Embed Size (px)
Citation preview

Fakultät für Physik und AstronomieRuprecht–Karls–Universität Heidelberg
Staatsexamensarbeit
im Studiengang Physik
vorgelegt von
Julia Schwärzle
aus Ulm
– 2005 –


Spektroskopische Messung von Halogenoxiden
in der marinen atmosphärischen Grenzschicht
in Alcântara/Brasilien
Die Staatsexamensarbeit wurde ausgeführt am
Institut für Umweltphysik
der
Universität Heidelbergunter der Betreuung von
Herrn Prof. Dr. Ulrich Platt


Zusammenfassung
Der Photochemie reaktiver Halogenoxide (ClO, BrO, IO) wird erst seit der Entdeckung, dass Ha-
logene stratosphärisches Ozon sehr effizient abbauen können, größeres Interesse zuteil. Seither gab es
verschiedene Untersuchungen, um die Konzentration reaktiver Halogenverbindungen in unterschiedli-
chen Regionen der Atmosphäre zu bestimmen, dennoch sind die globale Verteilung, Quellen, Quell-
stärken, sowie die photochemischen Reaktionen reaktiver Halogenverbindungen noch nicht ausrei-
chend bekannt. Im Rahmen dieser Arbeit wurde deshalb versucht, die Halogenoxide Iodoxid (IO) und
Bromoxid (BrO) in der tropischen marinen Grenzschicht nachzuweisen. Von Dezember 2004 bis Ja-
nuar 2005 wurde daher eine Messkampagne in Alcântara, im Nordosten Brasiliens, durchgeführt. Die
Messung wurde mittels eines Langpfad–DOAS–Instrumentes (Differentielle Optische AbsorptionsS-
pektroskopie) unternommen, bei dem ein (offener) Lichtweg von 29 km durch die Atmosphäre geführt
wurde. Dabei wurde Iodoxid mittels seiner atmosphärischen Absorption im blauen Spektralbereich
(42–440 nm) nachgewiesen, mit Mischungsverhältnissen von bis zu (0.82 ± 0.31) ppt. Die Nachweis-
grenze lag dabei bei 0,28 ppt. Als Quelle des Iodoxides kommt einerseits die örtliche Produktion
durch den photochemischen Abbau kurzlebiger organischer Halogenverbindungen, die aus dem biolo-
gisch aktiven Schelfgebiet emittiert werden, in Frage, andererseits auch die Emission und der
Transport dieser Vorläuferverbindungen über den offenen Ozean. Bromoxid konnte mit Mischungs-
verhältnissen oberhalb der Nachweisgrenze von 2,4 ppt während der Kampagne nicht festgestellt
werden.
Abstract
Since the discovery of the destruction of stratospheric ozone, the atmospheric photochemistry of reac-
tive halogens has become of particular concern. Ever since, numerous investigations have been con-
ducted to unravel the importance of reactive halogen compounds in different atmospheric regions. To
date, however, the fate of reactive halogens species in the atmosphere is not yet totally clear. Within
the scope of this work, the mixing ratio of iodine oxide (IO) has unambiguously been detected in the
tropical marine boundary layer for the first time with mixing ratios of up to (0.82 ± 0.31) ppt, with a
detection limit of 0,28 ppt. Contrary, BrO could not be detected above the detection limit of 2.4 ppt.
These measurements were conducted during a field campaign held in Alcântara/Northeastern Brazil
from December 2004 until January 2005, using a 29 km long lightpath by Differential Optical Ab-
sorption Spectroscopy (DOAS). Possible sources of the inorganic iodine in marine boundary layer are
the photochemical decay of short–lived iodine organics emitted from the biologically active oceanic
shelf region, or at lower concentrations by long–range transport of these species from the open ocean.


Inhaltsverzeichnis
1 Einleitung...............................................................................................................................................1
2 Physik und Chemie der Troposphäre.....................................................................................................3
2.1 Troposphäre und maritime atmosphärische Grenzschicht............................................................3
2.2 Bisherige Arbeiten und Erkenntnisse über Halogenoxide in der Troposphäre.............................6
2.3 Chemie der Halogenoxidradikale in der Troposphäre..................................................................9
2.3.1 Quellen der RHS ..................................................................................................................9
2.3.2 Senken der RHS..................................................................................................................11
2.4 Reaktionen in der Troposphäre...................................................................................................11
2.4.1 Reaktives Brom ..................................................................................................................13
2.4.2 Reaktives Iod.......................................................................................................................14
2.4.2.1 Reaktionszyklen von IO..............................................................................................16
2.4.3 Das Leighton–Verhältnis.....................................................................................................18
2.4.4 Der HOx–NOx–Zyklus und Ozon: Produktion – Abbau....................................................20
3 DOAS-Grundlagen..............................................................................................................................22
3.1 Lambert–Beer–Gesetz.................................................................................................................24
3.2 Prinzipien der DOAS-Methode...................................................................................................26
3.3 Prinzipien der Auswertung..........................................................................................................29
4 Beschreibung des DOAS-Geräts.........................................................................................................30
4.1 Gesamtaufbau..............................................................................................................................30
4.2 Lichtquelle...................................................................................................................................31
4.3 Optik ...........................................................................................................................................31
4.4 Quarzfaser–Modenmischer..........................................................................................................33
4.5 Spektrograph................................................................................................................................34
4.6 Photodiodenzeile.........................................................................................................................34
4.7 Kurzschlusssystem......................................................................................................................35
5 Grundlagen der Auswertung................................................................................................................37
5.1 Offset...........................................................................................................................................37
5.2 Dunkelstrom................................................................................................................................38
5.3 Hintergrund..................................................................................................................................38
5.4 Kanal–Wellenlängen–Zuordnung...............................................................................................40
5.5 Korrektur eines Messspektrums..................................................................................................40
5.6 Rauschen......................................................................................................................................41

5.7 Faltung der Absorptionswirkungsquerschnitte............................................................................42
5.8 Berechnung des Mischungsverhältnisses....................................................................................43
5.9 Fehlerbestimmung ......................................................................................................................43
6 Kampagne............................................................................................................................................44
6.1 Geographie des Messorts.............................................................................................................44
6.2 Die Messbedingungen.................................................................................................................46
6.3 Der Messablauf............................................................................................................................47
6.4 Die Auswertung...........................................................................................................................49
6.4.1 Einstellungen in Windoas....................................................................................................49
6.4.2 Zuordnung des Wellenlängenbereichs................................................................................49
6.4.3 Wellenlängenbereich 320 nm – 350 nm .............................................................................51
6.4.4 Wellenlängenbereich 418 nm – 438 nm..............................................................................51
6.4.5 Wellenlängenbereich 418 nm – 450 nm..............................................................................53
7 Ergebnisse und Diskussion..................................................................................................................54
7.1 Auswertebereich 320 nm – 350 nm.............................................................................................55
7.1.1 Bromoxid und Formaldehyd...............................................................................................55
7.1.2 Ozon....................................................................................................................................57
7.2 Auswertebereich 418 nm – 438 nm.............................................................................................58
7.2.1 Wasser.................................................................................................................................58
7.2.2 Stickstoffdioxid...................................................................................................................59
7.2.3 Iodoxid.................................................................................................................................60
7.3 Analyse der Luftprobenbehälter..................................................................................................63
8 Zusammenfassung und Ausblick.........................................................................................................65
9 Literaturangaben..................................................................................................................................67
10 Anhang...............................................................................................................................................75

1 Einleitung 1
1 Einleitung
Halogene sind sehr reaktive chemische Elemente, die beim katalytischen Abbau von Ozon sowohl in
der Stratosphäre als auch in der Troposphäre beteiligt sind. Diese Chemikalien rückten besonders seit
der Entdeckung des stratosphärischen Ozonlochs über der Antarktis im Frühjahr [Farman et al.,
1985] ins Blickfeld des allgemeinen Interesses, denn die stratosphärische Ozonschicht schützt das
Leben auf der Erde vor höherenergetischer, für Organismen schädliche UV–Strahlung der Sonne. In
der Troposphäre hingegen ist Ozon ein für Menschen und Pflanzen giftiges Gas, das zudem noch
durch Absorption im infraroten Bereich zum Treibhauseffekt beiträgt.
Vorläuferverbindungender Halogene in der Stratosphäre sind vor allem organische Halogenver-
bindungen die entweder natürlich oder durch den Menschen in die Atmosphäre freigesetzt werden
[WMO, 2002]. Gelangen diese Verbindungen in die Stratosphäre, so werden sie vornehmlich durch
kurzwellige UV–Strahlung – die noch bis in die Stratosphäre eindringt – photochemisch gespalten,
wodurch Chlor und Brom freigesetzt wird. Diese reaktiven Halogene (RHS = Reactive Halogene Spe-
cies) reagieren mit Ozon und bauen es in sogenannten katalytischen Zyklen ab. Hochenergetische
elektromagnetische Strahlung dringt jedoch nicht bis in die Troposphäre durch, wodurch die photoly-
tische Spaltung der vollständig halogenierter Kohlenwasserstoffe nicht stattfinden kann. Die Photoly-
se der stabilen organischen Halogenverbindungen in der Troposphäre ist daher nicht entscheidend für
den Abbau troposphärischen Ozons. Dort werden allerdings die weniger stabilen teilhalogenierten
organischen Halogenverbindungen wie z.B. (CH3X, CX3H, etc., mit X = Cl, Br, I) entweder direkt
photolysiert oder durch Reaktionen mit dem OH–Radikal abgebaut.
So fand man ausgelöst durch die Entdeckung von Barrie et al. [1988], dass in der Arktis im Frühjahr
in Episoden auftretendes Bromoxid bodennahes Ozon nach Sonnenaufgang vollständig zerstört,
nennenswerte Vorkommen an BrO (bis zu 220 ppt) nicht nur in der Arktis, sondern auch nahe von
Salzseen wie in Israel am Toten Meer [Hebestreit et al., 1999], oder auch in Abgasfahnen von aktiven
Vulkanen [Bobrowski et al., 2003]. Weiterhin wurde bei Feldmesskampagnen die in Israel, als auch in
Mace Head, Irland und auf Teneriffa, Spanien, stattfanden, auch das Radikal Iodoxid mit Mischungs-
verhältnissen von bis zu 10 ppt nachgewiesen [Zingler und Platt, 2005; Alicke et al., 1999; McFig-
gans et al., 2000]. Ausgelöst durch diese Messungen quantifizierbarer Mengen an Halogenoxiden
rückte zunehmend auch die Frage nach der globalen Bedeutung der Halogenoxide für die Chemie tro-
posphärischen Ozons in den Mittelpunkt.
So widmeten sich einige Untersuchungen den möglichen Quellen organischer Vorläufergase wie CH3I
und CHBr3 im Bereich des tropischen offenen Ozeans zu [Richter und Wallace, 2004; Quack et al.,
2004]. Dabei geht man davon aus, dass durch Algen und mikrobiologische Aktivität im Ozean [Quack
et al., 2004] und auch durch besondere Algen in Küstenregionen, wie z.B. Laminaria digitata und
saccharina [Carpenter et al., 1999], organisch gebundenes Brom und Iod erzeugt wird. Diese
organischen teilhalogenierten Verbindungen werden in der Troposphäre relativ rasch abgebaut

2 Einleitung
(Lebensdauern liegen bei einigen Stunden bis Wochen), wobei die dabei freiwerdenden reaktiven Ha-
logene in der Atmosphäre effizient Ozon abbauen [WMO, 2002]. Weiterhin haben die Halogenoxide
durch ihre Reaktion mit den sogenannten Radikalfamilien HOx (OH, HO2, H2O2) und NOx (NO, NO2)
einen starken Einfluss auf die Oxidationskapazität der Atmosphäre.
Insbesondere stellt sich auch die Frage nach der Oxidationskapazität der Troposphäre bzw. welche
Gebiete ozonerzeugend bzw. –vernichtend sind. Die NO2–Konzentrationen unterhalb derer O3 photo-
chemisch zerstört wird wurde von Carpenter et al. [1997] mit ~55±30 ppt für Mace Head, Irland, und
mit 23±20 ppt für Cape Grim, Tasmanien, angegeben.
Ziel der durchgeführten Messkampagne in Alcântara/Brasilien, einer nicht bis gering verschmutzten
Gegend in den marienen Tropen, war es daher, das vorhandene Mischungsverhältnis von Bromoxid
und Iodoxid zu bestimmen. Um zusätzlich noch den potentiellen Einfluss auf Ozon zu untersuchen
wurden auch in situ die Ozonwerte ermittelt, um eine mögliche Korrelationen zwischen Halogenoxid-
konzentration mit der Ozonkonzentration festzuhalten.
Der Kampagnemessort und die Auswertung der Messdaten werden in Kapitel 6 erläutert, Ergebnisse
werden in Kapitel 7 der Arbeit dargelegt und in Kapitel 8 bezüglich ihrer Bedeutung für das Gebiet
des Messortes diskutiert. Doch zuerst wird der Begriff der Troposphäre erläutert und dann auf die
Chemie der Halogene eingegangen.

2 Physik und Chemie der Troposphäre 3
2 Physik und Chemie der Troposphäre
2.1 Troposphäre und maritime atmosphärische Grenzschicht
Die Troposphäre (von griech. trepein: wenden, kehren) ist der unterste Bereich der Atmosphäre, die
die Erde umgibt. In dieser Schicht nimmt die Temperatur mit der Höhe um etwa 0,65°C/100m ab, die
Luft wird stark vertikal vermischt: „das Oberste [wird] zuunterst und das Unterste zuobert gekehrt“
[Kraus, 2000]. Die starke vertikale Durchmischung in der bodennahen Atmosphäre, die Strahlungs-
absorption und –emission in der Atmosphäre und die Strahlungsübergangsprozesse in der Atmosphäre
stehen in engem Zusammenhang mit dem Temperaturprofil.
Die Grenze zwischen der Troposphäre und der über ihr liegenden Stratosphäre wird durch die Tropo-
pause (von griech. pauein: aufhören) gebildet, einer isothermen Region die sich je nach geo-
graphischer Breite in unterschiedlichen Höhenlagen befindet:
Tabelle 2.1: Tropopausenhöhe in verschiedenen Breiten [Kraus, 2000]
Erdoberfläche Lufttempera-tur in °C
Tropopause Lufttemperaturin °C
Tropopausenhöhe in km
Erde (Mittel) +15 -56 11Tropen +26 -80 16Arktis, Januar -25 -58 8Arktis, Juli 0 -46 10
Die Troposphäre teilt sich in zwei Schichten:
Die Atmosphärische Grenzschicht, auch Planetare (oder auch planetarische [Kraus, 2000]) Grenz-
schicht genannt, ist die erdnahe Schicht in der Troposphäre, die durch Effekte die von der Erdoberflä-
che ausgehen beeinflußt wird. Die Effekte sind dabei die dynamische Wirkung (z.B. Reibung) und
thermodynamische Prozesse (z.B. Erwärmung, Abkühlung). Die Grenzschicht reicht von Dicken von
nur wenigen Dekametern in den polaren Gebieten, bis hin zu 3 km in Gebieten mit sehr starker Erwär-
mung.
Der Bereich der marinen atmosphärischen Grenzschicht (im Folgenden "MBL" abgekürzt, von
engl: marine boundary layer) ist die unterste, ca. 500–1000 m dicke Schicht der Troposphäre. Sie
steht in direktem Austausch mit der Meeresoberfläche. Ozonrelevante Prozesse in der MBL besitzen
aufgrund des großen Flächenanteils der Ozeane (ca. 70% der Erdoberfläche) globale Bedeutung für
Ozonhaushalt und Atmosphärenchemie.
Die über der Grenzschicht liegende freie Troposphäre ist der nicht mehr direkt von der Erdoberflä-
che beeinflußte Teil der Troposphäre.

4 Physik und Chemie der Troposphäre
In der äquatorialen Zone steigt durch die starke
Sonneneinstrahlung und die daraus resultie-
rende Konvektion die Luft nach oben. Dies er-
klärt, dass die Tropopause in dieser Zone höher
liegt als in Polnähe (vgl. Abb. 2.3). Dabei
strömt bodennahe Luft in die innertropische
Konvergenzzone (ITCZ) nach, wird durch die
Corioliskraft abgelenkt und es kommt zu einem
Nordostpassatwind auf der Nordhalbkugel und
zu einem Südostpassatwind auf der Südhalb-
kugel. Die Luftmassen kehren bei etwa 30°
Breite wieder zur Erdoberfläche zurück. Diesen
Zirkulation nennt man auch die Hadley–Zelle.
Durch die stärkere Erwärmung der Erdoberfläche gegenüber der Meeresoberfläche verschiebt sich die
ITCZ jedoch jahreszeitbedingt mehr nach Norden bzw. nach Süden (vgl. Abb. 2.2). Während der
Kampagnendauer lag der Messort im Gebiet des Passats, daher kommen die untersuchten Luftmassen
sehr wahrscheinlich vom offenen Atlantik.
In die marinen atmosphärischen Grenzschicht kommen halogenierte Verbindungen vor allem durch
Freisetzung aus Seesalz oder durch biogene Freisetzung durch Algen (vgl. Kap. 2.3.1).
Es wird vermutet, dass einige sehr kurzlebige Halogenverbindungen aus der planetaren Grenzschicht
bis in die obere Troposphäre bzw. in die untere Stratosphäre gelangen können. Kurzlebige Quellgase
(VSLS) sind in diesem Zusammenhang Gase mit einer Lebensdauer unter einem halben Jahr, bei-
Abb. 2.2: Lage der ITCZ, der rote Punkt gibt die Lage des Messorts Alcântarader durchgeführten Kampagne an
Abb. 2.1: Luftzyklen über der Erdoberfläche
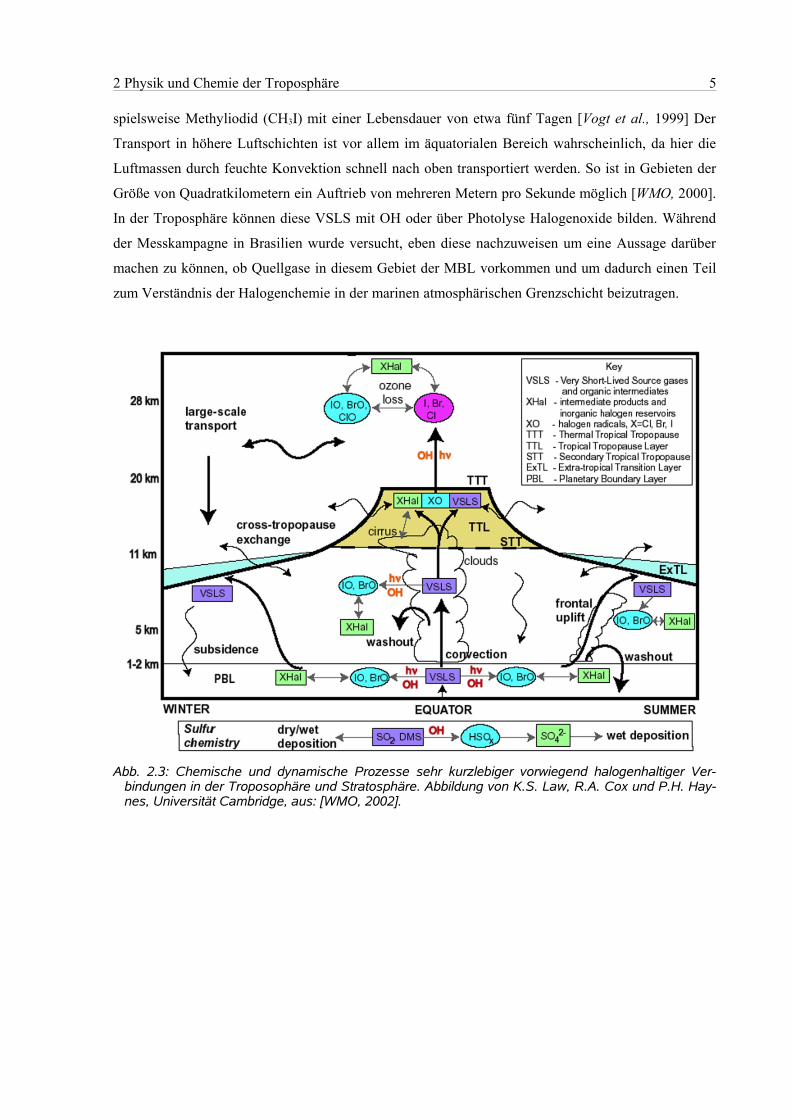
2 Physik und Chemie der Troposphäre 5
spielsweise Methyliodid (CH3I) mit einer Lebensdauer von etwa fünf Tagen [Vogt et al., 1999] Der
Transport in höhere Luftschichten ist vor allem im äquatorialen Bereich wahrscheinlich, da hier die
Luftmassen durch feuchte Konvektion schnell nach oben transportiert werden. So ist in Gebieten der
Größe von Quadratkilometern ein Auftrieb von mehreren Metern pro Sekunde möglich [WMO, 2000].
In der Troposphäre können diese VSLS mit OH oder über Photolyse Halogenoxide bilden. Während
der Messkampagne in Brasilien wurde versucht, eben diese nachzuweisen um eine Aussage darüber
machen zu können, ob Quellgase in diesem Gebiet der MBL vorkommen und um dadurch einen Teil
zum Verständnis der Halogenchemie in der marinen atmosphärischen Grenzschicht beizutragen.
Abb. 2.3: Chemische und dynamische Prozesse sehr kurzlebiger vorwiegend halogenhaltiger Ver-bindungen in der Troposophäre und Stratosphäre. Abbildung von K.S. Law, R.A. Cox und P.H. Hay-nes, Universität Cambridge, aus: [WMO, 2002].

6 Physik und Chemie der Troposphäre
2.2 Bisherige Arbeiten und Erkenntnisse über Halogenoxide in der
Troposphäre
Schon frühe Arbeiten hatten sich mit Halogenen befasst: Cauer stellte [1939] in Zentral– und West-
europa eine Iodverschmutzung mit atmosphärischen, gasförmigen Konzentrationen von bis zu 0.5 µg
m-3 oder größer fest, die durch ineffiziente Verbrennung von Makroalgen verursacht wurde. Eriksson
[1959a, b] fasste zusammen, dass die Hauptquelle für atmosphärische Halogene Seesalz sei, das durch
Zerplatzen von Luftblasen freigesetzt werde, welche durch Wellen oder andere Prozesse entstehen.
Später wurde gezeigt, dass Chlor und Brom von Seesealzpartikeln abgereichert [Duce et al., 1963],
Iod auf diesen jedoch stark angereichert wird.
Das stratosphärische Ozonloch über der Antarktis im Frühjahr konnte durch die von Molina und Row-
land [1974] postulierten These, dass industriell produzierte halogenierte Kohlenwasserstoffe, vor
allem Fluorchlorkohlenwasserstoffe (CFCs, CFCl3, CF2Cl2), stratosphärisches Ozon stark abbauen
können, nicht vollständig erklärt werden. Solomon et al. [1986] vervollständigten die Theorie durch
den Vorschlag, dass Chlorverbindungen auf der Oberfläche sogenannter polarer stratosphärischer
Wolken (PSC, von engl.: polar statospheric clouds) reagieren. Die PSC treten in der Antarktis nur bei
extrem tiefen Temperaturen (-80°C und kälter) während der Polarnacht auf, wenn aufgrund des
polaren Vortex kein Austausch mit anderen Luftmassen möglich ist. Als „polarer Vortex“ wird das
Zirkulieren der Luftmassen in der Stratosphäre bei sehr tiefen Temperaturen um ein Zentrum mit re-
lativer unbewegter Luft bezeichnet. 1986 schlug McElroy einen zusätzlichen Zyklus des Ozonabbaus
vor, in dem reaktives Brom und Chlor mitwirken.
Erst durch Barrie et al. [1988] wurde entdeckt, dass auch Halogene, die aus teilhalogenierten
organischen Verbindungen freigesetzt werden können, in der troposphärischen Chemie eine Rolle
spielen. Sie stellten fest, dass im Frühling in der Arktis starker Ozonabbau mit hohen Konzentrationen
von filterbarem (=über Filter detektierbares) Brom korreliert und dieser Effekt in Episoden auftritt.
Seitdem folgten viele Untersuchungen, die sich mit Halogenen in der freien Troposphäre und der
planetaren Grenzschicht beschäftigen, um ihre Rolle in der Chemie der Atmosphäre, vor allem ihren
Einfluß auf Ozon, besser verstehen zu können.
Mit DOAS-Messungen in Kanada wurde 1994 von Hausmann und Platt zum ersten Mal Bromoxid
(BrO) in der Troposphäre nachgewiesen. Weitere boden- und satellitengestützte Messungen in der
Arktis [Platt und Lehrer, 1997; Richter et al., 1998; Wagner und Platt, 1998] bestätigten, dass es im
Frühjahr zu polaren Ereignissen kommt, bei denen hohe troposphärische Mischungsverhältnisse von
BrO („Brom–Explosion“) mit starkem Ozonabbau korrelieren. Diese Ereignisse nennt man auch
Ozone–Depletion–Events (ODE). Quelle für das Brom bei der Brom–Explosion sind, neuesten Unter-
suchungen zufolge [Kaleschke et al., 2004], Regionen bedeckt von jungem Eis, das möglicherweise
mit Frostblumen bedeckt ist. Frostblumen sind Eiskristalle mit hoher Salzkonzentration, die nach
wenigen Tagen von Schnee bedeckt sind oder von Winden verweht werden und Aerosole bilden. Ihre

2 Physik und Chemie der Troposphäre 7
Form und Anzahl ist abhängig von Temperatur und Luftfeutigkeit. Die südlichste arktische Reagion in
der im Frühjahr Ozonabbau bedingt durch reaktive Halogene beobachtet wurde ist die Hudson Bay in
Kanada [Hönninger et al., 2004b].
Doch nicht nur in eisbedeckten Regionen sondern auch auf einer Kampagne am Toten Meer wurden
BrO–Ereignisse detektiert, die zu starkem Ozonabbau führten. Dabei wurden Mischungverhältnisse
von bis zu 0.8 ppb BrO gemessen [Stutz et al., 1999]. Hebestreit et al. [1999] vermuten, dass Brom
durch heterogene, autokatalytische Prozesse aus der Oberfläche der Salzpfannen im Tal des Toten
Meers freigesetzt wird. Auch im tropischen offenen Ozean wird Brom freigesetzt, hauptsächlich in
Form von Bromoform (CHBr3), das z.B. im tiefen Chlorophyll-Maximum der tropischen Thermokline
gebildet wird. Von dort kann es durch den äquatorialen Auftrieb an die Oberfläche gelangen und zu
lokal erhöhten Konzentrationen in der Atmosphäre führen [Quack et al., 2004].
Troposphärisches Iodoxid wurde erstmals in Mace Head, Irland, nachgewiesen, die hauptsächlichen
Quellen vor Ort sind Makroalgen und Phytoplankton [Carpenter et al., 1999], die organische iod-
haltige Verbindungen bilden und freisetzen. Diese Quellen sind auch die Hauptursache für Iodoxid in
Cape Grim, Tasmanien, und an der Atlantikküste in Frankreich. Flüchtige freigesetzte organische Ha-
logenverbindungen sind jedoch nicht in allen Gebieten als Hauptquelle für Iodoxid anzusehen. Saiz-
Lopez und Plane hatten 2004 auch gezeigt, dass zusätzlich zu Alkyliodid auch im Meer freigesetztes
molekulares Iod (I2) eine weitere große Quelle für atmosphärisches Iod ist. Der gesamte Fluss wurde
von ihnen mit 0,5 Tg Iod pro Jahr abgeschätzt.
Auch auf Teneriffa wurden von Allan et al. [2000] bis zu 3,5 ppt Iodoxid gemessen, dort kann vor Ort
eine genügend große biologisch Quelle ausgeschossen werden, weshalb Allan et al. die Vermutung
anstellten, dass die dort gemessenen Konzentrationen aufgrund von berechneten Rückwärtstrajektori-
en repräsentativ für die Grenzschicht des offenen Ozeans im subtropischen Nordatlantik seien.
Auch am Toten Meer wurden IO-Konzentrationen von bis zu 10 ppt gemessen. Aufgrund des hohen
Salzgehaltes des Wassers werden aber fast alle Organismen abgetötet, eine biologische Quelle ist da-
her nicht sehr wahrscheinlich. Deshalb schlagen Zingler und Platt [2005] eher eine abiotische Frei-
setzung von Iodoxid aus Meersalzoberflächen vor.
Die Ergebnisse der zahlreichen Kampagnen, auf denen versucht wurde mittels der DOAS–Technik IO
und BrO sowie auch andere reaktive Iodverbindungen nachzuweisen, werden in Tabelle 2.2 darge-
stellt:
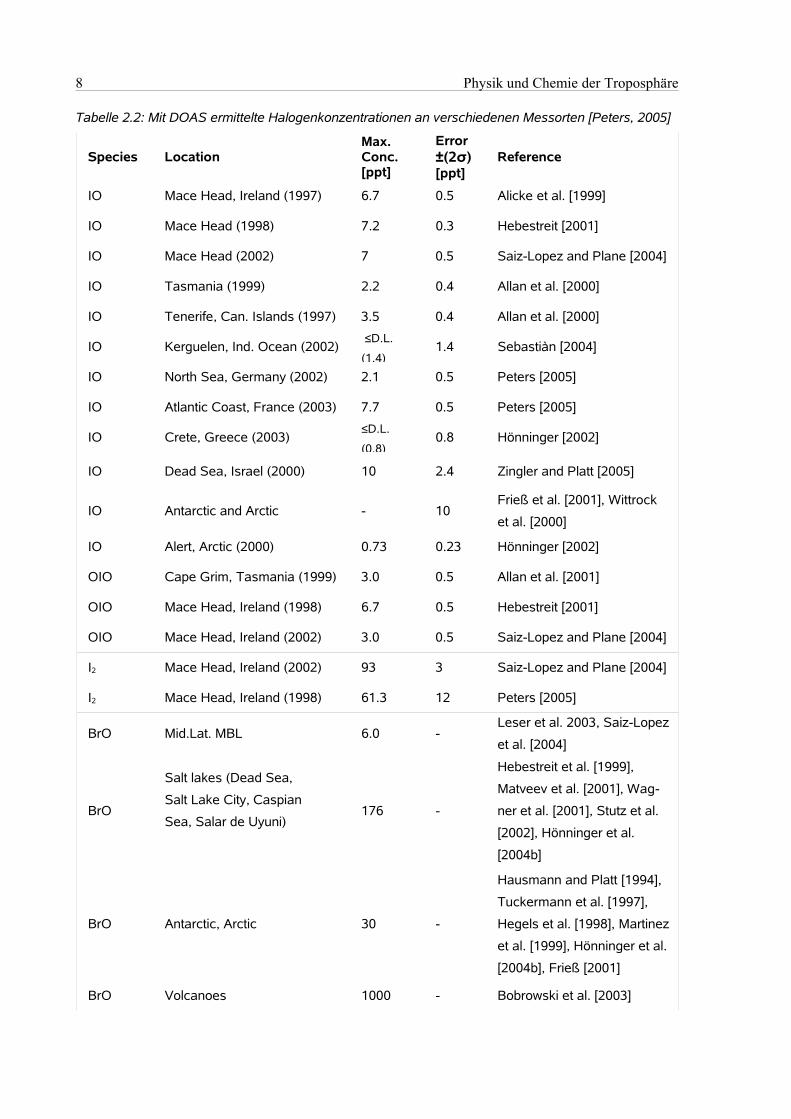
8 Physik und Chemie der Troposphäre
Tabelle 2.2: Mit DOAS ermittelte Halogenkonzentrationen an verschiedenen Messorten [Peters, 2005]
Species LocationMax.Conc.[ppt]
Error±(2σ)[ppt]
Reference
IO Mace Head, Ireland (1997) 6.7 0.5 Alicke et al. [1999]
IO Mace Head (1998) 7.2 0.3 Hebestreit [2001]
IO Mace Head (2002) 7 0.5 Saiz-Lopez and Plane [2004]
IO Tasmania (1999) 2.2 0.4 Allan et al. [2000]
IO Tenerife, Can. Islands (1997) 3.5 0.4 Allan et al. [2000]
IO Kerguelen, Ind. Ocean (2002) ≤D.L.(1.4)
1.4 Sebastiàn [2004]
IO North Sea, Germany (2002) 2.1 0.5 Peters [2005]
IO Atlantic Coast, France (2003) 7.7 0.5 Peters [2005]
IO Crete, Greece (2003) ≤D.L.(0.8)
0.8 Hönninger [2002]
IO Dead Sea, Israel (2000) 10 2.4 Zingler and Platt [2005]
IO Antarctic and Arctic - 10Frieß et al. [2001], Wittrocket al. [2000]
IO Alert, Arctic (2000) 0.73 0.23 Hönninger [2002]
OIO Cape Grim, Tasmania (1999) 3.0 0.5 Allan et al. [2001]
OIO Mace Head, Ireland (1998) 6.7 0.5 Hebestreit [2001]
OIO Mace Head, Ireland (2002) 3.0 0.5 Saiz-Lopez and Plane [2004]
I2 Mace Head, Ireland (2002) 93 3 Saiz-Lopez and Plane [2004]
I2 Mace Head, Ireland (1998) 61.3 12 Peters [2005]
BrO Mid.Lat. MBL 6.0 -Leser et al. 2003, Saiz-Lopezet al. [2004]
BrO
Salt lakes (Dead Sea,Salt Lake City, CaspianSea, Salar de Uyuni)
176 -
Hebestreit et al. [1999],Matveev et al. [2001], Wag-ner et al. [2001], Stutz et al.[2002], Hönninger et al.[2004b]
BrO Antarctic, Arctic 30 -
Hausmann and Platt [1994],Tuckermann et al. [1997],Hegels et al. [1998], Martinezet al. [1999], Hönninger et al.[2004b], Frieß [2001]
BrO Volcanoes 1000 - Bobrowski et al. [2003]

2 Physik und Chemie der Troposphäre 9
2.3 Chemie der Halogenoxidradikale in der Troposphäre
In den folgenden Kapiteln wird auf die Quellen und Senken, mögliche Kreuz– und Selbstreaktionen,
sowie auf die verschiedenen Hauptreaktionen der Halogene in denen Ozon abgebaut wird, einge-
gangen.
Prinzipiell verlaufen die Reaktionsschemen für die verschiedenen Halogene gleich, unterscheiden sich
jedoch in ihren Reaktionskonstanten. Um die wiederholte Aufzählung der Halogene im Folgenden zu
vermeiden, wird „X“ für die Halogene Cl, Br und I geschrieben. Sollten in einem Reaktionsschema
zwei Halogene auftreten, so wird das erste X und das zweite Y genannt. Fluor ist bei der Betrachtung
der Troposphäre unrelevant, da stabiles und daher sehr unreaktives HF gebildet wird (z.B. über die
Reaktion F + H2O → HF + OH [von Glasow und Crutzen, 2003]). Jedoch existieren einige antropogen
erzeugte, fluorhaltige Gase, die, aufgrund ihrer starken Absorption im Infraroten, zum Treibhauseffekt
beitragen.
Doch wie gelangen die reaktiven Halogene (X, X2, XY, XO, XnOm , HOX) in die Atmosphäre?
Hierfür gibt es verschiedene mögliche Erklärungen:
2.3.1 Quellen der RHS
• Biogene Quellen
Einige Arten von Makroalgen und Phytoplankton können Halogenalkane produzieren und
sind dadurch vor allem eine Quelle für Iod und Brom: in den Buchten bei Mace Head gab es
große Vorkommen von iodhaltigem Seetang wie z.B. Laminaria digitata und saccharina
[Carpenter et al., 1999]. Algenarten wie z. B. Fucus vesiculosos und Ulva lactuca setzen ver-
stärkt organische Bromverbindungen (v.a. CHBr3 und CH2Br2) frei [Schwarz, 2003].
Bromoform (CHBr3) kann Quack et al. [2004] zufolge eine mögliche Quelle für atmosphä-
risches, reaktives Brom (Br, BrO) sein. So tragen Makroalgen in Küstengebieten und Übersät-
tigung des Wassers im offenen Ozean zu einer CHBr3–Emission bei. Durch örtliche und zeitli-
che Variabilität in der Produktion und aufgrund seiner kurzen Lebensdauer von 4 Wochen
stellt man eine stark variierende CHBr3–Verteilung in der Atmosphäre fest [Quack und
Wallace, 2003].
CH3I war bis in die 90er Jahre der einzige iodierte Kohlenwasserstoff, der in der Atmosphäre
bestimmt wurde. Dies heißt jedoch nicht, dass CH3I die größte biologische Iodquelle ist. Der
Grund dafür, dass vor allem CH3I detektiert wurde, ist vermutlich eher dadurch zu erklären,
dass schwerere Kohlenstoff–Iod–Verbindungen wie z.B. CH2ICl, CH2I2, C2H5I und C3H7I
weniger flüchtig sind, eine kürzere Lebensdauer und einen niedrigeren Meer–zu–Luft–Fluß
haben, als durch niedrigere Produktionsraten im Ozean [Carpenter et al., 1999]. Es ist daher

10 Physik und Chemie der Troposphäre
möglich, dass die oben genannten Verbindungen eine größere Quelle für Iod darstellen als
CH3I.
Neuere Untersuchungen [Richter und Wallace, 2004] haben ergeben, dass biologische Aktivi-
tät vermutlich nur über organische Zwischenprodukte indirekt zur CH3I–Produktion beiträgt.
Ihren Ergebnissen zufolge spielen photochemische Reaktionen im offenen tropischen Ozean
eine größere Rolle. So wurde für die oberflächennahe Schicht bis zu einer Tiefe von 30 m
eine mittlere Produktion von 25,5 nmol m-2 Tag-1 CH3I abgeschätzt.
• Freisetzung aus Seesalz
In Seesalz sind nach Gewicht 55.7% Cl-, 0.19% Br- und 0.00002% I- enthalten [Holland,
1978]. Schon Duce et al. [1963] zeigten, dass Chlor und Brom aus Seesalzaerosolen frei-
gesetzt werden, wohingegen Iod darauf angereichert wird. Seesalzaerosole entstehen, indem
sich durch brechende Wellen und biologische Prozesse Luftblasen im Wasser formen. Bre-
chen diese Blasen auf, so bilden sich aus dem Film der Oberfläche kleine und größere Tröpf-
chen, wobei letztere sofort wieder an die Wasseroberfläche zurückkehren. Murphy et al.
[1998] zeigten, dass nahezu alle diese Aerosole, mit Trockendurchmesser von 0,13 µm und
größer, Meersalz enthalten. Keene et al. [2002] stellten fest, dass alle Teilchen mit mittlerem
Außendurchmesser bis hin zu 25 µm starke Säuren (HNO3, H2SO4) aufnehmen können. Da-
durch werden Halogene in der Form HX aus den Aerosolen freigesetzt. Diesen Vorgang wird
als acid displacement bezeichnet.
• anthropogene Quellen
Durch Verbrennung von Biomasse und organischer Stoffe werden halogenierte organische
Verbindungen freigesetzt. So stellte Cauer [1939] fest, dass uneffizientes Verbrennen von
Meeresalgen in West– und Mitteleuropa eine mittlere Konzentration von etwa 0.5 µg m-3 von
gasförmigem Iod in die Atmosphäre freisetzt.
Auch durch die Industrie gelangen Halogenverbindungen in die Atmosphäre, die Quellstärke
• Vulkane
Bobrowski et al. [2003] stellten fest, dass in der Abgasfahne von Vulkanen Bromoxid nach-
weisbar ist. Mit Mischungsverhältnissen von bis zu 1 ppb BrO stellt auch diese Quelle
vermutlich einen nicht zu vernachlässigenden Anteil am globalen Budget dar, der sowohl in
der Stratosphäre als auch in der Troposphäre von Bedeutung sein könnte. Iodoxid wurde bis-
her in diesen Abgasfahnen nicht nachgewiesen.

2 Physik und Chemie der Troposphäre 11
2.3.2 Senken der RHS
Damit sich reaktive Halogene wieder aus der Troposphäre entfernen, müssen sie zuerst mit Kohlen-
wasserstoffen (RH) oder Peroxidradikalen (HO2 und RO2) reagieren, wobei z.B. HX gebildet wird.
HX ist hydrophil, daher ist es wahrscheinlich, dass es durch nasse oder trockene Deposition irreversi-
bel aus der Atmosphäre entfernt wird [Platt, 2000]. Nasse Deposition bedeutet, dass das HX sich an
Partikel absetzt, z.B. in Wassertropfen von Wolken oder Nebel gelöst wird und durch Niederschlag
aus der Atmosphäre entfernt wird. Bei trockener Deposition lagert sich HX direkt in gasförmiger
Form im Ozean, am Erdboden, sowie auch an Gras, Bäumen, Gebäuden, etc. ab.
Die einzig mögliche Reaktivierung von HX hin zu einem reaktiven Halogen in der Gasphase ist die
Reaktion mit einem OH–Radikal:
HX + OH → X + H2O (R 2.1)
Die Bildung von HX ist vor allem für X = F, Cl sehr wahrscheinlich, da diese reaktiver sind als Brom
und Iod. Deshalb wird im Folgenden nicht weiter auf die Chemie von Fluor und Chlor eingegangen,
sondern auf die Reaktionen von Brom und Iod, die eng mit der Frage verbunden sind, welche Re-
aktionszyklen mit RHS in der Troposphäre möglich sind.
2.4 Reaktionen in der Troposphäre
Die besondere Bedeutung der Halogenchemie liegt darin, dass Halogen in der Lage sind, in kataly-
tischen Reaktionszyklen ozonabbauend zu wirken. In diesem Kapitel wird zunächst auf die Re-
aktionen von Brom und Iod näher eingegangen, dann auf den Einfluß der Halogenchemie auf das
Leighton–Verhältnis (NO2/NO) und den HOx–NOx–Zyklus, sowie auf die Bildung und Zerstörung von
Ozon in der Troposphäre.
Die wichtigsten Reaktionen der Halogene mit Ozon sind jene, in denen ein Halogenatom mit Ozon
reagiert und das daraus resultierende Halogenoxid schnell wieder photolysiert wird.
X + O3 → XO + O2 (R 2.2)
XO + hν → X + O (R 2.3)
Die Reaktionsrate der Reaktion R 2.2 wird von Atkinson et al. [2004] mit 1,2·10-12 cm3 Moleküle-1 s-1
(X = Br, I), bzw. 1,2·10-11 cm3 Moleküle-1 s-1 für X = Cl angegeben.

12 Physik und Chemie der Troposphäre
Dies ist jedoch noch nicht ausschlaggebend für den Ozonabbau und man kann einen solchen Kreislauf
einen „Nullzyklus“ bezeichnen. Die Ozonkonzentration erfährt bezüglich des Kreislaufs keine Ver-
änderung, da aus O2 und O wieder ein Ozonmolekül gebildet werden kann:
O2 + O + M → O3 + M (R 2.4)
Es gibt jedoch zwei Hauptzyklen in denen netto Ozon durch Halogene zerstört wird und die daher für
den Ozonabbau mitverantwortlich sind:
In Zyklus I reagieren Halogenoxide mit Hydroperoxyl:
XO + HO2 → HOX+ O2 (R 2.5
HOX+ hν → X + OH (R 2.6)
O3 + X → XO + O2 (R 2.7)
OH+ O3 → HO2 + O2 (R 2.8)
netto: 2 O3→ 3 O2
Durch die Reaktion mit HO2 wird verhindert, dass das XO photolysiert wird und sich wieder Ozon
bildet. Statt dessen werden durch den gesamten Zyklus II zwei Ozonmoleküle gespalten: eines durch
die Bildung von XO durch ein X–Radikal, und das andere durch die Reaktion von OH mit Ozon zur
Bildung des HO2.
In Zyklus II reagieren Halogenoxide entweder mit sich selbst oder mit anderen Halogenoxiden:
X + O3 → XO + O2 (R 2.9)
Y + O3 → XO + O2 (R 2.10)
XO + YO → Produkt (R 2.11)
netto: 2 O3→ 3 O2
Abb. 2.4: Reaktion von XO mit HO2
Abb. 2.5: Kreuzreaktion von Ha-logenoxiden

2 Physik und Chemie der Troposphäre 13
Die am wahrscheinlichsten gebildeten Produkte sind X + Y + O2 oder auch XY + O2, wobei XY
wieder zu X + Y photolysiert werden kann.
Selbstreaktion von BrO ist in der Troposphäre ein starker katalytischer Ozonabbau–Kreislauf, wenn
hohe Brom–Konzentrationen vorhanden sind (z.B. in polaren Regionen).
Bei der Selbstreaktion von IO können als Produkte auch OIO oder I2 entstehen. Das Produkt OIO kann
auch durch BrO–IO–Reaktion entstehen.
2.4.1 Reaktives Brom
Abb. 2.6 zeigt mögliche Reaktionen von Brom. Die in der Arktis und am Toten Meer entscheidende
Selbstreaktion von BrO (Brom–Explosion) ist in Küstengebieten der marinen Grenzschicht von
geringerer Bedeutung, da nur eine sehr geringe Konzentration an BrO erwartet wird. Untersuchungen
von Quack et al. [2004] ergaben, dass im Ozean produziertes Bromoform (CHBr3) die Hauptquelle für
organisches Brom (~10 Gmol Br Jahr-1) in der Atmosphäre ist. Die erhobenen Daten legen nahe, dass
der gesamte tropische offene Ozean, der in der Thermokline ein Chlorophyll–Maximum hat, eine
Quelle von CHBr3 darstellt. Dieses besitzt gegenüber Photooxidation eine Lebendauer von etwa vier
Wochen. Eine verstärkte CHBr3–Konzentration im Oberflächenwasser des offenen Ozeans wird übli-
cherweise mit dem Vorhandensein von Kieselalgen in Verbindung gebracht [Baker et al., 1999], es
gibt jedoch noch andere Möglichkeiten der lokalen Produktion von CHBr3, z.B. durch verstärktes
Vorhandensein von photosynthetisch aktiven Pigmenten. Makroalgen in Küstenregionen und Übersät-
tigung des Wassers des offenen Ozeans tragen zur CHBr3–Emission bei. Aufgrund der kurzen Lebens-
dauer kommt es zu regionalen und zeitlichen Schwankungen in der Produktion. Es wird eine stark va-
riierende atmosphärische CHBr3–Verteilung erzeugt.
Abb. 2.6: Überblick über die troposphärische Bromchemie. [Hönninger, 2002]

14 Physik und Chemie der Troposphäre
2.4.2 Reaktives Iod
Eine geringe anthropogene Quelle reaktiven Iods stellt die Verbrennung iodhaltiger organischer Ver-
bindungen dar, wie sie z.B. in Meeresalgen, aber auch in Kohle und Öl [Chameides und Davis, 1980]
vorkommen können. Die industrielle Quelle flüchtigen Iods wird auf weniger als 10% der natürlichen
biologischen Quellen geschätzt [Davis et al., 1996].
Eine biologische Quelle für das atmosphärische Iod ist gasförmiger Iodkohlenwasserstoff (CH3I,
CH2I2, C2H5I, CH2ClI, CH2IBr, etc.), der im Ozean durch Makroalgen und Phytoplankton freigesetzt
wird. So ist z.B. CH3I metabolisches Nebenprodukt vieler Arten mariner Algen und wurde als globale
Iodquelle mit 1–2 Tg/Jahr abgeschätzt [Davis et al., 1996]. Aufgrund ihrer geringen Löslichkeit kann
Ozeanwasser an Iodkohlenwasserstoffen übersättigt werden, was zu einem Fluss dieser Komponenten
vom Ozean in die Atmosphäre führt [Singh et al., 1983]. In der Troposphäre werden die Iodver-
bindungen rasch photolysiert und reaktives anorganisches Iod gebildet. Die photochemische Lebens-
dauer liegt dabei im Bereich von Minuten (für CH2I2, [WMO, 2002]) bis hin zu etwa 5 Tagen (CH3I,
[Vogt et al., 1999]). Sie ist deutlich kürzer als die der lichtempfindlichsten Bromverbindung: CHBr3
hat eine Lebensdauer von etwa 26 Tagen [WMO, 2002].
Die photochemische Umwandlung von organischem Iod kann auch Quelle für die Anreicherung von
anorganischem Iod auf Aerosolen und der Gasphase gegenüber Seesalz sein. Während Brom und Clor
in Meersalzaerosolen abgereichert werden, wird Iod in diesen stark angereichert, so dass, im Ver-
gleich zu Meerwasser, 100 bis 1000 mal mehr Iod nachgewiesen werden kann [Seto und Duce, 1972].
Daher können Aerosole (sowohl Seesalz– als auch Sulfataerosole) eher als Senke für Iod angesehen
werden denn als Quelle [Vogt et al., 1999; von Glasow und Crutzen, 2003].
Es ist nicht ganz geklärt, in welcher Form Iod im Aerosol vorliegt. Man unterscheidet zwischen den
beiden wichtigsten Salzen von Iod:
Iodat (IO3-) wird als inert angesehen und sollte daher, einmal gebildet, aus den ozonzerstörenden Zy-
klen entfernt werden, was impliziert, dass sich Iodate auf alternden marinen Aerosolen ablagern und
dadurch eine Senke für Iod darstellt. Diese Theorie steht bislang aber noch nicht im Einklang mit dem
Experiment, da Baker [2004] Iodat in diesen Aerosolen nur unterhalb der Nachweisgrenze finden
konnte, bzw. dieses auf der Südhalbkugel überhaupt nicht detektieren konnte.
Iodid (I-) kann mit HOX (X = Cl, Br, I) flüchtiges IX bilden [Baker, 2004]:
I- + HOX+ H+ → IX + H2O (R 2.12)
Diese Reaktion ist vom pH–Wert des Aerosols abhängig. Sie führt zusätzlich zur Reaktivierung des
Iods auch zur Verflüchtigung von Chlor und Brom. Nicht nur OIO sondern auch die anderen temporä-
ren Iodreservoirmoleküle wie HI, HOI, IONO2, I2O2 und INO2, die aus der Reaktion mit NOx und HOx
oder durch Selbstreaktion von IO entstehen, werden von marinen Aerosolen aufgenommen. Der Auf-
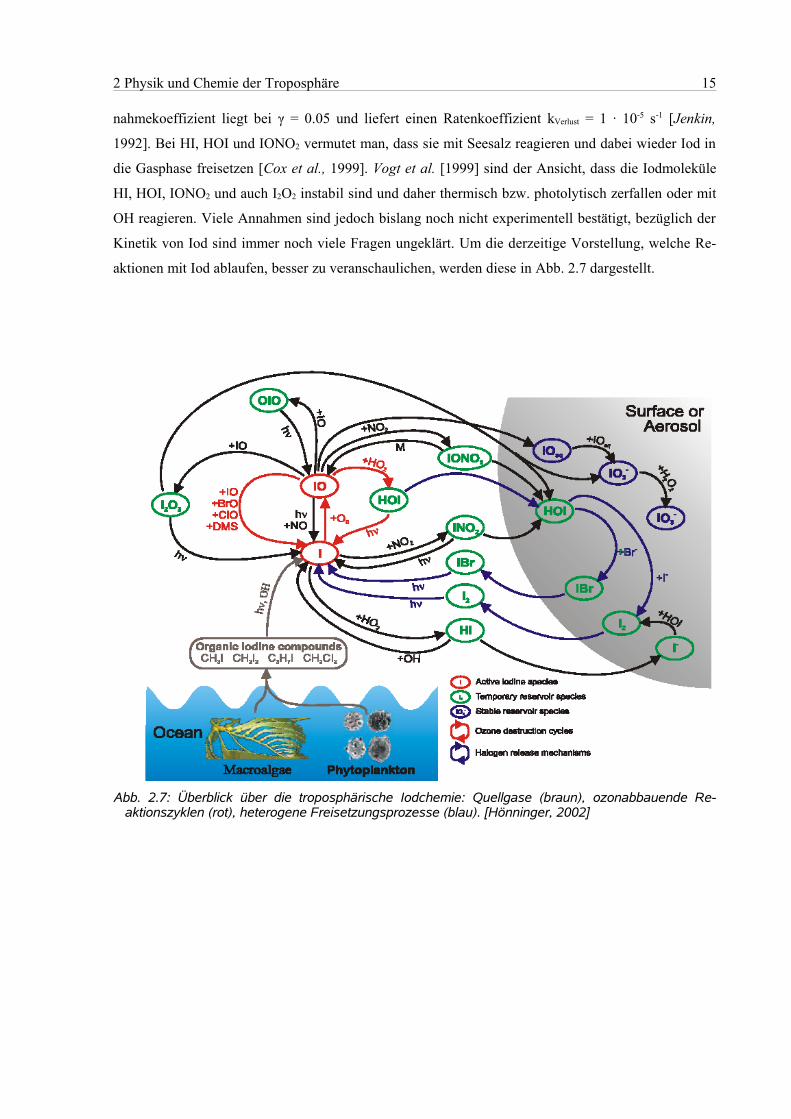
2 Physik und Chemie der Troposphäre 15
nahmekoeffizient liegt bei γ = 0.05 und liefert einen Ratenkoeffizient kVerlust = 1 · 10-5 s-1 [Jenkin,
1992]. Bei HI, HOI und IONO2 vermutet man, dass sie mit Seesalz reagieren und dabei wieder Iod in
die Gasphase freisetzen [Cox et al., 1999]. Vogt et al. [1999] sind der Ansicht, dass die Iodmoleküle
HI, HOI, IONO2 und auch I2O2 instabil sind und daher thermisch bzw. photolytisch zerfallen oder mit
OH reagieren. Viele Annahmen sind jedoch bislang noch nicht experimentell bestätigt, bezüglich der
Kinetik von Iod sind immer noch viele Fragen ungeklärt. Um die derzeitige Vorstellung, welche Re-
aktionen mit Iod ablaufen, besser zu veranschaulichen, werden diese in Abb. 2.7 dargestellt.
Abb. 2.7: Überblick über die troposphärische Iodchemie: Quellgase (braun), ozonabbauende Re-aktionszyklen (rot), heterogene Freisetzungsprozesse (blau). [Hönninger, 2002]

16 Physik und Chemie der Troposphäre
2.4.2.1 Reaktionszyklen von IO
Im Gegensatz zu Chlor und Brom reagiert Iod nicht mit Kohlenwasserstoffen, um HX zu bilden. Es
reagiert in erster Linie zusammen mit Ozon zu IO, kann jedoch durch Photolyse wieder in ein I–Atom
zurückgeführt werde:
I + O3 → IO + O2 (R 2.13)
IO + hν → I + O(3P) (R 2.14)
Der Ratenkoeffizient der Reaktion 2.13 liegt bei k = 1.2 · 10-12 cm3 Moleküle-1 s-1 [Atkinson et al.,
2004]. Die Reaktion läuft somit relativ langsam ab.
IO wird nicht nur photolysiert und reagiert wie in Zyklus I+II beschrieben, sondern kann auch mit IO
(„Selbstreaktion“), ClO und BrO („Kreuzreaktion“) reagieren. Für die Bildung von XO wird (über
Reaktion R 2.13) Ozon zerstört. Im Laufe der Zyklen wird kein einzelnes Sauerstoffatom gebildet, das
mit O2 wieder zu Ozon reagieren könnte, daher wirken alle diese Reaktionen ozonzerstörend.
Allgemeiner spricht man von Ozon–zerstörenden Reaktionen wenn im Produkt die Anzahl der „odd
oxygens“ (= O3, O(1D), XO, NO3, ...) geringer ist als im Edukt. Im gegenteiligen Fall spricht man von
einer ozonbildenden Reaktion.
IO–Selbstreaktion
IO + IO → 2 I + O2 (~ 0) (R 2.15)
→ I2 + O2 (~ 0) (R 2.16)
→ OIO + I (~ 0.4) (R 2.17)
IO + IO + M → I2O2 + M (~ 0.6) (R 2.18)
Die Reaktionen sind nicht gleich wahrscheinlich. Die jeweiligen Verzweigungsverhältnisse kr/kges sind
in der rechten Spalte angegeben [Crowley, 2004], wobei kr der Ratenkoeffizient der jeweiligen Re-
aktion ist. kges wird mit 8.2 · 10-11 cm3 Moleküle-1 s-1 angegeben.
I2 kann durch Photolyse wieder zu zwei I–Atomen gespalten werden, diese können erneut mit Ozon zu
IO reagieren. Daher tragen die beiden Reaktionen (R 2.15) und (R 2.16) direkt zur weiteren Ozonzer-
störung bei. Dabei kann I2 nicht nur durch die Reaktion (R 2.16) entstehen, es kann auch über weitere
Reaktion der anderen Produkte der Selbstreaktion gebildet werden. In Mace Head wurde I2 mit Mi-
schungsverhältnissen von bis zu 93 ppt bei Nacht und 25 ppt bei Tag gemessen [Saiz-Lopez und
Plane, 2004].
OIO wurde mit Hilfe der DOAS-Technik schon in der MBL in Mace Head [Hebestreit et al., 2000]
und in Tasmanien [Allan et al., 2001] detektiert. Hoffmann et al. [2001] schlagen jedoch vor, dass

2 Physik und Chemie der Troposphäre 17
OIO möglicherweise auf Teilchen kondensiert, was eine Erklärung für das große Vorkommen von Iod
in marinen Aerosolen mit kleinen Durchmessern liefern würde. Sie schlagen außerdem vor, dass die
Selbstreaktion von OIO Quelle für Partikelbildung in Küstenregionen sei. Die kinetischen und photo-
chemischen Eigenschaften des OIO–Radikals sind derzeit jedoch noch weitestgehend unbekannt [Pe-
ters, 2005] so dass nicht mit Klarheit beantwortet werden kann, welche Reaktionen OIO nun wirklich
eingeht.
Die Kreuzreaktionen mit ClO ist aus Bedjanian et al. [1997] entnommen, wieder mit den jeweiligen
Verzweigungsverhältnissen:
IO–ClO–Kreuzreaktion
IO + ClO → I + OClO (0.55±0.03) (R 2.19)
→ I + Cl + O2 (0.25±0.02) (R 2.20)
→ ICl + O2 (0.20±0.02) (R 2.21)
Der Ratenkoeffizient für die Reaktion von IO + ClO → Produkte beträgt kIO+ClO = (1.1 ± 0.2) · 10-11
cm3 Moleküle-1 s-1 bei 298 K. Die erste Reaktion führt zu einem Nullzyklus für den Ozonabbau, da
OClO unter anderem die Reaktion X + OClO → XO + ClO mit X = Cl, I eingeht und somit zur Bil-
dung der XO–Radikale kein Ozon zerstört werden muss.
Reaktion (R 2.21) hat den gleichen Effekt wie Reaktion (R 2.20), da ICl zu I + Cl photolysiert wird.
IO–BrO–Kreuzreaktion
IO + BrO → Br + OIO (kr/kges= 0.65–0.93) (R 2.22)
→ Br + I + O2 (kr/kges≤ 0.3) (R 2.23)
→ I + OBrO (kr/kges≤ 0.15) (R 2.24)
→ IBr + O2 (kr/kges≤ 0.05) (R 2.25)
Für das Verzweigungsverhältnis kr/kges wird der Ratenkoeffizient kges mit 8.5 · 10-11 cm3 Moleküle-1 s-1
angegeben [Rowley, 2001]. kr entspricht wieder dem Ratenkoeffizient der jeweiligen Reaktion.
Auch die Reaktion mit HO2 (vgl. Zyklus I) und in stärker belasteten Gebieten die Reaktion mit NO
und NO2 kann eine Senke für IO bilden. Um dies besser zu verdeutlichen wird zuerst auf das
NO2/NO–Verhältnis näher eingegangen, sowie auf den Zusammenhang zwischen den NOx– und HOx–
Zyklen. Dann wird der Einfluss von Iod auf diese Zyklen dargelegt, sowie die Ozonproduktion als
auch –zerstörung in diesem Zusammenhang.

18 Physik und Chemie der Troposphäre
2.4.3 Das Leighton–Verhältnis
NOx ist als Summe von NO und NO2 definiert [Finlyson-Pitts und Pitts, 2000]. Um eine Vorstellung
der Größenordnungen zu bekommen, in denen NOx normalerweise vorliegt, wird in Tabelle 2.3 zu-
nächst ein kleiner Überblick über Konzentrationen gegeben, wie sie abhängig von der jeweiligen Um-
gebung in der Troposphäre anzutreffen sind [Finlayson-Pitts und Pitts, 2000].
Tabelle 2.3: Typische NOx–Werte [Finlayson–Pitts und Pitts, 2000]
Ort Charakterisierung NOx-Werteentlegene Gebiete in Alaska Reinluftgebiet 25 pptKanada ländliches Gebiet 2.3 ppb (im Mittel)Paris, Los Angeles urban, starkt verschmutzt 200 ppb (Maximalwert)
Die bei der im Rahmen der Arbeit durchgeführten Kampagne traten NO2 Werte auf, die im Mittel bei
0.2 ppb lagen, mit Spitzenwerten bis zu 0.8 ppb. Das Messgebiet ist also in den Bereich wenig
verschmutzer Luft einzuordnen.
In der Grenzschicht sind die NOx–Arten (NO, NO2) in einen Nullzyklus mit Ozon eingebunden:
NO + O3 → NO2 + O2 (R 2.26)
NO2 + hν → NO + O (λ≤420 nm) (R 2.27)
O + O2 → O3 (R 2.28)
Daher stellt sich ein Gleichgewicht zwischen den Reaktionspartnern ein, das Verhältnis der Kon-
zentration von NO2 zu der von NO nennt man auch Leighton-Verhältnis:
Mit kNO+O3 = 1,8 · 10-14 cm3 Moleküle-1 s-1 [DeMore et al., 1997], JNO2 ≈ 10-2 [von Glasow, 2005] und
einem Ozonmischungsverhältnis, wie es auf der Kampagne zu messen war (≈ 20 ppb), also
[O3] = 4.8 · 1011 cm-3, ergibt sich ein Leighton-Verhältnis von etwa L = 1,16. Nachts wird, da keine
Photolyse auftritt, alles NO in NO2 umgewandelt, so dass [NOx] = [NO2].
Dieses Verhältnis wird jedoch durch einige eingreifende Reaktionen gestört:
NO reagiert auch mit HO2 und RO2, bei der Reaktion wird Ozon gebildet, jedoch nicht abgebaut:
(Gl. 2.1)L =[NO2][NO]
=kNOO3
⋅[O3]JNO2

2 Physik und Chemie der Troposphäre 19
HO2 + NO → OH + NO2 (R 2.29)
RO2 + NO → RO + NO2 (R 2.30)
mit kHO2+NO = 8,1 · 10-12 cm3 Moleküle-1 s-1 sowie kRO2+NO= 7,7 · 10-12 cm3 Moleküle-1 s-1 für RO2 = CH3O2
bzw. kRO2+NO= 8,7· 10-12 cm3 Moleküle-1 s-1 für RO2 = C2H5O2 [DeMore et al., 1997].
Ebenso reagieren, falls in der Troposphäre vorhanden, Halogenoxide mit NO. Dadurch wird sowohl
NO2 gebildet, das Ozon produzieren kann, als auch ein X–Radikal, das wiederum Ozon abbaut. Ty-
pische Ratenkoeffizienten der Reaktion
XO + NO → X + NO2 (R 2.31)
sind kClO+NO = 1.7 · 10-11 cm3 Moleküle-1 s-1, kBrO+NO = 2.1 · 10-11 cm3 Moleküle-1 s-1, kIO+NO = 2.0 · 10-11
cm3 Moleküle-1 s-1.
Durch die Reaktionen R 2.29, R 2.30 und R 2.31 verändert sich das Leighton–Verhältnis zu:
Berechnet man nun das Leighton–Verhältnis mit dieser Formel, so erhält man für typische Kampa-
gnenwerte ([O3] = 10 ppb, [IO] = 0,5 ppt) ein Verhältnis von L = 0,97. Nimmt man an, dass kein IO
vorhanden ist, so verschiebt sich das Verhältnis zu L = 0,95.
Für kleinere Ozonkonzentrationen nimmt auch das Leighton–Verhältnis ab, da über die Reaktion von
Ozon mit NO weniger NO2 gebildet wird. So erhält man für ein Ozonmischungsverhältnis [O3] = 16
ppb das Leighton–Verhältnis L = 0,80 (für [IO] = 0,5 ppt) bzw. L = 0,78 (für [IO] = 0 ppt). Wie man
sieht bewirkt bereits eine so kleine Menge an IO eine Veränderung des Leighton–Verhältnisses um
2%. Bei der Berechnung dieser Daten geht die Korrelation zwischen IO–Konzentration und O3–Kon-
zentration über eine Reaktion allerdings nicht ein.
(Gl. 2.2)
L =[NO2][NO]
=kNOO3
⋅[O3]kHO2NO⋅[HO2]kRO2NO⋅[RO2]kXONO⋅[XO]JNO2

20 Physik und Chemie der Troposphäre
2.4.4 Der HOx–NOx–Zyklus und Ozon: Produktion – Abbau
Dieser HOx–NOx–IO–Zyklus lässt sich auch graphisch wie in
Abb. 2.8 darstellen. Ob im gesamten Zyklus Ozon produziert
oder abgebaut wird hängt von den Größen der ozonprodu-
zierenden Zyklen (P) und der ozonabbauenden Zyklen (L) ab.
Mit Zyklus I und II wurden in Kapitel 2.4 schon die ersten
beiden ozonabbauenden Zyklen L1 und L2 vorgestellt. Das HO2
dieser Reaktionen wird aus CH4 über die Oxidation durch ein
OH–Radikal gebildet:
CH4 + OH → CH3 + H2O (R 2.32)
CH3 + O2 + M→ CH3O2 (R 2.33)
CH3O2 + NO → CH3O + NO2 (R 2.34)
CH3O + O2 → CH2O + HO2 (R 2.35)
Ozonabbauende Reaktion im Zyklus L3 ist die Reaktion von HO2 mit Ozon
HO2 + O3 → OH + 2 O2 (R 2.36)
OH + O3 → HO2 + O2 (R 2.37)
netto: 2 O3→ 3 O2
und auch die Photolyse von Ozon (L4):
O3 + hν → O2 + O(1D) (R 2.38)
Die Photolyserate von Ozon in den Tropen wurde von von Glasow [2005] mit JO3 = 5,5 · 10-5 s-1 be-
rechnet (0° Breite, Sonnenhöchststand), dies entspricht einem maximalen Ozonverlust um die Mit-
tagszeit von 4 ppb/h.
Die Reaktion von HO2 mit NO zu NO2 ist der ozonbildende Prozess im Zyklus P1:
HO2 + NO → OH + NO2 (R 2.39)
NO2 + hν → NO + O (λ≤420 nm) (R 2.40)
netto: O + O2 → O3 (R 2.41)
Abb. 2.8: Der HOx-NOx-Zyklus, undder Einfluss von Iod auf diesen(gepunktet).
CO, CH4 CO2, H2O
O2
I IO
HOI
OH HO2
I IONO2 NO
O O3
O2

2 Physik und Chemie der Troposphäre 21
Zusätzlich wird auch über (R 2.34) und die anschließende Photolyse von NO2 Ozon gebildet (P2).
Die geschwindigkeitsbestimmenden Reaktion in Produktionszyklus P1 und P2 ist jedoch nicht die Pho-
tolyse von NO2, die sehr schnell ist (JNO2 = 0,01 s-1), sondernd die Reaktion von NO mit HO2 bzw.
RO2. Diese sind nicht ganz so schnell, für typische Kampagnenwerte ergeben sich so mit den oben
angegebenen Reaktionskoeffizienten Ozonproduktionsraten von RP1 = 0,6 ppb/h und RP2 = 40 ppt/h.
Ob Ozon produziert oder abgebaut wird lässt sich dann durch die Raten der in den Zyklen geschwin-
digkeitsbestimmenden Reaktionen berechnen:
In Abb. 2.8 sind die Reaktionen, in denen Iod in den Prozess eingreift, gepunktet eingezeichnet. Mit
L1 reagiert HO2 mit reaktivem Iod (IO) verstärkt zu OH. Die HO2–Konzentration nimmt ab und die
OH–Konzentration nimmt zu, dadurch nimmt auch das Verhältnis OH/HO2 zu.
Im Falle von NO und NO2 ist es genau umgekehrt: über die Reaktion von IO mit NO2 wird verstärkt-
NO2 gebildet, das Leighton–Verhältnis nimmt ab. Diese letzte Reaktion hat keinen Einfluss auf Ozon,
da NO2 zu Ozon photolysiert werden kann, das I–Radikal jedoch mit Ozon reagiert und dieses abbaut.
Der Kampagnenmessort ist mit seinen typischen Konzentrationen von 0,2 ppb NO2 in Abb. 2.9 in den
Bereich der schon leicht verschmutzten marinen Grenzschicht einzuordnen. Sowohl Ozonproduktion
als auch Ozonabbau ist dort möglich. Diese Eigenschaft der Atmosphäre wird mit den Ergebnissen
des Ozonmonitors, der während der Kampagne betrieben wurde, in Kapitel 7.1.2 diskutiert.
(Gl. 2.3)d [O3]dt
= RP1RP2
RL1RL2
RL3RL4
Abb. 2.9: Beobachtete NOx-Werte in verschiedenen Gebieten der Troposphäre. Die Isopleten gebendie Ozon–Produktion über Mittag in ppb h-1 an. [Finlayson–Pitts und Pitts, 2002]

22 DOAS-Grundlagen
3 DOAS-Grundlagen
Zur Messung von Spurengasen in der Atmosphäre wurde erstmals von U. Platt, D. Perner und J. No-
xon [Platt et al., 1979] die differentielle optische Absorptionsspektroskopie (kurz: DOAS) eingeführt.
Diese Methode dient zur Ermittlung von Konzentrationen bzw. Mischungsverhältnissen von Stoffen
in der Atmosphäre bis in den ppt–Bereich (parts per trillion, also 1/1012) mit einer Genauigkeit von
90–99%.
Physikalische Grundlage der Methode ist, dass Atome und Moleküle Licht absorbieren. Dadurch
werden die Moleküle angeregt und auf höhere Rotations–, Vibrations– und elektronische Energienive-
aus angehoben. Da die Moleküle verschiedener Stoffe bei jeweils unterschiedlichen Wellenlängen un-
terschiedlich stark absorbieren, hat jeder Stoff charakteristische Absorptionslinien (i.A. Spektrallinien
genannt, da sie bei diesen Wellenlängen nicht nur absorbieren, sondern auch emittieren), die im Falle
der untersuchten Spurenstoffe so dicht liegen, dass ganze Absorptionsbanden beobachtbar sind. Aus
der charakteristischen Absorption eines jeden Stoffs kann man Rückschlüsse ziehen, welcher
Spurenstoff sich im Zeitraum der Messung im Lichtstrahl befand. Ist zudem noch die Länge des
Lichtwegs bekannt, so ist es möglich, auf diesem die mittlere Konzentration des Stoffes zu berechnen.
Dies hat den Vorteil, dass keine Proben genommen werden müssen, bei deren Probennahme z.B.
durch Ansaugen der Luft in die örtlichen Begebenheiten eingegriffen wird. Hochreaktive Spurenstoffe
reagieren beispielsweise während des Transports, wodurch sie später im Labor nicht mehr nachge-
wiesen werden können. So wurden mit DOAS schon die Konzentration sowohl sehr reaktiver Stoffe
wie die freien Radikale OH, NO3, ClO, BrO und IO bestimmt, als auch weitere für die Atmosphä-
renchemie bedeutenden Stoffe (SO2, CS2, O3, NO, NO2, HONO, NH3, CH2O und monozyklische
aromatische Kohlenwasserstoffe [Platt, 2000a]).
Ein weiterer Vorteil ist, dass aus einer Messung die Konzentration mehrerer Stoffe gleichzeitig be-
stimmt werden kann, vorausgesetzt die jeweiligen Absorptionswirkungsquerschnitte sind bekannt und
die Stoffe absorbieren im selben Längenwellenbereich über den auch gemessen wurde.
Beim ersten DOAS–Gerät 1979 wurde eine künstliche Lichtquelle benutzt („aktives“ DOAS), für
Messungen in der Stratosphäre dient z.B. das Sonnenlicht als Lichtquelle [Solomon et al., 1987] oder
Streulicht („passives“ DOAS). Auch von Satelliten aus wird das von der Erde zurückgestrahlte Licht
detektiert und auf die Absorptionen der Spurengase hin untersucht. So konnte 2004 mit Hilfe des auf
dem Europäischen Umweltsatelliten Envisat installierten Spektrometer Sciamachy (SCanning Imaging
Absorption spectroMeter for Atmospheric CHartographY) eine globale Verteilung der mittleren
vertikalen Säulendichte von troposphärischem NO2 bestimmt und die Hauptquellen der Abgase identi-
fiziert werden: die Industriezentren in Nordamerika, Europa und Asien, aber auch die Schifffahrts-
wege nach China sind deutlich sichtbar. Zusätzlich zur anthropogenen Erzeugung und der Erzeugung
durch Verbrennen von Biomasse tragen auch Blitze mit etwa 2,8 (0,8–14) Tg [N] pro Jahr [Beirle et
al., 2004a] zur NO2–Konzentration in der Atmosphäre bei.

3 DOAS-Grundlagen 23
Es existieren zahlreiche unterschiedliche DOAS–Messgeräte. Um einen Einblick zu bekommen,
werden hier einige Messgeräte, wie sie am Institut für Umweltphysik verwendet werden bzw. wurden,
kurz vorgestellt:
Passives DOAS (mit "passiver" Lichtquelle):
• MAX–DOAS: (Multi–Axis–Doas). Spektrograph mit Detektor, der mit Hilfe eines Motors in ver-
schiedenen Winkeln Aufnahmen machen kann. Durch Messung in verschiedenen Winkeln kann
vom Boden aus die Konzentration von Spurengasen in der Troposphäre als auch in der Stratosphä-
re, also in Abhängigkeit der Höhe bestimmt werden. Als Lichtquelle dient zumeist nicht das direk-
te Sonnenlicht, sondern Streulicht, das aus verschiedenen Zenit– und Azimutwinkeln ins Teleskop
einfällt. Für eine nähere Beschreibung sei auf Hönninger [2004a] verwiesen.
• MiniMAXDOAS: kompaktes und leicht transportables MAX–DOAS–Gerät, das derzeit verstärkt
zur Beobachtung der Abgasfahnen von Vulkanen genutzt wird [Bobrowski, 2002].
• IDOAS: (Imaging–DOAS). Diese Variante eines DOAS–Messgeräts hat als Detektor keine Photo-
diodenzeile (kurz: PDZ) sondern einen CCD–Chip, durch den man eine zweidimensionale Auflö-
sung erreicht. Durch die zweite Dimension erreicht man eine zusätzliche Ortsauflösung. Das Gerät
wurde 2004 von Lohberger et al. entwickelt.
• Ballongestützes DOAS: der DOAS–Aufbau ist auf einem Ballon installiert und misst in verschie-
denen Sichtgeometrien.
Aktives DOAS (mit "aktiver" Lichtquelle):
• LP–DOAS : (Langpfad–DOAS). Das von einer künstlichen Lichtquelle emittierte Licht wird mit
Hilfe einer Teleskopoptik parallelisiert und über einen Lichtweg gesandt, an dessen Ende es detek-
Abb. 3.1: NO2 in der Troposphäre, [Beirle et al., 2004b]

24 DOAS-Grundlagen
tiert wird oder durch Retroreflektoren zurückreflektiert wird um am Ausgangspunkt detektiert zu
werden. Vorteil der Variante mit Retroreflektoren ist, dass nur auf einer Seite des Lichtwegs eine
Stromquelle verfügbar sein muss. Für eine nähere Erläuterung der Messmethode sei auf diese
Arbeit, Kap. 4, oder auch auf Stutz [1996] verwiesen.
• Multi–Beam–DOAS: Von der Grundstruktur mit dem normalen Langpfad–DOAS vergleichbar,
allerdings durchlaufen mehrere Lichtstrahlen das Messgebiet, wodurch man eine räumliche Vertei-
lung der zu bestimmenden Spurengase bekommt.
3.1 Lambert–Beer–Gesetz
Elektromagnetische Strahlung wird beim Durchgang durch Materie von dieser absorbiert oder auch
durch Streuung abgeschwächt. Dabei ist die Änderung der Intensität dI proportional der ursprüngli-
chen Intensität I0 die von der Lichtquelle emittiert wird und der Wegstrecke dl.
Proportionalitätskonstanten sind die auf dem Lichtweg variierende Teilchenzahldichte ci(l) und der
Wirkungsquerschnitt σi des Gases i:
dI = I0 cil i ,Tdl
Das negative Vorzeichen gibt dabei an, dass die Lichtintensität beim Durchlaufen des Lichtwegs ab-
nimmt.
Integriert man nun entlang der Wegstrecke l und summiert über die Absorption der i Gase, so erhält
man das Lambert–Beer–Gesetz. Die Strahlungsintensität nach Durchlaufen der Wegstrecke l, auf der
die Konzentration ci(l) des i–ten Spurengases vorhanden ist lässt sich dann wie folgt berechnen:
I , L = I0e∫
0
L
∑i
i , pl,T lci dl
Über die Absorption der Spurengase muss aufsummiert werden, da sich bei der Feldmessung auf dem
Lichtweg nicht nur ein Gas, sondern viele verschiedene Spurengase befinden. Zudem ist der Ab-
sorptionswirkungsquerschnitt abhängig von Druck und Temperatur. Auch die Konzentration über den
Lichtweg schwankt, deshalb wird das Integral über den gesamten Lichtweg L gebildet.
(Gl. 3.3)
(Gl. 3.2)
dI ∝ I0 dl (Gl. 3.1)

3 DOAS-Grundlagen 25
Üblich verwendete Einheiten der obigen Größen sind:
[I], [I0] = Photonen cm-2 s-1 [L] = cm[λ] = nm[c] = Moleküle cm-3
[σ] = cm2 Moleküle-1
Der Absorptionswirkungsquerschnitt eines Spurengases lässt sich im Laborexperiment bestimmen, in-
dem man den Lichtweg kurz hält und auf den gesamten Lichtweg, z.B. mit Hilfe einer Küvette, das zu
bestimmende Gas in einer bekannten Konzentration einbringt. Die charakteristische Absorption des
Gases wird durch dessen Absorptionswirkungsquerschnitt erzeugt, der dann anhand der bekannten
Konzentration des Gases normiert werden kann.
Die zu bestimmende (über den Lichtweg gemittelte) Konzentration des Gases erhält man durch Um-
formen des Lambert–Beerschen–Gesetzes:
ci =ln I0 ,T
I i ,TL
=i
i ,TL
τ ist die optische Dichte. Sie ist definiert als der Logarithmus aus dem Verhältnis der Intensitäten vor
und nach Durchgang durch den Absorber:
i , L = lnI0
I , L= i⋅∫
0
L
cildl
Typische optische Dichten von atmosphärischen Spurengasen liegen bei 10-4 –10-2.
(Gl. 3.4)
(Gl. 3.5)
26 DOAS-Grundlagen
3.2 Prinzipien der DOAS-Methode
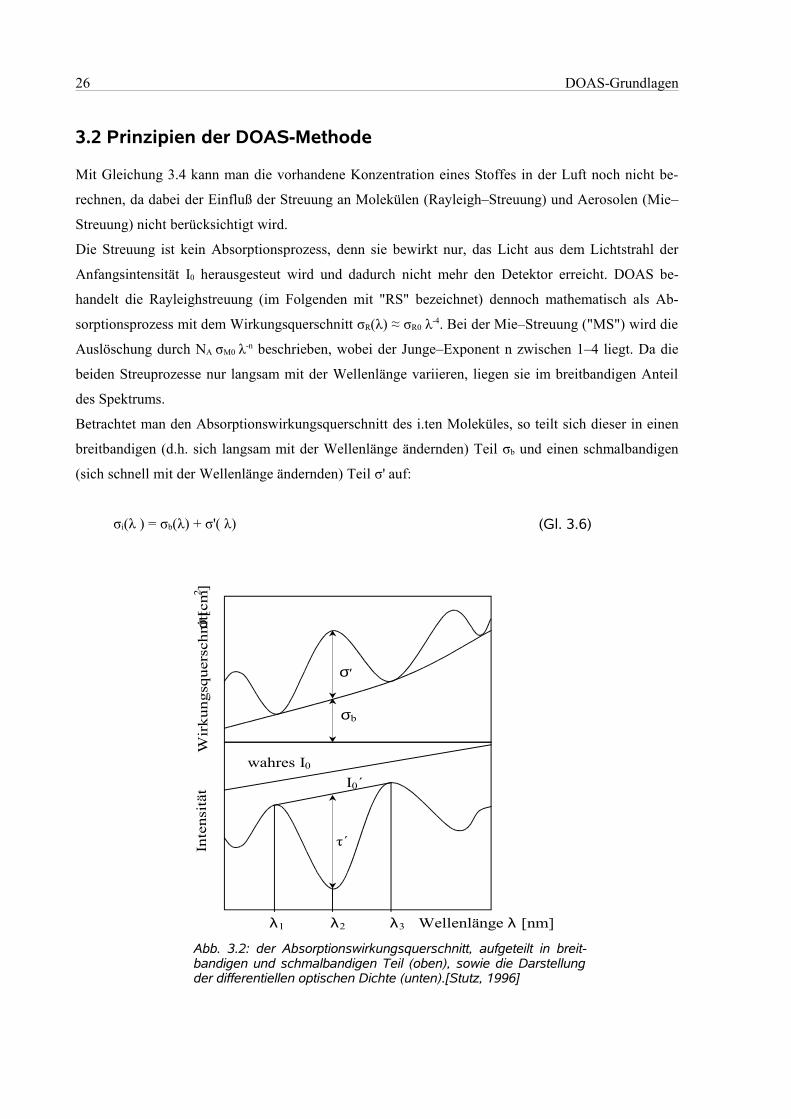
Mit Gleichung 3.4 kann man die vorhandene Konzentration eines Stoffes in der Luft noch nicht be-
rechnen, da dabei der Einfluß der Streuung an Molekülen (Rayleigh–Streuung) und Aerosolen (Mie–
Streuung) nicht berücksichtigt wird.
Die Streuung ist kein Absorptionsprozess, denn sie bewirkt nur, das Licht aus dem Lichtstrahl der
Anfangsintensität I0 herausgesteut wird und dadurch nicht mehr den Detektor erreicht. DOAS be-
handelt die Rayleighstreuung (im Folgenden mit "RS" bezeichnet) dennoch mathematisch als Ab-
sorptionsprozess mit dem Wirkungsquerschnitt σR(λ) ≈ σR0 λ-4. Bei der Mie–Streuung ("MS") wird die
Auslöschung durch NA σM0 λ-n beschrieben, wobei der Junge–Exponent n zwischen 1–4 liegt. Da die
beiden Streuprozesse nur langsam mit der Wellenlänge variieren, liegen sie im breitbandigen Anteil
des Spektrums.
Betrachtet man den Absorptionswirkungsquerschnitt des i.ten Moleküles, so teilt sich dieser in einen
breitbandigen (d.h. sich langsam mit der Wellenlänge ändernden) Teil σb und einen schmalbandigen
(sich schnell mit der Wellenlänge ändernden) Teil σ' auf:
σi(λ ) = σb(λ) + σ'( λ) (Gl. 3.6)
I0´
σb
σ′
wahres I0
Inte
nsit
ät
λ1 λ2 λ3 Wellenlänge λ [nm]
Wir
kung
sque
rsch
nitt
σ
[cm2 ]
τ´
Abb. 3.2: der Absorptionswirkungsquerschnitt, aufgeteilt in breit-bandigen und schmalbandigen Teil (oben), sowie die Darstellungder differentiellen optischen Dichte (unten).[Stutz, 1996]

3 DOAS-Grundlagen 27
Somit besteht das gesamte Spektrum der Intensität I aus einem breitbandigen Anteil, der vor allem
durch die Schwarzkörperstrahlung von Lampe und Sonne, die Erdatmosphäre (Rayleigh– und Mie–
Streuung) und andere Absorber erzeugt wird. Dieses wird vom schmalbandigen Spektrum mit den
charakteristischen schmalen Absorptionsbanden der gasförmigen Spurenstoffe als auch den Lampene-
missionsbanden überlagert.
Um die Konzentration der Spurenstoffe berechnen zu können konzentriert man sich bei der Aus-
wertung auf die schmalen Absorptionsbanden. Deshalb definiert man ein neues I'0 und den differenti-
ellen Absorptionsquerschnitt σ'. Dafür formt man Gleichung 3.3 um:
I =I0 exp[∫0
L
∑i
'icidl]exp[∫0
L
∑ibciR Mdl]A
=I0 exp[∫0
L
∑i
'icidl]⋅ f ⋅A
Die erste Exponentialfunktion beschreibt dabei den Effekt der differentiellen Absorption des
Spurengases, die zweite Exponentialfunktion den langsam variierenden Teil der Absorption des
Spurengases sowie die Effekte von RS und MS (hier beschrieben durch die Extinktionskoeffizienten
εR(λ) = σR0 λ-4 cAIR und εM(λ) = NA σM0 λ-n ). Mit dem Faktor A(λ) wird die Abschwächung des
optischen Systems, die langsam von der Wellenlänge abhängig ist, berücksichtigt. [Platt, 2000a]
Fasst man I0 mit der zweiten Exponentialfunktion zusammen, so erhält man die Intensität I'0, die der
Intensität in Abwesenheit der gesuchten Absorptionsstrukturen entspricht und Gleichung 3.7 lässt sich
umschreiben zu:
I , L = I0 ´e∫
0
L
∑i
i ´ , pl ,T lci dl
mit I0 ´ = I0⋅e∫
0
L
∑ bciR Mdl⋅A
Man beachte, dass in Gleichung 3.8 nur noch der differentielle Wirkungsquerschnitt (z.B. durch La-
boruntersuchungen) bekannt sein muss. Ebenso lässt sich nun auch die differentielle optische Dichte
τ' definieren:
´ , L = lnI0 ´I , L
= ∑i
i ´⋅∫0
L
cildl
(Gl. 3.7)
(Gl. 3.9)
(Gl. 3.8)

28 DOAS-Grundlagen
Da die Rayleigh– und Mie–Streuung auf die differentiellen Größen keinen Einfluss haben, kann man
bei der Auswertung die über den Lichtweg L gemittelte Konzentration c eines atmosphärischen
Spurenstoffs berechnen, indem man Gleichung 3.9 umschreibt zu:
ci =ln I0 ´ ,T
I ´i ,T⋅L
= ´
i ´ ,T⋅L
Auch im Messprozess durchläuft das Messspektrum verschiedene Veränderungen, die im Folgenden
konkret am Beispiel der verwendeten Messapparatur beschrieben wird (vgl. Abb. 3.3):
Das bei der Aufnahme eines Messspektrums aufgenommene Licht der Intensität I(λ,L) wird im Spek-
trograph zerlegt. Dieser hat nur ein endliches Auflösungsvermögen, was mathematisch einer Faltung
mit der Instrumentenfunktion gleich kommt. Der Detektor bestimmt pro Pixel die Zählrate, die pro-
portional zur detektierten Lichtintensität ist. Schlussendlich werden bei der Auswertung wie eben be-
schrieben nur noch die differentiellen Strukturen analysiert (das Argument der Exponentialfunktion in
Gleichung 3.8 und über die optische Dichte, die der Logarithmus aus dem Quotienten des Lampen-
referenzspektrums und dem Messspektrum ist, die Konzentration des Spurestoffs auf der Messstrecke
bestimmt.
(Gl. 3.10)
Abb. 3.3: Prinzipieller Aufbau eines DOAS-Geräts. Im rechten Teil wird ge-zeigt, wie sich die Intensität eines Absorptionspektrums (hier: Absorptiondurch Formaldehyd) durch den Messvorgang verändert.

3 DOAS-Grundlagen 29
3.3 Prinzipien der Auswertung
Zur Auswertung der Spektren wurde mit dem Programm WinDOAS [van Roozendael und Fayt, 2000]
gearbeitet. Das Prinzip dieses Programmes besteht darin, mit Hilfe der aus der Literatur bekannten
Absorptionswirkungsquerschnitte das gemessene Spektrum nachzumodellieren: Einer Linearkombina-
tion F der (ggf. gefalteten) Absorptionswirkungsquerschnitte σi werden Parameter ai bestmöglichst
angepasst, die breitbandige Struktur des Spektrums wird durch ein Polynom P 4.ten Grades berück-
sichtigt:
Nach der Gaußschen Minimierung der Fehlerquadrate [Bevington, 1969] werden die Parameter so ge-
wählt, dass die Quadrate der Fehler zwischen Modell und Messung minimiert werden:
Die zu bestimmende differentielle optische Dichte eines Spurenstoffs ist dann:
Mit Gleichung 3.10 lässt sich die Konzentration des i–ten Spurenstoffs berechnen.
Diese Methode kann jedoch nicht eine Verschiebung der Absorptionswirkungsquerschnitte gegenüber
einem Spektrum berechnen, da der hierfür anzupassende Parameter nicht linear von der Modellfunkti-
on abhängt. Deshalb muss das Modell dem Messspektrum numerisch angeglichen werden. Dies ge-
schieht nach der nichtlinearen Methode der Anpassung nach Levenberg-Marquart, die die Newton-
Gauß-Methode mit der Gradientenmethode verbindet. Diese Anpassroutine wird in Stutz [1996] ge-
nauer beschrieben.
Durch die mathematische Beschriebung des Anpassprozesses können auch negative Werte auftreten.
Dies geschieht dann, wenn keine detektierbar hohe Konzentration eines Spurenstoffes angepasst wird.
Negative Werte werden daher bei der Auswertung nicht berücksichtigt.
(Gl. 3.11)
(Gl. 3.13)
F ,T = P∑i
ai⋅ ´i ,T
´ =ai
´i
∑ ,T
I0 ,TF ,T2=∑
, TRes ,T = 2 =
! minimal (Gl. 3.12)

30 Beschreibung des DOAS-Geräts
4 Beschreibung des DOAS-Geräts
4.1 Gesamtaufbau
Abbildung 4.1 zeigt einen schematischen Aufbau der aktiven Langpfad–DOAS–Messapparatur. Aus
der Lichtquelle (Hochdruck–Xe–Bogenlampe) wird das Licht über Planspiegel umgelenkt und über
einen Hohlspiegel parallelisiert. Dann wird der Lichtstrahl in Richtung der Retroreflektoren gelenkt,
dort reflektiert und nach abermaligem Durchlaufen der Messstrecke am Eingang der Quarzfaser fo-
kussiert. Von dort gelangt er über den Modenmischer zum Spektrographen, in dem er spektral zerlegt
und mit einer Photodiodenzeile detektiert wird. Diese Daten werden im Analog–digital–Wandler
(ADC) digitalisiert und im PC gespeichert.
Abb. 4.1: schematischer Aufbau der Langpfad–DOAS–Messapparatur.
Quarzfaser mit Modenmischer
Photodiodenzeile Czerny-Turner Spectrograph
Lichtweg: 2 x 14,5 km
20 cm30 cm
paralleler Lichtstrahl
Lampenhaus für Xenon-Kurzbogenlampe
Langpfad koaxiales Newton Teleskop
Schrittmotor für Blende
Schrittmotor für Hg–Emissionslampe
Schrittmotor für 360° horizontale und vertikaleJustage
Haupt-spiegel
Analog–Digital–Wandler
Computer
DOASKontroller

4 Beschreibung des DOAS-Geräts 31
4.2 Lichtquelle
Als Lichtquelle dient eine Xe–Kurzbogenlampe vom Typ PLI SX5002.
Das von der Lampe emittierte Spektrum setzt sich aus der Überlagerung der Xe-Emissionslinien und
der thermischen Strahlung des zwischen den Elektroden erzeugten Plasmas zusammen. Da die Be-
triebstemperatur der Lampe sehr hoch ist (~6000 K) erhält man annähernd einen Schwarzen Strahler,
das Spektrum entspricht daher einem Kontinuum. Durch den hohen Druck in der Lampe kommt es zur
Verbreiterung der Xe–Linien. Diese schwankt jedoch mit der Zeit, ebenso wie die Emissionsstärke
der Lampe, die zusätzlich noch davon abhängt, aus welchem Teilbereich des Plasmas das Licht
stammt.
4.3 Optik
Bei dem Langpfad-Teleskop handelt es sich um ein koaxiales Newton–Teleskop, das sowohl Licht
sendet, als auch empfängt. Der Teleskoptubus besteht aus einem rechteckförmigen, länglichen
Stangengehäuse, an dessen einem Ende der Hohlspiegel angebracht ist, der das Licht parallel durch
das andere (offene) Ende des Tubus in Richtung der Retroreflektoren schickt und das reflektierte
Licht auch wieder empfängt. Die Xenonlampe ist seitlich angebracht, befindet sich jedoch im Brenn-
punkt des Hohlspiegels. Ihr Licht wird über einen in der Teleskopachse stehenden elliptischen Plan-
spiegel, den sog. Einkoppelspiegel, in Richtung des Hohlspiegels gelenkt. Dieser schickt den par-
allelen Lichtstrahl in Richtung der Retroreflektoren. Nach Durchlaufen der Lichtstrecke zu und von
den Retroreflektoren wird das Licht über den Hohlspiegel über einen weiteren elliptischen Plan-
spiegel (Auskoppelspiegel) im Eingang der seitlich am Tubus angebrachten Quarzfaser fokussiert.
Abb. 4.2: Strahlengang und Bauform von Retroprismen. [Linos, 2003]

32 Beschreibung des DOAS-Geräts
Da zwischen Einkoppelspiegel und Hohlspiegel der Auskoppelspiegel steht, reduziert sich das auf den
Lichtweg geschickte Licht auf einen Lichtring mit Außendurchmesser von 30 cm und Innendurch-
messer von 20 cm. Nach Durchlauf der 14,5 Kilometer zur Insel beleuchtet der Strahl aufgrund von
Streuung die gesamten Retroreflektoren, die den noch immer noch parallelen Lichtstrahl zurückschi-
cken. Das rückreflektierte Licht beleuchtet den Hohlspiegel ganz, bis auf einen kleinen Kreis von ca.
5 cm Durchmesser in der Mitte, der durch den auf der Teleskopachse angebrachten Einkoppelspiegel
abgeschattet bleibt. Somit wird der äußere Bereich des Hohlspiegels zum Senden, und der Ring mit
Außendurchmesser 20 cm und Innendurchmesser 5 cm zum Empfangen des Lichts genutzt. Abb. 4.3
ist ein Foto des Hohlspiegels während des Messbetriebs. Deutlich zu sehen ist der Ring, über den das
Licht gesendet wird. Der Schatten in der Mitte des Spiegels kommt durch den Auskoppelspiegel zu-
stande. Das von den Retroreflektoren zurückgesendete Licht ist von deutlich geringerer Intensität,
deshalb für das Auge nicht sichtbar, und wird daher blau schraffiert eingezeichnet. In der Mitte bleibt
der durch den Einkoppelspiegel geworfene Schatten.
Der gesamte Teleskoptubus ist auf einem horizontal und vertikal beweglichen Schrittmotor ange-
bracht, damit der Strahl gut auf die Retroreflektoren justiert werden kann.
Um Hintergrundspektren aufzunehmen, wird mit einem Filterrad, das direkt vor dem Lampenausgang
angebracht ist, die Öffnung verdeckt.
Die zur Kanal–Wellenlängen–Zuordnung (Dispersion) benötigten Referenzspektren einer Queck-
silberlampe nimmt man auf, indem man die an einer direkt vor dem Fasereingang an einem Filterrad
angebrachten Quecksilberlampe vor die Faser fährt.
Abb. 4.4: aufgebaute RetroreflektorenAbb. 4.3: Blick auf den Hohlspiegel, blau ein-gezeichnet der zurückkehrende Lichtstrahl

4 Beschreibung des DOAS-Geräts 33
4.4 Quarzfaser–Modenmischer
Nach Abziehen der Spurengasabsorption von den aufgenommenen Spektren bleiben typischerweise
Reststrukturen der Ordnung 1·10-4–2·10-4 (1σ), die vom Rauschen, der Instrumentenstruktur und unbe-
kannten Absorbern erzeugt werden. Wird bei der Langpfadmessung als Detektor eine Photodiodenzei-
le benutzt, so ergeben sich nach Auswertung der Spektren jedoch Residuumstrukturen der Ordnung
2·10-3 (1σ). Der Grund für die Vergrößerung der Strukturen ist, dass die PDZ eine von der Ausleuch-
tungsrichtung abhängige Empfindlichkeit hat, die wiederum durch Staub auf der PDZ, einer mögli-
chen Unregelmäßigkeit der Winkelabhängigkeiten der Dioden und vor allem durch eine unregelmäßig
dicke Schutzschicht zustande kommen kann. Dieser Effekt kann jedoch mit Hilfe des von Stutz und
Platt [1997] entwickelten Quartzfaser–Modenmischer verringert werden, da man damit die PDZ
gleichmäßig ausleuchtet.
Im Modenmischer wird die Quarzfaser zwischen zwei Aluminiumblöcken leicht gequetscht, die mit
Moosgummi ausgelegt sind, um die Faser nicht zu beschädigen. Dann wird sie in Schlaufen gewi-
ckelt, um sie später durch einen Ventilator in Bewegung zu versetzen. Dadurch werden thermische
und mechanische Änderungen der Faser gemittelt. Die Stabilität und Reproduzierbarkeit der
Messungen wird so deutlich gesteigert. Um kein Streulicht in die Faser kommen zu lassen, wird die
gesamte Apparatur des Modenmischers in einem Aluminiumgehäuse untergebracht. Die Faser wird
außerhalb des Gehäuses schwarz ummantelt.
Vorteil des Modenmischers gegenüber anderen Diffusoren wie z.B. Streuscheiben ist, dass kein hoher
Verlust an Lichtintensität auftritt.
Abb. 4.5: Skizze des Modenmischers. [Ackermann, 1997]

34 Beschreibung des DOAS-Geräts
4.5 Spektrograph
Für alle Messungen wurde ein Czerny–Turner–Spektrograph verwendet, der auf dem von Czerny und
Turner [1930] entwickelten Aufbau beruht. Die Quarzfaser übermittelt dabei das Licht vom Teleskop
zum Spektrographen, das Ende der Faser endet im Eingangsspalt, der mit dem Fokus des ersten kon-
vexen Spiegels übereinstimmt. Das Licht wird über einen ebenen Planspiegel auf diesen konvexen
Spiegel gelenkt, der davon ausgehende parallele Lichtstrahl läuft zu einem ebenen Reflektionsgitter,
an dem er spektral zerlegt wird und über einen weiteren konvexen Spiegel, der den Strahl wieder fo-
kussiert, direkt auf den Detektor.
Um thermisch bedingte Schwankungen minimal zu halten wird der Spektrograph auf eine konstante
Temperatur oberhalb der Umgebungstemperatur thermostatisiert und außen isoliert. Auf der Kampa-
gne wurde eine Spektrographentemperatur von 37°C gewählt. Zusätzliches Problem beim Spektro-
graphen ist dessen Streulichtverhalten. Im Spektrograph sowie an der Photodiodenzeile und der
Quarzscheibe des Detektorgehäuses wird Licht gestreut bzw. reflektiert. Stutz [1996] gibt an, dass das
Streulichtverhalten im Spektrograph mit 3% den größten systematischen Fehler des Geräts darstellt.
Tabelle 2.3: Eigenschaft des Spektrographs
Eigenschaften des verwendeten Spektrographen ACTON Spectra Pro 500:
Öffnung des Eingangsspalts 200 µm
Brennweite f 500 mmÖffnungsverhältnis f/6,9 (Spiegeldurchmesser durch Brennweite)Gitter 600 Striche pro mmDispersion 0,078 nm pro Kanal
4.6 Photodiodenzeile
Der Detektor besteht aus einer Photodiodenzeile (PDZ) des Typs Hamamatsu S5931–1024S. Die
lichtempfindliche Fläche beträgt 25,6mm × 2,5mm, bestehend aus 1024 Silizium–Halbleiterdioden
der Größe 25µm × 2,5mm und befindet sich in der Brennebene des Spektrographen.
Zu Beginn eines Auslesevorgangs wird an den Dioden in Sperrrichtung eine Spannung von 2,06 V
angelegt und die Sperrschichtkapazität wird mit Elektronen aufgeladen. Dann wird die Spannungs-
quelle abgetrennt und einfallende Photonen heben in der Verarmungszone Elektronen ins Leitungs-
band des Halbleiters, wodurch die Kapazität entladen wird. Diese Entladung verläuft proportional zur
aufgenommenen Lichtintensität und wird zusätzlich durch den Dunkelstrom verstärkt. Der Dunkel-
strom ist dabei ein thermischer Rekombinationsstrom in der Sperrschicht der Dioden, der zusätzlich
zur Temperatur auch noch vom Ladungszustand der Photodiodenzeile abhängig ist [Stutz, 1996]. Das

4 Beschreibung des DOAS-Geräts 35
Maß der gesamten Entladung entspricht der Ladungsmenge, die zur Wiederherstellung der Vor-
spannung benötigt wird. Anschließend wird die Photodiodenzeile ausgelesen, das elektronische Signal
wird verstärkt und es wird ein Offsetsignal addiert. Letzteres dient dazu, bei schwacher Ausleuchtung
negative Signale zu vermeiden. Ansonsten läge die volle Aussteuerung, die mit Offset bei 216 = 65536
bit pro Auslesevorgang liegt, aufgrund der zusätzlichen negativen Werte nur bei 32768 bit pro Aus-
lesevorgang. Über den 16–bit Analog–digital–Wandler wird das Signal digitalisiert und über den Con-
troller an den PC übertragen.
Die PDZ wird mit Hilfe eines Peltierelements auf konstante -20°C gekühlt um das Signal des Dunkel-
stroms, das man auch ohne Lichteinfall durch thermisches Rauschen erhält, möglichst gering zu hal-
ten. Das Detektorgehäuse und die Kühlung sind vakuumdicht und mit Argon gefüllt bzw. werden
ständig mit einer Pumpe auf Vakuum gehalten. Dadurch wird vermieden, dass Wasser auf der PDZ
gefriert, was zum Fabry–Perot–Étalon-Effekt, der Interferenz mehrfach reflektierter Lichtstrahlen,
führen würde. Dieser kann auch durch die SiO2–Oberflächenschicht der PDZ auftreten, wird jedoch
mit Hilfe des Modenmischers vermindert [Stutz und Platt, 1997] .
4.7 Kurzschlusssystem
Lampenreferenzspektren sind Spektren der Lichtquelle, die noch keine Absorption durch Moleküle in
der Luft aufweisen. Vergleicht man sie mit den Messspektren, so erkennt man, bei welchen Wellen-
längen Licht absorbiert wurde. Die Internsitäten des Lampenreferenzspektrums entsprechen der
ursprünglichen Intensität I0 in Gleichung 3.3. Zur Aufnahme dieser Spektren stellt man eine Dreh-
scheibe, auf der drei Retroreflektoren angebracht sind, direkt vor das offene Ende des Teleskoptubus.
Die auf dieser kurzen Lichtstrecke auftretende Absorption ist dabei vernachlässigbar gering.
Die Xenonlampe sendet kein gleichmäßiges Licht aus. Um dies auszugleichen wird versucht, bei der
Aufnahme des Lampenreferenzspektrums über möglichst alle Bereiche der Lampe zu mitteln. Hierzu
sind drei Retroreflektoren versetzt in den angestrahlten Lichtring eingebaut. Über den parallelen Ver-
satz der Retroreflektoren (vgl. Abb. 4.2) von bis zu 6 cm wird das Licht aus dem äußeren Lichtring in
einen inneren Lichtring gespiegelt und gelangt über Hohlspiegel und Auskoppelspiegel in die Faser.
Indem sich die Scheibe über einen Motor drehen lässt, schöpft man nahezu den gesamten Lichtring
aus.

36 Beschreibung des DOAS-Geräts
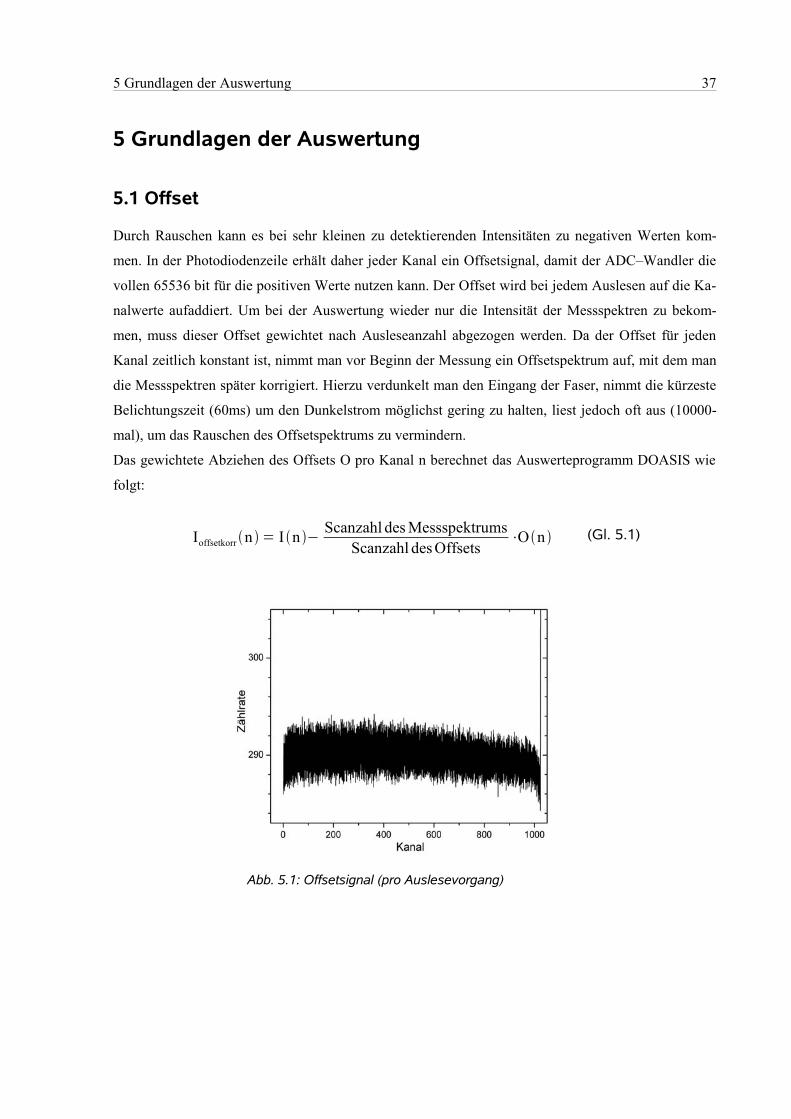
5 Grundlagen der Auswertung 37
5 Grundlagen der Auswertung
5.1 Offset
Durch Rauschen kann es bei sehr kleinen zu detektierenden Intensitäten zu negativen Werten kom-
men. In der Photodiodenzeile erhält daher jeder Kanal ein Offsetsignal, damit der ADC–Wandler die
vollen 65536 bit für die positiven Werte nutzen kann. Der Offset wird bei jedem Auslesen auf die Ka-
nalwerte aufaddiert. Um bei der Auswertung wieder nur die Intensität der Messspektren zu bekom-
men, muss dieser Offset gewichtet nach Ausleseanzahl abgezogen werden. Da der Offset für jeden
Kanal zeitlich konstant ist, nimmt man vor Beginn der Messung ein Offsetspektrum auf, mit dem man
die Messspektren später korrigiert. Hierzu verdunkelt man den Eingang der Faser, nimmt die kürzeste
Belichtungszeit (60ms) um den Dunkelstrom möglichst gering zu halten, liest jedoch oft aus (10000-
mal), um das Rauschen des Offsetspektrums zu vermindern.
Das gewichtete Abziehen des Offsets O pro Kanal n berechnet das Auswerteprogramm DOASIS wie
folgt:
(Gl. 5.1)Ioffsetkorr n = I n Scanzahl des MessspektrumsScanzahl des Offsets
⋅On
Abb. 5.1: Offsetsignal (pro Auslesevorgang)
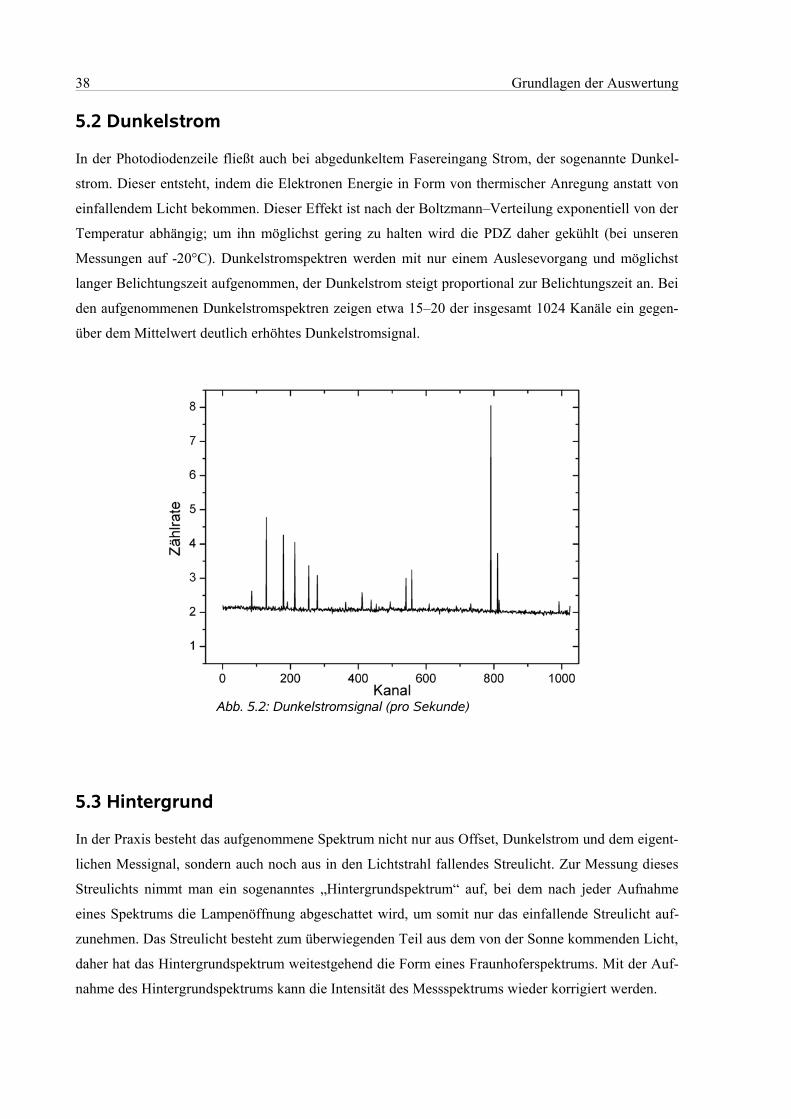
38 Grundlagen der Auswertung
5.2 Dunkelstrom
In der Photodiodenzeile fließt auch bei abgedunkeltem Fasereingang Strom, der sogenannte Dunkel-
strom. Dieser entsteht, indem die Elektronen Energie in Form von thermischer Anregung anstatt von
einfallendem Licht bekommen. Dieser Effekt ist nach der Boltzmann–Verteilung exponentiell von der
Temperatur abhängig; um ihn möglichst gering zu halten wird die PDZ daher gekühlt (bei unseren
Messungen auf -20°C). Dunkelstromspektren werden mit nur einem Auslesevorgang und möglichst
langer Belichtungszeit aufgenommen, der Dunkelstrom steigt proportional zur Belichtungszeit an. Bei
den aufgenommenen Dunkelstromspektren zeigen etwa 15–20 der insgesamt 1024 Kanäle ein gegen-
über dem Mittelwert deutlich erhöhtes Dunkelstromsignal.
5.3 Hintergrund
In der Praxis besteht das aufgenommene Spektrum nicht nur aus Offset, Dunkelstrom und dem eigent-
lichen Messignal, sondern auch noch aus in den Lichtstrahl fallendes Streulicht. Zur Messung dieses
Streulichts nimmt man ein sogenanntes „Hintergrundspektrum“ auf, bei dem nach jeder Aufnahme
eines Spektrums die Lampenöffnung abgeschattet wird, um somit nur das einfallende Streulicht auf-
zunehmen. Das Streulicht besteht zum überwiegenden Teil aus dem von der Sonne kommenden Licht,
daher hat das Hintergrundspektrum weitestgehend die Form eines Fraunhoferspektrums. Mit der Auf-
nahme des Hintergrundspektrums kann die Intensität des Messspektrums wieder korrigiert werden.
Abb. 5.2: Dunkelstromsignal (pro Sekunde)
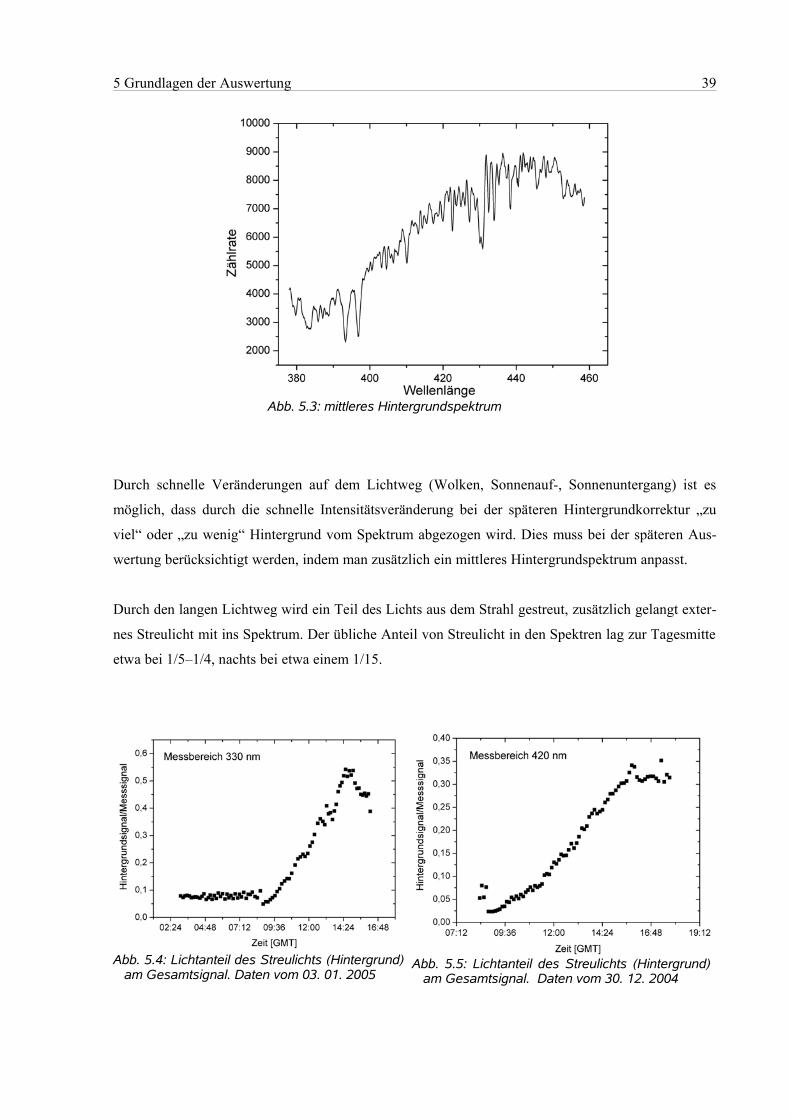
5 Grundlagen der Auswertung 39
Durch schnelle Veränderungen auf dem Lichtweg (Wolken, Sonnenauf-, Sonnenuntergang) ist es
möglich, dass durch die schnelle Intensitätsveränderung bei der späteren Hintergrundkorrektur „zu
viel“ oder „zu wenig“ Hintergrund vom Spektrum abgezogen wird. Dies muss bei der späteren Aus-
wertung berücksichtigt werden, indem man zusätzlich ein mittleres Hintergrundspektrum anpasst.
Durch den langen Lichtweg wird ein Teil des Lichts aus dem Strahl gestreut, zusätzlich gelangt exter-
nes Streulicht mit ins Spektrum. Der übliche Anteil von Streulicht in den Spektren lag zur Tagesmitte
etwa bei 1/5–1/4, nachts bei etwa einem 1/15.
Abb. 5.3: mittleres Hintergrundspektrum
Abb. 5.4: Lichtanteil des Streulichts (Hintergrund)am Gesamtsignal. Daten vom 03. 01. 2005
Abb. 5.5: Lichtanteil des Streulichts (Hintergrund)am Gesamtsignal. Daten vom 30. 12. 2004

40 Grundlagen der Auswertung
5.4 Kanal–Wellenlängen–Zuordnung
Die Photodiodenzeile nimmt nur die Zählrate pro Kanal auf, wobei die Zählrate proportional der
Lichtintensität ist. Man möchte die aufgenommenen Intensitäten in Abhängigkeit der jeweiligen
Wellenlänge und nicht die des jeweiligen Kanals bestimmen.
Nach jedem Messspektrum wird ein Referenzspektrum aufgenommen, wobei eine Quecksilberlampe
mit schmaler Linienbreite als Lichtquelle direkt vor den Fasereingang gefahren wird. Mit den Hg–
Linien und den aus der Literatur bekannten zugeordneten Wellenlängen wird den Kanälen die ent-
sprechende Wellenlänge zugeordnet. Da der Absorptionswirkungsquerschnitt temperatur– und luft-
druckabhängig ist, variiert die Zuordnung mit der Zeit. Um diese Schwankung auszugleichen und alle
Messspektren über den selben Wellenlängenbereich zu erhalten, wird jedes Messspektrum mit Hilfe
seines jeweiligen Referenzspektrums auf diesen so geeichten Wellenlängenbereich verschoben.
5.5 Korrektur eines Messspektrums
Das Rohspektrum A wird zunächst offsetkorrigiert, ebenso das Hintergrundspektrum (BG). Einer
Dunkelstromkorrektur (DC) bedarf es nicht, da sich dieser bei der Korrektur des Streulichts aufgrund
seiner Proportionalität zur Gesamtbelichtungszeit (= scans · etime) selbst korrigiert.
Der Faktor n beachtet dabei die verschiedene Anzahl wie oft ein Spektrum ausgelesen wird („scans“)
und die Belichtungszeit etime von A und BG. Er berechnet sich wie folgt:
Nach der Verschiebung der Messspektren anhand des zugehörigen Referenzspektrums auf den ge-
meinsamen Wellenlängenbereich werden so viele aufeinanderfolgende Spektren addiert, bis die Ge-
samtscanzahl 30 überschreitet. Durch das Aufaddieren verringert sich zwar die Anzahl der Spektren,
auf diese Weise kann jedoch das Rauschen vermindert werden, da dieses statistischen Schwankungen
unterliegt.
(Gl. 5.2)
(Gl. 5.3)
AoffsetkorrDCAn⋅BGoffsetkorrDCBG
n =scansA⋅etimeA
scansBG⋅etimeBG=
scansDCA⋅etimeDCA
scansDCBG⋅etimeDCBG
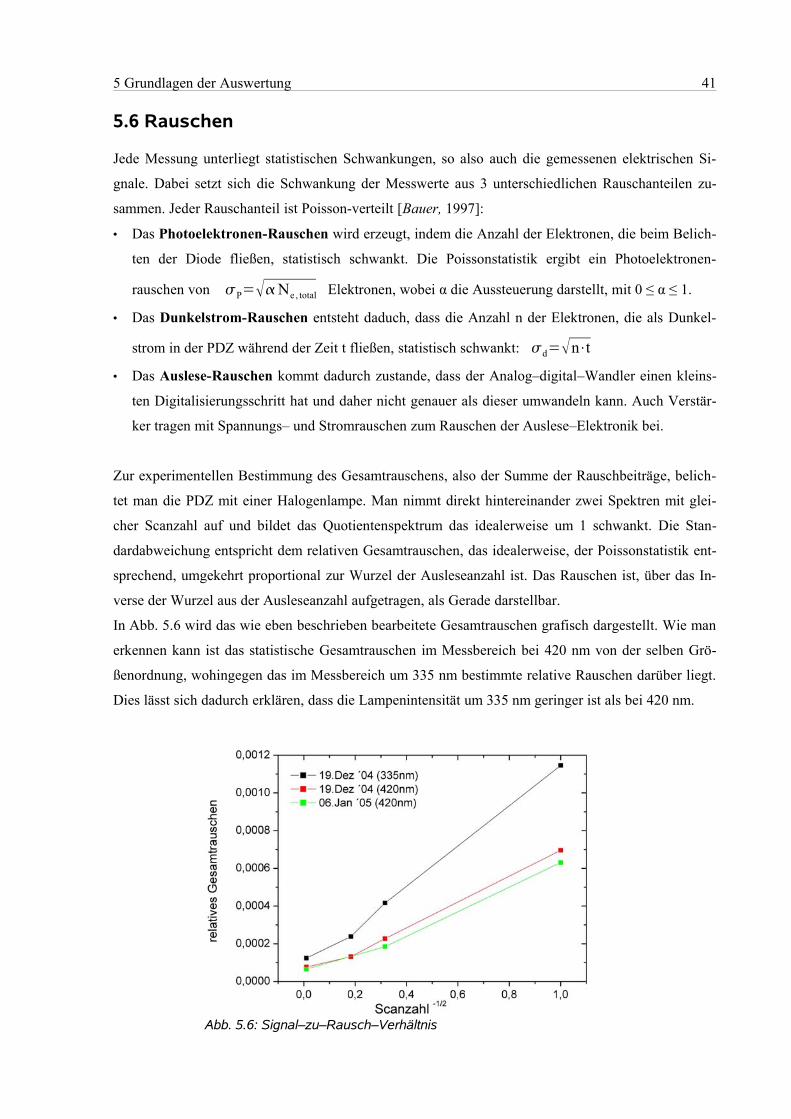
5 Grundlagen der Auswertung 41
5.6 Rauschen
Jede Messung unterliegt statistischen Schwankungen, so also auch die gemessenen elektrischen Si-
gnale. Dabei setzt sich die Schwankung der Messwerte aus 3 unterschiedlichen Rauschanteilen zu-
sammen. Jeder Rauschanteil ist Poisson-verteilt [Bauer, 1997]:
• Das Photoelektronen-Rauschen wird erzeugt, indem die Anzahl der Elektronen, die beim Belich-
ten der Diode fließen, statistisch schwankt. Die Poissonstatistik ergibt ein Photoelektronen-
rauschen von P= Ne, total Elektronen, wobei α die Aussteuerung darstellt, mit 0 ≤ α ≤ 1.
• Das Dunkelstrom-Rauschen entsteht daduch, dass die Anzahl n der Elektronen, die als Dunkel-
strom in der PDZ während der Zeit t fließen, statistisch schwankt: d=n⋅t
• Das Auslese-Rauschen kommt dadurch zustande, dass der Analog–digital–Wandler einen kleins-
ten Digitalisierungsschritt hat und daher nicht genauer als dieser umwandeln kann. Auch Verstär-
ker tragen mit Spannungs– und Stromrauschen zum Rauschen der Auslese–Elektronik bei.
Zur experimentellen Bestimmung des Gesamtrauschens, also der Summe der Rauschbeiträge, belich-
tet man die PDZ mit einer Halogenlampe. Man nimmt direkt hintereinander zwei Spektren mit glei-
cher Scanzahl auf und bildet das Quotientenspektrum das idealerweise um 1 schwankt. Die Stan-
dardabweichung entspricht dem relativen Gesamtrauschen, das idealerweise, der Poissonstatistik ent-
sprechend, umgekehrt proportional zur Wurzel der Ausleseanzahl ist. Das Rauschen ist, über das In-
verse der Wurzel aus der Ausleseanzahl aufgetragen, als Gerade darstellbar.
In Abb. 5.6 wird das wie eben beschrieben bearbeitete Gesamtrauschen grafisch dargestellt. Wie man
erkennen kann ist das statistische Gesamtrauschen im Messbereich bei 420 nm von der selben Grö-
ßenordnung, wohingegen das im Messbereich um 335 nm bestimmte relative Rauschen darüber liegt.
Dies lässt sich dadurch erklären, dass die Lampenintensität um 335 nm geringer ist als bei 420 nm.
Abb. 5.6: Signal–zu–Rausch–Verhältnis
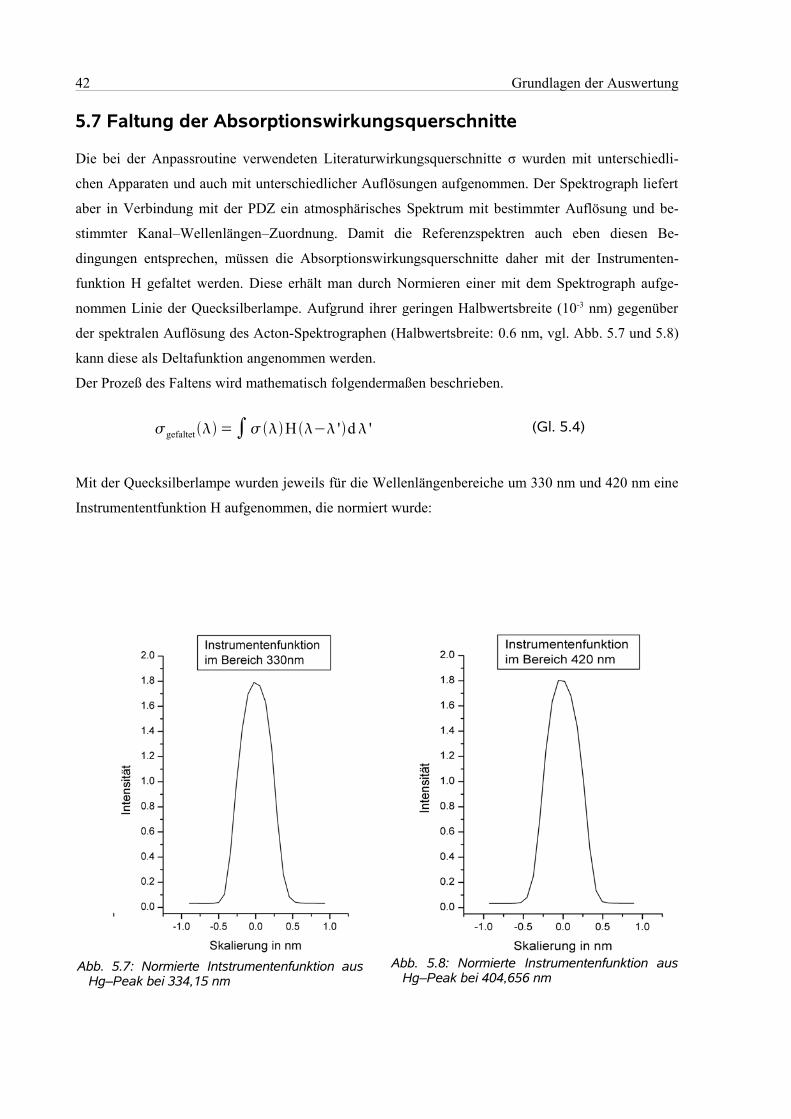
42 Grundlagen der Auswertung
5.7 Faltung der Absorptionswirkungsquerschnitte
Die bei der Anpassroutine verwendeten Literaturwirkungsquerschnitte σ wurden mit unterschiedli-
chen Apparaten und auch mit unterschiedlicher Auflösungen aufgenommen. Der Spektrograph liefert
aber in Verbindung mit der PDZ ein atmosphärisches Spektrum mit bestimmter Auflösung und be-
stimmter Kanal–Wellenlängen–Zuordnung. Damit die Referenzspektren auch eben diesen Be-
dingungen entsprechen, müssen die Absorptionswirkungsquerschnitte daher mit der Instrumenten-
funktion H gefaltet werden. Diese erhält man durch Normieren einer mit dem Spektrograph aufge-
nommen Linie der Quecksilberlampe. Aufgrund ihrer geringen Halbwertsbreite (10-3 nm) gegenüber
der spektralen Auflösung des Acton-Spektrographen (Halbwertsbreite: 0.6 nm, vgl. Abb. 5.7 und 5.8)
kann diese als Deltafunktion angenommen werden.
Der Prozeß des Faltens wird mathematisch folgendermaßen beschrieben.
Mit der Quecksilberlampe wurden jeweils für die Wellenlängenbereiche um 330 nm und 420 nm eine
Instrumententfunktion H aufgenommen, die normiert wurde:
(Gl. 5.4)gefaltet =∫H 'd '
Abb. 5.8: Normierte Instrumentenfunktion ausHg–Peak bei 404,656 nm
Abb. 5.7: Normierte Intstrumentenfunktion ausHg–Peak bei 334,15 nm

5 Grundlagen der Auswertung 43
5.8 Berechnung des Mischungsverhältnisses
Die Konzentration der Luftmoleküle Nluft (=Teilchenzahl pro Volumeneinheit) berechnet sich nach der
idealen Gasgleichung, die Konzentration ist abhängig vom Luftdruck p und der Temperatur T. Für
mittlere, auf der Kampagne ermittelte Werte der Temperatur und des Drucks ergibt sich eine Kon-
zentration der Luftmoleküle von 2,4·1019 Teilchen pro cm3:
5.9 Fehlerbestimmung
Sowohl durch die Messapparatur als auch bei der Auswertung ergeben sich Fehler für die Messergeb-
nisse. Im Folgenden wird zwischen systematischen und statistischen Fehlern unterschieden:
Systematische Fehler:
• Die Literaturwirkungsquerschnitte sind alle mit einem Fehler behaftet, der je nach Spurenstoff
etwa zwischen 1% und 15% liegt. Wasserwirkungsquerschnitte sind aufgrund der vielen, und im
Auswertebereich auch kleinen Absorptionslinien meist mit einem sehr großen Fehler behaftet.
• Durch im Spektrograph gestreutes oder an der PDZ reflektiertes Licht erhält man einen systema-
tischen Fehler, der von Stutz [1996] mit 3% angegeben wird und auch als größter systematischer
Fehler des Geräts abgeschätzt wurde.
• Zusätzlich kommt noch ein Fehler bei der Bestimmung des Lichtwegs hinzu. Die Standorte von
Teleskop und Retroreflektoren wurden mit GPS bestimmt, dabei ergibt sich auf der Lichtstrecke
von 2×14,5 km mit einer Ungenauigkeit von jeweils ±20m ein relativer Fehler von etwa 1o/oo und
ist somit gegenüber den anderen Fehlern vernachlässigbar.
Statistische Fehler:
Die Anpassungsroutine von WinDOAS gibt einen statistischen Fehler aus, der mit 3 multipliziert etwa
der 1–σ Standardabweichung entspricht. Der Faktor 3 wurde von Stutz und Platt [1996] vorge-
schlagen, da der von WinDOAS ausgegebene statistische Fehler nicht ganz dem realen statistischen
Fehler entspricht. Bei troposphärischen Messungen kann jedoch aufgrund von unbekannten Absor-
bern auf dem Lichtweg ein noch größerer Fehler angenommen werden.
Die Nachweisgrenze wird aus dem Residuum bestimmt. Es wird davon ausgegangen, dass Ab-
sorptionsstrukturen einer optischen Dichte, die der halben Peak–to–Peak–Entfernung entsprechen als
Absorption erkannt werden können. Diese optische Dichte rechnet man in das entsprechende Mi-
schungsverhältnis um.
NLuft T, p = N /V =kB⋅T
p= 1005 mbar
1,38⋅1023 JK
⋅ 303,14 K= 2,4⋅1019 cm3
(Gl. 5.5)

44 Kampagne
6 Kampagne
Die Messkampagne deren Daten dieser Arbeit zugrunde liegen fand vom 23.11.04 – 20.01.05 in Al-
cântara/Brasilien (2.39° Süd, 44.38° West) statt, wobei sich die Messungen aufgrund technischer Pro-
bleme auf den Zeitraum vom 22.12.04 – 15.01.05 beschränkten.
Ziel der Kampagne war es nachzuprüfen, ob vor Ort IO und BrO nachweisbar sind.
Zur Messung wurde ein Langpfad-DOAS eingesetzt, der gesamte Lichtweg, zu den Retroreflektoren
und wieder zurück, ging insgesamt über eine Länge von 29 km. Die Retroreflektoren wurden auf einer
Insel aufgestellt, so dass der Lichtweg größtenteils über Wasser führte.
IO konnte in geringem Maße mit einem Mischungsverhältnis von etwa 0,5 ppt nachgewiesen werden,
BrO konnte nicht oberhalb der Nachweisgrenze detektiert werden. Bevor auf die Auswertung der
Spektren eingegangen wird, werden zunächst die äußeren Bedingungen beschrieben.
6.1 Geographie des Messorts
Der Messort bei Alcântara befindet sich im Norden Brasiliens im Bundesstaat Maranhão an einer
Bucht des Atlantischen Ozean, bei 2,39°S und 44,38°W geographischer Lage, die Ortszeit war zur
Zeit der Kampagne drei Stunden gegenüber der GMT verschoben. Dem Ort gegenüber liegt die größe-
re Stadt São Luis.
Abb. 6.1: Der Nordosten Brasiliens

6 Kampagne 45
Um in diesem abgelegenen Gebiet eine einigermaßen stabile Stromversorgung vorzufinden, fand die
Kampagne auf dem Gelände der CLA (Centro do Lancamento de Alcântara) statt. Allerdings kam es
auch hier des öfteren zu Stromausfällen, welche der auf vier Autobatterien beschränkten Notstromver-
sorgung Probleme bereiteten.
Die Messgeräte (Teleskoptubus, Spektrographen, Mess–PC und Notstromversorgung) wurden unter
dem Vordach eines Wohnhauses aufgestellt. Der Teleskoptubus war auf die kurz vor dem Fähren-
hafen von Sao Luis gelegenen Insel Ilha do medo ausgerichtet. Auf dieser wurden die Retroreflekto-
ren aufgebaut.
Die Messstrecke betrug einfach 14,5 km, wovon nur wenige 100 m über Land verliefen. Höhenunter-
schied zwischen Land und Meeresspiegel lag bei etwa 10 m, die Retroreflektoren waren etwa 5 m
über dem Meeresspiegel angebracht. Der Tidenhub lag im Mittel bei etwa 3,30 m. Im Bereich des Ti-
denhubs gab es keine nennenswerten Algenvorkommen. Es kann daher eine starke biologische Aktivi-
tät, wie sie auf anderen Messkampagnen in Mace Head/Irland [Carpenter et al., 1999] oder in der
Bretagne [Peters, 2005] vorgefunden wurde, ausgeschlossen werden.
Die gegenüberliegende Stadt São Luis hat etwa 870.000 Einwohner, Hauptwirtschaftszweige sind
Handel, Industrie, Fischerei und Tourismus. Es gibt einen großen Hafen, dessen Hauptausfuhrgüter
Soja und Eisenerz sind. Um den Hafen zu erreichen, müssen die Frachter die Bucht und somit auch
Abb. 6.2: Messort: Standort der Messgeräte (schwarzer Pfeil), Lichtweg (gelbgepunktet), Schiffsroute (blau gepunktet)
10 km
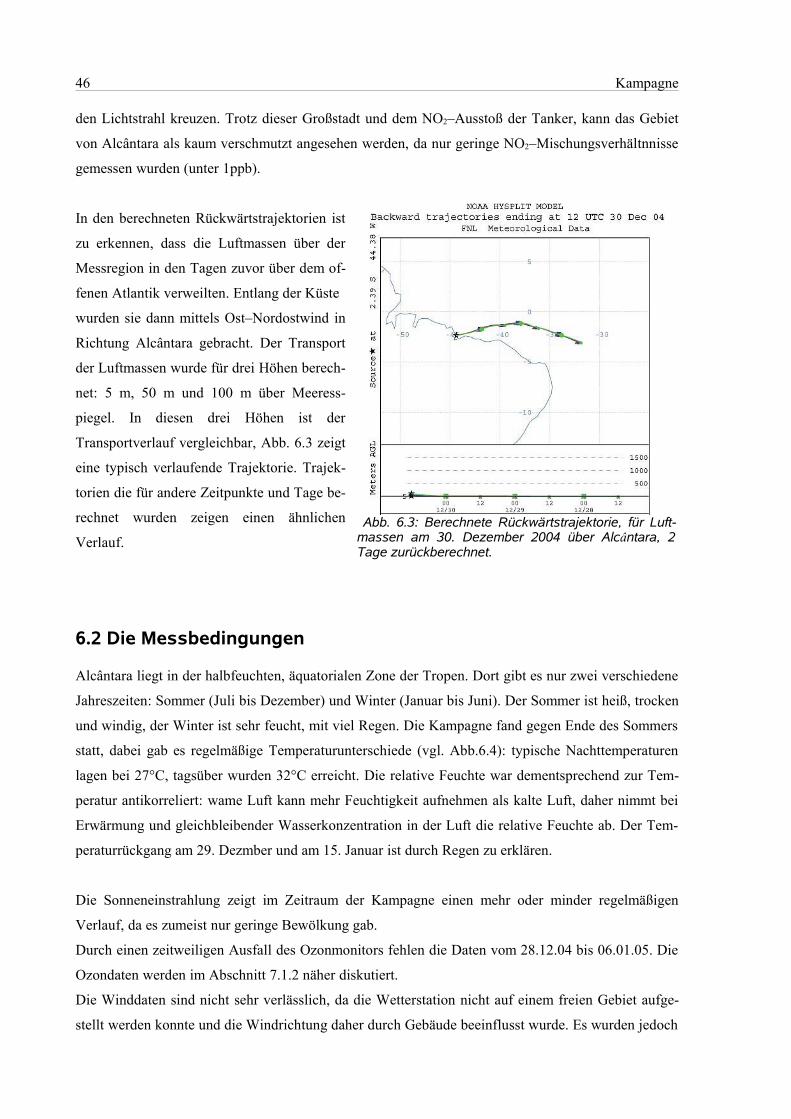
46 Kampagne
den Lichtstrahl kreuzen. Trotz dieser Großstadt und dem NO2–Ausstoß der Tanker, kann das Gebiet
von Alcântara als kaum verschmutzt angesehen werden, da nur geringe NO2–Mischungsverhältnnisse
gemessen wurden (unter 1ppb).
In den berechneten Rückwärtstrajektorien ist
zu erkennen, dass die Luftmassen über der
Messregion in den Tagen zuvor über dem of-
fenen Atlantik verweilten. Entlang der Küste
wurden sie dann mittels Ost–Nordostwind in
Richtung Alcântara gebracht. Der Transport
der Luftmassen wurde für drei Höhen berech-
net: 5 m, 50 m und 100 m über Meeress-
piegel. In diesen drei Höhen ist der
Transportverlauf vergleichbar, Abb. 6.3 zeigt
eine typisch verlaufende Trajektorie. Trajek-
torien die für andere Zeitpunkte und Tage be-
rechnet wurden zeigen einen ähnlichen
Verlauf.
6.2 Die Messbedingungen
Alcântara liegt in der halbfeuchten, äquatorialen Zone der Tropen. Dort gibt es nur zwei verschiedene
Jahreszeiten: Sommer (Juli bis Dezember) und Winter (Januar bis Juni). Der Sommer ist heiß, trocken
und windig, der Winter ist sehr feucht, mit viel Regen. Die Kampagne fand gegen Ende des Sommers
statt, dabei gab es regelmäßige Temperaturunterschiede (vgl. Abb.6.4): typische Nachttemperaturen
lagen bei 27°C, tagsüber wurden 32°C erreicht. Die relative Feuchte war dementsprechend zur Tem-
peratur antikorreliert: wame Luft kann mehr Feuchtigkeit aufnehmen als kalte Luft, daher nimmt bei
Erwärmung und gleichbleibender Wasserkonzentration in der Luft die relative Feuchte ab. Der Tem-
peraturrückgang am 29. Dezmber und am 15. Januar ist durch Regen zu erklären.
Die Sonneneinstrahlung zeigt im Zeitraum der Kampagne einen mehr oder minder regelmäßigen
Verlauf, da es zumeist nur geringe Bewölkung gab.
Durch einen zeitweiligen Ausfall des Ozonmonitors fehlen die Daten vom 28.12.04 bis 06.01.05. Die
Ozondaten werden im Abschnitt 7.1.2 näher diskutiert.
Die Winddaten sind nicht sehr verlässlich, da die Wetterstation nicht auf einem freien Gebiet aufge-
stellt werden konnte und die Windrichtung daher durch Gebäude beeinflusst wurde. Es wurden jedoch
Abb. 6.3: Berechnete Rückwärtstrajektorie, für Luft-massen am 30. Dezember 2004 über Alcântara, 2Tage zurückberechnet.
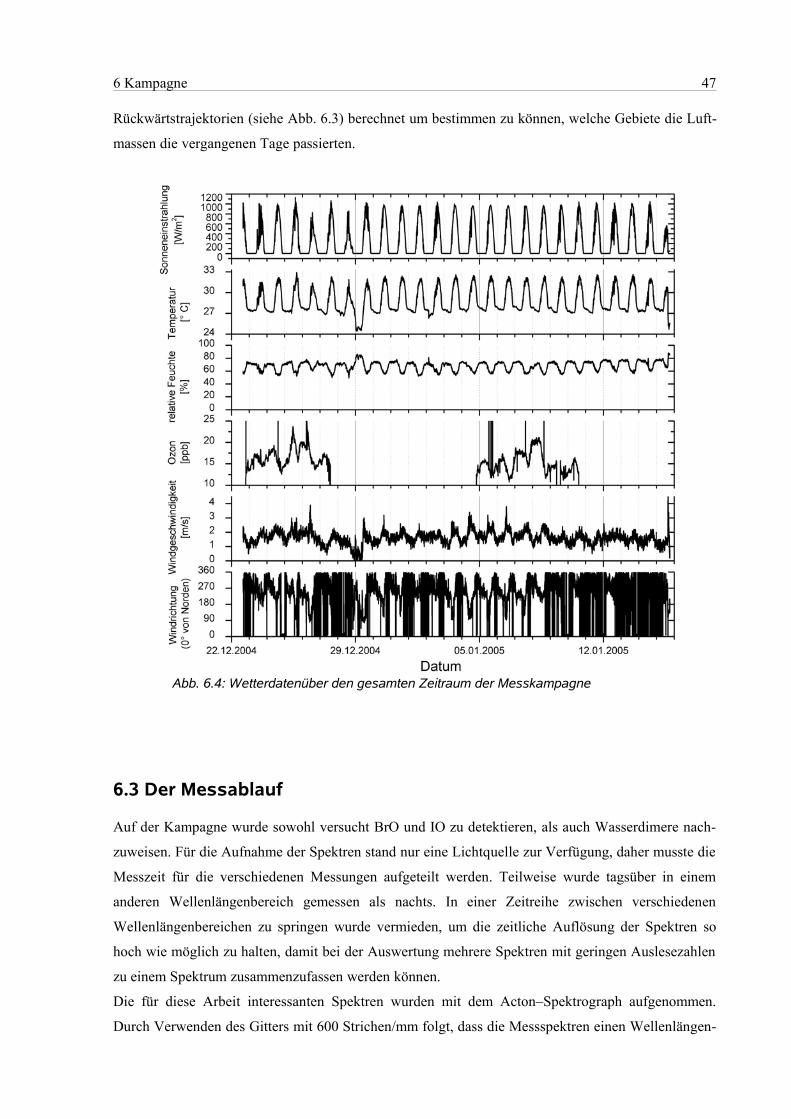
6 Kampagne 47
Rückwärtstrajektorien (siehe Abb. 6.3) berechnet um bestimmen zu können, welche Gebiete die Luft-
massen die vergangenen Tage passierten.
6.3 Der Messablauf
Auf der Kampagne wurde sowohl versucht BrO und IO zu detektieren, als auch Wasserdimere nach-
zuweisen. Für die Aufnahme der Spektren stand nur eine Lichtquelle zur Verfügung, daher musste die
Messzeit für die verschiedenen Messungen aufgeteilt werden. Teilweise wurde tagsüber in einem
anderen Wellenlängenbereich gemessen als nachts. In einer Zeitreihe zwischen verschiedenen
Wellenlängenbereichen zu springen wurde vermieden, um die zeitliche Auflösung der Spektren so
hoch wie möglich zu halten, damit bei der Auswertung mehrere Spektren mit geringen Auslesezahlen
zu einem Spektrum zusammenzufassen werden können.
Die für diese Arbeit interessanten Spektren wurden mit dem Acton–Spektrograph aufgenommen.
Durch Verwenden des Gitters mit 600 Strichen/mm folgt, dass die Messspektren einen Wellenlängen-
Abb. 6.4: Wetterdatenüber den gesamten Zeitraum der Messkampagne

48 Kampagne
bereich von etwa 80 nm abdecken. Um BrO zu detektieren wurde die mittlere Wellenlänge auf 330
nm eingestellt, für die Bestimmung von IO auf 420 nm.
Die Spektren wurden über das Programm DOASIS (DOAS Intelligent System) mit einer Messroutine
automatisiert aufgenommen:
Diese wurde nur für Lampenwechsel, zum Nachjustieren der Optik und zur Aufnahme der Lampen-
spektren mittels Kurzschlusssystem unterbrochen, bzw. zur Wahl anderer Messbereiche angehalten
und geändert.
Der Ablauf des Messroutine soll hier skizziert werden:
Die Belichtungszeit („etime“) eines Messspektrums wird bestimmt, indem ein Probespektrum aufge-
nommen, und dessen Belichtungszeit mit dem Faktor 0.8 oder 1.2 multipliziert wird, je nachdem ob
die ideale Sättigung der Kanäle von 48 000 counts über– oder unterschritten worden ist. Dieser
Optimierungsvorgang wird unterbrochen, sobald mit dem jeweils anderen Faktor mulitpliziert werden
müsste. Die minimale Belichtungszeit von 60 ms und die maximale Belichtungszeit von 180 000 ms
dürfen dabei nicht unter– bzw. überschritten werden.
Die Gesamtbelichtungszeit eines Messspektrums wird auf drei Minuten festgesetzt, da angenommen
wird, dass innerhalb von 3 min die Sichtbedingungen konstant sind. Nach diesen drei Minuten wird,
ausgehend von der vorherigen etime, die Belichtungszeit für das nächste Spektrum neu bestimmt. Ist
die etime festgelegt, so wird berechnet, wie oft für ein Messspektrum ausgelesen wird. Die Auslese-
zahl (Scanzahl) ist der Quotient aus Gesamtbelichtungszeit und etime.
Die Hintergrundspektren werden mit kürzerer Belichtungszeit (0.25 bzw. 0.5 · etime, bei Dunkelheit
nur 0.1 · etime) und nur der Hälfte der Scanzahl des Messspektrums aufgenommen.
Beim Referenzspektrum der Quecksilberlampe beträgt die Belichtungszeit 2 000 ms und es wird zwei
Mal ausgelesen.
Abb. 6.5: Ablauf der Messroutine
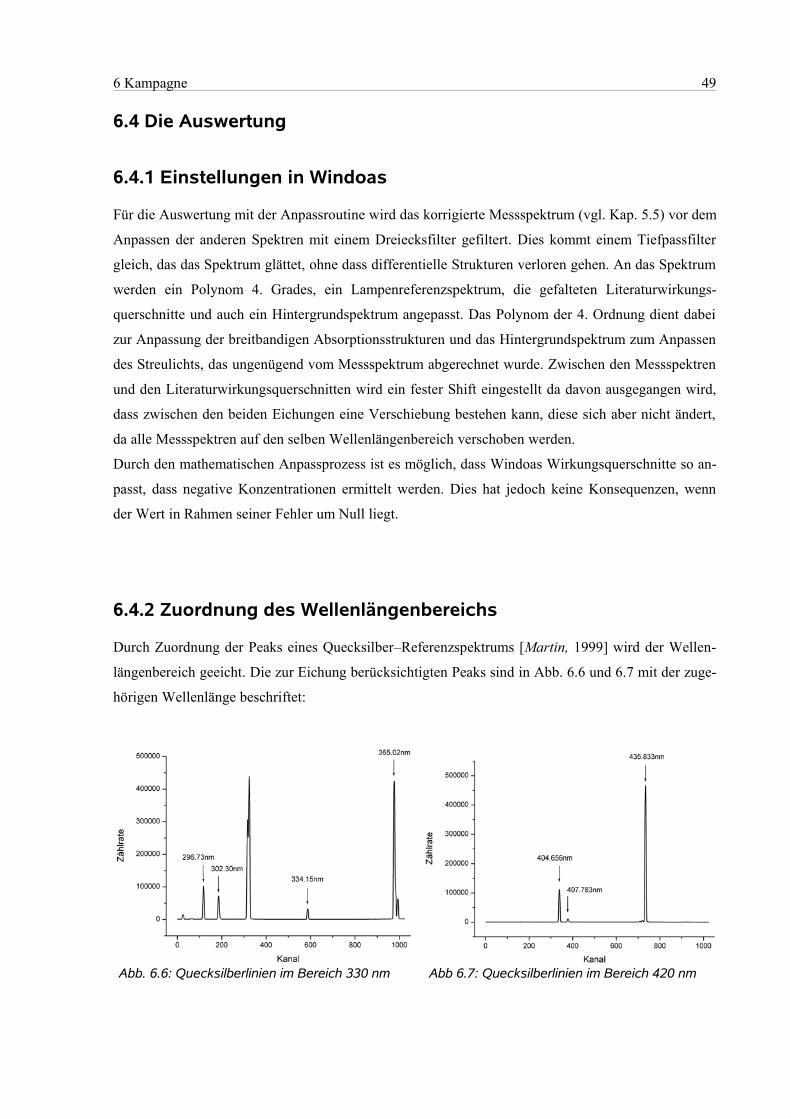
6 Kampagne 49
6.4 Die Auswertung
6.4.1 Einstellungen in Windoas
Für die Auswertung mit der Anpassroutine wird das korrigierte Messspektrum (vgl. Kap. 5.5) vor dem
Anpassen der anderen Spektren mit einem Dreiecksfilter gefiltert. Dies kommt einem Tiefpassfilter
gleich, das das Spektrum glättet, ohne dass differentielle Strukturen verloren gehen. An das Spektrum
werden ein Polynom 4. Grades, ein Lampenreferenzspektrum, die gefalteten Literaturwirkungs-
querschnitte und auch ein Hintergrundspektrum angepasst. Das Polynom der 4. Ordnung dient dabei
zur Anpassung der breitbandigen Absorptionsstrukturen und das Hintergrundspektrum zum Anpassen
des Streulichts, das ungenügend vom Messspektrum abgerechnet wurde. Zwischen den Messspektren
und den Literaturwirkungsquerschnitten wird ein fester Shift eingestellt da davon ausgegangen wird,
dass zwischen den beiden Eichungen eine Verschiebung bestehen kann, diese sich aber nicht ändert,
da alle Messspektren auf den selben Wellenlängenbereich verschoben werden.
Durch den mathematischen Anpassprozess ist es möglich, dass Windoas Wirkungsquerschnitte so an-
passt, dass negative Konzentrationen ermittelt werden. Dies hat jedoch keine Konsequenzen, wenn
der Wert in Rahmen seiner Fehler um Null liegt.
6.4.2 Zuordnung des Wellenlängenbereichs
Durch Zuordnung der Peaks eines Quecksilber–Referenzspektrums [Martin, 1999] wird der Wellen-
längenbereich geeicht. Die zur Eichung berücksichtigten Peaks sind in Abb. 6.6 und 6.7 mit der zuge-
hörigen Wellenlänge beschriftet:
Abb. 6.6: Quecksilberlinien im Bereich 330 nm Abb 6.7: Quecksilberlinien im Bereich 420 nm
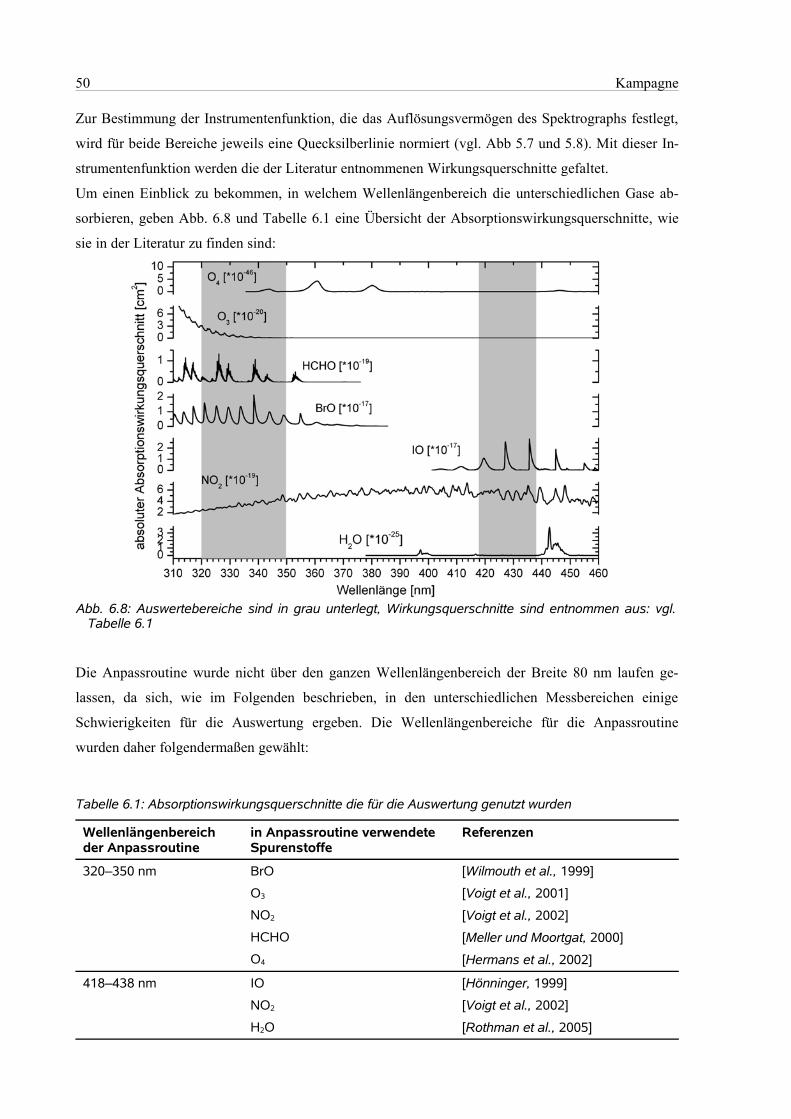
50 Kampagne
Zur Bestimmung der Instrumentenfunktion, die das Auflösungsvermögen des Spektrographs festlegt,
wird für beide Bereiche jeweils eine Quecksilberlinie normiert (vgl. Abb 5.7 und 5.8). Mit dieser In-
strumentenfunktion werden die der Literatur entnommenen Wirkungsquerschnitte gefaltet.
Um einen Einblick zu bekommen, in welchem Wellenlängenbereich die unterschiedlichen Gase ab-
sorbieren, geben Abb. 6.8 und Tabelle 6.1 eine Übersicht der Absorptionswirkungsquerschnitte, wie
sie in der Literatur zu finden sind:
Die Anpassroutine wurde nicht über den ganzen Wellenlängenbereich der Breite 80 nm laufen ge-
lassen, da sich, wie im Folgenden beschrieben, in den unterschiedlichen Messbereichen einige
Schwierigkeiten für die Auswertung ergeben. Die Wellenlängenbereiche für die Anpassroutine
wurden daher folgendermaßen gewählt:
Tabelle 6.1: Absorptionswirkungsquerschnitte die für die Auswertung genutzt wurden
Wellenlängenbereichder Anpassroutine
320–350 nm
in Anpassroutine verwendeteSpurenstoffe
BrOO3
NO2
HCHOO4
Referenzen
[Wilmouth et al., 1999][Voigt et al., 2001][Voigt et al., 2002][Meller und Moortgat, 2000][Hermans et al., 2002]
418–438 nm IONO2
H2O
[Hönninger, 1999][Voigt et al., 2002][Rothman et al., 2005]
Abb. 6.8: Auswertebereiche sind in grau unterlegt, Wirkungsquerschnitte sind entnommen aus: vgl.Tabelle 6.1
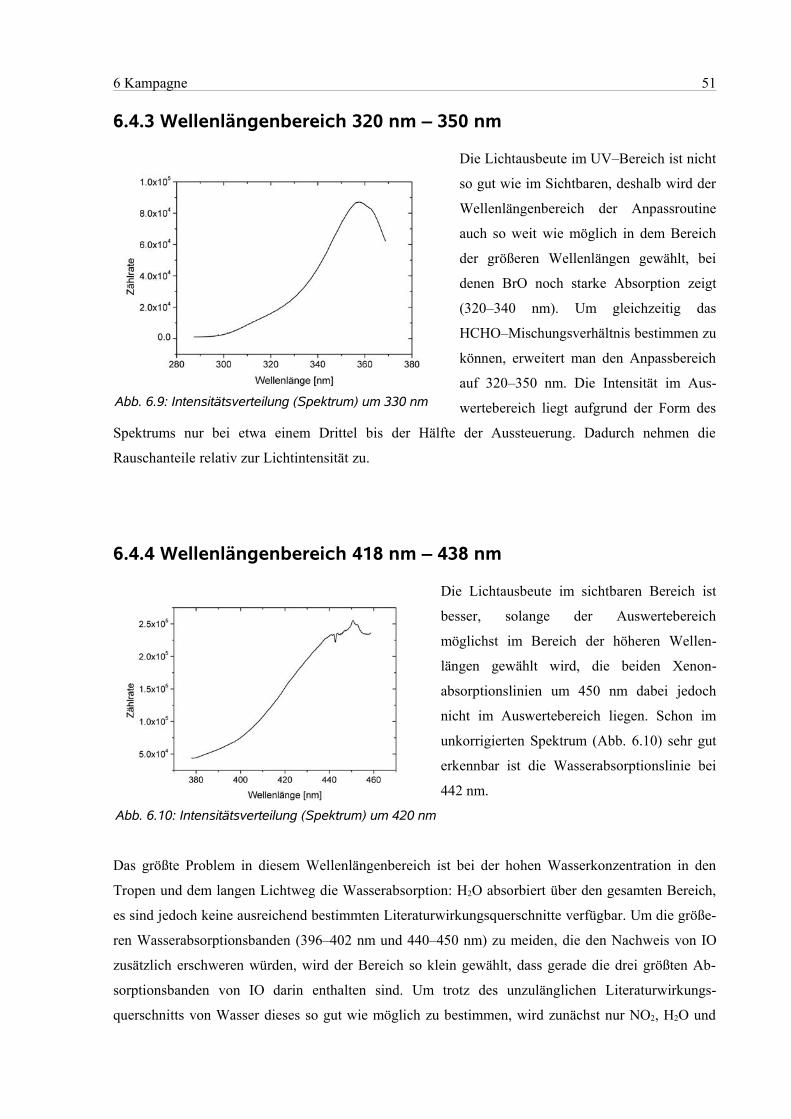
6 Kampagne 51
6.4.3 Wellenlängenbereich 320 nm – 350 nm
Die Lichtausbeute im UV–Bereich ist nicht
so gut wie im Sichtbaren, deshalb wird der
Wellenlängenbereich der Anpassroutine
auch so weit wie möglich in dem Bereich
der größeren Wellenlängen gewählt, bei
denen BrO noch starke Absorption zeigt
(320–340 nm). Um gleichzeitig das
HCHO–Mischungsverhältnis bestimmen zu
können, erweitert man den Anpassbereich
auf 320–350 nm. Die Intensität im Aus-
wertebereich liegt aufgrund der Form des
Spektrums nur bei etwa einem Drittel bis der Hälfte der Aussteuerung. Dadurch nehmen die
Rauschanteile relativ zur Lichtintensität zu.
6.4.4 Wellenlängenbereich 418 nm – 438 nm
Die Lichtausbeute im sichtbaren Bereich ist
besser, solange der Auswertebereich
möglichst im Bereich der höheren Wellen-
längen gewählt wird, die beiden Xenon-
absorptionslinien um 450 nm dabei jedoch
nicht im Auswertebereich liegen. Schon im
unkorrigierten Spektrum (Abb. 6.10) sehr gut
erkennbar ist die Wasserabsorptionslinie bei
442 nm.
Das größte Problem in diesem Wellenlängenbereich ist bei der hohen Wasserkonzentration in den
Tropen und dem langen Lichtweg die Wasserabsorption: H2O absorbiert über den gesamten Bereich,
es sind jedoch keine ausreichend bestimmten Literaturwirkungsquerschnitte verfügbar. Um die größe-
ren Wasserabsorptionsbanden (396–402 nm und 440–450 nm) zu meiden, die den Nachweis von IO
zusätzlich erschweren würden, wird der Bereich so klein gewählt, dass gerade die drei größten Ab-
sorptionsbanden von IO darin enthalten sind. Um trotz des unzulänglichen Literaturwirkungs-
querschnitts von Wasser dieses so gut wie möglich zu bestimmen, wird zunächst nur NO2, H2O und
Abb. 6.10: Intensitätsverteilung (Spektrum) um 420 nm
Abb. 6.9: Intensitätsverteilung (Spektrum) um 330 nm
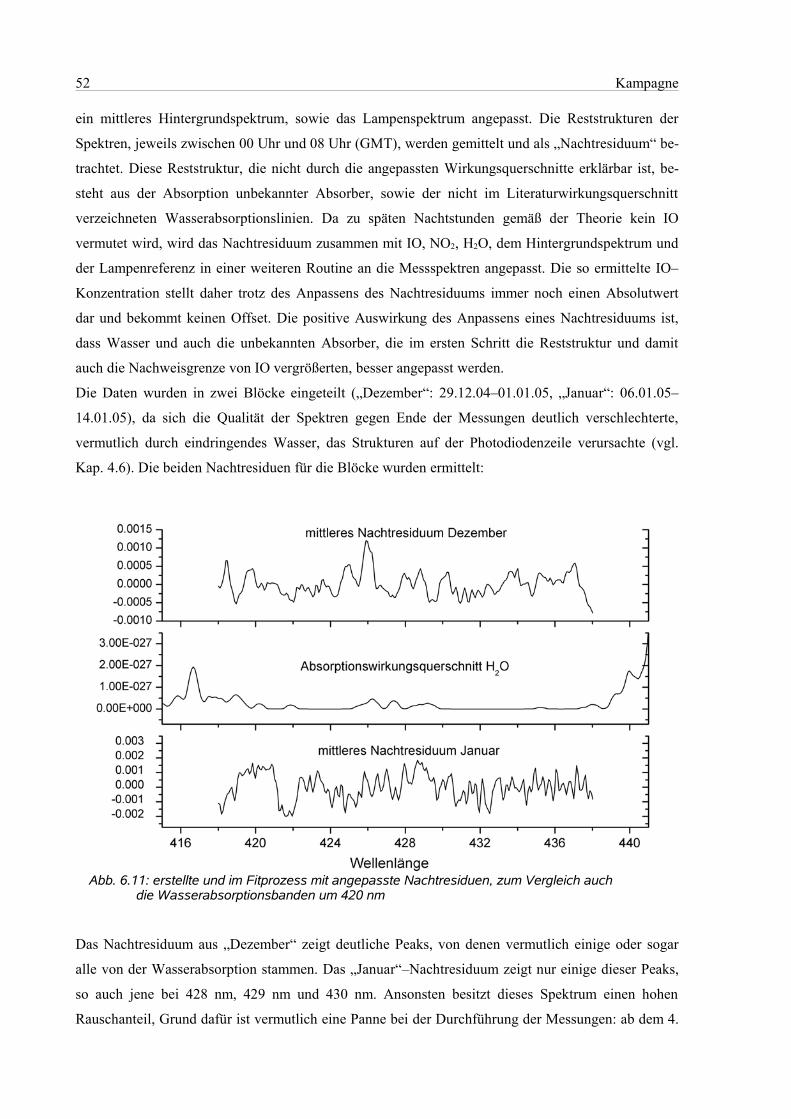
52 Kampagne
ein mittleres Hintergrundspektrum, sowie das Lampenspektrum angepasst. Die Reststrukturen der
Spektren, jeweils zwischen 00 Uhr und 08 Uhr (GMT), werden gemittelt und als „Nachtresiduum“ be-
trachtet. Diese Reststruktur, die nicht durch die angepassten Wirkungsquerschnitte erklärbar ist, be-
steht aus der Absorption unbekannter Absorber, sowie der nicht im Literaturwirkungsquerschnitt
verzeichneten Wasserabsorptionslinien. Da zu späten Nachtstunden gemäß der Theorie kein IO
vermutet wird, wird das Nachtresiduum zusammen mit IO, NO2, H2O, dem Hintergrundspektrum und
der Lampenreferenz in einer weiteren Routine an die Messspektren angepasst. Die so ermittelte IO–
Konzentration stellt daher trotz des Anpassens des Nachtresiduums immer noch einen Absolutwert
dar und bekommt keinen Offset. Die positive Auswirkung des Anpassens eines Nachtresiduums ist,
dass Wasser und auch die unbekannten Absorber, die im ersten Schritt die Reststruktur und damit
auch die Nachweisgrenze von IO vergrößerten, besser angepasst werden.
Die Daten wurden in zwei Blöcke eingeteilt („Dezember“: 29.12.04–01.01.05, „Januar“: 06.01.05–
14.01.05), da sich die Qualität der Spektren gegen Ende der Messungen deutlich verschlechterte,
vermutlich durch eindringendes Wasser, das Strukturen auf der Photodiodenzeile verursachte (vgl.
Kap. 4.6). Die beiden Nachtresiduen für die Blöcke wurden ermittelt:
Das Nachtresiduum aus „Dezember“ zeigt deutliche Peaks, von denen vermutlich einige oder sogar
alle von der Wasserabsorption stammen. Das „Januar“–Nachtresiduum zeigt nur einige dieser Peaks,
so auch jene bei 428 nm, 429 nm und 430 nm. Ansonsten besitzt dieses Spektrum einen hohen
Rauschanteil, Grund dafür ist vermutlich eine Panne bei der Durchführung der Messungen: ab dem 4.
Abb. 6.11: erstellte und im Fitprozess mit angepasste Nachtresiduen, zum Vergleich auch die Wasserabsorptionsbanden um 420 nm
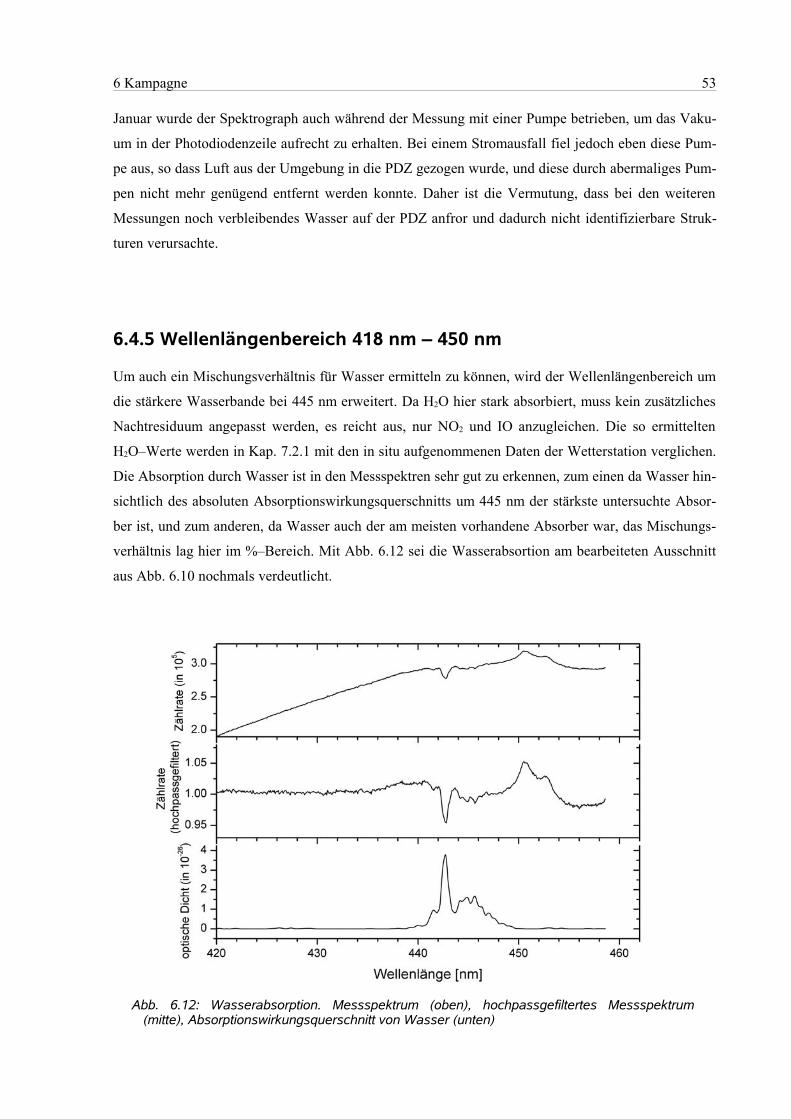
6 Kampagne 53
Januar wurde der Spektrograph auch während der Messung mit einer Pumpe betrieben, um das Vaku-
um in der Photodiodenzeile aufrecht zu erhalten. Bei einem Stromausfall fiel jedoch eben diese Pum-
pe aus, so dass Luft aus der Umgebung in die PDZ gezogen wurde, und diese durch abermaliges Pum-
pen nicht mehr genügend entfernt werden konnte. Daher ist die Vermutung, dass bei den weiteren
Messungen noch verbleibendes Wasser auf der PDZ anfror und dadurch nicht identifizierbare Struk-
turen verursachte.
6.4.5 Wellenlängenbereich 418 nm – 450 nm
Um auch ein Mischungsverhältnis für Wasser ermitteln zu können, wird der Wellenlängenbereich um
die stärkere Wasserbande bei 445 nm erweitert. Da H2O hier stark absorbiert, muss kein zusätzliches
Nachtresiduum angepasst werden, es reicht aus, nur NO2 und IO anzugleichen. Die so ermittelten
H2O–Werte werden in Kap. 7.2.1 mit den in situ aufgenommenen Daten der Wetterstation verglichen.
Die Absorption durch Wasser ist in den Messspektren sehr gut zu erkennen, zum einen da Wasser hin-
sichtlich des absoluten Absorptionswirkungsquerschnitts um 445 nm der stärkste untersuchte Absor-
ber ist, und zum anderen, da Wasser auch der am meisten vorhandene Absorber war, das Mischungs-
verhältnis lag hier im %–Bereich. Mit Abb. 6.12 sei die Wasserabsortion am bearbeiteten Ausschnitt
aus Abb. 6.10 nochmals verdeutlicht.
Abb. 6.12: Wasserabsorption. Messspektrum (oben), hochpassgefiltertes Messspektrum(mitte), Absorptionswirkungsquerschnitt von Wasser (unten)
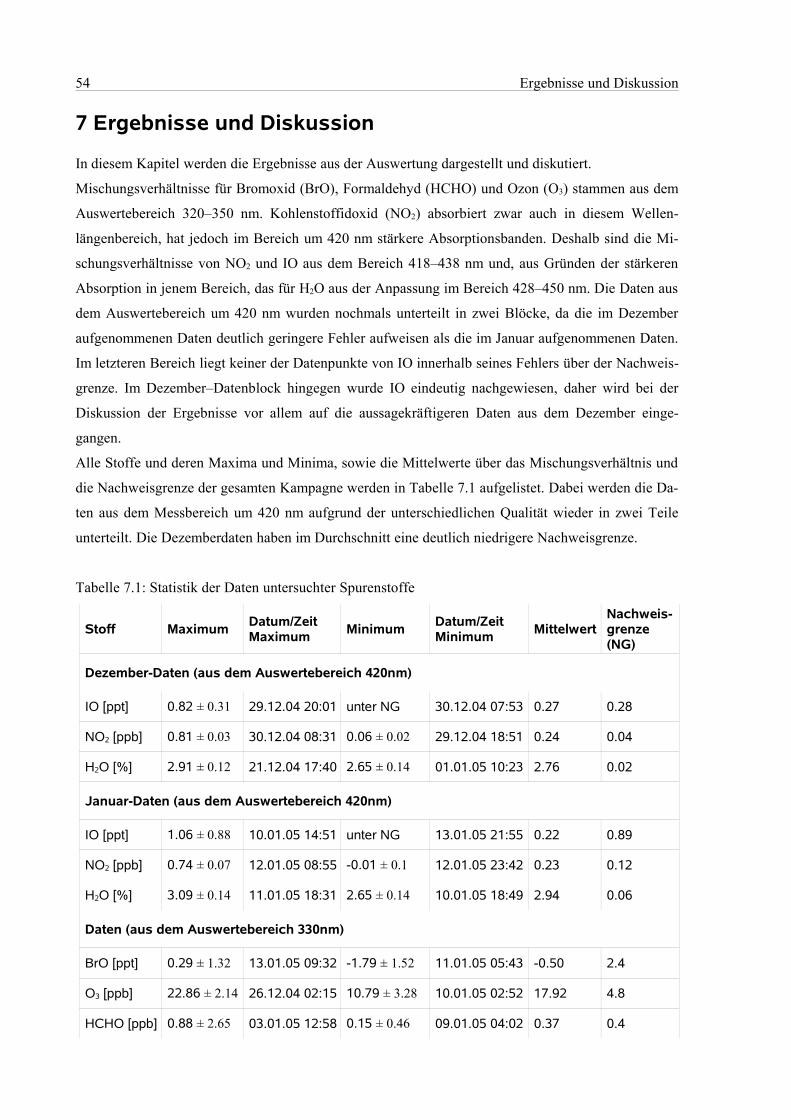
54 Ergebnisse und Diskussion
7 Ergebnisse und Diskussion
In diesem Kapitel werden die Ergebnisse aus der Auswertung dargestellt und diskutiert.
Mischungsverhältnisse für Bromoxid (BrO), Formaldehyd (HCHO) und Ozon (O3) stammen aus dem
Auswertebereich 320–350 nm. Kohlenstoffidoxid (NO2) absorbiert zwar auch in diesem Wellen-
längenbereich, hat jedoch im Bereich um 420 nm stärkere Absorptionsbanden. Deshalb sind die Mi-
schungsverhältnisse von NO2 und IO aus dem Bereich 418–438 nm und, aus Gründen der stärkeren
Absorption in jenem Bereich, das für H2O aus der Anpassung im Bereich 428–450 nm. Die Daten aus
dem Auswertebereich um 420 nm wurden nochmals unterteilt in zwei Blöcke, da die im Dezember
aufgenommenen Daten deutlich geringere Fehler aufweisen als die im Januar aufgenommenen Daten.
Im letzteren Bereich liegt keiner der Datenpunkte von IO innerhalb seines Fehlers über der Nachweis-
grenze. Im Dezember–Datenblock hingegen wurde IO eindeutig nachgewiesen, daher wird bei der
Diskussion der Ergebnisse vor allem auf die aussagekräftigeren Daten aus dem Dezember einge-
gangen.
Alle Stoffe und deren Maxima und Minima, sowie die Mittelwerte über das Mischungsverhältnis und
die Nachweisgrenze der gesamten Kampagne werden in Tabelle 7.1 aufgelistet. Dabei werden die Da-
ten aus dem Messbereich um 420 nm aufgrund der unterschiedlichen Qualität wieder in zwei Teile
unterteilt. Die Dezemberdaten haben im Durchschnitt eine deutlich niedrigere Nachweisgrenze.
Tabelle 7.1: Statistik der Daten untersuchter Spurenstoffe
Stoff Maximum Datum/ZeitMaximum Minimum Datum/Zeit
Minimum MittelwertNachweis-grenze(NG)
Dezember-Daten (aus dem Auswertebereich 420nm)
IO [ppt] 0.82 ± 0.31 29.12.04 20:01 unter NG 30.12.04 07:53 0.27 0.28
NO2 [ppb] 0.81 ± 0.03 30.12.04 08:31 0.06 ± 0.02 29.12.04 18:51 0.24 0.04
H2O [%] 2.91 ± 0.12 21.12.04 17:40 2.65 ± 0.14 01.01.05 10:23 2.76 0.02
Januar-Daten (aus dem Auswertebereich 420nm)
IO [ppt] 1.06 ± 0.88 10.01.05 14:51 unter NG 13.01.05 21:55 0.22 0.89
NO2 [ppb] 0.74 ± 0.07 12.01.05 08:55 -0.01 ± 0.1 12.01.05 23:42 0.23 0.12
H2O [%] 3.09 ± 0.14 11.01.05 18:31 2.65 ± 0.14 10.01.05 18:49 2.94 0.06
Daten (aus dem Auswertebereich 330nm)
BrO [ppt] 0.29 ± 1.32 13.01.05 09:32 -1.79 ± 1.52 11.01.05 05:43 -0.50 2.4
O3 [ppb] 22.86 ± 2.14 26.12.04 02:15 10.79 ± 3.28 10.01.05 02:52 17.92 4.8
HCHO [ppb] 0.88 ± 2.65 03.01.05 12:58 0.15 ± 0.46 09.01.05 04:02 0.37 0.4
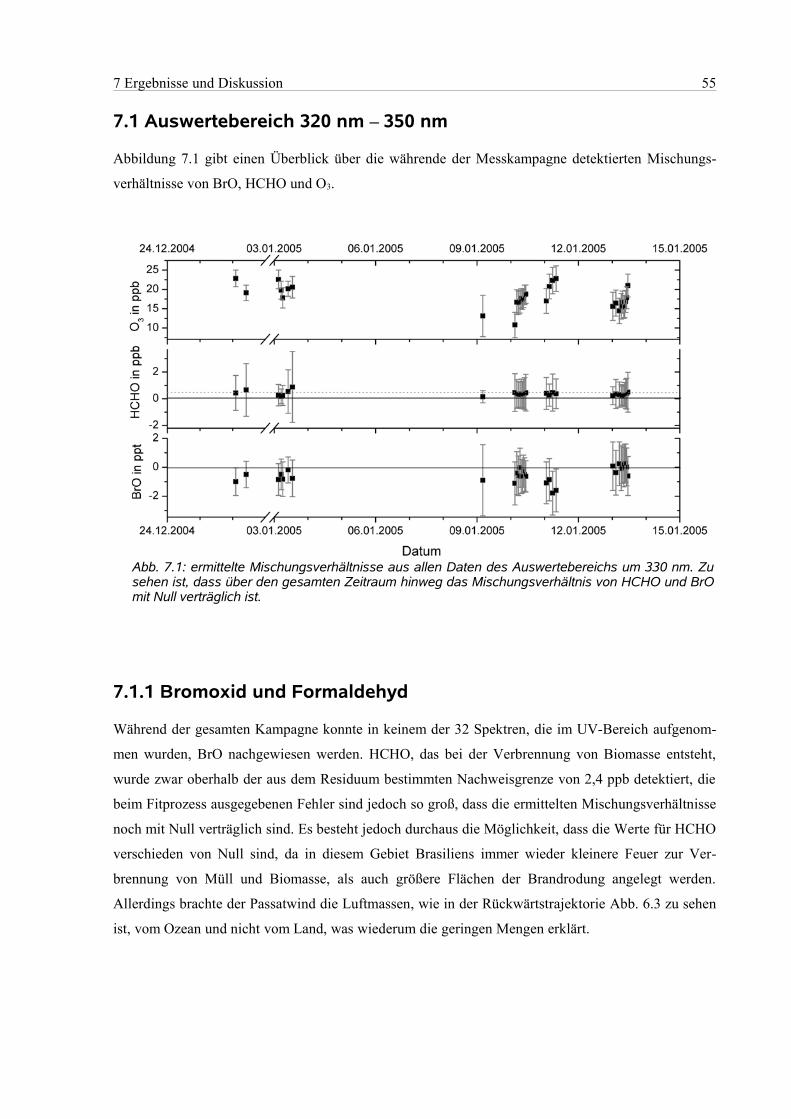
7 Ergebnisse und Diskussion 55
7.1 Auswertebereich 320 nm – 350 nm
Abbildung 7.1 gibt einen Überblick über die währende der Messkampagne detektierten Mischungs-
verhältnisse von BrO, HCHO und O3.
7.1.1 Bromoxid und Formaldehyd
Während der gesamten Kampagne konnte in keinem der 32 Spektren, die im UV-Bereich aufgenom-
men wurden, BrO nachgewiesen werden. HCHO, das bei der Verbrennung von Biomasse entsteht,
wurde zwar oberhalb der aus dem Residuum bestimmten Nachweisgrenze von 2,4 ppb detektiert, die
beim Fitprozess ausgegebenen Fehler sind jedoch so groß, dass die ermittelten Mischungsverhältnisse
noch mit Null verträglich sind. Es besteht jedoch durchaus die Möglichkeit, dass die Werte für HCHO
verschieden von Null sind, da in diesem Gebiet Brasiliens immer wieder kleinere Feuer zur Ver-
brennung von Müll und Biomasse, als auch größere Flächen der Brandrodung angelegt werden.
Allerdings brachte der Passatwind die Luftmassen, wie in der Rückwärtstrajektorie Abb. 6.3 zu sehen
ist, vom Ozean und nicht vom Land, was wiederum die geringen Mengen erklärt.
Abb. 7.1: ermittelte Mischungsverhältnisse aus allen Daten des Auswertebereichs um 330 nm. Zusehen ist, dass über den gesamten Zeitraum hinweg das Mischungsverhältnis von HCHO und BrOmit Null verträglich ist.
56 Ergebnisse und Diskussion
Tabelle 7.2: von WinDOAS ermittelte Mischungsverhältnisse des Spektrums X28
Spurenstoff Optische Dichte MischungsverhältnisO3 (1.07±0.07)·1018 (15.4±2.95) ppbNO2 (2.76±0.54)·1016 (0.40±0.23) ppbHCHO (1.76±0.71)·1016 (0.25±0.76) ppbBrO (2.14±0.03)·1011 (0.00±1.35) ppt
Abb: 7.2: Windoas–Auswertung eines typischen Spektrums (X28) im Auswertebereich zwischen320 und 350 nm. In schwarz sind die differentiellen Strukturen des Messspektrums, und in rotdie von WinDOAS angepasste optische Dichte eingezeichnet. Das Spektrum X28 setzt sich aus5 aufaddierten Einzelspektren zusammen, die am 13.01.05 zwischen 7 und 8 Uhr (GMT) aufge-nommen wurden.
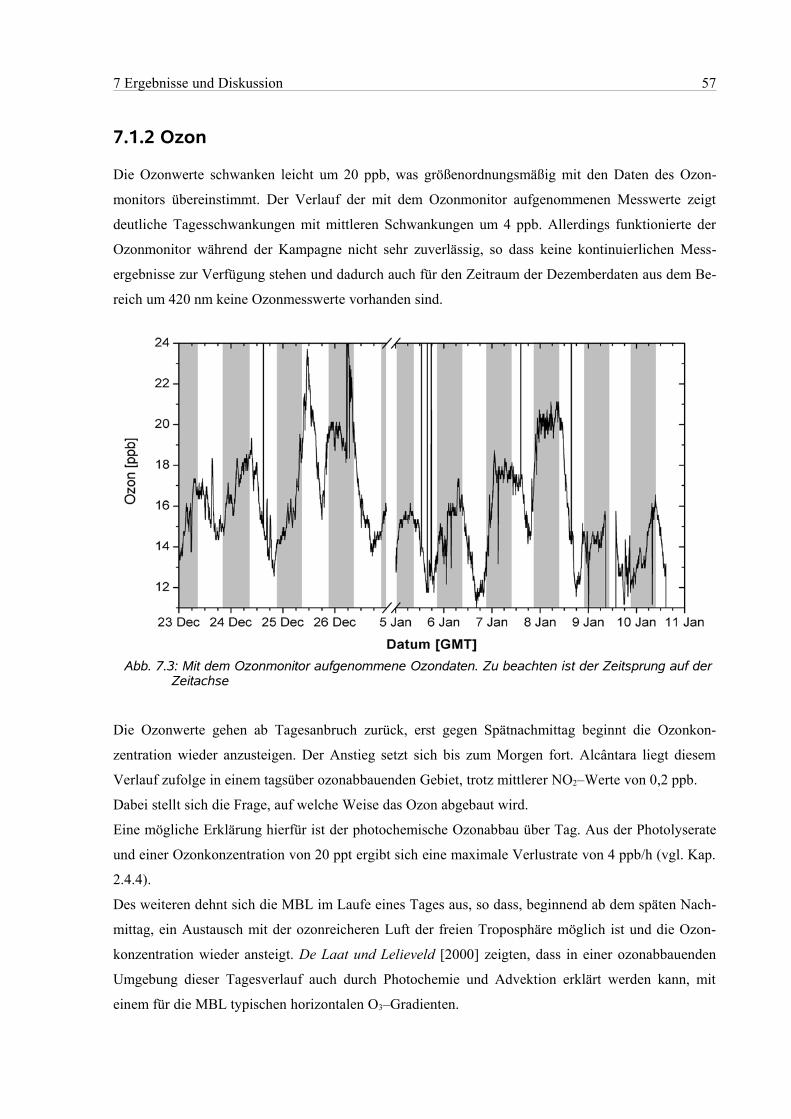
7 Ergebnisse und Diskussion 57
7.1.2 Ozon
Die Ozonwerte schwanken leicht um 20 ppb, was größenordnungsmäßig mit den Daten des Ozon-
monitors übereinstimmt. Der Verlauf der mit dem Ozonmonitor aufgenommenen Messwerte zeigt
deutliche Tagesschwankungen mit mittleren Schwankungen um 4 ppb. Allerdings funktionierte der
Ozonmonitor während der Kampagne nicht sehr zuverlässig, so dass keine kontinuierlichen Mess-
ergebnisse zur Verfügung stehen und dadurch auch für den Zeitraum der Dezemberdaten aus dem Be-
reich um 420 nm keine Ozonmesswerte vorhanden sind.
Die Ozonwerte gehen ab Tagesanbruch zurück, erst gegen Spätnachmittag beginnt die Ozonkon-
zentration wieder anzusteigen. Der Anstieg setzt sich bis zum Morgen fort. Alcântara liegt diesem
Verlauf zufolge in einem tagsüber ozonabbauenden Gebiet, trotz mittlerer NO2–Werte von 0,2 ppb.
Dabei stellt sich die Frage, auf welche Weise das Ozon abgebaut wird.
Eine mögliche Erklärung hierfür ist der photochemische Ozonabbau über Tag. Aus der Photolyserate
und einer Ozonkonzentration von 20 ppt ergibt sich eine maximale Verlustrate von 4 ppb/h (vgl. Kap.
2.4.4).
Des weiteren dehnt sich die MBL im Laufe eines Tages aus, so dass, beginnend ab dem späten Nach-
mittag, ein Austausch mit der ozonreicheren Luft der freien Troposphäre möglich ist und die Ozon-
konzentration wieder ansteigt. De Laat und Lelieveld [2000] zeigten, dass in einer ozonabbauenden
Umgebung dieser Tagesverlauf auch durch Photochemie und Advektion erklärt werden kann, mit
einem für die MBL typischen horizontalen O3–Gradienten.
Abb. 7.3: Mit dem Ozonmonitor aufgenommene Ozondaten. Zu beachten ist der Zeitsprung auf der Zeitachse
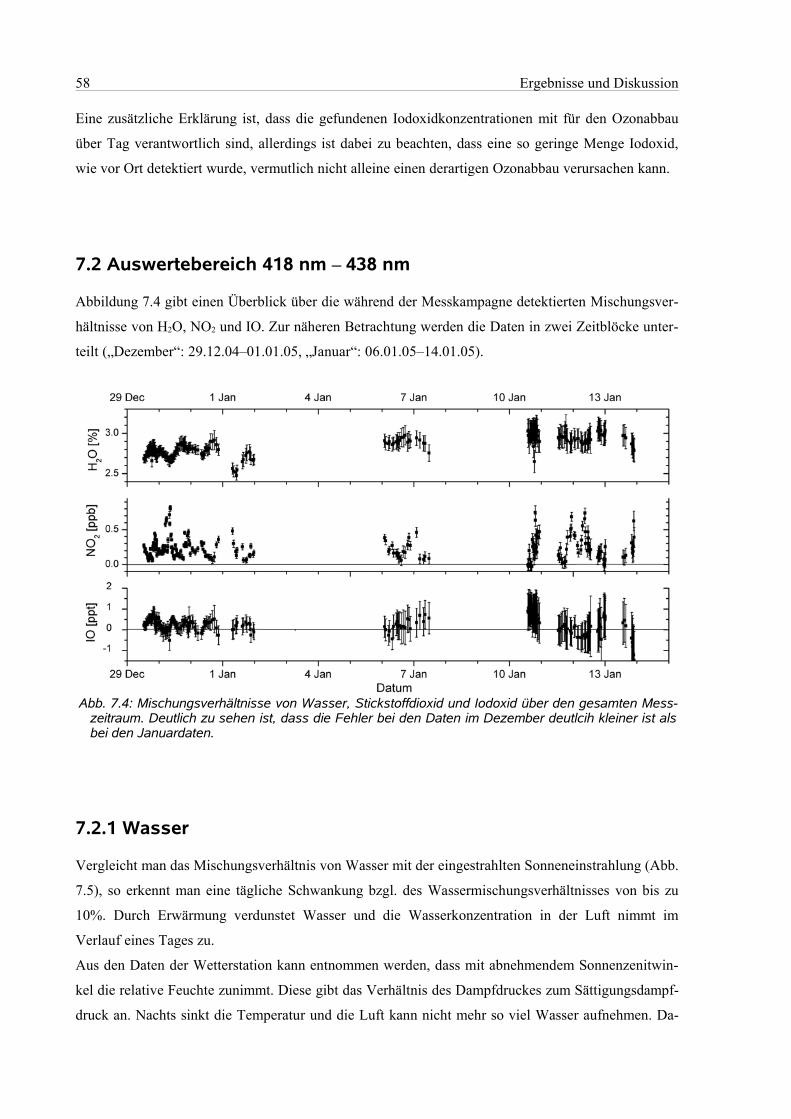
58 Ergebnisse und Diskussion
Eine zusätzliche Erklärung ist, dass die gefundenen Iodoxidkonzentrationen mit für den Ozonabbau
über Tag verantwortlich sind, allerdings ist dabei zu beachten, dass eine so geringe Menge Iodoxid,
wie vor Ort detektiert wurde, vermutlich nicht alleine einen derartigen Ozonabbau verursachen kann.
7.2 Auswertebereich 418 nm – 438 nm
Abbildung 7.4 gibt einen Überblick über die während der Messkampagne detektierten Mischungsver-
hältnisse von H2O, NO2 und IO. Zur näheren Betrachtung werden die Daten in zwei Zeitblöcke unter-
teilt („Dezember“: 29.12.04–01.01.05, „Januar“: 06.01.05–14.01.05).
7.2.1 Wasser
Vergleicht man das Mischungsverhältnis von Wasser mit der eingestrahlten Sonneneinstrahlung (Abb.
7.5), so erkennt man eine tägliche Schwankung bzgl. des Wassermischungsverhältnisses von bis zu
10%. Durch Erwärmung verdunstet Wasser und die Wasserkonzentration in der Luft nimmt im
Verlauf eines Tages zu.
Aus den Daten der Wetterstation kann entnommen werden, dass mit abnehmendem Sonnenzenitwin-
kel die relative Feuchte zunimmt. Diese gibt das Verhältnis des Dampfdruckes zum Sättigungsdampf-
druck an. Nachts sinkt die Temperatur und die Luft kann nicht mehr so viel Wasser aufnehmen. Da-
Abb. 7.4: Mischungsverhältnisse von Wasser, Stickstoffdioxid und Iodoxid über den gesamten Mess-zeitraum. Deutlich zu sehen ist, dass die Fehler bei den Daten im Dezember deutlcih kleiner ist alsbei den Januardaten.

7 Ergebnisse und Diskussion 59
durch sinkt der Wert der Sättigung, die relative Feuchte dagegen nimmt zu. Berechnet man aus den
Daten der Wetterstation über die Magnus-Formel [Kraus, 2000] das Wassermischungverhältnis, so er-
gibt sich ein vergleichbarer Verlauf, diese Daten liegen jedoch systematisch um 7 % unter den mit
DOAS ermittelten Werten. Dies ist vermutlich dadurch zu erklären, dass die Wetterstation Daten
einige hundert Meter vom Meer entfernt in situ aufzeichnete, wohingegen die DOAS–Messstrecke
den größten Teil der 14,5 km direkt über Wasser verlief. Dort haben Effekte wie Verdunstung oder
Seaspray (Tröpfchenbildung über der Wasseroberfläche, z.B. durch Wind) direkte Auswirkungen auf
die Wasserkonzentration in der Luft. Beim Transport ins Landesinnere kann die Wasserkonzentration
vermutlich durch Deposition gesenkt werden. Eine alternative Erklärung für den systematischen Un-
terschied der beiden Datensätze ist, dass der Absorptionswirkungsquerschnitt von Wasser im Bereich
320–350 nm zu klein angegeben und dadurch zu hohe Konzentrationen berechnet wurden.
Die gemessenen Januardaten von Wasser zeigen nur noch einen schwachen Tagesverlauf, was
vermutlich mit an ihren großen Fehlern liegt. Dafür zeigen hier die berechneten Wasserwerte einen
Tagesverlauf, wieder mit Mischungsverhältnissen unterhalb der gemessenen Werte. Die tageszeitli-
chen Schwankungen liegen bei etwa 4% des Wassermischungsverhältnisses.
7.2.2 Stickstoffdioxid
Das Mischungsverhältnis von NO2 lag im Mittel zwischen 50 und 400 ppt mit einigen Maximalwerten
von bis zu 800 ppt, der mittlere Wert der Daten liegt bei 240 ppt.
Grundsätzlich wird NO2 anthropogen erzeugt, es kann von einer geringen Verschmutzung an NO2
(„Hintergrund“) sowohl durch Einmischung von Luft, die aus São Luis antransportiert wird (die Ent-
fernung zwischen Alcântara und São Luis beträgt nur etwa 12 km), als auch durch die vielen vor São
Luis mehrere Tange lang ankernden Tanker, ausgegangen werden.
Abb. 7.5: Sonneneinstrahlung: Daten aus der Wetterstation, Wasserkonzentration: mit DOAS ermittelt(schwarz) und aus Daten der Wetterstation berechnet(blau).
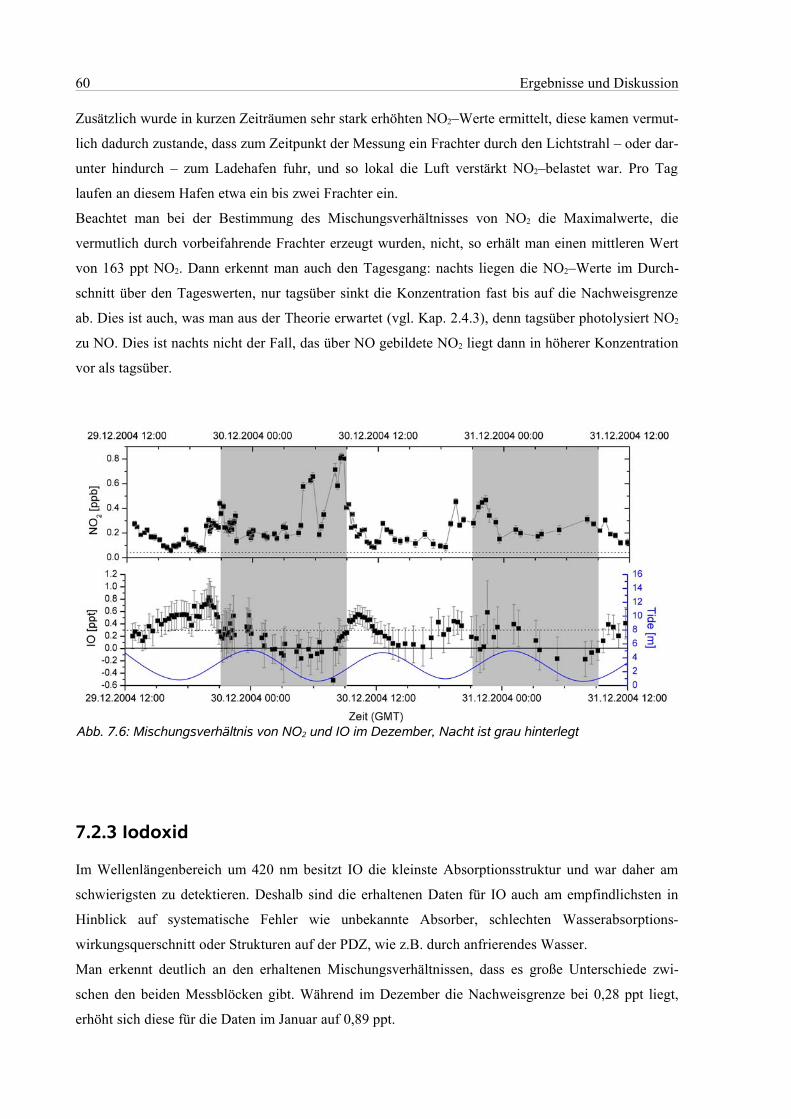
60 Ergebnisse und Diskussion
Zusätzlich wurde in kurzen Zeiträumen sehr stark erhöhten NO2–Werte ermittelt, diese kamen vermut-
lich dadurch zustande, dass zum Zeitpunkt der Messung ein Frachter durch den Lichtstrahl – oder dar-
unter hindurch – zum Ladehafen fuhr, und so lokal die Luft verstärkt NO2–belastet war. Pro Tag
laufen an diesem Hafen etwa ein bis zwei Frachter ein.
Beachtet man bei der Bestimmung des Mischungsverhältnisses von NO2 die Maximalwerte, die
vermutlich durch vorbeifahrende Frachter erzeugt wurden, nicht, so erhält man einen mittleren Wert
von 163 ppt NO2. Dann erkennt man auch den Tagesgang: nachts liegen die NO2–Werte im Durch-
schnitt über den Tageswerten, nur tagsüber sinkt die Konzentration fast bis auf die Nachweisgrenze
ab. Dies ist auch, was man aus der Theorie erwartet (vgl. Kap. 2.4.3), denn tagsüber photolysiert NO2
zu NO. Dies ist nachts nicht der Fall, das über NO gebildete NO2 liegt dann in höherer Konzentration
vor als tagsüber.
7.2.3 Iodoxid
Im Wellenlängenbereich um 420 nm besitzt IO die kleinste Absorptionsstruktur und war daher am
schwierigsten zu detektieren. Deshalb sind die erhaltenen Daten für IO auch am empfindlichsten in
Hinblick auf systematische Fehler wie unbekannte Absorber, schlechten Wasserabsorptions-
wirkungsquerschnitt oder Strukturen auf der PDZ, wie z.B. durch anfrierendes Wasser.
Man erkennt deutlich an den erhaltenen Mischungsverhältnissen, dass es große Unterschiede zwi-
schen den beiden Messblöcken gibt. Während im Dezember die Nachweisgrenze bei 0,28 ppt liegt,
erhöht sich diese für die Daten im Januar auf 0,89 ppt.
Abb. 7.6: Mischungsverhältnis von NO2 und IO im Dezember, Nacht ist grau hinterlegt

7 Ergebnisse und Diskussion 61
Bei den Dezemberdaten (Abb. 7.6) erkennt man deutlich zwei Maxima (29. Dezember nachmittags,
30. Dezember morgens), die über der Nachweisgrenze liegen. Iodoxid tritt tagsüber auf, nachts sind
die Werte gut mit Null verträglich. Dies wird auch aus der Theorie so erwartet, da biogene Quellen,
die tagsüber durch Photolyse Iod freisetzen, nachts nicht photolysiert werden können.
Auch eine Abhängigkeit von der Tide ist möglich: am 29. Dezember steigt die IO–Konzentration von
morgens bis zum Spätnachmittag an und geht dann zurück, der Tiefstand der Ebbe ist um etwa 15 Uhr
BRT erreicht. In der Nacht gehen die Werte wieder zurück, haben aber am Morgen des 30. Dezembers
erneut ein Maximum, das jedoch mit Wasserhöchststand wieder auf Null absinkt und erst am Spät-
nachmittag liegen wieder vereinzelte Messwerte über der Nachweisgrenze. Um eine klare Aussage
machen zu können, wie genau die IO–Konzentration mit dem Tidenstand schwankt bedarf es eines
Zeitraums von mehr als 2 Tagen detektierbaren Iodoxids.
Es stellt sich die Frage, woher das Iodoxid stammt.
Die berechneten Rückwärtstrajektorien zeigen, dass die untersuchten Luftmassen in den Tagen zuvor
über dem Atlantik verweilten. Eine mögliche Antwort ist demnach, dass aus dem Ozean emittierte
flüchtige Iodverbindungen (z.B.CH3I) mit den Luftmassen zum Messort transportiert wurden und so
stets Iod in geringen Mengen vorhanden ist.
Eine andere Erklärung ist, dass kurzlebige Iodverbindungen vor Ort freigesetzt werden, zum Beispiel
durch Mangrovenwälder. Dass Pflanzen für die Freisetzung hoher Iodkonzentrationen verantwortlich
sind ist bislang nur für Makroalgen und Phytoplankton bekannt [Carpenter et al., 1999], in den unter-
suchten Luftproben sind jedoch zum Teil erstaunlich hohe Konzentrationen an Methyliodid enthalten
(bis zu 12 pptv, vgl. Kap. 7.3), die für eine Freisetzung von Iodverbindungen vor Ort sprechen. Die
höchsten Konzentrationen wurden in den Luftproben ermittelt, die im Bereich der großflächigen
Mangrovenwälder lagen. Für eine örtliche Freisetzung spricht auch die mögliche Korrelation mit der
Tide, so wurden auch schon in Mace Head [Carpenter et al., 1999] und in der Bretagne [Peters,
2005] durch freiliegende Algen verstärkt flüchtige Iodverbindungen freigesetzt. Allerdings gab es im
Bereich der Messstrecke keinen nennenswerten Algenvorkommen oder Mangrovenwälder.
Ob dieses Iodoxid wirklich zum Ozonabbau beiträgt, kann jedoch nicht direkt belegt werden, zumal
im Zeitraum der Messungen durch den Ausfall des Ozonmonitors keine Ozondaten verfügbar sind.
Berechnet man den Anteil, den 0,5 ppt IO über die Reaktion mit HO2 und über Selbstreaktion zum
Ozonabbau beitragen (vgl Gl. 2.3), so erhält man eine Ozonabbaurate von nur 41 ppt/h, daher kommt
der größte Teil des Ozonabbaus über Tag vermutlich durch Photolyse von O3 (mit maximal 4 ppb/h,
vgl. Kap. 2.4.4). Dies soll jedoch nur eine grobe Abschätzung der Größenordnungen sein, denn die
Iodchemie ist viel komplexer als sie in Gleichung 2.3 berücksichtigt wird.
Das Ergebnis der Anpassung differentieller Stukturen bei der Auswertung mit WinDOAS ist in Abbil-
dung 7.7 dargestellt. Sie zeigt das Spektrum X78 (schwarze Linie) vom 30. Dez. 04, 10 Uhr, das sich
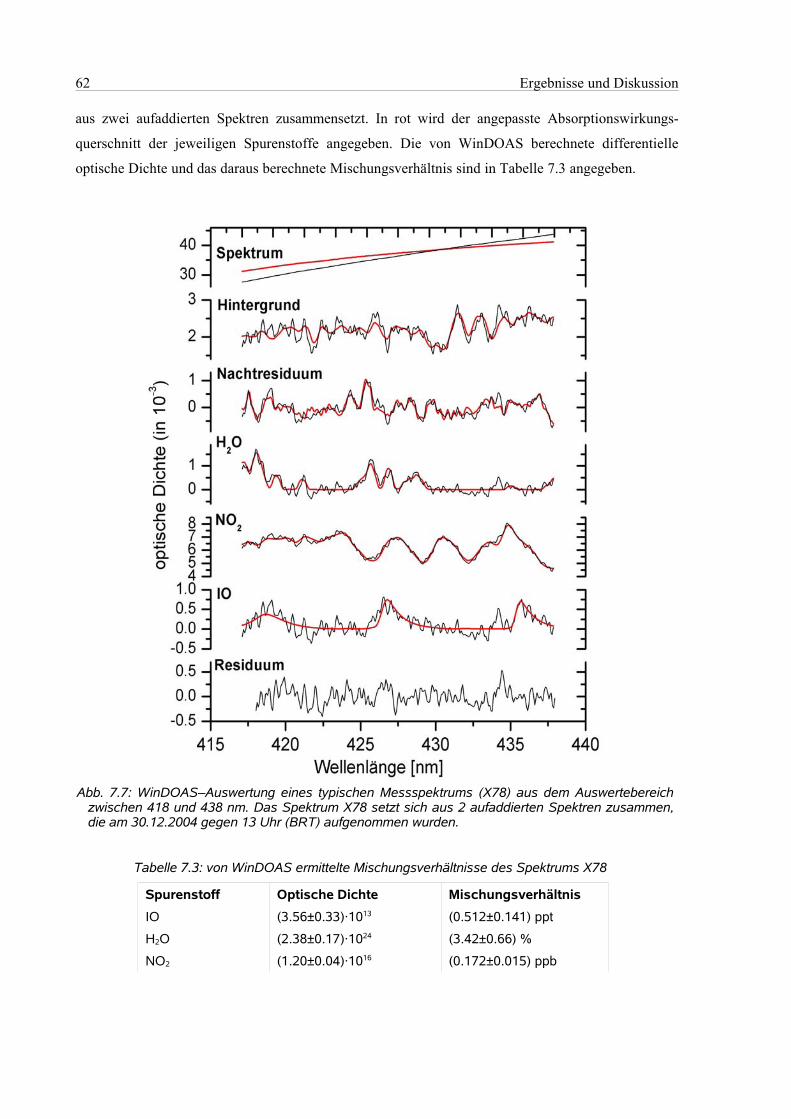
62 Ergebnisse und Diskussion
aus zwei aufaddierten Spektren zusammensetzt. In rot wird der angepasste Absorptionswirkungs-
querschnitt der jeweiligen Spurenstoffe angegeben. Die von WinDOAS berechnete differentielle
optische Dichte und das daraus berechnete Mischungsverhältnis sind in Tabelle 7.3 angegeben.
Tabelle 7.3: von WinDOAS ermittelte Mischungsverhältnisse des Spektrums X78
Spurenstoff Optische Dichte MischungsverhältnisIO (3.56±0.33)·1013 (0.512±0.141) pptH2O (2.38±0.17)·1024 (3.42±0.66) %NO2 (1.20±0.04)·1016 (0.172±0.015) ppb
Abb. 7.7: WinDOAS–Auswertung eines typischen Messspektrums (X78) aus dem Auswertebereichzwischen 418 und 438 nm. Das Spektrum X78 setzt sich aus 2 aufaddierten Spektren zusammen,die am 30.12.2004 gegen 13 Uhr (BRT) aufgenommen wurden.
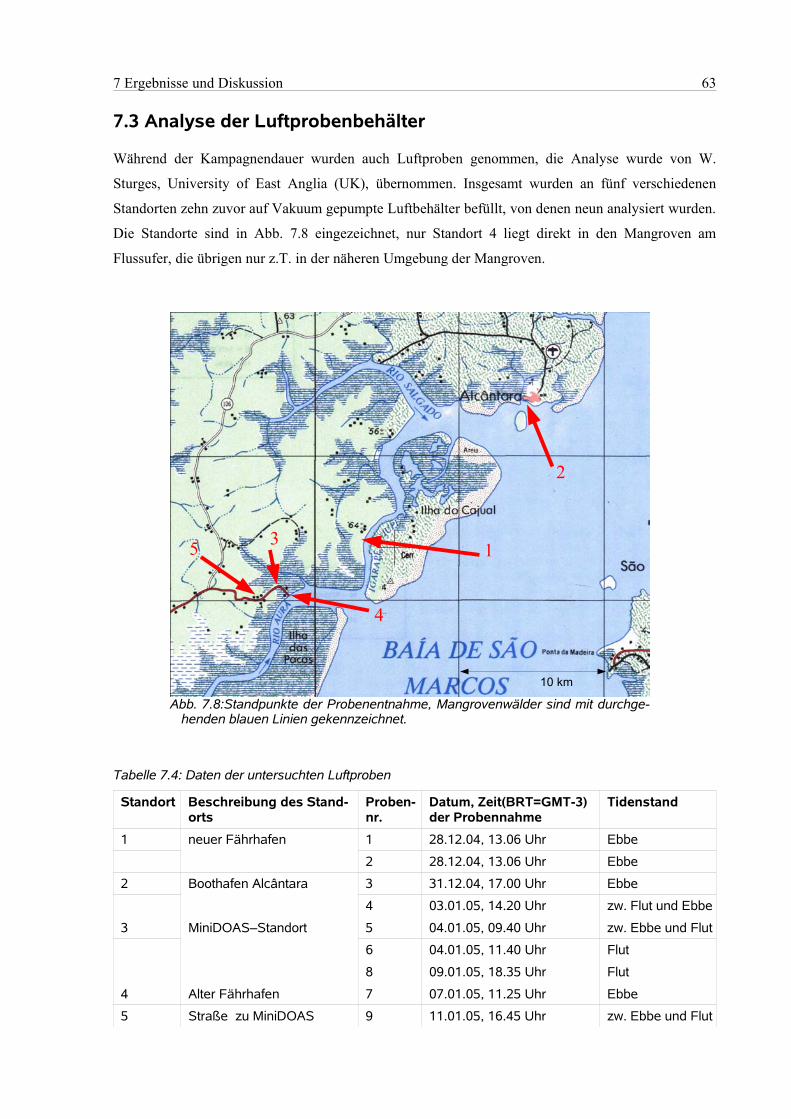
7 Ergebnisse und Diskussion 63
7.3 Analyse der Luftprobenbehälter
Während der Kampagnendauer wurden auch Luftproben genommen, die Analyse wurde von W.
Sturges, University of East Anglia (UK), übernommen. Insgesamt wurden an fünf verschiedenen
Standorten zehn zuvor auf Vakuum gepumpte Luftbehälter befüllt, von denen neun analysiert wurden.
Die Standorte sind in Abb. 7.8 eingezeichnet, nur Standort 4 liegt direkt in den Mangroven am
Flussufer, die übrigen nur z.T. in der näheren Umgebung der Mangroven.
Tabelle 7.4: Daten der untersuchten Luftproben
Standort Beschreibung des Stand-orts
Proben-nr.
Datum, Zeit(BRT=GMT-3)der Probennahme
Tidenstand
1 neuer Fährhafen 1 28.12.04, 13.06 Uhr Ebbe2 28.12.04, 13.06 Uhr Ebbe
2 Boothafen Alcântara 3 31.12.04, 17.00 Uhr Ebbe4 03.01.05, 14.20 Uhr zw. Flut und Ebbe
3 MiniDOAS–Standort 5 04.01.05, 09.40 Uhr zw. Ebbe und Flut6 04.01.05, 11.40 Uhr Flut8 09.01.05, 18.35 Uhr Flut
4 Alter Fährhafen 7 07.01.05, 11.25 Uhr Ebbe5 Straße zu MiniDOAS 9 11.01.05, 16.45 Uhr zw. Ebbe und Flut
Abb. 7.8:Standpunkte der Probenentnahme, Mangrovenwälder sind mit durchge-henden blauen Linien gekennzeichnet.
5
4
3
2
1
10 km
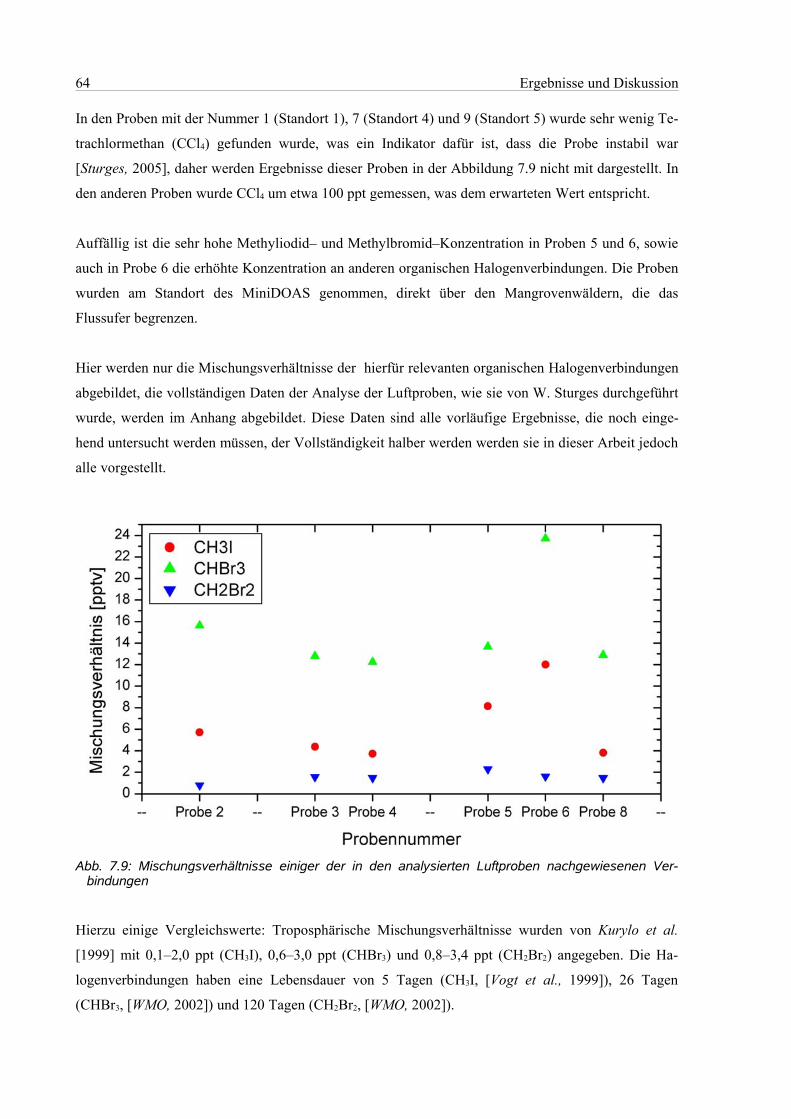
64 Ergebnisse und Diskussion
In den Proben mit der Nummer 1 (Standort 1), 7 (Standort 4) und 9 (Standort 5) wurde sehr wenig Te-
trachlormethan (CCl4) gefunden wurde, was ein Indikator dafür ist, dass die Probe instabil war
[Sturges, 2005], daher werden Ergebnisse dieser Proben in der Abbildung 7.9 nicht mit dargestellt. In
den anderen Proben wurde CCl4 um etwa 100 ppt gemessen, was dem erwarteten Wert entspricht.
Auffällig ist die sehr hohe Methyliodid– und Methylbromid–Konzentration in Proben 5 und 6, sowie
auch in Probe 6 die erhöhte Konzentration an anderen organischen Halogenverbindungen. Die Proben
wurden am Standort des MiniDOAS genommen, direkt über den Mangrovenwäldern, die das
Flussufer begrenzen.
Hier werden nur die Mischungsverhältnisse der hierfür relevanten organischen Halogenverbindungen
abgebildet, die vollständigen Daten der Analyse der Luftproben, wie sie von W. Sturges durchgeführt
wurde, werden im Anhang abgebildet. Diese Daten sind alle vorläufige Ergebnisse, die noch einge-
hend untersucht werden müssen, der Vollständigkeit halber werden werden sie in dieser Arbeit jedoch
alle vorgestellt.
Hierzu einige Vergleichswerte: Troposphärische Mischungsverhältnisse wurden von Kurylo et al.
[1999] mit 0,1–2,0 ppt (CH3I), 0,6–3,0 ppt (CHBr3) und 0,8–3,4 ppt (CH2Br2) angegeben. Die Ha-
logenverbindungen haben eine Lebensdauer von 5 Tagen (CH3I, [Vogt et al., 1999]), 26 Tagen
(CHBr3, [WMO, 2002]) und 120 Tagen (CH2Br2, [WMO, 2002]).
Abb. 7.9: Mischungsverhältnisse einiger der in den analysierten Luftproben nachgewiesenen Ver-bindungen

8 Zusammenfassung und Ausblick 65
8 Zusammenfassung und Ausblick
Ziel der Arbeit war es, in einem in den Tropen gelegenen Gebiet die dort vorliegende Konzentration
an den Halogenoxiden IO und BrO zu ermitteln. Hierzu wurde eine Messkampagne in Alcântara, im
Nordosten Brasiliens, durch geführt. Dort wurde mit einem bodengestützten Langpfad–DOAS–Gerät
über eine Absorptionsstrecke von 29 km gemessen. Der sehr lange Lichtweg machte im Falle von IO
eine Bestimmung des Mischungsverhältnisses bis in den sub–ppt–Bereich möglich, was bisher mit
DOAS noch nicht erreicht worden war. Nachteil eines langen Lichtwegs ist jedoch die Abhängigkeit
von atmosphärischen Bedingungen.
Es wurden Messspektren in zwei verschiedenen Wellenlängenbereichen aufgenommen:
Im UV–Bereich um 330 nm, wurden die Messspektren auf Absorption durch Ozon, Bromoxid und
Formaldehyd untersucht.
• O3 konnte mit Mischungsverhältnissen um 20 ppb nachgewiesen werden, womit die aufgenom-
menen Daten des Ozonmonitors gut übereinstimmen. Letztere zeigen einen deutlichen Tagesver-
lauf, in dem Ozon tagsüber um etwa 4 ppb abgebaut wird.
• BrO und HCHO waren über den gesamten Messzeitraum mit Null verträglich
Im blauen Spektralbereich (um 420 nm) wurden die Messspektren auf die Absorption von Iodoxid,
Stickstoffdioxid und Wasser hin untersucht.
• IO konnte im Dezember bei einer Zeitreihe, die über zwei Tagen ging, eindeutig über der Nach-
weisgrenze von 0,28 ppt nachgewiesen werden, mit einem Maximalwert von (0,82±0,31) ppt.
Eine Korrelation mit der IO–Maxima mit der Tide ist möglich. Im Januar waren aufgrund von
schlechteren Messbedingungen alle ermittelten Mischungsverhältnisse mit Null verträglich.
• Für NO2 wurde ein mittleres Mischungsverhältnis von 240 ppt ermittelt. Dabei wurden Maximal-
werte von bis zu 800 ppt gemessen, die vermutlich durch den Ausstoß von, den Lichtweg
kreuzenden, Tankern verursacht wurden.
• Das Mischungsverhältnis von H2O lag um 2,8 %, mit täglichen Schwankungen des Mischungsver-
hältnisses um etwa 10 %. Dies ist mit den aus der Wetterstation ermittelten Daten verträglich.
Mögliche Erklärungen, weshalb in diesem Gebiet IO nachweisbar ist, ist zum einen der Transport
kurzlebiger Iodverbindungen vom Ozean. Dies ist deshalb möglich, da die berechneten Rückwärtstra-
jektorien zeigen, dass die Luftmassen die Tage zuvor über dem Atlantik verweilten. Zum anderen ist
auch die lokale Freisetzung, z.B. durch Mangroven, möglich. Für letztere Möglichkeit spricht, dass
die IO–Konzentration vermutlich mit der Tide korreliert, was jedoch, mit nachgewiesenem IO an nur
zwei Tagen, nur vermutet, jedoch letztlich nicht bewiesen werden kann.

66 Zusammenfassung und Ausblick
Im Rahmen dieser Arbeit wurde IO nachgewiesen, wo und wodurch es jedoch tatsächlich freigesetzt
wurde, konnte nicht geklärt werden. Möglicherweise führt eine genauere Untersuchung und Diskussi-
on der Daten aus der Probenanalyse noch zu weiteren Erkenntnissen.
In den marinen Tropen sollten die nachfolgenden Fragen weitergehend untersucht werden:
• Haben die Mangroven eine Einwirkung auf die IO–Konzentrationen vor Ort? Hierzu könnte man
untersuchen, ob die IO–Konzentration tatsächlich mit der Tide korreliert, was aufgrund der Ergeb-
nissen dieser Arbeit vermutet wird.
• Welche Ozonwerte werden in einem Zeitraum, in dem IO nachgewiesen wird, tatsächlich ge-
messen? (Dies konnte auf der durchgeführten Kampagne aufgrund eines Ausfalls des Ozonmoni-
tors nicht durchgeführt werden.)

9 Literaturangaben 67
9 Literaturangaben
Ackermann (2000). Auswirkung von Kraftfahrzeugemission in der urbanen Atmosphäre. Dissertation
am Institut für Umweltphysik, Universität Heidelberg.
Alicke, B., K. Hebestreit, J. Stutz, U. Platt (1999). Iodine oxide in the marine boundary layer. Nature,
Vol. 397, 572–573.
Allan, B.J., G. McFiggans, J.M.C. Plane, C. Hugh (2000). Observation of iodine monoxide in the
remote marine boundary layer. J. Geophys. Res., Vol. 105, 14,363–14,369.
Allan, B.J., J.M.C. Plane, G. McFiggans (2001). Observations of OIO in the remote marine boundary
layer. Geophys. Res. Lett., Vol. 28, 10, 1945–1948.
Atkinson, R., D. Baulch, R. Cox, j. Crowley, R.F. Hampson, Jr., R.G. Hynes, M.E. Jenkin, J.A. Kerr,
M. Rossi, J. Troe (2004). Summary od Evaluated Kinetic and Photchemical Data for Atmospheric
Chemistry: Web Version, Juli 2004. J. Phys. Chem. Ref. Data.
http://www.iupac-kinetic.ch.cam.ac.uk/summary/IUPACsumm_web_latest.pdf
Baker, J.M., C.E. Reeves, P.D. Nightingale, S.A. Penkett, S.W. Gibb, A.D. Hatton (1999). Biological
production of methyl bromide in the coastal waters of the North Sea and open ocean of the
northeast Atlantic. Mar. Chem., Vol. 64, 267-285.
Baker, A.R. (2004). Inorganic iodine speciation in tropical Atlantic aerosol. Geophys. Res. Lett., Vol.
31, L23S02.
Barrie, L.A., J.W. Bottenheim, R.C. Schnell, P.J. Crutzen, R.A. Rasmussen (1988). Ozone destruction
and photochemical reactions at polar sunrise in the lower Arctic atmosphere. Nature, Vol. 334,
138–141.
Bauer, N. (1997). Charakterisierung des DOAS-Ballon_Spektrographen zur Bestimmung
stratosphärischer Spurenstoffe. Diplomarbeit am Institut für Umweltphysik, Universität
Heidelberg.
Bedjanian, Y., G. Le Bras, G. Poulet (1997). Kinetics and Mechanism of the IO + ClO Reaction. J.
Phys. Chem. A, Vol. 101, 4088–4096.

68 Literaturangaben
Beirle, S., U. Platt, M. Wenig, T. Wagner (2004a). NOx production by lightning estimated with
GOME. Advances in Space Research, Vol. 34, 793–797.
Beirle, S., U. Platt, T. Wagner (2004b). European Space Agency homepage, Heidelberg.
Bevington, P.R. (1969). Data reduction and error analysis for the physical science. McGraw–Hill,
New York
Bloss, W.J., D.M. Rowley, R.A. Cox, R.L. Jones (2001). Kinetics and Products of the IO Self–
Reaction. J. Phys. Chem. A, Vol. 105, 7840–7854.
Bobrowski, N. (2002). Volcanic gas studies by multi axis differential optical absorption spectroscopy.
Diplomarbeit am Institut für Umweltphysik, Universität Heidelberg.
Bobrowski, N., G. Hönninger, B. Galle, U. Platt. (2003). Detection of bromine monoxide in a
volcanic plume. Nature, Vol. 423, 273–276.
Carpenter, L.J., P.S. Monks, B.J. Bandy, S.A. Penkett, I.E. Galbally, C.P. Meyer (1997). A Study of
Peroxy Radicals and Ozone Photochemistry at Coastal Sites in the Northern and Southern
Hemispheres. J. Geophys. Res., Vol. 102, 25,417–25,427
Carpenter, L.J., W.T. Sturges, S.A. Penkett, P.S. Liss, B. Alicke, K. Hebestreit, U. Platt (1999).
Short–lived alkyl iodes and bromides at Mace Head, Ireland. J. Geophys. Res., Vol. 104, 1679–
1689.
Cauer, H. (1939). Schwankungen der Jodmenge der Luft in Mitteleuropa, deren Ursprung und deren
Bedeutung für den Jodgehalt unserer Nahrung. Angew. Chem., Vol. 52, 9–18.
Chameides, W.L., und D.D. Davis (1980). Iodine: Its possible role in tropospheric photochemistry. J.
Geophys. Res., Vol. 85, C 7383–7398.
Cox, R.A., W.J. Bloss, R.L. Jones, D.M. Rowley (1999). OIO and the Atmosoheric Cycle of Iodine.
Geophys. Res. Lett., Vol. 26, 13, 1857–1860.
Crowley, J. (2004). Annual Report ReHaTrop, http://www.afo-2000.de/001/afo2000.htm.

9 Literaturangaben 69
Crutzen, P.J. (1974). Photochemical reactions initiated by and influencing ozone in unpolluted
tropospheric air. Tellus, Vol. 26, 47–57.
Czerny, M., und A.F. Turner (1930). Z. Phys., Vol 61, 792.
Davis, D., J. Crawford, S. Liu, S. McKeen, A. Bandy, D. Thornton, F. Rowland, D. Blake (1996).
Potential impact of iodine on tropospheric levels of ozone and other critical oxidants. J. Geophys.
Res., Vol. 101, 2135–2147.
de Laat, A.T.J., und J. Lelieveld (2000). Diurnal ozone cycle in the tropical and subtropical marine
boundary layer. J. Geophys. Res., Vol. 105, 11,547–11,559.
DeMore, W.B., S.P. Sander, D.M. Golden, R.F. Hampson, M.J. Kurylo, C.J. Howard, A.R.
Ravishankara, C.E. Colc, M.J. Molina (1997). Chemical kinetics and photochemical data for uns in
stratospheric modeling. JPL Publication 97–4.
Duce, R.A., J.T. Wasson, J.W. Winchester, F. Burns (1963). Atmospheric iodine, bromine, and
chlorine. J. Geophys. Res., Vol. 68, 3943–3947.
Eriksson, E. (1959a). The yearly circulation of chloride and sulfur in nature; meteorological,
geochemical and pedological implications: Part I. Tellus, Vol. 11, 375–403.
Eriksson, E. (1959b). The yearly circulation of chloride and sulfur in nature; meteorological,
geochemical and pedological implications: Part II. Tellus, Vol. 12, 63–109.
Farman, J.C., B.G. Gardiner, J.D. Shanklin (1985). Large losses of total ozone in Antarcica
revealseasonal ClOx/NOx interaction. Nature, Vol. 315, 207–210.
Finlayson–Pitts, B.J. und J. N. Pitts Jr. (2000). Chemistry of the upper and lower atmosphere.
Academic Press, San Diego.
Hausmann, M., und U. Platt (1994). Spectroscopic measurement of bromine oxide and ozone in the
high Arctic during Polar Sunrise Experiment. J. Geophys. Res., Vol. 99, 25, 399–25,413.
Hebestreit, K., J. Stutz, D.R.V. Matveiv, M. Peleg, M. Luria, U. Platt (1999). DOAS Measurements of
Tropospheric Bromine Oxide in Mid–Latitudes. Science, Vol. 283, 55–57.

70 Literaturangaben
Hermans et al. (2002). Private Mitteilung. Für mehr Details siehe:
http://www.oma.be/BIRA-IASB/Scientific/Topics/Lower/LaboBase/Laboratory.html
Hoffmann, T., C. O'Dowd, J.H. Seinfeld (2001). Iodine oxide homogeneous nucleation: An
explanation for coastal new particle production. Geophys. Res. Lett., Vol. 28, 1949–1952.
Holland, H.D. (1978). The chemistry of the atmosphere and oceans. John Wiley & Sons, New York.
Hönninger, G. (1999). Referenzspektren reaktiver Halogenverbindungen dür DOAS-Messungen.
Diplomarbeit am Institut für Umweltphysik, Heidelberg.
Hönninger, G. (2002). Halogen Oxide Studies in the Boundary Layer by Multi Axis Differential
Optical Absorption Spectroscopy and Active Longpath–DOAS. Dissertation am Institut für
Umweltphysik, Heidelberg.
Hönninger, G., C. von Friedeburg, U. Platt (2004a). Multi axis differential optical absorption
spectroscopy (MAX-DOAS). Atmos. Chem. and Phys., Vol. 4, 1, 231–254.
Hönninger, G., H. Leser, O. Sebastián, U. Platt (2004b). Ground–based measurements of halogen
oxides at the Hudson Bay by active longpath DOAS and passive MAX–DOAS. Geophys. Res.
Lett., Vol. 31, Vol. 31, 4.
Jenkin, M.E. (1992). The photochemistry of iodine–containing compounds in the marine boundary
layer. AEA EE–0405.
Kaleschke, L., A. Richter, J. Burrows, O. Afe, G. Heygster, J. Notholt, A.M. Rankin, H.K. Roscoe, J.
Hollwedel, T. Wagner, H.W. Jacobi (2004). Frost flowers on sea ice as a source of sea salt and
their influence on tropospheric halogen chemistry. Geophys. Res. Lett., Vol. 31, L16114.
Keene, W.C., A.A.P. Pszenny, J.R. Maben, R. Sander (2002). Variation og marine aerosol acidity
with particle size. Geophys. Res. Lett., Vol. 29, GL013881.
Kraus, H. (2000). Die Atmosphäre der Erde. Kap. II.6. Vieweg Verlag, Braunschweig/Wiesbaden.
Kurylo, M.J., J.M. Rodríguez, M.O. Andreae, E.L. Atlas, D.R. Blake, J.H. Butler, S. Lal, D.J. Lary,
P.M. Midgley, S.A. Montzka, P.C. Novelli, C.E. Reeves, P.G. Simmonds, L.P. Steele, W.T.
Sturges, R.F. Weiss, Y. Yokouchi (1999). Shortlived ozone–related compounds, Kapitel 2 in

9 Literaturangaben 71
Scintific Assessment of Ozone Depletion: 1998, Global Ozone Research and Monitoring Project–
Report No. 44, World Meteorological Organization, Genf.
Linos (2003). LINOS Photonics Katalog, LINOS Photonics GmbH (Hrsg.), Göttingen.
Lohberger, F., G. Hönninger, U. Platt (2004). Ground–based imaging differential optical absorption
spectroscopy of atmospheric gases. Appl. Optics, Vol. 43, 24, 4711–4717.
Martin, W.C., J.R., Fuhr, D.E. Kelleher, A. Musgrove, J. Sugar, W.L. Wiese, P.J. Mohr, K. Ohlsen
(1999). NIST Atomic Spectra Database (version 2.0). Online verfügbar unter
http://physics.nist.gov/asd2. National Institute of Standards and Technology, Gaithersburg, MD.
McElroy, M.B., R.J., Salawitch, C.S. Wofsy, J.A. Logan (1986). Reductions of Antarctic ozone due to
synergistic interactions of chlorine and bromine. Nature, Vol. 321, 759-762.
McFiggans, G., J.M.C. Plane, B.J. Allan, L.J. Carpenter, H. Coe, C. O'Dowd (2000). A modeling
study of iodine chemistry in the marine boundary layer. J. Geophys. Res., Vol. 105, 14,371–
14,385.
Meller, R., und G.K. Moorgat (2000). Temperature dependance of the absorption cross section of
HCHO between 223 and 323K in the wavelength range 225–375 nm. J. Geophys. Res., Vol. 105,
7089–7102.
Molina, M.J. und F.S. Rowland (1974). Stratospheric sink for chlorofluoromethanes: chlorine–atom
catalysed destruction of ozone. Nature, Vol. 249, 810–812.
Murphy, D.M., J.R. Anderson, P.K. Quinn, L.M. McInnes, F.J. Brechtel, S.M. Kreidenweis, A.M.
Middlebrook, M. Pósfai, D.S. Thomson, P.R. Buseck (1998). Influence of sea–salt on aerosol
radiative properties in the Southern Ocean marine boundary layer. Nature, Vol. 392, 62–65.
Peters, C. (2005). Studies of Reactive Halogen Species (RHS) in the Marine and mid–Latitudinal
Boundary Layer by Acton Longpath Differential Optival Absorption Spectroscopy. Dissertation
am Institut für Umweltphysik, Universität Heidelberg.
Platt, U., D. Perner, H.W. Pätz (1979). Simultaneous Measurement of Atmospheric CH2O, O3 and
NO2 by Differential Optical Absorption. J. Geophys. Res., Vol. 84, 6329–6335.

72 Literaturangaben
Platt, U., und E. Lehrer (1997). Arctic Tropospheric Ozone Chemistry (ARCTOC) – Final Report to
the European Union, Heidelberg.
Platt, U. (2000). Reactive halogen species in the mid–latitude troposphere – recent discoveries.
Water, Air, and Soil Pollution, Vol. 123, 229–244.
Platt, U. (2000a). Differential Optical Absorption Spectroscopy (DOAS), in: Air Monitoring by
Spectroscopic Technique. Sigrist, M.W., Ed., Chemical Analysis Series, Vol. 127, John Wiley &
Sons, Inc.
Quack, B., und D.W.R. Wallace (2003). Air–sea flux of bromoform: Controls, rates, and implications.
Global Biogeochem. Cycles, Vol., 17(1), 1023, doi: 10.1029/2002GB001890.
Quack, B., E. Atlas, G. Petrick, V. Stroud, S. Schauffler, D.W.R. Wallace (2004). Oceanic bromoform
sources for tropical atmosphere. Geophys. Res. Lett., Vol. 31, L23S05.
Richter, A., F. Wittstock, M. Eisinger, J.P. Burrows (1998). GOME observations of tropospheric BrO
in northern hemispheric spring and summer 1997. Geophys. Res. Lett., Vol. 25, 2683–2686.
Richter, U., und D.W.R. Wallace (2004). Production of methyl iodine in the tropical Atlantic Ocean.
Geophys. Res. Lett., Vol. 31, L23S03.
Rothman, L.S., D. Jacyuemart, A. Barbe, D. Chris Benner, M. Birk, L.R. Brown, M.R. Carleer, C.
Cjackerian Jr., K. Chance, L.H., Coudert, V. Dana, V.M. Devi, J.–M. Flaud, R.R. Gamache, A.
Goldman, J.–M. Hartmann, K.W. Jucks, A.G. Maki, J.–Y. Mandin, S.T. Massie, J. Orphal, A.
Perrin, C.P. Rinsland, M.A.H. Smith, J. Tennyson, R.N. Tolchenov, R.A. Toth, J. Vander Auwera,
P. Varanasi, G. Wagner (2005). The HITRAN 2004 molecular spectroscopic database. J. Quant.
Spectrosc. Radiat. Transfer, doi: 10.1016/j.jqsrt.2004.10.008.
Rowley, D.M., W.J. Bloss, R.A. Cox, R.L. Jones (2001). Kinetics and Products of the IO+BrO
Reaction. J. of Phys. Chem. A, Vol. 105, 7855–7864.
Saiz-Lopez, A., und J.M.C. Plane (2004). Novel iodine chemistry in the marine boundary layer.
Geophys. Res. Lett., Vol. 31, L04112.
Schwarz, A. (2003). Entwicklung einer online Kopplungsmethode von GC mit ECD– und ICPMS–
Detektion zur Bestimmung biogener leichtflüchtiger bromierter und iodierter Kohlenwasserstoffe

9 Literaturangaben 73
in Meerwasser und der Atmosphäre. Dissertation am Institut für Anorganische und Analytische
Chemie, Universität Mainz.
Seto, F.Y.B, und R.A. Duce (1972). A laboratory study of iodine enrichment on atmospheric saéa-salt
particles produced by bubbles. J. Geophys. Res., Vol. 77, 5339–5349.
Singh, H.B., L.J. Salas, R.E. Stiles (1983). Methyl halides in and over the eastern pacific (40° N – 32°
S), J. Geophys. Res., Vol. 88, 3684–3590.
Solomon, S., R.R. Garcia, F.S. Rowland, D.J. Wuebbles (1986). On the depletion on Antarctic ozone.
Nature, Vol. 321, 755–758.
Solomon, S., H.G. Mount, R.W. Sanders, A.L. Schmeltekopf (1987). Visible spectroscopy at
McMurdo station, Antarctica 2. Observations of OClO. J. Geophys. Res., Vol. 92, 8329–8338.
Sturges, W. (2005). Persönliche Mitteilung.
Stutz, J., (1996). Messung der Konzentration troposphärischer Spurenstoffe mittels Differentieller–
Optischer–Absorptionsspektroskopie: Eine neue Generation von Geräten und Algorithmen.
Dissertation am Institut für Umweltphysik, Universität Heidelberg.
Stutz, J. und U. Platt (1996). Numerical analysis and estimation of the statistical error of differential
optical absorption spectroscopy measurement with least–squares methods. Appl. Opt., Vol. 35
(30), 6041–6053.
Stutz, J. und U. Platt (1997). Improving long-path differential optical absorption spectroscopy with a
quartz-fiber mode mixer. Appl. Opt., Vol. 36 (6), 1105–1115.
Stutz, J., K. Hebestreit, B. Alicke, U. Platt (1999). Chemistry of Halogen Oxides in the Troposphere:
Comparison of Model Calculation with Recent Field Data. J. Atm. Chem., Vol 34, 65–85.
van Roozendael, M., und C. Fayt (2000). WinDOAS 2.1 Software manual. Belgium Institute for
Space Aeronomie (BIRA/IASB).
Vogt, R., P.J. Crutzen, R. Sander (1996). A Mechanism for halogen release from sea-salt aerosol in
the remote boundary layer. Nature, Vol. 383, 327–330.

74 Literaturangaben
Vogt, R., R. Sander, R. von Glasow, P.J. Crutzen (1999). Iodine chemistry and its role in halogen
activation and ozone loss in the marine boundary layer: a model study. J. Atmos. Chem., Vol. 32,
375–395.
Voigt, S., J. Orphal, J.P. Burrows (2001). The temperature dependence (202–293 K) of the absorption
cross section of O3 in the 230–850 nm region measured by Fourier–transform spectroscopy.
Journal of Photochemistry and Photobiology, Vol. 143, 1–9.
Voigt, S., J. Orphal, J.P. Burrows (2002). The temperature and pressure dependence of the absorption
cross section of NO2 in the 250–800 nm region measured by Fourier–transform spectroscopy.
Journal of Photochemistry and Photobiology, Vol. 149, 1–7.
von Glasow, R., und P.J. Crutzen (2003). Tropospheric Halogen Chemistry. In: Treatise on
Geochemistry, Vol.4, The Atmosphere, Kapitel 2. Editiert von H.D. Holland, K.K. Turekian,
Elsevier, New York.
von Glasow, R. (2005). Persönliche Mitteilung. Photolyseraten berechnet nach dem Model von
Landgraf, J., and P. Crutzen, An efficient method for on-line calculations
of photolysis and heating rates, J. Atmos. Sci., 55, 863 878, 1998.
Wagner, T., und U. Platt (1998). Satellite mapping of enhanced BrO concentrations in the
troposphere. Nature, Vol. 395, 486–490.
Wilmouth, D.M., T.F., Hanisco, N.M. Donahue, J.G. Anderson (1999). Fourier Transform Ultraviolet
Spectroscopy of the A 2P3/2 X 2P2/3 Transition of BrO. J. Phys. Chem. A, Vol. 103, 45, 8935–
8945.
WMO, (2002). Scientific assessment of ozone depletion: 2002. World Meteorological Organisation
(WMO), Genf, Schweiz.
Zingler, J., und U. Platt (2005). Iodine oxide in the Dead Sea Valley: Evidence for inorganic source of
boundary layer IO. J. Geophys. Res., Vol. 110, D07307.
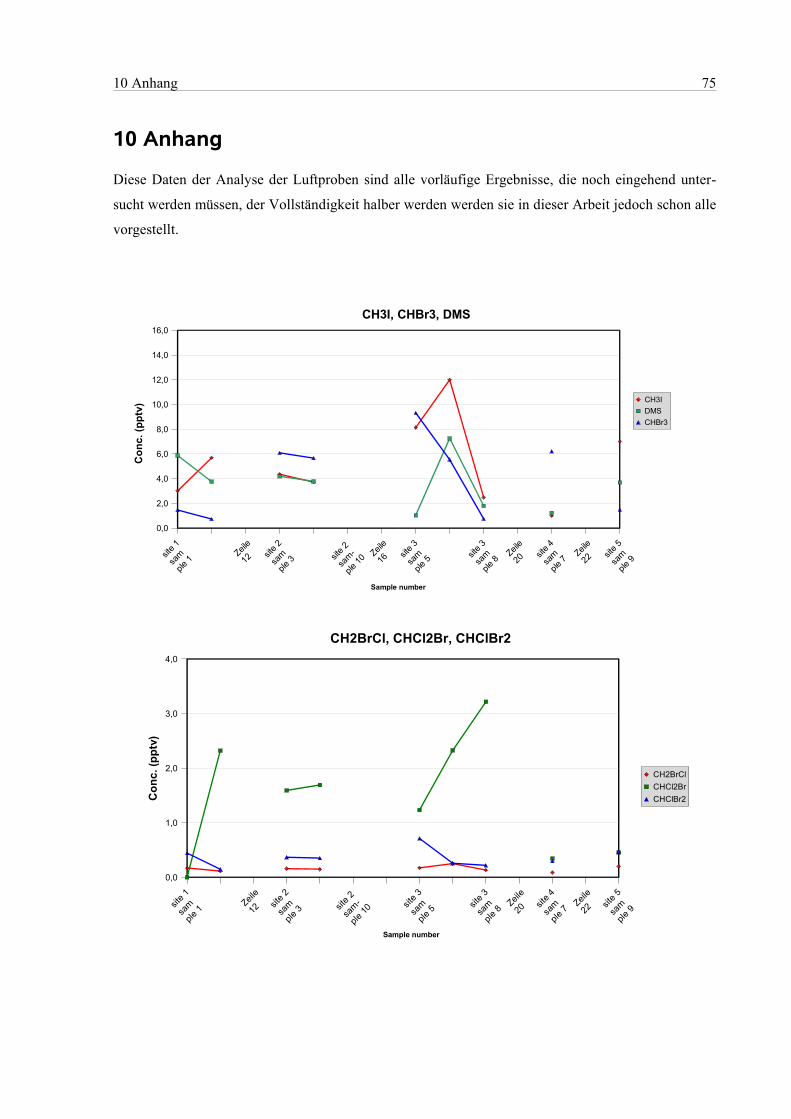
10 Anhang 75
10 Anhang
Diese Daten der Analyse der Luftproben sind alle vorläufige Ergebnisse, die noch eingehend unter-
sucht werden müssen, der Vollständigkeit halber werden werden sie in dieser Arbeit jedoch schon alle
vorgestellt.
site 1
sample
1Zeil
e
12 site 2
sample
3 site 2
sam-
ple 10
Zeile
16 site 3
sample
5sit
e 3
sample
8Zeil
e
20 site 4
sample
7Zeil
e
22 site 5
sample
9
0,0
2,0
4,0
6,0
8,0
10,0
12,0
14,0
16,0
CH3I, CHBr3, DMS
CH3IDMSCHBr3
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
3 site 2
sam-
ple 10
site 3
sample
5 site 3
sample
8Zeil
e
20 site 4
sample
7Zeil
e
22 site 5
sample
9
0,0
1,0
2,0
3,0
4,0
CH2BrCl, CHCl2Br, CHClBr2
CH2BrClCHCl2BrCHClBr2
Sample number
Con
c. (p
ptv)
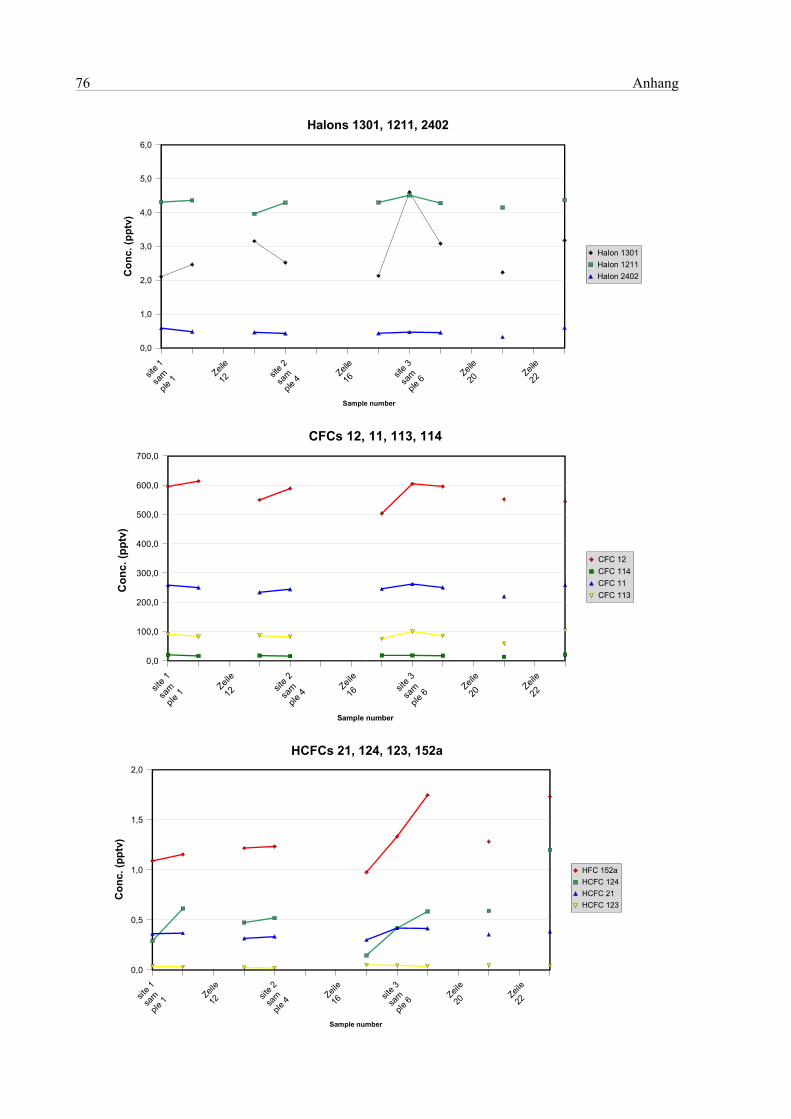
76 Anhang
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
100,0
200,0
300,0
400,0
500,0
600,0
700,0
CFCs 12, 11, 113, 114
CFC 12CFC 114CFC 11CFC 113
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
0,5
1,0
1,5
2,0
HCFCs 21, 124, 123, 152a
HFC 152aHCFC 124HCFC 21HCFC 123
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
1,0
2,0
3,0
4,0
5,0
6,0
Halons 1301, 1211, 2402
Halon 1301Halon 1211Halon 2402
Sample number
Con
c. (p
ptv)
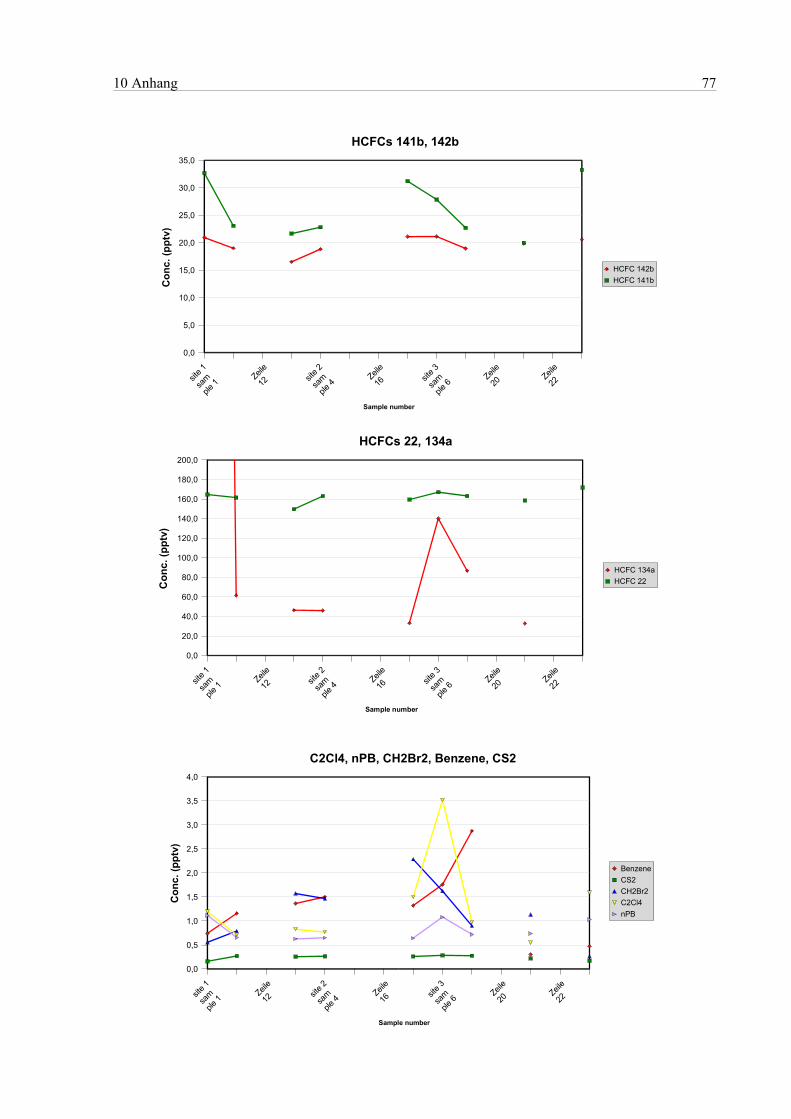
10 Anhang 77
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
5,0
10,0
15,0
20,0
25,0
30,0
35,0
HCFCs 141b, 142b
HCFC 142bHCFC 141b
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
20,0
40,0
60,0
80,0
100,0
120,0
140,0
160,0
180,0
200,0
HCFCs 22, 134a
HCFC 134aHCFC 22
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
C2Cl4, nPB, CH2Br2, Benzene, CS2
BenzeneCS2CH2Br2C2Cl4nPB
Sample number
Con
c. (p
ptv)
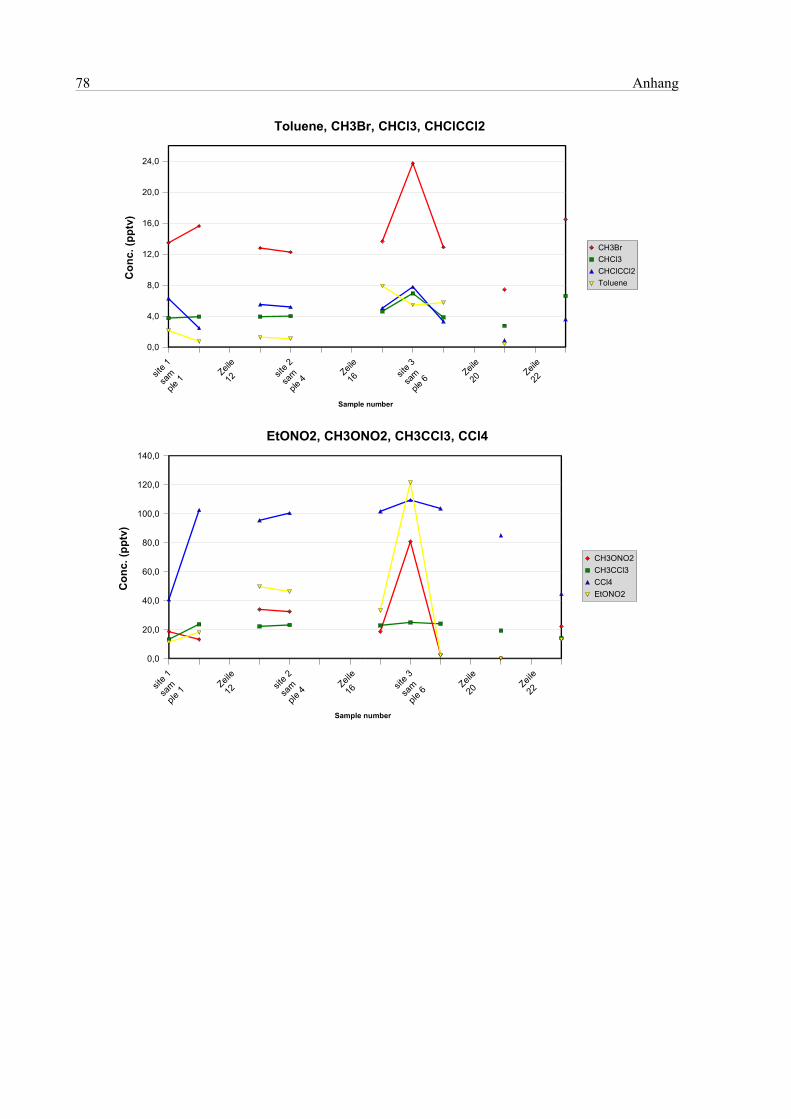
78 Anhang
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
4,0
8,0
12,0
16,0
20,0
24,0
Toluene, CH3Br, CHCl3, CHClCCl2
CH3BrCHCl3CHClCCl2Toluene
Sample number
Con
c. (p
ptv)
site 1
sample
1Zeil
e
12 site 2
sample
4Zeil
e
16 site 3
sample
6Zeil
e
20Zeil
e
22
0,0
20,0
40,0
60,0
80,0
100,0
120,0
140,0
EtONO2, CH3ONO2, CH3CCl3, CCl4
CH3ONO2CH3CCl3CCl4EtONO2
Sample number
Con
c. (p
ptv)

10 Danksagung
An dieser Stelle möchte ich mich bei all jenen Leuten bedanken, die direkt oder indirekt zum
Gelingen dieser Arbeit beigetragen haben.
An erster Stelle sei Herrn Prof. Dr. Ulrich Platt gedankt, dass ich an seinem Institut meine Staatsex-
amensarbeit schreiben durfte. Vielen Dank, dass ich – trotz der relativ kurzen Bearbeitungszeit – auch
an der Kampagne teilnehmen konnte, die mich sowohl an praktischer physikalischer Erfahrung als
auch durch viele neue persönliche Eindrücke bereichert hat.
Dankeschön auch an meinen Betreuer Priv.–Doz. Klaus Pfeilsticker, er hat mich in den vergangenen
Monaten sehr gut betreut und mich immer wieder motiviert. Vielen Dank für die – z.T. auch über das
Wissenschaftliche hinaus gehenden – Gespräche!
Mein herzlicher Danke geht auch an Andreas Lotter und Christina Peters, die bei der Kampagne dabei
waren und mir auch in späteren Auswerteproblemen mit Rat und Tat zur Seite standen. Tina, vielen
Dank, dass du die Zeit gefunden hast, die meine Arbeit nochmals durchzulesen.
Dankeschön an den ganzen Rest der Gruppe: André Butz, der die unglaubliche Geduld besaß, bereit-
willig auf wirklich alle meine Fragen einzugehen, Ulrike Reichl und Aaron Lindner für die sehr
angenehme Arbeitsatmosphäre in unserem Zimmer, Christoph Kern und Marcel Dorf fürs Zuhören
und Weiterhelfen in Auswertefragen. Vielen Dank auch Roland von Glasow, der mir beim Verstehen
der Halogenchemie auf die Sprünge half, immer ein offenes Ohr für mich hatte und mich mit Ideen
und Modellen unterstütze.
Danke – es war eine sehr schöne Zeit hier am Institut!!!
Zu guter Letzt auch ein Dankeschön an William Sturges, der die in Brasilien genommenen Luft-
proben so schnell analysierte, dass mir die Daten noch vor Abgabetermin zur Verfügung standen!

11 Erklärung
Ich erkläre, dass ich diese Arbeit selbständig und nur mit den angegebenen Hilfsmitteln angefertigt
habe und dass alle Stellen, die dem Wortlaut oder dem Sinne nach anderen Werken entnommen sind,
durch Angabe der Quellen als Entlehnung kenntlich gemacht worden sind.
Heidelberg, den 09. Juni 2005








