Embed Size (px)
Citation preview

JOURNAL OF POLYMER SCIENCE: PART C NO. 6, PP. 83-91
Kalorimetrische Messungen der Kristallisations- und Schmelzvorglnge an Polymeren
F. H. MULLER und H. MARTIN, Institut fiir Polymere, Phillips Universitat, Marburg/Lahn, West Germany
synopsis
Crystallizing and melting of polymers as a function of sample history is investigated by means of a differential thermal analytic method developed for quantitative evalua- tion. Crystallization and the subsequent melting of the same polymer occur at tem- peratures differing by 12 to 18OC. The melt temperature and the time of keeping the sample in the molten state, the cooling rate and the course of cooling have a reproducible effect on the subsequent melting. The crystttllization is followed at constant tempera- ture, and the crystallization rate is evaluated. Application of the Avrami method givee agreement with similar density and refractive index measurements.
Umwandlungserscheinungen in Polymeren, insbesondere Kristallisations- und Schmelzvorgange, werden heute vielfach mit einfacher Differential- thermoanalyse (DTA) untersucht. Die DTA-Methode kann jedoch im wesentlichen nur Informationen uber die Temperaturbereiche liefern, in denen Vorghge ablaufen, und nur gewisse qualitative Aussagen uber die Umwandlungswarmen.’ Fur quantitative Aussagen ist die Kenntnis der “spezifischen W arme,” die Warmeaufnahme, als Funktion der Temperatur erf orderlich .
Feste Polymere befinden sich eudem im allgemeinen nicht im thermo- dynamischen Gleichgewicht. Das bedeutet, das die “spezifische Warme” mit der Vorbehandlung variieren kann. Es tritt also das Problem auf, wie man quantitative c,-Messungen reproduzierbar erhalten kann. Dabei ist zu beachten, dass die Probe auch noch wahrend der Messung selbst einer Vorbehandlung unterliegt. Bei den Punkt-fur-Punkt-Messungen, etwa im adiabatischen Kalorimeter, besteht die Gefahr, dass wegen der Wartezeiten zum Temperaturausgleich an jedem Messpunkt die “Vorbe- handlung” in Hinblick auf das Zeitverhalten der Substanz verschieden ist.2
Dehierter, jedenfalls wegen dieser Zeiteff ekte leichter analysierbar, erscheinen uns daher Messungen mit zeitlich linear gefiihrter Aufheizung. Solche quasistatiunaren Messungen sind erreichbar, wenn man die Methode der Differentialthermoanalyse in quantitativer Richtung ausbaut. Das haben wir getan.a
Inzwischen wurde diese Apparatur in ihrer Empfindlichkeit und Eich- fahigkeit noch weiter ausgebaut.“ In einer theoretischen Untersuchung
83
84 F. 11. MULLER AND €1. MARTIN
0
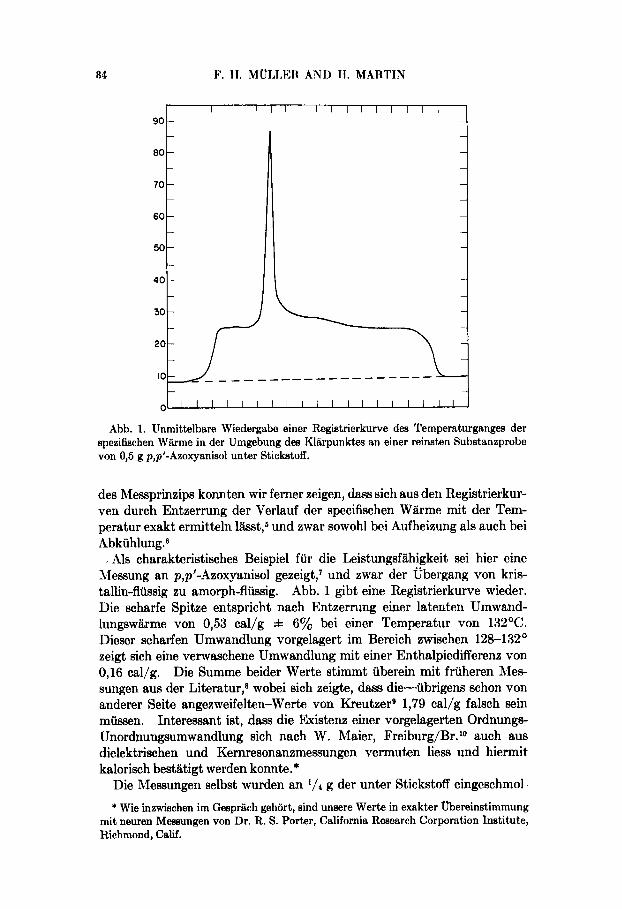
Abb. 1. Unmittelbare Wiedergabe einer Registrierkurve des Ternperaturganges der spezifischen Warrne in der Urngebung dea Klarpunktes an einer reinsten Substanzprobe von 0,5 g p,p'-Azoxyankol unter Stickstoff.
des Messprinzips konn ten wir ferner zeigen, dam sich aus den Registrierkur- ven durch Entzerrung der Verlauf der specifischen Warme mit der Tem- peratur exakt ermitteln lbst,S und zwar sowohl bei Aufheizung als auch bei Abkuhlung.6
Als charakteristisches Beispiel fur die Leistungsfahigkeit sei hier eine hlessung an p,p'-AzoxyanisoI gezeigt,' und zwar der Ubergang von kris- tallin-flussig zu amorph-flussig. Abb. 1 gibt eine Registrierkurve wieder. Die scharfe Spitze entspricht nach Entzerrung einer latenten Umwand- lungswarme von 0,53 cal/g f 6% bei einer Temperatur von 132OC. Dieser scharfen Umwandlung vorgelagert im Bereich zwischen 128-132' zeigt sich eine verwaschene Umwandlung mit einer Enthalpiedifferenz von 0,16 cal/g. Die Summe beider Werte stimmt uberein mit fruheren Ales- sungen aus der Literatur,* wobei sich zeigte, dass die-ubrigens schon von anderer Seite angezweifelten-Were von KreutzerB 1,79 cal/g falsch sein mussen. Interessant ist, dass die Existenx einer vorgelagerten Ordnungs- Unordnungsumwandlung sich nach W. Maier, Freiburg/Br. lo auch aus dielektrischen und Kernresonansmessungen vermuten liess und hiermit kalorisch bestatigt werden konnte.*
Die Messungen selbst wurden an ' /4 g der unter Stickstoff eingeschmol * Wie inzwitlchen im Gesprach gehort, sind unsere Werte in exakter Ubereinstimmung
niit neuren Messungen von Dr. R. S. Porter, California Research Corporation Institute, Richmond, Calif.
KALORIMETRISCIIE MESSUNGEN a5
250 360 O C
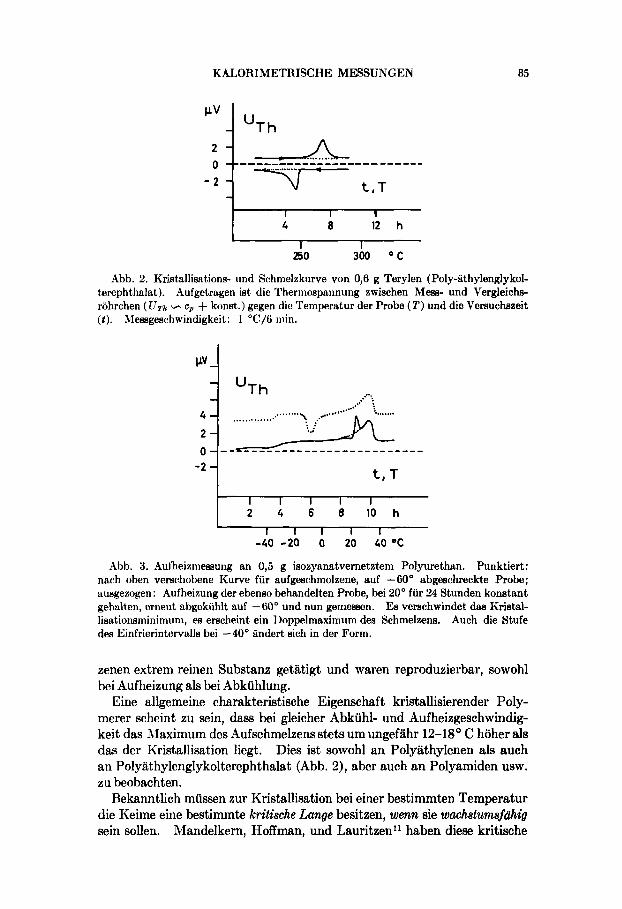
Abb. 2. Kristallisations- und Sc*hnielzkurve von 0,6 g Terylen (Poly-iithylenglykol- terephthalat). Aufgetragen ist die Therniospannung ewischen Mess- und Vergleichs- rohrchen ( [JTh - c p + konst.) gegen die Ternperatur der Probe (T) und die Versuchszeit ( 1 ) . Messgeschwindigkeit: 1 "C/6 niin.
2 4 l 0 -
-2 1 I I I I I
1 2 4 6 8 1 0 h
1 1 1 1 1 -40 -20 0 20 40 O C
Abb. 3. Aufheizmessung an 0,5 g isozyanatvernetztern Polyurethan. Punktiert : nach oben versohobene ICurve fur aufgeschrnolzene, auf - 60" abgeschreckte Probe; ausgexogen: Aufheizung der ebenso behandelten Probe, bei 20" fur 24 Stunden konstant gehalten, erneut abgekuhlt auf -60" und nun gemessen. Es verschwindet daa Kristal- lisationsminirnuni, es erscheint ein Doppelrnaximum des Schmelzens. Auch die Stufe des Einfrierintwvalls bei -40' andert sich in der Form.
zenen extrem reinen Substanz getatigt und waren reproduzierbar, sowohl bei Aufheizung als bei Abkuhlung.
Eine allgemeine charakteristische Eigenschaft kristallisierender Poly- merer scheint zu sein, dass bei gleicher Abkuhl- und Aufheizgeschwindig- keit das hlaximum des Aufschmelzens stets um ungefahr 12-18' C hoher als das der Nristallisation liegt. Dies ist sowohl an Polyathylenen als auch an Polyathylenglykolterephthalst (Abb. 2), aber auch an Polyamiden usw. zu beobachten.
Bekanntlich mussen zur Iiristrtllisation bei einer bestimmten Temperatur die Keime eine bestimmte kritische Lange besitzen, w a n sie wachstumsfdhig sein sollen. Mandelkern, Hoffman, und Lauritzen" haben diese kritische
86 F. H. MULLER AND H. MARTIN
Keimlange fur bundelformige Keime berechnet. Andererseits hangt die Schmelztemperatur des einzelnen Kristallits von seiner Kristullitlange ab, fur die Flory eine Besiehung abgeleitet hat. l2 Ein Vergleichbeider Formeln lilsst erkennen, dass unter bestimmten Voraussetsungen (vor allem die, dass der Schmelzpunkt der Kristalle nur von deren "Lange" abhangt) bei
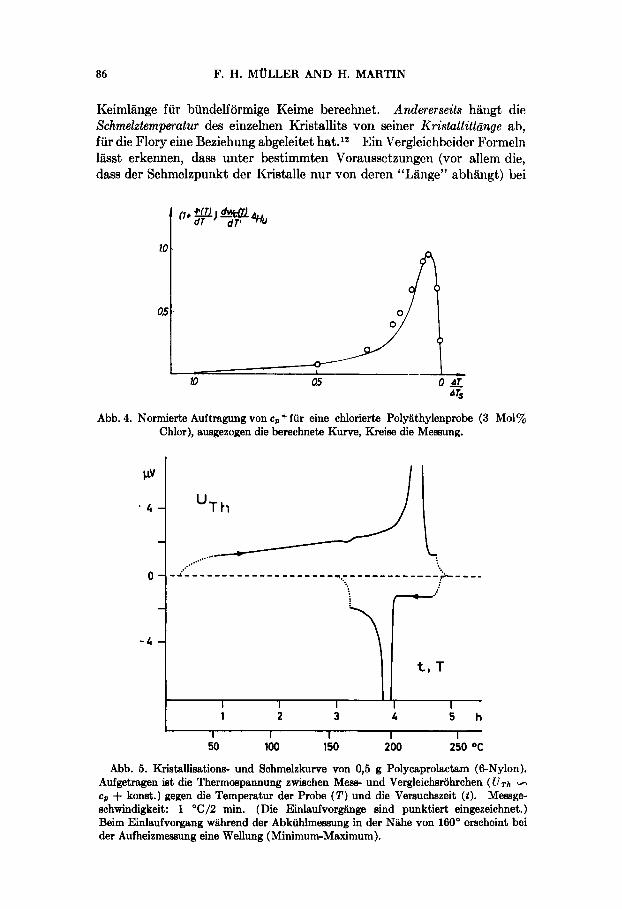
"G Abb. 4. Normierte Auftragung von cp+ fur eine chlorierte Polyiithylenprobe (3 Mol%
Chlor), ausgezogen die berechnete Kurve, Kreise die Meeaung.
0
-4
I I I I I I
I 1 2 3 4 5 h I I I I I
50 loo 150 200 250 O C
Abb. 5. Kristallisations- und Schmelakurve von 0,5 g Polycaprolactam (6Nylon). Aufgetragen ist die Thermospannung awischen Mess- und Vergleichsrohrchen ( U T h - cp + konst.) gegen die Temperatur der Probe (5") und die Versuchszeit ( t ) . Messge- schwindigkeit: 1 "C/2 min. (Die Einlaufvorgiinge sind punktiert eingeaeichnet.) Beim Einlaufvorgang wiihrend der Abkiihlmessung in der Niihe von 160' erscheint bei der Aufheizmessung eine Wellung (Minimum-Maximum).
KALORIMETRISCHE MESSUNGEN 87
l 2 4 6 8 h
I I I 120 160 200 oc
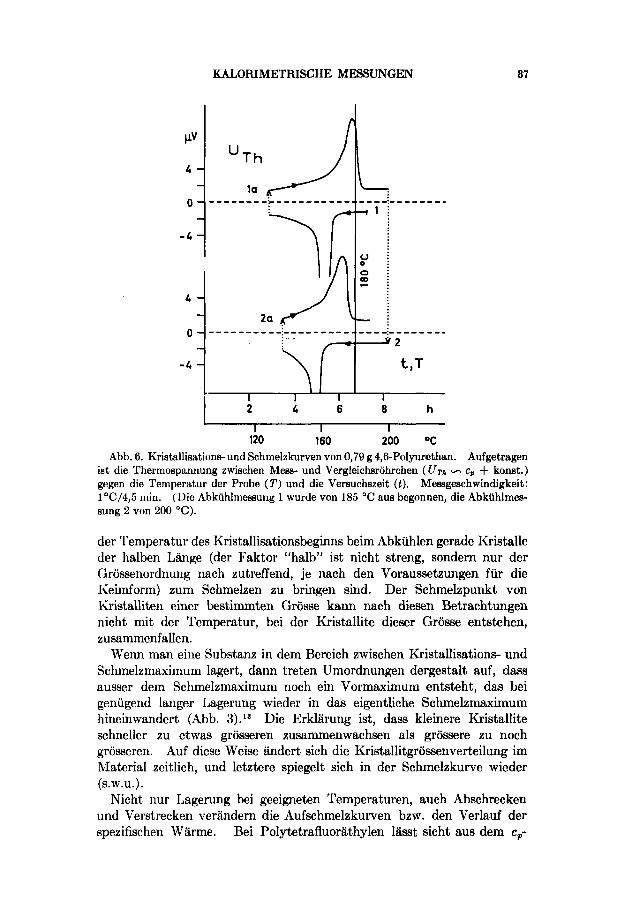
Abb. 6. Kristallisations- und Schmelzkurven von 0,79 g 4,BPolyurethan. Aufgetragen ist die Thermospannung zwischen Mess- und Vergleichsrohrchen ( UT,+ - cp + konst.) gegen die Temperatur der Probe (2') und die Versuchszeit ( 1 ) . Messgeschwindigkeit: 1 OC/4,5 min. (Die Abkiihlmessung 1 wurde von 185 "C aus begonnen, die Abkuhlmes- sung 2 von 200 "C).
der Temperatur des Kristallisationsbeginns beim Abkuhlen gerade Kristalle der halben Lange (der Faktor "halb" ist nicht streng, sondern nur der Grossenordnung nach zutreffend, je nach den Voraussetzungen fur die Keimform) zum Schmelzen zu bringen sind. Der Schmelzpunkt von Kristalliten einer bestimmten Grosse kann nach diesen Betrachtungen nicht mit der Temperatur, bei der Kristallite dieser Grosse entstehen, zusammenfallen.
Wenn man eitie Substanz in dem Bereich zwischen Kristallisations- und Schmelzmaximum lagert, dann treten Umordnungen dergestalt auf, dass ausser dem Schmelzmaximum noch ein Vormaximum entsteht, das bei genugend langer Lagerung wieder in das eigentliche Schmelzmaximum hineinwandert (Abb. 3). l3 Die Erklarung ist, dass kleinere Kristallite schneller zu etwas grosseren zusammenwachsen als grossere zu noch grosseren. Auf diese Weise andert sich die Kristallitgrossenverteilung im Material zeitlich, und letztere spiegelt sich in der Schmelzkurve wieder
Nicht nur Lagerung bei geeigneten Temperaturen, auch Abschrecken und Verstrecken veriindern die Aufschmelzkurven bzw. den Verlauf der spezifischen Warme. Bei Polytetrafluorathylen liisst sicht aus dem cp-
(S.W.U.).
88 F. 11. MULLEH AND 11. MARTIN
- 2 1
20 40 h
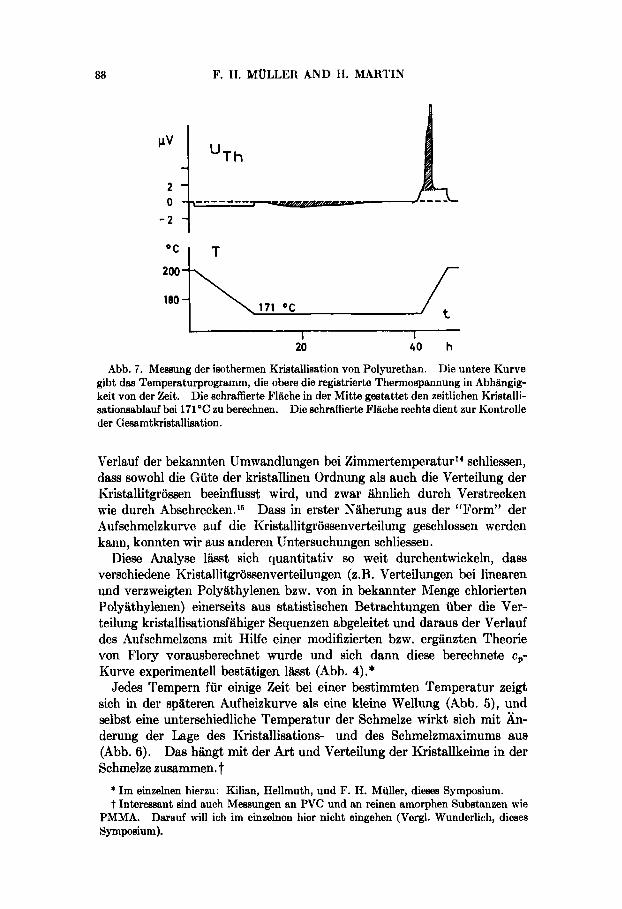
Abb. 7. Measung der isothermen Kristallisation von Polyurethan. Die untere Kurve gibt daa Temperaturprogramm, die obere die registrierte Thermospannung in Abhiingig- keit von der %it. Die schraffierte Fliiche in der Mitte gestattet den zeitlichen Kristalli- sationsablauf bei 171°C zu berechnen. Die schraffierte Fliiche rechta dient zur Kontrolle der Gesamtkristallisation.
Verlauf der bekannten Umwandlungen bei Zimmertemperatur" schliessen, dass sowohl die Gute der kristallinen Ordnung als auch die Verteilung der Kristallitgrossen beeinflusst wird, und zwar iihnlich durch Verstrecken wie durch Abschrecken.'s Dass in erster Naherung aus der "Form" der Aufschmelzkurve auf die Kristallitgrossenverteilung geschlossen werden kann, konnten wir aus anderen Untersuchungen schliessen .
Diese Analyse liLest sich quantitativ so weit durchentwickeln, dass verschiedene Iiristallitgrossenverteilungen (z.B. Verteilungen bei linearen und verzweigten Polyathylenen bzw. von in bekannter Menge chlorierten Polyathylenen) einerseits aus statistischen Betrachtungen uber die Ver- teilung kristallisationsfahiger Sequenzen abgeleitet und daraus der Verlauf des Aufschmelzens mit Hilfe einer modifizierten bzw. erganeten Theorie von Flory vorausberechnet wurde und sich dann diese berechnete cd Kurve experimentell bestatigen lflsst (Abb. 4).*
Jedes Tempern fur einige Zeit bei einer bestimmten Temperatur zeigt sich in der spateren Aufheizkurve als eine kleine Wellung (Abb. 5 ) , und selbst eine unterschiedliche Temperatur der Schmelze wirkt sich mit &I- derung der Lage des I~ristallisations- und des Schmelzmaximums aus (Abb. 6). Das hiingt mit der Art und Verteilung der Kristallkeime in der Schmelze zusammen. t
* Im einzelnen hierzu: Kilian, Hellmuth, und F. H. MUller, dieses Symposium. t Intermant sind auch Messungen an PVC und an reinen amorphen Substanzen wie
Darauf will ich im einaelnen hier nicht eingehen (Vergl. Wunderlich, dieses PMMA. Symposium).
KALORIMETRISCHE MESSUNGEN
1 . a
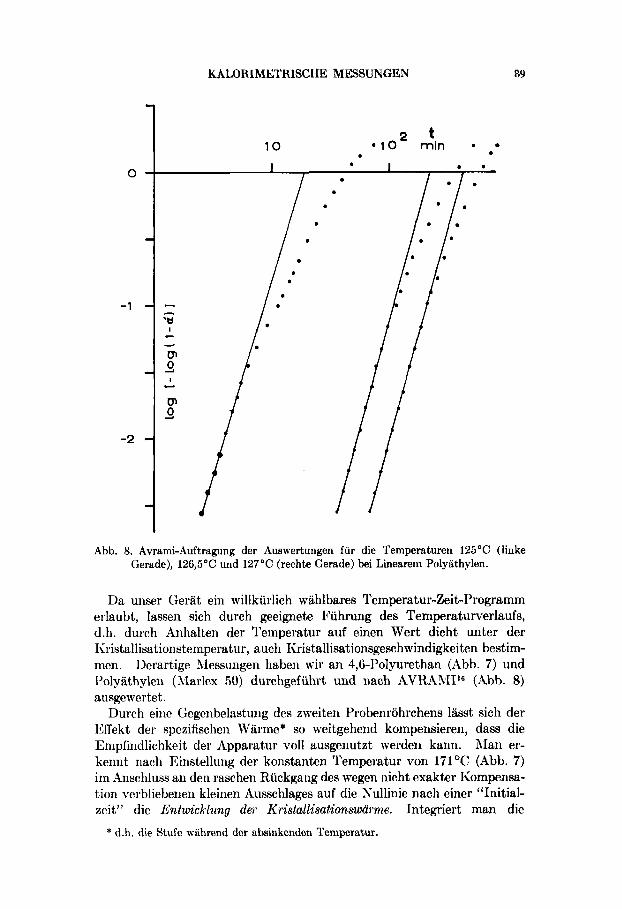
Abb. 8. Avrami-Auftragung der Auswertuugen fur die Ternperaturen 125OC (linke Gerade), 126,5OC und 127°C (rechte Gerade) bei Lineareni Polyathylen.
Da unser Gerat ein willkurlich wahlbares Temperatur-Zeit-Programm erlaubt, lassen sich durch geeignete Fuhrung des Temperaturverlaufs, d.h. durch Arihalten der Temperatur auf einen Wert dicht unter der Iiristallisationstemperatur, auch Kristallisationsgeschwindigkeiten bestim- men. Derartige AIessungen haben wir an 4,6-Polyurethan (Abb. 7) und Polyathylen (Rlarlex 50) durchgefuhrt und nach AVRARII116 (Abb. 8) ausgewertet.
Durch eine Gegenbelastung des zweiten Probenrohrchens lasst sich der Effekt der spezifischen Warme* so weitgehend kompensieren, dass die Empfiiidlichkeit der Apparatur voll ausgenutzt werden kann. Alan er- kenrit riach Rinstellung der konstanten Temperatur von 171 O C (Abb. 7) im Aiischluss an den raschen Ruckgang des wegen riicht exakter Iiompensa- tion verbliebenen kleinen Ausschlages auf die Nullinie nach einer “Initial- zeit” die Eniwich-lung der Kristallisationswa1.nle. Integriert man die
* d.h. die Stufe withrend der absinkenden Temperatur.

90 F. H. MULLER AND H. MARTIN
Flllchen jeweils bis zu verschiedenen Zeiten und vergleicht sie mit der Gesamtflache, so stellt diese Kurve F(t) /Fo gegen t die seitliche Ent- wicklung der Kristallisation dar. Die anschliessende Aufheizung dient lediglich dazu, die Gesamtkristallisation nochmals zu kontrollieren : beide schraff ierte FlLchen sollen ubereinstimmen.
Insbesondere die Messungen an Polyathylen ergaben weitgehende Be- statigung der Werte fur die Konstanten aus der Avrami-Formel mit den aus der Literatur auf Grund von Dichtemessungen bisher bekannten Werten. l7 Unsere Messungen zeigen die in der Avrami-Auftragung aus Dichten ebenfalls bekannten systematischen Abweichungen vom linearen Verlauf mit der Zeit (mit wachsendem Gehalt an Kristallisiertem), die einige Autoren ah Hohlraumbildung bei fortschreitender Kristallisation diskutiert haben. Unsere kalorischen Messungen bestatigen nun die allein durch Hohlraum- und Rissbildungen bedingt sind (was ja eine falsche Dichtebestimmung verursachen wiirde und damit eine nur vorgetauschte Abweichung bedeuten wurde), sondern dass sie offenbar spezifisch f i i r das Kristallwachstum Polymerer sind. Sie beginnen zudem schon bei relativ niedrigen Kristallisationsgraden, bei denen sich die Kristallite noch kaum beruhren durften, und sie treten bei umso kleineren Werten auf, je tiefer die gewahlte Kristallisationstemperatur ist. Der Vorteil der kalorimetrischen Methode scheint in ihrer hohen Empfindlichkeit vor allem im Beginn der Kristallisation zu liegen.
Literatur verzeichnis
1. Man vergleiche hiersu die verschiedensten Arbeiten auf diesem Symposium. 2. Die Frage der Wartereiten wird dbkutiert in den Arbeiten von M. Dole and Mitar-
3. Martin, H., und F. H. Mtiller, Kolloid-Z., 172.97 (1960). 4. Verbeeaerte Apparatur noch nicht veroffentlicht. 5. Adam, G., und F. H. Miiller, Kohid-Z., 192, in Erscheinen. 6. Martin, H., und F. H. Miiller, Kolloid-Z., 191,1(1963). 7. Mtiller, F. H., und H. Martin, Kolloid-Z., 187,107 (1963). 8. Schenk, R., Kristalline Flasigkeiten, pp. 84-89, Leipsig, Germany, 1905. 9. Kreutzer, H., Ann. Phys., 33,192 (1938).
beitern.
10. Maier, W., und Alfred Saupe, Zs. Naturforsch., 15A. 287 (1960); ibid., 16A. 816 (1961).
11. Mandelkern, L., J. Appl. Phys., 26, 443 (1955); L. Mandelkern, F. A. Quinn, und P. J. Flory, J. Appl. Phys., 25,830 (1954); J. I. Lauritsen und J. D. Hoffman, J. Rea. Natl. Bur. Res., 64A, 73 (1959). 12. Flory, P. J., J . Chem. Phys., 17,119 (1960). 13. Miiller, F. H., und H. Martin, Kolloid-Z., 171,119 (1960). 14. Muller, F. H., und H. Martin, Kolloid-Z., 188,19 (1963). 15. Kilian, H. G., und E. Jenckel, Zs. Elektrochem. 63,951 (1959). 16. Avrami, M., J. Chem. Phys., 7, 1103 (1939); ibid., 8, 212 (1940); ibid., 9, 177
(1941). 17. Rohleder, J., und H. A. Stuart, Makromol. Chem., 41,111 (1960); L. Mandelkern,
F. A. Quinn, und P. J. Flory, J. Appl. Phys., 25.830 (1954); J. Rabesiaka und A. J. Ko- vaca, J . Appl. Phys., 32,2314 (1961).

KALORIMETRISCHE MESSUNGEN 91
Zusammenfassung Kristallisieren und Schmelzen von Polymeren lLs t sich mit einer fur quantitative
Auswertungen weiterentwickelten Differentialtherrnoanalyse ah Funktion der Vorge- schichte untersuchen. Die Kristallisat.ion und daa anschliessend durchgefuhrte Schmel- sen desselben Polymeren erfolgt bei Temperaturen, die sich urn 12 bis 18°C unter- scheiden. Die Temperatur der Schmelze und die Zeitdauer des aufgeschmolzenen Zustandes, die Geschwindigkeit und der zeitliche Verlauf der Abkuhlung beeinflueaen den nachfolgend gemessenen Schmelzverlauf in reproduzierbarer Weise. Aus der Ver- folgung der Kristallisation bei konstanter Temperatur lrtssen sich Kristallisations- geschwindigkeiten ermitteln, deren Auswertung nach Avrami in hreinst imniung mit analogen Meeaungen an Dichte und Brechungsindex stehen.
Rhsum6 La cristallisation et la fusion dee polymbres peuvent &re btudidea comme une fonction
des traitements antkrieura Bur la base d’une analyse thermique diffbrentielle dbvelopp6e pour perniettre une 6valuation quantitative. La cristallisation et la fusion consecutive de ce r n h e polymbre ont lieu A dea temperatures qui se differencient par un &art de 12 ti 18°C. La temperature de la mame en fusion et la d u r b de vie de 1’6tat de fusion, la viteeae et le coura du refroidissement en fonction du temps influencent de manibre re- productible le dbroulement de la fusion mesurb par la suite. En suivant le deroulement de la cristallisation B temp6rature constante, on peut determiner des vitesses de criatalli- sation dont lea calcule suivant Avrami sou8 en accord avec des mesures analogues de densite e t d’indice de refraction.





























