Embed Size (px)
Citation preview

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 113
Nittleilungen aus dem chemischen Institiit der Universitat Heidelberg.
169. Einwirkung yon wasscrfreiem Hydrazh auf Nitrile;
Ernst Muller und Leonhard Herrdegen. l)
von
(Eingegangen am 12. Marz 1921.)
C u r t i u s und Dedichenz) erhielten beim Einleiten vou Cyan in Hydrazinhydrat eine weifie, krystaliinische Substanz, die sie als Carbohydrazimin bezeichneten, und die durch An- lagerung von 2 Mol. Hydrazin an die beiden Cyangruppen entstanden war:
CEZJ C P H 1 \NH . KH,
C' ANH
+ 2N,H, = , 1 NH.NH,
C"N
C u r t i u s und Dedichen3) haben weiter gezeigt, daB auch die Nitrile, wenn auch erst bei hoherer Temperatur und unter Druck , mit Hydrazinhydrat zu reagieren vermogen. Nach ihrer Auffassung verlauft die Einwirkuug voti Hydrazinhydrat auf Ace t on i t r i l folgenderma6en: Zuniichst lagert sich Hydrazin an das Nitril an unter Bildung von Methylcarbohydrazinlin. Von diesem vereinigen sich darauf bei der hohen Teriiperatur 2 Mol. unter Austritt yon Hytlrazin zu Methylhydrazicarbimin
N B I. (3Ef,.CN + N,H, = CH,.C/
\N€I.h'H, '
1) L c o n h a r d Her rdegen , ,,Einwirkung von wasserfreiem Hydr- azin auf Nitrile'<. 1naug.-Diss. Heidelberg 1918. Druck rnn J. Hiirning.
e) Dies. J u u m . [2] GO, 245 (1894); 32, 272 (1805). 8) Ebenda.

114 Muller u. Herrdegen: Einw. v. Hydrazin usw.
+ N,H, NH:NH, NH
h H \NH CH,. C/ CH,. C/
Ganz analog lieferte Hydrazinhydrat mit B en z o n i t ril Phenylhydrazicarbimin,
NH NH CeH6. C l b. C,H, .
\NH-NH/
Letzteres miiBte identisch sein mit dem zuerst von P i n n e r l ) aus Benzimidoather und Hydrazin erhaltenen Dibenzenylhydrazi- din, ist aber in Wirklichkeit Diphenyl-Isodihydrotetrazin a), das man jetzt nach dem Vorgang von Bulow3) als Diphenyl- N-amino triazol,
NIT,
betrachtet. Die Substanz enthalt somit 2 Wasserstoffatome weniger, als Cur t iu s und D e d i c h e n angenommen hatten. Tatsachlich hatten diese auch bei der Analyse weniger Wasser- stoff gefunden, als das vermeintliche Dihydrazidin verlaugt. 4,
Auch die aus Acetonitril dargestellte Verbindung gehort nach ihrem ganzen Verhalten in ,die Reihe der N-Aminotriazole.
Dedichen5) BuWerte spater die Vermutung, da8 bei der Einwirkung von Hydrazinhydrat auf Nitrile diese zunkchst unter Verseiflung und Bmmoniakabspaltung primare Saure- hydrazide liefern und letztere darauf, wie beim Erhitzen fiir
') Ber. 26, 2130 (1893). 2, Pinner u. Caro , Ber. Z7,3274,Anm. l (1894); vgl. auch Curt ius ,
dies. Journ. 121 $2, 272 (1895). 3, Ber. 89, 2618, 4106 (1906); vgl. dazu Curt ius , Darapsky u.
Miiller, Ber. 40, 1470 (1907); Sto118, dies. Journ. [ Z ] 75, 94, 416 (1907); B u s c h , Ber. 40, 2093 (1907).
4, Curt ius u. Dedichen, dies. Journ. [2] 60, 256 (1894). $) Ber. 39, 1855 (1906).

Miiller LL Herrdegen: Einw. v. Hydrazin usw. 115
sich nach Yel l izar i l ) und Stol le2) , in N-Aminotriazole iiber- gehen :
I. R.CN +- NH,.NH,, H,O = R.CO .NH.NH, + NH, . ,NH . NII, N--N
f 0 C . R = R . C j b . R $- 2H,O. \ N /
11. R.CO H/
XH, N H,
Es ist aber wahrscheinlicher anzunehmen, daB hierbei nach der urspriinglichen Ansicht von C u r t i u s und Dedichen8) nnd ganz, wie bei der Einwirkung von Hydrazinhydrat auf Cyan4), eine Anlagerung yon Hydrazin an die Cyangruppe stattfindet und ao zunachst ein Carbohydrazimin oder Mono- hydrazidin erhalten wird:
NH R.C-N +N,H, : R . C j
\NH . NH, *
Monohydrazidine wurden zuerst ron P i n n e r 6, dargestellt im Laufe seiner schonen und umfassenden Untersuchungen uber die Einwirkung von Hydrazin auf Imidoather. Bei dieser Reaktion entsteht wohl zunachst ein freilich nicht fagbares Additionsprodukt, aus dem sodann Alkohol austritt, wobei ent- weder ein Amidhydrazon oder das tautomere Imidhydrazid sich bildet:
.NH
I c
P i n n e r betrachtet die Monohydrazidine auf Grund ihres gesamten chemischen Terhaltens als Amidverbindungen, weist indessen auch auf die Mijglichkeit einer Tautomerie hin. Die Monohydrazidine, die einzigen direkten Einwirkungsprodukte
*) Gaze. chim. 26, IT, 430 (1896); Chem. Zentralbl. 1899, I, 1240. l ) Dies. Journ. [ 2 ] 6ci, 464 (1903). J, Dies. Journ. [a] 60, 246 (1894). 4, Ebenda S. 245. I) Ber. 27, 984, 3273 (1894); 28. 465 (1895); 30, 1871, 2010 (1897);
Ann. Chern. 287, 221 (1897); 298, 1 (1897).

116 Mul le r u. Herrdegen: Einw. v. Hydrazin usw.
von Hydrazin auf Imidoather, sind nun aber iiuJ3erst reaktions- fahig, so daB schon bei ihrer Entstehung ein Teil stets weiter umgewandelt wird. So bilden sich daraus mit unverandertem Imidoather unter Abspaltung von Alkohol die Dihydrazidine,
wahrend bei Gegenwart kleiner Mengen freien Hydrazins die ringfhrmigen Dihydrotetrazine erhalten werden, indem 2 Mol. Monohydrazidin mit 1 Mol. Hydrazin unter Austritt von 2 Mul. Ammoniak ein nicht existenzfahiges Zwischenprodukt liefern, das sofort wieder Hydrazin abgibt :
Die rStlichgelben Dihydrotetrazine, die als Hydrazoverbin- dungen schon durch den Sauerstoff der Luft zu den zugehorigen Azokorpern, den tief scharlach- bis blauroten Tetrazinen, oxydiert werden,
gehen weiter beim Erhitzen mit konzentrierten Sauren unter Ringverengerung einerseits in farblose, nicht mehr oxydations- fahige, isomere N - Aminotriazole (Iso-Dihydrotetrazine) uber, andererseits durch Austausch der Hydrazogruppe gegen Sauer- stoff in Furodiazole (Diazoxole):

Muller u. I’Terrdegen: Einw. Y. Hydrazin usw. 117 Eine besonders merkwiirdige freiwillige Zersetzung er-
hbren die Monohydrazidine endlich durch Kalilauge: Unter Aufnahme von Sauerstoff und Entwicklung von Stickstoff ent- ateht aus 1 Xol. Monohydrazidin ein nicht existenzfahiges Zwischenprodukt, das sich sofort mit einem zweiten Mol. un- veranderten Monohydrazidins vereinigt , diese Verbindung v0r- l i ed spontan Ammoniak und liefert ein Dihydrotriazol:
So stellt das Reaktionsprodukt aug salzsaurem Imidoather, Kalilauge und Hydrazinsulfat immer ein kompliziertes Gemimh von Monohydrazidinen , Dihydrazidinen, Dihydrotetrazinen, Tetrazinen und Dihydrotriazolen dar.
Nach diesen grundlegenden Untersuchungen von P i n n e r clurfte man auch in unserem Falle, bei der Einwirkung von Hydrazinhydrat auf Nitrile, vor aliem die Bildung von Di- hydrotetrazinen und Tetrazinen erwarten. DaB solche tat- sachlich hierbei entstehen, deutet schon eine Bemerkung in der alten Arbeit von C u r t i u s und D e d i c h e n an. Sie beob- achteten namlich, daB sich die waDrige Liisung des aus Aceto- nitril und Hydrazinhydrat im Rohr bei 150° erhaltenen Pro- duktes an der Luft violettrot farbte. ’) Diese Erscheinung laDt sich nur durch Oxydation von vorhandenem Dimethyl- dihydrotetrazin zum entsprechenden Tetrazin erklaren. Spl ter fanden K. A. Hofrnann und Ehrha r t2 ) , daB Benzonitril beim Stehen mit Hydrazinhydrat in alkoholischer Liisung bald starke Rotfarbung zeigte und beim Durchleiten von Luft Diphenyl- tetrazin sich daraus abschied.
Wir vermuten, daB die N-Aminotriazole auc‘n hier nicht unmittelbar aus den Monohydrazidinen sich bilden, sondern sekundair aus Dihydrotetrazinen durch Umlagerung infolge der angeffandten hohen Temperatur hervorgehen. So haben C u r t i u s , D a r a p s k y und Miil ler3) schon vor langerer Zeit gefimden,
I) Dies. Journ. [2] 60, 256 (1894). ’) Ber. 45, 2732 (1912). *) Ber. 40, 822 (1907).
Jezmnal f. prakt. Chemie [2 ] Bd. 1UB. 9

118 Miiller u. Herrdegen: Einw. v. Hlydr;izin U ~ W .
daS das unsubstituierte Dihydrotetrazin bereits beim Schmelzen sich in N-Aminotriazol umlagert; ein analoges Verhalten zeigt nach neueren Beobachtungen I) auch Dimethyldihydrotetrazin.
Einen besonderen Verlauf nimmt die von D a r a p s k y 4 untersuchte Einwirkung von Hydrazin auf b l ande l skure - n i t r i l ; man sollte aus diesem Oxysaurenitril eigentlich die Bildung eines Hydrazinosaurenitrils erwarten, in Wirklichkeii aber erhalt man schon bei gewohnlicher Temperatur unter A b s p a l t u n g von B l a u s a u r e und Ammoniak Dibenzenyl- hydrazidin und Diphenyldihydrotetrazin. Diese eigentumliche Reaktion ist vielleicht so zu erklaren, da6 das zuniiichst ent- stehende Hydrazinosaurenitril mit Hydrazin unter Austritt von Blausaure ein unbestandiges Zwischenprodukt liefert, das 40-
fort A mmoniak verliert uud S O Monobenzenj lbydra7idin liefert,
welch letzteres endlich weiter in Dibenzenylbyrlrazidin und Diphenyldihydrotetrazin ubergeht.
Dihydrotetrxzine entstehen ferner, wie J u n g h a h n B ) ge- funden hat, durch Einwirkung von Hydrazin auf Thianiidc nach der Gleichung:
Such bei dieser Reaktion kann man die Zwischenbildung VOII
Bfonohydrazidinen annehmen. 4,
Auf anderem Wege, von Saurehydraziden nusgehend, ge.- langte Stol ib5) zu Uibydrotetrazinen. Er erhielt zuniichst aus primaren Hydraziden beim Xrhitzen ueben N-Aminotriazolen (vgl. 5. 115) auch Dihydrotetrazine,
NH,.N N --N, + \C .R -=;L-+ R . C l 'h c . [t , \NH-XH/
R. C j " \NH.NH, HO/
*) C u r t i u s , Darapsky u. Muller , Ber. 48, 1624 (1:jlS). a) Dies. Journ. [2] 97, 186 (1918). 4, J u n g h a h n u. R u n i m o w i c z , Ber. 35, 3933 (1902). 5, Dies. Jonm. [2] 68, 464 (1903); 73, 277 (1906); 75: 416 (1907).
*) Ber. 31, 318 (1898).

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 119
somie weiter letztere allein aus Dihydraziden durch Uberfiihrung in Dihydrazidchloride und Umsetzung dieser mit Hydrazin- hydrat :
//N-N \ C . R R . C O . K H . N H . C O . R ---+ R . C \ci c1/
Dicarbonsfuren des Dihydrotetrazins, die wegen der eigen- tumlichen Bildung von Isomeren und deren Zerfall bei der Hydrolyse besmderes Interesse verdienen, gewannen C u r t i u s , D a r a p s k y und Miiller’) im Laufe ihrer Untersuchungen iiber die Einwirkiing von Basen auf Diazoessigester.
Mit konzentrierter Kalilauge, fliissigem Ammoniak oder primaren Aininen liefert Diazoessigester in der Kalte Verbin- dungen, die sich von der bimolekularen C,N-Dihydro-1,2,4,5- tetrazin-2,G-dicarbonsaure (Pseudodiazoessigsaure),
N-NH
N--N COOH. C< )CR. COOI-I,
ableiten ; erhitzt man dagegen den Diazoester mit konzen- triertem Alkali oder starkem Ammoniak, so bilden sich die Alkslisnlzo bzw. das Amid der N,N-1,2- bzw. N,N-l,$-Dihydro- 1,2,4,5 tetrazin.3,6-dicarbonsaure (Bisdiazoessigsiure):
Bei fortgcsetzter Xinwirkung starkster Kalilauge in der Wilrrne entstelien endlich unter Ringverengerung zwei weitere Sauren, n8mlich eine mit obigen Siuren isomere Dicarbon- sfure des N-Aminotriszols, sowie weiter un ter gleichzeitiger Abspaltung von 1 Mol. Kohlendioxyd eine Monocarbonsaure des C!-Aminotriazols:
3 - - N N--N b. COOH und NH, . CY b. COOH . \NH/
COOJ! .C,~ \ N /
NH,
I) Vgl. die zusammenfitssende Abhsndlung Ber. 41, 3161 (1908); vgl. fwner Rer. 42, 3284 (1909); Mii l ler , Ber. 42, 3270 (1909).
9 *

120 Muller u. Herrdegen: Einw. v. Hydrazin usw.
N,N-Dihydrotetrazindicarbonsaure (Bisdiazoessigsaure) l&Ot sich durch salpetrige Siiure leicht zur tiefroten 1,2,4,5-Tetrazin-
oxydieren. Aus dieser erhielten endlich Cur t iu s , Darapsky und Miiller') durch Erhitzen unter Abspaltung von 2 Mol. Kohlendioxyd die Stammsubstanz aller symmetrischen Tetrazinc, das 1,2,4,5-Tetrazin selbst, und durch dessen Reduktion das biaher gleichfalls noch unbekannte wahre Dihydrotetrazin:
Die Hydrolyse diarylierter Tetrazine wurde schon von P inne r2 ) naher studiert. Er fand, da6 Diphenyltetrazin beim Kochen rnit alkoholischer Kalilauge Stickstoff entwickelte und eine bei 206 * schmelzende Verbindung lieferte, die er selbst freilich als ein Isomeres des Benzalbenzhydrazids, namlich als s-Phenyloxymethylenbenzylidenhydrazon, amah. wahrend B am- berger3) sie zuerst als mit diesem identisch betrachtete:
+ C,R,.CII:N.NII.CO.C,IT,.
Eiagehend haben spater Cur t iu s , D s r a p s k y undMuller4) die hydrolytische Spaltung der von ihnen aus dem Diazoessig- ester dargestellten Dicarbonsauren des Tetrazins, N, N-Dihydro- und C, N-Dihydrotetrazins untersucht. Diese zerfallen zum Teil schon beim Erwarmen mit Wasser, vollig aber beirn Kochen mit verdiinnten Sauren. Dabei ergab sich die allgemeine Begel, daO eine im Molekul vorhandene Azogruppe --N=N- mit zwei doppelt untereinander gebundenen Stickstoffatomen als freier Stickstoff austritt, wahrend zwei einfach untereinander
l) Ber. 40, 84 (1907). g, Ber. 27, 1007 (1894); Ann. Chem. 297, 265 (1897). $) Ber. S, 3197, Anm. (1900). ') Ber. 41, 3165 (1908); vgl. auch Curt ius , Z. f. angew. Chem.
*&,I, 9 (1911).

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 121
gebundene Stickstoffatome, d. h. die Gruppen =N-N==, -NH-NH- und =N-NH, bei vollstindiger Hydrolyse a18 Hydrazin crhalten werden.
So liefert N, N-Dihy drotetrazindicarbonsaure (Bisdiazoessig- saure) beim Kochen mit verdiinnter Schwefelsiiure 2 Nol. Hy- drazinsalz und 2 Mol. Oxalsaure, die hierbei weiter in Ameisen- saure und Kohlensaure gespalten wird:
O H , H,O N - -N N- K,
COOH, G I b . C O O H -+ C 0 O H . d b. COOH. \N H - N lI/ NH--NH/
I 1 0 H H OH
C, N - Dihydrotetrazindicarbonsaure (Pseudodiazoessigsaure) dagegen zerfallt bei der Hydrolyse in 1 Idol. Stickstoff, 1 Mol. Hydrazin und 2 Mol. Glyoxylsaure nach dem Schema:
0 €1, €1 OH W-NH N-XH
\CH,COOH --t COOH.C/ ‘CH. COOR. \ K--N / COOH. (21 \N -N/
€1 OH
I m Jahre 1915 glaubte Lifschi tz’) , durch Einwirkuog von Hydrazinhydrat auf T e t r a z o l n i t r i l die Synthese von l’entazolen erreicht zu haben. Nach seiner Annahme verlauft die Reaktion in ahnlicher Weise wie die fruher von Cur t iu s , D a r a p s k y und Bockmuhl untersuchte EinwirkuDg von Hydrazin auf Diazoessigester. Wie dort unter Austritt von Ammoniak und Alkohol Azidoacethydrazid erhalten wurde,
N N m,~, + I ~ \ C H . C O O R --NTKzRRUI1+ ~\\N.cH,.co.NH.NH,,
N’ h/
sollte hier analog unter Entwicklung von Ammoniak des Kohlenstoffatom des Tetrazolrings durch StickstoE ersetzt werden und so unter gleichzeitiger Anlagerung von 1 Mol. Hydrazin an die Cyangruppe das Hydrazidin der Pentazido- essigsaure entstehen :
‘).Bee. &, 410 (1915). ’) Bee. 41, 944 (1908).

122 Muller u. Herrdegen: Einw. v. Hydrazin usw.
Wie sich aber durch die Untersuchungen von C u r t i u s , D a r a p s k y und Miil lerl) herausstellte, war das verineintliche Hydrazidin der Pentazidoessigsaure in Wirklichkeit dau Bis- Diammoniumsalz des Ditetruzyldihydrotetrazins,
Lc- CBN -N X&, 11 b - c L 9 / / , IU’JI,. N- NH/ \NH-XH/’ \NH-N
N--N N---.N
Auch die zahlreichen anderen daraus dargestellten angeblichen Pentazolverbindungen von Li fsch i t z erwiesen sich samtlich als Derivate dieses Tetrazins. Bei der Einwirkung von Hydrazin auf Tetrazolnitril tritt somit nur die Cyangruppe in normaler Weise mit Hydrazin in Reaktion , wahrend der Tetrazolring ganzlich unrerandert bleibt , wie dies iibrigens bei seiner gro6en Bestiindigkeit auch von vornherein zu ermarten war.
Es lag nahe zu priifen, wie weit iiberhaupt durch die Einwirkung yon Hydrazin auf Nitrile Dihydrotetrazine uud Tetrazine der verschiedensten Art erhalten werden konnen.
Das zuerst ron Cur t iu s und Dedichenz) studierte Ver- halten des einfachsten Alkylnitrils, des Acet on i t r i l s , gegcn Eydrazin hatten Cur t iu s , D a r a p s k y und Miil ler schon bei ihrer Widerlegung der Lifschitzschen Arbeit aufs neue in den Bereich ihrer Untersuchungen gezogen. 1)urch Kochen von Acetonitril mit H y d r a z i n h y d r a t wurde hauptsiichlich Dimethyl-N-aminotriazol neben wenig Dimethyldihydrotetrazin erhalten. Wurde dagegen Acetonitril mit wasser f re iem Ky- drazin mehrere Tage unter RuckfluS zum Sieden erhitzt, so entstand fast ausschliefilich Dimethyldihydrotetrazin.
Wir haben nunmelir die Einwirkung von Hydrazin auf die verschiedensten Alkylnitrile naher studiert und zunachst Dimethyltetrazin 3, in groBeren Mengen dargestellt. Die An- gaben uber die Eigenschaften des Dimethyltetrazins konnten wir durchaus bestatigen. Neu festgestellt wurden das Ab- sorptionsspektrum seiner atherischen Losung sowio die Zer- fallsprodukte bei der Hydrolyse.
*) Ber. 48, 1614 (1915); Li feeh i t z , Ber. 49, 489 (1916). 2, Dies. Journ. [2] 60, 246 (1894); 62, 272 (1895). *) Curtius, Darapsky und Mi i l l er , Ber. 48, 1633 (1915).

Xliiller 11. Herrdegen: Einw. v. Hydrazin usw. 123
Dimethyltetrazin sollte beim Kochen mit Sauren folgender- n,a8en zerfallen:
4. 2H,O - f CLT,.CH:S.NIT.CO.CH, + N, - f CH, . CHO + XH2. NEI, -I- HOCO. CH, f N9 .
Nun erhieltcn wir aber an Stelle von einem Nol. Stickstoff nur annahernd ’ I 2 Nol., statt des zu erwartenden einen Mol. Hydrazins dagegen nahezu 11/4 Idol. Die Menge der Essig- eaure wnr nur die Halfte der berechneten. Aldehyd konnte nicht nachgewiesen werden. Auch Ammoniak, das sich durch die oxydierende Wirkung von Hydrazin auf Acetaldehyd hatte bilden kiinnen I ) , wurde nicht gefunden.
Deli wirklichen Verlauf der Hydrolyse kann man sich folgenilermaBen erklaren: Dimethyltetrazin zerfallt zunachst in g-eringer Nenge nach obigem Schema. Der so entstehende Acetaldehyd reduziert sodann noch unzersetztes Tetrazin zum Dihydrotetrazin, welch letzteres bei der Hydrolyse ke in e n Stickstoff, dafur aber z wei 3101. Hydrazin liefert. Der Verlauf der Reaktion entspricht somit ganz der schon von C n r t i u s , D a r a p s k y und Ni i l le r2) naher untersuchten Hydrolyse des Tetrazins selbst, wobei gleichfalls weniger Stickstoff, aber 111 e h P Hydrazin und ke in Formaldehyd erhalten wurde.
Die Hydrolyse konnte aber auch so vor sich gehen. daS dtts Dimethyltetrazin zunachst in Diacethydrazid und das UII-
Iwstandige L)iimid gespalten wird, welch letzteres unter Stick- htoffentwicklung seinen Wasserstoff an noch unverandertes ‘Cetrazir: abgibt und dieses so zum Dihydrotetrazin reduziert:
--f CB,.CO.PU’H.NH.CO.C~S, + N, + ’i’ rr,o - Hs -----1_) C€T,.COOH + NH,.NEfs + HOCO.CH, + N, .
Nach dieser Auffassung wurde uberhaupt kein Aldehyd, ouch nicht vorubergehend , gebildet, nach beiden Erklarungs-
3 , Vgl. Curtius, D a r a p e k y und M i i l l e r , Rer. 39, 3413 (1906). ’) k r . 40, 1178 (1907).

124 Miiller u. Herrdegea: Einw. v. Kydrazin UBW.
weisen aber entsteht intermediar ein Dihydrotetrazin, dtbs Cur t ius , D a r a p s k y und Miiller') bei der Zersetzung von Ditetrazyltetrazin mit waBrjger oder alkoholischer Salzfiiiure auch als solches isolieren konnten.
Wir haben weiter die Einwirliung von Hydrazin arrf P r o p i o n i t r i l untersueht. Durch mehrtagiges Sieden des Gemisches beider Verbindungen entstand ein fltissiges Reak- tionsprodukt, das nach dem Eindunsten im Vakuum neben ge- ringen Mengen von Diathyldihydrotetrazin groBere Mengen von Diathyl-N-aminotriazol enthielt. Diathyldihydrotetrazin ward durcll Oxydation rnit salpetriger Saure in Diiithyltetrazin tibergefiuhrt, welch letzteres durch Reduktion mit Schwefel- wasserstoff in atheriseher Losung in das Diiithyldihydrotetrazin zuriickverwandelt wurde.
Die Hydrolyse von Diiithyltetrazin vcrlief der yon Di- methyltetrazin analog. Unter Austritt von 1 Mol. Stickstoff und Abspaltung von 1 Mol. Hydraziu sollten neben 1 Bfol. Propionaldehyd 1 Mol. Propionsiiure entstehen. Das Experi- ment ergab jedoch nur etwa '1, Mol. Stickstoff, neben etwa j e 1 Yol. Hydrazin und Propionsaure. Propionaldehyd war nictit nachzuweisen.
Durch mehrtagiges Erhitzen von n - B u t y r o n i t r i l iind Hydrazin unter RiickfluB und nachheriges Eindunsten im Vakuum eotstand eine weiBe, feste Masse. Diese enthielt nur Spuren des erwarteten Dihydrotetrazins, denn bei der Oxy- dation mit salpetriger Saure trat unter Gasentwicklung in der waBrigen Liisung des Dihydrokorpers nur geringe, bald wieder verschwindende Rotfarbung ein, FOII entstandenem Di-n-butgryl- tetrazin herruhrend.
Auf I s o - T a l e r on i t ri 1 wirkt wasserfreieu Hydraziu beim Erhitzen nicht ein. Es b i l d e t s ich ke in D i h y d r o t e t r a z i n mehr.
Es zeigt sich somit, daS bei der Einwirkung von Hydrazin auf Nitrile Dihydrotetrazine in um so geringerem MaBe ent- stehen, j e weiter man in der homologen Reihe der Nitrile zu den hoheren Gliedern aufuteigt. Da die Versuche mit den aliphatischen Nitrilen wenig befriedigende Resultate gaben,
Auch Ammoniak wurde nicht gefunden.
Durch Oxydation erhalt man keine Rotfkrbung.
') Ber. 45, 1619 (1918).

Miiller 11. Herrdegen: Einw. v. Hydrazin usw. 125
haben wir weiter die Einwirkung von wasserfreiem Hydrazin anf Nitrile der a r o m a t i s c h e n Reihe untersucht.
Beim Zusammenbringen von Benzon i t r i l mit wasger- f r e i e m Hydrazin und Stehenlassen bei geivijhnlicher Tempera- tur tritt erst nach einigen Nonaten Reaktion ein; unter Am- moniakentwicklung krystaliisieren aus dem Fiussigkeitsgemisch gelhe Nadeln von Diphenyldihydrotetrazin aus. Der geringste Luftzutritt genugt zur Oxydation des Diphenyldihydrotetrazins zum blaustichig rot gefarbten, in prachtigen Nadeln krystalli- sierenden Diphenyltetrazin. Rascher gelangt man zum Ziele durch mehrtagiges Erhitzen von Benzonitril mit wasserfreiem Hydrazin am RuckfluBkiihler. Es entsteht ausschliefilich Di- phenyldihydrotetrazin, das durch Oxydation mit salpetriger Saure, ja schon durch Luftsauerstoff, in Diphenyltetrazin uber- gefuhrt wird, welches P i n n e r I) zuerst durch Einwirkung von Hydrazin auf Benzimidoather gewonnen hatte.
DaR Benzonitril auch mit Hydrazinhydrat in alkoholi- scher Lijsung und zwar schon in der Kalte Diphenyldihydro- tetrazin und weiter Diphenyltetrazin licfert 9. wurde bereits 8. 117 erwahnt.
Die Aufspaltung des Diphenyltetrazins beim Erhitzen rnit dkoholischem Kali zu Benzalbenzhydrazid nach P i n n ers) wnrde gleichfalls bereits S. 120 erwahnt. Wir haben die Hydrolyse, wie auch sonst, durch Kochen mit Sauren aus- gefuhrt. Man sollte so bei normalem, viilligem Zerfall je 1 Mol. Stickstoff', Hydrazin, Benzaldehyd und Benzoesaure erwarten. Es zeigte sich jedoch auch hier, da6 die Spaltung teilweise in anderer Richtung verlguft. Nur je 1/2 Mol. Stickstoff und Hydrazin neben wenig Beneoesaure wurden erhalten. Geringe Mengen von Benzaldehyd konnten als m-Nitrobenzh ydrazon nachgewiesen werden. AuBerdem war noch Diphenylfurodiazol entstanden. Seine Bildung ist wohl so zu erklaren, dab Di- phenyltetrazin teilweise zunachst zu Diphenyldihydrotetrazin rcduziert wird; dieses liefert sodann, wie schon Pinner ') fand, mit Sauren unter Abspaltung von Hydrazin Diphenyldiazoxol oder Diphenylfurodiazol (vgl. auch S. 11 6).
I) Ber. 26, 2133 (1893). 2) K. A. I lofmann und Ehrhar t , Ber. 46, 2732 (1912). a) Ann. Chem. 291, 266 (1897). 4, Ebenda, S. 263.

126 Miiller u. Herrdegen: Einw. v. Hydraziri usw.
Benzylcyanid gab, mit wasserfreieni Hydrazin mehrere Tage uuter Ruckflub zum Sieden erhitzt, durch die eintretendo Rotung zu erkennen, dab sich ein Tetrazin gehildet hatte. Es gelang aber nicht, dieses aus der Fliissigkeit abzuscheiden. Dagegen konnte das Dibenzylaminotriazol l), das beim Erkalten der Flussigkeit krystallin sich ausschied, gefaBt und als solcbes durch die Analyse identifiziert werden. Wurde Benzylcyanid mit wasserfreiem Hydrazin bei gewohnlicher Temperatur sich selbst uberlassen, so trat selbst nach Monate langem Stehen Ireine Reaktion ein. Auch beim Erhitzen von Benzylcyanid mit wasserfreiem Hpdrazin im Rohr entstand nur das Amino- triazol.
Wir haben weiter die drei T o l u n i t r i l e in den Kreis der Untersuchung gezogen.
Auf O r t h o-Tolunitril wirkte wasserfreies Hydrazin ubcr- haupt nicht ein.
Aus Para-Tolunitril und wasserfreiem Hydrazin schiedeii sich bei mehrtagigem Kochen unter IliickfiuB schon in der Warme gelbe Krystalle von Di-p-tolyldihytfrotetrazin2) aua. Letzteres la& sich durch salpetrige SLure zu Di-p-tolyltetrazin 3,
oxydieren. Die Oxydation kann auch durch Luft sowie durch Brom in Eisessig ausgefuhrt werden.
Besonders glatt verlauft die Reaktion zwischen Met& - Tolunitril und Hydrazin. Wird m-Tolunitril mit wasserfreiein Hydrazin mehrere Stunden unter RuckfiuB zum Sieden er- hitzt, so erhiilt man beim Erkalten ausschlieBIich gelbe Niidel- chen von Di-m-tolyldihydrotetrazin. Dieses wird durch sd- petrige Sinre zu dem schon roten Di-m-tolyltetrazin oxydiert. Letzteres wird umgekehrt durch Reduktion niit Zinkstaub und ganz verdiinnter Schwefelsaure in iitherischcr Losung in dss Dihydrotctrazin zuriickverwandelt.
Bei eintagigem Erhitzen von wasserlreiem Hydrazin init m-Tolunitril wird kein Di-m-tolyldihydrotetrazin mehr erhalten, sondern dessen Umlagerungsprodukt, das in weii3en Blattchen krystallisiereude Di-m-tolylaminotriazol.
Die Hydrolyse des Di-m-tolyldihydrotetrazins verlief in
I ) Pinnep, Ann. Chem. 29S, 22 (1897). *) Ebenda S. 14. J, Ebenda 5. 17.

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 127
zwei Phasen. Beim Kochen mit konzentrierter Salzsaure ent- stand zunachst neben Hydrazinchlorid Di-m-tolylfurodiazol. Beim Erhitzen mit konzentrierter Salzsaure unter Druck warden dagegen, wie es der vollige Zerfall verlaDgt, j e 2 Mol. llydrazin und ru-‘l’oluylsaure erhalten.
Bei der Hydralyse des Di-m-tolyltetrazins waren zu er- warten : J e 1 Mol. m-Tolylaldehyd, Stickstoff, Hydrazin und m-‘l’oluylsaure. Der Aldehyd konnte nicht sicher nachgewiesen werden. Die iibrigen Spaltungsprodukte wurden dagegen er- halten.
8 - N a p h t o n i t r i l reagiert mit wasserfreiem Hydrazin in gleicher Weise wie p-Toluuitril. Das erhaltene Di-P-naphtyl- dihydrotetrazin gab bei der Oxydation rotes Di-P-naphtyl- tetrazin.
Di-P-naphtyldihydrotetr~zin wird beim Kochen rnit Sauren nur zum kleinen Teil in P-Naphtoesaure und Hydrazin, zum groBeren Teil aber in Di-@’-naphtylfurodiazol und Hydrazin gespalten.
Die Einxirkurig von Hydrazin auf das denkbar einfachste Sitril, die Blaus i iure , rnu8te zur Synthese des Dihydro- tetrazins selbst fiihren, und daraus muBte da5 Tetrazin, die Starnmsubstanz aller dieser Korper, gewonnen werden konnen.
E’ranzen und Lucking‘) haben bereits versucht, aus Hydraziiihydrat und wasserfreier Blausaure das Diammonium- a d z dar/,ustellen, konnten es aber nicht in fester Form iso- iieren. Wir erhielten die s e h r u n b e s t a n d i g e Verbindung beim Zusanimengeben YOU absoluter Blausaure und wasser- f re iem Hydrazin unter Turbinieren in der Kalte als schnee- weiSe Krystallmasse. Das Salz zersetzte sicli rnsch spontan und konnte deshalb nicht abgewogen und analysiert werden. Indessen IieB sich das Mengefiverhaltnis vou Hydrazin zu Blausiiure ermitteln. Es entsprach der erwarteten Formel N,H, .CN.
Beim Schmelzen und darauffolgenden Erwarrnen des wei6en Sttlzes bis zu 55O crhalt man unter lebhafter Am- moniakentwickluug eine hellgelbe, olige Flussigkeit, die im Fakuuni zu einer hellgelben, festen Masse erstarrt. Die Ana-
‘1 Z. f. morg. Chem. 70, 147 (1911).

128
fyse lieferte die Bruttoformel C,H,N,. der gelben Verbindung kommen zwei Formeln in Frage:
Muller u. Herrdegen: Einw. v. Hydrazin usw.
Fur die Konstitution
Die Entstehung eines Kiirpers von Formel I kann folgender- maBen erklart werden: Das Diammoniumsalz der Blausaiure lagert sich beim Schmelzen teilweise in das entsprechende Hydrazidin urn, von welchem sich nunmehr zwei Nol. unter Abspaltung von Amrnoniak zu Dihydrotetrazin vereinigen. Der sich nicht umlagernde Teil des Diammoniumsalzes ist beim Schmelzen hochst wahrscheinlich in seine beiden Komponenten, Blausiiure und Hydrazin, wieder zerfallen. Letzteres konnte sich an das freie Dihydrotetrazin anlagern unter Bildung eines sauren Diammoniumsalzes:
f H C / N H <NH.NH,
N,H,.CN ---
Nach obiger Formel 11 w5re der erhaltene gelbe Korper Dimethinhydrazodihydrazon. Seine Entstehung ware 80 zu deuten: 1 Mol. Hydrazidin tritt mit Hydrazin unter Abspaltung von einem $101. Ammoniak zu einer labilen Verbindung zu- sammen, die sofort mit einem zweiten Mol. Hydrazidin unter erneutem Verliist von einem Mol. Ammoniak reagiert :
Bei genauerer Untersuchung der gelben Substanz warde gefunden , daS sie mit Benzaldehyd ein Kondensationsprodukt lieferte, das annahernd die Zusammensetzung des DibenzaL Dimethin hgdrazodihydrazons,

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 129
C,H, . CH CH . C,H, II I1
hatte. Allerdings 11 urden daneben auch groBere Mengen von Benzaldazin erhalten.
Somit durfte die Formel I1 der wahrscheinlichste Aus- h c k fur die Konstitution der gelben Verbindung sein. Wie nach dieser Formel zu erwarten, zerfillt die Verbindung beim Auflosen in verdunnter SBure leicht unter RingschluB in Di- Zlydrotetrazin und Hydrazin:
Wird ferner die Substanz in verdiinnter Essigsaure gelost und die Losung mit Nitrit versetzt, so entsteht sofort das prachtig rote Tetrazin l), das mit Ather ausgezogen werden kann. Durch Reduktion der atherischen Losung mit Zink- staub wurde nach Verdunsten des Athers Dihydrotetrazin2) er- Zlalten. Eine kleine Probe davon gab leicht mit Nitrit und etwas verdiinnter Essigslure wieder Tetrazin.
Durch Erhitzen obigen Kijrpers C,H,N, bis zum Auf- horen der Ammoniakentwicklung entstand endlich N-Amino- triazol", das zur weiteren Charakterisierung mit salpetriger Saure in Triazo14) iibergefuhrt wurde. Man wird auch hier, wie bei der Einwirkung von verdiinnter Skure, zunachst Spal- tung in Dihydrotetrazin und Hydrazin anzunehmen hahen, von denen ersteres sich sodann in N-Aminotriazol umlagert.
In seinen Untersuchungeii uber die Einwirkung Ton Hydrazin auf Imidoather war P i n n e r 5, anfangs der Aleinuug, deB der Bildung der Dihydrotetrazine die von Dihydrazidinen vorausgeht, gab aber spater diese Ansicht auf, da f e r t i g e s Dibenzenylhydrazidin nicht rnit Hydrazin reagierte, und nahm statt dessen, wie bereits S. 116 naher ausgefiihrt wurde, ein anderes, freilich nur hypothetisclies Zwischenprodukt 6, an. Auch
l) Curtius, Darapsky und M u l l e r , Ber. 4.0, 84 (1907). *) Ebenda S. 836. <) Ebenda S. 827, 836. 5, Ber. 27, 986 (1894). 6 , Ber. 30, 1873 (1897); Ann. Chem. 297, 230 (1897).
3, Ebenda S. 837.
130 Miiller u. Herrdegen: Einw. v. Hydrazin USW.
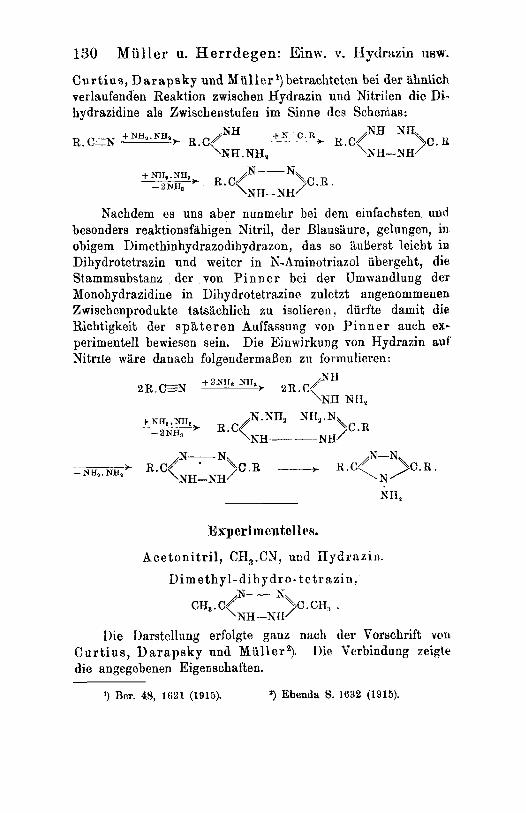
Curtius, Darapaky und Miiller I) betrachteten bei der iihnlich verlaufenden Reaktion zwischen Hydrazin und Nitrilen die Di- hydrazidine als Zwischenstufen im Sinne des Schemas:
Nachdem es uns aber nunmehr bei dem einfachsten uud besonders reaktionsfahigen Nitril, der Blausaure, gelungen, in obigem Dimethinhydrazodihydrazon, das so BuBerst leicht i.ta Dihydrotetrazin und weiter in N-Aminotriazol iibergeht, die Stammsubstanz der von P i n n e r bei der Umwandlung der Monohydrazidine in Dihydrotetrazine zuletzt angenommeneu Zwischenprodukte tatsachlich zu isolieren durfte damit die Richtigkeit der s p a t e r e n Auffassung von P i n n e r auch ex- perimentell bewiesen sein. Die Einwirkung von Hydrazin auf NitnIe ware danach folgendermaaen zii formulieren :
NH,
Exporimmtelles.
Acetoni t r i l , CH,.CN, uutl Xydraz in .
D ime t h y1- d i h y d ro - t e t r xzi n,
CH,.C/N-N'? )c.CH, . \NH--NFf
Die Darstellung erfolgte ganz nach der Vorschrift von Die Verbindung zeigte Cur t iu s , D a r a p s k y und Miillera).
die angegebenen Eigenschaften.
i, Rcr. 48, 1621 (1915). *) Ebends S. 1632 (1915).

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 131 Dime t h y l - t e t r a z i n ,
Wurde aus obigem Dimethyldihydrotetrazin nach den An- guben von Cur t iu s , D a r a p s k y und Muller') durch Oxy- dation mit salpetriger Saure gewonnen.
S p e k t r u m. Die atherische Liisung von Dimethyltetrazin zeigte im sichtbaren Spektrum 4 Absorptionsstreifen, die sich auf folgende Wellenlangen verteilen:
1. Streifen bei Wellenlange A 530--540 2. 1 , ,, 7 ,) 542-552 3. ,, ,, 1 1 ,, 5FO-570 4. . l ,, 1 ,, 574-584
Dimethyltetrazin wurde durch Kochen mit verdunnter Salzsaure zersetzt. Zur Bestimmung des e n t weichenden S t i cks to f f s benutzten w i r den von dem einen von uns 2) bei der Zersetzung des Dikaliumsalzes der Pseudodiazo- essigsaure angewandten Apparat. Dieser besteht aus einem kleinen Rundkolbchen, das die zu zersetzende Substanz enthalt, und das mit einem doppelt durchbohrten Gummistopfen ver- schlossen werden kann. Durch die eine Bohrung geht ein Tropftrichter, dessen Rohr durch ein seitlich angesetztes schrag nach oben fuhrendes Ansatzstuck mit einem Kohlensaure- apparat nach K r e u s l e r in Verbindung steht. I n der zweiten Hohrung steckt das untere Ende eines kleinen RiickfluEkiihlers, dcssen oberes Ende mit einem Schiffschen Azotometer ver- bunden ist, Sobald die im Apparat befindliche Luft durch die Kohlensaure sertrieben ist, wird durch den l'ropftrichter die zur Hydrolyse niitige Menge Sa!zsaure (zirka 25-30 ccm; spez. Gea. I ,]) eingeblasen; dann erwarmt man, bis die Gas- elitwicklung voriiber ist; der Rest des im Apparat noch zuriick- gebliebenen Stickstoffs wird durch einen Kohleoskurestrom vnllstaudig in das Azotometer iihergefiuhrt. Es wurde nahezu (lie Hiilfto der theoretischen Menge Stickstoff erhalten.
Hydrolyse .
0,2392 g gaben 24,09 ccrn N bei 12O und 752 mm. Auatritt von N, ails 1 Mol. C,H,W, (110):
Berechnet: 25,45"/,, N. Gefunden: 11,77 N.
1 ) Rer. 48, 1633 (1915). *) Mii l ler , Rer. 41, 3129 (1909).

132 Miiller u. Herrciegen: Einw. v. Hydrazin USW.
Nach kurzem Erhitzen schlug die Farbe von blaustichig Rot in helles Gelbgriin urn. Die Zersetzung war innerhalb 10 bis 15 Minuten beendet. B u s der salzsauren Losung schieden sich beim Abkuhlen feine weiBe Krystallchen aus, die sich beim Einstellen der Flussigkeit in Eis noch vermehrten; sie wurden als salzsaures Hydrazin identifiziert. Der Hauptteil des Hydraz ins , der sich noch in Losung befand, wurde mit Benzaldehyd als Benzaldazin ausgeschuttelt. Dieses wurde auf einem Filter gesammelt, getrocknet und gewogen. Ammonsalze waren nicht nachzuweisen.
0,2392 g Dimethyltetrazin lieferten 0,57 10 g Benzaldazin; theoretisch wiireu fur 1 Mol. Hydrazin 0,4525g Benzaldazin x u erwarten.
Zur Bestimmung der Ess igs i inre wurde das Tetrazin mit verdiinnter Schwefelsiiure (spez. Gew. 1,22) gespalten und die entstandene Essigsiiure in etwas Wasser destilliert. Nach dem Neutralisieren mit verdunntem Ammoniak wurde die Essigsaure mit Silbernitrat als essigsaures Silber gefallt. Dieses gab, aus wenig heiBem Wasser umkrystallisiert , bei der Analyse folgende Zahleu :
Dimethyltetrazin loste sich spielend in der Slure.
0,0574 g hinterlieBen beim Gluhon 0,0372 g Ag. Berechnet fur C,H,O,Ag (166,9): Gefunden : Ab‘ 64,64 64,90 ’/”
0,1391 g Dimethyltetrazin liefertcn 0,0574 g essigsaures Silber. Be- rechnet fur 1 Mol. essigsaures Silber 0,1091 g . Sornit wurdo nur etwa die Hllfte der theoretisch ni6glichen Menge Essigsaure erhalten.
Als besonders charakteristisctt ist noch zu bemerken, daR beim Kochen von Oimethyltetrazin somohl mit verdunnter Salz- saure wie mit verdiinnter Schwefelsaure ein eigentumlicher, sii5- lich aromatischer, etwas den Atem nehmender Geruch auftrrzt.
Pr opioni t r i l , CH, . CH, , CN , und H y d r a z in.
D i a t h y 1-di h y dr o - t e t r a z i n , C,H, . Cps-”\ \C. C,H, \NH--NFI/
N--S und D i a t h y 1 -N - amino- t r i az 01, C,H, . C<,N >C. C,H, .
NH, Eine Mischung von 33,03 g Propionitril (600 MM.), 19,2 g
wasserfreiem Hydrazin (600 MM.) und 14,l ccm absolutem Al-

Muller u. Herrdegen: Einw. v. Hydrazin usw. 133 kohol wurde 4l/, Tagc am RiickfluBkiihler zum Sieden erhitzt. Der Alkohol wurde zugegeben, damit Nitril und Hydrazin eine homogene Fliissigkeit bildeten. Es entwickelte sich Ammoniak. Die Losung, die sich beim Erkalten schwach rotete, wurde beim Stehen im Vakuum fest. Das Rohprodukt bildete schwach gelb gefiirbte Nadelchen, die sich in Wasser leicht lijsten. Die Ausbeute betrug nur 2,6 g.
Beim Umkrystallisieren der Substanz aus Benzol wurden weiBe, seidenglanzende Niidelchen erhalten. Diese fkrbten sich beim Rehandeln mit Nntriumnitrit und Eisessig nicht mehr rot und waren bereits das . Umlagerungsprodukt von Diathyl- dihydrotetrazin, das Di a t h y l am i no t r i az o 1. Es ist leicht loslich in Wasser und Aikohol, schwer loslich in Ather unti schmilzt bei 1609 Dedichen'), der die gleiche Verbindung aus Propionitril und Hydrazinhydrat im Rohr bei 180" dar- gestellt hat , fand nach mehrmaligem Umkrystallisieren aus Essigester den Schmp. 167 9
0,1084 g gaben 0,2054 g CO, und 0,0849 g H,O. 0,1036 g gaben 36,O ccrn N bei 14" und 752,5 mm.
Berechnet fur C,H,,h", (140,14): Gefunden : C 51,38 51,68 "lo
N 3 9 9 9 40,24 ,, . Ii 8,63 8,76 7,
Die Benzollosung enthielt noch D i a t h y l d i hydro t e t r azin. Sie zeigte namlich bei der Oxydation mit Luft oder Stickstoff- trioxyd die schijn rote Tetrazinfarbe.
Ui i i thy l - te t raz in ,
Die wiiBrige Losung obigen Rohproduktes wurde mit Nitrit versetzt und mit verdiinnter Essigsaure angesauert. Die Fliissigkeit fiirbte sich sofort rot. Durch wiederholtes Aus- ziehen mit Ather wurde der wiiI3rigen Losung das entstandene Tetrazin entzogen. Beim Verdunsten der vereinigten atheri- schen Auszuge hinterblieb ein rotes, ziihflussiges 01. Es gelang nicht , durch Sublimation, durch Einstellen in Kaltemischung
l) Be:.. 39, 1855 (1006). journal f . prakt. Chemie [27 Rd. 102. 10

136 Miiller u. Herrdegen: Einw. v. Hydra& UHW.
I s o -Val e r o n i t r i l , (CH,),CH. CH, . CN, u n d H y d r az in.
Eine Mischung von 4,15 g Iso-Valeronitril (50 MM.), 1.6 g wasserfreiem Hydrazin (50 NU.) und 1,5 ccm absolntem Alkohol wurde 4 "age am RuckfluBkdhler gekocht. Es entwickelte sich -4mmoniak. Die Reabtionsflussigkeit wurde beim Stehen im Vakuum fest. Die erhaltene feste Substanz gab mit, Natrium- nitrit und Eisessig keine Rotfirbung mehr.
B e n z o n i t r i l , C,H,.CN, u n d H y d r a z i u .
D i p h e n y 1- d i 11 y d r o - t e t r a z i n 3,
Xine ;2lischung von 15g Benzonitril (145 MM.) und 4,65 g wasserfreiem Hydrazin (145 MM.) wurde auf dem siedenden Wasserbade 3l/, Tage erhitzt. Es entwickelte sich Ammoniak. Gegen Ende des Erhitzens verwandelte sich die Flussigkeit in einen aus gelben Nadelchen bestehenden Krystallbrei. Dieser wurde abfiltriert, mit etwas absolutem Alkohol ausgewaschen und im Vakuum getrocknet. Diphenyldihydrotetrazin ist leicht loslich in Ather und Benzol; schwer loslich in absolutem Al- kohol und in kaltem wie warmem Wasser. Es wurde aus ab- solutem A41kohol umkrystallisiert. Schmp. 19 1 O.
0,1520 g gaben 0,3959 g CO, uud 0,0709 g H,O. 0,1175 g gaben 24,O ccm N bei 17O und 751 mm.
Berechnet fur C,,H,,NI (236,14): Gefunden : c 71,16 71,04 Olio
IY ",73 23,s; ,, . 13 6,12 5,22 ,,
Beim Durchsaugen von Luft durch die alkoholische Xutter- lauge des Diphenyldihydrotetrazins schieden sicli infolge Oxy- dation blaustichig rote Prismen von Diphenyltetrazin ab.
Diphenyldihydrotetrazin bildete sich auch schoa in der Kalte aus einer Mischung von Benzonitril und Hydrazin. 3 g Benzonitril (29 NM.) uod 0,93 g wasserfreies Hydrazin wurden zusammengegeben und bei gewohnlicher Temperator stehen gelassen. Nach ca. 6 mochigem Stehen entwickelte sich Am-
') P i n n e r , Ann. Chem. 297, 258 (1897).

Miiller u. Her rdegen: Einw. v. Hydrazin iisw. t37
moniak. Zwecks besserer Einwirkung des Hydrazins auf das Nitril wurtle das Gemisch mehrere Tage auf der Schiittel- maschine geschiittelt. Nach weiterem 6 Wochen langem Stehen schieden sich gelbe Krystalle ab, die sich schon durch die geringste Luftzufuhr infolge Oxgdation ziim Diphenyltetrazin schSn blaustichig rot fkrbten.
,N - N, Dip h en y 1- t e t 1: a xin I), C,H,. C 4' *C . C,,H,
\N=N/
Die wa5rige Liisurrg von LXphenyldihydrotetrazia wurde niit Satriumnitiit versetzt und mit Eisessig Ltngesauert. Es trat sofort Rotfarbung ein. Mit Ather wurde drts entstandeue Tetrazin der Flussigkeit entzogen. Beim Verdunsten des &hers blieben blaustichig rote Prismen von Diphenyltetrazin zuruck. Dieses ist leioht lijslich in Ather und Benzol, schwer liislich in absolutem Alkohol, unloslich in Wasser. Es schmolz, aus absolutern Alkohol umkrystallisiert, bci 195 ".
0,1321 g gahen 0.3484 g CO, nnd 0,0527 g H,O. 0,1520 g gaben 32,l ccm X' bei 19" und 760 mm.
C 71,77 71,93
n- 23,97 24,17 ,, .
Berechilet fiir C,,H,,N, (234,12): Gefunden :
H 4,31 4146 , l
Die Ltlkoholische Losung von Diphenyltetrazin gab im sichtbaren Teil des Spektrums ein ganz verschwommenes Ab- sorptionsband, in dem nur zaei Streifen bei den Wellenlangen il 575-585 und I 570-400 etwas deutlicher hervortraten.
Hydrolyse . 0,2006 g Diphenyltetrazin wurden durch Kocheii mit 20 ccm cines Gemisches gleicher Teile konzen- trierter und rerdunnter SchwefeleHure in dem auf S. 131 be- schriebenen Apparat zersetzt. Nach kurzem Erhitzen war die rote Farbe des Tetrazins verschwuuden und die Substanz in Losuug gegangen. Der abgespaltene S t i cks tof f wurde auf- gefangen und gemessen.
0,2006 g gaben 10,O ccin N bei 13,0° und 762 mm. luatr i t t von N, aus 1 Mol. CI4HION4 (234,lZ).
Berechnet: 11,98 O/, N. Gefunden: 5,88 O1, N.
') l ' i i iner, Ann. Uhem. 297, 261 jlS97): K. 9. Hofmann 11. E h r - h a r t , Ber. 45, 2732 (1912).

140 Muller u. Herrdegen: Einw. $7. Hydrazin usw.
der Rotfhrbung infolge Oxydation des bei der Reaktion ent- standenen Dibenzyldihydrotetrazins zum zugehorigen Tetrazin. Letzteres konnte nicht krystallin erhalten werden.
10 g Benzylcyanid wurden mit 2," g wasserfreiem Hydrazin irn Rohr 3 Tage auf 100O erhitzt. Beim Offnen der Bombe war ziemlich starker Druck und Ammoniakgeruch vorhanden. Bus der Mischung krystallisierte Dibenzyl-N-aminotriazol u1s wei6e Substanz aus. Die Flussigkeit zeigte bei der Oxydation mit salpetriger SBure Rotfarbung. Auch hier gelang es nicht, das Dibenzyltetrazin zu fassen.
Or tho- , Meta- u n d P a r a - T o l u n i t r i l , CH3.C,H,.CN, u n d Hydraz in .
o-Tolunitril reagierte selbst bei noch so langem Erhitzeu nnter RuckfluB nicht mit wasserfreiem Hydrazin.
D i - p - t o 1 y 1 - d i h y d r o - t e t r a z i n l:,
35,l g p-Tolunitril (300 MM.j wurden mit 9,ti g wasser- freiem Hydrazin (300 MM.) 2l/, Tage unter RuckfluB im Paraffin- bad zum Sieden erhitzt. Es entwickelte sich Ammoniak. Nock wiihrend des Erhitzens erstarrte das Reaktionsprodukt zu einer gelben, krystallinen Masse. Diese wurde abfiltriert, mit Ather gewaschen, auf Ton gestrichen und im Vakuum getrocknet. Die Ausbeute betrug 27,39 g. Di-p - tolyldihydrotetrazin kry- staliisiert aus Benzol in feinen, langen , hellgelben , seiden- glanzenden Nadeln vom Schmp, 2234 Eine aus Eisessig um- krystallisierte Probe schmolz in Ubereinstimmung mit den Angaben von Pinne r2 ) erst bei 235,5O. Leicht liislich in warmem Eisessig, schwer in Alkohol und Beuzol, fast unliislich in Athe:. und Wasser.
0,1088 g gaben 0,2909 g CO, und 0,0669 g H,O. 0,1214 g gaben 23,O ccm N bei 16,0° uiid 748 mni.
Berechnet fur C16Hl,N, (264,17): Gefunden : C 72,08 72,92 O/,,
H 6,11 5,85 > )
N tL1,21 21,51 ,. .
I) Pinner, Ann. Chein. '298, 1 3 (1897). p, Ebeiida S. 14.

Miiller LI. Herrdegen: Einw. v. Hydraziri usw. 141
L) i - p - t 0 1 y 1 - t e t r az in l), ,N-N
C,H,.C” \C.C,H, . \N=N/
Kine wiifirige Suspension von Di - p - tolyldihydrotetrazin wurde mit Natriumnitrit versetzt und mit Eisessig angesiiuert. Ks trat Bot€arbung ein. Das entstandene Tetrazin wurde mit lhther aufgenommen. Beim Verdunsten des Athers schiedeii sich blaustichig rote, lange, schmale Prismen init rhombischer Begrenzung aus; die Ausliischung bildct mit der Langsrichtung der Krystalle einen Winkel yon 45O. Der Korper lijste sich lpicht in Ather und Benzol. Er schmolz bei 232O unzersetzt.
0,1052 g gaben 80 cein N bei 16,OO uud 748 mrn.
Uerechnet fur C,,H,,N, (262,15): Orfunden : N 21,37 21,68 o / o .
Die alkoholische Lijsung von Di-1)-tolyltetrazin zeigte im sichtbaren Spektrum ein einziges Absorptionsband POU der Wellenlange 1 518-585.
Di-13 - tolyldihydrotetrazin wurcle in Blkoiiol unter Er- n gymen gelijst. Durch die Losuug wurde unter fortgesetztem Krwgrmen Luft gesaugt; dabei ging die gelbe Farbe der Losung in Rot uber, und nach kurzer Zeit begann die Ausscheidung yon Di- p-tolyltetrazin.
Auch mit Salpetersaure oder mit Brom uud Eisessig lie8 4ch Di-p- tolyldihydrotetrazin zum Tetrazin oxydieren.
D i - m - t ol y 1 - dil ly d r o - t e t r a z i n .
Eine Mischung voii 5,S5 g ni-‘i’olunitril (50 MM.) und 1,6 g wasserfreiem Hydrazin (50 MM.) wurde 5 Stunden im Paraftin- bad am RiickfluBkuhler ZUM Sieden erhitzt. Die AuRentempe- ratur wurde zwischen 100-107 O gehalten. Es entwickelte sich Ammoniak. Beim Erkalten entstand ein gelber Krystall- brei, der abgesaugt und im Vakuum getrocknet wurde. Die Ausbeute an Di-m-tolyldihydrotetrazin betrug 1,25 g ; die Mutter-
‘) P i n n e r , Ann. Chem. 298, 17 ( !89i ) .

143 Muller u. Herrdegcn: Einw. v. Hydrazin usw. 0,0983 g gaben 0,2630 g CO, u1.d 0,0524 g H,O. 0,1102 g gaben 21,O ccm N bei H , O 0 und 758 mm.
Berechnet fur C,,H,,N, (264,lV: Gefuudeu: C 72,68 72,97 O l 0
11 6,11 5 9 6 ,, N 21,21 21,41 ., .
Di-m-to ly l - t e t r a z i n ,
Eine waBrige Suspension von Di-m - tolyldihydrotetrctzin wurde mit Natriumnitrit versetzt und mit Eisessig angesiiuert. Es trat sofort Rotfarbung ein. Die waBrige Suspension wurde mehrmals rnit hither ausgezogen. Beim Verdunsten des athe- rischen Auszugs blieben schone, rote Nadeln zuruck. Sie liisten sich spielend in Ather, wenig in lraltem absoiuten Al- kohol, leicht in warmem; in Wasser waren sie unloslich. Das Tetrazin schmolz hei 150-152 O. 0,5 g Dihydrotetrazin gaben O,4 g Tetrazin.
0,0977 g gaben 0,2635 g CO, und 0,0457 g H,O. 0,1120 g gaben 21,2 ccm N bei 22,O' iind 756 mm.
Berechnet fur C,,H,4N, (262,15): Gefonden: C 73,2P 73,54 'I,, H 5,38 5123 ) i
N 21,37 21,24 .. . Die alkoholische Losung des Ui-m- tolyltetrazim gab im
sichtbaren Spektrum ein verschwommenes Absorptionsband zwischen den Wellenlangen A 520-580.
Redukt ion . 0,2 g Tetrazin wurden in 50 ccm Ather gelost, mit Zinkstaub versetzt und die Atherlosung mit ca. 20 ccm 5 prozentiger Schwefelssure angesauert. Nach kurzem, kraftigem Umschiitteln der atherischen Lijsung war die rote Farbe des Tetrazins in die gelbe des Dihydrotetrazins um- geschlagen. Die Atherliisung wurde von der Saure getrennt und erstere im Vakuum zur Trockne gebracht. Es bBeb ein in gelben Nadelchen krystallisiertes Produkt zuruck, das bei 195-196O schmolz und mit salpetriger Saure wieder Di-m- tolyltetrazin zurucklieferte.

Niiller u. Herrdegen: Einw. v. Hydrazin usw. 145
Hydrolyse. Di-m-tolyltetrazin wurde mit einer Mischung gleicher Teile konzentrierter Schwefelsaure und Wasser durch Kochen zersetzt und der entwoichende St icks tof f quantitativ bestimmt.
I. 11.
0,862i g gaben l5,4 ccm h’ bei 19,0° und 751 m111
0,3308 g gaben 15,5 ccm N bei 16,0° und 760 mm. Berechnet fiir Gefundcn :
C,,H, ,N, (f?62,15) : I. 11. s 10,69 6,63 5,44 Qi0 .
Das Tetrazin 1oste sich beim Erwkmen in der Schwefel- saure zunachst nicht; war die Temperatur hoch genug, so schmolz es zu Oltriipfchen, die auf der Oberflache der Saure schwammen. I m Verlanf der Hydrolyse verschwand die schon rote Farbe, und die Substanz liiste sich in der Siiure auf. Die heiBe, hellgelbe schwefelsaure Losung war klar und durch- sichtig ; beim Erkalten triibte sie sich unter Abscheidong einee weiBen Niederschlags, der abfiltriert, mit Wasser gewaschen, getrocknet und gewogen wurde. Er erwies sich durch seinen Schmp. 110,5O und durch die Analyse als m-Toluyls%ure.
0,0717 g gaben 0,1859 g CO, und 0,0371 g H,O. Berechnet fur C,H,O, (136): Gefunden: C 70,59 70,71 O/,,
H 5,88 5979 1, *
1. 0,2627 g Tetrazin lieferten 0,1346 g m-Toluylsaure; berechnet
11. 0,3308 g Tetrazin lieferten 0,0864 g m-Toluyleaure; berechnet
Zur Bestimmung des H y d r a z i n s wurde bei Versuch I das schwefelsaure Filtrat von der m-Toluylsaure auf ein ldeines Volumen eingeengt und das Hydrazinsulfat mit absolutem Al- kohol ausgefallt, auf einem Filter gesammelt, getrocknet und gewogen. Bei Versnch I1 wurde das Hydrazin als Renzaldazin zur ’UTagung gebracht.
I. 0,2627 g Tetrazin gaben 0,1711 g Hydrazinsulfat; berechnet fiir 1 Mol. Hydrazin: 0,1171 g Hydrazinsulfat.
11. 0,3308 g Tetrazin gaben 0,1696 g Benzaldaziu; berechnet fur 1 Mol. Hydrazin: 0,2629 g Benzaldazin.
Bei der Priifung auf m-Toly la ldehyd wurde n u r mit m-Nitrobenzhydrazid eine geringe Fallung erhalten, deren Menge
fur 1 Mol. m-Toluylsaure: 0,1363 g.
fur 1 Mol. m-Toluylslure: 0,1716 g.

148 Miiller u. Herrdegen: Einw. v. Hydrazin usw.
2. Eine zweite Probe Tab rnit N e E l e r s Reagens den fur Ammon- salze chsrakteristischen Niederschlag.
3. Eine dritte Probe murde in einem Recherglaschen mit Calcium- hydrosyd iiberschichtet und mit einem Uhrglas bedeckt, an dcm ein Streifen rates Lackniuspapier befestigt war. Dieser bliiute sich stark in- folge Arnrnoniakentwicklung.
4. Die wlBrige LBsiing der Yubstanz wurde niit Yilbernitrat ver- setst, woranf ein meiBer , kiisiger Niedersclilag ausfiel. Er wurde ab- filtriert, getrocknet. iin Reagensglas erhitzt und die entweichenden Gase angeziindet. Die pfirpichblutrote Flamrne zeigte Cycngas an.
Die (fasentwicklung und Selbsterwarmung war in 3-4 Stun- den beendet. Das flussige Reaktionsprodukt wurde beim Stehen im Vakuum unter nochrnaliger, lebhafter Gasentwicklang fest,. E5 hinterblieb eine hellgelbe Masse, die beim Eiulassen von Luft in den Exsiccator rasch dunkelbraun wurde und ver- schmierte; sie wurde auf Ton gestrichen und im Vakuum ge- trocknot. Dieses Produkt sinterte bei 102-103O und schmolz bei 11 1 -1 12 O unter Schaarzwerden nnd Gasentwicklung. Eine Probe der Substanz wurde in verdunnter Essigsaure gelost und einige Krystallchen Natriuinnitrit zugegehen. Rasch auftretende Rotfarbung, die von Ather nufgenommen wurde, zeigte Tetrazin- bildung an.
Um die beim Zusammeabringen von Blnusaure und Hy- drazin zunachst entstehende weiBe Krystallmasse naher unter- suchen zu konnen, wurde wie folgt verfahren:
Zu einer Mischung von 20 CCM Hydrazin und 40 ccni kaltem, absolute& Alkohol lieB man unter Turbinieren uncl Kuhlen rnit Kiiltemischung ein Gemisch von 24 ccm wasser- freier Blausaiure und 24 ccm nbsolutem Alkohol allmahlich zu- flieBen. J3s entstanci eine schneeweif3e Krystallmasse , die unter Eiskuhlung abgesaugt und mit kaltem, absolutem Alkohol ausgewaschen wurde. Wegen ihrer Unbestandigkeit konnte die erhaltene Sulostanz nicht analysiert werden. Nur das Ver- haltnis von Hydrazin zu Blausaure lietj sich ermitteln. Zu diesem Zweck wurde eine kleine Menge der schneeweiflen Massc in einem 100 ccm-NaBkolbchen mit 35 ccm verdiinnter Schwefelsaure versetzt und bis zur Marke mit destilliertem Wasser aufgefullt. In j e 50 ccm dieser Losung wurde dav Hydrazin als Hydrazinsulfat und die BlausBure als Cyansilber

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 149
bestimmt. Beide Niederschlage wurden auf j e einem Filter gesammelt, getrocknet und gewogen. Erhalten wurden :
1. 3,6710 g NpH,, H,SO, -= 0,9100 g N,H,, entsprechend 24,79 Ot,, N,H,.
4,5480 g AgCS = 0,9179 g HCN, entsprechend 20,25 o/o HCN.
2 . 17,8243 g R,H,, H,SO, =3 4,3944 g N,H4, entsprechend
'12.6924 g AgCN = 4,6593 g HCN, entsprechend
24,66 N,H,.
20,18 (',/0 HCN.
Fiir (N,H,), 2 FICN wurden sich berechnen. 37,2 o/O KtH4 und 62,79 O l i o HCS
Fiir (N,H,), HCN wiirden sich berechnen: 54,4 O I i , N,H, und 45,76°,'0 HCS.
Wie aus diesm Bnalysen ersichtlich, kam auf 1 Mol. Hydrazin 1 Mol. Blausaure. Die weiRe Substanz war also das e i n f a c h e D i a m m o n i u r n s s l z der B l a u s a u r e , N,H,, BCN.
Die Hauptmenge der weiBen Substanz wurrle in einem weiten und langen Reagensglase im Wasserbade langsam er- wsrmt. Hei 1 7 O schmolz die Substanz, und es bildeten sich zwei iibereinander geschichtete Fliissigkeiten, von derien die obere diinnfliissiger war als die untere. Kiihlte man sie in einer Kaltemischung ab, so erstarrte itie untere lilige Schicht wieder vollkommen zu einer wei6en Krystallmasse. Aus der oberen Scbicht krystallisierte gleichfalls eine weiDe Substanz aus. Ein Tell nber blieb fliissig; er wurde abgcgossen und destilliert. Durch (leu Siedepurikt und die Bildung von Cyan- silber beim Versetzen des Destillates mit Silbernitratliisnrig wurde diese Fliissigkeit als Blausaure identifiziert. Der Rest der Fliissigkeit enthielt nach dem Abdestillieren der Blaussure noch etwas Alkohol.
Beim Erwiirmen auf 55O mischten sich die beiden Schichten vollstiindig, und man erhielt eine homogene Fliissigkeit, die beim Abkiihlen sich wieder in zwei Pchichten trennte. Aus der gelben, homogenen Fliissigkeit cntwich bei 55 Bmmoniak in Stromen. Nan sorgte durch Umriihren dafur, d a ~ die Tem- poratur nicht durch Selbsterwarmung hoher stieg als 55 O. Obne
Journal 1. prekt Chemie [2] Rd. 1013. 11

150 Miiller u. Herrdegen: Einw. v. Hydrazin usw.
weiter zu erwarmen, bewegte sich die Temperatur zwischen 55-57 O. Nach beendeter Ammoniakentwicklung wurde die Fliissigkeit in eine Schale gegossen. Nach etwa zehnstiindigem Stehen im Vakuum war der Inhalt der Schale zu einer festen, gelben Masse erstarrt.
Wurde die schneeweiBe Masse, aus Hydrazin und Blau- saure, nach dem Auswaschen rnit absolutem Alkohol noch mit kaltem, trockenem Ather gewaschen, so entstanden beim Schmelzen keine zwei Schichten. Durch den Ather war die obere Schicht vollstandig entfernt worden. Die Schmelze blieb immer klar und zeigte nicht die Triibungen, wie dies bei der aus zwei Schichten bestehenden Schmelze der Fall war.
Die weif3e Substanz wurde in einem nicht evakuierten Exsiccator sich selbst uberlassen. Sie war bald spontan zu einer gelbbraunen, oligen Flussigkeit geschmolzen, die lebhaft Ammoniak entwickelte. Kach mehrtagigem Steheu fing die Fliissigkeit allmiihlich an, zu einer hellgelben, festen Masse zu erstarren. Diese wurde abfiltriert, die Mutterlauge im Vakuum zur Troche gebracht und so nochmals etwas hellgelbe, feste Substanz erhalten.
Un te r suchung d e r hel lgelben Subs tanz . Die beim Stehen im Vakuum von der gelben, digen
Fliissigkeit zuruckgebliebene feste Substanz wurde durch Ab- pressen auf Ton vollig getrocknet. Dann wurde sie mit abso- lutem Alkohol zu einem dunnen Brei angeruhrt, wobei ein kleioer Teil in Losung ging; der Hauptteil blieb ungelost. Dieser wurde abgesaugt, mit absolutem Alkohol, dann mit trockenem Ather gewaschen und im Vakuum iiber Schwefel- siiure getrocknet. Die so praparierte Substanz schmolz bei 132-124O und war von ganz hellgelber Farbe.
Zur Analyse ward sie im Achatmorser HuBerst fein ge- pulvert, nochmals mit absolutem Alkohol ausgewaschen und wieder uber Schwefels%ure im Vakuum getrocknet. Die Ana- lyse gab folgende Werte:
I.
11.
111.
0,1205 g gaben 0,0932 g CO, und 0,0756 g H,O. 0,0998 g gaben 63,4 ccm N bei 19O und 758 mm. 0,1246 g gaben 0,0976 g CO, und 0,0792 g H,O. 0,0930 g gaben 57,5 ccm N bei 15,OO und 758 mm. 0,1024 g gaben 0,0601 g GO, und 0,0688 g H,O.

Miiller u. Herrdegen: Einw. v. Hydrazin usw. 151 Berechnet f i r Gefunden :
C 20,69 21,09 21,36 21,34 o/o H 6,89 7,03 7,11 7,30 ,)
C,H,N, (116): I. 11. 111.
N 72,42 72,50 72,80 - 7,
In der hellgelben Substanz lag somit ein Korper von der Bruttoformel C,H,N, Tor. Wahrscheinlich war die Verbindung (vgl. S. 128)
Dimethin-hydrazo-dihydrazon, NH, NH,
Die gelbe Substanz zersetzte sich im Licht spontan, und selbst beim Aufbewahren in einem geschlossenen Wageglas farbte sie sich unter starker Ammoniakentwicklung schmutzig braun. Diese Beobachtung veranlaBte uns zu folgendem Ver- such. In ein Wageglas wurde eine abgewogene Menge gelber Substanz gebracht, dann wurde auf das oflene Wageglaschen ein Glasdreieck und in dieses ein kleines, mit konzentrierter Schwefelsaure gefiilltes, gewogenes Tiegelchen gestellt. Das Ganze wurde nun in einen nicht evnlruierten Exsiccator ge- bracht. Am Rand des die Schwefelsaure enthaltenden Tiegels schieden sich weiBe Krystalle aus, die sich bei der Unter- suchung als ein Gemisch von Hydrazinsulfat und Ammon- sulfat erwiesen.
4,21 g Subatanz verloren 0,29 g an Gewicht, davon wurden 0,lS g von der konzentrierten Sehwefelszure angenommen. I m Wageglaschen blieben 3,92 g Subatanz von gelbhrauner Farbe und etwas schmieriger Besehaffeuheit zuruck. Durch Digerieren mit etwas verduuntem Alkohol wurde die braune Schmiere gelost und nach dem Absaugen und Aus- waachen mit absolutem Alkohol blieben 3,71 g der hellgelben, ursprung- lichen Substanz vom Schmp. 124O zuruck.
Die gelbe Verbindung zersetzte sich offenbar spoutan unter Abgabe von Hydrazin und Ammoniak, welche Zerfalls- produkte dann wieder weiter zersetzend auf die Substanz ein- wirkten. Urn daher die Verbindung unveriindert anfbewahren zu konnen, mufiten die Zersetzungsprodukte sofort entfernt werden, was am einfachsten dadurch geschah, daB man die Substanz im evakuierten Exsiccator uher Schwefelsaure auf-
11’

152 Mul le r u. Herrdegen: Einw. v. Hydrazin usw.
bewahrte. Die Verbindung kiWt sich nicht unzersetzt um- krystallisieren. Sie bildet ein an der Luft rasch verschmiereii- des Pulver, das sich leicht in Wasser liist; in den iibrigen gebrauchlichen Losungsmitteln ist es unlijslich. Die wiil3rige Liiclung reagiert stark alkalisch.
Benza lve rb indung . 1 g der gelben Substanz wurde in der 20 fachen Menge Wasser geltist, die alkalische Liisung bis eben zur sauren Reaktion mit verdiinnter SchwefelsLure an- gesauert, mit Benzaldehyd versetzt und kriiftig geschuttelt. Die Benzalverbindung schied sich hierbei als gelbe, anfangs etwas schmierige Afassc an den Wandungen des GefaBes ab. Beim Loskratzen mit einem Glasstabe wurde sie vollkommen fest. Sie wurde abgesaugt und das Filtrat abermals mit Benz- aldehyd versetzt, die erneut gebi1det.e Benzalverbindung wieder abfiltriert und dies Verfahren so lange fortgesetzt, bis auf weiteren Zusatz Ton Benzaldehyd zum Filtrat kein Nieder- schlag mehr entstand, Die Benzalverbindung wurde auf t' m e m Tonteller getrocltnet und zur Entfernung von Benzaldazin so lange mit trockenem Ather ausgezogen, bis dieser vollkommeii farblos ablief. Sie begann danach beim Erhitzen im Schmelz- riihrchen bei 94O zu erweichen und schmolz bei 99O zu oiner klaren Fliissigkcit zusammen.
1 g hellgelhe Subvtanz gaben 2.24 .g Benzalprodukt; von letzterem waren 1,9 g in Ather 16slich. 0,32 g blieben ungelost. Die Ausbeute an
D i b c n z u 1 - d i m c t h i n - 11 y d P BZ o - d i h y d P B 1; o n , C,H,. CH: ?i JS :CH. C,H,
war somit sehr gerivg.
der Benzaiverbindung nicht ganz sicher zu ermitteln. Aus den erhaltenen Analysenresultaten war die Formel
I. 0,0971 g gaben 0,2198 g CO, und 0,0518 g H,O. 0,1101 g gaben 25,3 ccm N bei 18,O" und 743 mni.
11. 0,1002 g gaben 0,2269 g CO, und 0,0536 g H,O. 0,1256 g gaben 28,9 ccm h' bei 21,0° und 762 mm.
111. 0,1052 g gaben 0,2416 g CO, und 0,0564 g H,O. 0,1032 g gaben 25,l cem N bei 13,0° und 757 mm.
1V. 0,0988 g gaben 0,2346 g CO, und 0,0552 g H,O.

Miiller u. Herrdegen: Eiiiw. v. Hydraziii usvv. 153 Berechuet fur Qefunden :
cJJ18NL3: 1. TI. 111. IV. C 65,75 61,74 61,76 62,64 64,76 O / ,
H 5,48 5,97 5,9S .5,99 6,25 ,, s 28,77 25,82 26,15 25,49 - ,,
X- Amino - t r i az 01.
Die schiieeweiBe Krystallmasse aus Hydritzin snd Blau- siiure wurde geschmolzen und so hoch erhitzt, bis samtliches hiiimoniak entwichen war und ein eingetauchtes Thermometer 120° zeigte. Die hellgelbe Flussigkeit wurde im Vakuum er- kalten gelassen. D a m wurde die feste Substanz auf Ton ge- strichen, um sie vollkommen trocken zu erhalten. Hierauf wurde sie in wenig absolutem Alkohol gclijst. Reim Versetzen der alkalischen Losung mit trockenem Ather trat zuerst niilchige Trubung ein, und nach kurzer Zeit schieden sich feine! weiBe , seidenglanzencle, zu Buscheln vereiuigte Nadeln aus, die sich mit dem von Cur t iu s , D a r a p s k y und Mul1er1) fruher dargestellten N- Aminotriazol als identisch eraiesen. Schmp. 83O.
1) itr s t e 11 u n g d e s D i h y dr o t e t r a z i n s.
Die hellgelbe Substanz wnrde iu verdunnter Essigskure gelast und mit Natriumnitrit rersetzt, worauf Kotfkrbung in- folge Bildung von Tetrazin eintrat. Uieses wurde der essig- sauren Losung durch funfzehnnialiges husziehen mit Ather entzogen; die Btherische Lijbung wurde rnit Wasser gewaschen und danii niit weuig Kaliumcarbonat geschuttelt. In die so behandelte Btherische Losung wurde nach Zngabe yon 5 ccm Wasser so lange Schwefelwasserstoffgas eingeleitet bis Ent- farbung eingetreten war. Die entfarbte L6sung wurtle im Vakuum iiber Schwefelsiiure eingedunstet, der Ruckstand mit wenig Wasser aufgenommen und von neuem im Vakuum zur Trockne gebracht. Es hinterblieb ein hellgelbes, zahes 61, das nicht fest wurde. Diesem nwrde mit Benzol das Dihydro- tetrazin entzogea, das nur in Spuren rorlianden war. Auf Zugabe yon etwas Natriumnitrit und cerdunnter Essigsiiure
’) Ber. 40, 837 (1907).

164 Muller u. Herrdegen: Einw. v. Hydrazin usw.
zur Benzollijsung trat sofort wenn auch schwache Rotfarbung, infolge Bildung von Tetrazin, ein.
Der in Benzol unliisliche Teil des hellgelben Oles gab beim Behandeln rnit Phenylisocyanat eine feste, schwach gelb- lich gefarbte Verbindung. Da wir auf dem soeben beschrie- benen Weg das gewiinschte Dihydrotetrazin in fester Form nicht erhalten konnten, versuchten wir, auf folgende Weise zu ihm zu gelangen:
Die hellgelbe Substanz wurde in ganz verdunnter Essig- saure (1 : 5) gelost, 80 dai3 die Losung eben sauer reagierte und etwas Natriumnitrit zugegeben. Die Liisung farbte sich durch entstandenes Tetrazin sofort rot. Durch Ausschiitteln mit &her wurde es der Pliissigkeit entzogen. Nun wurde neues Natriumnitrit zugefugt , wobei erneut Rotfarbung eintrat, die wieder mit Ather aufgenommen wurde. Dieses Verfahren wurde so lange fortgesetzt, bis sich der Ather nur noch schwach rosa farbte. Die schwach saure atherische Liisung wurde nun mit Zinkstaub kraftig durchgeschiittelt , bis Entfarbung ein- getseten war. Die Saure wurde durch Behandeln der atheri- schen Losung mit Zinkoxyd abgestumpft und schlie5lich die Atherlosung mit Chlorcalcium getrocknet. Die so behandelte atherische Flussigkeit wurde im Vakuum eingedunstet. Es blieben hellgelbe Krystillchen von Dihydrotetrazin zuruck, die sich mit Natriumnitrit und verdiinnter Essigsaure wieder in Tetrazin l) zuruckverwandeln lieBen.
Die atherische Losung des freien Tetrazins zeigte im sichtbaren Spektrum 3 Absorptionsbanden im Grun von den Wellenlangen :
1. Rande: Wellenliingen I 510-5'20. 2. ,. ! 1 530-543. 3. 3 , ,, 2.553-665.
Das Dihpdro te t r az in wurde in Benzol gelost und die Lasnng im Vakuum eingedunstet. Zur Analyse wurden die Krystallchen auf Ton gestrichen und noch kurze Zeit ins Vakuum gebracht.
I. 11.
0,0781 g gaben 45,5 ccm N bei 19,OO und 753 mm. 0,005986 g gaben 1,190 ccm S hei lti,O0 11. 754 mm (nach Pregl ' .
') C u r t i n s , Darapsky und M u l l e r , Rer. 40, 84 [1907).

Muller u. Herrdegen: Einw. v. Hydmzin usw. 155 Berechnet fur Gefuund en : C,H,N, (84): I. 11.
s 66,66 66,14 66,78 o/:o . Obiges Dihydrotetrazin schmolz zwischen 1 17-1 19 ", Cur-
t i u s und seine Mitarbeiter fanden 125-126O. Sehr wahr- scheinlich war das von uns dargestellte Dihydroprodukt nicht vollkommen rein; die Ausbeute war ebenfalls sehr gering, was vermuten lieB, daB die Rildung des Dihydrotetrazins nur eine Nebenreaktion darstellte, die Hauptreaktion aber in ganz an- derer Richtung verlief.
Die waBrige essigsaure Losung, der das Dihydrotetrazin entzogen ward, wurde mit Natriumnitrit neutralisiert und auf dem Wasserbade zur Trockne gebracht. Der Eindampfruck- stand wurde mehrfach mit Renzol ausgezogen und die ver- einigten Benzolauszuge im Vakuum eingedunstet. Hierbei blieben weiSe, seidenglanzende Nadelchen zuruck, die sich mit dem sclion fruher in gleicher Weisel) erhnltenen T r i a z o l als identisch erwiesen. Schmp. 120,5 ".
Dws .von uns dargestellte Dihydrotetrazin zeigte die v m Cur t iu s , Darapsky und Miil ler angegebenen Eigenschaften 9. Die w&Brige Losung wirkte stark reduzierend, indem sie leicht unter Sauerstoffaufnahme in Tetrazin verwandelt wurde. Silber- nitratlosung wurde rasch unter Rotfarbung der Losung re- duziert.
I) C u r t i u s , Darapsky u. Miiller, Ber. 40, 836 (1907). 2, Ber. 40, 837 (1907).






![Electrosynthesis and Mechanism of Copper(I) Nitrile Complexes5.3.1.4. Synthesis of trans-[Cu2(µ-CN)(PPh3)2(bipy)2][BF4]·THF (4) Malononitrile as starting material ... Since the well-known](https://img.pdfslide.org/doc/110x75/5fdb86ebda8ea5648f6f4af4/electrosynthesis-and-mechanism-of-copperi-nitrile-complexes-5314-synthesis.jpg)












![Einfluss von Wasser auf die Kohlenstofffaser/Epoxid ...elib.suub.uni-bremen.de/edocs/00103654-1.pdf · auf der Oberfläche der C-Faser hervorgerufen werden [10–13]. Die Einwirkung](https://img.pdfslide.org/doc/110x75/5e15453b4e4920504e4f7328/einiuss-von-wasser-auf-die-kohlenstoifaserepoxid-elibsuubuni-auf-der.jpg)









