Embed Size (px)
Citation preview

1975 H. Quust und A . GeNPri 929 Liebigs Ann. Chem. 1975, 929-938
Heterocyclische Ylide, IV 1)
Decarboxylierung von 1-Methylchinolinium-2-carboxylat in Abwesenheit elektrophiler Reagenzien Helmut Quast *) und Andrcis Gellkri" * )
Institut fur Organische Chemie der Universitiit. D-8700 Wurzburg, Am Hubland
Eingegangen am 23. April 1974
Die Decarboxylierung von 1-Methylchinolinium-2-carboxylat in aprotischen Losungsmitteln oder in 2-Propanol bei Abwesenheit von elektrophilen Reagenzien lieferte ein Produkt- gemisch, dessen Hauptkomponente 14,14a-Dihydro-5-methylbenz[5,6]indolizino[l,~-~]- chinolinium-6-carboxylat (5) ist. Der Bildungsmechanismus von 5 wird diskutiert.
Heterocyclic Ylides, IW). - Decarboxylation of 1-Methylquinolinium-2-carboxylate in the Absence of Electrophilic Reagents The decarboxylation of 1 -methylquinolinium-2-carboxylate in aprotic solvents or in 2-propanol in the absence of electrophiles yields a mixture of products, whose major component has been shown to be 14,14a-dihydro-5-methylbenz[5,6]indolizino[l,2-c]quinolinium-6-carboxylate (5). The mechanism of the formation of 5 is discussed.
In vorangegangenen Mitteilungenl) konnten wir zeigen, da13 bei der Decarboxylie- rung der Betaine 1 in aprotischen Losungsmitteln primar die Ylide 2 entstehen, die durch Elektrophile Em abgefangen werden konnen.
Eo G C O F iu' (I) @/:Q - Q-
I I I c H, c H, CH3 1 2 3
Heterocycl i sche R e s t e be i a: 2-Pyr idyl , b: 2-Chinolyl, c: I -1sochinoly1, d: 3-Isochinolyl
Eo = Diazoniumionen, e lek t rophi le Diazoverbindungen o d e r Azide, Benzaldehyd, H'
Wahrend die leichte Decarboxylierung heterocyclischer a-Carbonsauren und die Struktur der entstehenden Grundkorper vielfach belegt ist 2.3), gibt es nur wenige Arbeiten, in denen Decarboxylierungsprodukte 3 (E= H) von quaternierten 2-Pyridin-
*) Korrespondenz bitte an diesen Autor richten. * *) Gegenwartige Anschrift: Zentralforschungsinstitut der Ungarischen Akademie der
Wissenschaften, Budapest 11, Ungarn. 1) I.-111. Mitteilung: 13) H. Quust und E. Frunke/?feid, Angew. Chem. 77, 680, 876 (1965);
Angew. Chem., Int. Ed. Engl. 4, 691 (1965). -- Ib) H . Quust und E. Schmitt, Liebigs Ann. Chem. 732, 43 (1970). - Ic) H. Quust und E. Schmitt, Liebigs Ann. Chem. 732, 64 (1970).
2) J . A . Zoltewicz, C . L. Smith und J . D . Meyer, Tetrahedron 24, 2269 (1968). 3) Zusammenfassungen: B. R . Brown, Quart. Rev. Chem. SOC. 5, 131 (1951); G. S. Punde,
J . Sci. Ind. Res. 26, 393 (1967). [C. A. 68, 21 333e (1968)l; L. W. Clurk in The Chemistry of Carboxylic Acids and Esters (S. Putai), S. 589, Interscience Publ., New York 1969.

930 H. Quast und A. Gelle'ri 1975
carbonsauren (la.H@)4-6) oder des Betains 1 b aus saurem Milieulb) isoliert wurden. Noch unbekannt ist das Verhalten der Ylide 2 in protischem Medium geringer Aciditat oder unter aprotischen Bedingungen ohne Zusatz eines Elektrophils.
Entsprechend der thermischen Empfindlichkeit waDriger oder Bthanolischer Losungen der Betdine l a , b7) entstehen z. B. beim Erwarmen von l a unter ,,tiefgreifender Zersetzung" 8)
,,dunkle Losungen"9). Selbst in einer kinetischen Studie der Decarboxylierung von Homarin (la) wurde nicht nach der Struktur der Decarboxylierungsprodukte gefragtlo). Dabei erscheint diese Frage nicht uninteressant, da Homarin in zahlreichen Invertebraten 10,11) sowie als Metabolit von 2-[(Hydroxyimino)1nethyl]-l-methylpyridiniumjodid (2-PAM) in Ratten12) auftritt und seine biochemische Funktion in Invertebraten13) mit der leichten Decarboxylie- rung in Zusammenhang gebracht wurdelo). Beim Erwarmen von l b in Athanol erhalt man rote Losungenl4). Noch leichter zersetzt sich die N-Athylverbindung unter Bildung ,,dunkel gefarbter Produkte" 14).
Die vorliegende Untersuchung sollte am Beispiel von 1 b klaren, welches Schicksal die Ylide 2 in Abwesenheit von Elektrophilen erleiden.
Nach orientierenden Versuchen Ib) mit 1 b entsteht bei der Decarboxylierung in aprotischen Losungsmitteln ein Gemisch tieffarbiger, zum groaten Teil intensiv fluoreszierender Substanzen, das mit zugesetztem Diazoniumsalz kein Ylidabfangpro- dukt 3b mehr lieferte. Erhitzte man 1 b in trockenem Acetonitril, in Nitromethan, in Dimethylsulfoxid oder in 1,2-Dichloriithan, so lieBen sich mindestens 5 Zersetzungs- produkte dunnschichtchrornatographisch nachweisen. Uberraschenderweise entstand bei der Thermolyse in siedendem trockenem 2-Propanol das gleiche Produktgemisch wie in Acetonitril. Ein N-Methylchinoliniumsalz 3b (E = H) konnte nicht nachgewie- sen werden, auch nicht beim Versuch in 2-Propanol.
Die Thermolyse in 2-Propanol besitzt praparative Vorteile, da das orange, orange- gelb fluoreszierende Hauptprodukt A (Ausbeute 24 y ; ; Schmp. 202 -203 "C) auskristal- lisiert. Durch Chromatographie der Mutterlauge wurde eine Verbindung B als metallisch blauglanzende Kristalle vom Schmp. 215°C isoliert. AuBerdem konnten noch rasch wandernde, hellblau fluoreszierende Zonen abgetrennt werden, die sich als Folgeprodukte von A herausstellten (s. unten).
4) A . Kirpal, Monatsh. Chem. 22, 361 (1901). 5 ) G. Goldschmidt und 0. HlinigscAmidt, Monatsh. Chem. 24, 703 (1903). 6 ) K . U'. R u f f s , R. K. Hone und W. G.Phillips, J. Amer. Chem. SOC. 91, 6115 (1969). 7 ) E. S. Could, Mechanismus und Struktur in der organischen Chemie, 1 . Aufl., S. 413,
8) A. Huntzsch, Ber. Deut. Chem. Ges. 19, 31 (1886). 9) E. M . Kosower und J . W. farron, J . Org. Chem. 26, 1318 (1961).
10) P . Huake und J. Mantecon, J. Amer. Chem. SOC. 86, 5230 (1964). 11) E. L. Gasfeiger, P . C. ffaake und J . A. Gergen, Ann. N. Y . Acad. Sci. 90, 622 (1960);
Zusarnmenfassung bei S. Brodzicki, Kosmos (Warschau) Ser. A 16, 431 (1967), [C. A. 68, 18617v (1968)J.
12) J. Enander, A. Sundwall und B. Sorho, Biochem. Pharmacol. 11, 377 (1962) [C. A. 57, 7841 h (1962)l.
13) Vgl. R . A . Levy, Comp. Biochem. Physiol. 23, 631 (1967) [C. A. 68, 37 181 g (1968)l. 14) W. H . Mills und F. M. Humer, J. Chem. SOC. 121, 2008 (1922). 15) Synthese und Redoxverhalten des 4-Dikations: D . Scheutzow, Dissertation Univ. Wurz-
burg 1966.
Verlag Chemie, Weinheim 1962.
1975 Heterocyclische Ylide, IV 93 1
Zur Struktur von A
Ylids 2b oder urn eine der hoheren Oxidationsstufen von 415).
Bei A handelt es sich nicht urn das hochst oxidationsempfindliche Dimere 4 des
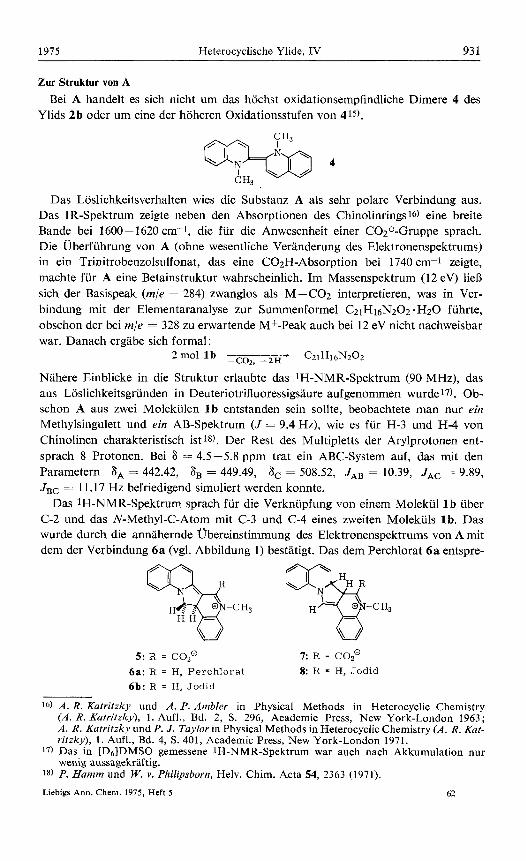
Das Loslichkeitsverhalten wies die Substanz A als sehr polare Verbindung aus. Das IR-Spektrum zeigte neben den Absorptionen des Chinolinrings 16) eine breite Bande bei 1600-1620 cm-1, die fur die Anwesenheit einer C02e-Gruppe sprach. Die Uberfuhrung von A (ohne wesentliche Veranderung des Elektronenspektrums) in ein Trinitrobenzolsulfonat, das eine COzH-Absorption bei 1740 cm-1 zeigte, machte fur A eine Betainstruktur wahrscheinlich. Im Massenspektrum (12 eV) lieB sich der Basispeak (m/e = 284) zwanglos als M-CO2 interpretieren, was in Ver- bindung mit der Elementaranalyse zur Summenformel C21H16N202. H20 fuhrte, obschon der bei m/e = 328 zu erwartende M+-Peak auch bei 12 eV nicht nachweisbar war. Danach ergabe sich formal:
Nahere Einblicke in die Struktur erlaubte das 1H-NMR-Spektrum (90 MHz), das aus Loslichkeitsgrunden in Deuteriotrifluoressigsaure aufgenommen wurde17). Ob- schon A aus zwei Molekulen 1 b entstanden sein sollte, beobachtete man nur ein Methylsingulett und ein AB-Spektrum ( J = 9.4 Hz), wie es fur H-3 und H-4 von Chinolinen charakteristisch ist 18). Der Rest des Multipletts der Arylprotonen ent- sprach 8 Protonen. Bei 8 = 4.5-5.8 ppm trat ein ABC-System auf, das mit den Parametern S A = 442.42, 8, = 449.49, SC = 508.52, JAB = 10.39, JAc = 9.89, JBc = 11.17 Hz befriedigend simuliert werden konnte.
Das 1H-NMR-Spektrum sprach fur die Verknupfung von einem Molekul 1 b uber C-2 und das N-Methyl-C-Atom mit C-3 und C-4 eines zweiten Molekuls l b . Das wurde durch die annahernde Ubereinstimmung des Elektronenspektrums von A mit dem der Verbindung 6a (vgl. Abbildung 1) bestatigt. Das dem Perchlorat 6a entspre-
2mol lb -COz, - 2 H + CziH16Nz02
% % H T \ I @N-CH3 H - 0 -CH,
- H H - \ / \ /
5: R = CO,' I: R = COzo 8: R = H, Jodid 6a: R = H, Perchlorat
6b: R = H, Jodid
16) A. R . Katritzky und A. P. Ambler in Physical Methods in Heterocyclic Chemistry (A. R. Katritzky), 1 . Aufl., Bd. 2, S. 296, Academic Press, New York-London 1963; A. R. Katritzky und P. J. Taylor in Physical Methods in Heterocyclic Chemistry (A. R. Kat- ritzky), 1 . Aufl., Bd. 4, S. 401, Academic Press, New York-London 1971.
17) Das in [D6]DMSO gemessene 1H-NMR-Spektrum war auch nach Akkumulation nur wenig aussagekraf tig.
18) P. Hamm und W. v. Philipsborn, Helv. Chim. Acta 54, 2363 (1971).
Liebigs Ann. Chem. 1975, Heft 5 62
932 H. Quast und A. Gelliri 1975
chende Jodid 6b hatte Krohnke 19) bei der Reaktion von N-Methylchinoliniumjodid mit KOH in Methanol in geringer Menge erhalten und dafur das Benz[5,6]indolizino- [1,2-c]chinolin-Grundgerust durch oxidativen Abbau eines substituierten Analogen gesichert. Seinerzeit konnte aber nicht zwischen den Isomeren 6b und 8 experimentell entschieden werden. Die Kopplungskonstanten des ABC-Systems im 1H-NMR- Spektrum von A sind aber nur mit der Struktur 5, nicht jedoch mit 7 vereinbar. Damit ist die Struktur 5 fur A gesichert und zugleich Krohnkes Formulierung des ,,roten Vor- produkts" 6b bewiesen.
333 LOO 500 nm , I 1 - 7 - 200 250
15
4 0
3 5
3 0
2 5
I
50000 ltoooo 30000 cm-' 20000
Abbildung 1. Vergleich der Elektronenspektren von Substanz A (entsprechend 5) und 6a in Methanol
Das Spektrum von 6a ist urn 0.5 log-Einheiten nach unten verschoben.
-1
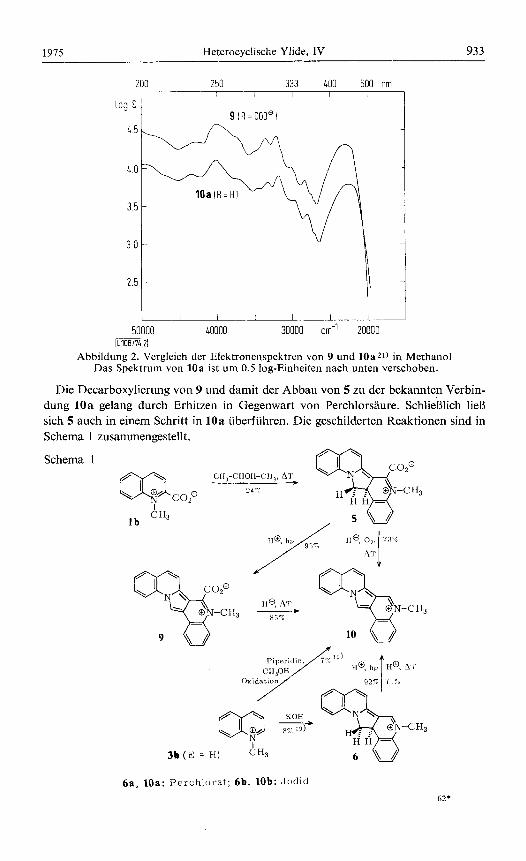
Wie der Grundkorper 6 ist 5 in Losung lichtempfindlich. Eine Photooxidation des 6 entsprechenden N-Benzyl-14-phenylkations erwahnten bereits Krohnke und Mit- arbeiterls). Uns gelang jetzt die praparative Umwandlung von 6a in 10a durch Luft- oxidation von 6a in Trifluoressigsaure oder durch Bestrahlen mit Licht von Wellen- Iangen oberhalb 400 nm in Anwesenheit von Essigsaure. Ganz analog verhielt sich 5. Wahrend durch diffuses Tageslicht oder durch Bestrahlung in neutralem Medium nur ein schwer trennbares Substanzgemisch entstand, erhielt man nach Einwirkung von Licht von Wellenlangen oberhalb 400 nm in schwach saurem Milieu ein einheit- liches Produkt (9). Die Struktur 9 des Photooxidationsproduktes wurde durch Ana- lyse, IR- und Massenspektrum 20) sowie den Vergleich des Elektronenspektrums mit dem von 10a gesichert (Abbildung 2). Die Verbindung 10b war bereits aus N- Methylchinoliniumjodid mit Piperidin in Methanol in Anwesenheit von o-iVitrobenz- aldehyd als Oxidationsmittel erhalten worden 19).
19) F. Krohnke, H. Dickhauser und 1. Vogt, Liebigs Ann. Chem. 644, 93 (1961). 20) Aus Loslichkeitsgrunden konnte kein 1H-NMR-Spektrum erhalten werden. 21) Elektronenspektrum yon l o b : G. Niederdellmann und F. Krohnke, Liebigs Ann. Chern.
688, 196 (1965).
1975 Heterocyclische Ylide, IV 93 3
log &
15
4 0
3 5
3 0
2 5
200 250 333 400 500 nm -7 I I I 1
9 i R = C O O Q l
I
I I I I I I
50000 40000 30000 cm-' 20000 1110817411
Abbildung 2. Vergleich der Elektronenspektren von 9 und 1Oazl) in Methanol Das Spektrum von 10a ist urn 0.5 log-Einheiten nach unten verschoben.
Die Decarboxylierung von 9 und damit der Abbau von 5 zu der bekannten Verbin- dung 10a gelang durch Erhitzen in Gegenwart von Perchlorsaure. SchlieRlich lieR sich 5 auch in einern Schritt in 10a iiberfiihren. Die geschilderten Reaktionen sind in Schema 1 zusammengestellt.
6a, 10a: Perchlorat; 6b, lob: .Todid h2*
934 H. Quast und A . Gelliri 1975
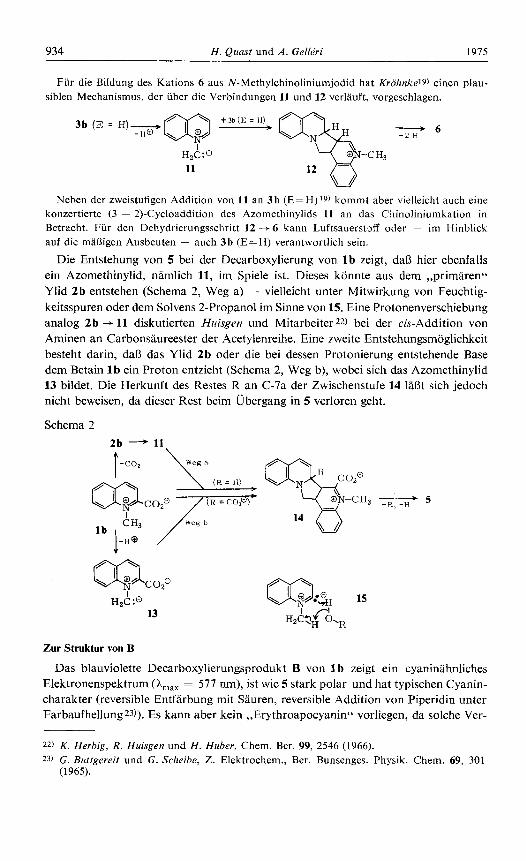
Fur die Bildung des Kations 6 aus N-Methylchinoliniumjodid hat Krohnkel9) einen plau- siblen Mechanisrnus, der uber die Verbindungen 11 und 12 verlauft, vorgeschlagen.
Neben der zweistufigen Addition von 11 an 3 b (E=H)J9) kommt aber vielleicht auch eine konzertierte (3 + 2)-Cycloaddition des Azomethinylids 11 an das Chinoliniurnkation in Betracht. Fur den Dehydrierungsschritt 12 --f 6 kann Luftsauerstoff oder ~ irn Hinblick auf die maRigen Ausbeuten - auch 3 b (E=H) verantwortlich sein.
Die Entstehung von 5 bei der Decarboxylierung von l b zeigt, daB hier ebenfalls ein Azomethinylid, namlich 11, im Spiele ist. Dieses konnte aus dem ,,primairen'< Ylid 2b entstehen (Schema 2, Weg a) - vielleicht unter Mitwirkung von Feuchtig- keitsspuren oder dem Solvens 2-Propanol im Sinne von 15. Eine Protonenverschiebung analog 2b + 11 diskutierten Huisgen und Mitarbeiter22) bei der cis-Addition von Aminen an Carbonsaureester der Acetylenreihe. Eine zweite Entstehungsmoglichkeit besteht darin, da8 das Ylid 2b oder die bei dessen Protonierung entstehende Base dem Betain 1 b ein Proton entzieht (Schema 2, Weg b), wobei sich das Azomethinylid 13 bildet. Die Herkunft des Restes R an C-7a der Zwischenstufe 14 1aBt sich jedoch nicht beweisen, da dieser Rest beim Ubergang in 5 verloren geht.
Schema 2 2b -+ 11
pJJ \ Q' cozo 15 I
H2C
13 H2&fJgA,R
Zur Struktur von B
Das blauviolette Decarboxylierungsprodukt B von 1 b zeigt ein cyaninahnliches Elektronenspektrum (A,,, = 577 nm), ist wie 5 stark polar und hat typischen Cyanin- charakter (reversible Entfarbung mit Sauren, reversible Addition von Piperidin unter Farbaufhellung23)). Es kann aber kein ,,Erythroapocyanin" vorliegen, da solche Ver-
22) K. Herbig, R . Huisgen und H. Huber, Chern. Ber. 99, 2546 (1966). 23) G. Buttgereit und G. Scheibe, 2. Elektrochem., Ber. Bunsenges. Physik. Chem. 69, 301
(1 965).

1975 Heterocyclische Ylide, I V 935
bindungen (z. B. 16) wesentlich kiirzerwellig absorbieren. Aus dem gleichen Grunde ist es unwahrscheinlich, daB B sich von einem Benzologen des Farbstoffs 17 ableitet. Eine Strukturaufklarung von B gelang bisher nicht.
&€I3 J' I
CH3 J o
16"): 1 mBy = 514 n m ( log& = 4.53) 1724': h in Methanol in Methanol
= 496 n m ( log& = 4.04)
Wie die beschriebenen Ergebnisse zeigen, lost die Decarboxylierung von 1 b in Abwesenheit zusatzlicher elektrophiler Reagenzien komplexe Reaktionen aus, deren wichtigste die uberraschende Bildung eines Azomethinylids (11 und/oder 13) und dessen Addition an unverandertes Betain l b ist. Es ist zu vermuten, daB unter ver- gleichbaren Bedingungen ahnliche Reaktionen auch bei der Decarboxylierung der anderen Betaine 1 im Spiele sind.
Wir danken Frau H. Heinze fur die Aufnahme der IH-NMR-Spektren und Herrn Dip1.- Chem. N . Pelz fur die Messung der Massenspektren. Der Stiftung Volkswagenwerk, die das NMR-Spektrometer HFX-90 (Fa. Bruker-Physik AG) und das Massenspektrometer SM 1 -BH (Fa. Varian-MAT) zur Verfugung stellte, sind wir sehr zu Dank verpflichtet. Fur groBzugige finanzielle Unterstutzung danken wir der Deutschen Forschungsgerneinschaft. A. GellCri hat besonders der Alexander-von-Hurnboldt-Stiftung fur ein einjahriges Forschungsstipendium (1971/1972) zu danken.
Experimenteller Teil Die Schmelzpunkts wurden nach Kofler bestimmt (Apparat der Fa. Reichert). - Auf-
genommen wurden die IR-Spektren mit dem Beckman-Gerat IR-10 (Eichung mit Polystyrol), die UV-Spektren mit einem Gerat Cary 17, die 90-MHz-1H-NMR-Spektren rnit dem Gerat HFX-90 der Fa. Bruker-Physik AG und die 60-MHz-1H-NMR-Spektren mit dem Varian- Gerat T 60 [(CH&Si als innerer Standard, 6 = 0 ppm]. Zur Spektrenakkumulation wurde ein Mittelwertrechner Nicolet 1074 verwendet. Die Parameter des ABC-Systems im 1H-NMR- Spektrum von 5 wurden mit Hilfe des Programms LAOCOON 111 auf einer Electrologica EL X8 optimiert. Zur Registrierung der Massenspektren diente das Varian-MAT-Gerat SM 1-BH rnit Datanorm; die Spektren wurden synchron analog und digital registriert. - Die Elementaranalysen wurden im Mikroanalytischen Laboratorium I. Beetz sowie im Ana- lytischen Laboratorium des Instituts fur Organische Chemie der Universitat Wurzburg angefertigt. - Acetonitril wurde wie beschriebenzs) gereinigt und vor der Verwendung iiber wenig P205 destilliert. 2-Propanol wurde durch fraktionierende Destillation getrocknet.
Ausgangs- und Vergleichssubstanzen
I-Methylchinolinium-2-carboxylat (1 b). - Es wurde nach den Angaben bei Lit. 1 C) dar- gestellt.
24) F. KrZihnke und K . F. Gross, Chem. Ber. 92, 22 (1959); H . Dickhauser, Dissertation Univ.
2s) If. Quast und S . Hiinig, Chem. Ber. 99, 2017 (1966). GieRen 1960, dort S. 60.

936 H. Quust und A. Gelliri 1975
I-Methylchinoliniumperchlorat26). - Man erhalt es aus I-Methylchinolinium(methylsu1fat) und waRriger NaC104-Losung als farblose Nadeln vom Schmp. 117.5 - 118.5"C (aus Athanol). - IH-NMR (CFsCOzH, 60 MHz): 8 = 4.82 (s; 3H), 7.95---8.65 (m; 5H), 9.08-9.35ppm (m; 2H).
14,14a-Dihydro-5-methylbenzi5,6!'indolizino~1,2-cichinoliniurnjodid (6 b). ~ Es wurde nach Krohnkelg) dargestellt.
14,I4a-Dihydro-5-methylbenz[5,6Jindolizitiu~l,2-r~chinoliniutr~perchloruf (6a) 27). - Wegen der Saureempfindlichkeit des Kations und ahnlicher Loslichkeit von Jodid und Perchlorat konnte dieses nicht aus dem Jodid erhalten werden. Die Vorschrift'g) fur das Jodid 6b wurde dahermodifiziert: 1.51 g (6.2 mmol) 1-Methylchinoliniumperchlorat in 24 ml heiBem Methanol wurden mit 0.75 ml 2 N methanol. KOH versetzt und 15 min unter RuckfluR erhitzt. Nach Zugabe von weiteren 0.82 ml 2 N methanol. KOH hielt man noch 1.5 h unter RiickfluR. Man lief3 abkuhlen, saugte das Rohprodukt ab und verruhrte es dreimal2 h mit je 10 ml Acetonitril. Der orangerote, kristalline Ruckstand wurde in 10 ml Dimethylformamid gelost, an 30 g A1203 (neutral, Aktiv.-Stufe 111) chromatographiert und die orangerote Fraktion mit Chloro- form/Methanol (9: 1) eluiert. Nach Einengen des Eluats auf ca. 5 ml erhielt man 71 mg (6 %) 6a, die aus 3 ml Dimethylformamid/H20 umkristallisiert wurden. Rote Kristalle vom Schmp. oberhalb 300°C. - IR (KBr): 1642, 1610, 1598, 1558, 1532 (C=C, C=N), 1070-1110cm-1 ((2104); bis auf die Absorption des Anions identisch mit dem des Jodids 6b. - UV (Methanol: s. Abb. 1): A,,, (log E ) = 472 (4.41), 353 (3.89). 311 (4.03), 256 (Schulter, 4.19), 232 nm (4.46); bis auf den Bereich unterhalb 250 nm identisch mit dem des Jodids 6b.
C20H17ClN204 (384.8) Ber. CI 9.21 N 7.27 Gef. CI 9.38 N 7.33
5-Methylbenr[5,6Jindolizino[1,2-cjchinoliniuniperchlorut (lOa) a) Aus dem entsprechenden Jodidlg) in heiBem Dimethylformamid/Wasser und wenig
70proz. Perchlorsaure erhielt man 1Oa als gelbe Kristalle vom Schmp. oberhalb 350°C (aus Dimethyl formamid/ Wasser).
C20H15CIN204 (382.8) Ber. C 62.77 H 3.95 CI 9.26 N 7.32 Gef. C 62.13 H 3.94 CI 9.32 N 7.29
b) Durch Photooxidufion von 6a: 200 mg (0.52 mmol) 6a in 70 ml Dimethylformamid/ Eisessig/Athanol (2:4: 1 ) wurden in einem offenen Kolben aus GWV-Glas28) unter Ruhren und Wasserkuhlung mit einer Osram Ultra-Vitalux-Lampe (300 Watt) 20 h belichtct. Nach Eindampfen und Behandeln des Ruckstandes rnit Athanol erhielt man 183 mg (92%), nach Umkristallisieren aus Dimethylformamid/Wasser (6: 4) 153 mg (77 %) 10a als gelbe Kristalle vom Schmp. oberhalb 350°C. - Laut IR-Spektrum identisch mit dem nach a) gewonnenem Produkt.
c) Durch Luftoxidation von 6a: 166 mg (0.43 mmol) 6a wurden in 16 ml Trifluoressigsiiure 8 h unter RuckfluR erhitzt. Die Losung wurde i. Vak. eingedampft und der Ruckstand mit Methanol behandelt. Erhalten wurden 124 mg (75 %), die, aus Dimethylformamid/Wasser umkristallisiert, 117 mg (71 %) 10a ergaben. Laut IR-Spektrum identisch mit dem nach a) dargestellten Produkt.
26) Ohne nahere Angaben erwlhnt von R . 5. Moodie, K. SchoJield und M. J. Williurnson, Chem. Ind. (London) 1963, 1283; R . Epple und Th. Forsrer, Z. Elektrochem., Ber. Bunsen- ges. Physik. Chem. 58, 783 (1954).
27) Inzwischen fanden wir eine wesentlich ergiebigere Darstellung dieser und analoger Ver- bindungen: H. Quasf und A . Gellgri Liebigs Ann. Chem. 1975, 939, nachstehend.
28) Fur die Uberlassung von GWV-Glasrohr danken wir Herrn Prof. E. A. Kuerner von Gustorf, Max-Planck-Institut fur Kohlenforschung, Muhlheim (Ruhr). Zur Lichtdurch- Iissigkeit siehe J. Streith, J. P. Luttringer und M. Nastusi, J. Org. Chem. 36, 2962 (1971).

1975 Heterocyclische Ylide, IV 931
Decarboxylierung von 1-Methylchinolinium-2-carboxylat (lb) 14,14u-Dihydro-5-1nethylbe1iz~5,6]indolizino[l,2-c~chinolinium-6-cnrboxylat-monohydrat (5)
a) 936 mg (5 mmol) l b wurden in 10 ml trockenem Acetonitril unter Riihren und RiickfluD 0.5 h erhitzt. Nach 15 h bei 20°C wurde abgesaugt. Man erhielt 224 mg rotviolette Kristalle vom Schmp. 183-185°C. Bei der Chromatographie an 180 g Kieselgel (0.2-0.5 mm) mit Methanol wurden die beiden ersten, hellblau fluoreszierenden und die langsam nachlaufende violettc Zone verworfen. Die orange, orangegelb fluoreszierende Hauptfraktion wurde i . Vak. auf 3 ml eingeengt und rnit Ather versetzt. Ausb. 32 mg (4%) 5 als hellrote Kristalle vom Schmp. 190-191°C. Laut IR- und UV-Spektrum identisch mit dem nach c) dargestelltem Produkt.
b) 5.62 g (30 mmol) l b wurden in 250 ml trockenem 1,2-Dichlorathan 0.5 h unter Stick- stoff und RuckfluR erhitzt. Die rote Losung wurde i. Vak. auf 40 ml eingeengt, mit 500 ml heiRem Methanol behandelt und heil3 filtriert. Die ungelosten Kristalle wurden rnit heiI3em Methanol gewaschen. Chromatographie des Filtrats an 800 g Kieselgel (0.2-0.5 mm) rnit Methanol ergab 12 mg (0.2 %) 5 (IR-Spektrum).
c) 3.75 g (20 mmol) fein gepulvertes l b wurden in 38 ml trockenem 2-Propanol unter Ruhren und Stickstoff 12-15 min zum Sieden erhitzt. Wahrend 15 h wurde auf -20°C abgekiihlt. Man erhielt 671 mg (19%) 5 als hellrote Kristalle vom Schmp. 201-202°C. Chromatographie der Mutterlauge an 260 g Kieselgel (0.2-0.5 mm) rnit Methanol ergab 4 Fraktionen: gelb (0.1 Liter), rot (0.2 Liter), orangerot (0.55 Liter) und blauviolett (2 Liter). Nach Eindampfen der orangeroten 3. Fraktion und Behandeln des Riickstandes rnit 15 ml Methanol erhielt man weitere 177 mg (5 %) 5 vom Schmp. 196°C. Umkristallisieren aus 70 ml Athanol ergab 623 mg (18%) hellrote Kristalle vom Schmp. 202-203°C. - IR (KBr): 2920,2845 (CH3), 1620-1600 (breit), 1595, 1562, 1552, 1530 cm-1 (C020, Chinolinringl6)). - UV (Methanol: s. Abb. 1): A,,, (log E) = 478 (4.32) 352 (3.82), 312 (3.98), 267 (Schulter, 4.10), 234.5 nm (4.40). - 1H-NMR (90 MHz; [D6]DMSO; 64 Durchkufe): 8 = 3.48 (NCHj), 4.2-5.0, 5.4-5.8 (m), 7.2-8.4 (m), 8.49 ppm (A-Teil eines AB-m, J- 9.5 Hz). I H - N M R ( ~ O M H Z ; C F ~ C O ~ D ) : S = 3.63 (s;3H),4.56-5.20,5.51-5.82(ABC-m),7.18-8.64 (m), 7.92, 8.54 ppm (AB, J = 9.4 Hz). - MS (12 eV, Tiegeltemp. 165"C, Temp. der Ionen- quelle 220°C): rnje (%) = 298 (3), 284 (100; M - COz), 283 (83), 268 (4), 44 (5, COz).
C21H16Nz02 .H20 (346.4) Ber. C 72.82 H 5.24 N 8.09 Gef. C 72.54 H 4.82 N 7.88 Gef. C 72.45 H 5.74 N 7.78
Die blauviolette 4. Fraktion wurde i. Vak. eingedampft und die metallisch blauglanzenden Kristalle aus Methanol/Ather umkristallisiert. Ausb. 208 mg; Schmp. 215°C. - UV (Metha- nol): A,,, : 577, 540 (Schulter), 302, 242, 227 nm. - Gef. C 65.64, 66.25; H 5.88, 5.98; N 7.37.
2,4,6-Trinitrobenzolsu(fonat von 5 : 410 mg (1.18 mmol) 5 in 30 ml Eisessig wurden mit 300 mg (1.19 mmol) Trinitrobenzolsulfonsaure, gelost in 2 ml Eisessig, und danach mit 15 ml Athanol versetzt. Nach 15 h bei -20°C erhielt man 565 mg (77%) Derivat, das, aus 100 ml Eisessig umkristallisiert, als rote Kristalle vom Schmp. oberhalb 300°C vorlag. - IR (KBr): 1740 cm-1 (C02H). - UV (Methanol): A,,, (log E) = 477 (4.59), 350 (Schulter, 3.92), 313 (4.13), 260 (Schulter, 4.39), 233 nm (4.63).
C ~ ~ H J ~ N S O ~ ~ S (621.5) Ber. N 11.28 S 5.16 Gef. N 11.55 S 5.15
Reaktionen von 5 Photooxidation zu 5-Methylbenz[5,6]indolizino[1,2-c]chinolinium-6-cnrboxylat (9). - 433 mg
(1.25 mmol) 5 in 90 ml Dimethylformamid/Eisessig/Athanol (2:4: 1) wurden in einem offenen Kolben a m GWV-Glas28) unter Riihren und Wasserkuhlung mit einer Osram Ultra-Vitalux-

938 H. Quast und A . Gelliri 1975
Lampe (300 Watt) 30 h bestrahlt. Dann wurde i. Vak. bei 40°C auf 25 ml eingeengt, mit 10 ml Wasser versetzt und erneut eingeengt. Es schieden sich 41 1 mg (95 %) gelbe Kristalle vom Schmp. 169-172°C ab. Nach Umkristallisieren aus 40 ml Eisessig/Athanol (2: 1) lagen 344 mg (80%) 9 vom Schmp. 174--175°C vor. - I R (KBr): 1635, 1593, 1547, 1522 cm-1 (COz0, Chinolinringls)). - UV (Methanol; s. Abb. 2): A,,, (log E) = 434 (4.31), 364 (Schul- ter, 3.63), 352 (3.83), 330 (4.01), 310.5 (4.40), 297.5 (4.38), 266 (Schulter, 4.41), 249.5 (4.57), 234 nm (4.33). - MS (70 eV, Tiegeltemp. 2OO0C, Temp. der Ionenquelle 220°C): m/e (%) =
298 (6; M - CO), 283 (3; M - CO - CH3), 268 (3); doppelt geladene Ionen zwischen m/e = 110-150 und 44 (100%; CO2).
C Z I H ~ ~ N Z O ~ . H ~ O (344.4) Ber. C 73.24 H 4.68 N 8.13 Gef. C 73.46 H 4.69 N 8.04
Oxidation und Decarboxylierung zu 5-Methylbenzl5,6]indolizino[I ,2-clchinoliniumperchlorat
a) Die Losung von 110 mg (0.32 mmol) 5 in 5 ml Trifluoressigsaure wurden 8 h bei Raum- temp. geriihrt, 10 min unter RiickfluB erhitzt und dann i. Vak. eingedampft. Der Ruckstand wurde in 10 ml Athanol gelost und n i t 0.5 ml 70proz. Perchlorsaure versetzt. Man erhielt 90 mg (73 %) 10a. Umkristallisieren aus Dimethylformamid/Wasser (6:4) ergab 81 mg (66%) verfilzte, gelbe Nadeln vom Schmp. oberhalb 360°C. Laut I R - und UV-Spektrum identisch mit authent. 10a. - IR (KBr): 1635, 1612, 1556, 1531 (Chinolinring16)), 1110-1075 cm-1 (Clod). - UV (Methanol; s. Abb. 2): Amax (log E) = 441 (4.31), 355 (3.91), 333 (4.08), 312 (4.41), 299 (4.34), 288 (4.25), 268 (4.37), 248.5 (4.61), 231 nm (4.43). - 1H-NMR (90 MHz; CFJCOZD): 8 = 4.91 (s; 3H), 7.7-8.9 (m), 9.21 (d, J = 9 Hz), 10.14 ppm (s; 1 H).
C ~ O H ~ ~ C I N ~ O ~ (382.8) Ber. C1 9.26 N 7.32 Gef. (39.19 N 7.37
(10 a)
b) Das Salz 10a entsteht auch durch Decarboxylierung von 9, wenn man dessen Losung in Dimethylformamid/Wasser (6: 4) bei Gegenwart von wenig Perchlorsaure 1 min zum Sieden erhitzt; Ausb. 85 ”/,. Identifizierung durch IR-Spektrenvergleich mit authent. 10a.
[108/74]







![Heterocyclische Siebenring-Verbindungen, XXX [1] Synthese ...zfn.mpdl.mpg.de/data/Reihe_B/42/ZNB-1987-42b-0217.pdf · -1, im 'H-NMR-Spektrum das Singulett der O CH3-Gruppe bei ö](https://img.pdfslide.org/doc/110x75/5e1de407443159751c398534/heterocyclische-siebenring-verbindungen-xxx-1-synthese-zfnmpdlmpgdedatareiheb42znb-1987-42b-0217pdf.jpg)