Embed Size (px)
Citation preview

F1-Praktikum
Genetik 08. März bis 26. März 2010
Allgemeine Einleitung
Als genetischer Modellorganismus hat die Fliege Drosophila einen zentralen Platz in der
modernen Biologie. Ihr Genom mit seinen ca. 13.000 Genen steuert und reguliert unter
anderem die Entwicklung und Funktionsweisen ihres Gehirns, die beide deswegen mit Hilfe
der klassischen und molekularen Genetik untersucht werden können. Mit ihrem Gehirn als
Informations- und Steuerzentrum kontrolliert die Fliege ihr Verhalten in ihrem ökologischen
Umfeld. Die Funktionen des Gehirns beruhen auf den spezifischen Wechselwirkungen
zwischen den sehr zahlreichen Bauelementen, den Neuronen und Gliazellen. Am Lehrstuhl
für Genetik und Neurobiologie wird am Beispiel der Fliege Drosophila die Frage bearbeitet:
Wie funktioniert dieses komplexe 3-dimensionale Gebilde? Zu dieser Frage trägt die Genetik
dadurch bei, dass sie die Methoden zur Untersuchung der Gehirnanatomie und des Verhaltens
zur Verfügung stellt. Besonders die nicht-invasiven Interventionsmethoden stellen einen
bedeutenden Fortschritt in der experimentellen Gehirn-Funktionsforschung dar.
Bei Drosophila liegt seit dem Frühjahr 2000 das Genom praktisch vollständig sequenziert vor.
Damit stellt sich die Frage, wie Gene, die für die Funktion des Gehirns wichtig sind,
identifiziert werden können. Am Lehrstuhl werden unterschiedliche Ansätze verwendet. Im
Rahmen des ersten, seit Mendel immer wieder erfolgreich angewendeten, Ansatzes werden
Gene durch ihre Wirkungen, d.h. durch erbliche Variationen der Erscheinungsform (des
Phänotyps) identifiziert. Der Phänotyp kann dabei auch das Verhalten sein. Zur Identifikation
muss das Gen zunächst mit klassisch-genetischen Methoden möglichst genau auf einem der 4
Chromosomen lokalisiert werden. In dieser Phase kommt es darauf an, mit P-Element-
Insertionen oder strukturellen Chromosomenaberrationen den Bereich des Gens auf der DNA-
Karte einzugrenzen. Anschließend kann man durch Keimbahntransformation mit
genomischen DNA-Fragmenten oder cDNA von Kandidaten-Genen den Phänotyp zu retten
(transgenic rescue), oder mittels RNA-interferenz den Effekt der Mutation nachzuahmen
(transgenic knock-down) versuchen (siehe http://nwg.glia.mdc-berlin.de/media/ppt/gerber-
Gal4.ppt). Diese Methode erlaubt es dann insbesondere auch, den zellulären Ort, an dem das
Gen seine Funktion ausübt, einzukreisen. Grundlage dieser Experimente ist, dass sich bei
Drosophila klonierte Gene mit Hilfe von P-Element-Vektoren, die in Embryonen injiziert
werden, stabil in die Keimbahn integrieren. Die Transformationsprozedur ist allerdings zu
langwierig, als dass sie im Rahmen dieses Praktikums durchgeführt werden könnte. Ein
weiterer Weg zur Identifikation eines Gens in einem durch Chromosomen-Bruchpunkte
1

eingegrenzten Bereich ist die Suche nach der relevanten Mutation in den Kandidaten-Genen
der Mutante durch Sequenzierung .
Der zweite Ansatz zur Identifizierung von 'Gehirngenen' geht von der Überlegung aus, dass
die molekulare Grundlage für die funktionelle Differenzierung der einzelnen Zellen eines
Organismus in der selektiven Expression bestimmter Gene liegen muss. Umgekehrt ist dann
zu erwarten, dass Gene, die selektiv nur im Gehirn exprimiert werden, für die spezifische
Funktion dieser Zellen wichtig sind. In diesem Zusammenhang muss man bedenken, dass das
Gehirn nicht nur strukturell sondern auch aufgrund seiner molekularen Vielfalt ein sehr
komplexes Organ ist. Man schätzt, dass etwa 30% der ca. 30.000 Gene des Menschen (bzw.
ca. 13.000 der Fliege) nur im Nervensystem exprimiert werden. Die Mehrzahl dieser Gene
kodiert für Proteine, deren Funktion noch völlig unbekannt ist, da sie keine Ähnlichkeit zu
bekannten Proteinen zeigen. Aufgrund der Konservierung der meisten Proteine in der
Evolution vom Wurm bis zum Menschen ist es sinnvoll, neue Proteine zunächst an einem
experimentell gut zugänglichen Modellsystem wie Drosophila zu analysieren, bevor man das
homologe menschliche Protein untersucht. Die Genprodukte (Kursteil Proteine) von solchen
unbekannten, zellspezifisch exprimierten Genen können über monoklonale Antikörper
immunhistochemisch lokalisiert werden. Einige monoklonale Antikörper, die an bestimmte
zellspezifische Antigene binden, lassen sich zur Charakterisierung von Gehirnmutanten
einsetzen. Es ist jedoch unter Umständen auch möglich, mit einem solchen Antikörper aus
einer Expressionsbibliothek einen cDNA Klon zu isolieren, dessen Gen für das Antikörper-
spezifische Antigen kodiert. Gegenwärtig werden am Lehrstuhl zwei Gene näher
charakterisiert, die mit Hilfe von Antikörpern kloniert worden sind (Synapsin und SAP47).
Die Verbindung zur klassischen Genetik kann hergestellt werden, indem mit der Sequenz der
cDNA das Gen auf dem Chromosom durch Vergleich mit den Drosophila-Datenbanken
lokalisiert wird. Man wird dann nachsehen, ob für das entsprechende Gen bereits Mutanten
beschrieben sind. Ein dritter Weg 'Gehirngene' zu identifizieren, ist durch die "Enhancer-
Trap"-Technik eröffnet worden (O'Kane und Gehring, 1987) die sich durch alle drei Kursteile
zieht und in Kursteil Molekulargenetik ausführlich erklärt ist.
Mit der Identifizierung eines Gens erhält man heute aus den einschlägigen Datenbanken
dessen Nukleotidsequenz bzw. die Aminosäurensequenz seines Genprodukts. Aus dem
Vergleich mit bekannten Proteinen ergeben sich oft erste Hinweise auf mögliche Funktionen
des Genprodukts auf molekularer und zellulärer Ebene. Mit der Frage: "Was ist die Funktion
der 'Gehirngene' bzw. ihrer Genprodukte?" sind wir am Kern des Kurses angelangt. Wir
werden sehen, dass hier die spezifischen Vorteile des Versuchstiers Drosophila in
besonderem Maße zum Tragen kommen, da der erste Schritt zur Funktionsanalyse eines Gens
bzw. Genproduktes in der Regel die Isolierung von Mutanten beinhaltet.
2

Ist die Isolierung einer Nullmutation des untersuchten Gens gelungen (also einer Mutation,
der des Genprodukt vollständig fehlt), so eröffnen sich bei Drosophila zahlreiche interessante
Perspektiven. Zum einen muss natürlich der Phänotyp der Mutanten auf allen Ebenen, der
molekularen durch Protein-biochemische Experimente, der zellulären durch
Elektrophysiologie und der systemischen durch anatomische und Verhaltensanalyse,
charakterisiert werden. Beispiele für solche Charakterisierungen werden im Kurs diskutiert.
Klonierte Gene kann man, um die o.a. transgenen Rettungs- und knock-down Experimente
weiter zu verfeinern, auch in-vitro verändern und durch Keimbahn-Transformation in das
Genom der Nullmutanten einführen. Dadurch ist es möglich, die Funktion von gezielten
Veränderungen einzelner Abschnitte, ggf. des Austausches nur einzelner Aminosäuren, des
entsprechenden Proteins zu untersuchen. Wir werden diese faszinierenden Perspektiven der
in-vivo Molekularbiologie im Kurs ausführlich diskutieren.
Zudem hat sich die Anatomie des Drosophila-Gehirns in jüngster Zeit durch die Entdeckung
neuer mikroskopischer und histologischer Methoden sowie neuer funktioneller
Fragestellungen rasch entwickelt. Einige von Ihnen werden Gelegenheit haben selbst Gehirne
zu präparieren und im LSC-Mikroskop zu untersuchen. Manche funktionelle Hypothesen über
die Eigenschaften des Gehirns lassen sich bereits aus der Struktur der Nervenzellen und ihren
Verknüpfungen ableiten. (Auf Wunsch können Präparate im Mikroskop angeschaut werden.
Mehrere besonders gute sind im Internet in einer Datenbank für Drosophila Neuroanatomie
unter << http://flybrain.neurobio.arizona.edu/>> zu sehen.)
Häufig möchte man jedoch nicht nur die Struktur des Gehirns untersuchen, sondern auch die
Verteilung von bestimmten Molekülen wie Neurotransmittern oder insbesondere endogenen
oder transgenen Proteinen, also Genprodukten darstellen. Als Beispiel für "chemische"
Neuroanatomie führen wir immunhistochemische Färbungen mit verschiedenen Antikörpern
durch, um Genexpressionsmuster in transgenen Tieren zu analysieren. Die Anatomie des
Fliegengehirns wird dabei eingehend besprochen.
Schliesslich wird mit dem Verhalten das wesentliche funktionelle Korrelat des Gehirn
untersucht. Das Verhalten ist auf das Engste mit der Physiologie des Tieres verwoben. Wir
versuchen mit Hilfe der oben skizzierten Methoden der modernen Neurogenetik
Verhaltensleistungen als Eigenschaften von Nervennetzen im Gehirn zu verstehen. In den
Verhaltensexperimenten versuchen wir Ihnen zunächst zu zeigen, wie sich Mutationen in
zwei Genen, Synapsin und SAP47, auf die Lernfähigkeit auswirken, und welche
Möglichkeiten diese Beobachtungen für eine weitere neurogenetische Analyse eröffnen, um
bestimmte Neuronengruppen bestimmten Verhaltensleistungen zuzuordnen.
3

Wir wollen darüber hinaus diskutieren, was man über die Wirkung der Gene im Gehirn sagen
kann und wie die gewaltige Menge phylogenetischer Erfahrung, die im Gehirn steckt, in den
Genen kodiert ist. Die Diskussion soll auch Gelegenheit bieten, die Fragestellung auf
Wirbeltiere und den Menschen auszudehnen. Die Beziehungen des Fliegenverhaltens zur
Psychologie des Menschen wird besprochen.
4

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Versuchsteil 1
PCR und Sequenzieren (V1)
1. Drosophila
Bestimmte Fliegenstämme (per01 und per
s ) zeigen ein vom Wildtyp (CantonS oder Ala)
abweichendes Wach/Schlaf Verhalten. Diese Fliegenstämme sollen auf ihren Genotyp hin
untersucht werden. Dazu ist es notwendig DNA zu isolieren, mit Hilfe von PCR spezifisch zu
amplifizieren und letztendlich aufzureinigen und zu sequenzieren.
Das period Gen
DNA Isolierung:
Die DNA wird aus einzelnen Fliegen isoliert. Unterschiedliche Gruppen isolieren DNA aus
unterschiedlichen Stämmen – A, B, C, D. Die Aufteilung erfolgt am Kurstag. Am letzten
Kurstag erfolgt die Analyse, welche Fliege welcher Stamm gewesen ist.
Genotyp:
1. Einzelne Fliege wird in ein vorgekühltes Eppendorf Cup gegeben.
2. 40 μl squishing buffer in eine gelbe Spitze geben, damit die Fliege im Cup
zerquetschen (gründlich, 20-30 sec). Erst dann die 50 μl Puffer in das Gefäß geben.
3. 10 μl ProteinaseK zu der Lösung geben. Dann bei 30-37°C 30 min inkubieren lassen.
4. Reaktion bei 95 °C, 2` abstoppen. DNA kann bei -20°C aufgehoben werden.
PCR :
Die isolierte DNA wird als template für die nachfolgende PCR benutzt.
PCR Ansatz (50 μl) :
Primerpaar 1:
period Gen Per1
Komponente Volumen/Reaktion √
10x Reaktionspuffer 5 μl
dNTP-Mix (40mM) 10 μl
Upstream Primer 10μl
Downstream Primer 10μl
Taq-DNA Polymerase 10μl
Template DNA 5μl
Gesamt 50μl
5

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
PCR-Programm
Temperatur Dauer Zyklen
1: 95°C “pause”
2: 95°C 5min
3: 95°C 30sec
55°C 30sec 34x
72°C 45sec
4: 72°C 10min
5: 4°C “pause”
Auf Grund der zu erwartenden Größe wird das PCR Produkte auf ein 1,2% Agarosegel
(1x TAE) aufgetragen. Der gesamte PCR Ansatz wird, nach dem er mit 8μl Ladepuffer
versehen wurde, auf das Gel aufgetragen.
Das Gel wird entweder bei 20 Volt über Nacht oder bei 80 Volt, 60` laufen gelassen. Eine
Spur muss mit dem Größenmarker (10μl) geladen werden.
Reinigung der DNA:
1. Die PCR Produkte werden anschließend im UV Licht auf ihre Größe und
Verwendbarkeit überprüft und ausgeschnitten.
2. Nach abwiegen des ausgeschnittenen Gelstückes wird die DNA aus diesem mit Hilfe
des PeqLab Gel Extraction Kits gereinigt.
3. Zu dem abgewogenem Gelstück wird das gleiche Volumen an Binding Buffer
gegeben, gemischt und dieses bei 55°C bis zum Lösen des Gels inkubiert (ca. 10`).
Gelegentliches Vortexen (alle 2-3`) verbessert das Lösen des Gels.
4. Max. 750μl der Lösung auf die Säule geben. Nach einer Minute Zentrifugation bei
10k g den Durchfluss verwerfen
5. 750μl CG Wash Buffer auf die Säule geben und 1 Min. bei 10k g zentrifugieren.
Durchfluss verwerfen.
6. Schritt 5 Wiederholen.
7. Die leere Säule wird erneut bei 10k g 1 Min zentrifugiert. (Trocknen)
8. Säule nun in ein sauberes Eppendorf Cup stellen. In die Mitte der Säule wird 30 μl
H2O pipettiert und für eine Minute bei 5000g zentrifugiert.
Test und Sequenzierung:
5 μl der DNA werden mit 2μl Ladepuffer und 5μl H2O versehen und auf ein 1,2 % Gel
aufgetragen. Bei 100 V wird das Gel 30 Minuten laufen gelassen.
Für die Sequenzierung werden 100ng DNA und 10pmol des Primers (2μl) in ein 1.5 ml Cup
gegeben.
6

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Mit Hilfe des DNA Standards soll die Menge bestimmt
werden.
Da sequenziert wird, natürlich nur einen Primer
verwenden. Die Auswertung erfolgt am letzten Kurstag
mit Hilfe des BioEdit Programms (bitte Laptop mitbringen)
2. Mensch
Wir Menschen weisen kleinere Unterschiede (z.B. eher Morgen/Abend Typ) in unserem
Aktivitätsverhalten auf. Weiterhin ist bekannt, dass im humanen Period3 Gen bestimmte
Polymorphismen vorkommen. Diese sollen nun mit Hilfe der PCR untersucht werden und
anschließend überprüft werden, ob ein bestimmter Polymorphismus mit einem
Verhaltenstyp (Morgen/Abend) korreliert.
DNA Isolierung
Die DNA soll aus den Epithelzellen der Mundschleimhaut (Wange) isoliert werden.
1. Mit Hilfe eines Wattestäbchens/Mundspatels werden Zellen der Mundschleimhaut
entnommen.
2. Die Spitze des Zahnstochers, bzw. Zellen vom Mundspatel werden in ein Eppendorf
Cup gegeben und dazu 200μl der Chelex Lösung (vorher gut vortexen) pipettiert.
3. 10μl ProteinaseK dazu geben.
4. Die Proben werden kurz gevortext und dann 30-60` bei 40°C inkubiert.
5. Anschließend die Probe erneut vortexen und dann für 8 Minuten auf 100°C gegeben.
6. Nach erneutem Vortexen wird die Probe bei 14k rpm 3` zentrifugieren.
7. Vom Überstand 150 μl mit abnehmen und in neues, beschriftetes 1,5 ml Eppendorf
Cup überführen; beim Abnehmen des Überstandes darauf achten, dass keine
Chelex-Kügelchen mit abgenommen werden.
7

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
PCR :
Die isolierte DNA wird als template für die nachfolgende PCR benutzt.
PCR Ansatz (50 μl) :
Primerpaar :
hPer3 Sense
hPer3 Antisense
PCR-Programm
Temperatur Dauer Zyklen
1: 95°C “pause”
2: 95°C 2min
3: 95°C 30sec
50°C 20sec 34x
72°C 30sec
4: 72°C 5min
5: 4°C “pause”
Auf Grund der zu erwartenden Größe wird das PCR Produkte auf ein 2% Agarosegel
(1x TAE) aufgetragen. Der gesamte PCR Ansatz wird, nach dem er mit 8μl Ladepuffer
versehen wurde, auf das Gel aufgetragen.
Das Gel wird entweder bei 20 Volt über Nacht oder bei 80 Volt, 60` laufen gelassen. Eine
Spur muss mit dem Größenmarker (10μl) geladen werden.
Das Gelphoto wird im Anschluss zusammen ausgewertet und diskutiert.
Komponente Volumen/Reaktion √
10x Reaktionspuffer 5 μl
dNTP-Mix (40mM) 10 μl
Upstream Primer 10 μl
Downstream Primer 10 μl
Taq-DNA Polymerase 10 μl
Template DNA 5 μl
Gesamt 50μl
8

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
RNA
qPCR (V2)
1. Quantitative PCR
Im Kurs soll untersucht werden, ob sich die Expression verschiedener mRNAs im
Tagesverlauf ändert oder diese konstant exprimiert werden.
Dazu wird die cryptochrome (cry) und period (per) mRNA-Konzentration in Fliegen
gemessen, die sich drei Tage lang in einem 12h:12h LD-Rhythmus befanden, bevor sie zu
vier (bzw. 8) verschiedenen Zeitpunkten (ZT3, ZT9, ZT15, ZT21) mit flüssigem Stickstoff
tiefgefroren wurden. Genotypen: Wildtyp CantonS, Wildtyp Ala, per01 und eine Syn
Mutante.
Isolierung der RNA
Zunächst muss gesamt RNA aus Fliegenköpfen isoliert werden. Bearbeitet werden die vom
Betreuer zugewiesenen Zeitpunkte und Genotypen:
Fliegenstamm: Zeitpunkt:
1. Das Eppendorf Cup mit 20 Köpfen aus dem Trockeneis/Stickstoff nehmen und auf 4°C Eis
geben.
2. 200µl RNAPure (vorgekühlt auf 4°C) zugeben und für 20sec homogenisieren
3. Homogenisat für 5` ruhen lassen und dann 5` bei 13.000rpm und 4°C zentrifugieren
9

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
4. Den Überstand (überall gleiche Menge, circa 180µl) in neues Epi überführen
5. 40µl Chloroform zugeben und 20sec vortexen
6. 3min bei 4°C stehen lassen, dann 10min bei 13.000rpm und 4°C zentrifugieren
7. Überstand in ein frisches Epi geben (auch hier wieder überall die gleiche Menge, ca.
80µl) (Achtung: nur die obere Phase abnehmen, auf gar keinen Fall die RNAse-
enthaltende Interphase berühren)
8. zum Überstand 1:1 (= gleiche Menge, ca. 80µl) Isopropanol geben
9. RNA und Isopropanol vermischen (nicht vortexen!) und bei -20°C fällen (z.B.
Mittagspause)
10. 15min bei 4°C und 11.000rpm zentrifugieren
11. Überstand verwerfen, das Pellet (ist gut sichtbar!) in 75% DEPC-EtOH, 500μl waschen
(evtl. 2x)
12. kurz zentrifugieren (circa 5min), damit Pellet sich nicht löst
13. EtOH ganz abpipettieren und kurz lufttrocknen. Ein vollständig getrocknetes Pellet löst
sich sehr schlecht…
14. Die RNA wird in 10μl H2O(DEPC) gelöst.
Reverse Transkription:
Nun wird die isolierte RNA in cDNA umgeschrieben. Um spezifisch die mRNA anzusprechen
wird mit einem oligo (dT)18 primer gearbeitet.
1. Den Reaktionsansatz auf Eis vorbereiten:
2. Die Lösung bei 65°C für 10 Minuten inkubieren.
3. Abkühlen auf Eis für 2`.
4. Zu dem Ansatz folgende Komponenten pipettieren:
5. Bei 37-50°C 50` inkubieren
6. Bei 70°C 15` Reaktion inaktivieren. Lösung auf Eis geben.
Test PCR (fakultativ)
Mittels PCR wird getestet, ob die cDNA richtig hergestellt wurde.
Komponente Volumen √
RNA 8μl
Oligo(dT) 2μl
10mM dNTP 1μl
Komponente Volumen √
5x SensiMix RT Puffer 4μl
RNase Inhibitor 1μl
SensiMix RT 0.25μl
mit H2O(DEPC) auf füllen auf 20μl
10

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
PCR Ansatz (25 μl) :
PrimerPaar:
tubulin qPCR
PCR-Programm
Temperatur Dauer Zyklen
1: 95°C “pause”
2: 95°C 5min
3: 95°C 30sec
56°C 30sec 34x
72°C 30sec
4: 72°C 5min
5: 4°C “pause”
Auf Grund der zu erwartenden Größe wird das PCR Produkte auf ein 1,5% Agarosegel (1x
TAE) aufgetragen. Der gesamte PCR Ansatz wird, nach dem er mit 4μl Ladepuffer versehen
wurde, auf das Gel aufgetragen.
Das Gel wird entweder bei 20 Volt über Nacht oder bei 120 Volt, 30` laufen gelassen. Eine
Spur muss mit dem Größenmarker (5μl) geladen werden.
quantitative PCR
Die Gene period und cryptochrome sollen untersucht werden. Als housekeeping Gene
fungiert tubulin.
1. Folgende Komponenten sollen für die qPCR pipettiert werden:
Der 2 x SYBR Mix soll vor
Gebrauch gevortext werden (30``)
Komponente Volumen/Reaktion √
10x Reaktionspuffer 2.5 μl
dNTP-Mix (40mM) 5 μl
Upstream Primer 5 μl
Downstream Primer 5 μl
Taq-DNA Polymerase 5 μl
Template DNA 2 μl
Gesamt 24,5
Komponente Volumen/Reaktion √
cDNA Template 2.0 μl
2x Sensi Mix 10 μl
Upstream Primer 4μl
Downstream Primer 4μl
Gesamt 20 μl
11

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Die Verteilung der verschiedenen Primerpaare, Genotypen und Zeitpunkte erfolgt am
Kurstag:
Primer: Genotyp: Zeitpunkt:
PCR-Programm:
Die Polymerase muss zunächst hitzeaktiviert werden. Im
Anschluss an die Amplifikation kann eine
Schmelzkurvenbestimmung durchgeführt werden.
Die Auswertung
Das Grundprinzip ist eine normale PCR-Reaktion. Im SybrGreen Reaktionsansatz befindet
sich jedoch der fluoreszente Farbstoff SYBR® Green I, der nur an doppelsträngige DNA
bindet, d.h. nur an dem zu amplifizierenden DNA-Stück.
Dann, und nur dann, kann er mit einer Wellenlänge von 494nm angeregt werden und
emittiert Licht bei 521nm. Dadurch lässt sich jede Amplifikationsrunde einzeln detektieren.
Die Detektion erfolgt einmal pro PCR-Zyklus am Ende des 72°C Schritts.
Der resultierende Graph (s. Abb.) kann grob in 3
Teile unterteilt werden. Im Baseline-Bereich ist
das Signal noch zu schwach, um eine
vernünftige Aussage zuzulassen. In der Log-
linearen Phase kann man die Samples (hier
zwei) auf Höhe des Thresholds vergleichen.
In der stationären Phase haben beide Samples
wieder ein ähnliches Niveau erreicht und
können somit nicht mehr unterschieden
werden (s. Abb.). Jedes Sample erreicht den
Threshold-Level bei einer bestimmten
Zyklenzahl (CT, crossing time), je nach vor-
liegender cDNA-Konzentration. Die cDNA-
Konzentration ist wiederum von der RNA-Konzentration abhängig, die bei der
vorausgehenden RT-PCR eingesetzt wurde. Der Unterschied beläuft sich auf nZyklendifferenz. Im
gezeigten Beispiel (Abb.) also auf circa 210.
Um Fehler zu vermeiden, die bei den verschiedenen Proben auf Pipettierungenauigkeiten,
Abweichungen in der RT-PCR oder Differenzen in der Menge der ursprünglich eingesetzten
RNA zurückzuführen sind, werden sogenannte Housekeeping-Gene benutzt, die immer in
einer konstanten Menge in der Zelle vorliegen. Diese sind z.B. RP49, Tubulin, Synaptobrevin
und CAP binding protein 20.
Temperatur Dauer Zyklen
1: 95°C 2 min
2: 95°C 15sec
55°C 30sec 40x
72°C 15sec
Zyklen
Fluo
resze
nz
Baseline
Log-Lineare
Phase
Stationäre Phase
Amplifikations-Graphik. Die DNA-Konzentration in Sample A ist größer als in Sample B.
12

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Die Berechnung erfolgt nach der ΔΔCT-Methode der relativen Quantifizierung:
15. zuerst wird der ΔCT-Wert bestimmt (CT = crossing time):
ΔCT1 (Probe1) = CT (Ziel-Gen) – CT (Housekeeping-Gen)
ΔCT2 (Probe2) = CT (Ziel-Gen) – CT (Housekeeping-Gen)
16. dann bestimmt man den Unterschied zwischen den beiden Proben, den ΔΔCT:
ΔΔCT= ΔCT1- ΔCT2
Besitzen die Effizienzwerte des Ziel-Gens und des Housekeeping-Gens die gleiche
Größenordnung, kann das Verhältnis nach der Formel 2- ΔΔCT berechnet werden.
Vektoren und Klonieren (V3)
Im Kurs soll das genetische Konstrukt LuciferaseRED
-Cry aus einem für Zellkultur
verwendeten Vektor – pAc5.1 – in ein Drosophila Vektor –pUAST- umkloniert werden, um
dann damit transgene Tiere herzustellen.
Cryptochrome:
Name Length (nt) Transcript Length (nt) CDS Length (aa) Predicted MW (kD)
Cry-PA 1859 bp 1629 bp 542 62,5 kDa
200 400 600 800 1000 1200 1400 1600
CryptochromeCDS
13

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Da die LuciferaseRED
-Cry CDS zwischen der EcoRI und XbaI Schnittstelle im pAc5.1 Vektor
inseriert ist, werden diese beiden Restriktionsenzyme benutzt um die CDS
herauszuschneiden. Auch der leere pUAST PLasmid muss mit diesen beiden Enzymen
geschnitten werden.
Folgender Ansatz sollte für den Testverdau pipettiert werden.
Dieser Ansatz soll anschließend mind. 60 Minuten bei 37°C inkubiert werden.
1. Anschließend wird der pAc und pUAST Vector auf ein 1,2% Agarosegel (1x TAE)
aufgetragen. Der gesamte Verdau wird, nach dem er mit 8μl Ladepuffer versehen
wurde, auf das Gel aufgetragen. Das Gel wird entweder bei 20 Volt über Nacht oder
bei 80 Volt, 60` laufen gelassen. Eine Spur muss mit dem Größenmarker (10μl)
geladen werden.
Reinigung der DNA:
1. Die PCR Produkte werden anschließend im UV Licht auf ihre Größe und
Verwendbarkeit überprüft und ausgeschnitten.
2. Nach abwiegen des ausgeschnittenen Gelstückes wird die DNA aus diesem mit Hilfe
des PeqLab Gel Extraction Kits gereinigt.
3. Zu dem abgewogenem Gelstück wird das gleiche Volumen an Binding Buffer
gegeben, gemischt und dieses bei 55°C bis zum Lösen des Gels inkubiert (ca. 10`).
Gelegentliches Vortexen (alle 2-3`) verbessert das Lösen des Gels.
4. Max. 750μl der Lösung auf die Säule geben. Nach einer Minute Zentrifugation bei
10k g den Durchfluss verwerfen
5. 750μl CG Wash Buffer auf die Säule geben und 1 Min. bei 10k g zentrifugieren.
Durchfluss verwerfen.
6. Schritt 5 Wiederholen.
7. Die leere Säule wird erneut bei 10k g 1 Min zentrifugiert. (Trocknen)
Säule nun in ein sauberes Eppendorf Cup stellen. In die Mitte der Säule wird 30 μl H2O
pipettiert und für eine Minute bei 5000g zentrifugiert.
Komponente Volumen √
DNA Vector pUAST 10 μl
EcoRI 10 μl
XbaI 10 μl
Puffer 5 μl
H2O auf füllen auf 50 μl
Komponente Volumen √
DNA Vektor pAc 15 μl
EcoRI 10 μl
XbaI 10 μl
Puffer 5 μl
H2O auf füllen auf 50 μl
14

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Ligation:
Nun muss das PCR Fragment in den geschnittenen Vector gegeben werden.
1. Folgender Ansatz soll pipettiert werden.
Der Reaktions Puffer muss vor Benutzung
gevortext werden.
Die Ligation soll circa 60‘ bei RT erfolgen.
Das so gewonnene Plasmid wird nun für eine Transformation in E.Coli Zellen benutzt.
Hitzeschock Transformation der Plasmide:
Die Hitzeschock kompetenten Zellen werden zunächst von -80°C auf Eis gegeben um sie
aufzutauen (10 Min).
1. Zu den Zellen werden vorsichtig 10 μl des Transformationsansatzes gegeben und
diese ganz leicht gemischt (Vorsichtiges GegenSchnippen - Achtung Zellen sind
zerbrechlich..)
2. Die Zellen werden anschließend wieder auf Eis gestellt und 20` lange inkubiert.
3. Die Zellen werden nun einem Hitzeschock ausgesetzt, i.e. 45-50 Sekunden bei 42°C
(relativ genau!)
4. Anschließend werden die Zellen mit 500μl LB Medium (ohne AMP!!) beschichtet. Die
Zellen sollen dann 60-90` bei 37°C geschüttelt werden.
5. Die Zellen werden dann kurz herunter zentrifugiert, der Überstand verworfen
(Abschütten) und die letzten 50-100μl auf einer LBamp Platte ausplattiert
(Bunsenbrenner).
6. Die Platten werden über Nacht bei 37°C inkubiert.
- nächster Tag –
7. Am Nachmittag/Abend des folgenden Tages werden Minipreps angesetzt. Dazu
werden Reagenzgläser mit jeweils 2-3 ml LBamp Medium versehen. Mit einer gelben
Pipettenspitze wird anschließend eine Kolonie von der Platte genommen und diese
in das vorbereitete Reagenzglas gegeben. Die Reagenzgläser werden über Nacht
unter Schütteln bei 37°C inkubiert.
- nächster Tag –
Komponente Volumen √
5X Ligase Reaction Buffer 5 μl
Insert 3x molar x μl
Vector 1x molar x μl
T4 DNA Ligase 2,5 μl
H2O auf füllen auf 20 μl
15

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Mini Preps
Hierbei sollen die Plasmide aus den Bakterien isoliert werden .
1.Die Bakterien werden in ein 1,5ml Eppi überführt. Dann 2’ bei 13k rpm zentrifugiert.
2. Der Überstand wird verworfen und das Pellet in 250µl STET Buffer und 20µl Lysozym
(20mg/ml) durch starkes vortexen gelöst.
3. Die Zellen werden nun 2’ bei 95°C gekocht, kurz auf Eis gegeben und anschließend 10’
bei 13k rpm zentrifugieren (RT).
4. Der Überstand wird in ein neues Eppi überführt. Dann 250µl IsoPropanol/10M NH4Acetat
(AmmoniumAc) 2:1 Mix hinzufügen und mischen.
5. Zum pelletieren 10’ bei 13k rpm zentrifugiern (RT)
6. Der Überstand wird verworfen und zur DNA mit 500 μl 70% EtOH hinzufügen/waschen.
7. Erneut kurz 5‘ bei 13k rpm zentrifugieren
8. EtOH entfernen und bei RT trocknen.
9. DNA in 40µl H2O(Rnase, 50ml/50µl RNAse) resuspendieren.
Nun muss überprüft werden ob ein Plasmid mit dem richtigen Insert isoliert werden konnte.
DNA TestVerdau
Hierzu wird überprüft, ob auch das richtige Fragment in den Vector inseriert ist.
1. Folgender Ansatz sollte für den Testverdau pipettiert werden.
Dieser Ansatz soll anschließend 30-60 Minuten
bei 37°C inkubiert werden.
Auf Grund der zu erwartenden Größe wird das PCR Produkte auf ein 1% Agarosegel (1x TAE)
aufgetragen. Der gesamte Verdau wird, nach dem er mit 3μl Ladepuffer versehen wurde,
auf das Gel aufgetragen. Das Gel wird entweder bei 20 Volt über Nacht oder bei 120 Volt,
30` laufen gelassen. Eine Spur muss mit dem Größenmarker (5μl) geladen werden.
Die positiven Zellen könnten nun für eine Plasmid Produktion im größeren Maßstab
benutzt werden.
Komponente Volumen √
DNA 10 μl
EcoRI 1 μl
XbaI I 1 μl
Puffer 2 μl
H2O auf füllen auf 20 μl
16

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Versuch Bioluminescence (V4)
Theorie:
Die in-vivo Messung der Genexpression erfolgte mittels Reporterkonstrukten, bei denen das
Luciferase-Gen aus dem Glühwürmchen (Photynus pyralis) an ein zu untersuchendes Gen
bzw. einen Genbereich fusioniert ist und unter dessen Kontrolle exprimiert wird. Es ist somit
möglich die Messung am lebenden Organismus durchzuführen und aufgrund der kurzen
Halbwertszeit der Luciferaseaktivität (Brandes et al., 1996; Stanewsky et al.,1997b)
rasche Schwankungen im Expressionslevel zu detektieren.
Um die molekularen Oszillationen der Uhrkomponenten erforschen zu können, werden
unterschiedliche Reporterkonstrukt des period-Gens verwendet werden. Das period Gen
spielt sowohl beim Menschen als auch bei der Fliege eine zentrale Komponente der
inneren Uhr.
Die period-Konstrukte können in zwei Gruppen eingeteilt werden:
A) nur die 5’-UTR und die Promotorsequenz (plo) sind enthalten und reflektiert somit die
Transkription des period Gens.
B) neben der flankierenden 5’-UTR-Sequenz, dem Promotor und nicht-kodierenden
Bereichen ist zudem noch ⅔ der kodierenden Sequenz enthalten (BG-luc),d.h. das PER
Protein wird betrachtet.
Alle Reporterkonstrukte bestehen aus Teilen der period gDNA, die im Leseraster mit 2
kb luciferase cDNA verbunden ist (Stanewsky et al., 1997b, Veleri et al., 2003).
17

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
Ein weiteres wichtiges Gen für die innere Uhr – vor allem bei der Lichtrezeption – stellt
cryptochrome dar. Um die lichtabhängige Degradierung des Cryptochrome Proteins zu
untersuchen, soll ein Fusionsprotein aus Cryptochrome und Luciferase verwendet werden.
Bei der Durchführung einer Messung werden nun die Fliegen mit luciferinhaltigem
Medium gefüttert und in verschiedenen künstlichen Tagesregimes (LD-Zyklen,
Temperaturzyklen) gehalten. Die emittierten Photonen jeder einzelnen Fliege protokolliert
18

Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
dabei stündlich ein automatisierter Szintillationszähler (LumiStar, BMG).
Zur Biolumineszenzmessung werden Fliegen in Microtiterplatten einsortiert, die
luziferinhaltiges Futter enthalten . Abhängig von der Testreihe werden bis zu neun
Platten in einem Fach des Szintillationszählers vermessen.
Durchführung:
Zu untersuchende Genotypen:
Bg-Luc plo Tim-Luc-Cry
Test adulter Fliegen
Mikrotiterplatten mit 96-wells (Packard OptiPlate ®) werden alternierend mit 100 µl eines
Luciferinmediums befüllt.
Herstellung Luciferinmedium:
1% Bacto-Agar, 5% Saccharose, 10mM Luziferin Biotium (aus 100mM Stock). Um die
Agarose aufzuschmelzen muss diese zunächst in der Mikrowelle erhitzt werden.
Wichtig: Luciferin erst nach dem abkühlen dazu geben, da etwas Hitzelabil.
Darin werden mit Diethylether oder CO2 betäubte Fliegen einsortiert (2-5 Tage alt) und
mit Plastikkappen bedeckt (200 µl PCR-Tube-Deckel, mit Luftlöchern), um die Fliegen
während der Messung in der richtigen Position der Z-Achse zu halten. Jede Platte
wird mit einer Plastikschutzfolie versiegelt und mit Luftlöchern versehen.
Semikulturen verschiedener Fliegengewebe
Für die Gewebekulturen wurden die wells einer Mikrotiterplatte mit 100 µl eines
Zellkulturmediums befüllt (88% Insect X-Press, 10% hitzeinaktiviertes fötales
Rinderserum, 1% Penizillin-Streptomycin-Mix, 2mM (aus 100mM Stock) Biosynth-
Luziferin, 0.5 % Insulin; Ivanchenko et al., 2001). Nach deren Transfer in die Wells der
Platte wurde diese mit einer Plastikfolie ohne Luftlöcher versiegelt. Eine wichtige Rolle bei
19
Kursteil I: Nukleinsäuren Mo, 08. März - Fr, 12. März 2010 (Erste Woche)
der Präparation scheinen dabei Schnelligkeit, Unversehrtheit der Gewebe und eine sterile
Zubereitung der Lösungen zu spielen. Es sollen Köpfe, Flügel und Beine untersucht werden.
Auftrageschema:
20

Kursteil II: Proteinanalyse Mo, 15. März - Fr, 19. März 2010
(Zweite Woche)
Die Forschung in Genetik hat in den letzten Jahrzehnten die grundlegenden Mechanismen
aufgeklärt, wie genetische Information gespeichert wird, wie sie von Zelle zu Zelle und
Generation zu Generation weitergegeben wird, und wie sie verwendet wird, um die zellulären
Funktionen zu realisieren und aus einer befruchteten Eizelle einen komplexen Organismus zu
entwickeln. Auf diesen Kenntnissen beruhen letzten Endes auch die Techniken, mit denen
Zellen oder Organismen gentechnisch manipuliert werden können, wie Sie es hier im Kurs
kennen lernen. Das Forschungsinteresse des Lehrstuhls für Genetik und Neurobiologie liegt
auf dem Einsatz dieser genetischen Techniken zur Untersuchung der Funktion des Gehirns.
Neben „Genomics“, dem Forschungsansatz, bei dem ganze Genome sequenziert und
analysiert werden, und „Transkriptomics“, wobei durch Chip-Technologie alle Transkripte
eines Gewebes identifiziert werden, spielt für die biologische Funktionsanalyse vor allem
„Proteomics“ eine zentrale Rolle, ein Ansatz, bei dem das „Proteom“, d.h. die Gesamtheit
aller Proteine aus einem Organismus betrachtet wird. Wichtige Techniken für diesen Ansatz
sind Gel-Elektrophorese (1- und 2-dimensional, oft in Verbindung mit Massenspektroskopie)
und Antikörper. Antikörper sind darüber hinaus auch ein wichtiges Werkzeug bei der
Charakterisierung der Expression von Genen, da man mit ihrer Hilfe das Genprodukt im
sowohl im Homogenat identifizieren als auch im Gewebe mit hoher Selektivität,
Empfindlichkeit und räumlicher Auflösung lokalisieren kann. Mit den beiden entsprechenden
Techniken, dem Western Blot und der Immunhistochemie einschließlich
Confokalmikroskopie, wollen wir in diesem Kursteil Fragen zur Abundanz und Lokalisation
von endogenen und transgenen Genprodukten im Gehirn von Drosophila untersuchen.
Weitere hochaktuelle Methoden, die durch die Gentechnik erst möglich geworden sind,
erlauben die Beobachtung von Fluoreszenz-markierten Proteinen am lebenden Präparat („live
imaging“), die optische Registrierung der Nervenzellaktivität („functional imaging“), und die
optische Reizung von genetisch definierten Nervenzellen im intakten Gehirn. Wichtiges Ziel
des Praktikums ist, Ihnen am Beispiel der Gehirnforschung einen Einblick zu vermitteln, was
man mit den Methoden der modernen Genetik an molekularer, struktureller und funktioneller
Information gewinnen kann.
21

Arbeitsprogramm Gruppe I Immunpräzipitation und Western Blot, larvale
whole mounts, confocale Mikroskopie (Alice, Mandy)
Gruppe II Immunpräzipitation, 2D-gele, Western Blot,
ELISA (Tulip, Bea)
Gruppe III Enhancer trap, confokale Mikroskopie, TRPA1
(Kirsa, Itsaso)
Gruppe IV Calcium Imaging, cAMP Imaging, walking ball,
Chop (Nidhi, Alex)
Gruppe I Immunpräzipitation (IP) und Western Blot, larvale whole mounts,
confocale Mikroskopie (Alice, Mandy)
Proteinisolierung aus adulten Fliegenköpfen mittels Immunpräzipitation
Um die Fliegen auf Proteinebene untersuchen zu können, wird eine Proteinisolierung mittels
einer Immunpräzipitation durchgeführt. Präzipitierte Proteine werden einerseits mit einem
Western Blot, andererseits mit einer Silberfärbung des Gels nachgewiesen. Wichtig bei allen
Experimenten sind Kontrollen. Diskutieren Sie für die IP die Vor- und Nachteile von
Kontrollen mit Präimmunserum, Weglassen des Antikörpers, und Nullmutanten für das zu
präzipitierende Protein.
Immunpräzipitation
Es werden 1ml Fliegen in einem 15ml Falcon abgemessen und in flüssigem Stickstoff
eingefroren. Dann werden sie mittels feinmaschiger Siebe dekapitiert. Die Fliegenköpfe
werden in 800µl gekühltem Lysispuffer homogenisiert, in Eppendorf caps überführt und
abzentrifugiert. Der Überstand wird in ein neues Reaktionsgefäß überführt und für 40
Minuten bei 4°C auf dem Nutator lysiert. Nach einer 30minütigen Zentrifugation wird der
Überstand in ein neues, gekühltes Reaktionsgefäß überführt. Zu diesem Ansatz werden 300µl
von dem Gemisch aus Lysispuffer mit Proteaseinhibitoren und einer complete Mini Tablette
zugegeben. Ebenfalls wird der Antikörper (nc46) in das Reaktionsgemisch pipettiert und für
10 Minuten auf dem Nutator inkubiert. Anschließend werden 100µl Protein G-Agarose
22

zugegeben und über Nacht auf dem Nutator rotiert. Die Protein G-Agarose bindet an die
Immunglobuline, an denen das Antigen gebunden vorliegt. Am nächsten Tag werden die
Agarose-Beads dreimal mit Lysispuffer gewaschen. Zum Schluss wird das Pellet in 40µl 2x
Laemmli-Puffer aufgenommen. Zur Proteindenaturierung wird der Ansatz fünf Minuten bei
96°C aufgekocht, und ein Western Blot kann durchgeführt werden. Die Proben werden in die
Taschen der Fertig-Gele pipettiert und für 1h bei 125 mA laufen gelassen.
Western Blot
Bei einem Western Blot werden Proteine auf eine Nitrocellulosemembran transferiert und
durch eine Immundetektion visualisiert. Dadurch kann man das Molekulargewicht und die
Expressionsstärke eines bestimmten Proteins untersuchen. Für das Blotten wird das Gel aus
den Plastikplatten entnommen und mit Whatmanpaper und einer Membran in Transferpuffer
befeuchtet. Die Blot-Apparatur wird, wie von BioRad angegeben, zusammengebaut. Das
Blotten erfolgt für 1h bei 100V, dadurch werden die Proteine aus dem Gel auf die Membran
übertragen.
Um Proteine auf der Nitrocellulosemembran nachzuweisen, verwendet man den Enhanced
Chemiluminescence Nachweis, mit welchem auch kleine Proteinmengen detektiert werden.
Nach dem Blotten wird die Membran über Nacht bei 4°C in einer 5%igen Milchpulver-
Lösung geblockt, um so noch freie Bindungsstellen abzusättigen. Am nächsten Tag wäscht
man die Membran 3x 10 Minuten mit 1x TBST. Nun wird die Membran mit dem ersten
Antikörper für eine Stunde inkubiert (3c11 oder nc46). Die unspezifisch gebundenen oder
überschüssigen Antikörper werden durch die Waschschritte mit 1x TBST entfernt. Es erfolgt
die Inkubation mit dem zweiten Antikörper, der gegen die Fc-Domäne des ersten Antikörpers
gerichtet ist (HRP anti-mouse 1:7500). Die ECL-Lösung von Millipore wird im Verhältnis
1:1 zusammenpipettiert, und damit wird die Membran für eine Minute überschichtet. An dem
zweiten Antikörper ist das Enzym HRP (horseradish peroxidase) gekoppelt, welches die
Umsetzung von Luminol in die oxidierte Form katalysiert. Bei dieser Reaktion wird Licht
emittiert, und dieses schwärzt den aufgelegten Röntgenfilm.
23

Immunfluoreszenzfärbungen an Larven
Vor Beginn des Versuchs werden die Larven auf Eis betäubt. Um Muskelkontraktionen
vorzubeugen, erfolgt die Präparation der Larven in Ca2+-freier Saline. Dabei wird die Larve
am anterioren und posterioren Pol mit je einer Minutie festgesteckt, dorsal aufgeschnitten und
mit vier weiteren Minutien festgesteckt und die inneren Organe entnommen. Die larvalen
Proteine werden durch das im Fixativ enthaltene Paraformaldehyd für 15 Minuten bei
Raumtemperatur fixiert. Anschließend wird 2x 15 Minuten bei Raumtemperatur mit 0,1%
PBST gewaschen. Die unspezifischen Bindungen werden durch das Aufbringen der
Blocklösung für eine Stunde bei Raumtemperatur besetzt. Danach kann der erste Antikörper
(nc82) in die Blocklösung (0,1mg BSA, 250µl Pferdeserum, 5ml PBST) pipettiert und über
Nacht bei 4°C inkubiert werden. Um überschüssigen Antikörper zu entfernen, wird am
nächsten Tag 4x 30 Minuten bei Raumtemperatur mit 0,1x PBST gewaschen. Im Anschluss
daran können die Larven mit dem zweiten, fluoreszenzgekoppelten Antikörper (anti-mouse
Alexa488, HRP-Cy3) für eine Stunde inkubiert werden. Alle folgenden Schritte werden
abgedunkelt durchgeführt, da die Fluoreszenzfarbstoffe lichtempfindlich sind und ausbleichen
können. Dann wird 4x 30 Minuten mit 0,1x PBST gewaschen. Nun können die fertigen
24

Larven in Vectashield eingebettet und bei 4°C aufbewahrt werden. Mit Hilfe eines konfokalen
Laserscanning-Mikroskops können die Präparate angeschaut werden.
Bestimmung der Flugfähigkeit nach Benzer (Benzer, 1973)
Um das Flugvermögen eines Fliegenstamms zu untersuchen, wird der Versuchsaufbau nach
Benzer gewählt. Hierbei dient ein mit Paraffinöl beschichteter 500ml Zylinder als Flugarena
(siehe Abbildung). Es werden jeweils 2x 50 männliche Fliegen jedes Genotyps (Wildtyp und
Nullmutante) getestet. Die Fliegen werden mit Hilfe eines Trichters in den Zylinder geklopft
und bleiben, sobald sie anfangen zu fliegen, an den beschichteten Wänden kleben. Nun kann
man die fixierten Fliegen auszählen. Anschließend wird anhand einer Excel-Tabelle die
Flugfähigkeit bzw. Flugunfähigkeit berechnet.
Abbildung: Versuchsaufbau zur Ermittlung der Flugfähigkeit (Benzer et al., 1973).
Präferenz Versuch
Bei diesem Versuch untersuchen wir, ob Fliegen auf bestimmte Alkoholwerte reagieren
können. Hierbei wird Mangosaft mit 5% oder 23% Ethanol versetzt. Es werden je 50
männliche Fliegen (Wildtyp und Nullmutante) gesammelt und über Nacht ruhen gelassen. Am
nächsten Tag werden diese Fliegen in große Bechergläser geschubst und über Nacht auf
Lichttischen belassen. Am nächsten Morgen wird ausgewertet.
2-D-Gelelektrophorese
Die 2D-Gelelektrophorese dient dazu, Proteingemische aufzutrennen. Für die Auflösung der
Proteingemische werden die Proteine durch isoelektrischen Fokussierung getrennt und danach
erfolgt eine SDS-Polyacrylamidgelelektrophorese. Die isolelektrische Fokussierung erfolgt
anhand der Zusammensetzung des Proteins aus sauren und basischen Aminosäuren (1.
25
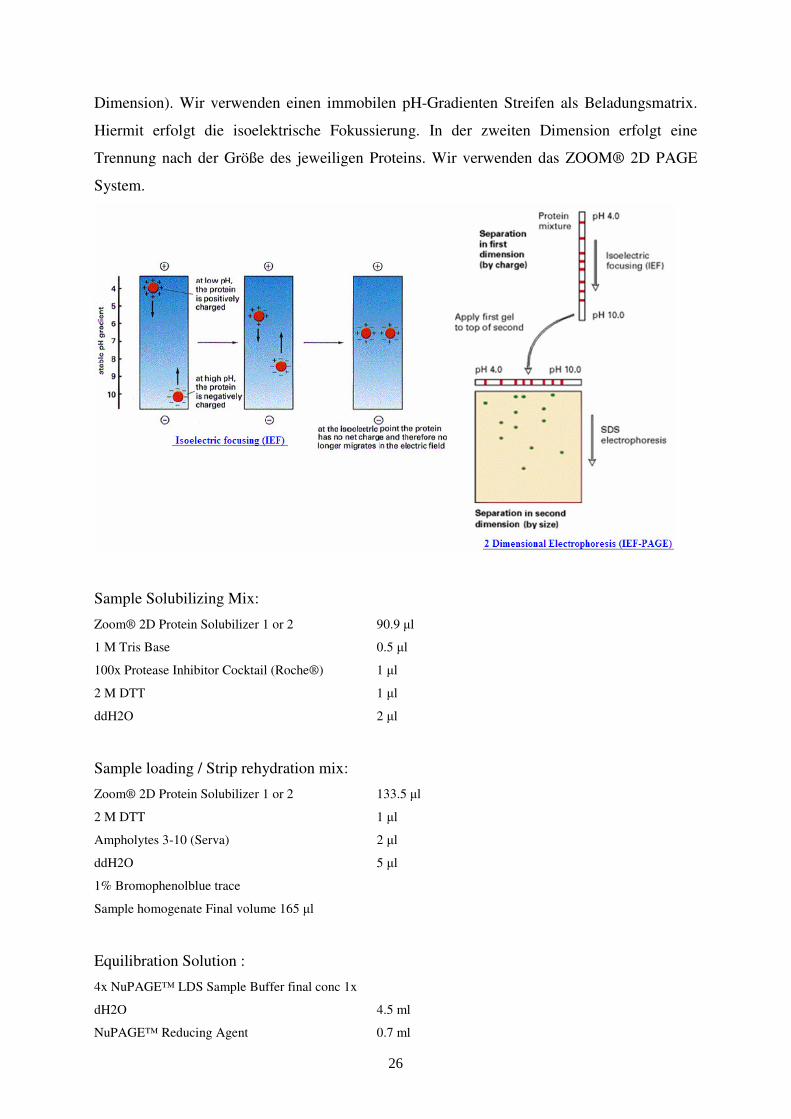
Dimension). Wir verwenden einen immobilen pH-Gradienten Streifen als Beladungsmatrix.
Hiermit erfolgt die isoelektrische Fokussierung. In der zweiten Dimension erfolgt eine
Trennung nach der Größe des jeweiligen Proteins. Wir verwenden das ZOOM® 2D PAGE
System.
Sample Solubilizing Mix:
Zoom® 2D Protein Solubilizer 1 or 2 90.9 µl
1 M Tris Base 0.5 µl
100x Protease Inhibitor Cocktail (Roche®) 1 µl
2 M DTT 1 µl
ddH2O 2 µl
Sample loading / Strip rehydration mix:
Zoom® 2D Protein Solubilizer 1 or 2 133.5 µl
2 M DTT 1 µl
Ampholytes 3-10 (Serva) 2 µl
ddH2O 5 µl
1% Bromophenolblue trace
Sample homogenate Final volume 165 µl
Equilibration Solution :
4x NuPAGE™ LDS Sample Buffer final conc 1x
dH2O 4.5 ml
NuPAGE™ Reducing Agent 0.7 ml
26

Alkylation Solution:
1x NuPAGE™ LDS Sample Buffer 400 ml
Iodoacetamide 0.0093 g
Vorgehensweise:
Die Wildtypfliegen werden gesammelt, in Stickstoff tiefgefroren und dekapitiert.
Die Köpfe werden in 95 µl Solubilisierungsmix homogenisiert und 2x für 15 min bei 13000
rpm bei 4°C zentrifugiert.
Dann wird der Rehydratationsmix hergestellt und in die ZOOM® IPGRunner® Kassette
geladen. (Endvolumen 165 µl).
Die Streifen werden in die Kassette hineingeführt und Luftblasen entfernt. Die Streifen
bleiben über Nacht auf 18 °C.
Auf die ZOOM® IPGRunner® Kassette werden Wattepads geklebt und diese werden mit 600
µl Wasser angefeuchtet. Danach wird die Kassette in die Gelkammer eingespannt und die
äußere Kammer wird mit 600ml Wasser gefüllt. Die Fokussierung erfolgt bei 1500Vh.
Danach wird die Kassette entnommen und bei -80°C tiefgefroren.
Am nächsten Tag werden die Streifen in Alkylierungslösung für 15 min unter Schütteln
behandelt. Danach werden die Streifen in die fertigen 4-12% Bis-Tris NuPAGE™ 2DE Gele
positioniert und mit 400 µl der 0.5% Agarose bedeckt und eine SDS-PAGE wird
durchgeführt.
Gruppe III Enhancer trap, confokale Mikroskopie, TRPA1
(Kirsa, Itsaso)
Einleitung
In den letzten Jahrzehnten wurden viele molekularbiologische Färbemethoden, wie
zum Beispiel: Antikörperfärbung, in situ RNA Hybridisierung entwickelt. Diese bieten die
Möglichkeit Neurotransmitter oder endogene bzw. transgene Proteine oder deren genetische
Vorläufer (mRNAs) in Geweben zu lokalisieren.
Wir werden uns in diesem Versuchsteil primär darauf konzentrieren die Expression von so
genannten GAL4-Linien zu bestimmen, welche für unsere Forschung essentiell sind. Dabei
handelt es sich um genetisch veränderte Fliegen, in deren Genom ein P-Element inseriert
wurde, welches den Hefe-Transkriptionsfaktor GAL4 trägt. Je nachdem unter Kontrolle
27

welchen Promotors oder Enhancers dieses P-Element liegt, wird GAL4 an bestimmten Stellen
im Gewebe bzw. Gehirn exprimiert. Dadurch kann bestimmt werden „Wo“ Expression in der
Fliege stattfindet. Die Insertion des P-Elementes geschieht dabei meist jedoch zufällig.
Deswegen muss im Nachhinein bestimmt werden in welchen Zellen der GAL4-
Transkriptionsfaktor exprimiert wird. Dies könnte direkt mit einer Färbung mit Antikörpern
gegen GAL4 geschehen. Eine höhere Empfindlichkeit erreicht man jedoch, wenn man das
Produkt eines Gens nachweist, dessen Expression durch GAL4 verstärkt (enhanced) wird.
Dafür werden nun die GAL4-Fliegen mit anderen transgen veränderten Fliegen, den sog.
UAS-GFP-Fliegen gekreuzt. In das Genom der UAS-Fliegen wurde ein P-Element inseriert,
das mehrere Kopien des UAS-Enhancers (upstream activating Sequence) vor einem
beliebigen Gen mit schwachem Promotor beinhaltet. Dieser Enhancer wird bei Bindung des
GAL4-Transkriptionsfaktors aktiviert und treibt dann die Expression des gekoppelten Gens
(„Was“), hier des Gens für GFP (grün fluoreszierendes Protein) als „Reporter“. Im Kurs
werden wir Färbungen mit Antikörpern gegen GFP an ganzen Gehirnen und Gehirnschnitten
durchführen und somit das GAL4-Expressionsmuster sichtbar machen.
Außerdem werden wir zwei Verhaltensversuche durchführen, die u.a. dazu dienen die Rolle
bestimmter Proteine aufzuklären.
A) Immunhistologie an „Whole mounts“ – Konfokale Mikroskopie
Konfokale Laser-Scanning Mikroskopie
Das Prinzip eines konfokalen Mikroskops wurde von Marvin Minsky in den 1950er Jahren
entwickelt.
Es beruhte auf der Fokussierung von Licht auf einen einzelnen Punkt, mit dem das Präparat
abgescannt wird. Dieses Prinzip macht das Besondere eines konfokalen Mikroskops aus.
Denn im Gegensatz zur konventionellen Lichtmikroskopie wird nicht das gesamte Präparat
beleuchtet, sondern zu jedem Zeitpunkt nur ein Bruchteil davon, in den meisten Fällen nur ein
beugungsbegrenzter Lichtfleck. Mit diesem kleinen Lichtfleck wird das Präparat Punkt für
Punkt abgerastert. Im Mikroskop entsteht also zu keinem Zeitpunkt ein vollständiges Bild.
Die Lichtintensitäten des emittierten Lichtes werden nacheinander an allen Orten des
Präparats gemessen, so dass eine anschließende Konstruktion des Bildes möglich ist. Die
28

Besonderheit des Konfokalmikroskops besteht darin, dass im Strahlengang des detektierten
Lichts eine Lochblende (englisch: Pinhole) angebracht ist, die Fluoreszenz-Licht von
außerhalb der Schärfeebene weitgehend blockiert. Dadurch verbessert sich die Tiefenschärfe
erheblich, wodurch wiederum die Auflösung entlang der optischen Achse (z-Richtung) steigt.
Die Konfokaltechnik umfasst eine Reihe von optischen Messverfahren die auf dem
Konfokalprinzip basieren: zwei optische Systeme sind konfokal, wenn sie einen gemeinsamen
Brennpunkt besitzen. Der Beleuchtungsstrahlengang und der Beobachtungsstrahlengang sind
somit konfokal.
Das Konfokale Grundprinzip: Eine Lichtquelle beleuchtet eine sehr kleine Lochblende. Das
Bild der Lochblende wird beugungsbegrenzt auf das Objekt in fokussiert. Das reflektierte und
gestreute Licht (oder Fluoreszenzlicht) von der Probe wird über einen Strahlteiler auf eine
zweite Lochblende, hinter der sich ein Detektor befindet, abgebildet. Diese Anordnung sorgt
dafür, dass Licht, das von der Probe außerhalb der Fokusebene stammt, weitgehend
ausgeblendet wird. Dadurch misst der Sensor eine erhöhte Lichtintensität, wenn das Objekt
im Fokus ist, detektiert aber nahezu keine Intensität, wenn sich das Objekt außerhalb des
Fokus befindet. Dieses Prinzip kann grundsätzlich auch mit Weißlicht zum Einsatz kommen,
wird heute aber in der Regel für Fluoreszenzmikroskopie unter Anregung mit einem
monochrmatischen Laser verwendet.
Der Punktsensor muss in allen drei Raumrichtungen über das Objekt geführt werden um eine
vollständige 3D-Abbildung zu erhalten. Die Bewegung entlang der optischen Achse (z-
Achse) erfolgt in der Regel durch Verschieben der Probe (Mikroskoptisch), das Scannen in
der x-y-Ebene aber durch einen schnell schwingenden Spiegels im Strahlengang.
29

Welche Genotypen?
Im Praktikum werden wir uns die Expression verschiedener GAL4-Linien anschauen, welche
Teil unserer aktuellen Forschung sind. Das heißt, dieser Versuch wird nicht nur zum Spaß
oder zum Lernen durchgeführt, sondern trägt bei Gelingen tatsächlich zu unserem Tagewerk
bei. Dabei werden im Prinzip zwei große Themengebiete im Focus stehen.
Zum einen soll das Expressionsmuster von GAL4-Linien aufgedeckt werden, welche
GAL4 in Neuronen eines der großen Neuropilkomplexe des Zentralhirns exprimieren, dem
Zentralkomplex. Der Zentralkomplex liegt in der Mitte des Zentralhirns und besteht aus 4
Kompartimenten, der Protocerebralbrücke (pb), dem Fächerförmigen Körper (fb), den Noduli
(no) und dem Ellipsoidkörper (eb) (Reihenfolge nach der Lage im Zentralhirn von posterior
nach anterior).
Der Zentralkomplex ist ein prominenter Teil des Zentralhirns, der sich durch alle
Insektenarten zieht. In Grashüpfern zum Beispiel wurden Neurone des Zentralkomplexes
gefunden, die bei der Orientierung nach polarisiertem Licht eine Rolle spielen. Außerdem
spielt der Zentralkomplex eine wesentliche Rolle bei der Lokomotion von Insekten. Mutante
Fliegen, die eine Strukturveränderung des Zentralkomplexes aufweisen, laufen zum Teil
langsamer, sind weniger aktiv oder werden schnell „faul“, laufen in Kreisen oder machen
kleinere Schritte. Aber auch bei der Bildung verschiedener Gedächtnisformen spielt der
Zentralkomplex eine Rolle. So konnte gezeigt werden, dass Neurone bestimmter Schichten
des Fächerförmigen Körpers notwendig und hinreichend sind, um verschiedene
Objektparameter bei aversiver Konditionierung im Flug zu diskriminieren. Dem
Ellipsoidkörper hingegen werden Rollen bei der Bildung des aversiven olfaktorischen
Gedächtnisses und des räumlichen Gedächtnisses (siehe, Versuchsteil D) zugeschrieben.
Ziel ist es hier GAL4 Linien zu detektieren, die aufgrund ihrer Expression in der
Protocerebralbrücke für zukünftige Lauf- und Lernversuche (Buchner Ball, Imaging s.a.:
Gruppe IV , Betreuer: Nidhi und Alex) verwendet werden können.
fb
no
pb
eb
30

Zum anderen, soll das Expressionsmuster einer GAL4-Linie bestimmt werden, bei der
das P-Element in die 5`Region des Gens CG12214 inseriert ist. Dieses Gen, das hohe
Homologie zum Tubulin-bindenden Chaperon Kofaktor E besitzt, wurde in einem Yeast-
Two-Hybrid Experiment aufgrund seiner Interaktion mit Sap47 für uns interessant (s.a.:
Gruppe II, Betreuer: Tulip und Bea)
Um die Expression der GAL4-Linien zu detektieren, müssen diese mit einem
transgenen Reporterstamm gekreuzt werden, der ein Zielgen trägt, welches die Visualisierung
der GAL4 positiven Neurone erlaubt. Dabei gibt es verschiedene Möglichkeiten, wie zum
Beispiel UAS-lacZ als Reporter mit anschließender lacZ Färbung. Die mittlerweile
prominenteste Art Neurone in Drosophila melanogaster sichtbar zu machen beinhaltet das
grün fluoreszierende Protein (GFP) aus der Qualle Aequora victoria. Das mittels UAS-GFP
(im Kurs verwenden wir UAS-mCD8-GFP, eine membranständige GFP-Form) exprimierte
GFP kann mithilfe eines Fluoreszenzmikroskops (z.B. Konfokales Mikroskop) bei passender
Anregungswellenlänge beobachtet werden. Wir werden das Fluoreszenzsignal durch
zusätzliche Antikörperfärbung (1.ab: anti-GFP; 2.ab: Alexa488) noch verstärken.
Dafür wurden Jungfrauen folgender GAL4-Linien:
a) w+/w+; +/+; 007Y-GAL4/TM3Sb
b) w+/w+; +/+; c232-GAL4/c232-GAL4
c) w-,NP2370-GAL4/FM7c; +/+; +/+
d) w-,NP5075-GAL4/FM7c; +/+; +/+
e) w-,MZ0604-GAL4/FM7c; +/+; +/+
f) w-/ w-; NP4786-GAL4/CyO; +/+
mit Männchen folgenden Genotyps gekreuzt:
a) yw,UAS-mCD8::GFP/Y; UAS-mCD8::GFP/CyO; UAS-mCD8::GFP/UAS-
mCD8::GFP
Zur Vorbereitung auf das Praktikum möchte ich Euch bitten, Kreuzungsschemata
aufzustellen, mit allen möglichen Genotypen der F1 Generation, welche wir dann am 1.
Praktikumstag besprechen werden. [FM7c, TM3Sb, CyO sind Balancer]
Protokoll:
Präparation: Fliegen auf Eis kühlen
31

Mit Minutiennadeln in Thorax und Abdomen fixieren, mit Ringerlösung
überdecken
Rüssel raus, Kopfkapsel abziehen, Tracheen entfernen
Freigelegte Gehirne in Eppis mit Fixans überführen
20 Köpfe/ml Fixans 0,3% Triton X-100
1 x PBS
2% Paraformaldehyd
Paraformaldehyd (4%ig)
2g Paraformaldehyd in 17.5 ml H2O bis 60°C (Thermometer) erhitzen,
15 min rühren, mit 100 µl frischer 1N NaOH klären, abkühlen lassen
+ 20 ml 1/15 M Na2HPO4 (Merck-Lösung)
+ 1/15 M KH2PO4 (Merck-Lösung) auf pH 7,4 einstellen, mit
beiden Merck Lösungen auf 50 ml, pH 7,4 auffüllen.
Fixierung : 2h / Taumler
Waschen: 3 x 10min mit 0,3% PBT / RT / Taumler / pro Eppi 600µl/ PBT
Blocken: 1,5h / 10% normal (goat) serum in 0,3% PBT / 4°C / Taumler
Prim. Antikörper: α-GFP: 1:2000 in 0,3% PBT + 3% normal (goat) serum / 200 µl/Eppi/
4°C / Taumler / über Nacht
Waschen: 6 x 10min mit 0,3% PBT / RT / Taumler
Sek. Antikörper: Alexa 488: 1:2000 in 0,3% PBT + 3% normal (goat) serum / 4°C /
Taumler / über Nacht
Waschen: 6 x 10min mit 0,3% PBT / RT / Taumler
Eindeckeln: Deckgläser ( Menzel-Glaser 20x20mm #00) doppelt mit Lochverstärkern
bekleben, in die Mitte einen kleinen Tropfen VECTOR Vectashield Mounting Medium H-
1000 geben. Dort hinein die Köpfe einbetten. Nagellack in die Ecken des Deckglases geben
und eindeckeln.
Literatur:
Kei Ito, Ryuichi Okada, Nobuaki Tanaka Nobuaki, and Takeshi Awasaki; Cautionary
Observations on Preparing and Interpreting Brain Images Using Molecular Biology-Based
Staining Techniques; Microscopy Research and Technique. 2003, 62:170-186
32

Aso Y; Grübel K, Busch S, Friedrich AB, Siwanowicz I, Tanimoto H; The mushroom
body of adult Drosophila characterized by GAL4 drivers. J Neurogenet. 2009; 23(1-2):156-72
Brand AH and Perrimon N; Targeted gene expüression as a means of altering cell fates
and generating dominant phenotypes, Development. 1993, 188: 401-415
Strauss R, The central complex and the genetic dissection of locomotor behaviour;
Current Opinion in Neurobiology. 2002, 12: 633-638
Liu G, Seiler H, Wen A, Zars T, Ito K, Wolf R, Heisenberg M, Liu L., Distinct
memory traces for two visual features in the Drosophila brain. Nature. 2006 Feb
2;439(7076):551-6.
Pan Y, Zhou Y, Guo C, Gong H, Gong Z, Liu L. Differential roles of the fan-shaped
body and the ellipsoid body in Drosophila visual pattern memory. Learn Mem. 2009 Apr
23;16(5):289-95
Neuser K, Triphan T, Mronz M, Poeck B, Strauss R. Analysis of a spatial orientation
memory in Drosophila. Nature. 2008 Jun 26;453(7199):1244-7
Yeh E, Gustafson K, Boulianne GL. Green flourescent protein as a vital marker and
reporter of gene expression in Drosophila. PNAS 1995, 92:7036-7040
Dissertation Natalja Funk, 2003, The Sap47 gene of Drosophila melanogaster:
mutagenesis and identification of interaction partners; University Würzburg
B) Immunhistologie auf Kryoschnitten
Um die Expression einer GAL4-Linie zu bestimmen oder die Expression einzelner Proteine,
zum Beispiel synaptischer Proteine, Neurotransmitter oder transgen exprimierter Proteine
kann man u.a. Gefrierschnitte des zu untersuchenden Gewebes mittels eines Kryostat-
Mikrotoms anfertigen. Diese Methode erlaubt es besonders dünne Gewebeschnitte zu
bekommen, um anschließend mittels Antikörperfärbung Expressionen sichtbar zu machen.
Im Praktikum werden wir die Expression einer GAL4-Linie bestimmen, welche in den
Ringneuronen des Ellipsoidkörpers färben sollte. Die Ringneurone arborisieren ringförmig
auf den Ellipsoidkörper - daher ihr Name. Dort bilden sie Präsynapsen aus und gehören somit
zu den Input-Neuronen des Zentralkomplexes, den sog. Großfeldneuronen.
Als erstes muss das Gewebe (hier: das Fliegengehirn) fixiert werden, um alle molekularen
Strukturen und Proteine intakt zu halten, damit später durch die Antikörperfärbung die
entsprechende Lokalisation stattfinden kann. Danach wird das Gewebe eingebettet und in
flüssigem Stickstoff gefroren. Nun kann es im Kryostaten geschnitten werden. Die Schnitte
werden auf beschichtete Objektträger überführt und bis zur folgenden Antikörperfärbung im
33

Kryostaten bei –20°C aufgehoben. Für die Antikörperfärbung werden wir ein sogenanntes
ABC-Kit (ABC=Avidin-biotin-complex) verwenden. Die ABC Methode basiert auf der
Bildung eines Avidin-Biotin-Komplexes, welcher letztendlich dazu führt, dass die
Empfindlichkeit der Antikörper erhöht wird und die Deutlichkeit der Nachweisreaktion
zunimmt. Dabei bindet der Primärantikörper an das zu detektierende Antigen. Dieser könnte
nun selbst mit einem Nachweismolekül markiert sein, was aber in der Herstellung sehr
aufwendig und damit kostenintensiv ist. Aus diesem Grund wird im Allgemeinen ein
Sekundärantikörper benutzt, welcher gegen die konstante Region des Primärantikörpers
gerichtet ist. Der sekundäre Antikörper ist bei Verwendung des ABC Systems biotinyliert.
An dieses Biotin kann nun im Folgenden der zugegebene Avidin-Biotin Komplex binden,
wobei Avidin (tetrameres Glycoprotein aus Hühnereiweiß) mit sehr hoher Affinität an das
Biotin bindet. Da an einen sekundären Antikörper mehrere Biotinmolkelüle konjugiert sind,
können auch mehrere AB-Komplexe an einen sek. Antikörper binden. Dies resultiert in der
Bildung makromolekularer Komplexe, welche die Empfindlichkeit der Nachweisreaktion
deutlich erhöhen. Der Avidin-Biotin-Komplex ist seinerseits mit HRP (horseradisch
peroxidase) markiert, welche als Nachweisenzym dient. Als Nachweissubstrat werden wir
DAB (Diaminobenzidin tetrahydrochlorid) verwenden, welches eine Braunfärbung des
Prezipitats bewirkt.
Genotypen:
G0:
virgins yw,UAS-mCD8::GFP/ yw,UAS-mCD8::GFP; UAS-mCD8::GFP/CyO; UAS-
mCD8::GFP/UAS-mCD8::GFP x w+/Y; +/+; c232-GAL4/c232-GAL4 males
Protokoll:
• Fixative: (Prepared freshly as described below)
34

o 4% PFA (2g in 25ml dH2O) heated to 60ºC with constant stirring o 100 µl of 1M NaOH was added and the solution turned clear o On complete dissolution it was cooled to 20ºC o 19 ml of (1/15)M Na2HPO4 and 5 ml of (1/15)M K2HPO4 were added o pH adjusted to 7.4 by (1/15)M K2HPO4 and final volume made to 50 ml o Solution was kept on ice till used.
• Drosophila Ringer NaCl 7.48g
KCl 0.35g CaCl2 0.2g Na2HPO4 0.105g KH2PO4 0.048g Final volume made upto 1l with dH2O
• Wash and Cryoprotectant 25% Saccharose (25g in 84 ml of above Ringer solution)
• Embedding medium 16% Carboxymethyl Cellulose (1.6g in 10ml
dH2O)
• PBS (10X) 14.8 g Na2HPO4 4.3 g KH2PO4 72.0 g NaCl pH adjusted to 7.4 and final volume made to 1l by dH2O Protocol:
o 5 adult, large ♀ CS flies were anesthetized by CO2 and placed on a glass petri dish kept on
ice bath.
o The flies were glued by their dorsal thoraces to a thin fiber stick using nail polish, which
was allowed to dry for 10 mins.
o Fiber sticks with immobilized flies were dipped in EtOH for 3 seconds and jerked to shed
off hairs, bristles etc and excess EtOH was soaked away.
o The flies immobilized on the fiber stick were then dipped in 5ml of freshly prepared 4%
PFA solution (taken in a small glass tube) and incubated for 30 mins on ice.
o Thereafter the fiber stick with flies was mounted under a stereomicroscope and the
proboscis and ventral air sacs were removed from each fly head under visual control with
fine tweezers.
o The flies (on fiber) were again incubated in 4% PFA solution for 3hrs at 4ºC.
o They were then incubated in 25% Saccharose solution at 4ºC overnight.
o Next morning the heads of the flies (on fiber) were cut off under a microscope.
o The heads were embedded in a small drop of 16% CMC placed on a special holder.
o After adjusting the head to the correct orientation, the holder was slowly dipped into
liq.N2, to gradually freeze it bottom to top.
35

o The holder with the frozen head embedded in CMC was then attached to a cryotome
(2800 Frigocut E, Reichert Jung®) and 10 µm thick cryosections were cut at -26ºC.
o The sections were collected on a subbed microscope slide one beside the other, briefly
thawed, refrozen and then air dried.
o The sections on the slide were blocked with Vectastain® normal serum (same species in
which the biotinylated 2º Ab was generated) diluted 50 µl in 3.3 ml 1x PBST for 2hrs at
RT in a humid chamber.
o Thereafter the sections were incubated with 1º MAb (na21 diluted 1:3 in 1x PBST) at 4ºC
overnight in a humid chamber.
o Next morning the excess 1º MAb was drained off and the slide was washed once briefly
and twice for 10 min each with 1x PBST.
o Biotinylated 2º Ab (α-mouse IgG) was added to the sections (5 µl/ml in 1x PBST)
and incubated at 37ºC for 1 hr.
o Just 5 mins before the end of the 2º Ab incubation, reagent A (avidin) and reagent B
(biotin-HRP) both diluted to 10 µl/ml were mixed to form the ABC.
o The slide was washed once briefly and twice with 1x PBST as described above.
o ABC was added to the slide and incubated at 37ºC for 1 hr.
o Excess ABC was discarded and the peroxidase substrate DAB diluted 2 drops in 1 ml
Buffer1 (both from Linaris®) was added to the sections and observed under a microscope
to check the development of the coloured precipitate.
o The reaction was stopped upon development of suitable colour intensity by dipping the
slides sequentially in dH2O and 1x PBST.
o The slides were then air dried and overlayed with Kaiser’s Glycerol Gelatin (Merck®)
prewarmed to 60ºC.
o Then a cover slip was laid upon it to permanently mount them.
o The slides were allowed to dry overnight and then observed next day.
o The slides can be stored at RT.
Literatur:
Hsu SM, Raine L, Fanger H., Use of Avidin-Biotin-Peroxidase Complex (ABC) in
Immunoperoxidase Techniques. J Histochem Cytochem. 1981 Apr;29(4):577-80.
C) Verhaltensversuch A: Expression eines Wärmesensors
In diesem Versuch wird in Fliegen ein Ionenkanal exprimiert, dessen Funktion es ist Wärme
zu detektieren. Dieser TrpA1-Kanal gehört zur Familie der TRPs (transient receptor
36

potential), welche die Temperaturwahrnehmung in Insekten, Säugetieren und Menschen
vermitteln. In Drosophila melanogaster ist dieser Kanal normalerweise in einer bestimmten
Zellgruppe (AC Neurone) notwendig, um Wärme wahrzunehmen. Exprimiert man TrpA1
mithilfe des GAL4/UAS Systems nun zusätzlich in anderen Neuronen, werden auch diese auf
Wärme reagieren. Wir benutzen im Kurs eine GAL4-Linie, die Expression panneural treibt
(elav-GAL4), um den TrpA1-Kanal (UAS-TrpA1) in allen Neuronen zu exprimieren.
Dadurch werden alle Neurone temperatursensitiv und wir wollen nun schauen, welchen Effekt
dies auf die Fliegen hat.
Genotypen:
G0: elav-GAL4/FM7a; +/+; +/+ x w/Y; +/+; UAS-TrpA1/Uas-TrpA1
(Jungfrauen) (Männchen)
Protokoll:
# Fliegen werden in leere, beschriftete Gläser überführt
# Gläser für 60s in ein 37°C warmes Wasserbad oder in eine 37°C warme Klimakammer
stellen
# Gläser auf Raumtemperatur bringen und Fliegen erholen lassen
Die gesamte Prozedur soll mit einer Digitalkamera festgehalten werden.
Literatur:
Hamada FN, Rosenzweig M, Kang K, Pulver SR, Ghezzi A, Jegla TJ, Garrity PA. An
internal thermal sensor controlling temperature preference in Drosophila. Nature. 2008 Jul
10;454(7201):217-20
D) Verhaltensversuch B: Das räumliche Gedächtnis
Drosophila melanogaster Fliegen besitzen ein räumliches Gedächtnis, d.h. sie sind in der
Lage sich die Position eines Objektes im Raum zu merken, sich daran zu erinnern und die
positionelle Information in zielgerichtetes Orientierungsverhalten zu integrieren. Um diese
Form des Gedächtnisses zu untersuchen, wurde von uns das sogenannte „detour-paradigma“
(s. Abbildung unten) entwickelt. Einzelne Fliegen, denen ein Tag vor dem Experiment die
Flügel gestutzt wurden, um ein Wegfliegen zu verhindern, werden zu Beginn des Experiments
in eine zylindrische LED-Arena gesetzt. Diese Arena ermöglicht es eine virtuelle Realität zu
erzeugen. Das Experiment umfast nun drei Phasen. In der ersten Phase werden der Fliege
37

zwei schwarze Streifen gegenüber voneinander präsentiert. Wildtypische Fliegen patrolieren
zwischen diesen beiden Streifen hin und her. Bülthoff et al. 1982 haben gezeigt, dass Fliegen
dies enorm lang stereotyp immer weiter machen, nach ca. acht Stunden wurde das Experiment
von den Experimentatoren abgebrochen. Im Detour Paradigma wird der Fliege erlaubt einmal
zwischen den Streifen zu patrolieren, überquert die Fliege dann wieder die Mittellinie, beginnt
Phase zwei des Experiments. Die beiden Streifen verschwinden und gleichzeitig erscheint
lateral ein neuer Streifen, der sog. Ablenkungsbalken. Wildtypische Fliegen drehen sich dann
meist sofort zu dem Ablenkungsbalken hin. Dieser wird, sobald die Fliege diesen fixiert hat
für weitere 500ms präsentiert und verschwindet dann ebenfalls. In der dritten Phase wird nun
aufgenommen, ob sich die Fliege wieder in die Richtung des zuvor frontalen Streifens
bewegt, also ihren initialen Anlauf beenden will.
Wildtypische Fliegen drehen sich tatsächlich in die initiale Richtung zurück und nehmen
wieder ihren Anlauf zu der Position des zuvor frontalen, jetzt nicht sichtbaren Streifens auf.
Mit anderen Worten, die Fliegen sind in der Lage sich die Position eines visuellen Objektes
zu merken, sich daran zu erinnern und die Information in ihr Verhalten zu integrieren- sie
besitzen ein räumliches Gedächtnis. Im Praktikum werden wir zum einen wildtypische
Fliegen des Genotyps Canton-S und transgene Fliegen testen, bei denen ein Gen, welches für
die Proeinkinase S6Kii codiert durch P-Element jump out deletiert wurde. Diese Fliegen
exprimieren keine S6KII Proteinkinase, sie sind also Nullmutanten. Die S6Kii Kinase gehört
zur Familie der ribosomalen serin kinasen (RSK), welche mit dem mitogen-aktivierten
Proteinkinase Signalweg interagieren. Im Praktikum soll nun die Rolle der S6KII Kinase an
der Bildung des räumlichen Gedächtnisses untersucht werden.
Genotypen:
# Canton-S (Wildtyp): w+/w+; +/+; +/+
# ign58/1: w+ ign58/1 / w+ ign
58/1; +/+; +/+
Literatur:
Neuser K., Triphan T., Mronz M., Poeck B., Strauss R. (2008). Analysis of a
38

spatial orientation memory in Drosophila. Nature 453, 1244-1247
Bülthoff H, Götz KG, Herre M. Recurrent Inversion of Visual Orientation
in the Walking Fly, Drosophila melanogaster. J Comp Physiol (1982) 148:471~481
Strauss R, Pichler J, Persistence of orientation toward a temporarily invisible landmark
in Drosophila melanogaster. J Comp Physiol A (1998) 182: 411±423
Putz G, Bertolucci F, Raabe T, Zars T, Heisenberg M., The S6KII (rsk) gene of
Drosophila melanogaster differentially affects an operant and a classical learning task. J
Neurosci. 2004 Nov 3;24(44):9745-51.
Gruppe II Immunpräzipitation, 2D-Gels, Western Blot,
ELISA (Tulip, Bea)
The techniques of immunoprecipitation, 2D-gels and Western blot have been described above,
the method of “enzyme-linked immunosorption assay” (ELISA) is outlined below. We will
use these techniques to characterize synapsin, SAP47, and TBCE-L, three proteins presently
under study. Details will be explained during the course.
ELISA Objective
ELISA (Enzyme -linked immunosorbent assay)
1. To quantitatively determine a neuronal protein in head homogenates of different genotypes of D.melanogaster.
2. Detect a possible difference in Synapsin content in SAP47 null mutant flies when compared to wild-type CS.
Materials
· PBS (10X) 14.8 g Na2HPO4 4.3 g KH2PO4 72.0 g NaCl Added 1 l H2O and adjusted the pH to 7.4 · EDTA 1%
39

Primary antibody Dilution
3c11 1:50
nc46 1:200
· Blocking buffer 1.0 g Bovine Serum Albumin (BSA) (1X) 100 ml of 1X PBS · Detection buffer 12.11 g Tris/HCl 100mM (1X) 0.2 g MgCl2 Added 1 l H2O and adjusted the pH to 9.5 · Antibodies
Secondary antibody : Anti Mouse Biotin conjugate 1:400
Procedure
ELISA is widely used to detect antibody or antigen quantitatively and/or
qualitatively as per the need. In this case the technique will be used to detect the
presence of antigen (protein) . The various steps involved in the technique are as
follows: The antigen is obtained in PBS buffer by homogenizing the fly heads in
PBS buffer using mortar and pestle. About 50 µl of antigen is coated or loaded
onto each well of the ELISA plate. The plate is kept at room temperature on a
shaker for 2 hours. After 2 hours the plate is washed once with water. Blocking
solution is added to each of the wells (300 µl / well). The blocking solution is
prepared by making a 1% solution of BSA in PBS. The plate is then kept on a
shaker for about 2 hours at room temperature. On completion of the blocking
step the primary antibody is added to each well (100 µl / well) and incubated at
4°C on a shaker for 12 hours. The primary antibody solution is prepared by
diluting the antibody in blocking solution.
40

ELISA procedure
PBS washes (3 times) are performed for 10 minutes duration each. In the
next step the secondary antibody (anti-mouse biotin conjugate) is added to each
well (100 µl / well) and the plate is incubated at room temperature for 2 hours.
The secondary antibody solution is prepared by diluting the antibody in blocking
solution (1:400). PBS washes (3 times) are performed for 10 minutes duration
each. In the next step, avidin alkaline phosphatase is added to each of the wells
(100 µl / well) and the plate is incubated at room temperature for 1 hour. The
solution is prepared by diluting avidin alkaline phosphatase in blocking solution
(1:10000). PBS washes (2 times) are performed for 10 minutes duration each
followed by the third wash using detection buffer (Tris/HCl, MgCl2 pH 9.5) for
10 minutes duration. The substrate (pNPP) is added (100 µl / well) and the plate
is kept in the dark until colour development is observed. The developed colour is
measured using the ELISA reader and the data is analysed using statistical
software (Origin Version 7.5).
Gruppe IV Calcium und cAMP Imaging, walking ball, Chop
(Nidhi, Alex)
OPTOGNETIC TOOLS:
41

Channelrhodopsin-2 is a light activated protein (~480nm) which forms a 7 trans-
membrane ion channel. This protein is normally present in green algae Chlamydomonas
reinhardtii and has been cloned in Drosophila (Schroll et al. 2006). The GAL4-UAS system
allows for the expression of Channelrhodopsin-2 in the desired subset of neuronal
populations.
Fig1. Shows the pictorial sketch of working of Channelrhodopsin-2 (depolarising cation channel) and light activated inhibitory protein called halorhodopsin (hyperpolarising).
These tools have been used extensively for optogenetic manipulations because they have
advantages over electrical or chemical stimulation of neuronal populations as the electrical or
chemical stimulation usually leads to non-specific activation or inhibition of undesired
neuronal populations. Using these optogentic tools we can achieve the precise spatial and
temporal manipulation of desired neuronal populations with little or no injury to the brain of
the animal.
.
42
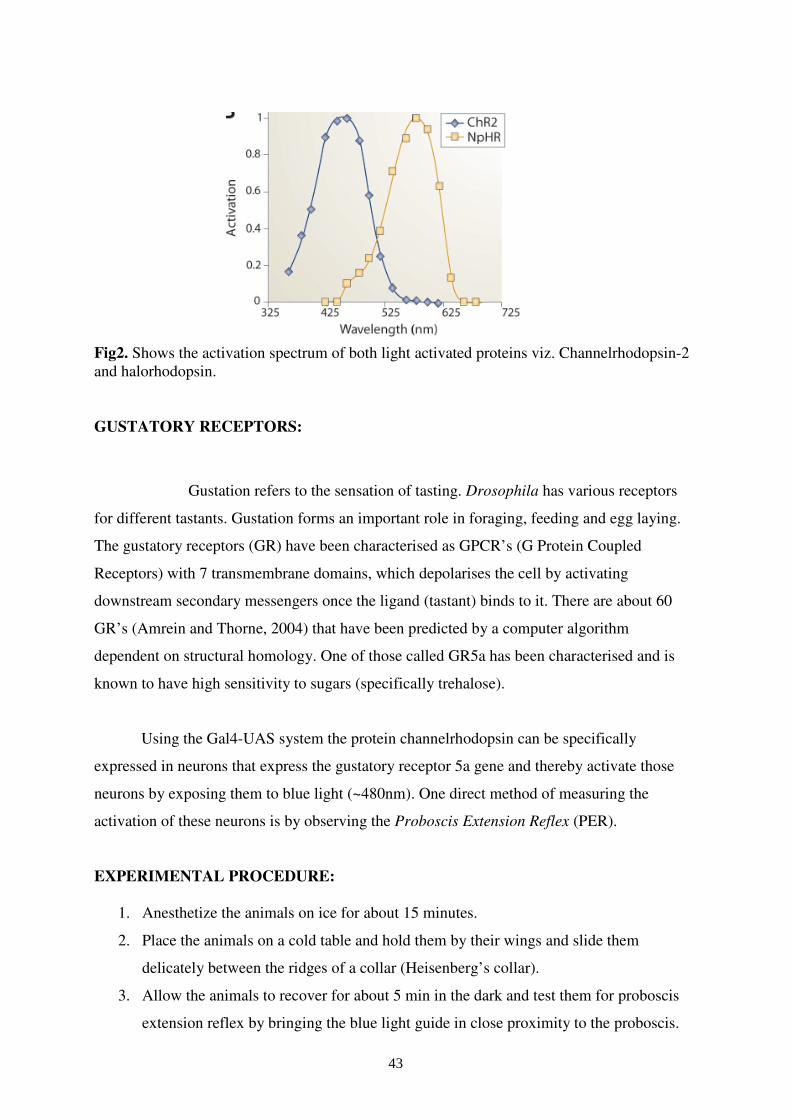
Fig2. Shows the activation spectrum of both light activated proteins viz. Channelrhodopsin-2 and halorhodopsin.
GUSTATORY RECEPTORS:
Gustation refers to the sensation of tasting. Drosophila has various receptors
for different tastants. Gustation forms an important role in foraging, feeding and egg laying.
The gustatory receptors (GR) have been characterised as GPCR’s (G Protein Coupled
Receptors) with 7 transmembrane domains, which depolarises the cell by activating
downstream secondary messengers once the ligand (tastant) binds to it. There are about 60
GR’s (Amrein and Thorne, 2004) that have been predicted by a computer algorithm
dependent on structural homology. One of those called GR5a has been characterised and is
known to have high sensitivity to sugars (specifically trehalose).
Using the Gal4-UAS system the protein channelrhodopsin can be specifically
expressed in neurons that express the gustatory receptor 5a gene and thereby activate those
neurons by exposing them to blue light (~480nm). One direct method of measuring the
activation of these neurons is by observing the Proboscis Extension Reflex (PER).
EXPERIMENTAL PROCEDURE:
1. Anesthetize the animals on ice for about 15 minutes.
2. Place the animals on a cold table and hold them by their wings and slide them
delicately between the ridges of a collar (Heisenberg’s collar).
3. Allow the animals to recover for about 5 min in the dark and test them for proboscis
extension reflex by bringing the blue light guide in close proximity to the proboscis.
43

2. Walking ball paradigm:
We have developed a new behavioural paradigm where the flies are hooked to a
manipulator by their thorax but can walk on a styrofoam ball which is suspended on an air
cushion. The movements of the ball are electronically monitored and thus the preferred
walking direction of the animal can be ascertained. To establish operant conditioning using
this paradigm we have employed heat punishment which has been operantly linked with a
walking direction. The animal thus is punished when it walks in a particular direction and
now requires to operantly choose between the punished and unpunished direction. From our
experiments we know that animals associate the punishment to a particular direction and
avoid walking in the punished direction. Having established this paradigm with heat
punishment we are currently using channelrhodopsin-2 as a tool to activate populations of
modulatory neurons (octopaminergic and dopaminergic) and sensory neurons (gustatory and
olfactory). We have been successful in activating a large subset of dopaminergic neurons and
we find that animals avoid situations that lead to activation of those neurons. During the
practical course we will test Gal-4 lines that label a small number of dopaminergic neurons
and see if those few neurons suffice to induce the avoidance behaviour seen by activation of
the larger subset of dopaminergic neurons.
44

Operant Conditioning (from Wikipedia):
Operant conditioning is the use of consequences to modify the occurrence and form of
behavior. Operant conditioning is distinguished from classical conditioning (also called
respondent conditioning, or Pavlovian conditioning) in that operant conditioning deals with
the modification of "voluntary behavior" or operant behavior. Operant behavior "operates" on
the environment and is maintained by its consequences, while classical conditioning deals
with the conditioning of respondent behaviors which are elicited by antecedent conditions.
45

Kurzinfo: GAL4-UAS System:
(Brand and Perrimon., 1993)
46
Kursteil III: Verhalten Mo, 22. März - Fr, 26. März 2010
(Dritte Woche)
Im Kursteil Verhalten wird ein Hauptprojekt zur Neurogenetik des assoziativen
Lernens bei Drosophila Larven durchgeführt; zudem werden vier verschiedene
weitere “Stationen” durchlaufen (Fig. 1):
Figure 1
Overview of timetable
47

Hauptprojekt
Larvales Lernen
Stationen:
i) Fressverhalten
ii) Adultes Lernen
iii) Literatur und Fliegen
iv) Operantes Lernen
Hauptprojekt
Larvales Lernen
There are more things in the world than there are possible behaviours. Thus, in order
to fulfill the needs of life, the things in the outside world need to be ‘funneled’ into
much fewer behavioural matters of concern. Integrating the sensory such with the
biological needs to come up with appropriate behaviour is what brains have evolved
for and what neurobiology needs to understand.
Notably, it cannot be known in advance which sensory-motor match would be
the most fitting one; thus, both during evolution and during learning, possible
matches need to be tried out, by taking chances, and the ones with the relatively best
fit be stabilized. As a study case, we focus on odour-taste associative learning in
larval Drosophila (review in Gerber & Stocker, 2007; Gerber et al. 2007).
Clearly, changes in behaviour can come about only by changes in the nervous
system. One of the key ideas as to how this “works” is that the connections between
nerve cells (synapses) are altered (see e.g. Dudel et al.). Here, we will investigate
48

whether and which role one particular synaptic protein plays in this regard: The
“Synapse Associated Protein of 47 kDa” (SAP47) (Funk et al., 2004). For this protein,
homologues are found in C. elegans, fish, mouse, and man. Mutant Drosophila
larvae which lack SAP47 (SAP47156; Funk et al. 2004) show a learning ability of only
about 50 % of normal, wild-type levels (Saumweber, 2007), but are normal in terms
of the sensory and motor faculties needed for the learning task. Within this course,
we seek to confirm and extend this initially reported learning defect of the
SAP47156mutant larvae.
These experiments are performed in larval Drosophila melanogaster because
these animals combine the genetic techniques available for Drosophila with a simple
nervous system: they have about 10 million times fewer neurons than humans, and
about ten to a hundred times fewer neurons as compared to adult flies. Still, larval
Drosophila can learn well and thus can be used a simple model for associative
learning research (Gerber & Stocker, 2007; Gerber et al. 2007).
Behavioural experimental protocol
Flies are kept in plastic vials with corn meal upon which they lay eggs for one day.
Larvae that hatch from these eggs develop in the food medium. For experiments a
spoonful of food is taken from a vial of 5 day old (3rd instar) larvae. Animals are
picked from the food with a brush and washed in tap water before experiments. They
are then placed onto an agarose substrate prepared in a petri dish and can move
about these dishes. They can be rewarded by the presence of sugar within the
agarose substrate; odours are presented by little containers from which odour can
evaporate.
We train groups of 30 larvae and compare olfactory choice performance after
either of two reciprocal training regimen: one group receives n-amylacetate (AM) with
49

a positive reinforcer and 1-octanol (OCT) without such reinforcer (AM+/ OCT); the
second group is trained reciprocally (AM/ OCT+).
After training, animals are tested for their odour choice. The larvae are placed
in the middle of a fresh, pure agarose assay plate with a container of AM on one side
and one of OCT on the other side to create a choice situation. After 3 min, the
number of animals on the “AM” or “OCT” side is counted. After this test is completed,
the next group of animals is trained reciprocally.
EXPERIMENTAL DESIGN
TRAIN TEST
Regimen 1 AM+ OCT AM+ OCT AM+ OCT Choice: AM or OCT ?
Regimen 2 AM OCT+ AM OCT+ AM OCT+ Choice: AM or OCT?
For both groups, we calculate an odour preference ranging from –1 to 1. We
determine the number of animals observed on the AM side minus the number of
animals observed on the OCT side, divided by the total number of animals:
(1) PREF = (#AM– #OCT)/ #TOTAL
Associative learning is indicated by systematic differences in choice test performance
between the groups from either training condition (specifically, animals rewarded with
AM should have a higher preference for AM than animals rewarded with OCT). This
50

conclusion is compelling as during training animals from both training regimes
experience identical exposure to both odorants and the reward- what differs between
groups is exclusively the contingency between these stimuli. To quantify associative
function, we determine to which extent these preferences are different depending on
training regimen, and analyze the paired data from the alternately run, reciprocally
trained groups and calculate a learning index ranging from –1 to 1 as:
(2) LI = (PREFAM+/ OCT– PREFAM/ OCT+)/ 2
A comparison of LIs against random levels is made by a one-sample sign test;
comparisons between LIs from three or more groups are performed with the Kruskal-
Wallis test; LIs between two genotypes are compared with the U-test.
Genotypes
As experimental genotypes we use a deletion mutant for the gene coding for SAP47:
The Sap47156 mutant is characterized by a 1.7 kb deletion, which includes the
promoter region, the first exon and a small part of the first intron of the Sap47 gene,
and is consequently is lacking the SAP47 protein (Funk et al. 2004; Saumweber
2007). Given that each mutant fly strain needs to be compared to its “own” wild-type
control strain (see de Belle and Heisenberg, 1996), we use the Canton-SNF strain for
comparison. This strain had been used for repeated outcrossing to adjust genetic
background.
51

Further Reading
Brookes M Drosophila: Die Erfolgsgeschichte der Fruchtfliege. Rowohlt Verlag.
De Belle JS, Heisenberg M (1996) Expression of Drosophila mushroom body
mutations in alternative genetic backgrounds: a case study of the mushroom
body miniature gene (mbm). Proceedings of the National Academy of
Sciences (USA), 93:9875-9880.
Dudel J, Menzel R, Schmidt RF Neurowissenschaft. Springer Verlag.
Funk N, Becker S, Huber S, Brunner M, Buchner E. (2004) Targeted mutagenesis of
the Sap47 gene of Drosophila: flies lacking the synapse associated protein of
47 kDa are viable and fertile. BMC Neuroscience 5:16.
Gerber B, Wegener S, Hendel T (2007). Riechen, Schmecken, Lernen:
Verhaltensneurogenetik der Drosophila Larve. Neuroforum 3:80-92.
Gerber B, Stocker RF (2007) The Drosophila larva as a model for studying
chemosensation and chemosensory learning: a review. Chemical Senses
32:65-89.
Gerber B, Tanimoto H, Heisenberg M (2004) An engram found? Evaluating the
evidence from fruit flies. Current Opinion in Neurobiology 14:737-744.
Godenschwege TA, Reisch D, Diegelmann S, Eberle K, Funk N, Heisenberg M,
Hoppe V, Hoppe J, Klagges BRE, Martin JR, Nikitina EA, Putz G, Reifegerste
R, Reisch N, Rister J, Schaupp M, Scholz H, Schwärzel M, Werner U, Zars T,
Buchner S, Buchner E (2004) Flies lacking all synapsins are unexpectedly
healthy but are impaired in complex behaviour. European Journal of
Neuroscience 20:611-622.
Heisenberg M (2003) Mushroom body memoir: From maps to models. Nature
Reviews Neuroscience 4:266-275.
52

Michels B, Diegelmann S, Tanimoto H, Schwenkert I, Buchner E, Gerber B (2005) A
role of synapsin for associative learning: The Drosophila larva as a study case.
Learning & Memory 12:224-231.
Sachs L (2004) Angewandte Statistik. Springer Verlag.
Saumweber T (2007) Associative learning is impaired upon lack of the presynaptic
protein SAP47. Diplomarbeit Universität Würzburg.
http://nwg.glia.mdc-berlin.de/media/ppt/gerber-Anleitung.ppt
Questions
Suppose the Sap47156 mutant shows a learning index which is significantly below
Canton-SNF levels. Does this mean that these animals are impaired in
learning?
Suppose the Sap47156 mutant were indeed impaired in learning. Does this mean that
the SAP47 protein participates in the learning process?
Do you think memory (or at least memory traces) can be localized to parts of the
brain? What would be reasonable criteria?
Do you think memory is possible without any change at any synapse?
Would it be possible to use t-tests and an ANOVA for an analysis of the PREF
values? Of the LI values?
Have another look at the “EXPERIMETAL DESIGN” and comment on the statement:
“…during training animals from both training regimes experience identical
exposure to both odorants and the reward- what differs between groups is
exclusively the contingency between these stimuli.”
53
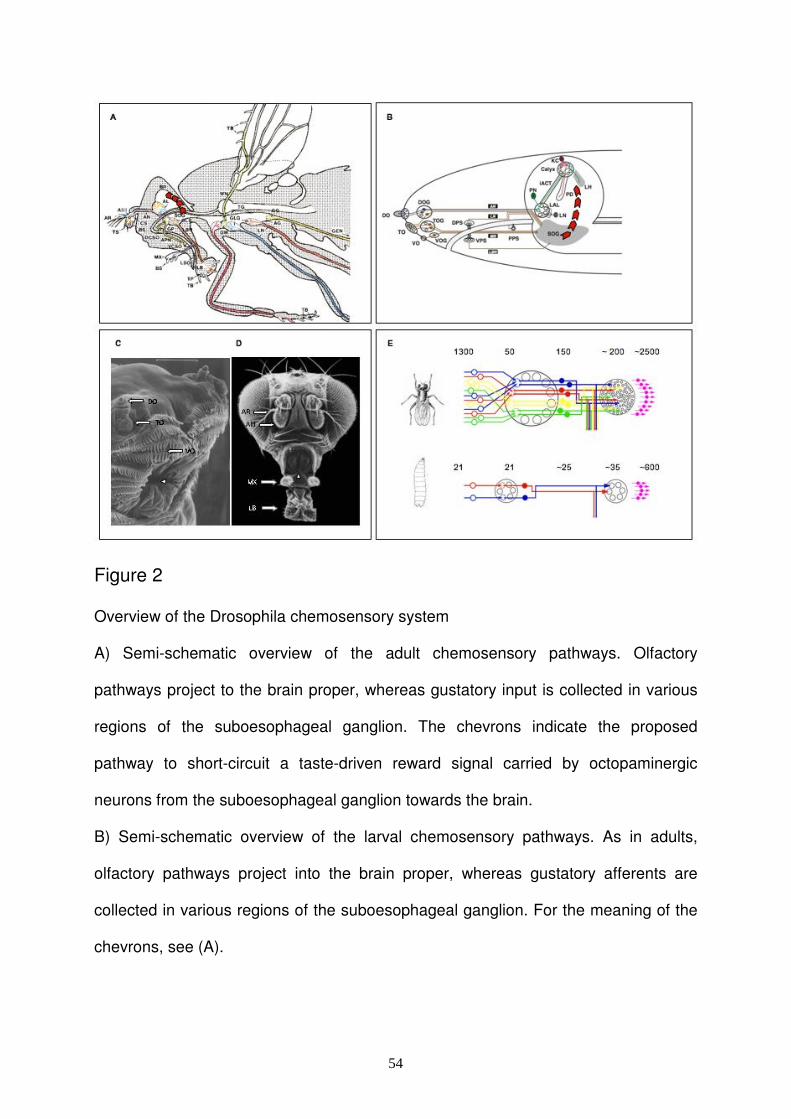
Figure 2
Overview of the Drosophila chemosensory system
A) Semi-schematic overview of the adult chemosensory pathways. Olfactory
pathways project to the brain proper, whereas gustatory input is collected in various
regions of the suboesophageal ganglion. The chevrons indicate the proposed
pathway to short-circuit a taste-driven reward signal carried by octopaminergic
neurons from the suboesophageal ganglion towards the brain.
B) Semi-schematic overview of the larval chemosensory pathways. As in adults,
olfactory pathways project into the brain proper, whereas gustatory afferents are
collected in various regions of the suboesophageal ganglion. For the meaning of the
chevrons, see (A).
54

C) SEM-overview of the larval head. One can discern the dome-shaped dorsal organ,
and the wart-like terminal organ. The cirri surround the mouth opening (triangle) and,
in the third row of cirri, cover the tiny ventral organ.
D) SEM-overview of the adult head and appendages in labellum-opened state.
Medial from the large complex eyes, one can discern the arista and the third antennal
segment, as well as the maxillary palps and the labellum; the mouth opening is
indicated by a triangle.
E) Comparison of the approximate number of, from left to right, olfactory sensory
neurons, antennal lobe glomeruli, projection neurons, calycal glomeruli in the
mushroom bodies, and mushroom body Kenyon cells. Note that the local
interneurons in the antennal lobe, which shape olfactory activity, are present in both
larva and adult, but are omitted in this figure.
A) from Stocker (1994); B) from Stocker (2006); C) by K Neuser, Universität
Würzburg; D) from The Interactive Fly (sdbonline.org/fly/aimain/1aahome.htm); E)
from Ramaekers et al. (2005).
55
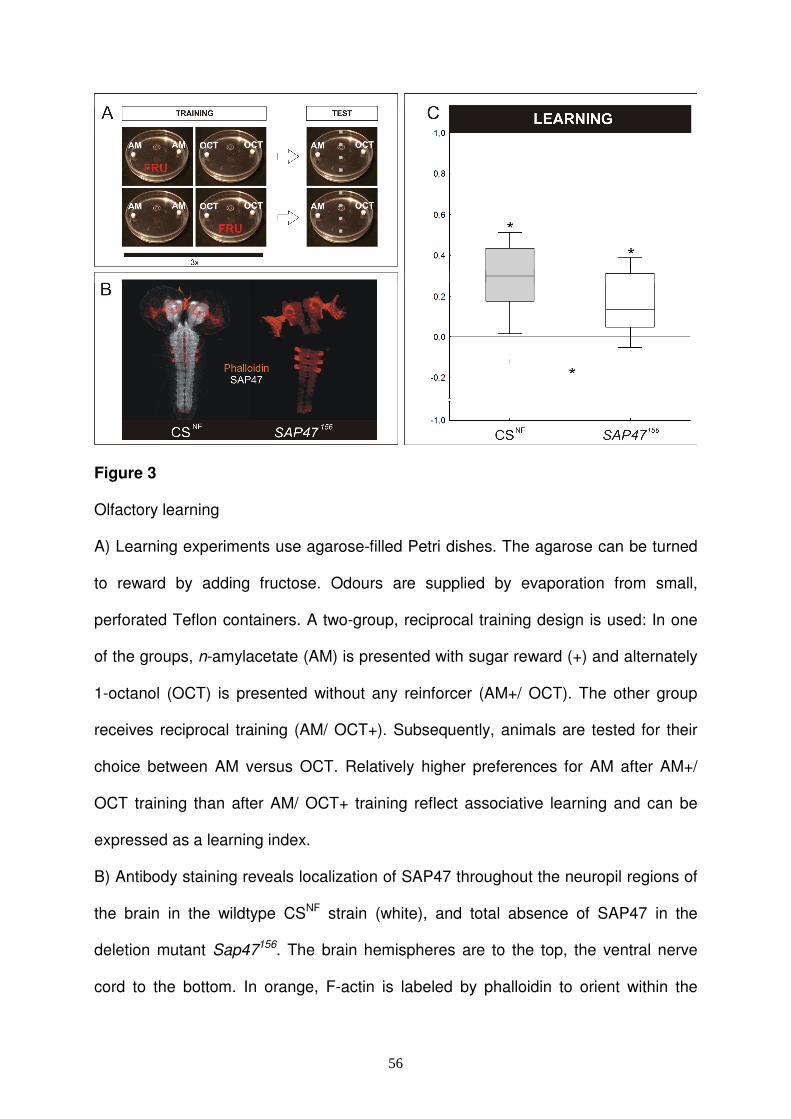
Figure 3
Olfactory learning
A) Learning experiments use agarose-filled Petri dishes. The agarose can be turned
to reward by adding fructose. Odours are supplied by evaporation from small,
perforated Teflon containers. A two-group, reciprocal training design is used: In one
of the groups, n-amylacetate (AM) is presented with sugar reward (+) and alternately
1-octanol (OCT) is presented without any reinforcer (AM+/ OCT). The other group
receives reciprocal training (AM/ OCT+). Subsequently, animals are tested for their
choice between AM versus OCT. Relatively higher preferences for AM after AM+/
OCT training than after AM/ OCT+ training reflect associative learning and can be
expressed as a learning index.
B) Antibody staining reveals localization of SAP47 throughout the neuropil regions of
the brain in the wildtype CSNF strain (white), and total absence of SAP47 in the
deletion mutant Sap47156. The brain hemispheres are to the top, the ventral nerve
cord to the bottom. In orange, F-actin is labeled by phalloidin to orient within the
56

preparations; towards the top one can discern the F-actin rich fibre bundles of the
developing adult eyes.
C) Sap47156 show a reduction of appetitve learning scores by appr. 50 %. Control
experiments testing for sensory or motor defects have revealed no difference
between Sap47156 and the wildtype CSNF strain (Saumweber 2007). Box plots
represent the median as middle line and 25, 75 as well as 10, 90 % quantiles as box
boundaries and whiskers, respectively.
A) by B Michels, Universität Würzburg; B) and C) from Saumweber (2007).
57

Station
i) Fressverhalten
Drosophila larvae live on their food, and feed continuously. We use an quantitative
feeding assay to see whether this behaviour can be modulated by salt.
We use wild-type, third-instar feeding-stage larvae aged 5 days (+/- 12 hrs)
after egg laying. To measure feeding, 10 larvae are placed on a Petri dish filled with a
solution containing 1 % agarose, 2 M salt (NaCl) and 30 % red food dye (type
strawberry; http://www.backfun.de). For comparison, we also use plates with dye-
only, but without salt. On these substrates the animals are allowed to feed for 15 min;
then they are washed in tap water. Next, we take a randomly chosen individual larva
from the dye-only Petri dish, briefly put it into boiling water and place it under a
binocular coupled to a digital camera; we then place a randomly chosen companion
larva from the salty Petri dish, place it next to the larva from the dye-only dish, and
take a picture of both of them together. Using an ImageJ-based, custom-written
software we determine for each larva both the area of its body (B) and the area of red
colour (R), if any, in its gut. From these data, we calculate a Feeding Index (FI) as:
FI = RSALT / BSALT – RDYE-ONLY / BDYE-ONLY
Thus, if larvae feed as much in the presence of salt as they do in its absence, feeding
scores are zero; if they eat more or less than in the absence of salt, respectively
positive and negative feeding scores result.
58
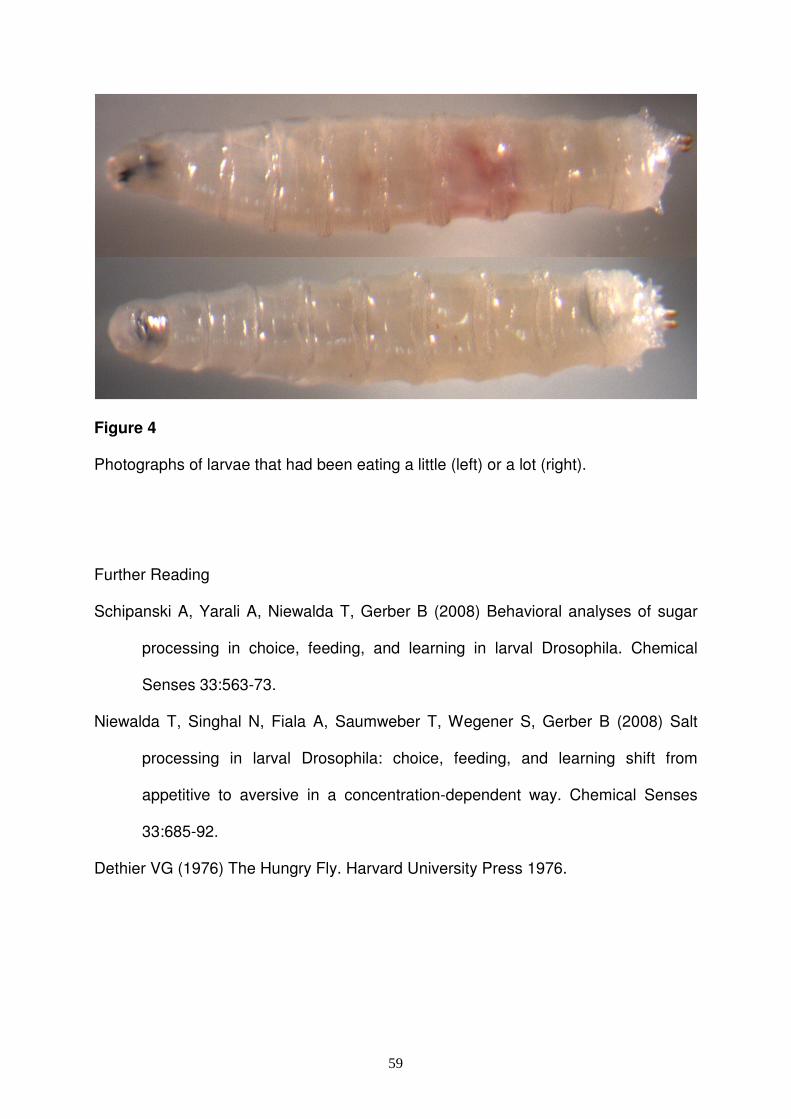
Figure 4
Photographs of larvae that had been eating a little (left) or a lot (right).
Further Reading
Schipanski A, Yarali A, Niewalda T, Gerber B (2008) Behavioral analyses of sugar
processing in choice, feeding, and learning in larval Drosophila. Chemical
Senses 33:563-73.
Niewalda T, Singhal N, Fiala A, Saumweber T, Wegener S, Gerber B (2008) Salt
processing in larval Drosophila: choice, feeding, and learning shift from
appetitive to aversive in a concentration-dependent way. Chemical Senses
33:685-92.
Dethier VG (1976) The Hungry Fly. Harvard University Press 1976.
59

Station
ii) Adultes Lernen
We use associative learning between odours and electric shock to study classical
conditioning in fruit flies. During training, one group of flies is trained with two kinds of
odours: one odour is presented together with electric shock (Punished Odour). A
second odour is presented alone (Control Odour).
In a subsequent test, flies are allowed to choose between these two odours. If they
avoid the Punished Odour relative to the Control Odour, we can conclude that this
odour indeed has been learned as a predictor of punishment.
Specifically, we use wild type flies aged 1 to 4 days after eclosure. Flies are kept in
mass culture at 25 °C temperature, 60-70 % relative humidity and 16h/8h light/dark
cycle. On the day prior to experiments, we transfer flies to fresh food vials and keep
them over-night at 18°C temperature.
Learning experiments are performed at 22-25 °C temperature and 70-85 % relative
humidity. Flies are trained and tested in groups of about 100. The training and testing
is done as follows (Fig. 5):
Flies are loaded to the experimental set-up. When loading is completed, time
counting begins (t:=0 min). The control odour is presented for 60 s from 1:00 to 2:00
min. The punished odour is then presented from 3:00 to 4:00 min, also for 60 s. From
3:15 to 4:10 min, the electric shock is delivered. The shock consists of 12 pulses of
100 V, each 1.2 s long and followed by the next pulse after an onset-onset interval of
5 s. At 5:00 the set-up is prepared for test. The flies are transferred to the choice
point of a T-maze, where they can choose between the control odour and the trained
60

odour. Test starts at 8:00. After 2 min, at 10:00, the arms of the maze are closed and
the number of animals (in the following denoted #) within each arm is counted. A
preference index (PI) is calculated as:
PI = (#Punished Odour - #Control Odour)*100 / # Total
Within each group, one subgroup is trained using 3-octanol (OCT) as the Control
Odour and benzaldehyde (BA) as the Punished Odour (OCT/ BA-punished), while a
second subgroup is trained reciprocally (BA/ OCT-punished). PIs from the two
reciprocal measurements are then averaged to obtain one learning index (LI) (Fig. 5):
LI = (PIBA + PIOCT) / 2
Positive LIs thus indicate conditioned approach to the trained odour, whereas
negative values reflect conditioned avoidance.
Note that within each subgroup, in half of the cases the sequence of events in
training is as indicated (i.e.: OCT/ BA-punished or BA/ OCT-punished), while in the
other half of the cases the sequences are reversed (i.e.: BA-punished/ OCT or OCT-
punished/ BA).
61
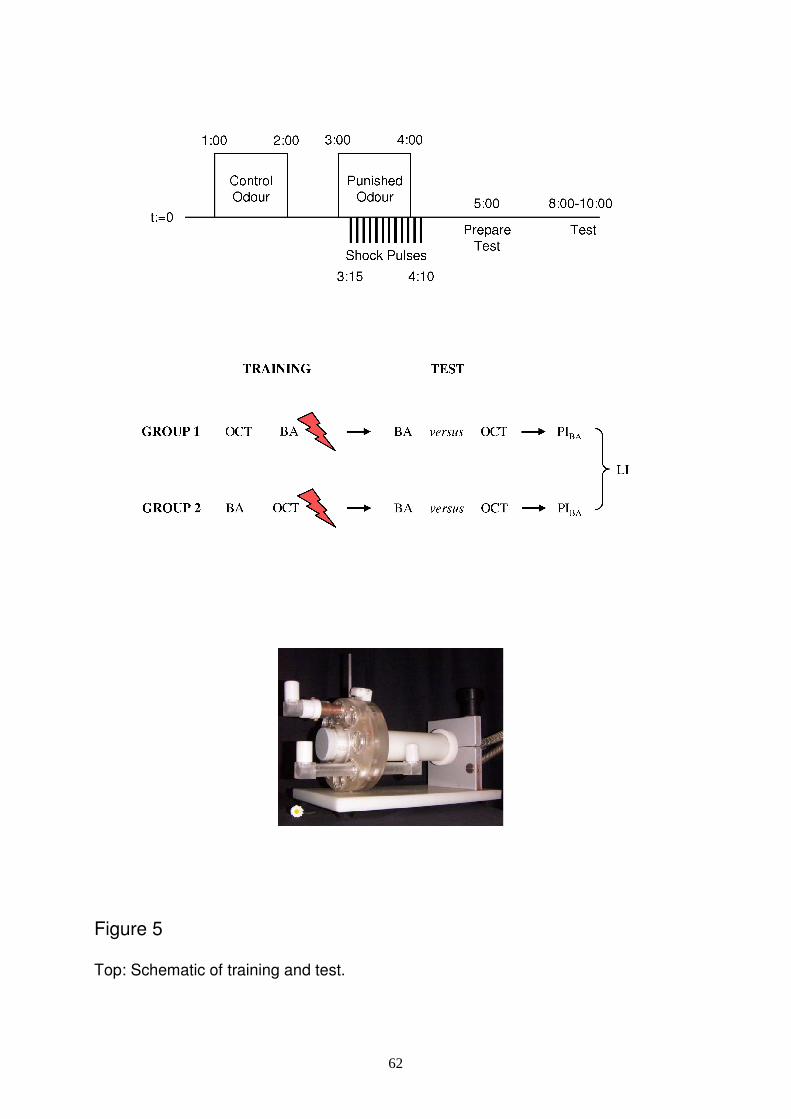
Figure 5
Top: Schematic of training and test.
62

Middle: Sketch of the reciprocal nature of the experiment. Note that within each
subgroup, in half of the cases the sequence of events in training is as indicated (i.e.:
OCT/ BA-punished or BA/ OCT-punished), while in the other half of the cases (not
depicted here) the sequences are reversed (i.e.: BA-punished/ OCT or OCT-
punished/ BA).
Bottom: Photograph of the training apparatus. The inner surface of the plastic tubings
is covered with a copper grid that can be electrified; at their ends, white, cylindrical
odorant containers can be attached. At eight sites of the apparatus, flies can be
trained simultaneously. The daisy serves as scale. Image courtesy of Ayse Yarali.
Further Reading
Tanimoto H, Heisenberg M, Gerber B (2004) Event-timing turns punishment to
reward. Nature 430:983.
Questions
1) Why do we calculate the learning index using the preferences of two reciprocally
trained groups?
2) Why do the cups for punished odour and control odour have different diameter?
3) Why does the training apparatus stand in a plexiglas box that can be heated?
4) If we would test our flies not with the trained pair of odours (benzaldehyde, 3-
octanol), but with two other odours that the flies have never been exposed to in the
experiment: do you think there could possibly be a significant learning index?
63

Station
iii) Literatur und Fliegen
Im Rahmen dieser Station sollen grundlegende Tatsachen über Drosophila vermittelt
werden, als wichtigstes erstens, Männchen von Weibchen zu unterscheiden (Fig. 6A-
C). Männchen sind generell etwas kleiner als Weibchen. Die schwarze Bänderung
des Hinterleibes ist beim Männchen in den letzten beiden Segmenten einheitlich
schwarz, im Gegensatz zum gestreiften Hinterleib des Weibchens. Zudem ist der
Hinterleib beim Männchen abgerundet, während er beim Weibchen eher zugespitzt
endet. Auch sind die dunklen, äußeren Geschlechtsorgane des Männchens deutlich
erkennbar. Das sieht man besonders gut, wenn es auf dem Rücken liegt. Da diese
Merkmale allerdings bei sehr schwach pigmentierten Tieren (das ist bei sehr jungen
Tieren der Fall sowie bei einer Reihe von Mutanten) nicht immer hinreichend deutlich
sind, sollte man sich insbesondere auf den sog. Geschlechtskamm konzentrieren: die
Männchen besitzen Halteborsten am vordersten Beinpaar, mit denen sie sich beim
Geschlechtsakt am Weibchen festhalten.
Zweitens soll gezeigt werden, wie man noch nicht verpaarte (jungfräuliche: rechts in
Fig. 6 B) von bereits verpaarten Weibchen (Mitte in Fig. 6 B) unterscheiden kann.
Dies ist für das Ansetzen von definierten Paarungen entscheidend, für die natürlich
nur Jungfrauen eingesetzt werden dürfen. Für die Bestimmung des jungfräulichen
Status macht man sich die Beobachtung zu Nutze, dass frisch geschlüpfte, nur
wenige Stunden alte Weibchen eine Paarung noch nicht zulassen. Man bestimmt
also eigentlich das Alter, und damit den Jungfrauenstatus nur indirekt. Solche sehr
jungen, jungfäulichen Weibchen sind an der noch sehr hellen Körperfärbung und-
besonders eindeutig - am sog. Meconium zu erkennen, einem unter der Kutikula
sichtbaren dunklen Fleck.
64

Drittens sollen eine Reihe klassischer Drosophila Mutanten vorgestellt werden: white
– weiße Augenfarbe; curly – gebogene Flügel; ebony – schwarzer Körper; sine oculis
– keine Augen; yellow – gelber Körper.
Schliesslich sollen an einem Literaturbeispiel die experimentellen Möglichkeiten des
Gal4-UAS Systems aufgezeigt werden: Es wird diskutiert, ob und in ggf. welchem
Sinn man bei Drosophila eine Gedächtnisspur 'lokalisieren' kann (Gerber et al 2004.
An engram found? Evaluating the evidence from fruit flies. Current Opinion in
Neurobiology 14:737-744. Download: #. Username: F1; Password: Genetik).
65

A)
B)
C)
66

D)
Drosophila Wildtyp
white – weiße Augenfarbe
curly – gebogene Flügel
ebony – schwarzer Körper
sine oculis – keine Augen yellow – gelber Körper
Figure 6
A) Gesamtansicht einer männlichen (links) und einer weiblichen (rechts) Drosophila.
B) Photographien von, von links nach rechts, männlichen, weiblichen und
jungfräulich-weiblichen Drosophila.
C) Der Geschlechtskamm der Männchen (rechts) fehlt bei den Weibchen (links).
D) Eine Reihe wichtiger Drosophila Mutanten.
67

Leseempfehlungen
Brookes M (2002) Die Fliege: Die Erfolgsgeschichte eines Labortiers. Rowohlt
Verlag.
Gerber B, Wegener S, Hendel T (2007) Riechen, Schmecken, Lernen:
Verhaltensneurogenetik der Drosophila-Larve. Neuroforum 03:80-92.
Greenspan R (2007) An Introduction to Nervous Systems. CSHL Press.
Greenspan R (2004) Fly Pushing. CSHL Press.
Schwarzmaier W (2007) Einfache Versuche zu Entwicklung und Verhalten:
Drosophila zum 100. Jubiläum" Biologie in unserer Zeit 02:112-118.
WWW sites
Fly Brain
http://flybrain.neurobio.arizona.edu/
Fly Base
http://flybase.org/
Pub Med
http://www.ncbi.nlm.nih.gov/sites/entrez?db=pubmed
Gal4 System
http://nwg.glia.mdc-berlin.de/media/ppt/gerber-Gal4.ppt
Präparation Larvengehirn
http://www.jove.com/index/details.stp?ID=85
68

Station
iv) Operantes Lernen
Beim „operanten Lernen“ lernen Tiere über die Konsequenzen ihrer eigenen
Handlungen: Welche Tat hat welche Konsequenz? So können die Tiere durch
Versuch, Irrtum und Erfolg dafür sorgen, möglichst viel angenehmes zu erleben, und
unangenehmes möglichst zu vermeiden.
In unserem Labor haben wir eine Apparatur entwickelt, die ein solches operantes
Lernen bei der Drosophila Larve zu studieren erlaubt: Wir setzen eine Larve in eine
mit Agarose ausgegossene Petrischale und filmen sie mit einer Kamera. Diese
Kamera sendet ihre Bilder zu einem Computer, der online bestimmte Parameter der
Bewegung der Larve berechnet, so beispielsweise die Richtung der Bewegung. Es
zeigt sich, dass die Larven, wie übrigens auch erwachsene Fliegen und Mücken, im
Zick-Zack laufen. Dabei gibt es in dieser Laborsituation nichts, was das Verhalten der
Tiere von außen bestimmen würde- die Larven machen einfach, was sie wollen. Wir
bestrafen dann die Tiere jedes Mal, wenn sie linksherum laufen (alternierend werden
andere Larven für rechtsherum Laufen bestraft). Das sollte die Larven dazu bringen,
lieber nach rechts zu laufen. Zur Bestrafung verwenden wir einen Lautsprecher: Eine
Petrischale wird auf diesen Lautsprecher gestellt (Figure 7); sobald die Larve
linksherum läuft, wird der Lautsprecher angeschaltet. Dieses Geräusch, bzw. die
Vibration des Untergrundes, ist ein für die Larven unangenehmes Ereignis .
In Zukunft wollen wir daran gehen, die wirklich spannenden Fragen zu stellen: Wann,
wo, und wie sich etwas am Schaltplan des Gehirns verändert, wenn die Larven durch
Versuch, Irrtum, und Erfolg diese operante Lernaufgabe lösen.
69

Figure 7
Prinzip des Experiments zum operanten Lernen. Links sieht man eine der typischen
larvalen Zick-zack Bewegungen, rechts den Aufbau zum operanten Lernen.
Leseempfehlungen
Brembs B (2003) Operant conditioning in invertebrates. Current Opinion in
Neurobiology 13:710-7.
Wolf R, Heisenberg M (1991) Basic organization of operant behavior as revealed in
Drosophila flight orientation. Journal of Comparative Physiology [A] 6:699-705.
Questions
Can larvae control their behaviour themselves (e.g. not-move, not-move in a
particular direction)?
How does the locomotion of a fruit fly larva look like? Do they walk curves?
How much turns does a larva do per minute? What is their speed of straight-walk?
70

Do you think that a larva can differentiate between its ‘right’ and its ‘left’?
What does ‘reflex’ and ‘voluntary action’ mean?
What is associated in PAVLOVian conditioning? What is associated in Operant
conditioning?
What is the role of chance (“Zufall”) for behaviour, in particular for trial-and-error
learning?
71





























