Embed Size (px)
Citation preview

260 W. I - I w ~ :
Neuentwicklungen bei Titrationen in nichtw~il~rigen Liisungsmitteln
W. HUBER
Badische Anilin- und Soda-Fabrik, Ludwigshafen
Eingegangen am 4. Oktober 1965
Summary. The acidimetrie titration of very weak bases, as for example carboxylic acid amides, with perchloric acid is so far only possible if the solvent, at least partly, consists of acetic anhydride. Many determinations, however, cannot be performed because of the aeetylating and leve]ling effect of this compound. I t is shown that acetonitrile can be used instead of acetic anhydride provided that no buffering substances, like glacial acetic acid or dioxane, are carried in together with the titrant. Thus, aliphatic and aromatic amines together with acid amides could be titrated in presence of each other for the first time. The applicability of the combination of catalytic hydrogenation and titration has been extended. In addition to nitriles nitro and azo compounds and also pyrrole derivatives can be converted to amines by hydrogenation in glacial acetic acid and titrated with perchloric acid. Among the pyrrole compounds benzopyrroles (indole, carbazole) can be selectively determined by direct potentiometric titration with tributylmethylammonium hydroxide in pyridine; the acidity of simple pyr- ro]es is not sufficient for such a titration. Mixtures of aliphatic sulphonic acids and primary and secondary sulphonic acid amides have been analysed by alkalimetry. The differentiation between secondary sulphonic acid amides and sulphonic acids has only been possible by the use of glacial acetic acid as solvent. Sulphonie acids may be selectively determined by titration with sodium acetate solution in glacial acetic acid; when titrating poten- tiometrically with tributylmethylammonium hydroxide in tert.-butanol they are determined together with the secondary sulphonic acid amides; the second potential jump corresponds to the primary sulphonic acids amides.
Die potentiometrische Titration sehr schwacher Basen in Acetonitril
Wenn man die NeutrMisation zwischen einer gelSsten Base B u n d einer gelSsten Sgure HS allgemein formuliert durch die Gleiehung
B -t- t IS ~- BH+ ~- S- ,
dann gibt diese Gleiehnng bekanntl ich den Vorgang nur formal wieder. I n Wirklichkeit liegt als weiterer t~eaktionsteilnehmer das L6sungsmit tel vor, was h&ufig zu einschneidenden Xonsequenzen ffihrt. Die An- wendung niehtwggriger L6sungsmit te l an Stelle yon Wasser stellt den Versueh dar, dem ideMen Neutral isat ionsprozeg, wie er der obigen Re- aktionsgleichung entsprieht, n&herzukommen. Ganz erreieht kann er bei Anwendung yon L6sungsmit teln nieht werden, da der Begriff des LSsens sehon eine gewisse Solvatat ion einschliegt, aber man kann ibm doeh

Titrationen in nichtw~Brigen LSsungsmi~teln 261
erheblich mehr niiherriicken, als wenn man Wasser als L6sungsmittel verwendet. Betrachtet man die potentiometrische Titration sehr schwacher Basen, wie I~arnstoffe, Carbons/iureamide, o-Nitranilin usw., so stellt man fest, dab sich diese Verbindungen infolge LSsungs~dtteleinflug in Wasser gar nicht und auch in Eisessig nicht befriedigend titrieren lassen. Die Basizit/~ anch des Eisessigs ist schon zu hoch und verhindert eine scharfe Endpunktsbestimmung. Verwendet man start des Eisessigs Essigsaureanhydrid, so lassen sieh Titrationen derartig schwacher Basen im Prinzip m/ihelos durchffihren6), i~. DaG der Eisessig tats~ehlieh nut eine Pufferwirkung ausiibt, li~Gt sich ersehen, wenn Misehungen yon Essigsgureanhydrid und Eisessig verwendet werden. Hierbei nimmt die Sehiirfe des Potentialsprungs mit zunehmender Essigs~urekonzentration stark ab i~. Essigsgureanhydrid hat zwei unangenehme Nebenwirkungen. Einmal aeetyliert es prim~re und sekundare Amine und kann aueh auf Harn- stoffe einwirken. Zweitens verwiseht es Unterschiede zwisehen stark und sehwach basischen Aminen dutch Nivellierung. Seine Anwend- barkeit unterliegt daher Beschrgnkungen. Die vorgesehlagene Ver- wendung yon Trifluoressigsi~ure li bedeutet keine entseheidende Ver- besserung, da hierdureh nur die Aeylierung verhindert wird, ganz ab- gesehen yon den praktisehen Schwierigkei~en der Anwendung. Es ersehien daher lohnend, gebr~uchliehe aprotisehe L6sungsmittel auf ihre Verwendbarkeit n~her zu untersuchen. Die Anwendung yon Nitro- methan is~ schon mehrfaeh besehrieben wordenS, i~ besonders mit einer Beimischung yon Essigsgureanhydrid, das als Absorptionsmittel ffir Wasserspuren gedaeht ist. Ohne diesen Zusatz, der ja prinzipiell un- erwfinseht ist, sind aber die Ergebnisse ziemlich nnbefriedigend. Aeeto- nitril verhielt sieh nach unseren Versuehen etwas g/instiger, wobei jedoeh bemerkenswert war, dab der Potentialsprung um so ansgepr/igter war, je weniger Substanz eingesetzt wurde (Abb. 1). Dies ffihr~e uns zu der Vermutung, dag das Dioxan, in dem unsere Perehlorsgure gel6s~ war, die st6rende Pnffersubstanz sein k6nnte. Dutch Zusitze yon Dioxan nnd Eisessig zu Aeetonitril konnte aueh belegt werden, dab die Pufferwirkung dieser Verbindungen in Aeetonitril weir ausgeprggter als in Essigsgnre- anhydrid ist. So ist z. B. Dimethytformamid in Acetonitril nur bis zu einem Eisessiggehal~ yon ca. 50/0 titrierbar, darfiber hinaus kommt es zu einer starken Verflaehung des Potentialsprungs. In Essigs/~ureanhydrid dagegen bewirkt sogar ein Zusatz yon 10~ fast noeh keinen Effekt. Wir haben ftir diese Versuehe eine L6sung yon Perehlorsgure in Aeetonitril verwendet. Mit dieser LSsung konnte eine betrgeh~liche Verbesserung bei der Titration sehwaeher Basen erzielt werden. Leider ist sie jedoeh nicht stabil, da das Aeetonitril in relativ kurzer Zeit zu Aeetamid verseift wird.
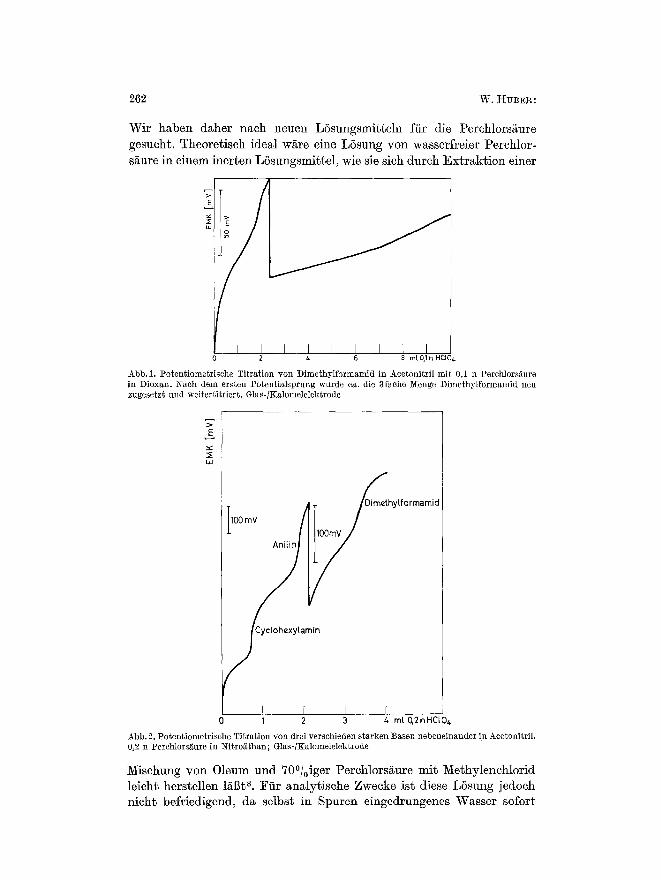
262 W. HvB~:
Wir haben daher nach neuen LSsungsmitteln fiir die Perchlors~ure gesucht. Theoretisch ideal wiire eine LSsung yon wasserfreier Perchlor- s~ure in ehlem inerten LSsungsmit~el, wie sie sich durch Ext r~kt ion einer
L q I I ~ I I I I I 0 2 4 6 8 mL 0,1n HC[O
Abb. 1. Potentiome~rische Titration yon Dimethylformamid in Acetonitril mit 0,1 n Perchlors~ture in Dioxan. Nach dem ersten Potentialsprung wurde ca. die 3fache ~[enge Dimethylformamid neu zugesetzt und weiter~itriert. Glas-/KalomeMektrode
E
2g
W
Ilo~ AI
I f I f 0 1 2 3 4 ml 0,2nHC[0~
Abb. 2. Potentiometrlsche TiLra~ion yon drei versohieden starken Basen nebeneinander in Acetonitril. 0,2 n Perchlors~ure in Nitro~than; Glas-/Kalomelelektrode
Mischung yon Oleum und 70~ Perchlors/~ure mit Methylenchlorid leieht herstellen lgl~t s. ~'iir analytisehe Zweeke ist diese LSsung jedoeh nieht befriedigend, da selbst~ in Spuren eingedrungenes Wasser sofort
Titrationen in niehtwgBrigen LSsungsmitteln 263
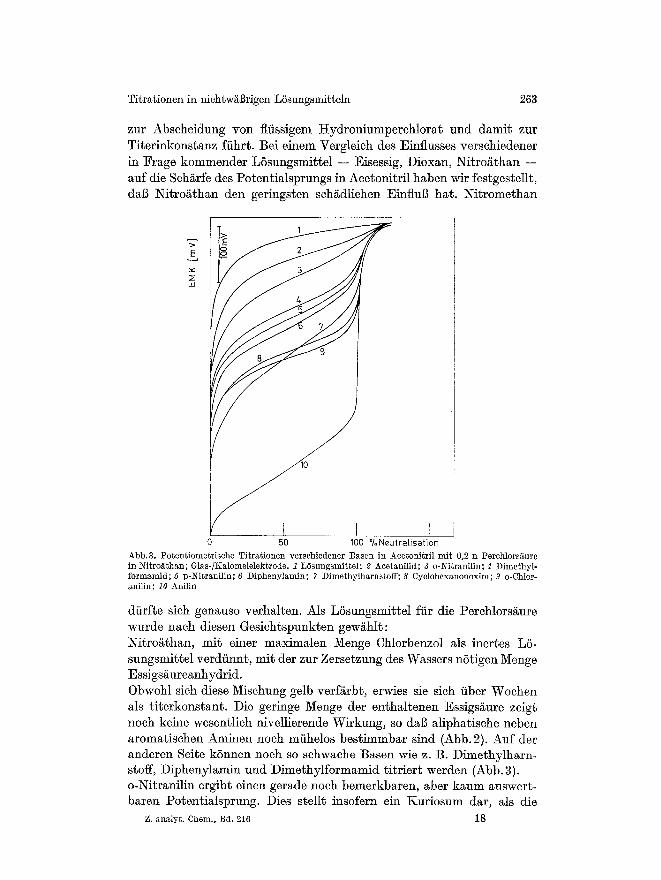
zur Abscheidung yon fliissigem ttydroniumperehlorat und damit zur Titerinkonstanz fiihr~. Bei einem Vergleieh des Einflusses versehiedener in tSrage kommender L6sungsmittel -- Eisessig, Dioxan, Nitro/~than -- auf die Seh/~rfe des Potentialsprungs in Aeetonitril haben Mr festgestellt, dab Nitro/~than den geringsten sehgdiiehen Einflug hat. Nitromethan
2~
0 50 100 ~ Neutralisalion
Abb. 3, Potentiometrische Titrationen verschiedener I~sen in Acetonitril mit 0,2 n Perchlors~ure in Nitrofi, than; Glas-/Kalomelelektrode. 1 LOsungsmittel; 2 Acetanilid; 3 o-Nitranilin; g Dimethyl- formamid; 5 p-Nitranilin; 6 Diphenylamin; 7 Dimethylharnstoff; 8 Cyclohexanonoxim; 9 o-Chlor- anilin; 10 Anilin
diirfte sieh genauso verhalten. Als L6sungsmittel ffir die Perchlorsi~ure wurde nach diesen Gesichtspunkten gewahlt: Nitroathan, mit einer maximalen Menge Chlorbenzol als inertes LS- sungsmit~el verdfinnt, mit der zur Zersetzung des Wassers nStigen Menge Essigsaureanhydrid. Obwohl sich diese Mischung gelb verf&rbt, erwies sie sieh fiber Wochen als ~iterkonstant. Die geringe Menge der enthaltenen Essigsi~ure zeigt noch keine wesentlich nivellierende Wirkung, so dab aliphatische neben aromatisehen Aminen noch mfihelos bestimmbar sind (Abb. 2). Auf der anderen Sei~e kSnnen noch so sehwache Basen wie z. B. Dimethylharn- stoff, Diphenylamin und Dimethylform~mid titriert warden (Abb. 3). o-Nitranilin ergibt einen gerade noch bemerkbaren, aber kaum auswert- baren Potentialsprung. Dies stellt insofern ein Kuriosum dar, als die
Z. analyt. Chem., Bd. 216 18
2 6 4 W . HIYBER:
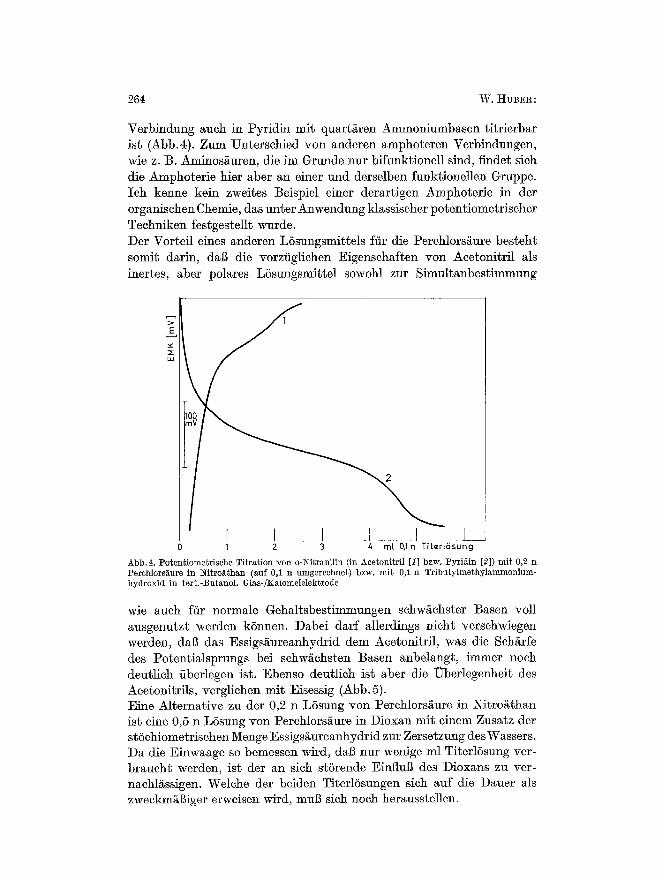
Verbindnng auch in Pyridin mit quart/~ren Ammoniumbasen titrierbar ist (Abb.4). Zum Unterschied yon anderen amphoteren Verbindnngen, wie z. B. Aminos/~uren, die im Grunde nur bifunktionell sind, finder sieh die Amphoterie hier aber an einer und derselben funktionellen Gruppe. Ich kenne kein zweites Beispiel einer derartigen Amphoterie in der organischen Chemie, das unter Anwendung klassischer potentiometrischer Techniken festgestellt wurde. Der Vorteil eines anderen L6sungsmitte]s ffir die Perchlorsgure besteht somit darin, dag die vorziiglichen Eigenschaften yon Acetonitril als inertes, aber polares L6sungsmittel sowohl zur Simultanbestimmung
I ' I I I I I I 0 1 2 3 4 mt 0,1n Titert6sung
Abb. 4. Poten~iome~rische Titration Yon o-Nitranilin (in Acetoni~ril [I] bzw. Pyridin [2]) mi t 0,2 n Perchlors~iure in Nitro~than (auf 0,1 n umgerechnet) bzw. mib 0, l n Tribu~ylmethylaramonium- hydroxid in ter~.-t~u~anol. Glas-lKalomelelektrode
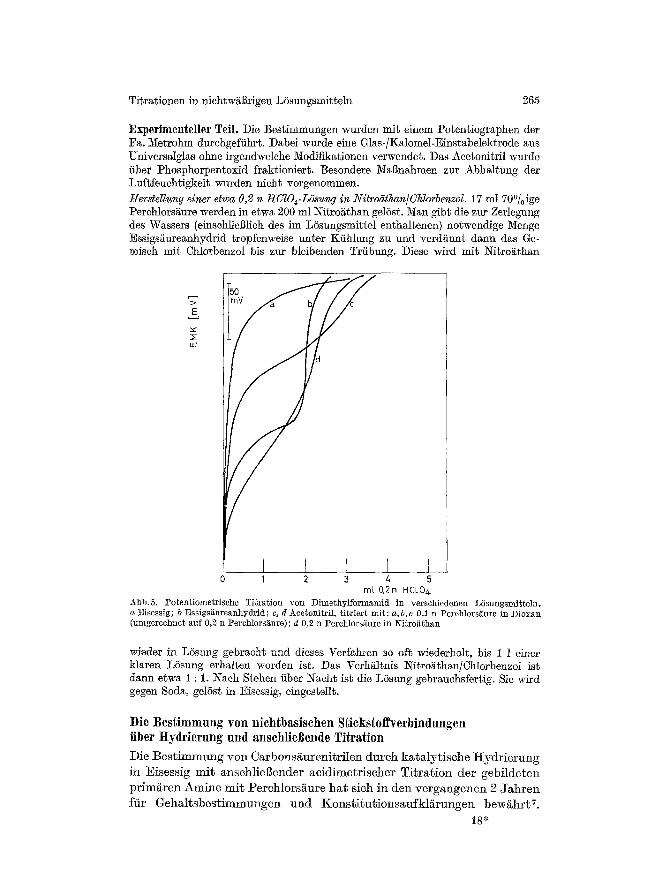
wie auch fiir normale Gehaltsbestimmungen schw/ichster Basen roll ausgenutzt werden k6nnen. Dabei darf allerdings nicht verschwiegen werden, dal~ das Essigs/~ureanhydrid dem Acetonitril, was die ScMrfe des Potentialsprungs bei schw~chsten Basen anbelangt, immer noch deutlich iiberlegen ist. Ebenso deutlich ist aber die Uberlegenheit des Acetonitrils, verglichen mit Eisessig (Abb. 5). Eine Alternative zu der 0,2 n L6sung yon Perchlors/~ure in Nitroathan ist eine 0,5 n L6sung yon Perchlors~ure in Dioxan mit einem Zusatz der stSchiometrischen Menge Essigs~ureanhydrid zur Zersetzung desWassers. Da die Einwaage so bemessen Mrd, dag nur wenige ml TiterlSsung ver- braucht werden, ist der an sich stSrende Einflug des Dioxans zu ver- nachlassigen. Welche der beiden Titerl6sungen sich auf die Dauer als zweckms erweisen wird, muB sich noch herausste]]en.
Titrationen in niehtw~grigen LSsungsmitteln 265
Experimenteller Teil. Die Bes~immungen wurden mit einem Potentiographen der Fa./~{etrohm durchgeffihrt. Dabei wurde eine Glas-/Kalome]-Einstabelektrode aus Universalglas ohne irgendwelche Modifikationen verwendet. Das Acetonitril wurde fiber Phosphorpentoxid fraktionierfa Besondere Nagnahmen zur Abhaltung der Luftfeuehtigkeit wurden nich~ vorgenommen.
Herstellung einer etwa 0,2 n HClO4-LSsung iu 2titroiithan/Chlorbenzol. 17 ml 70v/oige Perchlorsiiure werden in etwa 200 ml Nitrogthan gelSst. Mart gib~ die zur Zerlegung des Wassers (einsehliel31ieh des im LSsungsmittel enthaltenen) notwendige Menge Essigsgureanhydrid tropfenweise unter Kfihlung zu und verdfinnt dann alas Ge- miseh mi~ Chlorbenzol bis zur bleibenden Trfibung. Diese wird mit Nitrogthan
E
2~ LIJ
0 1 2 3 4 5 mt o.2n HcI0/+
Abb.5. 72otentiometrische Titration yon Dimethylforrnamid in verschiedonen L6sungsmi~teln. a Eisessig; b Essigsiiureanhydrld; c, d Acetonitril, titriert mi~: a,b,c 0,1 n Perchlors~ure in Dioxan (umgerechnet auf 0,2 n Perchlorsi~ure); d 0,2 n Perchlors~ure in l\'itroathan
wieder in LSsung gebraeh~ und dieses VerfM~ren so oft wiederholt, bis i 1 einer klaren /,6sung erhalten worden ist. Das Verh/~l~nis Ni~ro~than/Chlorbenzol is~ darm e~wa 1 : 1. Nach Stehen fiber Nacht is~ die LSsung gebrauchsfertig. Sie wird gegea Soda, gelSs~ in Eisessig, eingesteilt.
Die Bestimmung yon niehtbasisehen Stiekstoffverbindungen fiber Hydrierung und anschlie]lende Titration Die ] ~ e s t i m m u n g y o n Carbons / tu ren i t r i l en d u r c h k a t a l y t i s c h e I - Iydr ie rung
in Eisess ig m i t ansch l i eBende r a c i d i m e t r i s c h e r T i t r a t i o n de r g e b i l d e t e n
p r i m ~ r e n A m i n e m i t Pe r ch lo r s~u re h a t sich in d e n v e r g a n g e n e n 2 J a h r e n ffir G e h a l t s b e s t i m m u n g e n u n d K o n s t i t u t i o n s a u f k l / ~ r u n g e n b e w a h r t L
18"

266 W.I-I~B~R:
Es wurde daher versucht, die Methode auf andere neutrale Stiekstoff- verbindungen, die bei der Hydrierung Amine bflden, zu fibertragen.
Stic~stoMverbindungen, die bei der Hydrierung Basen bilden
I~--C--N -~ R--CH~--NH 2 I~--N0~ -~ R--NH 2 P~--N0 --~ R--NH~ I~--N=N--R'-~ R--NH~-~I~'--NK~ P~-- ONO 2 --~ ROI-I-F NH 3 Pyrrole --> Pyrrolidine
Dabei erwies es sich als notwendig, an Stelle des frfiher verwendeten Palladiummohrs a]s Kontakt Platinoxid nach ADAMS ZU verwenden. Die Hydrierung ] ~ t sich dadureh wesentlieh besser durchfiihren, spezie]l bei aromatischen PoIynitroverbindungen. Nach unseren frtiheren Untersuchungen enth~lt aber Platinoxid basisehe Verunreinigungen, die einen betr~ehtliehen Blindwert verursachen. Es gelang jetzt, dutch Aus- kochen des Platinoxids mit verdiinnter Salpeters~ure diesen Blindwer~ so weir zu verkleinern, dab die Kontaktmenge nicht genau eingewogen werden muB. Die Hydrierung aromatischer Nitroverbindungen erfolgt moist glatt zu den eycloaliphatisehen Aminen, was dureh den Wasserstoffverbraueh dokumentiert wird. Eine Ausnahme maeht die Pikrins~ure, die trgge und anscheinend nicht ganz vollsts hydriert. Nach der Hydrierung noeh vorhandene aromatische Aminoverbindungen zeigen eine sehr starke Autoxydation unter Bildung eines blauen Farbstoffes. Die Endbestim- mung lgBt sieh potentiometrisch aber ohne Sehwierigkeiten durch- fiihren. Aliphatisehe Nitroverbindungen kSnnen vorl~ufig nicht bestimmt werden. Der Grund ist ganz offensiehtlieh eine unvollstgndige Hydrie- rung. Die gefundenen Wasserstoffverbr~uehe sind zu tier, und der Um- satz erfo]gt sehr tr~ge, auch bei Erw~rmung auf 80 ~ C. Wenn ein bes- serer Kontakt gefunden werden kSnnte, ware diese Bestimmung sicher ebenfalls durchfiihrbar. Dasselbe gilt vom Nitrosodimethylanilin, das unerklgrlicherweise eben- falls nieht befriedigend hydriert werden konnte. Dagegen ist das a-Ni- troso-fl-naphthol ohne weiteres bestimmbar, wobei es allerdings sehwierig, aber aueh unnStig ist, den Kern ganz durchzuhydrieren. Cyelohexylnitrat als Vertreter der Salpeters~ureester hydriert trgge, ist aber naeh einer Verls der Hydrierzeit auf etwa 6 Std bestimm- bar. Aromatisehe Azo- und Hydrazoverbindungen hydrieren dagegen sehr raseh unter Aufspaltung tier N-N-Bindung. Nut beim Dinitrophenyl- hydrazin scheint diese Aufspaltung Schwierigkeiten zu bereiten, was auf Grund der Itydrier- und Titrierwerte zu schlieBen ist.

Titrationen in nichtwiigrigen LSsungsmitteln 267
Die Hydrierung yon Pyrrolderivaten erfolgt glatt. Gerade bei Pyrrol- verbindungen hat sieh diese Analysenmethode ffir Konsti tut ionsermitt- lungen bew/~hrt. Liegt eine ungef/irbte C,H,N-Verbindung ohne nach- weisbare Basizit~t vor, liegt der Verdacht auf Pyrrole oder Nitrile nahe. Der Naehweis kann, abgesehen yon spektroskopisehen Methoden, aueh in der Weise erfo]gen, dab die hydrierte L5sung qualitativ auf sekund/ire Amine untersueht wird. Dazu wird am besten der Umsatz sekund/irer Amine in ammoniakalischer L6sung mit Sehwefelkohlenstoff zu den Dithiocarbamids&uren benutzt, die mit CuH-Ionen braun gefgrbte, benzo]16sliche Komplexsalze geben. Nitrile ergeben bei der I tydr ierung prim~re Amine, welehe diese Reaktion nicht zeigen.
AuBer solehen Farbreaktionen besteht aueh die M6gliehkeit, die yore Kon tak t abfiltrierte LSsung mit etwas Salzs~ure zur Trockne abzu- dampfen. Die zuriiekbleibenden Aminhydroehloride kSnnen sehr gut nach bekannten Methoden papierehromatographiseh nntersucht werden. Diese M6glichkeit ist besonders bei Gemisehen bzw. bei Verdaeht auf vorliegende Gemische interessant. Dazu kann man aueh unsymmetrisehe Azo- und Hydrazoverbindungen zghlen, die sieh in die entspreehenden Amine spalten lassen. Eine andere Kombinat ion mi t einer bekannten Methode wgre eine ti tanometrische Titration. Das Anwendungsgebiet dieser Methode umfagt ebenfalls die Nitro-, Nitroso-, Azo- und Hydrazo- verbindungen, jedoeh ist die Aussage eine andere, da Reduktions- s erfagt werden. ]~ei Gemisehen versehiedener funktioneller Gruppen ist daher keine Bestimmung mSglieh. II ier kSnnte eine Kom- bination mit Hydrierung ~- Titration, welche die Anzahl der reduzier- baren N-Atome liefert, weiterhelfen und eine zweite BestimmungsgrSge liefern.
Eine Ubersicht fiber Analysenergebnisse der versehiedenen Verbindungs- gruppen gibt die Tabelle.
Ex-perimenteller Teil. Die Hydriertechnik wurde in einer frfiheren Arbeit beschrie- ben 7. An Stelle yon Palladiummohr wurde jetzt abet Platinoxid nach ADAMS ver- wendet, das vor der Benutzung mit 2 n Salpetersgure ausgekocht wurde. 100 mg Kontakt zeigen dann einen Blindwert yon etwa 0,1 ml 0,1 n Perchlorsgure. Die Einwgage betrug 0,5--2 reVal Substanz. Bei hSheren Einwaagen und stark ungesgttigten Verbindungen ist es vorteilhaft, eine Gasbiirette yon ca. 300 m] FassungsvermSgen anzuwenden, t/ei tr~,ger I{ydrierung kann die Schtittelente durch eine elektrische ~eizung a~f ca. 80 ~ C erwgrmt werden. Die gefundenen tIydrierjodzahlen stimmten mit den theoretisch geforderten Werten sehr gut iiberein, abgesehen yon den erw~hnten Ausnahmen. Sie kSnnen daher zu eventuellen Bereehnungen mit herangezogen werden. Als Titrationsmittel wurde eine 0,1 bzw. 0,05 n PercMorsgure in Eisessig benutztL Der Konta.kt braueht vor der Titration nicht abfiltriert zu werden. Die Titration k~nn potentiometriseh (Anwendung eines Potentiographen) oder mit Indieatoren durehgeftihrt werden. Dazu wurde entweder eirb Tropfen einer 0,05~ igen KrlstMtviolettlSsung in Eisessig benutzt (seharfer, uber nieht besonders gut
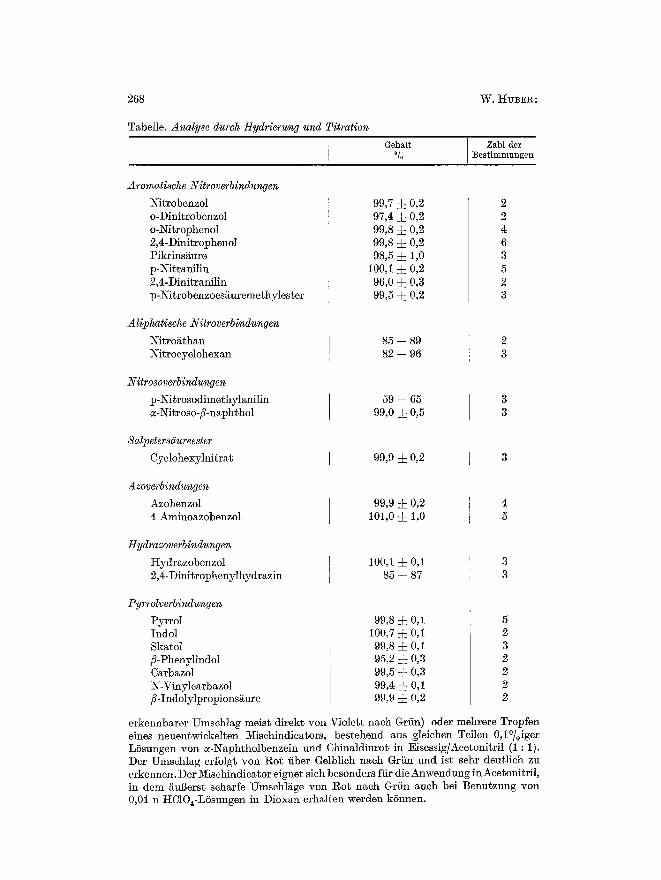
268 W. HUBE~ :
Tabelle. Analyse dutch Hydrierung und Titration GehaR
~ Zahl der
:Bestimmungen
Aromatisehe Nitroverbindungen Nitrobenzol o-Dinitrobenzol o-Nitrophenol 2,4-Dinitrophenol Pikrinsi~ure p-Nitranilin 2,4-Dinitranilin p-Nitrobenzoesguremethylester
99,7 ~ 0,2 97,4 :J: 0,2 99,8 ~ 0,2 99,8 ~ 0,2 98,5 4- 1,0
100,1 ~ 0,2 96,0 ~ 0,3 99,5 ~ 0,2
Aliphatische Nitroverbindungeu ~Nitro~th~n Nitrocyclohexan
85 -- 89 82 -- 96
Nitrosoverbindungen p-Nitrosodime~hylanilin ~-Nitroso-fi-n~phthol
59 -- 65 99,0 4- 0,5
Sal2~eters~iureester Cyclohexylnitrat 99,9 4- 0,2
Azoverbindungen Azobenzol 4-Aminoazobenzol
99,9 4- 0,2 101,0 4- 1,0
Hydrazoverbindungen I-Iydrazobenzol 2,4-Dinitrophenylhydrazin
100,1 ~ 0,1 85 -- 87
Pyrrolverbindungen Pyrrol Indol Skatol fi-Phenylindol Carbazol N-Viny]carbazo] fl-Indolylpropionsgure
99,8 • 0,1 100,7 • 0,1 99,8 4- 0,1 95,2 4- 0,3 99,5 4- 0,3 99,4 4- 0,1 99,9 4- 0,2
erkennbarer Umschlag meis~ direkt yon Violett nach Griin) oder mehrere Tropfen eines neuentwickelten Mischindicators, bestehend aus gleichen Teilen 0,1~ LSsungen yon ~-Naphtholbenzein und Chinaldinrot in Eisessig/Acetonitril (1 : 1). Der Umschlag erfolgt yon ~o~ fiber Gelblich nach Griin und ist sehr deutlich zu erkennen. Der Mischindicator eignet sich besonders fiir die Anwendung in Acetonitril, in dem ~u~erst scharfe Umschlgge yon Rot nach Grfin such bei Benutzung yon 0,01 n HClO~-LSsungen in Diox~n erhalten werden kSnnen.
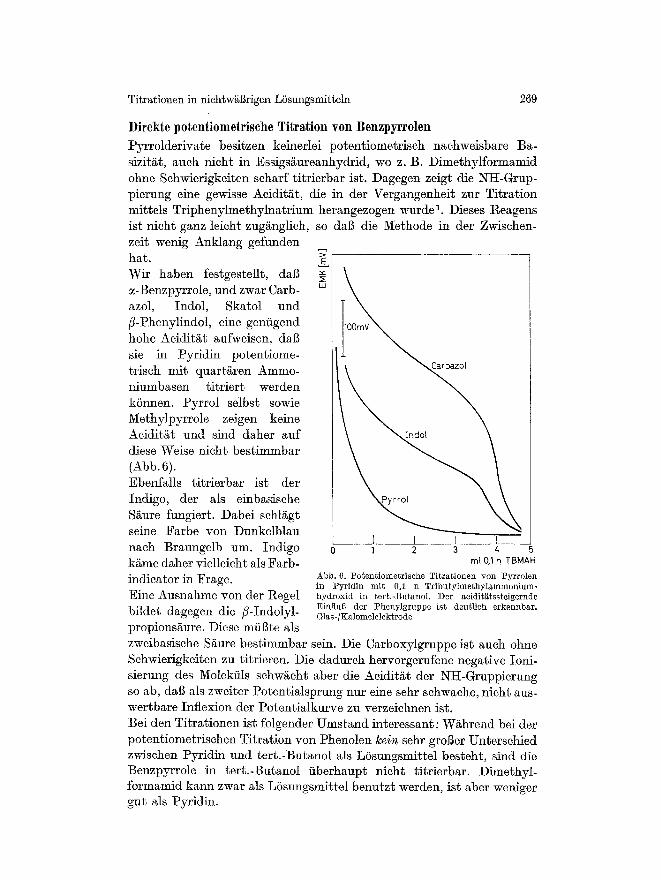
Titrationen in niehtwggrigen LSsungsmitte]n 269
D i r e k t e p o t e n t i o m e t r i s c h e T i t r a t i o n y o n B e n z p y r r o l e n
Pyrrolderivate besitzen keinerlei potentiometriseh nachweisbare Ba- sizit/~t, auch nieht in Essigs/~ureanhydrid, we z. B. Dimcthylformamid ohne Schwicrigkeiten scharf titrierbar ist. Dagegen zeigt die NI~-Grup- pierung eine gewisse Aciditgt, die in der Vergangenheit zur Titration mittels Triphenylmethylnatrium herangezogen wurde 1. Dieses t~eagens ist ~ficht ganz leicht zug~nglieh, so dag die Nethode in der Zwischen- zeit wenig Anklang gefunden hat. Wir haben festgestellt, dab ~-Benzpyrrole, und zwar Carb- azol, Indol, Skatol und ~-Phcnylindol, eine gentigend hohe Aeidit/~t aufweisen, dag sie in Pyridin potentiome- trisch mit quartgren Ammo- niumbasen titriert werden kSnnen. Pyrrol selbst sowie Methylpyrro]e zeigen keine Aeiditgt und sind daher auf diese Weise nieht bestimmbar (Abb. 6). Ebenfalls titrierbar ist der Indigo, der als einbasische S~ure fungiert. Dabei sehl/~gt seine Farbe yon Dunkelblau nach Braungelb am. Indigo kgme daher vielleicht als Farb- indicator in Frage. Eine Ausnahme yon der Regel bildet dagegen die /?-Indolyl- propions/~ure. Diese mfil3te a]s
I i I I 0 1 2 3 4
ml 0,1 n TBMAH
Abb. 6. Po~en~iometrlsche Titrationea yon Pyrrolen in Pyridin mit 0,1 n Tributylmethylammonium- hydroxid in terb.-Bu~anoI. Der acidlt~;~ssteigernde Einftug der Phenylgruppe ist deutlich erkemtbar. Glas-/Kalomelelektrode
zweibasische Ss bestimmbar sein. Die Carboxylgruppe ist such ohne Schwierigkeiten zu titrieren. Die dadurch hervorgerufene negative Ioni- sierung des Molekiils schws aber die Aeidit/~t der NII-Gruppierung so ab, dab als zweiter Potentialsprung nur eine sehr sohwache, nicht aus- wertbare Inflexion der Potentialkurve zu verzeichnon ist. Bei den Titrationen ist folgender Umstand interessant: W~hrend bei der potentiometrisohen Titration yon Phenolen l~ein sehr groger Untersehied zwischen Pyridin und tert.-Butanol als L6sungsmittel besteht, sind die Benzpyrrole in tert.-Butanol fiberhaupt nieht titrierbar. Dimethyl- formamid kann zwar als L6sungsmittel benutzt werden, ist aber weniger gut sis Pyridin.
270 W. HUBER:
Bei einer KombinaHon dieser alkalime~rischen Direktmethode mit der aeidimetrischen Titration naeh vorangegangener I-Iydrierung (siehe oben) ergibt sieh eine Methode zur Analyse yon Pyrrol/Benzpyrrol-Gemisehen. Derartige Verbindungen kommen besonders in RohSlen vor, wo sie den Grogteil des sogenannten nich%basischen Stickstoffs darstellen.
Experimenteller Tell. Die Bes~immungen wurden mi~ dem Potentiographen dureh- geffihrt. Dabei wurde eine unmodifizierte Glas-/Kalomel-EinstabmeBkette aus Universalg]as benu~zg. Die Titer]Ssung wurde aus kguflieher 40~ Tributyl- met~hylammoniumhydroxidl6sung in Wasser (1%. Fluka, Buchs, St. Gallen, Schweiz) durch Verdfinnung mit ter~.-Butanol (5% Benzol enthalgend) hergeste]lt. Die Ein- s~ellung erfolgte gegen Benzoesgure, gelSst in Isopropanol. Isg die LSsung carbonat- haltig, was durch Titration yon egwas Salzsiiure in Pyridin leicht fesgzusgellen ist (Auftregen yon 2 Potentialsprfingen), sollge gegen reinsges Indol in Pyridin ein- gesgellg werden. Die L6sung muB sorgfgltig vor Luftkohlensgure geschiitzg werden. Der Tiger hgl~ sich fiber Monate konstant. Das verwendete Pyridin wurde fraktionierg und war fast blindwertfrei. Die erziel- bare Genauigkeit lag bei =l= 1~ relativ.
Bestimmung yon aliphatischen prim~iren und sekund~iren SulIons~iureamiden sowie Sulfons~iuren nebeneinander
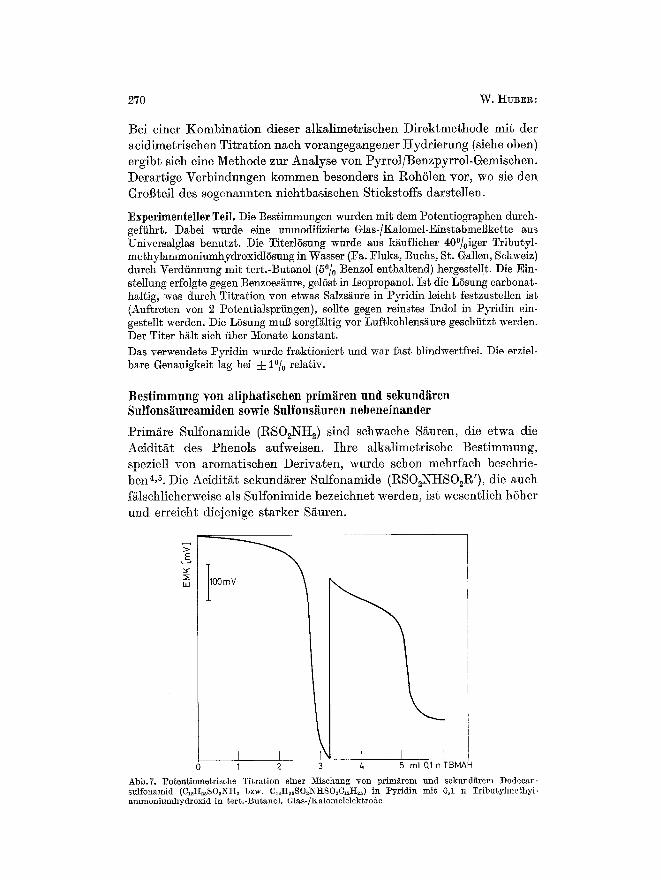
Primgre Sulfonamide (RS02Ntt2) sind schwache Si~uren, die etwa die Aeiditgt des Phenols aufweisen. Ihre alkalimetrisehe Bestimmung, speziell yon aromatisehen Derivaten, wurde sehon mehrfaeh besehrie- bent, ~. Die Aeiditgt sekundgrer Sulfonamide (RSO2NHSO~R'), die aueh fglschlicherweise als Sulfonimide bezeiehnet werden, ist wesentlieh h6her und erreicht diejenige starker S/~uren.
.E
hi 100 mV " ~
0 1 2 3 4 r
5 ml 0,1 n TBNAH Abb.7. PotenHometrlsche Titration einer :Mischung ~r prim~rem und sekund~trem Dodecan- sulfonamid (C12H2~SO2NH~ bzw. CI~tt~sSO~I~THSO~CI~H~) in Pyridin mit 0,1 n Tributylmethyl- amrnoniumhydroxid in tert.-Butanol. Glas-/Kalomelelektrode

Titrationen in nichtw~Brigen L6sungsmi%eln 271
Eine Simultanbestimmung dieser beiden Verbindungen kann daher keine Schwierigkeiten bereiten und ist potentiometrisch in verschiedenen LS- sungsmitteln, die nur eine ausreichend kleine Acidit/it aufweisen mfissen, durchffihrbar.
Die besten Kurven wurden bei der Titration in Pyridin mit 0,1 n Tri- butylmethylammoninmhydroxidl6sung erhalten (Abb.7). IIier ergibt sich aber folgende Schwierigkeit: Als Nebenkomponente enthalten die Amidgemische h/iufig freie Sulfon- sguren. Diese kSnnen in Pyridin neben sekund~ren Amiden nieht erfal~t werden, da sie gegenfiber diesen Verbindungen nivelliert sind. Der erste Potentialsprung erfagt also die Summe yon Sulfons/~nren und sekund/tren Sulfonamiden. Eine Trennung der beiden Kompo- nenten mul3 in einem L6sungsmittel yon extrem geringer Basizit/tt erfol- gen. Am besten bewiihrt hat sieh Eis- essig. Als Titrationsmittel wurde 0,1 n Natriumacetatl6sung in Eis- essig benutzt. Unter diesen Bedin- gungen k6nnen Sulfonsi~nren selek- t iv erfal3t werden (Abb.8). Beide
E
lOOmY
I I 2 3
mt 0.1 n Natriumacetat
Abb. 8. ~otengiometr i sehe T i t r a t i o n yon / )odecansulfonsi ture in Nisessig m i t 0 , / n N a t r i u m a e e t a t in Nisessig. Glas-/lt2alomeI- e lek t rode
Sulfonamide verhalten sich in diesem Medium nicht als Sguren. Ein Gemisch aller drei Komponenten kann somit fiber zwei Titrationen be- stimmt werden.
Experimenteller Teil. Die Ausfiihrung der Titration und Darstellung der 0,1 n Tributylmethylammoniumhydroxidl6sung erfolgte, wie bei den Benzpyrrolen be- schrieben. Ein Carbonatgehalt der L6sung ist hier besonders stSrend und sollte durch Ionenaustausch beseitigt werden 2. Die 0,1 n NatriumacetatlSsung ~arde durch L6sen der theoretischen Menge Soda in Eisessig hergestellt.
Zusammenfassung Die acidimetrische Titration schw~chster Basen, wie z. B. Carbons/~ure- amide, mit Perchlorsgure ist bis jetzt an die mindestens teilweise An- wendung yon Essigs/~ureanhydrid als L6sungsmittel gebunden. Infolge der acetylierenden und nivellierenden Wirkung dieser Verbindung k6nnen viele Bestimmungen jedoch nicht ausgeffihrt werden. Es wird gezeigt, dab Acetonitril das Essigss ersetzen kann, wenn man daftir sorgt, dab mit dem Titrationsmittel keine puffernden Sub-

272 W. I-Iv~g: TRrationen in nichtw~Brigen LSsungsmitteln
s tanzen wie Eisessig oder Dioxan e ingeschleppt werden. Dadurch war es e rs tmals mSglich, a l iphat i sche und a romat i sehe Amine m i t S~ureamiden zusammen nebene inander zu t i t r ieren. Die K o m b i n a t i o n yon ka t a ly t i s ehe r t t y d r i e r u n g und T i t r a t i on wurde wel te r ausgedehnt . AuBer Ni t r i l en kSnnen Ni t ro - und Azoverb indungen sowie P y r r o l d e r i v a t e durch H y d r i e r u n g in Eisessig in Amine i iberf i ihr t und mi t Perehlors~ure t i t r i e r t werden. I nne rha lb der Py r ro lve rb indungen lassen sieh Benzpyr ro le (Indol, Carbazo]) se lekt iv dureh d i rek te po ten t iomet r i sehe T i t r a t i on m i t Tri- b u t y l m e t h y l a m m o n i u m h y d r o x i d in P y r i d i n bes t immen. Die Ac id i t~ t e infacher Py r ro l e re ieht hierffir n i eh t aus. Misehungen yon a l ipha t i sehen Sulfons~uren sowie primi~ren und se- kundi~ren Sulfonsi~ureamiden wurden a lka l imet r i sch analys ier t . Die Dif- ferenzierung sekunds Sulfonss yon den Sulfonsi~uren gelang erst dureh Anwendung yon Eisessig als LSsungsmit te l . Durch T i t r a t i on m i t Na t r i umace t a t lS sung in Eisessig lassen sich die Sulfonsi~uren se lekt iv bes t immen. Bei der po ten t iomet r i sehen T i t r a t i on mi t T r i b u t y l m e t h y l - a m m o n i u m h y d r o x i d in t e r t . -Bu tano l werden sie zusammen mi t den sekundi~ren Sul fonss erfaBt. Der zweite Po ten t i a l sp rung erg ib t die primi~ren Sulfons~ureamide.
Fiir die Uberlassung yon Testsubstanzen danke ich ~ I-Ierrn Dr. R. FIsc~nn, ffir die Ausfiihrung der Versuche Fr]. E. K~DLER und den Herren H. LnI~m und G. H t o ~ .
Literatur 1 Co~w~, A.H., and •. C. ELLI~GSO~: J. Am. Chem. Soc. 64, 2098 (1942). --
CU~D~F, R. H., and P. C. MA~KV~AS: Anal Chem. 30, 1450 (1958); vgl. diese Z. 168, 123 (1959). -- 8 F~ITZ, J. S., and M. 0. FULDA: Anal. Chem. 25, 1837 (1953); vg]. diese Z. 147, 36 (1955). -- a F~ITZ, J. S., and R. T. K ~ : Anal Chem. 24, 308 (1952); vgl. diese Z. 137, 298 (1952/53). -- 5 F~ITZ, J. S., and S. S. Y~A~um~: Anal. Chem. 29, 1079 (1957); vgl. diese Z. 161, 48 (1958). -- 6 GR]~LION, A. F.: Anal Chem. 27, 133 (1955); vgl. diese Z. 148, 140 (1955/56). -- ~ ~IuB~, W.: diese Z. 197, 236 (1963). -- s I~AG~S, F., u. P. I-IEG~B~G: Angew. Chem. 74, 902 (1962). -- 9 S~EvLI, C. A.: Anal. Chem. 30, 997 (1958); vgl. diese Z. 168, 219 (1959). -- lo ST~VLI, C.A.: Anal Chem. 31, 1652 (1959); 32, 985 (1960); vgl. diese Z. 176, 199 (1960); 180, 129 (1961). -- 11 VRIES, J. ]~. I)E, J. 14. EnLIo~r, and 1%. T. HALL: Anal Chem. 20, 784 (1948). -- 1~ WLv[~I~, D. C.: Anal. Chem. 30, 77 (1958); vgl. diese Z. 165, 370 (1959).
Dr. W. HVBE~ in Fa. Badische Anilin- und Soda-Fabrik, A.G. Ammoniaklaboratorium, 6700 Ludwigshafen/Rhein





























