Embed Size (px)
Citation preview

Eine vollst~indige Racemattrennung durch Elutions-Chromatographie an Cellulose-tri-acetat
G. Hesse* / R. Hagel [1]
Institut for Organische Chemie tier Universit~t Erlancjen-NiJrnberg, D-852 Erlangen, Henkestr. 42, Bundesrepublik Deutschland
Summary
o A Complete Separation of a Racemic Mixture by Elution Chromatography on Cellulose Triacetate
Racemic Tr&ger's Base is conveniently and completely separated into its enantiomers by l iquid chromatography on cellulose triacetate using either columns or TL C-plates Dissolving the cellulose for acetylation or purification has to be avoided, since the material loses its adsorptive powers by such treatment and the residual adsorptive properties may even show reversed stereoselectivity.
This leads to the conclusion that sorption is not achieved by Tr6ger's Base simply adhering to one glucose ester moiety, but by insertion between two such moieties which are at appropriate positions in the secondary structure of the natural polymer.
Zusammenfassu ng
Das Racemat der Tr6gerschen Base I~lYt sich durch Fliissig- Chromatographie in der Sdule oder auf DSnnschichtplatten an Cellulose-tri-acetat leicht und vollstgndig in die Enantio- meren trennen. Die Cellulose darf jedoch bei der Acetylie- rung nicht in L6sung gehen. Durch Umf~llen aus orga- nischen L 6sungsmitteln verliert das Tri-ace tat sein A dsorp- tionsverm6gen weitgehend und die Stereoselektivitgt der Restadsorption kann umgekehrt sein. Daraus wird geschlos- sen, dab die Sorption nicht durch Auflage auf eine Glucose- estereinheit erfolgt, sondern eher durch Einklemmen zwischen zwei solchen Einheiten, die in der Sekund#rstruk- tur eine geeignete Lage einnehmen.
Seit den ersten eindeutigen Erfolgen yon Henderson und Rule [2] ist das Ringen um eine pr~iparativ befriedigende Racemat-Trennung in der chromatographischen Trenns~iule nicht mehr zur Ruhe gekommen [3]. Als Sorptionsmittel haben sich Kohlehydrate am besten bew/ihrt, besonders Milchzucker [2, 4], St~irke [5-8] und Cellulose [9]. In einigen F~llen konnten optisch reine Anfangs- und End- fraktionen erhalten werden, aber die Ausbildung von zwei getrennten Peaks ftir die beiden Antipoden im Chromato- gramm wurde unseres Wissens bisher nicht erreicht.
* Meincrn lieben Freund und Studiengenossen Prof. Dr. Th. Wieland zu seinem 60. Geburtstag gewidmct
A. Liittringhaus ftihrte das zur Herstellung yon Acetat- seide gebr~iuchliche Cellulose-2.5-acetat als Sorbens ein [10] und machte die wichtige Feststellung [11, 12], dat~ es vor dem Einf011en in die Trenns~iule im Elutionsmittel (Xthanol oder Benzol) gequollen werden mug. Dieses Material, das in geeignet vorbehandelter Form yon der Firma M. Woelm (Eschwege) in den Handel gebracht wird, scheint eine besonders grol~e Anwendungsbreite und gute Stereoselektivit~it zu besitzen. Daher haben wir unsere Arbeiten damit begonnen. Als Substrat wurden verschie- dene leicht zug~ingliche Racemate verwendet, doch soil hier nur yon der Tr6gerschen Base [4] die Rede sein.
A. Antipodentrennung durch Siiulen-Chromatographie
1. Sorptionsmittel
Die Tr6gersche Base gibt an Cellulose-2.5-acetat (Woelm) aus Athanol nur einen Peak, dessen Front vorwiegend das
Enantiomere enthiilt. In der kleinsten zur Messung aus- reichenden Menge des Durchlaufs betrug die Anreiche- rung ca. 60 %. An einer Acetylcellulose mit 40 % Acetyl- gehalt (Macherey und Nagel, Dtiren), ebenfalls einem Cellulose-2.5-acetat, wurde unter entsprechenden Bedin- gungen das linksdrehende Enantiomere zu 80 % ange- reichert. Auch bier wurde nur ein Peak, jedoch mit deut- lichem tailing, erhalten. Bei weiteren Versuchen wurde allein durch 5 sttindiges Mahlen des gleichen Cellulose- acetats in einer KugelmiJhle eine Umkehrung der Selektivi- t~it beobachtet. Acetylcellulosen mit geringerem Acetylge- halt der gleichen Firma gaben iiberhaupt keine nachweis- bare Enantiomerentrennung.
Die wechselnden Ergebnisse an unvollstfindig veresterter Cellulose lassen sich damit erkl~iren, dat~ diese notwendig verschiedene Strukturelemente enth~ilt, also Glucose- einheiten mit 0 3 Estergruppen nebeneinander. Unsere Versuche haben gezeigt, daft sie nicht nur verschiedene, sondern zurn Teil sogar entgegengesetzte Spezifit~ten haben. Die einzige Abhilfe kann die vollst~indige Veresterung aller Hydroxylfunktionen sein.
Solche peracetylierten Cellulose-ester mit einem Acetyl- gehalt von 45 % sind bekannt und nach verschiedenen Methoden zug~nglich [ 13]. Die besten Erfahrungen haben wir mit der Acetylierung in Benzolsuspension nach dem Schering-Verfahren gemacht, bei der die Acetylcellulose nur quillt, abet in keinem Augenblick eine echte L6sung erfolgt. Wit nehmen an, dal~ bei dieser Acetylierung ,,in situ" auch das Netzwerk der nattirlichen Cellulosefaser
Chrornatographia, Vol. 6. No. 6, June 1973 Originals 277
mit ihren Sekundarstrukturen erhalten bleibt. Die Sorp- tion der chiralen Molekttle erfolgt zweifellos durch Wasser- stoffbindungen, wie auch Krebs [5, 7] und andere Auto- ren annehmen. Jedoch braucht es sich dabei nicht um die Auflage auf eine Cellulose-esterfunktion zu handeln, son- dern eher um das ,,Einklemmen" zwischen zwei solchen Einheiten in benachbarten Ketten oder Schichten. Damit wird auch die Forderung yon Ltittringhaus verst~indlich, dab das Sorbens mit dem L6sungsmittel gequollen sein mu6. Die gleichen Verh/iltnisse liegen bei den erfolgreich- sten Racemattrennungen an St~ke [6] und an Cellulose- pulver [9] vor.
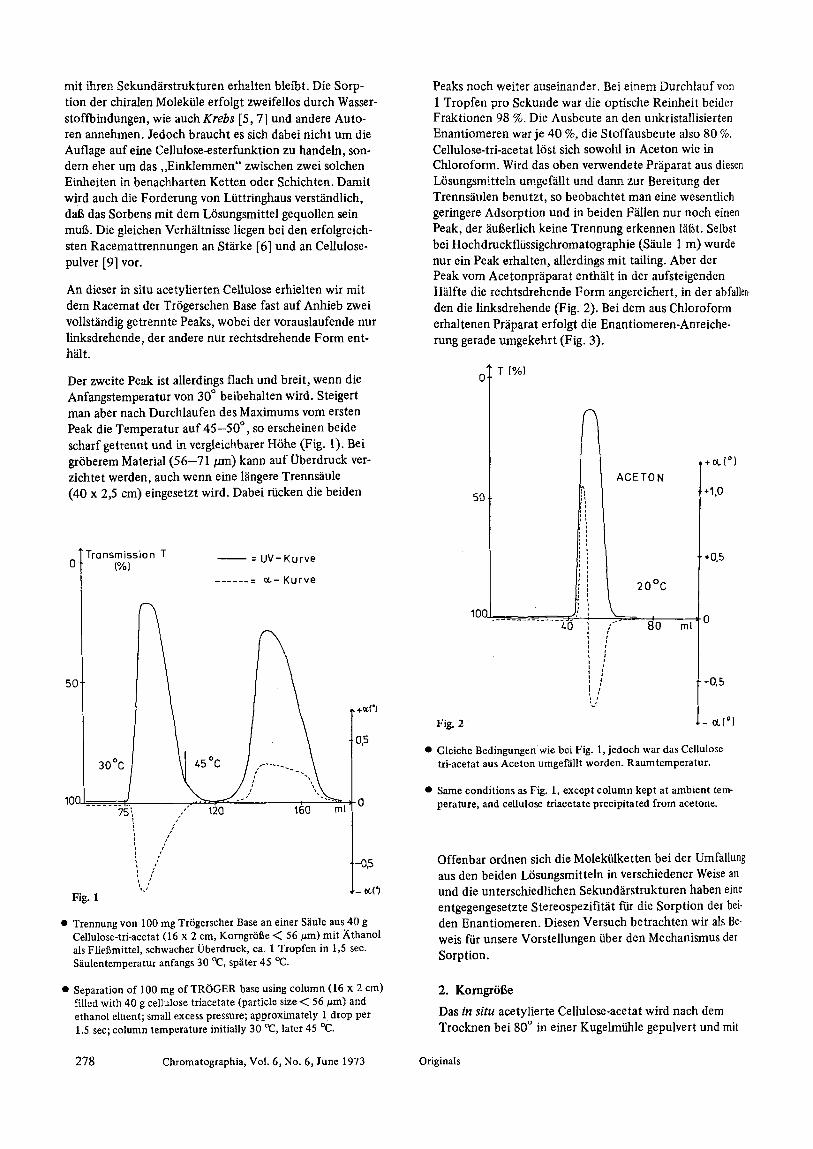
An dieser in situ acetylierten Cellulose erhielten wit mit dem Racemat der Tr6gerschen Base fast auf Anhieb zwei vollst~indig getrennte Peaks, wobei der vorauslaufende nur linksdrehende, der andere nur rechtsdrehende Form ent- h~lt.
Der zweite Peak ist allerdings flach und breit, wenn die Anfangstemperatur yon 30 ~ beibehalten wird. Steigert man aber nach Durchlaufen des Maximums vom ersten Peak die Temperatur auf 4 5 - 5 0 ~ so erscheinen beide scharf getrermt und in vergleichbarer H6he (Fig. 1). Bei gr6berem Material (56 -71 /am) kann auf Oberdruck ver- ziehtet werden, auch wenn eine l~ingere Trenns~iule (40 x 2,5 cm) eingesetzt wird. Dabei riicken die beiden
0
50
' T r a n s m i s s i o n T - - = U V - K u r v e (%1
. . . . . . = ~c- K u r v e
3 0 ~
100 . . . . . :. I 75",
,i
, , " 120 160 m[
/ /
/
r
/
, +~('1
0,5
-0,5
- ~(~ F i g . 1
�9 Trennung yon 100 mg Tr6gerscher Base an einer S~iule aus 40 g Cellulose-tri-acetat (16 x 2 cm, Korngr6t~e -< 56 #m) mit Xthanol als Fliegmittel, schwacher Oberdruck, ca. 1 Tropfen in 1,5 sec. S~iulentemperatur anfangs 30 ~ sp~iter 45 0(3.
�9 Separation of 100 mg of TROGER base using column (16 x 2 cm) filled with 40 g celtulose triacetate (particle size ~ 56 ~m) and ethanol eluent; small excess pressure; approximately 1. drop per 1.5 see; column temperature initially 30 ~ later 45 ~
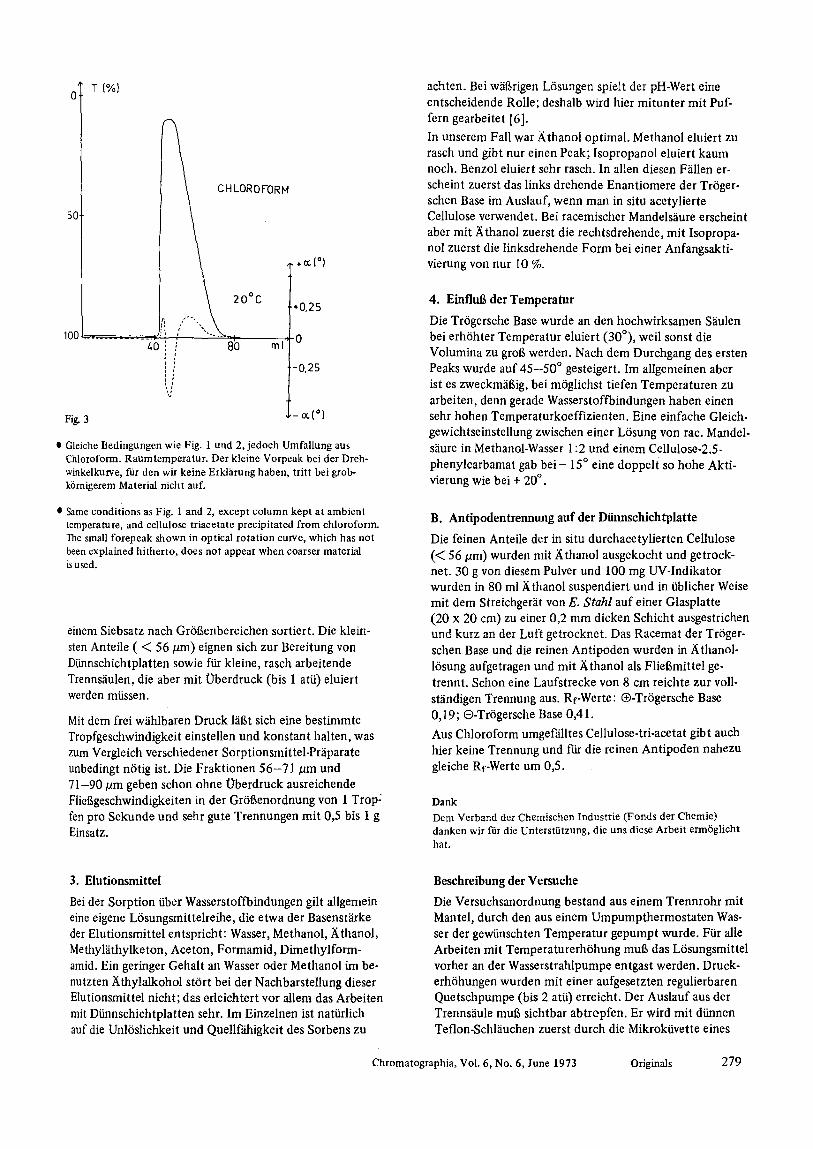
Peaks noch welter auseinander. Bei einem Durchlauf yon 1 Tropfen pro Sekunde war die optische Reinheit beider Fraktionen 98 %. Die Ausbeute an den umkristallisierten Enantiomeren war je 40 %, die Stoffausbeute also 80 %. Cellulose-tri-acetat 16st sich sowohl in Aceton wie in Chloroform. Wird das oben verwendete Pdiparat aus diesen L6sungsmitteln umgef~illt und dann zur Bereitung der Trenns~iulen benutzt, so beobachtet man eine wesentlich geringere Adsorption und in beiden F~llen nur noch einen Peak, der ~iu6erlich keine Trennung erkennen lfifit. Selbst bei HochdmckfliJssigchromatographie (S~iule 1 m) wurde nur ein Peak erhalten, allerdings mit tailing. Aber der Peak vom Acetonpr@arat enth~ilt in der aufsteigenden H~ilfte die rechtsdrehende Form angereichert, in der abfallen den die linksdrehende (Fig. 2). Beidem aus Chloroform erhaltenen Pr~iparat erfolgt die Enantiomeren-Anreiche- rung gerade umgekehrt (Fig. 3).
Fig. 2
Oi T (%)
5O
100 _=__= . . . . . . . . . = ~ 40
A C E T O N
2 0 ~
l I f
i ! !
J
ml
+~.(~
+I,0
*0.5
- - 0 , 5
_ r 1 7 6
�9 Gleiche Bedingungenwie bei Fig. 1, jedoch war das Cellulose- tri-acetat aus Aceton umgef'allt worden. Raumtemperatur.
�9 Same conditions as Fig. 1, except column kept at ambient tem- perature, and cellulose triacetate precipitated from acetone.
Offenbar ordnen sich die Molektilketten bei der Umfallung aus den beiden L6sungsmitteln in verschiedener Weise an und die unterschiedlichen Sekund~irstrukturen haben eine entgegengesetzte Stereospezifit~it ftir die Sorption der bei- den Enantiomeren. Diesen Versuch betrachten wir als Be- weis fiir unsere Vorstellungen fiber den Mechanismus der Sorption.
2. Korngr66e
Das in situ acetylierte Cellulose-acetat wird nach dem Trocknen bei 80 ~ in einer Kugelmiihle gepulvert und mit
278 Chromatographia, Vol. 6, No. 6, June 1973 Originals
O'
50
T [%}
100 ............. -.~r, 40
CHLOROFORM
.,.,~ 20.....~ OC f t p~
8b rnl
Fig. 3
+~(~
+0.25
0
-0,25
�9 Gleiche Bedingungen wie Fig. 1 und 2, jedoch Umf~iUung aus Chloroform. Raumtemperatur. Der kleine Vorpeak bei der Dreh- winkelkurve, far den wir keine Erkliimng haben, tritt bei grob- k6rnigerem Material nicht auf.
�9 Same conditions as Fig. 1 and 2, except column kept at ambient temperature, and cellulose triacetate precipitated from chloroform. The small forepeak shown in optical rotation curve, which has not been explained hitherto, does not appear when coarser material is used.
einem Siebsatz nach Gr66enbereichen sortiert. Die klein- sten Anteile ( < 56 gm) eignen sich zur Bereitung yon Dtinnschichtplatten sowie ftir kleine, rasch arbeitende Trenns/iulen, die aber mit Oberdruck (bis 1 atti) eluiert werden mtissen.
Mit dem frei w~hlbaren Druck lii6t sich eine bestimmte Tropfgeschwindigkeit einstellen und konstant halten, was zum Vergleich verschiedener Sorptionsmittel-Priiparate unbedingt n6tig ist. Die Fraktionen 56-71 /am und 71-90/am geben schon ohne Oberdruck ausreichende Flie6geschwindigkeiten in der Gr66enordnung yon 1 Trop- fen pro Sekunde und sehr gute Trennungen mit 0,5 bis 1 g Einsatz.
achten. Bei wafbrigen L6sungen spielt der pH-Wert eine entscheidende Rolle; deshalb wird hier mitunter mit Puf- fern gearbeitet [6].
In unserem Fall war Athanol optimal. Methanol eluiert zu rasch und gibt nur einen Peak; Isopropanol eluiert kaum noch. Benzol eluiert sehr rasch. In allen diesen F/illen er- scheint zuerst das links drehende Enantiomere der Tr6ger- schen Base im Auslauf, wenn man in situ acetylierte Cellulose verwendet. Bei racemischer Mandels/iure erscheint aber mit Athanol zuerst die rechtsdrehende, mit Isopropa- nol zuerst die linksdrehende Form bei einer Anfangsakti- vierung yon nur i0 %.
4. Einflug der Temperatur
Die Tr6gersche Base wurde an den hochwirksamen S~iulen bei erh6hter Temperatur eluiert (30~ weil sonst die Volumina zu grofi werden. Nach dem Durchgang des ersten Peaks wurde auf 45 -50 ~ gesteigert. Im allgemeinen aber ist es zweckm~gig, bei m6glichst tiefen Temperaturen zu arbeiten, denn gerade Wasserstoffbindungen haben einen sehr hohen Temperaturkoeffizienten. Eine einfache Gleich- gewichtseinstellung zwischen einer L6sung von rac. Mandel- siiure in Methanol-Wasser 1:2 und einem Cellulose-2.5- phenylcarbamat gab bei - 15 ~ eine doppelt so hohe Akti- vierung wie bei + 20 ~
B. Antipodentrennung auf der Diinnschichtplatte
Die feinen Anteile der in situ durchacetylierten Cellulose (< 56 tim) wurden mit Jtthanol ausgekocht und getrock- net. 30 g yon diesem Pulver und 100 mg UV-lndikator wurden in 80 ml Athanol suspendiert und in tiblicher Weise mit dem Streichger~it yon E. Stahl auf einer Glasplatte (20 x 20 cm) zu einer 0,2 mm dicken Schicht ausgestrichen und kurz an der Luft getrocknet. Das Racemat der Tr6ger- schen Base und die reinen Antipoden wurden in Xthanol- 16sung aufgetragen und mit Athanol als Flie6mittel ge- trennt. Schon eine Laufstrecke von 8 cm reichte zur roll- st/indigen Trennung aus. Rf-Werte: | Base 0,19; | Base 0,41.
Aus Chloroform umgefalltes Cellulose-tri-acetat gibt auch hier keine Trennung und fiir die reinen Antipoden nahezu gleiche Rf-Werte um 0,5.
Dank Dora Verband der Chemisehen Industrie (Fends der Chemie) danken wit fiir die Unterstiitzung, die uns diese Arbeit erm6glicht hat.
3. Elutionsmittel
Bei der Sorption tiber Wasserstoffbindungen gilt allgemein eine eigene L6sungsmittelreihe, die etwa der Basenstiirke der Elutionsmittel entspricht: Wasser, Methanol, Xthanol, Methyliithylketon, Aceton, Formamid, Dimethylform- amid. Ein geringer Gehalt an Wasser oder Methanol im be- nutzten ~.thylalkohol st6rt bei der Nachbarstellung dieser Elutionsmittel nieht; das erleiehtert vor aUem das Arbeiten mit Dtinnschichtplatten sehr. Im Einzelnen ist natiirlich auf die Unl6sliehkeit und Quellfahigkeit des Sorbens zu
Beschreibung der Versuche
Die Versuchsanordnung bestand aus einem Trennrohr mit Mantel, durch den aus einem Umpumpthermostaten Was- ser der gewiinschten Temperatur gepumpt wurde. FOr alle Arbeiten mit Temperaturerh6hung muff das L6sungsmittel vorher an der Wasserstrahlpumpe entgast werden. Druck- erh6hungen wurden mit einer aufgesetzten regulierbaren Quetschpumpe (bis 2 atti) erreicht. Der Auslauf aus der Trennsiiule mu6 sichtbar abtrepfen. Er wird mit dtinnen Teflon-Schlauchen zuerst durch die Mikroktivette eines
Chromatographia, Vol. 6, No. 6, June 1973 Originals 279

Polarimeters Perkin-Elmer 141, dann durch einen UV- Detektor LKB Uvicord II (beide mit Schreiber) geleitet und schliet~lich einem Fraktionensammler mit Wechsel- markierung zugeftihrt.
Cellulose-tri-acetat (in Anlehnung an Lit. [ 13]: 200 g Cellulosepulver (z.B. Schleicher u. Schiill Nr. 123 oder 144), 4 1 Benzol, 800 rnl Eisessig, 6 ml HCIO 4 60 % und 800 ml Acetanhydrid werden in der genannten Reihen- folge zusammengegeben und 3 Tage bei 35 ~ geriJhrt. Die Suspension wird durchscheinend und ffirbt sich dunkel. Dann wird abzentrifugiert, in Methanol aufgeschliimmt, wieder zentrifugiert und bei 30 ~ getrocknet. Es entstehen hellbraune, spr6de Klumpen (230 g), die sich leicht zer- kleinern lassen.
Fiillen der Trenns~iulen
a) Grobk6rniges Material (> 56/~rn): 130 g lufttrockenes Pulver wird in 220 rnl ~,thanol suspendiert, 1 rnin. an der Wasserstrahlpurnpe entgast, in einem GuI~ in die Trennsaule gefiiUt und aus einem aufgesetztem Tropf- trichter rnit entgastem )~thanol gewaschen. Nachdem ca. 500 ml durchgelaufen sind, ist die endgtiltige Pak- kung erreicht.
b) Feine Pulver (in Anlehnung an Lit. [14]: 40 g lufttrocke- nes Pulver (< 56 #m) werden in 60 ml Ethanol suspen- diert, an der Wasserstrahlpumpe entgast, in die Trenn- s~ule eingef'tillt und vorsichtig rnit Ethanol iiberschichtet. Dann wird die Quetschpurnpe aufgesetzt und so einge- stellt, daf~ 3 - 4 Tropfen pro Sekunde durchlaufen. Je
nach der Feinheit des Pulvers braucht man in der 2-cm- S~iule im Endzustand einen Oberdruck von 0,5 bis 1,5 atii, urn einen konstanten Durchlauf von t Tropfen] Sekunde zu erreichen; die BetthOhe ist dann ca. 16 cm.
Literatur
[1] Teil der Dissertation yon R. Hagel, Univ. Erlangen-Niirnberg [2] G.M. Henderson u.H.G. Rule, J. chem. Soc. London
1939, 1568 13] Vergl. G. Hesse in Methodicum Chimicum, Bd. 1/1, 92.
G. Thieme Verlag, Stuttgart 1973 [4] V. Prelog u.P. Wieland, Heir. chim. Acta 27, 1127 (1944) [51 H. Krebs, J .A. Wagneru. J. DiewaM, Chem. Ber. 89,1875
(1956) [61 11. Musso, Chem. Ber. 91,349 (1958) [71 11. Krebs u. W. Schumacher, Chem. Ber. 99, 1341 (1966) [8t W. Steckelberg, 3/1. Bloch u. H. Musso, Chem. Bet. 101,
1519 (1968) 191 Ir Mayer u. F. Merger, Liebigs Ann. Chem. 644, 65 (1961)
I10] Zuerst mitgeteilt von A. Liittringhaus auf der Biirgenstock- Konferenz fiir Stereochemie 1965
1111 A. L(ittringhaus, U. Hess u. H. J. Rosenbaum. Z. Natur- forsch. 22b, 1296 (1967)
[12] A. Liittringhaus u. K. C. Peters, Angew. Chem. 78,603 (1966)
[13] Houben.lCeyl-Miiller, Methoden der Organischen Chemie, 4. Aufl., Bd. 14/2, 875 (1963)
[14] W. Strubert, Chromatographia 6, 50 (1973)
Received: May 2, 1973 Accepted: May 10, 1973
280 Chromatographia Vol. 6, No. 6, June 1973 Originals





























