Ansatz uber der Stickstofftetroxydkugel in den Apparat geleitet, wobei er stets etwas Stickstofftetroxyd mit sich fuhrt, das ja schon bei Zimmertemperatur einen genugend hohen Dampfdruck zeigt oder, wenn notig, durch einen Mikrobrenner auf geeignet hohe Temperatur gebracht wurde. Das Sauerstoff-Stickstofftetroxydgasgemisch wird in einem P,O,-Rohr nochmals von allen Spuren Feuchtigkeit befreit und tritt dann durch einen Staubfanger in den eigentlichen Oxy- dationsapparat, um ihn nach dem Passieren von zwei ahnlichen Staubfangern durch ein P,O,-Rohr zu verlassen, Die beiden letzten Staubfanger haben den Zweck, das durch einen zu heftigen Gasstrom aufgewirbelte Selen und Dioxyd festzuhalten, wahrend der erste Staubfanger das bei etwizigem Abkuhlen der Stickstofftetroxydkugel und beim plotzlichen Versagen des Sauerstoffstromes zuriickgesaugte Selendioxyd usw. auffangt. Bei vorsichtigem Arbeiten und bei gut regulierter Gaszufuhr werden diese Ansiitze kaum in Anspruch genommen. Gelegentlich ist im mittleren Staubfanger ein ganz schwacher Beschlag zu sehen. Das zweite P,O,-Rohr verhindert das Kindringen von Luftfeuchtigkeit in den Apparat.
Bei dem ersten, nach dieser Skizze angefertigten Apparat war das HauptgefaB mit den Staubfangern aus Hartglas das iibrige System aus gewiihnlichem Glase hergestellt. Da sich aber das Ge- wicht des mit Glasstopseln verschlieBbaren Apparates unter dem EinfluB des Stickstofftetroxyds und infolge des haufigen Ausdiimpfens und Waschens fortwahrend anderte, muBte er als fur den vorliegen- den Zweck unbrauchbar angesehen werden. Bei zweistundigem Er- hitzen des leeren Apparates mit N,O,-Dampfen nahm sein Gewicht um 0.00050 g ZLI, nach dem nun folgenden Auswaschen mit reinem Wasser um 0.00076 g ab. Bei Versuchen, bei denen Selen oxydiert wurde, war nach dem Auswaschen haufig eine noch vie1 bedeutendere Gewichtsabnahrne zu konstatieren, gelegentlich aber war sie auch fast konstant geblieben. DaB die Gewichtszu- und -abnahmen jehsmal verschieden groB und deshalb ganz unkontrollierbar waren, beweisen auch die unter ganz gleichen Bedingungen angestellten Oxydationen dcs Selens, die zu Atomgewichtswerten zwischen 79.046 und 79.733 fiihrten. Bei sechs sonst einwandfrei durchgefiihrten Restimmungen waren nicht zwei auch nur einigermaBen iibereinstimmende Werte erhalten worden. DaB die Methode aber an sich einwandfrei sein musse, zeigte ein Versuch, hei dem das Selendioxyd mit Hilfe des spater zu beschreibenden Reduktionsapparates wieder zu Selen

Eiiae ueue Besti~nnwr~g des Atoiw!yewichtes des Seleris. 85
reduziert wurde und wobei das Gewicht der ursprunglich angewendeten Selenmenge genau zuruckerhalten wurde.
0.45879 g Selen lieferten 0.64405 g Selendioxyyd (miter der Annshme, daB das Gewicht des Apparates konstant geblieben war); die nach der Reduktion rnit Kydrazinhydrat erhaltene Selenmenge wog 0.45879 g. Das Atomgewicht wiirde sich daraus zu 79.250 un- korrigiert besechnen.
Fig. 3.
Da sich Glas als unbrauchbar erwiesen hatte, wurde zu den endgiiltigen Bestimmungen ein Apparat aus durchsichtigem, englischem Quarzglas verwendet ; denn Qusrzglas ist in Wasser vollstandig un- liislich und\wird auch durch Sauren und Stiuredtimpfe nicht ange- griffen. Die Teile des Apparates, die nicht zur Wtigung gebracht wurden, also die P,O,-Rohren und die N,O,-Kugel, waren BUS ge- wohnlichem Glase. Zum VerschlieBen des Apparates wahrend des Wtigens clienten zwei sorgfaltig eiiigeschliffene Qunrzglasstopfen. Das Arbeiten mit dem recht zerbrechlichen Apparate erforderte ein ungewohnliches MaB von Vorsicht und Ubung ; doch bot dieser Apparat sndererseits den groBen Vorteil, daI3 die Bruchstellen ver- haltnismafiig leicht repariert werden konnten und daB jede ver- dachtige, unsaubere Stelle, selbst an den Schliffen, mit der Stich- flamme ohne Miihe beseitigt werden konnte.
Prufung des Quarzapparates auf Gewichtskonstanz. Bei der .Behandlung des Quarzapparates mit Stickstofftetroxyd-
dampfen und darauf mit Wasser zeigte sich ein iiberraschend regel-

86
maBiges Verhalten in den Xnderungen des Gewichtes. Nachdem der Apparat mit Sauren und Wasser sorgfiltig gereinigt und rnittels liingerem Durchleiten trockener Luft im HeiDlnftbade getrocknet worden war, wurde er zur Gewiclitskonstanz gebracht und gewogen. Dann wurde er wieder im HeiBluftbade erhitzt, wahrend 1-2 Stunden lang trockene N,O,-Diimpfe hindurchgeleitet wurden. Nachdem die Stickoxyddiimpfe durch trockene Luft ersetzt waren, wurde gewogen, mit reinstem, aus Quarz destilliertem Wassgr ausgewaschen, getrocknet und wietler gewogen. Diese Operation wurde zu ganz verschiedenen Zeiten, narlidem der Apparat schon Iingere Zeit in Benutzung ge- weseii war, wieclerholt, im ganzen fiinfmal. Es zeigte sich hierbei, daB die Behandlung mit Tetroxyddiimpfcn eine regelmiBige Gewichts- zunelinie hervorrief, die imnier diclit bei 0.00011 g lag. iUach dern Auswaschen hingegen wurde eine ebenso regelmaaige Gewichtsver- minderung von 0.00006 g gegeniiber dem nrspriinglichen Gewichte festgestellt. Die Gewichtszunahme ist hochstwahrscheinlich auf die Rildung eines auBerst diinnen Hautchens von kondensiertem N,O, auf der Quarzoberflache zuruckzufiihren , da sich die auffallende Konstanz dieser Nrscheinung kaum anders erkliren laBt und mit Beobachtungen, die man an Kohlendioxyd und Glasoberflachen ge- macht hat, im Einklang steht. Da die Oxydation des Selens spater uiiter denselbeii Versuchsbedingungen erfolgt, so miissen von dem Gewichte cles Selendioxyds stets 0.0001 1 g abgezogen werden. Es sei iiochmals hervorgehoben, daB die Gcwichtsanderungen ganz regel- mliBig waren und auch nach 1Bngerem Gebrauch des Apparates wiedererhalten wurden.
.1. pJuniieX; u i ~ d ,J. JIeyei*.
Beschreibung einer Oxydation. Der sorgfaltig gesauberte und zuletzt mit unserm reinsten, aus
QuarzgefaDen destillierten Wasser ausgewaschene, an Platindraht aufgehangte Quarzapparat wurde zunachst mit freier Flamme ge- trocknet. Dann wurden die beiden P,O,-Rohren angesetzt und ein trockener Luftstrom, der bereits zwei Schwefelsaurewaschflaschen passiert hatte, langsam hindurchgesaugt. Zwischen der Wasser- strahlpumpe und dem zweiten P,O,-Rohr befand sich ebenfalls eine H,SO,-Waschflasche. Iler Quarzapparat hing wahrend des Luft- durchleitens in einem Nickellnftbade, das oben durch eine passend durchlocherte Asbestscheibe abgeschlossen war und mit voller Flamme auf ca. 400O erhitzt wurde. Das Erhitzen und Durchleiten trockener Luft dauerte ungefihr eine Stunde. Nach dem Erlralten

Eine 9Eeue Bestiinrnung cles Atowtgeioieldes des Selens. 87
im Luftstrome wurde der Apparat mit den Quarzstopfen verschlossen. in die Wage gehangt und nach einer Stunde gewogen. Darauf wurde in derselben Weise nochmals eine Stunde lang erhitzt und wiederum gewogen. Falls noch keine geniigende Konstanz erreicht war (die fur unsere Versuche zulassige hochste Differenz betrug 0.03 mg) wurde das Trocknen wiederholt. Nachdem Gewichts- konstanz (meistens bereits bei der zweiten Wagung), erreicht war, wurde mit Hilfe eines trockenen Trichterrohres reines, pulverformjges Selen, das vorher langere Zeit bei ca. 150O getrocknet worden war, auf den Boden des Apparates gebracht, worauf der mit den P,O,- Rijhren versehene Apparat in ein Anilinluftbad van ca. 170O gehiingt und das Selen mittels Durchleiten trockener Luft wiihrend 1-2 Stunden von den letzten, etwa noch am Selen haftenden Spuren Feuchtigkeit befreit wurde. Nach dem Erkalten wurde zugestopselt und nach einstundigem Verweilen in der Wage gewogen. Das Trocknen und Wagen wurde bis zur erforderlichen Gewichtskonstanz wiederholt. Bei sorgfaltig vorgetrocknetem Selen wurde die ge- wiinschte Konstanz meistens schon bei der zweiten Wagung erreicht. Sodann wurde der mit Selen beschickte Apparat in ein Nickelluft- bad gehangt, dessen Temperatur bis dicht an den Schmelzpunkt des Selens, also bis ungefahr 215O gebracht wurde. Es wurden zwei P,O,-Rohren und die N,O,-Kugel angesetzt nnd ein sorgfiiltig regu- lierter Sauerstoffstrom mit ungefahr 100 Blaschen in der Minute durch den Apparat geleitet. Die hier benutzten Pentoxyd-Trocken- rohren diirfen beim Durchleiten trockener Luft nicht verwendet werden. Denn das Phosphorpentoxyd absorbiert N,O,, halt es hart- nackig fest und gibt es beim Durchleiten voii Luft nur lnngsam ab, so daB die daruber hinweggegangene Luft stets etwas stickoxydhaltig ist, wie wir zu unserin Nachteile erst spater bemerkten. Die Te- troxydkugel wird anfangs mit der Hand etwas erwarmt, um einen geniigend hohen Dampfdruck zu erzeugen, und dann sich selbst iiberlassen. Falls die Zimmertemperatur sehr tief steht, muB durch ein sehr kleines Plammchen fur fortdauerndes Erwarmen gesorgt werden. Der Apparat fiallt sich rasch mit roten Dampfen, die auf der andern Seite nur sehr langsam wieder entweichen diirfen. All- mahlich verwandelt sich die obere Schicht des Selens in ein grau- weiBes Pulver, wahrend in dem unteren Teile der Masse glitzernde Kristalle von SeO, auftreten. Wknn alles Selen oberflachlich oxy- aiert ist, wird die Temperatur allmahlich gesteigert, ohne daB aber Schmelzen eintreten darf. Dann verwandelt sich das gesamte Selen

88 J. Jmnek wid J. JIeyev.
in rein weiBes Dioxyd, das allmahlich ein Stiickchen nach oben hin- aufsublimiert. Waihrend des Sublimierens mu8 sehr sorgfditig darauf geachtet werden, daI3 der Sauerstoffstrom iiicht zu rasch geht. Eine Unterbrechung des Gasstromes wiederum mu8 vermieden werden, da sonst sehr leicht eine Verstopfung der Gaszufiihrungkapillare durch Selendioxyd eintritt. Nach beendeter Osydation - der Boden mu8 ganz sauber und blank sein - la8t man den Appsrat erkalten und saugt nach Entfernung der Stickstofftetroxydkugel einen langeren trockenen Luftstrom hindurch, um die Stickoxyde zu ent- fernen. Dann werden auch clie Stickoxyd-Trockenrohren gegen die Luft-Trockenriihren vertauscht, worauf nochmals trockene Luft hindurchgesaugt wird. Dttnn wird der Apparat mit dem schnee- wei8en Selendioxyd in die Wage gehangt und nach der iihlichen Zeit gewogen. Um etwa eingeschlossene Stickoxyde vollstandig zu entfernen, lost man das hinaufsublimierte Selenoxyd durch Er- hitzen des Apparates mit der Stichflamme von den Wandungen 10s und la8t es durch gelindes Klopfen auf den Boden herabfallen, worauf man es im HeiBiuftbad wiederum nach oben subiimiert und rnit trocbener Luft behandelt. I m allgemeinen haben wir bei der nun folgenden zweiten Wagung noch eine geringe Gewichtsabnahme beobachtel, die eben auf die Entfernung eingeschlossener Stickoxyde zuriickzufiihren ist. Nach der dritten Sublimation und Wiigung war aber fast immer Gewichtskonstanz erreicht.
DaB das mehrfach umsublimierte Selendioxyd frei von Stickstoff- tetroxyd ist, zeigte ein Versuch, bei dem eine wasserige Losung dieses Dioxyds mit Diphenylamin und konzentrierter Schwefelsiiure behandelt wurde, aber eine nur auljerst schwache Blaufarbung er- gab. Grevimetrich ware die aus dem Tetroxyd stammende Salpeter- und salpetrige Saure dernnach analytisch bei weitem nicht nachzu- weiseii gewesen. Auch durch den Geruch konnte in dem Selen- dioxyd kein Stickstofftetroxyd mehr nachgewiesen werden.
Versuche zur Redubtion von Selendioxyd mit Kilfe trockener Gase. Nachdem es uns gelungen war, Selen quantitativ in absolut
wasserfreies Selendioxyd zu verwandeln, versuchten wir, diese Ver- suche dadurch zu kontrollieren, daI3 wir das erhaltene Selendioxyd wiederum zu Seleu reduzierten. Ein Vergleich der angewendeten und am SchluB wiedererhaltenen gelenmenge hatte dann den besten Beweis dafiir geliefert, daB bei dem ganzen Versuche kein Verlust einge tre t en war.

Eine neue Bestimmung des Atoingewichtes des Selens. 89
Wir beabsichtigten zuerst, die Reduktion des Dioxyds in dem- selben Quarzapparate auszufiihren, in dem die Oxydation vor sich gegangen war, und wir wandten verschiedene Gase, allerdings ohne Erfolg, an.
1. Im trockenen Wasserstoffstrom wird trockenes Selendioxyd, selbst beim Erhitzen bis zum Sublimieren, nicht reduziert. Diese Beobachtung steht im Widerspruch mit Angaben von A. KLAGES,~ nach dessen Befund bei Einfiihrung trockenen Wasserstoffes in ein Reagenzglas mit etwas ,,seleniger Saure" unter lebhafter Licht- erscheinung Abscheidung von Selen stattfindet.
2. Trockenes Schwefeldioxydgas wirkt ebensowenig auf trockenes Selendioxyd ein, im Gegensatz zu dem Verhalten der w&sserigen Losungen, wie auch schon von anderer Seite bemerkt worden ist.,
3. Auck beim Kohlenmonoxyd konnte keine Reduktionswirkung festgestellt werden.
4. Ammoniakgas, das in wasseriger Lasung mit seleniger Siiure Ammoniumselenite bildet, erweist sich trockenem Selendioxyd gegen- iiber als starkes Reduktionsmittel. Bei gewohnlicher Temperatur ist keine Einwirkung zu beobachten. Erst bei l ooo tritt im lebhaften Ammoniakstrom eine sehr heftige Reaktion unter erheblicher Warme- entwickelung ein, durch die das Selen bis zum Schmelzen gebracht werden kann. Dabei sind gelegentlich kleine Flammchen zu bemerken. Nach MICHAELIS verlauft diese Reaktion nach der Gleichung:
3Se0, -I- 4NH3 = 3Se + 4 N + 6H,O. Fuhrt man die Reduktion vorsichtig in einem senkrecht stehen-
den Probierrohrchen aus, auf dessen Boden etwas trockenes SeO, liegt und in das ein Kapillarrohr taucht, durch welches ein gilt regulierter NH,-Strom eingefuhrt wird, so wird das SeO, bei ge- niigend hoher Temperatur, von etwa looo an, sehr ruhig reduziert. Da aber bei dieser Reaktion Wasser entsteht und das ubrige trockene Dioxyd anfeuchtet, so hort die Reaktion bald auf, bevor das ge- samte SeO, reduziert worden ist. Erst wenn man die Temperatur so hoch steigert, dab die gebildete selenige Siiure wieder zerfallt und das Wasser verdampft (siehe Tension der selenigen SBure H,Se03 --t H,O + SeO,), geht die Reduktion weiter. Die Verwendung des Ammoniaks zur Reduktion war daher nicht zu empfehlen, weil die Temperatur dabei so hocli getrieben werden mu6, da8 gegebenenfalls Selendioxyd
Chem. Ztg. 25, 449. LADENBURG, Handwiirterbuch der Chemie 10, 607.

90 J. Jnnnek zind cJ. illeyer.
verclampft. AuBerdem ist es noch nicht vollig sichergestellt, ob die Reaktion zwischeri NH, und SeO, nach der oben angegebenen Gleichung verlauft und ob sich nicht noch andere Produkte wie Selennitride usw. bilden.
Da die Reduktionsversuche mit trockenen Gasen nicht ausfuhr- bar erschienen, sollte das Dioxyd direkt im Quarzapparat mit Hydrazinhydrat reduziert werden. Indessen bedingt die lebhafte Stick- stoffentwickelung leicht Verluste an dem fein verteilten Selen. Daher wurde die Beduktion in einem anderen Apparate vorgenommen.
Reduktion des Selendioxyds durch Kydrazinhydrat. Das Selendioxyd wird im Quarzapparate in Wasser aufgelost
und cliirch das Trichterrohr A in den Reduktionskolben B (Fig. 4)
Fig. 4.
gespiilt. Die Konstruktion dieses Re- duktionsapparates, der mit zwei Glas- schliffen zum Auseinandernehmen und bequemen Reinigen versehen ist, be- zweckt,einVerstauben undverfliichtigen der selenigen SBure bei der Beduktion zu verhindern. Wenn das Selendioxyd durch das Trichterrohr A quantitativ in den Reduktionskolben gespiilt ist, lBBt man durch das Trichterrohr sehr langsam 10 O l 0 ige Hydrazinhydratlo- sung in die siedend heiBe Losung ein8ieBen. Die Geschwindigkeit des EinflieBens wird durch die Stellung des Schliffes geregelt. Die Gase, die sich beimKochen und bei derstickstoff-
entwickelung infolge der Einwirkung des Hydrazinhydrats auf die selenige Saure bilden, konnen durch das Trichterrohr nicht ent- weichen, sondern miissen ihren Weg durch das Kugelventil C nehmen, um clann die mit etwas Wasser beschickte Vorlage D zu passieren, die auBerdem auch von auBen durch Eintauchen in kaltes Wasser gekuhlt wird. Ein Ehtweichen von Wasserdampf aus dieser Vor- lage, clurch das moglicherweise Selenverluste verursacht werden konnten, kann so mit Leichtigkeit vermieden werden. Erkaltet der Apparat wahrend eines Versuches, so wird die in der Vorlage befindliche Fliissigkeit zuriickgesaugt und spult dabei gleichzeitig hochgespritztes Selen und Selendioxyd aus dem Kugelventil wieder

Eino neue Bestirnmzcng des Atowagewiclites des Selens. 91
nach unten. Zur Reduktion muB verdiinntes Hydrazinhydrat ver- wendet werden; denn die Anwendung eines hochkonzentrierten Praparates bewirkt die Abscheidung des Selens in Form eines dicken porosen Kuchens. Durch verdunnte Hydrazinhydratliisungen hingegen wird das Selen zuerst in der roten amorpheii Form ausgefallt, die sich d a m beim Sieden bald zu einem grauschwarzen pulverformigen Niederschlage zusammenballt. Tritt auf erneuten Zusatz von Hydrazin- hydrat zu der siedenden Losung keine Rotfarbung mehr auf, so ist die Reduktion beendigt. In einem groBeren Uberschusse von Hydrazinhydrat ist das ausgeschiedene Selen wieder loslich, und zwar, wie oben ausgefuhrt worden ist, unter Bildung von Polyseleniden mit dnnkelroter Farbe, die auch durch liingeres Kochen nicht zerstort wird. Durch Zusatz von wenig Salzsaure tritt aber augenblicklich Entf'arbung unter Selenabscheidung ein, und man erhalt eine vollig wasserklare Flussigkeit rnit abgeschiedenem, pulverformigem, grau- echwarzem Selen. Das abgeschiedene Selen wird dann in einen bis zur Konstanz gewogenen Neubauerplatintiegel hineingespult und mehrfach mit heiEem Wasser gewaschen. Die Filtrate diirfen mit Jodkalium- und Starkelosung keine Reaktion mehr geben, was mit Leichtigkeit zu erreichen ist. Der Neubauertiegel wurde dann in einem Anilinbad bei ungefahr 170° bis zur Konstanz getrocknet und gewogen.
Die Wagung.
Zur Ausfuhrung der Wagungen diente eine von Bunge-Ham- burg bezogene Wage, die schon von A. LAD EN BURG^ und von JUL. MEYER~ zu den Wggungen bei ihren Atomgewichtsbestimmungen gedient hatte. Mit Benutzung einer Fernrohrablesung konnen an dieser Wage Gewichtsdifferenzen von 0.00001 g bestimmt werden. Die Grarnmgewichte des benutzten Gewichtssatzes waren aus Messing und gut platiniert. Die Dezi- und Zentigrammstucke waren aus durchsichtigem Quarze. Die Gewichtsstiicke von 5 g abwarts wurden nach einer von TH. w. RICHARDS^ mitgeteilten und unserem Ge- wichtssatze (1, 2, 2, 5 ) angepaBten Methode kalibriert. Die zu wagenden Apparate waren durch eine Tara yon demselben Materiale bis zur vierten Dezimale austariert , der Quarzapparat durch ein Reagenzglas aus Quarz, das mit der erforderlichen Menge von - __ _-
BerZ. Ber. 35 (1902), 2278.
Zeitschr. phys. Chem. 33 (1900), 603. a 2. amorg. Chem. 36 (1903), 313; 4b (1905), 251; 46 (1905), 45.

92
Quarzglasstucken ausgefullt war. Auf beiden Seiten war auch das- selbe Gewicht von Platindraht, der zum Aufhangen der Apparate in der Wage diente, zur Verwendung gelangt. Auch der Neubauer- tiegel, der bei der Reduktion gebraucht wurde, war durch einen etwas leichteren und durch einige Platinstuckcbeu austariert worden. Auf diese Weise war die Menge der bei den Wagungen zur An- wendung gelangenden Gewichtsstucke niemals groB und Fehler in- folge der hde rungen der meteorologischen Verhaltnisse wurden ganzlich ausgeschaltet.
Zum Reduzieren der Gewichte iil Luft auf Vakuumwerte wurde
die bekannte Formel B = KJZ l + -2) verwendet, wo 111 das ge-
suchte Vakuumgewicht, rn das scheinbare, in Luft bestimmte, i. die Dichte der Luft (gleich 0.00120 im Mittel), s das spezifische Ge- wicht des gewogenen Korpers und d das spezifische Gewicht der Gewichtsstucke ist. Das spezifische Gewicht des grauen kristalli- sierten Selens betragt nach SAUNDERS 4.80, das des Selendioxyds ist nach CLAUPNIZER~ 3.95. Die Dichte des Quarzes ist 2.65, die des Messings wurde zu 8.40 angenommen.
Aus diesen Werten berechnet sich die Korrektion fur 1 g Selen, mit Messinggewichten gewogen, zu + 0.000107, mit Quarzgewichten gewogen, zu - 0.000294; fur das Selendioxyd sind die entsprechen- den Zahlen + 0.000161 und - 0.000242.
I m folgenden ist ein Versuch, in der SchluBtabelle als Nr. 4 bezeichnet, ausfuhrlich wiedergegeben. Der leere Quarzapparat besafi, nachdem er gegen eine genugende Menge von Quarzglas aua- tariert worden war, ein Ubergewicht, yon 0.00138 g. Die Nullpunkts- korrektion betrug + 0.00002 g, so daB das wahre Ubergewicht 0.00140 g war. Nachdem der leere Quarzapparat wiederum eine Stunde lang in1 Nickelluftbade unter Durchleiten trockener Luft ge- trocknet worden war, betrug sein Ubergewicht + 0.00148. Die Null- punktskorrektion war jetzt - 0.00004, so daB das wahre Ujher- gewicht + 0.00144 g, und das mittlere aus beiden Wagungen + 0.00142 g betrug. Der Apparat wurde nun mit vorgetrocknetem Selen beschickt, getrocknet und gewogen. Die Trocknung wurde bis zur Gewichtskonstanz wiederholt:
J. ,Jannek und ,T. i i eyer .
L d
J O Z L ~ Z . phys. Chern. 4, 423. CLAUSNIZER, Lieb. Ann. 106, 265.

Eine nezie Beslimmung des Alo~ngewichtes des Selens.
Nullpunktskorrektion . . . . . . . . . . . + 4
Nullpunktskorrektion . . . . . . . . . . . - 5
93
1. Ubergewicht + Selen . . . . . . . . . . . 3.29780 g
Korrigiertes Gewicht . . . . . . . . . . . 3.29784 2. Wagung. . , . . . . . . . . . . . . . 3.29780
Korrigiertes Gewicht . . . . . . . . . . . 3.29775 3. Wagung . . . . . . . . . . . . . . . . 3.29773
Nullpunktskorrektion . . . . . . . . . . . - 6 Korrigiertes Gewicht . . . . . . . . . . . 3.29767
4. Wagung . . . . . . . . . . . . . . . . 3.29774
Korrigiertes Gewicht . . * . . . . . . . . 3.29769 Mittel aus 3. und 4. Wagung . . . . . . . . 3.29768 Gewicht des Selens nach Abzug des Ubergewkhtes 3.29626
Gewicht des Selens in Luft . . . . . . . 3.29608 ? > ?, ,, im Vakuum . . . . . . . 3.29634
Nullpunktskorrektion . . . . . . . . . . . - 5
Korrektion des Gewichtssatzes . . . . . . . . - 18
Diese Selenmenge wurde nun in der oben beschriebenen Weise durch das Gemisch von Sauerstoff und Stickstofftetroxyd in Selen- dioxyd verwandelt, das mehrere Male umsublimiert und dann zur Wiigung gebracht wurde ;
Ubergewicht + Selendioxyd . . . . . . . . . Nullpunktskorrektion . . . , . . . . . . . Korrigiertes Gewicht . . . . . . . . . . . Nullpunktskorrektion . . . . . . . . . . . Korrigiertes Gewicht . . . . . . . . . . . Gewicht des Dioxyds nach Abzug des Ubergewjchtes Nach Abzug der Konstanten . . . ~ . . . .
2. WHgung . . . . . . . . . . . . . . .
Korrektion des Gewichtsatzes . . . . . . . . Gewicht des Selendioxyds in Luft . . . . .
9 7 7 9 97 imVakuum . . . . .
4.63078 - 8
4.63070 4.63070
- 3 4.63067 4.62925 - 1 1
4.62914 - 38
4.62876 4.62932
Setzen wir das gesuchte Atomgewicht des Selens gleich x, so verhiilt sich x : IC + 32.000 = 3.29608 : 4.62876, wenn das Atom- gewicht des Selens auf Luft bezogen wird. Fu r die Vakuumherech- nung sind die Zahlen 3.29634 und 4.62932 einzusetzen. Aus der
32.000 ' 3'29608 ergibt sich das Atomgewicht des Auswertung x = 1.33268
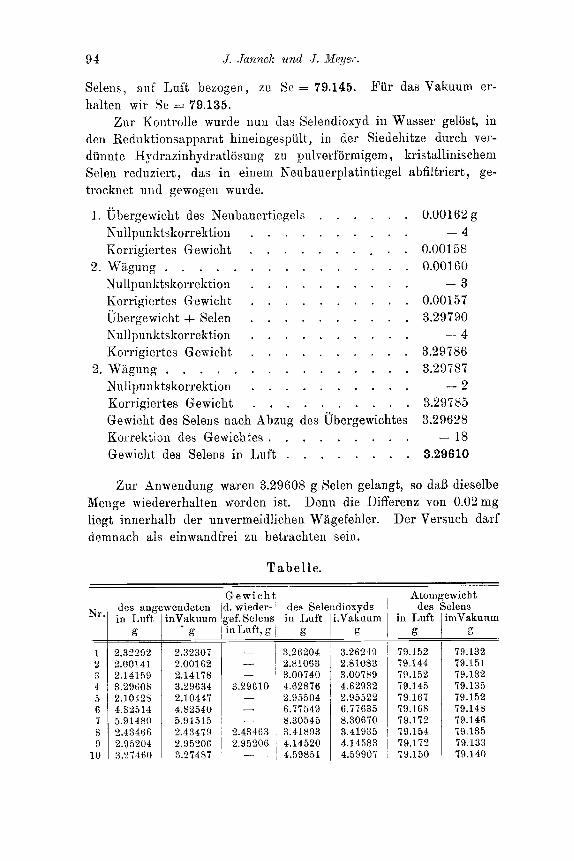
94 J. Junnek ztnd .T. M q e i . .
Selens, a d Luft bozogen, zu Se = 79.145. Fur das Vakuum er- halten wir Se = 79.135.
Zur Kontrolle wurde nun das Selendioxyd in Wasser gelost, in den Reduktionsapparat hiiieingespult, in der Siedehitze durch ver- dunnte Hyclrazinhydratliisunfi zii pulverformigem, kristallinischem Seleii reduziert , das in eineni Keubauerplatintiegel abfiltriert , ge- trocknet urid gewogen -iVuI.de.
1. Ubergewicht des Neuhaurrtiegels . . . . . . 0.00162g nTuilpnnlitskorrektiori . . . . . . . . . . - 4
2. W!igung . . . . . . . . . . . . . . . 0.00160 Sullpuiiktskorrelition . . . . . . . . . . - 3 Korrigiertes Gewicht . . . . . . . . . . 0.00157
Nullpunlrtskorrektion . . . . . . . . . . -- 4
2. Wiigung . . . . . . . . . . . . . . . 3.29787 Nnllpunktskorrektiori . . . . . . . . . . - 2
Korrigiertes Gewicht . . . . . . . . . . 0.00158
Uhergewicht + Selen . . . . . . . . . . 3.29790
Korrigiertes Gewicht . . . . . . . . . . 3.29786
Korrigiertes Gewicht . . . . . . . . . . 3.29785 Gewicht des Selens nach Bbzug des Ubergewichtes 3.29628 Korrektion des Gewichtes . . . . . . . . . - 18 Gewicht des Selens in Luft . . . . . . . . 3.29610
Nr'
1 3 3 4 5 6 7 5 9
10
Zur Anwendung waren 3.29608 g Selen gelangt, so daB dieselbe Denn die Differenz von 0.02 mg
Der Versuch darf Meiige wiedererhalten worden ist. liegt innerhalb der unvermeidlichen Wagefehler. demnach als einwandfrei zu betrachten sein.
in Luf t invdkuum 'gef. Sclens in Luf t 1 i.Vakuum g - g ~ inLuft , g 1 g I g
2.32292 2.32307 - ' 3.26204 3.36249 1 79.152 79.132 3.00141 2 00162 - j 2.81063 2.81053 79.144 79.151 2,14159 2.14178 - i 3.00740 3.00789 79.152 79.132 3.29608 3.29634 3.29610 1 4 62876 4.62932 79.145 79.135 2.10428 2.10447 - ~ 2.95504 2.95522 79167 79.152 4.82514 4.82540 I - 1 6.77549 6.77635 79.168 79.148 5.91480 5.!)1515 ~ - 8.30545 5.30670 I 79.172 79.146 2.43466 2.43479 2.43463 3.41893 3.41935 1 79.154 79.135 2.95204 2.95206 1 2.95206 4.14520 4.14583 ~ 79.172 79.133 3.27460 3.27487 I - 4.59851 4.59907 79.150 '79.140

Einine neuc Bestim?nimq des Atoniyeu~!ichtes des Selens. 95
I n vorstehender Tabelle sind die Ergebnisse von 10 Versuchen wiedergegeben. Die Reduktionsversuche, die ursprunglich als Kon- trolle bei jedem Oxydationsversuch ausgefuhrt werden sollten , ver- liefen nur in den drei Versuchen 4, 8 und 9 vollig einwandfrei. Bei den ubrigen Reduktionen traten geringe Verluste durch Verspritzen beim UmgieBen usw. ein.
Aus unseren Bestimmungen ergibt sich demnach fur das Atom- gewicht des Selens ein Wert, der erheblich geringer ist, als die meisten in den letzten Jahren gefundenen. Das arithmetische Mittel samtlicher Werte ist 70.158 und 79.140. Bewertet man die einzelnen Versuche nach ihren Gewichtsmengen, indem man die Summe des angewendeten Selens 31.26752 g, auf das Vakuuni bezogen 31.26955 g, und die Summe des erhaltenen Selendioxyds 43.90745 g, auf das Vakuum bezogen 43.91305 g, in Rechnung zieht, so erhalt man das Atomgewicht Se = 79.159, oder auf den leeren Raum bezogen 79.141.
Zusammenfassung. Nach einer kritisch-historischen Zusammenstellung der bisher
veriiffentlichten Untersuchungen iiber das Atomgewicht des Selens wurden zur Aufklarung des Verhaltens des Selendioxyds beim Sub- limieren die Dampfdruckkurven der selenigen Saure und des Selen- dioxyds bestimmt, woraus sich die groBe Schwierigkeit, wenn nicht Unmiiglichkeit der Herstellung absolut wasserfreien Selendioxyds durch Umsublimieren der selenigen Saure ergibt. Es wurde weiter- hin gefunden, daf3 das graue, kristallinische Selen in heif3em Wasser unloslich ist und in der Hohe seines Schmelzpunktes 217 einen verschwindend kleinen Dampfdruck besitzt.
Zur Darstellung absolut wasserfreien Selendioxyds wurde ein Verfahren ausgearbeitet, bei dem das pulverformige Selen in einem besonderen Apparate durch ein Gemisch von Sauerstoff und Stick- stofftetrosyd quantitativ in Selendioxyd iibergefuhrt wird.
Selensaure zerfallt bei genugend grof3er Hitze grBBtenteils, wenn geringe Mengen Salzsaure zugegen sind, aber vollstandig in Wasser, Sauerstoff und Selendioxyd.
Selenige Saure wird durch verdunntes Hydrazinhydrat in der Siedehitze zu Selen reduziert, durch konzentriertes Hydrazinhydrat wird aber die Bildung einer roten Losung bewirkt, die wahrschein- lich Hydrazoniumselenit und Hydrazoniumpolyselenide enthalt. Beirn starken Verdiinnen dieser Losung durch vie1 Wasser erhalt man

96 ,I. Jannek u. J. Uqer. Eine neue Bestimmuizg des Atomgewichtes usw.
eine sehr bestandige kolloidale Selenlosung. Schwefel verhalt sich ganz ahnlich, wghrend Tellur durch Hydrazinhydrat nicht gelost wird.
Die quantitative Uberfiihrung von Selen in absolut wasserfreies Selendioxyd durch Oxydation mittels Sauerstoffes und Stickstoff- tetroxyds in einem Quarzapparate ergab das Atomgewicht Se = 79.141, auf das Vakuum bezogen. Reduktionen des Selendioxyds durch Hydrazinhydrat zeigten, da6 bei diesem Verfahren keine Selenver- luste eintreten.
Brcsluzc, Chemisehes Inskilut der Unicersifiit.
Bei der Redaktion eingegangn am 14. Juli 1913.





























