Embed Size (px)
Citation preview

Instrumentelle Arzneistoffanalytik - WS 2016(Wolfgang Holzer)
empfehlenswerte Literatur:
Hesse - Meier - Zeeh; Spektroskopische Methoden in der organischen Chemie, Georg Thieme Verlag, Stuttgart, 9. Auflage, 2016
G. Rücker, M. Neugebauer, G. G. Willems; Instrumentelle pharmazeutische Analytik, Wissenschaftliche Verlagsgesellschaft mbH Stuttgart, 5. Auflage, 2013
A. Dominik, D. Steinhilber; Instrumentelle Analytik, Deutscher Apotheker Verlag, Stuttgart, 2002
J. B. Lambert, S. Gronert, H. F. Shurvell, D. A. Lightner; Spektroskopie - Strukturaufklärung in der organischen Chemie, Pearson, München, 2. Auflage, 2012


Instrumentelle Analytik
●●●● Spektroskopische und optische Methoden
●●●● Elektrochemische Methoden
●●●● Chromatographische Methoden

Pharmazeutische Analytik
●●●● Überprüfung und Charakterisierung von Arzneistoffen,Arzneizubereitungen, Ausgangsmaterialien, Zwischen-produkten mit dem Ziel der ‚Arzneimittelsicherheit‘
Beschreibende Qualitätsmerkmale:
Identität - Reinheit - Gehalt (= Konzentration)
●●●● Arzneistoffsynthese
●●●● Strukturaufklärung
●●●● Pharmakon-Rezeptor Wechselwirkungen
u.a.

Beurteilung (Validierung) analytischer Methoden
●●●● Präzision (Grad der Reproduzierbarkeit)
●●●● Richtigkeit (Abweichung wahrer Wert – Mittelwert)
●●●● Nachweisgrenze (Grenzkonzentration)
●●●● Selektivität/Spezifität (auch in für Bestimmung in einer ‚Matrix‘geeignet?)
●●●● Linearität
●●●● Empfindlichkeit (Anzeigen möglichst geringer Konz.änderungen)
●●●● Bestimmungsbereich
●●●● Robustheit (Störanfälligkeit)
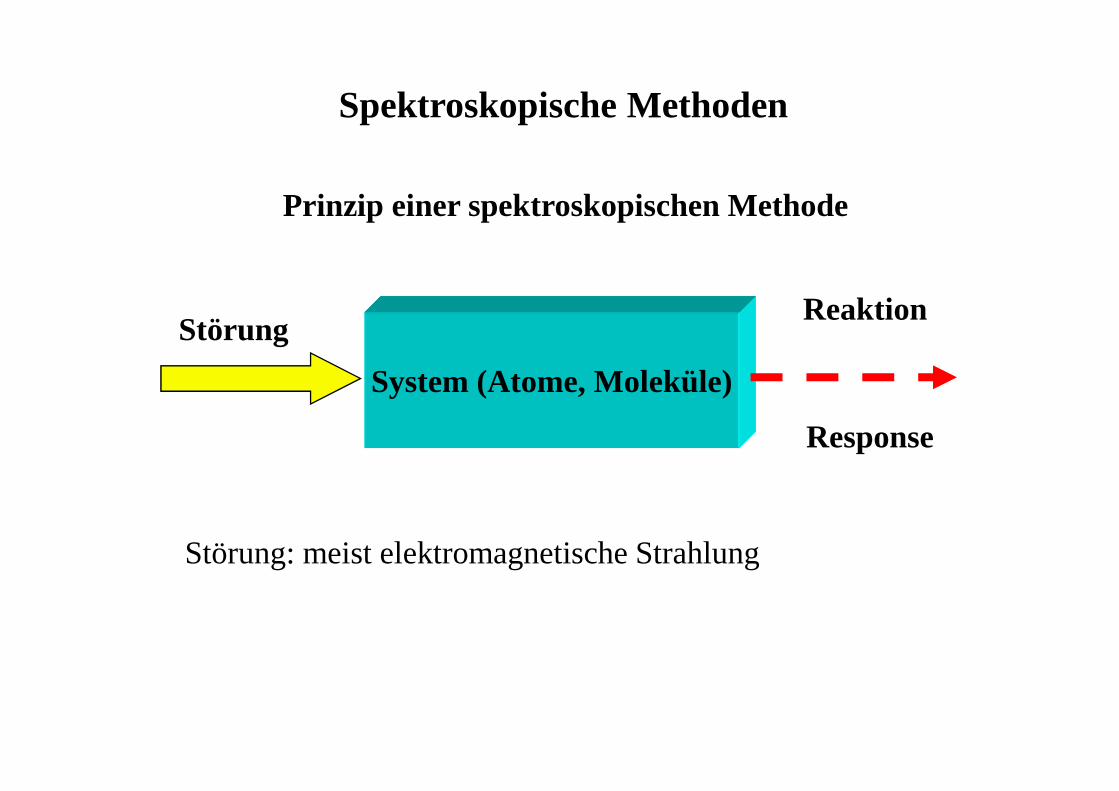
System (Atome, Moleküle)
Störung
Prinzip einer spektroskopischen Methode
Reaktion
Response
Störung: meist elektromagnetische Strahlung
Spektroskopische Methoden
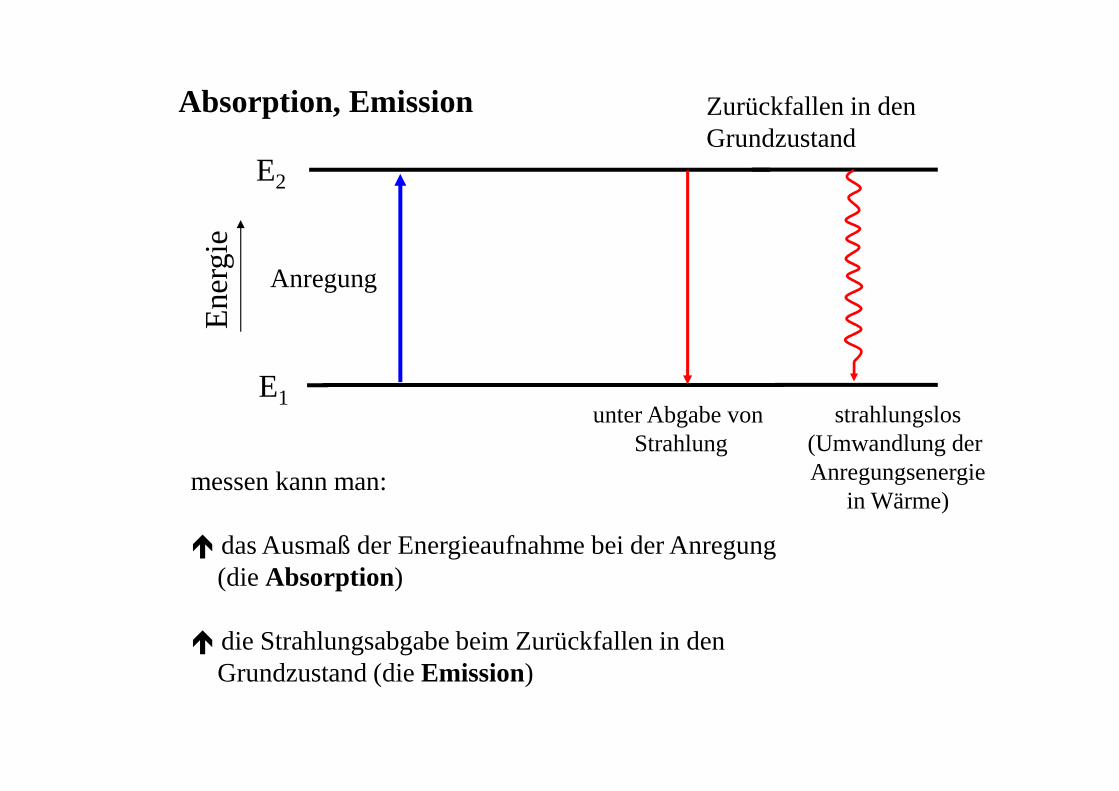
Anregung
E1
E2
Ene
rgie
Zurückfallen in denGrundzustand
unter Abgabe von Strahlung
strahlungslos(Umwandlung der Anregungsenergie
in Wärme)messen kann man:
� das Ausmaß der Energieaufnahme bei der Anregung (die Absorption)
� die Strahlungsabgabe beim Zurückfallen in denGrundzustand (die Emission)
Absorption, Emission

Klassifizierung spektroskopischer Methoden
Absorptionsspektroskopie:
●●●● AAS (Atomabsorptionsspektroskopie, atomic absorption)
●●●● UV/VIS (Ultraviolett-Sichtbar Sp., ultraviolet / visible)
●●●● IR (Infrarot Sp., infrared)
●●●● NMR (Kernresonanzspektroskopie, nuclear magnetic resonance)
Emissionsspektroskopie:
●●●● Fluorimetrie (Fluoreszenzspektroskopie, fluorescence sp.)
●●●● AES (Atomemissionssp. = Flammenphotometrie, atomic emission)
Sonderstellung: MS (Massenspektrometrie, mass spectrometry)
Röntgenstrukturanalyse (X-ray structure analysis)

Zusammenhang zwischen Energie, Wellenlänge und Frequenz
E: Energie (J) h: Planck'sches Wirkungsquantum (6.625•10-34 Js)
νννν: Frequenz (s-1 = Hz (Hertz) = cps (cycles per second))
c: Lichtgeschwindigkeit (2.9979 •108 ms-1)
λλλλ: Wellenlänge (m)
νhE •=λ
cν =
λ
chE
•=
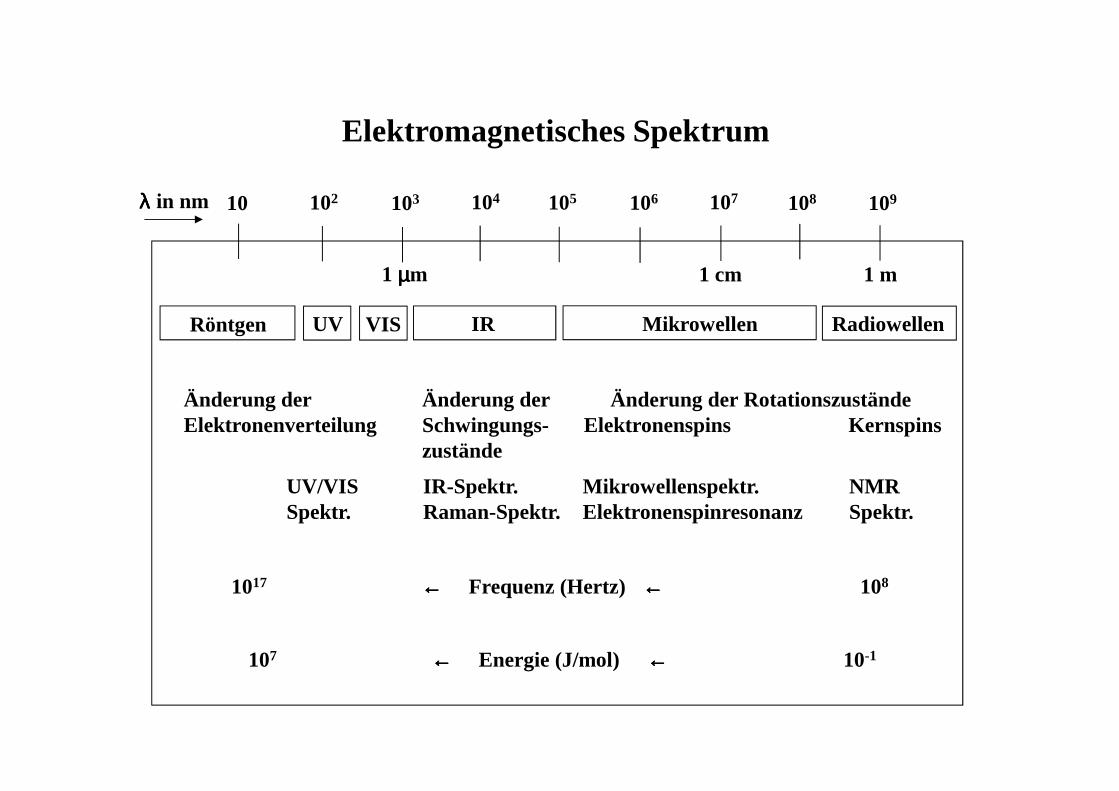
Elektromagnetisches Spektrum
10 102 103 104 105 106 107 108 109λλλλ in nm
1 m1 µµµµm 1 cm
Röntgen UV VIS IR Mikrowellen Radiowellen
Änderung der Elektronenverteilung
Änderung der Schwingungs-zustände
Änderung der Rotationszustände Elektronenspins Kernspins
UV/VISSpektr.
IR-Spektr.Raman-Spektr.
Mikrowellenspektr.Elektronenspinresonanz
NMR Spektr.
1017 ←←←← Frequenz (Hertz) ←←←← 108
107 ←←←← Energie (J/mol) ←←←← 10-1
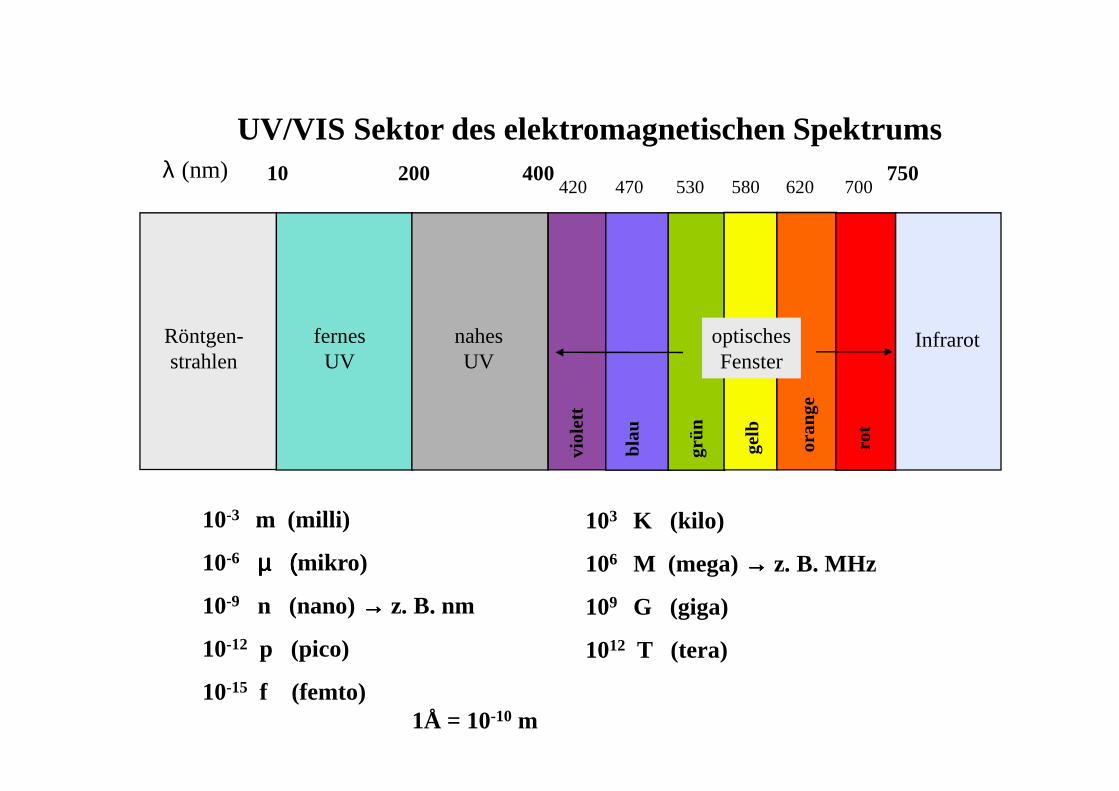
λ (nm) 10 200 400420 470 530 580 620 700
750
UV/VIS Sektor des elektromagnetischen Spektrums
Röntgen-strahlen
fernesUV
nahesUV
optischesFenster
Infrarot
10-3 m (milli)
10-6 µ (µ (µ (µ (mikro)
10-9 n (nano) →→→→ z. B. nm
10-12 p (pico)
10-15 f (femto)1Å = 10-10 m
103 K (kilo)
106 M (mega) →→→→ z. B. MHz
109 G (giga)
1012 T (tera)
viol
ett
blau
grün
gelb
oran
ge
rot
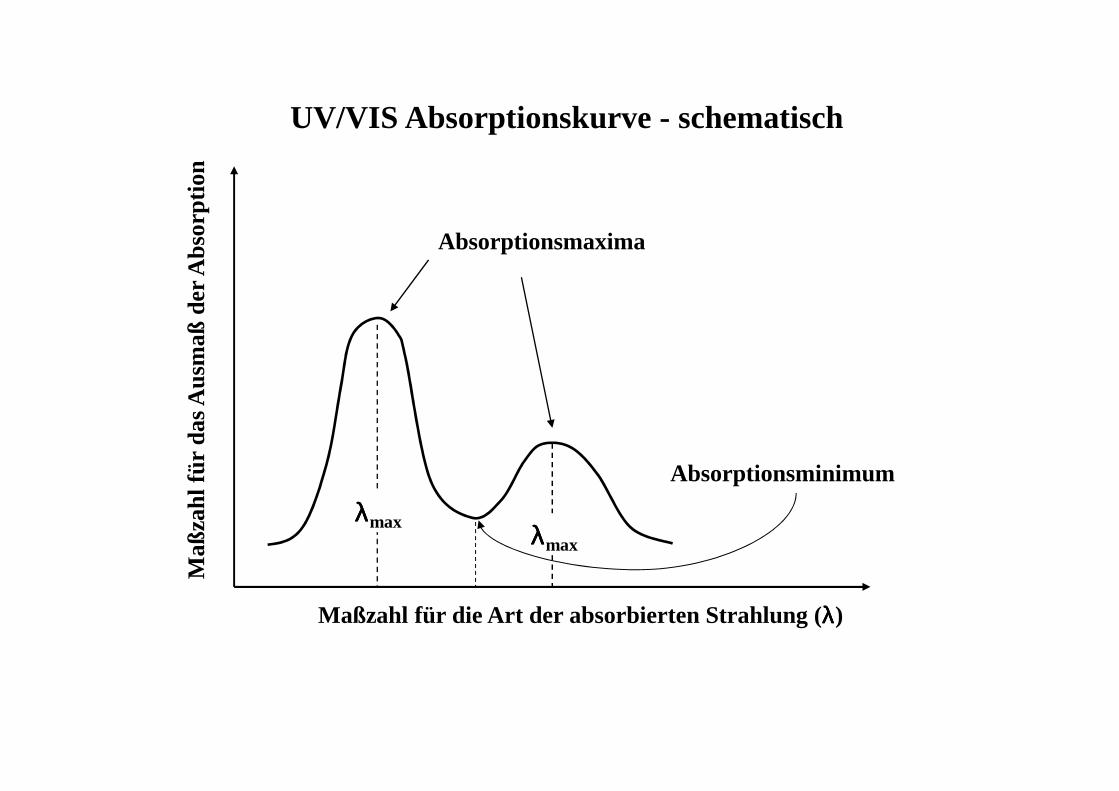
Maß
zahl
für
das
Aus
maß
der
Abs
orpt
ion
Maßzahl für die Art der absorbierten Strahlung (λλλλ)
Absorptionsmaxima
λλλλmax λλλλmax
UV/VIS Absorptionskurve - schematisch
Absorptionsminimum
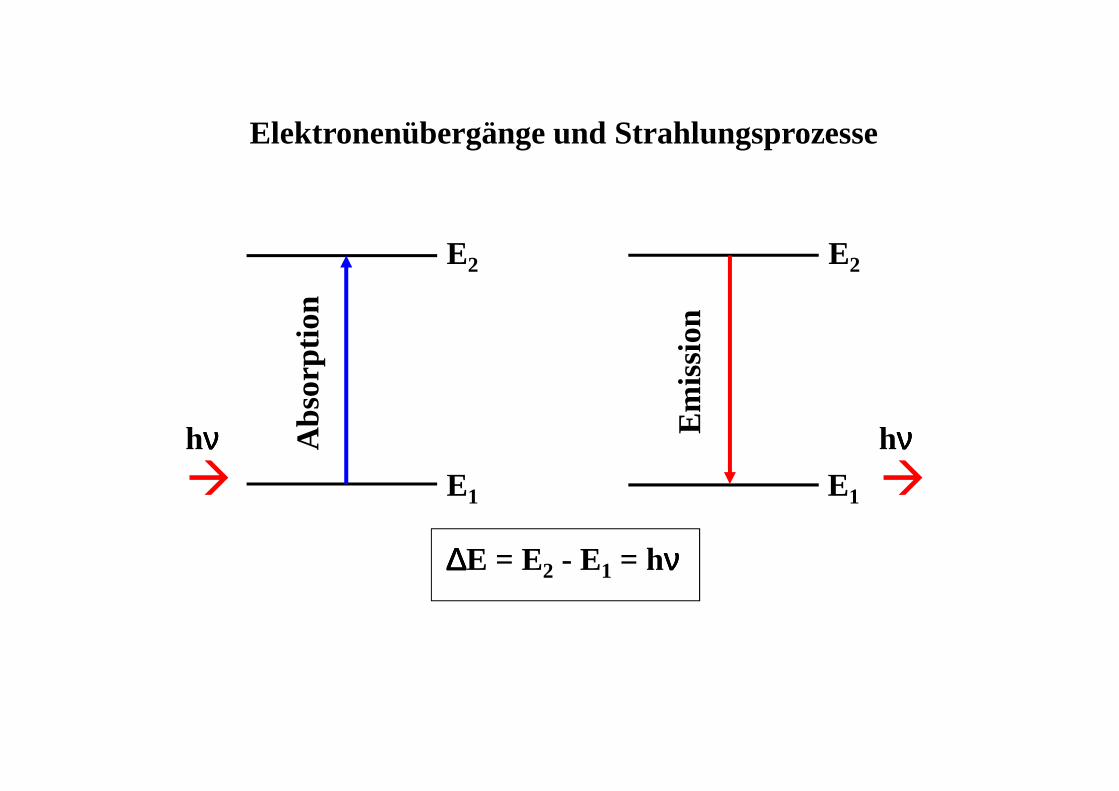
Elektronenübergänge und Strahlungsprozesse
E1
E2
E1
E2
Abs
orpt
ion
Em
issi
on
hνννν hνννν� �
∆∆∆∆E = E2 - E1 = hνννν
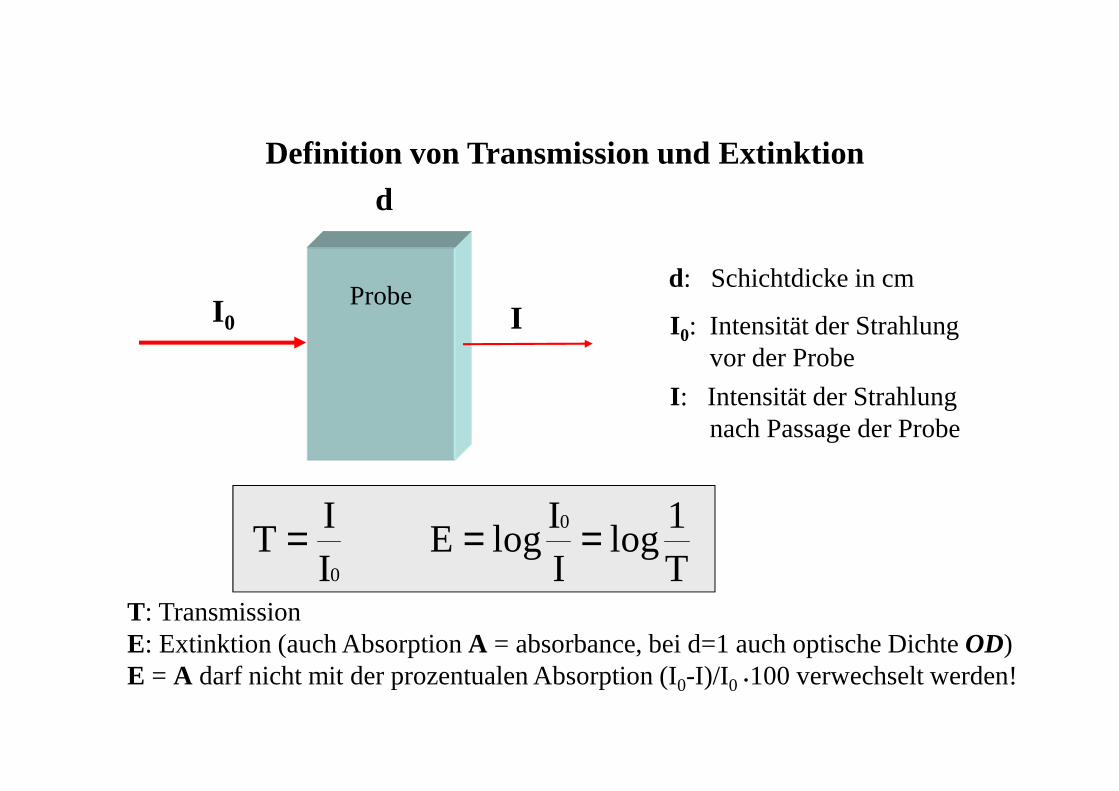
T: TransmissionE: Extinktion (auch Absorption A = absorbance, bei d=1 auch optische Dichte OD)E = A darf nicht mit der prozentualen Absorption (I0-I)/I0 •100 verwechselt werden!
Definition von Transmission und Extinktion
Probe
d
I 0 Id: Schichtdicke in cm
I 0: Intensität der Strahlung vor der Probe
I : Intensität der Strahlung nach Passage der Probe
0II
T =T1
logII
logE0 ==
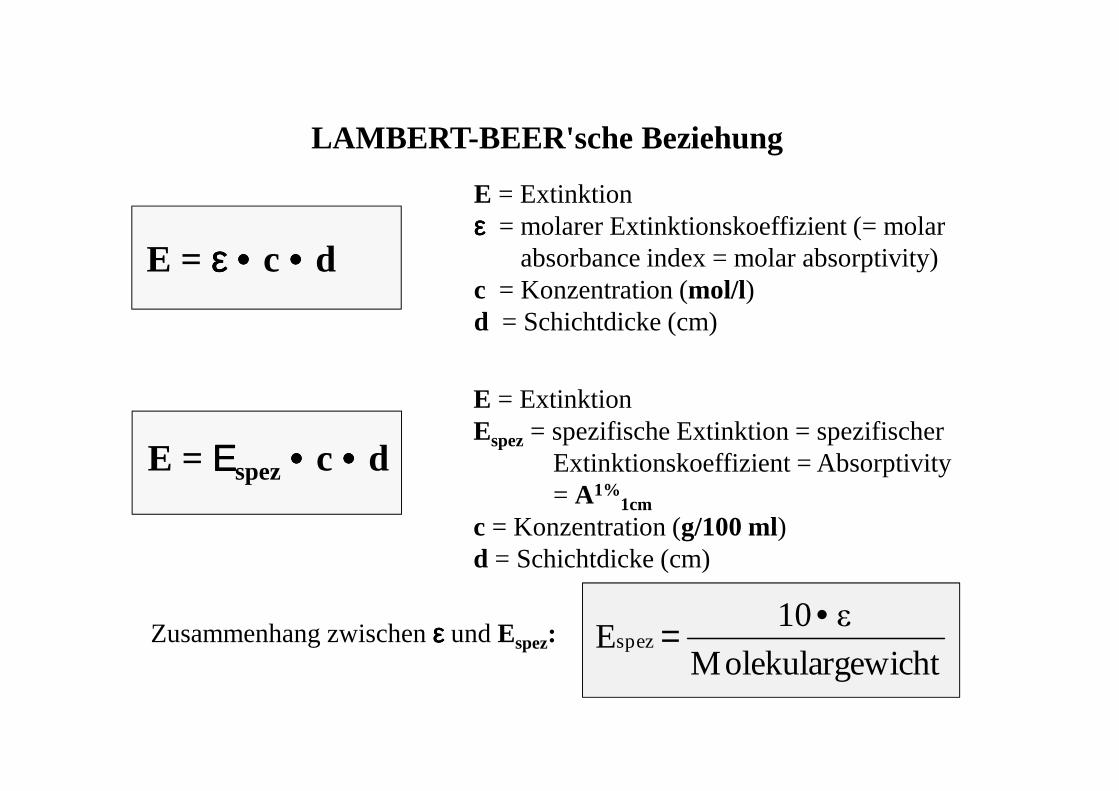
LAMBERT-BEER'sche Beziehung
E = εεεε •••• c •••• d
E = Extinktionεεεε = molarer Extinktionskoeffizient (= molar
absorbance index = molar absorptivity)c = Konzentration (mol/l)d = Schichtdicke (cm)
E = ΕΕΕΕspez•••• c •••• d
E = ExtinktionEspez= spezifische Extinktion = spezifischer
Extinktionskoeffizient = Absorptivity = A1%
1cmc = Konzentration (g/100 ml)d = Schichtdicke (cm)
Zusammenhang zwischen εεεε und Espez:ewichtMolekularg
ε10Espez
•=

Lambert – Beer:
• Gilt streng nur für monochromatisches Licht
• Gilt streng nur für verdünnte Lösungen (c ≤ 10-2 mol/l) – bei höheren Konzentrationen erfolgt Abweichung von der Linearität
• Lösung muss klar sein → keine Suspensionen oder Kolloide, die Schwächung der Strahlung muss ausschließlich durch Absorption verursacht sein
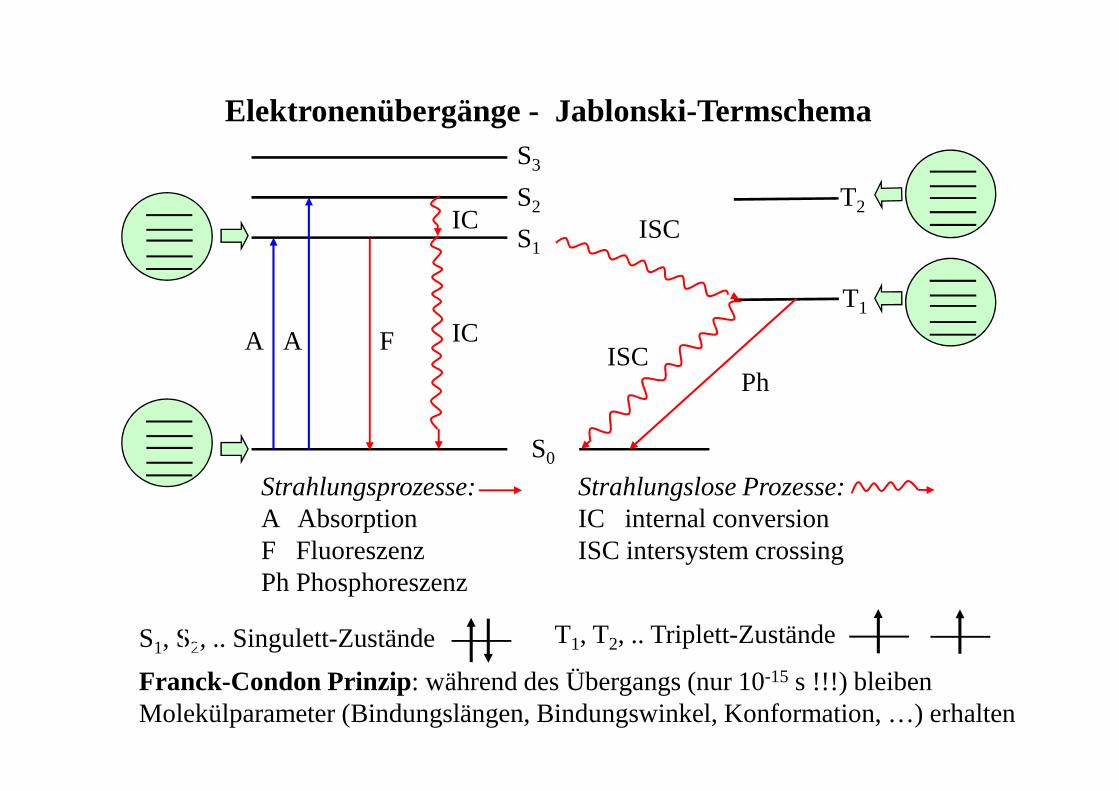
Elektronenübergänge - Jablonski-Termschema
Strahlungsprozesse:A AbsorptionF FluoreszenzPh Phosphoreszenz
Strahlungslose Prozesse:IC internal conversionISC intersystem crossing
S0
S1
S2
S3
T1
T2
A A F
PhISC
ISC
IC
IC
S1, S2, .. Singulett-Zustände T1, T2, .. Triplett-Zustände
Franck-Condon Prinzip: während des Übergangs (nur 10-15 s !!!) bleiben Molekülparameter (Bindungslängen, Bindungswinkel, Konformation, …) erhalten
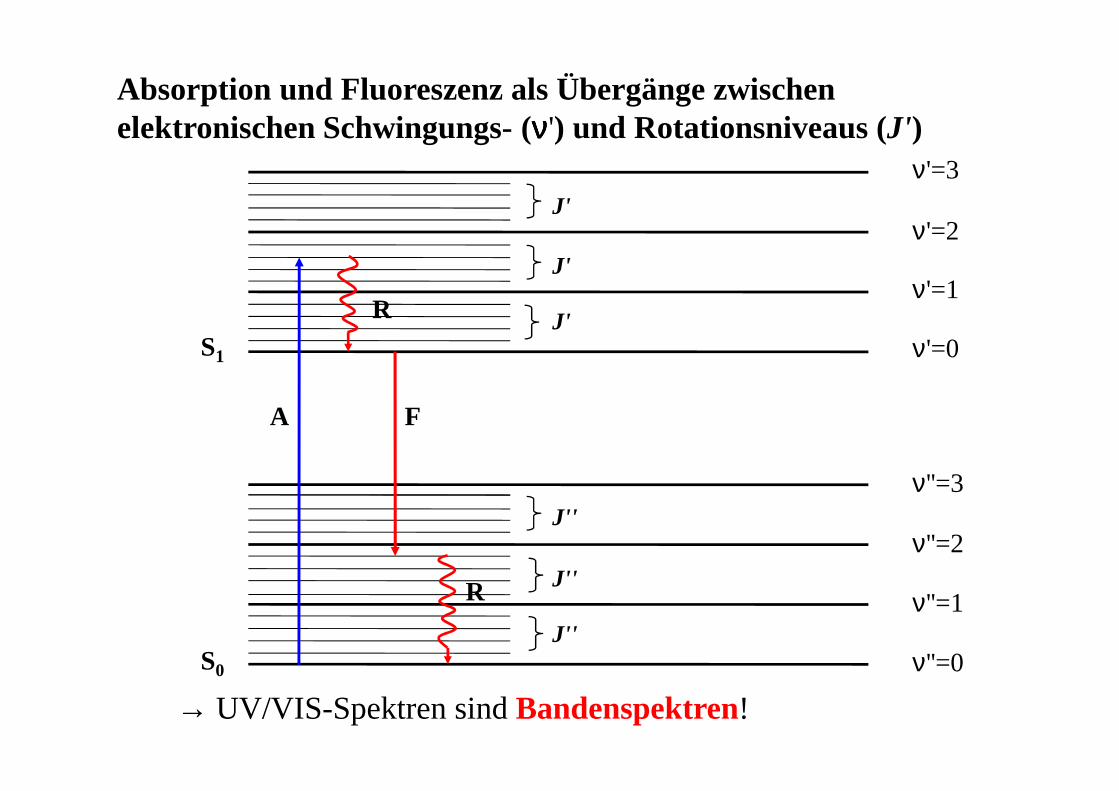
Absorption und Fluoreszenz als Übergänge zwischen elektronischen Schwingungs- (νννν') und Rotationsniveaus (J')
S0
S1
ν''=0
ν''=1
ν''=2
ν''=3
ν'=0
ν'=1
ν'=2
ν'=3
J'
J'
J'
J''
J''
J''
A F
R
R
→ UV/VIS-Spektren sind Bandenspektren!

Molekülorbitale•••• wie in Atomen befinden sich in Molekülen die Elektronen auf genau
festgelegten Bahnen (Molekülorbitale, MO)
•••• core-Orbitale (innere e-): sind für UV/VIS nicht interessant (E zu hoch), bei UV/VIS werden Bindungselektronen angeregt
•••• σσσσ-Orbitale: enthalten e- der Einfachbindungen zwischen den Atomen, durch Überlappung von Atomorbitalen entlang der Bindungsachse
•••• σσσσ∗-Orbitale: antibindendes σσσσ-Orbital, hohe Energie
•••• ππππ-Orbitale: enthalten e- der Doppel- und Mehrfachbindungen zwischenden Atomen, durch Überlappung von Atomorbitalen senkrecht zurBindungsachse; ππππ∗-Orbitale: antibindende ππππ-Orbitale
•••• n-Orbitale: enthalten e- der freien, nicht bindenden Elektronenpaare(lone-pairs), z.B. O, N, S Atome
•••• HOMO: h ighest occupied molecular orbital LUMO: l owestunoccupiedmolecularorbitalHOMO-LUMO Übergang ist energieärmster (= längstwelliger) Übergang
•••• Quantenmechanische Auswahlregeln legen fest, ob ein Übergang 'erlaubt'(hohe Wahrscheinlichkeit) oder 'verboten' (niedrige Wahrscheinlichkeit) ist

Quantenmechanische Auswahlregeln
Die meisten der prinzipiell möglichen Elektronenanregungen finden nicht statt → ‚verbotene Übergänge‘ welche Übergänge verboten sind legen die Auswahlregeln fest
Spin-Verbot: Multiplizität darf sich während der Anregung nicht ändern, d. h. nur S → S bzw T → T möglich, nicht aber S → T oder T → S
Überlappungsverbot: bei der Anregung eines Elektrons von einem Orbital in ein anderes müssen sich die betreffenden Orbitale überlappen, sind sie zu weit voneinander entfernt kann der Elektronen-Transfer nicht erfolgen
Paritätsverbot (Regel von Laporte): bei Molekülen mit Punktsymmetrie (z.B. Benzol, Ethen) sind nur Übergänge zwischen solchen Orbitalen möglich, die unterschiedliche Symmetrie-Eigenschaften bzgl. des Symmetriezentrums aufweisen
Symmetrieverbot: bei nicht punktsymmetrischen Molekülen gilt die Regel von Laporte zwar nicht, aber auch hier aufgrund von Symmetrie-Eigenschaften Verbote möglich
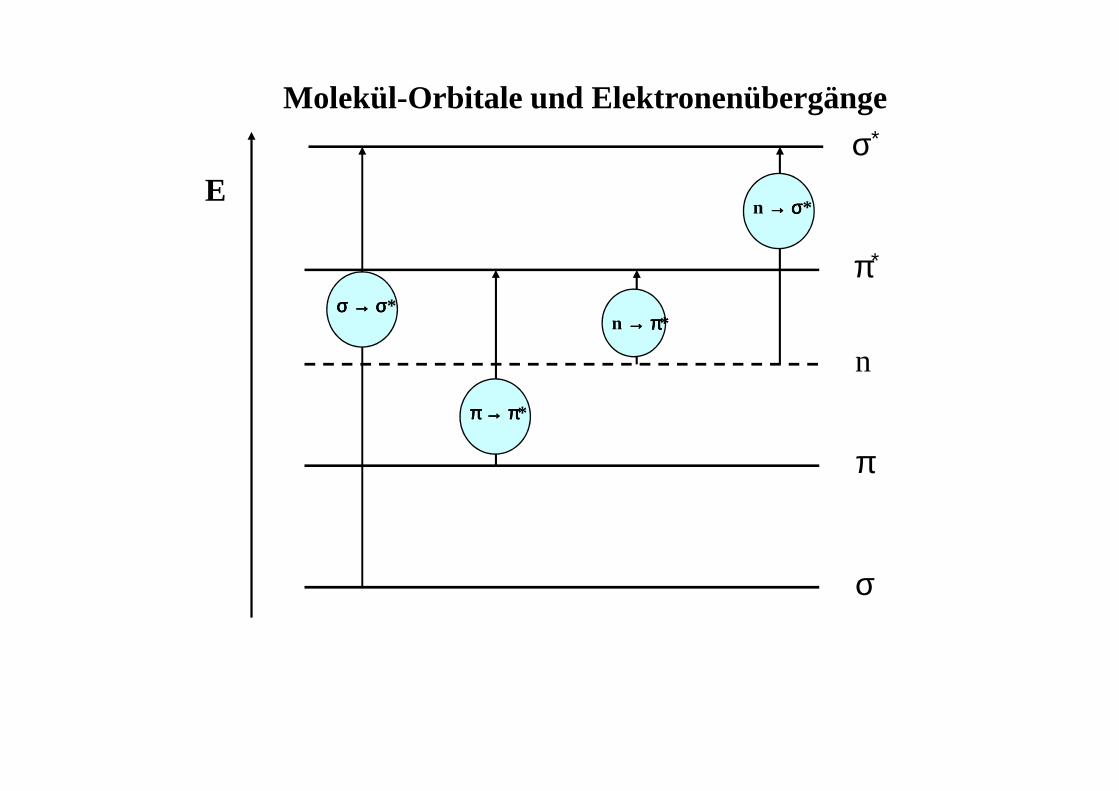
Molekül-Orbitale und Elektronenübergänge
E
σ
π
n
π∗
σ∗
σ σ σ σ →→→→ σσσσ*
n →→→→ σσσσ*
π π π π →→→→ ππππ*
n →→→→ ππππ*
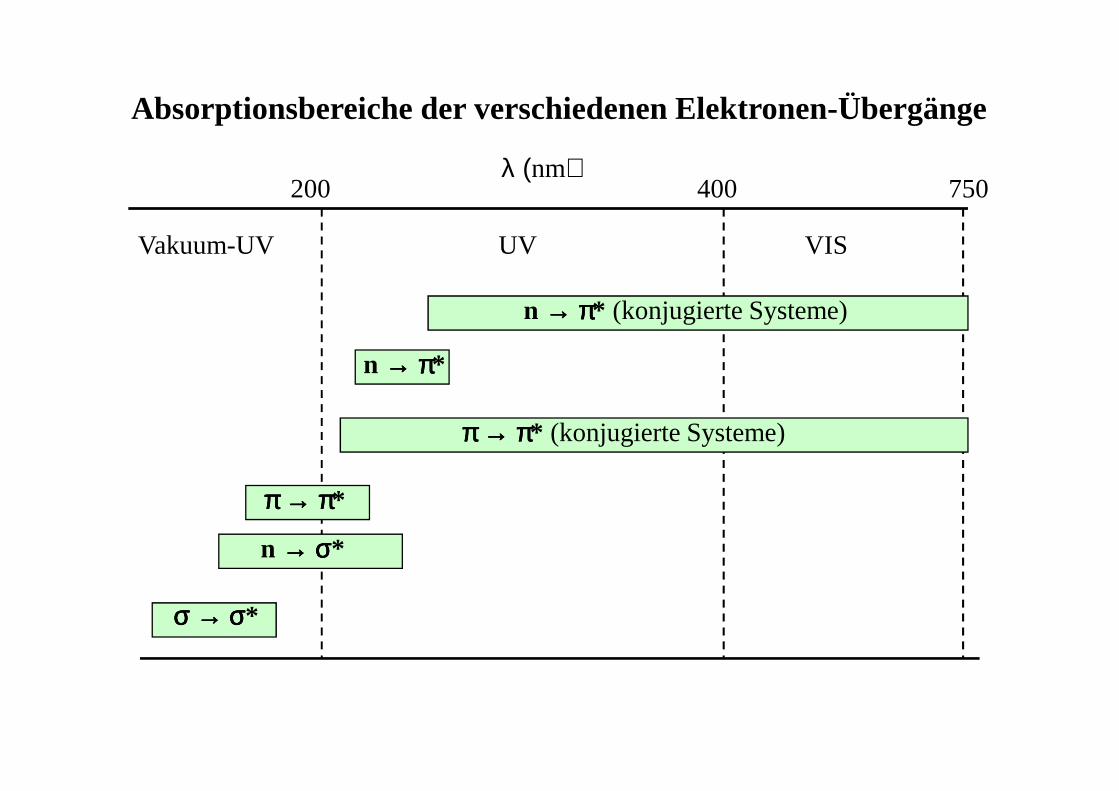
λ (nm)
Absorptionsbereiche der verschiedenen Elektronen-Übergänge
Vakuum-UV UV VIS
σσσσ →→→→ σσσσ*
n →→→→ σσσσ*
ππππ →→→→ ππππ*
ππππ →→→→ ππππ* (konjugierte Systeme)
n →→→→ ππππ*
n →→→→ ππππ* (konjugierte Systeme)
200 400 750

Absorption einfacher, nicht-konjugierter Chromophore
ChromophorBezeichnungdes Überganges λmax (nm)
σ-Elektronen
einsame Elektronenpaare (lone-pairs, non-bonding)
C C und C H
O
N
S
C O
C C (isoliert)
π-Elektronen
C O
σ → σ*
n → σ*
n → σ*
n → σ*
n → σ*
n → π*
π → π*
≤ 150
~ 185~ 195
~ 195
~ 300
~ 190
~ 190
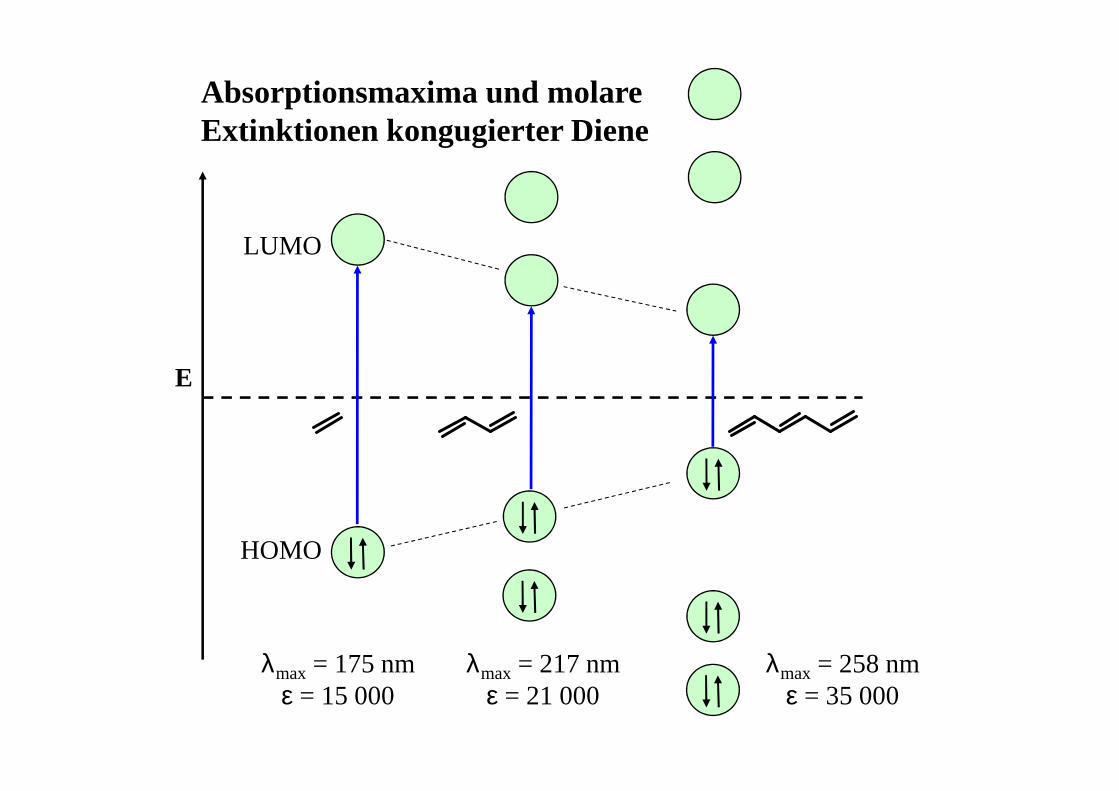
E
HOMO
LUMO
λmax = 175 nmε = 15 000
λmax = 217 nmε = 21 000
λmax = 258 nmε = 35 000
Absorptionsmaxima und molare Extinktionen kongugierter Diene
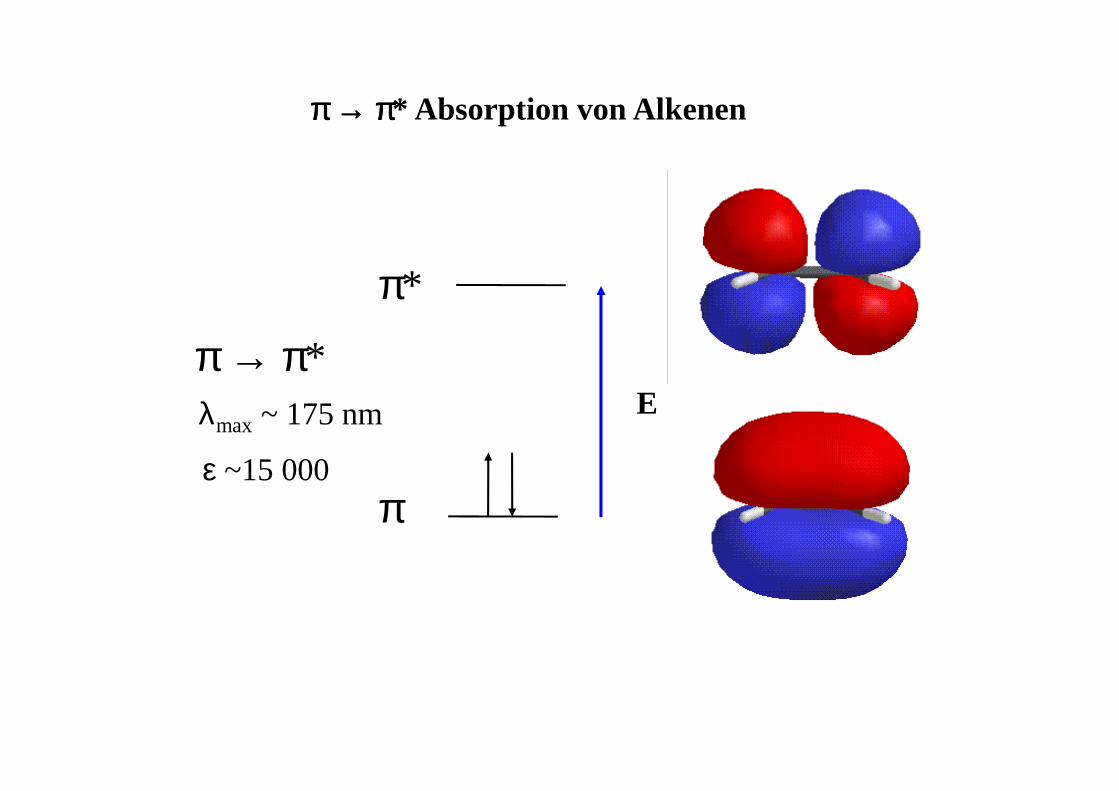
π
π*
λmax ~ 175 nm
ε ~15 000
E
ππππ →→→→ ππππ* Absorption von Alkenen
π → π*
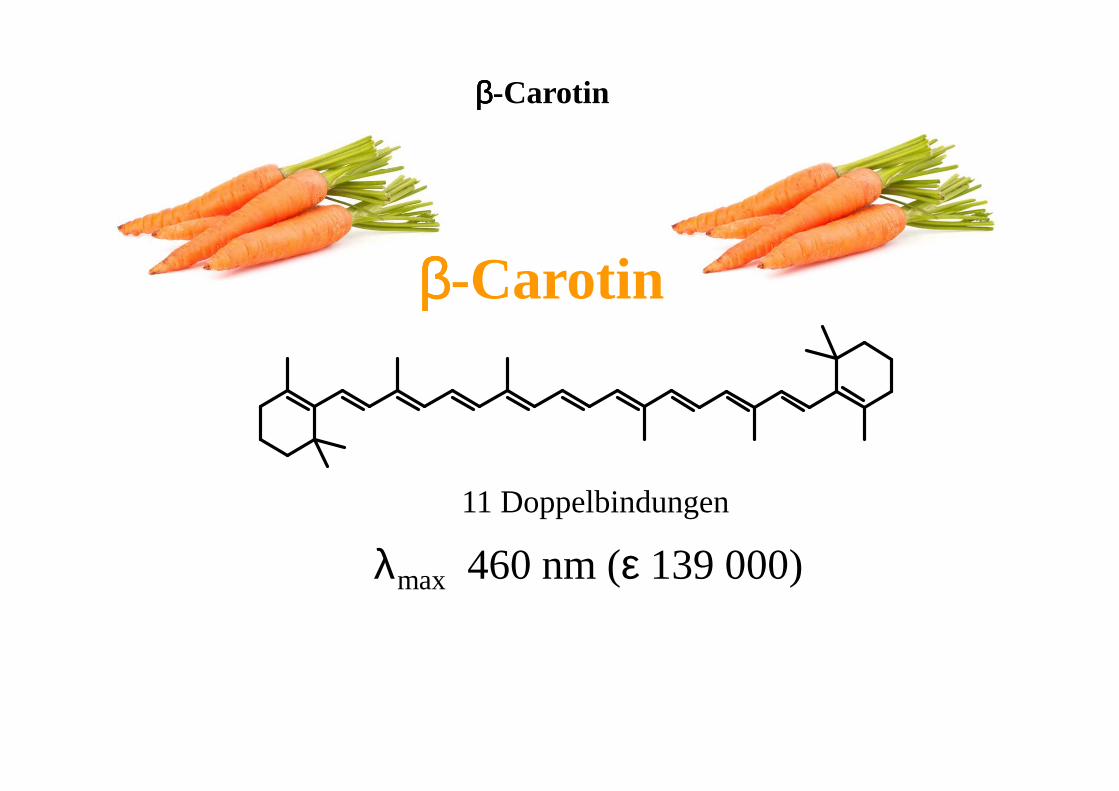
ββββ-Carotin
11 Doppelbindungen
λmax 460 nm (ε 139 000)
ββββ-Carotin
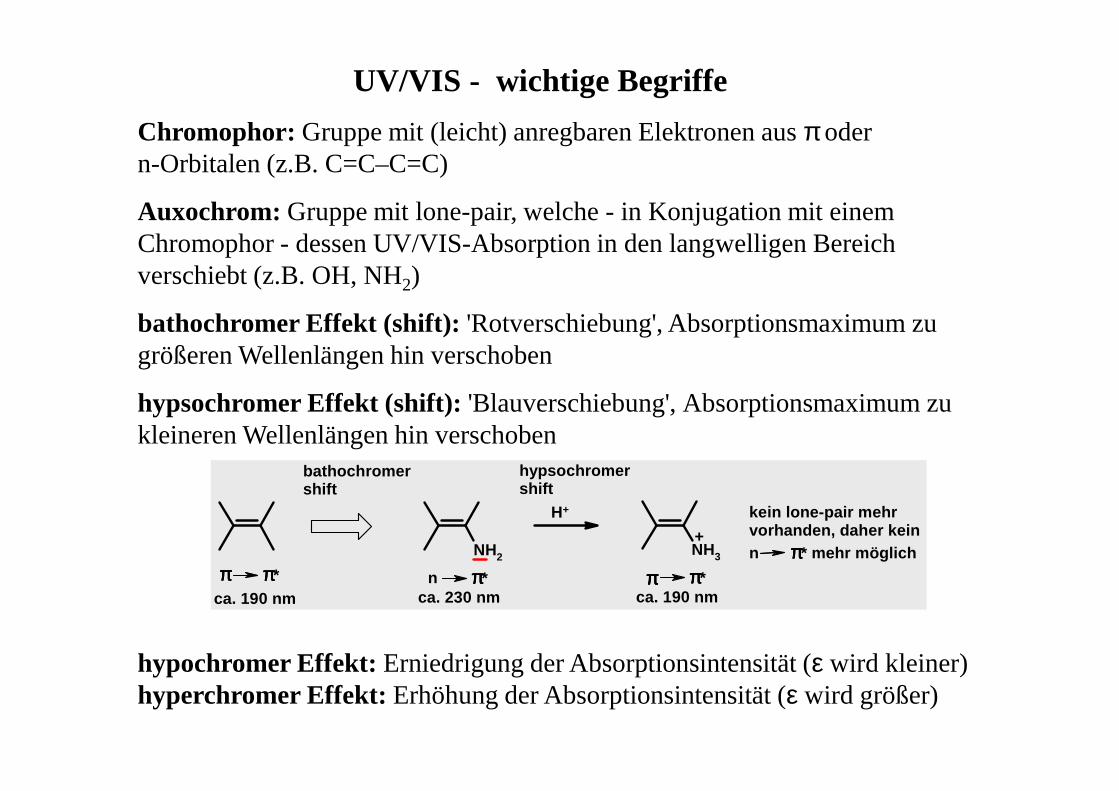
UV/VIS - wichtige Begriffe
Chromophor: Gruppe mit (leicht) anregbaren Elektronen aus π oder n-Orbitalen (z.B. C=C–C=C)
Auxochrom: Gruppe mit lone-pair, welche - in Konjugation mit einem Chromophor - dessen UV/VIS-Absorption in den langwelligen Bereich verschiebt (z.B. OH, NH2)
bathochromer Effekt (shift): 'Rotverschiebung', Absorptionsmaximum zu größeren Wellenlängen hin verschoben
hypsochromer Effekt (shift): 'Blauverschiebung', Absorptionsmaximum zu kleineren Wellenlängen hin verschoben
NH2
ππππ ππππ* n ππππ*ca. 190 nm ca. 230 nm
H+
NH3
ππππ ππππ*ca. 190 nm
+
bathochromershift
hypsochromershift
kein lone-pair mehr vorhanden, daher kein n ππππ* mehr möglich
hypochromer Effekt: Erniedrigung der Absorptionsintensität (ε wird kleiner) hyperchromer Effekt: Erhöhung der Absorptionsintensität (ε wird größer)
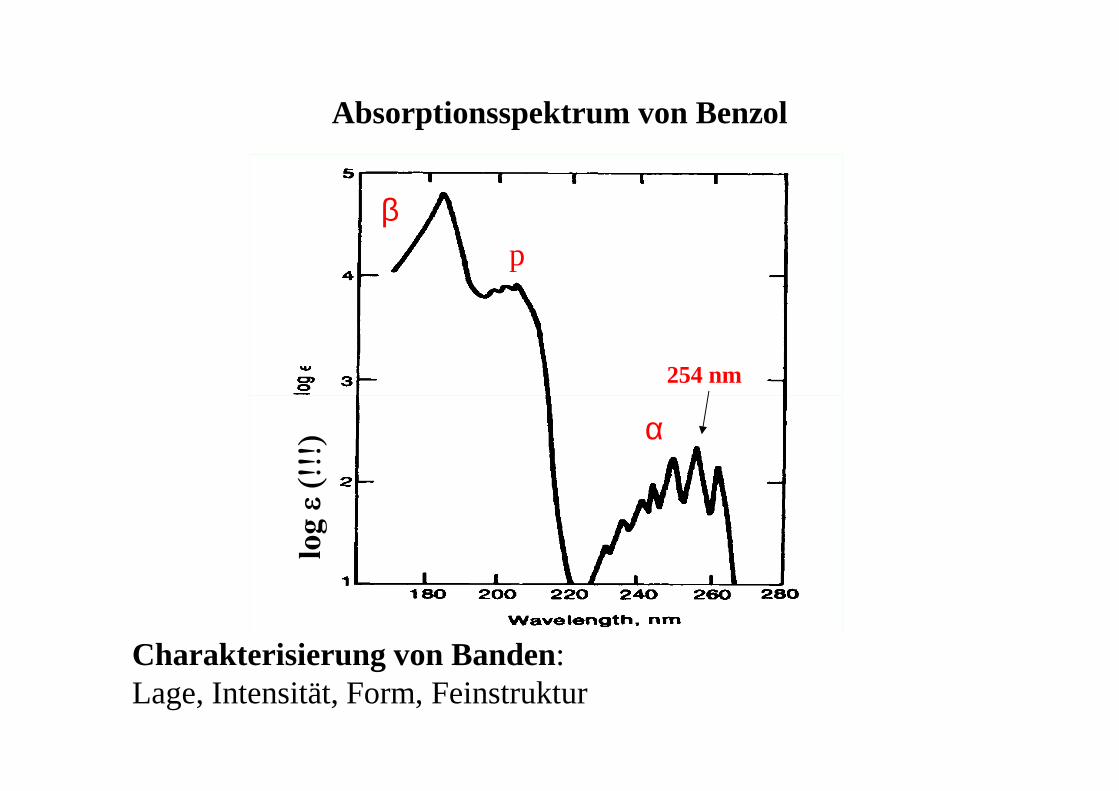
Absorptionsspektrum von Benzol
254 nm
βp
α
Charakterisierung von Banden: Lage, Intensität, Form, Feinstruktur
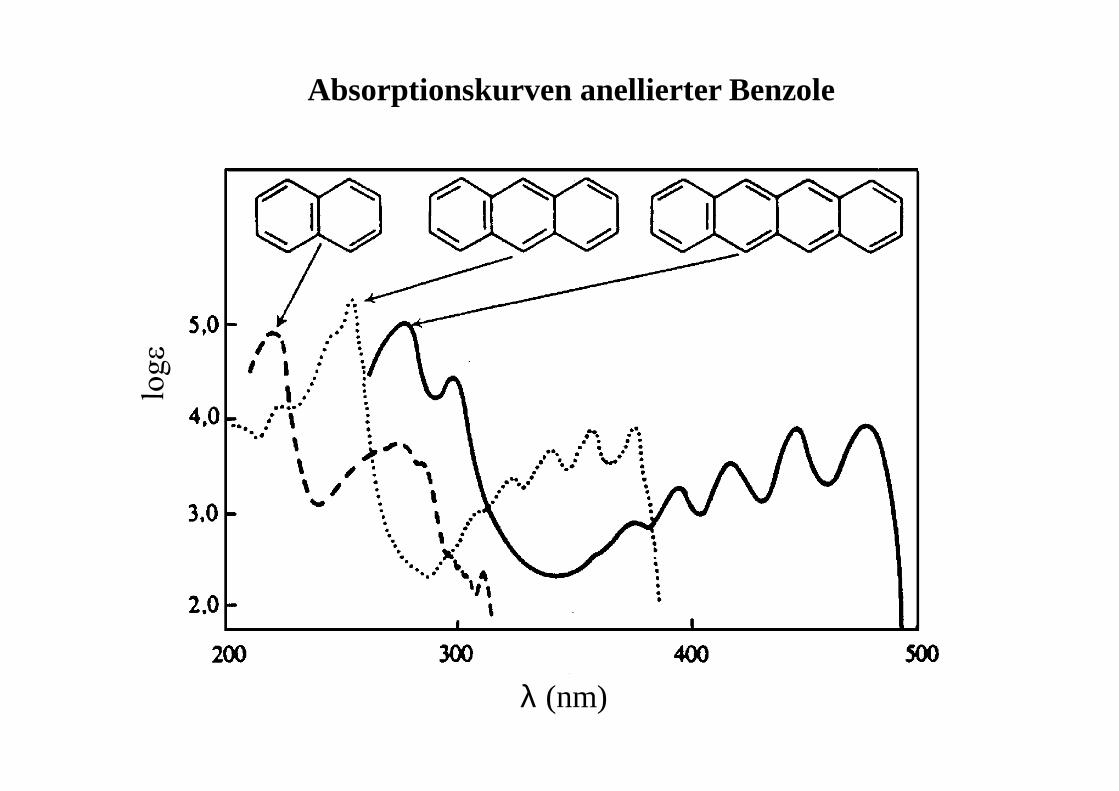
λ (nm)
Absorptionskurven anellierter Benzole
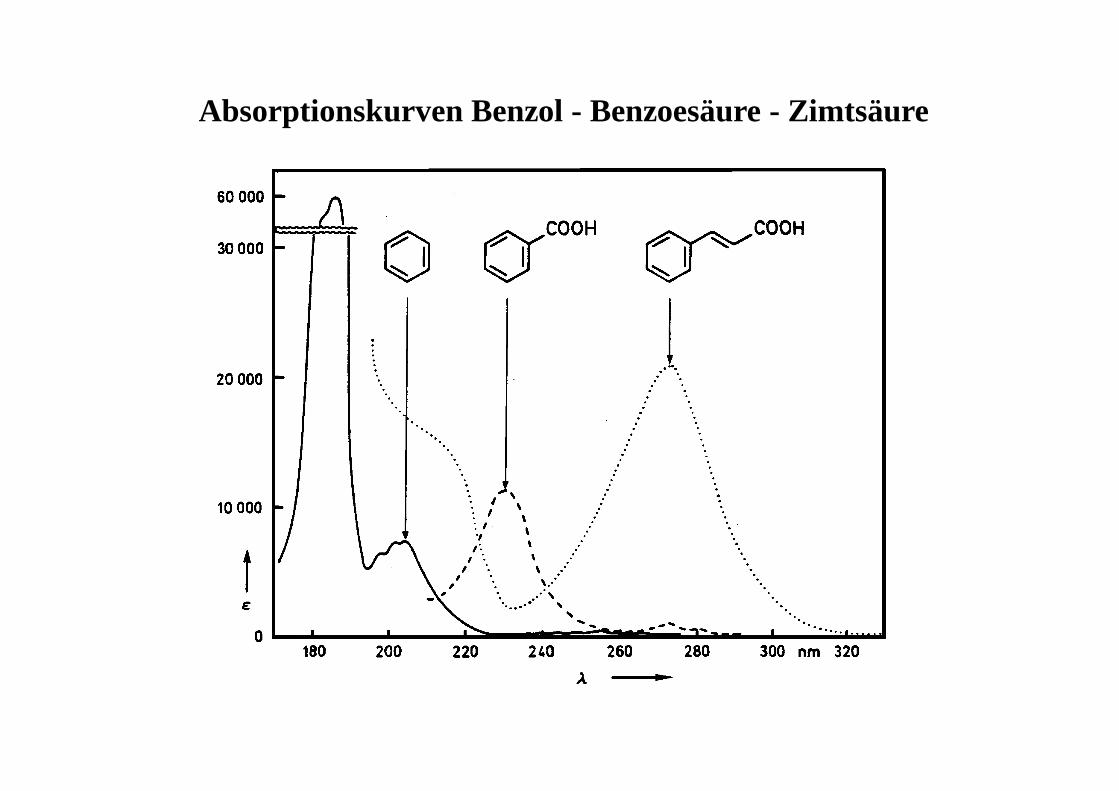
Absorptionskurven Benzol - Benzoesäure - Zimtsäure
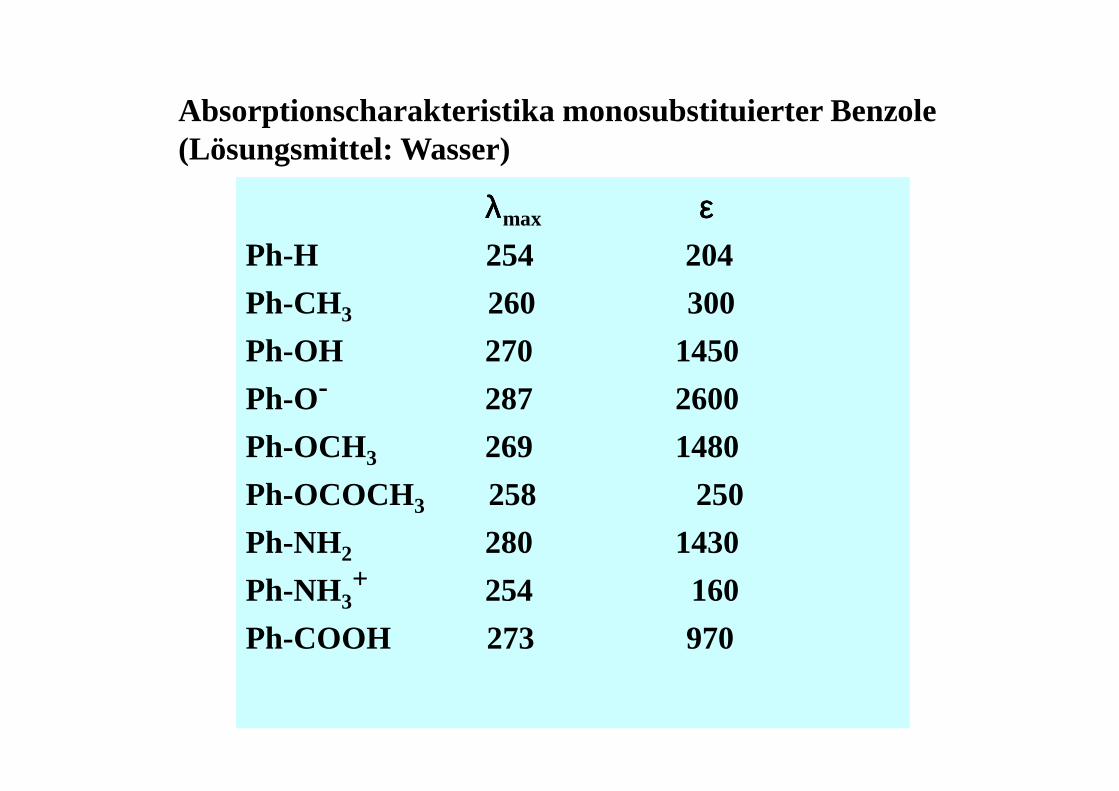
Absorptionscharakteristika monosubstituierter Benzole(Lösungsmittel: Wasser)
λλλλmax εεεεPh-H 254 204
Ph-CH3 260 300
Ph-OH 270 1450
Ph-O- 287 2600
Ph-OCH3 269 1480
Ph-OCOCH3 258 250
Ph-NH2 280 1430
Ph-NH3+ 254 160
Ph-COOH 273 970
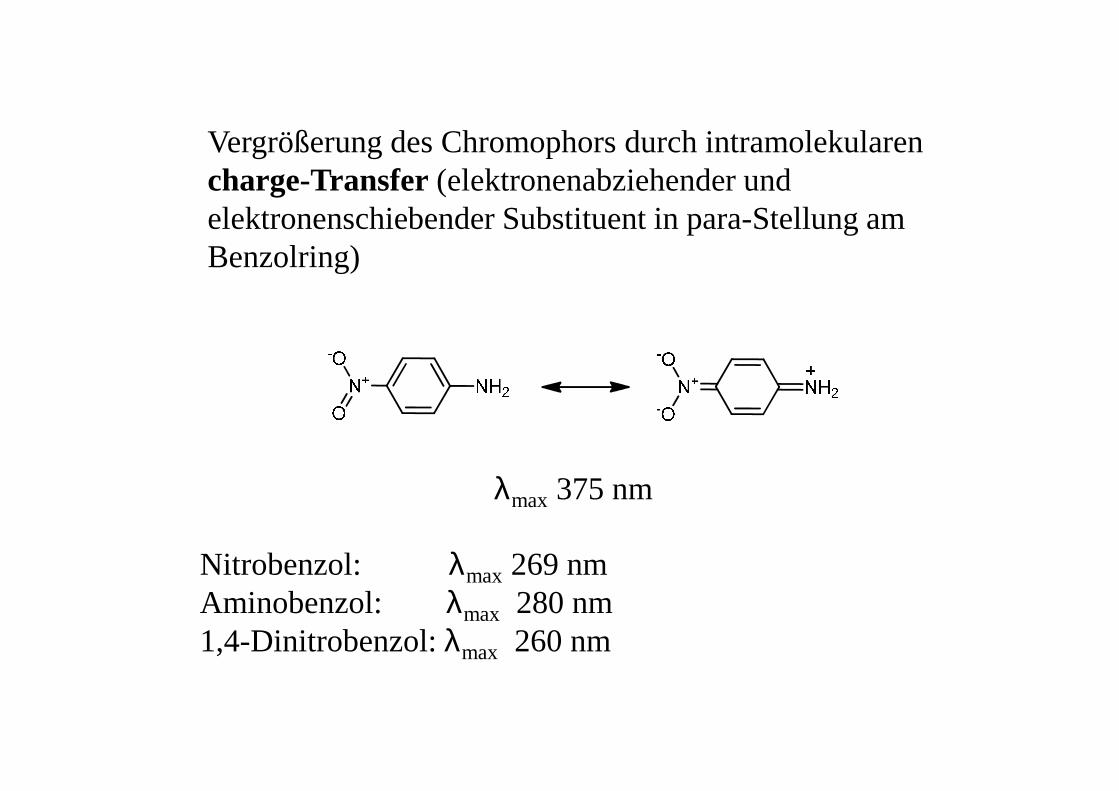
Vergrößerung des Chromophors durch intramolekularen charge-Transfer (elektronenabziehender und elektronenschiebender Substituent in para-Stellung am Benzolring)
λmax 375 nm
Nitrobenzol: λmax 269 nmAminobenzol: λmax 280 nm1,4-Dinitrobenzol: λmax 260 nm
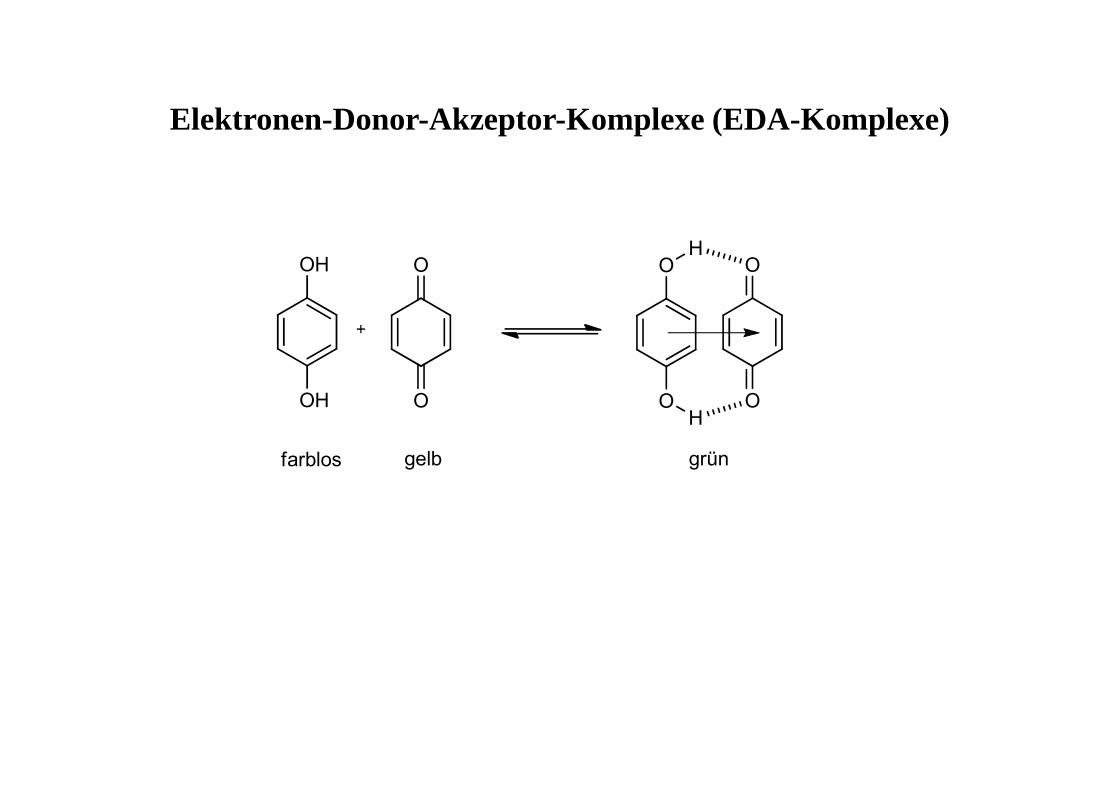
Elektronen-Donor-Akzeptor-Komplexe (EDA-Komplexe)
OH
OH
O
O
O
O
O
O
H
H
farblos gelb grün
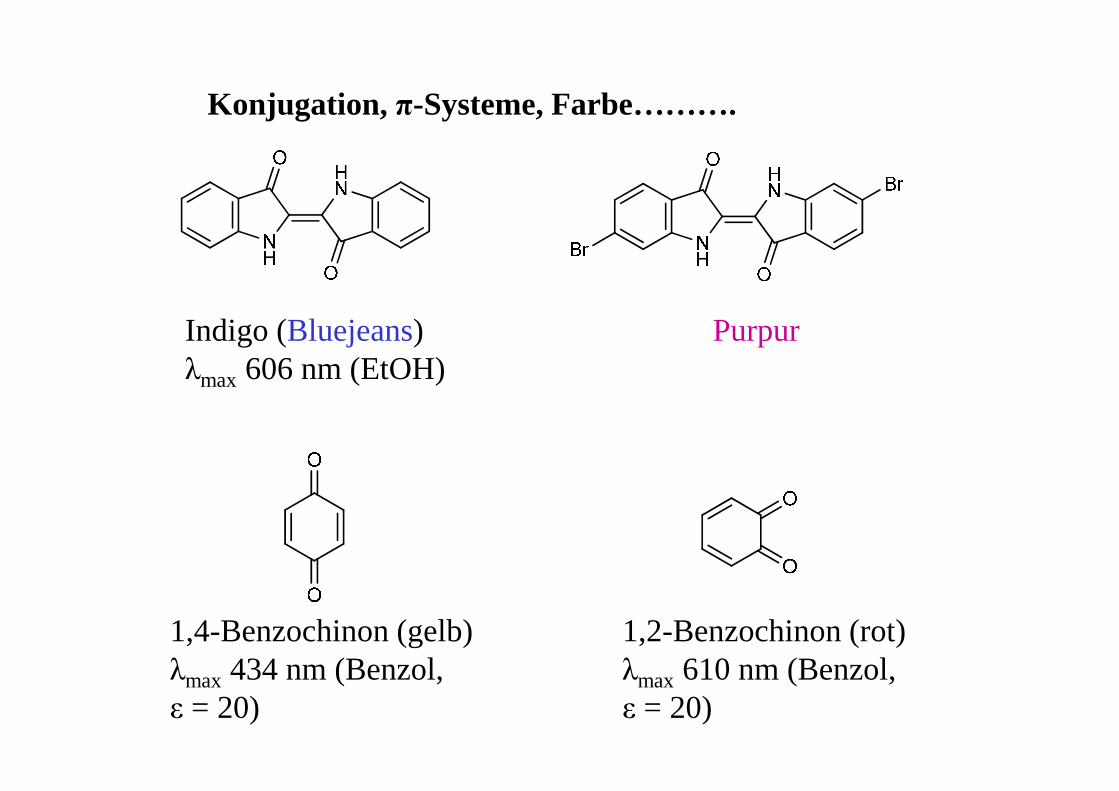
Konjugation, π-Systeme, Farbe……….
Indigo (Bluejeans)λmax 606 nm (EtOH)
Purpur
1,4-Benzochinon (gelb)λmax 434 nm (Benzol, ε = 20)
1,2-Benzochinon (rot) λmax 610 nm (Benzol, ε = 20)
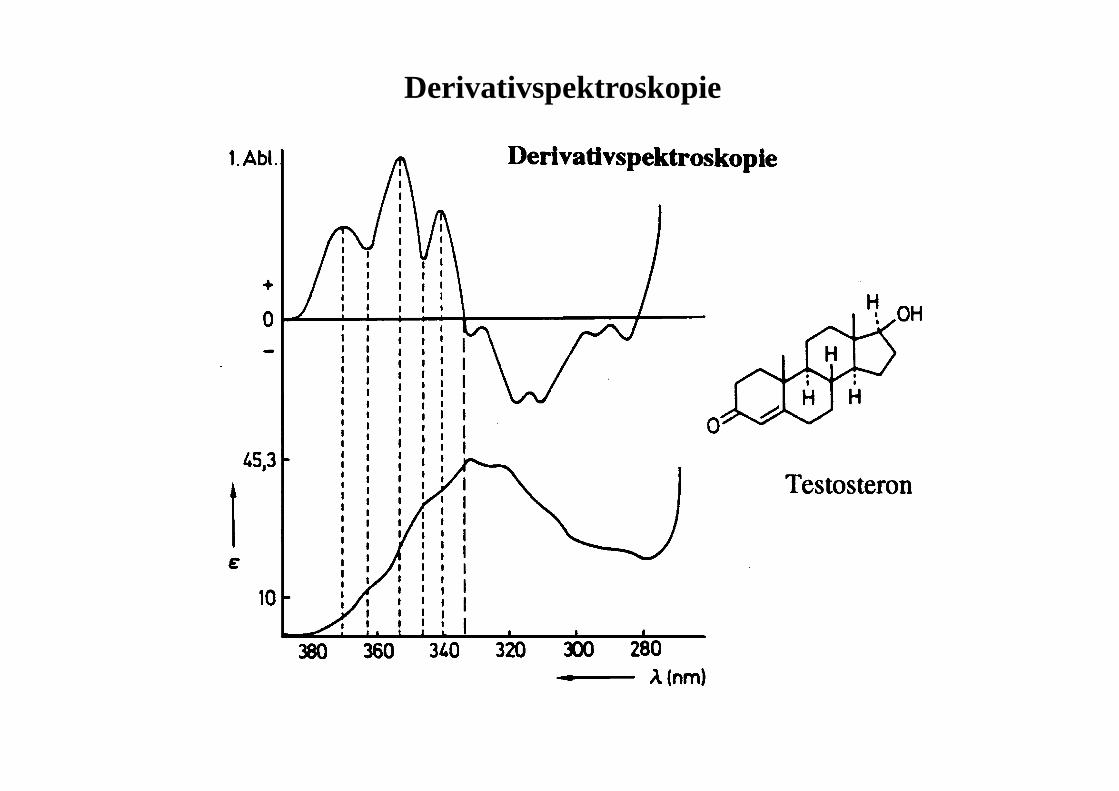
Derivativspektroskopie
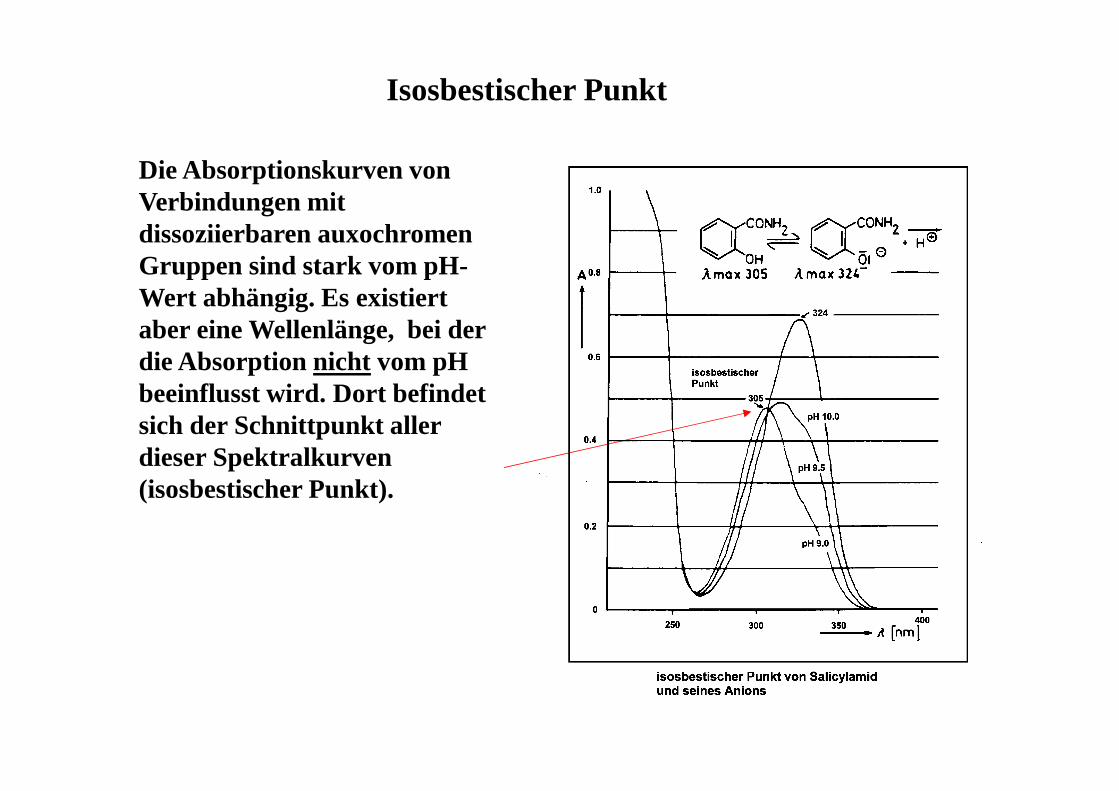
Isosbestischer Punkt
Die Absorptionskurven von Verbindungen mit dissoziierbaren auxochromen Gruppen sind stark vom pH-Wert abhängig. Es existiert aber eine Wellenlänge, bei der die Absorption nicht vom pH beeinflusst wird. Dort befindet sich der Schnittpunkt aller dieser Spektralkurven (isosbestischer Punkt).
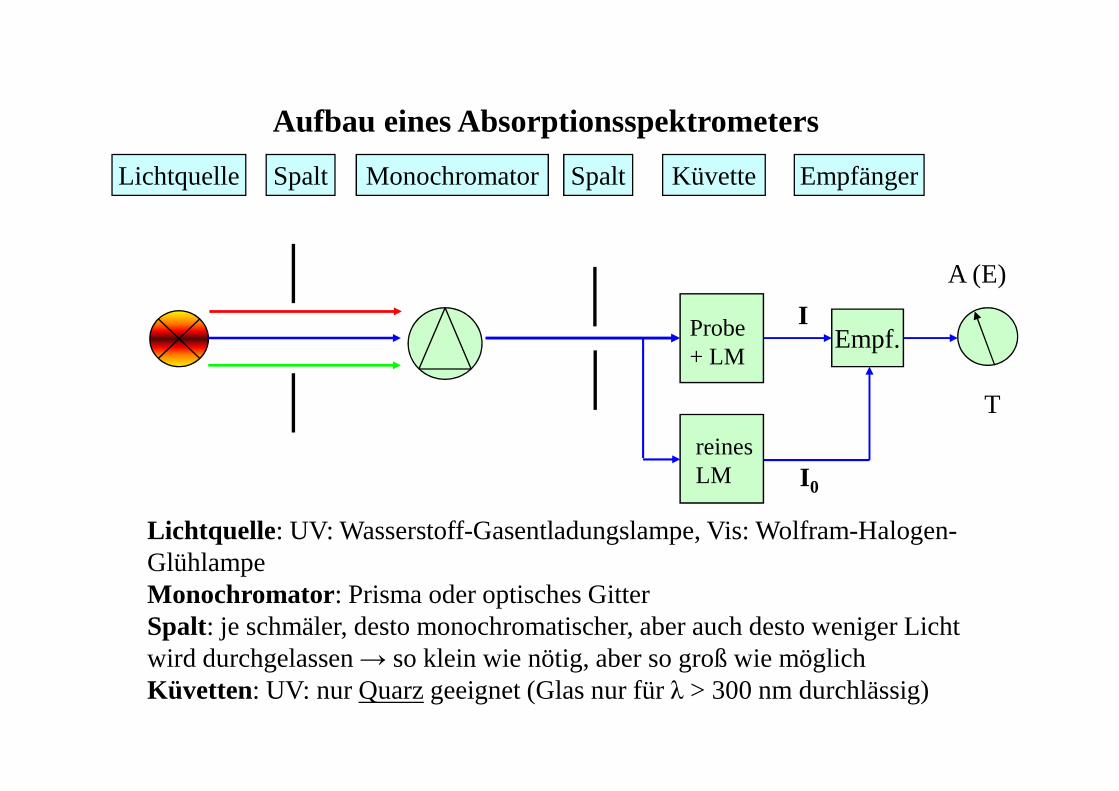
Aufbau eines Absorptionsspektrometers
Empf.I
I 0
A (E)
T
Lichtquelle Spalt Monochromator KüvetteSpalt Empfänger
Probe+ LM
reinesLM
Lichtquelle: UV: Wasserstoff-Gasentladungslampe, Vis: Wolfram-Halogen-GlühlampeMonochromator: Prisma oder optisches GitterSpalt: je schmäler, desto monochromatischer, aber auch desto weniger Licht wird durchgelassen → so klein wie nötig, aber so groß wie möglichKüvetten: UV: nur Quarz geeignet (Glas nur für λ > 300 nm durchlässig)
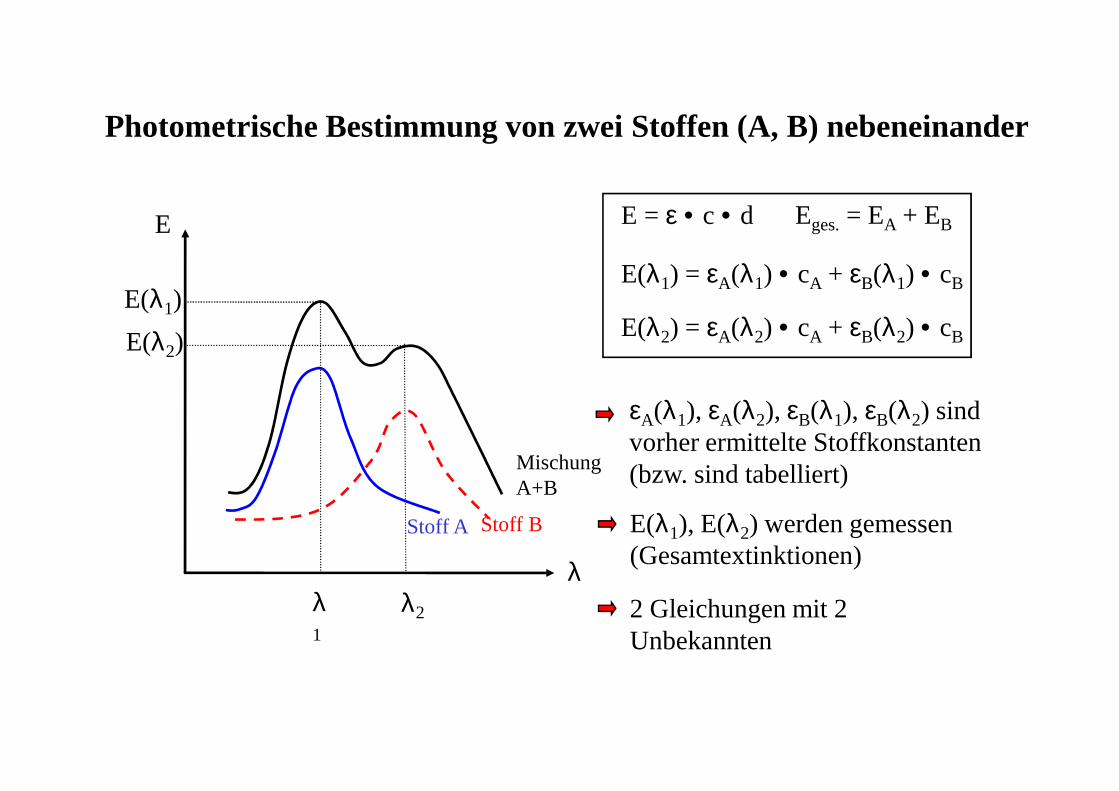
Photometrische Bestimmung von zwei Stoffen (A, B) nebeneinander
E
λλ2
E(λ1)
E(λ2)
Stoff A Stoff B
Mischung A+B
E = ε • c • d Eges.= EA + EB
E(λ1) = εA(λ1) • cA + εB(λ1) • cB
E(λ2) = εA(λ2) • cA + εB(λ2) • cB
εA(λ1), εA(λ2), εB(λ1), εB(λ2) sindvorher ermittelte Stoffkonstanten(bzw. sind tabelliert)
E(λ1), E(λ2) werden gemessen (Gesamtextinktionen)
2 Gleichungen mit 2 Unbekannten
λ1

Probenvorbereitung und Spektrenaufnahme
• Aufnahme in Lösung, c ~ 10-4 mol/l
• Verwendung optisch reiner Lösungsmittelab 195 nm: Pentan, Hexan, Heptan, Cyclohexan, Wasser, Acetonitrilab 210 nm: Methanol, Ethanol, DiethyletherDichlormethan (>220 nm), Chloroform (>240 nm), Tetrachlormethan (>260 nm)ab 280 nm: Benzol, Toluolab 310 nm: Pyridin
• Lösungsmittel mit Eigenabsorption im Messbereich sind ungeeignet!
• Das Lösungsmittel kann erheblichen Einfluss auf die Lage und das Aussehen der Banden haben (z.B. beim Wechsel unpolares LM → polares LM: bathochrome Verschiebung von π→π*, hypsochrome Verschiebung von n→π*)
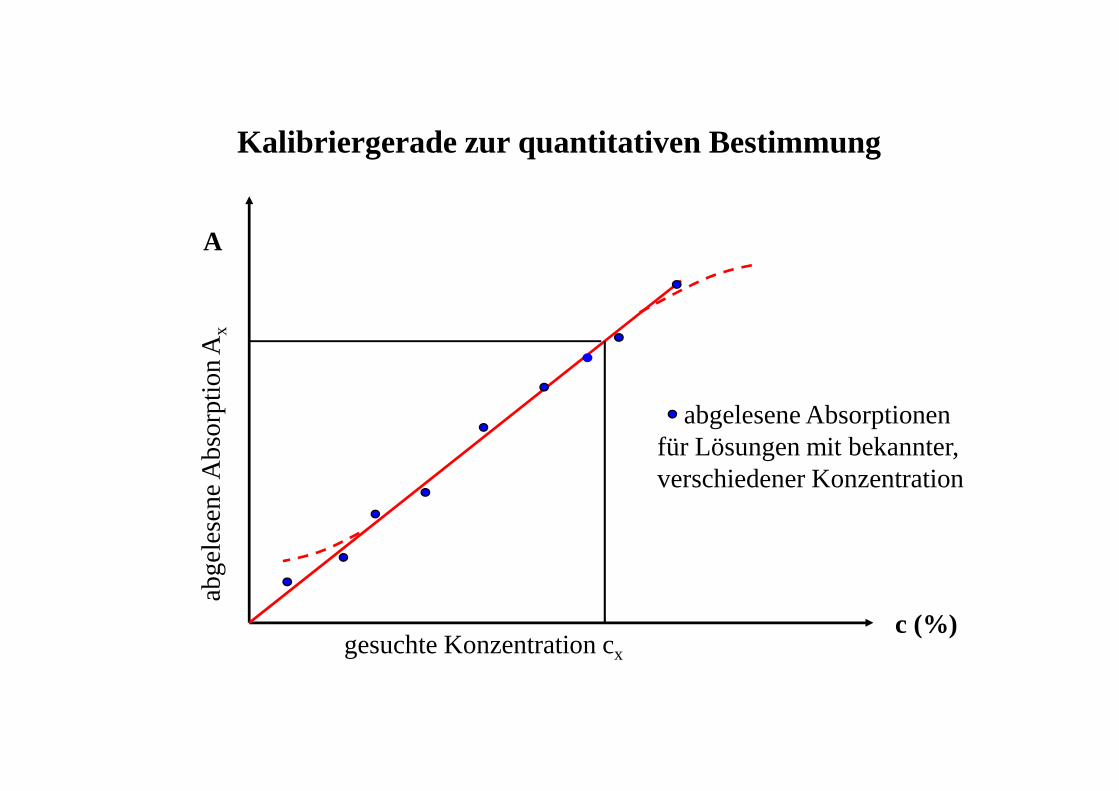
Kalibriergerade zur quantitativen Bestimmung
A
c (%)gesuchte Konzentration cx
abg
eles
ene
Ab
sorp
tion
A x
abgelesene Absorptionen für Lösungen mit bekannter, verschiedener Konzentration
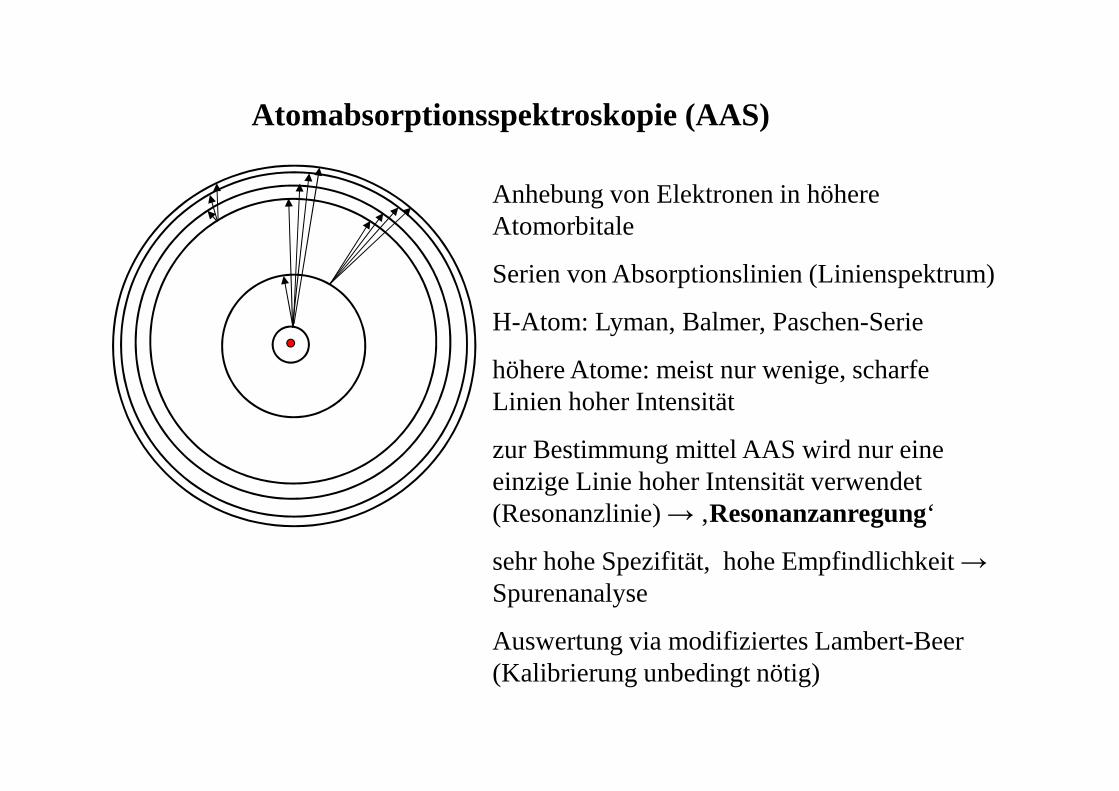
Atomabsorptionsspektroskopie (AAS)
Anhebung von Elektronen in höhere Atomorbitale
Serien von Absorptionslinien (Linienspektrum)
H-Atom: Lyman, Balmer, Paschen-Serie
höhere Atome: meist nur wenige, scharfe Linien hoher Intensität
zur Bestimmung mittel AAS wird nur eine einzige Linie hoher Intensität verwendet (Resonanzlinie) → ‚Resonanzanregung‘
sehr hohe Spezifität, hohe Empfindlichkeit → Spurenanalyse
Auswertung via modifiziertes Lambert-Beer (Kalibrierung unbedingt nötig)
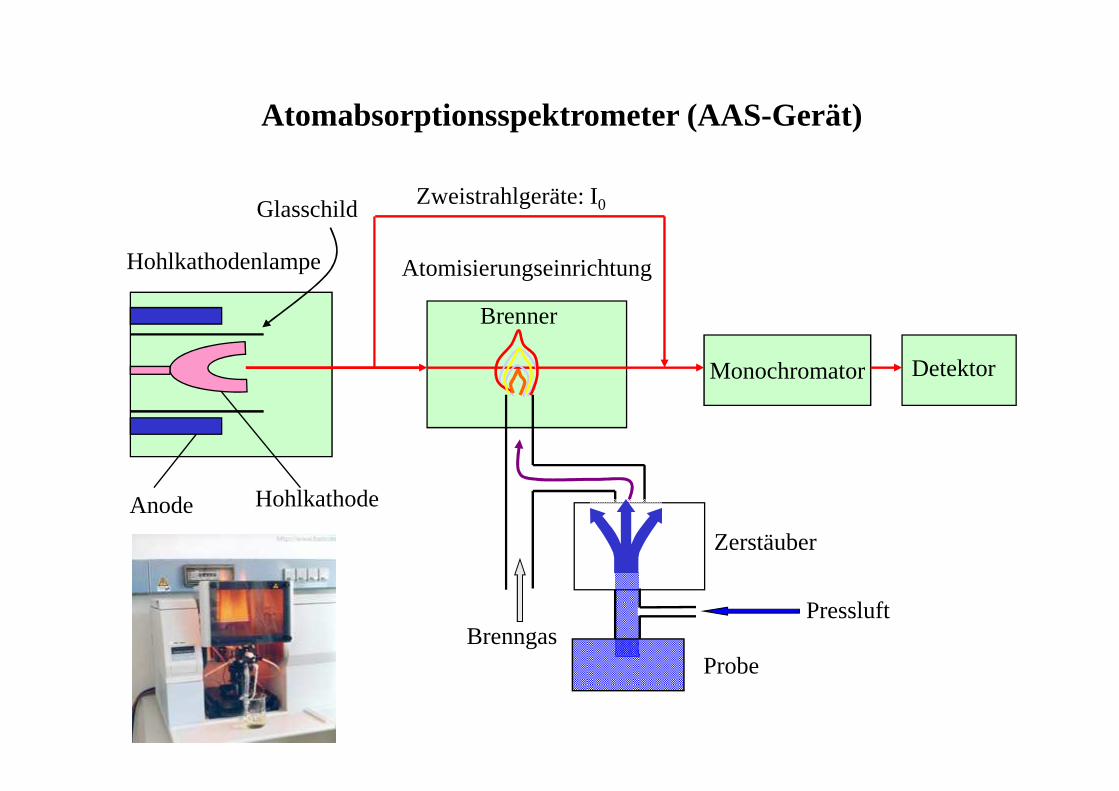
Atomabsorptionsspektrometer (AAS-Gerät)
Monochromator Detektor
Hohlkathodenlampe Atomisierungseinrichtung
Brenner
Anode Hohlkathode
Zweistrahlgeräte: I0Glasschild
Pressluft
Probe
Zerstäuber
Brenngas

Atomabsorptionsspektroskopie (AAS)
E = εεεε • c • l • f
E Extinktionεεεε Extinktionskoeffizientc Atomdampfkonzentrationl Atomreservoirlänge = Länge der Flammef Wirkungsgrad des Atomisierungsprozesses =
Ausmaß, in dem aus dem entsprechenden Salz Atome frei werden (abhängig von der Flammen-temperatur, von der Zusammensetzung des Brenngases u.v.a.)
es gehen viele Gerätekonstanten einEichkurve notwendig!
mittels AAS werden häufig bestimmt:
Al, As, Pb, Cd, Cr, Cu
Li, Na, Ni, Hg, Zn
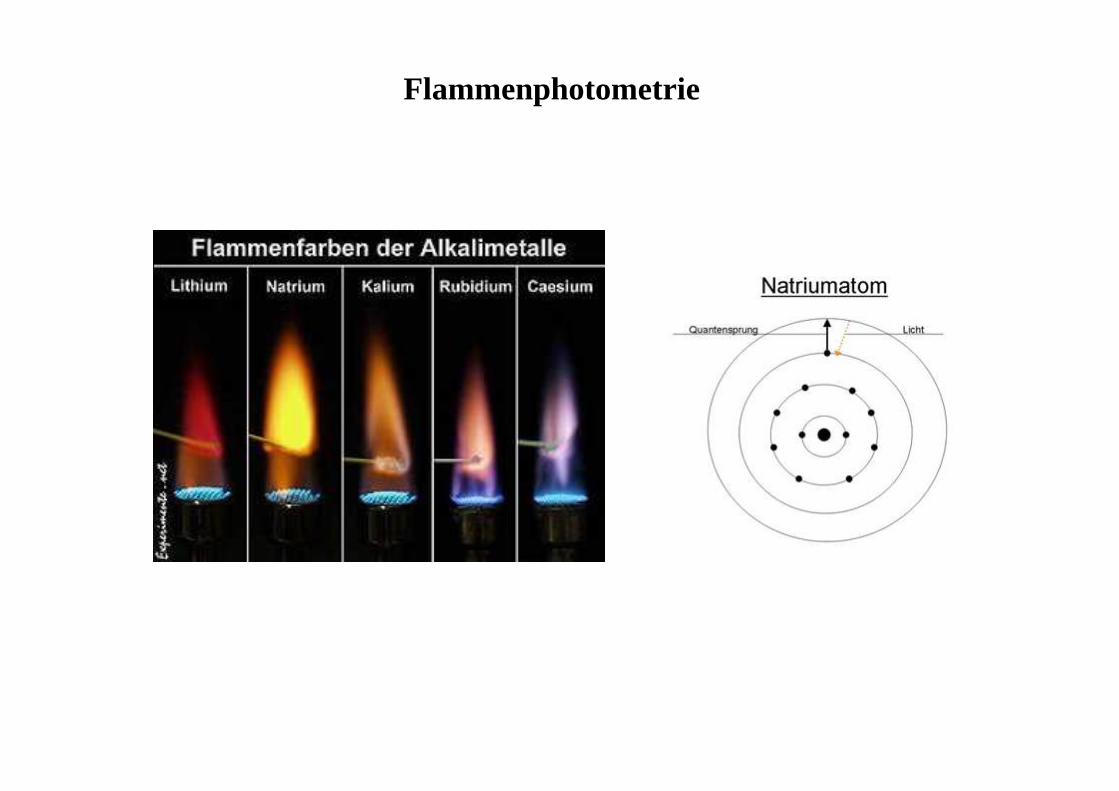
Flammenphotometrie
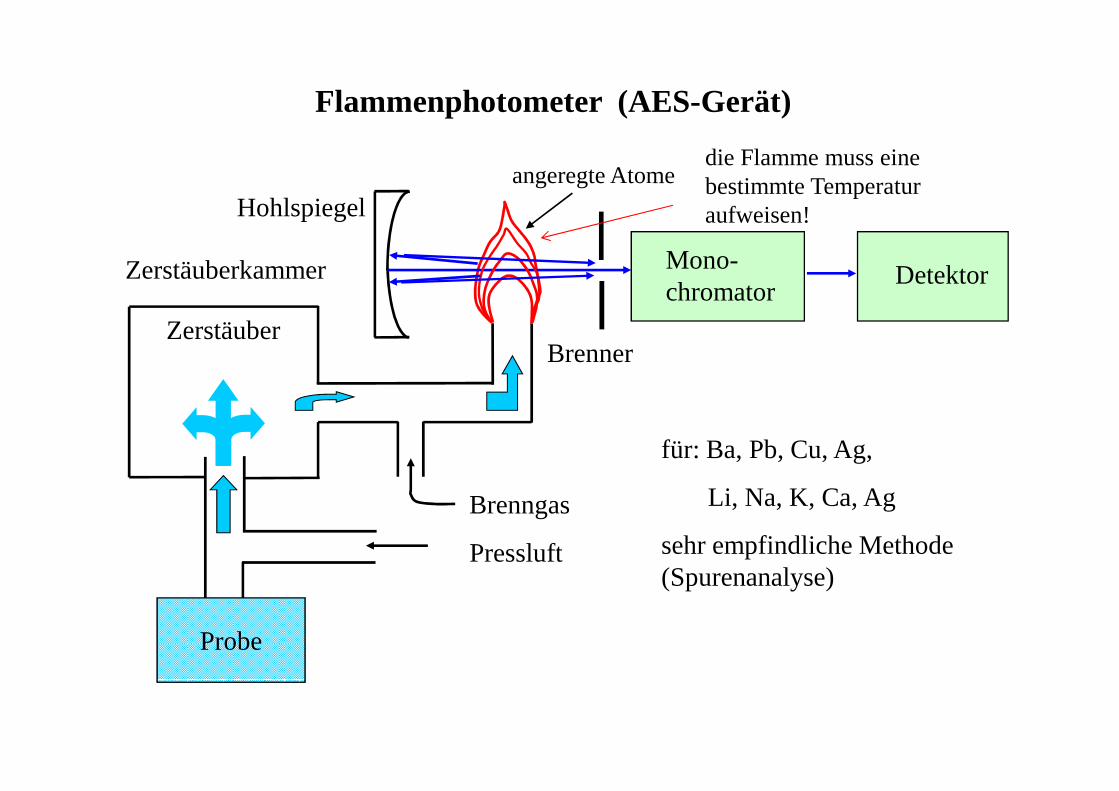
Flammenphotometer (AES-Gerät)
Probe
Pressluft
Brenngas
Zerstäuberkammer
Zerstäuber
Hohlspiegel
Mono-chromator
Detektor
angeregte Atome
Brenner
für: Ba, Pb, Cu, Ag,
Li, Na, K, Ca, Ag
sehr empfindliche Methode (Spurenanalyse)
die Flamme muss eine bestimmte Temperatur aufweisen!
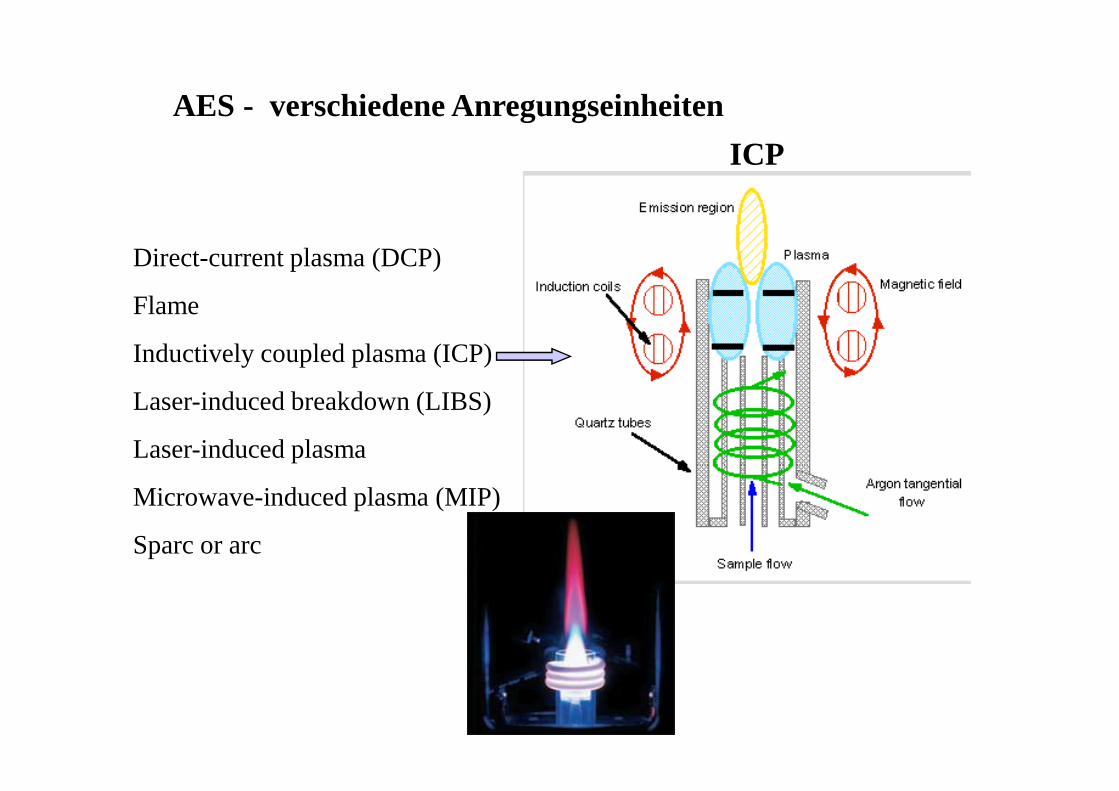
AES - verschiedene Anregungseinheiten
Direct-current plasma (DCP)
Flame
Inductively coupled plasma (ICP)
Laser-induced breakdown (LIBS)
Laser-induced plasma
Microwave-induced plasma (MIP)
Sparc or arc
ICP
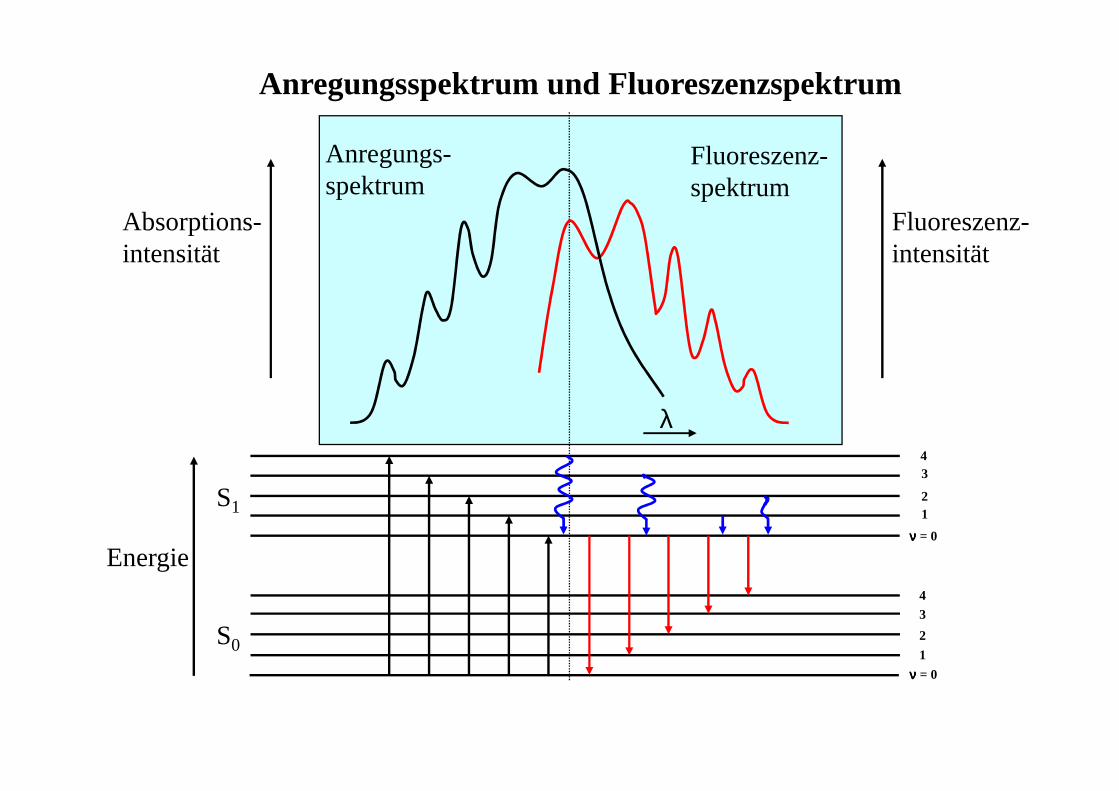
Anregungsspektrum und Fluoreszenzspektrum
S1
S0
Energie
Absorptions-intensität
Fluoreszenz-intensität
Anregungs-spektrum
Fluoreszenz-spektrum
λ
νννν = 0
1
43
2
1
43
2
νννν = 0

Definitionen - Fluorimetrie
F Anzahl der emittierten Photonen ϕ = = —————————————— ≤ 1
Iabs Anzahl der absorbierten Photonen
ϕϕϕϕ: Quantenausbeute der Fluoreszenz F: Intensität der FluoreszenzstrahlungI abs: Intensität der absorbierten Strahlung
F = k • I0 • ϕϕϕϕ • εεεε • c • d
k: Gerätekonstante (Eichkurve notwendig!) I 0: Intensität der Anregungsstrahlung εεεε: molarer Extinktionskoeffizient bei λ der Anregung (Absorptionsspektrum!) d: Schichtdicke c: Konzentration

• fluoreszenzfähig sind meist starre, planare Moleküle (größere Doppelbindungssysteme, (Hetero)Aromaten, Carbonylverbindungen)
• Fluoreszenzstrahlung ist immer langwelliger alsAnregungsstrahlung
• Fluoreszenzstrahlung ist immer viel schwächer als Anregungsstrahlung
• Einfluss des Lösungsmittels, von Gegenionen etc.
• oft Verwendung von Fluoreszenzmarkern (z.B. aromatische Sulfonsäurechloride – reagieren mit Aminen)
• Fluoreszenzdetektoren in der HPLC!
• sehr empfindliche Methode, zu quantitativen Bestimmungenin sehr niedrigen Konzentrationsbereichen geeignet (ppb-Bereich, 1 mg Substanz / Tonne Probe!)
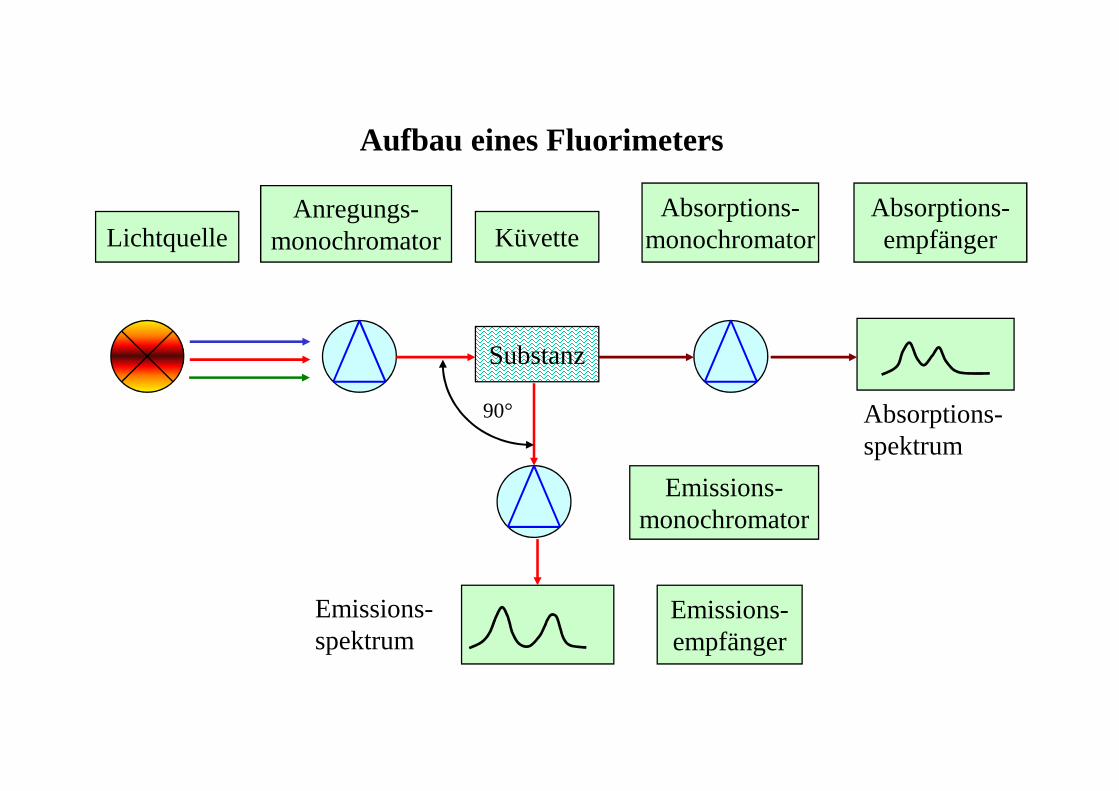
Aufbau eines Fluorimeters
LichtquelleAnregungs-
monochromator Küvette
Emissions-monochromator
Absorptions-empfänger
Absorptions-monochromator
Emissions-empfänger
Substanz
Absorptions-spektrum
Emissions-spektrum
90°

IR-Spektroskopie (Schwingungsspektroskopie)
• die IR-Spektroskopie erfasst Molekülschwingungen
• IR-aktiv sind Schwingungen, bei denen sich das Dipolmoment
des schwingenden Moleküls verändert (vollständig symmetrische
Schwingungen sind IR-inaktiv → Raman Spektroskopie)
• je größer die Polaritätsunterschiede der Schwingungspartner desto
intensiver die damit verbundenen Banden (z. B. C-O, C-N)
• 'normaler' IR-Bereich: 2.5 - 50 µm ⇒ 4000 - 200 cm-1
Wellenzahl ν: reziproker Wert der in cm ausgedrückten Wellenlänge (Vorteil: direkt proportional zu Frequenz und Energie)z. B.: λ = 20 µm = 20 • 10-6 m = 20 • 10-4 cm =
500 cm-1
∼
==⇒= 2
1000 ν cm
10002
cm 000 10
20 ∼
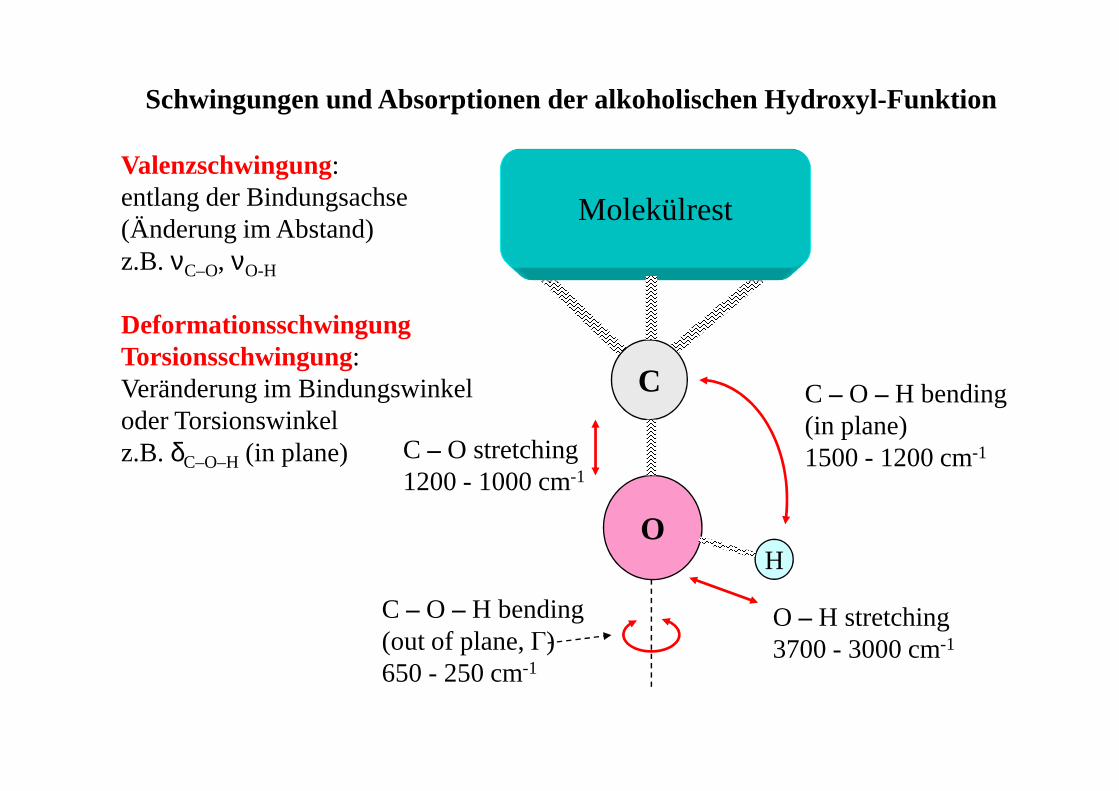
Molekülrest
C
OH
Schwingungen und Absorptionen der alkoholischen Hydroxyl-Funktion
C – O – H bending (in plane)1500 - 1200 cm-1C – O stretching
1200 - 1000 cm-1
O – H stretching3700 - 3000 cm-1
C – O – H bending (out of plane, Γ)650 - 250 cm-1
Valenzschwingung:entlang der Bindungsachse(Änderung im Abstand)z.B. νC–O, νO-H
DeformationsschwingungTorsionsschwingung:Veränderung im Bindungswinkeloder Torsionswinkelz.B. δC–O–H(in plane)
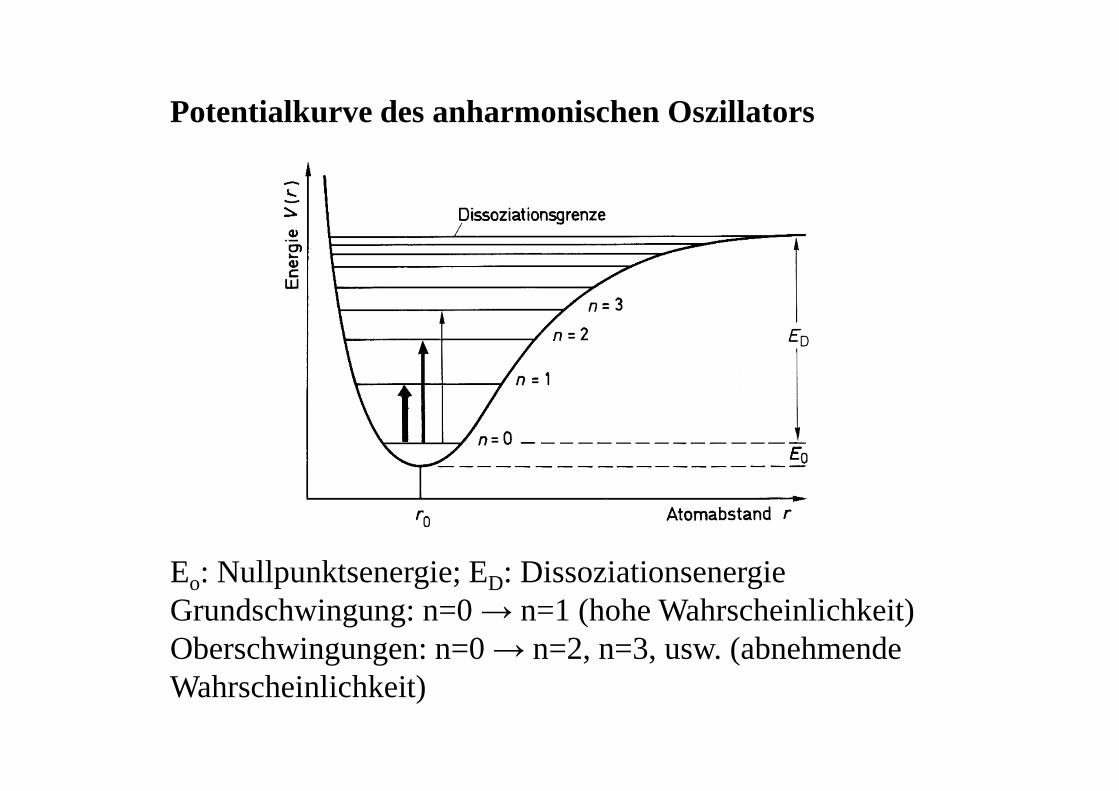
Potentialkurve des anharmonischen Oszillators
Eo: Nullpunktsenergie; ED: DissoziationsenergieGrundschwingung: n=0 → n=1 (hohe Wahrscheinlichkeit)Oberschwingungen: n=0 → n=2, n=3, usw. (abnehmende Wahrscheinlichkeit)
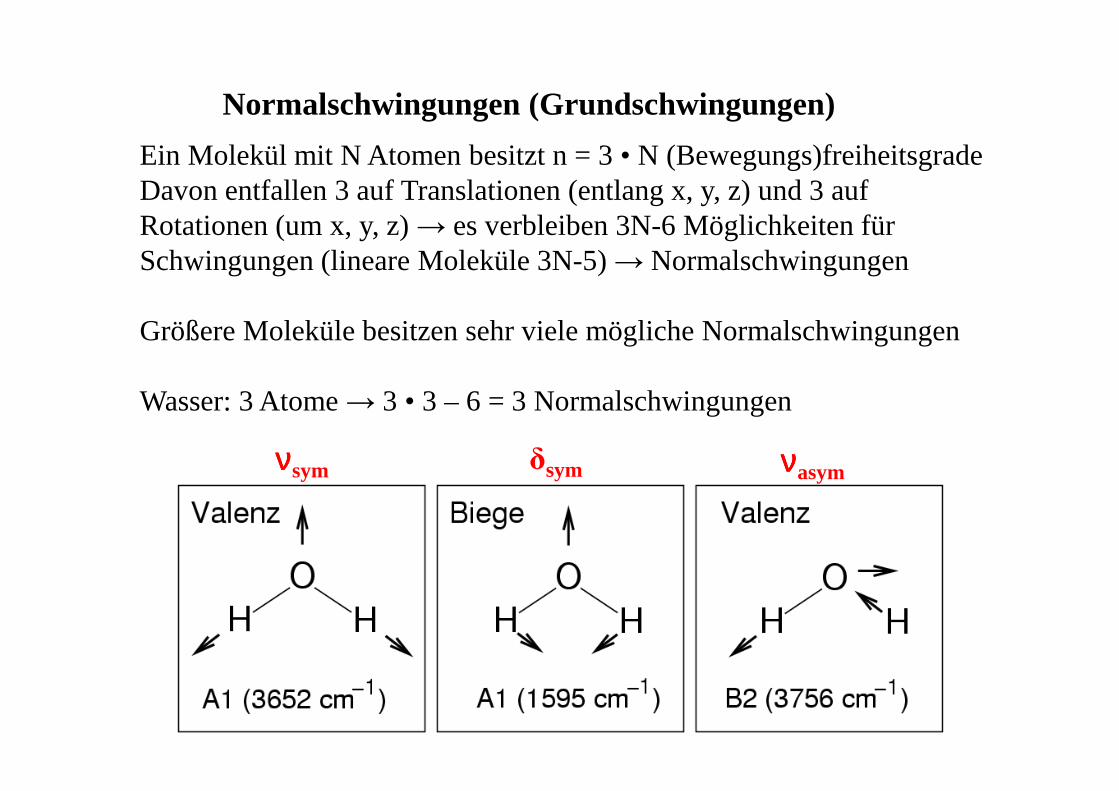
Ein Molekül mit N Atomen besitzt n = 3 • N (Bewegungs)freiheitsgradeDavon entfallen 3 auf Translationen (entlang x, y, z) und 3 auf Rotationen (um x, y, z) → es verbleiben 3N-6 Möglichkeiten für Schwingungen (lineare Moleküle 3N-5) → Normalschwingungen
Größere Moleküle besitzen sehr viele mögliche Normalschwingungen
Wasser: 3 Atome → 3 • 3 – 6 = 3 Normalschwingungen
Normalschwingungen (Grundschwingungen)
ννννsym ννννasymδsym
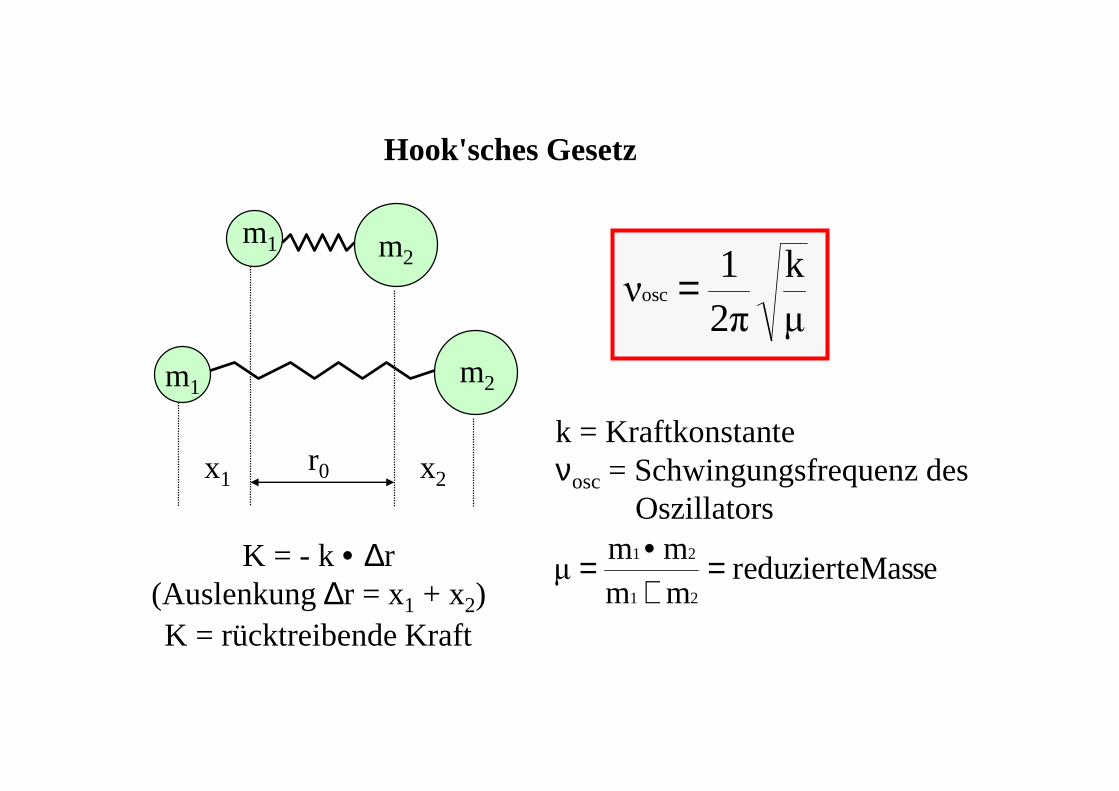
Hook'sches Gesetz
m1
m2
r0x1 x2
m1
m2
K = - k • ∆r(Auslenkung ∆r = x1 + x2)
µ
k2π1
νosc =
k = Kraftkonstanteνosc= Schwingungsfrequenz des
Oszillators
Massereduziertemmmm
µ21
21 =+•=
K = rücktreibende Kraft

Abhängigkeit der Schwingungsfrequenz von der Bindungsstärke:
Abhängigkeit der Schwingungsfrequenz von der Masse der Atome:
C – C 1000 cm-1
C = C 1640 cm-1
C ≡ C 2200 cm-1
C – H 3000 cm-1
C – Br 650 cm-1
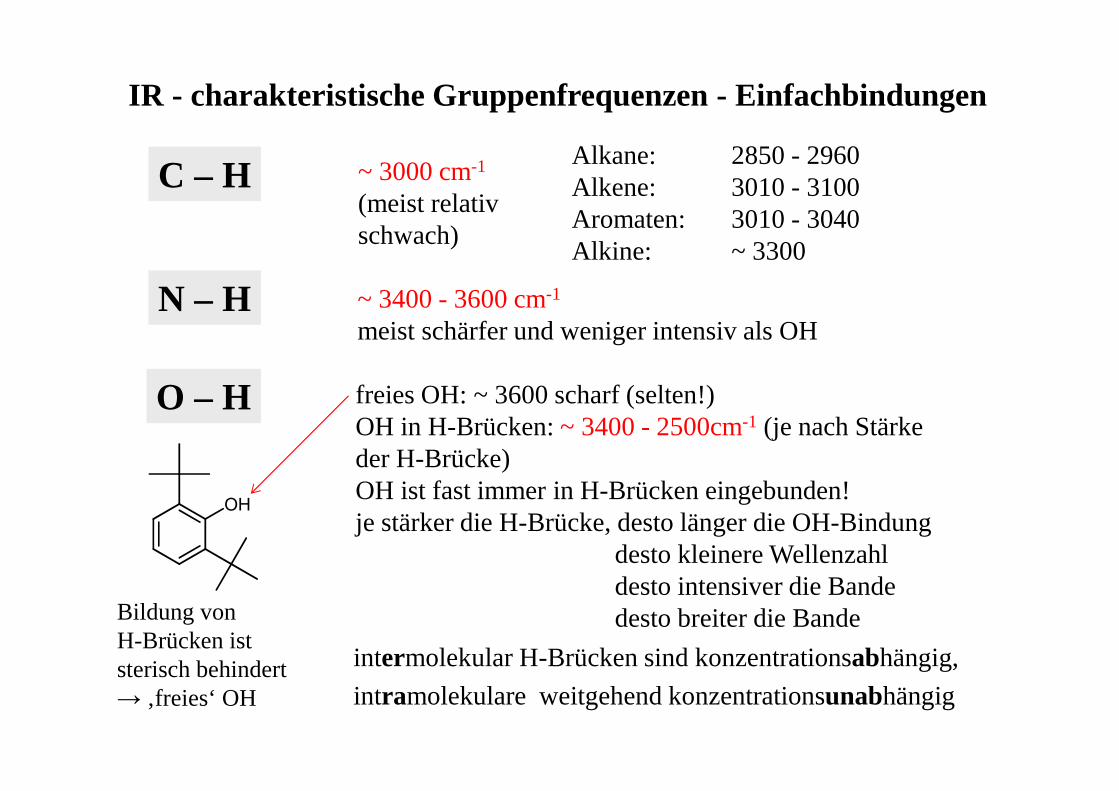
IR - charakteristische Gruppenfrequenzen - Einfachbindungen
C – HAlkane: 2850 - 2960Alkene: 3010 - 3100Aromaten: 3010 - 3040Alkine: ~ 3300
N – H ~ 3400 - 3600 cm-1
meist schärfer und weniger intensiv als OH
O – H freies OH: ~ 3600 scharf (selten!)OH in H-Brücken: ~ 3400 - 2500cm-1 (je nach Stärke der H-Brücke)OH ist fast immer in H-Brücken eingebunden!je stärker die H-Brücke, desto länger die OH-Bindung
desto kleinere Wellenzahldesto intensiver die Bandedesto breiter die Bande
intermolekular H-Brücken sind konzentrationsabhängig,
intramolekulare weitgehend konzentrationsunabhängig
~ 3000 cm-1
(meist relativ schwach)
OH
Bildung von H-Brücken ist sterisch behindert → ‚freies‘ OH

IR - charakteristische Gruppenfrequenzen - Dreifachbindungen
C ≡≡≡≡ C
C ≡≡≡≡ N
~ 2100 - 2300(Intensität variabel, abhängig von derStruktur des restlichen Moleküls)
– C ≡ C – 2150 - 2260– C ≡ C –H 2100 - 2140
~ 2200 - 2300(manchmal schwach oder abwesend)
Im Bereich 2100-2300 cm-1 auch kumulierte Doppelbindungen: Isocyanate (R-N=C=O), Isothiocyanate (R-N=C=S), Ketene (R1R2C=C=O), Carbodiimide (R-N=C=N-R‘), Azide (R-N3), Diazoalkane (R2CN2), usw.

IR - charakteristische Gruppenfrequenzen - Doppelbindungen
C = O ~ 1620 - 1850 cm-1, sehr wichtig!
1) abhängig von der Elektronegativität von Substituenten X ander Carbonylfunktion - je größer EN von X, desto größer νC=O
Aldehyd R-CO-H ~ 1720 - 1740Keton R-CO-R' ~ 1705 - 1725Carbonsäureester R-CO-OR' ~ 1735 - 1750Carbonsäure R-CO-OH ~ 1760 (Monomer, sehr selten!)Carbonsäurechlorid R-CO-Cl ~ 1790 - 1815
2) abhängig von Konjugationseffekten - in α,β-ungesättigtenC=O Funktionen ist νC=O um ca. 20 - 40 cm-1 kleiner
gesättigtes Keton R-CO-R' ~ 1705 - 1725 α,β-ungesättigtes Keton R-CO-CH=CH-R' ~ 1665 - 1685Aryl-Keton R-CO-Aryl ~ 1680 - 1700
R = jeweils Alkyl!!!
IR - charakteristische Gruppenfrequenzen - Doppelbindungen
C = O
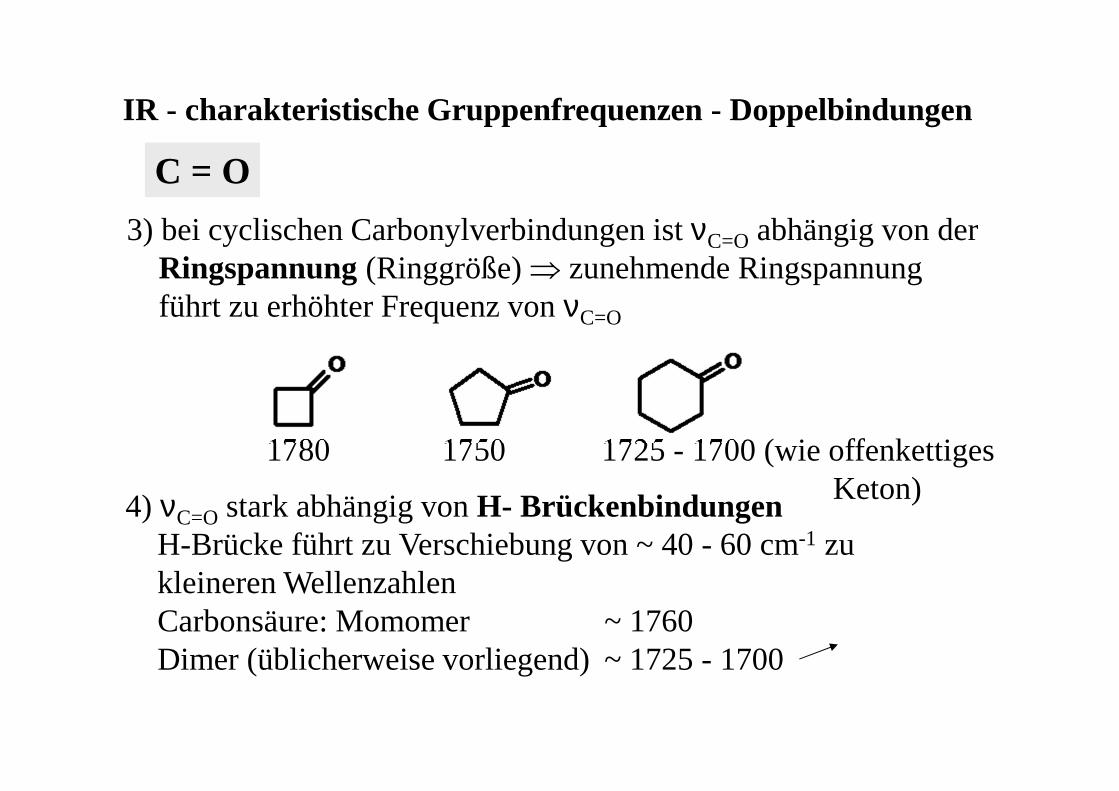
3) bei cyclischen Carbonylverbindungen ist νC=O abhängig von derRingspannung(Ringgröße) ⇒ zunehmende Ringspannungführt zu erhöhter Frequenz von νC=O
1780 1750 1725 - 1700 (wie offenkettigesKeton)4) νC=O stark abhängig von H- Brückenbindungen
H-Brücke führt zu Verschiebung von ~ 40 - 60 cm-1 zu kleineren Wellenzahlen Carbonsäure: Momomer ~ 1760 Dimer (üblicherweise vorliegend) ~ 1725 - 1700

weitere Doppelbindungen
C=C: Alkene (nicht konjugiert): 1620-1680 cm-1
α,β-ungesättigte Carbonylverbindungen: 1590-1640 cm-1
Diene, Triene: 1600, 1650 cm-1
Aromaten: 2-3 Banden 1500-1600 cm-1
C=N: 1640-1690 cm-1
C=S: 1050-1200 cm-1
NO2: 1350 und 1560 cm-1 (sym. und asym. Valenzschwingung)
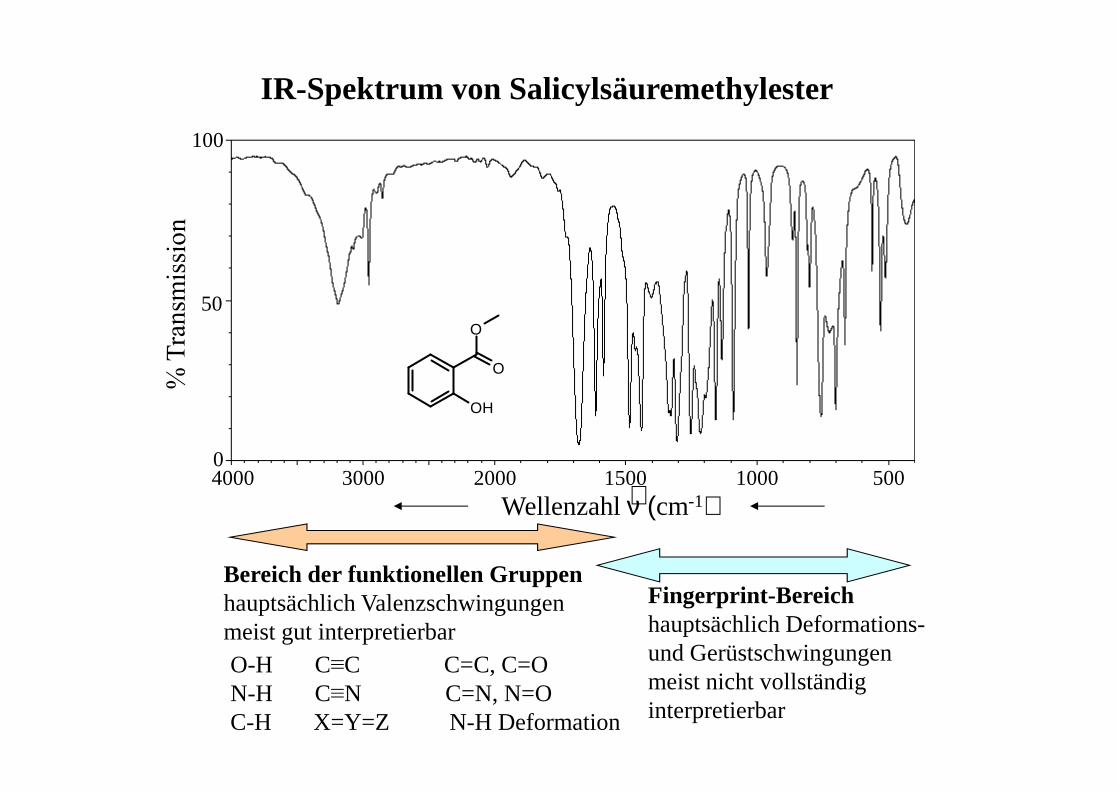
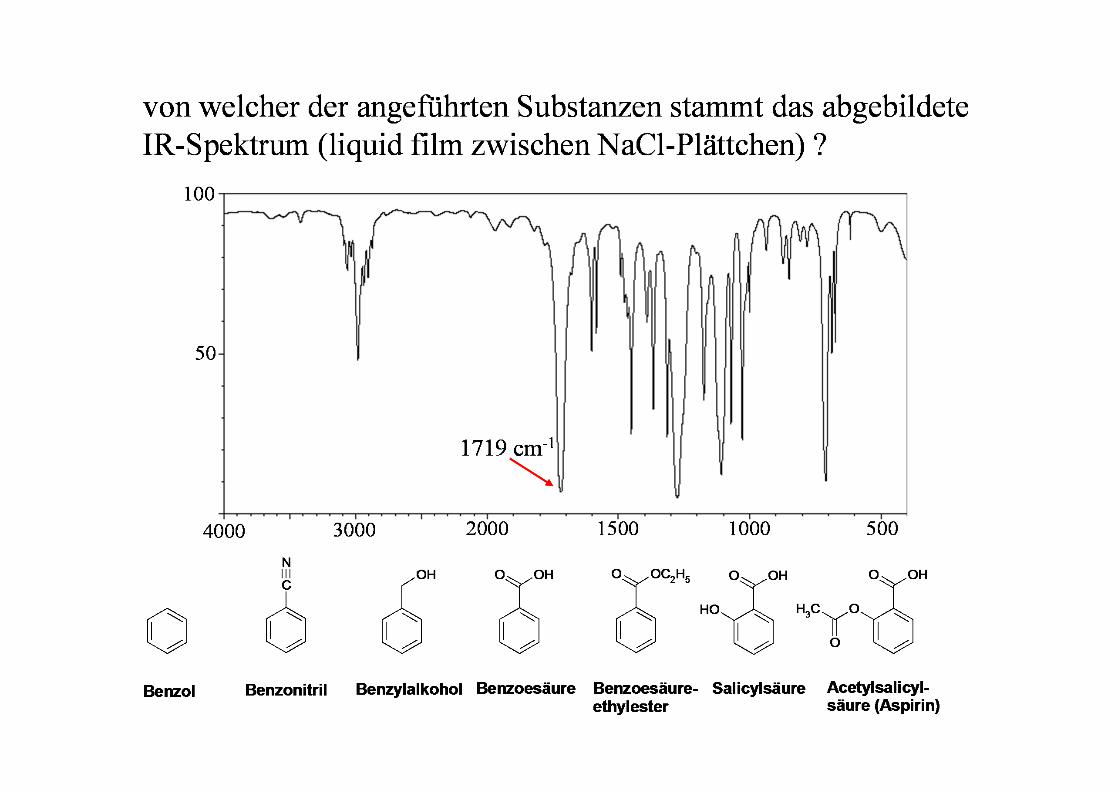
IR-Spektrum von Salicylsäuremethylester
Wellenzahl ν (cm-1)∼0
50
100
4000 3000 2000 1000 5001500
Bereich der funktionellen Gruppenhauptsächlich Valenzschwingungen meist gut interpretierbar
Fingerprint-Bereichhauptsächlich Deformations-und Gerüstschwingungen meist nicht vollständig interpretierbar
O
O
OH
O-H C≡C C=C, C=ON-H C≡N C=N, N=OC-H X=Y=Z N-H Deformation
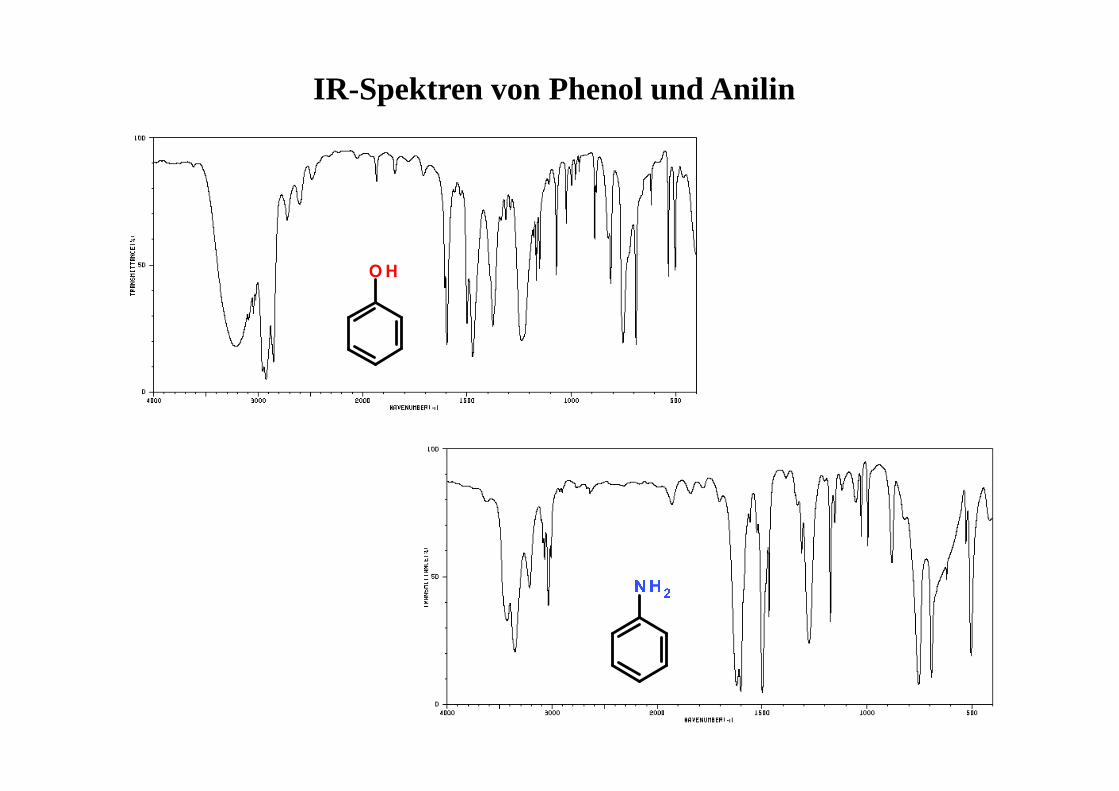
IR-Spektren von Phenol und Anilin
OH
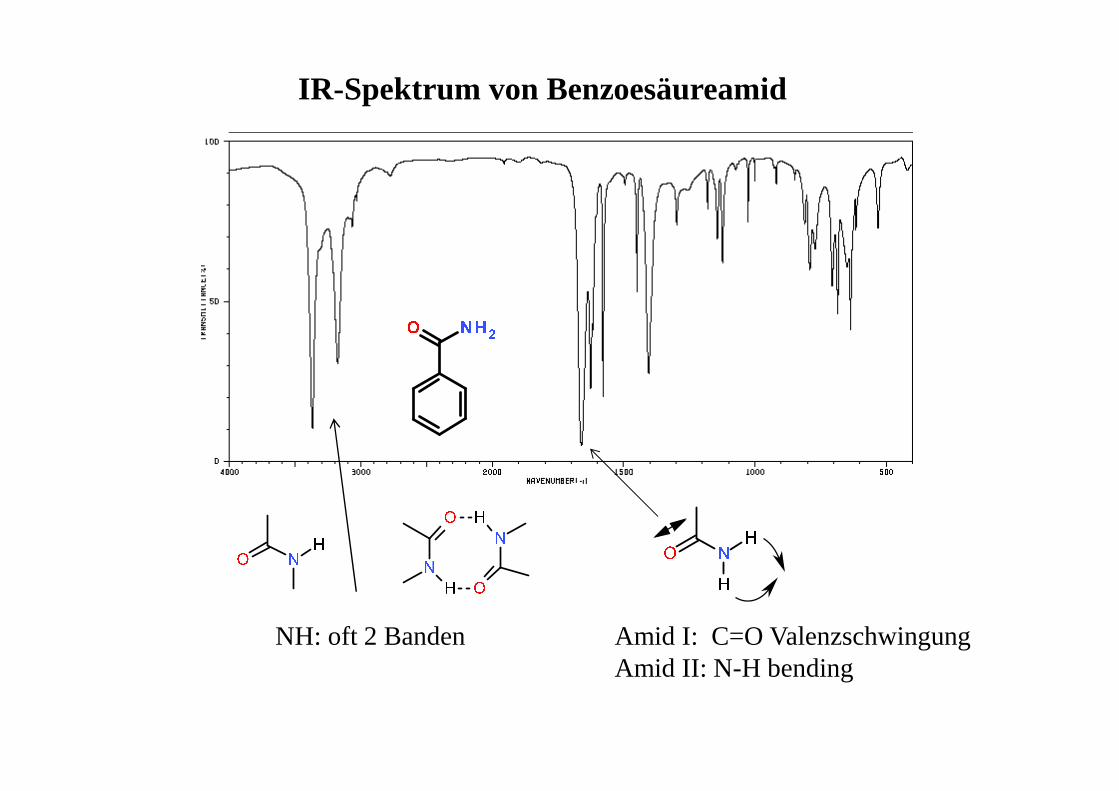
IR-Spektrum von Benzoesäureamid
NH: oft 2 Banden Amid I: C=O ValenzschwingungAmid II: N-H bending
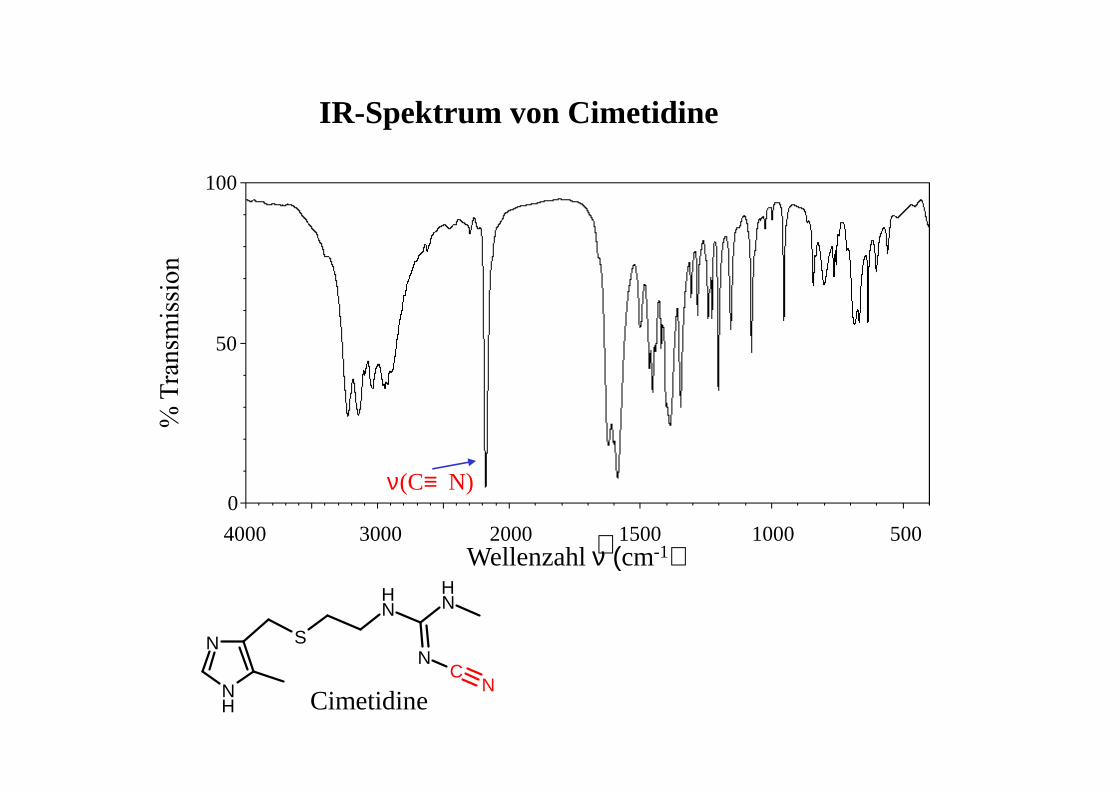
IR-Spektrum von Cimetidine
Wellenzahl ν (cm-1)∼0
50
100
4000 3000 2000 1000 5001500
NH
N S
NH N
H
NC
NCimetidine
ν(C≡ N)

IR - Aufnahmetechniken Probenvorbereitung für Transmissionsmessungen
• KBr (KCl) Pressling (KBr-disc): häufig verwendet, polar, enthält immer Spuren von H2O → νO-H ∼ 3450 cm-1
• Nujol-Technik (Paraffinöl): apolar, C-H (C-C) Bereich vom Medium überlagert (für Luft- bzw.feuchtigkeitsempfindliche Substanzen)
• in Lösung (NaCl-Zelle): Lösungsmittel z. B. CHCl3, CCl4(Eigenabsorption des LM, Zweistrahlgerät!)
• flüssige (nicht wässrige) Proben: direkt zwischen NaCl Plättchen (liquid film)
• gasförmige Proben in Spezialzellen (Umweltanalytik, GC-IR)
Das Aussehen des IR-Spektrums ein und derselben Substanz ist sehr stark von der verwendeten Aufnahmetechnik abhängig!!!
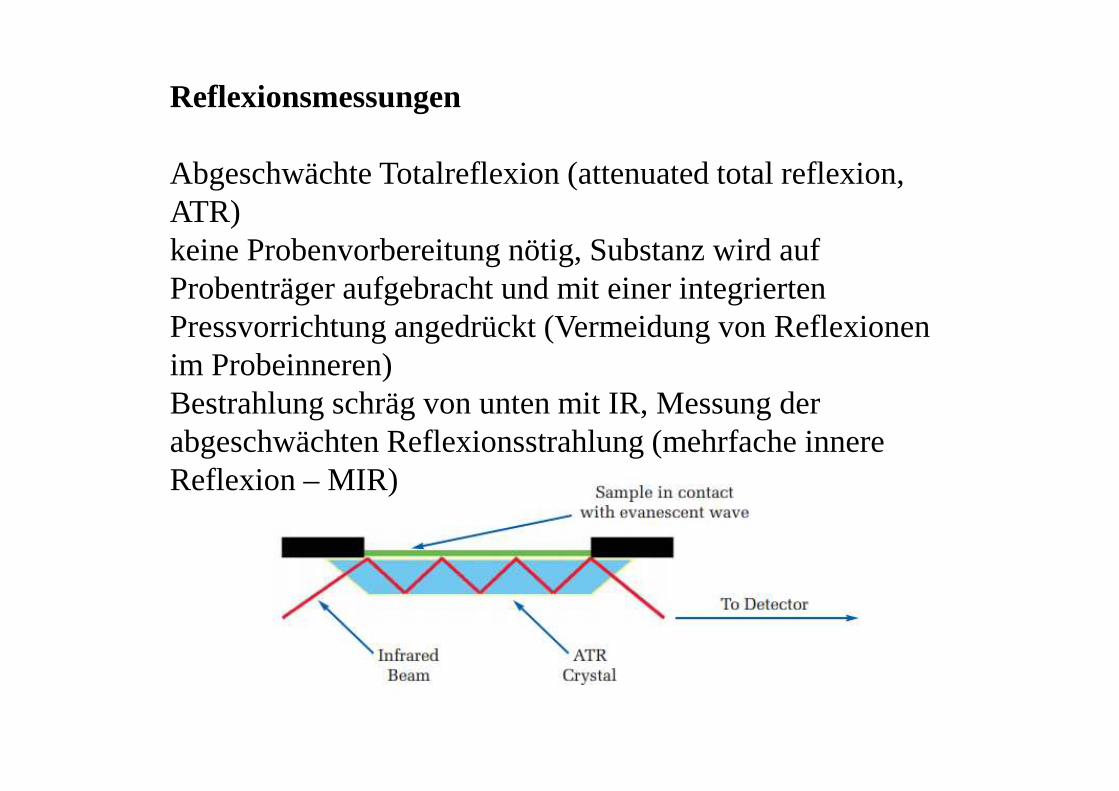
Reflexionsmessungen
Abgeschwächte Totalreflexion (attenuated total reflexion, ATR)keine Probenvorbereitung nötig, Substanz wird auf Probenträger aufgebracht und mit einer integrierten Pressvorrichtung angedrückt (Vermeidung von Reflexionen im Probeinneren)Bestrahlung schräg von unten mit IR, Messung der abgeschwächten Reflexionsstrahlung (mehrfache innere Reflexion – MIR)

IR - Spektrometer
• Absorptionsspektrometer(ähnlich UV, strahlungsdurch-lässige Bauteile dem IR-Bereich angepasst → Halogenid-Kristalle!)
• FTIR -Spektrometer (Fourier-Transform)• alle Wellenlängen werden gleichzeitig angeregt• man erhält ein Interferogramm (Überlagerung vonSchwingungen, 'Zeitdomäne')
• aus diesem wird durch eine mathematische Operation (Fourier-Transformation) das gewohnte IR-Spektrum erzeugt('Frequenzdomäne' = Frequenz (Wellenzahl) gegen Intensität)
• Vorteil: viel schneller (Kopplung mit chromatographischenVerfahren möglich), empfindlicher (besseres Signal-RauschVerhältnis), hohe Präzision (Eichung mittels Laser)
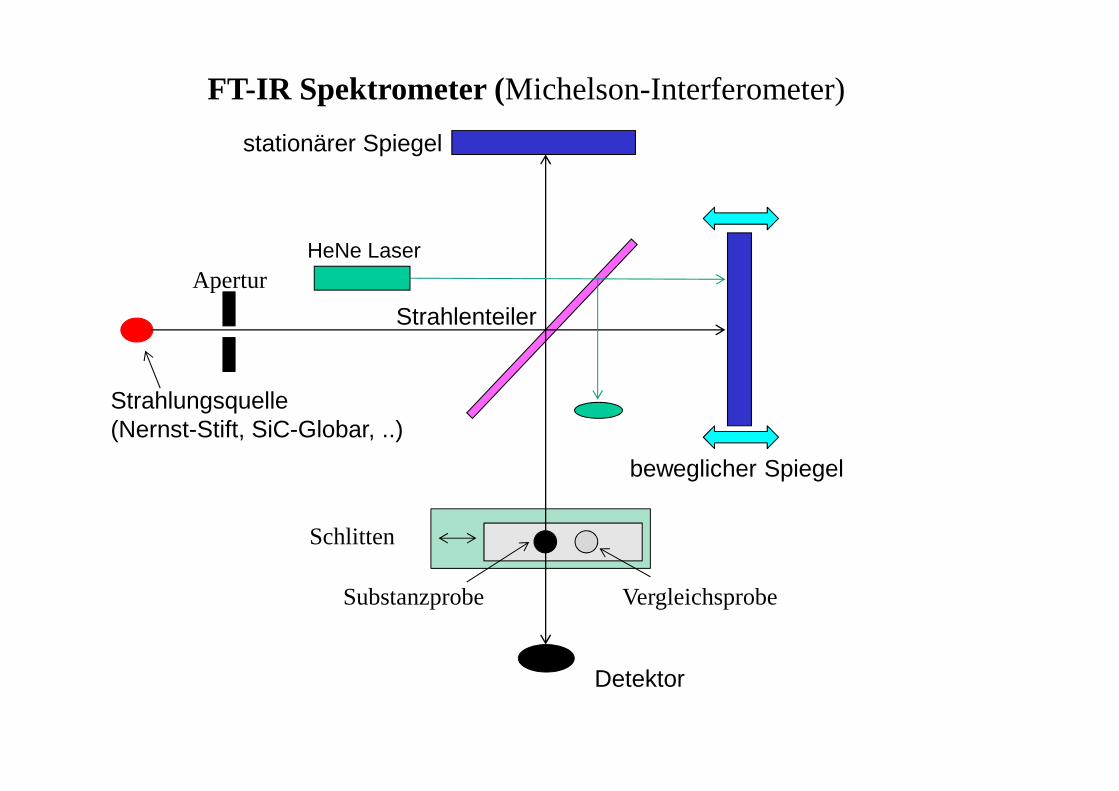
Strahlungsquelle(Nernst-Stift, SiC-Globar, ..)
stationärer Spiegel
beweglicher Spiegel
Detektor
Strahlenteiler
HeNe LaserApertur
Schlitten
Substanzprobe Vergleichsprobe
FT-IR Spektrometer (Michelson-Interferometer)
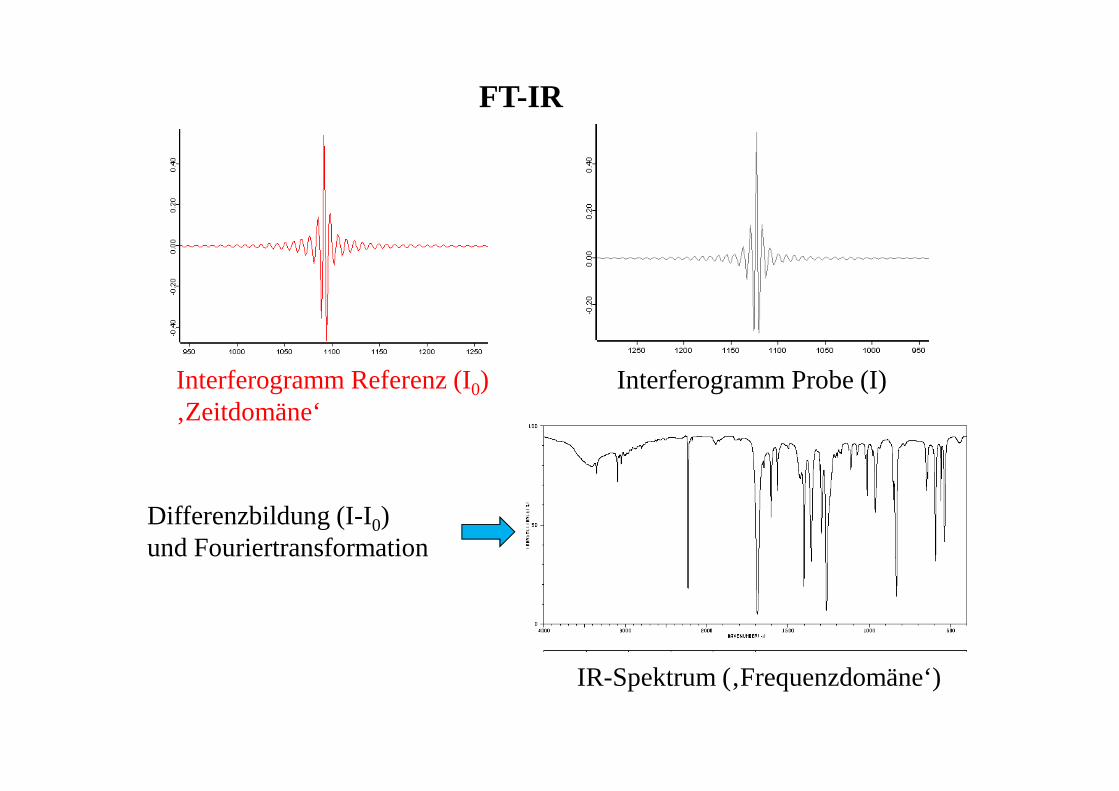
FT-IR
Interferogramm Referenz (I0)‚Zeitdomäne‘
Interferogramm Probe (I)
Differenzbildung (I-I0) und Fouriertransformation
IR-Spektrum (‚Frequenzdomäne‘)
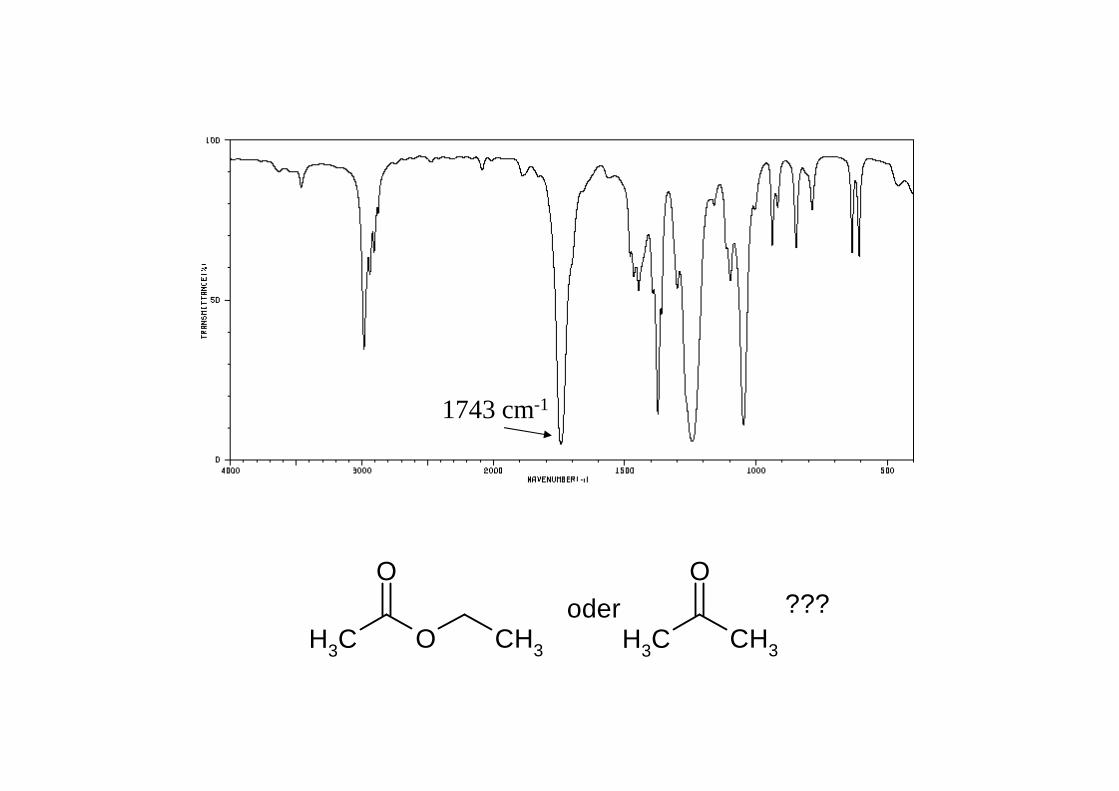
1743 cm-1
CH3 O
O
CH3
oder CH3 CH3
O???
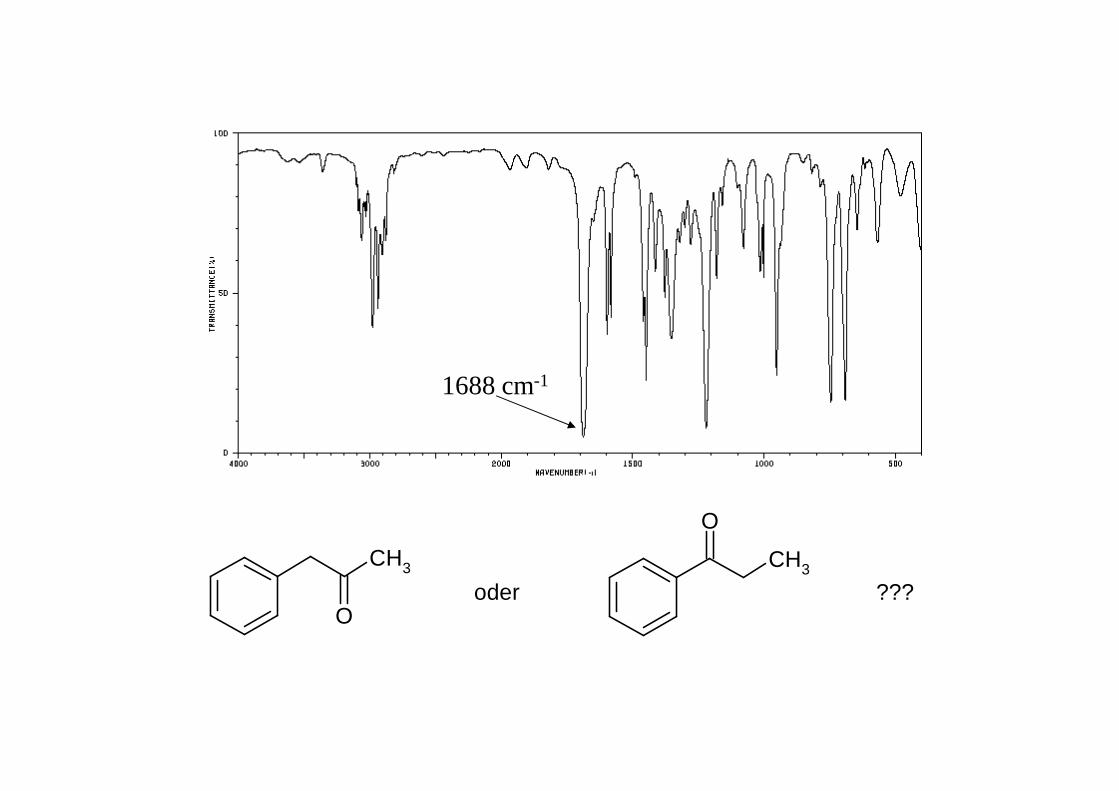
CH3
O
O
CH3
oder ???
1688 cm-1
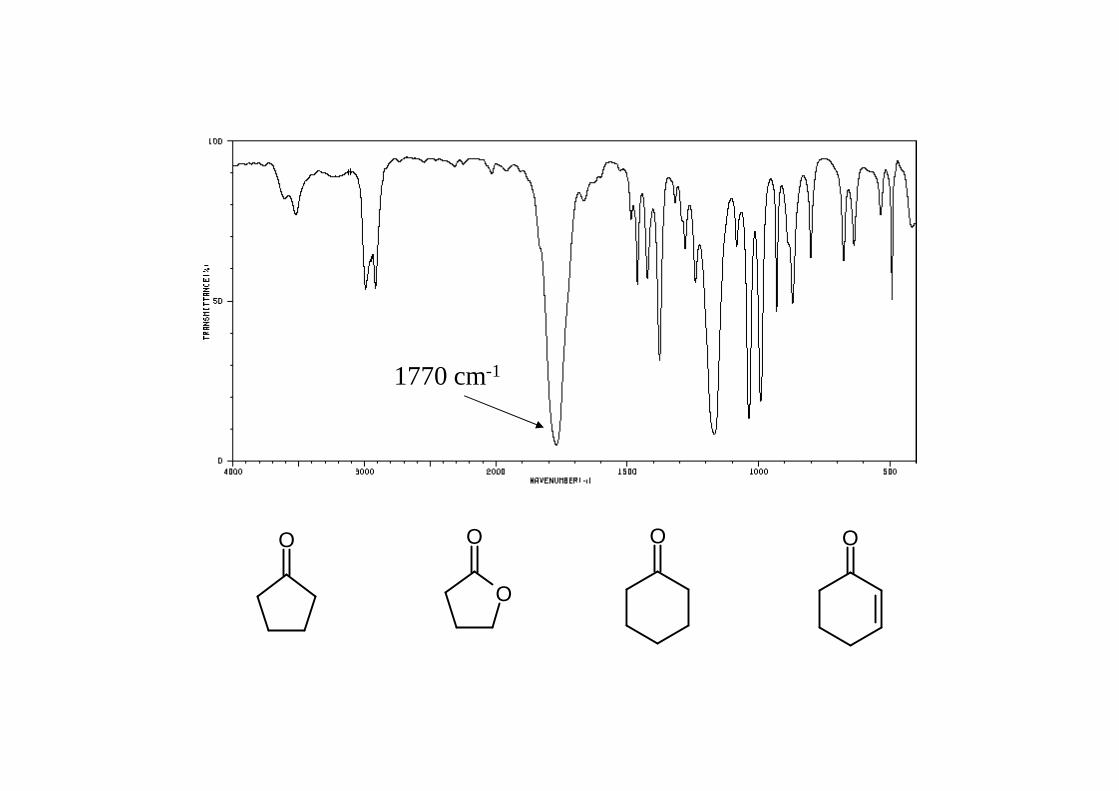
OO
O
O O
1770 cm-1

IR – Resümee
• Nachweis bestimmter funktioneller Gruppen (z.B. C=O)
• routinemäßig bei der Strukturaufklärung
• Pharmazeutische Analytik: Identifizierung von Arzneistoffen (Identitätsprüfung), Reinheitskontrolle
• meist nur qualitativ
• quantitative Bestimmungen zwar möglich, aber ungenauer und mühsamer als bei UV/vis

Massenspektrometrie
• im Massenspektrometer werden Moleküle, die sich im Gaszustand befinden, in energiereiche positive (bzw.negative) Ionen übergeführt → Ionisierung
• die gebildeten energiereichen Ionen zerfallen (zumindestzum Teil) in geladene und ungeladene Bruchstücke → Fragmentierung
• die geladenen Teilchen werden in einem elektrischen Feldbeschleunigt, nach ihrer Massenzahl (m/z) aufgetrennt(meist in einem Magnetfeld) und registriert
• im resultierenden Massenspektrum sind die Massenzahlender (Fragment)-Ionen gegen ihre relative Intensitätdargestellt
• das Massenspektrum gibt meist wertvolle Hinweise auf dieStruktur der untersuchten Substanz
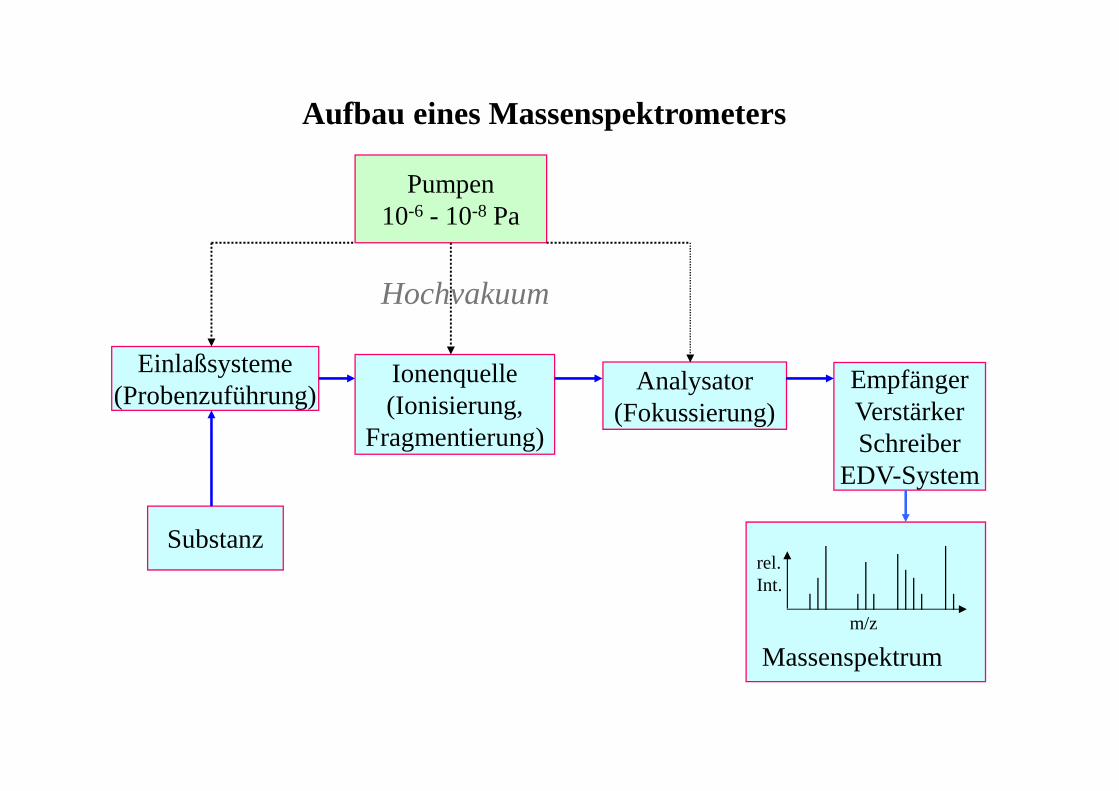
Aufbau eines Massenspektrometers
Einlaßsysteme(Probenzuführung)
Ionenquelle(Ionisierung,
Fragmentierung)
Analysator(Fokussierung)
EmpfängerVerstärkerSchreiber
EDV-System
Substanz
Pumpen10-6 - 10-8 Pa
m/z
rel.Int.
Massenspektrum
Hochvakuum

MS – Probenzuführung – Einlass-Systeme
• Direkt-Einlass ('Schubstange'): beheizbarer Tiegel für festeoder flüssige Proben, wird über Schleusenkammer in dasMassenspektrometer eingeführt, dann wird die Probeverdampft (kombinierbar mit EI, CI, MALDI)
• Direkt-Infusion : Probe wird aus einem Vorratsbehälter über eine Kapillare dosiert und kontinuierlich in das Massenspektrometer eingeführt (besonders für Proben in Lösung bzw. bereits im gasförmigem Zustand)kombinierbar mit EI, CI, Spray-Methodenerlaubt die Kopplung mit chromatographischen Methoden (GC-MS, HPLC-MS)
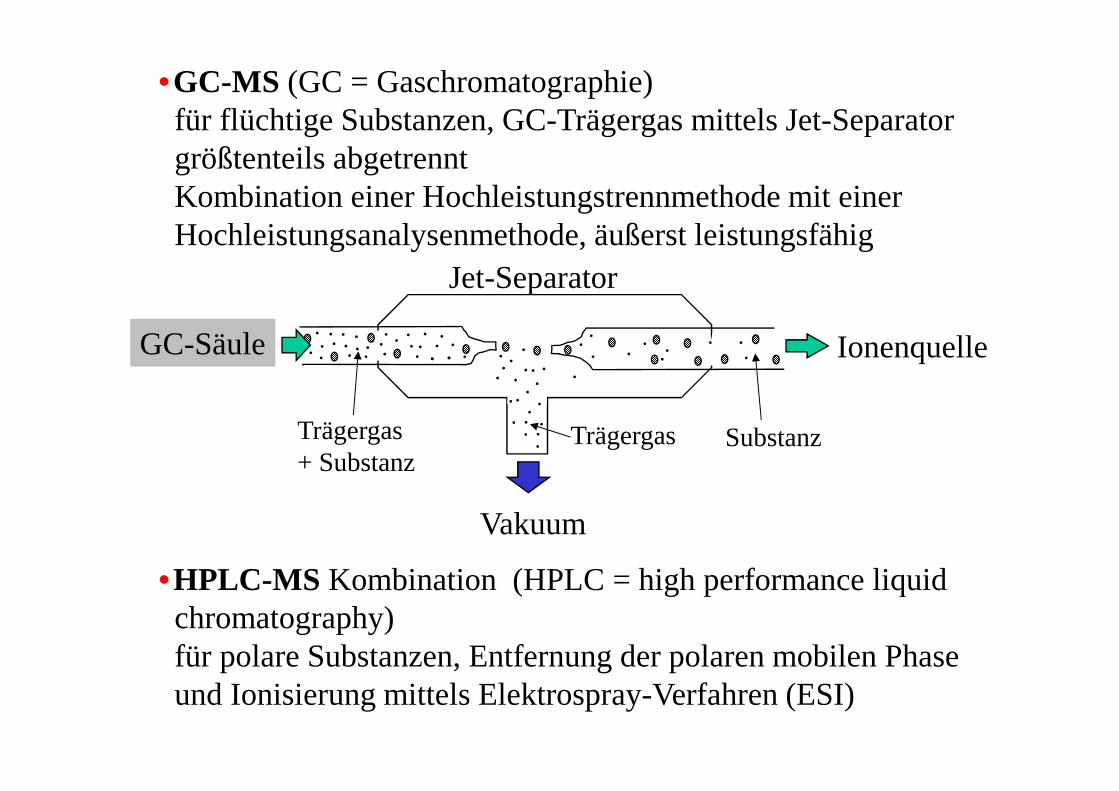
•GC-MS (GC = Gaschromatographie)für flüchtige Substanzen, GC-Trägergas mittels Jet-Separatorgrößtenteils abgetrennt Kombination einer Hochleistungstrennmethode mit einerHochleistungsanalysenmethode, äußerst leistungsfähig
•HPLC-MS Kombination (HPLC = high performance liquidchromatography)für polare Substanzen, Entfernung der polaren mobilen Phaseund Ionisierung mittels Elektrospray-Verfahren (ESI)
Ionenquelle
Vakuum
GC-Säule
Trägergas+ Substanz
SubstanzTrägergas
Jet-Separator

Ionisierung
• Entreißen eines Elektrons→ es entsteht ein Radikal-Kation der Ladung +1 (Molekül-Ion, M+•), sehr energiereich, fragmentiert stark, hauptsächlich bei EI
• Anlagerung eines Elektrons→ es entsteht ein Radikal-Kation der Ladung -1, weniger wichtig, nur für Moleküle mit hoher Elektronenaffinität (z.B. Halogenverbindungen)
• Protonierung→ durch Anlagerung eines Protons entsteht ein Kation [M+1]+
vorzugsweise an ‚basischen‘ Stellen des Moleküls (Atome mit freien Elektronenpaaren wie Amine, O-Verbindungen), bei CI, ESI, APCI, MALDI (Positiv-Modus); oft auch [M+Na]+
• Deprotonierung→ durch Entfernen eines Protons entsteht ein Anion [M-1]-, bei sauren Verbindungen (Carbonsäuren, Phenole), bei CI, ESI, APCI, MALDI (Negativ-Modus)

Ionisierungstechniken
Auswahl abhängig von • gewünschter Information• den Eigenschaften der Probe
leicht verdampfbare Substanzen(Verdampfungsmethoden)
schwer verdampfbareSubstanzen(Desorptionsmethoden)
Elektronenstoß-Ionisation(electron impact, EI)Chemische Ionisation (CI) → 'weiche' Ionisierungs-technik
Feld-Desorption (FD)Fast Atom Bombardement (FAB)matrix-assisted Laser-Desorption (MALDI)Sekundärionen-MS (SIMS)
Das Aussehen eines Massenspektrums hängt sehr stark von der jeweils verwendeten Ionisierungstechnik ab!
Substanzen in Lösung (Zerstäubungsmethoden)
Elektrospray-Ionisation (ESI)Atmospheric Pressure CI (APCI)Inductive Coupled Plasma (ICP)Desorption Electrospray Ionization (DESI)Atmospheric Pressure Photo Ionization (APPI)
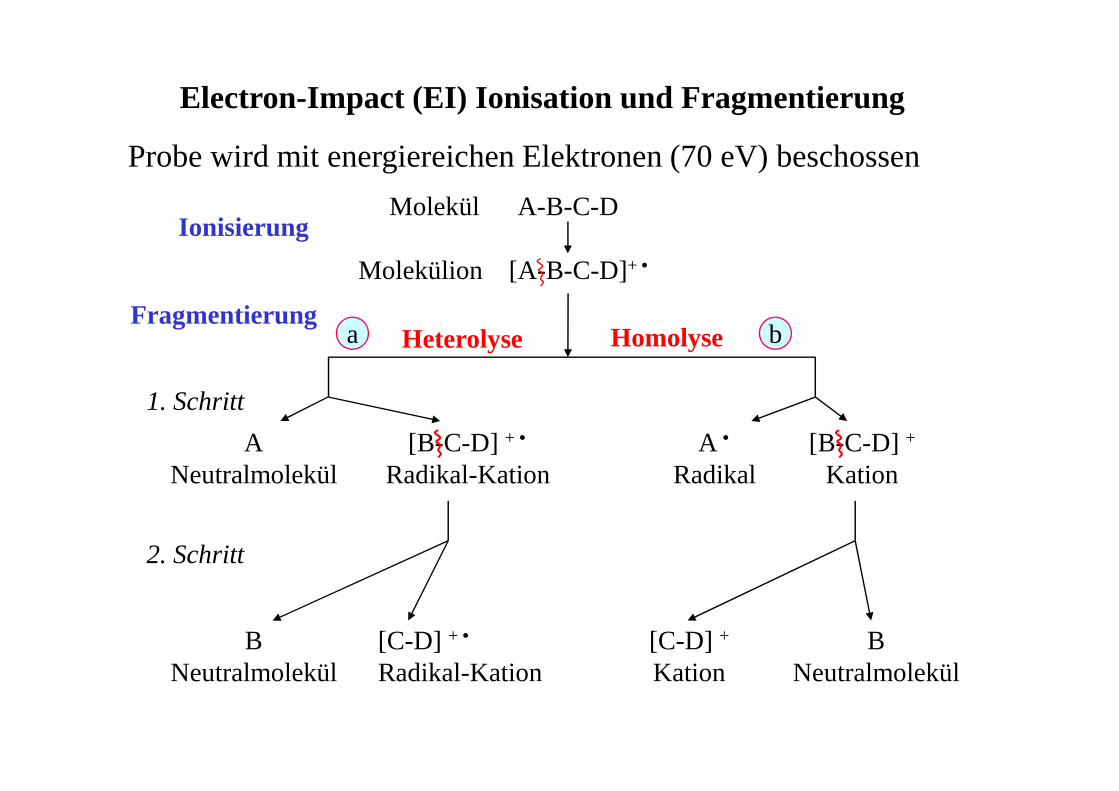
Electron-Impact (EI) Ionisation und Fragmentierung
Molekül A-B-C-D
Molekülion [A-B-C-D]+ •
Ionisierung
Fragmentierung
[B-C-D] + •
Radikal-KationA
NeutralmolekülA •
Radikal[B-C-D] +
Kation
1. Schritt
2. Schritt
[C-D] +
KationB
NeutralmolekülB
Neutralmolekül[C-D] + •
Radikal-Kation
a bHomolyseHeterolyse
Probe wird mit energiereichen Elektronen (70 eV) beschossen
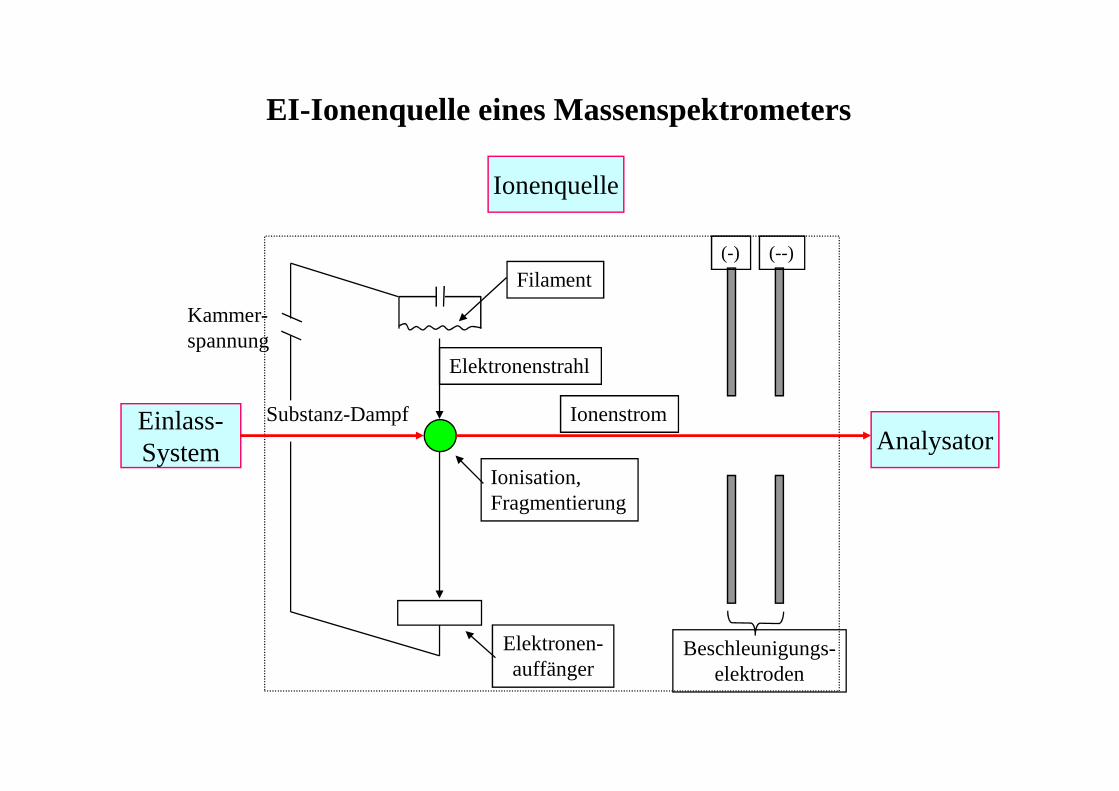
EI-Ionenquelle eines Massenspektrometers
Einlass-System
Ionenquelle
AnalysatorSubstanz-Dampf Ionenstrom
(-) (--)
Beschleunigungs-elektroden
Elektronenstrahl
Filament
Kammer-spannung
Elektronen-auffänger
Ionisation,Fragmentierung

Chemische Ionisation
1) CH4 →→→→ CH4+•••• + e- (Aktivierung des Gases)
2) CH4+•••• + CH4 →→→→ CH5
+ + CH3 ••••
3) CH5+ + M| →→→→ [M+H] + + CH4
Verwendung eines Hilfsgases, z. B. Methan, Isobutan, NH3
150 eV
[M+H] + ist kein Radikalkation → stabiler!
für saure Verbindungen: Verwendung eines basischen Hilfsgases (z.b. NH3) → negativ geladene Ionen [M-H]-
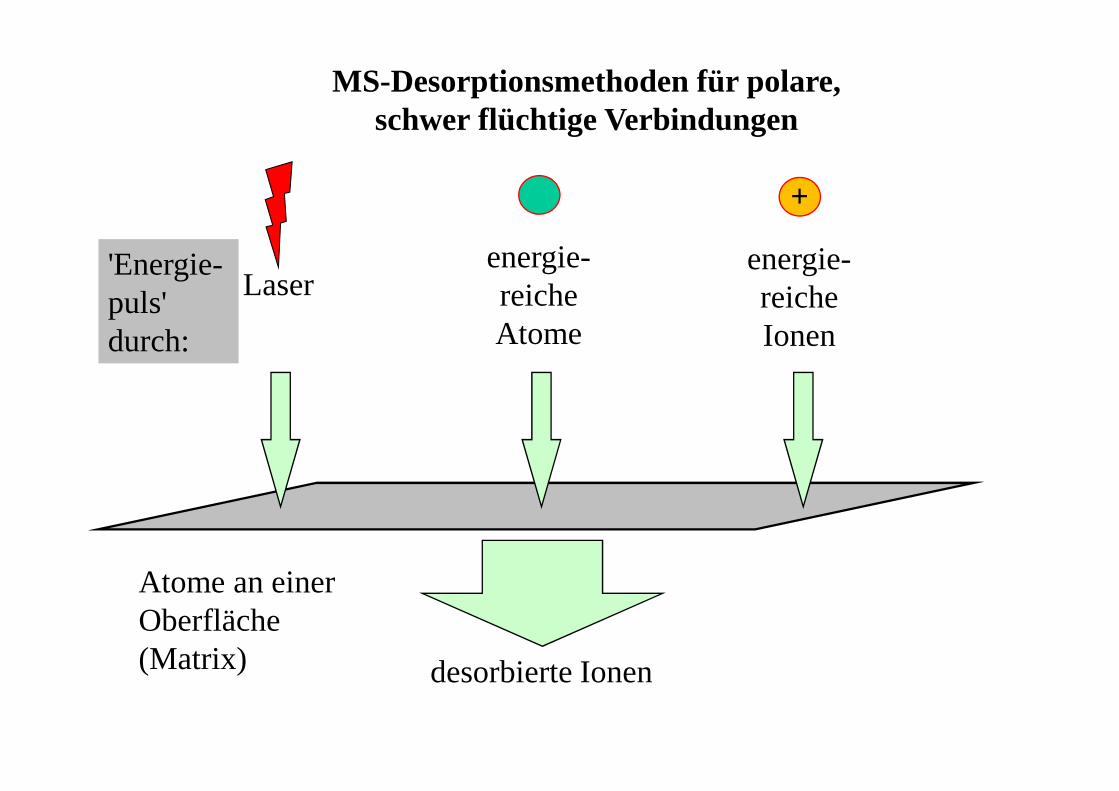
MS-Desorptionsmethoden für polare, schwer flüchtige Verbindungen
Laserenergie-reicheAtome
desorbierte Ionen
Atome an einer Oberfläche(Matrix)
'Energie-puls'durch:
energie-reicheIonen
+
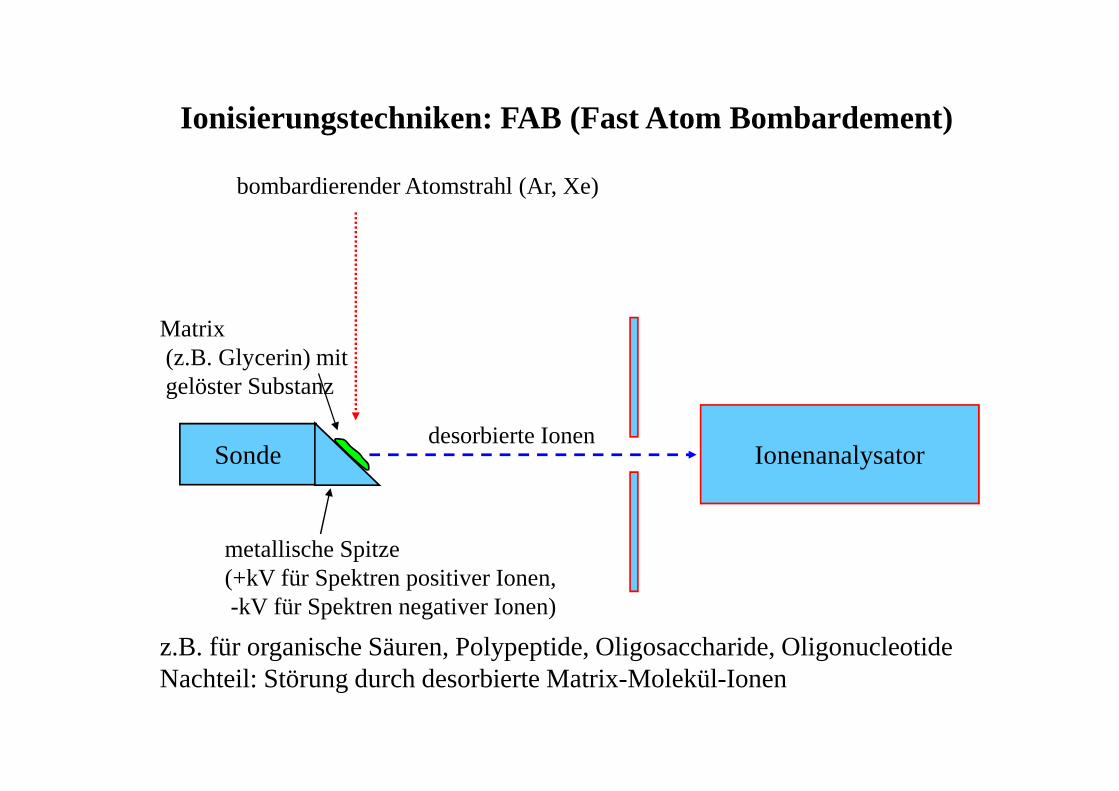
Ionisierungstechniken: FAB (Fast Atom Bombardement)
IonenanalysatorSonde
bombardierender Atomstrahl (Ar, Xe)
desorbierte Ionen
Matrix(z.B. Glycerin) mitgelöster Substanz
metallische Spitze(+kV für Spektren positiver Ionen,-kV für Spektren negativer Ionen)
z.B. für organische Säuren, Polypeptide, Oligosaccharide, OligonucleotideNachteil: Störung durch desorbierte Matrix-Molekül-Ionen
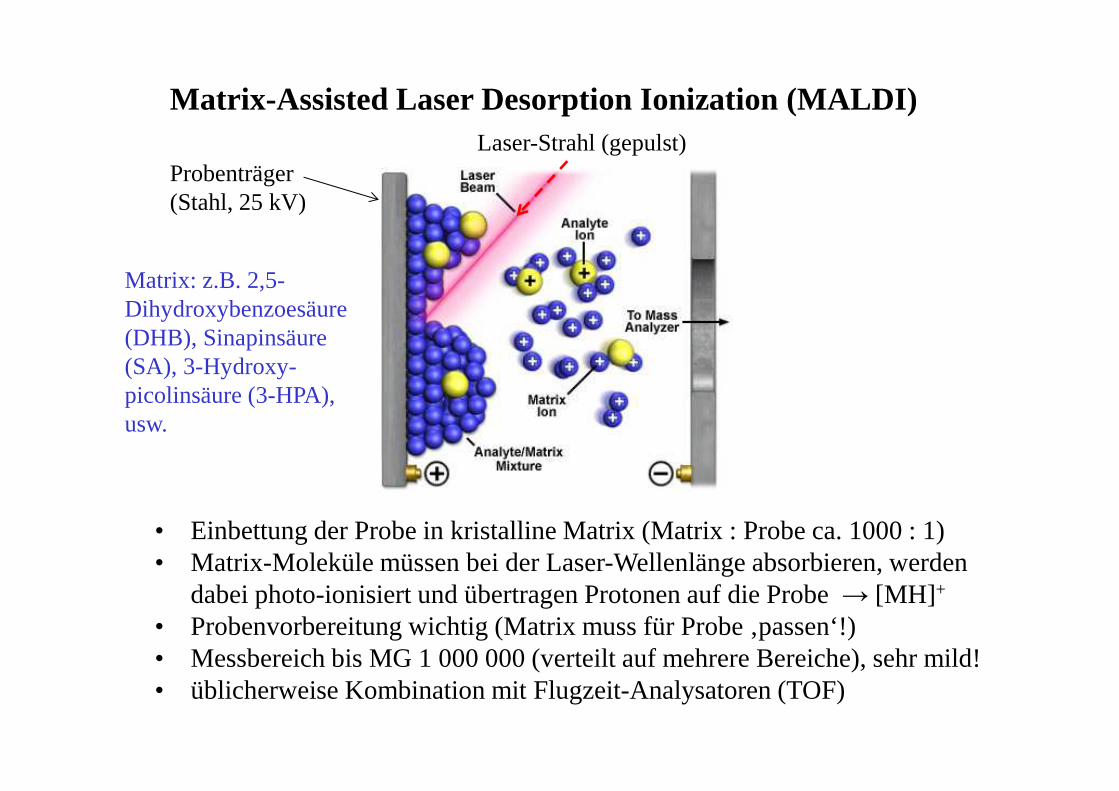
Matrix-Assisted Laser Desorption Ionization (MALDI)
Probenträger (Stahl, 25 kV)
Laser-Strahl (gepulst)
• Einbettung der Probe in kristalline Matrix (Matrix : Probe ca. 1000 : 1)• Matrix-Moleküle müssen bei der Laser-Wellenlänge absorbieren, werden
dabei photo-ionisiert und übertragen Protonen auf die Probe → [MH] +
• Probenvorbereitung wichtig (Matrix muss für Probe ‚passen‘!)• Messbereich bis MG 1 000 000 (verteilt auf mehrere Bereiche), sehr mild!• üblicherweise Kombination mit Flugzeit-Analysatoren (TOF)
Matrix: z.B. 2,5-Dihydroxybenzoesäure (DHB), Sinapinsäure (SA), 3-Hydroxy-picolinsäure (3-HPA), usw.

weitere Desorptionsmethoden
Sekundärionen-MS (SIMS)Durch Beschuss mit energiereichen Ionen (Cs+, Ar+, Ga+) werden aus der Probe, die sich auf einer Metalloberfläche befindet, positive und negative Ionen erzeugt; für Peptide und Oligosaccharide, zur Analyse von Kontaminationen auf Oberflächen
Feld-Desorption: Probe wird von einem aktivierten Draht (beschichtet mit feinsten Kohlenstoff-Nadeln) unter Einfluss starker elektrischer Felder desorbiert
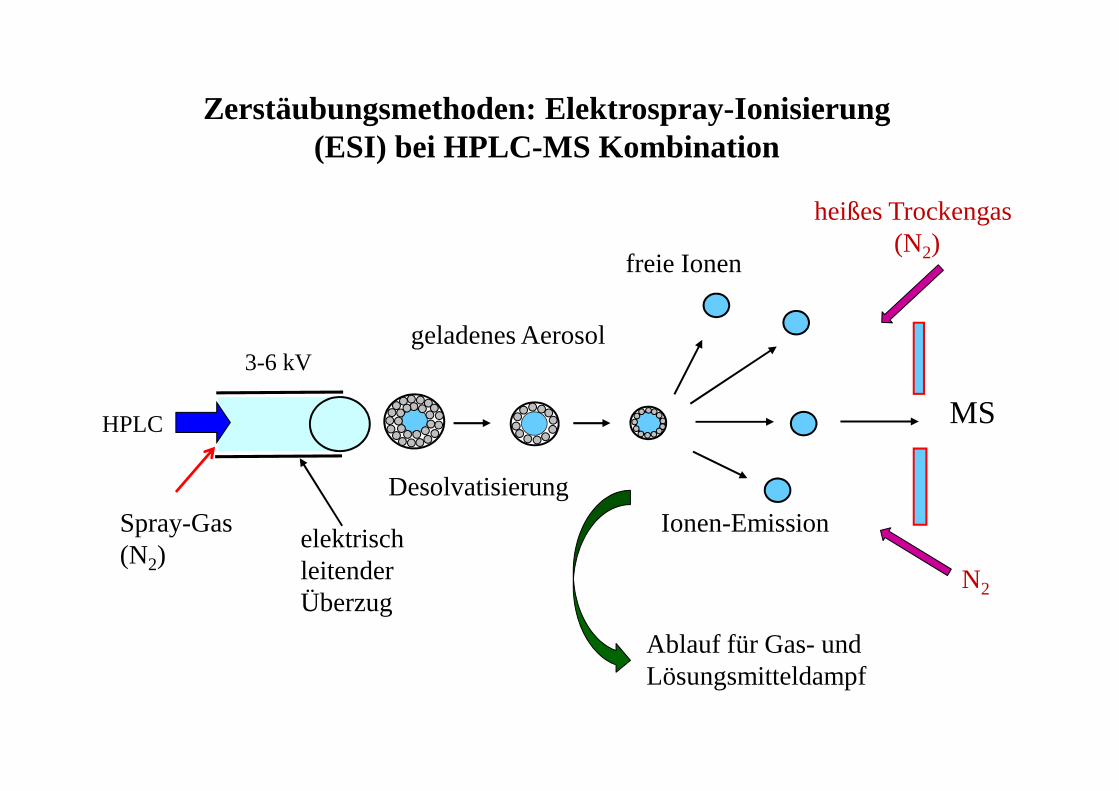
Zerstäubungsmethoden: Elektrospray-Ionisierung(ESI) bei HPLC-MS Kombination
3-6 kV
elektrisch leitender Überzug
MS
geladenes Aerosol
DesolvatisierungIonen-Emission
freie Ionen
HPLC
Spray-Gas (N2)
heißes Trockengas(N2)
Ablauf für Gas- und Lösungsmitteldampf
N2

weitere ZerstäubungsmethodenAtmospheric Pressure Chemical Ionization (APCI)Ähnlich ESI, aber keine Spannung auf der Spraykapillare, Ionisierung des Dampfgemisches (Probe plus N2) via Entladungsnadel Vorteil gegenüber ESI: auch für apolare Verbindungen; Nachteil: höhere thermische Belastung
Inductive Coupled Plasma (ICP)für Bestimmung von Elementen, Eluat trifft unter Inertgas auf ca. 10 000 °C heiße Fackel, Zersetzung in atomare Bestandteile, Bildung von Atom-Ionen
Desorption Electrospray Ionization (DESI) Oberflächenanalyse (z.B. DC-Platten); geladene LM-Tröpfchen werden auf Versuchsfläche gesprayt
Atmospheric Pressure Photo Ionization (APPI)ähnlich APCI, Ionen werden aus dem Spray durch Bestrahlung mit UV-Lampe erzeugt, für lipophile, unpolare Substanzen
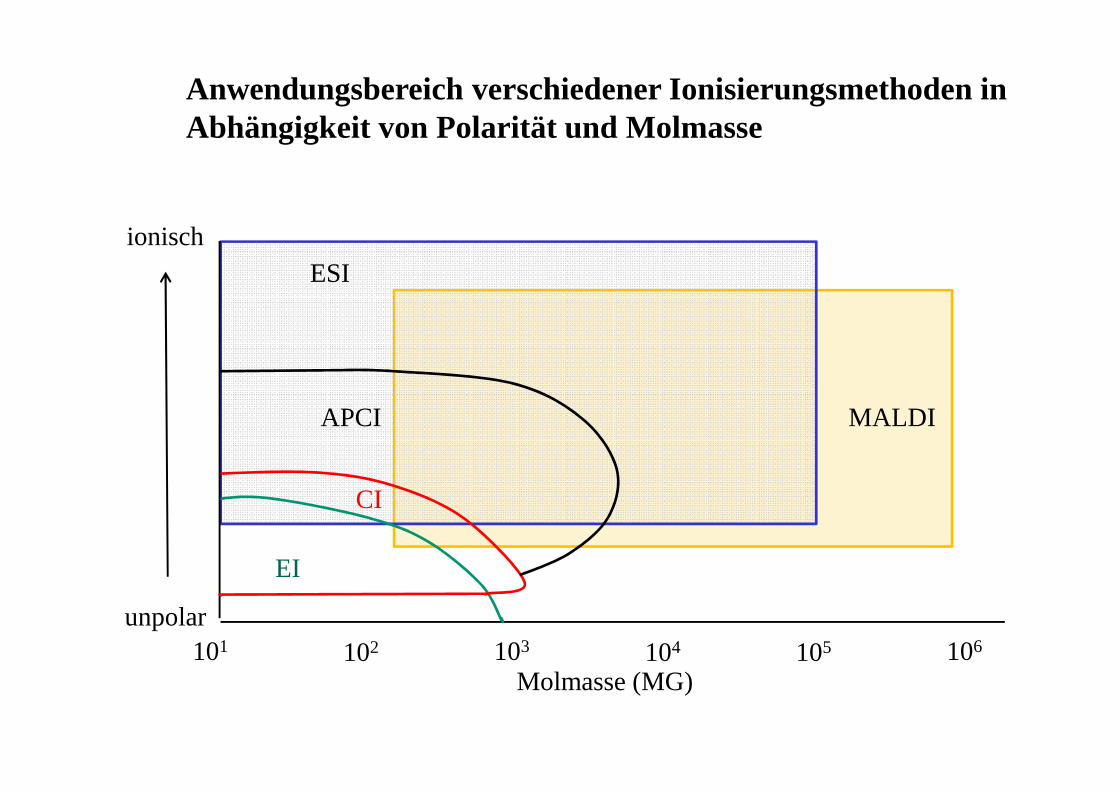
101 102 103 104 105 106
ionisch
unpolar
EI
CI
APCI MALDI
ESI
Molmasse (MG)
Anwendungsbereich verschiedener Ionisierungsmethoden in Abhängigkeit von Polarität und Molmasse
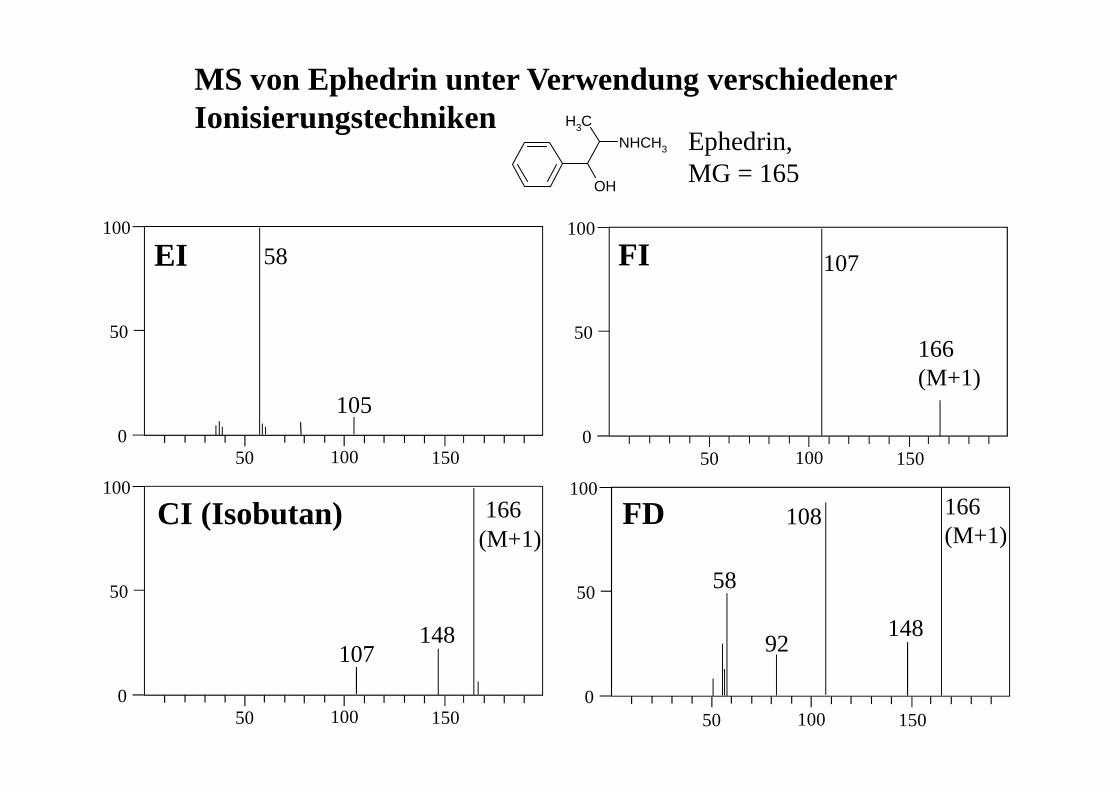
MS von Ephedrin unter Verwendung verschiedener Ionisierungstechniken
50 100 1500
50
100
OH
NHCH3
CH3
50 100 1500
50
100
50 100 1500
50
100
50 100 1500
50
100
Ephedrin,MG = 165
EI FI
CI (Isobutan) FD
105
58 107
166 (M+1)
107148
166(M+1)
166(M+1)
58
92
108
148

MS - Analysatoren
• magnetischer Sektor (Sektorfeldgeräte)
• Quadrupol-Analysator
• Ionenfalle (QIT) und Orbitrap
• time of flight (TOF)
• Fourier transform Ionencyclotronresonanz (FT-ICR)
Kenngrößen eines Analysators:• Massenbereich (Messbereich, z.B. zwischen m/z 50-2000)• Scangeschwindigkeit (Geschwindigkeit, mit dem der
Massenbereich durchgescannt werden kann, z.B. 2000 m/z s-1)• Genauigkeit (Abweichung von der berechneten Masse des
Ions, ∆m/m)• Auflösung: siehe 10% TAL-Definition
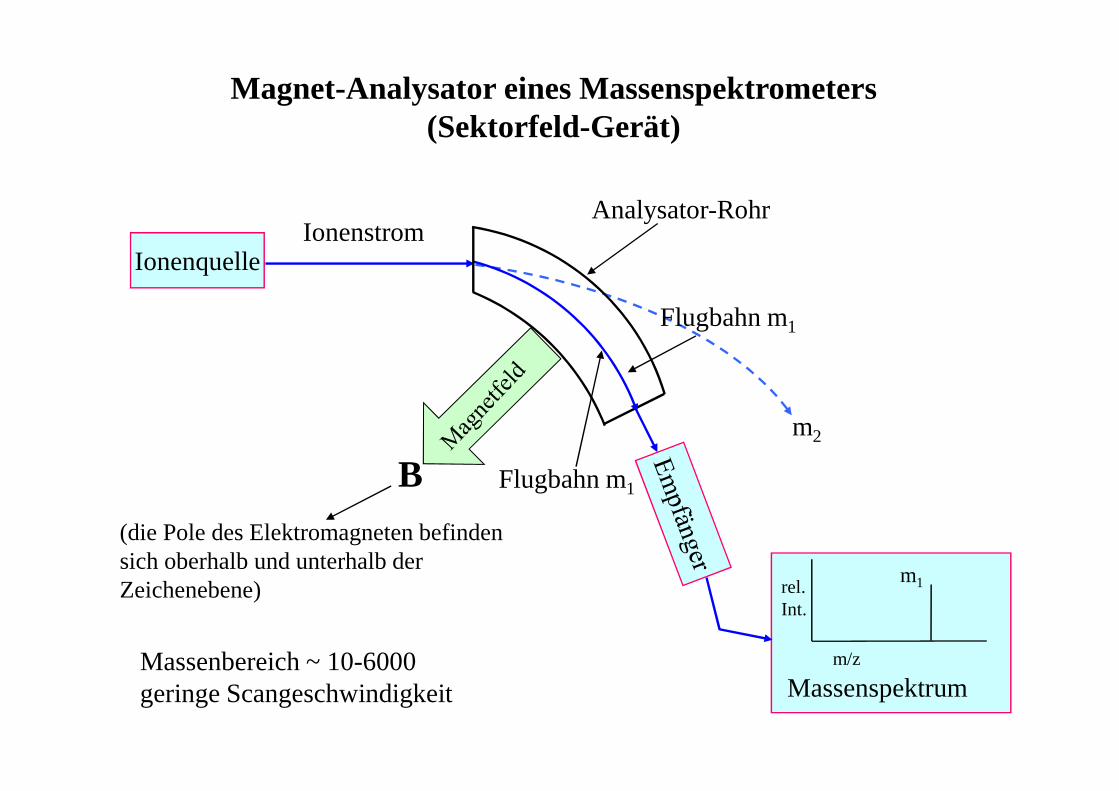
Magnet-Analysator eines Massenspektrometers(Sektorfeld-Gerät)
Ionenquelle
m/z
rel.Int.
Massenspektrum
m1
m2
IonenstromAnalysator-Rohr
Flugbahn m1
Flugbahn m1
B(die Pole des Elektromagneten befindensich oberhalb und unterhalb derZeichenebene)
Massenbereich ~ 10-6000geringe Scangeschwindigkeit

Auftrennung von Ionen im Magnetfeld(Sektorfeld-Gerät)
r FlugbahnradiusB MagnetfeldstärkeU Beschleunigungsspannungm Masse des Ionsz Ladung des Ions (meist 1)
z
mU2
B
1r
⋅⋅=
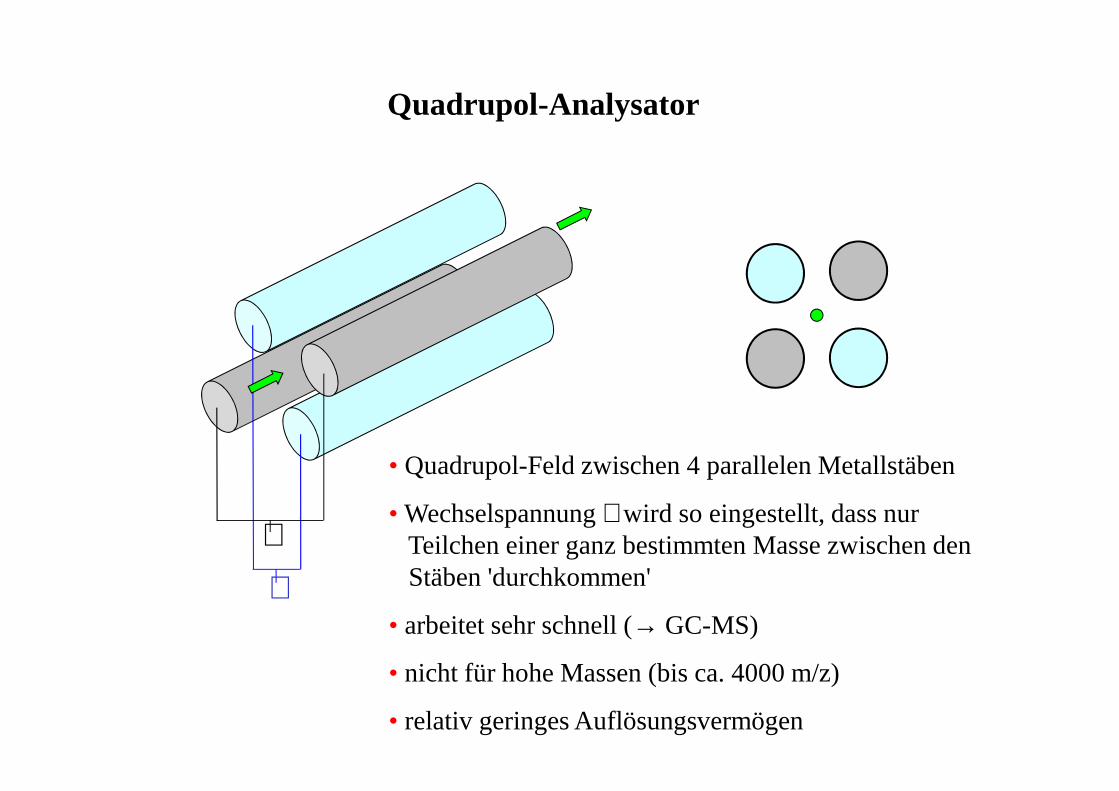
Quadrupol-Analysator
∼
∼
• Quadrupol-Feld zwischen 4 parallelen Metallstäben
• Wechselspannung ∼ wird so eingestellt, dass nur Teilchen einer ganz bestimmten Masse zwischen denStäben 'durchkommen'
• arbeitet sehr schnell (→ GC-MS)
• nicht für hohe Massen (bis ca. 4000 m/z)
• relativ geringes Auflösungsvermögen

Quadrupol-Ionenfalle (Ion Trap, QIT, Paul-Falle)
• Ionenquelle und Massenanalysator befinden sich in einer gemeinsamenKammer
• positive Ionen werden durch ein Radiofrequenzfeld (Ring-Elektrode) ineinem zylinderförmigen Behältnis 'gefangen'
• nach einem Stablilisierungsschritt wird die Spannung an derRingelektrode sukzessive erhöht und die Ionen nach steigender Massefreigesetzt und detektiert
• Massenbereich 50-6000, hohe Scangeschwindigkeit

Orbitrap-Analysatoren
•Ionen werden in elektrostatischem Feld gefangen, umkreisen die spindelförmige Zentralelektrode und schwingen gleichzeitig entlang der z-Achse in Abhängigkeit von m/z•Die Schwingungen erzeugen in den äußeren, gespaltenen Elektroden Signale, die mittels Fourier-Transformation in m/z-Werte umgewandelt werden•Vorteile: hohe Genauigkeit, hohe Auflösung, Massenbereich bis 6000 m/z,technisch viel weniger aufwendig als FT-ICR (keine Hochfeld-Magnete nötig)
Zentralelektrode
äußere Elektrode
Schwingung
m/zt ∝TOF
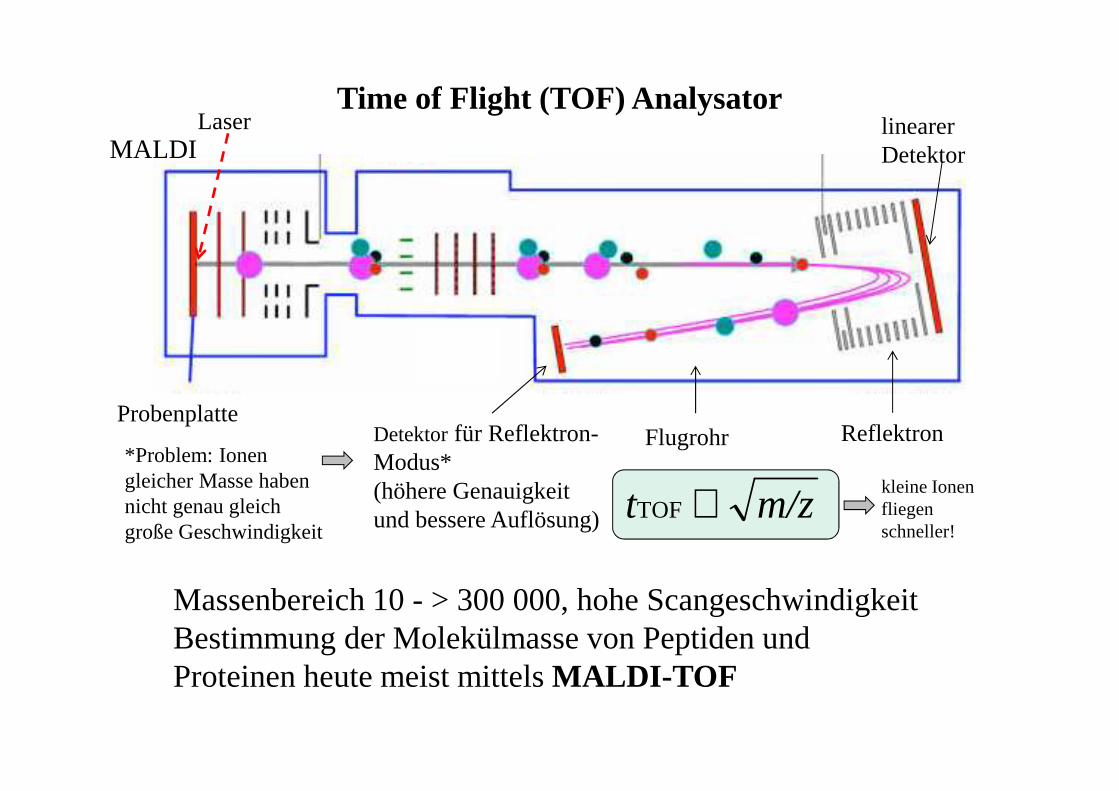
Time of Flight (TOF) Analysator
Massenbereich 10 - > 300 000, hohe ScangeschwindigkeitBestimmung der Molekülmasse von Peptiden und Proteinen heute meist mittels MALDI-TOF
ProbenplatteReflektron
linearerDetektor
Detektorfür Reflektron-Modus*(höhere Genauigkeit und bessere Auflösung)
Laser
Flugrohr
MALDI
kleine Ionen fliegen schneller!
*Problem: Ionen gleicher Masse haben nicht genau gleich große Geschwindigkeit
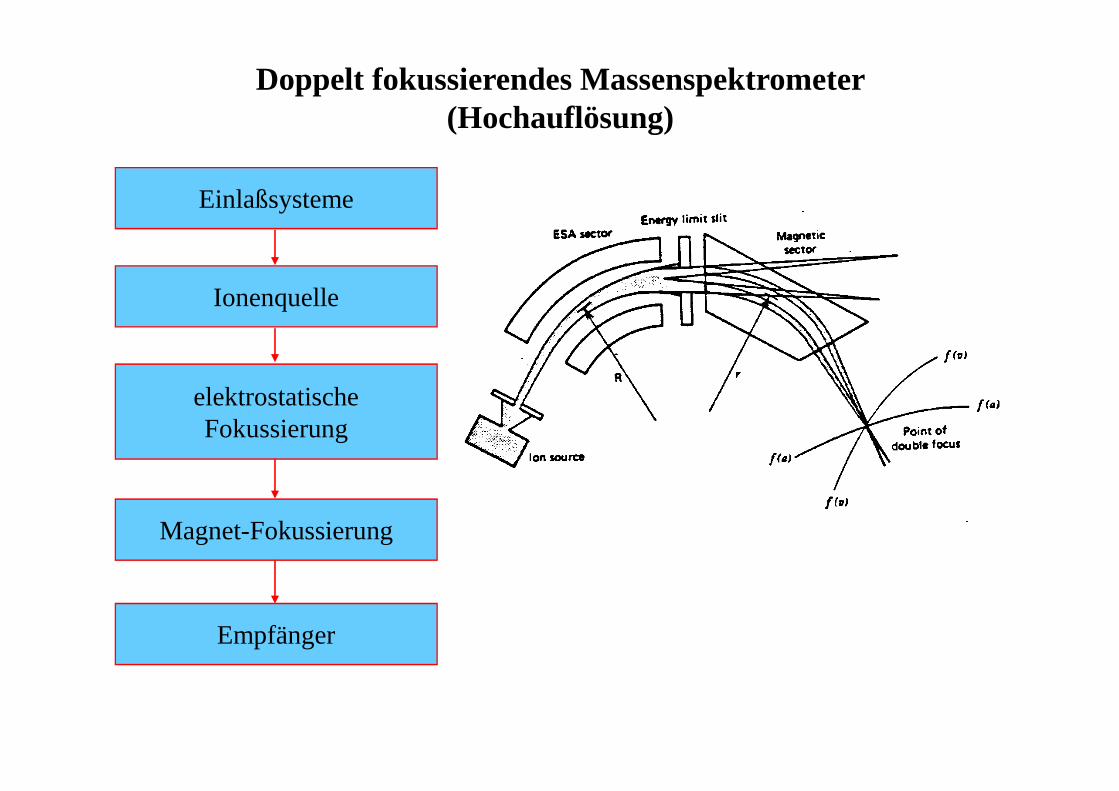
Doppelt fokussierendes Massenspektrometer (Hochauflösung)
Einlaßsysteme
Ionenquelle
elektrostatischeFokussierung
Magnet-Fokussierung
Empfänger
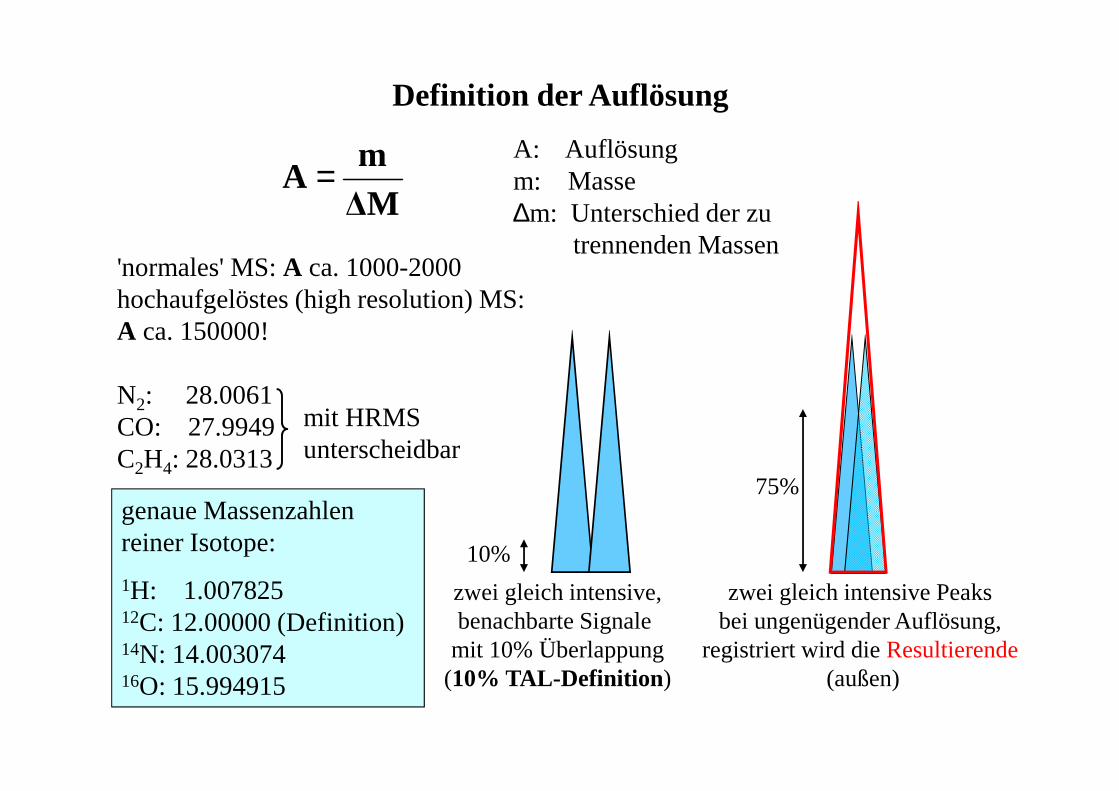
Definition der Auflösung
A: Auflösungm: Masse∆m: Unterschied der zu
trennenden Massen
10%
75%
zwei gleich intensive,benachbarte Signale
mit 10% Überlappung(10% TAL-Definition )
zwei gleich intensive Peaksbei ungenügender Auflösung,
registriert wird die Resultierende(außen)
'normales' MS: A ca. 1000-2000hochaufgelöstes (high resolution) MS:A ca. 150000!
N2: 28.0061CO: 27.9949C2H4: 28.0313
mit HRMS unterscheidbar
genaue Massenzahlen reiner Isotope:
1H: 1.00782512C: 12.00000 (Definition)14N: 14.00307416O: 15.994915
∆Mm
A =
Periodensystem1H1.0079
2He4.0026
3Li6.939
4Be9.0122
5B10.811
6C12.011
7N14.007
8O15.999
9F18.998
10Ne20.179
11Na20.99
12Mg24.305
13Al26.982
14Si28.086
15P30.974
16S32.06
17Cl35.453
18Ar39.948
19K39.098
20Ca40.08
21Sc44.956
22Ti47.90
23V50.942
24Cr51.996
25Mn54.938
26Fe55.847
26Co58.933
28Ni58.71
29Cu63.546
30Zn65.38
31Ga69.735
32Ge72.59
33As74.922
34Se78.96
35Br79.904
36Kr83.80
37Rb85.47
38Sr87.62
39Y88.905
40Zr91.22
41Nb92.906
42Mo95.94
43Tc98.906
44Ru101.07
45Rh102.91
46Pd106.4
47Ag107.87
48Cd112.4
49In114.82
50Sn118.69
51Sb121.75
52Te127.60
53J126.90
54Xe131.30
55Cs132.91
56Ba137.34
57La138.91
72Hf178.49
73Ta180.95
74W183.85
75Re186.2
76Os190.2
77Ir192.2
78Pt195.09
79Au196.97
80Hg200.59
81Tl204.37
82Pb207.19
83Bi208.98
84Po(209)
85At(210)
86Rn(222)
87Fr(223)
88Ra226.03
89Ac(227)
104Rf(261)
105Db(262)
106Sg(263)
107Bh(262)
108Hs(265)
109Mt(266)
58Ce140.12
59Pr140.91
60Nd144.24
61Pm(145)
62Sm150.35
63Eu151.96
64Gd157.25
65Tb158.93
66Dy162.50
67Ho164.93
68Er167.26
69Tm168.93
70Yb173.04
71Lu174.97
90Th232.04
91Pa231.04
92U238.03
93Np237.05
94Pu(224)
95Am(243)
96Cm(247)
97Bk(247)
98Cf(251)
99Es(254)
100Fm(257)
101Md(258)
102No(259)
103Lw(260)

Massenanalyse – Begriffe
Einheit: [u] ‚unit‘: 1/12 der Masse des Isotops 12C (synonym: Da (Dalton), amu)
nominale Masse: Summe der zu ganzen Zahlen gerundeten Atommassen einer Verbindung. Verbindungen gleicher nominaler Masse bezeichnet man als isobar(auch wenn sich diese in ihrer exakten Masse unterscheiden) z.B. N2, CO, C2H4
– alle 28
exakte Masse: die mit hoher Genauigkeit ( ≥4 Nachkommastellen, doppelt fokussierende Geräte) gemessene Masse: z.B. N2: 28.0061
isotopische Masse: exakte Masse eines Isotops
monoisotopischeMasse: Masse jenes Teilchens, bei dem nur die häufigsten Isotope der jeweiligen Elemente zur Gesamtmasse beitragenKohlenstoff: 12C, 13C, (14C); Wasserstoff: 1H = H, 2H = D, (3H=T)Methan: Mmi entspricht exakter Masse von 12CH4; nicht: 13CH4, 12CDH3, 12CD2H2, 12CD3H, 12CD4; 13CH4, 13CDH3, 13CD2H2, 13CD3H, 13CD4, 14CH4…usw……(14CT4).
‚Molekulargewicht‘: jener Massenwert, der sich aus der Summe der mittleren Massen der Elemente (über deren Isotopenverteilung gemittelt) ergibt – kann nicht direkt gemessen werden!
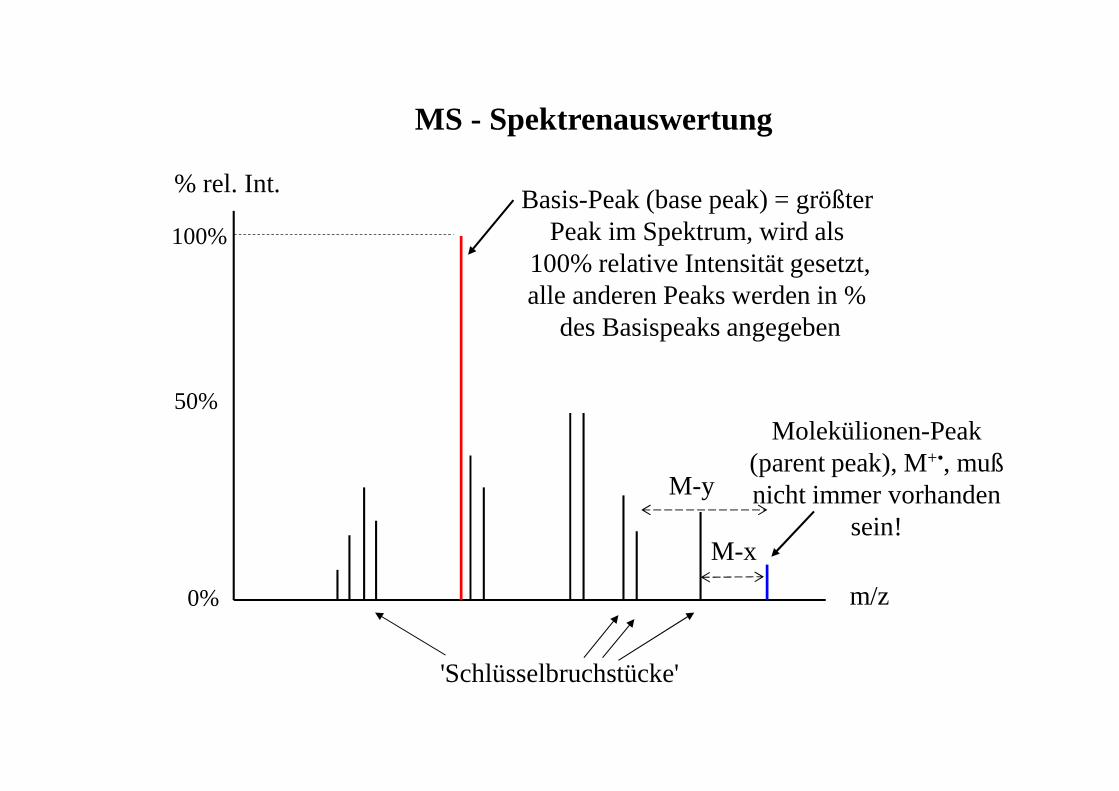
MS - Spektrenauswertung
m/z
% rel. Int.
100%
50%Molekülionen-Peak
(parent peak), M+•, mußnicht immer vorhanden
sein!
Basis-Peak (base peak) = größter Peak im Spektrum, wird als
100% relative Intensität gesetzt,alle anderen Peaks werden in %
des Basispeaks angegeben
0%
M-x
M-y
'Schlüsselbruchstücke'

Isotopenpeaks - Nachweis bestimmter Elemente - Brom
Brom: Gemisch aus 2 Isotopen 79Br : 81Br 1 : 1
1 Brom-Atom im Molekül (z.B. HBr) :
2 Brom-Atome im Molekül (z.B. Br2):
79Br–79Br → MG 158 W = 179Br–81Br → MG 160 81Br–79Br → MG 16081Br–81Br → MG 162 W = 1
W = 1+1 = 2
3 Brom-Atome im Molekül: → 1 : 3 : 3 : 1aaa X 1aab aba baa X+2 1 + 1 + 1 = 3abb bab bba X+4 1 + 1 + 1 = 3bbb X+6 1
80 82
1 : 1
HBr
158 160
1 : 2 : 1
Br2
162
X X+2
1 : 3 : 3 : 1
3 Br im Molekül-bzw. Fragmention
X+4 X+6

Isotopenpeaks - Nachweis bestimmter Elemente - Chlor
Chlor: Gemisch aus 2 Isotopen 35Cl : 37Cl 3 : 1
1 Chlor-Atom im Molekül (z.B. HCl) :
2 Chlor-Atome im Molekül (z.B. Cl2):
35Cl–35Cl → MG 70 W = 3•3 = 935Cl–37Cl → MG 72 W = 3•1 = 3 37Cl–35Cl → MG 72 W = 1•3 = 337Cl–37Cl → MG 74 W = 1•1 = 1
3+3 = 6
3 Chlor-Atome im Molekül: → 27 : 27 : 9 : 1aaa X W = 3•3•3 = 27aab aba baa X+2 W = 3•3•1 + 3•1•3 + 1•3•3 = 27abb bab bba X+4 W = 3•1•1 + 1•3•1 + 1•1•3 = 9bbb X+6 W = 1•1•1 = 1
36 38
3 : 1
HCl
70 72
9 : 6 : 1Cl2
74

Isotopenpeaks - allgemeine Formel
(a + b)n
binomischer Lehrsatz:
(a + b)1 = a + b
(a + b)2 = a2 + 2ab + b2
(a + b)3 = a3 + 3a2b + 3ab2 + b3
die Zahl der Glieder des Binoms gibt die Anzahl der Isotopenpeaks an, die Zahlen selbst das Verhältnis der Intensitäten
z.B. für 2 Cl-Atome im Molekül:(3 + 1)2 = 9 + 2•3•1 + 1 = 9 + 6 +1
a: leichteres Isotop b: schwereres Isotopn: Anzahl der Atome des isotopen Elementsa/b: Mengenverhältnis der Isotopen
Isotopenpeaks treten auch z.B. bei C auf, allerdings kaum auswertbar (12C : 13C ∼ 99 : 1) → sehr kleine 'Nebenpeaks'→ 'Schotter'

wichtig für Strukturaufklärung (Aufstellen einer Strukturformel) mittels MS:
• Elemente O, C, S: geradeWertigkeit, geradesAtomgewicht
• Elemente H, F, Cl, Br , I : ungeradeWertigkeit, ungeradesAtomgewicht
→ Verbindungen, die nur diese Elemente enthalten, habenimmer eine geradzahligeMolekülmasse (Molekulargewicht)
Ausnahme: N ungeradeWertigkeit, geradesAtomgewicht
→ Verbindungen mit einer ungeradenAnzahl an N-Atomen haben stets ein ungerades Molekulargewicht!
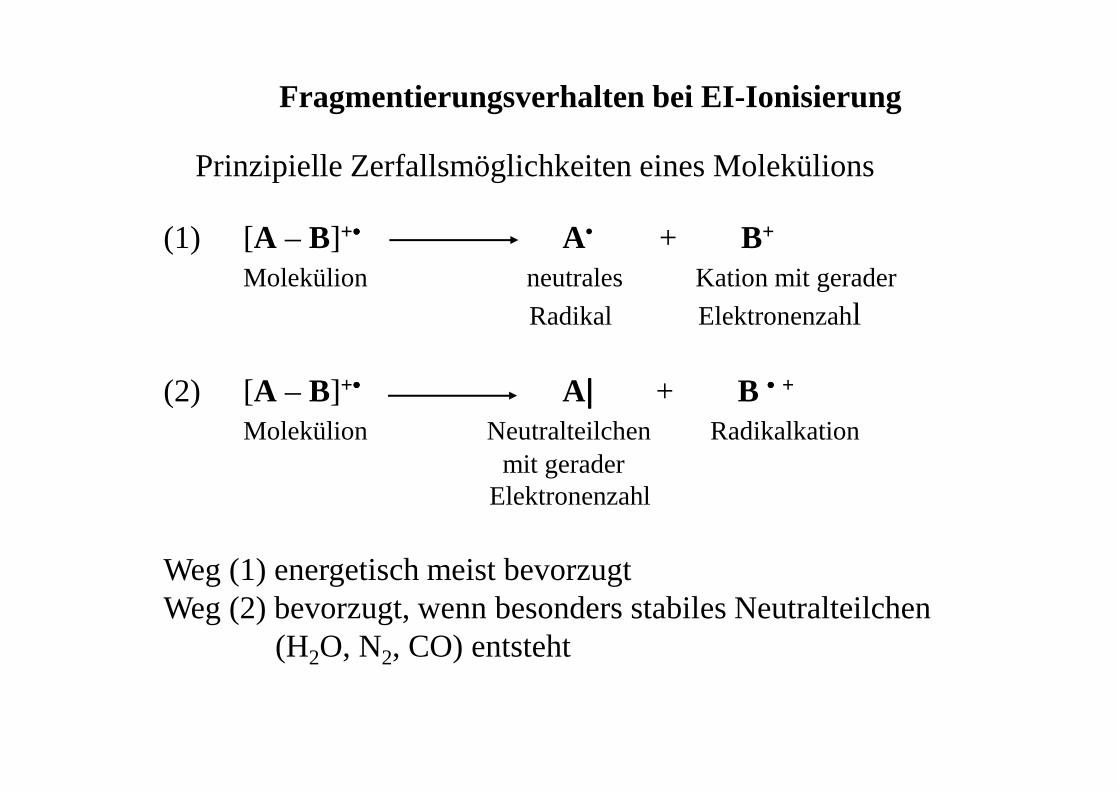
Prinzipielle Zerfallsmöglichkeiten eines Molekülions
(1) [A – B]+•••• A• + B+
Molekülion neutrales Kation mit gerader
Radikal Elektronenzahl
(2) [A – B]+•••• A |||| + B •••• +
Molekülion Neutralteilchen Radikalkationmit gerader
Elektronenzahl
Weg (1) energetisch meist bevorzugtWeg (2) bevorzugt, wenn besonders stabiles Neutralteilchen
(H2O, N2, CO) entsteht
Fragmentierungsverhalten bei EI-Ionisierung

Einige charakteristische Neutralteilchen, die aus M+• abgespalten werden
Neutralteilchen aus führt zu Ion mit m/z
H Aldehyden M-1
CH3 Methylverbindungen M-15
H2O Alkoholen M-18
C2H4
CO ex McLafferty M-28
N2
CHO Aldehyden M-29
C2H5 Ethylverbindungen M-29
OCH3 Methoxyverbindungen M-31
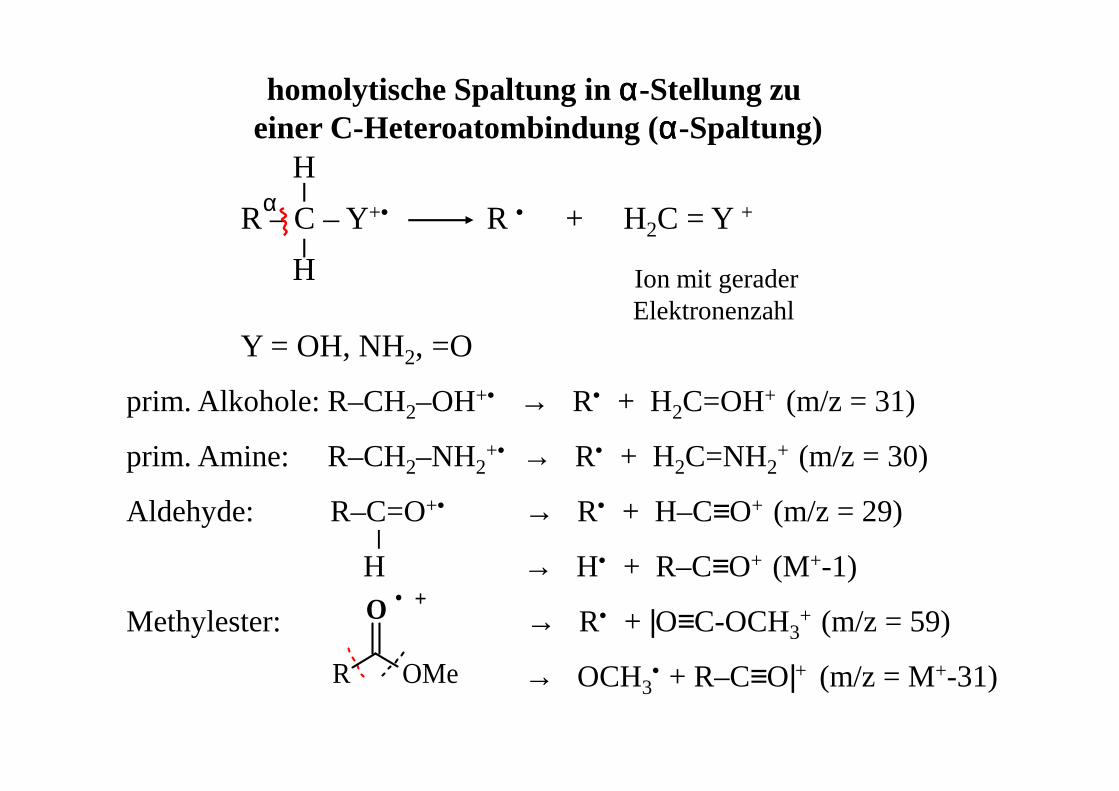
homolytische Spaltung in αααα-Stellung zu einer C-Heteroatombindung (αααα-Spaltung)
R – C – Y+• R • + H2C = Y +
Ion mit geraderElektronenzahl
Y = OH, NH2, =O
H
H
prim. Alkohole: R–CH2–OH+• → R• + H2C=OH+ (m/z = 31)
prim. Amine: R–CH2–NH2+• → R• + H2C=NH2
+ (m/z = 30)
Aldehyde: R–C=O+• → R• + H–C≡O+ (m/z = 29)
H → H• + R–C≡O+ (M+-1)
Methylester: → R• + |O≡C-OCH3+ (m/z = 59)
→ OCH3• + R–C≡O|+ (m/z = M+-31)
α
OMe
O · +
R
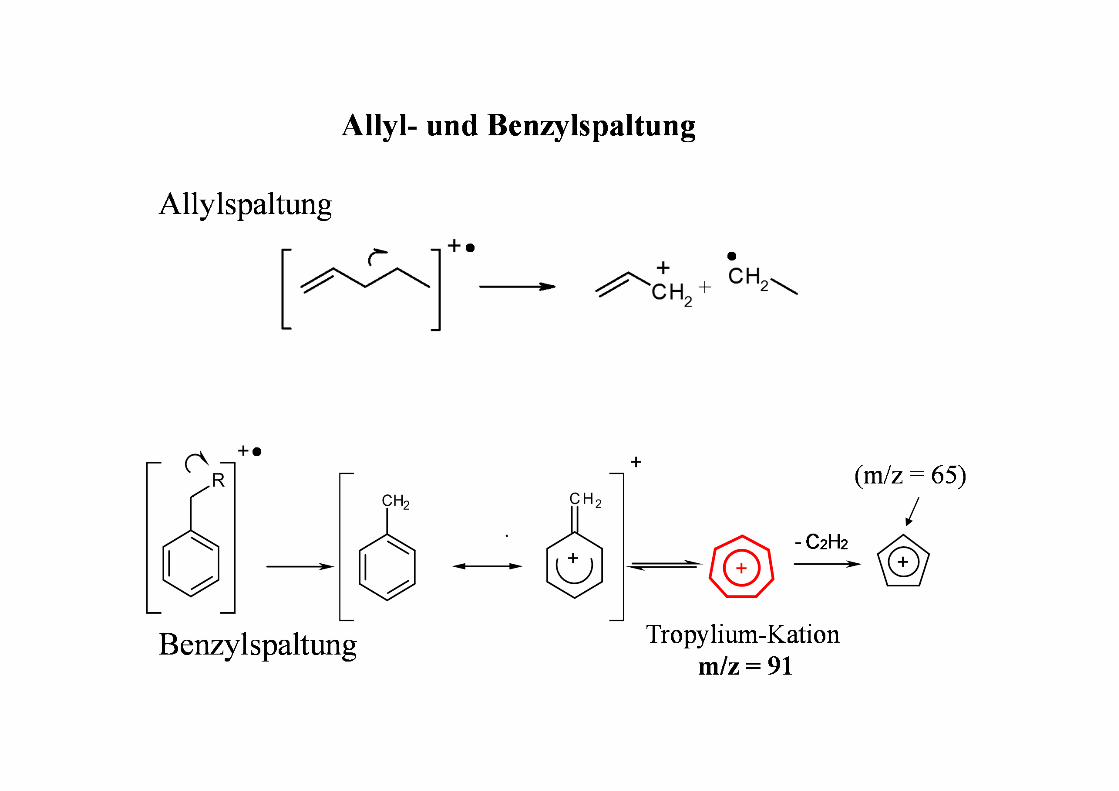
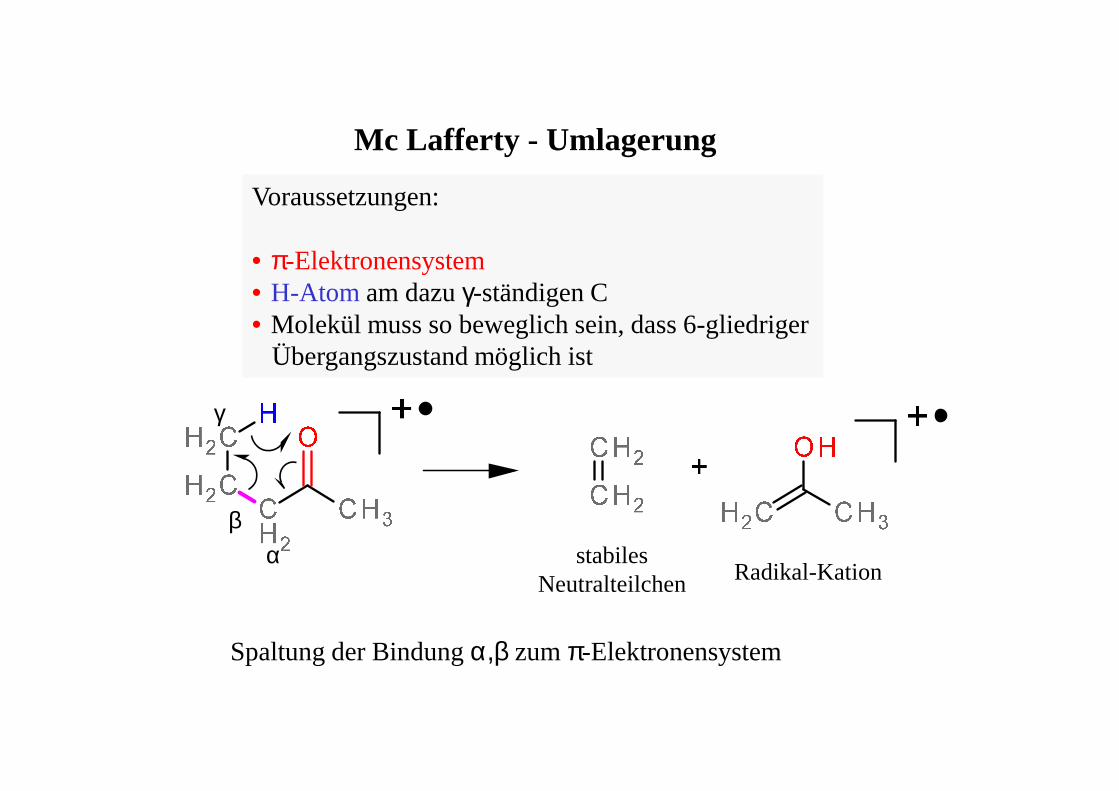
Mc Lafferty - Umlagerung
Voraussetzungen:
• π-Elektronensystem• H-Atom am dazu γ-ständigen C• Molekül muss so beweglich sein, dass 6-gliedriger
Übergangszustand möglich ist
Spaltung der Bindung α,β zum π-Elektronensystem
αβ
γ
stabilesNeutralteilchen Radikal-Kation
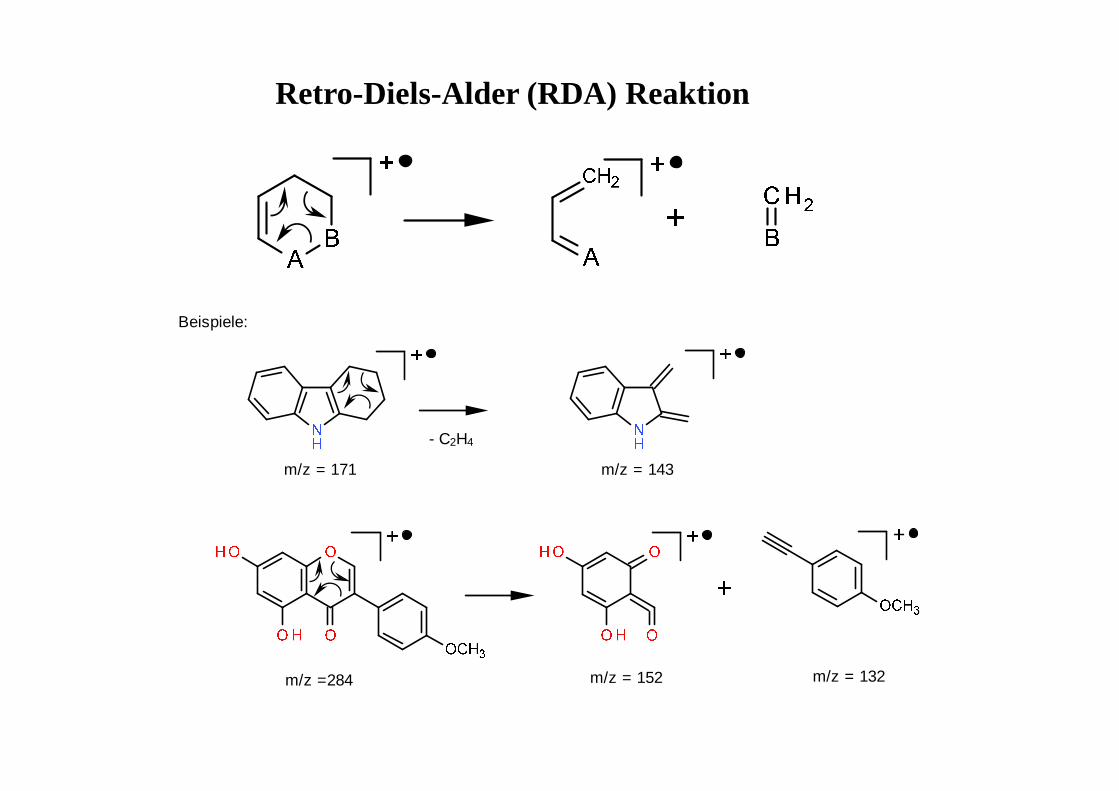
Retro-Diels-Alder (RDA) Reaktion
- C2H4
m/z = 171 m/z = 143
Beispiele:
m/z =284 m/z = 152 m/z = 132
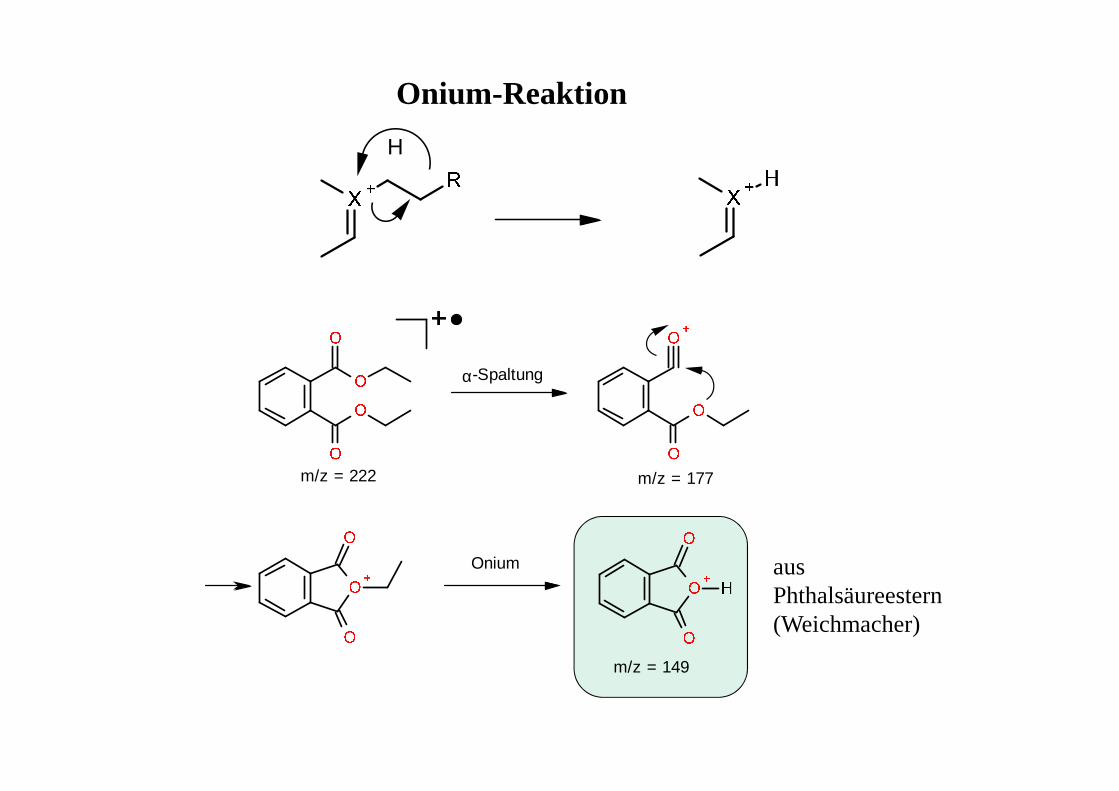
α-Spaltung
Onium
m/z = 222 m/z = 177
m/z = 149
Onium-Reaktion
H
aus Phthalsäureestern (Weichmacher)

charakteristische Fragmentionen
m/z Ion aus
—————————————————————————
29 HCO+ Aldehyden
C2H5+ Ethyl-Verbindungen
30 CH2NH2+ prim. Amin
31 CH2OH+ prim. Alkohol
OCH3+ Methoxy-Verbindungen
43 C3H7+ Propyl-Verbindungen
CH3CO+ CH3-CO-X
59 CH3OCO+ Methylester
77 C6H5+ Phenyl-Verbindungen
91 Tropylium+ Benzyl-Verbindungen
105 C6H5CO+ Benzoyl-Verbindungen

Übergangssignale (metastabile Ionen)
mT: Masse des Tochterions
mM: Masse des Mutterions
zM: Ladung des Mutterions (meist 1)
zT: Ladung des Tochterions (meist 1)
TM
M2
T
zm
zmm
⋅⋅=∗
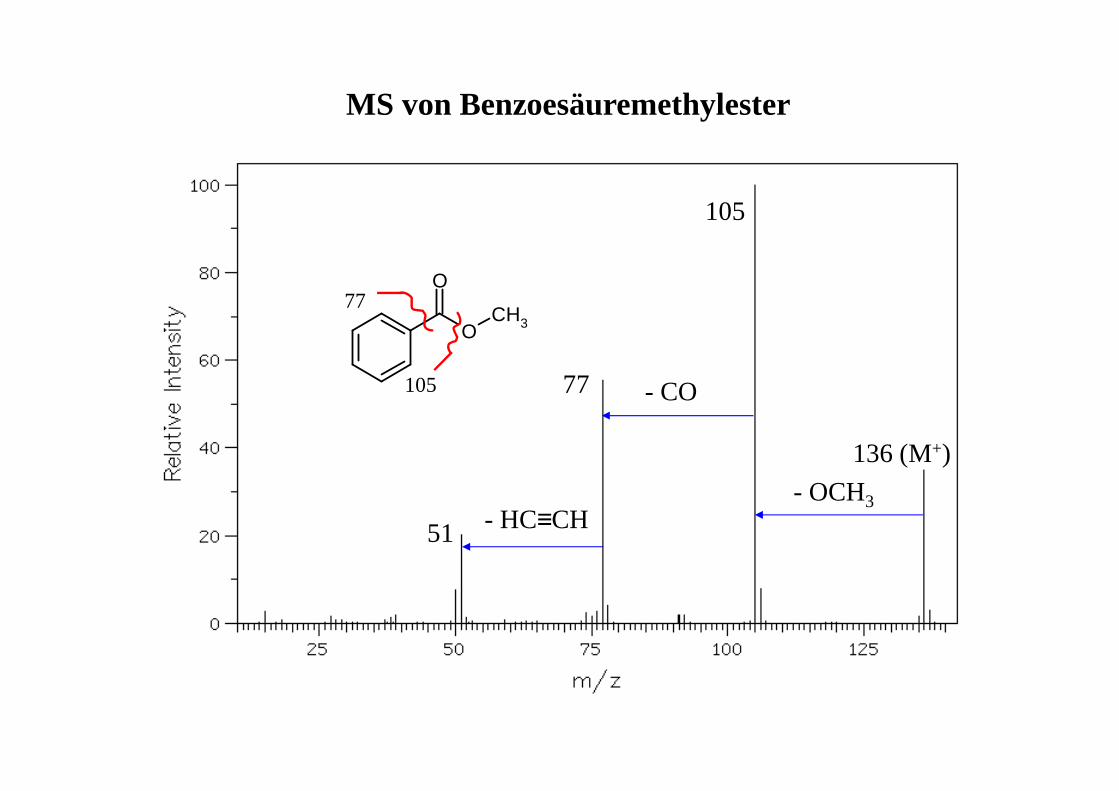
MS von Benzoesäuremethylester
51
77
105
136 (M+)
- HC≡CH
- CO
- OCH3
O
O
CH3
77
105
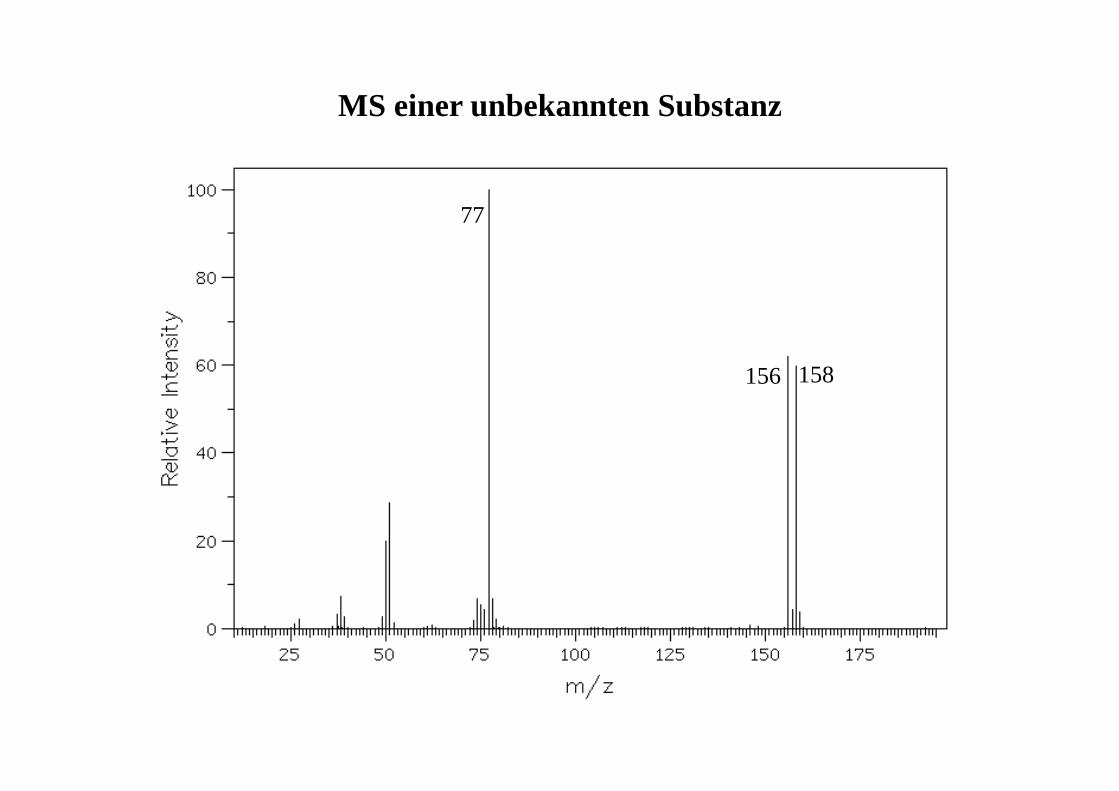
77
156 158
MS einer unbekannten Substanz
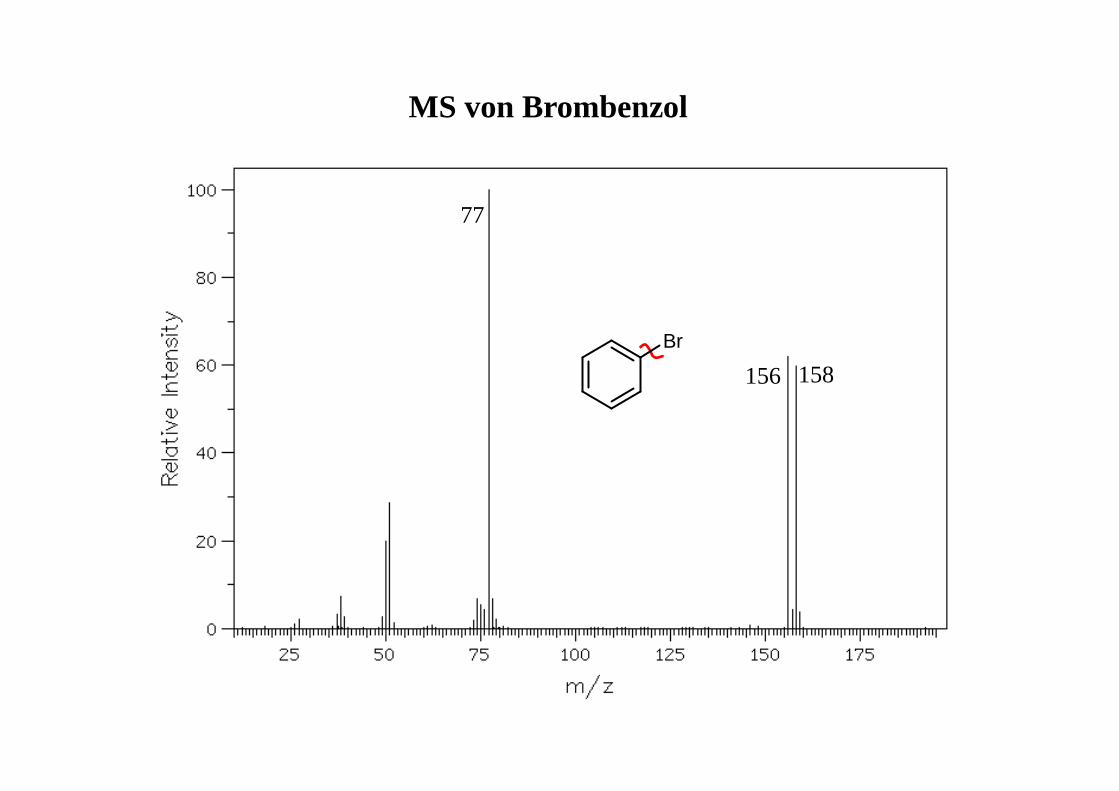
77
156 158
MS von Brombenzol
Br
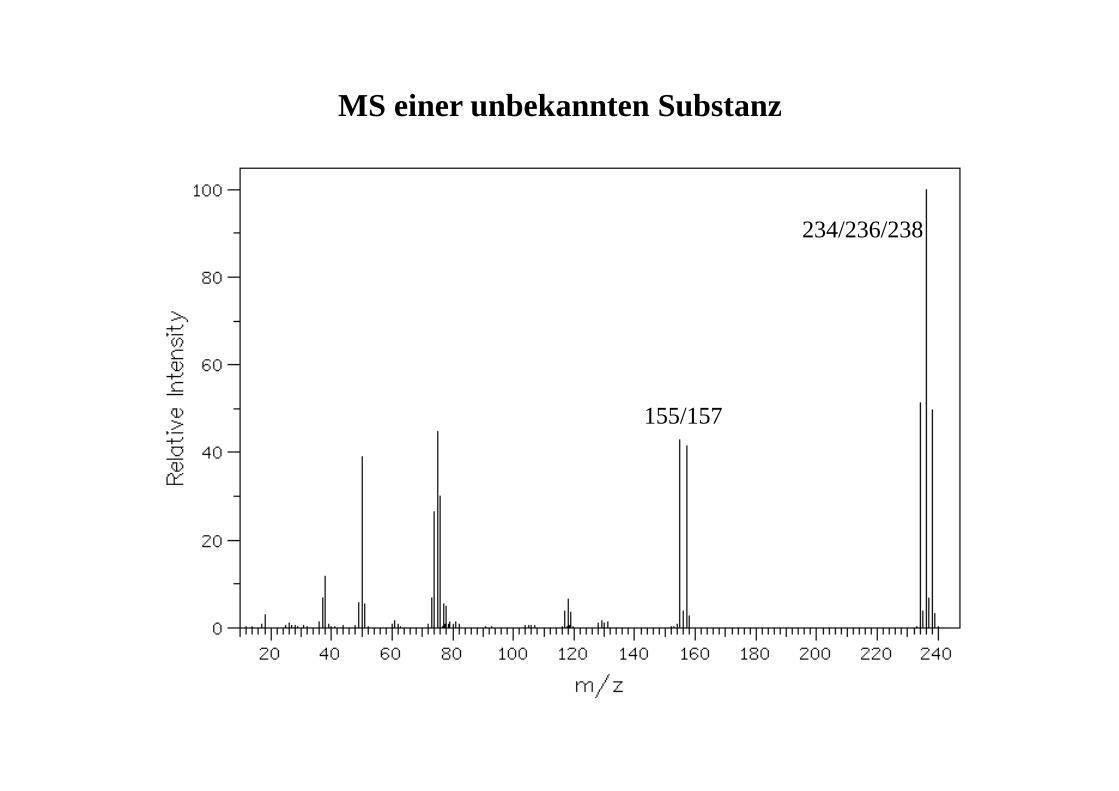
155/157
234/236/238
MS einer unbekannten Substanz
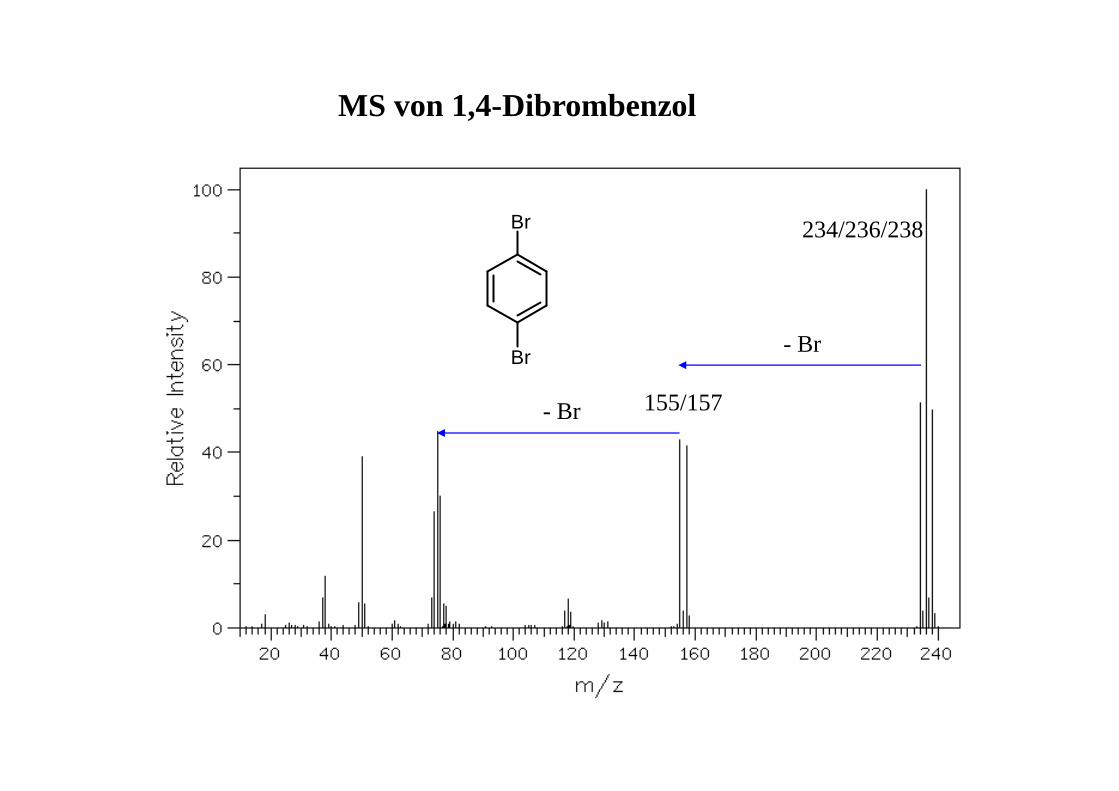
155/157
234/236/238
MS von 1,4-Dibrombenzol
Br
Br- Br
- Br
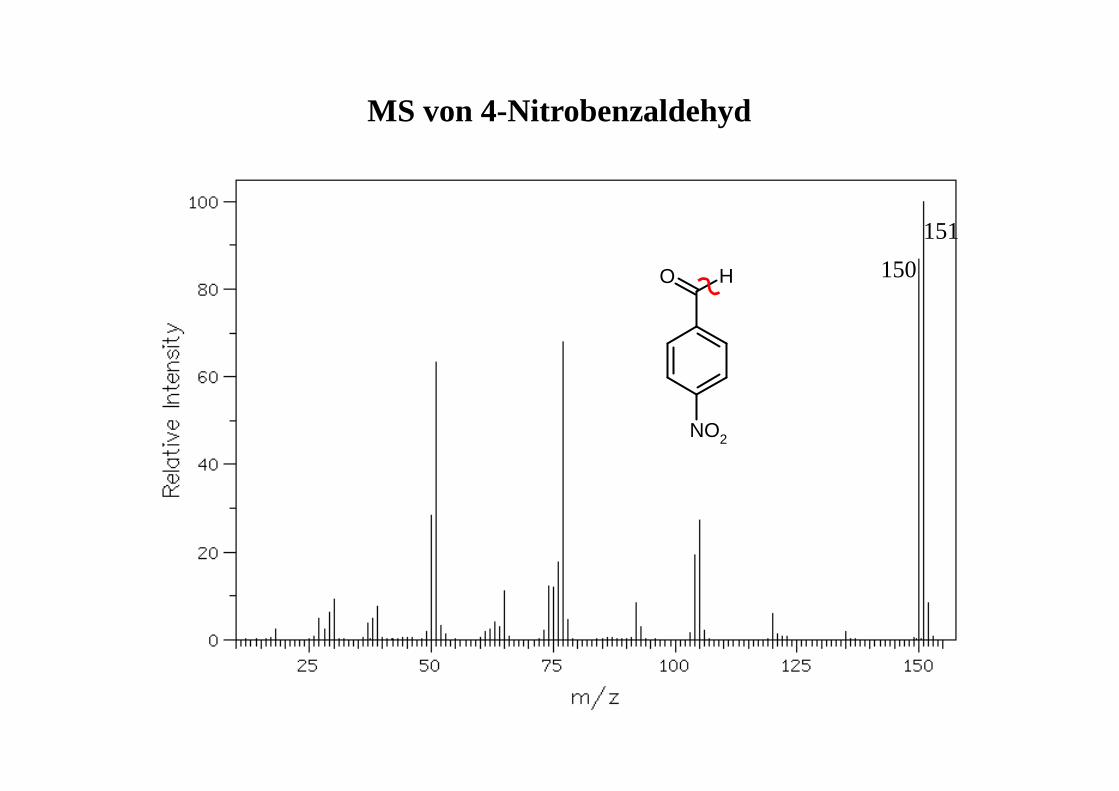
MS von 4-Nitrobenzaldehyd
150
151
NO2
HO
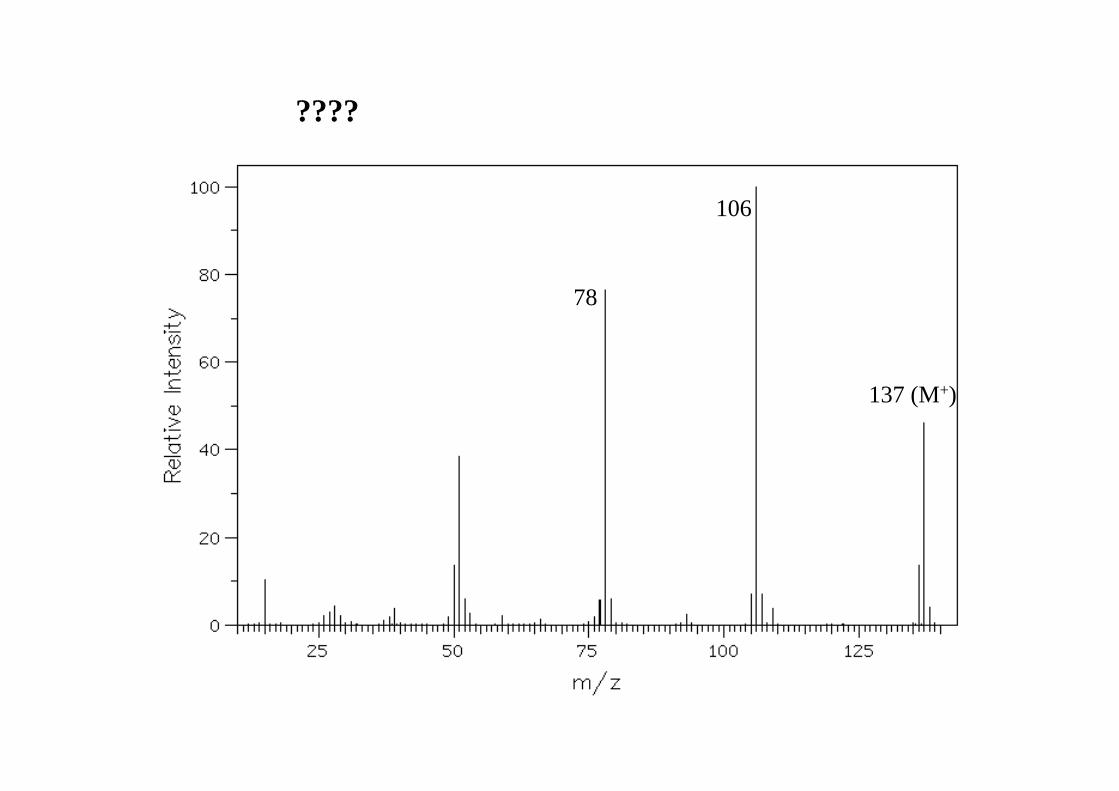
????
78
106
137 (M+)
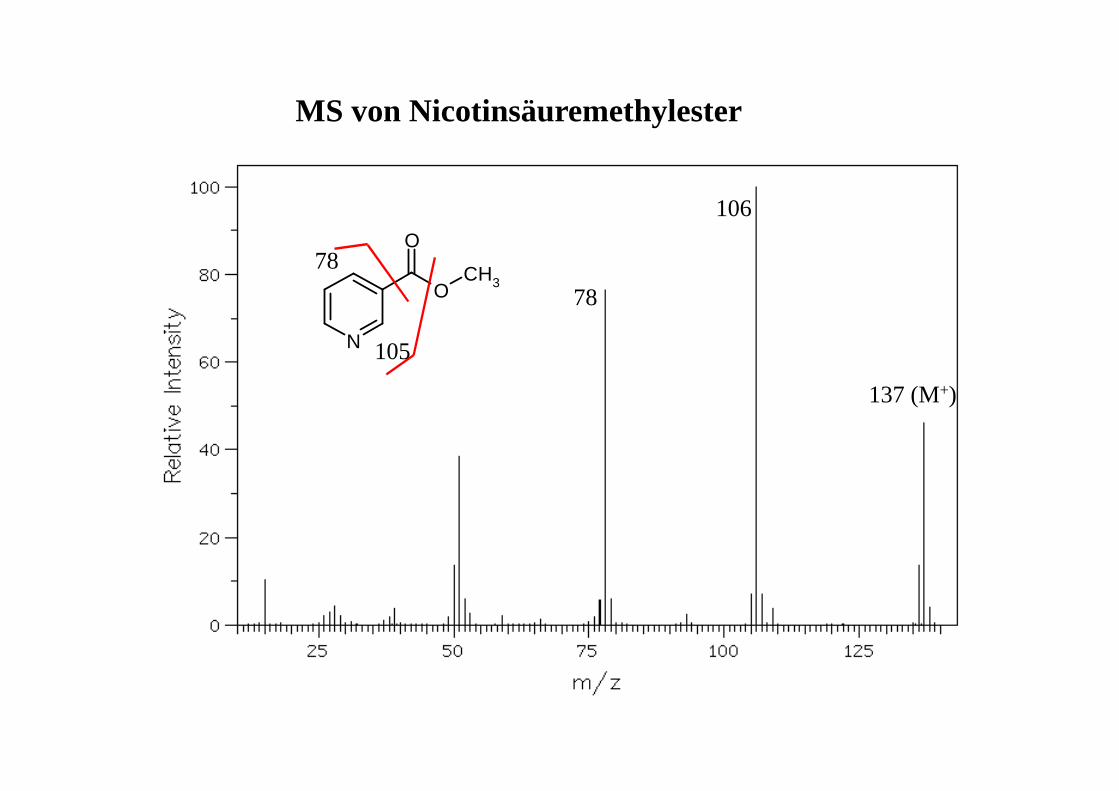
MS von Nicotinsäuremethylester
78
106
137 (M+)
N
O
O
CH378
105
M+ = 121
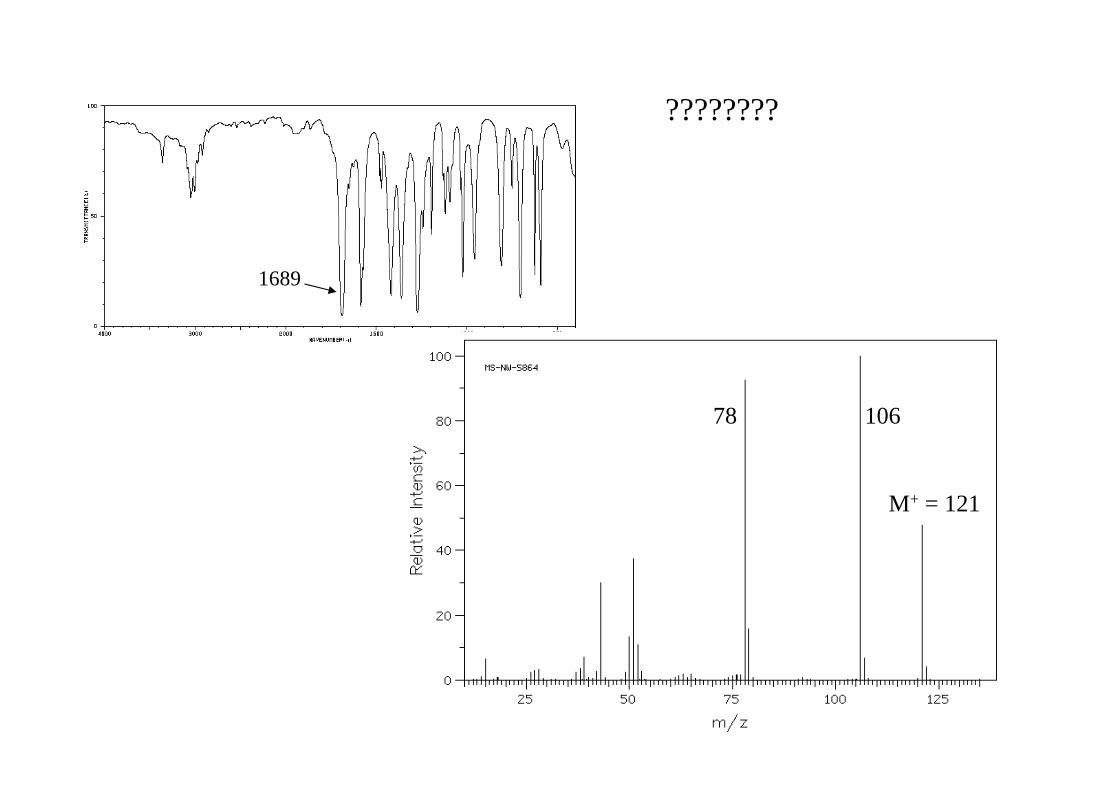
10678
1689
????????
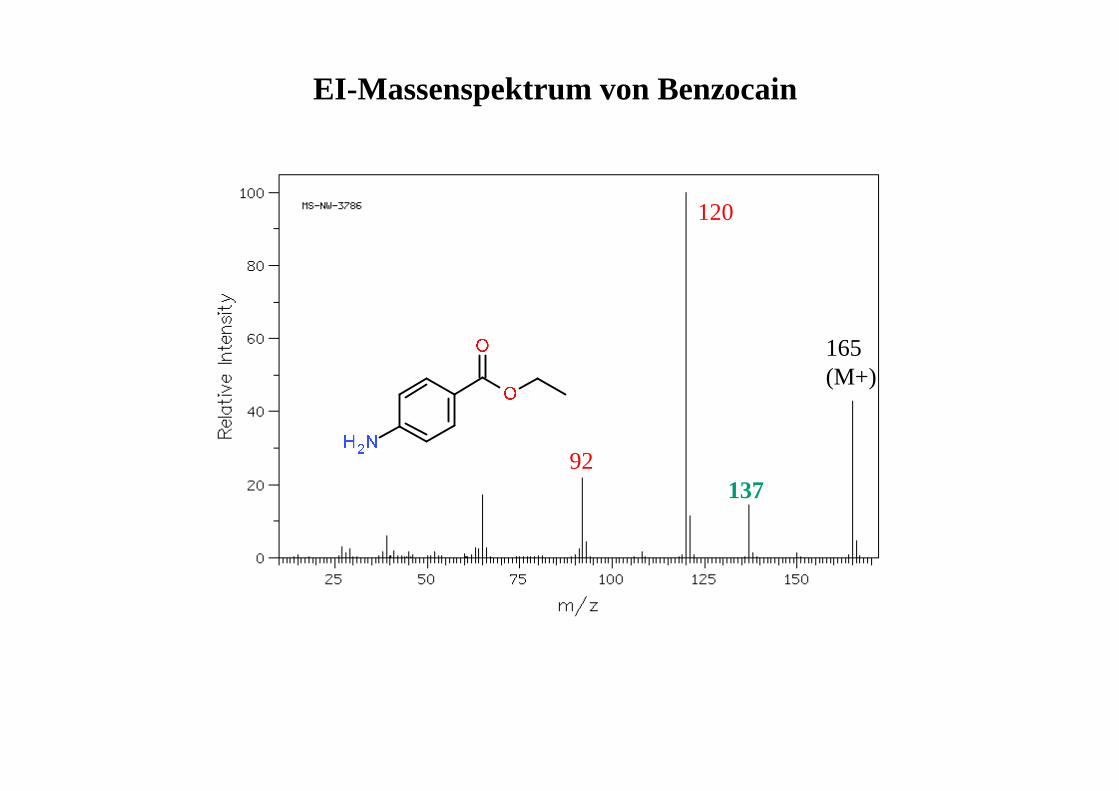
EI-Massenspektrum von Benzocain
165(M+)
120
92137
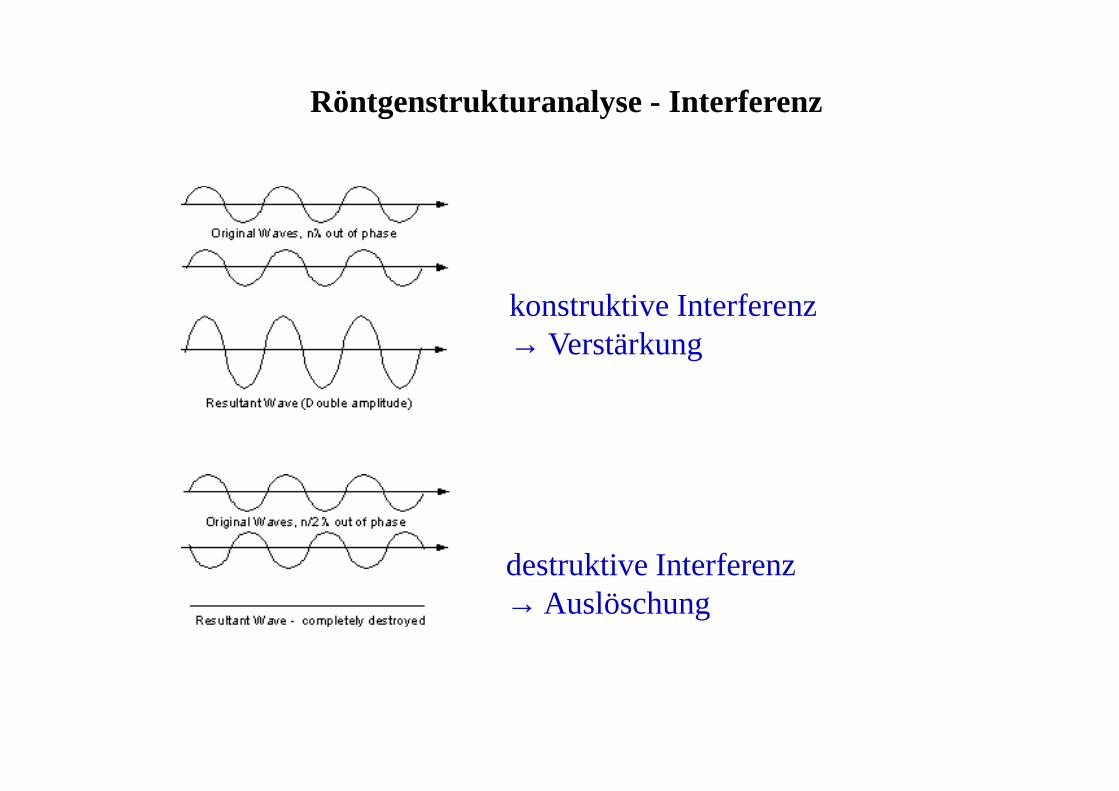
Röntgenstrukturanalyse - Interferenz
konstruktive Interferenz→ Verstärkung
destruktive Interferenz→ Auslöschung
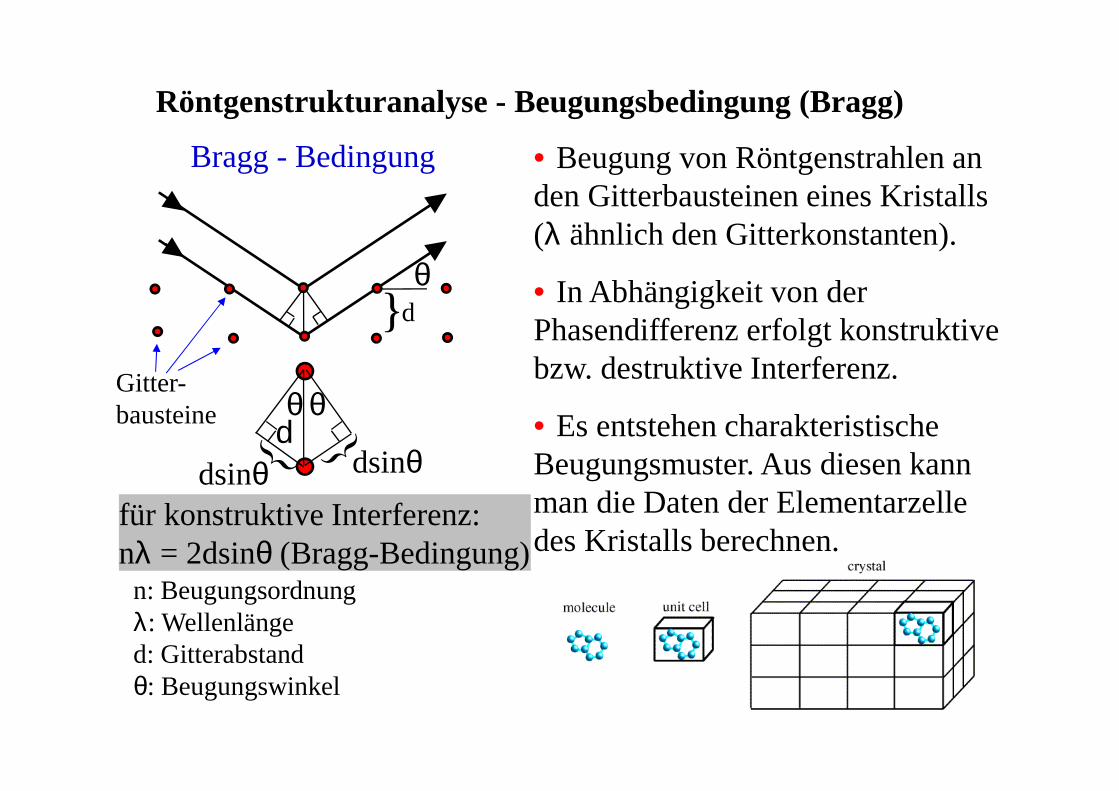
Bragg - Bedingung
für konstruktive Interferenz:nλ = 2dsinθ (Bragg-Bedingung)
} dθ
θ θd
Röntgenstrukturanalyse - Beugungsbedingung (Bragg)
dsinθdsinθ
Gitter-bausteine
n: Beugungsordnungλ: Wellenlänged: Gitterabstandθ: Beugungswinkel
• Beugung von Röntgenstrahlen an den Gitterbausteinen eines Kristalls (λ ähnlich den Gitterkonstanten).
• In Abhängigkeit von der Phasendifferenz erfolgt konstruktive bzw. destruktive Interferenz.
• Es entstehen charakteristische Beugungsmuster. Aus diesen kann man die Daten der Elementarzelle des Kristalls berechnen.
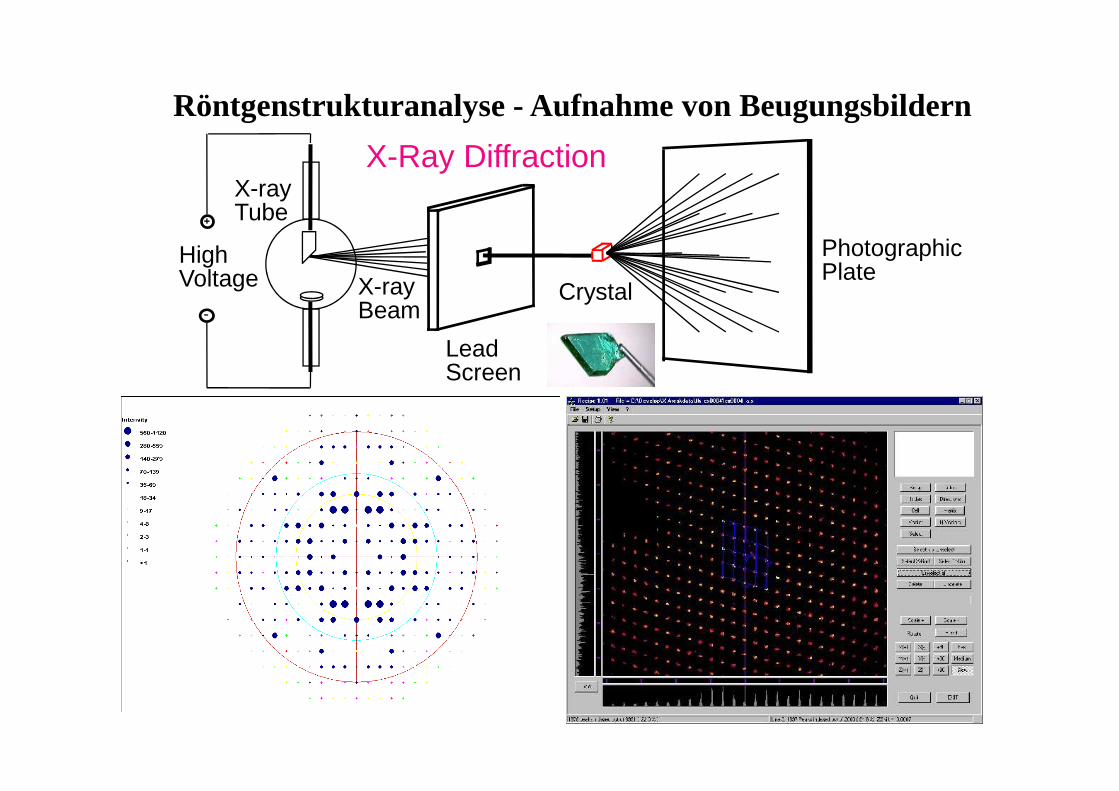
High Voltage
X-Ray DiffractionX-ray Tube
Lead Screen
X-ray Beam
Crystal
Photographic Plate
Röntgenstrukturanalyse - Aufnahme von Beugungsbildern
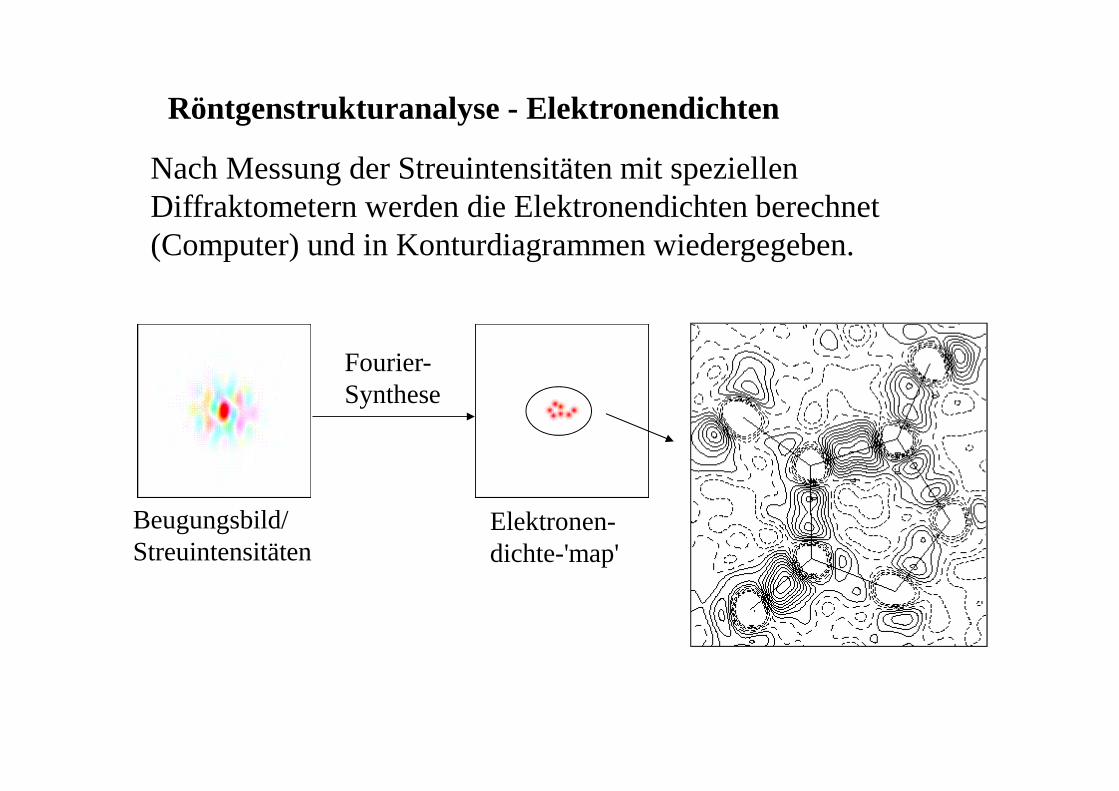
Röntgenstrukturanalyse - Elektronendichten
Nach Messung der Streuintensitäten mit speziellen Diffraktometern werden die Elektronendichten berechnet (Computer) und in Konturdiagrammen wiedergegeben.
Fourier-Synthese
Beugungsbild/Streuintensitäten
Elektronen-dichte-'map'
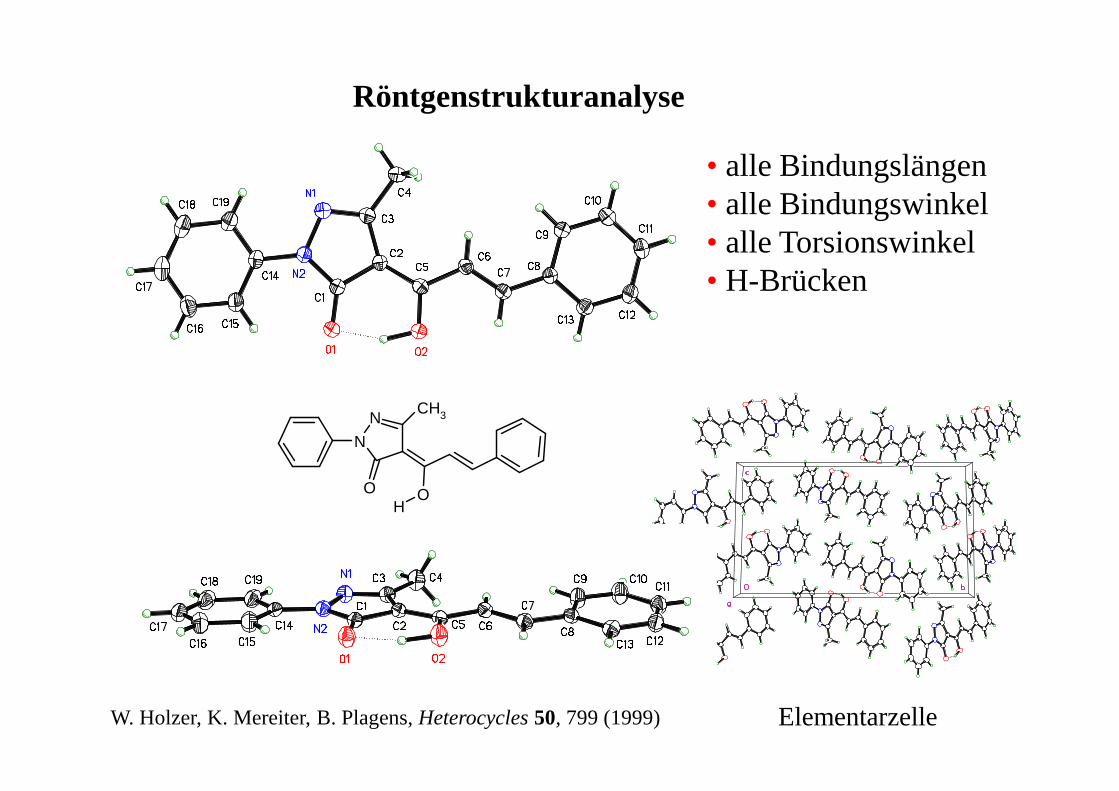
Röntgenstrukturanalyse
N
NCH3
O OH
W. Holzer, K. Mereiter, B. Plagens, Heterocycles 50, 799 (1999)
• alle Bindungslängen• alle Bindungswinkel• alle Torsionswinkel• H-Brücken
Elementarzelle

RöntgenstrukturanalyseVorteile:• liefert genaue 3-dimensionale Struktur (alle Bindungs-
längen, Bindungswinkel, Torsionswinkel)
• Bestimmung der absoluten Konfiguration bei chiralenVerbindungen (wenn schwereres Atom vorhanden)
Nachteile:• Beschränkung auf den festen Zustand
• Bio-Moleküle können im physiologischen Milieu andersvorliegen als im Kristall
• genügend große Einkristalle der Probe erforderlich (vieleSubstanzen kristallisieren nicht oder nicht gut genug)
• Position der H-Atome muß nachträglich berechnet werden,da deren Röntgenbeugung nur sehr schwach ist

Struktur eines Enzyms - ermittelt via X-ray

Kernresonanz-Spektroskopie (NMR) Einteilung der Atomkerne
Nuklid ist charakterisiert durch Ordnungszahl (OZ) und Massenzahl (MZ)
N (ug) N (uu) C (gu) C (gg)14
7
15
7
13
6
12
6OZ
MZ
ungerade (u)
gerade (g)
ug, gu, uu - Kerne: Spinquantenzahl I > 0 (1/2, 1, 3/2, 2, ...), besitzen magnetisches Moment µ
gg-Kerne: Spinquantenzahl I = 0, kein magnetisches Moment µ, dem NMR-Experiment nicht zugänglich! → z.B. 12C, 16O !
wichtige NMR-aktive Kerne: 1H (99.985%), 2H (0.015%), 13C (1.1%), 15N (0.36%), 19F (100%), 31P (100%), [17O (0.037%)]
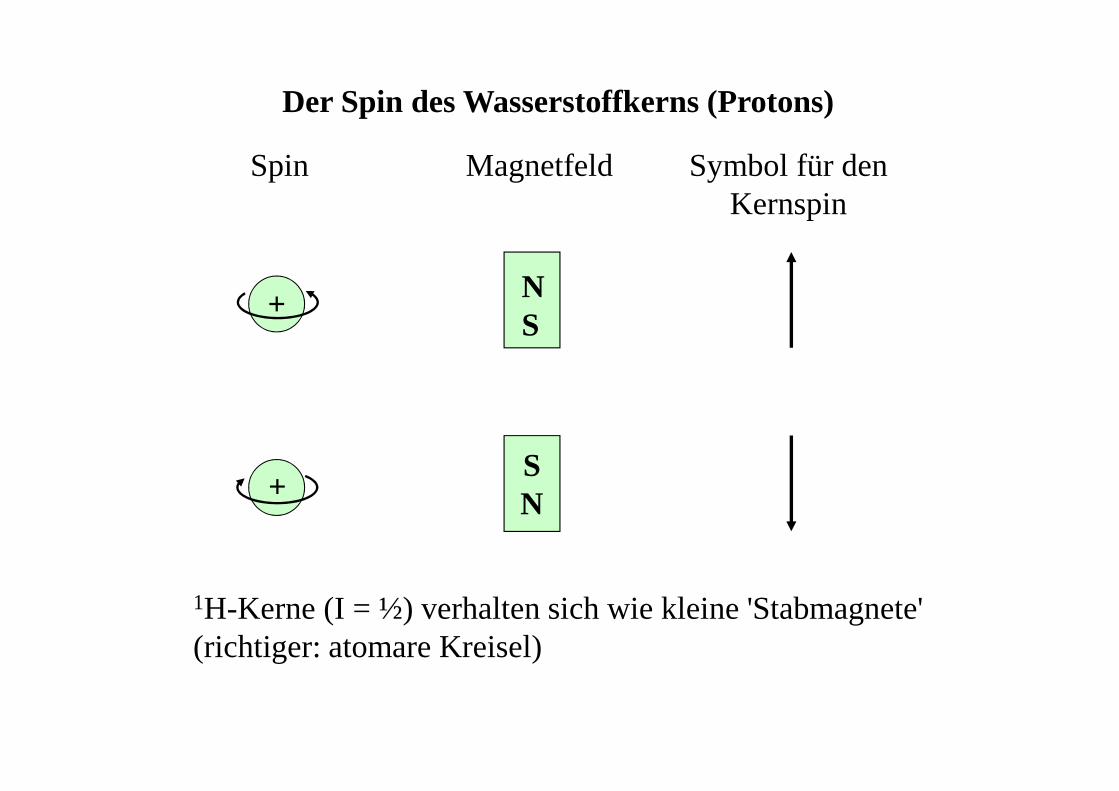
+
+SN
Der Spin des Wasserstoffkerns (Protons)
Spin Magnetfeld Symbol für denKernspin
NS
1H-Kerne (I = ½) verhalten sich wie kleine 'Stabmagnete' (richtiger: atomare Kreisel)
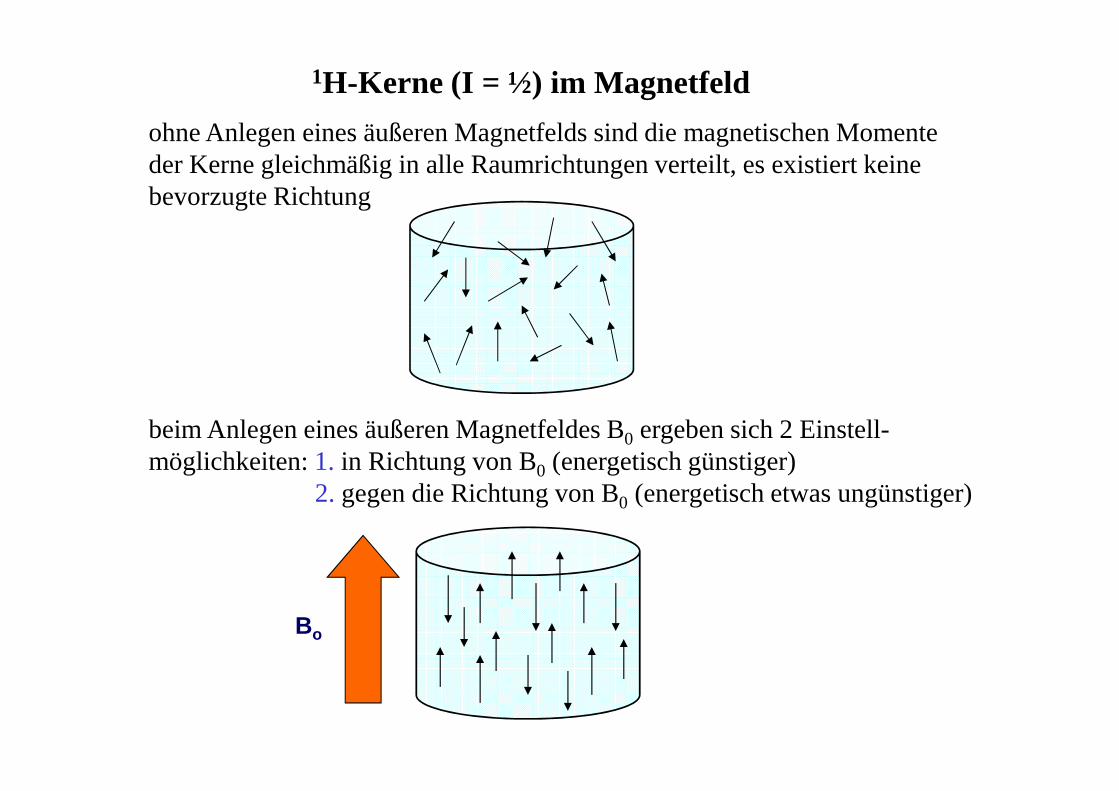
ohne Anlegen eines äußeren Magnetfelds sind die magnetischen Momente der Kerne gleichmäßig in alle Raumrichtungen verteilt, es existiert keinebevorzugte Richtung
beim Anlegen eines äußeren Magnetfeldes B0 ergeben sich 2 Einstell-möglichkeiten: 1. in Richtung von B0 (energetisch günstiger)
2. gegen die Richtung von B0 (energetisch etwas ungünstiger)
Bo
1H-Kerne (I = ½) im Magnetfeld
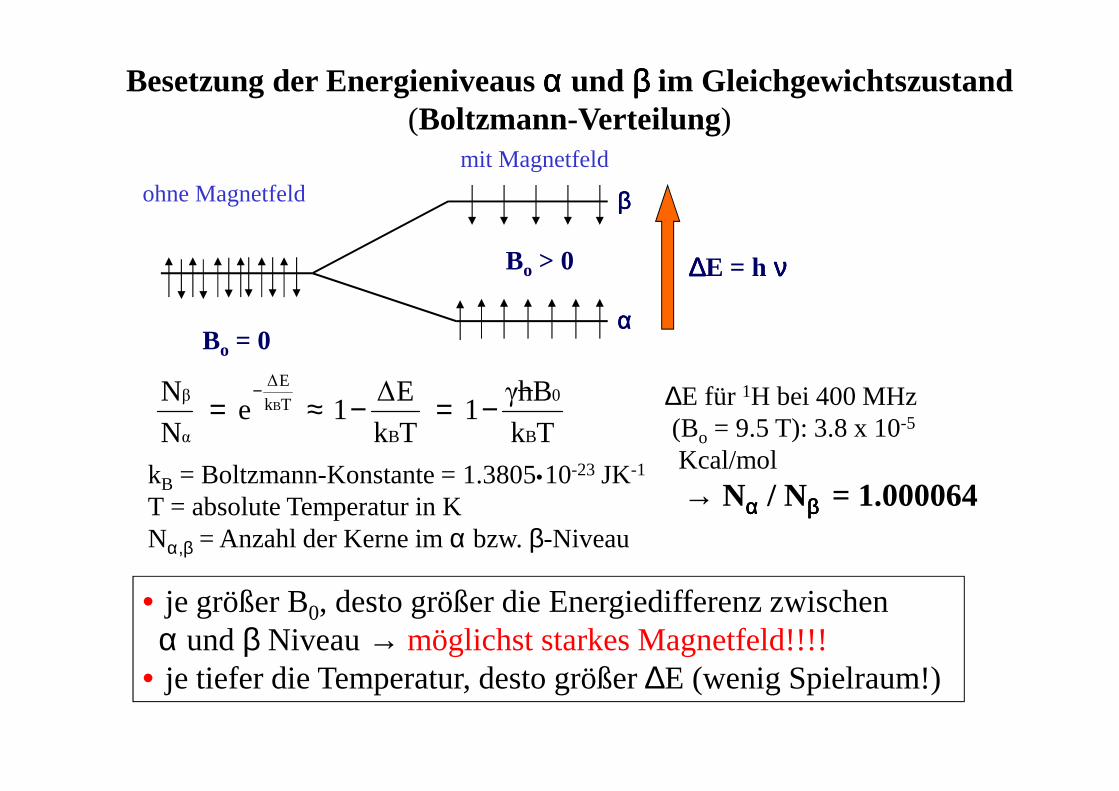
Bo = 0
Bo > 0 ∆∆∆∆E = h νννν
αααα
ββββ
Besetzung der Energieniveaus αααα und ββββ im Gleichgewichtszustand(Boltzmann-Verteilung)
kB = Boltzmann-Konstante = 1.3805•10-23 JK-1
T = absolute Temperatur in KNα,β = Anzahl der Kerne im α bzw. β-Niveau
ohne Magnetfeld
mit Magnetfeld
• je größer B0, desto größer die Energiedifferenz zwischen α und β Niveau → möglichst starkes Magnetfeld!!!!
• je tiefer die Temperatur, desto größer ∆E (wenig Spielraum!)
∆E für 1H bei 400 MHz (Bo = 9.5 T): 3.8 x 10-5
Kcal/mol
→ Nαααα / Nββββ = 1.000064
Tk
Bhγ1
Tk
∆E1e
N
N
B
0
B
Tk
∆E
α
βB −=−≈=
−
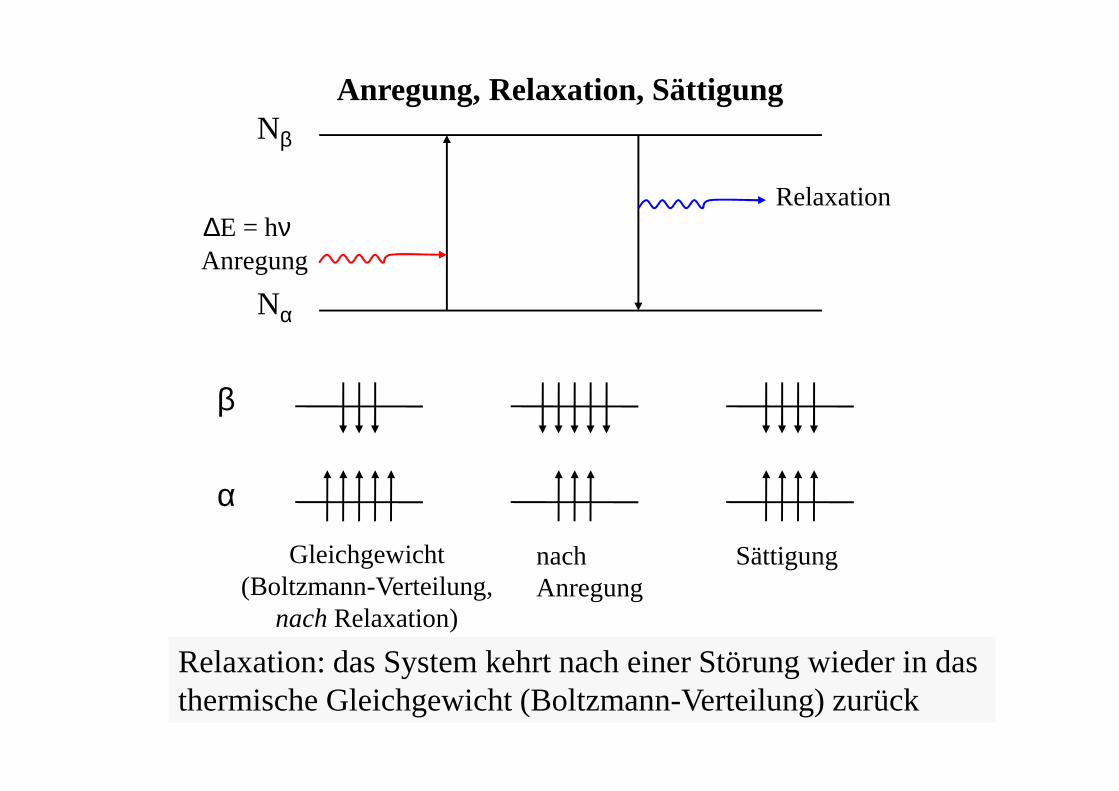
Nβ
Nα
Anregung
Relaxation
Anregung, Relaxation, Sättigung
Gleichgewicht(Boltzmann-Verteilung,
nach Relaxation)
nachAnregung
Sättigung
α
β
Relaxation: das System kehrt nach einer Störung wieder in das thermische Gleichgewicht (Boltzmann-Verteilung) zurück
∆E = hν
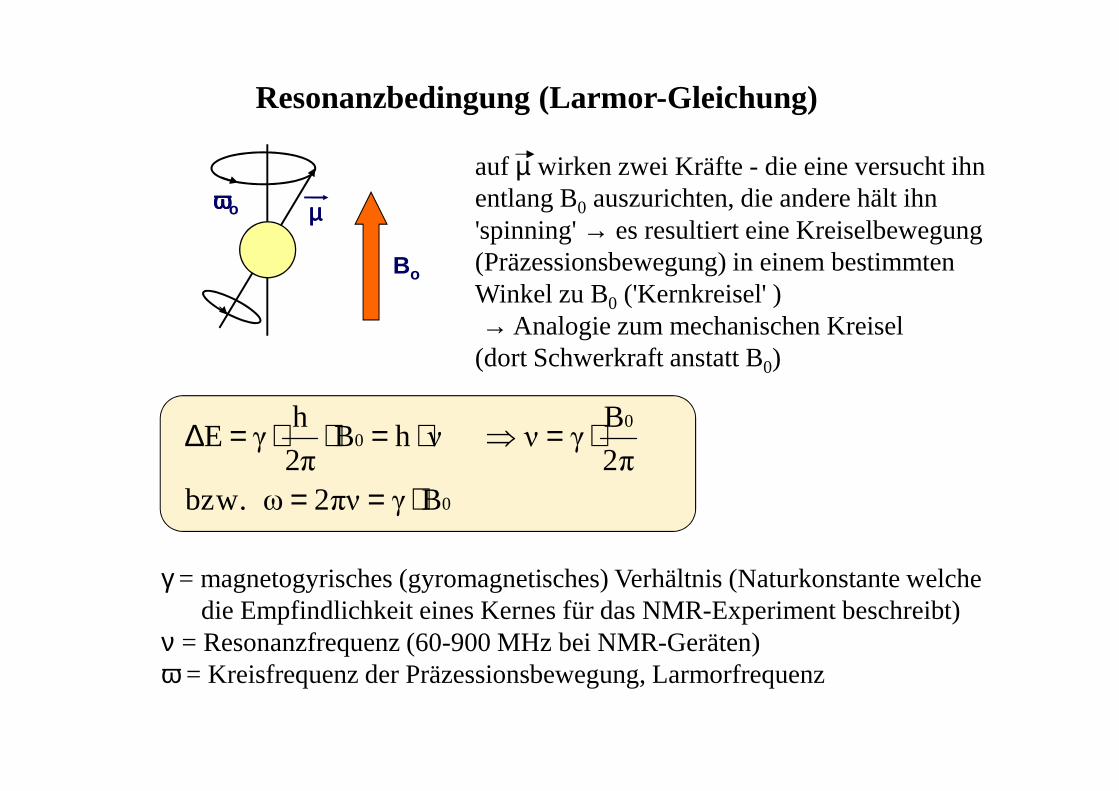
0
00
Bγπν2ω bzw.π2
Bγν νhB
2π
h γ E
⋅==
⋅=⇒⋅=⋅⋅=∆
Resonanzbedingung (Larmor-Gleichung)
γ = magnetogyrisches (gyromagnetisches) Verhältnis (Naturkonstante welchedie Empfindlichkeit eines Kernes für das NMR-Experiment beschreibt)
ν = Resonanzfrequenz (60-900 MHz bei NMR-Geräten) ω = Kreisfrequenz der Präzessionsbewegung, Larmorfrequenz
Bo
ωωωωo µµµµ
auf µ wirken zwei Kräfte - die eine versucht ihn entlang B0 auszurichten, die andere hält ihn 'spinning' → es resultiert eine Kreiselbewegung (Präzessionsbewegung) in einem bestimmten Winkel zu B0 ('Kernkreisel' )→ Analogie zum mechanischen Kreisel (dort Schwerkraft anstatt B0)
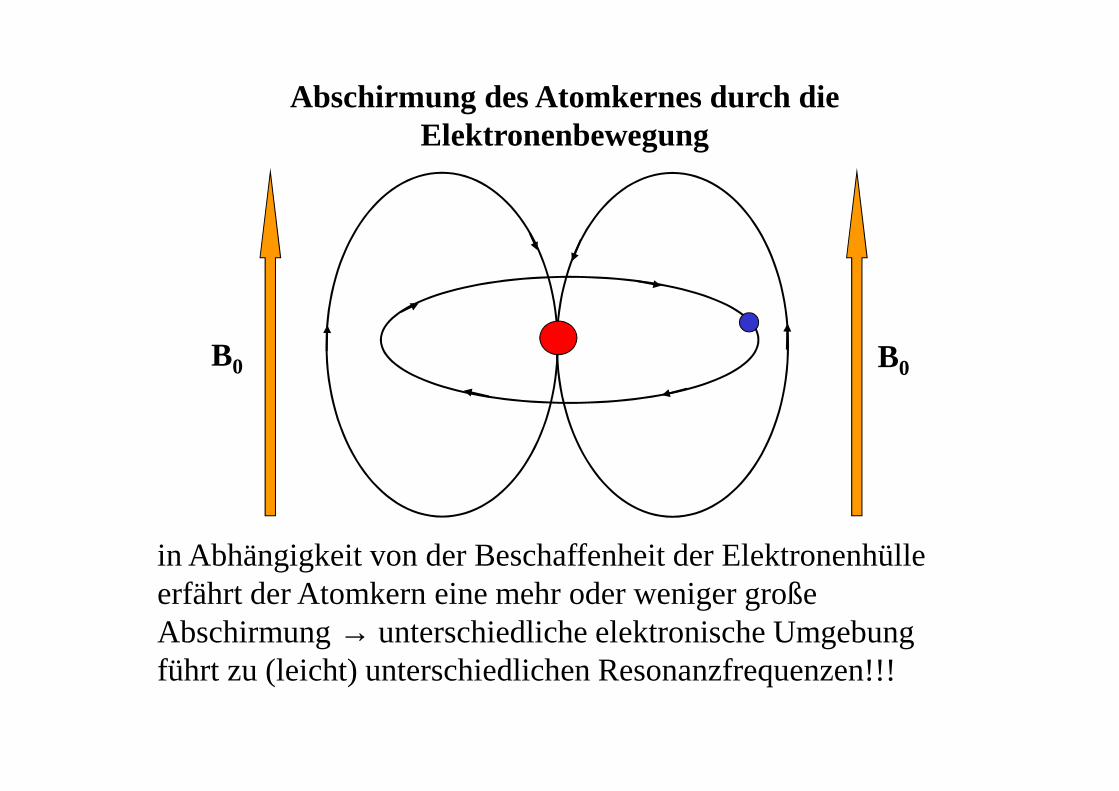
Abschirmung des Atomkernes durch die Elektronenbewegung
B0B0
in Abhängigkeit von der Beschaffenheit der Elektronenhülle erfährt der Atomkern eine mehr oder weniger große Abschirmung → unterschiedliche elektronische Umgebung führt zu (leicht) unterschiedlichen Resonanzfrequenzen!!!
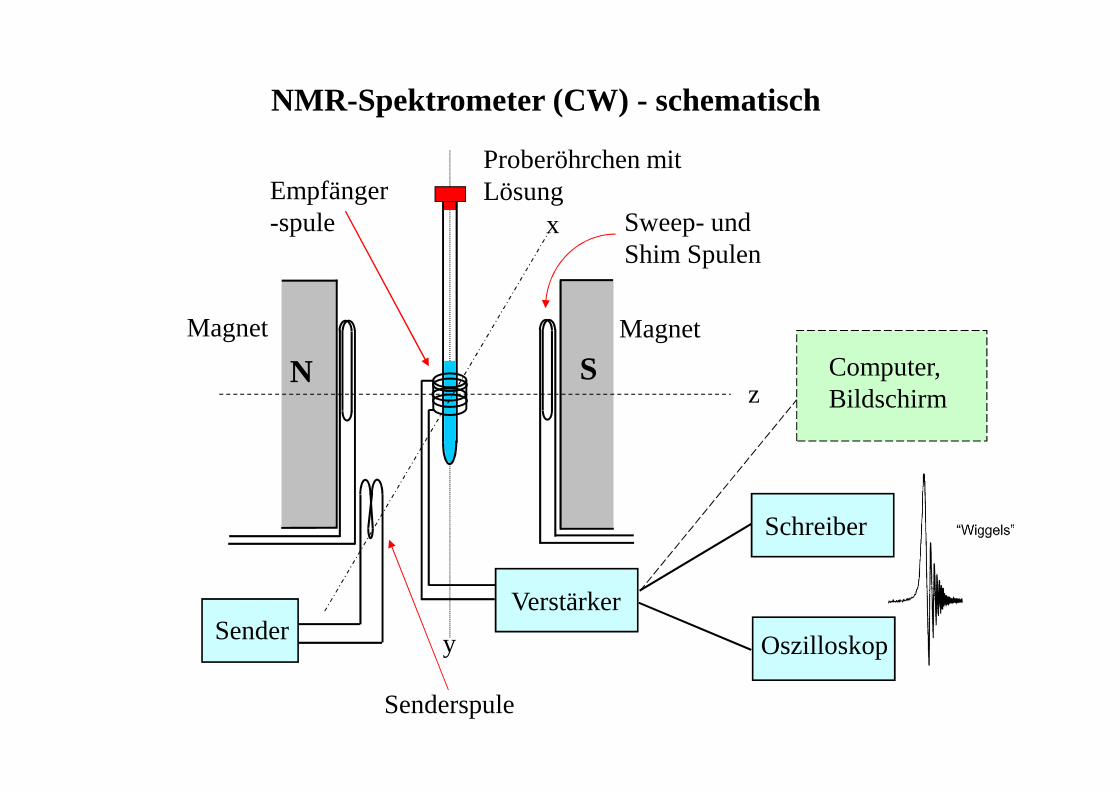
NMR-Spektrometer (CW) - schematisch
N S
x
y
z
SenderVerstärker
Empfänger-spule
Schreiber
Oszilloskop
MagnetMagnet
Proberöhrchen mitLösung
Senderspule
Sweep- und Shim Spulen
Computer,Bildschirm

NMR - Meßverfahren1) CW-Technik (continous wave)
die einzelnen Resonanzen werden nacheinander durch kontinuier-liche Veränderung von ν (Frequenz-sweep) oder B0 (Feld-sweep)unter Erfüllung der Resonanzbedingung ν = γ/2π•B0 erfaßt→ veraltet, langsam, unempfindlich (nur für empfindliche Kerne),keine modernen NMR-Experimente möglich
2) Puls-Fourier-Transform Technik (PFT)alle Resonanzen werden durch einen RF-Puls gleichzeitig angeregtdas erhaltene Interferogramm (Überlagerung aller Abklingkurven = free induction decay - FID) wird durch eine mathematischeOperation (Fourier-Transformation) in das NMR-Spektrumumgewandelt→ schnell, empfindlich (Akkumlierung von FIDs üblich), besserbearbeitbar; moderne NMR-Spektroskopie verwendet 'Pulsfolgen' =Kombination von mehreren Pulsen verschiedener Richtung undGröße, getrennt durch genau definierte Zeitintervalle

Kryomagnet (supraleitende Spule)heute praktisch ausschließlich verwendet (Permanent- oder Elektromagnete nur bis 100 MHz, Kryomagnet bis 1GHz)

Benchtop NMR Spektrometer
Spezieller Permanentmagnet (hier 60 MHz 1H-Frequenz)Für einfache Anwendungen wie Analyse unkomplizierter Moleküle, Reaktionskontrolle, Qualitätskontrolle, usw. Ausbildung (Lehrzwecke)
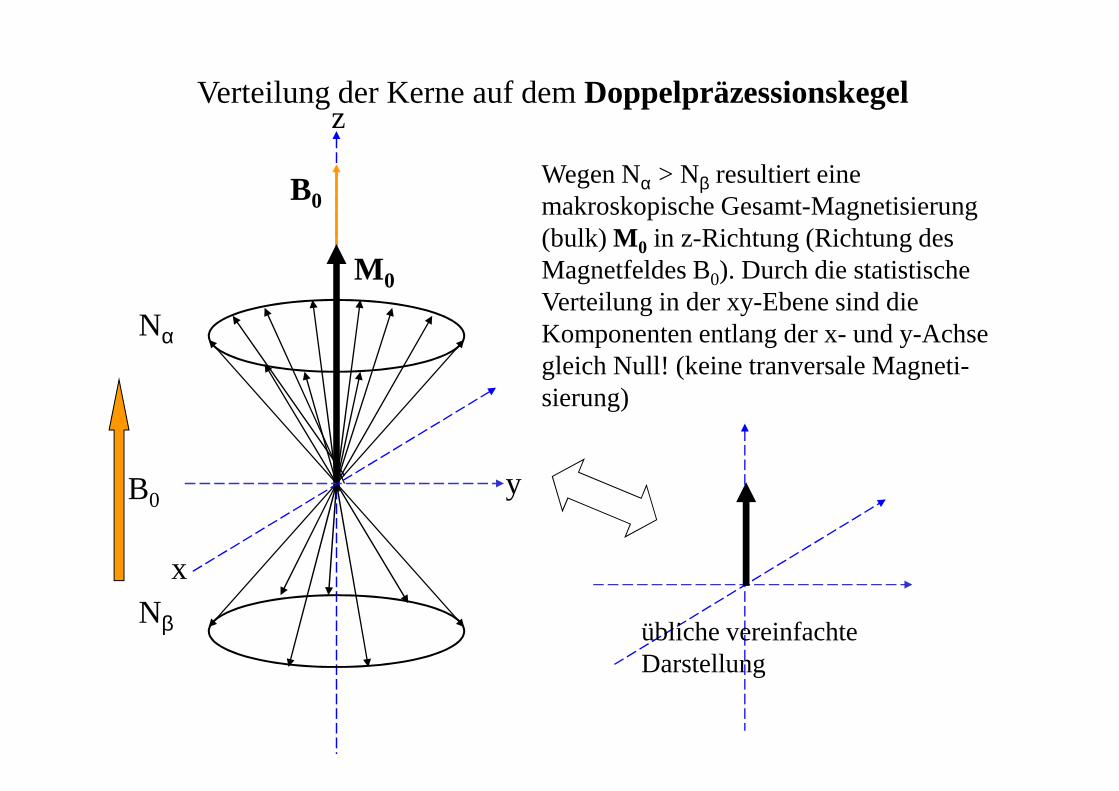
Verteilung der Kerne auf dem Doppelpräzessionskegelz
y
x
M 0
Nα
Nβ
Wegen Nα > Nβ resultiert eine makroskopische Gesamt-Magnetisierung(bulk) M 0 in z-Richtung (Richtung desMagnetfeldes B0). Durch die statistischeVerteilung in der xy-Ebene sind dieKomponenten entlang der x- und y-Achsegleich Null! (keine tranversale Magneti-sierung)
B0
B0
übliche vereinfachte Darstellung
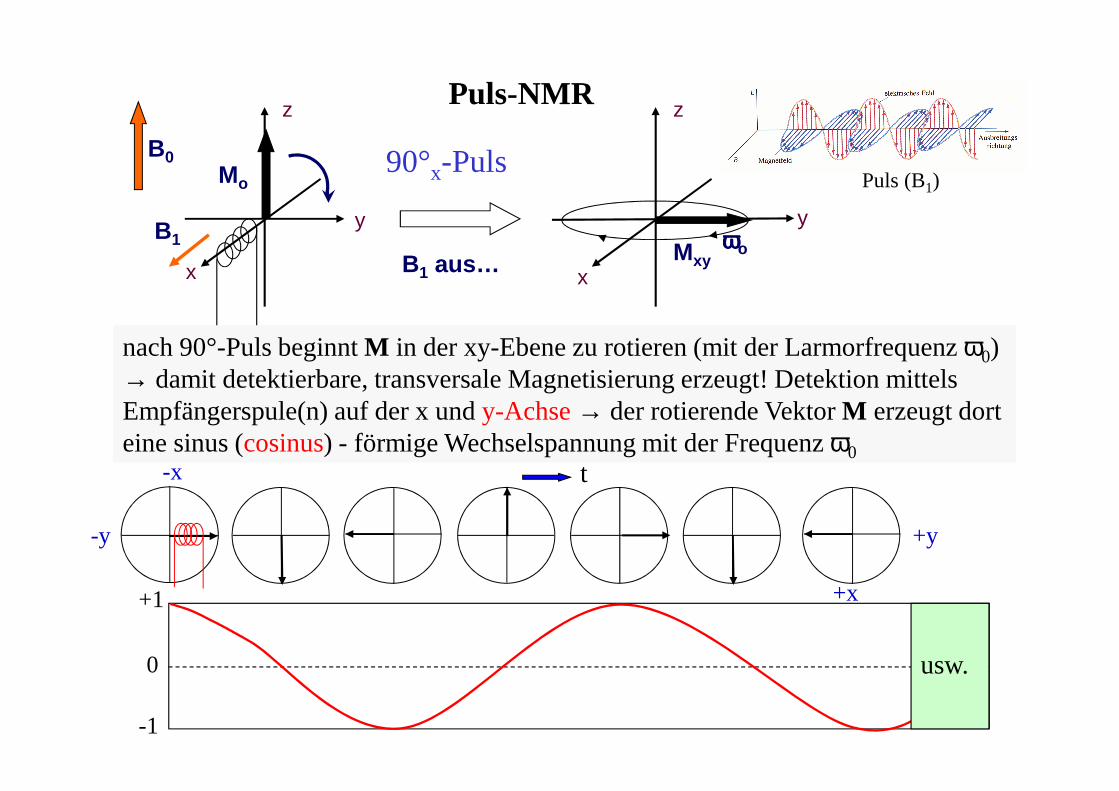
B1 aus…
Mo
z
x
B1
z
y
Mxy
y
x
ωωωωo
B0
Puls-NMR
90°x-Puls
nach 90°-Puls beginnt M in der xy-Ebene zu rotieren (mit der Larmorfrequenz ω0) → damit detektierbare, transversale Magnetisierung erzeugt! Detektion mittels Empfängerspule(n) auf der x und y-Achse→ der rotierende Vektor M erzeugt dort eine sinus (cosinus) - förmige Wechselspannung mit der Frequenz ω0
usw.
+1
-1
0
t
+y-y
+x
-x
Puls (B1)
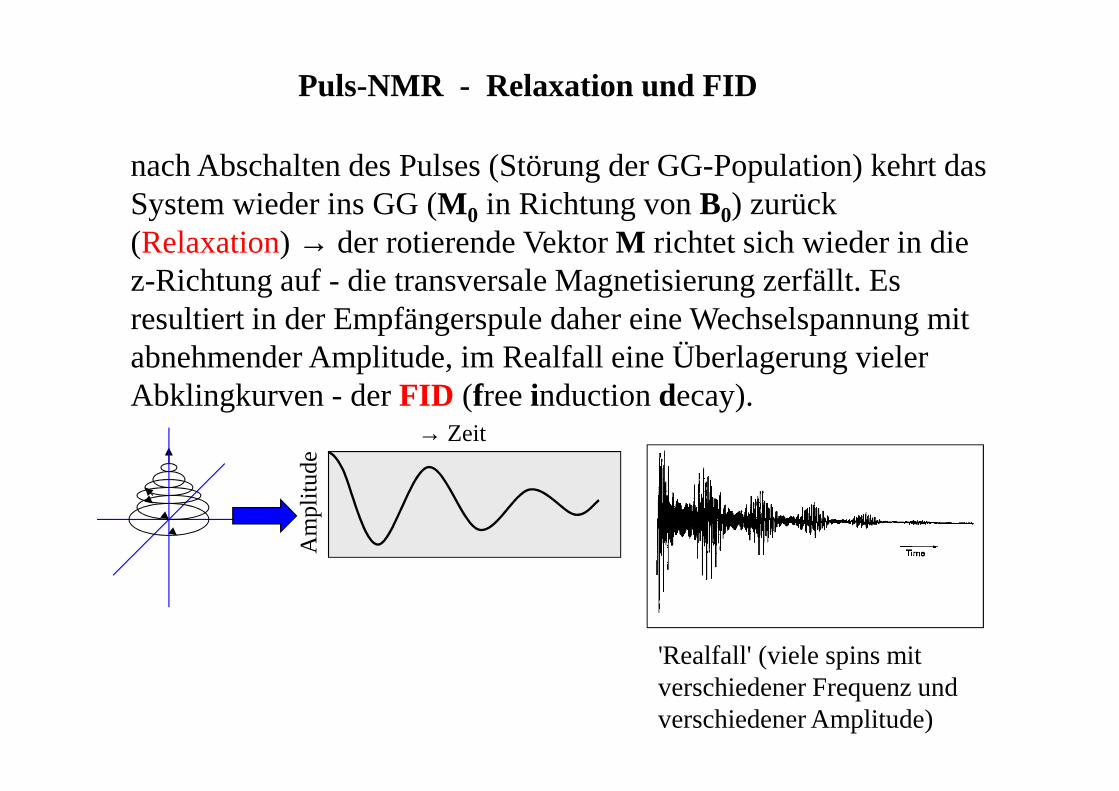
Puls-NMR - Relaxation und FID
nach Abschalten des Pulses (Störung der GG-Population) kehrt das System wieder ins GG (M 0 in Richtung von B0) zurück (Relaxation) → der rotierende Vektor M richtet sich wieder in die z-Richtung auf - die transversale Magnetisierung zerfällt. Es resultiert in der Empfängerspule daher eine Wechselspannung mit abnehmender Amplitude, im Realfall eine Überlagerung vieler Abklingkurven - der FID (free induction decay).
Am
plitu
de
→ Zeit
'Realfall' (viele spins mit verschiedener Frequenz und verschiedener Amplitude)
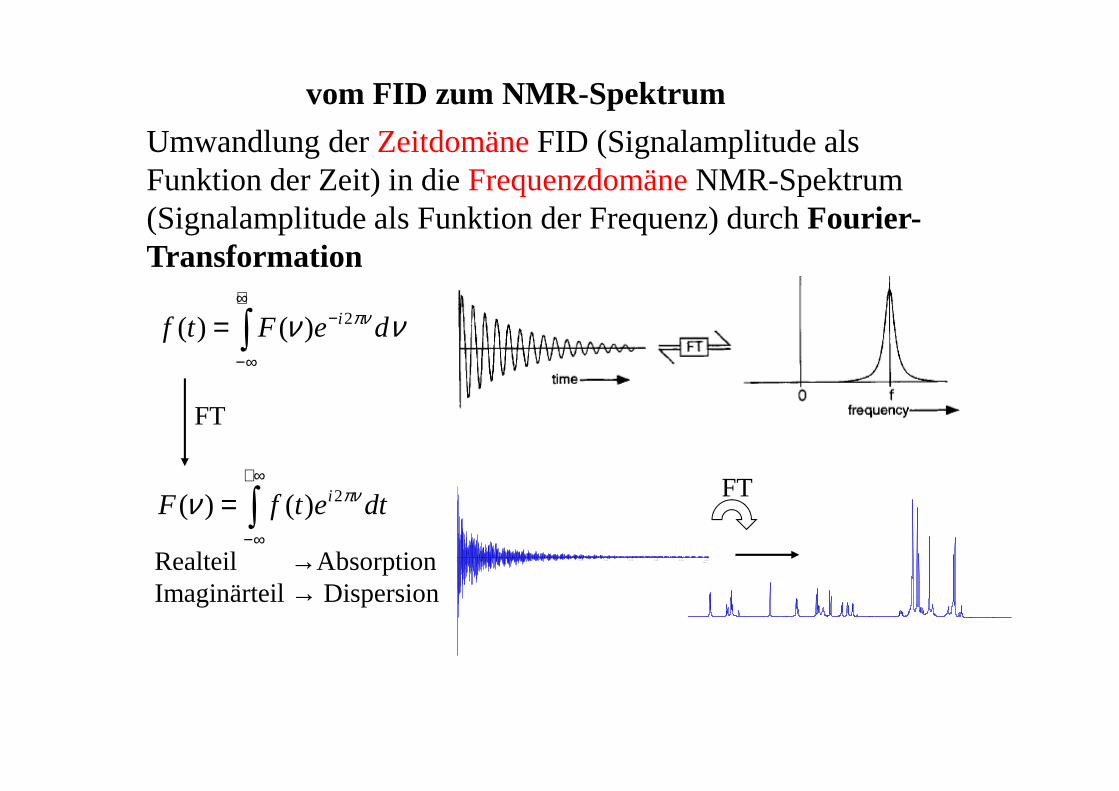
vom FID zum NMR-Spektrum
Umwandlung der ZeitdomäneFID (Signalamplitude als Funktion der Zeit) in die FrequenzdomäneNMR-Spektrum (Signalamplitude als Funktion der Frequenz) durch Fourier-Transformation
dtetfF
deFtf
i
i
∫
∫
∞+
∞−
+∞
∞−
−
=
=
πν
πν
ν
νν
2
2
)()(
)()(
FT
Realteil →AbsorptionImaginärteil → Dispersion
0 0.10 0.20 0.30 0.40 0.50 0.60 0.70 0.80 0.90 1.00t1 sec
FT

1H-NMR - Abschirmung und chemische Verschiebung
• 1H wichtigster NMR aktiver Kern – natürliche Häufigkeit ist99.985% I = ½ γ groß → sehr empfindlich
• Abschirmung: auf 1H-Kern wirkt nicht exakt B0, sondern –bedingt durch die elektronische Abschirmung – ein etwaskleineres Feld Beff
• Beff = B0 – σB0 = (1 –σ) B0 (σ: Abschirmungskonstante)→ Resonanzbedingung: ν = γ/2π (1 –σ) B0
• Protonen mit unterschiedlicher elektronischer Umgebung habenunterschiedliche Resonanzfrequenzen. Das 1H-NMR Spektrumeiner Substanz mit unterschiedlichen Sorten von Protonen bestehtdaher aus so vielen Signalen, wie es verschieden Typen vonProtonon im Molekül gibt
• Signale zueinander verschoben → chemische Verschiebung

Chemische Verschiebung
• Es wird nicht die absolute Lage eines Resonanzsignales gemessen (z.B. 500 000 423 Hz) sondern die relative Lagezum Signal einer Standardsubstanz (Tetramethylsilan = TMS). Die Lage des Standardsignales wird definitionsgemäß 0 gesetzt.
• Angabe der chemischen Verschiebung nicht in Hz weil dann Aufnahmen bei verschiedenen Feldstärken nicht vergleichbar sind (ν ist von B0 abhängig – siehe Resonanzbedingung)
• δδδδ-Skala: chem. Verschiebung als dimensionslose Zahl angegeben, damit unabhängig von B0
equenzBetriebsfr
)TMS(ν)Signal(νδ
−=Gerätes des MHz
ν(TMS)ν(Signal)(ppm) δ
−=oder
Si CH3CH3
CH3
CH3
TMS
z.B. 500 MHz-Gerät, ν(Signal) = 2000 Hz → δ (ppm) = 2000/500 = 4ppm = parts per million = 1/1 000 000 !!! (keine physikalische Einheit!)
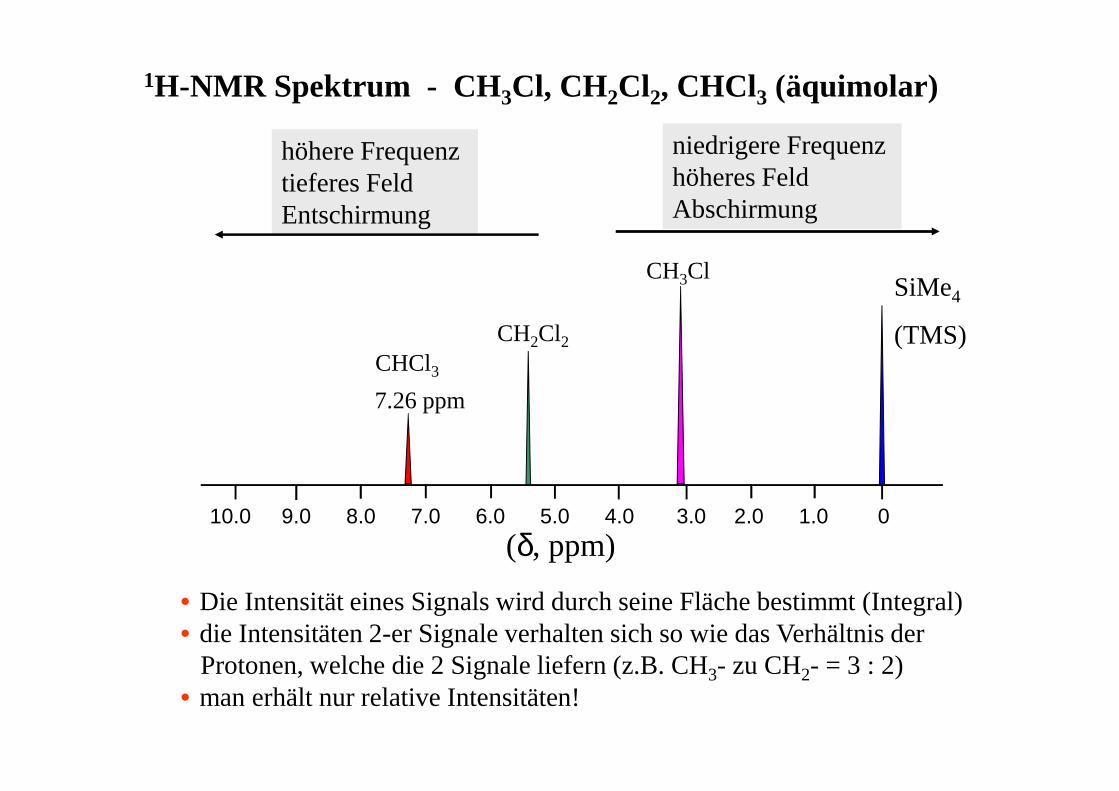
01.02.03.04.05.06.07.08.09.010.0
(δ, ppm)
SiMe4
(TMS)CHCl37.26 ppm
CH2Cl2
CH3Cl
höhere Frequenztieferes FeldEntschirmung
niedrigere Frequenzhöheres FeldAbschirmung
1H-NMR Spektrum - CH 3Cl, CH2Cl2, CHCl3 (äquimolar)
• Die Intensität eines Signals wird durch seine Fläche bestimmt (Integral)• die Intensitäten 2-er Signale verhalten sich so wie das Verhältnis der
Protonen, welche die 2 Signale liefern (z.B. CH3- zu CH2- = 3 : 2)• man erhält nur relative Intensitäten!

1H-NMR chemische Verschiebung
Beeinflussung hauptsächlich durch:
• Induktive Substituenteneffekteelektronegative Substituenten in der Nähe des betrachteten Protons erniedrigen die Elektronendichte → Entschirmung, Tieffeldverschiebung, δ großelektropositive Substituenten erhöhen die Abschirmung →Hochfeldverschiebung, δ klein
• Mesomerieffektepositive Partialladung in (relevanten) mesomeren Grenzstrukturen →Elektronenmangel, Entschirmung, δ großnegative Partialladung → hohe Elektronendichte, Abschirmung, δ klein
• Anisotropieeffektedurch B0 wird ein sekundäres Magnetfeld erzeugt, welches nicht in allen Raumrichtungen gleich groß ist (anisotrop). Je nach räumlicher Lage des betreffenden Protons zu diesem Sekundärfeld kann Entschirmung oder Abschirmung erfolgen
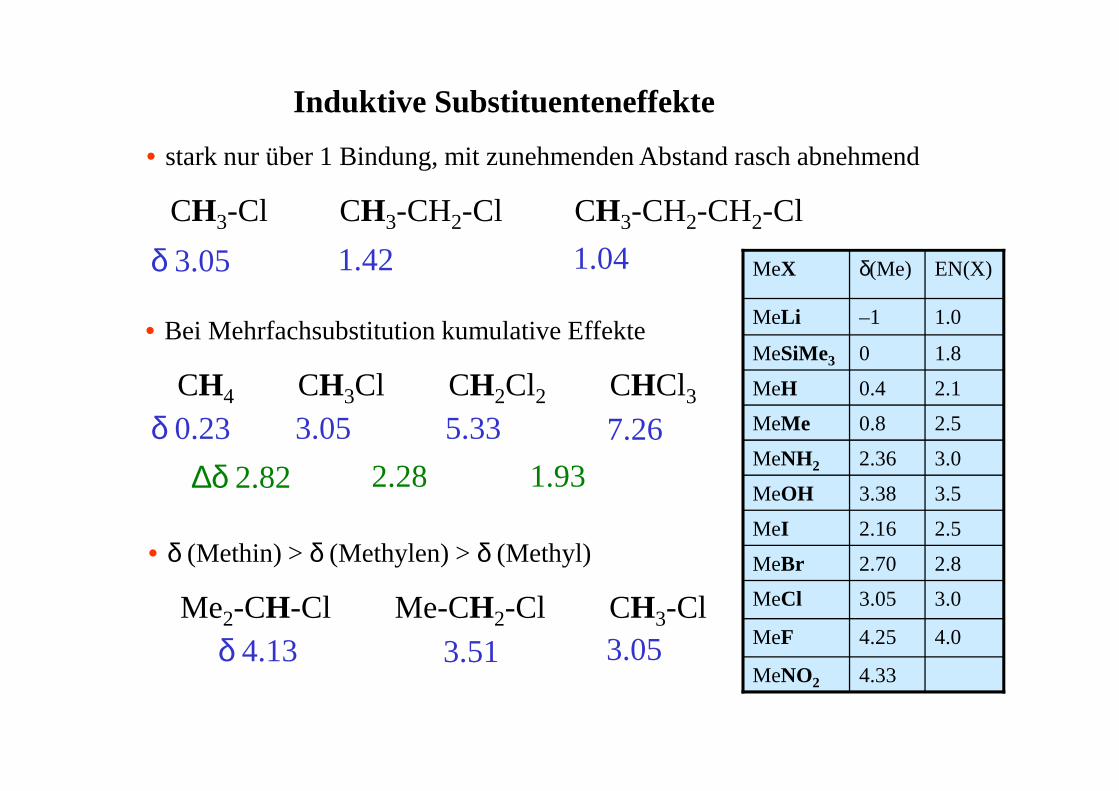
Induktive Substituenteneffekte
• stark nur über 1 Bindung, mit zunehmenden Abstand rasch abnehmend
CH3-Cl CH3-CH2-Cl CH3-CH2-CH2-Cl
• Bei Mehrfachsubstitution kumulative Effekte
CH4 CH3Cl CH2Cl2 CHCl3
• δ (Methin) > δ (Methylen) > δ (Methyl)
Me2-CH-Cl Me-CH2-Cl CH3-Cl
δ 3.05 1.42 1.04
δ 0.23 3.05 5.33 7.26
∆δ 2.82 2.28 1.93
δ 4.13 3.51 3.054.33MeNO2
4.04.25MeF
3.03.05MeCl
2.82.70MeBr
2.52.16MeI
3.53.38MeOH
3.02.36MeNH2
2.50.8MeMe
2.10.4MeH
1.80MeSiMe3
1.0–1MeLi
EN(X)δ(Me)MeX
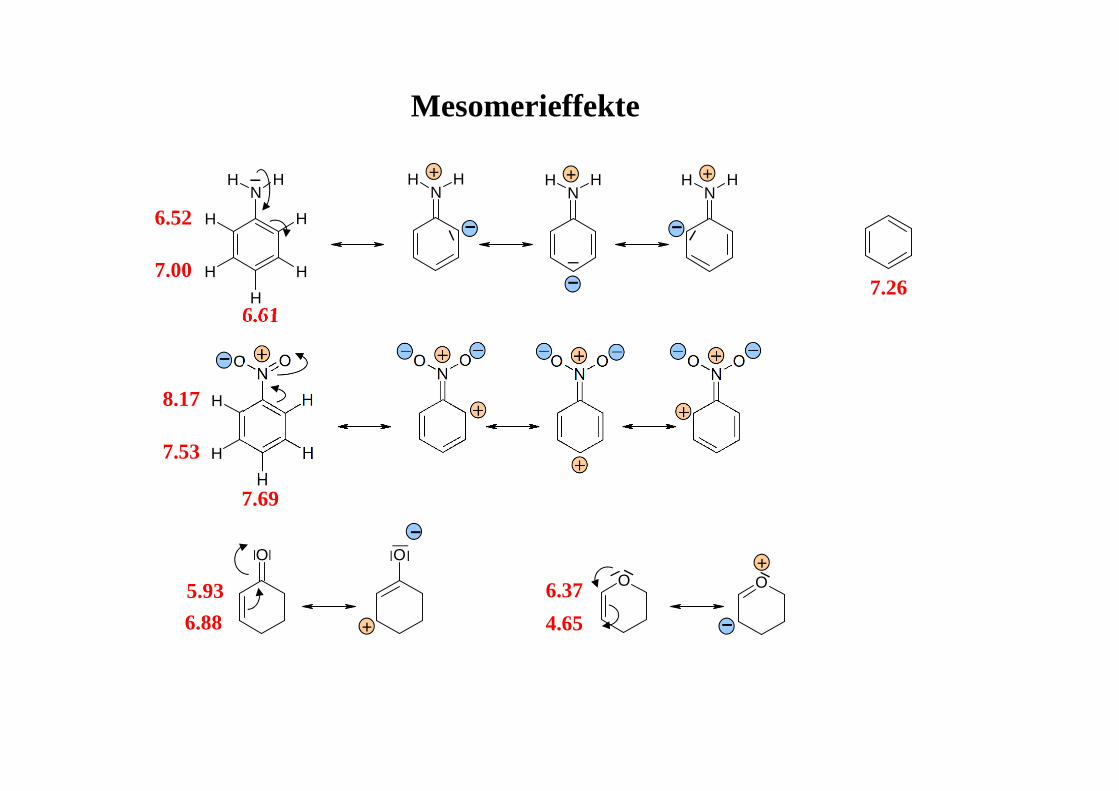
Mesomerieffekte
N
H
H
H
H
H
HHN
HHN
HHN
HH
N
H
H
H
H
H
OON
OON
OON
OO
O O
O O
+
−−−−
+ +
+ +
−−−−
−−−−
−−−− −−−− ++−−−− −−−− −−−− −−−− −−−−
+
+ +
+
+
−−−−
−−−−
6.52
7.00
6.617.26
8.17
7.53
7.69
6.88
5.93 6.37
4.65
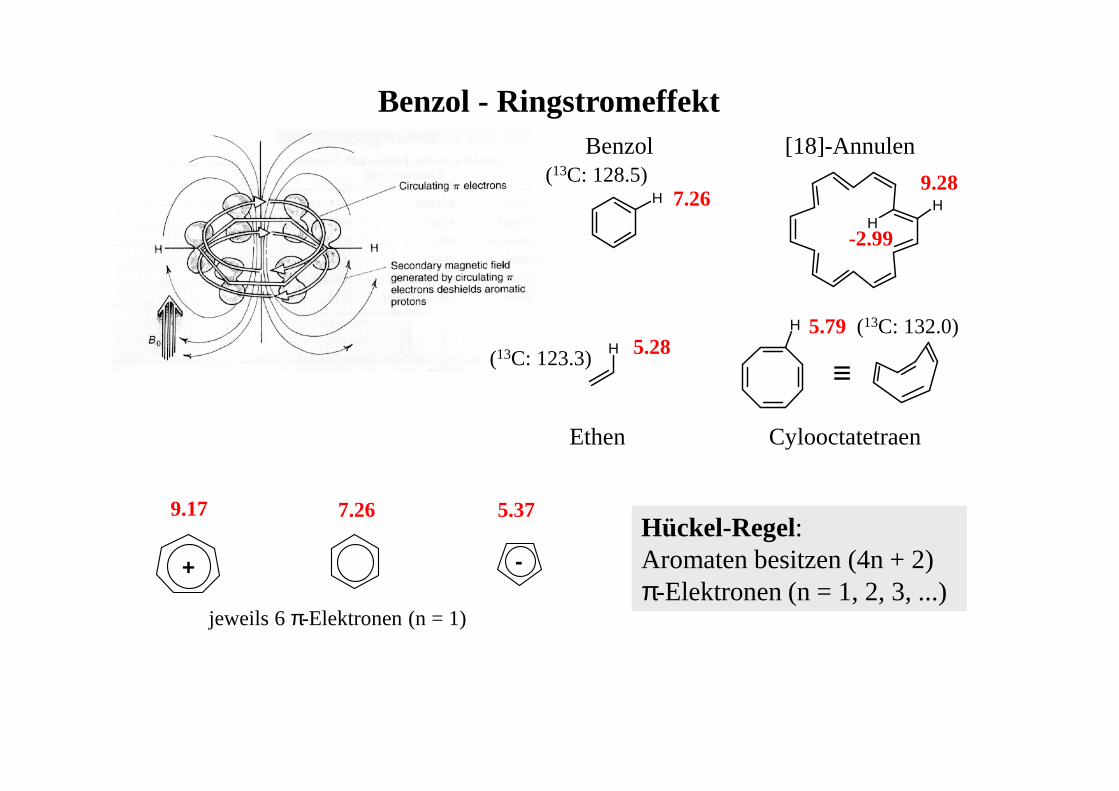
Benzol - Ringstromeffekt
H
H HH
H
[18]-Annulen
7.26(13C: 128.5) 9.28
-2.99
5.795.28(13C: 123.3) ≡
Benzol
Ethen Cylooctatetraen
Hückel-Regel:Aromaten besitzen (4n + 2) π-Elektronen (n = 1, 2, 3, ...)
+ -
9.17 7.26 5.37
jeweils 6 π-Elektronen (n = 1)
(13C: 132.0)
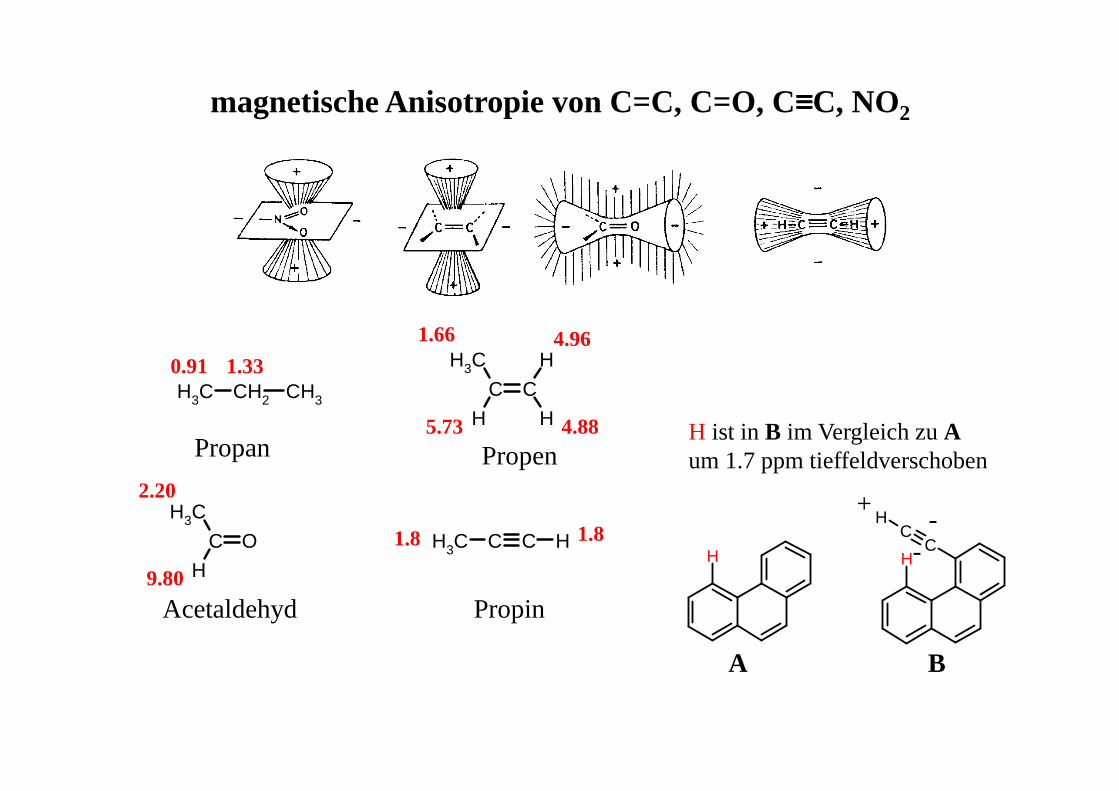
H HC
CH
C C
CH3
H H
H
CH3 CH2 CH3
C O
H
CH3
C C HCH3
magnetische Anisotropie von C=C, C=O, C≡≡≡≡C, NO2
Propan Propen
PropinAcetaldehyd
0.91 1.33
1.66
5.73
4.96
4.88
2.20
9.80
1.81.8
A B
H ist in B im Vergleich zu Aum 1.7 ppm tieffeldverschoben
+-
-
magnetische Anisotropie in Cyclohexanderivaten
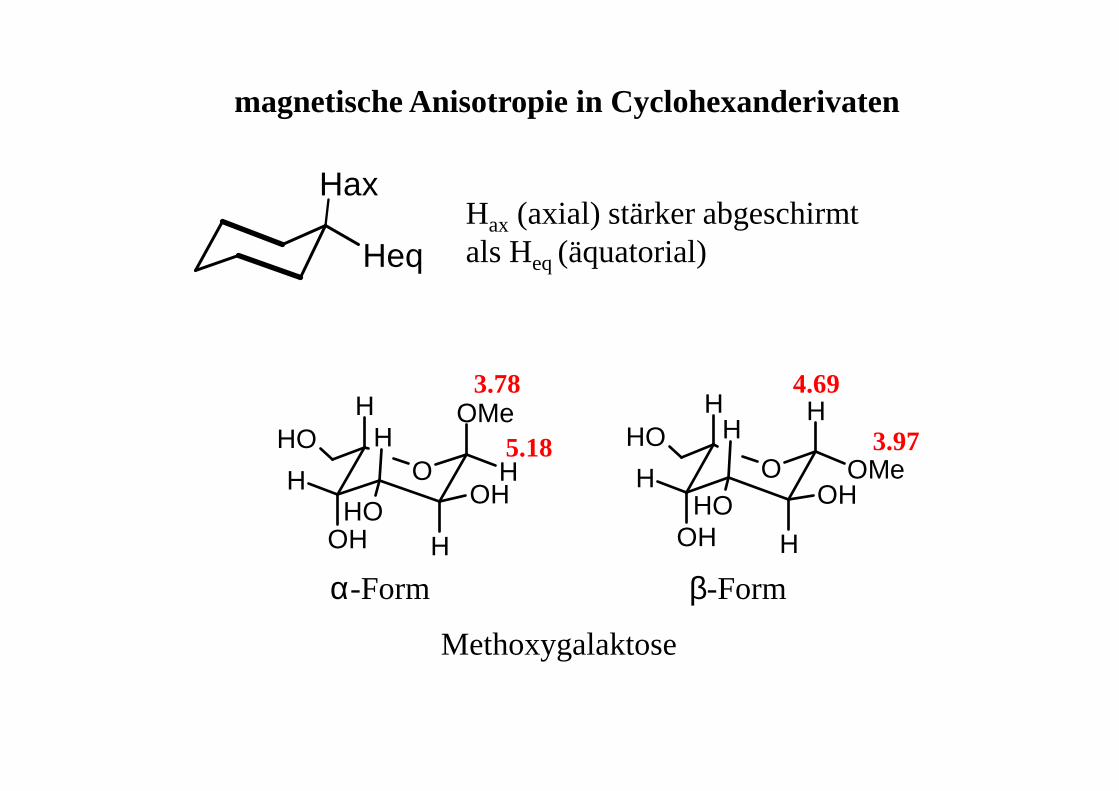
Hax
HeqHax (axial) stärker abgeschirmt als Heq (äquatorial)
OH
HH
H
H HOH
OH
OOH
OMe
OH
HH
H
HOHOH
OOH
H
OMe
Methoxygalaktose
α-Form β-Form
3.78
5.18
4.69
3.97
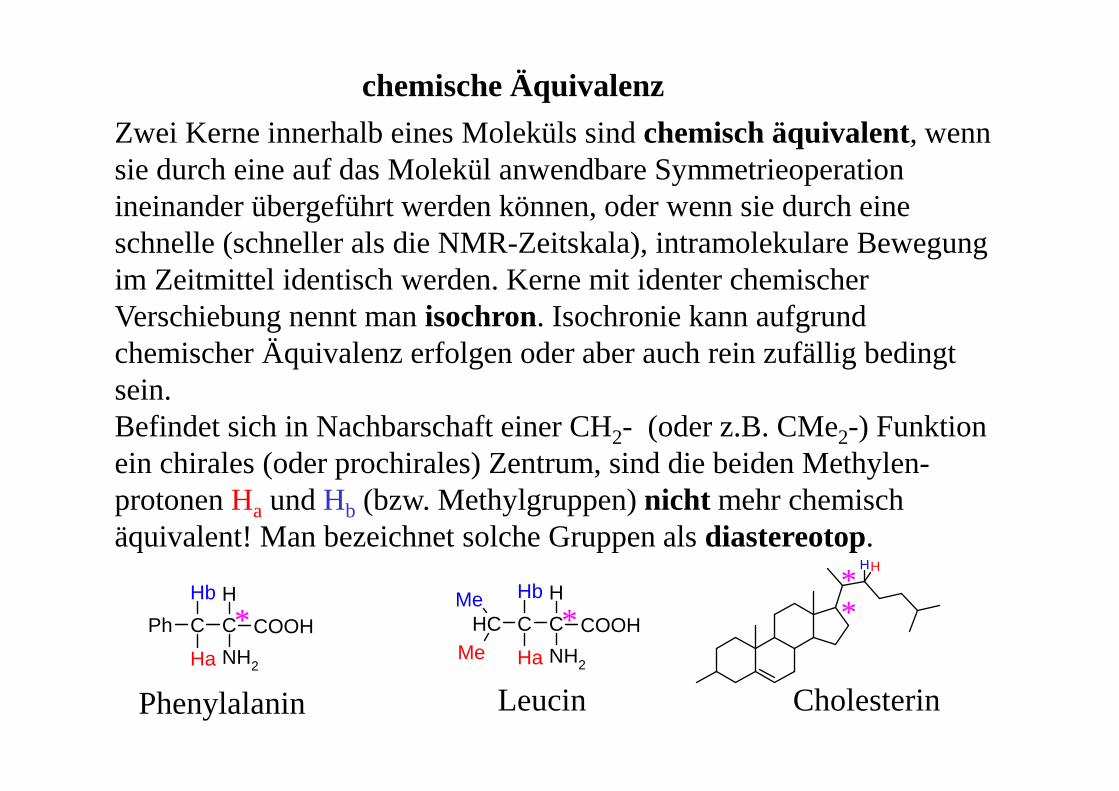
Zwei Kerne innerhalb eines Moleküls sind chemisch äquivalent, wenn sie durch eine auf das Molekül anwendbare Symmetrieoperation ineinander übergeführt werden können, oder wenn sie durch eine schnelle (schneller als die NMR-Zeitskala), intramolekulare Bewegung im Zeitmittel identisch werden. Kerne mit identer chemischer Verschiebung nennt man isochron. Isochronie kann aufgrund chemischer Äquivalenz erfolgen oder aber auch rein zufällig bedingt sein.Befindet sich in Nachbarschaft einer CH2- (oder z.B. CMe2-) Funktion ein chirales (oder prochirales) Zentrum, sind die beiden Methylen-protonen Ha und Hb (bzw. Methylgruppen) nicht mehr chemisch äquivalent! Man bezeichnet solche Gruppen als diastereotop.
chemische Äquivalenz
C C
H
NH2
Hb
Ha
Ph COOH C C
H
NH2
Hb
Ha
CH COOHMe
Me
HH
Phenylalanin Leucin Cholesterin
* * **
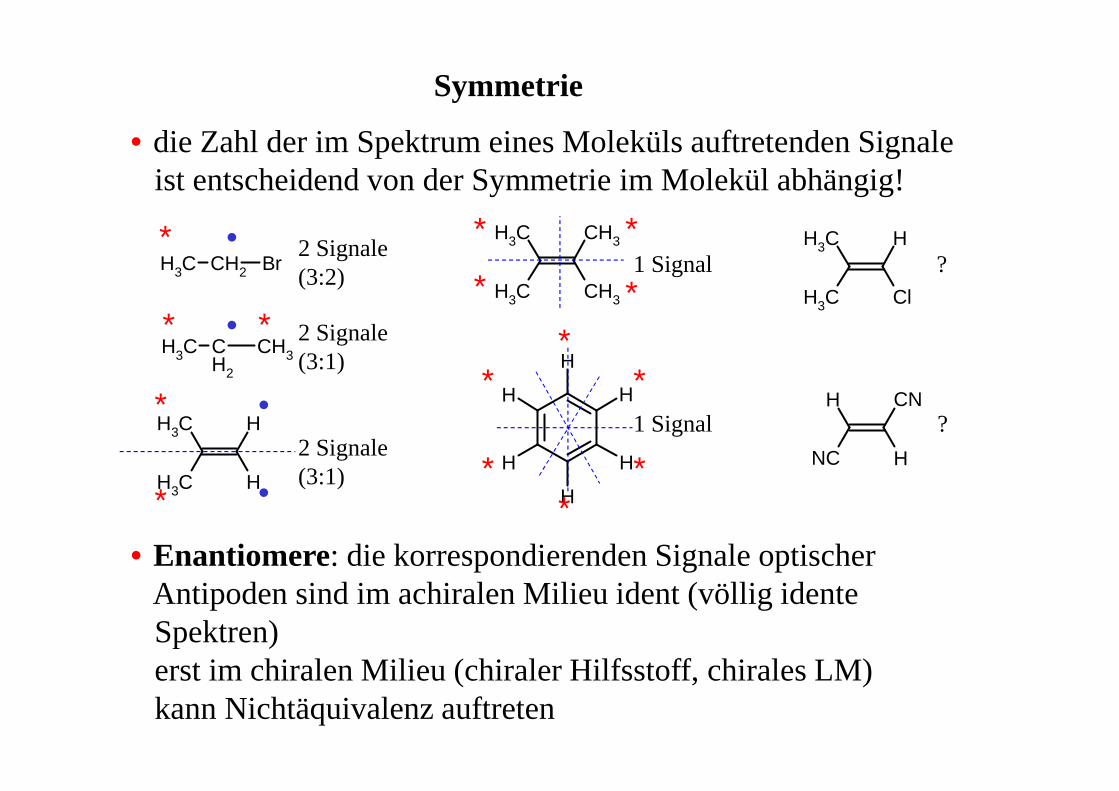
Symmetrie
• die Zahl der im Spektrum eines Moleküls auftretenden Signaleist entscheidend von der Symmetrie im Molekül abhängig!
• Enantiomere: die korrespondierenden Signale optischerAntipoden sind im achiralen Milieu ident (völlig identeSpektren)erst im chiralen Milieu (chiraler Hilfsstoff, chirales LM)kann Nichtäquivalenz auftreten
CH3
CH3
H
H
CH3
CH3
CH3
CH3
CH2 BrCH3
CH2
CH3CH3 H
H
H
H
H
H
CH3
CH3
Cl
H
H
H
CN
NC
∗∗
∗
∗
∗
∗
∗
∗
∗
∗
∗∗
∗∗
∗
2 Signale (3:2)
2 Signale(3:1)
2 Signale(3:1)
1 Signal
1 Signal ?
?•
•
•
•
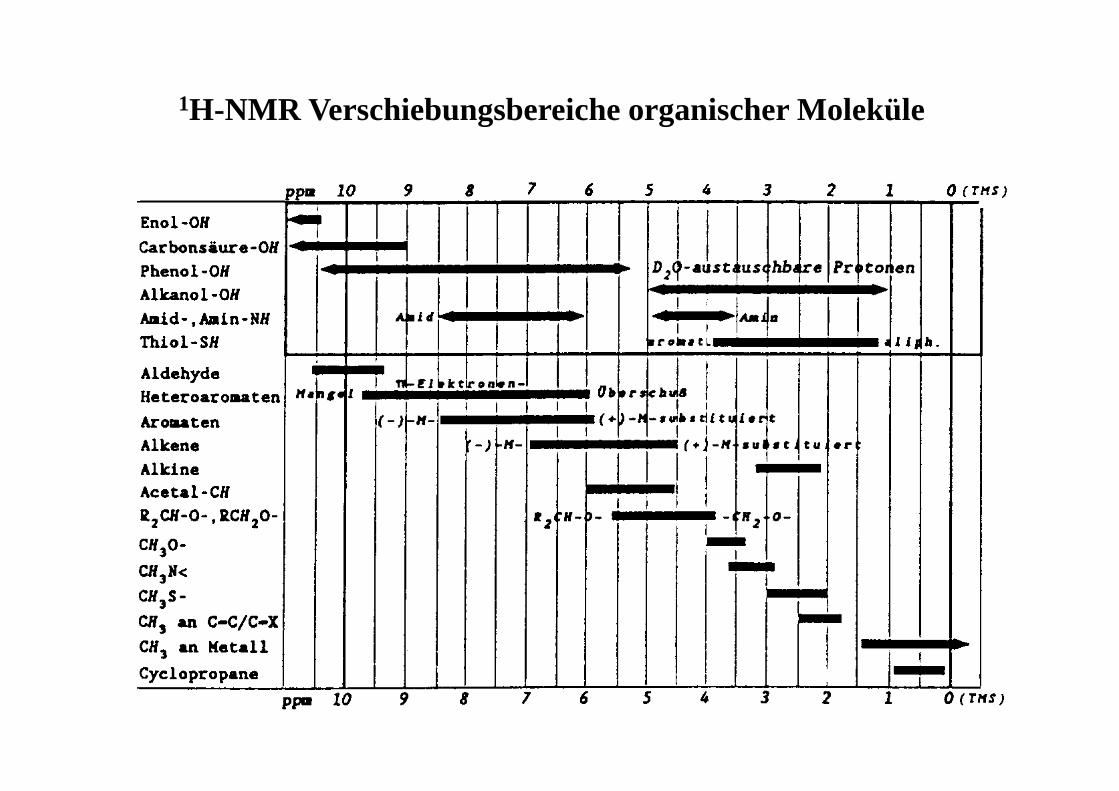
1H-NMR Verschiebungsbereiche organischer Moleküle
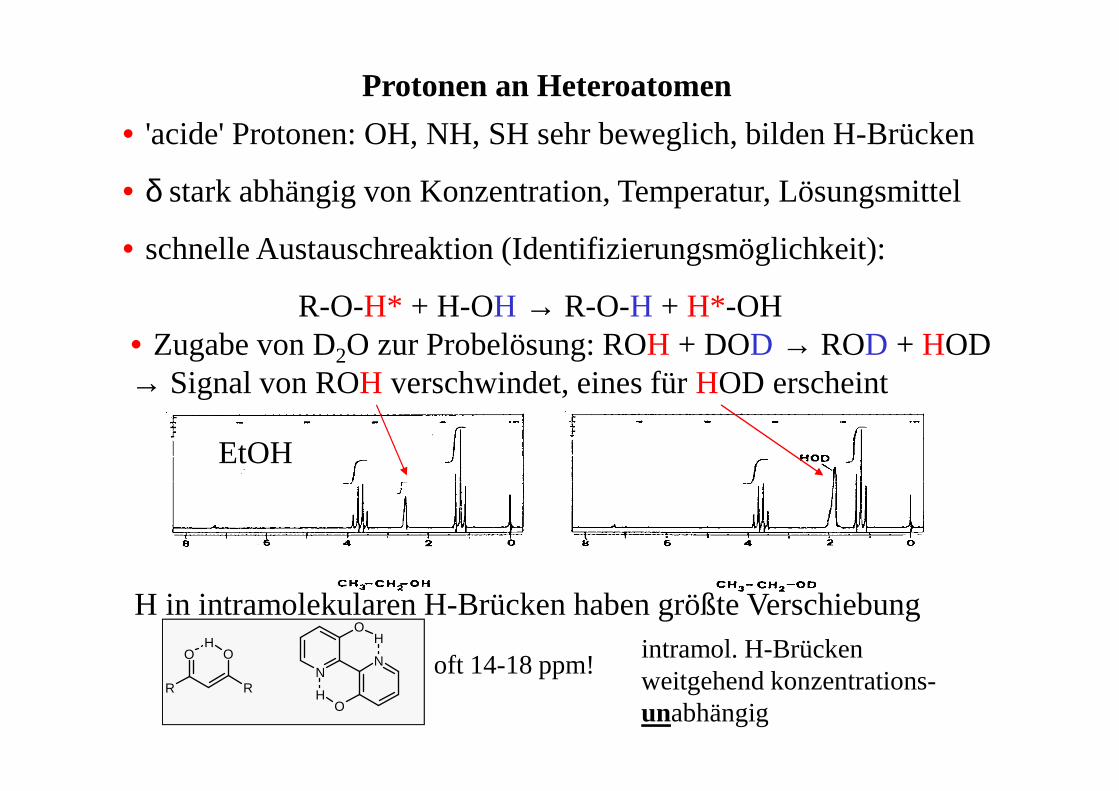
Protonen an Heteroatomen
• 'acide' Protonen: OH, NH, SH sehr beweglich, bilden H-Brücken
• δ stark abhängig von Konzentration, Temperatur, Lösungsmittel
• schnelle Austauschreaktion (Identifizierungsmöglichkeit):
R-O-H* + H-OH → R-O-H + H*-OH• Zugabe von D2O zur Probelösung: ROH + DOD → ROD + HOD→ Signal von ROH verschwindet, eines für HOD erscheint
H in intramolekularen H-Brücken haben größte Verschiebung
R
O
R
OH
NN
O
OH
H
oft 14-18 ppm!intramol. H-Brücken weitgehend konzentrations-unabhängig
EtOH

Spin-Spin Kopplung
C–HA
C-HB
B0
HA entweder ↑↑↑↑ (in Richtung B0 → Verstärkung von B0)oder ↓↓↓↓ (entgegen B0 → Abschwächung von B0)
• Wenn HB einen Kern HA der Sorte ↑↑↑↑ ‘spürt ‘ kommt es bei (geringfügig‘) anderer Frequenz zur Resonanz als wenn HA der Sorte ↓↓↓↓ angehört:
• da beide Sorten aber statistisch gleich verteilt sind (geringe Besetzungsunterschiedehier vernachlässigbar) kommt es zu einer 1:1 Aufspaltung des Signals von HB. Der
Abstand der beiden Signalkomponenten ist die Kopplungskonstante J.
• J kann ein positives oder negatives Vorzeichen haben (aus dem Spektrum meist nicht direkt ablesbar)
• Kopplungen werden durch Bindungselektronen übertragen (skalare Spin-SpinKopplung)
• Kopplung erfolgt gegenseitig → analoge Situation für HA: J(B,A) = J(A,B)
• die Größe von J ist unabhängigvon der Stärke des äußeren Magnetfeldes B0 !!!!!
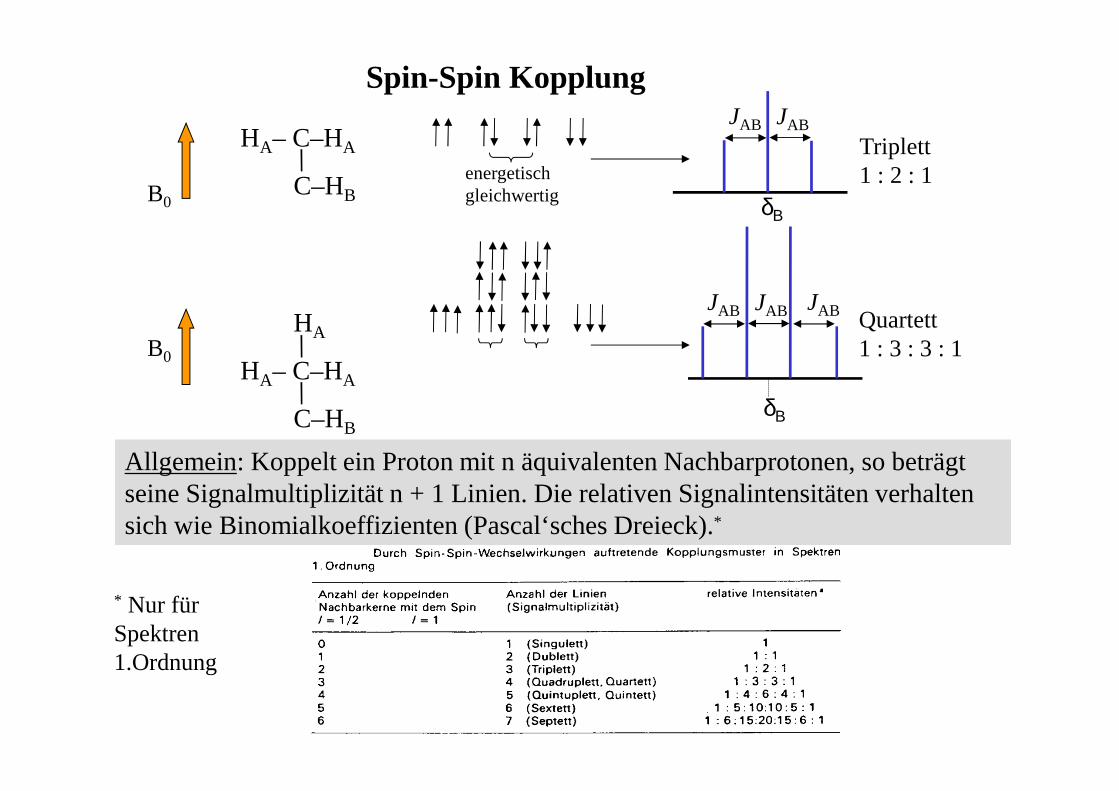
HA– C–HA
C–HBB0
energetischgleichwertig
Spin-Spin Kopplung
δΒ
JAB
JAB
JAB
HA
HA– C–HA
C–HB
B0
δΒ
JAB JAB
Triplett1 : 2 : 1
Quartett1 : 3 : 3 : 1
Allgemein: Koppelt ein Proton mit n äquivalenten Nachbarprotonen, so beträgt seine Signalmultiplizität n + 1 Linien. Die relativen Signalintensitäten verhalten sich wie Binomialkoeffizienten (Pascal‘sches Dreieck).*
* Nur für Spektren 1.Ordnung
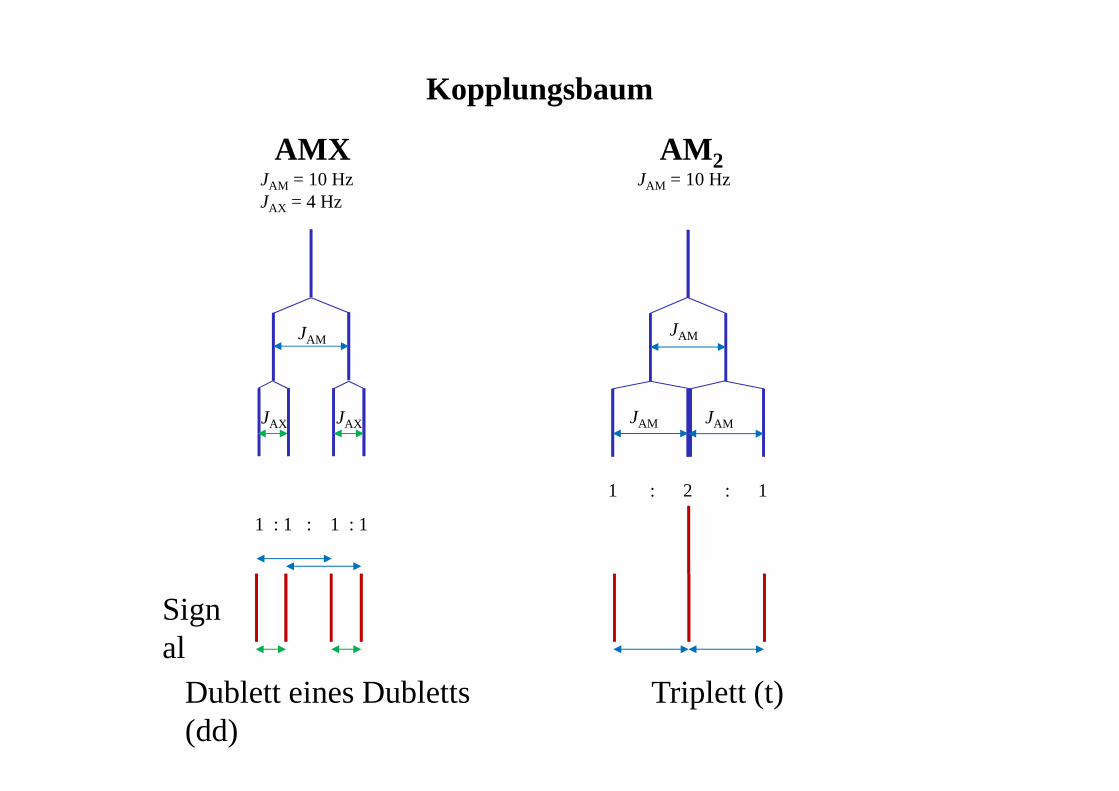
Kopplungsbaum
AMXJAM = 10 HzJAX = 4 Hz
AM 2JAM = 10 Hz
JAM
JAX JAX
1 : 1 : 1 : 1
1 : 2 : 1
JAM
JAM JAM
Signal
Dublett eines Dubletts (dd)
Triplett (t)
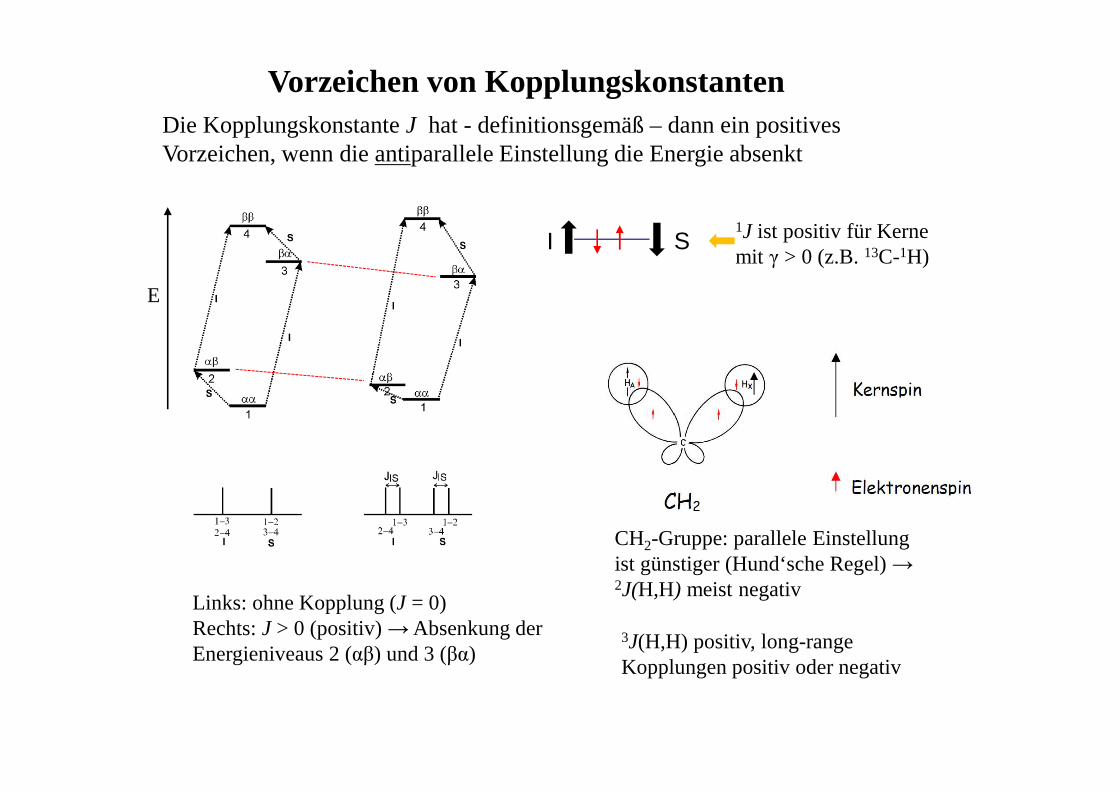
Vorzeichen von Kopplungskonstanten
Links: ohne Kopplung (J = 0)Rechts: J > 0 (positiv) → Absenkung der Energieniveaus 2 (αβ) und 3 (βα)
I S
Die Kopplungskonstante J hat - definitionsgemäß – dann ein positives Vorzeichen, wenn die antiparallele Einstellung die Energie absenkt
1J ist positiv für Kerne mit γ > 0 (z.B. 13C-1H)
CH2-Gruppe: parallele Einstellung ist günstiger (Hund‘sche Regel) → 2J(H,H) meist negativ
3J(H,H) positiv, long-range Kopplungen positiv oder negativ
E
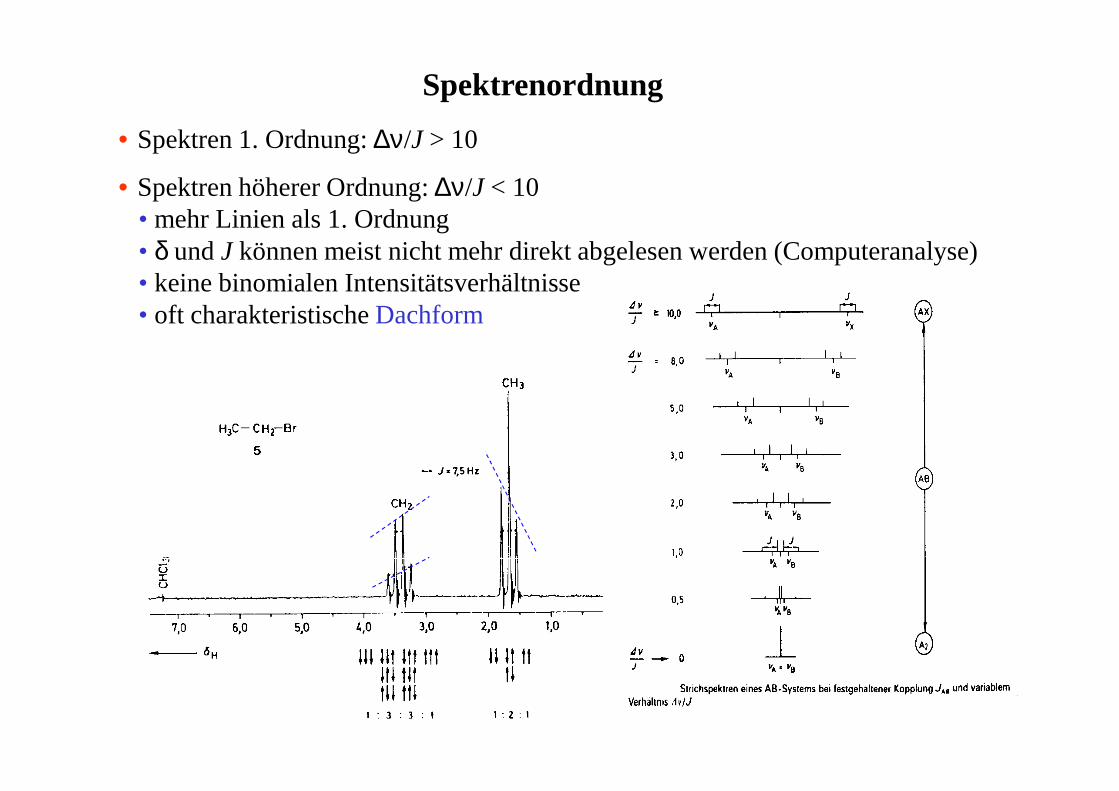
Spektrenordnung
• Spektren 1. Ordnung: ∆ν/J > 10
• Spektren höherer Ordnung: ∆ν/J < 10• mehr Linien als 1. Ordnung• δ und J können meist nicht mehr direkt abgelesen werden (Computeranalyse)• keine binomialen Intensitätsverhältnisse• oft charakteristische Dachform
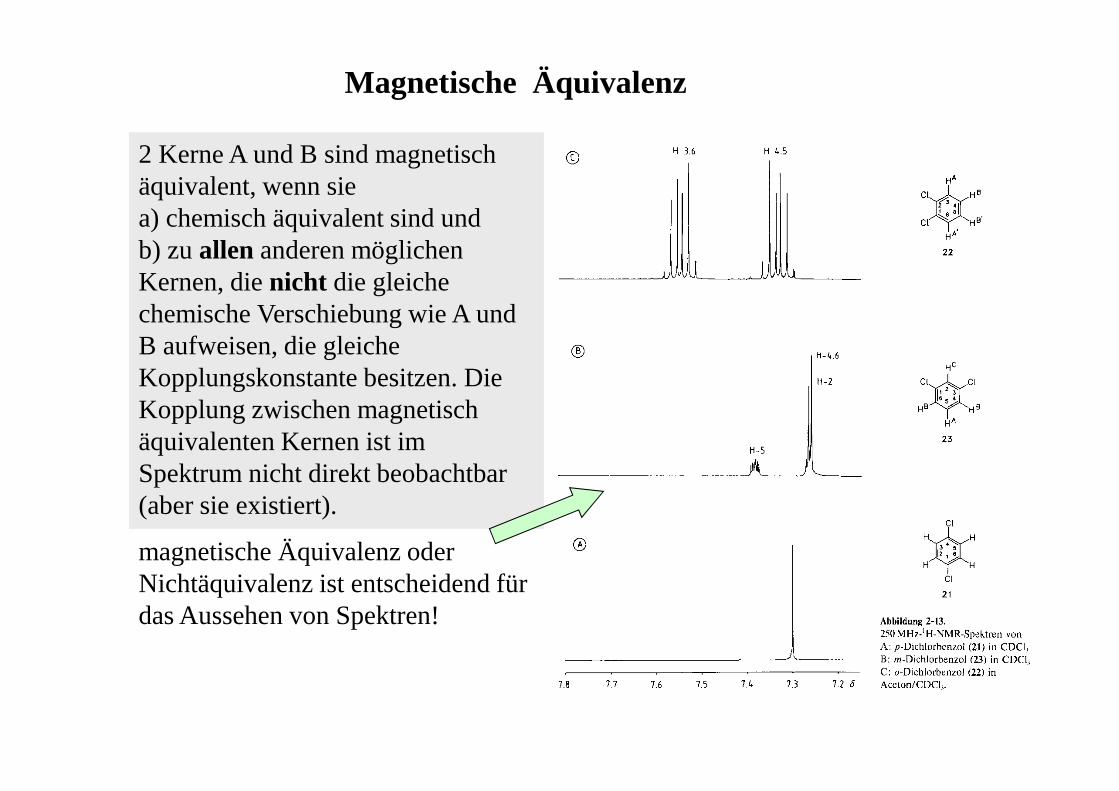
Magnetische Äquivalenz
2 Kerne A und B sind magnetisch äquivalent, wenn sie a) chemisch äquivalent sind undb) zu allen anderen möglichen Kernen, die nicht die gleiche chemische Verschiebung wie A und B aufweisen, die gleiche Kopplungskonstante besitzen. Die Kopplung zwischen magnetisch äquivalenten Kernen ist im Spektrum nicht direkt beobachtbar (aber sie existiert).
magnetische Äquivalenz oder Nichtäquivalenz ist entscheidend für das Aussehen von Spektren!
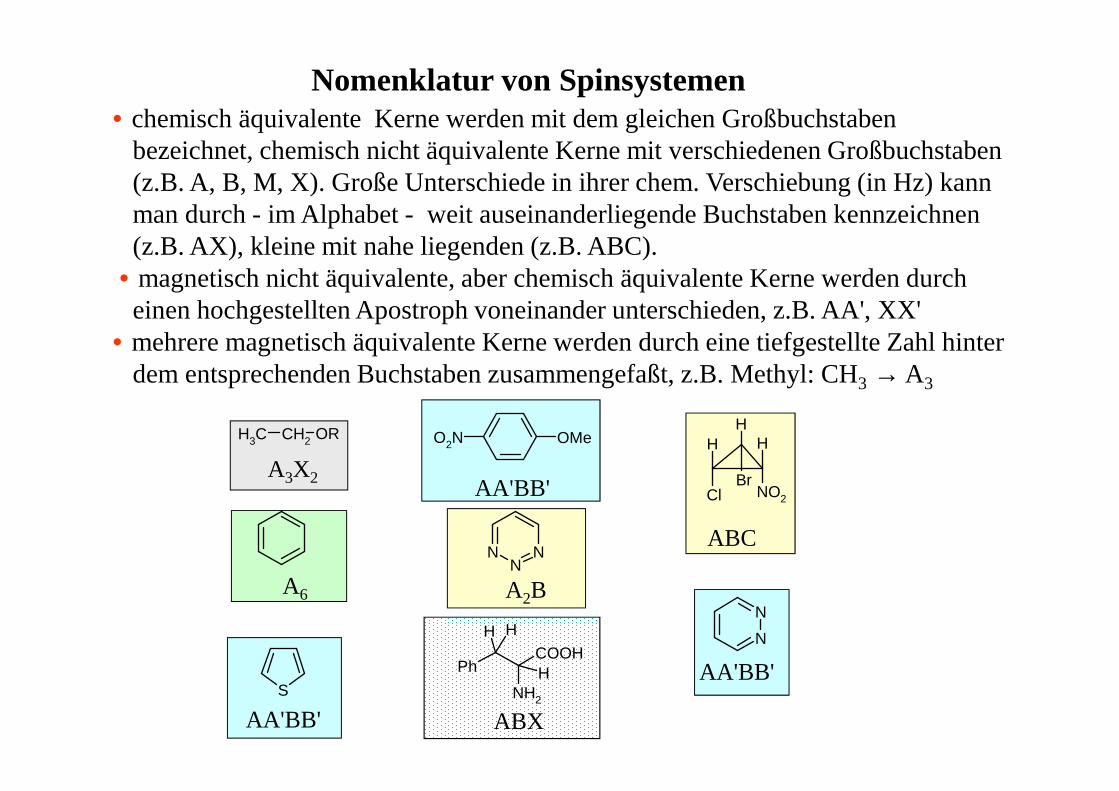
Nomenklatur von Spinsystemen• chemisch äquivalente Kerne werden mit dem gleichen Großbuchstaben
bezeichnet, chemisch nicht äquivalente Kerne mit verschiedenen Großbuchstaben(z.B. A, B, M, X). Große Unterschiede in ihrer chem. Verschiebung (in Hz) kannman durch - im Alphabet - weit auseinanderliegende Buchstaben kennzeichnen (z.B. AX), kleine mit nahe liegenden (z.B. ABC).
• magnetisch nicht äquivalente, aber chemisch äquivalente Kerne werden durcheinen hochgestellten Apostroph voneinander unterschieden, z.B. AA', XX'
• mehrere magnetisch äquivalente Kerne werden durch eine tiefgestellte Zahl hinterdem entsprechenden Buchstaben zusammengefaßt, z.B. Methyl: CH3 → A3
A3X2
A6
AA'BB'
AA'BB'
A2B
ABX
ABC
AA'BB'
CH3 CH2 OR
S
O2N OMe
NN
N
Ph
NH2
COOHH H
H
Cl
H HH
BrNO2
N
N
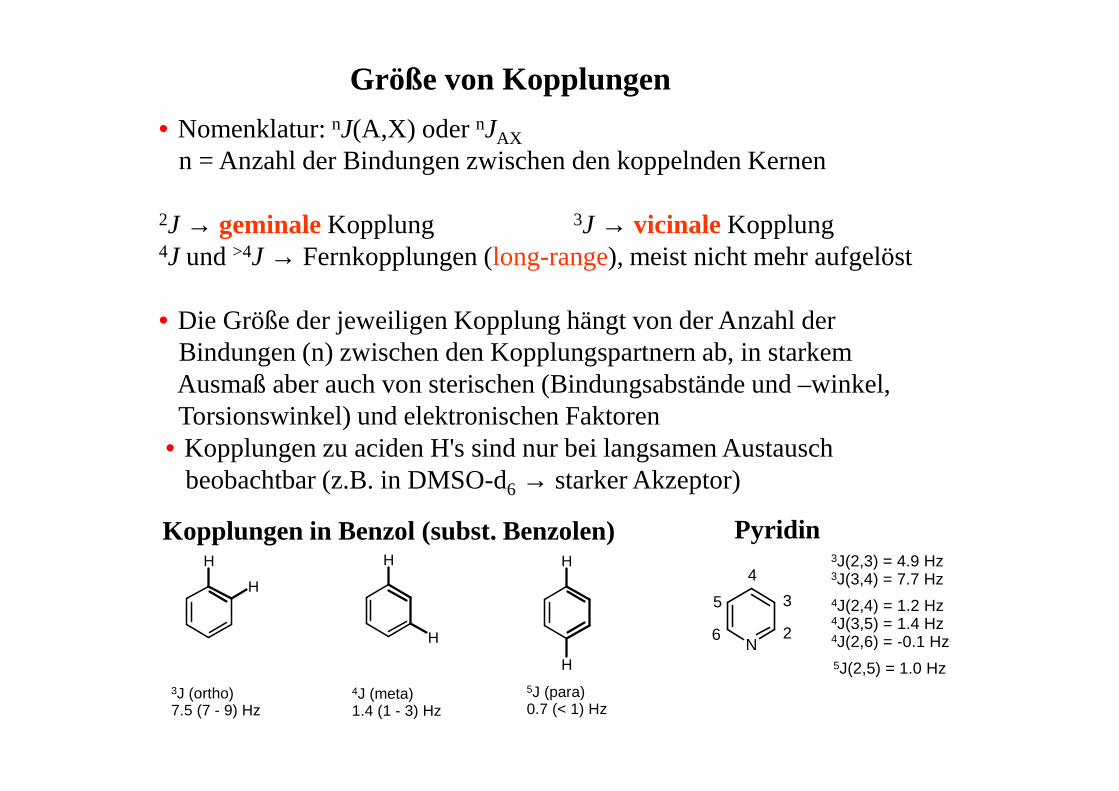
Größe von Kopplungen
• Nomenklatur: nJ(A,X) oder nJAX
n = Anzahl der Bindungen zwischen den koppelnden Kernen
2J → geminaleKopplung 3J → vicinale Kopplung4J und >4J → Fernkopplungen (long-range), meist nicht mehr aufgelöst
• Die Größe der jeweiligen Kopplung hängt von der Anzahl derBindungen (n) zwischen den Kopplungspartnern ab, in starkemAusmaß aber auch von sterischen (Bindungsabstände und –winkel,Torsionswinkel) und elektronischen Faktoren
• Kopplungen zu aciden H's sind nur bei langsamen Austauschbeobachtbar (z.B. in DMSO-d6 → starker Akzeptor)
H
H
H
H
H
H
3J (ortho)7.5 (7 - 9) Hz
4J (meta)1.4 (1 - 3) Hz
5J (para)0.7 (< 1) Hz
Kopplungen in Benzol (subst. Benzolen)
N2
3
4
5
6
3J(2,3) = 4.9 Hz3J(3,4) = 7.7 Hz4J(2,4) = 1.2 Hz4J(3,5) = 1.4 Hz4J(2,6) = -0.1 Hz5J(2,5) = 1.0 Hz
Pyridin
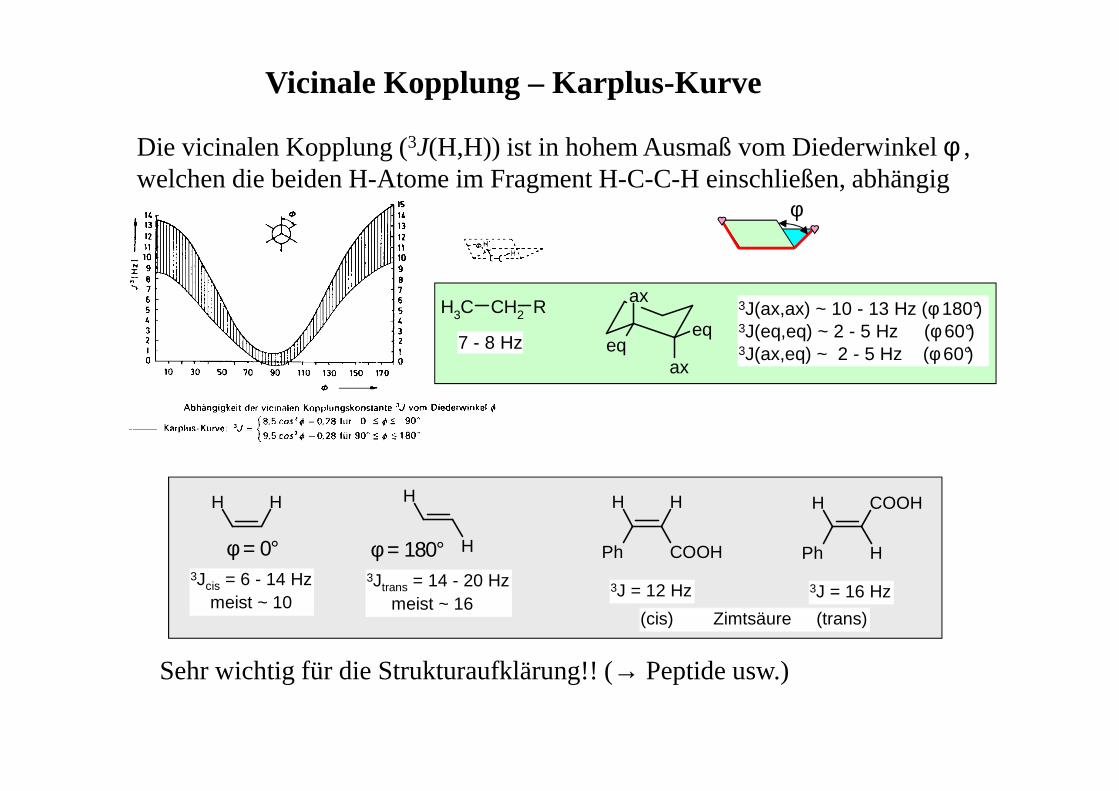
Vicinale Kopplung – Karplus-Kurve
Die vicinalen Kopplung (3J(H,H)) ist in hohem Ausmaß vom Diederwinkel φ , welchen die beiden H-Atome im Fragment H-C-C-H einschließen, abhängig
CH3 CH2 Rax
ax
eqeq7 - 8 Hz
3J(ax,ax) ~ 10 - 13 Hz (φ 180°)3J(eq,eq) ~ 2 - 5 Hz (φ 60°)3J(ax,eq) ~ 2 - 5 Hz (φ 60°)
H H H
H
H H
Ph COOH
H
HPh
COOH
3Jcis = 6 - 14 Hz meist ~ 10
3Jtrans = 14 - 20 Hz meist ~ 16
3J = 12 Hz 3J = 16 Hz
(cis) Zimtsäure (trans)
Sehr wichtig für die Strukturaufklärung!! (→ Peptide usw.)
φ = 0° φ = 180°
φ
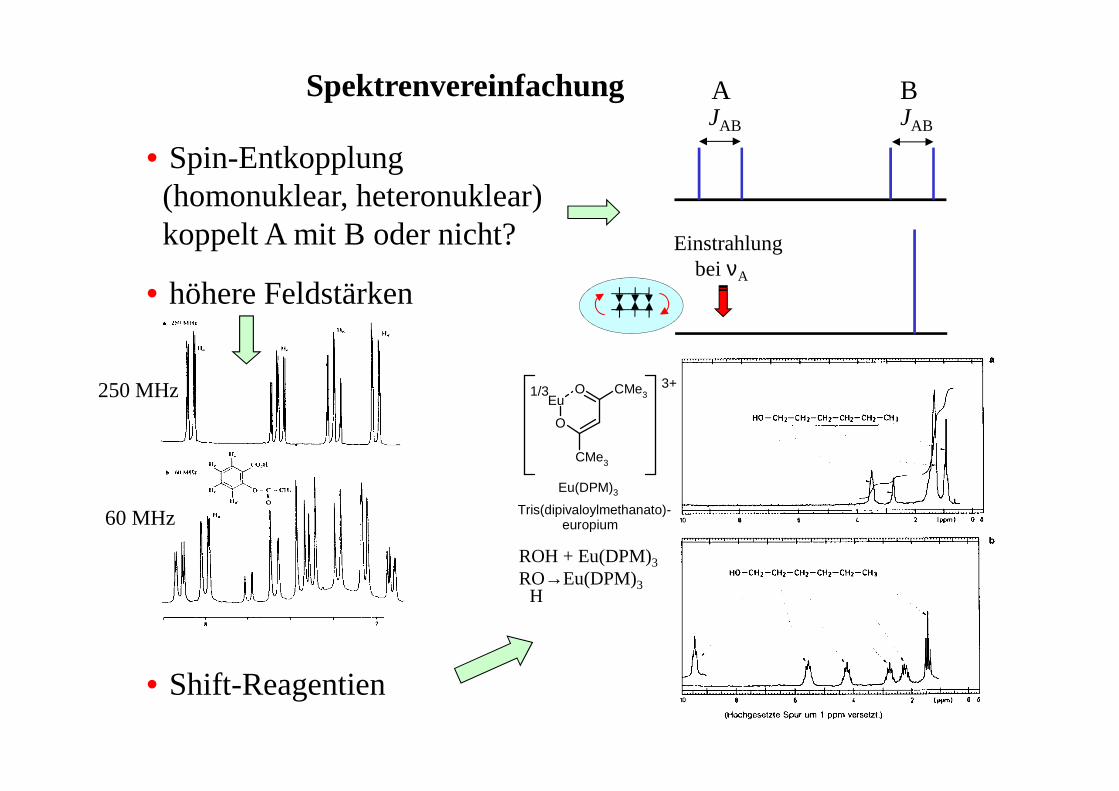
Spektrenvereinfachung
• Spin-Entkopplung(homonuklear, heteronuklear)koppelt A mit B oder nicht?
JAB JAB
A B
Einstrahlungbei νA
• höhere Feldstärken
• Shift-Reagentien
250 MHz
60 MHz
CMe3O
O
CMe3
Eu
1/33+
Eu(DPM)3
Tris(dipivaloylmethanato)- europium
ROH + Eu(DPM)3RO→Eu(DPM)3
H
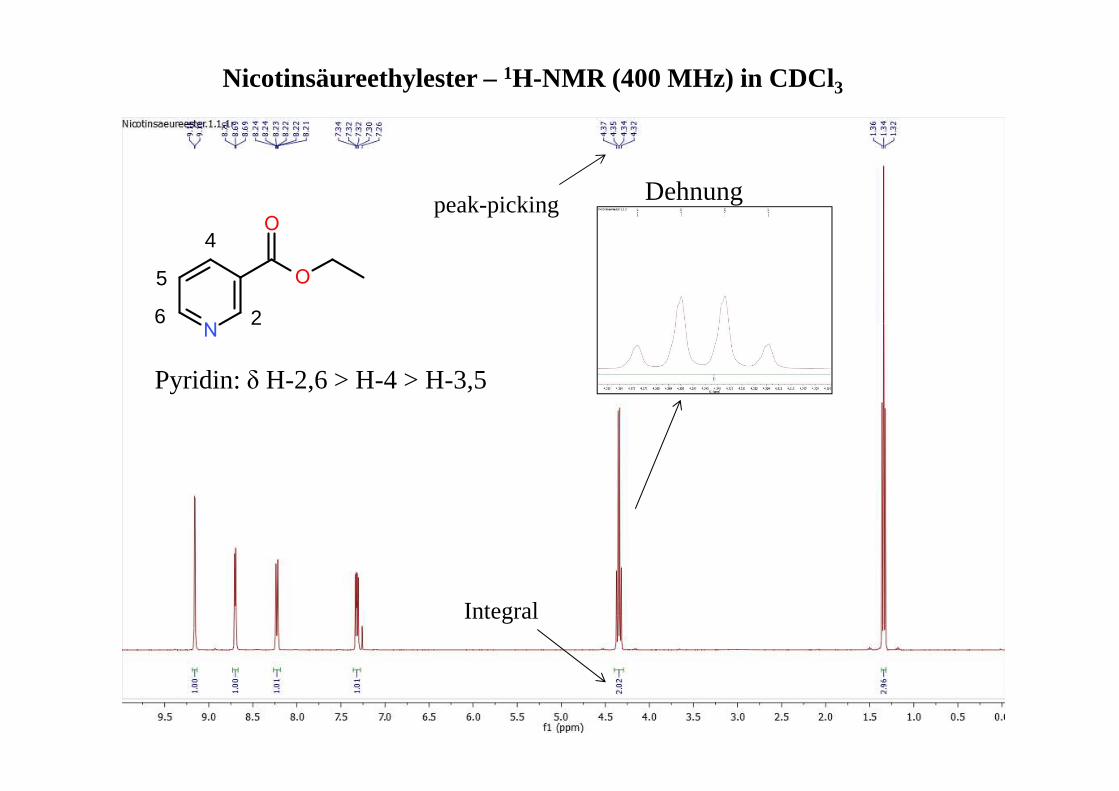
Nicotinsäureethylester –1H-NMR (400 MHz) in CDCl3
peak-picking
Integral
2
4
5
6
O
O
N
Pyridin: δ H-2,6 > H-4 > H-3,5
Dehnung
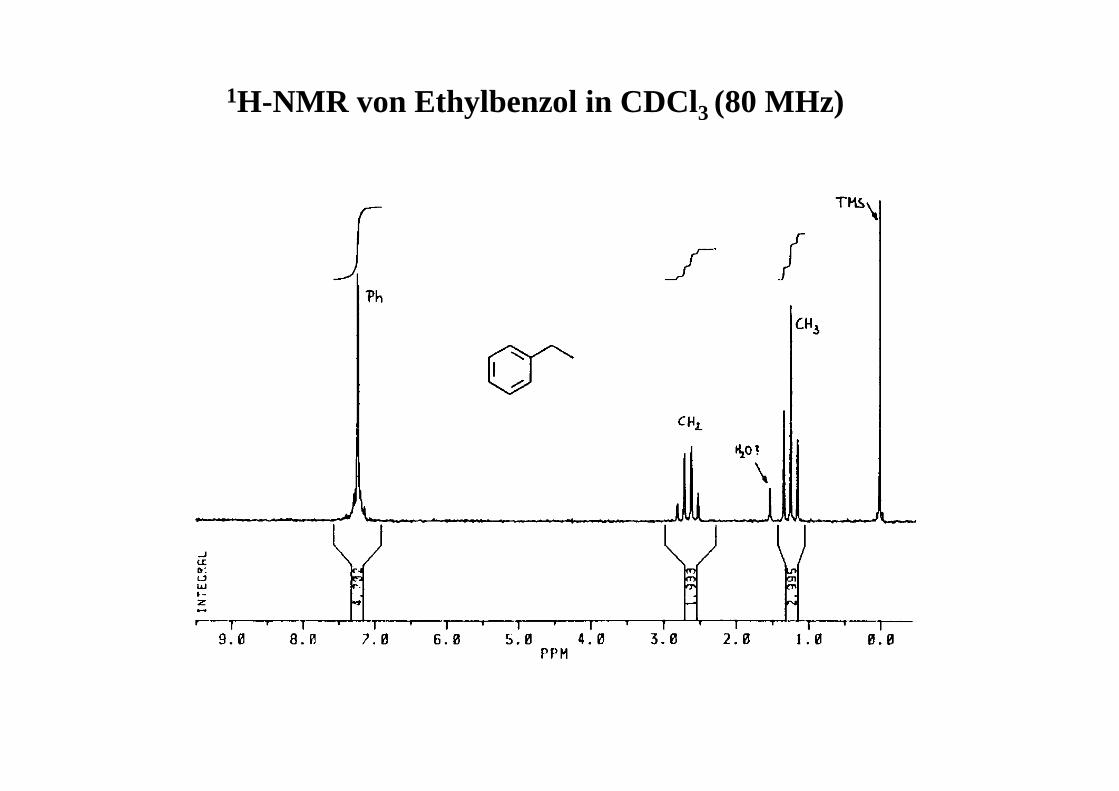
1H-NMR von Ethylbenzol in CDCl3 (80 MHz)
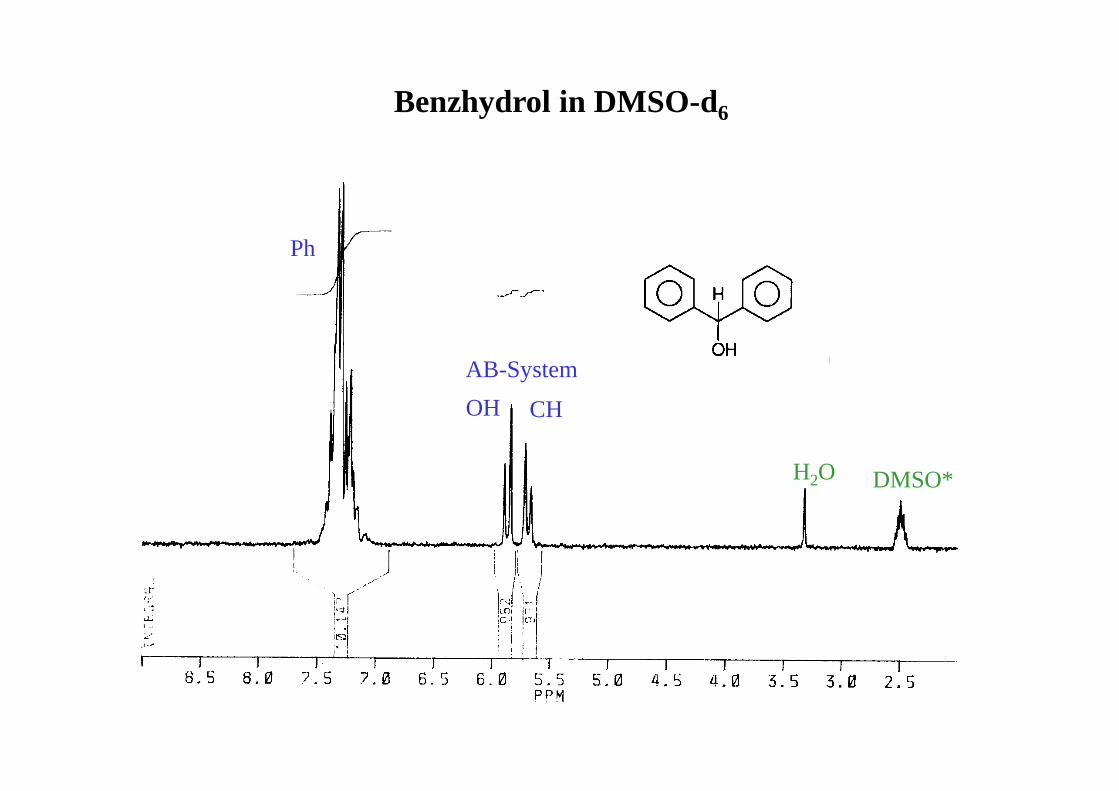
Benzhydrol in DMSO-d6
Ph
OH CH
DMSO*H2O
AB-System
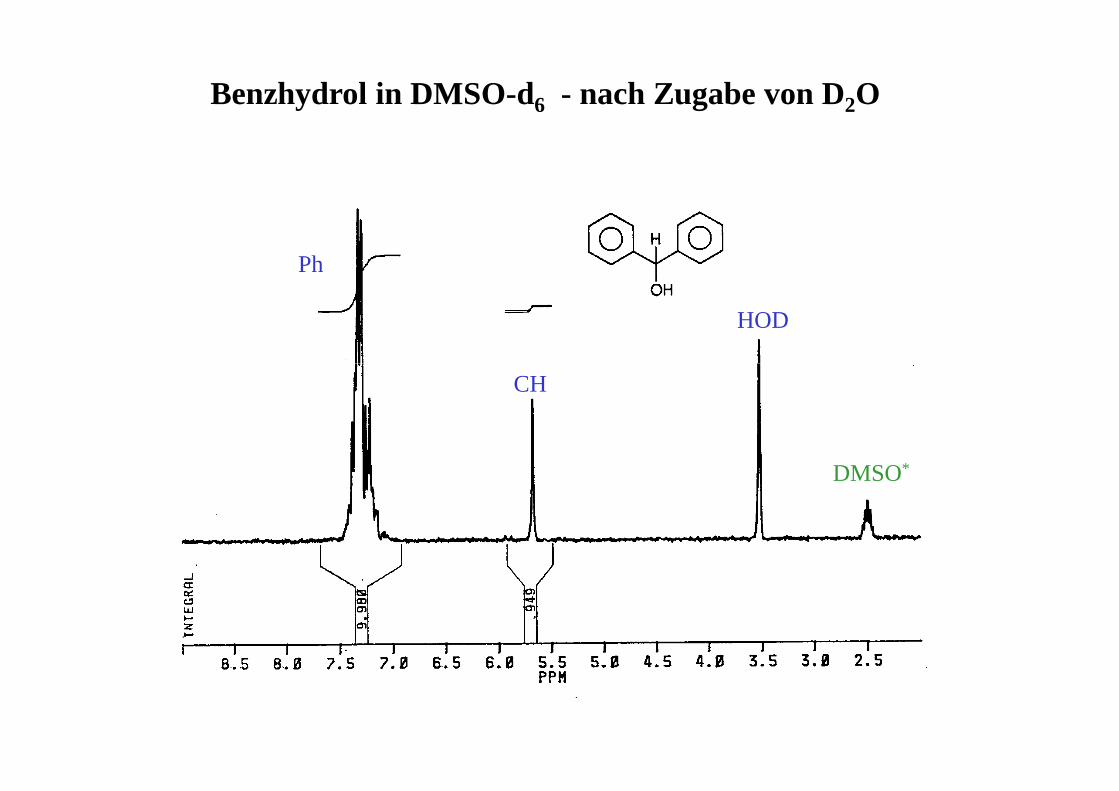
Benzhydrol in DMSO-d6 - nach Zugabe von D2O
HOD
DMSO*
Ph
CH
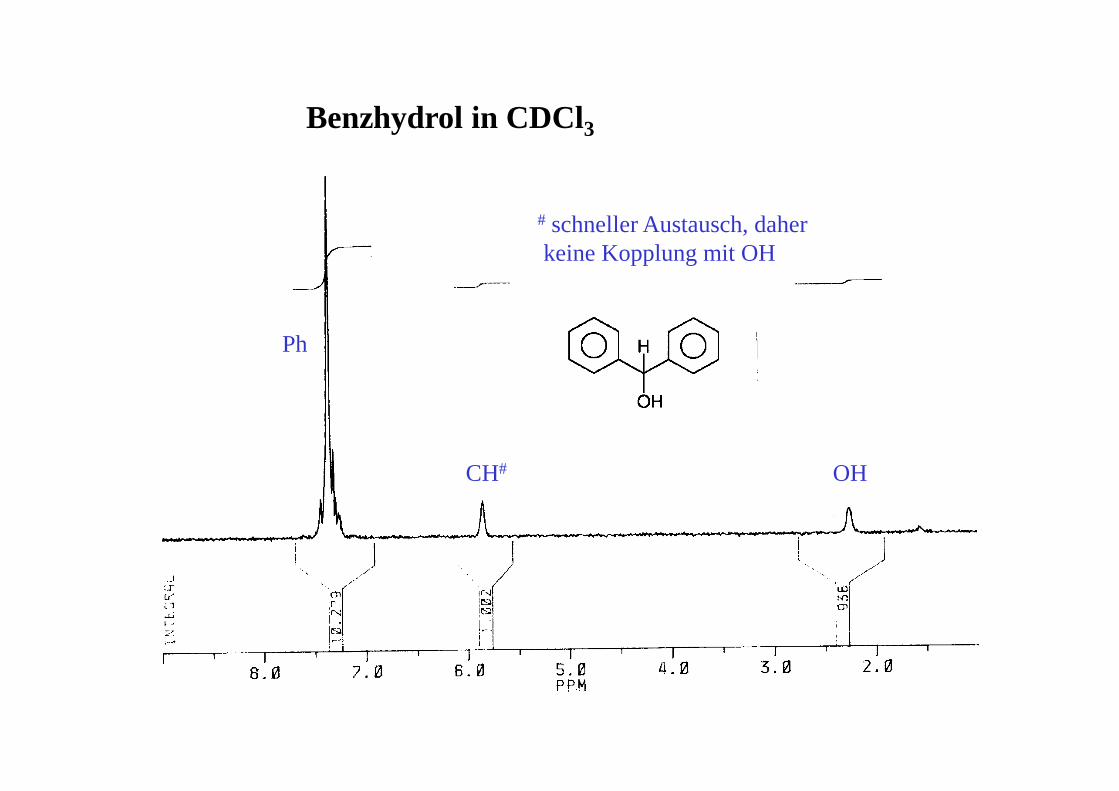
Benzhydrol in CDCl3
Ph
CH# OH
# schneller Austausch, daher keine Kopplung mit OH
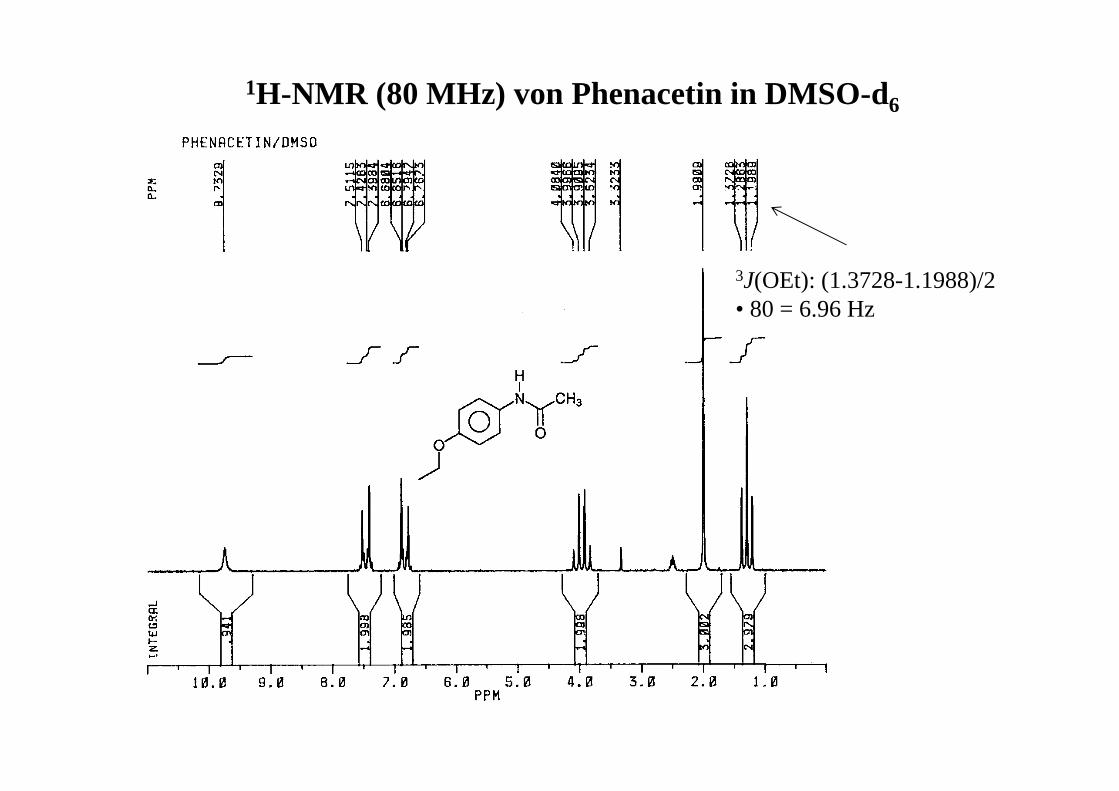
1H-NMR (80 MHz) von Phenacetin in DMSO-d6
3J(OEt): (1.3728-1.1988)/2 • 80 = 6.96 Hz
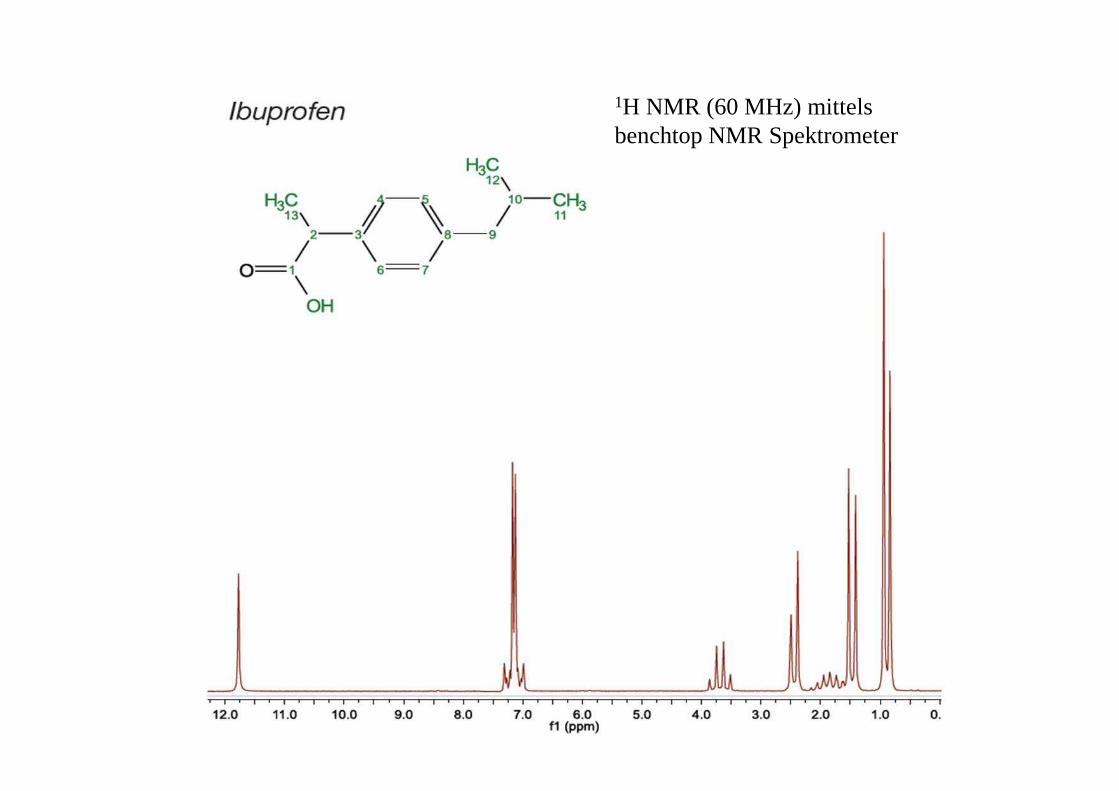
1H NMR (60 MHz) mittelsbenchtop NMR Spektrometer

13C: I = ½, natürliche Häufigkeit 1.1%, γ(13C) nur ¼ von γ(1H) → wesentlich weniger empfindlich als 1H (PFT-Spektrometer, Spektrenakkumulation, spezielle Meßtechniken)
13C ist 'diluted spin':C—C—C—C ca. 96%, dem NMR Experiment nicht zugänglich (I = 0)
C—C—C—C ca. 1% C— C—C—C ca. 1% C—C—C—C ca. 1% C—C—C—C ca. 1%
C—C—C—C ca. 1/100%C—C—C—C ca. 1/100%.......usw.........C—C—C—C ca. 1/106 %
13C-NMR Spektroskopie
diese Isotopomerewerden beim 'normalen' 13C-NMR Experiment gemessen
geht beim 'normalen' 13C-Experiment im Rauschen unter; mit speziellen Verfahren (INADEQUATE) meßbar
(C = 13C, C = 12C)

13C-NMR - Besonderheiten
• Das 'normale' 13C-NMR Spektrum stammt nicht von einereinheitlichen Substanz, sondern von einem Gemischverschiedener Isotopomere
• 13C,13C-Kopplungen sind im 'normalen' 13C-NMR Spektrumnicht sichtbar (die Wahrscheinlichkeit für 2 benachbarte 13C-Kerne ist äußerst gering)
• Kopplungen mit Protonen sind sichtbar. Daher meist sehr vieleLinien (wegen der – meist – vielen Kopplungen mit 1H).Vorteil: viel 'Nachbarschaftsinformation', Nachteil: oftunübersichtlich bis nicht mehr interpretierbar
• → durch verschiedene Maßnahmen (Entkopplung)Kopplungen zu 1H ganz oder teilweise eliminiert

Beff = (1 –σσσσ) B0 (σ: Abschirmungskonstante)
σ = = = = σσσσdia + σσσσpara + σσσσaniso
σσσσdia : beschreibt Abschirmung durch Elektronen mit sphärischer Symmetrie (s-Elektronen), bei 1H dominierendσσσσpara : beschreibt Einfluss von Elektronen mit nicht-sphärischer Ladungsverteilung, negatives Vorzeichen (→ Entschirmung), dominiert bei allen Kernen außer Wasserstoff, enthält Term 1/∆E (∆E ~ mittlere Anregungsenergie), d.h. geringere Anregungsenergien führen zu stärkerer Entschirmungσσσσaniso: beschreibt Einfluss der Anisotropie-Effekte von Nachbargruppen, weniger wichtig bei 13C als bei 1H
Ursachen von 13C chemischen Verschiebungen
δC=O = 215.6 ppmλmax (n → π*) = 292 nm∆E = 401.5 kJ/mol
δC=S = 269.0 ppmλmax (n → π*) = 492 nm∆E = 234.5 kJ/mol
Campher Thiocampher
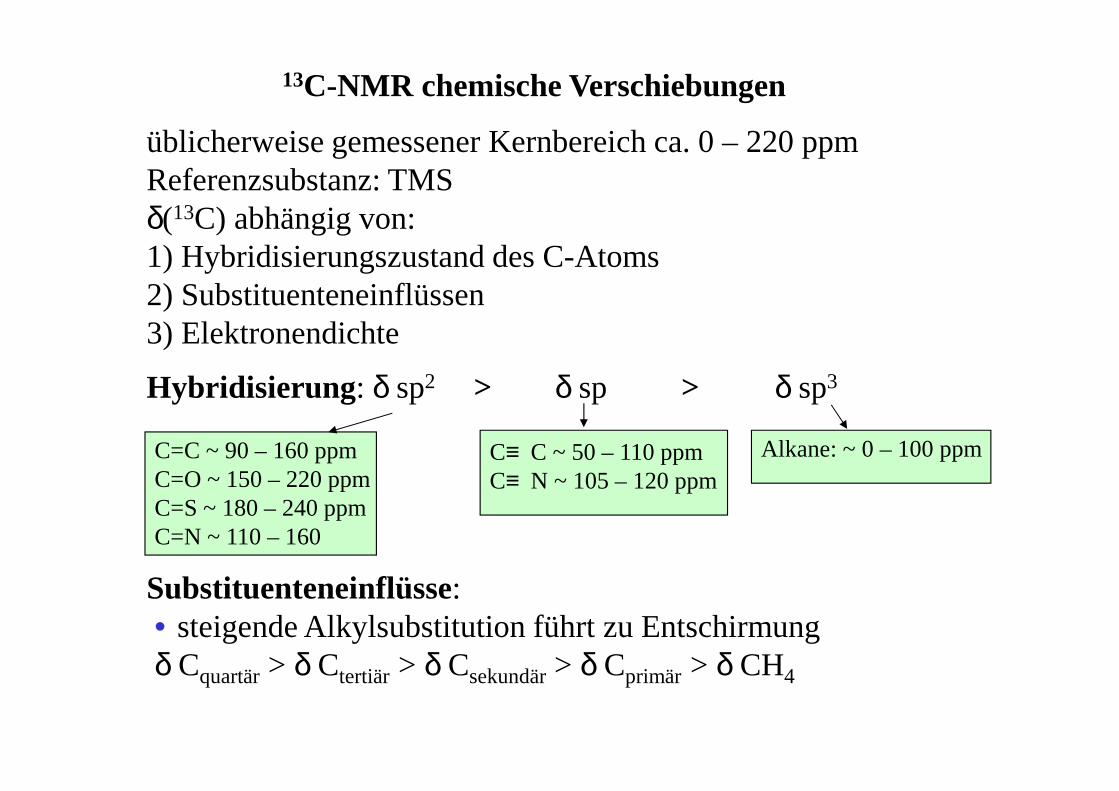
Hybridisierung : δ sp2 δ sp δ sp3
13C-NMR chemische Verschiebungen
üblicherweise gemessener Kernbereich ca. 0 – 220 ppmReferenzsubstanz: TMS δ(13C) abhängig von:1) Hybridisierungszustand des C-Atoms2) Substituenteneinflüssen3) Elektronendichte
Alkane: ~ 0 – 100 ppmC≡ C ~ 50 – 110 ppm C≡ N ~ 105 – 120 ppm
C=C ~ 90 – 160 ppmC=O ~ 150 – 220 ppmC=S ~ 180 – 240 ppmC=N ~ 110 – 160
> >
Substituenteneinflüsse: • steigende Alkylsubstitution führt zu Entschirmungδ Cquartär> δ Ctertiär > δ Csekundär> δ Cprimär > δ CH4
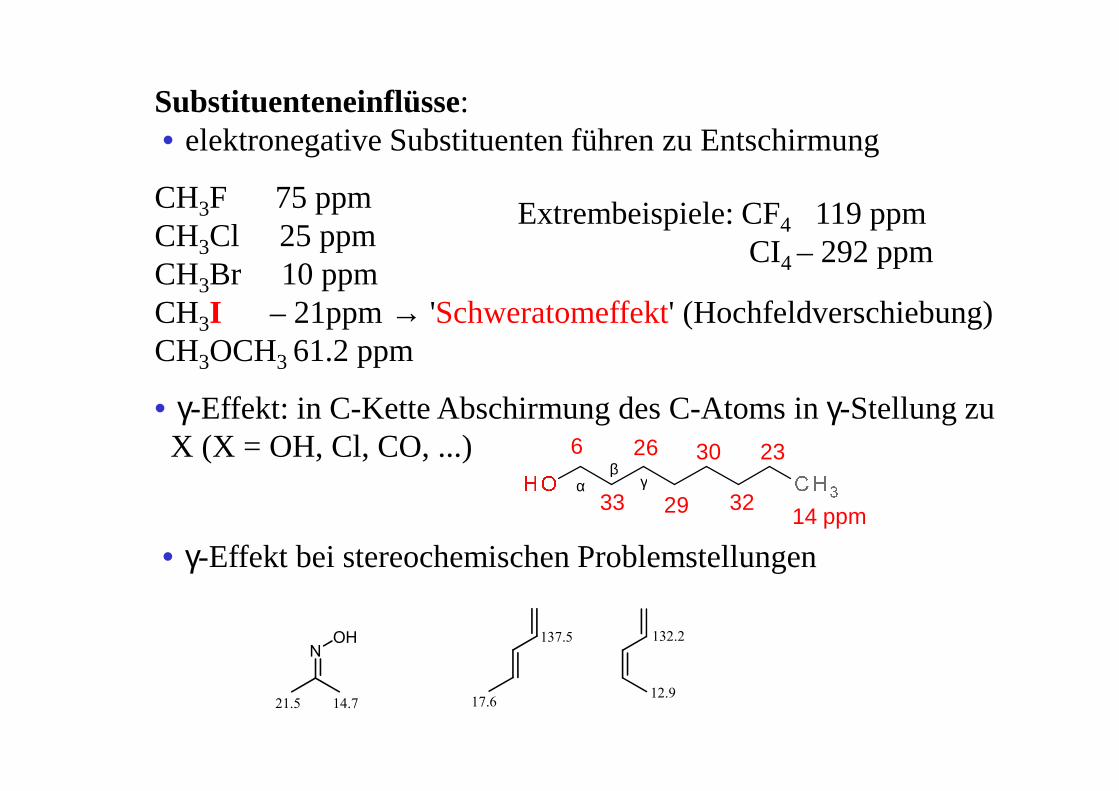
Substituenteneinflüsse: • elektronegative Substituenten führen zu Entschirmung
CH3F 75 ppmCH3Cl 25 ppmCH3Br 10 ppmCH3I – 21ppm → 'Schweratomeffekt' (Hochfeldverschiebung)CH3OCH3 61.2 ppm
• γ-Effekt: in C-Kette Abschirmung des C-Atoms in γ-Stellung zu X (X = OH, Cl, CO, ...)
Extrembeispiele: CF4 119 ppmCI4 – 292 ppm
6
33
26 30
29 32
23
14 ppm
αβ
γ
• γ-Effekt bei stereochemischen Problemstellungen
NOH
21.5 14.7 17.6
137.5
12.9
132.2
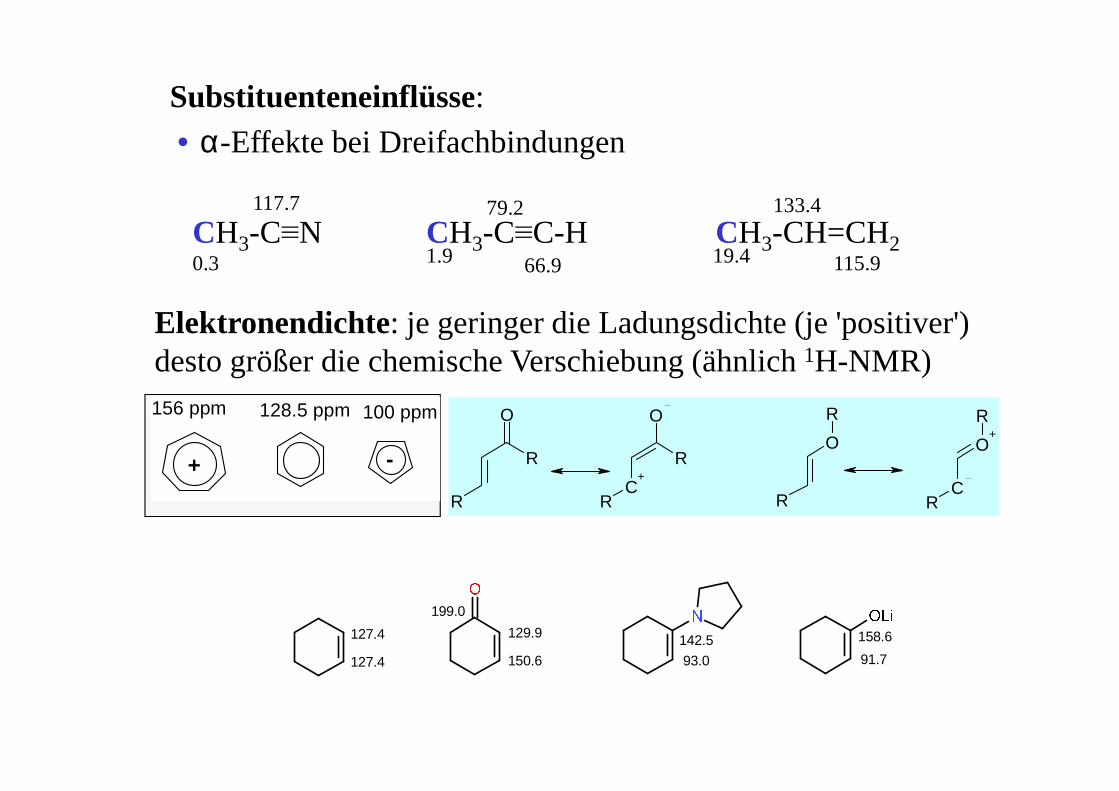
+ -
156 ppm 128.5 ppm 100 ppm
R
O
RC
+R
O
R
O
R
R
C
O+
R
R
Elektronendichte: je geringer die Ladungsdichte (je 'positiver') desto größer die chemische Verschiebung (ähnlich 1H-NMR)
Substituenteneinflüsse:• α-Effekte bei Dreifachbindungen
CH3-C≡N CH3-C≡C-H CH3-CH=CH20.3
117.7
1.9 19.4
133.4
115.9
79.2
66.9
127.4
127.4
199.0
129.9
150.6
142.5
93.0
158.6
91.7
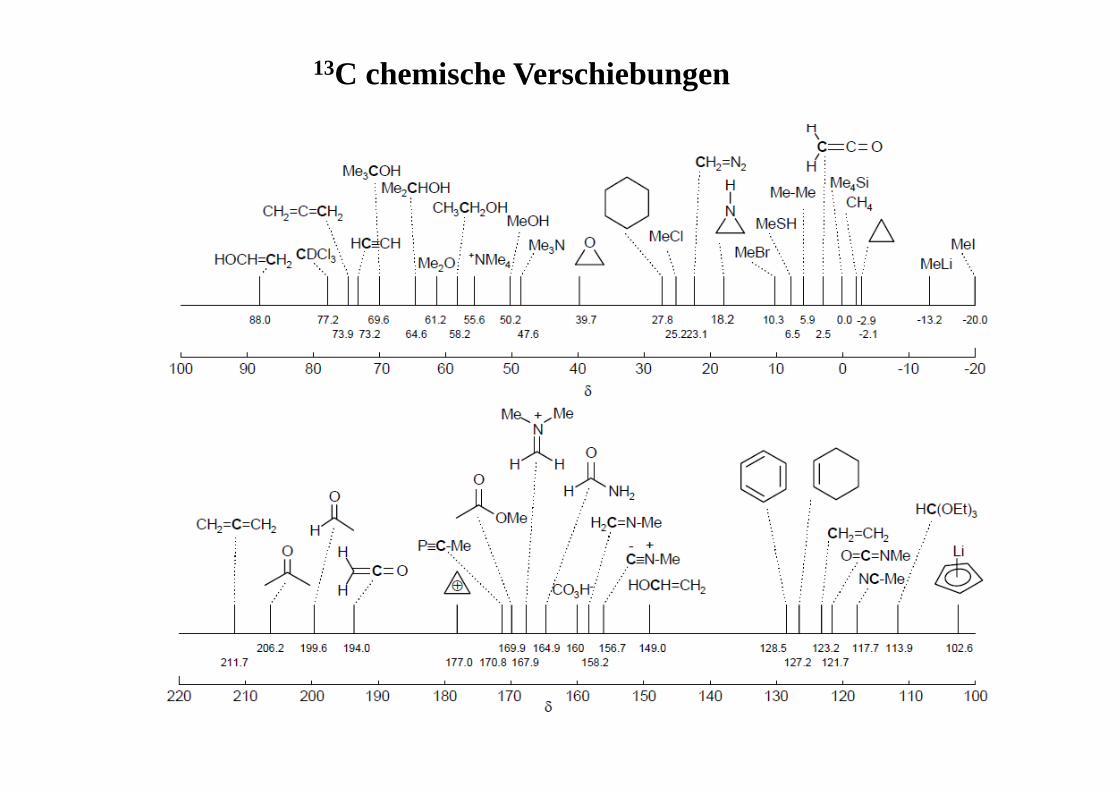
13C chemische Verschiebungen

Signalintensität: 13C-NMR Spektren unter üblichen Aufnahmebedingungen nicht integrierbar da sehr große Unterschiede im Relaxationsverhalten der einzelnen C-Atome; quartäre C-Atome meist geringere Signalintensität
13C,1H Spin-Spin Kopplung*
1J: direkte Kopplung 100 – 300 Hz, meist 120 – 200 abhängig von der Hybridisierung: sp > sp2 > sp3
(Ethan: 1J = 125 Hz, Ethen : 1J = 156 Hz, Ethin: 1J = 248 Hz)elektronegative Substituenten am C-Atom vergrößern 1J
2J, 3J, 4J ..... – 10 bis 60 Hz, meist 0 – 20 Hz (Absolutbetrag)
Nachteil 1H-gekoppelter Spektren: massive Signalüberlagerungen, zusätzlichVerschlechterung des Signal/Noise (S/N) durch Aufsplitten der Resonanzen insehr viele Linien (z.B. R-CH2-CH2-CH3 → 36 Linien!
⇒ Entkopplung von 1H !!
* Aufspaltungsregeln wie bei 1H
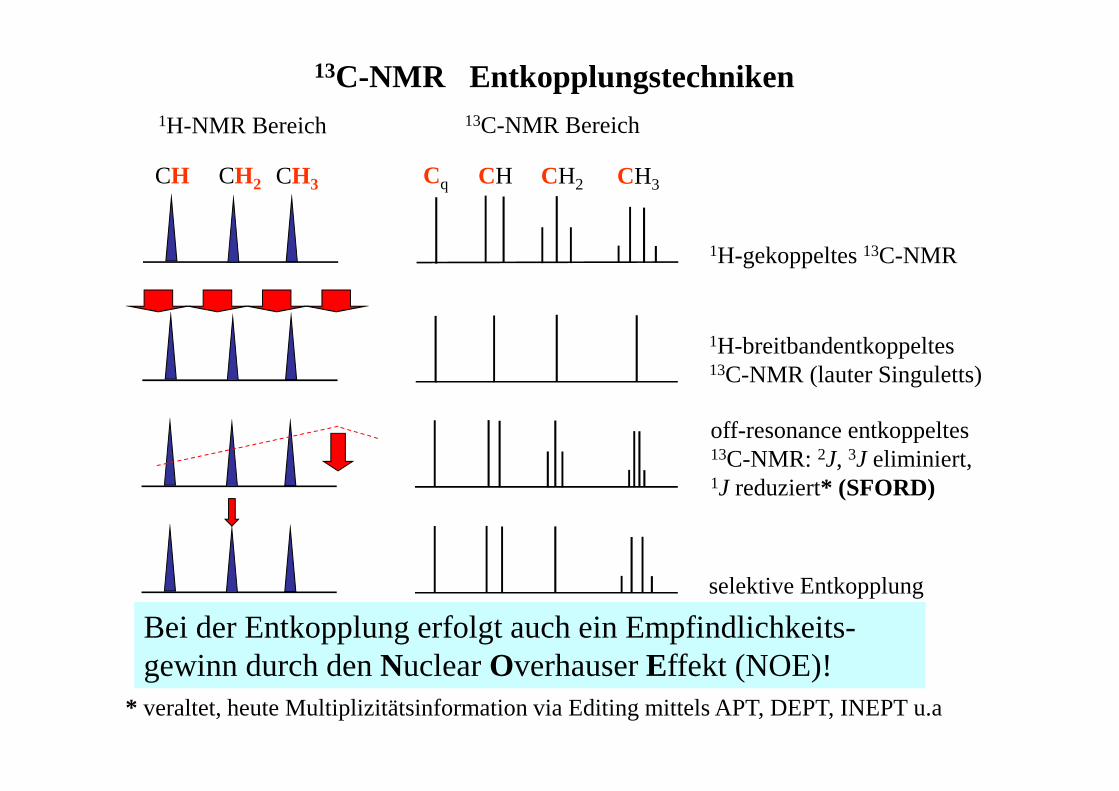
13C-NMR Entkopplungstechniken
Bei der Entkopplung erfolgt auch ein Empfindlichkeits-gewinn durch den Nuclear Overhauser Effekt (NOE)!
1H-gekoppeltes 13C-NMR
1H-NMR Bereich 13C-NMR Bereich
1H-breitbandentkoppeltes13C-NMR (lauter Singuletts)
off-resonance entkoppeltes 13C-NMR: 2J, 3J eliminiert, 1J reduziert* (SFORD)
selektive Entkopplung
Cq CH CH2 CH3CH CH2 CH3
* veraltet, heute Multiplizitätsinformation via Editing mittels APT, DEPT, INEPT u.a
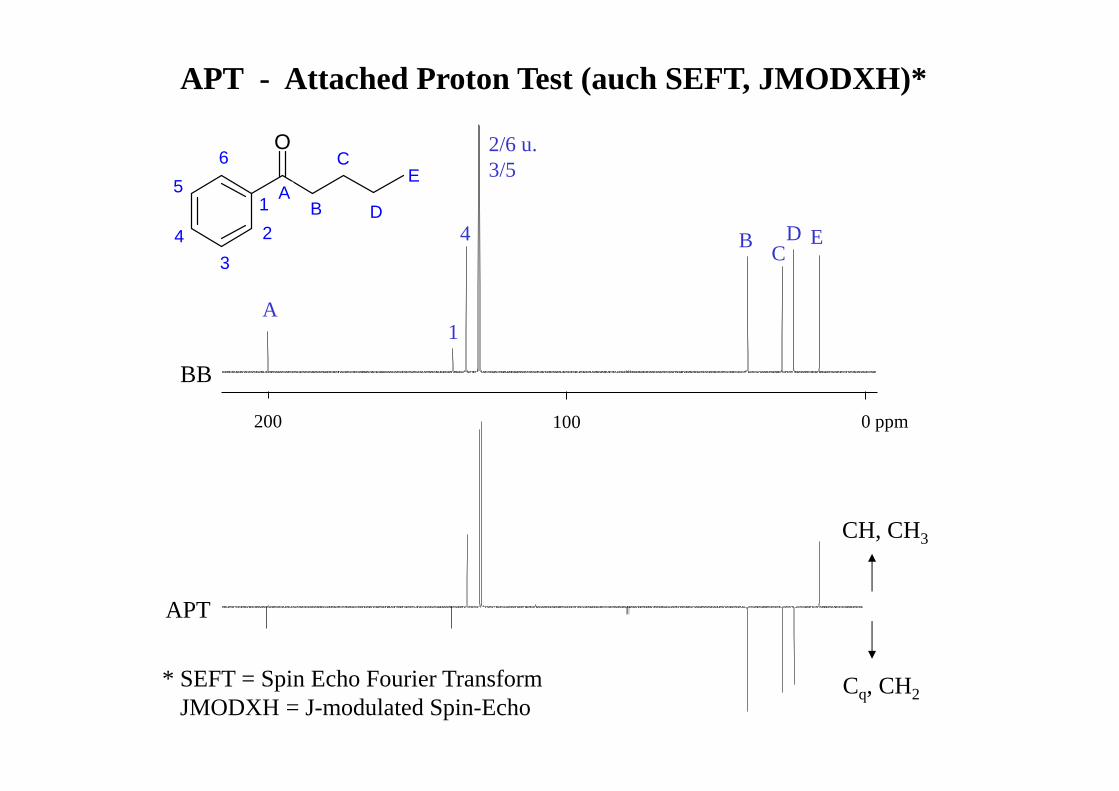
APT - Attached Proton Test (auch SEFT, JMODXH)*
BB
APT
CH, CH3
Cq, CH2
O
1
2
3
4
5
6
AB
CE
D
A
BC
D E
1
4
2/6 u. 3/5
* SEFT = Spin Echo Fourier TransformJMODXH = J-modulated Spin-Echo
200 100 0 ppm
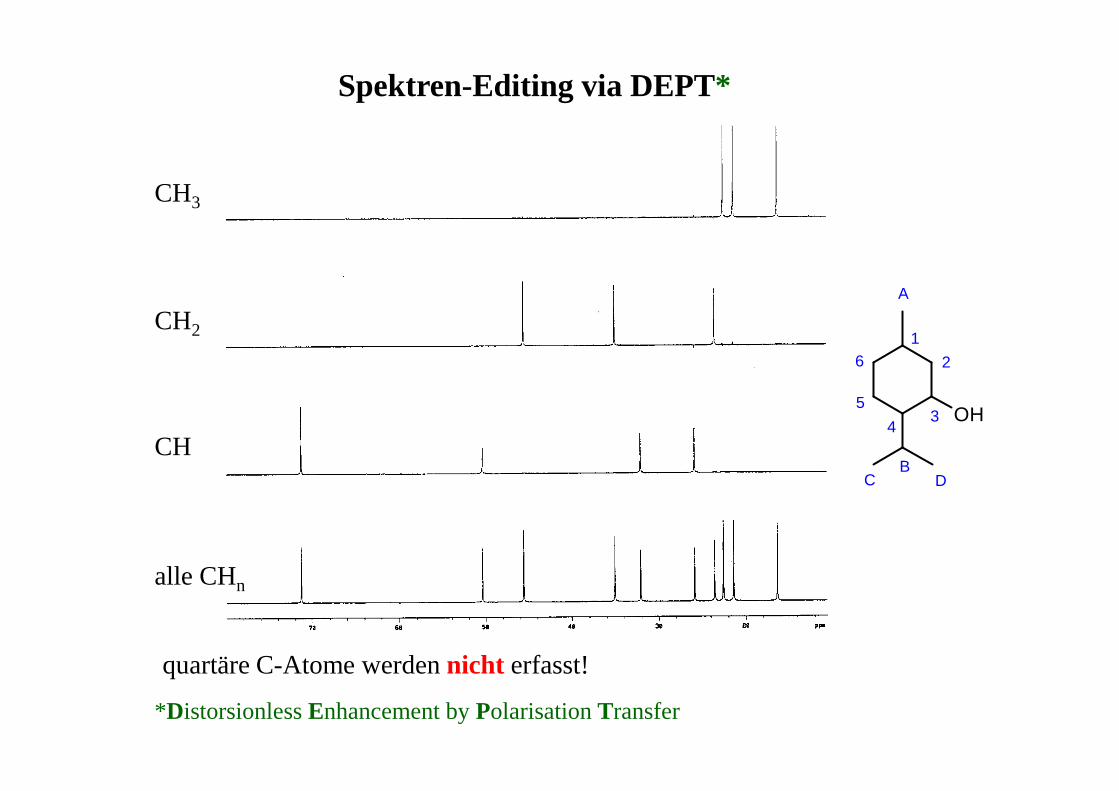
Spektren-Editing via DEPT*
*Distorsionless Enhancement by Polarisation Transfer
OH
1
2
34
5
6
A
BC D
CH
CH2
CH3
alle CHn
quartäre C-Atome werden nicht erfasst!
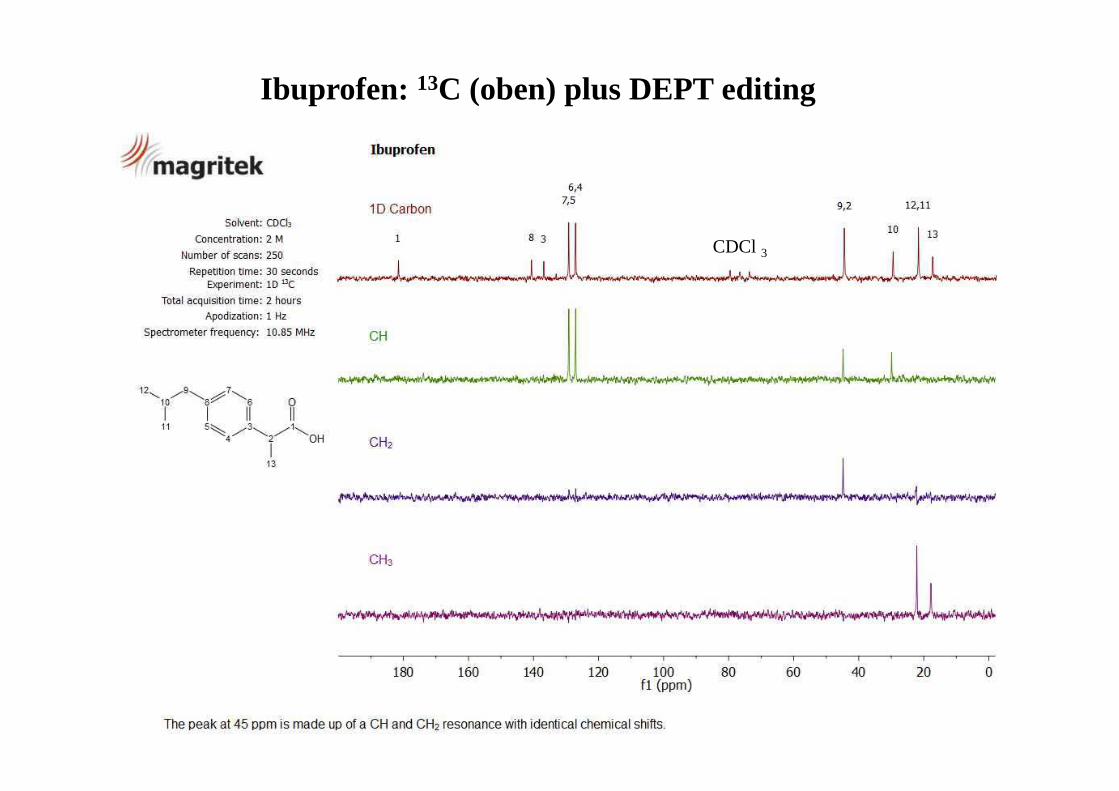
CDCl 3
Ibuprofen: 13C (oben) plus DEPT editing
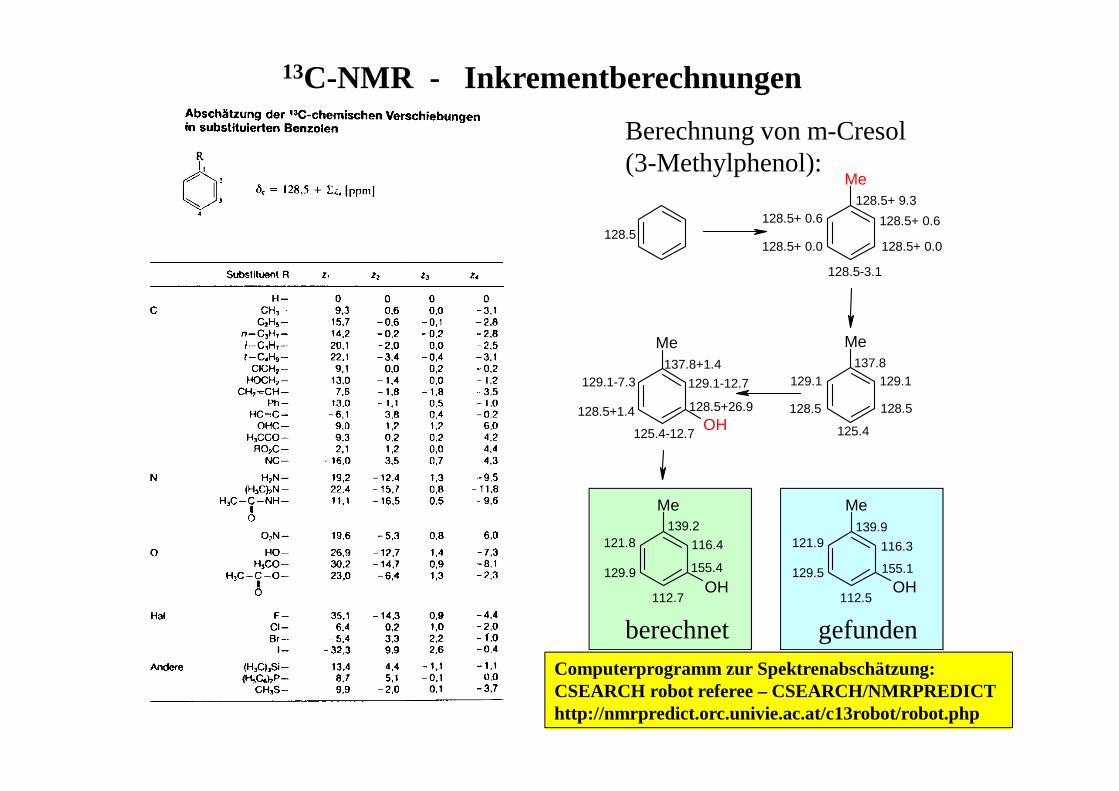
13C-NMR - Inkrementberechnungen
Me
MeMe
OH
Me
OH
Me
OH
128.5+ 9.3
128.5+ 0.6
128.5+ 0.0
128.5-3.1
128.5+ 0.6
128.5+ 0.0
137.8129.1
128.5
125.4
129.1
128.5
137.8+1.4129.1-7.3
128.5+26.9
125.4-12.7
129.1-12.7
128.5+1.4
139.2121.8
155.4
112.7
116.4
129.9
139.9121.9
155.1
112.5
116.3
129.5
128.5
Berechnung von m-Cresol (3-Methylphenol):
berechnet gefundenComputerprogramm zur Spektrenabschätzung:CSEARCH robot referee – CSEARCH/NMRPREDICThttp://nmrpredict.orc.univie.ac.at/c13robot/robot.php
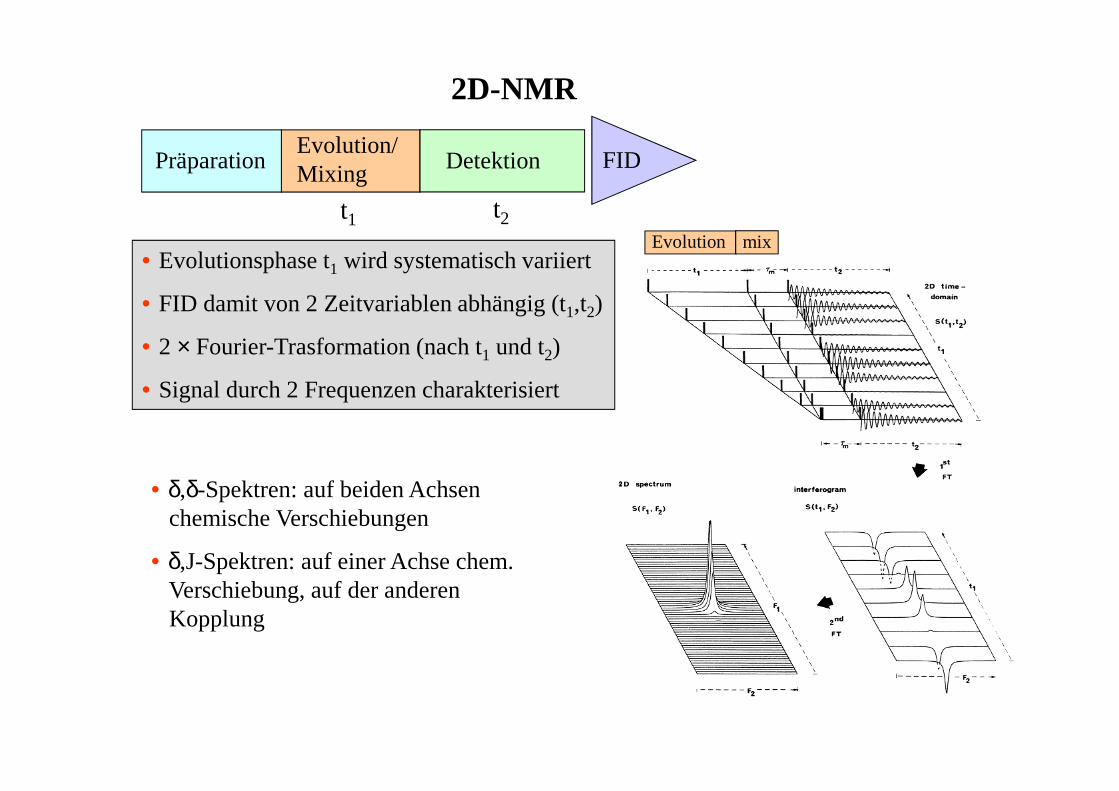
2D-NMR
PräparationEvolution/Mixing Detektion FID
• Evolutionsphase t1 wird systematisch variiert
• FID damit von 2 Zeitvariablen abhängig (t1,t2)
• 2 × Fourier-Trasformation (nach t1 und t2)
• Signal durch 2 Frequenzen charakterisiert
t1 t2
• δ,δ-Spektren: auf beiden Achsenchemische Verschiebungen
• δ,J-Spektren: auf einer Achse chem.Verschiebung, auf der anderenKopplung
Evolution mix
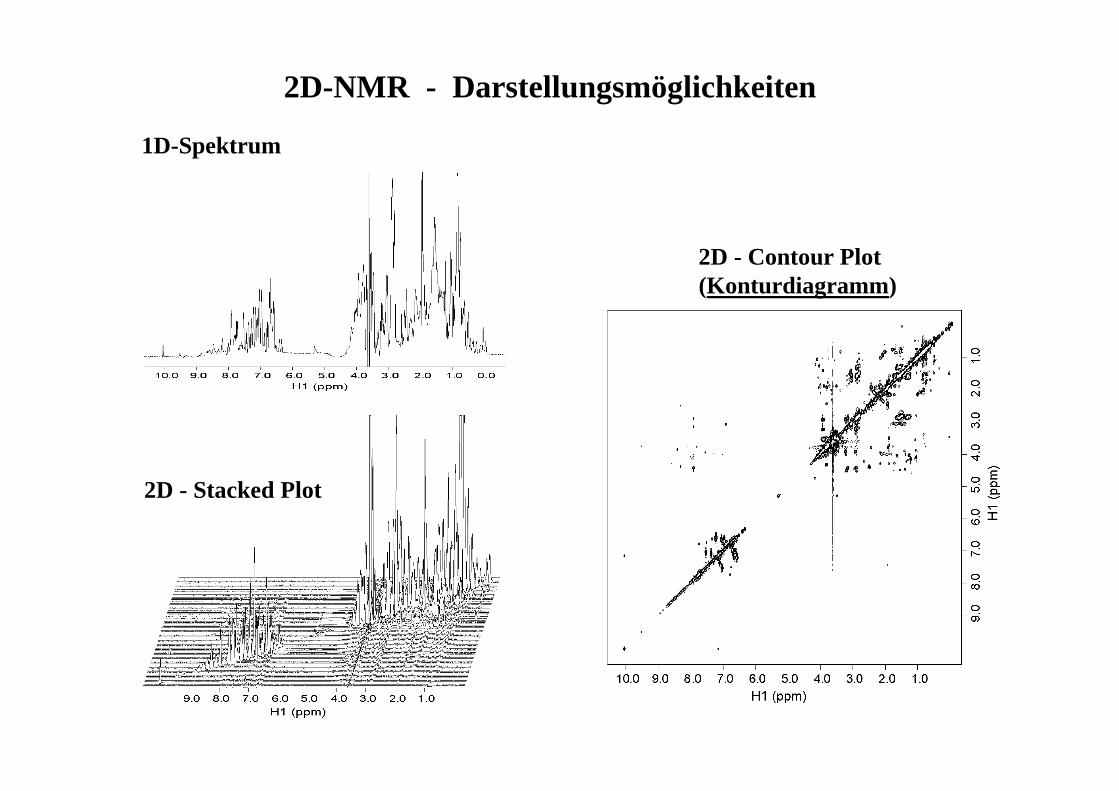
1D-Spektrum
2D - Contour Plot (Konturdiagramm)
2D-NMR - Darstellungsmöglichkeiten
2D - Stacked Plot
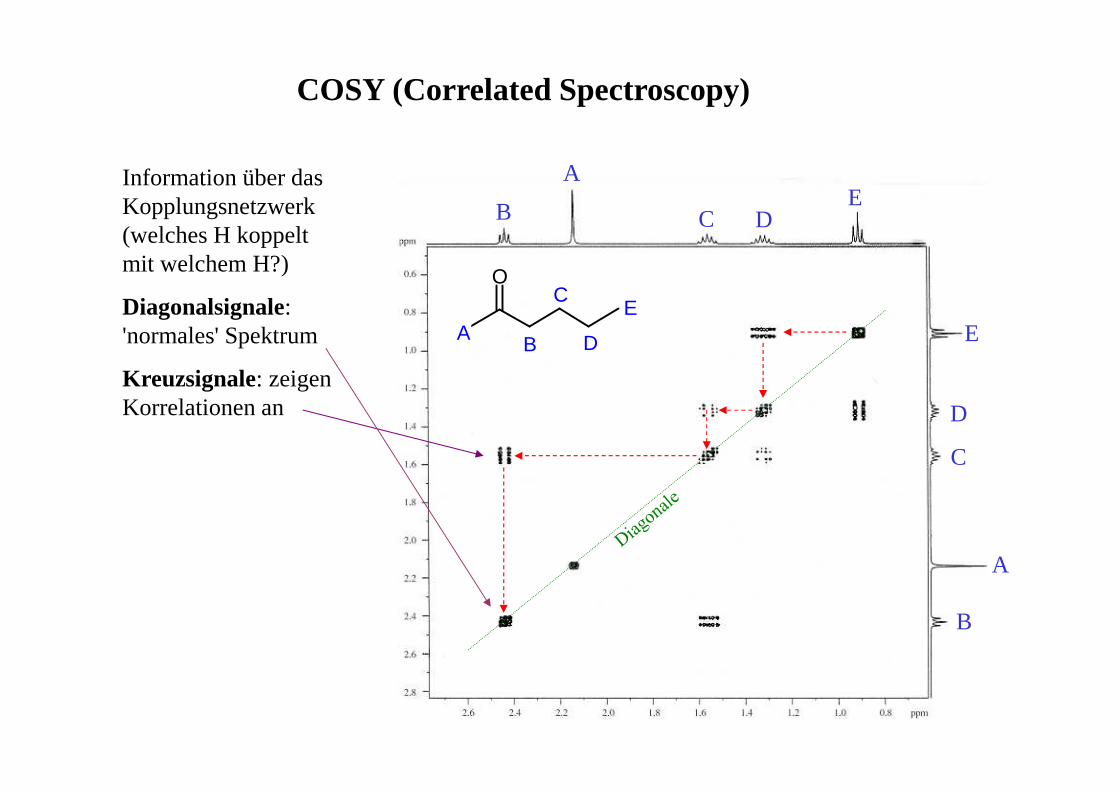
COSY (Correlated Spectroscopy)
Information über das Kopplungsnetzwerk (welches H koppelt mit welchem H?)
Diagonalsignale: 'normales' Spektrum
Kreuzsignale: zeigen Korrelationen an
O
A B
C
D
E
A
B
C
D
E
A
B
C
D
E
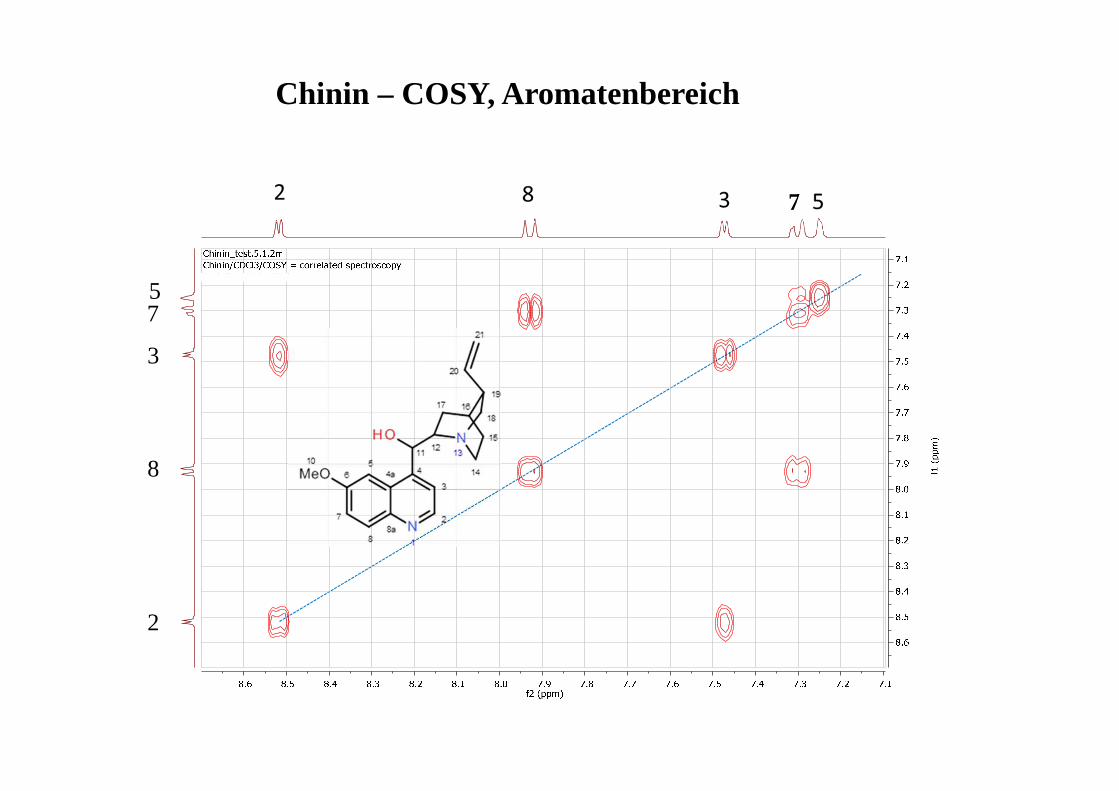
2
3
5
7
8
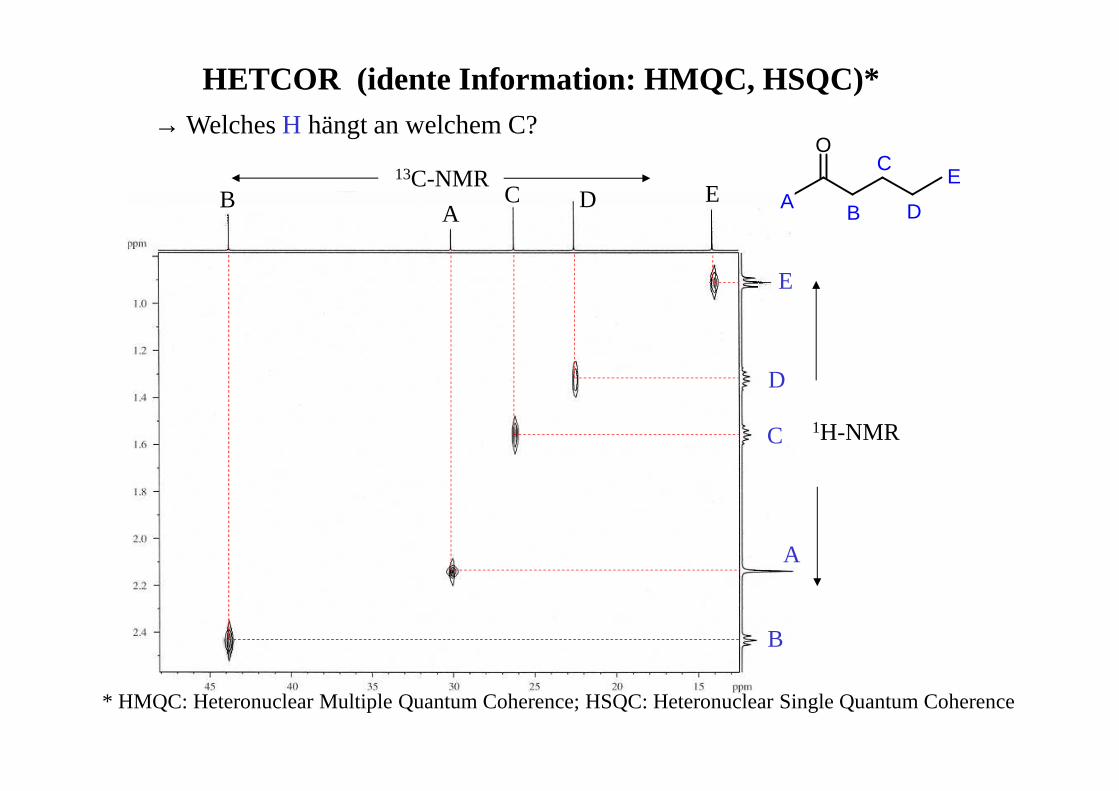
Chinin – COSY, Aromatenbereich
7
HETCOR (idente Information: HMQC, HSQC)*→ Welches H hängt an welchem C?
13C-NMR
1H-NMR
O
A B
C
D
E
A
E
B
C
D
AB C D E
* HMQC: Heteronuclear Multiple Quantum Coherence; HSQC: Heteronuclear Single Quantum Coherence
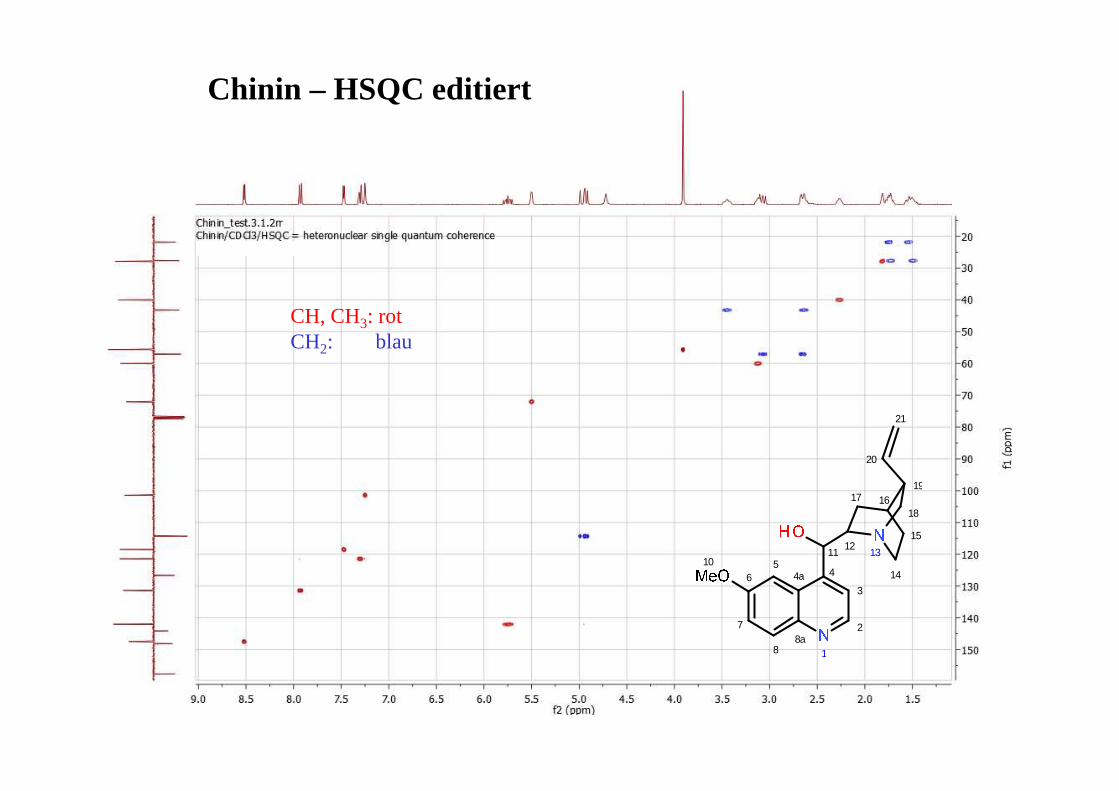
1013
14
1617
18
19
2
3
45
6
7
88a
1
4a
1112
21
20
15
Chinin – HSQC editiert
CH, CH3: rotCH2: blau
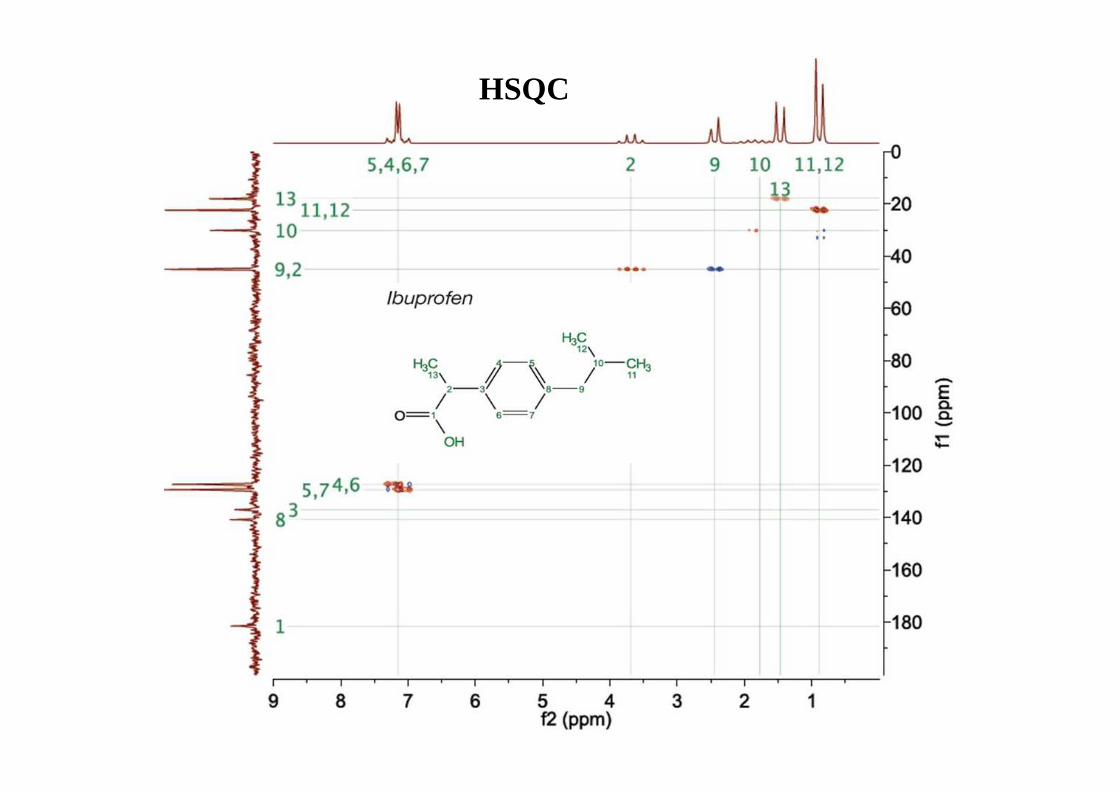
HSQC
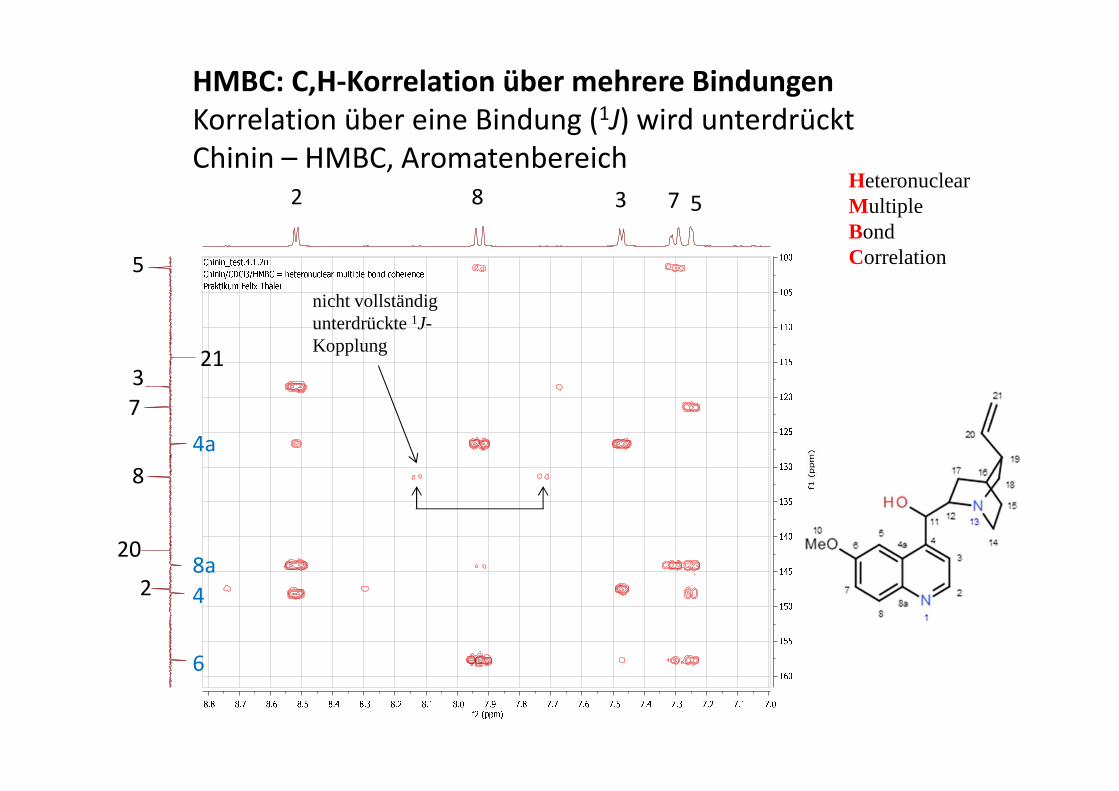
HMBC: C,H-Korrelation über mehrere Bindungen
Korrelation über eine Bindung (1J) wird unterdrückt
Chinin – HMBC, Aromatenbereich
2 8 3 7 5
2
8
3
7
5
208a
6
4a
4
21
nicht vollständig unterdrückte 1J-Kopplung
HeteronuclearMultipleBondCorrelation
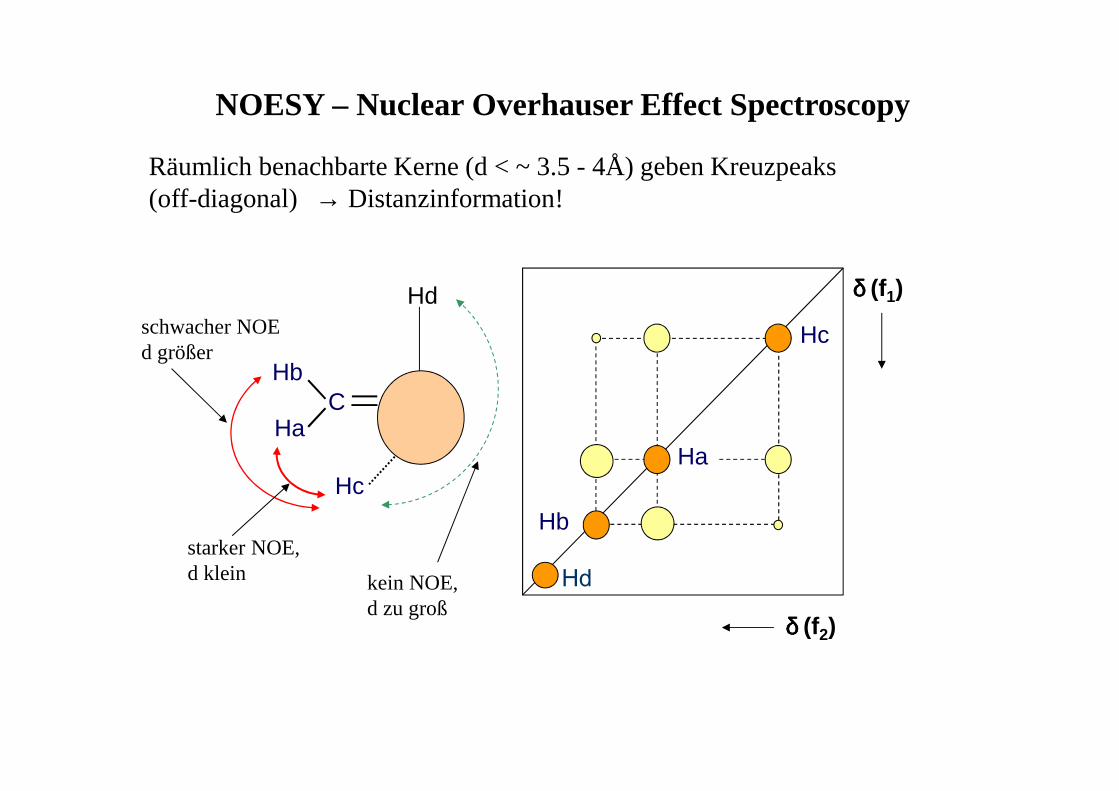
NOESY – Nuclear Overhauser Effect Spectroscopy
Hb
Ha
Hc
C
Hb
Ha
Hc
δ δ δ δ (f2)
δ δ δ δ (f1)
Räumlich benachbarte Kerne (d < ~ 3.5 - 4Å) geben Kreuzpeaks (off-diagonal) → Distanzinformation!
starker NOE,d klein
schwacher NOEd größer
Hd
kein NOE,d zu groß
Hd
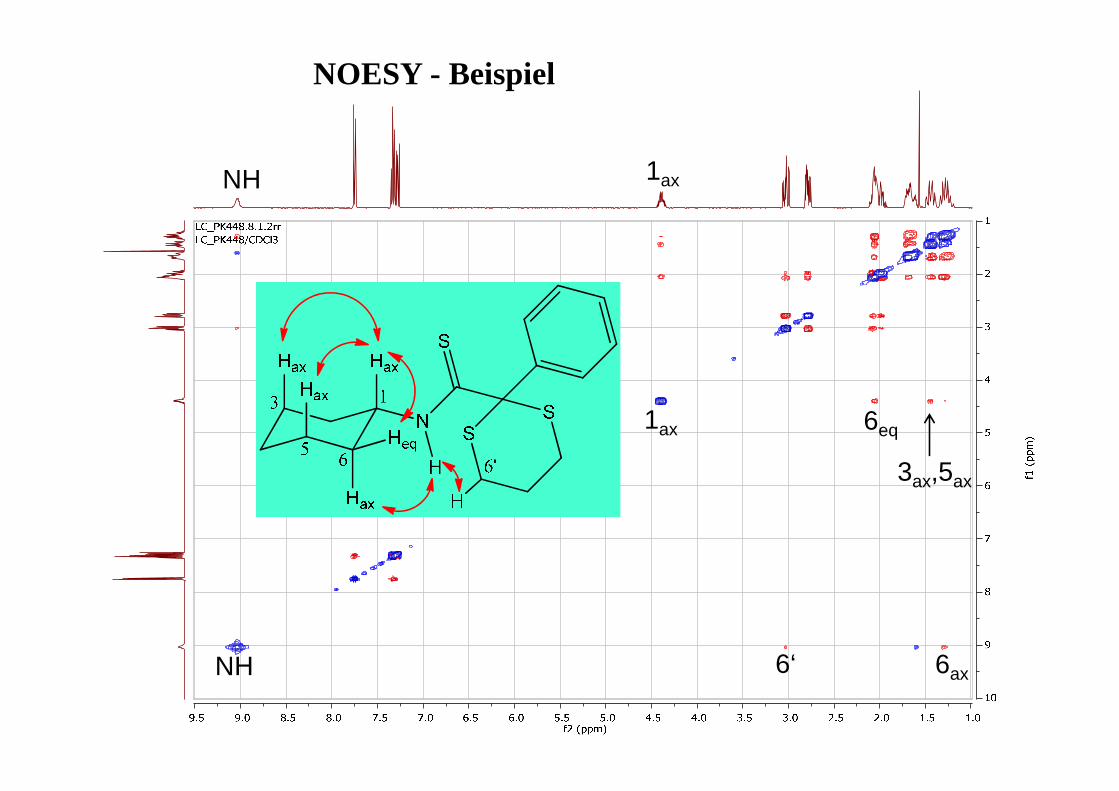
NH
NH 6‘ 6ax
1ax
3ax,5ax
6eq
NOESY - Beispiel
1ax
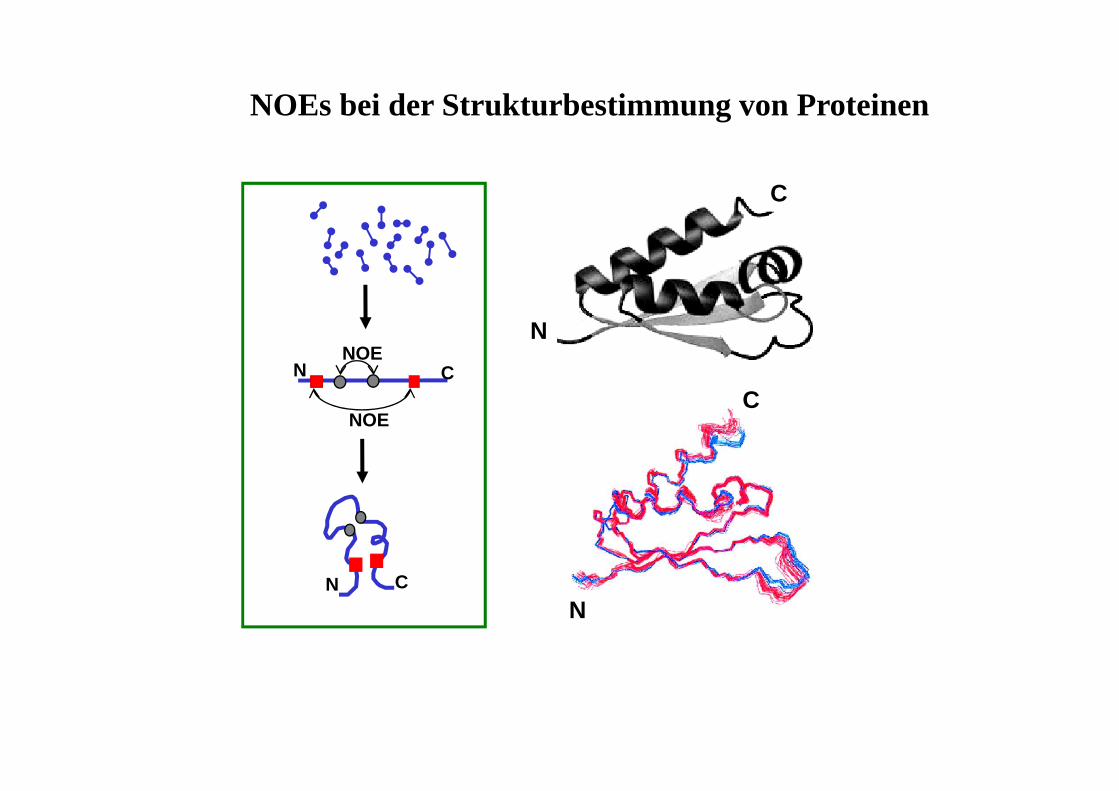
N C
N C
NOE
NOE
C
N
C
N
NOEs bei der Strukturbestimmung von Proteinen
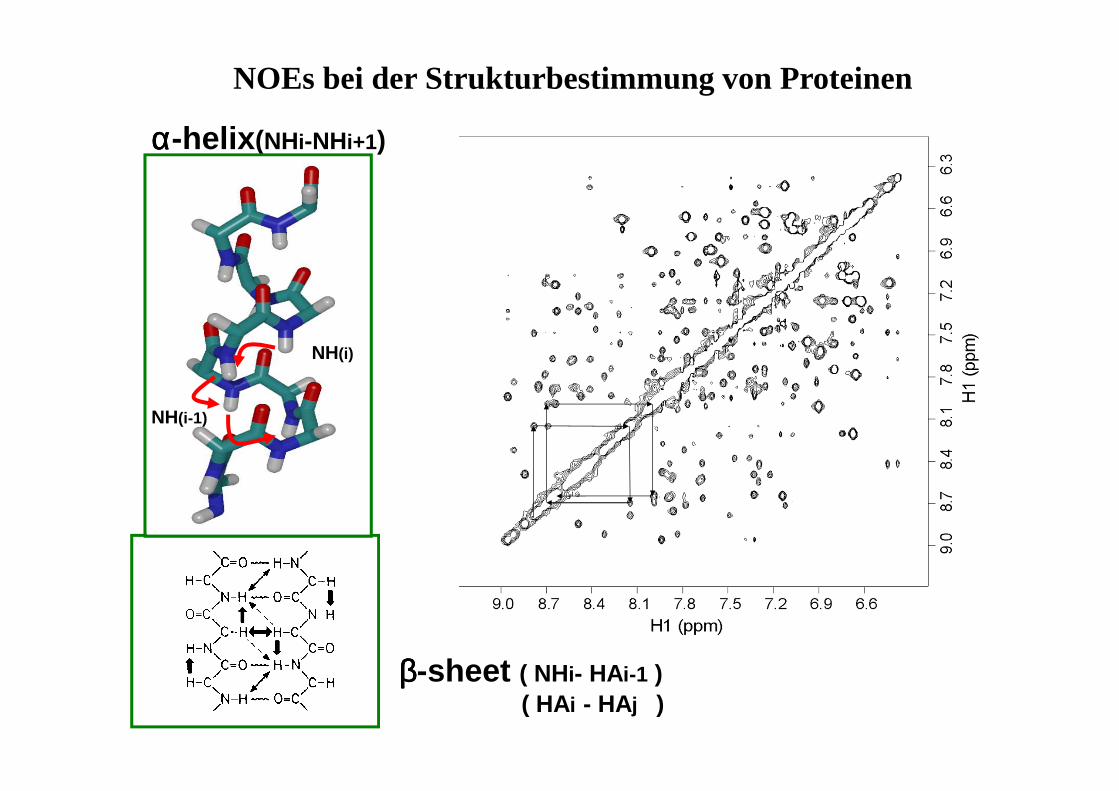
NOEs bei der Strukturbestimmung von Proteinen
αααα-helix (NHi-NHi+1)
ββββ-sheet ( NHi- HAi-1 )( HAi - HAj )
NH(i)
NH(i-1)

Praktische Durchführung von Messungen
• Probe wird in deuteriertem Lösungsmittel gelöst; häufig verwendet: CDCl3, d6-DMSO, d6-Aceton, d6-Benzol, D2O
• Lösung wird in zylindrisches Röhrchen (NMR-tube, Durchmesser meist 5 mm) gefüllt und dieses mit Teflonkappe verschlossen
• Röhrchen wird mit Spinnerturbine versehen (richtige Position wird mit Schublehre eingestellt – siehe Bild)
• Röhrchen wird in der Bohrung des Magneten (oder im Autosampler) platziert und mittels Pressluftstrom in den Probenkopf befördert
• ‚Lock‘ (Konstanthalten des Feld/Frequenz-Verhältnisses via Deuterium-Signal)• ‚Shim‘ (Feinhomogenisierung des Magnetfeldes via Shimspulen)• Abstimmung der Receiver-Empfindlichkeit• Messung durch Acquisition und Addition mehrerer Pulse (scans)→ FID
• Prozessieren (FT, Phasenkorrektur) meist dezentral

Resümee NMR - wichtigste Anwendungen in der pharm. Analytik/organ. Chemie
• Analytik in der Synthese: Kontrolle präparativen Arbeitens(Konstitution, Einheitlichkeit, Verunreinigungen)
• Strukturbestimmung in Lösung: Konstitution und Stereochemie, vonSyntheseprodukten, Naturstoffen, Biomolekülen; auch für größereMoleküle (z.B. Proteine zur Zeit bis ca. 30 kD ~ 250 Aminosäuren, allerdings erst nach Isotopenanreicherung)
• Untersuchung intermolekularer Wechselwirkungen(Enzym-Inhibitor, DNA-Interkalator oder Repressor, Rezeptor-Substrat, Rezeptor-Arzneistoff, Antikörper-Antigen usw.)
• Untersuchung der molekularen Dynamik(via Relaxationsparameter), z.B. Proteinfaltung, Proteinfunktion
• Bestimmung von Diffusionsparametern

DANKE!

Prüfungsmodalitäten für ‚Instrumentelle pharmazeutische Analytik‘
• Die Prüfung erfolgt schriftlich (ausnahmslos!)• 10 Fragen (darunter können unter anderem auch Rechenbeispiele,
multiple-choice Fragen, Skizzen, usw. sein)• Zur Beantwortung der Fragen steht genau 1 h Zeit zur Verfügung• Die Fragen müssen mit einem nicht-radierbaren Permanent-
Schreibgerät (Kugelschreiber, Faserschreiber, Füllfeder, NICHT mit Bleistift) beantwortet werden
• Die Verwendung von Taschenrechnern, Mobiltelephonen und anderen Hilfsmitteln ist nicht gestattet. Es wird empfohlen, ein Lineal zur Prüfung mitzubringen
• Für jede richtig beantwortete Frage wird 1 Punkt vergeben, dafür muss die Frage vollständig richtig beantwortet werden (es werden keine halben Punkte vergeben)
• Notenschlüssel: 10P = 1, 9P = 2, 8P = 3, 7P = 3, 6P = 4, ≤ 5P = 5 • Laut einer Richtlinie der Studienprogrammleitung werden komm.
Prüfung gleichzeitig mit den ‚normalen‘ Prüfungen unter den selben Rahmenbedingungen und mit den selben Fragen durchgeführt





























