Embed Size (px)
Citation preview

Entwicklung eines bakteriellen Stammes zur Produktion
des kompatiblen Solutes Mannosylglycerat
Dissertation zur
Erlangung des Doktorgrades (Dr. rer. nat.)
der
Mathematisch-Naturwissenschaftlichen Fakultät
der
Rheinischen Friedrich-Wilhelms-Universität Bonn
vorgelegt von
Amal Burdziak aus
Jerusalem
Bonn 2006

Angefertigt mit Genehmigung der Mathematisch-Naturwissenschaftlichen
Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn
1. Referent: Prof. Dr. Erwin A. Galinski
2. Referent: PD Dr. Hans Jörg Kunte
Tag der Promotion: 3. November 2006
Diese Dissertation ist auf dem Hochschulschriftenserver der ULB Bonn
http://hss.ulb.uni-bonn.de/diss_online elektronisch publiziert.
Erscheinungsjahr: 2006

FÜR DANIEL
9`n«Ç †« Ôr −ü
„A mind that has been stretched will never return to its original dimension.” - Albert Einstein

Abkürzungen und Trivialnamen
Abb. Abbildung
ABC ATP-Binding-Cassette
As Aminosäuren
Amp Ampicillin
AmpR Ampicillin-Resistenz(gen), Ampicillin-resistent
APS Ammoniumpersulfat
ATP Adenosintriphosphat
BCA 4,4’-Bichinolin-2,2’-Dicarbonsäure
BCC-Transporter Betain/Carnitin/Cholin-Transporter
BCIP 5-Bromo-4-chloro-3-indolyhosphat
Betain Glycin-Betain
bp Basenpaar / Basenpaare
BSA Bovine serum albumine (Rinderserumalbumin)
Carb Carbenicillin
CarbR Carbenicillin-Resistenz(gen), Carbenicillin-resistent
cBPG zyklisches 2,3-Bisphospho-Glycerat
cDNA komplementäre DNA
CDP* Dinatrium-4-chloro-3-[methoxyspiro{1,2-dioxetan-3,2´-
(5´-chloro) tricyclo[3.3.1.13,7] decan}-4-yl]-phenylphosphat
CIAP (Kalbs-) intestinale alkalische Phosphatase
Cm Chloramphenicol
CmR Chloramphenicol-Resistenz(gen), Chloramphenicol-resistent
CmS Chloramphenicol-sensitiv
DABA L-2,4-Diaminobuttersäure
DC Dünnschichtchromatographie
DEPC Diethylpyrocarbonat
DGP Di-Glycerol-Phosphat
DIG Digoxigenin
DIP Di-myo-Inositol-1,1’-Phosphat
DMSO Dimethylsulfoxid
dNTP Desoxynukleotidtriphosphat
D2O deuteriertes Wasser
DSM Deutsche Stammsammlung für Mikroorganismen
DTT Dithiothreitol
Ectoin 1,4,5,6-Tetrahydro-2-methyl-pyrimidin-4-carbonsäure
EDTA Ethylendiaminotetraessigsäure
g Fallbeschleunigung
h Stunde
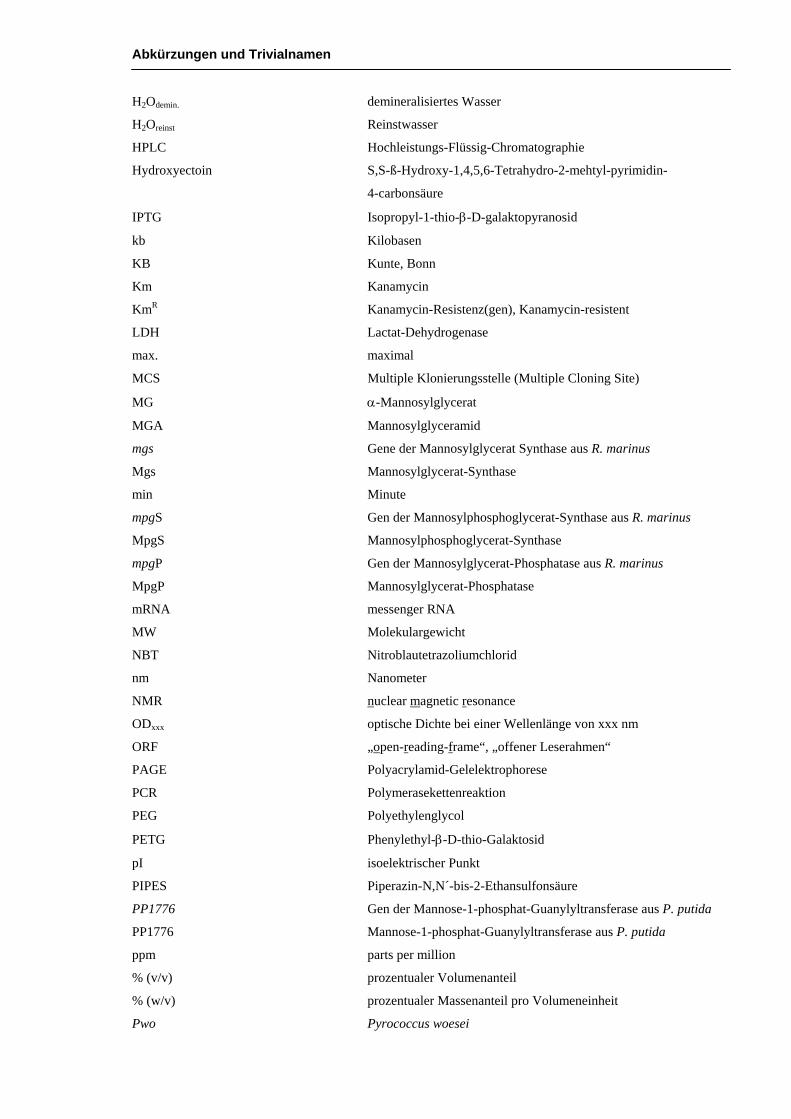
Abkürzungen und Trivialnamen
H2Odemin. demineralisiertes Wasser
H2Oreinst Reinstwasser
HPLC Hochleistungs-Flüssig-Chromatographie
Hydroxyectoin S,S-ß-Hydroxy-1,4,5,6-Tetrahydro-2-mehtyl-pyrimidin-
4-carbonsäure
IPTG Isopropyl-1-thio-β-D-galaktopyranosid
kb Kilobasen
KB Kunte, Bonn
Km Kanamycin
KmR Kanamycin-Resistenz(gen), Kanamycin-resistent
LDH Lactat-Dehydrogenase
max. maximal
MCS Multiple Klonierungsstelle (Multiple Cloning Site)
MG α-Mannosylglycerat
MGA Mannosylglyceramid
mgs Gene der Mannosylglycerat Synthase aus R. marinus
Mgs Mannosylglycerat-Synthase
min Minute
mpgS Gen der Mannosylphosphoglycerat-Synthase aus R. marinus
MpgS Mannosylphosphoglycerat-Synthase
mpgP Gen der Mannosylglycerat-Phosphatase aus R. marinus
MpgP Mannosylglycerat-Phosphatase
mRNA messenger RNA
MW Molekulargewicht
NBT Nitroblautetrazoliumchlorid
nm Nanometer
NMR nuclear magnetic resonance
ODxxx optische Dichte bei einer Wellenlänge von xxx nm
ORF „open-reading-frame“, „offener Leserahmen“
PAGE Polyacrylamid-Gelelektrophorese
PCR Polymerasekettenreaktion
PEG Polyethylenglycol
PETG Phenylethyl-β-D-thio-Galaktosid
pI isoelektrischer Punkt
PIPES Piperazin-N,N´-bis-2-Ethansulfonsäure
PP1776 Gen der Mannose-1-phosphat-Guanylyltransferase aus P. putida
PP1776 Mannose-1-phosphat-Guanylyltransferase aus P. putida
ppm parts per million
% (v/v) prozentualer Volumenanteil
% (w/v) prozentualer Massenanteil pro Volumeneinheit
Pwo Pyrococcus woesei
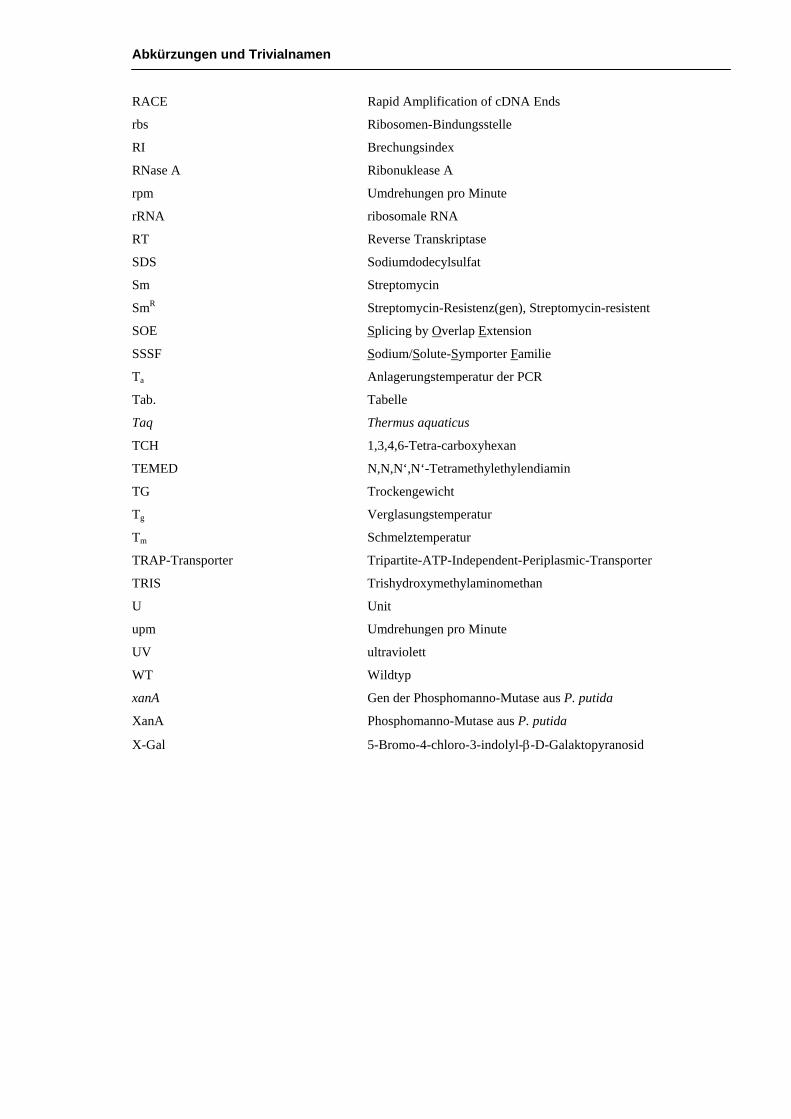
Abkürzungen und Trivialnamen
RACE Rapid Amplification of cDNA Ends
rbs Ribosomen-Bindungsstelle
RI Brechungsindex
RNase A Ribonuklease A
rpm Umdrehungen pro Minute
rRNA ribosomale RNA
RT Reverse Transkriptase
SDS Sodiumdodecylsulfat
Sm Streptomycin
SmR Streptomycin-Resistenz(gen), Streptomycin-resistent
SOE Splicing by Overlap Extension
SSSF Sodium/Solute-Symporter Familie
Ta Anlagerungstemperatur der PCR
Tab. Tabelle
Taq Thermus aquaticus
TCH 1,3,4,6-Tetra-carboxyhexan
TEMED N,N,N‘,N‘-Tetramethylethylendiamin
TG Trockengewicht
Tg Verglasungstemperatur
Tm Schmelztemperatur
TRAP-Transporter Tripartite-ATP-Independent-Periplasmic-Transporter
TRIS Trishydroxymethylaminomethan
U Unit
upm Umdrehungen pro Minute
UV ultraviolett
WT Wildtyp
xanA Gen der Phosphomanno-Mutase aus P. putida
XanA Phosphomanno-Mutase aus P. putida
X-Gal 5-Bromo-4-chloro-3-indolyl-β-D-Galaktopyranosid

Abbildungsverzeichnis
Abb. 1: Kompatible Solute thermophiler und hyperthermophiler Organismen S. 9
Abb. 2: Postulierte Biosynthesewege für Mannosylglycerat in R. marinus S. 11
Abb. 3: Zuckernachweis nach (Molisch & Biebl, 1955) S. 32
Abb. 4: Schematisch dargestellter Ablauf der DDRT-PCR S. 61
Abb. 5: Abhängigkeit der Wachstumsrate von R. marinus von der M162-Mediensalinität S. 77
Abb. 6: Wachstumsraten von R. marinus in MM162-Medium mit und ohne Zusatz von Soluten S. 79
Abb. 7: Abhängigkeit des zytoplasmatischen Mannosylglycerat-Gehalts von steigender Mediensalinität S. 80
Abb. 8: Restriktionskarte des Plasmids pThia1 und der Subklone pThiaEco, pThiaPst und pThiaSma S. 83
Abb. 9: Funktionelle Komplementierung der Thiamin-Auxotrophie von E. coli DH5α S. 83
Abb. 10: Strategie zur Sequenzierung der Plasmide pThiaEco und pThia1 S. 84
Abb. 11: Genetische Organisation der Thiamin-Gene aus R. marinus S. 85
Abb. 12: Sequenzvergleich ausgesuchter ThiG-ähnlicher Proteinsequenzen S. 86
Abb. 13: GA-reiche Sequenzen mit möglichen Ribosomen-Bindungsstellen S. 87
Abb. 14: Genetische Organisation des otsAB-Operons in E. coli MKH13 und E. coli BKA-13 S. 90
Abb. 15: Ableitung degenerierter Primer zur Verwendung in einer heterologen PCR S. 93
Abb. 16: Sequenzierungsstrategie des hypothetischen SSSF-Transporters aus R. marinus S. 95
Abb. 17: Hydropathie-Plot von SSSF aus R. marinus nach Kyte & Doolittle (1982) S. 97
Abb. 18: Sequenzvergleich von Natrium/Glucose-Symportern S. 99
Abb. 19: Elektrophoretische Auftrennung der DDRT-PCR-Produkte S. 101
Abb. 20: Sequenzierungsstrategie für das DDRT-PCR-Produkt dd2 S. 101
Abb. 21: Wachstumsraten von H. elongata KB1 und KB18.1 in Mineralsalzmedium S. 104
Abb. 22: 13C-NMR Spektrum des Zellextrakts von H. elongata KB1 S. 105
Abb. 23: Genetische Organisation der mgs-Region von R. marinus DSM 4252T und H. elongata KB10 S. 107
Abb. 24: Wachstum von H. elongata KB10 unter Zusatz unterschiedlicher C-Quellen S. 108
Abb. 25: Nachweisgrenze von Mannosylglycerat in einer Dünnschichtchromatographie (DC) S. 109
Abb. 26: Untersuchung der Zellextrakte von KB10 auf Vorhandensein von Mannosylglycerat S. 109
Abb. 27: Northern-Hybridisierung mit mgs- und ectB-Gensonden S. 111
Abb. 28: Nachweis der mgs-Transkription auf H. elongata KB10 (∆ectA::mgs) RNA mittels RT-PCR S. 112
Abb. 29: Gelelektrophoretische Auftrennung des RACE-PCR Produkts auf H. elongata KB10 cDNA S. 113
Abb. 30: Identifizierung eines Transkriptionsstartpunkts stromaufwärts von mgs in H. elongata KB10 S. 114
Abb. 31: mgs-Promotorregion von H. elongata KB10 S. 115
Abb. 32: Verlängerte DNA- und Aminosäuresequenz von mgs aus R. marinus S. 117
Abb. 33: Klonierungsstrategie zur Herstellung des Expressionsvektors pABB2 S. 118
Abb. 34: Nachweis von Mgs-(His)6 in Zellen von H. elongata Wildtyp und KB1 S. 119
Abb. 35: Sequenzänderungen in der Insertionsmutante H. elongata KB10.1 im Vergleich zum Wildtyp S. 122
Abb. 36: Genetische Organisation der PP1776xanA-Region S. 123
Abb. 37: Northern-Hybridisierung mit mgs, PP1776 und xanA-RNA-Sonden S. 125
Abb. 38: Untersuchung der mgsPP1776xanA-Transkription mittels RT-PCR S. 127
Abb. 39: Wachstum von H. elongata Stämmen bei Nutzung unterschiedlicher Kohlenstoffquellen S. 129
Abb. 40: Nachweis von Mannosylglycerat in Zellextrakten von H. elongata KB10.1 mittels DC S. 129
Abb. 41: Chromatogramm der Auftrennung von Mannosylglycerat S. 130
Abb. 42: Mannosylglycerat-Gehalt von H. elongata KB10.1 bei Nutzung unterschiedlicher C-Quellen S. 131

Abbildungsverzeichnis
Abb. 43: Akkumulation von Mannosylglycerat durch H. elongata KB10.1 S. 132
Abb. 44: Wachstum von H. elongata KB10.1 bei 40 °C S. 133
Abb. 45: 13C-NMR-Spektrum des Zellextrakts von H. elongata KB10.1 S. 134
Abb. 46: Regulationsmodell der Mannosylglycerat-Synthese von Rhodothermus marinus Wildtyp (a)
und einer Mannosylglycerat-Transport defekten Variante (b) S. 137
Abb. 47: Vergleich der Primersequenzen mit der Sequenz ihrer Bindungsstelle S. 145
Abb. 48: Etablierung einer Mannosylglycerat-Synthese in einem E. coli Stamm S. 151
Abb. 49: „Codon usage“ von R. marinus im Vergleich zu E. coli und H. elongata S. 154
Abb. 50: Sequenzvergleiche der identifizierten Promotorelemente S. 161
Abb. 51: Fructose- und Mannose-Metabolismus von P. putida KT2440 S. 164

Inhaltsverzeichnis
I. Einleitung .........................................................................................................................1 1. Extremophile Organismen................................................................................................................1 2. Thermophilie.....................................................................................................................................2
2.1 Halotolerante thermophile und hyperthermophile Mikroorganismen.........................................2 2.2 Anpassung an ein Leben bei hohen Temperaturen ..................................................................3
3. Akkumulation osmoregulatorischer Solute ......................................................................................4 3.1 Strategien der Osmoregulation..................................................................................................4
3.1.1 Akkumulation anorganischer osmoregulatorischer Solute .............................................5 3.1.2 Akkumulation organischer osmoregulatorischer Solute .................................................5
3.2 Osmoregulation des halophilen Stamms Halomonas elongata.................................................6 3.3 Osmoregulation thermophiler Mikroorganismen........................................................................7
3.3.1 Akkumulation spezieller kompatibler Solute ...................................................................7 3.3.2 Biosynthese von α-Mannosylglycerat in Rhodothermus marinus ..................................9
4. Kompatible Solute zur Stabilisierung von Biomolekülen................................................................11 5. Kompatible Solute: Nutzen und wirtschaftliche Bedeutung ...........................................................13 6. Ziel der Arbeit .................................................................................................................................15
II. Material und Methoden ..................................................................................... 17 1. Verwendete Bakterienstämme und Plasmide................................................................................17 2. Nährmedien....................................................................................................................................19
2.1 Medien zur Anzucht von R. marinus ........................................................................................19 2.2 Medien für Experimente mit E. coli und H. elongata................................................................20 2.3 Medien zur Transformation von E. coli Stämmen...................................................................21 2.4 Medien für die Herstellung chromosomaler Mutationen bei H. elongata.................................21 2.5 Medium zur Anzucht von Pseudomonas putida ......................................................................22 2.6 Medienzusätze.........................................................................................................................22
3. Puffer und Lösungen......................................................................................................................25 4. Kultivierungsverfahren ...................................................................................................................27
4.1 Stammhaltung von Bakterienstämmen....................................................................................27 4.2 Bakterienanzucht .....................................................................................................................28
4.2.1 Allgemeine Bakterienanzucht und Zellernte .................................................................28 4.2.2 Anzucht von Rhodothermus marinus............................................................................28 4.2.3 Anzucht von H. elongata ...............................................................................................29 4.2.4 Anzucht von E. coli Stämmen .......................................................................................30
5. Analytik und Nachweis kompatibler Solute ....................................................................................30 5.1 Gefriertrocknung ......................................................................................................................30 5.2 Dünnschichtchromatographie zum Zuckernachweis...............................................................30
5.2.1 Anzucht der Kulturen und Probenaufbereitung.............................................................31 5.2.2 Auftrennung von Zuckern und Mannosylglycerat .........................................................31
5.3 Hochleistungsflüssigkeitschromatographie (HPLC) ................................................................32 5.3.1 Probenaufbereitung für die HPLC-Analyse...................................................................32 5.3.2 Isokratische Hochleistungsflüssigkeitschromatographie ..............................................33
5.4 Kernresonanz-Spektroskopie („nuclear magnetic resonance“) NMR......................................34 5.4.1 Probenaufbereitung für 13C- NMR-Spektroskopie ........................................................35 5.4.2 NMR-Messung ..............................................................................................................35
6. Isolierung, Reinigung und Quantifizierung von DNA .....................................................................35 6.1 Isolierung von Gesamt-DNA ....................................................................................................35
6.1.1 Minipräparation von Gesamt-DNA (Marmur, 1961)........................................................35 6.1.2 Isolierung genomischer DNA mit Genomic-tip 100/G (Qiagen) .....................................37
6.2 Isolierung von Plasmid-DNA....................................................................................................37 6.2.1 Plasmid-Mini-Prep (Qiagen) ...........................................................................................37 6.2.2 Plasmid-Midi-Kit (Qiagen)...............................................................................................37
6.3 Reinigung, Konzentrierung und Quantifizierung von DNA ......................................................37

Inhaltsverzeichnis
6.3.1 Reinigung von DNA aus Agarosegelen (QIAquick Gelextraktionskit, Qiagen) ..............37 6.3.2 Reinigung von DNA durch Phenol/Chloroform-Extraktion..............................................38 6.3.3 Reinigung und Konzentrierung von DNA mit Microcon-Säulchen..................................38 6.3.4 Aufreinigung von PCR-Produkten ..................................................................................38 6.3.5 Konzentrations- und Reinheitsbestimmung von DNA ....................................................39
7. Enzymatische Modifikation von DNA .............................................................................................39 7.1 Restriktionsverdau von DNA....................................................................................................39 7.2 Dephosphorylierung von Vektor-DNA......................................................................................40 7.3 Abbau überhängender 3’-Enden, Herstellung glatter Enden...................................................40 7.4 Ligation.....................................................................................................................................41
8. Transformation von E. coli .............................................................................................................41 8.1 Herstellung superkompetenter Zellen von E. coli (Inoue et al., 1990).....................................41 8.2 Transformation superkompetenter Zellen von E. coli ..............................................................42
9. Polymerasekettenreaktion (PCR) ..................................................................................................42 9.1 Standard-PCR-Reaktion ..........................................................................................................43 9.2 Spezielle PCR-Techniken........................................................................................................43
9.2.1 In situ oder Kolonie-PCR ................................................................................................43 9.2.2 Ligase-vermittelte bzw. inverse-PCR .............................................................................44 9.2.3 Verschachtelte (nested)-PCR.........................................................................................44 9.2.4 Heterologe PCR unter Einsatz von degenerierten Primern............................................45 9.2.5 SOE-PCR (splicing by overlap extention).......................................................................45 9.2.6 Herstellung DIG-markierter DNA-Sonden mittels PCR ..................................................46
9.3 Verwendete Primer ..................................................................................................................46 10. Elektrophoretische Methoden zur Auftrennung von DNA.............................................................49
10.1 Agarosegelelektrophorese.....................................................................................................49 10.2 Southern-Hybridisierung ........................................................................................................50
11. RNA-Arbeitstechniken...................................................................................................................52 11.1 RNA-Isolierung und Aufreinigung ..........................................................................................52 11.2 Bestimmung der RNA-Konzentration.....................................................................................53 11.3 Gelelektrophoretische Auftrennung von RNA........................................................................53 11.4 Northern-Hybridisierung.........................................................................................................53
11.4.1 RNA Agarosegelelektrophorese - Denaturierung mit Glyoxal und DMSO...................54 11.4.2 Northern-Transfer .........................................................................................................55 11.4.3 Hybridisierung und immunologischer Nachweis der RNA-Hybride..............................55 11.4.4 In vitro-Transkription zur Herstellung einer Digoxigenin-markierten RNA-Sonde........56
11.5 Reverse Transkriptase-vermittelte Methoden........................................................................57 11.5.1 Reverse Transkriptase-vermittelte PCR.......................................................................57 11.5.2 5’-Race-PCR (rapid amplification of cDNA ends).........................................................57 11.5.3 Entwicklung einer Differential Display-RT-PCR Strategie für R. marinus ....................59
12. Konjugation ...................................................................................................................................62 13. Herstellung chromosomaler Mutationen .......................................................................................62
13.1 Mutagenese an H. elongata...................................................................................................63 13.2 Mutagenese an E. coli MKH13 ..............................................................................................63
14. Erstellung einer partiellen Genbank mit R. marinus DNA.............................................................65 13.1 Vektor pHSG575....................................................................................................................65 13.2 Vektor pXMJ19.......................................................................................................................65
15. Proteinbiochemische Methoden....................................................................................................66 15.1 Verwendetes Expressionssystem..........................................................................................66 15.2 Zellaufschluss und Isolierung von Zellfraktionen...................................................................66
15.2.1 Anzucht plasmidtragender H. elongata Stämme..........................................................66 15.2.2 Gewinnung des Gesamtzellproteins.............................................................................66 15.2.3 Gewinnung der periplasmatischen Fraktion durch osmotischen Schock.....................67 15.2.4 Isolierung von löslicher und unlöslicher zytoplasmatischer Fraktion............................68
15.3 Proteinbestimmung ................................................................................................................68 15.4 Diskontinuierliche Polyacrylamid-Gelelektrophorese unter denaturierenden........................
Bedingungen (SDS-PAGE)....................................................................................................69 15.5 Western-Hybridisierung .........................................................................................................71
16. Sequenzierung von DNA...............................................................................................................72 17. Sequenzanalysen..........................................................................................................................72 18. Verwendete Chemikalien ..............................................................................................................73

Inhaltsverzeichnis
III. Ergebnisse ..................................................................................................................76
1. Physiologische Untersuchungen an Rhodothermus marinus ........................................................77 1.1 Untersuchungen zum salinitätsabhängigen Wachstum ..........................................................77 1.2 Untersuchung des Substratspektrums von R. marinus ...........................................................78 1.3 Wachstum in Gegenwart externer kompatibler Solute ............................................................79 1.4 Abhängigkeit des Mannosylglycerat-Gehalts von der Mediensalinität ....................................80
2. Molekularbiologische Untersuchungen an Rhodothermus marinus ..............................................81 2.1 Erstellung einer Genbank von R. marinus ...............................................................................81 2.2 Identifizierung der Thiamin-Gene aus R. marinus durch funktionelle Komplementierung
von E. coli DH5α ......................................................................................................................81 2.2.1 Komplementierung von E. coli DH5α .............................................................................81 2.2.2 Subklonierung von pThia1 ..............................................................................................82 2.2.3 Sequenzierung der Inserts von pThiaEco und pThia1 ...................................................84 2.2.4 Analyse der DNA-Sequenz.............................................................................................84 2.2.5 Analyse der Expression von R. marinus Genen in E. coli ..............................................87
2.3 Untersuchung zur Identifizierung von Solutetransportern durch funktionelle Komplementierung transportdefizienter E. coli Stämme .........................................................88 2.3.1 Untersuchungen zur Komplementierung von E. coli MKH13 .........................................88 2.3.2 Untersuchungen zur Komplementierung von E. coli BKA-13.........................................89
2.3.2.1 Herstellung und Charakterisierung der otsB-Deletionsmutante E. coli BKA-13 89 2.3.2.2 Untersuchungen zur Komplementierung von E. coli BKA-13 ............................91
2.4 Isolierung eines DNA-locus mit Ähnlichkeiten zu Zuckertransportern mittels heterologer PCR..........................................................................................................................................92 2.4.1 Ableitung von degenerierten PCR-Primer mittels eines Sequenzvergleichs .................92 2.4.2 Heterologe PCR und Sequenzierung .............................................................................94 2.4.3 Analyse der identifizierten offenen Leserahmen ............................................................95
2.5 Differential Display RT-PCR an Gesamt-RNA R. marinus ......................................................100 III. Ergebnisse (Teil II).....................................................................................................102 3. Etablierung eines H. elongata Stamms für die Produktion des kompatiblen Soluts
Mannosylglycerat ...........................................................................................................................102 3.1 Wachstum von H. elongata KB1 und KB18.1 unter Zusatz von Mannosylglycerat.................103 3.2 Insertion des mgs-Gens in das Genom von H. elongata DSM2581T ......................................105
3.2.1 Herstellung der Insertionsmutante H. elongata KB10 ....................................................105 3.2.2 Charakterisierung der Insertionsmutante H. elongata KB10..........................................106
3.2.2.1 Physiologische Untersuchung von H. elongata KB10........................................107 3.2.2.2 Transkriptionsanalysen mittels Northern-Hybridisierung und RT-PCR..............110 3.2.2.3 Identifizierung möglicher Transkriptionsstartpunkte stromaufwärts von mgs ....113
3.2.3 Untersuchung zur heterologen Expression des mgs-Gen aus R. marinus in H. elongata Wildtyp und KB1 unter der Kontrolle des ectABC Promotors .................114
3.3 Integration hypothetischer P. putida Gene in H. elongata KB10 .............................................120 3.3.1 Herstellung von H. elongata KB10.1 ..............................................................................120 3.3.2 Charakterisierung von H. elongata KB10.1 ....................................................................122
3.3.2.1 Transkriptionsanalysen an KB10.1 mittels Northern-Hybridisierungen und RT-PCR..............................................................................................................123
3.3.2.2 Physiologische Charakterisierung von H. elongata KB10.1...............................128 IV. Diskussion ..................................................................................................................135 1. Strategie zur Produktion von Mannosylglycerat mittels Rhodothermus marinus ..........................135 2. Physiologische Charakterisierung von R. marinus ........................................................................138 3. Komplementierung von E. coli Stämmen mittels Genbänken von Rhodothermus marinus ..........139
3.1 Identifizierung der Thiamin-Gene von Rhodothermus marinus ...............................................140 3.2 Untersuchung zur Identifizierung von Mannosylglycerat-Transportern
mittels funktioneller Komplementierung transportdefekter E. coli Stämme.............................141 3.2.1 Komplementierungsexperimente mit E. coli MKH13 ......................................................141 3.2.2 Komplementierungsexperimente mit E. coli BKA-13......................................................142
4. Isolierung eines DNA-locus von R. marinus mit Ähnlichkeiten zu Natrium/Glucose-Symportern .144

Inhaltsverzeichnis
5. Amplifikation der 16S rDNA von R. marinus mittels Differential Display RT-PCR ........................147 6. Abschluss der Arbeiten mit Rhodothermus marinus......................................................................149 7. Halomonas elongata als Produktionsstamm für Mannosylglycerat ...............................................150 8. Charakterisierung der rekombinanten H. elongata Stämme .........................................................155
8.1 H. elongata KB10 (∆ectA::mgs) ...............................................................................................155 8.1.1 Physiologische Untersuchungen an H. elongata KB10..................................................155 8.1.2 Analyse der Transkription von mgs in H. elongata KB10 ...............................................157 8.1.3 Struktur des ectA-Promotors stromaufwärts von mgs in H. elongata KB10...................159 8.1.4 Heterologe Expression von mgs in H. elongata Wildtyp und KB1..................................161
8.2 H. elongata KB10.1 (∆ectABC::mgsPP1776xanA) ..................................................................162 8.2.1 Herstellung von H. elongata KB10.1 ..............................................................................162 8.2.2 Transkription der Fremdgene mgs, PP1776, und xanA in KB10.1 ................................165 8.2.3 Physiologische Charakterisierung von KB10.1 ..............................................................166
8.2.3.1 Nachweis der Mannosylglycerat-Synthese ........................................................166 8.2.3.2 Mannosylglycerat-Synthese bei Nutzung unterschiedlicher Substrate..............166 8.2.3.3 Mannosylglycerat-Gehalt in Abhängigkeit von der Wachstumsphase...............167 8.2.3.4 Ein Vergleich: H. elongata Wildtyp, KB1 und KB10.1 ........................................168 8.2.3.5 Energetische Bilanz der Solutesynthese............................................................170 8.2.3.6 Mannosylglycerat als kompatibles Solut für H. elongata....................................171 8.2.3.7 Vor- und Nachteile der Produktion von Mannosylglycerat in
H. elongata KB10.1 ............................................................................................172 9. Ausblick ..........................................................................................................................................174 V. Zusammenfassung.....................................................................................................177 VI. Literatur .......................................................................................................................179 Anhang

I. Einleitung
1. Extremophile Organismen
Als extremophil (philos: griechisch für liebend) bezeichnet werden Organismen, die sich sol-
chen Umweltbedingungen angepasst haben, die im Allgemeinen als lebensfeindlich betrach-
tet werden. Der Begriff extremophil geht jedoch über eine einfache Anpassungsfähigkeit hin-
aus. Extremophile Organismen benötigen diese unwirtlichen Umweltbedingungen für ihr
Wachstum. Um die Eigenschaften der Extremophilen genauer zu beschreiben, werden u.a.
die Begriffe
halophil – angepasst an eine hohe Salzkonzentration
thermophil – angepasst an eine hohe Temperatur
psychrophil – angepasst an eine niedrige Temperatur
alkaliphil – angepasst an einen hohen pH-Wert
acidophil – angepasst an einen niedrigen pH-Wert
barophil oder piezophil – angepasst an einen hohen Druck
verwendet. Diese Eigenschaften machen spezielle Modifikationen der zellulären Strukturen
und des Stoffwechsel erforderlich. Auch Mischformen dieser Gruppen sind möglich. So han-
delt es sich bei Halomonas campisalis str. 4A um einen halo-alkaliphilen Stamm (Mormile et
al., 1999), der gleichzeitig hohe Salzkonzentrationen und einen hohen pH-Wert für ein
optimales Wachstum benötigt. Unterschieden werden muss von dieser Extremophilie jedoch
die vorübergehende Anpassung an unterschiedliche Stressbedingungen. So kann ein extre-
mophiler Organismus gleichzeitig zu einem gewissen Grad auch an andere widrige Umwelt-
bedingungen angepasst sein. Das thermophile Bakterium Rhodothermus marinus
(Alfredsson et al., 1988) weist ein Temperaturoptimum von 65 °C auf und ist daher als
thermophil zu bezeichnen. Gleichzeitig toleriert es Salinitäten bis über 6 % NaCl in Komplex-
medium. Es kann jedoch nur als halotolerant, nicht jedoch halophil bezeichnet werden, da es
kein Salzbedürfnis von 0,5 M NaCl aufweist. Nach Reed (1986) besitzt ein halotoleranter
Organismus kein Salzbedürfnis von mehr als 0,5 M NaCl, kann aber auch bei einer Salinität
von 1 M NaCl noch wachsen. Oren (2002) bestimmt als Voraussetzung für die Halophilie
eines Organismus die Fähigkeit, bei einer Salinität von mindestens 10 % NaCl noch gut
wachsen zu können, auch wenn das Optimum deutlich niedriger liegen kann.
Ähnliches gilt für das Gram-negative γ-Proteobakterium Halomonas elongata DSM 2581T.
Hierbei handelt es sich um einen halophilen Organismus, der neben der hohen Salinität auch
erhöhte Temperaturen an seinem natürlichen Standort toleriert. Isoliert wurde H. elongata
aus einer Saline (solar saltern) auf den niederländischen Antillen (Vreeland et al., 1980). An

I. Einleitung 2
diesem Standort ist aufgrund unterschiedlich starker Sonneneinstrahlung auch mit Tempera-
turschwankungen zu rechnen. Dabei muss jedoch zwischen einer bestehenden Toleranz
gegenüber Temperaturerhöhungen und einer wirklichen Thermophilie unterschieden werden.
2. Thermophilie
2.1 Halotolerante thermophile und hyperthermophile Mikroorganismen Thermophile und hyperthermophile Organismen wurden aus verschiedenen thermalen
Standorten isoliert. Während sich unter den Thermophilen Pro- und Eukaryonten finden,
bleiben hyperthermophile Organismen auf die Prokaryonten beschränkt. Diese Mikroorganis-
men gehören den Domänen der Bacteria und Archaea an, wobei die Anzahl thermophiler
Archaea höher ist. Der Begriff thermophil wird bei Organismen genutzt, welche ein Wachs-
tumsoptimum zwischen 65 und 80 °C aufweisen, während hyperthermophil Organismen mit
einem Wachstumsoptimum oberhalb von 80 °C beschreibt (Blöchl et al., 1995). Im Tempe-
raturbereich von 75-90 °C kommen sowohl Archaea als auch Bacteria vor, der Bereich
oberhalb von 95 °C bleibt jedoch den Archaea vorbehalten (Daniel & Cowan, 2000). Die
Mehrzahl thermophiler Organismen wurde aus kontinentalen geo-thermalen oder künst-
lichen thermalen Standorten isoliert. Andere wurden in marinen hydro-thermalen Standorten
entdeckt. Die bekanntesten marinen thermophilen Bakterien sind Rhodothermus marinus
und Thermus thermophilus (Alfredsson et al., 1988; Da Costa et al., 2001). Das sauerstoff-
reiche Wasser kontinentaler heißer Quellen ist im Allgemeinen arm an Natrium-Ionen. Daher
können die meisten Isolate dieser Standorte eine Salzkonzentration von über 1 % NaCl (w/v)
kaum überleben. Dennoch gibt es auch an diesen Standorten halotolerante Stämme, die wie
T. thermophilus 4-6 % NaCl tolerieren können (Da Costa et al., 2001). Der marine Mikro-
organismus R. marinus weist ein Salzoptimum von 2 % auf und kann ohne NaCl-Zusatz nicht
wachsen (Alfredsson et al., 1988).
Die meisten hyperthermophilen Organismen stammen aus abyssischen Tiefseebecken, wie
z.B. Vulkanschloten, wo das Wasser für gewöhnlich eine höhere Salinität aufweist. Diese
Organismen sind daher generell leicht halophil, d.h. sie benötigen für das Wachstum NaCl.
Ihre optimale Salzkonzentration liegt zwischen 0,5 und 2 % NaCl (Blöchl et al., 1995). Im
Allgemeinen weisen moderat halophile Stämme ihre höchsten Wachstumsraten in Medien
auf, die 0,5-2,5 M NaCl enthalten (Ventosa et al., 1998). Bisher wurden jedoch keine
Stämme isoliert, die moderat oder extrem halophil sind und gleichzeitig an sehr heißen
Standorten vorkommen. Ein Grund dafür könnte sein, dass solche Standorte nicht existieren.
Bis zum heutigen Tag sind Halothermothrix orenii (Wachstumsoptimum: 60 °C; Salinitäts-
bereich: 4-20 % NaCl, (Cayol et al., 1994)) und Thermohalobacter berrensis (Wachstums-
optimum: 65 °C, Salinitätsbereich: 2-15 % NaCl, (Cayol et al., 2000)) die einzigen bekannten
halo-thermophilen Mikroorganismen.

I. Einleitung 3
2.2 Anpassung an ein Leben bei hohen Temperaturen Um an sehr heißen Standorten überleben zu können, haben Organismen verschiedene Stra-
tegien und Schutzmechanismen entwickelt. Nach in vitro-Untersuchungen sind viele lebens-
wichtige Metabolite, wie NAD+, ATP und Acetyl-CoA, bei hohen Temperaturen instabil. Diese
Instabilität scheint durch unterschiedliche Mechanismen aufgehoben werden zu können. So
ist es bekannt, dass ATP durch Variation des pH-Wertes und metallische Kationen stabilisiert
werden kann (Ramirez et al., 1980). Auch NAD+ ist bei höherem pH-Wert weitaus stabiler als
bei neutralem (Lowry et al., 1961). In manchen Mikroorganismen kommen möglicherweise
Bereiche im Zytoplasma vor, sog. „Metabolische Kanäle“, in denen für bestimmte Metabolite
stabilere Bedingungen vorherrschen. Innerhalb dieser Kanäle sorgen physikalisch assozi-
ierte Enzyme eines Stoffwechselweges für höhere Umsatzraten und geringere zytoplasma-
tische Konzentrationen der instabilen Intermediate (Kholodenko et al., 1996). Andere thermo-
phile Organismen verwenden Eisenproteine anstelle von NAD(P)+. So oxidiert Pyrococ-
cus furiosus Glucose mit Hilfe einer speziellen Oxidoreduktase, welche mit Ferredoxin ver-
knüpft ist, über den Entner-Doudoroff-Weg zu Pyruvat (Munkund & Adams, 1991). Ein
derartiger nicht-phosphorylierter Entner-Doudoroff-Weg ist ein typisches Merkmal thermo-
philer Archaea. Die nicht-phosphorylierten Intermediate weisen eine erhöhte Stabilität als
phosphorylierte Intermediate auf. Es ist bekannt, dass ATP bei 95 °C eine Halbwertszeit von
nur wenigen Minuten besitzt. Daher werden bei vielen thermophilen Enzymen anstelle von
ATP oft Pyrophosphat oder ADP verwendet (Kengan et al., 1994).
Im Gegensatz zu Enzymen mesophiler Organismen weisen viele Enzyme thermophiler Her-
kunft höhere Halbwertszeiten bei Temperaturen oberhalb von 100 °C auf. Diese Stabilität
beruht auf dem Austausch nur weniger Aminosäuren. Dieser Austausch hat in der Regel
jedoch keinen Einfluss auf die dreidimensionale Struktur der Proteine (Grutter et al., 1979).
Dennoch ist eine Verdichtung der Tertiärstruktur der Proteine zu beobachten. Daniel et al.
(1996) vermuteten, dass ein reziproker Zusammenhang zwischen thermodynamischer Stabi-
lität eines Enzyms und seiner spezifischen Stabilität besteht, welche durch molekulare Flexi-
bilität erreicht wird. Im Allgemeinen sind Enzyme thermophiler Herkunft bei allen Tempera-
turen stabiler, dafür jedoch weniger flexibel und weniger aktiv als verwandte mesophile
Enzyme. Die hohe Stabilität scheint Degradierungen entgegenzuwirken. Zudem verfügen die
meisten thermophilen Organismen über eine große Anzahl von Hitzeschockproteinen, wel-
che Polypeptide stabilisieren und ihnen bei beginnender Denaturierung die Möglichkeit zur
Rückfaltung bieten.
Ein weiteres Problem bei hohen Temperaturen sind Degradierung sowie Duplexdestabili-
sierung von Nukleinsäuren. In vitro kann DNA durch Zugabe von Salzen stabilisiert werden.
Das Vorhandensein hoher intrazellulärer Salzkonzentrationen (z.B. K+-Ionen) bei einigen
thermophilen Organismen bestätigt diesen stabilisierenden Effekt auch in vivo (Scholz et al.,

I. Einleitung 4
1992). Andere thermophile Organismen, wie z.B. Sulfolobus-Arten, akkumulieren anstelle
von Ionen polykationische Polyamine (z.B. Spermidin oder Thermospermin), welche die
Schmelztemperatur der DNA erhöhen und die Ribosomen vor Inaktivierung schützen
(Friedman, 1986). Außerdem kann die Schmelztemperatur der DNA geringfügig durch das
Heraufsetzen des GC-Gehalts erhöht werden. Bisher gibt es jedoch keine Beweise, dass
diese Strategie von Thermophilen benutzt wird. Dennoch stellte Grogan (1998) fest, dass der
GC-Gehalt in der 16S rRNA vieler hyperthermophiler Arten mit 63-69 % ziemlich hoch liegt.
Mechanismen wie das Supercoiling der DNA in Kombination mit kationischen Proteinen,
welche die DNA verpacken und dadurch die Schmelztemperatur erhöhen, scheinen jedoch
verbreiteter zu sein. Besonders durch die Entdeckung der Topoisomerase I in allen hyper-
thermophilen Archaea und einigen thermophilen Bacteria wurde die Theorie der DNA-Stabili-
sierung durch Supercoiling bestätigt (De La Tour et al., 1990). Nicht zuletzt wurden post-
transkriptionale Modifikationen, wie die Methylierung von Nukleosiden, insbesondere bei der
tRNA vieler hyperthermophiler Arten beobachtet (Edmonds et al., 1991).
Auch die Lipide und Membranen thermophiler Mikroorganismen erweisen sich als wider-
standsfähiger gegenüber hohen Temperaturen, extremen pH-Werten und oxidativem Stress.
Die Membranen verfügen über thermostabilere Glycerollipide, welche anstelle von Fett-
säuren in Esterbindung Isopren-Einheiten in Etherbindung enthalten (Konings et al., 2002).
Besonders bei Archaea aber auch in einigen thermophilen Bacteria wurde dies beobachtet
(Forterre, 1998). Zusätzlich wurde bei vielen Archaea die Lipiddoppelschicht der Membran
durch eine einfache Lipidschicht aus membrandurchspannenden C40-Phytanylketten ersetzt
(De Rosa et al., 1986).
3. Akkumulation osmoregulatorischer Solute
3.1 Strategien der Osmoregulation Wie mesophile Organismen sind (hyper)thermophile Organismen ständig mit Fluktuationen
der Wasseraktivität konfrontiert, welche auf einen unterschiedlichen Gehalt gelöster Sub-
stanzen zurückzuführen sind (Santos & Da Costa, 2002). Im Gegensatz zu ionischen (z.B.
NaCl) bzw. nicht-ionischen Soluten (z.B. Saccharose) ist Wasser frei über die Zytoplasma-
membran permeabel. Daher erniedrigen Mikroorganismen zur Anpassung an ein hyper-
osmotisches Medium das intrazelluläre chemische Potential des Wassers, bis sich ein osmo-
tisches Gleichgewicht zwischen Zytoplasma und umgebendem Medium einstellt, um so
einen Wasserausstrom zu verhindern oder rückgängig zu machen (Csonka, 1989). Die Er-
niedrigung des intrazellulären chemischen Potentials des Wassers erfolgt durch Akkumulati-
on osmoregulatorischer Solute. Dabei gibt es zwei unterschiedliche Strategien. Eine Gruppe
von Mikroorganismen akkumuliert vorwiegend anorganische Ionen, die andere Gruppe orga-
nische osmoregulatorische Solute im Zytoplasma.

I. Einleitung 5
3.1.1 Akkumulation anorganischer osmoregulatorischer Solute
Bei dieser Strategie werden in der Regel K+-Ionen in molarer Konzentration in der Zelle akku-
muliert, auch wenn im Außenmedium hauptsächlich NaCl vorliegt. Abhängig von der Wachs-
tumsphase werden auch Na+-Ionen akkumuliert. Die Akkumulation der K+-Ionen führt zu
einem Ausgleich des Konzentrationsgradienten über der Membran, so dass ein Verlust des
Wassers aus der Zelle verhindert wird (Galinski, E A, 1995; Da Costa et al., 1998) . Vertreter
dieser Strategie finden sich bei den Archaea und Bacteria. Beispiele sind die aeroben
Archaea der Familie Halobacteriaceae, die anaeroben Bacteria der Ordnung Halanaero-
biales sowie das aerobe halophile Bakterium Salinibacter ruber (Galinski, E A & Trüper,
1994; Anton et al., 2002; Oren et al., 2002). Einige Halobacterium-Arten akkumulieren K+-
Ionen bis zu einer Konzentration von 5 M, einem Wert, welcher die Löslichkeit des KCl in der
Zelle sogar übersteigt (Martin et al., 1999). Die Akkumulation hoher zytoplasmatischer Kon-
zentrationen von geladenen Ionen macht eine Anpassung zellulärer Strukturen erforderlich.
Neben den Enzymen weisen auch Kofaktoren und Ribosomen im Vergleich zu den nicht-
halophilen Komponenten vermehrt negative Ladungen auf und können daher K+-Ionen
binden (Galinski, E A, 1995). Dies wird durch einen hohen Anteil saurer (Glutamat und
Aspartat) und einen niedrigen Anteil basischer Aminosäuren (Lysin und Arginin) erreicht.
Zudem ist die Hydrophobizität dieser Proteine herabgesetzt (Lanyi, 1974). Die maximale
enzymatische Aktivität der Proteine wird nur bei Erreichen bestimmter zytoplasmatischer K+-
Konzentrationen erzielt, da die hohe Zahl negativer Ladungen vorher eine native Faltung
verhindert. Die geringere Hydrophobizität verhindert durch Steigerung der enzymatischen
Flexibilität ein Aussalzen der Enzyme bei hohen K+-Konzentrationen, bei geringen K+-Kon-
zentrationen wirkt sie jedoch zusätzlich destabilisierend. Aus den genannten Gründen sind
die Vertreter dieser Anpassungsstrategie weniger flexibel und an ein Leben in hochsalinem
Milieu gebunden (Martin et al., 1999).
3.1.2 Akkumulation organischer osmoregulatorischer Solute Die Akkumulation organischer osmoregulatorischer Solute zur Absenkung der intrazellulären
Wasseraktivität und zur Aufrechterhaltung des osmotischen Gleichgewichts ist bei nicht-halo-
toleranten, halotoleranten und halophilen Bacteria, halophilen methanogenen Archaea und
eukaryontischen Mikroorganismen, wie marinen Algen, verbreitet (Severin, 1993; Galinski, E
A & Trüper, 1994; Galinski, E A, 1995; Ventosa et al., 1998). Die weite Verbreitung dieser
Strategie ist ein Zeichen für ihren Erfolg. Die organischen Osmolyte werden in hoher Kon-
zentration (über 0,5 M) im Zytoplasma akkumuliert, ohne den Stoffwechsel der Zellen negativ
zu beeinflussen. Sie werden daher als kompatible Solute bezeichnet (Brown, 1976).
Kompatible Solute werden durch de novo-Synthese akkumuliert, können jedoch bei Anwe-
senheit im umgebenden Medium auch durch selektive Aufnahme angereichert werden, was

I. Einleitung 6
einen energetischen Vorteil bietet (Galinski, E A & Trüper, 1994). Bei den verwendeten
Substanzen handelt es um niedermolekulare, polare, wasserlösliche organische Verbin-
dungen, welche bei neutralem pH-Wert zumeist keine Nettoladung aufweisen (Galinski, E A
& Trüper, 1994; Galinski, E A, 1995). Kompatible Solute gehören verschiedenen Stoffklassen
an: Aminosäuren (Prolin, Glutamat), Aminosäurederivate (Glycin-Betain, Ectoin und
Hydroxyectoin), Polyole (Glycerol, Arabitol, Mannitol), Zucker (Trehalose, Saccharose,
Glucosylglycerol) und Schwefelverbindungen (Dimethylsulfoniopropionat) (Da Costa et al.,
1998). Einige kompatible Solute, wie Trehalose, Glycin-Betain und α-Glutamat, sind weit
verbreitet, andere dagegen sind spezifisch für bestimmte Organismengruppen. So kommen
Polyole bei Hefen und Algen vor, sind bei Bacteria eher selten und bei Archaea unbekannt.
Ectoin und Hydroxyectoin findet man nur in Bacteria.
3.2 Osmoregulation des halophilen Stamms Halomonas elongata Das moderat halophile, Gram-negative Bakterium Halomonas elongata DSM 2581T wurde
aus einem Salzgewinnungsbecken auf den niederländischen Antillen (Insel Bonaire) isoliert.
H. elongata (γ-Proteobakterium) toleriert in Mineralsalzmedium eine Salinität zwischen 0,3
und 25 % NaCl (Vreeland et al., 1980). Um dem osmotischen Ungleichgewicht zu begegnen,
synthetisiert und akkumuliert H. elongata die kompatiblen Solute Ectoin und Hydroxyectoin
(Wohlfarth et al., 1990). Aufgrund dieser Eigenschaften und der Tatsache, dass er genetisch
sehr gut zugänglich ist (Kunte & Galinski, 1995; Göller, 1999), rückte H. elongata in den
letzten Jahren zunehmend in den Mittelpunkt bei der Erforschung der verschiedenen Stress-
anpassungsmechanismen halophiler Mikroorganismen.
Das für H. elongata typische kompatible Solut Ectoin wird ausgehend von Aspartatsemi-
aldehyd unter Katalyse durch eine Diaminobuttersäure-Transaminase (EctB), eine Diamino-
buttersäure-Acetyltransferase (EctA) und eine Ectoin-Synthase (EctC) synthetisiert. Die
kodierenden Gene (ectABC) liegen hintereinander angeordnet im Genom vor (Göller et al.,
1998). Nach Untersuchungen von Stumpfe (2003) sind die Gene in einem Operon organi-
siert, werden jedoch zusätzlich auch als Einzeltranskripte exprimiert. Die hydroxylierte Form
des Ectoins wird vor allem auch durch Gram-positive Bacteria verwendet (Severin et al.,
1992). Durch Grammel (1999) wurde in Streptomyces chrysomallus ein Enzym (ThpD) iden-
tifiziert, welches die α-Ketoglutarat-abhängige Hydroxylierung des Ectoins zu Hydroxyectoin
katalysiert. Gene mit hohen Sequenzähnlichkeiten zu thpD wurden kürzlich auch in
H. elongata DSM 2581T und Chromohalobacter salexigens identifiziert (Ures, 2005; Garcia-
Estepa et al., 2006).
H. elongata zieht der Eigensynthese des Ectoins die Aufnahme kompatibler Solute aus dem
Medium vor. So wird Betain über mehrere Transportsysteme in die Zelle aufgenommen. Die
bisher identifizierten Transporter gehören der Gruppe der sekundären BCC-Transporter

I. Einleitung 7
(Betain/Carnitin/Cholin-Transporter) an und tragen die Bezeichnung BetH und BetG
(Kraegeloh, 2003). Für die Aufnahme des Ectoins nach einem hyperosmotischen Schock
wird einzig das hochaffine TRAP-Transportersystem (tripartite ATP Independend Periplasmic
Transporter, (Rabus et al., 1999)) TeaABC, verwendet (Grammann et al., 2002). Es handelt
sich dabei um eine neue Klasse osmoregulierter sekundärer Solutetransporter mit Ähnlich-
keiten zu Transportern aus Rhodobacter capsulatus. Das Transportsystem ist aus einem
großen und einem kleinen Membranprotein sowie einem periplasmatischen Protein aufge-
baut. Anders als bei ABC-Transportern ist der Transport nicht an eine ATP-Hydrolyse
sondern an einen Ionengradienten gekoppelt.
3.3 Osmoregulation thermophiler Mikroorganismen
3.3.1 Akkumulation spezieller kompatibler Solute
Thermophile und hyperthermophile Mikroorganismen akkumulieren organische kompatible
Solute, die bei mesophilen Organismen nicht vorkommen. Dies führte zu der Annahme, dass
diese kompatiblen Solute speziell mit dem Leben bei hohen Temperaturen assoziiert sind.
Auch die kompatiblen Solute thermophiler Mikroorganismen gehören verschiedenen Stoff-
klassen an: Zucker-Polyhydrat-Alkohole (Di-myo-Inositol-1,1’-Phosphat, Sulfotrehalose und
Mannosylglycerat), α- und β-Aminosäuren, Aminosäurederivate und gemischte Osmolyte
(z.B. cBPG). Im Gegensatz zu den kompatiblen Soluten mesophiler Organismen, die bei
physiologischem pH-Wert ungeladen bzw. zwitterionisch sind, weisen die meisten kompati-
blen Solute thermophiler Organismen eine negative Ladung auf. Die Autoren vermuteten
daher einen Zusammenhang zwischen der negativen Ladung dieser Solute und der Verbrei-
tung an heißen Standorten (Martins et al., 1996; Santos & Da Costa, 2002).
Nicht alle thermophilen Mikroorganismen akkumulieren „thermophile Solute“. So akkumuliert
das thermophile Bakterium T. thermophilus und das hyperthermophile Archaeon Pyro-
baculum aerophilum bevorzugt das „mesophile kompatible Solut“ Trehalose (Nunes et al.,
1995; Martins et al., 1997). Demgegenüber synthetisieren die halo-alkaliphilen Archaea
Natronococcus und Natronobacterium mit steigender Salinität eine negativ geladene Form
der Trehalose, 2-Sulfotrehalose, bei der eine Sulfat-Gruppe an eines der C2-Atome einer
Glucose-Einheit gebunden ist. In Natronobacterium zweigt die Biosynthese der Sulfotreha-
lose vermutlich von der Biosynthese der Glykolipide ab, denn in den Glykolipiden halophiler
Archaea sind oft Schwefelkohlenhydratverbindungen zu finden (Upasani et al., 1994).
Das häufigste Solut hyperthermophiler Archaea ist Di-myo-Inositol-1,1’-Phosphat (DIP,
Abb. 1). Dies wurde zuerst in Pyrococcus woesei identifiziert (Scholz et al., 1992). Die
Konzentration des DIP in der Zellen scheint im Zusammenhang mit steigender Temperatur
zu stehen und steigt bei P. furiosus an, wenn die Wachstumstemperatur oberhalb des Opti-
mums liegt (Martins & Santos, 1995). Dies führte zu der Vermutung, dass DIP auch als

I. Einleitung 8
Schutz gegen Hitzestress wirkt (Martins et al., 1999). Auch bei hyperthermophilen Bacteria,
wie Thermotoga neapolitana und T. maritima, steigt die zelluläre Konzentration des DIP bei
Osmo- und Hitzestress an. Zusätzlich wurden bei einigen Thermotoga-Arten zwei weitere
kompatible Solute entdeckt, Di-Mannosyl-di-myo-Inositol-Phosphat und Di-myo-Inositol-1,3’-
Phosphat (Martins et al., 1996). Diese spielen bei Osmostress jedoch eine untergeordnete
Rolle. Das ungewöhnliche kompatible Solut Di-Glycerol-Phosphat (DGP, Abb. 1) kommt
bisher ausschließlich bei Archaeoglobus fulgidus vor, wo es mit steigender Salinität und
Temperatur akkumuliert wird (Martins et al., 1997).
α-Mannosylglycerat (MG, Abb. 1) ist ein typisches Solut thermophiler Organismen. Seine
Biosynthese-Gene wurden bei vielen thermophilen Bacteria, wie Thermus thermophilus,
Rhodothermus marinus und Rubrobacter xylanophilus, sowie bei hyperthermophilen
Archaea, wie Aeropyrum pernix, Pyrococcus- und Thermococcus-Arten gefunden (Santos &
Da Costa, 2002). Vor kurzem wurde in dem mesophilen, halotoleranten, strikt anaeroben
Bakterium Dehalococcoides ethenogenes ein putatives Gen für die Biosynthese von Man-
nosylglycerat identifiziert (Empadinhas et al., 2004). Demnach kodiert das mgsD-Gen für ein
bifunktionales Enzym, welches Mannosylglycerat über ein Phosphat-Intermediärprodukt
synthetisiert. Empadinhas et al. (2004) vermuteten einen lateralen Gentransfer, da D. ethe-
nogenes mit keinem der thermophilen, zur Mannosylglycerat-Synthese fähigen Stämmen
verwandt ist. Das Vorkommen von Mannosylglycerat ist jedoch nicht unbedingt ein thermo-
philes prokaryontisches Merkmal, da dieses Solut erstmals in einer Rotalge der Gattung
Ceramiales als Nebensolut zu Mannitol identifiziert wurde (Bouveng et al., 1955). Die
meisten thermophilen Organismen akkumulieren Mannosylglycerat bei Osmo- und Hitze-
stress. In R. marinus kommt Mannosylglycerat gemeinsam mit seinem Amid-Derivat, Man-
nosylglyceramid, vor (Nunes et al., 1995; Silva et al., 1999, Abb. 1). Mannosylglyceramid
wurde bisher allein in R. marinus gefunden. Seine Konzentration steigt offenbar mit zuneh-
mender Mediensalinität an. Mannosylglycerat wird bei Steigerung des osmotischen Stresses
bei gleichzeitiger Erhöhung der Umgebungstemperatur akkumuliert.
Die einzige Aminosäure, die in den meisten Organismen generell als kompatibles Solut ge-
nutzt wird, ist α-Glutamat. β-Glutamat dagegen kommt hauptsächlich bei thermophilen aber
auch mesophilen methanogenen Archaea vor (Robertson et al., 1992). Als weitere β-Amino-
säuren werden β-Glutamin und Nε-Acetyl-β-Lysin von methanogenen halotoleranten und
thermophilen Archaea als Antwort auf Osmostress akkumuliert. Die β-Aminosäuren sind wie
ihre α-Isomere wasserlöslich, werden jedoch nicht metabolisiert. Die Stabilität der β-Amino-
säuren in wachsenden Zellen wurde durch NMR-Analysen bestätigt (Robertson et al., 1992).
Zuletzt wurden viele ungewöhnliche Verbindungen in hohen Konzentrationen bei thermo-
philen Archaea gefunden. Diese Polyanionen dienen zumeist als Gegenion für Kalium-Ionen.
So stellen das zyklische 2,3-Bisphospho-Glycerat (cBPG, Abb. 1) zusammen mit der Verbin-

I. Einleitung 9
dung 1,3,4,6-Tetra-carboxyhexan (TCH, Abb. 1) die Hauptsolute von Methanobacterium
thermoautotrophicum ∆H dar. TCH weist bei einem neutralen pH-Wert vier negative La-
dungen auf, bei cBPG sind es drei negative Ladungen. Daher liegen beide Polyanionen im
Vergleich zu den Kalium-Ionen in der Zelle in niedriger Konzentration vor (Martin et al.,
1999).
Mannosylglycerat Mannosylglyceramid
1,3,4,6-Tetracarboxyhexan (TCH) 2,3-Bisphospho-Glycerat (cBPG)
Di-myo-Inositol-1,1’-Phosphat (DIP)
Di-Glycerol-Phosphat (DGP)
Abb. 1: Kompatible Solute thermophiler und hyperthermophiler Organismen Dargestellt sind die häufigsten kompatiblen Solute thermophiler und hyperthermophiler Bacteria bzw. Archaea. Die meisten dieser Solute sind bei physiologischem pH-Wert negativ geladen. cBPG bzw. TCH weisen sogar drei bzw. vier negative Ladungen auf. Es wird vermutet, dass diese Ladung mit dem Vorkommen an heißen Standorten zusammenhängt. (Abbildung modifiziert nach Santos & Da Costa, 2002)
3.3.2 Biosynthese von α-Mannosylglycerat in Rhodothermus marinus
Bei Rhodothermus marinus handelt es sich um ein thermophiles, halotolerantes, Gram-
negatives Bakterium, das aus einer in Meeresnähe gelegenen heißen Quelle in
Isafjardardjup auf Island isoliert wurde (Alfredsson et al., 1988). Nach Analysen der 16S
rRNA gehört R. marinus phylogenetisch zur Cytophaga-Flexibacter-Bacteroides-Gruppe
(CFB) (Andresson & Fridjonsson, 1994), zur Gattung Sphingobacteria und zur Familie
Crenotrichacea (Garrity et al., 2004). Salinibacter ruber ist der nächste genetische, jedoch
nicht-thermophile Verwandte (Anton et al., 2002). Die nächsten verwandten thermophilen
Stämme sind Thermonema lapsum und T. rossianum, die ebenfalls zur CFB-Gruppe gehö-
ren (Patel et al., 1994; Tenreiro et al., 1997). R. marinus weist einen aeroben, chemo-
organotrophen Stoffwechsel auf, sein Temperaturoptimum liegt bei 65 °C (Alfredsson et al.,
1988). Zellen von R. marinus sind durch Karotinoide, die in der Zytoplasmamembran an
Proteine gebunden sind, rötlich gefärbt. Das Genom von R. marinus weist bei einem GC-

I. Einleitung 10
Gehalt von 64 % eine Länge von 3,3-3,6 Mbp auf (Moreira et al., 1996). Zur Anpassung an
hohe Salinität und Hitze produziert R. marinus, neben geringen Mengen an α-Glutamat und
Trehalose, die kompatiblen Solute α-Mannosylglycerat und Mannosylglyceramid (Nunes et
al., 1995; Silva et al., 1999, Abb. 1). Während Mannosylglycerat ein verbreitetes Solut
thermophiler Mikroorganismen darstellt, wird Mannosylglyceramid nur durch R. marinus
synthetisiert. Die Mannosylglyceramid-Konzentration steigt bei Salinitäten zwischen 3-5 %
NaCl an, bei höheren Temperaturen und höheren Salinitäten hingegen wird hauptsächlich
Mannosylglycerat synthetisiert. Mannosylglycerat wird über zwei alternative Stoffwechsel-
wege synthetisiert (Martins et al., 1999). GDP-Mannose ist das gemeinsame Ausgangs-
substrat (Abb. 2). Bei dem ersten Syntheseweg (Einschrittsyntheseweg) kondensiert GDP-
Mannose unter Einwirkung der Mannosylglycerat-Synthase (mgs-Gen) mit D-Glycerat zu
Mannosylglycerat. Dieser Weg konnte bisher nur bei R. marinus nachgewiesen werden,
während der Zweischrittsyntheseweg auch bei den übrigen thermophilen, Mannosylglycerat
produzierenden Stämmen, wie P. horikoshii und T. thermophilus, nachgewiesen wurde
(Empadinhas et al., 2001; Empadinhas et al., 2003). Bei diesem Syntheseweg wird die Um-
wandlung von GDP-Mannose und D-3-Phosphoglycerat zu Mannosylphosphoglycerat durch
die Mannosylphosphoglycerat-Synthase (mpgS-Gen) katalysiert. Anschließend wird unter
Einwirkung einer Mannosylglycerat-Phosphatase (mpgP-Gen) Mannosylglycerat freigesetzt.
Der Zweischrittsyntheseweg verläuft über ein phosphoryliertes Intermediat und ähnelt der
Biosynthese der kompatiblen Solute Trehalose (z.B. Giaever et al., 1988) und Glucosyl-
glycerol (Hagemann & Erdmann, 1994).
Die Tatsache, dass R. marinus über zwei alternative Mannosylglycerat-Biosynthesewege
verfügt, führte zu der Annahme, dass dieser eine hohe Flexibilität bei der Regulation der
Mannosylglycerat-Synthese aufweist (Santos & Da Costa, 2002). Abhängig von der vorherr-
schenden Stressbedingung (Osmo- oder Thermostress) werden unterschiedliche Biosynthe-
segene aktiviert. Diese Vermutung wurde durch Untersuchungen von Borges et al. (2004)
bekräftigt. Demnach wird die Mannosylglycerat-Synthase hauptsächlich bei Hitzestress
induziert und ist salzunabhängig. Dagegen benötigen die Mannosylphosphoglycerat-Syn-
thase und Mannosylphosphoglycerat-Phosphatase NaCl bzw. KCl für eine optimale Expres-
sion und Aktivität. In Salzschockexperimenten von 2 auf 6 % NaCl konnten Borges et al.
(2004) eine Verdreifachung der Mannosylphosphoglycerat-Synthase-Konzentration beobach-
ten.

I. Einleitung 11
Mannosyl-3-P-glycerat
Mannosylglycerat
Pi
D-3-P-Glycerat
Glucose-6-phosphat
Fructose-6-phosphat
Mannose-6-phosphat
Mannose-1-phosphat
GDP-Mannose
Mannosylglycerat
GTP
PPi
D-Glycerat
GDP GDP
1
2
3
4
5 6
7
Abb. 2: Postulierte Biosynthesewege für Mannosylglycerat in R. marinus Nach Martins et al. (1999) wird Mannosylglycerat in R. marinus ausgehend von Glucose-6-phosphat durch zwei alternative Biosynthesewege synthetisiert. Für nähere Erläuterungen sei auf den Text verwiesen. 1: Phospho-glucose-Isomerase, 2: Phosphomannose-Isomerase, 3: Phosphomanno-Mutase, 4: Mannose-1-phosphat-Guanylyltransferase, 5: Mannosylglycerat-Synthase, 6: Mannosyl-3-phosphoglycerat-Synthase, 7: Mannosyl-3-phosphoglycerat-Phosphatase. (Abbildung modifiziert nach Martins et al., 1999)
4. Kompatible Solute zur Stabilisierung von Biomolekülen Die stabilisierende Wirkung kompatibler Solute auf Biomoleküle, wie DNA und Proteine aber
auch auf ganze Zellen, ist seit langem bekannt. Voß (1998) und Karla (1997) beobachteten
bei Zusatz von kompatiblen Soluten, wie Trehalose und Ectoin, eine gesteigerte Überlebens-
rate getrockneter Zellen bei Lagerung und Rehydrierung. Dies wurde auf eine Stabilisierung
des flüssigkristallinen Zustands der Zellmembranen zurückgeführt (Nakagaki et al., 1992;
Pereira et al., 2004).
Malin et al. (1999) stellten fest, dass sich die DNA-Struktur durch Zusatz von kompatiblen
Soluten derart verändert, dass diese nicht mehr durch Restriktionsendonukleasen erkannt
und enzymatisch abgebaut werden kann. Außerdem werden kompatible Solute häufig in der

I. Einleitung 12
Polymerasekettenreaktion (PCR) zur Absenkung der Schmelztemperatur von GC-reichen
Regionen verwendet. Dies führt zu einer Amplifikation von sonst nur schwer amplifizierbaren
Regionen (Henke et al., 1997; Schnoor et al., 2004).
Die stabilisierende Wirkung kompatibler Solute auf viele Enzyme konnte in vitro in verschie-
denen Untersuchungen gezeigt werden. So konnten Lippert & Galinski (1992) und Knapp et
al. (1999) anhand von Modellversuchen mit Phosphofructokinase, Lactat-Dehydrogenase
(LDH) und Ribonuklease A (RNase A) zeigen, dass kompatible Solute gegen Stressfaktoren,
wie Hitze und Gefriertrocknung, schützen. Kompatible Solute thermophiler Herkunft, wie
Mannosylglycerat, DIP und DGP, weisen ähnliche stabilisierende Wirkungen auf Proteine
auf. So konnten Ramos et al. (1997) zeigen, dass Mannosylglycerat viele Enzyme sowohl
mesophiler Herkunft, wie die Lactat-Dehydrogenase (Kaninchen-Muskel) und die Alkohol-
Dehydrogenase (Bäckerhefe), als auch aus thermophiler Herkunft, wie die Glutamat-
Dehydrogenase aus Thermotoga maritima, gegen Hitzestress schützt. Zudem verleiht
Mannosylglycerat Schutz bei Gefriertrocknung. Melo et al. (2001), Faria et al. (2003) und
Faria et al. (2004) zeigten stabilisierende Effekte des Mannosylglycerats auf Enzyme wie die
RNase A. Die Auffaltungsenergie bzw. Schmelztemperatur (Tm) dieses Enzyms steigt in
Gegenwart von Mannosylglycerat an und ist höher als unter Zusatz von Trehalose. Die
Autoren gingen davon aus, dass die negative Ladung des Mannosylglycerats eine große
Rolle bei der Stabilisierung des jeweiligen Enzyms spielt. Dieser Effekt scheint pH-Wert
abhängig zu sein, da Faria et al. (2003) bei einem pH-Wert von 7, bei dem die Carboxyl-
gruppe des Mannosylglycerats vollständig ionisiert vorliegt, einen Anstieg der Tm der RNase
A von 3,2 °C verzeichneten. Außerdem stellten sie fest, dass Mannosylglycerat zum größten
Teil die Aggregation des denaturierten Proteins unterbindet.
Trotz der zahlreichen in vitro-Untersuchungen kann die stabilisierende Wirkungsweise des
Mannosylglycerats und der übrigen Soluten auf molekularer Ebene nicht vollständig geklärt
werden, da verschiedenen Solute bei unterschiedlichen Stressfaktoren die einzelnen Prote-
ine unterschiedlich gut schützen. Zur stabilisierenden Wirkung der kompatiblen Solute
wurden viele Wirkmechanismen, wie das Modell der „preferential exclusion“ von Arakawa &
Timasheff (1985), die „Water Replacement“- Hypothese von Clegg et al. (1982) und die
„Vitrification“- Hypothese von Sun & Leopold (1994), postuliert. Beim „preferential exclusion“-
Modell steigert das Solut die Oberflächenspannung der Lösung und wird so aus den Hydrat-
hüllen der Proteine ausgeschlossen. Eine Auffaltung und damit Denaturierung des Proteins
wird erschwert, da dies thermodynamisch ungünstig wäre. Der native Zustand der Proteine
wird somit stabilisiert. Dieses Modell kann in Teilen die Untersuchungen von Lippert &
Galinski (1992) erklären. Dennoch versagt es bei der Erklärung der stabilisierenden Wirkung
kompatibler Solute bei der Trocknung von Enzymen bzw. ganzen Zellen. Hier werden die
Modelle des „Water Replacement“ bzw. der „Vitrification” zur Erklärung vorgezogen. Viele

I. Einleitung 13
kompatible Solute verfügen über mindestens eine Hydroxylgruppe (z.B. Polyole, Trehalose,
Hydroxyectoin und Mannosylglycerat) und können somit Wechselwirkungen mit Zellbe-
standteilen eingehen. Das „Water Replacement“- Modell nimmt an, dass die Solute in der
Lage sind bei trocknungsbedingtem Wasserentzug die Hydrathüllen zu ersetzen und so die
flüssigkristalline Lipiddoppelschicht zu stabilisieren. Die Erhaltung dieses Membranzustan-
des sichert die Permeabilitätsbarriere und so das Überleben der Zelle. Beim „Vitrification”-
Modell stellen kompatible Solute die Fluidität der Membran sicher, indem sie bei bestimmten
stoffspezifischen Temperaturen (Tg) in einen glasartigen Zustand übergehen und so sämt-
liche Konformationsänderungen, die in der Zytoplasmamembran aufgrund der Trocknung
stattfinden, reduzieren bzw. unterdrücken (Pereira et al., 2004). Dabei muss jedoch die
Verglasungstemperatur (Tg) des Soluts höher liegen als die kritische Übergangstemperatur
(Tm), bei der die Lipide von der flüssigkristallinen Phase in die Gelphase übergehen (Koster
et al., 2000).
5. Kompatible Solute: Nutzen und wirtschaftliche Bedeutung
Die beschriebenen protektiven Eigenschaften kompatibler Solute ermöglichen ein so breites
Anwendungsspektrum, dass viele industrielle Firmen großes Interesse zeigen, diese im
großen Maßstab zu gewinnen. Die großtechnische Produktion von Ectoin wurde in einem
ersten Schritt durch das von Sauer & Galinski (1998) entwickelte Bakterienmelkverfahren
(hypoosmotischer Schock) wesentlich vereinfacht. Dabei setzt H. elongata nach der Anzucht
unter Hochsalzbedingungen durch mehrere osmotische Wechselschocks die im Zytoplasma
akkumulierten kompatiblen Solute frei. Die Aufreinigung des Ectoins erfolgt durch Kationen-
austauschchromatographie. Die Firma Bitop AG (Witten) ist ein Vorreiter auf diesem Gebiet.
In Zusammenarbeit mit Merck (Darmstadt) stellt Bitop großtechnische Mengen an Ectoin her.
Ectoin ist seit dem Jahr 2000 ein fester Bestandteil kosmetischer Produkte der Firma Merck
(Hautcreme RonaCareTM). Mittlerweile setzen jedoch auch andere Firmen Ectoin in ihren
Kosmetikprodukten, wie Hautcremes (Firma Marbert, Düsseldorf; Firma Sebapharm GmbH &
Co. KG, Bad Salzig) und Sonnenschutzmitteln (Firma Shiseido, Japan; Bioderma,
Frankreich), ein. Neuere Produkte sind das Anti-Aging Serum der Firma Annemarie Börlind
(Frick, 2005) sowie das Ectoin Anti-Aging Fluid der Firma DADO-cosmed GmbH (Calw).
Untersuchungen von Bünger (1999; 2000, Merck) und Bünger & Driller (2004, Merck)
zeigten, dass Ectoin ein breites Wirkungsspektrum besitzt. So schützt Ectoin gegen UV-
Strahlung, Hautrötung sowie Faltenbildung und unterstützt gleichzeitig die zelleigenen
Reparatur- und Schutzmechanismen. Bitop plant, verschiedene kompatible Solute in den
Bereichen Dermatologie, Diagnostik und Onkologie einzusetzen. Bisher gelang es schon, auf
der Basis kompatibler Solute gebrauchsfertige Stabilisierungslösungen für Enzyme, Anti-
körper und Nukleinsäuren für die Anwendungsforschung in Form der BioStab-Serie anzu-

I. Einleitung 14
bieten. In einer aktuellen Studie am Klinikum der Universität Köln untersucht Bitop den Ein-
satz von kompatiblen Soluten zur Toxizitätsminderung von Krebstherapeutika bei der
Immuntoxintherapie. Bei Verwendung von Immuntoxinen kommt es häufig als schwer-
wiegende Nebenwirkung zur Entwicklung des sog. „vascular leak syndrome“, welches zu
einer Permeabilisierung der Endothelschicht und so zur Bildung von Ödemen führt. Dies
schränkt den Einsatz dieser wirkungsvollen Therapie erheblich ein. Die Reduktion solcher
Nebenwirkungen eröffnet neue therapeutische Möglichkeiten und ermöglicht dem Patienten
eine höhere Lebensqualität. Andere Krankheiten, wie Kreuzfeld-Jakob, Parkinson und
Alzheimer, werden durch Fehlfaltung von Proteinen und Proteinaggregate verursacht.
Kanapathipillia et al. (2005) zeigten, dass Ectoin und Hydroxyectoin die Aggregation
verhindern und dadurch die Neurotoxizität, welche z.B. das Alzheimer β-Amyloid-Peptid
verursacht, herabsetzt. Basierend auf diesen und anderen Untersuchungen entwickelt Bitop
mittelfristig ein Konzept, gezielt neurodegenerativen Erkrankungen vorzubeugen und diese
zu behandeln.
Neben Ectoin hat auch Trehalose mittlerweile ein breites Anwendungspotential. So wird
dieses zur hypothermischen Aufbewahrung von Zellen und Organen von Säugetieren ge-
nutzt (Crowe et al., 2003). Außerdem läuft seit 2002 eine Studie, welche eine positive
Wirkung der Trehalose bei der Behandlung des sog. „dry eye syndrome“ beim Menschen
bestätigen soll (Matsuo et al., 2002). Sogar in der Landwirtschaft scheint der Einsatz von
kompatiblen Solute vielversprechend zu sein. So werden derzeit die Biosynthesegene
kompatibler Solute in salzempfindlichen Nutzpflanzen wie Reis (Flowers, 2004) und Tomaten
(Moghaieb et al., 2000) eingebracht. Ziel ist eine erhöhte Resistenz der Pflanzen gegenüber
Salzstress. Erfolgreiche Versuche liefen bereits an Modellpflanzen, wie Tabak und
Arabidopsis (Alia et al., 1998; Nakayama et al., 2000).
Die vielfältigen Möglichkeiten bei der Nutzung kompatibler Solute eröffnen einen immer
größer werdenden wirtschaftlichen Markt. Gerade im pharmazeutischen Bereich lassen sich
mit Neu- und Fortentwicklung von Produkten große Gewinne erzielen. Bedingt dadurch ver-
suchen die in diesem Feld tätigen Firmen ihre wirtschaftlichen Interessen zu schützen. Dies
erreichen sie u.a. durch das Anmelden von Patenten, die sich auf die Anwendung kompati-
bler Solute beziehen. Unter anderem gibt es Patente, welche die Gewinnung von kompati-
blen Soluten aus Zellen bzw. aus Organismen betreffen. Zu nennen sind in diesem Zusam-
menhang das Bakterienmelkverfahren (DE 04244580, Galinski, E A et al., 1994) und die
Herstellung einer Ausscheidermutante (WO 02002077254, Kunte et al., 2002). Andere
Patente schützen die Anwendung kompatibler Solute als Inhibitoren gegen einen
enzymatischen Abbau von Enzymen sowie Zellkomponenten (WO 01058446, Galinski, E A
et al., 2001) oder ihren Einsatz bzw. ihre pharmazeutische Formulierung in Arzneimitteln,

I. Einleitung 15
Impfstoffen und Kosmetika (z.B. WO 02002015868, Schwarz, 2002a; DE 010330243,
Kurtmann, 2005).
Des weiteren sind einige Patente, welche eine kosmetische Anwendung von Mannosyl-
glycerat beschreiben (z.B. WO 02002015867, Schwarz, 2002b) oder die Verwendung von
Mannosylglycerat zum Schutz von Zellkomponenten und Enzymen gegen Hitze-, Osmo-
stress und enzymatischen Abbau belegen (EP 0816509, Santos et al., 1998), angemeldet.
Patentanmeldung (EP 01526180, Da Costa et al., 2005) beschreibt eine Strategie für die
Produktion von Mannosylglycerat, die sich eines modifizierten Saccharomyces cerevisiae
Stamms bedient. In diesem Stamm wurde das plasmidkodierte Gen mgsD (kodiert für eine
bifunktionale Mannosylglycerat-Synthase) aus dem mesophilen Organismus Dehalococco-
ides ethenogenes heterolog exprimiert. Dadurch konnte eine Mannosylglycerat-Synthese in
Saccharomyces cerevisiae erreicht werden.
6. Ziel der Arbeit Ziel der vorliegenden Arbeit ist die Konstruktion eines bakteriellen Stamms, der eine indus-
trielle Gewinnung des kompatiblen Soluts Mannosylglycerat in größeren Mengen (> 10 g/l)
ermöglicht. Der natürliche Produktionsstamm Rhodothermus marinus genügt aus einigen
Gründen den industriellen Anforderungen nicht vollständig. So werden mit R. marinus in der
Fermentation zu geringe Zelldichten erreicht und die bis dato notwendige Anwendung des
Bakterienmelkverfahrens zur Gewinnung der kompatiblen Solute macht eine aufwendige
Abtrennung verunreinigender Substanzen erforderlich. Als Vorbild für den bakteriellen
Produktionsstamm dient Halomonas elongata KB2, der aufgrund einer Deletion in seinem
Ectoin-Transportsystem nicht mehr in der Lage ist, Ectoin aus dem Medium aufzunehmen,
und daher als Ectoin-Überproduzent bereits in der industriellen Produktion eingesetzt wird.
Die Nutzung dieses Stamms macht die Verwendung des Bakterienmelkverfahrens über-
flüssig, da dieser ständig Ectoin ins Außenmedium verliert.
Analog zu H. elongata sollte in einem ersten Projekt in R. marinus ein mögliches Transport-
system für Mannosylglycerat identifiziert sowie charakterisiert und schließlich durch mole-
kulargenetische Methoden ausgeschaltet werden. Resultat wäre ein R. marinus Stamm,
welcher Mannosylglycerat kontinuierlich ins Außenmedium abgibt. Zur Verwirklichung des
Projekts war zunächst die Etablierung der Methoden zur genetischen Manipulation von
R. marinus erforderlich. Entsprechende Arbeiten waren bei der Firma Prokaria (Island),
welche mittlerweile auch das Genom von R. marinus sequenziert hat, in Vorbereitung. Erste
erfolgreiche Versuche auf dem Gebiet der gentechnischen Manipulation von R. marinus
waren bereits durchgeführt und hatten zur Etablierung eines geeigneten Gentransfersystems
geführt (Ernstsson et al., 2003; Bjornsdottir et al., 2005). Die Identifizierung des hypothe-

I. Einleitung 16
tischen Mannosylglycerat-Transporters sollte mit Hilfe verschiedenster molekularbiolo-
gischer Methoden erfolgen.
Für den Fall, dass R. marinus nicht über einen Mannosylglycerat-Transporter verfügt oder
sich die Deletion desselben als schwierig erweisen sollte, wurde ein zweites Projekt zur
Etablierung einer bakteriellen Mannosylglycerat-Produktion geplant. Hier sollte zunächst das
zentrale Enzym der Mannosylglycerat-Synthese, die Mannosylglycerat-Synthase (mgs-Gen),
aus R. marinus (Abb. 2) in das Genom des halophilen Stamms H. elongata integriert werden.
Dieser Stamm schien u.a. durch die für ihn etablierten Fermentationsverfahren und seine
genetische Zugänglichkeit besonders geeignet. Als Insertionsort wurden die Ectoin-Gene
ausgewählt, da das Fremdgen so unter Kontrolle der Promotoren der Ectoin-Synthesegene
exprimiert wird und gleichzeitig die Ectoin-Synthese ausgeschaltet wird. Für den Fall eines
Ausbleibens der Mannosylglycerat-Synthese in H. elongata war die Integration weiterer
Gene eines geeigneten Spenders geplant. Sollte H. elongata nicht über ein Transportsystem
für Mannosylglycerat verfügen, wäre nach der Etablierung der Mannosylglycerat-Synthese
auch hier mit einem Verlust des Mannosylglycerats in das Außenmedium zu rechnen, wie es
bei H. elongata KB2 für Ectoin der Fall ist.
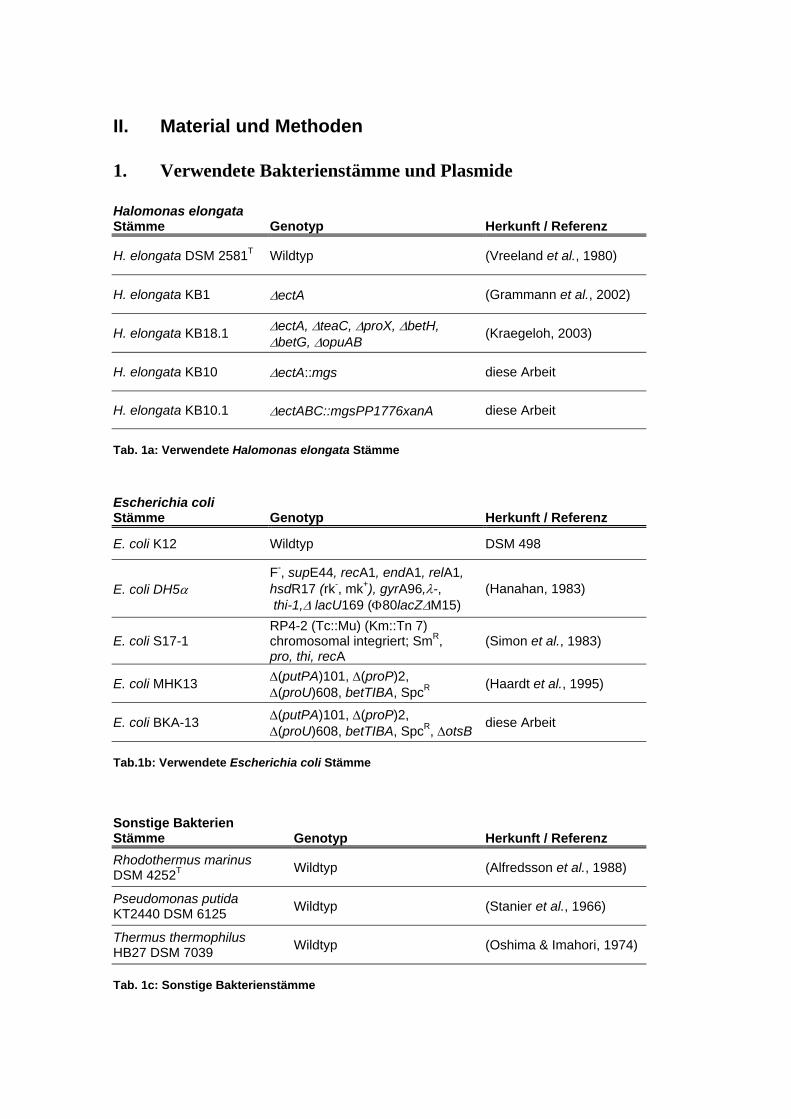
II. Material und Methoden
1. Verwendete Bakterienstämme und Plasmide
Halomonas elongata Stämme Genotyp Herkunft / Referenz
H. elongata DSM 2581T Wildtyp (Vreeland et al., 1980)
H. elongata KB1 ∆ectA (Grammann et al., 2002)
H. elongata KB18.1 ∆ectA, ∆teaC, ∆proX, ∆betH, ∆betG, ∆opuAB (Kraegeloh, 2003)
H. elongata KB10 ∆ectA::mgs diese Arbeit
H. elongata KB10.1 ∆ectABC::mgsPP1776xanA diese Arbeit
Tab. 1a: Verwendete Halomonas elongata Stämme Escherichia coli Stämme Genotyp Herkunft / Referenz
E. coli K12 Wildtyp DSM 498
E. coli DH5α F-, supE44, recA1, endA1, relA1, hsdR17 (rk-, mk+), gyrA96,λ-, thi-1,∆ lacU169 (Φ80lacZ∆M15)
(Hanahan, 1983)
E. coli S17-1 RP4-2 (Tc::Mu) (Km::Tn 7) chromosomal integriert; SmR, pro, thi, recA
(Simon et al., 1983)
E. coli MHK13 ∆(putPA)101, ∆(proP)2, ∆(proU)608, betTIBA, SpcR (Haardt et al., 1995)
E. coli BKA-13 ∆(putPA)101, ∆(proP)2, ∆(proU)608, betTIBA, SpcR, ∆otsB diese Arbeit
Tab.1b: Verwendete Escherichia coli Stämme Sonstige Bakterien Stämme Genotyp Herkunft / Referenz Rhodothermus marinus DSM 4252T Wildtyp (Alfredsson et al., 1988)
Pseudomonas putida KT2440 DSM 6125 Wildtyp (Stanier et al., 1966)
Thermus thermophilus HB27 DSM 7039 Wildtyp (Oshima & Imahori, 1974)
Tab. 1c: Sonstige Bakterienstämme
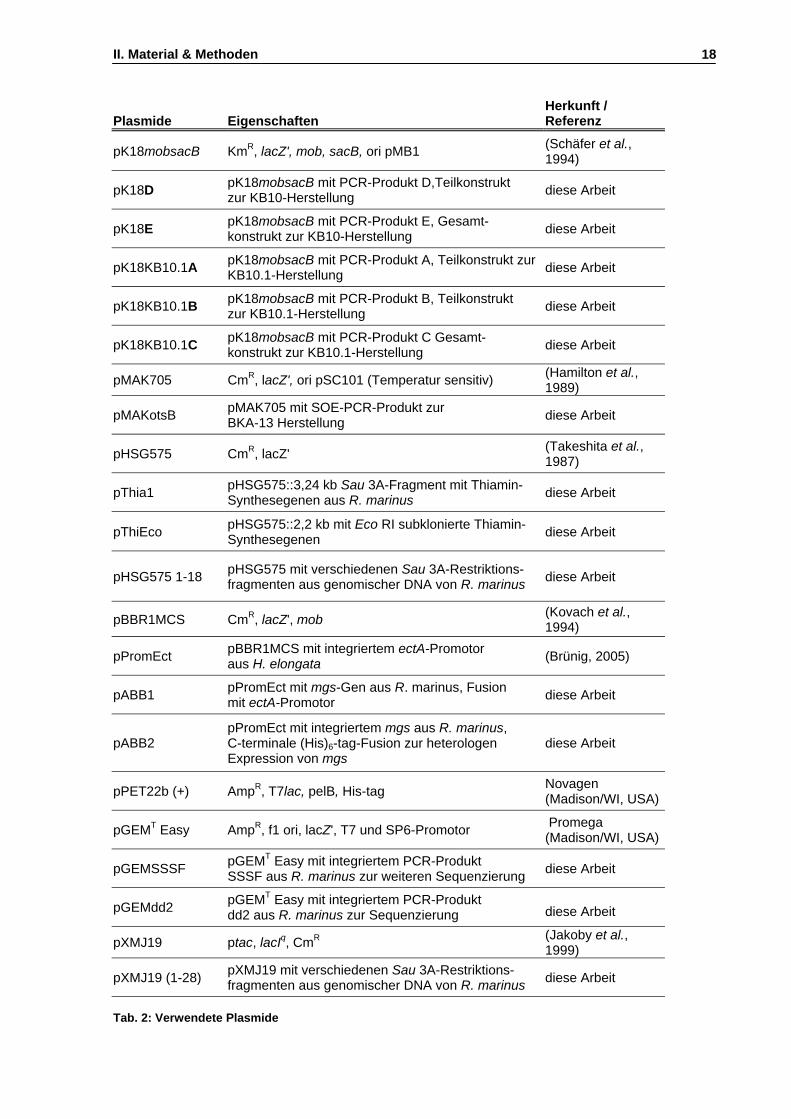
II. Material & Methoden 18
Plasmide Eigenschaften Herkunft / Referenz
pK18mobsacB KmR, lacZ', mob, sacB, ori pMB1 (Schäfer et al., 1994)
pK18D pK18mobsacB mit PCR-Produkt D,Teilkonstrukt zur KB10-Herstellung diese Arbeit
pK18E pK18mobsacB mit PCR-Produkt E, Gesamt- konstrukt zur KB10-Herstellung diese Arbeit
pK18KB10.1A pK18mobsacB mit PCR-Produkt A, Teilkonstrukt zur KB10.1-Herstellung diese Arbeit
pK18KB10.1B pK18mobsacB mit PCR-Produkt B, Teilkonstrukt zur KB10.1-Herstellung diese Arbeit
pK18KB10.1C pK18mobsacB mit PCR-Produkt C Gesamt- konstrukt zur KB10.1-Herstellung diese Arbeit
pMAK705 CmR, lacZ', ori pSC101 (Temperatur sensitiv) (Hamilton et al., 1989)
pMAKotsB pMAK705 mit SOE-PCR-Produkt zur BKA-13 Herstellung diese Arbeit
pHSG575 CmR, lacZ' (Takeshita et al., 1987)
pThia1 pHSG575::3,24 kb Sau 3A-Fragment mit Thiamin-Synthesegenen aus R. marinus diese Arbeit
pThiEco pHSG575::2,2 kb mit Eco RI subklonierte Thiamin-Synthesegenen diese Arbeit
pHSG575 1-18 pHSG575 mit verschiedenen Sau 3A-Restriktions-fragmenten aus genomischer DNA von R. marinus diese Arbeit
pBBR1MCS CmR, lacZ', mob (Kovach et al., 1994)
pPromEct pBBR1MCS mit integriertem ectA-Promotor aus H. elongata (Brünig, 2005)
pABB1 pPromEct mit mgs-Gen aus R. marinus, Fusion mit ectA-Promotor diese Arbeit
pABB2 pPromEct mit integriertem mgs aus R. marinus, C-terminale (His)6-tag-Fusion zur heterologen Expression von mgs
diese Arbeit
pPET22b (+) AmpR, T7lac, pelB, His-tag Novagen (Madison/WI, USA)
pGEMT Easy AmpR, f1 ori, lacZ', T7 und SP6-Promotor Promega (Madison/WI, USA)
pGEMSSSF pGEMT Easy mit integriertem PCR-Produkt SSSF aus R. marinus zur weiteren Sequenzierung diese Arbeit
pGEMdd2 pGEMT Easy mit integriertem PCR-Produkt dd2 aus R. marinus zur Sequenzierung
diese Arbeit
pXMJ19 ptac, lacIq, CmR (Jakoby et al., 1999)
pXMJ19 (1-28) pXMJ19 mit verschiedenen Sau 3A-Restriktions-fragmenten aus genomischer DNA von R. marinus diese Arbeit
Tab. 2: Verwendete Plasmide

II. Material & Methoden 19
2. Nährmedien
Je nach Bedarf wurden die Nährmedien mit unterschiedlichem NaCl-Gehalt hergestellt. Die
Prozentangabe (w/v) der zugefügten NaCl-Menge ist der Medienbezeichnung angehängt,
z.B. steht LB-0,5 für LB-Medium mit 0,5 % NaCl. Für die Herstellung von Agarplatten wurde
pro 100 ml Medium 2 g Agar eingewogen.
2.1 Medien zur Anzucht von R. marinus
Komplexmedium M162 (nach DSM 630, l-1) Hefeextrakt 2,5 g Trypton 2,5 g Nitrilotriessigsäure 0,1 g CaSO4 x 2 H2O 0,04 g MgCl2 x 6 H2O 0,2 g 0,01 M FeCitrat 0,5 ml Spurenelemente 0,5 ml Phosphatpuffer 100 ml N
aCl 1-6 %
pH 7,2 mit NaOH (1 M) Phosphatpuffer (l-1) KH2PO4 5,44 g N
a2HPO4 x 12 H2O 43,00 g
pH 7,2 Der Phosphatpuffer wurde getrennt autoklaviert und dann dem Medium zugegeben. Spurenelementlösung (l-1) Nitrilotriessigsäure 12,8 g FeCl2 x 4 H2O 1,00 g MnCl2 x 4 H2O 0,50 g CoCl2 x 4 H2O 0,30 g CuCl2 x 2 H2O 50,00 mg Na2MoO4 x 2 H2O 50,00 mg H3BO3 20,00 mg NiCl2 x 6 H2O 20,00 mg Zunächst wurde Nitrilotriessigsäure in H2Odemin. gelöst, dann der pH-Wert mit KOH (8-9 g)
auf 7,0 eingestellt. Die restlichen Salze wurden der Reihe nach gelöst und der pH schließlich
mit NaOH bzw. H2SO4 auf 6,8 eingestellt.

II. Material & Methoden 20
Modifiziertes Mineralsalzmedium MM162 (Alfredsson et al., 1985), (l-1) CaSO4 x 2 H2O 0,04 g MgCl2 x 6 H2O 0,2 g Nitrilotriessigsäure 0,1 g (NH4)SO4 0,26 g 0,01 M Fe Citrat 0,5 ml N
aCl 20 g
p
H 7,2
C
-Quelle 12,5-50 mM
Spurenelemente 0,5 ml Phosphatpuffer 100 ml Die C-Quelle wurde in einem Teilvolumen getrennt autoklaviert.
2.2 Medien für Experimente mit E. coli und H. elongata
AB-Medium( Antibiotic broth medium No.3, l-1 ) Antibiotic broth Medium 17,5 g N
aCl variabel
pH 7,2 Zur Stammhaltung auf Agarplatten wurde das Medium mit 2 % Agar eingesetzt.
LB-Medium (Miller, 1972), (l-1) Caseinhydrolysat (Trypton) 10 g Hefeextrakt 5,0 g NaCl variabel pH 7,2
Dieses Medium wurde zur Anzucht von Übernachtkulturen, nach der Zugabe von 2 % Agar
auch zum Ausplattieren von Transformationsansätzen genutzt.
Mineralsalzmedium MM63 (Larsen et al., 1987), (l-1) KH2PO4 13,61 g KOH 4,21 g (NH4)2SO4 1,98 g MgSO4 x 7 H2O 0,25 g F eSO4 x 7 H2O 0,0011 g NaCl
variabel
p H 7,2 mit KOH (1M) eingestellt.
Glucose x H2O 5,0 g Agar 2,0 %
Das Medium wurde für Wachstumsexperimente, aber auch zur Selektion einer Konjugation
und für RNA-Isolierungsexperimente verwendet. Für die Herstellung von Flüssigmedium
wurde Glucose getrennt von der Salzlösung sterilisiert und dem Medium nach dem Abkühlen
zugegeben. Zur Herstellung von Agarplatten wurden die Mineralsalze in 0,7 l H2Odemin. gelöst

II. Material & Methoden 21
und getrennt autoklaviert. Glucose wurde zusammen mit 2 % Agar in dem Restvolumen
(0,3 l) gelöst. Beim Wachstum auf unterschiedlichen C-Quellen, wurden diese jeweils aus
einer bereits sterilen Stammlösung der Mineralsalzlösung zugesetzt.
2.3 Medien zur Transformation von E. coli Stämmen
SOB-Medium (Inoue et al., 1990), (l-1) Caseinhydrolysat (Trypton) 20,0 g Hefeextrakt 5,0 g NaCl 0,585 g KCl 0,187 g MgCl2 x 6 H2O 2,033 g MgSO4 x 7 H2O 0,493 g (wurde getrennt sterilfiltriert) pH 6,7
Zur Herstellung superkompetenter Zellen wurde E. coli in diesem Medium über Nacht kulti-
viert.
SOC-Medium (Inoue et al., 1990), (l-1)
SOB-Medium + 20 mM Glucose
Das Medium wurde nach Zugabe von Glucose sterilfiltriert, aliquotiert und bei 4 °C gelagert.
Es wurde zur Regeneration der E. coli Zellen nach der Transformation verwendet.
2.4 Medien für die Herstellung chromosomaler Mutationen bei H. elongata
LBG- Medium (l-1)
Caseinhydrolysat 10 g Hefeextrakt 5,0 g Glucose x H2O 2,0 g NaCl 2,0 g pH 7,2
Bei der Herstellung von H. elongata Mutanten wurde dieses Medium in Schüttelkulturen ein-
gesetzt. Hier erfolgt spontan der zweite Rekombinationsprozess (Cross-Over), der zur Aus-
gliederung des Vektors aus dem Genom führt.
LBG-Saccharose-Medium (l-1) LBG-Medium + Saccharose 220 g Agar 2 % pH 7,2
Dieses Medium dient zur Selektion der H. elongata Stamme, bei denen der zweite Rekom-
binationsprozess stattgefunden hat.
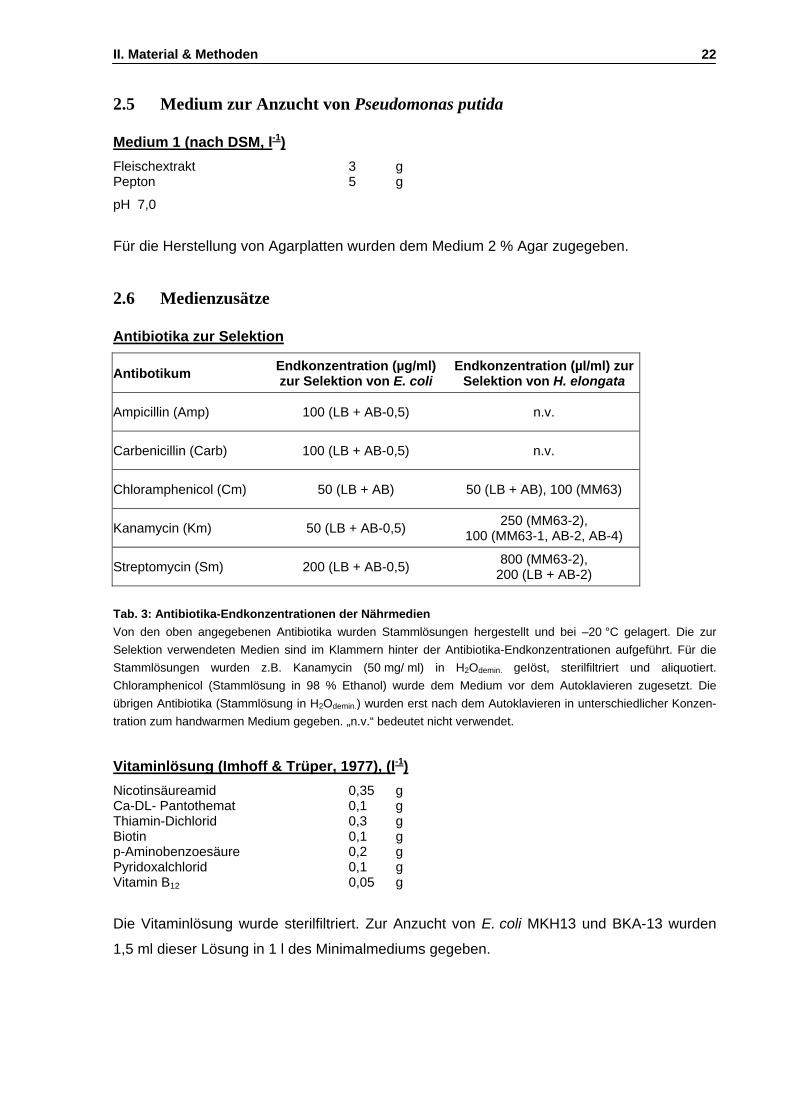
II. Material & Methoden 22
2.5 Medium zur Anzucht von Pseudomonas putida
Medium 1 (nach DSM, l-1) Fleischextrakt 3 g P
epton 5 g
pH 7,0
Für die Herstellung von Agarplatten wurden dem Medium 2 % Agar zugegeben.
2.6 Medienzusätze
Antibiotika zur Selektion
Antibotikum Endkonzentration (µg/ml) zur Selektion von E. coli
Endkonzentration (µl/ml) zur Selektion von H. elongata
Ampicillin (Amp) 100 (LB + AB-0,5) n.v.
Carbenicillin (Carb) 100 (LB + AB-0,5) n.v.
Chloramphenicol (Cm) 50 (LB + AB) 50 (LB + AB), 100 (MM63)
Kanamycin (Km) 50 (LB + AB-0,5) 250 (MM63-2), 100 (MM63-1, AB-2, AB-4)
Streptomycin (Sm) 200 (LB + AB-0,5) 800 (MM63-2), 200 (LB + AB-2)
Tab. 3: Antibiotika-Endkonzentrationen der Nährmedien Von den oben angegebenen Antibiotika wurden Stammlösungen hergestellt und bei –20 °C gelagert. Die zur Selektion verwendeten Medien sind im Klammern hinter der Antibiotika-Endkonzentrationen aufgeführt. Für die Stammlösungen wurden z.B. Kanamycin (50 mg/ ml) in H2Odemin. geIöst, sterilfiltriert und aliquotiert. Chloramphenicol (Stammlösung in 98 % Ethanol) wurde dem Medium vor dem Autoklavieren zugesetzt. Die übrigen Antibiotika (Stammlösung in H2Odemin.) wurden erst nach dem Autoklavieren in unterschiedlicher Konzen-tration zum handwarmen Medium gegeben. „n.v.“ bedeutet nicht verwendet.
Vitaminlösung (Imhoff & Trüper, 1977), (l-1) Nicotinsäureamid 0,35 g Ca-DL- Pantothemat 0,1 g Thiamin-Dichlorid 0,3 g Biotin 0,1 g p-Aminobenzoesäure 0,2 g Pyridoxalchlorid 0,1 g Vitamin B12 0,05 g
Die Vitaminlösung wurde sterilfiltriert. Zur Anzucht von E. coli MKH13 und BKA-13 wurden
1,5 ml dieser Lösung in 1 l des Minimalmediums gegeben.

II. Material & Methoden 23
Kompatible Solute Von den kompatiblen Soluten Mannosylglycerat, Mannosylglyceramid und Trehalose wurden
Stammlösungen mit Konzentrationen von 0,05 M oder 1 M in H2Odemin. hergestellt und steril-
filtriert. Die kompatiblen Solute wurden den Medien nach dem Autoklavieren zugesetzt.
X-Gal und IPTG zur Blau/Weiß-Selektion Viele der verwendeten Plasmide (pHSG575, pGEMT Easy, pK18mobsacB, pBBR1MCS)
tragen ein lacZ’-Fragment, das für das α-Peptid der β-Galaktosidase kodiert. Das entspre-
chende Gen ist in E. coli DH5α deletiert. Die o.g. Plasmide tragen das lacZ’-Fragment inner-
halb der multiplen Klonierungsstelle (MCS), die Schnittstellen für unterschiedliche Restrik-
tionsenzyme enthält. Transformiert man E. coli DH5α mit einem dieser Vektoren, kommt es
aufgrund der sog. α-Komplementierung zur Bildung einer funktionellen β-Galaktosidase. Eine
Insertion von Fremd-DNA innerhalb der MCS bewirkt eine Inaktivierung des lacZ’-Fragments
und so der β-Galaktosidase. Durch Zugabe des Induktors IPTG (Isopropyl-1-thio-β-D-
Galaktopyranosid) und von X-Gal (5-Bromo-4-chloro-3-indolyl-β-Galaktopyranosid), das von
der β-Galaktosidase als Substrat erkannt und zu einem blauen Indolfarbstoff umgesetzt wird,
kann man aufgrund der Blaufärbung zwischen nicht-rekombinanten (blau) und rekombinan-
ten (weiß) Klonen unterscheiden. Für die Blau/Weiß-Selektion wurden pro Agarplatte je 10 µl
einer IPTG-Stammlösung (0,1 M) und 50 µl einer X-Gal-Stammlösung (0,05 M in DMSO)
zusammen mit den Zellen ausplattiert.
Selektion von H. elongata Doppel-Cross-Over Mutanten mit Saccharose Zur Herstellung von H. elongata Deletions- bzw. Insertionsmutanten, die das zuvor über
homologe Rekombination ins Genom integrierte rekombinante Derivat des Vektors pK18
mobsacB nach einem Doppel-Cross-Over wieder verloren haben, wurden auf LBG-Medium
mit 22 % Saccharose selektioniert. Das Plasmid pK18mobsacB trägt ein modifiziertes sacB-
Gen aus Bacillus subtilis, das für eine Laevan-Saccharase kodiert (Schäfer et al., 1994). Die
Expression dieses Enzyms ist für H. elongata ab einer Saccharose-Konzentration von 20 %
letal und kann daher für den positiven Nachweis von Doppel-Cross-Over-Mutanten genutzt
werden (Göller, 1999). Durch den Verlust des Plasmids entsteht dabei entweder der Ur-
sprungsstamm oder die gewünschte Mutante.
C-Quellen zur Charakterisierung des Substratspektrums von R. marinus
Zur Charakterisierung des Substratspektrums von R. marinus, wurden dem Anzuchtmedium
verschiedene C-Quellen zugesetzt. Als Anzuchtmedium wurde das Minimalmedium MM162
verwendet, das von Alfredsson et al. (1985) für Thermus Arten beschrieben wurde. Da für
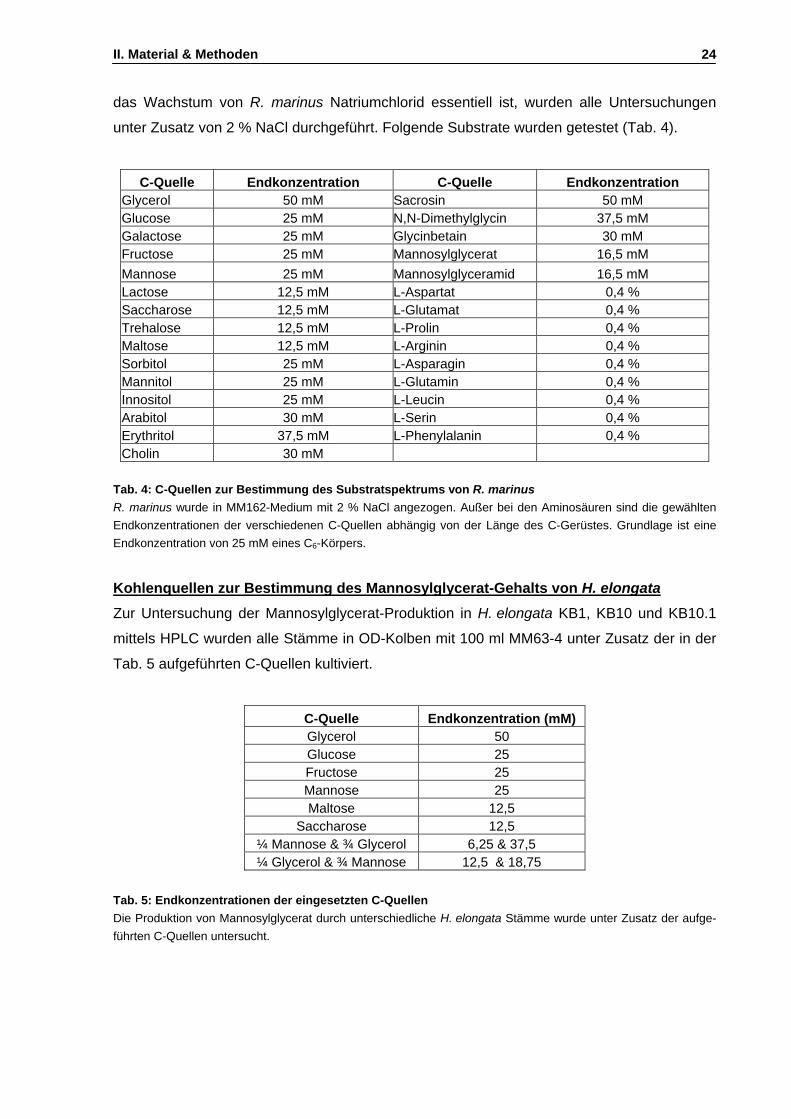
II. Material & Methoden 24
das Wachstum von R. marinus Natriumchlorid essentiell ist, wurden alle Untersuchungen
unter Zusatz von 2 % NaCl durchgeführt. Folgende Substrate wurden getestet (Tab. 4).
C-Quelle Endkonzentration C-Quelle Endkonzentration Glycerol 50 mM Sacrosin 50 mM Glucose 25 mM N,N-Dimethylglycin 37,5 mM Galactose 25 mM Glycinbetain 30 mM Fructose 25 mM Mannosylglycerat 16,5 mM Mannose 25 mM Mannosylglyceramid 16,5 mM Lactose 12,5 mM L-Aspartat 0,4 % Saccharose 12,5 mM L-Glutamat 0,4 % Trehalose 12,5 mM L-Prolin 0,4 % Maltose 12,5 mM L-Arginin 0,4 % Sorbitol 25 mM L-Asparagin 0,4 % Mannitol 25 mM L-Glutamin 0,4 % Innositol 25 mM L-Leucin 0,4 % Arabitol 30 mM L-Serin 0,4 % Erythritol 37,5 mM L-Phenylalanin 0,4 % Cholin 30 mM
Tab. 4: C-Quellen zur Bestimmung des Substratspektrums von R. marinus R. marinus wurde in MM162-Medium mit 2 % NaCl angezogen. Außer bei den Aminosäuren sind die gewählten Endkonzentrationen der verschiedenen C-Quellen abhängig von der Länge des C-Gerüstes. Grundlage ist eine Endkonzentration von 25 mM eines C6-Körpers.
Kohlenquellen zur Bestimmung des Mannosylglycerat-Gehalts von H. elongata Zur Untersuchung der Mannosylglycerat-Produktion in H. elongata KB1, KB10 und KB10.1
mittels HPLC wurden alle Stämme in OD-Kolben mit 100 ml MM63-4 unter Zusatz der in der
Tab. 5 aufgeführten C-Quellen kultiviert.
C-Quelle Endkonzentration (mM) Glycerol 50 Glucose 25 Fructose 25 Mannose 25 Maltose 12,5
Saccharose 12,5 ¼ Mannose & ¾ Glycerol 6,25 & 37,5 ¼ Glycerol & ¾ Mannose 12,5 & 18,75
Tab. 5: Endkonzentrationen der eingesetzten C-Quellen Die Produktion von Mannosylglycerat durch unterschiedliche H. elongata Stämme wurde unter Zusatz der aufge-führten C-Quellen untersucht.

II. Material & Methoden 25
3. Puffer und Lösungen
Puffer und Lösungen für DNA-Arbeiten (Isolierung und Lagerung) EB-Puffer 10 mM Tris-HCl, pH 8,5 TE-Puffer 10 mM Tris-HCl, 1 mM EDTA, pH 7,5 TES-Puffer 50 mM Tris-HCl, 5 mM EDTA, 50 mM NaCl, pH 8,0 Natriumacetat-Puffer 3 M Na-Acetat, pH 4,8 SDS-Lösung 20 % (w/v) SDS in H2O reinst Puffer und Lösungen für die Agarosegelelektrophorese von DNA TA-Puffer (10x) 2 M Tris-HCl, 1 M Eisessig, 50 mM EDTA, pH 8,0 TB-Puffer (10x) 0,9 M Tris-HCl, 0,9 M Borsäure, 25 mM EDTA, pH 8,3 Ladepuffer (6x) 60 % (v/v) Glycerol, 60 mM EDTA und 0,09 % (w/v) Brom- phenolblau, pH 8,0 Puffer und Lösungen für RNA-Isolierung Puffer A 50 mM Na-Acetat x 3 H2O, 10 mM EDTA, pH 4,5-5 Phenol Aqua-Roti-Phenol mit Puffer A überschichtet, pH 4,5- 5 Chloroform/Isoamylalkohol 24 ml Chloroform, 1 ml Isoamylalkohol Phenol:Chlorof./Isoamylalk. Phenol:Chloroform/Isoamyalkohol-Gemisch (1:2) Na-Acetat 3 M in H2Oreinst SDS-Lösung 10 % in H2Oreinst Puffer und Lösungen für Agarosegelelektrophoresen von RNA Phosphatpuffer 10x 0,1 M KH2PO4/ K2HPO4, pH 6,8
Bei der Herstellung des Puffers wurden zunächst Stammlösungen (0,2 M) beider Salze an-
gesetzt. Dann wurden 255 ml der KH2PO4-Lösung mit 245 ml der K2HPO4-Lösung gemischt
und auf ein Endvolumen von 1 l mit H2Odemin. aufgefüllt.
Ladepuffer 50 % (v/v) Glycerol, 0,05 % (w/v) Bromphenolblau, in 1x Phosphat-Puffer, pH 6,8 NaOH 0,1 M in H2O reinst.
Natrium-Acetat 0,2 M in H2O reinst. pH 5,2 Glyoxal 6 M DMSO 100 % DEPC 0,1 % in H2Oreinst
Puffer und Lösungen für Southern-Hybridisierungen Denaturierungslösung 0,4 M NaOH Neutralisierungslösung 0,5 M Tris-HCl, 3 M NaCl, pH 7,5 SDS-Lösung 10 % (w/v) SDS in H2Odemin.
20x SSC 3 M NaCl, 0,3 M NaCitrat x 2 H2O, pH 7,5 6x SSC 75 ml 20x SSC ad. 250 ml H2Odemin.

II. Material & Methoden 26
SSC-A 2x SSC, 0,1 % SDS SSC-B 0,1x SSC , 0,1 % SDS Vorhybridisierungslsg. 25 ml 20x SSC, 0,3 ml (35 %-ig) Lauroylsarcosin, 0,2 ml 10 %
(w/v) SDS, 0,5 g Blockingreagenz, ad. 100 ml Hybridisierungslsg. 10 ml Vorhybridisierungslösung mit (2-25 ng/ml) denaturierter
Digoxigenin (DIG) markierter Sonden-DNA Puffer 1 100 mM Tris-HCl, 150 mM NaCl, pH 7,5 (bis 10x) Puffer 2 0,5 % (w/v) Blockingreagenz in Puffer 1 Puffer 3 100 mM Tris-HCl, 100 mM NaCl, 50 mM MgCl2, pH 9,5
(nicht autoklavieren, nur kurz aufkochen) Puffer 4 10 mM Tris-HCl, 1 mM EDTA, pH 8,0 (bis 10x) Färbelösung 35 µl X-Phosphat und 33,7 µl NBT in 10 ml Puffer 3 Puffer und Lösungen für Northern-Hybridisierungen 20x SSC Puffer 3 M NaCl, 0,3 M Na-Citrat, pH 7,5 2x SSC Puffer 2x SSC, 0,1 % SDS 0,1x SSC Puffer 0,1x SSC, 0,1 % SDS Maleinsäure-Puffer 0,1 M Maleinsäure, 0,15 M NaCl, pH 7,5 mit NaOH (fest) Waschpuffer 0,3 % (v/v) Tween 20 in Maleinsäure-Puffer Blockierungslösung 1 % Blockingreagenz in Maleinsäure-Puffer Vorhybridisierungslösung DIG-EASY-Hyb. (Roche) 20 ml Hybridisierungslösung 100 ng RNA-Sonde in 10 ml DIG-EASY-Hyb. Antikörperlösung 1 µl Anti-DIG-AP Konjugat in 20 ml Blockierungslösung Detektionspuffer 0,1 M Tris-HCl, 0,1 M NaCl, pH 9,5 CDP-StarTM (Roche) 10 µl CDP-StarTM in 1 ml Detektionspuffer Puffer und Reagenzien für die Proteinisolierung 10x PBS-Puffer 1,37 M NaCl, 27 mM KCl, 19 mM KH2PO4, 82 mM, Na2HPO4,
pH 7,2 Resuspensionspuffer I 20 mM Tris-HCl, 0,5 M NaCl, pH 7,5 Resuspensionspuffer II 20 mM Tris-HCl, 0,5 M NaCl, pH 8,0 Lysozym-Stammlösung I 10 mg/ml Lysozym in H2Oreinst
Lysozym-Stammlösung II 100 mg/ml Lysozym in H2Oreinst
SDS-Lösung (10 %) 10 % SDS in H2Oreinst
SDS-Lösung (1 %) 1 % SDS in H2Oreinst
MgCl2-Stammlösung 50 mM MgCl x 6 H2O DNAse-Stammlösung 10 mg/ml DNAse in H2O Schockpuffer 30 mM Tris-HCl, 20 % Saccharose, 20 % NaCl, pH 8,0 EDTA-Lösung 0,5 M Na-EDTA in H2Oreinst
TCA-Lösung 100 % TCA in H2Oreinst
Puffer und Lösungen für Polyacrylamid-Gelelektrophorese Sammelgelpuffer 0,5 M Tris-HCl, pH 6,8 Trenngelpuffer 1,5 M Tris-HCl, pH 8,8 Laufpuffer SDS-PAGE 50 mM Tris-HCl, 0,37 M Glycin, 0,1 % (w/v) SDS, pH 8,3-8,6 den./red. Probenpuffer (2x) 0,5 M Tris-HCl, 20 % (v/v) Glycerol, 4 % SDS, 1,25 %

II. Material & Methoden 27
β-Mercaptoethanol, Spatelspitze Bromphenolblau Coomassie-Färbelösung 50 % (v/v) Methanol, 10 % (v/v) Eisessig, 0,115 %
Coomassie-brilliant-blue R250 (ÜN rühren, dann filtern) Coomassie-Entfärbelösung 10 % (v/v) Ethanol, 10 % (v/v) Eisessig, 0,8 l H2Odemin.
SDS-Lösung 10 % (w/v) H2Odemin.
Puffer und Lösungen für Western-Hybridisierung Towbin-Puffer 25 mM Tris-HCl, 192 mM Glycin, 20 % (v/v) Methanol,
pH 8,3 (nicht titrieren) TBS 10 mM Tris-HCl, 150 mM NaCl, pH 7,5 TBSTT 20 mM Tris-HCl, 0,5 M NaCl, pH 7,5, dann 0,05 % (v/v)
Tween 20, 0,2 % (v/v) TritonX-100 AP-Puffer 100 mM Tris-HCl, 100 mM NaCl, 1 mM MgCl2, pH 9,4 Blockierungreagenz TBS mit 1 % (w/v) Casein, 3 % (w/v) BSA Fraktion V,
pH auf 7,5 nachtitrieren Lsg. sekundärer Antikörper TBS mit 1 % (w/v) Casein, 10 % (w/v) Magermilchpulver Puffer für die Transformation TFB-Puffer 10 mM PIPES, 15 mM CaCl2, 250 mM KCl, 55 mM MnCl2
(sterilfiltriert zugesetzt), pH 6,7 Lösungen und Reagenzien für die Dünnschichtchromatographie TCA-Lösung 12 % TCA-Lösung in H2Oreinst
Laufmittel Ammoniak (25 %-ig) und n-Propanol im Verhältnis 1:1 Färbereagenz 10,5 ml 15 %-iges α-Naphtol (in Ethanol) wurden mit 4 ml
H2Oreinst gemischt. 40,5 ml Ethanol (99 %) und 6,5 ml Schwefelsäure (95-97 %) wurden hinzugegeben. Alle Bestandteile wurden frisch gemischt und im Dunkeln aufbewahrt.
Lösungen für die HPLC Bligh & Dyer-Lsg. 10 Teile Methanol, 5 Teile Chloroform, 4 Teile H2Oreinst
Laufmittel (Mannosylglycerat) 0,01 N H2SO4 in H2Oreinst
Laufmittel (Ectoin) Acetonitril & H2Oreinst im Volumenverhältnis 80:20
4. Kultivierungsverfahren
4.1 Stammhaltung von Bakterienstämmen Zur kurzfristigen Stammhaltung wurde R. marinus DSM 4252T in M162-2-Medium ange-
zogen und dann als Glycerolkultur bei –70 °C gelagert. Zur langfristigen Lagerung von
R. marinus wurde das Mikrobank-System (Prolab, Diagnostics, Kanada) verwendet.
Stammkulturen von H. elongata wurden abwechselnd auf Agarplatten mit AB-Medium (2-
6 % NaCl) und MM63 (2-6 % NaCl) angezogen. Die Stammkulturen wurden im Kühlschrank

II. Material & Methoden 28
(4 °C) bis zu 2 Monaten gelagert. Alle Stämme mit einer ectA-Deletion wurden ausschließlich
auf AB-2 Medium gehalten. Zur langfristigen Lagerung aller Stämme wurde das Mikrobank-
System verwendet.
Alle E. coli Stämme wurden zur Stammhaltung auf Agarplatten mit AB-Medium (0,5 % NaCl)
angezogen. Stämme, die über einen längeren Zeitraum aufbewahrt werden sollten, wurden
nach Anzucht in AB-Flüssigmedium als Glycerolkultur bei –20 °C gelagert.
Zur Herstellung von Glycerolkulturen wurden die Stämme in 25 ml Flüssigmedium (M162-
Medium für R. marinus bzw. AB-Medium für H. elongata und E. coli) angezogen. Zu jeweils
850 µl der Kultur wurden 150 µl Glycerol (Endkonzentration 15 % v/v) gegeben. Die Kultur
wurde anschließend gut gemischt und bei –20 °C gelagert.
4.2 Bakterienanzucht
4.2.1 Allgemeine Bakterienanzucht und Zellernte
Die Anzucht der Bakterienstämme erfolgte standardmäßig in sog. OD-Kolben (250 ml
Volumen). Diese weisen ein seitlich angebrachtes Glasrohr auf. Es dient der direkten
Verfolgung der optischen Dichte, die in einem Novaspec II Photometer (Pharmacia, Uppsala,
Schweden) bei einer Wellenlänge von 600 nm (R. marinus und H. elongata) bzw. 540 nm
(E. coli) gemessen wurde. Die Wachstumsraten der Kulturen wurde in der exponentiellen
Wachstumsphase bestimmt.
Größere Volumina (bis 250 ml) von Zellkulturen wurden in einer Zentrifuge Typ Sorvall RC
5B (DuPont, Bad Homburg) bei 10425x g (20 min, RT) in einem GS-3 Rotor bzw. in einer
Sorvall AvantiTM J-20 XP Zentrifuge (Beckman Coulter, Krefeld) bei 17700x g (20 min, RT) in
einem JA-10 Rotor geerntet. Kleinere Volumina (bis 50 ml) wurden bei 11325x g in einem J-
25.25 Rotor (15 min) abgeerntet. Bei jeder Zellernte wurde das restliche Zellzwischenwasser
durch Ausstreichen des Zellpellets auf Filterpapier entfernt. Anschließend wurde das Pellet
für mindestens 1 Stunde eingefroren (-20 °C) und durch Gefriertrocknung getrocknet.
4.2.2 Anzucht von Rhodothermus marinus
Die Anzucht von R. marinus erfolgte im Wasserbad bei 65 °C und 120 upm. Für die Anzucht
ist stets eine Vorkultur in Komplexmedium (M162-2) erforderlich. Dafür werden je 0,5 ml
einer Glycerolkultur zu 30 ml vorgewärmtem (65 °C) M162-2 Medium gegeben und über
Nacht angezogen. Zur Charakterisierung des Substratspektrums von R. marinus, wurden
dem Anzuchtmedium M162-2 verschiedene C-Quellen zugesetzt (Tab. 4) .
Um die Zellinhaltsstoffe von R. marinus NMR-spektroskopisch zu vermessen, wurde der
Organismus in MM162-3,5 angezogen. Die Vorkultur (50 ml MM162-3,5 mit Glucose) wurde
bei Erreichen einer OD600 von 1,0 in die Hauptkulturen (Start OD600 = 0,1) überimpft. Es

II. Material & Methoden 29
wurden sechs Hauptkulturen mit je 100 ml MM162-3,5 im Wasserbad angezogen. Beim
Erreichen einer OD600 von 0,4 wurde die gesamte Bakterienkultur geerntet.
Zur Analyse des Mannosylglycerat-Gehalts mittels HPLC wurde R. marinus in je 100 ml
M162- (1-4 % NaCl) bzw. MM162-Medium (1-4 % NaCl) angezogen. Die Zellernte erfolgte in
der spätexponentiellen Phase bei einer OD600 zwischen 0,6 und 0,8.
4.2.3 Anzucht von H. elongata
Die Anzucht von H. elongata erfolgte standardmäßig bei 30 °C, im Falle von Flüssigkulturen
bei 180 upm. Die Anzucht für die Isolierung genomischer DNA und für Konjugationsexperi-
mente erfolgte in 100 ml LB-3 oder AB-3-Medium im 250 ml OD-Kolben.
Für Wachstumsexperimente wurde Mineralsalzmedium verwendet. Die Hauptkulturen
wurden mit den entsprechenden Vorkulturen angeimpft (Start OD600 = 0,05-0,1) für die
dasselbe Medium verwendet wurde. Die Wachstumsratenbestimmung erfolgte unter Zugabe
verschiedener C-Quellen, bei unterschiedlichen Temperaturen.
Zur Bestimmung des Mannosylglycerat-Gehalts von H. elongata KB1, KB10 und KB10.1
wurden die Stämme in OD-Kolben mit 100 bzw. 250 ml MM63-4 unter Zusatz verschiedener
C-Quellen (Tab. 5) kultiviert. Die Hauptkultur wurde mit einer Vorkultur auf eine Ausgangs-
OD600 von 0,1 angeimpft und bei 37 °C angezogen. Der Wachstumsverlauf der Kulturen
wurde im OD-Kolben (250 ml) direkt gemessen. Für die Bestimmung des Mannosylglycerat-
Gehalts wurden die Kulturen bei Erreichen einer OD600 von 1,0 geerntet und anschließend
gefriergetrocknet.
Bei H. elongata KB10.1 wurde die Produktion von Mannosylglycerat sowohl in Abhängigkeit
von ausgewählten C-Quellen (Mannose bzw. Glucose) als auch verschiedenen Inkubations-
temperaturen (37 und 40 °C) bestimmt.
Zusätzlich wurde eine Abhängigkeit der Mannosylglycerat-Produktion von der Wachstums-
phase untersucht. Dafür wurde KB10.1 in AB-4-Vorkultur (30 ml) über Nacht bei 37 °C
angezogen. Die zweite Vorkultur (100 ml MM63-4, mit Mannose bzw. Glucose) wurde 2 %-ig
angeimpft und bei 37 °C bzw. 40 °C inkubiert. Die Hauptkulturen (250 ml MM63-4 im 1 l OD-
Kolben) wurden auf eine OD600 von 0,1 angeimpft und wiederum bei 37 °C bzw. 40 °C
angezogen. Die OD600-Werte wurden ständig verfolgt. Bei Erreichen verschiedener Wachs-
tumsphasen, wurden die Kolben abgeerntet und für eine HPLC-Analyse vorbereitet.
Um die Zellinhaltsstoffe der H. elongata Stämme KB1 und KB10.1 NMR-spektroskopisch zu
vermessen, wurden die Stämme in MM63-4 (KB10.1 mit Mannose, KB1 mit Glucose als C-
Quelle) angezogen. Die Vorkulturen wurden in 50 ml MM63-4 Medium bei 30 °C (KB1) bzw.
40 °C (KB10.1) und 180 upm angezogen. Bei Erreichen einer OD600 von 1,0 wurden die
Vorkulturen in die Hauptkulturen (Ausgangs-OD600 0,1) überimpft. Von den H. elongata
Stämmen wurden jeweils sechs 1 l OD-Kolben (je 250 ml MM63-4) bei 160 upm im Schüttler

II. Material & Methoden 30
angezogen. Beim Erreichen einer OD600 von 1,4 (KB1) bzw. 0,4 (KB10.1) wurde jeweils die
gesamte Bakterienkultur geerntet.
4.2.4 Anzucht von E. coli Stämmen
Die Anzucht von E. coli zur Isolierung genomischer DNA oder Plasmid-DNA sowie zur Her-
stellung von superkompetenten Zellen und Konjugationsexperimenten erfolgte in 30-50 ml
Flüssigmedium in einen 100 ml Erlenmeyerkolben mit Schikane (37 °C und 180 upm). Zur
Bestimmung der Salztoleranz von E. coli MKH13 und BKA-13 wurden MM63-Agarplatten
steigender Salinität (1 %, 2 %, 2,5 %, 2,7 %, 2,8 %, 3,0 % und 4,0 %) angefertigt. MKH13
und BKA-13 wurde in 20 ml LB-0,5 bis zu einer OD540 von 1,2 angezogen. 1 ml dieser
Kulturen wurden entnommen und fünfmal mit 1 ml MM63-0,5-Medium (ohne C-Quelle) ge-
waschen. Die OD540 wurde auf 0,1 eingestellt (in MM63-0,5). Anschließend wurden 50–
100 µl der Zellsuspension auf Agarplatten ausplattiert.
5. Analytik und Nachweis kompatibler Solute
5.1 Gefriertrocknung Das Zellmaterial der Bakterien wurde nach der Ernte gefriergetrocknet. Bei dieser Methode
wird das Zellzwischenwasser der gefrorenen Probe durch Sublimation schonend entfernt.
Zur Durchführung wurden die mit durchlöchertem Parafilm verschlossenen Probengefäße in
die Gefriertrocknungsanlage (Christ Alpha-I-6, Heraeus) gestellt und für 24 Stunden bei
einem Druck unter 0,05 mbar und 25-30 °C Heizflächentemperatur getrocknet. Das getrock-
nete Zellmaterial wurde in geschlossenen Reaktionsgefäßen bei Raumtemperatur gelagert.
5.2 Dünnschichtchromatographie zum Zuckernachweis Bei der Dünnschichtchromatographie strömt eine mobile Phase (Lösungsmittelgemisch) in
Folge der Kapillarwirkung über eine stationäre Phase (meistens aus polaren Materialien wie
z.B. Kieselgel oder Aluminiumoxid), die mit der mobilen Phase nicht mischbar ist. Die in der
mobilen Phase gelösten aufzutrennenden Substanzen haben unterschiedliche Affinität zur
mobilen bzw. stationären Phase. Hierdurch wird das Trenngemisch in seine Einzelkompo-
nenten zerlegt. Die einzelnen Komponenten zeigen unterschiedliche als Flecken sichtbare
Lauffronten, die alle in einer Linie zwischen dem Startpunkt und der Lösungsmittelfront
liegen. Daraus resultieren unterschiedliche Laufstrecken der einzelnen gelösten Substanzen,
die in Relation zur Laufmittelfront gesetzt werden (Rf-Wert, retarding factor).
Rf-Wert = (Weg der Substanz vom Start) / (Weg der Fließmittelfront vom Start)

II. Material & Methoden 31
Der Rf-Wert dient zur Beschreibung des Laufverhaltens einer aufgetrennten Substanz. Es
handelt sich beim ihm jedoch nur um einen Richtwert, der durch unterschiedliche Trenn-
bedingungen variieren kann. Bei einem Vergleich zwischen zwei Substanzen muss daher
immer mit einem Standard verglichen werden, wobei beide auf derselben Platte aufgetragen
werden müssen. Nach der Auftrennung können die einzelnen Komponenten entweder an
ihrer Eigenfarbe im Tageslicht erkannt werden oder sie müssen durch Reagenzien oder
andere Hilfsmittel sichtbar gemacht werden.
5.2.1 Anzucht der Kulturen und Probenaufbereitung
H. elongata KB10.1 wurde bei 37 °C in MM63-4 (100 ml) unter Zusatz verschiedener C-
Quellen angezogen. Bei einer OD600 von 1 wurde 1 ml Kultur entnommen und in einer Tisch-
zentrifuge 1 min bei 8000x g zentrifugiert (Eppendorf 5415). Der Überstand wurde verworfen
und das Pellet in 5 µl 12 %-igen TCA-Lösung resuspendiert. Die Proben wurden dann 10 min
auf Eis gestellt und wiederum 1 min zentrifugiert, der Überstand wurde bis zur Durchführung
der Dünnschichtchromatographie bei 4 °C gelagert. Alternativ wurden die Proben wie in Ab-
schnitt II 5.3.1 aufgearbeitet.
5.2.2 Auftrennung von Zuckern und Mannosylglycerat
Für die chromatographische Auftrennung wurde je 30 µl Probe eingesetzt. Die Proben
wurden ca. 1 cm vom unteren Rand entfernt auf eine Aluminium Dünnschichtchromato-
graphieplatte (Silica 60, Merck) aufgetragen und mit einem handelsüblichen Haartrockner
getrocknet. Zur Auftrennung der Zucker (z.B. Mannose) bzw. Zuckerderivate (z.B. Mannosyl-
glycerat) wurde als mobile Phase ein n-Propanol/Ammoniak (1:1, v/v) Gemisch eingesetzt.
Die Laufzeit der Chromatographie betrug 1-2 h. Sie wurde in einer geschlossenen Glaskam-
mer durchgeführt, in der die Atmosphäre mit dem n-Propanol/Ammoniak Gemisch gesättigt
war.
Die Visualisierung der substanzspezifischen Banden erfolgte nach Molisch & Biebl (1955).
Dazu wurden die Platten mit einem α-Naphtol-Schwefelsäurereagenz besprüht und
anschließend für 5 min bei 120 °C inkubiert. Zur Dokumentation wurden die Platten
eingescannt. In Gegenwart von konzentrierter Schwefelsäure (H2SO4 95-97 %) werden
Zucker unter Bildung von Furfuryl-Derivaten dehydratisiert. Bei der Dehydratisierung von
Pentosen entsteht Furfural, während aus Hexosen Hydroxymethylfurfural gebildet wird. α-
Naphtol reagiert mit diesen zyklischen Aldehyden und bildet pupurviolett gefärbte Konden-
sationsprodukte (Furfuryl-diphenylmethan-Farbstoffe, Abb. 3). Di-, Oligo- und Poly-
saccharide werden durch die Säure hydrolysiert, wodurch man die Monomere erhält. Somit
stellt diese Nachweismethode einen allgemeinen Test zum Nachweis von Kohlenhydraten
dar.

II. Material & Methoden 32
OO
HOH
H
H
OH
OH
OOHH
H
O
OH
-H2O+
5-Hydroxymethyl-furfural Naphthol-(1) Furfuryl-diphenyl-methan-Farbstoff
Abb. 3: Zuckernachweis nach (Molisch & Biebl, 1955) Die aldehydischen Furfuryl-Derivate kondensieren mit α-Naphtol zu einem pupurvioletten Komplexfarbstoff.
5.3 Hochleistungsflüssigkeitschromatographie (HPLC) Zur quantitativen und qualitativen Analyse komplexer Substanzgemische bedient man sich
der HPLC. Bei dieser werden die in der mobilen Phase gelöste Substanzen unter hohem
Druck über eine Säule (stationäre Phase) gepumpt. Dabei interagieren die gelösten Subs-
tanzen, z.B. abhängig von ihrer Hydrophobizität, verschieden stark mit der stationäre Phase,
so dass ihr Laufverhalten unterschiedlich verzögert wird, was eine Auftrennung der Subs-
tanzen bewirkt. Die Elution der Substanzen wird über einen nachgeschalteten Detektor
verfolgt, der die Absorptionsveränderungen der eluierten Lösung misst. Anhand der Retenti-
onszeiten (Zeit zwischen Start des Programms und der Elution der Substanz) und Absorpti-
onseigenschaften können die aufgetrennten Substanzen schließlich über Vergleich mit eben-
falls aufgetrennten Standardsubstanzen bekannter Konzentration in dem Chromatogramm
identifiziert werden.
5.3.1 Probenaufbereitung für die HPLC-Analyse
(Bligh & Dyer, 1959; modifiziert nach Galinski & Herzog, 1990)
Bei diesem Extraktionsverfahren wird mittels eines Chloroform/Methanol/Wasser-Gemischs
(10:5:4) und durch Zentrifugation eine Phasentrennung erzielt. Dabei sammeln sich die
denaturierten Proteine an der Phasengrenze als kompakte Masse, während sich die übrigen
Zellbestandteile abhängig von ihrer Löslichkeit auf die obere polare Methanol/Wasser-Phase
oder die untere unpolare Chloroform-Phase verteilen. Auf diese Weise werden die wasser-
löslichen kompatiblen Solute einer qualitativen und quantitativen Analyse durch HPLC
zugänglich gemacht.
Durchführung:
• 30 mg gefriergetrocknetes Zellmaterial wurden eingewogen, mit 500 µl Bligh & Dyer
Lösung versetzt und 30 min auf einem Schüttler (IKA Vibrax, Janke & Kunkel, Staufen)
bei Raumtemperatur inkubiert.

II. Material & Methoden 33
• Nach Zugabe von 130 µl Chloroform und 130 µl HPLC-Wasser wurde der Ansatz für
weitere 10 min bei RT geschüttelt.
• Zur Phasentrennung wurde die Proben 5 min in einer Tischzentrifuge bei 8000x g
zentrifugiert und die obere Phase (Methanol/Wasser) in ein neues Eppendorf-Reakti-
onsgefäß überführt und bis zu weiteren Verwendung bei -20 °C gelagert.
5.3.2 Isokratische Hochleistungsflüssigkeitschromatographie
Die verwendete isokratische HPLC erlaubt eine Analyse der in den Zellextrakten enthalte-
nen ungeladenen und geladenen kompatiblen Solute verschiedener Stoffklassen. Die Detek-
tion erfolgt entweder mittels eines Brechungsindex- oder eines UV-Detektors. Mit dem Bre-
chungsindexdetektor können unterschiedliche Substanzen, wie Zucker und Zuckerderivate
(z.B. Trehalose, Mannosylglycerat), organische Säure (z.B. Citrat, Malat), Polyalkohole (z.B.
Glycerol) und Tetrahydropyrimidine (Ectoin und Hydroxyectoin) nachgewiesen werden, wäh-
rend der UV-Detektor zu Detektion der Tetrahydropyrimidine auch in sehr geringer Konzen-
tration genutzt wird.
Zum Nachweis von Tetrahydropyrimidinen wurde eine HPLC-Säule verwendet, deren stati-
onäre Phase aus einem Umkehrphasenmaterial, das mit Aminogruppen modifiziert ist und
als Kationenaustauscher fungierte, bestand. Als mobile Phase diente ein Acetonitril/Wasser-
Gemisch mit 80 % (v/v) Acetonitril. Es wurde eine Flussrate von 1 ml/min gewählt. Als
Standard wurde Ectoin in Konzentrationen von 0,5-1 mM eingesetzt. Die HPLC-Apparatur
setzte sich aus folgenden Einzelkomponenten zusammen:
Software: ChromQuest Version 2.51, Thermo Quest Cooperation
Pumpe: Spectra System P100, Firma Thermo Seperation Products
Entgaser: SCM Vacuum Membrane Degasser, (Firma Thermo Seperation Products)
Probenaufgabe: Rheodyne Injektor Nr. 7125, Probenschleife 20 µl (Firma Rheodyne Inc.)
Trennsäule: Grom-Sil Amin-1PR, 3 µm, 125x4 mm, LiChrocart-System, Refill Alltech Grom
GmbH
Detektor: UV-Detektor Spectrasystem UV 1000, 210 nm, Firma Thermo Seperation
Products
Für den Nachweis von Zuckern (z.B. Mannose) und Zuckerderivaten (Mannosylglycerat),
bzw. Polyalkoholen (z.B. Glycerol) wurde eine Trennsäule Aminex HPX-87H (BioRad), die
speziell für die Trennung von organischen Säuren und Kohlenhydraten geeignet ist,
herangezogen. Die stationäre Phase der Säule bestand aus einer polymerbasierten Matrix,
die als Umkehrphase fungierte. Als mobile Phase wurde 0,01 N H2SO4 in Reinstwasser
eingesetzt. Als Flussrate für die Trennung der Substanzen wurde 0,6 ml/min gewählt. Als
Standardsubstanzen wurden Mannosylglycerat, Trehalose und alle Fütterungssubstanzen

II. Material & Methoden 34
(Tab. 5) in Konzentrationen von 5 mM und 10 mM eingesetzt. Die verwendete HPLC-
Apparatur bestand aus folgenden Einzelkomponenten:
Software: Eurochrom 2000, Version 2.05, Firma Knauer GmbH
Pumpe: K-120, Pumpenkopf 10 ml/min, Keramik–Inlays, Firma Knauer GmbH
Entgaser: SCM Vacuum Membrane Degasser, Firma Thermo Seperation Products
Probenaufgabe: Rheodyne Injektor Nr. 7125, Probenschleife 20 µl ,Firma Rheodyne Inc.
Trennsäule: Aminex HPX-87H, 300 x 7,8 mm, Firma BioRad
Detektor: Brechungsindexdetektor refractoMonitor IV, Firma LDC, Milton Roy
5.4 Kernresonanz-Spektroskopie („nuclear magnetic resonance“) NMR Die Kernresonanz-Spektroskopie basiert auf dem Kernmagnetismus. Einige Atomkerne rotie-
ren um ihre eigene Achse. Dieser Eigendrehimpuls wird als Kernspin bezeichnet. Mit dem
Drehimpuls (I) ist ein magnetisches Drehmoment (µ) verknüpft. Beide sind zueinander pro-
portional und führen mit einer bestimmten Frequenz (Larmor-Frequenz) eine Präzisions-
bewegung um die Richtung des Magnetfelds aus. Protonen und Neutronen tragen auch zum
Kernspin bei. Gerade Massen und Atomzahlen ergeben einen Kernspin von I = 0 und sind
somit für die NMR ungeeignet. Alle natürlich vorkommenden oder substituierten Isotope mit
einem von Null verschiedenem Kernspin (I) sind für die NMR geeignet.
Durch Anlegen eines äußeren Magnetfelds werden die Kernspins in eine bestimmte Aus-
richtung gebracht. Die Energie der eingestrahlten Radiowellen wird absorbiert und die Kern-
spins so vom energiearmen in einen energiereicheren Zustand angeregt. Der energiereiche
Spinzustand relaxiert anschließend, indem er die entsprechende Energiemenge in Form
einer bestimmten Frequenz abgibt. Diese Resonanzfrequenz wird gemessen. Es kommt
immer dann zur Resonanzabsorption, wenn die Frequenz eines Atomkerns mit der Mess-
frequenz übereinstimmt. Die Resonanzfrequenz ist abhängig von der chemischen Umge-
bung des Kerns, da alle Atome unterschiedliche lokale Magnetfelder besitzen und somit die
Larmor-Frequenz des untersuchten Atoms stark von deren chemischen Umgebung und
deren Bindung abhängt und aufgrund verschieden starker Abschirmung verändert wird.
Bei der angewendeten Methode wird der Anteil des natürlich vorkommenden 13C (etwa 1,1 %
der C-Atome) vermessen. Hier ergibt sich eine chemische Verschiebung (in Bezug auf den
Standard Na-Trimethylsilylpropansulfonsäure (TMSP)), die nur von Kernumgebung sowie
vom Lösungsmittel abhängt. Diese Verschiebung wird in ppm (parts per million) angegeben
und beruht auf dem Frequenzunterschied zum Signal des internen Standards, dessen
Verschiebung des Methylgruppensignals als 0 ppm definiert wird. Das sich ergebende
Signalmuster kann mit zuvor gemessenen Standardsubstanzen verglichen werden oder es
können über die chemischen Verschiebungen funktionelle Gruppen ermittelt werden.

II. Material & Methoden 35
5.4.1 Probenaufbereitung für 13C- NMR-Spektroskopie
Die Probenvorbereitung für die NMR-Spektroskopie entspricht der Extraktion wie sie für die
HPLC-Analyse beschrieben wurde (vgl. Abschnitt II, 5.3.1). Hier sollte jedoch ein Proben-
trockengewicht von mindestens 1 g verwendet werden. Nach Bestimmung des Trocken-
gewichts, wurde den Proben eine entsprechenden Menge an Bligh & Dyer-Lösung zugege-
ben und das Gemisch für 1,5 h auf einem Magnetrührer gerührt. Anschließend wurden
Chloroform und HPLC-Wasser zum Ansatz gegeben und dieser für weitere 30 min gerührt.
Die anschließende Phasentrennung erfolgte in einer Zentrifuge (Laborfuge AE, Heraeus) bei
2500x g für 30 min. Der wässrige Überstand wurde abgenommen und in ein Becherglas
überführt. Für die erneute Gefriertrocknung der Probe musste das verbliebene Methanol
entfernt werden. Dies geschah, indem die Probe für 1-2 Tage offen unter einen Abzug
gestellt wurde. Nach der Gefriertrocknung wurde die Probe in 1 ml D2O gelöst und mit 5 mg
des internen NMR-Standards Na-TMSP (3-Trimethylsily-propionsäure-d4-Natriumsalz), 10 µl
Acetonitril 100 % (v/v) und 10 µl Methanol versetzt. Anschließend wurde das Gemisch
abzentrifugiert und der Überstand in ein spezielles NMR-Messgefäß überführt.
5.4.2 NMR-Messung
Die NMR-Messung wurde am Pharmazeutischen Institut (Bonn) mit einem NMR-Spektro-
meter (Bruker Avance 300DPX) durchgeführt. Für die Messung wurden Messfrequenzen von
75,46 MHz (Transmitter) und 300 MHz (1H-Entkoppler) eingestrahlt. Acetonitril und Methanol
dienten als Kontrollen. Die Auswertung der Daten erfolgte mit dem Programm WIN-NMR
(Bruker).
6. Isolierung, Reinigung und Quantifizierung von DNA
6.1 Isolierung von Gesamt-DNA
6.1.1 Minipräparation von Gesamt-DNA (Marmur, 1961)
Die nachfolgend beschriebene Methode nach Marmur dient der schnellen Gewinnung klein-
erer Mengen von Gesamt-DNA aus Pseudomonas putida, Thermus thermophilus, Rhodo-
thermus marinus, E. coli und Halomonas elongata Stämmen.
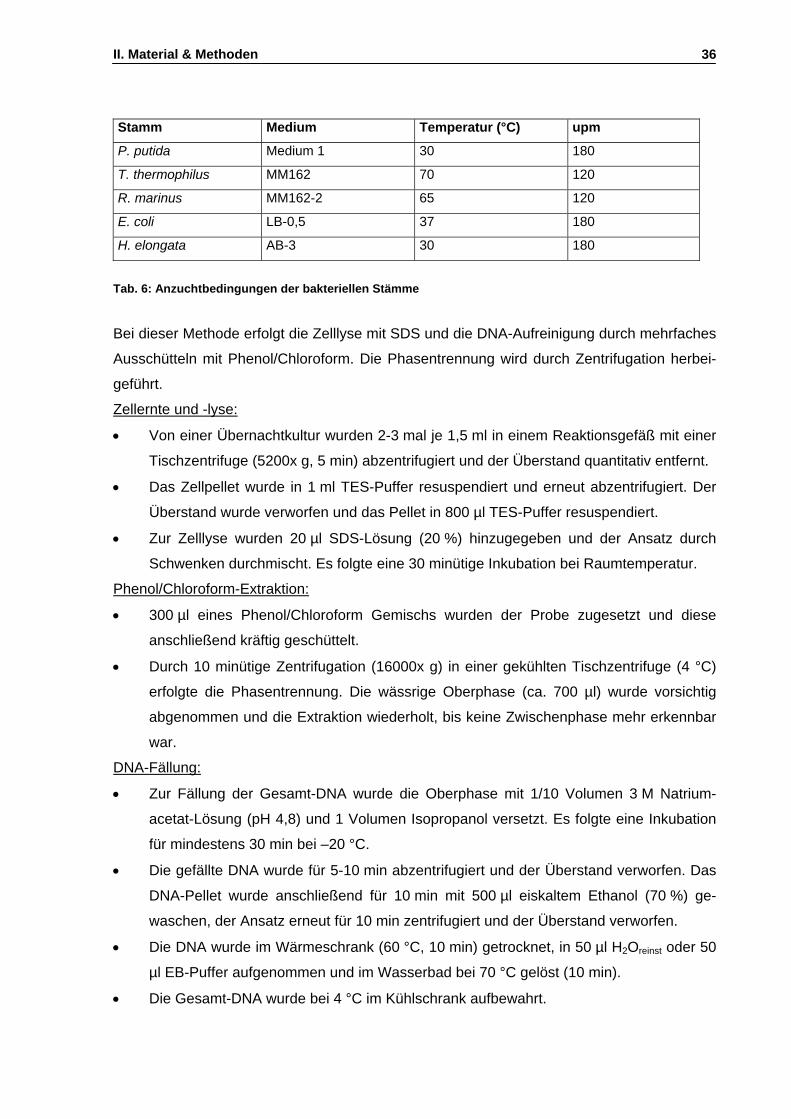
II. Material & Methoden 36
Stamm Medium Temperatur (°C) upm
P. putida Medium 1 30 180
T. thermophilus MM162 70 120
R. marinus MM162-2 65 120
E. coli LB-0,5 37 180
H. elongata AB-3 30 180
Tab. 6: Anzuchtbedingungen der bakteriellen Stämme
Bei dieser Methode erfolgt die Zelllyse mit SDS und die DNA-Aufreinigung durch mehrfaches
Ausschütteln mit Phenol/Chloroform. Die Phasentrennung wird durch Zentrifugation herbei-
geführt.
Zellernte und -lyse:
• Von einer Übernachtkultur wurden 2-3 mal je 1,5 ml in einem Reaktionsgefäß mit einer
Tischzentrifuge (5200x g, 5 min) abzentrifugiert und der Überstand quantitativ entfernt.
• Das Zellpellet wurde in 1 ml TES-Puffer resuspendiert und erneut abzentrifugiert. Der
Überstand wurde verworfen und das Pellet in 800 µl TES-Puffer resuspendiert.
• Zur Zelllyse wurden 20 µl SDS-Lösung (20 %) hinzugegeben und der Ansatz durch
Schwenken durchmischt. Es folgte eine 30 minütige Inkubation bei Raumtemperatur.
Phenol/Chloroform-Extraktion:
• 300 µl eines Phenol/Chloroform Gemischs wurden der Probe zugesetzt und diese
anschließend kräftig geschüttelt.
• Durch 10 minütige Zentrifugation (16000x g) in einer gekühlten Tischzentrifuge (4 °C)
erfolgte die Phasentrennung. Die wässrige Oberphase (ca. 700 µl) wurde vorsichtig
abgenommen und die Extraktion wiederholt, bis keine Zwischenphase mehr erkennbar
war.
DNA-Fällung:
• Zur Fällung der Gesamt-DNA wurde die Oberphase mit 1/10 Volumen 3 M Natrium-
acetat-Lösung (pH 4,8) und 1 Volumen Isopropanol versetzt. Es folgte eine Inkubation
für mindestens 30 min bei –20 °C.
• Die gefällte DNA wurde für 5-10 min abzentrifugiert und der Überstand verworfen. Das
DNA-Pellet wurde anschließend für 10 min mit 500 µl eiskaltem Ethanol (70 %) ge-
waschen, der Ansatz erneut für 10 min zentrifugiert und der Überstand verworfen.
• Die DNA wurde im Wärmeschrank (60 °C, 10 min) getrocknet, in 50 µl H2Oreinst oder 50
µl EB-Puffer aufgenommen und im Wasserbad bei 70 °C gelöst (10 min).
• Die Gesamt-DNA wurde bei 4 °C im Kühlschrank aufbewahrt.

II. Material & Methoden 37
6.1.2 Isolierung genomischer DNA mit Genomic-tip 100/G (Qiagen)
Zur Isolierung großer Mengen sehr reiner DNA von R. marinus (Herstellung der Genbank)
wurde das DNA-Isolierungskit (Midi-Präparation, Tip100-Säulen) der Firma Qiagen (Hilden)
verwendet. Dabei wird nach der Zelllyse mit Lysozym, dem Abbau von Proteinen (Protein-
ase K) und RNA (RNase) die genomische DNA in Anwesenheit einer geringen Salzkonzen-
tration spezifisch an ein Anionen-Austauscher gebunden. Die DNA-Isolierung und Aufreini-
gung erfolgte nach Angaben des Herstellers. Anschließend wurde die DNA mit einem vorge-
wärmten (50 °C) Puffer hoher Salzkonzentration eluiert, mittels Isopropanol-Präzipitation
konzentriert und entsalzt. Zuletzt wurde die genomische DNA in 150-200 µl EB-Puffer gelöst
(60 °C).
6.2 Isolierung von Plasmid-DNA
6.2.1 Plasmid-Mini-Prep (Qiagen)
Das Plasmidisolierungskit der Firma Qiagen (Hilden) wurde verwendet, um kleiner Mengen
sehr reine Plasmid-DNA für nachfolgende Ligationen, Sequenzierungen und Restriktions-
analysen zu gewinnen. Für die Isolierung wurde 1,5-8 ml Kultur eingesetzt. Das Prinzip der
Plasmid-Präparation beruht auf der alkalische Lyse der Zellen (Birnboim & Doly, 1979) und
einer Bindung der Plasmid-DNA an eine Silikamembran in Gegenwart hoher Salzkonzen-
tration. Die Mini-Präparation wurde nach Angaben des Herstellers durchgeführt und die
Plasmid-DNA mit 50 µl EB-Puffer (50 °C) von der Säule eluiert.
6.2.2 Plasmid-Midi-Kit (Qiagen)
Das Midi-Kit wurde zur Gewinnung größerer Mengen von Plasmid-DNA z.B. des low-copy-
Plasmids pHSG575 verwendet. Die Isolierung erfolgte aus 100 ml Kulturvolumen (LB-0,5 mit
Cm100) einer Übernachtkultur. Die Isolierung beruht auf der alkalischen Lyse der Zellen
(Birnboim & Doly, 1979) und der Bindung und Aufreinigung der DNA mit Hilfe einer Silika-
säule. Das Zellpellet wurde in 10 ml Puffer P1 resuspendiert und die Zellen mit je 10 ml
Puffer 2 und Puffer 3 lysiert. Die Präparation wurde anschließend mit einer Tip-100 Säule
entsprechend den Herstellerangaben für eine Midi-Präparation durchgeführt. Nach der
Isopropanol-Fällung wurde die Plasmid-DNA in 100-250 µl EB-Puffer gelöst (60 °C Wasser-
bad, 30 min).
6.3 Reinigung, Konzentrierung und Quantifizierung von DNA
6.3.1 Reinigung von DNA aus Agarosegelen (QIAquick Gelextraktionskit, Qiagen)
Zur Aufreinigung hydrolysierter DNA (z.B. Restriktionsverdau von Vektoren) bzw. von PCR-
Produkten (vor einer Ligation bzw. Sequenzierung) wurden die Proben zunächst elektro-

II. Material & Methoden 38
phoretisch im Agarosegel aufgetrennt. Nach der Färbung mit Ethidiumbromid wurden die
entsprechenden DNA-Banden unter schwachem UV-Auflicht aus dem Gel ausgeschnitten.
Die präparierte DNA wurde mit Hilfe des QIAquick Gelextraktionskits (Qiagen, Hilden) nach
Herstellerangaben aufgereinigt. Dabei bindet die DNA nach Auflösung des Gelblöckchens
mittels chaotroper Salze an die Silikamatrix des Aufreinigungssäulchens. Nach mehreren
Waschschritten wurde die DNA in 30 µl EB-Puffer (50 °C) eluiert.
6.3.2 Reinigung von DNA durch Phenol/Chloroform-Extraktion
Zur Aufreinigung von DNA Proben nach einem Restriktionsverdau oder zum Abstoppen einer
Hydrolyse genomischer DNA mit dem Restriktionsenzym (Sau 3A) wurde eine Phenol/
Chloroform-Extraktion durchgeführt. Dazu wurden die DNA-Proben (>100 µl) mit gleichem
Volumen Phenol/Chloroform versetzt, kräftig geschüttelt und 5 min bei 13600x g in einer
Tischzentrifuge zentrifugiert. Der wässrige, DNA-haltige Überstand wurde abgenommen.
Anschließend wurde die Reinigung wie bei der Minipräparation zur Gewinnung von
genomischer DNA fortgesetzt. Vor Einsatz derart aufgereinigter DNA in Ligationen wurden
die Proben mittels Microcon-Säulchen konzentriert.
6.3.3 Reinigung und Konzentrierung von DNA mit Microcon-Säulchen
Zum Pufferwechsel (z.B. vor Ligationen oder Sequenzierungen), zur Aufreinigung und zur
Konzentrierung von DNA-Proben wurden Microcon-Säulchen (Amicon, Beverly MA, USA)
verwendet. Die in den Säulchen enthaltenen Filter (Microcon YM 50) bestehen aus einer
hydrophilen Zellulosemembran, die für doppelsträngige DNA ab einer Länge von 100 bp un-
durchlässig, für Verunreinigungen bis zu einem Molekulargewicht von 50 Da jedoch durch-
lässig ist. Durch Umdrehen der Säulchen nach der ersten Zentrifugation und erneuter Zentri-
fugation mit niedrigerer Drehzahl löst sich die DNA wieder vom Molekularsieb und wird in
einem Reaktionsgefäß aufgefangen. Die Durchführung erfolgte entsprechend den Herstel-
lerangaben. Die DNA wurde dabei in 17-25 µl Reinstwasser aufgenommen.
6.3.4 Aufreinigung von PCR-Produkten
Zur Entfernung von Primern, Nukleotiden oder Enzymen aus Reaktionsansätzen und zur
Durchführung eines Pufferwechsels bei DNA-haltigen Proben wurde das QIAquick PCR-
Aufreinigungskit (Qiagen) verwendet. Das Kit ist für die Reinigung von Fragmenten mit einer
Größe von 0,1-10 kb geeignet und wurde nach der Anleitung des Herstellers verwendet. Die
Elution der DNA-Probe von der Membran erfolgte mit 30 µl EB-Puffer.

II. Material & Methoden 39
6.3.5 Konzentrations- und Reinheitsbestimmung von DNA
Die Basen der Nukleinsäuren weisen bei 260 nm ein Absorptionsmaximum auf, das zur
spektralphotometrischen Konzentrationsbestimmung DNA- und RNA-haltiger Lösungen her-
angezogen wird. Eine Lösung mit 50 µg/ml doppelstängiger DNA weist bei 260 nm und einer
Schichtdicke von 1 cm einem Absorptionswert von 1 auf.
Zur Bestimmung der Verunreinigung durch Proteine wird die Absorption bei 280 nm, dem
Absorptionsmaximum aromatischer Aminosäuren, gemessen und der Quotient A260/A280 er-
mittelt. Reine DNA besitzt einen Quotienten zwischen 1,7-1,9. Ein Quotient über 1,9 deutet
auf Verunreinigung mit RNA , ein Quotient unter 1,7 auf Verunreinigung mit Proteinen hin.
Die Absorptionsmessungen wurden in Quarzglas-Halbmikroküvetten mittels eines UV-Spek-
tralphotometers (Dioden-Array-Spektralphotometer, Agilent 8453) durchgeführt. Die DNA-
Lösung wurde mit TE-Puffer verdünnt, so dass A260 zwischen 0,1 und 0,3 lag.
Die DNA-Konzentration einer Probe kann auch im Agarosegel durch Vergleich mit DNA-
Proben bekannter Konzentration abgeschätzt werden. Diese Methode eignet sich vor allem,
wenn nur geringe Mengen der zu untersuchenden DNA vorliegen. Als Referenz-DNA wurde
Hin dIII-hydrolysierte λ-DNA eingesetzt (0,125 µg, 0,25 µg und 0,375 µg). Nach Ethidium-
bromid-Färbung der im Gel aufgetrennten Proben wurde die Bandenhelligkeit der zu be-
stimmenden DNA-Probe mit der Helligkeit der Referenz-Banden verglichen.
7. Enzymatische Modifikation von DNA
7.1 Restriktionsverdau von DNA Für die enzymatische Hydrolyse von DNA wurden je nach Bedarf unterschiedliche Enzyme
verwendet. Dabei wurden zur Wahl der optimalen Reaktionsbedingungen, wie Puffer und
Inkubationstemperatur, die Angaben des Herstellers berücksichtigt. Hydrolysen für Restrikti-
onsanalysen wurden in einem Gesamtvolumen von 20 µl durchgeführt. Es wurde eine
Enzymmenge zwischen 5-10 U, 2 µl Reaktionspuffer (10x) und jeweils etwa 500-1000 ng
DNA eingesetzt. Die Ansätze wurden für 2 bis 3 h im Wasserbad inkubiert und anschließend
in einem Agarosegel analysiert. Der Verdau von Vektor- und Insert-DNA für Ligationsansätze
erfolgte in einem Gesamtvolumen von 40 µl. Hier wurden bis zu 10 µg DNA, 4 µl Reaktions-
puffer (10x) und 10-20 U Restriktionsenzym eingesetzt. Die Ansätze wurden über Nacht im
Wasserbad inkubiert. Die Reaktion wurde am nächsten Tag durch Erhitzen auf 65 °C (10-
15 min) abgestoppt. Konnte das Enzym nicht durch Hitze inaktiviert werden, wurde die
Hydrolysereaktion mit Phenol/Chloroform abgestoppt und ein Pufferwechsel mit Microcon
Säulchen (Amicon) durchgeführt (Abschnitt II, 6.3.3).
Zur Erstellung der Genbank von R. marinus wurde die Gesamt-DNA partiell hydrolysiert. Bei
dieser Methode wird DNA mittels eines Restriktionsenzyms (hier Sau 3A) unvollständig

II. Material & Methoden 40
hydrolysiert, so dass die durchschnittliche Fragmentlänge der DNA optimiert werden kann.
Das Restriktionsenzym Sau 3A weist eine Erkennungssequenz von vier Basenpaaren
(↓GATC) auf. Die Gesamt-DNA von R. marinus wurde zunächst kurze Zeit (5-10 min) mit
unterschiedlichen Mengen von Sau 3A inkubiert. Dabei wurden zunächst mehrere gleiche
Reaktionsansätze ohne Restriktionsenzym erstellt. In den ersten Ansatz mit doppeltem
Reaktionsvolumen wurde z.B. 2 U des Enzyms hinzugegeben, der Ansatz wurde gemischt
und ein Volumen in den nächsten Reaktionsansatz pipettiert. Die Probe wurde wiederum
gemischt und das Teilvolumen weiterpipettiert. Auf diese Weise wurde jeweils eine
Halbierung der Enzymmenge erreicht, die nach festgelegter Inkubationszeit zu unterschied-
lich starker Hydrolyse führte. Nach Optimierung der Reaktionszeit wurde die gewünschte
durchschnittliche Fragmentgröße von 5-10 kb durch Einsatz von 4,8 µg genomischer
R. marinus DNA und 0,125 U Sau 3A bei einer Inkubationszeit von 5 bis 7,5 min erreicht. Die
Reaktion wurde durch Zugabe gleicher Volumen Phenol/Chloroform abgestoppt (Abschnitt II,
6.3.2).
7.2 Dephosphorylierung von Vektor-DNA Um eine Religation des Vektors zu verhindern und die Klonierung der Insert-DNA zu begüns-
tigen, wurde die Vektor-DNA vor der Ligationsreaktion dephosphoryliert. Die Reaktion wurde
während der präparativen Hydrolyse der Vektor-DNA durch Zugabe von 2 U CIAP (calf
intestine alkaline phosphatase, Zugabe 2 h nach Start der Hydrolyse) gestartet. Danach
wurde der Ansatz für weitere 3-4 h bei 37 °C inkubiert. Die Reaktion wurde durch Hitze-
inaktivierung des Enzyms abgestoppt (10 min, 65 °C).
7.3 Abbau überhängender 3’-Enden, Herstellung glatter Enden Bei der Amplifikation von PCR-Produkten mit Hilfe des GC-Rich-PCR-Systems, das neben
Tgo-Polymerase auch Taq-Poymerase enthält, entstehen am 3’-Ende der PCR-Produkte
überhängende A-Enden. Um daraus für den Einsatz in weiteren PCR-Reaktionen glatte
Enden herzustellen, wurden die PCR-Produkte mit T4–DNA–Polymerase inkubiert. Diese
verfügt neben der Polymeraseaktivität über eine Exonuklease-Funktion, die bei geringer
dNTP-Konzentration überwiegt und die überstehende 3’-Enden hydrolysiert sowie überhän-
gende 5’-Enden durch Polymerisation zugesetzter Nukleotide zu glatten Enden auffüllt. Pro
Ansatz wurden 2-5 µg DNA in einem Gesamtvolumen von 25 µl eingesetzt. Der Ansatz
enthielt 5 U T4-DNA-Polymerase, Reaktionspuffer (1x) und 0,12 mM dNTP-Gemisch. Die
Inkubation erfolgte für 5-7 min bei 37 °C und wurde durch 10 minütige Inkubation bei 65 °C
abgestoppt. Anschließend wurde die DNA aufgereinigt (Abschnitt II, 6.3.4).

II. Material & Methoden 41
7.4 Ligation Je nach verwendetem Restriktionsenzym wurden überhängende oder glatte DNA-Enden
nach ihrer zufälligen Zusammenlagerung durch Verwendung eine T4-DNA-Ligase miteinan-
der verknüpft. Das Enzym verknüpft benachbarte 5’-Phosphat- und 3’-OH-Gruppen über die
Bildung einer Phosphodiesterbindung. Der Ligationsansatz enthielt dephosphorylierte
Vektor-DNA und Fremd-DNA in einem molaren Verhältnis von 1:2 bis 1:4. Die Ligation wurde
in dem mitgelieferten Puffer (1x) in einem Gesamtvolumen von 20 µl durchgeführt. Bei der
Ligation überhängender Enden wurden 100-200 ng Vektor-DNA und 2 U T4-DNA-Ligase
eingesetzt. Bei der Ligation glatter Enden wurde dagegen die doppelte Menge an T4-DNA-
Ligase (4 U) und 2 µl PEG 4000 (1/10 des Volumens) zupipettiert. Die Ansätze wurden für
16 h bei 16 °C in einem Thermocycler (Primus 25, MWG) inkubiert und durch Hitzeinakti-
vierung bei 65 °C (10 min) abgestoppt. Die Ligationsansätze wurden direkt in PCR-Reak-
tionen oder zur Transformation von E. coli DH5α eingesetzt.
8. Transformation von E. coli Der Vorgang der DNA-Aufnahme wird als Transformation bezeichnet. E. coli erlangt durch
eine Behandlung mit CaCl2 und Kälteinkubation bzw. durch andere Salze (z.B. RbCl)
während eines Hitzeschocks eine Kompetenz zur Aufnahme freier DNA (z.B. Plasmid-DNA)
aus dem Medium (Cohen et al., 1972). Der Mechanismus der Wirkung von CaCl2 bzw.
anderer Salze ist weitgehend unbekannt. Möglicherweise verändert CaCl2 die Zellwand.
Andererseits könnte es auch für eine Bindung der DNA an die Zelloberfläche verantwortlich
sein (Old & Primrose, 1992). Die Effizienz der DNA-Aufnahme kann durch schockartiges
Gefrieren in flüssigem Stickstoff und durch Lagerung der superkompetente Zellen (Inoue et
al., 1990) bei -70 °C in Dimethysulfoxid (DMSO) noch gesteigert werden.
8.1 Herstellung superkompetenter Zellen von E. coli (Inoue et al., 1990) Als Vorkultur wurde 15 ml LB-0,5-Medium mit E. coli beimpft und über Tag bei 37 °C (180
upm) angezogen.
• Mit der Vorkultur wurde abends 100 ml SOB-Medium 0,5-1 %-ig animpft und bei Raum-
temperatur (22–23 °C) über Nacht bei 180 upm inkubiert.
• Bei Erreichen einer OD540 von 0,6-0,8 wurde die Kultur 10 min auf Eis gelagert und
anschließend abgeerntet (4 °C, GSA-Rotor, 5000x g, 15 min).
• Das Zellpellet wurde in 64 ml Transformationspuffer (TFB) resuspendiert, 10 min auf
Eis inkubiert und erneut für 10 min abzentrifugiert.
• Die Zellen wurden dann in 16 ml TFB-Puffer resuspendiert, portionsweise mit 1,12 ml
DMSO versetzt und der Ansatz erneut für 10 min auf Eis inkubiert.

II. Material & Methoden 42
• Nach Aliquotierung (1,5-2 ml) wurden die kompetenten Zellen in flüssigem Stickstoff
eingefroren und bei –70 °C gelagert.
8.2 Transformation superkompetenter Zellen von E. coli • Für die Transformation wurde ein Aliquot superkompetenter Zellen langsam auf Eis
aufgetaut.
• 200 µl Zellsuspension wurden mit einem Ligationsansatz (20 µl) bzw. 2 bis 4 µl zu
transformierender Plasmid-DNA versetzt und 30 min im Eiswasserbad inkubiert.
• Die Zellen wurden 2,5 min bei 43 °C inkubiert, anschließend wurde 800 µl vorgewär-
mtes SOC-Medium zugegeben.
• Zur Regeneration der Zellen wurden diese für 60 min bei 37 °C im Wasserbad inku-
biert. Zwischenzeitlich wurde der Ansatz durch Invertieren des Reaktionsgefäßes ge-
mischt.
• Es wurden Teilvolumen des Transformationsansatzes auf Selektionsmedium ausplat-
tiert und bei 37 °C über Nacht inkubiert.
9. Polymerasekettenreaktion (PCR) Mittels PCR können DNA-Bereiche definierter Länge und Sequenz selektiv in vitro amplifi-
ziert werden (Mullis & Faloona, 1987). Eine PCR untergliedert sich in mehrere zyklisch
wiederholte Phasen. In der ersten Phase eines PCR-Zyklus wird die doppelsträngige DNA
zunächst durch Hitzeeinwirkung zu Einzelsträngen denaturiert. Nach dem Absenken der
Temperatur hybridisieren zwei dem Reaktionsansatz zugesetzte komplementäre Oligo-
nukleotid-Primer mit der Zielsequenz (Anlagerungsphase). Sie dienen als Startpunkt der
Strangsynthese für die Taq-Polymerase. Diese komplettiert in Gegenwart von dNTPs die
Einzelstränge zum Doppelstrang, wobei sie an die freien 3’-OH-Gruppen immer neue
komplementäre Basen anhängt (Elongationsphase). Dadurch entstehen zwei neue, doppel-
strängige DNA-Moleküle. Das gewünschte Produkt wird somit exponentiell vermehrt, wenn
sich dieser Zyklus wiederholt. Die eingesetzte Anlagerungstemperatur lag etwa 2-4 °C unter
der Schmelztemperatur der eingesetzten Primer.
Für die PCR wurden unterschiedliche thermostabile DNA-Polymerasen eingesetzt. Für
Standardreaktionen wurde die Taq-Polymerase (Thermus aquaticus) verwendet. Für Klon-
ierungen und Mutagenese wurde die Pwo-Polymerase (Pyrococcus woesei) eingesetzt.
Dieses Enzym enthält im Gegensatz zur Taq-Polymerase eine 3’-5’-Exonukleaseaktivität.
Diese wird auch als proof-reading-Aktivität bezeichnet, da dieses Enzym den Einbau von
falschen Nukleotiden korrigiert. Bei einem hohen GC-Gehalt (60-65 %) der zu amplifi-
zierenden DNA-Matrize, wurde das GC-Rich-PCR-System (Roche) verwendet. Dieses ent-
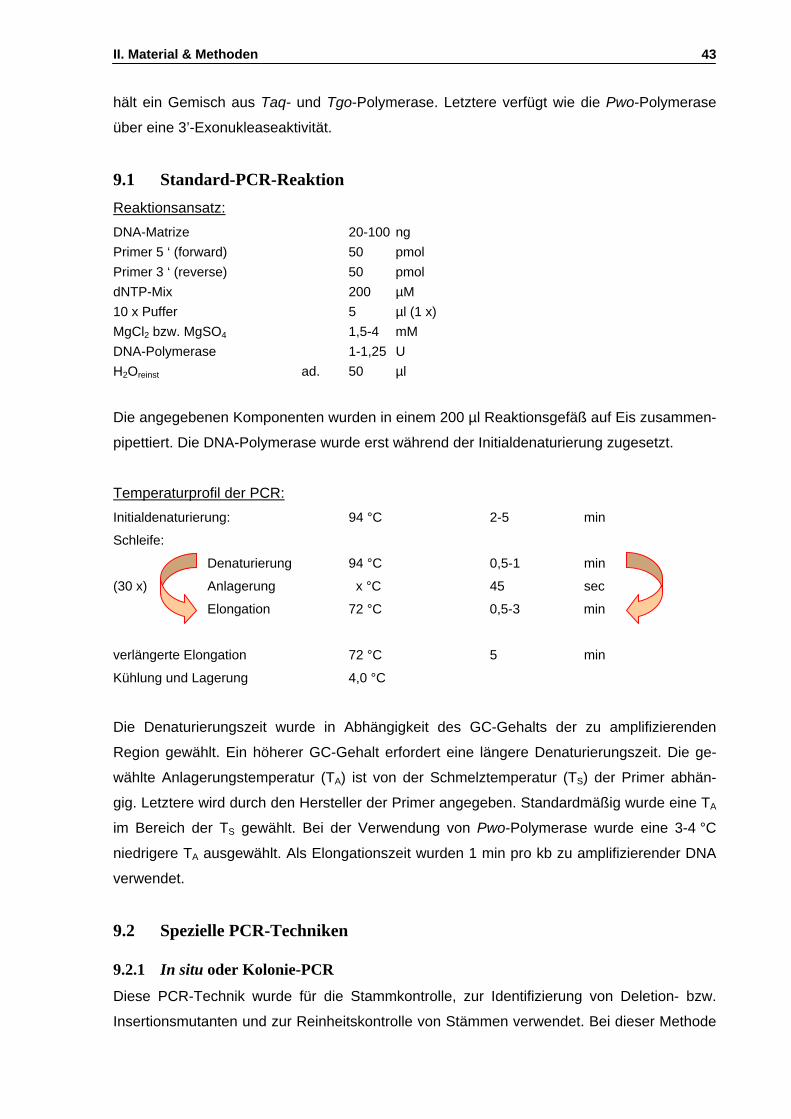
II. Material & Methoden 43
hält ein Gemisch aus Taq- und Tgo-Polymerase. Letztere verfügt wie die Pwo-Polymerase
über eine 3’-Exonukleaseaktivität.
9.1 Standard-PCR-Reaktion Reaktionsansatz: DNA-Matrize 20-100 ng Primer 5 ‘ (forward) 50 pmol Primer 3 ‘ (reverse) 50 pmol dNTP-Mix 200 µM 10 x Puffer 5 µl (1 x) MgCl2 bzw. MgSO4 1,5-4 mM DNA-Polymerase 1-1,25 U H2Oreinst ad. 50 µl
Die angegebenen Komponenten wurden in einem 200 µl Reaktionsgefäß auf Eis zusammen-
pipettiert. Die DNA-Polymerase wurde erst während der Initialdenaturierung zugesetzt.
Temperaturprofil der PCR: Initialdenaturierung: 94 °C 2-5 min
Schleife:
Denaturierung 94 °C 0,5-1 min
(30 x) Anlagerung x °C 45 sec
Elongation 72 °C 0,5-3 min
verlängerte Elongation 72 °C 5 min
Kühlung und Lagerung 4,0 °C
Die Denaturierungszeit wurde in Abhängigkeit des GC-Gehalts der zu amplifizierenden
Region gewählt. Ein höherer GC-Gehalt erfordert eine längere Denaturierungszeit. Die ge-
wählte Anlagerungstemperatur (TA) ist von der Schmelztemperatur (TS) der Primer abhän-
gig. Letztere wird durch den Hersteller der Primer angegeben. Standardmäßig wurde eine TA
im Bereich der TS gewählt. Bei der Verwendung von Pwo-Polymerase wurde eine 3-4 °C
niedrigere TA ausgewählt. Als Elongationszeit wurden 1 min pro kb zu amplifizierender DNA
verwendet.
9.2 Spezielle PCR-Techniken
9.2.1 In situ oder Kolonie-PCR
Diese PCR-Technik wurde für die Stammkontrolle, zur Identifizierung von Deletion- bzw.
Insertionsmutanten und zur Reinheitskontrolle von Stämmen verwendet. Bei dieser Methode

II. Material & Methoden 44
werden intakte Zellen anstelle isolierter DNA als Template verwendet (Joshi et al., 1991). Die
Zellen wurden zunächst mit einer sterilen Pipettenspitze von der Agarplatte in 10 µl H2Oreinst
überführt. Von dieser Suspension wurden nach einer Verdünnung (1:2) 0,5 µl in die PCR
eingesetzt. Um eine größere Anzahl von Kolonien gleichzeitig zu überprüfen, wurde Zell-
material von bis zu 10 Kolonien in 50 µl H2Oreinst überführt und gleichermaßen verfahren. Die
Dauer der Initialdenaturierung wurde auf 10 min erhöht, um einen Zellaufschluss sicherzu-
stellen.
9.2.2 Ligase-vermittelte bzw. inverse-PCR
Mittels Ligase-vermittelter PCR kann ein DNA-Fragment amplifiziert werden, bei dem nur
eine flankierende Sequenz bekannt ist. Dies war besonders bei der Sequenzierung von
DNA-Sequenzen stromabwärts einer bekannten Sequenz notwendig. Es kamen zwei Variati-
onen dieser Methode zum Einsatz.
Bei der ersten Variante wurde nach Hydrolyse der genomischen DNA mit einem geeigneten
Restriktionsenzym die DNA mit einem entsprechend linearisierten Vektor (hier pHSG575)
ligiert. Mit einem Primer, der innerhalb der MCS des Vektors bindet, und einem Primer, der
komplementär zum bekannten DNA-Bereich ist, wurde in einer PCR auf dem Ligationsansatz
der unbekannte Sequenzbereich amplifiziert. In die PCR wurden 1-3 µl des Ligationsan-
satzes eingesetzt.
Die zweite Variante der Methode wird auch als inverse-PCR bezeichnet. Hierbei erfolgt nach
einer enzymatischen Hydrolyse der genomischen DNA mit einer Restriktionsenzym, das
nicht im bekannten DNA-Bereich schneidet, eine Selbstligation der geschnittenen DNA-
Fragmente ohne Zusatz von Vektor-DNA. So entstehen zirkuläre DNA-Moleküle, die als
Matrize in der PCR dienten. Durch Zusatz von Primern, die jeweils an den beiden Enden der
bekannten DNA-Sequenz abgeleitet sind und eine entgegengesetzte Orientierung auf-
weisen, kann der unbekannte DNA-Bereich amplifiziert und schließlich sequenziert werden.
In der inversen-PCR wurden 2-3 µl der Ligationsansätze eingesetzt.
9.2.3 Verschachtelte (nested)-PCR
Bei der nested-PCR werden die Produkte einer vorangegangenen PCR als Matrizen-DNA
eingesetzt. Diese Methode wurde angewendet, wenn während einer ersten PCR nicht
genügend spezifisches PCR-Produkt amplifiziert werden konnte. Es wurden Primer verwen-
det, die jeweils am 3’-Ende von der Position der zuerst verwendeten Primer hybridisieren.
Die Produktspezifität lässt sich hierbei deutlich steigern, weil sich unspezifische Neben-
produkte der ersten PCR in der Regel nicht mehr bilden. Die nested-PCR kann auch als
Variante mit nur einem eingerückten Primer angewendet werden. Es wurden 1-5 µl des

II. Material & Methoden 45
vorangegangenen aufgereinigten PCR-Produkts als DNA-Matrize in der nested-PCR
eingesetzt.
9.2.4 Heterologe PCR unter Einsatz von degenerierten Primern
Degenerierte PCR-Primer werden eingesetzt, um unbekannte DNA-Bereiche zu ampli-
fizieren, zu denen es homologe oder ähnliche Sequenzbereiche gibt. Zum Einsatz kommt
diese Methode, wenn z.B. ein Genabschnitt in einer Spezies amplifiziert werden soll, dessen
genaue Sequenz nur aus anderen Arten bekannt ist.
Bei der Ableitung dieser Primer hilft eine Homologievergleich (DNA- und Proteinebene) der
bekannten Sequenzen. Abgeleitet werden Primer, die unter Berücksichtigung der „Codon-
usage“ der jeweiligen Spezies Nukleotidvariationen enthalten. Durch Beachtung der „Codon-
usage“ kann in vielen Fällen der Degenerationsgrad gemindert werden. Der Degenerations-
grad entspricht der Anzahl der unterschiedlichen Primermoleküle und errechnet sich aus der
Multiplikation der mehrfachen Nukleotide. Es ist wichtig darauf zu achten, dass jede Er-
höhung des Degenerationsgrades die tatsächliche Anzahl der einzelnen Primermoleküle im
Pool vermindert. In einer PCR muss die eingesetzte Primermenge dementsprechend erhöht
werden. Als Alternative kann auch ein Nukleotid mit neutraler Base wie z.B. Inosin (Base:
Hypoxanthin) verwendet werden, das stabile Wasserstoffbrückenbindungen zu Cytosin,
Thymin und Adenin aufbaut (dI-dC> dI-dA > dI-dG= dI-dT). Ein prinzipieller Nachteil dieser
Methode ist, dass die Gefahr unspezifischer Amplifikationen umso größer ist, je degener-
ierter die eingesetzten Primer sind. In der vorliegenden Arbeit wurde dieser Methode ver-
wendet, um mit Hilfe degenerierter Primer auf R. marinus DNA die Sequenz des Substrat-
bindeproteins eines möglichen α-Glucosid- bzw. Mannosylglycerat-Transporters zu amplifi-
zieren.
9.2.5 SOE-PCR (splicing by overlap extention)
Mit Hilfe der SOE-PCR lassen sich DNA-Moleküle ohne Verwendung von Ligase miteinan-
der verknüpfen (Horton et al., 1989). Die Methode wurde für die Gewinnung von DNA-Kon-
strukten, die für die Herstellung von „in-frame“ Mutanten benötigt wurden, eingesetzt. Hierfür
wurden die DNA-Bereiche, die das zu deletierende Gen flankieren, amplifiziert und mitein-
ander fusioniert. Die zu verbindenden Fragmente werden zunächst in zwei getrennte PCR-
Ansätzen vervielfältigt. Dabei werden an den zu fusionierenden Enden der Einzelprodukte
kurze zueinander komplementäre Sequenzen angefügt. So entstehen als Produkte der
ersten beiden PCR-Ansätze verlängerte DNA-Fragmente. In einer nachfolgenden dritten
PCR werden diese Produkte als Matrizen-DNA eingesetzt. Die komplementären DNA-
Bereiche hybridisieren und dienen als Primer für die Polymerase. Ausgehend von der Über-
lappungsstelle werden zunächst beide Stränge an ihrem 3’-Ende durch die DNA-Polymerase

II. Material & Methoden 46
verlängert. Das entstehende Produkt stellt schließlich eine Fusion der beiden einzelnen
DNA-Moleküle dar. Dieses wird schließlich durch die beiden äußeren Primer amplifiziert. Zur
Herstellung eines SOE-PCR-Produkts wurden jeweils vier Primer verwendet. Die Primer-
paare wurden so gewählt, dass beide Flanken dieselbe Länge aufweisen. Die inneren Primer
setzen sich aus zwei Teilen DNA-Sequenzen zusammen. Ein Teil liegt stromaufwärts, der
andere stromabwärts der zu deletierenden Sequenz. Sie sind somit zueinander komple-
mentär. Die äußeren Primer enthalten Restriktionsschnittstellen, die für die Klonierung des
SOE-PCR-Produkts verwendet wurden. In der SOE-PCR wurden ca. 30 ng der in den ersten
PCR-Reaktionen amplifizierten DNA-Fragmente eingesetzt. Die SOE-PCR wurde mit der
Pwo-Polymerase durchgeführt.
9.2.6 Herstellung DIG-markierter DNA-Sonden mittels PCR
Für Detektion und Nachweis einer spezifischen DNA-Sequenz in einem Southern-Hybridi-
sierungsexperiment wurde eine Digoxigenin-Desoxyuridintriphosphat (DIG-dUTP) markierte
DNA-Sonde eingesetzt. Die Sonde wurde mittels PCR hergestellt. Der eingesetzte dNTP-Mix
enthielt folgende Nukleotidkonzentrationen: je 100 µM dATP, dGTP und dCTP, 70 µM dTTP
und 30 µM DIG-11-dUTP. Die Durchführung der PCR entsprach dem Standardprotokoll. Zur
Kontrolle wurde parallel eine PCR mit unmarkiertem dNTP-Mix durchgeführt. Beide PCR-
Produkte wurden gelelektrophoretisch überprüft. Das markierte PCR-Produkt wandert im
Vergleich zum unmarkierten Produkt langsamer im Gel. Das markierte Produkt wurde aus
dem Gel herausgeschnitten, nach Aufreinigung in 30 µl H2Oreinst eluiert, vollständig in 5 ml
Hybridisierungslösung überführt und bis zur Verwendung bei –20 °C gelagert.
9.3 Verwendete Primer
Primer Sequenz (5' → 3' ) Tm (°C) 3' Pos./Strang/ Org. Merkmal
E. coli BKA-13
SOEotsB1 gtc cca gtc gac gcc acc ttc att aat atg aaa 68,2 6212/ - /Ec K12 Sal I
SOEotsB2 tct ggt ggt gca atc cgg ccg gtt ctc ctc ctt ct > 75 5232/ + /Ec K12 SOEotsB3 aga agg agg aga acc ggc cgg att gca cca cca ga > 75 4397/ - /Ec K12 SOEotsB4 ccc acg gtc gac tag cgg aat att ttc atc agt 69,5 3409/+ /Ec K12 Sal I
R. marinus Thiamingene
Thirev1 cgc tta tcg atg gtg caat 54,5 1611/ - /Rm Thioforxrevcomp1 cga acg acg atc cga tcac 58,8 635/ + /Rm Thirev2 gtc ggg ctt cag cag cgg ata 63,7 1981/ - /Rm Thifor2 atc gat aag cgg cca tga acc ga 62,4 1641/ + /Rm
II. Material & Methoden 47
pThio 1 down tgc ggc tgg cta aga taa gga 59,8 2534/ - /Rm
Differential Display
Poly A-C-Stop aaa aaa aaa aaa aaa Cin nin nin nin nin CTA 50,6 Poly A-G-Stop aaa aaa aaa aaa aaa Gin nin nin nin nin TTA 49,4 Poly A-T-Stop aaa aaa aaa aaa aaa Tin nin nin nin nin TCA 49,4 Oligo-A-dC aaa aaa aaa aaa aaa C 20,3 Oligo-A-dG aaa aaa aaa aaa aaa G 20,3 Oligo-A-dT aaa aaa aaa aaa aaa T 17,7 AP-Primer ggc cac gcg tcg act agt act ttt ttt ttt ttt ttt t 71,1 Sal I
AUAP ggc cac gcg tcg act agt a 61 Sal I
dd 2 for 1 gaa acc cat aga ggc ccc aag ca 64,2 1437/ + /Rm dd 2 rev 1 gac aat ttt cgg cgc cgg gcc gct 69,6 978/ - /Rm dd 2 for 2 aag cgt cgc agg ttg tct tc 59,4 1470/ + /Rm dd2 rev 2 gct cga cca gtg agc tgt ta 59,4 959/ - /Rm dd 2 for 3 Seq ttc gat gct acg cga gga ac 59,4 2287/ + /Rm dd 2 for 4 Seq taa ctc cac cac gcc tcc ac 61,4 3190/ + /Rm
Degenerierte Primer
TTC1627 for 1 gac ct(gc) ct(ag) (ag)a(ag) aag ta(ct) gg(ct) ta 59,8 TTC1627 rev 1 tgc ca(gt) cc(gt) cc(agc) ag(gct) gt(agc) gc(gct) gc 70,7 TTC1627 rev 2 gcg tag ggc ca(ag) ttg cgc (at)(ct)g aa 66
SSSF-Transporter R.marinusSSF for2 ggt tga ttc aca ttc cga cgg ac 62,4 513/ + /Rm SSF Sonde for 1 gtg agc gca acg caa tta atg t 58,4 28/ + /Rm SSF rev 1 aga tgc cca cga tcg gcg agg aa 66 309/ - /Rm SSF Sonde rev gac gac atc aat gcc gaa agc ag 62,4 604/ - /Rm SSF Sonde for 2 gtt gcg ctc gaa cag tga 56 278/ + /Rm
H. elongata KB10 mgsHelo1 gta gca agc ttc gac gat gcc agc gac gtg gtg gt > 75 25/ + /He Hin dIII
blau = 667/ - /Rm, mgsHelo2 aag gga aaa acg acc agg ctc att gtc gtg gtt
cgc tgt agc gaa t > 75 grün =505/ - /He
blau = 1860/ + /Rm,
mgsHelo3 ggg cgg cac tgt cga ccg cct gag ccg gga cgc cgc ctg gcc ggc ccg gta > 75
grün = 1134/+/He Sal I
mgsHelo4 Bgl act tgc aga tct cgg tgg tcg cca gcg cgc cca tgg tga cgc ca > 75 1605/ - /He Bgl II
grün = 528 / +/ He, mgsHelo5 aca gcg aac cac gac aat gag cct ggt cgt ttt
ccc tt 73,7 blau = 689 / +/ Rm
II. Material & Methoden 48
grün = 1107/ - /He, mgsHelo6 cag gcg gcg tcc cgg ctc agg cgg tcg aca
gtg ccg ccc > 75 blau = 1838/ - /Rm
Sal I
H. elongata
KB10.1 KB10 XanA 1 tgt agc gct agc tcg acg atg cca gcg acg tgg
tgg tcg gcg ctc aca tgg t > 75 40/ + /KB10 Nhe I
KB10 XanA 2 gcg ttc aag ctt ctg ggt ctg cat atg gac ctc ctg tgg ggg gaa agt gat aaa acga > 75 1770/ - /KB10 Hin dIII,
Nde I
KB10 XanA 3 gtt aag gac tca cat atg gaa ttg atc ccg gta a 67,1 1887 / + /Pp Nde I
KB10 XanA 4 tgc caa aag ctt tta acc tct gat taa gcc ggc ga 69,5 4780 / + /Pp Hin dIII
KB10 XanA 5 ctg taa aag ctt gca gta ttc tgc cgt ctc gca cga aga gc 74,4 2995/ + /He Hin dIII
KB10 XanA 6 cgt cta gaa gtc gac ctg aga cca tga tac cgc tct cct 74,7 4523/ - /He Xba I
Mgs in
H. elongata
mgs for 4 aac cag ctg aat cgt cat gag cct ggt cgt ttt tc 70,6 685/ + /Rm Pag I
mgs rev 4 tgc gtc cgt gtt gga tcc ggc ggt cga cga cag tgc cg > 75 1841/ - /Rm Bam HI
His-tag for 1 gtc cca taa gct tat ccg gat ata gtt cct cct ttc a 69,5 25/ + /pET22b (+) Hin dIII
His-tag rev 1 cga gct ccg tcg acg gat ccg cgg ccg cac tcg agc a > 75 156/ - /pET22b (+) Bam HI
Race / RT- PCR
AAP ggc cac gcg tcg act agt acg ggi igg gii ggg iig 70,8 Sal I
AUAP ggc cac gcg tcg act agt a 61 Sal I
Race mgs-3 cca gtc gct cct gca acc gga 65,7 721 bzw. 854/ - /KB10.1 bzw.Rm Race mgs 2 tcc cac tgc gtc tct tcc agg aaa ta 64,8 789 bzw. 922/ - /KB10.1 bzw. Rm Race mgs 1 tgc cgc acc agc ccg tag ccg aaa t 69,5 889 bzw. 1022/ - /KB10.1 bzw. Rm mgs rev 1 agg cca tct cgt cca tga agt t 60,3 1464 bzw. 1597/ - /KB10.1 bzw. Rm mgs for 3 ggc tac gac tac gcg cag cag tat ct 68 1633 bzw. 1766/ + /KB10.1 bzw. Rm mgs for1 gac gca atg atc act tgg atg at 58,9 958 bzw. 1091/ - /KB10.1 bzw. Rm gsp2 ect B gcg agc ctt gcg ctg cac gtc acg ct 72,7 2150 bzw. 2765 / - /He bzw. KB10 gsp 1 ect B cga agg ctt ggt ggg tga tct tgt 64,4 2294 bzw. 2909 / - /He bzw. KB10 PP rev 1 ttc tgc ttg ttg atg gtg cga t 58,4 2018 bzw. 2077/ - /KB10.1 bzw. Pp PP gsp 1 gaa cat gcc ggc gttc cag aaa 62,1 2394 bzw. 2453/ - /KB10.1 bzw. Pp PP gsp 2 tcg gaa aaa gcc gct tcg ttc t 60,3 2174 bzw. 2233/ - /KB10.1 bzw. Pp Xan gsp 2 ggt cga tgt aag cgc ttt tgt cgg a 64,6 3835 bzw. 3894/ - /KB10.1 bzw. Pp CoXan rev tca tca atg ctt ggg aaa gag tcg ga 63,2 3543 bzw. 3602/ - /KB10.1 bzw. Pp
mgs RNA-Sonde
mgs for- 2 agc ctg gtc gtt ttt ccc tt 57,3 556 bzw. 689/ + /KB10.1 bzw. Rm

II. Material & Methoden 49
mgs T7 Sonde gga tcc taa tac gac tca cta tag gga gag cgg cct gat agc ggt agc gt > 75 1643 bzw. 1782/ - /KB10.1 bzw. Rm
ectB RNA-Sonde ectB-F aac gca tgg agt ccg acg tt 59,4 1226 bzw. 1841/ + /He T 7 ectB-rev gga tcc taa tac gac tca cta tag ggg gca ctt
gac cac ttc 74,3 2356 bzw. 2971/ - / He bzw. KB10
PP1776 RNA-Sonde PPfor 3 cat atg gaa ttg atc ccg gta a 56,5 1828 bzw.1887/ + /KB10.1 bzw. Pp T 7 PPrev gga tcct aat acg act cac tat agg ggc aac gac
cgt agt tat cct cg > 75 3222 bzw. 3281/ - /KB10.1 bzw. Pp
XanA RNA-Sonde Xan for Sonde att cag acc cgc tgc ttc aaa gcc t 64,6 3418 bzw. 3477/ + /KB10.1 bzw. Pp T 7 Xan rev gga tcc taa tac gac tca cta tag ggt taa gcc
ggc gat ttg tgc aac ct > 75 4709 bzw. 4768/ - /KB10.1 bzw. Pp H. elongata KB1 ectA- up 5' gta gca agc ttc gac gat gcc agc gac 69,5 1/ + /He ectB '3' agc ata agc ttc cat cga acg gca tga 65 1178/ - /He Tab. 7: Verwendete Primer Von den Primeranbindungsstellen ist jeweils das 3’-Ende angegeben. Die Positionen beziehen sich auf die DNA-Sequenzen von E. coli (EcK12), H. elongata (He), R. marinus (Rm) bzw. P. putida (Pp). „+“ bzw. „-“ geben an, von welchem DNA-Strang der Primer abgeleitet wurde. „+“ bezieht sich auf den „sense“ Strang, „-“ auf den „antisense“ Strang. Die DNA-Sequenzen sind auf der NCBI Homepage erhältlich: E. coli Acc. Nr.: AE000283 (Region 960-7320), P. putida Acc. Nr.: AE016780 (Region 174969-177902), R. marinus Acc. Nr.: AF173987 (Region 667-1860) und H. elongata Wildtyp Acc. Nr.: AF031489. Die DNA-Sequenz von H. elongata KB10 und KB10.1 sind im Anhang der Arbeit aufgeführt. Komplementäre Sequenzbereiche zusammengehöriger Primer-paare sind gleichfarbig dargestellt, Restriktionsschnittstellen sind rot hervorgehoben und zusätzlich erwähnt. Die aufgeführten Primer zur Reinheitskontrolle von H. elongata KB1 (Grammann et al., 2002) binden in der Sequenz der Ectoin-Synthesegene.
10. Elektrophoretische Methoden zur Auftrennung von DNA
10.1 Agarosegelelektrophorese Zur gelelektrophoretischen Auftrennung von DNA-Fragmenten wurden Gelkammern der
Firmen Gibco BRL (Eggenstein, Gelvolumen 26 ml) und Owl (Portsmouth, Gelvolumen
35 ml) verwendet. Je nach Größe der aufzutrennenden DNA-Moleküle lag die Agarose-
konzentration zwischen 0,8 und 3 %. Die Agarose wurde durch Erhitzen in 1x TB-Puffer

II. Material & Methoden 50
gelöst und nach dem Abkühlen in den Gelträger gegossen. Nach dem Erstarren wurde das
Gel mit 1x TB-Puffer überschichtet. Die Proben wurden vor dem Auftragen mit 6x Ladepuffer
versetzt. Zur Größenbestimmung der aufgetrennten DNA-Fragmente wurde ein DNA-Marker
(1 kb DNA-Marker, 100 bp-DNA-Leiter oder Hin dIII hydrolysierte λ-DNA) verwendet. Wäh-
rend der Elektrophorese wurde eine Spannung von 65-85 V angelegt.
Zur Dokumentation der Elektrophorese wurde das Gel zunächst 5 min in Ethidiumbromid-
Lösung (10 µg/ml), danach für 5 min in H2Odemin. inkubiert. Anschließend wurden die DNA-
Banden im UV-Durchlicht photographiert (Videosystem Intas, Göttingen). Bei präparativen
Gelen wurden zunächst Photos unter schwachem UV-Auflicht (365 nm) festgehalten. An-
schließend wurde die gewünschte DNA-Bande mit einem Skalpell aus dem Gel geschnitten
und die DNA mit dem Gelextraktionskit von Qiagen aufgereinigt (Abschnitt II, 6.3.1).
10.2 Southern-Hybridisierung Bei dieser Methode nach Southern (1975) wird die zu untersuchende DNA mit Restriktions-
enzymen hydrolysiert, gelelektrophoretisch aufgetrennt und unter Beibehaltung des Banden-
musters auf eine Nylonmembran übertragen. Eine komplementäre, sequenzspezifische,
markierte DNA-Sonde kann mit der gesuchten DNA auf der Membran hybridisieren und
durch eine Färbereaktion nachgewiesen werden. Als DNA-Sonde wurde hier Digoxigenin-
markierte PCR-Sonde verwendet. Bei dem immunologischen Nachweis der DNA-Sonde
dient Digoxigenin als Antigen, das durch polyklonale DIG-spezifische Antikörper (Fab-Frag-
ment) detektiert wird. An den Anti-DIG-Antikörper ist eine alkalische Phosphatase gekoppelt
(Antikörperkonjugat). Diese setzt die Substrate NBT (Nitroblau-Tetrazoliumchlorid) und BCIP
(5-Bromo-4-chloro-3-indolylphosphat) um. Dabei kommt es zur Ausbildung eines dunkel-
blauen Niederschlags auf der Membran, der die Lage der gesuchten DNA-Moleküle anzeigt.
Elektrophorese und Transfer der DNA:
Die hydrolysierte DNA wurde zunächst in einem Agarosegel aufgetrennt, mit Ethidium-
bromid gefärbt und unter UV-Licht photographiert. Anschließend wurde ein passendes Stück
positiv geladene Nylonmembran (Sartolon 0,45 µM, Sartorius) zurechtgeschnitten mit
H2Odemin. befeuchtet und entsprechend den Herstellerangaben auf die Blottingapparatur
(Model 785 Vacuum Blotter, BioRad) aufgebracht. Nach dem Auflegen der Gelmaske und
des Gels auf die Blottingapparatur wurde der DNA-Transfer durch Anlegen des Vakuums
gestartet. Dazu wurde zunächst Depurinierungslösung auf das Gel getropft, später Denatur-
ierungslösung, die als Transferlösung diente. Der Transfer erfolgte für 60-90 min. Durch den
alkalischen pH der Transferlösung wurde die DNA kovalent mit der Membran vernetzt. Nach
der Neutralisierung der Membran (Neutralisierungslösung) wurde die Membran 3 min bei

II. Material & Methoden 51
60 °C getrocknet. Zur Überprüfung des Transfers wurde das Agarosegel nach Ethidium-
bromid-Färbung im UV-Licht kontrolliert.
Vorhybridisierung und Hybridisierung:
Ein Southern-Hybridisierungsexperiment wurde im Rahmen der Sequenzierung des hypo-
thetischen Natrium/Glucose-Symporters (SSSF) durchgeführt. Für die Hybridisierung wurde
eine DIG-markierte PCR-Sonde verwendet, die mit den Primern „SSFfor2“ und „SSFrev“
amplifiziert worden war. Die Sonde diente zum Nachweis des SSSF-tragenden Pst I-
Restriktionsfragments und zur Abschätzung der Größe des in der Ligase-vermittelten bzw.
inversen-PCR zu erzeugenden PCR-Produkts.
Vor der Hybridisierung der DNA-Sonde mit der fixierten DNA wurde eine Vorhybridisierung
durchgeführt. Diese erfolgte bei 65 °C für 1-3 h mit 20 ml vorgewärmter Vorhybridisierungs-
lösung (65 °C). Sie dient dazu, unspezifische Wechselwirkung zwischen Membran und
Sonde zu minimieren. Zur Herstellung der Hybridisierungslösung wurden ca. 150 ng DNA-
Sonde in 10 ml Vorhybridisierungslösung gegeben, 10 min in kochendem Wasser denaturiert
und auf Eis abgekühlt. Die Vorhybridisierungslösung wurde gegen die Hybridisierungs-
lösung ausgetauscht. Die Hybridisierung erfolgte über Nacht bei 65 °C in einem Hybridi-
sierungsofen (Uni-Equip, Martinsried). Nach Ende der Hybridisierung wurde die Lösung mit
der DNA-Sonde in ein steriles Röhrchen überführt und sofort eingefroren. Sie konnte so
mehrfach verwendet werden.
Immunologischer Nachweis der DNA-Hybride:
Nach der Hybridisierung wurde die Membran zwei stringenten Waschschritten unterzogen,
um unspezifisch gebundene Sonde von der Membran zu entfernen. Zuerst wurde die
Membran zweimal 5 min mit SSC-A Lösung (60 °C), anschließend zweimal 15 min mit SSC-
B Lösung (60 °C) gewaschen (RT, Schüttler, IKA Vibrax, VXR). Für den immunologischen
Nachweis wurde die Membran 1 min mit Puffer 1, danach 30 min mit Puffer 2 und wiederum
für 1 min mit Puffer 1 gewaschen. Die Antikörperanbindung erfolgte mit 20 ml Antikörper-
lösung (4 µl Antikörperkonjugat in 20 ml Puffer 2) im Hybridisierungsofen (30 min). Anschlie-
ßend wurde die Membran 2 min mit Puffer 3 äquilibiert. Danach erfolgte die Färbereaktion
durch Zugabe von 10 ml Färbereagenz (10 ml Puffer 3 versetzt mit 35 µl BCIP und 37 µl
NBT) im Dunkeln (RT, 5-10 min). Die Färbereaktion wurde durch Zugabe von Puffer 4
gestoppt. Die Membran wurde aus der Hybridisierungsröhrchen genommen, in Plastikfolie
eingeschweißt und zur Dokumentation eingescannt.

II. Material & Methoden 52
11. RNA-Arbeitstechniken Soweit nicht anders vermerkt, wurden alle Lösungen für das Arbeiten mit RNA mit doppelt
autoklaviertem und Diethylpyrocarbonat (DEPC, 0,1 %) behandeltem Wasser angesetzt (mit
Ausnahme von Tris-haltigen Lösungen). Die fertigen Lösungen und Puffer wurden erneut
autoklaviert. Glasflaschen, Glasgefäße, Messzylinder, Rührfische und Spatel wurden für
mindestens 6 h bei 160 °C gebacken. Um Arbeitsflächen, Arbeitsmaterialien und Gelkam-
mern zu reinigen, wurden diese mit „RNase-away“ behandelt.
11.1 RNA-Isolierung und Aufreinigung Zur RNA-Isolierung aus verschiedenen H. elongata Stämmen (WT, KB1, KB10 und KB10.1)
wurde eine entsprechende MM63-4 Übernachtkultur mit frischem Medium auf eine OD600 von
0,4 verdünnt (Gesamtvolumen: 100 ml). Die Zellernte erfolgte in der spätexponentiellen
Wachstumsphase. Entsprechend wurde P. putida in 100 ml Medium 1 angezogen. Bei der
RNA-Isolierung aus R. marinus wurde M162-Medium mit 2 % bzw. 6 % NaCl für die Anzucht
verwendet. Die Hauptkultur von R. marinus (100 ml) wurde 2 %-ig mit der Vorkultur beimpft.
Die Zellernte erfolgte hier bei einer OD600 von 0,5.
RNA-Isolierung:
Die Ernte erfolgte im GSA-Rotor bei 11000x g und 4 °C für 15 min.
• Es wurden maximal 100 mg des Zellpellets in ein Falkon-Röhrchen überführt und durch
Vortexen in 4 ml Puffer A (vorgekühlt) resuspendiert.
• Die Zellsuspension wurde mit 0,5 ml 10 %-igem SDS versetzt, kurz geschüttelt und
sofort mit 5 ml Phenol (65 °C) überschichtet. Anschließend wurde der Ansatz 4 min bei
65 °C unter mehrmaligen Invertieren inkubiert.
• Der Ansatz wurde nach Schockgefrieren in flüssigem Stickstoff (2 min) bei 37 °C im
Wasserbad aufgetaut. Für die Phasentrennung wurde die Probe bei 2710x g für 10 min
zentrifugiert (Laborfuge AE, Heraeus).
• Je 400 µl der Oberphase wurde auf 2 ml Eppendorf-Reaktionsgefäße aufgeteilt und mit
je 400 µl Phenol/Chloroform/Isoamylalkohol überschichtet, geschüttelt und weitere 5
min bei 8000x g zentrifugiert (4 °C, Biofuge, Fresco, Heraeus).
• Die RNA-haltige Oberphase wurde in 400 µl Aliquots in 2 ml Caps überführt. Zur Fäl-
lung der RNA wurde jeweils 1/10 des Volumens 3 M Natriumacetat-Lösung und 2,5
Volumen Ethanol (100 %) zugegeben und die Ansätze bis zum Gebrauch bei –70 °C
gelagert.
RNA-Reinigung:
• Je zwei gefällte RNA-Proben wurden für 10 min in einer Kühlzentrifuge (4 °C) bei
8000x g zentrifugiert und der Überstand verworfen. Die Pellets wurden in je 50 µl

II. Material & Methoden 53
H2ODEPC aufgenommen, vereinigt und mit dem RNeasy Kit (Qiagen) nach Anleitung des
Herstellers aufreinigt.
Für die Verwendung der RNA in RT-, DDRT- oder RACE-PCR wurde eine Reinigung unter
gleichzeitiger DNA-Hydrolyse (60 min) auf RNeasy-Säulen (Qiagen) durchgeführt. Mittels
PCR wurden anschließend die RNA-Ansätze auf DNA-Verunreinigungen überprüft. In diese
PCR wurden unterschiedliche Primer eingesetzt, mit denen kurze DNA-Fragmente (150-500
bp) amplifiziert werden können. Bei Vorhandensein eines PCR-Produkts im anschließenden
Agarosegel wurde die DNA-Hydrolyse auf den RNeasy-Säulen sooft wiederholt, bis kein
Produkt mehr amplifiziert werden konnte.
11.2 Bestimmung der RNA-Konzentration Die RNA-Konzentration in einer Probe wurde durch Messung der Absorption (Diodenarray-
Spektralphotometer, Agilent 8453) bei einer Wellenlänge von 260 nm bestimmt. Eine Lösung
mit 40 µg/ml RNA weist bei 260 nm und einer Schichtdicke von 1 cm eine Absorption von 1
auf. Die Reinheit der RNA wurde wie bei der DNA (Abschnitt II, 6.3.5) über den Quotient
A260/A280 abgeschätzt. Reine RNA weist einen Quotient von 1,9-2 auf. Die RNA wurde zur
Konzentrationsbestimmung 1:100 und 1:200 mit Tris-HCl (10 mM, pH 7) verdünnt, so dass
die gemessene Absorption zwischen 0,1 und 1 lag.
11.3 Gelelektrophoretische Auftrennung von RNA Aufgrund der hohen Neigung zur Ausbildung von Sekundärstrukturen durch intra- und inter-
molekulare Basenpaarungen zeigen einzelsträngige RNA-Aggregate ein nicht reproduzier-
bares Laufverhaltens im Agarosegel. Um RNA in Abhängigkeit von ihrem Molekulargewicht
aufzutrennen, wird daher eine Gelelektrophorese unter denaturierenden Bedingungen durch-
geführt. In der vorliegenden Arbeit wurden zwei Variationen angewandt (Abschnitt II,11.4.1).
RNA Agarosegelelektrophorese - Denaturierung mit Formamid:
Um vor Northern-Hybridisierungsexperimenten die Qualität der präparierten RNA zu über-
prüfen, wurde diese zunächst zu Kontrollzwecken in einem Agarosegel aufgetrennt. Dazu
wurden 3 µg RNA zunächst mit einem Formamid haltigen Ladepuffer (2x, Fermentas) 15 min
bei 70-85 °C denaturiert und anschließend in einem Agarosegel (0,8 %, 1x TB-Puffer) bei
einer Spannung von 40 V aufgetrennt (2 h). Der Ladepuffer enthielt Ethidiumbromid, so dass
das Gel direkt unter UV-Licht photographiert werden konnte.
11.4 Northern-Hybridisierung Die Northern-Hybridisierung dient dazu, die Expression bestimmter Gene auf Ebene der
Transkription zu untersuchen. Analog zur Southern-Hybridisierung wird die gelelektropho-

II. Material & Methoden 54
retisch aufgetrennte RNA auf einer Nylonmembran immobilisiert. Die Hybridisierung der zu
untersuchenden Transkripte erfolgte mit einer sequenzspezifischen Digoxigenin markierten
„antisense“ RNA-Sonde.
11.4.1 RNA Agarosegelelektrophorese - Denaturierung mit Glyoxal und DMSO
Für die Durchführung eines Northern-Hybridisierungsexperiments wurde die RNA mit Glyoxal
und DMSO denaturiert. Dazu wurde sie 50 min bei 50 °C mit Glyoxal und DMSO in
Phosphat-Puffer (Denaturierungslösung) inkubiert. Durch hohe Temperatur und DMSO
werden die inter- und intramolekularen Wasserstoffbrücken aufgebrochen und so die
Guanin-Reste für die kovalente Modifikation mit Glyoxal freigelegt.
RNA-Denaturierungsansatz:
Gesamt RNA 2-5 µg
deionisiertes Glyoxal 4 µl
10x Phosphatpuffer 2 µl
DMSO 10 µl
H2ODEPC ad 20 µl
Glyoxal oxidiert leicht zu Glyoxalsäure, welche zur Hydrolyse von RNA führt. Um die entstan-
dene Glyoxalsäure zu entfernen wurde das Glyoxal daher vor der Nutzung mit einem Ionen-
austauscherharz (AG501-X8, BioRad) behandelt.
Nach der Denaturierung wurden die Proben kurz auf Eis gestellt, sofort mit 5 µl Ladepuffer
versetzt und bei 40-50 V im Agarosegel (0,8 % mit 1x Phosphatpuffer, Gelkammer von Owl,
Portsmouth) aufgetrennt. Zur Längenbestimmung der durch Northern-Hybridisierung detek-
tierten RNA-Banden wurde neben den RNA-Proben ein RNA-Molekulargewichtsstandard
(RNA Ladder, High Range, MBI Fermentas) im Gel aufgetrennt.
Die Verbindung zwischen Glyoxal und Guanin ist nur bei neutralem und saurem pH stabil
und wird durch alkalische Bedingungen rückgängig gemacht. Damit es während der Gelelek-
trophorese mit Phosphatpuffer nicht zur Ausbildung eines pH-Gradienten und so zur
Deglyoxylierung der RNA kommt, wurde der Laufpuffer in der Gelkammer stetig von der
Anode zur Kathode gepumpt. Für die gleichmäßige Durchmischung sorgten zudem zwei
Rührfische und ein Magnetrührer (Ika-Combimag RCD), auf den die Gelkammer gestellt
wurde. Beide Vorrichtungen wurden erst nach dem Einwandern der Proben in das Gel ange-
schlossen.
Zur Dokumentation der Elektrophorese wurden alle RNA-Proben mit Ausnahme des RNA-
Leiters doppelt aufgetragen. Nach der Elektrophorese wurde der eine Teil des Gels zur
Kontrolle mit Ethidiumbromid angefärbt und photographiert, der andere Teil in den Transfer
eingesetzt. Vor der Färbung mit Ethidiumbromid musste das Glyoxal entfernt werden. Dafür

II. Material & Methoden 55
wurde das Gel 15 min in 0,1 M NaOH inkubiert und anschließend zweimal 15 min in 0,2 M
Na-Acetat neutralisiert. Die Ethidiumbromid-Färbung erfolgte wie für DNA-Gele beschrieben.
11.4.2 Northern-Transfer
Für den RNA-Transfer wurde das Turboblotting-System (Kappilarblot) der Firma Schleicher
& Schuell gemäß der Herstellerangaben verwendet. Zwischen Nylonmembran (S&S Nytran®
SuPerCharge) und Gel wurde eine Maske aus Parafilm gelegt. Der Transfer erfolgte über 5 h
mit 20x SSC. Nach dem Transfer wurden die Taschen des Gels mit einem weichen Bleistift
auf der Membran markiert. Die RNA wurde mittels eines UV-Crosslinkers (UV-Stratalinker®
18000, für 1 min, 120000 µJ, Wellenlänge 254 nm) kovalent mit der Membran verbunden.
Zum Entsalzen der Membran wurde diese kurz in 2x SSC gelegt. Die nachfolgende Hybridi-
sierungsreaktion erfolgte in einem RNAse-freie Glasröhrchen.
11.4.3 Hybridisierung und immunologischer Nachweis der RNA-Hybride
Vor der eigentliche Hybridisierung wird die Membran einer Vorhybridisierung unterzogen, um
unspezifische Wechselwirkung zwischen Sonde und Membran zu minimieren. Während der
Hybridisierung lagert sich die DIG-markierte RNA-Sonde spezifisch an komplementäre
Bereiche in mRNA-Transkripten an. Unspezifisch gebundene Sonde wird durch Waschen
unter stringenten Bedingungen (hohe Temperatur, niedrige Salzkonzentration) entfernt. Die
Detektion der RNA-Hybride erfolgt mit dem Digoxigenin-Anti-Digoxigenin Antikörper System.
Der Antikörper bindet spezifisch an das Digoxigenin der RNA-Sonde. Kovalent mit dem
Antikörper ist eine alkalische Phosphatase verknüpft, die ihrerseits das Substrat CDP*
dephosphoryliert. Unter Bildung eines metastabilen Dioxetan Phenolat-Anions, welches in
gepufferter Lösung zerfällt, wird Licht mit einer Wellenlänge von 466 nm emittiert. Das emit-
tierte Licht wird mittels Röntgenfilm aufgefangen.
Hybridisierung:
• Die Membran wurde 1 h bei 68 °C mit Vorhybridisierungslösung inkubiert.
• In H2Oreinst verdünnte RNA-Sonde (1 µg) wurde 5 min denaturiert (95 °C), auf Eis abge-
kühlt und zu 10 ml vorgewärmter (68 °C) Hybridisierungslösung gegeben. Die Hybridi-
sierung erfolgte über Nacht (14-16 h) bei 68 °C.
• Die Membran wurde 2x 5 min mit je 100 ml 2x SSC-Lösung (63 °C) gewaschen.
• Es folgten zwei Waschschritte mit vorgewärmter 0,1x SSC-Lösung (je 15 min).
Immunologischer Nachweis und Detektion:
• Die Membran wurde kurz mit Waschpuffer gespült, dann 50 min mit 100 ml Puffer 2
geblockt.

II. Material & Methoden 56
• Das Antikörperkonjugat wurde in 20 ml Puffer 2 1:20.000 verdünnt und die Membran
darin 30 min bei RT inkubiert (Hybridisierungsofen).
• Die Membran wurde zweimal 15 min mit Waschpuffer gewaschen und anschließend
5 min in 20 ml Detektionspuffer äquilibiert.
• Zur Detektion der RNA-Hybride wurden 10 µl CDP* (25 mM) (Roche, Mannheim) in
1 ml Detektionspuffer auf den Rand der Membran pipettiert. Anschließend wurde die
Membran 5 min im Dunkeln in einem Hybridisierungsbeutel inkubiert.
• Die Membran wurde in Plastikfolie eingeschweißt und die Chemilumineszens mittels
Röntgenfilm (Kodak X-omat, Sigma) erfasst. Die Belichtungszeit des Röntgenfilms be-
trug 5-30 min.
11.4.4 In vitro-Transkription zur Herstellung einer Digoxigenin-markierten RNA-Sonde
RNA-Sonden werden eingesetzt, um besonders geringe Mengen einer Zielsequenz (z.B.
RNA-Transkript) zu detektieren oder um eine Hybridisierung unter sehr stringenten Bedin-
gungen durchführen zu können und so unspezifische Hybridisierungssignale zu vermeiden.
RNA:RNA-Hybride weisen zudem eine höhere Stabilität auf als DNA:RNA-Hybride.
Für die Sonden-Synthese wurde zunächst ein PCR-Fragment amplifiziert, das an einem
Ende mit dem T7-Promotor (5’- TAA TAC GAC TCA CTA TAG G / Xmer 3´) ausgestattet ist.
Stromaufwärts des T7-Promotors wurde ein kurzer Überhang (Xmer= 5’- GGA TCC) ein-
gebaut, damit die T7-RNA-Polymerase an den Promotor binden kann. Bei der Primeraus-
wahl muss darauf geachtet werden, dass eine „antisense“ RNA-Sonde hergestellt wird, damit
die „sense“ mRNA des Gens detektiert werden kann. Die Promotorsequenz ist daher im
reversen PCR-Primer enthalten. Die Länge des PCR-Produkts erstreckte sich über das
vollständige Gen, dessen Transkript nachgewiesen werden sollte. Das PCR-Produkt wurde
im Gel überprüft und anschließend mit Hilfe des PCR-Purificationkits (Qiagen) aufgereinigt
(Abschnitt II, 6.3.4). Die DNA-Konzentration wurde auf 50 ng/µl eingestellt.
Reaktionsansatz für RNA-Sonden-Herstellung: DNA-Matrize 200 ng Transkriptionspuffer (10x) 2 µl DIG-RNA Labeling Mix (10x) 2 µl RNAse-Inhibitor 40 U T7-RNA-Polymerase 40 U H2ODEPC ad. 20 µl
Der Reaktionsansatz wurde für 2 h bei 37 °C inkubiert und die DNA anschließend durch
Zugabe von 20 U DNAse I (RNase frei) hydrolysiert (15 min, 37 °C). Die Hydrolyse wurde
durch Zugabe von 2 µl EDTA (0,2 mM) abgestoppt. Die RNA-Konzentration wurde photo-

II. Material & Methoden 57
metrisch bestimmt. Für den Einsatz als RNA-Sonde wurden 100 ng RNA pro ml Hybridi-
sierungslösung verwendet.
11.5 Reverse Transkriptase-vermittelte Methoden
11.5.1 Reverse Transkriptase-vermittelte PCR
Mit Hilfe der RT-PCR können spezifische RNA-Sequenzen vervielfältigt und damit nach-
gewiesen werden. Die Methode ist wesentlich sensitiver als ein Northern-Hybridisierungs-
experiment. Die Methode wurde u.a. eingesetzt, um mRNA-Transkriptlängen in den Inser-
tionsmutanten H. elongata KB10 und KB10.1 zu untersuchen.
Bei der RT-PCR erfolgt in einem vorgelagerten Schritt zunächst eine cDNA-Erststrang-
synthese mit DNA-freier RNA als Matrize, wobei genspezifische reverse-Primer (vom 3’-
Ende der Gene) eingesetzt werden. Um eine Kontamination mit DNA auszuschließen, wird
als Negativkontrolle ein cDNA-Syntheseansatz ohne die Zugabe der reversen Transkriptase
angesetzt. In der nachfolgenden RT-PCR wird ein forward-Primer und ein eingerückter
(nested) reverse-Primer eingesetzt. Durch Variation des forward-Primers kann die 5´-Aus-
dehnung der cDNA und somit der mRNA-Transkripte abgeschätzt werden. In der Negativ-
kontrolle der cDNA-Synthese darf kein PCR-Produkt nachgewiesen werden, da die Taq-
Polymerase RNA nicht erkennt. Die cDNA-Synthese wurde wie in dem nächsten Abschnitt II,
11.5.2 beschrieben durchgeführt.
11.5.2 5’-Race-PCR (rapid amplification of cDNA ends)
Diese Methode wurde entwickelt, um komplette 5’-Enden einer nicht vollständig bekannten
cDNA-Sequenz klonieren zu können (Tillett et al., 2000). In dieser Arbeit wurde sie angewen-
det, um für eine Promotoranalyse den Transkriptionsstartpunkt stromaufwärts des mgs-Gens
in H. elongata KB10 zu identifizieren.
Wie bei der RT-PCR wird mittels eines genspezifischen reverse-Primers die cDNA entlang
eines mRNA-Strangs bis zum 5’-Ende des Transkripts synthetisiert. Es folgt der hydro-
lytische Abbau der eingesetzten RNA und eine definierte Verlängerung des cDNA-Strangs
an seinem unbekannten 3’-Ende durch das Anhängen von dCTP. Diese als „tailing“ („C-
tailing“) bezeichnete Reaktion wird durch eine terminale Desoxynukleotid-Transferase (TdT)
katalysiert. Um die Zweitstrangsynthese zu ermöglichen, bindet in einer nachfolgenden PCR
ein „tail“-spezifischer Primer an diesen Überhang (AAP-Primer). Mittels eines zweiten
reverse-Primers, der in der bekannten cDNA-Sequenz bindet, wird das Fragment amplifiziert.

II. Material & Methoden 58
A) Ansatz für cDNA-Synthese bzw. Negativkontrolle:Gesamt RNA 5 µg dNTP-Mix (je Nukleotid) 500 nmol Primer 2 pmol H2ODEPC ad. 22 µl
B) Reaktionsmixe: cDNA-Synthese: Negativkontrolle: RT-Puffer (10x ) 1x 1x MgCl2 125 125 nmol DTT 500 500 nmol RNAse Out 100 100 U Superskript III 200 0 U H2ODEPC ad. 28 28 µl
Die RNA/Primer-Ansätze wurden zur Denaturierung 10 min bei 65 °C inkubiert und eine
Minute auf Eis gekühlt. Anschließend wurde der jeweilige Reaktionsmix hinzupipettiert und
die Reverse Transkriptase-Reaktion im Thermocycler gestartet.
Temperaturprofil: 10 min bei 25 °C Anlagerung der genspezifischen Primer 50 min bei 50 °C Synthesereaktion 5 min bei 80 °C Abstoppen der Reaktion
Die RNA in der Probe wurde nach Ablauf der Reaktion mit einem Gemisch aus RNaseH
(2 U) und RNaseA (Qiagen, 25 µg/µl) hydrolysiert (37 °C, 30 min). Die Ansätze wurden mit-
tels Qiaquick PCR-Purificationkit von Nukleotiden, Primern und Enzymen gereinigt und mit
50 µl H2ODEPC (65 °C) eluiert.
Für die Durchführung einer RACE-PCR wurde nach der cDNA-Synthese der „C-tail“ an das
5’-Ende der cDNA angehängt. Zusätzlich zur eigentlichen Reaktion wurde ein Ansatz als
Negativkontrolle pipettiert.
“C-tailing“-Reaktion: RT-Puffer 0,5x dCTP 5,0 nmol MgCl2 37 nmol cDNA gereinigt 10 µl H2ODEPC ad. 24 µl
Die cDNA wurde 3 min bei 95 °C in einem Thermocycler denaturiert und anschließend auf
37 °C abgekühlt. Bei Erreichen der Temperatur wurden dem Ansatz 15 U TdT zugegeben,
zum Kontrollansatz wurde die entsprechende Menge H2ODEPC pipettiert. Beide Ansätze
wurden 10 min bei 37 °C inkubiert und die Reaktion bei 65 °C abgestoppt (10 min).

II. Material & Methoden 59
Die eigentliche RACE-PCR erfolgte in einer PCR-Ansatz mit einem sequenzspezifischen
nested-Primer (20 pmol) und einem für den „C-tail“ spezifischen 5’-Race Abridged Anchor-
Primer (AAP, 20 pmol, Invitrogen) sowie je 5 µl cDNA bzw. Negativkontrolle aus der „C-
tailing“-Reaktion.
Reaktionszyklus:
95 °C 2 min
95 °C 0,5 min
55 °C 1 min 30x
72 °C 1 min
72 °C 5 min
11.5.3 Entwicklung einer Differential Display-RT-PCR Strategie für R. marinus
Differential Display RT-PCR ist eine Methode, die den Nachweis von differentiell exprimier-
ten mRNA-Transkripten ermöglicht. So können z.B. Gene identifiziert werden, die unter
wechselnden Anzuchtbedigungen (z.B. Salinität) einer Kultur oder Zellpopulation unter-
schiedlich stark exprimiert werden (Liang & Pardee, 1992).
Die Methode beruht auf der Amplifikation exprimierter Gensequenzen einer Zelle durch
reverse Transkription der mRNA (Abschnitt II, 11.5.2), einer anschließende PCR und einer
gelelektrophoretischen Auftrennung der PCR-Produkte. Durch Vergleich des Bandenmusters
werden differentiell exprimierte Gene identifiziert. Ein solcher Vergleich wird jedoch durch
zwei Faktoren erschwert. Erstens ist die mRNA-Population einer Zelle zu komplex, um alle
Transkripte im Gel voneinander zu trennen. Zweitens, liegen die Kopien der einzelnen Trans-
kripte in sehr unterschiedlicher Zahl vor, so dass unterrepräsentierte Transkripte schwer zu
erfassen sind. Die größte Schwierigkeit dieser Methode besteht daher darin, ein reproduzier-
bares Bandenmusters zu erhalten, aus dem man differentielle Banden mit einiger Verläss-
lichkeit isolieren kann. Um diese Faktoren zu minimieren, wird für die Amplifikation der
Transkripte auf spezielle Primer zurückgegriffen.
Bei der Durchführung der DDRT-PCR wird üblicherweise auf sog. „arbitrary“-Primer (10-11
bp) zurückgegriffen. Die Sequenzen dieser Primer werden mittels eines speziellen Compu-
terprogramms entsprechend der Genomsequenz des untersuchten Organismus ermittelt.
Verwendet werden Sequenzen, die mit großer Häufigkeit im Genom vertreten sind, jedoch
eine gewisse Differenzierung der Transkriptpopulationen ermöglichen. Da die Genom-
sequenz von R. marinus zum Zeitpunkt der Versuchsdurchführung nicht zugänglich war,
musste eine alternative Strategie erarbeitet werden.
Ziel dieser Strategie war es, die Anzahl der Amplifikate, die bei der DDRT-PCR gebildet
werden, so weit zu reduzieren, dass einzelne Produkte als distinkte DNA-Banden in einem
Agarosegel erkennbar sind und näher charakterisiert werden können. Im Gegensatz zu

II. Material & Methoden 60
Eukaryonten ist die mRNA in Prokaryonten nur zu einem geringen Teil mit einem „Poly-A-
tail“ ausgestattet, der ein spezifisches Umschreiben in cDNA mittels eines Poly-T-Primers
ermöglicht. Reine Random-Primer führen in der cDNA-Synthese zu einer Umschreibung der
Gesamt-RNA und somit zu einer Vielzahl späterer Amplifikate, die in einem Agarosegel nicht
aufgelöst werden können. Als Ansatzpunkt der cDNA-Synthese wurden daher die 3 unter-
schiedlichen Stopkodons ausgewählt. So sollte die cDNA-Population hin zu Protein kodieren-
den Sequenzen verschoben werden. Für jedes der Stopkodons wurde ein eigener cDNA-
Synthese-Primer entworfen (Abb. 4):
PolyA-C-Stop: AAA AAA AAA AAA AAA CIN NIN NIN NIN NIN CTA PolyA-G-Stop: AAA AAA AAA AAA AAA GIN NIN NIN NIN NIN TTA PolyA-T-Stop: AAA AAA AAA AAA AAA TIN NIN NIN NIN NIN TCA
Die Primer bestehen an ihrem 3´-Ende aus zu den Stopkodons komplementären Sequenzen.
Diesen Sequenzen ist eine Inosin reiche Random-Sequenz vorgelagert, die aufgrund ihrer
Basenzusammensetzung nahezu jeden Sequenzbereich zu binden vermag. Den 5´-Bereich
des Primers bildet eine Poly-A15-Sequenz. Zwischen dieser Poly-A-Sequenz und der
Random-Sequenz ist jeweils eine spezifische Base eingefügt (C, G oder T). Mit Hilfe dieser
spezifischen Base und einem gegen sie gerichteten Oligo-A-Primer besteht die Möglichkeit,
nach einer cDNA-Synthese mit einem Primergemisch in einer PCR-Reaktion die cDNA-
Populationen und ihre spezifischen PCR-Produkte zu unterscheiden (Abb. 4).
Oligo-A-dC: AAA AAA AAA AAA AAA C
Oligo-A-dG: AAA AAA AAA AAA AAA G
Oligo-A-dT: AAA AAA AAA AAA AAA T
Durchführung:
Nach der cDNA-Synthese und RNA-Hydrolyse, die wie in Abschnitt II, 11.5.2 beschrieben
durchgeführt wurde, wurde mit Hilfe des TdT-Enzyms ein A-Überhang („A-tail“) an das 3’-
Ende der cDNA gehängt. Das „A-tailing“ erfolgte entsprechend des in Abschnitt II, 11.5.2
beschriebenen „C-tailings“. Hier wurde jedoch dATP anstelle von dCTP als Nukleotid einge-
setzt. Die „A-tail“ tragende cDNA wurde einer ersten, einseitigen, asymmetrische PCR
unterzogen (Abb. 4). Als Primer wurde in diesen Reaktionen ein spezieller Poly-T-Primer
(AP-Primer) eingesetzt, der um Restriktionsschnitten erweitert ist. Es wurde ein Standard-
PCR-Reaktionsansatz angesetzt, in den 100 pmol des AP-Primers eingesetzt wurden. In der
eigentlichen DDRT-PCR (Standard-PCR-Ansatz) wurden obige Oligo-A-Primer sowie ein
sog. AUAP-Primer verwendet (Abb. 4). Letzterer ist zum 5´-Bereich des AP-Primers komple-
mentär.
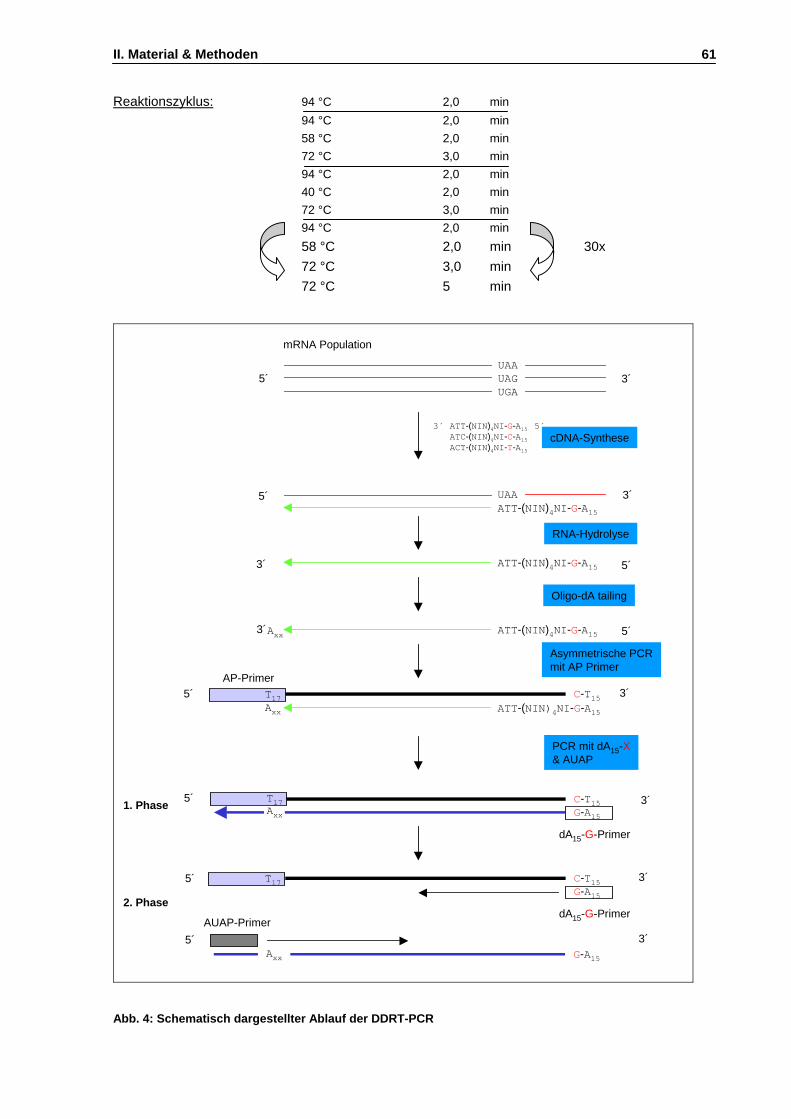
II. Material & Methoden 61
Reaktionszyklus: 94 °C 2,0 min 94 °C 2,0 min 58 °C 2,0 min 72 °C 3,0 min 94 °C 2,0 min 40 °C 2,0 min 72 °C 3,0 min 94 °C 2,0 min 58 °C 2,0 min 30x 72 °C 3,0 min 72 °C 5 min
UAAUAGUGA
3´ ATT-(NIN)4NI-G--C--T-
A15 5´ATC-(NIN)4NI A15ACT-(NIN)4NI A15
mRNA Population
cDNA-Synthese
UAAATT-(NIN)4NI A15-G-
ATT-(NIN)4NI A15-G-
Oligo-dA tailing
ATT-(NIN)4NI A15-G-Axx
RNA-Hydrolyse
Asymmetrische PCRmit AP Primer
ATT-(NIN)4NI A15-G-
AP-PrimerC-T15
C-G-T15A15
PCR mit dA15& AUAP
-X
T17 C-G-T15A15
G-A15
T17
Axx
AUAP-Primer
1. Phase
2. Phase
T17Axx
Axx
dA15 rimer
dA15 rimer
5´ 3´
5´ 3´
3´ 5´
3´
3´
5´
5´
5´
5´
5´
3´
3´
3´
-G-P
-G-P
Abb. 4: Schematisch dargestellter Ablauf der DDRT-PCR

II. Material & Methoden 62
12. Konjugation Als bakterielle Konjugation wird der Austausch genetischen Materials durch die Übertragung
eines Plasmids von einer Donor- zu einer Rezipienten-Zelle bezeichnet. In dieser Arbeit
wurden für die Konjugation Derivate zweier mobilisierbarer Plasmide eingesetzt
(pK18mobsacB und pPromEct), welche die Erkennungsstelle für die Mobilisierung (mob-site)
aufweisen. Die für die Konjugation notwendigen tra-Gene sind auf den Plasmiden nicht
vorhanden und müssen durch den Spenderorganismus bereitgestellt werden. Diese Komple-
mentierung wird durch das konjugative Plasmid RP4 gewährleistet, dessen tra-Gene
chromosomal im Donorstamm E. coli S17-1 integriert sind (Simon et al., 1983). In der
vorliegenden Arbeit erfolgte der Plasmidtransfer von E. coli S17-1 nach H. elongata nach der
von Kunte & Galinski (1995) entwickelten Methode. Zur Expression des mgs-Gens in
H. elongata wurde das Plasmid pPromEct verwendet (Brünig, 2005).
Durchführung:
• E. coli S17-1 wurde über Nacht auf einer AB-1 Agarplatte (Antibiotikum) bei 37 °C
angezogen. H. elongata wurde 24 h in 50 ml AB-1-Medium (250 ml OD-Kolben)
kultiviert.
• Die Zellen des Donors wurden mit einer Impföse von der Agarplatte entnommen und in
4 ml AB-1-Flüssigmedium resuspendiert (bis OD540 = 0,8). Je 0,5 ml Donor und
Rezipient wurden in Reaktionsgefäß gemischt und bei 15800x g (3 min) abgeerntet.
• Der Überstand wurde verworfen, die Zellen wurden im Rücklauf resuspendiert und auf
sterile hydrophile Nitrozellulosefilter (0,45 µm) pipettiert, die auf vorgewärmten Inkubati-
onsplatten (AB-1) lagen. Die Inkubation erfolgte bei 37 °C über Nacht.
• Nach 24 h wurde der Filter mit einer sterilen Pinzette von der Agaroberfläche genom-
men und in 1,5 ml Reaktionsgefäß überführt. Die Zellen wurden mit 1 ml NaCl-Lösung
(2 %) vom Filter gespült und resuspendiert. Die Zellsuspension wurde anschließend
10-1 bis 10-4 verdünnt. Je Verdünnungsstufe wurden 50-100 µl auf Selektionsmedium
ausplattiert.
Die erhaltenen Transkonjuganten weisen eine plasmidvermittelte Antibiotikaresistenz auf, die
ihnen das Wachstum auf MM63-2 mit Antibiotikum ermöglicht. Aufgrund einer Prolin-Auxo-
trophie kann E. coli S17-1 nicht auf Mineralsalzmedium wachsen, was eine Gegenselektion
nach erfolgter Konjugation ermöglicht.
13. Herstellung chromosomaler Mutationen Für die Herstellung von Mutationen in H. elongata bzw. E. coli MKH13 wurden zwei unter-
schiedliche Vektoren verwendet (pK18mobsacB bzw. pMAK705). Die DNA-Konstrukte, die
für die Mutagenese verwendet wurden, bestehen aus Vektor-DNA und inserierter rekom-

II. Material & Methoden 63
binanter DNA aus H. elongata, R. marinus, Pseudomonas putida bzw. E. coli. Die rekom-
binante DNA wurden mittels PCR-Techniken, Restriktionsbehandlungen und Klonierungs-
techniken hergestellt. Rekombinante Plasmide wurden durch Blau/Weiß-Selektion in E. coli
DH5α identifiziert. Die gesuchten DNA-Konstrukte wurde nach einer Plasmidisolierung
mittels Restriktionsanalyse erkannt.
13.1 Mutagenese an H. elongata Zur Herstellungen von Mutationen in H. elongata (KB10 und KB10.1) wurde der Vektor
pK18mobsacB verwendet (Schäfer et al., 1994). Bei diesem Vektor handelt es sich um ein
pBR322 Derivat, das neben einer Mobilisierungsregion (mob-site), die für den Transfer
notwendige Replikationsstelle oriT trägt. Das pMB1 Replicon, das zu einer engen Wirts-
spezifität führt (Sutcliffe, 1979), ist der Grund dafür, dass H. elongata pK18mobsacB durch
homologe Rekombination chromosomal integrieren muss, um die kodierte Km-Resistenz
(aus Tn5) auszuprägen. Nach dem konjugativen Transfer wurden die Transkonjuganten auf
Km-haltigem (250 µg/ml) MM63-2-Medium selektioniert. Diese Kolonien haben in einem
ersten Prozess homologer Rekombination pK18mobsacB mit der integrierten Fremd-DNA in
das Genom integriert. Das sacB-Gen (Laevan-Saccharase) aus Bacillus subtilis stellt den
negativen Selektionsmarker dar (Schäfer et al., 1994). Die Bildung dieses Enzyms ist für
H. elongata ab einer Saccharose-Konzentration von 20 % letal und wurde daher zur Selekti-
on von „Doppel-Cross-Over“ Mutanten genutzt. Auf Saccharose haltigem Medium können
nur Zellen überleben, die zuvor das Plasmid durch einen zweiten „Cross-Over“-Prozess aus
dem Genom entfernt haben. Dieser Prozess stellt entweder den Wildtyp wieder her, oder
führt zum Verbleib des rekombinanten DNA-Konstrukts im Genom. Wildtyp und Mutanten
können durch Kolonie-PCR (Abschnitt II, 9.2.1) unterschieden werden.
Zur Durchführung wurden KmR Kolonien von H. elongata über Nacht bei 30 °C in LBG-2-
Medium angezogen und anschließend mit frischem Medium verdünnt (10-1 bis 10-8). Von
allen Verdünnungsstufen wurden 100 µl auf LBG-2-Medium mit 22 % Saccharose
ausplattiert. Die angewachsenen Kolonien wurden zur Überprüfung in eine Kolonie-PCR
eingesetzt.
13.2 Mutagenese an E. coli MKH13 Zur Herstellung einer otsB-Deletionsmutante von E. coli MKH13 wurde der Vektor pMAK705
verwendet (Hamilton et al., 1989). Zur Selektion der Transformanten vermittelt dieser Vektor
eine Chloramphenicol-Resistenz (aus pBR325). Zudem ermöglicht der Vektor eine Blau/
Weiß-Selektion zur Identifizierung rekombinanter Transformanten. pMAK705 besitzt ein
temperatursensitives Replicon (Derivat des Vektors pSC101), welches ihm eine Replikation
nur bei einer Temperatur von ≤ 30 °C ermöglicht. Eine freie Vektorreplikation bei höheren

II. Material & Methoden 64
Temperaturen (z.B. 43 °C) ist nicht möglich. Folglich können E. coli Stämme, die diesen
Vektor tragen, bei erhöhter Temperatur die Chloramphenicol-Resistenz nur ausprägen, wenn
sie den Vektor über homologe Rekombination in das Genom integriert haben.
Die DNA-Konstrukte wurden in den Vektor pMAK705 ligiert und in E. coli DH5α trans-
formiert. Aufgrund des temperatursensitiven Replicons wurde bei der Transformation ein ver-
längerter Hitzeschock (5 min) bei geringerer Temperatur (37 °C) durchgeführt. Die Regene-
rationszeit der Zellen verlängerte sich auf 90 min bei 30 °C. Die Agarplatten wurden bei 30
°C inkubiert. Anschließend wurden die plasmidtragenden Klone mittels Restriktionsanalyse
identifiziert.
Bildung des Kointegrats in E. coli MKH13 :
Nach der Identifizierung des gesuchten DNA-Konstrukts wurde der rekombinante Vektor
entsprechend der obigen Protokolländerungen in E. coli MKH13 transformiert und bei 30 °C
bebrütet. Damit der Vektor nach der Transformation über homologe Rekombination in das
Genom von E. coli MKH13 integriert,
• wurde 3x 20 ml LB-0,5-Medium (Cm 50) mit je einer transformierten E. coli MHK13
Kolonie (CmR) beimpft und bei 30 °C und 180 upm für 24 h kultiviert.
• Nach dieser Zeit wurde die Kultur bis zu 10-5 verdünnt. Die Verdünnungsstufen 10-3 bis
10-5 wurden auf LB-0,5 (Cm50) ausplattiert und bei 43 °C bebrütet.
• Große, CmR Kolonien wurden mehrmals auf LB-0,5 (Cm50) überpickt und bei 43 °C
bebrütet.
Auflösung des Kointegrats:
Um otsB in E. coli MKH13 zu deletieren, musste der Vektor im nächsten Schritt durch
„Doppel-Cross-Over“ wieder aus dem Genom entfernt werden. Dabei entsteht entweder der
Wildtyp oder die gewünschte Deletionsmutante. Um die Ausgliederung des Plasmids zu
erreichen,
• wurden 30 ml LB-0,5 (ohne Cm) mit einer Kolonie eines Kointegrat haltigen Stamms
beimpft und bei 37 °C (180 upm) inkubiert.
• Nach einer Kultivierungszeit von etwa 12 h wurde die Kultur 2 %-ig in neues Medium
überimpft und erneut bei 37 °C inkubiert.
• Dieser Prozess wurde zwei weitere Male wiederholt.
• Nach dieser Zeit wurde die Kultur verdünnt. Von den Verdünnungsstufen 10-2- 10-10
wurde je 2x 100 µl auf LB-0,5 ausplattiert und bei 37 °C bebrütet.
• Am nächsten Tag wurden Einzelkolonien auf neue Platten LB-0,5 (ohne Cm) und LB-
0,5 (Cm50) überpickt und für 24 h bei 37 °C inkubiert.

II. Material & Methoden 65
• Die erhaltenen CmS E. coli Kolonien wurden durch Kolonie-PCR auf die otsB-Deletion
hin überprüft (Abschnitt II, 9.2.1).
14. Erstellung einer partiellen Genbank mit R. marinus DNA
14.1 Vektor pHSG575 Mit Hilfe einer R. marinus Genbank und E. coli Defektmutanten sollten Transportsysteme für
das kompatible Solut Mannosylglycerat identifiziert werden. Zur Konstruktion dieser Gen-
bank wurde der low-copy-number-Vektor pHSG575 verwendet (Takeshita et al., 1987).
Dieser weist ein Chloramphenicol-Resistenzgen (CmR) zur Selektion sowie ein lacZ’-Frag-
ment mit einer MCS auf.
Für die Erstellung der Genbank wurde pHSG575 mit Bam HI linearisiert, dephosphoryliert
und mit partiell Sau 3A hydrolysierter genomischer R. marinus DNA ligiert. Nach der
Transformation superkompetenter E. coli DH5α Zellen und Selektion weiß gefärbter
Kolonien, wurden diese Klone chargenweise vereinigt. Die pHSG575 basierte Genbank
umfasst etwa 17113 Klone in 18 Chargen mit jeweils 750-1000 Klonen. Die daraus isolierte
Plasmid-DNA wurde zur Transformation der E. coli Defektstämme (MKH13 bzw. BKA-13)
verwendet.
Zur Überprüfung der Methode wurde zunächst ein Experiment zur Komplementierung der
Thiamin-Auxotrophie von E. coli DH5α durchgeführt. Zum Nachweis der Komplementierung
wurden CmR Kolonien mit der Impföse von den Transformationsplatten abgenommen, mit
MM63-0,5 gewaschen und nach Verdünnung auf Mineralsalzmedium (MM63-0,5) ohne
Thiamin ausplattiert.
14.2 Vektor pXMJ19 Um eine Expression von R. marinus Genen in E. coli sicherzustellen, wurde eine zusätzliche
Genbank mit dem Vektor pXMJ19 angelegt (Jakoby et al., 1999). Diese R. marinus Genbank
umfasst 9705 Klone in 28 Chargen mit jeweils etwa 300-400 Klonen. Bei dem Vektor
pXMJ19 handelt es sich um ein Derivat des Plasmids pMMB67HE (Fürste et al., 1986) mit
einem eingebauten Chloramphenicol-Resistenzgen. Der Vektor weist ein weites Wirtsspek-
trum auf und repliziert sich in verschiedenen Gram-negativen Bakterien (z.B. Pseudomonas
aeruginosa und Serratia marcescens). Der tacP-Promotor (Hybrid-Promotor aus der -35 trp-
Promotorregion und der -10 lacUV5 Promotorregion aus E. coli, (De Boer et al., 1983) und
der lacIq-Repressors bilden ein durch IPTG induzierbares System, mit dessen Hilfe die
heterologe Expression verschiedener Gene in Gram-negativen Bakterien untersucht werden
kann (Bagdasarian et al., 1983). Dabei unterliegt der tacP-Promotor bis zur Induzierung
durch IPTG der Kontrolle des lac-Repressor. Um die Expression der inserierten Gene aus

II. Material & Methoden 66
R. marinus in E. coli zu induzieren, wurden die Zellen auf den Selektionsplatten zusammen
mit jeweils 50 µl IPTG (0,2 mM Endkonzentration) ausplattiert.
15. Proteinbiochemische Methoden
15.1 Verwendetes Expressionssystem Für die heterologe Expression des mgs-Gens aus R. marinus in H. elongata Wildtyp und
KB1 wurde das bereits von Brünig (2005) hergestellt pBBRMCS1-Derivat pPromEct
verwendet. Dieser Vektor trägt die Promotorregion der Ectoin-Gene von H. elongata. Ur-
sprung dieses Plasmids ist der für eine Chloramphenicol-Resistenz kodierende Vektor
pBBR1MCS (Kovach et al., 1994). Dabei handelt es sich um ein Derivat des natürlich vor-
kommenden Vektors pBBR1, welcher aus Bordetella bronchiseptica isoliert wurde (Antoine &
Locht, 1992). pBBR1 und seine Derivate besitzen ein breites Wirtsspektrum und wurden
schon zuvor erfolgreich für eine heterologe Expression in H. elongata genutzt (Schnoor,
2001; Burdziak, 2004; Brünig, 2005). Der Vektor pPromEct enthält die stromaufwärts des
ectA-Gens gelegene Promotorregion aus H. elongata (Brünig, 2005). Das mgs-Gen aus
R. marinus wurde stromabwärts dieser Promoterregion in den Vektor integriert und zusätz-
lich mit einer (His)6-tag kodierenden Sequenz versehen. Das entstandene Plasmid wurde
(pABB2) wurde durch Konjugation auf H. elongata Wildtyp und KB1 übertragen.
15.2 Zellaufschluss und Isolierung von Zellfraktionen
15.2.1 Anzucht plasmidtragender H. elongata Stämme
Die plasmidtragenden H. elongata Stämme (pPromEct bzw. pABB2) wurden in je 100 ml
MM63-4 und MM63-10 Medium unter Zusatz von Chloramphenicol angezogen. Die Anzucht
erfolgte in 250 ml Erlenmeyerkolben (37 °C, 180 upm) bis zu einer OD600 von 1. Diesen
Kulturen wurden mit LB-Vorkulturen gleicher Salinität beimpft.
15.2.2 Gewinnung des Gesamtzellproteins
Zur Gewinnung des Gesamtzellproteins wurden zwei unterschiedliche Vorgehensweisen an-
gewendet.
Gesamtzellprotein I:
1 ml Kultur (OD600 = 1) wurde 2 min bei 13600x g abzentrifugiert und der Überstand
verworfen. Das Zellpellet wurde in 100 µl 1x PBS-Puffer resuspendiert und mit 100 µl 2x
RSB-Puffer versetzt. Der Zellaufschluss erfolgte durch mehrmaliges Aufziehen der Probe mit
einer 26G-Kanüle (∅0,4 x 20 mm). Anschließend wurden die Proteine durch Erhitzen dena-
turiert (3 min, 70 °C).

II. Material & Methoden 67
Gesamtzellprotein II: (konzentrierte Proben, Brünig, 2005)
Bei dieser Methode wurde die DNA hydrolysiert und die Zelltrümmer weitestgehend abge-
trennt, so dass die Proben leichter auf das Gel aufzutragen waren und vorher noch eine
Proteinbestimmung durchgeführt werden konnte.
10-20 ml Kultur wurden durch mehrmalige Zentrifugation (13600x g, 5 min, RT) abgeerntet.
Die Zellpellets wurden über Nacht (ca. 16 h) eingefroren (–20 °C). Die Pellets wurden auf Eis
aufgetaut, in 200 µl Resuspensionspuffer II resuspendiert, mit 2 µl Lysozym (100 mg/ml)
versetzt und kräftig durchmischt. Die Proben wurden unter mehrmaligem Vortexen auf Eis
inkubiert (30 min). 10 µl SDS Lösung (10 %) wurden zugegeben und die Proben weitere 30
min auf Eis inkubiert. Dabei wurden die Proben schleimig bis fest. 2 µl DNase (10 mg/ml)
und 1 µl MgCl2 (50 mM) wurden zugegeben und die Ansätze erneut 1 h auf Eis inkubiert.
Dabei verloren die Proben ihre feste Konsistenz. Anschließend wurden die Proben
abzentrifugiert (13600x g, 30 min, 4 °C) und der proteinhaltigen Überstand abgenommen.
30-50 µl der Proben wurden für einen Proteintest aufbewahrt, der Rest wurde 1:1 mit 2x
RSB-Puffer versetzt und 3 min auf 75 °C erhitzt.
15.2.3 Gewinnung der periplasmatischen Fraktion durch osmotischen Schock
40 ml Bakterienkultur (aus Abschnitt II, 15.2.2) wurden abgeerntet und zur Gewinnung der
zytoplasmatischen Fraktion einem periplasmatischen Schock unterzogen (Nossal & Heppel,
1966). Dazu wurden die Zellen durch Zusatz einer 20 %-ige Saccharose-Lösung zunächst
einem hyperosmotischen Schock ausgesetzt. Dabei geht die Zelle in den Zustand der
Plasmolyse über. Die Zugabe von EDTA destabilisiert die äußere Membran durch Bindung
zweiwertiger Kationen zusätzlich. Im Schockpuffer enthaltenes TRIS, welches ebenfalls
zweiwertige Kationen der äußeren Membran bindet, führt zu einer Schwächung der Wech-
selwirkungen zwischen den einzelnen Lipopolysacchariden und einer gegenseitigen Absto-
ßung aufgrund ihrer negativen Ladungen. Nach Überführung der Zellen in ein hypotonisches
Milieu, dringt Wasser in das Periplasma ein, während Saccharose durch Porine nach außen
entlassen wird. Der Wassereinstrom ist schneller als der Saccharose-Ausstrom. Es entsteht
ein Überdruck, der die geschwächte äußere Membran aufbricht. Dadurch werden peri-
plasmatische Proteine ins umgebende Milieu freigesetzt (Nikaido & Vaara, 1985).
Durchführung:
Das Zellpellet wurde in 30 ml Schock-Puffer resuspendiert und mit 60 µl 0,5 M EDTA
versetzt. Die Zellen wurden 10 min gerührt (RT), abzentrifugiert (10 min, 11000x g, 4 °C), in
30 ml kalter MgSO4-Lösung (5 mM) resuspendiert und für weitere 10 min auf Eis gerührt.
Anschließend wurden die Zellen erneut zentrifugiert (10 min, 4 °C, 11000x g). Der Überstand

II. Material & Methoden 68
mit periplasmatischen Proteinen wurde aufbewahrt und das Pellet zur Gewinnung der zyto-
plasmatischen Fraktion verwendet.
15.2.4 Isolierung von löslicher und unlöslicher zytoplasmatischer Fraktion
Lösliche Fraktion:
Das Pellet aus Abschnitt II, 15.2.3 wurde in 5 ml kaltem Resuspensionspuffer aufgenommen,
50 µl Lysozym (10 mg/ml) wurden zugegeben und der Ansatz 15 min bei 30 °C inkubiert. Die
Zellen wurden mittels Ultraschallgerät (Branson Sonifier Cell Disruptor, Branson Ultrasonics
Corporation, Danbury/CT, USA) unter Kühlung aufgeschlossen (10 min). 1,5 ml des Zell-
lysats wurde in einer Tischzentrifuge pelletiert (10 min, 13600x g), der Überstand abgenom-
men und aufbewahrt (lösliche Fraktion). 30 µl wurden für Proteinbestimmung verwendet. Der
Rest wurde mit 2x RBS-Puffer versetzt und 3 min bei 75 °C erhitzt.
Unlösliche Fraktion:
Das Pellet (unlösliche Fraktion) enthält nach dem Zentrifugationsschritt nur noch unlösliche
Proteine (sog. inclusion bodies) und Trümmer von Membran und Zellwand. Es wurde 2x mit
750 µl Resuspensionspuffer gewaschen (Zentrifugation je bei 9700x g, 5 min), in 1,5 ml 1 %-
ig SDS-Lösung aufgenommen und durch Erhitzen und Schütteln (Thermomix, 65 °C) resus-
pendiert. Die Suspension wurde mit 2x RSB-Puffer versetzt und 3 min auf 75 °C erhitzt.
15.3 Proteinbestimmung Die Proteinbestimmung der Proben erfolgte mit dem BCA-Protein-Assay-Kit der Firma Pierce
(Rockford, II, USA) nach der Methode von Smith et al. (1985) in Mikrotiterplatten. Die
Methode beruht auf der Reduktion von proteingebundenem Cu2+ zu Cu1+ im alkalischen
Milieu. Cu1+ bildet spezifisch mit BCA (4,4’-Bichinolin-2,2’-Dicarbonsäure) einen violett ge-
färbten Komplex. Dieser Komplex weist ein Absorptionsmaximum bei 562 nm auf, das für
eine Quantifizierung herangezogen werden kann, da die Farbbildung proportional zur
Proteinkonzentration ist. Für die Reduktion von Cu2+ sind hauptsächlich die Peptidbindungen
verantwortlich (Wiechelman et al., 1988). Die Seitenketten von Cystein, Tryptophan und
Tyrosin spielen bei der Farbreaktion eine untergeordnete Rolle. Dadurch ist dieser Protein-
test relativ unabhängig von der Aminosäurezusammensetzung der zu bestimmenden
Proteine und kann gut auf Proteingemische angewendet werden.
Durchführung:
• Das Arbeitsreagenz wurde kurz vor Verwendung nach Herstellerangaben angesetzt.
• Die Proteinproben wurden entsprechend der Vorversuche mit H2Odemin.verdünnt.
• Die Proteineichreihe (0-500 µg BSA/ml H2Odemin.) wurde vorbereitet.

II. Material & Methoden 69
• Je 25 µl des Proteinstandards bzw. der Probe wurden in der Mikrotiterplatte mit 200 µl
Arbeitsreagenz gemischt und 30 min bei 37 °C inkubiert.
• Die Absorption wurde bei 550 nm in einem vollautomatischen Photometer bestimmt
(ATTC, SLT-Labinstruments, Salzburg).
15.4 Diskontinuierliche Polyacrylamid-Gelelektrophorese unter denaturie-
renden Bedingungen (SDS-PAGE) Mittels Polyacrylamid-Gelelektrophorese (PAGE) werden Proteine entsprechend ihres Mole-
kulargewichts im elektrischen Feld aufgetrennt. Die Proteine wandern durch die Poren des
Gels, deren Größe durch den Vernetzungsgrad vorgegeben ist. Abhängig von ihrer Eigen-
ladung und ihrer Tertiärstruktur, wandern die Proteine unterschiedlich schnell im Gel. Um
diese Einflüsse zu minimieren, kann die PAGE unter denaturierenden (Zusatz von SDS) und
bei Bedarf unter reduzierenden Bedingungen (Zusatz von β-Mercaptoethanol) durchgeführt
werden. (1970) wurde dieses Verfahren von Laemmli entwickelt und trägt den Namen SDS-
PAGE.
Das anionische Detergenz SDS maskiert die Eigenladung der Proteine, da gleiche Mengen
von Aminosäuren in etwa gleiche Mengen SDS an sich binden. Die Laufgeschwindigkeit
während der Elektrophorese ist damit von der Masse des aufzutrennenden Proteins abhän-
gig. Da die Höhe der eingeführten Ladung proportional zur Molekülmasse (1 SDS/2 Amino-
säuren) ist, lässt sich ein Protein anhand seiner Laufstrecke im Gel einem Molekulargewicht
zuordnen. Dabei handelt es sich um ein apparentes Molekulargewicht, da sich manche
Proteine nach diesem Protokoll nicht ihrem Molekulargewicht gemäß auftrennen lassen.
Inter- und intramolekulare Disulfidbrücken stabilisieren die Tertiärstruktur von Proteinen und
verknüpfen Polypeptide, was zu einem Laufverhalten der Proteine führt, das nicht dem Mole-
kulargewicht entspricht. Durch Einsatz von Reduktionsmitteln, wie z.B. β-Mercaptoethanol,
werden Disulfidbrücken reduktiv gespalten. Die unter diesen Bedingungen (denaturierend
und reduzierend) durchgeführte PAGE wird als SDS-PAGE bezeichnet. Die diskontinuier-
liche SDS-PAGE, als Verfeinerung der SDS-PAGE, wird verwendet, um einzelne Protein-
banden sehr scharf fokussieren zu können. Hierbei werden die Proteine zuerst in einem
Sammelgel (pH 6,8) bei hoher Feldstärke konzentriert und anschließend in einem höher
konzentrierten Trenngel (pH 8,8) gemäß ihres Molekulargewichts aufgetrennt.
Durchführung:
Das in den Gelen enthaltene Acrylamid reagiert bei Kontakt mit dem Radikalbildner
Ammoniumpersulfat (APS) in einer radikalischen Polymerisation zu langen Ketten, die durch
N,N’-Methylen-Bisacrylamid über Methylgruppen quervernetzt werden. TEMED dient bei
dieser Reaktion als Katalysator. Sowohl die Gesamtkonzentration des Acrylamids und des
Bisacrylamids (% T), als auch der Anteil des Quervernetzers an der Gesamtkonzentration
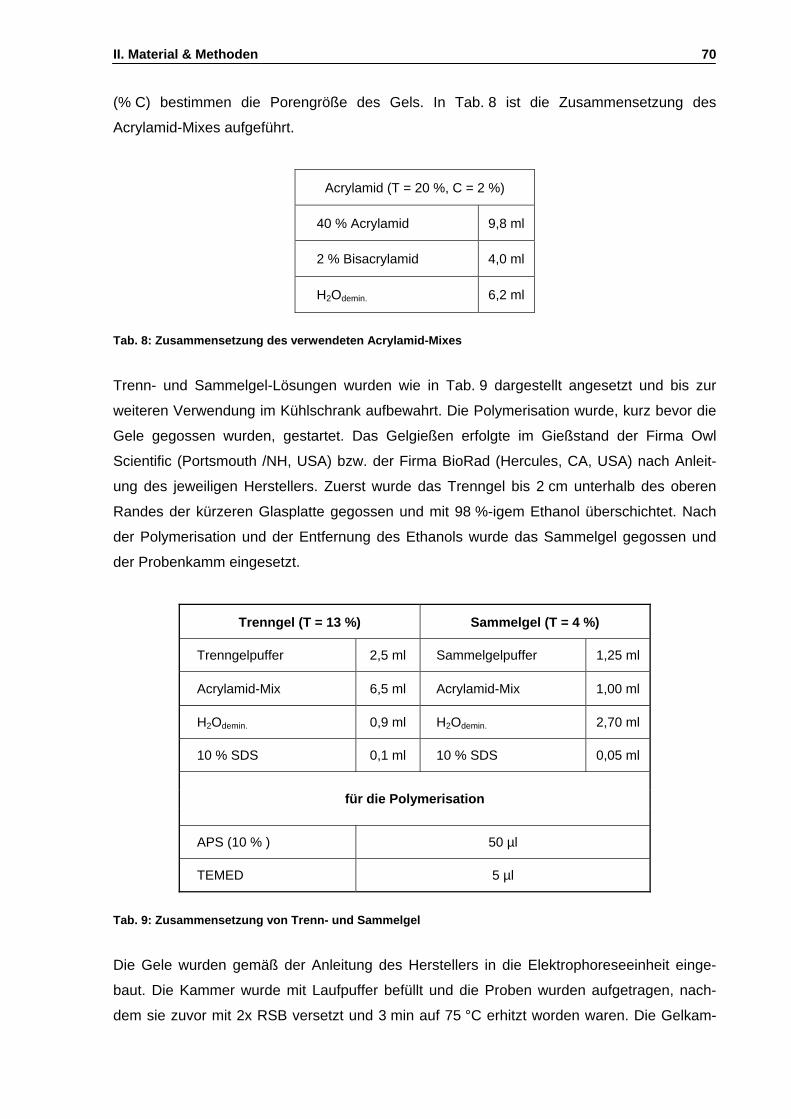
II. Material & Methoden 70
(% C) bestimmen die Porengröße des Gels. In Tab. 8 ist die Zusammensetzung des
Acrylamid-Mixes aufgeführt.
Acrylamid (T = 20 %, C = 2 %)
40 % Acrylamid 9,8 ml
2 % Bisacrylamid 4,0 ml
H2Odemin. 6,2 ml
Tab. 8: Zusammensetzung des verwendeten Acrylamid-Mixes
Trenn- und Sammelgel-Lösungen wurden wie in Tab. 9 dargestellt angesetzt und bis zur
weiteren Verwendung im Kühlschrank aufbewahrt. Die Polymerisation wurde, kurz bevor die
Gele gegossen wurden, gestartet. Das Gelgießen erfolgte im Gießstand der Firma Owl
Scientific (Portsmouth /NH, USA) bzw. der Firma BioRad (Hercules, CA, USA) nach Anleit-
ung des jeweiligen Herstellers. Zuerst wurde das Trenngel bis 2 cm unterhalb des oberen
Randes der kürzeren Glasplatte gegossen und mit 98 %-igem Ethanol überschichtet. Nach
der Polymerisation und der Entfernung des Ethanols wurde das Sammelgel gegossen und
der Probenkamm eingesetzt.
Trenngel (T = 13 %) Sammelgel (T = 4 %)
Trenngelpuffer 2,5 ml Sammelgelpuffer 1,25 ml
Acrylamid-Mix 6,5 ml Acrylamid-Mix 1,00 ml
H2Odemin. 0,9 ml H2Odemin. 2,70 ml
10 % SDS 0,1 ml 10 % SDS 0,05 ml
für die Polymerisation
APS (10 % ) 50 µl
TEMED 5 µl
Tab. 9: Zusammensetzung von Trenn- und Sammelgel
Die Gele wurden gemäß der Anleitung des Herstellers in die Elektrophoreseeinheit einge-
baut. Die Kammer wurde mit Laufpuffer befüllt und die Proben wurden aufgetragen, nach-
dem sie zuvor mit 2x RSB versetzt und 3 min auf 75 °C erhitzt worden waren. Die Gelkam-

II. Material & Methoden 71
mer wurde an eine Spannungsquelle (Power Supply EPS 500/400, Pharmacia,
Uppsala/Schweden) angeschlossen. Der Einlauf der Proben in das Sammelgel erfolgte bei
60 V, beim Übergang ins Trenngel wurde die Spannung auf 120 V erhöht.
Um die aufgetrennten Proteine im Gel sichtbar zu machen, wurden die Gele 30 min in
Coomassie-Färbelösung gefärbt. Coomassie-brilliant-blue R250 bindet im sauren Milieu irre-
versibel an Proteine und macht diese als blaue Banden sichtbar. Mit einer Nachweisgrenze
von 1,5 µg Protein pro Bande ist diese Färbung nicht sehr sensitiv, aber ausreichend. Die
Proteinbanden werden erst nach Entfärbung des Gels (Entfärberlösung, Schüttler) über
Nacht sichtbar. Zur Konservierung und Dokumentation der Gele wurden diese in einem
Vakuumtrockner (Aldo Xer Geltrockner, Fa. Schütt, Göttingen) ca. 60 min bei 50 °C
getrocknet. Hinterher wurden sie in Folien eingeschweißt und eingescannt.
15.5 Western-Hybridisierung
Analog zur Southern- und Northern-Hybridisierung, in denen ausgewählte Nukleinsäure-
sequenzen nachgewiesen werden, erfolgt bei der Western-Hybridisierung der Nachweis
einzelner Proteine eines Gemischs. Die Methode umfasst eine elektrophoretische Auftren-
nung der Proteine, einen Transfer der Proteine auf eine Nitrozellulosemembran, eine
Immunohybridisierung und einen kolorimetrischen Nachweis. Der Transfer der Proteine nach
der PAGE erfolgt per Elektroblot. Anschließend bindet ein spezifischer Antikörper an das
nachzuweisende Protein und wird selbst von einem sekundären Antikörper, der mit einer
alkalischen Phosphatase gekoppelt ist, gebunden. Mit geeigneten Nachweisreagenzien kann
die Lokalisation des Antikörper-AP-Konjugats sichtbar gemacht werden. In dieser Arbeit
wurde als primärer Antikörper ein gegen den „(His)6-tag“ gerichteter monoklonaler Maus Anti-
körper verwendet.
Western-Transfer (Towbin et al., 1979):
Für den Transfer wurde ein Elektroblotter der Fa. Owl Scientific (Portsmouth, USA) verwen-
det. Zwei Blotting-Papiere (Schleicher & Schuell, Dassel) und ein Stück Nitrozellulosemem-
bran wurden auf Gelgröße zurechtgeschnitten und 1 h in Towbin-Puffer inkubiert. Das Gel
wurde nach der Elektrophorese ebenfalls 15 min in Towbin-Puffer äquilibiert. Anschließend
wurde das Gel seitenverkehrt auf einem Stück Blotting-Papier auf die Kathode gelegt. Es
folgte die Nitrozellulosemembran und eine weitere Lage Blotting-Papier, die einen Kontakt
zur Anode herstellt. Der Aufbau sollte blasenfrei erfolgen. Nach Verschließen des Blotters
wurde für 45 min eine Spannung von 15 V angelegt. Die Proteine wandern aufgrund ihrer
negativen Ladung zur Anode und haften auf der Nitrozellulosemembran. Nach dem Transfer

II. Material & Methoden 72
wurde das Gel in Coomassie gefärbt und getrocknet. Die Membran wurde in die Immuno-
hybridisierung eingesetzt.
Immunohybridisierung:
Die Membran wurde über Nacht auf einem Schüttler in 80 ml Blockierungslösung inkubiert
und anschließend 2x 10 min mit 50 ml TBSTT und 1x 10 min mit TBS gewaschen. 3 µg
monoklonaler Maus-Anti-His6-tag Antikörper (Novagen) wurden in 25 ml Blockingreagenz
gelöst und für 1-2 h auf die Membran gegeben. Der Antikörper wurde entfernt und die
Membran erneut 2x 10 min mit TBSTT und 1x 10 min mit TBS gewaschen. Der sekundäre
Antikörper (polyklonaler Ziegen-Anti-Maus Antikörper, Novagen) wurde 1:5000 in 25 ml TBS
(1 % Casein, 10 % Magermilchpulver) verdünnt und die Membran darin 60 min inkubiert.
Anschließend wurde die Membran 5x 10 min mit TBSST gewaschen.
Kolorimetrischer Nachweis:
Als Substrat der alkalischen Phosphatase (an sek. Antikörper gekoppelt) dient BCIP,
welches nach seiner Dephosphorylierung oxidativ in einen blauen, wasserunlöslichen
Farbstoff überführt wird. Zur Intensivierung der Farbe wurde als Oxidationsmittel NBT
eingesetzt. 20 ml AP-Puffer wurden mit 66 µl NBT-Stammlösung (0,33 mg/ml) und 68 µl
BCIP-Stammlösung (0,17 mg/ml) versehen und die Membran darin unter leichtem
Schwenken 2-5 min inkubiert (bis gefärbte Proteinbande erkennbar). Anschließend wurde
die Membran mit H2Odemin. gewaschen, noch feucht in Folie eingeschweißten und zur
Dokumentation eingescannt.
16. Sequenzierung von DNA Die Sequenzierungen von Plasmidkonstrukten und PCR-Produkten wurden von der Firma
SequiServe (Vatterstetten) nach der Didesoxymethode (Sanger et al., 1977) durchgeführt.
17. Sequenzanalysen • Agilent: Biochemical-Analysis Software, Agilent-Technologies
• Amplify 1.2: (1999) William Engels, Genetics Department, Universtiy of Wisconsin, 445 Henry Mall, Madison, WI 53706
• Blast: (Basic Local Alignment Serach Tool) http://www.ncbi.nlm.nih.gov/BLAST/ (Altschul et al., 1990)
• CD-Search: (Conserved domain Search): http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml (Marchler-Bauer et al., 2003)
• Clone-Manager 5.0: Scientific & Educational Software, Durham,USA
• ClustalW: http://www.ebi.ac.uk/clustalw/; WWW Service at the European Bioinformatics Institute, Rodrigo Lopez, Service Programme (Thompson et al., 1994)

II. Material & Methoden 73
• COG: (Cluster of orthologous genes) NCBI-Homepage; http://www.ncbi.nlm.nih.gov/COG/ (Tatusov et al., 1997; Tatusov et al., 2001)
• DAS: (Dense Alignment Surface method) http://www.sbc.su.se/~miklos/DAS/ (Cserzo et al., 1997)
• DNA-Strider 1.2: Christian Marck, Service de Biochemie et de Genetique Moleculaire, Bat. 143 Centre d’Etudes de Saclay, 91191 GIF-Sur-Yuette Cedex, France
• GCUA: http://www.gcua.de/index.html; (Fuhrmann et al., 2004)
• HMMTOP: http://www.enzim.hu/hmmtop/ (Tusnady & Simon, 1998, 2001)
• KEGG : http://www.genome.jp/kegg/; (Kanehisa & Goto, 2000; Kanehisa et al., 2006) • KYTE DOOLITTLE HYDROPATHY PLOT: http://gcat.davidson.edu/rakarnik/kyte-
doolittle.htm, (Kyte & Doolittle, 1982)
• Mfold : http://www.bioinfo.rpi.edu/applications/mfold/; (Zucker, 2003)
• Neural Network Promoter Prediction: (NNPP) Berkely Drosophila Genome Project; http://www.fruitfly.org/seq_tools/promoter.htm; (Reese, 2001)
• ORF-Finder: http//www.ncbi.nlm.nih.gov/gorf/gorf.html; National Center for Biotechnology Information, Webseite
• Plasmid Map Enhancer, Version 3.1: Scientific & Educational Software (Durham, NC, USA)
• Primer Designer, Version 3.0: Scientific & Educational Software (Durham, NC, USA)
• PROSITE: us.expasy.org/tools/scanprosite/ (Bairoch et al., 1997; Falquet et al., 2002)
• ProtParam tool: http ://us.expasy.org/cgi-bin/protparam ; ExPASy Molecular Biology Servers
• PSORT-B: Version 1.1.2; http://www.psort.org/psortb/index.html; (Gardy et al., 2003)
• SignalP : http//www.cbs.dtu.dk/services/SignalP; (Bendtsen et al., 2004)
• SOSUI-Signal : http://www.proteome.bio.tuat.ac.jp/sosuisignal/sosuisignal_submit.html; (Gomi et al., 2000)
• Webcutter 2.0©: (1997) Max Heiman ; http://rna.lundberg.gu.se/cgi-bin/cutter2/cutter
18. Verwendete Chemikalien Nährböden und Medienzusätze: Mannosylglycerat, Mannosylglyceramid Bitop (Witten) Bacto Agar, Hefeextrakt, Trypton Difco (Detroid, USA) Sarcosin EGA-Chemie (Steinheim/Albuch) IPTG, X-Gal Fermentas (Vilinus, Litauen) Fructose, Mannose, Sorbitol, Nitrilotriessigsäure Fluka (Buchs, Schweiz) Betain, N,N-Dimethylglycin, Trehalose Fluka Fleischextrakt, Trypton, Glycerol Merck (Darmstadt) Glucose-Monohydrat, Saccharose Merck Galactose, Maltose, Merck Antibiotic broth medium No.3 Oxoid (Wesel) Arabitol, Innositol, Erythritol,Choline, DMSO Sigma (Deisenhofen) Antibiotika Merck (Darmstadt), Sigma (Deisenhofen) Vitamine und Aminosäuren Fluka (Buchs, Schweiz), Merck
(Darmstadt), Sigma (Deisen- hofen)

II. Material & Methoden 74
Mineralsalze MgSO4 x H2O und H3BO3 Fluka (Buchs, Schweiz) (NH4)2SO4, NaCl, KCl, CaCl2, Merck (Darmstadt) NiCl2, NaMoO4, CoCl2, KOH, NaOH, Merck KH2PO4 und K2HPO4 Merck FeSO4 x 7 H2O, CuCl2 Merck MnCl2, FeCl2, FeCitrat Sigma (Deisenhofen) Chemikalien für Chromatographische Verfahren Methanol, Chloroform, Schwefelsäure Merck (Darmstadt) Ammoniak (25 %), n-Propanol, Acetonitril Merck α-Naphtol Sigma (Deisenhofen) Chemikalien für molekularbiologischen Methoden Agarose Appligene β-Mercaptoethanol BioRad (Hercules, CA, USA) 100bp- und 1 kb- DNA -Leiter Fermentas (Vilnius, Litauen) dNTP-Mix, λ-DNA, RNA-Leiter Fermentas Ethidiumbromid, Glyoxal, Fluka (Buchs, Schweiz) Isoamylalkohol, Ethanol, Isopropanol Merck (Darmstadt) Borsäure, Bromphenolblau, Maleinsäure Merck Na-Acetat, Na3-Citrat x 2H2O Merck dATP, dCTP, dGTP, dTTP Roche (Mannheim) DIG-EASY-Hyb, DIG-RNA-labeling-Mix, Roche Anti Digoxigenin AP, Blockierungsreagenz, Roche CDP*, Dig-11-dUTP, NBT/ BCIP Stammlösung Roche RNAse Inhibitor Roche Aqua-Roti-Phenol, RNAse away, DEPC Roth (Karlsruhe) EDTA, SDS Serva (Heidelberg) Fixierer & Entwickler, Ficoll, Sigma (Deisenhofen) N-Lauroylsacrosin, PIPES Sigma Proteinchemie Amoniumpersulfat, APS, TEMED BioRad (Hercules, CA, USA) Proteinmarker Fermentas (Vilnius, Litauen) Triton X-100 Fluka (Buchs, Schweiz) Acrylamid, Bisacrylamid, Merck (Darmstadt) Casein, Glycin, Tween20 Merck Coomassie-brilliant-blue Merck Monoklonaler Maus anti-His-tag-Antikörper Novagen (Madison, WI, USA) Ziege-anti-Maus-IgG-AP-Konjugat Novagen Eisessig Roth (Karlsruhe) BSA, Glycerol Serva (Heidelberg) Magermilchpulver Uelzena (Uelzen)

II. Material & Methoden 75
Enzyme Restriktionsendonukleasen Promega (Madison, WI, USA), Roche, Fermentas Alkalische Phosphatase (CIAP), Fermentas (Vilnius, Litauen) T4-DNA-Polymerase, T4-DNA-Ligase und Fermentas Taq-Polymerase Fermentas Terminale Transferase Invitrogen (Calsbad, CA, USA) Lysozym Merck (Darmstadt) RNaseA Qiagen (Hilden) Pwo-Polymerase, DNAse (RNase-frei) Roche (Mannheim) T7-RNA-Polymerase Roche Kits Microcon Amicon (Beverly, USA) Superskript III-first-strand cDNA Synthese Kit Invitrogen (Calsbad, CA, USA) BCA-Proteintest Pierce (BA Oud, Beijerland, Niederlande) DNA-Isolierung, Plasmidisolierung Qiagen (Hilden) Midi- und Miniprep Kit, RNA-Isolierung Qiagen GC-Rich-PCR-System Roche (Mannheim)

III. Ergebnisse
Ziel der vorliegenden Arbeit ist die Etablierung eines bakteriellen Produktionsstamms für das
kompatible Solut Mannosylglycerat. Die Arbeit untergliedert sich in zwei Hauptteile. Der erste
Teil beschäftigt sich mit den Arbeiten an Rhodothermus marinus. Durch die Synthese von
Mannosylglycerat und Mannosylglyceramid ist dieser Stamm befähigt, Salzkonzentrationen
von über 6 % und Temperaturen bis 77 °C zu tolerieren. In Anlehnung an Arbeiten an
Halomonas elongata, bei denen durch Inaktivierung des Ectoin-Transportsystems ein indus-
trieller Produktionsstamm für die Synthese von Ectoin konstruiert wurde, sollte in R. marinus
ein Transporter für Mannosylglycerat identifiziert und inaktiviert werden.
Da die Suche nach einem Transportsystem in R. marinus trotz verschiedener angewendeter
Techniken nicht zum Ziel führte, wurde im zweiten Teil der Arbeit mit H. elongata gearbeitet.
Hier sollte durch Integration geeigneter Gene eine Mannosylglycerat-Synthese etabliert
werden.
Zu Beginn dieser Arbeit wurde untersucht, ob R. marinus über ein Transportsystem für das
kompatible Solut Mannosylglycerat verfügt. Nach den Untersuchungen verschiedener Ar-
beitsgruppen darf vermutet werden, dass ein thermophiles, halotolerantes Bakterium wie
R. marinus, das zum Schutz vor erhöhter Salinität, Osmolarität und Hitze die kompatiblen
Solute Mannosylglycerat und Mannosylglyceramid synthetisiert und im Zytoplasma akkumu-
liert, ein spezifisches Transportsystem für diese kompatiblen Solute besitzt. Diese spezi-
fischen Transportsysteme dienen dazu, aus der Zelle verlorene, selbst-synthetisierte Solute
in die Zelle zurückzutransportieren. Grundlage dieser Vermutung ist die Beobachtung, dass
die Ausschaltung dieser spezifischer Transportsysteme zu einem stetigen Verlust kompa-
tibler Solute über die Zytoplasmamembran führt. Dies wurde anhand des halotoleranten
Cyanobakteriums Synechocystis bzw. anhand von Bacillus subtilis für die kompatiblen Solute
Glucosylglycerol bzw. Prolin beschrieben (Hagemann et al., 1997; Von Blohn et al., 1997).
Schon vorher zeigte Lamark et al. (1992) eine Zyklisierung des Betains über der Zytoplasma-
membran von E. coli. Neuere Untersuchungen an dem halophilen Bakterium Halomonas
elongata lassen sogar vermutet, dass die Synthese einiger kompatibler Solute durch ihre
Abgabe und Aufnahme über die zytoplasmatische Membran reguliert wird (Grammann et al.,
2002).
Sollte in R. marinus ein derartiger Transporter identifiziert werden können, war geplant
diesen zu inaktivieren. Nach den Untersuchungen der verschiedenen Arbeitsgruppen wäre
das Resultat ein bakterieller Stamm, welcher permanent Mannosylglycerat produziert und ins
Außenmedium ausscheidet, d.h. ein überproduzierender Stamm. Dieses Ziel setzt vielseitige
gentechnische und molekularbiologische Techniken voraus, die bisher für R. marinus noch
nicht etabliert sind.
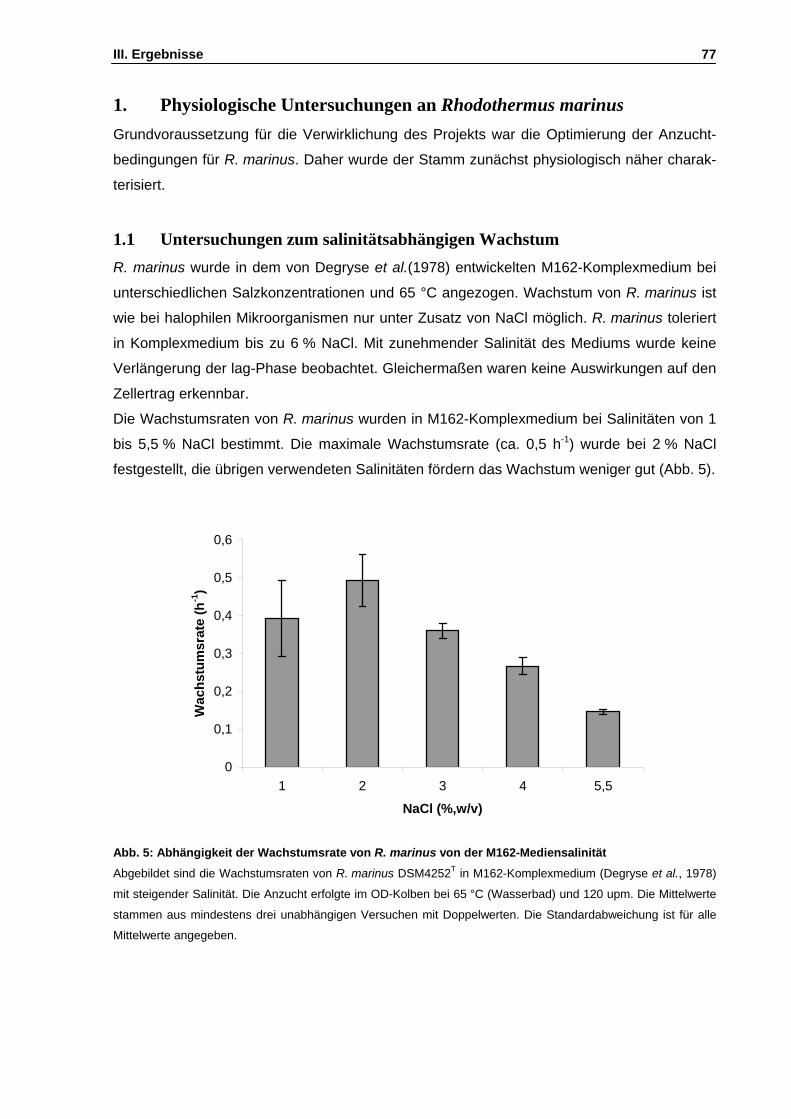
III. Ergebnisse 77
1. Physiologische Untersuchungen an Rhodothermus marinus Grundvoraussetzung für die Verwirklichung des Projekts war die Optimierung der Anzucht-
bedingungen für R. marinus. Daher wurde der Stamm zunächst physiologisch näher charak-
terisiert.
1.1 Untersuchungen zum salinitätsabhängigen Wachstum R. marinus wurde in dem von Degryse et al.(1978) entwickelten M162-Komplexmedium bei
unterschiedlichen Salzkonzentrationen und 65 °C angezogen. Wachstum von R. marinus ist
wie bei halophilen Mikroorganismen nur unter Zusatz von NaCl möglich. R. marinus toleriert
in Komplexmedium bis zu 6 % NaCl. Mit zunehmender Salinität des Mediums wurde keine
Verlängerung der lag-Phase beobachtet. Gleichermaßen waren keine Auswirkungen auf den
Zellertrag erkennbar.
Die Wachstumsraten von R. marinus wurden in M162-Komplexmedium bei Salinitäten von 1
bis 5,5 % NaCl bestimmt. Die maximale Wachstumsrate (ca. 0,5 h-1) wurde bei 2 % NaCl
festgestellt, die übrigen verwendeten Salinitäten fördern das Wachstum weniger gut (Abb. 5).
0
0,1
0,2
0,3
0,4
0,5
0,6
1 2 3 4 5,5
NaCl (%,w/v)
Wac
hstu
msr
ate
(h-1
)
Abb. 5: Abhängigkeit der Wachstumsrate von R. marinus von der M162-Mediensalinität
Abgebildet sind die Wachstumsraten von R. marinus DSM4252T in M162-Komplexmedium (Degryse et al., 1978)
mit steigender Salinität. Die Anzucht erfolgte im OD-Kolben bei 65 °C (Wasserbad) und 120 upm. Die Mittelwerte
stammen aus mindestens drei unabhängigen Versuchen mit Doppelwerten. Die Standardabweichung ist für alle
Mittelwerte angegeben.
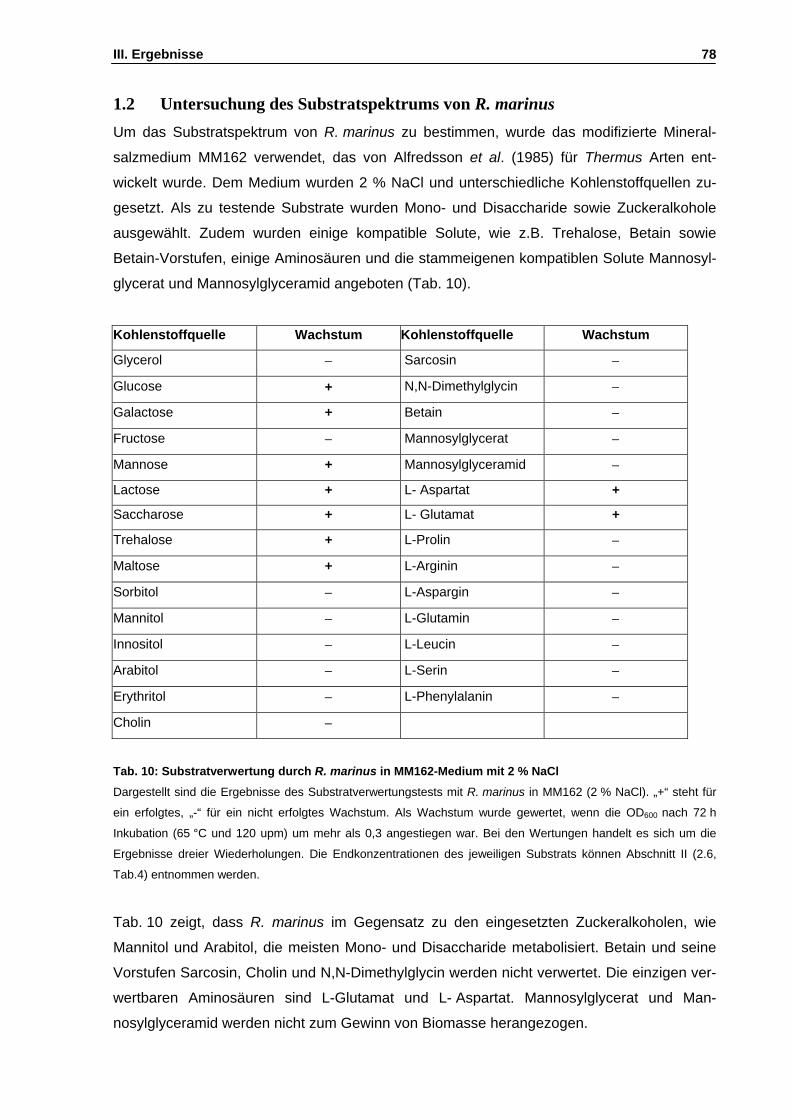
III. Ergebnisse 78
1.2 Untersuchung des Substratspektrums von R. marinus Um das Substratspektrum von R. marinus zu bestimmen, wurde das modifizierte Mineral-
salzmedium MM162 verwendet, das von Alfredsson et al. (1985) für Thermus Arten ent-
wickelt wurde. Dem Medium wurden 2 % NaCl und unterschiedliche Kohlenstoffquellen zu-
gesetzt. Als zu testende Substrate wurden Mono- und Disaccharide sowie Zuckeralkohole
ausgewählt. Zudem wurden einige kompatible Solute, wie z.B. Trehalose, Betain sowie
Betain-Vorstufen, einige Aminosäuren und die stammeigenen kompatiblen Solute Mannosyl-
glycerat und Mannosylglyceramid angeboten (Tab. 10).
Kohlenstoffquelle Wachstum Kohlenstoffquelle Wachstum
Glycerol − Sarcosin −
Glucose + N,N-Dimethylglycin −
Galactose + Betain −
Fructose − Mannosylglycerat −
Mannose + Mannosylglyceramid −
Lactose + L- Aspartat +
Saccharose + L- Glutamat +
Trehalose + L-Prolin −
Maltose + L-Arginin −
Sorbitol − L-Aspargin −
Mannitol − L-Glutamin −
Innositol − L-Leucin −
Arabitol − L-Serin −
Erythritol − L-Phenylalanin −
Cholin −
Tab. 10: Substratverwertung durch R. marinus in MM162-Medium mit 2 % NaCl
Dargestellt sind die Ergebnisse des Substratverwertungstests mit R. marinus in MM162 (2 % NaCl). „+“ steht für
ein erfolgtes, „-“ für ein nicht erfolgtes Wachstum. Als Wachstum wurde gewertet, wenn die OD600 nach 72 h
Inkubation (65 °C und 120 upm) um mehr als 0,3 angestiegen war. Bei den Wertungen handelt es sich um die
Ergebnisse dreier Wiederholungen. Die Endkonzentrationen des jeweiligen Substrats können Abschnitt II (2.6,
Tab.4) entnommen werden.
Tab. 10 zeigt, dass R. marinus im Gegensatz zu den eingesetzten Zuckeralkoholen, wie
Mannitol und Arabitol, die meisten Mono- und Disaccharide metabolisiert. Betain und seine
Vorstufen Sarcosin, Cholin und N,N-Dimethylglycin werden nicht verwertet. Die einzigen ver-
wertbaren Aminosäuren sind L-Glutamat und L- Aspartat. Mannosylglycerat und Man-
nosylglyceramid werden nicht zum Gewinn von Biomasse herangezogen.
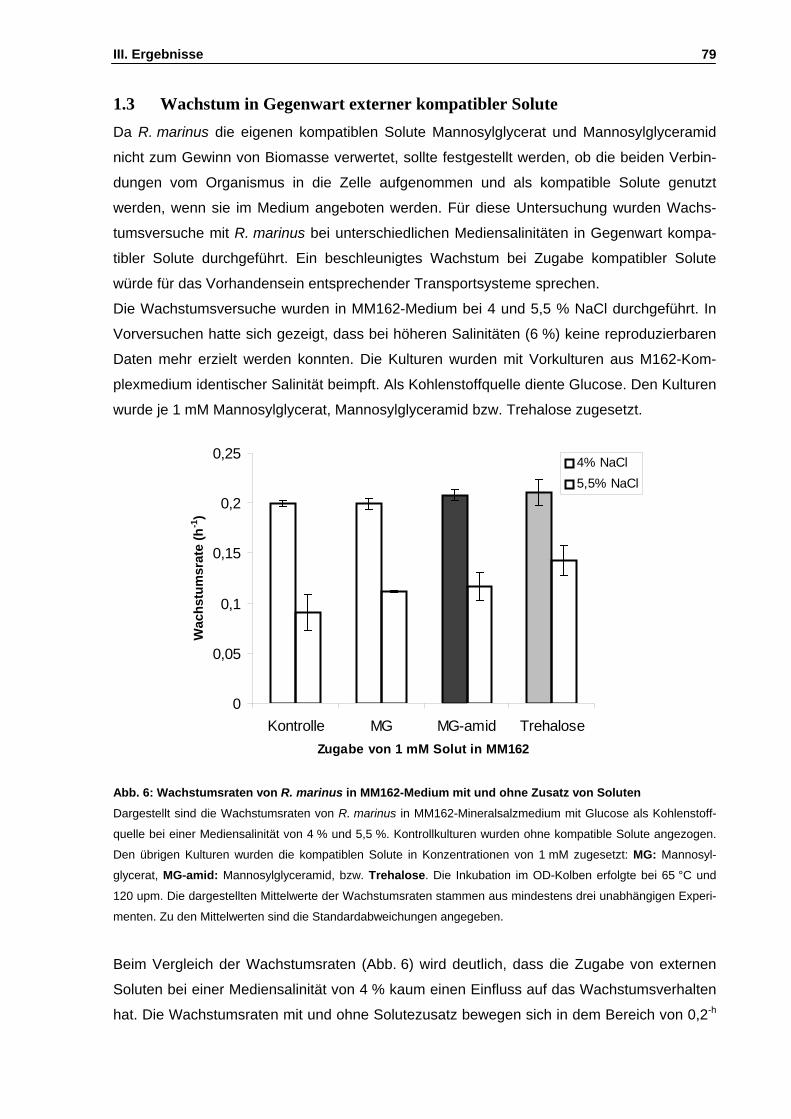
III. Ergebnisse 79
1.3 Wachstum in Gegenwart externer kompatibler Solute Da R. marinus die eigenen kompatiblen Solute Mannosylglycerat und Mannosylglyceramid
nicht zum Gewinn von Biomasse verwertet, sollte festgestellt werden, ob die beiden Verbin-
dungen vom Organismus in die Zelle aufgenommen und als kompatible Solute genutzt
werden, wenn sie im Medium angeboten werden. Für diese Untersuchung wurden Wachs-
tumsversuche mit R. marinus bei unterschiedlichen Mediensalinitäten in Gegenwart kompa-
tibler Solute durchgeführt. Ein beschleunigtes Wachstum bei Zugabe kompatibler Solute
würde für das Vorhandensein entsprechender Transportsysteme sprechen.
Die Wachstumsversuche wurden in MM162-Medium bei 4 und 5,5 % NaCl durchgeführt. In
Vorversuchen hatte sich gezeigt, dass bei höheren Salinitäten (6 %) keine reproduzierbaren
Daten mehr erzielt werden konnten. Die Kulturen wurden mit Vorkulturen aus M162-Kom-
plexmedium identischer Salinität beimpft. Als Kohlenstoffquelle diente Glucose. Den Kulturen
wurde je 1 mM Mannosylglycerat, Mannosylglyceramid bzw. Trehalose zugesetzt.
0
0,05
0,1
0,15
0,2
0,25
Kontrolle MG MG-amid TrehaloseZugabe von 1 mM Solut in MM162
Wac
hstu
msr
ate
(h-1
)
4% NaCl5,5% NaCl
Abb. 6: Wachstumsraten von R. marinus in MM162-Medium mit und ohne Zusatz von Soluten
Dargestellt sind die Wachstumsraten von R. marinus in MM162-Mineralsalzmedium mit Glucose als Kohlenstoff-
quelle bei einer Mediensalinität von 4 % und 5,5 %. Kontrollkulturen wurden ohne kompatible Solute angezogen.
Den übrigen Kulturen wurden die kompatiblen Solute in Konzentrationen von 1 mM zugesetzt: MG: Mannosyl-
glycerat, MG-amid: Mannosylglyceramid, bzw. Trehalose. Die Inkubation im OD-Kolben erfolgte bei 65 °C und
120 upm. Die dargestellten Mittelwerte der Wachstumsraten stammen aus mindestens drei unabhängigen Experi-
menten. Zu den Mittelwerten sind die Standardabweichungen angegeben.
Beim Vergleich der Wachstumsraten (Abb. 6) wird deutlich, dass die Zugabe von externen
Soluten bei einer Mediensalinität von 4 % kaum einen Einfluss auf das Wachstumsverhalten
hat. Die Wachstumsraten mit und ohne Solutezusatz bewegen sich in dem Bereich von 0,2-h
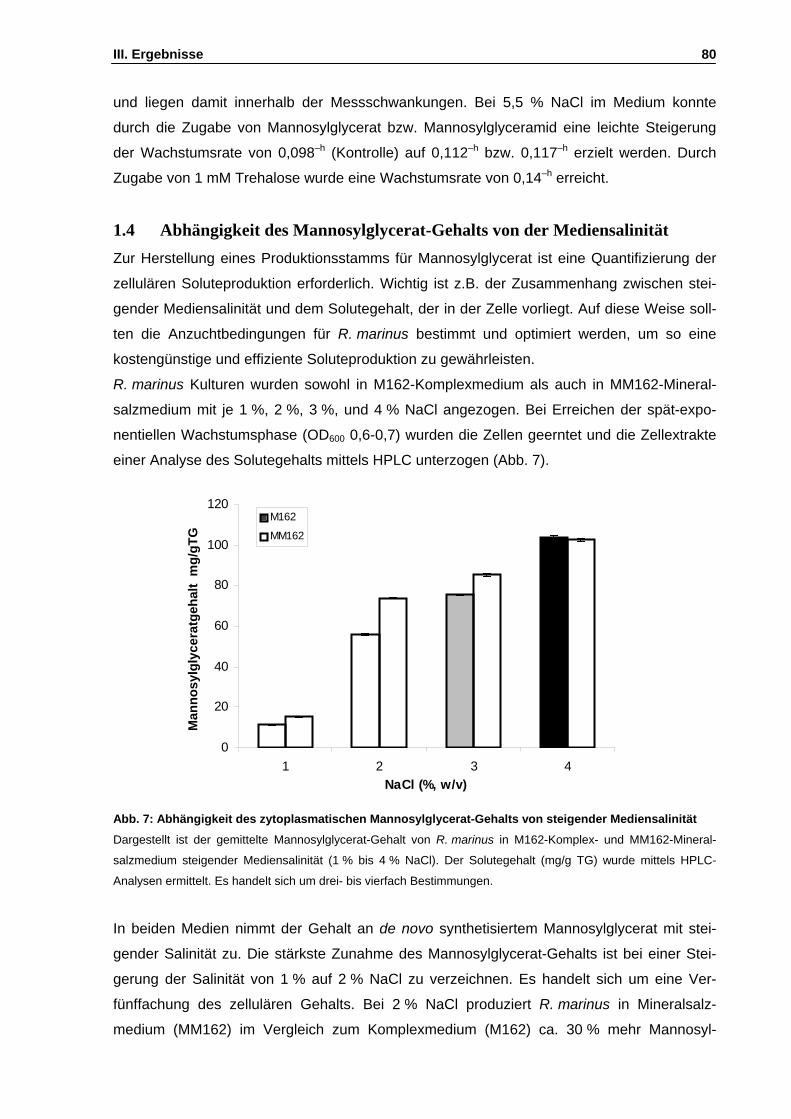
III. Ergebnisse 80
und liegen damit innerhalb der Messschwankungen. Bei 5,5 % NaCl im Medium konnte
durch die Zugabe von Mannosylglycerat bzw. Mannosylglyceramid eine leichte Steigerung
der Wachstumsrate von 0,098–h (Kontrolle) auf 0,112–h bzw. 0,117–h erzielt werden. Durch
Zugabe von 1 mM Trehalose wurde eine Wachstumsrate von 0,14–h erreicht.
1.4 Abhängigkeit des Mannosylglycerat-Gehalts von der Mediensalinität Zur Herstellung eines Produktionsstamms für Mannosylglycerat ist eine Quantifizierung der
zellulären Soluteproduktion erforderlich. Wichtig ist z.B. der Zusammenhang zwischen stei-
gender Mediensalinität und dem Solutegehalt, der in der Zelle vorliegt. Auf diese Weise soll-
ten die Anzuchtbedingungen für R. marinus bestimmt und optimiert werden, um so eine
kostengünstige und effiziente Soluteproduktion zu gewährleisten.
R. marinus Kulturen wurden sowohl in M162-Komplexmedium als auch in MM162-Mineral-
salzmedium mit je 1 %, 2 %, 3 %, und 4 % NaCl angezogen. Bei Erreichen der spät-expo-
nentiellen Wachstumsphase (OD600 0,6-0,7) wurden die Zellen geerntet und die Zellextrakte
einer Analyse des Solutegehalts mittels HPLC unterzogen (Abb. 7).
0
20
40
60
80
100
120
1 2 3 4NaCl (%, w/v)
Man
nosy
lgly
cera
tgeh
alt
mg/
gTG
M162
MM162
Abb. 7: Abhängigkeit des zytoplasmatischen Mannosylglycerat-Gehalts von steigender Mediensalinität
Dargestellt ist der gemittelte Mannosylglycerat-Gehalt von R. marinus in M162-Komplex- und MM162-Mineral-
salzmedium steigender Mediensalinität (1 % bis 4 % NaCl). Der Solutegehalt (mg/g TG) wurde mittels HPLC-
Analysen ermittelt. Es handelt sich um drei- bis vierfach Bestimmungen.
In beiden Medien nimmt der Gehalt an de novo synthetisiertem Mannosylglycerat mit stei-
gender Salinität zu. Die stärkste Zunahme des Mannosylglycerat-Gehalts ist bei einer Stei-
gerung der Salinität von 1 % auf 2 % NaCl zu verzeichnen. Es handelt sich um eine Ver-
fünffachung des zellulären Gehalts. Bei 2 % NaCl produziert R. marinus in Mineralsalz-
medium (MM162) im Vergleich zum Komplexmedium (M162) ca. 30 % mehr Mannosyl-

III. Ergebnisse 81
glycerat. Bei 3 % NaCl beträgt der Unterschied nur noch ca. 13 %. Bei Wachstum mit 4 %
NaCl ist kein Unterschied im Mannosylglycerat-Gehalt mehr zu beobachten.
2. Molekularbiologische Untersuchungen an Rhodothermus marinus
2.1 Erstellung einer Genbank von R. marinus Die vollständige Genbank eines Organismus enthält die Gesamtheit seiner genetischen
Information in Form klonierter DNA-Fragmente. Eine Genbank erlaubt so die Identifizierung
von Zielgenen mittels Komplementierung geeigneter Defektmutanten. In der vorliegenden
Arbeit sollte eine Genbank von R. marinus genutzt werden, um Transportsysteme für das
kompatible Solut Mannosylglycerat im Genom von R. marinus zu identifizieren. Als Träger
der Genbank wurde zunächst der low-copy-Vektor pHSG575 (Takeshita et al., 1987), später
der Expressionsvektor pXMJ19 (Jakoby et al., 1999) verwendet. Die durchschnittliche Größe
der klonierten DNA-Fragmente wurde mittels partieller Hydrolyse (Sau 3A) auf 5-10 kb
eingestellt. Die pHSG575 basierte Genbank umfasst 17113 Klone in 18 Chargen (je ca. 750-
1000 Klone). Die R. marinus Genbank in pXMJ19 umfasst 9705 Klone in 28 Chargen (je ca.
300-400 Klone).
2.2 Identifizierung der Thiamin-Gene aus R. marinus durch funktionelle
Komplementierung von E. coli DH5α Vor der vorliegenden Arbeit waren keinerlei Erfahrungen im Umgang mit Genbänken aus
R. marinus DNA vorhanden. So gab es keine Untersuchungen zur heterologen Expression
von R. marinus Genen unter Kontrolle der eigenen Promotoren in E. coli Stämmen. Genauso
war die Funktionalität thermophiler Proteine in E. coli unklar. Beides ist jedoch zwingende
Voraussetzung für die erfolgreiche Verwendung einer Genbank. Um die funktionelle Expres-
sion von R. marinus Genen in E. coli zu überprüfen, wurde die Thiamin-Auxotrophie von
E. coli DH5α mittels der plasmidkodierten Genbank von R. marinus komplementiert. Diese
erfolgreiche Komplementierung deutet die Funktionalität der Promotoren von R. marinus in
E. coli an. Außerdem zeigt sich, dass R. marinus Gene auch in E. coli in einen enzymatisch
aktiven Zustand exprimiert werden können.
2.2.1 Komplementierung von E. coli DH5α
Je 500-1000 Klone von E. coli DH5α mit plasmidkodierter Genbank wurden in LB-0,5-Flüs-
sigmedium (Cm50) angezogen. 1 ml dieser Kulturen wurde dreimal mit 1 ml MM63-0,5-Medi-
um gewaschen und auf MM63-0,5-Agarplatten ohne Vitaminzusatz ausplattiert. E. coli DH5α
pHSG575 diente als Negativkontrolle. Nach zwei Tagen Inkubation waren zwei große
Kolonien sichtbar. Die Negativkontrolle E. coli DH5α pHSG575 zeigte keinerlei Wachstum. In

III. Ergebnisse 82
einer Restriktionsanalyse (Eco RI und Hin dIII) der isolierten Plasmide zeigten beide
Kolonien ein identisches Restriktionsmuster. Beide Plasmide wurden erneut in E. coli DH5α
transformiert. Die nachfolgende Selektion bestätigte, dass die Komplementierung von E. coli
DH5α auf eine Eigenschaft der in die Plasmide inserierten R. marinus DNA zurückzuführen
war. Einer der Klone, E. coli DH5α pThia1, wurde für die weitere Charakterisierung und Se-
quenzierung der R. marinus DNA ausgewählt.
2.2.2 Subklonierung von pThia1
Da die in pThia1 inserierte R. marinus DNA zunächst für eine vollständige Sequenzierung zu
groß erschien, wurde das Plasmid subkloniert. Zur Durchführung dieser Subklonierung
wurde pThia1 zuvor einer Restriktionsanalyse unterzogen:
Fragmente
Eco RI ca. 1.0 & 5.9 kb
Hin dIII ca. 6,9 kb
Eco RI & Hin dIII ca. 1.0, 2.3 & 3.6 kb
Sma I ca. 0.6 & 6.3 kb
Pst I ca. 0.34, 1.15 & 5.41 kb
Nach der Restriktionsanalyse ergaben sich zwei mögliche vorläufige Restriktionskarten
(Abb. 8), von denen sich in der späteren Sequenzierung Orientierung A als die richtige her-
ausstellte.
Für die Subklonierung wurde pThia1 mit den Enzymen Eco RI, Pst I bzw. Sma I hydrolysiert.
Die hydrolysierte DNA wurde gelelektrophoretisch aufgetrennt. Das 5.9 kb Eco RI, das
5.41 kb Pst I und das 6,3 kb Sma I-Fragment wurden aus dem Agarosegel aufgereinigt. Alle
drei gereinigten Fragmente bestanden aus pHSG575-DNA und R. marinus DNA. Durch die
Religation der gereinigten DNA-Fragmente entstanden die Subklone pThiaEco (religiertes
Eco RI-Fragment), pThiaPst (religiertes Pst I-Fragment) und pThiaSma (religiertes Sma I-
Fragment) (Abb. 8).
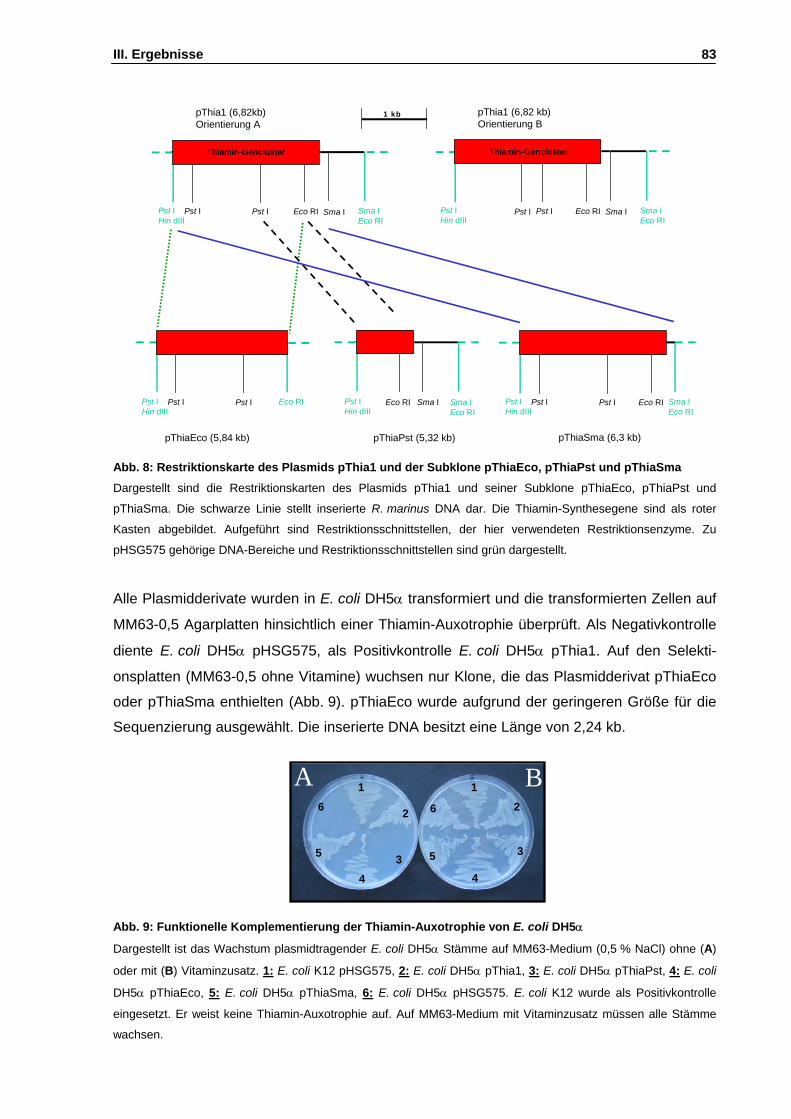
III. Ergebnisse 83
1 kb
Pst IHin dIII
Pst I Pst I Eco RI Sma I Sma IEco RI
pThia1 (6,82kb)Orientierung A
Pst IHin dIII
Pst I Pst I Eco RI
pThiaEco (5,84 kb)
Pst IHin dIII
Eco RI Sma I Sma IEco RI
pThiaPst (5,32 kb)
Pst IHin dIII
Pst I Pst I Eco RI Sma I Sma IEco RI
pThia1 (6,82 kb)Orientierung B
Pst IHin dIII
Pst I Pst I Eco RI Sma IEco RI
pThiaSma (6,3 kb)
Thiamin-Gencluster Thiamin-Gencluster
Abb. 8: Restriktionskarte des Plasmids pThia1 und der Subklone pThiaEco, pThiaPst und pThiaSma
Dargestellt sind die Restriktionskarten des Plasmids pThia1 und seiner Subklone pThiaEco, pThiaPst und
pThiaSma. Die schwarze Linie stellt inserierte R. marinus DNA dar. Die Thiamin-Synthesegene sind als roter
Kasten abgebildet. Aufgeführt sind Restriktionsschnittstellen, der hier verwendeten Restriktionsenzyme. Zu
pHSG575 gehörige DNA-Bereiche und Restriktionsschnittstellen sind grün dargestellt.
Alle Plasmidderivate wurden in E. coli DH5α transformiert und die transformierten Zellen auf
MM63-0,5 Agarplatten hinsichtlich einer Thiamin-Auxotrophie überprüft. Als Negativkontrolle
diente E. coli DH5α pHSG575, als Positivkontrolle E. coli DH5α pThia1. Auf den Selekti-
onsplatten (MM63-0,5 ohne Vitamine) wuchsen nur Klone, die das Plasmidderivat pThiaEco
oder pThiaSma enthielten (Abb. 9). pThiaEco wurde aufgrund der geringeren Größe für die
Sequenzierung ausgewählt. Die inserierte DNA besitzt eine Länge von 2,24 kb.
1
2
3
4
5
6
1
2
34
5
6
A B
Abb. 9: Funktionelle Komplementierung der Thiamin-Auxotrophie von E. coli DH5α
Dargestellt ist das Wachstum plasmidtragender E. coli DH5α Stämme auf MM63-Medium (0,5 % NaCl) ohne (A)
oder mit (B) Vitaminzusatz. 1: E. coli K12 pHSG575, 2: E. coli DH5α pThia1, 3: E. coli DH5α pThiaPst, 4: E. coli
DH5α pThiaEco, 5: E. coli DH5α pThiaSma, 6: E. coli DH5α pHSG575. E. coli K12 wurde als Positivkontrolle
eingesetzt. Er weist keine Thiamin-Auxotrophie auf. Auf MM63-Medium mit Vitaminzusatz müssen alle Stämme
wachsen.

III. Ergebnisse 84
2.2.3 Sequenzierung der Inserts von pThiaEco und pThia1
Die inserierte R. marinus DNA des Plasmids pThiaEco wurde ausgehend der Standard-
Primer M13-forward und M13-reverse nach der Kettenabbruch-Methode (Sanger et al.,
1977) ansequenziert. Von den erhaltenen DNA-Sequenzen wurden zwei weitere Primer
(Thirev1 und Thiforxrevcomp1) abgeleitet, mit denen nach zwei weiteren Sequenzierungs-
reaktionen eine Überlappung der sequenzierten DNA-Bereiche erzielt wurde (Abb. 10). Die
Analyse der erhaltenen DNA-Sequenz (Blast, NCBI-Homepage, Altschul et al., 1990) ergab
signifikante Ähnlichkeiten zu den Thiamin-Synthesegenen thiS, thiG, thiE und thiD verschie-
dener Organismen. Aufgrund dieser ersten Analysen wurde der R. marinus DNA-Bereich,
welcher zusätzlich in pThia1 enthalten ist, sequenziert (Abb. 10). Hierzu wurde zunächst
wiederum der Primer M13-reverse verwendet. Ein weiterer Primer (Thiodown1) wurde aus
der erhaltenen Sequenz abgeleitet. Insgesamt wurde ein zusammenhängender DNA-Bereich
von 3240 bp sequenziert (siehe Anhang).
1 kb
pThiaEco
Pst IHin dIII
Eco RI
M13rev (765 bp)
Thirev1 (765 bp)
M13for (758 bp)
Thiforxrevcomp1 (760 bp)
M13rev (950 bp)
Pst IHin dIII
Eco RI Sma I Sma IEco RI
pThia1
Thiodown 1 (450 bp)
Abb. 10: Strategie zur Sequenzierung der Plasmide pThiaEco und pThia1
Abgebildet sind die Sequenzierungsstrategien für die Plasmide pThiaEco und pThia1. Die zum Vektor pHSG575
gehörigen DNA-Bereiche und Restriktionsschnittstellen sind grün markiert, die inserierte DNA ist als schwarze
Linie und roter Pfeile (Thiamin-Gencluster) dargestellt. Die schwarzen Pfeile stellen die mit dem jeweiligen Primer
sequenzierten DNA-Bereiche (Länge (bp) in Klammern) dar.
2.2.4 Analyse der DNA-Sequenz
Mit Hilfe des Programms ORF-Finder (NCBI-Homepage) konnten vier offene Leserahmen
(ORF) identifiziert werden, deren abgeleitete Aminosäuresequenzen signifikante Ähnlich-

III. Ergebnisse 85
keiten zu den Thiamin-Syntheseenzymen ThiS, ThiG, ThiE und ThiD aufweisen (Abb. 11).
Ein fünfter ORF weist Ähnlichkeiten zu einer Helikase auf.
321823
153
172
996
1000
1626
1633 2490 2795
thiG thiE thiD
130 bp 824 bp 626 bp 858 bp
orf5thiS
Abb. 11: Genetische Organisation der Thiamin-Gene aus R. marinus
Dargestellt ist die Anordnung der hypothetischen Thiamin-Gene in R. marinus. Die vier identifizierten Gene
(thiSGED) liegen alle im gleichen Leseraster vor. thiS ist nicht vollständig sequenziert, der fehlende Sequenz-
bereich ist schraffiert dargestellt. Auch der Leserahmen von orf5 ist unvollständig. Die Zahlen unterhalb der Gene
geben die Länge der ORFs an, die Zahlen über den senkrechten Linien bezeichnen die Start- und Endpunkte der
jeweiligen ORFs. Die vollständige Sequenz findet sich im Anhang der Arbeit.
ORF thiS:
Der Start des Leserahmens thiS konnte nicht identifiziert werden, da dieser Bereich nicht
innerhalb der sequenzierten Region liegt. Die abgeleitete Aminosäuresequenz (42 As) zeigt
60 % Identität zu ThiSRP aus Rhodopseudomonas palustris, welches eine Gesamtlänge von
65 Aminosäuren aufweist. Außerdem zeigte die Sequenz 52 % Identität zu ThiSEC aus E. coli
K12 (66 As). Die Aminosäuresequenzen der thiS-Gene variieren in der Länge zwischen 65
und 74 Aminosäuren. Bei ThiS handelt es sich um ein schwefelübertragendes Protein,
welches an seinem C-Terminus ein Schwefelmolekül (ThiS-COSH) trägt (Begley et al.,
1999). Die Übertragung des Schwefelmoleküls ist Teil der Synthese eines Thiazol-Rings,
welcher eine Hälfte von Vitamin B1 (Thiamin) bildet.
ORF thiG:
thiG hat eine Länge von 824 bp und kodiert für ein potentielles 29,68 kDa Protein. Der Lese-
rahmen von thiG beginnt mit dem Startkodon ATG für Methionin. Die von thiG abgeleitete
Aminosäuresequenz weist eine Länge von 274 Aminosäuren auf. Sie ist zu 56 % mit einem
an der Thiazol-Biosynthese beteiligten Protein des δ-Proteobakteriums Geobacter metalli-
reducens (296 As) identisch. Zu ThiGEC aus E. coli K12 (256 As) beträgt die Identität 46 %
(Abb. 12). Bei ThiG handelt es sich um eine Thiazol-Synthase, die in E. coli die Bildung des
Thiazol-Rings aus Tyrosin, Deoxy-D-Xylulose und Cystein katalysiert (Vander Horn et al.,
1993; Begley et al., 1999).

III. Ergebnisse 86
Rhodothermus MSMQVVDGKGWQVQVDDEPLRIGPLELHTRLLMGSSGYPNVQTMLDALEAAGAELVTVAI 60 Streptomyces --------------MADDPFVLGDTPFSSRLIMGTGGAPSLDILERALVASGTELTTVAM 46 Geobacter ------------MSTTNDKLIIAGREFSSRLMVGTGKYASNEQMVAALEASGAEIITVAV 48 Pelobacter --------------MAEDKLVIAGREFNSRLMVGTGKYASFPQMAQALEASGAEIVTVAV 46 Escherichia ------------------MLRIADKTFDSHLFTGTGKFASSQLMVEAIRASGSQLVTLAM 42 Rhodothermus RRINLKDRSEESLLALLERHGYRVLPNTAGCYTAREAVLVAQLAREALETNLIKLEVIGD 120 Streptomyces RRVDAG--VHGSVLSVLDRLGIRVLPNTAGCFTAGEAVLTARLAREALGTDLIKLEVIAD 104 Geobacter RRVNITDRSKESLLDFIDTRKYTLLPNTAGCYTADDAVRTCRLAREAGMSDMVKLEVLGN 108 Pelobacter RRVNISDRSQESLLDYIDPTKYTLLPNTAGCYTADDAVRTCRLAREAGLSDFVKLEVLGD 106 Escherichia KRVDLR-QHNDAILEPLIAAGVTLLPNTSGAKTAEEAIFAAHLAREALGTNWLKLEIHPD 101 Rhodothermus DQTLLPDVEQLLKAARELINDGFVVLAYTNDDPITCRKLADLGCAAVMPLASPIGSGMGL 180 Streptomyces ERTLLPDPIELLDAAETLVDDGFTVLPYTNDDPVLARKLEDVGCAAVMPLGSPIGSGLGI 164 Geobacter ETTLYPDNEELLKAAKILVKDGFTVLPYTSDDPIICKKLEDIGCAAVMPLGAPIGSGLGI 168 Pelobacter EKTLFPDNEELLKAAAILIKDGFTVLPYCSDDVILCRKLEDLGCAAVMPLGAPIGSGLGI 166 Escherichia ARWLLPDPIETLKAAETLVQQGFVVLPYCGADPVLCKRLEEVGCAAVMPLGAPIGSNQGL 161 Rhodothermus VNPYNLHLIREMIDDVPLIVDAGIGTASDAAMAMELGYDGVLVNSAISQAEHPVLMARAV 240 Streptomyces RNPHNFQLIVEHAR-VPVILDAGAGTASDVALAMELGCAGVMLASAVTRAQEPVLMAEGM 223 Geobacter RNPYTIRIIMETVK-VPVIVDAGVGTASDAAIAMELGVDGVLMNTGIAGAQNPIAMAEAM 227 Pelobacter RNPYNIRIIMENVK-VPVIVDAGVGSASDAAIAMELGCDGVLMNTAIAGAQDPIAMAEAM 225 Escherichia ETRAMLEIIIQQAT-VPVVVDAGIGVPSHAAQALEMGADAVLVNTAIAVADDPVNMAKAF 220 Rhodothermus RHAVEAGRLAFRAGRMPRRFYARPSSPMEGRVGH------- 274 Streptomyces RHAVDAGRLAYRAGRIPRRHFAEASSPPEGRAHLDPERPAF 264 Geobacter NLAVRAGRLAYRAGRIPKKLYATASSPIEGMIE-------- 260 Pelobacter NLAVRAGRLSYKAGRIPRKLYANASSPLDGMIG-------- 258 Escherichia RLAVEAGLLARQSGPGSRSYFAHATSPLTGFLEASA----- 256
Abb. 12: Sequenzvergleich ausgesuchter ThiG-ähnlicher Proteinsequenzen
Beispielhaft dargestellt ist ein Sequenzvergleich der abgeleiteten Aminosäuresequenz von thiG aus R. marinus
mit bekannten ThiG-Sequenzen anderer Bakterien: Streptomyces avermitilis (Streptomyces; NP_ 626366.1),
Geobacter metallireducens (Geobacter; YP_ 385877.1), Pelobacter propionicus (Pelobacter; ZP_ 00676002.1)
und E. coli (Escherichia; NP_ 418418.2). Identische Aminosäuren (mindestens drei) sind schwarz unterlegt.
Konservative Aminosäureaustausche sind grau unterlegt. Als konservativ wurden nach Needleman & Wunsch
(1970) folgende Austausche definiert: G-A, I-L-V-M, S-T, R-K-H, D-E, N-Q, F-Y-W. Die Sequenzvergleiche
wurden mit dem Programm CLUSTAL W (Thompson et al., 1994) durchgeführt.
ORF thiE:
Der Leserahmen thiE besitzt eine Länge von 626 bp und kodiert für ein hypothetisches 208
As umfassendes Protein, mit einem berechneten Molekulargewicht von 22,12 kDa. Die abge-
leitete Aminosäuresequenz von thiERM weist 42 % Identität zu ThiERX aus Rubrobacter
xylanophilus, 39 % Identität zu ThiEEC aus E. coli und 44 % Identität zu ThiEMT aus Moorella
thermoacetica auf. Bei ThiE handelt sich um ein essentielles Genprodukt des Thiamin-Bio-
synthesewegs, die Thiaminphosphat-Synthase. Dieses Enzym katalysiert die Kopplung der
Pyrimidinpyrophosphat-Untereinheit an den Thiazolphosphat-Ring. Dabei entsteht die Vor-
stufe des aktiven Thiamins, Thiaminphosphat (Vander Horn et al., 1993; Begley et al., 1999).
ORF thiD:
Der Leserahmen thiD mit einer Länge von 858 bp beginnt mit dem Startkodon ATG (Methio-
nin) und kodiert für ein potentielles 30,27 kDa Protein. Die abgeleitete Aminosäuresequenz

III. Ergebnisse 87
(285 As) zeigt signifikante Ähnlichkeit (60 % Identität) zur Phosphomethylpyrimidin-Kinase
(HMP-P Kinase, 266 As) aus Chloroflexus aurantiacus. Die Identität mit ThiDSF (283 As) aus
Syntrophobacter fumarodidans beträgt 54 %, mit ThiDEC aus E. coli 46 % (266 As). ThiD
katalysiert die Phosphorylierung von Pyrimidin (Begley et al., 1999; Mizote et al., 1999).
ORF orf5:
Stromabwärts von thiD konnte der unvollständige Leserahmen orf5 identifiziert werden, des-
sen 3’-Ende nicht innerhalb der sequenzierten Region liegt. orf5 verfügt über ein ATG-Start-
kodon an Position 2795 der DNA-Region. Die abgeleitete Aminosäuresequenz besitzt Ähn-
lichkeiten zu konservierten Domäne eines sog. DEXDc-Proteins. Diese Proteine gehören zur
DEAD-Helikase Superfamilie und spielen eine Rolle bei der DNA-Entwindung, der
Ribosomen-Biosynthese, der Translation und dem RNA-Abbau. Die Aminosäuresequenzen
dieser Gene variiert zwischen 643 Aminosäuren bei Thiobacillus denitrificans (NCBI-Acc.Nr.:
YP_314755) und 238 Aminosäuren bei Vibrio cholerae (NCBI-Acc.Nr.: NP_933841).
2.2.5 Analyse der Expression von R. marinus Genen in E. coli
Die Komplementierung der Thiamin-Auxotrophie von E. coli DH5α zeigt die generelle Mög-
lichkeit der funktionellen Expression von R. marinus Genen in E. coli an. Die Funktionalität
von R. marinus Promotoren ließ sich nicht bestätigen, da stromaufwärts aller identifizierten
Leserahmen keine potentiellen Promoterregionen identifiziert werden konnten. Andererseits
ließen sich unmittelbar stromaufwärts der Startkodons von thiSGED und orf5 GA-reiche
Sequenzen identifizieren, welche Ähnlichkeiten zu Ribosomen-Bindungsstellen von E. coli
aufweisen (Abb. 13) und bei der funktionellen Komplementierung daher vermutlich als
Ribosomen-Bindungsstellen gedient haben.
thiG 154 5’-aaacgcacacgagaggctATGAGCATGCAGGTGGTTGACGGC-3’ M S M Q V V D G thiE 982 5’–cgcgtcggacactgaggcATGCTGCCCCGTTTGCTGCTGATC–3’ M L P R L L L I thiD 1616 5’-caccatcgataagcggccATGAACCGACAGATTCCACCGGTG-3’ M N R Q I P P V orf5 2776 5’-cggcccggccagcgccagATGGCCGAGACGGTCGTCCGGGCG-3’ M A E T V V R A
Abb. 13: GA-reiche Sequenzen mit möglichen Ribosomen-Bindungsstellen
Abgebildet sind GA-reiche Regionen mit möglichen Ribosomen-Bindungsstellen stromaufwärts der identifizierten
Leserahmen. Die angegebenen Zahlen bezeichnen die Positionen der aufgeführten Sequenzbereiche innerhalb
der DNA-Region (Anhang). Dunkelgrau schattierte Bereiche kennzeichnen die GA-reichen Regionen, an denen
vermutlich die Ribosomen-Anbindung erfolgt. Großbuchstaben repräsentieren die abgeleitete Aminosäurese-
quenzen, fette Großbuchstaben repräsentieren die DNA-Sequenzen innerhalb der Leserahmen.

III. Ergebnisse 88
2.3 Untersuchung zur Identifizierung von Solutetransportern durch
funktionelle Komplementierung transportdefizienter E. coli Stämme
Da die Komplementierung der Thiamin-Auxotrophie von E. coli DH5α durch eine plasmid-
kodierte Genbank aus R. marinus erfolgreich verlaufen war, sollte im nächsten Schritt ein
möglicher Mannosylglycerat-Transporter aus R. marinus durch Komplementierung der
Stämme E. coli MKH13 (Haardt et al., 1995) bzw. BKA-13 ausfindig gemacht werden.
2.3.1 Untersuchungen zur Komplementierung von E. coli MKH13
Von E. coli K12 ist bekannt, dass ihm ein Wachstum in Mineralsalzmedium ohne Zusatz von
kompatiblen Soluten nur bis zu einer Salinität von 3,8 % NaCl möglich ist (Larsen et al.,
1987). Die Zugabe kompatibler Solute erhöht die Salztoleranz bis auf über 5 % NaCl
(Gouesbet et al., 1994). E. coli MKH13 ist aufgrund von Deletionen verschiedener Solute-
transportergene (putPA, proP, proU und betT) nicht in der Lage kompatible Solute aus dem
umgebenden Medium aufzunehmen und kann daher in Mineralsalzmedium auch bei Zusatz
externer Solute nicht bei einer Salinität oberhalb 3,8 % wachsen. Darüber hinaus kann dieser
Stamm Cholin nicht zu Glycin-Betain oxidieren (∆betIBA) (siehe Abschnitt IV, 3.2.1). In eige-
nen Vorversuchen bestätigte sich dieses Wachstumsverhalten. Auch der Zusatz von Man-
nosylglycerat und Mannosylglyceramid (1 mM) ermöglichte E. coli MKH13 keine Wachstum
oberhalb einer Salinität von 3,8 % NaCl. MKH13 verfügt damit nicht über endogene Man-
nosylglycerat- bzw. Mannosylglyceramid-Transporter, eine zwingende Voraussetzung, um
mit diesem Stamm entsprechende R. marinus Gene zu identifizieren. Im Falle der Komple-
mentierung der Transportdefiziens von MKH13 durch plasmidkodierte Transportergene aus
R. marinus wurde mit einer Erhöhung der Salztoleranz des entsprechenden Klons bei
Zugabe von Mannosylglycerat bzw. Mannosylglyceramid gerechnet, zumal E. coli K12 bei
Zusatz externer Solute bis zu 5 % NaCl im Mineralsalzmedium toleriert.
Nach der Transformation von E. coli MKH13 mit der plasmidkodierten Genbank von
R. marinus wurden die Zellen auf MM63-4-Medium mit 1 mM Mannosylglycerat bzw. Man-
nosylglyceramid ausplattiert. Die Mannosylglycerat haltigen Platten wurden bei 43 °C inku-
biert, da dieses Solut sowohl bei erhöhter Mediensalinität als auch bei erhöhter Temperatur
durch R. marinus akkumuliert wird. Die Inkubation der Mannosylglyceramid haltigen Platten
erfolgte bei 37 °C.
Nach vier Tagen wuchsen in Charge 3 der pHSG575 kodierten Genbank auf den MM63-4
Platten mit 1 mM Mannosylglyceramid 13 unterschiedlich große Kolonien heran. Der Kon-
trollstamm E. coli MKH13 pHSG575 wuchs unter diesen Bedingungen nicht. Die Isolierung
der Plasmide ergab drei unterschiedliche Restriktionsmuster. Die unterschiedlichen Plasmide
wurden in E. coli MKH13 retransformiert und erneut selektioniert (MM63-4, 1 mM Mannosyl-

III. Ergebnisse 89
glyceramid). Auch nach 10 Tagen war kein Wachstum zu verzeichnen. Mehrfache Wieder-
holungen von Transformation und Retransformation ergaben vergleichbare Ergebnisse.
Diese Ergebnisse verdeutlichten, dass ein Wachstum von E. coli MKH13 Klonen auf den
Selektionsplatten keinen verlässlichen Hinweis auf eine Soluteaufnahme darstellt. Um ein
Wachstum solcher falsch-positiver Klone zu unterbinden, wurde ein neuer, auf E. coli MKH13
basierender Selektionsstamm hergestellt und für die weiteren Komplementierungsexperi-
mente eingesetzt.
2.3.2 Untersuchungen zur Komplementierung von E. coli BKA-13
2.3.2.1 Herstellung und Charakterisierung der otsB-Deletionsmutante E. coli BKA-13
Das Fehlschlagen der Komplementierungsexperimente mit E. coli MKH13 wurde auf ver-
schiedene durchführungsbedingte Parameter, wie die endogene Trehalose-Synthese,
zurückgeführt. So wurde vermutet, dass die endogene Trehalose-Synthese von E. coli
MKH13 diesem über eine Zeitspanne von mehreren Tagen ein Wachstum bei 4 % NaCl
erlaubt. Außerdem bestand die Möglichkeit, dass die Salinität von 4 % NaCl, welche für die
Komplementierungsexperimente gewählt wurde, für die Transportaktivität eines möglichen
R. marinus Transporterproteins zu hoch war und aus diesem Grund kein entsprechender
Klon identifiziert werden konnte. Eine geringere Salinität des Selektionsmediums konnte bei
Nutzung von E. coli MKH13 bedingt durch die endogene Trehalose-Synthese jedoch nicht
gewählt werde.
Trehalose wird in osmotisch gestressten E. coli Zellen de novo synthetisiert. Die Enzyme der
osmoregulatorischen Biosynthese von Trehalose werden durch die Gene otsA (Trehalose-6-
phosphat-Synthase) und otsB (Trehalose-6-Phosphatase) kodiert (Kaasen et al., 1992;
Strøm & Kaasen, 1993). Eine Inaktivierung dieser Gene sollte die Trehalose-Synthese unter-
binden und eine Absenkung Salinität des Selektionsmediums ermöglichen, da für den resul-
tierenden Stamm von einer verminderten Salztoleranz ausgegangen wurde. Dadurch sollten
einerseits weniger falsch-positive Klone auftreten, zum anderen die Transporteraktivität mög-
licher R. marinus Transporterproteine erhöht werden.
Die Trehalose-Synthese in E. coli MKH13 wurde ausgeschaltet, indem otsB vollständig im
Genom von E. coli MKH13 deletiert wurde. Stromaufwärts von otsB befindet sich das Gen
otsA, welches um 26 bp mit otsB überlappt. Bei der Deletion von otsB wurde somit auch die
Anfangssequenz des otsA-Gens deletiert (Abb. 14). Der resultierende Stamm wird als E. coli
BKA-13 bezeichnet.
Das für die Deletion benötigte rekombinante DNA-Konstrukt wurde mittels SOE-PCR ampli-
fiziert und in den Vektor pMAK705 kloniert (Tab. 11). Das resultierende Plasmid trägt die
Bezeichnung pMAKotsB.

III. Ergebnisse 90
E. coli Stamm Primerpaare Produkt-
größe kb Plasmid-
Bezeichnung
SOEotsB1 & SOEotsB2 1,049
SOEotsB3 & SOEotsB4 1,057 BKA-13 (∆otsB)
Fusionsprodukt: SOEotsB1 & SOEotsB4 2,072
pMAKotsB
Tab. 11: PCR-Produkte und Vektorkonstrukt zur Erstellung der ∆otsB-Mutante E. coli BKA-13
Aufgeführt sind die Primer, die zur Herstellung von BKA-13 benötigt wurden, sowie die entsprechende Plasmid-
bezeichnung. Das Konstrukt wurde mittels SOE-PCR mit E. coli MKH13 DNA amplifiziert. Das Fusionsprodukt
wurde über Restriktionsschnittstellen (Sal I) in den Vektor pMAK705 ligiert.
Zur Identifizierung der otsB Deletionsmutanten von E. coli MKH13 wurden CmS Klone in eine
Kolonie-PCR mit den Primern SOEotsB1 und SOEotsB4 eingesetzt. Mit Zellen von MKH13
wurde ein 2,867 kb großes PCR-Produkt amplifiziert, bei der ∆otsB Deletionsmutante E. coli
BKA-13 ein 2,072 kb großes Produkt (Ta= 56 °C) (Abb.14).
1 kb
Nde I
otsA
2106 3504
1399 bp
Nde I
E. coliBKA-13
otsA
801 bp 1425 bp
Nde I5‘ ATG
2906
2881
43052106
otsB
Nde I
E. coliMKH13
Abb. 14: Genetische Organisation des otsAB-Operons in E. coli MKH13 und E. coli BKA-13
Dargestellt ist die genetische Organisation des otsAB-Operons in E. coli MKH13 und der ∆otsB-Deletionsmutante
E. coli BKA-13: grün: otsB (Trehalose-6-phosphat-Phosphatase), rot: otsA (Trehalose-6-phosphat-Synthase)
(Kaasen et al., 1992; Strøm & Kaasen, 1993). Die ORFs von otsB und otsA überlappen um 26 bp. Der dar-
gestellte DNA-Bereich entspricht den Basen 900-7200 der E. coli K12 Sequenz mit der NCBI-Acc.Nr.: AE000283
in reverse komplementärer Orientierung. Zur Orientierung sind die Basenpositionen der ORF-Anfänge und
-Enden angegeben.
Mit Hilfe eines Plattentests wurde untersucht, wie sich das Ausbleiben der Trehalose-Syn-
these bei verschiedenen Salinitäten auf das Wachstum der Deletionsmutante E. coli BKA-13

III. Ergebnisse 91
(∆otsB) auswirkt. Für den Wachstumstest wurden Agarplatten mit 2 %, 2,5 %, 2,7 %, 2,8 %
und 3 % ohne Zusatz von kompatiblen Soluten verwendet. E. coli MKH13 und BKA-13
wurden über Nacht in MM63-1 Flüssigkultur angezogen. Am nächsten Tag wurde die OD540
auf 0,1 eingestellt. Je 1 ml der Kultur wurde dann zweimal mit 2 %, 2,5 %, 2,7 %, 2,8 % bzw.
3 % NaCl gewaschen. Schließlich wurden 50-100 µl der Zellsuspensionen auf entspre-
chenden MM63-Agarplatten ausplattiert.
Schon nach zwei Tagen wuchs E. coli MKH13 bei allen getesteten Salinitäten. Im Vergleich
dazu zeigte die ∆otsB-Mutante E. coli BKA-13 einen salzsensitiveren Phänotyp. Sie wuchs
deutlich langsamer und benötigte etwa 5-6 Tage, um bei einer Mediensalinität von 2,7 %
Kolonien auszubilden. Bei einer Mediensalinität von 2,8 % war auch nach 6 Tagen kein
Wachstum zu beobachten. In HPLC-Analysen der Zellextrakte von E. coli BKA-13 konnte
zudem keine Trehalose nachgewiesen werden.
2.3.2.2 Untersuchungen zur Komplementierung von E. coli BKA-13
Aufgrund des salzsensitiven Phänotyps von E. coli BKA-13 wurden die Selektionsbedingung
für die Komplementierung durch die plasmidkodierte Genbank von R. marinus geändert.
Dem Selektionsmedium wurden nur noch 3 % NaCl zugesetzt. Die pHSG575 kodierte
Genbank wurde in E. coli BKA-13 transformiert. Wiederum wurden je 500-1000 Klone von
CmR E. coli BKA-13 in LB-0,5 Flüssigmedium mit 50 µg/ml Cm angezogen. 1 ml der Flüssig-
kulturen wurden fünfmal mit 1 ml MM63-0,5 Flüssigmedium gewaschen und anschließend
auf MM63-3-Agarplatten mit Vitaminzusatz und 1 mM Mannosylglycerat bzw. Mannosyl-
glyceramid ausplattiert. Die Inkubation erfolgte bei 37 °C (1 mM Mannosylglyceramid) bzw.
bei 43 °C (1 mM Mannosylglycerat). E. coli BKA-13 pHSG575 diente als Negativkontrolle.
Nach vier Tagen waren sehr kleine Kolonien (1-2 mm ∅) am Rande der Mannosylglyceramid
supplementierten Platten erkennbar. Nach der Plasmidisolierung aus einigen dieser Kolo-
nien, wurden unterschiedliche Restriktionsmuster nachgewiesen. Plasmide aus sieben Kolo-
nien wurden in E. coli BKA-13 retransformiert. Keine der retransformierten Klone wuchs
erneut auf MM63-3 mit Vitaminen und Mannosylglyceramid. Der Versuch wurde mehrfach
mit einer unterschiedlichen Anzahl von Waschschritten wiederholt. In keinem Fall konnte
jedoch ein Klon identifiziert werden, welcher nach der Retransformation zu einem Wachstum
auf Selektionsmedium fähig war.
Um nach dem negativen Verlauf der Komplementierungsexperimente mögliche Probleme bei
der Expression hypothetischer R. marinus Transportergene zu umgehen, wurde eine neue
R. marinus Genbank in dem Expressionsvektor pXMJ19 erstellt (Jakoby et al., 1999). Dieser
besitzt einen induzierbaren tacP-Promotor. So sollten Probleme bei der Erkennung thermo-
philer Promotoren oder das Fehlen spezieller regulatorischer Elemente umgangen werden.

III. Ergebnisse 92
Die Genbank wurde in E. coli BKA-13 transformiert. Jeweils 500-1000 Klone wurden wie
oben dargestellt behandelt. Als Selektionsmedium dienten MM63-3-Agarplatten mit Vita-
minen und Mannosylglycerat bzw. Mannosylglyceramid (1 mM). Zur Induktion des tacP-Pro-
motors wurden die Zellen zusammen mit IPTG (0,2 mM Endkonzentration) ausplattiert.
Wiederum wuchsen nur sehr kleine Kolonien auf den Selektionsplatten mit 1 mM Mannosyl-
glyceramid. Nach der Retransformation der isolierten Plasmide konnte auch hier keiner der
Klone auf MM63-3-Agarplatten mit Mannosylglyceramid wachsen.
Aufgrund dieser Ergebnisse wurde die Suche nach Mannosylglycerat-Transportern mittels
einer Genbank vermittelten funktionellen Komplementierung transportdefizienter E. coli
Stämme eingestellt.
2.4 Isolierung eines DNA-locus mit Ähnlichkeiten zu Zuckertransportern
mittels heterologer PCR Ziel der vorliegenden Arbeit ist die Herstellung eines Produktionsstamms für das kompatible
Solut Mannosylglycerat. Da die Arbeiten zu Identifizierung eines möglichen Solutetrans-
porters mittels einer Genbank fehlschlugen (siehe Abschnitt IV, 3.2.2), wurde die Identifi-
zierung mittels einer heterologen PCR unter Verwendung degenerierter Primer versucht.
In der Literatur hatten sich für Thermus thermophilus und Thermococcus litoralis Hinweise
auf Ähnlichkeiten von Mannosylglycerat-Transporter und Zuckertransportern (Trehalose/
Maltose) ergeben (Abschnitt IV, 4., Xavier et al., 1996; Sampaio et al., 2003b). Bei all diesen
Verbindungen handelt es sich um α-Glucoside, zugehörige Transportproteine sollten daher
strukturelle Ähnlichkeiten aufweisen. Bekannte α-Glucosid-Transporter gehören zu der
Gruppe der ABC-Transporter und sind in vielen Genomsequenzen thermophiler Organis-
men zu finden (Horlacher et al., 1998; Koning et al., 2002). ABC-Transporter sind primäre
Transporter, die ihre Energie aus der Hydrolyse von ATP gewinnen. Sie sind insgesamt aus
fünf Proteinkomponenten aufgebaut. Dazu zählen ein periplasmatisches Substratbinde-
protein, ein Dimer bildendes integrales Membranprotein und eine im Zytoplasma lokalisierte
ATPase, die ebenfalls als Dimer vorliegt (Boos & Lucht, 1996).
2.4.1 Ableitung von degenerierten PCR-Primer mittels eines Sequenzvergleichs
Um die degenerierten Primer für die Amplifikation eines möglichen α-Glucosid oder Zucker-
transporter abzuleiten, wurden DNA- und Proteinsequenzen aus T. thermophilus und ander-
en Organismen, von denen einige auch zur Synthese von Mannosylglycerat fähig sind,
herangezogen. Zur Ableitung der Primer wurden die Sequenzen von Substratbindeproteinen
genutzt, da sich die membranintegralen Komponenten von ABC-Transportern mit unter-
schiedlichsten Substratspektren erfahrungsgemäß aufgrund der hydrophoben Sequenz-
bereiche sehr stark ähneln und so mit einer Vielzahl falscher Amplifikate zu rechnen ist

III. Ergebnisse 93
wurde. Die DNA-Sequenzen verschiedener Substratbindeproteine von Trehalose/Maltose-
Transportern wurden in Alignments verglichen. Besonders konservierte DNA-Bereiche
wurden für die Ableitung der Primer ausgewählt. Hier wurden die Aminosäurensequenzen in
ihren verschiedenen Variationen abgeleitet und mit ihrer Hilfe schließlich die Primersequenz
bestimmt. Beispielhaft ist hier die Verwendung einer Gruppe von Sequenzen dargestellt
(Abb. 15).
In Bereichen konservierter Aminosäuren jedoch unterschiedlicher genutzter Kodons wurde
die „Codon usage“ von R. marinus (siehe Abb. 49) in die Ableitung der Primer einbezogen.
Diese berücksichtigt die im Genom bevorzugt verwendeten Basentripletts für Aminosäuren,
welche durch verschiedenen Kodons kodiert werden können. Bei der Primerwahl wurde auf-
grund der „Codon usage“ in einigen Fällen statt eines bestimmten Kodons, ein Kodon mit
„Wobble Basen“ eingefügt. Dies war jeweils von der Häufigkeit der Nutzung des jeweiligen
Kodons durch R. marinus abhängig. Zu seltene Kodons wurden nicht einbezogen.
DR1438 414 CTGTACTACCGCACCGACCTGCTGAAAAAGTACGGTTATAATGCGCCCCCCAAGACCTGG 474 TTC1627 444 CTTTACTACCGCAAGGACCTCCTGGAGAAGTACGGCTATACGAGCCCGCCCCGGACCTGG 504 PF1739 498 TTGTATTATAGGAAAGACCTACTAGAAAAGTATGGCTATAGCAAACCCCCAGAAACTTGG 558 * ** ** * * ***** ** * ***** ** **** ** ** ** *** D L L K K Y G Y E Primer TTC1627for1 5’ gacctsctrraraagtayggyta 3’ DR1438 762 GCGGGCAACTCGGCCTTCATGCGCAACTGGCCCTACGCCTACGCGGCGGGTCAAAAGGAG 822 TTC1627 795 CAGGGCAACAGCCTCTTCATGCGCAACTGGCCCTACGCCTACGCCTTGGGCCAGGCGGAG 855 PF1739 861 CAGGGCAATGCTGCATTCGAGAGAAATTGGCCATACGCGTGGGGACTACACAATGCTGAT 921 ****** *** * * ** ***** ***** * * * * F M R N W P Y A E Primer TTC1627rev2 3’ aagywcgcgttraccgggatgcg 5’ DR1438 873 GGGGGCAAGCCC---GCGGCGACGCTCGGCGGCTGGCAGCTCGCGGTCAACGCCTACTCC 930 TTC1627 906 GCGGACGCCCCCAACGCCGCCACCCTGGGCGGCTGGCAGCTCATGGTCTCCGCCTACAGC 966 PF1739 972 GGTCATAAGAGT---GCAGCTACACTTGGAGGATGGCACATTGGGATAAGCAAGTACTCA 1029 * ** ** ** ** ** ** ***** * * * * *** A A T L G G W Q H Primer TTC1627rev1 3’ cgbcgvtgbgavcckcckaccgt 5’ Abb. 15: Ableitung degenerierter Primer zur Verwendung in einer heterologen PCR
Dargestellt sind drei Abschnitte aus einem Sequenzvergleich, in dem die DNA-Sequenz des periplasmatischen
Substratbindeproteins (TTC1627, NCBI-Acc.Nr.: NC_005835) eines Trehalose/Maltose Transporters aus Ther-
mus thermophilus HB27 homologen Sequenzen aus Pyrococcus furiosus (PF1739, NCBI-Acc.Nr.: AAL81863)
und Deinococcus radiodurans (DR1438, NCBI-Acc.Nr.: AAF11010) gegenübergestellt wurde. Die Basenangaben
beziehen sich auf den ORF des entsprechenden Gens. Grau unterlegt sind besonders konservierte DNA-
Bereiche, in denen die Primer für die heterologe PCR abgeleitete wurden. Die Aminosäuresequenz ist im
Einbuchstaben-Code angegeben. Die abgeleiteten Primersequenzen sind unterhalb der konservierten Regionen
aufgeführt. „Wobble Basen“ sind gelb unterlegt hervorgehoben: b: G, T oder C; k: G oder T; r: A oder G; s: G oder
C; v: G, A oder C; w: A oder T; y: T oder C. Bei Primer TTC1627for1 handelt es sich um einen forward-Primer, bei
TTC1627rev1 und TTC1627rev2 um zwei alternative reverse-Primer.

III. Ergebnisse 94
2.4.2 Heterologe PCR und Sequenzierung
Unter Verwendung der Primer TTC1627for1 und TTC1627rev2 wurde auf T. thermophilus
DNA das erwartete PCR-Produkt mit einer Größe von ca. 375 bp amplifiziert. Auf R. marinus
DNA wurde bei einer Anlagerungstemperatur von 50 °C und einer Primerkonzentration von
60 pmol/50 µl ein PCR-Produkt amplifiziert, welches nach Analyse im Agarosegel eine
Größe von ca. 450 bp aufwies. Das PCR-Produkt wurde in den pGEMT Easy Vektor kloniert
(pGEMSSSF) und mittels des T7-Promotor-Primers sequenziert. Die Sequenzierung ergab
nach Entfernung der Anbindungsbereiche der heterologen Primer 644 bp R. marinus DNA-
Sequenz (Abb. 16).
Die Analysen der abgeleiteten Aminosäuresequenz mit dem Programm Blast (Altschul et al.,
1990) ergaben große Sequenzähnlichkeiten zu Transportern der Sodium/Solute-Symporter
Familie (SSSF-Transporter). Nach diesen Analysen war das Transportergen nicht vollständig
sequenziert. Zur Vervollständigung der DNA-Sequenz wurde die Methode der inversen-PCR
angewendet (Abb. 16).
Zur Abschätzung der zu erwartenden PCR-Produktgrößen wurde R. marinus DNA zunächst
mit den Restriktionsenzymen (Pst I, Bam HI, Eco RI, Sal I, Hin dIII) hydrolysierte und in einer
Southern-Hybridisierung analysiert. Zum Nachweis des gesuchten DNA-Fragments diente
eine DIG-markierte PCR-Sonde, die mit den Primern SSFSondefor2 und SSFSonderev
amplifiziert worden war (Abb. 16). Es wurden DNA-Banden unterschiedlicher Größe nach-
gewiesen (Pst I: ca. 1,0 & 2,5 kb; Bam HI: ca. 2,0 & 3,5 kb; Eco RI: ca. 6,0 kb; Sal I: ca. 12,0
& 8,0 kb; Hin dIII: ca. 13 kb). Da alle Enzyme nicht innerhalb der Sondensequenz schneiden,
deutet das Auftreten zweier Banden in einem Southern-Hybridisierungsexperiment auf das
Vorhandensein von zwei ähnlichen DNA-Sequenzen im Genom von R. marinus hin (siehe
Abschnitt IV, 4.).
Als geeignetes Restriktionsenzym für die inverse-PCR wurde Pst I ausgewählt. Beide mög-
lichen Fragmentgrößen lassen sich aufgrund der geringen Größe problemlos auch bei
geringer Templatekonzentrationen in einer inversen-PCR amplifizieren. In der PCR mit den
Primern SSFrev1 und R.marinusSSFfor2 wurden zwei Produkte mit Größen von ca. 1,0 und
2,5 kb amplifiziert. Das größere Produkt lag in höherer Konzentration vor und wurde nach
der Aufreinigung mit dem Primer R.marinusSSFfor2 ansequenziert. Sequenzanalysen er-
gaben Übereinstimmungen mit dem ersten PCR-Produkt und zeigten die Vervollständigung
des ORFs des hypothetischen SSSF-Transporters an. Eine vollständige Sequenzierung des
PCR-Produkts wurde nicht durchgeführt. Insgesamt wurde eine 1467 bp umfassende Region
der R. marinus DNA sequenziert (siehe Anhang).
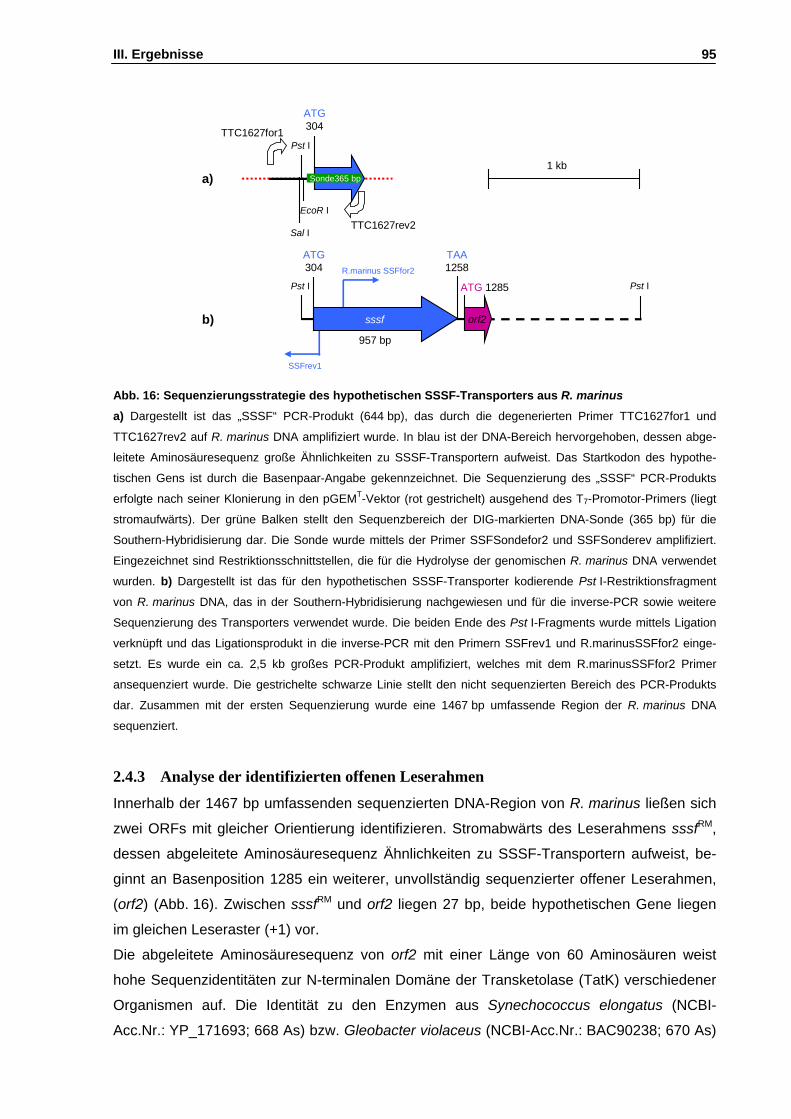
III. Ergebnisse 95
1 kb
TTC1627rev2
ATG304
Sal I
Pst I
EcoR I
TTC1627for1
a) Sonde365 bp
SSFrev1
Pst IR.marinus SSFfor2
957 bp
ATG 1285
ATG304
TAA1258
Pst I
b) orf2sssf
Abb. 16: Sequenzierungsstrategie des hypothetischen SSSF-Transporters aus R. marinus a) Dargestellt ist das „SSSF“ PCR-Produkt (644 bp), das durch die degenerierten Primer TTC1627for1 und
TTC1627rev2 auf R. marinus DNA amplifiziert wurde. In blau ist der DNA-Bereich hervorgehoben, dessen abge-
leitete Aminosäuresequenz große Ähnlichkeiten zu SSSF-Transportern aufweist. Das Startkodon des hypothe-
tischen Gens ist durch die Basenpaar-Angabe gekennzeichnet. Die Sequenzierung des „SSSF“ PCR-Produkts
erfolgte nach seiner Klonierung in den pGEMT-Vektor (rot gestrichelt) ausgehend des T7-Promotor-Primers (liegt
stromaufwärts). Der grüne Balken stellt den Sequenzbereich der DIG-markierten DNA-Sonde (365 bp) für die
Southern-Hybridisierung dar. Die Sonde wurde mittels der Primer SSFSondefor2 und SSFSonderev amplifiziert.
Eingezeichnet sind Restriktionsschnittstellen, die für die Hydrolyse der genomischen R. marinus DNA verwendet
wurden. b) Dargestellt ist das für den hypothetischen SSSF-Transporter kodierende Pst I-Restriktionsfragment
von R. marinus DNA, das in der Southern-Hybridisierung nachgewiesen und für die inverse-PCR sowie weitere
Sequenzierung des Transporters verwendet wurde. Die beiden Ende des Pst I-Fragments wurde mittels Ligation
verknüpft und das Ligationsprodukt in die inverse-PCR mit den Primern SSFrev1 und R.marinusSSFfor2 einge-
setzt. Es wurde ein ca. 2,5 kb großes PCR-Produkt amplifiziert, welches mit dem R.marinusSSFfor2 Primer
ansequenziert wurde. Die gestrichelte schwarze Linie stellt den nicht sequenzierten Bereich des PCR-Produkts
dar. Zusammen mit der ersten Sequenzierung wurde eine 1467 bp umfassende Region der R. marinus DNA
sequenziert.
2.4.3 Analyse der identifizierten offenen Leserahmen
Innerhalb der 1467 bp umfassenden sequenzierten DNA-Region von R. marinus ließen sich
zwei ORFs mit gleicher Orientierung identifizieren. Stromabwärts des Leserahmens sssfRM,
dessen abgeleitete Aminosäuresequenz Ähnlichkeiten zu SSSF-Transportern aufweist, be-
ginnt an Basenposition 1285 ein weiterer, unvollständig sequenzierter offener Leserahmen,
(orf2) (Abb. 16). Zwischen sssfRM und orf2 liegen 27 bp, beide hypothetischen Gene liegen
im gleichen Leseraster (+1) vor.
Die abgeleitete Aminosäuresequenz von orf2 mit einer Länge von 60 Aminosäuren weist
hohe Sequenzidentitäten zur N-terminalen Domäne der Transketolase (TatK) verschiedener
Organismen auf. Die Identität zu den Enzymen aus Synechococcus elongatus (NCBI-
Acc.Nr.: YP_171693; 668 As) bzw. Gleobacter violaceus (NCBI-Acc.Nr.: BAC90238; 670 As)
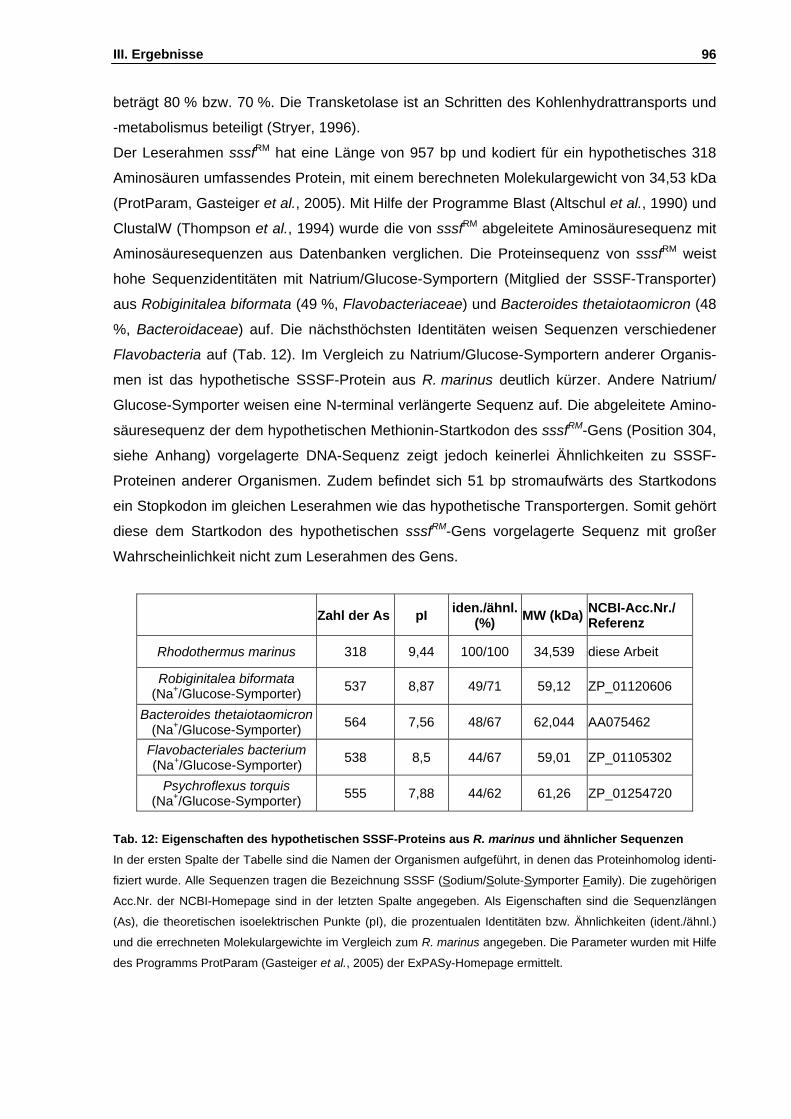
III. Ergebnisse 96
beträgt 80 % bzw. 70 %. Die Transketolase ist an Schritten des Kohlenhydrattransports und
-metabolismus beteiligt (Stryer, 1996).
Der Leserahmen sssfRM hat eine Länge von 957 bp und kodiert für ein hypothetisches 318
Aminosäuren umfassendes Protein, mit einem berechneten Molekulargewicht von 34,53 kDa
(ProtParam, Gasteiger et al., 2005). Mit Hilfe der Programme Blast (Altschul et al., 1990) und
ClustalW (Thompson et al., 1994) wurde die von sssfRM abgeleitete Aminosäuresequenz mit
Aminosäuresequenzen aus Datenbanken verglichen. Die Proteinsequenz von sssfRM weist
hohe Sequenzidentitäten mit Natrium/Glucose-Symportern (Mitglied der SSSF-Transporter)
aus Robiginitalea biformata (49 %, Flavobacteriaceae) und Bacteroides thetaiotaomicron (48
%, Bacteroidaceae) auf. Die nächsthöchsten Identitäten weisen Sequenzen verschiedener
Flavobacteria auf (Tab. 12). Im Vergleich zu Natrium/Glucose-Symportern anderer Organis-
men ist das hypothetische SSSF-Protein aus R. marinus deutlich kürzer. Andere Natrium/
Glucose-Symporter weisen eine N-terminal verlängerte Sequenz auf. Die abgeleitete Amino-
säuresequenz der dem hypothetischen Methionin-Startkodon des sssfRM-Gens (Position 304,
siehe Anhang) vorgelagerte DNA-Sequenz zeigt jedoch keinerlei Ähnlichkeiten zu SSSF-
Proteinen anderer Organismen. Zudem befindet sich 51 bp stromaufwärts des Startkodons
ein Stopkodon im gleichen Leserahmen wie das hypothetische Transportergen. Somit gehört
diese dem Startkodon des hypothetischen sssfRM-Gens vorgelagerte Sequenz mit großer
Wahrscheinlichkeit nicht zum Leserahmen des Gens.
Zahl der As pI iden./ähnl. (%) MW (kDa) NCBI-Acc.Nr./
Referenz
Rhodothermus marinus 318 9,44 100/100 34,539 diese Arbeit
Robiginitalea biformata (Na+/Glucose-Symporter) 537 8,87 49/71 59,12 ZP_01120606
Bacteroides thetaiotaomicron (Na+/Glucose-Symporter) 564 7,56 48/67 62,044 AA075462
Flavobacteriales bacterium (Na+/Glucose-Symporter) 538 8,5 44/67 59,01 ZP_01105302
Psychroflexus torquis (Na+/Glucose-Symporter) 555 7,88 44/62 61,26 ZP_01254720
Tab. 12: Eigenschaften des hypothetischen SSSF-Proteins aus R. marinus und ähnlicher Sequenzen In der ersten Spalte der Tabelle sind die Namen der Organismen aufgeführt, in denen das Proteinhomolog identi-
fiziert wurde. Alle Sequenzen tragen die Bezeichnung SSSF (Sodium/Solute-Symporter Family). Die zugehörigen
Acc.Nr. der NCBI-Homepage sind in der letzten Spalte angegeben. Als Eigenschaften sind die Sequenzlängen
(As), die theoretischen isoelektrischen Punkte (pI), die prozentualen Identitäten bzw. Ähnlichkeiten (ident./ähnl.)
und die errechneten Molekulargewichte im Vergleich zum R. marinus angegeben. Die Parameter wurden mit Hilfe
des Programms ProtParam (Gasteiger et al., 2005) der ExPASy-Homepage ermittelt.
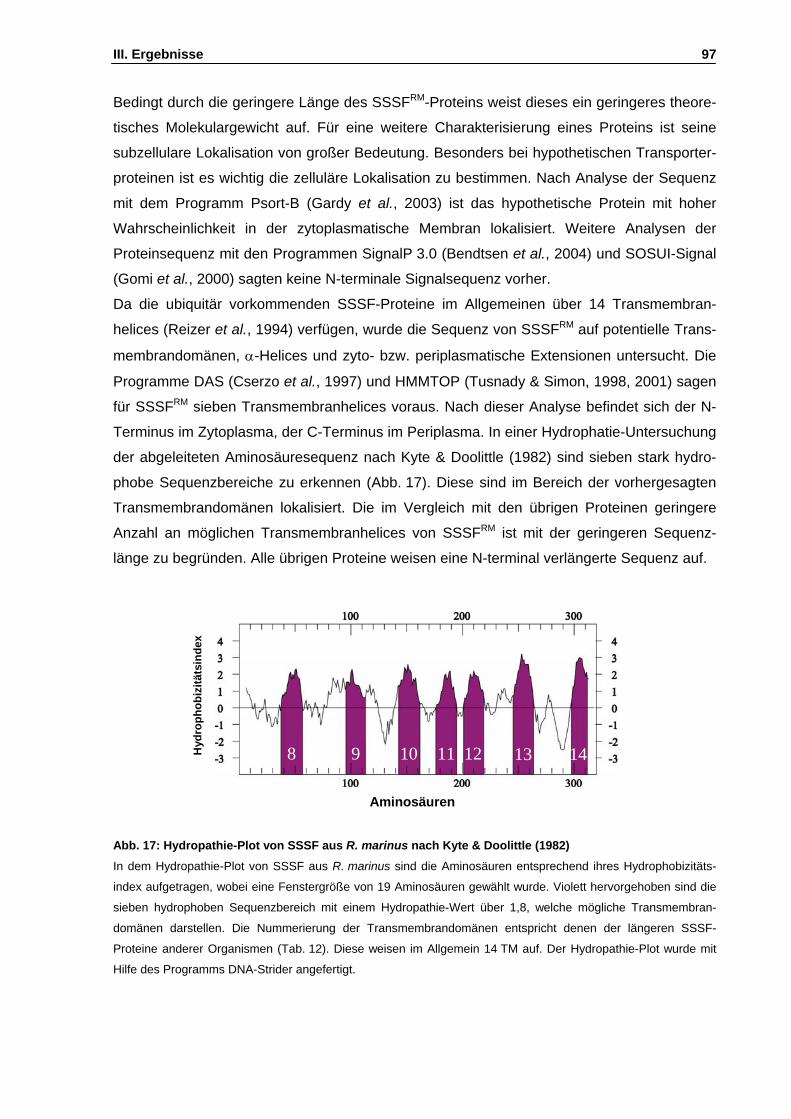
III. Ergebnisse 97
Bedingt durch die geringere Länge des SSSFRM-Proteins weist dieses ein geringeres theore-
tisches Molekulargewicht auf. Für eine weitere Charakterisierung eines Proteins ist seine
subzellulare Lokalisation von großer Bedeutung. Besonders bei hypothetischen Transporter-
proteinen ist es wichtig die zelluläre Lokalisation zu bestimmen. Nach Analyse der Sequenz
mit dem Programm Psort-B (Gardy et al., 2003) ist das hypothetische Protein mit hoher
Wahrscheinlichkeit in der zytoplasmatische Membran lokalisiert. Weitere Analysen der
Proteinsequenz mit den Programmen SignalP 3.0 (Bendtsen et al., 2004) und SOSUI-Signal
(Gomi et al., 2000) sagten keine N-terminale Signalsequenz vorher.
Da die ubiquitär vorkommenden SSSF-Proteine im Allgemeinen über 14 Transmembran-
helices (Reizer et al., 1994) verfügen, wurde die Sequenz von SSSFRM auf potentielle Trans-
membrandomänen, α-Helices und zyto- bzw. periplasmatische Extensionen untersucht. Die
Programme DAS (Cserzo et al., 1997) und HMMTOP (Tusnady & Simon, 1998, 2001) sagen
für SSSFRM sieben Transmembranhelices voraus. Nach dieser Analyse befindet sich der N-
Terminus im Zytoplasma, der C-Terminus im Periplasma. In einer Hydrophatie-Untersuchung
der abgeleiteten Aminosäuresequenz nach Kyte & Doolittle (1982) sind sieben stark hydro-
phobe Sequenzbereiche zu erkennen (Abb. 17). Diese sind im Bereich der vorhergesagten
Transmembrandomänen lokalisiert. Die im Vergleich mit den übrigen Proteinen geringere
Anzahl an möglichen Transmembranhelices von SSSFRM ist mit der geringeren Sequenz-
länge zu begründen. Alle übrigen Proteine weisen eine N-terminal verlängerte Sequenz auf.
Aminosäuren
Hyd
roph
obiz
itäts
inde
x
8 9 10 11 12 13 14
Abb. 17: Hydropathie-Plot von SSSF aus R. marinus nach Kyte & Doolittle (1982)
In dem Hydropathie-Plot von SSSF aus R. marinus sind die Aminosäuren entsprechend ihres Hydrophobizitäts-
index aufgetragen, wobei eine Fenstergröße von 19 Aminosäuren gewählt wurde. Violett hervorgehoben sind die
sieben hydrophoben Sequenzbereich mit einem Hydropathie-Wert über 1,8, welche mögliche Transmembran-
domänen darstellen. Die Nummerierung der Transmembrandomänen entspricht denen der längeren SSSF-
Proteine anderer Organismen (Tab. 12). Diese weisen im Allgemein 14 TM auf. Der Hydropathie-Plot wurde mit
Hilfe des Programms DNA-Strider angefertigt.

III. Ergebnisse 98
Ein Sequenzvergleich der einzelnen Natrium/Glucose-Symporter zeigte mehrere konser-
vierte Bereiche. Hier ist vor allem das stark konservierte Motiv (R-x-T-x4-F-L-A-G-x4-W-W-x2-G-A-S-L) nahe des N-Terminus hervorzuheben (Abb. 18, Motiv 1). Dieses Motiv tritt
häufig bei Mitgliedern der Natrium/Glucose-Symporter bzw. Mitgliedern der Natrium/Myo-
inositol-Symporter auf (Wright et al., 2004). Mittels des PROSITE-Servers (Falquet et al.,
2002) wurde im Bereich der vierten Transmembranhelix ein weiteres konserviertes Motiv
vorhergesagt (Abb. 18, Motiv 2). Beide Motive sind in der R. marinus Sequenz aufgrund ihrer
geringeren Größe nicht vorhanden. Ein häufig vorhandenes Natrium-Bindemotiv (SOB:
Sodium ion Binding Motif) (Reizer et al., 1994) mit der Sequenzabfolge (G-xn-A-x4-L-x3-G-R)
konnte nur in der Sequenz von Psychroflexus torquis (As 340-382) identifiziert werden.
Rob.biformata ------------MLDSLDWVTISIYFAILLGIAVWVILKKKD---NTEDYFLAGRNVGWF 45 Fla.bacterium ---------MDSILEKADWLVLGVYFAALIVVAIWVILQKNK---DTADYFLAGRNVGWF 48 Bac.thetaiot. -------------MEALDWLVIGVFFLALIGIIVWVVRQKQN---DSADYFLGGRDATWL 44 Psy.torquis -------------MEFLDIAIIVLYFGIILWIAQWASKGKKDAAFSSVDYFLAGKNQGWF 47 Rho.marinus ------------------------------------------------------------
Rob.biformata VVGASIFASNIGSEHVVGLAGTGASNKLPMLIYEIHAWIVLLL-GWVFLPFYARAGVFTM 104
Motiv 1
Fla.bacterium VIGASIFASNIGSEHVVGLAGTGAESGMPMAHYELHAWIVLLL-GWLFLPFYFRSNAFTM 107 Bac.thetaiot. AIGASIFASNIGSEHLIGLAGAGASSGMAMAHWEIQGWMILIL-GWVFVPFYSRSMVYTM 103 Psy.torquis VIGASLFASNIGSEIILGVSGAGARGSMPMANFEILASLVLILFGWVFVPFYLRTGVYTM 107 Rho.marinus ------------------------------------------------------------
Rob.biformata PEFLEKRFD-ARARWILSVFSIVAYVLTKISVTIYAGGVVVSALLGID-------FWTGA 156 Motiv 2
Fla.bacterium PEFLEKRFD-SRSRWFLSVFSLAGYVITKVSVTIYAGGIVVSELLGIP-------FWYGA 159 Bac.thetaiot. PEFLERRYN-PQSRTILSVISLVSYVLTKVAVTVYAGGLVFQQVFGIKELWGIDFFWIAA 162 Psy.torquis PEFLEKRYS-KACRNYLSVISVLAYIITKISLIIFAGALVF-EVLGIP-------FWTGA 158 Rho.marinus ------------------------------------------------------------
Rob.biformata IATVVLTGIYTVLGGMRAVVYTETLQAIILVLGAAALTVIGLDRVGGWESMRETVSPEYL 216 Fla.bacterium IGIVLFTGAYTVIGGMKAVIYTETLQTVVLIFGSVTITFLGLREVGGWDMLKETVGTEHF 219 Bac.thetaiot. IGLVVLTALYTIFGGMKSVLYTSVLQTPILLLGSLIILVLGFKELGGWDEMMRVCGAVTV 222 Psy.torquis IITVVATGFYTVLGGLKAVIYTDMAQAFILILGTMGVVFFGLYELGGWNEMIRTIDMAAN 218 Rho.marinus ------------------------------------------------------------ Rob.biformata N-----------MWRSATDPDFPWPWLIISSTIVGIWYWCTDQYIVQRALTAKNIKEGRR 265 Fla.bacterium N-----------MWRPMTDPDFPWTGMLFGGTIVGLWYWCTDQYIVQRTLAANNIKIGRR 268 Bac.thetaiot. NDYG---DTMTNLIRSNDDANFPWLGALIGSAIIGFWYWCTDQFIVQRVLSGKNEKEARR 279 Psy.torquis TSGNPPVDQFFNLWRSTDDTEYPWTGMLFGAPILGVWYWCTDQFIVQRALSAKDVSNARK 278 Rho.marinus ---------------------------MISSPIVGIWYWCTDQYIVQRTLAARSLKDARR 33 * * ******* **** * *
Rob.biformata GTIFGALLKLMPVFLFLIPGVIALTLKMRGELHWD-------------NPDEAFPVLMSN 312 Fla.bacterium GAIFGAYLKILPIFIFLIPGIIAFALAKQGVLSYD-------------KADEVFPVLVKT 315 Bac.thetaiot. GTIFGAYLKLLPVFLFLIPGMIAFALHQKYIGAGGEGFLPMLA-NGTANADAAFPTLVAK 338 Psy.torquis GALFAGYLKLLPVFLFFLPGVIAYALMQKGMISFS-----------MENADQALPTMITE 327 Rho.marinus GALWGGFLKVWPVFIFLVPGMIGYALHQKGLIHIPTDATG------KIQGDMVFPTLVAE 87 * ** * * * ** * * * * Rob.biformata LLPSGLRGLVAAGLLAALMSSLASVFNSCSTLFTVDIYKKLRPGTPEKKLVRTGQIATAI 372 Fla.bacterium LLPVGLKGLVAGGLMAALMSSLASVFNSCSTIFTIDIYKKLKPNTPERLLVNIGKFATTI 375 Bac.thetaiot. LLPAGVKGLVVCGILAALMSSLASLFNSSAMLFTIDFYKRFRPETPEKKLVGIGQIATVV 398 Psy.torquis FLPTGLKGLAIAGLLAALMSSLSSAFNSSSTLLTIDFYQKFKPEASEKDLVRFGRIATIV 387 Rho.marinus LLPLGLRGLVVGGLLSALMSSLSSLFNSCATLFTVDIYEKLRPGRPEPELVRVGRIATAV 147 ** * ** * ****** * *** * * * * * ** * **

III. Ergebnisse 99
Rob.biformata VVVIGIIWIPIMANISG---VLYEYLQSVQSYIAPPIAAVFLLGIFMKRVNAQGAFVTLI 429 Fla.bacterium VVILGIIWIPIMEKIGGG--TLYQYLQSVQSYIAPPIVAVFLLGILWKRVNSKAAIVTLF 433 Bac.thetaiot. IVILGILWIPIMRSVGD---VLYTYLQDVQSVLAPGIAAAFLLGICWKRTSAQGGMWGLI 455 Psy.torquis LVLLSLGWIPFMNALMGG--GIFHYLQSIQAYIAPPIAAVFLLGLFYKWINARAAIVTLW 445 Rho.marinus VVGLGLLWIPIMKVMAESKAGLYDYLQNVQGFLAPPITAVFLLGLASRRINAKGAVWGLG 207 * *** * *** * ** * * **** *
Rob.biformata VGFLGGALRISLELAKD----HLEPGSWLYTLGAVNYLIFAAWFFLFCIVVLVGVSLMTP 485 Fla.bacterium SGLIVATLRILAEIYKD----SLS--GVLLEFATINFAHMAIFMFILSVMICISISLATD 487 Bac.thetaiot. AGMIIGLTRLGAKVYYSN-AGEVADSTFKYLFYDMNWLFFCGWMFLFCIIVVIVVSLATE 514 Psy.torquis TGFVIGMFRLVAEFMVNEGSLLLTDDSALSYFLDINFLHFALFLFLFCSAELMIISKLGT 505 Rho.marinus VGFVLGLLKLTLQSMVQN--GALDPNTLLGAIGAFNGYYFSGLLFFVSVAIVVGVSLLTE 265 * * * * Rob.biformata APEPEKIANLTYATITREE----------KERNKASYNWKDIVVSVGILVIVIGIMIWFN 535 Fla.bacterium PPNYAAIAGLSFGTLSDED----------KRINKQSIAAIDIVLSLVLVAIVIFTLSYFV 537 Bac.thetaiot. APTAEKIQGLVFGTATKEQ----------KAATRASWDHWDIIHTVIILAITGAFYWYFW 564 Psy.torquis PQSLLLLEAVTFQKKKRTS----------FKWST------DVILTVVLIVLVLIIWFIFS 549 Rho.marinus APPEEKVRGLTFGTVSASD----------RSESRKTWDWRDVTLTVVVLGLVLAIYLYFS 315 * * Rob.biformata GK---- 537 Fla.bacterium G----- 538 Bac.thetaiot. ------ Psy.torquis PLGIGG 555 Rho.marinus FWV--- 318 Abb. 18: Sequenzvergleich von Natrium/Glucose-Symportern
Dargestellt ist ein Vergleich der abgeleiteten Aminosäuresequenz von sssfRM mit Natrium/Glucose-Symportern
unterschiedlicher Organismen. Die zugehörigen Acc.Nr. der NCBI-Homepage sind in Klammern angegeben:
Rob.biformata: Robiginitalea biformata (Acc.Nr.: ZP_01120606), Fla.bacterium: Flavobacteriales bacterium
HTCC2170 (Acc.Nr.: ZP_01105302), Bac.thetaiot.: Bacteroides thetaiotaomicron (Acc.Nr.: AA075462),
Psy.torquis: Psychroflexus torquis (Acc.Nr.: ZP_01254720), Rho.marinus: Rhodothermus marinus. Identische
Aminosäuren (mindestens drei) sind schwarz unterlegt. Konservative Aminosäureaustausche sind grau unterlegt.
Als konservativ wurden nach Needleman & Wunsch (1970) folgende Austausche definiert: G-A, I-L-V-M, S-T, R-K-H, N-Q, F-Y-W. Identische Aminosäuren sind in allen Sequenzen zusätzlich mit einem Sternchen (*) markiert.
Die Sequenzvergleiche wurden mit Hilfe des Programms ClustalW (Thompson et al., 1994) durchgeführt.
Bekannte Sequenzmotive von SSSF-Proteinen sind rot markiert. Die Motive 1 & 2 sind in der R. marinus Sequenz
nicht vorhanden.
Die heterologe PCR mit degenerierten Primern, welche mit Hilfe bekannter Sequenzen von
Trehalose/Maltose-Transportern abgeleitet worden waren, führte zur Identifizierung eines
hypothetischen R. marinus Gens, dessen abgeleitete Aminosäuresequenz Ähnlichkeiten zu
Natrium/Glucose-Symportern (Mitglied der SSSF-Transporter) aufweist. Ziel der vorliegen-
den Arbeit war jedoch, einen spezifischen Mannosylglycerat-Transporter zu lokalisieren, um
durch seine Ausschaltung einen Mannosylglycerat überproduzierenden R. marinus Stamm
zu etablieren. Diesem Ziel konnte sich nach eigenen Einschätzungen mit der Isolierung eines
möglichen Natrium/Glucose-Symporters nicht angenähert werden, auch wenn es sich bei
den Mitgliedern dieser Familie häufig um unspezifische Transportsysteme mit einem weiten
Substratspektrum handelt (Saier Jr, 2000). Eine physiologische Charakterisierung des hypo-
thetischen Natrium/Glucose-Symporters wurde nicht vorgenommen, da hierin kein Fortkom-
men für die Etablierung eines Mannosylglycerat überproduzierenden R. marinus Stamms
gesehen wurde.

III. Ergebnisse 100
2.5 Differential Display RT-PCR an Gesamt-RNA R. marinus Da mit der heterologen PCR keine potentieller Mannosylglycerat-Transporter identifiziert
werden konnte, wurde eine weitere molekularbiologische Methode, die Differential Display
RT-PCR, angewendet. Mit dieser Methode können bekannte, aber auch unbekannte Gene
untersucht werden, welche in sich unterscheidenden Zellpopulationen differentiell exprimiert
werden. So ist auch eine Identifizierung unbekannter Gene möglich.
Bei der Versuchsplanung wurde davon ausgegangen, dass ein Mannosylglycerat-Transpor-
ter bei einer erhöhten Mediensalinität verstärkt exprimiert wird, da unter diesen Bedingungen
höhere Konzentrationen Mannosylglycerat durch R. marinus synthetisiert werden und so
auch vermehrt Mannosylglycerat aus der Zelle „heraussickert“ (Abschnitt IV, 5.). Der Aus-
strom des kompatiblen Soluts Ectoin wurde anhand der Ausscheidermutante H. elongata
KB2 gezeigt (Volke, 2001). Im Falle einer differentiellen Expression eines Transporters sollte
auch ein differentielles Bandenmuster in der Differential Display RT-PCR erkennbar sein.
Für die DDRT-PCR wurde Gesamt-RNA aus Zellen von R. marinus isoliert, die zuvor in
M162-Medium mit 2 % und 6 % NaCl kultiviert worden waren (Abschnitt II, 11.1). Die cDNA-
Synthese erfolgte mit den drei Primern Poly A-C-Stop, Poly A-G-Stop und Poly A-T-Stop
(Abschnitt II, 9.3, Tab. 7). Nach Anhängen des A-Überhanges („A-tail“) wurde eine einseitige,
„asymmetrische“ PCR mit dem Poly-T-Primer (AP-Primer, gegen „A-tail“ gerichtet) durchge-
führt (Ta= 59 °C, 1,5 mM MgCl2). Damit sollten die Anzahl der vorliegenden unterrepräsen-
tierten cDNA-Produkte vermehrt werden. Anschließend wurden die PCR-Produkte gereinigt
(Qiaquick PCR-Purification) und als Template in der DDRT-PCR eingesetzt. Die eigentliche
DDRT-PCR wurde mit dem AUAP-Primer (gegen AP-Primer gerichtet) und einem der Primer
Oligo-A-dC, Oligo-A-dG bzw. Oligo-A-dT durchgeführt (Ta= 58 °C, 1,5 mM MgCl2). Dabei
ergab sich bei Vergleich beider Salinitäten (2 % und 6 % NaCl) für alle Primerkombinationen
ein ähnliches differentielles Bandenmuster (Abb. 19).
Die PCR-Produkte, die mit den Primern AUAP und Oligo-A-dT amplifiziert worden waren,
wurden als erste für eine Analyse herangezogen. Nach der Reinigung der ausgewählten
DDRT-PCR Produkte (700 bp: dd2; 920 bp: dd3; 450 bp: dd1) in einem Agarosegel wurden
diese in den T-Überhang Vektor pGEMT Easy kloniert. Der Vektor pGEMdd2 wurde als erster
mittels des T7-Promotor-Primers ansequenziert. Die exakte Sequenzlänge des Produkts dd2
lag bei 619 bp. Analysen mit den Programmen ORF-Finder (NCBI-Homepage) und Blast
(Proteinsearch, Altschul et al., 1990) ergaben keine ähnlichen Sequenzen. Daher wurde eine
weitere Sequenzierung unter Anwendung der inversen-PCR durchgeführt (Abschnitt II, 9.2.2,
Abb. 20). Dazu wurde genomische DNA von R. marinus mittels Pst I hydrolysiert und mit sich
selbst religiert. Die Amplifikation des gesuchten Ligationsprodukts erfolgte mit den Primern
dd2for2 und dd2rev2. Das Produkt der inversen-PCR enthält die Pst I-Schnittstelle, an der
die Ligation stattgefunden hat, und weist eine Länge von ca. 3,3 kb auf. Es wurde mit den
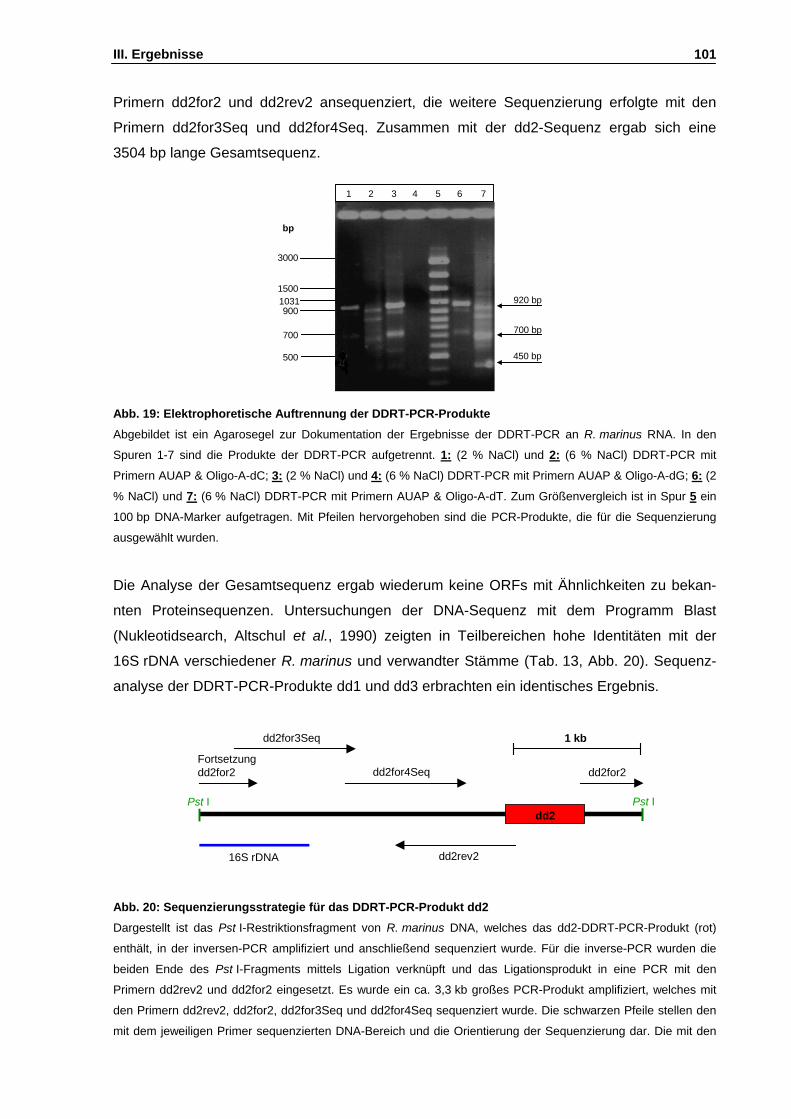
III. Ergebnisse 101
Primern dd2for2 und dd2rev2 ansequenziert, die weitere Sequenzierung erfolgte mit den
Primern dd2for3Seq und dd2for4Seq. Zusammen mit der dd2-Sequenz ergab sich eine
3504 bp lange Gesamtsequenz.
920 bp
700 bp
450 bp
3000
15001031900
700
500
bp
1 2 3 4 5 6 7
Abb. 19: Elektrophoretische Auftrennung der DDRT-PCR-Produkte Abgebildet ist ein Agarosegel zur Dokumentation der Ergebnisse der DDRT-PCR an R. marinus RNA. In den
Spuren 1-7 sind die Produkte der DDRT-PCR aufgetrennt. 1: (2 % NaCl) und 2: (6 % NaCl) DDRT-PCR mit
Primern AUAP & Oligo-A-dC; 3: (2 % NaCl) und 4: (6 % NaCl) DDRT-PCR mit Primern AUAP & Oligo-A-dG; 6: (2
% NaCl) und 7: (6 % NaCl) DDRT-PCR mit Primern AUAP & Oligo-A-dT. Zum Größenvergleich ist in Spur 5 ein
100 bp DNA-Marker aufgetragen. Mit Pfeilen hervorgehoben sind die PCR-Produkte, die für die Sequenzierung
ausgewählt wurden.
Die Analyse der Gesamtsequenz ergab wiederum keine ORFs mit Ähnlichkeiten zu bekan-
nten Proteinsequenzen. Untersuchungen der DNA-Sequenz mit dem Programm Blast
(Nukleotidsearch, Altschul et al., 1990) zeigten in Teilbereichen hohe Identitäten mit der
16S rDNA verschiedener R. marinus und verwandter Stämme (Tab. 13, Abb. 20). Sequenz-
analyse der DDRT-PCR-Produkte dd1 und dd3 erbrachten ein identisches Ergebnis.
Pst IPst I
dd2for3Seq
dd2for4Seq
dd2rev2
Fortsetzungdd2for2 dd2for2
dd2
1 kb
16S rDNA
Abb. 20: Sequenzierungsstrategie für das DDRT-PCR-Produkt dd2 Dargestellt ist das Pst I-Restriktionsfragment von R. marinus DNA, welches das dd2-DDRT-PCR-Produkt (rot)
enthält, in der inversen-PCR amplifiziert und anschließend sequenziert wurde. Für die inverse-PCR wurden die
beiden Ende des Pst I-Fragments mittels Ligation verknüpft und das Ligationsprodukt in eine PCR mit den
Primern dd2rev2 und dd2for2 eingesetzt. Es wurde ein ca. 3,3 kb großes PCR-Produkt amplifiziert, welches mit
den Primern dd2rev2, dd2for2, dd2for3Seq und dd2for4Seq sequenziert wurde. Die schwarzen Pfeile stellen den
mit dem jeweiligen Primer sequenzierten DNA-Bereich und die Orientierung der Sequenzierung dar. Die mit den
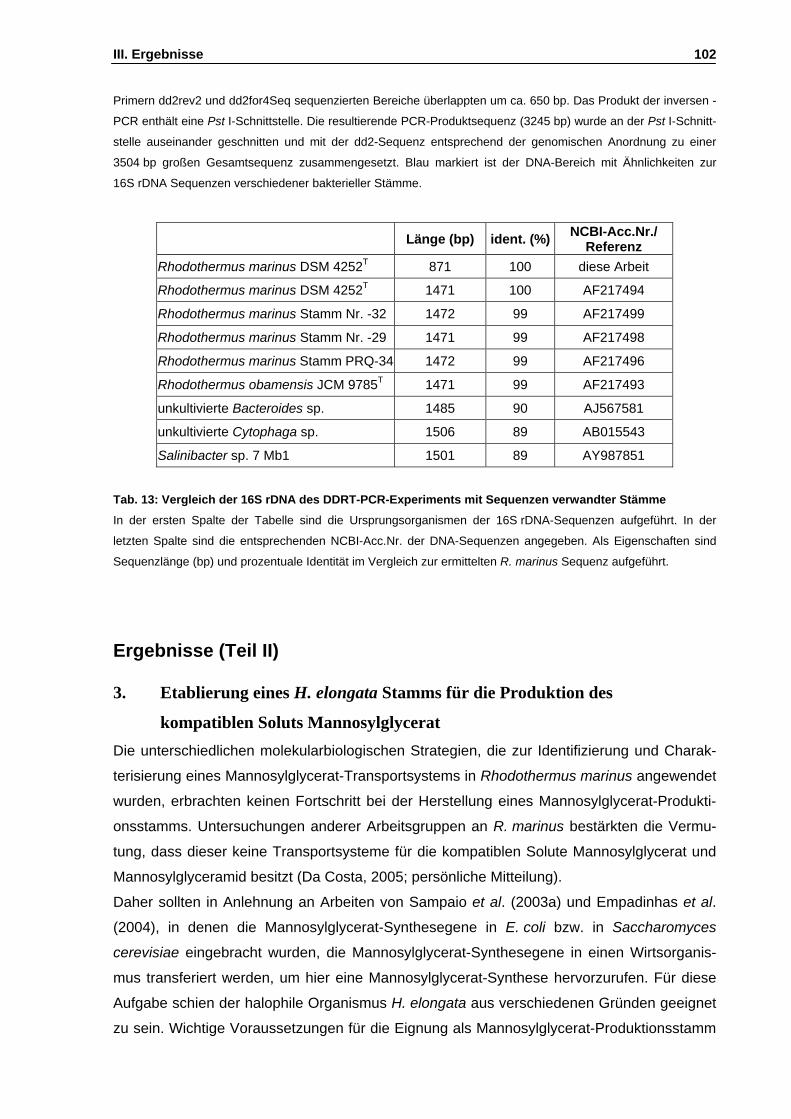
III. Ergebnisse 102
Primern dd2rev2 und dd2for4Seq sequenzierten Bereiche überlappten um ca. 650 bp. Das Produkt der inversen -
PCR enthält eine Pst I-Schnittstelle. Die resultierende PCR-Produktsequenz (3245 bp) wurde an der Pst I-Schnitt-
stelle auseinander geschnitten und mit der dd2-Sequenz entsprechend der genomischen Anordnung zu einer
3504 bp großen Gesamtsequenz zusammengesetzt. Blau markiert ist der DNA-Bereich mit Ähnlichkeiten zur
16S rDNA Sequenzen verschiedener bakterieller Stämme.
Länge (bp) ident. (%) NCBI-Acc.Nr./ Referenz
Rhodothermus marinus DSM 4252T 871 100 diese Arbeit
Rhodothermus marinus DSM 4252T 1471 100 AF217494
Rhodothermus marinus Stamm Nr. -32 1472 99 AF217499
Rhodothermus marinus Stamm Nr. -29 1471 99 AF217498
Rhodothermus marinus Stamm PRQ-34 1472 99 AF217496
Rhodothermus obamensis JCM 9785T 1471 99 AF217493
unkultivierte Bacteroides sp. 1485 90 AJ567581
unkultivierte Cytophaga sp. 1506 89 AB015543
Salinibacter sp. 7 Mb1 1501 89 AY987851 Tab. 13: Vergleich der 16S rDNA des DDRT-PCR-Experiments mit Sequenzen verwandter Stämme In der ersten Spalte der Tabelle sind die Ursprungsorganismen der 16S rDNA-Sequenzen aufgeführt. In der
letzten Spalte sind die entsprechenden NCBI-Acc.Nr. der DNA-Sequenzen angegeben. Als Eigenschaften sind
Sequenzlänge (bp) und prozentuale Identität im Vergleich zur ermittelten R. marinus Sequenz aufgeführt.
Ergebnisse (Teil II)
3. Etablierung eines H. elongata Stamms für die Produktion des
kompatiblen Soluts Mannosylglycerat Die unterschiedlichen molekularbiologischen Strategien, die zur Identifizierung und Charak-
terisierung eines Mannosylglycerat-Transportsystems in Rhodothermus marinus angewendet
wurden, erbrachten keinen Fortschritt bei der Herstellung eines Mannosylglycerat-Produkti-
onsstamms. Untersuchungen anderer Arbeitsgruppen an R. marinus bestärkten die Vermu-
tung, dass dieser keine Transportsysteme für die kompatiblen Solute Mannosylglycerat und
Mannosylglyceramid besitzt (Da Costa, 2005; persönliche Mitteilung).
Daher sollten in Anlehnung an Arbeiten von Sampaio et al. (2003a) und Empadinhas et al.
(2004), in denen die Mannosylglycerat-Synthesegene in E. coli bzw. in Saccharomyces
cerevisiae eingebracht wurden, die Mannosylglycerat-Synthesegene in einen Wirtsorganis-
mus transferiert werden, um hier eine Mannosylglycerat-Synthese hervorzurufen. Für diese
Aufgabe schien der halophile Organismus H. elongata aus verschiedenen Gründen geeignet
zu sein. Wichtige Voraussetzungen für die Eignung als Mannosylglycerat-Produktionsstamm

III. Ergebnisse 103
sind z.B. das Vermögen zur intrazellulären Akkumulation organischer, niedermolekularer
Substanzen unter osmotischen Stressbedingungen, die generelle Fähigkeit des Stamms
betreffende Fremdgene zu exprimieren, das Vorhandensein gentechnischer Methoden und
bestehende Anzuchtmethoden für die großtechnische Produktion (siehe Abschnitt IV, 7.). All
diese Bedingungen scheint neben dem üblicherweise verwendeten E. coli auch H. elongata
zu erfüllen. Einen wichtigen Vorteil gegenüber E. coli bietet jedoch die „Codon usage“ von
H. elongata, die stark derjenigen von R. marinus ähnelt (Abschnitt IV, 7., Abb. 49). Sollte
H. elongata darüber hinaus keinen Mannosylglycerat-Transporter besitzen, wäre mit einem
Verlust des synthetisierten Soluts in das umgebende Medium zu rechnen, wie es bei der
Ectoin-Ausscheidermutante H. elongata KB2 beobachtet wurde (Volke, 2001).
3.1 Wachstum von H. elongata KB1 und KB18.1 unter Zusatz von Mannosyl-
glycerat Um Hinweise auf das Vorhandensein von Mannosylglycerat-Transportern zu erhalten,
wurden Voruntersuchungen mit den Stämmen H. elongata KB1 (∆ectA, Grammann, 2000)
und KB18.1 (∆ectA, ∆betG, ∆betH, ∆teaC, ∆proX, ∆opuAB, Kraegeloh, 2003) durchgeführt.
KB1 und KB18.1 sind durch Deletion des ectA-Gens (kodiert für Diaminobuttersäure-Acetyl-
transferase) nicht in der Lage Ectoin zu synthetisieren und sind so in Mineralsalzmedium mit
einer Mediensalinität von ≥ 3 % NaCl ohne zugesetzte kompatible Solute nur zu einem
verlangsamten Wachstum fähig. Außerdem sind bei KB18.1 alle bisher bei H. elongata
bekannten Solutetransportsysteme ausgeschaltet. Bei Anzucht in einem Mannosylglycerat
haltigem Mineralsalzmedium würde eine salinitätsabhängige intrazelluläre Akkumulation von
Mannosylglycerat durch H. elongata das Vorhandensein eines geeigneten Transporters
anzeigen. Gleichzeitig könnte eine Steigerung der Wachstumsrate von KB1 und KB18.1
durch Zusatz von Mannosylglycerat dessen Eignung als kompatibles Solut auch für
H. elongata bestätigen.
Zur Charakterisierung des Wachstumsverhaltens wurden KB1 und KB18.1 bei 30 °C in
MM63-Medium mit 3 % NaCl und Glucose als Kohlenstoffquelle angezogen. Den Kulturen
wurde je 1 mM Mannosylglycerat zugesetzt. Ein Vergleich der Wachstumsraten mit und ohne
Mannosylglycerat zeigte, dass sowohl KB1 als auch KB18.1 mit Mannosylglycerat erkennbar
höhere Wachstumsraten aufweisen. Bei KB1 betrug die Erhöhung der Wachstumsrate ca.
23 %, bei KB18.1 ca. 18 % (Abb. 21).
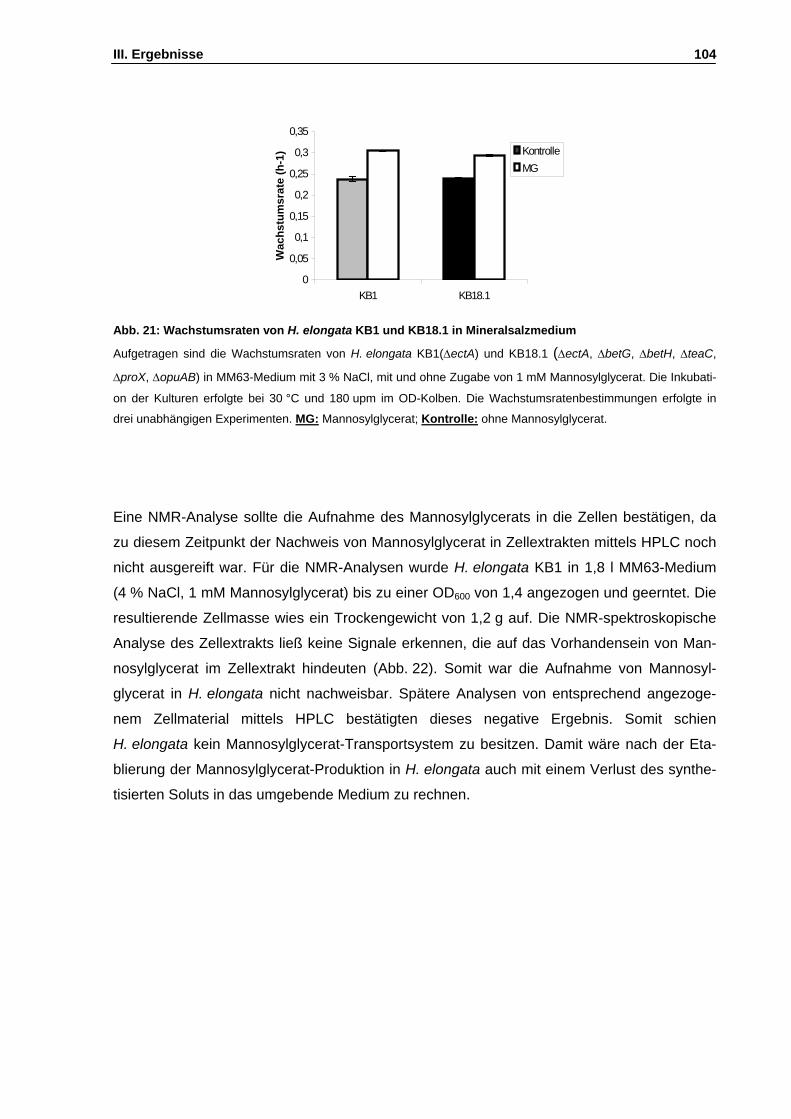
III. Ergebnisse 104
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
KB1 KB18.1W
achs
tum
srat
e (h
-1) Kontrolle
MG
Abb. 21: Wachstumsraten von H. elongata KB1 und KB18.1 in Mineralsalzmedium
Aufgetragen sind die Wachstumsraten von H. elongata KB1(∆ectA) und KB18.1 (∆ectA, ∆betG, ∆betH, ∆teaC,
∆proX, ∆opuAB) in MM63-Medium mit 3 % NaCl, mit und ohne Zugabe von 1 mM Mannosylglycerat. Die Inkubati-
on der Kulturen erfolgte bei 30 °C und 180 upm im OD-Kolben. Die Wachstumsratenbestimmungen erfolgte in
drei unabhängigen Experimenten. MG: Mannosylglycerat; Kontrolle: ohne Mannosylglycerat.
Eine NMR-Analyse sollte die Aufnahme des Mannosylglycerats in die Zellen bestätigen, da
zu diesem Zeitpunkt der Nachweis von Mannosylglycerat in Zellextrakten mittels HPLC noch
nicht ausgereift war. Für die NMR-Analysen wurde H. elongata KB1 in 1,8 l MM63-Medium
(4 % NaCl, 1 mM Mannosylglycerat) bis zu einer OD600 von 1,4 angezogen und geerntet. Die
resultierende Zellmasse wies ein Trockengewicht von 1,2 g auf. Die NMR-spektroskopische
Analyse des Zellextrakts ließ keine Signale erkennen, die auf das Vorhandensein von Man-
nosylglycerat im Zellextrakt hindeuten (Abb. 22). Somit war die Aufnahme von Mannosyl-
glycerat in H. elongata nicht nachweisbar. Spätere Analysen von entsprechend angezoge-
nem Zellmaterial mittels HPLC bestätigten dieses negative Ergebnis. Somit schien
H. elongata kein Mannosylglycerat-Transportsystem zu besitzen. Damit wäre nach der Eta-
blierung der Mannosylglycerat-Produktion in H. elongata auch mit einem Verlust des synthe-
tisierten Soluts in das umgebende Medium zu rechnen.
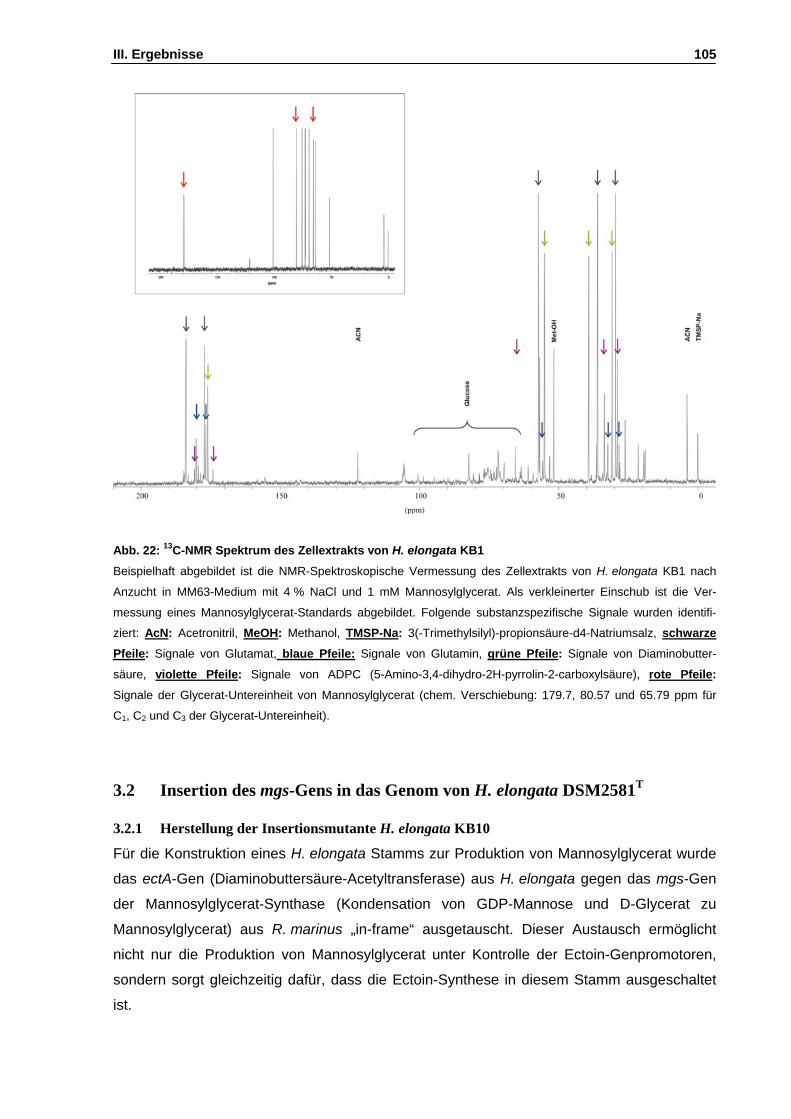
III. Ergebnisse 105
Abb. 22: 13C-NMR Spektrum des Zellextrakts von H. elongata KB1
Beispielhaft abgebildet ist die NMR-Spektroskopische Vermessung des Zellextrakts von H. elongata KB1 nach
Anzucht in MM63-Medium mit 4 % NaCl und 1 mM Mannosylglycerat. Als verkleinerter Einschub ist die Ver-
messung eines Mannosylglycerat-Standards abgebildet. Folgende substanzspezifische Signale wurden identifi-
ziert: AcN: Acetronitril, MeOH: Methanol, TMSP-Na: 3(-Trimethylsilyl)-propionsäure-d4-Natriumsalz, schwarze Pfeile: Signale von Glutamat, blaue Pfeile: Signale von Glutamin, grüne Pfeile: Signale von Diaminobutter-
säure, violette Pfeile: Signale von ADPC (5-Amino-3,4-dihydro-2H-pyrrolin-2-carboxylsäure), rote Pfeile:
Signale der Glycerat-Untereinheit von Mannosylglycerat (chem. Verschiebung: 179.7, 80.57 und 65.79 ppm für
C1, C2 und C3 der Glycerat-Untereinheit).
3.2 Insertion des mgs-Gens in das Genom von H. elongata DSM2581T
3.2.1 Herstellung der Insertionsmutante H. elongata KB10
Für die Konstruktion eines H. elongata Stamms zur Produktion von Mannosylglycerat wurde
das ectA-Gen (Diaminobuttersäure-Acetyltransferase) aus H. elongata gegen das mgs-Gen
der Mannosylglycerat-Synthase (Kondensation von GDP-Mannose und D-Glycerat zu
Mannosylglycerat) aus R. marinus „in-frame“ ausgetauscht. Dieser Austausch ermöglicht
nicht nur die Produktion von Mannosylglycerat unter Kontrolle der Ectoin-Genpromotoren,
sondern sorgt gleichzeitig dafür, dass die Ectoin-Synthese in diesem Stamm ausgeschaltet
ist.
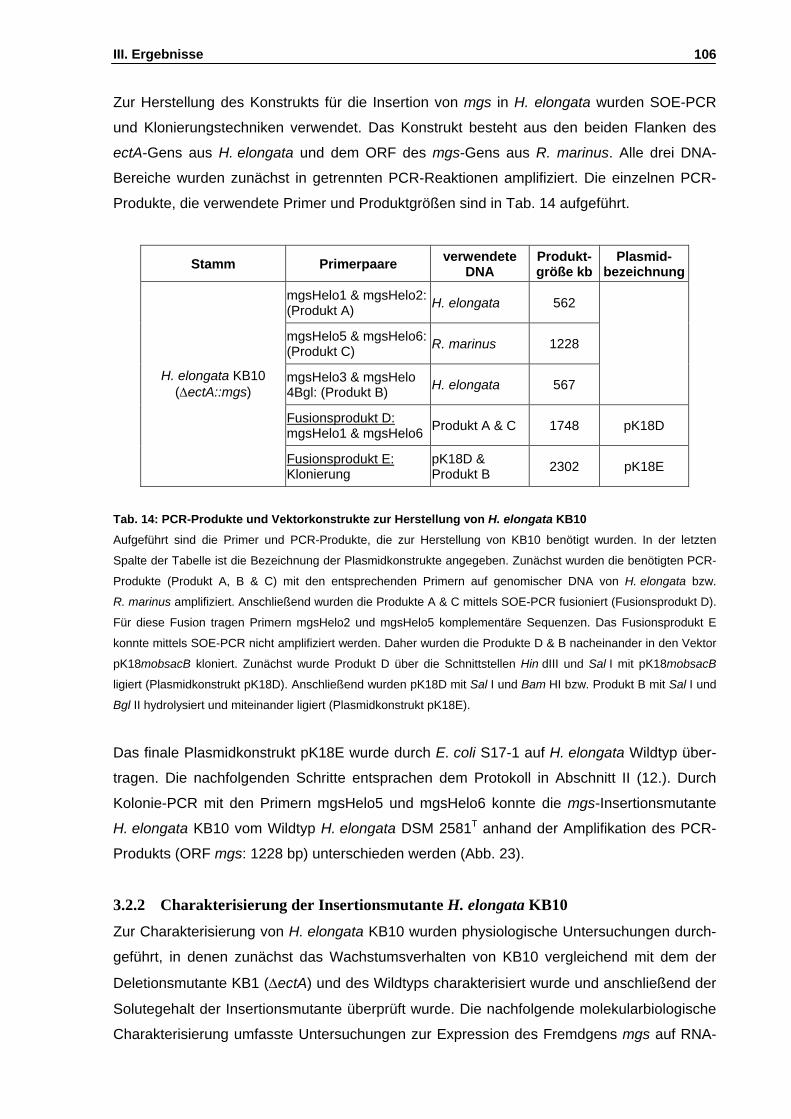
III. Ergebnisse 106
Zur Herstellung des Konstrukts für die Insertion von mgs in H. elongata wurden SOE-PCR
und Klonierungstechniken verwendet. Das Konstrukt besteht aus den beiden Flanken des
ectA-Gens aus H. elongata und dem ORF des mgs-Gens aus R. marinus. Alle drei DNA-
Bereiche wurden zunächst in getrennten PCR-Reaktionen amplifiziert. Die einzelnen PCR-
Produkte, die verwendete Primer und Produktgrößen sind in Tab. 14 aufgeführt.
Stamm Primerpaare verwendete DNA
Produkt- größe kb
Plasmid-bezeichnung
mgsHelo1 & mgsHelo2: (Produkt A) H. elongata 562
mgsHelo5 & mgsHelo6: (Produkt C) R. marinus 1228
mgsHelo3 & mgsHelo 4Bgl: (Produkt B) H. elongata 567
Fusionsprodukt D: mgsHelo1 & mgsHelo6 Produkt A & C 1748 pK18D
H. elongata KB10 (∆ectA::mgs)
Fusionsprodukt E: Klonierung
pK18D & Produkt B 2302 pK18E
Tab. 14: PCR-Produkte und Vektorkonstrukte zur Herstellung von H. elongata KB10 Aufgeführt sind die Primer und PCR-Produkte, die zur Herstellung von KB10 benötigt wurden. In der letzten
Spalte der Tabelle ist die Bezeichnung der Plasmidkonstrukte angegeben. Zunächst wurden die benötigten PCR-
Produkte (Produkt A, B & C) mit den entsprechenden Primern auf genomischer DNA von H. elongata bzw.
R. marinus amplifiziert. Anschließend wurden die Produkte A & C mittels SOE-PCR fusioniert (Fusionsprodukt D).
Für diese Fusion tragen Primern mgsHelo2 und mgsHelo5 komplementäre Sequenzen. Das Fusionsprodukt E
konnte mittels SOE-PCR nicht amplifiziert werden. Daher wurden die Produkte D & B nacheinander in den Vektor
pK18mobsacB kloniert. Zunächst wurde Produkt D über die Schnittstellen Hin dIII und Sal I mit pK18mobsacB
ligiert (Plasmidkonstrukt pK18D). Anschließend wurden pK18D mit Sal I und Bam HI bzw. Produkt B mit Sal I und
Bgl II hydrolysiert und miteinander ligiert (Plasmidkonstrukt pK18E).
Das finale Plasmidkonstrukt pK18E wurde durch E. coli S17-1 auf H. elongata Wildtyp über-
tragen. Die nachfolgenden Schritte entsprachen dem Protokoll in Abschnitt II (12.). Durch
Kolonie-PCR mit den Primern mgsHelo5 und mgsHelo6 konnte die mgs-Insertionsmutante
H. elongata KB10 vom Wildtyp H. elongata DSM 2581T anhand der Amplifikation des PCR-
Produkts (ORF mgs: 1228 bp) unterschieden werden (Abb. 23).
3.2.2 Charakterisierung der Insertionsmutante H. elongata KB10
Zur Charakterisierung von H. elongata KB10 wurden physiologische Untersuchungen durch-
geführt, in denen zunächst das Wachstumsverhalten von KB10 vergleichend mit dem der
Deletionsmutante KB1 (∆ectA) und des Wildtyps charakterisiert wurde und anschließend der
Solutegehalt der Insertionsmutante überprüft wurde. Die nachfolgende molekularbiologische
Charakterisierung umfasste Untersuchungen zur Expression des Fremdgens mgs auf RNA-

III. Ergebnisse 107
Ebene. Dazu gehören Northern-Hybridisierungs- und RT-PCR-Experimente, sowie die Identi-
fizierung der Transkriptionsstartpunkte stromaufwärts des inserierten mgs-Gens in
H. elongata KB10 mittels RACE-PCR.
H. elongata KB10(∆ectA::mgs)
ectB ectC
5‘ ATG
mgs
Sma I Sal I
ectRP1-P3
Sac I
H. elongataDSM 2581T ectA
Xho I5‘ ATG
ectB ectC
Sac I
ectRP1-P3
R. marinusDSM 4252T orfR orfD lig
Sma I Sal I
mgs
5‘ ATG
1 kb
Abb. 23: Genetische Organisation der mgs-Region von R. marinus DSM 4252T und H. elongata KB10 Dargestellt ist die genetische Organisation der mgs-Region von R. marinus und der Insertionsmutante
H. elongata KB10 im Vergleich zum H. elongata Wildtyp. Die eingezeichneten Gene kodieren für folgende
Produkte: mgs: Mannosylglycerat-Synthase aus R. marinus (Acc.Nr.: AF173987), orfR & orfD: hypothetische
Gene, lig: hypothetische DNA-Ligase, ectA: Diaminobuttersäure-Acetyltransferase, ectB: Diaminobuttersäure-
Transaminase, ectC: Ectoin-Synthase (EctABC katalysieren die Ectoin-Synthese in H. elongata, Acc. Nr.:
AF031489), ectR: potentieller Regulator der Ectoin-Synthese (Stumpfe, 2003; Schilz, 2005), symbolisiert
potentielle Promotorelemente der Ectoin-Gene (σ70 / σS & "gearbox", Göller, 1999; Stumpfe, 2003, diese Arbeit).
Die betreffende DNA-Sequenz von H. elongata KB10 findet sich im Anhang der Arbeit.
3.2.2.1 Physiologische Untersuchung von H. elongata KB10
Zur ersten Charakterisierung des Wachstums von H. elongata KB10 wurde dieser in Mineral-
salzmedium mit Glucose bei unterschiedlichen Mediensalinitäten angezogen. H. elongata
KB10 war wie die Deletionsmutante KB1 (∆ectA) lediglich bis zu einer Salzkonzentration von
maximal 5 % NaCl zu einem Wachstum fähig. Falls H. elongata KB10 zu einer Mannosyl-
glycerat-Synthese fähig wäre, deutete dies schon jetzt auf einen nur sehr geringen Gehalt
hin.
Die weiteren Untersuchungen des Wachstumsverhaltens von H. elongata KB10 wurden bei
einer Mediensalinität von 4 % NaCl durchgeführt (Abb. 24). Ziel war die Identifizierung einer
Kohlenstoffquelle, mit der KB10 zur Synthese von Mannosylglycerat fähig ist. Im Fall einer
erfolgten Mannosylglycerat-Synthese wurde mit einer deutlichen Verbesserung des Wachs-
tums von KB10 im Vergleich zu der ectA-Deletionsmutante KB1 (Negativkontrolle) gerech-
net. Sampaio et. al (2003a) hatten bei 37 °C in einem rekombinanten E. coli Stamm eine
geringe enzymatische Aktivität der Mannosylglycerat-Synthase nachweisen können. Daher
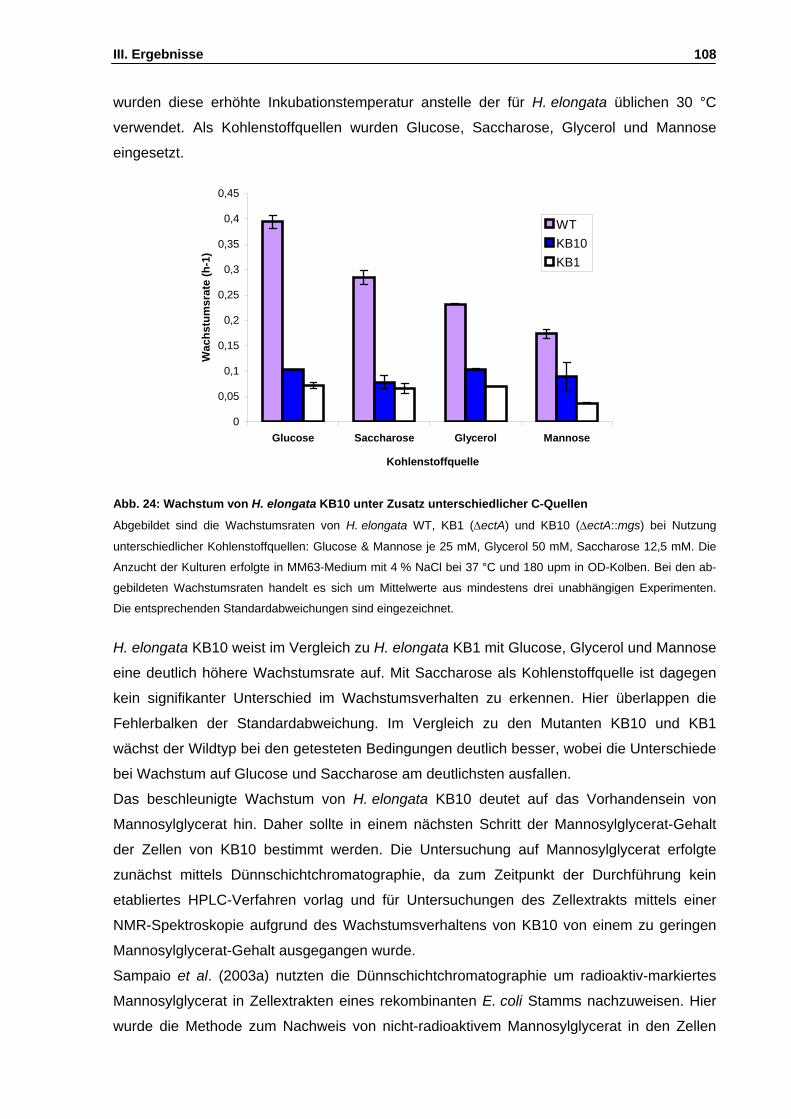
III. Ergebnisse 108
wurden diese erhöhte Inkubationstemperatur anstelle der für H. elongata üblichen 30 °C
verwendet. Als Kohlenstoffquellen wurden Glucose, Saccharose, Glycerol und Mannose
eingesetzt.
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
Glucose Saccharose Glycerol Mannose
Kohlenstoffquelle
Wac
hstu
msr
ate
(h-1
)
WTKB10KB1
Abb. 24: Wachstum von H. elongata KB10 unter Zusatz unterschiedlicher C-Quellen
Abgebildet sind die Wachstumsraten von H. elongata WT, KB1 (∆ectA) und KB10 (∆ectA::mgs) bei Nutzung
unterschiedlicher Kohlenstoffquellen: Glucose & Mannose je 25 mM, Glycerol 50 mM, Saccharose 12,5 mM. Die
Anzucht der Kulturen erfolgte in MM63-Medium mit 4 % NaCl bei 37 °C und 180 upm in OD-Kolben. Bei den ab-
gebildeten Wachstumsraten handelt es sich um Mittelwerte aus mindestens drei unabhängigen Experimenten.
Die entsprechenden Standardabweichungen sind eingezeichnet.
H. elongata KB10 weist im Vergleich zu H. elongata KB1 mit Glucose, Glycerol und Mannose
eine deutlich höhere Wachstumsrate auf. Mit Saccharose als Kohlenstoffquelle ist dagegen
kein signifikanter Unterschied im Wachstumsverhalten zu erkennen. Hier überlappen die
Fehlerbalken der Standardabweichung. Im Vergleich zu den Mutanten KB10 und KB1
wächst der Wildtyp bei den getesteten Bedingungen deutlich besser, wobei die Unterschiede
bei Wachstum auf Glucose und Saccharose am deutlichsten ausfallen.
Das beschleunigte Wachstum von H. elongata KB10 deutet auf das Vorhandensein von
Mannosylglycerat hin. Daher sollte in einem nächsten Schritt der Mannosylglycerat-Gehalt
der Zellen von KB10 bestimmt werden. Die Untersuchung auf Mannosylglycerat erfolgte
zunächst mittels Dünnschichtchromatographie, da zum Zeitpunkt der Durchführung kein
etabliertes HPLC-Verfahren vorlag und für Untersuchungen des Zellextrakts mittels einer
NMR-Spektroskopie aufgrund des Wachstumsverhaltens von KB10 von einem zu geringen
Mannosylglycerat-Gehalt ausgegangen wurde.
Sampaio et al. (2003a) nutzten die Dünnschichtchromatographie um radioaktiv-markiertes
Mannosylglycerat in Zellextrakten eines rekombinanten E. coli Stamms nachzuweisen. Hier
wurde die Methode zum Nachweis von nicht-radioaktivem Mannosylglycerat in den Zellen
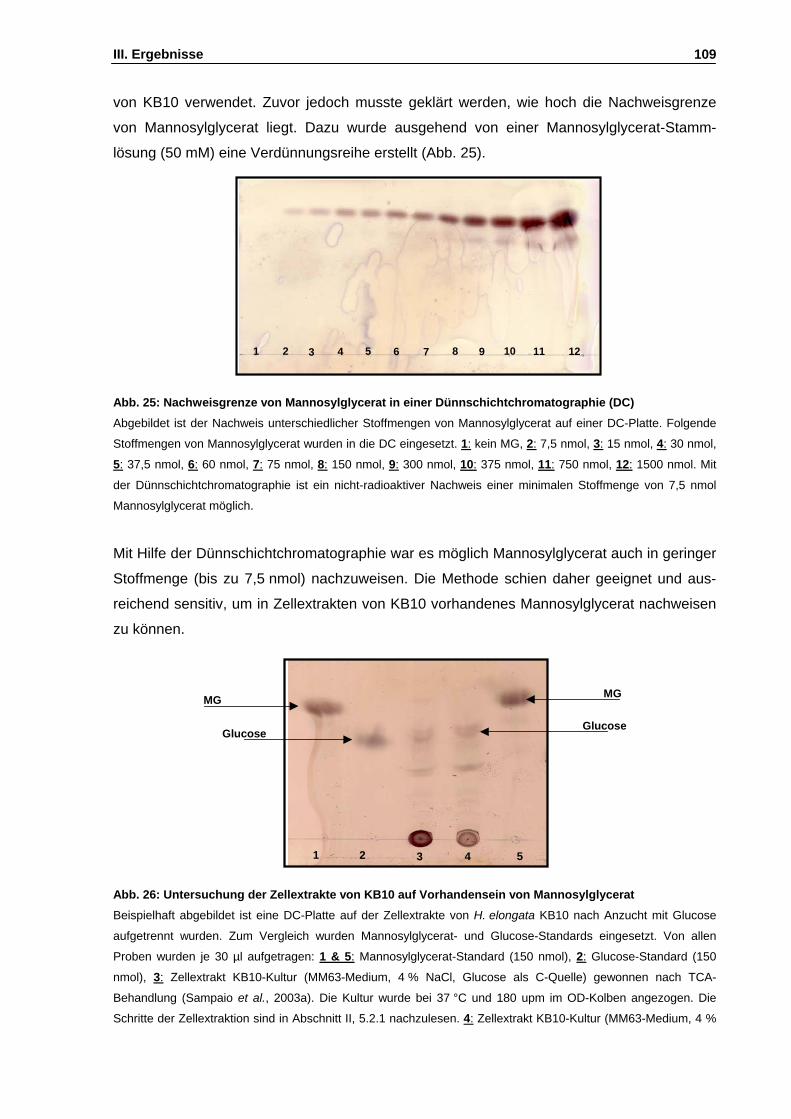
III. Ergebnisse 109
von KB10 verwendet. Zuvor jedoch musste geklärt werden, wie hoch die Nachweisgrenze
von Mannosylglycerat liegt. Dazu wurde ausgehend von einer Mannosylglycerat-Stamm-
lösung (50 mM) eine Verdünnungsreihe erstellt (Abb. 25).
1 2 3 4 5 6 87 9 10 11 12
Abb. 25: Nachweisgrenze von Mannosylglycerat in einer Dünnschichtchromatographie (DC) Abgebildet ist der Nachweis unterschiedlicher Stoffmengen von Mannosylglycerat auf einer DC-Platte. Folgende
Stoffmengen von Mannosylglycerat wurden in die DC eingesetzt. 1: kein MG, 2: 7,5 nmol, 3: 15 nmol, 4: 30 nmol,
5: 37,5 nmol, 6: 60 nmol, 7: 75 nmol, 8: 150 nmol, 9: 300 nmol, 10: 375 nmol, 11: 750 nmol, 12: 1500 nmol. Mit
der Dünnschichtchromatographie ist ein nicht-radioaktiver Nachweis einer minimalen Stoffmenge von 7,5 nmol
Mannosylglycerat möglich.
Mit Hilfe der Dünnschichtchromatographie war es möglich Mannosylglycerat auch in geringer
Stoffmenge (bis zu 7,5 nmol) nachzuweisen. Die Methode schien daher geeignet und aus-
reichend sensitiv, um in Zellextrakten von KB10 vorhandenes Mannosylglycerat nachweisen
zu können.
1 2 3 4 5
MG
Glucose
MG
Glucose
Abb. 26: Untersuchung der Zellextrakte von KB10 auf Vorhandensein von Mannosylglycerat Beispielhaft abgebildet ist eine DC-Platte auf der Zellextrakte von H. elongata KB10 nach Anzucht mit Glucose
aufgetrennt wurden. Zum Vergleich wurden Mannosylglycerat- und Glucose-Standards eingesetzt. Von allen
Proben wurden je 30 µl aufgetragen: 1 & 5: Mannosylglycerat-Standard (150 nmol), 2: Glucose-Standard (150
nmol), 3: Zellextrakt KB10-Kultur (MM63-Medium, 4 % NaCl, Glucose als C-Quelle) gewonnen nach TCA-
Behandlung (Sampaio et al., 2003a). Die Kultur wurde bei 37 °C und 180 upm im OD-Kolben angezogen. Die
Schritte der Zellextraktion sind in Abschnitt II, 5.2.1 nachzulesen. 4: Zellextrakt KB10-Kultur (MM63-Medium, 4 %

III. Ergebnisse 110
NaCl, Glucose als C-Quelle) gewonnen nach Bligh & Dyer (1959). Die Schritte der Zellextraktion sind in Abschnitt
II, 5.3.1 nachzulesen. Nach beiden Extraktionsmethoden ließ sich in H. elongata KB10 kein Mannosylglycerat
nachweisen.
Für den Mannosylglycerat-Nachweis wurden Zellen bzw. Zellextrakte von H. elongata KB10
wie in Abschnitt II, 5.2.1 & 5.3.1 beschrieben behandelt. Der nach TCA-Behandlung
(Sampaio et al., 2003a) bzw. nach Bligh & Dyer (1959) gewonnene Zellextrakt wurde auf
DC-Platten aufgetragen und für 2 Stunden mit n-Propanol und 25 % Ammoniak (1:1, v/v)
unter Kammersättigung aufgetrennt (Abb. 26). Weder nach der Extraktionsmethode von
Sampaio et al. (2003a) noch nach der Extraktionsmethode von Bligh & Dyer (1959) ließen
sich in H. elongata KB10 nach Anzucht mit Glucose, Mannose, Glycerol bzw. Saccharose
Spuren von Mannosylglycerat nachweisen. Die später durchgeführten HPLC-Analysen ent-
sprechender Zellextrakte von H. elongata KB10 bestätigten dieses Ergebnis.
3.2.2.2 Transkriptionsanalysen mittels Northern-Hybridisierung und RT-PCR
Nach dem Ausbleiben der Mannosylglycerat-Synthese in H. elongata KB10 wurde in einer
Northern-Hybridisierung und RT-PCR überprüft, ob eine Transkription des mgs-Gens erfolgt.
So sollte der Grund für das Ausbleiben der Mannosyglycerat-Synthese eingegrenzt werden.
Gleichzeitig wurde damit überprüft, ob mgs gemeinsam mit ectB und ectC transkribiert wird.
Wichtig wurden derartige Untersuchungen auch für die Integration weiterer Gene zur Eta-
blierung der Mannosylglycerat-Synthese.
In die Northern-Hybridisierungsexperimente wurde Gesamt-RNA von H. elongata Wildtyp,
H. elongata KB1 (∆ectA) und H. elongata KB10 (∆ectA::mgs) eingesetzt. Alle Stämme
wurden für die RNA-Isolierung in MM63-4-Medium kultiviert und in der exponentiellen
Wachstumsphase geerntet. Die RNA wurde nach gelelektrophoretischer Auftrennung durch
einen Northern-Transfer auf eine Nylonmembran übertragen und dort immobilisiert. Die
Hybridisierung erfolgte mit mgs- und ectB-spezifischen, DIG-markierten RNA-Sonden. Die
Sonden erstreckten sich über die Gesamtlänge des jeweiligen Gens.
Northern-Hybridisierung mit der mgs-Sonde:
In der Northern-Hybridisierung mit einer mgs-Sonde (1164 bp), die das Transkript der
Mannosylglycerat-Synthase nachweist, wurden RNA aus H. elongata Wildtyp, H. elongata
KB1 (∆ectA) (beide als Negativkontrolle) sowie die RNA aus H. elongata KB10 (∆ectA::mgs)
eingesetzt. Auf der Kontroll-RNA aus Wildtyp und KB1 konnte kein Signal nachgewiesen
werden, auf der RNA von KB10 wurden dagegen eine Vielzahl von Banden detektiert (Abb.
27a). Die erste Bande weist eine Länge von ca. 1,2 kb auf und entspricht in der Länge dem
mgs-Einzeltranskript. Die zweite Bande auf der RNA von H. elongata KB10 weist eine Frag-
mentgröße von etwa 2,5 kb auf, was der Länge eines mgsectB-Kotranskripts entsprechen
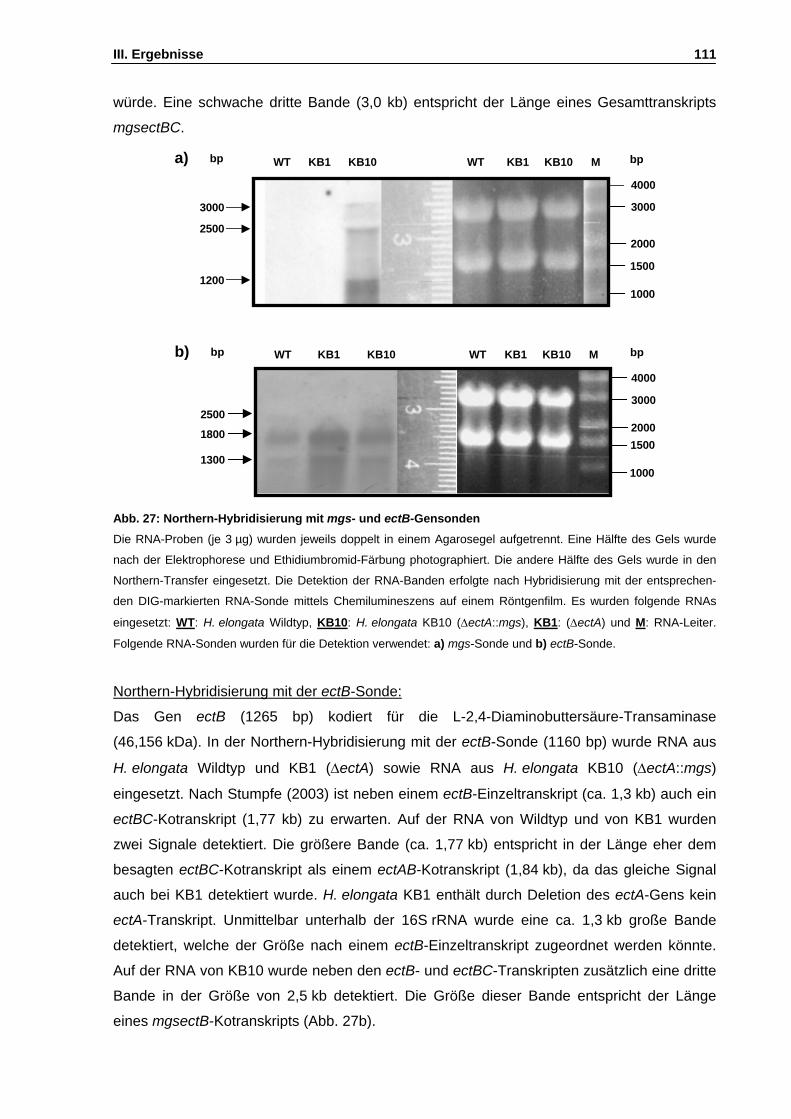
III. Ergebnisse 111
würde. Eine schwache dritte Bande (3,0 kb) entspricht der Länge eines Gesamttranskripts
mgsectBC.
a)
1200
2500
WTWT KB1 KB1 KB10KB10 M
4000
3000 3000
2000
1500
1000
bpbp
WTWT KB1 KB1 KB10KB10 M
3000
20001500
10001300
2500
1800
b) bpbp
4000
Abb. 27: Northern-Hybridisierung mit mgs- und ectB-Gensonden Die RNA-Proben (je 3 µg) wurden jeweils doppelt in einem Agarosegel aufgetrennt. Eine Hälfte des Gels wurde
nach der Elektrophorese und Ethidiumbromid-Färbung photographiert. Die andere Hälfte des Gels wurde in den
Northern-Transfer eingesetzt. Die Detektion der RNA-Banden erfolgte nach Hybridisierung mit der entsprechen-
den DIG-markierten RNA-Sonde mittels Chemilumineszens auf einem Röntgenfilm. Es wurden folgende RNAs
eingesetzt: WT: H. elongata Wildtyp, KB10: H. elongata KB10 (∆ectA::mgs), KB1: (∆ectA) und M: RNA-Leiter.
Folgende RNA-Sonden wurden für die Detektion verwendet: a) mgs-Sonde und b) ectB-Sonde.
Northern-Hybridisierung mit der ectB-Sonde:
Das Gen ectB (1265 bp) kodiert für die L-2,4-Diaminobuttersäure-Transaminase
(46,156 kDa). In der Northern-Hybridisierung mit der ectB-Sonde (1160 bp) wurde RNA aus
H. elongata Wildtyp und KB1 (∆ectA) sowie RNA aus H. elongata KB10 (∆ectA::mgs)
eingesetzt. Nach Stumpfe (2003) ist neben einem ectB-Einzeltranskript (ca. 1,3 kb) auch ein
ectBC-Kotranskript (1,77 kb) zu erwarten. Auf der RNA von Wildtyp und von KB1 wurden
zwei Signale detektiert. Die größere Bande (ca. 1,77 kb) entspricht in der Länge eher dem
besagten ectBC-Kotranskript als einem ectAB-Kotranskript (1,84 kb), da das gleiche Signal
auch bei KB1 detektiert wurde. H. elongata KB1 enthält durch Deletion des ectA-Gens kein
ectA-Transkript. Unmittelbar unterhalb der 16S rRNA wurde eine ca. 1,3 kb große Bande
detektiert, welche der Größe nach einem ectB-Einzeltranskript zugeordnet werden könnte.
Auf der RNA von KB10 wurde neben den ectB- und ectBC-Transkripten zusätzlich eine dritte
Bande in der Größe von 2,5 kb detektiert. Die Größe dieser Bande entspricht der Länge
eines mgsectB-Kotranskripts (Abb. 27b).

III. Ergebnisse 112
RT-PCR zur Nachweis der Transkripte:
Zur Bestätigung der Ergebnisse der Northern-Hybridisierung wurde eine RT-PCR durch-
geführt. Diese sehr sensitive Methode weist im Gegensatz zur Northern-Hybridisierung auch
Transkripte nach, die in nur sehr geringer Kopienzahl vorliegen.
Mit Gesamt-RNA von KB10, welcher bei 4 % NaCl angezogen worden war, wurde aus-
gehend des Primers gsp1ectB cDNA synthetisiert. Primer gsp1ectB bindet im Endbereich
(ca. 160 bp stromaufwärts des Stopkodons) des ectB-Gens. Die cDNA wurde anschließend
in zwei unterschiedliche RT-PCRs eingesetzt. Dabei wurde mit den Primern mgsfor2 und
Racemgs1 (beide binden innerhalb von mgs) ein 376 bp großes PCR-Produkt amplifiziert,
mit den Primern mgsfor1 und gsp2ectB ein 1891 bp großes PCR-Produkt (Abb. 28). Dadurch
konnte gezeigt werden, das die ausgehend des ectB-Gens synthetisierte cDNA die
Sequenzen von mgs und ectB umfasst.
RT- PCR
1 2 3 4 5 6 7
3000
20001500
1031
400
600
800
a)
380
1891
bp
b)mgs ectB
mgsfor2
Racemgs1 gsp2ectB
gsp1ectB
mgsfor1
mRNA
cDNA
1 kb
mgsfor2
Racemgs1
RT PCR 1
mgsfor1
gsp2ectB
RT PCR 2
Abb. 28: Nachweis der mgs-Transkription auf H. elongata KB10 (∆ectA::mgs) RNA mittels RT-PCR
a) Dargestellt ist die gelelektrophoretische Auftrennung der RT-PCR-Produkte. b) Vorgehen bei der Herstellung
der RT-PCR-Produkte. Die cDNA wurde ausgehend des Primers gsp1ectB synthetisiert. Mit den mgs-Primern
mgsfor2 und Racemgs1 wurde ein 380 bp großes Produkt amplifiziert (1). Spur (2) stellt die entsprechende
Negativkontrolle ohne cDNA-Synthese dar. Mit den Primern mgsfor1 und gsp2ectB wurde auf der cDNA ein 1891
bp großes PCR-Produkt amplifiziert (3). Spur (4) stellt die entsprechende Negativkontrolle ohne cDNA-Synthese
dar. In den Spuren (5) und (6) wurde Wasser anstelle von cDNA in die PCR-Reaktionen eingesetzt. Mit Hilfe der
beiden amplifizierten PCR-Produkte konnte gezeigt werden, das die ausgehend des ectB-Gens synthetisierte
cDNA die Sequenzen von mgs und ectB umfasst.
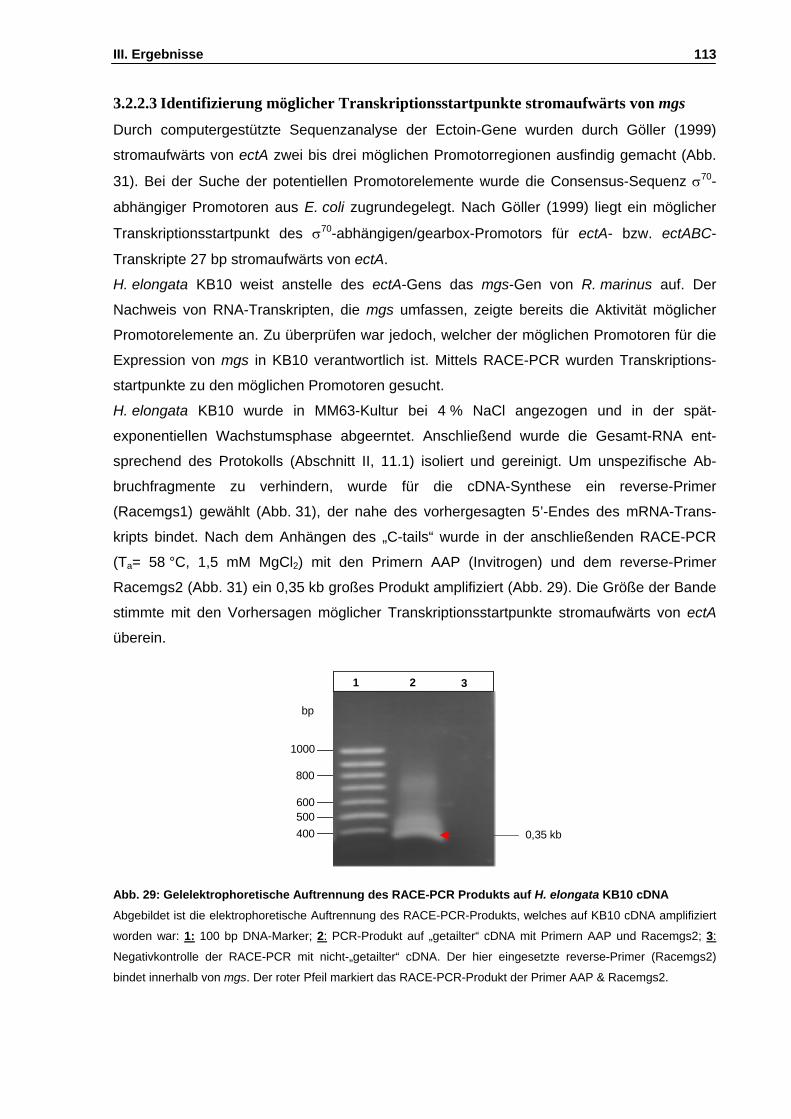
III. Ergebnisse 113
3.2.2.3 Identifizierung möglicher Transkriptionsstartpunkte stromaufwärts von mgs
Durch computergestützte Sequenzanalyse der Ectoin-Gene wurden durch Göller (1999)
stromaufwärts von ectA zwei bis drei möglichen Promotorregionen ausfindig gemacht (Abb.
31). Bei der Suche der potentiellen Promotorelemente wurde die Consensus-Sequenz σ70-
abhängiger Promotoren aus E. coli zugrundegelegt. Nach Göller (1999) liegt ein möglicher
Transkriptionsstartpunkt des σ70-abhängigen/gearbox-Promotors für ectA- bzw. ectABC-
Transkripte 27 bp stromaufwärts von ectA.
H. elongata KB10 weist anstelle des ectA-Gens das mgs-Gen von R. marinus auf. Der
Nachweis von RNA-Transkripten, die mgs umfassen, zeigte bereits die Aktivität möglicher
Promotorelemente an. Zu überprüfen war jedoch, welcher der möglichen Promotoren für die
Expression von mgs in KB10 verantwortlich ist. Mittels RACE-PCR wurden Transkriptions-
startpunkte zu den möglichen Promotoren gesucht.
H. elongata KB10 wurde in MM63-Kultur bei 4 % NaCl angezogen und in der spät-
exponentiellen Wachstumsphase abgeerntet. Anschließend wurde die Gesamt-RNA ent-
sprechend des Protokolls (Abschnitt II, 11.1) isoliert und gereinigt. Um unspezifische Ab-
bruchfragmente zu verhindern, wurde für die cDNA-Synthese ein reverse-Primer
(Racemgs1) gewählt (Abb. 31), der nahe des vorhergesagten 5’-Endes des mRNA-Trans-
kripts bindet. Nach dem Anhängen des „C-tails“ wurde in der anschließenden RACE-PCR
(Ta= 58 °C, 1,5 mM MgCl2) mit den Primern AAP (Invitrogen) und dem reverse-Primer
Racemgs2 (Abb. 31) ein 0,35 kb großes Produkt amplifiziert (Abb. 29). Die Größe der Bande
stimmte mit den Vorhersagen möglicher Transkriptionsstartpunkte stromaufwärts von ectA
überein.
0,35 kb
1000
800
600500400
1 2 3
bp
Abb. 29: Gelelektrophoretische Auftrennung des RACE-PCR Produkts auf H. elongata KB10 cDNA
Abgebildet ist die elektrophoretische Auftrennung des RACE-PCR-Produkts, welches auf KB10 cDNA amplifiziert
worden war: 1: 100 bp DNA-Marker; 2: PCR-Produkt auf „getailter“ cDNA mit Primern AAP und Racemgs2; 3:
Negativkontrolle der RACE-PCR mit nicht-„getailter“ cDNA. Der hier eingesetzte reverse-Primer (Racemgs2)
bindet innerhalb von mgs. Der roter Pfeil markiert das RACE-PCR-Produkt der Primer AAP & Racemgs2.

III. Ergebnisse 114
Das ca. 0,35 kb große RACE-PCR-Produkt wurde in einem präparativen Agarosegel
gereinigt und anschließend sequenziert. Die exakte Sequenzlänge beträgt 337 bp. Für die
Sequenzierung wurde der Primer Racemgs3 (Abb. 31) verwendet. Zur Bestimmung des
Transkriptionsstartpunkts wurde die Sequenz des Produkts nach dem „C-tail“ abgesucht
(Abb. 30).
RACE-Produkt: 3’ CCCCCCCCCCCCCCCTTTAAGCGAT... 5’
-+---++++++++++
Genom: 3’ ...GCTAGTTTAAGCGAT... 5’ Pos. 525
⏐⏐⏐⏐⏐⏐⏐⏐⏐⏐
Tr1: Pos. 503 5’ AAATTCGCTA... 3’ (kodierender Strang)
+1 Abb. 30: Identifizierung eines Transkriptionsstartpunkts stromaufwärts von mgs in H. elongata KB10
Dargestellt ist die DNA-Sequenz des RACE-PCR-Produkts im Bereich des „C-tails“. Zum Vergleich ist die ent-
sprechende genomische DNA-Sequenz angegeben. Die Positionsangaben beziehen sich auf die H. elongata
KB10 DNA-Sequenz im Anhang und in Abb. 31. „+“ deutet auf Übereinstimmungen, „- “auf Abweichungen von der
genomischen Sequenz hin. Für das RACE-PCR-Produkt ist der zugehörige auf dem kodierenden Gegenstrang
der DNA liegenden Transkriptionsstartpunkt angegeben (Tr1). Dieser ist unterstrichen und durch „+1“ markiert.
„⏐“ verdeutlicht die komplementäre Bereiche der DNA-Stränge.
Das RACE-PCR-Produkt verwies auf einen Transkriptionsstartpunkt, der 25 bp (Position
503) stromaufwärts des Startkodons von mgs liegt. Somit liegt dieser nur 2 bp entfernt von
dem mittels Computeranalysen vorhergesagten Transkriptionsstartpunkts (27 bp stromauf-
wärts von ectA) des vermutlichen σ70-abhängigen/gearbox Promotors (Abb. 31).
3.2.3 Untersuchung zur heterologen Expression des mgs-Gen aus R. marinus in
H. elongata Wildtyp und KB1 unter der Kontrolle des ectABC Promotors
Durch H. elongata KB10 wurde trotz der nachgewiesenen Transkription von mgs kein Man-
nosylglycerat synthetisiert. Verschiedene Gründe konnten dafür verantwortlich sein (Ab-
schnitt IV, 8.1.1). Ein schwerwiegendes Problem könnte die ungenügende Expression der
Mannosylglycerat-Synthase in H. elongata Stämmen sein. So könnte das Protein aufgrund
einer fehlerhaften Faltung inaktiv sein und darüber hinaus sogar „inclusion bodies“ ausbilden
(Hartely & Kane, 1988; Mitraki & King, 1989; Hart et al., 1990; King et al., 1996). Zur
Überprüfung dieses Sachverhaltes wurde die heterologe Expression des mgs-Gens in
H. elongata Wildtyp und KB1 analysiert. Die Expression eines löslichen Proteins wurde als
Indikator für die korrekte Faltung des Proteins gewertet. Der lösliche Charakter der Man-
nosylglycerat-Synthase sollte durch immunologischen Nachweis des Enzyms in der löslichen
zytoplasmatischen Zellfraktion von H. elongata Wildtyp und KB1 in einer Western-Analyse

III. Ergebnisse 115
bestätigt werden. Antigen für die Antikörper war eine der Mannosylglycerat-Synthase ange-
hängte His6-tag Sequenz.
σS/σ70-Element CCGCTACATACGCGGCCTGGGGAGTGGGCTATAATTTTCTATTATGGAATTCAGCAAGCA 453 -35 σ70 gearbox –10 σ70 +1 +1 AGATAACCTGGTTTTTGAAATGACCATAAGCGGCTGTTATGATGCCGATCAAATTCGCTA 513 rbs Start mgs CAGCGAACCACGACAATGAGCCTGGTCGTTTTTCCCTTCAAGCACGAACACCCCGAGGTG 573 CTGCTGCACAACGTGCGCGTGGCGGCTGCTCATCCCCGGGTGCACGAGGTGCTCTGCATC 633 GGATACGAGCGCGACCAGACCTACGAGGCCGTCGAACGGGCCGCGCCCGAAATCTCGCGG 693 GCCACAGGGACGCCGGTGTCGGTCCGGTTGCAGGAGCGACTGGGCACGCTGCGGCCCGGC 753 Racemgs3 AAGGGCGACGGCATGAACACCGCGCTCCGGTATTTCCTGGAAGAGACGCAGTGGGAGCGC 813 Racemgs2 ATCCACTTCTACGATGCCGACATCACCAGCTTCGGGCCGGACTGGATCACCAAGGCCGAA 873 GAGGCGGCCGATTTCGGCTACGGGCTGGTGCGGCACTACTTCCCGCGGGCCTCAACCGAC 933 Racemgs1 Abb. 31: mgs-Promotorregion von H. elongata KB10
Dargestellt ist der kodierende DNA-Strang aus der mgs-Region der Insertionsmutante H. elongata KB10. Ange-
geben sind die Transkriptionsstartpunkte (+1), Ribosomen-Bindungsstellen (rbs), möglichen Promotoren (σS/σ70
und σ70) sowie ein gearbox-Element. Der durch RACE-PCR identifizierter Transkriptionsstartpunkt ist eingerahmt
und gelb markiert, der durch computergestützte Analysen vorhergesagte Transkriptionsstartpunkt ist eingekreist
dargestellt (Göller, 1999). Beide liegen in unmittelbarer Nachbarschaft zu dem möglichen σ70-abhängigen Promo-
tor. Für die übrigen Promotorelemente konnten keine Transkriptionsstartpunkte identifiziert werden. Die Bin-
dungsstellen der in die cDNA-Synthese und RACE-PCR eingesetzten Primer sind mit Pfeilen gekennzeichnet.
Für die heterologe Expression von mgs in H. elongata wurde ein Derivat des pBBRMCS1-
Vektors, pPromEct, verwendet (Brünig, 2005). Der Vektor pBBRMCS1 (Kovach et al., 1994)
basiert auf dem natürlich vorkommenden Vektor pBBR1 aus Bordetella bronchiseptica
(Antoine & Locht, 1992). Bereits Schnoor (2001), Burdziak (2004) und Brünig (2005) hatten
mit pBBRMCS1 und Derivaten erfolgreich unterschiedliche Proteine in H. elongata expri-
miert. Brünig (2005) hatte durch Einfügen der ectA-Promoterregion aus H. elongata in die
multiple Klonierungsstelle von pBBRMCS1 das Plasmid pPromEct hergestellt. Dabei wurde
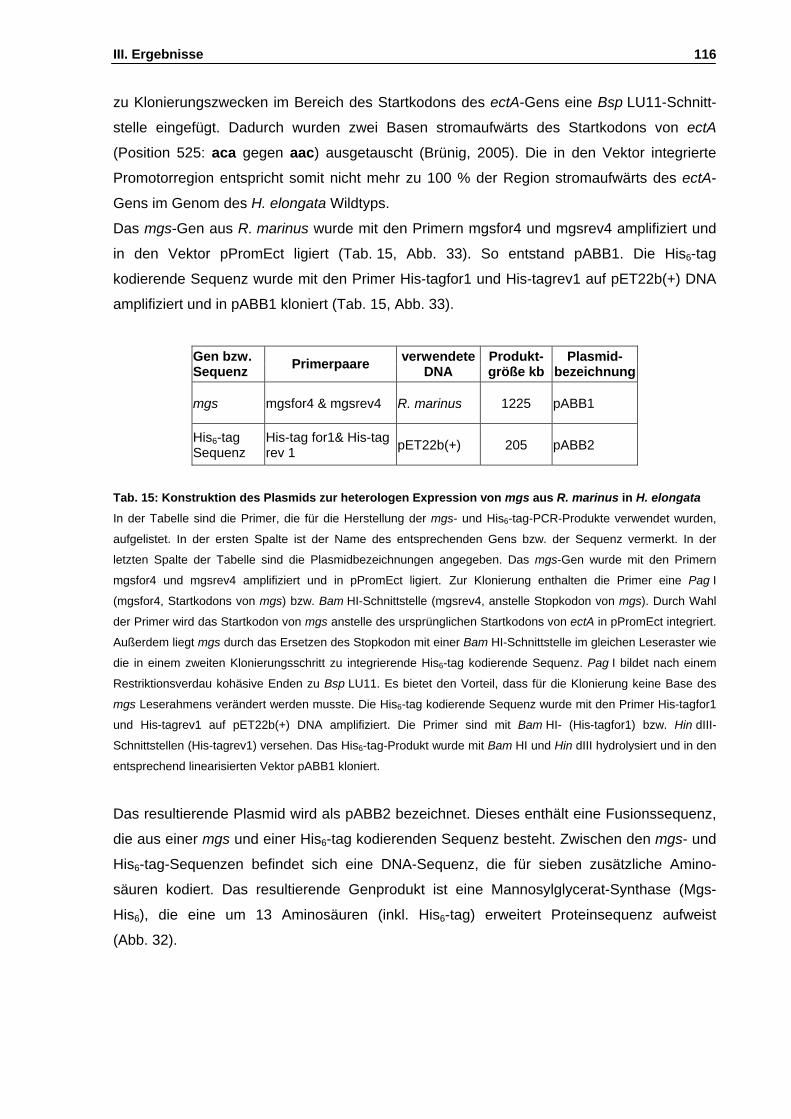
III. Ergebnisse 116
zu Klonierungszwecken im Bereich des Startkodons des ectA-Gens eine Bsp LU11-Schnitt-
stelle eingefügt. Dadurch wurden zwei Basen stromaufwärts des Startkodons von ectA
(Position 525: aca gegen aac) ausgetauscht (Brünig, 2005). Die in den Vektor integrierte
Promotorregion entspricht somit nicht mehr zu 100 % der Region stromaufwärts des ectA-
Gens im Genom des H. elongata Wildtyps.
Das mgs-Gen aus R. marinus wurde mit den Primern mgsfor4 und mgsrev4 amplifiziert und
in den Vektor pPromEct ligiert (Tab. 15, Abb. 33). So entstand pABB1. Die His6-tag
kodierende Sequenz wurde mit den Primer His-tagfor1 und His-tagrev1 auf pET22b(+) DNA
amplifiziert und in pABB1 kloniert (Tab. 15, Abb. 33).
Gen bzw. Sequenz Primerpaare verwendete
DNA Produkt- größe kb
Plasmid-bezeichnung
mgs mgsfor4 & mgsrev4 R. marinus 1225 pABB1
His6-tag Sequenz
His-tag for1& His-tag rev 1 pET22b(+) 205 pABB2
Tab. 15: Konstruktion des Plasmids zur heterologen Expression von mgs aus R. marinus in H. elongata
In der Tabelle sind die Primer, die für die Herstellung der mgs- und His6-tag-PCR-Produkte verwendet wurden,
aufgelistet. In der ersten Spalte ist der Name des entsprechenden Gens bzw. der Sequenz vermerkt. In der
letzten Spalte der Tabelle sind die Plasmidbezeichnungen angegeben. Das mgs-Gen wurde mit den Primern
mgsfor4 und mgsrev4 amplifiziert und in pPromEct ligiert. Zur Klonierung enthalten die Primer eine Pag I
(mgsfor4, Startkodons von mgs) bzw. Bam HI-Schnittstelle (mgsrev4, anstelle Stopkodon von mgs). Durch Wahl
der Primer wird das Startkodon von mgs anstelle des ursprünglichen Startkodons von ectA in pPromEct integriert.
Außerdem liegt mgs durch das Ersetzen des Stopkodon mit einer Bam HI-Schnittstelle im gleichen Leseraster wie
die in einem zweiten Klonierungsschritt zu integrierende His6-tag kodierende Sequenz. Pag I bildet nach einem
Restriktionsverdau kohäsive Enden zu Bsp LU11. Es bietet den Vorteil, dass für die Klonierung keine Base des
mgs Leserahmens verändert werden musste. Die His6-tag kodierende Sequenz wurde mit den Primer His-tagfor1
und His-tagrev1 auf pET22b(+) DNA amplifiziert. Die Primer sind mit Bam HI- (His-tagfor1) bzw. Hin dIII-
Schnittstellen (His-tagrev1) versehen. Das His6-tag-Produkt wurde mit Bam HI und Hin dIII hydrolysiert und in den
entsprechend linearisierten Vektor pABB1 kloniert.
Das resultierende Plasmid wird als pABB2 bezeichnet. Dieses enthält eine Fusionssequenz,
die aus einer mgs und einer His6-tag kodierenden Sequenz besteht. Zwischen den mgs- und
His6-tag-Sequenzen befindet sich eine DNA-Sequenz, die für sieben zusätzliche Amino-
säuren kodiert. Das resultierende Genprodukt ist eine Mannosylglycerat-Synthase (Mgs-
His6), die eine um 13 Aminosäuren (inkl. His6-tag) erweitert Proteinsequenz aufweist
(Abb. 32).

III. Ergebnisse 117
mgs
5’...CGGGCGGCACTGTCGACCGCCTGAC... 3’
R A A L S T A *
mgs-his6
Bam HI
5’...CGGGCGGCACTGTCGACCGCCGGATCCGCGGCCGCACTCGAGCACCACCACCACCACCACTGAGA...3’
R A A L S T A G S A A A L E H H H H H H *
Abb. 32: Verlängerte DNA- und Aminosäuresequenz von mgs aus R. marinus
Dargestellt ist die modifizierte DNA-Sequenz von mgs in Vektor pABB2 im Vergleich zu der ursprünglichen DNA-
Sequenz. Angegeben ist auch die um 13 Aminosäuren verlängerte Proteinsequenz von Mgs-His6. Diese Sequenz
wurde zur Überprüfung einer nativen Expression der Mannosylglycerat-Synthase in H. elongata verwendet. Das
Stopkodon TGA von mgs ist in Fettbuchstaben, die integrierte Bam HI-Schnittstelle ist umrahmt dargestellt. Grau
unterlegt ist die DNA-Sequenz der zusätzlich angehängten Aminosäuren, rot hervorgehoben sind die Histidin-
Reste des His6-tags.
Der Vektor pABB2 wurde mittels Konjugation von E. coli S17-1 auf H. elongata Wildtyp und
KB1 übertragen. Als Negativkontrolle wurde pPromEct in H. elongata Wildtyp und KB1 ein-
gebracht.
Um die Expression von mgs zu charakterisieren, wurden H. elongata Wildtyp und KB1 (mit
pABB2 und pPromEct als Negativkontrolle) in je 100 ml MM63-Medium mit Glucose als
Kohlenstoffquelle angezogen. Die Anzucht der Kulturen erfolgte bei 37 °C und 180 upm bis
zur einer OD600 von 1. Die Expression von mgs im H. elongata Wildtyp wurde bei einer
Mediensalinität von 10 % untersucht. Zur Untersuchung der mgs-Expression in einem
Ectoin-freien System wurde H. elongata KB1 verwendet. Dieser wurde bei einer Medien-
salinität von 4 % angezogen. Aufgrund der höheren Salinität wurde beim Wildtyp mit einer
stärkeren Expression von mgs gerechnet.
Bei der Mannosylglycerat-Synthase von R. marinus handelt es sich um ein zytoplasma-
tisches Protein (Martins et al., 1999). Weil es sich jedoch bei H. elongata um ein heterologes
Expressionssystem handelt, wurde das Vorkommen der Mannosylglycerat-Synthase neben
dem Gesamtzellprotein und der löslichen zytoplasmatischen Fraktion auch in der periplas-
matischen Fraktion untersucht. Zusätzlich wurde Zellmaterial nach der Methode zur Gewin-
nung möglicher „inclusion bodies“ aufgearbeitet (Abschnitt II, 15.2.4).
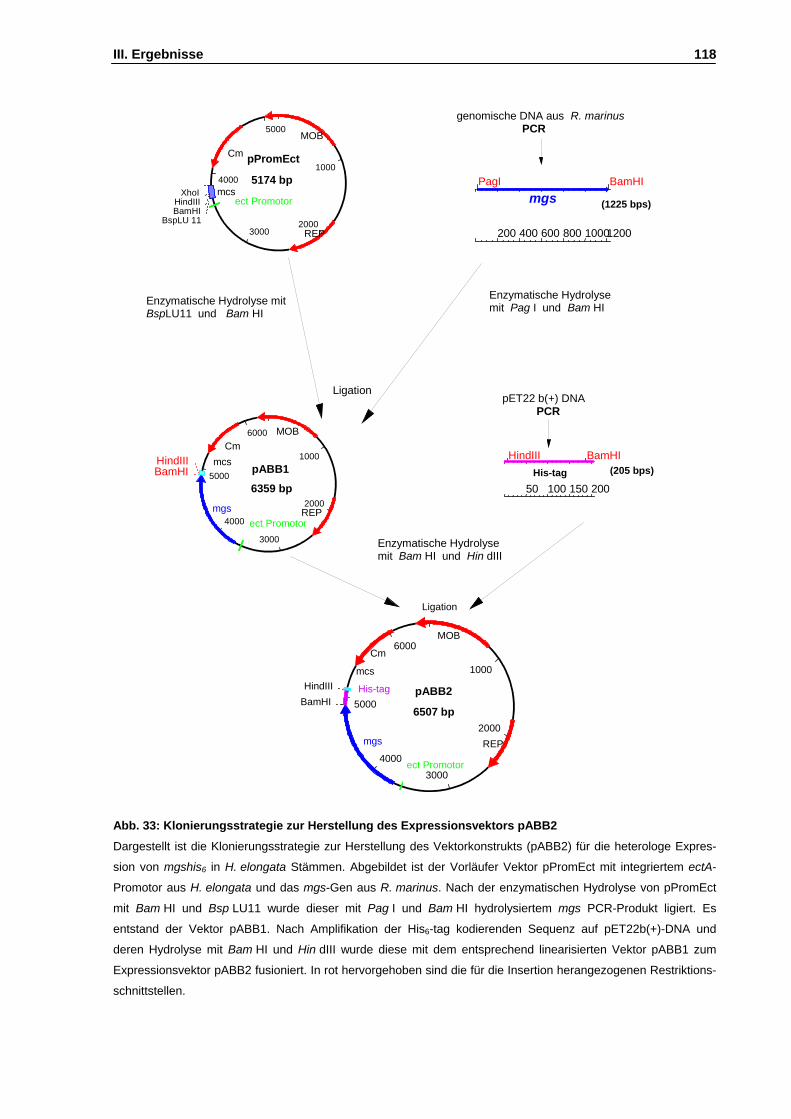
III. Ergebnisse 118
pPromEct
5174 bp1000
20003000
4000
5000
BspLU 11BamHIHindIII
XhoI
MOB
REP
ect Promotor mcs
Cm
6359 bp
1000
2000
3000
4000
5000
6000
BamHIHindIII
MOB
REPect Promotor
mgs
mcsCm
(1225 bps)
200 400 600 800 10001200
PagI BamHImgs
(205 bps)
50 100 150 200
HindIII BamHIHis-tag
6507 bp
1000
2000
30004000
5000
6000
BamHIHindIII
MOB
REP
ect Promotor
mgs
His-tag
mcs
Cm
Ligation
pABB1
Ligation
pET22 b(+) DNAPCR
pABB2
genomische DNA aus R. marinusPCR
Enzymatische Hydrolyse mit Pag I und Bam HIEnzymatische Hydrolyse mit
BspLU11 und Bam HI
Enzymatische Hydrolyse mit Bam HI und Hin dIII
Abb. 33: Klonierungsstrategie zur Herstellung des Expressionsvektors pABB2
Dargestellt ist die Klonierungsstrategie zur Herstellung des Vektorkonstrukts (pABB2) für die heterologe Expres-
sion von mgshis6 in H. elongata Stämmen. Abgebildet ist der Vorläufer Vektor pPromEct mit integriertem ectA-
Promotor aus H. elongata und das mgs-Gen aus R. marinus. Nach der enzymatischen Hydrolyse von pPromEct
mit Bam HI und Bsp LU11 wurde dieser mit Pag I und Bam HI hydrolysiertem mgs PCR-Produkt ligiert. Es
entstand der Vektor pABB1. Nach Amplifikation der His6-tag kodierenden Sequenz auf pET22b(+)-DNA und
deren Hydrolyse mit Bam HI und Hin dIII wurde diese mit dem entsprechend linearisierten Vektor pABB1 zum
Expressionsvektor pABB2 fusioniert. In rot hervorgehoben sind die für die Insertion herangezogenen Restriktions-
schnittstellen.
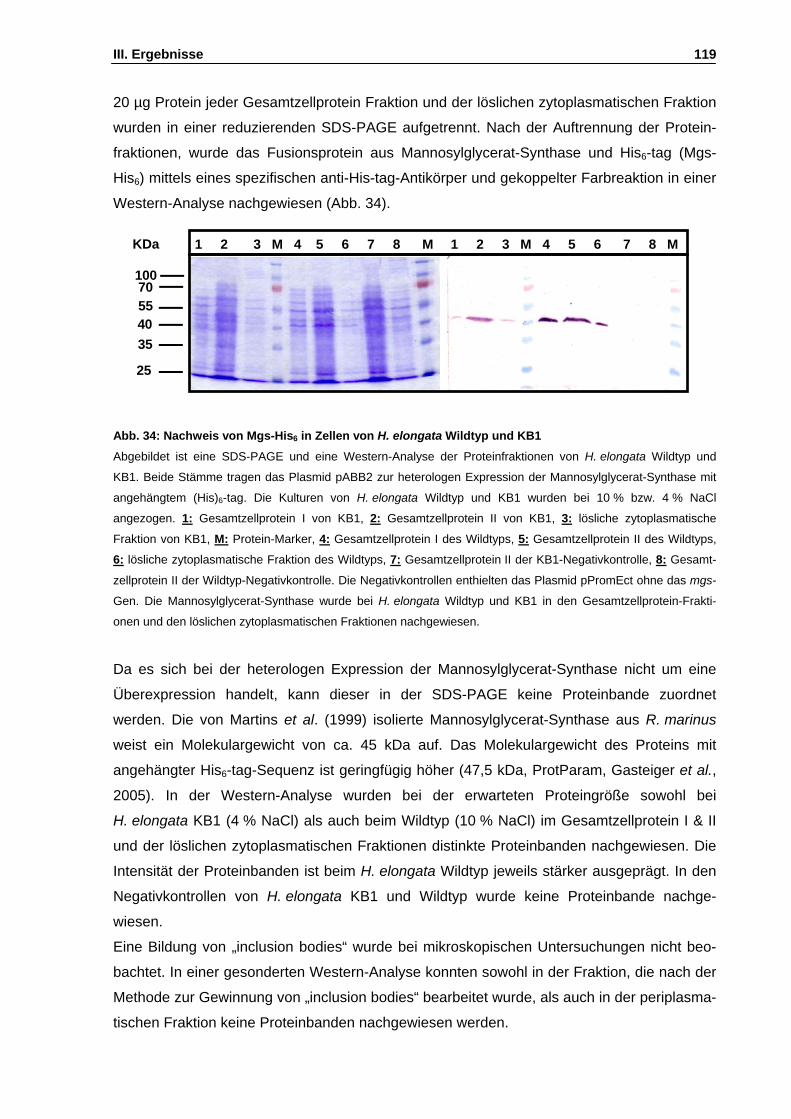
III. Ergebnisse 119
20 µg Protein jeder Gesamtzellprotein Fraktion und der löslichen zytoplasmatischen Fraktion
wurden in einer reduzierenden SDS-PAGE aufgetrennt. Nach der Auftrennung der Protein-
fraktionen, wurde das Fusionsprotein aus Mannosylglycerat-Synthase und His6-tag (Mgs-
His6) mittels eines spezifischen anti-His-tag-Antikörper und gekoppelter Farbreaktion in einer
Western-Analyse nachgewiesen (Abb. 34).
100
5570
40
25
35
KDa 1 2 3 M 4 5 6 7 8 M 1 2 3 M 4 5 6 7 8 M
Abb. 34: Nachweis von Mgs-His6 in Zellen von H. elongata Wildtyp und KB1
Abgebildet ist eine SDS-PAGE und eine Western-Analyse der Proteinfraktionen von H. elongata Wildtyp und
KB1. Beide Stämme tragen das Plasmid pABB2 zur heterologen Expression der Mannosylglycerat-Synthase mit
angehängtem (His)6-tag. Die Kulturen von H. elongata Wildtyp und KB1 wurden bei 10 % bzw. 4 % NaCl
angezogen. 1: Gesamtzellprotein I von KB1, 2: Gesamtzellprotein II von KB1, 3: lösliche zytoplasmatische
Fraktion von KB1, M: Protein-Marker, 4: Gesamtzellprotein I des Wildtyps, 5: Gesamtzellprotein II des Wildtyps,
6: lösliche zytoplasmatische Fraktion des Wildtyps, 7: Gesamtzellprotein II der KB1-Negativkontrolle, 8: Gesamt-
zellprotein II der Wildtyp-Negativkontrolle. Die Negativkontrollen enthielten das Plasmid pPromEct ohne das mgs-
Gen. Die Mannosylglycerat-Synthase wurde bei H. elongata Wildtyp und KB1 in den Gesamtzellprotein-Frakti-
onen und den löslichen zytoplasmatischen Fraktionen nachgewiesen.
Da es sich bei der heterologen Expression der Mannosylglycerat-Synthase nicht um eine
Überexpression handelt, kann dieser in der SDS-PAGE keine Proteinbande zuordnet
werden. Die von Martins et al. (1999) isolierte Mannosylglycerat-Synthase aus R. marinus
weist ein Molekulargewicht von ca. 45 kDa auf. Das Molekulargewicht des Proteins mit
angehängter His6-tag-Sequenz ist geringfügig höher (47,5 kDa, ProtParam, Gasteiger et al.,
2005). In der Western-Analyse wurden bei der erwarteten Proteingröße sowohl bei
H. elongata KB1 (4 % NaCl) als auch beim Wildtyp (10 % NaCl) im Gesamtzellprotein I & II
und der löslichen zytoplasmatischen Fraktionen distinkte Proteinbanden nachgewiesen. Die
Intensität der Proteinbanden ist beim H. elongata Wildtyp jeweils stärker ausgeprägt. In den
Negativkontrollen von H. elongata KB1 und Wildtyp wurde keine Proteinbande nachge-
wiesen.
Eine Bildung von „inclusion bodies“ wurde bei mikroskopischen Untersuchungen nicht beo-
bachtet. In einer gesonderten Western-Analyse konnten sowohl in der Fraktion, die nach der
Methode zur Gewinnung von „inclusion bodies“ bearbeitet wurde, als auch in der periplasma-
tischen Fraktion keine Proteinbanden nachgewiesen werden.

III. Ergebnisse 120
Da es aufgrund dieser Ergebnisse offensichtlich zu einer löslichen Expression von mgs in
H. elongata kommt; ist die fehlende Mannosylglycerat-Synthese (Abschnitt III, 3.2.2.1)
entweder auf eine zu geringe Aktivität des Enzyms oder einen Mangel an Synthesevor-
stufen zurückzuführen (Abschnitt IV, 8.1.1). Letzteres Problem sollte durch die Integration
weiterer Gene des Kohlenhydratstoffwechsels behoben werden.
3.3 Integration hypothetischer P. putida Gene in H. elongata KB10
3.3.1 Herstellung von H. elongata KB10.1
Die Insertionsmutante H. elongata KB10 war nicht in der Lage Mannosylglycerat zu synthe-
tisieren. Als ein möglicher Grund hierfür wurde die Bereitstellung der Synthesevorstufen
angesehen (siehe Abschnitt IV, 8.2.1). Die Synthese von Mannosylglycerat erfolgt in
R. marinus in mehreren Schritten ausgehend von Glucose-6-phosphat (Abschnitt I, 3.3.2,
Abb. 2). An der Synthese ist unter anderem eine Phosphomanno-Mutase (wandelt Mannose-
6-phosphat in Mannose-1-phosphat um) und eine Mannose-1-phosphat-Guanylyltransferase
(setzt Mannose-1-phosphat unter Abspaltung von GTP in GDP-Mannose um) beteiligt. Die
Gensequenzen von Enzymen gleicher Funktion sind in H. elongata, welcher als Produktions-
stamm für Mannosylglycerat dienen soll, nicht bekannt. Daher wurde auf entsprechende
Gene von Pseudomonas putida KT2440 zurückgegriffen. Dieser schien aus verschiedenen
Gründen, z.B. seines mesophilen Charakters, seiner „Codon usage“, seines vielfältigen
Zuckerstoffwechsels sowie seiner Einstufung als Sicherheitsstamm für biologisches Arbeiten,
gut als Spenderorganismus geeignet (Abschnitt IV, 8.2.1). Bei den ausgewählten DNA-
Sequenzen PP1776 (kodiert für mögliche Mannose-1-phosphat-Guanylyltransferase) und
xanA (kodiert für mögliche Phosphomanno-Mutase) handelt es sich um zwei benachbarte
hypothetische Gene. Sie wurden mit Hilfe der KEGG-Datenbank (Kanehisa & Goto, 2000;
Kanehisa et al., 2006) identifiziert.
Die Ectoin-Synthesegene ectBC von H. elongata wurden durch homologe Rekombination
gegen PP1776 und xanA aus P. putida ausgetauscht (Abb. 36). Das pK18mobsacB
Konstrukt zur Herstellung von H. elongata KB10.1 wurden durch eine spezielle Klonierungs-
strategie erstellt. Das Konstrukt besteht aus drei Produkten: der Flanke stromaufwärts von
ectB (bestehend aus der ectA-Promotorregion und dem mgs-Gen aus H. elongata KB10:
Produkt 1), den ORFs PP1776 und xanA sowie der zwischenliegenden DNA-Region aus
P. putida (Produkt 2) und der Flanke ectR (stromabwärts von ectC) aus H. elongata Wildtyp
(Produkt 3). Alle drei DNA-Bereiche wurden in getrennten PCR-Reaktionen amplifiziert und
nacheinander in den Vektor pK18mobsacB kloniert (Tab. 16).

III. Ergebnisse 121
Stamm Primerpaare verwendete DNA
Produkt- größe kb
Plasmid-bezeichnung
KB10XanA1 (Nhe I) & KB10XanA2 (Nde I & Hin dIII) (Produkt 1)
H. elongata KB10 1869 pK18KB10.1A
(Prod. 1)
KB10XanA3 (Nde I) & B10XanA4 (Hin dIII) K
(Produkt 2) P. putida 2963 pK18KB10.1B
(Prod. 1 & 2) H. elongata KB10.1
(∆ectABC::mgsPP1776xanA)
KB10XanA5 (Hin dIII) & KB10XanA6 (Xba I) (Produkt 3)
H. elongata 1609 pK18KB10.1C (Prod. 1, 2 & 3)
Tab. 16: PCR-Produkte und Vektorkonstrukte zur Herstellung von H. elongata KB10.1
In der Tabelle sind die Primer, die zur Herstellung von H. elongata KB10.1 benötigt wurden, aufgeführt. In der
letzten Spalte der Tabelle sind die Bezeichnungen der Plasmidkonstrukte angegeben. Die drei benötigten Einzel-
PCR-Produkte (Produkt 1, 2 & 3) wurden mit den entsprechenden Primern auf genomischer DNA von H. elongata
Wildtyp und KB10 bzw. P. putida amplifiziert. Zunächst wurde Produkt 1 amplifiziert. Zur Erleichterung der
Klonierungsstrategie ist Primer KB10XanA2 in seiner Sequenz um zwei Restriktionsschnittstellen (Nde I und
Hin dIII) erweitert. KB10XanA1 trägt eine Nhe I-Schnittstelle. Produkt 1 wurde über die Schnittstellen Nhe I und
Hin dIII in die MCS von pK18mobsacB kloniert (pK18KB10.1A). Nach Hydrolyse von Produkt 2 mit den
Restriktionsenzymen Nde I und Hin dIII wurde dieses in den entsprechend behandelten Vektor pK18KB10.1A
ligiert. Dabei entstand pK18KB10.1B. Um Produkt 3 stromabwärts von Produkt 2 in pK18KB10.1B zu
integrieren, wurde Produkt 3 mit Hin dIII und Xba I hydrolysiert und mit dem entsprechend behandelten
pk18KB10.1B Vektor verknüpft. Dabei entstand pk18KB10.1C.
Die Gesamtlänge der in pK18mobsacB inserierten DNA beträgt 6441 bp. Der Vektor
pK18KB10.1C wurde mittels konjugativem Plasmidtransfers auf H. elongata KB10 über-
tragen. Der resultierende Stamm trägt die Bezeichnung H. elongata KB10.1 (Abb. 36).
Dieser wurde in einer Kolonie-PCR mit den Primer mgsfor3 und CoXanrev identifiziert. Das
1991 bp großes Produkt kann nur auf H. elongata KB10.1 DNA amplifiziert werden, da
Primer CoXanrev spezifisch innerhalb von xanA bindet. Bei H. elongata KB10.1 sind die
Ectoin-Gene ectABC durch die Gene mgs, PP1776 und xanA ersetzt.
Bedingt durch die Klonierungsstrategie bei der Konstruktion der rekombinanten DNA weist
H. elongata KB10.1 im Vergleich zu H. elongata Wildtyp zusätzliche einige Sequenz-
änderungen auf, die außerhalb der ausgetauschten ORFs liegen (Abb. 35). Darüber hinaus
ist die zwischen ectB und ectC liegende DNA-Region von H. elongata gegen die Sequenz
zwischen PP1776 und xanA von P. putida ausgetauscht (Abb. 36).

III. Ergebnisse 122
H. elongata Wildtyp: RBS ectB
cag gag gtc gca ATG CAG ACC CAG ATT CTG
M Q T Q I L H. elongata KB10.1: RBS Nde I PP1776
cag gag gtc cat ATG GAA TTG ATC CCG GTA
M E L I P V
H. elongata Wildtyp: ectC
GAC CAG AAG CCG CTG TAA ccc ggc gac gta
D Q K P L * H. elongata KB10.1:
xanA Hin dIII
GGC TTA ATC AGA GGT TAA aag ctt gca gta
G L I R G *
Abb. 35: Sequenzänderungen in der Insertionsmutante H. elongata KB10.1 im Vergleich zum Wildtyp
Dargestellt sind die Sequenzänderungen stromaufwärts von PP1776 bzw. ectB und stromabwärts von xanA bzw.
ectC. Diese sind durch das Einfügen von Schnittstellen (Nde I bzw. Hin dIII, dunkelgrau unterlegt) entstanden.
Der Anfang bzw. das Ende der im Austausch gegen die ectBC-Region inserierten Gene PP1776 und xanA ist
hellgrau unterlegt. „RBS“ kennzeichnet eine potentielle Ribosomen-Bindungsstelle.
3.3.2 Charakterisierung von H. elongata KB10.1
Bei der Charakterisierung von H. elongata KB10.1 wurden ähnliche Untersuchungen wie bei
H. elongata KB10 durchgeführt. Zunächst wurde die Transkription der Fremdgene mgs,
PP1776 und xanA auf RNA-Ebene mittels Northern-Hybridisierungen und RT-PCR verifiziert.
Hinzu kamen physiologische Untersuchungen, welche das Wachstumsverhalten von KB10.1
vergleichend mit dem von H. elongata KB1 (∆ectA), KB10 (∆ectA::mgs) und dem Wildtyp-
stamm untersuchten. Zuletzt wurde der Mannosylglycerat-Gehalt von H. elongata KB10.1
unter wechselnden Wachstumsbedingungen bestimmt.

III. Ergebnisse 123
H. elongataDSM 2581T ectA
Xho I5‘ ATG
ectB ectC
Sac I
ectRP1-P3
H. elongata KB10(∆ectA::mgs)
ectB ectC
5‘ ATG
mgs
Sma I Sal I
ectRP1-P3
Sac I
P. putida KT2440 PP1776PP1775 PP1778xanA
1 kb
H. elongata KB10.1(∆ectABC::mgsPP1776xanA)
ectR
5‘ ATG
mgs
Sma I Sal I
PP1776 xanA
Hin dIII
NdeI
P1-P3
Abb. 36: Genetische Organisation der PP1776xanA-Region
Dargestellt ist die genetische Organisation der PP1776xanA-Region von P. putida und verschiedener H. elongata
Stämme. Die Gene kodieren für folgende Produkte: PP1775: hypothetische Metallo-β-Lactamase, PP1776: hypo-
thetische Mannose-1-phosphat-Guanylyltransferase (NCBI-Acc.Nr.: AAN67396), xanA: hypothetische Phospho-
manno-Mutase (Acc.Nr.: AAN67397), PP1778: hypothetischer Lipopolysaccharid-Transporter, ectA: Diamino-
buttersäure-Acetyltransferase, ectB: Diaminobuttersäure-Transaminase, ectC: Ectoin-Synthase (EctABC
katalysieren die Ectoin-Synthese in H. elongata), ectR: potentieller Regulator der Ectoin-Synthese (Stumpfe,
2003; Schilz, 2005), mgs: Mannosylglycerat-Synthase aus R. marinus. symbolisiert potentielle Promotorele-
mente der Ectoin-Gene (σ70 / σS & "gearbox", Göller, 1999; Stumpfe, 2003, dieser Arbeit). Die betreffenden DNA-
Sequenzen von H. elongata KB10 und KB10.1 finden sich im Anhang der Arbeit.
3.3.2.1 Transkriptionsanalysen an KB10.1 mittels Northern-Hybridisierungen und
RT-PCR
Bei H. elongata KB10 (∆ectA::mgs) konnte gezeigt werden, dass eine mgsectB-spezifische
cDNA synthetisiert worden war, d.h. ein polycistronisches mgsectB-Kotranskript vorliegt.
Gleichermaßen wurde das Vorhandensein von Einzeltranskripten (mgs und ectB) nachge-
wiesen. Die Transkriptionsanalysen an H. elongata KB10.1 hatten eine entsprechende
Untersuchung der Expression auch der zusätzlich eingefügten hypothetischen P. putida
KT2440 Gene, PP1776 und xanA zum Ziel.
In das Northern-Hybridisierungsexperiment wurde die Gesamt-RNA der Stämme H. elongata
Wildtyp, H. elongata KB10 (∆ectA::mgs), KB10.1 (∆ectABC::mgsPP1776xanA), P. putida
KT2440 und R. marinus DSM 4252T eingesetzt. Zur RNA-Isolierung wurden die H. elongata
Stämme in MM63-4-Medium, P. putida in Medium 1 und R. marinus in M162-2-Medium an-
gezogen. Alle Stämme wurden in der spät-exponentiellen Wachstumsphase geerntet. Die

III. Ergebnisse 124
RNA aus H. elongata Wildtyp wurde als Negativkontrolle eingesetzt. Die RNAs von P. putida
bzw. R. marinus dienten als Positivkontrollen, da es sich bei diesen Stämmen um die
Donatoren für PP1776 und xanA bzw. mgs handelt. Die Hybridisierung erfolgte mit DIG-
markierten RNA-Sonden, welche für die Gene mgs, PP1776 und xanA spezifisch waren. Die
RNA-Sonden erstrecken sich über die vollständige Länge des jeweiligen Gens.
Hybridisierung mit der mgs-Sonde:
Wie bei H. elongata KB10 wurden bei KB10.1 durch Hybridisierung mit einer mgs-Sonde
(1164 bp) die Transkripte der Mannosylglycerat-Synthase nachgewiesen. In diesem Experi-
ment wurden neben KB10.1 RNA zusätzlich RNA aus H. elongata Wildtyp (Negativkontrolle)
sowie RNA aus R. marinus (Positivkontrolle) und H. elongata KB10 (∆ectA::mgs) eingesetzt.
Auf der H. elongata Wildtyp RNA wurde kein Signal nachgewiesen. Auf der R. marinus RNA
wurden eine Bande, der H. elongata KB10 RNA zwei und der KB10.1 RNA drei Banden
detektiert (Abb. 37a). Die kleinste RNA-Bande, welche in allen RNAs nachgewiesen wurde,
weist eine Länge von ca. 1,2 kb auf und entspricht damit der Größe des erwarteten mgs-
Einzeltranskripts. Die zweite RNA-Bande, die auf H. elongata KB10 RNA detektiert wurde
(ca. 2,5 kb) entspricht der Länge nach einem mgsectB-Kotranskript (siehe Abschnitt III,
3.2.2.2). Die ähnlich große Bande auf der KB10.1 RNA (ca. 2,6 kb) könnte einem
mgsPP1776-Kotranskript entsprechen. Ein mgsPP1776xanA-Gesamttranskript, welches mit
einer Größe von ca. 4,0 kb deutlich oberhalb der 23S rRNA-Bande liegen müsste, wurde nur
sehr schwach angefärbt.
Hybridisierung mit der PP1776-Sonde:
Die Mannose-1-phosphat-Guanylyltransferase wird durch das hypothetische Gen PP1776
(1442 bp) kodiert. In die Hybridisierung mit der PP1776-Sonde (1415 bp) wurde neben RNA
aus H. elongata KB10.1 (∆ectABC::mgsPP1776xanA) RNA aus H. elongata Wildtyp und
KB10 (∆ectA::mgs) als Negativkontrollen sowie RNA aus P. putida KT2440 als Positivkon-
trolle eingesetzt. Auf beiden Negativkontrollen erfolgte keine Hybridisierung der RNA-Sonde
(Abb. 37b). Auf der Positivkontrolle wurde zwei RNA-Banden angefärbt (ca. 1,4 kb und ca.
2,8 kb). Die kürzere Bande entspricht der Länge eines PP1776-Einzeltranskripts, bei der
größeren Bande könnte es sich um ein PP1776xanA-Kotranskript handeln. Entsprechende
Banden wurden auch auf der KB10.1 RNA nachgewiesen. Zusätzlich wurde hier jedoch eine
schwache Bande, die der Größe nach einem Gesamttranskript entspricht (ca. 4,0 kb), nach-
gewiesen.
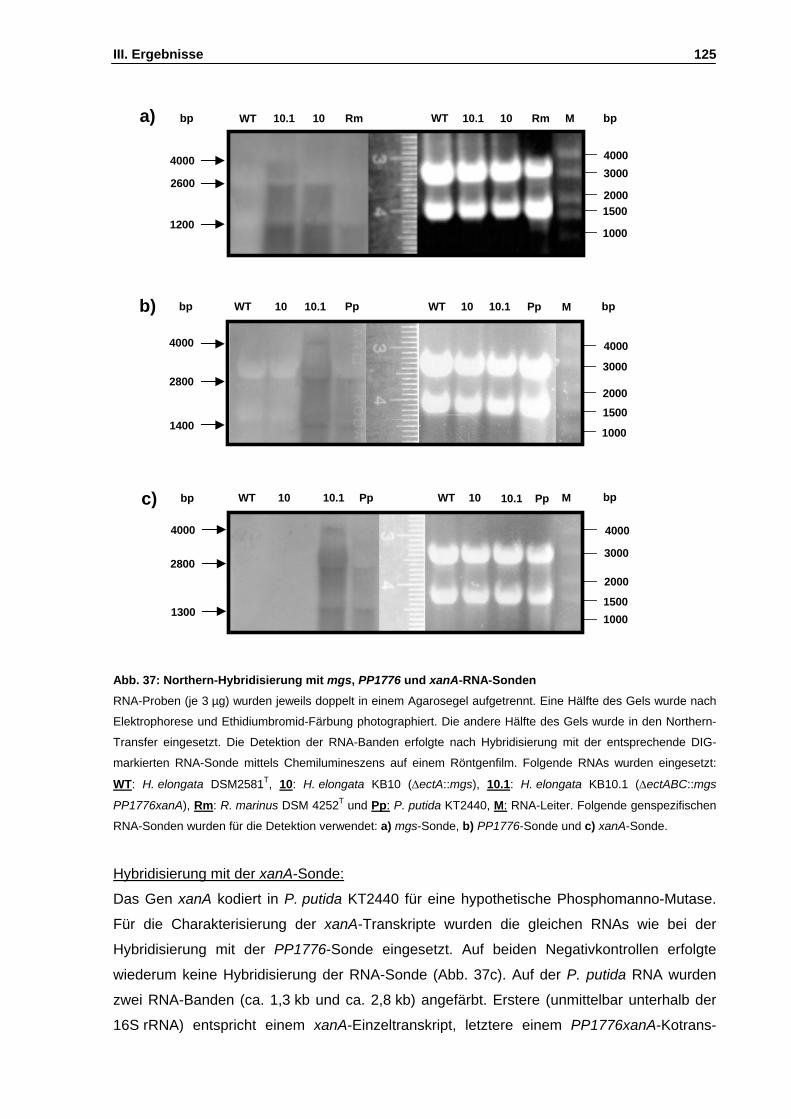
III. Ergebnisse 125
40003000
20001500
10001200
2600
4000
WT 10.1 10 Rm WT 10.1 10 Rm Ma) bpbp
WT 10.110 Pp WT 10.110 Pp M
1300
2800
4000
c)4000
3000
2000
15001000
bp bp
4000
3000
20001500
10001400
2800
4000
Pp10.110 WT 10.110 MWT Ppb) bp bp
Abb. 37: Northern-Hybridisierung mit mgs, PP1776 und xanA-RNA-Sonden
RNA-Proben (je 3 µg) wurden jeweils doppelt in einem Agarosegel aufgetrennt. Eine Hälfte des Gels wurde nach
Elektrophorese und Ethidiumbromid-Färbung photographiert. Die andere Hälfte des Gels wurde in den Northern-
Transfer eingesetzt. Die Detektion der RNA-Banden erfolgte nach Hybridisierung mit der entsprechende DIG-
markierten RNA-Sonde mittels Chemilumineszens auf einem Röntgenfilm. Folgende RNAs wurden eingesetzt:
WT: H. elongata DSM2581T, 10: H. elongata KB10 (∆ectA::mgs), 10.1: H. elongata KB10.1 (∆ectABC::mgs
PP1776xanA), Rm: R. marinus DSM 4252T und Pp: P. putida KT2440, M: RNA-Leiter. Folgende genspezifischen
RNA-Sonden wurden für die Detektion verwendet: a) mgs-Sonde, b) PP1776-Sonde und c) xanA-Sonde.
Hybridisierung mit der xanA-Sonde:
Das Gen xanA kodiert in P. putida KT2440 für eine hypothetische Phosphomanno-Mutase.
Für die Charakterisierung der xanA-Transkripte wurden die gleichen RNAs wie bei der
Hybridisierung mit der PP1776-Sonde eingesetzt. Auf beiden Negativkontrollen erfolgte
wiederum keine Hybridisierung der RNA-Sonde (Abb. 37c). Auf der P. putida RNA wurden
zwei RNA-Banden (ca. 1,3 kb und ca. 2,8 kb) angefärbt. Erstere (unmittelbar unterhalb der
16S rRNA) entspricht einem xanA-Einzeltranskript, letztere einem PP1776xanA-Kotrans-

III. Ergebnisse 126
kript. Dieses wurde auch mit der PP1776-Sonde nachgewiesen (siehe oben). Auf der KB10.1
RNA wurden entsprechende Signale angefärbt. Zusätzlich wurde hier eine weitere schwache
Bande detektiert, welche der Größe nach wiederum einem mgsPP1776xanA-Gesamt-
transkript zugeordnet werden kann (ca. 4,0 kb).
Die Ergebnisse der Northern-Hybridisierungsexperimente wiesen in H. elongata KB10.1 den
Austausch von ectABC gegen mgs, PP1776 und xanA und gleichzeitig die erfolgreiche
Transkription derselben nach. Ein mögliches mgsPP1776xanA-Gesamttranskript (ca. 4,0 kb)
wurde jeweils nur sehr schwach angefärbt. Möglicher Grund hierfür könnte die geringe Zahl
der vorhandenen mRNA-Kopien sein. Zur Bestätigung eines Gesamttranskripts wurde die
RT-PCR genutzt.
Transkriptionsanalysen an KB10.1 mittels RT- PCR:
Für den Nachweis eines mgsPP1776xanA-Gesamttranskripts wurde ausgehend des
Xangsp2 Primers cDNA synthetisiert (Abb. 38b). Zur Herstellung der cDNA wurde die
Gesamt-RNA einer KB10.1 Kultur, welche in MM63-Medium bei 4 % NaCl angezogenen
worden war, genutzt. In einer anschließenden RT-PCR wurde durch Einsatz eines in mgs
und PP1776 bindenden Primerpaars (mgsfor2 und PPrev1, Abb. 38a/b, RT-PCR 1) auf der
cDNA ein 1502 bp großes PCR-Produkt amplifiziert. Darüber hinaus wurde auf der gleichen
cDNA durch Verwendung eines in PP1776 und xanA bindenden Primerpaars (PPfor3 und
CoXanrev, Abb. 38a/b, RT-PCR 2) ein 1761 bp großes Produkt amplifiziert. Das Produkt der
Primer mgsfor2 und CoXanrev (Abb. 38a/b, RT-PCR 3) wies eine Länge von 3031 bp auf.
Durch die cDNA-Synthese und die drei anschließenden RT-PCR-Reaktionen konnte gezeigt
werden, dass die Gene mgs, PP1776 und xanA transkribiert werden und u.a. auch auf einem
gemeinsamen polycistronischen mgsPP1776xanA-Gesamttranskript vorliegen. Damit ist die
erste Voraussetzung für eine Mannosylglycerat-Synthese erfüllt. Eine Abgrenzung von
mgsPP1776 oder PP1776xanA-Kotranskripten vom Gesamttranskript kann mit dieser
Methode nicht erfolgen, da die vorliegende cDNA-Synthese nicht zwischen den unterschied-
lichen Transkriptpopulationen unterscheiden kann.
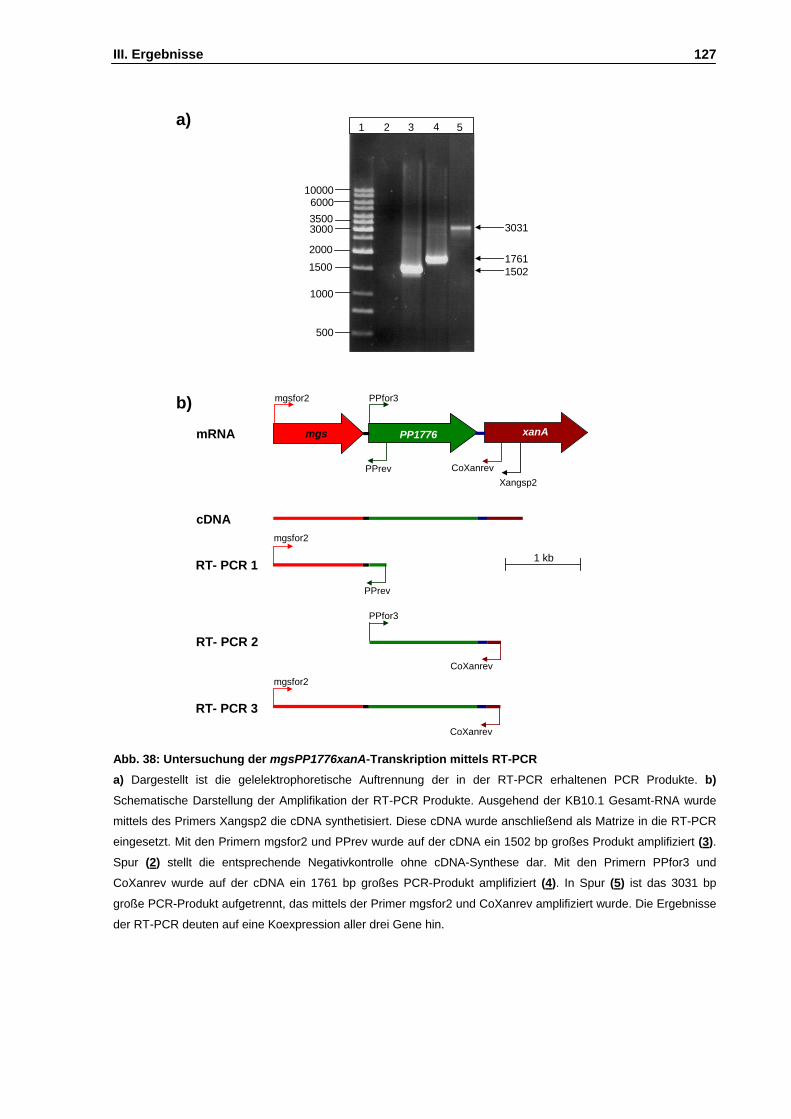
III. Ergebnisse 127
RT- PCR 1
mgsfor2
PPrev
b)
cDNA
mgs PP1776 xanAmRNA
mgsfor2
PPrevXangsp2
CoXanrev
PPfor3
1 kb
PPfor3
RT- PCR 2
CoXanrev
RT- PCR 3
mgsfor2
CoXanrev
1 2 3 4 5
100006000 3500
20001500
1000
500
3000 3031
17611502
a)
Abb. 38: Untersuchung der mgsPP1776xanA-Transkription mittels RT-PCR a) Dargestellt ist die gelelektrophoretische Auftrennung der in der RT-PCR erhaltenen PCR Produkte. b)
Schematische Darstellung der Amplifikation der RT-PCR Produkte. Ausgehend der KB10.1 Gesamt-RNA wurde
mittels des Primers Xangsp2 die cDNA synthetisiert. Diese cDNA wurde anschließend als Matrize in die RT-PCR
eingesetzt. Mit den Primern mgsfor2 und PPrev wurde auf der cDNA ein 1502 bp großes Produkt amplifiziert (3). Spur (2) stellt die entsprechende Negativkontrolle ohne cDNA-Synthese dar. Mit den Primern PPfor3 und
CoXanrev wurde auf der cDNA ein 1761 bp großes PCR-Produkt amplifiziert (4). In Spur (5) ist das 3031 bp
große PCR-Produkt aufgetrennt, das mittels der Primer mgsfor2 und CoXanrev amplifiziert wurde. Die Ergebnisse
der RT-PCR deuten auf eine Koexpression aller drei Gene hin.

III. Ergebnisse 128
3.3.2.2 Physiologische Charakterisierung von H. elongata KB10.1
Nach dem positiv verlaufenen Nachweis der Transkription der drei Fremdgene wurde bei
H. elongata KB10.1 ähnlich wie bei H. elongata KB10 die Fähigkeit, unter Salzstress zu
wachsen, untersucht. Hierzu wurde der Stamm in Mineralsalzmedium mit steigender Salinität
und Glucose als Kohlenstoffquelle bei 37 °C angezogen. H. elongata KB10.1 wies im
Vergleich zu KB10 ein nur geringfügig verbessertes Wachstum auf. Mit einer verlängerten
lag-Phase von bis zu zwei Tagen wuchs H. elongata KB10.1 auch bei einer Mediensalinität
von 5 % NaCl. Oberhalb dieser Salinität wurde kein Wachstum von KB10.1 beobachtet.
Die weiteren Untersuchungen zum Wachstumsverhalten von H. elongata KB10.1 wurden
bei 4 % NaCl und 37 °C durchgeführt. Analysiert wurde das Wachstum bei Nutzung unter-
schiedlicher Kohlenstoffquellen (Abb. 39). Sinn war der Nachweis von Mannosylglycerat und
gleichzeitig die Identifizierung der optimalen Kohlenstoffquelle zur Synthese desselben.
H. elongata Wildtyp, KB1, KB10 und KB10.1 wurden unter identischen Bedingungen ange-
zogen und für alle Stämme die Wachstumsraten vergleichend ermittelt. Für KB10.1 wurde im
Falle einer Mannosylglycerat-Synthese mit einer deutlichen Steigerung der Wachstumsrate
gegenüber H. elongata KB1 und KB10 gerechnet.
Die Wachstumsraten von H. elongata KB10.1 sind bei nahezu allen eingesetzten Kohlen-
stoffquellen dreimal so hoch wie die von H. elongata KB1. Im Vergleich zu KB10 fallen die
Steigerungen der Wachstumsraten jedoch gering aus. Beides deutete dennoch auf eine neu
erworbene biologische Fitness von H. elongata KB10.1 hin, und ließ vermuten, dass KB10.1
in der Lage ist, Mannosylglycerat zu synthetisieren und daher höhere Wachstumsraten als
KB1 und KB10 aufweist. Die Wachstumsraten des Wildtyps werden jedoch nicht erreicht.
Diese sind durchgehend 1,5 bis 2 mal so hoch wie die von KB10.1. Bei Nutzung von
Fructose als Kohlenstoffquelle weist der Wildtyp im Vergleich zu KB10.1 eine beinahe fünf-
fache Wachstumsrate auf.
Aufgrund dieser vielversprechenden Ergebnisse wurde der Mannosylglycerat-Gehalt von
H. elongata KB10.1 bestimmt. Der erste Nachweis von Mannosylglycerat erfolgte mittels
Dünnschichtchromatographie (Abb. 40). KB10.1 wurde in MM63-4-Medium bei 37 °C unter
Zusatz der oben genannten Kohlenstoffquellen bis zu einer OD600 von 1 angezogen und ge-
erntet. Die Zellextrakte von H. elongata KB10.1 wurden nach Bligh & Dyer (1959) gewonnen
(Abschnitt II, 5.3.1). Jeweils 30 µl Zellextrakt wurden auf eine DC-Platte aufgetragen und für
2 Stunden mit n-Propanol und 25 % Ammoniak (1:1, v/v) unter Kammersättigung aufge-
trennt. Die Detektion der Zuckerverbindungen erfolgte durch Einsprühen der DC-Platte mit
einem α-Naphtolschwefelsäure-Reagenz (Abb. 40).
In den Zellextrakten von KB10.1-Kulturen, welche mit Glucose, Mannose, Glycerol/Mannose,
Mannose/Glycerol bzw. Maltose angezogen worden waren, konnten mittels Dünnschicht-
chromatographie Spuren von Mannosylglycerat nachgewiesen werden (Abb. 40). Die Ergeb-
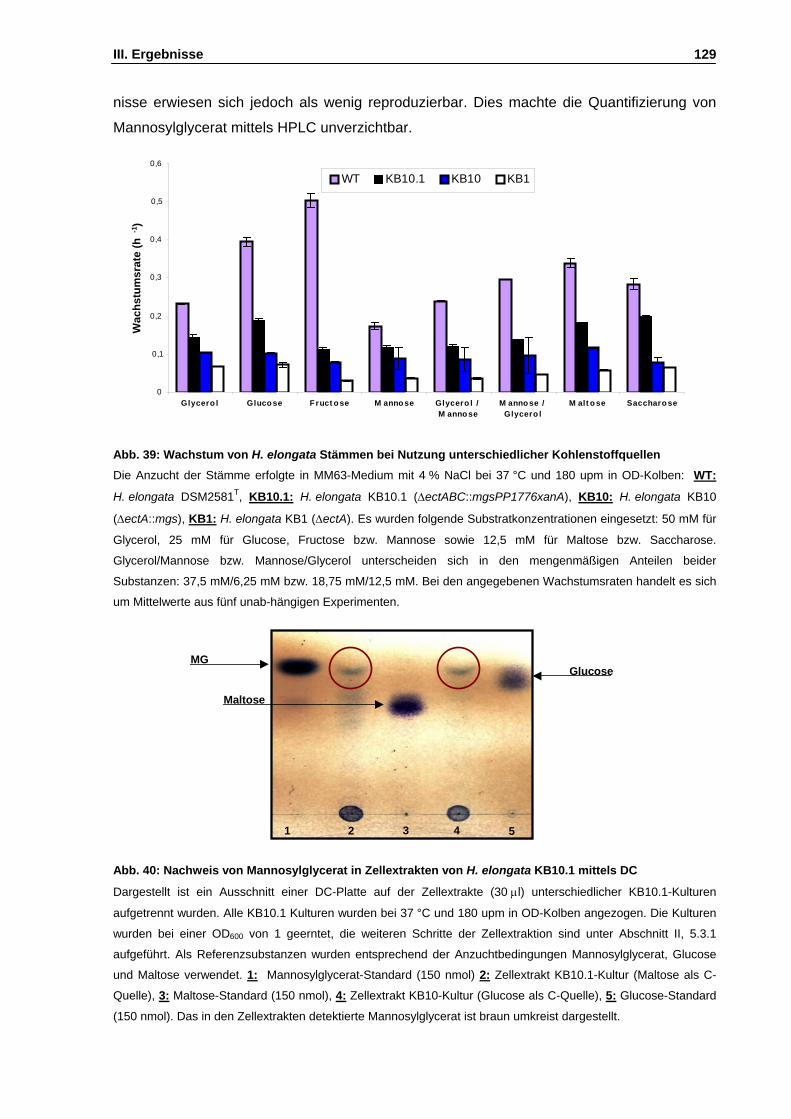
III. Ergebnisse 129
nisse erwiesen sich jedoch als wenig reproduzierbar. Dies machte die Quantifizierung von
Mannosylglycerat mittels HPLC unverzichtbar.
0
0,1
0,2
0,3
0,4
0,5
0,6
Glycero l Gluco se F ruct o se M anno se Glycero l /M anno se
M anno se /Glycero l
M alt o se Saccharo se
Wac
hstu
msr
ate
(h-1)
WT KB10.1 KB10 KB1
Abb. 39: Wachstum von H. elongata Stämmen bei Nutzung unterschiedlicher Kohlenstoffquellen
Die Anzucht der Stämme erfolgte in MM63-Medium mit 4 % NaCl bei 37 °C und 180 upm in OD-Kolben: WT:
H. elongata DSM2581T, KB10.1: H. elongata KB10.1 (∆ectABC::mgsPP1776xanA), KB10: H. elongata KB10
(∆ectA::mgs), KB1: H. elongata KB1 (∆ectA). Es wurden folgende Substratkonzentrationen eingesetzt: 50 mM für
Glycerol, 25 mM für Glucose, Fructose bzw. Mannose sowie 12,5 mM für Maltose bzw. Saccharose.
Glycerol/Mannose bzw. Mannose/Glycerol unterscheiden sich in den mengenmäßigen Anteilen beider
Substanzen: 37,5 mM/6,25 mM bzw. 18,75 mM/12,5 mM. Bei den angegebenen Wachstumsraten handelt es sich
um Mittelwerte aus fünf unab-hängigen Experimenten.
1 2 3 4 5
MG
Maltose
Glucose
Abb. 40: Nachweis von Mannosylglycerat in Zellextrakten von H. elongata KB10.1 mittels DC
Dargestellt ist ein Ausschnitt einer DC-Platte auf der Zellextrakte (30 µl) unterschiedlicher KB10.1-Kulturen
aufgetrennt wurden. Alle KB10.1 Kulturen wurden bei 37 °C und 180 upm in OD-Kolben angezogen. Die Kulturen
wurden bei einer OD600 von 1 geerntet, die weiteren Schritte der Zellextraktion sind unter Abschnitt II, 5.3.1
aufgeführt. Als Referenzsubstanzen wurden entsprechend der Anzuchtbedingungen Mannosylglycerat, Glucose
und Maltose verwendet. 1: Mannosylglycerat-Standard (150 nmol) 2: Zellextrakt KB10.1-Kultur (Maltose als C-
Quelle), 3: Maltose-Standard (150 nmol), 4: Zellextrakt KB10-Kultur (Glucose als C-Quelle), 5: Glucose-Standard
(150 nmol). Das in den Zellextrakten detektierte Mannosylglycerat ist braun umkreist dargestellt.
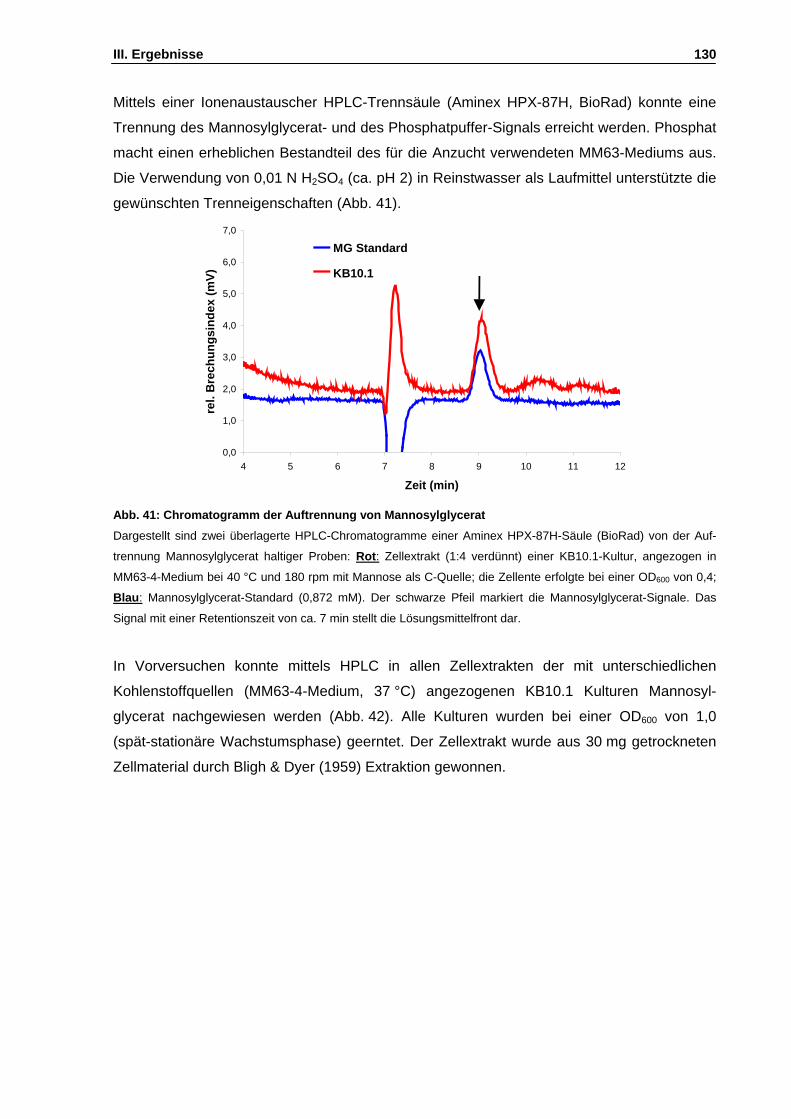
III. Ergebnisse 130
Mittels einer Ionenaustauscher HPLC-Trennsäule (Aminex HPX-87H, BioRad) konnte eine
Trennung des Mannosylglycerat- und des Phosphatpuffer-Signals erreicht werden. Phosphat
macht einen erheblichen Bestandteil des für die Anzucht verwendeten MM63-Mediums aus.
Die Verwendung von 0,01 N H2SO4 (ca. pH 2) in Reinstwasser als Laufmittel unterstützte die
gewünschten Trenneigenschaften (Abb. 41).
0,0
1,0
2,0
3,0
4,0
5,0
6,0
7,0
4 5 6 7 8 9 10 11 1
Zeit (min)
rel.
Bre
chun
gsin
dex
(mV)
2
Firoin
KB10.1
MG Standard
KB10.1
Abb. 41: Chromatogramm der Auftrennung von Mannosylglycerat
Dargestellt sind zwei überlagerte HPLC-Chromatogramme einer Aminex HPX-87H-Säule (BioRad) von der Auf-
trennung Mannosylglycerat haltiger Proben: Rot: Zellextrakt (1:4 verdünnt) einer KB10.1-Kultur, angezogen in
MM63-4-Medium bei 40 °C und 180 rpm mit Mannose als C-Quelle; die Zellente erfolgte bei einer OD600 von 0,4;
Blau: Mannosylglycerat-Standard (0,872 mM). Der schwarze Pfeil markiert die Mannosylglycerat-Signale. Das
Signal mit einer Retentionszeit von ca. 7 min stellt die Lösungsmittelfront dar.
In Vorversuchen konnte mittels HPLC in allen Zellextrakten der mit unterschiedlichen
Kohlenstoffquellen (MM63-4-Medium, 37 °C) angezogenen KB10.1 Kulturen Mannosyl-
glycerat nachgewiesen werden (Abb. 42). Alle Kulturen wurden bei einer OD600 von 1,0
(spät-stationäre Wachstumsphase) geerntet. Der Zellextrakt wurde aus 30 mg getrockneten
Zellmaterial durch Bligh & Dyer (1959) Extraktion gewonnen.
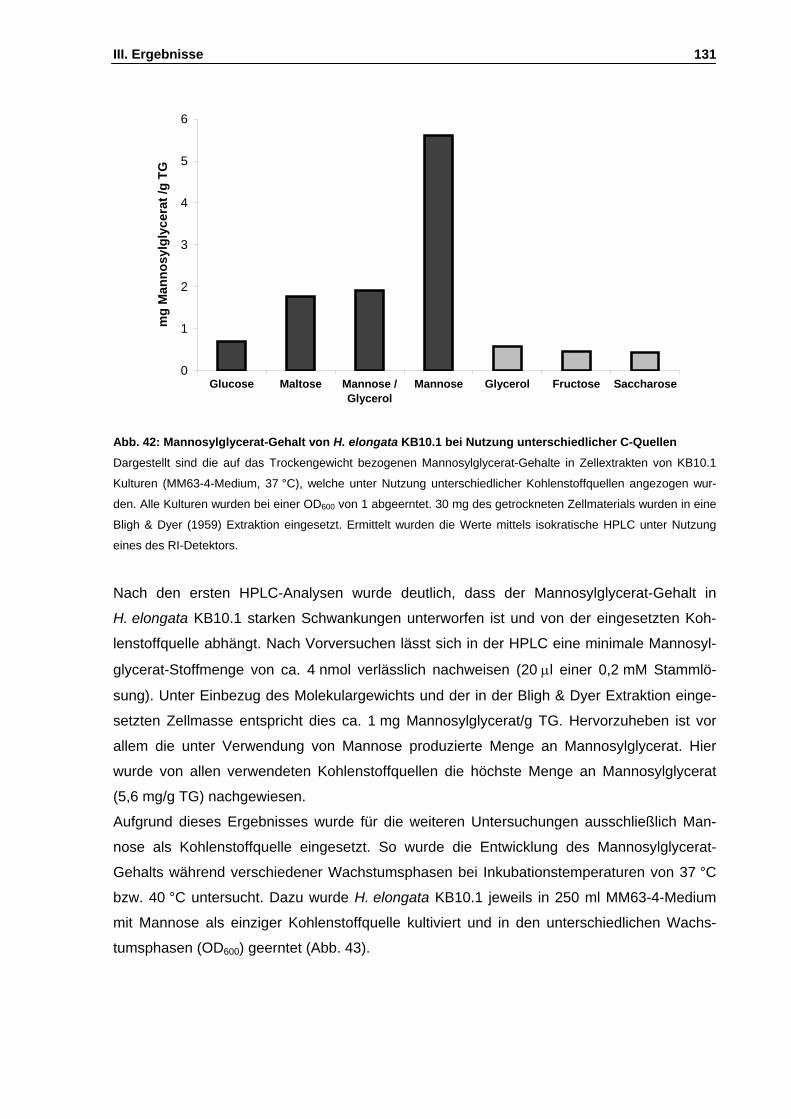
III. Ergebnisse 131
0
1
2
3
4
5
6
Glucose Maltose Mannose /Glycerol
Mannose Glycerol Fructose Saccharose
mg
Man
nosy
lgly
cera
t /g
TG
Abb. 42: Mannosylglycerat-Gehalt von H. elongata KB10.1 bei Nutzung unterschiedlicher C-Quellen
Dargestellt sind die auf das Trockengewicht bezogenen Mannosylglycerat-Gehalte in Zellextrakten von KB10.1
Kulturen (MM63-4-Medium, 37 °C), welche unter Nutzung unterschiedlicher Kohlenstoffquellen angezogen wur-
den. Alle Kulturen wurden bei einer OD600 von 1 abgeerntet. 30 mg des getrockneten Zellmaterials wurden in eine
Bligh & Dyer (1959) Extraktion eingesetzt. Ermittelt wurden die Werte mittels isokratische HPLC unter Nutzung
eines des RI-Detektors.
Nach den ersten HPLC-Analysen wurde deutlich, dass der Mannosylglycerat-Gehalt in
H. elongata KB10.1 starken Schwankungen unterworfen ist und von der eingesetzten Koh-
lenstoffquelle abhängt. Nach Vorversuchen lässt sich in der HPLC eine minimale Mannosyl-
glycerat-Stoffmenge von ca. 4 nmol verlässlich nachweisen (20 µl einer 0,2 mM Stammlö-
sung). Unter Einbezug des Molekulargewichts und der in der Bligh & Dyer Extraktion einge-
setzten Zellmasse entspricht dies ca. 1 mg Mannosylglycerat/g TG. Hervorzuheben ist vor
allem die unter Verwendung von Mannose produzierte Menge an Mannosylglycerat. Hier
wurde von allen verwendeten Kohlenstoffquellen die höchste Menge an Mannosylglycerat
(5,6 mg/g TG) nachgewiesen.
Aufgrund dieses Ergebnisses wurde für die weiteren Untersuchungen ausschließlich Man-
nose als Kohlenstoffquelle eingesetzt. So wurde die Entwicklung des Mannosylglycerat-
Gehalts während verschiedener Wachstumsphasen bei Inkubationstemperaturen von 37 °C
bzw. 40 °C untersucht. Dazu wurde H. elongata KB10.1 jeweils in 250 ml MM63-4-Medium
mit Mannose als einziger Kohlenstoffquelle kultiviert und in den unterschiedlichen Wachs-
tumsphasen (OD600) geerntet (Abb. 43).
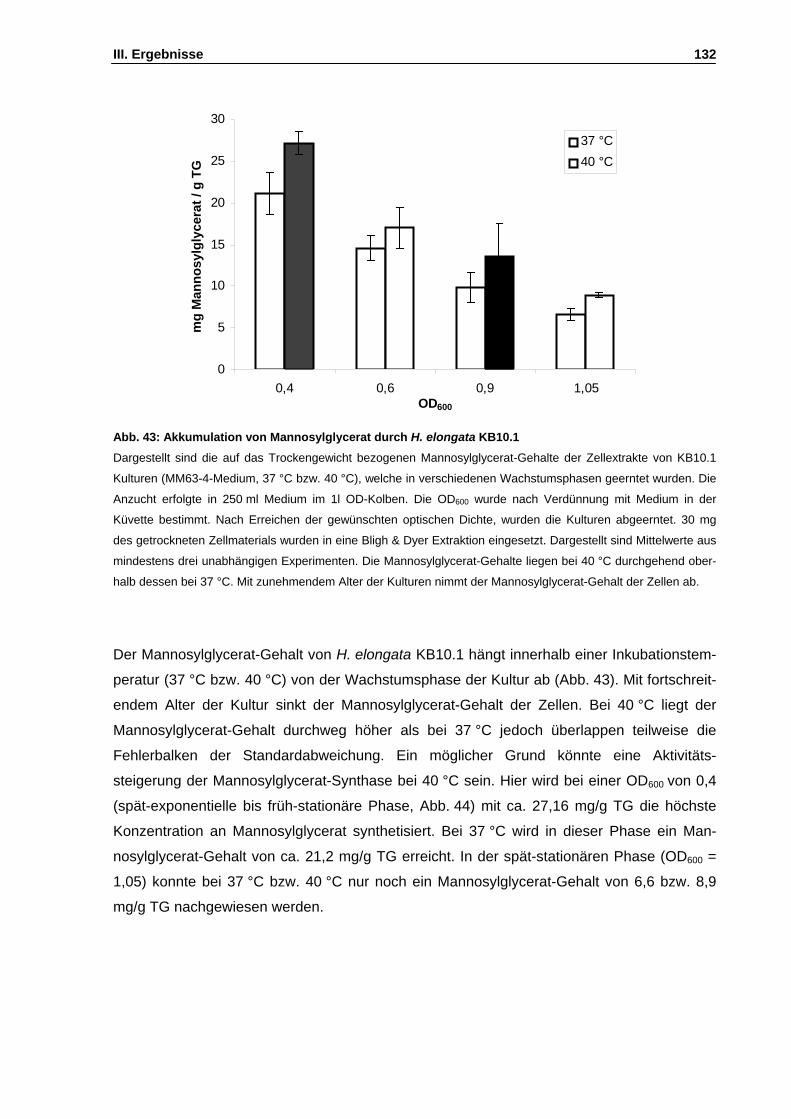
III. Ergebnisse 132
0
5
10
15
20
25
30
0,4 0,6 0,9 1,05OD600
mg
Man
nosy
lgly
cera
t / g
TG
37 °C40 °C
Abb. 43: Akkumulation von Mannosylglycerat durch H. elongata KB10.1 Dargestellt sind die auf das Trockengewicht bezogenen Mannosylglycerat-Gehalte der Zellextrakte von KB10.1
Kulturen (MM63-4-Medium, 37 °C bzw. 40 °C), welche in verschiedenen Wachstumsphasen geerntet wurden. Die
Anzucht erfolgte in 250 ml Medium im 1l OD-Kolben. Die OD600 wurde nach Verdünnung mit Medium in der
Küvette bestimmt. Nach Erreichen der gewünschten optischen Dichte, wurden die Kulturen abgeerntet. 30 mg
des getrockneten Zellmaterials wurden in eine Bligh & Dyer Extraktion eingesetzt. Dargestellt sind Mittelwerte aus
mindestens drei unabhängigen Experimenten. Die Mannosylglycerat-Gehalte liegen bei 40 °C durchgehend ober-
halb dessen bei 37 °C. Mit zunehmendem Alter der Kulturen nimmt der Mannosylglycerat-Gehalt der Zellen ab.
Der Mannosylglycerat-Gehalt von H. elongata KB10.1 hängt innerhalb einer Inkubationstem-
peratur (37 °C bzw. 40 °C) von der Wachstumsphase der Kultur ab (Abb. 43). Mit fortschreit-
endem Alter der Kultur sinkt der Mannosylglycerat-Gehalt der Zellen. Bei 40 °C liegt der
Mannosylglycerat-Gehalt durchweg höher als bei 37 °C jedoch überlappen teilweise die
Fehlerbalken der Standardabweichung. Ein möglicher Grund könnte eine Aktivitäts-
steigerung der Mannosylglycerat-Synthase bei 40 °C sein. Hier wird bei einer OD600 von 0,4
(spät-exponentielle bis früh-stationäre Phase, Abb. 44) mit ca. 27,16 mg/g TG die höchste
Konzentration an Mannosylglycerat synthetisiert. Bei 37 °C wird in dieser Phase ein Man-
nosylglycerat-Gehalt von ca. 21,2 mg/g TG erreicht. In der spät-stationären Phase (OD600 =
1,05) konnte bei 37 °C bzw. 40 °C nur noch ein Mannosylglycerat-Gehalt von 6,6 bzw. 8,9
mg/g TG nachgewiesen werden.
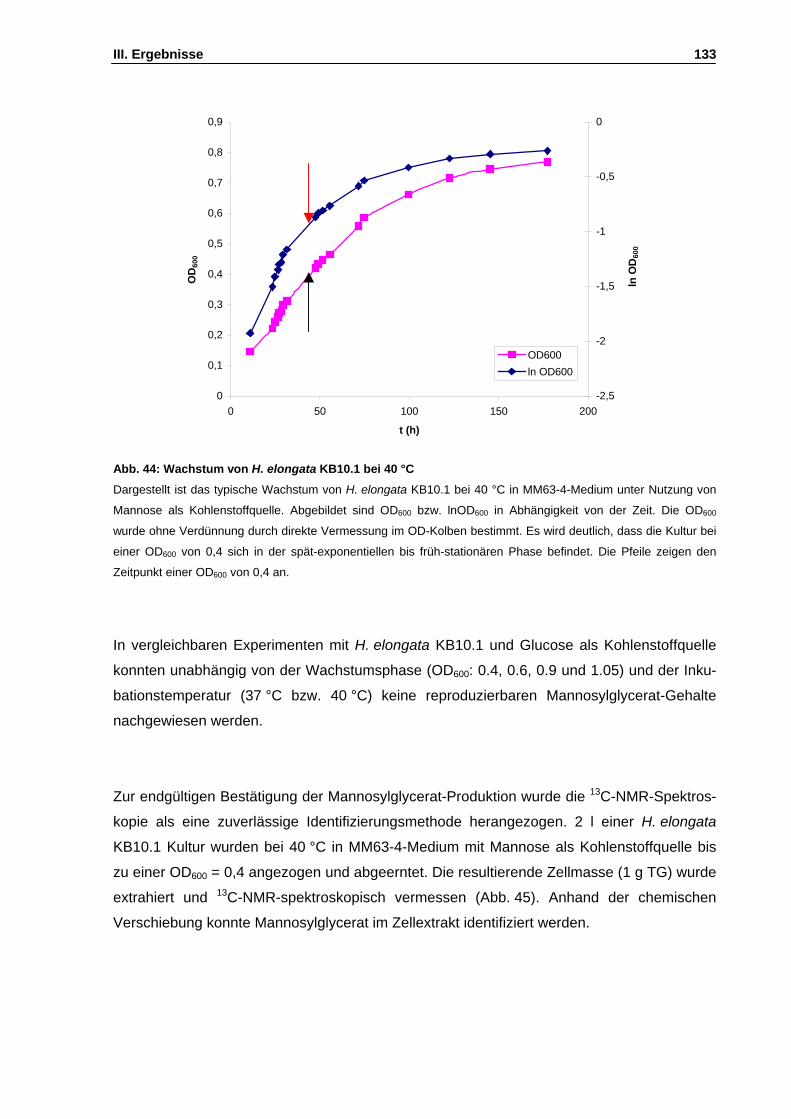
III. Ergebnisse 133
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
0 50 100 150 200
t (h)
ln O
D60
0
-2,5
-2
-1,5
-1
-0,5
0
OD
600
OD600ln OD600
Abb. 44: Wachstum von H. elongata KB10.1 bei 40 °C Dargestellt ist das typische Wachstum von H. elongata KB10.1 bei 40 °C in MM63-4-Medium unter Nutzung von
Mannose als Kohlenstoffquelle. Abgebildet sind OD600 bzw. lnOD600 in Abhängigkeit von der Zeit. Die OD600
wurde ohne Verdünnung durch direkte Vermessung im OD-Kolben bestimmt. Es wird deutlich, dass die Kultur bei
einer OD600 von 0,4 sich in der spät-exponentiellen bis früh-stationären Phase befindet. Die Pfeile zeigen den
Zeitpunkt einer OD600 von 0,4 an.
In vergleichbaren Experimenten mit H. elongata KB10.1 und Glucose als Kohlenstoffquelle
konnten unabhängig von der Wachstumsphase (OD600: 0.4, 0.6, 0.9 und 1.05) und der Inku-
bationstemperatur (37 °C bzw. 40 °C) keine reproduzierbaren Mannosylglycerat-Gehalte
nachgewiesen werden.
Zur endgültigen Bestätigung der Mannosylglycerat-Produktion wurde die 13C-NMR-Spektros-
kopie als eine zuverlässige Identifizierungsmethode herangezogen. 2 l einer H. elongata
KB10.1 Kultur wurden bei 40 °C in MM63-4-Medium mit Mannose als Kohlenstoffquelle bis
zu einer OD600 = 0,4 angezogen und abgeerntet. Die resultierende Zellmasse (1 g TG) wurde
extrahiert und 13C-NMR-spektroskopisch vermessen (Abb. 45). Anhand der chemischen
Verschiebung konnte Mannosylglycerat im Zellextrakt identifiziert werden.
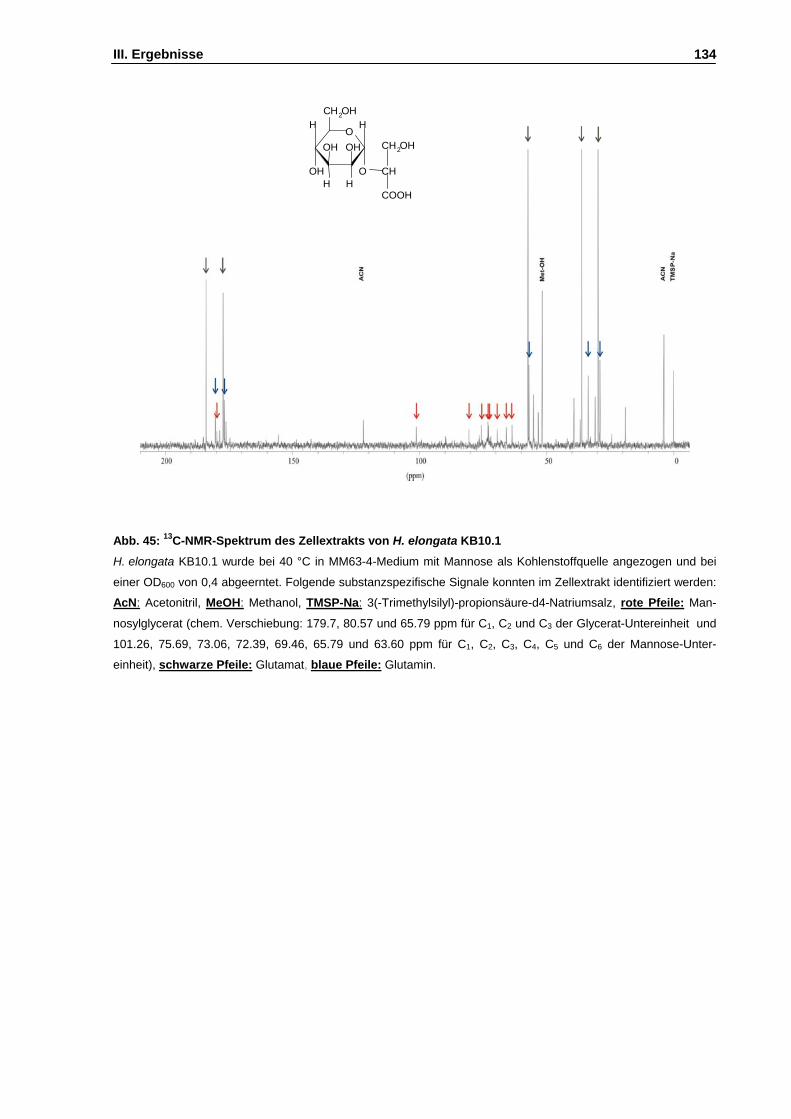
III. Ergebnisse 134
O
H
OH
H
OH
H
O CH
CH2OH
COOH
CH2OH
OH
H
Abb. 45: 13C-NMR-Spektrum des Zellextrakts von H. elongata KB10.1
H. elongata KB10.1 wurde bei 40 °C in MM63-4-Medium mit Mannose als Kohlenstoffquelle angezogen und bei
einer OD600 von 0,4 abgeerntet. Folgende substanzspezifische Signale konnten im Zellextrakt identifiziert werden:
AcN: Acetonitril, MeOH: Methanol, TMSP-Na: 3(-Trimethylsilyl)-propionsäure-d4-Natriumsalz, rote Pfeile: Man-
nosylglycerat (chem. Verschiebung: 179.7, 80.57 und 65.79 ppm für C1, C2 und C3 der Glycerat-Untereinheit und
101.26, 75.69, 73.06, 72.39, 69.46, 65.79 und 63.60 ppm für C1, C2, C3, C4, C5 und C6 der Mannose-Unter-
einheit), schwarze Pfeile: Glutamat, blaue Pfeile: Glutamin.

IV. Diskussion
1. Strategie zur Produktion von Mannosylglycerat mittels Rhodo-
thermus marinus Kompatible Solute wie Ectoin werden seit mehreren Jahren erfolgreich durch die kosmeti-
sche Industrie in Hautpflegeprodukten eingesetzt. Untersuchungen der Fa. Bitop AG zeig-
ten, dass die kompatiblen Solute Mannosylglycerat und Mannosylglyceramid bzgl. ihrer
Schutzfunktion ähnlich potente Substanzen sind. Als industrieller Produktionsstamm für
Mannosylglycerat und Mannosylglyceramid wurde Rhodothermus marinus ausgewählt, da
dieser als einziges untersuchtes thermophiles Bakterium in der Lage ist, beide kompatiblen
Solute in Abhängigkeit von der Salinität und Temperatur zu synthetisieren (Martins et al.,
1999; Silva et al., 1999). Bislang können Mannosylglycerat und Mannosylglyceramid durch
die Bitop AG nur in geringeren Mengen (1,85 g/l Fermentationsmedium) über das von Sauer
& Galinski (1998) entwickelte Bakterienmelkverfahren (hypoosmotischer Schock) aus Zell-
material von R. marinus gewonnen werden. Das generelle Funktionieren dieses Verfahrens
deutet an, dass die Regulation der Mannosylglycerat-Synthese ähnlich abläuft wie die Regu-
lation der internen Konzentration kompatibler Solute zur Osmoadaptation bei rein halophilen
Bakterien (Abb. 46). Nach Analysen von Silva et al. (1999) akkumuliert R. marinus Man-
nosylglycerat bis zu einem Gehalt von 1,17 µmol/mg Zellprotein, was nach ersten Abschät-
zungen einer berechneten zytoplasmatischen Solutekonzentration von 0,5 M entspricht
(Kunte, persönliche Mitteilung). Damit besteht ein ähnlich großer Solutegradient über der
zytoplasmatischen Membran wie er auch an halophilen Bakterien gemessen wurde.
Ziel dieser Arbeit ist die Entwicklung eines bakteriellen Produktionsstamms, der eine Ge-
winnung von Mannosylglycerat bzw. Mannosylglyceramid in größeren Mengen (> 10 g/l)
ermöglicht. Hierzu sollte nach dem Vorbild von Halomonas elongata KB2 (Volke, 2001),
einem Produktionsstamm für das kompatible Solute Ectoin, ein transportdefekter, Mannosyl-
glycerat überproduzierender Stamm von R. marinus hergestellt werden, der aufgrund einer
gestörten Regulation der Mannosylglycerat-Synthese Mannosylglycerat ins Medium abgibt
(Abb. 46). Die Nutzung von H. elongata KB2 als Produktionsstamm für Ectoin basiert auf
verschiedenen Untersuchungen, die zeigten, dass spezifische Solutetransporter nicht nur zur
Akkumulation externer Solute dienen, sondern auch für die Rückgewinnung aus der Zelle
verlorener Solute genutzt werden (Kunte, 2006). So führte bei Synechocystis sp. Stamm
PCC 6803 die Mutation der GgtA-Untereinheit des Glucosylglycerol spezifischen ABC-Trans-
porters zu einem Stamm, welcher Glucosylglycerol ins Außenmedium verliert (Hagemann et
al., 1997). Bei Bacillus subtilis führte die Deletion von opuE, welches für den alleinigen
osmoregulierten Prolin-Transporter kodiert, zu einem Prolin ausscheidenden Stamm (Von

IV. Diskussion 136
Blohn et al., 1997). Schon vorher zeigten Lamark et al. (1992) eine Zyklisierung des Betains
über der Zytoplasmamembran bei E. coli. Schließlich wiesen Arbeiten an dem halophilen
Bakterium H. elongata nach, dass das gebildete kompatible Solut Ectoin bei einer Aus-
schaltung des spezifischen Ectoin-Transporters TeaABC nicht nur kontinuierlich aus der
Zelle verloren geht, sondern der Stamm darüber hinaus Ectoin überproduziert (Grammann et
al., 2002). Abgabe und Aufnahme bilden daher vermutlich Teil eines Regulationsmechanis-
mus, um den intrazellulären Gehalt kompatibler Solute der Stoffkonzentration des Außen-
mediums anzupassen. Die Wiederaufnahme ausgeschiedener Solute durch die spezifischen
Transportsysteme dient vermutlich gleichzeitig als Signal, die Synthese dieser Verbindungen
zu verringern. Ein Ausfall der spezifischen Transportsysteme hat somit vermutlich zur Folge,
dass die Synthese des entsprechenden kompatiblen Soluts dereguliert ist (Grammann et al.,
2002; Kunte, 2006).
Mittlerweile wird durch die Bitop AG ein auf H. elongata KB2 (Volke, 2001; Grammann et al.,
2002) basierender Stamm erfolgreich für die industrielle Produktion von Ectoin eingesetzt.
Durch Mutation des spezifischen Ectoin-Transporters TeaABC ist H. elongata KB2 in der
Lage Ectoin überzuproduzieren und kontinuierlich in das Medium abzugeben. Dadurch
wurden einige Nachteile des Bakterienmelkverfahrens, wie eine zu geringe Produktkonzen-
tration in Rohlösung und die notwendige Abtrennung verunreinigender niedermolekularer
Zellbestandteile (z.B. Hydroxyectoin) überwunden. Hydroxyectoin wird durch H. elongata
KB2 nicht ins Außenmedium abgegeben, da es bei der für die Fermentation gewählten
geringen Salinität und Temperatur nicht synthetisiert wird (Wohlfarth, 1993; Volke, 2001).
Die Beobachtungen an E. coli, Synechocystis, B. subtilis und H. elongata lassen eine weite
Verbreitung des beschriebenen Mechanismus zur Regulierung des zytoplasmatischen
Solutegehalts bei Bakterien, so auch bei Rhodothermus marinus, vermuten (Abb. 46). Die
Modifikation diese Regulationsmechanismus sollte bei R. marinus nach dem Vorbild von
H. elongata KB2 zur Produktion von Mannosylglycerat genutzt werden. Dazu ist zunächst die
Identifizierung eines möglichen Transportsystems, mit dessen Hilfe R. marinus die kompati-
blen Solute Mannosylglycerat und Mannosylglyceramid aus dem Medium wiedergewinnt,
notwendig. Ein derartiger Transporter müsste nach dem Beispiel von H. elongata KB2 in
einem weiteren Schritt deletiert werden und schließlich ein industrielles Produktionsver-
fahren entwickelt werden.
Zur Identifizierung des möglichen Mannosylglycerat-Transporters wurden unterschiedliche
molekularbiologische Ansatzpunkte gewählt. Auf eine direkte Mutagenese von R. marinus
mittels Transposon mit einer anschließenden Selektion von Defektmutanten wurde verzich-
tet. Dies hätte die Etablierung gleich zweier Mutageneseverfahren erforderlich gemacht, da
vor der zu etablierenden Transposonmutagenese zunächst die ortspezifische Mutagenese
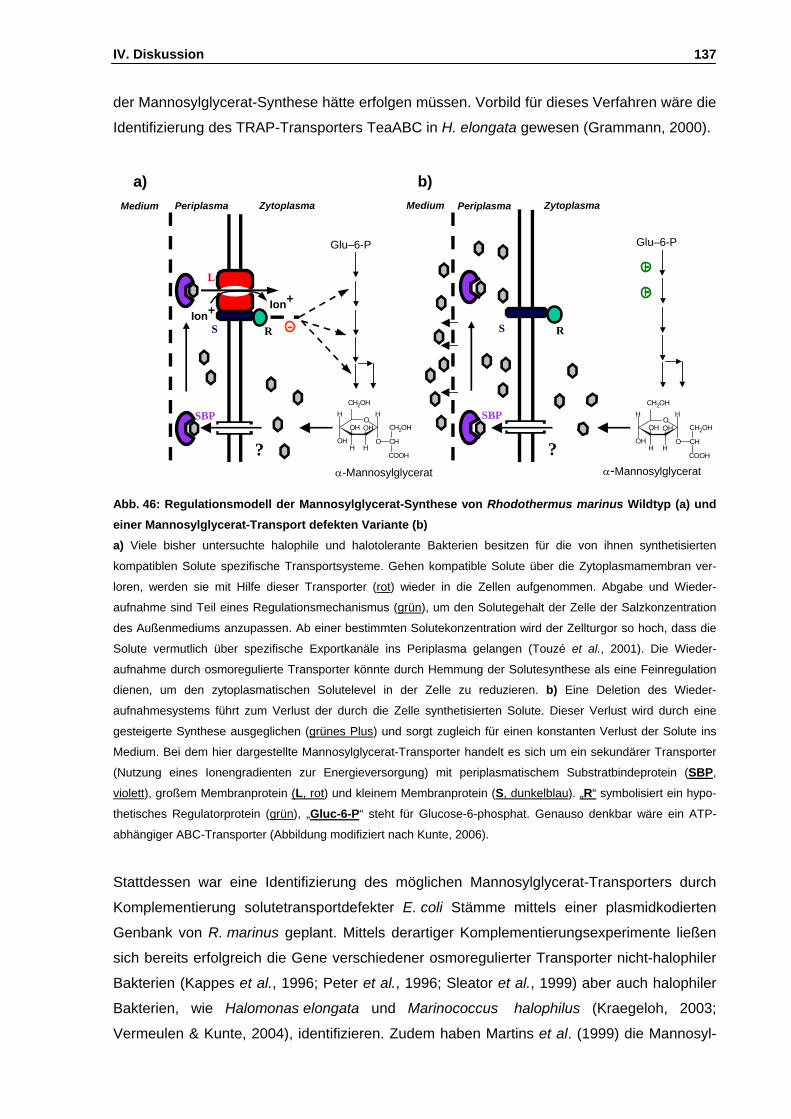
IV. Diskussion 137
der Mannosylglycerat-Synthese hätte erfolgen müssen. Vorbild für dieses Verfahren wäre die
Identifizierung des TRAP-Transporters TeaABC in H. elongata gewesen (Grammann, 2000).
Ion+ Ion+
Periplasma Zytoplasma
?
L
S R
SBP O
CH2OHH
OH
OH
H H
H
O CH
CH2OH
COOH
OH
Glu–6-P
α-Mannosylglycerat
Medium
a)Periplasma Zytoplasma
?
S R
SBP
Glu–6-P
α-Mannosylglycerat
Medium
O
CH2OHH
OH
OH
H H
H
O CH
CH2OH
COOH
OH
b)
Abb. 46: Regulationsmodell der Mannosylglycerat-Synthese von Rhodothermus marinus Wildtyp (a) und einer Mannosylglycerat-Transport defekten Variante (b) a) Viele bisher untersuchte halophile und halotolerante Bakterien besitzen für die von ihnen synthetisierten
kompatiblen Solute spezifische Transportsysteme. Gehen kompatible Solute über die Zytoplasmamembran ver-
loren, werden sie mit Hilfe dieser Transporter (rot) wieder in die Zellen aufgenommen. Abgabe und Wieder-
aufnahme sind Teil eines Regulationsmechanismus (grün), um den Solutegehalt der Zelle der Salzkonzentration
des Außenmediums anzupassen. Ab einer bestimmten Solutekonzentration wird der Zellturgor so hoch, dass die
Solute vermutlich über spezifische Exportkanäle ins Periplasma gelangen (Touzé et al., 2001). Die Wieder-
aufnahme durch osmoregulierte Transporter könnte durch Hemmung der Solutesynthese als eine Feinregulation
dienen, um den zytoplasmatischen Solutelevel in der Zelle zu reduzieren. b) Eine Deletion des Wieder-
aufnahmesystems führt zum Verlust der durch die Zelle synthetisierten Solute. Dieser Verlust wird durch eine
gesteigerte Synthese ausgeglichen (grünes Plus) und sorgt zugleich für einen konstanten Verlust der Solute ins
Medium. Bei dem hier dargestellte Mannosylglycerat-Transporter handelt es sich um ein sekundärer Transporter
(Nutzung eines Ionengradienten zur Energieversorgung) mit periplasmatischem Substratbindeprotein (SBP,
violett), großem Membranprotein (L, rot) und kleinem Membranprotein (S, dunkelblau). „R“ symbolisiert ein hypo-
thetisches Regulatorprotein (grün), „Gluc-6-P“ steht für Glucose-6-phosphat. Genauso denkbar wäre ein ATP-
abhängiger ABC-Transporter (Abbildung modifiziert nach Kunte, 2006).
Stattdessen war eine Identifizierung des möglichen Mannosylglycerat-Transporters durch
Komplementierung solutetransportdefekter E. coli Stämme mittels einer plasmidkodierten
Genbank von R. marinus geplant. Mittels derartiger Komplementierungsexperimente ließen
sich bereits erfolgreich die Gene verschiedener osmoregulierter Transporter nicht-halophiler
Bakterien (Kappes et al., 1996; Peter et al., 1996; Sleator et al., 1999) aber auch halophiler
Bakterien, wie Halomonas elongata und Marinococcus halophilus (Kraegeloh, 2003;
Vermeulen & Kunte, 2004), identifizieren. Zudem haben Martins et al. (1999) die Mannosyl-

IV. Diskussion 138
glycerat-Synthase aus R. marinus erfolgreich in E. coli exprimiert. Dies wurde als Hinweis
gedeutet, dass solche Experimente prinzipiell auch mit R. marinus DNA und E. coli möglich
sind.
2. Physiologische Charakterisierung von R. marinus Grundvoraussetzung für ein erfolgreiches Arbeiten mit R. marinus ist die nähere physio-
logische Charakterisierung des Stamms. In den ersten physiologischen Charakterisierungen
von Rhodothermus marinus bestätigten sich die Angaben der Erstbeschreibung von
Alfredsson et al. (1988). So liegt das auf die maximale Wachstumsrate (ca. 0,5 h-1) bezogene
Salinitätsoptimum von R. marinus bei einer Anzuchttemperatur von 65 °C in M162-Medium
bei 2 % NaCl. Bei höheren Salinitäten sinkt die Wachstumsrate kontinuierlich ab (0,145 h-1
bei 5,5 % NaCl). Bei einer Mediensalinität von 1 % erreicht R. marinus eine Wachstumsrate
von 0,39 h-1.
Die Untersuchung des Substratsspektrums von R. marinus stimmt gleichermaßen mit den
Ergebnissen der Erstbeschreibungen überein. Hierzu wurde das modifiziertes Mineralsalz-
medium MM162 (Alfredsson et al., 1985) verwendet. Die Versuche wurden bei 65 °C und
einer Mediensalinität von 2 % durchgeführt. R. marinus ist in der Lage die meisten einge-
setzten Mono- und Disaccharide (z.B. Trehalose) zur Gewinnung von Biomasse zu nutzen,
während dies bei Betain und seinen Vorstufen, aber auch Zuckeralkoholen wie Mannitol und
Arabitol nicht der Fall ist. Bis auf L-Glutamat und L-Aspartat wurden keine der eingesetzten
Aminosäuren zur Gewinnung von Biomasse herangezogen. Möglicherweise fehlen
R. marinus die erforderlichen Transporter, um diese Aminosäuren ins Zytoplasma zu trans-
portieren und anschließend als Kohlenstoffquelle zu nutzen. Auch die zelleigenen kompa-
tiblen Solute Mannosylglycerat und Mannosylglyceramid wurden durch R. marinus nicht ver-
wertet. Dies könnte darauf hinweisen, dass R. marinus entweder nicht in der Lage ist, seine
Solute zur Energiegewinnung heranzuziehen, wie es z.B. H. elongata beim eigenen Solut
Ectoin vermag, oder er keine entsprechenden Transportersysteme besitzt.
Ein vager Hinweis auf das Vorhandensein möglicher Mannosylglycerat-Transportsysteme in
R. marinus könnte eine Wachstumsverbesserung des Stamms verursacht durch dem
Medium zugesetzte kompatible Solute sein. Dazu wurden Wachstumsraten in Gegenwart
von je 1 mM Mannosylglycerat, Mannosylglyceramid bzw. Trehalose im Vergleich zur
Kontrolle (ohne Solutezusatz) bei Salinitäten von 4 und 5,5 % NaCl ermittelt. Eine gering-
fügige Steigerung der Wachstumsraten gegenüber der Kontrolle war nur bei einer Medien-
salinität von 5,5 % und Zusatz der Solute zu beobachten. Durch Zugabe von Mannosyl-
glycerat bzw. Mannosylglyceramid stieg die Wachstumsrate von 0,090 h-1 (Kontrolle) auf
0,1116 h-1 bzw. 0,1168 h-1. Allerdings überlappen die Bereich der Standardabweichungen,
was die Signifikanz der Wachstumsverbesserung in Frage stellt. Gerade bei Aufnahme eines

IV. Diskussion 139
„energetisch teuren“ Soluts wie Mannosylglycerat (Abschnitt IV, 8.2.2.5) wäre mit einer
deutlicheren Steigerung der Wachstumsrate zu rechnen, denn hierdurch böte sich ein
energetischer Vorteil, da die Eigensynthese entfällt. Das Vorhandensein osmoregulatorischer
Transportsysteme für die kompatiblen Solute Mannosylglycerat und Mannosylglyceramid
erscheint somit fraglich. Anders sieht es bei Zusatz von Trehalose aus. Hier ist eine
Steigerung der Wachstumsrate auf 0,142 h-1 zu beobachten. Allerdings könnte im Falle der
Trehalose auch eine Nutzung als Kohlenstoffquelle einen positiven Effekt auf das Wachstum
ausüben. Außerdem wurden für Trehalose durch Welsh & Herbert (1999) membranstabili-
sierende Effekte beobachtet, welche gleichermaßen eine Beschleunigung des Wachstum
von R. marinus zur Folge haben könnten.
Bei den Untersuchungen zur Abhängigkeit des Mannosylglycerat-Gehalts von der Medien-
salinität wurde R. marinus sowohl in M162-Komplexmedium als auch in MM162-Mineral-
salzmedium mit je 1–4 % NaCl angezogen. In beiden Medien wurde mittels HPLC ein fünf-
facher Anstieg der Mannosylglycerat-Produktion bei Erhöhung der Mediensalinität von 1 %
auf 2 % NaCl nachgewiesen. Nunes et al. (1995) konnten bei einer Mediensalinität von 1%
im Zellextrakt kein Mannosylglycerat nachweisen. Darüber hinaus liegen die übrigen Man-
nosylglycerat-Gehalte im Allgemeinen um einen Faktor 1,4 bis 1,9 niedriger als die in der
vorliegenden Arbeit bestimmten Gehalte. Der Unterschied könnte in der Verwendung unter-
schiedlicher Quantifizierungsmethoden begründet sein, zumal Nunes et al. (1995) mittels
NMR-Spektroskopie quantifizierten. Bei Mediensalinitäten von 1–3 % NaCl wird in MM162-
Mineralsalzmedium mehr Mannosylglycerat akkumuliert als in M162-Komplexmedium
(Faktor 1,3). Dieser Unterschied verringert sich bei 4 % NaCl. Grund für den in Komplex-
medium geringeren Mannosylglycerat-Gehalt könnte die im Medium enthaltene Trehalose
sein. Diese wird möglicherweise als kompatibles Solute akkumuliert. Betain hingegen wird
durch R. marinus nicht aufgenommen. Bei steigender Salinität kann Trehalose dann mög-
licherweise nicht mehr als effektives Solute dienen und wird zunehmend durch Mannosyl-
glycerat ersetzt. Trehalose dient ausschließlich bei nicht-halophilen und nicht-halotoleranten
Organismen als kompatibles Solut (Galinski, 1995). Möglicherweise spielt auch das höhere
spezifische Gewicht der Trehalose oder die Zunahme der Viskosität des Zytoplasmas eine
Rolle, wie es durch Oren (1999) für Disaccharide-Solute vermutet wurde.
3. Komplementierung von E. coli Stämmen mittels Genbänken von
Rhodothermus marinus Erstes Ziel der Arbeit war die Identifizierung eines R. marinus Transporters zur Aufnahme
von Mannosylglycerat und Mannosylglyceramid mittels Komplementierung eines solutetrans-
portdefekten E. coli Stamms durch eine plasmidkodierte Genbank von R. marinus. Grund-
voraussetzung für ein solches Verfahren ist die Verfügbarkeit von Defektmutanten, welche

IV. Diskussion 140
für die jeweiligen Komplementierungen geeignet sind. Darüber hinaus muss die Fremd-DNA
in diesen Stämmen auch funktionell exprimiert werden. Da es sich bei dem geplanten
Verfahren um die heterologe Expression eines Gens in einem physiologisch unterschied-
lichen Wirt handelt, kann es aufgrund verschiedener Faktoren zu Problemen kommen. So ist
es wichtig, dass die Promotersequenzen und Ribosomen-Bindungsstellen von R. marinus in
E. coli erkannt werden. Zudem müssen sich die kodierten Proteine in E. coli in einen aktiven
Zustand befinden, was voraussetzt, dass eine geeignete zelluläre Zielsteuerung, korrekte
Faltung und posttranslationale Modifikation der Proteine erfolgt. Das Protein kann außerdem
aufgrund unterschiedlicher pH-, Temperatur- bzw. Salzoptima eine verminderte Aktivität auf-
weisen. Ein weiteres wichtiges Problem bei der heterologen Expression könnte die sog.
„Codon usage“ des Wirts darstellen. Beinhaltet z.B. die mRNA eines Proteins eine große
Zahl des Kodons „AUA“ (kodiert für Isoleucin und ist eines der fünf seltenen Kodons in der
mRNA von E. coli) muss mit Problemen bei der Translation des Proteins gerechnet werden
(Spanjaard et al., 1990; Rosenberg et al., 1993; Del Tito Jr et al., 1995).
3.1 Identifizierung der Thiamin-Gene von Rhodothermus marinus Um vorab die generelle Möglichkeit einer funktionellen Expression von R. marinus Genen in
E. coli Defektmutanten zu überprüfen, wurde zunächst die Thiamin-Auxotrophie von E. coli
DH5α durch eine plasmidkodierte Genbank von R. marinus komplementiert. Dies bestätigte
als erste wichtige Voraussetzung die Funktionalität der Ribosomen-Bindungsstellen und
möglicherweise auch der Promotoren von R. marinus in E. coli. Gleichzeitig zeigte sich, dass
thermophile Enzyme in E. coli bei 37 °C in einem enzymatisch aktiven Zustand exprimiert
werden können.
Bei der Komplementierung wurden vier hypothetische Gene (thiSGED) identifiziert, deren
hypothetische Genprodukte unmittelbar an der Thiamin-Biosynthese von R. marinus beteiligt
zu sein scheinen. Thiaminpyrophosphat ist ein essentieller Kofaktor verschiedener Enzyme,
wie z.B. der Transketolase, Pyruvat-Dehydrogenase und α-Ketoglutarat-Dehydrogenase
(Vander Horn et al., 1993; Petersen & Downs, 1997). Die Thiamin-Gene bilden häufig
Cluster, die jedoch verstreut im Genom vorliegen. So wurden z.B. in E. coli fünf Thiamin-
Gene (thiCEFGH) identifiziert, die gemeinsam transkribiert werden, weitere Gene liegen
verstreut vor (Vander Horn et al., 1993). In B. subtilis wurden zwei Thiamin-Biosynthese-
operons und ein einzelner Genlocus identifiziert (Zhang, Y & Begley, 1997). Zumeist werden
die Transkriptionseinheiten durch Thiamin reguliert (Vander Horn et al., 1993; Petersen &
Downs, 1997; Morett et al., 2003).
Bei der Subklonierung des Plasmids pThai1, welches die Thiamin-Gene (thiSGED) enthält,
entstand Plasmid pThiaEco (Abschnitt III, 2.2.2). Auch dieses komplementiert den Thiamin-
Synthesedefekt. Die Sequenz von thiS ist in den Vektoren pThia1 und pThiaEco nur unvoll-

IV. Diskussion 141
ständig enthalten. thiS kann daher nicht für die Komplementierung von E. coli DH5α verant-
wortlich sein. Letzteres gilt auch für thiD, welches durch die Subklonierung in pThiaEco
unterbrochen wurde. pThiaEco enthält die vollständigen Sequenzen von thiG und thiE.
Daher ist davon auszugehen, dass eines dieser Gene für diese Komplementierung verant-
wortlich ist. Die Produkte beider Gene weisen hohe Sequenzidentitäten (ThiG 46 %, ThiE
39 %) mit E. coli Proteinen auf. Das Funktionieren einer derartigen Komplementierung
fehlender Thiamin-Gene bei E. coli durch die Gene eines thermophilen Bakteriums, zeigten
auch die Untersuchungen von Morett et al. (2003). Hier wurde ein Fehlen des thiE Gens von
E. coli durch die Gene thiDN aus Thermotoga maritima komplementiert, obwohl die beiden
Genprodukte ThiD und ThiN erst bei 72 °C ihre höchste enzymatische Aktivität erweisen.
Die erfolgreiche funktionelle Komplementierung des Thiamin-Synthesedefekts von E. coli
DH5α durch die Thiamin-Gene von R. marinus wurde als Hinweis gewertet, dass ein solches
Verfahren auch für die Identifizierung eines möglichen Mannosylglycerat-Transporters mittels
der Transportdefektmutante E. coli MKH13 genutzt werden kann.
3.2 Untersuchung zur Identifizierung von Mannosylglycerat-Transportern
mittels funktioneller Komplementierung transportdefekter E. coli Stämme
3.2.1 Komplementierungsexperimente mit E. coli MKH13
Durch funktionelle Komplementierung der Thiamin-Auxotrophie von E. coli DH5α wurden vier
Thiamin-Gene in R. marinus identifiziert. Die gleiche Genbank von R. marinus wurde in die
solutetransportdefekte Mutante E. coli MKH13 transformiert, um einen Mannosylglycerat-
Transporter zu identifizieren. Dieser Stamm zeichnet sich dadurch aus, dass er durch das
Fehlen der Transportsysteme BetT, PutP, ProP und ProU nicht in der Lage ist, extern
zugesetzte kompatible Solute aus dem Medium aufzunehmen. Zudem kann er Glycin-Betain
nicht aus der Vorstufe Cholin (∆betT) synthetisieren (Haardt et al., 1995). Nach Larsen et al.
(1987) kann E. coli K12 mit seinem eigens synthetisierten kompatiblen Solut Trehalose in
Mineralsalzmedium nur bis zur einer Mediensalinität von 3,8 % NaCl wachsen, wenn dem
Medium keine Solute halophiler Organismen zugesetzt sind. Vorversuche an E. coli MKH13
bestätigen dieses Wachstumsverhalten. Medienzusatz von Mannosylglycerat und Mannosyl-
glyceramid steigerten die Salztoleranz von E. coli K12 auf 5 % NaCl, von MKH13 jedoch
nicht.
Die Selektion plasmidtragender MKH13 Klone auf Mineralsalzmedium mit 4 % NaCl und
1 mM Mannosylglycerat bzw. Mannosylglyceramid führte nicht zur Identifizierung möglicher
Transporter. Daher wurde vermutet, dass die endogene Trehalose-Synthese von E. coli
MKH13 diesem über eine Zeitspanne von mehreren Tagen ein Wachstum bei 4 % NaCl
erlaubt. Darüber hinaus wurde vermutet, dass die Salinität des Selektionsmediums die Aktivi-

IV. Diskussion 142
tät des möglichen Transporters negativ beeinflusst. So haben Peter et al. (1996) bei der
heterologen Expression des osmoregulierten Glycin-Betain-Transporters BetP aus
C. glutamicum in E. coli MKH13 eine Verschiebung des Aktivitätsoptimums des Transporters
in Richtung einer geringeren Salzkonzentration beobachtet. Bei einer Mediensalinität ober-
halb von 3 % NaCl wurde in E. coli MKH13 keine Transporteraktivität mehr nachgewiesen,
während im Ursprungsorganismus (C. glutamicum) noch bei über 8 % NaCl Aktivität vor-
handen war. Unter der Annahme ähnlicher Auswirkungen wurde beschlossen eine Selektion
bei geringerer Mediensalinität durchzuführen. Voraussetzung dafür war jedoch die Her-
stellung von E. coli BKA-13.
3.2.2 Komplementierungsexperimente mit E. coli BKA-13
In osmotisch gestressten E. coli und S. typhimurium Zellen wird Trehalose de novo synthe-
tisiert (Kaasen et al., 1992; Strøm & Kaasen, 1993). Der osmoregulierte Trehalose-Syn-
theseweg beginnt mit der Kondensation von UDP-Glucose und Glucose-6-phosphat zu
Trehalose-6-phoshphat (Trehalose-6-phosphat-Synthase, kodiert durch otsA). Die Synthase
wird durch hohen Konzentrationen von Kaliumglutamat aktiviert, die in E. coli als erste Ant-
wort auf Osmostress akkumuliert werden. Trehalose entsteht dann unter Abspaltung des
Phosphat-Rests von Trehalose-6-phosphat durch Einwirkung einer Phosphatase (kodiert
durch otsB, Kaasen et al., 1992).
Durch Deletion von otsB wurde die Trehalose-Biosynthese in E. coli MKH13 ausgeschaltet.
Der resultierende Stamm trägt die Bezeichnung E. coli BKA-13 und weist bis maximal 2,8 %
NaCl ein Wachstum auf Mineralsalzmedium auf. Die plasmidtragenden Klone von E. coli
BKA-13 wurden auf Mineralsalzmedium mit 3 % NaCl und 1 mM Mannosylglycerat bzw.
Mannosylglyceramid selektioniert. Wiederum war keine funktionelle Komplementierung zu
beobachten, d.h. es wurde kein Mannosylglycerat-Transportssystem identifiziert. Das nega-
tive Ergebnis führte zu der Vermutung, dass die Expression des Mannosylglycerat-
Aufnahmesystems durch unbekannte thermophile Promotoren und Regulationselemente
gesteuert wird, welche in E. coli Stämme nicht aktiv sind. Um dies zu umgehen, wurde eine
zusätzliche Genbank aus R. marinus DNA in Plasmid pXMJ19 (Jakoby et al., 1999) erstellt.
Bei pXMJ19 handelt es sich um ein Derivat des Plasmids pMMB67HE (Fürste et al., 1986).
Es besitzt einen induzierbaren tacP-Hybrid Promotor (De Boer et al., 1983) und ist sowohl in
Gram-positiven Bakterien als auch in Gram-negativen Bakterien wie E. coli replizierbar
(Jakoby et al., 1999, Abschnitt II, 14.2). Der tacP-Promotor unterliegt dem lac-Repressor und
wird durch IPTG induziert. Auch mit diesem Expressionssystem war bei Induktion mit IPTG
keine funktionelle Komplementierung von E. coli BKA-13 zu beobachten.
Hinter dem Fehlschlagen der funktionellen Komplementierung wurden Gründe vermutet, die
mit dem geplanten Durchführungskonzept nicht umgangen werden konnten. So könnte es

IV. Diskussion 143
sich bei einem Mannosylglycerat-Aufnahmesystem um ein Mehrkomponenten-System han-
deln, dessen Strukturgene über das Genom von R. marinus verstreut vorliegen. Obwohl die
meisten Dreikomponenten-Transportsysteme (ABC-Transporter), wie z.B. die osmoregulier-
ten Betain-Transporter OpuA aus B. subtilis (Kempf & Bremer, 1998) und BusA aus Lacto-
coccus lactis (Obis et al., 1999) in einem Operon organisiert vorliegen, sind die Komponen-
ten einiger Zuckertransporter extremophiler Mikroorganismen, wie z.B. der hochaffinen
Maltose/Trehalose ABC-Transporter aus Thermus thermophilus (Silva et al., 2005) und
Thermococcus litoralis (Horlacher et al., 1998) oder des Maltose/Maltodextrin ABC-Trans-
porter aus dem thermo-acidophilen Gram-positiven Bakterium Alicyclobacillus acidocaldarius
(Hülsmann et al., 2000) über das Genom verteilt. Das Gen des ATP-Bindeproteins liegt
jeweils nicht mit den restlichen Komponenten in einem Operon organisiert vor. Auch die
unterschiedliche Physiologie von Spender- und Empfängerorganismus könnte ein Rolle bei
der Aktivität des Aufnahmesystems spielen. Offenbar hängt die Aktivität eines Transporters
unter anderem von der Zusammensetzung der umgebenden Lipiddoppelschicht ab (Peter et
al., 1996). Die Phospholipid-Zusammensetzung variiert nicht nur zwischen Gram-positiven
und Gram-negativen Bakterien, sondern wird auch durch äußere Umwelteinflüsse wie Aus-
trocknung, osmotischen Stress, Temperatur und pH-Wert stark beeinflusst (Miller, K J, 1985;
Kaye & Baross, 2004; Schiller et al., 2006). Die unterschiedliche Membranzusam-
mensetzung von E. coli und R. marinus macht sich besonders beim Vergleich der Fettsäuren
bemerkbar. Während bei E. coli hauptsächlich gesättigte (Palmitinsäure C16, 45 %) und
ungesättigte Fettsäuren (Palmitoleinsäure C16:1∆9, 35 % bzw. Vaccensäure C18:1∆11, 18 %)
vorliegen (Lengeler et al., 1999), beträgt der Anteil an gesättigter Palmitinsäure (C16) bei
R. marinus nur 5,4 %, während die verzweigten Fettsäuren wie Iso-Margarinsäure (iC17,
26,4 %) und Iso-Palmitinsäure (iC16, 13,0 %) den überwiegenden Anteil der Fettsäuren aus-
machen (Nunes et al., 1992; Moreira et al., 1996). Auch die Phospholipid-Zusammensetzung
unterscheidet sich in beiden Organismen. Den Hauptanteil von Phospholipiden bilden in
R. marinus das anionische Diphosphatidylglycerol (DPG) und das ungeladene Phosphatidyl-
ethanolamin (PE, Tindall, 1991). In E. coli hingegen überwiegt der Anteil an ungeladenem
Phosphatidylethanolamin (PE, 75 %). Hinzu kommt ein Anteil des anionischen Phosphatidyl-
glycerols (PG, 18 %) (Lengeler et al., 1999). Gleichermaßen können unterschiedliche
Turgorverhältnisse zu einer eingeschränkten bzw. veränderten Aktivität eines Transportsys-
tems führen (Peter et al., 1996).

IV. Diskussion 144
4. Isolierung eines DNA-locus von R. marinus mit Ähnlichkeiten zu
Natrium/Glucose-Symportern
Nachdem mittels funktioneller Komplementierung eine Identifizierung eines Mannosylgly-
cerat-Transporters nicht möglich war, wurde nach alternativen Strategien Ausschau gehal-
ten. In einer Arbeit von Sampaio et al. (2003b) ergaben sich Hinweise, dass die beiden 14C-
markierten kompatiblen Solute Mannosylglycerat und Trehalose (beide Substrate sind
α-Glucoside) mittels eines ABC-Transportsystems in die Zellen des thermophilen Bakteriums
Thermus thermophilus aufgenommen werden. Bei dem untersuchten System handelte es
sich um einen Trehalose/Maltose-Transporter. Eine nähere Charakterisierung des Transpor-
ters durch Silva et al. (2005) zeigte, dass dieser große Ähnlichkeiten zu bereits beschrie-
benen Trehalose/Maltose ABC-Aufnahmesysteme aus Thermococcus litoralis (Xavier et al.,
1996) und Pyrococcus furiosus (Koning et al., 2002) aufweist. Bemerkenswert war, dass
auch diese (hyper-) thermophilen Mikroorganismen in der Lage sind Mannosylglycerat de
novo zu synthetisieren (Martins & Santos, 1995; Nunes et al., 1995; Lamosa et al., 1998).
Daher lag auch das Vorhandensein von Mannosylglycerat-Transportern nahe. Weil darüber
hinaus ABC-Transporter bei (Hyper-) Thermophilen, die am häufigsten vertretene Transpor-
tergruppe darstellen (Albers et al., 2001), wurde beschlossen, die DNA-Sequenzen ver-
schiedener Substratbindeproteine bekannter α-Glucosid-ABC-Transporter (z.B. Trehalose/
Maltose) aus z.T. thermophilen Mikroorganismen untereinander zu vergleichen, um mittels
einer heterologen PCR einen potentiellen Mannosylglycerat-Transporter in R. marinus zu
identifizieren.
Ein ABC-Transporter wurde auf diese Weise jedoch nicht gefunden. Obwohl die Primer, die
zur Amplifikation eines Substratbindeproteins auf R. marinus DNA eingesetzt wurden, aus
hoch-konservierten Regionen abgeleitet worden waren (Abschnitt III, 2.4.1, Abb. 15), zeigte
sich, dass die Ähnlichkeit auf DNA-Ebene für die Anbindung der Primer nicht ausreichte. Im
Gegenteil, durch den Einsatz dieser Primer wurde ein DNA-Abschnitt amplifiziert, dessen
abgeleiteten Aminosäuresequenz Ähnlichkeiten zu einem Natrium/Glucose-Symporter der
Sodium/Solute-Symporter-Familie (SSSF-Transporter) aufweist. Die Amplifikation des Pro-
dukts erfolgte, da die 3’-Bereiche der Primer eine recht hohe Übereinstimmung zur Ziel-
sequenz aufweisen (Abb. 47). Ein Vergleich der Aminosäuresequenz der Region, in der die
Primer abgeleitet worden waren, mit der Aminosäuresequenz des Natrium/Glucose-Sympor-
ters zeigte keinerlei Ähnlichkeiten.
Die abgeleitete Aminosäuresequenz des Gens sssfRM ähnelt den Sequenzen von Trans-
portern der Sodium/Solute-Symporter-Familie in hohem Maße. Das hypothetische Protein
von sssf aus R. marinus wurde daher dieser Transporter-Familie zugeordnet. Die höchsten
Anteil identischer Aminosäuren zeigt SSSFRM im Vergleich zu Natrium/Glucose-Symporter
aus Robiginitalea biformata (49 %, Flavobacteriaceae) und Bacteroides thetaiotaomicron

IV. Diskussion 145
(48 %, Bacteroidaceae). Auch die Anteile identischer Aminosäuren bei Vergleich mit
Natrium/Glucose-Symportern aus Flavobacteriales bacterium (44 %) und Psychroflexus
torquis (44 %) sind sehr hoch, was auf eine stark konservierte Transporterfamilie mit ähn-
lichem Hydrophobizitätsprofil und Membrantopologie hindeutet.
Primer TCC1627for1 5’-GAC CTS CTR RAR AAG TAY GGY TA ⏐ ⏐ ⏐ ⏐ ⏐⏐ ⏐⏐ ⏐⏐ SSSF-Region Pos.30 5’-CGC AAC GCA ATT AAT GTG AGT TA Primer TCC1627 rev2 5’-GCG TAG GGC CAR TTG CGC WYG AA ⏐⏐ ⏐ ⏐ ⏐ ⏐ ⏐⏐ ⏐⏐⏐ ⏐⏐ SSSF-Region Pos.645 5’-CCG TGA AAA GCG TCG CGC ACG AA Abb. 47: Vergleich der Primersequenzen mit der Sequenz ihrer Bindungsstelle
Dargestellt sind die Primer TCC1627for1 und TCC1627rev2 sowie ihre Bindungsstelle auf R. marinus DNA. Die
Primer wurden in konservierten DNA-Bereichen innerhalb der DNA-Sequenz von Substratbindeproteinen
verschiedener Trehalose/Maltose-Transporter abgeleitet. Auf R. marinus DNA bindet TCC1627for1 in einer nicht
kodierenden Region stromaufwärts von sssfRM. Die Bindungsregion von Primer TCC1627rev2 kodiert für einen
Abschnitt zwischen den Transmembranhelices II und III des hypothetischen SSSF-Proteins. Die Zahlen be-
zeichnen die Positionen der Primerbindungsstellen innerhalb der sequenzierten sssfRM-Region (siehe Anhang).
Senkrechte Linien geben identische Basen an, handelt es sich um „Wobble Basen“ sind die senkrechten Linien
rot hervorgehoben. Als „Wobble Basen“ sind S: G oder C, W: A oder T; R: A oder G und Y: T oder C definiert. Bei
Primer TCC1627for1 handelt es sich um einen forward-Primer, bei TCC1627rev2 um einen reverse-Primer.
Sodium/Solute-Symporter sind sekundäre Transporter. Die Substrate werden gemeinsam mit
Natrium aufgenommen und so ein elektrochemischer Natrium-Gradient über der Membran
als treibende Kraft genutzt (Reizer et al., 1994). Es handelt sich um eine sehr große Familie,
die über 450 prokaryontische und eukaryontische Mitglieder umfasst (Wright et al., 2004).
Die meisten der bisher charakterisierten Sodium/Solute-Symporter haben ein ungewöhnlich
breites Substratspektrum (Saier Jr, 2000). Je nach Transportsystem werden durch diese
Familie Substrate wie Zucker (z.B. Glucose), Aminosäuren, Vitamine, Nucleoside, Inositole,
Jodid und Harnstoff aufgenommen (Reizer et al., 1994; Sarker et al., 1997; Prasad et al.,
1998; Wright et al., 2004). Die Aufnahme dieser Substrate ist Teil des katabolischen Stoff-
wechsels, d.h. die Substrate dienen als Kohlenstoff- und/oder Stickstoffquelle (Jung, 2001).
Einige Sodium/Solute-Symporter, wie z.B. OpuE (Prolin-Aufnahme) aus B. subtilis bzw. BetM
und EctM (Betain/Ectoin-Aufnahme) aus M. halophilus, sind in die Osmoadaptation involviert
(Von Blohn et al., 1997; Spiegelhalter & Bremer, 1998; Vermeulen & Kunte, 2004).
Die Sequenzlänge der Sodium/Solute-Symporter variiert zwischen 477 und 830 Amino-
säuren, wobei die eukaryontischen Proteine in der Regel 100-200 mehr Aminosäuren auf-
weisen. Sie bestehen wie alle sekundären Transporter aus einem Polypeptid, das die Mem-
bran 11 bis 15 mal durchspannt. Ein Kern aus 13 Transmembrandomänen ist den meisten

IV. Diskussion 146
Transporter gemeinsam (Saier Jr, 2000; Jung et al., 2002). Mitglieder dieser Familie
befinden sich in den drei Domänen, der Bacteria (z.B. Natrium/Glucose-Symporter SlgS aus
Vibrio parahaemolyticus), der Archaea (z.B. Prolin-Transporter PutP aus Halobacterium sp.
NRC-11) und der Eukarya (z.B. Natrium/Glucose-Symporter SGLT1 aus H. sapiens). Der
menschliche Natrium/Glucose-Symporter (hSGLT1) ist das erste und bisher am besten
charakterisierte Mitglied dieser Familie (Reizer et al., 1994; Panayotova-Heiermann et al.,
1999; Wright et al., 2004). Unter anderem wurde anhand dieses Transporters die Aufnahme-
kinetik der SSSF-Transporter aufgeklärt. Danach binden zunächst zwei Na+-Ionen an den
Transporter. Diese induzieren eine Konformationsänderung, was zu einem Anstieg der Affini-
tät zu Glucose auf das 25-fache führt. Anschließend dissoziieren im Zytoplasma zunächst
der Zucker, dann die Na+-Ionen (Wright et al., 2004). Nach Jung et al. (2002), Pirch et al.
(2002) und Pirch et al. (2003), die zur Aufklärung funktioneller Bereiche der Sodium/Solute-
Symporter als Modellsystem PutP (Prolin-Transporter) aus E. coli untersuchten, gilt, dass die
in der zweiten Transmembrandomäne liegenden Aminosäuren entscheidend für die Bindung
der Na+-Ionen sind.
Ein Hydrophobizitätsplot der von sssfRM abgeleiteten Aminosäuresequenz nach Kyte & Do-
olittle (1982) zeigt sieben stark hydrophobe Bereiche, die aus jeweils 16-19 Aminosäuren be-
stehen. Dies bestärkte die Vermutung, dass es sich bei SSSFRM um ein Transmembran-
protein mit möglicher Transporterfunktion handelt. Auch die Topologieprogramme DAS
(Cserzo et al., 1997) und HMMTOP (Tusnady & Simon, 1998, 2001) berechneten für
SSSFRM sieben Transmembrandomänen, wobei der N-Terminus ins Zytoplasma und der C-
Terminus ins Periplasma ragt. Im Gegensatz dazu besitzen die meisten bisher charakteri-
sierten Sodium/Solute-Symporter einen periplasmatischen N-Terminus und einen zytoplas-
matischen C-Terminus (Turk & Wright, 1997). Dies kann jedoch mit der Anzahl der Trans-
membranhelices variieren. Auch die Anzahl der Transmembrandomänen von SSSFRM stimmt
nicht mit der Anzahl der Transmembrandomänen bei Proteinen der SSS-Familie überein.
Diese liegt in der Regel zwischen 11 und 15 Transmembrandomänen. Natrium/Glucose-
Transporter weisen in der Regel 14 Transmembrandomänen auf (Saier Jr, 2000; Jung et al.,
2002). Zudem befindet sich der C-Terminus genauso wie der N-Terminus zumeist im Peri-
plasma (Wright et al., 2004). Nach Analyse der verkürzten Aminosäuresequenz könnte es
sich bei dem hypothetischen Sodium/Solute-Symporter aus R. marinus um einen sog.
„truncated“ (verkürzten) Natrium/Glucose-Symporter handeln (Tailor et al., 2001; Wright,
2001). Laut der Sequenzvergleiche mit anderen Natrium/Glucose-Transportern fehlen
SSSFRM die sieben N-terminalen Transmembrandomänen. Damit würde jedoch die Orien-
tierung des C-Terminus, dem der übrigen Natrium/Glucose-Transporter entsprechen. Ein
künstlich verkürzter menschlicher Natrium/Glucose-Symporter (hSGLT1), von dem nur die
fünf C-terminalen Transmembrandomänen (TM 10 bis 14) vorhanden sind, zeigte einen Na+-

IV. Diskussion 147
unabhängigen gering-affinen Glucose-Transport (Panayotova-Heiermann et al., 1999). Dies
könnte dafür sprechen, dass auch der Natrium/Glucose-Symporter aus R. marinus in der
Lage ist, Glucose mit geringe Affinität zu transportieren.
Viele Organismen besitzen mehrere Sodium/Solute-Symporter ähnlicher Sequenz, die in der
Lage sind, in der Zelle auch andere Aufgaben zu übernehmen. Bei Abwesenheit von
Zuckern wirken sie als Uniporter für Na+-Ionen oder als Harnstoff- und Wasserkanäle. Ande-
re, wie der menschliche Symporter SGLT3, dienen als Glucose-Sensor, transportieren keine
Glucose mehr und bauen stattdessen einen Na+-Gradienten auf (Wright et al., 2004). Der
Nachweis zweier DNA-Banden in der Southern-Hybridisierung (Abschnitt III, 2.4.2) könnte
daher auf das Vorkommen zweier ähnlicher Sodium/Solute-Symportergene unterschiedlicher
Funktion im Genom von R. marinus hindeuten.
5. Amplifikation der 16S rDNA von R. marinus mittels Differential
Display RT-PCR Da die Identifizierung eines potentiellen Mannosylglycerat-Transporters mittels funktioneller
Komplementierung solutetransportdefekter E. coli Stämme und heterologer PCR gescheitert
war, sollte die Lokalisation nun mittels Differential Display RT-PCR erfolgen. Diese Methode
wurde erstmals von Liang & Pardee (1992) erfolgreich an Eukaryonten angewendet, um
differentiell exprimierte Gene nachzuweisen. Die Methode der Differential Display RT-PCR
umfasst eine Folge mehrerer Schritte. Zunächst wird die Gesamt-RNA der zu untersuch-
enden Zellkultur isoliert. Diese wird anschließend mittels reverser Transkriptase in cDNA
umgeschrieben. Dies geschieht bei Eukaryonten üblicherweise unter Einsatz von Poly-T-
Primern. Zuletzt werden durch Einsatz sog. „arbitrary“-Primer und eines Poly-T-Primers
spezifische Anteile der cDNA amplifiziert. Die Amplifikate werden gelelektrophoretisch aufge-
trennt und entsprechend der Fragestellung miteinander verglichen.
Hintergrund des durchgeführten DDRT-Experiments ist die Tatsache, dass ein Organismus
im Zuge der Osmoadaptation mit steigender Salinität höhere Konzentrationen seines kompa-
tiblen Soluts produziert. Gleiches wird auch für R. marinus und seine kompatiblen Solute
Mannosylglycerat und Mannosylglyceramid gelten. Die steigende intrazelluläre Konzentration
führt nach dem oben dargestellten Regulationsmodell (Abschnitt IV, 1. Abb. 46, Kunte, 2006)
zu einem zunehmenden Verlust an kompatiblem Solut. Aus Gründen einer Energieersparnis
und Syntheseregulation transportiert ein solcher Organismus die verlorenen Solute wieder-
um in die Zelle. Bei dem Ectoin transportdefekten Stamm H. elongata KB2 kommt es zur An-
häufung von Ectoin im Medium, während dies beim Wildtyp nicht der Fall ist (Volke, 2001).
Für den vorliegenden Fall wurde angenommen, dass bei zunehmender Salinität und einem
drohenden steigenden Verlust von Mannosylglycerat mehr Transporterproteine erforderlich
sind und daher die entsprechenden Gene wesentlich stärker exprimiert werden. Als differen-

IV. Diskussion 148
tielle Anzuchtbedingungen wurden das Wachstum von R. marinus bei 2 % und 6 % NaCl
ausgewählt. Hier wurde damit gerechnet, dass sich unter den differentiell stärker exprimier-
ten DNA-Banden bei 6 % NaCl auch Amplifikate von Mannosylglycerat-Transporter
mRNA/cDNA befinden.
Zwar konnte unter den gewählten Bedingungen ein differentielles Bandenmuster amplifiziert
werden (Abschnitt III, 2.5, Abb. 19), jedoch ergab die Sequenzierung und Charakterisierung
der Banden keine differentiell exprimierten proteinkodierenden Sequenzen. Die Sequenzana-
lysen zeigten 100 %-ige Identität mit der 16S rDNA von R. marinus, dem Gen für die 16S
rRNA von R. marinus. Einer der Gründe für dieses Ergebnis ist, dass die verwendete
Gesamt-RNA prokaryontischer Herkunft hauptsächlich aus ribosomaler RNA besteht. Ent-
sprechend häufig treten derlei Probleme in der Literatur auf (Abu Kwaik & Pederson, 1996;
Brzostowicz et al., 2000; Lakey et al., 2002). Die Autoren betrachten die Amplifikation von
rRNA in der DDRT-PCR als ein häufig unvermeidbares Phänomen. Zu diesen „falsch-posi-
tiven“ Sequenzen zählen neben der rRNA auch DNA-Gyrasen und die Ribonucleotid-Reduk-
tase (Brzostowicz et al., 2000). Hintergrund für dieses Problem ist die Tatsache, dass die
meisten für diese Methode ausgearbeiteten Protokolle sich eher für die Identifizierung von
differentiell exprimierten Genen eukaryontischer Herkunft eignen (Liang & Pardee, 1992;
Sarkar, 1996; Matz & Lukyanov, 1998). Bisher wurde nur eine begrenzte Anzahl differentiell
exprimierter prokaryontischer Gene unter Verwendung dieser Methode identifiziert (Abu
Kwaik & Pederson, 1996; Amara & Vijaya, 1997; Fleming et al., 1998; Tsai & Shi, 2000).
Dies liegt im wesentlichen daran, dass die Prokaryonten nur über einen geringen Teil poly-
adenylierter mRNA verfügen, welche eine spezifische Isolierung und Umscheibung in cDNA
mittels eines Poly-T-Primers ermöglicht (Brzostowicz et al., 2000). Zur Durchführung von
DDRT-PCR bei Prokaryonten wird sowohl bei cDNA-Synthese als auch bei der Amplifikation
auf sog. „arbitrary“-Primer (10-11 bp) zurückgegriffen, deren Sequenz mittels eines speziel-
len Computerprogramms entsprechend der Genomsequenz des untersuchten Organismus
ermittelt wird (Fislage et al., 1997; Brzostowicz et al., 2000). Auf „arbitrary“-Primer konnte in
der vorliegenden Arbeit nicht zurückgegriffen werden, da die Genomsequenz von R. marinus
nicht zugänglich war. Als Ansatzpunkt der cDNA-Synthese wurden die drei unterschiedlichen
Stopkodons ausgewählt. So sollte die synthetisierte cDNA-Population hin zu proteinkodieren-
den Sequenzen verschoben werden. Die Primer bestehen an ihrem 3´-Ende aus Sequenzen,
die zu den Stopkodons komplementär sind. Diesen Sequenzen ist eine Inosin reiche
Random-Sequenz vorgelagert. Den 5’-Bereich bildet eine PolyA15-Sequenz. Zwischen
beiden Sequenzen ist jeweils eine spezifische Base eingefügt (C,G oder T). Nach der cDNA-
Synthese wurde die cDNA am 3´-Ende mit einer Poly-A-Extension versehen, die in der
Amplifikation der cDNA als Anlagerungsbereich für einen Poly-T-Primer dient. Die Amplifi-
kation der cDNA erfolgte mit einem Poly-T-Primer und einem der Primer Poly A-C-Stop,

IV. Diskussion 149
Poly A-G-Stop und Poly A-T-Stop. Die durchgeführte Strategie führte zwar zu einem differ-
entiellen Bandenmuster, das jedoch von Sequenzen der 16S rRNA dominiert wurde.
Die einzige, jedoch recht komplexe und anfällige Möglichkeit reine mRNA eines Prokary-
onten zu gewinnen, ist die Isolierung der mRNA aus intakten Polysomen. So gelang es
Amara & Vijaya (1997) die mRNA von E. coli nach einer enzymatischen Modifikation aus
intakten Polysomen zu isolieren. Die Autoren polyadenylierten die in Polysomen freiliegen-
den 3´-Bereiche der mRNA mittels einer Poly-(A)-Polymerase. Nach dem Aufbrechen der
Polysomen konnte die polyadenylierte mRNA dann mittels Poly-T-Primer in cDNA umge-
schrieben werden. Die Isolierung der Polysomen ist ein kompliziertes Verfahren und muss je
nach Bedarf modifiziert werden, da für die Integrität der Polysomen Mg2+-Ionen unentbehrlich
sind, während für eine optimale Aktivität des Poly-(A)-Polymerase Mn2+-Ionen gebraucht
werden. Beide Ionen müssen sorgfältig gegeneinander austitriert werden, um ein optimales
Ergebnis zu erzielen. Übrig bliebe für das vorliegende Projekt dennoch das Problem der
cDNA-Amplifikation. Hierfür müssten noch immer geeignete Primer gefunden werden. Eine
Identifizierung eines Mannosylglycerat-Transporters mittels DDRT-PCR erscheint vor diesem
Hintergrund auch in naher Zukunft unwahrscheinlich.
6. Abschluss der Arbeiten mit Rhodothermus marinus Alle bisherigen Verfahren, die funktionelle Komplementierung, die heterologe PCR und die
DDRT-PCR, hatten nicht zur Identifizierung eines Mannosylglycerat-Transporters in
R. marinus geführt. Die Möglichkeit zur kurzfristigen Optimierung dieser Verfahren wurde
nicht gesehen. Darüber hinaus ließen Arbeiten von Da Costa (pers. Mitteilung, 2005) darauf
schließen, dass R. marinus kein Mannosylglycerat-Transportsystem besitzt. Ein möglicher
Grund hierfür wäre, dass R. marinus aufgrund des Fehlens eines spezifischen Exportkanals
keine Solute ins Außenmedium verliert und daher auch keine entsprechenden Transport-
systeme besitzt. Ähnliches wird auch bei Corynebacterium glutamicum vermutet. Dieses
besitzt kein spezifisches Transportsystem für das kompatible Solut Prolin und verliert den-
noch kein Prolin ins Außenmedium (Kunte, pers. Mitteilung, 2006). Die Etablierung einer ver-
stärkten Mannosylglycerat-Produktion in R. marinus nach dem Vorbild von H. elongata KB2
(Volke, 2001) erschien somit unmöglich, da dann die postulierten Regulationsmechanismen,
die in KB2 vermutlich zu der verstärkten Ectoin-Produktion führen, ebenfalls nicht vorhanden
sind. Zudem war zum Erreichen des Projektziels noch immer die vorherige Etablierung
gentechnologischer Methoden für R. marinus erforderlich, ein Projekt, das durch Fa. Prokaria
in Zusammenarbeit mit der Firma Bitop AG verwirklicht werden sollte.
Aufgrund dieser Bedingungen wurde beschlossen, die Arbeiten an R. marinus einzustellen
und eine alternative Strategie zur Etablierung einer Mannosylglycerat-Produktion in einem
bakteriellen Stamm zu entwickeln.

IV. Diskussion 150
7. Halomonas elongata als Produktionsstamm für Mannosylglycerat In Arbeiten von Sampaio et al. (2003a) und Empadinhas et al. (2004) konnte durch die
Expression von Mannosylglycerat-Synthesegenen aus dem thermophilen Stamm R. marinus
(mgs, Mannosylglycerat-Synthase) bzw. dem mesophilen Stamm Dehalococcoides etheno-
genes (mgsD, Mannosylglycerat-Synthase) in E. coli bzw. Saccharomyces cerevisiae eine
Mannosylglycerat-Synthese hervorgerufen werden.
Zur Etablierung der Mannosylglycerat-Synthese verwendeten Sampaio et al. (2003a) einen
genetisch veränderten E. coli Stamm, der nicht in der Lage ist, extern zugesetzte Mannose
zu verstoffwechseln. Dieser Stamm schleust Mannose direkt in die Synthese von Mannosyl-
glycerat ein (Abb. 48). Durch Deletion des Gens manA (Phosphomannose-Isomerase; kata-
lysiert Umwandlung von Mannose-6-phosphat in Fructose-6-phosphat) wurde erreicht, dass
Mannose nicht in der Glykolyse metabolisiert wird. Eine Mutation von galU (kodiert für ein
Enzym der UDP-Glucose-Synthese) führte zu einem Ausfall der de novo Synthese von
Trehalose. Zusätzlich wurden die Gene cpsG und cpsB des cps-Genclusters deletiert, um
die Synthese von Colansäure (Kapselbildung, Andrianopoulos et al., 1998) zu unterbinden.
Das Gen cpsG kodiert für eine Phosphomanno-Mutase (katalysiert die Umwandlung von
Mannose-6-phosphat zu Mannose-1-phosphat), cpsB für eine Mannose-1-phosphat-
Guanylyltransferase (katalysiert die Bildung von GDP-Mannose aus GTP und Mannose-1-
phosphat). Da beide Genprodukte für die Bereitstellung der Mannsylglycerat-Synthese-
vorstufe GDP-Mannose erforderlich sind, wurden sie unter Kontrolle eines IPTG-induzier-
baren Promotors in einen Vektor eingebracht. In einen weiteren Expressionsvektor wurde
das mgs-Gen (kodiert für die Mannosylglycerat-Synthase aus R. marinus) inseriert. Nach
Induktion mit IPTG war in dem E. coli Stamm eine geringe Menge von Mannosylglycerat
nachweisbar.
Sampaio et al. (2003a) nannten für die geringe Ausbeute an Mannosylglycerat mehrere
Gründe, wie eine unzureichenden Expression bzw. eine zu geringe Aktivität der Mannosyl-
glycerat-Synthase (Mgs). Die Mannosylglycerat-Synthase stammt aus dem thermophilen
Stamm R. marinus und weist daher bei 37 °C eine nur geringe enzymatische Aktivität auf
(optimale Aktivität bei 85-90 °C, Martins et al., 1999). Darüber hinaus schien die Bildung von
Mannosylglycerat aus GDP-Mannose und D-Glycerat limitierend zu wirken. Jedoch führte
eine externe Zugabe von D-Glycerat nicht zu einer Steigerung der Mannosylglycerat-Syn-
these. Die Autoren gehen daher davon aus, dass der interne D-Glycerat-Pool nicht limi-
tierend wirkt. Andere Vermutungen gingen in Richtung der intrazellulären GTP-Konzentrati-
on, da die Syntheserate von GDP-Mannose gering war, obwohl die Mannose-1-phosphat-
Guanylyltransferase aufgrund ihrer Plasmidkodierung in hoher Konzentration in der Zelle
vorliegen müsste (Sampaio et al., 2003a).

IV. Diskussion 151
Glucose-6-P
Fructose-6-P
Mannose-6-P
Mannose-1-P
GDP-Mannose
Mannosylglycerat
mgs
cpsB
cpsG
GTP
PPi
Glucose
Mannose
ptsGX
XmanA
ptsM
D-Glycerat
GDP
Glykolyse
Abb. 48: Etablierung einer Mannosylglycerat-Synthese in einem E. coli Stamm
Dargestellt ist der Mannosylglycerat-Syntheseweg in einem rekombinanten E. coli Stamm. Durch die Deletion des
manA-Gens (kodiert für Phosphomannose-Isomerase), kann der Stamm Mannose nicht metabolisieren. Daher
wird Mannose direkt in die Produktion von Mannosylglycerat eingespeist. Die Einbringung der Gene cpsG (kodiert
für Phosphomanno-Mutase) und cpsB (kodiert für Mannose-1-phosphat-Guanylyltransferase) stellt die Bereit-
stellung der Synthesevorstufen sicher. mgs (kodiert für Mannosylglycerat-Synthase aus R. marinus) führt zu
Synthese von Mannosylglycerat. Alle Gene, deren Genprodukte an der Synthese von Mannosylglycerat beteiligt
sind, sind rot markiert. P: Phosphatgruppe, PPi: Pyrophosphat, PtsG & PtsM: Aufnahmesysteme für Glucose
bzw. Mannose. Der verwendete E. coli MS4601 kann aufgrund einer Tn10-Insertion im ptsG-Gen Glucose nur
durch das PtsM-System in die Zelle aufnehmen (Abbildung modifiziert nach Sampaio et al., 2003a).
Neben dem Temperaturoptimum könnte auch die im Vergleich zu R. marinus unterschied-
liche „Codon usage“ von E. coli für eine eingeschränkte Effektivität der heterologen Expres-
sion verantwortlich sein (Yamao et al., 1991; Zhang, S P et al., 1991; Del Tito Jr et al., 1995).
Enthält die mRNA eines nativen Proteins z.B. fünf aufeinander folgenden „AGG“ Kodons
(„AGG“ kodiert für die Aminosäure Arginin und ist ein seltenes Kodon in der mRNA von
E. coli), kann die Translation des Proteins in E. coli zum Stillstand kommen, weil der
tRNAsAGG-Gehalt in der Zelle zur Erschöpfung kommt (Rosenberg et al., 1993). Nach
Ikemura (1981) besteht zwischen der Häufigkeit eines bestimmten Kodons und der Verfüg-
barkeit seiner tRNA ein Zusammenhang. Vergleicht man die „Codon usage“ von E. coli und
R. marinus stellt man fest, dass es deutliche Unterschiede in der Häufigkeit der Nutzung
verschiedener Kodons gibt. Das Programm GCUA (graphical codon usage analyser,
http://gcua.schoedl.de/) bestimmt den durchschnittlichen Unterschied der „Codon usage“

IV. Diskussion 152
zwischen E. coli und R. marinus auf 18,17 % (Abb. 49). Die größten Unterschiede sind in der
Nutzung der Kodons für die Aminosäure Isoleucin und Lysin zu beobachten. In der Sequenz
der Mannosylglycerat-Synthase kommt die Aminosäure Isoleucin 14 mal vor. Sie wird 11 mal
durch das Kodon „AUC“ kodiert. R. marinus nutzt dieses Kodon mit einer Häufigkeit von
77 %, während E. coli es nur mit einer Häufigkeit von 42 % verwendet. Die Aminosäure
Lysin, die fünfmal in der Sequenz der Mannosylglycerat-Synthase vorkommt, wird viermal
durch das Kodon „AAG“ und nur einmal durch das Kodon „AAA“ kodiert. E. coli verwendet
„AAG“ mit einer Häufigkeit von 24 % während es bei R. marinus viel häufiger auftritt (72 %).
Dieser Unterschied in der Nutzung bestimmter Kodons könnte zu Problemen bei der hetero-
logen Expression führen. Liegt in der Zelle der Kodon spezifische tRNA-Levels einer Amino-
säure sehr niedrig, würden die Ribosomen bei der Translation zum Stillstand kommen,
während sie auf das richtige tRNA-Molekül warten. Dieses Blockieren der Ribosomen kann
verschiedene Translationsfehler, wie eine Verschiebung des Leserasters („frameshifting“)
oder ein Abbrechen der Translation, herbeiführen (Sorensen et al., 1989; Spanjaard et al.,
1990; Yamao et al., 1991).
Vergleicht man mittels des Programms GCUA dagegen die „Codon usages“ von R. marinus
und H. elongata, stellt man fest, dass sich diese sehr ähneln. Der durchschnittliche Unter-
schied der „Codon usage“ beträgt hier nur 6,42 %. Nach Auswertung der hinterlegten
Proteinsequenzen von H. elongata nutzt dieser das Kodon „AUC“ (Isoleucin, 79 %) fast
genauso häufig wie R. marinus (77 %). Das Kodon „AAG“ (Lysin) wird durch H. elongata
sogar deutlich öfters genutzt (81 %) als durch R. marinus (72 %, Abb. 49). Diese Beobach-
tungen lieferten die ersten Hinweise, dass eine Expression der Mannosylglycerat-Synthase
aus R. marinus in einem halophilen Bakterium wie H. elongata erfolgreich verlaufen könnte.
H. elongata schien jedoch aus weiteren Gründen gut als Expressionssystem geeignet zu
sein. So akkumuliert H. elongata unter hyperosmotischen Bedingungen verschiedenste
Zucker, Zuckerderivate, Polyole, Betaine und Aminosäuren als Osmolyte ohne diese zu
metabolisieren (Vreeland et al., 1980; Severin, 1993). Damit besteht die Möglichkeit, dass
auch Mannosylglycerat als Solut im Zytoplasma angereichert wird. Der halophile Charakter
von H. elongata erlaubt diesem ein Wachstum bei höheren Salinitäten (bis 25 % NaCl im
Mineralsalzmedium). Wachstum bei höherer Salinität bedeutet immer auch die Akkumulation
höherer Konzentrationen kompatibler Solute, was eine der wichtigsten Voraussetzungen für
einen geeigneten Produktionsstamm ausmacht. Wie bei E. coli sind bei H. elongata die
gentechnischen Methoden etabliert (Kunte, 1995; Kunte & Galinski, 1995; Göller, 1999),
welche eine Stammoptimierung erst ermöglichen. Bei H. elongata ist die genomische Region
der Ectoin-Gene bekannt (Göller, 1999; Stumpfe, 2003). Hier könnten die Gene der Man-
nosylglycerat-Synthese integriert werden. Dabei würden die integrierten Gene unter Kontrolle
des osmoregulierten Ectoin-Genpromotors gestellt werden. Resultat wäre eine salzabhän-

IV. Diskussion 153
gige Expression der Mannosylglycerat-Synthesegene bei gleichzeitiger Unterbrechung der
Ectoin-Synthese. Mannosylglycerat würde als alleiniges Solut synthetisiert. Eine genomische
Expression hätte gegenüber der plasmidkodierten Expression von Sampaio et al. (2003a)
darüber hinaus den Vorteil, dass auf die Nutzung von Antibiotika verzichtet werden kann.
Dies würde die Kosten reduzieren, zudem müssten keine Antibiotika bei der Produktaufrei-
nigung abgetrennt werden. Störende Einflüsse einer Vektorreplikation oder erhöhten Vektor-
kopienzahl auf die Expression der Mannosylglycerat-Synthase wären ausgeschlossen.
Um festzustellen, ob H. elongata ein Transportsystem für Mannosylglycerat aufweist und ob
Mannosylglycerat H. elongata ein verbessertes Wachstum ermöglicht, wurden Wachstums-
experimente mit den H. elongata Stämmen KB1 und H. elongata KB18.1 durchgeführt. Beide
können keine Ectoin synthetisieren, KB18.1 fehlen zudem alle bekannten Solutetransporter.
Die Zugabe von 1 mM Mannosylglycerat führte bei beiden Stämmen zu einer Steigerung der
Wachstumsraten (KB1: 23,5 %; KB18.1: 18,2 %). Dies deutete zunächst darauf hin, dass
H. elongata Mannosylglycerat als kompatibles Solut akkumuliert. Die Analysen der Zellex-
trakte (mittels NMR und später HPLC) zeigten jedoch, dass Mannosylglycerat durch beide
Stämme nicht in die Zelle aufgenommen wurde. Die Tatsache, dass in den Zellextrakten
beider H. elongata Stämme kein Mannosylglycerat nachgewiesen werden konnte, ließ ver-
muten, dass H. elongata weder ein spezifisches noch ein unspezifisches Transportsystem
für Mannosylglycerat besitzt. Dies würde bedeuten, dass ein Mannosylglycerat produzieren-
der H. elongata Stamm das Solut nach dem Vorbild des Ectoin-Produktionsstamms KB2
kontinuierlich ins Außenmedium verliert (Volke, 2001), ein weiteres Argument für die
Verwendung von H. elongata als Mannosylglycerat-Produktionsstamm.
Das beschleunigte Wachstum von H. elongata KB1 und KB18.1 in Gegenwart von Mannosyl-
glycerat beruht vermutlich auf dem membranstabilizierenden Effekt kompatibler Solute. Die-
ser Effekt wurde besonders bei extern zugesetzter Trehalose beobachtet. Bei salz- und
hitzegestressten E. coli Zellen wirkt die Akkumulation von Trehalose besonders stabilisierend
auf die Zellmembran. Trehalose geht Wasserstoffbrückenbindungen mit den Phospholipid-
Kopfgruppen ein und ersetzt so 10-12 Wasserstoffbrückenbindungen verlorener Wasser-
moleküle. Dies erhält nach Welsh & Herbert (1999) den Normalzustand der Membran (Lα--
Zustand, lamellarer Bilayer-Zustand) aufrecht. Dadurch wird der Übergang zur kristallinen
Doppelschicht (Gelphase, Lβ-Zustand) oder einer nicht-lamellaren Struktur der Membran
(invertierter Hexagonal-II-Zustand) vermieden bzw. verzögert. Beim Phasenübergang der
Membranlipide vom flüssigen in den festen Zustand, nimmt die Permeabilität der Zytoplas-
mamembran zu. Die Erhaltung des Lα-Zustands stellt somit die Funktion der Membran als
Permeabilitätsbarriere sicher. Fehlende Membranintegrität führt zum Zusammenbruch wichti-
ger Ionengradienten, was zu einem Absterben der Zelle führt (Crowe & Crowe, 1992; Leslie
et al., 1995; Welsh & Herbert, 1999).
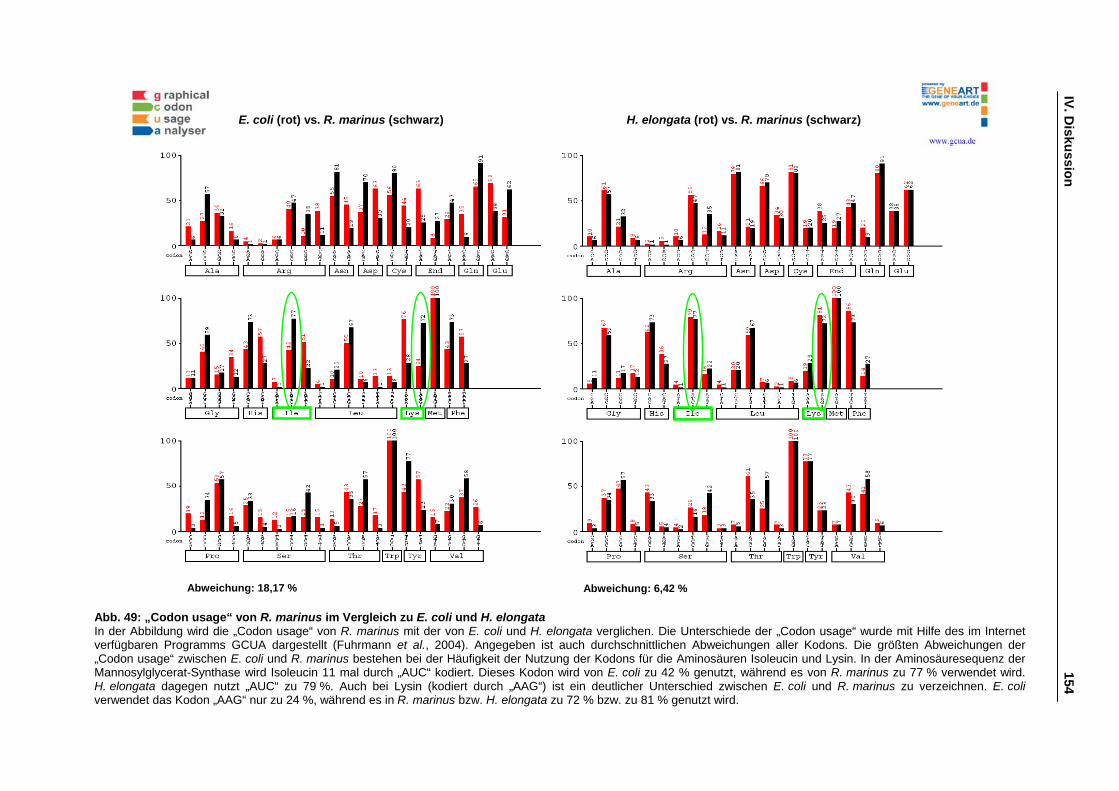
IV. Diskussion 153
Abweichung: 6,42 %Abweichung: 18,17 %
E. coli (rot) vs. R. marinus (schwarz) H. elongata (rot) vs. R. marinus (schwarz)
IV. Diskussion
154
Abb. 49: „Codon usage“ von R. marinus im Vergleich zu E. coli und H. elongata In der Abbildung wird die „Codon usage“ von R. marinus mit der von E. coli und H. elongata verglichen. Die Unterschiede der „Codon usage“ wurde mit Hilfe des im Internet verfügbaren Programms GCUA dargestellt (Fuhrmann et al., 2004). Angegeben ist auch durchschnittlichen Abweichungen aller Kodons. Die größten Abweichungen der „Codon usage“ zwischen E. coli und R. marinus bestehen bei der Häufigkeit der Nutzung der Kodons für die Aminosäuren Isoleucin und Lysin. In der Aminosäuresequenz der Mannosylglycerat-Synthase wird Isoleucin 11 mal durch „AUC“ kodiert. Dieses Kodon wird von E. coli zu 42 % genutzt, während es von R. marinus zu 77 % verwendet wird. H. elongata dagegen nutzt „AUC“ zu 79 %. Auch bei Lysin (kodiert durch „AAG“) ist ein deutlicher Unterschied zwischen E. coli und R. marinus zu verzeichnen. E. coli verwendet das Kodon „AAG“ nur zu 24 %, während es in R. marinus bzw. H. elongata zu 72 % bzw. zu 81 % genutzt wird.

IV. Diskussion 155
8. Charakterisierung der rekombinanten H. elongata Stämme
8.1 H. elongata KB10 (∆ectA::mgs) Als Insertionsort für die Etablierung der Mannosylglycerat-Synthese in H. elongata wurden
die Ectoin-Synthesegene ausgewählt. Mit dieser Strategie wurde zum einen das neu inse-
rierte Gen unter die Kontrolle der Ectoin-Genpromotoren gestellt, gleichzeitig wurde die
Ectoin-Synthese ausgeschaltet. Zur Etablierung der Mannosylglycerat-Synthese wurde zu-
nächst mittels homologer Rekombination in H. elongata Wildtyp der offenen Leserahmen des
ectA-Gens (kodiert für Diaminobuttersäure-Acetyltransferase) gegen den offenen Leserah-
men des Mannosylglycerat-Synthase Gens mgs aus R. marinus „in-frame“ ausgetauscht.
Der resultierende Stamm H. elongata KB10 wurde mit der von Grammann (2000) herge-
stellte Ectoin defizienten Deletionsmutante KB1 verglichen. Bei diesem Stamm ist das ectA-
Gen deletiert.
8.1.1 Physiologische Untersuchungen an H. elongata KB10
Als Bedingungen für eine vergleichende Wachstumsanalyse von H. elongata KB1 und KB10
wurde eine Mediensalinität von 4 % NaCl und eine Inkubationstemperatur von 37 °C ge-
wählt. Beide Stämme sind in Mineralsalzmedium bei einer Mediensalinität von 5 % nur zu
sehr beschränktem Wachstum fähig, darüber hinaus nicht mehr. Sampaio et al. (2003a)
hatten in E. coli bei einer Temperatur von 37 °C eine wenn auch geringe Enzymaktivität der
Mannosylglycerat-Synthase nachweisen können, was als Hinweis gewertet wurde, dass
diese Temperatur ausreichen sollte, um die enzymatische Aktivität der Mannosylglycerat-
Synthase sicherzustellen, wenn diese in H. elongata exprimiert wird.
In Wachstumsexperimenten zeigte sich jedoch, dass H. elongata KB10 bei Nutzung aller
Kohlenstoffquellen nur geringfügig höhere Wachstumsraten aufweist wie KB1. Besonders bei
Verwertung der Saccharose ist kaum eine Verbesserung der Wachstumsrate zu verzeich-
nen. Der Wildtyp erreicht stets die höchsten Wachstumsraten (Abb. 24). Besonders deutliche
Unterschiede im Vergleich zum Wildtyp treten beim Wachstum mit Glucose und Saccharose
auf. Eine signifikante Steigerung der Wachstumsraten von H. elongata KB10 im Vergleich zu
KB1 hätte für eine Akkumulation des kompatiblen Soluts Mannosylglycerat im Zytoplasma
von KB10 hingedeutet, die nur geringfügige Wachstumsratensteigerung ließ jedoch einen
nur geringen Mannosylglycerat-Gehalt erwarten. Entsprechend konnte mittels einer nicht-
radioaktiven Dünnschichtchromatographie kein Mannosylglycerat im Zellextrakt nachgewie-
sen werden. Nach eigenen Analysen liegt die Nachweisgrenze dieser Methode bei einer
Stoffmenge 7,5 nmol Mannosylglycerat. Auf eine Vermessung der Zellextrakte mittels
Standard-NMR-Spektroskopie wurde aufgrund der geringeren Sensitivität der zur Verfügung
stehenden Methode verzichtet. Auch die spätere Analyse der Zellextrakte mittels HPLC be-

IV. Diskussion 156
stätigte dieses Ergebnis. In der HPLC lässt sich eine Stoffmenge von ca. 4 nmol Mannosyl-
glycerat mittels Brechungsindexdetektor nachweisen.
Für das Ausbleiben der Mannosylglycerat-Synthese in H. elongata KB10 konnten mehrere
Gründe verantwortlich sein. Erstes Hemmnis könnte eine unzureichende Transkription des
inserierten Fremdgens mgs sein. Die geringe Anzuchttemperatur des mesophilen Wirts
könnte darüber hinaus zu Probleme bei der Translation des thermophilen Proteins führen.
Denkbar sind in diesem Zusammenhang auch Probleme bei der Proteinfaltung. Eine fehler-
hafte Proteinfaltung kann zur Inaktivierung eines Proteins und Bildung von unlöslichen Prote-
inaggregaten, sog. „inclusion bodies“, führen (Hartely & Kane, 1988; Mitraki & King, 1989;
Hart et al., 1990; King et al., 1996). In „inclusion bodies“ finden sich meist nicht ausgereifte,
zum Teil falsch gefaltete Proteinintermediate (Hart et al., 1990). Das Ausmaß einer „inclusion
body“-Bildung variiert bei einer heterologen Expression und ist hauptsächlich von der Stabili-
tät und Löslichkeit des gebildeten Proteins abhängig (Chrunyk et al., 1993).
Andere Gründe für das Ausbleiben der Mannosylglycerat-Synthese stehen im Zusammen-
hang mit der enzymatischen Aktivität der Mannosylglycerat-Synthase. Nach Untersuchungen
von Martins et al. (1999), welche die Mannosylglycerat-Synthase von R. marinus aufgereinigt
und charakterisiert hatten, weist diese zwischen 35 und 95 °C eine enzymatische Aktivität
auf, wobei die höchste Aktivität bei 85-90 °C erreicht wird. Die Wachstumsversuche und
Untersuchungen des Mannosylglycerat-Gehalts erfolgten mit H. elongata KB10 Kulturen,
welche bei 37 °C angezogen wurden. Hier ist eine nur geringe enzymatische Aktivität zu
erwarten. Eine Steigerung der Inkubationstemperatur wird jedoch durch den mesophilen
Charakter von H. elongata begrenzt. Andere wichtige Faktoren könnten die ionischen
Verhältnisse im Zytoplasma des Expressionsstamms sein. So stellten Martins et al. (1999)
fest, dass die maximale Aktivität der Mannosylglycerat-Synthase in vitro nur durch Zugabe
von 50 mM Mg2+-Ionen erreicht werden kann. Das Fehlen dieses divalenten Ions führt zu
einer 2,5-fachen Abnahme der Aktivität der Mannosylglycerat-Synthase. Inwieweit die Mg2+-
Konzentration auch in vivo Auswirkungen auf die Enzymaktivität zeigt, ist nicht bekannt.
Möglicherweise ist jedoch die zytoplasmatische Mg2+-Konzentration von H. elongata KB10
bei der verwendeten Mediensalinität von 4 % für eine enzymatische Aktivität nicht aus-
reichend. Vreeland et al. (1983) hatten in Abhängigkeit von der Mediensalinität in H. elongata
bei einen Mg2+-Gehalt des Außenmediums von 24 mM eine zelluläre Mg2+-Konzentration
zwischen 9 und 23 mM gemessen. Eine NaCl- bzw. KCl-Konzentration von 50-500 mM hatte
keinen direkten Effekt auf die Aktivität der Mannosylglycerat-Synthase. Daher gingen Martins
et al. (1999) von einer Salzunabhängigkeit des Enzyms aus (Abschnitt I, 3.3.2).
Ein weiterer gewichtiger Grund für das Ausbleiben der Mannosylglycerat-Synthese könnte
eine zu geringe Konzentration von Synthesevorstufen wie D-Glycerat und GDP-Mannose

IV. Diskussion 157
sein. Martins et al. (1999) untersuchten die Substratspezifität der Mannosylglycerat-Synthase
anhand der Zugabe verschiedener Mannose-Donatoren, wie GDP-Mannose, ADP-Mannose,
UDP-Mannose, Mannose-1-phosphat und Mannose-6-phosphat aber auch anderer Zucker-
donatoren, wie GDP-Glucose, UDP-Glucose und ADP-Glucose, sowie verschiedene Zucker-
akzeptoren, wie D-Glycerat, L-Glycerat, D-3-Phosphoglycerat und Glycerol-3-phosphat. Das
Enzym weist eine absolute Stereospezifität für GDP-Mannose und D-Glycerat auf, d.h. hohe
zytoplasmatische Konzentrationen anderer Zuckerdonatoren und -akzeptoren führen nicht zu
einer Mannosylglycerat-Produktion.
8.1.2 Analyse der Transkription von mgs in H. elongata KB10
Nach der Analyse der Zellextrakte von KB10 wurde die Transkription des mgs-Gens analy-
siert, um die Ursachen für das Ausbleiben der Mannosylglycerat-Synthese einzugrenzen.
Einen möglichen Grund stellt eine fehlerhafte Transkription der Mannosylglycerat-Synthase
in KB10 dar.
Die Untersuchungen zur Expression des mgs-Gens mittels Northern-Hybridisierung und RT-
PCR wiesen mgs-Transkripte unterschiedlicher Längen nach (Abb. 27). Der Nachweis des
Gesamttranskripts mittels mgs-Sonde macht deutlich, dass in H. elongata KB10 eine
gemeinsame Transkription von mgs und den verbliebenen Ectoin-Synthesegenen erfolgt. Die
Bildung dieses Transkripts ist vor allem für die Integration weiterer Gene zur Etablierung der
Mannosylglycerat-Synthese von großer Bedeutung. So kann davon ausgegangen werden,
dass auch diese Gene nach ihrer Integration gemeinsam mit mgs transkribiert werden.
Mit der mgs-Sonde konnten darüber hinaus ein mgs-Einzeltranskript und ein mgsectB-Ko-
transkript visualisiert werden (Abb. 27a). Entsprechend dem mgs-Einzeltranskript wurde
durch Stumpfe (2003) ein ectA-Einzeltranskript nachgewiesen. Die starken Hintergrund-
signale deuten darauf hin, dass außerdem Bruchstücke der längeren Transkripte vorhanden
sind. Möglicherweise spielen hier Instabilitäten der Transkripte oder auch die Dauer der
RNA-Aufreinigung eine Rolle. Ein dem schwachen mgsectBC-Gesamttranskript (2,9 kb) ent-
sprechendes ectABC-Gesamttranskript hatten Egler (2001) und Stumpfe (2003) im
H. elongata Wildtyp in Northern-Analysen nicht nachweisen können. Dies könnte daran
liegen, dass sich Transkripte mit einer Größe ähnlich der ribosomalen RNA (23S rRNA: ca.
2,9 kb) nur schwer nachweisen lassen. Die rRNA macht den größten Anteil der isolierten
Gesamt-RNA aus (ca. 80 %, Stryer, 1996) Ihre Banden sind daher stark verbreitert und
überdecken ähnlich lange mRNA-Transkripte.
Mit der ectB-Sonde gelang der Nachweis eines ectB-Einzeltranskripts (1,3 kb), eines
mgsectB-Kotranskripts (2,5 kb) und eines ectBC-Kotranskripts (1,8 kb), der Nachweis des
mgsectBC-Gesamttranskripts gelang nicht (Abb. 27b). Ursachen für das Fehlen des Gesamt-
transkripts könnten die Qualität und Sensitivität der synthetisierten ectB-Sonde in Kombi-

IV. Diskussion 158
nation mit der Qualität der isolierten KB10 mRNA sein. Eine zu geringe Sensitivität der ectB-
Sonde würde zu einem Ausbleiben des mit der mgs-Sonde nachgewiesenen schwachen
mgsectBC-Signals führen. Darüber hinaus spielt auch die Kopienzahl eines Transkripts eine
entscheidende Rolle, die wiederum von der Stabilität eines Transkripts abhängt. Aufgrund
der begrenzten Sensitivität der Methode können sich Transkripte mit geringer Kopienzahl
einer Detektion mittels Chemilumineszenz in einer Northern-Analyse entziehen. So konnte
Stumpfe (2003) das ectABC-Gesamttranskript erst mittels einer RT-PCR nachweisen.
Mit den mgs- und den ectB-Sonden konnte auf H. elongata KB10 eine 2,5 kb große Bande,
die der Länge nach einem mgsectB-Kotranskript entspricht, detektiert werden. Eine derartige
Koexpression hatte durch Stumpfe (2003) anhand des ectAB-Transkripts nicht nachge-
wiesen werden können. Aufgrund des Fehlens des ectAB-Kotranskripts ging Stumpfe (2003)
davon aus, dass eine potentielle starke, jedoch nur mittels Computeranalysen vorhergesagte
Terminationsschleife stromabwärts von ectA die Synthese von Transkripten, die über ectA
hinauslaufen, stark einschränkt. Für einen möglichen Abbruch der Transkription an dieser
Stelle spricht auch die freie Energie der möglichen Terminationsschleife (Anhang). Nach
Analysen des Programms „mfold“ (Zucker, 2003) beträgt die freie Energie zur Bildung der
Schleife ∆G0 = -107 kJ/mol. Damit handelt es sich um eine für die Transkriptionstermination
ausreichend stabile Schleifenstruktur (Kunte, persönliche Mitteilung). Allerdings konnte
stromaufwärts von ectB bisher keine Promotorsequenz nachgewiesen werden, welche dann
die Existenz des ectB-Einzeltranskripts und des ectBC-Kotranskripts (ca. 1,8 kb) hätten
erklären können. Diese wurden auch schon durch Stumpfe (2003) nachgewiesen. Genauso
kann es sich bei den ectA- bzw. mgs-Einzeltranskripten um präparationsbedingte Artefakte
handeln. Anhand des Nachweises des mgsectBC-Gesamttranskripts und des mgsectB-
Kotranskripts konnte gezeigt werden, dass die Transkription zumindest teilweise über die
potentielle Terminationsschleife stromabwärts von ectA bzw. mgs hinausläuft. Ein Nachweis
eines Promotors stromaufwärts von ectB würde auch diese wahrscheinlicher wirken lassen,
zumal die stoffwechselphysiologische Bedeutung eines ectA- bzw. mgs-Einzeltranskripts
nicht erklärbar ist. Gleiches gilt für ein ectB-Einzeltranskript. Im Fall des TRAP-Trans-
portergens dctP aus Rhodobacter capsulatus vermuteten Kelly & Thomas (2001) als
Ursache für eine durchlaufende Transkription das Vorhandensein eines an die Terminations-
schleife angrenzenden CTTT-Motivs. Ein derartiges Motiv ist auch unmittelbar stromabwärts
des TRAP-Transportergens teaA aus Halomonas elongata (Grammann, 2004) und strom-
abwärts von ectA (H. elongata Wildtyp) bzw. mgs (H. elongata KB10) zu finden. In den
Untersuchungen von Kelly & Thomas (2001) und Grammann (2004) wurde vermutet, dass
dieses Motiv ein teilweise Durchlesen der RNA-Polymerase und damit die Synthese eines
Gesamttranskripts erlaubt. Es ist daher denkbar, dass die Transkription stromabwärts von
ectA (H. elongata Wildtyp) bzw. mgs (H. elongata KB10) nicht konsequent terminiert wird.

IV. Diskussion 159
8.1.3 Struktur des ectA-Promotors stromaufwärts von mgs in H. elongata KB10
Nach Computeranalysen liegt bei H. elongata Wildtyp ein möglicher Transkriptionsstartpunkt
von ectA 27 bp stromaufwärts des Startkodons (Göller, 1999). Bei KB10 wurde ectA durch
mgs ersetzt. 5’-RACE-PCR Analysen auf Gesamt-RNA aus H. elongata KB10 identifizierten
stromaufwärts von mgs ein Transkriptionsstartpunkt, der 25 bp stromaufwärts des Start-
kodons und damit nahe des Bereichs der Computervorhersagen liegt. Dieser Transkriptions-
startpunkt konnte somit einem σ70-abhängigen Promotor zugeordnet werden und bestätigte
so die Analysen von Göller (1999). Ein σ70-Promotor ist durch konservierte –35-und –10-
Regionen gekennzeichnet, die 17 ± 2 bp voneinander entfernt liegen. Im vorliegenden Fall
beträgt der Abstand 17 Nukleotide. Die –10-Region eines σ70-Promotors liegt in der Regel
maximal 10 bp vom zugehörigen Transkriptionsstartpunkt entfernt, die –35-Region entspre-
chend. Die Abstände beider Regionen zum Transkriptionsstartpunkt betragen im vorliegen-
den Fall 7 bzw. 31 Nukleotide. Die –35-Region σ70-abhängiger Promotoren ist essentiell für
die Aktivität des Promotors. Große Abweichungen in der Consensus-Sequenz führen zu
einer erheblichen Verringerung der Promotoraktivität. Abweichungen der –10-Region von der
Consensus-Sequenz verringern die Transkriptionseffizienz nur geringfügig. Beide Regionen
weichen mit nur jeweils einem Nukleotid von denen als Consensus-Sequenzen festgelegten
Hexanukleotidsequenzen ab (Abb. 50). Nach Auswertung aller Parameter handelt es sich
nach Vorgaben von Miller (1992) daher um einen starken σ70-abhängigen Promotor.
σ70 ist ein primärer Sigmafaktor und daher für das Überleben der Zellen essentiell (Wösten,
1998). Gene mit σ70-abhängigen Promotoren werden vorwiegend während der exponen-
tiellen Wachstumsphase und nicht als Antwort auf einen bestimmten Stressfaktor transkri-
biert. Der σ70-Faktor erkennt jedoch nicht nur Standardpromotoren, die mit den üblichen –35-
und –10-Region versehen sind, sondern auch davon abweichende Sequenzen. Dazu ge-
hören Promotoren denen ein –35-Element fehlt, sofern sie ein erweitertes –10-Element
besitzen (Consensus Sequenz: TGnTATAAT) oder σS-abhängige Promotoren (Kumar et al.,
1993; Dombroski, 1997). Eine derartige Promotorregion wurde durch Göller (1999) 101 bp
stromaufwärts von ectA bzw. mgs (H. elongata KB10) vermutet, konnte in der vorliegenden
Arbeit jedoch durch RACE-PCR-Analysen nicht bestätigt werden.
Als weiteres mögliches Promotorelement stromaufwärts von ectA, ein gearbox-Element, war
durch Göller (1999) benachbart zum σ70-abhängigen Promotor identifiziert worden (Abb. 31).
Diese Promotoren zeichnen sich dadurch aus, dass sich die Transkriptionsrate der von ihnen
gesteuerten Gene umgekehrt proportional zur Wachstumsrate verhält (Aldea et al., 1990).
Die Consensus-Sequenzen dieses Promotorentyps weichen von σ70- oder σS-abhängigen
Promotorsequenzen ab. Diese sind nach Aldea et al. (1990) in den Bereichen –10
(Consensus-Sequenz: CTGCAA) und –35 (Consensus-Sequenz: CGGCNAGTA) des Trans-
kriptionsstartpunkts lokalisiert. Verglichen mit der Consensus-Sequenz weicht die –35-

IV. Diskussion 160
Region in zwei von sechs Nukleotiden ab, die –10-Region ist in sieben von neun Nukleotiden
mit der Consensus-Sequenz identisch (Abb. 50). Nach den vorliegenden RACE-PCR-Ana-
lysen beträgt der Abstand der –10-Region zu dem identifizierten Transkriptionsstartpunkt 11
Nukleotide und liegt damit über den üblichen 10 bp. Noch höhere Abweichungen waren
durch Burdziak (2004) bei der Identifizierung eines σ70-abhängigen Promotors stromaufwärts
des stressassoziierten Gens yhgI von H. elongata festgestellt worden. Außerdem beträgt der
Abstand zwischen –10- und –35-Region nur 10 Nukleotide und entspricht daher nicht den in
E. coli gefundenen Abständen (17 ± 2). Allerdings können gearbox-Promotoren sowohl σ70-
als auch σS-abhängig sein (Nguyen & Burgess, 1997; Ballesteros et al., 1998). Somit könnte
diese Region der Kontrolle von σS unterliegen, bei denen eine –35-Region nicht notwendiger-
weise vorhanden sein muss. Kennzeichen von σS-Promotoren sind häufig eine fehlende
konservierte –35-Region (Tanaka et al., 1995).
Trotz der Abweichungen ist der identifizierte Transkriptionsstartpunkt geeignet, die Existenz
eines σ70-abhängigen Promotors und eines gearbox-Promotors zu bestätigen. Dadurch
werden die Analysen von Göller (1999) zur Promotorregion von ectA zum Teil bekräftigt. Ein
weiterer Transkriptionsstartpunkt stromabwärts des vermuteten σS-abhängigen Promotors,
101 bp stromaufwärts von ectA bzw. mgs konnte nicht bestätigt werden. Gründe hierfür
könnten methodischen Ursprungs sein. So hatte Burdziak (2004) bei der Durchführung der
RACE-PCR an H. elongata eine im Vergleich zu E. coli schlechtere mRNA Qualität fest-
gestellt, was sich in unklareren Produktbanden der RACE-PCR widerspiegelte (siehe
Abb. 29). Als Ursache wurde ein häufiger Abbruch der cDNA-Synthese oder eine allgemeine
Instabilität der H. elongata mRNA vermutet.
Die Transkriptionsanalysen mittels RACE-PCR bestätigten somit die Eignung des Ectoin-
Genlocus als Insertionsort für die Etablierung der Mannosylglycerat-Synthese. Die identifi-
zierten Promotoren (σ70/ gearbox) sollten sowohl während des exponentiellen Wachstums
als auch in der Phase einer Wachstumsverlangsamung unter hyperosmotischen Bedin-
gungen aktiv sein.

IV. Diskussion 161
-35 -10 σ70 Consensus-Sequenz TTGACA TATAAT
σ70-abhängiger Promotor 25 bp vor mgs TTGAaA TATgAT gearbox Consensus-Sequenz CTGCAA CGGCNAGTA gearbox-Element 36 bp vor mgs tTGaAA CGGCTgtTA Abb. 50: Sequenzvergleiche der identifizierten Promotorelemente
Dargestellt sind Sequenzvergleiche der Consensus-Sequenzen σ70-abhängiger und gearbox-gekennzeichneter
E. coli Promotoren mit möglichen Promotorregionen stromaufwärts von mgs in H. elongata KB10. Diese
Promotoren entsprechen den Promotoren stromaufwärts von ectA in H. elongata Wildtyp. Von den Consensus-
Sequenzen abweichende Basen sind als Kleinbuchstabe dargestellt und grau unterlegt .
8.1.4 Heterologe Expression von mgs in H. elongata Wildtyp und KB1
Um nach dem erfolgreichen Nachweis der mgs-Transkription die möglichen Ursachen für
das Ausbleiben der Mannosylglycerat-Synthese weiter einzugrenzen, wurde die heterologe
Expression der Mannosylglycerat-Synthase in H. elongata Stämmen mit Hilfe des pPromEct-
Vektors analysiert. Dieser Vektor weist stromaufwärts seiner multiplen Klonierungsstelle die
Promotorregion des ectA-Gens auf (Brünig, 2005). Bei der Konstruktion des Plasmids wurde
das Basentriplett stromaufwärts des ectA-Startkodons ausgetauscht (von aca in aac,
Position 525). Daher entspricht diese Region nicht zu 100 % der Region stromaufwärts des
mgs-Gens in KB10. Aufgrund der Positionierung dieses Basenaustauschs betrifft dieser
jedoch nicht die mutmaßliche Promotorregion stromaufwärts von mgs in KB10 und mit
großer Wahrscheinlichkeit auch nicht die dem Startkodon vorgelagerte Ribosomen-Bin-
dungsstelle.
Das Fusionsprotein aus Mannosylglycerat-Synthase und (His)6-tag konnte in einer Western-
Analyse in den löslichen zytoplasmatischen Fraktionen von H. elongata Wildtyp und KB1
nachgewiesen werden (Abb. 34). „Inclusion bodies“ waren in beiden Stämmen nicht nach-
weisbar. Damit bestätigte sich, dass eine heterologe Expression der Mannosylglycerat-
Synthase in H. elongata Stämmen möglich ist. Dabei spielt es keine Rolle, ob in den Zellen
Ectoin vorhanden ist oder nicht, da die Expression in H. elongata KB1 in einem Ectoin freien
Hintergrund erfolgte. Die mutmaßlich stabilisierende Wirkung des Ectoins zeigte bei der
vorliegenden heterologen Expression keine positiven Auswirkungen auf die Bildung des
löslichen Fusionsproteins der Mannosylglycerat-Synthase. Somit sollte die Expression auch
in H. elongata KB10, der wie KB1 nicht zur Ectoin-Synthese fähig ist, möglich sein. In den
Negativkontrollen der Western-Analyse (H. elongata Wildtyp und KB1 mit nicht-rekombinan-

IV. Diskussion 162
tem Plasmid) wurde keine Proteinbande detektiert, was bestätigte, dass es sich bei den
übrigen Banden nicht um unspezifische Kreuzreaktionen handelt.
Die Expression der Mannosylglycerat-Synthase wurde unter Zusatz von 4 % (KB1) bzw.
10 % NaCl (Wildtyp) untersucht. Anhand dessen sollte die osmoregulierte Expression der
Gesamtmenge des gebildeten Fusionsproteins abgeschätzt werden. So beobachtete Brünig
(2005) mit dem gleichen Expressionssystem eine Steigerung der gebildeten GFPuv-Menge
bei Erhöhung der Mediensalinität. Dies wurde auf die osmoregulierende Wirkung der
Promotorregion des Ectoin-Genclusters zurückgeführt. Auch für das Fusionsprotein der
Mannosylglycerat-Synthase konnte eine Konzentrationszunahme mit steigender Medien-
salinität beobachtet werden (Abb. 34).
Die lösliche Expression des Fusionsproteins der Mannosylglycerat-Synthase deutet an, dass
das Ausbleiben einer Mannosylglycerat-Produktion in H. elongata KB10 nicht auf eine
unzulängliche Expression zurückzuführen ist. Vielmehr verstärkte sich der Verdacht, dass
die Mannosylglycerat-Synthase aufgrund einer niedrigen Anzuchttemperatur von H. elongata
eine geringe enzymatische Aktivität aufweist und/oder dass benötigte Synthesevorstufen
nicht in ausreichender Menge bereitgestellt werden.
8.2 H. elongata KB10.1 (∆ectABC::mgsPP1776xanA)
8.2.1 Herstellung von H. elongata KB10.1
Da die Expression des Fusionsproteins der Mannosylglycerat-Synthase in H. elongata KB10
sowohl auf Transkriptions- als auch auf Translationsebene mittels Northern- und Western-
Analysen bestätigt wurde, verstärkte sich der Verdacht, dass das Fehlen von Mannosyl-
glycerat entweder mit einer zu niedrigen enzymatischen Aktivität der Mannosylglycerat-
Synthase oder mit einer mangelnden Bereitstellung von Synthesevorstufen zu begründen ist.
Die Mannosylglycerat-Synthase entfaltet ihre optimale Aktivität erst bei Temperaturen
zwischen 85 und 90 °C. Die KB10 Kulturen wurden jedoch bei nur 37 °C angezogen. An der
Synthese von Mannosylglycerat sind die Vorstufen Mannose-1-phosphat und GDP-Mannose
beteiligt (Abschnitt I, 3.3.2, Abb. 2). Diese werden durch eine Phosphomanno-Mutase
(wandelt Mannose-6-phosphat in Mannose-1-phosphat um) und eine Mannose-1-phosphat-
Guanylyltransferase (kondensiert D-Glycerat und Mannose-1-phosphat zu GDP-Mannose)
bereitgestellt. Die Gene dieser Enzyme sind in R. marinus bis dato nicht charakterisiert, die
Aktivität der Enzyme wurde jedoch durch Messung an zellfreien Extrakten von R. marinus
mittels 13C-markierter Substrate nachgewiesen (Martins et al., 1999). Um die Bereitstellung
der Synthesevorstufen zu gewährleisten, sollten die Gene entsprechender Enzyme in das
Genom von H. elongata KB10 im Austausch gegen ectBC integriert und dort zusammen mit
dem mgs-Gen in einem Operon exprimiert werden. Bei der Auswahl der Gene wurde auf die

IV. Diskussion 163
Sequenzen mesophiler Organismen zurückgegriffen werden, da bei thermophilen Enzymen
wie bei der Mannosylglycerat-Synthase mit einer zu geringen enzymatischen Aktivität ge-
rechnet wurde. Zudem sind entsprechende Gensequenzen bei R. marinus nicht zugänglich.
Gleiches gilt für entsprechenden Sequenzen von H. elongata. So mussten für die Etablierung
der Mannosylglycerat-Produktion orthologe/homologe Gene verwandter Stämmen von
H. elongata ausfindig gemacht werden. Pseudomonaden besitzen einen sehr vielseitigen
Zuckerstoffwechsel, der sie zur Nutzung vieler Kohlenstoffquellen befähigt (Lessie & Phibbs,
1984). So nutzt Pseudomonas aeruginosa Mannose-1-phosphat und GDP-Mannose als
Synthesevorstufe für Alginat, einem Exopolysaccharid. Die Vorstufen werden durch eine
Phosphomanno-Mutase (kodiert durch algB) und eine Mannose-1-phosphat-Guanylyl-
transferase (kodiert durch algC) bereitgestellt (Sa-Correia et al., 1987). Orthologe Gen-
sequenzen ließen sich auch in der Genomsequenz (NCBI-Acc.Nr. NC_002947) von P. putida
KT2440 nachweisen (Nelson et al., 2002). Dieser Stamm schien aus mehreren Gründen
geeignet als Spenderorganismus für die gesuchten Gensequenzen zu dienen. Wie
H. elongata gehört P. putida KT2440 zu den Gram-negativen γ-Proteobakterien. Darüber
hinaus handelt es sich bei P. putida KT2440 im Gegensatz zu vielen anderen Pseudo-
monaden um einen Sicherheitsstamm für gentechnisches Arbeiten. Auch der GC-Gehalt von
H. elongata (60,5 %) ist mit dem von P. putida KT2440 (61,6 %) vergleichbar (Vreeland et
al., 1980; Weinel et al., 2002). Zudem weicht die „Codon usage“ beider Organismen um
durchschnittlich nur 4,76 % voneinander ab (Analyse auf http://gcua.schoedl.de/).
Mit Hilfe der KEGG-Datenbank (Kanehisa & Goto, 2000; Kanehisa et al., 2006) wurde der
Zuckerstoffwechsel von P. putida KT2440 analysiert und so die orthologen Sequenzen xanA
(kodiert für eine mögliche Phosphomanno-Mutase) und PP1776 (kodiert für eine mögliche
Mannose-1-phosphat-Guanylyltransferase) identifiziert. Es handelt bei diesen um zwei
benachbarte, hypothetische Gene, die mutmaßlich am Fructose- und Mannose-Stoffwechsel
von P. putida KT2440 beteiligt sind (Abb. 51). Die Benennung des xanA-Gens von P. putida
KT2440 erfolgte nach Computervorhersagen in Analogie zu dem ursprünglich identifizierten
xanA-Gen des Gram-negativen pflanzenpathogenen Bakteriums Xanthomonas campestris
(Köplin et al., 1992). Dieses Bakterium wurde durch die Bildung von Xanthan bekannt. Dabei
handelt es sich um ein Exopolysaccharid, dessen Bildung mit der Besiedlung von Pflanzen-
oberflächen gekoppelt ist (Chatterjee & Vidaver, 1985). Köplin et al. (1992) isolierten xanA
und bestätigten die Phosphomanno-Mutase Funktion seines Genprodukts.
Nach Angaben der KEGG-Datenbank, der NCBI-Homepage (Acc.Nr.: NP_743932) und CD-
Search Datenbank (Marchler-Bauer et al., 2003) kodiert PP1776 für ein hypothetisches bi-
funktionales Enzym, welches neben seiner Mannose-1-phosphat-Guanylyltransferase Aktivi-
tät eine Mannose-6-phosphat-Isomerase-Aktivität aufweist. Nach diesen Analysen sollte der
N-terminale Bereich des Proteins als Guanylyltransferase fungieren, während der C-Termi-

IV. Diskussion 164
nus die Isomerase-Aktivität ausprägt. Letztere würde für eine Umwandlung von Fructose-6-
phosphat in Mannose-6-phosphat sorgen. Dies könnte von Vorteil sein, wenn dem Stamm
Fructose als kostengünstige Kohlenstoffquelle zugesetzt wird. Einen Nachweis der enzyma-
tischen Aktivität beider Genprodukte (XanA und PP1776) gab es für P. putida KT2440 nicht.
Es handelte sich lediglich um orthologe Sequenzen mit gewissen Sequenzähnlichkeiten zu
den Genprodukten der Gene algBC aus P. aeruginosa.
Zur Herstellung von H. elongata KB10.1 (∆ectABC::mgsPP1776xanA) wurden in H. elongata
KB10 die Gene ectBC mittels homologer Rekombination gegen die Gene PP1776 und xanA
aus P. putida KT2440 ausgetauscht.
xanA
PP1776
PP1776
Abb. 51: Fructose- und Mannose-Metabolismus von P. putida KT2440 Dargestellt ist ein Ausschnitt des Zuckermetabolismus von P. putida KT2440 (KEGG-Datenbank,
http://www.genome.jp/kegg/, Kanehisa & Goto, 2000; Kanehisa et al., 2006). Alle für P. putida KT2440 relevanten
Stoffwechselreaktionen sind blau dargestellt. Die eingerahmten Zahlen stehen für eine spezifische Stoffwechsel-
reaktion. Die hypothetischen Gene xanA bzw. PP1776 kodieren für eine hypothetische Phosphomanno-Mutase
(NCBI-Acc.Nr.: AAN67397) bzw. eine hypothetische Mannose-1-phosphat-Guanylyltransferase (NCBI-Acc.Nr.:
AAN67396). Die Aktivität der Mannose-1-phosphat-Guanylyltransferase ist in Abhängigkeit von der Substrat-
konzentration vermutlich reversible. Außerdem katalysieren zu PP1776 orthologe Enzyme die Umwandlung von
Fructose-6-phosphat in Mannose-6-phosphat und umgekehrt.

IV. Diskussion 165
8.2.2 Transkription der Fremdgene mgs, PP1776 und xanA in KB10.1
Die Northern-Analysen mit den genspezifischen Sonden von mgs, PP1776 und xanA wiesen
das Vorhandensein der Einzeltranskripte aller drei Gene und die Kotranskripte mgsPP1776
sowie PP1776xanA in H. elongata KB10.1 nach. Außerdem wurde mit allen genannten RNA-
Sonden eine schwache ca. 4 kb große Bande detektiert. Die Größe dieser Bande stimmt mit
der Länge eines mgsPP1776xanA-Gesamttranskripts überein. Damit bestätigte sich die
Expression der drei Fremdgene in einem Operon. Dieses Ergebnis wurde mittels RT-PCR
bestätigt. In KB10 hatte mit der ectB-Sonde kein mgsectBC-Gesamttranskript nachgewiesen
werden können. Ein direkter Vergleich der Ergebnisse mit den Transkriptionsanalysen an
KB10 (Abschnitt III, 3.2.2.2) und Wildtyp (Stumpfe, 2003) ist jedoch nicht möglich, da
H. elongata KB10.1 auch in den nicht-kodierenden DNA-Bereichen Fremd-DNA von
P. putida KT2440 enthält. Hier wurde der DNA-Bereich zwischen ectB und ectC mit seinen
möglichen Terminationssignalen gegen Sequenz aus P. putida KT2440 ausgetauscht.
Wie schon mit der mgs-Sonde, wurden auch mit den genspezifischen Sonden von PP1776
und xanA jeweils Banden detektiert, die auf die Synthese von Einzeltranskripten hinweisen.
Diese Tatsache könnte auf unmittelbar stromabwärts von PP1776 und xanA lokalisierte
Transkriptionsterminationssignale hindeuten. Die freie Energie der möglichen Terminations-
schleife stromabwärts von PP1776 bzw. xanA beträgt nach „mfold“ ∆G0 = -98 kJ/mol bzw.
-92 kJ/mol. Beide Terminationsschleifen scheinen nur unwesentlich schwächer ausgeprägt
als diejenige stromabwärts von mgs bzw. ectA (Abschnitt IV, 8.1.2). Allerdings folgt bei
charakteristischen Terminationssignalen auf die palindrome, GC-reiche Region in der Regel
eine AT-reiche Sequenz, die aufgrund der schwachen Basenpaarung zum Ablösen der
mRNA von der Matrize beiträgt (Stryer, 1996). Die unmittelbar an das Terminationssignal
angrenzende Sequenz stromabwärts von PP1776 ist jedoch eher GC-reich (TCGCTCGGC,
siehe Anhang). Dies könnte erklären, warum die Transkription stromabwärts von PP1776
sowohl bei H. elongata KB10.1 als auch bei der Positivkontrolle P. putida KT2440 nicht
zwingend zum Abbruch kommt. Dies wiederum würde die Entstehung eines PP1776xanA-
Kotranskripts ermöglichen, welches in KB10.1 und P. putida KT2440 nachgewiesen werden
konnte. Anderseits könnte es sich stromabwärts von PP1776 auch um eine Rho-abhängige
Termination handelt. Zudem könnte es sich bei den Einzeltranskripten auch um präparati-
onsbedingte Artefakte handeln.
Auch wenn bezüglich der Regulation der Expression der Fremdgene viele Fragen offen
bleiben, konnte gezeigt werden, dass von allen Genen Transkripte in den Zellen von
H. elongata KB10.1 vorliegen und somit die ersten Voraussetzungen für die Etablierung der
Mannosylglycerat-Synthese erfüllt sind. Eine genauere Analyse der Expression scheint vor
diesem Hintergrund nicht sinnvoll, zumal es sich um ein „künstlich“ etabliertes System
handelt, bei dem die Auswirkungen auf die Stoffwechselregulation nicht absehbar sind.

IV. Diskussion 166
8.2.3 Physiologische Charakterisierung von KB10.1
8.2.3.1 Nachweis der Mannosylglycerat-Synthese
Die nachgewiesenen Expression der Gene mgs, PP1776 und xanA in H. elongata KB10.1
führte unabhängig von der verwerteten Kohlenstoffquelle im Vergleich zu H. elongata KB10
und KB1 zu einem wenn auch geringfügig beschleunigten Wachstum. Die Wachstumsraten
des Wildtypstamms H. elongata DSM 2581T werden jedoch bei weitem nicht erreicht
(Abb. 39). HPLC-Analysen zeigten bei allen verwendeten Kohlenstoffquellen das Vorhanden-
sein von Mannosylglycerat an. Damit bestätigten sich indirekt auch die vorhergesagten
Enzymaktivitäten, der durch PP1776 und xanA kodierten hypothetischen Proteine. Zudem
deutet sich an, dass die Mannosylglycerat-Synthase in H. elongata nicht nur in löslicher,
sondern auch in aktiver Form gebildet wird und dass ihre Aktivität mit steigender Temperatur
zunimmt. Die Steigerung der Inkubationstemperatur von 37 °C auf 40 °C führt zu einer
Zunahme des Mannosylglycerat-Gehalts (Abb. 43). Gleichzeitig könnte die Erhöhung der
Anzuchttemperatur auch in einer Aktivitätssteigerung des von Göller (1999) vermuteten σS-
Promoters resultieren. So wird in E. coli die allgemeine Stressantwort durch den Sigmafaktor
σS vermittelt (Loewen & Hengge-Aronis, 1994). Dieser Faktor kontrolliert eine große Anzahl
von Genen, die unter anderem für Osmoschutz, Thermoschutz, Säure und Ethanol-Toleranz
verantwortlich sind (Lee et al., 1995; Farewell et al., 1998). Auch wenn durch die in dieser
Arbeit durchgeführten RACE-PCR die Existenz eines solches Promotors nicht bestätigt
werden konnte, kann seinen Einfluss auf die Ectoin-Synthesegene (hier Mannosylglycerat-
Synthese) nicht völlig ausgeschlossen werden. Eine höhere Aktivität σS-abhängiger Promo-
toren führt zu einer gesteigerten Expression der so regulierten Gene und damit möglicher-
weise zu einer gesteigerten Menge der Mannosylglycerat-Synthase. Gleichermaßen kann
das gearbox-Element zu einer gesteigerten Expression führen, da die Transkriptionsrate der
durch die gearbox-Elemente gesteuerten Gene umgekehrt proportional zur Wachstumsrate
ist (Aldea et al., 1990).
Nach diesen Analysen ist davon auszugehen, dass als Grund für das Ausbleiben einer
Mannosylglycerat-Synthese in H. elongata KB10 nur das Fehlen von Synthesevorstufen in
Frage kommt.
8.2.3.2 Mannosylglycerat-Synthese bei Nutzung unterschiedlicher Substrate
Die höchsten Wachstumsraten von KB10.1 werden mit den Kohlenstoffquellen Saccharose,
Maltose und Glucose erzielt. Trotzdem liegt der mittels HPLC-Analyse ermittelte Mannosyl-
glycerat-Gehalt bei Anzucht mit diesen Substraten unterhalb dessen bei Anzucht mit Man-
nose (spät-stationäre Phase, Abb. 42). Auch eine Anzucht von H. elongata KB10.1 mit
Fructose, einer kostengünstigen Kohlenstoffquelle, erbrachte einen nur sehr geringen
Mannosylglycerat-Gehalt. Offensichtlich weist das hypothetische Genprodukt von PP1776

IV. Diskussion 167
nicht die vermutete Mannose-6-phosphat-Isomerase-Aktivität auf, die zur Umwandlung von
Fructose-6-phosphat in Mannose-6-phosphat und so zu einer verstärkten Einspeisung in die
Mannosylglycerat-Synthese geführt hätte.
H. elongata ist zum Wachstum mit Mannose bzw. Glycerol fähig (Vreeland et al., 1980).
Beide wurden u.a. auch in Kombination in zwei unterschiedlichen Mengenverhältnissen als
Kohlenstoffquelle eingesetzt (Abschnitt III, 3.2.2.1). Hierdurch sollte überprüft werden, ob die
Kombination beider Substrate einen positiven Einfluss auf das Wachstum und die Produktion
von Mannosylglycerat hat. Glycerol sollte als Kosubstrat zu einer Steigerung des intrazellu-
lären D-Glycerat-Gehalts führen. Intrazellulär wird vermutlich Glycerol durch die Glycerol-
Kinase zu Glycerol-3-phosphat phosphoryliert. Die Glycerolphosphat-Dehydrogenase oxi-
diert dieses zu Dihydroxyacetonphosphat. Glycerolaldehyd-3-phosphat wird schließlich durch
eine Triosephosphat-Isomerase gebildet. Ausgehend von Glycerolaldehyd-3-phosphat ent-
steht ein 3-Phosphoglycerat, welches katalysiert durch eine Glycerat-Kinase in D-Glycerat
umgewandelt werden kann (Stryer, 1996). Eine Steigerung des Mannosylglycerat-Gehalts im
Vergleich zur Anzucht mit Mannose als alleiniger Kohlenstoffquelle war nicht zu beobachten
(Abb. 42). Allerdings wurde deutlich, dass bei Kombination von Mannose und Glycerol ein
höherer Anteil von Mannose zu einer höheren Wachstumsrate führt (Abb. 39). Damit
bestätigt sich auch für H. elongata die Analysen von Sampaio et al. (2003a), nach denen bei
E. coli die intrazelluläre D-Glycerat-Konzentration bei der Mannosylglycerat-Synthese nicht
limitierend wirkt.
8.2.3.3 Mannosylglycerat-Gehalt in Abhängigkeit von der Wachstumsphase
Nach Analyse der Wachstumskurve von H. elongata KB10.1 geht dieser Stamm bei Anzucht
mit Mannose schon bei geringen optischen Kulturdichten in die stationäre Wachstumsphase
über (Abb. 44). Die bei Anzucht mit Mannose für H. elongata KB10.1 ermittelte Mannosyl-
glycerat-Konzentrationen stehen in Abhängigkeit von der Wachstumsphase aber auch der
Temperatur. So wurde bei einer Inkubationstemperatur von 40 °C und einer Salinität von 4 %
bei einer OD600 von 0,4, bei der sich KB10.1 in der früh-stationären Phase befindet (Abb. 44),
die höchste intrazellulären Mannosylglycerat-Konzentration nachzuweisen (27,16 mg / g TG;
Abb. 43). Beim Übergang in die spät-stationäre Phase, nimmt der Mannosylglycerat-Gehalt
kontinuierlich ab (9 mg / g TG bei OD600 = 1,05). Ein ähnlicher Verlauf des Mannosylglycerat-
Gehalts ist auch bei einer Anzuchttemperatur von 37 °C zu beobachten. Hier liegen die
Mannosylglycerat-Gehalte jedoch durchweg niedriger. Ein Abnehmen des Solutegehalts
beschrieben Wohlfarth et al. (1990) und Frings et al. (1995) für den Gesamtsolutegehalt
verschiedener moderat halophiler Bakterien wie H. elongata. Das Absinken des Solute-
gehalts deutet auf eine Metabolisierung desselben hin. Wird Mannosylglycerat dem Wildtyp
als Kohlenstoffquelle angeboten, erfolgt ein nur sehr langsames Wachstum, das sich über

IV. Diskussion 168
einen Zeitraum von zwei Wochen erstreckt, wobei die gewonnene Biomasse sehr gering
bleibt. Dies bestärkt wiederum die Annahme, dass H. elongata kein spezifisches Mannosyl-
glycerat-Transportsystem besitzt. Ist Mannosylglycerat z.B. durch Eigensynthese in den
Zellen vorhanden, kann es, nach der Entwicklung des Mannosylglycerat-Gehalts zu urteilen,
dennoch metabolisiert werden.
Im Zusammenhang mit einem absinkenden Mannosylglycerat- (KB10.1) bzw. Ectoin-Gehalt
(Wildtyp) erscheint die durch Göller (1999) vermutete „duale Kontrolle“ der Ectoin-Gene
fraglich. Danach erfolgt die Expression der Ectoin-Gene vermutlich nicht nur unter osmo-
tischer Kontrolle, sondern auch in Abhängigkeit von der Wachstumsphase. Da die Expres-
sion der integrierten Fremdgene zur Mannosylglycerat-Synthese in der stationäre Phase
anscheinend jedoch abnimmt, was an der Abnahme der Mannosylglycerat-Konzentration
während dieser Phase zu ersehen ist, stellt sich die Frage, ob die vermutete positive Regula-
tion durch das gearbox-Element (stromaufwärts von ectA bzw. mgs) tatsächlich erfolgt.
8.2.3.4 Ein Vergleich: H. elongata Wildtyp, KB1 und KB10.1
Zur besseren Verdeutlichung der Mannosylglycerat-Produktion ist eine Bestimmung der
intrazellulären Mannosylglycerat-Konzentration von KB10.1 hilfreich. Dazu muss jedoch der
zelluläre Mannosylglycerat-Gehalt auf das Zellvolumen bezogen werden. Da in der vorlie-
genden Arbeit keine Volumenbestimmungen durchgeführt wurden, wurden die von Miguelez
& Gilmour (1994) für H. elongata ermittelten Zellvolumina zugrunde gelegt. Die ermittelten
Werte beziehen sich auf Zellen in der spät-exponentiellen bis früh-stationäre Wachstums-
phase, der Phase in der sich auch die Zellen von KB10.1 mit dem höchsten bestimmten
Mannosylglycerat-Gehalt zum Zeitpunkt der Probennahme befanden (Abb. 43 & 44). Geht
man davon aus, dass Zellen von H. elongata KB10.1 das gleiche Volumen wie der Wildtyp
aufweisen, entspricht das Zellvolumen von H. elongata KB10.1 bei einer Mediensalinität von
4 % einem Wert von 2,15 µl/mg Protein. Geht man davon aus, dass der Proteinanteil je nach
Mediensalinität 40-50 % des Trockengewichts von H. elongata ausmacht (Schlegel, 1972),
würde dieser Wert einem Volumen von 0,86-1,075 µl/mg TG entsprechen. Das zelluläre
Volumen variiert abhängig vom Salzgehalt, der Wachstumsphase und von der Anzucht-
temperatur. So nimmt das Zellvolumen mit steigender Temperatur und fortschreitender
Wachstumsphase ab (Matheson et al., 1976; Baldwin et al., 1988; Wohlfarth, 1993). Auf-
grund der erhöhten Inkubationstemperatur von 40 °C wurde daher mit einem Zellvolumen
von 0,86 µl/mg TG weitergearbeitet. Bezogen darauf ergibt sich bei einer OD600 = 0,4 (40 °C)
eine intrazelluläre Mannosylglycerat-Konzentration von 103 mM (MW: 307 g/mol). Verglichen
damit weist der H. elongata Wildtyp bei einer Mediensalinität von 4 % und einer Anzucht-
temperatur von 40 °C eine intrazelluläre Ectoin-Konzentration von 302 mM auf (Wohlfarth,
1993). Da die Ectoin-Konzentration anders als bei KB10.1 jedoch in der spät-stationäre

IV. Diskussion 169
Phase ermittelt wurde, kann dieser Wert nur als Nährungswert betrachtet werden und liegt
vermutlich in der früh-stationären Phase höher. Beim Übergang in die spät-stationäre Phase,
nimmt der Mannosylglycerat-Gehalt von KB10.1 kontinuierlich ab (34,1 mM bei OD600 = 1,05)
(Abb. 43).
H. elongata Wildtyp, KB1 und KB10.1 akkumulieren u.a. intrazellulär Glutamat. Glutamat ist
bei physiologischen pH-Wert negativ geladen und spielt als Gegenion zu K+-Ionen bei der
Osmoregulation Gram-negativer Bakterien eine wichtige Rolle. Bis zu einer Salinität von 3 %
NaCl stellt K+-Glutamat das Hauptsolut von H. elongata dar (Kraegeloh, 1998). In angepas-
sten Zellen und nach hyperosmotischem Schock liegen Kalium und Glutamat in einem
konstanten Verhältnis von 2,4:1 vor (Kraegeloh, 2003). Bei höheren Salinitäten produziert
H. elongata vermehrt Ectoin, der intrazelluläre Kalium-Level bleibt relativ konstant im Bereich
von über 0,5 M (Kraegeloh, 1998). Dies entspricht bei gleichbleibendem Mengenverhältnis
einer zellulären Glutamat-Konzentration von ca. 0,2 M. Der Glutamat-Level von H. elongata
KB1, welcher kein Ectoin mehr produzieren kann, liegt bei 4 % NaCl mit ca. 0,377 M deutlich
höher (Grammann, 2000). Entsprechend hoch wird der Kalium-Level sein, da sich der Gluta-
mat-Gehalt bei H. elongata Wildtyp in Abhängigkeit vom Kalium-Level ändert (Kraegeloh,
2003). Genauso wie H. elongata KB1 produziert KB10.1 kein Ectoin mehr, dafür jedoch
aufgrund der integrierten Fremdgene neben Glutamat zusätzlich Mannosylglycerat. Wie
Glutamat weist Mannosylglycerat bei physiologischem pH-Wert eine negative Ladung auf
und dient aufgrund dessen möglicherweise als Gegenion für Kalium. Nach ersten Abschätz-
ungen bleibt der Glutamat-Gehalt von KB10.1 über die gesamten Wachstumsphasen auf
gleichem Niveau (ca. 22 mg/gTG bzw. 0,174 M) und steigt auch nicht an, um den dras-
tischen Abfall des Mannosylglycerat-Gehalts in der spät-stationäre Phase auszugleichen.
Der intrazelluläre Glutamat-Level liegt damit deutlich unter dem von KB1 (30 °C). Mit dem
Glutamat-Level (0,2 M) des Wildtyps bei 4 % NaCl und 30 °C ist der für KB10.1 bestimmte
Wert vergleichbar. Nach Vermutungen von Grammann (2000) akkumuliert KB1 vermehrt
Kalium-Ionen zum Ausgleich des osmotischen Ungleichgewichts. Ähnliches wird auch
H. elongata KB10.1 tun. Beide benötigen jedoch Gegenionen, um den Ladungsausgleich
sicherzustellen. In H. elongata KB10.1 erscheint der intrazelluläre Glutamat-Level dafür
jedoch zu niedrig. Neben Glutamat akkumuliert KB10.1 jedoch Mannosylglycerat, welches
einfach negativ geladen ist und daher vermutlich nur ein Kalium zu binden vermag. Nach den
obigen Konzentrationsbestimmungen können die Konzentrationen von Mannosylglycerat (ca.
0,1 M) und Glutamat (ca. 0,174 M) jedoch nicht zu einem Ladungsausgleich führen.
Durch viele halophile Mikroorganismen werden beim Übergang in die stationäre Phase
vermehrt anorganische Solute (z.B. NaCl) akkumuliert, um die organischen Solute zu ersetz-
en und Energie einzusparen (Oren, 1999). Untersuchungen von Sadler et al. (1980) an
Halomonas halodenitrificans (vormals Paracoccus halodenitrificans) hatten gezeigt, dass

IV. Diskussion 170
beim Übergang in die stationäre Phase die ATP-Konzentration in der Zelle sinkt und die
Aktivitäten der membrangebundenen ATPase und der NADH-Oxidase abnehmen. ATP-
Mangel bedeutet Energiemangel und eine eingeschränkte Syntheseleistung, was auch
kompatible Solute betreffen kann. So wurde gemutmaßt, dass die Abnahme beider Enzym-
aktivitäten zu einem Zusammenbruch des Protonengradienten führt, was letztendlich mit
einer Verringerung der Aktivität des Na+/H+-Antiporters einhergeht. Folglich kämme es zu
dem gemessenen intrazellulären Anstieg der Na+-Konzentration. Sadler et al. (1980) ver-
muteten, dass die Anhäufung der Na+-Ionen zusätzlich zu einem ladungsbedingten Ausstrom
der K+-Ionen führt. Kurz (2003) hatte in H. elongata DSM2581T einen potentiellen Na+/H+-
Antiporter identifiziert, was ähnliche Prozesse auch in H. elongata nahe legt.
Bei H. elongata KB10.1 treten derartige Ereignisse möglicherweise schon sehr früh auf, da
er sich schon bei geringer OD600 in der stationäre Phase befindet. Resultat ist vermutlich eine
hohe Konzentration von K+-Ionen mit anorganischem Gegenionen, was das Wachstum
negativ beeinflusst und einen möglicherweise wachstumsfördernden Einfluss des Mannosyl-
glycerats schmälert. Grund für die zu niedrige Glutamat-Konzentration könnte darüber hin-
aus der künstlich etablierte Stoffwechselweg zur Synthese von Mannosylglycerat sein. Zum
einen wird übermäßig viel Energie in die Synthese von Mannosylglycerat verwandt
(Abschnitt IV, 8.2.2.5) zum anderen werden permanent indirekte Synthesevorstufen des
Glutamats (Glycerolaldehyd-3-phosphat) vor dem Eintritt in den Trikarbonsäurezyklus abge-
zogen. Es entsteht eine energetische Konkurrenz zwischen Energiegewinn, Glutamat- und
Mannosylglycerat-Synthese. Das Resultat sind Stoffwechselengpässe, die sowohl die Bio-
synthese von Mannosylglycerat als auch von Glutamat limitieren können.
8.2.3.5 Energetische Bilanz der Solutesynthese
Bei Ectoin, dem kompatiblen Solut von H. elongata, handelt es sich energetisch betrachtet
um ein kostengünstiges Osmolyt (Oren, 1999; Maskow & Babel, 2001). Nach Oren (1999)
kostet die Biosynthese von Ectoin (C6-Körper) genau eine Glucose-Einheit und damit 38
ATP-Äquivalente. Grundlage für diese Berechnung ist, dass eine NADH-Einheit drei ATP-
Äquivalenten entspricht. Bei der Synthese von Ectoin aus Mannose sieht die energetische
Bilanz gleich aus.
Mannosylglycerat (C9-Körper) besteht aus einer Mannose- und eine Glycerat-Einheit. Für die
Biosynthese von Mannosylglycerat muss H. elongata KB10.1 neben einer vollständigen
Mannose-Einheit eine weitere halbe Mannose-Einheit zur Bereitstellung von Glycerat inves-
tieren. Damit fällt der Verlust an ATP-Äquivalenten bei der Synthese von Mannosylglycerat
wesentlich höher aus. Nach eigenen Analysen kann H. elongata aus der zweiten Mannose-
Einheit nur 22 oder 23 der 38 ATP-Äquivalente gewinnen, weil 15 oder 16 ATP verloren-
gehen, um ausgehend von Glycerolaldehyd-3-phosphat ein Molekül Glycerat bereitzustellen.

IV. Diskussion 171
Glycerolaldehyd-3-phosphat wird zunächst zu 3-Phosphoglycerat oxidiert, welches dann
durch die Einwirkung einer 3-Phosphoglycerat-Phosphatase oder einer Glycerat-Kinase zu
D-Glycerat umgewandelt wird. Die Aktivität der Glycerat-Kinase führt zum Gewinn eines
ATP-Moleküls. Welches der beiden Enzyme letztlich in H. elongata aktiv ist, ist bisher unbe-
kannt. Abhängig davon gewinnt H. elongata aus dieser Mannose-Einheit 22 bzw. 23 ATP-
Äquivalente. Der Rest der Mannose-Einheit wird zu Pyruvat oxidiert und zur Energie-
gewinnung in den Trikarbonsäurezyklus eingeschleust. Ein zusätzliches ATP-Äquivalent wird
bei der Umwandlung von Mannose-1-phosphat zu GDP-Mannose verbraucht (Abschnitt I,
Abb. 2 & Abschnitt IV, Abb. 48). Zusammengefasst ergibt dies insgesamt 54 bzw. 55 ATP-
Äquivalente, welche H. elongata KB10.1 bei der Biosynthese von Mannosylglycerat verloren-
gehen. Danach kostet eine Mannosylglycerat-Einheit energetisch betrachtet soviel wie 1,4
Ectoin-Einheiten. Im Vergleich dazu kostet die Biosynthese von Glucosylglycerol, einem
kompatiblen Solut halophiler Cyanobakterien, mehr als 60 ATP-Äquivalente (Oren, 1999).
Dieses Solut weist bei physiologischem pH-Wert jedoch keine Nettoladung auf.
Die energetische Bilanz der Mannosylglycerat-Synthese ist sicherlich ein gewichtiger Grund
für das Absinken der intrazellulären Mannosylglycerat-Konzentration in der spät-stationären
Phase. In dieser Phase, in welcher die Substratkettenphosphorylierung die einzige Möglich-
keit des Energiegewinns darstellt, ist keine Energie für die Synthese derartig „teuerer“ Solute
übrig. Vorhandene Solute werden darüber hinaus zur Energiegewinnung herangezogen.
8.2.3.6 Mannosylglycerat als kompatibles Solut für H. elongata
Mannosylglycerat hatte in verschiedenen Untersuchungen sein Potential bei der Stabili-
sierung verschiedener Enzyme gezeigt (Ramos et al., 1997; Melo et al., 2001; Faria et al.,
2003; Faria et al., 2004). Dennoch weist H. elongata KB10.1 verglichen mit H. elongata
Wildtyp nur sehr niedrige Wachstumsraten auf. Gegenüber KB1 ist das Wachstum beschleu-
nigt. Gleichzeitig scheint die durch Mannosylglycerat verursachte Wachstumsverbesserung,
nicht unbedingt konzentrationsabhängig zu sein. So ist die deutlichste Steigerung der
Wachstumsraten bei Verwendung von Saccharose als Kohlenstoffquelle zu verzeichnen
(Abb. 39). Der höchste Mannosylglycerat-Gehalt der Kulturen bei einer OD600 = 1 (stationäre
Phase) wurde jedoch bei Verwendung von Mannose als Kohlenstoffquelle erzielt (Abb. 42).
Gleiches wird auch in der exponentiellen Phase gelten, in der definitionsgemäß die Bestim-
mung der Wachstumsraten erfolgt. In dieser Wachstumsphase werden durch H. elongata
KB10.1 vermutlich die höchsten nachgewiesenen Mannosylglycerat-Konzentrationen ak-
kumuliert. So wurde bei Anzucht mit Mannose als Kohlenstoffquelle bei einer OD600 von 0,4,
bei der sich der Stamm gerade in der früh-stationären Phase befindet (Abb. 44), die
höchsten intrazellulären Mannosylglycerat-Konzentrationen nachgewiesen (Abb. 43). Trotz-
dem fällt die Steigerung der Wachstumsraten im Vergleich zu KB1 und KB10 nur gering aus.

IV. Diskussion 172
Dies wirft zumindest für H. elongata die Frage nach der „Qualität“ des synthetisierten
Mannosylglycerats bei der Erfüllung der Funktion eines kompatibles Soluts bzw. Osmolyts
auf. Gründe für die nur beschränkte Funktion als kompatibles Solut werden die Ladung des
Mannosylglycerats bei physiologischem pH-Wert, die hohen Synthesekosten und eine
mögliche Konkurrenz zur Glutamat-Synthese sein. Die negative Ladung von Mannosyl-
glycerat wird eine physiologische Bedeutung bei thermophilen Organismen besitzen, zumal
viele der thermophilen Solute negativ geladen sind (Martins et al., 1996; Santos & Da Costa,
2002).
8.2.3.7 Vor- und Nachteile der Produktion von Mannosylglycerat in H. elongata KB10.1
Mannosylglycerat wird durch die Fa. Bitop AG derzeit aus Rhodothermus marinus bF07 ge-
wonnen. Die Anzucht des Stamms erfolgt nach einem nicht veröffentlichten Produktions-
verfahren in einem modifizierten Degryse-Medium (Nunes et al., 1992), welches zusätzlich
Malz sowie Ammoniumchlorid und -sulfat sowie final 7 % NaCl enthält. Die Gewinnung des
Mannosylglycerats erfolgt durch Anwendung des Bakterienmelkverfahrens (Sauer, 1995;
Sauer & Galinski, 1998). Die Ausbeute an Mannosylglycerat beträgt schließlich 1,85 g/l.
Bezogen auf das Zelltrockengewicht entspricht dies einer Ausbeute von 36,3 g/kg TG
(Schwarz Fa. Bitop, pers. Mitteilung). In Schüttelkolbenversuchen mit H. elongata KB10.1
konnte in der früh-stationären Wachstumsphase (OD600 = 0,4) in Zellextrakten ein maximaler
Mannosylglycerat-Gehalt von 27 g/kg TG ermittelt werden (MM63-Medium, 4 % NaCl, 40 °C,
Abb. 43). Diese Werte liegen auf den ersten Blick in der gleichen Größenordnung, sind
jedoch nicht direkt miteinander vergleichbar, da es sich einmal um eine wirkliche Ausbeute,
im andern Fall um einen zellulären Gehalt handelt und sich außerdem die Mediensalinitäten
unterscheiden. Nach eigenen HPLC-Analysen liegt der intrazelluläre Mannosylglycerat-
Gehalt bei Anzucht von R. marinus in MM162-Medium (4 % NaCl) bei etwa 100 g/kg TG und
damit etwa viermal so hoch wie bei H. elongata KB10.1. Entsprechend werden sich auch die
Ausbeuten bei der Aufreinigung unterscheiden.
Bei der Gewinnung von Mannosylglycerat müssen Verluste bei der Durchführung des Bak-
terienmelkens und der weiteren Produktaufreinigung berücksichtigt werden. Allerdings ent-
fällt bei Produktion durch KB10.1 die aufwendige Abtrennung von Mannosylglyceramid, wel-
ches neben Mannosylglycerat durch R. marinus synthetisiert wird. Weniger Aufreinigungs-
schritte machen eine höhere Produktausbeute möglich.
Vorbild für den Mannosylglycerat-Produktionsstamm H. elongata KB10.1 ist der Ectoin-Pro-
duktionsstamm H. elongata KB2 (Volke, 2001). Es handelt sich dabei um eine Ectoin-Aus-
scheidermutante, die durch Deletion von Komponenten des zelleigenen Ectoin-Transport-
systems nicht mehr in der Lage ist, ins Medium verlorenes Ectoin in die Zelle aufzunehmen.
Dieser Stamm scheidet nur Ectoin ins Medium ab, während bei der klassischen Ectoin-

IV. Diskussion 173
Gewinnung aus H. elongata Wildtyp mittels Bakterienmelken zusätzlich Hydroxyectoin ins
Medium abgegeben wird. Hydroxyectoin wird durch H. elongata KB2 nicht ins Außenmedium
abgegeben, da es bei der für die Fermentation gewählten geringen Salinität und Temperatur
nicht synthetisiert wird (Wohlfarth, 1993; Volke, 2001). Bei einer Gewinnung von Ectoin
mittels KB2 entfällt somit die verlustreiche Trennung von Ectoin und Hydroxyectoin. Im
Medienüberstand von H. elongata KB10.1 konnte keine Mannosylglycerat nachgewiesen
werden. Damit handelt es sich bei KB10.1 auf den ersten Blick nicht um einen Mannosyl-
glycerat ausscheidenden Stamm. Zu Bedenken ist jedoch, dass die intrazelluläre Mannosyl-
glycerat-Konzentration verglichen mit der Ectoin-Konzentration von H. elongata Wildtyp und
KB2 geringer ausfällt und daher auch der Konzentrationsgradient über der zytoplasma-
tischen Membran weniger steil ausgeprägt ist. Resultat ist eine minimale extrazelluläre
Mannosylglycerat-Konzentration, die nicht mittels der bestehenden analytischen Methoden
nachgewiesen werden kann. Ein optimiertes Anzuchtverfahren mit hohen Zelldichten, würde
vermutlich jedoch zum Nachweis von Mannosylglycerat im Außenmedium führen. Anderer-
seits handelt es sich bei Mannosylglycerat nicht um das Solut, das von H. elongata natür-
licherweise synthetisiert wird. Möglicherweise fehlt ihm daher ein spezifischer Exportkanal
(Touzé et al., 2001) und das notwendige osmoregulierte Aufnahmesystem.
Stämme von H. elongata sind gut für die großtechnische Produktion geeignet, da sie sich bis
zu hohen Zelldichten (OD600 = 80, Sauer, 1995) anziehen lassen. Im Gegensatz dazu
erreicht R. marinus Zelldichten von maximal 8-9 und erweist sich daher für die groß-
technische Produktion von Mannosylglycerat als problematisch (Glomb Fa. Bitop, pers.
Mitteilung). Eine höhere Kulturdichte bedeutet in der Regel auch eine höhere Produkt-
ausbeute.
Sollte H. elongata KB10.1 das produzierte Mannosylglycerat nicht ins Medium abgeben, böte
seine Verwendung dennoch einige Vorteile. Im Gegensatz zu H. elongata Stämmen ist
R. marinus nicht zu einem Wachstum auf festen Medien fähig (Glomb Fa. Bitop, pers.
Mitteilung). Die Anzucht auf festen Medien erleichtert erheblich die Stammhaltung und Rein-
heitskontrolle der Kulturen. Das Bakterienmelkverfahren ist für H. elongata Stämme seit
Jahren etabliert und wurde bisher für die Ectoin-Produktion eingesetzt. Die Gewinnung von
Mannosylglycerat aus R. marinus erfolgt bei einer finalen Mediensalinität von 7 % NaCl,
während die Mediensalinität im Falle von H. elongata KB10.1 nur 4 % beträgt. Eine geringere
Salinität macht das Aufreinigungsverfahren kostengünstiger und verringert darüber hinaus
den Stress, der beim Bakterienmelken (hypoosmotischer Schock) auf die Zellen einwirkt und
zu Zelllyse und Proteinfreisetzung führen kann (Tschichholz & Trüper, 1990). Außerdem
werden mit hoher Salinität einhergehende Entsorgungsprobleme gelöst. Der Verzicht auf
hohen Salzkonzentrationen würde zusätzlich Korrosionserscheinungen am Bioreaktor ver-
mindern. Korrosion nimmt auch mit steigender Anzuchttemperatur zu. Eine Nutzung von

IV. Diskussion 174
H. elongata als Produktionsstamm würde zur Senkung der Betriebstemperatur (von 65 auf
40 °C) und somit Verminderung von Korrosionsschäden und Energieersparnis führen.
Dennoch dürfen einige wenige im Moment noch vorhandene Nachteile der Mannosyl-
glycerat-Produktion durch H. elongata KB10.1 nicht verschwiegen werden. Aufgrund des
zellulären Metabolismus ist KB10.1 bisher nur in der Lage höhere Konzentrationen von Man-
nosylglycerat unter Nutzung von Mannose als Kohlenstoffquelle zu synthetisieren. Im Ver-
gleich zu Glucose (25 g: 1,43 Euro), Saccharose (25 g: 1,9 Euro) bzw. Fructose (25 g: 1,92
Euro) kosten 25 g Mannose 22,9 Euro (Fluka) und damit fast das 12 bis 16-fache. Allerdings
sind solche Vergleiche mit Vorsicht zu genießen, weil es sich bei den Preisen um Endver-
braucherpreise handelt, die unabhängig von den Herstellungskosten sein können. Ist die
Nachfrage sehr gering, wird ein in der Herstellung günstiges Produkt zumeist sehr teuer
angeboten. Nicht zuletzt stößt die Vermarktung von Produkten, welche durch gentechnisch
veränderte Organismen gewonnen werden, durch Fehlinformation häufig auf fehlende
Akzeptanz in der Bevölkerung. Darüber hinaus ist die Vermarktung durch Genehmigungs-
verfahren und Sicherheitsbestimmungen mit einem erheblichen bürokratischen Aufwand
verbunden.
Beim Betrachten aller genannten Punkte wird deutlich, dass die Mannosylglycerat-Produkti-
on mittels H. elongata KB10.1 eine Menge Potential und Möglichkeit zur weiteren Optimie-
rung bietet. Darunter fällt z.B. die Nutzung kostengünstiger Kohlenstoffquellen und die
Etablierung eines Fermentationsverfahrens. Eine geschickte Vermarktung eines derartigen
Produkts kann durchaus zur einer allgemeinen Akzeptanz, wie sie z.B. bei rekombinant
hergestelltem Insulin vorherrscht, führen.
9. Ausblick In der vorliegenden Arbeit zeigte sich, dass der höchste Mannosylglycerat-Gehalt von
KB10.1 bei Anzucht mit Mannose akkumuliert wird. Daher ist es für die Etablierung eines
geeigneten Mannosylglycerat-Produktionsverfahrens zunächst einmal wichtig, sicherzustel-
len, dass H. elongata KB10.1 Mannosylglycerat auch unter Nutzung „kostengünstiger“
Kohlenstoffquellen in größeren Mengen synthetisiert. Denkbar wären hierbei Substrate wie
Glucose, Saccharose oder Fructose. Bei Anzucht von KB10.1 mit diesen hatten nur geringe
Mannosylglycerat-Konzentrationen nachgewiesen werden können. Offensichtlich wurde
durch KB10.1 nur ein Bruchteil dieser Substrate in die Mannosylglycerat-Synthese einge-
lenkt. Denkbar wäre es, eine Steigerung der Mannosylglycerat-Ausbeute durch das Einfügen
von Stoffwechselgenen, welche eine Nutzung von Glucose, Saccharose bzw. Fructose er-
lauben, zu erreichen. Dies ist in der vorliegenden Arbeit bereits im Zusammenhang mit der

IV. Diskussion 175
Bereitstellung von Mannosylglycerat-Synthesevorstufen gelungen (PP1776 und xanA aus
P. putida). So könnte das Einbringen einer Phosphomannose-Isomerase (z.B. PP5369,
NCBI-Acc.Nr.: AAN70934) aus P. putida KT2440 die Verwendung von Saccharose (Glucose-
α-(1,2)-Fructose) bzw. Fructose als geeignete Kohlenstoffquellen ermöglichen. Die Phospho-
mannose-Isomerase katalysiert die Umwandlung von Fructose-6-phosphat in Mannose-6-
phosphat, wodurch eine Steigerung des zellulären Mannose-6-phosphat-Gehalts erzielt
werden könnte. Mannose-6-phosphat dient als Vorstufe für die Mannosylglycerat-Synthese.
Darüber hinaus könnte durch die Einbringung einer Glycerat-Kinase (z.B. PP3178, NCBI-
Acc.Nr.: AAN68786), welche 3-Phosphoglycerat unter Abspaltung von ATP in D-Glycerat
umsetzt, der zelluläre Gehalt von D-Glycerat erhöht werden und der Metabolismus mög-
licherweise in Richtung des Mannosylglycerats gezwungen werden. Als geeigneter Inserti-
onsort für diese Fremdgene könnte der locus des ectR-Gens im Genom von H. elongata
KB10.1 dienen. Die Deletion dieses Gens beim H. elongata Wildtyp führte nicht zu einem
eindeutigen Phänotyp, der auf eine allgemeine Beeinflussung des Stoffwechsels hindeuten
ließ (Schilz, 2005).
Analysen des Mannosylglycerat-Gehalts hatten gezeigt, dass dieser in der spät-stationären
Phase dramatisch absinkt. Hinter diesem Phänomen wird u.a. eine abnehmende Expression
der Synthesegene vermutet. Eine Optimierung der Expression durch Veränderung der
Promotorstrukturen liegt daher nahe. Die Optimierung könnte dadurch erreicht werden, dass
in H. elongata KB10.1 die Stärke der identifizierten σ70-abhängigen bzw. gearbox-Promo-
toren stromaufwärts von mgs erhöht wird. Ein starker Promotor bedeutet eine höhere
Transkriptionsrate und dadurch vermutlich auch einen höheren Level der Genprodukte.
Möglich wäre dies durch Mutagenese der –35-Region des σ70-abhängigen Promotors, der in
nur einer Base von der Consensus-Sequenz entsprechender E. coli Promotoren abweicht.
Basierend auf Untersuchungen von Miller (1992), die sich mit der lac-Promotorregion von
E. coli beschäftigen, wird die Promotoraktivität vor allem durch Sequenzabweichungen in der
–35-Region stark verringert. Die Herstellung der Consensus-Sequenz sollte demnach zu
einer Verstärkung der Expression der integrierten Fremdgene führen. Entsprechende Verän-
derungen des gearbox-Promotors könnten die Expression der Gene in der spät-stationären
Phase verstärken.
Außerdem wäre eine Erhöhung der Mannosylglycerat-Syntheserate auch durch die Eta-
blierung eines alternativen Mannosylglycerat-Synthesewegs in H. elongata denkbar. So kön-
nten anstelle des mgs-Gens (kodiert für die Mannosylglycerat-Synthase, Mgs) auch zwei
andere R. marinus Gene (mpgS und mpgP, NCBI-Acc.Nr.: AY271294), welche für die
Enzyme eines alternativen Mannosylglycerat-Synthesewegs kodieren (Abschnitt I, 3.3.2,
Abb. 2), genutzt werden. Die Mannosylphosphoglycerat-Synthase (mpgS) katalysiert die
Umwandlung von GDP-Mannose und 3-Phosphoglycerat in Mannosylphosphoglycerat,

IV. Diskussion 176
welches dann durch Einwirkung der Mannosyl-3-phosphoglycerat-Phosphatase (mpgP) in
Mannosylglycerat umgesetzt wird (Martins et al., 1999). Bogers et al. (2004) hatten fest-
gestellt, dass die enzymatische Aktivität der Mpgs im Vergleich zur Mgs vor allem Salz
abhängig ist, wobei das Temperaturoptimum ähnlich wie bei der Mgs zwischen 70 und 80 °C
liegt. Western-Analysen zeigten außerdem, dass sich bei einem osmotischen Schock von 2
auf 6 % NaCl der zelluläre Gehalt an Mpgs-Protein verdreifacht. Die Salzabhängigkeit der
enzymatischen Aktivität könnte sich bei einer Mediensalinität von 4 % NaCl positiv auf die
Mannosylglycerat-Synthese in H. elongata auswirken. Vielversprechender erscheint jedoch
die Nutzung des mgsD-Gens (NCBI-Acc.Nr.: CS075488) aus dem mesophilen, halotoler-
anten, Gram-negativen Bakterium Dehalococcoides ethenogenes (Abschnitt I, 3.3.1). Das
Produkt dieses Gens vereinigt die enzymatischen Aktivitäten der Mannosylphosphoglycerat-
Synthase sowie der Mannosyl-3-phosphoglycerat-Phosphatase und weist ein Temperatur-
optimum von 50 °C auf, ist jedoch zwischen 10 und 60 °C enzymatisch aktiv. Empadinhas et
al. (2004) hatten mgsD erfolgreich in E. coli und Saccharomyces cerevisiae überexprimiert
und in der Hefe auch Mannosylglycerat nachweisen können. Eine erfolgreiche Expression
des mgsD-Gens wäre somit auch in H. elongata denkbar. Der mesophile Charakter des
Enzyms sollte auch hier zur Produktion von Mannosylglycerat führen.
Um eine optimale Mannosylglycerat-Ausbeute durch H. elongata KB10.1 zu erreichen, ist die
Kultivierung in einem Fermenter unentbehrlich. Dabei müssen die Anzuchtbedingungen nach
den bisherigen Wachstumscharakteristika von KB10.1 so gewählt werden, dass die Kultur
möglichst lange in der exponentiellen Wachstumsphase verweilt, da sich die Synthese von
Mannosylglycerat durch H. elongata KB10.1 nach den vorliegenden Analysen vermutlich auf
diese Phase beschränkt. Dies wäre mit Hilfe einer gezielten Fütterungsstrategien in einer
sog. Fed-Batch-Fermentation möglich. Andererseits könnte man auch durch hohe Zell-
dichten (z.B. in einer Hochzelldichte-Fermentation) eine hohe Mannosylglycerat-Ausbeute
erreichen. So konnte Sauer (1995) mit H. elongata Wildtyp durch dieses Verfahren eine
maximale Zelltrockenmasse von 48 g/l erreichen und somit auch die auf das Kulturvolumen
bezogene Ectoin-Ausbeute erhöhen. Eine alternative Strategie wäre es, die Kultur in einer
Fed-Batch-Fermentation sehr langsam unter kontinuierlicher Zugabe eines limitierenden
Nährstofffaktors (z.B. Stickstoff) bis zu einer hohen Kulturdichte wachsen zu lassen, ohne
dass die Zellen in die stationäre Phase eintreten. Durch die Limitierung sollten die Zellen ein
nur sehr reduziertes Wachstum durchführen können, so dass die angebotene Kohlenstoff-
quelle möglichst vollständig in Mannosylglycerat umgesetzt wird und nur einen Bruchteil in
Biomasse umgewandelt wird (Biokonversion).

V. Zusammenfassung Bislang können die kompatiblen Solute Mannosylglycerat bzw. Mannosylglyceramid durch die Fa. Bitop AG nur in geringen Mengen (bis 1,85 g/l Fermentationsmedium) über das Bakterienmelkverfahren aus Rhodothermus marinus gewonnen werden. Ziel der vorliegen-den Arbeit war die Entwicklung eines bakteriellen Produktionsstamms, der eine Gewinnung von Mannosylglycerat in größeren Mengen ermöglicht. Hierzu sollte nach dem Vorbild von H. elongata KB2 (Volke, 2001), einem Produktionsstamm für das kompatible Solute Ectoin, ein transportdefekter Mannosylglycerat überproduzierender Stamm von R. marinus herge-stellt werden, der aufgrund einer gestörten Regulation der Mannosylglycerat-Synthese Man-nosylglycerat ins Medium abgibt. Zur Identifizierung des Mannosylglycerat-Transporters wurden unterschiedliche molekularbiologische Methoden, die funktionelle Komplementierung solutetransportdefizienter E. coli Stämme, die heterologe PCR und die DDRT-PCR ange-wendet. Zunächst wurde die Möglichkeit einer funktionellen Expression von R. marinus Genen mittels
des Thiamin-auxotrophen Stamms E. coli DH5α überprüft. So konnten vier Genorte (thiSGED) von R. marinus identifiziert werden, die laut Sequenzanalysen an der Thiamin-Synthese beteiligt sind. Eine funktionelle Komplementierung des in der Aufnahme kompa-tibler Solute defekten E. coli Stamms MKH13 wurde nicht erreicht. Das Fehlschlagen dieser Versuche wurde auf die endogene Trehalose-Synthese von E. coli MKH13 zurückgeführt. Daher wurde der Trehalose-Synthese defiziente, auf E. coli MKH13 basierende Stamm E. coli BKA-13 hergestellt. Auch mittels dieses Stamms konnte kein Mannosylglycerat-Trans-porter identifiziert werden. Gleichermaßen konnte durch den Einsatz degenerierter Primer kein Mannosylglycerat-Transporter in R. marinus identifiziert werden. Stattdessen wurde ein hypothetischer, verkürzter Natrium/Glucose-Symporter gefunden. Ein Hydropathie-Plot seiner Aminosäure-sequenz zeigte das Vorhandensein von nur sieben hydrophoben Sequenzbereichen an. Zur Durchführung der Differential Display RT-PCR auf R. marinus RNA wurde zunächst eine Strategie für die cDNA-Synthese etabliert. Als Ansatzpunkt hierfür wurden die 3 unterschied-lichen Stopkodons ausgewählt. Für jedes dieser Stopkodons wurde ein eigener cDNA-Synthese Primer entworfen. Die Gesamt-RNA aus R. marinus Kulturen, welche bei 2 bzw. 6 % NaCl angezogen worden waren, wurde in cDNA umgeschrieben. Anschließend wurde eine DDRT-PCR durchgeführt, die ein differentielles Bandenmuster hervorbrachte. Die Analyse der differentiell exprimierten DNA-Banden zeigte hohe Identitäten mit der 16S rDNA verschiedener R. marinus und verwandter Stämme. Keine der angewandten molekularbiologischen Methoden führte zur Identifizierung eines Mannosylglycerat-Transporters in R. marinus. Als alternative Strategie zur Etablierung eines bakteriellen Produktionsstamms für das kompatible Solut Mannosylglycerat wurde daher das Einbringen der Synthesegene in H. elongata erdacht. Hier wurden die Gene für die Mannosylglycerat-Synthese unter Kontrolle der Promotoren der osmoregulierten Ectoin-

V. Zusammenfassung 178
Gene gestellt. So konnte eine salzabhängige Expression erfolgen, gleichzeitig wurde die Ectoin-Synthese ausgeschaltet. Das ectA-Gen (Diaminobuttersäure-Acetyltransferase) aus H. elongata wurde „in-frame“ gegen das mgs-Gen (Mannosylglycerat-Synthase) aus R. marinus ausgetauscht. Der resul-
tierende Stamm H. elongata KB10 weist im Vergleich zu KB1 (∆ectA) eine nur geringfügig höhere Wachstumsrate auf. Mannosylglycerat konnte in KB10 mittels Dünnschichtchromato-graphie und HPLC-Analysen nicht nachgewiesen werden. Northern-Analysen und RT-PCR wiesen jedoch verschiedene mgs-Transkripte nach. In Promotoranalysen mittels RACE-PCR wurde in KB10 außerdem ein Transkriptionsstartpunkt 25 bp stromaufwärts des inserierten
mgs-Gens identifiziert. Dieser konnte einem σ70-abhängigen Promotor bzw. einem über-lappenden gearbox-Element zugeordnet werden. Um einen Grund für das Ausbleiben der Mannosylglycerat-Synthese zu finden, wurde die Expression der Mannosylglycerat-Synthase in H. elongata analysiert. Dabei wurde die lösliche Expression des Fusionsproteins aus Mannosylglycerat-Synthase (Mgs) und His6-tag im Zytoplasma von H. elongata Wildtyp (10 % NaCl) und KB1 (4 % NaCl) durch Western-Analysen bestätigt. Dies deutete darauf hin, dass das Fehlen von Mannosylglycerat in KB10 nicht auf eine fehlerhafte Expression zurück-zuführen war. Vermutet wurde, dass Mgs aufgrund eines zu hohen Temperaturoptimums in H. elongata nicht aktiv ist oder dass die benötigten Vorstufen der Mannosylglycerat-Synthese in H. elongata KB10 nicht in ausreichender Menge synthetisiert werden. Zur Bereitstellung der Mannosylglycerat-Synthesevorstufen in H. elongata wurden die Gene PP1776 (Mannose-1-phosphat-Guanylyltransferase) und xanA (Phosphomanno-Mutase) aus Pseudomonas putida KT2440 in das Genom von H. elongata KB10 integriert. Die Ectoin-Synthesegene ectBC von H. elongata KB10 wurden mittels homologer Rekombination gegen die beiden P. putida Gene ausgetauscht. In dem resultierenden Stamm H. elongata KB10.1 wurden in RT-PCR und Northern-Analysen Transkripte der drei Fremdgene mgs, PP1776 und xanA, darunter ein Gesamttranskript, identifiziert. Die Wachstumsraten von KB10.1 sind bei fast allen eingesetzten C-Quellen dreimal so hoch wie die von H. elongata KB1. Den ersten Hinweis auf eine Synthese von Mannosylglycerat in KB10.1 ergab die Analyse von Zellextrakten in einer Dünnschichtchromatographie. Durch HLPC-Analysen wurde die Ab-hängigkeit der Mannosylglycerat-Synthese von der verwendeten Kohlenstoffquelle und der Anzuchttemperatur festgestellt. Mit Mannose als Kohlenstoffquelle wurde in der früh-stati-onären Wachstumsphase mit ca. 27,16 mg/g TG die höchste nachgewiesene Menge von Mannosylglycerat synthetisiert. In der vorliegenden Arbeit konnte gezeigt werden, dass grundsätzlich eine Produktion von Mannosylglycerat mittels H. elongata möglich ist. Diese ersten Erfolge konnten mit einem für industrielle Prozesse relativ geringen Aufwand realisiert werden. Die weitere Optimierung zur Etablierung eines industriellen Mannosylglycerat-Produktionsverfahrens erscheint daher sowohl zeitlich als auch ökonomisch lohnend.

VI. Literatur Abu Kwaik, Y and Pederson, L L (1996) The use of differential display-PCR to isolate and characterize a Legionella pneumophila locus induced during the intracellular infection of macrophages. Mol Microbiol 21: 543-556.
Albers, S V; Van De Vossesberg, J L C M; Driessen, A J and Konings, W N (2001) Bioenergetics and solute uptake under extreme conditions. Extremophiles 5: 285-294.
Aldea, M; Garrido, T; Pla, J and Vicente, M (1990) Division genes in Eschrichia coli are expressed coordinately to cell septum requierments by gearbox promoters. EMBO J 8: 3923-3931.
Alfredsson, G A; Baldursson, S and Kristjansson, J K (1985) Nutritional diversity among Thermus spp. isolated from Icelandic hot springs. Syst Appl Microbiology 6: 308-311.
Alfredsson, G A; Kristjansson, J K ; Hjörleifsdottir, S and Stetter, K O (1988) Rhodothermus marinus, gen. nov., sp. nov., a thermophilic, halophilic bacterium from submarine hot springs in Iceland. J Gen Microbiol 134: 299-306.
Alia, H H; Sakamoto, A and Murata, N (1998) Enhancement of the tolerance of Arabidopsis to high temperatures by genetic engineering of the synthesis of glycinebetaine. Plant J 16: 155-161.
Altschul, S; Gish, W; Miller, W; Myers, E and Lipman, D (1990) Basic local alignment search tool. J Mol Biol 215: 403-410.
Amara, R and Vijaya, S (1997) Specific polyadenylation and purification of total messenger RNA from Escherichia coli. Nucleic Acids Res 25: 3465-3470.
Andresson, O S and Fridjonsson, O H (1994) The sequence of the single 16S rRNA gene of the thermophilic eubacterium Rhodothermus marinus reveals a distant relationship to the group containing Flexibacter, Bacteroides, and Cytophaga species. J Bacteriol 176: 6165-6169.
Andrianopoulos, K; Wang, L and Reeves, P R (1998) Identification of the Fucose Synthetase Gene in the Colanic Acid Gene Cluster of Escherichia coli K-12. J Bacteriol 180: 998-1001.
Antoine, R and Locht, C (1992) Isolation and molecular characterization of a novel broad-host-range plasmid from Bordetella bronchiseptica with sequence similarities to plasmids from gram-positive organisms. Mol Microbiol 6: 1785-1799.
Anton, J; Oren, A; Benlloch, S; Rodriguez-Valera, F; Amann, R and Rossello-Mora, R (2002) Salinibacter ruber gen. nov., sp. nov., a novel, extremely halophilic member of the Bacteria from saltern crystallizer ponds. Int J Syst Evol Microbiol 52: 485-491.
Arakawa, T and Timasheff, S N (1985) The stabilization of proteins by osmolytes. Biochemistry 47: 411-414.
Bagdasarian, M M; Lurz, A; Rückert, R B and Bagdasarian, M (1983) Activity of the hybrid trp-lac(tac) promoter of Escherichia coli in Pseudomonas putida. Construction of broad-host-range, controlled-expression vectors. Gene 26: 273-282.
Bairoch, A; Bucher, P and Hofmann, K (1997) The PROSITE database, its status in 1997. Nucleic Acids Res 25: 217-221.
Baldwin, W W; Sheu, M J T; Bankston, P W and Woldringh, C L (1988) Changes in buoyant density and cell size of Escherichia coli in response to osmotic shocks. J Bacteriol 170: 452-455.
Ballesteros, M; Kusano, S; Ishihama, A and Vicente, M (1998) The ftsQ1p gearbox promotor of Escherichia coli is a major σ S-dependent promotor in the ddIB-ftsA region. Mol Microbiol 30: 419-430.
Begley, T P; Downs, D M; Ealick, S E; Mclafferty, F W; Van Loon, A P G M; Taylor, S, et al. (1999) Thiamin biosynthesis in prokaryotes. Archives of Microbiology 171: 293-300.
Bendtsen, J D; Nielsen, H; Von Heijne, G and Brunak, S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783-795.

VI. Literatur 180
Birnboim, H C and Doly, J (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7: 1513-1523.
Bjornsdottir, S H; Thorbjarnardottir, S H and Eggertsson, G (2005) Establishment of a gene transfer system for Rhodothermus marinus. Appl Microbiol Biotechnol 66: 675-682.
Bligh, E G and Dyer, W J (1959) A rapid method of lipid extraction and purification. Canadian Journal of Biochemistry and Physiology 37: 911-917.
Blöchl, E; Burggraf, S; Fiala, G; Lauerer, G; Huber, G and Huber, R (1995) Isolation, taxonomy and phylogeny of hyperthermophilic microorganisms. World J Microbiol Biotechnol 11: 9-16.
Boos, W and Lucht, J M (1996) Periplasmic binding protein-dependent ABC-Transporters. In Escherichia coli and Salmonella. Neidhardt, F.C. (ed). Washington, DC: ASM press, pp. 1175-1290.
Borges, N; Marugg, J D; Empadinhas, N; Da Costa, M S and Santos, H (2004) Specialized Roles of the Two Pathways for the Synthesis of Mannosylglycerate in Osmoadaptation and Thermoadaptation of Rhodothermus marinus. J Biol Chem 279: 9892-9898.
Bouveng, H; Lindberg, B and Wickberg, B (1955) Lowmolecular carbohydrates in algae. Structure of the glyceric acid mannoside from red algae. Acta Chem Scand 9: 807-809.
Brown, A (1976) Microbial water stress. Microbiol Mol Biol Rev 40: 803-846.
Brünig, A (2005) Molekulargenetische und physiologische Studien zur Entwicklung eines Expressionssystems in Halomonas elongata DSMZ 2581T. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Brzostowicz, P C; Gibson, K L; Thomas, S M; Blasko, M S and Rouviere, P (2000) Simultaneous Identification of Two Cyclohexanone Oxidation Genes from an Environmental Brevibacterium Isolate using mRNA Differential Display. J Bacteriol 182: 4241-4248.
Bünger, J (1999) Ectoine added protection and care for the skin. Eurocosm 7: 22-24.
Bünger, J (2000) Neue Rohstoffe. Mehr Schutz für die Haut mit einem neuen Wirkstoff für die Kosmetikindustrie. SÖFW-Journal 126: 90.
Bünger, J and Driller, H (2004) Ectoin: An effective natural substance to prevent UVA-induced premature photoaging. Skin Pharmacol Physio 17: 232-237.
Burdziak, D (2004) Untersuchungen zur Bedeutung des Gens yhgI für die Stressanpassung von Halomonas elongata DSM 2581T und Escherichia coli K-12. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Cayol, J L; Ollivier, B; Patel, B K; Prensier, G; Guezennec, J and Garcia, J L (1994) Isolation and characterization of Halothermothrix orenii gen. nov., sp. nov., a halophilic, thermophilic, fermentative, strictly anaerobic bacterium. Int J Syst Bacteriol 44: 534-540.
Cayol, J L; Ducerf, S; Patel, B K C; Garcia, J L; Thomas, P and Ollivier, B (2000) Thermohalobacter berrensis gen. nov., sp. nov., a thermophilic, strictly halophilic bacterium from a solar saltern. Int J Syst Evol Microbiol 50: 559-564.
Chatterjee, A and Vidaver, A (1985) Genetics of pathogenicity factors: application to phytopathogenic bacteria. In Advances in plant pathology. Williams, I.D.S.I.a.P.H. (ed). London: Academic press, Inc (London), pp. 1-224.
Chrunyk, B A; Evans, J; Lillquist, J; Young, P and Wetzel, R (1993) Inclusion body formation and protein stability in sequence variants of interleukin-1 beta. J Biol Chem 268: 18053-18061.
Clegg, J S; Seitz, P; Seitz, W and Hazlewood, C F (1982) Cellular response to extreme water loss: The water replacement hypothesis. Cryobiol 19: 306-316.
Cohen, S; Chang, A and Hsu, L (1972) Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. PNAS 69: 2110-2114.
Crowe , H J; Tablin, F; Wolkers, W F; Gousset, K; Tsvetkova, N M and Ricker, J (2003) Stabilization of membranes in human platelets freeze-dried with trehalose. Chem Phys Lipids 122: 41-52.

VI. Literatur 181
Crowe, L M and Crowe, J H (1992) Stabilization of dry liposomes by carbohydrates. Dev Biol Stand 74: 285-294.
Cserzo, M; Wallin, E; Simon, I; Von Heijene, G and Elofsson, A (1997) Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Engineering 10: 673-676.
Csonka, L N (1989) Physiological and genetic responses of bacteria to osmotic stress. Microbiol Mol Biol Rev 53: 121-147.
Da Costa, M S; Santos, H and Galinski, E A (1998) An overview of the role and diversity of compatible solutes in Bacteria and Archaea. Adv Biochem Eng Biotechnol 61: 117-153.
Da Costa, M S; Nobre, M F and Rainey, F A (2001) The genus Thermus. In In Bergey 's Manual of Systematic Bacteriology. Boone, D.R., and Castenholtz, R W (ed). New York: Springer, pp. 404-414.
Da Costa, M S; Empadinhas, N; Da Costa, A P and Santos, H (2005) Bifunctional gene for mannosylglycerate synthesis.EP 01526180 A1
Daniel, R M; Dines, M and Petach, H H (1996) The denaturation and degradation of stable enzymes at high temperatures. Biochem J 317: 1-11.
Daniel, R M and Cowan, D A (2000) Biomolecular stability and life at high temperatures. Cell Mol Life Sci 57: 250-264.
De Boer, H A; Comstock, L J and Vasser, M (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci USA 80: 21-25.
De La Tour, C B; Portemer, C; Nadal, M; Stetter, K O; Forterre, P and Duguet, M (1990) Reverse gyrase, a hallmark of the hyperthermophilic archaebacteria. J Bacteriol 172: 6803-6808.
De Rosa, M; Gambacorta, A and Gliozzi, A (1986) Structure, biosynthesis and physical properties of archaebacterial lipids. Microbiol Rev 50: 70-80.
Degryse, E; Glansdorff, N and Pierard, A (1978) A comparative analysis of extreme thermophilic bacteria belonging to the genus Thermus. Arch Microbiol 117: 189-196.
Del Tito Jr, B J; Ward, J M; Hodgson, J; Gershater, C J; Edwards, H; Wysocki, L A, et al. (1995) Effects of a minor isoleucyl tRNA on heterologous protein translation in Escherichia coli. J Bacteriol 177: 7086-7091.
Dombroski, A (1997) Recognition of the -10 Promoter sequence by a partial polypeptide of σ 70 in vitro. J Biol Chem 272: 3487-3494.
Edmonds, C G; Crain, P F; Gupta, R; Hashizume, T; Hocart, C H; Kowalak, J A , et al. (1991) Posttranscriptional modification of tRNA in thermophilic archaea (archaebacteria). J Bacteriol 173: 3138-3148.
Egler, C (2001) Nachweis und Quantifizierung von Transkripten des Ectoin-Genclusters in Halomonas elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Münster: Westfälische Wilhelms-Universität, Münster.
Empadinhas, N; Marugg, J D; Borges, N; Santos, H and Da Costa, M S (2001) Pathway for the Synthesis of Mannosylglycerate in the Hyperthermophilic Archaeon Pyrococcus horikoshii. BIOCHEMICAL AND GENETIC CHARACTERIZATION OF KEY ENZYMES. J Biol Chem 276: 43580-43588.
Empadinhas, N; Albuquerque, L; Henne, A; Santos, H and Da Costa, M S (2003) The Bacterium Thermus thermophilus, Like Hyperthermophilic Archaea, Uses a Two-Step Pathway for the Synthesis of Mannosylglycerate. Appl Envir Microbiol 69: 3272-3279.
Empadinhas, N; Albuquerque, L; Costa, J; Zinder, S H; Santos, M ; Santos, H and Da Costa, M S (2004) A Gene from the Mesophilic Bacterium Dehalococcoides ethenogenes Encodes a Novel Mannosylglycerate Synthase. J Bacteriol 186: 4075-4084.
Ernstsson, S; Bjornsdottir, S H; Jonsson, Z O; Thorbjarnardottir, S H; Eggertsson, G and Palsdottir, A (2003) Identification and nucleotide sequence analysis of a cryptic plasmid, pRM21, from Rhodothermus marinus. Plasmid 49: 188-191.
Falquet, L; Pagni, M; Bucher, P; Hulo, N; Sigrist, C; Hofmann, K and Bairoch, A (2002) The PROSITE database, its status in 2002. Nucleic Acids Res 30: 235-238.

VI. Literatur 182
Farewell, A; Kvint, K and Nyström, T (1998) uspB, a new σ S -regulated gene in Escherichia coli which is required for stationary-phase resistance to ethanol. J Bacteriol 180: 6140-6147.
Faria, T Q; Knapp, S; Ladenstein, R; Macanita, A and Santos, H (2003) Protein stabilisation by compatible solutes: effect of mannosylglycerate on unfolding thermodynamics and activity of ribonuclease A. Chembiochem 4: 734-741.
Faria, T Q; Lima, J C; Bastos, M; Macanita, A and Santos, H (2004) Protein Stabilization by Osmolytes from Hyperthermophiles: EFFECT OF MANNOSYLGLYCERATE ON THE THERMAL UNFOLDING OF RECOMBINANT NUCLEASE A FROM STAPHYLOCOCCUS AUREUS STUDIED BY PICOSECOND TIME-RESOLVED FLUORESCENCE AND CALORIMETRY. J Biol Chem 279: 48680-48691.
Fislage, R; Berceanu, M; Humboldt, Y; Wendt, M and Oberender, H (1997) Primer design for a prokaryotic differential display RT-PCR. Nucleic Acids Res 25: 1830-1835.
Fleming, J T; Yao, W H and Sayler, G (1998) Optimization of Differential Display of Prokaryotic mRNA: Application to Prue Culture and Soil Microcosms. Appl Environ Microbiol 64: 3698-3706.
Flowers, T J (2004) Improving crop salt tolerance. Journal of Experimental Botany 55: 307-319.
Forterre, P (1998) Were our ancestors actually hyperthermophiles ? Viewpoint of a devil's advocate. In In: Thermophiles: The Key to Molecular Evolution and the Origins of Life. W, W.J.a.A.M.W. (ed). London: Taylor and Francis., pp. 137-146.
Frick, F (2005) Der Natur auf der Spur. Spektrum der Wissenschaft 02/05: 86-88.
Friedman, S M (1986) Properties of the translational machinery from a sulfur-dependent archaebacterium. System Appl Microbiol 7: 325-329.
Frings, E; Sauer, T and Galinski, E A (1995) Production of hydroxyectoine: High cell-density cultivation and osmotic downshock of Marinococcus strain M52. J Biotech 43: 53-61.
Fuhrmann, M ; Hausherr, A ; Ferbitz, L; Schödl, T; Heitzer, M and Hegemann, P (2004) Monitoring dynamic expression of nuclear genes in Chlamydomonas reinhardtii by using a synthetic luciferase reporter gene. Plant Molecular Biology 55: 869-881.
Fürste, J P; Pansegrau, W; Frank, R; Blöcker, H; Scholz, P; Bagdasarian, M and Lanka, E (1986) Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48: 119-131.
Galinski, E A and Herzog, R M (1990) The role of trehalose as a substitute for nitrogen containig compatible solutes (Ectothiorhodospira halochloris). Arch Microbiol 153: 607-613.
Galinski, E A and Trüper, H G (1994) Microbial behaviour in salt-stressed ecosystems. FEMS Microbiol Rev 15: 95-108.
Galinski, E A; Trüper, H G and Sauer, T (1994) Verfahren zur in vivo Gewinnung von Inhaltsstoffen aus Zellen. DE 04244580 A1
Galinski, E A (1995a) Osmoadaptation in bacteria. Adv Microb Physiol 37: 272-328.
Galinski, E A (1995b) Halophile und halotolerante Eubakterien. In Mikroorganismen in ausgefallenen Lebensräumen. Hausmann, K., and Kremer, B. (eds): VCH Weinheim.
Galinski, E A; Kaufmann, M; Schwarz, T and Vangala, M D (2001) Use of compatible solutes as inhibitors of the enzymatic decomposition of macromolecular biopolymers. WO 01058446 A1
Garcia-Estepa, R; Argandona, M; Reina-Bueno, M; Capote, N; Iglesias-Guerra, F; Nieto, J J and Vargas, C (2006) The ectD Gene, Which Is Involved in the Synthesis of the Compatible Solute Hydroxyectoine, Is Essential for Thermoprotection of the Halophilic Bacterium Chromohalobacter salexigens. J Bacteriol 188: 3774-3784.
Gardy, J L; Spencer, C; Wang, K; Ester, M; Tusnady, G E; Simon, I , et al. (2003) PSORT-B: improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res 31: 3613-3617.
Garrity, G M; Bell, J A and Lilburn, T G (2004) Taxonomic outline of the prokaryotes release 5.0 May 2004. In In: Bergey's manual of systematic bacteriology. New York: Springer-Verlag.

VI. Literatur 183
Gasteiger, E ; Hoogland, C; Gattiker, A ; Duvaud, S ; Wilkins, M R; Appel, R D and Bairoch, A (2005) Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook. Walker, J.M. (ed): Humana Press, pp. 571-607.
Giaever, H M; Styrvold, O B; Kaasen, I and Strøm, A (1988) Biochemical and genetic characterization of osmoregulatory trehalose synthesis in Escherichia coli. J Bacteriol 170: 2841-2849.
Göller, K; Ofer, A and Galinski, E A (1998) Construction and characterization of an NaCl-sensitive mutant of Halomonas elongata impaired in ectoine biosynthesis. FEMS Microbiol Lett 161: 293-300.
Göller, K (1999) Identifizierung und Charakterisierung des Ectoin-Genclusters in Halomonas elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Gomi, M; Akazawa, F and Shigeki, M (2000) SOSUIsignal: Software System for Prediction of Signal Peptide and Membrane Protein. Genome Informatics 11: 414-415.
Gouesbet, G; Jebbar, M; Talibart, R; Bernard, T and Blanco, C (1994) Pipecolic acid is an osmoprotectant for Escherichia coli taken up by the general osmoporters ProU und ProP. Microbiology 140: 2415-2422.
Grammann, K (2000) Identifizierung und Charakterisierung eines osmoregulierten Solutetransporters in einer ∆ectA Mutante von H. elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Grammann, K; Volke, A and Kunte, H J (2002) New Type of Osmoregulated Solute Transporter Identified in Halophilic Members of the Bacteria Domain: TRAP Transporter TeaABC Mediates Uptake of Ectoine and Hydroxyectoine in Halomonas elongata DSM 2581T. J Bacteriol 184: 3078-3085.
Grammann, K (2004) Molekularbiologische Charakterisierung des osmoregulierten TRAP-Transportsystem TeaABC und seines potentiellen Regulatorproteins TeaD aus Halomonas elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Grammel, N (1999) Molekulargenetische und biochemische Analyse der Biosynthese von 2-Methyl-4-carboxy-3,4,5,6-tetrahydropyrimidin und seinem 5-Hydroxyderivat, zwei salzstreßinduzierbaren Osmolyten, in Streptomyces chrysomallus. In. Berlin: Technischen Universität Berlin.
Grogan, D W (1998) Hyperthermophiles and the problem of DNA stability. Mol Microbiol 28: 1043-1049.
Grutter, M G; Hawkes, R B and Matthews, B W (1979) Molecular basis of thermostability in the lysozyme from bacteriophage T4. Nature 277: 667-669.
Haardt, M; Kempf, B; Faatz, E and Bremer, E (1995) The osmoprotectant proline betaine is a major substrate for the binding-protein-dependent transport system ProU of Escherichia coli K-12. Mol Gen Genet 246: 783-786.
Hagemann, M and Erdmann, N (1994) Activation and pathway of glucosylglycerol synthesis in the cyanobacterium Synechocystis sp. PCC 6803. Microbiol 140: 1427-1431.
Hagemann, M; Richter, S and Mikkat, S (1997) The ggtA gene encodes a subunit of the transport system for the osmoprotective compound glucosylglycerol in Synechocystis sp. strain PCC 6803. J Bacteriol 179: 714-720.
Hamilton, C M; Aldea, M; Washburn, B K; Babitzke, P and Kushner, S R (1989) New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 171: 4617-4622.
Hanahan, D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166: 557-580.
Hart, R A; Rinas, U and Bailey, J E (1990) Protein composition of Vitreoscilla hemoglobin inclusion bodies produced in Escherichia coli. J Biol Chem 265: 12728-12733.
Hartely, D and Kane, J (1988) Properties of inclusion bodies from recombinat Escherichia coli. Biochem Soc Trans 16: 101-102.
Henke, W; Herdel, K; Jung, K; Schnorr, D and Loening, S A (1997) Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Res 25: 3957-3958.
Horlacher, R; Xavier, K B; Santos, H; Diruggiero, J; Kossmann, M and Boos, W (1998) Archaeal Binding Protein-Dependent ABC Transporter: Molecular and Biochemical Analysis of the Trehalose/Maltose Transport System of the Hyperthermophilic Archaeon Thermococcus litoralis. J Bacteriol 180: 680-689.

VI. Literatur 184
Horton, R; Hunt, H; Ho, S; Pullen, J and Pease, L (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61-68.
Hülsmann, A; Lurz, R; Scheffel, F and Schneider, E (2000) Maltose and Maltodextrin Transport in the Thermoacidophilic Gram-Positive Bacterium Alicyclobacillus acidocaldarius Is Mediated by a High-Affinity Transport System That Includes a Maltose Binding Protein Tolerant to Low pH. J Bacteriol 182: 6292-6301.
Ikemura, T (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol 146: 1-21.
Imhoff, J F and Trüper, H G (1977) Ectothiorhodospira halochloris sp. nov., a new extremely halophilic phototrophic bacterium containing bacteriochlorophyll b. Arch Microbiol 114: 115-121.
Inoue, H; Nojima, H and Okayama, H (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96: 23-28.
Jakoby, M; Ngouoto-Nkili, C E and Burkovski, A (1999) Construction and application of new Corynebacterium glutamicum vectors. Biotechnology Techniques 13: 437–441.
Joshi, a K; Baichwal, V and Ames, G F-I (1991) Rapid polymerase chain reaction amplification using intact bacterial cells. Biotechniques 10: 42-45.
Jung, H (2001) Towards the molecular mechanism of Na+/ solute symport in prokaryotes. Biochim Biophys Acta 1505 (1): 131-143.
Jung, H; Rübenhagen, R; Tebbe, S; Leifker, K; Tholema, N; Quick, M and Schmid, R (2002) Topology of Na+/ Proline Transporter of Escherichia coli. J Biol Chem 273: 26400-26407.
Kaasen, I; Falkenberg, P; Styrvold, O B and Strøm, A (1992) Molecular cloning and physical mapping of the otsBA genes, which encode the osmoregulatory trehalose pathway of Escherichia coli: evidence that transcription is activated by katF (AppR). J Bacteriol 174: 889-898.
Kanapathipillai, M; Lentzen, G; Sierks, M and Park, C B (2005) Ectoine and hydroxyectoine inhibit aggregation and neurotoxicity of Alzheimer's beta-amyloid. FEBS Lett 579: 4775-4780.
Kanehisa, M and Goto, S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28: 27-30.
Kanehisa, M; Goto, S; Hattori, M; Aoki-Kinoshita, K F; Itoh, M; Kawashima, S, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34: D354-357.
Kappes, R M; Kempf, B and Bremer, E (1996) Three transport systems for the osmoprotectant glycine betaine operate in Bacillus subtilis: characterization of OpuD. J Bacteriol 178: 5071-5079.
Karla, K (1997) Untersuchungen zur Trockenstabilität von Vibrio fischeri kompatible Solute. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Kaye, J Z and Baross, J A (2004) Synchronous Effects of Temperature, Hydrostatic Pressure, and Salinity on Growth, Phospholipid Profiles, and Protein Patterns of Four Halomonas Species Isolated from Deep-Sea Hydrothermal-Vent and Sea Surface Environments. Appl Envir Microbiol 70: 6220-6229.
Kelly, D J and Thomas, G H (2001) The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol Rev 25: 405-424.
Kempf, B and Bremer, E (1998) Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch Microbiol 170: 319-330.
Kengan, S W M; De Bok, F A M; Van Loo, N D; Dijkema, C; Stams, A J M and De Vos, W M (1994) Evidence for the operation of a novel Emden-Meyerhof pathway that involves ADP-dependent kinases during sugar fermentation by Pyrococcus furiosus. J Biol Chem 269: 17537-17541.
Kholodenko, B M; Sakamoto, N; Puigjaner, J; Westerhoff, H V and Cascante, M (1996) Strong control on the transit time in metabolic channelling. FEBS Lett 389: 123-125.

VI. Literatur 185
King, J; Haase-Pettingell, C; Robinson, A ; Speed, M and Mitraki, A (1996) Thermolabile folding intermediates: inclusion body precursors and chaperonin substrates. FASEB J 10: 57-66.
Knapp, S; Ladenstein, R and Galinski, E A (1999) Extrinsic protein stabilization by the naturally occurring osmolytes beta-hydroxyectoine and betaine. Extremophiles 3: 191-198.
Koning, S M; Albers, S V; Konings, W N and Driessen, A J (2002) Sugar transport in (hyper)thermophilic archaea. Res Microbiol 153: 61-67.
Konings, W N; Albers, S V; Koning, S and Driessen, A J (2002) The cell membrane plays a crucial role in survival of bacteria and archaea in extreme environments. Antonie Van Leeuwenhoek 81: 61-72.
Köplin, R; Arnold, W; Hötte, B; Simon, R; Wang, G and Pühler, A (1992) Genetics of Xanthan Produktion in Xanthomonas campestris: the xanA and xanB Genes Are Involoved in UDP-Glucose and GDP-Mannose Biosynthesis. J Bacteriol 174: 191-199.
Koster, K L; Lei, Y P; Anderson, M; Martin, S and Bryant, G (2000) Effects of Vitrified and Nonvitrified Sugars on Phosphatidylcholine Fluid-to-Gel Phase Transitions. Biophys J 78: 1932-1946.
Kovach, M E; Phillips, R W; Elzer, P H; Roop, R M and Peterson, K M (1994) pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16: 800-802.
Kraegeloh, A (1998) Die Bedeutung von Kalium für die Osmoadaptation von Halomonas elongata. In Mathematisch-Naturwissenschaftlichen Fakultät. Bonn: Universität Bonn.
Kraegeloh, A (2003) Untersuchungen zur Osmoregulation von Halomonas elongata: Identifizierung und Charakterisierung von Aufnahmesystemen für Kalium und organische Solute. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Kumar, A; Malloch, R A; Fujita, N; Smillie, D A; Ishihama, A and Hayward, R S (1993) The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initiation of transcription at an "extended minus 10" promoter. J Mol Biol 232: 406-418.
Kunte, H J (1995) Grundlagen zur molekularbiologischen Untersuchung halophiler Eubakterien: Aufbau eines Insertionsmutagenese-Systems für Halomonas elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Kunte, H J and Galinski, E A (1995) Transposon mutagenesis in halophilic eubacteria: conjugal transfer and insertion of transposon Tn5 and Tn1732 in Halomonas elongata. FEMS Microbiol Lett 128: 293-299.
Kunte, H J; Galinski, E A; Grammann, K; Volke, A and Bestvater, T (2002) Method for obtaining useful products from organisms. WO 02002077254 A2
Kunte, H J (2006) Osmoregulation in Bacteria: Compatible Solute Accumulation and Osmosensing. Environ Chem 3: 94-99.
Kurtmann, J (2005) Verwendung von aus extremophilen Bakterien gewonnenen Osmolyten zur Herstellung von Arzneimitteln zur äusserlichen Behandlung von Neurodermitis.DE 010330243 A1
Kurz, M (2003) Natrium/Protonen-Antiporter und mechanosensitive Kanäle von Halomonas elongata: Überlebensstrategien bei osmotischem Stress. In Mathematisch-Naturwissenschaftliche Fakultät. Münster: Westfälische Wilhelms-Universität, Münster.
Kyte, J and Doolittle, R F (1982) A simple method or displaying the hydropathic character of a protein. J Mol Biol 157: 105-132.
Laemmli, U (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685.
Lakey, D; Zhang, Y; Talaat, A ; Samten, B; Desjardin, L E; Eisenach, K D, et al. (2002) Priming reverse transcription with oligo(dT) does not yield representative samples of Mycobacterium tuberculosis cDNA. Microbiology 148: 2567-2572.
Lamark, T; Styrvold, O B and Strøm, A (1992) Efflux of choline and glycine betaine from osmoregulating cells of Escherichia coli. FEMS Microbiol Lett 75: 149-154.

VI. Literatur 186
Lamosa, P; Martins, L O; Da Costa, M S and Santos, H (1998) Effects of Temperature, Salinity, and Medium Composition on Compatible Solute Accumulation by Thermococcus spp.. Appl Envir Microbiol 64: 3591-3598.
Lanyi, J (1974) Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol Rev 38: 272-290.
Larsen, P; Sydnes, L; Landfald, B and Strøm, A (1987) Osmoregulation in Escherichia coli by accumulation of organic osmolytes: betaines, glutamic acid, and trehalose. Arch Microbiol 147: 1-7.
Lee, I S; Lin, J; Hall, H K; Bearson, B and W, Foster J (1995) The sationary-phase sigma factor S (rpoS) is required for a sustained acid tolerance response in virulent Salmonella typhimurium. Mol Microbiol 17: 155-167.
Lengeler, J W; Drews, G and Schlegel, H G (1999) In: Lengeler, JW, Drews, G und Schlegel, HG. Biology of the Prokaryotes. Stuttgart, New York: Thieme Verlag.
Leslie, S B; Israeli, E; Lighthart, B; Crowe, J H and Crowe, L M (1995) Trehalose and sucrose protect both membranes and proteins in intact bacteria during drying. Appl Envir Microbiol 61: 3592-3597.
Lessie, T G and Phibbs, P V (1984) Alternative pathways of carbohydrate utilization in pseudomonads. Annu Rev Microbiol 38: 359-388.
Liang, P and Pardee, A (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257: 967-971.
Lippert, K and Galinski, E A (1992) Enzyme stabilization by ectoine-type compatible solutes: protection against heating, freezing and drying. Appl Microbiol Biotechnol 37: 61-65.
Loewen, P C and Hengge-Aronis, R (1994) Regulation in the rpoS regulon of Escherichia coli. Can J Microbiol 44: 707-717.
Lowry, O H; Passonneau, J V and Rock, M K (1961) The stability of pyridine nucleotides. J Biol Chem 236: 2756-2759.
Malin, G; Iakobashvili, R and Lapidot, A (1999) Effect of tetrahydropyrimidine derivatives on protein-nucleic acid interactions. J Biol Chem 274: 6920 - 6929.
Marchler-Bauer, A; Anderson, J; Deweese-Scott, C; Fedorova, N; Geer, L; He, S, et al. (2003) CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res 31: 383-387.
Marmur, J (1961) A procedure for the isolation of desoxyribonucleic acid from microorganism. J Mol Biol 3: 208-218.
Martin, D D; Ciulla, R A and Roberts, M F (1999) Osmoadaptation in Archaea. Appl Envir Microbiol 65: 1815-1825.
Martins, L O and Santos, H (1995) Accumulation of Mannosylglycerate and Di-myo-Inositol-Phosphate by Pyrococcus furiosus in Response to Salinity and Temperature. Appl Envir Microbiol 61: 3299-3303.
Martins, L O; Carreto, L S; Da Costa, M S and Santos, H (1996) New compatible solutes related to Di-myo-inositol-phosphate in members of the order Thermotogales. J Bacteriol 178: 5644-5651.
Martins, L O; Huber, R; Huber, H; Stetter, K O; Da Costa, M S and Santos, H (1997) Organic Solutes in Hyperthermophilic Archaea. Appl Envir Microbiol 63: 896-902.
Martins, L O; Empadinhas, N; Marugg, J D; Miguel, C; Ferreira, C; Da Costa, M S and Santos, H (1999) Biosynthesis of Mannosylglycerate in the Thermophilic Bacterium Rhodothermus marinus. BIOCHEMICAL AND GENETIC CHARACTERIZATION OF A MANNOSYLGLYCERATE SYNTHASE. J Biol Chem 274: 35407-35414.
Maskow, T and Babel, W (2001) Calorimetrically obtained information about the efficiency of ectoine synthesis from glucose in Halomonas elongata. Biochim Biophys Acta 1527: 4-10.
Matheson, A ; Sprott, G D; Mcdonald, I J and Tessier, H (1976) Some properties of an unidentified halophile: growth characteristics, internal salt concentration, and morphology. Can J Microbiol 22: 780-786.

VI. Literatur 187
Matsuo, T; Tsuchida, Y and Morimoto, N (2002) Trehalose eye drops in the treatment of dry eye syndrome. Ophthalmology 109: 2024-2029.
Matz, M V and Lukyanov, S A (1998) Different strategies of differential display: areas of application. Nucleic Acids Res 26: 5537-5543.
Melo, E P; Faria, Q T ; Martins, L O; Goncalves, A and Cabral, J M S (2001) Cutinase Unfolding and Stabilization by Trehalose and Mannosylglycerate. Proteins: Structure, Function and Genetics. 42: 542-552.
Miguelez, E and Gilmour, D J (1994) Regulation of cell volume in the salt tolerant bacterium Halomonas elongata. Lett Appl Microbiol 19: 363-365.
Miller, J H (1972) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for E. coli and Related Bacteria. Los Angeles: Cold Spring Harbor Laboratory Press.
Miller, J H (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for E. coli and Related Bacteria. Los Angeles: Cold Spring Harbor Laboratory Press.
Miller, K J (1985) Effects of temperature and sodium chloride concentration on the phospholipid and fatty acid compositions of a halotolerant Planococcus sp.. J Bacteriol 162: 263-270.
Mitraki, A and King, J (1989) Protein folding intermediates and inclusion body formation. BioTechnology 7: 690-697.
Mizote, T; Tsuda, M; Smith, D D; Nakayama, H and Nakazawa, T (1999) Cloning and characterization of the thiD/J gene of Escherichia coli encoding a thiamin-synthesizing bifunctional enzyme, hydroxymethylpyrimidine kinase/phosphomethylpyrimidine kinase. Microbiology 145: 495-501.
Moghaieb, R E A; Tanaka, N; Saneoka, H; Hussein, H A ; Yousef, S S; Ewada, M A , et al. (2000) Expression of betaine aldehyde dehydrogenase gene in transgenic tomato hairy roots leads to the accumulation of glycine betaine and contributes to the maintenance of the osmotic potencial under salt stress. Soil Science and Plant Nutrition 46: 873-883.
Molisch, H and Biebl, R (1955) Botanische Versuche und Beobachtungen ohne Apparate. Ein Experimentierbuch für jeden Pflanzenfreund. Stuttgart: Fischer.
Moreira, L; Nobre, M F; Sa-Correia, I and Da Costa, M S (1996) Genomic Typing and Fatty acid Composition of Rhodothermus marinus. Syst Appl Microbiol 19: 83-90.
Morett, E; Korbel, J O; Rajan, E; Saab-Rincon, G; Olvera, L; Olvera, M, et al. (2003) Systematic discovery of analogous enzymes in thiamine biosynthesis. Nat Biotechnol 21: 790-795.
Mormile, M R; Romine, M F; Garcia, M T; Ventosa, A; Bailey, T J and Peyton, B M (1999) Halomonas campisalis sp. nov., a denitrifying, moderately haloalkaliphilic bacterium. Syst Appl Microbiol 22: 551-558.
Mullis, K B and Faloona, F A (1987) Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol 155: 335-350.
Munkund, S and Adams, M W W (1991) The novel tungsten-iron-sulfur protein of the hyperthermophilic archaebacterium, Pyrococcus furiosus, is an aldehyde ferredoxin oxidoreductase; evidence for its participation in a unique glycolytic pathway. J Biol Chem 266: 14208-14216.
Nakagaki, M; Nagase, H and Ueda, H (1992) Stabilization of the lamellar structure of phosphatidylcholine by complex-formation with trehalose. J Membr Sci 73: 173-180.
Nakayama, H; Yoshida, K; Ono, H; Murooka, Y and Shinmyo, A (2000) Ectoine, the compatible solute of Halomonas elongata, confers hyperosmotic tolerance in cultured tobacco cells. Plant Physiol 122: 1239-1247.
Needleman, S and Wunsch, C (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443-453.
Nelson, K; Paulsen, I; Weinel, C; Dodson, R; Hilbert, H; Fouts, D, et al. (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4: 799-808.

VI. Literatur 188
Nguyen, L H and Burgess, P R (1997) Comparative analysis of the interactions of Escherichia coli σ S and σ 70 RNA polymerase holoenzyme with the stationary-phase-specific bolAp1 promoter. Biochemistry 36: 1748-1754.
Nikaido, H and Vaara, M (1985) Molecular basis of bacterial outer membrane permeability. Microbiol Rev 49: 1-32.
Nossal, N G and Heppel, L A (1966) The release of enzymes by osmotic shock from Escherischia coli in exponential phase. J Biol Chem 241: 3055-3062.
Nunes, O C; Donato, M M; Manaia, C M and Da Costa, M S (1992) The Polar Lipid and Fatty Acid Composition of Rhodothermus Strains. Syst Appl Microbiol 15: 59-62.
Nunes, O C; Manaia, C M; Da Costa, M S and Santos, H (1995) Compatible Solutes in the Thermophilic Bacteria Rhodothermus marinus and Thermus thermophilus. Appl Envir Microbiol 61: 2351-2357.
Obis, D; Guillot, A; Gripon, J-C; Renault, P; Bolotin, A and Mistou, M-Y (1999) Genetic and Biochemical Characterization of a High-Affinity Betaine Uptake System (BusA) in Lactococcus lactis Reveals a New Functional Organization within Bacterial ABC Transporters. J Bacteriol 181: 6238-6246.
Old, R and Primrose, S (1992) Gentechnologie- Eine Einführung. Stuttgart: Thieme Verlag.
Oren, A (1999) Bioenergetic Aspects of halophilism. Microbiol Mol Biol Rev 63: 334-348.
Oren, A (2002) Halophilic Microorganisms and their Environments. Dodrecht/Boston/London: Kluwer Academic Publishers.
Oren, A; Heldal, M; Norland, S and Galinski, E A (2002) Intracellular ion and organic solute concentrations of the extremely halophilic bacterium Salinibacter ruber. Extremophiles 6: 491-498.
Oshima, T and Imahori, K (1974) Description of Thermus thermophilus (Yoshid and Oshima) comb. nov., a nonsporulating thermophilic bacterium from a Japanese thermal spa. Int J Syst Bact 24: 102-112.
Panayotova-Heiermann, M; Leung, D W; Hirayama, B A and Wright, E M (1999) Purification and functional reconstitution of a truncated human Na (+)/ glucose cotransporter (SGLT1) expressed in E. coli. FEBS Lett 459: 386-390.
Patel, B K; Saul, D S; Reeves, R A; Williams, L C; Cavanagh, J E; Nichols, P D and Bergquist, P L (1994) Phylogeny and lipid composition of Thermonema lapsum, a thermophilic gliding bacterium. FEMS Microbiol Lett 115: 313-317.
Pereira, C S; Lins, R D; Chandrasekhar, I; Freitas, L C G and Hunenberger, P H (2004) Interaction of the Disaccharide Trehalose with a Phospholipid Bilayer: A Molecular Dynamics Study. Biophys J 86: 2273-2285.
Peter, H; Burkovski, A and Kramer, R (1996) Isolation, characterization, and expression of the Corynebacterium glutamicum betP gene, encoding the transport system for the compatible solute glycine betaine. J Bacteriol 178: 5229-5234.
Petersen, L A and Downs, D M (1997) Identification and characterization of an operon in Salmonella typhimurium involved in thiamine biosynthesis. J Bacteriol 179: 4894-4900.
Pirch, T; Quick, M; Nietschke, M; Langkamp, M and Jung, H (2002) Sites Important for Na+ and Substrate Binding in the Na+/Proline Transporter of Escherichia coli, a Member of the Na+/Solute Symporter Family. J Biol Chem 277: 8790-8796.
Pirch, T; Landmeier, S and Jung, H (2003) Transmembrane Domain II of the Na+/Proline Transporter PutP of Escherichia coli forms part of a conformationally flexible, cytoplasmic exposed aqueous cavity within the membrane. J Biol Chem 278: 42942-42949.
Prasad, P D; Wang, H; Kekuda, R; Fujita, T; Fei, Y J; Devoe, L D, et al. (1998) Cloning and functional expression of a cDNA encending a mammalian sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin and lipoate. J Biol Chem 273: 7501-7506.
Rabus, R; Jack, D L; Kelly, D J and Saier Jr, M H (1999) TRAP transporters: an ancient family of extracytoplasmic solute- receptor-dependent secondary active transporters. Microbiology 145: 3431-3445.

VI. Literatur 189
Ramirez, F; Marecek, J F and Szamosi, J (1980) Magnesium and calcium ion effects on hydrolysis rates of adenosine-5'-triphosphate. J. Org. Chem. 45: 4748-4752.
Ramos, A; Raven, N D H; Sharp, R J; Bartolucci, S; Rossi, M; Cannio, R, et al. (1997) Stabilization of Enzymes against Thermal Stress and Freeze-Drying by Mannosylglycerate. Appl Envir Microbiol 63: 4020-4025.
Reed, R H (1986) Halotolerant and halophilic microbes. In Microbes in extreme enviroments. Herbert, R., CODD, G A, Academic Press (ed). London: Special publications for the Society for General Microbiology, pp. 51-81.
Reese, M (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem 26: 51-56.
Reizer, J; Reizer, A and Saier Jr, M H (1994) A functional superfamily of sodium/ solute symporters. Biochim Biophys Acta 1197 (2): 133-166.
Robertson, D E; Noll, D and Roberts, M F (1992) Free amino acid dynamics in marine methanogens. beta-Amino acids as compatible solutes. J Biol Chem 267: 14893-14901.
Rosenberg, A; Goldman, E; Dunn, J J; Studier, F W and Zubay, G (1993) Effects of consecutive AGG codons on translation in Escherichia coli, demonstrated with a versatile codon test system. J Bacteriol 175: 716-722.
Sa-Correia, I; Darzins, A; Wang, S K; Berry, A and Chakrabarty, A (1987) Alginate Biosynthetic Enzymes in Mucoid and Nonmocuid Pseudomonas aeruginosa: Overproduktion of Phosphomannose Isomerase, Phosphomannomutase, and GDP-Mannose Pyrophosphorylase by Overexpression of the Phosphomannose Isomerase (pmi) Gene. J Bacteriol 169: 3224-3231.
Sadler, M; Mcaninch, M; Alico, R and Hochstein, L I (1980) The intracellular Na+ and K+ composition of the moderately halophilic bacterium, Paracoccus halodenitrificans. Can J Microbiol 26: 496-502.
Saier Jr, M H (2000) A Functional-Phylogenetic Classification System for Transmembrane Solute Transporters. Microbiol Mol Biol Rev 64: 354-411.
Sampaio, M-M; Santos, H and Boos, W (2003a) Synthesis of GDP-Mannose and Mannosylglycerate from Labeled Mannose by Genetically Engineered Escherichia coli without Loss of Specific Isotopic Enrichment. Appl Envir Microbiol 69: 233-240.
Sampaio, M-M; Silva, Z; Da Costa, M S; Boos, W and Santos, H (2003b) Transport of Mannosylglycerate and Trehalose in the Thermophilic Bacterium Thermus thermophilus. In Thermophiles.
Sanger, F; Nicklen, S and Golson, A (1977) DNA sequencing with chain-terminating inhibitors. PNAS 74: 5463-5467.
Santos, H; Martins, A and Da Costa, M S (1998) Thermostabiliziation, osmoprotection and protection against desiccation of enzymes, cell components and cells by mannosylglycerate. EP 0186509 A2
Santos, H and Da Costa, M S (2002) Compatible solutes of organisms that live in hot saline environments. Environ Microbiol 4: 501-509.
Sarkar, N (1996) Polyadenylation of mRNA in bacteria. Microbiology 142: 3125-3133.
Sarker, R I; Ogawa, W; Shimamoto, T and Tsuchiya, T (1997) Primary structure and properties of Na+/ glucose symporter (SglS) of Vibrio parahaemolyticus. J Bacteriol 179: 1805-1808.
Sauer, T (1995) Untersuchungen zur Nutzung von Halomonas elongata für die Gewinnung kompatibler Solute. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Sauer, T and Galinski, E A (1998) Bacterial milking: a novel bioprocess for production of compatible solutes. Biotechnol Bioeng 57: 306-313.
Schäfer, A; Tauch, A; Jäger, W; Kalinowski, J; Thierbach, G and Pühler, A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145: 69-73.
Schiller, D; Ott, V; Kramer, R and Morbach, S (2006) Influence of Membrane Composition on Osmosensing by the Betaine Carrier BetP from Corynebacterium glutamicum. J Biol Chem 281: 7737-7746.

VI. Literatur 190
Schilz, Y (2005) Untersuchung zur Bedeutung des Gens ectR in Halomonas elongata DSM 2581T. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Schlegel, H G (1972) Allgemeine Mikrobiologie. Stuttgart: Georg Thieme Verlag.
Schnoor, M (2001) Konstruktion eines Expressionsvektors für Halomonas elongata. In Mathematisch-Naturwissenschaftliche Fakultät. Münster: Universität Münster.
Schnoor, M ; Voß, P; Cullen, P ; Boking, T; Galla, H J ; Galinski, E A and Lorkowski, S (2004) Characterization of the synthetic compatible solute homoectoine as a potent PCR enhancer. Biochem Biophys Res Commun 322: 867-872.
Scholz, S; Sonnenbichler, J; Schafer, W and Hensel, R (1992) Di-myo-inositol-1,1'-phosphate: a new inositol phosphate isolated from Pyrococcus woesei. FEBS Lett 306: 239-242.
Schwarz, T (2002a) Cosmetic Formulations. WO 0215868 A2
Schwarz, T (2002b) Use of beta-mannosylglycerate and derivatives in cosmetic and dermatological formulations. WO 0200201587 A1
Severin, J; Wohlfarth, A and Galinski, E A (1992) The predominant role of recently discovered tetrahydropyrimidines for the osmoadaptation of halophilic eubacteria. J Gen Microbiol 138: 1629-1638.
Severin, J (1993) Kompatible Solute und Wachstumskinetik bei halophilen aeroben heterotrophen Eubakterien. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Silva, Z; Borges, N; Martins, L O; Wait, R; Da Costa, M S and Santos, H (1999) Combined effect of the growth temperature and salinity of the medium on the accumulation of compatible solutes by Rhodothermus marinus and Rhodothermus obamensis. Extremophiles 3: 163-172.
Silva, Z; Sampaio, M-M; Henne, A; Bohm, A; Gutzat, R; Boos, W, et al. (2005) The High-Affinity Maltose/Trehalose ABC Transporter in the Extremely Thermophilic Bacterium Thermus thermophilus HB27 Also Recognizes Sucrose and Palatinose. J Bacteriol 187: 1210-1218.
Simon, R; Priefer, U and Pühler, A (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Biotechnology 1: 784-791.
Sleator, R D; Gahan, C G M; Abee, T and Hill, C (1999) Identification and Disruption of BetL, a Secondary Glycine Betaine Transport System Linked to the Salt Tolerance of Listeria monocytogenes LO28. Appl Envir Microbiol 65: 2078-2083.
Smith, P; Krohn, R; Hermanson, G; Mallia, A; Gartner, F; Provenzano, M, et al. (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85.
Sorensen, M A; Kurland, C G and Pedersen, S (1989) Codon usage determines translation rate in Escherichia coli. J Mol Biol 207: 365-377.
Southern, E (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 503-517.
Spanjaard, R A; Chen, K; Walker, J R and Van Duin, J (1990) Frameshift suppression at tandem AGA and AGG codons by cloned tRNA genes: assigning a codon to argU tRNA and T4 tRNAArg. Nucleic Acids Res 18: 5031-5036.
Spiegelhalter, F and Bremer, E (1998) Osmoregulation of the opuE proline transport gene from Bacillus subtilis: contributions of the sigmaA- and sigmaB-dependent stress-responsive promoters. Molecular Microbiology 29: 285-296.
Stanier, R Y; Palleroni, N J and Doudoroff, M (1966) The aerobic pseudomonads: a taxonomic study. J Gen Microbiol 43: 159-271.
Strøm, A and Kaasen, I (1993) Trehalose metabolism in Escherichia coli: stress protection and stress regulation of gene expression. Mol Microbiol 8: 205-210.
Stryer, L (1996) Biochemie. Heidelberg/Berlin/Oxford: Spektrum Akademischer Verlag.

VI. Literatur 191
Stumpfe, D (2003) Aufbau und Regulation der Ectoin-Synthesegene ectABC aus Halomonas elongata DSM2581T. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Sun, W Q and Leopold, A (1994) Glassy state and seed storage stability: a viability equation analysis. Ann Bot 74: 601-604.
Sutcliffe, J G (1979) Complete nucleotide sequence of the Escherichia coli plasmid pBR322. Cold Spring Harbor Symposium of Quantitative Biology 43: 77-90.
Tailor, C S; Marin, M; Nouri, A; Kavanaugh, M P and Kabat, D (2001) Truncated Forms of the Dual Function Human ASCT2 Neutral Amino Acid Transporter/Retroviral Receptor Are Translationally Initiated at Multiple Alternative CUG and GUG Codons. J Biol Chem 276: 27221-27230.
Takeshita, S; Sato, M; Toba, M; Masahashi, W and Hashimoto-Gotoh, T (1987) High-copy-number and low-copy-number plasmid vectors for lacZ alpha-complementation and chloramphenicol- or kanamycin-resistance selection. Gene 61: 63-74.
Tanaka, K; Kusano, S; Fujita, N; Ishihama, A and Takahashi, H (1995) Promoter determinants for Eschrichia coli RNA polymerase holoenzyme containig σ 38 (the rpoS gene product). Nucleic Acids Res 23: 827-834.
Tatusov, R; Koonin, E and D, Lipman (1997) A Genomic Prespective on Protein Families. Science 278: 631-637.
Tatusov, R; Natale, D; Garkavtsev, I; Tatusova, T; Shankavaram, U; Rao, B, et al. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29: 22-28.
Tenreiro, S; Nobre, M F; Rainey, F A; Miguel, C and Da Costa, M S (1997) Thermonema rossianum sp. nov., a new thermophilic and slightly halophilic species from saline hot springs in Naples, Italy. Int J Syst Bacteriol 47: 122-126.
Thompson, J D; Higgins, D G and Gibson, T J (1994) CLUSTAL W : improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673-4680.
Tillett, D; Burns, B and Neilan, B (2000) Optimized rapid amplification of cDNA ends (RACE) for mapping bacterial mRNA transcripts. Biotechniques 28: 448-456.
Tindall, B J (1991) Lipid composition of Rhodothermus marinus. FEMS Microbiol Lett 80: 65-68.
Touzé, T; Gouesbet, G; Boiangiu, C; Jebbar, M; Bonnassie, S and Blanco, C (2001) Glycine betaine loses its osmoprotective activity in a bspA strain of Erwinia chrysanthemi. Mol Microbiol 42: 87-99.
Towbin, H; Staehelin, T and Gordon, J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. PNAS 76: 4350-4354.
Tsai, J P and Shi, W (2000) Analysis of gene expression in Treponema denticola with differential display polymerase chain reaction. Oral Microbiol Immunol 15: 305-308.
Tschichholz, I and Trüper, H G (1990) Fate of compatible solutes during dilution stress in Ectothiorhodospira halochloris. FEMS Microbiol Ecol 73: 181-186.
Turk, E and Wright, E M (1997) Membrane topology motifs in the SGLT cotransporter family. J Membr Biol 159: 1-20.
Tusnady, G E and Simon, I (1998) Principles Govering Amino Acids Composition of Integral Membrane Proteins: Application to Topology Prediction. J Mol Biol 283: 489-506.
Tusnady, G E and Simon, I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849-850.
Upasani, V N; Desai, S G; Moldoveanu, N and Kates, M (1994) Lipids of extremely halophilic archaeobacteria from saline environments in India: a novel glycolipid in Natronobacterium strains. Microbiology 140: 1959-1966.
Ures, A (2005) Identifizierung und Charakterisierung des Ectoin-Hydroxylasegens ectD aus Halomonas elongata DSM2581T. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.

VI. Literatur 192
Vander Horn, P B; Backstrom, A ; Stewart, V and Begley, T P (1993) Structural genes for thiamine biosynthetic enzymes (thiCEFGH) in Escherichia coli K-12. J Bacteriol 175: 982-992.
Ventosa, A; Nieto, J J and Oren, A (1998) Biology of Moderately Halophilic Aerobic Bacteria. Microbiol Mol Biol Rev 62: 504-544.
Vermeulen, V and Kunte, H J (2004) Marinococcus halophilus DSM 20408T encodes two transporters for compatible solutes belonging to the betaine-carnitine-choline transporter family: identification and characterization of ectoine transporter EctM and glycine betaine transporter BetM. Extremophiles 8: 175-184.
Volke, A (2001) Konstruktion des Produktionsstammes Halomonas elongata KB2 für die Synthese von Ectoin durch Mutation des Ectointransporters TeaABC. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Von Blohn, C; Kempf, B; Kappes, R M and Bremer, E (1997) Osmostress response in Bacillus subtilis: characterization of a proline uptake system (OpuE) regulated by high osmolarity and alternative transcription factor sigma B. Molecular Microbiology 25: 175-187.
Voß, P (1998) Nutzung kompatibler Solute zur Verbesserung der Trockenstabilisierung von Vibrio fischeri. In Mathematisch-Naturwissenschaftliche Fakultät. Münster: Universität Münster.
Vreeland, R H; Litchfield, C D; Martin, E L and Elliot, E (1980) Halomonas elongata, a New Genus and Species of Extremely Salt-Tolerant Bacteria. Int J Syst Bact 30: 485-495.
Vreeland, R H; Mierau, B D; Litchfield, C D and Martin, E L (1983) Relationship of the internal solute composition to salt tolerance of Halomonas elongata. Can J Microbiol 29: 407-414.
Weinel, C; Nelson, K E and Tümmler, B (2002) Global features of the Pseudomonas putida KT2440 genome sequence. Environ Microbiol 4: 809-818.
Welsh, D T and Herbert, R A (1999) Osmotically induced intracellular trehalose, but not glycine betaine accumulation promotes desiccation tolerance in Escherichia coli. FEMS Microbiol Lett 174: 57-63.
Wiechelman, K J; Braun, R D and Fitzpatrick, J F (1988) Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal Biochem 175: 231-237.
Wohlfarth, A; Severin, J and Galinski, E A (1990) The spectrum of compatible solutes in heterotrophic halophilic eubacteria of the family Halomonadaceae. J Gen Microbiol 136: 705-712.
Wohlfarth, A (1993) Neue Naturstoffe aus halophilen heterotrophen Eubakterien. In Mathematisch-Naturwissenschaftliche Fakultät. Bonn: Universität Bonn.
Wösten, M (1998) Eubacterial sigma-factors. FEMS Microbiol Rev 22: 127-150.
Wright, E M (2001) Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol 280: F10-18.
Wright, E M; Loo, D D; Hirayama, B A and Turk, E (2004) Surprising Versatility of Na+-Glucose Cotransporters: SLC5. Physiology 19: 370-376.
Xavier, K B; Martins, L O; Peist, R; Kossmann, M; Boos, W and Santos, H (1996) High-affinity maltose/trehalose transport system in the hyperthermophilic archaeon Thermococcus litoralis. J Bacteriol 178: 4773-4777.
Yamao, F; Andachi, Y; Muto, A; Ikemura, T and Osawa, S (1991) Levels of tRNAs in bacterial cells as affected by amino acid usage in proteins. Nucleic Acids Res 19: 6119-6122.
Zhang, S P; Zubay, G and Goldman, E (1991) Low-usage codons in Escherichia coli, yeast, fruit fly and primates. Gene 105: 61-72.
Zhang, Y and Begley, T P (1997) Cloning, sequencing and regulation of thiA, a thiamine biosynthesis gene from Bacillus subtilis. Gene 198: 73-82.
Zucker, M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406-3415.

Anhang
Rhodothermus marinus DSM 4252T
Thiamin-Gene HindIII PstI SalI BamHI | | | | GCCAAGCTTGGCTGCAGGTCGACGGATCCGGAAACGGCGCGCGGCATTGCCGTGGCGGTAAACGAGGAGGTCGTGCGCCGTTCGCAATGGGCCGAACTCCGGCTCCAGGCAGGCGATCGC 120 CGGTTCGAACCGACGTCCAGCTGCCTAGGCCTTTGCCGCGCGCCGTAACGGCACCGCCATTTGCTCCTCCAGCACGCGGCAAGCGTTACCCGGCTTGAGGCCGAGGTCCGTCCGCTAGCG +1 rbs GTGGAAGTGGTAACGGCCCGTCAGGGCGGATAAAAACGCACACGAGAGGCTATGAGCATGCAGGTGGTTGACGGCAAGGGCTGGCAGGTGCAGGTGGACGACGAGCCGCTGCGCATCGGG 240 CACCTTCACCATTGCCGGGCAGTCCCGCCTATTTTTGCGTGTGCTCTCCGATACTCGTACGTCCACCAACTGCCGTTCCCGACCGTCCACGTCCACCTGCTGCTCGGCGACGCGTAGCCC CCGCTGGAACTGCACACGCGGCTGCTGATGGGTTCGAGCGGCTATCCGAACGTGCAGACCATGCTCGATGCGCTGGAGGCGGCCGGGGCCGAGCTGGTGACGGTGGCCATCCGGCGCATC 360 GGCGACCTTGACGTGTGCGCCGACGACTACCCAAGCTCGCCGATAGGCTTGCACGTCTGGTACGAGCTACGCGACCTCCGCCGGCCCCGGCTCGACCACTGCCACCGGTAGGCCGCGTAG AACCTGAAGGATCGTTCCGAGGAGAGCCTGCTGGCGCTGCTCGAACGGCACGGCTATCGGGTGCTGCCTAACACGGCTGGCTGCTACACGGCCCGCGAAGCTGTGCTCGTGGCGCAACTG 480 TTGGACTTCCTAGCAAGGCTCCTCTCGGACGACCGCGACGAGCTTGCCGTGCCGATAGCCCACGACGGATTGTGCCGACCGACGATGTGCCGGGCGCTTCGACACGAGCACCGCGTTGAC GCCCGCGAGGCGCTCGAAACGAACCTGATCAAGCTGGAGGTCATCGGCGACGACCAGACGCTTTTGCCCGACGTGGAGCAACTGCTGAAGGCCGCGCGGGAGCTGATTAACGACGGCTTC 600 CGGGCGCTCCGCGAGCTTTGCTTGGACTAGTTCGACCTCCAGTAGCCGCTGCTGGTCTGCGAAAACGGGCTGCACCTCGTTGACGACTTCCGGCGCGCCCTCGACTAATTGCTGCCGAAG GTAGTGCTGGCCTACACGAACGACGATCCGATCACCTGCCGCAAGCTGGCCGATCTGGGTTGTGCGGCCGTCATGCCGCTGGCCTCACCAATCGGAAGCGGCATGGGACTGGTCAATCCC 720 CATCACGACCGGATGTGCTTGCTGCTAGGCTAGTGGACGGCGTTCGACCGGCTAGACCCAACACGCCGGCAGTACGGCGACCGGAGTGGTTAGCCTTCGCCGTACCCTGACCAGTTAGGG NcoI | TACAACCTGCACCTGATCCGGGAAATGATCGACGACGTGCCGCTCATCGTGGATGCCGGGATCGGGACGGCCAGCGACGCGGCAATGGCCATGGAGCTGGGCTACGACGGCGTGCTGGTC 840 ATGTTGGACGTGGACTAGGCCCTTTACTAGCTGCTGCACGGCGAGTAGCACCTACGGCCCTAGCCCTGCCGGTCGCTGCGCCGTTACCGGTACCTCGACCCGATGCTGCCGCACGACCAG AACTCGGCCATCTCGCAGGCGGAGCATCCGGTGCTGATGGCCCGCGCCGTCCGCCATGCGGTCGAGGCCGGCCGCCTGGCCTTCAGGGCCGGACGCATGCCGCGGCGCTTCTACGCACGG 960 TTGAGCCGGTAGAGCGTCCGCCTCGTAGGCCACGACTACCGGGCGCGGCAGGCGGTACGCCAGCTCCGGCCGGCGGACCGGAAGTCCCGGCCTGCGTACGGCGCCGCGAAGATGCGTGCC rbs CCGTCCTCGCCGATGGAAGGGCGCGTCGGACACTGAGGCATGCTGCCCCGTTTGCTGCTGATCGCCGATCGCTTTACCGATCCGCATCGGGCCAGGGTAGTGCGCCGGGCAGTAACCGCC 1080 GGCAGGAGCGGCTACCTTCCCGCGCAGCCTGTGACTCCGTACGACGGGGCAAACGACGACTAGCGGCTAGCGAAATGGCTAGGCGTAGCCCGGTCCCATCACGCGGCCCGTCATTGGCGG PstI | GGCGTGCCCTGGGTGCAGCTCCGTGACCACCGGGTCGATCCCGACACGTTCGCCCGTGCGGCCGAAGCGCTCGTGCCGGTCCTGCAGGCCGAAAATCCAAACCTGCTGCTCTCGGTCAAC 1200 CCGCACGGGACCCACGTCGAGGCACTGGTGGCCCAGCTAGGGCTGTGCAAGCGGGCACGCCGGCTTCGCGAGCACGGCCAGGACGTCCGGCTTTTAGGTTTGGACGACGAGAGCCAGTTG

ACGCACCTGGAAGTGGCGCAGCGTCTGGGGCTGGGACTGCACGTCGGGCGACGCGGTCCCTCGGTGGCCGAAGCCCGGCGGCGACTCGGGTCCGGGACGTTGCTGGGCTACTCGGCGCAC 1320 TGCGTGGACCTTCACCGCGTCGCAGACCCCGACCCTGACGTGCAGCCCGCTGCGCCAGGGAGCCACCGGCTTCGGGCCGCCGCTGAGCCCAGGCCCTGCAACGACCCGATGAGCCGCGTG GATCTGGACGCGGCCCGTCGGGCTGCTGATGAAGGGGCCGATTATCTGCTCTTCAGCCCGGTTTTTCCCACGGCCAGCAAGCCCGGCGTACCGGCAGTCGGTCTGGAGGCGCTGGCGGCG 1440 CTAGACCTGCGCCGGGCAGCCCGACGACTACTTCCCCGGCTAATAGACGAGAAGTCGGGCCAAAAAGGGTGCCGGTCGTTCGGGCCGCATGGCCGTCAGCCAGACCTCCGCGACCGCCGC PstI | GTCTGTCGAGCGGTTCCCATTCCCGTACTGGCGCTGGGCGGCATCACGCCCGAGCGCGTCTCGGCCTGCCTGCAGCAGGGTGCCCACGGCGTGGCCGTCGTCTCGGCCATTCTGGACGCT 1560 CAGACAGCTCGCCAAGGGTAAGGGCATGACCGCGACCCGCCGTAGTGCGGGCTCGCGCAGAGCCGGACGGACGTCGTCCCACGGGTGCCGCACCGGCAGCAGAGCCGGTAAGACCTGCGA BamHI | rbs TCGGATCCTGAAGGGGCCGTTCGGCAGTTTTTAGCACGTACCGAAAACGCATTGCACCATCGATAAGCGGCCATGAACCGACAGATTCCACCGGTGGCACTGACGATCGCCGGAAGCGAC 1680
Anhang II
AGCCTAGGACTTCCCCGGCAAGCCGTCAAAAATCGTGCATGGCTTTTGCGTAACGTGGTAGCTATTCGCCGGTACTTGGCTGTCTAAGGTGGCCACCGTGACTGCTAGCGGCCTTCGCTG TCGGGCGGGGGCGCCGGCATCCAGGCCGACATCAAGGCGATGCAGGCCAACGGCGTTTTTGCCGCCTCGGTCATCACGGCCGTGACGGCGCAGAACACGCGGGCCGTGACGGCCGCCTTC 1800 AGCCCGCCCCCGCGGCCGTAGGTCCGGCTGTAGTTCCGCTACGTCCGGTTGCCGCAAAAACGGCGGAGCCAGTAGTGCCGGCACTGCCGCGTCTTGTGCGCCCGGCACTGCCGGCGGAAG GAGCTACCGCTGGAGCTGATCGAAGCACAGATCGACGCGGTTTTTGAAGACCTGCCGGTGGCGGCCGTCAAGACGGGCATGCTTTCGTCGGCGGCCATCATCGAACTGGTGGCGCGTAAG 1920 CTCGATGGCGACCTCGACTAGCTTCGTGTCTAGCTGCGCCAAAAACTTCTGGACGGCCACCGCCGGCAGTTCTGCCCGTACGAAAGCAGCCGCCGGTAGTAGCTTGACCACCGCGCATTC GCCGAACAATGGCAGATGCGACCGCTCGTGGTCGATCCGGTGATGATTTCCAAAAGTGGCTATCCGCTGCTGAAGCCCGACGCCGTTGCCACGTTGCGCGAGGCGCTGTTGCCGCTCGCC 2040 CGGCTTGTTACCGTCTACGCTGGCGAGCACCAGCTAGGCCACTACTAAAGGTTTTCACCGATAGGCGACGACTTCGGGCTGCGGCAACGGTGCAACGCGCTCCGCGACAACGGCGAGCGG NcoI | ACGCTGGTGACGCCCAATGCGCACGAGGCCGCTCATCTGACCGGCCTGAAGATCGAAACGGTCGAAGATCTCTACGAGGCGGCCCGGCGCATCAAAGCCATGGGGCCGCAGGCCGTGCTG 2160 TGCGACCACTGCGGGTTACGCGTGCTCCGGCGAGTAGACTGGCCGGACTTCTAGCTTTGCCAGCTTCTAGAGATGCTCCGCCGGGCCGCGTAGTTTCGGTACCCCGGCGTCCGGCACGAC EcoRI KpnI | | GTGAAGGGCGGGCACCTGAGCCGTGAGGCCGTGGCGGTCGATGTGCTTTTCGACGGCGAGCGCTTCGAGGAATTCCGCGCGCCCCGGATCGACACGCCGCACACGCACGGTACCGGCTGC 2280 CACTTCCCGCCCGTGGACTCGGCACTCCGGCACCGCCAGCTACACGAAAAGCTGCCGCTCGCGAAGCTCCTTAAGGCGCGCGGGGCCTAGCTGTGCGGCGTGTGCGTGCCATGGCCGACG ACCTACGCTTCGGCCATCGCCGCCAACCTGGCGCGGGGATTTTCGCTGGTCGAGGCCGTGCGTCGCGCCAAGCGGTACGTGACCGAGGCGATCCGCCATGCATTGCCCATCGGCGGCGGC 2400 TGGATGCGAAGCCGGTAGCGGCGGTTGGACCGCGCCCCTAAAAGCGACCAGCTCCGGCACGCAGCGCGGTTCGCCATGCACTGGCTCCGCTAGGCGGTACGTAACGGGTAGCCGCCGCCG CACGGCCCCACGCACCACTTCTATTTTCTGGAATCCTTCAAAGGATTTCCCATCGGCGCCGAAGAACCCGAAAAAACGGCCGGCGTATAAGCGGATGCTTTCCAAGGTTGGCGCTTGCGT 2520 GTGCCGGGGTGCGTGGTGAAGATAAAAGACCTTAGGAAGTTTCCTAAAGGGTAGCCGCGGCTTCTTGGGCTTTTTTGCCGGCCGCATATTCGCCTACGAAAGGTTCCAACCGCGAACGCA SmaI +1 | CCGAAGCGACGCATCCTTATCTTAGCCAGCCGCAGTAAGCGTGCCCGTAGCTCAGCTGGATAGAGCGTCTGACTACGGATCAGAAGGTCGGGGGTTCGAATCCTCCCGGGCACGCCACCT 2640 GGCTTCGCTGCGTAGGAATAGAATCGGTCGGCGTCATTCGCACGGGCATCGAGTCGACCTATCTCGCAGACTGATGCCTAGTCTTCCAGCCCCCAAGCTTAGGAGGGCCCGTGCGGTGGA

Anhang III
GGCCCGCACCTTCCACGGTGCGGGCTTTTCTGTTAGAAACCAGTGAGCCTGCGTGGAGAGCCTTTCACTCCGAGCGCTGTGGGGGCGCGTACGGGAGTTGCTGGCCCGTCTGGAGCAGCA 2760 CCGGGCGTGGAAGGTGCCACGCCCGAAAAGACAATCTTTGGTCACTCGGACGCACCTCTCGGAAAGTGAGGCTCGCGACACCCCCGCGCATGCCCTCAACGACCGGGCAGACCTCGTCGT rbs ACCCGGTTTTGAGCTGCGGCCCGGCCAGCGCCAGATGGCCGAGACGGTCGTCCGGGCGATCGCCGAGGGGCTTCCCGCGGCAATCGAGGCCGGAACCGGCACGGGTAAGACGTTCGGCTA 2880 TGGGCCAAAACTCGACGCCGGGCCGGTCGCGGTCTACCGGCTCTGCCAGCAGGCCCGCTAGCGGCTCCCCGAAGGGCGCCGTTAGCTCCGGCCTTGGCCGTGCCCATTCTGCAAGCCGAT CCTGATTCCGGCGCTCTGCGCAGAGGCGCGGGTGCTCGTCTCCACCAGCACAAAGGCTCTTCAGGAACAGCTCGTTCACAAGGATCTGGAACGTCCCGAATCCCAGCAGGGGCATTTCGA 3000 GGACTAAGGCCGCGAGACGCGTCTCCGCGCCCACGAGCAGAGGTGGTCGTGTTTCCGAGAAGTCCTTGTCGAGCAAGTGTTCCTAGACCTTGCAGGGCTTAGGGTCGTCCCCGTAAAGCT NcoI | CCCCGTTGTTCAGGGTTACTTTTCGCATGGCCCATTCAGGTTATGGCTCCAGCATGACTTTGATGGCCCGGCGTTCGTCCATGGCCCGGTAGCCCTCGGCCACTCGGTCCAGCGGCAGCG 3120 GGGGCAACAAGTCCCAATGAAAAGCGTACCGGGTAAGTCCAATACCGAGGTCGTACTGAAACTACCGGGCCGCAAGCAGGTACCGGGCCATCGGGAGCCGGTGAGCCAGGTCGCCGTCGC XhoI EcoRI | | TCAGGTCGAAGACCCGGCCCGGCTCGATGCGGCCCTCGAGCACGTCGGGCACAAGCTCCGGCATGTAGGCGCGCGTGGGGGCCGGCCCGCCGGCCACGATCCCCGGAATTCGTAATCATG 3240 AGTCCAGCTTCTGGGCCGGGCCGAGCTACGCCGGGAGCTCGTGCAGCCCGTGTTCGAGGCCGTACATCCGCGCGCACCCCCGGCCGGGCGGCCGGTGCTAGGGGCCTTAAGCATTAGTAC Enzym Schnittstelle Anzahl BamHI G^GATCC 2 EcoRI G^AATTC 2 HindIII A^AGCTT 1 KpnI GGTAC^C 1 NcoI C^CATGG 3 NdeI CA^TATG 0
Abb. 1: DNA-Sequenz der Thiamin-Generegion von R. marinus Dargestellt ist die DNA-Sequenz von R. marinus aus der Region der Gene thiS, thiG, thiE, thiD undorf5. Angegeben sind wichtige Restriktionsschnittstellen und Sequenzmerkmale, wie Transkriptions-startpunkte (+1) und Ribosomen-Bindungsstellen (rbs).
NotI GC^GGCCGC 0 PstI CTGCA^G 3 SalI G^TCGAC 1 SmaI CCC^GGG 1 XbaI T^CTAGA 0 XhoI C^TCGAG 1

Anhang IV
Rhodothermus marinus DSM 4252T
Natrium/Glucose-Symporter (SSSF) -35 σ70 -10 σ70 +1 -35 σ70 CAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCACACAGG 120 GTCACTCGCGTTGCGTTAATTACACTCAATCGAGTGAGTAATCCGTGGGGTCCGAAATGTGAAATACGAAGGCCGAGCATACAACACACCTTAACACTCGCCTATTGTTAAAGTGTGTCC BstXI SacI NdeI SalI PstI EcoRI -10 σ70 | | | | | | AAACAGCTATGACCATGATTACGCCAAGCTATTTAGGTGACACTATAGAATACTCAAGCTATGCATCCAACGCGTTGGGAGCTCTCCCATATGGTCGACCTGCAGGCGGCCGCGAATTCA 240 TTTGTCGATACTGGTACTAATGCGGTTCGATAAATCCACTGTGATATCTTATGAGTTCGATACGTAGGTTGCGCAACCCTCGAGAGGGTATACCAGCTGGACGTCCGCCGGCGCTTAAGT | NotI rbs CTAGTGATTGCGTAGGGCCAGTTGCGCTCGAACAGTGATCCGGTGTTTCCGTGGCTGGGCGTGATGATTTCCTCGCCGATCGTGGGCATCTGGTACTGGTGCACGGACCAGTACATTGTG 360 GATCACTAACGCATCCCGGTCAACGCGAGCTTGTCACTAGGCCACAAAGGCACCGACCCGCACTACTAAAGGAGCGGCTAGCACCCGTAGACCATGACCACGTGCCTGGTCATGTAACAC CAGCGGACGCTGGCGGCGCGTTCGCTCAAGGATGCACGTCGGGGCGCGCTCTGGGGTGGTTTTCTGAAGGTGTGGCCGGTGTTCATTTTTCTGGTGCCGGGGATGATCGGCTATGCGCTG 480 GTCGCCTGCGACCGCCGCGCAAGCGAGTTCCTACGTGCAGCCCCGCGCGAGACCCCACCAAAAGACTTCCACACCGGCCACAAGTAAAAAGACCACGGCCCCTACTAGCCGATACGCGAC CACCAGAAGGGGTTGATTCACATTCCGACGGACGCCACGGGGAAGATTCAGGGCGACATGGTCTTTCCGACGCTGGTGGCGGAGTTGTTGCCGCTGGGCCTGCGTGGGCTGGTCGTCGGC 600 GTGGTCTTCCCCAACTAAGTGTAAGGCTGCCTGCGGTGCCCCTTCTAAGTCCCGCTGTACCAGAAAGGCTGCGACCACCGCCTCAACAACGGCGACCCGGACGCACCCGACCAGCAGCCG HindIII | GGCCTGCTTTCGGCATTGATGTCGTCGCTTTCGTCGCTGTTCAATTCGTGCGCGACGCTTTTCACGGTGGACATTTACGAGAAGCTTCGTCCGGGTCGCCCGGAGCCGGAGCTGGTGCGT 720 CCGGACGAAAGCCGTAACTACAGCAGCGAAAGCAGCGACAAGTTAAGCACGCGCTGCGAAAAGTGCCACCTGTAAATGCTCTTCGAAGCAGGCCCAGCGGGCCTCGGCCTCGACCACGCA BspHI | GTGGGACGGATTGCGACGGCGGTGGTGGTGGGGCTGGGCTTGCTGTGGATTCCGATCATGAAGGTGATGGCCGAGTCGAAGGCGGGGTTGTACGACTATTTGCAGAACGTGCAGGGTTTT 840 CACCCTGCCTAACGCTGCCGCCACCACCACCCCGACCCGAACGACACCTAAGGCTAGTACTTCCACTACCGGCTCAGCTTCCGCCCCAACATGCTGATAAACGTCTTGCACGTCCCAAAA CTGGCGCCGCCGATCACGGCGGTATTTCTGCTGGGATTGGCCAGTCGTCGCATCAATGCGAAGGGTGCGGTGTGGGGGCTTGGTGTTGGTTTTGTGCTGGGGCTGTTGAAGTTGACGTTG 960 GACCGCGGCGGCTAGTGCCGCCATAAAGACGACCCTAACCGGTCAGCAGCGTAGTTACGCTTCCCACGCCACACCCCCGAACCACAACCAAAACACGACCCCGACAACTTCAACTGCAAC

BamHI | -35 σ70 -10 σ70 +1 CAGTCGATGGTGCAGAACGGGGCGCTGGATCCGAATACGCTCCTGGGCGCAATCGGTGCGTTTAATGGCTATTACTTCTCCGGACTGTTATTTTTCGTGAGCGTGGCGATTGTGGTGGGT 1080 GTCAGCTACCACGTCTTGCCCCGCGACCTAGGCTTATGCGAGGACCCGCGTTAGCCACGCAAATTACCGATAATGAAGAGGCCTGACAATAAAAAGCACTCGCACCGCTAACACCACCCA | XhoII BseRI | GTGTCGTTGCTGACGGAGGCGCCGCCGGAGGAGAAGGTTCGGGGGTTGACGTTTGGTACGGTGTCGGCGTCGGAC GTCGGAAGACGTGGGACTGGCGGGATGTGACGTTG 1200 CACAGCAACGACTGCCTCCGCGGCGGCCTCCTCTTCCAAGCCCCCAACTGCAAACCATGCCACAGCCGCAGCCTG CAGCCTTCTGCACCCTGACCGCCCTACACTGCAAC ACGGTGGTGGTGCTGGGGCTGGTGCTGGCGATTTATCTGTACTTCTCCTTCTGGGTGTAACCTGAAAGACATAAA TGCAGGAACAGAAGGCGACGGAGGCGATTCGGGAA 1320 TGCCACCACCACGACCCCGACCACGACCGCTAAATAGACATGAAGAGGAAGACCCACATTGGACTTTCTGTATTT ACGTCCTTGTCTTCCGCTGCCTCCGCTAAGCCCTT ACCGACTTTTCGCCGGGTGAACTGGACAATCTGTGCATCAATACGATTCGATTTCTGGCCGTCGATGCCGTCGAG CCGGCCATCCAGGCATGCCGATGGGCGCGGCGCCA 1440 TGGCTGAAAAGCGGCCCACTTGACCTGTTAGACACGTAGTTATGCTAAGCTAAAGACCGGCAGCTACGGCAGCTC GGCCGGTAGGTCCGTACGGCTACCCGCGCCGCGGT ATGGCCTACGTGCTCTGGACCCGGCAT 1467
Anhang V
TACCGGATGCACGAGACCTGGGCCGTA Enzym Schnittstelle Anzahl BamHI G^GATCC 1 BglII A^GATCT 0 BspHI T^CATGA 0 BstXI CCANNNNN^NTGG 1 BseRI GAGGAG 1 EcoRI G^AATTC 1 Eco47III AGC^GCT 0 HindIII A^AGCTT 1 KpnI GGTAC^C 0 NcoI C^CATGG 0 NdeI CA^TATG 1 Abb. 2: DNA-Sequenz der sssf-Regio arinus
Dargestellt ist die DNA-Sequenz von R us der Region von sssf RM und orf2. Angegebensind wichtige Restriktionsschnittstellen nzmerkmale, wie Transkriptionsstartpunkte (+1),
Ribosomen-Bindungsstellen (rbs) und motorregionen (σ70).
NotI GC^GGCCGC 1 PstI CTGCA^G 1 SacI GAGCT^C 1 SalI G^TCGAC 1 SmaI CCC^GGG 0 XbaI T^CTAGA 0 XhoII R^GATCY 1 XmaI C^CCGGG 0
CGGTCTGAGAGCCAGACTCT
rbs CCAGAAACCAGGTCTTTGGT
CAGGCGAAGTGTCCGCTTCA
n von R m. marinus a
und Seque
mögliche Pro

Halomonas elongata KB10 TCGACGATGCCAGCGACGTGGTGGTCGGCGCTCACATGGTGGGCGAGGAGGCCGGCGAGATCATCCAGGGCATCGCCATTGCCGTACGGGCCGGGCTGACCAAGGCACAGTTCGACCAGA 120
Anhang VI
AGCTGCTACGGTCGCTGCACCACCAGCCGCGAGTGTACCACCCGCTCCTCCGGCCGCTCTAGTAGGTCCCGTAGCGGTAACGGCATGCCCGGCCCGACTGGTTCCGTGTCAAGCTGGTCT EcoRI XhoI | | CGGTGGGCATTCATCCCACCGGCGCCGAAGAATTCGTGACCATGCGTACTCCGACACGCCGCTGACGACCTTCGGGCAATCCTGTCACGGCGATGTCATTCGGACGGGCCACCCTCGAGG 240 GCCACCCGTAAGTAGGGTGGCCGCGGCTTCTTAAGCACTGGTACGCATGAGGCTGTGCGGCGACTGCTGGAAGCCCGTTAGGACAGTGCCGCTACAGTAAGCCTGCCCGGTGGGAGCTCC TGGCCCGTTGCGTTATGCGTATGGCCCGAATGAATGTGGACTAAACTGGGTCTGTGCGAACGTCCTGCATTCGATGCCTTTTCGTACAAGACCTGCCGGGGGACATCAGCCGGTACGAGC 360 ACCGGGCAACGCAATACGCATACCGGGCTTACTTACACCTGATTTGACCCAGACACGCTTGCAGGACGTAAGCTACGGAAAAGCATGTTCTGGACGGCCCCCTGTAGTCGGCCATGCTCG EcoRI σS/σ70-Element | -35 σ70
GCCGGTGGTCGATGTCGCAAGAATGGGGAGAGCCGCTACATACGCGGCCTGGGGAGTGGGCTATAATTTTCTATTATGGAATTCAGCAAGCAAGATAACCTGGTTTTTGAAATGACCATA 480 CGGCCACCAGCTACAGCGTTCTTACCCCTCTCGGCGATGTATGCGCCGGACCCCTCACCCGATATTAAAAGATAATACCTTAAGTCGTTCGTTCTATTGGACCAAAAACTTTACTGGTAT gearbox –10 σ70 +1 rbs AGCGGCTGTTATGATGCCGATCAAATTCGCTACAGCGAACCACGACAATGAGCCTGGTCGTTTTTCCCTTCAAGCACGAACACCCCGAGGTGCTGCTGCACAACGTGCGCGTGGCGGCTG 600 TCGCCGACAATACTACGGCTAGTTTAAGCGATGTCGCTTGGTGCTGTTACTCGGACCAGCAAAAAGGGAAGTTCGTGCTTGTGGGGCTCCACGACGACGTGTTGCACGCGCACCGCCGAC SmaI | CTCATCCCCGGGTGCACGAGGTGCTCTGCATCGGATACGAGCGCGACCAGACCTACGAGGCCGTCGAACGGGCCGCGCCCGAAATCTCGCGGGCCACAGGGACGCCGGTGTCGGTCCGGT 720 GAGTAGGGGCCCACGTGCTCCACGAGACGTAGCCTATGCTCGCGCTGGTCTGGATGCTCCGGCAGCTTGCCCGGCGCGGGCTTTAGAGCGCCCGGTGTCCCTGCGGCCACAGCCAGGCCA TGCAGGAGCGACTGGGCACGCTGCGGCCCGGCAAGGGCGACGGCATGAACACCGCGCTCCGGTATTTCCTGGAAGAGACGCAGTGGGAGCGCATCCACTTCTACGATGCCGACATCACCA 840 ACGTCCTCGCTGACCCGTGCGACGCCGGGCCGTTCCCGCTGCCGTACTTGTGGCGCGAGGCCATAAAGGACCTTCTCTGCGTCACCCTCGCGTAGGTGAAGATGCTACGGCTGTAGTGGT GCTTCGGGCCGGACTGGATCACCAAGGCCGAAGAGGCGGCCGATTTCGGCTACGGGCTGGTGCGGCACTACTTCCCGCGGGCCTCAACCGACGCAATGATCACTTGGATGATCACGCGCA 960 CGAAGCCCGGCCTGACCTAGTGGTTCCGGCTTCTCCGCCGGCTAAAGCCGATGCCCGACCACGCCGTGATGAAGGGCGCCCGGAGTTGGCTGCGTTACTAGTGAACCTACTAGTGCGCGT CGGGCTTTGCCCTGCTCTGGCCGCATACCGAACTGTCGTGGATCGAGCAGCCGCTGGGCGGCGAGCTGCTCATGCGGCGCGAAGTGGCCGCCATGCTTTACGAGGATGAACGGGTGCGAC 1080 GCCCGAAACGGGACGAGACCGGCGTATGGCTTGACAGCACCTAGCTCGTCGGCGACCCGCCGCTCGACGAGTACGCCGCGCTTCACCGGCGGTACGAAATGCTCCTACTTGCCCACGCTG GGCGGAGCGACTGGGGCATCGACACACTCTACACGTTCGTTACCGTGCAGCAGGGCGTTTCGATCTACGAGTGCTACATTCCTGAGGGCAAGGCGCATCGCCTCTACGGCGGGCTCGACG 1200 CCGCCTCGCTGACCCCGTAGCTGTGTGAGATGTGCAAGCAATGGCACGTCGTCCCGCAAAGCTAGATGCTCACGATGTAAGGACTCCCGTTCCGCGTAGCGGAGATGCCGCCCGAGCTGC ACCTGCGCACCATGCTGGTCGAGTGTTTTGCCGCCATCCAGAGTCTGCAACATGAGGTCGTTGGCCAGCCGGCCATTCATCGCCAGGAGCACCCGCACCGCGTGCCGGTCCACATTGCCG 1320 TGGACGCGTGGTACGACCAGCTCACAAAACGGCGGTAGGTCTCAGACGTTGTACTCCAGCAACCGGTCGGCCGGTAAGTAGCGGTCCTCGTGGGCGTGGCGCACGGCCAGGTGTAACGGC

AACGCGTGGGCTACGACGTGGAGGCCACGCTGCATCGCCTCATGCAGCACTGGACGCCCCGGCAGGTGGAGCTGCTGGAGCTTTTTACCACACCCGTGCGCGAAGGACTGCGCACCTGTC 1440 TTGCGCACCCGATGCTGCACCTCCGGTGCGACGTAGCGGAGTACGTCGTGACCTGCGGGGCCGTCCACCTCGACGACCTCGAAAAATGGTGTGGGCACGCGCTTCCTGACGCGTGGACAG XhoI | AGCGTCGCCCGGCTTTCAACTTCATGGACGAGATGGCCTGGGCGGCCACCTATCACGTGCTGCTCGAGCACTTTCAGCCGGGCGATCCGGACTGGGAGGAGCTGCTCTTCAAACTCTGGA 1560 TCGCAGCGGGCCGAAAGTTGAAGTACCTGCTCTACCGGACCCGCCGGTGGATAGTGCACGACGAGCTCGTGAAAGTCGGCCCGCTAGGCCTGACCCTCCTCGACGAGAAGTTTGAGACCT CGACCCGCGTGCTCAACTACACGATGACGGTCGCGCTGCGGGGCTACGACTACGCGCAGCAGTATCTCTACCGGATGCTGGGACGCTACCGCTATCAGGCCGCTCTGGAAAACGGCCGTG 1680 GCTGGGCGCACGAGTTGATGTGCTACTGCCAGCGCGACGCCCCGATGCTGATGCGCGTCGTCATAGAGATGGCCTACGACCCTGCGATGGCGATAGTCCGGCGAGACCTTTTGCCGGCAC SalI | hp hp AT-reich rbs GTCATCCGGTCCCGCCGCGGGCGGCACTGTCGACCGCCTGAGCCGGGACGCCGCCTGGCCGGCCCGGTACGGGCCGGCAACCCGTCTTTTCGTTTTATCACTTTCCCCCCACAGGAGGTC 1800
Anhang VII
CAGTAGGCCAGGGCGGCGCCCGCCGTGACAGCTGGCGGACTCGGCCCTGCGGCGGACCGGCCGGGCCATGCCCGGCCGTTGGGCAGAAAAGCAAAATAGTGAAAGGGGGGTGTCCTCCAG GCAATGCAGACCCAGATTCTCGAACGCATGGAGTCCGACGTTCGGACCTACTCCCGCTCCTTCCCGGTCGTCTTCACCAAGGCGCGCAATGCCCGCCTGACCGACGAGGAAGGGCGCGAG 1920 CGTTACGTCTGGGTCTAAGAGCTTGCGTACCTCAGGCTGCAAGCCTGGATGAGGGCGAGGAAGGGCCAGCAGAAGTGGTTCCGCGCGTTACGGGCGGACTGGCTGCTCCTTCCCGCGCTC TACATCGACTTCCTGGCCGGTGCCGGCACCCTGAACTACGGCCACAACAACCCGCACCTCAAGCAGGCGCTGCTCGACTATATCGACAGCGACGGCATCGTCCACGGCCTGGACTTCTGG 2040 ATGTAGCTGAAGGACCGGCCACGGCCGTGGGACTTGATGCCGGTGTTGTTGGGCGTGGAGTTCGTCCGCGACGAGCTGATATAGCTGTCGCTGCCGTAGCAGGTGCCGGACCTGAAGACC ACTGCGGCCAAGCGCGACTATCTGGAAACCCTGGAAGAGGTGATCCTCAAGCCGCGCGGTCTCGACTACAAGGTGCATCTGCCCGGACCGACTGGCACCAACGCCGTCGAGGCGGCCATT 2160 TGACGCCGGTTCGCGCTGATAGACCTTTGGGACCTTCTCCACTAGGAGTTCGGCGCGCCAGAGCTGATGTTCCACGTAGACGGGCCTGGCTGACCGTGGTTGCGGCAGCTCCGCCGGTAA SmaI | CGCCTGGCCCGGGTCGCCAAGGGGCGCCACAATATCGTCTCCTTCACCAACGGCTTTCATGGCGTCACCATGGGCGCGCTGGCGACCACCGGTAACCGCAAGTTCCGCGAGGCCACCGGT 2280 GCGGACCGGGCCCAGCGGTTCCCCGCGGTGTTATAGCAGAGGAAGTGGTTGCCGAAAGTACCGCAGTGGTACCCGCGCGACCGCTGGTGGCCATTGGCGTTCAAGGCGCTCCGGTGGCCA GGCGTGCCGACCCAGGCTGCTTCCTTCATGCCGTTCGATGGCTACCTCGGCAGCAGCACCGACACCCTCGACTACTTCGAGAAGCTGCTCGGCGACAAGTCCGGCGGCCTGGACGTGCCC 2400 CCGCACGGCTGGGTCCGACGAAGGAAGTACGGCAAGCTACCGATGGAGCCGTCGTCGTGGCTGTGGGAGCTGATGAAGCTCTTCGACGAGCCGCTGTTCAGGCCGCCGGACCTGCACGGG XhoI | GCGGCGGTGATCGTCGAGACAGTGCAGGGCGAGGGCGGTATCAATGTCGCCGGCCTGGAGTGGCTCAAGCGCCTCGAGAGCATCTGCCGCGCCAATGACATCCTGCTGATCATCGACGAC 2520 CGCCGCCACTAGCAGCTCTGTCACGTCCCGCTCCCGCCATAGTTACAGCGGCCGGACCTCACCGAGTTCGCGGAGCTCTCGTAGACGGCGCGGTTACTGTAGGACGACTAGTAGCTGCTG ATCCAGGCGGGCTGCGGCCGGACCGGCAAGTTCTTCAGCTTCGAGCATGCCGGCATCACGCCGGATATCGTGACCAACTCCAAGTCGCTGTCCGGTTACGGCCTGCCGTTCGCTCACGTC 2640 TAGGTCCGCCCGACGCCGGCCTGGCCGTTCAAGAAGTCGAAGCTCGTACGGCCGTAGTGCGGCCTATAGCACTGGTTGAGGTTCAGCGACAGGCCAATGCCGGACGGCAAGCGAGTGCAG SacI | CTGATGCGCCCCGAGCTCGACAAGTGGAAGCCCGGTCAGTACAACGGCACCTTCCGCGGCTTCAACCTGGCTTTCGCCACTGCTGCTGCCGCCATGCGCAAGTACTGGAGCGACGACACC 2760 GACTACGCGGGGCTCGAGCTGTTCACCTTCGGGCCAGTCATGTTGCCGTGGAAGGCGCCGAAGTTGGACCGAAAGCGGTGACGACGACGGCGGTACGCGTTCATGACCTCGCTGCTGTGG TTCGAGCGTGACGTGCAGCGCAAGGCTCGCATCGTCGAGGAACGCTTCGGCAAGATCGCCGCCTGGCTGAGCGAGAACGGCATCGAGGCCTCCGAGCGCGGCCGCGGGCTGATGCGGGGC 2880 AAGCTCGCACTGCACGTCGCGTTCCGAGCGTAGCAGCTCCTTGCGAAGCCGTTCTAGCGGCGGACCGACTCGCTCTTGCCGTAGCTCCGGAGGCTCGCGCCGGCGCCCGACTACGCCCCG
Anhang VIII
ATCGACGTGGGTTCCGGCGATATCGCCGACAAGATCACCCACCAAGCCTTCGAGAACGGGTTGATCATCGAAACCAGCGGTCAGGACGGCGAAGTGGTCAAGTGCCTGTGCCCGCTGACC 3000 TAGCTGCACCCAAGGCCGCTATAGCGGCTGTTCTAGTGGGTGGTTCGGAAGCTCTTGCCCAACTAGTAGCTTTGGTCGCCAGTCCTGCCGCTTCACCAGTTCACGGACACGGGCGACTGG XhoI | -35 hp ATTCCCGACGAAGACCTGGTCGAGGGACTCGACATCCTCGAGACCAGCACCAAGCAGGCCTTTAGCTGATCGCCTGAGGTGCGCCATCGGGCCTGTCCATGGCATCCTGTATCGGTCGGC 3120 TAAGGGCTGCTTCTGGACCAGCTCCCTGAGCTGTAGGAGCTCTGGTCGTGGTTCGTCCGGAAATCGACTAGCGGACTCCACGCGGTAGCCCGGACAGGTACCGTAGGACATAGCCAGCCG hp –10 gearbox AT-reich rbs CGTGCGCGGCCGGCCAGTCATTGATTCACTGGAGAATCGACATGATCGTTCGCAATCTCGAAGAAGCGCGCCAGACCGACCGTCTGGTCACCGCCGAAAACGGCAACTGGGACAGCACCC 3240 GCACGCGCCGGCCGGTCAGTAACTAAGTGACCTCTTAGCTGTACTAGCAAGCGTTAGAGCTTCTTCGCGCGGTCTGGCTGGCAGACCAGTGGCGGCTTTTGCCGTTGACCCTGTCGTGGG KpnI | GCCTGTCGCTGGCCGAAGATGGTGGCAACTGCTCCTTCCACATCACCCGCATCTTCGAGGGTACCGAGACCCACATCCACTATAAGCATCACTTCGAGGCTGTTTATTGCATCGAAGGCG 3360 CGGACAGCGACCGGCTTCTACCACCGTTGACGAGGAAGGTGTAGTGGGCGTAGAAGCTCCCATGGCTCTGGGTGTAGGTGATATTCGTAGTGAAGCTCCGACAAATAACGTAGCTTCCGC BglII | AGGGCGAAGTGGAAACCCTGGCCGATGGCAAGATCTGGCCCATCAAGCCGGGTGACATCTACATCCTCGACCAGCACGACGAGCACCTGCTGCGCGCCAGCAAGACCATGCACCTGGCCT 3480 TCCCGCTTCACCTTTGGGACCGGCTACCGTTCTAGACCGGGTAGTTCGGCCCACTGTAGATGTAGGAGCTGGTCGTGCTGCTCGTGGACGACGCGCGGTCGTTCTGGTACGTGGACCGGA GCGTGTTCACGCCGGGCCTGACCGGCAACGAAGTGCACCGCGAAGACGGTTCCTACGCACCTGCCGACGAAGCCGACGACCAGAAGCCGCTGTAACCCGGCGCAGTATTCTGCCGTCTCG 3600 CGCACAAGTGCGGCCCGGACTGGCCGTTGCTTCACGTGGCGCTTCTGCCAAGGATGCGTGGACGGCTGCTTCGGCTGCTGGTCTTCGGCGACATTGGGCCGCGTCATAAGACGGCAGAGC hp AT-reich -35 σ70 -10 σ70 CACGAAGAGCCCCCGGTCACGATCGGGGGCTCTTTCGTTGTTCGCAGCGGCACGGGTGCCAGTAGCTGGACGCCTCGGACGTTTCATGCCTACTATTGCGTCATGATTGATTCATCGTCC 3720 GTGCTTCTCGGGGGCCAGTGCTAGCCCCCGAGAAAGCAACAAGCGTCGCCGTGCCCACGGTCATCGACCTGCGGAGCCTGCAAAGTACGGATGATAACGCAGTACTAACTAAGTAGCAGG rbs AGAAACCGTCAGCCCGTCGAGGAAGGGGCAGTCGAGGAAGGGGCATTGCCCAACGACCTGGTCAGCGAACTGCTGCTCGGCATGCGTCTAAGCGGCATCCAGTATCGCCGCATACAGGCG 3840 TCTTTGGCAGTCGGGCAGCTCCTTCCCCGTCAGCTCCTTCCCCGTAACGGGTTGCTGGACCAGTCGCTTGACGACGAGCCGTACGCAGATTCGCCGTAGGTCATAGCGGCGTATGTCCGC SmaI XhoI | | GTTCCTCCCTTCGGCATCGGGGGCTTCGGTGCCAGCCCGGGGTGGGCCCACTTTCACTTCATCGCGCGAGGGCCAGTATATCTGCGCAGTCCCGGCGGGGCCGTGCACCGGCTCGAGGTC 3960 CAAGGAGGGAAGCCGTAGCCCCCGAAGCCACGGTCGGGCCCCACCCGGGTGAAAGTGAAGTAGCGCGCTCCCGGTCATATAGACGCGTCAGGGCCGCCCCGGCACGTGGCCGAGCTCCAG GGTGACGCAGTGCTTCTGCCGCGCGGCGGACCGCATGAGCTGCTGTCGTCGCCGGAGCAATCCGCCAGTCGTGATATCGCCAGCTTCACGACCGCTCCGCTCTGCAAGGCCGTCAGTGCG 4080 CCACTGCGTCACGAAGACGGCGCGCCGCCTGGCGTACTCGACGACAGCAGCGGCCTCGTTAGGCGGTCAGCACTATAGCGGTCGAAGTGCTGGCGAGGCGAGACGTTCCGGCAGTCACGC GTGCGCAACGGTTCCCCAGAAGTTTGCCAGGAGAGTGGGGCCGTCATCTTCAGCGGCTGCATGGAGTTCGATCTCGGCGGCATGCATCCGCTCGTCGGCTTGATGCCCGAAGTGATGCGT 4200 CACGCGTTGCCAAGGGGTCTTCAAACGGTCCTCTCACCCCGGCAGTAGAAGTCGCCGACGTACCTCAAGCTAGAGCCGCCGTACGTAGGCGAGCAGCCGAACTACGGGCTTCACTACGCA GTCGATACGCTTCAGGATCGCCACCCGGAGATACTGCCGATCCTCCAGGCGATGGAACGCGAAGCGCAGGCCGAGCGGGCGGGCTTCGCCAGCATTCTGTCGCGACTCGCCGATGTCGTT 4320 CAGCTATGCGAAGTCCTAGCGGTGGGCCTCTATGACGGCTAGGAGGTCCGCTACCTTGCGCTTCGCGTCCGGCTCGCCCGCCCGAAGCGGTCGTAAGACAGCGCTGAGCGGCTACAGCAA

Anhang IX
TCCGCCTGCATCGTGCGTGGCTGGGTCGAGTGCGGCTGTGGCGACGCTTCCGGTTGGGTCGAGGCCTTGCGTGACCCACGGCTGGGACGTGTCATCGCCGCCATGCATCGGGAGCCCGGC 4440 AGGCGGACGTAGCACGCACCGACCCAGCTCACGCCGACACCGCTGCGAAGGCCAACCCAGCTCCGGAACGCACTGGGTGCCGACCCTGCACAGTAGCGGCGGTACGTAGCCCTCGGGCCG SacI | CGTCATTGGACGGTCGCTGAACTGGCGAAACAGATGGGGAGCTCGCGCTCGGTCTTCGCCGAGCGCTTCCTGACAGTCACCGGACTGACTCCGCATCGTTACATGACCGAGTTGCGCATG 4560 GCAGTAACCTGCCAGCGACTTGACCGCTTTGTCTACCCCTCGAGCGCGAGCCAGAAGCGGCTCGCGAAGGACTGTCAGTGGCCTGACTGAGGCGTAGCAATGTACTGGCTCAACGCGTAC CGGCTGGCCACCCAGTGGATCGGACGGGACAGGCAGCCCATCGATGCCGTCGCCCAACGCCTTGGTTATGGTTCCCAGGCCGCCTTCAGCCGTGCCTACAAGCGTGTGACCGGCCGTTCG 4680 GCCGACCGGTGGGTCACCTAGCCTGCCCTGTCCGTCGGGTAGCTACGGCAGCGGGTTGCGGAACCAATACCAAGGGTCCGGCGGAAGTCGGCACGGATGTTCGCACACTGGCCGGCAAGC hp CCCGGTGCGGTGCGTGCCGAAGCGAGTCGCGAGGGACTTCGGGCATGAGGACCTGAGCCCATTCAGCAGACGTGATATCCGCCCACAATGGAAAAAGGAGAGGTGCCGTTGCATGGAATA 4800 GGGCCACGCCACGCACGGCTTCGCTCAGCGCTCCCTGAAGCCCGTACTCCTGGACTCGGGTAAGTCGTCTGCACTATAGGCGGGTGTTACCTTTTTCCTCTCCACGGCAACGTACCTTAT GCTCCCTCGACAATGCACCATGCCCAGATGCTCTGGGACTTCATGAGTCTCCGGCAGCCTTGCACAGCGGCGGACGCCATTCTTTGCCTGGGCAGCAACGACATTCAAGTGCCGCGAGTT 4920 CGAGGGAGCTGTTACGTGGTACGGGTCTACGAGACCCTGAAGTACTCAGAGGCCGTCGGAACGTGTCGCCGCCTGCGGTAAGAAACGGACCCGTCGTTGCTGTAAGTTCACGGCGCTCAA GGGGCACGCCTATGGCGGGAGAAGTGGGCACCTTATCTGATCATGAGCGGAGGGTTGGCCCACCAGCATGACCTGGCAGCTACCGGCTGGTCGCAACCTGAGGCCGATGTATTTTCCCGA 5040 CCCCGTGCGGATACCGCCCTCTTCACCCGTGGAATAGACTAGTACTCGCCTCCCAACCGGGTGGTCGTACTGGACCGTCGATGGCCGACCAGCGTTGGACTCCGGCTACATAAAAGGGCT SmaI | GAAGCCCGCCATGCGGGCGTTCCCCGGGATGCCATATTATGTGAGCGTGCCGCGCGTCATACCGGCGACAATTTCCGCCTGACACGATGTCTGGCGGAGGAGAGCGGTATCATGGTCTCA 5160 CTTCGGGCGGTACGCCCGCAAGGGGCCCTACGGTATAATACACTCGCACGGCGCGCAGTATGGCCGCTGTTAAAGGCGGACTGTGCTACAGACCGCCTCCTCTCGCCATAGTACCAGAGT SalI | GGTCGACTCATCATCG 5176 CCAGCTGAGTAGTAGC Enzym Schnittstelle Anzahl BamHI G^GATCC 0 BglII A^GATCT 1 BspHI T^CATGA 1 BstXI CCANNNNN^NTGG 1 Eco47III AGC^GCT 0 EcoRI G^AATTC 2 HindIII A^AGCTT 0 KpnI GGTAC^C 1 NcoI C^CATGG 5 NdeI CA^TATG 0 Abb. 3: DNA-Sequenz der mgsectBC-Region von H elongata KB10
Dargestellt ist die DNA-Sequenz von H. elongata KB10 aus der Region von mgs, ectB, ectC undectR. Angegeben sind wichtige Restriktionsschnittstellen und Sequenzmerkmale, wie Transkripti-onsstartpunkte (+1), Ribosomen-Bindungsstellen (rbs), Promotorregionen (σS/ σ70 und gearbox-Element), palindromische Bereiche möglicher Terminationsschleifen (hp) und die darauf folgendeAT-reiche Region.
NotI GC^GGCCGC 0 PstI CTGCA^G 0 SacI GAGCT^C 2 SalI G^TCGAC 2 SmaI CCC^GGG 4 XbaI T^CTAGA 0 XhoI C^TCGAG 5

Anhang X
Halomonas elongata KB10.1 TCGACGATGCCAGCGACGTGGTGGTCGGCGCTCACATGGTGGGCGAGGAGGCCGGCGAGATCATCCAGGGCATCGCCATTGCCGTACGGGCCGGGCTGACCAAGGCACAGTTCGACCAGA 120 AGCTGCTACGGTCGCTGCACCACCAGCCGCGAGTGTACCACCCGCTCCTCCGGCCGCTCTAGTAGGTCCCGTAGCGGTAACGGCATGCCCGGCCCGACTGGTTCCGTGTCAAGCTGGTCT EcoRI XhoI | | CGGTGGGCATTCATCCCACCGGCGCCGAAGAATTCGTGACCATGCGTACTCCGACACGCCGCTGACGACCTTCGGGCAATCCTGTCACGGCGATGTCATTCGGACGGGCCACCCTCGAGG 240 GCCACCCGTAAGTAGGGTGGCCGCGGCTTCTTAAGCACTGGTACGCATGAGGCTGTGCGGCGACTGCTGGAAGCCCGTTAGGACAGTGCCGCTACAGTAAGCCTGCCCGGTGGGAGCTCC TGGCCCGTTGCGTTATGCGTATGGCCCGAATGAATGTGGACTAAACTGGGTCTGTGCGAACGTCCTGCATTCGATGCCTTTTCGTACAAGACCTGCCGGGGGACATCAGCCGGTACGAGC 360 ACCGGGCAACGCAATACGCATACCGGGCTTACTTACACCTGATTTGACCCAGACACGCTTGCAGGACGTAAGCTACGGAAAAGCATGTTCTGGACGGCCCCCTGTAGTCGGCCATGCTCG EcoRI σS/σ70-Element | -35 σ70 GCCGGTGGTCGATGTCGCAAGAATGGGGAGAGCCGCTACATACGCGGCCTGGGGAGTGGGCTATAATTTTCTATTATGGAATTCAGCAAGCAAGATAACCTGGTTTTTGAAATGACCATA 480 CGGCCACCAGCTACAGCGTTCTTACCCCTCTCGGCGATGTATGCGCCGGACCCCTCACCCGATATTAAAAGATAATACCTTAAGTCGTTCGTTCTATTGGACCAAAAACTTTACTGGTAT gearbox –10 σ70 +1 rbs AGCGGCTGTTATGATGCCGATCAAATTCGCTACAGCGAACCACGACAATGAGCCTGGTCGTTTTTCCCTTCAAGCACGAACACCCCGAGGTGCTGCTGCACAACGTGCGCGTGGCGGCTG 600 TCGCCGACAATACTACGGCTAGTTTAAGCGATGTCGCTTGGTGCTGTTACTCGGACCAGCAAAAAGGGAAGTTCGTGCTTGTGGGGCTCCACGACGACGTGTTGCACGCGCACCGCCGAC SmaI | CTCATCCCCGGGTGCACGAGGTGCTCTGCATCGGATACGAGCGCGACCAGACCTACGAGGCCGTCGAACGGGCCGCGCCCGAAATCTCGCGGGCCACAGGGACGCCGGTGTCGGTCCGGT 720 GAGTAGGGGCCCACGTGCTCCACGAGACGTAGCCTATGCTCGCGCTGGTCTGGATGCTCCGGCAGCTTGCCCGGCGCGGGCTTTAGAGCGCCCGGTGTCCCTGCGGCCACAGCCAGGCCA TGCAGGAGCGACTGGGCACGCTGCGGCCCGGCAAGGGCGACGGCATGAACACCGCGCTCCGGTATTTCCTGGAAGAGACGCAGTGGGAGCGCATCCACTTCTACGATGCCGACATCACCA 840 ACGTCCTCGCTGACCCGTGCGACGCCGGGCCGTTCCCGCTGCCGTACTTGTGGCGCGAGGCCATAAAGGACCTTCTCTGCGTCACCCTCGCGTAGGTGAAGATGCTACGGCTGTAGTGGT GCTTCGGGCCGGACTGGATCACCAAGGCCGAAGAGGCGGCCGATTTCGGCTACGGGCTGGTGCGGCACTACTTCCCGCGGGCCTCAACCGACGCAATGATCACTTGGATGATCACGCGCA 960 CGAAGCCCGGCCTGACCTAGTGGTTCCGGCTTCTCCGCCGGCTAAAGCCGATGCCCGACCACGCCGTGATGAAGGGCGCCCGGAGTTGGCTGCGTTACTAGTGAACCTACTAGTGCGCGT CGGGCTTTGCCCTGCTCTGGCCGCATACCGAACTGTCGTGGATCGAGCAGCCGCTGGGCGGCGAGCTGCTCATGCGGCGCGAAGTGGCCGCCATGCTTTACGAGGATGAACGGGTGCGAC 1080 GCCCGAAACGGGACGAGACCGGCGTATGGCTTGACAGCACCTAGCTCGTCGGCGACCCGCCGCTCGACGAGTACGCCGCGCTTCACCGGCGGTACGAAATGCTCCTACTTGCCCACGCTG GGCGGAGCGACTGGGGCATCGACACACTCTACACGTTCGTTACCGTGCAGCAGGGCGTTTCGATCTACGAGTGCTACATTCCTGAGGGCAAGGCGCATCGCCTCTACGGCGGGCTCGACG 1200 CCGCCTCGCTGACCCCGTAGCTGTGTGAGATGTGCAAGCAATGGCACGTCGTCCCGCAAAGCTAGATGCTCACGATGTAAGGACTCCCGTTCCGCGTAGCGGAGATGCCGCCCGAGCTGC ACCTGCGCACCATGCTGGTCGAGTGTTTTGCCGCCATCCAGAGTCTGCAACATGAGGTCGTTGGCCAGCCGGCCATTCATCGCCAGGAGCACCCGCACCGCGTGCCGGTCCACATTGCCG 1320 TGGACGCGTGGTACGACCAGCTCACAAAACGGCGGTAGGTCTCAGACGTTGTACTCCAGCAACCGGTCGGCCGGTAAGTAGCGGTCCTCGTGGGCGTGGCGCACGGCCAGGTGTAACGGC

Anhang XI
AACGCGTGGGCTACGACGTGGAGGCCACGCTGCATCGCCTCATGCAGCACTGGACGCCCCGGCAGGTGGAGCTGCTGGAGCTTTTTACCACACCCGTGCGCGAAGGACTGCGCACCTGTC 1440 TTGCGCACCCGATGCTGCACCTCCGGTGCGACGTAGCGGAGTACGTCGTGACCTGCGGGGCCGTCCACCTCGACGACCTCGAAAAATGGTGTGGGCACGCGCTTCCTGACGCGTGGACAG XhoI | AGCGTCGCCCGGCTTTCAACTTCATGGACGAGATGGCCTGGGCGGCCACCTATCACGTGCTGCTCGAGCACTTTCAGCCGGGCGATCCGGACTGGGAGGAGCTGCTCTTCAAACTCTGGA 1560 TCGCAGCGGGCCGAAAGTTGAAGTACCTGCTCTACCGGACCCGCCGGTGGATAGTGCACGACGAGCTCGTGAAAGTCGGCCCGCTAGGCCTGACCCTCCTCGACGAGAAGTTTGAGACCT CGACCCGCGTGCTCAACTACACGATGACGGTCGCGCTGCGGGGCTACGACTACGCGCAGCAGTATCTCTACCGGATGCTGGGACGCTACCGCTATCAGGCCGCTCTGGAAAACGGCCGTG 1680 GCTGGGCGCACGAGTTGATGTGCTACTGCCAGCGCGACGCCCCGATGCTGATGCGCGTCGTCATAGAGATGGCCTACGACCCTGCGATGGCGATAGTCCGGCGAGACCTTTTGCCGGCAC SalI | hp hp AT-reich rbs GTCATCCGGTCCCGCCGCGGGCGGCACTGTCGACCGCCTGAGCCGGGACGCCGCCTGGCCGGCCCGGTACGGGCCGGCAACCCGTCTTTTCGTTTTATCACTTTCCCCCCACAGGAGGTC 1800 CAGTAGGCCAGGGCGGCGCCCGCCGTGACAGCTGGCGGACTCGGCCCTGCGGCGGACCGGCCGGGCCATGCCCGGCCGTTGGGCAGAAAAGCAAAATAGTGAAAGGGGGGTGTCCTCCAG NdeI BspHI | | CATATGGAATTGATCCCGGTAATTTTATCCGGTGGCGTTGGTAGCCGTCTGTGGCCAGTATCACGTGAGGCTCATCCCAAGCCGTTCATGACCCTGCCAGACGGTCAGAACCTCATCCAG 1920 GTATACCTTAACTAGGGCCATTAAAATAGGCCACCGCAACCATCGGCAGACACCGGTCATAGTGCACTCCGAGTAGGGTTCGGCAAGTACTGGGACGGTCTGCCAGTCTTGGAGTAGGTC AAGACTTTCCTGCGAGCCGCCGACCTGAACGGTGTGGTTGAAATCCTTACGGTCACCAACCGCGAATTGCTGTTCAAGACCGAGGACGAATATCGCACCATCAACAAGCAGAACCTCAGC 2040 TTCTGAAAGGACGCTCGGCGGCTGGACTTGCCACACCAACTTTAGGAATGCCAGTGGTTGGCGCTTAACGACAAGTTCTGGCTCCTGCTTATAGCGTGGTAGTTGTTCGTCTTGGAGTCG CAGGGTTACATTCTCGAACCCTTCGGTCGCAATACCGCCGCAGCCGTGGCCGCTGCCGCATTGCAGCTTCTGGAGTCCCACGGCCCTCAAGTGCACATGCTCGTGCTGGCGGCCGATCAT 2160 GTCCCAATGTAAGAGCTTGGGAAGCCAGCGTTATGGCGGCGTCGGCACCGGCGACGGCGTAACGTCGAAGACCTCAGGGTGCCGGGAGTTCACGTGTACGAGCACGACCGCCGGCTAGTA CTGATCCAGAACGAAGCGGCTTTTTCCGAAGCAGTCAGCAAGGCCGTTCAGCTGGCAGGCGAGGGTTGGCTGGTCACATTCGGCATTAAACCCCAATACCCTGAAACCGGCTTTGGCTAC 2280 GACTAGGTCTTGCTTCGCCGAAAAAGGCTTCGTCAGTCGTTCCGGCAAGTCGACCGTCCGCTCCCAACCGACCAGTGTAAGCCGTAATTTGGGGTTATGGGACTTTGGCCGAAACCGATG ATCGAGGCTGCGGCTGGCGGTGTGCTCGAAGGTGGGCTAAGAGTCGAGCGTTTCGTCGAAAAACCCGACGCCAAAACCGCTGAGGCCTATGTCGCTGCTGGCAACTATTTCTGGAACGCC 2400 TAGCTCCGACGCCGACCGCCACACGAGCTTCCACCCGATTCTCAGCTCGCAAAGCAGCTTTTTGGGCTGCGGTTTTGGCGACTCCGGATACAGCGACGACCGTTGATAAAGACCTTGCGG GGCATGTTCTGCTTCCAGGTTGGCACTGTGATCGAGCAGTTCAGGGCTTACGCTCCTGATGTGCTTGAAGCTGTCGAGCGTACCCTCGAAGCCTCGCGCCGCTCTACCTCCAAAGGCTAT 2520 CCGTACAAGACGAAGGTCCAACCGTGACACTAGCTCGTCAAGTCCCGAATGCGAGGACTACACGAACTTCGACAGCTCGCATGGGAGCTTCGGAGCGCGGCGAGATGGAGGTTTCCGATA BspXI XhoI | | AGCTGCCTGGCTCTGGATGCAGAGTGCTTCGCCAGCGTGCCGGACATCTCCATCGATTACGCCTTGATGGAACGCTCGAGTAAAGTGGCGACCATTCCGTGCGATATCGGGTGGAGCGAT 2640 TCGACGGACCGAGACCTACGTCTCACGAAGCGGTCGCACGGCCTGTAGAGGTAGCTAATGCGGAACTACCTTGCGAGCTCATTTCACCGCTGGTAAGGCACGCTATAGCCCACCTCGCTA ATTGGCTCGTGGAATGCTGTCAGCGAACTGACCCTGCCGGATGAGCATGGCAACCGCTTCGACGGCGAAGTGATGGCTTACGGGGCGAGCAACAACTATGTCAGCACCGAAGACCGCCTG 2760 TAACCGAGCACCTTACGACAGTCGCTTGACTGGGACGGCCTACTCGTACCGTTGGCGAAGCTGCCGCTTCACTACCGAATGCCCCGCTCGTTGTTGATACAGTCGTGGCTTCTGGCGGAC GCTGCGCTGGTAGGTGTTCAGGACTTGCTGGTGGTAGACACGCCGGATGCACTGCTGATTGCTCATAAAGACCACGCTCAGGACGTGAAACATATCGTCAAGCGCTTGAAGAATGATGGT 2880 CGACGCGACCATCCACAAGTCCTGAACGACCACCATCTGTGCGGCCTACGTGACGACTAACGAGTATTTCTGGTGCGAGTCCTGCACTTTGTATAGCAGTTCGCGAACTTCTTACTACCA CACACCGCCCATTTACTGCATCAAACAGTGCACCGCCCCTGGGGTACTTACACCACCTTGGAAGATGGTGAGCGCTTCAAGATCAAACGCATTGTGGTCAAGCCCAAGGCGTCGCTGTCC 3000 GTGTGGCGGGTAAATGACGTAGTTTGTCACGTGGCGGGGACCCCATGAATGTGGTGGAACCTTCTACCACTCGCGAAGTTCTAGTTTGCGTAACACCAGTTCGGGTTCCGCAGCGACAGG

Anhang XII
PstI | CTGCAGATGCATCACCACCGCAGTGAGCACTGGATTGTGGTGAGCGGTATGGCCGTGGTGGTCAATGATGATCAGGAACTGATGCTCAACACGAACGAATCCACGTTCATTCGGGCGGGT 3120 GACGTCTACGTAGTGGTGGCGTCACTCGTGACCTAACACCACTCGCCATACCGGCACCACCAGTTACTACTAGTCCTTGACTACGAGTTGTGCTTGCTTAGGTGCAAGTAAGCCCGCCCA SmaI | CACAAGCACCGTTTGCAGAACCCGGGTGTAATCGACCTAGTGCTGATCGAAGTTCAAAGCGGCGACTACCTGGGCGAAGACGATATCGTTCGCTTCGAGGATAACTACGGTCGTTGCGAC 3240 GTGTTCGTGGCAAACGTCTTGGGCCCACATTAGCTGGATCACGACTAGCTTCAAGTTTCGCCGCTGATGGACCCGCTTCTGCTATAGCAAGCGAAGCTCCTATTGATGCCAGCAACGCTG SalI | hp hp AT-reich GCCTGATCCAGCCTCTCCAGTCGACCCCGTGGGCCTGATCCAAGCGCGGGGTCGATTTCGCTCGGCAAGCCTGTCGCCGACTTCCCCTTTTTCGAATGGTGTTCGCTCAAGCGCCGCCGT 3360 CGGACTAGGTCGGAGAGGTCAGCTGGGGCACCCGGACTAGGTTCGCGCCCCAGCTAAAGCGAGCCGTTCGGACAGCGGCTGAAGGGGAAAAAGCTTACCACAAGCGAGTTCGCGGCGGCA ATACAAGTGAGATGTATGTACCCACCCATTCAGACCCGCTGCTTCAAAGCCTATGACATTCGCGGCCAAGTGCCGGCTGATCTCAATGAGGATATTGCCTATCGTATTGGTCGTGCCTTG 3480 TATGTTCACTCTACATACATGGGTGGGTAAGTCTGGGCGACGAAGTTTCGGATACTGTAAGCGCCGGTTCACGGCCGACTAGAGTTACTCCTATAACGGATAGCATAACCAGCACGGAAC GTGGCTGAGCTGGGTGGGCGCAGCTACGTGGTAGGTCGTGACATGCGCTTGGAAAGTCCGACTCTTTCCCAAGCATTGATGAGGGGCCTTACTGAAGGCGGCGCCGACGTTATAGATATT 3600 CACCGACTCGACCCACCCGCGTCGATGCACCATCCAGCACTGTACGCGAACCTTTCAGGCTGAGAAAGGGTTCGTAACTACTCCCCGGAATGACTTCCGCCGCGGCTGCAATATCTATAA GGCCTTTGTGGCACCGAGGAAGTGTACTTCGCTACCAGCCACTATCAGGCCGACGGTGGCATCATGGTAACCGCCAGCCATAATCCCAAAGGCTATAACGGCATGAAACTGGTGCGCGAG 3720 CCGGAAACACCGTGGCTCCTTCACATGAAGCGATGGTCGGTGATAGTCCGGCTGCCACCGTAGTACCATTGGCGGTCGGTATTAGGGTTTCCGATATTGCCGTACTTTGACCACGCGCTC CAGTCGCGACCTATCAGTGGTGACACGGGCCTCAAGGATATTCGCCAGCGGGTGGAATCCGGTGATTTGGGGACCGCAGCCCCGGCCGCAGGCAGTGTTCGCCTGGCGTCCGACAAAAGC 3840 GTCAGCGCTGGATAGTCACCACTGTGCCCGGAGTTCCTATAAGCGGTCGCCCACCTTAGGCCACTAAACCCCTGGCGTCGGGGCCGGCGTCCGTCACAAGCGGACCGCAGGCTGTTTTCG BamHI | GCTTACATCGACCACCTGCTGACCTATGTGGACCTGGCTACCCTCAAGCCATTGAAAGTTCTGGCGGATCCGGGTAATGGTGCTGTCGGCCCGGTGCTGGAGTTGTTGAAGGCGCGCCTG 3960 CGAATGTAGCTGGTGGACGACTGGATACACCTGGACCGATGGGAGTTCGGTAACTTTCAAGACCGCCTAGGCCCATTACCACGACAGCCGGGCCACGACCTCAACAACTTCCGCGCGGAC CCACTGAAGCTCACTGTTATCAATGGGGAACCAGATGGCAACTTCCCTAATGGCATCCCCAACCCGCTGCTACCGGAAAACCGTGACCTTACACGCCAAGCTGTCCTGGATACCGGTGCA 4080 GGTGACTTCGAGTGACAATAGTTACCCCTTGGTCTACCGTTGAAGGGATTACCGTAGGGGTTGGGCGACGATGGCCTTTTGGCACTGGAATGTGCGGTTCGACAGGACCTATGGCCACGT GACATGGGTATTGCCTTCGATGGTGATTTCGATCGCTGCTTCTTCTTCGACAATCAGGGGCGTTTCATCGAGGGCTACTATGTAGTTGGCCTGTTGGCTGAAATGCTGCTGGCCAAGCAC 4200 CTGTACCCATAACGGAAGCTACCACTAAAGCTAGCGACGAAGAAGAAGCTGTTAGTCCCCGCAAAGTAGCTCCCGATGATACATCAACCGGACAACCGACTTTACGACGACCGGTTCGTG CCAGGCAGCAAAATCATTCACGACCCGCGCCTGACCTGGAACACCATCAAACAGGTCGAAGAAGCAGGGGGAATGGCAATCCAGAGCAAGACCGGCCATGCCTTCATAAAAGAGCGTATG 4320 GGTCCGTCGTTTTAGTAAGTGCTGGGCGCGGACTGGACCTTGTGGTAGTTTGTCCAGCTTCTTCGTCCCCCTTACCGTTAGGTCTCGTTCTGGCCGGTACGGAAGTATTTTCTCGCATAC CGCGCCGAAGATGCTGTATATGGCGGCGAAATGAGCGCCCACCATTACTTCCGTGACTTTGCCTACTGCGACAGCGGTAACATCCCTTGGCTCCTGGTCGCTCAGTTGATGAGCGTAACG 4440 GCGCGGCTTCTACGACATATACCGCCGCTTTACTCGCGGGTGGTAATGAAGGCACTGAAACGGATGACGCTGTCGCCATTGTAGGGAACCGAGGACCAGCGAGTCAACTACTCGCATTGC GGCAAAAGCCTGGGGCAACTGGTGGACGAACGTATTGCAGCGTTCCCGTGCAGTGGCGAAATCAACTACGAAGTGGAGGATGTTAAAGACACTATCGAGAAGATTCTTGCTCACTACTTG 4560 CCGTTTTCGGACCCCGTTGACCACCTGCTTGCATAACGTCGCAAGGGCACGTCACCGCTTTAGTTGATGCTTCACCTCCTACAATTTCTGTGATAGCTCTTCTAAGAACGAGTGATGAAC CCGCGAAACCCCAGCATCGACCGCACCGACGGTATCAGCGTAGAGTTCGCTGACTGGCGTTTCAGCCTGCGTGGGTCCAACACCGAGCCATTGCTGCGCCTCAATGTCGAGAGCCGAGGC 4680 GGCGCTTTGGGGTCGTAGCTGGCGTGGCTGCCATAGTCGCATCTCAAGCGACTGACCGCAAAGTCGGACGCACCCAGGTTGTGGCTCGGTAACGACGCGGAGTTACAGCTCTCGGCTCCG

HindIII | hp AT-reich GATCAGGCTTTGGTTGAGCGCCAGGTTGCACAAATCGCCGGCTTAATCAGAGGTTAAAAGCTTGCAGTATTCTGCCGTCTCGCACGAAGAGCCCCCGGTCACGATCGGGGGCTCTTTCGT 4800 CTAGTCCGAAACCAACTCGCGGTCCAACGTGTTTAGCGGCCGAATTAGTCTCCAATTTTCGAACGTCATAAGACGGCAGAGCGTGCTTCTCGGGGGCCAGTGCTAGCCCCCGAGAAAGCA BspHI -35 σ70 -10 σ70 | rbs
Anhang XIII
TGTTCGCAGCGGCACGGGTGCCAGTAGCTGGACGCCTCGGACGTTTCATGCCTACTATTGCGTCATGATTGATTCATCGTCCAGAAACCGTCAGCCCGTCGAGGAAGGGGCAGTCGAGGA 4920 ACAAGCGTCGCCGTGCCCACGGTCATCGACCTGCGGAGCCTGCAAAGTACGGATGATAACGCAGTACTAACTAAGTAGCAGGTCTTTGGCAGTCGGGCAGCTCCTTCCCCGTCAGCTCCT SmaI | AGGGGCATTGCCCAACGACCTGGTCAGCGAACTGCTGCTCGGCATGCGTCTAAGCGGCATCCAGTATCGCCGCATACAGGCGGTTCCTCCCTTCGGCATCGGGGGCTTCGGTGCCAGCCC 5040 TCCCCGTAACGGGTTGCTGGACCAGTCGCTTGACGACGAGCCGTACGCAGATTCGCCGTAGGTCATAGCGGCGTATGTCCGCCAAGGAGGGAAGCCGTAGCCCCCGAAGCCACGGTCGGG XhoI | GGGGTGGGCCCACTTTCACTTCATCGCGCGAGGGCCAGTATATCTGCGCAGTCCCGGCGGGGCCGTGCACCGGCTCGAGGTCGGTGACGCAGTGCTTCTGCCGCGCGGCGGACCGCATGA 5160 CCCCACCCGGGTGAAAGTGAAGTAGCGCGCTCCCGGTCATATAGACGCGTCAGGGCCGCCCCGGCACGTGGCCGAGCTCCAGCCACTGCGTCACGAAGACGGCGCGCCGCCTGGCGTACT GCTGCTGTCGTCGCCGGAGCAATCCGCCAGTCGTGATATCGCCAGCTTCACGACCGCTCCGCTCTGCAAGGCCGTCAGTGCGGTGCGCAACGGTTCCCCAGAAGTTTGCCAGGAGAGTGG 5280 CGACGACAGCAGCGGCCTCGTTAGGCGGTCAGCACTATAGCGGTCGAAGTGCTGGCGAGGCGAGACGTTCCGGCAGTCACGCCACGCGTTGCCAAGGGGTCTTCAAACGGTCCTCTCACC GGCCGTCATCTTCAGCGGCTGCATGGAGTTCGATCTCGGCGGCATGCATCCGCTCGTCGGCTTGATGCCCGAAGTGATGCGTGTCGATACGCTTCAGGATCGCCACCCGGAGATACTGCC 5400 CCGGCAGTAGAAGTCGCCGACGTACCTCAAGCTAGAGCCGCCGTACGTAGGCGAGCAGCCGAACTACGGGCTTCACTACGCACAGCTATGCGAAGTCCTAGCGGTGGGCCTCTATGACGG GATCCTCCAGGCGATGGAACGCGAAGCGCAGGCCGAGCGGGCGGGCTTCGCCAGCATTCTGTCGCGACTCGCCGATGTCGTTTCCGCCTGCATCGTGCGTGGCTGGGTCGAGTGCGGCTG 5520 CTAGGAGGTCCGCTACCTTGCGCTTCGCGTCCGGCTCGCCCGCCCGAAGCGGTCGTAAGACAGCGCTGAGCGGCTACAGCAAAGGCGGACGTAGCACGCACCGACCCAGCTCACGCCGAC TGGCGACGCTTCCGGTTGGGTCGAGGCCTTGCGTGACCCACGGCTGGGACGTGTCATCGCCGCCATGCATCGGGAGCCCGGCCGTCATTGGACGGTCGCTGAACTGGCGAAACAGATGGG 5640 ACCGCTGCGAAGGCCAACCCAGCTCCGGAACGCACTGGGTGCCGACCCTGCACAGTAGCGGCGGTACGTAGCCCTCGGGCCGGCAGTAACCTGCCAGCGACTTGACCGCTTTGTCTACCC SacI | GAGCTCGCGCTCGGTCTTCGCCGAGCGCTTCCTGACAGTCACCGGACTGACTCCGCATCGTTACATGACCGAGTTGCGCATGCGGCTGGCCACCCAGTGGATCGGACGGGACAGGCAGCC 5760 CTCGAGCGCGAGCCAGAAGCGGCTCGCGAAGGACTGTCAGTGGCCTGACTGAGGCGTAGCAATGTACTGGCTCAACGCGTACGCCGACCGGTGGGTCACCTAGCCTGCCCTGTCCGTCGG BspXI | CATCGATGCCGTCGCCCAACGCCTTGGTTATGGTTCCCAGGCCGCCTTCAGCCGTGCCTACAAGCGTGTGACCGGCCGTTCGCCCGGTGCGGTGCGTGCCGAAGCGAGTCGCGAGGGACT 5880 GTAGCTACGGCAGCGGGTTGCGGAACCAATACCAAGGGTCCGGCGGAAGTCGGCACGGATGTTCGCACACTGGCCGGCAAGCGGGCCACGCCACGCACGGCTTCGCTCAGCGCTCCCTGA TCGGGCATGAGGACCTGAGCCCATTCAGCAGACGTGATATCCGCCCACAATGGAAAAAGGAGAGGTGCCGTTGCATGGAATAGCTCCCTCGACAATGCACCATGCCCAGATGCTCTGGGA 6000 AGCCCGTACTCCTGGACTCGGGTAAGTCGTCTGCACTATAGGCGGGTGTTACCTTTTTCCTCTCCACGGCAACGTACCTTATCGAGGGAGCTGTTACGTGGTACGGGTCTACGAGACCCT

Anhang XIV
BspHI | CTTCATGAGTCTCCGGCAGCCTTGCACAGCGGCGGACGCCATTCTTTGCCTGGGCAGCAACGACATTCAAGTGCCGCGAGTTGGGGCACGCCTATGGCGGGAGAAGTGGGCACCTTATCT 6120 GAAGTACTCAGAGGCCGTCGGAACGTGTCGCCGCCTGCGGTAAGAAACGGACCCGTCGTTGCTGTAAGTTCACGGCGCTCAACCCCGTGCGGATACCGCCCTCTTCACCCGTGGAATAGA BspHI SmaI | | GATCATGAGCGGAGGGTTGGCCCACCAGCATGACCTGGCAGCTACCGGCTGGTCGCAACCTGAGGCCGATGTATTTTCCCGAGAAGCCCGCCATGCGGGCGTTCCCCGGGATGCCATATT 6240 CTAGTACTCGCCTCCCAACCGGGTGGTCGTACTGGACCGTCGATGGCCGACCAGCGTTGGACTCCGGCTACATAAAAGGGCTCTTCGGGCGGTACGCCCGCAAGGGGCCCTACGGTATAA SalI | ATGTGAGCGTGCCGCGCGTCATACCGGCGACAATTTCCGCCTGACACGATGTCTGGCGGAGGAGAGCGGTATCATGGTCTCAGGTCGACTCATCATCG 6338 TACACTCGCACGGCGCGCAGTATGGCCGCTGTTAAAGGCGGACTGTGCTACAGACCGCCTCCTCTCGCCATAGTACCAGAGTCCAGCTGAGTAGTAGC Enzym Schnittstelle Anzahl BamHI G^GATCC 1 BglII A^GATCT 0 BspHI T^CATGA 4 BspXI AT^CGAT 2 Eco47III AGC^GCT 0 EcoRI G^AATTC 1 HindIII A^AGCTT 1 KpnI GGTAC^C 0 NcoI C^CATGG 0 NdeI CA^TATG 1 Abb. 4: DNA-Sequenz der mgsPP1776xanA-Region von H. elongata KB10.1
Dargestellt ist die DNA-Sequenz von H. elongata KB10.1 aus der Region von mgs, PP1776, xanAund ectR. Angegeben sind wichtige Restriktionsschnittstellen und Sequenzmerkmale, wie Trans-kriptionsstartpunkte (+1), Ribosomen-Bindungsstellen (rbs), Promotorregionen (σS/σ70 und gearbox-Element), palindromische Bereiche möglicher Terminationsschleifen (hp) und die darauf folgendeAT-reiche Region.
NotI GC^GGCCGC 0 PstI CTGCA^G 1 SacI GAGCT^C 1 SalI G^TCGAC 3 SmaI CCC^GGG 4 XbaI T^CTAGA 1 XhoI C^TCGAG 4

Danksagung
• Ich danke Herrn Prof. Dr. Erwin A. Galinski für sein reges Interesse am Fortgang dieser
Arbeit und die stete Diskussionsbereitschaft insbesondere in Fragen der NMR-Analysen,
für die Möglichkeit in zahlreichen Kursen und Lehrveranstaltungen mitzuwirken und
dadurch Erfahrungen zu sammeln, nicht zuletzt für die Bereitstellung des Arbeitsplatzes
und für die Übernahme des Referats.
• Herrn PD Dr. Hans Jörg Kunte danke ich herzlich für die Überlassung des interessanten
Themas, seine Diskussionsbereitschaft und für die Übernahme des Korreferats.
• Ein herzlicher Dank gilt auch Herrn Prof. Dr. Dr. Dr. h.c. Hans G. Trüper für sein offenes
Ohr, seine Güte und nicht zuletzt für „die Rettung des Genetivs“.
• Frau Marlene Stein möchte ich meinen besonderen Dank aussprechen für ihre Unter-
stützung bei den HPLC-Analysen. Ebenso danke ich herzlich „der Seele des Hauses“,
Frau Birgit Amendt. Ihre ausgeprägte Hilfsbereitschaft, ihre ständige Aufmunterung und
ihr großes Herz hat „trübe“ Gemütszustände weggezaubert.
• Frau Andrea Ures, Frau Yvonne Schilz und Frau Elizabeth Witt sei ganz herzlich gedankt
für die schöne, unvergessliche Zeit und die willkommene Abwechslung im Labor. Herrn
Dennis Raschke danke ich außerdem für seine hilfreichen „Tipps“ in Computerfragen.
• Frau Dr. Katrin Grammann danke ich für die gute Zusammenarbeit im Labor, ihre Freund-
schaft, die lebhaften Telefonate und nicht zuletzt für ihre Zeit bei der Durchsicht dieses
Manuskripts.
• Herrn Jens Rudat danke ich für die abwechslungsreiche Zeit im Labor, seine Freund-
schaft und nicht zuletzt für seine virtuose musikalische Begleitung. Gemeinsam mit Frau
Dr. Annette Kraegeloh hatten wir unvergessliche Auftritte bei zahlreichen Instituts-Weih-
nachtsfeiern. Ein dickes Dankeschön geht an Herrn Elmar Kopp für seine Freundschaft
und für die schönen gemeinsamen Feiern.
• Gedankt sei auch Herrn Dr. Matthias Kurz für seine Hilfsbereitschaft. Der gesamten AG
Biotechnologie, allen früheren und jetzigen Mitarbeitern, danke ich für eine gute Zeit. Den
Mitarbeitern der AG Fakoussa, AG Dahl, AG Sahl, AG Krämer und AG Klemme & Kaspari
sei gedankt für die Möglichkeit zur Nutzung ihrer Geräte.
• Außerdem möchte ich der Firma Bitop AG (Witten) für die Bereitstellung der kompatiblen
Solute Mannosylglycerat und Mannosylglyceramid und dafür, dass sie diese Arbeit
mehrere Monate finanziell unterstützt hat, danken.

• Meinem lieben, wunderbaren Mann, Dr. Daniel Burdziak, gilt mein tief empfundener Dank
für seine fachlichen Denkanstöße, seine wertvollen Anregungen bei der Durchsicht des
Manuskripts sowie seine Geduld und nicht zuletzt für seine warmherzige Liebe, die mein
andauerndes Heimweh während des Zusammenschreibens linderte.
Was würde ich ohne Dich tun? 1ÜÃ{k Nœ¨.„
• Mein abschließender Dank geht an meine liebe Familie in Bethlehem und in Deutschland.
Besonders danken möchte ich meiner geliebten Mutter May, die mir aus der Ferne eine
ständige moralische Stütze ist, zusammen mit meinem Vater Dipl.-Ing. Ghasub Nasser
Rashed, der mir schon zu Kindeszeiten ein Studium in Deutschland schmackhaft gemacht
hat. Mein besonderer Dank gilt meinen lieben Schwestern Marwa, Sundus, Hiba und Hind
für ihre uneingeschränkte Unterstützung. Meiner lieben Schwiegermutter Heidrun
Burdziak danke ich für ihr Verständnis, ihre aufmunternden Worte und nicht zuletzt für ihre
Kochkünste.
• Mein heimlicher Dank gilt der „deutschen Elf“, insbesondere Poldi, Klose, Ballack und
Schweini für Ihre Fußballkünste. Ihr fabelhaftes Spiel hat mich in den letzten Wochen des
Zusammenschreibens beflügelt.
$/9]z:«[ /²½˜^/ +±:b

![Bakterielle Resistenzmechanismen – Spezielle Formen der Resistenz · 2019. 8. 20. · Carbapeneme, Monobaktame], Glykopeptide wie Vancomycin) • Inhibierung der bakteriellen Proteinsynthese](https://img.pdfslide.org/doc/110x75/60c9a1343afb1115981d336b/bakterielle-resistenzmechanismen-a-spezielle-formen-der-resistenz-2019-8-20.jpg)



















![Literaturverzeichnis - link.springer.com978-3-8348-2080-8/1.pdfLiteraturverzeichnis [Car03] Carter, Nicholas P.: Computerarchitektur. mitp-Verlag, 2003. ISBN 3–8266–0907–7 [Dar10]](https://img.pdfslide.org/doc/110x75/5b4a429f7f8b9a5c278c11da/literaturverzeichnis-link-978-3-8348-2080-81pdfliteraturverzeichnis-car03.jpg)







