Embed Size (px)
Citation preview

Vollständig und teilweise fluorierte Triarylborane
als LEWIS-Säure-Katalysatoren: Synthese und Anwendungen
in der H‒H- und Si‒H-Bindungsaktivierung
vorgelegt von
Master of Science
Jens Mohr
geb. in Otterndorf
von der Fakultät II – Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
Dr. rer. nat.
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. Andreas Grohmann
Gutachter: Prof. Dr. Martin Oestreich
Gutachter: Prof. Dr. Jan Paradies
Tag der wissenschaftlichen Aussprache: 16. Juni 2016
Berlin 2016


Die vorliegende Dissertation wurde am Institut für Chemie der Technischen Universität
Berlin in der Zeit von November 2012 bis April 2016 unter der Anleitung von Prof. Dr.
Martin Oestreich angefertigt.
Prof. Dr. Martin Oestreich danke ich für das entgegengebrachte Vertrauen und die
exzellente Betreuung, die ein freies und kreatives Arbeiten ermöglichte.
Prof. Dr. Jan Paradies danke ich für die Übernahme des Zweitgutachtens und Prof. Dr.
Andreas Grohmann für die Übernahme des Prüfungsvorsitzes.
Den analytischen Abteilungen der TU Berlin gebührt Dank für ihre Hilfe: Dr. Sebastian
Kemper und Samantha Voges von der NMR-Abteilung, Dr. Maria Schlangen-Ahl und
Marc Griffel für massenspektrometrische Charakterisierungen sowie Dr. Elisabeth Irran
und Paula Nixdorf für röntgenographische Charakterisierungen. Ebenso gilt mein Dank
dem Bunkerteam für die alltäglichen Dinge.
Für die konstruktive Zusammenarbeit möchte ich mich bei allen ehemaligen und
aktuellen Mitgliedern des Arbeitskreises OESTREICH und des Arbeitskreises TEICHERT
bedanken. Einige haben zudem auf besondere Weise zu dieser Arbeit beigetragen. Für
die direkte Zusammenarbeit bei verschiedenen Projekten danke ich zunächst Dr.
Kristine Müther, Dr. Mustafa Durmaz, Digvijay Porwal und Dr. Indranil Chatterjee. Banjo
Single-Liertz danke ich für die Durchführung orientierender Experimente zur
Hydrazonreduktion. Dr. Andreas Weickgenannt gebührt Dank für die Einführung in die
Welt des AK OE, auch wenn dies weit vor Beginn dieser Arbeit geschah. Für die
hilfreichsten Diskussionen zeichneten Dr. Toni „naiß“ Metsänen (Kiitos!), Dr. Thomas
„Schrifiröfer“ Fallon (Thanks!) und Dr. Antoine „la barbe française“ Simonneau (Merci!)
verantwortlich. Für die immens wichtige Bereitstellung der Hydrieranlage und die stete
Hilfsbereitschaft danke ich Dr. Johannes „Happy hydrogenating!“ Teichert mit seinen
Teichboys, Felix „H zwei!“ Pape und Niklas „H zwei!“ Thiel. Bei Sebastian Keeß und
Lars Süße („Team ß“) möchte ich mich für das Korrekturlesen bedanken. Carolin
„Doppelmett“ Fopp gilt ein spezieller Dank, nicht nur für ihre Rolle als Nummerngirl.
Alexander „Was ist mit denen?“ Hensel sei für das gemeinsame Bestreiten eines
prägenden Lebensabschnitts gedankt, Stephanie „vier Getränke“ Krombach für ihr
offenes Ohr und die Hilfe bei bürokratischen Hürden. Meinen Labor-
abschnittsgefährtinnen Dr. Julia Hermeke und Susanne Bähr danke ich für eine
angenehme Laboratmosphäre und allerlei Blödsinn.

Für fachfremdes Interesse und Motivation gilt mein Dank Sandra Kola, Dr. Till Proeger,
Henrik Sadatzki, Sina Rose, Dorit Mohr, Dirk Mohr und vor allem Birgit Mohr und Klaus
Mohr.

Teile dieser Arbeit wurden bereits veröffentlicht:
[1] „Silylium Ion Promoted Reduction of Imines with Hydrosilanes”,
K. Müther, J. Mohr, M. Oestreich,
Organometallics 2013, 32, 6643–6646.
[2] „Tris(5,6,7,8-tetrafluoronaphthalen-2-yl)borane, a Partially Fluorinated Boron
Lewis Acid with Fluorination Distal to the Boron Atom”,
J. Mohr, M. Durmaz, E. Irran, M. Oestreich,
Organometallics 2014, 33, 1108–1111.
[3] „B(C6F5)3-Catalyzed Hydrogenation of Oxime Ethers without Cleavage of the
N–O Bond”,
J. Mohr, M. Oestreich,
Angew. Chem. 2014, 126, 13494–13497; Angew. Chem. Int. Ed. 2014, 53,
13278–13281.
[4] „Extending the Scope of the B(C6F5)3-Catalyzed C=N Bond Reduction:
Hydrogenation of Oxime Ethers and Hydrazones”,
J. Mohr, D. Porwal, I. Chatterjee, M. Oestreich,
Chem. Eur. J. 2015, 21, 17583–17586.

ZUSAMMENFASSUNG
Die vorliegende Dissertation widmet sich der Entwicklung übergangsmetallfreier
Reduktionsmethoden durch die Verwendung von Bor-LEWIS-Säuren und der Darstellung
neuartiger Bor-LEWIS-Säure-Katalysatoren.
Die chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen lässt sich durch
übergangsmetallkatalysierte Hydrierungsreaktionen nicht verlässlich erreichen, da diese
im Regelfall mit der Spaltung der N–O-Bindung einhergehen und daher Amine erhalten
werden. Durch den Einsatz der Bor-LEWIS-Säure B(C6F5)3 als Katalysator wurde diese
Überreduktion umgangen. Sterisch anspruchsvolle Substituenten am Sauerstoffatom
(tert-Butyl oder Triisopropyl) wurden benötigt, um die Oximether in die entsprechenden
geschützten Hydroxylamine zu überführen. Während repräsentative Ketoximether
bereits bei Raumtemperatur in Toluol unter einem H2-Druck von 100 bar mit einer
Katalysatorbeladung von 5 Mol-% umgesetzt wurden, erforderte die Reduktion der
weniger basischen Aldoximether einen höhere Katalysatorbeladung, erhöhte
Temperaturen und einen Lösungsmittelwechsel zu 1,4-Dioxan. Durch anschließende
Abspaltung der Triisopropylsilylschutzgruppe lassen sich ungeschützte Hydroxylamine
erhalten. Strukturell verwandte Hydrazone wurden unter ähnlichen Bedingungen zu den
entsprechenden Hydrazinen hydriert. Hierbei eigneten sich phthaloylgeschützte
Substrate zur nachfolgenden Freisetzung ungeschützter Hydrazine. Bei der C=N-
Reduktion von Hydrazonen zeigte sich derselbe Reaktivitätsunterschied zwischen
keton- und aldehydabgeleiteten Substraten wie im Falle der Oximether. Im Zuge der
Entwicklung hauptgruppenelementkatalysierter C=N-Reduktionen wurde zudem eine
silyliumionvermittelte Iminhydrosilylierungsmethode ausgearbeitet.
Ein weiterer Schwerpunkt der vorliegenden Dissertation liegt auf der Darstellung von
Derivaten der eingesetzten Bor-LEWIS-Säure B(C6F5)3. Obschon eine Reihe von
Boranen mit unterschiedlichem Fluorierungsgrad der Phenylsubstituenten bekannt sind,
war bisher nicht untersucht, ob eine Fluorierung der nicht direkt an das Boratom
gebundenen Arylsubstituenten eine ausreichende LEWIS-Säure-Stärke bewirkt. Es
wurde ein partiell fluoriertes β-naphthylbasiertes Boran synthetisiert und charakterisiert,
welches zur Klärung dieser Frage geeignet schien. Durch GUTMANN–BECKETT-Analyse
wurde eine LEWIS-Acidität von 98% relativ zu B(C6F5)3 festgestellt und in typischen Si–
H-Bindungsaktivierungskatalysen erwies sich das neuartige Boran als ähnlich effizient
wie B(C6F5)3. Ein Beitrag zur Entwicklung chiraler Bor-LEWIS-Säuren gelang durch die
Darstellung eines Boranvorläufers zur Synthese eines chiralen B(C6F5)3-Derivats,
welches chirale Substituenten anstatt der Fluoratome in para-Position aufweist.
Hierdurch wird im Gegensatz zu bisher bekannten chiralen B(C6F5)3-Abkömmlingen, bei
denen mindestens einer der perfluorierten Arylsubstituenten durch einen chiralen
Substituenten ersetzt ist, ein weitgehender Erhalt der LEWIS-Acidität erwartet.

ABSTRACT
This dissertation is devoted to the development of transition metal-free reduction
methods by employing boron LEWIS acids and to the synthesis of novel boron LEWIS acid
catalysts.
The chemoselective C=N reduction of oxime ethers to hydroxylamines cannot be reliably
achieved by transition metal-catalyzed hydrogenations. These are usually accompanied
by N–O bond cleavage, resulting in the formation of amines. This overreduction was
circumvented by employing the boron LEWIS acid B(C6F5)3 as catalyst. Sterically
demanding substituents at the oxygen atom (tert-butyl or triisopropylsilyl) were required
to convert oxime ethers into the corresponding protected hydroxylamines.
Representative ketoxime ethers already reacted at room temperature in toluene under
100 bar dihydrogen pressure with a catalyst loading of 5 mol% whereas the reduction of
less basic aldoxime ethers required higher catalyst loading, higher temperatures, and
the use of 1,4-dioxane as solvent. Unprotected hydroxylamines can be obtained by
subsequent removal of the triisopropylsilyl protecting group. Structurally related
hydrazones were hydrogenated under similar conditions to yield the corresponding
hydrazines. Phthaloyl-protected substrates were suitable to subsequently liberate
unprotected hydrazines. In the C=N reduction of hydrazones, the same reactivity
difference of ketone- and aldehyde-derived substrates as in the case of oxime ethers
was observed. As part of the development of main group element-catalyzed C=N
reductions, a silylium ion-promoted imine hydrosilylation method was established.
This dissertation also focuses on the preparation of derivatives of the boron LEWIS acid
B(C6F5)3. Although a number of boranes with various degrees of fluorination of the
phenyl substituents is known, the effect of fluorination of aryl substituents not directly
attached to the boron atom on Lewis acidity had not been explored. To address this
question, a partially fluorinated β-naphthyl-based borane was synthesised and
characterised. GUTMANN–BECKETT analysis revealed a LEWIS acidity of 98% relative to
B(C6F5)3, and the novel borane showed similar activity to B(C6F5)3 in typical catalyses
involving Si–H bond activation. A contribution to the development of chiral boron LEWIS
acids was accomplished by the preparation of a borane precursor for the synthesis of a
chiral B(C6F5)3 congener bearing chiral substituents instead of fluorine atoms in the para
positions. In contrast to other chiral B(C6F5)3 congeners where at least one
perfluorinated aryl group is replaced by the chiral substituent, the LEWIS acidity is
expected to be mostly preserved.


INHALTSVERZEICHNIS


INHALTSVERZEICHNIS
THEORETISCHER TEIL
1 EINLEITUNG 1
1.1 Mechanistische Betrachtung von C=X-Reduktionen durch katalytisch
generierte Borhydride
3
1.2 C=X-Reduktionen durch katalytisch generierte Borhydride 8
1.2.1 Reduktionen von C=O- und C=S-Bindungen 8
1.2.2 Reduktionen von C=N- und C≡N-Bindungen 13
1.2.3 Reduktionen von C=C- und C≡C-Bindungen 19
1.3 Problemstellung und Zielsetzung 24
1.3.1 Katalytische C=N-Reduktion von Oximen und Hydrazonen 24
1.3.2 Darstellung elektronenarmer Borane für katalytische Si–H- und H–H-
Bindungsaktivierungen
27
2 B(C6F5)3-KATALYSIERTE CHEMOSELEKTIVE C=N-REDUKTION VON
OXIMETHERN ZU HYDROXYLAMINEN
30
2.1 Versuche zur C=N-Reduktion von Oximethern durch
borankatalysierte Hydrosilylierung
30
2.2 C=N-Reduktion von Oximethern durch borankatalysierte Hydrierung 31
2.3 Entfernung der Silylschutzgruppe zur Freisetzung ungeschützter
Hydroxylamine
40
2.4 Fazit 41
3 B(C6F5)3-KATALYSIERTE C=N-REDUKTION VON HYDRAZONEN
ZU HYDRAZINEN
42
3.1 Versuche zur C=N-Reduktion von Hydrazonen durch
borankatalysierte Hydrosilylierung
42
3.2 C=N-Reduktion von Hydrazonen durch borankatalysierte Hydrierung 44
3.3 Entfernung der Phthaloylschutzgruppe 48
3.4 Fazit 49

4 EXKURS: SILYLIUMIONVERMITTELTE C=X-REDUKTIONEN MIT HYDROSILANEN 50
4.1 Silyliumionvermittelte C=N-Reduktion von Iminen mit Hydrosilanen 53
4.2 Fazit 58
5 SYNTHESE, LEWIS-ACIDITÄT UND KATALYTISCHE ANWENDUNGEN EINES
ELEKTRONENARMEN β-NAPHTHYLBASIERTEN BORANS
60
5.1 Synthese des Borans 108 60
5.2 Bestimmung der LEWIS-Acidität des Borans 108 65
5.3 Aktivität des Borans 108 in Reaktionen mit katalytischer Si–H-und H–H-
Bindungsaktivierung
67
5.4 Fazit 69
6 EINLEITENDE UNTERSUCHUNGEN ZUR SYNTHESE EINES B(C6F5)3-DERIVATS
MIT CHIRALEN SUBSTITUENTEN IN DEN PARA-POSITIONEN
71
6.1 Darstellung des chiralen Binaphthylbausteins (R)-261 71
6.2 Fazit 73
7 ZUSAMMENFASSUNG 75
7.1 Hauptgruppenelementkatalysierte C=N-Reduktionen 75
7.2 Darstellung elektronenarmer Borane 76
EXPERIMENTELLER TEIL
1 ALLGEMEINE ARBEITSWEISE 81
2 ALLGEMEINE ARBEITSVORSCHRIFTEN 87
2.1 Allgemeine Arbeitsvorschrift zur Darstellung O-alkylsubstituierter
Oxime (AAV 1)
87
2.2 Allgemeine Arbeitsvorschrift zur Darstellung ungeschützter
Oxime (AAV 2)
87

2.3 Allgemeine Arbeitsvorschrift zur Darstellung O-silylsubstituierter
Oxime (AAV 3)
87
2.4 Allgemeine Arbeitsvorschrift zu Hydrierungsreaktionen in
Toluol (AAV 4)
88
2.5 Allgemeine Arbeitsvorschrift zu Hydrierungsreaktionen in
1,4-Dioxan (AAV 5)
88
2.6 Allgemeine Arbeitsvorschrift zur silyliumionvermittelten Iminreduktion
mit Hydrosilanen (AAV 6)
88
3 BESCHREIBUNG DER EXPERIMENTE 90
3.1 Darstellung von Oximethern 90
3.2 Darstellung von Hydroxylaminen 113
3.3 Darstellung von Hydrazonen 128
3.4 Darstellung von Hydrazinen 130
3.5 Silyliumionvermittelte Iminreduktion mit Hydrosilanen 132
3.6 Darstellung von Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108) 133
3.7 Boran/Phosphinoxid-Addukte 138
3.8 Durch Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108) katalysierte
Reaktionen
142
3.9 Darstellung von Boranvorläufer (R)-261 145
ANHANG
A1 ABKÜRZUNGSVERZEICHNIS 159
A2 LITERATURVERZEICHNIS 163


THEORETISCHER TEIL


1 Einleitung 1
1 EINLEITUNG
Borhydride sind als stöchiometrisches Reduktionsmittel in der synthetischen Chemie
vielseitig einsetzbar.[1] Ihr wohl bedeutendster Vertreter, Natriumborhydrid (NaBH4), ein
Nebenprodukt der Kriegsforschung, wurde 1953 von SCHLESINGER und BROWN vorgestellt[2]
und fand rasch zahlreiche Anwendungen in Industrie und Forschung. Die Entwicklung von
Reduktionsmethoden durch katalytisch generierte Borhydride ist eng verknüpft mit den
Forschungsgebieten der Bor-LEWIS-Säuren[3] und der frustrierten LEWIS-Paare („frustrated
LEWIS pairs“ = FLPs).[4]
PIERS und Mitarbeiter zeigten 1996, dass das bereits mehr als 30 Jahre zuvor erstmals
synthetisierte[5] elektronenarme Triarylboran Tris(pentafluorphenyl)boran [B(C6F5)3 (1),
Abbildung 1.1, links] als übergangsmetallfreier LEWIS-Säure-Katalysator für die
Hydrosilylierung von Carbonylverbindungen geeignet ist.[6,7] STEPHAN und Mitarbeiter stellten
[1] P. Rittmeyer, U. Wietelmann in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH,
Weinheim, 2002, S. 103–131. [2]
H. I. Schlesinger, H. C. Brown, B. Abraham, A. C. Bond, N. Davidson, A. E. Finholt, J. R.
Gilbreath, H. Hoekstra, L. Horvitz, E. K. Hyde, J. J. Katz, J. Knight, R. A. Lad, D. L. Mayfield,
L. Rapp, D. M. Ritter, A. M. Schwartz, I. Sheft, L. D. Tuck, A. O. Walker J. Am. Chem. Soc.
1953, 75, 186–190. [3]
Übersichten zur Chemie elektronenarmer Borane: a) R. L. Melen, Chem. Commun. 2014, 50,
1161–1174; b) G. Erker, Dalton Trans. 2005, 1883–1890; c) W. E. Piers, Adv. Organomet.
Chem. 2004, 52, 1–76; d) W. E. Piers, G. J. Irvine, V. C. Williams, Eur. J. Inorg. Chem. 2000,
2131–2142; e) W. E. Piers, T. Chivers, Chem. Soc. Rev. 1997, 26, 345–354. [4]
Übersichten zur Chemie der FLPs: a) D. W. Stephan, J. Am. Chem. Soc. 2015, 137, 10018–
10032; b) D. W. Stephan, Acc. Chem. Res. 2015, 48, 306–316; c) D. W. Stephan, G. Erker,
Angew. Chem. Int. Ed. 2015, 54, 6400–6441; d) S. A. Weicker, D. W. Stephan, Bull. Chem.
Soc. Jpn. 2015, 88, 1003–1016; e) D. W. Stephan, G. Erker, Chem. Sci. 2014, 5, 2625–2641;
f) Topics in Current Chemistry, Vol. 332 (Hrsg.: G. Erker, D. W. Stephan), Springer, Berlin,
Heidelberg, 2013; g) Topics in Current Chemistry, Vol. 334 (Hrsg.: G. Erker, D. W. Stephan),
Springer, Berlin, Heidelberg, 2013; h) D. W. Stephan, G. Erker, Angew. Chem. Int. Ed. 2010,
49, 46–76; i) D. W. Stephan, Org. Biomol. Chem. 2008, 6, 1535–1539. [5]
a) A. G. Massey, A. J. Park, F. G. A. Stone, Proc. Chem. Soc. 1963, 212; b) A. G. Massey, A.
J. Park, J. Organomet. Chem. 1964, 2, 245–250; c) A. G. Massey, A. J. Park, J. Organomet.
Chem. 1966, 5, 218–225. [6]
D. J. Parks, W. E. Piers, J. Am. Chem. Soc. 1996, 118, 9440–9441. [7]
Vereinzelte Berichte über B(C6F5)3 (1) als LEWIS-Säure-Katalysator in nicht verwandten
Reaktionen waren bereits zuvor bekannt: a) K. Ishihara, N. Hananki, H. Yamamoto, Synlett
1993, 577–579; b) K. Ishihara, M. Funahashi, N. Hanaki, M. Miyata, H. Yamamoto, Synlett
1994, 963–964; c) K. Ishihara, N. Hanaki, H. Yamamoto, Synlett 1995, 721–722; d) K.
Ishihara, N. Hanaki, M. Funahashi, M. Miyata, H. Yamamoto, Bull. Chem. Soc. Jpn. 1995, 68,
1721–1730. Von größerer Bedeutung war B(C6F5)3 (1) bis dato lediglich als Co-Katalysator für
metallocenkatalysierte Polymerisationsreaktionen. Für frühe Beispiele siehe: e) X. Yang, C. L.
Stern, T. J. Marks, J. Am. Chem. Soc. 1991, 113, 3623–3625; f) X. Yang, C. L. Stern, T. J.

2 THEORETISCHER TEIL
ein Jahrzehnt später das intramolekulare LEWIS-Säure/Base-Paar 2 (Abbildung 1.1, rechts)
vor.[8] Die erste katalytische Anwendung dieses sogenannten frustrierten LEWIS-Paares, bei
dem die LEWIS-Säure- und LEWIS-Base-Komponenten aus sterischen Gründen kein LEWIS-
Säure/Base-Addukt ausbilden können,[9] stellte die Hydrierung von Iminen dar.[10]
Abbildung 1.1: Typische Vertreter elektronenarmer Borane (links) und frustrierter LEWIS-Paare
(rechts).
Seit den wegweisenden Arbeiten der Gruppen um PIERS und STEPHAN wurden eine Vielzahl
von Reaktionen gefunden, die die katalytische Aktivierung von Hydrosilanen oder
Diwasserstoff durch elektronenarme Borane wie B(C6F5)3 (1) oder FLPs wie 2 nutzen.[11] Von
besonderer Bedeutung für die vorliegende Arbeit sind dabei Reduktionen von C=X-
Bindungen (I→II, Schema 1.1), welche sowohl von Bor-LEWIS-Säuren (BR3) als auch von
LEWIS-Paaren mit Bor-LEWIS-Säure-Komponente katalysiert werden.[12] Als
stöchiometrisches Reduktionsmittel Y–H (III) werden typischerweise Hydrosilane (Y = R3Si)
oder Diwasserstoff (Y = H) eingesetzt, der eigentliche Reduktionsschritt erfolgt aber in allen
Marks, J. Am. Chem. Soc. 1994, 116, 10015–10031; g) M. Bochmann, S. J. Lancaster
Organometallics 1994, 13, 2235-2243; h) B. Temme, G. Erker, J. Karl, H. Luftmann, R.
Fröhlich, S. Kotila, Angew. Chem. Int. Ed. Engl. 1995, 34, 1755–1757. Eine Übersicht bietet: i)
E. Y.-X. Chen, T. J. Marks, Chem. Rev. 2000, 100, 1391–1434. [8]
G. C. Welch, R. R. S. Juan, J. D. Masuda, D. W. Stephan, Science 2006, 314, 1124–1126. [9]
Ironischerweise wurde über die allgemein als frühestes Beispiel eines FLPs angesehene
Mischung aus 2,6-Lutidin und BMe3 vor der Beschreibung von NaBH4 berichtet. Zudem ist
diese Entdeckung derselben Gruppe zuzuschreiben: H. C. Brown, H. I. Schlesinger, S. Z.
Cardon, J. Am. Chem. Soc. 1942, 64, 325–329. [10]
P. A. Chase, G. C. Welch, T. Jurca, D. W. Stephan, Angew. Chem. Int. Ed. 2007, 46, 8050–
8053. [11]
Übersichten: a) M. Oestreich, J. Hermeke, J. Mohr, Chem. Soc. Rev. 2015, 44, 2202–2220; b)
L. Shi, Y.-G. Zhou, ChemCatChem 2015, 7, 54–56; c) M. Alcarazo, Synlett 2014, 1519–1520;
d) X. Feng, H. Du, Tetrahedron Lett. 2014, 55, 6959–6964; e) L. J. Hounjet, D. W. Stephan,
Org. Process Res. Dev. 2014, 18, 385–391; f) J. Paradies, Angew. Chem. Int. Ed. 2014, 53,
3552–3557; g) J. Paradies, Synlett 2013, 777–780; h) D. W. Stephan, Org. Biomol. Chem.
2012, 10, 5740–5746; i) D. W. Stephan, Chem. Commun. 2010, 46, 8526–8533; j) D. W.
Stephan, Dalton Trans. 2009, 3129–3136. [12]
Für eine Übersicht zu hauptgruppenelementkatalysierten Reduktionen siehe: K. Revunova, G.
I. Nikonov, Dalton Trans. 2015, 44, 840–866.

1 Einleitung 3
Fällen aus einer katalytisch generierten Borhydridspezies. Im Folgenden sollen die zugrunde
liegenden Mechanismen vorgestellt und auf die Unterschiede zwischen Borankatalyse und
LEWIS-Paar-Katalyse eingegangen werden.
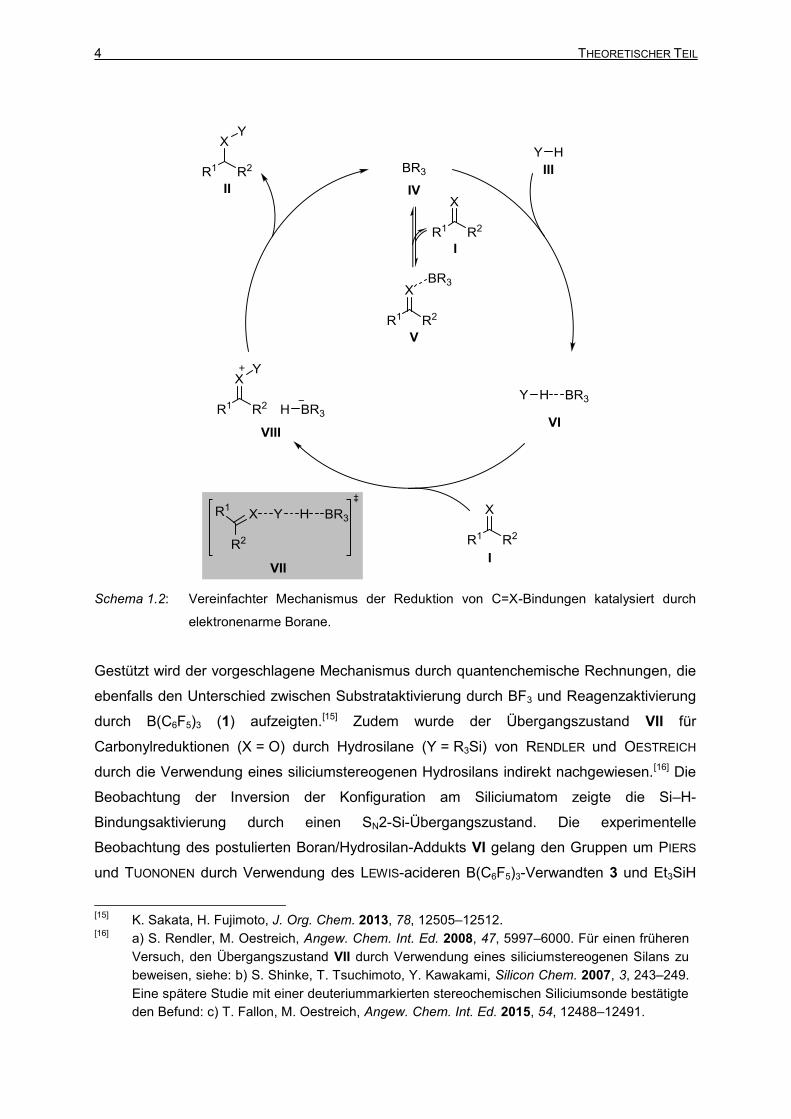
Schema 1.1: Reduktion von C=X-Bindungen katalysiert durch elektronenarme Borane oder FLPs.
1.1 Mechanistische Betrachtung von C=X-Reduktionen durch
katalytisch generierte Borhydride
Die Gruppe um PIERS beobachtete in ihren Arbeiten zur B(C6F5)3-katalysierten
Hydrosilylierung von Carbonylverbindungen (X = O, Y = R3Si, Schema 1.2),[6,13] dass eine
Erniedrigung der LEWIS-Basizität des Substrats I zu einer Erhöhung der
Reaktionsgeschwindigkeit zum Reduktionsprodukt II führt. Eine höhere
Substratkonzentration ging mit einer Verringerung der Reaktionsgeschwindigkeit einher.
Dies ließ die Autoren vermuten, dass eine klassische LEWIS-Säure-Aktivierung des
Substrats, wie beispielsweise für Carbonylhydrosilylierungen mit BF3 als LEWIS-Säure
angenommen,[14] nicht zur Produktbildung führt, sondern eine reversible „Sackgasse“ im
Katalysezyklus darstellt (I+IV⇌V). Stattdessen wurde ein Mechanismus vorgeschlagen, der
über eine Aktivierung des Hydrosilans III (Y = R3Si) durch η1-Koordination des Borans IV
verläuft. Die Bildung des Boran/Hydrosilan-Addukts VI erhöht die Elektrophilie des
Siliciumatoms, welches daraufhin von dem LEWIS-basischen Substrat I nukleophil
angegriffen wird. Im Übergangszustand VII wird die Si–H-Bindung unter Bildung des
Ionenpaares VIII gebrochen. Dieses besteht aus einem Silylcarboxoniumion und einem
Borhydrid. Der Reduktionsschritt zum Produkt II erfolgt schließlich durch einen
Hydridtransfer vom katalytisch generierten Borhydrid auf das elektrophile Kohlenstoffatom
des Carboxoniumions unter Zurückbildung des Borankatalysators IV.
[13] D. J. Parks, J. M. Blackwell, W. E. Piers, J. Org. Chem. 2000, 65, 3090–3098.
[14] a) M. P. Doyle, C. T. West, S. J. Donnelly, C. C. McOsker, J. Organomet. Chem. 1976, 117,
129–140; b) J. L. Fry, M. Orfanopoulos, M. G. Adlington, W. R. Dittman, Jr., S. B. Silverman, J.
Org. Chem. 1978, 43, 374–375.
4 THEORETISCHER TEIL
Schema 1.2: Vereinfachter Mechanismus der Reduktion von C=X-Bindungen katalysiert durch
elektronenarme Borane.
Gestützt wird der vorgeschlagene Mechanismus durch quantenchemische Rechnungen, die
ebenfalls den Unterschied zwischen Substrataktivierung durch BF3 und Reagenzaktivierung
durch B(C6F5)3 (1) aufzeigten.[15] Zudem wurde der Übergangszustand VII für
Carbonylreduktionen (X = O) durch Hydrosilane (Y = R3Si) von RENDLER und OESTREICH
durch die Verwendung eines siliciumstereogenen Hydrosilans indirekt nachgewiesen.[16] Die
Beobachtung der Inversion der Konfiguration am Siliciumatom zeigte die Si–H-
Bindungsaktivierung durch einen SN2-Si-Übergangszustand. Die experimentelle
Beobachtung des postulierten Boran/Hydrosilan-Addukts VI gelang den Gruppen um PIERS
und TUONONEN durch Verwendung des LEWIS-acideren B(C6F5)3-Verwandten 3 und Et3SiH
[15] K. Sakata, H. Fujimoto, J. Org. Chem. 2013, 78, 12505–12512.
[16] a) S. Rendler, M. Oestreich, Angew. Chem. Int. Ed. 2008, 47, 5997–6000. Für einen früheren
Versuch, den Übergangszustand VII durch Verwendung eines siliciumstereogenen Silans zu
beweisen, siehe: b) S. Shinke, T. Tsuchimoto, Y. Kawakami, Silicon Chem. 2007, 3, 243–249.
Eine spätere Studie mit einer deuteriummarkierten stereochemischen Siliciumsonde bestätigte
den Befund: c) T. Fallon, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54, 12488–12491.

1 Einleitung 5
(4, Schema 1.3).[17,18] Das Gleichgewicht der Adduktbildung wurde durch
temperaturabhängige NMR-Spektroskopiestudien verfolgt und das Addukt 5 zudem
röntgenographisch charakterisiert.
Schema 1.3: Adduktbildung zwischen dem elektronenarmen Boran 3 und dem Hydrosilan 4.
Bei der verwandten borankatalysierten Hydrierung (Y = H, Schema 1.2) ungesättigter
Substrate wird von einem der Hydrosilylierung ähnlichen Mechanismus ausgegangen.[19] Die
Produktbildung erfolgt hier durch einen Hydridübertrag von einem katalytisch generierten
Borhydrid auf das protonierte Substrat (Ionenpaar VIII→II). Ob der H–H-Bindungsaktivierung
durch das LEWIS-basische Substrat I und den LEWIS-aciden Borankatalysator IV im
Übergangszustand VII die Existenz eines Boran/Diwasserstoff-Addukts VI vorausgeht, ist
allerdings fragwürdig. Zwar ist das Boran 3 in der Lage, in Abwesenheit von LEWIS-Basen
Diwasserstoff zu aktivieren,[20] was eine zuvor erfolgte Adduktbildung impliziert.
Quantenchemische Rechnungen favorisieren für kooperative H–H-Bindungsaktivierungen
durch Bor-LEWIS-Säuren in Kombination mit LEWIS-Basen jedoch Modelle ohne
Präkoordination des Diwasserstoffs.[21] Es sollte zudem darauf hingewiesen werden, dass
[17] A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers, H. M. Tuononen, Nat. Chem.
2014, 6, 983–988. [18]
Ein entsprechendes Hydrosilanaddukt des Alans Al(C6F5)3 wurde ebenfalls charakterisiert: J.
Chen, E. Y.-X. Chen, Angew. Chem.Int. Ed. 2015, 54, 6842–6846. [19]
Am Beispiel der borankatalysierten Hydrierung von Iminen (X = NR): a) P. A. Chase, T. Jurca,
D. W. Stephan, Chem. Commun. 2008, 1701–1703; b) T. A. Rokob, A. Hamza, A. Stirling, I.
Pápai, J. Am. Chem. Soc. 2009, 131, 2029–2036. Für einen vergleichenden Artikel zur
Aktivierung von Si–H- und H–H-Bindungen durch elektronenarme Borane siehe: c) W. E.
Piers, A. J. V. Marwitz, L. G. Mercier, Inorg. Chem. 2011, 50, 12252–12262. [20]
a) A. Y. Houghton, V. A. Karttunen, W. E. Piers, H. M. Tuononen, Chem. Commun. 2014, 50,
1295–1298. Für weitere Beispiele von Diwasserstoffaktivierung durch elektronenarme Borane
ohne externe LEWIS-Base siehe: b) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen, M.
Parvez, J. Am. Chem. Soc. 2010, 132, 9604–9606; c) A. Y. Houghton, V. A. Karttunen, C. Fan,
W. E. Piers, H. M. Tuononen, J. Am. Chem. Soc. 2013, 135, 941–947. [21]
Für Beispiele siehe: Lit 19b; a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós, I. Pápai, Angew.
Chem. Int. Ed. 2008, 47, 2435–2438; b) Y. Guo, S. Li, Inorg. Chem. 2008, 47, 6212–6219; c)
T. A. Rokob, A. Hamza, I. Pápai, J. Am. Chem. Soc. 2009, 131, 10701–10710; d) A. Hamza,
A. Stirling, T. A. Rokob, I. Pápai, Int. J. Quantum Chem. 2009, 109, 2416–2425; e) B.

6 THEORETISCHER TEIL
die Möglichkeit einer autoinduziert katalytischen Reaktion besteht, bei der das reduzierte
Produkt II anstatt des Substrats I als LEWIS-Base-Komponente am H–H-
Bindungsaktivierungsschritt teilnimmt. Diese wurde von CHEN und KLANKERMAYER für die
B(C6F5)3-katalysierte Hydrierung von Iminen (X = NR) vorgeschlagen[22] und im Folgenden
durch quantenchemische Rechnungen bestätigt.[19b,23] Experimentelle Befunde zeigten, dass
ein solcher Reaktionsverlauf abhängig von der LEWIS-Säure-Stärke des Borankatalysators IV
ist.[24] Der autoinduziert katalytische Reaktionsweg entspricht der Katalyse durch LEWIS-
Paare, die im Folgenden vorgestellt wird.
Schema 1.4: Vereinfachter Mechanismus der Reduktion von C=X-Bindungen katalysiert durch
elektronenarme Borane in Kombination mit LEWIS-Basen (LB).
Schirmer, S. Grimme, Chem. Commun. 2010, 46, 7942–7944; f) S. Grimme, H. Kruse, L.
Goerigk, G. Erker, Angew. Chem. Int. Ed. 2010, 49, 1402–1405; g) L. L. Zeonjuk, N. Vankova,
A. Mavrandonakis, T. Heine, G.-V. Röschenthaler, J. Eicher, Chem. Eur. J. 2013, 19, 17413–
17424; h) T. A. Rokob, I. Bakó, A. Stirling, A. Hamza, I. Pápai, J. Am. Chem. Soc. 2013, 135,
4425–4437; i) M. Pu, T. Privalov, ChemPhysChem 2014, 15, 2936–2944; j) M. Pu, T. Privalov,
ChemPhysChem 2014, 15, 3714–3719. [22]
D. Chen, J. Klankermayer, Chem. Commun. 2008, 2130–2131. [23]
T. Privalov, Eur. J. Inorg. Chem. 2009, 2229–2237. [24]
a) S. Tussing, L. Greb, S. Tamke, B. Schirmer, C. Muhle-Goll, B. Luy, J. Paradies, Chem. Eur.
J. 2015, 21, 8056–8059; b) S. Tussing, K. Kaupmees, J. Paradies, Chem. Eur. J. 2016, 22,
7422–7426.

1 Einleitung 7
Die katalytische Reduktion ungesättigter Substrate I lässt sich nicht nur durch LEWIS-saure
Borane IV katalysieren (siehe Schema 1.2), sondern auch durch die Kombination derselben
mit LEWIS-Basen IX (Schema 1.4).[11] Die Wahl des (inter- oder intramolekularen) LEWIS-
Base-Partners IX ist dabei nicht beschränkt auf sterisch anspruchsvolle LEWIS-Basen, die mit
dem Boran IV ein FLP bilden, sondern schließt auch solche ein, die die Bildung eines LEWIS-
Säure/Base-Addukts X eingehen, sofern diese reversibel ist und genügend freier Katalysator
für die gewünschte Transformation zur Verfügung steht. Die Bindungsaktivierung der Y–H-
Bindung des Reduktionsmittels III erfolgt kooperativ durch die LEWIS-Säure IV und die
LEWIS-Base IX im Übergangszustand XI. Das entstehende Ionenpaar XII, bestehend aus
einem Borhydrid und einer protonierten (Y = H) bzw. silylierten (Y = R3Si) LEWIS-Base,
überträgt daraufhin Proton bzw. Silylgruppe (XII+I→XIII+IX) und Hydrid (XIII→II) auf das
Substrat I, wodurch das Reduktionsprodukt II gebildet und das Katalysatorsystem IV/IX
zurückerhalten wird. Der Erhalt eines Ionenpaares XII durch kooperative H–H-
Bindungsaktivierung einer LEWIS-Säure und einer LEWIS-Base wurde von STEPHAN gezeigt
(Schema 1.5).[8] Das FLP 6 bildet bei Behandlung mit Diwasserstoff bei Raumtemperatur das
Phosphoniumborat 2. Die Diwasserstoffaktivierung ist reversibel. Bei erhöhten Temperaturen
wird in Abwesenheit eines Substrats wieder Diwasserstoff frei.
Schema 1.5: Reversible Diwasserstoffaktivierung durch das FLP 6.
Der Einsatz einer zusätzlichen externen LEWIS-Base IX ermöglicht folglich die Reduktion von
Substraten I, deren eigene LEWIS-Basizität nicht ausreichend ist, um selbst als LEWIS-Base
in der kooperativen Y–H-Bindungsaktivierung (siehe Übergangszustand VII, Schema 1.2) zu
fungieren. Nachfolgend sollen die zahlreichen Anwendungen elektronenarmer Borane bzw.
Boran/LEWIS-Base-Systeme in Reduktionsreaktionen von C=X-Bindungen, die den
beschriebenen Mechanismen im wesentlichen folgen, vorgestellt und verglichen werden.

8 THEORETISCHER TEIL
1.2 C=X-Reduktionen durch katalytisch generierte Borhydride
1.2.1 Reduktion von C=O- und C=S-Bindungen
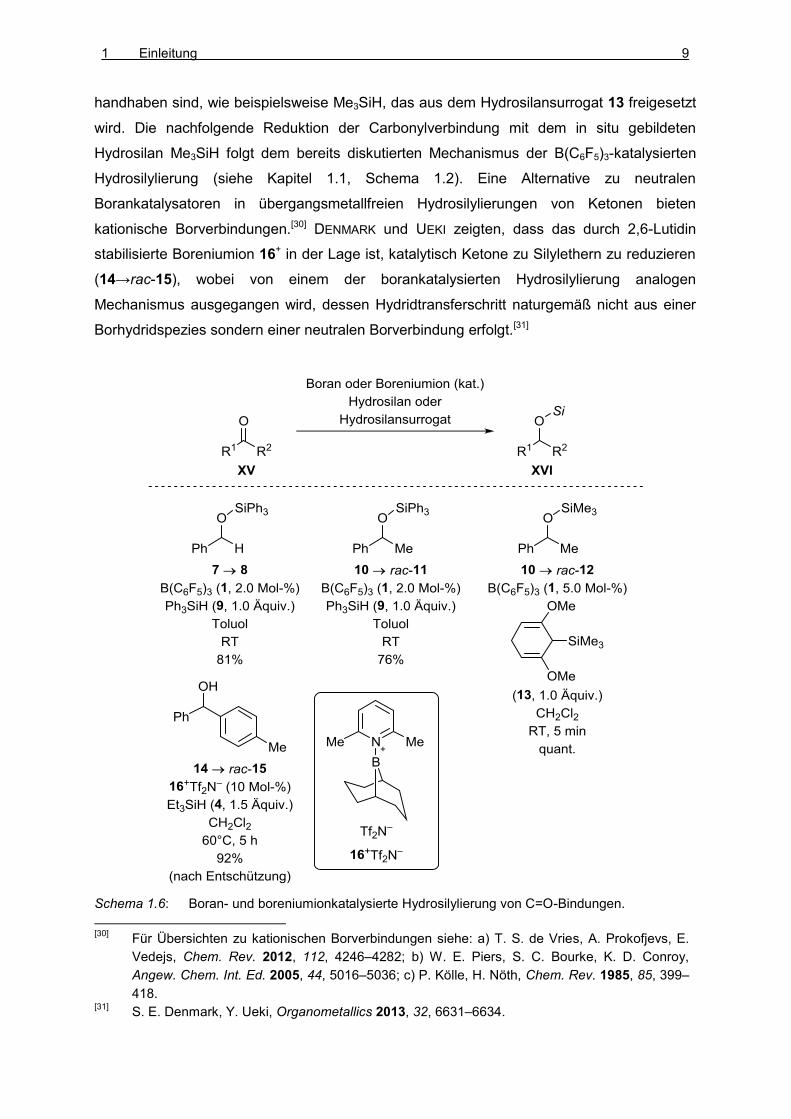
PARKS und PIERS zeigten zunächst, dass sich sowohl Aldehyde (7→8, Schema 1.6) als auch
Ketone (10→rac-11) durch Behandlung mit Ph3SiH (9) in Gegenwart katalytischer Mengen
B(C6F5)3 (1) unter milden Bedingungen zu den entsprechenden Silylethern reduzieren
lassen.[6,25,26,27] Eine verwandte Methode stellt die borankatalysierte Transferhydrosilylierung
von OESTREICH und Mitarbeitern dar (10→rac-12).[28,29] Aus einem mit einer Silylgruppe
versehenen Cyclohexa-1,4-dien wird das gewünschte Hydrosilan in situ unter Freisetzung
eines Äquivalents des entsprechenden Benzolderivats gebildet. Diese Vorgehensweise
bietet die Möglichkeit, Hydrosilane einzusetzen, die als Reinsubstanz flüchtig und schwer zu
[25]
Für ein diastereoselektives Beispiel einer B(C6F5)3-katalysierten Carbonylhydrosilylierung
siehe: a) M. K. Skjel, A. Y. Houghton, A. E. Kirby, D. J. Harrison, R. McDonald, L. Rosenberg,
Org. Lett. 2010, 12, 376–379. Für B(C6F5)3-katalysierte konjugierte Reduktionen von
Carbonylverbindungen mit Hydrosilanen siehe: b) J. M. Blackwell, D. J. Morrison, W. E. Piers,
Tetrahedron 2002, 58, 8247–8254; c) S. Chandrasekhar, G. Chandrashekar, M. S. Reddy, P.
Srihari, Org. Biomol. Chem. 2006, 4, 1650–1652; d) L. Greb, P. Oña-Burgos, A. Kubas, F. C.
Falk, F. Breher, K. Fink, J. Paradies, Dalton Trans. 2012, 41, 9056–9060; e) Y. Kim, S. Chang,
Angew. Chem. Int. Ed. 2016, 55, 218–222. Für die Reduktion von Estern zu Acetalen durch
B(C6F5)3-katalysierte Hydrosilylierung siehe: Lit. 6, Lit. 13. Für die chemoselektive Reduktion
von Carbonsäuren zu Aldehyden siehe: f) D. Bézier, S. Park, M. Brookhart, Org. Lett. 2013,
15, 496–499. [26]
Für Boran- und FLP-katalysierte Reduktionsmethoden von CO2 mit Hydrosilanen siehe: a) A.
Berkefeld, W. E. Piers, M. Parvez, J. Am. Chem. Soc. 2010, 132, 10660–10661; b) J. Chen, L.
Falivene, L. Caporaso, L. Cavallo, E. Y.-X. Chen, J. Am. Chem. Soc. 2016, 138, 5321–5333;
c) D. Mukherjee, D. F. Sauer, A. Zanardi, J. Okuda, Chem. Eur. J. 2016, 22, 7730–7733. [27]
Einige der nachfolgend vorgestellten Hydrosilylierungen und Hydrierungen lassen sich
ebenfalls durch Phosphoniumkationen katalysieren. Für Hydrosilylierungsreaktionen wird
ebenfalls von einer Hydrosilanaktivierung durch η1-Koordination des Katalysators
ausgegangen. Für Beispiele siehe: a) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky,
D. W. Stephan, J. Am. Chem. Soc. 2013, 135, 18308–18310; b) M. H. Holthausen, M. Mehta,
D. W. Stephan, Angew. Chem. Int. Ed. 2014, 53, 6538–6541 (Alken- und
Alkinhydrosilylierung); c) T. vom Stein, M. Peréz, R. Dobrovetsky, D. Winkelhaus, C. B.
Caputo, D. W. Stephan, Angew. Chem. Int. Ed. 2015, 54, 10178–10182 (Alkenhydrierung); d)
M. Pérez, Z.-W. Qu, C. B. Caputo, V. Podgorny, L. J. Hounjet, A. Hansen, R. Dobrovetsky, S.
Grimme, D. W. Stephan, Chem. Eur. J. 2015, 21, 6491–6500 (Keton-, Imin- und
Nitrilhydrosilylierung). Einen Überblick bietet: e) J. M. Bayne, D. W. Stephan, Chem. Soc. Rev.
2016, 45, 765–774. [28]
S. Keess, A. Simonneau, M. Oestreich, Organometallics 2015, 34, 790–799. [29]
Für eine Übersicht zu Transferhydrosilylierungen siehe: a) M. Oestreich, Angew. Chem. Int.
Ed. 2016, 55, 494–499. Für eine B(C6F5)3-katalysierte Carbonyltransferhydroaluminierung
siehe: S. Dagorne, I. Janowska, R. Welter, J. Zakrzewski, G. Jaouen, Organometallics 2004,
23, 4706–4710.
1 Einleitung 9
handhaben sind, wie beispielsweise Me3SiH, das aus dem Hydrosilansurrogat 13 freigesetzt
wird. Die nachfolgende Reduktion der Carbonylverbindung mit dem in situ gebildeten
Hydrosilan Me3SiH folgt dem bereits diskutierten Mechanismus der B(C6F5)3-katalysierten
Hydrosilylierung (siehe Kapitel 1.1, Schema 1.2). Eine Alternative zu neutralen
Borankatalysatoren in übergangsmetallfreien Hydrosilylierungen von Ketonen bieten
kationische Borverbindungen.[30] DENMARK und UEKI zeigten, dass das durch 2,6-Lutidin
stabilisierte Boreniumion 16+ in der Lage ist, katalytisch Ketone zu Silylethern zu reduzieren
(14→rac-15), wobei von einem der borankatalysierten Hydrosilylierung analogen
Mechanismus ausgegangen wird, dessen Hydridtransferschritt naturgemäß nicht aus einer
Borhydridspezies sondern einer neutralen Borverbindung erfolgt.[31]
Schema 1.6: Boran- und boreniumionkatalysierte Hydrosilylierung von C=O-Bindungen.
[30]
Für Übersichten zu kationischen Borverbindungen siehe: a) T. S. de Vries, A. Prokofjevs, E.
Vedejs, Chem. Rev. 2012, 112, 4246–4282; b) W. E. Piers, S. C. Bourke, K. D. Conroy,
Angew. Chem. Int. Ed. 2005, 44, 5016–5036; c) P. Kölle, H. Nöth, Chem. Rev. 1985, 85, 399–
418. [31]
S. E. Denmark, Y. Ueki, Organometallics 2013, 32, 6631–6634.
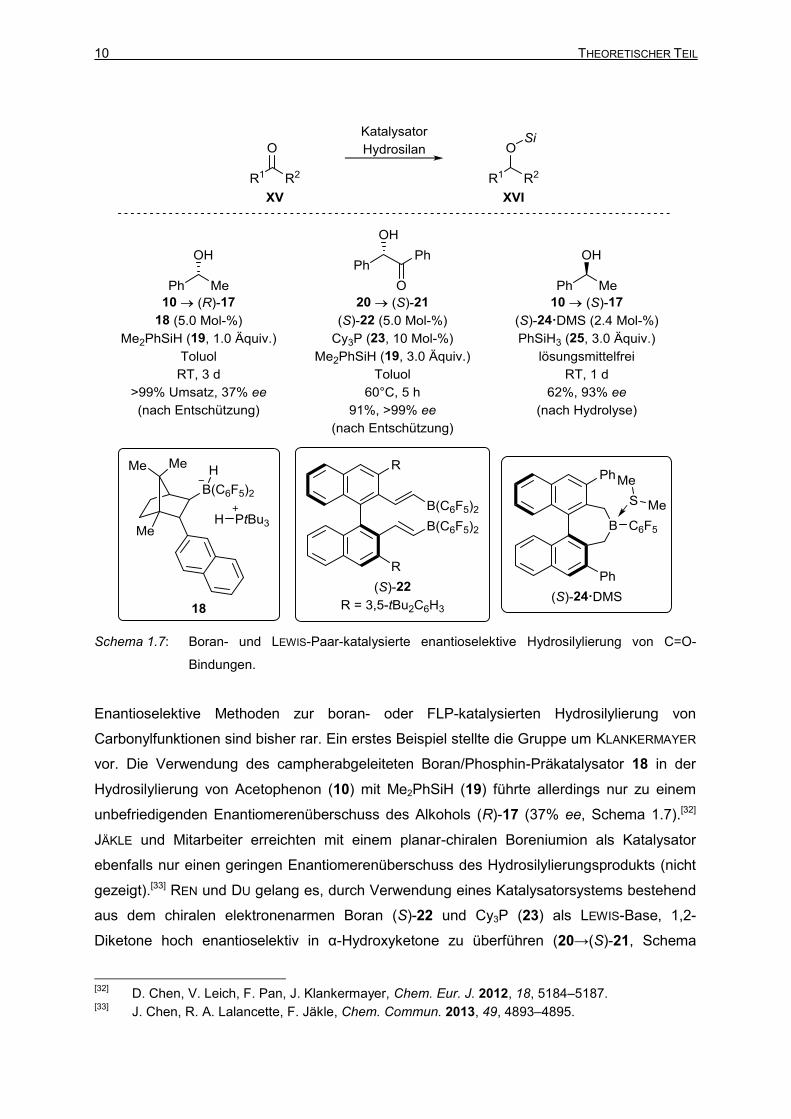
10 THEORETISCHER TEIL
Schema 1.7: Boran- und LEWIS-Paar-katalysierte enantioselektive Hydrosilylierung von C=O-
Bindungen.
Enantioselektive Methoden zur boran- oder FLP-katalysierten Hydrosilylierung von
Carbonylfunktionen sind bisher rar. Ein erstes Beispiel stellte die Gruppe um KLANKERMAYER
vor. Die Verwendung des campherabgeleiteten Boran/Phosphin-Präkatalysator 18 in der
Hydrosilylierung von Acetophenon (10) mit Me2PhSiH (19) führte allerdings nur zu einem
unbefriedigenden Enantiomerenüberschuss des Alkohols (R)-17 (37% ee, Schema 1.7).[32]
JÄKLE und Mitarbeiter erreichten mit einem planar-chiralen Boreniumion als Katalysator
ebenfalls nur einen geringen Enantiomerenüberschuss des Hydrosilylierungsprodukts (nicht
gezeigt).[33] REN und DU gelang es, durch Verwendung eines Katalysatorsystems bestehend
aus dem chiralen elektronenarmen Boran (S)-22 und Cy3P (23) als LEWIS-Base, 1,2-
Diketone hoch enantioselektiv in α-Hydroxyketone zu überführen (20→(S)-21, Schema
[32]
D. Chen, V. Leich, F. Pan, J. Klankermayer, Chem. Eur. J. 2012, 18, 5184–5187. [33]
J. Chen, R. A. Lalancette, F. Jäkle, Chem. Commun. 2013, 49, 4893–4895.

1 Einleitung 11
1.7).[34] Das Boran (S)-22 wird hierbei in situ durch Hydroborierung des entsprechenden
Diins mit HB(C6F5)2 (PIERS Boran) gebildet. Eine Verwendung des Borans (S)-22 als
alleiniger Katalysator für diese Reaktion zeigte nur geringen Umsatz und niedrige
Enantioselektivität. Eine enantioselektive Ketonhydrosilylierungsmethode katalysiert durch
ein elektronenarmes Boran ohne externe LEWIS-Base wurde von der Gruppe um OESTREICH
entwickelt.[35] Das chirale Boran (S)-24 weist lediglich eine elektronenziehende C6F5-Gruppe
am Boratom auf, ist aber dennoch in der Lage, die Hydrosilylierung von Acetophenon (10)
mit PhSiH3 (25) zu katalysieren und liefert zudem einen hohen Enantiomerenüberschuss des
nach Hydrolyse erhaltenen Alkohols (S)-17.
Analog zu C=O-Bindungen lassen sich auch C=S-Bindungen B(C6F5)3-katalysiert mit
Hydrosilanen reduzieren wie von ROSENBERG und Mitarbeitern gezeigt, wobei extrem
niedrige Katalysatorbeladungen ausreichten (26→27, Schema 1.8).[36] In Gegenwart eines
Ketons und eines Thioketons wird selektiv das Keton hydrosilyliert (nicht gezeigt).
Schema 1.8: Borankatalysierte Hydrosilylierung von C=S-Bindungen.
Die Verwendung von Diwasserstoff als Reduktionsmittel für die boran- oder LEWIS-Paar-
katalysierte Carbonylreduktion blieb lange ohne Erfolg. Frühe Versuche in donoratomfreien
Lösungsmitteln scheiterten an der Nukleophilie der entstandenen Alkoholprodukte
gegenüber des LEWIS-Säure-Katalysators, die in dessen Zersetzung resultierte.[37] Erst die
Durchführung der Reaktion in Ethern als Lösungsmitteln ermöglichte den Gruppen um
STEPHAN[38] und ASHLEY
[39] die B(C6F5)3-katalysierte übergangsmetallfreie Hydrierung[40] von
[34]
X. Ren, H. Du, J. Am. Chem. Soc. 2016, 138, 810–813. [35]
L. Süsse, J. Hermeke, M. Oestreich, J. Am. Chem. Soc. 2016, 138, 6940–6943. [36]
a) D. J. Harrison, R. McDonald, L. Rosenberg, Organometallics 2005, 24, 1398–1400; b) P. T.
K. Lee, M. K. Skjel, L. Rosenberg, Organometallics 2013, 32, 1575–1578. [37]
a) M. Lindqvist, N. Sarnela, V. Sumerin, K. Chernichenko, M. Leskelä, T. Repo, Dalton Trans.
2012, 41, 4310–4312; b) L. E. Longobardi, C. Tang, D. W. Stephan, Dalton Trans. 2014, 43,
15723–15726. [38]
T. Mahdi, D. W. Stephan, J. Am. Chem. Soc. 2014, 136, 15809–15812. [39]
D. J. Scott, M. J. Fuchter, A. E. Ashley, J. Am. Chem. Soc. 2014, 136, 15813–15816. [40]
Für übergangsmetallfreie basenkatalysierte Hydrierungen siehe: a) C. Walling, L. Bollyky, J.
Am. Chem. Soc. 1961, 83, 2968–2969; b) C. Walling, L. Bollyky, J. Am. Chem. Soc. 1964, 86,
3750–3752; c) A. Berkessel, T. J. S. Schubert, T. N. Müller, J. Am. Chem. Soc. 2002, 124,
8693–8698.

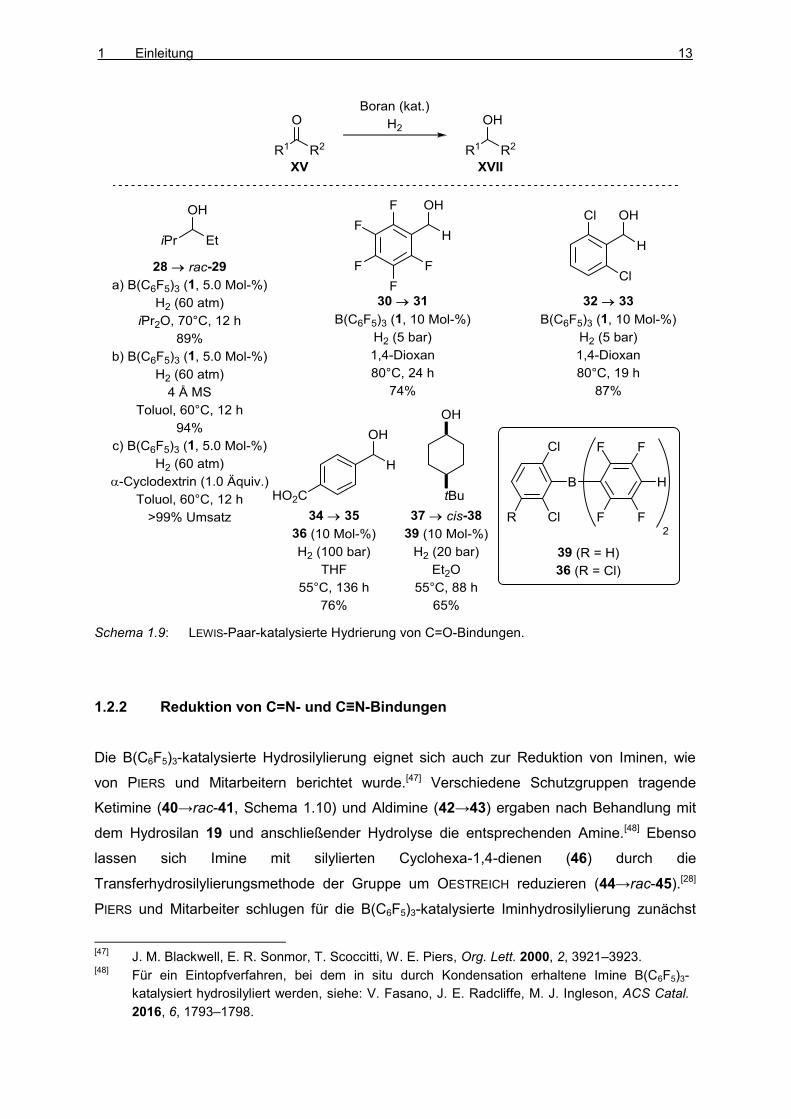
12 THEORETISCHER TEIL
Aldehyden (30→31 und 32→33, Schema 1.9) und Ketonen (28→rac-29, Methode a).[41] Das
Lösungsmittel, dessen LEWIS-Paar-Bildung mit dem Borankatalysator reversibel ist, nimmt in
diesen Fällen die Rolle der externen LEWIS-Base im H–H-Bindungsaktivierungsschritt ein.
Diese Reaktionen zeigten eine merkliche Wassertoleranz und können bei Erhöhung des
Diwasserstoffdrucks auch in ungetrockneten Lösungsmitteln durchgeführt werden.[42,43,44]
Denselben Vorteil bietet der Einsatz der von SOÓS und Mitarbeitern vorgestellten sterisch
anspruchsvolleren B(C6F5)3-verwandten Borane 36 und 39, die sogar eine freie
Carboxylgruppe (34→35) tolerieren und mit der Reduktion des Ketons 37 zum Alkohol cis-38
ein diastereoselektives Beispiel einer borankatalysierten Carbonylhydrierung
ermöglichten.[45] MAHDI und STEPHAN zeigten später, dass sich die B(C6F5)3-katalysierte
Hydrierungsreaktionen von Carbonylfunktionen auch in nichtkoordinierendem Toluol
durchführen lassen, wenn Molekularsieb (28→rac-29, Methode b) oder α-Cyclodextrin
(28→rac-29, Methode c) als externe LEWIS-Base hinzugegeben wird.[46] Eine in letzterem
Fall erhoffte Enantioselektivität wurde jedoch nicht beobachtet.
[41]
Für eine stöchiometrische Hydrierung von CO2 durch ein FLP siehe: A. E. Ashley, A. L.
Thompson, D. O'Hare, Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [42]
D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter, A. E. Ashley, ACS
Catal. 2015, 5, 5540–5544. [43]
Für die Verwendung von Katalysatorregeneratoren in boran- und FLP-katalysierten
Hydrierungen siehe: J. W. Thomson, J. A. Hatnean, J. J. Hastie, A. Pasternak, D. W. Stephan,
P. A. Chase, Org. Process Res. Dev. 2013, 17, 1287–1292. [44]
Für Untersuchungen von B(C6F5)3/Wasser-Addukten siehe: a) C. Bergquist, B. M.
Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner, G. Parkin, J. Am. Chem. Soc. 2000,
122, 10581–10590; b) T. Beringhelli, D. Maggioni, G. D'Alfonso, Organometallics 2001, 20,
4927–4938. [45]
Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján, T. Soós, ACS Catal. 2015, 5, 5366–
5372. [46]
T. Mahdi, D. W. Stephan, Angew. Chem. Int. Ed. 2015, 54, 8511–8514.
1 Einleitung 13
Schema 1.9: LEWIS-Paar-katalysierte Hydrierung von C=O-Bindungen.
1.2.2 Reduktion von C=N- und C≡N-Bindungen
Die B(C6F5)3-katalysierte Hydrosilylierung eignet sich auch zur Reduktion von Iminen, wie
von PIERS und Mitarbeitern berichtet wurde.[47] Verschiedene Schutzgruppen tragende
Ketimine (40→rac-41, Schema 1.10) und Aldimine (42→43) ergaben nach Behandlung mit
dem Hydrosilan 19 und anschließender Hydrolyse die entsprechenden Amine.[48] Ebenso
lassen sich Imine mit silylierten Cyclohexa-1,4-dienen (46) durch die
Transferhydrosilylierungsmethode der Gruppe um OESTREICH reduzieren (44→rac-45).[28]
PIERS und Mitarbeiter schlugen für die B(C6F5)3-katalysierte Iminhydrosilylierung zunächst
[47]
J. M. Blackwell, E. R. Sonmor, T. Scoccitti, W. E. Piers, Org. Lett. 2000, 2, 3921–3923. [48]
Für ein Eintopfverfahren, bei dem in situ durch Kondensation erhaltene Imine B(C6F5)3-
katalysiert hydrosilyliert werden, siehe: V. Fasano, J. E. Radcliffe, M. J. Ingleson, ACS Catal.
2016, 6, 1793–1798.

14 THEORETISCHER TEIL
einen analogen Mechanismus zu dem der entsprechenden Carbonylhydrosilylierung vor
(siehe Kapitel 1.1, Schema 1.2 mit Y = R3Si und X = NR bzw. X = O).[47] Mechanistische
Untersuchungen von OESTREICH und Mitarbeitern zeigten später, dass für enolisierbare
Imine weitere Reaktionswege möglich sind.[49] Der Hydridtransferschritt durch das gebildete
Borhydrid kann demzufolge auf verschiedene Iminiumionen erfolgen, was eine
enantioselektive Variante dieser Reaktion erschwert.
Schema 1.10: B(C6F5)3-katalysierte Hydrosilylierung von C=N-Bindungen.
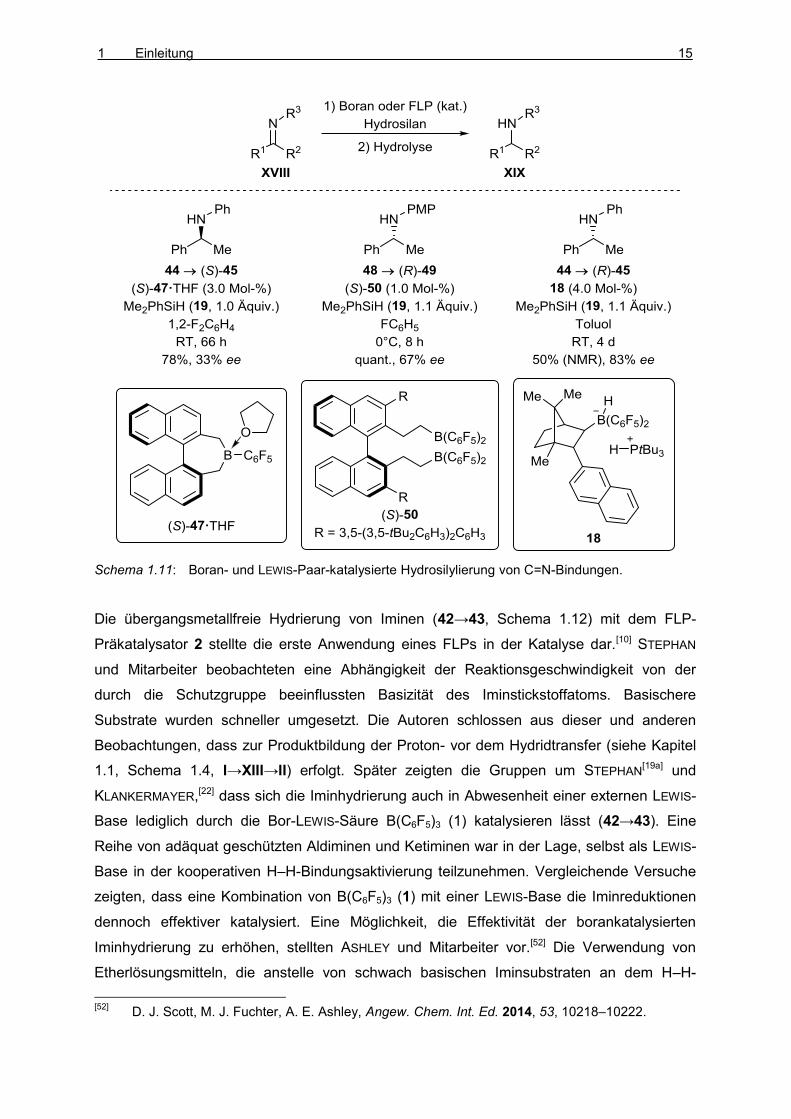
Bei der Verwendung des chiralen Borans (S)-47 in der Reduktion von Ketiminen (44→(S)-
45, 33% ee, Schema 1.11) mit dem Hydrosilan 19 beobachteten MEWALD und OESTREICH
dementsprechend nur geringe Enantiomerenüberschüsse der Amine.[50] ZHU und DU
erreichten bei Einsatz des chiralen Borans (S)-50 als Katalysator, welches in situ durch
Hydroborierung des entsprechenden Diens mit HB(C6F5)2 generiert wurde, ebenfalls nur
moderate Enantioselektivitäten (48→(R)-49, 67% ee).[51] Höhere Enantioselektivitäten
(44→(R)-45, 83% ee) wurden mit dem campherabgeleiteten Boran/Phosphin-Präkatalysator
18 erzielt.[32] KLANKERMAYER und Mitarbeiter stellten dabei fest, dass die Verwendung des
entsprechenden chiralen Borans als alleiniger Katalysator nur eine unbefriedigende
Enantioinduktion zur Folge hatte (nicht gezeigt).
[49]
J. Hermeke, M. Mewald, M. Oestreich, J. Am. Chem. Soc. 2013, 135, 17537–17546. [50]
M. Mewald, M. Oestreich, Chem. Eur. J. 2012, 18, 14079–14084. [51]
X. Zhu, H. Du, Org. Biomol. Chem. 2015, 13, 1013–1016.
1 Einleitung 15
Schema 1.11: Boran- und LEWIS-Paar-katalysierte Hydrosilylierung von C=N-Bindungen.
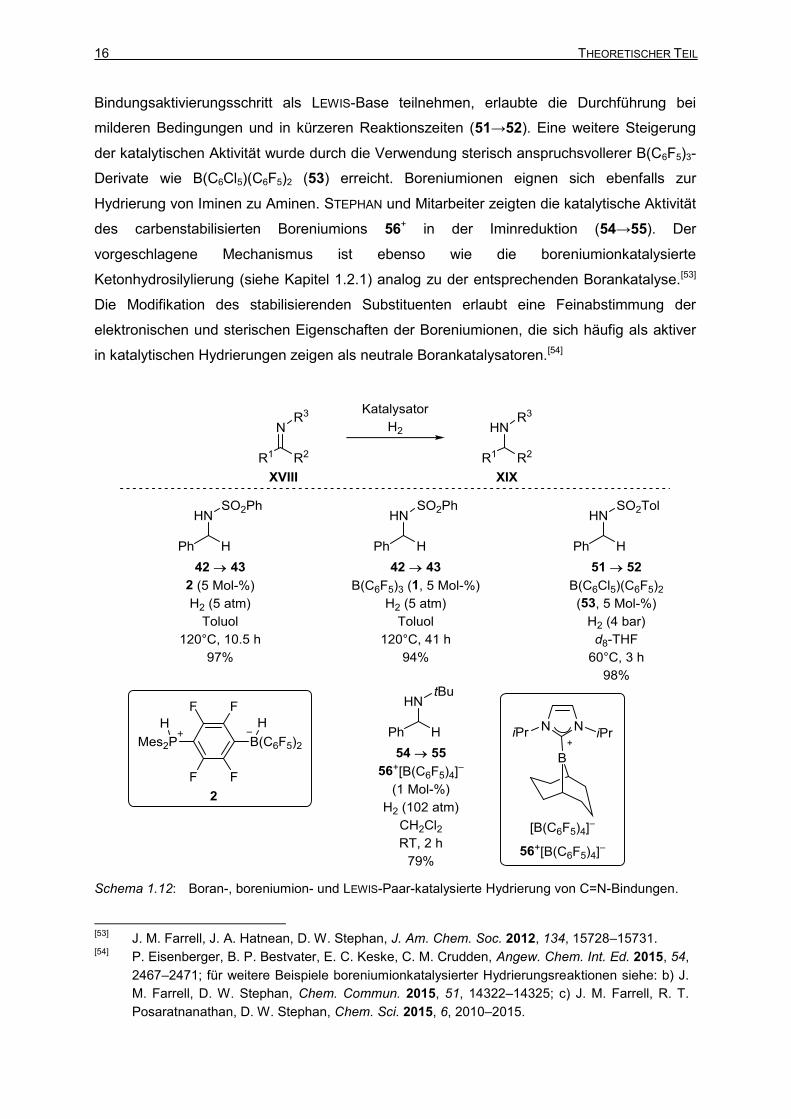
Die übergangsmetallfreie Hydrierung von Iminen (42→43, Schema 1.12) mit dem FLP-
Präkatalysator 2 stellte die erste Anwendung eines FLPs in der Katalyse dar.[10] STEPHAN
und Mitarbeiter beobachteten eine Abhängigkeit der Reaktionsgeschwindigkeit von der
durch die Schutzgruppe beeinflussten Basizität des Iminstickstoffatoms. Basischere
Substrate wurden schneller umgesetzt. Die Autoren schlossen aus dieser und anderen
Beobachtungen, dass zur Produktbildung der Proton- vor dem Hydridtransfer (siehe Kapitel
1.1, Schema 1.4, I→XIII→II) erfolgt. Später zeigten die Gruppen um STEPHAN[19a] und
KLANKERMAYER,[22] dass sich die Iminhydrierung auch in Abwesenheit einer externen LEWIS-
Base lediglich durch die Bor-LEWIS-Säure B(C6F5)3 (1) katalysieren lässt (42→43). Eine
Reihe von adäquat geschützten Aldiminen und Ketiminen war in der Lage, selbst als LEWIS-
Base in der kooperativen H–H-Bindungsaktivierung teilzunehmen. Vergleichende Versuche
zeigten, dass eine Kombination von B(C6F5)3 (1) mit einer LEWIS-Base die Iminreduktionen
dennoch effektiver katalysiert. Eine Möglichkeit, die Effektivität der borankatalysierten
Iminhydrierung zu erhöhen, stellten ASHLEY und Mitarbeiter vor.[52] Die Verwendung von
Etherlösungsmitteln, die anstelle von schwach basischen Iminsubstraten an dem H–H-
[52]
D. J. Scott, M. J. Fuchter, A. E. Ashley, Angew. Chem. Int. Ed. 2014, 53, 10218–10222.
16 THEORETISCHER TEIL
Bindungsaktivierungsschritt als LEWIS-Base teilnehmen, erlaubte die Durchführung bei
milderen Bedingungen und in kürzeren Reaktionszeiten (51→52). Eine weitere Steigerung
der katalytischen Aktivität wurde durch die Verwendung sterisch anspruchsvollerer B(C6F5)3-
Derivate wie B(C6Cl5)(C6F5)2 (53) erreicht. Boreniumionen eignen sich ebenfalls zur
Hydrierung von Iminen zu Aminen. STEPHAN und Mitarbeiter zeigten die katalytische Aktivität
des carbenstabilisierten Boreniumions 56+ in der Iminreduktion (54→55). Der
vorgeschlagene Mechanismus ist ebenso wie die boreniumionkatalysierte
Ketonhydrosilylierung (siehe Kapitel 1.2.1) analog zu der entsprechenden Borankatalyse.[53]
Die Modifikation des stabilisierenden Substituenten erlaubt eine Feinabstimmung der
elektronischen und sterischen Eigenschaften der Boreniumionen, die sich häufig als aktiver
in katalytischen Hydrierungen zeigen als neutrale Borankatalysatoren.[54]
Schema 1.12: Boran-, boreniumion- und LEWIS-Paar-katalysierte Hydrierung von C=N-Bindungen.
[53]
J. M. Farrell, J. A. Hatnean, D. W. Stephan, J. Am. Chem. Soc. 2012, 134, 15728–15731. [54]
P. Eisenberger, B. P. Bestvater, E. C. Keske, C. M. Crudden, Angew. Chem. Int. Ed. 2015, 54,
2467–2471; für weitere Beispiele boreniumionkatalysierter Hydrierungsreaktionen siehe: b) J.
M. Farrell, D. W. Stephan, Chem. Commun. 2015, 51, 14322–14325; c) J. M. Farrell, R. T.
Posaratnanathan, D. W. Stephan, Chem. Sci. 2015, 6, 2010–2015.
1 Einleitung 17
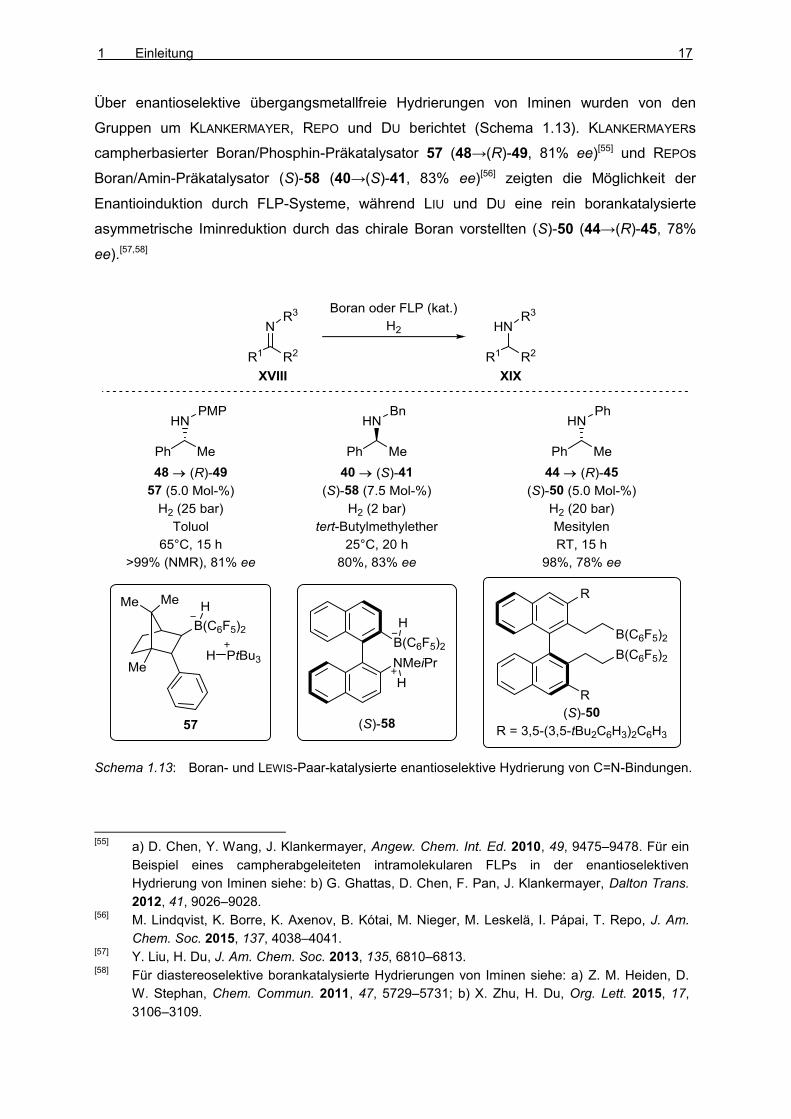
Über enantioselektive übergangsmetallfreie Hydrierungen von Iminen wurden von den
Gruppen um KLANKERMAYER, REPO und DU berichtet (Schema 1.13). KLANKERMAYERs
campherbasierter Boran/Phosphin-Präkatalysator 57 (48→(R)-49, 81% ee)[55] und REPOs
Boran/Amin-Präkatalysator (S)-58 (40→(S)-41, 83% ee)[56] zeigten die Möglichkeit der
Enantioinduktion durch FLP-Systeme, während LIU und DU eine rein borankatalysierte
asymmetrische Iminreduktion durch das chirale Boran vorstellten (S)-50 (44→(R)-45, 78%
ee).[57,58]
Schema 1.13: Boran- und LEWIS-Paar-katalysierte enantioselektive Hydrierung von C=N-Bindungen.
[55]
a) D. Chen, Y. Wang, J. Klankermayer, Angew. Chem. Int. Ed. 2010, 49, 9475–9478. Für ein
Beispiel eines campherabgeleiteten intramolekularen FLPs in der enantioselektiven
Hydrierung von Iminen siehe: b) G. Ghattas, D. Chen, F. Pan, J. Klankermayer, Dalton Trans.
2012, 41, 9026–9028. [56]
M. Lindqvist, K. Borre, K. Axenov, B. Kótai, M. Nieger, M. Leskelä, I. Pápai, T. Repo, J. Am.
Chem. Soc. 2015, 137, 4038–4041. [57]
Y. Liu, H. Du, J. Am. Chem. Soc. 2013, 135, 6810–6813. [58]
Für diastereoselektive borankatalysierte Hydrierungen von Iminen siehe: a) Z. M. Heiden, D.
W. Stephan, Chem. Commun. 2011, 47, 5729–5731; b) X. Zhu, H. Du, Org. Lett. 2015, 17,
3106–3109.

18 THEORETISCHER TEIL
Borankatalysierte formale Hydrierungen lassen sich durch die Verwendung von H2-
Transferreagenzien erreichen (Schema 1.14). STEPHAN und Mitarbeiter nutzten die Tatsache
aus, dass der Hydridübertragungsschritt durch Borhydride auf Iminumiumionen reversibel ist
(siehe Kapitel 1.1, Schema 1.2, VIII→II+IV mit X = NR und Y = H). Durch Verwendung eines
Überschusses des Amins 59 und einer katalytischen Menge B(C6F5)3 (1) wurden aus
Aldiminen (54→55) und Ketiminen (44→rac-45) die entsprechenden Reduktionsprodukte
erhalten.[59] OESTREICH und Mitarbeiter entwickelten eine weitere Methode zur B(C6F5)3-
katalysierten Transferhydrierung von Iminen (51,44→52,rac-45) und fanden, dass sich
Cyclohexa-1,4-diene (60) als Diwasserstoffquelle eignen.[60] Das Reduktionsmittel
Diwasserstoff liegt dabei im Gegensatz zum Hydrosilan in der Transferhydrosilylierung mit
Cyclohexa-1,4-dienen (siehe Kapitel 1.2) nicht oder nur in geringen Mengen frei vor.
Stattdessen nahmen die Autoren an, dass nach Hydridabstraktion durch B(C6F5)3 (1) das
aus dem Cyclohexa-1,4-dien gebildete WHELAND-Intermediat als BRØNSTED-Säure direkt das
Iminsubstrat zum Iminiumion protoniert und auf diese Weise das kinetisch stabile
Borhydrid/Iminiumion-Ionenpaar gebildet wird, aus dem daraufhin das Amin hervorgeht.
Schema 1.14: Borankatalysierte Transferhydrierung von C=N-Bindungen.
[59]
J. M. Farrell, Z. M. Heiden, D. W. Stephan, Organometallics 2011, 30, 4497–4500. [60]
I. Chatterjee, Z.-W. Qu, S. Grimme, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54, 12158–
12162.

1 Einleitung 19
Die borankatalysierte Hydrosilylierung von C≡N-Bindungen wurde von CHANG und
Mitarbeitern untersucht.[61] Bei Verwendung eines Äquivalents des Monohydrosilans 4 in
Gegenwart katalytischer Mengen von B(C6F5)3 (1) wurde eine monoselektive
Hydrosilylierung von Nitrilen zu den entsprechenden N-silylierten Iminen beobachtet
(61→62, Schema 1.15). Wurden die Nitrile mit einem Überschuss an Dihydrosilan 64 oder
67 behandelt, erfolgte eine vollständige Reduktion zu den entsprechenden Aminen
(61,65,68→63,66,69).
Schema 1.15: Borankatalysierte Hydrosilylierung von C≡N-Bindungen.
1.2.3 Reduktion von C=C- und C≡C-Bindungen
GEVORGYAN und Mitarbeiter zeigten, dass sich nicht nur σ-basische Verbindungen wie
Carbonylverbindungen oder Imine, sondern auch π-basische Alkene B(C6F5)3-katalysiert
hydrosilylieren lassen (Schema 1.16).[62,63] Terminale (70→71) und interne (72→cis-73)
[61]
a) N. Gandhamsetty, J. Jeong, J. Park, S. Park, S. Chang, J. Org. Chem. 2015, 80, 7281–
7287. Für die B(C6F5)3-katalysierte Reduktion konjugierter Nitrile mit Dihydrosilanen siehe: b)
N. Gandhamsetty, J. Park, J. Jeong, S.-W. Park, S. Park, S. Chang, Angew. Chem. Int. Ed.
2015, 54, 6832–6836. [62]
M. Rubin, T. Schwier, V. Gevorgyan, J. Org. Chem. 2002, 67, 1936–1940. Für intramolekulare
B(C6F5)3-katalysierte C=C-Hydrosilylierungen siehe: R. Shchepin, C. Xu, P. Dussault, Org.
Lett. 2010, 12, 4772–4775. [63]
Für formale Hydrierungen von C=C-Bindungen bei Verwendung von Hydrosilanen als
Reduktionsmittel siehe: a) M. Tan, Y. Zhang, Tetrahedron Lett. 2009, 50, 4912–4915; b) L. D.
Curless, E. R. Clark, J. J. Dunsford, M. J. Ingleson, Chem. Commun. 2014, 50, 5270–5272; c)
L. Greb, S. Tamke, J. Paradies, Chem. Commun. 2014, 50, 2318–2320.

20 THEORETISCHER TEIL
Alkene waren gleichermaßen geeignete Substrate. In Übereinstimmung mit dem
schrittweisen Mechanismus B(C6F5)3-katalysierter Hydrosilylierungen (Silyltransfer gefolgt
von Hydridtransfer) wurde eine trans-Selektivität als Folge des von der sterisch weniger
gehinderten Seite erfolgenden Hydridtransfers festgestellt. SIMONNEAU und OESTREICH
berichteten von der entsprechenden B(C6F5)3-katalysierten Transferhydrosilylierung (70→74
bzw. 76) mit silylsubstituerten Cyclohexa-1,4-dienen (75,77).[64] Die Hydrosilylierung von
Styrol (70) mit dem Surrogat 77 für das explosive und toxische Gas SiH4 zeigte sich dabei
selektiv für das dreifach hydrosilylierte Produkt 76.
Schema 1.16: Borankatalysierte Hydrosilylierung von C=C-Bindungen.
Die katalytische Hydrierung von C=C-Bindungen durch LEWIS-Paare eignet sich besonders
zur Reduktion elektronenreicher Doppelbindungen. ERKER und Mitarbeiter entwickelten
Boran/Phospin-Systeme, um die Hydrierung von Silylenolethern (78→rac-12)[65] und
Enaminen (82→rac-83)[66] zu katalysieren (Schema 1.14). WEI und DU gelang die
enantioselektive Hydrierung von Silylenolethern mit dem chiralen Boran (S)-80 in
Kombination mit tBu3P (81) als LEWIS-Base, um nach Entschützung
[64]
a) A. Simonneau, M. Oestreich, Angew. Chem. Int. Ed. 2013, 52, 11905–11907; b) A.
Simonneau, M. Oestreich, Nat. Chem. 2015, 7, 816–822. Für eine mechanistische
Betrachtung durch quantenchemische Rechnungen siehe: K. Sakata, H. Fujimoto,
Organometallics 2015, 34, 236–241. [65]
H. Wang, R. Fröhlich, G. Kehr, G. Erker, Chem. Commun. 2008, 5966–5968. [66]
P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich, G. Erker, Angew. Chem. Int. Ed.
2008, 47, 7543–7546.
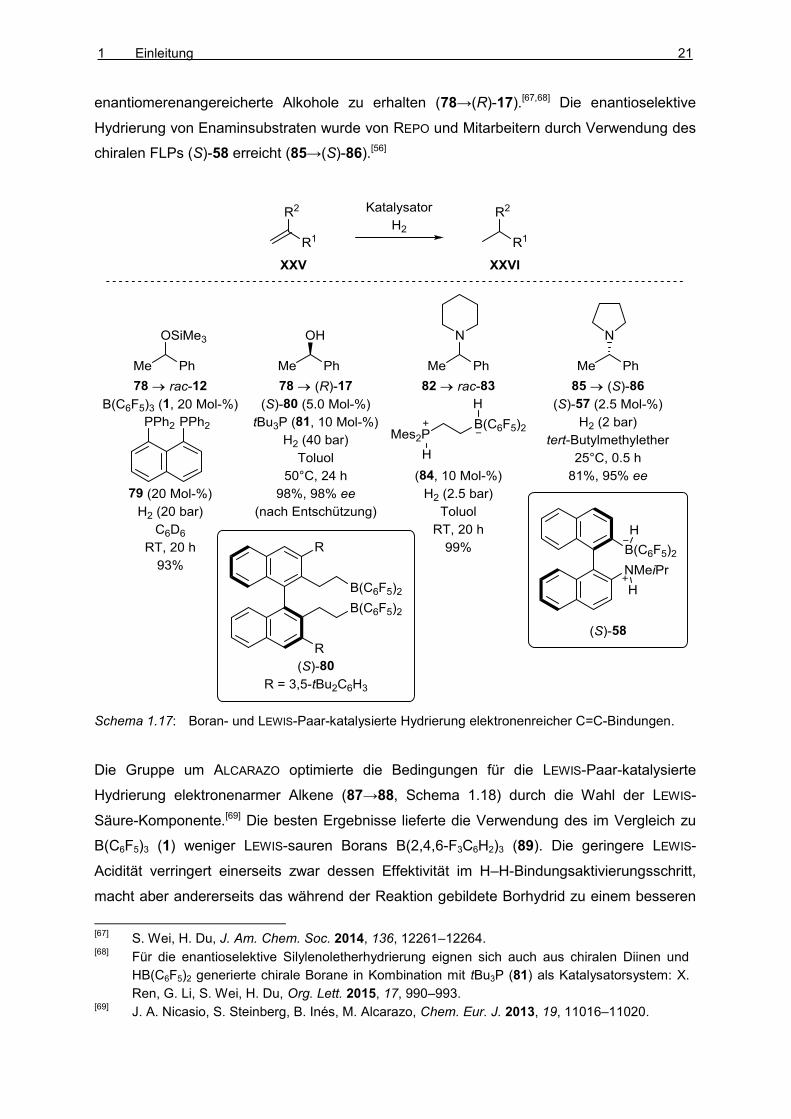
1 Einleitung 21
enantiomerenangereicherte Alkohole zu erhalten (78→(R)-17).[67,68] Die enantioselektive
Hydrierung von Enaminsubstraten wurde von REPO und Mitarbeitern durch Verwendung des
chiralen FLPs (S)-58 erreicht (85→(S)-86).[56]
Schema 1.17: Boran- und LEWIS-Paar-katalysierte Hydrierung elektronenreicher C=C-Bindungen.
Die Gruppe um ALCARAZO optimierte die Bedingungen für die LEWIS-Paar-katalysierte
Hydrierung elektronenarmer Alkene (87→88, Schema 1.18) durch die Wahl der LEWIS-
Säure-Komponente.[69] Die besten Ergebnisse lieferte die Verwendung des im Vergleich zu
B(C6F5)3 (1) weniger LEWIS-sauren Borans B(2,4,6-F3C6H2)3 (89). Die geringere LEWIS-
Acidität verringert einerseits zwar dessen Effektivität im H–H-Bindungsaktivierungsschritt,
macht aber andererseits das während der Reaktion gebildete Borhydrid zu einem besseren
[67]
S. Wei, H. Du, J. Am. Chem. Soc. 2014, 136, 12261–12264. [68]
Für die enantioselektive Silylenoletherhydrierung eignen sich auch aus chiralen Diinen und
HB(C6F5)2 generierte chirale Borane in Kombination mit tBu3P (81) als Katalysatorsystem: X.
Ren, G. Li, S. Wei, H. Du, Org. Lett. 2015, 17, 990–993. [69]
J. A. Nicasio, S. Steinberg, B. Inés, M. Alcarazo, Chem. Eur. J. 2013, 19, 11016–11020.

22 THEORETISCHER TEIL
Hydriddonor und sorgt damit für eine schnellere Reduktion.[70] Alkenhydrierungen, die von
einem elektronenarmen Boran ohne externe LEWIS-Base katalysiert werden, sind bisher
nicht bekannt. STEPHAN und Mitarbeiter zeigten jedoch, dass die Verwendung einer
Kombination von B(C6F5)3 (1) mit ebenfalls katalytischen Mengen an Diethylether als LEWIS-
Base-Komponente ausreicht, um ausgewählte Substrate (91→92, Methode a) zu
hydrieren.[71] Eine Alternative bietet die Transferhydrierung mit Cyclohexa-1,4-dienen der
Gruppe um OESTREICH (91→92, Methode b).[72] Der aus dem Cyclohexa-1,4-dien 93 durch
Hydridabstraktion mit B(C6F5)3 (1) gebildete WHELAND-Komplex ist BRØNSTED-acide genug,
um einfache Alkene zu protonieren, bevor der Hydridtransfer durch die Borhydridspezies
H–B(C6F5)3– das Alkan liefert.
Schema 1.18: Boran- und LEWIS-Paar-katalysierte Hydrierung und Transferhydrierung von C=C-
Bindungen.
C≡C-Bindungen lassen sich bei der Verwendung eines Äquivalents an Hydrosilan
monoselektiv borankatalysiert hydrosilylieren (Schema 1.19). Das erste Beispiel stellten
CURLESS und INGLESON vor, die bei der Hydrosilylierung des Alkins 94 mit dem Dihydrosilan
[70]
Für eine vergleichende Studie zur Hydriddonorstärke gängiger Hauptgruppenelementhydride
durch quantenchemische Rechnungen siehe: Z. M. Heiden, A. P. Lathem, Organometallics
2015, 34, 1818–1827. [71]
L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme, D. W. Stephan, Angew.
Chem. Int. Ed. 2013, 52, 7492–7495. [72]
I. Chatterjee, Z.-W. Qu, S. Grimme, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54, 12158–
12162.

1 Einleitung 23
64 eine moderat selektive trans-Addition zum (Z)-Alken 95 feststellten.[73] OESTREICH und
Mitarbeiter untersuchten diese Reaktion ebenfalls und beobachteten für die Addition von
Monohydrosilanen 4 bzw. aus ihren Surrogaten 75 freigesetzte Monohydrosilane an interne
Alkine perfekte trans-Selektivitäten (96→(Z)-97 bzw. 96→(Z)-98).[28]
Schema 1.19: Borankatalysierte Reduktion von C≡C-Bindungen mit Hydrosilanen.
SCHWIER und GEVORGYAN stellten eine B(C6F5)3-katalysierte Alkinreduktionsmethode vor, bei
der Hydrogermane statt Hydrosilanen als Reduktionsmittel dienten (Schema 1.20).[74] An
einfache Alkine erfolgt die Hydrogerman- ebenso wie die Hydrosilanaddition trans-selektiv
(94+100→(Z)-99 und 101+103→(Z)-102), weshalb von einem Mechanismus ausgegangen
wird, der dem der B(C6F5)3-katalysierten Hydrosilylierung entspricht. Estersubstituierte Alkine
führten hingegen zu einer cis-Addition des Hydrogermans (104+103→(E)-105), was durch
einen abweichenden Mechanismus erklärt wurde, der nicht über die
Hydrogermanaktivierung durch B(C6F5)3 (1) verläuft, sondern durch die Koordination der
LEWIS-basischen Estergruppe an den Katalysator initiiert wird. Boran- und FLP-katalysierte
cis-selektive Hydrierungen von Alkinen,[75] B(C6F5)3-katalysierte Hydrostannylierungen von
[73]
L. D. Curless, M. J. Ingleson, Organometallics 2014, 33, 7241–7246. [74]
T. Schwier, V. Gevorgyan, Org. Lett. 2005, 7, 5191–5194. [75]
a) B.-H. Xu, G. Kehr, R. Fröhlich, B. Wibbeling, B. Schirmer, S. Grimme, G. Erker, Angew.
Chem. Int. Ed. 2011, 50, 7183–7186; b) K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M.
Leskelä, T. Repo, Nat. Chem. 2013, 5, 718–723; c) Y. Liu, L. Hu, H. Chen, H. Du, Chem. Eur.
J. 2015, 21, 3495–3501; d) K. C. Szeto, W. Sahyoun, N. Merle, J. L. Castelbou, N. Popoff, F.
Lefebvre, J. Raynaud, C. Godard, C. Claver, L. Delevoye, R. M. Gauvin, M. Taoufik, Catal.
Sci. Technol. 2016, 6, 882–889.

24 THEORETISCHER TEIL
C=C- und C≡C-Bindungen[76] sowie B(C6F5)3- und boreniumionvermittelte Hydroborierungen
von C≡C-Bindungen[77] folgen ebenfalls abweichenden Mechanismen (nicht gezeigt).
Schema 1.20: Borankatalysierte Hydrogermylierung von C≡C-Bindungen.
1.3 Problemstellung und Zielsetzung
1.3.1 Katalytische C=N-Reduktion von Oximen und Hydrazonen
Die funktionelle Gruppe der Hydroxylamine ist in Molekülen mit biologischer Aktivivität von
Bedeutung[78] und deren Darstellung durch C=N-Reduktion aus einfach zugänglichen
Oximen erstrebenswert (XXXIII→XXXIV, Schema 1.21). Die C=N-Reduktion von Oximen zu
Hydroxylaminen ist jedoch eine anspruchsvolle Transformation, da eine Überreduktion
infolge eines N–O-Bindungsbruchs zu Aminen führen kann (XXXIII→XXXIV→XXXV). Die
heterogen katalysierte Hydrierung von Oximen XXXIII zu Hydroxylaminen XXXIV wurde
bereits vor beinahe einem Jahrhundert entwickelt,[79] wobei jedoch nur einzelne Substrate
[76]
a) V. Gevorgyan, J.-X. Liu, Y. Yamamoto, J. Org. Chem. 1997, 62, 2963–2967; b) V.
Gevorgyan, J.-X. Liu, Y. Yamamoto, Chem. Commun. 1998, 37–38; c) M. S. Oderinde, M. G.
Organ, Angew. Chem. Int. Ed. 2012, 51, 9834–9837. [77]
J. S. McGough, S. M. Butler, I. A. Cade, M. J. Ingleson, Chem. Sci. 2016, 7, 3384–3389. [78]
a) A. Melman in The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, Part 1
(Hrsg.: Z. Rappoport, J. F. Liebman), Wiley-VCH, Chichester, 2009, S. 117–161; b) D.
Geffken, M. A. Köllner in Science of Synthesis, Vol. 40 (Hrsg.: E. Schaumann, D. Enders),
Thieme, Stuttgart, 2008, S. 973–1082. [79]
VAVON und Mitarbeiter berichteten von der heterogenen platinkatalysierten Hydrierung von
Oximen. N-Monosubstituierte Hydroxylamine wurden bei der Hydrierung von aliphatischen und
alizyklischen Ketoximen erhalten, während aromatische Ketoxime zu primären Aminen
reduziert wurden. Die Hydrierung von Aldoximen ergab N,N-disubstituierte Hydroxylamine: a)

1 Einleitung 25
chemoselektiv reduziert wurden und auch die Reproduzierbarkeit in Zweifel gezogen
wurde.[80] Homogen übergangsmetallkatalysierte Methoden lieferten fast ausschließlich
primäre Amine XXXV infolge von Überreduktion.[81] Es ist lediglich ein Patent bekannt, in
welchem eine homogen katalytische Oximetherreduktion zu den Hydroxylaminen XXXIV
führte.[82]
Schema 1.21: Reduktion von Oximen (XXXIII) zu Hydroxylaminen (XXXIV) und Überreduktion zu
Aminen (XXXV).
In Ermangelung einer generellen Methode war ein Ziel dieser Dissertation die Entwicklung
einer homogen katalytischen chemoselektiven C=N-Reduktion von Oximen oder Oximethern
zu den entsprechenden Hydroxylaminen. Ein Großteil der etablierten Methoden zur
chemoselektiven C=N-Reduktion von Oximen und Oximethern basieren auf der
stöchiometrischen Verwendung von Borhydriden als Reduktionsmittel.[78,79,83,84,85] Um eine
G. Vavon, A. L. Berton, Bull. Soc. Chim. Fr. 1925, 37, 296–305; b) G. Vavon, P. Anziani, Bull.
Soc. Chim. Fr. 1927, 41, 1638–1649; c) G. Vavon, N. Krajcinovic, Bull. Soc. Chim. Fr. 1928,
43, 231–237. Für die heterogene platinkatalysierte Hydrierung von Ketoximethern siehe: d) L.
W. Jones, R. T. Major, J. Am. Chem. Soc. 1930, 52, 669–679. [80]
H. Feuer, B. F. Vincent, Jr., J. Am. Chem. Soc. 1962, 84, 3771–3772. [81]
a) M. Murakami, J.-W. Kang, Bull. Chem. Soc. Jpn. 1963, 36, 763–769 (Cobaltkatalyse); b) P.
Krasik, H. Alper, Tetrahedron: Asymmetry 1992, 3, 1283–1288 (Rutheniumkatalyse); c) A. S.
C. Chan, C.-C. Chen, C.-W. Lin, Y.-C. Lin, M.-C. Cheng, S.-M. Peng, J. Chem. Soc., Chem.
Commun. 1995, 1767–1768 (Rhodiumkatalyse); d) Y. Xie, A. Mi, Y. Jiang, H. Liu, Synth.
Commun. 2001, 31, 2767–2771 (Iridiumkatalyse); e) K. Huang, S. Li, M. Chang, X. Zhang,
Org. Lett. 2013, 15, 484–487 (Rhodiumkatalyse). [82]
Eine homogene übergangsmetallkatalysierte Hydrierung von O-Alkyloximethern zu O-
Alkylhydroxylaminen wurde beschrieben in: R. Kadyrov, T. Riermeier, J. Almena, A. Monsees,
P. Groß, K. Rossen (Evonik Degussa GmbH), EP 1 862 446 A2, 2007. Dieses Patent enthält
auch enantioselektive Beispiele. [83]
a) H. Feuer, B. F. Vincent, Jr., R. S. Bartlett, J. Org. Chem. 1965, 30, 2877–2880 (BH3∙THF);
b) R. F. Borch, M. D. Bernstein, H. D. Durst, J. Am. Chem. Soc. 1971, 93, 2897–2904
(NaCNBH3); c) Y. Kikugawa, M. Kawase, Chem. Lett. 1977, 1279–1280 (BH3∙Pyridin); d) G.
W. Gribble, R. W. Leiby, M. N. Sheehan, Synthesis 1977, 856–858 (NaBH4). [84]
Für asymmetrische Oxim-zu-Hydroxylamin-Reduktionen mit chiralen Boran/Oxazaborolidin-
Addukten siehe: a) J. T. Dougherty, J. R. Flisak, J. Hayes, I. Lantos, L. Liu, L. Tucker,
Tetrahedron: Asymmetry 1997, 8, 497–499; b) E. Fontaine, C. Namane, J. Meneyrol, M.
Geslin, L. Serva, E. Roussey, S. Tissandié, M. Maftouh, P. Roger, Tetrahedron: Asymmetry

26 THEORETISCHER TEIL
chemoselektive katalytische C=N-Reduktion von Oximen zu erreichen, könnte demnach eine
Reaktion, die über eine katalytisch generierte Borhydridspezies verläuft, ebenso geeignet
sein. Durch Verwendung katalytischer Mengen eines elektronenarmen Borans oder eines
FLPs und stöchiometrischen Mengen eines Reduktionsmittels III (Hydrosilan, Y = R3Si oder
Diwasserstoff, Y = H) sollte eine generelle Methode zur Durchführung dieser Transformation
gefunden werden (XXXIII→XXXIV, Schema 1.22).
Schema 1.22: Entwicklung einer homogen katalytischen Methode zur chemoselektiven C=N-
Reduktion von Oximen.
Eine strukturell eng verwandte funktionelle Gruppe der Oxime stellen Hydrazone dar. Deren
C=N-Reduktion zu Hydrazinen lässt sich ebenfalls durch Behandlung mit stöchiometrischen
Mengen von Borhydriden erreichen.[86] Übergangsmetallkatalysierte Hydrierungsreaktionen
zur Erreichung derselben Transformation sind ebenfalls bekannt.[87] Eine
übergangsmetallfreie katalytische Methode existiert hingegen noch nicht. Ein weiteres Ziel
2001, 12, 2185–2189; c) M. P. Krzemiński, M. Zaidlewicz, Tetrahedron: Asymmetry 2003, 14,
1463–1466. [85]
Eine alternative Methode bietet die Verwendung von Hydrosilanen als Reduktionsmittel in
Trifluoressigsäure: a) D. D. Sternbach, W. C. L. Jamison, Tetrahedron Lett. 1981, 22, 3331–
3334; b) M. Fujita, H. Oishi, T. Hiyama, Chem. Lett. 1986, 837–838. Oximether lassen sich
ebenso mit Bu3SnH als Reduktionsmittel vermittelt durch BF3∙OEt2 zu den entsprechenden
Hydroxylaminen reduzieren: M. Ueda, H. Miyabe, M. Namba, T. Nakabayashi, T. Naito,
Tetrahedron Lett. 2002, 43, 4369–4371. [86]
a) G. N. Walker, M. A. Moore, B. N. Weaver, J. Org. Chem. 1961, 26, 2740‒2747 (NaBH4); b)
J. A. Blair, R. J. Gardner, J. Chem. Soc. C 1970, 1714–1717 (BH3∙Bis(2-methoxyethyl)ether);
c) Y. Kikugawa, M. Kawase, Synth. Commun. 1979, 9, 49–52 (BH3∙Pyridin). Für weitere
stöchiometrische Reduktionsmethoden mit Hydriddonoren siehe: d) R. L. Hinman, J. Chem.
Soc. 1957, 79, 414–417 (LiAlH4); e) P.-L. Wu, S.-Y. Peng, J. Magrath, Synthesis 1995, 435–
438 (Et3SiH/TFA). [87]
Für Beispiele siehe: a) H. L. Yale, K. Losee, J. Martins, M. Holsing, F. M. Perry, J. Bernstein,
J. Am. Chem. Soc. 1953, 75, 1933‒1942 (Platinkatalyse); b) A. Ebnöther, E. Jucker, A.
Lindenmann, E. Rissi, R. Steiner, R. Süess, A. Vogel, Helv. Chim. Acta 1959, 533–563
(Nickelkatalyse); c) M. J. Burk, J. E. Feaster, J. Am. Chem. Soc. 1992, 114, 6266‒6267
(Rutheniumkatalyse); d) Z.-P. Chen, S.-B. Hu, M.-W. Chen, Y.-G. Zhou, Org. Lett. 2016, 18,
2676–2679. Für eine nickelkatalysierte Transferhydrierung von Hydrazonen siehe: e) H. Xu, P.
Yang, P. Chuanprasit, H. Hirao, J. Zhou, Angew. Chem. Int. Ed. 2015, 54, 5112‒5116.

1 Einleitung 27
dieser Dissertation sollte die Entwicklung einer solchen boran- oder FLP-katalysierten
Hydrierungs- oder Hydrosilylierungsmethode sein (XXXVI→XXXVII, Schema 1.23).
Schema 1.23: Entwicklung einer übergangsmetallfreien katalytischen Methode zur C=N-Reduktion
von Hydrazonen.
1.3.2 Darstellung elektronenarmer Borane für katalytische Si–H- und H–H-
Bindungsaktivierungen
Für das Forschungsgebiet der übergangsmetallfreien Katalyse ist die Synthese neuer
elektronenarmer Borane mit unterschiedlicher Anzahl an in verschiedenem Maße fluorierten
Arylsubstituenten von Bedeutung.[88] Durch sterische und elektronische Modifikationen
lassen sich in systematischen Studien geeignete Kombinationen von Katalysator, Substrat
und Reduktionsmittel ermitteln.[28,69] Zur Einschätzung der potentiellen katalytischen Aktivität
für Si–H- und H–H-Bindungsaktivierungen wird häufig die relativ zu B(C6F5)3 (1) angegebene
LEWIS-Acidität der entsprechenden Borane herangezogen.[89] Das nicht fluorierte
Triarylboran 106 (Schema 1.24, links), das eine LEWIS-Acidität von 70% relativ zu B(C6F5)3
(1) aufweist, ist nicht in der Lage, ungesättigte Substrate effektiv katalytisch zu
hydrosilylieren.[28,90] Typische B(C6F5)3-Derivate für katalytische Hydrosilylierungen und
[88]
Für Beispiele von B(C6F5)3-Derivaten in katalytischen H–H-Bindungsaktivierungen siehe: Lit.
24; 45; 52; 55a; 57; 69; a) C. Jiang, O. Blacque, H. Berke, Chem. Commun. 2009, 5518–5520;
b) G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi, T. Soós, Angew. Chem. Int.
Ed. 2010, 49, 6559–6563; c) G. Erős, K. Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G.
Tárkányi, T. Soós, Chem. Eur. J. 2012, 18, 574–585. Für Beispiele von B(C6F5)3-Derivaten in
katalytischen Si–H-Bindungsaktivierungen siehe: Lit. 28; 32; 34; 54b; d) R. Roesler, B. J. N.
Har, W. E. Piers, Organometallics 2002, 21, 4300–4302; e) M. Mewald, R. Fröhlich, M.
Oestreich, Chem. Eur. J. 2011, 17, 9406–9414; f) D. Chen, V. Leich, F. Pan, J. Klankermayer,
Chem. Eur. J. 2012, 18, 5184–5187. [89]
Für eine Übersicht der LEWIS-Acidität von Borverbindungen siehe: I. B. Sivaev, V. I. Bregadze,
Coord. Chem. Rev. 2014, 270-271, 75–88. [90]
In Kombination mit sehr starken Basen können auch schwache LEWIS-Säuren wie das Boran
106 katalytische Aktivität in Hydrierungsreaktionen zeigen: S. Mummadi, D. K. Unruh, J. Zhao,
S. Li, C. Krempner, J. Am. Chem. Soc. 2016, 138, 3286–3289. Eine weitere Ausnahme zeigt
Lit. 26c.

28 THEORETISCHER TEIL
Hydrierungen weisen mindestens einen (partiell) fluorierten Phenylsubstituenten auf
(XXXVIII, Schema 1.24, Mitte).[88] Bereits ein geringer Grad an Fluorierung [beispielsweise in
B(2,6-F2C6H3)3 (107)] sorgt für eine ausreichende LEWIS-Acidität, um Hydrosilylierungen[28]
und Hydrierungen[24,69] bestimmter Substrate zu katalysieren. Die LEWIS-Acidität des Borans
(S)-47, welches lediglich einen perfluorierten C6F5-Substituenten aufweist, reicht ebenfalls
aus, um als Hydrosilylierungskatalysator zu dienen.[49,50,88e] Fluoratome in ortho-Position zum
Boratom wurden zudem aufgrund von aktivierenden Wechselwirkungen mit dem
Siliciumatom als vorteilhaft in der Si–H-Bindungsaktivierung identifiziert.[15,28] Eine bisher
nicht adressierte Fragestellung stellt der Effekt einer Fluorierung an nicht direkt an das
zentrale Boratom gebundenen aromatischen Kohlenstoffatomen dar. Zu diesem Zweck sollte
das partiell fluorierte Boran 108[91] (Schema 1.24, rechts) dargestellt werden, welches
Fluoratome nur an dem dem Borzentrum entfernt gelegenen aromatischen System trägt und
jegliche Fluoratome in ortho-Position zum Boratom vermissen lässt. Neben der Bestimmung
der relativen LEWIS-Acidität des Borans 108 sollte auch dessen Aktivität in katalytischen Si–
H- und H–H-Bindungsaktivierungen getestet werden.
Schema 1.24: Elektronenarme Borane: Einfluss von Fluoratomen auf die LEWIS-Acidität.
Um boran- oder FLP-katalysierte Transformationen asymmetrisch durchzuführen, ist die
Synthese chiraler Borane und FLPs von Bedeutung. Bisher bekannte chirale Borane oder
Borankomponenten von FLPs weisen das Strukturmotiv RchiralnB(C6F5)3-n (XXXIX mit n = 1
oder 2, Schema 1.25, links) auf, bei dem mindestens einer der elektronenarmen C6F5-
Substituenten des Borans B(C6F5)3 (1) formal durch einen oder mehrere die chirale
Information tragenden Substituenten ersetzt ist.[92] Ein Boran der Art XL (Schema 1.25,
[91]
Das Boran 108 wurde in einem Patent genannt, eine detaillierte Synthese desselben ist
allerdings nicht beschrieben worden: G. Rodriguez, F. C. Rix, P. S. Ravishankar, M. C.
Kuchta, PCT Int. Appl. WO 03/051892 A1, 2003. [92]
Für Beispiele chiraler Borane und boranbasierter FLPs in katalytischen H–H-
Bindungsaktivierungen siehe: Lit. 55a; 55b; 56; 57; 67; 68; a) V. Sumerin, K. Chernichenko, M.

1 Einleitung 29
rechts), welches den chiralen Substituenten in para-Position der Arylsubstituenten trägt,
stellt eine bisher nicht untersuchte Möglichkeit dar. Durch die Tatsache, dass Borane der Art
XL weiterhin drei fluorierte und somit elektronenarme Substituenten tragen, ist ein Erhalt der
LEWIS-Acidität und der elektronischen Situation in direkter Nähe des zentralen Boratoms im
Vergleich zu B(C6F5)3 (1) sichergestellt. Ein weiteres Ziel dieser Dissertation war folglich die
Identifizierung eines geeigneten Strukturmotivs zur Darstellung eines solchen Borans.
Schema 1.25: Strukturmotive chiraler elektronenarmer Borane.
Nieger, M. Leskelä, B. Rieger, T. Repo, Adv. Synth. Catal. 2011, 353, 2093–2110; b) X. Wang,
G. Kehr, C. G. Daniliuc, G. Erker, J. Am. Chem. Soc. 2014, 136, 3293–3303; c) Z. Zhang, H.
Du, Org. Lett. 2015, 17, 2816–2819; d) Z. Zhang, H. Du, Org. Lett. 2015, 17, 6266–6269; e) Z.
Zhang, H. Du, Angew. Chem. Int. Ed. 2015, 54, 623–626. Für Beispiele chiraler Borane und
boranbasierter FLPs in katalytischen Si–H-Bindungsaktivierungen siehe: Lit. 32; 34; 35; 88e.
Für chirale elektronenarme Borane und boranbasierte FLPs, die bisher weder in H–H- noch in
Si–H-Bindungsaktivierungen eingesetzt wurden, siehe: f) D. J. Morrison, W. E. Piers, M.
Parvez, Synlett 2004, 2429–2433; g) K. Venkatasubbaiah, M. Bolte, F. Jäkle, J. Fluor. Chem.
2010, 131, 1247–1251; h) J. Chen, K. Venkatasubbaiah, T. Pakkirisamy, A. Doshi, A.
Yusupov, Y. Patel, R. A. Lalancette, F. Jäkle, Chem. Eur. J. 2010, 16, 8861–8867; i) J. Chen,
R. A. Lalancette, F. Jäkle, Chem. Eur. J. 2014, 20, 9120–9129.

30 THEORETISCHER TEIL
2 B(C6F5)3-KATALYSIERTE CHEMOSELEKTIVE C=N-
REDUKTION VON OXIMETHERN ZU HYDROXYLAMINEN
Dieses Kapitel beschreibt die Identifizierung geeigneter homogen katalytischer Methoden für
die chemoselektive C=N-Reduktion von Oximethern zu den entsprechenden
Hydroxylaminen.[93,94] Die Methoden der katalytischen Generierung von Borhydriden aus der
Kombination elektronenarmer Borane mit Hydrosilanen bzw. Diwasserstoff werden auf ihre
Tauglichkeit im Hinblick auf diese Transformation untersucht. Neben der Bedeutung der
Schutzgruppe wird auf den Reaktivitätsunterschied von keton- und aldehydabgeleiteten
Substraten eingegangen. Die Ergebnisse der Aldoximreduktion wurden in Zusammenarbeit
mit DIGVIJAY PORWAL und Dr. INDRANIL CHATTERJEE erarbeitet.
2.1 Versuche zur C=N-Reduktion von Oximethern durch
borankatalysierte Hydrosilylierung
Entsprechend der Arbeit der Gruppe um PIERS zur B(C6F5)3-katalysierten Hydrosilylierung
der strukturell verwandten Substratklasse der Imine zu Aminen[47] (siehe Kapitel 1.2.2)
gedachten wir zunächst, diese Methode auf die Reduktion von Oximen und Oximethern zu
Hydroxylaminen zu übertragen. Wir waren uns allerdings bewusst, dass im Falle von
Oximen und Oximethern im Schritt der kooperativen Si–H-Bindungsspaltung eine
Konkurrenzsituation zweier LEWIS-basischer Atome des Substrats auftritt: Beteiligung des
Stickstoffatoms (XLI, Abbildung 2.1, links) führt nach Hydridtransfer des entstehenden
Borhydrids zum gewünschten C=N-Reduktionsprodukt, während dieses bei Beteiligung des
Sauerstoffatoms (XLII, Abbildung 2.1, rechts) nicht gebildet werden kann.
Abbildung 2.1: Konkurrenz LEWIS-basischer Atome in kooperativer Si–H-Bindungsspaltung durch
B(C6F5)3 (1) und Oxime. Si = SiR3.
[93]
J. Mohr, M. Oestreich, Angew. Chem. Int. Ed. 2014, 53, 13278–13281. [94]
J. Mohr, D. Porwal, I. Chatterjee, M. Oestreich, Chem. Eur. J. 2015, 21, 17583–17586.
Synthesevorschriften für und Charakterisierungen von Substanzen, die im experimentellen
Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.

2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 31
Bei der Behandlung verschieden substituierter acetophenonabgeleiteter Oxime XLII mit
diversen Hydrosilanen XLV in Gegenwart katalytischer Mengen von B(C6F5)3 (1) mit
anschließender Hydrolyse (Schema 2.1) zeigte sich, dass die Oxophilie des Siliciumatoms
entsprechend Szenario XLII (Abbildung 2.1, rechts) in der Tat die gewünschte Bildung der
Hydroxylamine XLIV verhindert. Auch bei sterisch anspruchsvollen Substituenten am
Sauerstoffatom der Oxime XLIII wurden lediglich Produktgemische erhalten, deren
massenspektrometrische Analyse die Bildung von Silylethern XLVI und somit den Bruch der
N–O-Bindung aufzeigte. Ebenfalls nachgewiesen wurde in einigen Fällen die Bildung von N-
Ethylanilin (109), das aus einer mit der BECKMANN-Umlagerung verwandten reduktiven
Umlagerung resultierte.[95]
Schema 2.1: Gescheiterte C=N-Reduktion von Oximethern durch B(C6F5)3-katalysierte
Hydrosilylierung. Si = SiR3, R‘ = H, Me, Bn, tBu.
2.2 C=N-Reduktion von Oximethern durch borankatalysierte
Hydrierung
Nach den erfolglosen Versuchen zur Oximetherreduktion durch B(C6F5)3-katalysierte
Hydrosilylierung entschieden wir uns, Diwasserstoff statt Hydrosilanen als Reduktionsmittel
zu verwenden. Auch in diesem Fall war die verwandte C=N-Reduktion von Iminen bereits
eine etablierte Methode (siehe Kapitel 1.2.2). Wir waren zudem zuversichtlich, dass bei
[95]
Zur Durchführung dieser Umlagerungsreaktion mit BH3 als Reduktionsmittel siehe: a) M. Ortiz-
Marciales, L. D. Rivera, M. De Jesús, S. Espinosa, J. A. Benjamin, O. E. Casanova, I. G.
Figueroa, S. Rodríguez, W. Correa, J. Org. Chem. 2005, 70, 10132–10134; b) M. Ortiz-
Marciales, D. Figueroa, J. A. López, M. De Jesús, R. Vega, Tetrahedron Letters 2000, 41,
6567–6570. Ebenfalls bekannt ist diese Reaktion für Oximreduktionen mit DIBAL–H, eine
Übersicht bietet: c) H. Cho, Tetrahedron 2014, 70, 3527–3544.

32 THEORETISCHER TEIL
adäquat geschützten Oximen die zu erwartende Konkurrenzsituation zwischen Stickstoff-
(XLVII, Abbildung 2.2, links) und Sauerstoffatom (XLVIII, Abbildung 2.2, rechts) des
Substrats bei dem Diwasserstoffspaltungsschlüsselschritt zugunsten des basischeren
Stickstoffatoms ausfallen und damit eine selektive C=N-Reduktion ermöglichen wird.
Abbildung 2.2: Konkurrenz basischer Atome in kooperativer H–H-Bindungsspaltung durch B(C6F5)3
(1) und Oxime.
Wir testeten zunächst, ob sich das ungeschützte acetophenonabgeleitete Oxim 110 in
Gegenwart katalytischer Mengen von B(C6F5)3 (1) in Toluol hydrieren lässt, konnten jedoch
keinen Umsatz beobachten (Tabelle 2.1, Eintrag 1). Als Konsequenz wandten wir uns O-
alkylierten Substraten zu. Das methylsubstituierte Oxim 111 zeigte unter einem
Diwasserstoffdruck von 100 bar bei Raumtemperatur mit 5.0 Mol-% B(C6F5)3 (1) zwar
Umsatz zum entsprechenden Hydroxylamin rac-119, jedoch wurde keine katalytische
Umsetzung erreicht (Eintrag 2). Dieses Ergebnis steht in Übereinkunft mit der Beobachtung,
die STEPHAN und Mitarbeiter bei der durch Boran/Phosphin-katalysierten Hydrierung von
Iminen mit sterisch wenig anspruchsvollen Schutzgruppen machten, welche aufgrund von
Amin/Boran-Adduktbildung nur eine stöchiometrische Reduktion erlaubten.[10] Durch
quantenchemische Rechnungen zeigten PÁPAI und Mitarbeiter, dass auch in B(C6F5)3-
katalysierten Iminhydrierungen der Katalysator durch stabile LEWIS-Paar-Bildung mit dem
sich im Laufe der Reaktion anreichernden Aminprodukt inhibiert werden kann.[19b] Durch
Hinzugabe der LEWIS-Base Mes3P (126) versuchten wir den Umsatz zu steigern (Eintrag 3),
da dieses Katalysatorsystem sich im Falle der Iminreduktion als überlegen gegenüber
B(C6F5)3 gezeigt hatte.[19a] Dies resultierte allerdings in einem völligen Ausbleiben eines
Umsatzes. Wir entschieden uns daraufhin, eine sterisch anspruchsvollere Schutzgruppe am
Sauerstoffatom zu wählen, um eine etwaige LEWIS-Paar-Bildung von Substrat oder Produkt
mit dem Katalysator zu unterdrücken. Das tert-butylsubstituierte Oxim 112 zeigte bereits bei
60 bar Diwasserstoffdruck und Raumtemperatur einen Umsatz von 95% (Eintrag 4), wobei
die Umsetzung hoch chemoselektiv verlief: Weder ein N–O-Bindungsbruch noch andere
Nebenprodukte wurden beobachtet, sondern lediglich das gewünschte Hydroxylamin rac-
120 und nicht umgesetztes Startmaterial. Durch Erhöhung des Diwasserstoffdrucks auf 100
bar wurde vollständiger Umsatz gewährleistet und das O-alkylierte Hydroxylamin rac-120 in
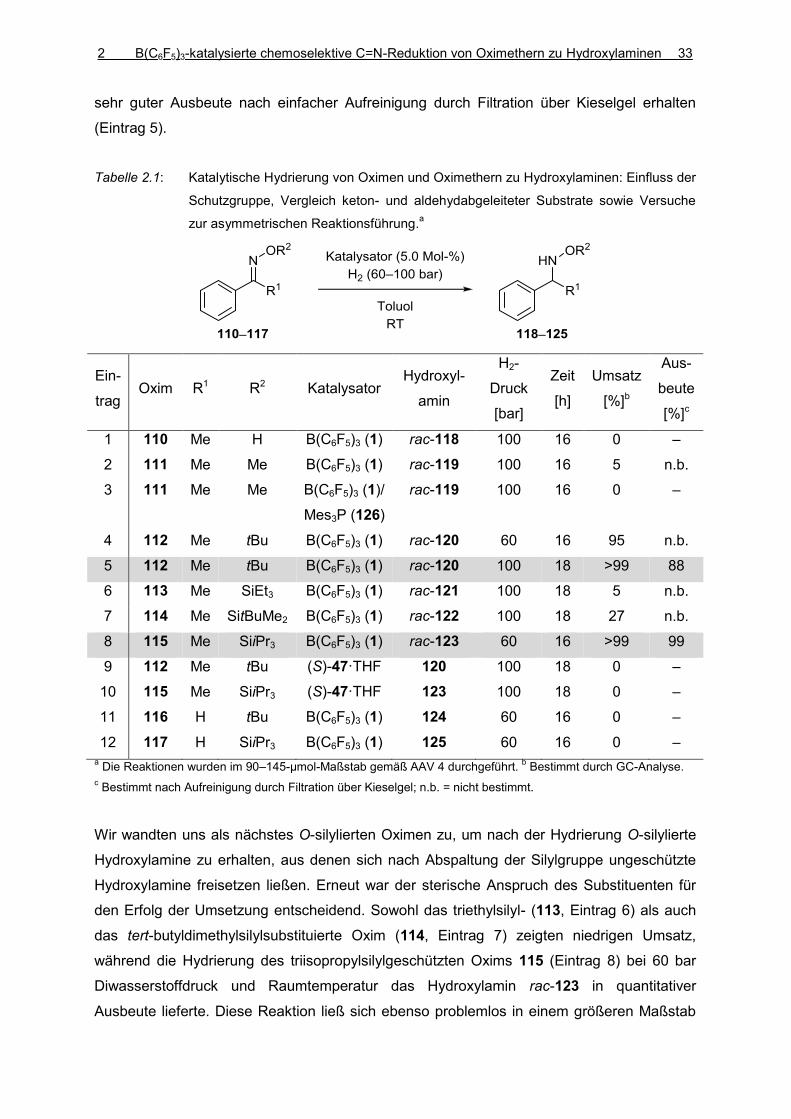
2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 33
sehr guter Ausbeute nach einfacher Aufreinigung durch Filtration über Kieselgel erhalten
(Eintrag 5).
Tabelle 2.1: Katalytische Hydrierung von Oximen und Oximethern zu Hydroxylaminen: Einfluss der
Schutzgruppe, Vergleich keton- und aldehydabgeleiteter Substrate sowie Versuche
zur asymmetrischen Reaktionsführung.a
Ein-
trag Oxim R1 R2 Katalysator
Hydroxyl-
amin
H2-
Druck
[bar]
Zeit
[h]
Umsatz
[%]b
Aus-
beute
[%]c
1 110 Me H B(C6F5)3 (1) rac-118 100 16 0 –
2 111 Me Me B(C6F5)3 (1) rac-119 100 16 5 n.b.
3 111 Me Me B(C6F5)3 (1)/
Mes3P (126)
rac-119 100 16 0 –
4 112 Me tBu B(C6F5)3 (1) rac-120 60 16 95 n.b.
5 112 Me tBu B(C6F5)3 (1) rac-120 100 18 >99 88
6 113 Me SiEt3 B(C6F5)3 (1) rac-121 100 18 5 n.b.
7 114 Me SitBuMe2 B(C6F5)3 (1) rac-122 100 18 27 n.b.
8 115 Me SiiPr3 B(C6F5)3 (1) rac-123 60 16 >99 99
9 112 Me tBu (S)-47·THF 120 100 18 0 –
10 115 Me SiiPr3 (S)-47·THF 123 100 18 0 –
11 116 H tBu B(C6F5)3 (1) 124 60 16 0 –
12 117 H SiiPr3 B(C6F5)3 (1) 125 60 16 0 –
a Die Reaktionen wurden im 90–145-µmol-Maßstab gemäß AAV 4 durchgeführt.
b Bestimmt durch GC-Analyse.
c Bestimmt nach Aufreinigung durch Filtration über Kieselgel; n.b. = nicht bestimmt.
Wir wandten uns als nächstes O-silylierten Oximen zu, um nach der Hydrierung O-silylierte
Hydroxylamine zu erhalten, aus denen sich nach Abspaltung der Silylgruppe ungeschützte
Hydroxylamine freisetzen ließen. Erneut war der sterische Anspruch des Substituenten für
den Erfolg der Umsetzung entscheidend. Sowohl das triethylsilyl- (113, Eintrag 6) als auch
das tert-butyldimethylsilylsubstituierte Oxim (114, Eintrag 7) zeigten niedrigen Umsatz,
während die Hydrierung des triisopropylsilylgeschützten Oxims 115 (Eintrag 8) bei 60 bar
Diwasserstoffdruck und Raumtemperatur das Hydroxylamin rac-123 in quantitativer
Ausbeute lieferte. Diese Reaktion ließ sich ebenso problemlos in einem größeren Maßstab

34 THEORETISCHER TEIL
(1.5 g, 5.1 mmol Startmaterial) durchführen. Eine asymmetrische Reaktionsführung der
zuvor mit B(C6F5)3 (1) erfolgreich reduzierten Substrate 112 und 115 unter Verwendung des
chiralen B(C6F5)3-Derivats (S)-47·THF scheiterte jedoch, es wurde in keinem der beiden
Fälle ein Umsatz der Oximether beobachtet (Einträge 9 und 10). Ebenfalls erfolglos verlief
die Hydrierung der O-tert-butyl- und O-triisopropylsilylsubstituierten Aldoxime 116 und 117
(Einträge 11 und 12). Da eine die Aktivität des Katalysators mindernde Adduktbildung der
Substrate 116 und 117 mit dem Katalysator 1 bei Raumtemperatur durch Analyse einer
äquimolaren Mischung aus Substrat und Katalysator in C6D6 mittels 11B- und 19F-NMR-
Spektroskopie ausgeschlossen wurde[96] (die Oximether 112, 114 und 115 zeigten ebenfalls
keine Adduktbildung mit Katalysator 1), gehen wir davon aus, dass aldehydabgeleitete
Oxime im Gegensatz zu ketonabgeleiteten Oximen eine zur Teilnahme an der kooperativen
Diwasserstoffaktivierung zu geringe LEWIS-Basizität am Stickstoffatom aufweisen.
Nachdem wir geeignete Substituenten am Sauerstoffatom identifiziert hatten, gingen wir
dazu über, die Substratbreite der Hydrierung zu ermitteln und setzten repräsentative O-tert-
butyl- und O-triisopropylgeschützte Ketoximether einem Wasserstoffdruck von 100 bar in
Gegenwart katalytischer Mengen B(C6F5)3 (1) aus (Tabelle 2.2). Sowohl elektronenarme
(126 und 127, Einträge 1 und 2) als auch elektronenreiche Oximether (128 und 129,
Einträge 3 und 4) wurden erfolgreich hydriert und die entsprechenden N-monosubstituierten
Hydroxylamine rac-144–147 in exzellenten Ausbeuten erhalten, wobei für die vollständige
Umsetzung der silylgeschützten Substrate 127 und 129 eine Erhöhung der
Reaktionstemperatur von Raumtemperatur auf 60°C erforderlich war. Im Gegensatz zur
Erhöhung der Temperatur hatte eine Verlängerung der Reaktionszeit generell keinen bis
wenig Einfluss auf den Umsatz. Eine Hydrierung gelang auch für halogenierte (130–133,
Einträge 5–8) und ortho-substituierte Oximether (134 und 135, Einträge 9 und 10). Eine
Dehalogenierung wurde weder für die chlorsubstituierten Substrate 130 und 131 noch für die
bromsubstituerten Substrate 132 und 133 beobachtet. Während der LEWIS-basische
Methoxysubstituent der Oximether 134 und 135 (Einträge 3 und 4) kein Problem darstellte,
wurde ein Imidazolylsubstituent im Oximether 136 nicht toleriert (Eintrag 11). Die
benzophenonabgeleiteten Oximether 137 und 138 (Einträge 12 und 13) zeigten sich weit
weniger reaktiv als arylalkylsubstituierte Substrate. Ein vollständiger Umsatz wurde weder
für das O-tert-butylsubstituierte 137 noch für das O-triisopropylsubstituierte Derivat 138
[96]
Eine Quarternisierung des Boratoms von B(C6F5)3 (1) würde in einem Hochfeldshift des
Signals im 11
B-NMR-Spektrum und einer charakteristischen Veränderung der chemischen
Verschiebung des Signals des para-Fluoratomsignals im 19
F-NMR-Spektrum resultieren. Bei
Koordination einer LEWIS-Base verschiebt sich dieses Resonanzsignal zu höherem Feld, was
häufig durch einen Δδ-Wert relativ zum Resonanzsignal des meta-Fluoratoms ausgedrückt
wird: a) T. Beringhelli, D. Donghi, D. Maggioni, G. D’Alfonso, Coord. Chem. Rev. 2008, 252,
2292–2313; Lit. 44b.

2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 35
erhalten. Letzteres konnte selbst mit erhöhter Temperatur und doppelter
Katalysatorbeladung lediglich zu 50% umgesetzt werden. Die aliphatischen Oximether 139,
140 und rac-141 reagierten langsam bei Raumtemperatur, zeigten jedoch vollständigen
Umsatz bei 60°C (Einträge 14–16). Verschiedene unabhängige Reaktionen ergaben das
Hydroxylamin 159 (Eintrag 16) in stark schwankender Diastereoselektivität. Eine
Erweiterung der Substratbreite auf zyklische Substrate gelang nicht, die Isoxazoline rac-142
(Eintrag 17) und rac-143 (Eintrag 18) zeigten unter den Reaktionsbedingungen keinen
Umsatz. Möglicherweise sind die LEWIS-basischen Atome dieser Substrate aufgrund der
Einbettung in ein Ringsystem zu exponiert, um eine Adduktbildung mit dem Katalysator 1 zu
verhindern.[97]
Tabelle 2.2: Substratbreite der B(C6F5)3-katalysierten Hydrierung von Ketoximethern zu O-
geschützten Hydroxylaminen.a
Ein-
trag Oximether Hydroxylamin
Kataly-
satorbe-
ladung
[Mol-%]
T
[°C]
Umsatz
[%]b
Ausbeute
[%]c
1
2
126 (R3 = tBu)
127 (R3 = SiiPr3)
rac-144 (R3 = tBu)
rac-145 (R3 = SiiPr3)
5.0
5.0
25
60
>99
>99
80
99
3
4
128 (R3 = tBu)
129 (R3 = SiiPr3)
rac-146 (R3 = tBu)
rac-147 (R3 = SiiPr3)
5.0
10
25
60
>99
>99
96
97
[97]
Die Hydrierung der Isoxazoline rac-142 und rac-143 war auch in 1,4-Dioxan erfolglos.
36 THEORETISCHER TEIL
Ein-
trag Oximether Hydroxylamin
Kataly-
satorbe-
ladung
[Mol-%]
T
[°C]
Umsatz
[%]b
Ausbeute
[%]c
5
6
130 (R3 = tBu)
131 (R3 = SiiPr3)
rac-148 (R3 = tBu)
rac-149 (R3 = SiiPr3)
5.0
10
25
60
>99
>99
99
99
7
8
132 (R3 = tBu)
133 (R3 = SiiPr3)
rac-150 (R3 = tBu)
rac-151 (R3 = SiiPr3)
5.0
5.0
25
25
>99
>99
96
95
9
10
134 (R3 = tBu)
135 (R3 = SiiPr3)
rac-152 (R3 = tBu)
rac-153 (R3 = SiiPr3)
8.2
5.0
60
25
95
>99
94d
99
11
136 (R3 = tBu)
rac-154 (R3 = tBu) 10 60 0 –
12
13
137 (R3 = tBu)
138 (R3 = SiiPr3)
155 (R3 = tBu)
156 (R3 = SiiPr3)
10
10
60
60
90
50
66d
n.b.
2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 37
Ein-
trag Oximether Hydroxylamin
Kataly-
satorbe-
ladung
[Mol-%]
T
[°C]
Umsatz
[%]b
Ausbeute
[%]c
14
15
139 (R3 = tBu)
140 (R3 = SiiPr3)
157 (R3 = tBu)
158 (R3 = SiiPr3)
5.0
5.0
60
60
>99
>99
n.b.e
99
16
rac-141 (R3 = SiiPr3)
159 (R3 = SiiPr3) 10 60 >99 89
17
rac-142
160 5.0 25 0 –
18
rac-143
161 5.0 25 0 –
a Die Reaktionen wurden im 0.12–0.14-mmol-Maßstab gemäß AAV 4 durchgeführt.
b Bestimmt durch GC-
Analyse. c Bestimmt nach Aufreinigung durch Filtration über Kieselgel; n.b. = nicht bestimmt.
d Berechnete
Ausbeute nach GC- und 1H-NMR-Analyse, enthält Edukt.
e Wegen Flüchtigkeit nicht isoliert.
Nach der erfolgreichen Entwicklung einer Methode für die C=N-Reduktion von
ketonabgeleiteten Oximethern wandten wir uns erneut dem Problem der Reduktion von
aldehydabgeleiteten Oximethern zu, deren Vertreter sich in der B(C6F5)3-katalysierten
Hydrierung in Toluol als unreaktiv erwiesen hatten (siehe Tabelle 2.1, Einträge 11 und 12).
Wir schrieben dies dem Mangel an LEWIS-Basizität am Stickstoffatom zu, welcher eine
Teilnahme des Substrats an der heterolytischen H–H-Bindungsspaltung verhindert (XLIX,
Abbildung 2.3, links). Bei der Durchführung der Hydrierung in Lösungsmitteln, die eine
Ethergruppe aufweisen (beispielsweise 1,4-Dioxan), könnte dieses Problem umgangen
werden, da das Lösungsmittel selbst in der Lage ist, die Rolle der LEWIS-Base-Komponente
in der Diwasserstoffspaltung einzunehmen (L, Abbildung 2.3, rechts oben). Das entstehende
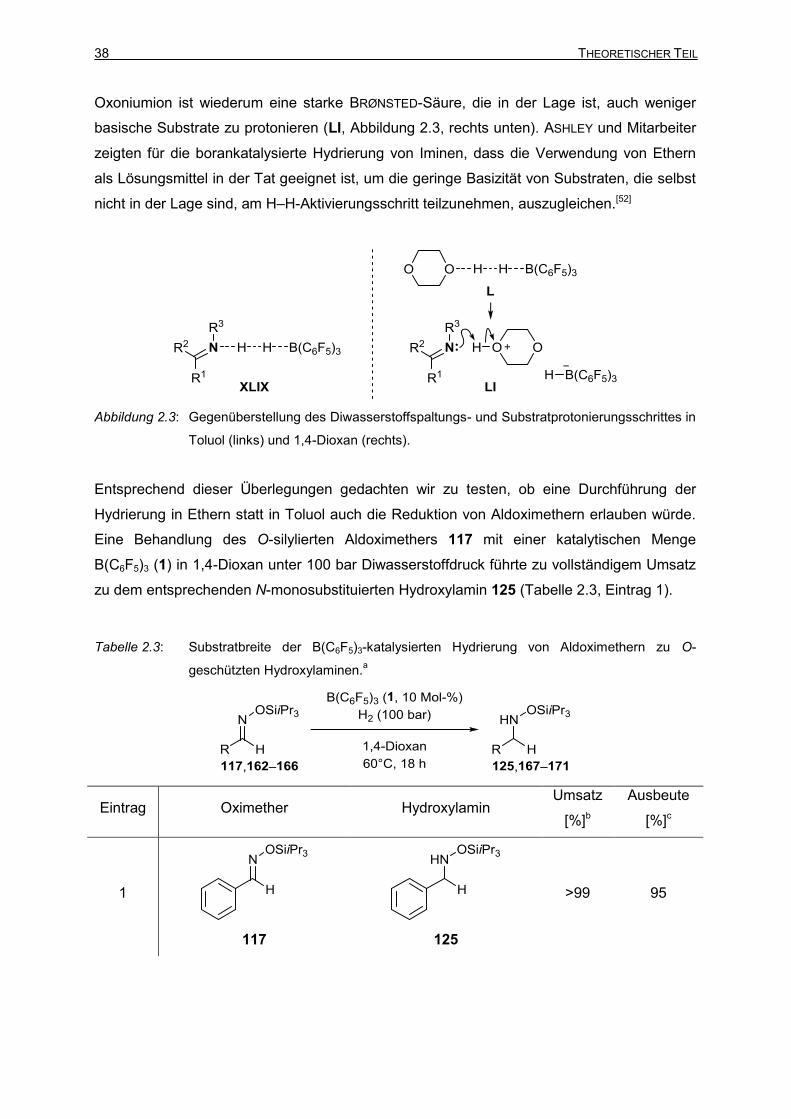
38 THEORETISCHER TEIL
Oxoniumion ist wiederum eine starke BRØNSTED-Säure, die in der Lage ist, auch weniger
basische Substrate zu protonieren (LI, Abbildung 2.3, rechts unten). ASHLEY und Mitarbeiter
zeigten für die borankatalysierte Hydrierung von Iminen, dass die Verwendung von Ethern
als Lösungsmittel in der Tat geeignet ist, um die geringe Basizität von Substraten, die selbst
nicht in der Lage sind, am H–H-Aktivierungsschritt teilzunehmen, auszugleichen.[52]
Abbildung 2.3: Gegenüberstellung des Diwasserstoffspaltungs- und Substratprotonierungsschrittes in
Toluol (links) und 1,4-Dioxan (rechts).
Entsprechend dieser Überlegungen gedachten wir zu testen, ob eine Durchführung der
Hydrierung in Ethern statt in Toluol auch die Reduktion von Aldoximethern erlauben würde.
Eine Behandlung des O-silylierten Aldoximethers 117 mit einer katalytischen Menge
B(C6F5)3 (1) in 1,4-Dioxan unter 100 bar Diwasserstoffdruck führte zu vollständigem Umsatz
zu dem entsprechenden N-monosubstituierten Hydroxylamin 125 (Tabelle 2.3, Eintrag 1).
Tabelle 2.3: Substratbreite der B(C6F5)3-katalysierten Hydrierung von Aldoximethern zu O-
geschützten Hydroxylaminen.a
Eintrag Oximether Hydroxylamin Umsatz
[%]b
Ausbeute
[%]c
1
117
125
>99 95
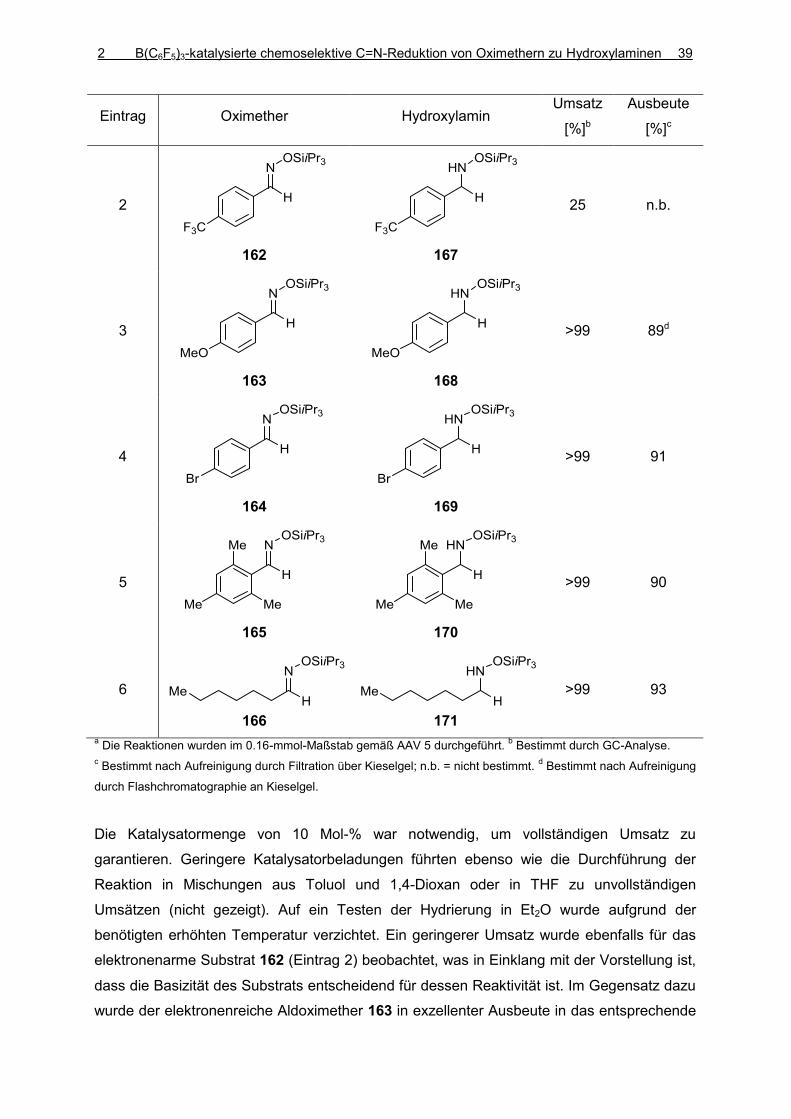
2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 39
Eintrag Oximether Hydroxylamin Umsatz
[%]b
Ausbeute
[%]c
2
162
167
25 n.b.
3
163
168
>99 89d
4
164
169
>99 91
5
165
170
>99 90
6
166 171
>99 93
a Die Reaktionen wurden im 0.16-mmol-Maßstab gemäß AAV 5 durchgeführt.
b Bestimmt durch GC-Analyse.
c Bestimmt nach Aufreinigung durch Filtration über Kieselgel; n.b. = nicht bestimmt.
d Bestimmt nach Aufreinigung
durch Flashchromatographie an Kieselgel.
Die Katalysatormenge von 10 Mol-% war notwendig, um vollständigen Umsatz zu
garantieren. Geringere Katalysatorbeladungen führten ebenso wie die Durchführung der
Reaktion in Mischungen aus Toluol und 1,4-Dioxan oder in THF zu unvollständigen
Umsätzen (nicht gezeigt). Auf ein Testen der Hydrierung in Et2O wurde aufgrund der
benötigten erhöhten Temperatur verzichtet. Ein geringerer Umsatz wurde ebenfalls für das
elektronenarme Substrat 162 (Eintrag 2) beobachtet, was in Einklang mit der Vorstellung ist,
dass die Basizität des Substrats entscheidend für dessen Reaktivität ist. Im Gegensatz dazu
wurde der elektronenreiche Aldoximether 163 in exzellenter Ausbeute in das entsprechende

40 THEORETISCHER TEIL
Hydroxylamin 168 überführt (Eintrag 3). Halogenierung (164→169, Eintrag 4) und sterischer
Anspruch (165→170, Eintrag 5) wurden gleichermaßen toleriert und auch ein
alkylsubstituierter Aldoximether wurde erfolgreich zum entsprechenden Hydroxylamin
hydriert (166→171, Eintrag 6).
2.3 Entfernung der Silylschutzgruppe zur Freisetzung ungeschützter
Hydroxylamine
Da die Reduktion des ungeschützten Oxims 110 erfolglos verlaufen war (siehe Tabelle 2.1,
Eintrag 1), entschieden wir uns, silylgeschützte Oxime zu hydrieren, um aus den
entstehenden O-Silylhydroxylaminen durch Abspaltung der Silylschutzgruppe die freien
Hydroxylamine zu erhalten. Wir untersuchten daher die Möglichkeit der Entschützung des O-
silylierten Hydroxylamins rac-123. Die Si–O-Bindung erwies sich als spaltbar unter sauren
und basischen Bedingungen. Allerdings verhinderten Folgereaktionen die Isolierung des
freien Hydroxylamins rac-118. Abhängig vom Lösungsmittel wurde Zersetzung oder In-situ-
Oxidation zurück zum Oxim 110[98] beobachtet (nicht gezeigt). Ebenso resultierte die
Verwendung von Tetra-n-butylammoniumfluorid (TBAF) in THF zwar in einem Si–O-
Bindungsbruch, ergab jedoch nur eine komplexe Produktmischung. Schlussendlich gelang
es, das freie Hydroxylamin rac-118 nach Behandlung des O-Silylhydroxylamins rac-123 mit
HF·Pyridin in THF bei 0°C in guter isolierter Ausbeute zu erhalten (Schema 2.2).
Schema 2.2: Entfernung der Silylschutzgruppe zur Freisetzung des ungeschützten Hydroxylamins
rac-118.
[98]
S. Horiyama, K. Suwa, M. Yamaki, H. Kataoka, T. Katagi, M. Takayama, T. Takeuchi, Chem.
Pharm. Bull. 2002, 50, 996–1000.

2 B(C6F5)3-katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen 41
2.4 Fazit
Im vorliegenden Kapitel wurde die erfolgreiche Entwicklung von Methoden für die homogen
katalysierte chemoselektive C=N-Reduktion von Oximethern zu Hydroxylaminen
beschrieben. Während der Einsatz von Hydrosilanen und katalytischer Mengen der
Hauptgruppen-LEWIS-Säure B(C6F5)3 (1) an der Oxophilie des Siliciumatoms scheiterte,
wurde mit Diwasserstoff als Reduktionsmittel die Überführung von O-Alkyl- und O-
Silyloximethern in die entsprechenden N-monosubstituierten Hydroxylamine ohne N–O-
Bindungsbruch ermöglicht. Hierbei war die Wahl der Schutzgruppe für den Erfolg der
Reaktion entscheidend; als geeignet wurden sterisch anspruchsvolle Gruppen identifiziert
(tert-Butyl und Triisopropyl). Die Verwendung von Diwasserstoff stellte zudem sehr saubere
Umsetzungen sicher, die in einfacher Aufreinigung der Reaktionsmischungen und generell
hohen Ausbeuten der Produkte resultierten. Ketoximether wurden bei Raumtemperatur oder
60°C in Toluol unter einem Diwasserstoffdruck von 100 bar reduziert, wofür eine
Katalysatorbeladung von 5.0 Mol-% in den meisten Fällen ausreichte. Elektronische und
sterische Eigenschaften der Substituenten am Oximkohlenstoffatom waren dabei für den
Erfolg der Reduktion weitgehend unbedeutend. Enantiomerenangereicherte Hydroxylamine
durch die Verwendung eines chiralen B(C6F5)3-Derivats konnten bislang allerdings nicht
erhalten werden. Für die Hydrierung von Aldoximethern erwies sich die Reduktionsmethode
in Toluol als ungeeignet. Die geringere Basizität aldehydabgeleiteter Substrate, welche
verhindert, dass diese an der kooperativen H–H-Bindungsspaltung mit dem LEWIS-Säure-
Katalysator teilnehmen, konnte durch Reaktionsführung in 1,4-Dioxan, welches selbst in der
Lage ist, als LEWIS-Base in ebendiesem Schritt zu fungieren, kompensiert werden.
Zusätzlich war eine im Vergleich zur Hydrierung von Ketoximethern höhere
Katalysatorbeladung von 10 Mol-% notwendig. Es wurde zudem gezeigt, dass sich
ungeschützte Hydroxylamine durch die Entfernung der Triisopropylsilylgruppe aus O-
silylierten Hydroxylaminen freisetzen lassen.

42 THEORETISCHER TEIL
3 B(C6F5)3-KATALYSIERTE C=N-REDUKTION VON
HYDRAZONEN ZU HYDRAZINEN
In diesem Kapitel wird die Erweiterung der metallfreien katalytischen C=N-Reduktion auf die
Substratklasse der Hydrazone beschrieben.[94] Es werden sowohl die Anwendung der durch
elektronenarme Borane katalysierten Hydrosilylierung als auch die der entsprechenden
Hydrierung dargestellt. Im Falle der Hydrierung wird analog zu den Ergebnissen in der
Oximetherreduktion (siehe Kapitel 2) ein Vergleich zwischen keton- und aldehydabgeleiteten
Substraten angestellt. Die Ergebnisse in den Kapiteln 3.2 und 3.3 wurden in
Zusammenarbeit mit DIGVIJAY PORWAL und Dr. INDRANIL CHATTERJEE erarbeitet.
3.1 Versuche zur C=N-Reduktion von Hydrazonen durch
borankatalysierte Hydrosilylierung
Wir testeten zunächst die Methode der B(C6F5)3-katalysierten Hydrosilylierung, die zur
Reduktion von Iminen geeignet ist (siehe Kapitel 1.2.2), in der Reduktion von Oximen jedoch
an der Oxophilie des Siliciums gescheitert war (siehe Kapitel 2.1). Anhand der Umsetzung
der dibenzylgeschützten Hydrazone 172 und 173 mit Me2PhSiH (19) und katalytischen
Mengen an B(C6F5)3 (1) bei Raumtemperatur erprobten wir die generelle Durchführbarkeit
genannter Reduktionsmethode für diese Substratklasse (Schema 3.1). Nach einer
Reaktionszeit von 18 h stellten wir für das ketonabgeleitete Substrat 172 wie auch für das
aldehydabgeleitete Substrat 173 einen Umsatz zu den Reduktionsprodukten rac-174 bzw.
175 fest, welcher sich jedoch durch eine Verlängerung der Reaktionszeit nicht bzw. nur
geringfügig steigern ließ (nicht gezeigt).
Schema 3.1: Versuch der B(C6F5)3-katalysierten Hydrosilylierung dibenzylgeschützter Hydrazone.
Wir gingen dazu über, weitere Testreaktionen an dem Hydrazon 176 durchzuführen,
welches eine einfach zu entfernende Phthaloylschutzgruppe trägt (Schema 3.2). Eine

3 B(C6F5)3-katalysierte C=N-Reduktion von Hydrazonen zu Hydrazinen 43
Behandlung mit Et3SiH (4, keine Produktbildung beobachtbar) bzw. Me2PhSiH (19, Spuren
des Produkts 177) verlief noch erfolgloser als im Falle der dibenzylgeschützten Substrate.
Die Verwendung von PhSiH3 (25) ergab zwar einen vollständigen Umsatz, das allerdings
erst bei erhöhter Temperatur und in einem Produktgemisch resultierend. Auch mit den
Cyclohexa-2,5-dien-1-yl-substituierten Surrogaten von Me3SiH (75) und SiH4 (77) konnte das
Hydrazin 177 nur als Teil einer Mischung erhalten werden.
Schema 3.2: Versuch der B(C6F5)3-katalysierten Hydrosilylierung eines phthaloylgeschützten
Hydrazons.
Die massenspektrometrische Analyse der erhaltenen Produktgemische zeigte, dass eine
Desoxygenierung des Phthaloylsubstituenten eine konkurrierende Reaktion zur
gewünschten C=N-Reduktion darstellte (nicht gezeigt). Wie für die Reduktion der Oxime
durch Hydrosilylierung beobachtet (siehe Kapitel 2) tritt hier ebenso eine Konkurrenz zweier
LEWIS-basischer Atome auf (LII vs. LIII, Abbildung 3.1), wenn auch nicht
substratklasseninhärent, sondern durch die Wahl der Schutzgruppe. Das Auftreten der
Desoxygenierung der Phthaloylschutzgruppe ist rückblickend als wenig überraschend
einzustufen, da B(C6F5)3-katalysierte Desoxygenierungsmethoden von Amiden mit
Hydrosilanen bereits zuvor entwickelt wurden.[99]
[99]
Lit. 63a; a) E. Blondiaux, T. Cantat, Chem. Commun. 2014, 50, 9349–9352; b) R. C.
Chadwick, V. Kardelis, P. Lim, A. Adronov, J. Org. Chem. 2014, 79, 7728–7733.

44 THEORETISCHER TEIL
Abbildung 3.1: Konkurrenz basischer Atome in kooperativer Si–H-Bindungsspaltung durch B(C6F5)3
(1) und phthaloylgeschützte Hydrazone.
3.2 C=N-Reduktion von Hydrazonen durch borankatalysierte
Hydrierung
Aufgrund der auftretenden Probleme in der Hydrazonreduktion durch borankatalysierte
Hydrosilylierung konzentrierten wir unsere Bemühungen im Folgenden auf die verwandte
Hydrierung, die sich bereits in der Oximetherreduktion bewährt hatte (siehe Kapitel 2.2).
Hierzu verglichen wir zunächst das Verhalten unterschiedliche Schutzgruppen tragender
acetophenon- und benzaldehydabgeleiteter Hydrazone in den im Kapitel 2.2 vorgestellten
katalytischen Hydrierungsmethoden in Toluol und 1,4-Dioxan (Tabelle 2.1). Im Gegensatz
zur Oximetherreduktion war ein hoher sterischer Anspruch der Schutzgruppe für den Erfolg
der Hydrazonreduktion nicht zwingend notwendig, wie die erfolgreiche Umsetzung des N,N-
dimethylgeschützten acetophenonabgeleiteten Hydrazons 178 in Toluol unter einem
Diwasserstoffdruck von 100 bar mit 5.0 Mol-% B(C6F5)3 (1) zeigte (Tabelle 3.1, Eintrag 1).
Die Reaktionsführung in 1,4-Dioxan ergab allerdings keine vollständige Umsetzung des
Hydrazons 178 (Eintrag 2). Durch einen Wechsel der Schutzgruppen zu sterisch
anspruchsvolleren Benzylsubstituenten wurde eine vollständige Umsetzung unabhängig vom
Lösungsmittel sichergestellt (172→rac-174, Einträge 3 und 4). Im Gegensatz dazu erreichte
das entsprechende benzaldehydabgeleitete Hydrazon 173 auch bei erhöhter
Katalysatorbeladung von 10 Mol-% keinen vollständigen Umsatz (Einträge 5 und 6). Wir
erprobten dann die Hydrierung der phthaloylgeschützten Substrate 179 und 176, um zu
leicht entschützbaren Hydrazinen zu gelangen. Das ketonabgeleitete Hydrazon 179
reagierte in beiden Lösungsmitteln chemoselektiv zu dem entsprechenden Hydrazin rac-181
(Einträge 7 und 8), während das weniger basische aldehydabgeleitete Hydrazon 176 erneut
schlechtere Ergebnisse lieferte. In Toluol wurde keine katalytische Umsetzung beobachtet
(Eintrag 9) und in 1,4-Dioxan war der Umsatz zwar höher, jedoch immer noch
unbefriedigend (Eintrag 10). Eine Verdopplung der Katalysatorbeladung auf 10 Mol-% war

3 B(C6F5)3-katalysierte C=N-Reduktion von Hydrazonen zu Hydrazinen 45
notwendig, um einen vollständigen Umsatz des Hydrazons 176 zum Hydrazin 177 zu
erhalten (Eintrag 11).
Tabelle 3.1: Katalytische Hydrierung von Hydrazonen zu Hydrazinen: Vergleich von
Schutzgruppen und Einfluss des Lösungsmittels.a
Ein-
trag
Hydra-
zon R1 R2 R3 Hydrazin
Katalysator-
beladung
[Mol-%]
Lösungs-
mittel
Umsatz
[%]b,c
Aus-
beute
[%]d
1 178 Me Me Me rac-180 5.0 Toluol >99 n.b.
2 178 Me Me Me rac-180 5.0 1,4-Dioxan 85 n.b.
3 172 Me Bn Bn rac-174 5.0 Toluol >99 89
4 172 Me Bn Bn rac-174 5.0 1,4-Dioxan >99 n.b.
5 173 H Bn Bn 175 10 Toluol 48 n.b.
6 173 H Bn Bn 175 10 1,4-Dioxan 77 n.b.
7 179 Me Phthaloyl rac-181 5.0 Toluol >99 95
8 179 Me Phthaloyl rac-181 5.0 1,4-Dioxan >99 n.b.
9 176 H Phthaloyl 177 5.0 Toluol 3 n.b.
10 176 H Phthaloyl 177 5.0 1,4-Dioxan 13 n.b.
11 176 H Phthaloyl 177 10 1,4-Dioxan >99 94
a Die Reaktionen wurden im 0.15–0.17-mmol-Maßstab gemäß AAV 4 (in Toluol) oder AAV 5 (in 1,4-Dioxan)
durchgeführt. b Bestimmt durch GC-Analyse.
c In Fällen von Reaktionen, die nicht vollständig abliefen, variierten
die Umsätze teilweise deutlich. Die angegebenen Umsätze repräsentieren die niedrigsten Umsätze, die erhalten
wurden. d Bestimmt nach Aufreinigung durch Filtration über Kieselgel; n.b. = nicht bestimmt.
Die in Tabelle 3.1 dargestellten Ergebnisse zeigen, dass analog zur Hydrierung von
Oximethern eine externe LEWIS-Base wie 1,4-Dioxan zur Reduktion von ketonabgeleiteten
Substraten nicht benötigt wird und diese bereits in nicht koordinierendem Toluol reduzierbar
sind. Wir entschieden uns daher, die B(C6F5)3-katalysierte Hydrierung weiterer
ketonabgeleiteter phthaloylgeschützter Hydrazone ausschließlich in Toluol durchzuführen
(Tabelle 3.2).
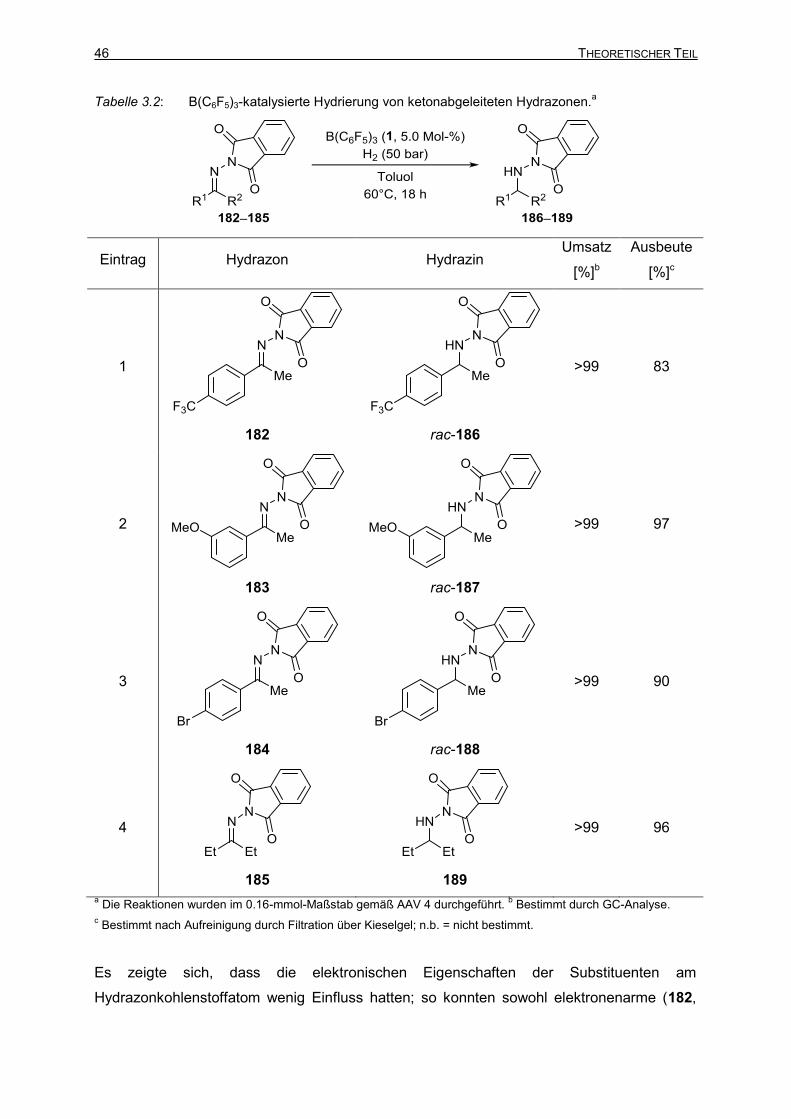
46 THEORETISCHER TEIL
Tabelle 3.2: B(C6F5)3-katalysierte Hydrierung von ketonabgeleiteten Hydrazonen.a
Eintrag Hydrazon Hydrazin Umsatz
[%]b
Ausbeute
[%]c
1
182
rac-186
>99 83
2
183
rac-187
>99 97
3
184
rac-188
>99 90
4
185
189
>99 96
a Die Reaktionen wurden im 0.16-mmol-Maßstab gemäß AAV 4 durchgeführt.
b Bestimmt durch GC-Analyse.
c Bestimmt nach Aufreinigung durch Filtration über Kieselgel; n.b. = nicht bestimmt.
Es zeigte sich, dass die elektronischen Eigenschaften der Substituenten am
Hydrazonkohlenstoffatom wenig Einfluss hatten; so konnten sowohl elektronenarme (182,
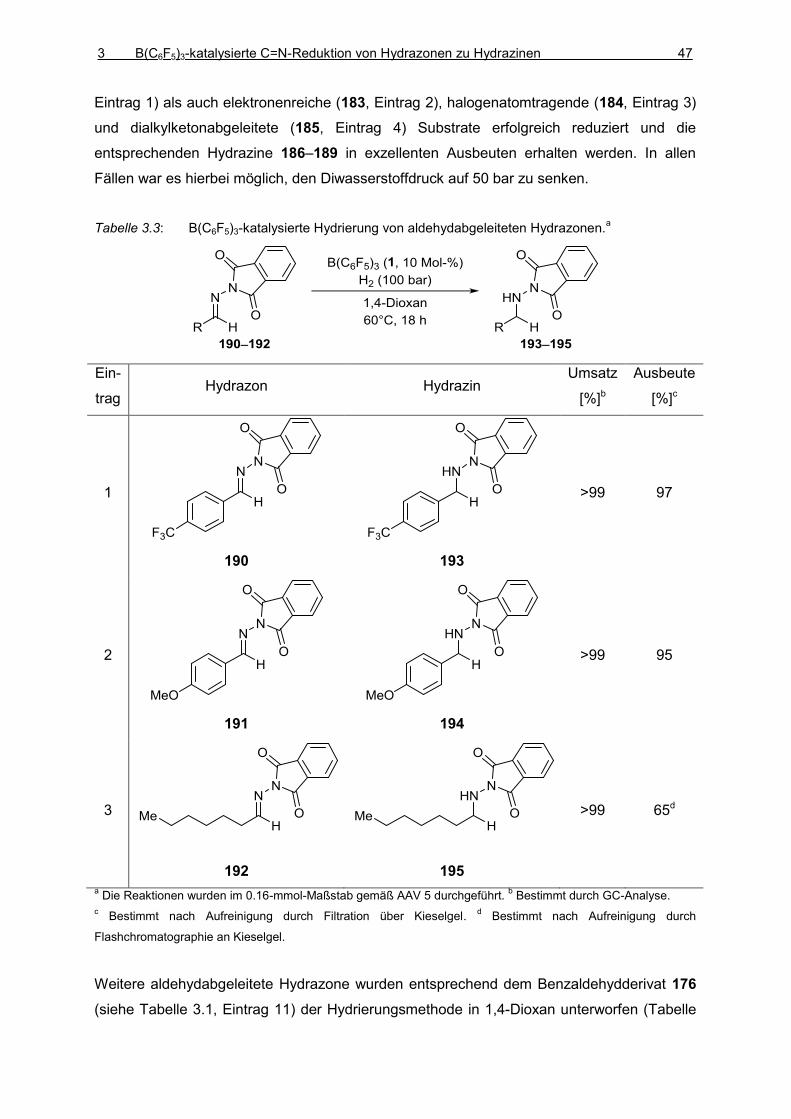
3 B(C6F5)3-katalysierte C=N-Reduktion von Hydrazonen zu Hydrazinen 47
Eintrag 1) als auch elektronenreiche (183, Eintrag 2), halogenatomtragende (184, Eintrag 3)
und dialkylketonabgeleitete (185, Eintrag 4) Substrate erfolgreich reduziert und die
entsprechenden Hydrazine 186–189 in exzellenten Ausbeuten erhalten werden. In allen
Fällen war es hierbei möglich, den Diwasserstoffdruck auf 50 bar zu senken.
Tabelle 3.3: B(C6F5)3-katalysierte Hydrierung von aldehydabgeleiteten Hydrazonen.a
Ein-
trag Hydrazon Hydrazin
Umsatz
[%]b
Ausbeute
[%]c
1
190
193
>99 97
2
191
194
>99 95
3
192
195
>99 65d
a Die Reaktionen wurden im 0.16-mmol-Maßstab gemäß AAV 5 durchgeführt.
b Bestimmt durch GC-Analyse.
c Bestimmt nach Aufreinigung durch Filtration über Kieselgel.
d Bestimmt nach Aufreinigung durch
Flashchromatographie an Kieselgel.
Weitere aldehydabgeleitete Hydrazone wurden entsprechend dem Benzaldehydderivat 176
(siehe Tabelle 3.1, Eintrag 11) der Hydrierungsmethode in 1,4-Dioxan unterworfen (Tabelle

48 THEORETISCHER TEIL
3.3). Auch hier ließen sich das elektronenarme Hydrazon 190 und das elektronenreiche
Hydrazon 191 problemlos zu den entsprechenden Hydrazinen 193 und 194 reduzieren
(Einträge 1 und 2). Bei der Hydrierung des alkylsubstituerten Hydrazons 192 wurden geringe
Mengen nicht identifizierter Nebenprodukte beobachtet, die eine Aufreinigung per
Flashchromatographie nötig machten und in einer im Vergleich geringeren Ausbeute des
Hydrazins 195 resultierten (Eintrag 3).
3.3 Entfernung der Phthaloylschutzgruppe
Nach der erfolgreichen Hydrierung phthaloylgeschützter keton- und aldehydabgeleiteter
Hydrazone galt es, die Möglichkeit der Freisetzung ungeschützter Hydrazine durch
Entfernung der Schutzgruppe aus den Reduktionsprodukten zu zeigen. Die Entschützung
des phthaloylsubstituierten Hydrazins 177 durch Behandlung mit Hydrazinhydrat (197,
Schema 3.3, oben) war bereits von der Gruppe um HEARN beschrieben worden.[100] Die
Entfernung der Phthaloylschutzgruppe aus dem Hydrazin rac-181 gelang uns ebenfalls
durch dessen Umsetzung mit Hydrazinhydrat (197, Schema 3.3, unten). Um die Isolierung
zu vereinfachen, wurde das freie Hydrazin in situ mit Essigsäure in das acetylsubstituierte
Hydrazin rac-198 überführt, welches in guter Ausbeute erhalten wurde.
Schema 3.3: Entfernung der Phthaloylgruppe geschützter Hydrazine.
[100]
M. J. Hearn, E. R. Lucero, J. Heterocycl. Chem. 1982, 19, 1537–1539.

3 B(C6F5)3-katalysierte C=N-Reduktion von Hydrazonen zu Hydrazinen 49
3.4 Fazit
Im vorangegangenen Kapitel wurden übergangsmetallfreie katalytische Methoden zur C=N-
Reduktion von Hydrazonen zu Hydrazinen beschrieben. Borankatalysierte
Hydrosilylierungen waren zwar prinzipiell möglich, aber aufgrund geringer Reaktivität oder
mangelnder Chemoselektivität infolge der Schutzgruppenwahl nicht praktikabel. Eine
effektive Hydrazonreduktion bot hingegen die B(C6F5)3-katalysierte Hydrierung. Wie im Falle
der Oximetherreduktion (siehe Kapitel 2) zeigte sich auch für die Substratklasse der
Hydrazone ein Reaktivitätsunterschied zwischen keton- und aldehydabgeleiteten Substraten.
Während aldehydabgeleitete phthaloylgeschützte Hydrazone die Verwendung eines
Etherlösungsmittels als externe LEWIS-Base zur Diwasserstoffaktivierung und höhere
Katalysatorbeladungen verlangten, konnten die entsprechenden LEWIS-basischeren
ketonabgeleiteten Substrate bereits in nicht koordinierendem Toluol bei geringeren
Diwasserstoffdrücken reduziert werden. Zudem wurde gezeigt, dass sich die gewählte
Phthaloylschutzgruppe aus den Reduktionsprodukten einfach entfernen ließ.

50 THEORETISCHER TEIL
4 EXKURS: SILYLIUMIONVERMITTELTE C=X-REDUKTIONEN
MIT HYDROSILANEN
Ebenso wie elektronenarme Borverbindungen lassen sich auch elektrophile
Siliciumverbindungen als LEWIS-Säure-Katalysatoren einsetzen.[101] Silyliumionen, dreifach
koordinierte tetravalente Siliciumkationen,[102] sind isovalenzelektronisch zu Boranen wie
B(C6F5)3 (1) und ihren neutralen, tetrakoordinierten Verwandten als Katalysatoren oftmals
überlegen.[103] Nachdem sich die Forschung der Silyliumionenchemie zunächst auf die
Isolierung und Charakterisierung dieser reaktiven Verbindungen in kondensierter Phase
konzentrierte, finden sich in der darauffolgenden Zeit vermehrt Anwendungen von
Silyliumionen in LEWIS-Säure-katalysierten Reaktionen.[104]
Ein frühes Beispiel einer silyliumionvermittelten C=C-Reduktion zeigten LAMBERT und
Mitarbeiter, die Diphenylethylen (199) mit Et3SiH (4) und einer katalytischen Menge an
Tritylborat Ph3C+[B(C6F5)4]
– (200+[B(C6F5)4]–) zum Hydrosilylierungsprodukt 201 umsetzten
(Schema 4.1).[105]
Schema 4.1: Silyliumionvermittelte C=C-Reduktion nach LAMBERT.
[101]
Eine Übersicht bietet: A. D. Dilman, S. L. Ioffe, Chem. Rev. 2003, 103, 733–772. [102]
Übersichtsartikel zu Silyliumionen: a) T. A. Kochina, D. V. Vrazhnov, I. S. Ignatyev, E. N.
Sinotova, M. G. Voronkov, J. Organomet. Chem. 2011, 696, 1331–1340; b) H. Čičak, Kem.
Ind. 2010, 59, 111–124; c) T. A. Kochina, D. V. Vrazhnov, E. N. Sinotova, M. G. Voronkov,
Russ. Chem. Rev. 2006, 75, 95–110; d) T. Müller, Adv. Organomet. Chem. 2005, 53, 155–
215; e) J. B. Lambert, Y. Zhao, S. M. Zhang, J. Phys. Org. Chem. 2001, 14, 370–379; f) C. A.
Reed, Acc. Chem. Res. 1998, 31, 325–332; g) J. B. Lambert, L. Kania, S. Zhang, Chem. Rev.
1995, 95, 1191–1201; h) R. J. P. Corriu, M. Henner, J. Organomet. Chem. 1974, 74, 1–28. [103]
Für eine vergleichende Studie siehe: K. Hara, R. Akiyama, M. Sawamura, Org. Lett. 2005, 7,
5621–5623. [104]
Für Übersichtsartikel zu Anwendungen von Silyliumionen in der Katalyse siehe: a) A. Schulz,
A. Villinger, Angew. Chem. Int. Ed. 2012, 51, 4526–4528; b) H. F. T. Klare, M. Oestreich,
Dalton Trans. 2010, 39, 9176–9184. [105]
J. B. Lambert, Y. Zhao, H. Wu, J. Org. Chem. 1999, 64, 2729–2736.

4 Exkurs: Silyliumionvermittelte C=X-Reduktionen mit Hydrosilanen 51
Aus dem Tritylkation 200+ und einer geringen Menge des Hydrosilans 4 wird durch
Hydridtransfer das Silyliumion Et3Si+ (202+) generiert,[106] welches zunächst durch
Koordination an das Lösungsmittel C6D6 stabilisiert vorliegt (Schema 4.2). Die so gebildete
katalytische Menge an Silyliumion 202+ wird durch Reaktion mit dem Alken 199 unter
Bildung des Kations 204+ verbraucht und anschließend im produktbildenden Schritt durch
Hydridtransfer des Hydrosilans 4 zurückgebildet.
Schema 4.2: Mechanismus der silyliumionvermittelten C=C-Reduktion nach LAMBERT. Das
Gegenanion [B(C6F5)4]– ist zur besseren Übersicht nicht dargestellt.
Die entsprechende trialkylsilyliumionvermittelte C=O-Reduktion von Carbonylverbindungen
wurde bereits zuvor von KIRA und Mitarbeitern untersucht, die jedoch nicht die erwarteten
Silylether, sondern defunktionalisierte Alkane oder Alkene in Folge von Deoxygenierung
erhielten (nicht gezeigt).[107,108] PIERS und Mitarbeiter zeigten, dass die für die
[106]
Dieser Silicium-zu-Kohlenstoff-Hydridtransfer wird gemeinhin als BARTLETT-CONDON-
SCHNEIDER-Hydridtransfer bezeichnet: a) P. D. Bartlett, F. E. Condon, A. Schneider, J. Am.
Chem. Soc. 1944, 66, 1531–1539. Nach den Arbeiten von COREY hat sich diese Technik zur
Generierung von Silyliumionen als Standardmethode etabliert: b) J. Y. Corey, J. Am. Chem.
Soc. 1975, 97, 3237–3238; c) J. Y. Corey, D. Gust, K. Mislow, J. Organomet. Chem. 1975,
101, C7–C8. Weitere Methoden zur Generierung sind der „allyl leaving group approach“: d) J.
B. Lambert, Y. Zhao, H. Wu, W. C. Tse, B. Kuhlmann, J. Am. Chem. Soc. 1999, 121, 5001–
5008; der „cyclohexadienyl leaving group approach“: e) A. Simonneau, T. Biberger, M.
Oestreich, Organometallics 2015, 34, 3927–3929; durch Substituentenaustausch zuvor
gebildeter Silyliumionen: f) A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase, T.
Müller, Angew. Chem. Int. Ed. 2011, 50, 12636–12638; g) M. Reißmann, A. Schäfer, S. Jung,
T. Müller, Organometallics 2013, 32, 6736–6744. [107]
M. Kira, T. Hino, H. Sakurai, Chem. Lett. 1992, 555–558.

52 THEORETISCHER TEIL
Desoxygenierung verantwortliche Bildung von Disiloxan als gute Abgangsgruppe in der
silyliumionvermittelten Hydrosilylierung im Gegensatz zur Situation in der B(C6F5)3-
katalysierten Carbonylhydrosilylierung bevorzugt ist.[13] In letzterem Fall erfolgt diese nur bei
Verwendung eines Überschusses an Hydrosilan.[109]
Erst durch den Einsatz des ferrocenstabilisierten
Silyliumions 205+ (Abbildung 4.1)[110] gelang die
Hydrosilylierung von Carbonylverbindungen zu Silylethern in
unserem Arbeitskreis (Schema 4.3).[111] Ebenso wie bei
LAMBERTs Arbeit wurde eine katalytische Menge des
Tritylborats 200+[B(C6F5)4]– zur Generierung des Silyliumions
205+ aus dem Hydrosilan 206 genutzt. Die Hydrosilylierung
von Ketonen (207, 208) zu Silylethern (209, 210) erfolgte in hohen Ausbeuten. Entscheidend
für die Unterdrückung der Desoxygenierung scheint die einzigartige intramolekulare
Stabilisierung des kationischen Siliciumzentrums des Silyliumions 205+ durch dessen
Ferrocenylsubstituenten und die sich hieraus ergebende Hydriddonorstärke des
entsprechenden Hydrosilans 206 zu sein.[112]
[108]
Für silyliumionvermittelte Reduktionen von CO2 mit Hydrosilanen als Reduktionsmittel siehe:
a) A. Schäfer, W. Saak, D. Haase, T. Müller, Angew. Chem. Int. Ed. 2012, 51, 2981–2984; b)
M. Khandelwal, R. J. Wehmschulte, Angew. Chem. Int. Ed. 2012, 51, 7323–7326. [109]
V. Gevorgyan, J.-X. Liu, M. Rubin, S. Benson, Y. Yamamoto, Tetrahedron Lett. 1999, 40,
8919–8922. [110]
Für die Beschreibung der Stabilisierung und Bindungsverhältnisse in Silyliumion 205+ siehe:
a) K. Müther, R. Fröhlich, C. Mück-Lichtenfeld, S. Grimme, M. Oestreich, J. Am. Chem. Soc.
2011, 133, 12442–12444; für weitere ferrocenstabilisierte Silyliumionen siehe: b) K. Müther, P.
Hrobárik, V. Hrobáriková, M. Kaupp, M. Oestreich, Chem. Eur. J. 2013, 19, 16579–16594; für
Anwendungen ferrocenstabilisierter Silyliumionen als Katalysatoren in DIELS–ALDER-
Reaktionen siehe: c) H. F. T. Klare, K. Bergander, M. Oestreich, Angew. Chem. Int. Ed. 2009,
48, 9077–9079; d) R. K. Schmidt, K. Müther, C. Mück-Lichtenfeld, S. Grimme, M. Oestreich, J.
Am. Chem. Soc. 2012, 134, 4421–4428; e) A. R. Nödling, K. Müther, V. H. G. Rohde, G. Hilt,
M. Oestreich, Organometallics 2014, 33, 302–308; f) R. K. Schmidt, H. F. T. Klare, R.
Fröhlich, M. Oestreich, Chem. Eur. J. 2016, 22, 5367–5383. [111]
K. Müther, M. Oestreich, Chem. Commun. 2011, 47, 334–336. [112]
Für eine Carbonylhydrosilylierungsmethode mit einem neutralen tetrakoordinierten Silicium-
LEWIS-Säure-Katalysator, in der Desoxygenierungsreaktionen ebenfalls unterdrückt wurden,
siehe: a) A. L. Liberman-Martin, R. G. Bergman, T. D. Tilley, J. Am. Chem. Soc. 2015, 137,
5328–5331. Für mechanistisch verwandte durch siliciumhaltige Metallkomplexe katalysierte
Carbonylhydrosilylierungen siehe: b) T. Muraoka, Y. Shimizu, H. Kobayashi, K. Ueno, H.
Ogino, Organometallics 2010, 29, 5423–5426; c) T. T. Metsänen, D. Gallego, T. Szilvási, M.
Driess, M. Oestreich, Chem. Sci. 2015, 6, 7143–7149.
Abbildung 4.1: Das ferrocen-
stabilisierte Silyliumion 205+.

4 Exkurs: Silyliumionvermittelte C=X-Reduktionen mit Hydrosilanen 53
Schema 4.3: Silyliumionvermittelte C=O-Reduktion nach OESTREICH. Fc = Ferrocenyl.
Silyliumionvermittelte Reduktionsmethoden mit Diwasserstoff als Reduktionsmittel sind im
Gegensatz zu den vorgestellten Hydrosilylierungen bisher nicht bekannt, obschon von
einigen Kombinationen aus Silyliumion und LEWIS-Base-Partnern berichtet wurde, die in der
Lage sind, Diwasserstoff heterolytisch zu spalten.[113]
Wir stellten uns die Frage, ob sich die silyliumionvermittelte Hydrosilylierung auch auf die
C=N-Reduktion von Iminen zu Aminen anwenden ließe. Nachfolgend wird die Entwicklung
einer solchen Methode beschrieben und die katalytische Aktivität des
ferrocenylsubstituierten Silyliumions 205+ mit der des Triethylsilyliumions 202+ verglichen.
Die vorgestellten Ergebnisse wurden in Zusammenarbeit mit Dr. KRISTINE MÜTHER
erarbeitet.[114]
4.1 Silyliumionvermittelte C=N-Reduktion von Iminen mit
Hydrosilanen
Zur Entwicklung einer silyliumionvermittelten Iminreduktionsmethode begannen wir damit,
repräsentative benzaldehydabgeleitete Imine mit verschiedenen Schutzgruppen am
Stickstoffatom zu testen (Tabelle 4.1). Wir behandelten die Aldimine (E)-51,(E)-211–(E)-216
mit einer Mischung aus durch Reaktion des Hydrosilans 206 mit Tritylkation 200+
präformiertem ferrocenstabilisierten Silyliumion 205+ und Hydrosilan 206, um nach
Hydrosilylierung und Hydrolyse die entsprechenden Amine 52,217–222 zu erhalten. Da wir
diese Reaktionen bei Raumtemperatur durchführten, entschieden wir uns statt CH2Cl2 (wie
in der Carbonylreduktion, siehe oben) 1,2-Cl2C6H4 oder 1,2-F2C6H4 als Lösungsmittel
einzusetzen. In diesen ist das Silyliumion 205+ bei Raumtemperatur stabil,[110a] in CH2Cl2
[113]
Lit. 106f, 106g; a) A. Schäfer, M. Reißmann, A. Schäfer, M. Schmidtmann, T. Müller, Chem.
Eur. J. 2014, 20, 9381–9386; b) T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J.
P. White, P. A. Hunt, A. E. Ashley, Chem. Commun. 2014, 50, 12753–12756. [114]
K. Müther, J. Mohr, M. Oestreich, Organometallics 2013, 32, 6643–6646.
Synthesevorschriften für und Charakterisierungen von Substanzen, die im experimentellen
Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.

54 THEORETISCHER TEIL
hingegen nicht.[110c] Diphenylphosphinoyl- [(E)-211, Eintrag 1], tert-butoxycarbonyl- [(E)-212,
Eintrag 2] und benzylgeschützte [(E)-213, Eintrag 3] Aldimine zeigten keinen Umsatz unter
den Reaktionsbedingungen. Eine langsame Hydrosilylierung wurde für das
benzhydrylsubstituierte Imin (E)-214 beobachtet und das Reduktionsprodukt 220 in 40%
Ausbeute erhalten (Eintrag 4). Der Einsatz des phenylgeschützten Imins (E)-215 resultierte
in einer sehr guten Ausbeute des entsprechenden Amins 221, allerdings nach langer
Reaktionszeit (Eintrag 5). Das tosylgeschütze Imin (E)-51 wurde schließlich vollständig
umgesetzt und das Amin 52 in 90% Ausbeute erhalten (Eintrag 6). Das entsprechende
tosylgeschützte acetophenonabgeleitete Ketimin (E)-216 ließ sich dagegen nur unvollständig
in sein Reduktionsprodukt rac-222 überführen (Eintrag 7).
Tabelle 4.1: Silyliumionvermittelte Reduktion von Iminen: Wahl der Schutzgruppe.a
Eintrag Imin R1 R2 Amin Zeit [h] Ausbeute [%]b
1 (E)-211 H P(O)Ph2 217 70 – (kein Umsatz)
2 (E)-212 H CO2tBu 218 70 – (kein Umsatz)
3 (E)-213 H Bn 219 70 – (kein Umsatz)
4 (E)-214 H CHPh2 220 142 40d
5 (E)-215 H Ph 221 46 84
6c (E)-51 H SO2Tol 52 16 90
7c (E)-216 Me SO2Tol rac-222 119 19d
a Die Reaktionen wurden im 0.200–0.252-mmol-Maßstab gemäß AAV 6 oder ähnlich durchgeführt.
b Bestimmt
nach Aufreinigung durch Flashchromatographie an Kieselgel. c Verwendung von 1,2-Cl2C6H4 als Lösungsmittel.
d Es wurde kein vollständiger Umsatz des Imins erreicht.
In Analogie zu den bekannten silyliumionvermittelten C=C-[105] und C=O-Reduktionen[111]
nehmen wir für die C=N-Reduktion einen ähnlichen Mechanismus an (Schema 4.4). Durch
Hydridtransfer des Hydrosilans LIV auf das Tritylkation 200+ wird zunächst eine katalytische
Menge an Silyliumion LV+ generiert. Das bisher eingesetzte ferrocenylsubstituierte
Silyliumion 205+ ist intramolekular bereits hinreichend stabilisiert, während das
Triethylsilyliumion (Et3Si+, 202+) koordiniert durch das Lösungsmittel oder durch η1-Si–H-
Koordination eines weiteren Hydrosilanmoleküls in Form eines Hydroniumions vorliegen

4 Exkurs: Silyliumionvermittelte C=X-Reduktionen mit Hydrosilanen 55
könnte (LVI+). Hydridverbrückte Kationen der Art LVI+ sind bereits von mehreren Gruppen
charakterisiert worden.[115] Daraufhin erfolgt die Bildung eines Silyliminiumions LVII+ aus
dem Iminsubstrat XVII und dem freien Silyliumion LV+ oder dem Silyliumion/Silan-Addukt
LVI+, wobei der letztere Fall dem Si–H-Bindungsaktivierungsschritt B(C6F5)3-katalysierter
Reduktionen ähnelt (siehe Kapitel 1.1). Ein Hydridtransfer vom Hydrosilan LIV auf das
Kohlenstoffatom des Silyliminiumions LVII+ regeneriert den Katalysator LVI+ und bildet das
silylierte Amin LVIII,[116] welches sich zum Amin XIX hydrolysieren lässt. Der Einfluss der
Schutzgruppe sollte sich vor allem in der Hydridaffinität des Intermediats LVII+ zeigen. Eine
elektronenziehende Schutzgruppe destabilisiert das Kation LVII+ und steigert damit dessen
Reaktivität im Hydridübertragungsschritt. Das lässt sich zur Erklärung der höheren
Reaktivität des tosylgeschützten Imins (E)-51 (Tabelle 4.1, Eintrag 6) im Vergleich zu den
Iminen (E)-213–(E)-215 (Einträge 3–5) heranziehen. Die ausbleibende Reaktivität der
ebenfalls eine elektronenziehende Schutzgruppe tragenden Imine (E)-211 und (E)-212
(Einträge 1 und 2) ist möglicherweise dadurch zu begründen, dass deren LEWIS-basischen
Atome zu stark mit dem Iminstickstoffatom um LEWIS-Paar-Bildung mit dem Silyliumion LV+
konkurrieren. Ein stöchiometrisches NMR-Experiment in 1,2-Cl2C6D4 zeigte, dass das Imin
(E)-212 durch das Silyliumion 205+ nicht zersetzt wird, sondern ein stabiles Addukt bildet
(29Si-NMR: δ = 33.0 gegenüber 114.6 ppm[110a] für freies 205+). Der zusätzliche Substituent
des Ketimins (E)-216 (Eintrag 7) sorgt dagegen für eine erhöhte Stabilität des Intermediats
LVII+ und bringt zudem einen erhöhten sterischen Anspruch mit sich, was einen
vollständigen Umsatz verhindert.
[115]
Intermolekulare Beispiele: a) S. P. Hoffmann, T. Kato, F. S. Tham, C. A. Reed, Chem.
Commun. 2006, 767–769; b) M. Nava, C. A. Reed, Organometallics 2011, 30, 4798–4800; c)
S. J. Connelly, W. Kaminsky, D. M. Heinekey, Organometallics 2013, 32, 7478–7481;
intramolekulare Beispiele: d) T. Müller, Angew. Chem. Int. Ed. 2001, 40, 3033–3036; e) R.
Panisch, M. Bolte, T. Müller, J. Am. Chem. Soc. 2006, 128, 9676–9682; f) A. Y. Khalimon, Z.
H. Lin, R. Simionescu, S. F. Vyboishchikov, G. I. Nikonov, Angew. Chem. Int. Ed. 2007, 46,
4530–4533. [116]
Um zu beweisen, dass Silan LIV als Hydriddonor dient, wurde die silyliumionvermittelte
Reduktion des Imins (E)-51 mit dem deuteriertem Silan 206-d durchgeführt und das Amin 52-d
in 90% Ausbeute mit einem Deuterierungsgrad >98% erhalten wurde.
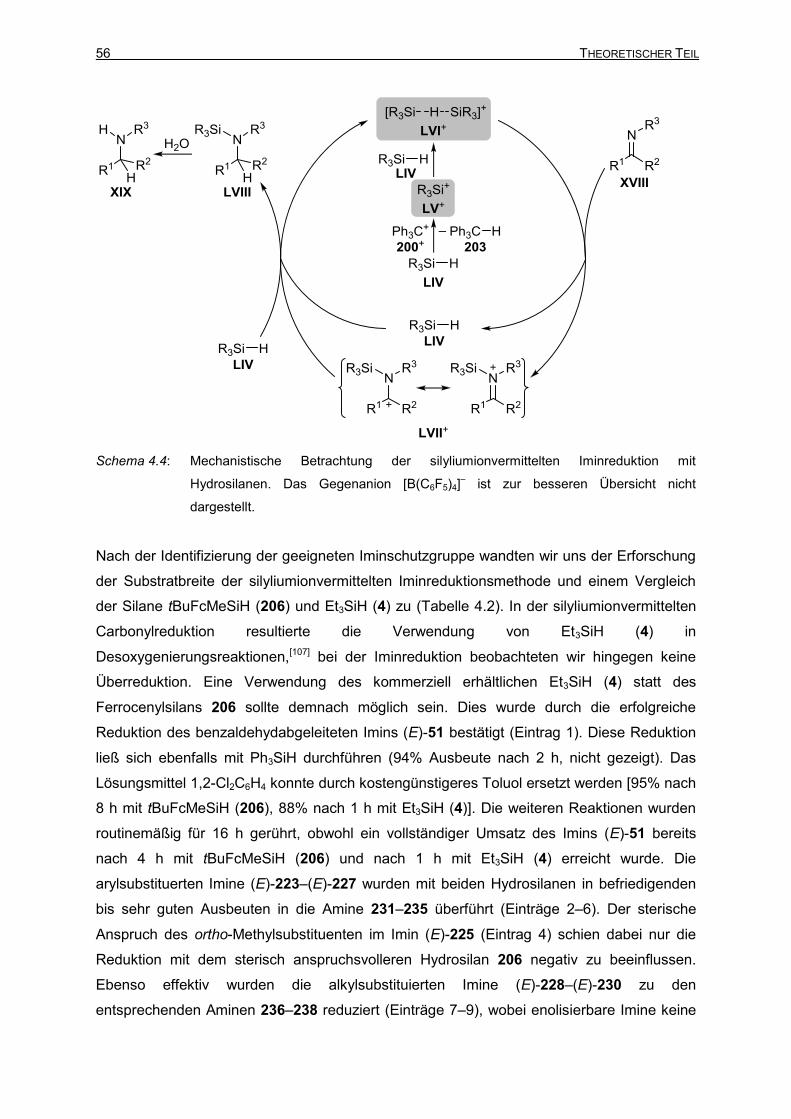
56 THEORETISCHER TEIL
Schema 4.4: Mechanistische Betrachtung der silyliumionvermittelten Iminreduktion mit
Hydrosilanen. Das Gegenanion [B(C6F5)4]– ist zur besseren Übersicht nicht
dargestellt.
Nach der Identifizierung der geeigneten Iminschutzgruppe wandten wir uns der Erforschung
der Substratbreite der silyliumionvermittelten Iminreduktionsmethode und einem Vergleich
der Silane tBuFcMeSiH (206) und Et3SiH (4) zu (Tabelle 4.2). In der silyliumionvermittelten
Carbonylreduktion resultierte die Verwendung von Et3SiH (4) in
Desoxygenierungsreaktionen,[107] bei der Iminreduktion beobachteten wir hingegen keine
Überreduktion. Eine Verwendung des kommerziell erhältlichen Et3SiH (4) statt des
Ferrocenylsilans 206 sollte demnach möglich sein. Dies wurde durch die erfolgreiche
Reduktion des benzaldehydabgeleiteten Imins (E)-51 bestätigt (Eintrag 1). Diese Reduktion
ließ sich ebenfalls mit Ph3SiH durchführen (94% Ausbeute nach 2 h, nicht gezeigt). Das
Lösungsmittel 1,2-Cl2C6H4 konnte durch kostengünstigeres Toluol ersetzt werden [95% nach
8 h mit tBuFcMeSiH (206), 88% nach 1 h mit Et3SiH (4)]. Die weiteren Reaktionen wurden
routinemäßig für 16 h gerührt, obwohl ein vollständiger Umsatz des Imins (E)-51 bereits
nach 4 h mit tBuFcMeSiH (206) und nach 1 h mit Et3SiH (4) erreicht wurde. Die
arylsubstituerten Imine (E)-223–(E)-227 wurden mit beiden Hydrosilanen in befriedigenden
bis sehr guten Ausbeuten in die Amine 231–235 überführt (Einträge 2–6). Der sterische
Anspruch des ortho-Methylsubstituenten im Imin (E)-225 (Eintrag 4) schien dabei nur die
Reduktion mit dem sterisch anspruchsvolleren Hydrosilan 206 negativ zu beeinflussen.
Ebenso effektiv wurden die alkylsubstituierten Imine (E)-228–(E)-230 zu den
entsprechenden Aminen 236–238 reduziert (Einträge 7–9), wobei enolisierbare Imine keine
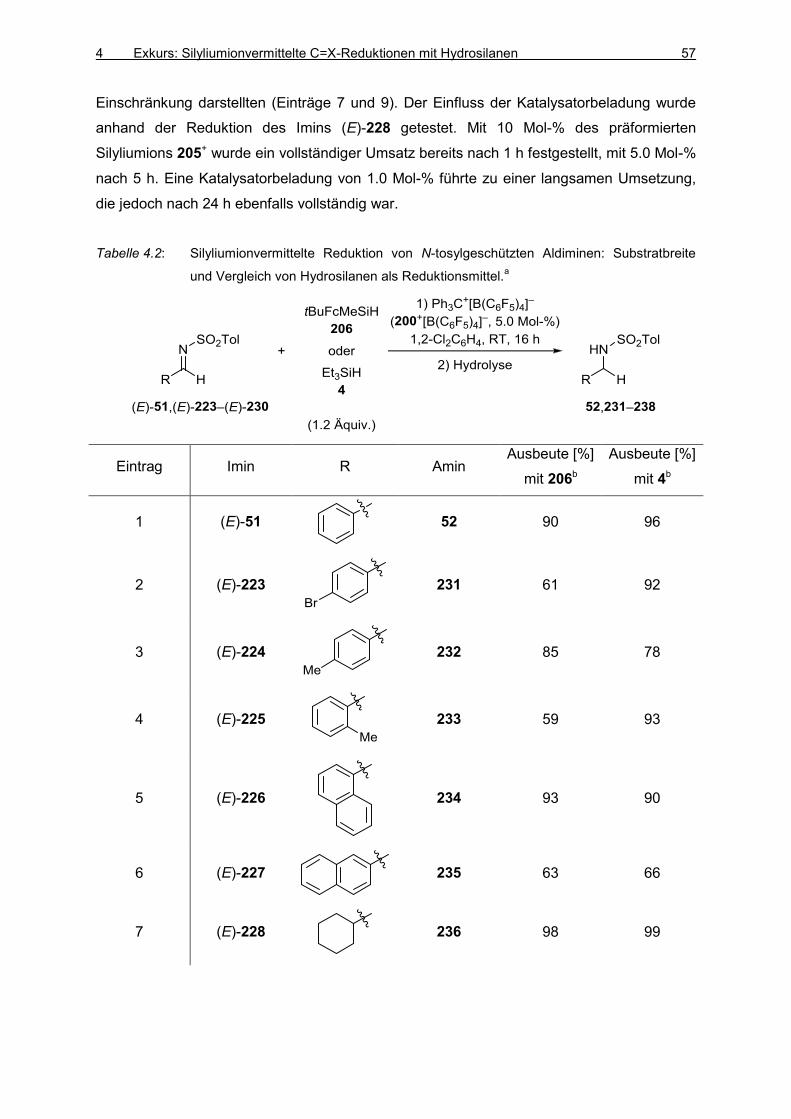
4 Exkurs: Silyliumionvermittelte C=X-Reduktionen mit Hydrosilanen 57
Einschränkung darstellten (Einträge 7 und 9). Der Einfluss der Katalysatorbeladung wurde
anhand der Reduktion des Imins (E)-228 getestet. Mit 10 Mol-% des präformierten
Silyliumions 205+ wurde ein vollständiger Umsatz bereits nach 1 h festgestellt, mit 5.0 Mol-%
nach 5 h. Eine Katalysatorbeladung von 1.0 Mol-% führte zu einer langsamen Umsetzung,
die jedoch nach 24 h ebenfalls vollständig war.
Tabelle 4.2: Silyliumionvermittelte Reduktion von N-tosylgeschützten Aldiminen: Substratbreite
und Vergleich von Hydrosilanen als Reduktionsmittel.a
Eintrag Imin R Amin Ausbeute [%]
mit 206b
Ausbeute [%]
mit 4b
1 (E)-51
52 90 96
2 (E)-223
231 61 92
3 (E)-224
232 85 78
4 (E)-225
233 59 93
5 (E)-226
234 93 90
6 (E)-227
235 63 66
7 (E)-228
236 98 99

58 THEORETISCHER TEIL
Eintrag Imin R Amin Ausbeute [%]
mit 206b
Ausbeute [%]
mit 4b
8 (E)-229 237 95 96
9 (E)-230 238 97 96
aDie Reaktionen wurden im 0.200–0.252-mmol-Maßstab gemäß AAV 6 oder ähnlich durchgeführt.
b Bestimmt
nach Aufreinigung durch Flashchromatographie an Kieselgel.
Die höhere Reaktivität der Reduktionsmethode mit Et3SiH (4) im Vergleich zu tBuFcMeSiH
(206) führten wir auf den geringeren sterischen Anspruch zurück. Das inspirierte uns, die
Reduktion des tosylgeschützten Ketimins (E)-216, die mit tBuFcMeSi+ (205+) als Katalysator
unbefriedigend verlief (siehe Tabelle 4.1, Eintrag 7), mit Et3Si+ (202+) als Katalysator erneut
zu untersuchen (Schema 4.5). Mit Et3SiH (4) als Reduktionsmittel war die Hydrosilylierung
des Ketimins (E)-216 erfolgreich und das Amin rac-222 wurde bereits nach einer
Reaktionszeit von 1 h in guter Ausbeute erhalten.
Schema 4.5: Silyliumionvermittelte Reduktion eines tosylgeschützten Ketimins.
4.2 Fazit
Die Reaktivität von Silyliumionen als LEWIS-Säure-Katalysatoren wurde ausgenutzt, um eine
katalytische C=N-Reduktionsmethode für Imine zu entwickeln. Die Verwendung des
ferrocenstabilisierten Silyliumions tBuFcMeSi+ (205+) war ebenso wie die des einfachen
Trialkylsilyliumions Et3Si+ (202+) geeignet, die Hydrosilylierung von Iminen zu vermitteln.
Insbesondere N-tosylgeschützte Aldimine eigneten sich als Substrate, wobei aryl- und
alkylsubstituierte Imine gleichermaßen zu den entsprechenden Aminen reduziert wurden.

4 Exkurs: Silyliumionvermittelte C=X-Reduktionen mit Hydrosilanen 59
Bei Verwendung des reaktiveren Systems Et3Si+ (202+)/Et3SiH (4) gelang auch die
Hydrosilylierung eines Ketimins.

60 THEORETISCHER TEIL
5 SYNTHESE, LEWIS-ACIDITÄT UND KATALYTISCHE
ANWENDUNGEN EINES ELEKTRONENARMEN β-NAPHTHYL-
BASIERTEN BORANS
Dieses Kapitel widmet sich der Darstellung des partiell fluorierten β-naphthylbasierten
Borans 108 (Abbildung 5.1), dessen Fluoratome sich ausschließlich an den dem Boratom
entfernt gelegenen C-5-, C-6-, C-7- und C-8-Positionen befinden.[117] Nach erfolgter
Synthese sollte untersucht werden, wie sich solch eine entfernte Fluorierung auf die LEWIS-
Acidität des Borans und die Reaktivität als LEWIS-Säure-Katalysator in Reaktionen mit Si–H-
und H–H-Bindungsaktivierungen auswirkt. Die Entwicklung der Synthese des Borans 108
erfolgte in Zusammenarbeit mit Dr. MUSTAFA DURMAZ.
Abbildung 5.1: Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108): ein partiell fluoriertes β-naphthyl-
basiertes Boran.
5.1 Synthese des Borans 108
Wir gingen davon aus, dass sich das Boran 108 aus einem entsprechenden Halogenid des
fluorierten β-Naphthylsubstituenten darstellen lässt und sahen die Synthese des Bromids
239 (Schemata 5.1 und 5.2) als zielführend an. Anschließend wäre das Arylbromid 239
durch Metallierung in ein Nukleophil überführbar, welches durch elektrophile Substitution mit
BX3 (X = Abgangsgruppe) das entsprechende Triarylboran 108 liefern sollte. Wir gedachten
zunächst, den Baustein 239 durch Bildung des thermisch stabilen[3e] GRIGNARD-Reagenzes
241 aus dem Aromaten 240 und anschließende Umsetzung desselben mit 3-Bromthiophen
(242) zu erhalten (240→241→239, Schema 5.1, links). Obwohl diese Methode
[117]
J. Mohr, M. Durmaz, E. Irran, M. Oestreich, Organometallics 2014, 33, 1108–1111.
Synthesevorschriften für und Charakterisierungen von Substanzen, die im experimentellen
Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.
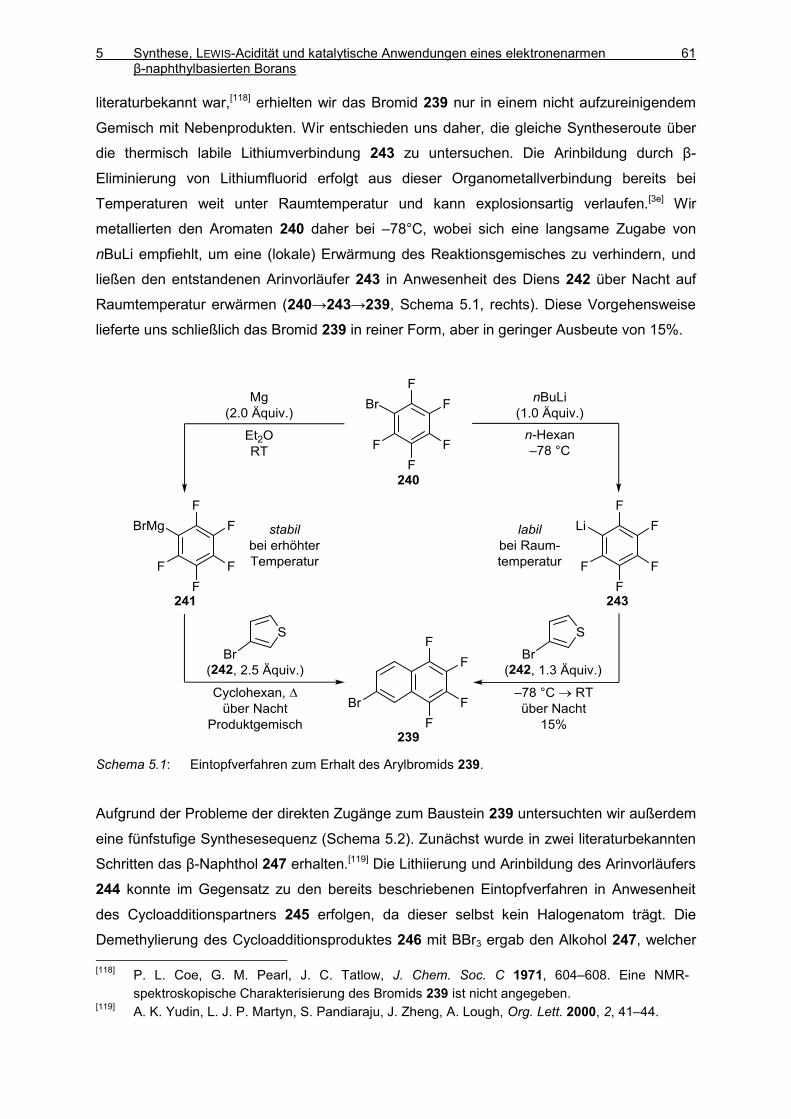
5 Synthese, LEWIS-Acidität und katalytische Anwendungen eines elektronenarmen 61 β-naphthylbasierten Borans
literaturbekannt war,[118] erhielten wir das Bromid 239 nur in einem nicht aufzureinigendem
Gemisch mit Nebenprodukten. Wir entschieden uns daher, die gleiche Syntheseroute über
die thermisch labile Lithiumverbindung 243 zu untersuchen. Die Arinbildung durch β-
Eliminierung von Lithiumfluorid erfolgt aus dieser Organometallverbindung bereits bei
Temperaturen weit unter Raumtemperatur und kann explosionsartig verlaufen.[3e] Wir
metallierten den Aromaten 240 daher bei –78°C, wobei sich eine langsame Zugabe von
nBuLi empfiehlt, um eine (lokale) Erwärmung des Reaktionsgemisches zu verhindern, und
ließen den entstandenen Arinvorläufer 243 in Anwesenheit des Diens 242 über Nacht auf
Raumtemperatur erwärmen (240→243→239, Schema 5.1, rechts). Diese Vorgehensweise
lieferte uns schließlich das Bromid 239 in reiner Form, aber in geringer Ausbeute von 15%.
Schema 5.1: Eintopfverfahren zum Erhalt des Arylbromids 239.
Aufgrund der Probleme der direkten Zugänge zum Baustein 239 untersuchten wir außerdem
eine fünfstufige Synthesesequenz (Schema 5.2). Zunächst wurde in zwei literaturbekannten
Schritten das β-Naphthol 247 erhalten.[119] Die Lithiierung und Arinbildung des Arinvorläufers
244 konnte im Gegensatz zu den bereits beschriebenen Eintopfverfahren in Anwesenheit
des Cycloadditionspartners 245 erfolgen, da dieser selbst kein Halogenatom trägt. Die
Demethylierung des Cycloadditionsproduktes 246 mit BBr3 ergab den Alkohol 247, welcher
[118]
P. L. Coe, G. M. Pearl, J. C. Tatlow, J. Chem. Soc. C 1971, 604–608. Eine NMR-
spektroskopische Charakterisierung des Bromids 239 ist nicht angegeben. [119]
A. K. Yudin, L. J. P. Martyn, S. Pandiaraju, J. Zheng, A. Lough, Org. Lett. 2000, 2, 41–44.
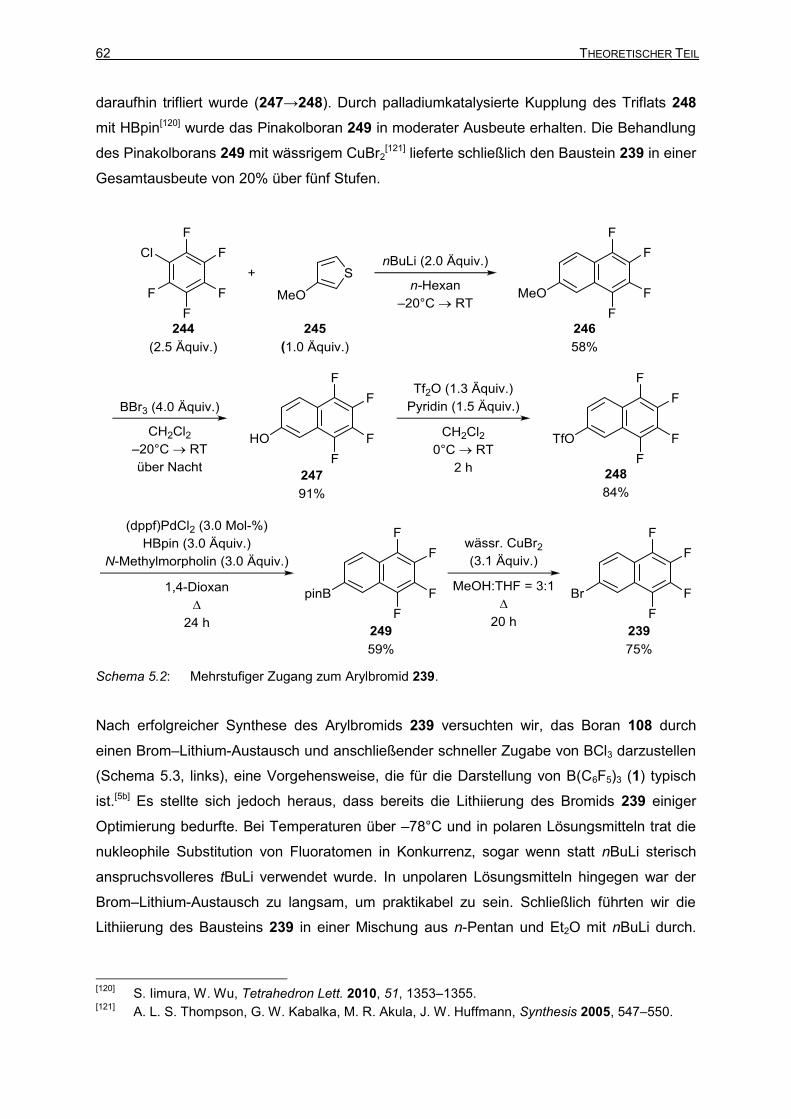
62 THEORETISCHER TEIL
daraufhin trifliert wurde (247→248). Durch palladiumkatalysierte Kupplung des Triflats 248
mit HBpin[120] wurde das Pinakolboran 249 in moderater Ausbeute erhalten. Die Behandlung
des Pinakolborans 249 mit wässrigem CuBr2[121] lieferte schließlich den Baustein 239 in einer
Gesamtausbeute von 20% über fünf Stufen.
Schema 5.2: Mehrstufiger Zugang zum Arylbromid 239.
Nach erfolgreicher Synthese des Arylbromids 239 versuchten wir, das Boran 108 durch
einen Brom–Lithium-Austausch und anschließender schneller Zugabe von BCl3 darzustellen
(Schema 5.3, links), eine Vorgehensweise, die für die Darstellung von B(C6F5)3 (1) typisch
ist.[5b] Es stellte sich jedoch heraus, dass bereits die Lithiierung des Bromids 239 einiger
Optimierung bedurfte. Bei Temperaturen über –78°C und in polaren Lösungsmitteln trat die
nukleophile Substitution von Fluoratomen in Konkurrenz, sogar wenn statt nBuLi sterisch
anspruchsvolleres tBuLi verwendet wurde. In unpolaren Lösungsmitteln hingegen war der
Brom–Lithium-Austausch zu langsam, um praktikabel zu sein. Schließlich führten wir die
Lithiierung des Bausteins 239 in einer Mischung aus n-Pentan und Et2O mit nBuLi durch.
[120]
S. Iimura, W. Wu, Tetrahedron Lett. 2010, 51, 1353–1355. [121]
A. L. S. Thompson, G. W. Kabalka, M. R. Akula, J. W. Huffmann, Synthesis 2005, 547–550.
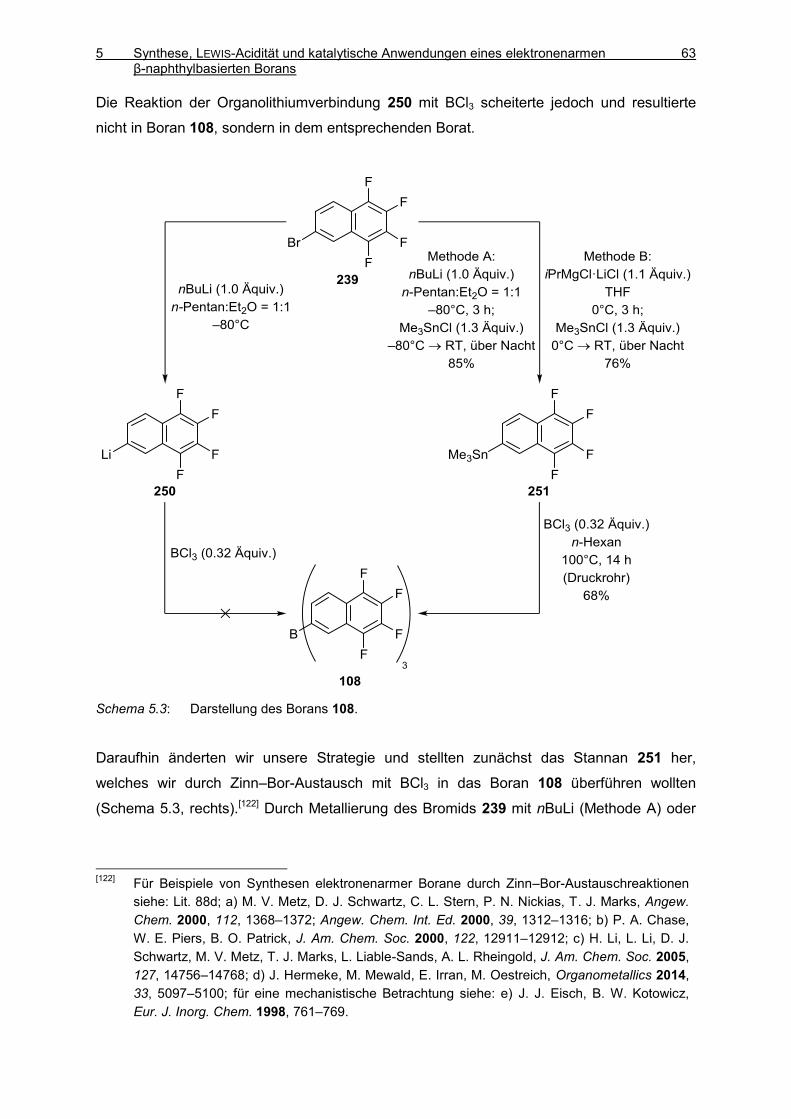
5 Synthese, LEWIS-Acidität und katalytische Anwendungen eines elektronenarmen 63 β-naphthylbasierten Borans
Die Reaktion der Organolithiumverbindung 250 mit BCl3 scheiterte jedoch und resultierte
nicht in Boran 108, sondern in dem entsprechenden Borat.
Schema 5.3: Darstellung des Borans 108.
Daraufhin änderten wir unsere Strategie und stellten zunächst das Stannan 251 her,
welches wir durch Zinn–Bor-Austausch mit BCl3 in das Boran 108 überführen wollten
(Schema 5.3, rechts).[122] Durch Metallierung des Bromids 239 mit nBuLi (Methode A) oder
[122]
Für Beispiele von Synthesen elektronenarmer Borane durch Zinn–Bor-Austauschreaktionen
siehe: Lit. 88d; a) M. V. Metz, D. J. Schwartz, C. L. Stern, P. N. Nickias, T. J. Marks, Angew.
Chem. 2000, 112, 1368–1372; Angew. Chem. Int. Ed. 2000, 39, 1312–1316; b) P. A. Chase,
W. E. Piers, B. O. Patrick, J. Am. Chem. Soc. 2000, 122, 12911–12912; c) H. Li, L. Li, D. J.
Schwartz, M. V. Metz, T. J. Marks, L. Liable-Sands, A. L. Rheingold, J. Am. Chem. Soc. 2005,
127, 14756–14768; d) J. Hermeke, M. Mewald, E. Irran, M. Oestreich, Organometallics 2014,
33, 5097–5100; für eine mechanistische Betrachtung siehe: e) J. J. Eisch, B. W. Kotowicz,
Eur. J. Inorg. Chem. 1998, 761–769.
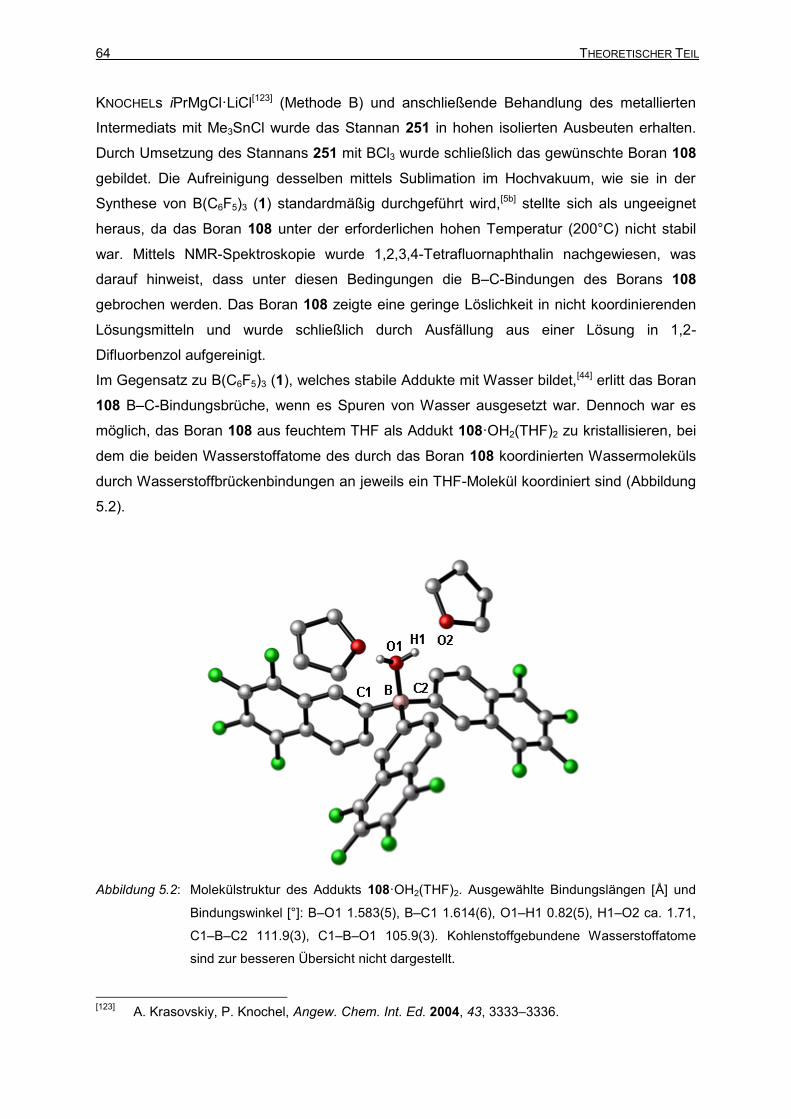
64 THEORETISCHER TEIL
KNOCHELs iPrMgCl·LiCl[123] (Methode B) und anschließende Behandlung des metallierten
Intermediats mit Me3SnCl wurde das Stannan 251 in hohen isolierten Ausbeuten erhalten.
Durch Umsetzung des Stannans 251 mit BCl3 wurde schließlich das gewünschte Boran 108
gebildet. Die Aufreinigung desselben mittels Sublimation im Hochvakuum, wie sie in der
Synthese von B(C6F5)3 (1) standardmäßig durchgeführt wird,[5b] stellte sich als ungeeignet
heraus, da das Boran 108 unter der erforderlichen hohen Temperatur (200°C) nicht stabil
war. Mittels NMR-Spektroskopie wurde 1,2,3,4-Tetrafluornaphthalin nachgewiesen, was
darauf hinweist, dass unter diesen Bedingungen die B–C-Bindungen des Borans 108
gebrochen werden. Das Boran 108 zeigte eine geringe Löslichkeit in nicht koordinierenden
Lösungsmitteln und wurde schließlich durch Ausfällung aus einer Lösung in 1,2-
Difluorbenzol aufgereinigt.
Im Gegensatz zu B(C6F5)3 (1), welches stabile Addukte mit Wasser bildet,[44] erlitt das Boran
108 B–C-Bindungsbrüche, wenn es Spuren von Wasser ausgesetzt war. Dennoch war es
möglich, das Boran 108 aus feuchtem THF als Addukt 108·OH2(THF)2 zu kristallisieren, bei
dem die beiden Wasserstoffatome des durch das Boran 108 koordinierten Wassermoleküls
durch Wasserstoffbrückenbindungen an jeweils ein THF-Molekül koordiniert sind (Abbildung
5.2).
Abbildung 5.2: Molekülstruktur des Addukts 108·OH2(THF)2. Ausgewählte Bindungslängen [Å] und
Bindungswinkel [°]: B–O1 1.583(5), B–C1 1.614(6), O1–H1 0.82(5), H1–O2 ca. 1.71,
C1–B–C2 111.9(3), C1–B–O1 105.9(3). Kohlenstoffgebundene Wasserstoffatome
sind zur besseren Übersicht nicht dargestellt.
[123]
A. Krasovskiy, P. Knochel, Angew. Chem. Int. Ed. 2004, 43, 3333–3336.

5 Synthese, LEWIS-Acidität und katalytische Anwendungen eines elektronenarmen 65 β-naphthylbasierten Borans
5.2 Bestimmung der LEWIS-Acidität des Borans 108
Nach der erfolgreichen Synthese des Borans 108 stellten wir uns die Frage, in welchem
Ausmaß die nicht vorhandene Fluorierung in der Nähe des Boratoms die LEWIS-Acidität des
Borans 108 im Vergleich zu B(C6F5)3 (1) verringert. Wir entschieden uns dafür, dies mittels
der GUTMANN–BECKETT-Methode[124] zu untersuchen, die sich als eine Standardmethode zur
Bestimmung der LEWIS-Acidität für elektronenarme Borane etabliert hat.[125] Diese Methode
beruht auf der Verwendung der LEWIS-Base Et3PO als NMR-Sonde. Ein Maß für die LEWIS-
Acidität ist die chemische Verschiebung des Signals des Boran/Et3PO-Addukts im 31P{1H}-
NMR-Spektrum im Vergleich zu freiem Et3PO (Tabelle 5.1). Zum Vergleich der LEWIS-
Acidität des Borans 108 mit der von B(C6F5)3 (1) bestimmten wir die chemischen
Verschiebungen der entsprechenden Boran/Et3PO-Addukte und des freien Phosphinoxids in
C6D6 (Eintrag 1) und CD2Cl2 (Eintrag 2). Es stellte sich heraus, dass die ∆δ-Werte des
Boranaddukts 108·Et3PO nur geringfügig niedriger waren als die des B(C6F5)3-Addukts
1·Et3PO und das unabhängig vom verwendeten Lösungsmittel.[126] Gegenüber Et3PO weist
das Boran 108 somit eine Lewis-Acidität von 98% (in C6D6, Eintrag 1) bzw. 99% (in CD2Cl2,
Eintrag 2) relativ zu B(C6F5)3 (1) auf.
[124]
a) U. Mayer, V. Gutmann, W. Gerger, Monatsh. Chem. 1975, 106, 1235–1257; b) V. Gutmann
Coord. Chem. Rev. 1976, 18, 225–255; c) M. A. Beckett, G. C. Strickland, J. R. Holland, K. S.
Varma, Polymer 1996, 37, 4629–4631; d) M. A. Beckett, D. S. Brassington, S. J. Coles, M. B.
Hursthouse, Inorg. Chem. Commun. 2000, 3, 530–533; e) M. A. Beckett, D. S. Brassington,
M. E. Light, M. B. Hursthouse, J. Chem. Soc., Dalton Trans. 2001, 1768–1772. [125]
Eine weitere experimentelle Methode zur LEWIS-Aciditätsbestimmung stellt die CHILDS-
Methode dar, bei der Crotonaldehyd als NMR-Sonde dient: a) R. F. Childs, D. L. Mulholland,
A. Nixon, Can. J. Chem. 1982, 60, 801–808; es können widersprüchliche Resultate bei der
Verwendung der GUTMANN–BECKETT- und CHILDS-Methoden erhalten werden: b) G. J. P.
Britovsek, J. Ugolotti, A. J. P. White, Organometallics 2005, 24, 1685–1691; c) T. J.
Herrington, A. J. W. Thom, A. J. P. White, A. E. Ashley, Dalton Trans. 2012, 41, 9019–9022;
für eine Diskussion dieses Sachverhalts siehe: d) G. C. Welch, L. Cabrera, P. A. Chase, E.
Hollink, J. D. Masuda, P. Wei, D. W. Stephan, Dalton Trans. 2007, 3407–3414;
quantenchemisch lassen sich LEWIS-Aciditäten für elektronenarme Borane durch Berechnung
von Anionenaffinitäten bestimmen: e) A. Y. Timoshkin, G. Frenking, Organometallics 2008,
27, 371–380; f) L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-
Quiñones, V. Brecht, I. Krossing, Angew. Chem. Int. Ed. 2008, 47, 7659–7663; g) H. Böhrer,
N. Trapp, D. Himmel, M. Schleep, I. Krossing, Dalton Trans. 2015, 44, 7489–7499. [126]
Eine charakteristische Veränderung der chemischen Verschiebung der Signale im 19
F-NMR-
Spektrum nach Adduktbildung von Boran 108 wurde nicht beobachtet. Für entsprechende
Beobachtungen in Addukten von B(C6F5)3 (1) siehe: Lit.96a; 124e; A. D. Horton, J. de With,
Organometallics 1997, 5424–5436.
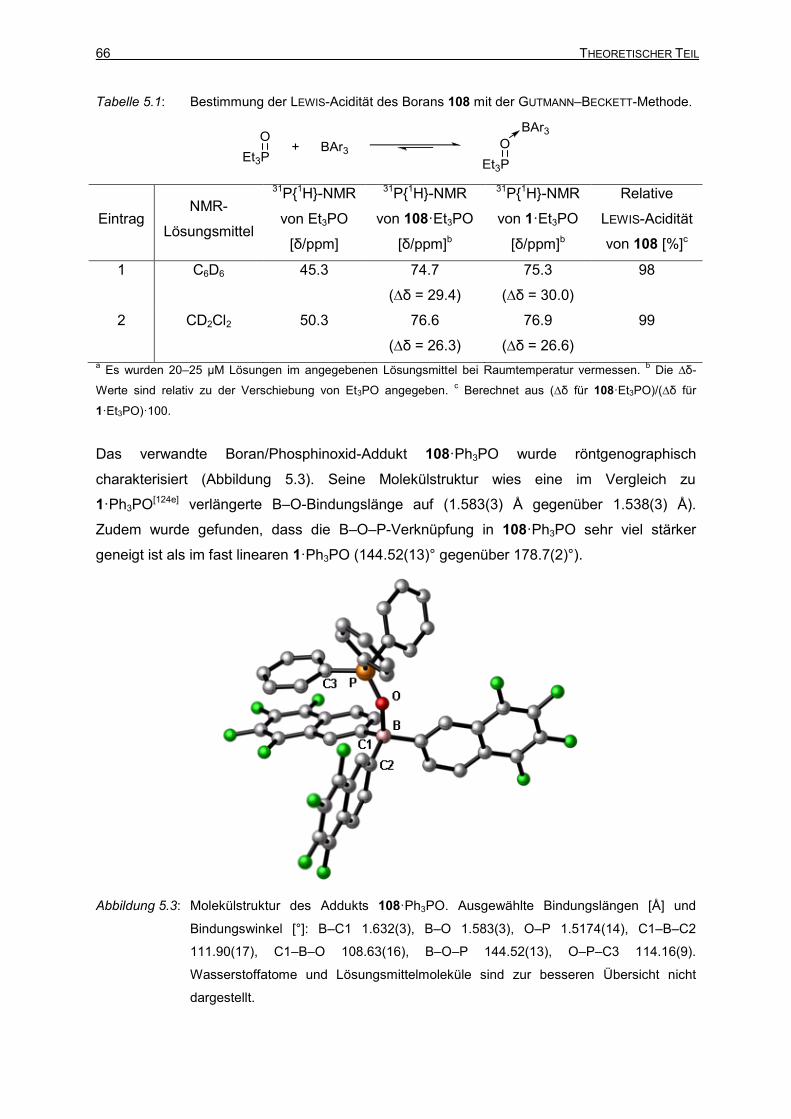
66 THEORETISCHER TEIL
Tabelle 5.1: Bestimmung der LEWIS-Acidität des Borans 108 mit der GUTMANN–BECKETT-Methode.
Eintrag NMR-
Lösungsmittel
31P{1H}-NMR
von Et3PO
[δ/ppm]
31P{1H}-NMR
von 108·Et3PO
[δ/ppm]b
31P{1H}-NMR
von 1·Et3PO
[δ/ppm]b
Relative
LEWIS-Acidität
von 108 [%]c
1 C6D6 45.3 74.7
(∆δ = 29.4)
75.3
(∆δ = 30.0)
98
2 CD2Cl2 50.3 76.6
(∆δ = 26.3)
76.9
(∆δ = 26.6)
99
a Es wurden 20–25 µM Lösungen im angegebenen Lösungsmittel bei Raumtemperatur vermessen.
b Die ∆δ-
Werte sind relativ zu der Verschiebung von Et3PO angegeben. c Berechnet aus (∆δ für 108·Et3PO)/(∆δ für
1·Et3PO)·100.
Das verwandte Boran/Phosphinoxid-Addukt 108·Ph3PO wurde röntgenographisch
charakterisiert (Abbildung 5.3). Seine Molekülstruktur wies eine im Vergleich zu
1·Ph3PO[124e] verlängerte B–O-Bindungslänge auf (1.583(3) Å gegenüber 1.538(3) Å).
Zudem wurde gefunden, dass die B–O–P-Verknüpfung in 108·Ph3PO sehr viel stärker
geneigt ist als im fast linearen 1·Ph3PO (144.52(13)° gegenüber 178.7(2)°).
Abbildung 5.3: Molekülstruktur des Addukts 108·Ph3PO. Ausgewählte Bindungslängen [Å] und
Bindungswinkel [°]: B–C1 1.632(3), B–O 1.583(3), O–P 1.5174(14), C1–B–C2
111.90(17), C1–B–O 108.63(16), B–O–P 144.52(13), O–P–C3 114.16(9).
Wasserstoffatome und Lösungsmittelmoleküle sind zur besseren Übersicht nicht
dargestellt.

5 Synthese, LEWIS-Acidität und katalytische Anwendungen eines elektronenarmen 67 β-naphthylbasierten Borans
5.3 Aktivität des Borans 108 in Reaktionen mit katalytischer Si–H-
und H–H-Bindungsaktivierung
Nachdem die Bestimmung der LEWIS-Acidität des Borans 108 nach der GUTMANN-BECKETT-
Methode ergeben hatte, dass dieses fast so LEWIS-acide wie B(C6F5)3 (1) ist (siehe Kapitel
5.2), waren wir zuversichtlich, dass das Boran 108 auch in der Lage ist, Reaktionen mit Si–
H- und H–H-Bindungsaktivierungen zu katalysieren, in denen typischerweise B(C6F5)3 (1) als
LEWIS-Säure-Katalysator eingesetzt wird. Wir testeten die Aktivität des Borans 108 folglich in
der C=N-Reduktion[47] des Imins (E)-44 mit dem Hydrosilan 19 (Schema 5.4, oben). Mit einer
Katalysatorbeladung von 3.7 Mol-% wurde das Reduktionsprodukt rac-45 nach 48 h und
Hydrolyse in exzellenter Ausbeute erhalten. Die verwandte C=O-Reduktion[6] von
Acetophenon (10) zum Silylether rac-252 verlief erwartungsgemäß schneller und bedurfte
einer geringen Katalysatormenge von 0.48 Mol-% (Schema 5.4, Mitte). Ebenso effektiv
konnte die dehydrierende Si–O-Kupplung[127] zwischen dem Alkohol rac-17 und dem
Hydrosilan 19 durch das Boran 108 katalysiert werden (Schema 5.4, unten). Diese
Ergebnisse zeigten, dass nicht nur die LEWIS-Acidität, sondern auch die Aktivität des Borans
108 als Katalysator in ausgewählten Reaktionen mit Si–H-Bindungsaktivierung mit der von
B(C6F5)3 (1) vergleichbar ist. Weiterführende systematische Studien in unserem Arbeitskreis
zeigten allerdings, dass das Boran 108 für C=C- und C≡C-Reduktionen mit Hydrosilanen
nicht geeignet ist (nicht gezeigt).[28] Als Ursache wurde das Fehlen von Fluoratomen in ortho-
Position zum Boratom vermutet. Dass ortho-Fluorierung von Bor-LEWIS-Säure-Katalysatoren
bei der Hydrosilanaktivierung einen Einfluss ausüben kann, wurde zudem durch
quantenchemische Rechnungen von SAKATA und FUJIMOTO aufgezeigt.[15] Für die Reduktion
von C=O-Bindungen zeigten diese, dass ein ortho-Fluoratom mit dem Siliciumatom des
Hydrosilans wechselwirkt und dieses durch die resultierende Pentakoordination zusätzlich
für den Angriff des LEWIS-basischen Substrats aktiviert wird.[128]
[127]
J. M. Blackwell, K. L. Foster, V. H. Beck, W. E. Piers, J. Org. Chem. 1999, 64, 4887–4892. [128]
Eine Übersicht zu LEWIS-Base-Aktivierungen von LEWIS-Säuren bietet: S. E. Denmark, G. L.
Beutner, Angew. Chem. Int. Ed. 2008, 47, 1560–1638.
68 THEORETISCHER TEIL
Schema 5.4: Reaktivität des Borans 108 in Reaktionen mit katalytischer Si–H-Bindungsaktivierung.
Um das Boran 108 als LEWIS-Säure-Katalysator für H–H-Bindungsaktivierungen zu testen,
entschieden wir uns, die C=N-Reduktionsmethode von Oximethern zu testen, deren
Entwicklung in Kapitel 2 beschrieben wurde. Die Hydrierung des O-tert-butylsubstituierten
Oximethers 112 unter Standardbedingungen gelang jedoch nicht, es wurde kein Umsatz
festgestellt (Schema 5.5). Möglicherweise wird die kooperative H–H-Bindungsspaltung durch
sterische Wechselwirkungen der β-Naphthylsubstituenten des Borans 108 mit dem Substrat
verhindert. Einen Hinweis hierauf liefert die Tatsache, dass sich der Oximether 112 unter
analogen Reaktionsbedingungen durch STEPHANs Boran B(2,3,5,6-F4C6H)3 (253),[129]
welches nach der GUTMANN–BECKETT-Methode eine ähnliche LEWIS-Acidität wie das Boran
108 aufweist, aber kleinere Arylsubstituenten trägt, mit vollständigem Umsatz in das
[129]
M. Ullrich, A. J. Lough, D. W. Stephan, J. Am. Chem. Soc. 2009, 131, 52–53.

5 Synthese, LEWIS-Acidität und katalytische Anwendungen eines elektronenarmen 69 β-naphthylbasierten Borans
Hydroxylamin rac-120 überführen ließ. Von der Untersuchung weiterer Hydrierungen mit
dem Boran 108 sahen wir daraufhin ab.
Schema 5.5: Vergleich der Aktivität der Borane 108 und B(2,3,5,6-F4C6H)3 (253) in der
katalytischen Oximetherreduktion.
5.4 Fazit
In diesem Kapitel wurde die Synthese des elektronenarmen Borans 108 beschrieben,
welches drei partiell fluorierte β-Naphthylsubstituenten trägt. Im Gegensatz zur etablierten
Bor-LEWIS-Säure B(C6F5)3 (1) und typischen B(C6F5)3-Derivaten weist dieses neuartige
Boran keine Fluoratome in direkter Umgebung des Boratoms auf. Es wurde der Frage
nachgegangen, inwiefern sich eine solche entfernte Fluorierung auf die LEWIS-Acidität und
die Reaktivität in Reaktionen mit katalytischer Si–H- und H–H-Bindungsaktivierung auswirkt.
Es zeigte sich, dass die nach der GUTMANN–BECKETT-Methode bestimmte LEWIS-Acidität
des Borans 108 im Vergleich mit B(C6F5)3 (1) kaum einen Unterschied aufweist. Auch in
typischerweise durch B(C6F5)3 (1) katalysierten Reaktionen mit Si–H-Bindungsaktivierung
(C=N- und C=O-Reduktion sowie dehydrierende Si–O-Kupplung) zeigte sich das Boran 108
reaktiv. Die in Kapitel 2 vorgestellte Oximetherreduktion mit katalytischer H–H-
Bindungsaktivierung wurde durch das Boran 108 nicht erreicht.

70 THEORETISCHER TEIL
Da die C1-Positionen der β-Naphthylsubstituenten des Borans
108 nicht durch Fluoratome besetzt sind, ließen sich diese
formal mit einer weiteren Napthyleinheit verknüpfen, um ein
chirales binaphthylbasiertes Boran zu erhalten. Dieses ließe sich
zudem in den C3-Positionen mit Substituenten versehen, da
auch diese für weitere Modifikationen verfügbar sind (Abbildung
5.4).
Abbildung 5.4

6 Einleitende Untersuchungen zur Synthese eines B(C6F5)3-Derivats mit chiralen 71 Substituenten in den para-Positionen
6 EINLEITENDE UNTERSUCHUNGEN ZUR SYNTHESE EINES
B(C6F5)3-DERIVATS MIT CHIRALEN SUBSTITUENTEN IN DEN
PARA-POSITIONEN
Bisher entwickelte chirale B(C6F5)3-Derivate für asymmetrische Reduktionen durch
katalytische Hydrierungen oder Hydrosilylierungen verdanken ihre Chiralität dem formalen
Austausch einer oder mehrerer elektronenarmer C6F5-Einheiten durch chirale Substituenten.
Dies resultiert zwangsläufig in einer Erniedrigung der LEWIS-Acidität. Ein chirales Boran, das
drei elektronenarme Binaphthylsubstituenten mit entfernter Fluorierung trägt und dessen
Synthese im vorigen Kapitel vorgeschlagen wurde, lässt hingegen die für einige
Umsetzungen entscheidenden Fluoratome in ortho-Position zum Boratom vermissen. In
einem Boran, welches drei fluorierte Phenylsubstituenten trägt, welche wiederum in para-
Position mit chiralen Substituenten versehen sind, bliebe die direkte Umgebung des
Boratoms im Vergleich zu B(C6F5)3 (1) erhalten und eine gleichermaßen hohe LEWIS-Acidität
wäre garantiert. Dieses Kapitel beschreibt einleitende Untersuchungen zur Synthese des
chiralen elektronenarmen Borans (R,R,R)-254 (Abbildung 6.1), welches drei
Binaphthylsubstituenten trägt, die sich jeweils in para-Position zum Boratom an einem
tetrafluorierten Aromaten befinden.
Abbildung 6.1: Das elektronenarme Boran (R,R,R)-254 mit chiralen Substituenten in para-Position
zum Boratom.
6.1 Darstellung des chiralen Binaphthylbausteins (R)-261
Wir gingen davon aus, dass das Boran (R,R,R)-254 aus dem chiralen Baustein (R)-261
(Schema 6.1) durch Metallierung desselben und anschließender Umsetzung mit einer
Borquelle zugänglich ist. Zur Darstellung des elektronenarmen Aromaten (R)-261 gingen wir
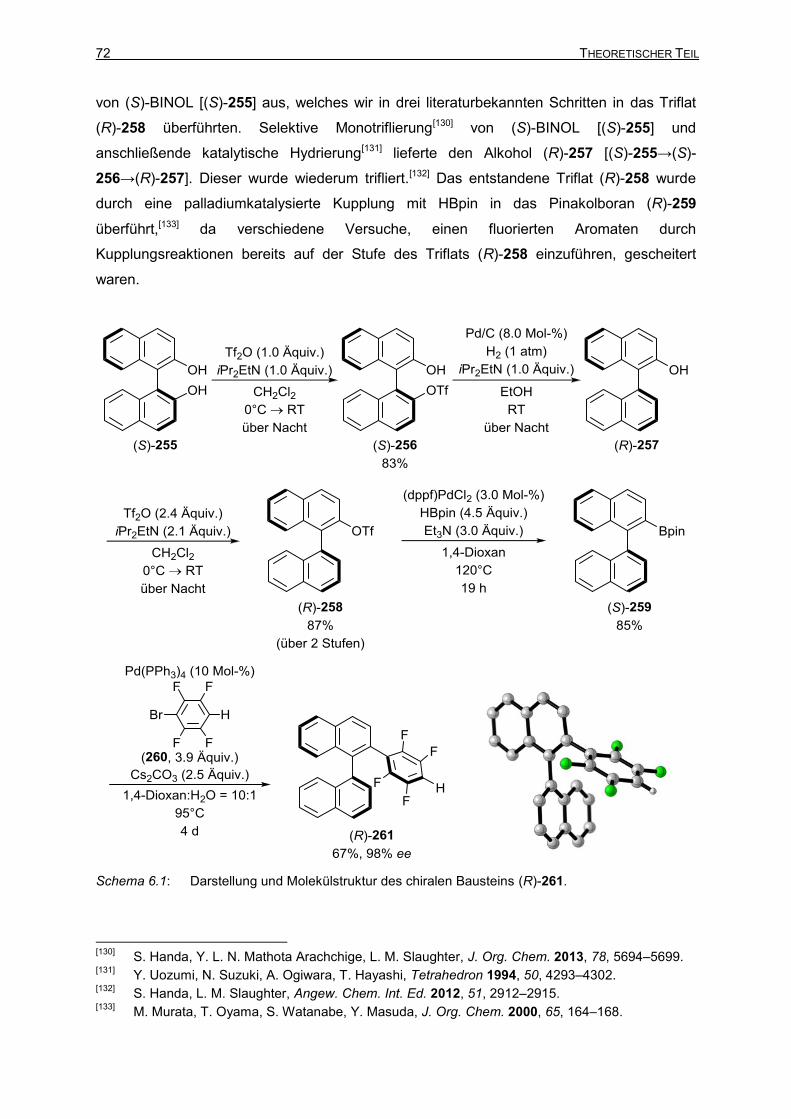
72 THEORETISCHER TEIL
von (S)-BINOL [(S)-255] aus, welches wir in drei literaturbekannten Schritten in das Triflat
(R)-258 überführten. Selektive Monotriflierung[130] von (S)-BINOL [(S)-255] und
anschließende katalytische Hydrierung[131] lieferte den Alkohol (R)-257 [(S)-255→(S)-
256→(R)-257]. Dieser wurde wiederum trifliert.[132] Das entstandene Triflat (R)-258 wurde
durch eine palladiumkatalysierte Kupplung mit HBpin in das Pinakolboran (R)-259
überführt,[133] da verschiedene Versuche, einen fluorierten Aromaten durch
Kupplungsreaktionen bereits auf der Stufe des Triflats (R)-258 einzuführen, gescheitert
waren.
Schema 6.1: Darstellung und Molekülstruktur des chiralen Bausteins (R)-261.
[130]
S. Handa, Y. L. N. Mathota Arachchige, L. M. Slaughter, J. Org. Chem. 2013, 78, 5694–5699. [131]
Y. Uozumi, N. Suzuki, A. Ogiwara, T. Hayashi, Tetrahedron 1994, 50, 4293–4302. [132]
S. Handa, L. M. Slaughter, Angew. Chem. Int. Ed. 2012, 51, 2912‒2915. [133]
M. Murata, T. Oyama, S. Watanabe, Y. Masuda, J. Org. Chem. 2000, 65, 164–168.

6 Einleitende Untersuchungen zur Synthese eines B(C6F5)3-Derivats mit chiralen 73 Substituenten in den para-Positionen
Das Pinakolboran (R)-259 diente anschließend als Kupplungspartner in einer SUZUKI–
MIYAURA-Kupplung mit dem Arylbromid 260, welche im Erhalt des Bausteins (R)-261
resultierte. Dieser finale Schritt der Synthesesequenz benötigte eine relativ hohe
Katalysatorbeladung, einen Überschuss des Bromids 260 und eine Reaktionszeit von
mehreren Tagen, kann aber bei sorgfältigem Sauerstoffausschluss verlässlich mit
annehmbaren Ausbeuten durchgeführt werden. Der chirale Baustein (R)-261 wurde in einer
Gesamtausbeute von 41% über fünf Stufen erhalten und zudem röntgenographisch
charakterisiert. Mittels HPLC-Analyse an chiraler stationärer Phase wurde ein weitgehender
Erhalt der Stereoinformation festgestellt.
Zur Darstellung des Borans (R,R,R)-254 bietet es sich an, den Baustein (R)-261
entsprechend der Synthese des Borans 108 (siehe Kapitel 5.1) nach Metallierung zunächst
in das entsprechende Stannan und daraufhin durch Umsetzung mit BCl3 in das Boran
(R,R,R)-254 zu überführen (Schema 6.2). Erste Testreaktionen zeigten, dass eine
Deprotonierung des Aromaten (R)-261 durch Behandlung mit tBuLi bei –78°C in Et2O
sauber verläuft.[88d] In der Folge gilt es nun, die weiteren Schritte zur Darstellung des
chiralen Borans (R,R,R)-254 durchzuführen, dieses zu charakterisieren und in LEWIS-Säure-
katalysierten Reaktionen auf seine Enantioinduktion zu testen. Aktuelle Arbeiten in unserem
Arbeitskreis sind diesen Problemstellungen gewidmet.
Schema 6.2: Strategie zur Darstellung des Borans (R,R,R)-254 aus dem chiralen Baustein (R)-261.
6.2 Fazit
In diesem Kapitel wurde die Synthese einer chiralen Bor-LEWIS-Säure vorgeschlagen,
welche sich von bisher bekannten chiralen B(C6F5)3-Derivaten dadurch abhebt, dass die
elektronenarmen Aromaten nicht durch chirale Substituenten ersetzt werden, sondern diese
in para-Position tragen. Hierdurch soll die direkte Umgebung des Boratoms im Vergleich zu
B(C6F5)3 (1) unverändert bleiben und ein weitestgehender Erhalt der LEWIS-Acidität

74 THEORETISCHER TEIL
gewährleistet werden. Der hierfür benötigte chirale Baustein wurde erfolgreich dargestellt.
Die geplante Überführung in das entsprechende Boran steht noch aus.

7 Zusammenfassung 75
7 ZUSAMMENFASSUNG
7.1 Hauptgruppenelementkatalysierte C=N-Reduktionen
Die katalytische chemoselektive C=N-Reduktion von Oximen und Oximethern zu den
entsprechenden Hydroxylaminen stellte ein letztlich ungelöstes Problem dar, da
metallkatalysierte Hydrierungen in der Regel zu Aminprodukten infolge reduktiven N–O-
Bindungsbruchs führen.[81] Die Verwendung des Bor-LEWIS-Säure-Katalysators B(C6F5)3 (1)
ermöglichte die Reduktion von adäquat geschützten Oximethern XXXIII zu N-
monosubstituierten Hydroxylaminen XXXIV ohne Überreduktion unter einem
Diwasserstoffdruck von 60–100 bar bei 60°C oder Raumtemperatur (Schema 7.1). Die
Hydrierung von Ketoximethern lässt sich in Toluol durchführen, während für die Reduktion
weniger basischer aldehydabgeleiteter Substrate die Verwendung eines Etherlösungsmittels
wie 1,4-Dioxan und eine höhere Katalysatorbeladung benötigt wird. Aus O-
triisopropylsilylierten Hydroxylaminen XXXIV lassen sich zudem ungeschützte
Hydroxylamine LIX freisetzen.
Schema 7.1: B(C6F5)3-katalysierte Hydrierung von Oximethern zu Hydroxylaminen.
Die B(C6F5)3-katalysierte Hydrierungsmethode wurde im Folgenden auf die C=N-Reduktion
von Hydrazonen übertragen. Nach Hydrierung von phthaloylgeschützten Hydrazonen XXXVI
zu den entsprechenden Hydrazinen XXXVII lassen sich auch aus diesen die ungeschützten
Reduktionsprodukte LX erhalten (Schema 7.2). Bei der Reduktion von Hydrazonen zeigte
sich derselbe Reaktivitätsunterschied zwischen keton- und aldehydabgeleiteten Substraten
wie im Falle der Oximether.

76 THEORETISCHER TEIL
Schema 7.2: B(C6F5)3-katalysierte Hydrierung von Hydrazonen zu Hydrazinen.
In einem Exkurs wurde zudem untersucht, ob sich analog der B(C6F5)3-katalysierten
Iminreduktion durch Hydrosilylierung[47] eine C=N-Reduktion auch silyliumionvermittelt
erreichen lässt. Es wurde eine entsprechende Methode zur Reduktion von N-
tosylgeschützten Iminen LXI zu den Aminprodukten LXII entwickelt (Schema 7.3). Als
Reduktionsmittel und Silyliumionvorläufer eigneten sich sowohl das Ferrocenylhydrosilan
206 als auch kommerziell erhältliches Triethylsilan (4).
Schema 7.3: Silyliumionvermittelte Hydrosilylierung von Iminen zu Aminen.
7.2 Darstellung elektronenarmer Borane
Ein weiterer Schwerpunkt der vorliegenden Dissertation lag auf der Darstellung neuartiger
elektronenarmer Borane. Obschon eine Reihe von Boranen mit unterschiedlicher Anzahl an
in verschiedenem Maße fluorierten Phenylsubstituenten bekannt sind, war bisher nicht
untersucht, ob eine Fluorierung an nicht direkt an das zentrale Boratom gebundene
Arylsubstituenten eine für katalytische Aktivität ausreichende LEWIS-Säure-Stärke bewirkt.
Wir synthetisierten daher das Boran 108, welches zur Klärung dieser Frage geeignet schien
(Schema 7.4). Durch GUTMANN–BECKETT-Analyse wurde eine LEWIS-Acidität von 98–99%
relativ zu B(C6F5)3 (1) festgestellt. In typischen Si–H-Bindungsaktivierungskatalysen erwies
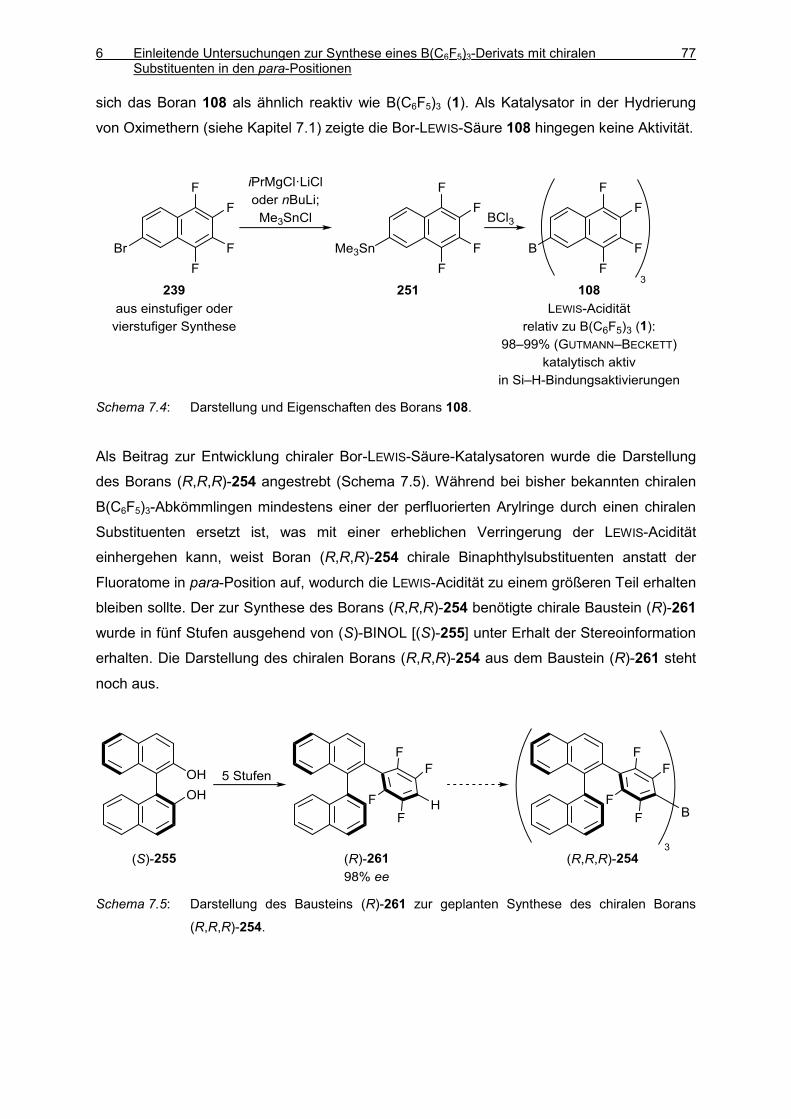
6 Einleitende Untersuchungen zur Synthese eines B(C6F5)3-Derivats mit chiralen 77 Substituenten in den para-Positionen
sich das Boran 108 als ähnlich reaktiv wie B(C6F5)3 (1). Als Katalysator in der Hydrierung
von Oximethern (siehe Kapitel 7.1) zeigte die Bor-LEWIS-Säure 108 hingegen keine Aktivität.
Schema 7.4: Darstellung und Eigenschaften des Borans 108.
Als Beitrag zur Entwicklung chiraler Bor-LEWIS-Säure-Katalysatoren wurde die Darstellung
des Borans (R,R,R)-254 angestrebt (Schema 7.5). Während bei bisher bekannten chiralen
B(C6F5)3-Abkömmlingen mindestens einer der perfluorierten Arylringe durch einen chiralen
Substituenten ersetzt ist, was mit einer erheblichen Verringerung der LEWIS-Acidität
einhergehen kann, weist Boran (R,R,R)-254 chirale Binaphthylsubstituenten anstatt der
Fluoratome in para-Position auf, wodurch die LEWIS-Acidität zu einem größeren Teil erhalten
bleiben sollte. Der zur Synthese des Borans (R,R,R)-254 benötigte chirale Baustein (R)-261
wurde in fünf Stufen ausgehend von (S)-BINOL [(S)-255] unter Erhalt der Stereoinformation
erhalten. Die Darstellung des chiralen Borans (R,R,R)-254 aus dem Baustein (R)-261 steht
noch aus.
Schema 7.5: Darstellung des Bausteins (R)-261 zur geplanten Synthese des chiralen Borans
(R,R,R)-254.


EXPERIMENTELLER TEIL


1 Allgemeine Arbeitsweise 81
1 ALLGEMEINE ARBEITSWEISE
Reaktionen wurden, sofern nicht anders angegeben, in zuvor im Ölpumpenvakuum
ausgeheizten Glasapparaturen unter Schutzgasatmosphäre (Stickstoff, getrocknet mit KC-
Trockenperlen® Orange oder Phosphorpentoxid) oder in einer Glovebox des Typs MB-
Labstar der Fa. MBraun unter Argonatmosphäre durchgeführt. Die verwendeten Glasgeräte
wurden zur Reinigung über Nacht in ein iPrOH/KOH-Bad eingelegt, anschließend mit
deionisiertem Wasser gespült und bei 120°C getrocknet. Nach Reaktionen mit
Übergangsmetallverbindungen wurden die Glasgeräte zuvor mit Königswasser
(konzentrierte wässrige HCl-Lösung und konzentrierte wässrige HNO3-Lösung im Verhältnis
3:1) gespült. Für die Zugabe von Reagenzien und Lösungsmitteln durch Septen wurden
mehrfach mit Schutzgas gespülte Einwegspritzen und -kanülen oder bei 120°C gelagerte
Glasspritzen und Edelstahlkanülen verwendet. Feststoffe wurden im Schutzgasgegenstrom
oder in Lösung zugegeben. Tieftemperaturreaktionen wurden entweder mit einem
Eintauchkühler TC100E der Fa. huber oder in einem Aceton- bzw. Ethanol-Trockeneisbad
durchgeführt.
Lösungsmittel
Diethylether (Et2O), Dichlormethan (CH2Cl2), 1,2-Difluorbenzol (1,2-F2C6H4), 1,2-
Dichlorbenzol (1,2-Cl2C6H4) und n-Pentan wurden unter Stickstoffatmosphäre über CaH2 am
Rückfluss erhitzt und abdestilliert. Tetrahydrofuran (THF) wurde über KOH abdestilliert,
anschließend unter Stickstoffatmosphäre über Kalium mit Benzophenon als Indikator am
Rückfluss erhitzt und bei Blaufärbung des Indikators abdestilliert. 1,4-Dioxan wurde unter
Stickstoffatmosphäre über Natrium mit Benzophenon als Indikator am Rückfluss erhitzt und
bei Blaufärbung des Indikators abdestilliert. n-Hexan wurde unter Stickstoffatmosphäre über
Kalium erhitzt und abdestilliert oder einer Lösungsmitteltrocknungsanlage des Typs MB-
SPS-800 der Fa. MBraun entnommen. Toluol wurde unter Stickstoffatmosphäre über
Natrium am Rückfluss erhitzt und abdestilliert. Für Reaktionen und Extraktionen wurde
deionisiertes Wasser (H2O) verwendet. Die zur Extraktion und Flashchromatographie
verwendeten technischen Lösungsmittel tert-Butylmethylether, Cyclohexan, Dichlormethan,
n-Pentan und Ethylacetat wurden zuvor unter vermindertem Druck destilliert. Zur
Flashchromatographie verwendetes Triethylamin (Et3N) und als Lösungsmittel für
Reaktionen verwendetes Ethanol wurden in technischer Qualität verwendet. Zur
Flashchromatographie verwendetes Toluol und Methanol als Lösungsmittel für Reaktionen
wurden in HPLC-Qualität verwendet. Für die Hochleistungsflüssigchromatographie (HPLC)
wurden Lösungsmittel entsprechenden Reinheitsgrades verwendet.

82 EXPERIMENTELLER TEIL
Physikalische Daten
Schmelzpunkte wurden mit einem Schmelzpunktbestimmungsgerät SMP20 der FA. Stuart
bestimmt und sind nicht korrigiert.
Chromatographie
Qualitative Dünnschichtchromatographie (DC) wurde auf mit Kieselgel 60 F254
beschichteten Glasplatten der Fa. Merck KGaA durchgeführt. Die Indikation der Analyten
erfolgte nach verschiedenen Methoden:
Bestrahlung der DC-Platte mit UV-Licht (λ = 254 nm) bei UV-Absorption durch die
Analyten.
Eintauchen der DC-Platte in eine Lösung von (NH4)3[P(Mo3O10)4] (100 g), Ce(SO4)2
(4.00 g) und konzentrierter H2SO4 (100 mL) in H2O (900 mL) und anschließendes
Erwärmen mit einem Heißluftgebläse.
Eintauchen der DC-Platte in eine Lösung von KMnO4 (3.0 g), K2CO3 (20 g) und KOH
(0.30 g) in H2O (300 mL) und anschließendes Erwärmen mit einem Heißluftgebläse.
Als stationäre Phase für die Flashchromatographie wurde Kieselgel Davisil LC60A
(Korngröße 40–63 µm, Porengröße 60 Å) der Fa. Grace GmbH verwendet. Die Angaben im
experimentellen Teil sind in der Form „(d × h, A:B = a:b, gegebenenfalls Vorlauf, C, #n–m)“
angegeben, wobei d der Säulendurchmesser und h die Füllhöhe an stationärer Phase sind.
A und B geben die als mobile Phase verwendeten Lösungsmittel und a:b deren
Volumenverhältnis an. C ist das Fraktionsvolumen in mL, #n–m entspricht den
Fraktionsnummern, in welchen sich die jeweilige Substanz befand.
Analytische Gaschromatographie (GC) von Reaktionsmischungen und Reinsubstanzen
wurde mit einem Gaschromatographen des Typs 7820A der Fa. Agilent Technologies
ausgestattet mit einer Quarzkapillarsäule des Typs FS-SE-54 der Fa. CS Chromatographie
Service (Länge: 30 m, Innendurchmesser: 0.32 mm; Filmdicke der kovalent gebundenen,
stationären Phase: 0.25 µm) durchgeführt. Zur Analyse wurden folgende Methoden
verwendet: Trägergas: N2, Injektortemperatur: 250°C, Detektortemperatur: 300°C, Fließrate:
1.7 mL/min, Starttemperatur: 40°C, Heizrate: 10°C/min, Endtemperatur: 280°C für 10 min.
Analytische Trennungen mittels Hochleistungsflüssigkeitschromatographie (HPLC)
wurden an einem Gerät Infinity 1260 der Fa. Agilent Technologies durchgeführt. Als chirale
stationäre Phase wurde die Säule Daicel Chiralcel OJ-RH (reversed phase) eingesetzt.
Kernspinresonanzspektroskopie (NMR-Spektroskopie)
Die Messung der NMR-Spektren erfolgte in der NMR-Abteilung des Instituts für Chemie der
Technischen Universität Berlin an den Geräten AV 400, AV 500 und AV 700 der Fa. Bruker.

1 Allgemeine Arbeitsweise 83
Als Lösungsmittel dienten CDCl3, C6D6, und CD2Cl2. Die angegebenen chemischen
Verschiebungen sind in den 1H-NMR-Spektren auf die Resonanzlinie des im CDCl3
enthaltenen CHCl3 (δ = 7.26 ppm), des im C6D6 enthaltenen C6D5H (δ = 7.16 ppm) und des
im CD2Cl2 enthaltenen CDHCl2 (δ = 5.32 ppm) kalibriert. Die 13C-NMR-Spektren sind auf die
Resonanz des CDCl3 (δ = 77.16 ppm), C6D6 (δ = 128.06 ppm) und CD2Cl2 (δ = 53.84 ppm)
kalibriert.[134] Alle anderen Kerne wurden relativ zur Resonanzlinie von Tetramethylsilan im
1H-NMR-Spektrum mit Hilfe der normierten Skala für chemische Verschiebungen (unified
chemical shift scale) geräteintern referenziert.[135] 29Si-DEPT-Messungen wurden auf eine
Kopplungskonstante von 7.0 Hz optimiert. Folgende Abkürzungen wurden zur Angabe der
Spinmultiplizitäten verwendet: s (Singulett), d (Dublett), t (Triplett), q (Quartett), sept
(Septett), m (Multiplett) und mc (zentrosymmetrisches Multiplett), wobei diese auch
kombiniert wurden. Breite Signale wurden durch ein der Abkürzung der Spinmultiplizität
vorangestelltes „br“ gekennzeichnet. Bei zentrosymmetrischen Signalen wurde der
Signalschwerpunkt und bei nicht zentrosymmetrischen Multipletts der Resonanzbereich
angegeben. Die Zuweisung der Signale bezieht sich auf die Bezifferung der Strukturen in
den Abbildungen und wurde durch Korrelationsspektren (1H/1H-COSY-, 1H/1H-NOESY-,
1H/13C-HMQC- und 1H/13C-HMBC-NMR-Spektren) unterstützt. 13C-, 11B-, 29Si-, 31P- und
119Sn-NMR-Messungen wurden wasserstoffentkoppelt durchgeführt. War eine eindeutige
Zuordnung der Signale nicht möglich, sind die zugewiesenen Atome mit Sternen (* oder **)
gekennzeichnet. Diastereotope Wasserstoff- bzw. Kohlenstoffatome wurden mit „A“ und „B“
bezeichnet. Die Bezeichnung „Ar“ bezeichnet nicht näher zugeordnete Wasserstoffatome
oder Kohlenstoffatome eines aromatischen Systems. Die Bezeichnung „Alkyl“ bezeichnet
nicht näher zugeordnete aliphatische Kohlenstoffatome oder an diese gebundene
Wasserstoffatome. Der Index „quart“ in der Auswertung der 13C-NMR-Spektren steht für
„quartär“ und bezeichnet nicht weiter zugeordnete quartäre Kohlenstoffatome.
Massenspektrometrie (MS)
Die massenspektrometrischen Analysen wurden von der Abteilung für Massenspektrometrie
am Institut für Chemie, Technische Universität Berlin durchgeführt. Exakte
Massenbestimmungen wurden durch Elektronenstoßionisation (EI) an dem Gerät Finnigan
MAT 95S (Elektronenenergie: 70 eV) sowie durch Elektrosprayionisation (ESI) bzw. durch
chemische Ionisation bei Atmosphärendruck (APCI) am Orbitrap LTQ XL der Fa. Thermo
[134]
a) H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512–7515; b) G. R.
Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw,
K. I. Goldberg, Organometallics 2010, 29, 2176–2179. [135]
R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow, P. Granger, Pure Appl.
Chem. 2001, 73, 1795–1818.

84 EXPERIMENTELLER TEIL
Scientific durchgeführt. Die jeweils verwendete Ionisationsmethode ist bei der Beschreibung
der Experimente angegeben. GC-MS-Messungen wurden an einem Gerät des Typs 5975C
der Fa. Agilent Technologies mit Elektronenstoßionisation (EI) durchgeführt. Die GC-Einheit
ist mit einer Quarzkapillarsäule des Typs HP-5MS der Fa. Agilent Technologies (Länge: 30
m; Innendurchmesser: 0.25 mm; Filmdicke der kovalent gebundenen stationären Phase:
0.25 µm) ausgestattet. Zur Analyse wurde folgende Methode verwendet: Trägergas: He,
Injektortemperatur: 300°C, Detektortemperatur: 300°C, Fließrate: 0.8 mL/min,
Starttemperatur: 40°C, Heizrate: 10°C/min, Endtemperatur: 280°C für 10 min.
Infrarotspektroskopie (IR-Spektroskopie)
Infrarotspektren wurden an einem Fourier-Transformations-Infrarotspektrometer des Typs
Cary 630FT-IR der Fa. Agilent gemessen. Der Analyt wurde hierbei direkt mittels einer ATR-
Einheit vermessen. In der Auswertung erfolgt die Angabe charakteristischer Banden.
Drehwerte
Drehwerte optisch aktiver Substanzen wurden mit einem Polarimeter Polartronic H532 der
Fa. Schmidt+Haensch ermittelt. Die Analyten wurden als Lösung in dem angegebenen
Lösungsmittel in 1-dm-Küvetten vermessen. Die spezifischen Drehwerte [α]λϑ wurden nach
folgender Formel berechnet:
[α]λϑ=
[α]×100
c × d
Hierbei ist λ die Wellenlänge in nm, ϑ die Messtemperatur in °C, [α] der am Polarimeter
ermittelte Drehwert, c die Konzentration in g/100 mL und d die Länge der Messküvette in
dm. Die Verwendung der Natrium-D-Linie (λ = 589 nm) als Lichtquelle ist mit „D“
gekennzeichnet.
Röntgenstrukturanalyse
Die Röntgenstrukturanalysen wurden von der Abteilung für Kristallstrukturbestimmung am
Institut für Chemie, Technische Universität Berlin durchgeführt. Hierbei wurde ein Oxford
Diffraction XCalibur Röntgendiffraktometer ausgestattet mit einem Sapphir CCD Detektor
und einem graphitmonochromatischer Mo-Kα-Strahlung (λ = 0.7107 Ǻ) oder ein Agilent
SuperNova Röntgendiffraktometer ausgestattet mit einem CCD Flächendetektor Atlas und
einem graphitmonochromatischer Cu-Kα-Strahlung (λ = 1.5418 Å) verwendet. Geeignete
Kristalle wurden mit Perfluorpolyalkylether-Öl auf einem Glasfaden befestigt und während
der Messung auf 150 K gekühlt.

1 Allgemeine Arbeitsweise 85
Software
Die Aufnahme und Auswertung von GC-Daten erfolgte mit dem Programm EZChrom Elite
Compact der Fa. Agilent Technologies. NMR-Daten wurden mit dem Programm Topspin der
Fa. Bruker ausgewertet. Für die Erstellung und Analyse der GC-MS-Daten diente Enhanced
ChemStation der Fa. Agilent Technologies. Für die Erstellung und Analyse der HPLC-Daten
diente Chemstation for LC 3D Systems B.04.03 der Fa. Agilent Technologies. Das
Programm Mass++ der Firmen Shimadzu Corporation und Eisai Co.,Ltd. wurde zur Analyse
von APCI-, EI- und ESI-Daten verwendet. Für die Aufnahme und Auswertung von IR-
Spektren wurden Microlab und Agilent Resolutions Pro 5.2.0 der Fa. Agilent Technologies
verwendet. Die Erstellung von Schemata für die vorliegende Dissertation erfolgte mit dem
Programm ChemBioDraw der Fa. CambridgeSoft. Abbildungen von Molekülstrukturen
wurden mit dem Programm Ortep von L. J. FARRUGIA (University of Glasgow) und dem
Programm CYLview von C. Y. LEGAULT (University of Sherbrooke) erstellt.
Literaturbekannte Synthesevorschriften und Verbindungen
Folgende Verbindungen wurden nach literaturbekannten Synthesevorschriften hergestellt:
Tris(pentafluorphenyl)boran (1),[136] Tris(2,3,5,6-tetrafluorphenyl)boran (253, hergestellt von
A. SIMONNEAU),[129] (S)-4-(Pentafluorphenyl)-4,5-dihydro-3H-dinaphtho[2,1-c:1‘,2‘-e]borepin-
Tetrahydrofuran-Addukt [(S)-47·THF, hergestellt von J. HERMEKE],[88e] 1-Phenylethan-1-
onoxim (110, hergestellt von J. HERMEKE),[137] rac-3,5-Diphenyl-4,5-dihydroisoxazol (rac-
142),[138] rac-5-Methyl-3,5-diphenyl-4,5-dihydroisoxazol (rac-143),[139] 1,1-Dimethyl-2-(1-
phenylethyliden)hydrazin (178, hergestellt von D. PORWAL),[140] 1,1-Dibenzyl-2-(1-
phenylethyliden)hydrazin (172),[141] (Benzylidenamino)isoindolin-1,3-dion (176, hergestellt
von D. PORWAL),[142] Tri(cyclohexa-2,5-dien-1-yl)silan (77, hergestellt von A. SIMONNEAU),[64b]
Cyclohexa-2,5-dien-1-yltrimethylsilan (75, hergestellt von A. SIMONNEAU),[64a] iPrMgCl·LiCl in
THF,[123] (E)-N,1-Diphenylethan-1-imin [(E)-44, hergestellt von J. HERMEKE],[143] (S)-2'-
Hydroxy-(1,1'-binaphthalin)-2-yltrifluormethansulfonat [(S)-256],[130] (R)-(1,1'-Binaphthalin)-2-
ol [(R)-257],[131] (R)-(1,1'-Binaphthalin)-2-yltrifluormethansulfonat [(R)-258],[132]
[136]
C. Wang, G. Erker, G. Kehr, K. Wedeking, R. Fröhlich, Organometallics 2005, 24, 4760–4773. [137]
M. J. Burk, G. Casy, N. B. Johnson, J. Org. Chem. 1998, 63, 6084–6085. [138]
B. A. Mendelsohn, S. Lee, S. Kim, F. Teyssier, V. S. Aulakh, M. A. Ciufolini, Org. Lett. 2009,
11, 1539–1542. [139]
Dargestellt gemäß Lit. 138, eine Charakterisierung findet sich in: X. Zhu, Y.-F. Wang, W. Ren
F.-L. Zhang, S. Chiba, Org. Lett. 2013, 15, 3214–3217. [140]
G. R. Newkome, D. L. Fishel, Org. Synth. 1970, 50, 102–104. [141]
G. Zhang, Y. Zhao, H. Ge, Angew. Chem. Int. Ed. 2013, 52, 2559–2563. [142]
J. Mohr, D. Porwal, I. Chatterjee, M. Oestreich, Chem. Eur. J. 2015, 21, 17583–17586. [143]
T. Imamoto, N. Iwadate, K. Yoshida, Org. Lett. 2006, 8, 2289–2292.

86 EXPERIMENTELLER TEIL
Ph3C+[B(C6F5)4]– (200+[B(C6F5)4]–, hergestellt von K. MÜTHER/A. SIMONNEAU),[144] tert-
Butylferrocenylsilan (206),[144] (E)-4-Methyl-N-(benzyliden)phenylsulfonamid [(E)-51,
hergestellt von O. ROLING],[144] (E)-4-Methyl-N-(4-brombenzyliden)phenylsulfonamid [(E)-
223, hergestellt von O. ROLING],[144] (E)-4-Methyl-N-(4-methylbenzyliden)phenylsulfonamid
[(E)-224, hergestellt von K. MÜTHER],[144] (E)-4-Methyl-N-(2-
methylbenzyliden)phenylsulfonamid [(E)-225, hergestellt von K. MÜTHER],[144] (E)-4-Methyl-
N-(naphthalin-1-ylmethylen)phenylsulfonamid [(E)-226, hergestellt von K. MÜTHER],[144] (E)-4-
Methyl-N-(naphthalin-2-ylmethylen)phenylsulfonamid [(E)-227, hergestellt von K.
MÜTHER],[144] (E)-4-Methyl-N-(cyclohexylmethylen) [(E)-228, hergestellt von K. MÜTHER],[144]
(E)-4-Methyl-N-(2,2-dimethylpropyliden)phenylsulfonamid [(E)-229, hergestellt von K.
MÜTHER],[144] (E)-4-Methyl-N-(2-ethylbutyliden)phenylsulfonamid [(E)-230, hergestellt von K.
MÜTHER],[144] (E)-tert-Butyl(benzyliden)carbamat [(E)-212],[145] (E)-4-Methyl-N-(1-
phenylethyliden)phenylsulfonamid [(E)-216].[146]
Nomenklatur und Nummerierung von Molekülstrukturen
Die Benennung der Verbindungen wurde sinngemäß vorgenommen und muss nicht den
IUPAC-Empfehlungen entsprechen, obschon diese weitgehend berücksichtigt wurden. Die
Nummerierung der Molekülstrukturen ist sinngemäß und folgt nicht der Nummerierung in der
Nomenklatur.
[144]
Kristine Müther, Dissertation, Technische Universität Berlin, 2014. [145]
A. S. Tsai, M. E. Tauchert, R. G. Bergman, J. A. Ellman, J. Am. Chem. Soc. 2011, 133, 1248‒
1250. [146]
J. L. García Ruano, J. Alemán, M. B. Cid, A. Parra, Org. Lett. 2005, 7, 179‒182.

2 Allgemeine Arbeitsvorschriften 87
2 ALLGEMEINE ARBEITSVORSCHRIFTEN
Die Substrat- und Lösungsmittelmengen für die Durchführung der Reaktionen sind den
einzelnen Reaktionsvorschriften zu entnehmen.
2.1 Allgemeine Arbeitsvorschrift zur Darstellung O-alkyl-
substituierter Oxime (AAV 1)
In einem 20-mL-Gläschen wird ein Gemisch aus Keton bzw. Aldehyd (1.00 Äquiv.) und O-
Alkylhydroxylaminhydrochlorid (1.00 Äquiv.) in Methanol (10 mL) bei Raumtemperatur für
mehrere Stunden ohne Ausschluss von Sauerstoff und Feuchtigkeit gerührt. Nach Entfernen
des Lösungsmittels unter vermindertem Druck wird der Rückstand mittels
Flashchromatographie an Kieselgel zum O-Alkyloxim aufgereinigt.
2.2 Allgemeine Arbeitsvorschrift zur Darstellung ungeschützter
Oxime (AAV 2)
In einem 20-mL-Gläschen wird ein Gemisch aus Keton bzw. Aldehyd (1.00 Äquiv.),
Hydroxylaminhydrochlorid (1.00 Äquiv.) und Natriumacetat (1.00 Äquiv.) in Methanol (10 mL)
über Nacht bei Raumtemperatur ohne Ausschluss von Sauerstoff und Feuchtigkeit gerührt.
Die Reaktionsmischung wird mit H2O (30 mL) verdünnt und mit CH2Cl2 (3 × 30 mL)
extrahiert. Die vereinigten organischen Phasen werden über Na2SO4 getrocknet. Entfernen
der Lösungsmittel unter vermindertem Druck liefert das ungeschützte Oxim, welches ohne
weitere Aufreinigung verwendet wird.
2.3 Allgemeine Arbeitsvorschrift zur Darstellung O-silyl-
substituierter Oxime (AAV 3)
Einer Vorschrift von ORTIZ-MARCIALES und Mitarbeitern[147] folgend werden das ungeschützte
Oxim (1.00 Äquiv.) und das entsprechende Chlorsilan (1.00–1.20 Äquiv.) in CH2Cl2 (5 mL)
vorgelegt. Eine Lösung von Imidazol (1.80–2.00 Äquiv.) in CH2Cl2 (5 mL) wird tropfenweise
hinzugegeben und die resultierende Mischung über Nacht bei Raumtemperatur gerührt.
Nach Entfernen des Lösungsmittels unter vermindertem Druck wird der Rückstand mittels
Flashchromatographie an Kieselgel zum O-Silyloxim aufgereinigt.
[147]
M. Ortiz-Marciales, L. D. Rivera, M. De Jesús, S. Espinosa, J. A. Benjamin, O. E. Casanova, I.
G. Figueroa, S. Rodríguez, W. Correa, J. Org. Chem. 2005, 70, 10132–10134.

88 EXPERIMENTELLER TEIL
2.4 Allgemeine Arbeitsvorschrift zu Hydrierungsreaktionen in Toluol
(AAV 4)
In einer Glovebox mit Argonatmosphäre werden in einem Gläschen (50 x 14 mm, Schütt)
das Oxim bzw. das Hydrazon (1.0 Äquiv.) und das angegebene Boran (5.0–10 Mol-%) in
Toluol (0.5–1 mL) vorgelegt. Das Gläschen wird mit einem Gummiseptum verschlossen, aus
der Glovebox genommen und unter Stickstoffgegenstrom in einen zuvor mit Stickstoff
gespülten Stahlautoklaven BR-100 oder BR-300 (inklusive des entsprechenden
Metallheizblocks, Berghof) gestellt. Unter anhaltendem Stickstoffgegenstrom wird eine Nadel
(0.90 x 50 mm, Braun) durch das Gummiseptum gestochen. Der Autoklav wird verschlossen
und zunächst mit Stickstoff (3 x) und dann mit Wasserstoff (3 x) gespült. Daraufhin wird der
angegebene Wasserstoffdruck bei der angegebenen Temperatur eingestellt. Nach Rühren
der Reaktionsmischung für 16–18 h wird diese über Kieselgel filtriert.
2.5 Allgemeine Arbeitsvorschrift zu Hydrierungsreaktionen in 1,4-
Dioxan (AAV 5)
In einer Glovebox mit Argonatmosphäre werden in einem Gläschen (50 x 14 mm, Schütt)
das Oxim bzw. das Hydrazon (1.0 Äquiv.) und das angegebene Boran (5.0–10 Mol-%)
vorgelegt. Das Gläschen wird mit einem Gummiseptum verschlossen, aus der Glovebox
genommen und 1,4-Dioxan (1 mL) wird mit einer Spritze hinzugegeben. Das Gläschen wird
unter Stickstoffgegenstrom in einen zuvor mit Stickstoff gespülten Stahlautoklaven BR-100
oder BR-300 (inklusive des entsprechenden Metallheizblocks, Berghof) gestellt. Unter
anhaltendem Stickstoffgegenstrom wird eine Nadel (0.90 x 50 mm, Braun) durch das
Gummiseptum gestochen. Der Autoklav wird verschlossen und zunächst mit Stickstoff (3 x)
und dann mit Wasserstoff (3 x) gespült. Daraufhin wird der angegebene Wasserstoffdruck
bei der angegebenen Temperatur eingestellt. Nach Rühren der Reaktionsmischung für 18 h
wird die Reaktionsmischung über Kieselgel filtriert.
2.6 Allgemeine Arbeitsvorschrift zur silyliumionvermittelten
Iminreduktion mit Hydrosilanen (AAV 6)
In einer Glovebox mit Argonatmosphäre wird zu einer Lösung von Ph3C+[B(C6F5)4]– (5.0 Mol-
%) in 1,2-Cl2C6H4 oder Toluol (0.3 mL) eine geringe Menge des entsprechenden Hydrosilans
(0.18–0.26 Äquiv.) gegeben und für 5–10 min gerührt. Eine Lösung des angegeben Imins
(1.00 Äquiv.) in 1,2-F2C6H4, 1,2-Cl2C6H4 oder Toluol (2–3 mL) und eine Lösung des

2 Allgemeine Arbeitsvorschriften 89
entsprechenden Hydrosilans in 1,2-F2C6H4, 1,2-Cl2C6H4 oder Toluol (0.7 mL) werden
nacheinander hinzugegeben und das Reaktionsgefäß mit einem Gummiseptum
verschlossen. Das Reaktionsgemisch wird außerhalb der Glovebox an einer Schlenkline mit
Stickstoffatmosphäre gerührt. Die Reaktion wird durch Zugabe von H2O (5 mL) beendet, die
Phasen getrennt und die wässrige Phase mit CH2Cl2 (3 x 5 mL) extrahiert. Die vereinigten
organischen Phasen werden über Na2SO4 getrocknet, die Lösungsmittel unter vermindertem
Druck entfernt und der Rückstand mittels Flashchromatographie an Kieselgel aufgereinigt.

90 EXPERIMENTELLER TEIL
3 BESCHREIBUNG DER EXPERIMENTE
3.1 Darstellung von Oximethern
3.1.1 1-Phenylethan-1-on-O-methyloxim (111)
Dargestellt aus Acetophenon (288 mg, 2.40 mmol, 1.00 Äquiv.) und O-
Methylhydroxylaminhydrochlorid (200 mg, 2.39 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 16 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #7–
14) lieferte die Titelverbindung 111 (259 mg, 72%) als farbloses Öl.
Rf = 0.21 (Cyclohexan).
GC (FS-SE-54): tR = 10.1 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 2.24 (s, 3H, H-1), 4.01 (s, 3H, OCH3), 7.33–7.41 (m,
3H, H-5, H-6), 7.62–7.69 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.8 (C-1), 62.0 (OCH3), 126.2 (C-4), 128.5 (C-5),
129.1 (C-6), 136.8 (C-3), 154.8 (C-2).
IR (ATR): ~ /cm–1 = 2935, 2897, 2816, 1494, 1442, 1367, 1311, 1185, 1045, 888, 757, 691.
HRMS (APCI) für C9H12NO+ [(M+H)+]: ber. 150.0913
gef. 150.0915

3 Beschreibung der Experimente 91
3.1.2 1-Phenylethan-1-on-O-(tert-butyl)oxim (112)
Dargestellt aus Acetophenon (143 mg, 1.19 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (150 mg, 1.19 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 18 cm, Cyclohexan:Ethylacetat = 50:1, 60 mL, #15–
20) lieferte die Titelverbindung 112 (201 mg, 88%) als farbloses Öl.
Rf = 0.21 (Cyclohexan).
GC (FS-SE-54): tR = 12.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.37 (s, 9H, OC(CH3)3), 2.21 (s, 3H, H-1), 7.30–7.39
(m, 3H, H-5, H-6), 7.66–7.71 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.4 (C-1), 27.9 (OC(CH3)3), 78.8 (OC(CH3)3), 126.0
(C-4), 128.4 (C-5), 128.7 (C-6), 137.8 (C-3), 152.5 (C-2).
IR (ATR): ~ /cm–1 = 2974, 1443, 1362, 1314, 1259, 1192, 995, 937, 758, 691.
HRMS (APCI) für C12H18NO+ [(M+H)+]: ber. 192.1383
gef. 192.1383

92 EXPERIMENTELLER TEIL
3.1.3 Benzaldehyd-O-(tert-butyl)oxim (116)
Dargestellt aus Benzaldehyd (169 mg, 1.59 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (200 mg, 1.59 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 19 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #20–
27) lieferte die Titelverbindung 116 (217 mg, 77%) als farbloses Öl.
Rf = 0.17 (Cyclohexan).
GC (FS-SE-54): tR = 11.1 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.38 (s, 9H, OC(CH3)3), 7.33–7.40 (m, 3H, H-4, H-5),
7.60 (mc, 2H, H-3), 8.06 (s, 1H, H-1).
13C-NMR (126 MHz, CDCl3): δ/ppm = 27.8 (OC(CH3)3), 79.3(OC(CH3)3), 127.0 (C-3), 128.7
(C-4), 129.4 (C-5), 133.4 (C-2), 147.3 (C-1).
IR (ATR): ~ /cm–1 = 2975, 2928, 1491, 1446, 1386, 1362, 1256, 1191, 1073, 1032, 956, 838,
753, 690.
HRMS (APCI) für C11H16NO+ [(M+H)+]: ber. 178.1226
gef. 178.1222

3 Beschreibung der Experimente 93
3.1.4 1-(4-(Trifluormethyl)phenyl)ethan-1-on-O-(tert-butyl)oxime (126)
Dargestellt aus 1-(4-(Trifluormethyl)phenyl)ethan-1-on (269 mg, 1.19 mmol, 1.00 Äquiv.) und
O-(tert-Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 16 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #18–
26) lieferte die Titelverbindung 126 (319 mg, 86%) als farbloses Öl.
Rf = 0.34 (Cyclohexan).
GC (FS-SE-54): tR = 12.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.36 (s, 9H, OC(CH3)3), 2.21 (s, 3H, H-1), 7.60 (d, 3J5,4
= 8.3 Hz, 2H, H-5), 7.78 (d, 3J4,5 = 8.3 Hz, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.4 (C-1), 28.0 (OC(CH3)3), 79.5 (OC(CH3)3), 124.5
(q, 1J7,F = 272 Hz, C-7), 125.4 (q, 3J5,F = 3.9 Hz, C-5), 126.3 (C-4), 130.6 (q, 2J6,F = 32 Hz; C-
6), 141.2 (C-3), 151.5 (C-2).
19F-NMR (471 MHz, CDCl3): δ/ppm = –62.7.
IR (ATR): ~ /cm–1 = 2978, 1605, 1407, 1364, 1322, 1261, 1165, 1123, 1063, 1001, 942, 840,
730.
HRMS (APCI) für C13H17F3NO+ [(M+H)+]: ber. 260.1257
gef. 260.1258

94 EXPERIMENTELLER TEIL
3.1.5 1-(4-Methoxyphenyl)ethan-1-on-O-(tert-butyl)oxim (128)
Dargestellt aus 1-(4-Methoxyphenyl)ethan-1-on (215 mg, 1.43 mmol, 1.00 Äquiv.) und O-
(tert-Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde für 4 h bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 17 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #20–
26) lieferte die Titelverbindung 128 (261 mg, 82%) als weißen Feststoff.
Rf = 0.52 (Cyclohexan:Ethylacetat = 9:1).
Smp.: 31°C (Cyclohexan).
GC (FS-SE-54): tR = 15.6 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.35 (s, 9H, OC(CH3)3), 2.18 (s, 3H, H-1), 2.82 (s, 3H,
OCH3), 6.88 (d, 3J5,4 = 8.7 Hz, 2H, H-5), 7.63 (d, 3J4,5 = 8.7 Hz, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.3 (C-1), 27.9 (OC(CH3)3), 55.5 (OCH3), 78.5
(OC(CH3)3), 113.8 (C-5), 127.3 (C-4), 130.5 (C-3), 152.1 (C-2), 160.2 (C-6).
IR (ATR): ~ /cm–1 = 2975, 2932, 2837, 1599, 1508, 1456, 1360, 1319, 1248, 1196, 1118,
1030, 998, 934, 839, 708.
HRMS (APCI) für C13H20NO2+ [(M+H)+]: ber. 222.1489
gef. 222.1490

3 Beschreibung der Experimente 95
3.1.6 1-(4-Chlorphenyl)propan-1-on-O-(tert-butyl)oxim (130)
Dargestellt aus 1-(4-Chlorphenyl)propan-1-on (241 mg, 1.43 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 15 cm, Cyclohexan:Ethylacetat:Toluol:Triethylamin
= 50:1:1:1, 60 mL, #3–5) lieferte die Titelverbindung 130 (251 mg, 73%) als farbloses Öl.
Rf = 0.31 (Cyclohexan).
GC (FS-SE-54): tR = 15.1 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.10 (t, 3J1,2 = 7.6 Hz, 3H, H-1), 1.34 (s, 9H, OC(CH3)3),
2.70 (q, 3H, 3J2,1 = 7.6 Hz, 2H, H-2), 7.29–7.34 (m, 2H, H-6), 7.58–7.63 (m, 2H, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm = 11.2 (C-1), 19.6 (C-2), 27.8 (OC(CH3)3), 78.8
(OC(CH3)3), 127.4 (C-5), 128.6 (C-6), 134.5 (C-7), 135.2 (C-4), 156.6 (C-3).
IR (ATR): ~ /cm–1 = 2974, 2933, 1603, 1491, 1461, 1361, 1259, 1191, 1090, 1030, 944, 885,
830, 717.
HRMS (APCI) für C13H19ClNO+ [(M+H)+]: ber. 240.1150
gef. 240.1153

96 EXPERIMENTELLER TEIL
3.1.7 1-(4-Bromphenyl)ethan-1-on-O-(tert-butyl)oxim (132)
Dargestellt aus 1-(4-Bromophenyl)ethan-1-on (285 mg, 1.43 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (6 × 15 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #7–
19) lieferte die Titelverbindung 132 (318 mg, 82%) als farbloses Öl.
Rf = 0.25 (Cyclohexan).
GC (FS-SE-54): tR = 15.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.36 (s, 9H, OC(CH3)3), 2.18 (s, 3H, H-1), 7.47 (mc, 2H,
H-5), 7.56 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.2 (C-1), 27.9 (OC(CH3)3), 79.1 (OC(CH3)3), 122.8
(C-6), 127.5 (C-4), 131.5 (C-5), 136.6 (C-3), 151.5 (C-2).
IR (ATR): ~ /cm–1 = 2974, 1607, 1486, 1362, 1315, 1259, 1190, 1084, 999, 938, 821, 751,
709.
HRMS (APCI) für C12H17BrNO+ [(M+H)+]: ber. 270.0488
gef. 270.0487

3 Beschreibung der Experimente 97
3.1.8 1-(2,4-Dimethylphenyl)ethan-1-on-O-(tert-butyl)oxim (134)
Dargestellt aus 1-(2,4-Dimethylphenyl)ethan-1-on (212 mg, 1.43 mmol, 1.00 Äquiv.) und O-
(tert-Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 15 cm, Cyclohexan:Ethylacetat:Toluol:Triethylamin
= 50:1:1:1, 50 mL, #3–5) lieferte die Titelverbindung 134 als Isomerengemisch (88:12,
bestimmt durch GC-Analyse, 249 mg, 79%) als farbloses Öl.
Rf = 0.08 (Cyclohexan).
GC (FS-SE-54): tR = 12.4 min (Mindermengenisomer), 14.1 min (Hauptmengenisomer).
1H-NMR (500 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 1.35 (s, 9H, OC(CH3)3), 2.16 (s,
3H, H-1), 2.33 (s, 3H, 6-CH3), 2.37 (s, 3H, 4-CH3), 7.02 (d, 3J7,8 = 7.5 Hz, 1H, H-7), 7.04 (s,
1H, H-5), 7.16 (d, 3J8,7 = 7.7 Hz, 1H, H-8).
13C-NMR (126 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 16.2 (C-1), 21.0 (4-CH3), 21.3
(6-CH3), 28.1 (OC(CH3)3), 78.3 (OC(CH3)3), 126.6 (C-7), 128.5 (C-8), 131.8 (C-5), 135.7 (C-
6), 136.1 (C-3), 137.9 (C-4), 154.9 (C-2).
IR (ATR): ~ /cm–1 = 2973, 2923, 1613, 1447, 1361, 1313, 1258, 1192, 998, 938, 913, 815,
729.
HRMS (APCI) für C14H22NO+ [(M+H)+]: ber. 220.1696
gef. 220.1695

98 EXPERIMENTELLER TEIL
3.1.9 1-(4-(1H-Imidazol-1-yl)phenyl)ethan-1-on-O-(tert-butyl)oxim (136)
Dargestellt aus 1-(4-(1H-Imidazol-1-yl)phenyl)ethan-1-on (148 mg, 0.795 mmol, 1.00 Äquiv.)
und O-(tert-Butyl)hydroxylaminhydrochlorid (100 mg, 0.795 mmol, 1.00 Äquiv.) gemäß AAV
1. Die Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung
mittels Flashchromatographie an Kieselgel (7 × 9 cm, Ethylacetat:Triethylamin = 20:1, 50
mL, #5–14) lieferte die Titelverbindung 136 (195 mg, 95%) als gelblichen Feststoff.
Rf = 0.36 (Ethylacetat).
Smp.: 76°C (Ethylacetat).
GC (FS-SE-54): tR = 21.6 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.37 (s, 9H, OC(CH3)3), 2.22 (s, 3H, H-1), 7.21 (mc, 1H,
H-8), 7.28 (mc, 1H, H-9), 7.36 (mc, 2H, H-5), 7.79 (mc, 2H, H-4), 7.86 (mc, 1H, H-7).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.2 (C-1), 27.9 (OC(CH3)3), 79.2 (OC(CH3)3), 118.2
(C-9), 121.2 (C-5), 127.4 (C-4), 130.7 (C-8), 135.6 (C-7), 136.9 (C-6), 137.4 (C-3), 151.3 (C-
2).
IR (ATR): ~ /cm–1 = 3142, 3119, 2973, 2926, 1608, 1519, 1474, 1417, 1366, 1302, 1245,
1188, 1108, 1055, 1031, 994, 935, 916, 898, 817, 739.
HRMS (APCI) für C15H20N3O+ [(M+H)+]: ber. 258.1601
gef. 258.1594

3 Beschreibung der Experimente 99
3.1.10 Diphenylmethanon-O-(tert-butyl)oxim (137)
Dargestellt aus Benzophenon (261 mg, 1.43 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (180 mg, 1.43 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde über Nacht bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 16 cm, Cyclohexan:Ethylacetat:Toluol:Triethylamin
= 100:5:1:1, 50 mL, #2–5) lieferte die Titelverbindung 137 (133 mg, 37%) als weißen
Feststoff.
Rf = 0.12 (Cyclohexan).
Smp.: 66°C (Cyclohexan).
GC (FS-SE-54): tR = 17.6 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.35 (s, 9H, OC(CH3)3), 7.30–7.44 (m, 8H, H-Ar), 7.51
(mc, 2H, H-Ar).
13C-NMR (126 MHz, CDCl3): δ/ppm = 27.9 (OC(CH3)3), 79.5 (OC(CH3)3), 128.0 (C-Ar), 128.1
(C-Ar), 128.3 (C-Ar), 128.6 (C-Ar), 129.0 (C-Ar), 130.0 (C-Ar), 134.0 (Cquart), 137.9 (Cquart),
154.9 (C-5).
IR (ATR): ~ /cm–1 = 2976, 2924, 1441, 1357, 1324, 1261, 1192, 1073, 954, 913, 865, 764,
690.
HRMS (APCI) für C17H20NO+ [(M+H)+]: ber. 254.1539
gef. 254.1540

100 EXPERIMENTELLER TEIL
3.1.11 Cyclohexanon-O-(tert-butyl)oxim (139)
Dargestellt aus Cyclohexanon (156 mg, 1.59 mmol, 1.00 Äquiv.) und O-(tert-
Butyl)hydroxylaminhydrochlorid (200 mg, 1.59 mmol, 1.00 Äquiv.) gemäß AAV 1. Die
Reaktionsmischung wurde für 9 h bei Raumtemperatur gerührt. Aufreinigung mittels
Flashchromatographie an Kieselgel (5 × 15 cm, Cyclohexan:Ethylacetat = 50:1, 50 mL, #14–
24) lieferte die Titelverbindung 139 (215 mg, 80%) als farbloses Öl.
Rf = 0.06 (Cyclohexan).
GC (FS-SE-54): tR = 8.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.26 (s, 9H, OC(CH3)3), 1.53–1.61 (m, 4H, H-3, H-4),
1.65 (mc, 2H, H-5), 2.19 (mc, 2H, H-6), 2.45 (mc, 2H, H-2).
13C-NMR (126 MHz, CDCl3): δ/ppm = 25.3 (C-2), 26.0 (C-Alkyl), 26.2 (C-Alkyl), 27.3 (C-
Alkyl), 27.7 (OC(CH3)3), 32.6 (C-6), 77.0 (OC(CH3)3), 158.9 (C-1).
IR (ATR): ~ /cm–1 = 2973, 2928, 2857, 1448, 1360, 1245, 1192, 990, 941, 877, 840, 790.
HRMS (APCI) für C10H20NO+ [(M+H)+]: ber. 170.1539
gef. 170.1538

3 Beschreibung der Experimente 101
3.1.12 1-Phenylethan-1-on-O-triethylsilyloxim (113)
Dargestellt aus 1-Phenylethan-1-onoxim (110, 654 mg, 4.84 mmol, 1.00 Äquiv.),
Chlortriethylsilan (0.811 mL, 729 mg, 4.84 mmol, 1.00 Äquiv.) und Imidazol (659 mg, 9.68
mmol, 2.00 Äquiv.) gemäß AAV 3. Aufreinigung mittels Flashchromatographie an Kieselgel
(7 × 9 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #3–10) lieferte die Titelverbindung 113 (894
mg, 74%) als farbloses Öl.
Rf = 0.82 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 16.4 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.78 (q, 3JSiCH2CH3,SiCH2CH3 = 8.0 Hz, 6H, SiCH2CH3),
1.03 (t, 3JSiCH2CH3,SiCH2CH3 = 8.0 Hz, 9H, SiCH2CH3), 2.28 (s, 3H, H-1), 7.33–7.40 (m, 3H, H-5,
H-6), 7.65–7.72 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 4.7 (SiCH2CH3), 6.9 (SiCH2CH3), 12.2 (C-1), 126.2 (C-
4), 128.4 (C-5), 129.1 (C-6), 137.2 (C-3), 158.7 (C-2).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 25.8.
IR (ATR): ~ /cm–1 = 2953, 2875, 1458, 1412, 1366, 1307, 1238, 991, 915, 849, 728, 689.
HRMS (APCI) für C14H24NOSi+ [(M+H)+]: ber. 250.1622
gef. 250.1626

102 EXPERIMENTELLER TEIL
3.1.13 1-Phenylethan-1-on-O-(tert-butyldimethylsilyl)oxim (114)
Dargestellt aus 1-Phenylethan-1-onoxim (110, 605 mg, 4.48 mmol, 1.00 Äquiv.), tert-
Butylchlordimethylsilan (675 mg, 4.48 mmol, 1.00 Äquiv.) und Imidazol (609 mg, 8.95 mmol,
2.00 Äquiv.) gemäß AAV 3. Aufreinigung mittels Flashchromatographie an Kieselgel (7 × 14
cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–15) lieferte die Titelverbindung 114 (791 mg,
71%) als farbloses Öl.
Rf = 0.81 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 15.3 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.23 (s, 6H, SiCH3), 0.99 (s, 9H, SiC(CH3)3), 2.27 (s,
3H, H-1), 7.33–7.40 (m, 3H, H-5, H-6), 7.66–7.71 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = –5.0 (SiCH3), 12.3 (C-1), 18.4 (SiC(CH3)3), 26.3
(SiC(CH3)3), 126.2 (C-4), 128.4 (C-5), 129.1 (C-6), 137.1 (C-3), 158.8 (C-2).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 26.6.
IR (ATR): ~ /cm–1 = 2928, 2855, 1469, 1364, 1308, 1249, 992, 866, 834, 778, 757, 690.
HRMS (APCI) für C14H24NOSi+ [(M+H)+]: ber. 250.1622
gef. 250.1623

3 Beschreibung der Experimente 103
3.1.14 1-Phenylethan-1-on-O-triisopropylsilyloxim (115)
Dargestellt aus 1-Phenylethan-1-onoxim (110, 763 mg, 5.65 mmol, 1.00 Äquiv.),
Chlortriisopropylsilan (1.21 mL, 1.09 g, 5.65 mmol, 1.00 Äquiv.) und Imidazol (769 mg, 11.3
mmol, 2.00 Äquiv.) gemäß AAV 3. Aufreinigung mittels Flashchromatographie an Kieselgel
(Cyclohexan:Ethylacetat) lieferte die Titelverbindung 115 (1.23 g, 75%) als farbloses Öl.
Rf = 0.78 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 19.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.12 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 18H, SiCH(CH3)2),
1.29 (sept, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 3H, SiCH(CH3)2), 2.28 (s, 3H, H-1), 7.31–7.40 (m,
3H, H-5, H-6), 7.65–7.72 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (C-1), 12.1 (SiCH(CH3)2), 18.2 (SiCH(CH3)2),
126.2 (C-4), 128.4 (C-5), 129.0 (C-6), 137.2 (C-3), 158.4 (C-2).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 20.2.
IR (ATR): ~ /cm–1 = 2941, 2864, 1461, 1366, 1307, 1248, 989, 922, 880, 844, 750, 687.
HRMS (APCI) für C17H30NOSi+ [(M+H)+]: ber. 292.2091
gef. 292.2094

104 EXPERIMENTELLER TEIL
3.1.15 Benzaldehyd-O-triisopropylsilyloxim (117)
Benzaldehyd (1.53 g, 14.4 mmol, 1.00 Äquiv.) und Hydroxylaminhydrochlorid (1.00 g, 14.4
mmol, 1.00 Äquiv.) wurden über Nacht bei Raumtemperatur in Methanol (10 mL) mit einem
Tropfen konz. HCl ohne Auschluss von Sauerstoff und Feuchtigkeit gerührt. Nach Entfernen
des Lösungsmittels unter vermindertem Druck wurde der Rückstand mittels
Flashchromatographie an Kieselgel (9 × 13 cm, Cyclohexan:Ethylacetat = 10:1, 50 mL, #34–
50) aufgereinigt und das ungeschützte Oxim (1.10 g, 63%) erhalten. Das ungeschützte Oxim
(632 mg, 522 mmol, 1.00 Äquiv.) wurde mit Chlortriisopropylsilan (1.12 mL, 1.01 g, 5.22
mmol, 1.00 Äquiv.) und Imidazol (710 mg, 10.4 mmol, 2.00 Äquiv.) gemäß AAV 3
umgesetzt. Aufreinigung mittels Flashchromatographie an Kieselgel (9 × 11 cm,
Cyclohexan:Ethylacetat = 9:1, 50 mL, #18–28) lieferte die Titelverbindung 117 (1.37 g, 95%)
als farbloses Öl.
Rf = 0.83 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 18.5 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.15 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.5 Hz, 18H,
SiCH(CH3)2), 1.31 (mc, 3H, SiCH(CH3)2), 7.36–7.40 (m, 3H, H-4, H-5), 7.59–7.63 (m, 2H, H-
3), 8.24 (s, 1H, H-1).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.2 (SiCH(CH3)2), 18.2 (SiCH(CH3)2), 127.2 (C-3),
128.9 (C-4), 129.9 (C-5), 133.2 (C-2), 153.2 (C-1).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 21.3.
IR (ATR): ~ /cm–1 = 2942, 2864, 1462, 1383, 1335, 1249, 1207, 997, 942, 881, 781, 753,
686.

3 Beschreibung der Experimente 105
HRMS (APCI) für C16H28NOSi+ [(M+H)+]: ber. 278.1935
gef. 278.1928
3.1.16 1-(4-(Trifluormethyl)phenyl)ethan-1-on-O-triisopropylsilyloxim (127)
Das ungeschützte Oxim wurde aus 1-(4-(Trifluormethyl)phenyl)ethan-1-on (1.00 g, 5.31
mmol, 1.00 Äquiv.), Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und
Natriumacetat (436 mg, 5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit
Chlortriisopropylsilan (1.36 mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6
mmol, 2.00 Äquiv.) gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels
Flashchromatographie an Kieselgel (8 × 13 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–
14) lieferte die Titelverbindung 127 (1.81 g, 95%) als farbloses Öl.
Rf = 0.74 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 18.6 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.12 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 18H, SiCH(CH3)2),
1.29 (mc, 3H, SiCH(CH3)2), 2.30 (s, 3H, H-1), 7.62 (mc, 2H, H-5), 7.79 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (C-1), 12.2 (SiCH(CH3)2), 18.2 (SiCH(CH3)2),
124.4 (q, 1J7,F = 272 Hz, C-7), 125.5 (q, 3J5,F = 3.8 Hz, C-5), 126.5 (C-4), 130.9 (q, 2J6,F = 32
Hz, C-6), 140.7 (C-3), 157.5 (C-2).
19F-NMR (471 MHz, CDCl3): δ/ppm = –62.7.
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 21.2.
IR (ATR): ~ /cm–1 = 2944, 2866, 1603, 1462, 1406, 1323, 1166, 1127, 1085, 993, 929, 881,
841, 786, 733, 681.

106 EXPERIMENTELLER TEIL
HRMS (APCI) für C18H29F3NOSi+ [(M+H)+]: ber. 360.1965
gef. 360.1964
3.1.17 1-(4-Methoxyphenyl)ethan-1-on-O-triisopropylsilyloxim (129)
Das ungeschützte Oxim wurde aus 1-(4-Methoxyphenyl)ethan-1-on (2.16 g, 14.4 mmol, 1.00
Äquiv.), Hydroxylaminhydrochlorid (1.00 g, 14.4 mmol, 1.00 Äquiv.) und Natriumacetat (1.18
g, 14.4 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit Chlortriisopropylsilan
(3.70 mL, 3.33 g, 17.3 mmol, 1.20 Äquiv.) und Imidazol (1.96 g, 28.8 mmol, 2.00 Äquiv.)
gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels Flashchromatographie
an Kieselgel (8 × 12 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–15) lieferte die
Titelverbindung 129 (3.86 g, 83%) als farbloses Öl.
Rf = 0.68 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 21.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.12 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.3 Hz, 18H, SiCH(CH3)2),
1.22–1.33 (m, 3H, SiCH(CH3)2), 2.25 (s, 3H, H-1), 3.83 (s, 3H, OCH3), 6.89 (mc, 2H, H-5),
7.63 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 11.8 (C-1), 12.0 (SiCH(CH3)2), 18.0 (SiCH(CH3)2), 55.3
(OCH3), 113.7 (C-5), 127.3 (C-4), 129.7 (C-3), 157.7 (C-2), 160.3 (C-6).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 19.8.
IR (ATR): ~ /cm–1 = 2941, 2864, 1608, 1511, 1461, 1365, 1314, 1247, 1176, 1034, 989, 917,
880, 829, 714, 679.

3 Beschreibung der Experimente 107
HRMS (APCI) für C18H32NO2Si+ [(M+H)+]: ber. 322.2197
gef. 322.2199
3.1.18 1-(4-Chlorphenyl)propan-1-on-O-triisopropylsilyloxim (131)
Das ungeschützte Oxim wurde aus 1-(4-Chlorphenyl)propan-1-on (896 mg, 5.31 mmol, 1.00
Äquiv.), Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und Natriumacetat (436
mg, 5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit Chlortriisopropylsilan
(1.36 mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6 mmol, 2.00 Äquiv.)
gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels Flashchromatographie
an Kieselgel (8 × 14 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–12) lieferte die
Titelverbindung 131 als Isomerengemisch (87:13, bestimmt durch GC-Analyse, 1.74 g, 96%)
als farbloses Öl.
Rf = 0.81 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 20.3 min (Mindermengenisomer), 21.4 min (Hauptmengenisomer).
1H-NMR (500 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 1.09–1.16 (m, 21H, SiCH(CH3)2,
H-1), 1.23–1.34 (m, 3H, SiCH(CH3)2), 2.79 (q, 3J2,3 = 7.6 Hz, 2H, H-2), 7.30–7.36 (m, 2H, H-
6), 7.58–7.63 (m, 2H, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 10.9 (C-1), 11.9 (SiCH(CH3)2),
18.0 (SiCH(CH3)2), 19.2 (C-2), 127.4 (C-5), 128.6 (C-6), 134.5 (C-4), 134.7 (C-7), 161.9 (C-
3).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 20.5.
IR (ATR): ~ /cm–1 = 2941, 2865, 1600, 1461, 1383, 1250, 1091, 1013, 930, 881, 830, 758,
675.

108 EXPERIMENTELLER TEIL
HRMS (APCI) für C18H31ClNOSi+ [(M+H)+]: ber. 340.1858
gef. 340.1858
3.1.19 1-(4-Bromphenyl)ethan-1-on-O-triisopropylsilyloxim (133)
Das ungeschützte Oxim wurde aus 1-(4-Bromphenyl)ethan-1-on (968 mg, 5.31 mmol, 1.00
Äquiv.), Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und Natriumacetat (436
mg, 5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit Chlortriisopropylsilan
(1.36 mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6 mmol, 2.00 Äquiv.)
gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels Flashchromatographie
an Kieselgel (8 × 16 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #10–25) lieferte die
Titelverbindung 133 (1.49 g, 76%) als farbloses Öl.
Rf = 0.46 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 22.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.11 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 18H, SiCH(CH3)2),
1.23–1.33 (m, 3H, SiCH(CH3)2), 2.25 (s, 3H, H-1), 7.48 (mc, 2H, H-5), 7.55 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 11.8 (C-1), 12.1 (SiCH(CH3)2), 18.1 (SiCH(CH3)2),
123.3 (C-6), 127.7 (C-4), 135.6 (C-5), 136.1 (C-3), 157.5 (C-2).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 20.7.
IR (ATR): ~ /cm–1 = 2941, 2864, 1603, 1461, 1392, 1311, 1250, 1069, 991, 922, 880, 820,
759.
HRMS (APCI) für C17H29BrNOSi+ [(M+H)+]: ber. 370.1196
gef. 370.1195

3 Beschreibung der Experimente 109
3.1.20 1-(2,4-Dimethylphenyl)ethan-1-on-O-triisopropylsilyloxim (135)
Das ungeschützte Oxim wurde aus 1-(2,4-Dimethylphenyl)ethan-1-on (787 mg, 5.31 mmol,
1.00 Äquiv.), Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und Natriumacetat
(436 mg, 5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit
Chlortriisopropylsilan (1.36 mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6
mmol, 2.00 Äquiv.) gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels
Flashchromatographie an Kieselgel (8 × 13 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–
13) lieferte die Titelverbindung 135 (1.60 g, 94%) als farbloses Öl.
Rf = 0.79 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 21.3 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.12 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 18H, SiCH(CH3)2),
1.29 (sept, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 3H, SiCH(CH3)2), 2.25 (s, 3H, H-1), 2.27 (s, 3H, 4-
CH3), 2.29 (s, 3H, 6-CH3), 7.12 (mc, 1H, H-5), 7.42 (mc, 1H, H-7), 7.45 (mc, 1H, H-8).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.1 (C-1), 12.1 (SiCH(CH3)2), 18.2 (SiCH(CH3)2), 19.7
(4-CH3)*, 20.1 (6-CH3)*, 123.7 (C-7), 127.4 (C-8), 129.7 (C-5), 134.9 (C-3), 136.5 (C-4),
137.6 (C-6), 158.5 (C-2).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 19.8.
IR (ATR): ~ /cm–1 = 2941, 2864, 1461, 1364, 1318, 1249, 986, 938, 876, 679.
HRMS (APCI) für C19H34NOSi+ [(M+H)+]: ber. 320.2404
gef. 320.2401

110 EXPERIMENTELLER TEIL
3.1.21 Diphenylmethanon-O-triisopropylsilyloxim (138)
Das ungeschützte Oxim wurde aus Benzophenon (968 mg, 5.31 mmol, 1.00 Äquiv.),
Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und Natriumacetat (436 mg,
5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit Chlortriisopropylsilan (1.36
mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6 mmol, 2.00 Äquiv.) gemäß
AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels Flashchromatographie an
Kieselgel (8 × 14 cm, Cyclohexan:Ethylacetat = 9:1, 60 mL, #6–10) lieferte die
Titelverbindung 138 (1.22 g, 65%) als farbloses Öl.
Rf = 0.78 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 23.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.07 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.4 Hz, 18H, SiCH(CH3)2),
1.20–1.30 (m, 3H, SiCH(CH3)2), 7.30–7.43 (m, 8H, H-Ar), 7.50–7.54 (m, 2H, H-Ar).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (SiCH(CH3)2), 18.1 (SiCH(CH3)2), 127.8 (C-Ar),
128.0 (C-Ar), 128.3 (C-Ar), 128.5 (C-1)*, 129.2 (C-9)*, 129.5 (C-Ar), 133.7 (C-4)**, 137.1 (C-
6)**, 160.4 (C-5).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 21.0.
IR (ATR): ~ /cm–1 = 2942, 2864, 1462, 1383, 1323, 1249, 1165, 943, 881, 805, 767, 690.
HRMS (APCI) für C22H32NOSi+ [(M+H)+]: ber. 354.2248
gef. 354.2247

3 Beschreibung der Experimente 111
3.1.22 Cyclohexanon-O-triisopropylsilyloxim (140)
Dargestellt aus Cyclohexanonoxim (800 mg, 7.07 mmol, 1.00 Äquiv.), Chlortriisopropylsilan
(1.51 mL, 1.36 g, 7.07 mmol, 1.00 Äquiv.) und Imidazol (866 mg, 12.7 mmol, 1.80 Äquiv.)
gemäß AAV 3. Aufreinigung mittels Flashchromatographie an Kieselgel (8 × 15 cm,
Cyclohexan:Ethylacetat = 9:1, 50 mL, #9–15) lieferte die Titelverbindung 140 (1.78 g, 93%)
als farbloses Öl.
Rf = 0.79 (Cyclohexan:Ethylacetat = 9:1).
GC (FS-SE-54): tR = 16.4 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.07 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 7.3 Hz, 18H, SiCH(CH3)2),
1.15–1.26 (m, 3H, SiCH(CH3)2), 1.54–1.68 (m, 6H, H-3, H-4, H-5), 2.20 (mc, 2H, H-6), 2.53
(mc, 2H, H-2).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.1 (SiCH(CH3)2), 18.2 (SiCH(CH3)2), 25.0 (C-6), 26.1
(C-Alkyl), 26.2 (C-Alkyl), 27.3 (C-Alkyl), 32.5 (C-2), 163.9 (C-1).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 18.0.
IR (ATR): ~ /cm–1 = 2931, 2862, 1462, 1382, 1251, 987, 905, 837, 766, 678.
HRMS (APCI) für C15H32NOSi+ [(M+H)+]: ber. 270.2248
gef. 270.2249

112 EXPERIMENTELLER TEIL
3.1.23 rac-4-(tert-Butyl)cyclohexanon-O-triisopropylsilyloxim (rac-141)
Das ungeschützte Oxim wurde aus rac-4-(tert-Butyl)cyclohexan-1-on (819 mg, 5.31 mmol,
1.00 Äquiv.), Hydroxylaminhydrochlorid (369 mg, 5.31 mmol, 1.00 Äquiv.) und Natriumacetat
(436 mg, 5.31 mmol, 1.00 Äquiv.) gemäß AAV 2 erhalten. Umsetzung mit
Chlortriisopropylsilan (1.36 mL, 1.23 g, 6.38 mmol, 1.20 Äquiv.) und Imidazol (724 mg, 10.6
mmol, 2.00 Äquiv.) gemäß AAV 3 ergab das silylgeschützte Oxim. Aufreinigung mittels
Flashchromatographie an Kieselgel (7 × 14 cm, Cyclohexan:Ethylacetat = 19:1, 50 mL, #17–
25) lieferte die Titelverbindung rac-141 (1.37 g, 79%) als farbloses Öl.
Rf = 0.12 (Cyclohexan).
GC (FS-SE-54): tR = 20.1 min.
1H-NMR (400 MHz, CDCl3): δ/ppm = 0.87 (s, 9H, 4-C(CH3)3), 1.07 (d, 3JSiCH(CH3)2,SiCH(CH3)2 =
7.0 Hz, 18H, SiCH(CH3)2), 1.11–1.30 (m, 6H, SiCH(CH3)2, H-4, H-Alkyl), 1.62 (dt, J = 13.9
Hz, J = 4.5 Hz, 1H, H-Alkyl), 1.92 (mc, 2H, H-Alkyl), 2.02 (dt, J = 13.4 Hz, J = 4.5 Hz, 1H, H-
Alkyl) 2.45 (mc, 1H, H-Alkyl), 3.49 (mc, 1H, H-Alkyl).
13C-NMR (101 MHz, CDCl3): δ/ppm = 12.1 (SiCH(CH3)2), 18.2 (SiCH(CH3)2), 24.8 (C-Alkyl),
26.7 (C-Alkyl), 27.7 (4-C(CH3)3), 28.0 (C-Alkyl), 32.2 (C-Alkyl), 32.6 (4-C(CH3)3), 47.9 (C-4),
164.1 (C-1).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 18.0.
IR (ATR): ~ /cm–1 = 2944, 2864, 1724, 1462, 1383, 1365, 1256, 986, 911, 881, 835, 778,
679.

3 Beschreibung der Experimente 113
HRMS (APCI) für C19H40NOSi+ [(M+H)+]: ber. 326.2874
gef. 326.2866
3.2 Darstellung von Hydroxylaminen
3.2.1 rac-O-(tert-Butyl)-N-(1-phenylethyl)hydroxylamin (rac-120)
Gemäß AAV 4 wurde eine Mischung von 1-Phenylethan-1-on-O-(tert-butyl)oxim (112, 27
mg, 0.14 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.6 mg, 7.0 µmol, 5.0 Mol-
%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei Raumtemperatur für 18 h gerührt.
Die Reaktionsmischung wurde über Kieselgel filtriert und mit Cyclohexan:Ethylacetat = 1:1
eluiert. Nach Entfernen der Lösungsmittel unter vermindertem Druck wurde die
Titelverbindung rac-120 (24 mg, 88%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 11.7 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.15 (s, 9H, OC(CH3)3), 1.39 (d, 3J1,2 = 6.6 Hz, 3H, H-
1), 4.04 (q, 3J2,1 = 6.6 Hz, 1H, H-2), 4.95 (br s, 1H, NH), 7.23–7.29 (m, 1H, H-6), 7.29–7.37
(m, 4H, H-4, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm = 20.1 (C-1), 27.1 (OC(CH3)3), 60.7 (C-2), 76.9
(OC(CH3)3), 127.5 (C-Ar), 127.7 (C-Ar), 128.5 (C-Ar), 143.3 (C-3).
IR (ATR): ~ /cm–1 = 2971, 2927, 1493, 1452, 1359, 1257, 1194, 1074, 992, 927, 875, 759,
697.
HRMS (APCI) für C12H20NO+ [(M+H)+]: ber. 194.1539
gef. 194.1540

114 EXPERIMENTELLER TEIL
3.2.2 rac-O-(tert-Butyl)-N-(1-(4-(trifluormethyl)phenyl)ethyl)hydroxylamin (rac-144)
Gemäß AAV 4 wurde eine Mischung von 1-(4-(Trifluormethyl)phenyl)ethan-1-on-O-(tert-
butyl)oxim (126, 36 mg, 0.14 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.6 mg,
7.0 µmol, 5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei Raumtemperatur
für 18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-144 (29 mg, 80%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 11.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.11 (s, 9H, OC(CH3)3), 1.37 (d, 3J1,2 = 6.7 Hz, 3H, H-
1), 4.09 (q, 3J2,1 = 6.7 Hz, 1H, H-2), 4.75 (br s, 1H, NH), 7.45 (d, 3J4,5 = 8.2 Hz, 2H, H-4), 7.57
(d, 3J5,4 = 8.2 Hz, 2H, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm = 20.0 (C-1), 27.0 (OC(CH3)3), 60.4 (C-2), 77.2
(OC(CH3)3), 124.5 (q, 1J7,F = 272 Hz, C-7), 125.4 (q, 3J5,F = 3.8 Hz, C-5), 128.1 (C-4), 129.8
(q, 2J6,F = 32 Hz, C-6), 147.5 (C-3).
19F-NMR (471 MHz, CDCl3): δ/ppm = –62.4.
IR (ATR): ~ /cm–1 = 2974, 2931, 1619, 1461, 1417, 1361, 1322, 1163, 1121, 1066, 1016,
875, 836, 751.
HRMS (APCI) für C13H19F3NO+ [(M+H)+]: ber. 262.1413
gef. 262.1416

3 Beschreibung der Experimente 115
3.2.3 rac-O-(tert-Butyl)-N-(1-(4-methoxyphenyl)ethyl)hydroxylamin (rac-146)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Methoxyphenyl)ethan-1-on-O-(tert-butyl)oxim
(128, 28 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.2 mg, 6.3 µmol,
5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei Raumtemperatur für 18 h
gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-146 (27 mg, 96%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 15.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.14 (s, 9H, OC(CH3)3), 1.36 (d, 3J1,2 = 6.6 Hz, 3H, H-
1), 3.80 (s, 3H, OCH3), 3.99 (q, 3J2,1 = 6.6 Hz, 1H, H-2), 4.86 (br s, 1H, NH), 6.86 (mc, 2H, H-
5), 7.26 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 20.0 (C-1), 27.0 (OC(CH3)3), 55.4 (OCH3), 59.9 (C-2),
76.7 (OC(CH3)3), 113.7 (C-5), 128.7 (C-4), 135.3 (C-3), 159.0 (C-6).
IR (ATR): ~ /cm–1 = 2970, 2929, 1611, 1511, 1458, 1359, 1301, 1242, 1195, 1099, 1033,
992, 925, 876, 827.
HRMS (APCI) für C13H22NO2+ [(M+H)+]: ber. 224.1645
gef. 224.1650

116 EXPERIMENTELLER TEIL
3.2.4 rac-O-(tert-Butyl)-N-(1-(4-chlorphenyl)propyl)hydroxylamin (rac-148)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Chlorphenyl)propan-1-on-O-(tert-butyl)oxim
(130, 31 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.3 mg, 6.4 µmol,
5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei Raumtemperatur für 18 h
gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-148 (31 mg, 99%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 15.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.79 (dd, 3J1,2A = 7.5 Hz, 3J1,2B = 7.5 Hz, 3H, H-1), 1.09
(s, 9H, OC(CH3)3), 1.57 (mc, 1H, H-2A), 1.89 (mc, 1H, H-2B), 3.73 (mc, 1H, H-3), 4.90 (br s,
1H, NH), 7.22 (mc, 2H, H-5), 7.28 (mc, 2H, H-6).
13C-NMR (126 MHz, CDCl3): δ/ppm = 10.8 (C-1), 26.5 (C-2), 27.0 (OC(CH3)3), 67.0 (C-3),
77.0 (OC(CH3)3), 128.4 (C-6), 129.6 (C-5), 133.1 (C-7), 140.4 (C-4).
IR (ATR): ~ /cm–1 = 2969, 2930, 2874, 1490, 1460, 1359, 1193, 1089, 1014, 880, 819, 752,
720.
HRMS (APCI) für C13H21ClNO+ [(M+H)+]: ber. 242.1306
gef. 242.1311

3 Beschreibung der Experimente 117
3.2.5 rac-N-(1-(4-Bromphenyl)ethyl)-O-(tert-butyl)hydroxylamin (rac-150)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Bromphenyl)ethan-1-on-O-(tert-butyl)oxim
(132, 35 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.3 mg, 6.4 µmol,
5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei Raumtemperatur für 18 h
gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-150 (34 mg, 96%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 15.2 min.
1H-NMR (400 MHz, CDCl3): δ/ppm = 1.11 (s, 9H, OC(CH3)3), 1.34 (d, 3J1,2 = 6.7 Hz, 3H, H-
1), 4.00 (q, 3J2,1 = 6.7 Hz, 1H, H-2), 4.86 (br s, 1H, NH), 7.21 (mc, 2H, H-4), 7.43 (mc, 2H, H-
5).
13C-NMR (101 MHz, CDCl3): δ/ppm = 19.9 (C-1), 27.0 (OC(CH3)3), 60.0 (C-2), 76.9
(OC(CH3)3), 121.1 (C-6), 129.4 (C-4), 131.4 (C-5), 142.4 (C-3).
IR (ATR): ~ /cm–1 = 2971, 2927, 1486, 1454, 1359, 1258, 1193, 1071, 1009, 926, 874, 819,
771, 716.
HRMS (APCI) für C12H19BrNO+ [(M+H)+]: ber. 272.0645
gef. 272.0642

118 EXPERIMENTELLER TEIL
3.2.6 rac-O-(tert-Butyl)-N-(1-(2,4-dimethylphenyl)ethyl)hydroxylamin (rac-152)
Gemäß AAV 4 wurde eine Mischung von 1-(2,4-Dimethylphenyl)ethan-1-on-O-(tert-
butyl)oxim (134, 26 mg, 0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 5.0 mg,
9.8 µmol, 8.2 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für 18 h
gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-152 als Mischung mit Startmaterial 134 (26 mg, rac-
152:134 = 95:5, bestimmt durch GC-Analyse, 94%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 14.3 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.19 (s, 9H, OC(CH3)3), 1.37 (d, 3J1,2 = 6.6 Hz, 3H, H-
1), 2.31 (s, 3H, 6-CH3), 2.37 (s, 3H, 4-CH3), 4.29 (q, 3J2,1 = 6.6 Hz, 1H, H-2), 4.87 (br s, 1H,
NH), 6.98 (s, 1H, H-5), 7.01 (d, 3J7,8 = 7.8 Hz, 1H, H-7), 7.28 (d, 3J8,7 = 7.8 Hz, 1H, H-8).
13C-NMR (126 MHz, CDCl3): δ/ppm = 19.5 (4-CH3), 19.6 (C-1), 21.2 (6-CH3), 27.1
(OC(CH3)3), 55.8 (C-2), 76.8 (OC(CH3)3), 126.3 (C-8), 126.9 (C-7), 131.2 (C-5), 135.9 (C-4),
136.6 (C-6), 138.2 (C-3).
HRMS (APCI) für C14H24NO+ [(M+H)+]: ber. 222.1852
gef. 222.1849

3 Beschreibung der Experimente 119
3.2.7 N-Benzhydryl-O-(tert-butyl)hydroxylamin (155)
Gemäß AAV 4 wurde eine Mischung von Diphenylmethanon-O-(tert-butyl)oxim (137, 31 mg,
0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 6.3 mg, 12 µmol, 10 Mol-%) in
Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für 18 h gerührt. Die
Reaktionsmischung wurde über Kieselgel filtriert und mit Cyclohexan:Ethylacetat = 1:1
eluiert. Nach Entfernen der Lösungsmittel unter vermindertem Druck wurde die
Titelverbindung 155 als Mischung mit Startmaterial 137 (23 mg, 155:137 = 90:10, bestimmt
durch GC-Analyse, 66%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 18.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.09 (s, 9H, OC(CH3)3), 5.11 (s, 1H, H-1), 5.35 (br s,
1H, NH), 7.21–7.25 (m, 2H, H-5), 7.28–7.32 (m, 4H, H-4), 7.35–7.39 (m, 4H, H-3).
13C-NMR (126 MHz, CDCl3): δ/ppm = 27.0 (OC(CH3)3), 69.6 (C-1), 77.1 (OC(CH3)3), 127.3
(C-5), 128.2 (C-3), 128.3 (C-4), 141.9 (C-2).
HRMS (APCI) für C17H22NO+ [(M+H)+]: ber. 256.1696
gef. 256.1692
3.2.8 rac-N-(1-Phenylethyl)-O-(triisopropylsilyl)hydroxylamin (rac-123)

120 EXPERIMENTELLER TEIL
Gemäß AAV 4 wurde eine Mischung von 1-Phenylethan-1-on-O-triisopropylsilyloxim (115,
35 mg, 0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.1 mg, 6.1 µmol, 5.0
Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (60 bar) bei Raumtemperatur für 16 h
gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-123 (35 mg, 99%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 18.5 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.05–1.10 (m, 18H, SiCH(CH3)2), 1.11–1.21 (m, 3H,
SiCH(CH3)2), 1.43 (d, 3J1,2 = 6.7 Hz, 3H, H-1), 4.11 (q, 3J2,1 = 6.7 Hz, 1H, H-2), 4.96 (br s,
1H, NH), 7.24–7.30 (m, 1H, H-Ar), 7.30–7.36 (m, 4H, H-Ar).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.1 (SiCH(CH3)2), 18.4 (SiCH(CH3)2), 18.4
(SiCH(CH3)2), 19.7 (C-1), 62.4 (C-2), 127.6 (C-Ar), 127.6 (C-Ar), 128.5 (C-Ar), 143.0 (C-3).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 17.9.
IR (ATR): ~ /cm–1 = 2940, 2864, 1459, 1382, 1248, 1072, 990, 926, 857, 815, 758, 676.
HRMS (APCI) für C17H32NOSi+ [(M+H)+]: ber. 294.2248
gef. 294.2251
3.2.9 rac-N-(1-(4-Trifluormethyl)phenylethyl)-O-(triisopropylsilyl)hydroxylamin
(rac-145)
Gemäß AAV 4 wurde eine Mischung von 1-(4-(Trifluormethyl)phenyl)ethan-1-on-O-
triisopropylsilyloxim (127, 47 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1,
3.3 mg, 6.4 µmol, 5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für
18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit

3 Beschreibung der Experimente 121
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-145 (47 mg, 99%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 18.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.00–1.08 (m, 18H, SiCH(CH3)2), 1.09–1.17 (m, 3H,
SiCH(CH3)2), 1.41 (d, 3J1,2 = 6.7 Hz, 3H, H-1), 4.15 (q, 3J2,1 = 6.7 Hz, 1H, H-2), 4.97 (br s,
1H, NH), 7.45 (d, 3J4,5 = 8.1 Hz, 2H, H-4), 7.58 (d, 3J5,4 = 8.1 Hz, 2H, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm = 11.9 (SiCH(CH3)2), 18.2 (SiCH(CH3)2), 18.3
(SiCH(CH3)2), 19.4 (C-1), 62.0 (C-2), 124.4 (q, 1J7,F = 272 Hz, C-7), 125.3 (q, 3J5,F = 3.7 Hz,
C-5), 127.8 (C-4), 129.7 (q, 2J6,F = 32 Hz, C-6), 147.2 (C-3).
19F-NMR (471 MHz, CDCl3): δ/ppm = –62.5.
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 18.5.
IR (ATR): ~ /cm–1 = 2942, 2865, 1619, 1462, 1417, 1323, 1164, 1124, 1068, 993, 930, 880,
837, 784, 737, 677.
HRMS (APCI) für C18H31F3NOSi+ [(M+H)+]: ber. 362.2122
gef. 362.2115
3.2.10 rac-N-(1-(4-Methoxyphenyl)ethyl)-O-(triisopropylsilyl)hydroxylamin (rac-147)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Methoxyphenyl)ethan-1-on-O-
triisopropylsilyloxim (129, 40 mg, 0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1,
6.4 mg, 12 µmol, 10 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für
18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit

122 EXPERIMENTELLER TEIL
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-147 (39 mg, 97%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 21.1 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.05–1.10 (m, 18H, SiCH(CH3)2), 1.12–1.21 (m, 3H,
SiCH(CH3)2), 1.41 (d, 3J1,2 = 6.6 Hz, 3H, H-1), 3.80 (s, 3H, OCH3), 4.06 (q, 3J2,1 = 6.6 Hz, 1H,
H-2), 4.86 (br s, 1H, NH), 6.86 (mc, 2H, H-5), 7.26 (mc, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (SiCH(CH3)2), 18.3 (SiCH(CH3)2), 19.4 (C-1), 55.4
(OCH3), 61.6 (C-2), 113.8 (C-5), 128.6 (C-4), 134.9 (C-3), 159.0 (C-6).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 17.8.
IR (ATR): ~ /cm–1 = 2940, 2864, 1610, 1511, 1462, 1366, 1285, 1244, 1177, 1087, 1036,
990, 921, 881, 829, 794, 725, 675.
HRMS (APCI) für C18H34NO2Si+ [(M+H)+]: ber. 324.2353
gef. 324.2349
3.2.11 rac-N-(1-(4-Chlorphenyl)propyl)-O-(triisopropylsilyl)hydroxylamin (rac-149)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Chlorphenyl)propan-1-on-O-
triisopropylsilyloxim (131, 44 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1,
6.6 mg, 13 µmol, 10 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für
18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung rac-149 (44 mg, 99%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 21.1 min.

3 Beschreibung der Experimente 123
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.81 (dd, 3J1,2A = 7.9 Hz, 3J1,2B = 7.9 Hz, 3H, H-1), 1.00–
1.06 (m, 18H, SiCH(CH3)2), 1.07–1.15 (m, 3H, SiCH(CH3)2), 1.54–1.64 (m, 1H, H-2A)*, 1.95
(mc, 1H, H-2B)*, 3.80 (dd, 3J3,2A = 5.4 Hz, 3J3,2B = 8.7 Hz, 1H, H-3), 4.95 (br s, 1H, NH), 7.21
(mc, 2H, H-5), 7.29 (mc, 2H, H-6).
13C-NMR (126 MHz, CDCl3): δ/ppm = 10.8 (C-1), 11.9 (SiCH(CH3)2), 18.3 (SiCH(CH3)2), 18.3
(SiCH(CH3)2), 26.2 (C-2), 68.7 (C-3), 128.5 (C-6), 129.4 (C-5), 133.1 (C-7), 140.3 (C-4).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 18.2.
IR (ATR): ~ /cm–1 = 2940, 2864, 1461, 1381, 1248, 1091, 1013, 880, 822, 784, 676.
HRMS (APCI) für C18H33ClNOSi+ [(M+H)+]: ber. 342.2014
gef. 342.2010
3.2.12 rac-N-(1-(4-Bromphenyl)ethyl)-O-(triisopropylsilyl)hydroxylamin (rac-151)
Gemäß AAV 4 wurde eine Mischung von 1-(4-Bromphenyl)ethan-1-on-O-
triisopropylsilyloxim (133, 48 mg, 0.13 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1,
3.3 mg, 6.4 µmol, 10 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei
Raumtemperatur für 18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und
mit Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter
vermindertem Druck wurde die Titelverbindung rac-151 (46 mg, 95%) als farbloses Öl
erhalten.
GC (FS-SE-54): tR = 21.6 min.
1H-NMR (400 MHz, CDCl3): δ/ppm = 1.00–1.08 (m, 18H, SiCH(CH3)2), 1.08–1.16 (m, 3H,
SiCH(CH3)2), 1.38 (d, 3J1,2 = 6.7 Hz, 3H, H-1), 4.05 (q, 3J2,1 = 6.7 Hz, 1H, H-2), 4.90 (br s,
1H, NH), 7.20 (mc, 2H, H-4), 7.44 (mc, 2H, H-5).

124 EXPERIMENTELLER TEIL
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (SiCH(CH3)2), 18.3 (SiCH(CH3)2), 18.3
(SiCH(CH3)2), 19.4 (C-1), 61.7 (C-2), 121.2 (C-6), 129.2 (C-4), 131.5 (C-5), 142.1 (C-3).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 18.5.
IR (ATR): ~ /cm–1 = 2940, 2863, 1461, 1382, 1248, 1153, 1073, 992, 926, 858, 823, 790,
676.
HRMS (APCI) für C17H30BrNOSi+ [(M+H)+]: ber. 372.1353
gef. 372.1351
3.2.13 rac-N-(1-(2,4-Dimethylphenylethyl)-O-(triisopropylsilyl)hydroxylamin
(rac-153)
Gemäß AAV 4 wurde eine Mischung von 1-(2,4-Dimethylphenyl)ethan-1-on-O-
triisopropylsilyloxim (135, 39 mg, 0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1,
3.1 mg, 6.1 µmol, 5.0 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei
Raumtemperatur für 18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und
mit Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter
vermindertem Druck wurde die Titelverbindung rac-153 (39 mg, 99%) als farbloses Öl
erhalten.
GC (FS-SE-54): tR = 20.5 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.07–1.12 (m, 18H, SiCH(CH3)2), 1.15–1.22 (m, 3H,
SiCH(CH3)2), 1.43 (d, 3J1,2 = 6.6 Hz, 3H, H-1), 2.26 (s, 3H, 4-CH3)*, 2.27 (s, 3H, 6-CH3)*,
4.06 (q, 3J2,1 = 6.6 Hz, 1H, H-2), 4.96 (br s, 1H, NH), 7.06–7.13 (m, 3H, H-5, H-7, H-8).

3 Beschreibung der Experimente 125
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.1 (SiCH(CH3)2), 18.4 (SiCH(CH3)2), 19.6 (C-Alkyl),
19.6 (C-Alkyl), 20.0 (C-Alkyl), 62.1 (C-2), 124.9 (C-Ar), 128.9 (C-Ar), 129.8 (C-Ar), 135.9 (C-
4)*, 136.6 (C-6)*, 140.2 (C-3).
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 17.9.
IR (ATR): ~ /cm–1 = 2940, 2864, 1505, 1460, 1382, 1083, 987, 907, 881, 848, 819, 718, 675.
HRMS (APCI) für C19H36NOSi+ [(M+H)+]: ber. 322.2561
gef. 322.2560
3.2.14 N-Cyclohexyl-O-(triisopropylsilyl)hydroxylamin (158)
Gemäß AAV 4 wurde eine Mischung von Cyclohexanon-O-triisopropylsilyloxim (140, 33 mg,
0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 3.1 mg, 6.1 µmol, 5.0 Mol-%) in
Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C für 18 h gerührt. Die
Reaktionsmischung wurde über Kieselgel filtriert und mit Cyclohexan:Ethylacetat = 1:1
eluiert. Nach Entfernen der Lösungsmittel unter vermindertem Druck wurde die
Titelverbindung 158 (33 mg, 99%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 16.9 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.99 (mc, 2H, H-2B), 1.06–1.10 (m, 18H, SiCH(CH3)2),
1.12–1.21 (m, 4H, SiCH(CH3)2, H-4A) 1.26 (mc, 2H, H-3A), 1.62 (mc, 1H, H-4B), 1.73 (mc,
2H, H-3B), 1.98 (mc, 2H, H-2A), 2.78 (tt, 3J1,2B = 11.1 Hz, 3J1,2A = 3.7 Hz, 1H, H-1), 4.66 (br s,
1H, NH).
13C-NMR (126 MHz, CDCl3): δ/ppm = 12.0 (SiCH(CH3)2), 18.4 (SiCH(CH3)2), 25.0 (C-3), 26.4
(C-4), 31.0 (C-2), 61.2 (C-1).

126 EXPERIMENTELLER TEIL
29Si-DEPT-NMR (99 MHz, CDCl3): δ/ppm = 17.2.
IR (ATR): ~ /cm–1 = 2926, 2861, 1461, 1365, 1251, 1074, 1012, 997, 881, 862, 780, 674.
HRMS (APCI) für C15H34NOSi+ [(M+H)+]: ber. 272.2404
gef. 272.2399
3.2.15 N-4-(tert-Butyl)cyclohexyl-O-(triisopropylsilyl)hydroxylamin (159)
Gemäß AAV 4 wurde eine Mischung von rac-4-(tert-Butyl)cyclohexanon-O-
triisopropylsilyloxim (rac-141, 39 mg, 0.12 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran
(1, 6.2 mg, 12 µmol, 10 Mol-%) in Toluol (0.5 mL) unter H2-Atmosphäre (100 bar) bei 60°C
für 18 h gerührt. Die Reaktionsmischung wurde über Kieselgel filtriert und mit
Cyclohexan:Ethylacetat = 1:1 eluiert. Nach Entfernen der Lösungsmittel unter vermindertem
Druck wurde die Titelverbindung 159 (35 mg, 89%, dr = 96:4,[148] bestimmt durch GC-
Analyse) als gelbes Öl erhalten.
GC (FS-SE-54): tR = 20.0 min (Mindermengenisomer), 20.8 min (Hauptmengenisomer).
1H-NMR (500 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 0.84 (s, 9H, 4-C(CH3)3), 0.95–
1.02 (m, 1H, H-4), 1.09 (d, 3JSiCH(CH3)2,SiCH(CH3)2 = 6.5 Hz, 18H, SiCH(CH3)2), 1.10–1.22 (m,
5H, SiCH(CH3)2, H-3A), 1.40 (mc, 2H, H-2A), 1.51 (mc, 2H, H-3B), 1.99 (mc, 2H, H-2B), 3.10
(s, 1H, H-1), 4.99 (br s, 1H, NH).
13C-NMR (126 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 12.1 (SiCH(CH3)2), 18.4
(SiCH(CH3)2), 21.7 (C-3), 27.6 (4-C(CH3)3), 28.3 (C-2), 32.7 (4-C(CH3)3), 48.2 (C-4), 56.2 (C-
1).
[148]
Die Diastereomerenverhältnisse unabhängiger Reaktionen unter den Standardbedingungen
variierten stark bis hin zur Umkehr der Selektivität.

3 Beschreibung der Experimente 127
1H/29Si-HMQC-NMR (500 MHz/99 MHz, CDCl3): δ/ppm (Hauptmengenisomer) = 1.12/17.4.
IR (ATR): ~ /cm–1 = 2940, 2863, 1515, 1462, 1365, 1239, 1090, 1055, 999, 972, 919, 881,
820, 786, 676.
HRMS (APCI) für C19H42NOSi+ [(M+H)+]: ber. 328.3030
gef. 328.3028
3.2.16 rac-N-(1-Phenylethyl)-hydroxylamin (rac-118)
Eine auf 0°C gekühlte Lösung von rac-N-(1-Phenylethyl)-O-(triisopropylsilyl)hydroxylamin
(rac-123, 211 mg, 0.719 mmol, 1.00 Äquiv.) in THF (10 mL) wurde mit HF·Pyridine (~70%
HF, ~40 mg, ~1.4 mmol, ~1.9 Äquiv.) versetzt. Nach Rühren für 3 h bei dieser Temperatur
wurden Wasser (30 mL) und CH2Cl2 (30 mL) hinzugefügt. Die Phasen wurden getrennt und
die wässrige Phase mit CH2Cl2 (3 × 30 mL) extrahiert. Die vereinigten organischen Phasen
wurden über Na2SO4 getrocknet und die Lösungsmittel unter vermindertem Druck entfernt.
Filtrieren über Kieselgel mit Cyclohexan:Ethylacetat = 1:1 als Eluent lieferte die
Titelverbindung rac-118 (76 mg, 77%) als weißen Feststoff.
Rf = 0.29 (Cyclohexan:Ethylacetat = 1:1).
Smp.: 70°C (Cyclohexan/Ethylacetat).
1H-NMR (500 MHz, CDCl3): δ/ppm = 321.38 (d, 3J1,2 = 6.7 Hz, 3H, H-1), 4.11 (q, 3J2,1 = 6.7
Hz, 1H, H-2), 5.67 (br s, 2H, NH, OH), 7.26–7.30 (m, 1H, H-6), 7.31–7.37 (m, 4H, H-4, H-5).
13C-NMR (126 MHz, CDCl3): δ/ppm = 19.5 (C-1), 61.9 (C-2), 127.3 (C-4), 127.7 (C-6), 128.7
(C-5), 142.4 (C-3).

128 EXPERIMENTELLER TEIL
IR (ATR): ~ /cm–1 = 3260, 3137, 3062, 2963, 2910, 2866, 1511, 1451, 1423, 1364, 1324,
1205, 1158, 1077, 1005, 931, 863, 807, 754, 695.
HRMS (APCI) für C8H12NO+ [(M+H)+]: ber. 138.0913
gef. 138.0913
3.3 Darstellung von Hydrazonen
3.3.1 1,1-Dibenzyl-2-benzylidenhydrazin (173)
Benzaldehyd (500 mg, 4.71 mmol, 1.00 Äquiv.) und N,N-Dibenzylhydrazin (1.00 g, 4.71
mmol, 1.00 Äquiv.) wurden in Methanol (25 mL) ohne Ausschluss von Sauerstoff und
Feuchtigkeit über Nacht am Rückfluss erhitzt. Nach Entfernen des Lösungsmittels unter
vermindertem Druck lieferte eine Aufreinigung mittels Flashchromatographie an Kieselgel (7
× 14 cm, Cyclohexan:Ethylacetat:CH2Cl2 = 50:1:1, 60 mL, #13–26) die Titelverbindung 173
(1.23 g, 79%) als weißen Feststoff.
Rf = 0.14 (Cyclohexan).
Smp.: 81°C (Cyclohexan/Ethylacetat).
GC (FS-SE-54): tR = 25.9 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 4.56 (s, 4H, H-1‘), 7.20–7.25 (m, 2H, H-Ar), 7.26–7.38
(m, 11H, H-Ar), 7.55 (mc, 2H, H-Ar). H-1 wurde nicht detektiert.
13C-NMR (126 MHz, CDCl3): δ/ppm = 58.0 (C-1‘), 125.7 (C-Ar), 127.3 (C-Ar), 127.8 (C-4‘)*,
128.5 (C-Ar), 128.7 (C-3‘)*, 131.9 (C-5), 137.1 (C-2), 137.8 (C-2‘). C-1 wurde nicht detektiert.

3 Beschreibung der Experimente 129
IR (ATR): ~ /cm–1 = 3020, 2930, 2900, 2845, 1586, 1559, 1490, 1447, 1346, 1316, 1264,
1155, 1123, 1069, 1023, 970, 913, 877, 850, 819, 793, 733, 693.
HRMS (APCI) für C12H21N2+ [(M+H)+]: ber. 301.1699
gef. 301.1693
3.3.2 2-((1-Phenylethyliden)amino)isoindolin-1,3-dion (179)
Acetophenon (741 mg, 6.17 mmol, 1.00 Äquiv.) und N,N-Aminophthalimid (1.00 g, 6.17
mmol, 1.00 Äquiv.) wurden in Ethanol (20 mL) ohne Ausschluss von Sauerstoff und
Feuchtigkeit über Nacht am Rückfluss erhitzt. Die Reaktionsmischung wurde mit CH2Cl2 (50
mL) und H2O (50 mL) verdünnt, die Phasen getrennt und die wässrige Phase mit CH2Cl2 (50
mL) extrahiert. Die vereinigten organischen Phasen wurden über Na2SO4 getrocknet und die
Lösungsmittel unter vermindertem Druck entfernt. Aufreinigung des Rückstandes mittels
Flashchromatographie an Kieselgel (7 × 14 cm, Cyclohexan:Ethylacetat = 6:1→1:2, 60 mL,
#27–38) lieferte die Titelverbindung 179 (1.16 g, 71%) als weißen Feststoff.
Rf = 0.25 (Cyclohexan:Ethylacetat = 6:1).
Smp.: 182°C (Cyclohexan/Ethylacetat).
GC (FS-SE-54): tR = 24.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 2.36 (s, 3H, H-1), 7.41–7.47 (m, 2H, H-5), 7.48–7.52
(m, 1H, H-6), 7.72–7.76 (m, 2H, H-4’), 7.86–7.91 (m, 2H, H-3’), 7.96–8.00 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 18.2 (C-1), 123.6 (C-3‘), 127.8 (C-4), 128.6 (C-5),
131.4 (C-2‘), 131.6 (C-6), 134.3 (C-4‘), 136.4 (C-3), 164.3 (C-1‘), 175.4 (C-2).

130 EXPERIMENTELLER TEIL
IR (ATR): ~ /cm–1 = 1784, 1708, 1604, 1464, 1369, 1284, 1171, 1129, 1079, 926, 880, 762,
692.
HRMS (APCI) für C16H13N2O2+ [(M+H)+]: ber. 265.0972
gef. 265.0966
3.4 Darstellung von Hydrazinen
3.4.1 rac-1,1-Dibenzyl-2-(1-phenylethyl)hydrazin (rac-174)
Gemäß AAV 4 wurde eine Mischung von 1,1-Dibenzyl-2-benzylidenhydrazin (172, 48 mg,
0.16 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 4.1 mg, 8.0 µmol, 5.0 Mol-%) in
Toluol (1 mL) unter H2-Atmosphäre (100 bar) bei 60°C für 18 h gerührt. Die
Reaktionsmischung wurde über Kieselgel filtriert und mit Cyclohexan:Ethylacetat = 1:1
eluiert. Nach Entfernen der Lösungsmittel unter vermindertem Druck wurde die
Titelverbindung rac-174 (43 mg, 89%) als farbloses Öl erhalten.
GC (FS-SE-54): tR = 24.3 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.18 (d, 3J1,2 = 6.5 Hz, 3H, H-1), 2.77 (br s, 1H, NH),
3.70 (mc, 4H, H-1‘), 3.81 (q, 3J2,1 = 6.5 Hz, 1H, H-2), 7.14–7.33 (m, 15H, H-4, H-5, H-6, H-3‘,
H-4‘, H-5‘).
13C-NMR (126 MHz, CDCl3): δ/ppm = 22.0 (C-1), 57.7 (C-2), 61.1 (C-1‘), 127.0 (C-Ar), 127.1
(C-Ar), 127.4 (C-Ar), 128.2 (C-Ar), 128.2 (C-Ar), 129.7 (C-Ar), 138.3 (C-2‘), 145.2 (C-3).
IR (ATR): ~ /cm–1 = 3060, 3026, 2969, 2923, 1601, 1492, 1450, 1361, 1069, 1026, 976, 908,
886, 745, 695.

3 Beschreibung der Experimente 131
HRMS (APCI) für C22H25N2+ [(M+H)+]: ber. 317.2012
gef. 317.2007
3.4.2 rac-2-((1-Phenylethyl)amino)isoindolin-1,3-dion (rac-181)
Gemäß AAV 4 wurde eine Mischung von 2-((1-Phenylethyliden)amino)isoindolin-1,3-dion
(179, 45 mg, 0.17 mmol, 1.0 Äquiv.) und Tris(pentafluorphenyl)boran (1, 4.4 mg, 8.6 µmol,
5.0 Mol-%) in Toluol (1 mL) unter H2-Atmosphäre (100 bar) bei 60°C für 18 h gerührt. Die
Reaktionsmischung wurde über Kieselgel filtriert und mit Cyclohexan:Ethylacetat = 1:1
eluiert. Nach Entfernen der Lösungsmittel unter vermindertem Druck wurde die
Titelverbindung rac-181 (43 mg, 95%) als gelbliches Öl erhalten.
GC (FS-SE-54): tR = 22.9 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.46 (d, 3J1,2 = 6.6 Hz, 3H, H-1), 4.53 (q, 3J2,1 = 6.6 Hz,
1H, H-2), 4.55 (br s, 1H, NH), 7.21–7.26 (m, 1H, H-6), 7.27–7.31 (m, 2H, H-5), 7.42–7.46 (m,
2H, H-4), 7.66–7.71 (m, 2H, H-4‘), 7.75–7.80 (m, 2H, H-3‘).
13C-NMR (126 MHz, CDCl3): δ/ppm = 20.9 (C-1), 59.4 (C-2), 123.5 (C-3‘), 127.5 (C-4), 128.1
(C-6), 128.6 (C-5), 130.1 (C-2‘), 134.3 (C-4‘), 141.4 (C-3), 166.9 (C-1‘).
IR (ATR): ~ /cm–1 = 3288, 2971, 1784, 1714, 1608, 1465, 1377, 1281, 1193, 1113, 1083,
1024, 988, 908, 881, 760, 699.
HRMS (APCI) für C16H15N2O2+ [(M+H)+]: ber. 267.1128
gef. 267.1124

132 EXPERIMENTELLER TEIL
3.5 Silyliumionvermittelte Iminreduktion mit Hydrosilanen
3.5.1 rac-4-Methyl-N-(1-phenylethyl)phenylsulfonamid (rac-222)
Unter Verwendung von tert-Butylferrocenylmethylsilan (206):
Dargestellt aus (E)-4-Methyl-N-(1-phenylethyliden)phenylsulfonamid [(E)-216, 45.7 mg,
0.167 mmol, 1.00 Äquiv.), tert-Butylferrocenylmethylsilan (206, 57.5 mg, 0.195 mmol, 1.16
Äquiv.) und Ph3C+[B(C6F5)4]– (200+[B(C6F5)4]–, 7.7 mg, 8.3 µmol, 5.0 Mol-%) in 1,2-Cl2C6H4 (3
mL) gemäß AAV 6. Die Reaktionsmischung wurde für 119 h gerührt. Die Aufreinigung
mittels Flashchromatographie an Kieselgel (3 × 20 cm, Cyclohexan:Ethylacetat:Triethylamin
= 24:4:1, 20 mL, #32–40) lieferte die Titelverbindung rac-222 (8.6 mg, 19%) als
orangefarbenes Öl.
Unter Verwendung von Triethylsilan (4):
Dargestellt aus (E)-4-Methyl-N-(1-phenylethyliden)phenylsulfonamid [(E)-216, 50.5 mg,
0.185 mmol, 1.00 Äquiv.), Triethylsilan (4, 25.8 mg, 0.219 mmol, 1.19 Äquiv.) und
Ph3C+[B(C6F5)4]– (200+[B(C6F5)4]–, 8.5 mg, 9.2 µmol, 5.0 Mol-%) in 1,2-Cl2C6H4 (4 mL)
gemäß AAV 6. Die Reaktionsmischung wurde für 1 h gerührt. Die Aufreinigung mittels
Flashchromatographie an Kieselgel (3 × 20 cm, Cyclohexan:Ethylacetat:Triethylamin =
24:4:1, 20 mL, #24–44) lieferte die Titelverbindung rac-222 (38.6 mg, 76%) als
orangefarbenes Öl.
Rf = 0.21 (Cyclohexan:Ethylacetat = 6:1).
GC (FS-SE-54): tR = 23.3 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.42 (d, 3J1,2 = 6.9 Hz, 3H, H-1), 2.38 (s, 3H, H-5‘), 4.60
(mc, 1H, NH), 4.86 (d, 3J2,1 = 6.3 Hz, 1H, H-2), 7.06–7.14 (m, 2H, H-4), 7.14–7.23 (m, 5H, H-
5, H-6, H-3‘), 7.62 (mc, 2H, H-2‘).

3 Beschreibung der Experimente 133
13C-NMR (126 MHz, CDCl3): δ/ppm = 21.6 (C-5‘), 23.7 (C-1), 53.7 (C-2), 126.2 (C-4), 127.2
(C-2‘), 127.6 (C-6), 128.7 (C-5), 129.6 (C-3‘), 137.8 (C-1‘), 142.2 (C-3), 143.3 (C-4‘).
IR (ATR): ~ /cm–1 = 3250, 2924, 2870, 1729, 1597, 1449, 1321, 1154, 1081, 1015, 956, 857,
811.
HRMS (ESI) für C15H17NO2SNa+ [(M+Na)+]: ber. 298.0872
gef. 298.0869
3.6 Darstellung von Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108)
3.6.1 6-Brom-1,2,3,4-tetrafluornaphthalin (239)
Zu einer auf –78°C gekühlten Lösung von Brompentafluorbenzol (240, 7.10 mL, 13.8 g, 55.8
mmol, 1.00 Äquiv.) in n-Hexan (80 mL) wurde tropfenweise nBuLi (1.50M in Hexanfraktion,
37.2 mL, 55.8 mmol, 1.00 Äquiv.) hinzugefügt. ACHTUNG: Höhere Temperaturen, die auch
durch eine zu schnelle Zugabe des nBuLi erreicht werden können, können zu Explosionen
führen! Nach Rühren für 1 h bei dieser Temperatur wurde 3-Bromthiophen (242, 6.79 mL,
11.8 g, 72.5 mmol, 1.30 Äquiv.) hinzugetropft und die Reaktionsmischung über einen
Zeitraum von 15 h auf Raumtemperatur erwärmen gelassen. Die Reaktionsmischung wurde
filtriert und unter vermindertem Druck konzentriert. Die Aufreinigung des Rückstandes
mittels Flashchromatographie an Kieselgel (9 × 16 cm, n-Pentan, 1 L Vorlauf, 100 mL, #1–7)
und anschließende Kristallisation aus Methanol lieferte die Titelverbindung 239 (2.30 g,
15%) als weißen Feststoff.
Rf = 0.57 (Cyclohexan).
Smp.: 65°C (n-Pentan).

134 EXPERIMENTELLER TEIL
GC (FS-SE-54): tR = 13.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 7.63 (d, 3J3,4 = 9.0 Hz, 1H, H-3), 7.86 (d, 3J4,3 = 9.0 Hz,
1H, H-4), 8.12 (s, 1H, H-1).
13C-NMR (126 MHz, CDCl3): δ/ppm = 118.2 (dd, 2J10,5-F = 14.9 Hz, 3J10,6-F = 3.8 Hz, C-10),
120.7 (dd, 2J9,8-F = 14.5 Hz, 3J9,7-F = 3.6 Hz, C-9), 121.9 (mc, C-4), 122.1 (C-2), 122.4 (mc, C-
1), 131.2 (C-3), 138.1 (dmc, 1J6,6-F = 253 Hz, C-6)*, 138.7 (mc,
1J7,7-F = 254 Hz, C-7)*, 141.4
(dm, 1J8,8-F = 253 Hz, C-8), 142.4 (dm, 1J5,5-F = 251 Hz, C-5).
19F-NMR (471 MHz, CDCl3): δ/ppm = –157.8 (mc), –156.6 (mc), –150.0 (mc), –149.4 (mc).
IR (ATR): ~ /cm–1 = 3108, 2124, 1921, 1753, 1666, 1635, 1601, 1509, 1482, 1452, 1408,
1362, 1283, 1248, 1200, 1125, 1067, 1029, 962, 879, 815, 751, 716, 669.
HRMS (EI) für C10H3BrF4+ [M+]: ber. 277.9348
gef. 277.9349
3.6.2 Trimethyl(5,6,7,8-tetrafluornaphthalin-2-yl)stannan (251)
Darstellung über Organolithiumverbindung:
Zu einer auf –80°C gekühlten Lösung von 6-Brom-1,2,3,4-tetrafluornaphthalin (239, 1.13 g,
4.05 mmol, 1.00 Äquiv.) in n-Pentan:Et2O (1:1, 40 mL) wurde tropfenweise nBuLi (1.21M in
Hexanfraktion, 3.35 mL, 4.05 mmol, 1.00 Äquiv.) hinzugefügt. Nach Rühren für 3 h bei
dieser Temperatur wurde eine Lösung von Me3SnCl (1.06 g, 5.32 mmol, 1.31 Äquiv.) in
Diethylether (5 mL) hinzugetropft und die Reaktionsmischung über Nacht auf
Raumtemperatur erwärmen gelassen. Die Lösungsmittel wurden unter vermindertem Druck
entfernt. Die Aufreinigung des Rückstandes mittels Flashchromatographie an Kieselgel (8 ×
13 cm, Cyclohexan, 50 mL, #11–21) und lieferte die Titelverbindung 251 (1.25 g, 85%) als
weißen Feststoff.

3 Beschreibung der Experimente 135
Darstellung über Grignardverbindung:
Zu einer auf –0°C gekühlten Lösung von 6-Brom-1,2,3,4-tetrafluornaphthalin (239, 1.98 g,
7.10 mmol, 1.00 Äquiv.) in THF (5 mL) wurde tropfenweise iPrMgCl·LiCl (0.38M in THF, 20.5
mL, 7.79 mmol, 1.10 Äquiv.) hinzugefügt. Die Reaktionsmischung wurde für 3 h bei
Raumtemperatur gerührt und erneut auf 0°C gekühlt. Eine Lösung von Me3SnCl (1.84 g,
9.23 mmol, 1.30 Äquiv.) in THF (5 mL) wurde hinzugefügt und die Reaktionsmischung über
Nacht auf Raumtemperatur erwärmen gelassen. H2O (20 mL) wurde hinzugefügt, die
Phasen getrennt, die wässrige Phase mit Diethylether (2 × 20 mL) extrahiert und die
vereinigten organischen Phasen über Na2SO4 getrocknet. Nach Entfernen der Lösungsmittel
unter vermindertem Druck lieferte die Aufreinigung des Rückstandes mittels
Flashchromatographie an Kieselgel (9 × 12 cm, Cyclohexan, 1.5 L Vorlauf, 50 mL, #1–11)
die Titelverbindung 251 (1.97 g, 76%) als weißen Feststoff.
Rf = 0.59 (Cyclohexan).
GC (FS-SE-54): tR = 16.5 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.42 (s, 3JH,Sn = 55 Hz, 9H, Sn(CH3)3), 7.71 (d, 3J3,4 = 8.2
Hz, 1H, H-3), 7.93 (d, 3J4,3 = 8.2 Hz, 1H, H-4), 8.13 (s, 1H, H-1).
13C-NMR (126 MHz, CDCl3): δ/ppm = –9.3 (Sn(CH3)3), 118.7 (mc, C-4), 119.2 (dd, 2J9,8-F =
14.3 Hz, 3J9,7-F = 4.5 Hz, C-9), 119.6 (dd, 2J10,5-F = 14.1 Hz, 3J10,6-F = 4.2 Hz, C-10), 127.3 (mc,
C-1), 134.0 (C-3), 143.0 (C-2). Aufgrund sich überlagernder Signale kann die chemische
Verschiebung der fluoratomtragenden Kohlenstoffatome nicht angegeben werden.
19F-NMR (471 MHz, CDCl3): δ/ppm = –159.6 (mc), –159.6 (mc), –151.6 (mc), –151.4 (mc).
119Sn-NMR (187 MHz, CDCl3): δ/ppm = –19.9.
HRMS (EI) für C13H12F4Sn+ [M+]: ber. 363.9892
gef. 363.9897

136 EXPERIMENTELLER TEIL
3.6.3 Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108)
In einem Druckrohr wurde Trimethyl(5,6,7,8-tetrafluornaphthalin-2-yl)stannan (251, 1.26 g,
3.47 mmol, 3.16 Äquiv.) in n-Hexan (5 mL) vorgelegt. BCl3 (1.00M in n-Heptan, 1.10 mL,
1.10 mmol, 1.00 Äquiv.) wurde hinzugegeben, das Druckrohr verschlossen und die
Reaktionsmischung für 14 h auf 100°C erhitzt. Die Reaktionsmischung wurde auf
Raumtemperatur abkühlen gelassen und der Großteil des Lösungsmittels und der flüchtigen
Nebenprodukte durch Erhitzen der Reaktionsmischung auf 50°C im Ölpumpenvakuum
entfernt. Das resultierende leicht gelbe Rohprodukt wurde portionsweise in einer Glovebox
in 1,2-F2C6H4 gelöst. Abkühlen der Mischungen auf –35°C ließ die Titelverbindung 108
ausfallen, welche nach Entfernen der überstehenden Lösung und Trocknen für mehrere
Stunden im Ölpumpenvakuum als weißer Feststoff (457 mg, 68%) erhalten wurde.
1H-NMR (500 MHz, CD2Cl2): δ/ppm = 7.87 (d, 3J3,4 = 8.5 Hz, 3H, H-3), 8.21 (d, 3J4,3 = 8.5 Hz,
3H, H-4), 8.37 (s, 3H, H-1).
13C-DEPT-NMR (126 MHz, CD2Cl2): δ/ppm = 119.4 (C-3), 132.0 (C-4), 135.2 (C-1).
19F/13C-HMQC-NMR (659 MHz/176 MHz, CD2Cl2): δ/ppm = –159.6/138.1 (5-F/C-5)*,
–156.5/138.9 (6-F/C-6)*, –150.9/142.2 (7-F/C-7)*, –149.7/142.9 (8-F/C-8)*.
C-2, C-9 und C-10 wurden nicht detektiert.
11B-NMR (161 MHz, CD2Cl2): δ/ppm = 71.0.
19F-NMR (471 MHz, CD2Cl2): δ/ppm = –159.3 (mc), –156.4 (mc), –150.9 (mc), –149.6 (mc).
HRMS (ESI) für C30H9BF12+ [M+]: ber. 608.0600
gef. 608.0570
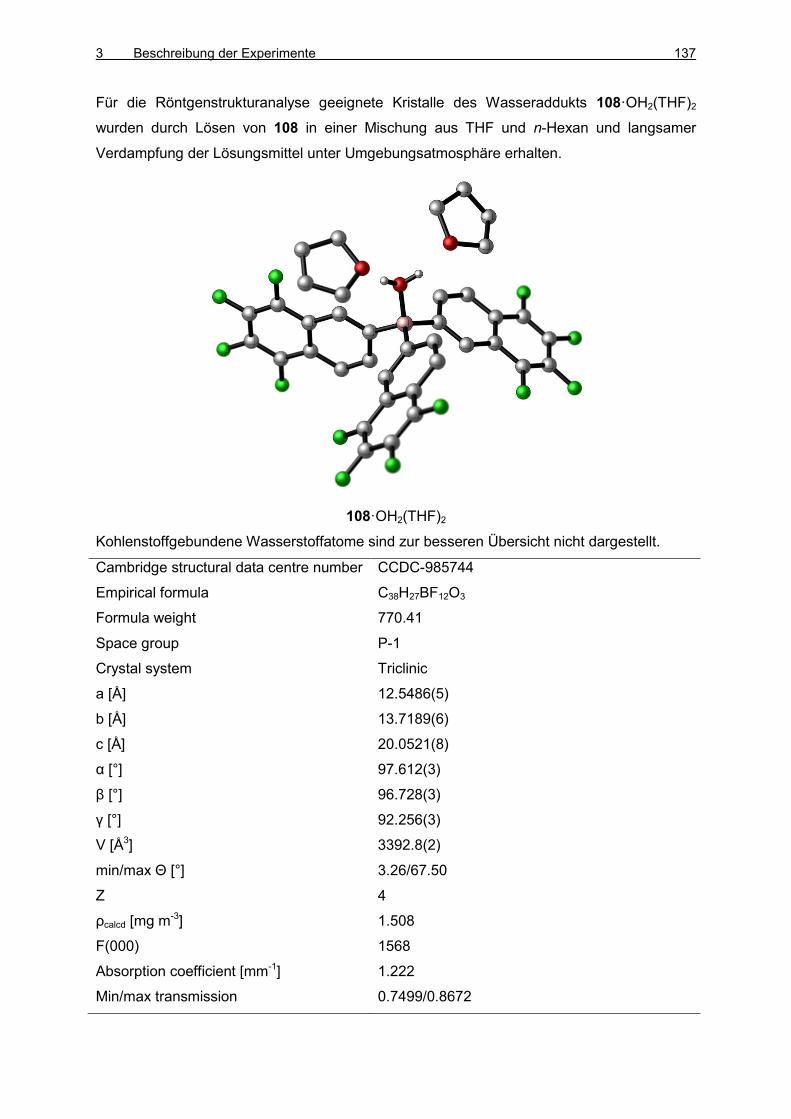
3 Beschreibung der Experimente 137
Für die Röntgenstrukturanalyse geeignete Kristalle des Wasseraddukts 108·OH2(THF)2
wurden durch Lösen von 108 in einer Mischung aus THF und n-Hexan und langsamer
Verdampfung der Lösungsmittel unter Umgebungsatmosphäre erhalten.
108·OH2(THF)2
Kohlenstoffgebundene Wasserstoffatome sind zur besseren Übersicht nicht dargestellt.
Cambridge structural data centre number CCDC-985744
Empirical formula C38H27BF12O3
Formula weight 770.41
Space group P-1
Crystal system Triclinic
a [Å] 12.5486(5)
b [Å] 13.7189(6)
c [Å] 20.0521(8)
α [°] 97.612(3)
β [°] 96.728(3)
γ [°] 92.256(3)
V [Å3] 3392.8(2)
min/max Θ [°] 3.26/67.50
Z 4
ρcalcd [mg m-3] 1.508
F(000) 1568
Absorption coefficient [mm-1] 1.222
Min/max transmission 0.7499/0.8672

138 EXPERIMENTELLER TEIL
Wavelength [Å] 1.54184
Temperature [K] 150(2)
Crystal size [mm3] and description 0.12 × 0.18 × 0.25, colourless block
Reflections collected 23231
Independent reflections 12217 (merging R = 0.0449)
Observed reflections 12217 [>2sigma(I)]
Refined parameters 985
R 0.0814
wR 0.1891
Goodness of fit 1.098
3.7 Boran/Phosphinoxid-Addukte
3.7.1 Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran/Triethylphosphinoxid-Addukt
(108·Et3PO)
In C6D6:
In einer Glovebox wurde Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108, 15 mg, 25 µmol,
1.0 Äquiv) in C6D6 (0.5 mL) mit Triethylphosphinoxid (3.3 mg, 25 µmol, 1.0 Äquiv) in C6D6
(0.5 mL) versetzt. Die resultierende Mischung wurde in ein NMR-Röhrchen überführt und
NMR-spektroskopisch untersucht.
1H-NMR (500 MHz, C6D6): δ/ppm = 0.47 (mc, 9H, H-2‘), 0.86 (mc, 6H, H-1‘), 7.73 (d, 3J3,4 =
8.5 Hz, 3H, H-3)*, 7.88 (d, 3J4,3 = 8.5 Hz, 3H, H-4)*, 8.37 (s, 3H, H-1).
11B-NMR (161 MHz, C6D6): δ/ppm = 6.3.
19F-NMR (471 MHz, C6D6): δ/ppm = –161.8 (mc), –161.1 (mc), –153.4 (mc), –151.8 (mc).

3 Beschreibung der Experimente 139
31P-NMR (203 MHz, C6D6): δ/ppm = 74.7.
In CD2Cl2:
In einer Glovebox wurde Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108, 13 mg, 21 µmol,
1.0 Äquiv) in CD2Cl2 (0.5 mL) mit Triethylphosphinoxid (2.9 mg, 22 µmol, 1.0 Äquiv) in
CD2Cl2 (0.5 mL) versetzt. Die resultierende Mischung wurde in ein NMR-Röhrchen überführt
und NMR-spektroskopisch untersucht.
1H-NMR (500 MHz, CD2Cl2): δ/ppm = 1.14 (mc, 9H, H-2‘), 1.73 (mc, 6H, H-1‘), 7.72 (d, 3J3,4 =
8.5 Hz, 3H, H-3)*, 7.90 (d, 3J4,3 = 8.6 Hz, 3H, H-4)*, 8.16 (s, 3H, H-1).
11B-NMR (161 MHz, CD2Cl2): δ/ppm = 5.9.
19F-NMR (471 MHz, CD2Cl2): δ/ppm = –163.1 (mc), –162.4 (mc), –153.1 (mc), –152.8 (mc).
31P-NMR (203 MHz, CD2Cl2): δ/ppm = 76.6.
3.7.2 Tris(pentafluorphenyl)boran/Triethylphosphinoxid-Addukt (1·Et3PO)
In C6D6:
In einer Glovebox wurde Tris(pentafluorphenyl)boran (1, 12 mg, 23 µmol, 1.0 Äquiv) in C6D6
(0.5 mL) mit Triethylphosphinoxid (3.1 mg, 23 µmol, 1.0 Äquiv) in C6D6 (0.5 mL) versetzt. Die
resultierende Mischung wurde in ein NMR-Röhrchen überführt und NMR-spektroskopisch
untersucht.
1H-NMR (500 MHz, C6D6): δ/ppm = 0.27 (dt, 3J2,P = 18.6 Hz, 3J2,1 = 7.7 Hz, 9H, H-2), 0.94
(dq, 2J2,P = 12.3 Hz, 3J1,2 = 7.7 Hz, 6H, H-1).

140 EXPERIMENTELLER TEIL
11B-NMR (161 MHz, C6D6): δ/ppm = –2.1.
19F-NMR (471 MHz, C6D6): δ/ppm = –164.0, –157.4, –133.9.
31P-NMR (203 MHz, C6D6): δ/ppm = 75.3.
In CD2Cl2:
In einer Glovebox wurde Tris(pentafluorphenyl)boran (1, 10 mg, 20 µmol, 1.0 Äquiv) in
CD2Cl2 (0.5 mL) mit Triethylphosphinoxid (2.7 mg, 20 µmol, 1.0 Äquiv) in CD2Cl2 (0.5 mL)
versetzt. Die resultierende Mischung wurde in ein NMR-Röhrchen überführt und NMR-
spektroskopisch untersucht.
1H-NMR (500 MHz, CD2Cl2): δ/ppm = 1.07 (dt, 3J2,P = 18.6 Hz, 3J2,1 = 7.7 Hz, 9H, H-2), 1.92
(dq, 2J2,P = 12.2 Hz, 3J1,2 = 7.7 Hz, 6H, H-1).
11B-NMR (161 MHz, CD2Cl2): δ/ppm = –2.4.
19F-NMR (471 MHz, CD2Cl2): δ/ppm = –165.0, –159.1, –134.4.
31P-NMR (203 MHz, CD2Cl2): δ/ppm = 76.9.
Die spektroskopischen Daten stimmen mit den Literaturangaben überein.[88e]
3.7.3 Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran-Triphenylphosphinoxid-Addukt
(108·Ph3PO)
Für die Röntgenstrukturanalyse geeignete Kristalle des Addukts 108·Ph3PO wurden durch
Kristallisation einer äquimolaren Mischung aus Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran
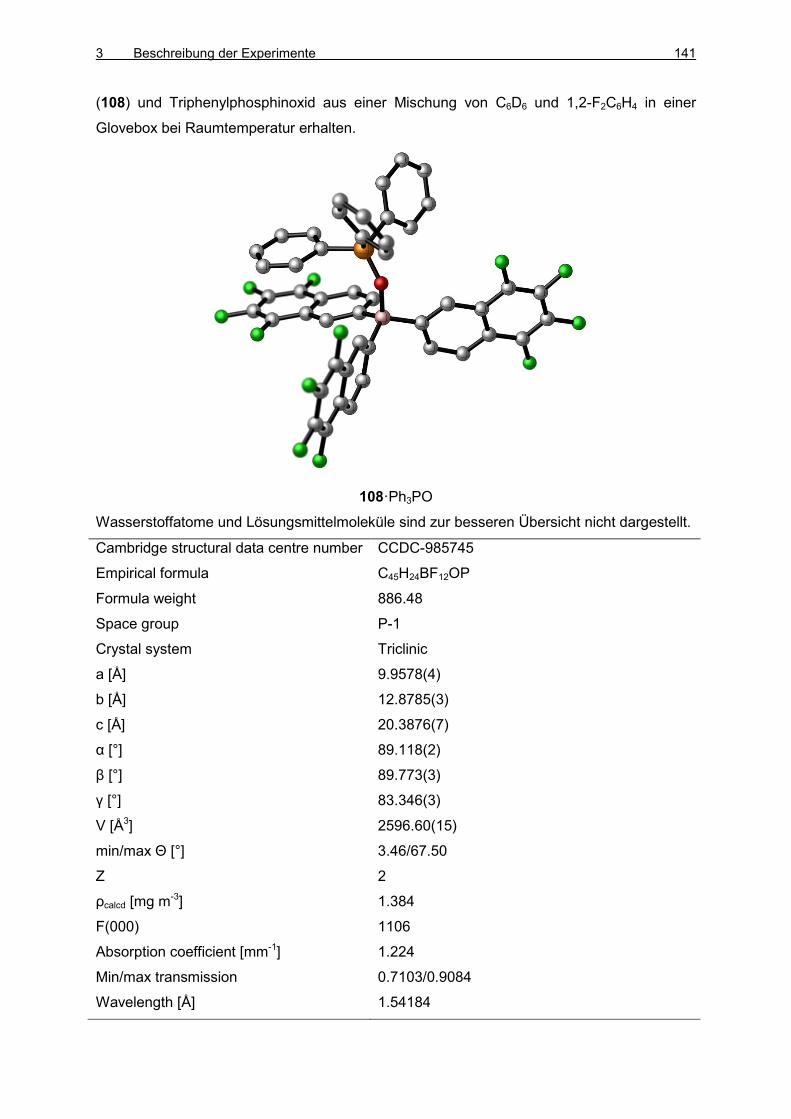
3 Beschreibung der Experimente 141
(108) und Triphenylphosphinoxid aus einer Mischung von C6D6 und 1,2-F2C6H4 in einer
Glovebox bei Raumtemperatur erhalten.
108·Ph3PO
Wasserstoffatome und Lösungsmittelmoleküle sind zur besseren Übersicht nicht dargestellt.
Cambridge structural data centre number CCDC-985745
Empirical formula C45H24BF12OP
Formula weight 886.48
Space group P-1
Crystal system Triclinic
a [Å] 9.9578(4)
b [Å] 12.8785(3)
c [Å] 20.3876(7)
α [°] 89.118(2)
β [°] 89.773(3)
γ [°] 83.346(3)
V [Å3] 2596.60(15)
min/max Θ [°] 3.46/67.50
Z 2
ρcalcd [mg m-3] 1.384
F(000) 1106
Absorption coefficient [mm-1] 1.224
Min/max transmission 0.7103/0.9084
Wavelength [Å] 1.54184

142 EXPERIMENTELLER TEIL
Temperature [K] 150(2)
Crystal size [mm3] and description 0.08 × 0.12 × 0.30, colourless rod
Reflections collected 19257
Independent reflections 9361 (merging R = 0.0300)
Observed reflections 9361 [>2sigma(I)]
Refined parameters 813
R 0.0468
wR 0.1364
Goodness of fit 1.036
3.8 Durch Tris(5,6,7,8-tetrafluornaphthalin-2-yl)boran (108)
katalysierte Reaktionen
3.8.1 rac-Dimethylphenyl(1-phenylethoxy)silan (rac-252)
Durch Hydrosilylierung:
Eine Mischung aus Acetophenon (10, 77.9 mg, 0.648 mmol, 1.00 Äquiv.),
Dimethylphenylsilan (19, 90.7 mg, 0.666 mmol, 1.03 Äquiv.) und Tris(5,6,7,8-
tetrafluornaphthalin-2-yl)boran (108, 1.9 mg, 3.1 µmol, 0.48 Mol-%) in Toluol (0.5 mL) wurde
in einem 2-mL-Gläschen in einer Glovebox für 2 h bei Raumtemperatur gerührt. Direkte
Aufreinigung der Reaktionsmischung mittels Flashchromatographie an Kieselgel (3.5 × 13
cm, Cyclohexan:Ethylacetat = 30:1, 20 mL, #4–8) lieferte die Titelverbindung rac-252 (150
mg, 90%) als farbloses Öl.
Durch dehydrierende Si–O-Kupplung:
Eine Mischung aus rac-1-Phenylethanol (rac-17, 61.6 mg, 0.504 mmol, 1.03 Äquiv.),
Dimethylphenylsilan (19, 67.0 mg, 0.492 mmol, 1.00 Äquiv.) und Tris(5,6,7,8-

3 Beschreibung der Experimente 143
tetrafluornaphthalin-2-yl)boran (108, 1.4 mg, 2.3 µmol, 0.47 Mol-%) in Toluol (0.5 mL) wurde
in einem 2-mL-Gläschen in einer Glovebox für 2 h bei Raumtemperatur gerührt. Direkte
Aufreinigung der Reaktionsmischung mittels Flashchromatographie an Kieselgel (3.5 × 13
cm, Cyclohexan:Ethylacetat = 30:1, 20 mL, #3–6) lieferte die Titelverbindung rac-252 (101
mg, 80%) als farbloses Öl.
Rf = 0.68 (Cyclohexan:Ethylacetat = 20:1).
GC (FS-SE-54): tR = 16.8 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.33 (s, 3H, SiCH3), 0.37 (s, 3H, SiCH3), 1.44–1.48 (m,
3H, H-1), 4.89 (mc, 1H, H-2), 7.21–7.44 (m, 8H, H-4, H-5, H-6, H-2’*, H-4’), 7.57–7.61 (m,
2H, H-3’)*.
13C-NMR (126 MHz, CDCl3): δ/ppm = –1.2 (SiCH3), –0.7 (SiCH3), 27.0 (C-1), 71.2 (C-2),
125.5 (C-4), 127.0 (C-6), 127.9 (C-2‘)*, 128.3 (C-5), 129.6 (C-4‘), 133.7 (C-3‘)*, 138.3 (C-1‘),
146.4 (C-3).
29Si-NMR (99 MHz, CDCl3): δ/ppm = 6.6.
IR (ATR): ~ /cm–1 = 2969, 1491, 1447, 1367, 1251, 1085, 1029, 957, 823, 784, 738, 696.
HRMS (EI) für C16H20OSi+ [M+]: ber. 256.1278
gef. 256.1269
Die analytischen Daten stimmen mit den Literaturangaben überein.[88e]
3.8.2 rac-N-(1-Phenylethyl)anilin (rac-45)

144 EXPERIMENTELLER TEIL
Eine Mischung aus (E)-N,1-Diphenylethan-1-imin [(E)-44, 39.7 mg, 0.203 mmol, 1.00
Äquiv.], Dimethylphenylsilan (19, 30.0 mg, 0.220 mmol, 1.08 Äquiv.) und Tris(5,6,7,8-
tetrafluornaphthalin-2-yl)boran (108, 4.6 mg, 7.6 µmol, 3.7 Mol-%) in Toluol (0.5 mL) wurde
in einem 2-mL-Gläschen in einer Glovebox für 48 h bei Raumtemperatur gerührt. Direkte
Aufreinigung der Reaktionsmischung mittels Flashchromatographie an Kieselgel (3.5 × 17
cm, Cyclohexan:Ethylacetat = 50:1, 20 mL, #10–23) lieferte die Titelverbindung rac-45 (34.4
mg, 86%) als farbloses Öl.
Rf = 0.41 (Cyclohexan:Ethylacetat = 20:1).
GC (FS-SE-54): tR = 17.2 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 1.53 (d, 3J1,2 = 6.8 Hz, 3H, H-1), 4.15 (br s, 1H, NH),
4.49 (q, 3J2,1 = 6.8 Hz, 1H, H-2), 6.53 (mc, 2H, H-2‘), 6.65 (tt, 3J4‘,3‘ = 7.3 Hz, 4J4‘,2‘ = 1.0 Hz,
1H, H-4‘), 7.10 (mc, 2H, H-3‘), 7.23 (tt, 3J6,5 = 7.3 Hz, 4J6,4 = 1.0 Hz, 1H, 1H, H-6), 7.30–7.35
(m, 2H, H-5), 7.35–7.40 (m, 2H, H-4).
13C-NMR (126 MHz, CDCl3): δ/ppm = 25.1 (C-1), 53.7 (C-2), 113.6 (C-2‘), 117.5 (C-4‘),
126.0 (C-4), 127.0 (C-6), 128.8 (C-5), 129.2 (C-3‘), 145.3 (C-3), 147.3 (C-1‘).
IR (ATR): ~ /cm–1 = 3406, 3020, 2964, 2865, 1598, 1501, 1447, 1314, 1255, 1204, 1138,
867, 754, 690.
HRMS (ESI) für C14H16N+ [(M+H)+]: ber. 198.1277
gef. 198.1276
Die analytischen Daten stimmen mit den Literaturangaben überein.[88e]

3 Beschreibung der Experimente 145
3.9 Darstellung von Boranvorläufer (R)-261
3.9.1 (S)-2-(1,1’-Binaphthalin)-2-yl-4,4,5,5-tetramethyl-1,3,2-dioxaborolan
[(S)-259]
Entsprechend einer modifizierten Vorschrift von MASUDA und Mitarbeitern,[133] wurde eine
Mischung von Triflat (R)-258 (873 mg, 2.17 mmol, 1.00 Äquiv.) und Triethylamin (0.90 mL,
0.65 g, 6.5 mmol, 3.0 Äquiv.) in 1,4-Dioxan (6 mL) zu einer Lösung von [1,1‘-
Bis(diphenylphosphino)ferrocen]dichlorpalladium(II) (47.6 mg, 65.1 µmol, 3.00 Mol-%) in 1,4-
Dioxan (3 mL) hinzugefügt. Pinakolboran (1.42 mL, 1.25 g, 9.79 mmol, 4.51 Äquiv.) wurde
hinzugetropft und die Reaktionsmischung für 19 h auf 120°C erhitzt. H2O (30 mL) wurde
hinzugegeben und die Phasen getrennt. Die wässrige Phase wurde mit CH2Cl2 (2 × 30 mL)
extrahiert und die vereinigten organischen Phasen über Na2SO4 getrocknet, bevor die
Lösungsmittel unter vermindertem Druck entfernt wurden. Die Aufreinigung des
Rückstandes mittels Flashchromatographie an Kieselgel (6 × 19 cm, Cyclohexan/Ethylacetat
= 30:1, 50 mL, #10–28) lieferte die Titelverbindung (S)-259 (698 mg, 85%) als weißen
Feststoff.
Rf = 0.42 (Cyclohexan/Ethylacetat = 20:1).
Smp.: 114°C (n-Pentan).
GC (FS-SE-54): tR = 29.0 min.
1H-NMR (500 MHz, CDCl3): δ/ppm = 0.80 (s, 6H, OCCH3), 0.93 (s, 6H, OCCH3), 7.24–7.35
(m, 3H, H-Ar), 7.41–7.53 (m, 4H, H-Ar), 7.56–7.61 (m, 1H, H-Ar), 7.87–7.99 (m, 5H, H-Ar).

146 EXPERIMENTELLER TEIL
13C-NMR (126 MHz, CDCl3): δ/ppm = 24.3 (OCCH3), 24.4 (OCCH3), 83.3 (OCCH3), 125.1
(C-Ar), 125.5 (C-Ar), 125.8 (C-Ar), 125.9 (C-Ar), 126.6 (C-Ar), 126.7 (C-Ar), 126.9 (C-Ar),
127.4 (C-Ar), 127.4 (C-Ar), 127.9 (C-Ar), 128.0 (C-Ar), 128.2 (C-Ar), 130.1 (C-Ar), 132.9
(Cquart), 133.6 (Cquart), 134.1 (Cquart), 134.7 (Cquart), 139.1 (Cquart), 144.9 (Cquart). C-2 wurde
nicht detektiert.
IR (ATR): ~ /cm–1 = 1471, 1378, 1354, 1301, 1264, 1134, 1109, 964, 850, 818, 800, 774,
745.
HRMS (APCI) für C26H26BO2+ [(M+H)+]: ber. 381.2020
gef. 381.2023
3.9.2 (R)-2-(2,3,5,6-Tetrafluorphenyl)-1,1’-binaphthalin [(R)-261]
Eine Mischung von (S)-2-(1,1’-Binaphthalin)-2-yl-4,4,5,5-tetramethyl-1,3,2-dioxaborolan [(S)-
259, 1.50 g, 3.94 mmol, 1.00 Äquiv.), Brom-2,3,5,6-tetrafluorbenzol (260, 1.87 mL, 3.52 g,
15.4 mmol, 3.90 Äquiv.) und Cäsiumcarbonat (3.16 g, 9.70 mmol, 2.46 Äquiv.) in 1,4-
Dioxan:Wasser (10:1, 22 mL) wurde für 4 d auf 95°C erhitzt. Die Reaktionsmischung wurde
über Celite filtriert und unter vermindertem Druck konzentriert. Die Aufreinigung des
Rückstandes mittels Flashchromatographie an Kieselgel (8 × 18 cm, Cyclohexan:Toluol =
10:1, 100 mL, #19–37) lieferte die Titelverbindung (R)-261 (1.07 g, 67%, >98 ee, bestimmt
durch HPLC an chiraler stationärer Phase)[149] als weißen Feststoff.
Rf = 0.30 (Cyclohexan).
Smp.: 158°C (n-Pentan).
[149]
Racemisches 261 wurde durch Erhitzen von (R)-261 in Diphenylether in einer Mikrowelle bei
200°C für 12 h und anschließende Aufreinigung mittels präparativer
Dünnschichtchromatographie (Cyclohexan) erhalten.

3 Beschreibung der Experimente 147
1H-NMR (500 MHz, CDCl3): δ/ppm = 6.77 (mc, 1H, H-1), 7.27–7.61 (m, 9H, H-Ar), 7.83–7.89
(m, 2H, H-Ar), 8.03 (mc, 1H, H-Ar), 8.10 (mc, 1H, H-Ar).
13C-NMR (126 MHz, CDCl3): δ/ppm = 105.0 (mc, C-1), 121.9 (mc, C-4), 125.1 (C-Ar), 125.7
(mc, C-2’), 126.0 (C-Ar), 126.1 (C-Ar), 126.2 (C-Ar), 126.2 (C-Ar), 126.8 (C-Ar), 127.0 (C-Ar),
127.2 (C-Ar), 127.3 (C-Ar), 128.1 (C-Ar), 128.1 (C-Ar), 128.2 (C-Ar), 128.4 (C-Ar), 132.5
(Cquart), 133.3 (Cquart), 133.4 (Cquart), 133.8 (Cquart), 135.5 (Cquart), 139.1 (Cquart), 142.4–143.0
(m, CF), 144.3–144.9 (m, 2C, CF), 146.3–146.6 (m, CF).
19F-NMR (471 MHz, CDCl3): δ/ppm = –140.0– –139.8 (m, 2F), –139.5– –139.3 (m, 1F),
–138.8– –138.6 (m, 1F).
IR (ATR): ~ /cm–1 = 1491, 1387, 1365, 1281, 1255, 1170, 968, 934, 871, 849, 821, 803, 781,
755, 711.
HRMS (APCI) für C26H14F4+ [M+]: ber. 402.1026
gef. 402.1033
HPLC (Daicel Chiralcel OJ-RH, MeCN:H2O = 60:40, Fließrate 0.40 mL/min, λ = 254 nm,
Säulentemperatur 20°C): tR = 52.0 min [(S)-261]: tR = 53.6 min [(R)-261].
[α]D20
= –1128 (c = 1.05, CHCl3).
Für die Röntgenstrukturanalyse geeignete Kristalle wurden durch Kristallisation aus einer
Lösung von (R)-261 in CH2Cl2 bei Umgebungsatmosphäre erhalten.
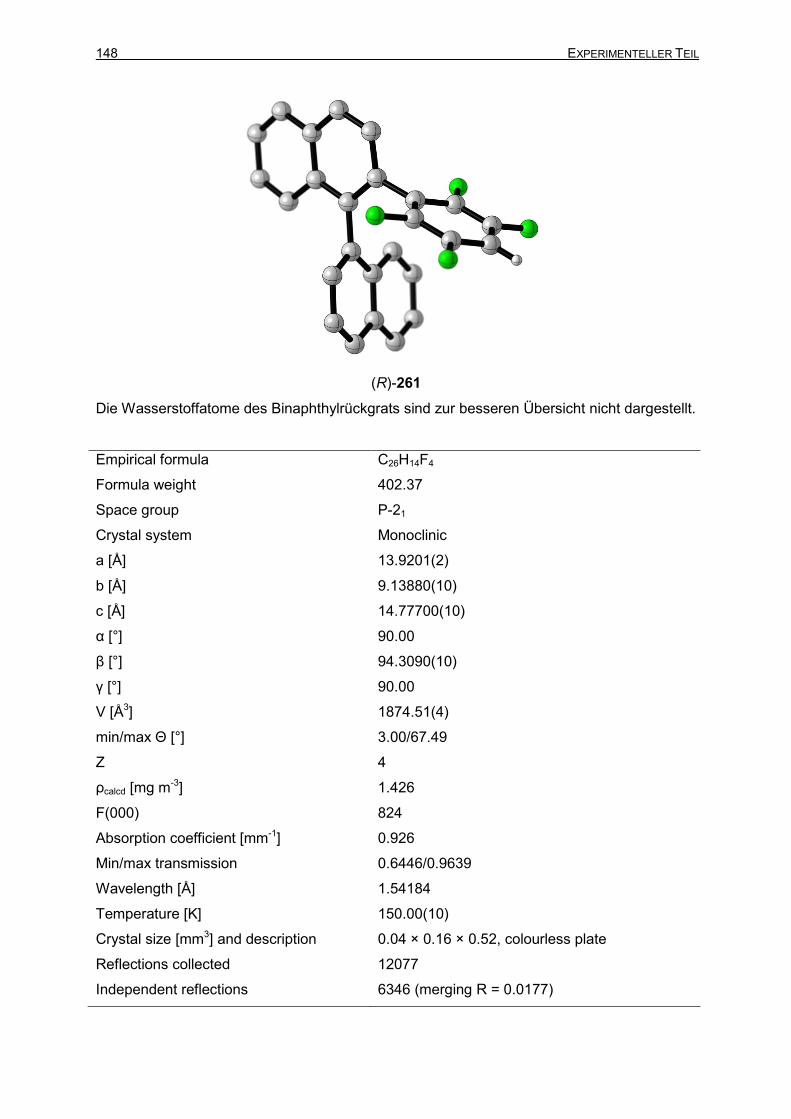
148 EXPERIMENTELLER TEIL
(R)-261
Die Wasserstoffatome des Binaphthylrückgrats sind zur besseren Übersicht nicht dargestellt.
Empirical formula C26H14F4
Formula weight 402.37
Space group P-21
Crystal system Monoclinic
a [Å] 13.9201(2)
b [Å] 9.13880(10)
c [Å] 14.77700(10)
α [°] 90.00
β [°] 94.3090(10)
γ [°] 90.00
V [Å3] 1874.51(4)
min/max Θ [°] 3.00/67.49
Z 4
ρcalcd [mg m-3] 1.426
F(000) 824
Absorption coefficient [mm-1] 0.926
Min/max transmission 0.6446/0.9639
Wavelength [Å] 1.54184
Temperature [K] 150.00(10)
Crystal size [mm3] and description 0.04 × 0.16 × 0.52, colourless plate
Reflections collected 12077
Independent reflections 6346 (merging R = 0.0177)
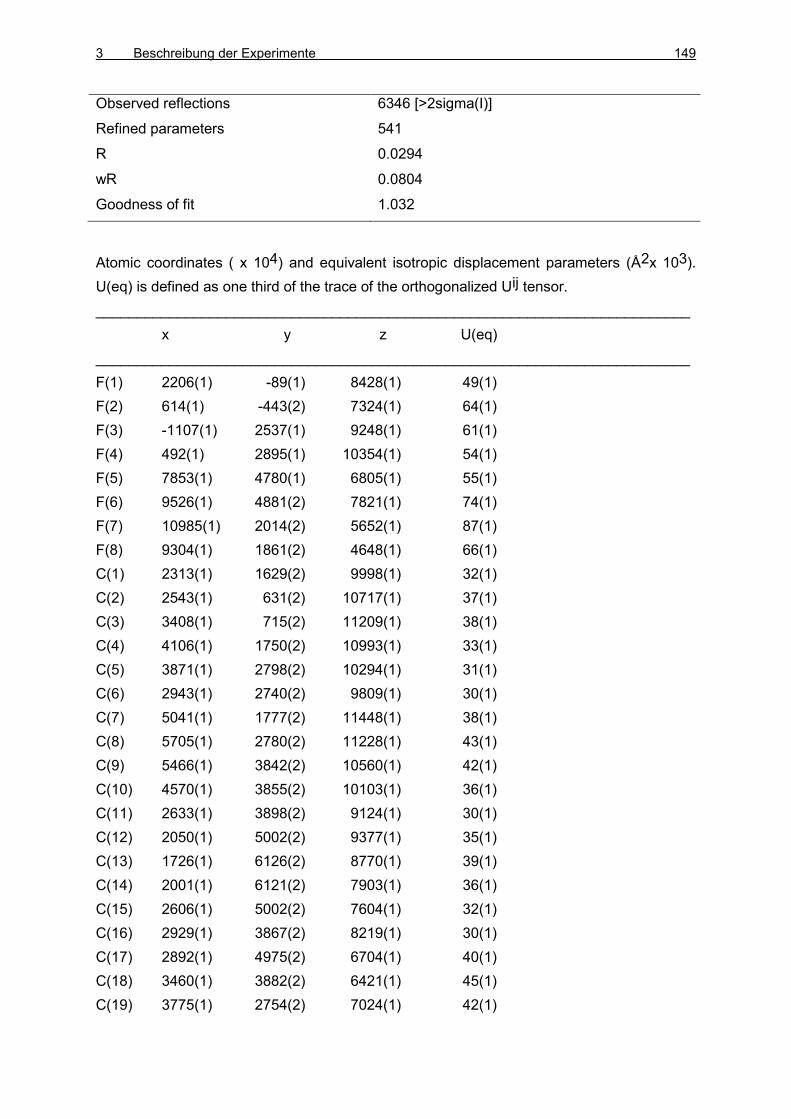
3 Beschreibung der Experimente 149
Observed reflections 6346 [>2sigma(I)]
Refined parameters 541
R 0.0294
wR 0.0804
Goodness of fit 1.032
Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103).
U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
_________________________________________________________________________
x y z U(eq)
_________________________________________________________________________
F(1) 2206(1) -89(1) 8428(1) 49(1)
F(2) 614(1) -443(2) 7324(1) 64(1)
F(3) -1107(1) 2537(1) 9248(1) 61(1)
F(4) 492(1) 2895(1) 10354(1) 54(1)
F(5) 7853(1) 4780(1) 6805(1) 55(1)
F(6) 9526(1) 4881(2) 7821(1) 74(1)
F(7) 10985(1) 2014(2) 5652(1) 87(1)
F(8) 9304(1) 1861(2) 4648(1) 66(1)
C(1) 2313(1) 1629(2) 9998(1) 32(1)
C(2) 2543(1) 631(2) 10717(1) 37(1)
C(3) 3408(1) 715(2) 11209(1) 38(1)
C(4) 4106(1) 1750(2) 10993(1) 33(1)
C(5) 3871(1) 2798(2) 10294(1) 31(1)
C(6) 2943(1) 2740(2) 9809(1) 30(1)
C(7) 5041(1) 1777(2) 11448(1) 38(1)
C(8) 5705(1) 2780(2) 11228(1) 43(1)
C(9) 5466(1) 3842(2) 10560(1) 42(1)
C(10) 4570(1) 3855(2) 10103(1) 36(1)
C(11) 2633(1) 3898(2) 9124(1) 30(1)
C(12) 2050(1) 5002(2) 9377(1) 35(1)
C(13) 1726(1) 6126(2) 8770(1) 39(1)
C(14) 2001(1) 6121(2) 7903(1) 36(1)
C(15) 2606(1) 5002(2) 7604(1) 32(1)
C(16) 2929(1) 3867(2) 8219(1) 30(1)
C(17) 2892(1) 4975(2) 6704(1) 40(1)
C(18) 3460(1) 3882(2) 6421(1) 45(1)
C(19) 3775(1) 2754(2) 7024(1) 42(1)
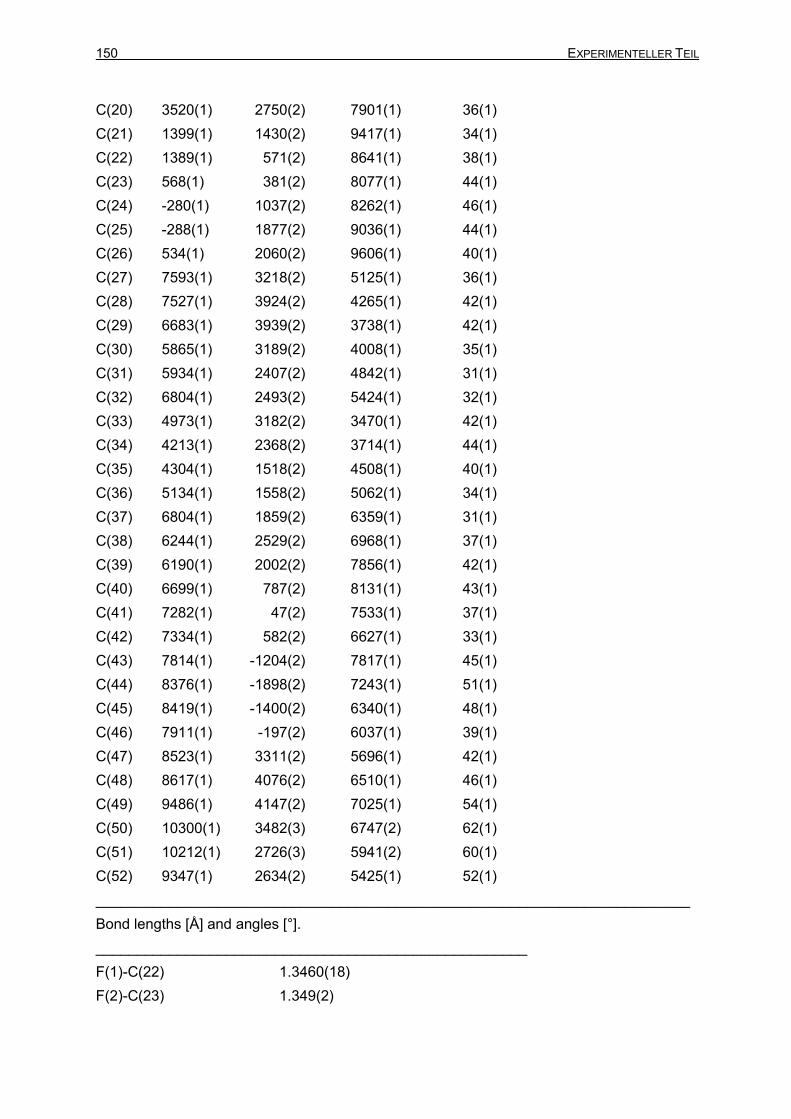
150 EXPERIMENTELLER TEIL
C(20) 3520(1) 2750(2) 7901(1) 36(1)
C(21) 1399(1) 1430(2) 9417(1) 34(1)
C(22) 1389(1) 571(2) 8641(1) 38(1)
C(23) 568(1) 381(2) 8077(1) 44(1)
C(24) -280(1) 1037(2) 8262(1) 46(1)
C(25) -288(1) 1877(2) 9036(1) 44(1)
C(26) 534(1) 2060(2) 9606(1) 40(1)
C(27) 7593(1) 3218(2) 5125(1) 36(1)
C(28) 7527(1) 3924(2) 4265(1) 42(1)
C(29) 6683(1) 3939(2) 3738(1) 42(1)
C(30) 5865(1) 3189(2) 4008(1) 35(1)
C(31) 5934(1) 2407(2) 4842(1) 31(1)
C(32) 6804(1) 2493(2) 5424(1) 32(1)
C(33) 4973(1) 3182(2) 3470(1) 42(1)
C(34) 4213(1) 2368(2) 3714(1) 44(1)
C(35) 4304(1) 1518(2) 4508(1) 40(1)
C(36) 5134(1) 1558(2) 5062(1) 34(1)
C(37) 6804(1) 1859(2) 6359(1) 31(1)
C(38) 6244(1) 2529(2) 6968(1) 37(1)
C(39) 6190(1) 2002(2) 7856(1) 42(1)
C(40) 6699(1) 787(2) 8131(1) 43(1)
C(41) 7282(1) 47(2) 7533(1) 37(1)
C(42) 7334(1) 582(2) 6627(1) 33(1)
C(43) 7814(1) -1204(2) 7817(1) 45(1)
C(44) 8376(1) -1898(2) 7243(1) 51(1)
C(45) 8419(1) -1400(2) 6340(1) 48(1)
C(46) 7911(1) -197(2) 6037(1) 39(1)
C(47) 8523(1) 3311(2) 5696(1) 42(1)
C(48) 8617(1) 4076(2) 6510(1) 46(1)
C(49) 9486(1) 4147(2) 7025(1) 54(1)
C(50) 10300(1) 3482(3) 6747(2) 62(1)
C(51) 10212(1) 2726(3) 5941(2) 60(1)
C(52) 9347(1) 2634(2) 5425(1) 52(1)
_________________________________________________________________________
Bond lengths [Å] and angles [°].
_____________________________________________________
F(1)-C(22) 1.3460(18)
F(2)-C(23) 1.349(2)
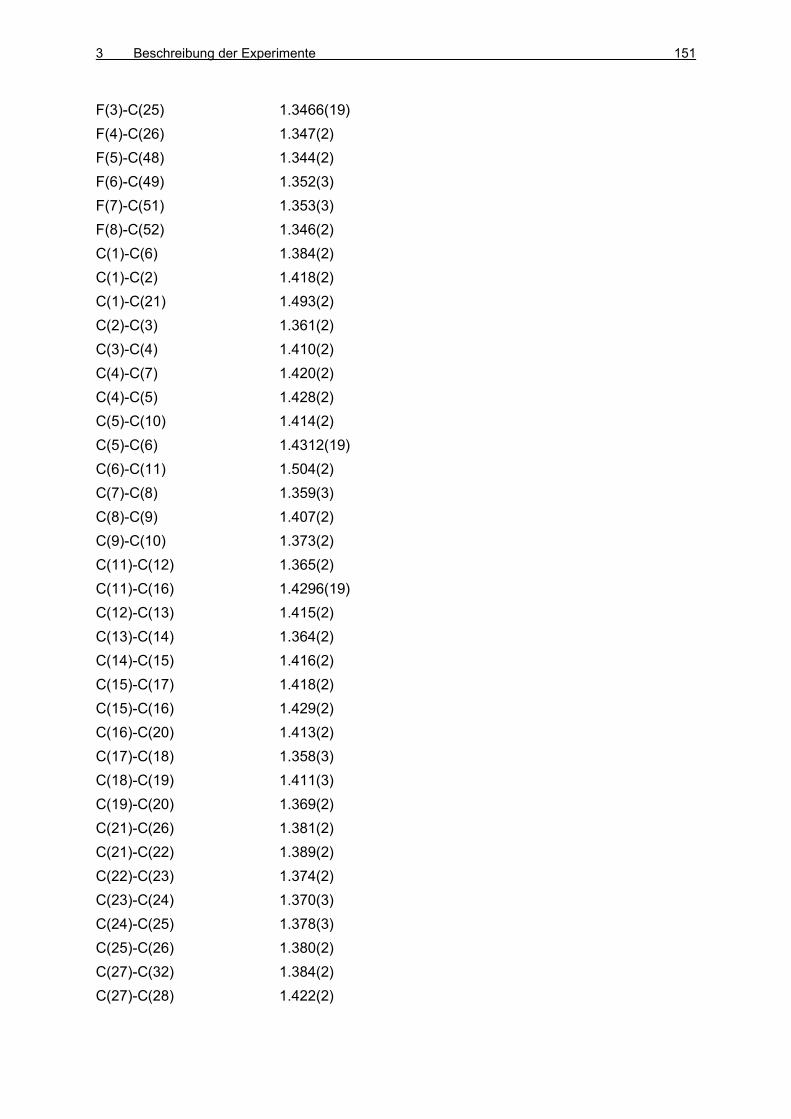
3 Beschreibung der Experimente 151
F(3)-C(25) 1.3466(19)
F(4)-C(26) 1.347(2)
F(5)-C(48) 1.344(2)
F(6)-C(49) 1.352(3)
F(7)-C(51) 1.353(3)
F(8)-C(52) 1.346(2)
C(1)-C(6) 1.384(2)
C(1)-C(2) 1.418(2)
C(1)-C(21) 1.493(2)
C(2)-C(3) 1.361(2)
C(3)-C(4) 1.410(2)
C(4)-C(7) 1.420(2)
C(4)-C(5) 1.428(2)
C(5)-C(10) 1.414(2)
C(5)-C(6) 1.4312(19)
C(6)-C(11) 1.504(2)
C(7)-C(8) 1.359(3)
C(8)-C(9) 1.407(2)
C(9)-C(10) 1.373(2)
C(11)-C(12) 1.365(2)
C(11)-C(16) 1.4296(19)
C(12)-C(13) 1.415(2)
C(13)-C(14) 1.364(2)
C(14)-C(15) 1.416(2)
C(15)-C(17) 1.418(2)
C(15)-C(16) 1.429(2)
C(16)-C(20) 1.413(2)
C(17)-C(18) 1.358(3)
C(18)-C(19) 1.411(3)
C(19)-C(20) 1.369(2)
C(21)-C(26) 1.381(2)
C(21)-C(22) 1.389(2)
C(22)-C(23) 1.374(2)
C(23)-C(24) 1.370(3)
C(24)-C(25) 1.378(3)
C(25)-C(26) 1.380(2)
C(27)-C(32) 1.384(2)
C(27)-C(28) 1.422(2)
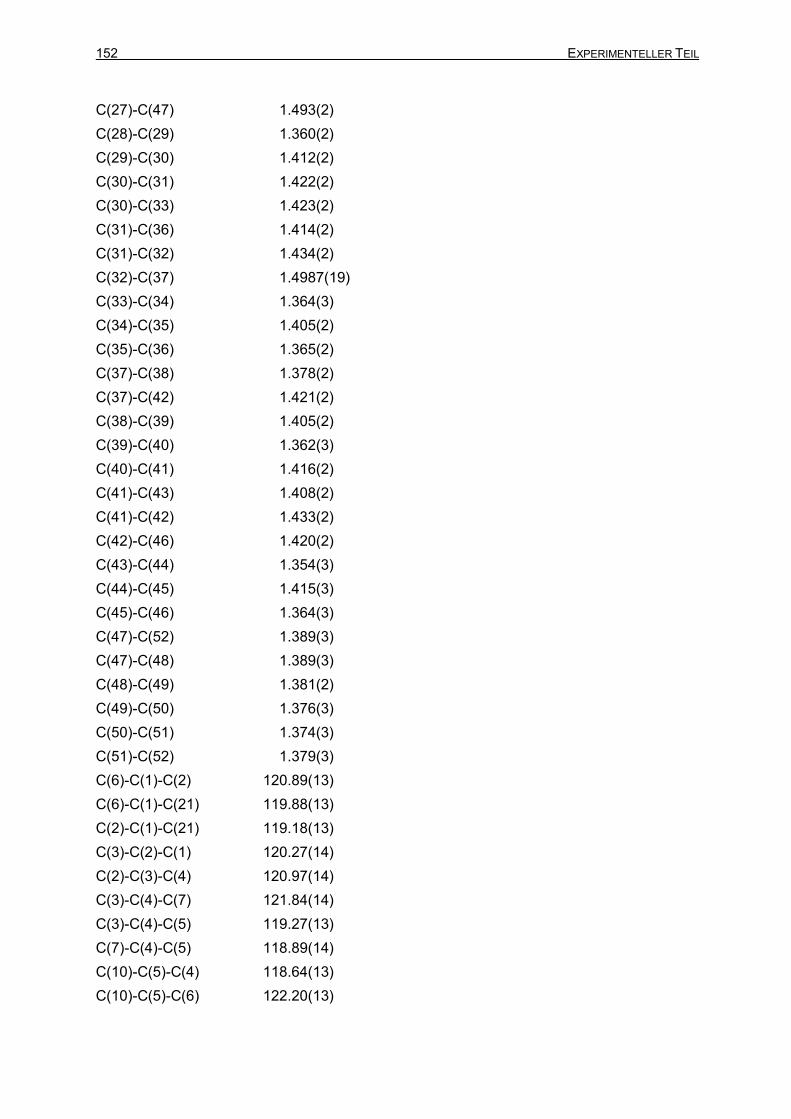
152 EXPERIMENTELLER TEIL
C(27)-C(47) 1.493(2)
C(28)-C(29) 1.360(2)
C(29)-C(30) 1.412(2)
C(30)-C(31) 1.422(2)
C(30)-C(33) 1.423(2)
C(31)-C(36) 1.414(2)
C(31)-C(32) 1.434(2)
C(32)-C(37) 1.4987(19)
C(33)-C(34) 1.364(3)
C(34)-C(35) 1.405(2)
C(35)-C(36) 1.365(2)
C(37)-C(38) 1.378(2)
C(37)-C(42) 1.421(2)
C(38)-C(39) 1.405(2)
C(39)-C(40) 1.362(3)
C(40)-C(41) 1.416(2)
C(41)-C(43) 1.408(2)
C(41)-C(42) 1.433(2)
C(42)-C(46) 1.420(2)
C(43)-C(44) 1.354(3)
C(44)-C(45) 1.415(3)
C(45)-C(46) 1.364(3)
C(47)-C(52) 1.389(3)
C(47)-C(48) 1.389(3)
C(48)-C(49) 1.381(2)
C(49)-C(50) 1.376(3)
C(50)-C(51) 1.374(3)
C(51)-C(52) 1.379(3)
C(6)-C(1)-C(2) 120.89(13)
C(6)-C(1)-C(21) 119.88(13)
C(2)-C(1)-C(21) 119.18(13)
C(3)-C(2)-C(1) 120.27(14)
C(2)-C(3)-C(4) 120.97(14)
C(3)-C(4)-C(7) 121.84(14)
C(3)-C(4)-C(5) 119.27(13)
C(7)-C(4)-C(5) 118.89(14)
C(10)-C(5)-C(4) 118.64(13)
C(10)-C(5)-C(6) 122.20(13)
3 Beschreibung der Experimente 153
C(4)-C(5)-C(6) 119.16(13)
C(1)-C(6)-C(5) 119.13(13)
C(1)-C(6)-C(11) 119.90(12)
C(5)-C(6)-C(11) 120.93(12)
C(8)-C(7)-C(4) 120.94(15)
C(7)-C(8)-C(9) 120.28(14)
C(10)-C(9)-C(8) 120.62(16)
C(9)-C(10)-C(5) 120.57(15)
C(12)-C(11)-C(16) 119.31(13)
C(12)-C(11)-C(6) 118.86(12)
C(16)-C(11)-C(6) 121.83(13)
C(11)-C(12)-C(13) 122.09(14)
C(14)-C(13)-C(12) 119.62(15)
C(13)-C(14)-C(15) 120.69(14)
C(14)-C(15)-C(17) 121.38(14)
C(14)-C(15)-C(16) 119.51(13)
C(17)-C(15)-C(16) 119.11(14)
C(20)-C(16)-C(15) 118.44(13)
C(20)-C(16)-C(11) 122.77(13)
C(15)-C(16)-C(11) 118.79(13)
C(18)-C(17)-C(15) 120.83(15)
C(17)-C(18)-C(19) 120.19(14)
C(20)-C(19)-C(18) 120.62(16)
C(19)-C(20)-C(16) 120.79(15)
C(26)-C(21)-C(22) 116.65(14)
C(26)-C(21)-C(1) 123.45(14)
C(22)-C(21)-C(1) 119.91(13)
F(1)-C(22)-C(23) 118.72(15)
F(1)-C(22)-C(21) 119.40(13)
C(23)-C(22)-C(21) 121.88(15)
F(2)-C(23)-C(24) 120.32(15)
F(2)-C(23)-C(22) 118.78(15)
C(24)-C(23)-C(22) 120.89(16)
C(23)-C(24)-C(25) 118.07(15)
F(3)-C(25)-C(24) 120.11(15)
F(3)-C(25)-C(26) 118.81(16)
C(24)-C(25)-C(26) 121.08(15)
F(4)-C(26)-C(25) 118.92(14)

154 EXPERIMENTELLER TEIL
F(4)-C(26)-C(21) 119.66(14)
C(25)-C(26)-C(21) 121.41(15)
C(32)-C(27)-C(28) 120.19(14)
C(32)-C(27)-C(47) 121.46(14)
C(28)-C(27)-C(47) 118.33(14)
C(29)-C(28)-C(27) 120.78(15)
C(28)-C(29)-C(30) 120.90(14)
C(29)-C(30)-C(31) 119.05(14)
C(29)-C(30)-C(33) 122.19(14)
C(31)-C(30)-C(33) 118.76(15)
C(36)-C(31)-C(30) 118.13(13)
C(36)-C(31)-C(32) 122.44(13)
C(30)-C(31)-C(32) 119.42(13)
C(27)-C(32)-C(31) 119.34(13)
C(27)-C(32)-C(37) 122.32(13)
C(31)-C(32)-C(37) 118.26(13)
C(34)-C(33)-C(30) 121.09(15)
C(33)-C(34)-C(35) 119.98(15)
C(36)-C(35)-C(34) 120.29(15)
C(35)-C(36)-C(31) 121.51(14)
C(38)-C(37)-C(42) 119.39(13)
C(38)-C(37)-C(32) 117.99(14)
C(42)-C(37)-C(32) 122.60(13)
C(37)-C(38)-C(39) 122.05(16)
C(40)-C(39)-C(38) 119.62(15)
C(39)-C(40)-C(41) 120.85(14)
C(43)-C(41)-C(40) 121.01(15)
C(43)-C(41)-C(42) 119.52(16)
C(40)-C(41)-C(42) 119.48(15)
C(46)-C(42)-C(37) 123.15(13)
C(46)-C(42)-C(41) 118.25(14)
C(37)-C(42)-C(41) 118.60(14)
C(44)-C(43)-C(41) 120.62(16)
C(43)-C(44)-C(45) 120.46(17)
C(46)-C(45)-C(44) 120.76(17)
C(45)-C(46)-C(42) 120.35(16)
C(52)-C(47)-C(48) 116.55(16)
C(52)-C(47)-C(27) 120.76(16)

3 Beschreibung der Experimente 155
C(48)-C(47)-C(27) 122.68(15)
F(5)-C(48)-C(49) 118.67(17)
F(5)-C(48)-C(47) 119.78(14)
C(49)-C(48)-C(47) 121.55(18)
F(6)-C(49)-C(50) 119.85(17)
F(6)-C(49)-C(48) 118.72(19)
C(50)-C(49)-C(48) 121.4(2)
C(51)-C(50)-C(49) 117.34(17)
F(7)-C(51)-C(50) 119.62(18)
F(7)-C(51)-C(52) 118.6(2)
C(50)-C(51)-C(52) 121.78(19)
F(8)-C(52)-C(51) 119.02(18)
F(8)-C(52)-C(47) 119.64(17)
C(51)-C(52)-C(47) 121.3(2)
_____________________________________________________________


ANHANG


A1 Abkürzungsverzeichnis 159
A1 ABKÜRZUNGSVERZEICHNIS
[α] spezifischer Drehwert
δ chemische Verschiebung
λ Wellenlänge
~ Wellenzahl
Δ Rückflusstemperatur
Å Ångström
AAV allgemeine Arbeitsvorschrift
APCI atmospheric pressure chemical ionization
ATR abgeschwächte Totalreflexion
Äquiv. Äquivalente
ber. berechnet
BINOL 1,1‘-Bi-2-naphthol
Bn Benzyl
Bu Butyl
bzw. beziehungsweise
°C Grad Celsius
c Konzentration
cm Zentimeter
COSY Korrelationsspektroskopie
Cy Cyclohexyl
d Dublett oder Tage
DABCO 1,4-Diazabicyclo[2.2.2]octan
DC Dünnschichtchromatographie
DEPT distortionless enhancement by polarization transfer
DIBAL–H Diisobutylaluminiumhydrid
dm Dezimeter
DMS Dimethylsulfid
dppf 1,1‘-Bis(diphenylphosphino)ferrocen
dr Diastereomerenverhältnis

160 ANHANG
ee Enantiomerenüberschuss
EI elektronische Ionisation
ESI electron spray ionization
Et Ethyl
Fa. Firma
Fc Ferrocenyl
FLP frustriertes LEWIS-Paar
g Gramm
Ge Germylgruppe
gef. gefunden
GC Gaschromatographie
h Stunde
HMBC heteronuclear multiple bond coherence
HMQC heteronuclear multiple quantum coherence
HPLC Hochleistungsflüssigkeitschromatographie
HRMS Hochauflösende Massenspektrometrie
Hrsg. Herausgeber
Hz Hertz
IR Infrarotspektroskopie
i iso
J Kopplungskonstante
K Kelvin
kat. katalytisch
LB LEWIS-Base
Lit. Literatur
M Molekülmasse
M molar
m Multiplett
mc zentrosymmetrisches Multiplett

A1 Abkürzungsverzeichnis 161
m/z Masse-zu-Ladung-Verhältnis
Me Methyl
Mes Mesityl
mg Milligramm
min Minute
mL Milliliter
mm Millimeter
mmol Millimol
Mol-% Molprozent
MS Massenspektrometrie oder Molekularsieb
n normal
n.b. nicht bestimmt
NMR Kernspinresonanz
NOESY nuclear OVERHAUSER effect spectroscopy
Ph Phenyl
pin Pinakolato
PMP para-Methoxyphenyl
ppm parts per million
Pr Propyl
q Quartett
quant. quantitativ
R organischer Rest oder wie definiert
Rf Retardierungsfaktor
rac racemisch
RT Raumtemperatur
s Singulett
S Seite
Si Silylgruppe
sept Septett
Smp. Schmelzpunkt
SN2 nukleophile Substitution zweiter Ordnung

162 ANHANG
t Triplett
t tertiär
tR Retentionszeit
TFA Trifluoressigsäure
THF Tetrahydrofuran
Tf Trifluormethansulfonyl
Tol Toluyl
UV Ultraviolett
vs. versus
wässr. wässrig

A2 Literaturverzeichnis 163
A2 LITERATURVERZEICHNIS
[1] P. Rittmeyer, U. Wietelmann in Ullmann's Encyclopedia of Industrial Chemistry,
Wiley-VCH, Weinheim, 2002, S. 103–131.
[2] H. I. Schlesinger, H. C. Brown, B. Abraham, A. C. Bond, N. Davidson, A. E. Finholt,
J. R. Gilbreath, H. Hoekstra, L. Horvitz, E. K. Hyde, J. J. Katz, J. Knight, R. A. Lad,
D. L. Mayfield, L. Rapp, D. M. Ritter, A. M. Schwartz, I. Sheft, L. D. Tuck, A. O.
Walker J. Am. Chem. Soc. 1953, 75, 186–190.
[3] Übersichten zur Chemie elektronenarmer Borane: a) R. L. Melen, Chem. Commun.
2014, 50, 1161–1174; b) G. Erker, Dalton Trans. 2005, 1883–1890; c) W. E. Piers,
Adv. Organomet. Chem. 2004, 52, 1–76; d) W. E. Piers, G. J. Irvine, V. C. Williams,
Eur. J. Inorg. Chem. 2000, 2131–2142; e) W. E. Piers, T. Chivers, Chem. Soc. Rev.
1997, 26, 345–354.
[4] Übersichten zur Chemie der FLPs: a) D. W. Stephan, J. Am. Chem. Soc. 2015, 137,
10018–10032; b) D. W. Stephan, Acc. Chem. Res. 2015, 48, 306–316; c) D. W.
Stephan, G. Erker, Angew. Chem. Int. Ed. 2015, 54, 6400–6441; d) S. A. Weicker,
D. W. Stephan, Bull. Chem. Soc. Jpn. 2015, 88, 1003–1016; e) D. W. Stephan, G.
Erker, Chem. Sci. 2014, 5, 2625–2641; f) Topics in Current Chemistry, Vol. 332
(Hrsg.: G. Erker, D. W. Stephan), Springer, Berlin, Heidelberg, 2013; g) Topics in
Current Chemistry, Vol. 334 (Hrsg.: G. Erker, D. W. Stephan), Springer, Berlin,
Heidelberg, 2013; h) D. W. Stephan, G. Erker, Angew. Chem. Int. Ed. 2010, 49, 46–
76; i) D. W. Stephan, Org. Biomol. Chem. 2008, 6, 1535–1539.
[5] a) A. G. Massey, A. J. Park, F. G. A. Stone, Proc. Chem. Soc. 1963, 212; b) A. G.
Massey, A. J. Park, J. Organomet. Chem. 1964, 2, 245–250; c) A. G. Massey, A. J.
Park, J. Organomet. Chem. 1966, 5, 218–225.
[6] D. J. Parks, W. E. Piers, J. Am. Chem. Soc. 1996, 118, 9440–9441.
[7] Vereinzelte Berichte über B(C6F5)3 (1) als LEWIS-Säure-Katalysator in nicht
verwandten Reaktionen waren bereits zuvor bekannt: a) K. Ishihara, N. Hananki, H.
Yamamoto, Synlett 1993, 577–579; b) K. Ishihara, M. Funahashi, N. Hanaki, M.
Miyata, H. Yamamoto, Synlett 1994, 963–964; c) K. Ishihara, N. Hanaki, H.
Yamamoto, Synlett 1995, 721–722; d) K. Ishihara, N. Hanaki, M. Funahashi, M.
Miyata, H. Yamamoto, Bull. Chem. Soc. Jpn. 1995, 68, 1721–1730. Von größerer
Bedeutung war B(C6F5)3 (1) bis dato lediglich als Co-Katalysator für
metallocenkatalysierte Polymerisationsreaktionen. Für frühe Beispiele siehe: e) X.
Yang, C. L. Stern, T. J. Marks, J. Am. Chem. Soc. 1991, 113, 3623–3625; f) X.
Yang, C. L. Stern, T. J. Marks, J. Am. Chem. Soc. 1994, 116, 10015–10031; g) M.

164 ANHANG
Bochmann, S. J. Lancaster Organometallics 1994, 13, 2235-2243; h) B. Temme, G.
Erker, J. Karl, H. Luftmann, R. Fröhlich, S. Kotila, Angew. Chem. Int. Ed. Engl. 1995,
34, 1755–1757. Eine Übersicht bietet: i) E. Y.-X. Chen, T. J. Marks, Chem. Rev.
2000, 100, 1391–1434.
[8] G. C. Welch, R. R. S. Juan, J. D. Masuda, D. W. Stephan, Science 2006, 314,
1124–1126.
[9] Ironischerweise wurde über die allgemein als frühestes Beispiel eines FLPs
angesehene Mischung aus 2,6-Lutidin und BMe3 vor der Beschreibung von NaBH4
berichtet. Zudem ist diese Entdeckung derselben Gruppe zuzuschreiben: H. C.
Brown, H. I. Schlesinger, S. Z. Cardon, J. Am. Chem. Soc. 1942, 64, 325–329.
[10] P. A. Chase, G. C. Welch, T. Jurca, D. W. Stephan, Angew. Chem. Int. Ed. 2007,
46, 8050–8053.
[11] Übersichten: a) M. Oestreich, J. Hermeke, J. Mohr, Chem. Soc. Rev. 2015, 44,
2202–2220; b) L. Shi, Y.-G. Zhou, ChemCatChem 2015, 7, 54–56; c) M. Alcarazo,
Synlett 2014, 1519–1520; d) X. Feng, H. Du, Tetrahedron Lett. 2014, 55, 6959–
6964; e) L. J. Hounjet, D. W. Stephan, Org. Process Res. Dev. 2014, 18, 385–391;
f) J. Paradies, Angew. Chem. Int. Ed. 2014, 53, 3552–3557; g) J. Paradies, Synlett
2013, 777–780; h) D. W. Stephan, Org. Biomol. Chem. 2012, 10, 5740–5746; i) D.
W. Stephan, Chem. Commun. 2010, 46, 8526–8533; j) D. W. Stephan, Dalton
Trans. 2009, 3129–3136.
[12] Für eine Übersicht zu hauptgruppenelementkatalysierten Reduktionen siehe: K.
Revunova, G. I. Nikonov, Dalton Trans. 2015, 44, 840–866.
[13] D. J. Parks, J. M. Blackwell, W. E. Piers, J. Org. Chem. 2000, 65, 3090–3098.
[14] a) M. P. Doyle, C. T. West, S. J. Donnelly, C. C. McOsker, J. Organomet. Chem.
1976, 117, 129–140; b) J. L. Fry, M. Orfanopoulos, M. G. Adlington, W. R. Dittman,
Jr., S. B. Silverman, J. Org. Chem. 1978, 43, 374–375.
[15] K. Sakata, H. Fujimoto, J. Org. Chem. 2013, 78, 12505–12512.
[16] a) S. Rendler, M. Oestreich, Angew. Chem. Int. Ed. 2008, 47, 5997–6000. Für einen
früheren Versuch, den Übergangszustand VIII durch Verwendung eines
siliciumstereogenen Silans zu beweisen, siehe: b) S. Shinke, T. Tsuchimoto, Y.
Kawakami, Silicon Chem. 2007, 3, 243–249. Eine spätere Studie mit einer
deuteriummarkierten stereochemischen Siliciumsonde bestätigte den Befund: c) T.
Fallon, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54, 12488–12491.
[17] A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers, H. M. Tuononen,
Nat. Chem. 2014, 6, 983–988.
[18] Ein entsprechendes Hydrosilanaddukt des Alans Al(C6F5)3 wurde ebenfalls
charakterisiert: J. Chen, E. Y.-X. Chen, Angew. Chem.Int. Ed. 2015, 54, 6842–6846.

A2 Literaturverzeichnis 165
[19] Am Beispiel der borankatalysierten Hydrierung von Iminen (X = NR): a) P. A. Chase,
T. Jurca, D. W. Stephan, Chem. Commun. 2008, 1701–1703; b) T. A. Rokob, A.
Hamza, A. Stirling, I. Pápai, J. Am. Chem. Soc. 2009, 131, 2029–2036. Für einen
vergleichenden Artikel zur Aktivierung von Si–H- und H–H-Bindungen durch
elektronenarme Borane siehe: c) W. E. Piers, A. J. V. Marwitz, L. G. Mercier, Inorg.
Chem. 2011, 50, 12252–12262.
[20] a) A. Y. Houghton, V. A. Karttunen, W. E. Piers, H. M. Tuononen, Chem. Commun.
2014, 50, 1295–1298. Für weitere Beispiele von Diwasserstoffaktivierung durch
elektronenarme Borane ohne externe LEWIS-Base siehe: b) C. Fan, L. G. Mercier,
W. E. Piers, H. M. Tuononen, M. Parvez, J. Am. Chem. Soc. 2010, 132, 9604–9606;
c) A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers, H. M. Tuononen, J. Am.
Chem. Soc. 2013, 135, 941–947.
[21] Für Beispiele siehe: Lit 19b; a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós, I. Pápai,
Angew. Chem. Int. Ed. 2008, 47, 2435–2438; b) Y. Guo, S. Li, Inorg. Chem. 2008,
47, 6212–6219; c) T. A. Rokob, A. Hamza, I. Pápai, J. Am. Chem. Soc. 2009, 131,
10701–10710; d) A. Hamza, A. Stirling, T. A. Rokob, I. Pápai, Int. J. Quantum
Chem. 2009, 109, 2416–2425; e) B. Schirmer, S. Grimme, Chem. Commun. 2010,
46, 7942–7944; f) S. Grimme, H. Kruse, L. Goerigk, G. Erker, Angew. Chem. Int. Ed.
2010, 49, 1402–1405; g) L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine,
G.-V. Röschenthaler, J. Eicher, Chem. Eur. J. 2013, 19, 17413–17424; h) T. A.
Rokob, I. Bakó, A. Stirling, A. Hamza, I. Pápai, J. Am. Chem. Soc. 2013, 135, 4425–
4437; i) M. Pu, T. Privalov, ChemPhysChem 2014, 15, 2936–2944; j) M. Pu, T.
Privalov, ChemPhysChem 2014, 15, 3714–3719.
[22] D. Chen, J. Klankermayer, Chem. Commun. 2008, 2130–2131.
[23] T. Privalov, Eur. J. Inorg. Chem. 2009, 2229–2237.
[24] a) S. Tussing, L. Greb, S. Tamke, B. Schirmer, C. Muhle-Goll, B. Luy, J. Paradies,
Chem. Eur. J. 2015, 21, 8056–8059; b) S. Tussing, K. Kaupmees, J. Paradies,
Chem. Eur. J. 2016, 22, 7422–7426.
[25] Für ein diastereoselektives Beispiel einer B(C6F5)3-katalysierten
Carbonylhydrosilylierung siehe: a) M. K. Skjel, A. Y. Houghton, A. E. Kirby, D. J.
Harrison, R. McDonald, L. Rosenberg, Org. Lett. 2010, 12, 376–379. Für B(C6F5)3-
katalysierte konjugierte Reduktionen von Carbonylverbindungen mit Hydrosilanen
siehe: b) J. M. Blackwell, D. J. Morrison, W. E. Piers, Tetrahedron 2002, 58, 8247–
8254; c) S. Chandrasekhar, G. Chandrashekar, M. S. Reddy, P. Srihari, Org.
Biomol. Chem. 2006, 4, 1650–1652; d) L. Greb, P. Oña-Burgos, A. Kubas, F. C.
Falk, F. Breher, K. Fink, J. Paradies, Dalton Trans. 2012, 41, 9056–9060; e) Y. Kim,
S. Chang, Angew. Chem. Int. Ed. 2016, 55, 218–222. Für die Reduktion von Estern

166 ANHANG
zu Acetalen durch B(C6F5)3-katalysierte Hydrosilylierung siehe: Lit. 6, Lit. 13. Für die
chemoselektive Reduktion von Carbonsäuren zu Aldehyden siehe: f) D. Bézier, S.
Park, M. Brookhart, Org. Lett. 2013, 15, 496–499.
[26] Für Boran- und FLP-katalysierte Reduktionsmethoden von CO2 mit Hydrosilanen
siehe: a) A. Berkefeld, W. E. Piers, M. Parvez, J. Am. Chem. Soc. 2010, 132,
10660–10661; b) J. Chen, L. Falivene, L. Caporaso, L. Cavallo, E. Y.-X. Chen, J.
Am. Chem. Soc. 2016, 138, 5321–5333; c) D. Mukherjee, D. F. Sauer, A. Zanardi, J.
Okuda, Chem. Eur. J. 2016, 22, 7730–7733.
[27] Einige der nachfolgend vorgestellten Hydrosilylierungen und Hydrierungen lassen
sich ebenfalls durch Phosphoniumkationen katalysieren. Für
Hydrosilylierungsreaktionen wird ebenfalls von einer Hydrosilanaktivierung durch η1-
Koordination des Katalysators ausgegangen. Für Beispiele siehe: a) M. Pérez, L. J.
Hounjet, C. B. Caputo, R. Dobrovetsky, D. W. Stephan, J. Am. Chem. Soc. 2013,
135, 18308–18310; b) M. H. Holthausen, M. Mehta, D. W. Stephan, Angew. Chem.
Int. Ed. 2014, 53, 6538–6541 (Alken- und Alkinhydrosilylierung); c) T. vom Stein, M.
Peréz, R. Dobrovetsky, D. Winkelhaus, C. B. Caputo, D. W. Stephan, Angew.
Chem. Int. Ed. 2015, 54, 10178–10182 (Alkenhydrierung); d) M. Pérez, Z.-W. Qu, C.
B. Caputo, V. Podgorny, L. J. Hounjet, A. Hansen, R. Dobrovetsky, S. Grimme, D.
W. Stephan, Chem. Eur. J. 2015, 21, 6491–6500 (Keton-, Imin- und
Nitrilhydrosilylierung). Einen Überblick bietet: e) J. M. Bayne, D. W. Stephan, Chem.
Soc. Rev. 2016, 45, 765–774.
[28] S. Keess, A. Simonneau, M. Oestreich, Organometallics 2015, 34, 790–799.
[29] Für eine Übersicht zu Transferhydrosilylierungen siehe: a) M. Oestreich, Angew.
Chem. Int. Ed. 2016, 55, 494–499. Für eine B(C6F5)3-katalysierte
Carbonyltransferhydroaluminierung siehe: S. Dagorne, I. Janowska, R. Welter, J.
Zakrzewski, G. Jaouen, Organometallics 2004, 23, 4706–4710.
[30] Für Übersichten zu kationischen Borverbindungen siehe: a) T. S. de Vries, A.
Prokofjevs, E. Vedejs, Chem. Rev. 2012, 112, 4246–4282; b) W. E. Piers, S. C.
Bourke, K. D. Conroy, Angew. Chem. Int. Ed. 2005, 44, 5016–5036; c) P. Kölle, H.
Nöth, Chem. Rev. 1985, 85, 399–418.
[31] S. E. Denmark, Y. Ueki, Organometallics 2013, 32, 6631–6634.
[32] D. Chen, V. Leich, F. Pan, J. Klankermayer, Chem. Eur. J. 2012, 18, 5184–5187.
[33] J. Chen, R. A. Lalancette, F. Jäkle, Chem. Commun. 2013, 49, 4893–4895.
[34] X. Ren, H. Du, J. Am. Chem. Soc. 2016, 138, 810–813.
[35] L. Süsse, J. Hermeke, M. Oestreich, J. Am. Chem. Soc. 2016, 138, 6940–6943.

A2 Literaturverzeichnis 167
[36] a) D. J. Harrison, R. McDonald, L. Rosenberg, Organometallics 2005, 24, 1398–
1400; b) P. T. K. Lee, M. K. Skjel, L. Rosenberg, Organometallics 2013, 32, 1575–
1578.
[37] a) M. Lindqvist, N. Sarnela, V. Sumerin, K. Chernichenko, M. Leskelä, T. Repo,
Dalton Trans. 2012, 41, 4310–4312; b) L. E. Longobardi, C. Tang, D. W. Stephan,
Dalton Trans. 2014, 43, 15723–15726.
[38] T. Mahdi, D. W. Stephan, J. Am. Chem. Soc. 2014, 136, 15809–15812.
[39] D. J. Scott, M. J. Fuchter, A. E. Ashley, J. Am. Chem. Soc. 2014, 136, 15813–
15816.
[40] Für übergangsmetallfreie basenkatalysierte Hydrierungen siehe: a) C. Walling, L.
Bollyky, J. Am. Chem. Soc. 1961, 83, 2968–2969; b) C. Walling, L. Bollyky, J. Am.
Chem. Soc. 1964, 86, 3750–3752; c) A. Berkessel, T. J. S. Schubert, T. N. Müller, J.
Am. Chem. Soc. 2002, 124, 8693–8698.
[41] Für eine stöchiometrische Hydrierung von CO2 durch ein FLP siehe: A. E. Ashley, A.
L. Thompson, D. O'Hare, Angew. Chem. Int. Ed. 2009, 48, 9839–9843.
[42] D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter, A. E.
Ashley, ACS Catal. 2015, 5, 5540–5544.
[43] Für die Verwendung von Katalysatorregeneratoren in boran- und FLP-katalysierten
Hydrierungen siehe: J. W. Thomson, J. A. Hatnean, J. J. Hastie, A. Pasternak, D.
W. Stephan, P. A. Chase, Org. Process Res. Dev. 2013, 17, 1287–1292.
[44] Für Untersuchungen von B(C6F5)3/Wasser-Addukten siehe: a) C. Bergquist, B. M.
Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner, G. Parkin, J. Am. Chem.
Soc. 2000, 122, 10581–10590; b) T. Beringhelli, D. Maggioni, G. D'Alfonso,
Organometallics 2001, 20, 4927–4938.
[45] Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján, T. Soós, ACS Catal. 2015, 5,
5366–5372.
[46] T. Mahdi, D. W. Stephan, Angew. Chem. Int. Ed. 2015, 54, 8511–8514.
[47] J. M. Blackwell, E. R. Sonmor, T. Scoccitti, W. E. Piers, Org. Lett. 2000, 2, 3921–
3923.
[48] Für ein Eintopfverfahren, bei dem in situ durch Kondensation erhaltene Imine
B(C6F5)3-katalysiert hydrosilyliert werden, siehe: V. Fasano, J. E. Radcliffe, M. J.
Ingleson, ACS Catal. 2016, 6, 1793–1798.
[49] J. Hermeke, M. Mewald, M. Oestreich, J. Am. Chem. Soc. 2013, 135, 17537–17546.
[50] M. Mewald, M. Oestreich, Chem. Eur. J. 2012, 18, 14079–14084.
[51] X. Zhu, H. Du, Org. Biomol. Chem. 2015, 13, 1013–1016.
[52] D. J. Scott, M. J. Fuchter, A. E. Ashley, Angew. Chem. Int. Ed. 2014, 53, 10218–
10222.

168 ANHANG
[53] J. M. Farrell, J. A. Hatnean, D. W. Stephan, J. Am. Chem. Soc. 2012, 134, 15728–
15731.
[54] P. Eisenberger, B. P. Bestvater, E. C. Keske, C. M. Crudden, Angew. Chem. Int. Ed.
2015, 54, 2467–2471; für weitere Beispiele boreniumionkatalysierter
Hydrierungsreaktionen siehe: b) J. M. Farrell, D. W. Stephan, Chem. Commun.
2015, 51, 14322–14325; c) J. M. Farrell, R. T. Posaratnanathan, D. W. Stephan,
Chem. Sci. 2015, 6, 2010–2015.
[55] a) D. Chen, Y. Wang, J. Klankermayer, Angew. Chem. Int. Ed. 2010, 49, 9475–
9478. Für ein Beispiel eines campherabgeleiteten intramolekularen FLPs in der
enantioselektiven Hydrierung von Iminen siehe: b) G. Ghattas, D. Chen, F. Pan, J.
Klankermayer, Dalton Trans. 2012, 41, 9026–9028.
[56] M. Lindqvist, K. Borre, K. Axenov, B. Kótai, M. Nieger, M. Leskelä, I. Pápai, T. Repo,
J. Am. Chem. Soc. 2015, 137, 4038–4041.
[57] Y. Liu, H. Du, J. Am. Chem. Soc. 2013, 135, 6810–6813.
[58] Für diastereoselektive borankatalysierte Hydrierungen von Iminen siehe: a) Z. M.
Heiden, D. W. Stephan, Chem. Commun. 2011, 47, 5729–5731; b) X. Zhu, H. Du,
Org. Lett. 2015, 17, 3106–3109.
[59] J. M. Farrell, Z. M. Heiden, D. W. Stephan, Organometallics 2011, 30, 4497–4500.
[60] I. Chatterjee, Z.-W. Qu, S. Grimme, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54,
12158–12162.
[61] a) N. Gandhamsetty, J. Jeong, J. Park, S. Park, S. Chang, J. Org. Chem. 2015, 80,
7281–7287. Für die B(C6F5)3-katalysierte Reduktion konjugierter Nitrile mit
Dihydrosilanen siehe: b) N. Gandhamsetty, J. Park, J. Jeong, S.-W. Park, S. Park,
S. Chang, Angew. Chem. Int. Ed. 2015, 54, 6832–6836.
[62] M. Rubin, T. Schwier, V. Gevorgyan, J. Org. Chem. 2002, 67, 1936–1940. Für
intramolekulare B(C6F5)3-katalysierte C=C-Hydrosilylierungen siehe: R. Shchepin, C.
Xu, P. Dussault, Org. Lett. 2010, 12, 4772–4775.
[63] Für formale Hydrierungen von C=C-Bindungen bei Verwendung von Hydrosilanen
als Reduktionsmittel siehe: a) M. Tan, Y. Zhang, Tetrahedron Lett. 2009, 50, 4912–
4915; b) L. D. Curless, E. R. Clark, J. J. Dunsford, M. J. Ingleson, Chem. Commun.
2014, 50, 5270–5272; c) L. Greb, S. Tamke, J. Paradies, Chem. Commun. 2014,
50, 2318–2320.
[64] a) A. Simonneau, M. Oestreich, Angew. Chem. Int. Ed. 2013, 52, 11905–11907; b)
A. Simonneau, M. Oestreich, Nat. Chem. 2015, 7, 816–822. Für eine
mechanistische Betrachtung durch quantenchemische Rechnungen siehe: K.
Sakata, H. Fujimoto, Organometallics 2015, 34, 236–241.
[65] H. Wang, R. Fröhlich, G. Kehr, G. Erker, Chem. Commun. 2008, 5966–5968.

A2 Literaturverzeichnis 169
[66] P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich, G. Erker, Angew.
Chem. Int. Ed. 2008, 47, 7543–7546.
[67] S. Wei, H. Du, J. Am. Chem. Soc. 2014, 136, 12261–12264.
[68] Für die enantioselektive Silylenoletherhydrierung eignen sich auch aus chiralen
Diinen und HB(C6F5)2 generierte chirale Borane in Kombination mit tBu3P (80) als
Katalysatorsystem: X. Ren, G. Li, S. Wei, H. Du, Org. Lett. 2015, 17, 990–993.
[69] J. A. Nicasio, S. Steinberg, B. Inés, M. Alcarazo, Chem. Eur. J. 2013, 19, 11016–
11020.
[70] Für eine vergleichende Studie zur Hydriddonorstärke gängiger
Hauptgruppenelementhydride durch quantenchemische Rechnungen siehe: Z. M.
Heiden, A. P. Lathem, Organometallics 2015, 34, 1818–1827.
[71] L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme, D. W. Stephan,
Angew. Chem. Int. Ed. 2013, 52, 7492–7495.
[72] I. Chatterjee, Z.-W. Qu, S. Grimme, M. Oestreich, Angew. Chem. Int. Ed. 2015, 54,
12158–12162.
[73] L. D. Curless, M. J. Ingleson, Organometallics 2014, 33, 7241–7246.
[74] T. Schwier, V. Gevorgyan, Org. Lett. 2005, 7, 5191–5194.
[75] a) B.-H. Xu, G. Kehr, R. Fröhlich, B. Wibbeling, B. Schirmer, S. Grimme, G. Erker,
Angew. Chem. Int. Ed. 2011, 50, 7183–7186; b) K. Chernichenko, Á. Madarász, I.
Pápai, M. Nieger, M. Leskelä, T. Repo, Nat. Chem. 2013, 5, 718–723; c) Y. Liu, L.
Hu, H. Chen, H. Du, Chem. Eur. J. 2015, 21, 3495–3501; d) K. C. Szeto, W.
Sahyoun, N. Merle, J. L. Castelbou, N. Popoff, F. Lefebvre, J. Raynaud, C. Godard,
C. Claver, L. Delevoye, R. M. Gauvin, M. Taoufik, Catal. Sci. Technol. 2016, 6, 882–
889.
[76] a) V. Gevorgyan, J.-X. Liu, Y. Yamamoto, J. Org. Chem. 1997, 62, 2963–2967; b) V.
Gevorgyan, J.-X. Liu, Y. Yamamoto, Chem. Commun. 1998, 37–38; c) M. S.
Oderinde, M. G. Organ, Angew. Chem. Int. Ed. 2012, 51, 9834–9837.
[77] J. S. McGough, S. M. Butler, I. A. Cade, M. J. Ingleson, Chem. Sci. 2016, 7, 3384–
3389.
[78] a) A. Melman in The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids,
Part 1 (Hrsg.: Z. Rappoport, J. F. Liebman), Wiley-VCH, Chichester, 2009, S. 117–
161; b) D. Geffken, M. A. Köllner in Science of Synthesis, Vol. 40 (Hrsg.: E.
Schaumann, D. Enders), Thieme, Stuttgart, 2008, S. 973–1082.
[79] Vavon und Mitarbeiter berichteten von der heterogenen platinkatalysierten
Hydrierung von Oximen. N-Monosubstituierte Hydroxylamine wurden bei der
Hydrierung von aliphatischen und alizyklischen Ketoximen erhalten, während
aromatische Ketoxime zu primären Aminen reduziert wurden. Die Hydrierung von

170 ANHANG
Aldoximen ergab N,N-disubstituierte Hydroxylamine: a) G. Vavon, A. L. Berton, Bull.
Soc. Chim. Fr. 1925, 37, 296–305; b) G. Vavon, P. Anziani, Bull. Soc. Chim. Fr.
1927, 41, 1638–1649; c) G. Vavon, N. Krajcinovic, Bull. Soc. Chim. Fr. 1928, 43,
231–237. Für die heterogene platinkatalysierte Hydrierung von Ketoximethern siehe:
d) L. W. Jones, R. T. Major, J. Am. Chem. Soc. 1930, 52, 669–679.
[80] H. Feuer, B. F. Vincent, Jr., J. Am. Chem. Soc. 1962, 84, 3771–3772.
[81] a) M. Murakami, J.-W. Kang, Bull. Chem. Soc. Jpn. 1963, 36, 763–769
(Cobaltkatalyse); b) P. Krasik, H. Alper, Tetrahedron: Asymmetry 1992, 3, 1283–
1288 (Rutheniumkatalyse); c) A. S. C. Chan, C.-C. Chen, C.-W. Lin, Y.-C. Lin, M.-C.
Cheng, S.-M. Peng, J. Chem. Soc., Chem. Commun. 1995, 1767–1768
(Rhodiumkatalyse); d) Y. Xie, A. Mi, Y. Jiang, H. Liu, Synth. Commun. 2001, 31,
2767–2771 (Iridiumkatalyse); e) K. Huang, S. Li, M. Chang, X. Zhang, Org. Lett.
2013, 15, 484–487 (Rhodiumkatalyse).
[82] Eine homogene übergangsmetallkatalysierte Hydrierung von O-Alkyloximethern zu
O-Alkylhydroxylaminen wurde beschrieben in: R. Kadyrov, T. Riermeier, J. Almena,
A. Monsees, P. Groß, K. Rossen (Evonik Degussa GmbH), EP 1 862 446 A2, 2007.
Dieses Patent enthält auch enantioselektive Beispiele.
[83] a) H. Feuer, B. F. Vincent, Jr., R. S. Bartlett, J. Org. Chem. 1965, 30, 2877–2880
(BH3∙THF); b) R. F. Borch, M. D. Bernstein, H. D. Durst, J. Am. Chem. Soc. 1971,
93, 2897–2904 (NaCNBH3); c) Y. Kikugawa, M. Kawase, Chem. Lett. 1977, 1279–
1280 (BH3∙Pyridin); d) G. W. Gribble, R. W. Leiby, M. N. Sheehan, Synthesis 1977,
856–858 (NaBH4).
[84] Für asymmetrische Oxim-zu-Hydroxylamin-Reduktionen mit chiralen
Boran/Oxazaborolidin-Addukten siehe: a) J. T. Dougherty, J. R. Flisak, J. Hayes, I.
Lantos, L. Liu, L. Tucker, Tetrahedron: Asymmetry 1997, 8, 497–499; b) E.
Fontaine, C. Namane, J. Meneyrol, M. Geslin, L. Serva, E. Roussey, S. Tissandié,
M. Maftouh, P. Roger, Tetrahedron: Asymmetry 2001, 12, 2185–2189; c) M. P.
Krzemiński, M. Zaidlewicz, Tetrahedron: Asymmetry 2003, 14, 1463–1466.
[85] Eine alternative Methode bietet die Verwendung von Hydrosilanen als
Reduktionsmittel in Trifluoressigsäure: a) D. D. Sternbach, W. C. L. Jamison,
Tetrahedron Lett. 1981, 22, 3331–3334; b) M. Fujita, H. Oishi, T. Hiyama, Chem.
Lett. 1986, 837–838. Oximether lassen sich ebenso mit Bu3SnH als
Reduktionsmittel vermittelt durch BF3∙OEt2 zu den entsprechenden Hydroxylaminen
reduzieren: M. Ueda, H. Miyabe, M. Namba, T. Nakabayashi, T. Naito, Tetrahedron
Lett. 2002, 43, 4369–4371.
[86] a) G. N. Walker, M. A. Moore, B. N. Weaver, J. Org. Chem. 1961, 26, 2740‒2747
(NaBH4); b) J. A. Blair, R. J. Gardner, J. Chem. Soc. C 1970, 1714–1717

A2 Literaturverzeichnis 171
(BH3∙Bis(2-methoxyethyl)ether); c) Y. Kikugawa, M. Kawase, Synth. Commun. 1979,
9, 49–52 (BH3∙Pyridin). Für weitere stöchiometrische Reduktionsmethoden mit
Hydriddonoren siehe: d) R. L. Hinman, J. Chem. Soc. 1957, 79, 414–417 (LiAlH4);
e) P.-L. Wu, S.-Y. Peng, J. Magrath, Synthesis 1995, 435–438 (Et3SiH/TFA).
[87] Für Beispiele siehe: a) H. L. Yale, K. Losee, J. Martins, M. Holsing, F. M. Perry, J.
Bernstein, J. Am. Chem. Soc. 1953, 75, 1933‒1942 (Platinkatalyse); b) A. Ebnöther,
E. Jucker, A. Lindenmann, E. Rissi, R. Steiner, R. Süess, A. Vogel, Helv. Chim. Acta
1959, 533–563 (Nickelkatalyse); c) M. J. Burk, J. E. Feaster, J. Am. Chem. Soc.
1992, 114, 6266‒6267 (Rutheniumkatalyse); d) Z.-P. Chen, S.-B. Hu, M.-W. Chen,
Y.-G. Zhou, Org. Lett. 2016, 18, 2676–2679. Für eine nickelkatalysierte
Transferhydrierung von Hydrazonen siehe: e) H. Xu, P. Yang, P. Chuanprasit, H.
Hirao, J. Zhou, Angew. Chem. Int. Ed. 2015, 54, 5112‒5116.
[88] Für Beispiele von B(C6F5)3-Derivaten in katalytischen H–H-Bindungsaktivierungen
siehe: Lit. 24; 45; 52; 55a; 57; 69; a) C. Jiang, O. Blacque, H. Berke, Chem.
Commun. 2009, 5518–5520; b) G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király,
G. Tárkányi, T. Soós, Angew. Chem. Int. Ed. 2010, 49, 6559–6563; c) G. Erős, K.
Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G. Tárkányi, T. Soós, Chem. Eur. J.
2012, 18, 574–585. Für Beispiele von B(C6F5)3-Derivaten in katalytischen Si–H-
Bindungsaktivierungen siehe: Lit. 28; 32; 34; 54b; d) R. Roesler, B. J. N. Har, W. E.
Piers, Organometallics 2002, 21, 4300–4302; e) M. Mewald, R. Fröhlich, M.
Oestreich, Chem. Eur. J. 2011, 17, 9406–9414; f) D. Chen, V. Leich, F. Pan, J.
Klankermayer, Chem. Eur. J. 2012, 18, 5184–5187.
[89] Für eine Übersicht der LEWIS-Acidität von Borverbindungen siehe: I. B. Sivaev, V. I.
Bregadze, Coord. Chem. Rev. 2014, 270-271, 75–88.
[90] In Kombination mit sehr starken Basen können auch schwache LEWIS-Säuren wie
das Boran 106 katalytische Aktivität in Hydrierungsreaktionen zeigen: S. Mummadi,
D. K. Unruh, J. Zhao, S. Li, C. Krempner, J. Am. Chem. Soc. 2016, 138, 3286–3289.
[91] Das Boran 108 wurde in einem Patent genannt, eine detaillierte Synthese desselben
ist allerdings nicht beschrieben worden: G. Rodriguez, F. C. Rix, P. S. Ravishankar,
M. C. Kuchta, PCT Int. Appl. WO 03/051892 A1, 2003.
[92] Für Beispiele chiraler Borane und boranbasierter FLPs in katalytischen H–H-
Bindungsaktivierungen siehe: Lit. 55a; 55b; 56; 57; 67; 68; a) V. Sumerin, K.
Chernichenko, M. Nieger, M. Leskelä, B. Rieger, T. Repo, Adv. Synth. Catal. 2011,
353, 2093–2110; b) X. Wang, G. Kehr, C. G. Daniliuc, G. Erker, J. Am. Chem. Soc.
2014, 136, 3293–3303; c) Z. Zhang, H. Du, Org. Lett. 2015, 17, 2816–2819; d) Z.
Zhang, H. Du, Org. Lett. 2015, 17, 6266–6269; e) Z. Zhang, H. Du, Angew. Chem.
Int. Ed. 2015, 54, 623–626. Für Beispiele chiraler Borane und boranbasierter FLPs

172 ANHANG
in katalytischen Si–H-Bindungsaktivierungen siehe: Lit. 32; 34; 35; 88e. Für chirale
elektronenarme Borane und boranbasierte FLPs, die bisher weder in H–H- noch in
Si–H-Bindungsaktivierungen eingesetzt wurden, siehe: f) D. J. Morrison, W. E.
Piers, M. Parvez, Synlett 2004, 2429–2433; g) K. Venkatasubbaiah, M. Bolte, F.
Jäkle, J. Fluor. Chem. 2010, 131, 1247–1251; h) J. Chen, K. Venkatasubbaiah, T.
Pakkirisamy, A. Doshi, A. Yusupov, Y. Patel, R. A. Lalancette, F. Jäkle, Chem. Eur.
J. 2010, 16, 8861–8867; i) J. Chen, R. A. Lalancette, F. Jäkle, Chem. Eur. J. 2014,
20, 9120–9129.
[93] J. Mohr, M. Oestreich, Angew. Chem. Int. Ed. 2014, 53, 13278–13281.
[94] J. Mohr, D. Porwal, I. Chatterjee, M. Oestreich, Chem. Eur. J. 2015, 21, 17583–
17586. Synthesevorschriften für und Charakterisierungen von Substanzen, die im
experimentellen Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.
[95] Zur Durchführung dieser Umlagerungsreaktion mit BH3 als Reduktionsmittel siehe:
a) M. Ortiz-Marciales, L. D. Rivera, M. De Jesús, S. Espinosa, J. A. Benjamin, O. E.
Casanova, I. G. Figueroa, S. Rodríguez, W. Correa, J. Org. Chem. 2005, 70,
10132–10134; b) M. Ortiz-Marciales, D. Figueroa, J. A. López, M. De Jesús, R.
Vega, Tetrahedron Letters 2000, 41, 6567–6570. Ebenfalls bekannt ist diese
Reaktion für Oximreduktionen mit DIBAL–H, eine Übersicht bietet: c) H. Cho,
Tetrahedron 2014, 70, 3527–3544.
[96] Eine Quarternisierung des Boratoms von B(C6F5)3 (1) würde in einem Hochfeldshift
des Signals im 11B-NMR-Spektrum und einer charakteristischen Veränderung der
chemischen Verschiebung des Signals des para-Fluoratomsignals im 19F-NMR-
Spektrum resultieren. Bei Koordination einer LEWIS-Base verschiebt sich dieses
Resonanzsignal zu höherem Feld, was häufig durch einen Δδ-Wert relativ zum
Resonanzsignal des meta-Fluoratoms ausgedrückt wird: a) T. Beringhelli, D.
Donghi, D. Maggioni, G. D’Alfonso, Coord. Chem. Rev. 2008, 252, 2292–2313; Lit.
44b.
[97] Die Hydrierung der Isoxazoline rac-142 und rac-143 war auch in 1,4-Dioxan
erfolglos.
[98] S. Horiyama, K. Suwa, M. Yamaki, H. Kataoka, T. Katagi, M. Takayama, T.
Takeuchi, Chem. Pharm. Bull. 2002, 50, 996–1000.
[99] Lit. 63a; a) E. Blondiaux, T. Cantat, Chem. Commun. 2014, 50, 9349–9352; b) R. C.
Chadwick, V. Kardelis, P. Lim, A. Adronov, J. Org. Chem. 2014, 79, 7728–7733.
[100] M. J. Hearn, E. R. Lucero, J. Heterocycl. Chem. 1982, 19, 1537–1539.
[101] Eine Übersicht bietet: A. D. Dilman, S. L. Ioffe, Chem. Rev. 2003, 103, 733–772.
[102] Übersichtsartikel zu Silyliumionen: a) T. A. Kochina, D. V. Vrazhnov, I. S. Ignatyev,
E. N. Sinotova, M. G. Voronkov, J. Organomet. Chem. 2011, 696, 1331–1340; b) H.

A2 Literaturverzeichnis 173
Čičak, Kem. Ind. 2010, 59, 111–124; c) T. A. Kochina, D. V. Vrazhnov, E. N.
Sinotova, M. G. Voronkov, Russ. Chem. Rev. 2006, 75, 95–110; d) T. Müller, Adv.
Organomet. Chem. 2005, 53, 155–215; e) J. B. Lambert, Y. Zhao, S. M. Zhang, J.
Phys. Org. Chem. 2001, 14, 370–379; f) C. A. Reed, Acc. Chem. Res. 1998, 31,
325–332; g) J. B. Lambert, L. Kania, S. Zhang, Chem. Rev. 1995, 95, 1191–1201;
h) R. J. P. Corriu, M. Henner, J. Organomet. Chem. 1974, 74, 1–28.
[103] Für eine vergleichende Studie siehe: K. Hara, R. Akiyama, M. Sawamura, Org. Lett.
2005, 7, 5621–5623.
[104] Für Übersichtsartikel zu Anwendungen von Silyliumionen in der Katalyse siehe: a)
A. Schulz, A. Villinger, Angew. Chem. Int. Ed. 2012, 51, 4526–4528; b) H. F. T.
Klare, M. Oestreich, Dalton Trans. 2010, 39, 9176–9184.
[105] J. B. Lambert, Y. Zhao, H. Wu, J. Org. Chem. 1999, 64, 2729–2736.
[106] Dieser Silicium-zu-Kohlenstoff-Hydridtransfer wird gemeinhin als Bartlett-Condon-
Schneider-Hydridtransfer bezeichnet: a) P. D. Bartlett, F. E. Condon, A. Schneider,
J. Am. Chem. Soc. 1944, 66, 1531–1539. Nach den Arbeiten von Corey hat sich
diese Technik zur Generierung von Silyliumionen als Standardmethode etabliert: b)
J. Y. Corey, J. Am. Chem. Soc. 1975, 97, 3237–3238; c) J. Y. Corey, D. Gust, K.
Mislow, J. Organomet. Chem. 1975, 101, C7–C8. Weitere Methoden zur
Generierung sind der „allyl leaving group approach“: d) J. B. Lambert, Y. Zhao, H.
Wu, W. C. Tse, B. Kuhlmann, J. Am. Chem. Soc. 1999, 121, 5001–5008; der
„cyclohexadienyl leaving group approach“: e) A. Simonneau, T. Biberger, M.
Oestreich, Organometallics 2015, 34, 3927–3929; durch Substituentenaustausch
zuvor gebildeter Silyliumionen: f) A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D.
Haase, T. Müller, Angew. Chem. Int. Ed. 2011, 50, 12636–12638; g) M. Reißmann,
A. Schäfer, S. Jung, T. Müller, Organometallics 2013, 32, 6736–6744.
[107] M. Kira, T. Hino, H. Sakurai, Chem. Lett. 1992, 555–558.
[108] Für silyliumionvermittelte Reduktionen von CO2 mit Hydrosilanen als
Reduktionsmittel siehe: a) A. Schäfer, W. Saak, D. Haase, T. Müller, Angew. Chem.
Int. Ed. 2012, 51, 2981–2984; b) M. Khandelwal, R. J. Wehmschulte, Angew. Chem.
Int. Ed. 2012, 51, 7323–7326.
[109] V. Gevorgyan, J.-X. Liu, M. Rubin, S. Benson, Y. Yamamoto, Tetrahedron Lett.
1999, 40, 8919–8922.
[110] Für die Beschreibung der Stabilisierung und Bindungsverhältnisse in Silyliumion
205+ siehe: a) K. Müther, R. Fröhlich, C. Mück-Lichtenfeld, S. Grimme, M.
Oestreich, J. Am. Chem. Soc. 2011, 133, 12442–12444; für weitere
ferrocenstabilisierte Silyliumionen siehe: b) K. Müther, P. Hrobárik, V. Hrobáriková,
M. Kaupp, M. Oestreich, Chem. Eur. J. 2013, 19, 16579–16594; für Anwendungen

174 ANHANG
ferrocenstabilisierter Silyliumionen als Katalysatoren in DIELS–ALDER-Reaktionen
siehe: c) H. F. T. Klare, K. Bergander, M. Oestreich, Angew. Chem. Int. Ed. 2009,
48, 9077–9079; d) R. K. Schmidt, K. Müther, C. Mück-Lichtenfeld, S. Grimme, M.
Oestreich, J. Am. Chem. Soc. 2012, 134, 4421–4428; e) A. R. Nödling, K. Müther,
V. H. G. Rohde, G. Hilt, M. Oestreich, Organometallics 2014, 33, 302–308; f) R. K.
Schmidt, H. F. T. Klare, R. Fröhlich, M. Oestreich, Chem. Eur. J. 2016, 22, 5367–
5383.
[111] K. Müther, M. Oestreich, Chem. Commun. 2011, 47, 334–336.
[112] Für eine Carbonylhydrosilylierungsmethode mit einem neutralen tetrakoordinierten
Silicium-LEWIS-Säure-Katalysator, in der Desoxygenierungsreaktionen ebenfalls
unterdrückt wurden, siehe: a) A. L. Liberman-Martin, R. G. Bergman, T. D. Tilley, J.
Am. Chem. Soc. 2015, 137, 5328–5331. Für mechanistisch verwandte durch
siliciumhaltige Metallkomplexe katalysierte Carbonylhydrosilylierungen siehe: b) T.
Muraoka, Y. Shimizu, H. Kobayashi, K. Ueno, H. Ogino, Organometallics 2010, 29,
5423–5426; c) T. T. Metsänen, D. Gallego, T. Szilvási, M. Driess, M. Oestreich,
Chem. Sci. 2015, 6, 7143–7149.
[113] Lit. 106f, 106g; a) A. Schäfer, M. Reißmann, A. Schäfer, M. Schmidtmann, T. Müller,
Chem. Eur. J. 2014, 20, 9381–9386; b) T. J. Herrington, B. J. Ward, L. R. Doyle, J.
McDermott, A. J. P. White, P. A. Hunt, A. E. Ashley, Chem. Commun. 2014, 50,
12753–12756.
[114] K. Müther, J. Mohr, M. Oestreich, Organometallics 2013, 32, 6643–6646.
Synthesevorschriften für und Charakterisierungen von Substanzen, die im
experimentellen Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.
[115] Intermolekulare Beispiele: a) S. P. Hoffmann, T. Kato, F. S. Tham, C. A. Reed,
Chem. Commun. 2006, 767–769; b) M. Nava, C. A. Reed, Organometallics 2011,
30, 4798–4800; c) S. J. Connelly, W. Kaminsky, D. M. Heinekey, Organometallics
2013, 32, 7478–7481; intramolekulare Beispiele: d) T. Müller, Angew. Chem. Int. Ed.
2001, 40, 3033–3036; e) R. Panisch, M. Bolte, T. Müller, J. Am. Chem. Soc. 2006,
128, 9676–9682; f) A. Y. Khalimon, Z. H. Lin, R. Simionescu, S. F. Vyboishchikov,
G. I. Nikonov, Angew. Chem. Int. Ed. 2007, 46, 4530–4533.
[116] Um zu beweisen, dass Silan LIV als Hydriddonor dient, wurde die
silyliumionvermittelte Reduktion des Imins (E)-50 mit dem deuteriertem Silan 206-d
durchgeführt und das Amin 51-d in 90% Ausbeute mit einem Deuterierungsgrad
>98% erhalten wurde.
[117] J. Mohr, M. Durmaz, E. Irran, M. Oestreich, Organometallics 2014, 33, 1108–1111.
Synthesevorschriften für und Charakterisierungen von Substanzen, die im
experimentellen Teil dieser Arbeit nicht aufgenommen wurden, finden sich dort.

A2 Literaturverzeichnis 175
[118] P. L. Coe, G. M. Pearl, J. C. Tatlow, J. Chem. Soc. C 1971, 604–608. Eine NMR-
spektroskopische Charakterisierung des Bromids 239 ist nicht angegeben.
[119] A. K. Yudin, L. J. P. Martyn, S. Pandiaraju, J. Zheng, A. Lough, Org. Lett. 2000, 2,
41–44.
[120] S. Iimura, W. Wu, Tetrahedron Lett. 2010, 51, 1353–1355.
[121] A. L. S. Thompson, G. W. Kabalka, M. R. Akula, J. W. Huffmann, Synthesis 2005,
547–550.
[122] Für Beispiele von Synthesen elektronenarmer Borane durch Zinn–Bor-
Austauschreaktionen siehe: Lit. 88d; a) M. V. Metz, D. J. Schwartz, C. L. Stern, P.
N. Nickias, T. J. Marks, Angew. Chem. 2000, 112, 1368–1372; Angew. Chem. Int.
Ed. 2000, 39, 1312–1316; b) P. A. Chase, W. E. Piers, B. O. Patrick, J. Am. Chem.
Soc. 2000, 122, 12911–12912; c) H. Li, L. Li, D. J. Schwartz, M. V. Metz, T. J.
Marks, L. Liable-Sands, A. L. Rheingold, J. Am. Chem. Soc. 2005, 127, 14756–
14768; d) J. Hermeke, M. Mewald, E. Irran, M. Oestreich, Organometallics 2014, 33,
5097–5100; für eine mechanistische Betrachtung siehe: e) J. J. Eisch, B. W.
Kotowicz, Eur. J. Inorg. Chem. 1998, 761–769.
[123] A. Krasovskiy, P. Knochel, Angew. Chem. Int. Ed. 2004, 43, 3333–3336.
[124] a) U. Mayer, V. Gutmann, W. Gerger, Monatsh. Chem. 1975, 106, 1235–1257; b) V.
Gutmann Coord. Chem. Rev. 1976, 18, 225–255; c) M. A. Beckett, G. C. Strickland,
J. R. Holland, K. S. Varma, Polymer 1996, 37, 4629–4631; d) M. A. Beckett, D. S.
Brassington, S. J. Coles, M. B. Hursthouse, Inorg. Chem. Commun. 2000, 3, 530–
533; e) M. A. Beckett, D. S. Brassington, M. E. Light, M. B. Hursthouse, J. Chem.
Soc., Dalton Trans. 2001, 1768–1772.
[125] Eine weitere experimentelle Methode zur LEWIS-Aciditätsbestimmung stellt die
CHILDS-Methode dar, bei der Crotonaldehyd als NMR-Sonde dient: a) R. F. Childs,
D. L. Mulholland, A. Nixon, Can. J. Chem. 1982, 60, 801–808; es können
widersprüchliche Resultate bei der Verwendung der GUTMANN–BECKETT- und
CHILDS-Methoden erhalten werden: b) G. J. P. Britovsek, J. Ugolotti, A. J. P. White,
Organometallics 2005, 24, 1685–1691; c) T. J. Herrington, A. J. W. Thom, A. J. P.
White, A. E. Ashley, Dalton Trans. 2012, 41, 9019–9022; für eine Diskussion dieses
Sachverhalts siehe: d) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D.
Masuda, P. Wei, D. W. Stephan, Dalton Trans. 2007, 3407–3414; quantenchemisch
lassen sich LEWIS-Aciditäten für elektronenarme Borane durch Berechnung von
Anionenaffinitäten bestimmen: e) A. Y. Timoshkin, G. Frenking, Organometallics
2008, 27, 371–380; f) L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery,
G. Santiso-Quiñones, V. Brecht, I. Krossing, Angew. Chem. Int. Ed. 2008, 47, 7659–

176 ANHANG
7663; g) H. Böhrer, N. Trapp, D. Himmel, M. Schleep, I. Krossing, Dalton Trans.
2015, 44, 7489–7499.
[126] Eine charakteristische Veränderung der chemischen Verschiebung der Signale im
19F-NMR-Spektrum nach Adduktbildung von Boran 108 wurde nicht beobachtet. Für
entsprechende Beobachtungen in Addukten von B(C6F5)3 (1) siehe: Lit.96a; 124e; A.
D. Horton, J. de With, Organometallics 1997, 5424–5436.
[127] J. M. Blackwell, K. L. Foster, V. H. Beck, W. E. Piers, J. Org. Chem. 1999, 64,
4887–4892.
[128] Eine Übersicht zu LEWIS-Base-Aktivierungen von LEWIS-Säuren bietet: S. E.
Denmark, G. L. Beutner, Angew. Chem. Int. Ed. 2008, 47, 1560–1638.
[129] M. Ullrich, A. J. Lough, D. W. Stephan, J. Am. Chem. Soc. 2009, 131, 52–53.
[130] S. Handa, Y. L. N. Mathota Arachchige, L. M. Slaughter, J. Org. Chem. 2013, 78,
5694–5699.
[131] Y. Uozumi, N. Suzuki, A. Ogiwara, T. Hayashi, Tetrahedron 1994, 50, 4293–4302.
[132] S. Handa, L. M. Slaughter, Angew. Chem. Int. Ed. 2012, 51, 2912‒2915.
[133] M. Murata, T. Oyama, S. Watanabe, Y. Masuda, J. Org. Chem. 2000, 65, 164–168.
[134] a) H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512–7515; b)
G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M.
Stoltz, J. E. Bercaw, K. I. Goldberg, Organometallics 2010, 29, 2176–2179.
[135] R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow, P. Granger,
Pure Appl. Chem. 2001, 73, 1795–1818.
[136] C. Wang, G. Erker, G. Kehr, K. Wedeking, R. Fröhlich, Organometallics 2005, 24,
4760–4773.
[137] M. J. Burk, G. Casy, N. B. Johnson, J. Org. Chem. 1998, 63, 6084–6085.
[138] B. A. Mendelsohn, S. Lee, S. Kim, F. Teyssier, V. S. Aulakh, M. A. Ciufolini, Org.
Lett. 2009, 11, 1539–1542.
[139] Dargestellt gemäß Lit. 138, eine Charakterisierung findet sich in: X. Zhu, Y.-F.
Wang, W. Ren F.-L. Zhang, S. Chiba, Org. Lett. 2013, 15, 3214–3217.
[140] G. R. Newkome, D. L. Fishel, Org. Synth. 1970, 50, 102–104.
[141] G. Zhang, Y. Zhao, H. Ge, Angew. Chem. Int. Ed. 2013, 52, 2559–2563.
[142] J. Mohr, D. Porwal, I. Chatterjee, M. Oestreich, Chem. Eur. J. 2015, 21, 17583–
17586.
[143] T. Imamoto, N. Iwadate, K. Yoshida, Org. Lett. 2006, 8, 2289–2292.
[144] Kristine Müther, Dissertation, Technische Universität Berlin, 2014.
[145] A. S. Tsai, M. E. Tauchert, R. G. Bergman, J. A. Ellman, J. Am. Chem. Soc. 2011,
133, 1248‒1250.
[146] J. L. García Ruano, J. Alemán, M. B. Cid, A. Parra, Org. Lett. 2005, 7, 179‒182.

A2 Literaturverzeichnis 177
[147] M. Ortiz-Marciales, L. D. Rivera, M. De Jesús, S. Espinosa, J. A. Benjamin, O. E.
Casanova, I. G. Figueroa, S. Rodríguez, W. Correa, J. Org. Chem. 2005, 70,
10132–10134.
[148] Die Diastereomerenverhältnisse unabhängiger Reaktionen unter den
Standardbedingungen variierten stark bis hin zur Umkehr der Selektivität.
[149] Racemisches 261 wurde durch Erhitzen von (R)-261 in Diphenylether in einer
Mikrowelle bei 200°C für 12 h und anschließende Aufreinigung mittels präparativer
Dünnschichtchromatographie (Cyclohexan) erhalten.





























