Embed Size (px)
Citation preview

ISBN 978-3-86345-286-5
Verlag: Deutsche Veterinärmedizinische Gesellschaft Service GmbH35392 Gießen · Friedrichstraße 17 · Tel. 0641 / 24466 · Fax: 0641 / 25375
E-Mail: [email protected] · Internet: www.dvg.de
Mu
ham
mad
Nad
eem
Han
no
ver
2015



Bibliografische Informationen der Deutschen Bibliothek
Die Deutsche Bibliothek verzeichnet diese Publikation in der
Deutschen Nationalbibliografie;
Detaillierte bibliografische Daten sind im Internet über http://dnb.ddb.de abrufbar.
1. Auflage 2015
© 2015 by Verlag: Deutsche Veterinärmedizinische Gesellschaft Service GmbH,
Gießen
Printed in Germany
ISBN 978-3-86345-286-5
Verlag: DVG Service GmbH
Friedrichstraße 17
35392 Gießen
0641/24466
www.dvg.de

University of Veterinary Medicine Hannover
Department of Pathology
Susceptibility of goats to the BSE agent with special emphasis on the neuropathogenesis
THESIS
Submitted in partial fulfillment of the requirements for the degree
DOCTOR OF PHILOSOPHY
(PhD)
Awarded by the University of Veterinary Medicine Hannover
By
Muhammad Nadeem
Born in Faisalabad, Punjab/Pakistan
Hannover, Germany 2015

Supervisor: Prof. Dr. Wolfgang Baumgärtner
Supervision Group: Prof. Dr. Wolfgang Baumgärtner
Prof. Dr. Kirsten Haastert-Talini
Prof. Dr. Gerd Bicker
1st Evaluation: Prof. Dr. Wolfgang Baumgärtner
Department of Pathology, University of Veterinary Medicine
Hannover, Germany
Prof. Dr. Kirsten Haastert-Talini
Institute of Neuroanatomy, Medical School Hannover, Germany
Prof. Dr. Gerd Bicker
Institute of Animal ecology and cell biology, University of
Veterinary Medicine Hannover, Germany
2nd
Evaluation: Prof. A. Gröne
Department of Pathobiology, University of Utrecht, The
Netherlands
Date of final exam: 6th
November 2015
Muhammad Nadeem has received financial support from Higher education commission
Islamabad, Pakistan and DAAD Germany

To my family

Parts of the thesis have been published in peer-reviewed
journals previously:
AGUILAR-CALVO, P., FAST, C., TAUSCHER, K., ESPINOSA, J.C., GROSCHUP, M.H.,
NADEEM, M., GOLDMANN, W., LANGEVELD, J., BOSSERS, A., ANDREOLETTI, O.,
and TORRES, J.M. (2015):
Effect of Q211 and K222 PRNP Polymorphic Variants in the Susceptibility of Goats to Oral
Infection with Goat Bovine Spongiform Encephalopathy.
J Infect Dis. DOI: 10.1093/infdis/jiv112.
Parts of the thesis have been presented as poster at
congresses:
MUHAMMAD NADEEM, VERENA HAIST, CHRISTINE FAST, MARTIN H.
GROSCHUP, WOLFGANG BAUMGÄRTNER:
Ultrastructural pathology of the peripheral nervous system in caprine prion diseases. 2nd
International Workshop of Veterinary Neuroscience, Hannover, Germany, March 20-22,
2014
MUHAMMAD NADEEM, VERENA HAIST, CHRISTINE FAST, MARTIN H.
GROSCHUP, WOLFGANG BAUMGÄRTNER:
Ultrastructural pathology of the peripheral nervous system in caprine prion diseases. 2nd
N-
RENNT Symposium on Neuroinfectiology, Hannover, Germany, February 16-17, 2015.

Contents I
CHAPTER 1 INTRODUCTION OF TRANSMISSIBLE SPONGIFORM
ENCEPHALOPATHIES (TSEs) ............................................................................... 2
1.1. DEFINITION .............................................................................................................. 2
1.2. HISTORY .................................................................................................................... 2
1.3. CAUSATIVE AGENT ............................................................................................... 5
1.3.1. THE PRION THEORY ................................................................................ 5
1.3.2. NOMENCLATURE ...................................................................................... 6
1.3.3. PrPSc
FORMATION ...................................................................................... 7
1.3.4. PATHOGENESIS OF TSEs ......................................................................... 9
1.3.5. PATHOLOGICAL CHARACTERISTICS OF TSEs .............................. 11
1.4. CLINICAL MANIFESTATIONS............................................................................ 12
1.5. RODENT MODELS OF PRION DISEASES AND BRAIN PATHOLOGY IN
HAMSTERS ............................................................................................................. 12
1.5.1. CONVENTIONAL RODENT MODELS .................................................. 12
1.5.2. TRANSGENIC RODENTS MODELS ...................................................... 14
1.5.2 .1. PrP KNOCK-OUT MODELS ....................................................... 17
1.5.3. BRAIN PATHOLOGY IN HAMSTERS .................................................. 17
1.5.3 .1. STRAIN VARIATIONS ................................................................. 18
1.6. SYNAPTIC PATHOLOGY IN NEURODEGENERATIVE DISEASES ............ 20
1.6.1. MECHANISMS OF SYNAPTIC DYSFUNCTION ................................. 21
1.6.1.1. ROLE OF MITOCHONDRIA IN SYNAPTIC
DEGENERATION ......................................................................... 22
1.7. AIMS OF THE STUDY ............................................................................................ 24
CHAPTER 2 EFFECT of Q211 and K222 PRNP POLYMORPHIC VARIANTS IN THE
SUSCEPTIBILTY OF GOATS TO ORAL INFECTION WITH GOAT
BOVINE SPONGIFORM ENCEPHALOPATHY................................................. 25
CHAPTER 3 BSE INFECTION OF GOATS ALTERS NEUROFILAMENT
PHOSPHORYLATION STATUS OF SPINAL CORD AXONS ......................... 29
CHAPTER 4 DISCUSSION ............................................................................................................ 43
CHAPTER 5 SUMMARY ................................................................................................................ 51
CHAPTER 6 ZUSAMMENFASSUNG ........................................................................................... 55
CHAPTER 7 REFERENCES .......................................................................................................... 61
CHAPTER 8 ACKNOWLEDGEMENTS ...................................................................................... 89

II List of tables and figures
Table 1 TYPICAL FEATURES OF TSEs INCLUDING YEAR OF FIRST
DESCRIPTION, ROUTE OF TRANSMISSION, CLINICAL SIGNS AND
OTHER FEATURES .................................................................................................. 3
Table 2 NOMENCLATURE OF DIFFERENT TRANSMISSIBLE
SPONGIFORM ENCEPHALOPATHIES (TSES) ................................................. 6
Table 3 ORIGIN AND DIVERSITY OF EXPERIMENTAL TSE STRAINS
IN RODENTS ........................................................................................................... 13
Table 4 MOUSE MODELS TO STUDY PRION DISEASES MECHANISMS ............... 16
Fig. 1 FUNCTIONS OF NORMAL CELLULAR PRPC ................................................... 7
Fig. 2 CONVERSION OF NORMAL PRPC INTO ABNORMAL MISFOLDED
PRPSC
........................................................................................................................... 9

Abbreviation list III
Abbreviation list
TSEs = transmissible spongiform encephalopathies
BSE = bovine spongiform encephalopathy
CWD = chronic wasting disease
TME = transmissible mink encephalopathy
FSE = feline spongiform encephalopathy
CJD = Creutzfeldt-Jakob disease
sCJD = sporadic Creutzfeldt-Jakob disease
vCJD = variant Creutzfeldt-Jakob disease
Prion = proteinaceous infectious particle
PrPC
= normal cellular prion protein (C for cellular)
PrPSc
= abnormal prion protein (Sc for Scrapie)
Ov = ovine
Mk = mink
MDe = mule and deer
Bov = bovine
Fe = feline
Nya = nyala
Hu = human
PRNP = prion protein gene in human
GPI = glycosylphosphatidylinositol
CNS = central nervous system
PK = proteinase kinase
LRS = lymphoreticular system
SNS = sympathetic nervous system
ENS = enteric nervous system
GMCG = ganglion mesenterium craniale/ganglion coeliacum complex
DMNV = dorsal motor nucleus of the vagus
SHa = Syrian hamsters
SSBP = sheep scrapie brain pool
Tg = transgenic
WT = wild type
HY = hyper
DY = drowsy
SNAP25 = synaptosomal-associated protein of 25kDa
DPI = days post infection
PSD = post synaptic density
NMDA-R = N-methyl-D-aspartate-receptor
AMPA-R = α-amino-3-hydroxy-5-methyl-isoxazoleproprionic acid receptor
mGluR = metabotropic glutamate receptor
CSP = synaptophysin cysteine string protein
SNARE = soluble NSF attachment protein receptor
COX = cytochrome c oxidase
β-APP = beta amyloid precursor protein
CNPase = 2',3'-cyclic-nucleotide 3'-phosphodiesterase
Iba 1 = ionized calcium-binding adapter molecule
MAP 2 = microtubule associated protein
MHC Class II = major histocompatibility complex
P75NTR
= low affinity neurotrophin receptor p75

IV Abbreviation list
pNF = phosphorylated neurofilament
nNF = non-phosphorylated neurofilament
Tau 1 = tau protein

Chapter 1 1
Chapter 1
Introduction

2 Chapter 1
CHAPTER 1
1. Introduction of transmissible spongiform encephalopathies (TSEs)
1.1. Definition
Transmissible spongiform encephalopathies (TSEs) are also known as prion diseases. This is
a group of progressive conditions/syndromes that affects the nervous system of humans and
various animal species. They include bovine spongiform encephalopathy (BSE), scrapie,
chronic wasting disease (CWD), transmissible mink encephalopathy (TME), feline
spongiform encephalopathy (FSE), and others (CHESEBRO, 2003; LIBERSKI, 2012; LEE et
al., 2013). In humans, among others, Creutzfeldt-Jakob disease (CJD) with a sporadic (sCJD),
a variant (vCJD, the term new variant CJD is also used, here we will use the term vCJD), an
iatrogenic and a familial subtype exists (HAÏK and BRANDEL, 2014). The characteristic
features of TSEs comprise a long incubation period, multifocal spongiform changes,
astrogliosis, neuronal loss, and absence of inflammatory reaction in brain tissue
(CHESEBRO, 2003; IULINI et al., 2012).
The cause of TSEs is still under debate but it has been widely accepted that a transformed
host protein called prion (proteinaceous infectious particle), a novel type of infectious agent,
represents the etiology of these progressive diseases (PRUSINER, 1982).
1.2. History
As far as history about TSE diseases is concerned, scrapie was the first naturally acquired
prion disease known to the globe up to the 18th century. Scrapie transmission to healthy
sheep and goats from an affected sheep was reported during the 20th century (CUILLE and
CHELLE, 1936; CUILLE and CHELLE, 1939; CHELLE, 1942; DETWILER, 1992).
Transmissible mink encephalopathy (closely resembles L-type bovine spongiform
encephalopathy) as a food-borne disease of mink was first reported in the USA (MARSH and
HADLOW, 1992) and was later on detected in several other parts of the world
(SIGURDSON and MILLER, 2003). Kuru in humans was for the first time reported in 1957
and infection occurred most likely due to ritualistic cannibalism (ingestion of brain tissue) in
Papua New Guinea (GAJDUSEK and ZIGAS, 1957). The first case of feline spongiform
encephalopathy (FSE) was reported in a domestic cat in early 1990s (WYATT et al., 1990;
Chapter 1 3
BENCSIK et al., 2009), Chronic wasting disease (CWD) was firstly reported in 1967 as a
clinical syndrome of unknown etiopathogenesis in a restrained mule deer, originating from a
free-ranging population in Colorado, USA. A similar syndrome was identified in Wyoming
(USA) in 1978, and a spongiform encephalopathy was observed in the same year in affected
animals from several wildlife facilities in Colorado (WILLIAMS and YOUNG, 1980).
Bovine spongiform encephalopathy (BSE) was first reported in the UK in 1986 in cattle.
Recycled meat and bone meal from sheep with scrapie may be the cause for BSE in cattle.
Cattle being fed the remains of other cattle in form of meat and bone meal caused the spread
of the infectious agent (WELLS et al., 1987; WILESMITH et al., 1988). As a result a large
epidemic occurred that affected more than 182,000 cattle in 13 European countries, Canada,
the USA and Japan (GAVIER-WIDEN et al., 2005). Due to the implementation of rapid
screening tests and active surveillance (ANONYMOUS, 2001), BSE was also identified in
those European countries that were previously supposed to be BSE disease free countries.
Recently, outbreaks of TSE have been reported in small ruminants in a wide range of
countries, with the exceptions of Australia and New Zealand (GAVIER-WIDEN et al., 2005).
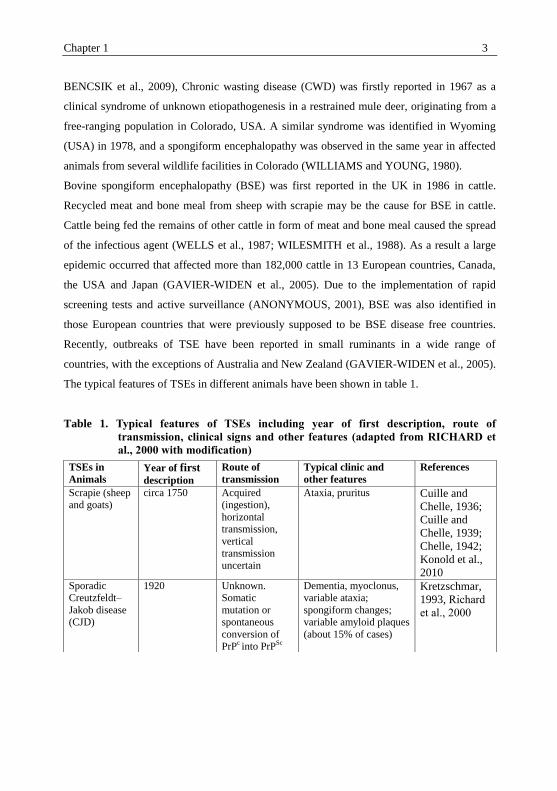
The typical features of TSEs in different animals have been shown in table 1.
Table 1. Typical features of TSEs including year of first description, route of
transmission, clinical signs and other features (adapted from RICHARD et
al., 2000 with modification)
TSEs in
Animals Year of first description
Route of
transmission
Typical clinic and
other features
References
Scrapie (sheep
and goats)
circa 1750 Acquired
(ingestion),
horizontal
transmission,
vertical
transmission
uncertain
Ataxia, pruritus Cuille and
Chelle, 1936;
Cuille and
Chelle, 1939;
Chelle, 1942;
Konold et al.,
2010
Sporadic
Creutzfeldt–
Jakob disease
(CJD)
1920 Unknown.
Somatic
mutation or
spontaneous
conversion of
PrPc into PrP
Sc
Dementia, myoclonus,
variable ataxia;
spongiform changes;
variable amyloid plaques
(about 15% of cases)
Kretzschmar,
1993, Richard
et al., 2000
4 Chapter 1
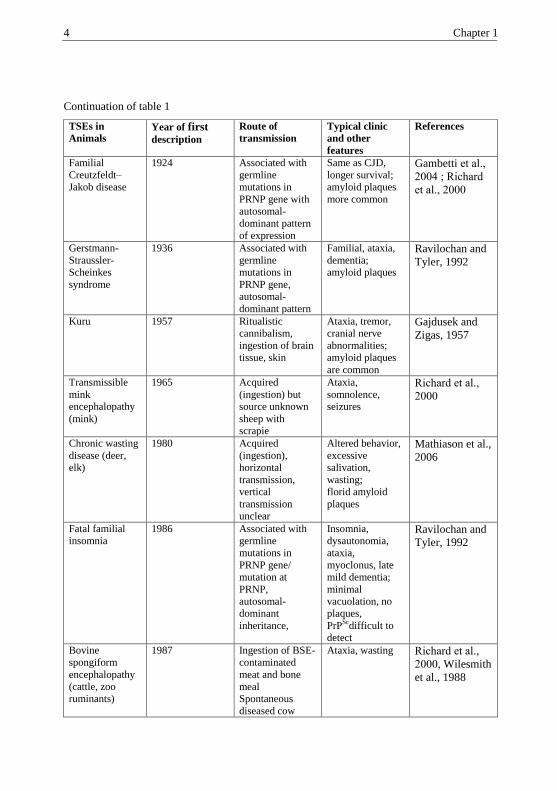
Continuation of table 1
TSEs in
Animals Year of first description
Route of
transmission
Typical clinic
and other
features
References
Familial
Creutzfeldt–
Jakob disease
1924 Associated with
germline
mutations in
PRNP gene with
autosomal-
dominant pattern
of expression
Same as CJD,
longer survival;
amyloid plaques
more common
Gambetti et al.,
2004 ; Richard
et al., 2000
Gerstmann-
Straussler-
Scheinkes
syndrome
1936 Associated with
germline
mutations in
PRNP gene,
autosomal-
dominant pattern
Familial, ataxia,
dementia;
amyloid plaques
Ravilochan and
Tyler, 1992
Kuru 1957 Ritualistic
cannibalism,
ingestion of brain
tissue, skin
Ataxia, tremor,
cranial nerve
abnormalities;
amyloid plaques
are common
Gajdusek and
Zigas, 1957
Transmissible
mink
encephalopathy
(mink)
1965 Acquired
(ingestion) but
source unknown
sheep with
scrapie
Ataxia,
somnolence,
seizures
Richard et al.,
2000
Chronic wasting
disease (deer,
elk)
1980 Acquired
(ingestion),
horizontal
transmission,
vertical
transmission
unclear
Altered behavior,
excessive
salivation,
wasting;
florid amyloid
plaques
Mathiason et al.,
2006
Fatal familial
insomnia
1986 Associated with
germline
mutations in
PRNP gene/
mutation at
PRNP,
autosomal-
dominant
inheritance,
Insomnia,
dysautonomia,
ataxia,
myoclonus, late
mild dementia;
minimal
vacuolation, no
plaques,
PrPSc
difficult to
detect
Ravilochan and
Tyler, 1992
Bovine
spongiform
encephalopathy
(cattle, zoo
ruminants)
1987 Ingestion of BSE-
contaminated
meat and bone
meal
Spontaneous
diseased cow
Ataxia, wasting Richard et al.,
2000, Wilesmith
et al., 1988
Chapter 1 5
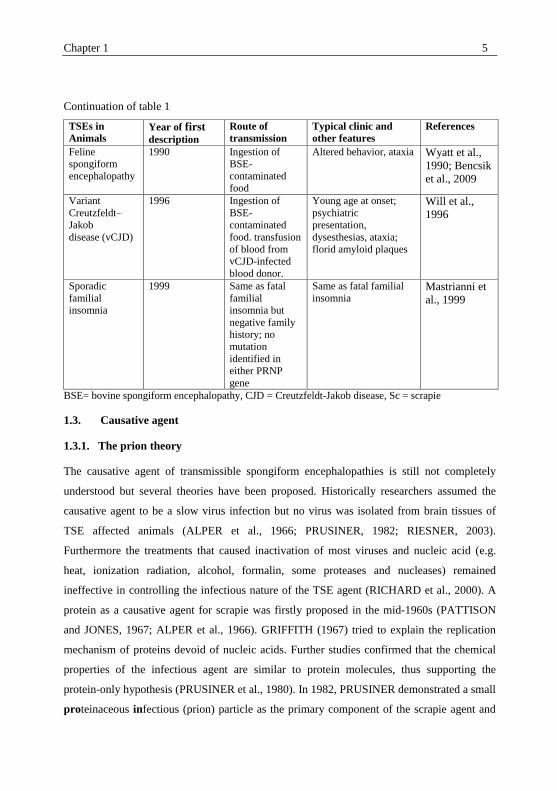
Continuation of table 1
TSEs in
Animals Year of first description
Route of
transmission
Typical clinic and
other features
References
Feline
spongiform
encephalopathy
1990 Ingestion of
BSE-
contaminated
food
Altered behavior, ataxia Wyatt et al.,
1990; Bencsik
et al., 2009
Variant
Creutzfeldt–
Jakob
disease (vCJD)
1996 Ingestion of
BSE-
contaminated
food. transfusion
of blood from
vCJD-infected
blood donor.
Young age at onset;
psychiatric
presentation,
dysesthesias, ataxia;
florid amyloid plaques
Will et al.,
1996
Sporadic
familial
insomnia
1999 Same as fatal
familial
insomnia but
negative family
history; no
mutation
identified in
either PRNP
gene
Same as fatal familial
insomnia Mastrianni et
al., 1999
BSE= bovine spongiform encephalopathy, CJD = Creutzfeldt-Jakob disease, Sc = scrapie
1.3. Causative agent
1.3.1. The prion theory
The causative agent of transmissible spongiform encephalopathies is still not completely
understood but several theories have been proposed. Historically researchers assumed the
causative agent to be a slow virus infection but no virus was isolated from brain tissues of
TSE affected animals (ALPER et al., 1966; PRUSINER, 1982; RIESNER, 2003).
Furthermore the treatments that caused inactivation of most viruses and nucleic acid (e.g.
heat, ionization radiation, alcohol, formalin, some proteases and nucleases) remained
ineffective in controlling the infectious nature of the TSE agent (RICHARD et al., 2000). A
protein as a causative agent for scrapie was firstly proposed in the mid-1960s (PATTISON
and JONES, 1967; ALPER et al., 1966). GRIFFITH (1967) tried to explain the replication
mechanism of proteins devoid of nucleic acids. Further studies confirmed that the chemical
properties of the infectious agent are similar to protein molecules, thus supporting the
protein-only hypothesis (PRUSINER et al., 1980). In 1982, PRUSINER demonstrated a small
proteinaceous infectious (prion) particle as the primary component of the scrapie agent and
6 Chapter 1
introduced the term prion. Prions consist of a misfolded prion protein (PrP) designated as
PrPSc
(Sc for scrapie); whereas the normal cellular prion protein is designated as PrPC
(C for
cellular prion protein). This protein-only model of the prion theory suggests that a molecular
mechanism is involved in replication of the TSE agent by which abnormally folded PrPSc
serves as a template or catalyst which recruits cellular PrP and transforms it to its 3-
dimensional infectious structure (BEEKES and McBRIDE, 2007; MORINET, 2014).
1.3.2. Nomenclature
The nomenclature of PrP species is complicated. The normal cellular isoform is designated as
PrPC whereas PrP
Sc stands for the proteinase K resistant, misfolded protein that remains
insoluble in denaturing detergent. However, there are exceptions in few diseases occasionally
the pathological isoform of PrP fails to show proteinase K resistance (GABIZON et al.,
1996). Recently, the term PrPTSE
has been introduced for diseases associated PrP from TSE
infected individuals to avoid the confusion with the complex PrP nomenclatures e.g. PrPCJD
,
PrPCWD
, PrPSEN
, PrPres
, PrPSc
and PrPBSE
. The currently used nomenclature for the different
prion isoforms is shown in table 2 (BROWN and CERVENAKOVA, 2005).
Table 2. Nomenclature of different transmissible spongiform encephalopathies (TSEs)
Disease name Natural host Prion name PrP isoform
Non-human mammals
Scrapie Sheep and goat Scrapie prion OvPrPSc
Transmissible mink
encephalopathy (TME) Mink TME prion MkPrP
Sc
Chronic wasting disease
(CWD)
Elk, White-tailed deer,
Mule Deer and Red Deer CWD prion MDePrP
Sc
Bovine spongiform
encephalopathy (BSE) Cattle BSE prion BovPrP
Sc
Feline spongiform
encephalopathy (FSE) Cat FSE prion FePrP
Sc
Exotic ungulate
encephalopathy (EUE) Nyala and Greater kudu EUE prion NyaPrP
Sc
Human diseases
Kuru
Humans
Kuru prion
HuPrPSc
Creutzfeldt-Jakob disease
(CJD) CJD prion
(New) Variant Creutzfeldt-
Jakob disease (vCJD,
nvCJD)
vCJD prion
Gerstmann-Sträussler-
Scheinker syndrome (GSS) GSS prion
Chapter 1 7
Ov = ovine, Mk = mink, MDe = mule and deer, Bov = bovine, Fe = feline, Nya = Nyala, Hu
= human, Sc = scrapie
1.3.3. PrPSc
formation
The normal cellular prion protein (PrPC) is a glycoprotein primarily present on the
membranes of neurons, glial cells and in various organs including uterus, placenta, thymus,
heart, lung, muscle and gastrointestinal tract (BUDKA, 2003). PrPC is encoded by the prion
protein gene (PRNP) and is highly conserved among different species (van RHEEDE et al.,
2003). PrPC is attached to the cell surface by using a glycosylphosphatidylinositol (GPI)
anchor (RIESNER, 2003). The normal cellular prion is an α-helical conformational copper-
binding protein with an approximately 220 amino acid residue (RIEK et al., 1996;
HORNEMANN et al., 1997; GAVIER-WIDEN et al., 2005). In the central nervous system
(CNS) PrPC
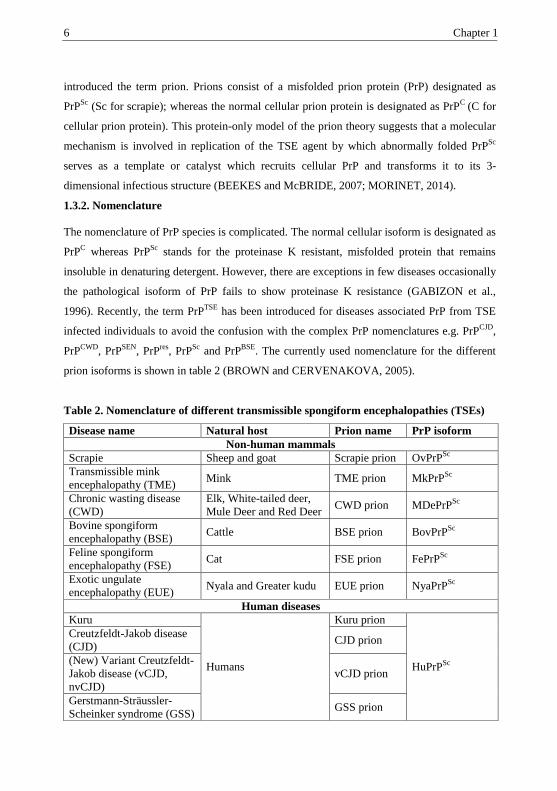
has several functions (Fig.1) including neuronal survival, neurite outgrowth,
synapse formation, maintenance of myelinated fibers and protection against apoptosis or
oxidative stress (WESTERGARD et al., 2007; AGUZZI et al., 2008).
Fig. 1. Functions of normal cellular PrPC (adapted with modifications from AGUZZI et al.,
2008 and learn.genetics.utah.edu)
The abnormal, comparatively protease-resistant, ß-pleated sheet-rich isoform of PrPC is
traditionally called PrP scrapie (GAVIER-WIDEN et al., 2005). The formation of the
pathological isoform of prion protein is thought to be activated either by a mutation of the
normal cellular prion protein gene or by the transmission of pathogenic prion isoforms
(DeMARCO and DAGGETT, 2004; AGUZZI et al., 2008).

8 Chapter 1
The posttranslational modification of PrPC into the abnormal pathological form occurs
through a process of conformational changes, whose mechanism remains elusive until now.
Studies using transgenic mouse models have shed some light on genetic and biochemical
mechanisms responsible for the conversion. According to these models, PrPC
is converted
into PrPSc
by the formation of a PrPC/PrP
Sc complex, but this complex has never been isolated
in pure form. Therefore, it remains unclear whether PrPC binds to one or more additional
macromolecules during the conversion process (PRUSINER et al., 1990; MEIER et al., 2003;
AGUZZI et al., 2008). During conformational changes, ß-pleated sheets become dominant
over the α-helical structure, resulting in a characteristic fibrillar aggregated structure in the
brain as seen in many TSE diseases (PRUSINER, 1998; FRASER, 2002; PRUSINER, 2013).
Spectroscopic measurements of PrPC from healthy hamster brains demonstrated that PrP
C is
mainly composed of α-helices (42%) with negligible amount of β-sheets (3%). On the other
hand, PrPSc
recovered from scrapie infected hamster brain consists of 43% β-sheets and 30%
α-helices (PAN, 1993).
PrPC is normally present on the neuronal cell surface in contrast; PrP
Sc is found in the
cytoplasm of affected cells and shows high resistance against common sterilization methods
(e.g. autoclaving, heat and radiation), proteolytic enzymes, and conventional desinfectants
including alcohol, formalin, and phenol (BELLINGER et al, 1987; BELL and IRONSIDE,
1993).
Once the abnormal isoform is formed or acquired (Fig. 2), it catalyzes the conversion of PrPC
molecules into PrPSc
through an autocatalytic process (CAUGHEY and RAYMON, 1991).
Breakage of these provides more PrPSc
templates for further conversion of the cellular prion
protein in neighbouring neuronal cells. Thus proteinase kinase (PK)-resistant, non-degradable
PrPSc
aggregates in the neuronal tissues are formed and serve as the most effective marker of
prion diseases (McKINLEY et al., 1983).

Chapter 1 9
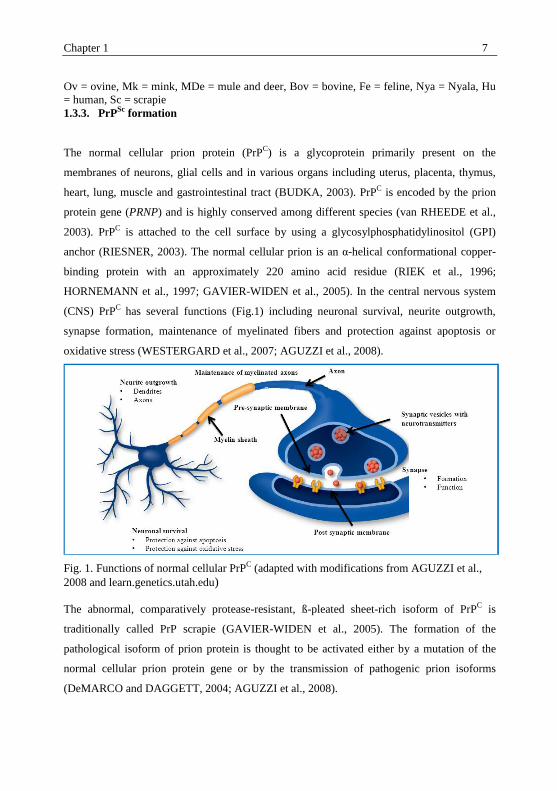
Fig. 2. Conversion of normal PrPC into abnormal misfolded PrP
Sc (adapted from AGUZZI
and POLYMENIDOU, 2004 with modification).
(1) The refolding or template assistance model highlights the interaction between an
externally introduced misfolded protein form (PrPSc
) and normal cellular prion protein (PrPC).
PrPSc
utilizes the normal cellular prion protein as template to transfer itself into further PrPSc
.
A high energy barrier may avoid further spontaneous conversion of the misfolded form. (2)
The seeding or nucleation-polymerization model proposes that the normal cellular prion
protein and the abnormal misfolded form are in a thermodynamic equilibrium. When several
monomeric PrPSc
molecules are mounted into a highly ordered and infectious seed, they can
recruit further PrPSc
and finally aggregate in the form of amyloid.
1.3.4. Pathogenesis of TSEs
The pathogenesis of prion diseases is also poorly understood. Among naturally acquired
prion diseases, scrapie and BSE are the most relevant forms because of their economic
importance. Futhermore, BSE plays an important role due to its transmission to humans and
by causing vCJD (SCOTT et al., 1999; BEEKES and McBride, 2007). Infection begins upon
ingestion of the TSE agent and subsequent invasion of the gut by the pathogen in the majority
of cases of scrapie, BSE, TME, CWD, vCJD and kuru (GAJDUSEK, 1977). Ritualistic
cannibalism seems to be one route of kuru transmission, and BSE contaminated foodstuff
ingestion served as a source of vCJD. In comparison to other TSEs, scrapie and CWD are the
only TSEs that are not only transmissible but also contagious. Infected placenta, abraded skin
Heterodimer Homodimer
Amyloid; not
essential for
replication
Template assisted refolding
Conversion
prevented
by energy
barrier
Equilibrium
between
both forms
PrPC
PrPSc
PrPC
PrPSc
Seed formation
(very slow) Recruitment of
PrPSc
(fast) Infectious
seed
Amyloid Fragmentation
into seeds
Seeding Nucleation
1
2

10 Chapter 1
and flesh of dead animals (in the form of meat and bone meal) are considered the major cause
of horizontal or vertical transmission of scrapie in sheep (BROWN and GAJDUSEK, 1991;
RACE et al., 1998; DETWILER and BAYLIS, 2003). Additionally mites, fly larvae and
pupae serve as living harbours of ingestible infectivity (WISNIEWSKI et al., 1996; POST et
al., 1999). Recently, prion agents were found in CWD infected cervid saliva (MATHIASON
et al., 2006). Along ingestion, scarification of skin or gums is also an important route of agent
entry into the body. In the case of kuru, transdermal/conjuctival invasion served as an
alternative natural source of infection (GOODFIELD, 1997). In early stages of disease
progression, infectious prion agents cross the mucous membrane barriers and can be detected
in tonsils, Peyer’s patches, and lymph nodes of the alimentary canal (ANDRÉOLOTTI et al.,
2000; SPRAKER et al., 2002; JEFFREY and GONZÁLEZ, 2004; WELLS et al., 2005). This
phenomenon of early lymphoid invasion has been demonstrated experimentally as early as 6
weeks after infection in CWD (SIGURDSON et al., 1999) and at 3 months of age in naturally
occurring scrapie in lambs (ANDRÉOLOTTI, 2004).
After invasion of the infectious agent, a replication period lasting from months to years takes
place in the lymphoreticular system (LRS) involving spleen and lymph nodes in most cases
of TSEs. However, in BSE and in some scrapie cases, there is little involvement of the LRS
(JEFFREY and GONZÁLEZ, 2004; WELLS et al., 2005). After incubation the infectious
prion agent spreads towards the brain, where it progressively aggregates, resulting in fatal
neurodegenerative alterations. The mechanisms involving the spread of prions from the
alimentary tract or tonsils to the brain are inadequately investigated. Hematogenous and
retrograde axonal routes, involving fibers innervating lymphoid tissues or the autonomic
nervous fibers of the digestive tract, have been implicated (SIGURDSON et al., 1999;
ANDRÉOLOTTI et al., 2000) resulting in a model of neuroimmune invasion that comprises
two phases. The first phase is characterized by the widespread colonization of
lymphoreticular organs by a mechanism that depends on B lymphocytes and follicular
dendritic cells. The second phase involves the expression of PrPSc
in the peripheral
sympathetic nervous system (SNS) nerves and results in the prion distribution in the CNS.
The neuronal spread of prion infectious agents from the enteric and peripheral nervous
system to the spinal cord after oral uptake of the TSE agent from the gut was first proposed
after an intra-gastric scrapie challenge to mice (KIMBERLIN and WALKER, 1989). Later,
hamster adapted 263K scrapie served as a model to observe the neuronal spread of the prion

Chapter 1 11
agent from the alimentary canal to the brain after oral uptake (BEEKES et al., 1998;
McBRIDE et al., 2001). It was shown that the N. splanchnicus and N. vagus of sympathetic
and parasympathetic systems; respectively, are the main routes of prion spread from the gut
to the CNS. Efferent and afferent nerve fibers are used to reach either to the thoracic spinal
cord (splanchnic nerves) or the solitary tract nucleus and the dorsal motor nucleus of the N.
vagus. Centripetal and centrifugal spread of the prion agent to the cervical and lumbal spinal
cord originating from the thoracic spinal cord (McBRIDE et al., 2001; BALKEMA-
BUSCHMANN et al., 2011; KAATZ et al., 2012; McGOVERN et al., 2015). Sheep and
goats naturally or experimentally infected with prion disease have shown a significant
propagation of the scrapie agent in lymphoid organs including Peyer´s patches, spleen and
lymphoid ganglions during the early stage of infection. With the progression of the disease
the agent is present in several tissue and fluids with high infectious titers in the brain. In the
case of cattle TSE, infectivity is mainly detected in different parts of the CNS, the peripheral
nervous system and autonomic nervous system.
1.3.5. Pathological characteristics of TSEs
Neuropathologically, TSEs are characterized by spongiosis or vacuolation in the neuropil
(vacuolation of neuronal processes), and/or neuronal bodies showing single or multiple
vacuoles in the perikarya of neurons (WELLS et al., 1987; WELLS et al., 1989; WILLIAMS
and YOUNG, 1993; SPRAKER et al., 2002). PrPSc
aggregation and accumulation in neurons
and glial cells in the brain is the characteristic feature of TSE and can be detected earlier than
vacuolar changes (JEFFREY and GONZÁLEZ, 2004; SPRAKER et al., 2004). Other
remarkable changes including neuronal cell death or loss, astrocyte proliferation and amyloid
plaque formation are variably seen in some forms of human and animal TSEs (BUDKA et al.,
1995; WELLS and WILESMITH, 1995; LIBERSKI et al., 1998; LIBERSKI and BUDKA,
1999; FRASER, 2002). Classical inflammatory responses against the infectious isoform of
PRPTSE
are not initated presumable since the faulty protein is not recognized as foreign
material by the immune system (GAVIER-WIDEN et al., 2005). The mechanism(s) causing
brain damage through accumulation of PrPSc
have not been fully elucidated. Although a
noticeable accumulation of the pathological isoform is also present in the lymphoid tissue, no
histological alterations in lymphoid tissues have been observed (GAVIER-WIDEN et al.,
2005).

12 Chapter 1
1. 4. Clinical Manifestations
Infected animals may develop signs of the disease slowly and many months and years after
primary exposure. In cattle it may take 2 to 8 years from the time an animal becomes infected
until it shows first signs of the disease. Signs include a change in attitude and behaviour,
gradual uncoordinated movements, trouble in standing and walking, weight loss despite
normal appetite, and decreased milk production. From the onset of signs, the animal
deteriorates until it either dies or is destroyed. This disease process may take from 2 weeks to
6 months after first initial clinical signs have been noticed. Similar symptoms consisting of
muscle spasms, lack of muscle control, deteriorating problems with memory may develop in
humans.
1.5. Rodent models of prion disease and brain pathology in hamsters
Due to the unavailability of cell culture systems for pathogenetic studies of prion diseases,
conventional or transgenic animal models provide an opportunity to study most aspects of
prion propagation and infectivity (WATTS and PRUSINER, 2014). Previous in vivo studies
on TSEs were carried out mostly in the natural host species. Rodent models expressing
cellular prion proteins from different species provide the opportunity to study the disease in a
more formalized manner. These models help to understand the neuropathological
mechanisms on the molecular level, normal functions of PrPC, species barrier mechanisms,
cell specificity, role of glycosylation, prion agent spread mechanism and interaction between
PrPC and PrP
Sc (GROSCHUP and BUSCHMANN, 2008). They also shed some light into
mapping of prion protein segments which are involved in prion conversion and replication
and helped to understand the role of the host prion gene in the genetic control of the disease
(BARON, 2002; GROSCHUP and BUSCHMANN, 2008).
1.5.1. Conventional rodent models
Since the first transmission of scrapie to mice, the use of animal models has laid the basis for
a more comprehensive understanding of prion diseases (CHANDLER, 1961). Over the years,
rats, golden hamsters and voles were also used as animal models (CHANDLER, 1971;
CHANDLER and FISHER, 1963; CHANDLER and TURFREY, 1972). These animal
species provide a great opportunity to study disease characteristics in more detail due to the
Chapter 1 13
short incubation time compared to the natural hosts (see table 3 for brief description of these
models and their origin). Initial experiments performed on Syrian hamsters (SHa) allowed the
prediction of the prion protein theory (PRUSINER et al., 1980). Successful experimental
transmission of prion isolates has also been reported to non-human primates including
monkeys (macaque and squirrel), lemurs and chimpanzees (GIBBS et al., 1994; WILLIAMS
et al., 2007; LASMÉZAS et al., 2005).
Table 3. Origin and diversity of experimental TSE strains in rodents (adapted from
BERINGUE et al., 2008).
Name/abbreviation
of strains
Origin Passage in
intermediate
species
Host References
ME7
(Prn-a)
Sheep scrapiea None Mouse Bruce, 1993; Bruce
et al., 2002
87A Sheep scrapiea None Mouse (Prn-
a)
Bruce and
Dickinson, 1987
221C Sheep scrapiea None Mouse (Prn-
a)
Bruce et al., 2002
87V Sheep scrapiea
SSBP/1b
None, goatsc Mouse (Prn-
b)
Bruce, 1993; Bruce
et al., 2002
79A SSBP/1b goats
c Mouse (Prn-
a)
Bruce, 1993
79V SSBP/1b goats
c Mouse (Prn-
b)
Bruce, 1993
139Ad SSBP/1b goats
c Mouse (Prn-
a)
Chandler and
Fisher, 1963
14 Chapter 1
Continuation of table 3
Name/abbreviation
of strains
Origin Passage in
intermediate
species
Host References
22C SSBP/1b None, goats
e Mouse (Prn-a) Bruce, 1993
22H Uncloned 22C None, goatse Mouse (Prn-b) Bruce, 1993
22L SSBP/1b None Mouse (Prn-a) Bruce, 1993
22A SSBP/1b None Mouse (Prn-b) Bruce, 1993
22F Cloned 22A None Mouse (Prn-a) Bruce, 1993
301Cf BSE Direct or not Mouse (Prn-a) Bruce et al.,
1994
301V BSE Direct or not Mouse (Prn-b) Bruce et al.,
1994
139H Cloned 139A None Syrian hamster Kimberlin et
al., 1987
263Kg SSBP/1b goats
c, mice,
rats
Syrian hamster Kimberlin and
Walker , 1977
ME7-H Cloned ME7 None Syrian hamster Kimberlin and
Walker, 1989
HYh TMEi None Syrian hamster Bessen and
Marsh, 1992
DY TMEi None Syrian hamster Bessen and
Marsh, 1992
a = Field isolate; b = SSBP/1: sheep scrapie brain pool 1; c = Passage of SSBP/1 through
goats: ‘drowsy’ goat source; d = also termed Chandler; e = Passage of SSBP/1 through goats:
‘scratching’ goat source; f = not known if 301C yields to 301V in Prn-b mice; g = Sc237 is a
subclone of 263K; h = similar to 263K?; i = TME: Stetsonville isolate, BSE: bovine
spongiform encephalopathy
1.5.2. Transgenic rodent models
Mouse transgenesis has made enormous contribution to prion research (table 4). SCOTT et
al. (1989) generated transgenic mice overexpressing hamster PrPC that proved to be
susceptible to 263K hamster prions in contrast to conventional mice. Nowadays a series of
transgenic mouse lines is available which express mink, human, caprine, bovine, ovine,

Chapter 1 15
cervid, and mouse PrPC (table 4). In addition, SHMERLING et al. (1998), generated mouse
models expressing transgenic PrPC with amino-proximal deletions at residues 32-121 or 32-
134. These mice showed severe ataxia along with neuronal death in the granular layer of the
cerebellum as early as 1-3 months after birth. This deficiency was recovered by introducing
one copy of a wild-type PrP gene. PrPC transgenic mice with deleted individual regions of the
putative secondary structure demonstrated that lacking of one of the C-terminal helices lead
to the incidence of CNS failures. This accumulation of PrP within neurons as cytoplasmic
inclusions (MURAMOTO et al., 1997) highlights the probable role of α-helix in protein
stability and normal trafficking. Glycosylphosphatidylinositol lacking transgenic (GPI–Tg)
mice, inoculated with scrapie prion exhibited susceptibility to infection but an altered clinical
disease manifestation and PrPSc
deposits were noticed (CHESEBRO et al., 2005). On the
other hand, the wild type mice inoculated with the scrapie prion generated the normal profile
of prion disease; thus, highlighting the possible role of the GPI anchors in disease outcome.
Transgenic mice expressing PRNP with point mutations, insertions, or deletions exhibited
phenotypically a similar spongiform diseases (SIGURDSON et al., 2009). It has been
observed that a moderate overexpression in transgenic mPrP (170N, 174T) mice (a mouse
PrP with two point mutations that affect the structure of its globular domain) resulted in the
generation of spongiform encephalopathy with cerebral PrPSc
plaques. This genetic disease
was restored by intracerebrally inoculation of brain homogenate to tga20 mice
overexpressing wild type (WT-PrP) PrP (SIGURDSON et al., 2009). Transgenic mice allow
to study the pathogenesis of several mutations related to different forms of genetic TSEs, the
transmission barrier phenomenon (AGUILAR-CALVO et al., 2014) and hence to assess the
relative risk of each TSE strain for humans. For example, tg650 mice expressing human PrP
Met129 were inoculated with field isolates of different forms of BSE. Unlike the classical
BSE agent, L-type BSE emerged to proliferate in these mice with no obvious transmission
barrier, whereas H-type prions were unable to infect these mice (BERINGUE et al., 2008).
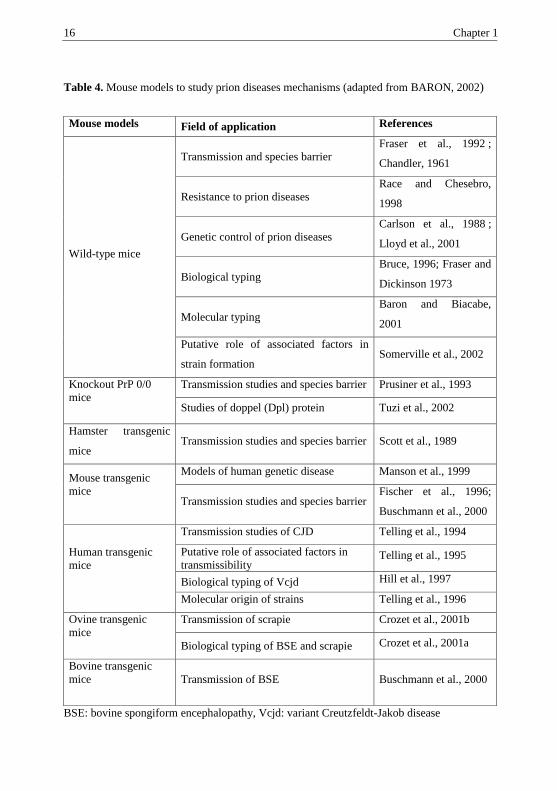
16 Chapter 1
Table 4. Mouse models to study prion diseases mechanisms (adapted from BARON, 2002)
BSE: bovine spongiform encephalopathy, Vcjd: variant Creutzfeldt-Jakob disease
Mouse models Field of application References
Wild-type mice
Transmission and species barrier
Fraser et al., 1992 ;
Chandler, 1961
Resistance to prion diseases
Race and Chesebro,
1998
Genetic control of prion diseases
Carlson et al., 1988 ;
Lloyd et al., 2001
Biological typing
Bruce, 1996; Fraser and
Dickinson 1973
Molecular typing
Baron and Biacabe,
2001
Putative role of associated factors in
strain formation Somerville et al., 2002
Knockout PrP 0/0
mice
Transmission studies and species barrier Prusiner et al., 1993
Studies of doppel (Dpl) protein Tuzi et al., 2002
Hamster transgenic
mice Transmission studies and species barrier Scott et al., 1989
Mouse transgenic
mice
Models of human genetic disease Manson et al., 1999
Transmission studies and species barrier Fischer et al., 1996;
Buschmann et al., 2000
Human transgenic
mice
Transmission studies of CJD Telling et al., 1994
Putative role of associated factors in
transmissibility Telling et al., 1995
Biological typing of Vcjd Hill et al., 1997
Molecular origin of strains Telling et al., 1996
Ovine transgenic
mice
Transmission of scrapie Crozet et al., 2001b
Biological typing of BSE and scrapie Crozet et al., 2001a
Bovine transgenic
mice
Transmission of BSE Buschmann et al., 2000

Chapter 1 17
1.5.2.1. PrP knock-out models
After development of the protein-only hypothesis, the generation of transgenic mouse strains
lacking PrPC
expression (PrP knockout mice) helped to understand the physiological function
of PrPC
and its role in neurodegenerative diseases in more detail. At least four lines of mice
lacking PrPC have been developed (WEISSMANN and FLECHSIG, 2003). Ablation of PrP
C
in these models did not result in major anatomical and developmental deficits; however, these
animals were resistant to scrapie challenge (BUELER et al., 1992; BUELER et al., 1993;
PRUSINER et al., 1993; SAILER et al., 1994; WEISSMANN and FLECHSIG, 2003). After
reintroduction of PrP transgenes in PrP knockout animals, the susceptibility to infection was
restored confirming a correlation between host PrPC and expression of TSE. Introduction of
multiple prion gene copies to the mouse genome leading to the overexpression of PrPC,
showed that the PrPC expression concentration plays an important factor for prion disease
susceptibility (PRUSINER et al., 1990; FISCHER et al., 1996; WEISSMANN and
FLECHSIG, 2003; UCHIYAMA et al., 2014).
1.5.3. Brain pathology in hamsters
Transmission of scrapie prions to golden hamsters was first reported by ZLOTNIK and
RENNIE (1965) using the ME7 strain of the scrapie agent. This was confirmed by
CHANDLER and TURFREY (1972), who successfully transmitted scrapie to Chinese
hamsters. Among the transmissible spongiform encephalopathies, the hamster 263K scrapie
prion model is a rapid and well characterized model (BOLTON et al., 1991). A low
concentration of prions can easily be detected with hamster prion models as compared to
other models which are less sensitive (BOLTON, 1998). Incubation periods of scrapie in
hamsters are remarkably short as compared to mouse models (KIMBERLIN and WALKER,
1977). Hamsters inoculated intracerebrally with a high dose of scrapie prions develop clinical
signs consisting of neurological dysfunction after 60-65 days. This is half of the incubation
time found in mice (BARINGER et al., 1983).
The prion burden remains stable in various CNS regions throughout the disease course and
precedes pathological changes. Histologically, the cerebrum shows minimal vacuolation in
the absence of astrogliosis prior to clinical signs. After the onset of clinical signs, severe
vacuolation with moderate astrogliosis has been observed in the cerebral cortex. Cerebellum,

18 Chapter 1
brain stem, and spinal cord display a moderate degree of vacuolation along with astrogliosis
(BARINGER et al., 1983).
1.5.3.1. Strain variations
Concerning the causative agents of TSEs some reservations remain. A foremost question is
whether the causative agents are exclusively composed of one specific abnormal isoform of
the normal cellular prion protein. A major problem for the protein-only hypothesis of prion
diseases has been how to explain the presence of multiple isolates or strains of prions. The
existence of different prion strains was first observed in goats after inoculation with sheep
brain homogenates (SSBP/1), which resulted in two different clinical disease phenotypes: a
scratching and a drowsy syndrome (PATTISON et al., 1959). Now several discrete strains of
naturally occurring sheep scrapie have been isolated in mice. Such strains are distinguished
by their biological properties including distinct incubation periods and lesion profiles in
defined inbred mouse lines (BRUCE et al., 1992). For instance, they can be serially
propagated in inbred mice with the same Prnp genotype. Moreover, strains can be re-isolated
in mice after passage in intermediate species with dissimilar PrP primary structures (BRUCE
et al., 1994). Usually, distinct strains of conventional pathogens including bacteria and
viruses are described by their difference in their nucleic acid genome. In the absence of such
a scrapie genome, alternate prospects must be considered. WEISSMANN`s (1991) “unified
hypothesis” suggested that strain characteristics could be encoded by a small cellular nucleic
acid, or “coprion.” According to this hypothesis the strain characteristics would be sensitive
to ultraviolet irradiation, but he failed to present such results. On the other hand, the protein-
only hypothesis proposed by GRIFFITH 1967 would have to explain how a single
polypeptide chain might encode multiple disease phenotypes. Evidently, understanding how a
protein-only infectious agent could convert such phenotypic information is of significant
biological importance (COLLINGE, 2001).
Strain specificity encoded by PrP itself was supported by the study of two distinct strains of
TME prions propagated in hamsters, designated as hyper (HY) and drowsy (DY). These
strains can be differentiated by differing biochemical properties produced by the accumulated
PrPSc
in the brains of affected hamsters (BESSEN and MARSH, 1992; COLLINGE, 2001).
With limited proteolysis, strain-specific migration patterns of PrPSc
were seen on
polyacrylamide gels. They were linked to different N-terminal ends of HY and DY PrPSc

Chapter 1 19
following protease treatment and involved differing conformations of HY and DY PrPSc
(BESSEN and MARSH 1994; COLLINGE, 2001). Several human PrPTSE
confirmations
related to different phenotypes of CJD have been identified (TELLING et al., 1996;
COLLINGE et al, 1996; COLLINGE, 2001). The different fragment sizes after proteinase K
treatment as seen on Western blots highlight the presence of different PrPSc
conformations
(SAFAR et al., 1998). Biochemically modified PrP served as candidates for the molecular
substrate of prion strain diversity. This aspect has been elaborated on studies with CJD
isolates. PrPTSE
fragment sizes and PrP glycoforms ratios (diglycosylated, monoglycosylated,
and unglycosylated PrP) were maintained in human PrP expression in transgenic mice
following passages. Additionally, transmission of human and bovine prions to wild type mice
results in murine PrPSc
with fragment sizes and glycoforms ratios corresponding to the
original inoculum (COLLINGE et al 1996; COLLINGE, 2001). Vcjd is distinct from
classical CJD on the basis of PrPSc
glycoforms ratios. Related ratios are also observed in BSE
in cattle and BSE transmitted to numerous other species. These observations intensely
support the protein-only hypothesis of infectivity and indicate that strain variation is
determined by the arrangement of PrP conformation and glycosylation. Moreover,
polymorphisms shown by the PrP sequence can affect the generation of specific PrPSc
conformers. As glycosylation happens before changing to PrPSc
, the diverse glycoforms ratios
may signify selection of specific PrPC glycoforms by PrP
Sc of diverse conformations.
Following such a hypothesis, PrP conformation would be the major factor determining the
strain type, with glycosylation as a secondary process. However, as it is observed that
different cell types glycosylate proteins differently, PrPSc
glycosylation forms might offer a
substrate for the neuropathological targeting that discriminates diverse prion strains
(COLLINGE et al 1996; COLLINGE., 2001). Specific PrPSc
glycoforms might replicate well
in neuronal populations expressing a similar PrP glycoforms on the cell surface. Such
targeting could also help to explain the different incubation periods that also allows
distinguishing strains. Subsequently, targeting of brain regions with higher levels of PrP
expression will likely yield shorter incubation periods (SAFAR et al., 1998). The results also
suggested that different conformations of PrPSc
could encipher properties of different prion
strains characterized by altered pathological behaviours (SAFAR et al., 1998). Furthermore,
it was shown that from a single source of a scrapie agent a mixture of strains could be
isolated (KIMBERLIN and WALKER, 1978). However, it is still unclear if these sub-strains
are stable in themselves or are dependent on the co-existence of their “partner strains”.

20 Chapter 1
1.6. Synaptic pathology in neurodegenerative diseases
Despite the significant importance of neuronal death in neuropathology of prion diseases, the
events and mechanism(s) that lead to neuronal dysfunction and ultimately neurodegeneration
remain inadequately understood. The suggestions of a possible correlation between cognitive
decline and synaptic loss in Alzheimer’s disease have opened new avenues in the prion field
too (TERRY et al., 1991). There is a growing body of evidence that indicates that synaptic
dysfunction plays an early and important role in the development and progression of prion
diseases and it may be an early key event in many neurodegenerative diseases (CLINTON et
al., 1993; CUNNINGHAM et al., 2003; JEFFREY et al., 2000; SISKOVA et al., 2009; REIS
et al., 2015).
Immunohistochemical, ultrastructural and cellular studies have demonstrated that pre-
synaptic terminals in brain synapses are enriched in PrPC
(FOURNIER et al., 1995;
HAEBERLE et al., 2000; BROWN, 2001). Early events in the development of prion diseases
involve synaptic loss associated with deposition of abnormal PrPSc
in synaptic boutons
especially in pre-synaptic terminals (JEFFREY et al., 2000). Neurotransmission and exosome
associated synaptic vesicle proteins e.g. synaptophysin and synapsin-I, and proteins of pre-
synaptic plasma membrane e.g. synaptosomal-associated protein of 25kDa (SNAP-25) and
syntaxin-I, are reduced in patients suffering from prion diseases (FERRER, 2002; FERRER
et al., 1999). These proteins play a vital role in exocytosis and neurotransmission, and some
of these proteins contribute for normal synaptic function. Therefore, it may be assumed that
pre-synaptic modulation is damaged in prion diseases (FERRER, 2002). However, reduction
in synaptic protein expression should not be considered as an exclusive cause of synaptic
loss. Impaired or abnormal protein synthesis or turnover may also represent a complementary
event in synaptic dysfunction (FERRER, 2002). Synaptic dysfunction is an essential and
constant feature of prion disease, irrespective of the existence or lack of spongiform changes,
neuronal loss and severe gliosis (CLINTON et al., 1993). In rodent models of
neurodegenerative diseases it is well documented that synaptic pathology precedes the
degeneration of neuronal cell bodies in the hippocampus (CUNNINGHAM et al., 2003;
JEFFREY et al., 2000; SISKOVA et al., 2009). Malformed electrophysiological recordings in
scrapie infected hamster hippocampal and cortical slices further substantiate the synaptic
alterations (BARROW et al 1999). The murine ME7 scrapie model was among the first to
present observations that allowed to distinct between synaptic dysfunction and neuronal cell

Chapter 1 21
death (JEFFREY et al., 2000; CUNNINGHAM et al., 2003). In this model, synaptic
degeneration within the stratum radiatum of the hippocampus is characterized by the
degeneration of the pre-synaptic terminal, proceeding to the loss or degeneration of the post-
synaptic dendritic spine. In addition, these changes occur in the absence of detectable
neuronal cell death (CUNNINGHAM et al., 2003; SISKOVA et al., 2009). Electron
microscopic studies in the murine model showed that synaptic dysfunction and loss
associated with PrPSc
preceded neuronal loss and clinical onset of disease. Scrapie infected
murine hippocampus revealed degenerated axon terminals at about 98 days post infection
(dpi), whereas definite clinical scrapie is apparent not before 226 dpi (JEFFREY et al., 2000;
SISKOVA et al., 2009). Intact synapses have pre-synaptic terminals packed with electron-
lucent cytoplasm, characteristic small round synaptic vesicles and opposing bar-like post-
synaptic densities. Degenerating synapses in prion disease are characterized by the presence
of electron dense pre-synaptic terminals, the loss of integrity of vesicles and other organelles.
The pre-synaptic membrane remains intact and the post-synaptic membrane appears to be
increased in the curvature and thickness. With the progression of the disease the post-synaptic
membrane progressively curves around degenerating pre-synaptic elements (SISKOVA et al.,
2009). In advanced stages of prion disease the pre-synaptic terminal appears to be completely
engulfed by a post synaptic density (SISKOVA et al., 2009).
1.6.1. Mechanisms of synaptic dysfunction
Apart from the obvious significance of synapse degeneration in neurodegenerative diseases
extremely little is known about the basic cellular and molecular events by which a misfolded
protein leads to synapse degeneration or dysfunction. Neuronal cell loss, spongiform
appearance and gliosis are prime features of prion diseases; however, the first noticeable
changes emerge to be related to synaptic dysfunction (JEFFREY et al., 2000; SISKOVA et
al., 2009; REIS et al., 2015). In the murine ME7 model of prion disease, early behavioral
deficits emerge in conjunction with PrPSc
deposition and synaptic dysfunction preceding
neuronal death (JEFFREY et al., 2000; RUSSELAKIS-CARNEIRO et al., 2004; SOTO and
SATANI, 2010). Studies performed on knock-out mice have also highlighted the role of
prion proteins in synaptic function (COLLINGE et al., 1994). PrPC enriched in the synapses
interacts with proteins participating in synaptic transmission e.g. synaptophysin (FOURNIER

22 Chapter 1
et al., 1995; HAEBERLE et al., 2000; BROWN, 2001). Immunohistologically, abnormal
PrPSc
staining is found in the region of neuronal cell bodies and dendrites, mimicking
synaptophysin distribution, also signifying abnormal PrPSc
accumulation in synaptic
structures (KITAMOTO et al., 1992; FOURNIER et al., 1995). During the initial stages of
the disease, PrPSc
accumulates in membrane lipid rafts. This accumulation leads to the
detachment of caveolin and synaptophysin from these membrane domains and probably
impacts synaptic function (RUSSELAKIS-CARNEIRO et al., 2004). Exocytosis and
neurotransmission linked proteins e.g. SNAP-25, syntaxins, synaptophysin cysteine string
protein (CSP), VAMP-2, synapsin and Rab3a have also been reported to be decreased in
prion disease in the CNS (FERRER et al., 2000; GRAY et al., 2009; HILTON et al., 2013).
Biochemical analysis highlighted the fact that loss of synaptic vesicle proteins, especially
CSP, VAMP-2, and synapsin precedes the changes of proteins in the post-synaptic division
(GRAY et al., 2009). CSPα, as an important synaptic protein, exists in pre-synaptic terminals
and forms a chaperone complex to maintain normal synapses (TOBABEN et al., 2001). It is
of particular interest that mice lacking CSP demonstrate a synaptic degenerative phenotype
(FERNÁNDEZ-CHACÓN et al., 2004). Depletion or reduction of CSPα results in an
abnormal SNAP-25 conformation that resists soluble NSF attachment protein receptor
(SNARE) complex formation, and is subject to ubiquitylation and proteasomal degradation
(SHARMA et al., 2011a; YI and EHLERS, 2007). An impairment of the SNARE complex
due to an alteration or reduction in SNAP-25 finally correlates to neurodegeneration
(SHARMA et al., 2011b; HE et al., 2003). Thus deletion or reduction of CSPα may result in a
massive neurodegeneration at the synaptic level that impairs survival in the ME7 model of
prion disease (FERNÁNDEZ-CHACÓN et al., 2004).
Summarized, these data indicate that conversion of normal PrPC to abnormal PrP
Sc affects the
strength and function of synapses, ultimately leading to neurological damage and finally
initiating the clinical onset of disease (HILTON et al., 2013).
1.6.1.1. Role of mitochondria in synaptic degeneration
Damage or dysfunctions of mitochondria are frequently associated with neurodegenerative
diseases and it is well documented that neuronal synaptic function and mitochondria are co-
dependent (CASTELLANI et al., 2002; LI et al., 2004; SISKOVA et al., 2010). However, the
exact mechanism of mitochondrial contribution to neurodegeneration has not been explored

Chapter 1 23
completely. A recent study in the ME7 model reported that synaptic pathology was
accompanied by alterations in mitochondria (SISKOVA et al., 2010). The phenomenon of
early involvement of neuronal mitochondria is further detailed by the finding that N-acetyl
aspartate (synthesized by neuronal mitochondria) level decreased in the thalamus and
hippocampus as well as in brain areas associated with the early onset of behavioral deficits
(SISKOVA et al., 2010). During the initial stage of prion disease, synaptic density remains
unchanged. This requires decreased respiration which leads to reduction or silencing of
mitochondrial function, followed by a withdrawal of the degenerated synaptic terminal from
the remaining axonal portion. Neuronal mitochondria exhibit various morphological changes
in the inner membrane following prion disease progression. Chronologically, succinate
dehydrogenase and cytochrome c oxidase (COX) activity assays showed structural changes in
mitochondria with functional impairment of complex IV activity in the initial stage of prion
disease. Impairment of complex IV activity leads to compromised mitochondrial respiratory
activity in prion disease and corresponds with the beginning of synaptic loss (SISKOVA et
al., 2010). In addition, up-regulation of nitric oxide can be accompanied by astrogliosis in
prion diseases (ALMER et al., 1999; GRAY et al., 2009). It is predicted that nitric oxide is an
effective mediator of brain damage and may directly disturb mitochondrial function by
interfering to the oxygen binding to complex IV (CLEETER et al., 1994; BOLANOS et al.,
1997; SISKOVA et al., 2010). In addition expression of nitric oxide may disturb
mitochondrial respiratory chain complex I and IV activity (SMITH and LASSMANN, 2002;
ZHANG et al., 2005; SISKOVA et al., 2010). During the initial stage of prion disease, a
significant increase in neuronal nitric oxide synthase in the hippocampus has been observed
(PICANCO-DINIZ et al., 2004; SISKOVA et al., 2010). The increased production of
neuronal nitric oxide takes place in the stratum radiatum and a decline in the late disease
stage are paralleling COX activity changes and hence justifying the idea of nitric oxide
involvement in the damage of mitochondria in the ME7 scrapie model (SISKOVA et al.,
2010). In ultrastructural studies of the ME7 murine model, neuronal mitochondria appeared
reduced in number, swollen, having significantly large diameter and poorly defined swollen
cristae as compared to healthy wild type mice (SISKOVA et al., 2010). Due to the presence
of morphological defects and complex IV activity dysfunction, the respiratory capability of
neuronal mitochondria in prion disease could be compromised in the initial stage of the prion
disease and may correspond to the initiation of synaptic dysfunction. A misbalance in

24 Chapter 1
reactive oxygen levels, along with other changes, could be induced and may contribute to the
intensification of neuropathological processes (SISKOVA et al., 2010).
1.7. Aims of the study
It is well-known that the gene encoding the prion protein (PRNP) critically influences the
susceptibility of small ruminants for certain forms of TSEs, which has contributed to the
development of selective breeding programs, for instance of sheep with a lower susceptibility
to scrapie (AGUILAR-CALVO et al., 2014). Moreover, transgenic mice expressing a certain
polymorphic variant of the goat PRNP gene are resistant to scrapie and BSE (AGUILAR-
CALVO et al., 2014). However, whether the genotype similarly has an impact on the
susceptibility of goats for BSE remained enigmatic so far. A detailed elucidation of this
question has important implications for the control of TSEs as it will contribute to identify
appropriate genotypes, which could selectively be chosen for targeted breeding programs of
goats.
While abundant data exist, which contributed to a detailed insight into the neuropathogenesis
of TSEs in the brain itself, comparatively less is known about the involvement of the
vegetative nervous system and the spinal cord, even though the sympathetic nervous system
has been demonstrated to play a pivotal role in the initial spread of prions in BSE-infected
cattle. Research upon BSE has mainly focused on cattle; however, the pathogenesis in goats
has been subjected to little research so far. Moreover, the cellular and molecular constituents
and mechanisms that facilitate the spread of the agent remain undetermined.
Thus, the present study aimed to (i) clarify the effect of certain PRNP genotypes in the oral
transmission of the BSE agent to goats, (ii) to detect axonal cytoskeletal and transport
disturbances during the course of BSE in the spinal cord and peripheral tissues of
experimentally infected goats, and lastly (iii) to unravel potential ultrastructural changes in
the superior cervical ganglion of experimentally BSE-infected goats with a special emphasis
upon synapse pathology.

Chapter 2 25
Chapter 2

26 Chapter 2
Chapter 2 EFFECT of Q211 and K222 PRNP
POLYMORPHIC VARIANTS IN THE
SUSCEPTIBILTY OF GOATS TO ORAL
INFECTION WITH GOAT BOVINE
SPONGIFORM ENCEPHALOPATHY
AGUILAR-CALVO, P., FAST, C., TAUSCHER, K., ESPINOSA, J-C., GROSCHUP, M. H.,
NADEEM, M., GOLDMANN, W., LANGEVELD, J., BOSSERS, A., ANDREOLETTI, O.
TORRES, J-M.
BACKGROUND: The prion protein-encoding gene (PRNP) is one of the major
determinants for scrapie occurrence in sheep and goats. However, its effect on bovine
spongiform encephalopathy (BSE) transmission to goats is not clear.
METHODS: Goats harboring wild-type, R/Q211 or Q/K222 PRNP genotypes were orally
inoculated with a goat-BSE isolate to assess their relative susceptibility to BSE infection.
Goats were killed at different time points during the incubation period and after the onset of
clinical signs, and their brains as well as several peripheral tissues were analyzed for the
accumulation of pathological prion protein (PrPSc
) and prion infectivity by mouse bioassay.
RESULTS: R/Q211 goats displayed delayed clinical signs compared with wild-type goats.
Deposits of PrPSc
were detected only in brain, whereas infectivity was present in peripheral
tissues too. In contrast, none of the Q/K222 goats showed any evidence of clinical prion
disease. No PrPSc
accumulation was observed in their brains or peripheral tissues, but very
low infectivity was detected in some tissues very long after inoculation (44-45 months)
CONCLUSIONS: These results demonstrate that transmission of goat BSE is genotype
dependent, and they highlight the pivotal protective effect of the K222 PRNP variant in the
oral susceptibility of goats to BSE.
KEYWORDS: BSE; PRNP polymorphisms; goats; susceptibility/resistance; transgenic mice
Published in Journal of infectious diseases 2015, DOI: 10.1093/infdis/jiv112.

Chapter 2 27
AUTHORS CONTRIBUTIONS
AGUILAR-CALVO, P., FAST, C., TAUSCHER, K., ESPINOSA, J-C., GROSCHUP, M. H.,
NADEEM, M., GOLDMANN, W., LANGEVELD, J., BOSSERS, A., ANDREOLETTI, O.
TORRES, J-M.
EFFECT of Q211 and K222 PRNP POLYMORPHIC VARIANTS IN THE SUSCEPTIBILTY
OF GOATS TO ORAL INFECTION WITH GOAT BOVINE SPONGIFORM
ENCEPHALOPATHY.
Published in The Journal of Infectious Diseases 2015, DOI: 10.1093/infdis/jiv112.
P Aguilar-Calvo and J.-C. Espinosa were involved in the study design, coordination of the
mouse bioassays, and drafted the manuscript.
C. Fast and K. Tauscher were involved in the study design, coordination of the goat
experiments, and drafted the manuscript.
M. H. Groschup was involved in the study design, the coordination of the goat experiments,
editing the manuscript, and in obtaining funding.
M. Nadeem has performed the immunohistochemistry and analyzed the data.
W. Goldmann was involved in the coordination of the study and in editing the manuscript.
J. Langeveld and A. Bossers were involved in in the study design, funding obtainment, and in
editing the manuscript.
O. Andreoletti was involved in the coordination of the study and in editing the manuscript.
J.-M. Torres was involved in the coordination of the animal experiments, coordination of
mouse bioassays, obtained funding, and edited the manuscript

28 Chapter 2

Chapter 3 29
Chapter 3

30 Chapter 3
Chapter 3 BSE INFECTION OF GOATS ALTERS
NEUROFILAMENT
PHOSPHORYLATION STATUS OF
SPINAL CORD AXONS
NADEEM, M., SPITZBARTH, I., HAIST, V., ROHN, K., TAUSCHER, K., ROHN, K.,
BOSSER, A., LANGEVELD, J., GROSCHUP, M.H., BAUMGÄRTNER, W., FAST, C.,
GERHAUSER, I.
Abstract
Transmissible spongiform encephalopathies (TSEs) represent a group of progressive diseases
that affect the nervous system of humans and various animal species. Recently reported
bovine spongiform encephalopathy (BSE) infections in goats in the United Kingdom and
France have brought small ruminant species into the focus of prion disease research.
Immunohistochemistry was performed to detect axonal cytoskeletal and transport
disturbances during the course of BSE in the spinal cord and autonomous ganglia of
experimentally infected goats. Interestingly, the present study demonstrated abnormal
expression of non-phosophorylated neurofilament (nNF) in axons of the white matter of the
spinal cord, which was restricted to goats with clinical BSE. The results indicate axonal
damage and disturbances in axonal transport during BSE in goats. However, whether
abnormal nNF accumulations are related to disturbed axonal transport mechanisms in TSEs,
the spread of TSE agents, and neuronal degeneration, has to be evaluated in future studies.
Moreover, the study reports for the first time that there is immunohistochemical evidence for
PrPSc
deposition in spinal cord white matter glial cells of BSE positive goats, indicating the
involvement of glial cells in the spread of the agent.
Submitted for publication

Chapter 3 31
AUTHORS CONTRIBUTIONS
NADEEM, M., SPITZBARTH, I., HAIST, V., ROHN, K., TAUSCHER, K., ROHN, K.,
BOSSER, A., LANGEVELD, J., GROSCHUP, M.H., BAUMGÄRTNER, W., FAST, C.,
GERHAUSER, I.
BSE INFECTION OF GOATS ALTERS NEUROFILAMENT PHOSPHORYLATION
STATUS OF SPINAL CORD AXONS.
Submitted for publication.
M. Nadeem performed immunohistochemistry and electron microscopy, analyzed the
data, and drafted the manuscript.
I. Spitzbarth was involved in the coordination of the immunohistochemical studies,
performed statistical analysis, and drafted the manuscript.
V. Haist was involved in the coordination of the ultrastructural studies and revised the
manuscript.
K. Rohn was involved in performing electron microscopy and generation of ultrastructural
photographs.
K. Tauscher performed the animal experiments and obtained tissue samples.
K. Rohn was involved in the statistical analysis of the data.
A. Bosser, J. Langeveld, and M. H. Groschup were involved in the study design, the
coordination of the animal experiments, revision of the manuscript, and in obtaining funding.
W. Baumgärtner was involved in the coordination of the immunohistochemical and
ultrastructural studies, edited the manuscript, and obtained parts of the funding.
C. Fast was involved in the study design, coordination of the animal experiments, the
conduction of immunohistochemistry, and edited the manuscript.
I. Gerhauser was involved in the coordination of the immunohistochemical studies,
performed statistical analyses, designed figures, and edited the manuscript.

32 Chapter 3
SHORT COMMUNICATION
BSE infection of goats alters the neurofilament phosphorylation status of spinal cord
axons
Muhammad Nadeem1,2,*
, Ingo Spitzbarth1,2,*
, Verena Haist1,
Kerstin Rohn1, Kerstin
Tauscher3, Karl Rohn
4, Alex Bossers
5, Jan Langeveld
5, Martin H. Groschup
3, Wolfgang
Baumgärtner1,2,#
, Christine Fast
3,+, Ingo Gerhauser
1,+
1 Department of Pathology, University of Veterinary Medicine, Hannover, Germany
2 Center for Systems Neuroscience, University of Veterinary Medicine, Hannover, Germany
3 Friedrich Loeffler Institute, Institute of Novel and Emerging Infectious Diseases,
Greifswald-Insel Riems, Germany
4 Department of Biometry, Epidemiology and Information Processing, University of
Veterinary Medicine, Hannover, Germany
5 Central Veterinary Institute, Wageningen UR, Lelystad, The Netherlands
* both authors contributed equally to this study and should be considered co-first authors
(first authors in alphabetical order)
+ both authors contributed equally to this study and should be considered co-last authors (last
authors in alphabetical order)
# Corresponding author:
Prof. Dr. Wolfgang Baumgärtner, Ph.D.
University of Veterinary Medicine Hannover, Department of Pathology
Bünteweg 17, D-30559 Hannover, Germany
Tel.: +49-(0)-511-953-8620; Fax: +49-(0)-511-953-8675
E-mail: [email protected]

Chapter 3 33
TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES (TSEs) including bovine
spongiform encephalopathy (BSE) are devastating neurodegenerative disorders caused by
conversion of the normal cellular prion protein (PrPC) into an abnormal isoform (PrP
Sc;
Prusiner, 1982; Chesebro, 2003). Following the discovery of two goat BSE cases in the UK
and France (Eloit and others 2005; Jeffery and others 2006) small ruminants were considered
to pose an BSE infection risk/source for cattle and humans in particular. In goats
experimental TSE susceptibility strongly depends on polymorphisms in the prion protein
gene (PRNP) (Aguilar-Calvo and others 2014; Aguilar-Calvo and others 2015). In particular,
goats with R/Q211 polymorphism (IQQ/IRQ) and Q/K222 polymorphism (IRK/IRQ) show
delayed or even absent clinical signs compared with wild-type goats (IRQ/IRQ) (Aguilar-
Calvo and others 2015). After oral infection of ruminants, the agent spreads via the
autonomous nervous system, ultimately resulting in manifest disease in the brain (Van
Keulen and others 2002; Hoffmann and others 2007; Kaatz and others 2012). However, the
cellular and molecular mechanisms that facilitate the spread of prions within the nervous
system and the involvement of the spinal cord in the pathogenesis of TSEs has been subjected
to little research so far, even though PrPSc
has been detected in the spinal cord (Flechsig and
others 2000; Fukuda and others 2012; Kaatz and others 2012).
The aim of the present study was to elucidate potential changes in the antigen expression of
various immunohistochemical markers in regions of the sympathetic infection route, which
have been demonstrated to represent key localizations for the spread of the BSE agent (Kaatz
and others 2012), including the celiac and mesenteric ganglion complex (CMGC) and the
spinal cord. Moreover, we sought to characterize ultrastructural changes in neurons of the
superior cervical ganglion (SCG).
Samples were collected during an oral BSE challenge (1g BSE-positive homogenized caprine
brain) and serial kill study of Alpine-Saanen-mixed breed goats approved by the local
authorities of the Federal State of Mecklenburg-Western Pomerania, Germany (LALLF
7221.3-2.5-001/05). The goats carried different PRNP genotypes with differing susceptibility.
Parts of the same population have been described in a previously published study (Aguilar-
Calvo and others, 2015; table 1). The animals were killed 6-45 months post infection (mpi;
table 1). Formalin-fixed tissue samples were incubated for 1 h in 98% formic acid prior to
paraffin embedding (Kaatz and others 2012). Three-micrometer sections were stained with
haematoxylin and eosin (HE).
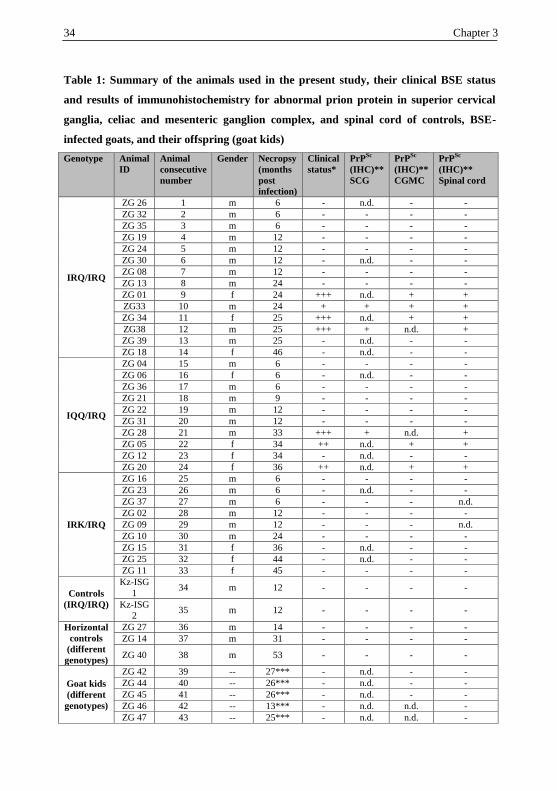
34 Chapter 3
Table 1: Summary of the animals used in the present study, their clinical BSE status
and results of immunohistochemistry for abnormal prion protein in superior cervical
ganglia, celiac and mesenteric ganglion complex, and spinal cord of controls, BSE-
infected goats, and their offspring (goat kids)
Genotype
Animal
ID
Animal
consecutive
number
Gender Necropsy
(months
post
infection)
Clinical
status*
PrPSc
(IHC)**
SCG
PrPSc
(IHC)**
CGMC
PrPSc
(IHC)**
Spinal cord
IRQ/IRQ
ZG 26 1 m 6 - n.d. - -
ZG 32 2 m 6 - - - -
ZG 35 3 m 6 - - - -
ZG 19 4 m 12 - - - -
ZG 24 5 m 12 - - - -
ZG 30 6 m 12 - n.d. - -
ZG 08 7 m 12 - - - -
ZG 13 8 m 24 - - - -
ZG 01 9 f 24 +++ n.d. + +
ZG33 10 m 24 + + + +
ZG 34 11 f 25 +++ n.d. + +
ZG38 12 m 25 +++ + n.d. +
ZG 39 13 m 25 - n.d. - -
ZG 18 14 f 46 - n.d. - -
IQQ/IRQ
ZG 04 15 m 6 - - - -
ZG 06 16 f 6 - n.d. - -
ZG 36 17 m 6 - - - -
ZG 21 18 m 9 - - - -
ZG 22 19 m 12 - - - -
ZG 31 20 m 12 - - - -
ZG 28 21 m 33 +++ + n.d. +
ZG 05 22 f 34 ++ n.d. + +
ZG 12 23 f 34 - n.d. - -
ZG 20 24 f 36 ++ n.d. + +
IRK/IRQ
ZG 16 25 m 6 - - - -
ZG 23 26 m 6 - n.d. - -
ZG 37 27 m 6 - - - n.d.
ZG 02 28 m 12 - - - -
ZG 09 29 m 12 - - - n.d.
ZG 10 30 m 24 - - - -
ZG 15 31 f 36 - n.d. - -
ZG 25 32 f 44 - n.d. - -
ZG 11 33 f 45 - - - -
Controls
(IRQ/IRQ)
Kz-ISG
1 34 m 12 - - - -
Kz-ISG
2 35 m 12 - - - -
Horizontal
controls
(different
genotypes)
ZG 27 36 m 14 - - - -
ZG 14 37 m 31 - - - -
ZG 40 38 m 53 - - - -
Goat kids
(different
genotypes)
ZG 42 39 -- 27*** - n.d. - -
ZG 44 40 -- 26*** - n.d. - -
ZG 45 41 -- 26*** - n.d. - -
ZG 46 42 -- 13*** - n.d. n.d. -
ZG 47 43 -- 25*** - n.d. n.d. -
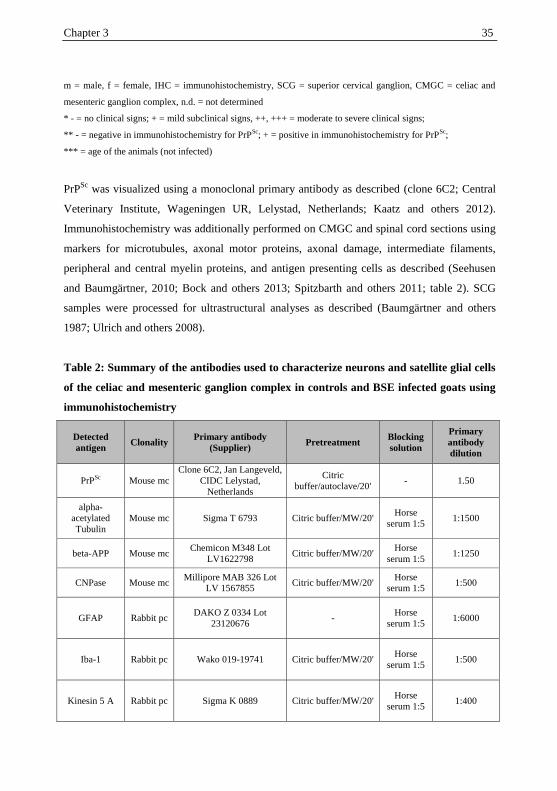
Chapter 3 35
m = male, f = female, IHC = immunohistochemistry, SCG = superior cervical ganglion, CMGC = celiac and
mesenteric ganglion complex, n.d. = not determined
* - = no clinical signs; + = mild subclinical signs, ++, +++ = moderate to severe clinical signs;
** - = negative in immunohistochemistry for PrPSc
; + = positive in immunohistochemistry for PrPSc
;
*** = age of the animals (not infected)
PrPSc
was visualized using a monoclonal primary antibody as described (clone 6C2; Central
Veterinary Institute, Wageningen UR, Lelystad, Netherlands; Kaatz and others 2012).
Immunohistochemistry was additionally performed on CMGC and spinal cord sections using
markers for microtubules, axonal motor proteins, axonal damage, intermediate filaments,
peripheral and central myelin proteins, and antigen presenting cells as described (Seehusen
and Baumgärtner, 2010; Bock and others 2013; Spitzbarth and others 2011; table 2). SCG
samples were processed for ultrastructural analyses as described (Baumgärtner and others
1987; Ulrich and others 2008).
Table 2: Summary of the antibodies used to characterize neurons and satellite glial cells
of the celiac and mesenteric ganglion complex in controls and BSE infected goats using
immunohistochemistry
Detected
antigen Clonality
Primary antibody
(Supplier) Pretreatment
Blocking
solution
Primary
antibody
dilution
PrPSc
Mouse mc
Clone 6C2, Jan Langeveld,
CIDC Lelystad,
Netherlands
Citric
buffer/autoclave/20' - 1.50
alpha-
acetylated
Tubulin
Mouse mc Sigma T 6793 Citric buffer/MW/20' Horse
serum 1:5 1:1500
beta-APP Mouse mc Chemicon M348 Lot
LV1622798 Citric buffer/MW/20'
Horse
serum 1:5 1:1250
CNPase Mouse mc Millipore MAB 326 Lot
LV 1567855 Citric buffer/MW/20'
Horse
serum 1:5 1:500
GFAP Rabbit pc DAKO Z 0334 Lot
23120676 -
Horse
serum 1:5 1:6000
Iba-1 Rabbit pc Wako 019-19741 Citric buffer/MW/20' Horse
serum 1:5 1:500
Kinesin 5 A Rabbit pc Sigma K 0889 Citric buffer/MW/20' Horse
serum 1:5 1:400
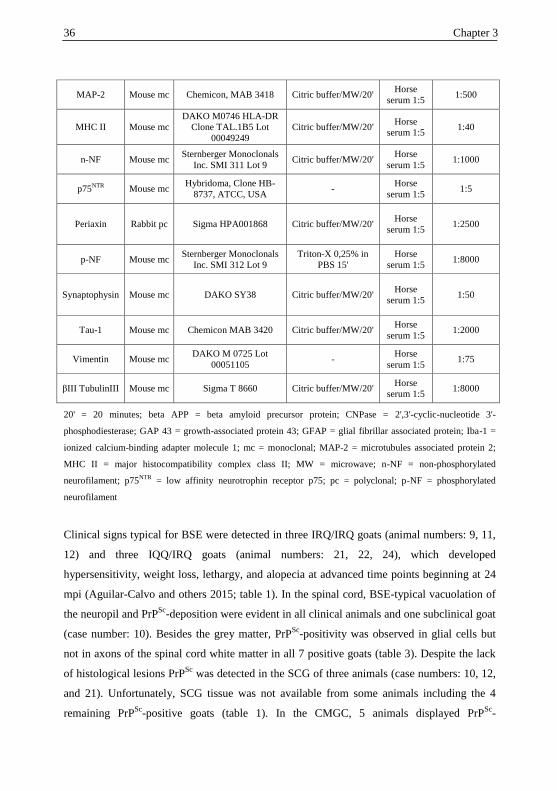
36 Chapter 3
MAP-2 Mouse mc Chemicon, MAB 3418 Citric buffer/MW/20' Horse
serum 1:5 1:500
MHC II Mouse mc
DAKO M0746 HLA-DR
Clone TAL.1B5 Lot
00049249
Citric buffer/MW/20' Horse
serum 1:5 1:40
n-NF Mouse mc Sternberger Monoclonals
Inc. SMI 311 Lot 9 Citric buffer/MW/20'
Horse
serum 1:5 1:1000
p75NTR
Mouse mc Hybridoma, Clone HB-
8737, ATCC, USA -
Horse
serum 1:5 1:5
Periaxin Rabbit pc Sigma HPA001868 Citric buffer/MW/20' Horse
serum 1:5 1:2500
p-NF Mouse mc Sternberger Monoclonals
Inc. SMI 312 Lot 9
Triton-X 0,25% in
PBS 15'
Horse
serum 1:5 1:8000
Synaptophysin Mouse mc DAKO SY38 Citric buffer/MW/20' Horse
serum 1:5 1:50
Tau-1 Mouse mc Chemicon MAB 3420 Citric buffer/MW/20' Horse
serum 1:5 1:2000
Vimentin Mouse mc DAKO M 0725 Lot
00051105 -
Horse
serum 1:5 1:75
βIII TubulinIII Mouse mc Sigma T 8660 Citric buffer/MW/20' Horse
serum 1:5 1:8000
20' = 20 minutes; beta APP = beta amyloid precursor protein; CNPase = 2',3'-cyclic-nucleotide 3'-
phosphodiesterase; GAP 43 = growth-associated protein 43; GFAP = glial fibrillar associated protein; Iba-1 =
ionized calcium-binding adapter molecule 1; mc = monoclonal; MAP-2 = microtubules associated protein 2;
MHC II = major histocompatibility complex class II; MW = microwave; n-NF = non-phosphorylated
neurofilament; p75NTR
= low affinity neurotrophin receptor p75; pc = polyclonal; p-NF = phosphorylated
neurofilament
Clinical signs typical for BSE were detected in three IRQ/IRQ goats (animal numbers: 9, 11,
12) and three IQQ/IRQ goats (animal numbers: 21, 22, 24), which developed
hypersensitivity, weight loss, lethargy, and alopecia at advanced time points beginning at 24
mpi (Aguilar-Calvo and others 2015; table 1). In the spinal cord, BSE-typical vacuolation of
the neuropil and PrPSc
-deposition were evident in all clinical animals and one subclinical goat
(case number: 10). Besides the grey matter, PrPSc
-positivity was observed in glial cells but
not in axons of the spinal cord white matter in all 7 positive goats (table 3). Despite the lack
of histological lesions PrPSc
was detected in the SCG of three animals (case numbers: 10, 12,
and 21). Unfortunately, SCG tissue was not available from some animals including the 4
remaining PrPSc
-positive goats (table 1). In the CMGC, 5 animals displayed PrPSc
-
Chapter 3 37
immunopositivity (case numbers: 9, 10, 11, 22, and 24) and three of them also neuronal
vacuolation (case numbers: 9, 11, and 22). No CGMC tissue was available in goats 12 and 21
(table 1). Strikingly, expression of non-phosphorylated neurofilament (n-NF) was not
detected in any spinal cord axon of the PrPSc
-negative animals, but was significantly up-
regulated in both swollen and normal appearing axons of the spinal cord white matter in 6
PrPSc
-positive animals (figure 1). In contrast, expression of all other investigated antigens
(table 2) was not influenced by the BSE status. Similarly, synapses were normal in SCG
neurons in both control and PrPSc
-positive goats at the ultrastructural level. Mitochondrial
swelling, cristolysis, and occasionally dilatation of rough endoplasmic reticulum was noted in
animals of each genotype, PrPSc
-status, and age, thus most probably indicating post-mortem
artifacts.
Table 3: Summary of the deposition of prion protein in spinal cord grey and white
matter of BSE-positive goats
Groups
(variants
of prion
genes)
Animal
consecutive
number
Animal
internal
number
PrPSc
deposition
grey matter
(IHC)
PrPSc
deposition white matter
(IHC)
Neuronal
vacuolation
ST AT DT Axons
IRQ/IRQ 9 ZG 01 +++ +++ +++ +++ - +++*
IRQ/IRQ 10 ZG 33 ++ + - - - +
IRQ/IRQ 11 ZG 34 +++ ++ ++ + - +
IRQ/IRQ 12 ZG 38 ++ + + + - +
IQQ/IRQ 21 ZG 28 +++ +++ +++ +++ - +
IQQ/IRQ 22 ZG 05 +++ + + + - ++*
IQQ/IRQ 24 ZG 20 +++ + - - - +++*
ST = spinal tract, AT = ascending tract, DT = descending tract,* = spongiform changes
+ - +++ mild to severe immunopositivity
The present study demonstrates for the first time that BSE infection of goats is associated
with glial PrPSc
-deposition in the spinal cord white matter along with unsuspected alterations
in the neurofilament phosphorylation status of axons. While absent in normal axons,
abnormal neurofilament expression has been shown in various neurologic diseases including
Creutzfeldt-Jakob disease (Gajdusek, 1985; Liberski and Budka, 1999; Tsunoda and
Fujinami, 2002; Seehusen and Baumgärtner, 2010; Bock and others 2013). Axonal
expression of n-NF in the present study is indicative of disturbances in axonal transport
processes. Underlining this hypothesis, some immunopositive axons had an increased
diameter (figure 1).
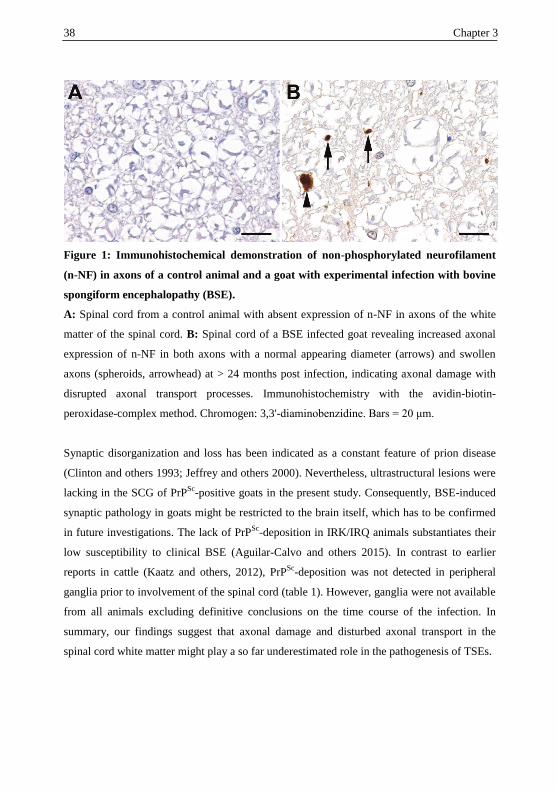
38 Chapter 3
Figure 1: Immunohistochemical demonstration of non-phosphorylated neurofilament
(n-NF) in axons of a control animal and a goat with experimental infection with bovine
spongiform encephalopathy (BSE).
A: Spinal cord from a control animal with absent expression of n-NF in axons of the white
matter of the spinal cord. B: Spinal cord of a BSE infected goat revealing increased axonal
expression of n-NF in both axons with a normal appearing diameter (arrows) and swollen
axons (spheroids, arrowhead) at > 24 months post infection, indicating axonal damage with
disrupted axonal transport processes. Immunohistochemistry with the avidin-biotin-
peroxidase-complex method. Chromogen: 3,3'-diaminobenzidine. Bars = 20 μm.
Synaptic disorganization and loss has been indicated as a constant feature of prion disease
(Clinton and others 1993; Jeffrey and others 2000). Nevertheless, ultrastructural lesions were
lacking in the SCG of PrPSc
-positive goats in the present study. Consequently, BSE-induced
synaptic pathology in goats might be restricted to the brain itself, which has to be confirmed
in future investigations. The lack of PrPSc
-deposition in IRK/IRQ animals substantiates their
low susceptibility to clinical BSE (Aguilar-Calvo and others 2015). In contrast to earlier
reports in cattle (Kaatz and others, 2012), PrPSc
-deposition was not detected in peripheral
ganglia prior to involvement of the spinal cord (table 1). However, ganglia were not available
from all animals excluding definitive conclusions on the time course of the infection. In
summary, our findings suggest that axonal damage and disturbed axonal transport in the
spinal cord white matter might play a so far underestimated role in the pathogenesis of TSEs.

Chapter 3 39
Acknowledgements
This study was in part supported by the Niedersachsen-Research Network on
Neuroinfectiology (N-RENNT) of the Ministry of Science and Culture of Lower Saxony and
in part by European Union projects (FOOD-CT-2006-36353, GoatBSE, and 219235 ERA-
NET EMIDA, GOAT-TSE-FREE). Muhammad Nadeem is thankful to Higher Education
Commission (HEC), Pakistan and Deutscher Akademischer Austauschdienst (DAAD),
Germany for providing a scholarship. The authors acknowledge the work of Dr. Susanne
Niedermeyer, Gesine Kreplin, Bettina Buck, Petra Grünig, Claudia Herrmann, Christiane
Namneck, Kerstin Schöne, Caroline Schütz, and Brigitte Behrens.
References
AGUILAR-CALVO, P., ESPINOSA, J.C., PINTADO, B., GUTIÉRREZ-ADÁN, A.,
ALAMILLO, E., MIRANDA, A., PRIETO, I., BOSSERS, A., ANDREOLETTI, O.
& TORRES, J.M. (2014) Role of the goat K222-PrPC polymorphic variant in prion
infection resistance. Journal of Virology 88, 2670-2676
AGUILAR-CALVO, P., FAST, C., TAUSCHER, K., ESPINOSA, J-C., GROSCHUP, M. H.,
NADEEM, M., GOLDMANN, W., LANGEVELD, J., BOSSERS, A.,
ANDREOLETTI, O. & TORRES, J-M. (2015) Effect of Q211 and K222PRNP
polymorphic variants in the susceptibility of goats to oral infection with Goat-BSE.
The Journal of Infectious Diseases 10, jiv112
BAUMGÄRTNER, W., KRAKOWKA, S. & BLAKESLEE, Z.R. (1987) Persistent infection
of Vero cells by paramyxoviruses a morphological and immunoelectron microscopic
investigation. Intervirology 27, 218-223
BOCK, P., SPITZBARTH, I., HAIST, V., STEIN, V.M., TIPOLD, A., PUFF, C., BEINEKE,
A. & BAUMGÄRTNER, W. (2013) Spatio-temporal development of axonopathy in
canine intervertebral disc disease as a translational large animal model for
nonexperimental spinal cord injury. Brain Pathology 23, 82-99
CHESEBRO, B. (2003) Introduction to the transmissible spongiform encephalopathies or
prion disease. British Medical Bulletin 66, 1-20
CLINTON, J., FORSYTH, C., ROYSTON, M.C. & ROBERTS, G.W. (1993) Synaptic
degeneration is the primary neuropathological feature in prion disease: a preliminary
study. Neuroreport 4, 65-68

40 Chapter 3
ELOIT, M., ADJOU, K., COULPIER, M., FONTAINE, J.J., HAMEL, R., LILIN, T.,
MESSIAEN, S., ANDREOLETTI, O., BARON, T., BENCSIK, A., BIACABE, A.G.,
BERINGUE, V., LAUDE, H., LE DUR, A., Vilotte, J.L., COMOY, E., DESLYS,
J.P., GRASSI, J., SIMON, S., LANTIER, F. & SARRADIN, P. (2005) BSE agent
signatures in a goat. The Veterinary Record 16, 523-524
FLECHSIG, E.D., SHMERLING, I., HEGYI, A., RAEBER, J., FISCHER, M., FISCHER,
M., COZZIO, A., von MERING, C., AGUZZI, A. & WEISSMANN C (2000) Prion
protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP
knockout mice. Neuron 27, 399-408
FUKUDA, S., ONOE, S., NIKAIDO, S., FUJII, K., KAGEYAMA, S., IWAMARU, Y.,
IMAMURA, M., MASUJIN, K., MATSUURA, Y., SHIMIZU, Y., KASAI, K.,
YOSHIOKA, M., MURAYAMA, Y., MOHRI, S., YOKOYAMA, T. & OKADA, H.
(2012) Neuroanatomical distribution of disease-associated prion protein in
experimental bovine spongiform encephalopathy in cattle after intracerebral
inoculation. The Journal of Infectious Diseases 65, 37-44
GAJDUSEK, D.C. (1985) Hypothesis: interference with axonal transport of neurofilaments
as a common pathogenetic mechanism in certain diseases of the central nervous
system. New England Journal of Medicine 312, 714-719
HOFFMANN, C., ZIEGLER, U., BUSCHMANN, A., WEBER, A., KUPFER, L.,
OELSCHLEGEL, A., HAMMERSCHMIDT, B. & GROSCHUP, M.H. (2007) Prions
spread via the autonomic nervous system from the gut to the central nervous system in
cattle incubating bovine spongiform encephalopathy. Journal of General Virology 88,
1048-1055
JEFFREY, M., HALLIDAY, W.G., BELL, J., JOHNSTON, A.R., MACLEOD, N.K.,
INGHAM, C., SAYERS, A.R., BROWN, D.A. & FRASER, J.R. (2000) Synapse loss
associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected
murine hippocampus. Neuropathology and Applied Neurobiology 26, 41-54
KAATZ, M., FAST, C., ZIEGLER, U., BALKEMA-BUSCHMANN, A.,
HAMMERSCHMIDT, B., KELLER, M., OELSCHLEGEL, A., MCINTYRE, L. &
GROSCHUP, M.H. (2012) Spread of classical BSE prions from the gut via the
peripheral nervous system to the Brain. Immunopathology and Infectious Diseases
181, 515-524

Chapter 3 41
LIBERSKI, P.P. & BUDKA, H. (1999) Neuroaxonal pathology in Creutzfeldt-Jakob disease.
Acta Neuropathologica 97, 329-334
MEDRANO, A.Z., BARMADA, S.J., BIASINI, E. & HARRIS, D.A. (2008) GFP-tagged
mutant prion protein forms intra-axonal aggregates in transgenic mice. Neurobiology
of Disease 31, 20-32
PRUSINER, S.B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216,
136-144
SEEHUSEN, F. & BAUMGÄRTNER, W. (2010) Axonal pathology and loss precede
demyelination and accompany chronic lesions in a spontaneously occurring animal
model of multiple sclerosis. Brain Pathology 20, 551-559
SPITZBARTH, I., BOCK, P., HAIST, V., STEIN, V.M., TIPOLD, A., WEWETZER, K.,
BAUMGÄRTNER, W. & BEINEKE, A. (2011) Prominent microglial activation in
the early proinflammatory immune response in naturally occurring canine spinal cord
injury. Journal of Neuropathology and Experimental Neurology 70,703-714
TSUNODA, I. & FUJINAMI, R.S. (2002) Inside-Out versus Outside-In models for virus
induced demyelination: axonal damage triggering demyelination. Springer Seminars
in Immunopathology 24, 105-125
ULRICH, R., SEELIGER, F., KREUTZER, M., GERMANN, P.G. & BAUMGÄRTNER, W.
(2008) Limited remyelination in Theiler's murine encephalomyelitis due to
insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)-positive
putative oligodendroglial progenitor cells. Neuropathology and Applied Neurobiology
34, 603-620
van KEULEN, L.J., VROMANS, M.E. & van ZIJDERVELD, F.G. (2002) Early and late
pathogenesis of natural scrapie infection in sheep. APMIS.: Acta pathologica,
microbiologica, et immunologica Scandinavica 110, 23-32

42 Chapter 3

Chapter 4 - Discussion 43
Chapter 4
Discussion

44 Chapter 4 - Discussion
Chapter 4: Discussion
Transmissible spongiform encephalopathies (TSEs) including bovine spongiform
encephalopathy (BSE) and Creutzfeldt-Jakob disease are fatal neurodegenerative diseases
caused by the abnormal isoform (PrPSc
) of the host encoded normal cellular glycoprotein
(PrPC) resulting in motor dysfunctions, dementia, spongiosis, astrogliosis, and neuronal loss
(PRUSINER, 1982; CHESEBRO, 2003; ERMOLAYEV et al., 2009). Though intracerebral
inoculation is the most efficient route for the propagation of TSE agents, the natural route of
exposure appears to be oral (WILLIAMS and MILLER, 2003). Recent studies strongly
support the importance of the lymphoreticular system as well as the peripheral nervous
system (PNS) for neuroinvasion (KLEIN et al., 1997; KÜNZI et al., 2002, KAATZ et al.,
2012). In addition, the sympathetic nervous system seems to play a major role in the initial
neural distribution (KAATZ et al., 2012). In cattle, the sympathetic route of BSE spread
consists of the celiac and mesenteric ganglion complex (CMGC), superior cervical ganglion,
splanchnic nerves, and the intermediolateral cell column of the thoracic spinal cord (KAATZ
et al., 2012). In recent years comprehensive research activities have achieved significant
progress in dissecting the spreading pathways of prion proteins of different TSEs, especially
BSE and scrapie, in naturally as well as in experimentally challenged animals via the oral
route. However, the exact subcellular mechanisms of transport and spread of the agent are
enigmatic so far. A detailed knowledge of prion dissemination in the nervous system might
not only specify new approaches for interfering with prion spread in the PNS, but might also
help to identify therapeutic targets to prevent disease progression.
The onset and distribution of cattle BSE disease specific prion protein accumulations in sheep
and goats are strongly influenced by the genetic predisposition mediated trough the gene
encoding the prion protein (AGUILAR-CALVO et al., 2014; McGOVERN et al., 2015). In

Chapter 4 - Discussion 45
particular, the risk of disease development by prion infection is dependent on the
polymorphism of the prion protein gene (PRNP). This has been observed in sheep with
different PRNP genotypes orally infected with cattle-derived BSE agent (McGOVERN et al.,
2015) and in transgenic mice expressing either the Q222 (wild type) or the K222 variant of goat
PrPC intracerebrally infected with the goat scrapie agent (AGUILAR-CALVO et al., 2014).
AGUILAR-CALVO et al. (2014) described that the K222 PRNP variant might affect PrPC-
PrPSc
interaction and the conversion rate of PrPSc
by the scrapie agent, ultimately resulting in
a delayed incubation period and/or lower susceptibility of Q/K222 compared to wild type-PrP
transgenic mice. The present study aimed to clarify whether the PRNP genotype also affects
the susceptibility of goats to the BSE agent. To achieve this goal, goats harboring different
PRNP genotypes including IRQ/IRQ (Wild type, abbreviated as WT), IQQ/IRQ (abbreviated
as R/Q211), and IRK/IRQ (abbreviated as Q/K222) were orally infected with caprine BSE.
WT goats developed the first clinical signs of TSE 24 months post infection (mpi), which
were consistent with clinical signs previously reported in sheep and goats infected with BSE
(FOSTER et al., 2001a; van KEULEN et al., 2008a). PrPSc
deposits and neurodegenerative
lesions were mainly detected at the level of the obex region in the brain of goats similar to
previous observations in BSE-infected sheep and goats (FOSTER et al., 2001b; GONZÁLEZ
et al., 2005; LEZMI et al., 2011). Prion infectivity, but with lower titers than in the brain, was
also detected in muscles and lymph node tissues, which is consistent with previous findings
in sheep and cattle orally infected with BSE (BUSCHMANN and GROSCHUP, 2005; van
KEULEN et al., 2008b).
In R/Q211 animals, PrPSc
infectivity was higher in brain as compared to peripheral tissue, thus
strongly supporting the results observed in WT goats. However, in contrast to WT goats
R/Q211 animals presented a delayed clinical onset of the disease, starting at 33 mpi, thus
indicating that the PRNP polymorphic variant Q211 might have an influence on the incubation

46 Chapter 4 - Discussion
period of BSE, but not on the general susceptibility. Therefore, R/Q211 polymorphism could
be linked with low resistance against goat BSE compared to the high resistance of this
genotype to scrapie as demonstrated in other field and experimental studies using goats
(BOUZALAS et al., 2010; CORBIÈRE et al., 2013; LACROUX et al., 2014). Consequently,
the prion strain represents a determining factor for TSE occurrence (AGUZZI et al., 2007).
In contrast to the similar susceptibility of WT and R/Q211 goats to goat BSE, none of the
Q/K222 goats examined in this study presented any evidence of disease. In all Q/K222 goats
neither clinical signs nor PrPSc
deposits were observed. Histopathological exanimation also
failed to demonstrate any neuropathological lesions. However, very low infectivity was
detected in the brain of 1 of the 2 Q/K222 goats euthanized at 44-45 mpi, suggesting that K222
PRNP variant drastically decreases the susceptibility of goats to goat BSE.
In cattle, the BSE agent is mainly deposited in the CNS and vegetative nervous system
(BUSCHMANN and GROSCHUP, 2005; KAATZ et al., 2012), whereas it is lymphotropic in
sheep and goats with PrPSc
deposits in several lymphoid tissues (FOSTER et al., 1996;
ANDRÉOLOTTI et al., 2000; BELLWORTHY et al., 2005; KUJALA et al., 2011). In
accordance with this, the present study indicates that the resistance of Q/K222 goats to oral
BSE infection might depend on a lesser capability of goat BSE to alter K222-PrPC in
peripheral tissues. In addition, lesser expression of K222-PrPC
along with a local inconsistency
of the K222-PrPC
isoforms in the structures involved in the major spreading route cannot be
excluded (BERINGUE et al., 2003). In fact, discrepancies in the susceptibility to prion agents
dependent on the inoculation route have been described earlier in R/H154, R/Q211, or Q/K222
goats, which showed complete resistance to the orally administered scrapie agent but were
susceptible to the scrapie agent after intracerebral inoculation (LACROUX et al., 2014).
R/H154 and R/Q211 goats presented 100% susceptibility to intracerebral inoculation of scrapie,
whereas only 2 out of 5 Q/K222 (40%) goats developed clinical disease with 4-5 times longer

Chapter 4 - Discussion 47
incubation periods compared to WT goats, indicating that the K222 variant of PRNP is
associated with less susceptibility of goats to scrapie infection compared to the Q211 variant
(LACROUX et al., 2014). Moreover, our data together with other data from several
experimental (ACUTIS et al., 2012; AGUILAR-CALVO et al., 2014; LACROUX et al.,
2014) and epidemiological studies (ACUTIS et al., 2006; VACCARI et al., 2006;
BARILLET et al., 2009; BOUZALAS et al., 2010; FRAGKIADAKI et al., 2011;
PAPASAVVA-STYLIANOU et al., 2011; ACIN et al., 2013; CORBIÈRE et al., 2013)
emphasize the low susceptibility of the Q/K222 polymorphism to TSE infection.
Consequently, the K222 variant might represent an interesting target to improve breeding
programs in order to control and ultimately eradicate TSE in goats.
The present study also aimed to characterize the role of the spinal cord and autonomic
ganglia in the spread of prion agents in goats. Immunohistochemistry was performed on the
spinal cord and celiac and mesenteric ganglion complex of goats from different genotypic
groups (WT, R/Q211and Q/K222), orally infected with caprine BSE. In addition, synapses were
visualized in the superior cervical ganglion using transmission electron microscopy. WT and
R/Q211 goats exhibited prion protein deposition in the spinal cord, whereas all Q/K222 goats
were negative indicating resistance or lower susceptibility to BSE, and thus substantiating the
previous study (AGUILAR-CALVO et al., 2014). PrPSc
positive goats demonstrated a strong
intra- and perineuronal staining of spinal cord grey matter neurons and glial cells with linear
staining of neuronal processes consistent with previously described prion protein expression
patterns in BSE infection (FUKUDA et al., 2012; VIDAL et al., 2006; JEFFREY and
GONZÁLEZ, 2007). PrPSc
deposition was also observed in spinal cord white matter glial
cells, which was irrespective of ascending or descending tracts, but not in white matter axons.
The present study is the first report describing PrPSc
deposition in spinal cord white matter of
BSE infected goats.

48 Chapter 4 - Discussion
TSEs target neurons and ultimately lead to neuronal loss. However, the role of glial cells in
the uptake, spread, and propagation of prion agents in the CNS during the course of prion
disease has received little attention so far. Previous studies have already suggested that glial
cells, especially astrocytes, may play a key role in the disease process (RAEBER et al., 1997:
MALLUCCI et al., 2003; CRONIER et al., 2004; PRITZKOW et al., 2011, HOLLISTER et
al., 2015). In 263K infected hamsters with clinical signs, abnormal prion agent was detected
in a high proportion of hippocampal astrocytes (YE et al., 1998). In sheep naturally infected
with the scrapie agent, PrPSc
even accumulates to higher levels in astrocytes and other glial
cells than in neurons (van KEULEN et al., 1995; GONZÁLEZ et al., 2002; GONZÁLEZ et
al., 2003). PrPSc
in glial cells has also been observed as early as 8 weeks after intracerebral or
cerebellar inoculation of the scrapie agent in mice, thus suggesting that glial cells, and here
mainly astrocytes, play a pivotal role in the spread of the infection (DIEDRICH et al., 1991).
However, the question how PrPSc
spreads between glial cells and neurons remains unsolved.
Recent in vitro studies of scrapie infected hamster neurons demonstrated that PrPSc
is
transported to late endosomal/lysosomal compartments and throughout cell bodies and
processes of astrocytes and neurons including their contacts (HOLLISTER et al., 2015).
Consequently, glial cells might facilitate the dissemination of PrPSc
towards the CNS and
from the CNS to peripheral tissues.
The PNS seems to be deeply involved in the spread of prion agents from the gut to the CNS
(KAATZ et al., 2012). Consequently, the potential neuropathological effects of goat BSE on
different neuronal, axonal, and satellite glial cell markers was investigated in the celiac and
mesenteric ganglion complex and the spinal cord. However, the PrPSc
status (BSE-positive
vs. BSE-negative goats) did not influence the expression of acetylated α tubulin, βIII tubulin,
β-APP, CNPase, GFAP, Iba1, Kinesin 5A, MAP2, MHC class II, p75NTR
, periaxin,
synaptophysin, pNF, Tau1, and Vimentin by neurons, glial cells, and axons. In addition, the

Chapter 4 - Discussion 49
ultrastructural examination of the superior cervical ganglion did not reveal any significant
changes in cytoplasmic organelles of neurons including mitochondria and the Golgi complex
as well as synapses, which is in contrast to previous findings in CNS lesions of TSEs in mice
(JEFFREY et al., 2000; CUNNINGHAM et al., 2003; SISKOVA et al., 2009). Consequently,
synaptic pathology in goats caused by the BSE agent might be restricted to the CNS itself;
however, this needs to be confirmed in future investigations.
Neurofilaments are a major component of the neuronal cytoskeleton, provide shape and
architecture to axons, and are involved in the relay of signals from the plasma membrane to
the nucleus (TOIVOLA et al., 2005). They are synthesized within neuronal perikarya and
transported to axons via slow axonal transport (MILLER et al., 2002). Within axons
neurofilaments are extensively phosphorylated. Interestingly, the phosphorylation of
neurofilaments seems to be a mechanism for regulating their transport properties, possibly by
promoting their detachment from motor proteins (MILLER et al., 2002). An interference of
neurofilaments with axonal transport has been hypothesized for a long time as a common
pathogenetic mechanism in several CNS diseases (GAJDUSEK, 1985). Similarly,
neuroaxonal pathology, characterized by neuritic swellings and dystrophic neurites seems to
play an important role in TSEs (LIBERSKI and BUDKA, 1999). The present study
documented for the first time that there is abnormal expression of nNF in spinal cord white
matter axons of PrPSc
-positive goats. The cause for the impaired phosphorylation of
neurofilaments yet remains to be elucidated. Nevertheless, nNF accumulations are likely to
compromise axonal transport. However, the influence of nNF on axonal transport
mechanisms and its potential role in the spread of TSE agents as well as neuronal
degeneration needs to be investigated in detail in future studies.

50

Chapter 5 - Summary 51
Chapter 5
Summary

52 Chapter 5 - Summary
Chapter 5: Summary
Susceptibility of goats to the BSE agent with special emphasis on the neuropathogenesis
Muhammad Nadeem
Transmissible spongiform encephalopathies (TSEs) represent a group of progressive diseases
that affect the nervous system of humans and various animal species. The characteristic
neuropathological features of TSEs comprise spongiform changes, astrogliosis, neuronal loss,
and accumulation of an abnormal isoform (PrPSc
) of the host-encoded cellular prion protein
(PrPC). Among animal diseases, bovine spongiform encephalopathy (BSE) is of particular
interest because of its transmissibility to humans causing a variant of Creutzfeldt-Jakob-
disease (vCJD). Recently reported BSE infections in goats in the United Kingdom and France
have brought small ruminant species into the focus of prion disease research. Among TSEs
scrapie affects small ruminants all around the world and its incidence is intensely modified by
the PRNP genotype. In goats several PRNP polymorphisms have been linked to lower
susceptibility to classical scrapie. Among them, the K222 PRNP variant produced most
favorable results, being related with resistance to scrapie in both epidemiological studies as
well in experimental infections in goats and transgenic mice. In contrast, little is known about
the factors defining the risk of goats to BSE.
Studies on orally infected TSEs strongly support the importance of the lymphoreticular
system as well as the peripheral nervous system (PNS) for neuroinvasion. Recent studies in
hamsters, sheep, and cattle demonstrated that TSE agents spread via the sympathetic and/or
parasympathetic nervous system to the central nervous system (CNS). However, the
sympathetic route consisting of the celiac and mesenteric ganglion complex (CMGC), the
superior cervical ganglion, splanchnic nerves, and the intermediolateral cell column of the

Chapter 5 - Summary 53
thoracic spinal cord seems to play the major role in the initial neuronal distribution in orally
BSE-infected cattle. The cellular and molecular constituents and mechanisms that facilitate
the centripetal spread of prions remain undetermined. Principally, prion proteins could use
axonal transport systems, spread cell-free or cell-associated in neural interspaces, sequentially
infect Schwann cells, and convert normal cellular prion protein to abnormal isoforms. The
sequence of events important for the clinical onset of the prion disease and knowledge of the
role of different areas of the CNS and PNS is still fragmentary.
Thus, the present study aimed to clarify the effect of certain PRNP genotypes in the oral
transmission of the goat BSE agent to goats. Immunohistochemistry was performed to detect
axonal cytoskeletal and transport disturbances during the course of BSE in the spinal cord
and peripheral tissues of experimentally infected goats and to elucidate possible pathogenetic
links between ultrastructural changes and PrPSc
accumulation in the superior cervical
ganglion of experimentally BSE-infected goats. Tissues from CNS and PNS from different
genotypes (IRQ/IRQ, IQQ/IRQ and IRK/IRQ) of control and BSE-infected goats were
investigated. IRQ/IRQ wild-type goats presented clinical signs after 24 months post infection
(mpi), while IQQ/IRQ (R/Q211) developed first clinical onset of disease at 33 mpi. In contrast
to the BSE susceptibility of IRQ/IRQ and IQQ/IRQ goats, none of IRK/IRQ (R/K222) goats
presented clinical signs or PrPSc
deposition with neuropathological lesions. However, very
low infectivity was noticed in the brain in 1 of 2 IRK/IRQ goats slaughtered at 44-45 mpi.
The data indicate that IRK/IRQ goats are less susceptible to the transmission of goat BSE
than IRQ/IRQ and IQQ/IRQ, highlighting that the K222 PRNP variant can protect goats
against BSE oral infection. In conclusion, goat BSE agent transmission is genotype-
dependent and the K222 PRNP variant represents a good candidate for better breeding
programs in order to control and eradicate TSEs in goats.

54 Chapter 5 - Summary
The present study did not detect any changes in the expression of various antigens (acetylated
α tubulin, βIII tubulin, β-APP, CNPase, GFAP, Iba1, Kinesin 5A, MAP2, MHC class II,
p75NTR
, periaxin, synaptophysin, pNF, Tau1, and vimentin ) in the spinal cord or the CMGC
of BSE positive compared to negative goats. Moreover, the superior cervical ganglion did not
exhibit ultrastructural changes, which might indicate that synaptic pathology is restricted to
the CNS itself. However, this needs to be confirmed in future studies.
Interestingly, the present study demonstrated abnormal expression of non-phosophorylated
neurofilament (nNF) in axons of the white matter of the spinal cord, which was restricted to
goats with clinical BSE. The results indicate axonal damage and disturbances in axonal
transport during BSE in goats. However, whether abnormal nNF accumulations are related to
disturbed axonal transport mechanisms in TSEs, the spread of TSE agents, and neuronal
degeneration, has to be evaluated in future studies. Moreover, the study reports for the first
time that there is immunohistochemical evidence for PrPSc
deposition in spinal cord white
matter glial cells of BSE positive goats, indicating the involvement of glial cells in the spread
of the agent.

Chapter 6 – Zusammenfassung 55
Chapter 6
Zusammenfassung

56 Chapter 6 - Zusammenfassung
Chapter 6: Zusammenfassung
Die Empfänglichkeit von Ziegen gegenüber BSE unter besonderer Berücksichtigung
der Neuropathogenese
Muhammad Nadeem
Transmissible spongiforme Enzephalopathien (TSEs) stellen eine Gruppe progressiver
Erkrankungen des Nervensystems von Tieren und Menschen dar, deren Neuropathologie
typischerweise durch spongiforme Veränderungen in der grauen Substanz, eine Astrogliose
sowie einen neuronalen Verlust charakterisiert ist. Im Hinblick auf die Pathogenese werden
diese Alterationen durch die Akkumulation einer abnormalen und infektiösen Isoform (PrPSc
)
des von Wirtszellen gebildeten Prionproteins (PrPC) initiiert. Seitdem die Übertragbarkeit der
bovinen spongiformen Enzephalopathie (BSE) auf den Menschen in Form einer Variante der
Creutzfeldt-Jakob-Krankheit (vCJK) bekannt ist, besteht ein großes, öffentliches Interesse an
der Erforschung dieser Erkrankung. Neben dem Rind besteht in der jüngsten Zeit zunehmend
ein wissenschaftliches Interesse an der Erforschung von BSE bei kleinen Wiederkäuern.
Beispielsweise wurde im Vereinigten Königreich und Frankreich eine Infektion von Ziegen
mit BSE nachgewiesen.
Unter den verschiedenen TSEs stellt die Scrapie (Traberkrankheit) die wichtigste Erkrankung
der kleinen Wiederkäuer dar, deren Ausprägung stark vom Genotyp des für das zelluläre
Prionprotein kodierenden Gens (PRNP) abhängt. Bei Ziegen wurden verschiedene PRNP-
Polymorphismen beschrieben, die mit einer differierenden Empfänglichkeit für die klassische
Scrapie einhergehen. Hierbei wiesen mehrere epidemiologische Studien, experimentelle
Infektionen von Ziegen sowie Untersuchungen an transgenen Mäusen auf den positiven

Chapter 6 – Zusammenfassung 57
Einfluss der K222 PRNP-Variante in Bezug auf die Widerstandsfähigkeit gegenüber der
Scrapie-Infektion hin. Dagegen ist der potenzielle Einfluss des PRNP-Genotyps auf eine
BSE-Erkrankung bei Ziegen bisher unbekannt.
Mehrere Studien an oral mit Prionen infizierten Tieren zeigten die große Bedeutung des
lymphoretikulären Systems und des peripheren Nervensystems (PNS) für die Neuroinvasion
des Agens. So gelangen TSE-Erreger bei Hamstern, Schafen und Rindern entlang des
sympathischen und parasympathischen Nervensystems in das zentrale Nervensystem (ZNS).
Bei oral mit BSE infizierten Rindern scheint dem sympathischen Nervensystem die größte
Rolle für die initiale Ausbreitung des Prion-Agens zuzukommen. Hierbei betrifft bei Rindern
eine Prionenausbreitung entlang des sympathischen Nervensystems die Ganglia coeliacum
und mesentericum craniale (GCMC), das Ganglion cervicale craniale, die Nervi splanchnici
und die Substantia intermedia im thorakolumbalen Bereich des Rückenmarks. Die zellulären
und molekularen Komponenten und Mechanismen, die an der zentripetalen Ausbreitung von
Prionen beteiligt sind, sind bisher weitgehend unbekannt. Hypothetisch könnten
Prionenproteine axonale Transportsysteme benutzen, sich im interneuralen Raum zellfrei und
zellgebunden ausbreiten, sequentiell Schwannzellen infizieren, um somit final die
Konformationsänderung des unveränderten zellulären Prionproteins in die abnormale Isoform
zu induzieren. Dennoch bleiben der genaue Zeitverlauf und die Rolle der verschiedenen
Regionen im ZNS und PNS für das klinische Auftreten einer Prionenerkrankung immer noch
unklar.
Die Ziele dieser Studie bestanden darin, den Einfluss bestimmter PRNP-Genotypen auf die
orale Empfänglichkeit für BSE bei Ziegen aufzuklären sowie pathologische Veränderungen
in ausgewählten Lokalisationen des PNS und im Rückenmark zu charakterisieren. Hierfür
wurde eine immunhistologische Untersuchung bei experimentell infizierten Ziegen
durchgeführt, um potenzielle Schäden am axonalen Zytoskelett und Störungen des axonalen

58 Chapter 6 - Zusammenfassung
Transports aufzudecken. Außerdem sollte geklärt werden, inwieweit es einen
pathogenetischen Zusammenhang zwischen ultrastrukturellen Veränderungen und PrPSc
-
Ablagerungen im Ganglion cervicale craniale bei experimentell mit BSE-infizierten Ziegen
gibt. Für diese Untersuchungen wurden verschiedene Gewebe des ZNS und PNS von
Kontrollziegen und BSE-infizierten Ziegen verschiedener Genotypen (IRQ/IRQ, IQQ/IRQ
und IRK/IRQ) entnommen. Die IRQ/IRQ-Wildtyp-Ziegen zeigten die ersten klinischen
Symptome 24 Monate post infectionem (p.i.), während diese bei IQQ/IRQ (R/Q211)-Ziegen
erst 33 Monate p.i. auftraten. Im Gegensatz zu den IRQ/IRQ- und IQQ/IRQ-Ziegen wurden
bei IRK/IRQ (R/K222)-Ziegen weder klinische Symptome noch PrPSc
-Ablagerungen mit
neuropathologischen Veränderungen festgestellt. Allerdings wies das Gehirn einer IRK/IRQ-
Ziege, welche 44 Monate p.i. getötet wurde, eine sehr geringe Infektiosität auf. Diese
Ergebnisse deuten auf eine geringere BSE-Empfänglichkeit von IRK/IRQ-Ziegen gegenüber
IRQ/IRQ- und IQQ/IRQ-Ziegen hin, so dass die K222 PRNP-Variante Ziegen möglicherweise
vor einer oralen BSE-Infektion schützen kann. Somit stellt die gezielte Zucht der K222 PRNP-
Variante möglicherweise eine geeignete Möglichkeit dar, natürliche Fälle von TSE bei
Ziegen auszumerzen.
In der vorliegenden Studie wurde die Expression zahlreicher Antigene (azetyliertes α-
Tubulin, βIII Tubulin, β-APP, CNPase, GFAP, Iba1, Kinesin 5A, MAP2, MHC class II,
p75NTR
, Periaxin, Synaptophysin, pNF, Tau1 und Vimentin) im Rückenmark und GCMC
immunhistologisch untersucht. Allerdings fanden sich keine Unterschiede in der
Antigenexpression zwischen BSE-positiven und -negativen Ziegen. Darüber hinaus zeigten
sich keine ultrastrukturellen Synapsenveränderungen im Ganglion cervicale craniale. Ob
Synapsenveränderungen bei Prionenerkrankungen auf das ZNS beschränkt sind, muss jedoch
in weiteren Studien bestätigt werden. Interessanterweise zeigte die vorliegende Studie eine
abnormale Expression des nicht-phosphorylierten Neurofilaments (nNF) in Axonen der

Chapter 6 – Zusammenfassung 59
weißen Substanz des Rückenmarks, welche auf klinisch an BSE erkrankte und PrPSc
-positive
Ziegen beschränkt war. Somit treten wahrscheinlich im Verlauf der BSE-Erkrankung von
Ziegen axonale Schäden und Störungen des axonalen Transports auf. Allerdings muss in
zukünftigen Untersuchungen abgeklärt werden, ob die abnormale Akkumulation von nNF in
einem kausalen Zusammenhang mit gestörten Transportmechanismen im Axon, der
Ausbreitung von Prionen und/oder einer neuronalen Degeneration steht. Des Weiteren wurde
mittels der durchgeführten immunhistologischen Untersuchungen erstmalig eine Ablagerung
von PrPSc
in Gliazellen der weißen Substanz des Rückenmarks bei an BSE erkrankten Ziegen
nachgewiesen, so dass Gliazellen möglicherweise eine wichtige Rolle in der Ausbreitung von
Prionen spielen.

60 Chapter 6 - Zusammenfassung

Chapter 7- References 61
Chapter 7
References

62 Chapter 7 - Reference
Chapter 7: References
ACIN, C., MARTIN-BURRIEL, I., MONLEON, E., LYAHYAI, J., PITARCH, J.L., SERRANO, C.,
MONZÓN, M., ZARAGOZA, P., and BADIOLA, J.J. (2013):
Prion protein gene variability in Spanish goats. inference through susceptibility to classical scrapie strains and
pathogenic distribution of peripheral PrPsc.
PLoS One. 8, e61118.
ACUTIS, P.L., BOSSERS, A., PRIEM, J., RIINA, M.V., PELETTO, S., MAZZA, M., CASALONE, C.,
FORLONI, G., RU, G., and CARAMELLI, M. (2006):
Identification of prion protein gene polymorphisms in goats from Italian scrapie outbreaks.
J. Gen. Virol. 87, 1029-1033.
ACUTIS, P.L., MARTUCCI, F., D’ANGELO, A., PELETTO, S., COLUSSI, S., MAURELLA, C.,
PORCARIO, C., IULINI, B., MAZZA, M., DELL'ATTI, L., ZUCCON, F., CORONA, C.,
MARTINELLI, N., CASALONE, C., CARAMELLI, M., and LOMBARDI, G. (2012):
Resistance to classical scrapie in experimentally challenged goats carrying mutation K222 of the prion protein
gene.
Vet. Res. 43, 8.
AGUILAR-CALVO, P., ESPINOSA, J.C., PINTADO, B., GUTIÉRREZ-ADÁN, A., ALAMILLO, E.,
MIRANDA, A., PRIETO, I., BOSSERS, A., ANDREOLETTI, O., and TORRES, J.M. (2014):
Role of the goat K222-PrPC polymorphic variant in prion infection resistance.
J. Virol. 88, 2670-2676.
AGUZZI, A., and POLYMENIDOU, M. (2004):
Mammalian prion biology: one century of evolving concepts.
Cell 116, 313-327.
AGUZZI, A., BAUMANN, F., and BREMER, J. (2008):
The prion’s elusive reason of being.
Annu. Rev. Neurosci. 341, 439-477.
AGUZZI, A., HEIKENWALDER, M., and POLYMENIDOU, M. (2007):
Insights into prion strains and neurotoxicity.
Nat. Rev. Mol. Cell Biol. 8, 552-561.
ALMER, G., VUKOSAVIC, S., ROMERO, N., and PRZEDBORSKI, S. (1999):

Chapter 7- References 63
Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral
sclerosis.
J. Neurochem. 72, 2415-2425.
ALPER, T., Haig, D.A., and Clarke, M.C. (1966):
The exceptionally small size of the scrapie agent.
Biochem. Biophys. Res. Commun. 22, 278-284.
ANDRÉOLOTTI, O. (2004):
PrPSc
Immunohistochemistry. In: Methods and tools in biosciences and medicine—techniques in prion research,
Eds. Lehmann S, Grassi J.
Birkhauser Verlag, Berlin.
Pp. 82–96.
ANDRÉOLOTTI, O., BERTHON, P., MARC, D., SARRADIN, P., GROSCLAUDE, J., van KEULEN, L.,
SCHELCHER, F., ELSEN, J.M., and LANTIER, F. (2000):
Early accumulation of PrPSc in gut-associated lymphoid and nervous tissues of susceptible sheep from a
Romanov flock with natural scrapie.
J. Gen. Virol. 81, 3115-3126.
ANONYMOUS, (2001):
Rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies (and
amendments).
European Parliament and the council of the European Union. Regulation (EC) No 999/2001.
BALKEMA-BUSCHMANN, A., FAST, C., KAATZ, M., EIDEN, M., ZIEGLER, U., MCINTYRE, L.,
KELLER, M., HILLS, B., and GROSCHUP, M.H. (2011):
Pathogenesis of classical and atypical BSE in cattle.
Prev. Vet. Med. 102, 112-117.
BARILLET, F., MARIAT, D., AMIGUES, Y., FAUGERAS, R., CAILLAT, H., MOAZAMI-
GOUDARZI, K., RUPP, R., BABILLIOT, J.M., LACROUX, C., LUGAN, S., SCHELCHER, F.,
CHARTIER, C., CORBIÈRE, F., ANDRÉOLETTI, O., and PERRIN-CHAUVINEAU, C. (2009):
Identification of seven haplotypes of the caprine PrP gene at codons 127, 142, 154, 211, 222 and 240 in French
Alpine and Saanen breeds and their association with classical scrapie.
J. Gen. Virol. 90, 769-776.
BARINGER, J.R., BOWMAN, K.A., and PRUSINER, S.B. (1983):
Replication of the scrapie agent in hamster brain precedes neuronal vacuolation.

64 Chapter 7 - Reference
J. Neuropathol. 42, 485-597.
BARON, T. (2002):
Identification of inter-species transmission of prion strains.
J. Neuropathol. Exp. Neurol. 61, 377-383.
BARON, T.M., and BIACABE, A.G. (2001):
Molecular analysis of the abnormal prion protein during coinfection of mice by the bovine spongiform
encephalopathy and a scrapie agent.
J. Virol. 75, 107-114.
BARROW, P.A., HOLMGREN, C.D., TAPPER, A.J., and JEFFERYS, J.G. (1999):
Intrinsic physiological and morphological properties of principal cells of the hippocampus and neocortex in
hamsters infected with scrapie.
Neurobiol. Dis. 6, 406-423.
BEEKES, M., and McBRIDE, P.A. (2007):
The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies.
FEBS J. 274, 588-605.
BEEKES, M., McBRIDE, P.A., and BALDAUF, E. (1998):
Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie.
J. Gen. Virol. 79, 601-607.
BELL, J.E., and IRONSIDE, J.W. (1993):
Neuropathology of spongiform encephalopathies in humans.
Br. Med. Bull. 49, 738-777.
BELLINGER, K., DIENER, T.O., MCKINLEY, M.P., GROTH, D.F., SMITH, D.R., and PRUSINER,
S.B. (1987):
Purified scrapie prions resist inactivation by procedures that hydrolyze, modify, or shear nucleic acids.
Virol. 160, 271-274.
BELLWORTHY, S.J., HAWKINS, S.A., GREEN, R.B., BLAMIRE, I., DEXTER, G., DEXTER, I.,
LOCKEY, R., JEFFREY, M., RYDER, S., BERTHELIN-BAKER, C., and SIMMONS, M.M. (2005):
Tissue distribution of bovine spongiform encephalopathy infectivity in Romney sheep up to the onset of clinical
disease after oral challenge.
Vet. Rec. 156, 197-202.

Chapter 7- References 65
BENCSIK, A., DEBEER, S., PETIT, T., and BARON, T. (2009):
Possible case of maternal transmission of feline spongiform encephalopathy in a captive cheetah.
PLoS ONE 4, 6929.
BERINGUE, V., MALLINSON, G., KAISAR, M., TAYEBI, M., SATTAR, Z., JACKSON, G., ANSTEE,
D., COLLINGE, J., and HAWKE, S. (2003):
Regional heterogeneity of cellular prion protein isoforms in the mouse brain.
Brain 126, 2065-2073.
BERINGUE, V., VILOTTE, J.L., and LAUDE, H. (2008):
Review article: Prion agent diversity and species barrier.
Vet. Res. 39, 47.
BESSEN, R.A., and MARSH, R.F. (1992):
Biochemical and physical properties of the prion protein from two strains of the transmissible mink
encephalopathy agent.
J. Virol. 66, 2096-2101.
BESSEN, R.A., and MARSH, R.F. (1994):
Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy.
J. Virol. 68, 7859-7868.
BOLANOS, J.P., ALMEIDA, A., FERNANDEZ, E., MEDINA, J.M., LAND, J.M., CLARK, J.B., and
HEALES, S.J. (1997):
Potential mechanisms for nitric oxide-mediated impairment of brain mitochondrial energy metabolism.
Biochem. Soc. Trans. 25, 944-949.
BOLTON, D.C. (1998):
Prion distribution in hamster lung and brain following intraperitoneal inoculation.
J. Gen. Virol. 79, 2557-2562.
BOLTON, D.C., RUDELLI, R.D., CURRIE, J.R., and BENDHEIM, P.E. (1991):
Copurification of Sp33-37 and scrapie agent from hamster brain prior to detectable histopathology and clinical
disease.
J. Gen. Virol. 72, 2905-2913.
BOUZALAS, I.G., DOVAS, C.I., BANOS, G., PAPANASTASOPOULOU, M., KRITAS, S.,
OEVERMANN, A., PAPAKOSTAKI, D., EVANGELIA, C., PAPADOPOULOS, O., SEUBERLICH, T.,
and KOPTOPOULOS, G. (2010):

66 Chapter 7 - Reference
Caprine PRNP polymorphisms at codons 171, 211, 222 and 240 in a Greek herd and their association with
classical scrapie.
J. Gen. Virol. 91, 1629-1634.
BROWN, D.R. (2001):
Prion and prejudice: normal protein and the synapse.
Trends Neurosci. 24, 85-90.
BROWN, P., and CERVENAKOVA, L. (2005):
A prion lexicon (out of control).
Lancet 365, 122.
BROWN, P., and GAJDUSEK, D.C. (1991):
Survival of scrapie virus after 3 years' interment.
Lancet 337, 269-270.
BRUCE, M. (1996):
Strain typing studies of scrapie and BSE. In methods in molecular medicine: Prion diseases (Baker, H. et al.,
Eds.),
Pp. 223-236.
BRUCE, M., CHREE, A., MCCONNELL, I., FOSTER, J., PEARSON, G., and FRASER, H. (1994):
Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier.
Philos. Trans. R. Soc. Lond. B. Biol. Sci. 343, 405-411.
BRUCE, M.E. (1993):
Scrapie strain variation and mutation.
Br. Med. Bull. 49, 822-838.
BRUCE, M.E., and DICKINSON, A.G. (1987):
Biological evidence that scrapie agent has an independent genome.
J. Gen. Virol. 68, 79-89.
BRUCE, M.E., BOYLE, A., COUSENS, S., MCCONNELL, I., FOSTER, J., GOLDMANN, W., and
FRASER, H. (2002):
Strain characterization of natural sheep scrapie and comparison with BSE.
J. Gen. Virol. 83, 695-704.

Chapter 7- References 67
BRUCE, M.E., FRASER, H., MCBRIDE, P.A., SCOTT, J.R., and DICKINSON, A.G. (1992):
The basis of strain variation in scrapie, in prion diseases of humans and animals (PRUSINER, S. B.,
COLLINGE, J., POWELL, J., AND ANDERTON, B., edS.), Ellis Horwood, Chichester,
Pp. 497-508.
BUDKA, H. (2003):
Neuropathology of prion diseases.
Br. Med. Bull. 66, 121-130.
BUDKA, H., AGUZZI, A., BROWN, P., BRUCHER, J.M., BUGIANI, O., GULLOTTA, F., HALTIA, M.,
HAUW, J.J., IRONSIDE, J.W., and JELLINGER, K. (1995):
Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform
encephalopathies.
Brain Pathol., 4, 459-466.
BUELER, H., AGUZZI, A., SAILER, A., GREINER,R.A., AUTENRIED, P., AGUET, M., and
WEISSMANN, C. (1993):
Mice devoid of PrP are resistant to scrapie.
Cell 73, 1339-1347.
BUELER, H., FISCHER, M., LANG, Y., BLUETHMANN, H., LIPP, H.P., DEARMOND, S.J.,
PRUSINER, S.B., AGUET, M., and WEISSMANN, C. (1992):
Normal development and behaviour of mice lacking the neuronal cell surface PrP protein. Nature 356, 577-582.
BUSCHMANN, A., and GROSCHUP, M.H. (2005):
Highly bovine spongiform encephalopathy sensitive transgenic mice confirm the essential restriction of
infectivity to the nervous system in clinically diseased cattle.
J. Infect. Dis. 192, 934-942.
BUSCHMANN, A., PFAFF, E., REIFENBERG, K., MÜLLER, H.M., and GROSCHUP, M.H. (2000):
Detection of cattle-derived BSE prions using transgenic mice overexpressing bovine PrPC.
Arch. Virol. Suppl. 16, 75-86.
CARLSON, G.A., GOODMAN, P.A., LOVETT, M., BENJAMIN, A., TAYLOR, B.A., MARSHALL,
S.T., PETERSON-TORCHIA, M., WESTAWAY, D., and PRUSINER, S.B. (1988):
Genetics and polymorphism of the mouse prion gene complex: control of scrapie incubation time.
Mol. Cell. Biol. 8, 5528-5540.

68 Chapter 7 - Reference
CASTELLANI, R., HIRAI, K., ALIEV, G., DREW, K.L., NUNOMURA, A., TAKEDA, A., CASH, A.D.,
OBRENOVICH, M.E., PERRY, G., and SMITH, M.A, (2002):
Role of mitochondrial dysfunction in Alzheimer’s disease.
J. Neurosci. Res. 70, 357-360.
CAUGHEY, B., and RAYMON, G. (1991):
The scrapie associated form of PrP is made from a cell surface precursor that is both protease and
phospholipase-sensitive.
J. Biol. Chem. 266, 18217-18223.
CHANDLER, R.L. (1961):
Encephalopathy in mice produced by inoculation with scrapie brain material.
Lancet 1, 1378-1379.
CHANDLER, R.L. (1971):
Intramammary inoculation of mice with scrapie.
Br. Vet. J. 127, 1-2.
CHANDLER, R.L., and FISHER, J. (1963):
Experimental transmission of scrapie to rats.
Lancet 2, 1165.
CHANDLER, R.L., and TURFREY, B.A. (1972):
Inoculation of voles, Chinese hamsters, gerbils and guinea-pigs with scrapie brain material.
Res. Vet. Sci. 13, 219-224.
CHESEBRO, B. (2003):
Introduction to the transmissible spongiform encephalopathies or prion disease.
Br. Med. Bull. 66, 1-20.
CHESEBRO, B., TRIFILO, M., RACE, R., MEADE-WHITE, K., TENG, C., LACASSE, R.,
RAYMOND, L., FAVARA, C., BARON, G., PRIOLA, S., CAUGHEY, B., MASLIAH, E., and
OLDSTONE, M. (2005):
Anchorless prion protein results in infectious amyloid disease without clinical scrapie.
Science 308, 1435-1439.
CLEETER, M.W., COOPER, J.M., DARLEY-USMAR, V.M., MONCADA, S., and SCHAPIRA, A.H.
(1994):

Chapter 7- References 69
Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by
nitric oxide. Implications for neurodegenerative diseases.
FEBS Lett. 345, 50-54.
CLINTON, J., FORSYTH, C., ROYSTON, M.C., and ROBERTS, G.W. (1993):
Synaptic degeneration is the primary neuropathological feature in prion disease: a preliminary study.
Neuroreport. 4, 65-68.
COLLINGE, J. (2001):
Prion diseases of humans and animals: Their Causes and Molecular Basis.
Annu. Rev. Neurosci. 24, 519-550.
COLLINGE, J., BECK, J., CAMPBELL, T., ESTIBEIRO, K., and WILL, R.G. (1996):
Prion protein gene analysis in new variant cases of Creutzfeldt-Jakob disease.
Lancet 348, 56.
COLLINGE, J., WHITTINGTON, M.A., SIDLE, K.C., SMITH, C.J., PALMER, M.S., CLARKE, A.R.,
and JEFFERYS, J.G. R. (1994):
Prion protein is necessary for normal synaptic function.
Nature 370, 295-297.
CORBIÈRE, F., PERRIN-CHAUVINEAU, C., LACROUX, C., COSTES, P., THOMAS, M., BRÉMAUD,
I., MARTIN, S., LUGAN, S., CHARTIER, C., SCHELCHER, F., BARILLET, F., and ANDREOLETTI,
O. (2013):
PrP-associated resistance to scrapie in five highly infected goat herds.
J. Gen. Virol. 94, 241-245.
CRONIER, S., LAUDE, H., and PEYRIN, J.M. (2004):
Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death.
Proc. Natl. Acad. Sci. USA 101, 12271-12276.
CROZET, C., BENCSIK, A., FLAMANT, F., LEZMI, S., SAMARUT, J., and BARON, T. (2001b):
Florid plaques in ovine PrP transgenic mice infected with an experimental ovine BSE.
EMBO Rep. 2, 952-956.
CROZET, C., FLAMANT, F., BENCSIK, A., AUBERT, D., SAMARUT, J., and BARON, T. (2001a):
Efficient transmission of two different sheep scrapie isolates in transgenic mice expressing the ovine PrP gene.
J. Virol. 75, 5328-5334.

70 Chapter 7 - Reference
CHELLE, P.L. (1942):
Un cas de tremblante chez la chevre.
Bull Acad. Vet. Fr. 15, 294-295.
CUILLE, J., and CHELLE, P.L. (1936):
Pathologie animale – La maladie dite tremblante du mouton est-elle inoculable ?
Comptes rendus hebdomadaires des seances de l’Academie des Sciences 203, 1552-1554.
CUILLE, J., and CHELLE, P.L. (1939):
Transmission experimentale de la tremblante a la chevre.
Comptes rendus hebdomadaires des siences de l’Academie des Sciences 208, 1058-1060.
CUNNINGHAM, C., DEACON, R., WELLS, H., BOCHE, D., WATERS, S., DINIZ, C.P., SCOTT, H.,
RAWLINS, J.N., and PERRY, V.H. (2003):
Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease.
Eur. J. Neurosci. 17, 2147-2155.
DeMARCO, M.L., and DAGGETT, V. (2004):
From conversion to aggregation: protofibril formation of the prion protein.
Proc. Natl. Acad. Sci. U.S.A. 101, 2293-2298.
DETWILER, L.A. (1992):
Scrapie.
Rev. Sci. Tech. 11, 491-537.
DETWILER, L.A., and BAYLIS, M. (2003):
The epidemiology of scrapie.
Rev. Sci. Tech. 22, 121-143.
DIEDRICH, J.F., BENDHEIM, P.E., KIM, Y.S., CARP, R.I., and HAASE, A.T. (1991):
Scrapie-associated prion protein accumulates in astrocytes during scrapie infection.
Proc. Natl. Acad. Sci. USA 88, 375-379.
ERMOLAYEV, V., CATHOMEN, T., MERK, J., FRIEDRICH, M., HÄRTIG, W., HARMS, G.S.,
KLEIN, M.A., and FLECHSIG, E. (2009):
Impaired axonal transport in motor neurons correlates with clinical prion disease.
Plos Pathog. 5, e1000558.

Chapter 7- References 71
FERNÁNDEZ-CHACÓN, R., WÖLFEL, M., NISHIMUNE, H., TABARES, L., SCHMITZ, F.,
CASTELLANO-MUÑOZ, M., ROSENMUND, C., MONTESINOS, M.L., SANES, J.R.,
SCHNEGGENBURGER, R., and SÜDHOF, T.C. (2004):
The synaptic vesicle protein CSP alpha prevents presynaptic degeneration.
Neuron 42, 237-251.
FERRER, I. (2002):
Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease.
Cerebellum 1, 213-222.
FERRER, I., PUIG, B., BLANCO, R., and MARTI, E. (2000):
Prion protein deposition and abnormal synaptic protein expression in the cerebellum in Creutzfeldt-Jakob
disease.
Neuroscience 97, 715-726.
FERRER, I., RIVERA, R., BLANCO, R., and MART, E. (1999):
Expression of proteins linked to exocytosis and neurotransmission in patients with Creutzfeldt-Jakob disease.
Neurobiol. Dis. 6, 92-100.
FISCHER, M., RULICKE1,T., RAEBER, A., SAILER, A., MOSER, M., OESCH, B., BRANDNER, S.,
AGUZZI, A., and WEISSMANN, C. (1996):
Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie.
EMBO J. 15, 1255-1264.
FOSTER, J.D., BRUCE, M., McCONNELL, I., CHREE, A., and FRASER, H. (1996):
Detection of BSE infectivity in brain and spleen of experimentally infected sheep.
Vet. Rec. 138, 546-548.
FOSTER, J.D., PARNHAM, D., CHONG, A., GOLDMANN, W., and HUNTER, N. (2001a):
Clinical signs, histopathology and genetics of experimental transmission of BSE and natural scrapie to sheep
and goats.
Vet. Rec. 148, 165-171.
FOSTER, J.D., PARNHAM, D.W., HUNTER, N., and BRUCE, M. (2001b):
Distribution of the prion protein in sheep terminally affected with BSE following experimental oral
transmission.
J. Gen. Virol. 82, 2319-2326.
FOURNIER, J.G., ESCAIG-HAYE, F., BILLETTE DE VILLEMEUR, T., and ROBAIN, O. (1995):

72 Chapter 7 - Reference
Ultrastructural localization of cellular prion protein (PrPC) in synaptic boutons of normal hamster hippocampus.
CR Acad. Sci. III 318, 339-344.
FRAGKIADAKI, E.G., VACCARI, G., EKATERINIADOU, L.V., AGRIMI, U., GIADINIS, N.D.,
CHIAPPINI, B., ESPOSITO, E., CONTE, M., and NONNO, R. (2011):
PRNP genetic variability and molecular typing of natural goat scrapie isolates in a high number of infected
flocks.
Vet. Res. 42, 104.
FRASER, H., and DICKINSON, A.G. (1973):
Scrapie in mice: agent-strain differences in the distribution and intensity of grey matter vacuolation.
J. Comp. Pathol. 83, 29-40.
FRASER, H., BRUCE, M.E., CHREE, A., MCCONNELL, I., and WELLS, G.A.H. (1992):
Transmission of bovine spongiform encephalopathy and scrapie to mice.
J. Gen. Virol. 73, 1891-1897.
FRASER, J.R. (2002):
What is the basis of transmissible spongiform encephalopathy induced neurodegeneration and can it be
repaired?
Neuropathol. Appl. Neurobiol. 28, 1-11.
FUKUDA, S., ONOE, S., NIKAIDO, S., FUJII, K., KAGEYAMA, S., IWAMARU, Y., IMAMURA, M.,
MASUJIN, K., MATSUURA, Y., SHIMIZU, Y., KASAI, K., YOSHIOKA, M., MURAYAMA, Y.,
MOHRI, S., YOKOYAMA, T., and OKADA, H. (2012):
Neuroanatomical distribution of disease-associated prion protein in experimental bovine spongiform
encephalopathy in cattle after intracerebral inoculation.
Jpn. J. Infect. Dis. 65, 37-44.
GABIZON, R., TELLING, G., MEINER, Z., HALIMI, M., KAHANA, I., and PRUSINER, S.B. (1996):
Insoluble wild-type and protease-resistant mutant prion protein in brains of patients with inherited prion
diseases.
Nat. Med. 2, 59-64.
GAJDUSEK, D.C. (1977):
Unconventional viruses and the origin and disappearance of kuru.
Science 197, 943-960.
GAJDUSEK, D.C. (1985):

Chapter 7- References 73
Hypothesis: interference with axonal transport of neurofilaments as a common pathogenetic mechanism in
certain diseases of the central nervous system.
N. Engl. J. Med. 312, 714-719.
GAJDUSEK, D.C., and ZIGAS, V. (1957):
Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native
population.
N. Engl. J. Med. 257, 974-978.
GAMBETTI, P., PETERSEN, R.B., PARCHI, P., CHEN, S.G., CAPELLARI, S., and GOLDFARB, L.
(2004):
Inherited prion diseases. In: Prusiner SB, eds. Prion Biology and Diseases.
Cold Spring Harbor Laboratory
Pp. 673-775.
GAVIER-WIDEN, D., STACK, M. J., BARON, T., BALACHANDRAN, A., and SIMMONS, M. (2005):
Diagnosis of transmissible spongiform encephalopathies in animals: a review.
J. Vet. Diagn. Invest. 17, 509-527.
GIBBS, C. J., ASHER, D.M., KOBRINE, A., AMYX, H.L., SULIMA, M.P., and GAJDUSEK, D.C.
(1994):
Transmission of Creutzfeldt-Jakob disease to a chimpanzee by electrodes contaminated during neurosurgery.
J. Neurol. Neurosurg. Psych. 57, 757-758.
GONZÁLEZ, L., MARTIN, S., and JEFFREY, M. (2003):
Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie and BSE-infected sheep: implications for
differential cell targeting and PrP processing. J Gen Virol 84: 1339–1350.
GONZÁLEZ, L., MARTIN, S., BEGARA-MCGORUM, I., HUNTER, N., HOUSTON, F., SIMMONS,
M., and JEFFREY, M. (2002):
Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally
affected with scrapie.
J. Comp. Pathol. 126, 17-29.
GONZÁLEZ, L., MARTIN, S., HOUSTON, F.E., HUNTER, N., REID, H.W., BELLWORTHY, S.J., and
JEFFREY, M. (2005):
Phenotype of disease-associated PrP accumulation in the brain of bovine spongiform encephalopathy
experimentally infected sheep.
J. Gen. Virol. 86, 827-838.

74 Chapter 7 - Reference
GOODFIELD, J. (1997)
Cannibalism and kuru.
Nature 387, 841.
GRAY, B.C., SISKOVA, Z., PERRY, V.H., and O’CONNOR, V. (2009):
Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology.
Neurobiol. Dis. 35, 63-74.
GRIFFITH, J.S. (1967):
Self replication and scrapie.
Nature 215, 1043-1044.
GROSCHUP, M.H., and BUSCHMANN, A. (2008):
Rodent models for prion diseases.
Vet. Res. 39, 32.
HAEBERLE, A.M., RIBAUT-BARASSIN, C., BOMBARDE, G., MARIANI. J., HUSSMANN, G.,
GRASSI, J., and BAILLY, Y. (2000):
Synaptic prion immunoreactivity in the rodent cerebellum.
Microsc. Res. Tech. 50, 66-76.
HAÏK, S., and BRANDEL, J.P. (2014):
Infectious prion diseases in humans: cannibalism, iatrogenicity and zoonoses.
Infect. Genet. Evol. 26, 303-312.
HE, L., LU, X.Y., JOLLY, A.F., ELDRIDGE, A.G.,WATSON, S.J., JACKSON, P.K., BARSH, G.S., and
GUNN, T.M. (2003):
Spongiform degeneration in mahoganoid mutant mice.
Science 299, 710-712.
HILL, A., DESBRUSLAIS, M., JOINER, S., KATIE, C., SIDLE, L., GOWLAND, I., COLLINGE, J.,
DOEY, L. J., and LANTOS, P. (1997):
The same prion strain causes vCJD and BSE.
Nature 389, 448-450.
HILTON, K.J., CUNNINGHAM, C., REYNOLDS, R.A., and PERRY, V.H. (2013):
Early Hippocampal Synaptic Loss Precedes Neuronal Loss and Associates with Early Behavioural Deficits in
Three Distinct Strains of Prion Disease.
PLoS One. 8, e68062.

Chapter 7- References 75
HOLLISTER, J.R., LEE, K.S., DORWARD, D.W., and BARON, G.S. (2015):
Efficient Uptake and Dissemination of Scrapie Prion Protein by Astrocytes and Fibroblasts from Adult Hamster
Brain.
PLoS One 10, e0115351.
HORNEMANN, S., KORTH, C., OESCH, B., RIEK, R., WIDER, G., WÜTHRICH, K., and
GLOCKSHUBER, R. (1997):
Recombinant full-length murineprion protein, mPrP(23–231): purification and spectroscopic characterization.
FEBS Lett. 413, 277-281.
IULINI, B., MAURELLA, C., PINTORE, M.D., VALLINO, C.E., CORBELLINI, D., PORCARIO, C.,
PAUTASSO, A., SALATA, C., GELMETTI, D., AVANZATO, T., PALÙ, G., D'ANGELO, A.,
CARAMELLI, M., CASALONE, C. (2012):
Ten years of BSE surveillance in Italy: neuropathological findings in clinically suspected cases.
Res. Vet. Sci. 93, 872-878.
JEFFREY, M., and GONZÁLEZ, L. (2004):
Pathology and pathogenesis of bovine spongiform encephalopathy and scrapie.
Curr. Topics Microbiol. Immunol. 284, 65-97.
JEFFREY, M., and GONZÁLEZ, L. (2007):
Classical sheep transmissible spongiform encephalopathies: pathogenesis, pathological phenotypes and clinical
disease.
Neuropath. Appl. Neurobiol. 33, 373-394.
JEFFREY, M., HALLIDAY, W.G., BELL, J., JOHNSTON, A.R., MACLEOD, N.K., INGHAM, C.,
SAYERS, A.R., BROWN, D.A., and FRASER, J.R. (2000):
Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine
hippocampus.
Neuropathol. Appl. Neurobiol. 26, 41-54.
KAATZ, M., FAST, C., ZIEGLER, U., BALKEMA-BUSCHMANN, A., HAMMERSCHMIDT, B.,
KELLER, M., OELSCHLEGEL, A., McINTYRE, L., and GROSCHUP, M. (2012):
Spread of classical BSE prions from the gut via the peripheral nervous system to the Brain.
Immunopathol. Infect. Dis. 181, 515-524.
KIMBERLIN, R.H., and WALKER, C.A. (1977):
Characteristics of a short incubation model of scrapie in the golden hamster.
J. Gen. Virol. 34, 295-304.

76 Chapter 7 - Reference
KIMBERLIN, R.H., and WALKER, C.A. (1978):
Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains
from a mixture.
J. Gen. Virol. 39, 487-496.
KIMBERLIN, R.H., and WALKER, C.A. (1989):
Pathogenesis of scrapie in mice after intragastric infection.
Virus Res. 12, 213-220.
KIMBERLIN, R.H., COLE, S., and WALKER, C.A. (1987):
Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and
hamsters.
J. Gen. Virol. 68, 1875-1881.
KITAMOTO, T., SHIN, R.W., DOH-URA, K., TOMOKANE, N., MIYAZONO, M., MURAMOTO, T.,
and TATEISHI, J. (1992):
Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in
patients with Creutzfeldt-Jakob disease.
Am. J. Pathol. 140, 1285-1294.
KLEIN, M.A., FRIGG, R., FLECHSIG, E., RAEBER, A.J., KALINKE, U., BLUETHMANN, H.,
BOOTZ, F., SUTER, M., ZINKERNAGEL, R.M., and AGUZZI, A. (1997):
A crucial role for B cells in neuroinvasive scrapie.
Nature 390, 687-690.
KONOLD, T., GOLDMANN, W., CAWTHRAW, S., HAWKINS, A.C., GEMMA., E.B., PHELAN, L.J.,
SIMMONS, M.M., GONZÁLEZ, L., and SISÓ, S. (2010):
Monitoring of clinical signs in goats with transmissible spongiform encephalopathies.
BMC Vet. Res. 6, 13.
KRETZSCHMAR, H.A. (1993):
Human prion diseases (spongiform encephalopathies).
Arch. Virol. Suppl. 7, 261-293.
KUJALA, P., RAYMOND, C.R., ROMEIJN, M., GODSAVE, S.F., van KASTEREN, S.I., WILLE, H.,
PRUSINER, S.B., MABBOTT, N.A., and PETERS, P.J. (2011):
Prion uptake in the gut: identification of the first uptake and replication sites.
PLoS Pathog. 7, e1002449.

Chapter 7- References 77
KÜNZI, V., GLATZEL, M., NAKANO, M.Y., GREBER, U.F., LEUVEN, F.V., and AGUZZI, A. (2002):
Unhampered Prion Neuroinvasion despite Impaired Fast Axonal Transport in Transgenic Mice Overexpressing
Four Repeat Tau.
J. Neurosci. 22, 7471-7477.
LACROUX, C., PERRIN-CHAUVINEAU, C., CORBIÈRE, F., ARON, N., AGUILAR-CALVO, P.,
TORRES, J.M., COSTES, P., BRÉMAUD, I., LUGAN, S., SCHELCHER, F., BARILLET, F., and
ANDRÉOLETTI, O. (2014):
Genetic resistance to scrapie infection in experimentally challenged goats.
J. Virol. 88, 2406-2413.
LASMÉZAS, C. I., COMOY, E., HAWKINS, S., HERZOG, C., MOUTHON, F., KONOLD, T., AUVRÉ,
F., CORREIA, E., LESCOUTRA-ETCHEGARAY, N., SALÈS, N., WELLS, G., BROWN, P., and
DESLYS, J.P. (2005):
Risk of oral infection with bovine spongiform encephalopathy agent in primates.
Lancet 365, 781-783.
LEE, J., KIM, S.Y., HWANG, K.J., JU, Y.R., and WOO, H.J. (2013):
Prion diseases as transmissible zoonotic diseases.
Osong Public Health Res. Perspect. 4, 57-66.
LEZMI, S., SEUBERLICH, T., OEVERMANN, A., BARON, T., and BENCSIK, A. (2011):
Comparison of brain PrPd distribution in ovine BSE and scrapie.
Vet. Pathol. 48, 1101-1108.
LI, Z., OKAMOTO, K., HAYASHI, Y., and SHENG, M. (2004):
The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses.
Cell 119, 873-887.
LIBERSKI, P.P. (2012):
Historical overview of prion diseases: a view from afar.
Folia Neuropathol. 50, 1-12.
LIBERSKI, P.P., and BUDKA, H. (1999):
Neuroaxonal pathology in Creutzfeldt-Jakob disease.
Acta Neuropathol. 97, 329-334.
LIBERSKI, P.P., KORDEK, R., BROWN, P., and GAJDUSEK, D.C. (1998):

78 Chapter 7 - Reference
Astrocytes in transmissible spongiform encephalopathies (prion diseases), in: Astrocytes in Brain Aging and
Neurodegeneration, edited by H. M. Schipper (R. G. Landes Company, Austin, Texas, USA)
pp. 127–164.
LLOYD, S.E., ONWUAZOR, O.N., BECK, J.A., MALLINSON, G., FARRALL, M., TARGONSKI, P.,
COLLINGE, J., and ELIZABETH, M.C.F. (2001):
Identification of multiple quantitative trait loci linked to prion disease incubation period in mice.
Proc. Natl. Acad. Sci. USA 98, 6279-6283.
MALLUCCI, G., DICKINSON, A., LINEHAN, J., KLOHN, P.C., BRANDNER, S., and COLLINGE, J.
(2003):
Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis.
Science 302, 871-874.
MANSON, J.C., JAMIESON, E., BAYBUTT, H., TUZI, N.L., BARRON, R., MCCONNELL, I.,
SOMERVILLE, R., IRONSIDE, J., WILL, R., SY, M.S., MELTON, D.W., HOPE, J., and BOSTOCK, C.
(1999):
A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of
transmissible spongiform encephalopathy.
EMBO J. 18, 6855-6864.
MARSH, R.F., and HADLOW, W.J. (1992):
Transmissible mink encephalopathy.
Rev. Sci. Tech. 11, 539-550.
MASTRIANNI, J.A., NIXON, R., LAYZER, R., TELLING, G.C., HAN, D., DEARMOND, S.J., and
PRUSINER, S.B. (1999):
Prion protein conformation in a patient with sporadic fatal insomnia.
NEJM 340, 1630-1638.
MATHIASON, C. K., POWERS, J.G., DAHMES. J.S, OSBORN, D.A., MILLER, K.V., WARREN, R.J.,
MASON, G.L. HAYS, S.A., HAYES-KLUG, J., SEELIG, D.M., WILD, M.A., WOLFE, L.L., SPRAKER,
T.R., MILLER, M.W., SIGURDSON, C.J., TELLING, G.C., and HOOVER., E.A. (2006):
Infectious Prions in the Saliva and Blood of Deer with Chronic Wasting Disease.
Science 314, 133-136.
McBRIDE, P.A., SCHULZ-SCHAEFFER, W.J., DONALDSON, M., BRUCE, M., DIRINGER, H.,
KRETZSCHMAR, H.A., and BEEKES, M. (2001):
Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of

Chapter 7- References 79
the splanchnic and vagus nerves.
J. Virol. 75, 9320-9327.
McGOVERN, G., MARTIN, S., JEFFREY, M., BELLWORTHY, S.J., SPIROPOULOS, J., GREEN, R.,
LOCKEY. R., VICKERY, C.M., THURSTON, L., DEXTER, G., HAWKINS, S.A., and GONZÁLEZ, L.
(2015):
Influence of Breed and Genotype on the Onset and Distribution of Infectivity and Disease-associated Prion
Protein in Sheep Following Oral Infection with the Bovine Spongiform Encephalopathy Agent.
J. Comp. Pathol. 152, 28-40.
McKINLEY, M.P., BOLTON, D.C., and PRUSINER S.B. (1983):
A protease resistant protein is a structural component of the scrapie prion.
Cell 35, 57-62.
MEIER, P., GENOUD, N., PRINZ, M., MAISSEN, M., RÜLICKE, T., ZURBRIGGEN, A., RAEBER,
A.J., and AGUZZI, A. (2003):
Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease.
Cell 113, 49-60.
MILLER, C.C., ACKERLEY, S., BROWNLEES, J., GRIERSON, A.J., JACOBSEN, N.J., and
THORNHILL, P. (2002):
Axonal transport of neurofilaments in normal and disease states.
Cell Mol. Life Sci. 59, 323-330.
MORINET, F. (2014):
Prions: a model of conformational disease?
Pathol. Biol. 62, 96-99.
MURAMOTO, T., DEARMOND, S.J., SCOTT, M., TELLING, G.C., COHEN, F.E., and PRUSINER,
S.B. (1997):
Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an
alpha-helix.
Nat. Med. 3, 750-755.
PAN, K.M., (1993):
Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins.
Proc. Natl. Acad. Sci. USA. 90, 10962-10966.

80 Chapter 7 - Reference
PAPASAVVA-STYLIANOU, P., WINDL, O., SAUNDERS, G., MAVRIKIOU, P., TOUMAZOS, P., and
KAKOYIANNIS, C. (2011):
PrP gene polymorphisms in Cyprus goats and their association with resistance or susceptibility to natural
scrapie.
Vet. J. 187, 245-250.
PATTISON, I.H., and JONES, K.M. (1967):
The possible nature of the transmissible agent of scrapie.
Vet. Rec. 80, 2-9.
PATTISON, I.H., GORDON, W.S., and MILLSON, G.C. (1959):
Experimental production of scrapie in goats.
J. Comp. Pathol. 69, 300-312.
PICANCO-DINIZ, C.W., BOCHE, D., GOMES-LEAL, W., PERRY, V.H., and CUNNINGHAM, C.
(2004):
Neuropil and neuronal changes in hippocampal NADPH-diaphorase histochemistry in the ME7 model of murine
prion disease.
Neuropathol. Appl. Neurobiol. 30, 292-303.
POST, K., RIESNER, D., WALLDORF, V., and MEHLHORN, H. (1999):
Fly larvae and pupae as vectors for scrapie.
Lancet. 354, 1969-1970.
PRITZKOW, S., WAGENFUHR, K., DAUS, M.L., BOERNER, S., LEMMER, K., THOMZIG, A.,
MIELKE, M., and BEEKES, M. (2011):
Quantitative detection and biological propagation of scrapie seeding activity in vitro facilitate use of prions as
model pathogens for disinfection.
PLoS ONE 6, e20384.
PRUSINER, S.B., GROTH, D.F., COCHRAN, S.P., MASIARZ, F.R., MCKINLEY, M.P., and
MARTINEZ, H.M. (1980):
Molecular properties, partial purification, and assay by incubation period measurements of the hamster scrapie
agent.
Biochem. 19, 4883-4891.
PRUSINER, S.B. (1982):
Novel proteinaceous infectious particles cause scrapie.
Science 216, 136-144.

Chapter 7- References 81
PRUSINER, S.B. (1998):
Prions.
Proc. Natl. Acad. Sci. USA 95, 13363-13383.
PRUSINER, S.B. (2013):
Biology and genetics of prions causing neurodegeneration.
Annu. Rev. Genet. 47, 601-623.
PRUSINER, S.B., GROTH, D., SERBAN, A., KOEHLER, R., FOSTER, D., TORCHIA, M., BURTON,
D., YANG, S-L., and DEARMOND, S.J. (1993):
Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP
antibodies.
Proc. Natl. Acad. Sci. 90, 10608-10612.
PRUSINER, S.B., SCOTT, M., FOSTER, D., PAN, K-M., GROTH, D., MIRENDA, TORCHIA, C., M.,
YANG S-L., SERBAN, D., CARLSON, G.A., HOPPE, P.C., WESTAWAY, D., and DEARMOND S.J.
(1990):
Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication.
Cell 63, 673-686.
RACE, R., and CHESEBRO, B. (1998):
Scrapie infectivity found in resistant species.
Nature 392, 770.
RACE, R., JENNY, A., and SUTTON, D. (1998):
Scrapie infectivity and proteinase K-resistant prion protein in sheep placenta, brain, spleen, and lymph node:
implications for transmission and antemortem diagnosis.
J. Infect. Dis. 178, 949-953.
RAEBER, A.J., RACE, R.E., BRANDNER, S., PRIOLA, S.A., SAILER, A., BESSEN, R.A., MUCKE, L.,
MANSON, J., AGUZZI, A., OLDSTONE, M.B., WEISSMANN, C., and CHESEBRO, B. (1997):
Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster
scrapie.
EMBO J. 16, 6057-6065.
RAVILOCHAN, K., and TYLER, K.L. (1992):
Human transmissible neurodegenerative diseases (prion diseases).
Semin. Neurol. 12, 178-192.

82 Chapter 7 - Reference
REIS, R., HENNESSY, E., MURRAY, C., GRIFFIN, É.W., and CUNNINGHAM, C. (2015):
At the centre of neuronal, synaptic and axonal pathology in murine prion disease: degeneration of
neuroanatomically linked thalamic and brainstem nuclei.
Neuropathol. Appl. Neurobiol. doi. 10.1111/nan.12232.
RICHARD, J., WHITLEY, M.D., NONI MACDONALD, M.D., DAVID, M., and ASHER, M.D. (2000):
Technical report: transmissible spongiform encephalopathies: A Review for pediatricians.
Pediatrics. 106, 160-165.
RIESNER, D., (2003):
Biochemistry and structure of PrP(C)
and PrP(Sc)
.
Br. Med. Bull. 66, 21-33.
RIEK, R., HORNEMANN, S., WIDER, G., BILLETER, M., GLOCKSHUBER, R., and WUTHRICH, K.
(1996):
NMR structure of the mouse prion protein domain PrP(121–321).
Nature 382, 180-182.
RUSSELAKIS-CARNEIRO, M., HETZ, C., MAUNDRELL, K., and SOTO, C. (2004):
Prion replication alters the distribution of synaptophysin and caveolin 1 in neuronal lipid rafts.
Am. J. Pathol. 165, 1839-1848.
SAFAR, J., WILLE, H., ITRRI, V., GROTH, D., SERBAN, H., TORCHIA, M., COHEN, F.E., and
PRUSINER, S.B. (1998):
Eight prion strains have PrPSc
molecules with different conformations.
Nat. Med. 4, 1157-1165.
SAILER, A., BUELER, H., FISCHER, M., AGUZZI, A., and WEISSMANN, C. (1994):
No propagation of prions in mice devoid of PrP.
Cell 77, 967-968.
SCOTT, M., FOSTER, D., MIRENDA, C., SERBAN, D., COUFAL, F., WIILCHLI, M., TORCHIA, M.,
GROTH, D., CARLSON, G., DEARMOND, S.J., WESTAWAY, D., and PRUSINER, S.B. (1989):
Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid
plaques.
Cell 59, 847-857.
SCOTT, M.R., WILL, R., IRONSIDE, J., NGUYEN, H.O., TREMBLAY, P., DeARMOND, S.J., and
PRUSINER, S.B. (1999):

Chapter 7- References 83
Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans.
Proc. Natl. Acad. Sci. U.S.A. 96, 15137-15142.
SHARMA, M., BURRÉ, J., and SÜDHOF, T.C. (2011b):
CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity.
Nat. Cell Bio. 13, 30-39.
SHARMA, M., BURRÉ, J., BRONK, P., ZHANG, Y., XU, W., and SÜDHOF, T.C. (2011a):
CSPα knockout causes neurodegeneration by impairing SNAP-25 function.
The EMBO J. 31, 829- 841.
SHMERLING, D., HEGYI, I., FISCHER, M., BLÄTTLER, T., BRANDNER, S., GÖTZ, J., RÜLICKE,
T., FLECHSIG, E., COZZIO, A., VON MERING, C., HANGARTNER, C., AGUZZI, A., and
WEISSMANN, C., (1998):
Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions.
Cell 93, 203-214.
SIGURDSON, C.J., and MILLER, M.W. (2003):
Other animal prion diseases.
Br. Med. Bull. 66, 199-212.
SIGURDSON, C.J., NILSSON, K.P., HORNEMANN, S., HEIKENWALDER, M., MANCO, G.,
SCHWARZ, P., OTT, D., RÜLICKE, T., LIBERSKI, P. P., JULIUS, C., FALSIG, J., STITZ, L.,
WÜTHRICH, K., and AGUZZI, A., (2009):
De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis.
Proc. Natl. Acad. Sci. USA 106, 304-309.
SIGURDSON, C.J., WILLIAMS, E.S., and MILLER, M.W. (1999):
Oral transmission and early tropism of chronic wasting disease PrPres
in mule deer fawns (Odocoileus
hemionus).
J. Gen. Virol. 80, 2757-2764.
SISKOVA, Z., MAHAD, D.J., PUDNEY, C., CAMPBELL, G., CADOGAN, M., ASUNI, A., O'CONNOR,
V., and PERRY, V.H. (2010):
Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion
disease.
Am. J. Pathol. 177, 1411-1421.
SISKOVA, Z., PAGE, A., O’CONNOR, V., and PERRY, V.H. (2009):

84 Chapter 7 - Reference
Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping.
Am. J. Pathol. 175, 1610-1621.
SMITH, K.J., and LASSMANN, H. (2002):
The role of nitric oxide in multiple sclerosis.
Lancet Neurol. 1, 232-241.
SOMERVILLE, R.A., OBERTHUR, R.C., HAVEKOST, U., MACDONALD, F., TAYLOR, D.M., and
DICKINSON, A.G. (2002):
Characterization of thermodynamic diversity between transmissible spongiform encephalopathy agent strains
and its theoretical implications.
J. Biol. Chem. 277, 11084-11089.
SOTO, C., and SATANI, N. (2010):
The intricate mechanisms of neurodegeneration in prion diseases.
Trends Mol. Med. 17, 1-22.
SPRAKER, T.R., BALACHANDRAN, A., ZHUANG, D., and O’ROURKE, K.I. (2004):
Variable patterns of distribution of PrPCWD
in the obex and cranial lymphoid tissues of Rocky Mountain elk
(Cervus elaphus nelsoni) with subclinical chronic wasting disease.
Vet. Rec. 155, 295-302.
SPRAKER, T.R., ZINK, R.R., and CUMMINGS, B.A. (2002):
Distribution of protease-resistant prion protein and spongiform encephalopathy in free-ranging mule deer
(Odocoileus hemionus) with chronic wasting disease.
Vet. Pathol. 39, 546-556.
TELLING, C.G., PARCHI, P., DEARMOND, S.J., CORTELLI, P., MONTAGNA, P., GABIZON, R.,
MASTRIANNI, J., LUGARESI, E., GAMBETTI, P., and PRUSINER, S.B. (1996):
Evidence for the conformation of the pathologic isoform of the prion enciphering and propagating prion
diversity.
Science 274, 2079-2082.
TELLING, G.C., SCOTT, M., HSIAO, K.K., FOSTER, D., YANG, S.L., TORCHIA, M., SIDLE, K.C.,
COLLINGE, J., DEARMOND, S.J., and PRUSINER, S.B. (1994):
Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse
prion protein.
Proc. Natl. Acad. Sci. USA 91, 9936-9940.

Chapter 7- References 85
TELLING, G.C., SCOTT, M., MASTRIANNI, J., GABIZON, R., TORCHIA, M., COHEN, F.E.,
DEARMOND, S.J., and PRUSINER S.B. (1995):
Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interactions of cellular
PrP with another protein.
Cell 83, 79-90.
TERRY, R.D., MASLIAH, E., SALMON, D.P., BUTTERS, N., DETERESA, R., HILL, R., HANSEN,
L.A., and KATZMAN, R. (1991):
Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive
impairment.
Ann. Neurol. 30, 572-580.
TOBABEN, S., THAKUR, P., FERNÁNDEZ-CHACÓN, R., SÜDHOF, T.C., RETTIG, J., and STAHL,
B. (2001):
A trimeric protein complex functions as a synaptic chaperone machine.
Neuron 31, 987-999.
TOIVOLA, D.M., TAO, G.Z., HABTEZION, A., LIAO, J., and OMARY, M.B. (2005):
Cellular integrity plus: organelle-related and protein-targeting functions of intermediate filaments.
Trends Cell Biol.15, 608-617.
TUZI, N.L., GALL, E., MELTON, D., and MANSON, J.C. (2002):
Expression of doppel in the CNS of mice does not modulate transmissible spongiform encephalopathy disease.
J. Gen. Virol. 83, 705-711.
UCHIYAMA, K., MIYATA, H., YANO, M., YAMAGUCHI, Y., IMAMURA, M., MURAMATSU, N.,
DAS, N.R., CHIDA, J., HARA, H., and SAKAGUCHI, S. (2014):
Mouse-hamster chimeric prion protein (PrP) devoid of N-terminal residues 23-88 restores susceptibility to 22L
prions, but not to RML prions in PrP-knockout mice.
PLoS One. 9, e109737.
VACCARI, G., DI-BARI, M.A., MORELLI, L., NONNO, R., CHIAPPINI, B., ANTONUCCI, G.,
MARCON, S., ESPOSITO, E., FAZZI, P., PALAZZINI, N., TROIANO, P., PETRELLA, A., DI-
GUARDO, G., and AGRIMI, U. (2006):
Identification of an allelic variant of the goat PrP gene associated with resistance to scrapie.
J. Gen. Virol. 87, 1395-1402.
van KEULEN, L.J., BOSSERS, A., and van ZIJDERVELD, F. (2008a):
TSE pathogenesis in cattle and sheep.

86 Chapter 7 - Reference
Vet. Res. 39, 24.
van KEULEN, L.J., VROMANS, M.E., DOLSTRA, C.H., BOSSERS, A., and van ZIJDERVELD, F.G.
(2008b):
Pathogenesis of bovine spongiform encephalopathy in sheep.
Arch. Virol. 153, 445-453.
van KEULEN, L.J.M., SCHREUDER, B.E.C., MELOEN, R.H., POELEN-VAN, D.B.M., MOOIJ-
HARKES, G., VROMANS, M.E., and LANGEVELD, J.P. (1995):
Immunohistochemical detection oand localization of prion protein in brain tissue of sheep with natural scrapie.
Vet. Pathol. 32, 299-308.
van RHEEDE, T., SMOLENAARS, N.M., MADSEN, O., and JONG, W.W. (2003):
Molecular evolution of the mammalian prion protein.
Mol. Biol. Evol. 20, 111-121.
VIDAL, E., MÁRQUEZ, M., TORTOSA, R., COSTA, C., SERAFÍN, A., and PUMAROLA, M. (2006):
Immunohistochemical approach to the pathogenesis of bovine spongiform encephalopathy in its early stages.
J. Virol. Methods. 134, 15-29.
WATTS, J.C., and PRUSINER, S.B. (2014):
Mouse models for studying the formation and propagation of prions.
J. Biol. Chem. 289, 19841-19849.
WEISSMANN C., and FLECHSIG, E. (2003):
PrP knock-out and PrP transgenic mice in prion research.
Br. Med. Bull. 66, 43-60.
WEISSMANN, C. (1991):
A “unified theory” of prion propagation.
Nature 352, 679-683.
WELLS, G.A., and WILESMITH, J.W. (1995):
The neuropathology and epidemiology of bovine spongiform encephalopathy.
Brain Pathol. 5, 91-103.
WELLS, G.A., HANCOCK, R.D., COOLEY, W.A., and RICHARDS, M.S. (1989):
Bovine spongiform encephalopathy: diagnostic significance of vacuolar changes in selected nuclei of the
medulla oblongata.

Chapter 7- References 87
Vet. Rec. 125, 521-524.
WELLS, G.A., SCOTT, A.C., JOHNSON, C.T., and GUNNING, R.F. (1987):
A novel progressive spongiform encephalopathy in cattle.
Vet. Rec. 121, 419-420.
WELLS, G.A., SPIROPOULOS, J., HAWKINS, S.A., and RYDER, S. (2005):
Pathogenesis of experimental bovine spongiform encephalopathy: preclinical infectivity in tonsil and
observations on the distribution of lingual tonsil in slaughtered cattle.
Vet. Rec. 156, 401-407.
WESTERGARD, L., CHRISTENSEN, H.M., and HARRIS, D.A. (2007):
The cellular prion protein (PrP(C)): its physiological function and role in disease.
Biochim Biophys Act. 1772, 629-644.
WILESMITH, J.W., WELLS, G.A., CRANWELL, M.P., and RYAN, J.B. (1988):
Bovine spongiform encephalopathy: epidemiological studies.
Vet. Rec. 123, 638-644.
WILL, R.G., IRONSIDE, J.W., ZEIDLER, M., COUSENS, S.N., ESTIBEIRO, K., and ALPEROVITCH,
A. (1996):
A new variant of Creutzfeldt-Jakob disease in the UK.
Lancet 347, 921-925.
WILLIAMS, E.S., and MILLER, M.W. (2003):
Transmissible spongiform encephalopathies in non-domestic animals: origin, transmission and risk factors.
Rev. Sci. Tech. 22, 145-156.
WILLIAMS, E.S., and YOUNG, S. (1980):
Chronic wasting disease of captive mule deer: a spongiform encephalopathy.
J. Wildl. Dis. 16, 89-98.
WILLIAMS, E.S., and YOUNG, S. (1993):
Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and elk (Cervus elaphus
nelsoni).
Vet. Pathol. 30, 36-45.
WILLIAMS, L., BROWN, P., IRONSIDE, J., GIBSON, S., WILL, R., RITCHIE, D., KREIL, T.R., and
ABEE, C. (2007):

88 Chapter 7 - Reference
Clinical, neuropathological and immunohistochemical features of sporadic and variant forms of Creutzfeldt-
Jakob disease in the squirrel monkey (Saimiri sciureus).
J. Gen. Virol. 88, 688-695.
WISNIEWSKI, H.M., SIGURDARSON, S., RUBENSTEIN, R., KASCSAK, R.J., and CARP, R.I. (1996):
Mites as vectors for scrapie.
Lancet 347, 1114.
WYATT, J.M., PEARSONG, R., SMERDOTN, N., GRUFFYDD-JONES, T. J., and WELLSG, A. H.
(1990):
Spongiform encephalopathy in a cat.
Vet. Rec. 126, 513.
YE, X., SCALLET, A.C., KASCSAK, R.J., and CARP, R.I. (1998):
Astrocytosis and amyloid deposition in scrapie-infected hamsters.
Brain Res. 809, 277-287.
YI, J.J., and EHLERS, M.D. (2007):
Emerging roles for ubiquitin and protein degradation in neuronal function.
Pharmacol. Rev. 59, 14-39.
ZHANG, J., JIN, B., LI, L., BLOCK, E.R., and PATEL, J.M. (2005):
Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial
cytochrome-c oxidase in lung endothelial cells.
Am. J. Physiol. Cell Physiol. 288, 840-849.
ZLOTNIK, K., and RENIE, J.C. (1965):
Experimental transmission of mouse passaged scrapie to goats, sheep, rats and hamsters.
J. Comp. Pathol. 75, 147-157.

Chapter 8 - Acknowledgements 89
Chapter 8
Acknowledgements

90 Chapter 8- Acknowledgements
Chapter 8: Acknowledgements
Finally, I would like to thank all those people who made this thesis possible and an
unforgettable experience for me.
First of all, I would like to express my deepest sense of Gratitude (with a capital and bold g)
to my supervisor Professor Dr. Wolfgang Baumgärtner, who offered his continuous advice
and encouragement throughout the course of this thesis. I thank him for the systematic
guidance and great effort he put into training me in the scientific field. Like a charm!
I would also like to thanks my co-supervisors, Prof. Dr. Kirsten Haastert-Talini and Prof.
Dr. Gerd Bicker for the constructive co-supervision and brilliant comments and suggestions.
I would like to express my very sincere gratitude to Dr. Verena Haist for the support and
encouragement whenever I was in need throughout the project.
I place on record, my sincere thanks to Dr. Ingo Gerhauser and Dr. Ingo Spitzbarth for
their excellent guidance, caring, patience, for correcting manuscripts, and constructive
discussions. Without their guidance and persistent help this dissertation would not have been
possible.
I would like to thanks Prof. Martin H. Groschup and Dr. Christine Fast from Institute for
Novel and Emerging Infectious Diseases, Friedrich-Loeffler-Institut, Südufer, Greifswald-
Insel Riems for marvellously supporting this project by providing the samples for electron
microscopy and immunohistochemistry, and providing me the opportunity to work in OIE
manual lab under their supervision. It was really a memorable part of my life.
My very sincere thanks to all members of the technical staff, without whose help this work
would definitely not have been possible, especially Kerstin Rohn for her selfless support,
encouragement, and love given to me during the course of this thesis. With her guidance and
courage, I was able to learn electron microscopy technique quite easily and she always helped
me in each and every step of electron microscopy. I would also like to thanks Brigitte

Chapter 8 - Acknowledgements 91
Behrens for continuously helping me in developing the electron microscopic images. I also
thank Bettina Buck, Petra Grünig, Danuta Waschke, Kerstin Schöne and Caroline Schütz for
their inestimable help during immunohistochemistry.
I would like to thank Muhammad Akram Khan, who as a good friend was always willing
to help and give his best suggestions. Many thanks to my office mates; Dr. Florian
Hansmann, Barbara Raddatz, Annika Lehmbecker and Adnan Fayyad for giving me
great atmosphere for working. I would also like to thanks all my colleagues including Ning,
Lin, Yimin, Yanyong for the great and unforgettable time that we spent together in the
Department of Pathology.
I am deeply thankful to Higher Education Commission (HEC), Pakistan and Deutscher
Akademischer Austauschdienst (DAAD), Germany for providing a scholarship and PMAS-
Arid Agriculture University, Rawalpindi, Pakistan for providing me leave opportunity to
complete my Ph.D.
I take this opportunity to express the profound gratitude from my deep heart to my beloved
parents (especially my late father Bashir Ahmad, that was the greatest lost I have during my
thesis work. May his soul rest in peace. Ameen), and my siblings for their love. I would like
to thank especially my brother and his wife, Mr. and Mrs. Dr. Tanveer Ahmad for their
continuous support – both spiritually and materially. I also like to thank my kids and niece
Ahmad Abdullah, Fatima-binte-Nadeem, Imsaal Tanvir and Wania Tanvir. I am sorry
that I missed your childhood.
Finally, I would like to thank my wife, Faiza Nadeem. She was always there cheering me up
and stood by me through the good times and bad.



ISBN 978-3-86345-286-5
Verlag: Deutsche Veterinärmedizinische Gesellschaft Service GmbH35392 Gießen · Friedrichstraße 17 · Tel. 0641 / 24466 · Fax: 0641 / 25375
E-Mail: [email protected] · Internet: www.dvg.de
Mu
ham
mad
Nad
eem
Han
no
ver
2015