Embed Size (px)
Citation preview

Modellierung und Analyse von Fehlern und Störungen in
der präparativen Flüssigchromatographie
vorgelegt von
Dipl.-Ing. Konstantin Lenz
von der Fakultät III
- Prozeßwissenschaften –
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Ingenieurwissenschaften
- Dr. Ing. –
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr.-Ing. Martin Kraume
Berichter: Prof. Dr.-Ing. Wolfgang Arlt
Berichter: Prof. Dr.-Ing. Günter Wozny
Tag der wissenschaftlichen Aussprache: 11. Juli 2003
Berlin 2004
D83

Danksagung
Diese Arbeit entstand während meiner Tätigkeit als wissenschaftlicher Mitarbeiter am Fachgebiet
Thermodynamik und thermische Verfahrenstechnik der Technischen Universität Berlin.
Meinem Doktorvater Prof. Dr.-Ing. Wolfgang Arlt möchte ich danken für die Möglichkeit bei Ihm zu
promovieren, für das anspruchsvolle Thema, die Freiheiten bei der Gestaltung der Arbeit und
dafür, daß er bei Problemen immer Zeit und ein offenes Ohr hatte.
Mein besonderer Dank gilt auch Prof. Dr.-Ing. Günter Wozny, nicht nur dafür, dass er sich bereit
erklärt hat als zweiter Gutachter zu fungieren, sondern insbesondere auch dafür, daß ich die
gPROMS und FLUENT-Lizenzen und viel Rechenzeit des Fachgebiets Dynamik und Betrieb
technischer Anlagen für meine Forschung in Anspruch nehmen durfte.
Herrn Prof. Dr.-Ing. M. Kraume danke ich für die Übernahme des Vorsitzes des Promotions-
ausschusses.
Für die exzellente Zusammenarbeit und die zahllosen fruchtbaren Diskussionen sowie seine Hilfe
inbesondere bei der Einarbeitung in das CFD-Gebiet und bei der Vorbereitung für die
Promotionsprüfung möchte ich Henning Boysen danken.
Ganz besonders möchte ich mich auch bei York Beste bedanken, der mir insbesondere in der
Anfangszeit mit seinem großen Erfahrungsschatz zur Seite stand und dessen Ideen und
Ratschläge immer sehr wertvoll waren.
Für die ausgezeichnete fachliche Zusammenarbeit und für zahlreiche Anregungen im Rahmen der
Chromatographiegruppe möchte ich Dirk-Uwe Astrath, Tobias Laiblin und Mark Lisso danken.
Ganz besonders bedanken möchte ich mich auch bei meinem Mitarbeiter Jürgen Scheer, der mir
nicht nur mit seinen exzellenten Programmierkenntnissen sondern auch im Labor bei manch nicht
so ganz einfachem Experiment zur Seite stand. Henning Boysen, Dirk-Uwe Astrath und Jürgen
Scheer möchte ich ferner für das Korrekturlesen meiner Arbeit danken.
Auch gilt mein Dank für so vieles, insbesondere das tolle Klima am Institut, meinen Kollegen
Andreas Böhme, Götz Fischer, Steffi Hiller, Carsten Jork, Matthias Seiler, Thomas Schneider, Irina
Smirnova, Oliver Spuhl, Supakij Suttiruengwong, Feelly Tumakaka und natürlich meinem

Inhaltsverzeichnis
ehemaligen Zimmerkollegen Marko Tischmeyer. Danken möchte ich auch ganz besonders
Susanne Hoffmann für ihre vielfache Unterstützung und ihre Nachsichtigkeit hinsichtlich meiner
Laborarbeitsplatzgestaltung und Herrn Schmidt für seine Hilfe und seine Betreuung der Versuche
mit der statischen Adsorptionsapparatur. Weiterhin gilt mein Dank Herrn Stübing, Christina und
Sylva in der Halle, Birgit und Manuela im Sekretariat sowie Lothar, Klaus, Shorty, Dietmar und
Uwe in der Werkstatt.
Es war eine unvergeßlich schöne Zeit...
III

Inhaltsverzeichnis
Inhaltsverzeichnis
Inhaltsverzeichnis ............................................................................................................................ IV
Kurzfassung ....................................................................................................................................VII
Notation......................................................................................................................................... VIII
1 Einleitung ....................................................................................................................................1
2 Grundlagen der Flüssigchromatographie ..................................................................................3 2.1 Begriffsbestimmung ........................................................................................................................ 3 2.2 Die Adsorption in der flüssigen Phase........................................................................................... 4 2.3 Der Adsorptionsexzeß ..................................................................................................................... 5 2.4 Die Anwendung der Flüssigchromatographie .............................................................................. 8
2.4.1 Das Chromatogramm ...................................................................................................................................9 2.4.2 Die Porosität ..............................................................................................................................................12 2.4.3 Die Adsorptionsisotherme ..........................................................................................................................14 2.4.4 Grundlagen der Kinetik ..............................................................................................................................19 2.4.5 Die Van Deemter Kurve .............................................................................................................................22
2.5 Die präparative HPLC.................................................................................................................. 23 2.6 Die Gegenstromchromatographie................................................................................................ 25
2.6.1 Die wirkliche Gegenstromchromatographie (TMB)...................................................................................26 2.6.2 Funktionsweise der Simulierten Gegenstromchromatographie (SMB) ......................................................28 2.6.3 Modellierung der Gegenstromchromatographie ........................................................................................31 2.6.4 Auslegung und Optimierung einer Trennung mit einer SMB-Anlage.........................................................40
2.7 Berechnung des Druckverlustes einer chromatographischen Anlage...................................... 45
3 Methoden zur Bestimmung von Adsorptionsisothermen.........................................................48 3.1 Statische Meßmethoden................................................................................................................ 48 3.2 Dynamische Meßmethoden .......................................................................................................... 50
3.2.1 Frontalanalyse............................................................................................................................................50 3.2.2 Die Perturbations- oder Pulsmethode........................................................................................................52 3.2.3 Peak-Fitting-Methode.................................................................................................................................53 3.2.4 Elution am charakteristischen Punkt..........................................................................................................54
3.3 Bestimmung der Porosität ............................................................................................................ 54 3.3.1 Bestimmung der Gesamtporosität ..............................................................................................................55 3.3.2 Bestimmung der externen Porosität............................................................................................................55 3.3.3 Bestimmung der internen Porosität ............................................................................................................55
4 Fehleruntersuchungen in der Literatur ...................................................................................57 4.1 Vergleiche von statischen und dynamischen Meßmethoden zur Bestimmung von
Adsorptionsisothermen............................................................................................................................. 57 4.2 Fehlerquellen in der Flüssigchromatographie............................................................................ 59
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen......................62 5.1 Durchführung der Experimente .................................................................................................. 62
5.1.1 Auswahl eines Testsystems .........................................................................................................................63
IV

Inhaltsverzeichnis
5.1.2 Durchführung der statischen Messungen ...................................................................................................65 5.1.3 Durchführung der dynamischen Messungen ..............................................................................................71
5.2 Vergleich der Meßergebnisse ....................................................................................................... 81 5.2.1 Umrechnung der statischen Exzeßisothermen............................................................................................81 5.2.2 Vergleich der Adsorptionsisothermen ........................................................................................................91
5.3 Fehlerbetrachtung......................................................................................................................... 94 5.4 Auswirkungen der Abweichungen auf simulierte Peaks ........................................................... 95
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen ..........................................97 6.1 Methodik des Vorgehens .............................................................................................................. 97 6.2 Modellierung und Untersuchung von Effekten verursacht durch ein abgesacktes Festbett.. 99
6.2.1 Experimentelle Untersuchung des abgesackten Festbettes ........................................................................99 6.2.2 Simulation des abgesackten Festbettes unter Verwendung von FLUENT und gPROMS.........................100
6.3 Modellierung und Untersuchung von Effekten verursacht durch eine partiell undurchlässige
Auslaßfritten............................................................................................................................................ 103 6.4 Modellierung und Untersuchung von Effekten verursacht durch radiale Heterogenität im
Festbett ..................................................................................................................................................... 105 6.4.1 Grundsätzliche Untersuchungen unter Verwendung von FLUENT .........................................................105 6.4.2 Theoretische Abschätzung der radialen Dispersion.................................................................................107 6.4.3 Simulationsstudien unter Verwendung von gPROMS und FLUENT........................................................109
6.5 Modellierung und Untersuchung von Effekten verursacht durch Kanalbildung ................. 112
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie .116 7.1 Beschreibung der beispielhaft zur Untersuchung herangezogenen SMB-Anlage................. 116 7.2 Klassifizierung von Fehlern bei der Simulierten Gegenstromchromatographie................... 119 7.3 Entwicklung eines vereinfachten integralen Bilanzmodells .................................................... 120
7.3.1 Komponententransport durch nur einen Abschnitt...................................................................................121 7.3.2 Komponententransport in zwei Richtungen..............................................................................................122 7.3.3 Transport einer Komponente durch alle Abschnitte in identische Richtung ............................................123
7.4 Untersuchung der Effekte ausgelöst durch globale Fehlfunktionen ...................................... 125 7.4.1 Vorgehen bei der Untersuchung von abweichenden Volumenströmen der Pumpen ................................126 7.4.2 Abweichender Volumenstrom der Eluentpumpe.......................................................................................129 7.4.3 Abweichender Volumenstrom der Extraktpumpe .....................................................................................134 7.4.4 Abweichender Volumenstrom der Feedpumpe .........................................................................................136 7.4.5 Abweichender Volumenstrom der Raffinatpumpe ....................................................................................137 7.4.6 Zusammenfassung der Charakteristika ....................................................................................................139
7.5 Modellierung und Untersuchung von lokalen Fehlfunktionen ............................................... 140 7.5.1 erhöhte axiale Dispersion in einer Säule..................................................................................................141 7.5.2 Simulation einer Leckage im System ........................................................................................................142 7.5.3 Simulation undichter Ventile ....................................................................................................................145 7.5.4 Simulation geschlossener Ventile .............................................................................................................147
7.6 Ansätze zur Fehlererkennung .................................................................................................... 152 7.6.1 Untersuchung der Fehler im Anfahr- und Übergangszustand .................................................................152 7.6.2 Möglichkeiten der Fehlererkennung.........................................................................................................154
8 Zusammenfassung und Ausblick............................................................................................156
Literatur ..........................................................................................................................................160
9 Anhang.....................................................................................................................................173
V

Inhaltsverzeichnis
A Versuchsergebnisse .................................................................................................................173 A.1 Versuchsergebnisse der statischen Bestimmung von Adsorptionsisothermen .......................... 173 A.2 Versuchsergebnisse der dynamischen Bestimmung von Adsorptionsisothermen..................... 178
B Vorgehensweise bei der Durchführung der Experimente mit der Zirkulationsapparatur...184
C Auswertung der Biegeschwingermeßwerte.............................................................................187
D Herleitung der Minka-Gleichung ...........................................................................................191
E Modellierung von globalen Fehlfunktionen ..........................................................................194 E.1 Modellierung eines Komponententransport durch nur einen Abschnitt ................................... 194 E.2 Modellierung des Transports einer Komponente in zwei Richtungen ....................................... 196 E.3 Modellierung des Transports einer Komponente durch alle Abschnitte in identische Richtung
................................................................................................................................................................... 199
VI

Kurzfassung
Kurzfassung
Die vorliegende Arbeit betrachtet die Auswirkungen möglicher Fehlfunktionen und Störungen auf
ausgewählte Systeme der präparativen Flüssigchromatographie. Diese Betrachtung konzentriert
sich dabei auf drei Komplexe:
Zum ersten auf den Vergleich der Daten, die bei Nutzung von statischen und dynamischen
Methoden zur Bestimmung von Adsorptionsisothermen gemessen wurden. Zum Einsatz kamen
dabei die Zirkulationsmethode, die Frontalanaylse und die Perturbationsmethode. Werden die
experimentellen Ergebnisse in einen identischen Bezugsrahmen überführt, so ist es möglich, die
Ergebnisse miteinander zu vergleichen. Führt man diese Prozedur für die Daten der beiden ver-
messenen Testsysteme durch, so kann unter Bestimmung und Einrechnung der eingeführten
effektiven Porosität festgestellt werden, daß die Differenzen in den experimentellen Ergebnissen
geringer sind als die berechneten Meßfehler.
Der zweite Komplex befaßt sich mit fehlerhaft gepackten Festbetten präparativer chromato-
graphischer Säulen. Von Interesse sind dabei die Auswirkungen bestimmter Packungsfehler auf
die die Säule durchlaufende Peaks. Unter Verwendung experimenteller Ergebnisse sowie der
Nutzung der Simulationsumgebungen gPROMS und FLUENT konnte dabei festgestellt werden,
daß sich die Auswirkungen eines Hohlraumes über einem abgesackten chromatographischen
Festbett mit Hilfe eines vorgeschalteten idealen Rührkessels beschreiben lassen. Weitere Punkte
betrafen eine teilweise undurchlässige Auslassfritte, die nur zu einer etwas geringeren Retentions-
zeit führt und den Peak nur in geringem Maße verändert. Bei den nur mittels Simulationen
untersuchten Phänomenen der radialen Heterogenität und der Kanalbildung im Festbett konnte
das Auftreten von Peakassymmetrien und Doppelpeakverhalten festgestellt werden.
Der dritte Komplex schließlich beleuchtet die Auswirkungen bestimmter Fehler beim Betrieb der
Simulierten Gegenstromchromatographie (SMB). Diese wurden mit Hilfe dynamischer Simu-
lationen unter Variation verschiedener Parameter erforscht, um Ihre Gesetzmäßigkeiten zu unter-
suchen. Dabei konnten zwei Klassen von Fehlern ausgemacht werden: Die Auswirkungen globaler
Fehler sind in jeden Takt identisch und können zusätzlich mit Hilfe eines Massenbilanzmodells
beschrieben werden. Die Auswirkungen sind dabei primär von den Konstellationen dominierender
Transportrichtungen abhängig. Lokale Fehler zeigen dagegen in den verschiedenenen Takten
unterschiedliche Charakteristika und sind nur mittels dynamischer Simulationen zu beschrieben.
Die Ergebnisse dieser Untersuchungen zeigen die Möglichkeit, ein System zur Fehlerdiagnose für
SMB-Anlagen zu entwickeln.
VII

Notation
Notation
Formelzeichen:
A m2/g spezifische Oberfläche
ai - Parameter des Redlich-Kister-Polynoms
A m Parameter der Van Deemter Kurve
A m2 Oberfläche
Ai - Parameter der Ching-Isotherme
B - Parameter der Geradengleichung
bi m3/kg Krümmungsparameter der Langmuir-Gleichung
bi mmol/g Beladungskapazität des Adsorbens
Bi m2/s Parameter der Van Deemter Kurve
Bij - Parameter der Ching-Isotherme
C g/l Konzentration
CDax m Konstante des axialen Dispersionskoeffizienten
Ci s Parameter der Van Deemter Kurve
D mm Durchmesser
Dax m2/s axiale Dispersionskoeffizient
Dm,i m2/s molekularer Diffusionskoeffizient
F N/m2 Fugazität
F - Phasenverhältnis
hk - Kozeny-Koeffizient
H - Heaviside-Koeffizient
HETP m Höhe einer theoretischen Trennstufe
keff,fl,I m/s effektiver Stoffübergangskoeffizient des Fluidfilmmodells
keff,fest,i m/s effektiver Stoffübergangskoeffizient des Festfilmmodells
K - Gleichgewichtskoeffizient, Selektivität
L m Länge
M g Masse
m& kg/h Massenstrom
mi - Parameter der Ching-Isotherme
M g/mol molare Masse
N mmol Stoffmenge
nij - Parameter der Ching-Isotherme
NTU - Anzahl der theoretischen Trennstufen
VIII

Notation
O m2 Oberfläche
Q g/l Beladung
Q* g/l Beladung der stationären Phase im Gleichgewicht mit ci
qs g Beladung der gesamten chromatographischen Säule
R - Verhältnis der molaren Volumina
RS - Auflösung
RMS - Wurzel der Summe der mittleren Fehlerquadrate
T s Zeit
t‘ mm Schichtdicke der adsorbierten Phase
t0 s Totzeit
tR s Retentionszeit
T s Schwingungsdauer
V m3/mol molares Volumen
vs m3/kg spezifisches Volumen
V m3 Volumen
V& m3/h Volumenstrom
W m/s Zwischenkorngeschwindigkeit
W0 m/s Leerrohrgeschwindigkeit
wc,i m/s Konzentrationswanderungsgeschwindigkeit
wrel m/s Relativgeschwindigkeit zwischen Flüssigkeit und Feststoff
W - Wahrscheinlichkeit
X mol/mol Molenbruch
Y - Parameter der Geradenglechung
Z m Koordinate in axiale Richtung
∆z m Ortsschrittweite (Länge einer Stützstelle)
griechische Formelzeichen
α - Trennfaktor
β - Stabilitätsfaktor
γ - Aktivitätskoeffizient
γ1, γ2 - Konstanten zur Berechnung von Dax
η Pa s dynamische Viskosität
ε - Porosität
Φ J/g spezifische Benetzungsenthalpie
IX

Notation
µ1,ι s erstes bezogenes Anfangsmoment
µ2, s2 zweites zentrales Moment
ρ kg/m3 Dichte
σ s2 Varianz
σ N/m Oberflächenspannung
τ s Taktzeit
ω s Basisweite
Indizes (hochgestellt)
0 Ausgangszustand
ad Adsorbens
aus aus einem Abschnitt austretend
E Exzeß
ein in einen Abschnitt eintretend
N n-tes Konzentrationsniveau
SMB für den Fall des simulierten Gegenstromprozesses
TMB für den Fall des wirklichen Gegenstromprozesses
` Adsorbat-Phase
I Konzentrationsniveau I (unteres Konzentrationsniveau)
II Konzentrationsniveau II (oberes Konzentrationsniveau)
Indizes (tiefgestellt)
1 Komponente 1
2 Komponente 2
A Komponente A
B Komponente B
D Desorbent
eff effektiv
ext extern
Ex Extrakt
Feed Feed
fest feste Phase, Feststoff
fl flüssige Phase, Flüssigkeit
X

Notation
ges gesamt
i, j Komponente i, j
int intern
k Laufindex für den Abschnitt
M Makeup
n Laufindex der Säule
p Produktstrom
P Partikel
PF Partikelfeststoff
Pr Laufindex für den Produktstrom
Raff Raffinat
Ref Referenz
S Säule
Z Zielkomponente
I, II, III, IV Abschnitte des SMB bzw. TMB-Prozesses
Abkürzungen
DCM Dichlormethan
FA Frontalanalyse
PM Perturbationsmethode
SMB Simulated Moving Bed
TMB True Moving Bed
XI

1 Einleitung
1
1 Einleitung
Seit der Russe Tswett Ende des 19.Jahrhundert das Prinzip der Chromatographie erkannte, wird
dieses Trennverfahren heute nicht mehr nur als Analyseverfahren verwendet, sondern auch immer
mehr als Methode zur Isolierung und Gewinnung von Wertstoffen im präparativen Maßstab
eingesetzt. Der Vorteil der Chromatographie gegenüber konventionellen Trennverfahren be-
gründet sich in einer hohen Selektivität aufgrund des Einsatzes zweier Hilfsstoffe und sowie der
schonenden Produktbehandlung aufgrund der Vermeidung einer klassischer Phasenumwandlung.
Durch die Entwicklung der Computertechnik und den damit verbundenen, gewachsenen
Ressourcen für die Simulationstechnik ist es möglich, immer komplexer gestaltete
chromatographische Systeme zu modellieren und vor allem zu simulieren, um sie in die Lage zu
versetzen, immer schwieriger werdende Trennprobleme mit steigender Effizienz lösen zu können.
Mit steigender Komplexität eines technischen Systems steigt aber oft auch seine Empfindlichkeit
gegenüber Störungen, und die Komplexität macht die Erkennung der Ursachen immer
schwieriger.
Neben den Problemen der Fehlererkennung verlangen aber auch steigende Anforderung an die
Sicherheit von Produkten vom Anwender wachsende Sorgfalt hinsichtlich der Qualität seiner
Produkte und damit natürlich auch steigende Qualitätsansprüche hinsichtlich seiner
Herstellungsverfahren.
Ziel dieser Arbeit ist es, die Auswirkungen bestimmter Fehler und Störungen auf
flüssigchromatographische Trennungen zu untersuchen. Dabei werden die Begriffe „Fehler“ bzw.
„Fehlfunktion“ und „Störung“ im folgenden interpretiert gemäß der Definiton in der Richtlinie
VDI/VDE 3542, wie sie von Isermann (1994) wiedergegeben und ausführlich erläutert werden: „Ein
Fehler ist eine unzulässige Abweichung mindestens eines Merkmals einer Betrachtungseinheit“,
eine Störung ein „nach Beanspruchungsbeginn entstandener vorübergehender Fehler“. Im
Rahmen dieser Arbeit werden also Phänomene untersucht, die eine Abweichung des
Betriebszustandes vom Sollzustand verursachen können.
Da solche Phänomene ebenso wie die angewendeten Verfahren mannigfaltiger Natur sein
können, konzentriert sich diese Arbeit zunächst in Kapitel 2 auf einen Überblick über die theore-
tischen Grundlagen der Flüssigchromatographie, Kapitel 3 erläutert die verwendeten Methoden
zur Bestimmung von Adsorptionsisothermen.

1 Einleitung
2
Nach einem Überblick über Literaturstellen, die sich mit ähnlichen Fragestellungen beschäftigt
haben konzentrieren sich die Untersuchungen von Fehlern und Störungen in der präparativen
Flüssigchromatographie im Rahmen dieser Arbeit auf drei Themenkomplexe: Kapitel 5 behandelt
zunächst den Vergleich von statischen und dynamischen Verfahren zur Bestimmung von
Adsorptionsisothermen. Ihre Kenntnis ist der Imperativ jeder präparativen, chromatographischen
Trennung. Daher ist die Frage, ob diese beiden Arten von Verfahren zu ähnlichen Ergebnissen
führen oder ob die Wahl der Meßmethode bereits einen Fehler in Auslegung einbringen kann,
essentiell. Danach erfolgt ein Übergang hin zu Fehlern, die im Betrieb chromatographischer
Systeme auftreten können. Kapitel 6 beschäftigt sich dabei mit den Auswirkungen von Fehlern,
wie sie speziell in präparativen Batch-Chromatographiesäulen auftreten können, Kapitel 7
betrachtet die Auswirkungen von Fehlfunktionen und Störungen, wie sie bei einem kontinuierlichen
Verfahren auftreten können, nämlich dem der simulierten Gegenstromchromatographie (SMB).

2 Grundlagen der Flüssigchromatographie
2 Grundlagen der Flüssigchromatographie
3
2.1 Begriffsbestimmung
Unter dem Begriff der Adsorption versteht man allgemein das Anhaften von Molekülen aus einer
fluiden Phase an einer Phasengrenze, das durch molekulare Anziehungskräfte verursacht wird.
Die fluide Phase kann dabei eine Flüssigkeit sein (Flüssigadsorption), sie kann aber auch
gasförmig sein (Gasadsorption). Als Oberfläche, die als Gegenpart für die Adsorption dient,
kommen sowohl Flüssigkeiten als auch Feststoffe in Frage
Im freibeweglichen Zustand wird die zu adsorbierende Komponente als Adsorptiv, im gebundenen
Zustand als Adsorbat bezeichnet. Die gesamte fluide Phase wird im folgenden als Bulk-Phase
bezeichnet, speziell bei der Chromatographie wird sie aufgrund des fließenden Charakters als
mobile Phase bezeichnet. Das Adsorptionsmittel, an dessen Oberfläche die Adsorption stattfindet,
wird als Adsorbens oder speziell in der Chromatographie als stationäre Phase bezeichnet (vgl.
Abbildung 2.1).
Phasentrennfläche
Bulk- oder mobilePhase
Adsorbat-Phase
stärker adsorbierende Komp.
schwächer adsorbierende Komp. Abbildung 2.1: Darstellung der Nomenklatur
Adsorbens oder stationäre Phase
Man unterscheidet zwischen der physikalischen Adsorption (Physiosorption) und der chemischen
Adsorption (Chemisorption). Bei der Physiosorption wird zwischen Adsorbens und Adsorbat nur
eine lockere Bindung eingegangen, die durch van-der-Waals-Kräfte ausgelöst werden. Diese
Bindung kann leicht wieder gelöst werden [Arlt (1999)]. Bei der Chemisorption wirken chemische
Bindungskräfte zwischen Adsorbens und Adsorbat. Diese Kräfte sind im allgemeinen stärker als
die physikalischen Bindungskräfte, so daß der Aufwand, die so gebundenen Moleküle wieder in
den frei beweglichen Zustand zurückzuversetzen, erheblich steigt.

2 Grundlagen der Flüssigchromatographie
4
Die vorliegende Arbeit beschäftigt sich ausschließlich mit der Flüssigchromatographie, bei der die
fluide Phase flüssig und das Adsorbens ein Feststoff ist. Weiterhin ist die Adsorption
physikalischer Natur.
2.2 Die Adsorption in der flüssigen Phase
Die Flüssigadsorption zeichnet sich in Unterscheidung zur Gasadsorption in zwei Punkten aus:
Zum einen wirken zwischen den Molekülen der Bulk-Phase ebenso Anziehungs- bzw.
Abstoßungskräfte wie zwischen bestimmten Molekülen der Bulk-Phase und dem Adsorbat. Bei der
Gasadsorption dagegen sind die Anziehungskräfte zwischen den Molekülen der Bulk-Phase
untereinander meist vernachlässigbar.
Adsorbens
Gibbs'sche Phasentrennflächexi [m ol/m ol]
Abstand von der Adsorbensoberfläche
xi
xi
Adsorbat-Phase
Bulk-Phase
realer Verlauf
0
Abbildung 2.2: realer Konzentrationsverlauf und Definition der Gibbs'schen Phasengrenzfläche [aus Hirsch (2000)]
Der für die thermodynamische Betrachtung relevante Unterschied zwischen den beiden Arten der
Adsorption besteht in der Natur der Phasengrenzfläche zwischen Bulk-Phase und Adsorbat. Eine
Phasengrenzfläche ist nach Schade (1989) so definiert, daß sich an ihr mindestens eine intensive
Größe sprunghaft ändert. Dies trifft im Bereich der Gasadsorption zu, da sich bei der adsorbierten
Phase, vergleichbar der Kondensation, die Packungsdichte der anhaftenden Moleküle sprunghaft
ändert. Bei der Flüssigadsorption dagegen ist eine solche sprunghafte Veränderung einer
intensiven Größe nicht festzustellen, vielmehr erfolgt hier die Veränderung bestimmter Größen
stetig. Für die Modellierung wurde daher eine Phasengrenzfläche angenommen, die allgemein als

2 Grundlagen der Flüssigchromatographie
Gibbs'sche Phasengrenzfläche bezeichnet wird. [Findenegg (1996)] Den Unterschied zwischen
einem realen und demjenigen Konzentrationsverlauf, der in der Theorie durch die Einführung der
Gibbs'sche Phasengrenzfläche entsteht, verdeutlicht Abbildung 2.2.
2.3 Der Adsorptionsexzeß
Um die Adsorption in der Flüssigphase mathematisch-physikalisch beschreiben zu können, wurde
von Oswald der Adsorptionsexzeß eingeführt und später von Kipling (1965) aufgegriffen. Dabei
erfolgt die Beschreibung der durch Adsorption veränderten Konzentration in der Bulk-Phase bei
Verwendung des in der Thermodynamik gängigen Molenbruchs xi als Konzentrationsmaß mittels
des Stoffmengenexzesses niE:
)xx(nn i0i
0Ei −= (T, P konstant). (2.1)
Dabei ist n0 die Stoffmenge in der Bulk-Phase vor Kontakt mit dem Adsorbens und xi0 deren
Zusammensetzung. xi ist die Zusammensetzung der Bulk-Phase nach der Einstellung eines
Adsorptionsgleichgewichtes wie Abbildung 2.3 veranschaulicht.
n x00, i
Abbildung 2.3: Veranschaulichung des A
Der Stoffmengenexzeß beschreibt also
einem realen System gegenüber einem
gleiche Stoffmenge n0 enthält und in
Systems vorliegt [Hirsch (2000)].
Heuchel und Bräuer (1986) zeigten, daß
Gibbs'schen Exzeß-Thermodynamik en
Beschreibung des Adsorptionseffektes is
Mit einer einfachen Massenbilanz über d
Gleichgewichts-
d
h
d
ts
t.
a
einstellung
5
n x, i
sorptionsexzesses
den Überschuß der Stoffmenge von Komponente i in
ypothetischen Referenzsystem ohne Adsorption, das die
em die Gleichgewichtszusammensetzung des realen
der mit Gleichung (2.1) definierte Stoffmengenexzeß der
pricht und damit die Basis für eine thermodynamische
s reale System mit

2 Grundlagen der Flüssigchromatographie
6
nnn0 ′+= , (2.2)
wobei n' die gesamte adsorbierte Stoffmenge und n die Stoffmenge in der Bulk-Phase ist, läßt sich
der Adsorptionsexzeß auch mit Größen der adsorbierten Phase ausdrücken:
)xx(nn iiEi −′′= , (2.3)
wobei xi' die Zusammensetzung der adsorbierten Phase darstellt. Gleichung (2.3) wird auch als
Ostwald-Izaguirre-Gleichung bezeichnet [Kipling (1965)]. Sie zeigt, daß der Stoffmengenexzeß die
Menge ist an adsorbierter Komponente i vermindert um die Menge von Komponente i, die
adsorbieren würde, wenn die Zusammensetzung der adsorbierten Phase identisch mit derjenigen
der Bulk-Phase wäre [Minka et al. (1973)].
Aus der Definition des Stoffmengenexzesses folgt bei isothermer und isobarer Zusammensetzung
und bei einer festen Zusammensetzung der Bulk-Phase:
∑ =i
Ei 0n . (2.4)
Für Reinstoffe gilt:
0nEi = . (2.5)
Um Meßergebnisse miteinander vergleichen zu können und um den Einfluß unterschiedlicher
Adsorbensmengen zu eliminieren, wird der Stoffmengenexzeß meist auf die Masse des
Adsorbens mad bezogen, was auf den Adsorptionsexzeß führt:
( )i
0i
ad
0
ad
Ei xx
mn
mn
−= . (2.6)
Neben dem Adsorptionsexzeß gibt es auch die Möglichkeit, den Stoffmengenexzeß auf andere
Größen zu beziehen, wie zum Beispiel als Oberflächenexzeß:
2 Grundlagen der Flüssigchromatographie
7
( )i
0i
adad
0
ad
Ei xx
amn
An
−= (2.7)
mit der gewichtsspezifischen Oberfläche aad und der Gesamtoberfläche Aad. Analog gibt es noch
den Porenvolumenexzeß mit dem spezifischen Porenvolumen vP,ad und dem Gesamtporen-
volumen Vp,ad:
( )i
0i
ad,pad
0
ad,p
Ei xx
vmn
Vn
−= . (2.8)
Beziehungen, die eine Umrechnung der verschiedenen Exzeßgrößen ineinander erlauben, sowie
deren geometrische und algebraische Interpretationen werden von Kiraly et al. (1988) dargestellt.
Betrachtet man den Verlauf von Exzeßisothermen eines binären Systems über den gesamten
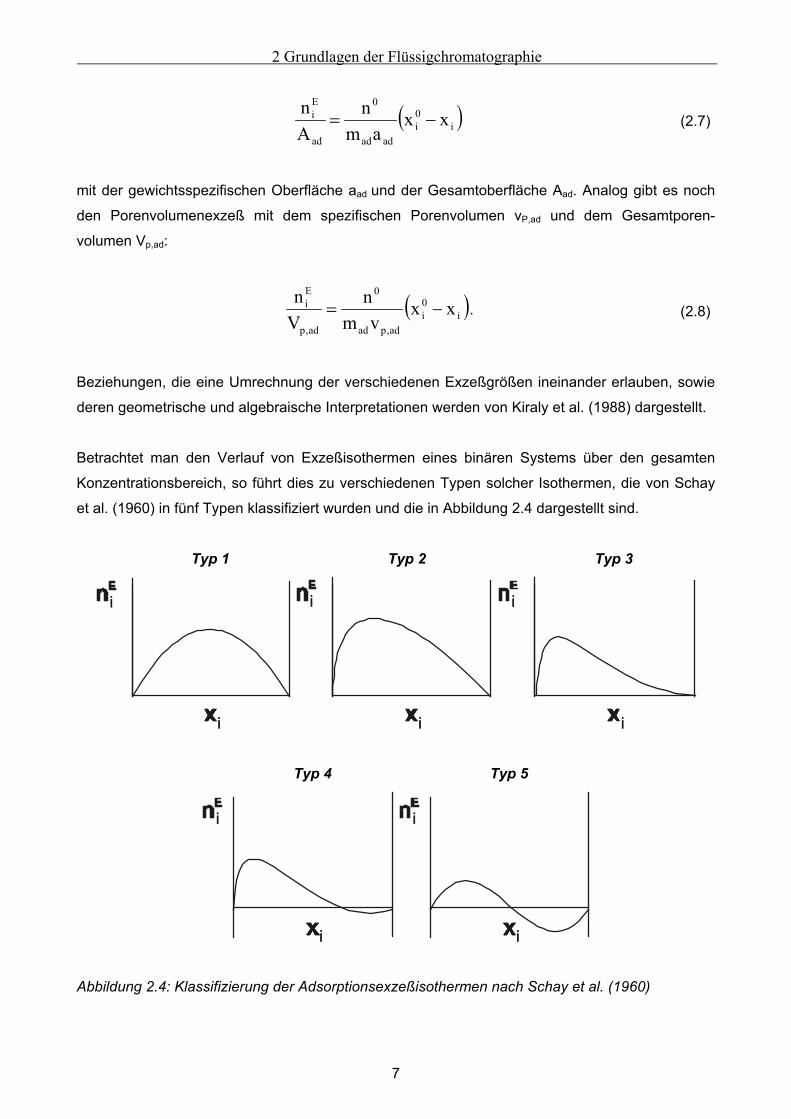
Konzentrationsbereich, so führt dies zu verschiedenen Typen solcher Isothermen, die von Schay
et al. (1960) in fünf Typen klassifiziert wurden und die in Abbildung 2.4 dargestellt sind.
Typ 1 Typ 2 Typ 3
ii
ii
ii
ii
ii
ii
Typ 4 Typ 5
ii
ii
ii
ii
Abbildung 2.4: Klassifizierung der Adsorptionsexzeßisothermen nach Schay et al. (1960)

2 Grundlagen der Flüssigchromatographie
8
Exzeßisothermen vom Typ 1 zeichnen sich durch ein ideales Verhalten beider Komponenten in
Bulk und Adsorbat-Phase sowie durch ein niedrigen Trennfaktor aus. Typ 2 zeigt eine starke
Bevorzugung der aufgetragenen Komponente i bei der Adsorption. Ein maximaler Effekt stellt sich
schon bei gering vorhandenen Mengen der Komponente i in der Bulk-Phase ein. Dies gilt auch für
die Isothermen des Typs 3, jedoch zeigen sie eine ausgeprägte Nichtlinearität für höhere
Gleichgewichtskonzentrationen der Komponente i.
Die Adsorptionsisothermen vom Typ 4 und Typ 5 besitzten ein sogenanntes Adsorptionsazeotrop.
Wird das Adsorbens mit einer Bulk-Phase bestimmter Zusammensetzung xi0 (nE=0) in Kontakt
gebracht, so stellt sich analog zu den Reinstoffen nach Kontakt mit dem Adsorbens keinerlei
Konzentrationsänderung und damit auch kein Adsorptionseffekt ein. Eine solche Situation tritt
meist bei Adsorbentien ein, die verschiedenartige Adsorptionszentren besitzen [Dabrowski et al.
(1987)]. Links dieses Adsorptionsazeotrops wird Komponente i bevorzugt adsorbiert, auf der
rechten Seite Komponente j.
2.4 Die Anwendung der Flüssigchromatographie
Bei der Flüssig- ebenso wie bei der Gaschromatographie nutzt man das Phänomen der
Adsorption, um bestimmte Komponenten eines flüssigen Gemisches abzutrennen. Bei der Säulen-
chromatographie wird zu diesem Zweck eine Trennsäule, die mit einem festen Hilfsstoff
(stationäre Phase) gefüllt ist, von einer flüssigen Hilfsphase (mobile Phase) durchströmt. Die
mobile Phase transportiert dabei die zu trennenden Komponenten durch die Säule. Dabei erfolgt
die Stofftrennung durch den Stoffaustausch zwischen stationärer und mobiler Phase. Aufgrund
der unterschiedlich starken Wechselwirkungen der einzelnen Komponenten mit der stationären
Phase kommt es zu einer unterschiedlich langen Verweildauer der verschiedenen Moleküle in der
Säule, so daß sie zeitlich versetzt die Säule verlassen [Deckert (1997)].
Neben der Trennung durch das Phänomen der Adsorption (Adsorptionschromatographie) gibt es
noch die Ionenaustauschromatographie, die Ausschlußchromatographie und die Affinitäts-
chromatographie [Meyer (1992)]. Bei der Ionenaustauschchromatographie enthält die stationäre
Phase ionische Gruppen, die mit den zu trennenden Komponenten in Wechselwirkung treten. Bei
der Ausschlußchromatographie werden die Moleküle durch unterschiedliche Größen getrennt.
Dabei ist die stationäre Phase derart beschaffen, daß sie nur Moleküle bis zu einer bestimmten
Größe in die Poren eindringen läßt und ihnen dadurch eine Adsorption ermöglicht. Die
Affinitätschromatographie schließlich ist das Prinzip mit der größten Selektivität, bei dem die
Wechselwirkungen biochemischer Natur sind (z. B. Schlüssel-Schloß-Wechselwirkungen). Diese
2 Grundlagen der Flüssigchromatographie
9
Art der Bindung ist am stärksten und kann meist nur durch Lösungsmittelwechsel oder pH-Wert
Änderung gelöst werden.
Bei der Anwendung der Flüssigkeitschromatographie wird zwischen zwei Zielsetzungen unter-
schieden: Man nutzt sie zum einen in der Analytik, um Gemischkomponenten aufzutrennen. Dabei
sind die aufgegebenen Mengen im allgemeinen gering. Bei der Chromatographie im präparativen
Maßstab dagegen steht die Gewinnung reiner Stoffe im Vordergrund. Hier wird meist mit höheren
Konzentrationen gearbeitet, um größere Durchsätze zu erzielen, da hierbei auch wirtschaftliche
Überlegungen eine Rolle spielen. Auch die Durchmesser der Trennsäulen und die Menge an
stationärer Phase sind hier wesentlich größer als bei der Analytik. Die Vergrößerung der
Probemenge wird als Überladung der Trennsäule bezeichnet.
Im folgenden sind die Grundlagen der Modellierung der Vorgänge bei der Flüssigchromatographie
erläutert.
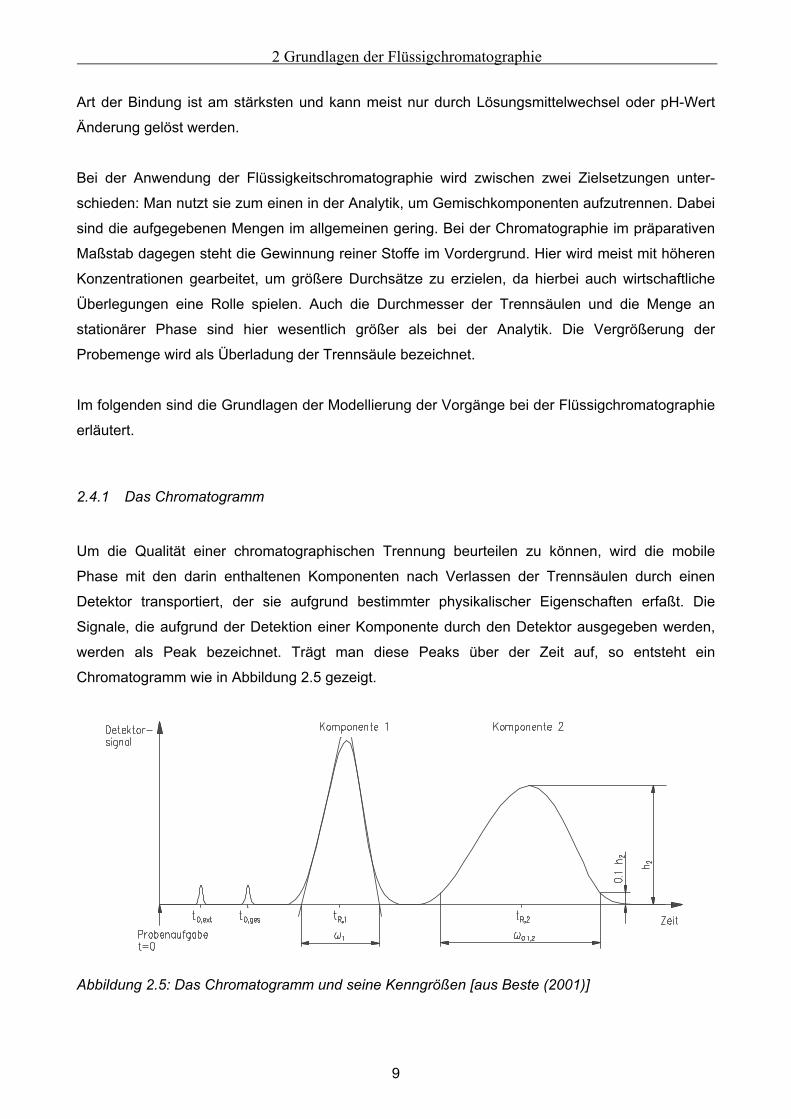
2.4.1 Das Chromatogramm
Um die Qualität einer chromatographischen Trennung beurteilen zu können, wird die mobile
Phase mit den darin enthaltenen Komponenten nach Verlassen der Trennsäulen durch einen
Detektor transportiert, der sie aufgrund bestimmter physikalischer Eigenschaften erfaßt. Die
Signale, die aufgrund der Detektion einer Komponente durch den Detektor ausgegeben werden,
werden als Peak bezeichnet. Trägt man diese Peaks über der Zeit auf, so entsteht ein
Chromatogramm wie in Abbildung 2.5 gezeigt.
Abbildung 2.5: Das Chromatogramm und seine Kenngrößen [aus Beste (2001)]

2 Grundlagen der Flüssigchromatographie
10
Ein Chromatogramm enthält verschiedene chromatographische Kenngrößen:
• Die Peakhöhe hi und die Fläche unter dem Peak sind proportional zur detektierten Menge
einer Komponente. Durch Kalibrierung des Detektors ist es möglich, die Menge einer
Komponente zu quantifizieren.
• Die Retentionszeit tR,i einer Substanz i ist bei einem symmetrischen Peak die Zeit, die
zwischen Peakaufgabe auf die Trennsäule zum Zeitpunkt t=0 und der Detektion des
Peakmaximums durch den Detektor liegt. Sie kann genutzt werden, um die Komponente zu
identifizieren.
• Die externe Totzeit t0,ext entspricht der Totzeit einer Substanz, die weder adsorbiert noch in die
Poren der stationären Phase eindringt. Findet dagegen eine nicht-adsorbierende Substanz
den Weg in die Poren, so wandert sie mit der Geschwindigkeit der mobilen Phase durch die
Säule. Aufgrund des größeren Flüssigkeitsvolumens, das ihr zugänglich ist, benötigt sie länger
durch die Säule als die nicht porengängige Substanz und erscheint nach der Gesamttotzeit
t0,ges .
• Die Basisweite ωi eines Peaks sowie die Peakweite ω0.1,i charakterisieren die
Peakverbreiterung im Verhältnis zu den Peakhöhen hi.
Mit Hilfe dieser Kenngrößen lassen sich Aussagen treffen über eine Trennsäule und die Güte
einer Trennung. Zu diesem Zweck wurden verschiedene Kennzahlen geschaffen, die diese als
Maß quantifizieren:
Ein Maß für die Eigenschaft eines chromatographischen Systems, zwei Stoffe zu trennen, also für
dessen Selektivität, ist der Trennfaktor α21. Dieser berechnet sich aus den Retentionszeiten und
der Totzeit, wobei entweder die externe Totzeit t0,ext oder die die Gesamttotzeit t0,ges verwendet
werden kann. Der Trennfaktor läßt sich ferner über die Adsorptionsgleichgewichtskoeffizienten Ki
(s. Abschnitt 2.4.3) berechnen, so daß sich formal ergibt:
1
2
01,R
02,R21 K
Ktttt
=−
−=α . (2.9)
Damit Gleichung (2.9) gilt, muß diejenige Totzeit benutzt werden, die auch zur Bestimmung der
Adsorptionsgleichgewichtskoeffizienten verwandt wurde.

2 Grundlagen der Flüssigchromatographie
11
Die Güte der Trennung wird durch die Auflösung RS beschrieben und ist definiert durch den
Quotienten aus dem zeitlichen Abstand zweier benachbarter Peakmaxima und dem
arithmetischen Mittel aus den Basisweiten ω1 und ω2:
( )21
1,R2,RS
21
ttR
ω+ω
−= .
(2.10)
Zur Charakterisierung der Effektivität einer Säule kann die Anzahl der theoretischen Trennstufen
NTUi herangezogen werden. Mit Hilfe des Chromatogramms läßt sich für Peaks, die in Form einer
Gaußkurve vorliegen, die Trennstufenzahl einer Säule NTUi nach Aced und Möckel (1991) wie
folgt bestimmen:
2
i
i,Ri
t16NTU
ω
= . (2.11)
In der Realität weisen chromatographische Peaks oft eine nicht vernachlässigbare Unsymmetrie
auf, die sich als Tailing (langgezogener Abfall) oder Fronting (langgezogener Anstieg) äußern
kann. In diesem Fall ist die zuverlässigste Methode zur Bestimmung der Trennstufenzahl und der
Retentionszeit die Momentenmethode. Die Retentionszeit entspricht dabei dem ersten bezogenen
Anfangsmoment µ‘1,i.
∑
∑
∫
∫
=
=
∞
∞
∆
∆≈=≡µ n
1jjj,i
n
1jjjj,i
0i
0i
i,R'
i,1
tc
ttc
dtc
dtctt (2.12)
Liegt das Chromatogramm in digitaler Form vor, so können die Integrale durch die Summen über
die n Datenpunkte ersetzt werden. Die Trennstufenzahl NTUi ist über das erste bezogene
Anfangsmoment µ1,i’ und das zweite zentrale Moment µ2,i (Varianz σ) definiert:
2
i,2
'i,1
iNTU
µ
µ= , (2.13)
wobei sich das zweite zentrale Moment berechnet über

2 Grundlagen der Flüssigchromatographie
12
j
n
1jj,i
n
1jji,Rjj,i
0i
0
2i,Ri
2ii,2
tc
t)tt(c
dtc
dt)tt(c
∆
∆⋅−≈
−=σ≡µ
∑
∑
∫
∫
=
=
∞
∞
. (2.14)
Je geringer die Trennstufenzahl NTUi einer Säule ist, desto höher muß die Selektivität α21 des
chromatographischen Systems sein, um das gleiche Trennergebnis, also die gleiche Auflösung
RS, zu erreichen.
Neben Peaks werden auch Konzentrationssprünge oder -stufen auf Säulen gegeben, um die
Säuleneigenschaften zu untersuchen. Aus den dabei entstehenden Durchbruchskurven können
die Retentionszeit
( )( )I
iIIi
n
1jjj,i
IIi
0
i
0
i
front,i,R cc
tcc
dtt
c
dttt
c
t−
∆⋅−≈
⋅∂
∂
⋅∂
∂
=∑
∫
∫=
∞
∞
(2.15)
und die Varianz
( )2
i,RIi
IIi
n
1jjjj,i
IIi
0
i
0i,R
i
2front,i t
cc
ttcc
dtt
c
dt)tt(t
c
−−
∆⋅−≈
⋅∂
∂
⋅−⋅∂
∂
=σ∑
∫
∫=
∞
∞
(2.16)
bestimmt werden.
2.4.2 Die Porosität
Für die Beschreibung chromatographischer Effekte spielt das Phasenverhältnis Feststoff-
Flüssigkeit in der Säule eine wesentliche Rolle. Zu deren Beschreibung wird die Porosität
ε benutzt, die allerdings aus dem Bezug auf unterschiedliche Teilvolumina unterschiedliche
Interpretationen beinhalten kann.

2 Grundlagen der Flüssigchromatographie
13
Ausgehend vom Gesamtvolumen einer chromatographichen Säule VS können drei Teilvolumina
beschrieben werden [Seidel-Morgenstern (1995)]
• Das externe Volumen Vext ist das Volumen der mobilen Phase außerhalb der Partikel:
sextext VV ε= . (2.17)
• Das Partikelvolumen VP umfaßt das Volumen der Partikel inklusive seines Porensystems:
( ) sextP V1V ε−= . (2.18)
• Das interne Volumen Vint gibt das Volumen der Poren in den Partikeln an und ist definiert als:
adsintint VV ε= . (2.19)
• Das Volumen des Partikelfeststoffs VPF ergibt sich damit aus Gleichung (2.17) und (2.19) mit:
( ) ( )( ) sextintextsintextsPF V1(VVVVV ε−ε+ε−=+−= (2.20)
Mit Hilfe dieser Volumina läßt sich das Gesamtvolumen Vges der mobilen Phase in der
chromatographischen Säule definieren:
intextsgesges VVVV +=ε= (2.21)
Die Porositäten können mit Hilfe der Totzeiten t0,ext und t0,ges sowie des Volumenstroms der
mobilen Phase V bestimmt werden durch &
s
ext,0
s
extext V
VtVV &
==ε (2.22)
bzw.

2 Grundlagen der Flüssigchromatographie
14
S
ges,0
S
intextges V
VtV
VV &=
+=ε . (2.23)
Im Rahmen dieser Arbeit wird für den Vergleich von statischen und dynamischen Methoden zur
Bestimmung von Adsorptionsisothermen im folgenden die Gesamtporosität verwendet; die Gründe
dafür werden in Kapitel 5 näher erläutert. Im Gegensatz dazu wird bei der Betrachtung der
präparativen HPLC und der Simulierten Gegenstromchromatographie die externe Porosität
benutzt, da diese auch in der strömungskinetischen Betrachtung Anwendung findet, weil im
allgemeinen davon ausgegangen wird, daß in Poren nur diffusiver Stofftransport stattfindet.
2.4.3 Die Adsorptionsisotherme
Die Adsorptionsisotherme, im folgenden zur besseren Unterscheidung vom Adsorptionsexzeß
auch als Beladungsisotherme bezeichnet, stellt den funktionalen Zusammenhang zwischen der
Konzentration eines Stoffes der mobilen Phase ci und der Beladung der stationären Phase qi in
einem thermodynamisch bestimmten Adsorptionsgleichgewicht dar. Eine Adsorptionsisotherme,
bei der die Beladung ausschließlich eine Funktion der Konzentration des Stoffes i in der mobilen
Phase ist, wird als Einkomponentenisotherme bezeichnet. Hierbei betrachtet man im Gegensatz
zur Exzeßbetrachtung das Eluent als inert und nimmt es deshalb auch in die Nomenklatur nicht
mit hinein.
Die einfachste Form einer Adsorptionsisotherme ist die lineare Einstoffisotherme, bei der die
Beladung qi einer Komponente linear von ihrer Konzentration in der mobilen Phase abhängt:
iii cKq = . (2.24)
Lineare Einstoffisothermen treten meist bei geringeren Konzentrationen , wie z. B. in der analy-
tischen Chromatographie, auf.
Da in der Flüssigchromatographie die Masse des Adsorbens, die sich in der Säule befindet,
unbekannt und im allgemeinen auch schwer zugänglich ist, wird die Beladung meist auf das
Volumen des Adsorbens bezogen. Das Volumen bzw. die Porosität, die man hier verwendet, also
entweder die externe oder die Gesamtporosität, geht als Bezugsgröße in die
Adsorptionsisotherme ein und wird somit ihr Bestandteil.

2 Grundlagen der Flüssigchromatographie
15
Wenn man annimmt, daß die Menge des Stoffes i, der sich in den Poren befindet, Bestandteil der
adsorbierten Phase ist, so ergibt sich die Gesamtmenge mi von Komponente i, die sich in der
equilibrierten Säule befindet, zu [Seidel-Morgenstern (1995)]:
ges,iadsiexti qVcVm += (2.25)
mit der Beladung qi,ges in der Einheit [g/l] bezogen auf das Partikelvolumen inklusive Poren.
Betrachtet man dagegen die Menge des Stoffes i, der sich in den Poren befindet, getrennt von der
Menge an Stoff i, die tatsächlich physikalisch an der stationären Phase adsorbiert, so ergibt eine
Massenbilanz
iPFPore,iintiexti qVcVcVm ++= . (2.26)
wobei ci,Pore die Konzentration der Komponente i in den Poren und qi die Menge i ist, die
tatsächlich adsorptiv an der stationären Phase anhaftet, ebenfalls in der Einheit [g/l], allerdings
bezogen auf das Volumen des Partikelfeststoffs ohne Poren. Der Unterschied zwischen den
beiden Ansätzen liegt in der Betrachtungsweise der Adsorption: Bei Betrachtung gemäß
Gleichung (2.25) wird die Substanzmenge, die eine Pore durchläuft, der Beladung qi zuge-
schlagen und damit quasi als adsorbiert betrachtet, auch wenn ein Teil von Komponente i im
Inneren der Pore nicht mit der Oberfläche der stationären Phase gemäß Betrachtung in
Unterkapitel 2.2 in Wechselwirkung tritt. Verwendet man dagegen bei einer porengängigen
Substanz die Gesamtporosität wie in Gleichung (2.26) geschehen, so beschränkt sich die
Beladung qi auf die Adsorption im tatsächlichen, physikalischen Sinne.
Bei Annahme eines schnellen Stoffaustausches, also ci=ci,Pore ist eine Umrechnung beider
Beladungen ineinander möglich:
( )( ) i
ext
gesiintges,i q
11
cqε−
ε−+ε= . (2.27)
Damit ergeben sich als lineare Adsorptionsisothermen
iges,iges,i cKq = (2.28)
bzw.

2 Grundlagen der Flüssigchromatographie
16
iii cKq = (2.29)
wobei Ki bzw. Ki,ges jeweils die Steigung der linearen Isotherme ist.
Mit Hilfe von Bilanzbetrachtungen läßt sich die Retentionszeit eines Peaks bei bekannter externer
Porosität und Adsorptionsisotherme ermitteln durch [Seidel-Morgenstern (1995)]:
ε
ε−+= ges,i
ext
extext,oiR K
11tt mit
VV
t Sextext,0 &
ε= (2.30)
bzw. bei Verwendung der Gesamtporosität:
ε
ε−+= i
ges
ges0iR K
11tt mit
VV
t Sges0 &
ε= . (2.31)
Bei höheren Konzentrationen, wie sie in der präparativen Chromatographie Anwendung finden, ist
der Gleichgewichtskoeffizient Ki meist nicht mehr konstant, sondern stellt eine Funktion der
Konzentration ci dar, so daß sich eine nicht-lineare Adsorptionsisotherme ergibt. Der Verlauf
solcher Adsorptionsisothermen kann z.B. durch die einfache Langmuir-Gleichung
ii
ii
ii
iii,Si cb1
cncb1
cbqq
+=
+= (2.32)
mathematisch beschrieben werden. ni entspricht dabei dem Anfangsanstieg der Isotherme und ist
das Produkt der Sättigungsbeladung qs,i und des Krümmungsparameters bi, da die Langmuir-
Isotherme für kleine Konzentrationen in eine lineare Isothermenform übergeht.
Der Verlauf der Adsorptionsisotherme hat entscheidenden Einfluß auf die Form der eluierten
Peaks, da bei einer nichtlinearen Adsorptionsisotherme die Wanderungsgeschwindigkeit wc,i und
damit auch die Retentionszeit einer Substanz nicht nur von der Zwischenkorngeschwindigkeit
w, sondern auch von der Konzentration der Komponente i abhängt:
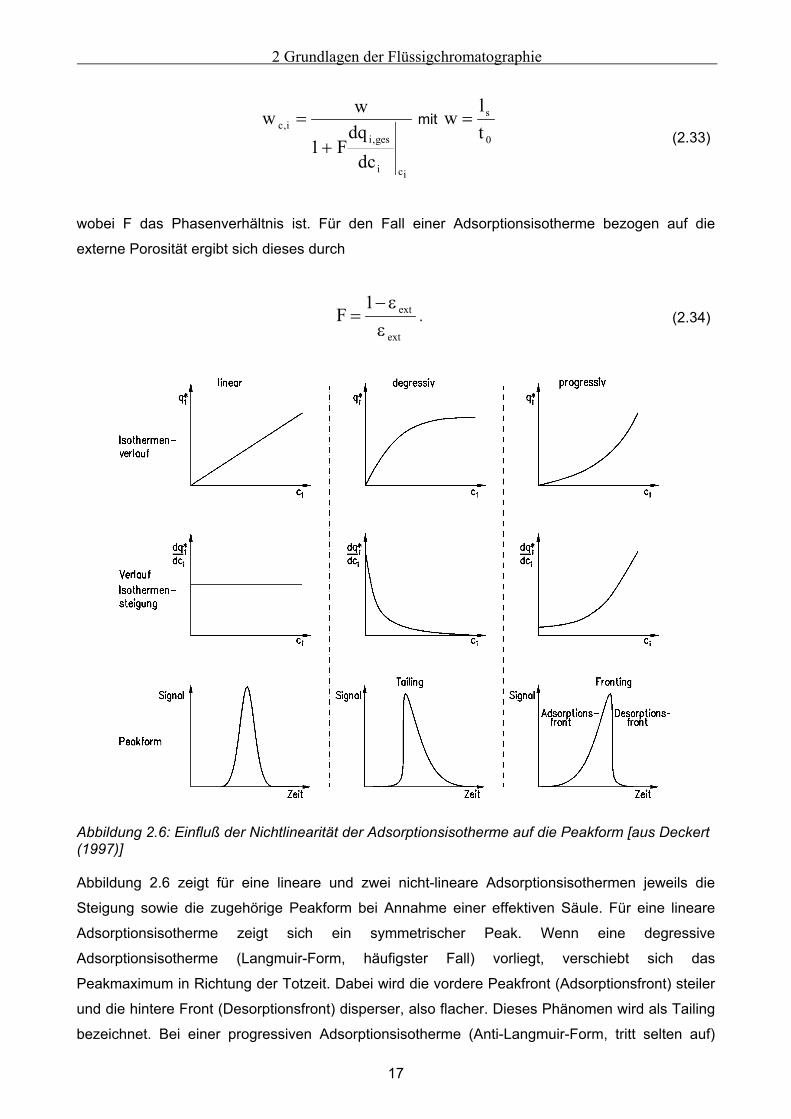
2 Grundlagen der Flüssigchromatographie
17
ici
ges,ii,c
dcdq
F1
ww+
= mit 0
s
tl
w = (2.33)
wobei F das Phasenverhältnis ist. Für den Fall einer Adsorptionsisotherme bezogen auf die
externe Porosität ergibt sich dieses durch
ext
ext1F
εε−
= . (2.34)
Abbildung 2.6: Einfluß der Nichtlinearität der Adsorptionsisotherme auf die Peakform [aus Deckert (1997)] Abbildung 2.6 zeigt für eine lineare und zwei nicht-lineare Adsorptionsisothermen jeweils die
Steigung sowie die zugehörige Peakform bei Annahme einer effektiven Säule. Für eine lineare
Adsorptionsisotherme zeigt sich ein symmetrischer Peak. Wenn eine degressive
Adsorptionsisotherme (Langmuir-Form, häufigster Fall) vorliegt, verschiebt sich das
Peakmaximum in Richtung der Totzeit. Dabei wird die vordere Peakfront (Adsorptionsfront) steiler
und die hintere Front (Desorptionsfront) disperser, also flacher. Dieses Phänomen wird als Tailing
bezeichnet. Bei einer progressiven Adsorptionsisotherme (Anti-Langmuir-Form, tritt selten auf)

2 Grundlagen der Flüssigchromatographie
18
dagegen kommt es zu einem Fronting. Das Peakmaximum verschiebt sich zu späteren
Elutionszeiten, die Adsorptionsfront wird flacher, die Desorptionsfront steiler [Guiochon et al.
(1994)].
Insbesondere bei der in der präparativen Chromatographie praktizierten Überladung der
Trennsäule ist die Beladung qi häufig nicht mehr ausschließlich eine Funktion der Konzentration ci,
sondern kann ebenfalls von der Konzentration eines anderen Stoffes cj in der mobilen Phase
abhängen. Dies kann dadurch erklärt werden, daß es zu einer Konkurrenzsituation um die
Adsorptionsplätze kommt. Solche Isothermen werden als Mehrkomponentenisothermen
bezeichnet.
Um solche Isothermen mathematisch beschreiben zu können, reichen die Modelle für Einstoff-
isothermen nicht mehr aus. Daher wurde eine Vielzahl von empirischen Gleichungen für
nichtlineare und gekoppelte Isothermen entwickelt, von denen im folgenden einige exemplarisch
erläutert werden [Seidel-Morgenstern (1995), Beste (2001)]:
Die Multi-Langmuir-Gleichung kann den Verlauf von realen Adsorptionsisothermen gut
wiedergeben, da diese meist einen degressiven Verlauf zeigen:
∑=
+= r
1jjj
iii
cb1
cnq .
(2.35)
Um einen konzentrationsabhängigen Trennfaktor αij zwischen zwei Komponenten beschreiben zu
können, wird Gleichung (2.35) additiv um einen linear konzentrationsabhängigen Term ergänzt,
was auf die Modifizierte Langmuir-Isotherme führt:
∑=
++= r
1jjj
iiii
cb1
cnkq .
(2.36)
Kombiniert man zwei Multi-Langmuir Terme miteinander, führt dies zur gekoppelten Bi-Langmuir-
Isotherme. Sie basiert auf der Annahme, daß zwei unterschiedliche Zentren vorhanden sind, an
denen jeweils eine konkurrierende Adsorption stattfindet. In der Praxis führt die Zunahme der
anpaßbaren Parameter zu einer größeren Flexibiltät der Gleichung [Beste (2001)]:

2 Grundlagen der Flüssigchromatographie
19
∑∑==
++
+= r
1jj2,j
i2,ir
1jj1,j
i1,ii
cb1
cn
cb1
cnq .
(2.37)
Eine empirische Gleichung, mit der sowohl progressive als auch degressive gekoppelte
Zweistoffisothermen beschrieben werden können, wird von Ching et al. (1993) vorgeschlagen:
( ) iijn
jijim
iiii ccBcAKq ++= . (2.38)
Dabei sind Ki, Ai und mi Reinstoffparameter. Die binären Parameter Bij und nij müssen aus
Gemischdaten ermittelt werden. Mit der Ching-Isotherme kann ebenfalls ein
konzentrationsabhängiger Trennfaktor αij beschrieben werden.
2.4.4 Grundlagen der Kinetik
Neben der beschriebenen Thermodynamik spielen beim Stofftransport durch chromatographische
Festbetten auch kinetische Effekte eine bedeutende Rolle. Diese bewirken bei einem das Festbett
durchlaufenden Peak eine zunehmende Verbreiterung, welche mit einer gleichzeitigen
Reduzierung der Peakhöhe einhergeht. Im folgenden wird auf diese Effekte näher eingegangen.
2.4.4.1 Axiale Dispersion
Die axiale Dispersion faßt Stofftransportphänomene zusammen, die in der mobilen Phase
zusätzlich zum konvektiven Transport auftreten. Dies sind im einzelnen:
• Strömungsungleichverteilungen: Aufgrund der Wandhaftung treten in der Nähe der
Säulenwand und auch zwischen den Partikeln Geschwindigkeitsunterschiede in radialer
Richtung auf [vgl. Tsotsas und Schlünder (1988)], so daß Moleküle, die sich in Wandnähe
befinden, eine niedrigere Geschwindigkeit gegenüber denjenigen im Kern der Strömung
aufweisen.
• Zu- und Abflußeffekte: An den Übergängen von kleinen (Zu- und Abflußleitungen) zu großen
Strömungsquerschnitten (Trennsäule) wird das Geschwindigkeitsprofil verändert. Mit
zunehmendem Säulenquerschnitt führt die radiale Verteilung zu einer Verzögerung der
Moleküle, die an den Säulenrand transportiert werden. Untersuchungen haben ergeben, daß

2 Grundlagen der Flüssigchromatographie
20
diese Effekte zu einem sehr großen Anteil zur Peakverbreiterung beitragen [Brandt (1997)],
[Lisso et al. (2000)], [Lisso (2002)].
• Eddy-Diffusion: Die Moleküle nehmen unterschiedliche Wege durch die Säule, da die
Flüssigkeit vielmals aufgeteilt und zusammengeführt wird. Daraus resultieren für sie
unterschiedliche Weglängen.
• molekulare Diffusion: Aufgrund der hervorgerufenen Konzentrationsunterschiede tritt
molekulare Diffusion sowohl in axialer als auch in radialer Richtung auf.
Diese Effekte, die die axiale Dispersion ausmachen, werden durch den axialen
Dispersionskoeffizienten Dax zusammengefaßt und charakterisiert. Er ist abhängig von der
Zwischenkorngeschwindigkeit w, der Konzentration ci und der Temperatur T. In der Literatur
finden sich zahlreiche empirische Bestimmungsgleichungen für die axiale Dispersion. Ruthven
(1984) gibt eine Gleichung an, die ursprünglich für Gase entwickelt wurde. Sie berücksichtigt den
Einfluß der molekularen Diffusion Dm,i und einen vom Partikeldurchmesser dp und der
Zwischenkorngeschwindigkeit w abhängigen Term:
wdγDγD P2m,i1ax += (2.39)
Die Konstanten γ1 und γ2 haben für poröse Partikel normalerweise Werte von 50 und 0,5. Bei der
Flüssigadsorption kann die molekulare Diffusion Dm,i im allgemeinen vernachlässigt werden, so
daß die axiale Dispersion für alle Komponenten gleich groß ist.
2.4.4.2 Stoffaustauschphänomene und Stofftransportwiderstand
Der Stoffaustausch zwischen mobiler und stationärer Phase wird durch den Stofftransport-
widerstand behindert, der zu einer zeitlichen Verzögerung der Einstellung eines
Adsorptionsgleichgewichtes führt. Durch die Adsorption wird ein Molekül einige Zeit an der
Feststoffoberfläche festgehalten. Die mit der Strömung transportierten Moleküle wandern den in
den Poren befindlichen voraus, so daß die Stoffaustauschphänomene (Adsorption und
Stofftransportwiderstand) mit zunehmender Fluidgeschwindigkeit verstärkt zu einer
Peakverbreiterung beitragen.
Dem Stofftransport in das Innere der Partikel werden in der frei strömenden Flüssigkeit und in den
Poren der Partikel Widerstände entgegengesetzt (siehe Abbildung 2.7). Bei der Zwei-

2 Grundlagen der Flüssigchromatographie
21
schichthypothese [Brauer (1971)] werden vereinfachend die Konzentrationsdifferenzen als
treibende Kraft für den Stofftransport angenommen. Eine Konzentrationsänderung tritt nur in den
hypothetischen Grenzschichten in der festen und der flüssigen Phase in der Nähe der
Phasengrenzfläche auf. An der hypothetischen Phasengrenzfläche herrscht ein Gleichgewicht,
welches durch die Adsorptionsisotherme vorgegeben ist. In der Praxis wird der Stofftrans-
portwiderstand häufig zusammenfassend über einen linearisierten effektiven Stoffüber-
gangskoeffizienten keff,i dargestellt. Es werden dann die in den beiden Phasen auftretenden
Widerstände zu einem effektiven Widerstand zusammengefaßt, der entweder der festen
(Festfilmmodell) oder der flüssigen Phase (Fluidfilmmodell) zugeordnet wird.
Abbildung 2.7: Konzentrationsverlauf nach der Zweischichthypothese [aus Jork (2001)]
Die zeitliche Änderung der über die Partikel gemittelten Beladung, d.h. der Stoffstrom in die
Partikel bezogen auf das Partikelvolumen, wird für das Festfilmmodell wie folgt berechnet:
)q(qk
tq
i*ieff,fest,i
i −=∂
∂. (2.40)
Für das Fluidfilmmodell gilt:
)c(c
VO
kt
q *ii
P
Peff,fl,i
i −=∂
∂. (2.41)
Der Stoffstrom, der durch beide Grenzschichten tritt, muß betragsmäßig gleich groß sein,
)c(cOk)c(cKV=km *iiPeff,fl,ii
*iiPeff,fest,ii −−=−& . (2.42)
2 Grundlagen der Flüssigchromatographie
22
so daß die effektiven Stoffübergangskoeffizienten beider Modelle ineinander überführt werden
können:
Pi
Peff,fl,ieff,fest,i VK
Okk = . (2.43)
Einige empirische Korrelationsgleichungen zur Berechnung der effektiven Stoffübergangs-
koeffizienten sind in Ruthven (1984) und Deckert (1998) aufgelistet.
Der effektive Stofftransportwiderstand ist im allgemeinsten Fall von der Konzentration, der
Geschwindigkeit und der Temperatur abhängig. Der Widerstand im Fluid hängt von der
Zwischenkorngeschwindigkeit ab, da sich die Dicke der Grenzschicht mit ihr ändert. Der
Widerstand in den Makroporen dagegen ist geschwindigkeitsunabhängig. Aus diesen Gründen
kann der effektive Widerstand als geschwindigkeitsunabhängig betrachtet werden, wenn der
Widerstand in den Partikeln dominiert.
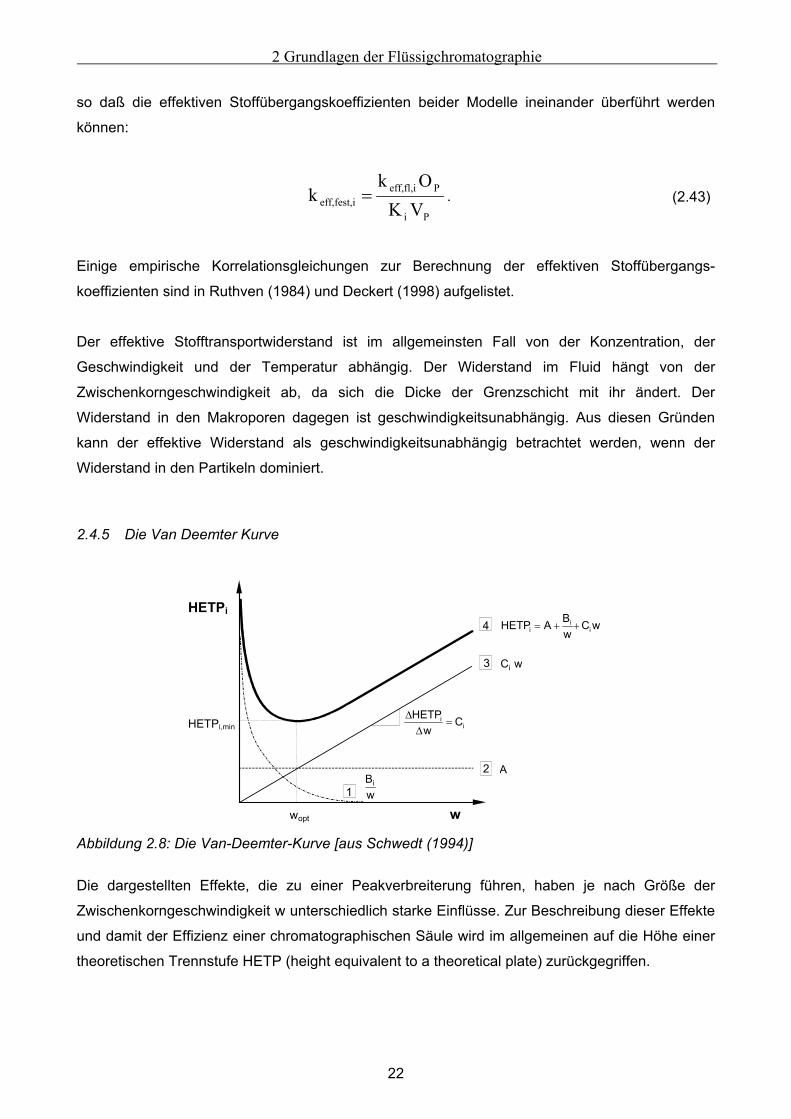
2.4.5 Die Van Deemter Kurve
2
3
1
4
w
HETPi
HETPi,min
wopt
Bw
i
wCwBAHETP i
ii ++=
ii C
wHETP
=∆
∆
C wi
A
Abbildung 2.8: Die Van-Deemter-Kurve [aus Schwedt (1994)]
Die dargestellten Effekte, die zu einer Peakverbreiterung führen, haben je nach Größe der
Zwischenkorngeschwindigkeit w unterschiedlich starke Einflüsse. Zur Beschreibung dieser Effekte
und damit der Effizienz einer chromatographischen Säule wird im allgemeinen auf die Höhe einer
theoretischen Trennstufe HETP (height equivalent to a theoretical plate) zurückgegriffen.

2 Grundlagen der Flüssigchromatographie
23
Je schmaler die Peaks bzw. je schärfer die Fronten sind, desto höher ist die Säuleneffizienz und
desto geringer ist die HETP. In Abhängigkeit von der Zwischenkorngeschwindigkeit tragen axiale
Dispersion und Stofftransportwiderstand unterschiedlich stark zur Peakverbreiterung bei. Diese
Abhängkeit wird durch die Van-Deemter-Kurve dargestellt, wie sie in Abbildung 2.8 dargestellt ist.
Die Höhe einer theoretischen Trennstufe HETPi summiert sich dabei aus drei Termen:
wC
wB
A+NTU
l=HETP i
i
i
Si += (2.44)
Die drei Parameter A, Bi und Ci dieser nach Van Deemter (1956) benannten Gleichung sind für ein
gegebenes System (Trennsäule, mobile Phase, zu trennende Komponente) und eine Temperatur
konstant.
• Kurve 1 erfaßt den Anteil der molekularen Diffusion in axialer Richtung, deren Einfluß mit
zunehmender Zwischenkorngeschwindigkeit abnimmt. In der Flüssigkeitschromatographie ist
der Anteil der molekularen Diffusion so gering, daß dieser Term meist vernachlässigt werden
kann.
• Kurve 2 beschreibt den Anteil der Eddy-Diffusion, der Zu- und Abflußeffekte und der
Strömungsungleichverteilung, welche unabhängig sind von der Zwischenkorngeschwindigkeit
und von der Molekülsorte. Der Wert der Konstante A wird durch die Partikelgröße und die
Packungsgüte der stationären Phase bestimmt.
• Kurve 3 beschreibt den Anteil der Stoffaustauschphänomene, die mit steigender Zwi-
schenkorngeschwindigkeit linear zunehmen [Meyer (1992)].
Vermißt man die Van-Deemter-Kurve eines Stoffes, der nicht adsorbiert, wodurch der Term Ci
wegfällt, so ist eine Bestimmung der Parameter A und Bi möglich. Die axiale Dispersion, die auch
zur Beschreibung der Güte der Packung dient, ist dadurch unabhängig von den Stoffaustausch-
phänomenen meßbar.
2.5 Die präparative HPLC
Die präparative HPLC (High Performance Liquid Chromatography) gehört zu den dis-
kontinuierlichen chromatographischen Verfahren und ist ein Chargenprozeß, in dem eine
definierte Probenmenge aufgegeben und getrennt wird. Sie wird auch als Batch-Chromatographie
bezeichnet.

2 Grundlagen der Flüssigchromatographie
24
Je nachdem, welche Probenmenge aufgegeben wird, unterscheidet man zwischen Elutions- und
Frontalchromatographie. Bei der Frontalchromatographie wird die Säule durch eine größere
Probenmenge vollständig gesättigt, wodurch es zur Ausbildung einer Plateaukonzentration und zu
einem Durchbruch der Substanz am Säulenende kommt. In der Elutionschromatographie
hingegen wird die Probenmenge so gewählt, daß die Konzentration am Ausgang der Säule nicht
die Eingangskonzentration erreicht. Die Untersuchungen der diskontinuierlichen Chromatographie
im Rahmen dieser Arbeit wurden ausschließlich für die Elutionschromatographie durchgeführt.
Ziel der präparativen Chromatographie ist es generell, größere Mengen einer reinen Substanz
herzustellen. Im Gegensatz zur analytischen Chromatographie werden dabei aus wirtschaftlichen
Überlegungen heraus pro Injektion wesentlich größere Mengen der zu trennenden Substanzen auf
die Säule aufgegeben. Aus diesen Gründen werden die Säulendurchmesser und die
Volumenströme des Eluenten wesentlich größer gewählt. Aber die Maßstabsvergrößerung allein
bringt noch keine Veränderung der Wirtschaftlichkeit einer chromatographischen Trennung [Brandt
(1997)]. Erst durch eine Erhöhung der Beladung der stationären Phase werden die Kosten bei der
Trennung größerer Substanzmengen deutlich reduziert. Die Erhöhung der Beladung über den
analytischen Bereich hinaus wird allgemein als Überladung bezeichnet. Eine solche Überladung
einer chromatographischen Säule führt im allgemeinen dazu, daß der lineare Bereich der
Adsorptionsisothermen verlassen wird, also eine Abhängigkeit der Wanderungsgeschwindigkeit
von der Konzentration vorliegt.
Der Aufbau einer präparativen HPLC-Anlage unterscheidet sich prinzipiell nur wenig von dem
einer Apparatur zur Analyse von Substanzgemischen. Um einen größeren Mengendurchsatz zu
ermöglichen, sind Säule und Verbindungsleitungen der präparativen Anlage größer dimensioniert.
Das Auffangen der einzelnen Komponenten erfolgt mit Hilfe eines Fraktionssammlers.
Wichtig beim Aufbau präparativer Säulen mit einem großen Durchmesser ist vor allem die
gleichmäßige Verteilung der Substanz auf den erweiterten Querschnitt der Säule. Zu diesem
Zweck ist vor und nach dem chromatographischen Festbett ein Verteiler eingebaut, der als Fritte
bezeichnet wird. Diese bestehen meist auf gesintertem Metall oder Metallgewebe, deren
Durchläßigkeit in radiale Richtung größer ist als in axiale Richtung. Diese können durch
Verteilerbleche ergänzt werden. Eine Übersicht über die gebräuchlichsten Systeme gibt Lisso
(2002).
Für die präparative Flüssigchromatographie existieren Weiterentwicklungen etwa in Hinblick auf
die Probenaufgabe (z.B. die Verdrängungschromatographie), die Pumprichtung (Flip-Flop-

2 Grundlagen der Flüssigchromatographie
25
Chromatographie) oder die Verschaltung der chromatographischen Anlage (Rezyklierungs-
techniken, wie z.B das Closed-Loop-Verfahren oder das Alternate-Pumping-Recycling).
Eine ausführliche Darstellung der technischen Verfahren zur Umsetzung der adsorptiven und
chromatographischen Trennprozesse findet sich z. B. bei Ganetsos und Barker (1993).
2.6 Die Gegenstromchromatographie
Der Nachteil der in Unterkapitel 2.5 behandelten Batch-Chromatographie liegt in ihrer
diskontinuierlichen Arbeitsweise, da hier ein Stoffgemisch injiziert und die beiden Komponenten
zeitlich versetzt die Säule verlassen (vgl. Abbildung 2.9a). Im Gegensatz dazu ist es vornehmliche
Eigenschaft kontinuierlich arbeitender Prozesse, eine kontinuierliche Zugabe des Feeds sowie
eine konstante Entnahme der Produkte zu ermöglichen. Zu diesem Zweck ist es notwendig, eine
relative Bewegung von Feststoff und fluider Phase zueinander zu erreichen.
Abbildung 2.9: Funktionsprinzip der a) diskontinuierlichen oder Batch-Chromatographie, b) der kontinuierlichen Gegenstromchromatographie (TMB) und c) der simulierten Gegenstrom-chromatographie (SMB) [Broughten (1984)]
Eine mögliche Realisierung ist die annulare Chromatographie, bei der Eluent und stationäre Phase
im Kreuzstrom zueinander geführt werden. Das Verfahren und seine Anwendungen ist z. B. bei
Bart et al. (1996) ausführlich beschrieben und soll an dieser Stelle nicht weiter behandelt werden.
Eine andere Art der Realisierung schlägt sich nieder in der Gegenstromchromatographie. Da diese
im Rahmen dieser Arbeit untersucht wird, wird deren Theorie im folgenden beschrieben. Zunächst
erfolgt in Abschnitt 2.6.1 eine Erläuterung der wirklichen Gegenstromchromatographie, die zumeist
zur Veranschaulichung und Auslegung des Prozesses dient. Darauf folgt in Abschnitt 2.6.2 eine

2 Grundlagen der Flüssigchromatographie
26
Erläuterung der Simulierten Gegenstromchromatographie und in Abschnitt 2.6.3 eine Erläuterung
über deren Modellierung. Da als Ausgangspunkt für die Fehlerbetrachtung ein optimierter Prozeß
dient, wird abschließend in Abschnitt 2.6.4 die Optimierung der Trennung behandelt.
2.6.1 Die wirkliche Gegenstromchromatographie (TMB)
Bei der wirklichen Gegenstromchromatographie (engl: True Moving Bed (TMB)) werden die mobile
Phase (im folgenden als Desorbent oder Eluent bezeichnet) und der Feststoff im Gegenstrom zu-
einander betrieben, wie Abbildung 2.9b verdeutlicht. Dabei tritt das Desorbent am Kopf der Säule
ein, während der Feststoff von unten nach oben transportiert wird. Das zu trennende Gemisch der
Komponenten A und B wird kontinuierlich als Feed in der Mitte der Säule aufgegeben. Bei
geeigneter Wahl der Flüssigkeits- und Feststoffströme werden die Komponenten A und B in
entgegengesetzte Richtungen transportiert, wobei die stärker adsorbierende Komponente A mit
dem Feststoff nach oben und die schwächer adsorbierende Komponente B mit der Flüssigkeit
nach unten wandert. Als Produkt werden zwei Teilströme entnommen: Das Extrakt, welches
vornehmlich die Komponente A enthält, und das Raffinat, in dem Komponente B überwiegt.
Abbildung 2.10: Funktionsweise der vier Abschnitte in der Gegenstromchromatographie
Durch die Zu- und Abfuhr der Ströme repräsentiert durch Makeup , Feed , Raffinat
und Extrakt wird die Säule in vier Abschnitte unterteilt, in denen die unterschiedliche
Volumenströme vorliegen (vgl. Abbildung 2.10). Der entgegengerichtete Feststoff-
strom ist in allen Abschnitten gleich groß, da mit den externen Strömen keinerlei Feststoff
MV& FeedV&
RaffV& ExV&
I,fl ...V& IV,flV&
festV&

2 Grundlagen der Flüssigchromatographie
27
abgezogen wird. Den Abschnitten I bis IV werden dabei unterschiedliche Funktionen zuteil, nach
denen sie auch benannt werden.
Abschnitt I, Feststoffreinigung:
Durch Zugabe von reinem Eluent am Kopf der Säule wird die Komponente A und, falls vorhanden,
die Komponente B desorbiert. Im Abschnitt I fließt der größte Volumenstrom, der groß genug sein
muß, um beide Komponenten mit der Flüssigkeit in den darunterliegenden Abschnitt II zu
transportieren. Der Feststoff, an dem am oberen Ende der Säule nur Eluent adsorbiert sein sollte,
wird entnommen und kreisläufig am unteren Ende von Abschnitt IV wieder zugeführt.
Abschnitt II und III: Trennzonen:
Zwischen diesen Abschnitten wird das Feed zugegeben, so daß der Volumenstrom im Abschnitt III
größer ist als derjenige in Abschnitt II. Da Komponente A eine stärkere Affinität zum Feststoff hat,
wandert sie langsamer in die Richtung der Flüssigkeitsströmung als Komponente B.
Bei gegebenem Feststoffstrom wird zur Erreichung einer Trennung der Volumenstrom im
Abschnitt II so gewählt, daß Komponente A nach oben zum Extrakt und Komponente B nach
unten in Abschnitt III gefördert wird. Auch Abschnitt III muß einen Volumenstrom vorweisen, der
Komponente A nach oben zum Abschnitt II befördert, während Komponte B zum Raffinat wandert.
Der Volumenstrom in den Abschnitten II und III darf aber nicht gleich groß sein, da sonst kein
Feed mehr zugeführt werden könnte.
Kommt es zu Wechselwirkungen zwischen den Komponenten A und B durch eine konkurrierende
Adsorption, so wird die Wanderungsgeschwindigkeit von Komponente B im Abschnitt III durch die
Verdrängung durch A erhöht. Dies muß bei der Wahl der Volumenströme mitberücksichtigt
werden.
Abschnitt IV: Desorbent-Reinigung:
Beim Durchströmen des Abschnittes IV adsorbiert der Feststoff die Komponente B, und falls noch
vorhanden, auch die Komponente A, und transportiert diese in den darüberliegenden Abschnitt III,
da hier die Fließgeschwindigkeit der Flüssigkeit am geringsten ist. Bei korrekter Funktion sollte
reines Desorbent das untere Ende der Säule verlassen, welches unter Zugabe des Makeup-
stroms wieder am Kopf der Säule zugeführt wird. MV&

2 Grundlagen der Flüssigchromatographie
28
Tabelle 2.1: Beziehung zwischen den äußeren Strömen und den Strömen in den vier Abschnitten an den Zulauf- und Abzugsstellen Zulauf- bzw.
Abzugsstelle
Beziehung zwischen
den Volumenströmen
Beziehung zwischen den
Konzentrationen
Makeup - Zulauf MIVI VVV &&& += MMi,IV
ausIVi,I
einIi, VcVcVc &&& +=
Extrakt - Abzug ExIII VVV &&& −= ein
IIi,aus
Ii,Exi, ccc ==
Feed - Zulauf FeedIIIII VVV &&& += FeedFeedi,II
ausIIi,III
einIIIi, VcVcVc &&& +=
Raffinat - Abzug RaffIIIIV VVV &&& −= ein
IVi,aus
IIIi,Ri, ccc ==
Das Aufstellen der Massen- und Komponentenbilanzen an den Zu- und Ablaufstellen zeigt den
Zusammenhang zwischen den Volumenströmen und den Konzentrationen in den äußeren
Strömen sowie in den Strömen in den vier Abschnitten, was in Tabelle 2.1 dargestellt ist. In
Unterkapitel 7.3 werden dann die Zusammenhänge für die Berechnung der Konzentrationen
dargestellt, wenn es durch Fehlfunktionen zu anderen Konstellationen dominierender Transport-
richtungen kommt als es vorgegeben wurde.
Entscheidend für die Trennung der Komponenten A und B ist, daß sie in jedem Abschnitt zu ihrer
Entnahmestelle hin transportiert werden, was durch die Wahl der Ströme in den einzelnen
Abschnitten sowie des entgegenfließenden Feststoffstroms erreicht wird. Charakteristisch für das
kontinuierliche Gegenstromverfahren ist das Vorliegen eines lokal fixierten Konzentrationsprofils in
einem stationären Zustand.
2.6.2 Funktionsweise der Simulierten Gegenstromchromatographie (SMB)
Die für eine kontinuierliche Gegenstromchromatographie notwendige Bewegung des Feststoffs
entgegen dem Flüssigkeitsstrom ist in der Praxis kaum umzusetzen, da sich eine gleichmäßige
Packung und die darauf beruhende hohe Effektivität nicht realisieren läßt. Es gibt allerdings die
Möglichkeit, den Feststoffgegenstrom durch Umsetzen der Zu- und Ablaufstellen in Richtung des
Flüssigkeitsstroms zu simulieren. Der Feststoff bleibt dabei in der Säule fixiert, die mindestens in
vier Festbetten unterteilt wird. In festen Zeitintervallen, gemeinhin als Takte bezeichnet, werden
die Zu- und Ablaufstellen von Makeup, Extrakt, Feed und Raffinat, wie in Abbildung 2.9c
beispielhaft durch ein Rotationsventil geschehen, genau um eine Unterteilung in Richtung des
Flüssigkeitsstroms umgesetzt. Zu jeder Zeit sind also vier Leitungen aktiv, deren Abstand

2 Grundlagen der Flüssigchromatographie
29
zueinander konstant bleibt. Sind die Zu- und Ablaufstellen wieder an ihrem Ausgangspunkt
angekommen, so wurde ein Taktzyklus vollendet.
Je mehr Unterteilungen der Gesamtsäule vorgenommen werden, desto mehr nähert sich das
SMB-Verfahren an das kontinuierliche Gegenstromverfahren an. In der Praxis ist am häufigsten
eine Unterteilung eines Abschnittes in zwei Einzelbetten anzutreffen, was bei SMB-Anlagen mit
vier Abschnitten folgerichtig zum Vorhandensein von acht Festbetten führt.
Abbildung 2.11: Prinzipskizze einer aus 12 Einzelsäulen bestehenden SMB-Anlage [Beste (2001)]
Die am häufigsten anzutreffende praktische Umsetzung eines SMB-Systems zeigt Abbildung 2.11:
Dabei wird das Festbett in einzelne chromatographische Säulen aufgeteilt und diese Säulen zu
einem Kreis verschaltet. Da die vier Abschnitte zusammen mit den Zu- und Ablaufstellen rotieren,
muß die Kreislaufpumpe, je nachdem, in welchem Abschnitt sie sich gerade befindet, einen
anderen Volumenstrom fördern. Es gibt allerdings auch die Alternative, die Kreislaufpumpe
außerhalb des Kreislaufes zu installieren, so daß die Verschaltung zu dem Ziel führt, daß sich die
Kreislaufpumpe immer zwischen Abschnitt I und IV befindet, was zwei Vorteile bietet: Zum einen
fördert die Pumpe immer den gleichen Volumenstrom, zum anderen wird das Mitpumpen der zu
trennenden Komponenten im Normalfall vermieden.
Im Gegensatz zum TMB-Verfahren, bei dem das Konzentrationsprofil stationär ist, wandern diese
beim SMB-Verfahren mit dem Umsetzen der Zu- und Ablaufstellen. Dabei wandert es im Laufe
eines Taktes jeweils genau um eine Unterteilung in Fließrichtung der Flüssigkeit. Dadurch variiert
innerhalb eines Taktes auch die Position der Ablaufstellen relativ zum Konzentrationsprofil, so daß

2 Grundlagen der Flüssigchromatographie
30
der zeitliche Konzentrationsverlauf ein SMB-typisches periodisches Verhalten aufweist, welches
Abbildung 2.12 zeigt.
Zeit
Kon
zent
ratio
n Maximum
Minimum
Amplitude
Integr.Konz.
Abbildung 2.12: Periodisches Signal einer SMB-Anlage im quasistationären Gleichgewicht
Bei der SMB hat sich genau dann ein quasistationärer Zustand eingestellt, wenn das
Konzentrationsprofil innerhalb eines Taktes in zwei aufeinanderfolgenden Taktzyklen identisch ist.
Weiterhin gibt es bei beiden Verfahren Unterschiede hinsichtlich der Flüssigkeitsströme in den vier
Abschnitten. Während die äußeren Ströme bei beiden Verfahren identisch sind, muß zur Erzielung
der selben Relativgeschwindigkeit zwischen Flüssigkeit und Feststoff beim SMB-Verfahren,
bei dem der Feststoff fixiert ist, in den vier Abschnitten jeweils eine um die
Feststoffgeschwindigkeit größere Flüssigkeitsgeschwindigkeit eingestellt werden, was sich
formell darstellen läßt als
relw
TMBfestw
TMBfest
TMBfl
SMBflk,rel wwww +== , (2.45)
wobei sich die Geschwindigkeiten mit dem allgemeinen, bekannten Zusammenhang
AwV ⋅=& (2.46)
über die Querschnittsfläche und den Volumenstrom darstellen lassen als
( ) Sext
fest
Sext
TMB
Sext
SMB
A1V
AV
AV
ε−+
ε=
ε
&&& (2.47)

2 Grundlagen der Flüssigchromatographie
31
bzw.
fest
ext
extTMBSMB V1
VV &&&ε−
ε+= . (2.48)
Die Taktzeit τ beim SMB-Verfahren ist die analoge Größe zum Feststoffstrom beim TMB-
Verfahren. Die beiden Größen sind unter Kenntnis des Feststoffvolumens VS einer Säule mit
( )fest
SSext
fest
P
VlA1
VV
&&ε−
==τ (2.49)
ineinander überführbar.
Für beide Verfahren kann die Realisierung von Abschnitten unterschiedlicher Länge sinnvoll sein,
nämlich wenn z. B. aufgrund nichtlinearer Adsorptionsisothermen disperse oder steile Fronten
auftreten. Konkret bedeutet dies für das TMB-Verfahren, daß Festbetten unterschiedlicher Länge
eingesetzt werden, und für das SMB-Verfahren, daß eine unterschiedliche Zahl von Einzelsäulen
gleicher Länge pro Abschnitt installiert werden.
2.6.3 Modellierung der Gegenstromchromatographie
Die Simulierte Gegenstromchromatographie (SMB) ist ein leistungsfähiges aber auch ein
komplexes Trennverfahren mit einer großen Anzahl sich gegenseitig beeinflussender Parameter.
Zur Optimierung aber auch zur Untersuchung bestimmter Betriebszustände ist es daher
unerläßlich, auf der Modellierung der Gegenstromchromatographie eine dynamische Simulation
aufzubauen, die das Verhalten der Anlage abbildet. Mit Ihrer Hilfe können die Einflüsse der
Modell- und Betriebsparameter, aber auch, wie im Rahmen dieser Arbeit geschehen, die
Auswirkungen auftretender Fehler und daraus resultierender Störungen auf das
Konzentrationsprofil untersucht werden. Dies kann ohne Chemikalien- und mit erheblich
geringerem Ressourcenverbrauch geschehen und ist schneller als die experimentelle Alternative.
Zur Abbildung einer SMB-Anlage ist die Kenntnis mehrerer Parameter-Arten notwendig:
• Zum einen die konstruktiven Parameter. Sie stellen die Eigenschaften der Trennapparatur dar,
also die Anzahl und Abmessungen der Säulen und Verbindungsrohrleitungen.

2 Grundlagen der Flüssigchromatographie
32
• Weiterhin die physiko-chemischen Parameter Porosität, Adsorptionsisothermen, die
Konstanten der axialen Dispersionskoeffizienten sowie die Stoffübergangskoeffizienten,
welche die zu trennenden Substanzen sowie die stationäre und mobile Phase beschreiben.
• Die Betriebsparameter schließlich sind die Feedkonzentration, die Volumenströme von Feed,
Abschnitt I, Extrakt und Raffinat sowie die Taktzeit, die den fiktiven Feststoffvolumenstrom
darstellt. Dieses sind die Stellgrößen für die Optimierung, wenn eine SMB-Apparatur mit
Stoffsystem gegeben ist und damit die Modellparameter feststehen.
Die Kenntnis aller genannten Parameter ist notwendig, um über Simulationsrechnungen die reale
Anlage abzubilden.
Um eine reale SMB-Anlage zu modellieren, gibt es zwei Modellansätze:
1. Zum einen über das TMB-Modell (True Moving Bed), das einen kontinuierlichen Gegenstrom
von Flüssigkeit und Feststoff annimmt.
2. Zum anderen über das SMB-Modell (Simulated Moving Bed), das den Gegenstrom wie in der
realen Anlage durch das Umsetzen der Zu- und Ablaufstellen abbildet.
Das chromatographische Festbett kann für beide Ansätze durch einen Rohrreaktor oder durch
eine Kaskade ideal durchmischter Rührkessel abgebildet werden [Seidel-Morgenstern (1995)].
Bei Nutzung des TMB-Modells bildet sich ein örtlich festes Konzentrationsprofil und zeitlich
konstante Produktkonzentrationen aus, während beim SMB-Modell das Konzentrationsprofil mit
den Zulauf- und Abzugsstellen entlang der Säule wandert und die Produktkonzentrationen
periodischen Änderungen unterworfen sind (siehe Abbildung 2.12). Die TMB kann also einen
stationären Zustand erreichen und als solcher simuliert werden, die SMB kann nur einen
instationären oder quasistationären Zustand einnehmen.
Die Beschreibung einer Anlage zur Simulierten Gegenstromchromatographie mit dem SMB-Modell
ist natürlich realistischer, da das Umsetzen der Zu- und Ablaufstellen und damit besagte Quasi-
stationärität des Prozesses berücksichtigt wird.
In der Literatur werden SMB-Modelle beschrieben u. a. von Storti et al. (1988a), Chu und Hashim
(1995), Hassan et al. (1995), Strube (1996) und Pais (1998); TMB-Modelle u. a. von Hashimoto et
al. (1983), Ruthven (1984), Storti (1988b) und Ching et al. (1992). Während die Modellgleichungen
keinen wesentlichen Veränderungen unterliegen, gibt es bei den numerischen Lösungsverfahren

2 Grundlagen der Flüssigchromatographie
33
laufend Verbesserungen mit dem Ziel, die Rechenzeit zu verringern [Gu (1995)], [Dünnebier und
Klatt (2000)].
2.6.3.1 Voraussetzungen und grundlegende Annahmen
Die stationäre Betrachtung mit Hilfe des TMB-Modells reicht meist für eine überschlägige
Ermittlung der Betriebsparameter und eine erste Beurteilung des Trennergebnisses. Allerdings
kommt es insbesondere bei die Simulation von effizienten stationären Phasen und nicht-linearen
Adsorptionsisothermen zu zunehmenden Abweichungen zwischen den Ergebnissen des TMB-
und des SMB-Modells. Auch die überwiegende Zahl möglicher Fehlfunktionen in einer SMB-
Anlage sind mit dem TMB-Modell nicht modellierbar. Insbesondere zur Ermittlung der
quasistationären Konzentrationsverläufe in den Produktströmen ist nur das SMB-Modell geeignet.
Zur Beschreibung des chromatographischen Festbettes gibt es mehrere Möglichkeiten [Seidel-
Morgenstern (1995)], von denen hier das Rohrreaktor-Modell ausgewählt wurde. Dieses hat den
Vorteil, daß die Einflüsse der kinetischen Effekte axiale Dispersion und Strofftransport getrennt
voneinander erfaßt werden können [Cziesla (1994)]. Bei Verwendung der Rührkesselkaskade, die
ein Stufenmodell ist, werden diese Effekte gemeinsam über die Trennstufenzahl NTUi beschrieben
(s. Gl. (2.11) und (2.13)), was mehrere Schwierigkeiten verursacht:
• Bei gekoppelten Adsorptionsisothermen ist eine unabhängige Beschreibung der kinetischen
Effekte mit einem Stufenmodell nicht mehr möglich.
• Wegen der Konzentrationsabhängigkeit der Trennstufenzahl müßten Stufen unterschiedlicher
Höhe eingeführt werden, deren Höhen für das SMB-Modell wegen der Wanderung des
Konzentrationsprofils zu jedem berechneten Zeitpunkt anzupassen wären.
• Die Anzahl der Trennstufen ist von der Geschwindigkeit der mobilen Phase abhängig, die bei
der Gegenstromchromatographie in jedem Abschnitt unterschiedlich ist. Für das SMB-Modell
würde dieses zu einem erheblichen Rechenaufwand führen, da durch das Umschalten die
Säulen durch die verschiedenen Abschnitte wandern. Das Konzentrationsprofil dieser Säulen
müßte auf eine andere Trennstufenzahl umgerechnet werden, indem Beladungen und
Konzentrationen in den einzelnen Stufen neu berechnet werden [Strube (1996)]
Sowohl dem TMB- als auch dem SMB-Modell liegen folgende Annahmen zugrunde:

2 Grundlagen der Flüssigchromatographie
34
• Es wird ein isothermes Temperaturfeld angenommen, d.h. thermische Effekte, die
beispielsweise aufgrund von Dissipation, Wärmeabgabe an die Umgebung sowie durch Ad-
und Desorption auftreten können, werden vernachlässigt. Unter dieser Annahme entfällt die
Notwendigkeit, die Energiebilanz zu lösen und die physiko-chemischen Parameter
temperaturabhängig zu beschreiben.
• Es findet eine Vernachlässigung des Druckgefälles in der Anlage statt, d.h. der auftretende
Druckverlust wird nicht berücksicht. Ausnahme ist die spätere Betrachtung undichter Ventile,
wo das Auftreten von Alternativströmen betrachtet werden muß, für deren Größe der
Druckverlust verantwortlich ist. Da ein Kolbenprofil angenommen wird, entfällt die
Notwendigkeit der Lösung der Impulsbilanz.
• Es wird eine vollständige radiale Durchmischung angenommen, das Auftreten von radialen
Konzentrationsunterschieden wird vernachlässigt, so daß nur ein eindimensionales, axiales
Konzentrationsprofil zu berechnen ist. Diese Annahme, die nach Deckwer (1973) bei
turbulenter Strömung und nicht zu großem Durchmesser des Strömungsrohres mit
aureichender Genauigkeit erlaubt ist, wird vereinfachend getroffen, obschon im Bereich
laminarer Strömung gearbeitet wird.
• Es wird eine homogene Verteilung der Schüttung in der Säule angenommen. Ausnahmen
davon werden im Rahmen der Fehlermodellierung gesondert betrachtet.
• Die Beschreibung des Stofftransport erfolgt über einen linearisierten Stoffübergang in der
festen Phase (Festfilmmodell, siehe Unterabschnitt 2.4.4.2). Der Stoffübergangskoeffizient
wird als unabhängig von der Konzentration und der Fluidgeschwindigkeit angenommen.
• Der axiale Dispersionskoeffizient wird als konzentrationsunabhängig aber linear von der
Relativgeschwindigkeit abhängig (Dax=CDax wrel) angenommen.
• Die Adsorption verläuft reversibel.
Da durch die Implementation des SMB-Modells in die Simulationsumgebung gPROMS (general
Process Modelling System) im Rahmen der Arbeit von Scheer (2003) die Rechenzeit gegenüber
dem alten Modell unter SPEEDUP um mehr als 90% reduziert werden konnte, ist der
Rechenzeitunterschied zwischen TMB- und SMB-Simulation auf wenige Minuten reduziert worden,
so daß im Rahmen dieser Arbeit nahezu vollständig auf das SMB-Modell zurückgegriffen wurde.
Daher soll in dieser Arbeit das TMB-Modell nur soweit beschrieben werden, wie es als Basis aber
auch als Verständnisgrundlage für das SMB-Modell dient. Ebenfalls wichtig ist das Aufzeigen der
Unterschiede. Auf eine detailliertere Beschreibung insbesondere der Numerik zur Lösung des
TMB-Modells wird an dieser Stelle verzichtet und auf die Arbeiten von Beste (2001), Cziesla
(1994) und Uysal (2000) verwiesen.

2 Grundlagen der Flüssigchromatographie
35
2.6.3.2 Modellierung der kontinuierlichen Gegenstromchromatographie (TMB)
Die dem TMB-Modell zugrundeliegenden Säulen werden als isothermer Rohrreaktor betrachtet, in
dem Flüssigkeit und Feststoff in Kolbenströmung gegeneinander strömen. In der Bilanzierung wird
neben dem konvektiven Transport ein Stofftransportwiderstand und in der flüssigen Phase eine
axiale Dispersion berücksichtigt.
Dem Feststoff wird eine fiktive Gewindigkeit wfest zugeordnet, die über die Säulenlänge und die
Takzeit τ einer SMB definiert ist:
τ= S
festl
w . (2.50)
Die Geschwindigkeit der flüssigen Phasen wfl in einem Abschnitt k ist vom Querschnitt der Säule
As abhängig:
extS
kk,fl A
Vw
ε=
&.
(2.51)
Für die Beschreibung mittels Differentialgleichungen entfallen für das stationäre TMB-Modell die
zeitlichen Differentialquotienten, so daß die differentiellen Stoffbilanzen am Volumenelement für
die beiden Komponenten in der flüssigen und festen Phase gewöhnliche Differentialgleichungen
sind. Die beiden Differentialgleichungen sind über einen Stoffaustauschterm gekoppelt [Hidajat
(1990)].
Flüssigkeit:
4444 34444 21434214434421 uschStoffausta
i*iifest,eff,ext
Konvektion
ikfl,ext
Dispersion.axiale
2i
2
kax,ext )q(qk)(1dzdc
wdz
cdD0 −ε−−ε−ε= (2.52)
Feststoff:
4444 34444 2144 344 21 uschStoffausta
i*iifest,eff,ext
Konvektion
ifestext )q(q)k1(
dzdq
)w1(=0 −ε−+ε− (2.53)
Die axiale Dispersion hängt linear von der Relativgeschwindigkeit ab:

2 Grundlagen der Flüssigchromatographie
36
relDaxax wCD = (2.54)
Als Adsorptionsisothermen können die entsprechenden Isothermenmodelle aus Abschnitt 2.4.3
verwendet werden.
Abbildung 2.13: Bilanzierung einer Säule (Rohrreaktor für den Gegenstromfall)
Die Randbedingungen wurden nach Danckwerts (1953) formuliert, wobei am Eintritt einer Säule n
(z=0) die Konzentration ci, n|z=0 bereits aufgrund der zugelassenen axialen Dispersion verringert ist.
Am Austritt (z=ls) wird angenommen, daß die Konzentrationsgradienten gleich Null sind.
Flüssigkeitseintritt (z=0):
0z
i
k,fl
k,ax
0zn,iein
ni, dzdc
wD
cc=
=⋅−=
(2.55)
Flüssigkeitsaustritt (z=ls):
0
dzdc
Slz
n,i ==
(2.56)
Neben den differentiellen Bilanzen müssen für jede Säule n die integralen Komponentenbilanzen
erfüllt sein.
( ) ( )einn,i
ausn,ifest
ausn,i
einn,ik,fl qqVccV −=− && (2.57)
2.6.3.3 Modellierung der simulierten Gegenstromchromatographie (SMB)
Die Modellierung und dynamische Simulation des SMB-Verfahrens wurde ursprünglich für die
Simulationsumgebung SPEEDUP [Lisso (1996)] durchgeführt. Basierend auf dieser Arbeit wurde

2 Grundlagen der Flüssigchromatographie
37
das SMB-Modell im Jahr 2000 im Rahmen einer Studienarbeit [Scheer (2003)] in gPROMS
implementiert. Eine Validierung des gPROMS-Modells erfolgte durch Beste (2001), eine
Validierung des SPEEDUP-Modells durch Deckert (1997).
Die dem dynamischen SMB-Rohrreaktor-Modell zugrundeliegenden partiellen Differential-
gleichungen unterscheiden sich von denen des TMB-Modell dahingehend, daß feststoffseitig kein
konvektiver Stofftransport stattfindet und sowohl für die flüssige als auch die feste Phase die
zeitliche Änderung der Konzentration bzw. der Beladung im differentiellen Volumenelement
berücksichtigt wird. Dies ergibt für die Flüssigkeit:
4444 34444 214444 34444 214342143421321
Dispersion radiale
i2
i2
axext
uschStoffausta
i*ii,fest,effext
Konvektion
irelext
Dispersionaxiale
2i
2
axext
gSpeicherun
iext r
cr1
rc
D)qq(k)1(zc
wzc
Dt
c
∂∂
+∂∂
ε+−ε−−∂∂
ε−∂∂
ε=∂
∂ε
(2.58)
und für den Feststoff:
4444 34444 2143421 uschStoffausta
i*ii,fest,effext
gSpeicherun
iext )qq(k)1(
tq)1( −ε−=∂
∂ε− .
(2.59)
Gleichung (2.58) enthält dabei den Term der radialen Dispersion, der im Rahmen der SMB-
Untersuchungen vernachlässigt wurde, der jedoch bei der Simulation präparativer Batch-Säulen
mit FLUENT in Kapitel 6 durchaus eine Rolle spielt.
Die Anfangsbedingungen werden so gewählt, daß zu Versuchsbeginn zum Zeitpunkt t=0 alle
Säulen über die gesamte Säulenlänge ls mit reinem Desorbent gefüllt sind und der Feststoff
unbeladen ist, also
0)l...0z,0t(q0)l...0z,0t(c
Sn,i
Sn,i
===
===. (2.60)
Die zur Lösung der partiellen Differentialgleichungen notwendigen Randbedingungen lassen am
Säuleneintritt keinerlei Dispersion zu, die Konzentration am Einlauf entspricht also der
Zulaufkonzentration. Am Säulenende wird wie beim TMB-Modell angenommen, daß keine
Änderung der Konzentration stattfindet. Es gilt am Flüssigkeitseintritt (z=0):

2 Grundlagen der Flüssigchromatographie
38
0zn,i
einni, cc
== (2.61)
und am Säulenende (z=ls):
0
dzdc
xSlz
n,i =+=
. (2.62)
Bis zu diesem Punkt handelt es sich um die Simulation einer einzelnen chromatographischen
Säule, wie sie auch im Rahmen dieser Arbeit verwendet wurde. Zur Darstellung einer SMB-Anlage
mittels Simulation ist also noch die Verknüpfung der Säulen miteinander sowie die Modellierung
der Umschaltvorgänge der Zu- und Ablußströme notwendig.
Zwischen den Säulen werden Verbindungsstücke angenommen, die ähnlich den chromato-
graphischen Säulen als Rohrreaktor modelliert wurden und deren jeweilige Längen der am Institut
vorhandenen Anlage angepaßt wurden. Diese Vorsäulen enthalten keinen Feststoff. Damit ergibt
sich folgende beschreibende Differentialgleichung:
{ 4342143421Konvektion
irel
Dispersionaxiale
2i
2
ax
gSpeicherun
i
zcw
zcD
tc
∂∂
−∂∂
=∂∂ .
(2.63)
Abbildung 2.14: Verschaltung der chromatographischen Hauptsäule mit Vorsäule und Zwischenstück
Um die SMB-Anlage in der Simulation abbilden zu können, müssen die Säulen sowie die
dazwischenliegenden Rohrstücke verschaltet werden. Dies geschieht durch das Hintereinander-
schalten von Strukturen wie sie in Abbildung 2.14 dargestellt sind. Dabei werden die Volumina der
Rohrleitungen zwischen den Säulen jeweils in einer Vorsäule zusammengefaßt, denen die
chromatographischen Hauptsäulen nachgeschaltet sind. Dazu kommt ein volumenloses, ideal
durchmischtes Zwischenstück, welches sowohl die Zu- und Abläufe der Anlage als auch den
Volumenstrom der vorgeschalteten Säule zusammenfaßt, die sich in der Simulation theoretisch für
jedes Zwischenstück für jeden Takt getrennt schalten lassen.

2 Grundlagen der Flüssigchromatographie
39
Für eine SMB-Anlage mit 8 Säulen, wie sie den Simulationen dieser Arbeit zugrunde gelegt wurde,
werden 8 der in Abbildung 2.14 gezeigten Strukturen hinteraneinandergeschaltet und mit einer
Rückführung von der achten zur ersten Säule versehen.
Das Umschalten der äußeren Prozeßströme nach Ablauf der Taktzeit wird durch das Multiplizieren
der vier äußeren Prozeßströme eines Verbindungsstückes entsprechend des aktuellen Taktes mit
0 oder 1 realisiert. Die mit 1 multiplizierten Ströme werden der Anlage zugeführt oder entnommen,
das Vorzeichen für die einzelnen Produkte liegt im Simulationssheet fest.
Zur Lösung des Differentialgleichungssystems stellt gPROMS die sogenannte „Method of Lines“
[Dieterich (1992)] zur Verfügung, die die nichtlineare partielle Differentialgleichung in eine
gewöhnliche Differentialgleichung nach der Zeit überführt. Dabei werden die örtlichen
Differentialquotienten erster und zweiter Ordnung durch zentrale Differenzenquotienten ersetzt
[Lisso (1996)]. Für eine Stützstelle l und eine Ortsschrittweite ∆z nimmt dies die folgende Form an:
21,i,i1,i
2,i
2
1,i1,i,i
zcc2c
zc
z2cc
zc
∆
+−≈
∂
∂
∆
−≈
∂
∂
−+
−+
lll
ll
l
l
(2.64)
Das Einsetzen dieser Differenzenquotienten in die beschreibenden Differentialgleichungen (2.58)
und (2.59) führt zu folgender Gleichung:
( )i*ii,fest,eff
ext
ext1l,i1l,irel2
1l,il,i1l,iax
l,i qqk1
z2cc
wz
cc2cD
tc
−ε
ε−−
∆
−−
∆
+−=
∂
∂ −+−+
(2.65)
Das Gleichungssystem enthält gewöhnliche, zeitabhängige Differentialgleichungen als
unabhängige Variablen und die Adsorptionsisothermen, die entweder lineare oder nicht-lineare
algebraische Gleichungen sein können. Es wird als Differential-Algebra-(DA)-System bezeichnet.
Dieses muß wegen der Kopplung der Stützstellen für einen Zeitpunkt simultan gelöst werden.
Dieses wird durch die Simulationsumgebung gPROMS erledigt, das über einen DA-
Gleichungslöser und eine automatische Schrittweitensteuerung der Zeitdiskretisierung verfügt.

2 Grundlagen der Flüssigchromatographie
40
2.6.4 Auslegung und Optimierung einer Trennung mit einer SMB-Anlage
2.6.4.1 Kriterien der Auslegung und Optimierung
Prinzipiell gibt es zwei verschiedene Aufgabenstellungen bei der Auslegung und Optimierung einer
SMB-Anlage:
1. Die Trennapparatur besteht bereits und nur die Betriebsparameter, also die einzelnen
Volumenströme und die Taktzeit, müssen für ein gegebenes System ermittelt und optimiert
werden.
2. Nur das Trennsystem ist gegeben, und neben den Betriebsparametern muß auch die Anlage,
also Anzahl, Geometrie und Segmentierung der Säulen ermittelt werden.
Innerhalb dieser Arbeit ist nur Fall Nr. 1 von Interesse. Das Ziel ist die Modellierung von Fehlern
und Störungen sowie das Studium ihrer Charakteristika. Zu diesem Zweck wird davon
ausgegangen, daß eine vorgegebene Anlage, die ein bestimmtes Trennproblem zu lösen hat, für
welches die Betriebsparameter bereits ermittelt wurden, einen dieser Betriebsparameter aufgrund
einer Fehlfunktion nicht einhalten kann, so daß es zu einer Abweichung vom Soll-Zustand kommt.
Da auf der Basis der Auslegungs- und Optimierungsstrategie, die im Rahmen der Arbeiten u. a.
von Nicoud (1992) und Chu (1995) entwickelt wurde, ein Modell zur Beschreibung bestimmter
Fehlfunktionen und Störungen aufgebaut wurde, welches in Unterkapitel 7.3 dargelegt wird, wird
dieses in diesem Unterabschnitt zunächst detailliert beschrieben. Da weiterhin auch das
Auslegungsdiagramm von Storti et. al. (1993) verwendet wird, wird auch dieses nachfolgend
erläutert.
Bevor eine Optimierung bei einem Trennproblem durchgeführt werden kann, sind
Optimierungskriterien festzulegen. Neben der Qualität einer Trennung ist auch die Stabilität des
Trennprozesses von Bedeutung. Aus ökonomischer Sicht ist es wichtig, die spezifischen
Produktionskosten bei vorgegebenen Produktanforderungen zu minimieren.
Um die Trennleistung verschiedener SMB-Anlagen miteinander vergleichen zu können, werden im
folgenden Größen definiert, die die Qualität der Trennung beschreiben.

2 Grundlagen der Flüssigchromatographie
41
• Die Reinheit eines Produktes wird als Verhältnis der Konzentration der Zielkomponente Z im
Produktstrom p und der Summe der Konzentrationen aller n Komponenten in diesem Strom
definiert.
%100
p,c
cRH n
1ii
p,Zp,Z ⋅=
∑=
. (2.66)
• Die Ausbeute AB ist das prozentuale Verhältnis des Massenstroms einer Komponente im
Produktstrom zum zugeführten Massenstrom dieser Komponente im Feedstrom:
%100
VcVc
ABFp,Z
pp,Zp,Z ⋅=
&
&. (2.67)
Diese beiden Größen sind über die Komponentenbilanzen verknüpft.
• Die Produktivität PD wird definiert als das Verhältnis der Masse einer Komponente im
Produktstrom bezogen auf die eingesetzte Feststoffmenge in der Anlage
%100
mVc
PDAd
pp,Zp,Z ⋅=
&. (2.68)
• Der Desorbentbedarf DB gibt das Verhältnis des Desorbentstroms zu einem Produktstrom
wieder:
%100
VV
DBp
DP ⋅=
&
&. (2.69)
2.6.4.2 Auslegung und Optimierung unter ausschließlicher Betrachtung der Thermodynamik
Bei vorgegebener Geometrie einer SMB-Anlage hängt die Auslegung nur von den physiko-
chemischen Parametern des Trennsystems ab, also von der Thermodynamik in Form der
Adsorptionsisothermen, vom Formparameter Porosität sowie von den kinetischen Parametern
axiale Dispersion und Stofftransportwiderstand. Dabei hat die Thermodynamik in der Regel den

2 Grundlagen der Flüssigchromatographie
42
größeren Einfluß, so daß die Kinetik bei der ersten Auslegung zunächst vernachlässigt wird. Für
effiziente chromatographische Systeme reicht dies meist sogar vollständig aus. Effizient bedeutet
dabei eine hohe Trennstufenzahl, die meist bei der Verwendung kleiner Partikel (<20µm) als
stationäre Phase erreicht wird.
.
Recycle
AB
AB
AB
AB
mI > KA mII > KB mIII < KA mIV < KB
Extrakt (A) Feed (A,B) Raffinat (B)Makeup Zone I
Feststoffstrom
Abbildung 2.15: Binäre TMB mit vier Zonen und gewünschten Transportbedingungen der Komponenten [aus Beste (2001)]
Die Auslegung sei für lineare und ungekoppelte Adsorptionsisothermen dargestellt, was bedeutet,
daß die Wanderungsgeschwindigkeit einer Komponente nicht von ihrer Konzentration abhängt.
Abbildung 2.15 zeigt die notwendigen Transportrichtungen der Komponenten für eine
2-Komponenten-Trennung mit vier Abschnitten. Zur verständlicheren Darstellung wurde die
Anlage dabei als TMB gezeichnet.
Um eine hundertprozentige Reinheit beider Komponenten in den Produktströmen zu erreichen,
muß die stärker adsorbierende Komponente A vollständig im Extrakt und die schwächer
adsorbierende Komponente B vollständig im Raffinat abgezogen werden. Dies ist der Fall, wenn,
wie in Abbildung 2.15 dargestellt, Komponente A immer in Richtung des Extraktes und
Komponente B immer in Richtung des Raffinats transportiert wird, wobei ein Transport einer
Komponente über den Recycle-Strom bzw. über die Rückführung des Feststoffstrom
auszuschließen ist.
Die Einhaltung der fett markierten Transportrichtungen ist hinreichend, da dann die anderen
Bedingungen auch eingehalten sind.
Ob eine Komponente mit dem Feststoffstrom oder mit der Flüssigkeit wandert, hängt von der
Thermodynamik, der Porosität und den Fließgeschwindigkeiten von Flüssigkeit und Feststoff ab.
Die Wanderungsrichtung läßt sich über das Massenstromverhältnis σi, K für eine Komponente i im
Abschnitt k ausdrücken, das definiert ist als
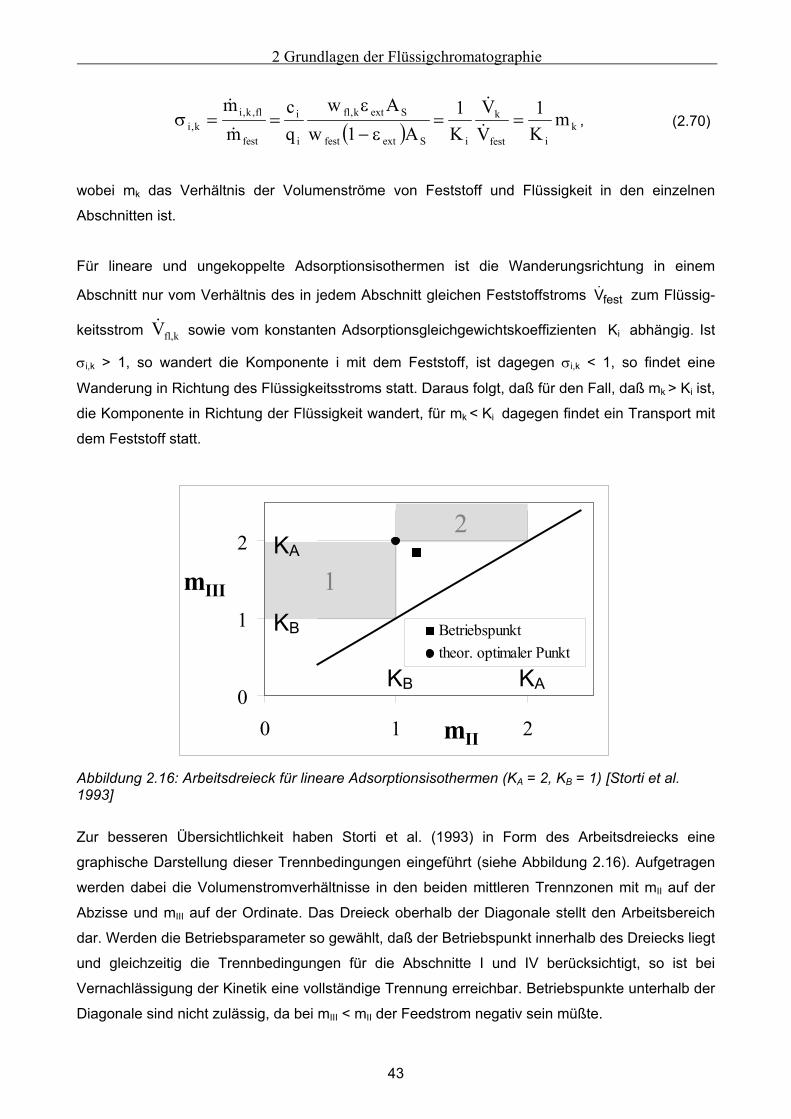
2 Grundlagen der Flüssigchromatographie
43
( ) kifest
k
iSextfest
Sextk,fl
i
i
fest
fl,k,ik,i m
K1
VV
K1
A1wAw
qc
mm
==ε−
ε==σ
&
&
&
&, (2.70)
wobei mk das Verhältnis der Volumenströme von Feststoff und Flüssigkeit in den einzelnen
Abschnitten ist.
Für lineare und ungekoppelte Adsorptionsisothermen ist die Wanderungsrichtung in einem
Abschnitt nur vom Verhältnis des in jedem Abschnitt gleichen Feststoffstroms zum Flüssig-
keitsstrom sowie vom konstanten Adsorptionsgleichgewichtskoeffizienten K
festV&
kfl,V& i abhängig. Ist
σi,k > 1, so wandert die Komponente i mit dem Feststoff, ist dagegen σi,k < 1, so findet eine
Wanderung in Richtung des Flüssigkeitsstroms statt. Daraus folgt, daß für den Fall, daß mk > Ki ist,
die Komponente in Richtung der Flüssigkeit wandert, für mk < Ki dagegen findet ein Transport mit
dem Feststoff statt.
1
2KA
KA
KB
KB0
1
2
0 1 2mII
mIII
Betriebspunkttheor. optimaler Punkt
Abbildung 2.16: Arbeitsdreieck für lineare Adsorptionsisothermen (KA = 2, KB = 1) [Storti et al. 1993]
Zur besseren Übersichtlichkeit haben Storti et al. (1993) in Form des Arbeitsdreiecks eine
graphische Darstellung dieser Trennbedingungen eingeführt (siehe Abbildung 2.16). Aufgetragen
werden dabei die Volumenstromverhältnisse in den beiden mittleren Trennzonen mit mII auf der
Abzisse und mIII auf der Ordinate. Das Dreieck oberhalb der Diagonale stellt den Arbeitsbereich
dar. Werden die Betriebsparameter so gewählt, daß der Betriebspunkt innerhalb des Dreiecks liegt
und gleichzeitig die Trennbedingungen für die Abschnitte I und IV berücksichtigt, so ist bei
Vernachlässigung der Kinetik eine vollständige Trennung erreichbar. Betriebspunkte unterhalb der
Diagonale sind nicht zulässig, da bei mIII < mII der Feedstrom negativ sein müßte.

2 Grundlagen der Flüssigchromatographie
44
Ist mIII > KA (Bereich 2), so führt dies zu einem mit Komponente A verunreinigtem Raffinat, ist
mII < KB (Bereich 1) so findet sich im Extrakt auch Komponente B. Der Punkt im Schnittpunkt der
beiden Katheten ist der Punkt maximaler Produktivität, weil hier der Feedstrom maximal ist. In der
Praxis wird meist ein Betriebspunkt gewählt, der gewisse Abstände zu den Grenzen des Dreiecks
besitzt, da sowohl kinetische Effekte als auch Instabilitäten den Arbeitsbereich verkleinern.
Zur Festlegung und zur Charakterisierung dieses Betriebspunktes wird der Stabilitätsfaktor β
definiert, der innerhalb der Grenzen von
B
A
KK
1 <β< (2.71)
gewählt werden muß, um eine vollständige Trennung zu erreichen. Mit Hilfe dieses
Stabilitätsfaktors ist es möglich, Gleichungen zum Festlegen der einzelnen Volumenströme
anzugeben, wobei die Größe eines Volumenstromes vorzugeben ist. Im folgenden wird die
Darstellung unter Annahme eines vorgegebenen Feedstroms zunächst für den TMB-Fall
dargestellt.
Der Feststoffstrom ist festgelegt durch:
β−β
=B
a
Feedfest
KKV
V&
& . (2.72)
Die Ströme in den 4 Abschnitten sind:
festA
TMBI VKV && ⋅β= , (2.73)
festB
TMBII VKV && ⋅β= , (2.74)
β= festATMB
III
VKV
&& , (2.75)

2 Grundlagen der Flüssigchromatographie
45
β= festBTMB
IV
VKV
&& . (2.76)
Neben den inneren Strömen können auch die äußeren Ströme berechnet werden, nämlich der
Extraktvolumenstrom
( ) festBAEx VKKV && −⋅β= , (2.77)
der Raffinatvolumenstrom
festBARaff V)KK(1V && −
β= (2.78)
und der Makeup-Strom
FeedRaffExM VVVV &&&& −+= . (2.79)
Da sich beim SMB-Verfahren der Feststoffstrom tatsächlich nicht bewegt, sondern in den Säulen
fixiert ist, unterscheiden sich die Volumenströme vom TMB-Fall. Eine Umrechnung für den
Abschnitt k erfolgt über:
fest
ext
extTMBk
SMBk V
1VV &&&
ε−ε
+= . (2.80)
Die Taktzeit ergibt sich aus:
fest
extS
fest
SS
V)1(V
V)1(LA
&&ε−
=ε−
=τ . (2.81)
2.7 Berechnung des Druckverlustes einer chromatographischen Anlage
Für spätere Untersuchungen ist die Kenntnis des Druckverlustes sowohl im chromatographischen
Festbett als auch in der restlichen Anlage erforderlich.

2 Grundlagen der Flüssigchromatographie
46
Der Druckverlust ∆p in einem chromatographischen Festbett steigt mit zunehmender
Fließgeschwindigkeit und abnehmender Partikelgröße des Adsorbens. Die Kenntnis des
Druckverlustes in einer chromatographischen Säule ist aus zwei Gründen von Relevanz: Zum
einen gibt er den maximalen Volumenstrom für ein System mit gegebener mobiler und stationärer
Phase vor, was wiederum die obere Grenze für die maximale Produktivität vorgibt. Zum anderen
ist er für die Einstellung unterschiedlicher Volumenströme von absoluter Relevanz, nämlich dann,
wenn entweder zwei parallel geschaltete Säulen unterschiedliche Porositäten haben, oder wenn
innerhalb eines chromatographischen Festbettes radiale Heterogenitäten der Porositäten
auftreten. Solche Fälle werden in Kapitel 6 und 7 untersucht.
Der Druckverlust in einer Schüttung läßt sich berechnen mit Hilfe der Kozeny-Carman-Gleichung
[Charton und Nicoud (1995)]:
w
1d36hlP
2
ext
ext2P
kS η
ε
ε−=∆ . (2.82)
Dabei ist hk der Kozeny-Koeffizient, der einen Wert von ca. 4,5 annimmt. Der Druckverlust kann
auch nach dem VDI-Wärmeatlas (1994) berechnet werden.
Relevant für den Druckverlust speziell in einer SMB-Anlage sind auch die Druckverluste in
Leitungen, sowie solche, die durch Einbauten wie z.B. Ventile oder durch Krümmer verursacht
werden. Auf das Formelwerk zur Berechnung solcher Druckverluste soll an dieser Stelle nicht
weiter eingegangen werden. Sie sind verfügbar z. B. bei Schade (1989) oder im VDI-Wärmeatlas
(1994).
Speziell die zur Untersuchung der Fehler in den Säulen verwendete CFD-Software FLUENT
benutzt zur Charakterisierung einer laminaren Strömung in einem porösen Medium die
Permeabilität κ, welche aus Darcy’s Gesetz [Fluent (1996)] stammt:
Pw 0 ∆
ηκ
−= . (2.83)
Hierbei ist w0 die Leerrohrgeschwindigkeit und η die dynamische Viskosität. Die Permeabilität läßt
sich einerseits über Gleichung (2.83) mit dem Druckverlust aus Gleichung (2.82) berechnen, die
Berechnung ist aber auch direkt über die Blake-Kozeny Gleichung [Bird et al. (1960)] möglich:

2 Grundlagen der Flüssigchromatographie
47
( )2ext
3ext
2P
ε1ε
150d
κ−
= , (2.84)
Die Anwendbarkeit des Gesetzes von Darcy wird durch den Strömungszustand in der
chromatographischen Säule gerechtfertigt. Für die Reynoldszahl in der Säule definiert durch
ηρ
= PwdRe , (2.85)
ergeben sich für grenzwertige Betrachtungen, also Kombinationen von hohen
Zwischenkorngeschwindigkeiten, großen Partikeldurchmessern, hohen Dichten und niedrigen
dynamischen Viskositäten Werte unter 1. Da für Rohr- und Zylinderströmungen der laminare
Bereich der Reynoldszahl zwischen 0 und 40 liegt [Schade und Kunz (1989)], ist die Annahme
einer Kriechströmung durch das Festbett gerechtfertigt.

3 Methoden zur Bestimmung von Adsorptionsisothermen
48
3 Methoden zur Bestimmung von Adsorptions-isothermen
Um Adsorptionsisothermen ermitteln zu können, wurden verschiedene Meßmethoden entwickelt,
die sich in statische und dynamische Meßmethoden unterteilen lassen. Im folgenden werden in
den Unterkapiteln 3.1 und 3.2 zunächst die Grundlagen von statischen und dynamischen
Methoden erläutert, wobei der Schwerpunkt auf die im Rahmen dieser Arbeit verwendeten
Verfahren Zirkulationsmethode, Frontalanalyse und Perturbationsmethode gelegt wird. Unter-
kapitel 3.3 erläutert mögliche Methoden zur Bestimmung der unterschiedlichen Porositäten. In
Kapitel 5 findet dann ein Vergleich der Ergebnisse statt, die unter Nutzung beider Arten von
Meßmethoden erzielt wurden.
3.1 Statische Meßmethoden
Statische Meßmethoden zur Bestimmung von Adsorptionsisothermen beruhen darauf, daß eine
Mischung mit bekannter Zusammensetzung mit einem Adsorbens in Kontakt gebracht wird. Nach
dem Erreichen eines Adsorptionsgleichgewichtes wird die Zusammensetzung der Bulk-Phase
ermittelt (siehe Abbildung 2.3). Wenn die Zusammensetzung der adsorbierten Phase xi' nicht
ermittelbar ist, geben die Ergebnisse der statischen Messung unter der Annahme, daß sich mehr
als eine Komponente in der adsorbierten Phase befindet, keine Auskunft über die adsorbierte
Menge ni' bzw. qi einer Komponente, sondern nur eine quantitative Aussage, welche Komponente
stärker adsorbiert wird. Daher wird zur Auswertung statischer Meßmethoden im allgemeinen der
Adsorptionsexzeß (vgl. Unterkapitel 2.3) herangezogen.
Die statischen Meßverfahren lassen sich folgendermaßen unterteilen:
• Tauchmethode oder Schüttelmethode:
Dies ist die älteste und auch einfachste statische Meßmethode. Bulk-Phase und Adsorbens
werden in einem thermostatisierten Gefäß in Kontakt gebracht, wobei die Gleichgewichts-
einstellung durch Rühren oder Schütteln beschleunigt werden kann. Die Konzentrations-
bestimmung erfolgt in der Regel über diverse Probennahmen während der Versuchszeit.
Diese Methode ist die Urform aller Batch-Methoden und findet in unterschiedlicher Form ihren
Niederschlag in den verschiedenen statischen Meßmethoden.

3 Methoden zur Bestimmung von Adsorptionsisothermen
49
• Zirkulationsmethode:
Sie arbeitet im Prinzip wie die Tauchmethode, allerdings wird die Bulk-Phase in einem
Kreislauf zwischen Adsorptionszelle und einer Analyseeinheit umgepumpt, so daß eine
Online-Analyse der Bulk-Phase möglich ist. Da das Umpumpen das Rühren oder Schütteln
ersetzt, wird ferner die mechanische Belastung des Adsorbens reduziert. Diese Methode
wurde im Rahmen dieser Arbeit verwendet, daher findet sich in Abschnitt 5.1.2 eine
detaillierte Beschreibung einer Anlage dieser Art.
• Schlammethode:
Diese Methode zeichnet sich durch einen Verzicht auf die konstruktive Trennung von Feststoff
und Bulk-Phase zur Analyse aus. Es wird eine Probe des Fest-Flüssig-Gemisches
entnommen, zentrifugiert und analysiert.
• Nullmethode:
Nach Vorbild der Zirkulationsmethode wird das Adsorbens mit der Bulk-Phase in Kontakt
gebracht und deren Zusammensetzung analysiert. Es wird dann allerdings nicht die
Gleichgewichtszusammensetzung bestimmt, sondern es erfolgt eine schrittweise Zudosierung
der in der Bulk-Phase vorhandenen Reinstoffe, bis die Ausgangszusammensetzung wieder
erreicht ist. Aus der zudosierten Stoffmenge läßt sich dann der Adsorptionsexzeß berechnen.
• Adsorptions/Desorptionsmethode:
Hierbei wird eine Adsorbensmenge in einer Säule mit einer Bulk-Phase konstanter Konzen-
tration so lange beaufschlagt, bis die Ausgangskonzentration der Eingangskonzentration
entspricht. Diese gesättigte Säule wird nun mit reinem Eluent gespült, bis am Austritt der
Säule nur noch reines Eluent detektiert wird. Der aufgefangene Austrittsstrom wird mengen-
mäßig erfaßt und seine Zusammensetzung analysiert, so daß eine Berechnung der
adsorbierten Menge möglich ist.
Für die statische Bestimmung der Adsorptionsisothermen wurde die Zirkulationsmethode ausge-
wählt, da sie zum einen genauer ist als die Tauchmethode und auch eine mehrfache Verwendung
des Adsorbens möglich ist, zum anderen ist sie gerade im Bereich geringerer Konzentrationen
genauer ist als die Adsorptions/Desorptionsmethode.
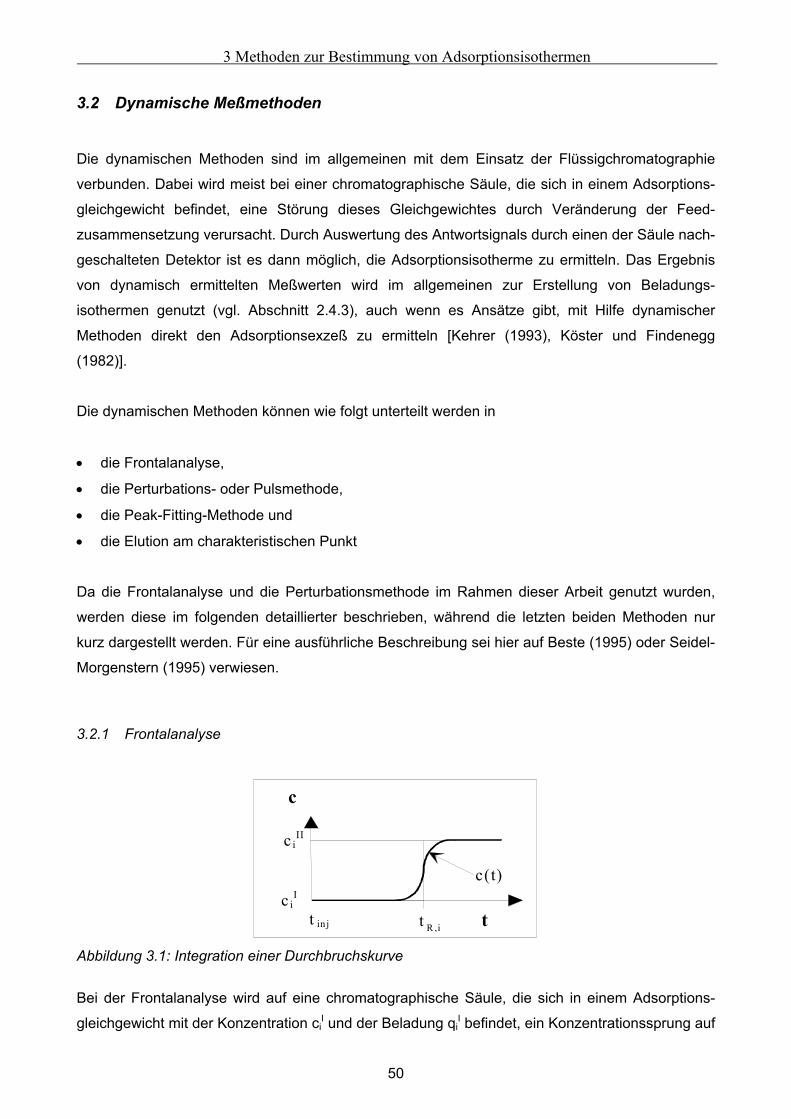
3 Methoden zur Bestimmung von Adsorptionsisothermen
50
3.2 Dynamische Meßmethoden
Die dynamischen Methoden sind im allgemeinen mit dem Einsatz der Flüssigchromatographie
verbunden. Dabei wird meist bei einer chromatographische Säule, die sich in einem Adsorptions-
gleichgewicht befindet, eine Störung dieses Gleichgewichtes durch Veränderung der Feed-
zusammensetzung verursacht. Durch Auswertung des Antwortsignals durch einen der Säule nach-
geschalteten Detektor ist es dann möglich, die Adsorptionsisotherme zu ermitteln. Das Ergebnis
von dynamisch ermittelten Meßwerten wird im allgemeinen zur Erstellung von Beladungs-
isothermen genutzt (vgl. Abschnitt 2.4.3), auch wenn es Ansätze gibt, mit Hilfe dynamischer
Methoden direkt den Adsorptionsexzeß zu ermitteln [Kehrer (1993), Köster und Findenegg
(1982)].
Die dynamischen Methoden können wie folgt unterteilt werden in
• die Frontalanalyse,
• die Perturbations- oder Pulsmethode,
• die Peak-Fitting-Methode und
• die Elution am charakteristischen Punkt
Da die Frontalanalyse und die Perturbationsmethode im Rahmen dieser Arbeit genutzt wurden,
werden diese im folgenden detaillierter beschrieben, während die letzten beiden Methoden nur
kurz dargestellt werden. Für eine ausführliche Beschreibung sei hier auf Beste (1995) oder Seidel-
Morgenstern (1995) verwiesen.
3.2.1 Frontalanalyse
c
ciII
c(t)c i
I
t in j t R ,i Abbildung 3.1: Integration einer Durchbruchskurve
t
Bei der Frontalanalyse wird auf eine chromatographische Säule, die sich in einem Adsorptions-
gleichgewicht mit der Konzentration ciI und der Beladung qi
I befindet, ein Konzentrationssprung auf
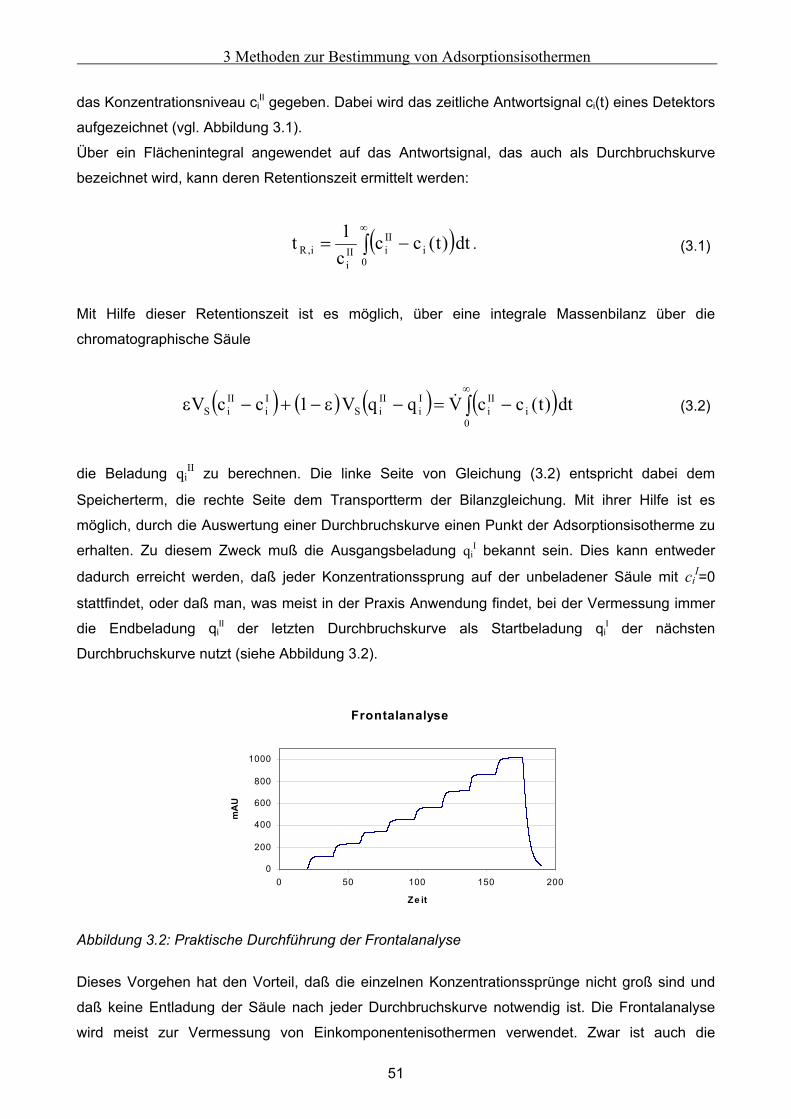
3 Methoden zur Bestimmung von Adsorptionsisothermen
51
das Konzentrationsniveau ciII gegeben. Dabei wird das zeitliche Antwortsignal ci(t) eines Detektors
aufgezeichnet (vgl. Abbildung 3.1).
Über ein Flächenintegral angewendet auf das Antwortsignal, das auch als Durchbruchskurve
bezeichnet wird, kann deren Retentionszeit ermittelt werden:
( )∫∞
−=0
iIIiII
ii,R dt)t(cc
c1t . (3.1)
Mit Hilfe dieser Retentionszeit ist es möglich, über eine integrale Massenbilanz über die
chromatographische Säule
( ) ( ) ( ) ( )dt)t(ccVqqV1ccV0
iIIi
Ii
IIiS
Ii
IIiS ∫
∞
−=−ε−+−ε & (3.2)
die Beladung qiII zu berechnen. Die linke Seite von Gleichung (3.2) entspricht dabei dem
Speicherterm, die rechte Seite dem Transportterm der Bilanzgleichung. Mit ihrer Hilfe ist es
möglich, durch die Auswertung einer Durchbruchskurve einen Punkt der Adsorptionsisotherme zu
erhalten. Zu diesem Zweck muß die Ausgangsbeladung qiI bekannt sein. Dies kann entweder
dadurch erreicht werden, daß jeder Konzentrationssprung auf der unbeladener Säule mit ciI=0
stattfindet, oder daß man, was meist in der Praxis Anwendung findet, bei der Vermessung immer
die Endbeladung qiII der letzten Durchbruchskurve als Startbeladung qi
I der nächsten
Durchbruchskurve nutzt (siehe Abbildung 3.2).
Frontalanalyse
0
200
400
600
800
1000
0 50 100 150 200
Ze it
mA
U
Abbildung 3.2: Praktische Durchführung der Frontalanalyse
Dieses Vorgehen hat den Vorteil, daß die einzelnen Konzentrationssprünge nicht groß sind und
daß keine Entladung der Säule nach jeder Durchbruchskurve notwendig ist. Die Frontalanalyse
wird meist zur Vermessung von Einkomponentenisothermen verwendet. Zwar ist auch die

3 Methoden zur Bestimmung von Adsorptionsisothermen
52
Vermessung von Mehrkomponentenisothermen möglich, dies erfordert jedoch den Einsatz eines
selektiven Detektors.
Für die in Gleichung (3.2) benutzte Porosität ε kann man gemäß Abschnitt 2.4.3 entweder die
externe oder die Gesamtporosität verwenden. Die Beladung hat die Einheit [g/l].
3.2.2 Die Perturbations- oder Pulsmethode
0
10
20
30
40
50
60
70
80
90
0 0.2 0.4 0.6 0.8
Zeit [h]
Kon
zent
ratio
n [g
/l]
Signal
injt i,Rt
injt i,Rt
Abbildung 3.3: Durchführung der Perturbationsmethode
Auf eine chromatographische Säule, die sich mit der Konzentration ciI in einem Adsorptions-
gleichgewicht befindet, wird eine kleine Konzentrationsstörung gegeben. Diese Störung, die zum
Zeitpunkt tinj aufgegeben wird, kann sowohl positiv als auch negativ sein [Blümel (1997)]. Die
wesentliche Meßgröße bei der Perturbationsmethode ist die Retentionszeit einer Konzentrations-
störung. Mit Hilfe der leicht abgewandelten Gleichung (2.31)
−+=
Ici
i0i,R dc
dq11ttε
ε (3.3)
ist es möglich, über die gemessene Retentionszeit die Tangentensteigung der
Adsorptionsisotherme zu bestimmen, was bedeutet, daß eine Linearisierung der Isothermen
entsprechend dem jeweiligen Konzentrationsniveau durchgeführt wird. Die Vermessung auf einem
Konzentrationsniveau ergibt also einen Punkt der Adsorptionsisotherme. Durch Messung der
Retentionszeiten von Störungen auf verschiedenen Konzentrationsniveaus erhält man mehrere
Tangentensteigungen. Durch eine Parameteranpassung der ersten Ableitung eines
Isothermenmodells, wie beispielsweise derjenigen der einfachen Langmuir-Isotherme

3 Methoden zur Bestimmung von Adsorptionsisothermen
53
( ) ( )2ii
i2ii,s
ii
ii,s
i
i
cb1
cbqcb1
bqdcdq
+−
+= (3.4)
an diese Tangentensteigungen (in diesem Fall qs,i und bi) ist es möglich, den Verlauf der
Adsorptionsisotherme zu ermitteln.
Wichtig bei der Anwendung dieser Methode ist, daß die injizierte Menge zur Erzeugung der
Perturbation nicht zu groß sein darf. Dies hat vornehmlich zwei Gründe [Beste (1995)]:
1. Zum einen entspricht die Konzentration im Maximum der Störung nicht genau derjenigen des
zu vermessenden Konzentrationsniveaus, sondern ist etwas kleiner bzw. größer, je nach Art
der Störung. Je größer diese Abweichung mit steigender Probemenge ist, desto größer wird
diese Konzentrationsabweichung. Bei einer gekrümmten Isotherme gibt eine andere
Konzentration jedoch eine andere Wanderungsgeschwindigkeit an, so daß nicht mehr nur ein
Punkt, sondern ein Bereich der Isotherme gemessen wird. Die Linearisierung durch Gleichung
(3.3) ist nicht mehr korrekt.
2. Der andere Grund liegt in der Peakform: Bei sehr kleinen Konzentrationsabweichungen ist die
Verformung des Peaks verursacht durch eine gekrümmte Isotherme schwach und annährend
symmetrisch. Demzufolge kann die Retentionszeit der Wanderungsgeschwindigkeit des
Peakmaximums gleichgesetzt werden. Bei größeren Abweichungen muß diese Verformung
unter Umständen durch Bestimmung der Retentionszeit des Peakmoments (vgl. Unterkapitel
2.4) korrigiert werden.
Die aufgegebene Störung darf aber auch nicht zu klein sein, da sie sonst im Rauschen des
Detektors untergeht.
Die Perturbationsmethode eignet sich sowohl für die Messung von Einkomponenten- als auch die
von Mehrkomponentenisothermen. Inbesondere für die Vermessung von gekoppelten Isothermen
ist die Perturbationsmethode besser geeignet als die Frontalanalyse, da eine Vermessung auch
mit einem unselektiven Detektor möglich ist.
3.2.3 Peak-Fitting-Methode
Bei dieser Methode vergleicht man bestimmte, experimentell ermittelte Chromatogramme mit
Chromatogrammen, die mit einem Simulationsprogramm errechnet wurden. Mit Hilfe einer

3 Methoden zur Bestimmung von Adsorptionsisothermen
54
Zielfunktion führt man eine Optimierung der thermodynamischen Parameter der
Adsorptionsisotherme durch, wobei vorher die kinetischen Parameter zu vermessen sind, damit
deren Einfluß bekannt ist. Ein Nachteil dieser Methode ist, daß das Adsorptionsisothermenmodell
vor der Simulation gewählt werden muß. Ob das jeweilige Gleichgewicht dabei gut beschrieben
wird, kann erst nach erfolgter Simulation festgestellt werden. Mittels der angepaßten Parameter
kann dann die Beladung berechnet werden.
3.2.4 Elution am charakteristischen Punkt
Bei der Elution am charakteristischen Punkt wird auf eine mit reinem Lösungsmittel gesättigte
Säule ein kurzer und hoher Konzentrationsstoß aufgegeben. Die Gleichgewichtsdaten werden
dann aus der hinteren, dispersen Front der Elutionskurve ermittelt, die in jedem Punkt analysiert
wird. Dabei wird in jedem charakteristischen Punkt einer Konzentration durch die Elutionskurve
eine Pseudoretentionszeit zugeordnet, woraus sich die Retentionszeit als Funktion der
Konzentration ergibt. Da die Retentionszeit über Gleichung (3.3) mit der Ableitung der
Adsorptionsisotherme verknüpft ist, ergibt sich hieraus die Möglichkeit, die Adsorptionisotherme zu
bestimmen.
Sowohl die Anwendung der Peak-Fitting-Methode als auch diejenige der Elution am
charakteristischen Punkt erfordern zum Erhalt genauer Ergebnisse den Einsatz sehr effizienter
Säulen mit einigen tausend Trennstufen, bei denen der Kurvenverlauf der dispersen Front
eindeutig vom Adsorptionsgleichgewicht dominiert wird. Da im Rahmen dieser Arbeit Adsorbens
aus dem präparativen Bereich eingesetzt wurde, bei denen aufgrund der größeren Korngröße
keine so große NTU zu erreichen ist wie bei Säulen aus dem analytischen Bereich, wurden als
Meßmethoden die Frontalanalyse und die Perturbationsmethode ausgewählt, da sie auch bei
weniger effizienten Säulen gut einsetzbar sind.
3.3 Bestimmung der Porosität
Wie bereits in Abschnitt 2.4.2 erwähnt wurde, ist die Porosität ein notwendiger Parameter zur
dynamischen Bestimmung von Adsorptionsisothermen. Je nach Annahme ist es hierbei
notwendig, entweder die externe Porosität εext oder die Gesamtporosität εges zu bestimmen. Auch
Möglichkeiten, die interne Porosität εint zu bestimmen, sind in der Literatur zu finden.

3 Methoden zur Bestimmung von Adsorptionsisothermen
55
Zu dieser Problematik gibt es eine Reihe von Veröffentlichungen: Einen sehr guten Überblick über
die Aufteilung des Lückenvolumens gibt Alhedai et al. (1989), eine thermodynamische
Betrachtung ist bei Kazakevich et al. (1993) nachzulesen, während Bidlingmeyer et al. (1991)
praktische Überlegungen in den Vordergrund stellen. Besonders intensiv wurde die Bestimmung
der Porosität in der Reversed-Phase-Chromatographie behandelt. Beispielhaft genannt seien hier
die Arbeiten von McCormick et al. (1980), Wätzig et al. (1991), Berendsen et al. (1980) und
insbesondere Krstulovic et al. (1982).
3.3.1 Bestimmung der Gesamtporosität
Die einfachste Methode, die Gesamtporosität zu bestimmen, ist diejenige, die Retentionszeit eines
nicht adsorbierenden und vollständig porengängigen Tracers zu messen [Aced und Möckel
(1991)] und daraus das Totvolumen zu berechnen. Dazu kommen vor allem Tracer in Betracht, die
das gleiche Wechselwirkungspotential haben wie das Lösungsmittel. Dies sind meist sehr ähnliche
Moleküle oder Isotope. Beispiele sind Wasser und D2O, das einen abweichenden Brechungsindex
besitzt und damit detektierbar ist. Engelhardt et al. (1984) geben als Kriterien für gute Tracer an,
daß er in Sorptionswärme und Molekülgröße dem Eluent möglichst ähnlich sein sollte. Eine
andere Möglichkeit, wie sie vor allem bei Reversed-Phase Materialien benutzt wird, führt über die
Retentionszeit verschiedener Alkane, wobei aus diesen die Retentionszeit eines Alkans mit der
Länge Null extrapoliert wird. Daneben gibt es noch andere Möglichkeiten, die z. B. bei Aldehai et
al. (1989) verzeichnet sind.
3.3.2 Bestimmung der externen Porosität
Bei der externen Porosität ist das zwischenpartikuläre Volumen von Interesse, also das verfügbare
Volumen zwischen den Partikeln. Die Bestimmung erfolgt mit Hilfe von Tracersubstanzen, die
aufgrund von Ausschlußeffekten nicht in das Porensystem eindringen. Verwendet werden hierbei
entweder sehr große Moleküle oder Salze. Eine weitere Möglichkeit besteht darin, die externe
Porosität durch Differenzbildung von Gesamt- und interner Porosität zu ermitteln.
3.3.3 Bestimmung der internen Porosität
Die interne Porosität wird aus dem Porenvolumen der Partikel ermittelt. Zu diesem Zweck existiert
eine Titrationsmethode, die erstmals von Innes (1956) erwähnt und von Mottlau und Fisher (1962)

3 Methoden zur Bestimmung von Adsorptionsisothermen
56
weiterentwickelt wurde. Dabei wird ein festgelegtes Volumen der stationären Phase in ein
verschließbares Becherglas gefüllt und im Vakuumofen getrocknet. Sobald keine Restbeladung
mehr vorliegt, wird ein porengängiges Lösungsmittel durch Magnetrührung in das Becherglas
titriert, bis sich das Erscheinungsbild der stationären Phase ändert: Solange das Lösungsmittel in
die Poren hineindiffundieren kann, bildet sich an der Partikeloberfläche kein Flüssigkeitsfilm aus
und die Partikel bewegen sich frei. Wenn jedoch die Porenkapazität ausgefüllt ist, entwickelt sich
ein Lösungsmittelfilm auf der Oberfläche und es kommt zur Agglomeration der Partikel, was sehr
gut sichtbar ist. Wenn sich bei Unterbrechung der Flüssigkeitszufuhr diese Agglomerate durch
Transport der Flüssigkeit in unbenetzte Partikel nicht schnell wieder auflösen, ist der
Umschlagpunkt tatsächlich erreicht und das hineintitrierte Lösungsmittelvolumen gibt Auskunft
über die interne Porosität.

4 Fehleruntersuchungen in der Literatur
4 Fehleruntersuchungen in der Literatur
57
Dieses Kapitel gibt als Abschluß des theoretischen Teils dieser Arbeit einen Überblick über
Literatur, die sich mit vergleichbaren Phänomenen beschäftigt hat, die auch Gegenstand der
vorliegenden Arbeit sind. Dabei faßt Unterkapitel 4.1 zunächst Literaturstellen zusammen, die sich
mit dem Vergleich der Ergebnisse von statischen und dynamischen Messung zur Bestimmung von
Adsorptionsisothermen beschäftigt haben. Unterkapitel 4.2 gibt Literaturstellen an, die sich mit
Fehlerquellen in präparativen Chromatographiesäulen auseinandersetzen
4.1 Vergleiche von statischen und dynamischen Meßmethoden zur Bestimmung
von Adsorptionsisothermen
Obwohl sowohl statische als auch dynamische Meßmethoden zur Beschreibung des Flüssig-
adsorptionsgleichgewichtes in der Literatur häufig verwendet werden, sind Vergleiche beider Arten
von Meßmethoden für das Gleiche Trennsystem selten und darüberhinausgehend auch wider-
sprüchlich:
Wang et al. (1978) führten einen Vergleich durch, indem sie das System Hexan-Hexanol auf
einem Silica-Gel sowohl mit Hilfe der Tauchmethode als auch mit der Frontalanalyse
untersuchten. Als Ergebnis erhielten eine Adsorptionsisotherme in klassischer Langmuir-Form
(siehe Abbildung 4.1)
Abbildung 4.1: Massenreduzierte Adsorptionsisothermen von n-Hexanol im System n-Hexanol / n-Hexan auf einem Silica-Gel (340 m2/g) bei 25°C. (Ο) Statische Messung mit absoluter Refraktometrie, (∆) statische Messung mit Differenzrefraktometrie, ( ) dynamische Messung [aus Wang et al. (1978)]
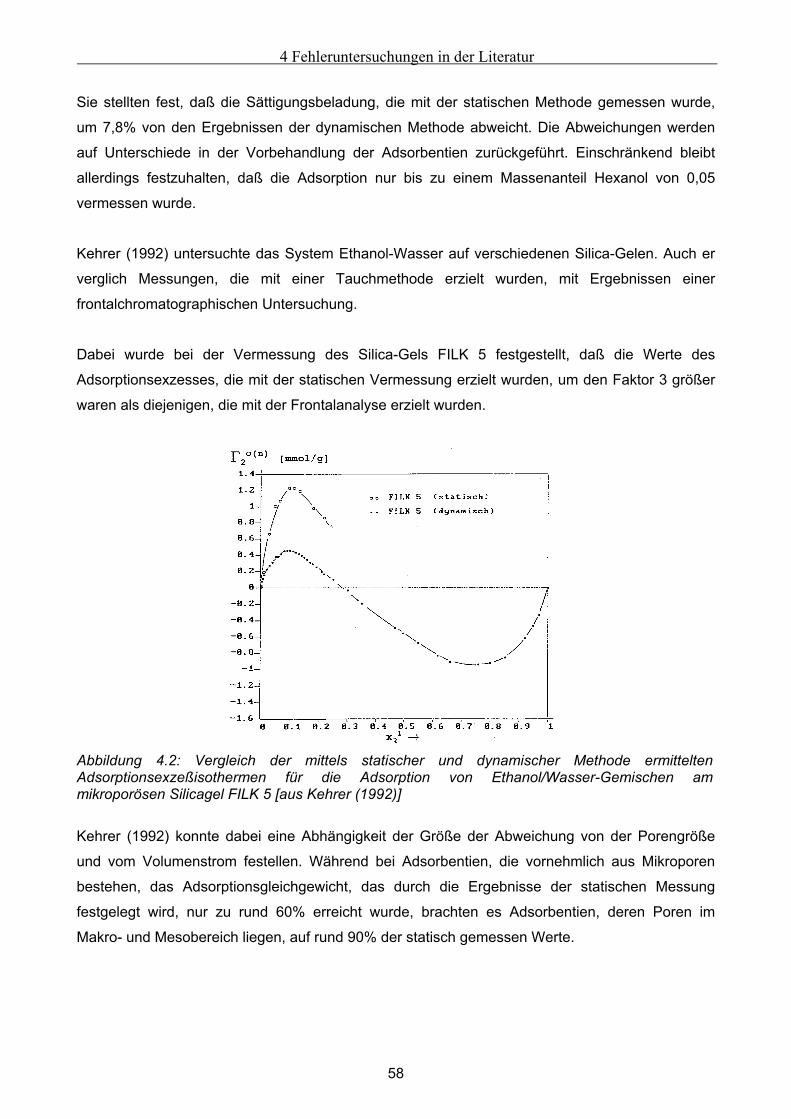
4 Fehleruntersuchungen in der Literatur
58
Sie stellten fest, daß die Sättigungsbeladung, die mit der statischen Methode gemessen wurde,
um 7,8% von den Ergebnissen der dynamischen Methode abweicht. Die Abweichungen werden
auf Unterschiede in der Vorbehandlung der Adsorbentien zurückgeführt. Einschränkend bleibt
allerdings festzuhalten, daß die Adsorption nur bis zu einem Massenanteil Hexanol von 0,05
vermessen wurde.
Kehrer (1992) untersuchte das System Ethanol-Wasser auf verschiedenen Silica-Gelen. Auch er
verglich Messungen, die mit einer Tauchmethode erzielt wurden, mit Ergebnissen einer
frontalchromatographischen Untersuchung.
Dabei wurde bei der Vermessung des Silica-Gels FILK 5 festgestellt, daß die Werte des
Adsorptionsexzesses, die mit der statischen Vermessung erzielt wurden, um den Faktor 3 größer
waren als diejenigen, die mit der Frontalanalyse erzielt wurden.
Abbildung 4.2: Vergleich der mittels statischer und dynamischer Methode ermittelten Adsorptionsexzeßisothermen für die Adsorption von Ethanol/Wasser-Gemischen am mikroporösen Silicagel FILK 5 [aus Kehrer (1992)]
Kehrer (1992) konnte dabei eine Abhängigkeit der Größe der Abweichung von der Porengröße
und vom Volumenstrom festellen. Während bei Adsorbentien, die vornehmlich aus Mikroporen
bestehen, das Adsorptionsgleichgewicht, das durch die Ergebnisse der statischen Messung
festgelegt wird, nur zu rund 60% erreicht wurde, brachten es Adsorbentien, deren Poren im
Makro- und Mesobereich liegen, auf rund 90% der statisch gemessen Werte.

4 Fehleruntersuchungen in der Literatur
59
Auch der Volumenstrom, bei dem die Frontalanalyse durchgeführt wurde, zeigte einen Einfluß auf
die Abweichung von den Ergebnissen der statischen Messung, wobei zwei gegenläufige Effekte
zu beobachten waren:
Bei Adsorbentien, die vornehmlich aus Mikroporen bestehen, führte eine Erhöhung des
Volumenstroms in der Frontalanalyse zu einer Zunahme der Exzeßwerte. Dagegen wurden bei
Adsorbentien mit Meso- und Makroporen die Exzeßwerte mit höherem Volumenstrom kleiner.
Kehrer (1992) erklärt dies dadurch, daß sich bei den mikroporösen Adsorbentien bei einer
Erhöhung des Volumenstroms und damit der Strömungsgeschwindigkeit die Dicke der laminaren
Grenzschicht verringert, wobei die Adsorbat-Phase unbeeinflußt bleibt. Durch die geringere Dicke
der laminaren Grenzschicht verbessert sich der Stoffübergang, wodurch die Exzeßwerte
ansteigen. Bei den meso- und makroporösen Partikeln wird zwar ebenfalls Stoffübergang durch
die Abnahme der laminaren Grenzschichtdicke begünstigt, jedoch reduziert sich gleichzeitig die
Menge der Adsorbat-Phase, da ein Teil der Flüssigphase nicht in den Poren verweilt, sondern
infolge des größeren Porendurchmessers von der Strömung mitgerissen wird. Wie die von Kehrer
(1992) beobachteten Effekte in Zusammenhang mit den in dieser Arbeit gewonnenen
Erkenntnisse erklärbar sind, wird in Unterabschnitt 5.2.2 erläutert.
4.2 Fehlerquellen in der Flüssigchromatographie
Im folgenden sei ein Überblick gegeben über Literatur, die sich mit defekten Chromatographie-
systemen und insbesondere mit fehlerbehafteten chromatographischen Festbetten befaßt.
Einen allgemeinen Überblick über Fehlerquellen in der Gas- sowie der analytischen
Flüssigchromatographie gibt Barwick (1999), wobei dieser sich allerdings hauptsächlich mit
Fehlern auseinandersetzt, die durch die Fehlfunktionen der Detektoren verursacht werden.
Nach Angabe von Kaminski (1992) kann eine schlechte Performance präparativer
chromatographischer Säulen auf folgende Ursachen zurückzuführen sein:
• Durch einen schlechten Plug-Flow der mobilen Phase verursacht durch eine ungleichmäßige
Verteilung von Partikeln unterschiedlicher Größe oder eine ungleichmäßige, radiale
Packungsgüte.
• Ein instabiles Festbett oder Ungleichmäßigkeiten wie größere Lücken zwischen den Partikeln,
ein Riß im Festbett oder eine größere Lücke zwischen Einlaßfritte und dem Festbett, die auf
ein Absacken während des Betriebs zurückzuführen ist.

4 Fehleruntersuchungen in der Literatur
60
• Ein schlechtes Design der Einlaßfritten, die z. B. einen zu großen Strömungswiderstand in
radiale Richtung.
Guiochon et al. (1997) werteten mehrere Untersuchungen hinsichtlich des Packens von
Chromatographiesäulen aus. Sie kamen zu dem Ergebnis, daß das häufigste Phänomen sich
dergestalt äußert, daß sich über den Säulendurchmesser ein parabolisches Geschwindigkeitprofil
entwickelt, wobei die Geschwindigkeit in Wandnähe geringer ist als im Zentrum. Gleichzeitig ist die
HETP am Rand größer, die Packung in Wandnähe also weniger effizient. Erklärt wird dieser Effekt
mit der Reibung des Bettes an der Säulenwand während des Packvorgangs: Diese Reibung
behindert das Herunterrutschen eines homogenen Bettes während des Packvorganges, so daß
der Kompressionsdruck entlang der Wand höher ist als im Zentrum der Säule. Da das Bett
kompressibel ist, wird das Bett entlang der Säulenwand zwar dichter, die externe Porosität also
kleiner, aber auch unregelmäßiger, was die Vergrößerung der HETP in der Nähe der Wand
verursacht.
Diese Erklärungen werden experimentell bestätigt durch die Untersuchungen von Farkas et al.
(1996). Diese untersuchten das radiale Strömungsfeld und die radiale Variation der HETP
mehrerer analytischer Säulen. Sie installierten dazu Glasfasern in der Auslaßfritte, die mit einem
optischen Meßsystem verbunden wurden. Sie konnten nicht nur feststellen, daß die
Fließgeschwindigkeit in Wandnähe ungefähr um ein bis zwei Prozent geringer ist, sondern auch,
daß die Höhe einer theoretischen Trennstufe in Wandnähe um den Faktor zwei größer und damit
die Säuleneffizienz geringer ist. Bei den Untersuchungen von Knox et al. (1976) war die HETP
sogar um den Faktor fünf größer.
Miyabe und Guichon untersuchten auf theoretischer Ebene den Einfluß dieser radialen
Heterogenitäten zum einen auf das Tailing (1999), zum anderen auf das Fronting (1999a) eines
Peaks. Die Untersuchung wurde dabei auf den linearen Konzentrationsbereich beschränkt, um
unsymmetrische Effekte durch die Thermodynamik auszuschließen. Die Simulation erfolgte mit
durch Modellierung mit 50 annularen Säulen, wobei radiale Dispersion vernachlässigt wurde.
Sie kamen zu dem Ergebnis, daß eine radiale Heterogenität in der Säule eine wichtige Ursache
für das Tailing eines Peaks sein kann. Dies wird erklärt durch eine geringere Geschwindigkeit und
insbesondere eine größere HETP in Wandnähe. Ein Fronting kann dann entstehen, wenn die
Geschwindigkeit in Wandnähe zwar größer ist, die HETP aber kleiner.

4 Fehleruntersuchungen in der Literatur
61
Tsotsas und Schlünder (1988) untersuchten den Einfluß von Strömungsungleichverteilungen in
gepackten Betten, wobei drei Typen definiert wurden, nämlich mikroskopische, mesoskopische
und makroskopische Strömungsungleichverteilungen.
Die mikroskopische Ungleichverteilung entsteht in jedem einzelnen Strömungskanal aufgrund der
Haftbedingungen an der Partikeloberfläche und des wechselnden Kanalquerschnittes.
Mesoskopische Ungleichverteilungen kommen zustande, wenn relativ dichte Partikel-Agglomerate
vom Fluid umgangen werden. Makroskopische Ungleichverteilungen schließlich entstehen durch
die Auflockerung der Schüttung in unmittelbarer Nähe der Wand. Die Charakterisierung der
Ursache für makroskopische Ungleichverteilungen widerspricht allerdings den oben erwähnten
Untersuchungen von Farkas et al. (1996) und Guiochon et al. (1997).
Nach Angaben von Oliveros und Smith (1982) ist Kanalbildung, also makroskopische
Ungleichverteilungen über einen längeren Weg, eine verbreitete Ursache für eine schlechte
Performance von Prozessen, die in gepackten Betten ablaufen. Sie untersuchten Kanaleffekte mit
Hilfe eines Heliumstracers in mit Luft durchströmten, gepackten Festbetten, das in zwei Annulli
unterteilt wurde, wobei der eine bewußt sehr schlecht gepackt wurde. Als Ergebnis erhielten sie
Peaks mit verschieden starken Assymmetrien.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
62
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
Die folgenden drei Kapitel untersuchen die Auswirkungen möglicher Fehler in der präparativen
Flüssigchromatographie. Dabei beschäftigt sich dieses Kapitel mit dem Vergleich der Ergebnisse
statischer und dynamischer Meßmethoden zur Bestimmung von Adsorptionsisothermen mit dem
Ziel, zu klären, inwiefern es möglich ist, mit diesen beiden Arten von Meßmethoden ähnliche
Ergebnisse zu erzielen. Dieses vor dem Hintergrund der Abschätzung, inwieweit die Wahl der
Meßmethode einen Fehler in die Auslegung einer chromatographischen Trennung einbringen
kann.
Im Anschluß an dieses Kapitel befaßt sich Kapitel 6 mit Fehlern in der gepackten Bettstruktur
präparativer chromatographischer Säulen. Von Interesse sind dabei die Auswirkungen bestimmter
Packungsfehler auf die die Säule durchlaufende Peaks. Kapitel 7 schließlich beleuchtet die Aus-
wirkungen bestimmter Fehler beim Betrieb der Simulierten Gegenstromchromatographie (SMB).
In Unterkapitel 5.1 werden zunächst die im Rahmen dieser Arbeit durchgeführten Experimente
zum Vergleich von statischen und dynamischen Methoden erläutert. In Unterkapitel 5.2 werden die
Ergebnisse des Vergleichs präsentiert und diskutiert. Nach einer Fehlerbetrachtung in Unterkapitel
5.3 wird in Unterkapitel 5.4 schließlich anhand eines hypothetischen Falles dargelegt, wie sich die
erarbeiteten Erkenntnisse in der chromatographischen Praxis auswirken können.
5.1 Durchführung der Experimente
Der Vergleich von statischen und dynamischen Meßmethoden zur Bestimmung von
Adsorptionsisothermen ist aus mehreren Gründen für die Fehlerbetrachtung in der präparativen
Chromatographie interessant:
Am wichtigsten ist die Frage, ob mit den dynamischen Meßmethoden tatsächlich eine Messung
der unverfälschten Thermodynamik möglich ist, oder ob es eine Verfälschung durch kinetische
Effekte gibt. Daraus ergibt sich die Problematik, ob es möglich und auch sinnvoll ist,
Adsorptionsisothermen, die für den Einsatz in der präparativen Flüssigchromatographie benötigt
werden, durch den Einsatz von statischen Meßmethoden zu ermitteln, um unter Umständen eine
höhere Genauigkeit zu erzielen oder Thermodynamik und Kinetik bei entsprechender Theorie zu
entkoppeln.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
63
Um diese Problematik experimentell zu untersuchen, wird in Abschnitt 5.1.1 zunächst dargelegt,
welche Kriterien bei der Auswahl eines Testsystems im Vordergrund standen. Darauf erfolgt eine
Beschreibung der Experimente, die durchgeführt wurden, um im Rahmen dieser Arbeit statische
(Abschnitt 5.1.2) und dynamische (Abschnitt 5.1.3) Meßmethoden miteinander zu vergleichen [vgl.
auch Lenz et al. (2002)]
5.1.1 Auswahl eines Testsystems
Um statische und dynamische Meßmethoden miteinander vergleichen zu können, sind zunächst
Testsysteme auszuwählen. Diese bestehen zum einen aus einer stationären Phase, zum anderen
aus zwei flüssigen Reinstoffen, die vollständig mischbar sein sollten. Um diese Testsysteme
möglichst unkompliziert zu gestalten, soll ein Stoff sehr viel stärker adsorbieren als der andere.
Die auszuwählenden Testsysteme müssen eine Reihe weiterer Anforderungen erfüllen, um eine
Vermessung sowohl mit statischen als auch mit dynamischen Meßmethoden zu ermöglichen und
gleichzeitig auch eine Vergleichbarkeit der Ergebnisse herzustellen:
Wichtig bei der Auswahl der stationären Phase ist es, daß keine Quellvorgänge stattfinden, um
eine konzentrationsunabgängige Bestimmung der Porosität zu ermöglichen. Zur Durchführung der
statischen Messungen ist es notwendig, daß das Adsorbens vor dem Experiment getrocknet
werden kann. Dies erfolgt durch Aufheizen und gleichzeitigem Anlegen eines Vakuums.
Stationäre Phasen, die diese beide Anforderungen erfüllen, sind u. a. Normal-Phase Silica-Gele.
Diese bestehen in ihrem chemischen Aufbau aus Siliziumatomen, die durch Sauerstoffatome
dreidimensional verbrückt sind und deren Endstellen durch OH-Gruppe (Silanolgruppen)
abgesättigt sind. Für die Experimente wurden zwei Normal-Phase Silica-Gele ausgewählt: Zum
einen Kromasil NP 10µm 100A der Firma EKA-Nobel, ein Normal-Phase Silica-Gel mit einer
Partikelgröße von 10µm und eine durchschnittlichen Porengröße von 100 Angström. Das zweite
Material hat Partikelgrößen zwischen 40µm und 63µm sowie eine durchschnittliche Porengröße
von 60 Angström. Es wurde von der Firma Uetikon Chemie hergestellt und trägt die Bezeichnung
C-Gel C560 40-63µm. Detaillierte Untersuchungen beider stationärer Phasen inklusive einer
elektronenmikroskopischen Untersuchung der Oberfläche sind Laiblin (2002) zu entnehmen.
Von Bedeutung bei der Auswahl dieser stationären Phasen ist neben der leichten Verfügbarkeit,
daß die stationären Phasen eine unterschiedliche Porengrößenverteilung haben, um u. U. deren
Einfluß auf das Ergebnis zu studieren. Der Adsorptionsmechanismus von Normal-Phase Silica-

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
64
Gelen beruht auf der Polarität der Moleküle. Je größer die Polarität ist, desto größer ist die zu
erwartende Retention dieser Substanz.
Neben den stationären Phasen müssen auch die Flüssigkeiten bestimmte Anforderungen erfüllen,
die sich aus den Erfordernissen der statischen und dynamischen Experimente ergeben.
Wichtig für die statische Messungen sind Substanzen, die sich leicht wieder vom Adsorbens durch
Ausheizen und Vakuum entfernen lassen, ohne daß die Wärmebelastung für das Adsorbens zu
groß wird. Um während der Experimente die Zusammensetzung der Bulk-Phase mittels eines
Biegeschwingers bestimmen zu können, sollten die beiden Substanzen möglichst verschieden in
ihren Dichten sein, um den Meßfehler hier möglichst gering zu halten.
Für die dynamischen Messungen ist es bedeutend, daß die stärker adsorbierende Komponente
ein besseres UV-Absorptionsvermögen besitzt als die schwächer adsorbierende, da die Detektion
im Rahmen dieser Arbeit mittels eines Diode-Array-Detektors erfolgt.
Weiterhin von Bedeutung bei der Auswahl der Substanzen für die Bulk-Phase war, daß beide eine
vollständige Porengängigkeit aufweisen sollten, es also nicht zu einem Größenausschluß kommen
sollte, um sich in der Betrachtung auf die Bestimmung nur einer Porosität, hier der Gesamt-
porosität, beschränken zu können.
Tabelle 5.1: Chemisch-physikalische Daten von Dichlormethan und n-Hexan Name Dichlormethan n-Hexan
Strukturformel CH2Cl2 C6H14
Molmasse 84,93 g/mol 86,18 g/mol
Dichte (20°C) 1,33 g/cm³ 0,66 g/cm²
Siedepunkt (1013 hPa) 40°C 69°C
Dampfdruck (20°C) 475 hPa 160 hPa
Polarität E0 (SiO2) [Meyer (1992)] 0,3234 0,00
Wie Tabelle 5.1 zeigt, verfügt das System Dichlormethan- n-Hexan über die erforderlichen
Eigenschaften. Dabei ist Dichlormethan diejenige Substanz, die von den Normal-Phase Silica-
Gelen stärker adsorbiert wird. Beide Chemikalien wurden von der Firma Merck KGaA in der
Qualitätsstufe LiChrosolv bezogen und so verwendet. Die Reinheit des Dichlormethans wurde mit
>99,9 % angegeben, diejenige des n-Hexans mit >98 %.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
65
5.1.2 Durchführung der statischen Messungen
Die statischen Messungen wurden mit Hilfe einer Zirkulationsapparatur durchgeführt, die im
Rahmen der Arbeit von Hirsch (2000) entwickelt und gebaut wurde. Eine sehr detaillierte
Beschreibung der Anlage und der Versuchsdurchführung kann Geißler (1996) entnommen
werden. Im folgenden wird zunächst der Aufbau der verwendeten Zirkulationsapparatur
beschrieben. Daran schließt sich eine Beschreibung der Versuchsdurchführung an.
5.1.2.1 Aufbau der Zirkulationsapparatur
Die Zirkulationsapparatur, deren Prinzipschaltbild in Abbildung 5.1 gezeigt wird, läßt sich formal in
eine Meßeinheit und in eine Analyseeinheit unterteilen, die durch einen Strömungskreislauf der zu
untersuchenden Bulk-Phase miteinander in Verbindung stehen.
Abbildung 5.1: Prinzip der verwendeten Zirkulationsapparatur
Die Meßeinheit ist in einem Luftbadthermostaten untergebracht. Dieser besteht aus einem
Metallgestell, das mit isolierten Seitenverkleidungen versehen ist. An der Vorderseite befindet sich
eine Tür, in die ein aus zwei temperaturbeständigen Glasscheiben bestehendes Sichtfenster
eingelassen ist. In der Rückwand sind ein Umluftventilator, eine Heizschlange sowie eine mit
Kühlwasser versorgte Kühlschlange eingebaut. Die Regelung der Temperatur im Luftbad erfolgt
mit Hilfe eines Pt-100-Widerstandsthermometers, eines Reglers sowie eines Thyristors. Mit diesen
Komponenten ist es möglich, eine Temperaturkonstanz von ±0,2K zu erreichen. Für den Fall, daß
die Adsorptionsmessungen bei höheren Temperaturen stattfinden sollen, ist der Thermostat mit
einer zusätzlichen Grundheizung versehen.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
66
Im Luftbadthermostat befinden sich als relevante Bauteile die Adsorptionszelle und die Befüllzelle.
• Die Adsorptionszelle:
In der Adsorbenszelle wie sie in Abbildung 5.2 (links) abgebildet ist, befindet sich das
Adsorbens, dessen Adsorptionseigenschaften untersucht werden. Sie hat einen
Innendurchmesser von 36mm und ein Volumen von ca. 100cm³. Sie wird an beiden Enden mit
Deckeln verschlossen, wobei graphitierte Metalldichtungen die Dichtheit gewährleisten. In
diesen ist jeweils ein Sintermetalfilter fixiert, der verhindert, daß Adsorbens aus der
Adsorptionszelle herausgetragen wird und die gleichzeitig noch eine Verteilungsfunktion
erfüllen. Je nach Partikelgröße des Adsorbens können Filter mit dazu passender Porengröße
ausgewählt und eingesetzt werden.
Abbildung 5.2: Die Adsorptionszelle (links) und die Befüll- und Mischzelle (rechts)
Um eine gute Flüssigkeitsverteilung innerhalb der Adsorptionsvolumens zu gewährleisten und
den Totraum im Zulauf möglichst gering zu halten, ist im Ein- und Auslaß jeweils ein
Verdrängungselement installiert.
Um das Ausheizen des Adsorbens zu ermöglichen, kann eine Heizmanschette mit einer
Heizleistung von 800 W um die Adsorptionszelle gelegt werden. Mittels zweier darin
angebrachter Pt100-Fühler und einer angeschlossenen Temperaturregelung ist es möglich,

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
67
das Adsorbens in der Zelle kontrolliert auszuheizen und dabei eine zu große Wärmebelastung
zu vermeiden.
• Die Befüllzelle:
Sie besteht aus einem innen geschliffenen Rohrstück, welches an beiden Enden mit
Flanschen versehen ist (siehe Abbildung 5.2 rechts). Gegen diese werden zwei Deckflansche
gesetzt, um die Zelle zu verschließen. Im unteren Deckflansch befindet sich ein Rührer, der
über Drehdurchführung mit einem Motor verbunden ist. Der Sinn dieses Rührers besteht zum
einen darin, eine Mischung aus zugegebenen Reinstoffen zu homogenisieren, zum anderen ist
auch eine Beschleunigung der Lösung eines vorher in die Zelle eingebrachten Feststoffs
möglich. Eine solche Einbringung ist durch Entfernung des oberen Deckflansches und des
Kolbens möglich.
In dem Rohrstück befindet sich eine aus zwei Anpreßscheiben und einer Teflonscheibe
bestehende Kolbenkonstruktion. In der V-förmigen Nase der Teflonscheibe befindet sich ein
O-Ring, der, selbst durch die Teflonscheiben vor Chemikalien geschützt, die Dichtheit durch
seine Flexibilität verstärkt. In der unteren Anpreßscheibe des Kolbens ist eine Gewindestange
eingeschweißt, so daß eine Optimierung von Beweglichkeit und Dichtheit des Kolbens möglich
ist, indem über die Mutter die Teflonlippen und der O-Ring entsprechend gegen die Rohrwand
gepreßt werden.
Durch die Präsenz des Kolbens ist es möglich, die Adsorptionsapparatur ausgehend von der
Befüllzelle mit der Bulk-Phase zu füllen. Dabei kann ein Kontakt mit Inertgasen sowie die
Bildung eine Dampfphase vermieden werden. Weiterhin ist es möglich, die Menge der Bulk-
Phase in der befüllten Apparatur zu vergrößern, um einen neuen Meßpunkt zu vermessen,
ohne die gesamte Anlage entleeren zu müssen.
Während eines Experiments wird der Kolben mit Druck aus einem Stickstoffsystem
beaufschlagt. Damit der Rührer nicht durch den Kolben blockiert oder beschädigt wird,
befindet sich dieser in einem Laufradkäfig.
Die Befüllung der Zelle mit den zu untersuchenden Stoffen erfolgt über eine absperrende
Schnellkupplung.
Die Verbindung dieser Einheiten mit der Verrohrung erfolgt über teflongedichtete Cayon-
Verbindungen, die eine extrem dichte aber plane Rohrverbindung erlauben. Dies ermöglicht einen

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
68
einfachen Ein- und Ausbau der Einheiten zu Reinigungszwecken und zur Vorbereitung der
Messungen.
Das Herzstück der Analyseeinheit ist ein Biegeschwinger zur Dichtemessung, hergestellt von der
Firma Anton Paar [Paar (1978)]. Da der Biegeschwinger aus einer empfindlichen Elektronik
besteht, die einer separatem Kühlung bedarf, ist eine räumliche Trennung von Analyseeinheit und
Luftbadthermostat geboten. Um eine Mindesttemperatur im Übergangsbereich sicherzustellen,
verlaufen die Rohrleitungen in einem mit Silikonöl gefüllten Thermostatbehälter, in dem sich
wiederum eine Kupfer-Rohrschlange befindet, die von einer Thermostatflüssigkeit durchströmt
wird. Die Thermostatflüssigkeit, je nach Einsatzbereich Wasser oder Silikonöl, die auch die
Meßzelle des Biegeschwingers temperiert, wird von einem Flüssigkeitsthermostat der Firma
Lauda bereitgestellt und durchläuft einen geschlossenen Kreislauf.
• Der Biegeschwinger:
Der Biegeschwinger besteht aus einer Meßzelle (Sonderausführung DMA 602 HP) und dem
Dichtemeßgerät DMA 60 als Auswerteeinheit. Die Meßzelle läßt dabei einen Betrieb bis
423,15K zu und garantiert einen zugelassen Druckbereich von 0 bis 50 bar. Sie wird, wie
bereits erwähnt, durch die Thermostatflüssigkeit temperiert. Die Temperatur der Bulk-Phase
wird durch zwei Pt100 W5-Meßfühler gemessen, wobei einer am Eintritt der Meßzelle
angebracht ist und der zweite im dafür vorgesehenen Rohr in der Meßzelle. Sie sind
verbunden mit einem Digitalmultimeter der Firma Prema (Typ 6001).
Das Prinzip ist die aus der Mechanik bekannte bekannte Ermittlung der Schwingungsdauer T
einer ungedämpft schwingenden Masse, die phasenrichtig - hier durch eine Elektronik - zu
Schwingungen angeregt wird. Eine ausführliche Beschreibung der Methode ist der
Herstellerbeschreibung von Paar (1978) zu entnehmen.
• Druckmessung:
Zur Druckmessung kommt ein piezoresistiver Druckmeßumformer zum Einsatz, welcher für
Temperaturen des Meßgutes bis 150°C geeignet ist. Durch eine spezielle Konstruktion wird
das Druckmittlermedium zwischen der Trennmembran zum Meßmedium und der
Siliziummembran mit Widerstandmeßbrücken durch Konvektion gekühlt, um die Meßelektronik
vor hohen Temperaturen zu schützen. Der Spannungsausgang (0-10V) wird mit einem
Digitalmultimeter der Firma Prema (Typ 6001) erfaßt.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
69
Eine Übersicht über die verwendeten Meßgeräte findet sich in Anhang A.1.
• Die Spritze:
Zur Veränderung der Zusammensetzung der Bulk Phase dient eine spezielle
Spritzenkonstruktion. Hierbei wurden zwei Swagelockkupplungen mit unterschiedlicher
Kopplungrichtung mit einer lösungsmittelbeständigen Spritze verbunden, wobei das ganze
Systeme luftdicht gestaltet wurde. Dabei kamen zwei Spritzen mit einem Hubvolumen von
20 ml und 50 ml zum Einsatz.
Die Durchführung der Experimente erfolgte wie in Anhang B beschrieben.
5.1.2.2 Auswertung der Meßergebnisse
Ziel der Messungen mit der statischen Adsorptionsapparatur ist die Ermittlung der
Zusammensetzung der Bulk-Phase vor und insbesondere nach der Einstellung eines
Adsorptionsgleichgewichtes, um daraus den Adsorptionsexzeß zu ermitteln. Die Ermittlung der
Gleichgewichtszusammensetzung erfolgt mit Hilfe des Biegeschwingers, da sich bei einer binären
Mischung aus der ermittelten Dichte der Bulk-Phase direkt die Zusammensetzung ermitteln läßt,
sofern die verwendeten Stoffe unterschiedliche Reinstoffdichten vorweisen.
Um die Meßwerte des Biegeschwingers zur Berechnung der Dichten und damit der
Zusammensetzung nutzen zu können, ist es zunächst notwendig, eine Kalibrierkurve zu
vermessen. Diese wurde ermittelt, indem die Biegeschwingerwerte aufgenommen wurden, die bei
verschiedenen, bekannten Bulk-Phasen-Zusammensetzungen ermittelt wurden. Zu diesem Zweck
wurde die Adsorptionsapparatur befüllt wie in Anhang B beschrieben, mit dem Unterschied, daß
statt der Adsorptionszelle nur ein Stück Rohr eingesetzt wurde, damit keine Adsorption im System
stattfindet. Durch Zuspritzen einer Komponente wurden die Zusammensetzung der Bulk-Phase
variiert, um eine Reihe von Meßpunkten zu erhalten, wobei mindestens zehn Meßpunkte
vorhanden sein sollten. Die Anpassung der Kalibrierkurve sowie das Vorgehen zur Berechnung
der Zusammensetzung der flüssigen Phase erfolgt wie bei Hirsch (2000) beschrieben und ist auch
in Anhang C erläutert.
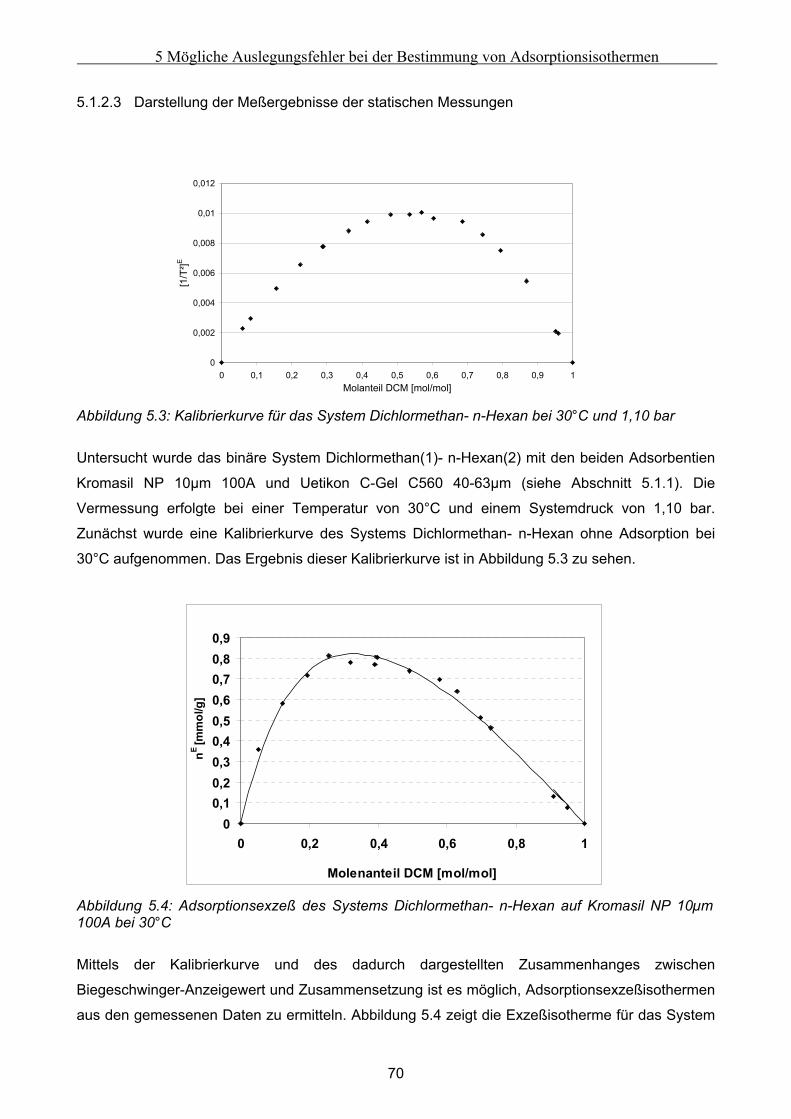
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
70
5.1.2.3 Darstellung der Meßergebnisse der statischen Messungen
0
0,002
0,004
0,006
0,008
0,01
0,012
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1Molanteil DCM [mol/mol]
[1/T
²]E
Abbildung 5.3: Kalibrierkurve für das System Dichlormethan- n-Hexan bei 30°C und 1,10 bar
Untersucht wurde das binäre System Dichlormethan(1)- n-Hexan(2) mit den beiden Adsorbentien
Kromasil NP 10µm 100A und Uetikon C-Gel C560 40-63µm (siehe Abschnitt 5.1.1). Die
Vermessung erfolgte bei einer Temperatur von 30°C und einem Systemdruck von 1,10 bar.
Zunächst wurde eine Kalibrierkurve des Systems Dichlormethan- n-Hexan ohne Adsorption bei
30°C aufgenommen. Das Ergebnis dieser Kalibrierkurve ist in Abbildung 5.3 zu sehen.
00,10,20,30,40,50,60,70,80,9
0 0,2 0,4 0,6 0,8 1
Molenanteil DCM [mol/mol]
nE [mm
ol/g
]
Abbildung 5.4: Adsorptionsexzeß des Systems Dichlormethan- n-Hexan auf Kromasil NP 10µm 100A bei 30°C
Mittels der Kalibrierkurve und des dadurch dargestellten Zusammenhanges zwischen
Biegeschwinger-Anzeigewert und Zusammensetzung ist es möglich, Adsorptionsexzeßisothermen
aus den gemessenen Daten zu ermitteln. Abbildung 5.4 zeigt die Exzeßisotherme für das System
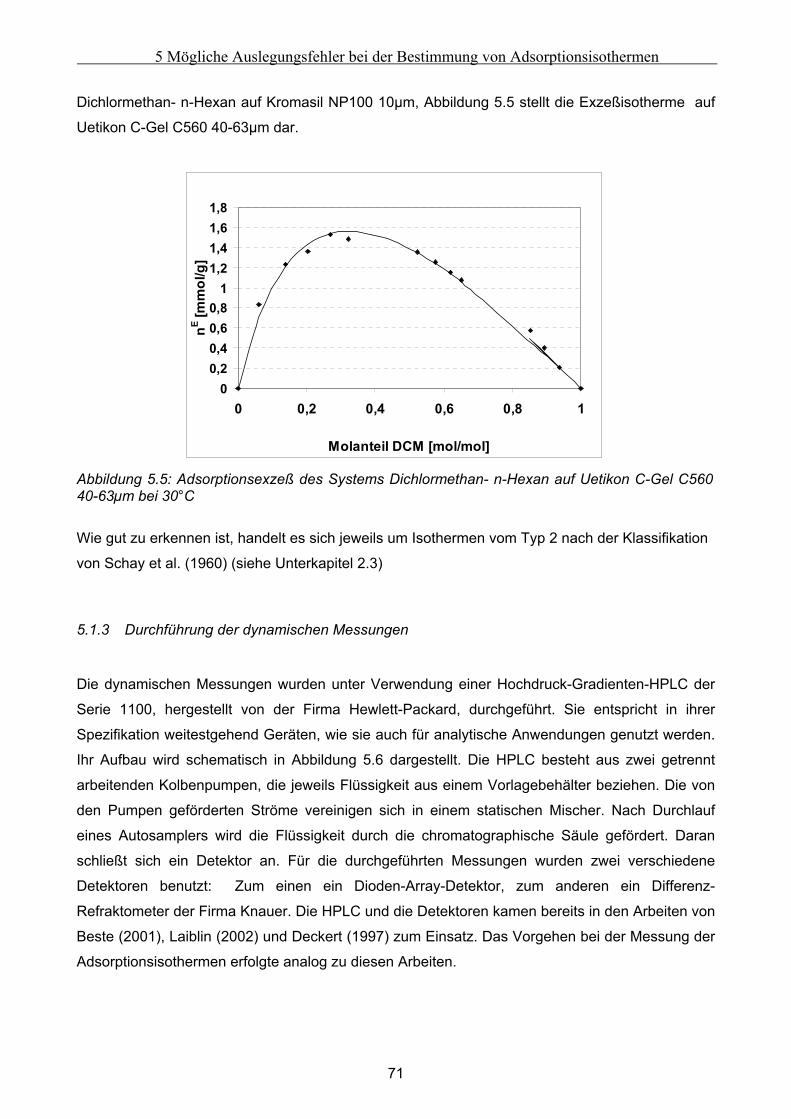
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
71
Dichlormethan- n-Hexan auf Kromasil NP100 10µm, Abbildung 5.5 stellt die Exzeßisotherme auf
Uetikon C-Gel C560 40-63µm dar.
00,20,40,60,8
11,21,41,61,8
0 0,2 0,4 0,6 0,8 1
Molanteil DCM [mol/mol]
nE [mm
ol/g
]
Abbildung 5.5: Adsorptionsexzeß des Systems Dichlormethan- n-Hexan auf Uetikon C-Gel C560 40-63µm bei 30°C
Wie gut zu erkennen ist, handelt es sich jeweils um Isothermen vom Typ 2 nach der Klassifikation
von Schay et al. (1960) (siehe Unterkapitel 2.3)
5.1.3 Durchführung der dynamischen Messungen
Die dynamischen Messungen wurden unter Verwendung einer Hochdruck-Gradienten-HPLC der
Serie 1100, hergestellt von der Firma Hewlett-Packard, durchgeführt. Sie entspricht in ihrer
Spezifikation weitestgehend Geräten, wie sie auch für analytische Anwendungen genutzt werden.
Ihr Aufbau wird schematisch in Abbildung 5.6 dargestellt. Die HPLC besteht aus zwei getrennt
arbeitenden Kolbenpumpen, die jeweils Flüssigkeit aus einem Vorlagebehälter beziehen. Die von
den Pumpen geförderten Ströme vereinigen sich in einem statischen Mischer. Nach Durchlauf
eines Autosamplers wird die Flüssigkeit durch die chromatographische Säule gefördert. Daran
schließt sich ein Detektor an. Für die durchgeführten Messungen wurden zwei verschiedene
Detektoren benutzt: Zum einen ein Dioden-Array-Detektor, zum anderen ein Differenz-
Refraktometer der Firma Knauer. Die HPLC und die Detektoren kamen bereits in den Arbeiten von
Beste (2001), Laiblin (2002) und Deckert (1997) zum Einsatz. Das Vorgehen bei der Messung der
Adsorptionsisothermen erfolgte analog zu diesen Arbeiten.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
72
SäulePumpenMischer
Detektor
Abbildung 5.6: Schema der benutzten HPLC
Für die Messungen kamen zwei chromatographische Säulen der Firma Merck (Modell Hibar,
250x10mm) zum Einsatz, wobei die eine mit Kromasil NP 10µm 100A und die andere mit Uetikon
C-Gel C560 40-63µm gefüllt wurde. Beide Säulen wurden vor Ihrer Benutzung slurry-gepackt,
wobei zum Packen jeweils das später verwendete Lösungsmittel n-Hexan zum Einsatz kam, um
eine Vorbeladung der stationären Phase mit einem anderen Lösungsmittel zu vermeiden. Da sich
die stationäre Phase während der Lagerung nicht unter Vakuum befand, wurde vor dem Packen
der Säulen das Adsorbens jeweils unter Vakuum ausgeheizt. Da die stationäre Phase für die
statischen und die dynamischen Messung in einem möglichst identischen Zustand vorliegen sollte,
wurde das Ausheizen mit identischen Randbedingungen durchgeführt, nämlich bei einem Druck
von 0,06 mbar und einer Temperatur von 105°C.
5.1.3.1 Bestimmung der Porosität
Wie bereits in den Abschnitten 3.2.1 und und 3.2.2 gezeigt wurde, ist zur Auswertung der
Meßdaten sowohl der Frontalanalyse als auch der Perturbationsmethode die Bestimmung der
Porosität zwingend erforderlich. Da sowohl Dichlormethan als auch n-Hexan vollständig
porenfüllend sind, wird zur Berechnung der Beladung die Gesamtporosität verwendet. Da die
Trennwirkung der verwendeten Normal-Phase-Silica-Gele auf der Polarität beruht, wurde n-
Heptan als Tracer verwendet, da es ebenso unpolar ist wie das Lösungsmittel n-Hexan (Meyer
(1992)). Damit durchläuft n-Heptan einerseits ohne Adsorption alle Poren, andererseits wird aber
auch keine zu geringe Porosität gemessen, was hervorgerufen werden könnte durch die Tatsache,
daß adsorbierte Moleküle des Lösungsmittels nicht vom Tracer verdrängt werden können und
dadurch das zu durchlaufende Volumen verkleinert wird.
Um genau jenen letztgenannten Effekt bei einer mit Dichlormethan beladenen Säule zu erfassen,
wurde eine Untersuchung der Porosität nicht nur der unbeladenen Säule durchgeführt, sondern
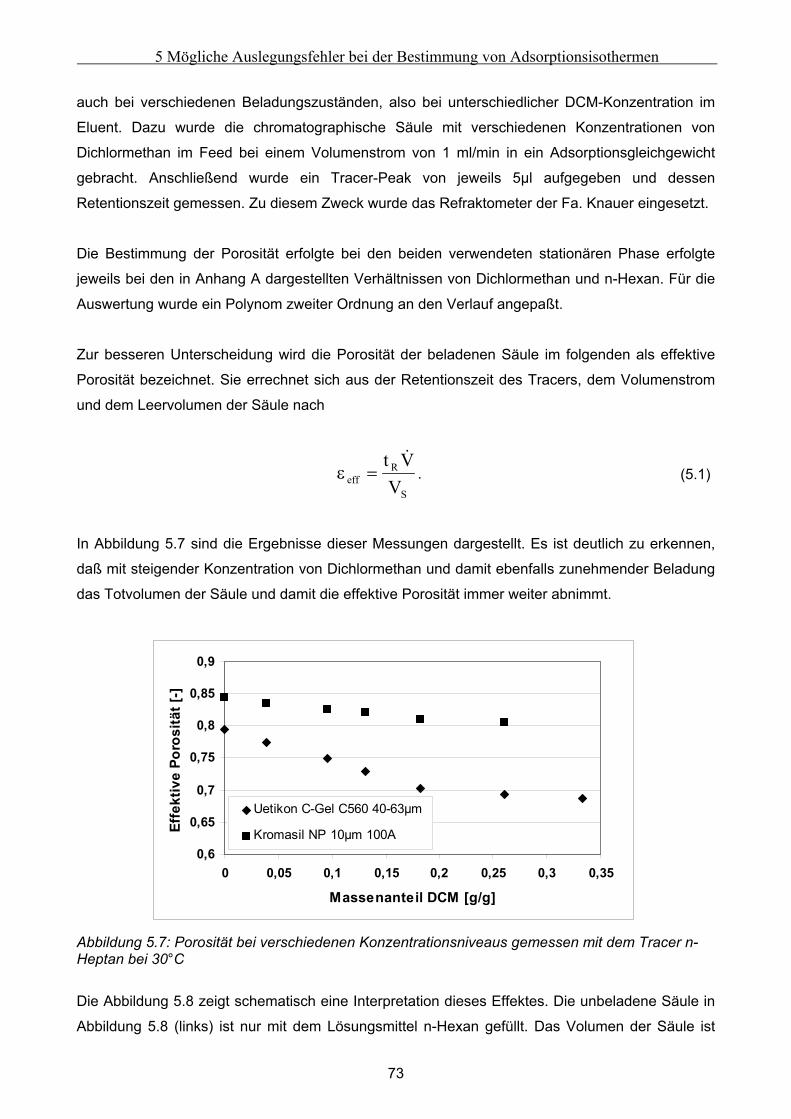
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
73
auch bei verschiedenen Beladungszuständen, also bei unterschiedlicher DCM-Konzentration im
Eluent. Dazu wurde die chromatographische Säule mit verschiedenen Konzentrationen von
Dichlormethan im Feed bei einem Volumenstrom von 1 ml/min in ein Adsorptionsgleichgewicht
gebracht. Anschließend wurde ein Tracer-Peak von jeweils 5µl aufgegeben und dessen
Retentionszeit gemessen. Zu diesem Zweck wurde das Refraktometer der Fa. Knauer eingesetzt.
Die Bestimmung der Porosität erfolgte bei den beiden verwendeten stationären Phase erfolgte
jeweils bei den in Anhang A dargestellten Verhältnissen von Dichlormethan und n-Hexan. Für die
Auswertung wurde ein Polynom zweiter Ordnung an den Verlauf angepaßt.
Zur besseren Unterscheidung wird die Porosität der beladenen Säule im folgenden als effektive
Porosität bezeichnet. Sie errechnet sich aus der Retentionszeit des Tracers, dem Volumenstrom
und dem Leervolumen der Säule nach
S
Reff V
Vt &=ε . (5.1)
In Abbildung 5.7 sind die Ergebnisse dieser Messungen dargestellt. Es ist deutlich zu erkennen,
daß mit steigender Konzentration von Dichlormethan und damit ebenfalls zunehmender Beladung
das Totvolumen der Säule und damit die effektive Porosität immer weiter abnimmt.
0,6
0,65
0,7
0,75
0,8
0,85
0,9
0 0,05 0,1 0,15 0,2 0,25 0,3 0,35
Massenanteil DCM [g/g]
Effe
ktiv
e Po
rosi
tät [
-]
Uetikon C-Gel C560 40-63µm
Kromasil NP 10µm 100A
Abbildung 5.7: Porosität bei verschiedenen Konzentrationsniveaus gemessen mit dem Tracer n-Heptan bei 30°C
Die Abbildung 5.8 zeigt schematisch eine Interpretation dieses Effektes. Die unbeladene Säule in
Abbildung 5.8 (links) ist nur mit dem Lösungsmittel n-Hexan gefüllt. Das Volumen der Säule ist

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
74
aufgeteilt in das Volumen von n-Hexan und das der stationären Phase. Nach der Adsorption von
Dichlormethan wird das für die mobile Phase zugängliche Volumen um das Volumen der
Adsorbat-Phase bestehend aus Dichlormethan-Molekülen verringert, was zu einer geringeren
Totzeit der Säule führt (Abbildung 5.8 (rechts)).
stat. Phase
Lückenvolumen
stat. Phase
Lückenvolumen adsorbierte Moleküle
Abbildung 5.8:Unbeladene (links) und beladene Säule (rechts)
Solche Effekte sind aus der Gasadsorption bekannt. Mit Hilfe der sog. T-Methode [Do (1998)]
versucht man dabei beispielsweise, die Dicke der Adsorbensschicht zu berechnen.
Die Bedeutung dieses Effektes für den Vergleich von statischen und dynamischen Meßmethoden
wird in Abschnitt 5.2.2 erläutert.
5.1.3.2 Auswertung der dynamischen Meßmethoden mit Hilfe der effektiven Porosität
Die Auswertung der dynamischen Meßmethoden unter Benutzung der Porosität der unbeladenen
Säule erfolgt mit Gleichung (3.2) für die Frontalanalyse bzw. Gleichung (3.3) für die Perturbations-
methode. Für die Auswertung über die effektive Porosität ist zur Erhaltung der Massenbilanz ein
etwas differenzierterer Ansatz notwendig, der im folgenden dargestellt wird.
Zur Auswertung der Frontalanalyse wird Gleichung (3.2) umgeschrieben als:
)cc(tV)qq(V)1()cc(V IIIR
Ieff
IIeffS
IIeff
IIIS
IIeff −=−⋅ε−+−⋅ε & (5.2)
wobei die zugehörige effektive Porosität zu dem jeweiligen Konzentrationsniveau darstellt.
ist die dazugehörige Beladung. Gleichung (5.2) wird umgestellt, um für jede Konzentrations-
stufe den Zuwachs an Beladung zu berechnen:
IIeffε
IIeffq
effq∆

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
75
( ) ( )( ) S
IIeff
IIIS
IIeff
IIIR
effIeff
IIeff V1
ccVcctVqqq
ε−−ε−−
=∆=−&
. (5.3)
Gleichung (5.3) gibt den Zuwachs an Beladung bezogen auf das Volumen der stationären Phase
an. Diese wird umgerechnet in den Zuwachs an Beladung effqs∆ für die gesamte chromato-
graphische Säule mit Hilfe von
S
IIeffeffeff V)1(qqs ε−∆=∆ (5.4)
Die Beladung der Säule ergibt sich für jedes Konzentrationsniveau aus:
eff
Ieff
IIeff qsqsqs ∆+= . (5.5)
Um die Ergebnisse mit den Resultaten der statischen Messung vergleichen zu können, ist eine
Umrechnung in eine molare, auf die Adsorbensmasse bezogene Beladungen mit
1ads
IIeff
1 Mmqs
n⋅
=′ (5.6)
notwendig, wobei mads die Masse Adsorbens in der Säule und M1 die Molmasse von
Komponente 1, im konkreten Fall von Dichlormethan, ist.
Die Auswertung der Perturbationsmethode erfolgte mit Hilfe von Gleichung (3.3) unter Ver-
wendung der effektiven Porosität spezifisch für jedes Konzentrationsniveau. Im Gegensatz zur
normalen Auswertung der Perturbationsmethode und analog zur Auswertung der Frontalanalyse
wird mit Hilfe des parameter-angepaßten Adsorptionsisothermenmodells, in diesem Fall einer ein-
fachen Langmuir-Gleichung, die Änderung der volumenbezogenen Beladung zwischen der (n-1)-
ten und der n-ten vermessenen Konzentration berechnet über
1neff
neff
neff qqq −−=∆ . (5.7)
Die Umrechnung dieses volumenbezogenen Beladungszuwachses auf die Zunahme der Beladung
für die gesamte Säule erfolgt analog zu Gleichung (5.4) mit
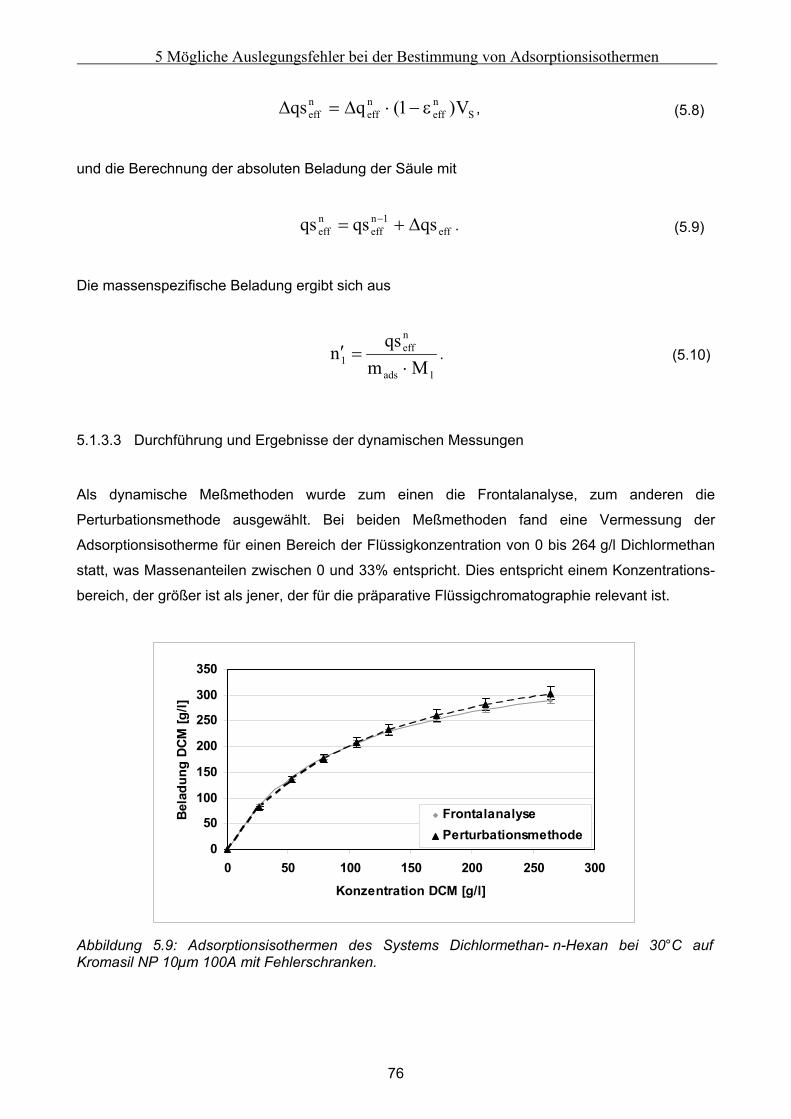
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
76
S
neff
neff
neff V)1(qqs ε−⋅∆=∆ , (5.8)
und die Berechnung der absoluten Beladung der Säule mit
eff
1neff
neff qsqsqs ∆+= − . (5.9)
Die massenspezifische Beladung ergibt sich aus
1ads
neff
1 Mmqs
n⋅
=′ . (5.10)
5.1.3.3 Durchführung und Ergebnisse der dynamischen Messungen
Als dynamische Meßmethoden wurde zum einen die Frontalanalyse, zum anderen die
Perturbationsmethode ausgewählt. Bei beiden Meßmethoden fand eine Vermessung der
Adsorptionsisotherme für einen Bereich der Flüssigkonzentration von 0 bis 264 g/l Dichlormethan
statt, was Massenanteilen zwischen 0 und 33% entspricht. Dies entspricht einem Konzentrations-
bereich, der größer ist als jener, der für die präparative Flüssigchromatographie relevant ist.
0
50
100
150
200
250
300
350
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bel
adun
g D
CM
[g/l]
FrontalanalysePerturbationsmethode
Abbildung 5.9: Adsorptionsisothermen des Systems Dichlormethan- n-Hexan bei 30°C auf Kromasil NP 10µm 100A mit Fehlerschranken.
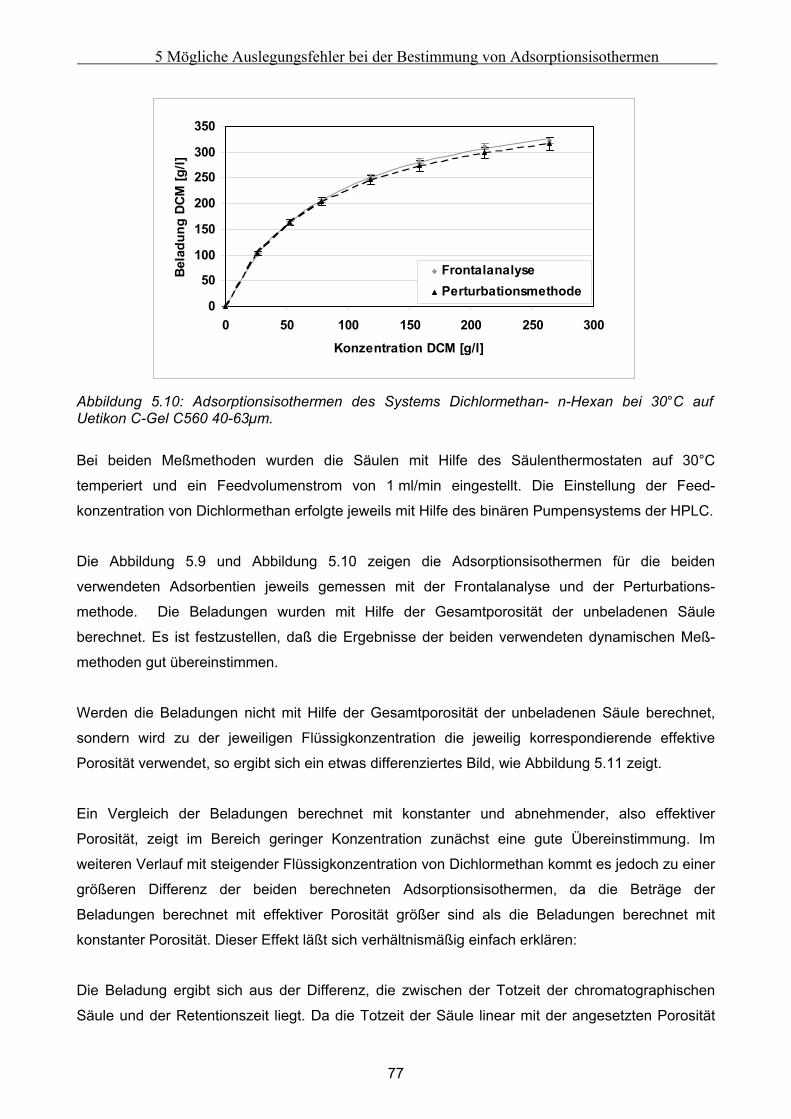
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
77
0
50
100
150
200
250
300
350
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bela
dung
DCM
[g/l]
FrontalanalysePerturbationsmethode
Abbildung 5.10: Adsorptionsisothermen des Systems Dichlormethan- n-Hexan bei 30°C auf Uetikon C-Gel C560 40-63µm.
Bei beiden Meßmethoden wurden die Säulen mit Hilfe des Säulenthermostaten auf 30°C
temperiert und ein Feedvolumenstrom von 1 ml/min eingestellt. Die Einstellung der Feed-
konzentration von Dichlormethan erfolgte jeweils mit Hilfe des binären Pumpensystems der HPLC.
Die Abbildung 5.9 und Abbildung 5.10 zeigen die Adsorptionsisothermen für die beiden
verwendeten Adsorbentien jeweils gemessen mit der Frontalanalyse und der Perturbations-
methode. Die Beladungen wurden mit Hilfe der Gesamtporosität der unbeladenen Säule
berechnet. Es ist festzustellen, daß die Ergebnisse der beiden verwendeten dynamischen Meß-
methoden gut übereinstimmen.
Werden die Beladungen nicht mit Hilfe der Gesamtporosität der unbeladenen Säule berechnet,
sondern wird zu der jeweiligen Flüssigkonzentration die jeweilig korrespondierende effektive
Porosität verwendet, so ergibt sich ein etwas differenziertes Bild, wie Abbildung 5.11 zeigt.
Ein Vergleich der Beladungen berechnet mit konstanter und abnehmender, also effektiver
Porosität, zeigt im Bereich geringer Konzentration zunächst eine gute Übereinstimmung. Im
weiteren Verlauf mit steigender Flüssigkonzentration von Dichlormethan kommt es jedoch zu einer
größeren Differenz der beiden berechneten Adsorptionsisothermen, da die Beträge der
Beladungen berechnet mit effektiver Porosität größer sind als die Beladungen berechnet mit
konstanter Porosität. Dieser Effekt läßt sich verhältnismäßig einfach erklären:
Die Beladung ergibt sich aus der Differenz, die zwischen der Totzeit der chromatographischen
Säule und der Retentionszeit liegt. Da die Totzeit der Säule linear mit der angesetzten Porosität
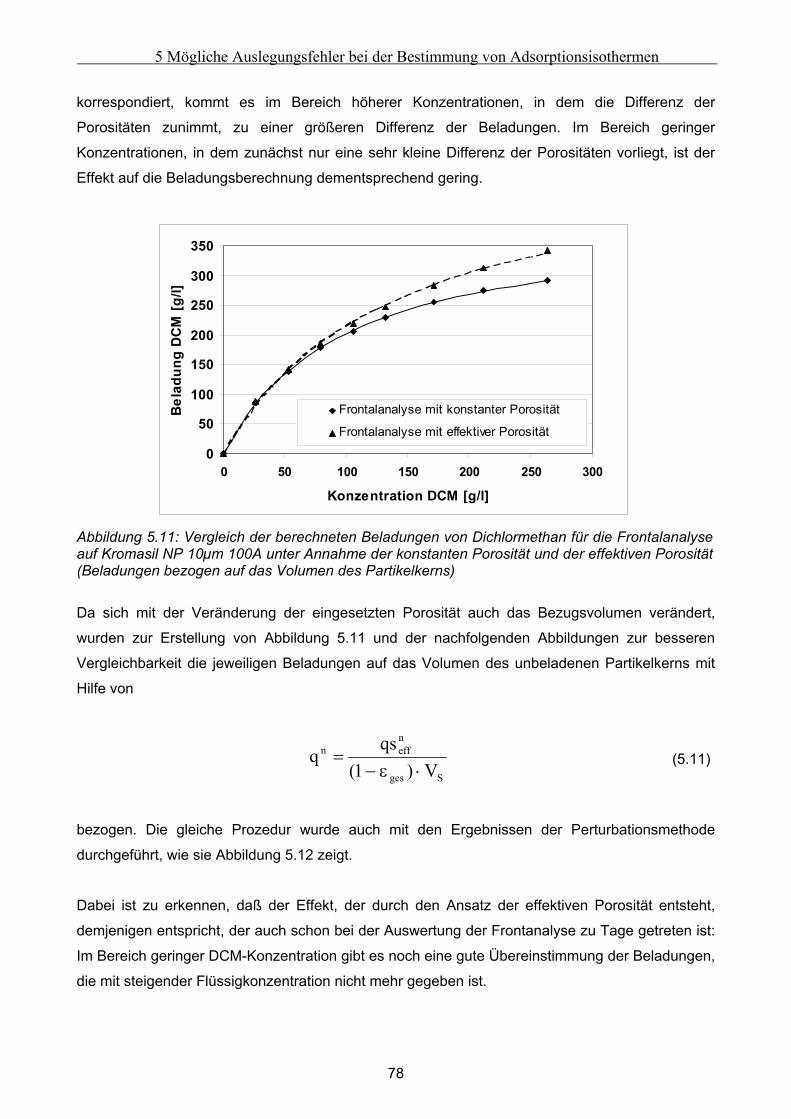
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
78
korrespondiert, kommt es im Bereich höherer Konzentrationen, in dem die Differenz der
Porositäten zunimmt, zu einer größeren Differenz der Beladungen. Im Bereich geringer
Konzentrationen, in dem zunächst nur eine sehr kleine Differenz der Porositäten vorliegt, ist der
Effekt auf die Beladungsberechnung dementsprechend gering.
0
50
100
150
200
250
300
350
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bel
adun
g D
CM
[g/l]
Frontalanalyse mit konstanter Porosität
Frontalanalyse mit effektiver Porosität
Abbildung 5.11: Vergleich der berechneten Beladungen von Dichlormethan für die Frontalanalyse auf Kromasil NP 10µm 100A unter Annahme der konstanten Porosität und der effektiven Porosität (Beladungen bezogen auf das Volumen des Partikelkerns)
Da sich mit der Veränderung der eingesetzten Porosität auch das Bezugsvolumen verändert,
wurden zur Erstellung von Abbildung 5.11 und der nachfolgenden Abbildungen zur besseren
Vergleichbarkeit die jeweiligen Beladungen auf das Volumen des unbeladenen Partikelkerns mit
Hilfe von
Sges
neffn
V)1(qs
q⋅ε−
= (5.11)
bezogen. Die gleiche Prozedur wurde auch mit den Ergebnissen der Perturbationsmethode
durchgeführt, wie sie Abbildung 5.12 zeigt.
Dabei ist zu erkennen, daß der Effekt, der durch den Ansatz der effektiven Porosität entsteht,
demjenigen entspricht, der auch schon bei der Auswertung der Frontanalyse zu Tage getreten ist:
Im Bereich geringer DCM-Konzentration gibt es noch eine gute Übereinstimmung der Beladungen,
die mit steigender Flüssigkonzentration nicht mehr gegeben ist.
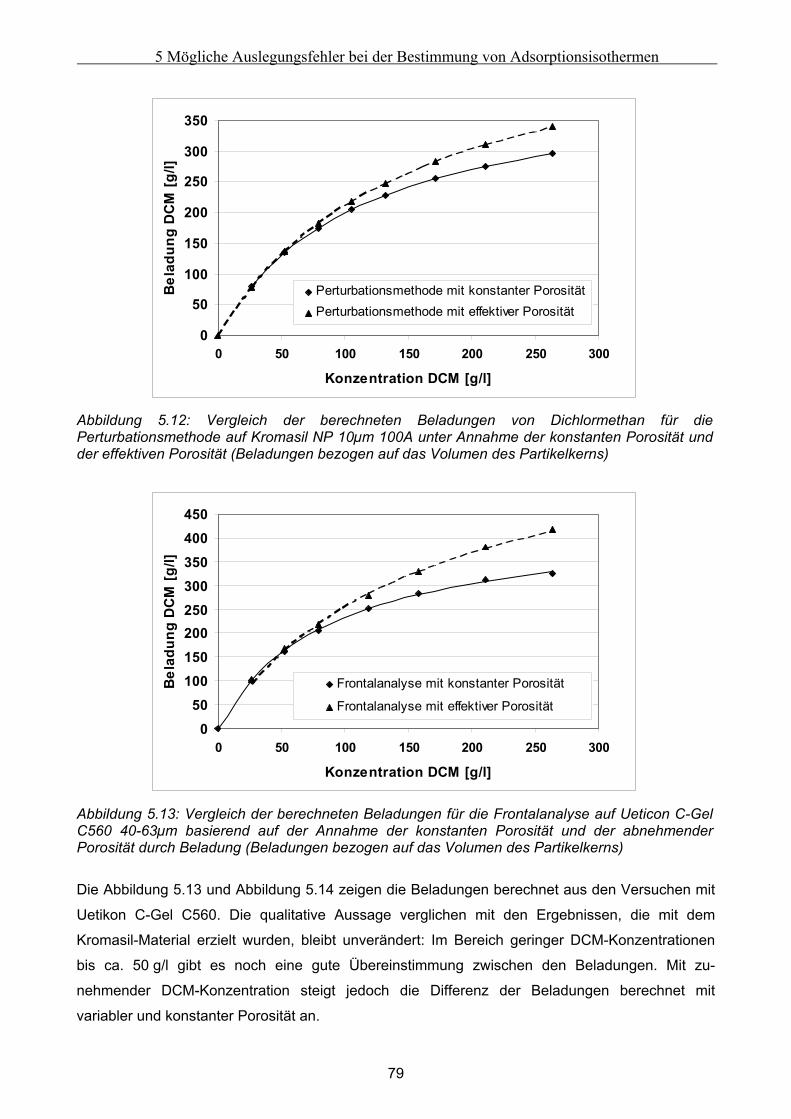
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
79
0
50
100
150
200
250
300
350
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bel
adun
g D
CM
[g/l]
Perturbationsmethode mit konstanter PorositätPerturbationsmethode mit effektiver Porosität
Abbildung 5.12: Vergleich der berechneten Beladungen von Dichlormethan für die Perturbationsmethode auf Kromasil NP 10µm 100A unter Annahme der konstanten Porosität und der effektiven Porosität (Beladungen bezogen auf das Volumen des Partikelkerns)
050
100150200250300350400450
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bel
adun
g D
CM
[g/l]
Frontalanalyse mit konstanter Porosität
Frontalanalyse mit effektiver Porosität
Abbildung 5.13: Vergleich der berechneten Beladungen für die Frontalanalyse auf Ueticon C-Gel C560 40-63µm basierend auf der Annahme der konstanten Porosität und der abnehmender Porosität durch Beladung (Beladungen bezogen auf das Volumen des Partikelkerns)
Die Abbildung 5.13 und Abbildung 5.14 zeigen die Beladungen berechnet aus den Versuchen mit
Uetikon C-Gel C560. Die qualitative Aussage verglichen mit den Ergebnissen, die mit dem
Kromasil-Material erzielt wurden, bleibt unverändert: Im Bereich geringer DCM-Konzentrationen
bis ca. 50 g/l gibt es noch eine gute Übereinstimmung zwischen den Beladungen. Mit zu-
nehmender DCM-Konzentration steigt jedoch die Differenz der Beladungen berechnet mit
variabler und konstanter Porosität an.
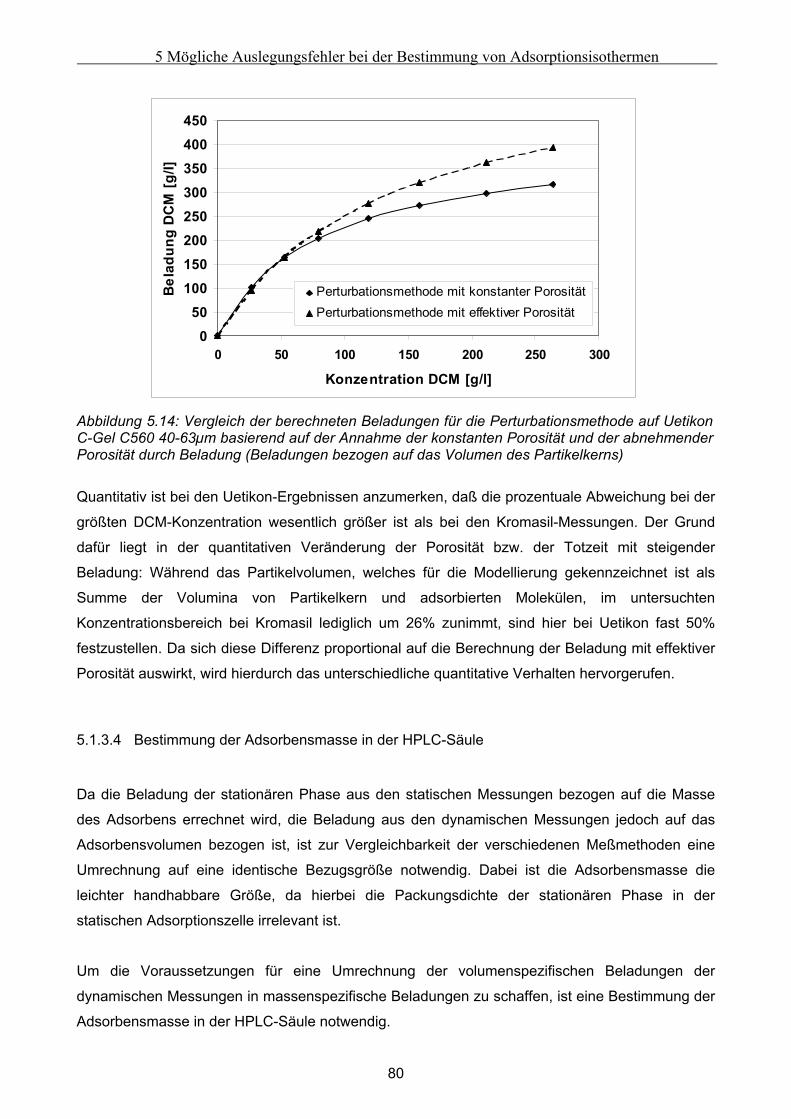
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
80
050
100150200250300350400450
0 50 100 150 200 250 300
Konzentration DCM [g/l]
Bel
adun
g D
CM
[g/l]
Perturbationsmethode mit konstanter PorositätPerturbationsmethode mit effektiver Porosität
Abbildung 5.14: Vergleich der berechneten Beladungen für die Perturbationsmethode auf Uetikon C-Gel C560 40-63µm basierend auf der Annahme der konstanten Porosität und der abnehmender Porosität durch Beladung (Beladungen bezogen auf das Volumen des Partikelkerns)
Quantitativ ist bei den Uetikon-Ergebnissen anzumerken, daß die prozentuale Abweichung bei der
größten DCM-Konzentration wesentlich größer ist als bei den Kromasil-Messungen. Der Grund
dafür liegt in der quantitativen Veränderung der Porosität bzw. der Totzeit mit steigender
Beladung: Während das Partikelvolumen, welches für die Modellierung gekennzeichnet ist als
Summe der Volumina von Partikelkern und adsorbierten Molekülen, im untersuchten
Konzentrationsbereich bei Kromasil lediglich um 26% zunimmt, sind hier bei Uetikon fast 50%
festzustellen. Da sich diese Differenz proportional auf die Berechnung der Beladung mit effektiver
Porosität auswirkt, wird hierdurch das unterschiedliche quantitative Verhalten hervorgerufen.
5.1.3.4 Bestimmung der Adsorbensmasse in der HPLC-Säule
Da die Beladung der stationären Phase aus den statischen Messungen bezogen auf die Masse
des Adsorbens errechnet wird, die Beladung aus den dynamischen Messungen jedoch auf das
Adsorbensvolumen bezogen ist, ist zur Vergleichbarkeit der verschiedenen Meßmethoden eine
Umrechnung auf eine identische Bezugsgröße notwendig. Dabei ist die Adsorbensmasse die
leichter handhabbare Größe, da hierbei die Packungsdichte der stationären Phase in der
statischen Adsorptionszelle irrelevant ist.
Um die Voraussetzungen für eine Umrechnung der volumenspezifischen Beladungen der
dynamischen Messungen in massenspezifische Beladungen zu schaffen, ist eine Bestimmung der
Adsorbensmasse in der HPLC-Säule notwendig.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
81
Zu diesem Zweck wird eine vergleichsweise einfache Wägemethode verwendet, wie sie von
Alhedai et al. (1989) beschrieben wurde. Dabei wird die chromatographische Säule mit
verschiedenen Flüssigkeiten bekannter Dichte vollständig gefüllt und anschließend gewogen. Bei
Abzug des Leergewichtes der Säule ist es nun möglich, eine Gerade der Form
by)mm( LeerSäule +ρ⋅=− (5.12)
durch die Meßpunkte zu legen und die Parameter y und b anzupassen. Da bei der Dichte ρ=0 nur
die Summe aus der Masse des Adsorbens und der leeren Säule zu messen wäre, entspricht der
Parameter b der Masse des Adsorbens, also
bm Ad = . (5.13)
Diese Prozedur wurde nach Beendigung der dynamischen Messungen durchgeführt. Die
verwendeten Stoffe waren n-Hexan, Dichlormethan und Toluol. Als Adsorbensmasse der
Kromasil-Säule ergab sich ein Wert von 9.6968 g, in der Uetikon-Säule waren 8.947 g stationäre
Phase.
5.2 Vergleich der Meßergebnisse
Dieses Unterkapitel zeigt den Vergleich der Ergebnisse von statischen und dynamischen
Messungen. Abschnitt 5.2.1 erläutert die notwendigen Umrechnungsschritte, um die Ergebnisse
beide Methoden miteinander vergleichen zu können. In Abschnitt 5.2.2 erfolgt ein Vergleich der
sich daraus ergebenden Resulte.
5.2.1 Umrechnung der statischen Exzeßisothermen
In diesem Abschnitt werden verschiedene Methoden, Exzeßisothermen in Beladungsisothermen
umzurechnen, zunächst entwickelt und anschließend auf die gemessenen Daten angewendet und
diskutiert. Dabei werden die Grundlagen der verwendeten Gleichungen nur soweit dargestellt, wie
sie zum Verständnis notwendig sind. Für eine vollständige Herleitung der verwendeten
Gleichungen wird auf die Arbeit von Hirsch (2000) sowie auf die Artikel von Larionov (1971) und
Minka (1973) verwiesen.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
82
5.2.1.1 Umrechnung mittels einer analytischen Gleichung
Ziel dieser Umrechnung ist es, die adsorbierte Stoffmenge von Komponente 1 (Dichlormethan) in
einem binären Gemisch zu berechnen. Diese definiert sich über
11 xnn ′⋅′=′ . (5.14)
Setzt man Gleichung (2.3) in Gleichung (5.14) ein, so erhält man
( )11
1E1
1 xxxn
n−′
′=′ . (5.15)
Diese Gleichung ist nur analytisch lösbar, wenn die Zusammensetzung der adsorbierten Phase
bekannt ist. Da diese jedoch nicht meßbar ist, führt hier nur eine Annahme über die
Zusammensetzung zum Ziel: Die einzige logische Annahme wäre diejenige, daß die schwächer
adsorbierende Komponente 2 nicht in der Adsorbatphase anzutreffen ist, sondern sich
ausschließlich im Adsorptiv befindet. Formal bedeutet dies die Annahme:
1x1 =′ . (5.16)
Diese Annahme ist natürlich umso realistischer, je größer der Unterschied der Adsorptionsaffinität
der beiden Komponenten ist. Dies kann bei dem verwendeten Testsystem als gegeben
angesehen werden.
Damit kann nun unter Umformung von Gleichung (2.1) und (2.3) die Menge von Komponente 1 im
Adsorbat formal berechnet werden durch:
2
0020
1 xnx
nn⋅
−=′ . (5.17)
5.2.1.2 Umrechnung mittels der Minka-Gleichung
Für den Fall, daß eine gleichzeitigen Adsorption der Komponente 2 modelliert werden soll, ist die
Anpassung bestimmter Parameter mittels Least-Square Methode an die Exzeßisotherme

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
83
notwendig. Wichtig ist dabei, daß die verwendeten Gleichungen eine Verbindung zwischen dem
Exzeß und der Beladung herstellen. In diesem Unterabschnitt sei zunächst eine Umrechnungs-
methode dargestellt, die auf der Arbeit von Minka und Myers (1973) basiert. Im folgenden
Unterabschnitt wird dann noch eine Methode vorgestellt, die auf der Everett-Schay-Gleichung
basiert.
Ausgangspunkt für das Modell von Minka und Myers (1973) sind zwei Annahmen: Zum einen die
Aufteilung der Flüssigkeit im Adsorptionsgleichgewicht in eine Adsorbatphase mit der Molmenge n‘
und eine Bulk-Phase mit der restlichen Stoffmenge n, was auf Gleichung (2.2) führt:
nnn 0 ′+= . (5.18)
Die zweite Annahme betrifft die Stoffmenge in der Adsorbatphase n‘, die sich abschätzen läßt über
∑
′=
′ i
i
bx
n1
, (5.19)
wobei bi die Beladungskapazität des Adsorbens in Molen der reinen flüssigen Komponente i ist.
An dieser Stelle sei auf den Nachteil der im folgenden beschriebenen Methoden zur Umrechnung
von Exzeß- in Beladungsisothermen hingewiesen, daß nämlich das Volumen der Adsorbatphase
über den gesamten Konzentrationsbereich konstant bleibt, eine Zu- oder Abnahme dieses
Volumens durch adsorptive Vorgänge also nicht berücksichtigt wird.
Aus der Isofugazitätsbedingung leitet Minka (1973) dann für den in dieser Arbeit betrachteten
binären Fall spezifisch für die Adsorbensmasse folgenden Zusammenhang her (siehe auch
Anhang D):
+
−=
122
2
1
1
1221
ad
E1
Kbx
bx
)K1(xxmn
. (5.20)
mit der vereinfachten Schreibweise von K12:
−=
1
1
2
212 b
aba
expK (5.21)

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
84
Diese Herleitung ist auch bei Hirsch (2000) oder Thelen (1993) nachzulesen.
K wird auch als Selektivität des Adsorbens bezeichnet [Everett (1981)] und gibt die Möglichkeit,
die Zusammensetzung von Bulk- und Adsorbat-Phase zu berechnen: 12
12
2112 xx
xxK
′′
= (5.22)
Die Zusammensetzung der Adsorbatphase läßt sich im binären Fall über [Minka (1973)]:
1221
11 Kxx
xx
+=′ (5.23)
ermitteln. Gleichung (5.20) wird an den gemessen Verlauf der Exzeßisotherme angepasst. Die
anzupassenden Parameter sind a1, a2, b1 und b2. Um eine eindeutige Lösung zu gewährleisten, ist
es notwendig, die Anzahl der anzupassenden Parameter auf drei zu reduzieren. Dies geschieht
unter Verwendung der Gurwitsch-Regel [Gurwitsch (1915)], die besagt, daß das Verhältnis der
Beladungskapazitäten des Adsorbens b1 und b2 umgekehrt proportional zum Verhältnis der
molaren Reinstoffvolumina v1 und v2 ist, also
1
2
2
1
vv
bb
≈ . (5.24)
Mit Hilfe des nun bekannten K12 ist es möglich, die Zusammensetzung der Adsorbatphase mittels
Gleichung (5.23) zu berechnen. Zur Berechnung der spezifischen, molaren Beladung von
Komponente 1 n1‘ ergibt sich unter Verwendung von Gleichung (2.3) die Beziehung
1
11
adE1
ad
1 xxx
mnmn ′
−′=
′. (5.25)

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
85
5.2.1.3 Umrechnung mittels Everett-Schay-Gleichung
Eine andere Möglichkeit der Umrechnung basiert auf der Veröffentlichung von Berger und Dekany
(1997). Danach dient als Ausgangspunkt die sogenannte Everett-Schay-Gleichung [Everett
(1979)]:
−−
+−
= 121
21
211E1
21 x1KrK
1Kr
b1
nxx
(5.26)
mit
12
21
1221 xx
xxK1K
′′
== . (5.27)
Ferner ist r das Verhältnis der molaren Volumina:
1
2
vv
r = . (5.28)
Bei diesem Vorgehen wird Gleichung (5.26) an den Verlauf der Exzeßisotherme angepaßt, wobei
die anzupassenden Parameter die Beladungskapazität b1 und der Adsorptionstrennfaktor K21 sind.
Die Zielfunktion lautet also:
x1KrK
1Kr
bxx.n
n1 n
1i
2
121
21
21
121Eexp∑
−−
+−
=
Die Berechnung der Zusammensetzung der Adsorbat-Ph
umgeformten Gleichung (5.27) oder über den im folgenden besch
Aus einfachen geometrischen Überlegungen heraus kann das
beschrieben werden über
112211 vbvnvnatV =′+′=′=′ ,
!
Min= (5.29)ase erfolgt dabei mittels der
riebenen Weg:
Volumen der adsorbierten Phase
(5.30)

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
86
wobei t‘ die Schichtdicke der Adsorbatphase und a die spezifische Oberfläche des Adsorbens ist.
Mit dem allgemeinen Zusammenhang
21 nnn ′+′=′ (5.31)
wird Gleichung (5.30) nach n‘ aufgelöst und in Gleichung (2.3) eingesetzt, was auf eine
Berechnung der Zusammensetzung der Adsorbatphase über folgenden Zusammenhang führt:
)1r(nvVvVxrn
x E11
11E1
1 −+′′+
=′ . (5.32)
Ob Gleichung (5.32) oder Gleichung (5.27) zur Berechnung der Zusammensetzung der Adsorbat-
Phase benutzt wird, die Berechnung der adsorbierten Menge n1‘ erfolgt mit Hilfe von Gleichung
(5.25).
5.2.1.4 Unterschiede zwischen den Umrechnungsmethoden
a) b)
Komponente 1Komponente 2
Bulk-Phase
Adsorbat-Phase Adsorbens
Phasengrenze
Komponente 1Komponente 2
Bulk-Phase
Adsorbat-Phase
Adsorbens
Phasengrenze
Abbildung 5.15: Hypothetische Phasengrenzflächen für die analytische (a) und die numerische Lösung (b)
Der Hauptunterschied zwischen den numerischen Umrechnungsmethoden basierend auf den
Arbeiten von Minka und Myers (1973) sowie von Berger und Dekany (1997) und der in
Unterabschitt 5.2.1.1 vorgestellten analytischen Lösung liegt in der Art der hypothetischen

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
87
Phasengrenzfläche zwischen Adsorbat- und Bulk-Phase, was schematisch in Abbildung 5.15
dargestellt ist.
Die Verwendung der analytischen Lösung beinhaltet die Annahme, daß die adsorbierte Phase
ausschließlich aus Molekülen der stärker adsorbierenden Komponente 1 besteht, während alle
Moleküle der schwächer adsorbierenden Komponente 2 Teil der Bulk-Phase bleiben. Daher ist
das Volumen der Adsorbat-Phase abhängig von der Beladung, damit abhängig von der
Zusammensetzung im gesamten System und folglich variabel (Abbildung 5.15a).
Im Gegensatz dazu bleibt das Volumen bei Nutzung der numerischen Lösungsmethoden über den
gesamten Konzentrationsbereich konstant, kann allerdings Moleküle beider Komponenten ent-
halten (Abbildung 5.15b). Die Anzahl der Moleküle in der Adsorbat-Phase ist nur abhängig vom
molaren Volumen des Gemisches in selbiger. Eine weitere Einschränkung der numerischen
Lösungsmethoden ist die Tatsache, daß über den gesamten Konzentrationsbereich ein konstanter
Trennfaktor angenommen wird.
Der Unterschied der Betrachtung läßt sich anhand der hypothetischen Phasengrenzfläche
zwischen Adsorbat- und Bulk-Phase verdeutlichen: Bei der analytischen Lösung in Abbildung
5.15a ist die hypothetische Phasengrenze flexibel, sie umfaßt nur die adsorbierten Moleküle der
Komponente 1, alle Moleküle der Komponente 2 werden in die Bulk-Phase ausgeschlossen. Im
Gegensatz dazu ist die Form der Phasengrenze in Abbildung 5.15b über den gesamten Kon-
zentrationsbereich fest, unabhängig davon, wieviele Moleküle von welcher Sorte durch sie der
Adsorbatphase zugeordnet werden. In Abbildung 5.15 wird dabei das Volumen der Adsorbatphase
durch das Volumen der Pore repräsentiert. Dies muß jedoch nicht zwingend so sein, es kann sich
z.B. auch nur um einen Teil der Porenstruktur des Adsorbens handeln. Ebenso können sich auch
Moleküle von Komponente 2 in der Pore befinden, sie sind jedoch Teil der fließenden Bulk-Phase.
Daraus ergibt sich schlüssig, daß die Umrechnung mit Hilfe der analytischen Lösung zu
geringeren Beladungen führt als dies bei Nutzung der numerischen Lösungen der Fall ist. Der
Grund dafür liegt in der Tatsache begründet, daß nur die Zusammensetzung der Bulk-Phase
meßtechnisch zugänglich ist. Je mehr Moleküle der schwächer adsorbierenden Komponente 2
aus der Bulk-Phase entfernt werden, desto mehr Moleküle von Komponente 1 müssen aus der
Bulk-Phase entfernt werden, um deren Zusammensetzung konstant zu halten.
Resümierend muß also festgehalten werden, daß beide Arten der Umrechnung einschränkende
Annahmen beinhalten. Die analytische Lösung, die nur unter der Voraussetzung funktioniert, daß
ausschließlich eine Komponente adsorbiert, scheint eher realistisch zu sein bei Systemen, bei
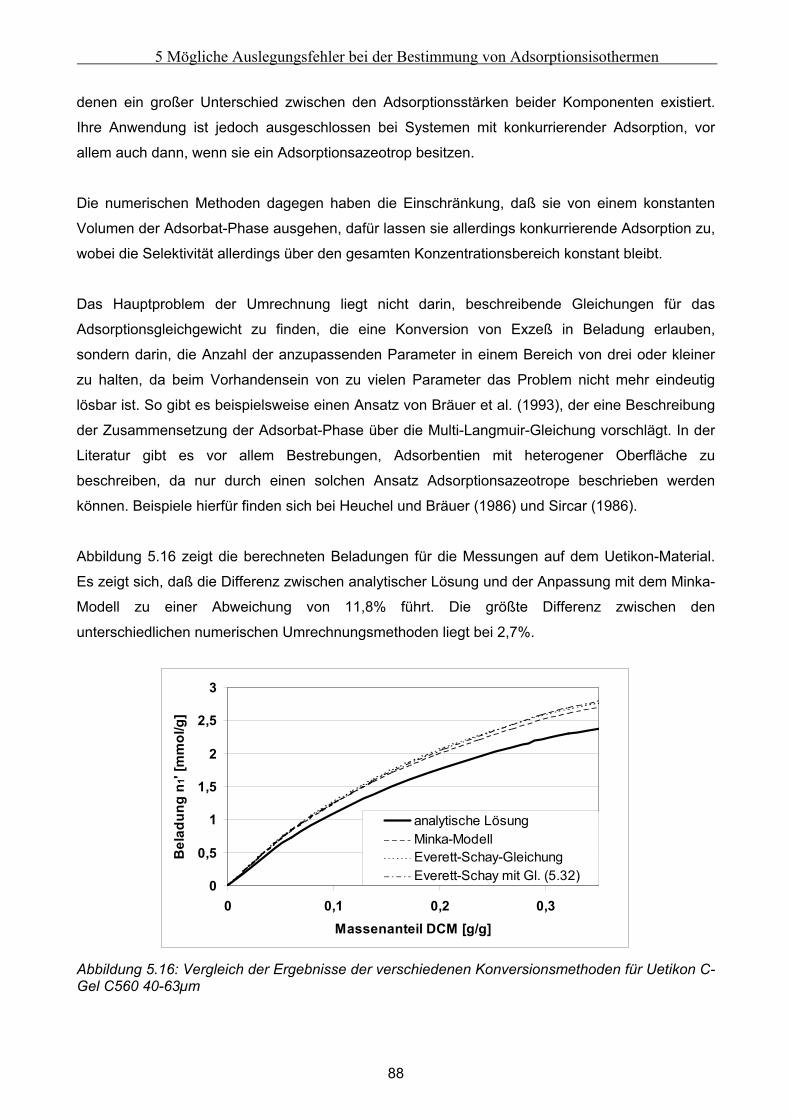
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
88
denen ein großer Unterschied zwischen den Adsorptionsstärken beider Komponenten existiert.
Ihre Anwendung ist jedoch ausgeschlossen bei Systemen mit konkurrierender Adsorption, vor
allem auch dann, wenn sie ein Adsorptionsazeotrop besitzen.
Die numerischen Methoden dagegen haben die Einschränkung, daß sie von einem konstanten
Volumen der Adsorbat-Phase ausgehen, dafür lassen sie allerdings konkurrierende Adsorption zu,
wobei die Selektivität allerdings über den gesamten Konzentrationsbereich konstant bleibt.
Das Hauptproblem der Umrechnung liegt nicht darin, beschreibende Gleichungen für das
Adsorptionsgleichgewicht zu finden, die eine Konversion von Exzeß in Beladung erlauben,
sondern darin, die Anzahl der anzupassenden Parameter in einem Bereich von drei oder kleiner
zu halten, da beim Vorhandensein von zu vielen Parameter das Problem nicht mehr eindeutig
lösbar ist. So gibt es beispielsweise einen Ansatz von Bräuer et al. (1993), der eine Beschreibung
der Zusammensetzung der Adsorbat-Phase über die Multi-Langmuir-Gleichung vorschlägt. In der
Literatur gibt es vor allem Bestrebungen, Adsorbentien mit heterogener Oberfläche zu
beschreiben, da nur durch einen solchen Ansatz Adsorptionsazeotrope beschrieben werden
können. Beispiele hierfür finden sich bei Heuchel und Bräuer (1986) und Sircar (1986).
Abbildung 5.16 zeigt die berechneten Beladungen für die Messungen auf dem Uetikon-Material.
Es zeigt sich, daß die Differenz zwischen analytischer Lösung und der Anpassung mit dem Minka-
Modell zu einer Abweichung von 11,8% führt. Die größte Differenz zwischen den
unterschiedlichen numerischen Umrechnungsmethoden liegt bei 2,7%.
0
0,5
1
1,5
2
2,5
3
0 0,1 0,2 0,3Massenanteil DCM [g/g]
Bel
adun
g n 1
' [m
mol
/g]
analytische LösungMinka-ModellEverett-Schay-GleichungEverett-Schay mit Gl. (5.32)
Abbildung 5.16: Vergleich der Ergebnisse der verschiedenen Konversionsmethoden für Uetikon C-Gel C560 40-63µm
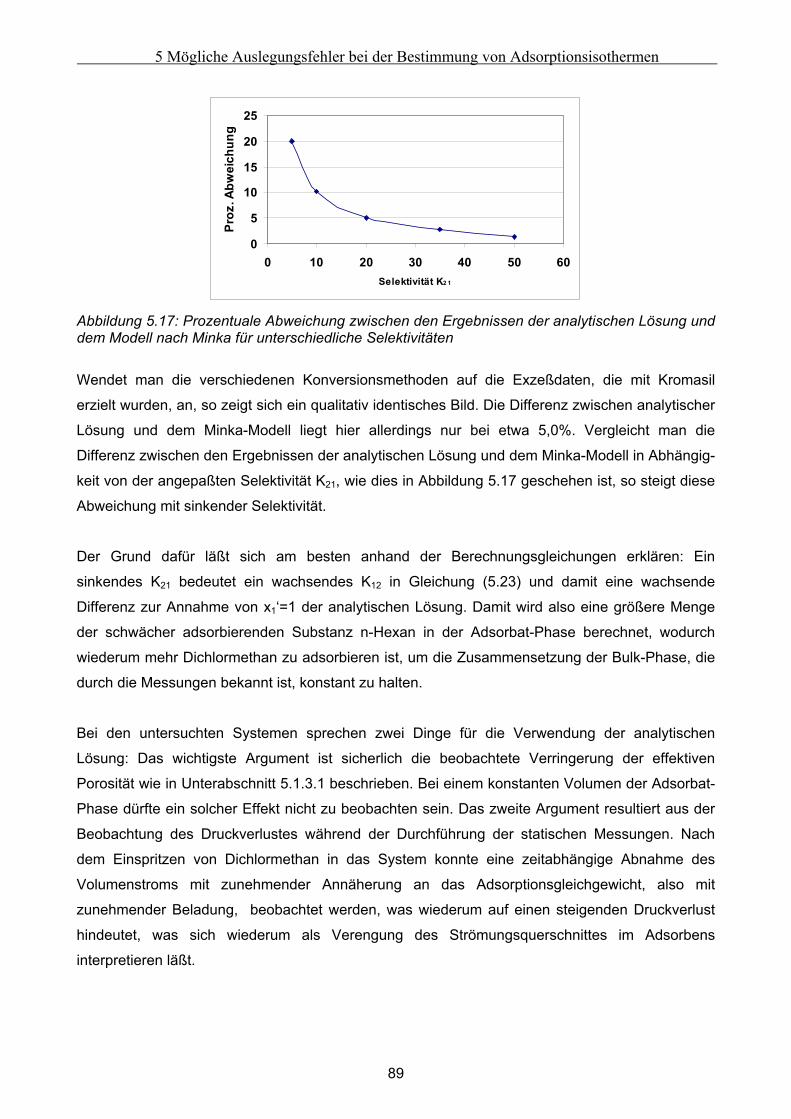
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
89
0
5
10
15
20
25
0 10 20 30 40 50 60Selektivität K2 1
Proz
. Abw
eich
ung
Abbildung 5.17: Prozentuale Abweichung zwischen den Ergebnissen der analytischen Lösung und dem Modell nach Minka für unterschiedliche Selektivitäten
Wendet man die verschiedenen Konversionsmethoden auf die Exzeßdaten, die mit Kromasil
erzielt wurden, an, so zeigt sich ein qualitativ identisches Bild. Die Differenz zwischen analytischer
Lösung und dem Minka-Modell liegt hier allerdings nur bei etwa 5,0%. Vergleicht man die
Differenz zwischen den Ergebnissen der analytischen Lösung und dem Minka-Modell in Abhängig-
keit von der angepaßten Selektivität K21, wie dies in Abbildung 5.17 geschehen ist, so steigt diese
Abweichung mit sinkender Selektivität.
Der Grund dafür läßt sich am besten anhand der Berechnungsgleichungen erklären: Ein
sinkendes K21 bedeutet ein wachsendes K12 in Gleichung (5.23) und damit eine wachsende
Differenz zur Annahme von x1‘=1 der analytischen Lösung. Damit wird also eine größere Menge
der schwächer adsorbierenden Substanz n-Hexan in der Adsorbat-Phase berechnet, wodurch
wiederum mehr Dichlormethan zu adsorbieren ist, um die Zusammensetzung der Bulk-Phase, die
durch die Messungen bekannt ist, konstant zu halten.
Bei den untersuchten Systemen sprechen zwei Dinge für die Verwendung der analytischen
Lösung: Das wichtigste Argument ist sicherlich die beobachtete Verringerung der effektiven
Porosität wie in Unterabschnitt 5.1.3.1 beschrieben. Bei einem konstanten Volumen der Adsorbat-
Phase dürfte ein solcher Effekt nicht zu beobachten sein. Das zweite Argument resultiert aus der
Beobachtung des Druckverlustes während der Durchführung der statischen Messungen. Nach
dem Einspritzen von Dichlormethan in das System konnte eine zeitabhängige Abnahme des
Volumenstroms mit zunehmender Annäherung an das Adsorptionsgleichgewicht, also mit
zunehmender Beladung, beobachtet werden, was wiederum auf einen steigenden Druckverlust
hindeutet, was sich wiederum als Verengung des Strömungsquerschnittes im Adsorbens
interpretieren läßt.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
90
5.2.1.5 Umrechnung mittels Langmuir-Gleichung
Ein weiterer Ansatz, diese Problematiken zu umgehen, führt über einen halbempirischen Weg. Der
Grundgedanke ist derjenige, die Zusammensetzung und die Gesamtmolmenge über eine Funktion
zu beschreiben und diese an den Exzeßverlauf anzupassen. Zur Beschreibung der
Zusammensetzung der Adsorbat-Phase in Gleichung (2.3) wird dazu die einfache Langmuir-
Gleichung benutzt:
( )1x1
1x1x
1,S1 xb1xb
qx+
=′. (5.33)
Diese Funktion ist monoton steigend, was unter der Voraussetzung der thermodynamischen
Stabilitätsbedingung [Findenegg (1996)]
0xx
Ti
i ≥∂
′∂ (5.34)
für die Existenz zweier koexistenter Phasen, in diesem Fall die Bulk- und die Adsorbat-Phase,
gerechtfertigt zu sein scheint. Da x1‘=1 bei x1=1 gilt, liegt damit das qS,1X bereits fest und die Zahl
der anzupassenden Parameter verringert sich um einen. Die Gesamtstoffmenge, die sich in der
Adsorbat-Phase befindet, wird ebenfalls über eine Gleichung in Langmuir-Form beschrieben:
( )1n1
1n1n
1,S xb1xb
qn+
=′ . (5.35)
Die monotone Steigung rechtfertigt sich hierbei zum einen durch durch die höhere Reinstoffdichte
von Dichlormethan, so daß selbst bei einem konstanten Volumen der Adsorbat-Phase eine Zu-
nahme der adsorbierten Gesamtstoffmenge stattfinden würde, zum anderen auch durch die beo-
bachteten Effekte wie die Abnahme der effektiven Porosität mit steigender Dichlormethan-
beladung.
Paßt man die Parameter qS,1n, b1
x und b1n an den Exzeßverlauf unter Verwendung von Gleichung
(2.3) mittels
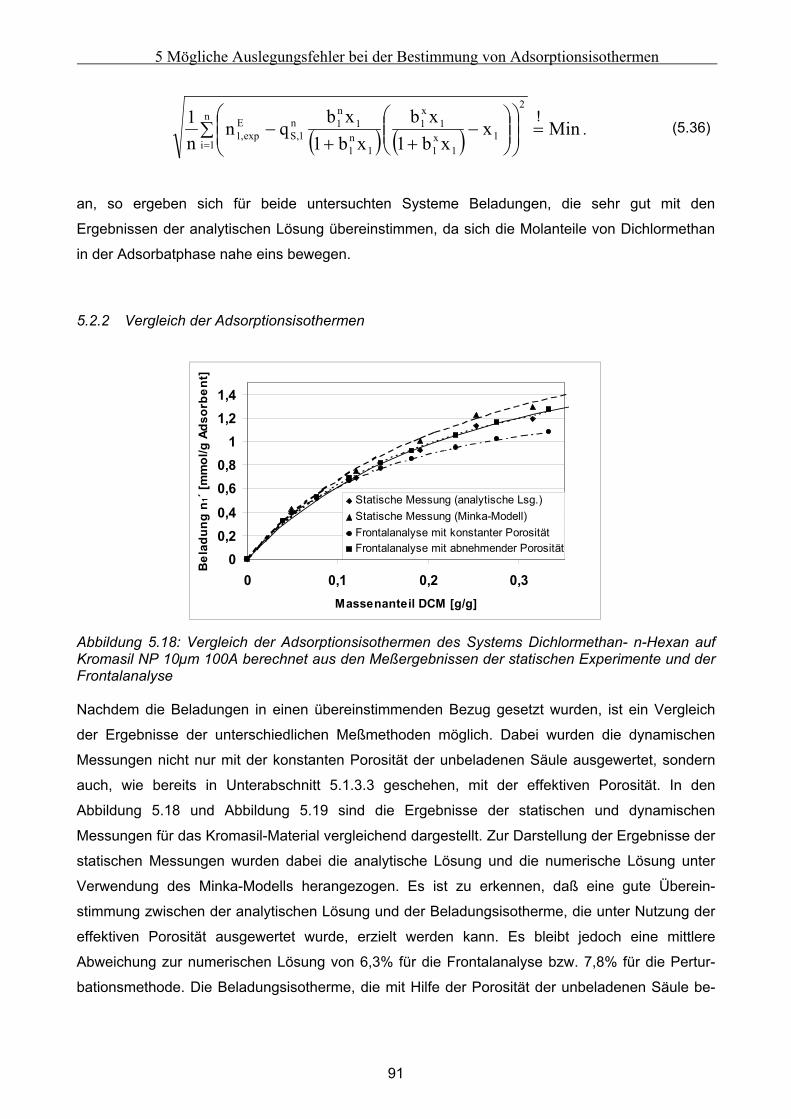
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
91
( ) ( ) xxb1
xbxb1
xbqn
n1
2n
1i1
1x1
1x1
1n1
1n1n
1,SE
exp,1∑
−
++−
=
an, so ergeben sich für beide untersuchten Systeme Beladung
Ergebnissen der analytischen Lösung übereinstimmen, da sich die M
in der Adsorbatphase nahe eins bewegen.
5.2.2 Vergleich der Adsorptionsisothermen
00,20,4
0,60,8
11,21,4
0 0,1 0,2 0,3Massenanteil DCM [g/g]
Bel
adun
g n 1
´ [m
mol
/g A
dsor
bent
]
Statische Messung (analytische LsStatische Messung (Minka-Modell)Frontalanalyse mit konstanter PoroFrontalanalyse mit abnehmender Po
Abbildung 5.18: Vergleich der Adsorptionsisothermen des Systems Kromasil NP 10µm 100A berechnet aus den Meßergebnissen der statFrontalanalyse Nachdem die Beladungen in einen übereinstimmenden Bezug gese
der Ergebnisse der unterschiedlichen Meßmethoden möglich. Dabe
Messungen nicht nur mit der konstanten Porosität der unbeladenen
auch, wie bereits in Unterabschnitt 5.1.3.3 geschehen, mit der e
Abbildung 5.18 und Abbildung 5.19 sind die Ergebnisse der st
Messungen für das Kromasil-Material vergleichend dargestellt. Zur Da
statischen Messungen wurden dabei die analytische Lösung und di
Verwendung des Minka-Modells herangezogen. Es ist zu erkenne
stimmung zwischen der analytischen Lösung und der Beladungsisoth
effektiven Porosität ausgewertet wurde, erzielt werden kann. Es
Abweichung zur numerischen Lösung von 6,3% für die Frontalanalys
bationsmethode. Die Beladungsisotherme, die mit Hilfe der Porosität
!
Min= . (5.36)en, die sehr gut mit den
olanteile von Dichlormethan
g.)
sitätrosität
Dichlormethan- n-Hexan auf ischen Experimente und der
tzt wurden, ist ein Vergleich
i wurden die dynamischen
Säule ausgewertet, sondern
ffektiven Porosität. In den
atischen und dynamischen
rstellung der Ergebnisse der
e numerische Lösung unter
n, daß eine gute Überein-
erme, die unter Nutzung der
bleibt jedoch eine mittlere
e bzw. 7,8% für die Pertur-
der unbeladenen Säule be-
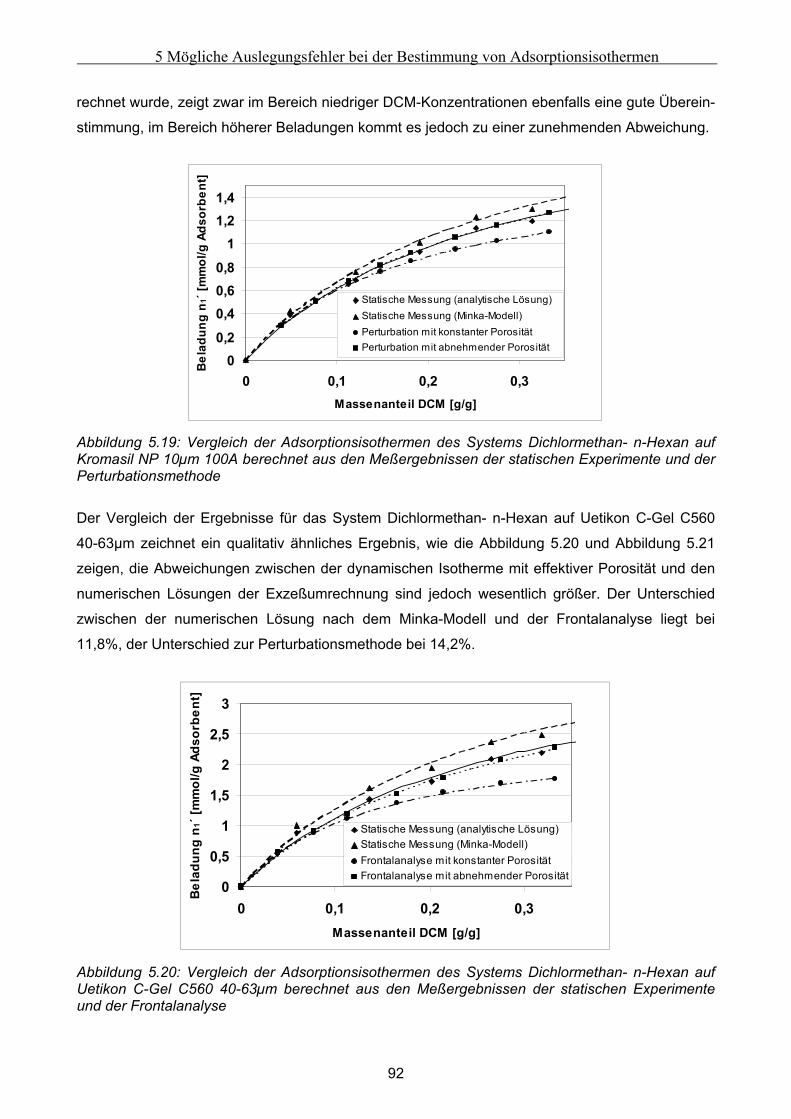
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
92
rechnet wurde, zeigt zwar im Bereich niedriger DCM-Konzentrationen ebenfalls eine gute Überein-
stimmung, im Bereich höherer Beladungen kommt es jedoch zu einer zunehmenden Abweichung.
00,2
0,40,60,8
11,21,4
0 0,1 0,2 0,3Massenanteil DCM [g/g]
Bel
adun
g n 1
´ [m
mol
/g A
dsor
bent
]
Statische Messung (analytische Lösung)Statische Messung (Minka-Modell)Perturbation mit konstanter PorositätPerturbation mit abnehmender Porosität
Abbildung 5.19: Vergleich der Adsorptionsisothermen des Systems Dichlormethan- n-Hexan auf Kromasil NP 10µm 100A berechnet aus den Meßergebnissen der statischen Experimente und der Perturbationsmethode
Der Vergleich der Ergebnisse für das System Dichlormethan- n-Hexan auf Uetikon C-Gel C560
40-63µm zeichnet ein qualitativ ähnliches Ergebnis, wie die Abbildung 5.20 und Abbildung 5.21
zeigen, die Abweichungen zwischen der dynamischen Isotherme mit effektiver Porosität und den
numerischen Lösungen der Exzeßumrechnung sind jedoch wesentlich größer. Der Unterschied
zwischen der numerischen Lösung nach dem Minka-Modell und der Frontalanalyse liegt bei
11,8%, der Unterschied zur Perturbationsmethode bei 14,2%.
0
0,5
1
1,5
2
2,5
3
0 0,1 0,2 0,3Massenanteil DCM [g/g]
Bel
adun
g n 1
´ [m
mol
/g A
dsor
bent
]
Statische Messung (analytische Lösung)Statische Messung (Minka-Modell)Frontalanalyse mit konstanter PorositätFrontalanalyse mit abnehmender Porosität
Abbildung 5.20: Vergleich der Adsorptionsisothermen des Systems Dichlormethan- n-Hexan auf Uetikon C-Gel C560 40-63µm berechnet aus den Meßergebnissen der statischen Experimente und der Frontalanalyse
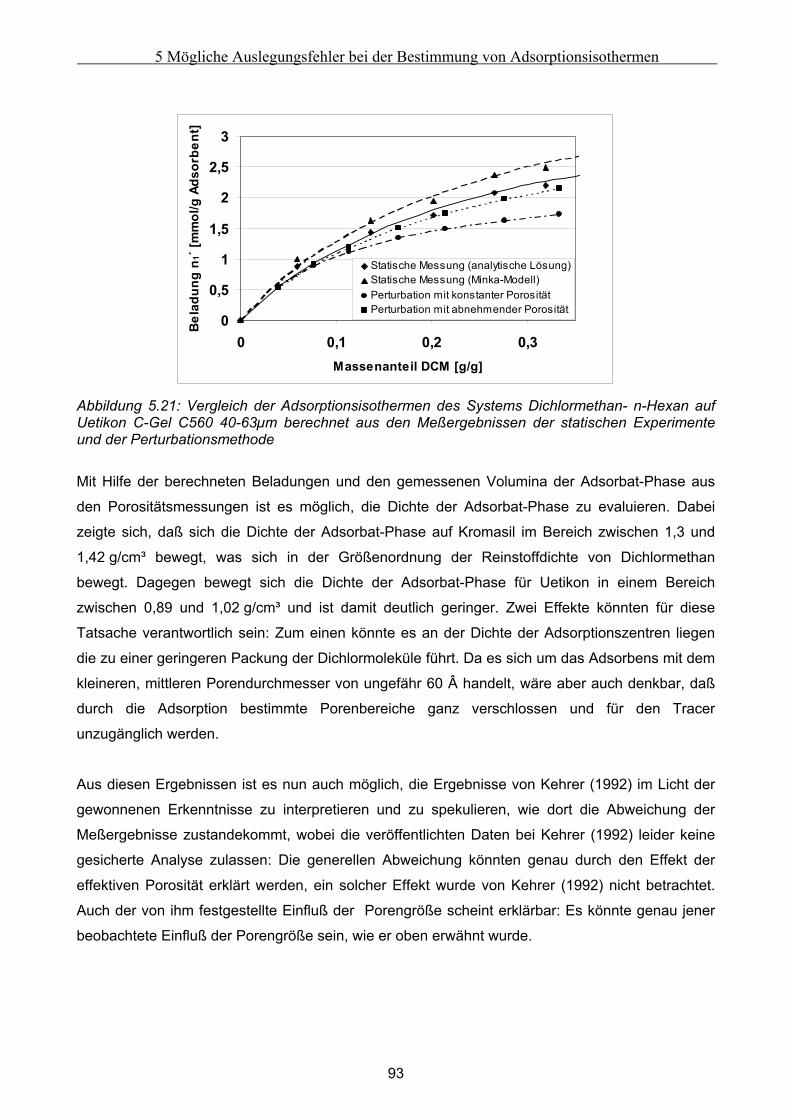
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
93
0
0,5
1
1,5
2
2,5
3
0 0,1 0,2 0,3Massenanteil DCM [g/g]
Bel
adun
g n 1
´ [m
mol
/g A
dsor
bent
]
Statische Messung (analytische Lösung)Statische Messung (Minka-Modell)Perturbation mit konstanter PorositätPerturbation mit abnehmender Porosität
Abbildung 5.21: Vergleich der Adsorptionsisothermen des Systems Dichlormethan- n-Hexan auf Uetikon C-Gel C560 40-63µm berechnet aus den Meßergebnissen der statischen Experimente und der Perturbationsmethode
Mit Hilfe der berechneten Beladungen und den gemessenen Volumina der Adsorbat-Phase aus
den Porositätsmessungen ist es möglich, die Dichte der Adsorbat-Phase zu evaluieren. Dabei
zeigte sich, daß sich die Dichte der Adsorbat-Phase auf Kromasil im Bereich zwischen 1,3 und
1,42 g/cm³ bewegt, was sich in der Größenordnung der Reinstoffdichte von Dichlormethan
bewegt. Dagegen bewegt sich die Dichte der Adsorbat-Phase für Uetikon in einem Bereich
zwischen 0,89 und 1,02 g/cm³ und ist damit deutlich geringer. Zwei Effekte könnten für diese
Tatsache verantwortlich sein: Zum einen könnte es an der Dichte der Adsorptionszentren liegen
die zu einer geringeren Packung der Dichlormoleküle führt. Da es sich um das Adsorbens mit dem
kleineren, mittleren Porendurchmesser von ungefähr 60 Â handelt, wäre aber auch denkbar, daß
durch die Adsorption bestimmte Porenbereiche ganz verschlossen und für den Tracer
unzugänglich werden.
Aus diesen Ergebnissen ist es nun auch möglich, die Ergebnisse von Kehrer (1992) im Licht der
gewonnenen Erkenntnisse zu interpretieren und zu spekulieren, wie dort die Abweichung der
Meßergebnisse zustandekommt, wobei die veröffentlichten Daten bei Kehrer (1992) leider keine
gesicherte Analyse zulassen: Die generellen Abweichung könnten genau durch den Effekt der
effektiven Porosität erklärt werden, ein solcher Effekt wurde von Kehrer (1992) nicht betrachtet.
Auch der von ihm festgestellte Einfluß der Porengröße scheint erklärbar: Es könnte genau jener
beobachtete Einfluß der Porengröße sein, wie er oben erwähnt wurde.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
94
5.3 Fehlerbetrachtung
Beim Vergleich der benutzten Meßverfahren ist auch eine Fehlerrechnung von Interesse.
Grundlage für die nachfolgende Betrachtung ist die Erfassung des in die Adsorptionsmessungen
eingehenden Fehlers mit Hilfe der quadratischen Fehlerfortpflanzung [siehe auch Lichten (1988)].
Solche Fehlerbetrachtungen wurden für die Zirkulationsmethode bereits von Geißler (1996), für
die Frontalanalyse von Beste (1997) durchgeführt. Im Gegensatz zur Arbeit von Beste (1997), der
eine Fehlerbetrachtung für die Zielgröße der volumenbezogenen Beladung qII durchführte, ist bei
den im Rahmen dieser Arbeit durchgeführten Messungen eine Betrachtung unter Beachtung der
Zielgröße n’1 notwendig, was bedeutet, daß neben den in Gleichung (3.2) vorhandenen Größen
noch die Masse des Adsorbens in die Betrachtung miteinfließt. Damit ergibt sich die Beladung n’1
aus den dynamischen Messungen als Funktion folgender Größen:
)m,V,q,V,,c,c,t,t(fn adsIiS
Ii
IIiextR1
&ε=′ (5.37)
Unter Ansatz der analytischen Exzeßumrechnung ergibt sich die Beladung aus den statischen
Messungen als Funktion folgender Größen:
)m,x,x,x,n(fn ads1101
01 ′=′ (5.38)
Eine solche Fehlerrechnung für die durchgeführten Messungen wurde mittels der quadratischen
Fehlerfortpflanzung durchgeführt. Die Fehler der einzelnen Meßgrößen für die statischen
Messungen wurden dabei Hirsch (2000) und Geißler (1996), diejenigen der dynamischen
Messungen Beste (1997) bzw. dem Referenzhandbuch der HPLC (1996) entnommen.
Dabei ergab sich bei den Messungen mit Kromasil für die statischen Messungen ein durch-
schnittlicher Fehler für den betrachteten Bereich von 1,20%, für die Frontalanalyse von 2,21% und
4,27% bei der Perturbationsmethode, wobei sich der Fehler der dynamischen Messungen
zusammensetzt aus dem Meßfehler bei der Bestimmung der Beladung und demjenigen bei der
Bestimmung der Adsorbensmasse. Demgegenüber steht eine mittlere Abweichung zwischen den
Ergebnissen der statischen Messung (analytische Lösung) und der Frontalanalyse von 2,83%,
zwischen denen von analytischer Lösung und Perturbationsmethode liegen 3,14%. Dies bedeutet,
daß die Abweichungen der Ergebnisse der einzelnen Meßmethoden untereinander innerhalb der
Meßfehler liegen.

5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
95
Die Fehlerrechnung für die Messungen mit dem Uetikon-Material ergeben im Durchschnitt 1,09%
für die statischen Messungen und 1,88% für die Frontalanalyse bzw. 3,81% für die Perturbations-
methode unter Einrechnung des Meßfehlers der Adsorbensmassenbestimmung. Demgegenüber
stehen mittlere Abweichungen zwischen analytischer Lösung und Frontalanalyse von 2,9 %, die
Perturbationsmethode weicht 4,3 % ab. Damit liegen auch hier die Abweichungen innerhalb der
Meßfehler.
Für die Fehlerbetrachtung bleibt noch die Betrachtung des Fehlers erwähnenswert, der sich für
Konzentrationen der stärker adsorbierenden Komponente aus der Umrechnung Exzeß in
Beladung ergeben kann. Dabei haben die partiellen Ableitungen von Gleichung (5.15)
11
10
ads01
1
xxxnm
xn
−′′
=∂
′∂
(5.39)
und
( )( )2
11
1101
0ads
11
10
ads
1
1
xxxxxnm
xxxnm
xn
−′
′−+
−′′
=∂
′∂
(5.40)
den weitaus größten Einfluß. Die Differenz zwischen der Zusammensetzung der Adsorbat-Phase
und der Bulkphase in den Nennern zeichnet dafür verantwortlich, daß im Bereich hoher
Konzentrationen von Komponente 1, wie sie hier nicht betrachtet wurden, der Fehler bei der
Bestimmung der Beladung steil ansteigt, da diese Differenz aufgrund des Ansatzes von x1’=1
immer kleiner wird. Da aber auch bei den untersuchten Systemen für die numerischen
Umrechnung bei hohen Konzentration von DCM x1’≈1 gilt, gilt hier ein nahezu identisches
Fehlerverhalten. Dies triff bei der Berechnung des Exzesses jedoch nicht zu [vgl. Geißler (1996)],
der Fehler entsteht hierbei nur durch die Umrechnung des Exzesses in die Beladung.
5.4 Auswirkungen der Abweichungen auf simulierte Peaks
Um die Frage zu beantworten, welche Auswirkungen diese Unterschiede zwischen statischen und
dynamischen Messungen inbesondere unter Berücksichtigung der Verwendung der effektiven
Porosität haben, kann folgender, hypothetischer Fall untersucht werden, der mit Hilfe von
gPROMS simuliert wurde:
Zur Auslegung einer chromatographischen Trennung auf dem Uetikon-Material wurde die
Adsorptionsisotherme von Dichlormethan mit dem Lösungsmittel n-Hexan mit Hilfe der
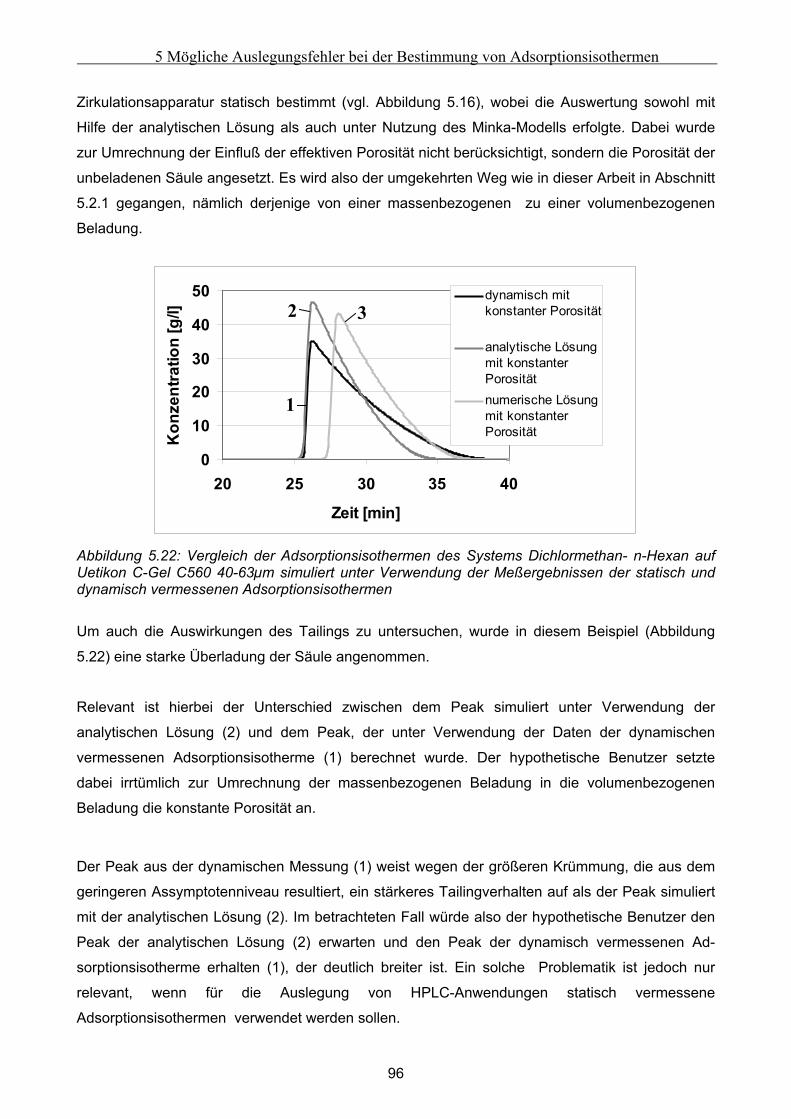
5 Mögliche Auslegungsfehler bei der Bestimmung von Adsorptionsisothermen
96
Zirkulationsapparatur statisch bestimmt (vgl. Abbildung 5.16), wobei die Auswertung sowohl mit
Hilfe der analytischen Lösung als auch unter Nutzung des Minka-Modells erfolgte. Dabei wurde
zur Umrechnung der Einfluß der effektiven Porosität nicht berücksichtigt, sondern die Porosität der
unbeladenen Säule angesetzt. Es wird also der umgekehrten Weg wie in dieser Arbeit in Abschnitt
5.2.1 gegangen, nämlich derjenige von einer massenbezogenen zu einer volumenbezogenen
Beladung.
0
10
20
30
40
50
20 25 30 35 40
Zeit [min]
Kon
zent
ratio
n [g
/l]
dynamisch mitkonstanter Porosität
analytische Lösungmit konstanterPorositätnumerische Lösungmit konstanterPorosität
1
2 3
Abbildung 5.22: Vergleich der Adsorptionsisothermen des Systems Dichlormethan- n-Hexan auf Uetikon C-Gel C560 40-63µm simuliert unter Verwendung der Meßergebnissen der statisch und dynamisch vermessenen Adsorptionsisothermen
Um auch die Auswirkungen des Tailings zu untersuchen, wurde in diesem Beispiel (Abbildung
5.22) eine starke Überladung der Säule angenommen.
Relevant ist hierbei der Unterschied zwischen dem Peak simuliert unter Verwendung der
analytischen Lösung (2) und dem Peak, der unter Verwendung der Daten der dynamischen
vermessenen Adsorptionsisotherme (1) berechnet wurde. Der hypothetische Benutzer setzte
dabei irrtümlich zur Umrechnung der massenbezogenen Beladung in die volumenbezogenen
Beladung die konstante Porosität an.
Der Peak aus der dynamischen Messung (1) weist wegen der größeren Krümmung, die aus dem
geringeren Assymptotenniveau resultiert, ein stärkeres Tailingverhalten auf als der Peak simuliert
mit der analytischen Lösung (2). Im betrachteten Fall würde also der hypothetische Benutzer den
Peak der analytischen Lösung (2) erwarten und den Peak der dynamisch vermessenen Ad-
sorptionsisotherme erhalten (1), der deutlich breiter ist. Ein solche Problematik ist jedoch nur
relevant, wenn für die Auslegung von HPLC-Anwendungen statisch vermessene
Adsorptionsisothermen verwendet werden sollen.

6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
97
6 Fehler in der Bettstruktur präparativer Chromato-graphiesäulen
Dieses Kapitel untersucht Fehler in der Bettstruktur chromatographischer Säulen. Im Vordergrund
steht dabei die phänomenologische Untersuchung der Auswirkungen solcher Fehler auf
chromatographische Peaks. Dabei erläutert Unterkapitel 6.1 zunächst die Methodik des
Vorgehens, die Unterkapitel 6.2 bis 6.5 zeigen die Auswirkungen der modellierten Fehler auf
Peaks adsorbierender und nicht-adsorbierender Substanzen.
6.1 Methodik des Vorgehens
Aus der in Unterkapitel 4.2 erwähnten Literatur und aus Erfahrungsberichten kristallisierten sich
vier mögliche Fehler in chromatographischen Säulen heraus, die im Rahmen dieser Arbeit
untersucht wurden:
• Das chromatographische Festbett sackt während des Betriebs ab.
• Die Fritte hinter dem Festbett, die zum einen das Bett fixiert und zum anderen als inverser
Verteiler dient, ist partiell undurchlässig.
• Im chromatographischen Festbett tritt eine relevante Kanalbildung auf.
• Im chromatographischen Festbett kommt es beim Packen zu einer radialen Heterogenität der
Porosität.
Die genannten Fehler wurden mit Hilfe der CFD-Simulationssoftware FLUENT (Version 4.5.6)
simuliert. Dazu wurde auf einem validierten Modell aufgebaut, das im Rahmen der Arbeit von
Lisso (2000) entwickelt und von Boysen et al. (2001) weiterentwickelt und und in Zusammenarbeit
mit Laiblin (2002) validiert wurde. Dieses Modell simuliert die Säule zweidimensional und
axialsymmetrisch. Es wurde entsprechend verändert, um die oben angegebenen Fehlerquellen zu
simulieren. Von diesen Fehlerquellen wurden drei auch experimentell untersucht. Eine
experimentelle Untersuchung der radialen Heterogenität unter definierten Bedingungen war nicht
möglich.
Effekte, die auf mangelhaftes Design der Verteilung am Kopf der Säule zurückzuführen sind,
wurden bereits ausführlich von Lisso (2001) oder auch von Broyles et al. (1999) behandelt und
werden aus diesem Grunde im Rahmen dieser Arbeit nicht weiter untersucht.

6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
98
Ziel dieser Untersuchungen ist es, Aussagen über die Auswirkungen solcher Fehler zu erhalten
und die dahinterstehenden Gesetzmäßigkeiten zu erkennen. Die Simulationen konzentrierten sich
auf eine definierte Säulengeometrie, die einer Säule entspricht, wie sie für präparative
Anwendungen benutzt wird. Dabei wurde eine Länge des Festbettes von 30 cm und ein
Durchmesser von 10 cm festgelegt. Zusätzlich dazu kommen zwei Fritten jeweils vor und nach
dem Festbett mit einer Dicke von jeweils 0,5 cm und einer Frittenqualität von FQ=50. Die
Frittenqualität beschreibt das Verhältnis der radialen zur axialen Permeabilität der Fritte [Lisso
(2001)]. Der Zulauf wurde mit einem Radius von 0,005555 cm vergrößert angenommen, um
numerische Probleme beim Auftreffen des Volumenstroms auf die Einlaßfritte zu vermeiden. Im
Referenzfall wurde eine Porosität von ε=0,4 sowohl für die Fritte als auch für das Festbett
angenommen.
Abbildung 6.1: Darstellung der simulierten, chromatographischen Säule
Weiterhin wurde Isothermie über die gesamte Säule angenommen und jeweils ein Volumenstrom
von 100 ml/h aufgegeben.
Es wurden gleichzeitig jeweils zwei Tracer aufgegeben. Ein nicht-adsorbierender Stoff und ein
Stoff, dessen Steigung der linearen Adsorptionsisotherme bei 0,5 liegt. Diese Tracer wurden
jeweils eine Sekunde lang aufgegeben, was einem Probenvolumen von jeweils 0,83 ml entspricht.
Für die Simulation wurde ein Gitter benutzt, das in axiale Richtung 200 Zellen und in radiale
Richtung 36 Zellen ausmachte, wobei für die Fritten jeweils 15 Zellen in axiale Richtung angesetzt
wurden. Für die Simulationen der abgesackten Festbetten wurden in axialer Richtung 250 Zellen
verwendet. Es wurden keine Parameter für die Dispersion in axiale oder radiale Richtung
vorgegeben. Die die Peaks verbreiternde Dispersion der simulierten Proben ist einzig das
Ergebnis numerischer Dispersion.
Die experimentellen Untersuchungen wurden durchgeführt mit Hilfe der bereits in Abschnitt 5.1.3
beschriebenen HPLC der Fa. Hewlett-Packard. Es wurde jeweils eine Säule (Modell Hibar,

6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
99
250x10mm) mit Glaskugeln slurry-gepackt, deren Größe zwischen 70-110 µm liegt. Diese
Glaskugeln kamen bereits im Rahmen der Arbeit von Laiblin (2002) zum Einsatz und deren
Eigenschaften sind dort detailliert beschrieben. Wie die jeweiligen Störungen in die Säulen
eingebaut wurden, wird in den folgenden Unterkapiteln beschrieben. Als mobile Phase wurde
Methanol verwendet, das bei 25°C mit 2 ml/min durch die Säule gefördert wurde. Es wurde jeweils
ein Peak von 1µl Toluol als Tracer aufgegeben.
6.2 Modellierung und Untersuchung von Effekten verursacht durch ein
abgesacktes Festbett
In der chromatographischen Praxis kommt es insbesondere nach dem Slurry-Packen von nicht
axial komprimierbaren Säulen vor, daß das Festbett im Betrieb z. B. durch einen hohen
Volumenstrom und dem damit verbundenen erhöhten Druckgradienten absackt, so daß sich
zwischen Einlaßfritte und Festbett eine Lücke bildet.
Abbildung 6.2: Darstellung der simulierten, chromatographischen Säule mit abgesacktem Festbett
6.2.1 Experimentelle Untersuchung des abgesackten Festbettes
Es war möglich, diese Art des Fehlers experimentell zu untersuchen, allerdings mit einem kleinen
Unterschied: Da ein kontrolliertes Absacken des Festbettes um eine bestimmte Länge nicht
möglich war, wurde jeweils nach einer Messung ein Teil der stationären Phase aus der Säule
entfernt. Dadurch erhöht sich die Gesamtporosität der Säule. Bei den Experimenten wurde das
Verhalten des Festbettes mit Lückenhöhen von einem, zwei und drei Zentimetern Länge zwischen
Festbett und Einlaßfritte untersucht. Die experimentellen Peaks zeigt Abbildung 6.2.
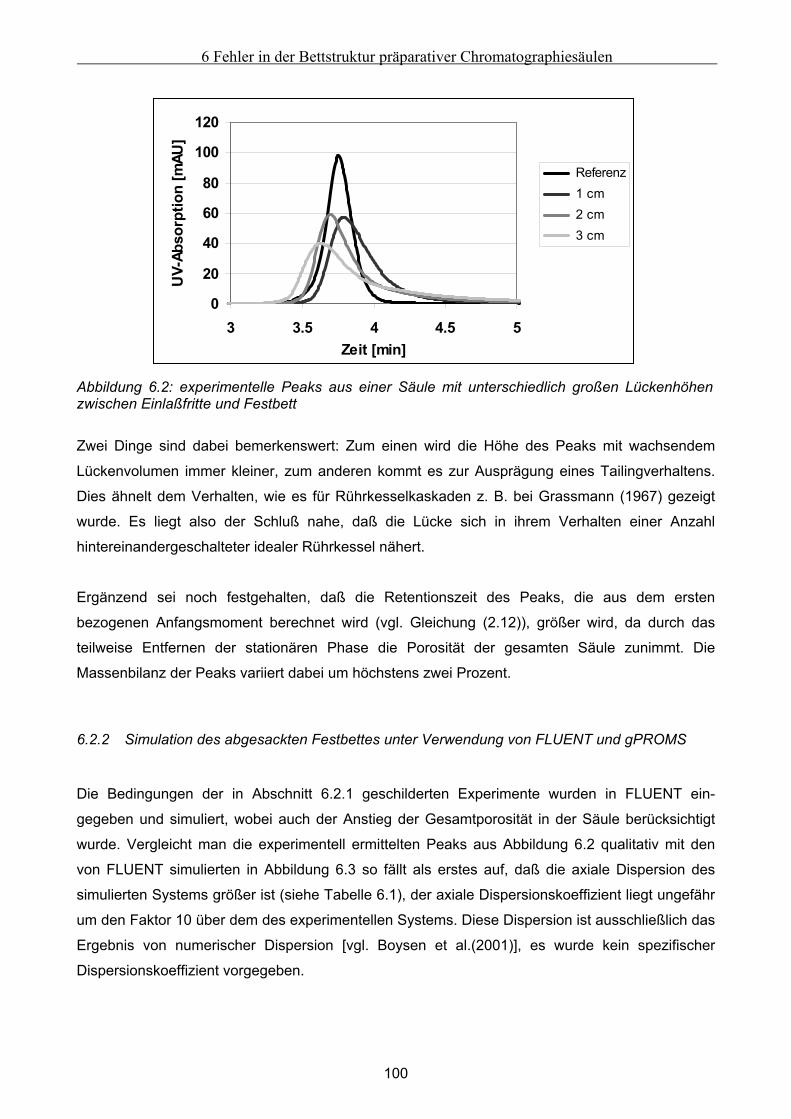
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
100
0
20
40
60
80
100
120
3 3.5 4 4.5 5Zeit [min]
UV-
Abso
rptio
n [m
AU]
Referenz 1 cm 2 cm 3 cm
Abbildung 6.2: experimentelle Peaks aus einer Säule mit unterschiedlich großen Lückenhöhen zwischen Einlaßfritte und Festbett
Zwei Dinge sind dabei bemerkenswert: Zum einen wird die Höhe des Peaks mit wachsendem
Lückenvolumen immer kleiner, zum anderen kommt es zur Ausprägung eines Tailingverhaltens.
Dies ähnelt dem Verhalten, wie es für Rührkesselkaskaden z. B. bei Grassmann (1967) gezeigt
wurde. Es liegt also der Schluß nahe, daß die Lücke sich in ihrem Verhalten einer Anzahl
hintereinandergeschalteter idealer Rührkessel nähert.
Ergänzend sei noch festgehalten, daß die Retentionszeit des Peaks, die aus dem ersten
bezogenen Anfangsmoment berechnet wird (vgl. Gleichung (2.12)), größer wird, da durch das
teilweise Entfernen der stationären Phase die Porosität der gesamten Säule zunimmt. Die
Massenbilanz der Peaks variiert dabei um höchstens zwei Prozent.
6.2.2 Simulation des abgesackten Festbettes unter Verwendung von FLUENT und gPROMS
Die Bedingungen der in Abschnitt 6.2.1 geschilderten Experimente wurden in FLUENT ein-
gegeben und simuliert, wobei auch der Anstieg der Gesamtporosität in der Säule berücksichtigt
wurde. Vergleicht man die experimentell ermittelten Peaks aus Abbildung 6.2 qualitativ mit den
von FLUENT simulierten in Abbildung 6.3 so fällt als erstes auf, daß die axiale Dispersion des
simulierten Systems größer ist (siehe Tabelle 6.1), der axiale Dispersionskoeffizient liegt ungefähr
um den Faktor 10 über dem des experimentellen Systems. Diese Dispersion ist ausschließlich das
Ergebnis von numerischer Dispersion [vgl. Boysen et al.(2001)], es wurde kein spezifischer
Dispersionskoeffizient vorgegeben.
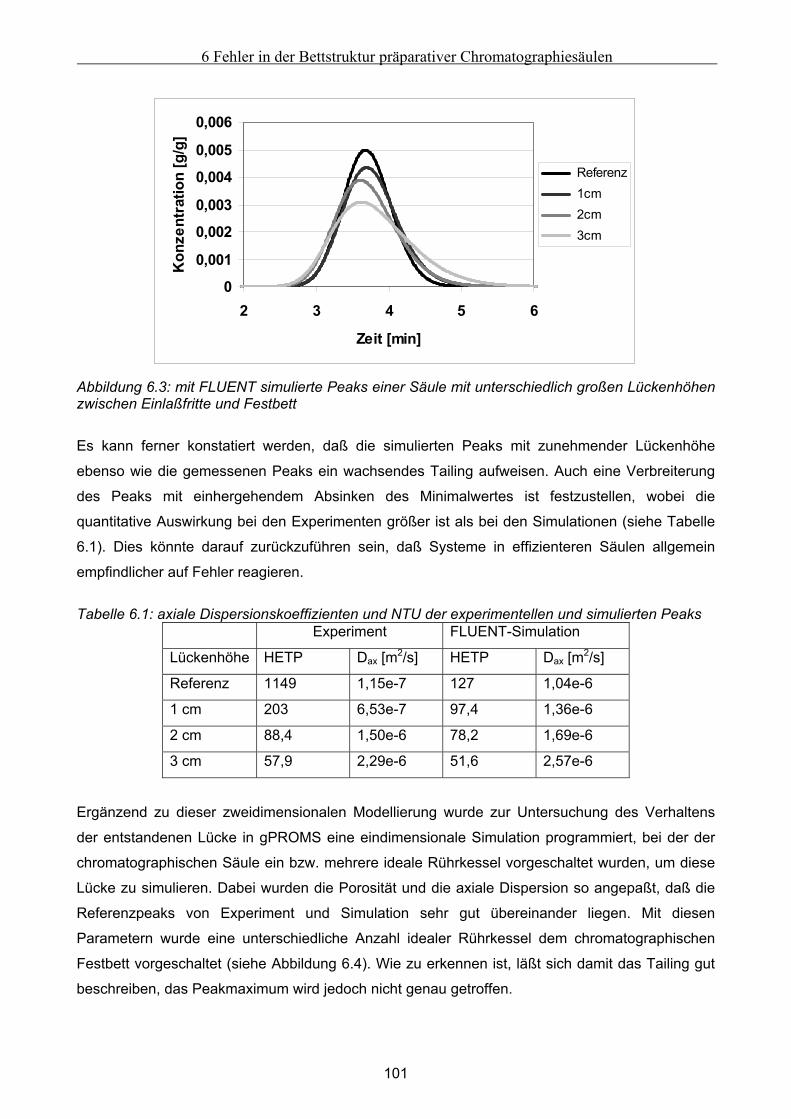
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
101
0
0,001
0,002
0,003
0,004
0,005
0,006
2 3 4 5 6
Zeit [min]
Kon
zent
ratio
n [g
/g]
Referenz 1cm 2cm 3cm
Abbildung 6.3: mit FLUENT simulierte Peaks einer Säule mit unterschiedlich großen Lückenhöhen zwischen Einlaßfritte und Festbett
Es kann ferner konstatiert werden, daß die simulierten Peaks mit zunehmender Lückenhöhe
ebenso wie die gemessenen Peaks ein wachsendes Tailing aufweisen. Auch eine Verbreiterung
des Peaks mit einhergehendem Absinken des Minimalwertes ist festzustellen, wobei die
quantitative Auswirkung bei den Experimenten größer ist als bei den Simulationen (siehe Tabelle
6.1). Dies könnte darauf zurückzuführen sein, daß Systeme in effizienteren Säulen allgemein
empfindlicher auf Fehler reagieren.
Tabelle 6.1: axiale Dispersionskoeffizienten und NTU der experimentellen und simulierten Peaks
Experiment FLUENT-Simulation
Lückenhöhe HETP Dax [m2/s] HETP Dax [m2/s] Referenz 1149 1,15e-7 127 1,04e-6
1 cm 203 6,53e-7 97,4 1,36e-6
2 cm 88,4 1,50e-6 78,2 1,69e-6
3 cm 57,9 2,29e-6 51,6 2,57e-6
Ergänzend zu dieser zweidimensionalen Modellierung wurde zur Untersuchung des Verhaltens
der entstandenen Lücke in gPROMS eine eindimensionale Simulation programmiert, bei der der
chromatographischen Säule ein bzw. mehrere ideale Rührkessel vorgeschaltet wurden, um diese
Lücke zu simulieren. Dabei wurden die Porosität und die axiale Dispersion so angepaßt, daß die
Referenzpeaks von Experiment und Simulation sehr gut übereinander liegen. Mit diesen
Parametern wurde eine unterschiedliche Anzahl idealer Rührkessel dem chromatographischen
Festbett vorgeschaltet (siehe Abbildung 6.4). Wie zu erkennen ist, läßt sich damit das Tailing gut
beschreiben, das Peakmaximum wird jedoch nicht genau getroffen.
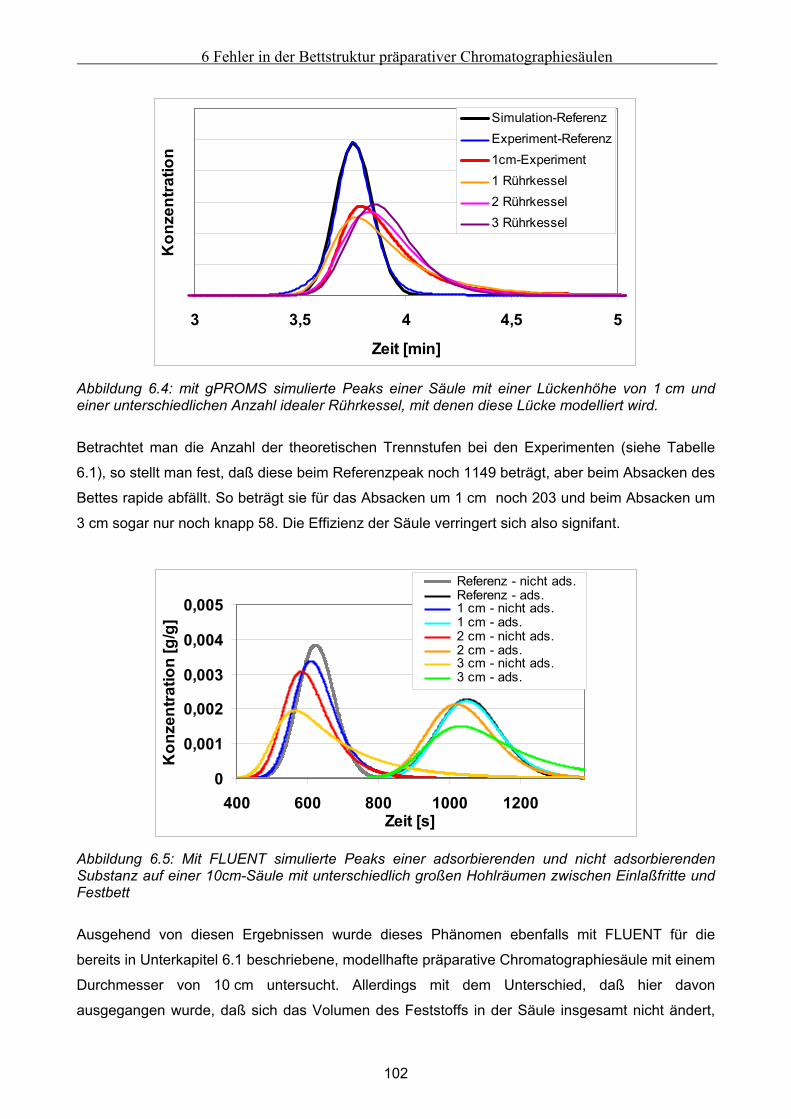
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
102
3 3,5 4 4,5 5
Zeit [min]
Kon
zent
ratio
n
Simulation-ReferenzExperiment-Referenz1cm-Experiment1 Rührkessel2 Rührkessel3 Rührkessel
Abbildung 6.4: mit gPROMS simulierte Peaks einer Säule mit einer Lückenhöhe von 1 cm und einer unterschiedlichen Anzahl idealer Rührkessel, mit denen diese Lücke modelliert wird.
Betrachtet man die Anzahl der theoretischen Trennstufen bei den Experimenten (siehe Tabelle
6.1), so stellt man fest, daß diese beim Referenzpeak noch 1149 beträgt, aber beim Absacken des
Bettes rapide abfällt. So beträgt sie für das Absacken um 1 cm noch 203 und beim Absacken um
3 cm sogar nur noch knapp 58. Die Effizienz der Säule verringert sich also signifant.
0
0,001
0,002
0,003
0,004
0,005
400 600 800 1000 1200Zeit [s]
Kon
zent
ratio
n [g
/g]
Referenz - nicht ads. Referenz - ads. 1 cm - nicht ads. 1 cm - ads. 2 cm - nicht ads. 2 cm - ads. 3 cm - nicht ads. 3 cm - ads.
Abbildung 6.5: Mit FLUENT simulierte Peaks einer adsorbierenden und nicht adsorbierenden Substanz auf einer 10cm-Säule mit unterschiedlich großen Hohlräumen zwischen Einlaßfritte und Festbett
Ausgehend von diesen Ergebnissen wurde dieses Phänomen ebenfalls mit FLUENT für die
bereits in Unterkapitel 6.1 beschriebene, modellhafte präparative Chromatographiesäule mit einem
Durchmesser von 10 cm untersucht. Allerdings mit dem Unterschied, daß hier davon
ausgegangen wurde, daß sich das Volumen des Feststoffs in der Säule insgesamt nicht ändert,
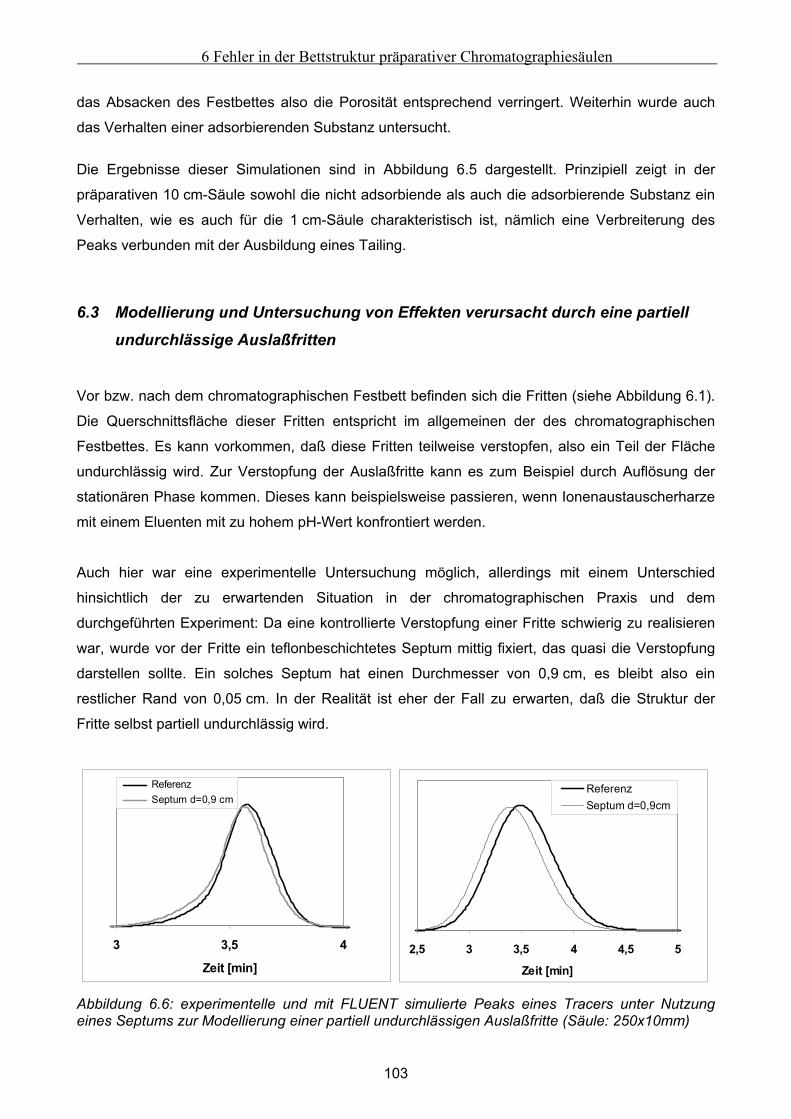
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
103
das Absacken des Festbettes also die Porosität entsprechend verringert. Weiterhin wurde auch
das Verhalten einer adsorbierenden Substanz untersucht.
Die Ergebnisse dieser Simulationen sind in Abbildung 6.5 dargestellt. Prinzipiell zeigt in der
präparativen 10 cm-Säule sowohl die nicht adsorbiende als auch die adsorbierende Substanz ein
Verhalten, wie es auch für die 1 cm-Säule charakteristisch ist, nämlich eine Verbreiterung des
Peaks verbunden mit der Ausbildung eines Tailing.
6.3 Modellierung und Untersuchung von Effekten verursacht durch eine partiell
undurchlässige Auslaßfritten
Vor bzw. nach dem chromatographischen Festbett befinden sich die Fritten (siehe Abbildung 6.1).
Die Querschnittsfläche dieser Fritten entspricht im allgemeinen der des chromatographischen
Festbettes. Es kann vorkommen, daß diese Fritten teilweise verstopfen, also ein Teil der Fläche
undurchlässig wird. Zur Verstopfung der Auslaßfritte kann es zum Beispiel durch Auflösung der
stationären Phase kommen. Dieses kann beispielsweise passieren, wenn Ionenaustauscherharze
mit einem Eluenten mit zu hohem pH-Wert konfrontiert werden.
Auch hier war eine experimentelle Untersuchung möglich, allerdings mit einem Unterschied
hinsichtlich der zu erwartenden Situation in der chromatographischen Praxis und dem
durchgeführten Experiment: Da eine kontrollierte Verstopfung einer Fritte schwierig zu realisieren
war, wurde vor der Fritte ein teflonbeschichtetes Septum mittig fixiert, das quasi die Verstopfung
darstellen sollte. Ein solches Septum hat einen Durchmesser von 0,9 cm, es bleibt also ein
restlicher Rand von 0,05 cm. In der Realität ist eher der Fall zu erwarten, daß die Struktur der
Fritte selbst partiell undurchlässig wird.
3 3,5 4
Zeit [min]
Referenz Septum d=0,9 cm
2,5 3 3,5 4 4,5 5
Zeit [min]
ReferenzSeptum d=0,9cm
Abbildung 6.6: experimentelle und mit FLUENT simulierte Peaks eines Tracers unter Nutzung eines Septums zur Modellierung einer partiell undurchlässigen Auslaßfritte (Säule: 250x10mm)
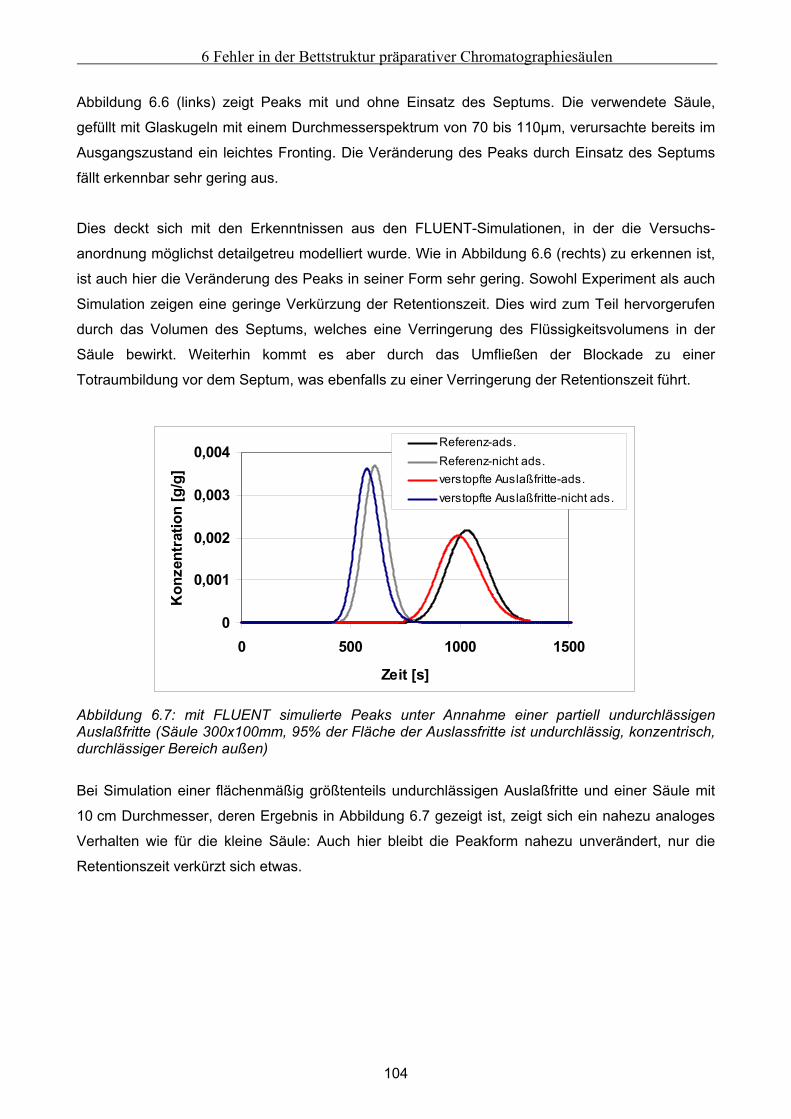
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
104
Abbildung 6.6 (links) zeigt Peaks mit und ohne Einsatz des Septums. Die verwendete Säule,
gefüllt mit Glaskugeln mit einem Durchmesserspektrum von 70 bis 110µm, verursachte bereits im
Ausgangszustand ein leichtes Fronting. Die Veränderung des Peaks durch Einsatz des Septums
fällt erkennbar sehr gering aus.
Dies deckt sich mit den Erkenntnissen aus den FLUENT-Simulationen, in der die Versuchs-
anordnung möglichst detailgetreu modelliert wurde. Wie in Abbildung 6.6 (rechts) zu erkennen ist,
ist auch hier die Veränderung des Peaks in seiner Form sehr gering. Sowohl Experiment als auch
Simulation zeigen eine geringe Verkürzung der Retentionszeit. Dies wird zum Teil hervorgerufen
durch das Volumen des Septums, welches eine Verringerung des Flüssigkeitsvolumens in der
Säule bewirkt. Weiterhin kommt es aber durch das Umfließen der Blockade zu einer
Totraumbildung vor dem Septum, was ebenfalls zu einer Verringerung der Retentionszeit führt.
0
0,001
0,002
0,003
0,004
0 500 1000 1500
Zeit [s]
Kon
zent
ratio
n [g
/g]
Referenz-ads.Referenz-nicht ads.verstopfte Auslaßfritte-ads.verstopfte Auslaßfritte-nicht ads.
Abbildung 6.7: mit FLUENT simulierte Peaks unter Annahme einer partiell undurchlässigen Auslaßfritte (Säule 300x100mm, 95% der Fläche der Auslassfritte ist undurchlässig, konzentrisch, durchlässiger Bereich außen)
Bei Simulation einer flächenmäßig größtenteils undurchlässigen Auslaßfritte und einer Säule mit
10 cm Durchmesser, deren Ergebnis in Abbildung 6.7 gezeigt ist, zeigt sich ein nahezu analoges
Verhalten wie für die kleine Säule: Auch hier bleibt die Peakform nahezu unverändert, nur die
Retentionszeit verkürzt sich etwas.
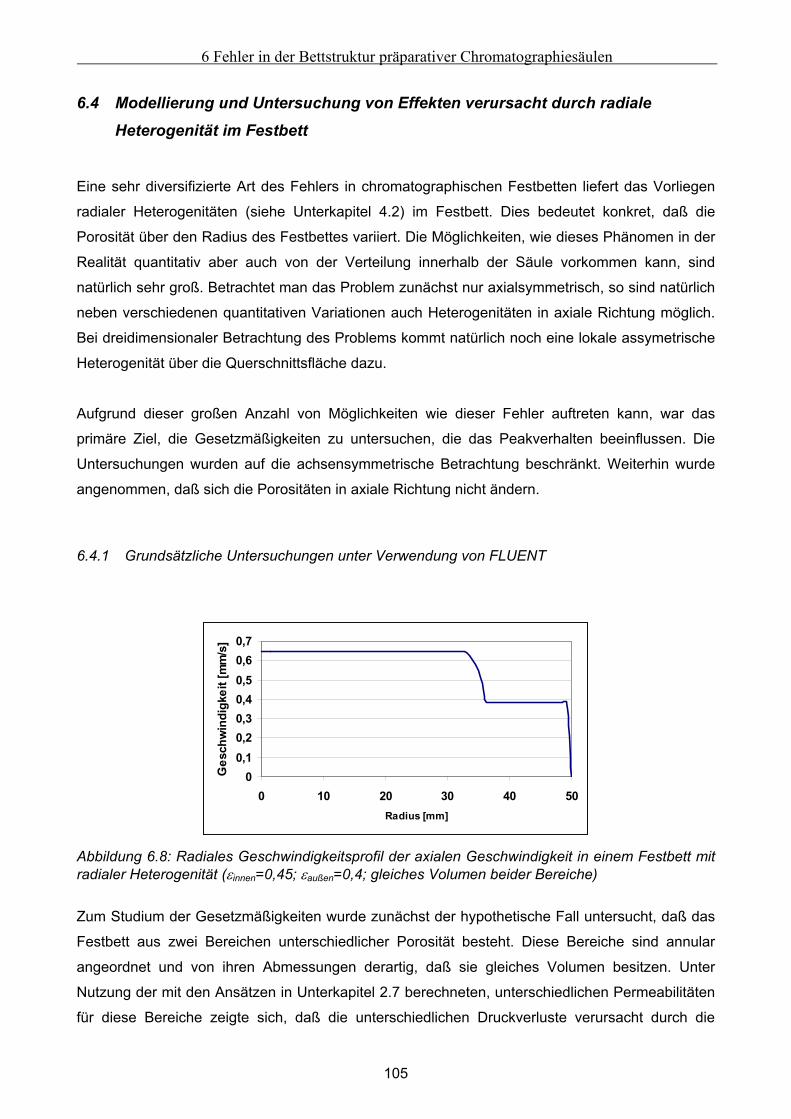
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
105
6.4 Modellierung und Untersuchung von Effekten verursacht durch radiale
Heterogenität im Festbett
Eine sehr diversifizierte Art des Fehlers in chromatographischen Festbetten liefert das Vorliegen
radialer Heterogenitäten (siehe Unterkapitel 4.2) im Festbett. Dies bedeutet konkret, daß die
Porosität über den Radius des Festbettes variiert. Die Möglichkeiten, wie dieses Phänomen in der
Realität quantitativ aber auch von der Verteilung innerhalb der Säule vorkommen kann, sind
natürlich sehr groß. Betrachtet man das Problem zunächst nur axialsymmetrisch, so sind natürlich
neben verschiedenen quantitativen Variationen auch Heterogenitäten in axiale Richtung möglich.
Bei dreidimensionaler Betrachtung des Problems kommt natürlich noch eine lokale assymetrische
Heterogenität über die Querschnittsfläche dazu.
Aufgrund dieser großen Anzahl von Möglichkeiten wie dieser Fehler auftreten kann, war das
primäre Ziel, die Gesetzmäßigkeiten zu untersuchen, die das Peakverhalten beeinflussen. Die
Untersuchungen wurden auf die achsensymmetrische Betrachtung beschränkt. Weiterhin wurde
angenommen, daß sich die Porositäten in axiale Richtung nicht ändern.
6.4.1 Grundsätzliche Untersuchungen unter Verwendung von FLUENT
00,10,20,30,40,50,60,7
0 10 20 30 40 5Radius [mm]
Ges
chw
indi
gkei
t [m
m/s
]
0
Abbildung 6.8: Radiales Geschwindigkeitsprofil der axialen Geschwindigkeit in einem Festbett mit radialer Heterogenität (εinnen=0,45; εaußen=0,4; gleiches Volumen beider Bereiche)
Zum Studium der Gesetzmäßigkeiten wurde zunächst der hypothetische Fall untersucht, daß das
Festbett aus zwei Bereichen unterschiedlicher Porosität besteht. Diese Bereiche sind annular
angeordnet und von ihren Abmessungen derartig, daß sie gleiches Volumen besitzen. Unter
Nutzung der mit den Ansätzen in Unterkapitel 2.7 berechneten, unterschiedlichen Permeabilitäten
für diese Bereiche zeigte sich, daß die unterschiedlichen Druckverluste verursacht durch die
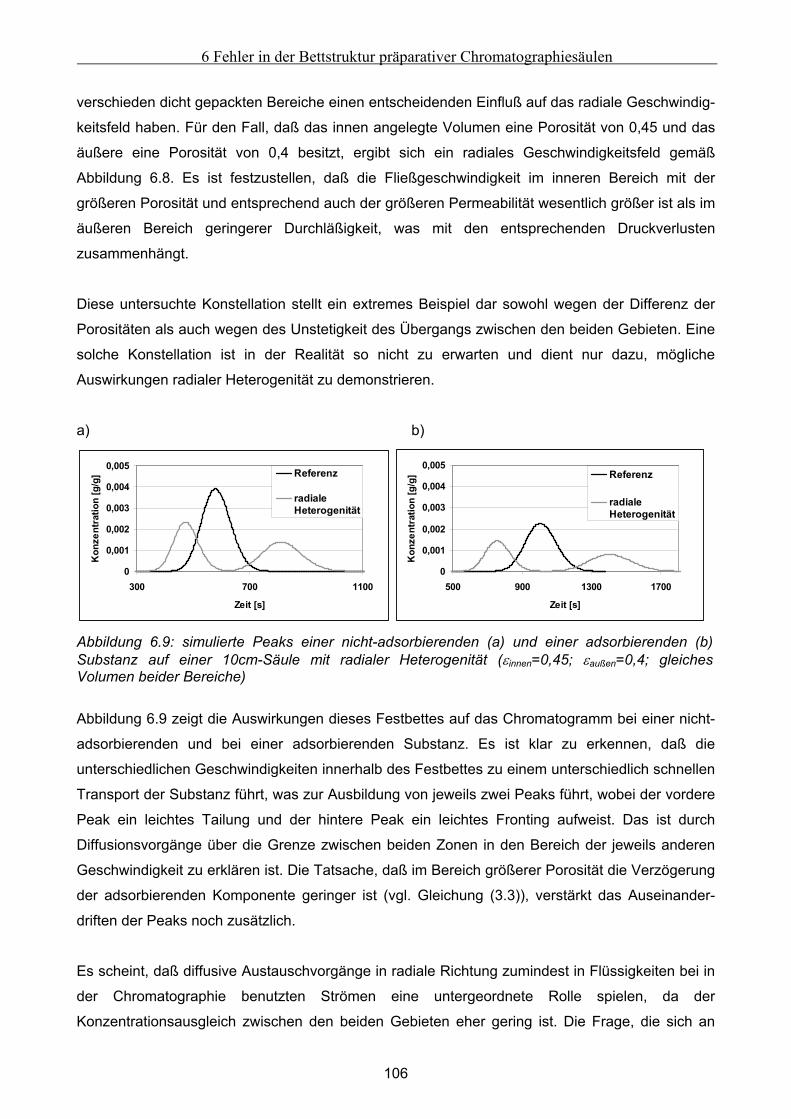
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
106
verschieden dicht gepackten Bereiche einen entscheidenden Einfluß auf das radiale Geschwindig-
keitsfeld haben. Für den Fall, daß das innen angelegte Volumen eine Porosität von 0,45 und das
äußere eine Porosität von 0,4 besitzt, ergibt sich ein radiales Geschwindigkeitsfeld gemäß
Abbildung 6.8. Es ist festzustellen, daß die Fließgeschwindigkeit im inneren Bereich mit der
größeren Porosität und entsprechend auch der größeren Permeabilität wesentlich größer ist als im
äußeren Bereich geringerer Durchläßigkeit, was mit den entsprechenden Druckverlusten
zusammenhängt.
Diese untersuchte Konstellation stellt ein extremes Beispiel dar sowohl wegen der Differenz der
Porositäten als auch wegen des Unstetigkeit des Übergangs zwischen den beiden Gebieten. Eine
solche Konstellation ist in der Realität so nicht zu erwarten und dient nur dazu, mögliche
Auswirkungen radialer Heterogenität zu demonstrieren.
a) b)
0
0,001
0,002
0,003
0,004
0,005
300 700 1100
Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
radialeHeterogenität
0
0,001
0,002
0,003
0,004
0,005
500 900 1300 1700
Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
radialeHeterogenität
Abbildung 6.9: simulierte Peaks einer nicht-adsorbierenden (a) und einer adsorbierenden (b) Substanz auf einer 10cm-Säule mit radialer Heterogenität (εinnen=0,45; εaußen=0,4; gleiches Volumen beider Bereiche)
Abbildung 6.9 zeigt die Auswirkungen dieses Festbettes auf das Chromatogramm bei einer nicht-
adsorbierenden und bei einer adsorbierenden Substanz. Es ist klar zu erkennen, daß die
unterschiedlichen Geschwindigkeiten innerhalb des Festbettes zu einem unterschiedlich schnellen
Transport der Substanz führt, was zur Ausbildung von jeweils zwei Peaks führt, wobei der vordere
Peak ein leichtes Tailung und der hintere Peak ein leichtes Fronting aufweist. Das ist durch
Diffusionsvorgänge über die Grenze zwischen beiden Zonen in den Bereich der jeweils anderen
Geschwindigkeit zu erklären ist. Die Tatsache, daß im Bereich größerer Porosität die Verzögerung
der adsorbierenden Komponente geringer ist (vgl. Gleichung (3.3)), verstärkt das Auseinander-
driften der Peaks noch zusätzlich.
Es scheint, daß diffusive Austauschvorgänge in radiale Richtung zumindest in Flüssigkeiten bei in
der Chromatographie benutzten Strömen eine untergeordnete Rolle spielen, da der
Konzentrationsausgleich zwischen den beiden Gebieten eher gering ist. Die Frage, die sich an

6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
107
diesem Punkt stellt ist, welche Rolle eine radiale Dispersion bei parallel zur Symmetrieachse
verlaufenden Stromlinien spielen könnte. Diese wird von FLUENT nicht erfaßt, da das poröse
Medium nicht wirklich als eine Ansammlung von Partikeln modelliert wird, sondern nur als Medium
mit geringerer Permeabilität.
6.4.2 Theoretische Abschätzung der radialen Dispersion
In der Literatur sind Aussagen hinsichtlich der Größe des radialen Dispersionskoeffizienten eher
selten anzutreffen: Baer (1972) beziffert das Verhältnis von axialer zu radialer Dispersion mit 5 bis
7 für ein porösen Medium, Knox et al. (1976) sowie Eon (1978) fanden heraus, daß die radiale
Dispersion in einer chromatographischen Säule mit Peclet-Zahlen von ca. 300 ungefähr 25 mal
kleiner ist als die axiale Dispersion. Allerdings berichtet Knox et al. (1976) von einer axialen und
radialen Heterogenität des Festbettes, was natürlich zu einer makroskopischen Geschwindigkeit in
radiale Richtung führen kann. Eine Messung der radialen Dispersion kann dadurch verfälscht
werden.
Abbildung 6.10: Prinzip der Berechnung der Wahrscheinlichkeitsverteilung für die Durchströmung des Festbettes
Bei der Frage welche Faktoren, neben der molekularen Diffusion und makroskopischen
Geschwindigkeiten, eine radiale Dispersion in einem chromatographischen Festbett beeinflussen
könnten, führt dies bei Betrachtung der Faktoren, die für die axiale Dispersion verantwortlich sind
(vgl. Unterabschnitt 2.4.4.1), nur zu einem Effekt, der vergleichbar ist mit der Eddy-Diffusion
Dieser resultiert also aus dem Weg, den die Moleküle um die Partikel zurücklegen müssen. Um
die Größe und damit den möglichen Einfluß eines solchen Effektes abschätzen zu können, sei
dieser im folgenden Gedankenmodell auf theoretischer Ebene zweidimensional und stark
vereinfacht betrachtet
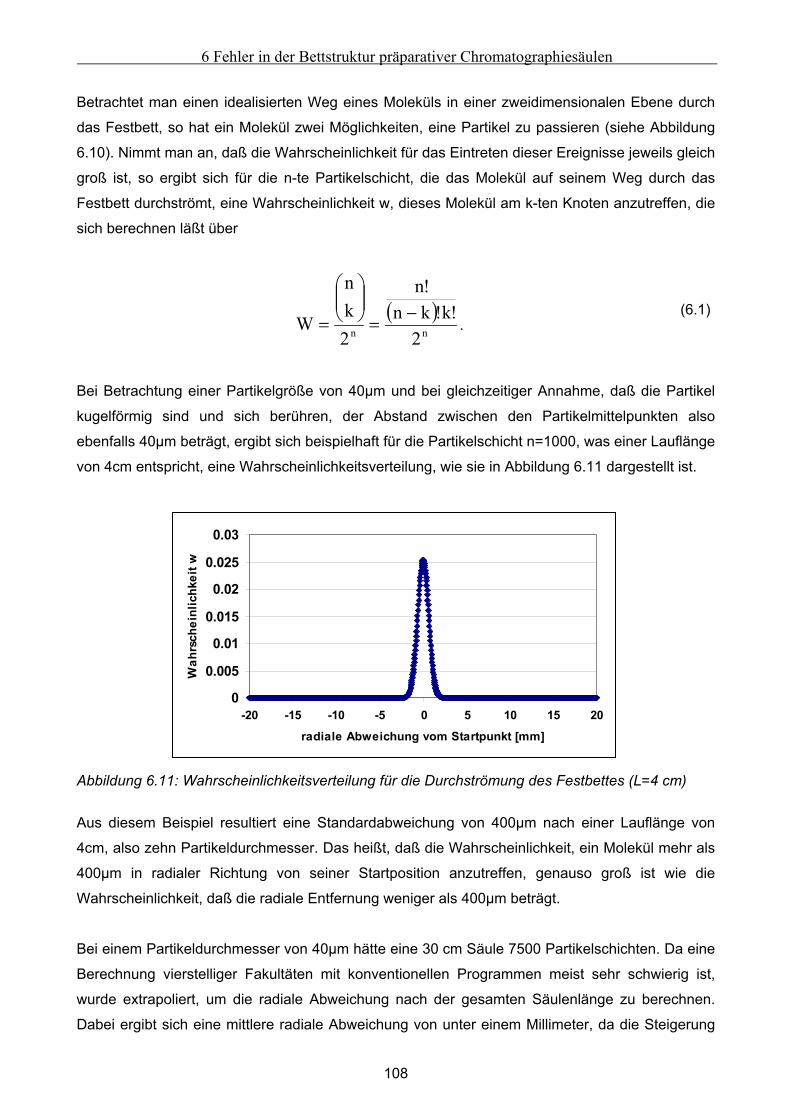
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
108
Betrachtet man einen idealisierten Weg eines Moleküls in einer zweidimensionalen Ebene durch
das Festbett, so hat ein Molekül zwei Möglichkeiten, eine Partikel zu passieren (siehe Abbildung
6.10). Nimmt man an, daß die Wahrscheinlichkeit für das Eintreten dieser Ereignisse jeweils gleich
groß ist, so ergibt sich für die n-te Partikelschicht, die das Molekül auf seinem Weg durch das
Festbett durchströmt, eine Wahrscheinlichkeit w, dieses Molekül am k-ten Knoten anzutreffen, die
sich berechnen läßt über
( )nn 2
!k!kn!n
2kn
W −=
= . (6.1)
Bei Betrachtung einer Partikelgröße von 40µm und bei gleichzeitiger Annahme, daß die Partikel
kugelförmig sind und sich berühren, der Abstand zwischen den Partikelmittelpunkten also
ebenfalls 40µm beträgt, ergibt sich beispielhaft für die Partikelschicht n=1000, was einer Lauflänge
von 4cm entspricht, eine Wahrscheinlichkeitsverteilung, wie sie in Abbildung 6.11 dargestellt ist.
0
0.005
0.01
0.015
0.02
0.025
0.03
-20 -15 -10 -5 0 5 10 15 20
radiale Abweichung vom Startpunkt [mm]
Wah
rsch
einl
ichk
eit w
Abbildung 6.11: Wahrscheinlichkeitsverteilung für die Durchströmung des Festbettes (L=4 cm)
Aus diesem Beispiel resultiert eine Standardabweichung von 400µm nach einer Lauflänge von
4cm, also zehn Partikeldurchmesser. Das heißt, daß die Wahrscheinlichkeit, ein Molekül mehr als
400µm in radialer Richtung von seiner Startposition anzutreffen, genauso groß ist wie die
Wahrscheinlichkeit, daß die radiale Entfernung weniger als 400µm beträgt.
Bei einem Partikeldurchmesser von 40µm hätte eine 30 cm Säule 7500 Partikelschichten. Da eine
Berechnung vierstelliger Fakultäten mit konventionellen Programmen meist sehr schwierig ist,
wurde extrapoliert, um die radiale Abweichung nach der gesamten Säulenlänge zu berechnen.
Dabei ergibt sich eine mittlere radiale Abweichung von unter einem Millimeter, da die Steigerung
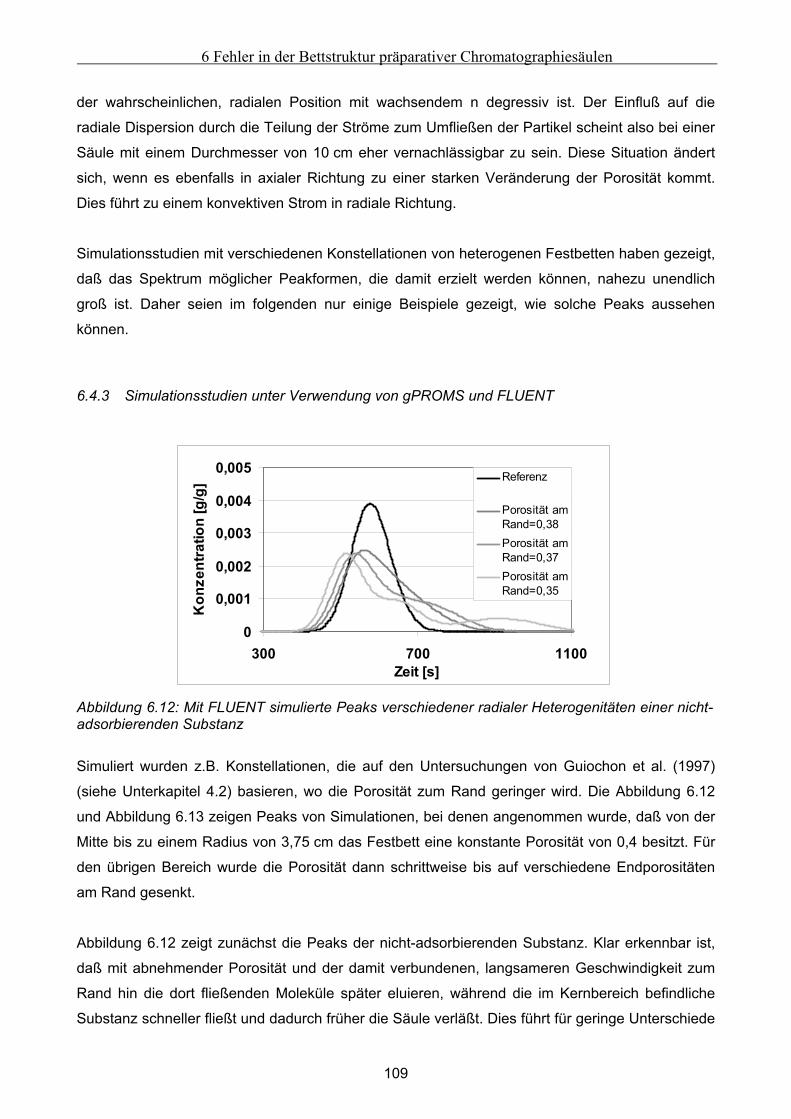
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
109
der wahrscheinlichen, radialen Position mit wachsendem n degressiv ist. Der Einfluß auf die
radiale Dispersion durch die Teilung der Ströme zum Umfließen der Partikel scheint also bei einer
Säule mit einem Durchmesser von 10 cm eher vernachlässigbar zu sein. Diese Situation ändert
sich, wenn es ebenfalls in axialer Richtung zu einer starken Veränderung der Porosität kommt.
Dies führt zu einem konvektiven Strom in radiale Richtung.
Simulationsstudien mit verschiedenen Konstellationen von heterogenen Festbetten haben gezeigt,
daß das Spektrum möglicher Peakformen, die damit erzielt werden können, nahezu unendlich
groß ist. Daher seien im folgenden nur einige Beispiele gezeigt, wie solche Peaks aussehen
können.
6.4.3 Simulationsstudien unter Verwendung von gPROMS und FLUENT
0
0,001
0,002
0,003
0,004
0,005
300 700 1100Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
Porosität amRand=0,38Porosität amRand=0,37Porosität amRand=0,35
Abbildung 6.12: Mit FLUENT simulierte Peaks verschiedener radialer Heterogenitäten einer nicht-adsorbierenden Substanz
Simuliert wurden z.B. Konstellationen, die auf den Untersuchungen von Guiochon et al. (1997)
(siehe Unterkapitel 4.2) basieren, wo die Porosität zum Rand geringer wird. Die Abbildung 6.12
und Abbildung 6.13 zeigen Peaks von Simulationen, bei denen angenommen wurde, daß von der
Mitte bis zu einem Radius von 3,75 cm das Festbett eine konstante Porosität von 0,4 besitzt. Für
den übrigen Bereich wurde die Porosität dann schrittweise bis auf verschiedene Endporositäten
am Rand gesenkt.
Abbildung 6.12 zeigt zunächst die Peaks der nicht-adsorbierenden Substanz. Klar erkennbar ist,
daß mit abnehmender Porosität und der damit verbundenen, langsameren Geschwindigkeit zum
Rand hin die dort fließenden Moleküle später eluieren, während die im Kernbereich befindliche
Substanz schneller fließt und dadurch früher die Säule verläßt. Dies führt für geringe Unterschiede
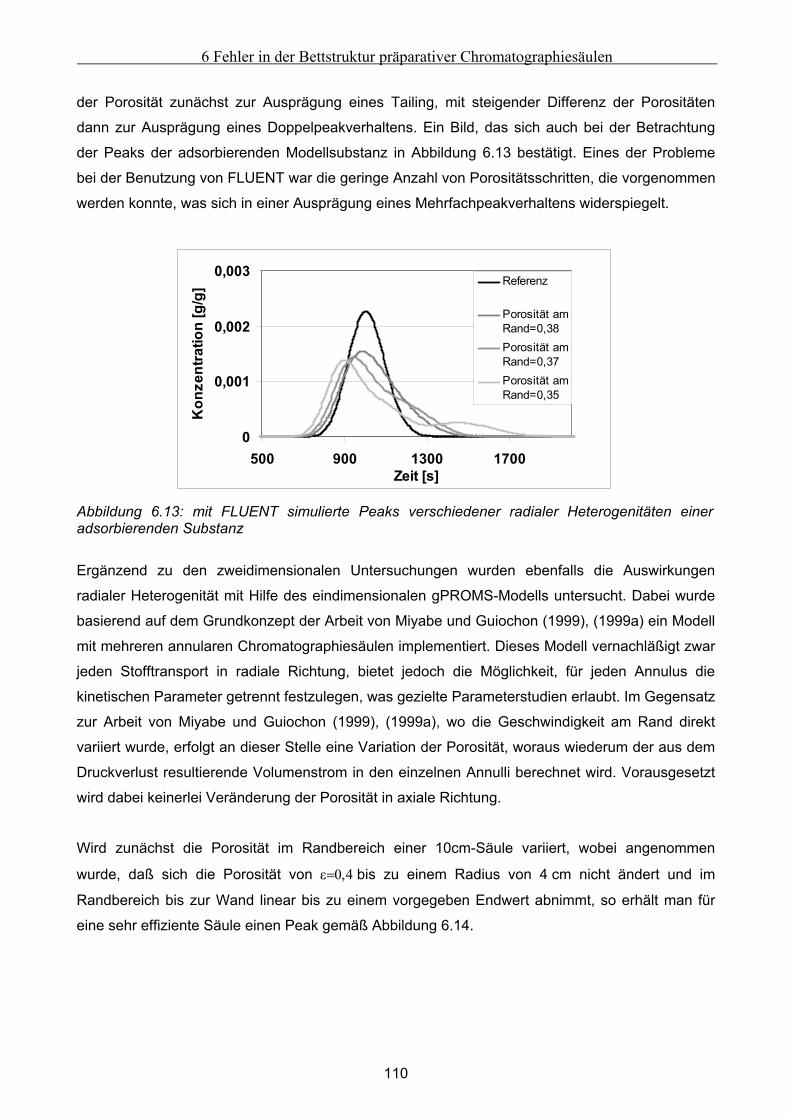
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
110
der Porosität zunächst zur Ausprägung eines Tailing, mit steigender Differenz der Porositäten
dann zur Ausprägung eines Doppelpeakverhaltens. Ein Bild, das sich auch bei der Betrachtung
der Peaks der adsorbierenden Modellsubstanz in Abbildung 6.13 bestätigt. Eines der Probleme
bei der Benutzung von FLUENT war die geringe Anzahl von Porositätsschritten, die vorgenommen
werden konnte, was sich in einer Ausprägung eines Mehrfachpeakverhaltens widerspiegelt.
0
0,001
0,002
0,003
500 900 1300 1700Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
Porosität amRand=0,38Porosität amRand=0,37Porosität amRand=0,35
Abbildung 6.13: mit FLUENT simulierte Peaks verschiedener radialer Heterogenitäten einer adsorbierenden Substanz
Ergänzend zu den zweidimensionalen Untersuchungen wurden ebenfalls die Auswirkungen
radialer Heterogenität mit Hilfe des eindimensionalen gPROMS-Modells untersucht. Dabei wurde
basierend auf dem Grundkonzept der Arbeit von Miyabe und Guiochon (1999), (1999a) ein Modell
mit mehreren annularen Chromatographiesäulen implementiert. Dieses Modell vernachläßigt zwar
jeden Stofftransport in radiale Richtung, bietet jedoch die Möglichkeit, für jeden Annulus die
kinetischen Parameter getrennt festzulegen, was gezielte Parameterstudien erlaubt. Im Gegensatz
zur Arbeit von Miyabe und Guiochon (1999), (1999a), wo die Geschwindigkeit am Rand direkt
variiert wurde, erfolgt an dieser Stelle eine Variation der Porosität, woraus wiederum der aus dem
Druckverlust resultierende Volumenstrom in den einzelnen Annulli berechnet wird. Vorausgesetzt
wird dabei keinerlei Veränderung der Porosität in axiale Richtung.
Wird zunächst die Porosität im Randbereich einer 10cm-Säule variiert, wobei angenommen
wurde, daß sich die Porosität von ε=0,4 bis zu einem Radius von 4 cm nicht ändert und im
Randbereich bis zur Wand linear bis zu einem vorgegeben Endwert abnimmt, so erhält man für
eine sehr effiziente Säule einen Peak gemäß Abbildung 6.14.
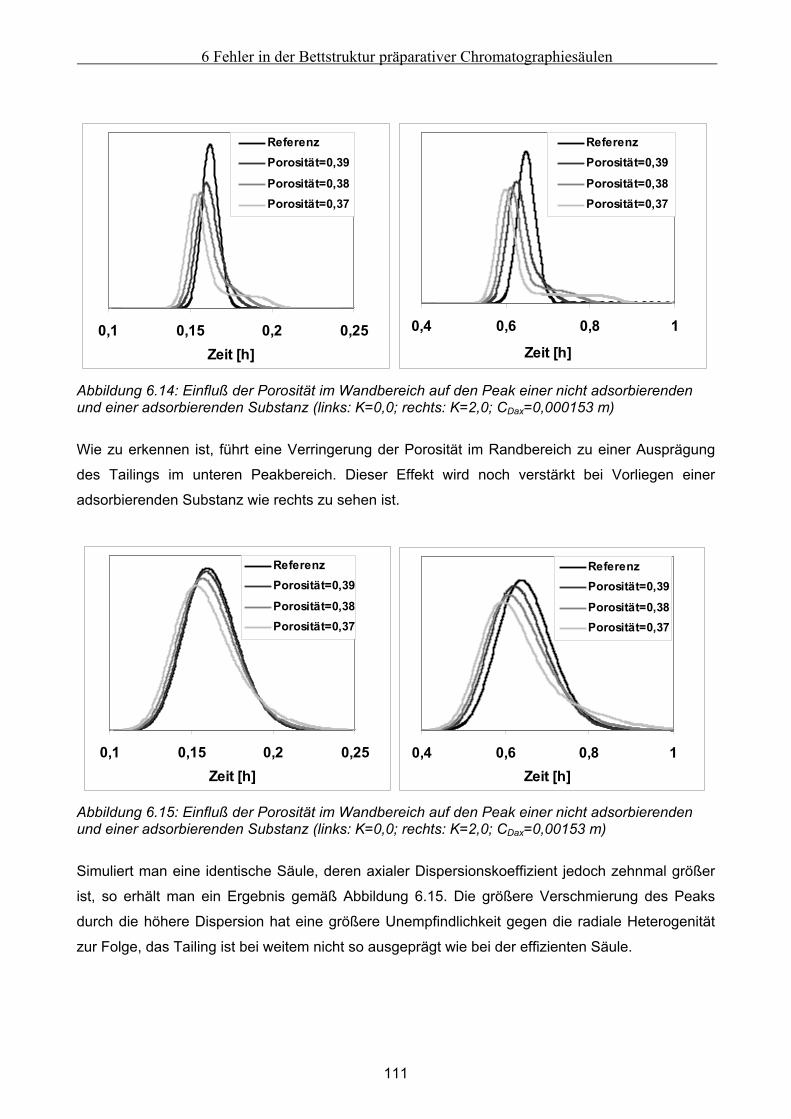
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
111
0,1 0,15 0,2 0,25Zeit [h]
ReferenzPorosität=0,39
Porosität=0,38Porosität=0,37
0,4 0,6 0,8 1
Zeit [h]
ReferenzPorosität=0,39
Porosität=0,38Porosität=0,37
Abbildung 6.14: Einfluß der Porosität im Wandbereich auf den Peak einer nicht adsorbierenden und einer adsorbierenden Substanz (links: K=0,0; rechts: K=2,0; CDax=0,000153 m)
Wie zu erkennen ist, führt eine Verringerung der Porosität im Randbereich zu einer Ausprägung
des Tailings im unteren Peakbereich. Dieser Effekt wird noch verstärkt bei Vorliegen einer
adsorbierenden Substanz wie rechts zu sehen ist.
0,1 0,15 0,2 0,25Zeit [h]
ReferenzPorosität=0,39
Porosität=0,38Porosität=0,37
0,4 0,6 0,8 1Zeit [h]
ReferenzPorosität=0,39
Porosität=0,38Porosität=0,37
Abbildung 6.15: Einfluß der Porosität im Wandbereich auf den Peak einer nicht adsorbierenden und einer adsorbierenden Substanz (links: K=0,0; rechts: K=2,0; CDax=0,00153 m)
Simuliert man eine identische Säule, deren axialer Dispersionskoeffizient jedoch zehnmal größer
ist, so erhält man ein Ergebnis gemäß Abbildung 6.15. Die größere Verschmierung des Peaks
durch die höhere Dispersion hat eine größere Unempfindlichkeit gegen die radiale Heterogenität
zur Folge, das Tailing ist bei weitem nicht so ausgeprägt wie bei der effizienten Säule.
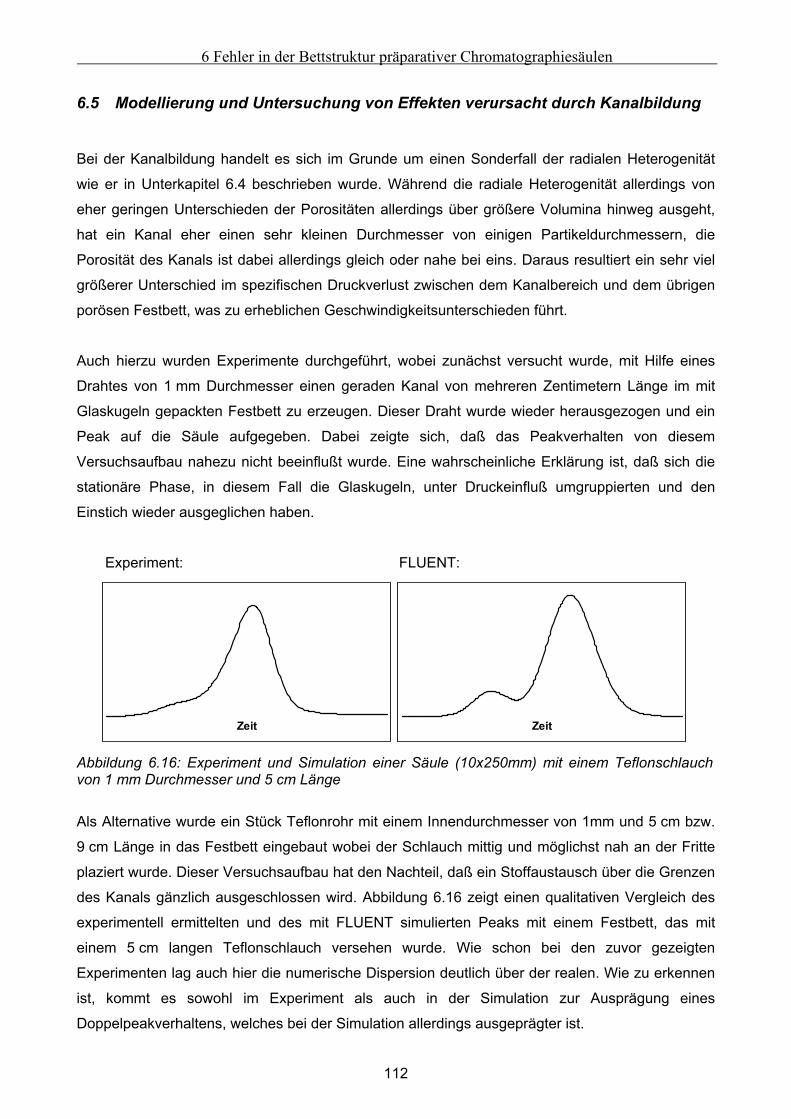
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
112
6.5 Modellierung und Untersuchung von Effekten verursacht durch Kanalbildung
Bei der Kanalbildung handelt es sich im Grunde um einen Sonderfall der radialen Heterogenität
wie er in Unterkapitel 6.4 beschrieben wurde. Während die radiale Heterogenität allerdings von
eher geringen Unterschieden der Porositäten allerdings über größere Volumina hinweg ausgeht,
hat ein Kanal eher einen sehr kleinen Durchmesser von einigen Partikeldurchmessern, die
Porosität des Kanals ist dabei allerdings gleich oder nahe bei eins. Daraus resultiert ein sehr viel
größerer Unterschied im spezifischen Druckverlust zwischen dem Kanalbereich und dem übrigen
porösen Festbett, was zu erheblichen Geschwindigkeitsunterschieden führt.
Auch hierzu wurden Experimente durchgeführt, wobei zunächst versucht wurde, mit Hilfe eines
Drahtes von 1 mm Durchmesser einen geraden Kanal von mehreren Zentimetern Länge im mit
Glaskugeln gepackten Festbett zu erzeugen. Dieser Draht wurde wieder herausgezogen und ein
Peak auf die Säule aufgegeben. Dabei zeigte sich, daß das Peakverhalten von diesem
Versuchsaufbau nahezu nicht beeinflußt wurde. Eine wahrscheinliche Erklärung ist, daß sich die
stationäre Phase, in diesem Fall die Glaskugeln, unter Druckeinfluß umgruppierten und den
Einstich wieder ausgeglichen haben.
Experiment: FLUENT:
Zeit Zeit
Abbildung 6.16: Experiment und Simulation einer Säule (10x250mm) mit einem Teflonschlauch von 1 mm Durchmesser und 5 cm Länge
Als Alternative wurde ein Stück Teflonrohr mit einem Innendurchmesser von 1mm und 5 cm bzw.
9 cm Länge in das Festbett eingebaut wobei der Schlauch mittig und möglichst nah an der Fritte
plaziert wurde. Dieser Versuchsaufbau hat den Nachteil, daß ein Stoffaustausch über die Grenzen
des Kanals gänzlich ausgeschlossen wird. Abbildung 6.16 zeigt einen qualitativen Vergleich des
experimentell ermittelten und des mit FLUENT simulierten Peaks mit einem Festbett, das mit
einem 5 cm langen Teflonschlauch versehen wurde. Wie schon bei den zuvor gezeigten
Experimenten lag auch hier die numerische Dispersion deutlich über der realen. Wie zu erkennen
ist, kommt es sowohl im Experiment als auch in der Simulation zur Ausprägung eines
Doppelpeakverhaltens, welches bei der Simulation allerdings ausgeprägter ist.
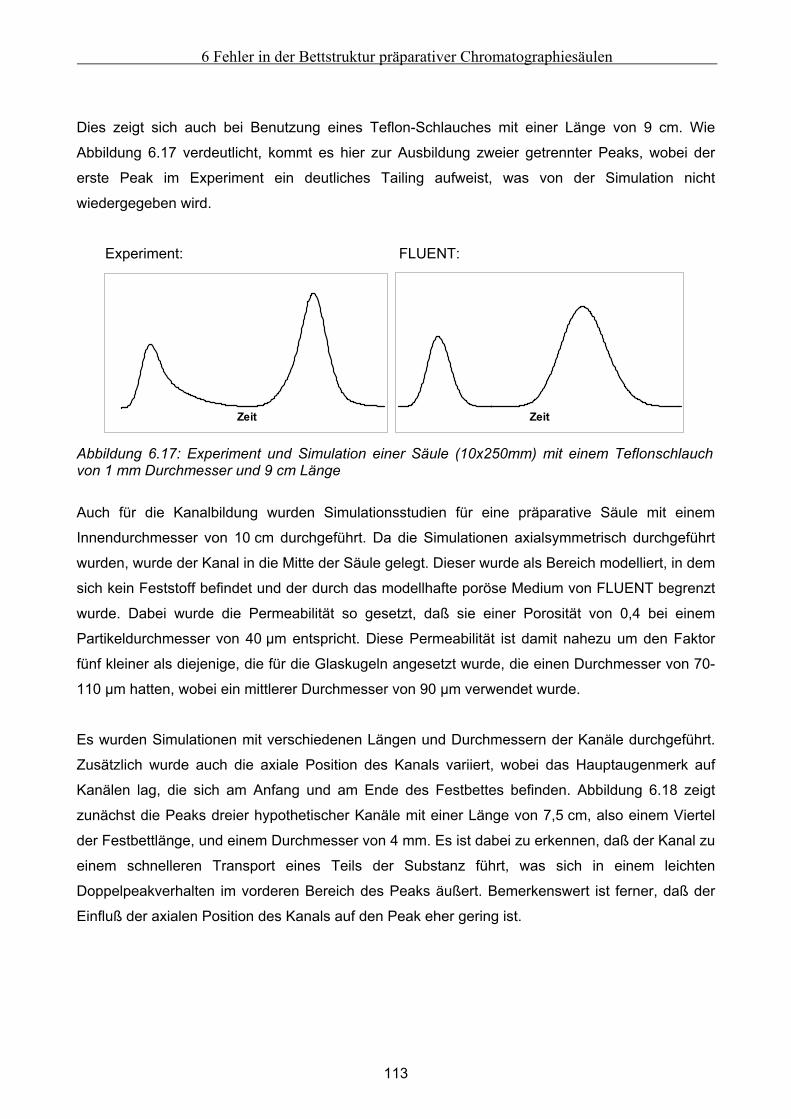
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
113
Dies zeigt sich auch bei Benutzung eines Teflon-Schlauches mit einer Länge von 9 cm. Wie
Abbildung 6.17 verdeutlicht, kommt es hier zur Ausbildung zweier getrennter Peaks, wobei der
erste Peak im Experiment ein deutliches Tailing aufweist, was von der Simulation nicht
wiedergegeben wird.
Experiment: FLUENT:
Zeit Zeit
Abbildung 6.17: Experiment und Simulation einer Säule (10x250mm) mit einem Teflonschlauch von 1 mm Durchmesser und 9 cm Länge
Auch für die Kanalbildung wurden Simulationsstudien für eine präparative Säule mit einem
Innendurchmesser von 10 cm durchgeführt. Da die Simulationen axialsymmetrisch durchgeführt
wurden, wurde der Kanal in die Mitte der Säule gelegt. Dieser wurde als Bereich modelliert, in dem
sich kein Feststoff befindet und der durch das modellhafte poröse Medium von FLUENT begrenzt
wurde. Dabei wurde die Permeabilität so gesetzt, daß sie einer Porosität von 0,4 bei einem
Partikeldurchmesser von 40 µm entspricht. Diese Permeabilität ist damit nahezu um den Faktor
fünf kleiner als diejenige, die für die Glaskugeln angesetzt wurde, die einen Durchmesser von 70-
110 µm hatten, wobei ein mittlerer Durchmesser von 90 µm verwendet wurde.
Es wurden Simulationen mit verschiedenen Längen und Durchmessern der Kanäle durchgeführt.
Zusätzlich wurde auch die axiale Position des Kanals variiert, wobei das Hauptaugenmerk auf
Kanälen lag, die sich am Anfang und am Ende des Festbettes befinden. Abbildung 6.18 zeigt
zunächst die Peaks dreier hypothetischer Kanäle mit einer Länge von 7,5 cm, also einem Viertel
der Festbettlänge, und einem Durchmesser von 4 mm. Es ist dabei zu erkennen, daß der Kanal zu
einem schnelleren Transport eines Teils der Substanz führt, was sich in einem leichten
Doppelpeakverhalten im vorderen Bereich des Peaks äußert. Bemerkenswert ist ferner, daß der
Einfluß der axialen Position des Kanals auf den Peak eher gering ist.
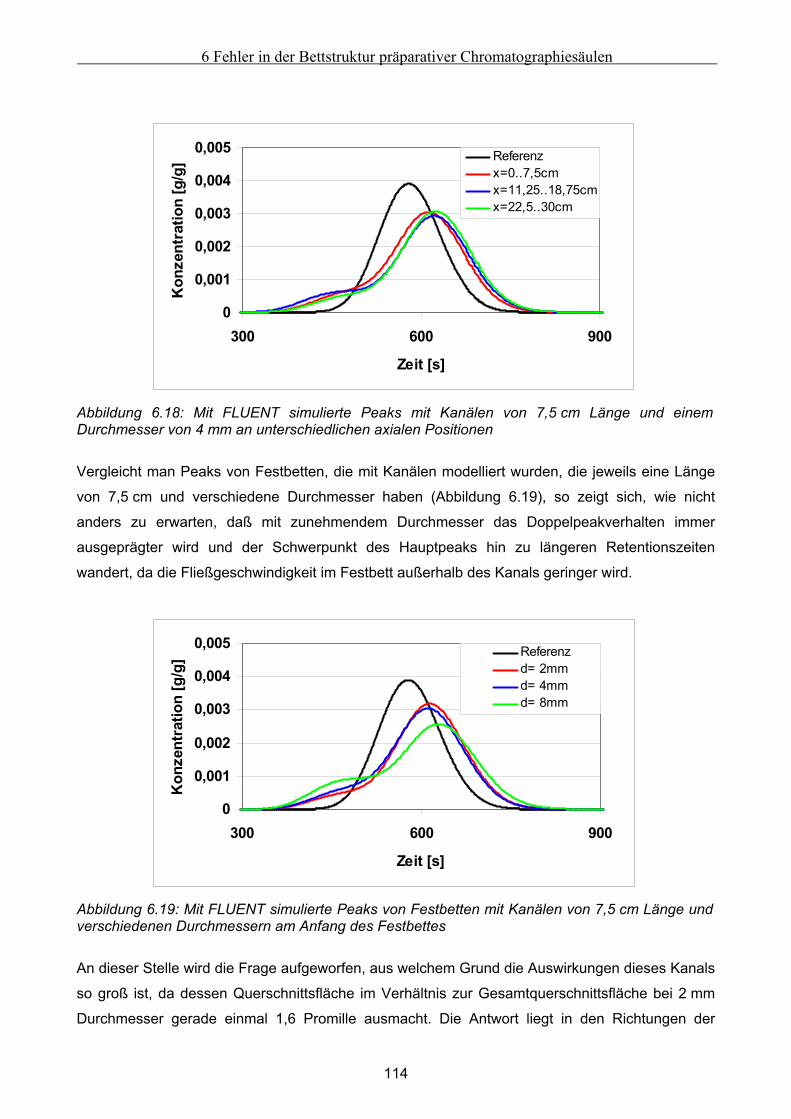
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
114
0
0,001
0,002
0,003
0,004
0,005
300 600 900
Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
x=0..7,5cmx=11,25..18,75cmx=22,5..30cm
Abbildung 6.18: Mit FLUENT simulierte Peaks mit Kanälen von 7,5 cm Länge und einem Durchmesser von 4 mm an unterschiedlichen axialen Positionen
Vergleicht man Peaks von Festbetten, die mit Kanälen modelliert wurden, die jeweils eine Länge
von 7,5 cm und verschiedene Durchmesser haben (Abbildung 6.19), so zeigt sich, wie nicht
anders zu erwarten, daß mit zunehmendem Durchmesser das Doppelpeakverhalten immer
ausgeprägter wird und der Schwerpunkt des Hauptpeaks hin zu längeren Retentionszeiten
wandert, da die Fließgeschwindigkeit im Festbett außerhalb des Kanals geringer wird.
0
0,001
0,002
0,003
0,004
0,005
300 600 900
Zeit [s]
Kon
zent
ratio
n [g
/g] Referenz
d= 2mmd= 4mmd= 8mm
Abbildung 6.19: Mit FLUENT simulierte Peaks von Festbetten mit Kanälen von 7,5 cm Länge und verschiedenen Durchmessern am Anfang des Festbettes
An dieser Stelle wird die Frage aufgeworfen, aus welchem Grund die Auswirkungen dieses Kanals
so groß ist, da dessen Querschnittsfläche im Verhältnis zur Gesamtquerschnittsfläche bei 2 mm
Durchmesser gerade einmal 1,6 Promille ausmacht. Die Antwort liegt in den Richtungen der
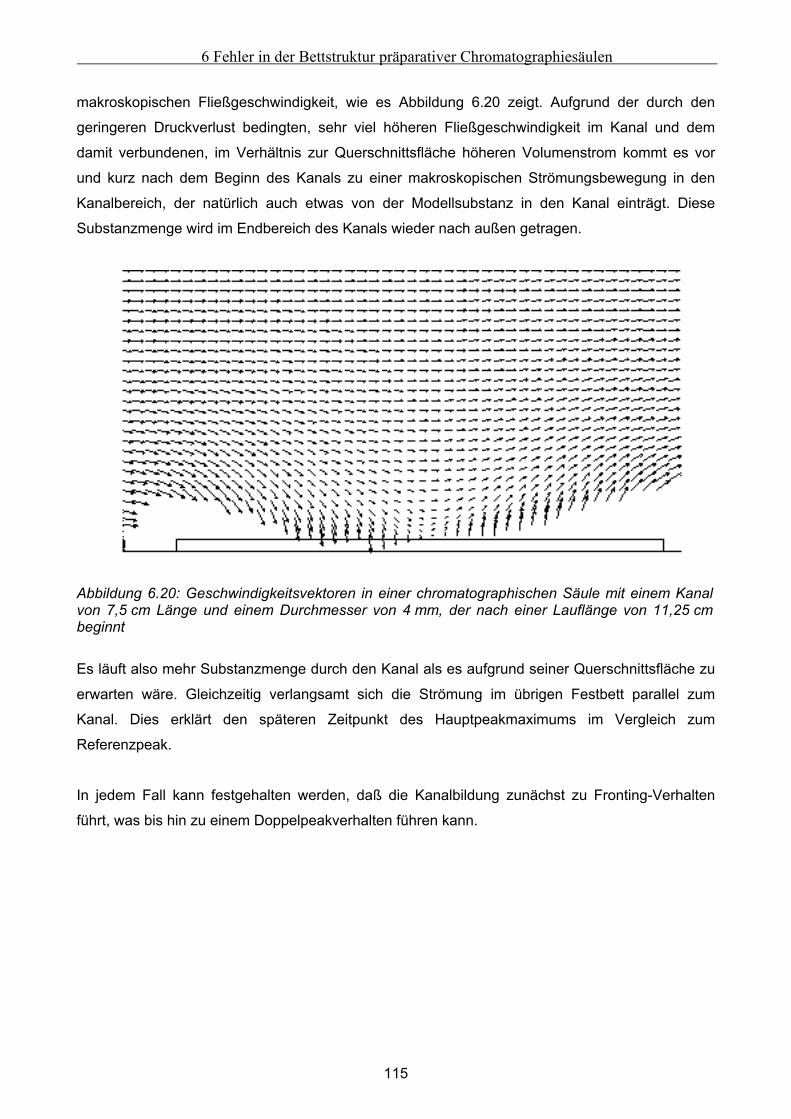
6 Fehler in der Bettstruktur präparativer Chromatographiesäulen
115
makroskopischen Fließgeschwindigkeit, wie es Abbildung 6.20 zeigt. Aufgrund der durch den
geringeren Druckverlust bedingten, sehr viel höheren Fließgeschwindigkeit im Kanal und dem
damit verbundenen, im Verhältnis zur Querschnittsfläche höheren Volumenstrom kommt es vor
und kurz nach dem Beginn des Kanals zu einer makroskopischen Strömungsbewegung in den
Kanalbereich, der natürlich auch etwas von der Modellsubstanz in den Kanal einträgt. Diese
Substanzmenge wird im Endbereich des Kanals wieder nach außen getragen.
Abbildung 6.20: Geschwindigkeitsvektoren in einer chromatographischen Säule mit einem Kanal von 7,5 cm Länge und einem Durchmesser von 4 mm, der nach einer Lauflänge von 11,25 cm beginnt
Es läuft also mehr Substanzmenge durch den Kanal als es aufgrund seiner Querschnittsfläche zu
erwarten wäre. Gleichzeitig verlangsamt sich die Strömung im übrigen Festbett parallel zum
Kanal. Dies erklärt den späteren Zeitpunkt des Hauptpeakmaximums im Vergleich zum
Referenzpeak.
In jedem Fall kann festgehalten werden, daß die Kanalbildung zunächst zu Fronting-Verhalten
führt, was bis hin zu einem Doppelpeakverhalten führen kann.

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
116
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
Wie bereits in Abschnitt 2.6 angedeutet, handelt es sich bei Anlagen zur Simulierten Gegen-
stromchromatographie im allgemeinen um relativ komplexe Systeme, die aus einer ganzen Reihe
von Komponenten bestehen, die natürlich auch, wie es in der technischen Natur der Dinge liegt,
partiell oder vollständig ihre Funktion versagen können.
Ziel dieses Kapitels ist es, die Auswirkungen des Versagens einzelner Komponenten auf das
Trennverhalten auf phänomenologischer Ebene darzustellen und daraus allgemeine Gesetz-
mäßigkeiten abzuleiten, wobei nicht nur die thermodynamischen Grundlagen der SMB, sondern
auch dynamische Simulationen der gesamten Anlage unter Verwendung von gPROMS genutzt
werden. Dabei ist zum einen natürlich von Interesse, wie sich bestimmte Fehler auf die axialen
Konzentrationsprofile der in der Anlage zu trennenden Komponenten auswirken, zum anderen
aber auch, wie sich diese Fehlfunktionen auf die online verfügbaren Antwortsignale auswirken,
nämlich die zeitlichen Konzentrationsverläufe in Extrakt und Raffinat.
Untersucht wurde dabei beispielhaft, wie sich Fehler auf die SMB-Anlage auswirken würden, die
sich am Fachgebiet Thermodynamik und thermische Verfahrenstechnik der TU Berlin befindet.
Dies hat den Grund, daß von dieser Anlage alle notwendigen Daten für die Simulationen
problemlos verfügbar waren. Daher wird in Unterkapitel 7.1 zunächst diese Anlage vorgestellt,
soweit es für das weitere Vorgehen relevant ist. Die Klassifizierung möglicher Fehlfunktionen
sowie deren Modellierung und deren charakteristische Auswirkungen auf das Verhalten der SMB
steht im Mittelpunkt von Unterkapitel 7.2, woran sich in Unterkapitel 7.3 die Erläuterung eines
vereinfachten TMB-Bilanzmodells zur Modellierung von globalen Fehlfunktionen anschließt.
Unterkapitel 7.4 vergleicht die Ergebnisse dieses Modells mit den dynamischen Simulationen und
zeigt Charakteristika der einzelnen Fehlerarten, Unterkapitel 7.5 zeigt Simulationen von lokalen
Fehlfunktionen. Schließlich werden in Unterkapitel 7.6 einige Überlegungen zur möglichen
Verwendung der Ergebnisse im Rahmen einer Fehlererkennung diskutiert.
7.1 Beschreibung der beispielhaft zur Untersuchung herangezogenen SMB-Anlage
Die hypothetische Anlage, die zur Simulation möglicher Fehler beispielhaft verwendet wurde,
orientiert sich weitgehend an der Pilotanlage am Fachgebiet Thermodynamik und thermische
Verfahrenstechnik, wie sie im Rahmen der Arbeit von Deckert (1997) und Deckert und Arlt (1997)

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
117
entwickelt und aufgebaut wurde, und deren Schaltbild in Abbildung 7.1 dargestellt ist. Es handelt
sich dabei um eine Anlage mit den 8 Säulen S1-S8 (ls=499 mm, ds=26 mm), die über Ver-
bindungsleitungen zu einem Kreis verschaltet sind. Die Thermostatisierung der Säulen erfolgt zum
einen über eine Doppelmantelung der Säulen, die über zwei Thermostate TH1 und TH2 ange-
schlossen ist, zum anderen aber auch über die den Säulen vorgeschalteten Wärmeübertrager, die
sicherstellen, daß das in die Säulen eintretende Fluid die gewünschte Temperatur besitzt.
Über vier Kolbenmembranpumpen P1-P4 werden die Prozeßströme Feed, Eluent in Abschnitt I,
Extrakt und Raffinat dosiert. Das Umsetzen der Zu- und Ablaufstellen der äußeren Ströme nach
Ablauf der Taktzeit erfolgt mit Hilfe von 2-Weg-Magnetventilen. Jeder der vier äußeren Ströme
kann zwischen zwei aufeinanderfolgenden Säulen zugeführt bzw. entnommen werden, indem
durch das Öffnen eines Magnetventils (VF, VD, VR, VE, je achtmal) eine Verbindung zwischen der
zentralen Verteilungsleitung und der Verbindungsleitung der Säulen geschaffen werden. Für das
Umsetzen der Ströme werden also entsprechend vier Magnetventile pro Säule benötigt.
Die hypothetische Anlage unterscheidet sich jedoch in zwei Punkten von der Pilotanlage: Zum
einen wird angenommen, daß alle Verbindungsleitungen zwischen den einzelnen Säulen exakt
gleich lang sind, die Säulen also quasi im Kreis angeordnet sind. Bei der Pilotanlage sind die
Säulen in Reihe angeordnet, so daß die Leitung zwischen Säule S8 und Säule S1 sehr viel länger
ist als die zwischen den übrigen Säulen, was zu kleinen Variationen der Minimalwerte des
zeitlichen Konzentrationsprofils in den Produktströmen während des Zyklus führt. Da der
Ausgangspunkt für die Untersuchungen jedoch die Identität aller Takte sein soll, ist diese
Divergenz von der Pilotanlage ein Imperativ.
Der zweite Unterschied zwischen Simulation und Pilotanlage resultiert gleichermaßen aus der
Reihenverschaltung der Säulen. Da sich der Auslaß der Produktströme hinter der Säule S8
befindet, die Verbindungsleitung also bei S1 startet, kommt es beim Umschalten der Produkt-
ströme zur nächsten Säule zu einer Konstellation, daß etwas vom Produktstrom in dem Leitungs-
stück zwischen aktuellem und vorherigem Produktstrom verbleibt, das sich dann im quasi toten
Ende der Leitung befindet. Beim Umschalten des Produktstromes von den Ventilen VE8 bzw. VR8
zu den Ventilen VE1 bzw. VR1 wird das gesamte Produkt, das sich vorher in der Leitung befand,
auf einen Schlag durch die Detektoren in die Auffangbehälter gefördert, was zu einem zusätz-
lichen Peak im Detektorsignal führt. Dieser Effekt wurde in den Simulationen ausgeschlossen.
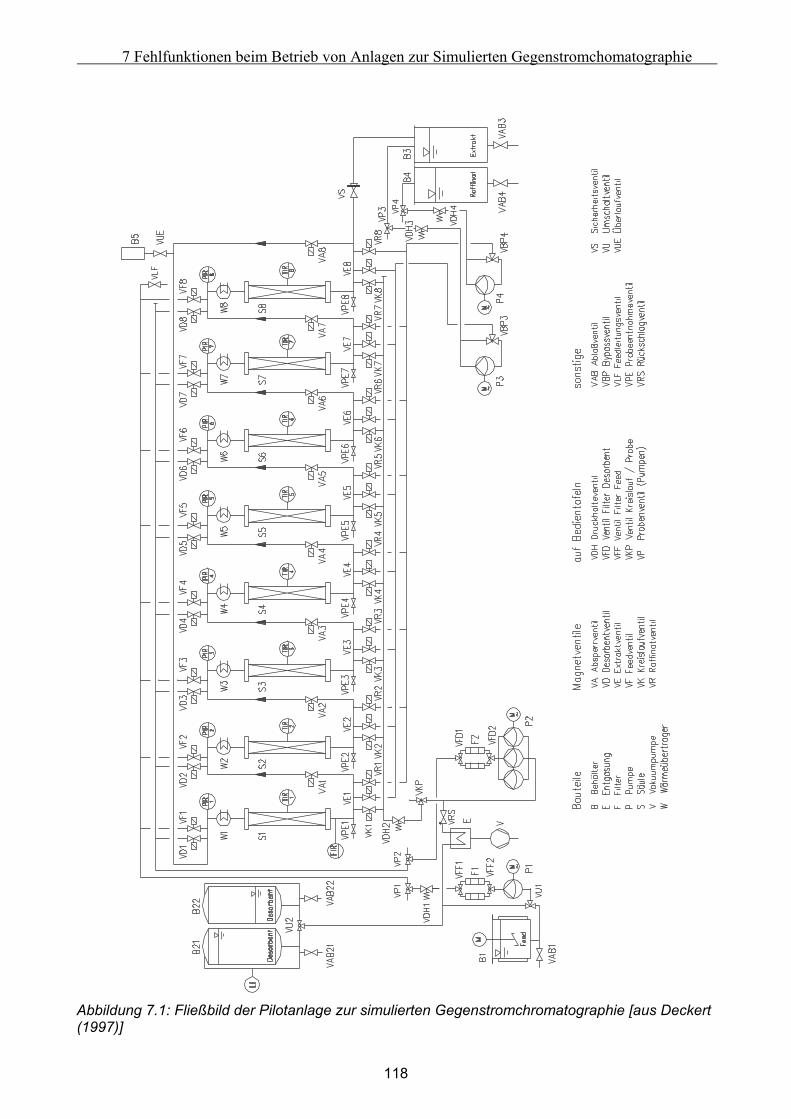
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
118
Abbildung 7.1: Fließbild der Pilotanlage zur simulierten Gegenstromchromatographie [aus Deckert (1997)]

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
119
7.2 Klassifizierung von Fehlern bei der Simulierten Gegenstromchromatographie
In den theoretischen und simulativen Untersuchungen konnten zwei Klassen auftretender
Fehlfunktionen ausgemacht werden: Zum einen gibt es Fehler, die sich mit dem TMB-Modell
beschreiben lassen, also bei einer hypothetischen TMB ebenso auftreten können. Diese Fehler
zeichnen sich dadurch aus, daß sie sich auf die gesamte Anlage und in jedem Takt gleichartig
auswirken, weil sie quasi beim Umtakten mit den Komponenten mitwandern und sich somit immer
im selben Abschnitt befinden. Aufgrund dieses Charakters werden diese Fehlfunktionen im
folgenden als globale Fehlfunktionen bezeichnet. Untersuchte Beispiele hierfür sind Fehl-
funktionen der Pumpen, die konstant vom Sollwert abweichende Volumenströme fördern. Weitere
Beispiele sind Auslegungsfehler resultierend aus fehlerhaft vermessen Adsorptionsisothermen
oder kinetischen Parametern.
Abbildung 7.2: Klassifizierung der untersuchten Fehlern bei der simulierten Gegenstromchroma-tographie
Im Gegensatz dazu stehen Fehler, die im umgetakteten Bereich lokal fest sind und sich durch das
Umtakten durch die verschieden Abschnitte bewegen. Dies bedingt, daß sich die Auswirkungen in
den unterschiedlichen Takten zum Teil erheblich unterscheiden können. Aufgrund der Tatsache,
daß diese Fehler physisch immer an der selben Stelle sind, werden sie nachfolgend als lokale
Fehler bezeichnet. Untersuchte Beispiele hierfür sind Fehlfunktionen der Ventile, was sowohl die
Nicht-Öffnung als auch Undichtigkeiten im geschlossenen Zustand beinhaltet. Dazu kommen
Leckagen in den Leitungen sowie Fehler in den Säulen, wobei in dieser Arbeit eine erhöhte
Dispersion in einer Säule modelliert wird.

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
120
Da bei der TMB diese Fehler aufgrund des fehlenden Umtaktens nicht vorkommen können, sind
diese mit den einfacheren Gleichungen für das TMB-Verhalten nicht zu beschreiben, sondern
müssen direkt dynamisch mit gPROMS simuliert werden.
Ziel sowohl der theoretischen Modellierung und der Simulationen ist die Darstellung allgemeiner
Gesetzmäßigkeiten für einen quasistationären Zustand auch unter dem Gesichtspunkt, wie diese
Ergebnisse für eine mögliche Fehlererkennung genutzt werden könnten. Dazu wird untersucht,
wie sich diese Fehler auf das Konzentrationprofil der Gesamtanlage auswirken, wobei sich die
Betrachtung auf lineare Adsorptionsisothermen beschränkt. Zusätzlich ist von Interesse, wie sich
diese Fehler auf den zeitlichen Konzentrationsverlauf in den Produktströmen auswirken.
7.3 Entwicklung eines vereinfachten integralen Bilanzmodells
Ziel dieses Unterkapitels ist es, die Grundlagen zur Untersuchung der Gesetzmäßigkeiten beim
Auftreten globaler Fehlfunktionen herauszuarbeiten. Da es für diese charakteristisch ist, daß sie
auch bei einer TMB auftreten können, können mittels einer integralen Komponentenbilanz an einer
TMB unter Vernachläßigung der kinetischen Einflußparameter Dispersion und Stofftransport-
widerstand die prinzipiellen Gesetzmäßigkeiten dargestellt werden. Da die durchgeführten
Simulationen sowohl die thermodynamischen als auch die kinetischen Einflüsse beinhaltet, kann
durch das nachfolgend vorgestellte Modell, welches aus einer integralen Massenbilanz um die
TMB resultiert und welches im folgenden der Kürze halber einfach als TMB -Bilanzmodell
bezeichnet wird, eine Entkoppelung von Thermodynamik und Kinetik durchgeführt werden. Dies ist
allerdings nur bei der Verwendung linearer Adsorptionsisothermen möglich.
Von Interesse für die Untersuchungen sind dabei die Konzentrationen in den Auslaßströmen
sowie die rechnerischen Maximal- bzw. Minimalkonzentrationen in den einzelnen Abschnitten, da
diese durch die Wanderung des Konzentrationsprofils über die Auslaßstellen im Verlauf ein Taktes
von Relevanz sind.
Für die TMB sind formell drei verschiedene Fälle dominierender Transportrichtungen zu unter-
scheiden, die in den folgenden Abschnitten dargestellt werden: Im ersten Fall wird die
Komponente vom Feed jeweils auschließlich zu einem der Produktströme transportiert (Abschnitt
7.3.1). Dies entspricht dem Fall, wie er in der Regel vom Anwender vorgesehen ist. Abschnitt 7.3.2
schildert die Konstellation, bei der eine Komponente vom Feed aus sowohl in Richtung des
Extraktes als auch des Raffinats transportiert wird, ohne daß es zu einem Transport der
Komponente über die Grenze zwischen Abschnitt I und IV hinweg kommt. Schließlich gibt es noch
den Fall, daß die Komponente in jedem Abschnitt in dieselbe Richtung und damit über die Grenze

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
121
zwischen Abschnitt I und IV fließt, was Abschnitt 7.3.3 beschreibt. Dabei werden nur Gleichungen
zur Konzentrationsbestimmung präsentiert, die Herleitung dieser Gleichungen findet man in
Anhang E.
7.3.1 Komponententransport durch nur einen Abschnitt
A A A AB B B B
ZS 2 ZS 3 ZS 4ZS 1
I II III IV
II,Ac
III,BcEx,Ac Raff,BcFeed,Ac
festV&
I,flV& II,flV& III,flV& IV,flV&
festV&festV&festV&
Abbildung 7.3: Komponententransport durch nur einen Abschnitt (Komponente A durch Abschnitt II, Komponente B durch Abschnitt III)
Abbildung 7.3 zeigt den Komponententransport in der TMB, wenn die dominierenden
Transportrichtungen so gegeben sind, daß die Komponenten vom Feed jeweils zu genau einem
Produktstrom transportiert wird. Im Regelfall ist dies der angestrebte Zustand. Leitet man eine
integrale Massenbilanz zur Berechnung der Konzentrationen in den Abschnitten und den Produkt-
strömen her, so erhält man für die Konzentration von Komponente A im Abschnitt II
AfestIl,fl
Feed,AFeedII,A KVV
cVc
&&
&
−= (7.1)
und für die Konzentration von Komponente B in Abschnitt III
BfestIII,fl
Feed,BFeedIII,B KVV
cVc
&&
&
−= .
(7.2)
Die Nenner entsprechen dabei den effektiven Transportvolumenströmen der Komponenten in der
TMB. Die Konzentration von Komponente A im Extrakt ergibt sich zu
Ex
Feed,AFeedEx,A V
cVc
&
&= ,
(7.3)
die von Komponente B im Raffinat zu

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
122
Raff
Feed,BFeedRaff,B V
cVc
&
&= .
(7.4)
Die Herleitung dieser Gleichungen findet man in Anhang E.1.
7.3.2 Komponententransport in zwei Richtungen
A A A AZS 2 ZS 3 ZS 4ZS 1
I II III IV
II,Ac
Ex,Ac
III,Ac
Raff,AcFeed,Ac
festV&festV&festV&festV&
I,flV& II,flV& III,flV& IV,flV&
Abbildung 7.4: Komponententransport von Komponente A durch die Abschnitte II und III
Erhöht sich der Flüssigvolumenstrom in Abschnitt III, tritt irgendwann der Fall ein, daß
Komponente A nicht nur in Abschnitt II mit dem Feststoff in Richtung des Extraktes, sondern auch
in Abschnitt III mit der Flüssigkeit in Richtung des Raffinates transportiert wird, was Abbildung 7.4
verdeutlicht. Dabei liegen die Volumenströme in den Abschnitten I und IV nach wie vor so, daß
kein Weitertransport durch diese Abschnitte stattfindet.
Möchte man die Konzentrationen von Komponente A in den Abschnitten II und III berechnen, so
ist mathematisch beweisbar, daß gilt:
Feed,AIII,AII,A ccc == . (7.5)
Die Konzentration von A im Extrakt berechnet mit:
Ex
II,AEx,A V
mc
&
&= , (7.6)
die im Raffinat mit
Raff
III,ARaff,A V
mc
&
&= . (7.7)
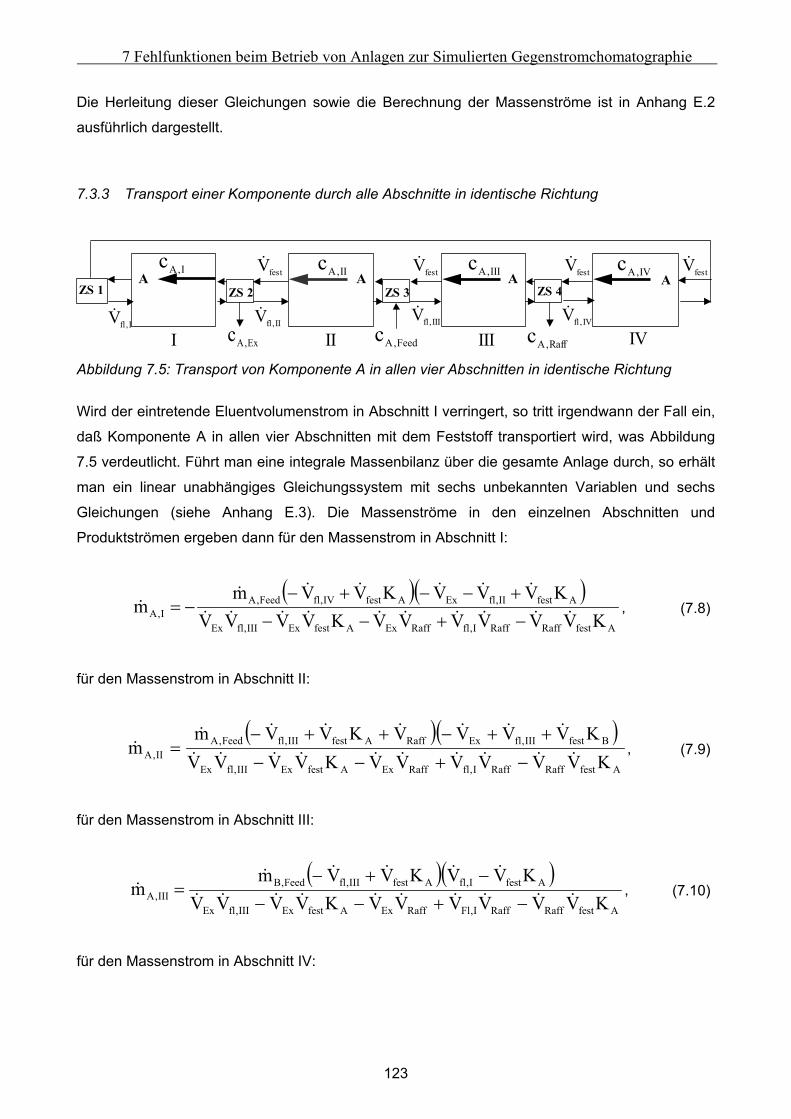
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
123
Die Herleitung dieser Gleichungen sowie die Berechnung der Massenströme ist in Anhang E.2
ausführlich dargestellt.
7.3.3 Transport einer Komponente durch alle Abschnitte in identische Richtung
A A A AZS 2 ZS 3 ZS 4ZS 1
I II III IV
II,Ac
Ex,Ac
I,AcIII,Ac IV,Ac
Raff,AcFeed,Ac
festV&festV&festV&festV&
I,flV& II,flV& III,flV& IV,flV&
Abbildung 7.5: Transport von Komponente A in allen vier Abschnitten in identische Richtung
Wird der eintretende Eluentvolumenstrom in Abschnitt I verringert, so tritt irgendwann der Fall ein,
daß Komponente A in allen vier Abschnitten mit dem Feststoff transportiert wird, was Abbildung
7.5 verdeutlicht. Führt man eine integrale Massenbilanz über die gesamte Anlage durch, so erhält
man ein linear unabhängiges Gleichungssystem mit sechs unbekannten Variablen und sechs
Gleichungen (siehe Anhang E.3). Die Massenströme in den einzelnen Abschnitten und
Produktströmen ergeben dann für den Massenstrom in Abschnitt I:
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
AfestII,flExAfestIV,flFeed,AI,A KVVVVVVKVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
−+−−+−−+−
−= , (7.8)
für den Massenstrom in Abschnitt II:
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
BfestIII,flExRaffAfestIII,flFeed,AII,A KVVVVVVKVVVV
KVVVVKVVmm
&&&&&&&&&&
&&&&&&&&
−+−−
++−++−= , (7.9)
für den Massenstrom in Abschnitt III:
( )( )AfestRaffRaffI,FlRaffExAfestExIII,flEx
AfestI,flAfestIII,flFeed,BIII,A KVVVVVVKVVVV
KVVKVVmm
&&&&&&&&&&
&&&&&&
−+−−−+−
= , (7.10)
für den Massenstrom in Abschnitt IV:
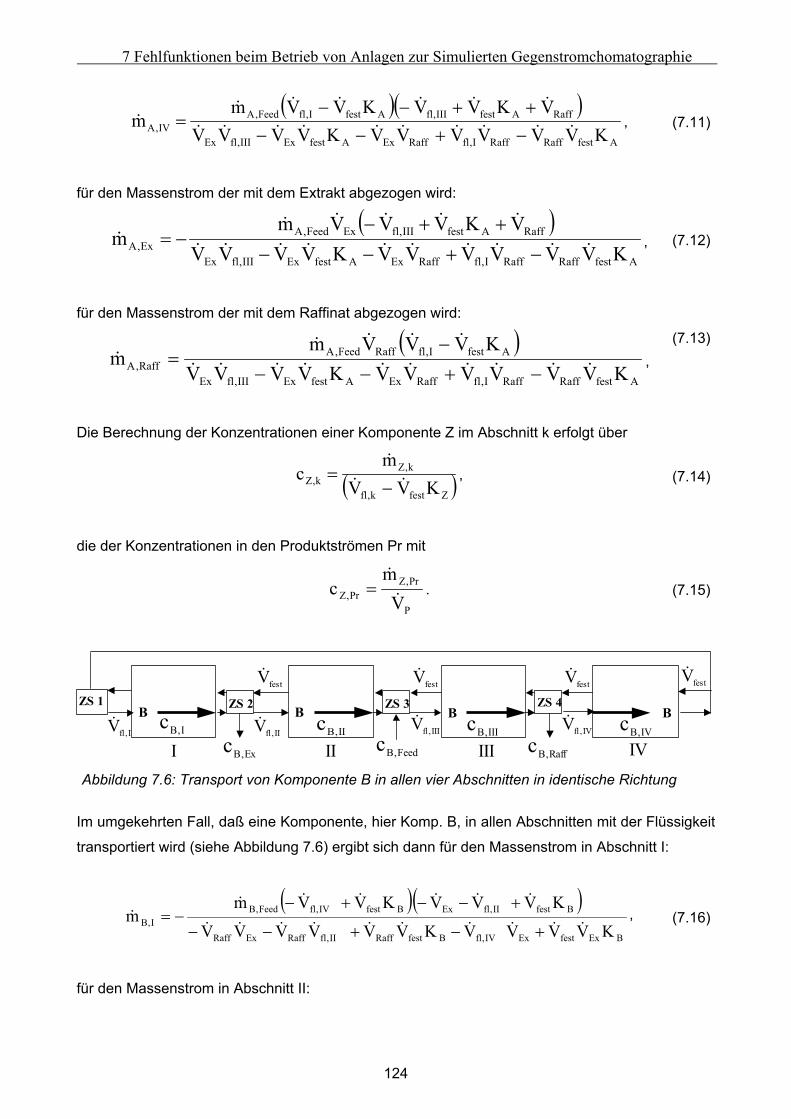
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
124
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
RaffAfestIII,flAfestI,flFeed,AIV,A KVVVVVVKVVVV
VKVVKVVmm
&&&&&&&&&&
&&&&&&&
−+−−++−−
= , (7.11)
für den Massenstrom der mit dem Extrakt abgezogen wird:
( )AfestRaffRaffI,flRaffExAfestExIII,flEx
RaffAfestIII,flExFeed,AEx,A KVVVVVVKVVVV
VKVVVmm
&&&&&&&&&&
&&&&&&
−+−−++−
−= , (7.12)
für den Massenstrom der mit dem Raffinat abgezogen wird:
( )AfestRaffRaffI,flRaffExAfestExIII,flEx
AfestI,flRaffFeed,ARaff,A KVVVVVVKVVVV
KVVVmm
&&&&&&&&&&
&&&&&
−+−−−
= , (7.13)
Die Berechnung der Konzentrationen einer Komponente Z im Abschnitt k erfolgt über
( )Zfestk,fl
k,Zk,Z KVV
mc
&&
&
−= , (7.14)
die der Konzentrationen in den Produktströmen Pr mit
P
Pr,ZPr,Z V
mc
&
&= . (7.15)
B B B BZS 2 ZS 3 ZS 4ZS 1
I II III IVIII,BcII,Bc IV,BcI,Bc
Ex,Bc Feed,Bc Raff,Bc
festV&festV&festV&festV&
I,flV& II,flV& III,flV& IV,flV&
Abbildung 7.6: Transport von Komponente B in allen vier Abschnitten in identische Richtung
Im umgekehrten Fall, daß eine Komponente, hier Komp. B, in allen Abschnitten mit der Flüssigkeit
transportiert wird (siehe Abbildung 7.6) ergibt sich dann für den Massenstrom in Abschnitt I:
( )( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flExBfestIV,flFeed,BI,B KVVVVKVVVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
+−+−−
+−−+−−= , (7.16)
für den Massenstrom in Abschnitt II:

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
125
( ) ( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flFeed,BBfestIV,flII,B KVVVVKVVVVVV
KVVmKVVm
&&&&&&&&&&
&&&&&&
+−+−−
+−+−= , (7.17)
für den Massenstrom in Abschnitt III:
( )( )BExfestExIV,flBfestRaffII,flRaffExRaff
RaffBfestIV,flBfestfl,IIExFeed,BIII,B KVVVVKVVVVVV
VKVVKVVVmm
&&&&&&&&&&
&&&&&&&&
+−+−−
−+−+−−−= , (7.18)
für den Massenstrom in Abschnitt IV:
( )( )BExfestExIV,flBfestRaffII,flRExRaff
BfestII,flExBfestIV,flFeed,BIVB KVVVVKVVVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
+−+−−+−−+−
−= . (7.19)
Für den Massenstrom von Komponente B, der mit dem Extrakt abgezogen wird:
( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestIV,flExFeed,BEx,B KVVVVKVVVVVV
KVVVmm
&&&&&&&&&&
&&&&&
+−+−−+−
= , (7.20)
und der Massenstrom, der mit dem Raffinat abgezogen wird:
( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flExRaffFeed,BRaff,B KVVVVKVVVVVV
KVVVVmm
&&&&&&&&&&
&&&&&&
+−+−−+−−
=
.
(7.21)
Die Umrechnung in Konzentrationen erfolgt mit den Gleichungen (7.14) bzw. (7.15).
7.4 Untersuchung der Effekte ausgelöst durch globale Fehlfunktionen
In diesem Unterkapitel wird untersucht, wie sich globale Fehlfunktionen auf das Konzentrations-
profil und den zeitlichen Verlauf der Konzentrationen in den Produktströmen einer SMB-Anlage
auswirken. Dabei werden die Ergebnisse, die mit Hilfe des TMB-Bilanzmodells aus Unterkapitel
7.3 berechnet werden, mit SMB-Simulationsergebnissen verglichen, die durch gPROMS mit unter-
schiedlich stark zugrunde gelegten, kinetischen Effekten gerechnet wurden. Das TMB-Bilanz-
modell vernachlässigt dabei die kinetischen Effekte Dispersion und Stofftransportwiderstand. Das
Ziel dabei ist vor allem die Effekte auf die Konzentrationen in den vier Abschnitten und den beiden
Produktströmen zu untersuchen, aber auch, welchen Einfluß die Größe der beiden kinetischen
Parameter hat.
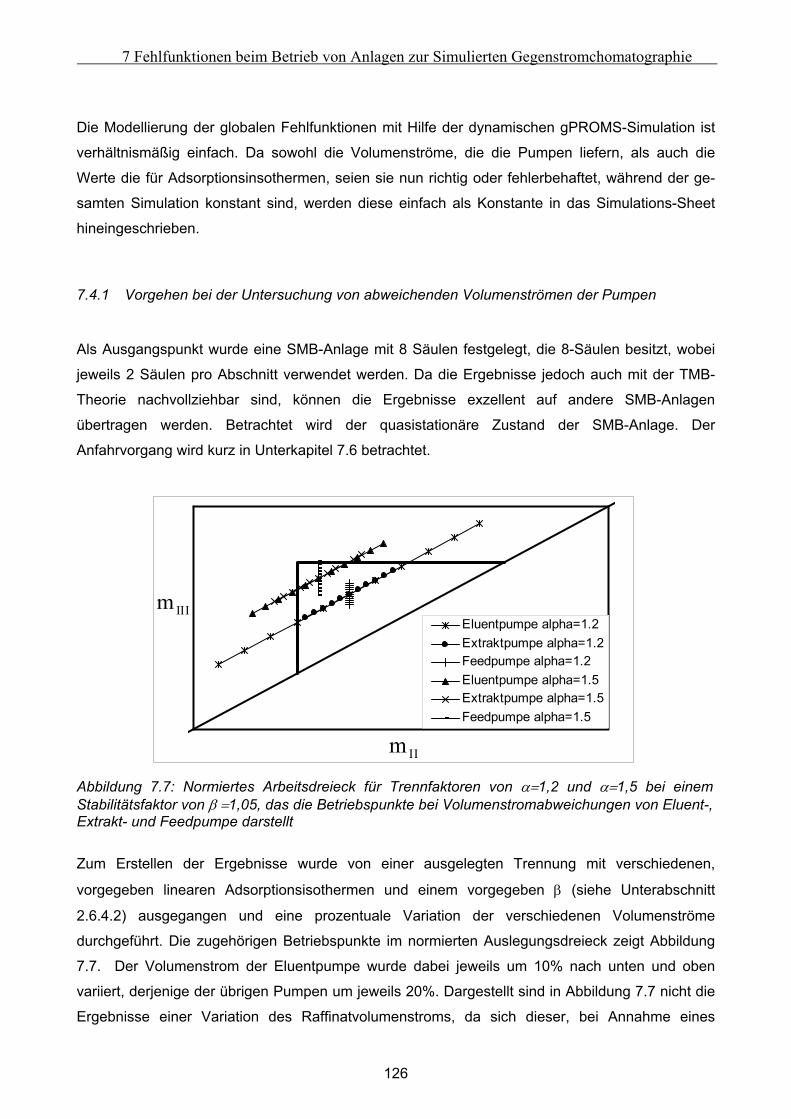
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
126
Die Modellierung der globalen Fehlfunktionen mit Hilfe der dynamischen gPROMS-Simulation ist
verhältnismäßig einfach. Da sowohl die Volumenströme, die die Pumpen liefern, als auch die
Werte die für Adsorptionsinsothermen, seien sie nun richtig oder fehlerbehaftet, während der ge-
samten Simulation konstant sind, werden diese einfach als Konstante in das Simulations-Sheet
hineingeschrieben.
7.4.1 Vorgehen bei der Untersuchung von abweichenden Volumenströmen der Pumpen
Als Ausgangspunkt wurde eine SMB-Anlage mit 8 Säulen festgelegt, die 8-Säulen besitzt, wobei
jeweils 2 Säulen pro Abschnitt verwendet werden. Da die Ergebnisse jedoch auch mit der TMB-
Theorie nachvollziehbar sind, können die Ergebnisse exzellent auf andere SMB-Anlagen
übertragen werden. Betrachtet wird der quasistationäre Zustand der SMB-Anlage. Der
Anfahrvorgang wird kurz in Unterkapitel 7.6 betrachtet.
Eluentpumpe alpha=1.2Extraktpumpe alpha=1.2Feedpumpe alpha=1.2Eluentpumpe alpha=1.5Extraktpumpe alpha=1.5Feedpumpe alpha=1.5
IIm
IIIm
Abbildung 7.7: Normiertes Arbeitsdreieck für Trennfaktoren von α=1,2 und α=1,5 bei einem Stabilitätsfaktor von β =1,05, das die Betriebspunkte bei Volumenstromabweichungen von Eluent-, Extrakt- und Feedpumpe darstellt
Zum Erstellen der Ergebnisse wurde von einer ausgelegten Trennung mit verschiedenen,
vorgegeben linearen Adsorptionsisothermen und einem vorgegeben β (siehe Unterabschnitt
2.6.4.2) ausgegangen und eine prozentuale Variation der verschiedenen Volumenströme
durchgeführt. Die zugehörigen Betriebspunkte im normierten Auslegungsdreieck zeigt Abbildung
7.7. Der Volumenstrom der Eluentpumpe wurde dabei jeweils um 10% nach unten und oben
variiert, derjenige der übrigen Pumpen um jeweils 20%. Dargestellt sind in Abbildung 7.7 nicht die
Ergebnisse einer Variation des Raffinatvolumenstroms, da sich dieser, bei Annahme eines
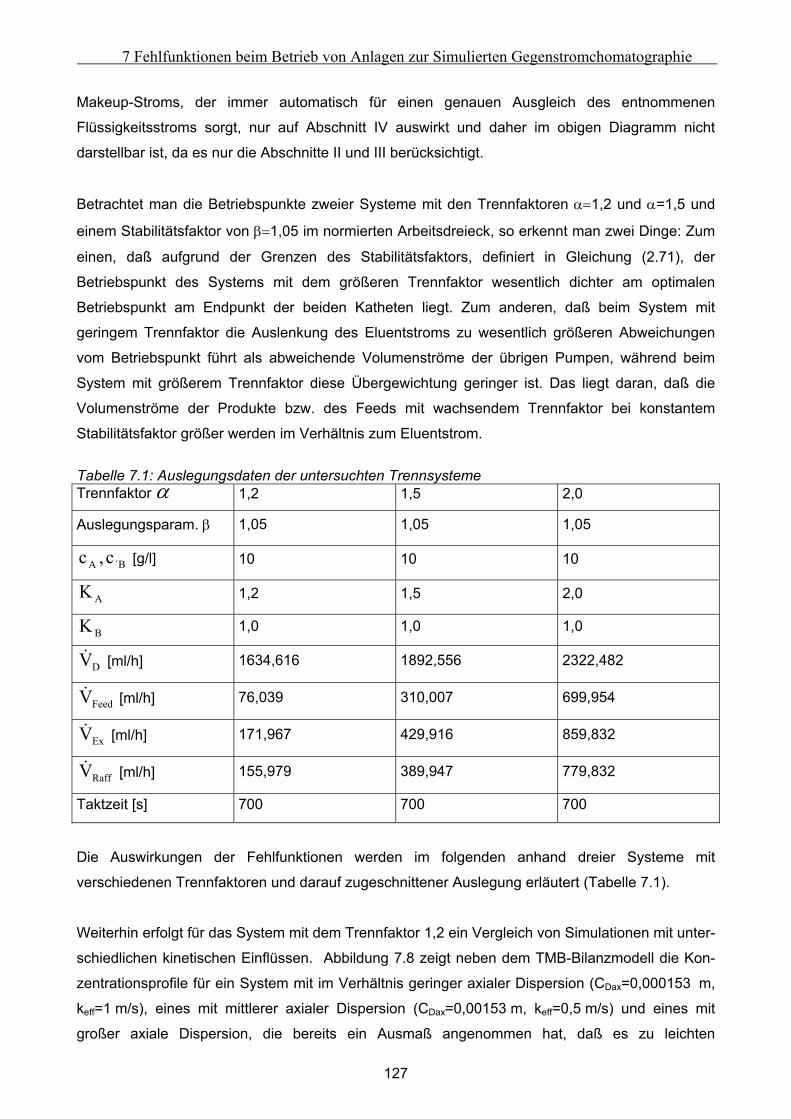
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
127
Makeup-Stroms, der immer automatisch für einen genauen Ausgleich des entnommenen
Flüssigkeitsstroms sorgt, nur auf Abschnitt IV auswirkt und daher im obigen Diagramm nicht
darstellbar ist, da es nur die Abschnitte II und III berücksichtigt.
Betrachtet man die Betriebspunkte zweier Systeme mit den Trennfaktoren α=1,2 und α=1,5 und
einem Stabilitätsfaktor von β=1,05 im normierten Arbeitsdreieck, so erkennt man zwei Dinge: Zum
einen, daß aufgrund der Grenzen des Stabilitätsfaktors, definiert in Gleichung (2.71), der
Betriebspunkt des Systems mit dem größeren Trennfaktor wesentlich dichter am optimalen
Betriebspunkt am Endpunkt der beiden Katheten liegt. Zum anderen, daß beim System mit
geringem Trennfaktor die Auslenkung des Eluentstroms zu wesentlich größeren Abweichungen
vom Betriebspunkt führt als abweichende Volumenströme der übrigen Pumpen, während beim
System mit größerem Trennfaktor diese Übergewichtung geringer ist. Das liegt daran, daß die
Volumenströme der Produkte bzw. des Feeds mit wachsendem Trennfaktor bei konstantem
Stabilitätsfaktor größer werden im Verhältnis zum Eluentstrom.
Tabelle 7.1: Auslegungsdaten der untersuchten Trennsysteme Trennfaktor α 1,2 1,5 2,0
Auslegungsparam. β 1,05 1,05 1,05
B´A c,c [g/l] 10 10 10
AK 1,2 1,5 2,0
BK 1,0 1,0 1,0
DV& [ml/h] 1634,616 1892,556 2322,482
FeedV& [ml/h] 76,039 310,007 699,954
ExV& [ml/h] 171,967 429,916 859,832
RaffV& [ml/h] 155,979 389,947 779,832
Taktzeit [s] 700 700 700
Die Auswirkungen der Fehlfunktionen werden im folgenden anhand dreier Systeme mit
verschiedenen Trennfaktoren und darauf zugeschnittener Auslegung erläutert (Tabelle 7.1).
Weiterhin erfolgt für das System mit dem Trennfaktor 1,2 ein Vergleich von Simulationen mit unter-
schiedlichen kinetischen Einflüssen. Abbildung 7.8 zeigt neben dem TMB-Bilanzmodell die Kon-
zentrationsprofile für ein System mit im Verhältnis geringer axialer Dispersion (CDax=0,000153 m,
keff=1 m/s), eines mit mittlerer axialer Dispersion (CDax=0,00153 m, keff=0,5 m/s) und eines mit
großer axiale Dispersion, die bereits ein Ausmaß angenommen hat, daß es zu leichten
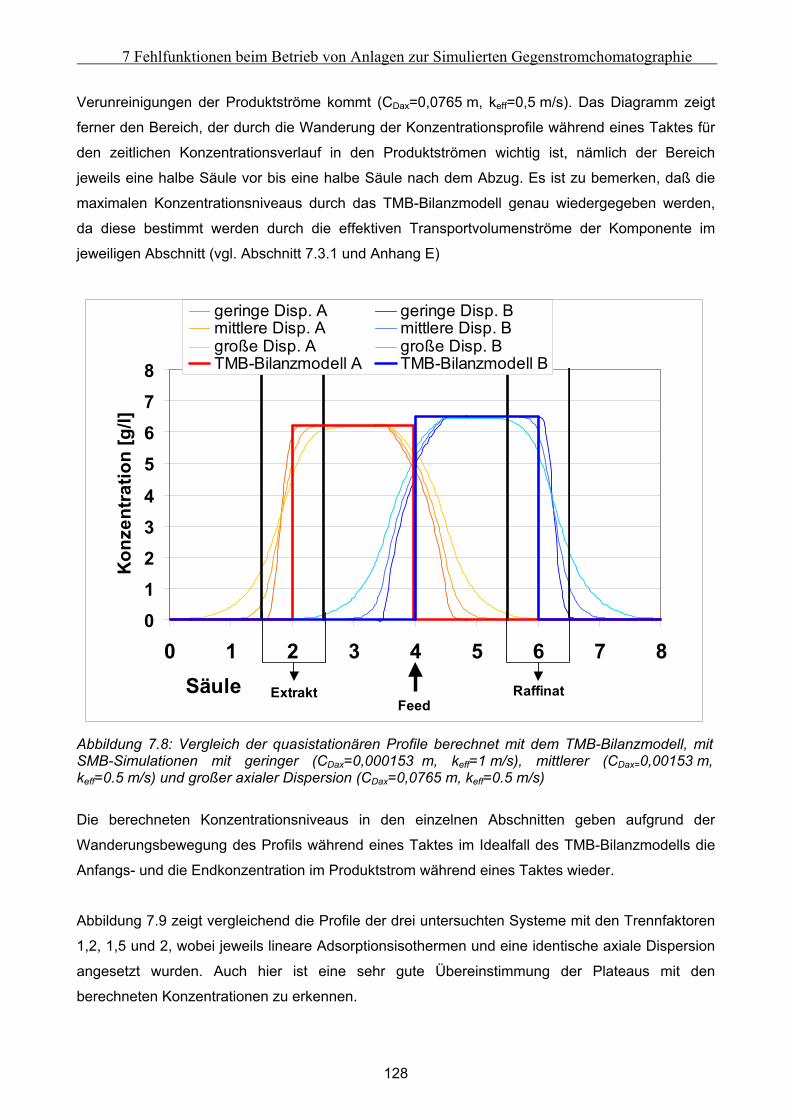
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
128
Verunreinigungen der Produktströme kommt (CDax=0,0765 m, keff=0,5 m/s). Das Diagramm zeigt
ferner den Bereich, der durch die Wanderung der Konzentrationsprofile während eines Taktes für
den zeitlichen Konzentrationsverlauf in den Produktströmen wichtig ist, nämlich der Bereich
jeweils eine halbe Säule vor bis eine halbe Säule nach dem Abzug. Es ist zu bemerken, daß die
maximalen Konzentrationsniveaus durch das TMB-Bilanzmodell genau wiedergegeben werden,
da diese bestimmt werden durch die effektiven Transportvolumenströme der Komponente im
jeweiligen Abschnitt (vgl. Abschnitt 7.3.1 und Anhang E)
01234
5678
0 1 2 3 4 5 6 7 8Säule
Kon
zent
ratio
n [g
/l]
geringe Disp. A geringe Disp. Bmittlere Disp. A mittlere Disp. Bgroße Disp. A große Disp. BTMB-Bilanzmodell A TMB-Bilanzmodell B
Extrakt RaffinatFeed
Abbildung 7.8: Vergleich der quasistationären Profile berechnet mit dem TMB-Bilanzmodell, mit SMB-Simulationen mit geringer (CDax=0,000153 m, keff=1 m/s), mittlerer (CDax=0,00153 m, keff=0.5 m/s) und großer axialer Dispersion (CDax=0,0765 m, keff=0.5 m/s)
Die berechneten Konzentrationsniveaus in den einzelnen Abschnitten geben aufgrund der
Wanderungsbewegung des Profils während eines Taktes im Idealfall des TMB-Bilanzmodells die
Anfangs- und die Endkonzentration im Produktstrom während eines Taktes wieder.
Abbildung 7.9 zeigt vergleichend die Profile der drei untersuchten Systeme mit den Trennfaktoren
1,2, 1,5 und 2, wobei jeweils lineare Adsorptionsisothermen und eine identische axiale Dispersion
angesetzt wurden. Auch hier ist eine sehr gute Übereinstimmung der Plateaus mit den
berechneten Konzentrationen zu erkennen.
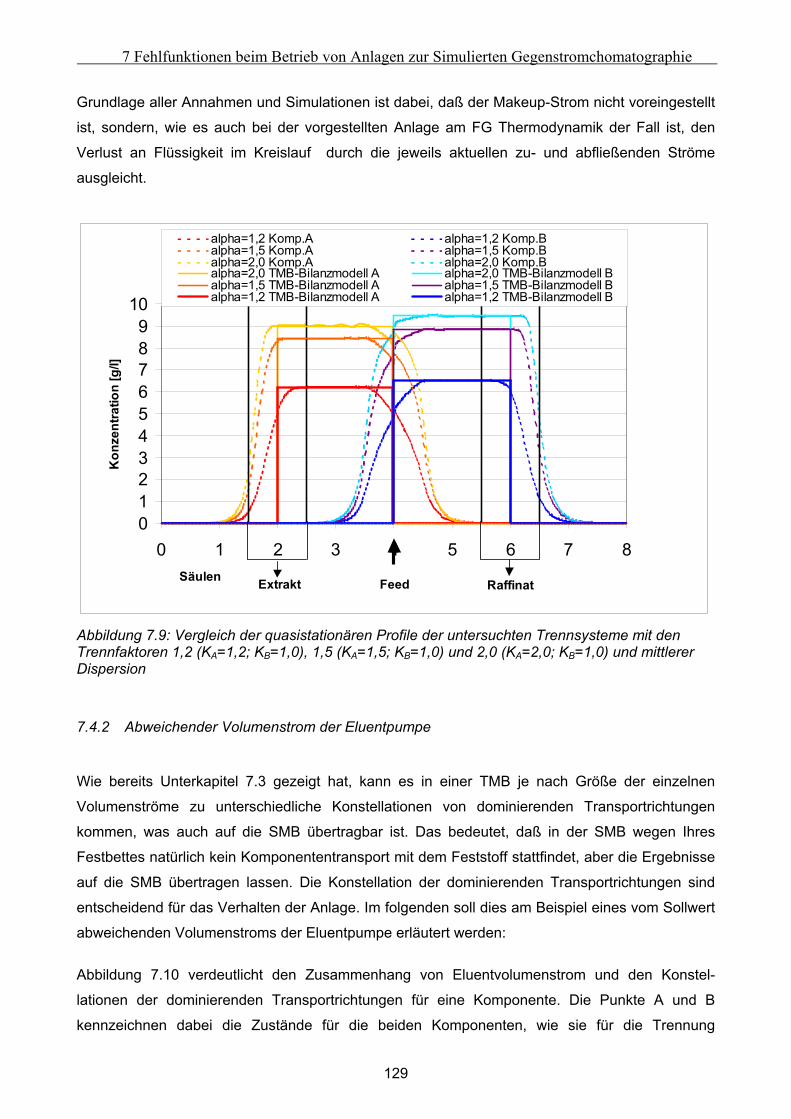
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
129
Grundlage aller Annahmen und Simulationen ist dabei, daß der Makeup-Strom nicht voreingestellt
ist, sondern, wie es auch bei der vorgestellten Anlage am FG Thermodynamik der Fall ist, den
Verlust an Flüssigkeit im Kreislauf durch die jeweils aktuellen zu- und abfließenden Ströme
ausgleicht.
0123456789
10
0 1 2 3 4 5 6 7 8Säulen
Kon
zent
ratio
n [g
/l]
alpha=1,2 Komp.A alpha=1,2 Komp.Balpha=1,5 Komp.A alpha=1,5 Komp.Balpha=2,0 Komp.A alpha=2,0 Komp.Balpha=2,0 TMB-Bilanzmodell A alpha=2,0 TMB-Bilanzmodell Balpha=1,5 TMB-Bilanzmodell A alpha=1,5 TMB-Bilanzmodell Balpha=1,2 TMB-Bilanzmodell A alpha=1,2 TMB-Bilanzmodell B
Extrakt RaffinatFeed
Abbildung 7.9: Vergleich der quasistationären Profile der untersuchten Trennsysteme mit den Trennfaktoren 1,2 (KA=1,2; KB=1,0), 1,5 (KA=1,5; KB=1,0) und 2,0 (KA=2,0; KB=1,0) und mittlerer Dispersion
7.4.2 Abweichender Volumenstrom der Eluentpumpe
Wie bereits Unterkapitel 7.3 gezeigt hat, kann es in einer TMB je nach Größe der einzelnen
Volumenströme zu unterschiedliche Konstellationen von dominierenden Transportrichtungen
kommen, was auch auf die SMB übertragbar ist. Das bedeutet, daß in der SMB wegen Ihres
Festbettes natürlich kein Komponententransport mit dem Feststoff stattfindet, aber die Ergebnisse
auf die SMB übertragen lassen. Die Konstellation der dominierenden Transportrichtungen sind
entscheidend für das Verhalten der Anlage. Im folgenden soll dies am Beispiel eines vom Sollwert
abweichenden Volumenstroms der Eluentpumpe erläutert werden:
Abbildung 7.10 verdeutlicht den Zusammenhang von Eluentvolumenstrom und den Konstel-
lationen der dominierenden Transportrichtungen für eine Komponente. Die Punkte A und B
kennzeichnen dabei die Zustände für die beiden Komponenten, wie sie für die Trennung

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
130
angestrebt sind. Ist der Eluentvolumenstrom zu gering (a), so reicht der Volumenstrom in keinem
Abschnitt aus, um die Komponente gegen den Feststoffstrom zu transportieren. Erhöht sich der
Volumenstrom, so kommt es als erstes im am stärksten durchströmten Abschnitt I zu einem
Transport mit dem Eluent (b). Es ist der Normalzustand für die stärker adsorbierende Komponente
erreicht. Wird der Eluentvolumenstrom weiter erhöht, so ändert sich im Abschnitt III ebenfalls die
dominierende Transportrichtung, es kommt zu einem konkurrierenden Transport vom Feed aus zu
den beiden Produktströmen (c). Erhöht sich der Volumenstrom weiter, so wird der Zustand (d)
erreicht, der ideal ist für die schwächer adsorbierende Komponente. Schließlich wird irgendwann
ein Zustand erreicht, daß auch der betragsmäßig geringste Volumenstrom im Abschnitt IV
ausreicht um die Komponente gegen den Feststoffstrom zu transportieren.
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
IIIIII
IVZunehmender Eluentvolumenstrom
A B
a) b) c) d) e)
untere Grenze von (für die TMB)
−
Ex
Fest
VVK&
&
+ RaffEx
FeedFest
VVVVK&&
&&
++
−
(7.22) (7.23) (7.24) (7.25) (7.26)
Abbildung 7.10: Konstellationen der dominierenden Transportrichtungen in einer TMB für eine Komponente in Abhängigkeit vom Eluentvolumenstrom
DV&
0FestVK &
Ex
FeedFest
VVVK
&
&&
+
Für die Fehleranalyse werden im folgenden Konzentrationen betrachtet, die bei SMB-Systemen
leicht verfügbar sind, nämlich diejenigen in den Produktströmen. Dabei werden zum einen die über
den Taktzyklus integrierten Konzentrationen der Komponenten betrachtet. Diese Konzentrationen
wurden mit Hilfe der SMB-Simulationen berechtet und entsprechen den Konzentrationen, die mit
Hilfe des TMB-Bilanzmodells errechnet werden. Dazu kommen die Konzentrationen jeweils am
Taktende und am Taktanfang, was bei linearen Adsorptionsisothermen der Maximal- und der
Minimalkonzentration eines Taktes entspricht (siehe Abbildung 2.12). Diese Konzentrationen
werden mit den Konzentrationen des TMB-Bilanzmodells in den dem Auslaß benachbarten
Abschnitten verglichen (siehe Abbildung 7.8).
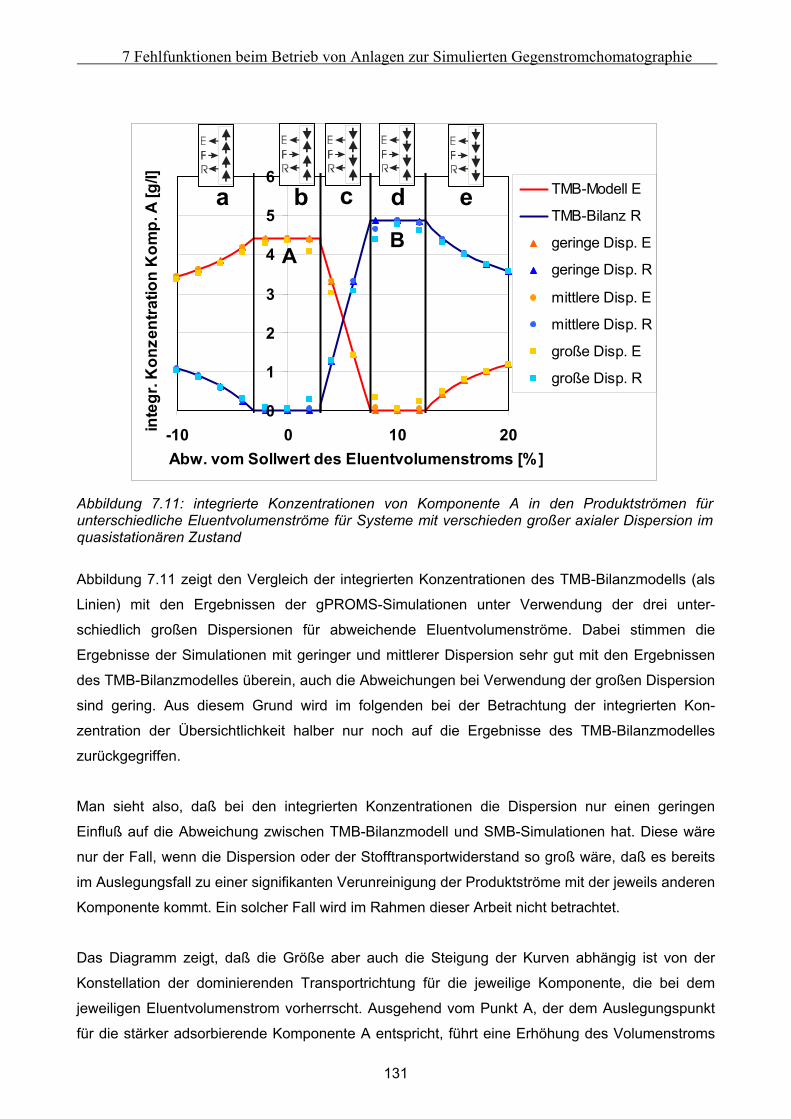
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
131
0
1
2
3
4
5
6
-10 0 10 20Abw. vom Sollwert des Eluentvolumenstroms [%]
inte
gr. K
onze
ntra
tion
Kom
p. A
[g/l]
TMB-Modell E
TMB-Bilanz R
geringe Disp. E
geringe Disp. R
mittlere Disp. E
mittlere Disp. R
große Disp. E
große Disp. R
AB
a b c d e
Abbildung 7.11: integrierte Konzentrationen von Komponente A in den Produktströmen für unterschiedliche Eluentvolumenströme für Systeme mit verschieden großer axialer Dispersion im quasistationären Zustand
Abbildung 7.11 zeigt den Vergleich der integrierten Konzentrationen des TMB-Bilanzmodells (als
Linien) mit den Ergebnissen der gPROMS-Simulationen unter Verwendung der drei unter-
schiedlich großen Dispersionen für abweichende Eluentvolumenströme. Dabei stimmen die
Ergebnisse der Simulationen mit geringer und mittlerer Dispersion sehr gut mit den Ergebnissen
des TMB-Bilanzmodelles überein, auch die Abweichungen bei Verwendung der großen Dispersion
sind gering. Aus diesem Grund wird im folgenden bei der Betrachtung der integrierten Kon-
zentration der Übersichtlichkeit halber nur noch auf die Ergebnisse des TMB-Bilanzmodelles
zurückgegriffen.
Man sieht also, daß bei den integrierten Konzentrationen die Dispersion nur einen geringen
Einfluß auf die Abweichung zwischen TMB-Bilanzmodell und SMB-Simulationen hat. Diese wäre
nur der Fall, wenn die Dispersion oder der Stofftransportwiderstand so groß wäre, daß es bereits
im Auslegungsfall zu einer signifikanten Verunreinigung der Produktströme mit der jeweils anderen
Komponente kommt. Ein solcher Fall wird im Rahmen dieser Arbeit nicht betrachtet.
Das Diagramm zeigt, daß die Größe aber auch die Steigung der Kurven abhängig ist von der
Konstellation der dominierenden Transportrichtung für die jeweilige Komponente, die bei dem
jeweiligen Eluentvolumenstrom vorherrscht. Ausgehend vom Punkt A, der dem Auslegungspunkt
für die stärker adsorbierende Komponente A entspricht, führt eine Erhöhung des Volumenstroms
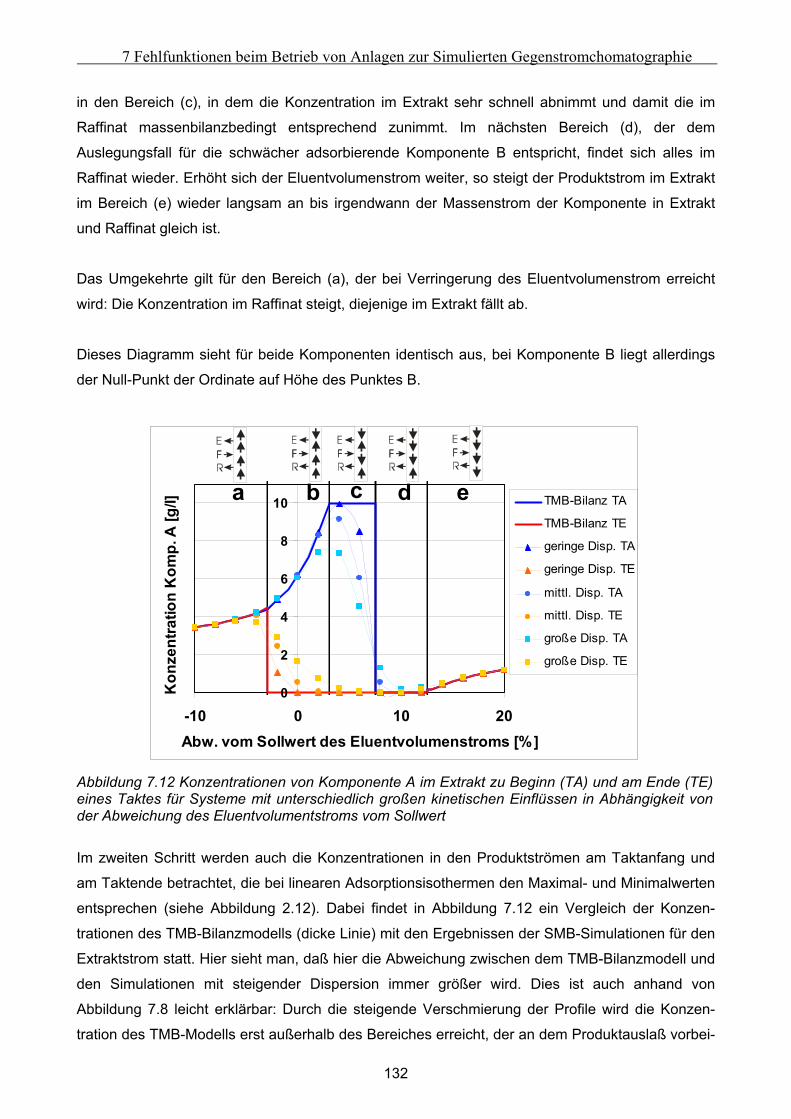
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
132
in den Bereich (c), in dem die Konzentration im Extrakt sehr schnell abnimmt und damit die im
Raffinat massenbilanzbedingt entsprechend zunimmt. Im nächsten Bereich (d), der dem
Auslegungsfall für die schwächer adsorbierende Komponente B entspricht, findet sich alles im
Raffinat wieder. Erhöht sich der Eluentvolumenstrom weiter, so steigt der Produktstrom im Extrakt
im Bereich (e) wieder langsam an bis irgendwann der Massenstrom der Komponente in Extrakt
und Raffinat gleich ist.
Das Umgekehrte gilt für den Bereich (a), der bei Verringerung des Eluentvolumenstrom erreicht
wird: Die Konzentration im Raffinat steigt, diejenige im Extrakt fällt ab.
Dieses Diagramm sieht für beide Komponenten identisch aus, bei Komponente B liegt allerdings
der Null-Punkt der Ordinate auf Höhe des Punktes B.
0
2
4
6
8
10
-10 0 10 20Abw. vom Sollwert des Eluentvolumenstroms [%]
Kon
zent
ratio
n K
omp.
A [g
/l] TMB-Bilanz TA
TMB-Bilanz TE
geringe Disp. TA
geringe Disp. TE
mittl. Disp. TA
mittl. Disp. TE
große Disp. TA
große Disp. TE
a b c d e
Abbildung 7.12 Konzentrationen von Komponente A im Extrakt zu Beginn (TA) und am Ende (TE) eines Taktes für Systeme mit unterschiedlich großen kinetischen Einflüssen in Abhängigkeit von der Abweichung des Eluentvolumentstroms vom Sollwert
Im zweiten Schritt werden auch die Konzentrationen in den Produktströmen am Taktanfang und
am Taktende betrachtet, die bei linearen Adsorptionsisothermen den Maximal- und Minimalwerten
entsprechen (siehe Abbildung 2.12). Dabei findet in Abbildung 7.12 ein Vergleich der Konzen-
trationen des TMB-Bilanzmodells (dicke Linie) mit den Ergebnissen der SMB-Simulationen für den
Extraktstrom statt. Hier sieht man, daß hier die Abweichung zwischen dem TMB-Bilanzmodell und
den Simulationen mit steigender Dispersion immer größer wird. Dies ist auch anhand von
Abbildung 7.8 leicht erklärbar: Durch die steigende Verschmierung der Profile wird die Konzen-
tration des TMB-Modells erst außerhalb des Bereiches erreicht, der an dem Produktauslaß vorbei-
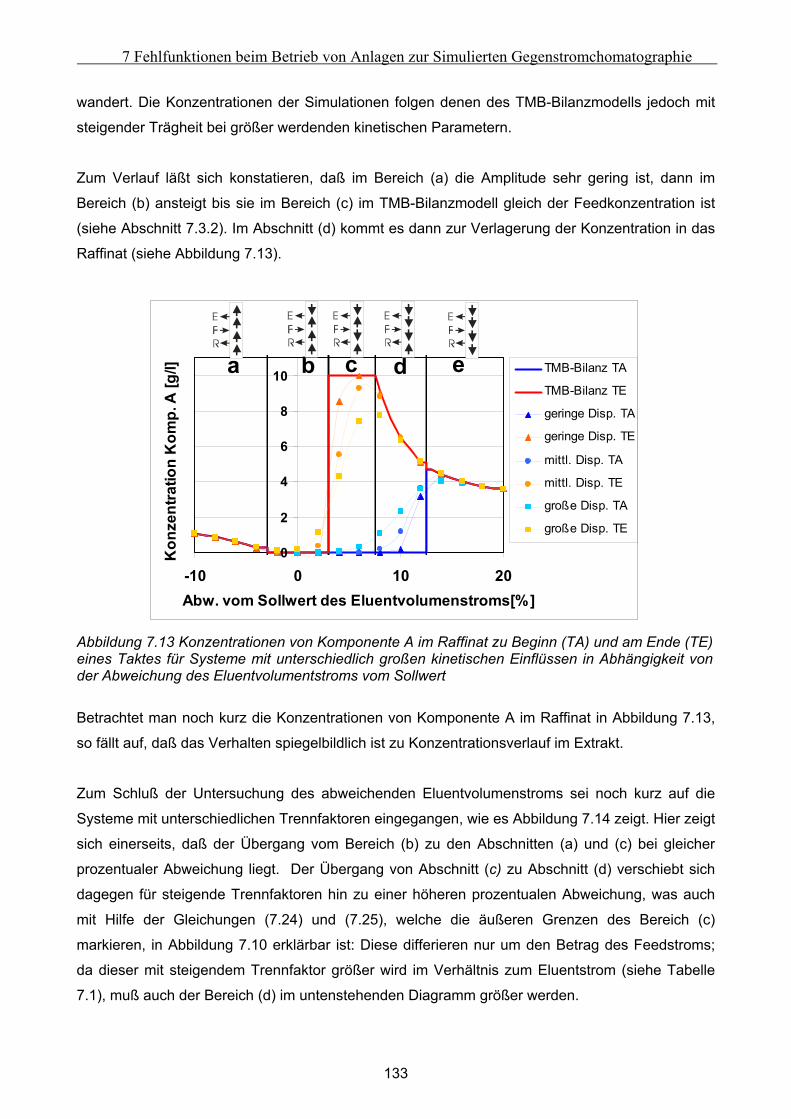
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
133
wandert. Die Konzentrationen der Simulationen folgen denen des TMB-Bilanzmodells jedoch mit
steigender Trägheit bei größer werdenden kinetischen Parametern.
Zum Verlauf läßt sich konstatieren, daß im Bereich (a) die Amplitude sehr gering ist, dann im
Bereich (b) ansteigt bis sie im Bereich (c) im TMB-Bilanzmodell gleich der Feedkonzentration ist
(siehe Abschnitt 7.3.2). Im Abschnitt (d) kommt es dann zur Verlagerung der Konzentration in das
Raffinat (siehe Abbildung 7.13).
0
2
4
6
8
10
-10 0 10 20Abw. vom Sollwert des Eluentvolumenstroms[%]
Kon
zent
ratio
n K
omp.
A [g
/l] TMB-Bilanz TA
TMB-Bilanz TE
geringe Disp. TA
geringe Disp. TE
mittl. Disp. TA
mittl. Disp. TE
große Disp. TA
große Disp. TE
a b c d e
Abbildung 7.13 Konzentrationen von Komponente A im Raffinat zu Beginn (TA) und am Ende (TE) eines Taktes für Systeme mit unterschiedlich großen kinetischen Einflüssen in Abhängigkeit von der Abweichung des Eluentvolumentstroms vom Sollwert
Betrachtet man noch kurz die Konzentrationen von Komponente A im Raffinat in Abbildung 7.13,
so fällt auf, daß das Verhalten spiegelbildlich ist zu Konzentrationsverlauf im Extrakt.
Zum Schluß der Untersuchung des abweichenden Eluentvolumenstroms sei noch kurz auf die
Systeme mit unterschiedlichen Trennfaktoren eingegangen, wie es Abbildung 7.14 zeigt. Hier zeigt
sich einerseits, daß der Übergang vom Bereich (b) zu den Abschnitten (a) und (c) bei gleicher
prozentualer Abweichung liegt. Der Übergang von Abschnitt (c) zu Abschnitt (d) verschiebt sich
dagegen für steigende Trennfaktoren hin zu einer höheren prozentualen Abweichung, was auch
mit Hilfe der Gleichungen (7.24) und (7.25), welche die äußeren Grenzen des Bereich (c)
markieren, in Abbildung 7.10 erklärbar ist: Diese differieren nur um den Betrag des Feedstroms;
da dieser mit steigendem Trennfaktor größer wird im Verhältnis zum Eluentstrom (siehe Tabelle
7.1), muß auch der Bereich (d) im untenstehenden Diagramm größer werden.
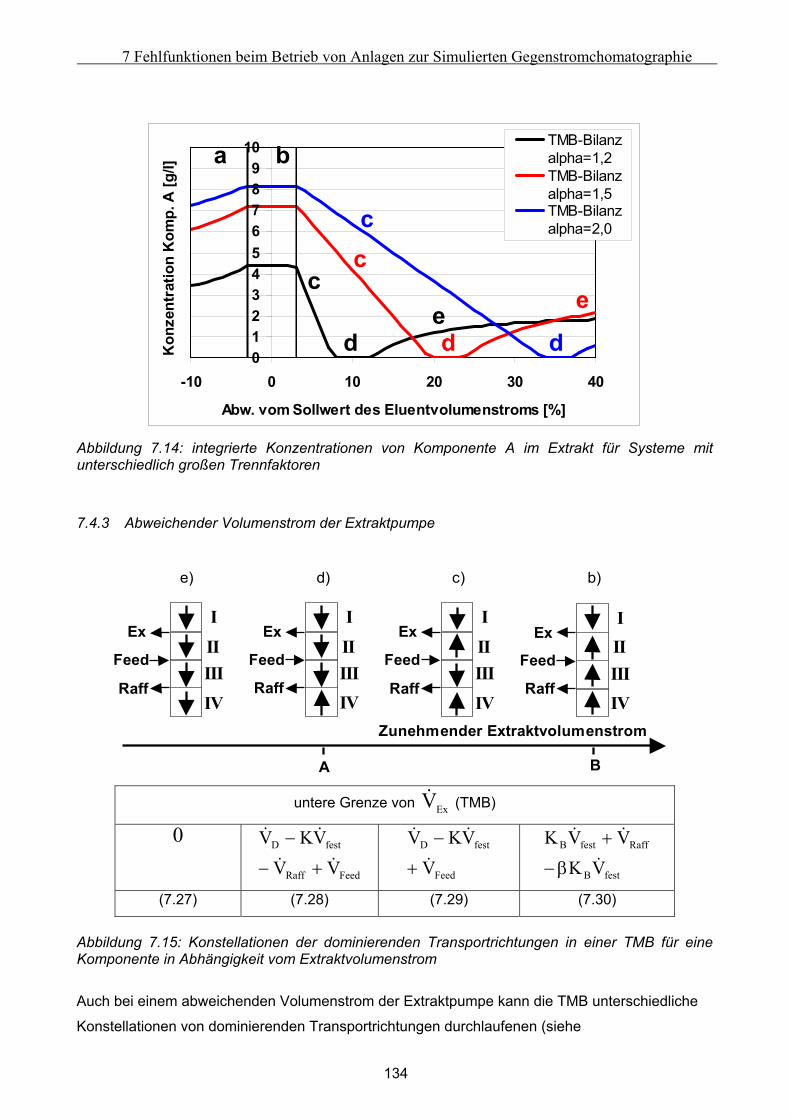
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
134
0123456789
10
-10 0 10 20 30 40
Abw. vom Sollwert des Eluentvolumenstroms [%]
Kon
zent
ratio
n K
omp.
A [g
/l]
TMB-Bilanzalpha=1,2TMB-Bilanzalpha=1,5TMB-Bilanzalpha=2,0
e
a
c
b
d
c
d
Abbildung 7.14: integrierte Konzentrationen von Komponente A im Extrakt für Systeme mit unterschiedlich großen Trennfaktoren
c
d
e
7.4.3 Abweichender Volumenstrom der Extraktpumpe
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
I
IIIII
IV
Ex
Feed
Raff
I
IIIIIIV
Ex
Feed
Raff
I
IIIII
IVZunehmender Extraktvolumenstrom
A B
b)c)d)e)
untere Grenze von V (TMB)
(7.27) (7.28) (7.29) (7.30) Abbildung 7.15: Konstellationen der dominierenden Transportrichtungen in einer TMB für eine Komponente in Abhängigkeit vom Extraktvolumenstrom
Ex&
0
FeedRaff
festD
VVVKV&&
&&
+−
−
Feed
festD
VVKV
&
&&
+
−
festB
RafffestB
VKVVK
&
&&
β−
+
Auch bei einem abweichenden Volumenstrom der Extraktpumpe kann die TMB unterschiedliche
Konstellationen von dominierenden Transportrichtungen durchlaufenen (siehe
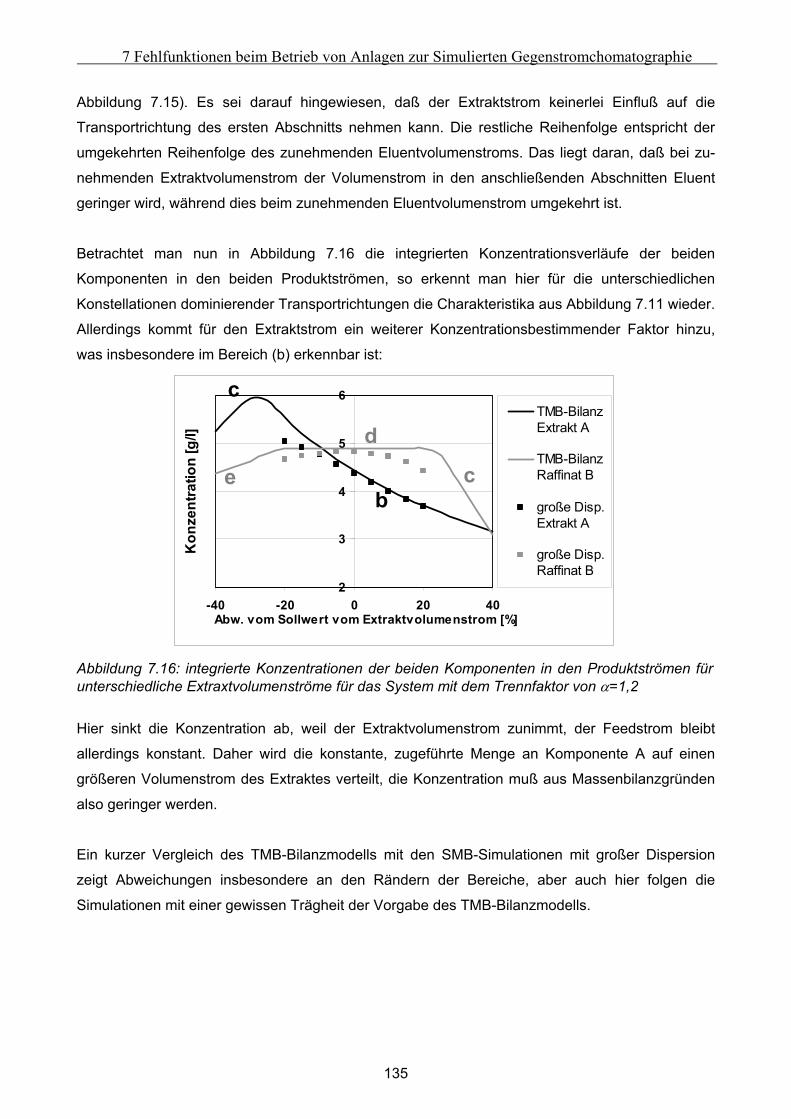
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
135
Abbildung 7.15). Es sei darauf hingewiesen, daß der Extraktstrom keinerlei Einfluß auf die
Transportrichtung des ersten Abschnitts nehmen kann. Die restliche Reihenfolge entspricht der
umgekehrten Reihenfolge des zunehmenden Eluentvolumenstroms. Das liegt daran, daß bei zu-
nehmenden Extraktvolumenstrom der Volumenstrom in den anschließenden Abschnitten Eluent
geringer wird, während dies beim zunehmenden Eluentvolumenstrom umgekehrt ist.
Betrachtet man nun in Abbildung 7.16 die integrierten Konzentrationsverläufe der beiden
Komponenten in den beiden Produktströmen, so erkennt man hier für die unterschiedlichen
Konstellationen dominierender Transportrichtungen die Charakteristika aus Abbildung 7.11 wieder.
Allerdings kommt für den Extraktstrom ein weiterer Konzentrationsbestimmender Faktor hinzu,
was insbesondere im Bereich (b) erkennbar ist:
2
3
4
5
6
-40 -20 0 20 40Abw. vom Sollwert vom Extraktvolumenstrom [%]
Kon
zent
ratio
n [g
/l]
TMB-BilanzExtrakt A
TMB-BilanzRaffinat B
große Disp.Extrakt A
große Disp.Raffinat B
b
c
d
ce
Abbildung 7.16: integrierte Konzentrationen der beiden Komponenten in den Produktströmen für unterschiedliche Extraxtvolumenströme für das System mit dem Trennfaktor von α=1,2
Hier sinkt die Konzentration ab, weil der Extraktvolumenstrom zunimmt, der Feedstrom bleibt
allerdings konstant. Daher wird die konstante, zugeführte Menge an Komponente A auf einen
größeren Volumenstrom des Extraktes verteilt, die Konzentration muß aus Massenbilanzgründen
also geringer werden.
Ein kurzer Vergleich des TMB-Bilanzmodells mit den SMB-Simulationen mit großer Dispersion
zeigt Abweichungen insbesondere an den Rändern der Bereiche, aber auch hier folgen die
Simulationen mit einer gewissen Trägheit der Vorgabe des TMB-Bilanzmodells.

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
136
7.4.4 Abweichender Volumenstrom der Feedpumpe
Bei der Betrachtung eines abweichenden Feedvolumenstroms kommen zwei Dinge zum Tragen:
Zum einen durchlaufen die beiden Komponenten jeweils unterschiedliche Konstellationen
dominierender Transportrichtungen, was auf die Tatsache zurückzuführen ist, daß wiederum der
Feedstrom nur Auswirkungen auf die nachfolgenden Abschnitte III und IV haben kann, die beiden
Komponenten aber im Ausgangszustand bereits in Abschnitt II eine unterschiedliche
Transportrichtung haben (siehe Abbildung 7.17).
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
I
IIIIIIV
Ex
Feed
Raff
I
IIIIIIV
Ex
Feed
Raff
I
IIIIIIV
Zunehmender Feedvolumenstrom
A B
b) c) f) e)
Ex
Feed
Raff
I
IIIIIIV
d)
untere Grenze von V (für die TMB) Feed&
Komponente A Komponente B
(7.31) (7.32) (7.33) (7.34) (7.35)
Abbildung 7.17: Konstellationen der dominierenden Transportrichtungen für eine Komponente in Abhängigkeit vom Feedvolumenstrom
0
β− B
AFest K
KV&
festB
RafffestA
VKVVK
&
&&
β−
+ 0
festB
RafffestB
VKVVK
&
&&
β−
+
Zum anderen kommt hier noch die Konstellation (f) hinzu, die allerdings im Rahmen dieser Arbeit
nicht weiter betrachtet wird, da sie nur bei extrem großen Abweichungen des Feedvolumenstroms
auftreten kann. Diese wäre aber auch mit einer Massenbilanz nach Vorbild von Abschnitt 7.3.3
abbildbar.
Zeichnet man auch hier die Konzentrationen mit steigendem Feedvolumenstrom wie in Abbildung
7.18, so erkennt man auch die typischen Charakteristika der einzelnen Konstellationen wie sie
bereits in Abschnitt 7.4.2 erläutert wurden, allerdings auch hier wiederum mit einem zusätzlichen,
verändernden Einflußfaktor: Da der Feedstrom zunimmt, erhöht sich auch die zugeführte Menge
an zu trennenden Komponenten, die mit den gleichbleibenden Produktströmen abgeführt werden.
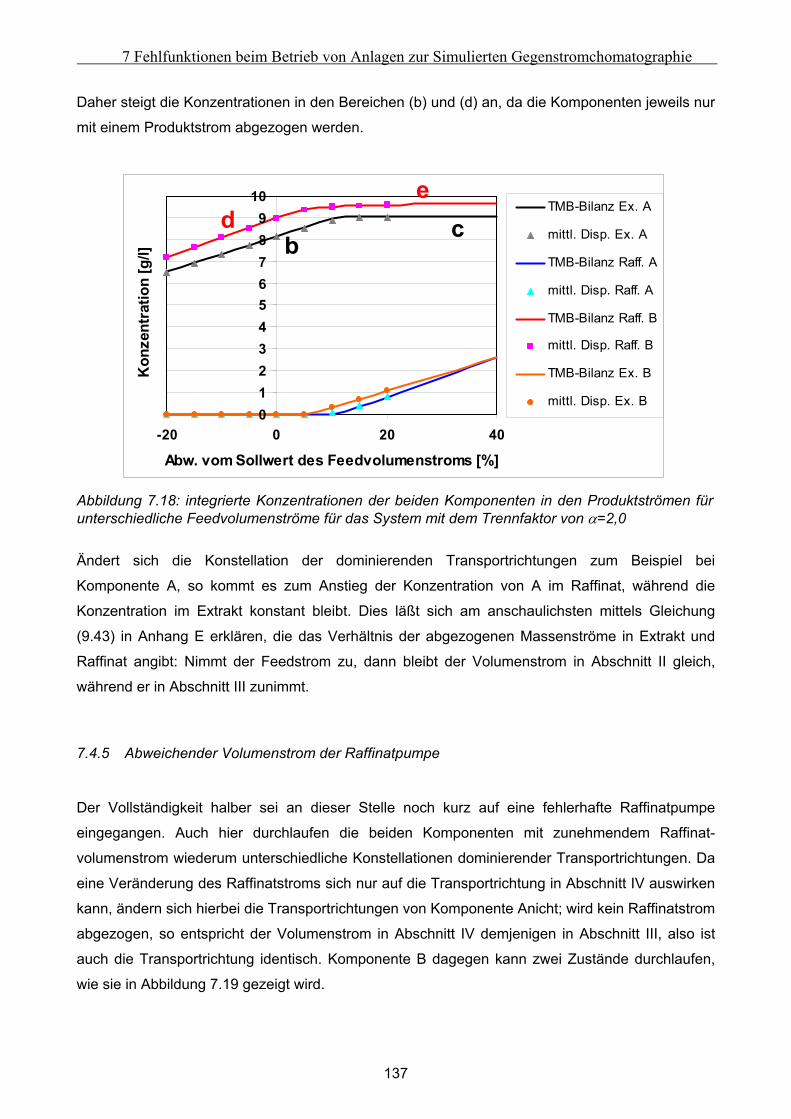
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
137
Daher steigt die Konzentrationen in den Bereichen (b) und (d) an, da die Komponenten jeweils nur
mit einem Produktstrom abgezogen werden.
0123456789
10
-20 0 20 40
Abw. vom Sollwert des Feedvolumenstroms [%]
Kon
zent
ratio
n [g
/l]
TMB-Bilanz Ex. A
mittl. Disp. Ex. A
TMB-Bilanz Raff. A
mittl. Disp. Raff. A
TMB-Bilanz Raff. B
mittl. Disp. Raff. B
TMB-Bilanz Ex. B
mittl. Disp. Ex. B
bc
Abbildung 7.18: integrierte Konzentrationen der beiden Komponenten in den Produktströmen für unterschiedliche Feedvolumenströme für das System mit dem Trennfaktor von α=2,0
de
Ändert sich die Konstellation der dominierenden Transportrichtungen zum Beispiel bei
Komponente A, so kommt es zum Anstieg der Konzentration von A im Raffinat, während die
Konzentration im Extrakt konstant bleibt. Dies läßt sich am anschaulichsten mittels Gleichung
(9.43) in Anhang E erklären, die das Verhältnis der abgezogenen Massenströme in Extrakt und
Raffinat angibt: Nimmt der Feedstrom zu, dann bleibt der Volumenstrom in Abschnitt II gleich,
während er in Abschnitt III zunimmt.
7.4.5 Abweichender Volumenstrom der Raffinatpumpe
Der Vollständigkeit halber sei an dieser Stelle noch kurz auf eine fehlerhafte Raffinatpumpe
eingegangen. Auch hier durchlaufen die beiden Komponenten mit zunehmendem Raffinat-
volumenstrom wiederum unterschiedliche Konstellationen dominierender Transportrichtungen. Da
eine Veränderung des Raffinatstroms sich nur auf die Transportrichtung in Abschnitt IV auswirken
kann, ändern sich hierbei die Transportrichtungen von Komponente Anicht; wird kein Raffinatstrom
abgezogen, so entspricht der Volumenstrom in Abschnitt IV demjenigen in Abschnitt III, also ist
auch die Transportrichtung identisch. Komponente B dagegen kann zwei Zustände durchlaufen,
wie sie in Abbildung 7.19 gezeigt wird.
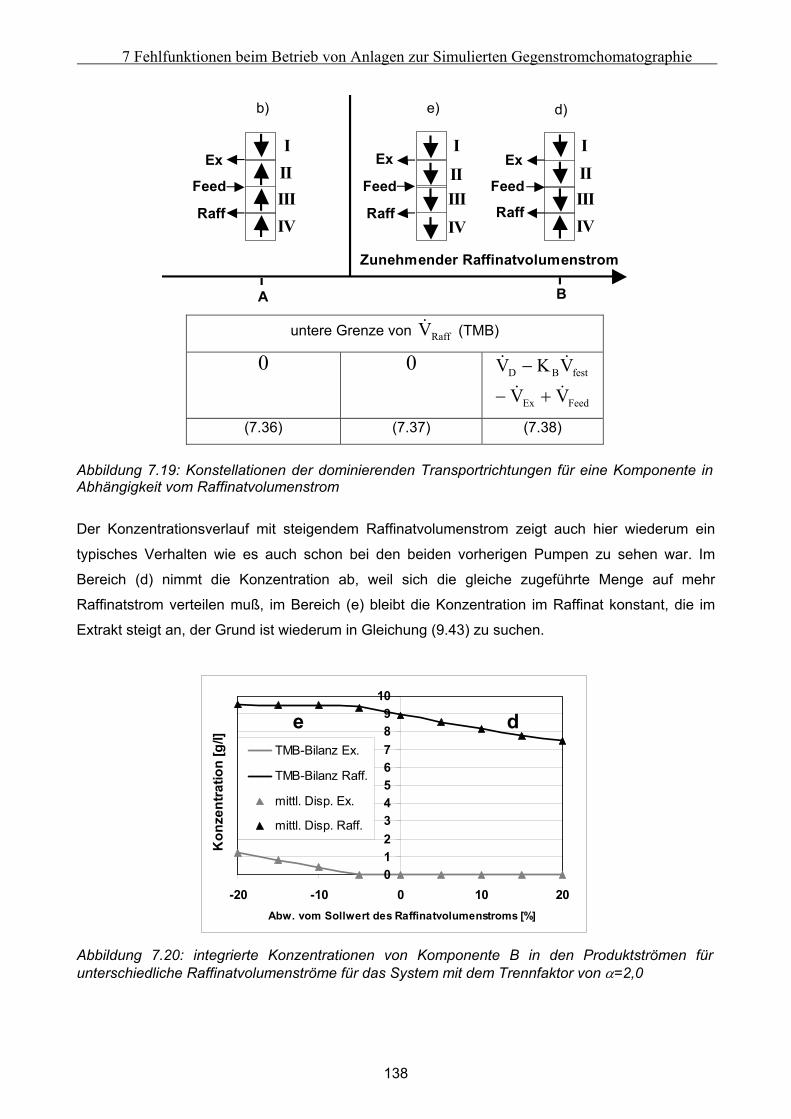
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
138
Ex
Feed
Raff
IIIIIIIV
Ex
Feed
Raff
I
IIIII
IV
Zunehmender Raffinatvolumenstrom
A B
b) e)
Ex
Feed
Raff
IIIIIIIV
d)
untere Grenze von (TMB)
(7.36) (7.37) (7.38)
Abbildung 7.19: Konstellationen der dominierenden Transportrichtungen für eine Komponente in Abhängigkeit vom Raffinatvolumenstrom
RaffV&
0 0
FeedEx
festBD
VVVKV&&
&&
+−
−
Der Konzentrationsverlauf mit steigendem Raffinatvolumenstrom zeigt auch hier wiederum ein
typisches Verhalten wie es auch schon bei den beiden vorherigen Pumpen zu sehen war. Im
Bereich (d) nimmt die Konzentration ab, weil sich die gleiche zugeführte Menge auf mehr
Raffinatstrom verteilen muß, im Bereich (e) bleibt die Konzentration im Raffinat konstant, die im
Extrakt steigt an, der Grund ist wiederum in Gleichung (9.43) zu suchen.
0123456789
10
-20 -10 0 10 20Abw. vom Sollwert des Raffinatvolumenstroms [%]
Kon
zent
ratio
n [g
/l]
TMB-Bilanz Ex.
TMB-Bilanz Raff.
mittl. Disp. Ex.
mittl. Disp. Raff.
e d
Abbildung 7.20: integrierte Konzentrationen von Komponente B in den Produktströmen für unterschiedliche Raffinatvolumenströme für das System mit dem Trennfaktor von α=2,0
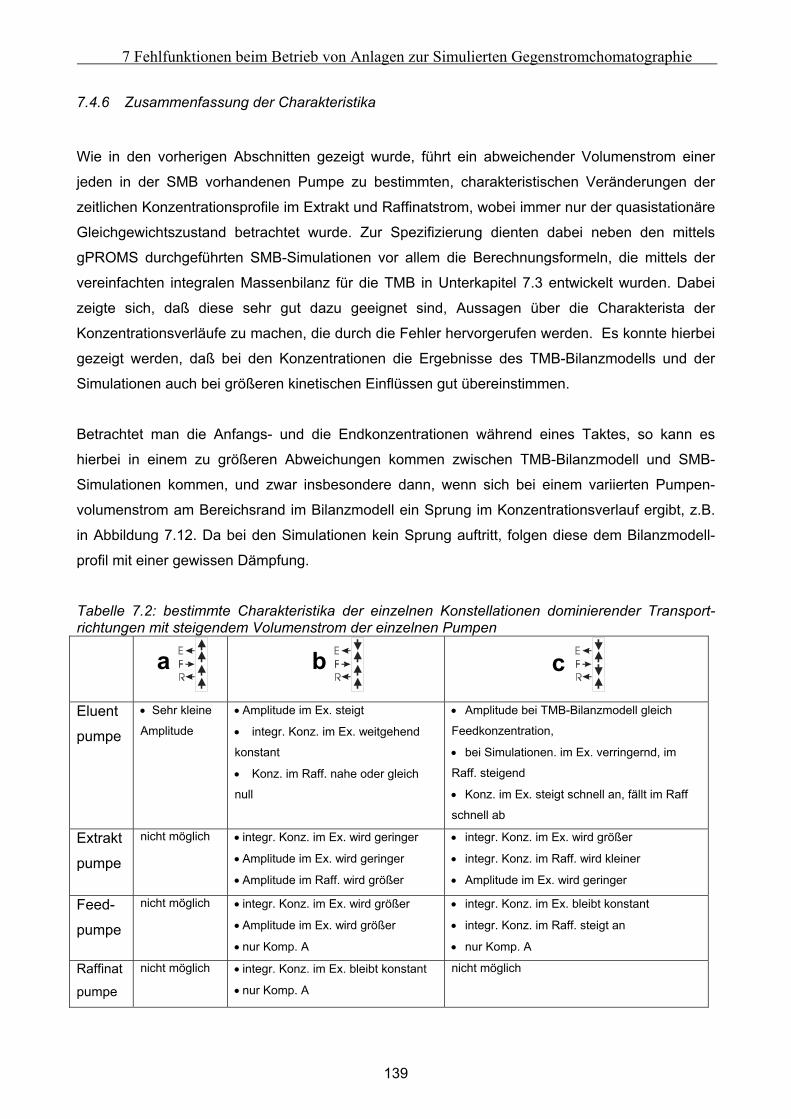
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
139
7.4.6 Zusammenfassung der Charakteristika
Wie in den vorherigen Abschnitten gezeigt wurde, führt ein abweichender Volumenstrom einer
jeden in der SMB vorhandenen Pumpe zu bestimmten, charakteristischen Veränderungen der
zeitlichen Konzentrationsprofile im Extrakt und Raffinatstrom, wobei immer nur der quasistationäre
Gleichgewichtszustand betrachtet wurde. Zur Spezifizierung dienten dabei neben den mittels
gPROMS durchgeführten SMB-Simulationen vor allem die Berechnungsformeln, die mittels der
vereinfachten integralen Massenbilanz für die TMB in Unterkapitel 7.3 entwickelt wurden. Dabei
zeigte sich, daß diese sehr gut dazu geeignet sind, Aussagen über die Charakterista der
Konzentrationsverläufe zu machen, die durch die Fehler hervorgerufen werden. Es konnte hierbei
gezeigt werden, daß bei den Konzentrationen die Ergebnisse des TMB-Bilanzmodells und der
Simulationen auch bei größeren kinetischen Einflüssen gut übereinstimmen.
Betrachtet man die Anfangs- und die Endkonzentrationen während eines Taktes, so kann es
hierbei in einem zu größeren Abweichungen kommen zwischen TMB-Bilanzmodell und SMB-
Simulationen kommen, und zwar insbesondere dann, wenn sich bei einem variierten Pumpen-
volumenstrom am Bereichsrand im Bilanzmodell ein Sprung im Konzentrationsverlauf ergibt, z.B.
in Abbildung 7.12. Da bei den Simulationen kein Sprung auftritt, folgen diese dem Bilanzmodell-
profil mit einer gewissen Dämpfung.
Tabelle 7.2: bestimmte Charakteristika der einzelnen Konstellationen dominierender Transport-richtungen mit steigendem Volumenstrom der einzelnen Pumpen
a
b
c
Eluent
pumpe
• Sehr kleine
Amplitude
• Amplitude im Ex. steigt
• integr. Konz. im Ex. weitgehend
konstant
• Konz. im Raff. nahe oder gleich
null
• Amplitude bei TMB-Bilanzmodell gleich
Feedkonzentration,
• bei Simulationen. im Ex. verringernd, im
Raff. steigend
• Konz. im Ex. steigt schnell an, fällt im Raff
schnell ab
Extrakt
pumpe
nicht möglich • integr. Konz. im Ex. wird geringer
• Amplitude im Ex. wird geringer
• Amplitude im Raff. wird größer
• integr. Konz. im Ex. wird größer
• integr. Konz. im Raff. wird kleiner
• Amplitude im Ex. wird geringer
Feed-
pumpe
nicht möglich • integr. Konz. im Ex. wird größer
• Amplitude im Ex. wird größer
• nur Komp. A
• integr. Konz. im Ex. bleibt konstant
• integr. Konz. im Raff. steigt an
• nur Komp. A
Raffinat
pumpe
nicht möglich • integr. Konz. im Ex. bleibt konstant
• nur Komp. A
nicht möglich
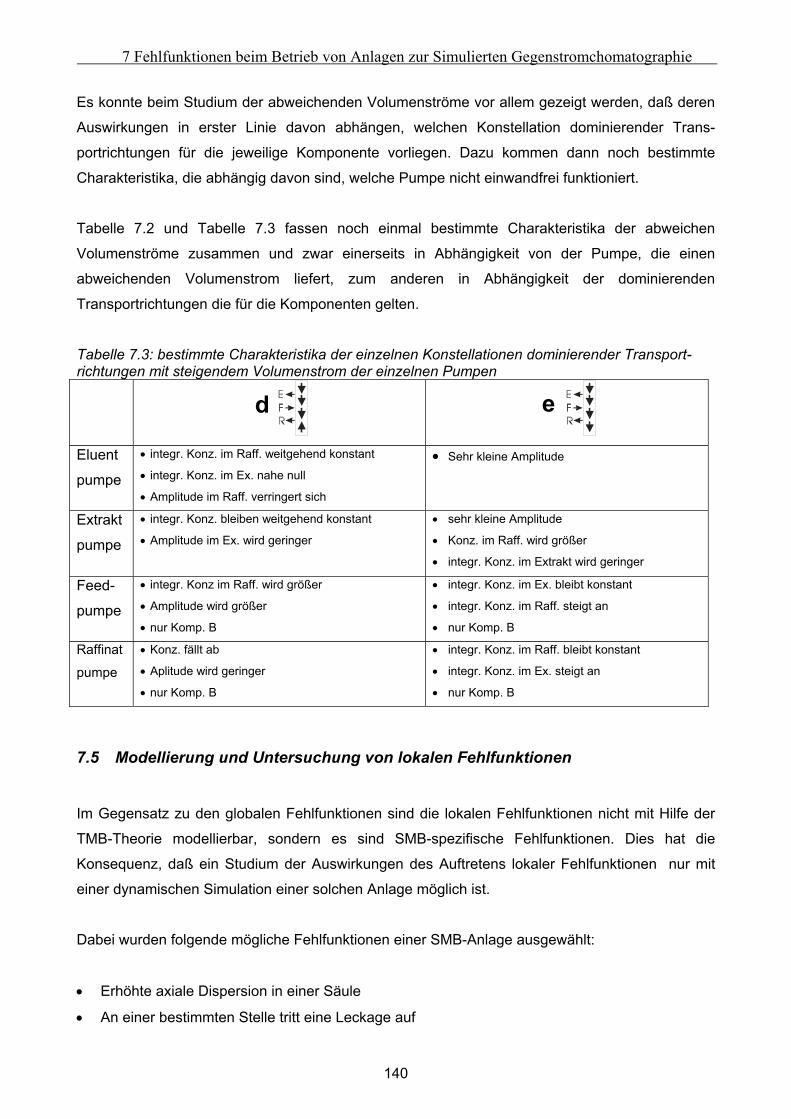
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
140
Es konnte beim Studium der abweichenden Volumenströme vor allem gezeigt werden, daß deren
Auswirkungen in erster Linie davon abhängen, welchen Konstellation dominierender Trans-
portrichtungen für die jeweilige Komponente vorliegen. Dazu kommen dann noch bestimmte
Charakteristika, die abhängig davon sind, welche Pumpe nicht einwandfrei funktioniert.
Tabelle 7.2 und Tabelle 7.3 fassen noch einmal bestimmte Charakteristika der abweichen
Volumenströme zusammen und zwar einerseits in Abhängigkeit von der Pumpe, die einen
abweichenden Volumenstrom liefert, zum anderen in Abhängigkeit der dominierenden
Transportrichtungen die für die Komponenten gelten.
Tabelle 7.3: bestimmte Charakteristika der einzelnen Konstellationen dominierender Transport-richtungen mit steigendem Volumenstrom der einzelnen Pumpen
d
e
Eluent
pumpe
• integr. Konz. im Raff. weitgehend konstant
• integr. Konz. im Ex. nahe null
• Amplitude im Raff. verringert sich
• Sehr kleine Amplitude
Extrakt
pumpe
• integr. Konz. bleiben weitgehend konstant
• Amplitude im Ex. wird geringer
• sehr kleine Amplitude
• Konz. im Raff. wird größer
• integr. Konz. im Extrakt wird geringer
Feed-
pumpe
• integr. Konz im Raff. wird größer
• Amplitude wird größer
• nur Komp. B
• integr. Konz. im Ex. bleibt konstant
• integr. Konz. im Raff. steigt an
• nur Komp. B
Raffinat
pumpe
• Konz. fällt ab
• Aplitude wird geringer
• nur Komp. B
• integr. Konz. im Raff. bleibt konstant
• integr. Konz. im Ex. steigt an
• nur Komp. B
7.5 Modellierung und Untersuchung von lokalen Fehlfunktionen
Im Gegensatz zu den globalen Fehlfunktionen sind die lokalen Fehlfunktionen nicht mit Hilfe der
TMB-Theorie modellierbar, sondern es sind SMB-spezifische Fehlfunktionen. Dies hat die
Konsequenz, daß ein Studium der Auswirkungen des Auftretens lokaler Fehlfunktionen nur mit
einer dynamischen Simulation einer solchen Anlage möglich ist.
Dabei wurden folgende mögliche Fehlfunktionen einer SMB-Anlage ausgewählt:
• Erhöhte axiale Dispersion in einer Säule
• An einer bestimmten Stelle tritt eine Leckage auf
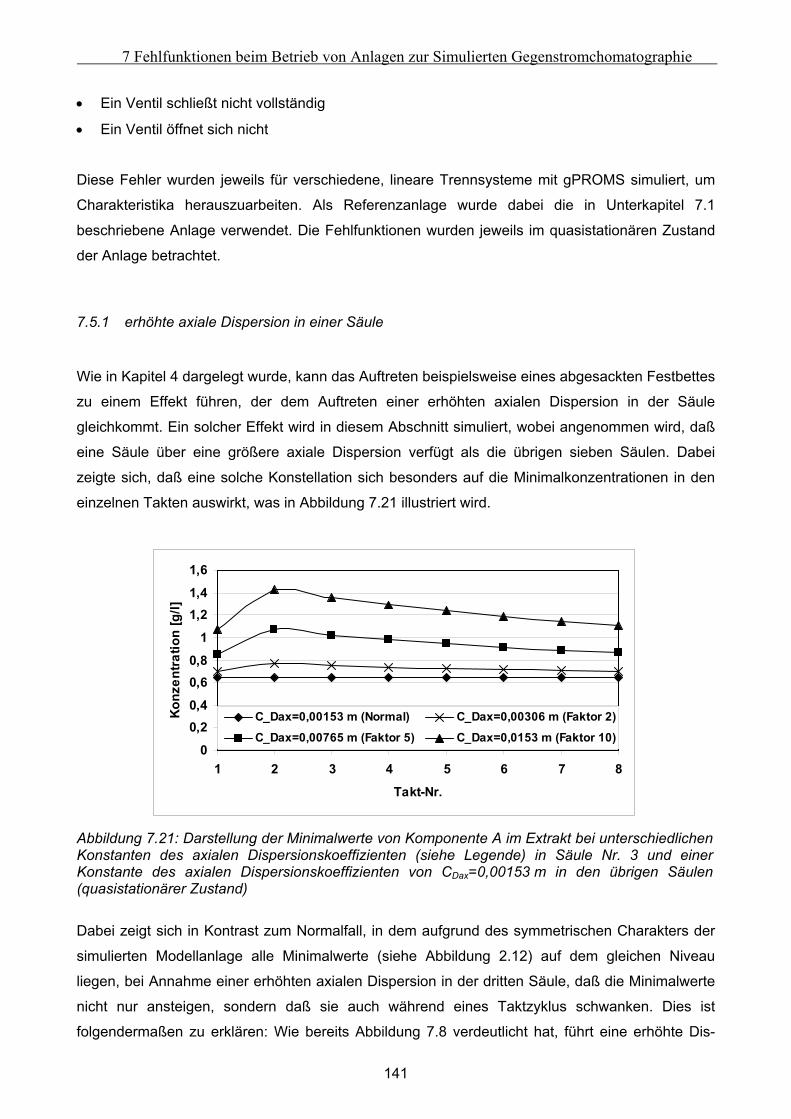
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
141
• Ein Ventil schließt nicht vollständig
• Ein Ventil öffnet sich nicht
Diese Fehler wurden jeweils für verschiedene, lineare Trennsysteme mit gPROMS simuliert, um
Charakteristika herauszuarbeiten. Als Referenzanlage wurde dabei die in Unterkapitel 7.1
beschriebene Anlage verwendet. Die Fehlfunktionen wurden jeweils im quasistationären Zustand
der Anlage betrachtet.
7.5.1 erhöhte axiale Dispersion in einer Säule
Wie in Kapitel 4 dargelegt wurde, kann das Auftreten beispielsweise eines abgesackten Festbettes
zu einem Effekt führen, der dem Auftreten einer erhöhten axialen Dispersion in der Säule
gleichkommt. Ein solcher Effekt wird in diesem Abschnitt simuliert, wobei angenommen wird, daß
eine Säule über eine größere axiale Dispersion verfügt als die übrigen sieben Säulen. Dabei
zeigte sich, daß eine solche Konstellation sich besonders auf die Minimalkonzentrationen in den
einzelnen Takten auswirkt, was in Abbildung 7.21 illustriert wird.
0
0,20,4
0,60,8
1
1,21,4
1,6
1 2 3 4 5 6 7 8
Takt-Nr.
Konz
entra
tion
[g/l]
C_Dax=0,00153 m (Normal) C_Dax=0,00306 m (Faktor 2)
C_Dax=0,00765 m (Faktor 5) C_Dax=0,0153 m (Faktor 10)
Abbildung 7.21: Darstellung der Minimalwerte von Komponente A im Extrakt bei unterschiedlichen Konstanten des axialen Dispersionskoeffizienten (siehe Legende) in Säule Nr. 3 und einer Konstante des axialen Dispersionskoeffizienten von CDax=0,00153 m in den übrigen Säulen (quasistationärer Zustand)
Dabei zeigt sich in Kontrast zum Normalfall, in dem aufgrund des symmetrischen Charakters der
simulierten Modellanlage alle Minimalwerte (siehe Abbildung 2.12) auf dem gleichen Niveau
liegen, bei Annahme einer erhöhten axialen Dispersion in der dritten Säule, daß die Minimalwerte
nicht nur ansteigen, sondern daß sie auch während eines Taktzyklus schwanken. Dies ist
folgendermaßen zu erklären: Wie bereits Abbildung 7.8 verdeutlicht hat, führt eine erhöhte Dis-
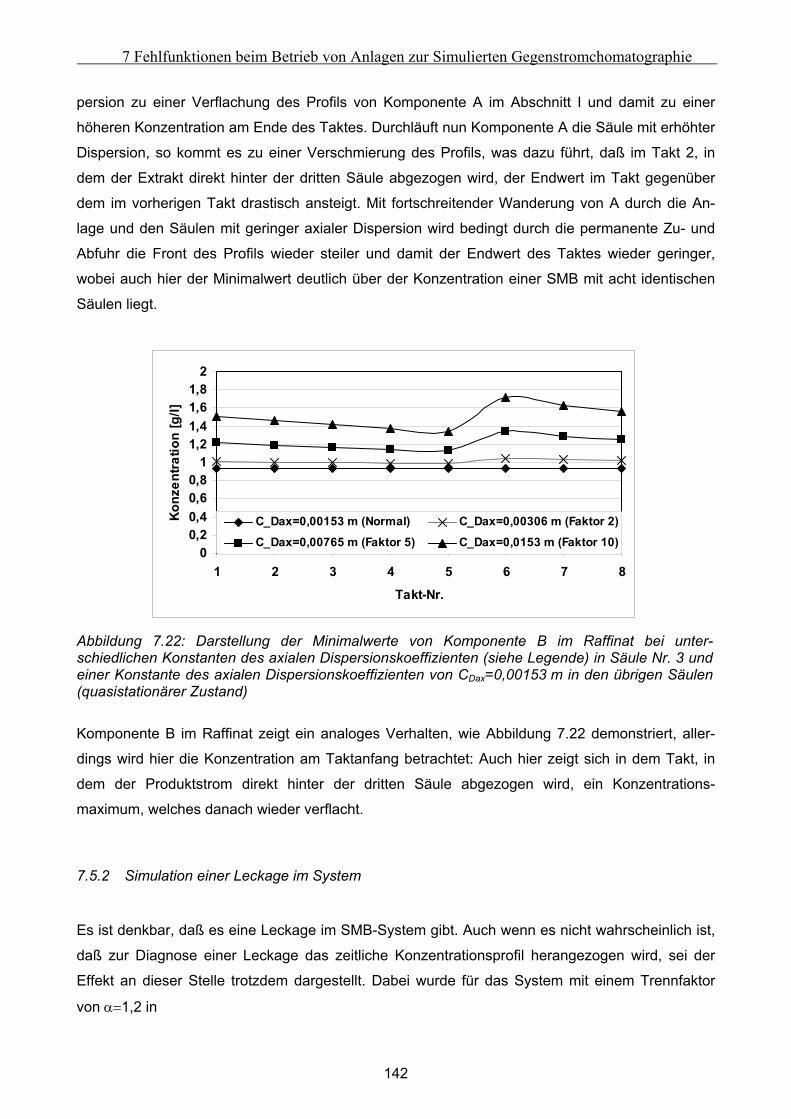
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
142
persion zu einer Verflachung des Profils von Komponente A im Abschnitt I und damit zu einer
höheren Konzentration am Ende des Taktes. Durchläuft nun Komponente A die Säule mit erhöhter
Dispersion, so kommt es zu einer Verschmierung des Profils, was dazu führt, daß im Takt 2, in
dem der Extrakt direkt hinter der dritten Säule abgezogen wird, der Endwert im Takt gegenüber
dem im vorherigen Takt drastisch ansteigt. Mit fortschreitender Wanderung von A durch die An-
lage und den Säulen mit geringer axialer Dispersion wird bedingt durch die permanente Zu- und
Abfuhr die Front des Profils wieder steiler und damit der Endwert des Taktes wieder geringer,
wobei auch hier der Minimalwert deutlich über der Konzentration einer SMB mit acht identischen
Säulen liegt.
00,20,40,60,8
11,21,41,61,8
2
1 2 3 4 5 6 7 8
Takt-Nr.
Konz
entr
atio
n [g
/l]
C_Dax=0,00153 m (Normal) C_Dax=0,00306 m (Faktor 2)
C_Dax=0,00765 m (Faktor 5) C_Dax=0,0153 m (Faktor 10)
Abbildung 7.22: Darstellung der Minimalwerte von Komponente B im Raffinat bei unter-schiedlichen Konstanten des axialen Dispersionskoeffizienten (siehe Legende) in Säule Nr. 3 und einer Konstante des axialen Dispersionskoeffizienten von CDax=0,00153 m in den übrigen Säulen (quasistationärer Zustand)
Komponente B im Raffinat zeigt ein analoges Verhalten, wie Abbildung 7.22 demonstriert, aller-
dings wird hier die Konzentration am Taktanfang betrachtet: Auch hier zeigt sich in dem Takt, in
dem der Produktstrom direkt hinter der dritten Säule abgezogen wird, ein Konzentrations-
maximum, welches danach wieder verflacht.
7.5.2 Simulation einer Leckage im System
Es ist denkbar, daß es eine Leckage im SMB-System gibt. Auch wenn es nicht wahrscheinlich ist,
daß zur Diagnose einer Leckage das zeitliche Konzentrationsprofil herangezogen wird, sei der
Effekt an dieser Stelle trotzdem dargestellt. Dabei wurde für das System mit einem Trennfaktor
von α=1,2 in
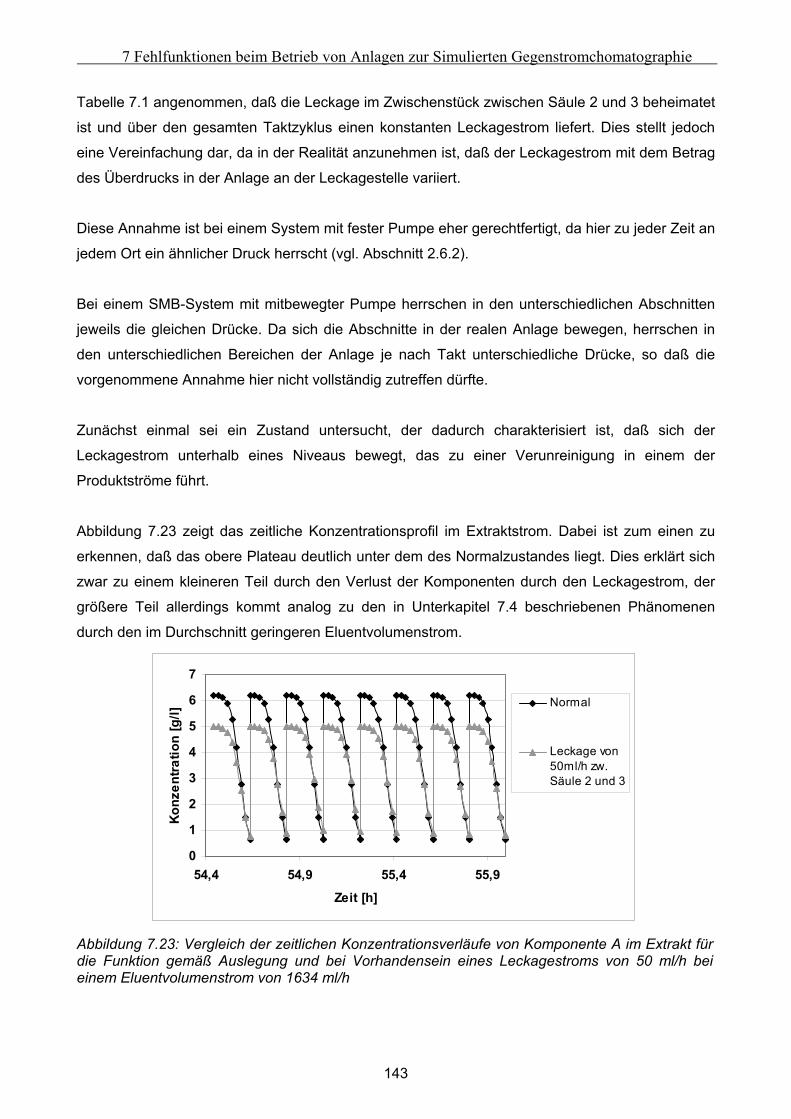
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
143
Tabelle 7.1 angenommen, daß die Leckage im Zwischenstück zwischen Säule 2 und 3 beheimatet
ist und über den gesamten Taktzyklus einen konstanten Leckagestrom liefert. Dies stellt jedoch
eine Vereinfachung dar, da in der Realität anzunehmen ist, daß der Leckagestrom mit dem Betrag
des Überdrucks in der Anlage an der Leckagestelle variiert.
Diese Annahme ist bei einem System mit fester Pumpe eher gerechtfertigt, da hier zu jeder Zeit an
jedem Ort ein ähnlicher Druck herrscht (vgl. Abschnitt 2.6.2).
Bei einem SMB-System mit mitbewegter Pumpe herrschen in den unterschiedlichen Abschnitten
jeweils die gleichen Drücke. Da sich die Abschnitte in der realen Anlage bewegen, herrschen in
den unterschiedlichen Bereichen der Anlage je nach Takt unterschiedliche Drücke, so daß die
vorgenommene Annahme hier nicht vollständig zutreffen dürfte.
Zunächst einmal sei ein Zustand untersucht, der dadurch charakterisiert ist, daß sich der
Leckagestrom unterhalb eines Niveaus bewegt, das zu einer Verunreinigung in einem der
Produktströme führt.
Abbildung 7.23 zeigt das zeitliche Konzentrationsprofil im Extraktstrom. Dabei ist zum einen zu
erkennen, daß das obere Plateau deutlich unter dem des Normalzustandes liegt. Dies erklärt sich
zwar zu einem kleineren Teil durch den Verlust der Komponenten durch den Leckagestrom, der
größere Teil allerdings kommt analog zu den in Unterkapitel 7.4 beschriebenen Phänomenen
durch den im Durchschnitt geringeren Eluentvolumenstrom.
0
1
2
3
4
5
6
7
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l]
Normal
Leckage von50ml/h zw.Säule 2 und 3
Abbildung 7.23: Vergleich der zeitlichen Konzentrationsverläufe von Komponente A im Extrakt für die Funktion gemäß Auslegung und bei Vorhandensein eines Leckagestroms von 50 ml/h bei einem Eluentvolumenstrom von 1634 ml/h
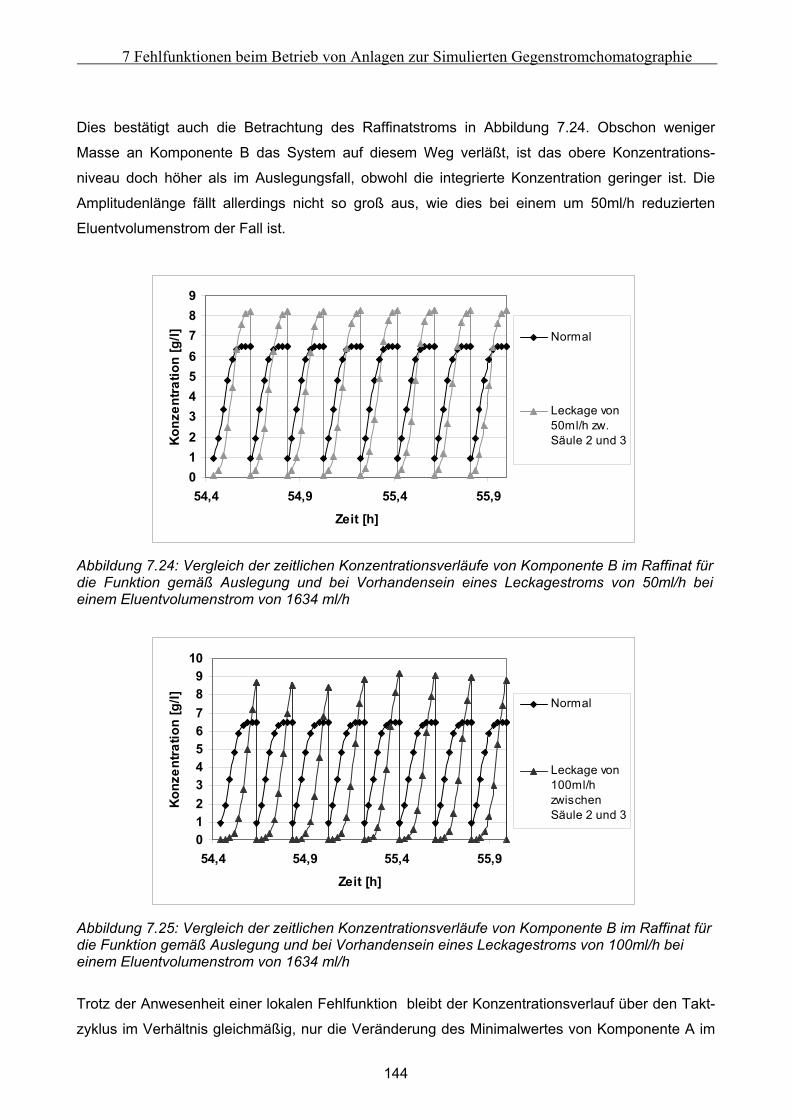
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
144
Dies bestätigt auch die Betrachtung des Raffinatstroms in Abbildung 7.24. Obschon weniger
Masse an Komponente B das System auf diesem Weg verläßt, ist das obere Konzentrations-
niveau doch höher als im Auslegungsfall, obwohl die integrierte Konzentration geringer ist. Die
Amplitudenlänge fällt allerdings nicht so groß aus, wie dies bei einem um 50ml/h reduzierten
Eluentvolumenstrom der Fall ist.
0123456789
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l] Normal
Leckage von50ml/h zw.Säule 2 und 3
Abbildung 7.24: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Raffinat für die Funktion gemäß Auslegung und bei Vorhandensein eines Leckagestroms von 50ml/h bei einem Eluentvolumenstrom von 1634 ml/h
0123456789
10
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l] Normal
Leckage von100ml/hzwischenSäule 2 und 3
Abbildung 7.25: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Raffinat für die Funktion gemäß Auslegung und bei Vorhandensein eines Leckagestroms von 100ml/h bei einem Eluentvolumenstrom von 1634 ml/h
Trotz der Anwesenheit einer lokalen Fehlfunktion bleibt der Konzentrationsverlauf über den Takt-
zyklus im Verhältnis gleichmäßig, nur die Veränderung des Minimalwertes von Komponente A im

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
145
Extrakt zeugt von der Assymmetrie der Volumenströme. Bei einer Verdoppelung des Leckage-
stroms (Abbildung 7.25) zeigt sich auch im Raffinat eine Variation des Maximalwertes des
zeitlichen Konzentrationsprofils.
Aus den Ergebnissen der weiterführenden Simulationen konnte die Erkenntnis gewonnen werden,
daß eine Leckage den gleichen Gesetzmäßigkeiten gehorcht wie die Verringerung des
Eluentvolumenstroms, allerdings reagiert das System auf ein bestimmtes Defizit an Eluent-
volumenstrom sehr viel empfindlicher als auf den gleichen Betrag als Leckagestrom. Das ist
dadurch zu erklären, daß von der Eluentpumpe bis zur Leckagestelle die Volumenströme dem Soll
entsprechen. Dieser Bereich ist je nach Takt zwischen null und sieben Säulen lang. Bei einer
defekten Eluentpumpe hingegen entspricht der Volumenstrom zu keiner Zeit dem Soll. Dadurch
schwächt sich der Effekt der Leckage insgesamt ab.
7.5.3 Simulation undichter Ventile
Im Normalzustand ist eine SMB-Anlage so geschaltet, daß es keine verzweigenden Ströme gibt,
die an anderer Stelle wieder zusammenfließen. Sind bestimmte Ventile jedoch undicht in dem
Sinne, daß Substanz durch das Ventil in den daran angrenzenden Rohrteil gelangt, obwohl dies
nicht vorgesehen ist, so kommt es zum Auftreten einer solchen Konstellation. Dies hat zur
Konsequenz, daß es zur Simulation der dadurch entstehenden Effekte notwendig ist zu wissen,
welcher Volumenstrom sich auf den beiden alternativen Strecken in jedem der 8 verschiedenen
Takte einstellt.
Abbildung 7.26: Strömungskonstellation bei Defekt des Raffinatventils VR2 im 8.Takt (Ausschnitt aus Abbildung 7.1)

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
146
Abbildung 7.26 als Ausschnitt aus Abbildung 7.1 verdeutlicht diese Problematik anhand der
Strömungskonstellation für ein undichtes Raffinatventil VR2 im achten Takt eines Taktzyklus.
Zwischen den Punkten A und B gibt es dabei zwei Wege für den Flüssgkeitsstrom. Zur
Berechnung der sich auf diese beiden Wege einstellenden Volumenströme ist es notwendig, die
Druckverluste der beiden alternativen Ströme zwischen Punkt A und B zu berechnen,
gleichzusetzen und die Volumenströme zu berechnen. Dabei ist es ein Unterschied, welcher
Ventiltyp defekt ist, also ob es ein Ventil ist, das den Eluent-, den Feed-, den Extrakt oder
Raffinatstrom absperrt. Da der spezifische Druckverlust in den Leitungen sehr viel geringer ist als
in den Schüttungen, kommt es schon bei kleinen Undichtigkeiten der Ventile zu durchaus
signifikanten Strömungen über den Rohrbereich.
Zur praktischen Durchführung der Simulationen ist erwähnenswert, daß jeweils ein undichtes
Eluentventil (VD3), ein Feed- (VF3), ein Extrakt- (VE2) und ein Raffinatventil (VR2) simuliert
wurde. Diese befinden sich jeweils zwischen Säule 2 und 3.
Diese Fehler unterscheiden sich von den anderen untersuchten Fehlern in der SMB insofern, als
daß deren Ausmaß sehr stark von der Bauart der Anlage aber auch von der Korngröße der
stationären Phase abhängt, die wiederum für den Druckverlust innerhalb der chromato-
graphischen Säulen verantwortlich zeichnet. Auch die Bauart der Ventile und ihr Druckverlust ist
von entscheidender Bedeutung für die Ausbildung der Ströme. Weiterhin sehr wichtig ist auch,
welche flächenmäßige Abmessungen die Undichtigkeit im Ventil hat.
Die Auswertung der Simulationen zeigte kein eindeutig identifizierbares Muster der
Konzentrationsverläufe in den Produktströmen für die untersuchten Systeme. Verkompliziert wird
die Sache auch dadurch, daß es bei einem während der gesamten Simulationszeit vollständig
geöffneten Ventil teilweise sogar zu Strömungen entgegen der vorgesehenen Fließrichtung
kommt.
Aus diesen Gründen wird an dieser Stelle nicht weiter auf die Konzentrationsprofile eingegangen,
sondern der Fokus soll vielmehr auf ein anderes Charakteristikum gelenkt werden, welches eher
zur Identifikation geeignet ist, nämlich dem Verhalten des Druckes an verschiedenen Stellen des
Systems. Ein solches Verhalten ist dann identifizierbar, wenn es Drucksensoren an verschiedenen
Stellen des Systems gibt.
Bei einer Verzweigung der Strömung wie in Abbildung 7.26 fließt je nach Größe der Undichtigkeit
im Ventil zum Teil sehr viel weniger Flüssigkeit durch die Säulen zwischen den Punkten A und B,
als dies bei einer einwandfrei arbeitenden Anlage der Fall wäre. Da der Druckverlust gemäß

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
147
Gleichung (2.82) proportional dem Volumenstrom ist, unterscheiden sich die Drücke an einzelnen
Stellen des Flüssigkeitskreislaufes von denen des normalen Laufes. Nimmt man an, daß sich das
Ventil gänzlich öffnet, sofern es gemäß Regelung geöffnet zu sein hat, so entsprechen die
Druckverluste während eines Taktes denen des normalen Verlaufes. Dadurch läßt sich der Fehler
auf die vier oder fünf Ventile, die in diesem Takt offen stehen, je nach Bauweise der SMB,
eingrenzen. Anhand der Drücke an den einzelnen Stellen läßt sich dann auch ermitteln, welches
der in Frage kommenden Ventile defekt ist. Dies wird ermöglicht durch die Tatsache, daß
zwischen Zu- und Abflußstelle signifikant weniger Flüssigkeit strömt als in den anderen Bereichen
bzw. im fehlerlosen Zustand. Durch die Druckverluste läßt sich darauf schließen, zwischen
welchen Stellen des Systems eine Abkürzungsströmung besteht.
Dieses Vorgehen setzt voraus, daß die Anlage über mehrere Drucksensoren möglichst vor oder
nach jeder Säule verfügt.
7.5.4 Simulation geschlossener Ventile
Neben der Möglichkeit undicht zu sein, ist auch annehmbar, daß ein bestimmtes Ventil sich nicht
öffnet, wenn es durch den elektrischen Impuls von der Regelung dazu aufgefordert wird. Hier
wurden wiederum jeweils Ventile im Zwischenstück zwischen Säule 2 und 3 zur Modellierung
herangezogen, wobei jeweils das Versagen eines Feed-, eines Extrakt- und eines Raffinatventils
simuliert wurde. Das Versagen eines Desorbentventils wurde nicht untersucht, da dies aufgrund
des völligen Ausbleibens des Desorbentstroms in einem Takt zu numerischen Schwierigkeiten
führte, die den Absturz der Simulationen zur Folge hatten.
7.5.4.1 Simulation eines geschlossenen Extraktventils
Abbildung 7.27 vergleicht das zeitliche Konzentrationsprofil von Komponente A im Extrakt mit dem
Profil, das beim Geschlossenbleiben des Extraktventils VR2 simuliert wurde. Besonders
charakteristisch ist dabei das Ausbleiben eines Konzentrationsprofils in dem Takt, in dem das
defekte Ventil geöffnet sein müßte. Die Simulation nimmt in diesem Fall die Konzentration von 0g/l
an. In einer realen Anlage wäre dabei zu erwarten, daß die Konzentration detektiert wird, die am
Ende des vorhergehenden Taktes im Extraktauslaß vorherrschte, da die Flüssigkeit mit dieser
Konzentration im Detektor steht.
Da pro Taktzyklus in einem vollen Takt kein Extrakt abgezogen wird, wird pro Taktzyklus nur 7/8
des eingestellten Extraktvolumenstroms abgezogen. Vergleicht man das Profil, das durch das
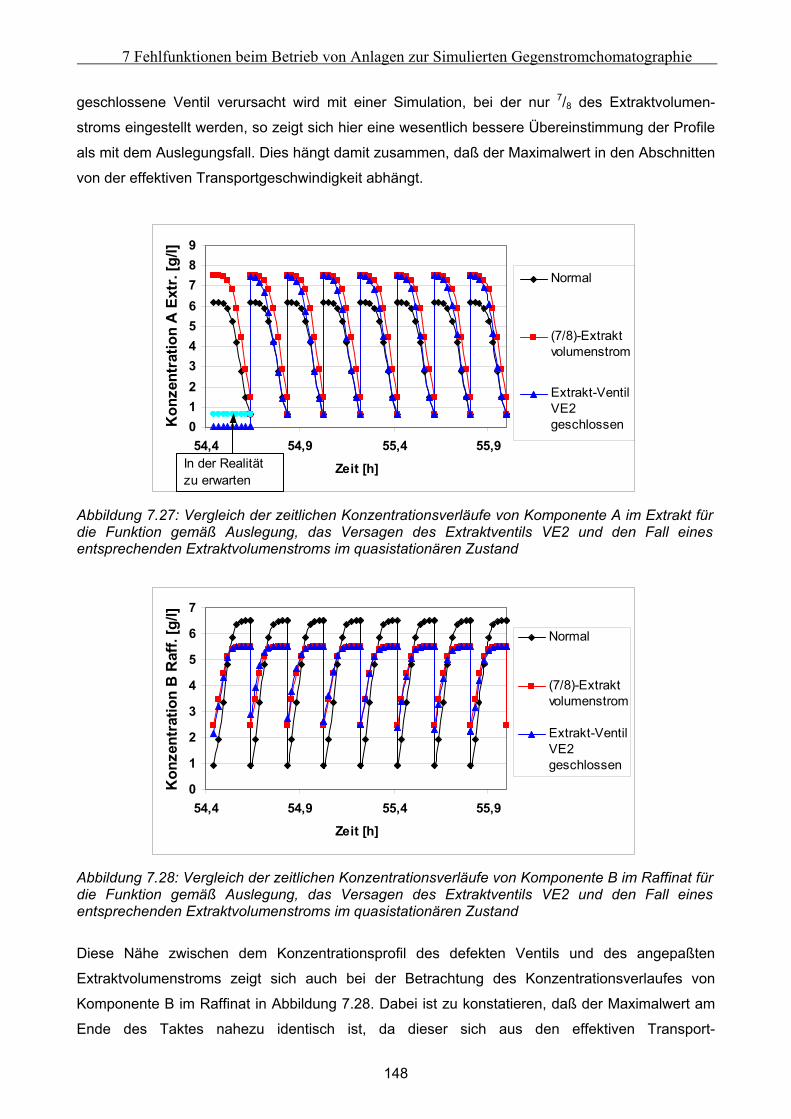
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
148
geschlossene Ventil verursacht wird mit einer Simulation, bei der nur 7/8 des Extraktvolumen-
stroms eingestellt werden, so zeigt sich hier eine wesentlich bessere Übereinstimmung der Profile
als mit dem Auslegungsfall. Dies hängt damit zusammen, daß der Maximalwert in den Abschnitten
von der effektiven Transportgeschwindigkeit abhängt.
0123456789
54,4 54,9 55,4 55,9
Zeit [h]
Kon
zent
ratio
n A
Ext
r. [g
/l]
Normal
(7/8)-Extraktvolumenstrom
Extrakt-VentilVE2geschlossen
In der Realität zu erwarten
Abbildung 7.27: Vergleich der zeitlichen Konzentrationsverläufe von Komponente A im Extrakt für die Funktion gemäß Auslegung, das Versagen des Extraktventils VE2 und den Fall eines entsprechenden Extraktvolumenstroms im quasistationären Zustand
0
1
2
3
4
5
6
7
54,4 54,9 55,4 55,9
Zeit [h]
Kon
zent
ratio
n B
Raf
f. [g
/l]
Normal
(7/8)-Extraktvolumenstrom
Extrakt-VentilVE2geschlossen
Abbildung 7.28: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Raffinat für die Funktion gemäß Auslegung, das Versagen des Extraktventils VE2 und den Fall eines entsprechenden Extraktvolumenstroms im quasistationären Zustand
Diese Nähe zwischen dem Konzentrationsprofil des defekten Ventils und des angepaßten
Extraktvolumenstroms zeigt sich auch bei der Betrachtung des Konzentrationsverlaufes von
Komponente B im Raffinat in Abbildung 7.28. Dabei ist zu konstatieren, daß der Maximalwert am
Ende des Taktes nahezu identisch ist, da dieser sich aus den effektiven Transport-
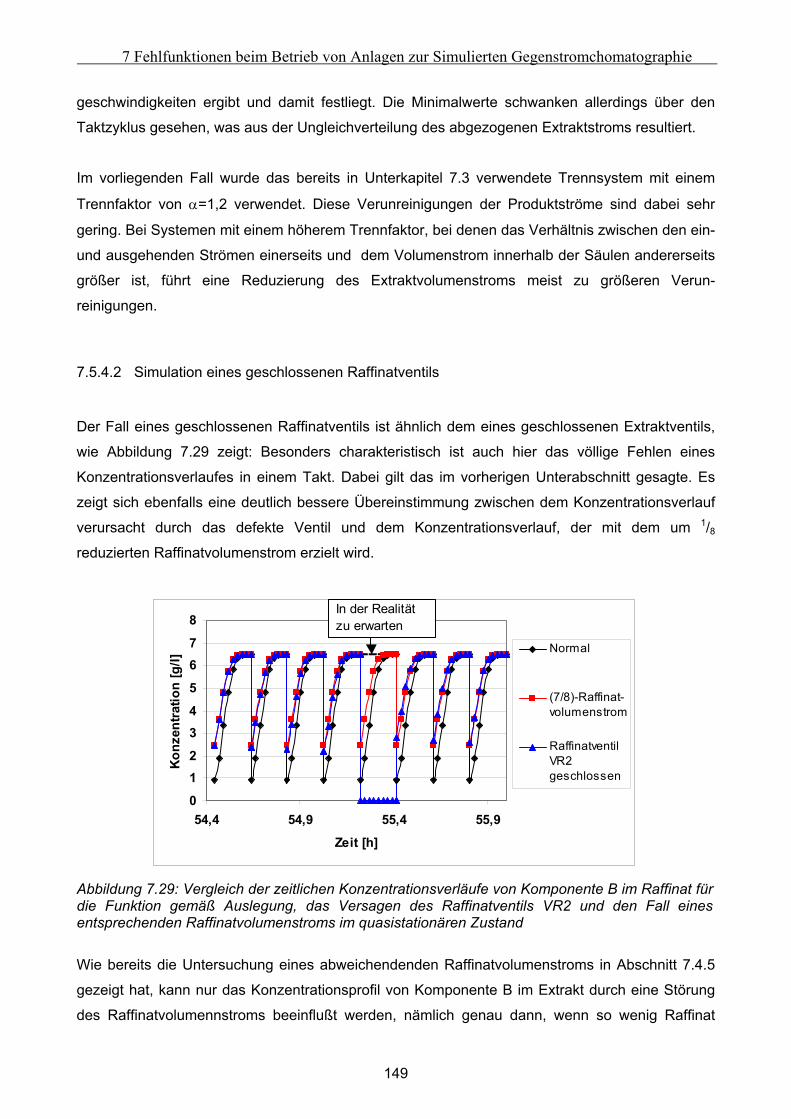
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
149
geschwindigkeiten ergibt und damit festliegt. Die Minimalwerte schwanken allerdings über den
Taktzyklus gesehen, was aus der Ungleichverteilung des abgezogenen Extraktstroms resultiert.
Im vorliegenden Fall wurde das bereits in Unterkapitel 7.3 verwendete Trennsystem mit einem
Trennfaktor von α=1,2 verwendet. Diese Verunreinigungen der Produktströme sind dabei sehr
gering. Bei Systemen mit einem höherem Trennfaktor, bei denen das Verhältnis zwischen den ein-
und ausgehenden Strömen einerseits und dem Volumenstrom innerhalb der Säulen andererseits
größer ist, führt eine Reduzierung des Extraktvolumenstroms meist zu größeren Verun-
reinigungen.
7.5.4.2 Simulation eines geschlossenen Raffinatventils
Der Fall eines geschlossenen Raffinatventils ist ähnlich dem eines geschlossenen Extraktventils,
wie Abbildung 7.29 zeigt: Besonders charakteristisch ist auch hier das völlige Fehlen eines
Konzentrationsverlaufes in einem Takt. Dabei gilt das im vorherigen Unterabschnitt gesagte. Es
zeigt sich ebenfalls eine deutlich bessere Übereinstimmung zwischen dem Konzentrationsverlauf
verursacht durch das defekte Ventil und dem Konzentrationsverlauf, der mit dem um 1/8
reduzierten Raffinatvolumenstrom erzielt wird.
0
12
34
5
67
8
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l]
Normal
(7/8)-Raffinat-volumenstrom
RaffinatventilVR2geschlossen
In der Realität zu erwarten
Abbildung 7.29: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Raffinat für die Funktion gemäß Auslegung, das Versagen des Raffinatventils VR2 und den Fall eines entsprechenden Raffinatvolumenstroms im quasistationären Zustand
Wie bereits die Untersuchung eines abweichendenden Raffinatvolumenstroms in Abschnitt 7.4.5
gezeigt hat, kann nur das Konzentrationsprofil von Komponente B im Extrakt durch eine Störung
des Raffinatvolumennstroms beeinflußt werden, nämlich genau dann, wenn so wenig Raffinat
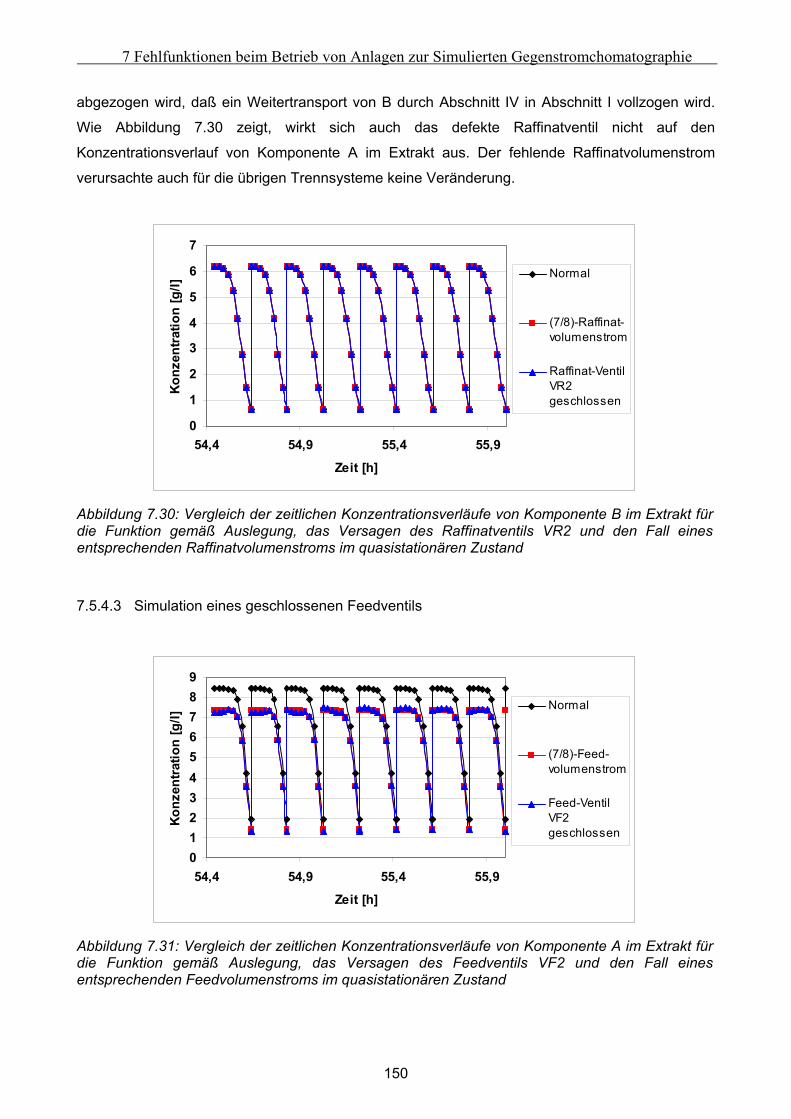
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
150
abgezogen wird, daß ein Weitertransport von B durch Abschnitt IV in Abschnitt I vollzogen wird.
Wie Abbildung 7.30 zeigt, wirkt sich auch das defekte Raffinatventil nicht auf den
Konzentrationsverlauf von Komponente A im Extrakt aus. Der fehlende Raffinatvolumenstrom
verursachte auch für die übrigen Trennsysteme keine Veränderung.
0
1
2
3
4
5
6
7
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l]
Normal
(7/8)-Raffinat-volumenstrom
Raffinat-VentilVR2geschlossen
Abbildung 7.30: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Extrakt für die Funktion gemäß Auslegung, das Versagen des Raffinatventils VR2 und den Fall eines entsprechenden Raffinatvolumenstroms im quasistationären Zustand
7.5.4.3 Simulation eines geschlossenen Feedventils
0123456789
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l]
Normal
(7/8)-Feed-volumenstrom
Feed-VentilVF2geschlossen
Abbildung 7.31: Vergleich der zeitlichen Konzentrationsverläufe von Komponente A im Extrakt für die Funktion gemäß Auslegung, das Versagen des Feedventils VF2 und den Fall eines entsprechenden Feedvolumenstroms im quasistationären Zustand
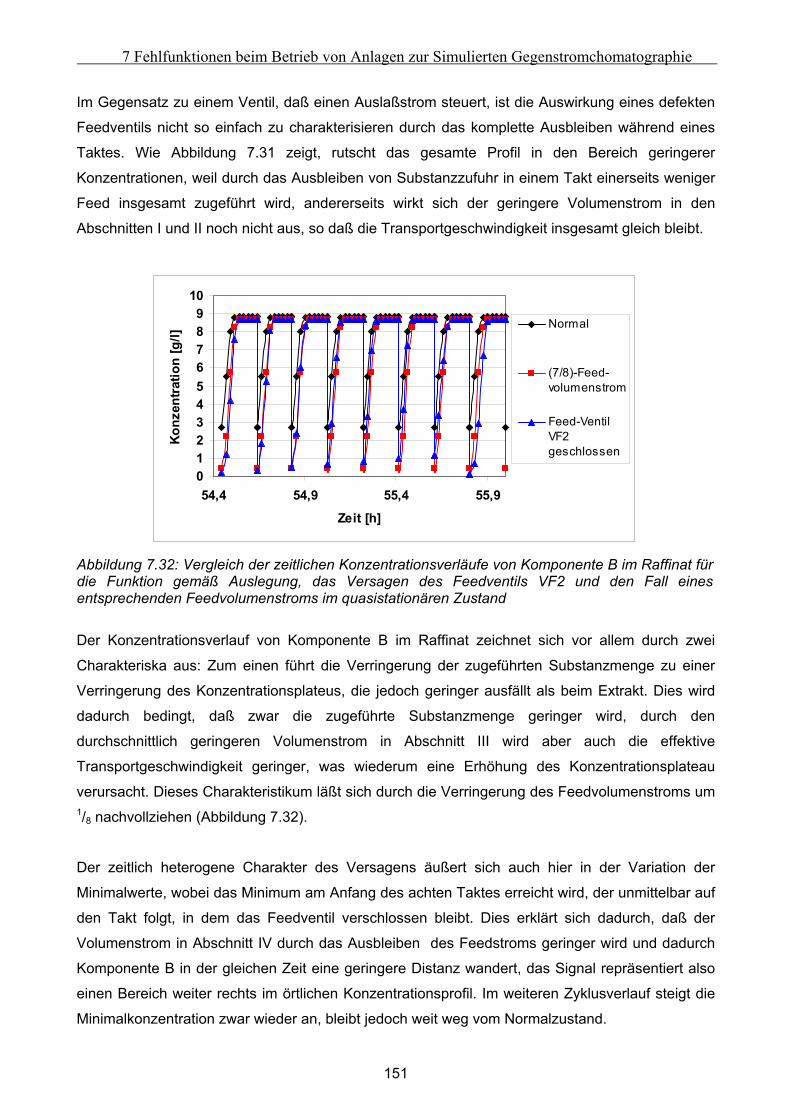
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
151
Im Gegensatz zu einem Ventil, daß einen Auslaßstrom steuert, ist die Auswirkung eines defekten
Feedventils nicht so einfach zu charakterisieren durch das komplette Ausbleiben während eines
Taktes. Wie Abbildung 7.31 zeigt, rutscht das gesamte Profil in den Bereich geringerer
Konzentrationen, weil durch das Ausbleiben von Substanzzufuhr in einem Takt einerseits weniger
Feed insgesamt zugeführt wird, andererseits wirkt sich der geringere Volumenstrom in den
Abschnitten I und II noch nicht aus, so daß die Transportgeschwindigkeit insgesamt gleich bleibt.
0123456789
10
54,4 54,9 55,4 55,9
Zeit [h]
Konz
entra
tion
[g/l]
Normal
(7/8)-Feed-volumenstrom
Feed-VentilVF2geschlossen
Abbildung 7.32: Vergleich der zeitlichen Konzentrationsverläufe von Komponente B im Raffinat für die Funktion gemäß Auslegung, das Versagen des Feedventils VF2 und den Fall eines entsprechenden Feedvolumenstroms im quasistationären Zustand
Der Konzentrationsverlauf von Komponente B im Raffinat zeichnet sich vor allem durch zwei
Charakteriska aus: Zum einen führt die Verringerung der zugeführten Substanzmenge zu einer
Verringerung des Konzentrationsplateus, die jedoch geringer ausfällt als beim Extrakt. Dies wird
dadurch bedingt, daß zwar die zugeführte Substanzmenge geringer wird, durch den
durchschnittlich geringeren Volumenstrom in Abschnitt III wird aber auch die effektive
Transportgeschwindigkeit geringer, was wiederum eine Erhöhung des Konzentrationsplateau
verursacht. Dieses Charakteristikum läßt sich durch die Verringerung des Feedvolumenstroms um 1/8 nachvollziehen (Abbildung 7.32).
Der zeitlich heterogene Charakter des Versagens äußert sich auch hier in der Variation der
Minimalwerte, wobei das Minimum am Anfang des achten Taktes erreicht wird, der unmittelbar auf
den Takt folgt, in dem das Feedventil verschlossen bleibt. Dies erklärt sich dadurch, daß der
Volumenstrom in Abschnitt IV durch das Ausbleiben des Feedstroms geringer wird und dadurch
Komponente B in der gleichen Zeit eine geringere Distanz wandert, das Signal repräsentiert also
einen Bereich weiter rechts im örtlichen Konzentrationsprofil. Im weiteren Zyklusverlauf steigt die
Minimalkonzentration zwar wieder an, bleibt jedoch weit weg vom Normalzustand.
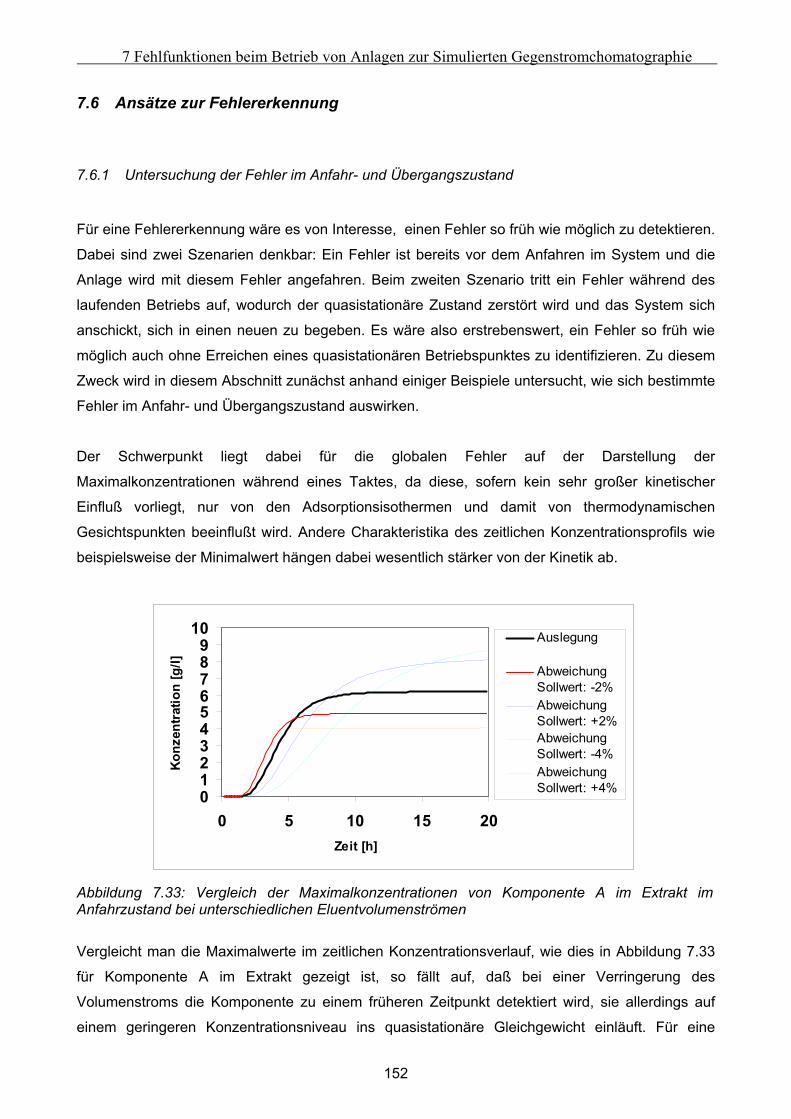
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
152
7.6 Ansätze zur Fehlererkennung
7.6.1 Untersuchung der Fehler im Anfahr- und Übergangszustand
Für eine Fehlererkennung wäre es von Interesse, einen Fehler so früh wie möglich zu detektieren.
Dabei sind zwei Szenarien denkbar: Ein Fehler ist bereits vor dem Anfahren im System und die
Anlage wird mit diesem Fehler angefahren. Beim zweiten Szenario tritt ein Fehler während des
laufenden Betriebs auf, wodurch der quasistationäre Zustand zerstört wird und das System sich
anschickt, sich in einen neuen zu begeben. Es wäre also erstrebenswert, ein Fehler so früh wie
möglich auch ohne Erreichen eines quasistationären Betriebspunktes zu identifizieren. Zu diesem
Zweck wird in diesem Abschnitt zunächst anhand einiger Beispiele untersucht, wie sich bestimmte
Fehler im Anfahr- und Übergangszustand auswirken.
Der Schwerpunkt liegt dabei für die globalen Fehler auf der Darstellung der
Maximalkonzentrationen während eines Taktes, da diese, sofern kein sehr großer kinetischer
Einfluß vorliegt, nur von den Adsorptionsisothermen und damit von thermodynamischen
Gesichtspunkten beeinflußt wird. Andere Charakteristika des zeitlichen Konzentrationsprofils wie
beispielsweise der Minimalwert hängen dabei wesentlich stärker von der Kinetik ab.
0123456789
10
0 5 10 15 20Zeit [h]
Konz
entr
atio
n [g
/l]
Auslegung
AbweichungSollwert: -2%AbweichungSollwert: +2%AbweichungSollwert: -4%AbweichungSollwert: +4%
Abbildung 7.33: Vergleich der Maximalkonzentrationen von Komponente A im Extrakt im Anfahrzustand bei unterschiedlichen Eluentvolumenströmen
Vergleicht man die Maximalwerte im zeitlichen Konzentrationsverlauf, wie dies in Abbildung 7.33
für Komponente A im Extrakt gezeigt ist, so fällt auf, daß bei einer Verringerung des
Volumenstroms die Komponente zu einem früheren Zeitpunkt detektiert wird, sie allerdings auf
einem geringeren Konzentrationsniveau ins quasistationäre Gleichgewicht einläuft. Für eine
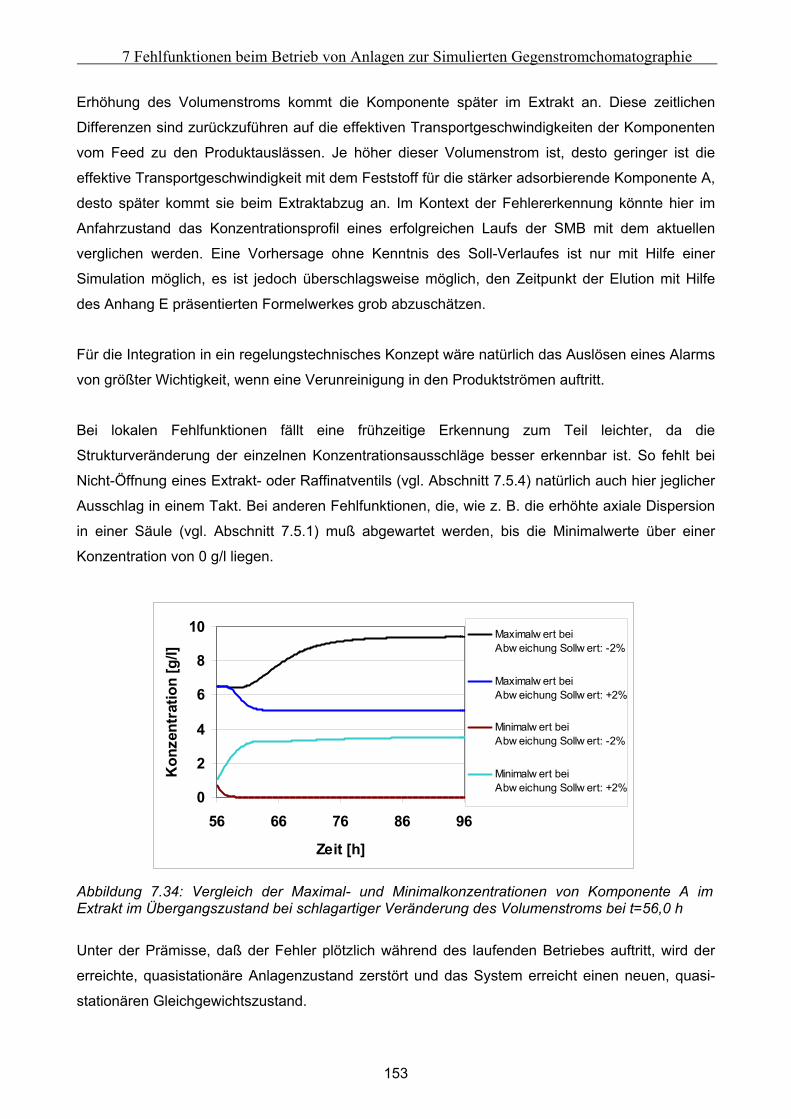
7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
153
Erhöhung des Volumenstroms kommt die Komponente später im Extrakt an. Diese zeitlichen
Differenzen sind zurückzuführen auf die effektiven Transportgeschwindigkeiten der Komponenten
vom Feed zu den Produktauslässen. Je höher dieser Volumenstrom ist, desto geringer ist die
effektive Transportgeschwindigkeit mit dem Feststoff für die stärker adsorbierende Komponente A,
desto später kommt sie beim Extraktabzug an. Im Kontext der Fehlererkennung könnte hier im
Anfahrzustand das Konzentrationsprofil eines erfolgreichen Laufs der SMB mit dem aktuellen
verglichen werden. Eine Vorhersage ohne Kenntnis des Soll-Verlaufes ist nur mit Hilfe einer
Simulation möglich, es ist jedoch überschlagsweise möglich, den Zeitpunkt der Elution mit Hilfe
des Anhang E präsentierten Formelwerkes grob abzuschätzen.
Für die Integration in ein regelungstechnisches Konzept wäre natürlich das Auslösen eines Alarms
von größter Wichtigkeit, wenn eine Verunreinigung in den Produktströmen auftritt.
Bei lokalen Fehlfunktionen fällt eine frühzeitige Erkennung zum Teil leichter, da die
Strukturveränderung der einzelnen Konzentrationsausschläge besser erkennbar ist. So fehlt bei
Nicht-Öffnung eines Extrakt- oder Raffinatventils (vgl. Abschnitt 7.5.4) natürlich auch hier jeglicher
Ausschlag in einem Takt. Bei anderen Fehlfunktionen, die, wie z. B. die erhöhte axiale Dispersion
in einer Säule (vgl. Abschnitt 7.5.1) muß abgewartet werden, bis die Minimalwerte über einer
Konzentration von 0 g/l liegen.
0
2
4
6
8
10
56 66 76 86 96
Zeit [h]
Kon
zent
ratio
n [g
/l]
Maximalw ert beiAbw eichung Sollw ert: -2%
Maximalw ert beiAbw eichung Sollw ert: +2%
Minimalw ert beiAbw eichung Sollw ert: -2%
Minimalw ert beiAbw eichung Sollw ert: +2%
Abbildung 7.34: Vergleich der Maximal- und Minimalkonzentrationen von Komponente A im Extrakt im Übergangszustand bei schlagartiger Veränderung des Volumenstroms bei t=56,0 h
Unter der Prämisse, daß der Fehler plötzlich während des laufenden Betriebes auftritt, wird der
erreichte, quasistationäre Anlagenzustand zerstört und das System erreicht einen neuen, quasi-
stationären Gleichgewichtszustand.

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
154
Einen solchen Fall zeigt Abbildung 7.34. Hier wurde der Volumenstrom bei t=56,0 h schlagartig
verändert. Es zeigt sich, daß die Zeit bis zur Einstellung eines neuen quasistationären Zustandes
sich in ähnlichen Dimensionen bewegt wie im Anfahrzustand, da auch hier die effektive
Transportgeschwindigkeit von entscheidender Bedeutung ist.
Für die lokalen Fehler gelten für den Übergangszustand die gleichen Aussagen wie für den
Anfahrvorgang. Auch hier kommt es sehr schnell zu einer Veränderung der Struktur der einzelnen
Konzentrationsausschläge.
7.6.2 Möglichkeiten der Fehlererkennung
In dieser Arbeit steht die phänomenologische Betrachtung der einzelnen Fehler im Vordergrund.
Im folgenden werden Hinweise zur Umsetzung entsprechender Diagnosekonzepte gegeben,
deren Realisierung in Folgearbeiten geplant ist.
Die herausgearbeiteten Charakteristika der einzelnen untersuchten Fehlfunktionen legen die
Möglichkeit einer Identifizierung bestimmter Fehler nahe. Die Simulationen mit gPROMS geben
die Möglichkeit, die Konzentrationsverläufe beider zu trennender Komponenten separat zu
erfassen und zu betrachten. Diese Möglichkeit besteht bei vielen realen Trennsystemen nicht.
Hier liegt häufig der Fall so, daß die Detektoren ein Summensignal für beide Komponenten bilden,
was zum Beispiel bei UV-Detektoren oder auch Refraktometern häufig der Fall ist. Erschwerend
kommt hier meist dazu, daß die Konzentrationen der beiden Komponenten mit unterschiedlicher
Gewichtung in das addierte Signal eingehen. Aber auch eine Substraktion der Konzentrationen im
Detektionssignal ist möglich, etwa bei Nutzung eines Polarimeters.
Es gäbe mehrere Möglichkeiten wie sich diese Detektorproblematik umgehen oder lösen ließe.
Eine arbeitsaufwendige Möglichkeit wäre diejenige, Proben aus der laufenden Anlage zu
analysieren. Dies könnte so aussehen, daß zur Identifizierung eines Fehlers Proben aus den
Produktströmen am Anfang, in der Mitte und am Ende eines Taktes gezogen werden. Da im
Regelfall eine analytische Säule zur Vermessung der Adsorptionsisothermen vorhanden ist,
könnte die Zusammensetzung der Proben mit Hilfe der HPLC analysiert werden, um damit aus
den geschilderten Charakteristika auf den vorliegenden Fehler zu schließen.
Neben der zu prüfenden Möglichkeit, mit Hilfe der Kalibrierung des Detektorsignals eine Auskunft
über die Zusammensetzung zu erhalten, könnte sich auch aus den Arbeiten von Mangold (1993)
und Mangold et al. (1994) eine weitere Möglichkeit ergeben. Diese entwickelten für ihr

7 Fehlfunktionen beim Betrieb von Anlagen zur Simulierten Gegenstromchomatographie
155
Regelungskonzept eine Beobachterstrategie, die in der Lage ist, mehr Informationen über den
Zustand der Anlage zu liefern als aus den bloßen Meßwerten ersichtlich ist.

8 Zusammenfassung und Ausblick
156
8 Zusammenfassung und Ausblick
Im Mittelpunkt dieser Arbeit steht die Untersuchung von Fehlern in der präparativen Flüssig-
chromatographie. Die Untersuchungen konzentrierten sich dabei auf drei unterschiedliche
Komplexe: Den Vergleich von ausgewählten statischen und dynamischen Meßmethoden zur
Bestimmung von Adsorptionsisothermen unter der Maßgabe festzustellen, inwieweit eine
Vergleichbarkeit der Ergebnisse dieser Methoden gegeben ist und welche Schritte dazu notwendig
sind. Der zweite Komplex konzentriert sich auf Fehler in der Bettstruktur von Säulen zur
präparativen Batch-Chromatographie. Im Vordergrund steht dabei die Frage, welche Aus-
wirkungen bestimmte Fehler im chromatographischen Festbett auf Peaks von das Festbett durch-
laufender Substanzen haben. Der dritte Komplex schließlich befaßt sich mit der Auswirkung von
Fehlern, die in der simulierten Gegenstromchromatographie auftreten. Der Schwerpunkt liegt
dabei insbesondere in der Betrachtung von Fehlfunktionen peripherer Systeme; studiert werden
inbesondere die Auswirkungen dieser Fehler auf das Konzentrationsprofil und die Konzentrations-
verläufe in den Produktströmen.
Beim Vergleich der Ergebnisse erzielt unter Verwendung der Zirkulationsmethode als
Repräsentant statischer Meßmethoden bzw. der dynamischen Meßmethoden Frontalanalyse und
Petrubationmethode konnte gezeigt werden, daß für die untersuchten Systeme unter bestimmten
Bedingungen Ergebnisse erzielt werden können, deren Differenzen voneinander geringer inner-
halb des Bereiches der berechneten Messfehler liegen. Diese Bedingungen sind im einzelnen,
• daß die Abnahme der Porosität mit zunehmender Beladung bei der Auswertung der
dynamisch erzielten Meßergebnissen berücksichtigt wird,
• daß die stationäre Phase für statische und dynamische Messungen in einer ähnlichen Art und
Weise vorbereitet wird. Eine besondere Rolle spielt dabei die notwendige Vakuumtrocknung
unter Wärmeeinfluß sowie die Vermeidung von Flüssigkeitskontakt mit Ausnahme der
vermessenen Substanzen.
• und daß die Umrechnung der Exzeßdaten mit Hilfe der analytischen Lösung aus Unter-
abschnitt 5.2.1.1 vollzogen wird, was die Annahme beinhaltet, daß die Adsorbatphase aus-
schließlich aus dem stärker adsorbierenden Dichlormethan besteht und alle n-Hexan-Moleküle
Teil der fließenden Bulk-Phase sind.
Daraus können zwei bedeutende Schlußfolgerungen gezogen werden:

8 Zusammenfassung und Ausblick
157
• Es ist möglich, sowohl mit Hilfe der verwendeten statischen als auch den dynamischen
Meßmethoden die reine Thermodynamik zu messen. Zu diesem Zweck muß bei den
untersuchten System für jeden Meßpunkt die entsprechende effektive Porosität berücksichtigt
werden.
• Wenn volumenbezogene Adsorptionsisothermen für eine chromatographische Anwendung
benötigt werden, hat ihre Bestimmung mit Hilfe statischer Methoden zwei signifikante
Nachteile:
Bei der Umrechnung der gemessenen Exzeßdaten in Beladungsisothermen müssen die
Unterschiede der verschiedenen Umrechnungsmethoden zumindest in die allgemeinen
Überlegungen miteinbezogen werden. Gerade für den Bereich von Exzeßisothermen mit
Adsorptionsazeotrop ist ist hier auf jeden Fall weiterer Forschungsbedarf gegeben. Ferner
muß für die Umrechnung der Exzeßdaten in volumenbezogene Isothermen die effektive
Porosität als Funktion der Beladung bekannt sein.
Zur Betrachtung von fehlerhaft gepackten Festbetten wurden vor allem eindimensionale gPROMS-
und zweidimensionale CFD-Simulationen durchgeführt, zusätzlich wurden auch experimentelle
Untersuchungen durchgeführt. Folgende Punkte sind dabei festzustellen:
• Hauptverantwortlich für die Strömungsverteilung in Festbetten mit einer radialen
Heterogenität, was die Kanalbildung mit einschließt, ist der Druckverlust, der verursacht wird
durch die unterschiedlichen Porositäten. Kleinere Strömungsunterschiede verursachen dabei
zunächst ein Tailing oder Fronting, was bis zu einem Doppelpeakverhalten auswachsen kann.
• Der Leerraum über einem abgesackten Festbett nähert sich in seinen Auswirkungen
demjenigen mehrerer hintereinandergeschalteter idealer Rührkessel an. Dies führt zu einem
Tailing verbunden mit einer Zunahme der axialen Dispersion.
• Die Auswirkungen einer verstopften Auslaßfritte sind auch bei einer flächenmäßig großen
Nichtdurchlässigkeit sehr gering, einzig der Peak eluiert nach etwas kürzerer Zeit.
Hier bietet sich noch weiterer Forschungsbedarf, besonders die Ausweitung der Simulationen auf
den dreidimensionalen Bereich bietet Potential; dieses Unterfangen scheiterte im Rahmen dieser
Arbeit insbesondere an den Kapazitäten von Rechenzeit und Speicher. Aber auch direkte
Maßnahmen zur praktischen Identifizierung sind denkbar, z.B. eine speziell konstruierte
Auslaßfritte, die in der Lage ist, die radiale Verteilung der Strömungsgeschwindigkeiten zur
Identifizierung insbesondere von Inhomogenitäten in der Säule zu messen. Eine solche
Spezialfritte könnte kurzzeitig die normale Fritte zur Fehleridentifikation ersetzen.

8 Zusammenfassung und Ausblick
158
Zur Untersuchung von Fehlfunktionen in der simulierten Gegenstromchromatographie wurde die
Modellierung und Simulation unter Verwendung der Simulationsumgebung gPROMS zur
Erarbeitung der Gesetzmäßigkeiten genutzt. Wie bereits in Unterkapitel 7.2 diskutiert, konnten
dabei zwei Klassen von Fehlern ausgemacht werden: Zum einen die globalen Fehler, zu deren
Untersuchung neben der dynamischen Simulation auch ein theoretisches Modell auf Basis einer
integralen TMB-Bilanz herangezogen werden konnte, zum anderen lokale Fehler, zu deren
Untersuchung aufgrund ihres SMB-spezifischen Charakters alleinig die dynamische gPROMS-
Simulation herangezogen werden konnte. Untersucht wurden dabei die Auswirkungen häufig
auftretender Fehlfunktionen bei hypothetischen Trennsystemen mit verschieden großen
Trennfaktoren und linearen Adsorptionsisothermen sowie unterschiedlich stark ausgeprägten
Kinetiken.
Die Untersuchung globaler Fehler zeigte, daß abhängig von der Ausprägung der von den unter-
schiedlichen Volumenströmen abhängigen dominierenden Transportrichtungen in den einzelnen
Abschnitten es zu einer sehr unterschiedlichen Verteilung der zu trennenden Komponenten in der
Anlage kommt. Dabei wurden fünf Konstellationen dominierender Transportrichtungen ausge-
macht, untersucht und diesen Charakteristika zugeordnet.
Wenn es unter Nutzung des TMB-Bilanzmodells zu einer abrupten Veränderung bei einer
Veränderung der Transportrichtungen kommt, folgen die Konzentration der Simulationen, die das
kinetische Verhalten berücksichtigen, zwar denen des Bilanzmodells, jedoch mit wachsender
Trägheit. Nichtsdestotrotz konnte gezeigt werden, daß für jeden Fehler in den Pumpen bestimmte
Charakteristika in Konzentration und Amplitude auszumachen sind.
Bei den lokalen Fehlern wurden insgesamt vier verschiedene Fehlerquellen untersucht, die sich
zum Teil noch weiter unterteilen lassen. Es konnte an Beispielen gezeigt werden, daß drei dieser
Fehlerquellen sich durch bestimmte Charakteristika in den zeitlichen Konzentrationsverläufen
auszeichnen. Einzig nicht korrekt geschlossene Ventile zeichnen sich aufgrund der besonderen
Problematik nicht durch eindeutig identifizierbare Charakteristika aus, hier bliebe zur Identifikation
nur der Umweg über Druckmeßdaten aus der Anlage.
Insgesamt bietet die Identifizierung von Fehlern noch einen großen Forschungsbedarf:
Fanden im Rahmen der Untersuchungen, die im Rahmen dieser Arbeit durchgeführt wurden, nur
eine Betrachtung von Systemen mit linearen Adsorptionsisothermen statt, so ist hier sicherlich
eine tiefergreifende Erforschung der Einflüsse des Vorliegens nicht-linearer und gekoppelter
Adsorptionsisothermen von Interesse. Mit Hilfe dieser Daten könnte dann ein Katalog von

8 Zusammenfassung und Ausblick
159
Charakteristika erstellt werden, der z.B. durch Integration in ein Beratungssystem dazu dient,
Fehler in einem SMB-System zu identifizieren. Eine Erweiterung dieses Konzeptes bestünde
darin, die gewonnene Erkenntnisse in ein Regelungskonzept zu integrieren, um eine
automatisches Reaktion der Regelung auf eine auftretende Fehlfunktion zu initieren.
In den bisherigen Überlegungen und Untersuchungen wurde nur vom Vorhandensein einer
Fehlfunktion ausgegangen. Da nach den zusammengestellten Erfahrungen von Bloch (1985)
jedoch grundsätzlich ein Unglück selten allein kommt, also mehr als ein Fehler im System ist,
wären sicherlich hier weitere Untersuchungen wünschenswert. Da jedoch dabei der
Simulationsaufwand quadratisch ansteigt, sind hier zunächst neue Strategien der Vorgehensweise
zu erarbeiten.

Literatur
Literatur
Aced, G.; Möckel, H. J.
Liquidchromatography
Verlag Chemie, Weinheim, 1991
Alhedai, A.; Martire, D. E.; Scott, R. P. W.
Column Dead Volume in Liquid Chromatography
Analyst, 1989, 114, 869-875
Arlt, W.
Grundlagen der Flüssigadsorption
Kurs im Haus der Technik am 1.2.1999
Baer, J.
Dynamics of fluids in porous media
Elsevier, New York, 1972
Barwick, V. J.
Sources of uncertainty in gas chromatography and high-performance liquid
chromatography
J. Chromatogr. A, 1999, 849, 13-33
Bart, H.J.; Messenböcl, R. C.; Byers, C. H.; Prior, A.; Wolfgang, J.
Continuous chromatographic separation of fructose, mannitol and sorbitol
Chem. Eng. Process., 1996, 35, 459-471
Berendsen, G. E.;Schoenmakers, J.; de Galan, L.; Vigh, G.; Varga-Puchony, Z.; Inczedy, J.
On the Determination of the Hold-Up Time in Reversed Phase Liquid Chromatography
J. Liq. Chromatogr., 1980, 3(11), 1669-1686
Berger, F.; Dekany, I.
Multilayer adsorption with variable thickness at solid-liquid interfaces.
Colloid Polym. Sci. 1997, 275, 876-882.
160

Literatur
Beste, Y. A.
Bestimmung der Modellparameter für die Gegenstromchromatographie
Diplomarbeit, TU Berlin, 1995
Beste, Y. A.
Fehlerbetrachtung bei der dynamischen Messung von Adsorptionsisothermen
Vortrag auf dem 25. TTTK, 1997
Beste, Y. A.; Lisso, M.; Wozny, G.; Arlt, W.
Optimization of simulated moving bed plants with low efficient stationary phases:
separation of fructose and glucose
J. Chromatogr. A, 2000, 868, 169-188
Beste, Y. A.
Anwendung der SMB-Chromatographie auf Systeme mit wesentlichen Stofftransport-
widerstand und ihre Erweiterung auf Mehrkomponententrennungen mittels zusätzlichem
Seitenstrom
Dissertation, TU Berlin, 2001
Bidlingmeyer, B. A.; Warren, F. V.; Weston, A.; Nugent, C.
Some Practical Considerations when Determing the Void Volume in HPLC
J. Chromatographic Sci., 1991, 29, 275-279
R.B. Bird, W.E. Steward, E.N. Lightfoot
Transport Phenomena,
Wiley, New York, 1960
Bloch, A.
Gesammelte Gründe, warum alles schiefgeht, was schiefgehen kann
Goldmann, 1985
Blümel, C.
Verwendung der Methode der kleinen Störungen im Kreislauf zur Messung von
Adsorptionsisothermen gelöster Stoffe an Chromatographieträgern
Dissertation, TU Berlin, 1997
161

Literatur
Boysen, H.; Wozny, G.; Arlt, W.; Laiblin, T.; Lisso, M.
Simulation of high pressure liquid chromatography (HPLC) columns with CFD
Vortrag auf der “Thermal Separation”, 5.-6. April 2001, Bamberg
Boysen, H.; Wozny, G.; Laiblin, T.; Arlt, W.
CFD-Simulation von präparativen Chromatographiesäulen unter Berücksichtigung von
nicht-linearen Adsorptions- und Desorptionsisothermen
Tagungsband der GVC-Fachausschußsitzung „Prozeß- und Anlagentechnik“, 2001a
Brandt, A.
Untersuchung der Temperaturabhängigkeit der Trennleistung in der
Flüssigchromatographie mit präparativer Zielsetzung
Dissertation, TU Berlin, 1997
Bräuer, P.; Heuchel, M.; Kind, T.; v. Szombathely, M.; Messow, U.; Kind, B.; Jaroniec, M.
Mathematische und numerische Probleme bei der thermodynamischen Auswertung von
Exzeßisothermen für die Adsorption binärer flüssiger Mischungen an Festkörpern
Chem. Technik, 1993, 45(1), 2-18
Broughton, D. B.
Production-Scale Adsorptive Separations of Liquid Mixtures by Simulated Moving-Bed
Technology
Sep. Sci. Technol., 1984, 19(11&12), 723-736
Broyles, B. S.; Shalliker, R. A.; Guiochon, G.
Visualization of sample introduction in liquid chromatography columns – The effect of the
frit diameter
J. Chromatogr. A, 1999, 855, 367-382
Charton, F.; Nicoud, R.-M.:
Complete Design of a Simulated Moving Bed
J. Chromatogr. A, 1995, 702, 97-112
Ching, C. B.; Ruthven, D. M.; Hidajat, K.
Experimental Study of a Simulated Counter Current Adsorption System-III. Sorbex
Operation
Chem. Eng. Sci, 1985, 40(8), 1411-1417
162

Literatur
Ching, C. B.; Chu, K. H.; Hidajat, K.; Uddin, M. S.:
Comparative Study of Flow Schemes for a Simulated Counter-Current Adsorption
Separation Process
AIChE J., 1992, 38(11), 1744-1750
Ching, C. B.; Chu, K. H.; Hidajat, K.:
Experimental Study of a Simulated Counter-Current Adsorption System-VII. Effects of Non-
Linear and Interacting Isotherms
Chem. Eng. Sci., 1993, 48(7), 1343-1351
Chu, K. H.; Hashim, M. A.
Simulated Counter-Current Adsorption Process: A Comparison of Modelling Strategies
The Chem. Eng. J., 1995, 56, 59-65
Cziesla, F.:
Auswahl und Programmierung eines Modells zur Vorausberechnung des
Konzentrationsprofils für die kontinuierliche Gegenstromchromatographie
Diplomarbeit, TU Berlin, 1994
Dabrowski, A.; Jaroniec, M.; Oscik, J.
Multilayer and Monolayer Adsorption from Liquid Mixtures of Nonelectrolytes on Solid
Surfaces
In: Matijevic, E. (Ed): Surface and Interface Science, Vol. 14, Plenum Press New York
1987, 83-214
Danckwerts, P.V.:
Continuous Flow Systems, Distribution of Residence Times
Chem. Eng. Sci., 1953, 2 (1), 1-13
Deckert, P.; Arlt, W.
Simulierte Gegenstromchromatographie
Chem. Ing. Tech., 1994, 66(10), 1334-1340
Deckert, P.
Aufbau und Inbetriebnahme einer Pilotanlage zur Simulierten Gegenstromchromatographie
Dissertation, TU Berlin, Shaker Verlag, 1997
163

Literatur
Deckwer, W.D.:
Modelle axial-durchmischter Einphasenreaktoren
Dissertation, TU Berlin, 1973
Dieterich, E.; Sorescu, G.; Eigenberger, G.:
Numerische Methoden zur Simulation verfahrenstechnischer Prozesse
Chem. Ing. Tech. 1992, 64(2), 136-147
Do, D. D.
Adsorption Analysis: Equilibrium and Kinetics
Imperial College Press, London, 1998
Dünnebier, G.; Klatt, K.-U.:
Modelling and Simulation of Nonlinear Chromatographic Separation Processes: A
Comparison of Different Modelling Approaches
Chem. Eng. Sci. 2000, 55, 373-380
Engelhardt, H.; Müller, H.; Dreyer, B.
Is There a „True“ Dead Volume for HPLC Columns?
Chromatographia, 1984, 19, 240-245
Eon, C.
Comparison of Broadening Patterns in Regular and Radially Compressed Large-Diameter
Columns
J. Chromatogr., 1978, 149, 29-42
Everett, D. H.
Adsorption at the Solid/Liquid Interface: Non-electrolyte Systems
Colloid Sci. 1979, 3, 63-147.
Everett, D. H.
Adsorption of Near-Ideal Binary Liquid Mixtures by Graphon
J. Colloid Interf. Sci. 1981, 82(1), 14-24.
Farkas, T.; Sepaniak, M. J.; Guiochon, G.
Column radial homogenity in high-performance liquid chromatography
J. Chromatogr. A, 1996, 740, 169-181
164

Literatur
Findenegg, G. H.
Thermodynamik von Grenzflächenerscheinungen
in Lehrbuch der Grenzflächenchemie, ed. Schweiger, M. J.
Georg Thieme Verlag, Stuttgart, 1996
Fluent Europe
Fluent User`s Guide, Volume 1
Fluent Incorporated, Release 4.4, August 1996
Ganetsos, G.; Barker, P. E.
Preparative and Production Scale Chromatography
Marcel Dekker, New York, 1993
Geißler, A.
Inbetriebnahme einer neu aufgebauten Apparatur zur hochgenauen Messung von
Adsorptionsexzeß-Isothermen aus der Flüssigphase
Diplomarbeit TU Berlin, 1996
Grassmann, P.
Einführung in die thermische Verfahrenstechnik
de Gruyter, Berlin, 1967
Gu, T:
Mathematical Modelling and Scale-up of Liquid Chromatography
Springer Verlag Berlin, Heidelberg, 1995
Guiochon, G.; Shirazi, S. G.; Katti, A. M.
Fundamentals of Preparative and Nonlinear Chromatography
Academic Press, New York, 1994
Guiochon, G.; Farkas, T.; Guan-Sajonz, H.; Koh, J.-H.; Sarker, M.; Stanley, B. J.; Yun, T.
Consolidation of particle beds and packing of chromatographic columns
J. Chromatogr. A, 1997, 762, 83-88
Gurvitsch, L.. J.
Phys. Chem. Soc. Russ. 1915, 47, 805.
165

Literatur
Hassan, M. M.; Shamsur Rahman, A. K. M.; Loughlin, K. F.
Modelling of Simulated Moving Bed Adsorption System: A More Precise Approach
Sep. Technol., 1995, 5, 77-89
Hashimoto, K.; Shirai, Y.; Adachi, S.
A Simulated Moving-Bed Adsorber for the Separation of Tricomponents
J. Chem. Eng. Jpn., 1993, 26(1), 52-56
Heuchel, M.; Bräuer, P.
Über den Einfluß der Oberflächenheterogenität und der realen Mischungseigenschaften
auf Adsorptionsexzeßisothermen binärer flüssiger Mischungen
Wiss. Z. Karl-Marx-Univ. Leipzig, Math.-Naturwiss. R., 1986, 35(4), 401-408
Heuchel, M.; Bräuer, P.; Messow, U.; Jaroniec, M.
Phenomenological Thermodynamics of Adsorption from Binary Non-electrolytic Liquid
Mixtures on Solid Surfaces
Chem. Sci., 1989, 29, 353-360
Hidajat, K.; Ching, C. B.:
Simulation of the Performance of a Continuous Counter-Current Adsorption System by the
Method of Orthogonal Collocation with Non-Linear and Interacting Adsorption Isotherms
Trans IChemE, 1990, 68A, 104-108
Hirsch, R.
Experimentelle Untersuchung der Mehrkomponentenadsorption aus der Flüssigphase
mittels einer Zirkulationsapparatur
Dissertation, TU Berlin 1999, Shaker Verlag Aachen 2000
Hoffmann, K.
Bestimmung von Modell- und Anlagenparametern einer simulierten Gegenstrom-
chromatographie Apparatur
Studienarbeit, TU Berlin, 1996
Innes, W. B.
Total Porosity and Particle Density of Fluid Catalysts by Liquid Titration
Anal. Chem., 1956, 28(3), 332-334
166

Literatur
Isermann, R. (Ed.)
Überwachung und Fehlerdiagnose
VDI Verlag, Düsseldorf, 1994
Jork, C.
Diplomarbeit, TU Berlin, 2001
Kaminski, M.
Simple test for determination of the degree of distortion of the liquid-phase flow profile in
columns for preparative liquid chromatography
J. Chromatography, 1992, 589, 61-70
Kazakevich, Y. V.; McNair, H. M.
Thermodynamic Definition of HPLC Dead Volume
J. Chromatogr. Sci., 1993, 31, 317-322
Kehrer, U.
Frontalchromatographische Untersuchungen des Adsorptionsverhaltens von
Ethanol/Wasser-Gemischen an Silicagelen
Diplomarbeit an der Universität Leipzig, Fachbereich Chemie, 1992
Kipling, J. J.
Adsorption from Solutions of Non-Electrolytes
Academic Press, New York, 1965
Kiraly, Z., Dekany, I.
Algebraic and geometric interpretations of adsorption excess quantities
Colloid Polym. Sci, 1988, 266, 663-671
Knox, J. H.; Laird, G. R.; Raven, P. A.
Interaction of Radial and Axial Dispersion in Liquid Chromatography in Relation to the
„Infinite Diameter Effect“
J. Chromatogr. 1976, 122, 129-145
Köster, F.; Findenegg, G. H.
Adsorption from Binary Solvent Mixtures onto Silica Gel by HPLC Frontal Analysis
Chromatographia, 1982, 15(12), 743-747
167

Literatur
Krstulovic, A. M.; Colin, H.; Guiochon, G.
Comparison of Methods Used for the Determination of Void Volume in Reversed-Phase
Liquid Chromatography
Anal. Chem, 1982, 54, 2438-2443
Kubota, K.; Hta, C.; Hayashi, S.
A Study of a Simulated Moving Bed Adsorber Based on the Axial Dispersion Model
Can. J. Chem. Eng. , December 1989, 67, 1025-1028
Laiblin, T.
Charakterisierung der Verteilungen in präparativen chromatographischen Säulen
Dissertation, TU Berlin, 2002
Larionov, O. G.; Myers, A. L.
Thermodynamics of adsorption from nonideal solutions of nonelectrolytes
Chem. Eng. Sci., 1971, 26, 1025-1030
Lenz, K.; Beste, Y. A.; Arlt, W.
Comparison of Static and Dynamic Measurements of Adsorption Isotherms
Separation Science and Technology, 2002, 37(7), 1611-1629
Lichten, W.
Skriptum Fehlerrechnung
Springer Verlag Berlin, 1988
Lisso, M.:
Modellierung und dynamische Simulation des SMB-Verfahrens
Diplomarbeit, TU Berlin, 1996
Lisso, M.; Wozny, G.; Arlt, W.; Beste, Y. A.:
Optimierung der HETP präparativer HPLC-Säulen
Chem. Ing. Tech., 2000, 72, 494-502
Lisso, M.
Optimierung der hydrodynamischen Verteilung in semipräparativen und präparativen
HPLC-Säulen
Dissertation, TU-Berlin, 2002
168

Literatur
Mangold, M.
Beobachter mit verteilten Parametern für Adsorptionsprozesse
Diplomarbeit Universität Stuttgart, 1993
Mangold, M. ; Lauschke, G. ; Schaffner, J. ; Zeitz, M. ; Gilles, E.-D.
State and parameter estimation for adsorption columns by nonlinear distributed parameter
state observers
J. Proc. Cont., 1994, 3(4), 163-172
McCormick, R. M.; Karger, B. L.
Distribution Phenomena of Mobile-Phase Components and Determination of Dead Volume
in Reversed-Phase Liquid Chromatography
Anal. Chem., 1980, 52, 2249-2257
Meyer, V. R.
Praxis der Hochleistungs-Flüssigchromatographie
Salle + Sauerländer, Frankfurt a. M., 1992
Minka, C.; Myers, A. L.
Adsorption from Ternary Liquid Mixtures on Solids
AIChE J. 1973, 19(3),453-459
Miyabe, K.; Guiochon, G.
Estimation of the column radial heterogenity from an analysis of the characteristics of
tailing peaks in linear chromatography
J. Chromatogr. A, 1999, 830, 29-39
Miyabe, K.; Guiochon, G.
Influence of column radial heterogenity on peak fronting in linear chromatography
J. Chromatogr. A, 1999a, 857, 69-87
Mottlau, A. Y.; Fisher, N. E.
Measurement of Pore Volume by a Titration Technique
Anal. Chem., 1962, 34(6), 714-715
169

Literatur
Nicoud, R. M.
The Simulated Moving-Bed: A Powerful Chromatographic Process
LC-GC INTL 1992, 5(5), 43-47
Oliveros, G.; Smith, J. M.
Dynamic Studies of Dispersion and Channeling in Fixed Beds
AIChE J., 1982, 28(5), 751-759
Paar, A. (Hrsg.)
Gebrauchsanleitung für externe Meßzellen vom Typ DMA eines U-Rohr Biegeschwingers
Graz, 1978
Pais, L. S.; Loureiro, J. M.; Rodrigues, A. E.
Modeling Strategies for Enantiomers Separation by SMB Chromatography
AIChE J., 1998, 44(3), 561-569
Reichl, A.
Messung und Korrellierung von Gaslöslichkeiten halogenierter Kohlenwasserstoffe
Dissertation, TU Berlin, Shaker Verlag, 1996
Referenzhandbuch der HPLC der Series 1100
Hewlett-Packard, 1996
Ruthven, D. M.
Principles of Adsorption and Adsorption Processes
John Wiley & Sons, New York, 1984
Schade, H.; Kunz, E.
Strömungslehre
de Gruyter, Berlin-New York, 1989
Schay, G.; Nagy, L. G.; Szekrényesy, T.;
Comparative Studies on the Determination of Specific Surface Areas by Liquid Adsorption
Periodica Polytechnica Ch., 1960, IV(2), 95-117
170

Literatur
Scheer, J.
Modellierung und dynamische Simulation einer Anlage zur Simulierten
Gegenstromchromatographie in der Simulationsumgebung gPROMS
Studienarbeit TU Berlin, 2003
Schwedt, G.:
Chromatographische Trennmethoden,
Georg Thieme Verlag, Stuttgart 1994
Seidel-Morgenstern, A.
Mathematische Modellierung der präparativen Flüssigchromatographie
DUV Wiesbaden 1995
Sircar, S.;
Thermodynamics of Adsorption from Binary Liquid Mixtures on Heterogenious Adsorbents
J. Chem. Soc., Faraday Trans. I, 1986, 82, 831-841
Storti, G.; Masi, M.; Paludetto, R.; Morbidelli, M.; Carra, S. (a)
Adsorption Separation Processes: Counter-Current and Simulated Counter-Current
Operations
Comput. Chem. Eng., 1988, 12(5), 475-482
Storti, G.; Masi, M.; Carra, S. (b)
Modelling and Design of Simulated Moving-Bed Adsorption Separation Units
Preparative Chromatography 1988, 1(1), 1-27
Storti, G.; Mazzotti, M.; Morbidelli, M.; Carra, S.
Robust Design of Binary Countercurrent Adsorption Separation Processes
AIChE J.; 1993, 39(3), 471-492
Strube, J.
Simulation und Optimierung kontinuierlicher Simulated-Moving-Bed (SMB)
Chromatographie-Prozesse
Dissertation, Universität Dortmund, 1996
171

Literatur
Thelen, H.
Thermodynamische Modelle zur Beschreibung der Adsorption aus Flüssigkeiten
Diplomarbeit, TU Berlin, 1993
Tsotsas, E.; Schlünder, E. U.
On Axial Dispersion in Packed Beds with Fluid Flow
Chem. Eng. Proc., 1988, 24, 15-31
Uysal, T.:
Entwicklung eines FORTRAN Programms für die Simulation der kontinuierlichen
Gegenstromchromatographie (TMB) von Mehrkomponentensystemen beliebiger
Anlagenkonfiguration
Studienarbeit, TU Berlin, 2000
van Deemter, J. J.; Zuiderweg, F. J.; Klinkenberg, A.:
Longitudinal Diffusion and Resistance to Mass Transfer as Causes of Nonideality in
Chromatography
Chem. Eng. Sci.,1956, 5, 271-289
VDI Wärmeatlas:
VDI Verlag Düsseldorf, 1994, 7. Auflage
Wang, H. L.; Duda, J. L.; Radke, C. J.
Solution Adsorption form Liquid Chromatography
J. Colloid Interf. Sci, 1978, 66(1), 153-165
Wätzig, H.; Ebel, S.
Estimation of Dead-Time in Liquid Chromatography from Retention Behaviour of
Homologous Series by Non-Linear Regression
Chromatographia, 1991, 31(11/12), 544-548
172
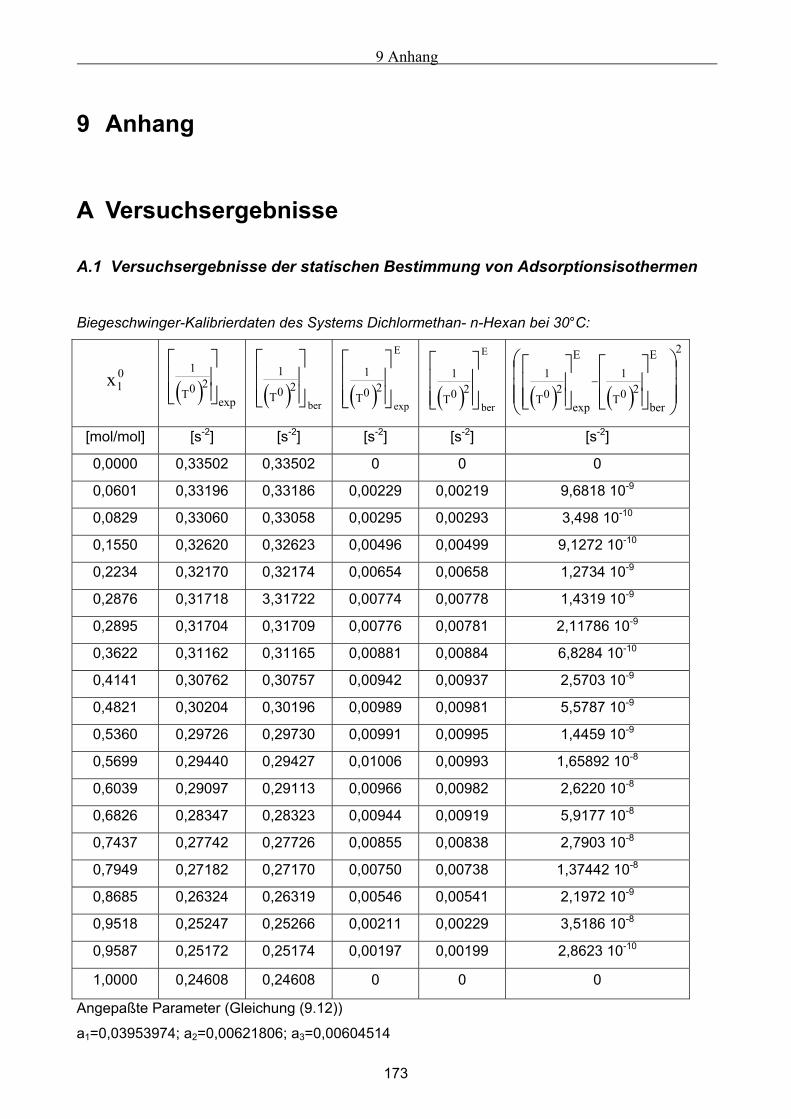
9 Anhang
9 Anhang
173
A Versuchsergebnisse
A.1 Versuchsergebnisse der statischen Bestimmung von Adsorptionsisothermen
Biegeschwinger-Kalibrierdaten des Systems Dichlormethan- n-Hexan bei 30°C:
01x ( ) exp
20T
1
( ) ber
20T
1
( )
E
exp
20T
1
( )
E
ber
20T
1
( ) ( )
2E
ber20T
1E
exp20T
1
−
[mol/mol] [s-2] [s-2] [s-2] [s-2] [s-2]
0,0000 0,33502 0,33502 0 0 0
0,0601 0,33196 0,33186 0,00229 0,00219 9,6818 10-9
0,0829 0,33060 0,33058 0,00295 0,00293 3,498 10-10
0,1550 0,32620 0,32623 0,00496 0,00499 9,1272 10-10
0,2234 0,32170 0,32174 0,00654 0,00658 1,2734 10-9
0,2876 0,31718 3,31722 0,00774 0,00778 1,4319 10-9
0,2895 0,31704 0,31709 0,00776 0,00781 2,11786 10-9
0,3622 0,31162 0,31165 0,00881 0,00884 6,8284 10-10
0,4141 0,30762 0,30757 0,00942 0,00937 2,5703 10-9
0,4821 0,30204 0,30196 0,00989 0,00981 5,5787 10-9
0,5360 0,29726 0,29730 0,00991 0,00995 1,4459 10-9
0,5699 0,29440 0,29427 0,01006 0,00993 1,65892 10-8
0,6039 0,29097 0,29113 0,00966 0,00982 2,6220 10-8
0,6826 0,28347 0,28323 0,00944 0,00919 5,9177 10-8
0,7437 0,27742 0,27726 0,00855 0,00838 2,7903 10-8
0,7949 0,27182 0,27170 0,00750 0,00738 1,37442 10-8
0,8685 0,26324 0,26319 0,00546 0,00541 2,1972 10-9
0,9518 0,25247 0,25266 0,00211 0,00229 3,5186 10-8
0,9587 0,25172 0,25174 0,00197 0,00199 2,8623 10-10
1,0000 0,24608 0,24608 0 0 0
Angepaßte Parameter (Gleichung (9.12))
a1=0,03953974; a2=0,00621806; a3=0,00604514
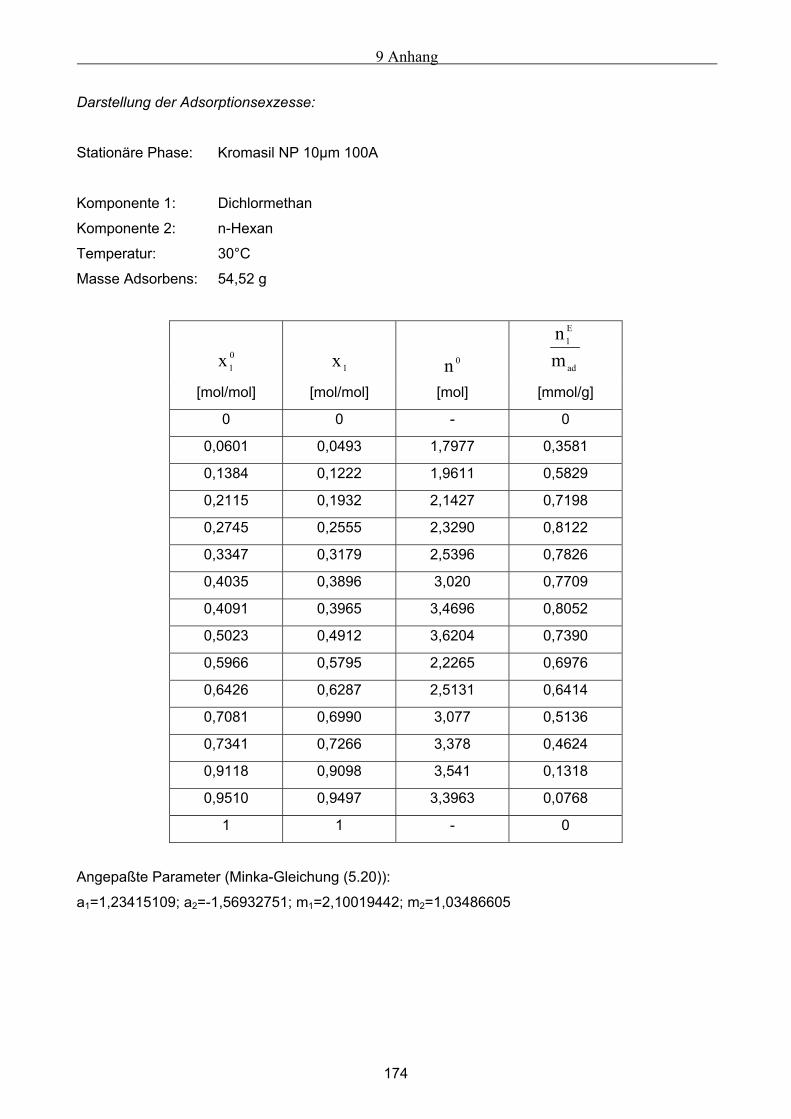
9 Anhang
174
Darstellung der Adsorptionsexzesse:
Stationäre Phase: Kromasil NP 10µm 100A
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Temperatur: 30°C
Masse Adsorbens: 54,52 g
01x
1x
0n ad
E1
mn
[mol/mol] [mol/mol] [mol] [mmol/g]
0 0 - 0
0,0601 0,0493 1,7977 0,3581
0,1384 0,1222 1,9611 0,5829
0,2115 0,1932 2,1427 0,7198
0,2745 0,2555 2,3290 0,8122
0,3347 0,3179 2,5396 0,7826
0,4035 0,3896 3,020 0,7709
0,4091 0,3965 3,4696 0,8052
0,5023 0,4912 3,6204 0,7390
0,5966 0,5795 2,2265 0,6976
0,6426 0,6287 2,5131 0,6414
0,7081 0,6990 3,077 0,5136
0,7341 0,7266 3,378 0,4624
0,9118 0,9098 3,541 0,1318
0,9510 0,9497 3,3963 0,0768
1 1 - 0
Angepaßte Parameter (Minka-Gleichung (5.20)):
a1=1,23415109; a2=-1,56932751; m1=2,10019442; m2=1,03486605
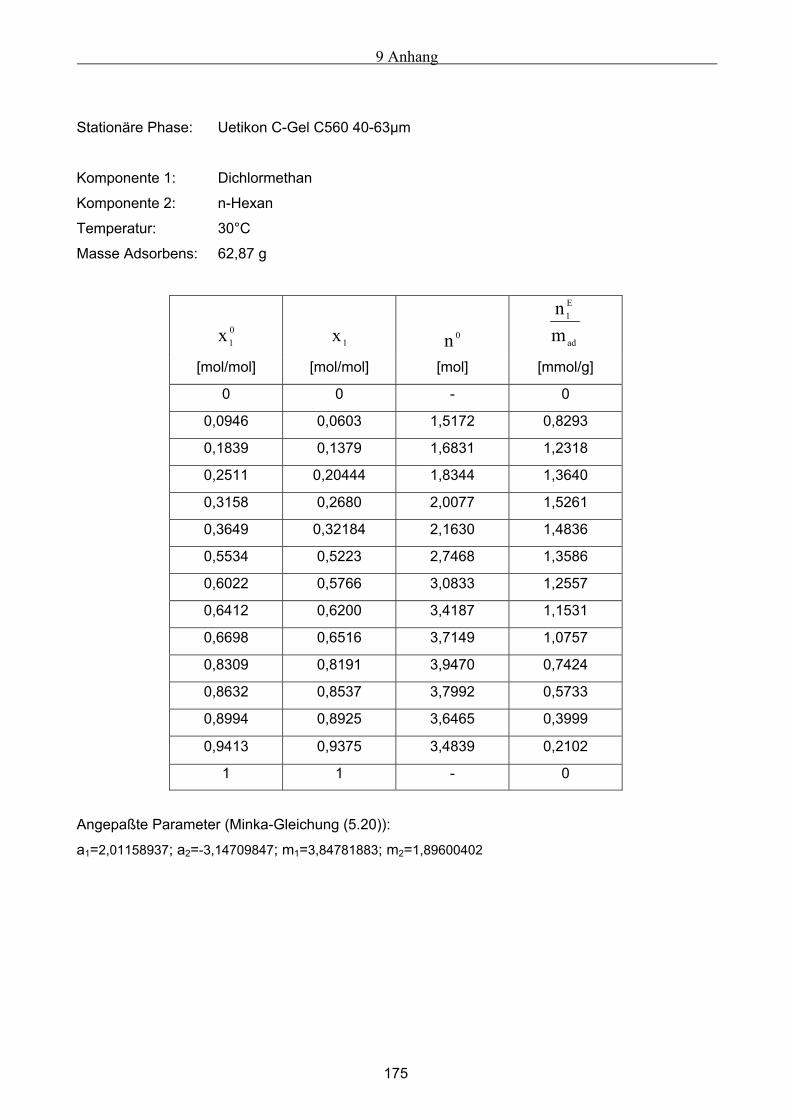
9 Anhang
175
Stationäre Phase: Uetikon C-Gel C560 40-63µm
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Temperatur: 30°C
Masse Adsorbens: 62,87 g
01x
1x
0n ad
E1
mn
[mol/mol] [mol/mol] [mol] [mmol/g]
0 0 - 0
0,0946 0,0603 1,5172 0,8293
0,1839 0,1379 1,6831 1,2318
0,2511 0,20444 1,8344 1,3640
0,3158 0,2680 2,0077 1,5261
0,3649 0,32184 2,1630 1,4836
0,5534 0,5223 2,7468 1,3586
0,6022 0,5766 3,0833 1,2557
0,6412 0,6200 3,4187 1,1531
0,6698 0,6516 3,7149 1,0757
0,8309 0,8191 3,9470 0,7424
0,8632 0,8537 3,7992 0,5733
0,8994 0,8925 3,6465 0,3999
0,9413 0,9375 3,4839 0,2102
1 1 - 0
Angepaßte Parameter (Minka-Gleichung (5.20)):
a1=2,01158937; a2=-3,14709847; m1=3,84781883; m2=1,89600402
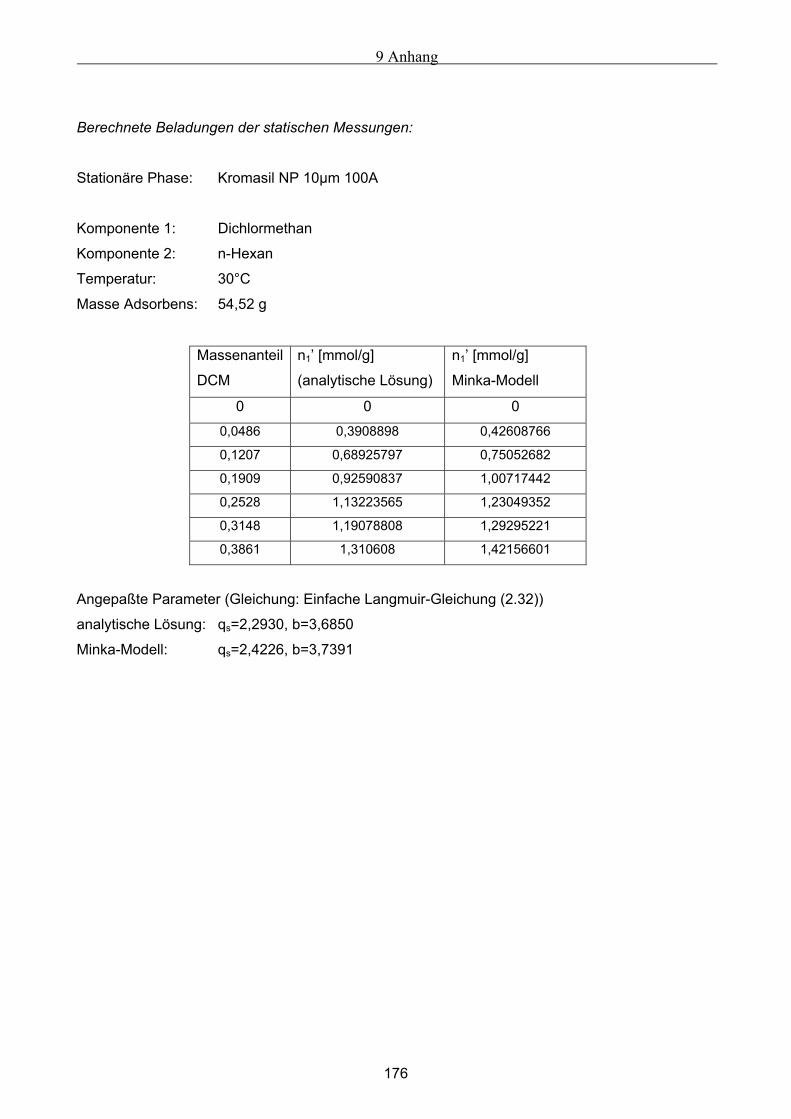
9 Anhang
176
Berechnete Beladungen der statischen Messungen:
Stationäre Phase: Kromasil NP 10µm 100A
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Temperatur: 30°C
Masse Adsorbens: 54,52 g
Massenanteil
DCM
n ’ [mmol/g] 1
(analytische Lösung)
n ’ [mmol/g] 1
Minka-Modell
0 0 0
0,0486 0,3908898 0,42608766
0,1207 0,68925797 0,75052682
0,1909 0,92590837 1,00717442
0,2528 1,13223565 1,23049352
0,3148 1,19078808 1,29295221
0,3861 1,310608 1,42156601
Angepaßte Parameter (Gleichung: Einfache Langmuir-Gleichung (2.32))
analytische Lösung: q =2,2930, b=3,6850 s
Minka-Modell: q =2,4226, b=3,7391 s
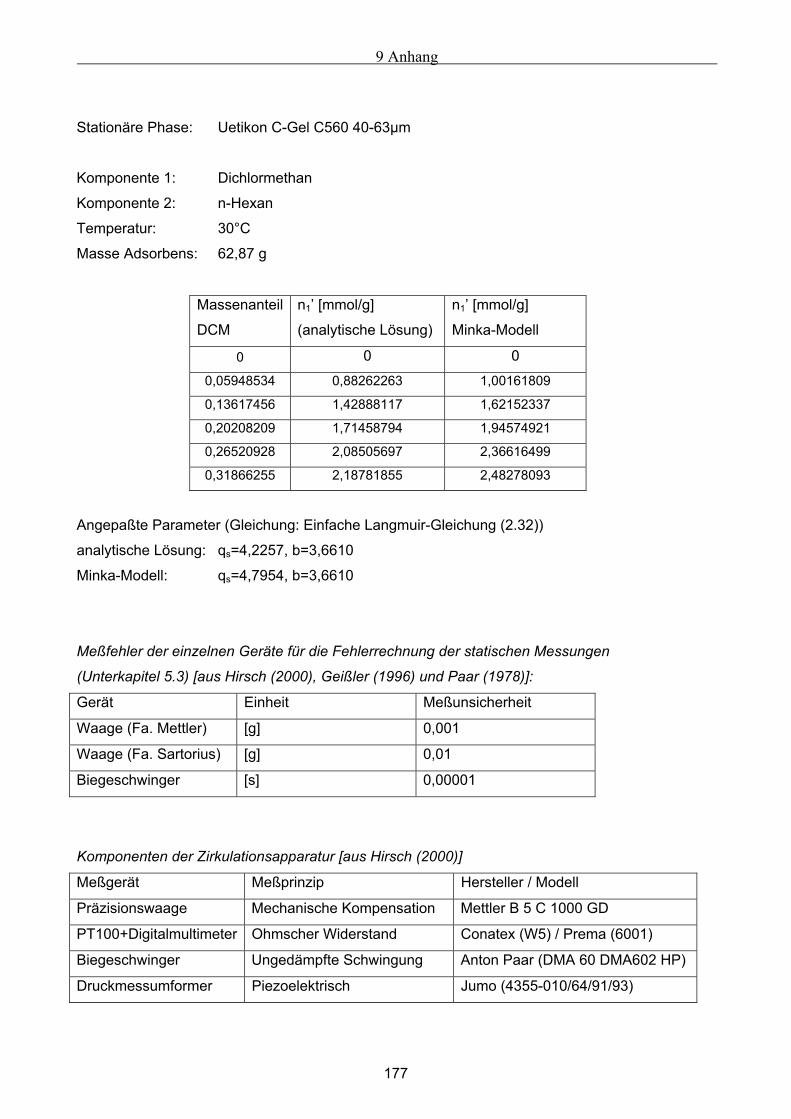
9 Anhang
177
Stationäre Phase: Uetikon C-Gel C560 40-63µm
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Temperatur: 30°C
Masse Adsorbens: 62,87 g
Massenanteil
DCM
n ’ [mmol/g] 1
(analytische Lösung)
n ’ [mmol/g] 1
Minka-Modell
0 0 0
0,05948534 0,88262263 1,00161809
0,13617456 1,42888117 1,62152337
0,20208209 1,71458794 1,94574921
0,26520928 2,08505697 2,36616499
0,31866255 2,18781855 2,48278093
Angepaßte Parameter (Gleichung: Einfache Langmuir-Gleichung (2.32))
analytische Lösung: q =4,2257, b=3,6610 s
Minka-Modell: q =4,7954, b=3,6610 s
Meßfehler der einzelnen Geräte für die Fehlerrechnung der statischen Messungen
(Unterkapitel 5.3) [aus Hirsch (2000), Geißler (1996) und Paar (1978)]:
Gerät Einheit Meßunsicherheit
Waage (Fa. Mettler) [g] 0,001
Waage (Fa. Sartorius) [g] 0,01
Biegeschwinger [s] 0,00001
Komponenten der Zirkulationsapparatur [aus Hirsch (2000)]
Meßgerät Meßprinzip Hersteller / Modell
Präzisionswaage Mechanische Kompensation Mettler B 5 C 1000 GD
PT100+Digitalmultimeter Ohmscher Widerstand Conatex (W5) / Prema (6001)
Biegeschwinger Ungedämpfte Schwingung Anton Paar (DMA 60 DMA602 HP)
Druckmessumformer Piezoelektrisch Jumo (4355-010/64/91/93)
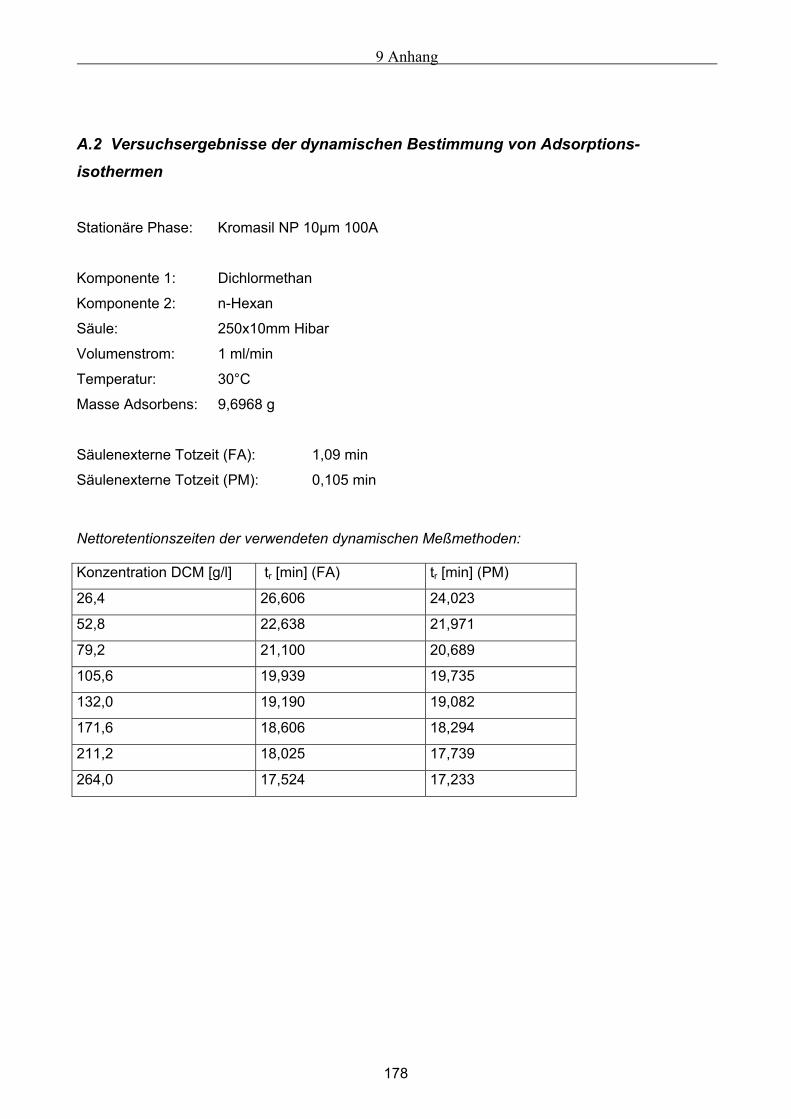
9 Anhang
178
A.2 Versuchsergebnisse der dynamischen Bestimmung von Adsorptions-
isothermen
Stationäre Phase: Kromasil NP 10µm 100A
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Säule: 250x10mm Hibar
Volumenstrom: 1 ml/min
Temperatur: 30°C
Masse Adsorbens: 9,6968 g
Säulenexterne Totzeit (FA): 1,09 min
Säulenexterne Totzeit (PM): 0,105 min
Nettoretentionszeiten der verwendeten dynamischen Meßmethoden:
Konzentration DCM [g/l] t [min] (FA) r t [min] (PM) r
26,4 26,606 24,023
52,8 22,638 21,971
79,2 21,100 20,689
105,6 19,939 19,735
132,0 19,190 19,082
171,6 18,606 18,294
211,2 18,025 17,739
264,0 17,524 17,233
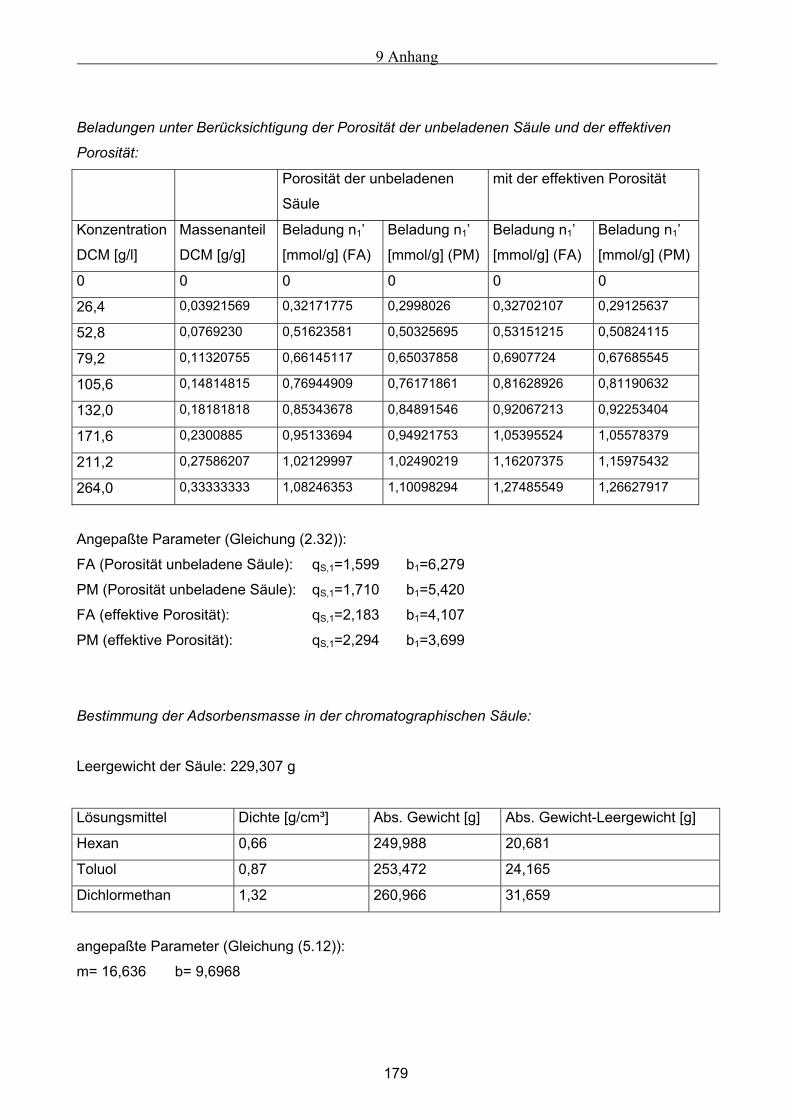
9 Anhang
179
Beladungen unter Berücksichtigung der Porosität der unbeladenen Säule und der effektiven
Porosität:
Porosität der unbeladenen
Säule
mit der effektiven Porosität
Konzentration
DCM [g/l]
Massenanteil
DCM [g/g]
Beladung n1’
[mmol/g] (FA)
Beladung n1’
[mmol/g] (PM)
Beladung n1’
[mmol/g] (FA)
Beladung n1’
[mmol/g] (PM)
0 0 0 0 0 0
26,4 0,03921569 0,32171775 0,2998026 0,32702107 0,29125637
52,8 0,0769230 0,51623581 0,50325695 0,53151215 0,50824115
79,2 0,11320755 0,66145117 0,65037858 0,6907724 0,67685545
105,6 0,14814815 0,76944909 0,76171861 0,81628926 0,81190632
132,0 0,18181818 0,85343678 0,84891546 0,92067213 0,92253404
171,6 0,2300885 0,95133694 0,94921753 1,05395524 1,05578379
211,2 0,27586207 1,02129997 1,02490219 1,16207375 1,15975432
264,0 0,33333333 1,08246353 1,10098294 1,27485549 1,26627917
Angepaßte Parameter (Gleichung (2.32)):
FA (Porosität unbeladene Säule): qS,1=1,599 b1=6,279
PM (Porosität unbeladene Säule): qS,1=1,710 b1=5,420
FA (effektive Porosität): qS,1=2,183 b1=4,107
PM (effektive Porosität): qS,1=2,294 b1=3,699
Bestimmung der Adsorbensmasse in der chromatographischen Säule:
Leergewicht der Säule: 229,307 g
Lösungsmittel Dichte [g/cm³] Abs. Gewicht [g] Abs. Gewicht-Leergewicht [g]
Hexan 0,66 249,988 20,681
Toluol 0,87 253,472 24,165
Dichlormethan 1,32 260,966 31,659
angepaßte Parameter (Gleichung (5.12)):
m= 16,636 b= 9,6968
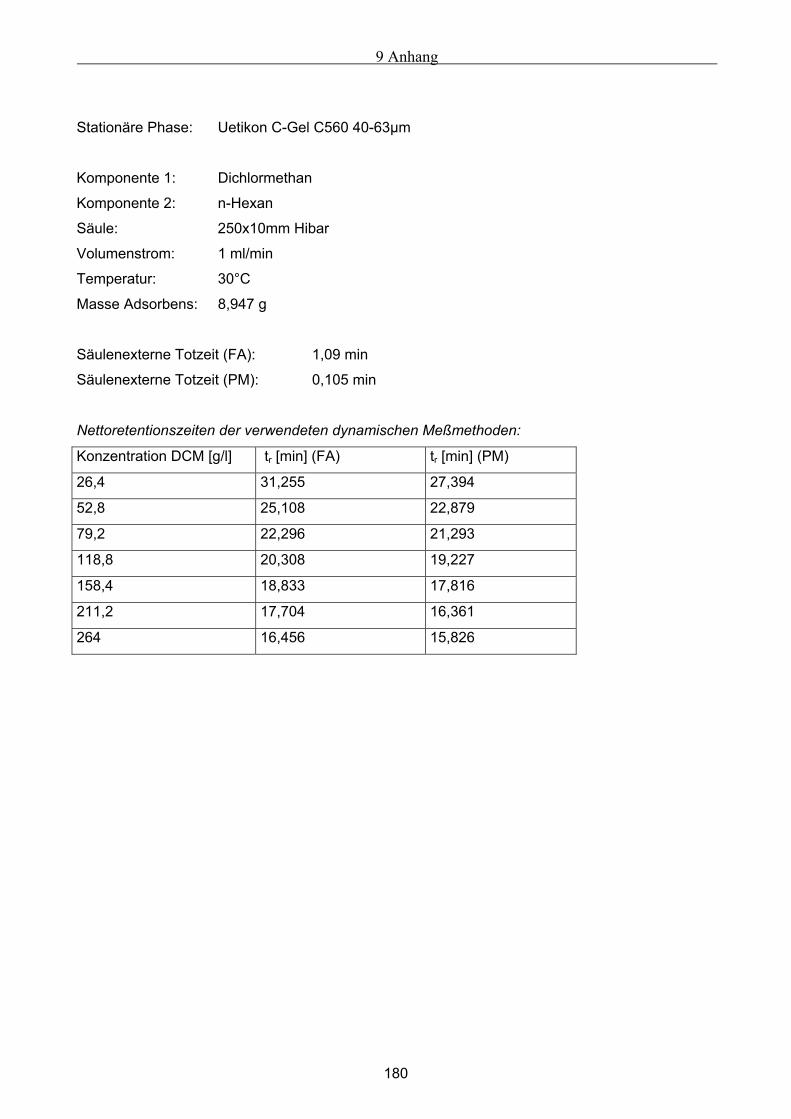
9 Anhang
180
Stationäre Phase: Uetikon C-Gel C560 40-63µm
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Säule: 250x10mm Hibar
Volumenstrom: 1 ml/min
Temperatur: 30°C
Masse Adsorbens: 8,947 g
Säulenexterne Totzeit (FA): 1,09 min
Säulenexterne Totzeit (PM): 0,105 min
Nettoretentionszeiten der verwendeten dynamischen Meßmethoden:
Konzentration DCM [g/l] tr [min] (FA) tr [min] (PM)
26,4 31,255 27,394
52,8 25,108 22,879
79,2 22,296 21,293
118,8 20,308 19,227
158,4 18,833 17,816
211,2 17,704 16,361
264 16,456 15,826
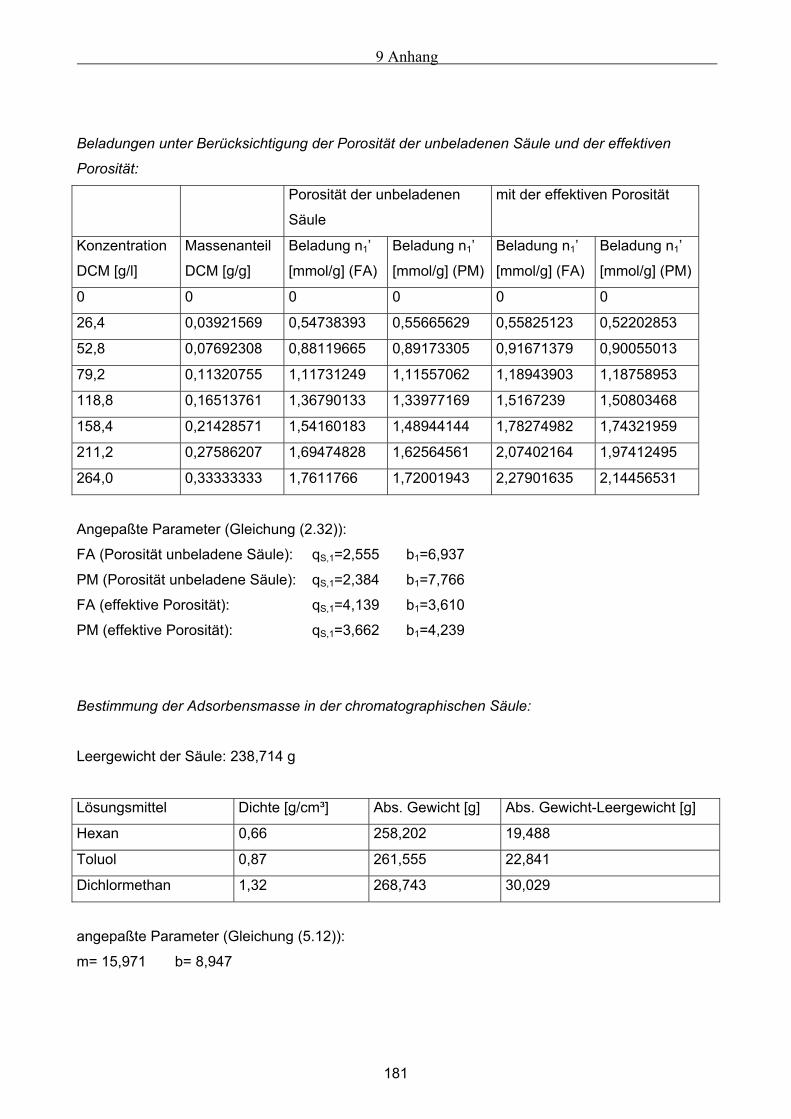
9 Anhang
181
Beladungen unter Berücksichtigung der Porosität der unbeladenen Säule und der effektiven
Porosität:
Porosität der unbeladenen
Säule
mit der effektiven Porosität
Konzentration
DCM [g/l]
Massenanteil
DCM [g/g]
Beladung n1’
[mmol/g] (FA)
Beladung n1’
[mmol/g] (PM)
Beladung n1’
[mmol/g] (FA)
Beladung n1’
[mmol/g] (PM)
0 0 0 0 0 0
26,4 0,03921569 0,54738393 0,55665629 0,55825123 0,52202853
52,8 0,07692308 0,88119665 0,89173305 0,91671379 0,90055013
79,2 0,11320755 1,11731249 1,11557062 1,18943903 1,18758953
118,8 0,16513761 1,36790133 1,33977169 1,5167239 1,50803468
158,4 0,21428571 1,54160183 1,48944144 1,78274982 1,74321959
211,2 0,27586207 1,69474828 1,62564561 2,07402164 1,97412495
264,0 0,33333333 1,7611766 1,72001943 2,27901635 2,14456531
Angepaßte Parameter (Gleichung (2.32)):
FA (Porosität unbeladene Säule): qS,1=2,555 b1=6,937
PM (Porosität unbeladene Säule): qS,1=2,384 b1=7,766
FA (effektive Porosität): qS,1=4,139 b1=3,610
PM (effektive Porosität): qS,1=3,662 b1=4,239
Bestimmung der Adsorbensmasse in der chromatographischen Säule:
Leergewicht der Säule: 238,714 g
Lösungsmittel Dichte [g/cm³] Abs. Gewicht [g] Abs. Gewicht-Leergewicht [g]
Hexan 0,66 258,202 19,488
Toluol 0,87 261,555 22,841
Dichlormethan 1,32 268,743 30,029
angepaßte Parameter (Gleichung (5.12)):
m= 15,971 b= 8,947
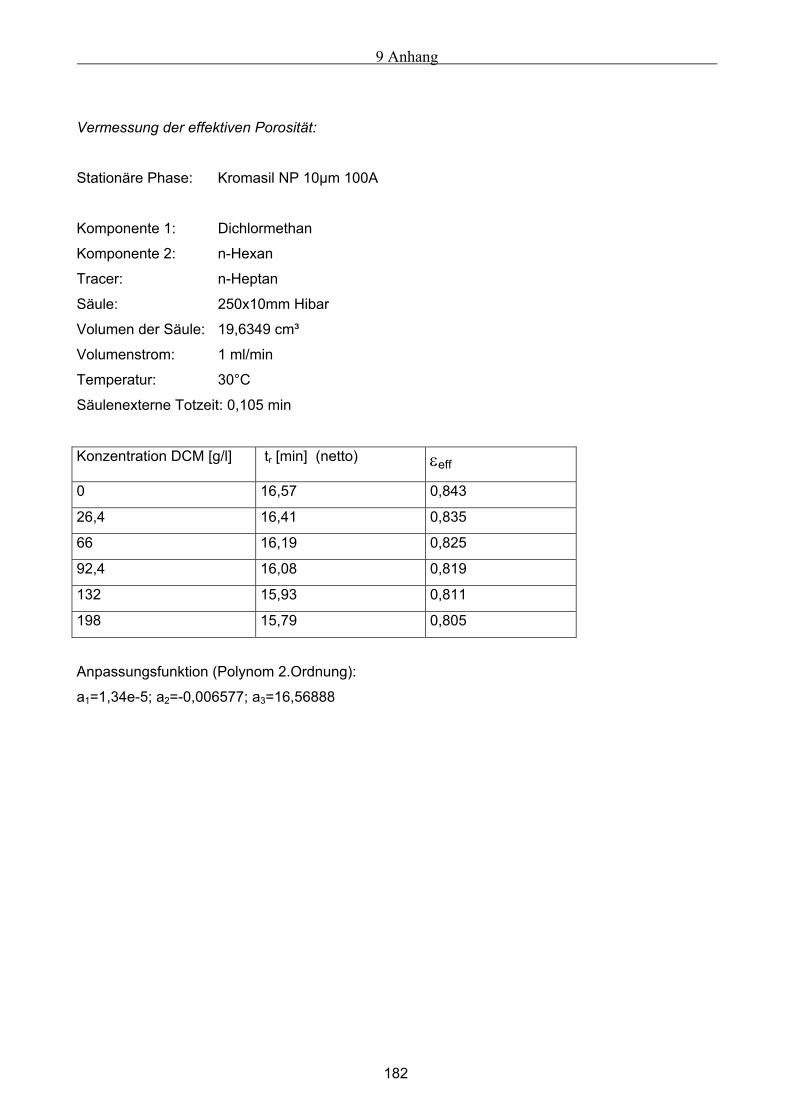
9 Anhang
182
Vermessung der effektiven Porosität:
Stationäre Phase: Kromasil NP 10µm 100A
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Tracer: n-Heptan
Säule: 250x10mm Hibar
Volumen der Säule: 19,6349 cm³
Volumenstrom: 1 ml/min
Temperatur: 30°C
Säulenexterne Totzeit: 0,105 min
Konzentration DCM [g/l] tr [min] (netto) εeff
0 16,57 0,843
26,4 16,41 0,835
66 16,19 0,825
92,4 16,08 0,819
132 15,93 0,811
198 15,79 0,805
Anpassungsfunktion (Polynom 2.Ordnung):
a1=1,34e-5; a2=-0,006577; a3=16,56888
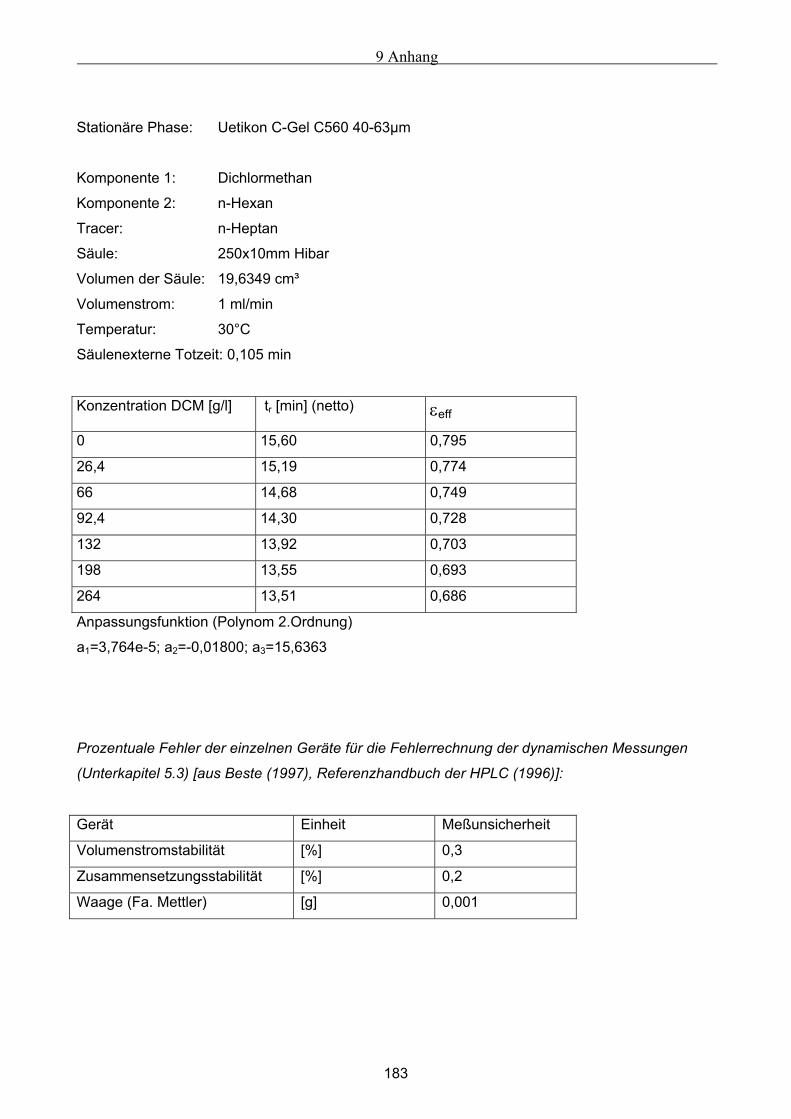
9 Anhang
183
Stationäre Phase: Uetikon C-Gel C560 40-63µm
Komponente 1: Dichlormethan
Komponente 2: n-Hexan
Tracer: n-Heptan
Säule: 250x10mm Hibar
Volumen der Säule: 19,6349 cm³
Volumenstrom: 1 ml/min
Temperatur: 30°C
Säulenexterne Totzeit: 0,105 min
Konzentration DCM [g/l] tr [min] (netto) εeff
0 15,60 0,795
26,4 15,19 0,774
66 14,68 0,749
92,4 14,30 0,728
132 13,92 0,703
198 13,55 0,693
264 13,51 0,686
Anpassungsfunktion (Polynom 2.Ordnung)
a1=3,764e-5; a2=-0,01800; a3=15,6363
Prozentuale Fehler der einzelnen Geräte für die Fehlerrechnung der dynamischen Messungen
(Unterkapitel 5.3) [aus Beste (1997), Referenzhandbuch der HPLC (1996)]:
Gerät Einheit Meßunsicherheit
Volumenstromstabilität [%] 0,3
Zusammensetzungsstabilität [%] 0,2
Waage (Fa. Mettler) [g] 0,001

9 Anhang
184
B Vorgehensweise bei der Durchführung der Experimente mit der Zirkulationsapparatur
Vor Beginn der eigentlichen Messungen werden die zum Einsatz kommenden Stoffe vorbehandelt.
Diese Vorbehandlung umfaßt sowohl die Flüssigkeiten als auch den zu untersuchenden Feststoff:
Vor ihrem Einsatz werden die Flüssigkeiten entgast. Zu diesem Zweck werden sie getrennt
voneinander jeweils in einen Glasrundkolben eingebracht. Es wird jeweils etwas mehr als die
später im Experiment benötigte Menge eingefüllt, um eventuelle Verluste durch das Entgasen
auszugleichen. Auf diesen Glaskolben ist ein Teflon-Ventil aufgesetzt. Eine Swagelock-Schnell-
kupplung, dichtet das System ab. Weiterhin befindet sich im Glaskolben ein magnetischer
Rührfisch.
Die Flüssigkeit in den beiden Kolben wird durch den Magnetrührer kräftig gerührt, um eine große
Phasengrenzfläche durch Erzeugung einer starken Trombe zu erzeugen. Sie werden dabei an
eine Entgasungsapparatur angeschlossen, die im Rahmen der Arbeit von Reichl (1996) aufgebaut
und dort auch detailliert beschrieben wurde. Die stufenweise Druckabsenkung wird in Form eines
Schleusenverfahrens durchgeführt.
Nach erfolgter Entgasung werden die Kolben mit den darin befindlichen Flüssigkeiten gewogen.
Zum Einsatz kam hierbei eine Balkenwaage der Fa. Mettler mit einer Meßgenauigkeit von 0,001g,
die in der Lage ist, Proben im Bereich 0 bis 1000g zu wiegen.
Parallel zum Entgasen der Flüssigkeiten wird das Adsorbens in die Adsorptionszelle gefüllt,
nachdem diese vorher im leeren Zustand gewogen wurde. Anschließend wird das Adsorbens aus-
geheizt, um sicherzustellen, daß sich keine Flüssigkeit mehr in der Poren befindet. Dabei wurde
bei den Normal-Phase Silica-Gelen auf Ratschlag der Fa. Muder & Wochele eine Temperatur von
105°C nicht überschritten. Das Ausheizen erfolgt, bis in der Zelle ein Druck von 0,06 mbar erreicht
ist, was dem maximalen Potential der angeschlossenen Vakuumpumpe entspricht. Anschließend
wird die Adsorptionszelle erneut gewogen, um mittels Differenzbildung die eingefüllte
Adsorbensmasse zu ermitteln. Für die Wägungen wird eine Waage der Fa. Sartorius verwendet,
die die über 4 kg schwere Adsorptionszelle mit einer Genauigkeit von 0,01g wiegt.
Vor dem Beginn der Experimente werden die Flüssigkeiten über die Schnellkupplung in die
Befüllzelle eingebracht, wobei im Regelfall die Flüssigkeit mit dem niedrigeren Dampfdruck als
erstes eingefüllt wird. Der Kolben, der als zweites zum Befüllen genutzt wird, wird komplett

9 Anhang
185
entleert, um eventuelle Rückstände durch Rückverdampfung zu vermeiden. Um die
Adsorptionsapparatur komplett zu füllen, sind mindestens 180 cm³ Flüssigkeit notwendig.
Nach erfolgter Befüllung werden die beiden Glaskolbensysteme erneut gewogen, um mittels
Differenzbildung die eingefüllten Mengen der beiden Flüssigkeiten zu ermitteln. Anschließend
werden die Befüllzelle und die Adsorptionszelle in die Apparatur eingebaut und es werden alle
Leitungen und die Adsorptionszelle evakuiert, bis ein Druck von 0,06mbar erreicht ist. Wichtig ist
dabei, daß die Flüssigkeiten in der Befüllzelle durchmischt werden. Dies kann durch den
eingebauten Rührer geschehen, für gut mischbare Flüssigkeiten erwies sich aber auch ein
manuelles Schütteln der Befüllzelle vor dem Einbau als ausreichende Gewähr für eine gute
Durchmischung.
Nachdem der Flüssig- und der Luftbadthermostat die vorgegebene Meßtemperatur erreicht haben,
werden die Ventile der Befüllzelle geöffnet und der Kolben mit Druck beaufschlagt, so daß der
Kolben nach unten rutscht und die Anlage mit Flüssigkeit füllt. Der Druck sollte dabei so gewählt
werden, daß er über den Dampfdrücken der verwendeten Reinstoffe liegt, um die Ausbildung einer
Dampfphase zu verhindern.
Vor dem Einschalten der Zahnradpumpe wird ein Anzeigewert des Biegeschwinger abgelesen. Da
zu diesem Zeitpunkt die Adsorption noch keinen Einfluß auf die Zusammensetzung der Flüssigkeit
in der Meßzelle hat, kann die abgelesene Dichte der Kalibrierkurve hinzugefügt werden. Die
Erstellung der Kalibrierkurve wird in Unterabschnitt 5.1.2.2 behandelt.
Nach dem Einschalten der Zahnradpumpe wird jede Stunde, sofern eine betreuende Person
anwesend ist, ein Biegeschwingerwert abgelesen. Dabei ist zu beachten, daß die Temperatur in
der Meßzelle auf 0,01K exakt eingestellt ist. Die Messung läuft, bis das Adsorptionsgleichgewicht
erreicht ist. Schwankt der angezeigte Biegeschwingerwert bei drei aufenanderfolgenden
Ablesungen nur noch um die normale Schwankungsbreite von 0,000003, ändert sich die Dichte
nicht mehr und das Adsorptionsgleichgewicht ist erreicht.
Um einen neuen Meßpunkt zu vermessen ohne die gesamte Anlage zu entleeren und neu zu
befüllen, wird mittels der in Unterabschnitt 5.1.2.1 beschriebenen Spritzenkonstruktion ein Rein-
stoff nachgespritzt, um die Zusammensetzung zu verändern. Zu diesem Zweck wird die Spritze,
nachdem sie evakuiert wurde, an die Kolbenkonstruktion mit der erwünschten Flüssigkeit ange-
schlossen. Dann wird mittels manueller Kraft die Flüssigkeit aus dem Behälter in die Spritze
gezogen und diese anschließend gewogen. Um die Flüssigkeit in die Adsorptionsapparatur einzu-
bringen, wird die Spritzenkonstruktion an die Kupplung der Befüllzelle angeschlossen und unter

9 Anhang
186
gleichzeitiger Entlastung des Kolbens eingespritzt. Um die nachgefüllte Menge zu bestimmen, wird
die Spritze dann erneut gewogen.
Diese Prozedur wurde maximal dreimal durchgeführt, bevor die Anlage komplett wieder entleert
und für die nächsten Messungen vorbereitet wurde. Dies geschieht, indem die gesamte Anlage
zunächst entleert und mit Stickstoff oder Preßluft gespült wird. Anschließend wird das gesamte
System einschließlich Adsorbens, Befüll- und Meßzelle ausgeheizt, bis sich wieder ein Vakuum
von 0,06 mbar eingestellt hat.

9 Anhang
187
C Auswertung der Biegeschwingermeßwerte
Die Schwingungsdauer T, die mit Hilfe des Biegeschwingers ermittelt wird, berechnet sich aus:
cm2T π= , (9.1)
wobei c die Federkonstante und m die schwingende Masse ist. Im Falle des schwingenden U-
Rohres des Biegeschwingers setzt sich die schwingende Masse zusammen aus der Masse des
Quarzglasrohres des Biegeschwingers und derjenigen des Fluids im Hohlraum V dieses Rohres,
so daß gilt:
Vmm FluidGlas ρ+= . (9.2)
Somit kann die Dichte eines Fluids aus der eines Referenzfluids mit bekannter Dichte ermittelt
werden über
( )2fRe
2FluidfReFluid TTK −=ρ−ρ . (9.3)
Dabei ist K eine Gerätekonstante, deren Abhängigkeit von der Temperatur aus Kalibrier-
messungen mit zwei Referenzmedien bekannter Dichte ermittelt wird [Paar (1978)].
Bei den Messungen, die im Rahmen dieser Arbeit durchgeführt wurden, steht aber nicht die
Ermittlung der Dichte im Vordergrund, sondern es wird vielmehr die Tatsache genutzt, daß die
Schwingungsdauer in eindeutigem Zusammenhang zur Dichte bzw. zum spezifischen Volumen
eines Fluids steht. Daraus ergibt sich die Möglichkeit, durch Kalibriermessungen eine Funktion
)x(T 1 (9.4)
aufzustellen, die dann aufgrund ihrer Temperaturabhängigkeit allerdings nur isotherm genutzt
werden kann. Diese Beziehung kann dann genutzt werden, um aus dem Biegeschwingerwert die
Zusammensetzung der Bulk-Phase zu ermitteln.

9 Anhang
188
Die Kalibrierkurve wurde ermittelt, indem die Biegeschwingerwerte aufgenommen wurden, die bei
verschiedenen, bekannten Bulk-Phasen-Zusammensetzungen ermittelt wurden. Zu diesem Zweck
wurde die Adsorptionsapparatur befüllt wie in Anhang B beschrieben, mit dem Unterschied, daß
statt der Adsorptionszelle nur ein Stück Rohr eingesetzt wurde, damit keine Adsorption im System
stattfindet. Durch Zuspritzen einer Komponente wurden die Zusammensetzung der Bulk-Phase
variiert, um eine Reihe von Meßpunkten zu erhalten, wobei mindestens zehn Meßpunkte
vorhanden sein sollten. Dieses Vorgehen entspricht gleichzeitig der Bestimmung des
Exzeßvolumens der Mischung.
Wie aus Gleichung (9.3) ersichtlich ist, besteht ein proportionaler Zusammenhang zwischen dem
Wert der Schwingungsdauer T und der Dichte ρ eines Fluids. Unter Berücksichtigung des
spezifischen Volumens ergibt sich dabei:
2S T
1~1vρ
= . (9.5)
Daraus ergibt sich die Möglichkeit, die Schwingungsdauer als 1/T² auszudrücken und mittels
bekannter Korrelationsgleichungen für das spezifische Volumen v bei konstanter Temperatur und
konstantem Druck darzustellen [Hirsch (2000)].
Das spezifische Volumen einer binären Mischung setzt sich dabei zusammen aus den
spezifischen Volumina der Reinstoffe v01 und v02 additiv ergänzt um das Exzeßvolumen vE gemäß
( ) ( )1E
0220111S xvvxvxxv ++= (T, P = const.) (9.6)
Für die Beschreibung der Konzentrationsabhängigkeit des Exzeßvolumens hat sich das Redlich-
Kister-Polynom bewährt:
( ) ( ) ( )∑=
−−−=k
1i
1i1i111
E 1x2ax1xxv . (9.7)
Für die innerhalb dieser Arbeit betrachteten Spezialfall der binären Mischung ergibt sich damit
unter Verwendung eines 3-Parameter Redlich-Kister-Polynoms

9 Anhang
189
( ) ( )[ ] ( ) ( )[ ]( ) ([ 11
213
11121111E
x1x1x2a
x1x1x2ax1xaxv
−−
+−−+−=
)]. (9.8)
Da im Rahmen dieser Arbeit nur binäre Mischungen betrachtet wurden, deren Reinstoffe bei
Raumtemperatur in flüssiger Form vorliegen, wird im folgenden nur das Vorgehen für diesen Fall
beschrieben. Wie vorzugehen ist, wenn ein Stoff in fester Form vorliegt, kann Hirsch (2000)
entnommen werden.
Unabdingbar für die Kalibrierung ist die Ermittlung der Schwingungsdauer des Biegeschwingers
für die beiden verwendeten Reinstoffe, die für das spezifische Volumen dargestellt werden durch
201T1
bzw. 202T1
. (9.9)
Neben diesen Schwingungsdauern für die Reinstoffe werden für verschieden zusammengesetzte
Bulk-Phasen die Werte
( )( )21xT
1 (9.10)
ermittelt. Damit ist dann folgende Auswertung möglich:
202
2201
12
E
2 T1x
T1x
T1
T1
−−=
. (9.11)
Unter Verwendung des 3-Parameter Redlich-Kister Polynoms aus Gleichung (9.8) erhält man
dann
( )[ ] ( ) ( )[ ]
( ) ([ ]112
13
1112111
E
2
x1x1x2a
x1x1x2ax1xaT1
−−
+−−+−=
), (9.12)
wobei die Parameter a1, a2 und a3 an den gemessenen Verlauf der Kalibrierkurve angepaßt
werden. Mit Hilfe dieser ermittelten Parameter ist es dann möglich, über die Beziehung

9 Anhang
190
( ) ( )[ ]
( ) ( )[ ] ( ) ([ ]112
131112
111202
1201
12
x1x1x2ax1x1x2a
x1xaT1x1
T1x
T1
−−+−−
+−+−+=
) (9.13)
mit Hilfe des Newton-Verfahrens aus dem Meßwert für die Schwingungsdauer T den Molanteil x1
in einer Bulk-Phase zu bestimmen, die mit dem zu untersuchenden Adsorbens in ein
Adsorptionsgleichgewicht gebracht wurde.
Die Güte der Anpassung der Kalibrierkurve wird durch die Wurzel der mittleren Fehlerquadrate
RMS (Routed Mean Square) beschrieben, wobei m die Anzahl der Meßpunkte darstellt:
∑=
−==
m
1i
2
i
2ber
2exp
2 T1
T1
m1RMS
T1d . (9.14)

9 Anhang
191
D Herleitung der Minka-Gleichung
Die Herleitung der von Gleichung (5.20) beginnt allgemein mit der Grundgleichung des Phasen-
gleichgewichtes, nämlich der Isofugazitätsbeziehung einer Komponente in der Adsorbat- und der
Bulkphase:
ii ff =′ (9.15)
Die Fugazität der Komponente i in der flüssigen Phase wird beschrieben durch:
( )
−γ=
RTPPv
expxPfLVi0
L)T(i0
iiLVi0i (9.16)
mit dem Standardzustand des flüssigen Reinstoffes bei Systemtemperatur T und Systemdruck P.
Für die Fugazität der Komponente i in der Adsorbat-Phase ergibt sich in analoger Art und Weise
( )
σ−σ−′γ′=′
RTba
expxpfi
i0ii
LVi0i ,
(9.17)
wobei ersetzt wurde durch a/bdpvLi0 i dσ, da bei der Adsorption Oberflächeneffekte auftreten, die
durch Hinzunahme der spezifischen Oberfläche a und der Oberflächenspannung σ beschrieben
werden. Unter Definition der freien Benetzungsenthalpie
σ=Φ a (9.18)
erhält man dann
( )
Φ−Φ−′γ′=′
RTbexpxpf
i
i0ii
LVi0i . (9.19)
Unter Eliminierung von n‘ und xi‘ aus Gleichung (2.3) mit Hilfe von Gleichung (5.19) und (9.19)
ergibt sich für den Adsorptionsexzeß [Thelen (1993)]:
( )∑
∑ −=
jijj
ijjiEi b/Kx
K1xxn
(9.20)

9 Anhang
192
mit
( ) ( )
Φ−Φ−
Φ−Φ
γγ′γγ′
=RTbRTb
expKi
i0
j
j0
ij
jiij ,
(9.21)
wobei Kij folgende Eigenschaften hat:
pjipij
jiijii KKK,
K1K,1K === .
(9.22)
In der vorliegenden Arbeit wird für die Aktivitätskoeffizienten vereinfachend angenommen, daß
1
ij
ji =γγ′γγ′
(9.23)
gesetzt werden kann [Hirsch (2000)]. Hieraus folgt jedoch nicht automatisch, daß ein ideales
Phasenverhalten bei der Betrachtung der Adsorption aus der Flüssigphase zugrunde gelegt wird.
Es ergibt sich aus Gleichung (9.20) für den in dieser Arbeit betrachteten binären Fall spezifisch für
die Adsorbensmasse:
+
−=
122
2
1
1
1221
ad
E1
Kbx
bx
)K1(xxmn
. (9.24)
mit der vereinfachten Schreibweise von K12:
−=
1
1
2
212 b
aba
expK (9.25)
mit
RTa i0
i
Φ−Φ= .
(9.26)

9 Anhang
193
K12 wird auch als Selektivität des Adsorbens bezeichnet [Everett (1981)] und gibt die Möglichkeit,
die Zusammensetzung von Bulk- und Adsorbat-Phase zu berechnen:
12
2112 xx
xxK
′′
= (9.27)
Die Zusammensetzung der Adsorbatphase läßt sich über [Minka (1973)]:
∑=′
ijj
ijjj Kx
Kxx (9.28)
ermitteln, was im binären Fall die Form
1221
11 Kxx
xx
+=′ (9.29)
annimmt. Es ist nun möglich, Gleichung (5.20) an den gemessen Verlauf der Exzeßisotherme
anzupassen, wobei die anzupassenden Parameter a1, a2, b1 und b2 sind. Um eine eindeutige
Lösung zu gewährleisten, ist es notwendig, die Anzahl der anzupassenden Parameter auf drei zu
reduzieren. Dies geschieht unter Verwendung der Gurwitsch-Regel [Gurwitsch (1915)], die besagt,
daß das Verhältnis der Beladungskapazitäten des Adsorbens b1 und b2 umgekehrt proportional
zum Verhältnis der molaren Reinstoffvolumina v1 und v2 ist, also
1
2
2
1
vv
bb
≈ . (9.30)
Mit Hilfe des nun bekannten K12 ist es möglich, die Zusammensetzung der Adsorbatphase mittels
Gleichung (5.23) zu berechnen. Zur Berechnung der spezifischen, molaren Beladung von
Komponente 1 n1‘ ergibt sich unter Verwendung von Gleichung (2.3) die Beziehung
111
adE1
ad
1 xxx
mnmn
′−′
=′
. (9.31)

9 Anhang
194
E Modellierung von globalen Fehlfunktionen
In diesem Teil des Anhangs werden die Gleichungen aus Unterkapitel 7.3 mittels einer integralen
Massenbilanz um eine TMB-Anlage hergeleitet. Die Gleichungen gelten dabei nur für lineare
Adsorptionsisothermen.
E.1 Modellierung eines Komponententransport durch nur einen Abschnitt
Zunächst sei der Fall betrachtet, daß der Transport der Komponenten mit linearen Adsorptions-
isothermen wie in Abbildung 2.15 dargestellt in die gewünschten Richtungen stattfindet.
Der Massenstrom der Komponente A im Abschnitt II wird berechnet über
II,AAfestII,AII,flII,AfestII,AII,flII,A cKVcVqVcVm &&&&& −=−= (9.32)
und analog dazu für Komponente B im Abschnitt III über
III,BBfestIII,BIII,flIII,BfestIII,BIII,flIII,B cKVcVqVcVm &&&&& −=−= (9.33)
wobei zu beachten ist, daß aufgrund der unterschiedlichen Transportrichtung m ein negatives
Vorzeichen besitzt. Zunächst sei der Fall betrachtet, daß Komponente A vom Feed ausschließlich
in Richtung des Extrakts und Komponente B vom Feed ausschließlich in Richtung des Raffinats
transportiert werden. Dies läßt sich formell durch folgende Ungleichungen ausdrücken, wobei es
im folgenden möglich ist, die Konzentrationen aufgrund der linearen Adsorptionsisothermen
auszuklammern:
IIA,&
0KVVKVV AfestIII,flAfestII,fl <−<− &&&& (9.34)
für Komponente A bzw.
0KVVKVV BfestII,flBfestIII,fl >−>− &&&& (9.35)
für Komponente B.

9 Anhang
195
Die Termi der Form
ifestk,fl KVV && − (9.36)
werden aufgrund ihrer Dimension im folgenden als effektive Transportvolumenströme bezeichnet.
Mit Hilfe der Gleichungen (9.32) bzw. (9.33) ist es möglich, die Konzentrationen der Komponenten
A und B in den Abschnitten II und III zu berechnen, was bei Einbeziehung von axialer Dispersion
und Stofftransport den maximalen Konzentrationen der jeweiligen Komponenten in den einzelnen
Abschnitten entspricht.
Um die Massenbilanz einzuhalten, dürfen die Massenströme bzw. nicht größer sein
als die mit dem Feed jeweils zugeführte Menge an Komponente A und B. Ein Gleichsetzen des
Feedmassenstroms mit den Gleichungen (9.32) und (9.33) führt hierbei zu den Beziehungen
II,Am& III,Bm&
AfestIl,fl
Feed,AFeedII,A KVV
cVc
&&
&
−=
(9.37)
und
BfestIII,fl
Feed,AFeedIII,B KVV
cVc
&&
&
−= ,
(9.38)
wobei der Nenner von Gleichung (9.37) aufgrund von Ungleichung (9.34) als Betrag verwendet
wird, um das Auftreten positiver Konzentrationen sicherzustellen.
Die Konzentrationen der Komponenten A und B in Extrakt und Raffinat folgen aus einer Massen-
bilanz um die gesamte TMB-Anlage, wobei angenommen wird, daß sich das System in einem
quasistationären Zustand befindet und es keine Verunreinigungen der jeweiligen Produktströmen
mit der jeweils anderen Komponente gibt. Komponente A wird also ausschließlich mit dem Extrakt
und Komponente B ausschließlich mit dem Raffinat abgezogen. Da also in den Produktströmen
die mit dem Feed zugeführte Menge abgezogen wird, folgt für die Konzentrationen:

9 Anhang
196
Ex
Feed,AFeedEx,A V
cVc
&
&=
(9.39)
und
Raff
Feed,AFeedRaff,B V
cVc
&
&= .
(9.40)
Festzuhalten bleibt, daß die Konzentrationen sowohl in den Produktströmen als auch in den
Abschnitten II und III sich aus den Massenbilanzen und Wanderungsgeschwindigkeiten ergeben .
und
charakterisiert wird, berechnet sich das Verhältnis der Massenströme von Komponente A in
Extrakt und Raffinat als Verhältnis der effektiven Transportvolumenströme in den Abschnitten II
und III zueinander:
E.2 Modellierung des Transports einer Komponente in zwei Richtungen
In diesem Abschnitt werden die Beziehungen für die Konzentrationen in den einzelnen
Abschnitten und Produktströmen hergeleitet, wenn es zu Verunreinigungen in den Produktströmen
kommt, weil die Bedingungen in den Ungleichungen (9.34) bzw. (9.35) nicht gelten. Dabei wird
zunächst nur der Fall betrachtet, daß es zu einer Verunreinigung des Raffinats mit Komponente A
durch Transport durch Abschnitt III kommt (Abbildung 7.4), wobei auch der analoge Fall einer
Verunreinigung des Extraktes mit Komponente B durch Transport durch Abschnitt II dargestellt
wird. Auf den komplexeren Fall der Verunreinigung durch Transport durch die Abschnitte I und IV
wird in Abschnitt 0 eingegangen.
Für den Fall, daß es zu einer Verunreinigung des Raffinats kommt, was durch die Ungleichungen
0KVV AfestII,fl <− && (9.41)
(9.42)0KVV AfestIII,fl >− &&

9 Anhang
197
(9.43)
AfestIII,fl
AfestII,fl
Raff,ARaff
Ex,AEx
Raff,A
Ex,A
KVV
KVVcVcV
mm
&&
&&
&
&
&
&
−
−== .
Aufgrund der unterschiedlichen Transportrichtungen und der damit verbundenen unterschiedlichen
Vorzeichen werden in Gleichung (9.43) Beträge verwendet, um in jedem Schritt positive Werte zu
erhalten. Mit der Massenbilanz für Komponente A über die gesamte, stationäre TMB-Anlage ist es
mit der Beziehung
Raff,AEx,AFeed,A mmm &&& += (9.44)
Auch die Maximalkonzentrationen von Komponente A in den Abschnitten A und B lassen sich
berechnen. Da in diesem Abschnitt der Fall betrachtet wird, daß es kein Transport der
Komponente A durch die Abschnitte I und IV gibt, muß gelten:
möglich, die Konzentrationen von Komponente A in Extrakt und Raffinat zu berechnen.
Raff,A
Ex,A
III,A
II,A
mm
m
m&
&
&
&= (9.45)
bzw.
Raff,ARaff
Ex,AEx
III,AAfestIII,AIII,fl
II,AAfestII,AII,fl
cVcV
cKVcV
cKVcV&
&
&&
&&=
−
−,
(9.46)
woraus sich zusammen mit Gleichung (9.44) die Konzentrationen und berechnen
lassen. Aus Gleichung (9.46) folgt zusammen mit Gleichung (9.43) die Beziehung
II,Ac III,Ac
AfestIII,fl
AfestII,fl
III,AAfestIII,AIII,fl
II,AAfestII,AII,fl
KVV
KVV
cKVcV
cKVcV&&
&&
&&
&&
−
−=
−
−
(9.47)
woraus sich die betragsmäßige Gleichheit der Konzentrationen von Komponente A in den
Abschnitten II und III ergibt:

9 Anhang
198
III,AII,A cc = . (9.48)
Löst man Gleichung (9.48) unter Berücksichtigung der unterschiedlichen Transportrichtungen
ohne Betragsstriche, so erhält man die Beziehung
0cc II,AIII,A >−= , (9.49)
was die Tatsache der unterschiedlichen Transportrichtung widerspiegelt. Wichtig ist, daß absolute
Konzentration in den Abschnitten II und III gleich ist. Diese sich einstellende Konzentration läßt
sich noch weiter herleiten. Da der mit dem Feed zugeführte Massenstrom in den Abschnitten II
und III im stationären Zustand vom Feedzulauf abzutransportieren ist, gilt:
(9.50))KVV(c)KVV(cmmm AfestIII,flIII,AAfestII,flII,AIII,AII,AFeed,A&&&&&&& −+−=+=
Mit dem Wissen um das Vorzeichen aus den Ungleichungen (9.41), (9.42) und (9.49) lassen sich
die Betragsstriche in Gleichung (9.50) eliminieren. so daß man erhält:
( ) ( )AfestIII,flIII,AAfestII,flII,AFeed,AFeed KVVcKVVccV &&&&& −+−= (9.51)
Unter Berücksichtigung von Gleichung (9.49) wird
(9.52)
II,AIII,AIII,II,A cc:c −==
definiert. Eingesetzt in Gleichung (9.51) und nach aufgelöst, ergibt sich III,II,Ac
II,flIII,fl
Feed,AFeedIII,II,A VV
cVc
&&
&
−= .
(9.53)
Da der Nenner genau dem Feedstrom entspricht, vereinfacht sich (9.53) zu
Feed,AIII,II,A cc = , (9.54)

9 Anhang
199
was bedeutet, daß die Konzentration in den Abschnitten II und III bei Anwesenheit von
Komponente A in beiden Produktströmen bei vernachläßigter Dispersion und Stofftransport-
widerstand genau der Feedkonzentration entspricht.
Das Vorgehen zur Berechnung der Konzentrationen von Komponente B in den Produktströmen
und in den Abschnitten II und III bei vergleichbarer Konstellation erfolgt genau analog und führt
ebenfalls auf die betragsmäßige Gleichheit der Konzentrationen in den Abschnitten II und III
III,BII,B cc = .
Weiterhin entspricht die Konzentration in den Abschnitten II und III genau der Feedkonzentration:
. (9.56)
E.3 Modellierung des Transports einer Komponente durch alle Abschnitte in
identische Richtung
(9.58)
(9.55)
Feed,BIII,II,B cc =
Bisher wurde nur der Fall betrachtet, daß die Komponenten A und B nur in den Abschnitten II und
III transportiert werden, ein Transport über die Grenze zwischen Abschnitt I und IV aufgrund der
gewählten Volumenströme nicht stattfindet. Dieser Fall soll nun in diesem Abschnitt untersucht
werden. Zunächst soll der Fall betrachtet werden, daß Komponente B durch Abschnitt IV in
Richtung Abschnitt I transportiert wird (Abbildung 7.6), was formell durch folgende Ungleichung
ausgedrückt wird:
0KVV BfestIV,fl >− && . (9.57)
Um die Konzentrationen in den Produktströmen sowie die Konzentrationen in den einzelnen
Abschnitten zu berechnen, wird zunächst eine integrale Bilanz der Massenströme für die
Zwischenstücke zwischen den vier Abschnitten durchgeführt. Dies ergibt
für die Grenze zwischen den Abschnitten I und II:
Ex,BI,BII,B mmm &&& −= ,

9 Anhang
200
,
.
Zusätzlich notwendig ist die Information, wie das Verhältnis der Massenströme an den
Entnahmestellen der Produktströme quantifizierbar ist. Es entspricht dem Verhältnis des effektiven
Transportvolumenstroms des abgehenden Stroms zum Volumenstrom des Produktstromes, also
für die Entnahmestelle des Extraktes
für die Grenze zwischen Abschnitt II und III:
Feed,BII,BIII,B mmm += && , (9.59)
für die Grenze zwischen Abschnitt III und IV:
(9.60)Raff,BIII,BIV,B mmm &&& −=
und für die Grenze zwischen Abschnitt IV und I:
(9.61)IV,BI,B mm && =
Ex
BfFestII,fl
Ex,B
II,B
VKVV
mm
&
&&
&
& −= (9.62)
bzw.
Raff
BfestIV,fl
Raff,B
IV,B
VKVV
mm
&
&&
&
& −= (9.63)
für die Entnahmestelle des Raffinates.
Dies sind sechs Gleichungen, denen mit den vier Konzentrationen in den vier Abschnitten sowie
den Konzentrationen in den beiden Produktströmen sechs unbekannte Variablen
gegenüberstehen. Es ist also eine analytische Lösung möglich, da die Gleichungen voneinander
linear unabhängig sind.
Die Lösungen für die Massenströme von Komponente B sehen dann folgendermaßen aus:
Für den Massenstrom in Abschnitt I:
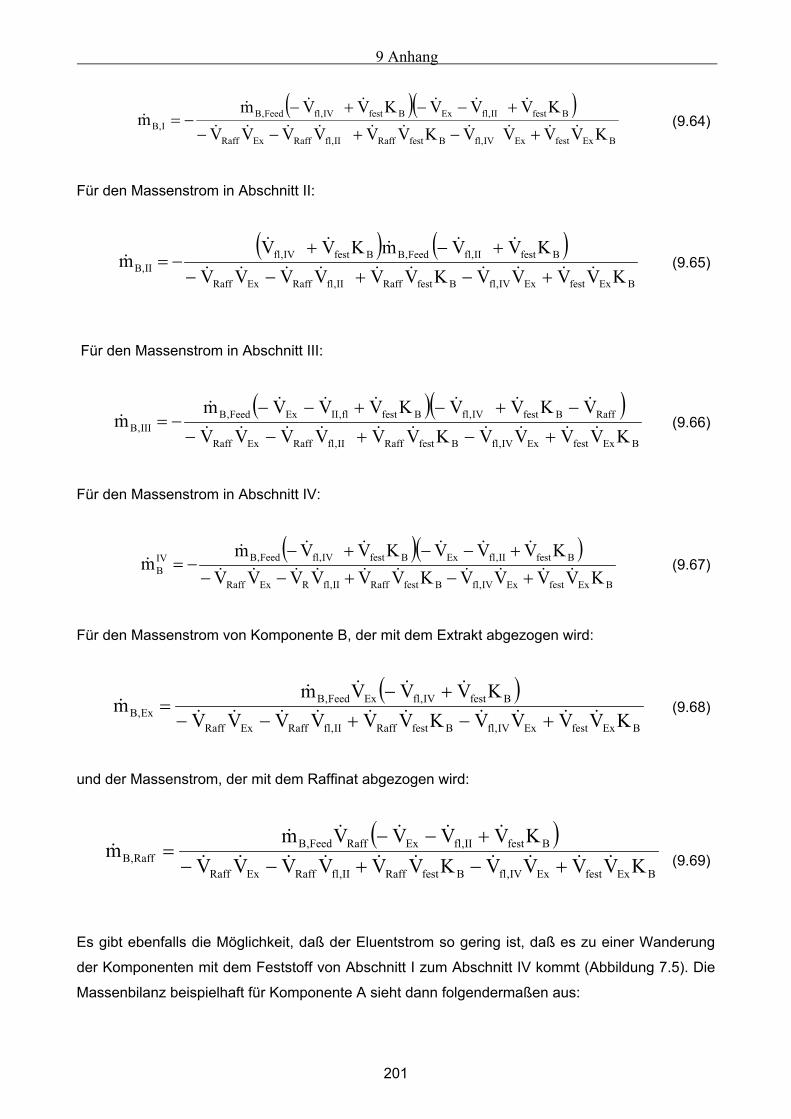
9 Anhang
201
( )( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flExBfestIV,flFeed,BI,B KVVVVKVVVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
+−+−−
+−−+−−= (9.64)
Für den Massenstrom in Abschnitt II:
( ) ( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flFeed,BBfestIV,flII,B KVVVVKVVVVVV
KVVmKVVm
&&&&&&&&&&
&&&&&&
+−+−−
+−+−= (9.65)
Für den Massenstrom in Abschnitt III:
( )( )BExfestExIV,flBfestRaffII,flRaffExRaff
RaffBfestIV,flBfestfl,IIExFeed,BIII,B KVVVVKVVVVVV
VKVVKVVVmm
&&&&&&&&&&
&&&&&&&&
+−+−−
−+−+−−−= (9.66)
Für den Massenstrom in Abschnitt IV:
( )( )BExfestExIV,flBfestRaffII,flRExRaff
BfestII,flExBfestIV,flFeed,BIVB KVVVVKVVVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
+−+−−+−−+−
−= (9.67)
Für den Massenstrom von Komponente B, der mit dem Extrakt abgezogen wird:
( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestIV,flExFeed,BEx,B KVVVVKVVVVVV
KVVVmm
&&&&&&&&&&
&&&&&
+−+−−+−
= (9.68)
und der Massenstrom, der mit dem Raffinat abgezogen wird:
( )BExfestExIV,flBfestRaffII,flRaffExRaff
BfestII,flExRaffFeed,BRaff,B KVVVVKVVVVVV
KVVVVmm
&&&&&&&&&&
&&&&&&
+−+−−+−−
=
(9.69)
Es gibt ebenfalls die Möglichkeit, daß der Eluentstrom so gering ist, daß es zu einer Wanderung
der Komponenten mit dem Feststoff von Abschnitt I zum Abschnitt IV kommt (Abbildung 7.5). Die
Massenbilanz beispielhaft für Komponente A sieht dann folgendermaßen aus:

9 Anhang
202
(9.71)
.
Analog zu den Gleichungen (9.62) und (9.63) ist auch hier die Information notwendig, wie das
Verhältnis der Massenströme an den Entnahmestellen der Produktströme quantifizierbar ist:
Für die Grenze zwischen den Abschnitten I und II:
Ex,AII,AI,A mmm &&& −= , (9.70)
für die Grenze zwischen Abschnitt II und III:
Feed,AIII,AII;A mmm += && ,
für die Grenze zwischen Abschnitt III und IV:
Raff,AIV,AIII,A mmm &&& −= , (9.72)
und für die Grenze zwischen Abschnitt IV und I:
(9.73)IV,AI,A mm && =
Ex
AfestII,fl
Ex,A
II,A
VKVV
mm
&
&&
&
& −= (9.74)
bzw.
Raff
AfestIV,fl
Raff,A
IV,A
VKVV
mm
&
&&
&
& −= (9.75)
für die Entnahmestelle des Raffinates.
Die Lösungen für die Massenströme von Komponente A sehen dann folgendermaßen aus:
Für den Massenstrom in Abschnitt I:

9 Anhang
203
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
AfestII,flExAfestIV,flFeed,AI,A KVVVVVVKVVVV
KVVVKVVmm
&&&&&&&&&&
&&&&&&&
−+−−+−−+−
−= (9.76)
Für den Massenstrom in Abschnitt II:
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
BfestIII,flExRaffAfestIII,flFeed,AII,A KVVVVVVKVVVV
KVVVVKVVmm
&&&&&&&&&&
&&&&&&&&
−+−−
++−++−= (9.77)
Für den Massenstrom in Abschnitt III:
( )( )AfestRaffRaffI,FlRaffExAfestExIII,flEx
AfestI,flAfestIII,flFeed,AIII,A KVVVVVVKVVVV
KVVKVVmm
&&&&&&&&&&
&&&&&&
−+−−
−+−= (9.78)
Für den Massenstrom in Abschnitt IV:
( )( )AfestRaffRaffI,flRaffExAfestExIII,flEx
RaffAfestIII,flAfestI,flFeed,AIV,A KVVVVVVKVVVV
VKVVKVVmm
&&&&&&&&&&
&&&&&&&
−+−−++−−
= (9.79)
Für den Massenstrom der mit dem Extrakt abgezogen wird:
( )AfestRaffRaffI,flRaffExAfestExIII,flEx
RaffAfestIII,flExFeed,AEx,A KVVVVVVKVVVV
VKVVVmm
&&&&&&&&&&
&&&&&&
−+−−++−
−= (9.80)
Für den Massenstrom der mit dem Raffinat abgezogen wird:
(9.81)( )AfestRaffRaffI,flRaffExAfestExIII,flEx
AfestI,flRaffFeed,ARaff,A KVVVVVVKVVVV
KVVVmm
&&&&&&&&&&
&&&&&
−+−−−
=
Die Berechnung der Konzentrationen einer Komponente Z im Abschnitt k erfolgt über
( )Zfestk,fl
k,Zk,Z KVV
mc
&&
&
−= , (9.82)

9 Anhang
204
die der Konzentrationen in den Produktströmen Pr mit
P
Pr,ZPr,Z V
mc
&
&= . (9.83)
Mit Hilfe der drei Ansätze aus den Unterabschnitten E.1, E.2 und E.3 ist es nun möglich, die
Konzentrationen bei konstanten Volumenstromabweichungen der Pumpen und auch bei einer
fehlerhaften Adsorptionsisotherme unter Ausschluß von kinetischen Effekten darzustellen.




![Effektiver Ressourceneinsatz mit [FIS]-Dienstleister-Plattform fileDienstleister im Netz- und Anlagenbetrieb Effektiver Ressourceneinsatz mit [FIS]-Dienstleister-Plattform Überblick](https://img.pdfslide.org/doc/110x75/5e00a72f1538476f422fdbd6/effektiver-ressourceneinsatz-mit-fis-dienstleister-im-netz-und-anlagenbetrieb.jpg)
























