Embed Size (px)
Citation preview

Nickelspeziesanalyse im Cytosol humaner intestinaler
Gewebeproben mittels online-Kopplung von
Kapillarelektrophorese und induktiv gekoppeltem
Plasma-Massenspektrometer
Der Fakultät für Naturwissenschaften
der Universität Duisburg-Essen
zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
genehmigte Dissertation
von
Sven Bonsack
aus
Bochum
Referent: Prof. Dr. A. Golloch
Koreferent: Prof. Dr. B. Neidhart
Tag der mündlichen Prüfung: 02.04.2003

Erklärung
Hiermit erkläre ich, daß ich die Arbeit selbständig verfaßt habe. Die verwendetenQuellen sowie die verwendeten Hilfsmittel sind vollständig angegeben.
Göttingen, den 06.01.2003

Herrn Priv.-Doz. Dr. Prange vom GKSS Forschungszentrum danke ich für die Überlassungdes interessanten Themas und für die aufregenden Diskussionen. Bei Herrn Prof. Dr. Gollochbedanke ich für die universitäre Betreuung meiner Arbeit.
Mein Dank gilt weiterhin Herrn Prof. Dr. Neidhart für die wissenschaftliche Betreuung vonSeiten des GKSS Forschungszentrums und für die Korrektur dieser Arbeit.
Bei Herrn Prof. Oellerich von der Abteilung Klinische Chemie der Georg-August-UniversitätGöttingen möchte ich mich für flexible Gestaltung meiner Arbeitszeit und für das mirentgegengebrachte Verständnis bei die Erstellung dieser Arbeit bedanken.
Besonders möchte ich mich bei Frau Dr. Evelin Denkhaus für Ihre hervorragende inhaltlicheund moralische Betreuung der Arbeit bedanken. Ohne Ihre ständige Diskussionsbereitschaftbzw. Beiträge und Ihr offenes Ohr wäre diese Arbeit nie zu dem geworden, was sie ist.
Weiterhin möchte ich mich bei Herrn Dr. Dirk Schaumlöffel vom GKSS Forschungszentrumfür die Einführung in die Tiefen der CE / ICP-MS Kopplung und seine Hilfestellung beiauftretenden Problemen der Kopplung herzlich bedanken.
Außerdem gilt mein Dank den Mitarbeitern der Abteilung KAE des Instituts fürKüstenforschung/Physikalische und Chemische Analytik des GKSS ForschungszentrumsGeesthacht für die Hilfsbereitschaft und für das angenehme Arbeitsklima.
Für die Durchführung der TXRF Messungen möchte ich meinen Dank Herrn Dr. Reus vomGKSS Forschungszentrum Geesthacht aussprechen.
Besonders bedanken möchte ich mich bei meiner Mutter und meinem Bruder, die durchständige moralische Unterstützung zum Gelingen dieser Arbeit beigetragen haben.
Am Ende möchte ich mich noch bei meiner Freundin Ines dafür bedanken, dass sie in der Zeitdes Zusammenschreibens meine Launen ausgehalten und mir immer Mut zugesprochen hat,wenn es nötig war. Vor allem aber bin ich Ihr für das Korrekturlesen meiner Arbeit dankbar.

Für meinen Großvater und meinen Vater, die, so glaube ich,
ein bisschen stolz auf mich wären. Ich verspreche Euch,
dass ich Eure Worte über „Doktoren der Chemie“ nie
vergessen werde.

Inhaltsverzeichnis
Inhaltsverzeichnis
1 Einleitung__________________________________________________ 1
2 Stand der Forschung ________________________________________ 4
2.1 Nickelspezies in biologischen Proben___________________________ 4
2.2 Induktiv-gekoppltes-Plasma / Massenspektrometrie (ICP-MS) _______ 92.2.1 Zerstäuber für chromatographische Trennverfahren ___________________ 10
2.2.2 HPLC / ICP-MS Kopplung _______________________________________ 11
2.2.2.1 Reversed-Phase-Chromatographie gekoppelt mit der ICP-MS ___________ 11
2.2.2.2 Ionenpaar-Chromatographie gekoppelt mit der ICP-MS ________________ 12
2.2.2.3 Ionenaustausch-Chromatographie gekoppelt mit der ICP-MS ____________ 13
2.2.2.4 Größenausschluss-Chromatographie gekoppelt mit der ICP-MS _________ 14
2.2.3 Kapillarelektrophorese gekoppelt mit der ICP-MS _____________________ 15
3 Problemstellung ___________________________________________ 20
4 Analysenverfahren _________________________________________ 21
4.1 Probenmaterial_____________________________________________ 21
4.2 Probenvorbereitung_________________________________________ 23
4.3 Eingesetzte Kopplungstechniken______________________________ 244.3.1 CE / ICP-MS Kopplung __________________________________________ 26
4.3.2 SEC-Chromatographie mit UV-Detektion ____________________________ 28
4.3.3 SEC / ICP-MS Kopplung_________________________________________ 29
4.4 Totalreflexionsröntgenfluoreszensanalyse (TXRF) _______________ 31
4.5 Gesamtproteinbestimmung nach Biuret-Lowry __________________ 31
5 Optimierung der Analysenmethoden __________________________ 32
5.1 Reduzierung von Nickelkontaminationen _______________________ 325.1.1 Reinigung der Gefäße __________________________________________ 32
5.1.2 Reinigung der Chemikalien_______________________________________ 33
5.1.3 Reduzierung der Nickelkontamination durch apparative Veränderungen____ 35
5.2 Optimierung der CE-Trennbedingungen mithilfe eines gemischten Metallothionein-Standards ___________________________________ 36
5.2.1 Optimierung der Kapillarentemperatur ______________________________ 38
5.2.2 Optimierung der Kapillarenlänge __________________________________ 39
5.2.3 Optimierung des Innendurchmessers der Kapillare ____________________ 40
5.2.4 Optimierung der Konzentration des Puffers __________________________ 41

Inhaltsverzeichnis
5.2.5 Optimierung des pH-Wertes des Puffers ____________________________ 42
5.2.6 Optimierte Messparameter für die CE / ICP-MS Kopplung_______________ 44
5.3 Vergleich unterschiedlicher Cytosolherstellungsverfahren ________ 45
5.4 Optimierung der Probenvorbehandlung ________________________ 46
5.5 Trennbedingungen der SEC-Trennung _________________________ 515.5.1 Einfluss der Temperatur auf die Stabilität des Cytosols während der
SEC-Trennung ________________________________________________ 51
5.5.2 Einfluss eines Antioxidants auf die SEC-Trennung ____________________ 52
5.5.3 Zusammenfassung der Trennbedingungen der SEC / ICP-MS Kopplung ___ 53
6 Nickelspeziesanalyse im Cytosol kolorektaler Human-
gewebeproben_____________________________________________ 54
6.1 Bestimmung der Gesamtnickelgehalte im Cytosol mithilfe der TXRF 546.1.1 Diskussion der Ergebnisse _______________________________________ 56
6.2 Gesamtproteinbestimmung der Cytosole der Proben 1-5 __________ 576.2.1 Diskussion der Ergebnisse _______________________________________ 57
6.3 Nickelspeziesanalyse mithilfe der CE / ICP-MS Kopplung__________ 586.3.1 Auswertung und Aussage der Speziesanalyse________________________ 60
6.4 Nickelspeziesanalyse mithilfe der SEC / ICP-MS Kopplung ________ 676.4.1 SEC mit UV-Detektion __________________________________________ 67
6.4.1.1 Ergebnis und Diskussion ________________________________________ 68
6.4.2 Nickelspeziesanalyse mithilfe der SEC / ICP-MS Kopplung________________
6.4.2.1 Auswertungen und Aussagen der Nickelspeziesanalyse ________________ 69
6.4.2.2 Fraktionierung mittels SEC und anschließende Speziesanalyse mithilfe der
CE / ICP-MS Kopplung__________________________________________ 75
6.4.2.3 Ergebnisse und Diskussion der Fraktionierung _______________________ 76
6.4.2.4 Semiquantitative Nickelspeziesanalyse _____________________________ 76
6.5 Verdrängungsreaktionen der Ni2+- durch Zn2+-Ionen ______________ 79
6.5.1 Durchführung der Verdrängungsreaktion __________________________79
6.5.2 Ergebnisse und Diskussion ______________________________________ 80
7 Zusammenfassung _________________________________________ 82
8 Ausblick__________________________________________________ 86
9 Literaturverzeichnis ________________________________________ 88
10 Anhang__________________________________________________ 101
10.1 Geräteliste________________________________________________ 101

Abbildungsverzeichnis
10.2 Verwendete Chemikalien____________________________________ 102
10.3 Verwendete Abkürzungen___________________________________ 103
10.4 Experimetelles ____________________________________________ 105
10.5 Optimierung ______________________________________________ 10610.5.1 Cytosolherstellung mit dem Potter Eveljhem Homogenisator____________ 106
10.5.2 Cytosolherstellung mit dem Ultraschalldesintegrator __________________ 106
10.6 Optimierung der Cytosolherstellung __________________________ 108
10.7 TXRF Messungen__________________________________________ 109
10.8 Gesamtproteinbestimmung _________________________________ 111
10.9 Berechnung statistischer Größen für die CE / ICP-MS Kopplung___ 112
10.10 Nickelspeziesanalyse mithilfe der CE / ICP-MS (Probe 1-2 und 10-15)11310.10.1 Nickel/Kupfer Korrelation bei der CE / ICP-MS_______________________ 120
10.11 Berechnung statistischer Größen für die SEC / ICP-MS Kopplung _ 128
10.12 Nickelspeziesanalyse mit Hilfe der SEC / ICP-MS Kopplung_______ 12910.12.1 Nickel/Kupfer Korrelation bei der SEC / ICP-MS _____________________ 131
AbbildungsverzeichnisAbbildung 1-1: Dosis-Wirkungs-Prinzip _________________________________________ 1
Abbildung 4.1-1: Entnahmestellen der Gewebeproben ______________________________ 22
Abbildung 4.2-1: Potter-Eveljhem-Homogenisator (links), Ultraschalldesintegrator (rechts) __ 23
Abbildung 4.3-1: Schematischer Aufbau eines Sektorfeld-Massenspektrometers__________ 25
Abbildung 4.3-2: Shield Torch System___________________________________________ 25
Abbildung 4.3-3: Schema der CE / ICP-MS Kopplung_______________________________ 26
Abbildung 4.3-4: Schematischer Aufbau des Interface für die CE / ICP-MS Kopplung ______ 27
Abbildung 4.3-5: Verwendetes Interface _________________________________________ 28
Abbildung 4.3-6: Schematischer Aufbau einer SEC mit einem UV-Detektor ______________ 29
Abbildung 4.3-7: Schematischer Aufbau einer SEC / ICP-MS Kopplung_________________ 30
Abbildung 4.3-8: Kopplung SEC mit ICP-MS (A), Sprühkammer (B) ____________________ 30
Abbildung 5.1-1: Dampfreinigungsapparatur zur Reinigung der PFA-Gefäße _____________ 33
Abbildung 5.1-2: Aufbau des Conensystems eines ICP-MS __________________________ 35
Abbildung 5.1-3: Gegenüberstellung von zwei Elektropherogrammen eines humanen Gewebe-
cytosols unter Einsatz verschiedener Conen; Detektion 60Ni ____________ 36
Abbildung 5.2-1: Vergleich eines Elektropherogramms eines Cd-beladenen Metallothionein-
gemisches mit einem Elektropherogramm eines Cytosols von einer malignen
Kolongewebeprobe unter optimierten Bedingungen __________________ 37

Abbildungsverzeichnis
Abbildung 5.2-2: Verlauf des Temperaturgradienenten innerhalb einer Kapillare __________ 38
Abbildung 5.2-3: Elektropherogramm des MT-Gemisches bei einer Pufferkonzentration
von 20 mmol/L _______________________________________________ 41
Abbildung 5.2-4: Elektropherogramm des MT-MIX Standards mit einer Pufferkonzentration
von 50 mmol/L _______________________________________________ 42
Abbildung 5.2-5: Einfluss des pH-Wertes des Puffers auf die elektrophoretische Trennung eines
Cadmium beladenen MT-MIX Standards __________________________ 43
Abbildung 5.3-1: Elektropherogramme von Nickelspezies in Abhängigkeit von der
Cytosolgewinnung ____________________________________________ 45
Abbildung 5.3-2: Vergleich der Gesamtnickelkonzentration im Cytosol in Abhängigkeit von der
verwendeten Aufschlussmethode (mechanisch und mittels Ultraschall) ___ 46
Abbildung 5.4-1: Elektropherogramme von zwei Cytosolen (NG und TG) vor der
Acetonitrilfällung______________________________________________ 47
Abbildung 5.4-2: Elektropherogramme von zwei Cytosolen (NG und TG) nach der
Acetonitrilfällung______________________________________________ 47
Abbildung 5.4-3: SEC / ICP-MS Chromatogramme eines Cytosols vor und nach
der Acetonirilfällung ___________________________________________ 49
Abbildung 5.4-4: Probenvorbehandlung zur Nickelspeziesanalyse mittels CE / ICP-MS und
SEC / ICP-MS Kopplung _______________________________________ 50
Abbildung 5.5-1: SE-Chromatogramme eines Cytosols in Abhängigkeit von der
Säulentemperatur_____________________________________________ 51
Abbildung 5.5-2: SE-Chromatogramme eines Cytosols in Abhängigkeit von der Zugabe eines
Antioxidants _________________________________________________ 52
Abbildung 6.1-1: Ergebnisse der TXRF-Messung für die Cytosole vor der Acetonitrilfällung _ 55
Abbildung 6.1-2: Ergebnisse der TXRF-Messung für die Cytosole nach der Acetonitrilfällung 55
Abbildung 6.2-1: Gesamtproteingehalte der Cytosole (Probe 1-5) _____________________ 57
Abbildung 6.3-1: Elektropherogramme der Cytosole (NG/TG) der Probe 3_______________ 59
Abbildung 6.3-2: Elektropherogramme der Cytosole (NG/TG) der Probe 5_______________ 59
Abbildung 6.3-3: Elektropherogramme der Cytosole (NG/entz.G) der Probe 4 ____________ 59
Abbildung 6.3-4: Elektropherogramme einer Ni-Standardlösung (20 µg/L) und eines Cytosols
der Probe NG 3 ______________________________________________ 60
Abbildung 6.3-5: Übereinstimmung der Migrationszeit der Nickelspezies I und der Kupferspezies
im Cytosol von NG 3 __________________________________________ 61
Abbildung 6.3-6: Elektropherogramme zur Identifizierung einer weiteren Nickelspezies_____ 64
Abbildung 6.3-7: Verhalten der Nickelspezies im Cytosol gegenüber niedrig konzentrierten
EDTA-Lösungen______________________________________________ 65
Abbildung 6.3-8: Verhalten der Nickelspezies im Cytosol gegenüber hoch konzentrierten EDTA-
Lösungen ___________________________________________________ 65
Abbildung 6.4-1: Massenkalibriergerade für die SEC-Säule __________________________ 67
Abbildung 6.4-2: SE-Chromatogramme der Cytosole von Normalgewebe und Tumorgewebe
der Probe 1(UV-Detektion bei 254 nm) ____________________________ 68

Abbildungsverzeichnis
Abbildung 6.4-3: Vergleich Chromatogramm eines Cytosol (TG) mit UV und
mit ICP-MS Detektion _______________________________________ 69
Abbildung 6.4-4: SE-Chromatogramme der Cytosole (NG/TG) der Probe 3 ___________ 70
Abbildung 6.4-5: SE-Chromatogramme der Cytosole (NG/TG) der Probe 5 ___________ 70
Abbildung 6.4-6: SE-Chromatogramme der Cytosole (NG/entz.G) der Probe 4 ________ 71
Abbildung 6.4-7: Zuordnung der Nickelspezies über eine Kupferspezies (Molmasse 6 kDa)
der Probe NG 1____________________________________________ 73
Abbildung 6.4-8: Zuordnung der Nickelspezies über eine Kupferspezies (Molmasse 6 kDa)
der Probe TG 1 ____________________________________________ 74
Abbildung 6.4-9: Fraktionierung des Cytosols der Probe TG 1 _____________________ 75
Abbildung 6.4-10: Elektropherogramme der SEC-Fraktionen (4 kDa und 9 kDa) ________ 76
Abbildung 6.5-1: SE-Chromatogramme nach Zusatz von Zn2+- und Ni2+-Ionen zum Cytosol
der Probe NG 10___________________________________________ 80
Abbildung 6.5-2: SE-Chromatogramme nach Zusatz von Zn2+- und Ni2+-Ionen zum Cytosol
der Probe TG 10 ___________________________________________ 80
Abbildung 10.6-1: Elektropherogramme von Kupferspezies in Abhängigkeit von der Cytosol-
herstellung_______________________________________________ 108
Abbildung 10.6-2: Elektropherogramme von Zinkspezies in Abhängigkeit von der Cytosol-
herstellung_______________________________________________ 108
Abbildung 10.6-3: Elektropherogramme von Bleispezies in Abhängigkeit von der Cytosol-
herstellung_______________________________________________ 108
Abbildung 10.8-1: Kalibriergerade zur Bestimmung des Gesamtproteingehaltes der
Cytosolproben____________________________________________ 111
Abbildung 10.10-1: Elektropherogramme der Cytosole (NG/TG) der Probe 1 ___________ 113
Abbildung 10.10-2: Elektropherogramme der Cytosole (NG/TG) der Probe 2 ___________ 114
Abbildung 10.10-3: Elektropherogramme der Cytosole (NG/TG) der Probe 10 __________ 114
Abbildung 10.10-4: Elektropherogramme der Cytosole (NG/TG) der Probe 11 __________ 114
Abbildung 10.10-5: Elektropherogramme der Cytosole (NG/TG) der Probe 12 __________ 115
Abbildung 10.10-6: Elektropherogramme der Cytosole (NG/TG) der Probe 13 __________ 115
Abbildung 10.10-7: Elektropherogramme der Cytosole (NG/TG) der Probe 14 __________ 115
Abbildung 10.10-8: Elektropherogramme der Cytosole (NG/TG) der Probe 15 __________ 116
Abbildung 10.10-9: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 1 ___________________________ 120
Abbildung 10.10-10: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 1 ___________________________ 121
Abbildung 10.10-11: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG2 ___________________________ 121
Abbildung 10.10-12: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG2____________________________ 121
Abbildung 10.10-13: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG3____________________________ 122

Abbildungsverzeichnis
Abbildung 10.10-14: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG4 ___________________________ 122
Abbildung 10.10-15: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von entz.G4 _________________________ 122
Abbildung 10.10-16: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 5 ___________________________ 123
Abbildung 10.10-17: Übereinstimmung der Migrationszeiten der Nickelspezies I und der __ 123
Kupferspezies im Cytosol von TG5____________________________ 123
Abbildung 10.10-18: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 10 __________________________ 123
Abbildung 10.10-19: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 10 __________________________ 124
Abbildung 10.10-20: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 11 __________________________ 124
Abbildung 10.10-21: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG11___________________________ 124
Abbildung 10.10-22: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 12 __________________________ 125
Abbildung 10.10-23: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 12 __________________________ 125
Abbildung 10.10-24: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 13 __________________________ 125
Abbildung 10.10-25: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 13 __________________________ 126
Abbildung 10.10-26: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 14 __________________________ 126
Abbildung 10.10-27: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 14 __________________________ 126
Abbildung 10.10-28: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 15 __________________________ 127
Abbildung 10.10-29: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 15 __________________________ 127
Abbildung 10.12-1: SE-Chromatogramme der Cytosole (NG/TG) der Probe 1 __________ 129
Abbildung 10.12-2: SE-Chromatogramme der Cytosole (NG/TG) der Probe 10 _________ 129
Abbildung 10.12-3: SE-Chromatogramme der Cytosole (NG/TG) der Probe 11 _________ 129
Abbildung 10.12-4: SE-Chromatogramme der Cytosole(NG/TG) der Probe 13__________ 130
Abbildung 10.12-5: SE-Chromatogramme der Cytosole (NG/TG) der Probe 14 _________ 130
Abbildung 10.12-6: SE-Chromatogramme der Cytosole (NG/TG) der Probe 15 _________ 130
Abbildung 10.12-7: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 3___________________________________________ 131

Tabellenverzeichnis
Abbildung 10.12-8: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 3 ___________________________________________ 131
Abbildung 10.12-9: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 4___________________________________________ 131
Abbildung 10.12-10: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe entzündetes G 4 __________________________________ 132
Abbildung 10.12-11: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 5___________________________________________ 132
Abbildung 10.12-12: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 5 ___________________________________________ 132
Abbildung 10.12-13: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 10__________________________________________ 133
Abbildung 10.12-14: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 10 __________________________________________ 133
Abbildung 10.12-15: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 11__________________________________________ 133
Abbildung 10.12-16: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 11 __________________________________________ 134
Abbildung 10.12-17: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 13__________________________________________ 134
Abbildung 10.12-18: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 13 __________________________________________ 134
Abbildung 10.12-19: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 14__________________________________________ 135
Abbildung 10.12-20: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 14 __________________________________________ 135
Abbildung 10.12-21: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe NG 15__________________________________________ 135
Abbildung 10.12-22: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa)
der Probe TG 15 __________________________________________ 136
TabellenverzeichnisTabelle 2.1-1: Molare Massen der Nickelspezies im Cytosol von Rattennieren_____________ 4
Tabelle 2.1-2: Ergebnisse der Nickelspeziesanalyse im Cytosol von unterschiedlichen
Gewebeproben der Ratte bzw. des Frosches ___________________________ 6
Tabelle 2.2-1: Zusammenfassung der CE / ICP-MS Literatur _________________________ 17
Tabelle 4.1-1: Kenndaten der analysierten Gewebeproben___________________________ 22
Tabelle 5.1-1: Konditionierungsschritte des Chelex 100 für die anschließende Reinigung des
Tris-HNO3-Puffers _______________________________________________ 34
Tabelle 5.2-1: Gewählte Messparameter für die Optimierung der Kapillarentemperatur _____ 39

Tabellenverzeichnis
Tabelle 5.2-2: Auflistung der optimierten Parameter der CE / ICP-MS Kopplung________ 44
Tabelle 5.5-1: Optimierte Messparameter für die SEC / ICP-MS Kopplung ____________ 53
Tabelle 6.3-1: Peakflächenverhältnisse der Nickelspezies I zu Spezies II _____________ 62
Tabelle 6.4-1: Proteinstandards für die Massenkalibrierung der SEC-Säule. ___________ 67
Tabelle 6.4-2: Molmassen der Nickelspeziesfraktionen ___________________________ 71
Tabelle 6.4-3: Peakflächenverhältnisse der Nickelspezies I (6 kDa) zu Nickelspezies II
(9 kDa); SEC / ICP-MS Messungen_______________________________ 77
Tabelle 10.7-1: Ergebisse der TXRF-Messung für die Cytosolproben
vor der Acetonitrilfällung_______________________________________ 109
Tabelle 10.7-2: Ergebisse der TXRF-Messung für die Cytosolproben
nach der Acetonitrilfällung _____________________________________ 110
Tabelle 10.8-1: Ergebnisse der Kalibrierung für die Gesamtproteinbestimmung (n=3) ___ 111
Tabelle 10.8-2: Ermittelte Extinktionen und die berechneten Gesamtproteingehalte (n=3) 112
Tabelle 10.9-1: Wiederholpräzision eines Cytosols für die CE / ICP-MS Kopplung3 _____ 112
Tabelle 10.9-2: Abschätzung der Nachweisgrenze und Bestimmungsgrenze der CE / ICP-MS
Kopplung __________________________________________________ 113
Tabelle 10.10-1: berechnete Flächen der Nickelspezies I und II für die CE / ICP-MS und
Flächenverhältnisse für die Proben 1-5 und 10-15 __________________ 116
Tabelle 10.10-2: Wertetabelle zur Durchführung des t-Test (auf der Ebene von 0,01) für die
Peakflächenverhältnisse (CE / ICP-MS) __________________________ 117
Tabelle 10.10-3: Ergebnisse des t-Test der Peakflächen ___________________________ 118
Tabelle 10.11-1: Wiederholpräzision (Rententionszeit) eines Cytosols für die SEC / ICP-MS
Kopplung (n=5)______________________________________________ 128
Tabelle 10.11-2: Wiederholpräzision (Peakflächen) eines Cytosols für die SEC / ICP-MS
Kopplung (n=5)______________________________________________ 128
Tabelle 10.12-1: berechnete Flächen der Nickelspezies I und II für die SEC / ICP-MS und
Flächenverhältnisse für die Proben 1-5 und 10-15 __________________ 136
Tabelle 10.12-2: Wertetabelle zur Durchführung des t-Test (Ebene 0,01) für die
Peakflächenverhältnisse (SEC / ICP-MS) _________________________ 137
Tabelle 10.12-3: Ergebnisse des t-Test der Peakflächen ___________________________ 137

Einleitung 1
1 EinleitungEvolutionsbedingt sind eine Vielzahl von Elementen des Periodensystems am humanen
Stoffwechsel beteiligt. Dabei wird zwischen Mineral- und Spurenelementen unterschie-
den. Zu den Mineralelementen zählen Kalium, Magnesium, Calcium und Natrium. Spu-
renelemente sind chemische Elemente, die der menschliche, tierische oder pflanzliche
Organismus nur in Spuren enthält und die oftmals lebenswichtige Aufgaben erfüllen.
Spurenelemente kommen im Bereich von Gramm (Zn: 2,3g) bis Milligramm (Mo: 5
mg) im humanen Organismus vor. Die Aufnahme geschieht über die Nahrung, wobei
als limitierende Faktoren sowohl der Gehalt in den Nahrungsmitteln als die
Bioverfügbarkeit des entsprechenden Spurenelementes berücksichtigt werden muss.
Sowohl der Magen als auch der Darm gelten als Resorptionsort. Ihre Wirkung entfalten
die Spuren-elemente nur in ionisch gelöster oder komplexgebundener Form. Sie können
in drei Gruppen eingeteilt werden.
1. essentielle Spurenelemente, deren Notwendigkeit für den Menschen erwiesen ist
und deren Mangel zur Manifestation von klinischen Symptomen führt.
2. Elemente, deren Funktion für den Menschen als noch nicht genügend gesichert gilt
3. Elemente, die in größeren Mengen beim Menschen toxisch wirken, z.B. Cadmium,
Blei, Quecksilber, Arsen usw.
Abbildung 1-1: Dosis-Wirkungs-Prinzip[2]

Einleitung 2
Abbildung 1-1 zeigt die Einteilung chemischer Elemente in essentielle, nicht essentielle
und toxische Elemente nach dem Dosis-Wirkungs-Prinzip. Nicht essentielle Elemente
sind bis zu einer bestimmten Elementkonzentration im Körper noch tolerierbar. Sobald
diese Konzentration überschritten wird, wirkt das Element toxisch. Ein als Gift bezeich-
netes Spurenelement wirkt schon in den kleinsten Konzentrationen toxisch. Der Verlauf
des Dosis-Wirkungs-Prinzip für essentielle Elemente zeigt ein breites Maximum bei
einer mittleren Elementkonzentration.
Da Spurenelemente ihre Wirkung im Organismus nur in ionisch gelöster oder komplex-
gebundener Form ausüben können, sind sie häufig Bestandteile von Makromolekülen,
wie z.B. Proteine und Enzyme. Daher sind qualitative und quantitative Informationen
über die Bindungsform (= Spezies) der Spurenelemente von großer Bedeutung, da so
Aussagen z.B. über die Toxizität der Spurenelementspezies möglich sind. Hierzu müs-
sen Analysenverfahren zur Speziesanalyse entwickelt werden, die sowohl die Molekül-
als auch die Speziesinformationen erhalten. Diesen Anspruch erfüllen z.B. online-
Kopplungen (Hyphenated Systems) von chromatographischen Trenntechniken mit
elementspezifischen Detektoren. Diese Kopplungen bilden heute die Grundlage der
Metallspeziesanalyse. Die Anforderung an ein solches analytisches Verfahren sind recht
hoch, so darf die Speziesinformation während der Probennahme, Lagerung und Analyse
nachweislich nicht verändert werden bzw. die Veränderungen müssen reproduzierbar
und bekannt sein [5]. Das eingesetzte Trennverfahren muss einerseits ein hohes
Trennvermögen aufweisen und das Nachweisverfahren andererseits eine sehr gute
Empfindlichkeit besitzen.
Diese Anforderungen gilt es durch die richtige Wahl des Trennverfahrens bzw. des
Nachweisverfahrens für eine Nickelspeziesanalyse zu erfüllen. Nickel zählt neben Zinn,
Vanadium und Mangan zu der 2. Kategorie der Spurenelemente, deren Funktion für den
Menschen noch nicht vollständig geklärt ist. Da Nickel in geringen Konzentrationen im
menschlichen Körper vorliegt, wurden bisher nur die Gesamtnickelkonzentration in
humanen Gewebeproben bestimmt, nicht aber der Gehalt einzelner Nickelspezies, da
die Konzentration für eine Nickelspeziesanalyse zu gering eingestuft wurde. Es konnten
also keine Aussagen über die Bindungsform des Nickels im humanen Organismus
gemacht werden. Mithilfe von tierexperimentellen Versuchen, in denen Versuchstieren
eine sehr hohe Dosis an 63NiCl2 oral verabreicht oder injiziert wurde, konnten erste Ein-
flüsse einer definierten Nickelspezies auf den Metabolismus des Tieres untersucht wer-
den. Dies widerspricht streng genommen einer Speziesanalyse, da es nicht nachweislich

Einleitung 3
gesichert ist, dass die Speziesinformationen durch das übersteigerte Angebot an 63NiCl2
unverändert bleiben. Die Nickelspeziesanalyse ist unter Erhalt des natürlichen
Verteilungsmuster der Nickelspezies in biologischen Realproben von besonderem
Interesse, um Informationen über die Rolle von Nickel im Körper bzw. im Metabolis-
mus zu erhalten.

Stand der Forschung 4
2 Stand der Forschung
Die folgenden beiden Unterkapitel beinhalten zwei Themenschwerpunkte. Zum einen
werden die bisher identifizierten Nickelspezies in biologischen Proben vorgestellt und
zum anderen die neusten Entwicklungen der Kopplung verschiedener Trennverfahren
(chromatographisch, elektrophoretisch) mit der induktiv gekoppelten Plasma Massen-
spektrometerie (ICP-MS) erläutert.
2.1 Nickelspezies in biologischen Proben
Schon zu Beginn der achtziger Jahre wurden die ersten Arbeiten mit dem Ziel einer
Nickelspeziesanalyse im Cytosol von Nagetieren nach Injektion von 63Nickelchlorid
(63Ni2+ als radioaktives Ion) durchgeführt [7-14/ 19]. Bis zu diesem Zeitpunkt waren nur
α2-Macroglobulin und Albumin als nickelbindende Serumproteine bekannt.
Sunderman et al. waren die Vorreiter auf dem Gebiet der Nickelspeziesanalyse im Cyto-
sol von Ratten nach Inkubation mit 63Nickelchlorid [8]. Sie fanden mithilfe chromato-
graphischer Methoden heraus, dass 68 Prozent von 63Nickel an Substanzen, deren Mol-
masse kleiner als 2 kDa war, gebunden wurde. Das restliche Nickel verteilte sich auf
fünf makromolekulare Verbindungen mit Molmassen von 130 kDa, 70 kDa, 55 kDa, 30
kDa und 10 kDa (Zuordnung der Fraktion zu möglichen Substanzen bzw. Substanz-
klassen siehe Tabelle 2.1-1). In der folgenden Tabelle 2.1-1 sind die Ergebnisse (molare
Masse der Nickelspezies) der ersten Arbeiten auf dem Gebiet der Nickelspeziesanalyse
im Cytosol von Rattennieren aufgelistet.
Tabelle 2.1-1: Molare Massen der Nickelspezies im Cytosol von Rattennieren [8]
molare Masse / kDa Substanz / Substanzklasse
130 Nickeloplasmin
70 Albumin
55 63Ni-Metalloprotein [16]
30 -----
10
Metallothionein ähnliches
Protein (MLP) [18]
Die elektrophoretische Mobilität des Proteins mit einer Molmasse von 130 kDa besaß
sehr große Ähnlichkeit mit der des Nickeloplasmins, sodass man davon ausging, dass es

Stand der Forschung 5
sich um dieses Protein handeln könnte. Aufgrund der Daten, die aus der Anwendung
verschiedener Trennmethoden gewonnen wurde, könnte es sich bei der 70 kDa Spezies
um Albumin handeln. Hinter der 55 kDa Spezies könnte sich das von Jacobsen be-
schriebene 63Ni-Metalloprotein verbergen, das er im Cytosol von humanen Drüsen
gefunden hatte [16]. Zu diesem Zeitpunkt konnte der Nickelspezies mit einer molaren
Masse von 30 kDa noch keine Substanz oder Substanzklasse zugeordnet werden (ein
Verdacht deutete auf eine nicht näher spezifizierte Nickelspezies hin, die Oskarsson [17]
im Cytosol der Lunge bzw. Niere von Mäusen gefunden hatte).
Diese Ergebnisse wurden von Herlant-Peers et al. [19] bestätigt, die im Cytosol von
Mäusen nach Inkubation mit 63Nickelchlorid eine Nickelspeziesanalyse mithilfe der
Gelelektrophorese (Gradienten SDS1 Page) durchgeführt hatten. Sie untersuchten so-
wohl die in vitro als auch die in vivo Einlagerung von 63Ni2+ in subzellare Lungen- und
Leberfraktionen von Mäusen und detektierten nickelbindende Proteine mit ähnlichen
molaren Massen wie die Gruppe um Sunderman [19].
Bis Ende der achtziger Jahre flachte das Interesse an der Nickelspeziesanalyse in biolo-
gischen Matrizes deutlich ab. Erst wiederum durch Arbeiten von Sunderman, die auf
seine bisher gewonnenen Erkenntnisse (siehe Tabelle 2.1-1) aufbauten, stieg das Inter-
esse an der Nickelspeziesanalyse wieder. Aufgrund der Weiterentwicklung der Trenn-
techniken war man nun in der Lage, die Spezies besser separieren zu können. Es
wurden im Vergleich zu den Anfängen eine deutlich höhere Anzahl an Nickelspezies im
Cytosol verschiedener Rattenorgane nachgewiesen. Diese Untersuchung beschränkte
sich aber nicht mehr auf das Cytosol von Ratten, sondern wurde auf Cytosole von
Froschorganen ausgeweitet [7]. Es wurden weitere Versuche unternommen, die
gefundenen Nickelspezies zu identifizieren. Die Tabelle 2.1-2 zeigt eine
Zusammenfassung der Nickelspezies-analyse in Cytosolen von verschiedenen Ratten-
und Froschorganen.
1 SDS = Natriumdodecylsulfat
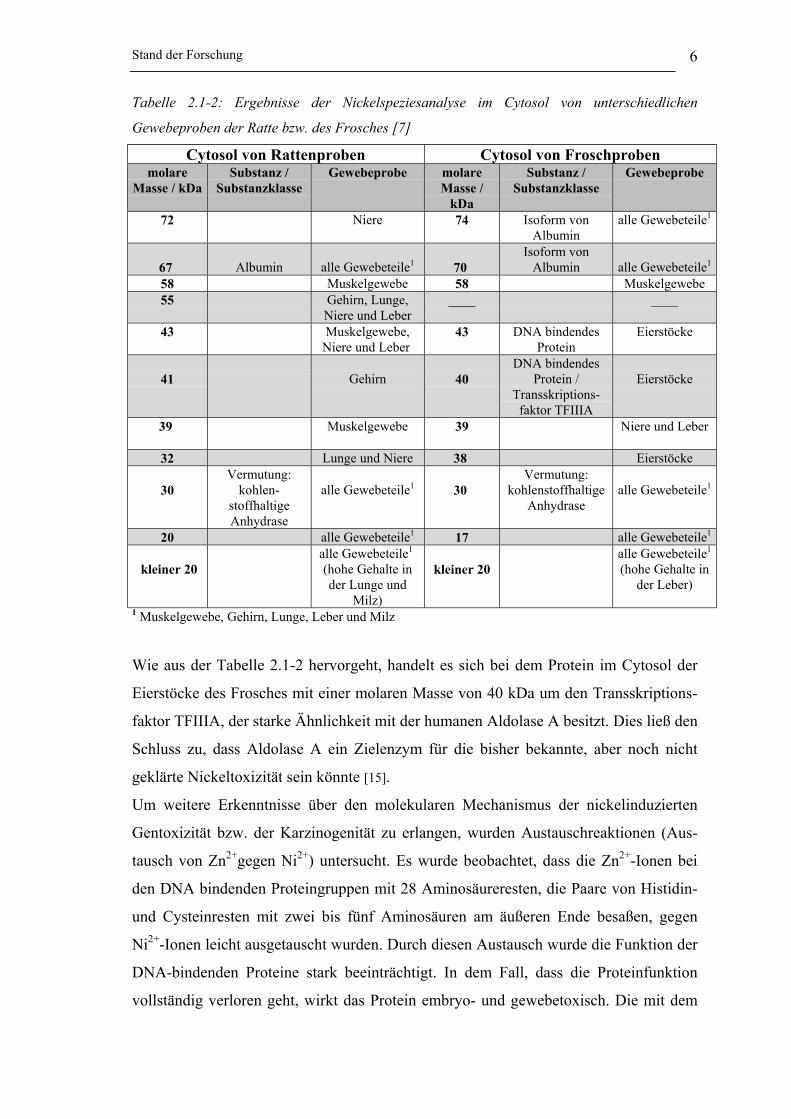
Stand der Forschung 6
Tabelle 2.1-2: Ergebnisse der Nickelspeziesanalyse im Cytosol von unterschiedlichen
Gewebeproben der Ratte bzw. des Frosches [7]
Cytosol von Rattenproben Cytosol von Froschprobenmolare
Masse / kDaSubstanz /
SubstanzklasseGewebeprobe molare
Masse /kDa
Substanz /Substanzklasse
Gewebeprobe
72 Niere 74 Isoform vonAlbumin
alle Gewebeteile1
67 Albumin
alle Gewebeteile1 70 Isoform von
Albumin alle Gewebeteile1
58 Muskelgewebe 58 Muskelgewebe55 Gehirn, Lunge,
Niere und Leber____ ____
43 Muskelgewebe,Niere und Leber
43 DNA bindendesProtein
Eierstöcke
41 Gehirn 40DNA bindendes
Protein /Transskriptions-
faktor TFIIIA
Eierstöcke
39 Muskelgewebe 39 Niere und Leber
32 Lunge und Niere 38 Eierstöcke
30Vermutung:
kohlen-stoffhaltigeAnhydrase
alle Gewebeteile1 30
Vermutung:kohlenstoffhaltige
Anhydrasealle Gewebeteile1
20 alle Gewebeteile1 17 alle Gewebeteile1
kleiner 20alle Gewebeteile1 (hohe Gehalte inder Lunge und
Milz)
kleiner 20
alle Gewebeteile1
(hohe Gehalte inder Leber)
1 Muskelgewebe, Gehirn, Lunge, Leber und Milz
Wie aus der Tabelle 2.1-2 hervorgeht, handelt es sich bei dem Protein im Cytosol der
Eierstöcke des Frosches mit einer molaren Masse von 40 kDa um den Transskriptions-
faktor TFIIIA, der starke Ähnlichkeit mit der humanen Aldolase A besitzt. Dies ließ den
Schluss zu, dass Aldolase A ein Zielenzym für die bisher bekannte, aber noch nicht
geklärte Nickeltoxizität sein könnte [15].
Um weitere Erkenntnisse über den molekularen Mechanismus der nickelinduzierten
Gentoxizität bzw. der Karzinogenität zu erlangen, wurden Austauschreaktionen (Aus-
tausch von Zn2+gegen Ni2+) untersucht. Es wurde beobachtet, dass die Zn2+-Ionen bei
den DNA bindenden Proteingruppen mit 28 Aminosäureresten, die Paare von Histidin-
und Cysteinresten mit zwei bis fünf Aminosäuren am äußeren Ende besaßen, gegen
Ni2+-Ionen leicht ausgetauscht wurden. Durch diesen Austausch wurde die Funktion der
DNA-bindenden Proteine stark beeinträchtigt. In dem Fall, dass die Proteinfunktion
vollständig verloren geht, wirkt das Protein embryo- und gewebetoxisch. Die mit dem

Stand der Forschung 7
Austausch verbundene Induzierung von Reaktionen freier Radikale wirkt sich
wiederum negativ auf die Onkogenese aus [7/9/12].
Bei der Nickelspeziesanalyse von humanen Cytosolproben der Niere konnten sowohl
peptid- als auch kohlenhydrathaltige Nickelkomplexe gefunden werden [20]. Die nickel-
bindenden Peptide wiesen einen hohen Gehalt an Glutamin- und Aspartamsäure auf,
aromatische und schwefelhaltige Aminosäuren konnten nicht nachgewiesen werden.
Ferner konnte Ni2+ unter physiologischen Bedingungen auch bei einem Überschuss an
Cu2+ nicht verdrängt werden, was auf eine starke Bindung von Nickel an die Peptide
schließen ließ [20].
Mit der Erkenntnis, dass Nickel bzw. Ni2+-Ionen bevorzugt an histidinreiche Proteine
binden [23/186], konnte Sunderman ein weiteres nickelbindendes Protein mit histidinrei-
chen Nickel(II)-Bindungsdomänen isolieren und identifizieren. Dabei handelt es sich
um ein Serin Proteinase Inhibitor, das er mit „pNiXa“ (43-45 kDa) bezeichnet [9-10/13].
Die Koordinierung des Nickel(II)-Ions im pNiXa kann auf drei verschiedene Weisen
erfolgen. Zum einen kann das Octadecapeptid mit seinen histidinreichen Domänen für
eine sechsfache Koordination des Nickel(II)-Ions sorgen, sodass ein oktaedrischer
Nickel(II)-Komplex resultiert, wenn der N-Term von pNiXa acetyliert ist. Fehlt diese
funktionelle Gruppe liegt ein quadratisch planer Nickel(II)-Komplex vor [9]. Er unter-
suchte zusätzlich die Interaktionen von pNiXa mit Chymotrypsin, wobei sich heraus-
stellte, dass pNiXa ein aktiver Chymotrypsin Inhibitor ist [10].
Neben pNiXa wurde auch das Protein Hpn entdeckt und charakterisiert [11]. Bei Hpn
handelt es sich wiederum um ein histidinreiches, metallbindendes (nickelbindendes)
Polypetid des Helicobacter pylori bzw. des Helicobacter mustulae. Helicobacter pylori
ist ein humanes gastrointestinales Pathogen, das mit Gastritis und anderen Darmerkran-
kungen in Verbindung gebracht wird. Diese Mikroorganismen produzieren eine hohe
Anzahl von Ureasen, die wie alle bekannten Ureasen, Ni2+ und Zn2+ binden. Das Protein
des Helicobacters pylori wurde nach klinischer Isolierung identifiziert und das identi-
sche Protein im Helicobacter mustulae wiedergefunden. Auch in diesem Fall handelte
es sich sowohl um ein Ni2+-bindendes als auch um Zn2+-bindendes Protein [11].

Stand der Forschung 8
Zum selben Ergebnis kamen auch Fu et al. in ihren Arbeiten [24]. Sie untersuchten das
metallbindende Protein HypB des Bradyrhizobium japonicum und konnten zeigen, dass
dieses Protein aufgrund des hohen Histidingehaltes 18 divalente Nickel-Ionen pro Di-
mer binden kann. Diese Eigenschaft nutzten sie zur Reinigung bzw. Isolierung des Pro-
teins aus, indem sie es mithilfe einer Ni2+-beladenen Metallchelat-Affinitätschromato-
graphie von der Matrix trennten. Neben Ni2+-Ionen bindet HypB noch Zn2+, Cu2+, Co2+,
Cd2+ und Mn2+.
2002 wurde ein Review von Denkhaus und Salnikow veröffentlicht [26], in dem auf die
Essentialität, Toxizität und Karzinogenität von Nickel eingegangen wurde; zusätzlich
wurden auch nickelbindende Proteine beschrieben. Nach Ihren Ausführungen geht
Nickel, obwohl es nicht signifikant mit der DNA reagiert, starke Interaktionen mit Pro-
teinen ein [26/27]. Neben den erwähnten nickelbindenden Proteinen führten sie noch die
Gruppe der DAN-Gene und Genprodukte auf. Diese können ebenfalls Nickel binden, da
sie (HX)n2
-Ketten aufweisen, die essentiell für eine Ni2+-Koordination sind. DAN-Gene
besitzen Tumorsuppressor Aktivitäten, was in vitro nachgewiesen wurde [28-29] und für
die vorliegende Arbeit von besonderem Interesse sein könnte.
2 (HX)n = Histidinhaltige Sequenz

Stand der Forschung 9
2.2 Induktiv-gekoppltes-Plasma / Massenspektrometrie (ICP-MS)
In den letzten Jahren haben gekoppelte Systeme (Hyphenated Systems) zur Element-
speziesanalyse in den Bereichen der Umweltforschung, der Lebensmitteluntersuchung
und der bio-medizinschen Forschung zunehmend an Bedeutung gewonnen, da Fragen
nach Metallspezies und deren Konzentration in den verschiedenen Probenmaterialien
immer mehr in den Vordergrund gerückt sind. Die Fragen der Beurteilung von Gefah-
renpotenzialen, die von verschiedenen Metallspezies ausgehen können, mussten und
müssen weiterhin geklärt werden. Dazu sollte eine Analysenmethode, die eine
Erhaltung der Speziesinformation garantiert, entwickelt werden. In den meisten Fällen
wurden dazu chromatographische (Gaschromatographie (GC), Flüssigchromatographie
(LC), Ionenchromatographie (IC)) oder elektrophoretische Trenneinheiten mit dem
ICP-MS gekoppelt. Der Gebrauch eines ICP-MS als chromatographischer Detektor
wurde erstmals in den späten achtziger Jahren beschrieben.
Der Vorteil der Kopplung eines Trennsystems mit einem ICP-MS liegt auf der Hand.
Das ICP ist eine energiereiche Ionenquelle, die einen hohen Grad an Ionisation für die
Elemente bei guter Empfindlichkeit bietet [30/157]. Es ist in der Lage die eluierten Spe-
zies bei gleichzeitigem Abbau von Matrix zu atomisieren. Das Massenspektrometer
zählt zu den atomspektroskopischen Detektoren, die in der Lage sind, mehrere Isotope
eines Analyten während eines chromatographischen oder elektrophoretischen Laufes
simultan zu bestimmen, da es zeitaufgelöst Signale liefern kann [162].
Im weiteren Verlauf der Arbeit werden die gängigen chromatographischen und elektro-
phoretischen online Kopplungen mit der ICP-MS beschrieben.
Die GC / ICP-MS Kopplung ist zwar eine äußerst leistungsfähige Methode zur
Elementspeziesanalyse, besitzt aber ein eingeschränktes Einsatzgebiet. Sie wird
ausschließlich im Umweltbereich und dort speziell in der Analyse von
metallorganischen Substanzen (z.B. Organo-Zinn-Verbindungen) eingesetzt, da sowohl
das Medium des Analyten wie auch der Analyt selbst gut verdampfbar sein muss. Da
diese Methode bei bio-medizinischen Untersuchungen keine Anwendung findet, wird
sie in den weiteren Ausführungen nicht weiter behandelt.
Dagegen haben sowohl die LC als auch die CE / ICP-MS Kopplung ein breites Anwen-
dungsspektrum, wobei der Fokus zunehmend auf Analysen metallhaltiger Biomoleküle
liegt. Aufgrund der kleinen Probenmengen wird der Einsatz der CE / ICP-MS Kopplung
in der bio-medizinischen Analytik bevorzugt (mikroanalytischer Ansatz).

Stand der Forschung 10
2.2.1 Zerstäuber für chromatographische Trennverfahren
Die Flüssigkeitschromatographie mit ihren verschiedenen Trennmodi wie die Reversed-
phase-Chromatographie (RPLC), die Ionenpaar-Chromatographie (IPC), die Ionenaus-
tausch-Chromatographie (IEC) und die Größenausschluss-Chromatographie (SEC) hat
einen bedeutenden Vorteil gegenüber der Kapillarelektrophorese (CE) und der
Gaschromatographie (GC). Da die Flussraten der genannten Trennmethoden im unteren
mL Bereich liegen, also keine Optimierung der Flussraten an den Zerstäuber erforder-
lich ist, ist kein spezielles Interface wie bei der CE und GC nötig, sondern das Elutat
kann sofort über einen Zerstäuber ins Plasma überführt werden. Es werden meist
kommerziell erhältliche Zerstäuber wie Cross-Flow oder konzentrische Zerstäuber mit
gekühlter Sprühkammer eingesetzt, sodass nur 1-3 % der Probe ins Plasma gelangt. Die
Transferleitung besteht aus einer inerten PEEK3 oder PTFE4 Kapillare [34]. Der Einsatz
eines hydraulischen Hochdruckzerstäubers zeigte eine Steigerung der Empfindlichkeit
für einige Elemente im Vergleich zu den herkömmlichen Zerstäubern [35]. Eine
Alternative fanden Koropchak et al. und Tomlinson et al. [36/37]. Sie setzten die
Thermospraytechnologie zur Zerstäubung ein, um mit einer Flussrate von 2 mL/min
arbeiten zu können. Ein anderer Ansatzpunkt war der Einsatz eines Ultraschall-
zerstäubers. Dieser verbesserte den Probentransport, sodass die generelle Effizienz der
Zerstäubung bei 10-30 % lag. Shum et al. entwickelten einen Direktinjektionszerstäuber
speziell für die LC / ICP-MS Kopplung. Der Zerstäuber befand sich direkt in der Fackel
des ICPs. Der theoretische Probentransport ins Plasma lag bei 100% und der Zerstäuber
arbeitete mit einer sehr geringen Flussrate von 30-120 µL/min [38-40].
Für die online-Kopplung von CE mit der ICP-MS würde sich ein Zerstäuber mit einer
sehr geringen Flußrate anbieten, da die CE mit solchen Flußraten arbeitet. Das Totvo-
lumen des eingesetzten Zerstäubers muss sehr klein sein, da sonst die scharfe Trennung
der CE durch das zu große Totvolumen innerhalb des Zerstäubers aufgehoben wird. Aus
diesen Gründen wäre ein modifizierter konzentrischer Zerstäuber für eine solche
Kopplung sehr gut geeignet.
3 Abk. für Polyetheretherketon, einen hochtemperaturbeständigen u. schlagzähen Thermoplasten4 Kurzz. (nach DIN 7728, Tl. 1, Jan. 1988) für Polytetrafluorethylene.

Stand der Forschung 11
2.2.2 HPLC / ICP-MS Kopplung
Die HPLC / ICP-MS Kopplung ist aufgrund des geringen instrumentellen Aufwandes
(kein spezielles Interface nötig) für die Speziesanalyse weit verbreitet [41-44]. In den
letzten Jahren sind viele Veröffentlichungen erschienen, die den Einsatz dieser Technik
dokumentieren. Das Einsatzgebiet liegt hauptsächlich in der Arsen- und Selenspezies-
analyse, z.B. in Pflanzenextrakten [45-52]. Gleichzeitig stieg auch das Interesse an
weiteren Metallspeziesanalysen in biologischen Proben mittels HPLC / ICP-MS
Kopplung. So führten Inagaki et al. eine Speziesanalyse von Spurenelementen, die an
Proteine gebunden sind, im humanen Blutserum durch [53]. Jakubowski et al. koppelten
nach der Einführung der ICP-MS Geräte mit eingebauter Kollisionszelle diese mit einer
HPLC-Anlage, um eine Selenspeziesanalyse im humanen Urinproben durchführen zu
können [54].
2.2.2.1 Reversed-Phase-Chromatographie gekoppelt mit der ICP-MS
Die Reversed-Phase-Chromatographie (RPLC) ist eine der am häufigsten benutzen Me-
thoden der Flüssigchromatographie, bei der die stationäre Phase weniger polar ist als
die mobile Phase. Als stationäre Phase werden häufig C8 oder C18 Materialien
eingesetzt. Das Einsatzgebiet der Reversed-Phase-Chromatographie liegt hauptsächlich
auf den Gebieten der Umwelt- [56-59] und klinischen Forschung [34/60-64]. Innerhalb der
Umweltforschung konnten so Butylzinnverbindungen sowohl in
Polyvinylchloridprodukten als auch in Fungiziden bzw. Insektiziden analysiert werden
[56]. Neben dem Einsatz dieser Kopplung auf dem Gebiet der Quecksilberanalytik unter
Verwendung eines Ultraschallzerstäubers (NWG5: 70-160 pg) wurden auch organische
Bleiverbindungen (Trimetyl-, Triethyl- und Triphenylverbindnungen) unter
isokratischen Bedingungen getrennt und quantifiziert [58-59].
In der klinischen Forschung wird die RPLC / ICP-MS Kopplung zur Analyse von Me-
tallkomplexen bzw. von Metallspezies in Biomolekülen eingesetzt [64]. So trennten und
analysierten Taktera et al. Iodid und fünf Iodoaminosäuren in Hormonen der Schild-
drüse, die sie auch im Urin und Blutplasma zur Früherkennung von Schilddrüsener-
krankungen bestimmen konnten (NWG: 35-130 pg) [60]. Ebenso konnten Metallopor-
phyrine sowohl im Blut als auch im Urin getrennt und quantifiziert werden [62]. Ein
5 NWG = Nachweisgrenze

Stand der Forschung 12
weiteres Anwendungsgebiet stellt die Analyse von platinhaltigen Chemotherapeutika
da. Hierzu benutzten Cairns et al. eine vorgeschaltete Desolvatationseinheit, um mit
100%igem organischem Lösungsmittel arbeiten zu können [63].
2.2.2.2 Ionenpaar-Chromatographie gekoppelt mit der ICP-MS
Eine weitere Möglichkeit der Kopplung einer Trenneinheit mit der ICP-MS ist die
Kombination Ionenpaar-Chromatographie (IPC) und ICP-MS. Mithilfe der IPC können
hydrophobe ionische Spezies getrennt werden, indem die gebildeten Ionenpaare auf-
grund ihres Adsorptionsverhalten an der unpolaren stationären Phase retardiert werden.
Hier werden die gleichen Anforderungen an die Zerstäubereinheit wie bei der RPLC /
ICP-MS Kopplung gestellt.
Die Hauptanwendungsgebiete dieser online-Kopplungstechnik liegen in der Analyse
von umwelt-relevanten bzw. biologischen Proben. Vor allem bei der Analyse von
Arsenspezies in biologischen Proben (Fisch, Urin, Blutserum) und in Umweltproben
(Wasser und Meerwasser) wird sehr häufig auf die Ionenpaar-Chromatographie als
Trenntechnik zurückgegriffen. Hierbei können absolute Nachweisgrenzen von bis zu
0,1 pg (auf Arsen bezogen) erreicht werden [65-71]. Beauchemine et al. setzten der
mobilen Phase 5% Methanol zu, um die Trennung der Arsenspezies zu verbessern.
Zugleich mussten sie die Generatorleistung des ICP-MS steigern, um eine vergleichbare
Nachweisstärke wie nach einer Trennung mit einer rein wässrigen mobilen Phase zu
erhalten [65-67].
Neben der Analyse von Organoquecksilber- und Organozinnverbindungen [77-80] haben
Yang und Jiang auf der Basis der IPC / ICP-MS Kopplung eine Selenspeziesanalyse im
Urin entwickelt, um so Nachweisgrenzen bis 0,17 ng/mL erreichen zu können. Brown et
al. modifizierten die bisher beschriebenen Kopplungen und führte eine Isotopenver-
dünnungsanalyse zur Bestimmung von Bleispezies durch. Im isokratischen Modus der
IPC konnten sie Bleispezies bis zu einer absoluten Nachweisgrenze von 0,37-3,9 pg je
nach Spezies (auf Blei bezogen) analysieren [76-77]. Jakubowski et al. verbesserten das
Kopplungssystem und setzten zur Chromspeziesanalyse in Umweltproben einen
hydraulischen Hochdruckzerstäuber ein [35]. Um die auftretenden Kohlenstoffinter-
ferenzen im Plasma zu unterdrücken, fügten sie dem Plasmagas Sauerstoff zu.

Stand der Forschung 13
Im Gegensatz zur RPLC / ICP-MS-Kopplung besitzt die IPC / ICP-MS-Kopplung nur
geringe Bedeutung in der klinischen Forschung. Ein Beispiel für die Anwendung liegt
in der Untersuchung von Reaktionen des cis-Platin mit verschiedenen Proteinen [81].
2.2.2.3 Ionenaustausch-Chromatographie gekoppelt mit der ICP-MS
Die Kopplung von ICP-MS und Ionenaustausch-Chromatographie (IEC) spielt eine
zentrale Rolle in der Elementspeziesanalytik.
Bei der IEC werden zwischen zwei Arten unterschieden, die Anionen-Austausch- und
die Kationen-Austausch-Chromatographie.
Ein typisches Packmaterial einer IEC Säule ist ein Copolymerisat aus Styren und Di-
vinylbenzen. Mithilfe dieser Packungsmaterialien können geladene und ungeladene
Spezies aufgrund unterschiedlicher Affinität zum Austauschermaterial getrennt werden.
Die Trenneffizienz hängt stark vom pH-Wert, von der Pufferkonzentration, von der
Konzentration des organischen Modifieres, der Temperatur und der Flussrate der mobi-
len Phase ab.
Es gibt drei große Anwendungsgebiete der Anionen-Austausch-Chromatographie-
Kopplung. Neben der schon von der IPC / ICP-MS Kopplung (es wird kein spezielles
Interface benötigt) bekannten Arsenspeziesanalyse in Urin wird sie neben der Chrom-
speziesanalyse auch in der klinischen Forschung eingesetzt [34].
Das Hauptproblem bei der Arsenspeziesanalyse mittels Massenspektrometrie stellt die
gleichzeitige Anwesenheit von Chlorid-Ionen in der Probe (z.B. Urin) dar. Die Interfe-
renzen von ArCl+ auf der Masse 75 (Arsen) führen zu Problemen bei der Quantifizie-
rung der Arsenspezies. Trotzdem konnten Nachweisgrenzen von 20-92 pg Arsen erzielt
werden [82-88]. Goosens et al. überführten alternativ zur Hydrid-Generierung die Arsen-
spezies (in Meerwasser, humanem Serum, Urin) mittels einer Dowex-IX 8 Säule in die
Nitratform und trennte sie so von den Chlorid-Ionen ab. Anschließend wurden die Säule
mit 0,03 molarer Salpetersäure gespült, wobei die Chlorid-Ionen zurückgehalten
wurden [90]. Sheppard et al. stellten fest, dass neben der Chloridabtrennung die
Zumischung von Helium ins Plasmagas die Nachweisgrenze deutlich verbessert.
Zusätzlich verdünnt Sheppard die Urinproben mit Wasser (1:5), um die Oxidation von
Arsenit zum Arsenat zu verhindern [83]. Ohne die Chlorid-Ionen abzutrennen,
versuchten Chen et al. den Metabolismus von Dimethylarsinsäure (DMA) bei Ratten
durch Urinanalytik, nach oraler Einnahme von mit DMA versetztes Trinkwasser, unter

Stand der Forschung 14
Einsatz der IEC / ICP-MS Kopplung zu klären [89]. Mithilfe der Ionen-Austausch-
Chromatographie auf Basis der Anionen wurde der Anstieg an Arsenit, DMA,
Trimethylarsinoxid und einer nicht definierten Verbindung im Urin beobachtet. Die
Autoren kamen zu dem Ergebnis, dass DMA durch ein intestinales Bakterium zu
anorganischem Arsen demethyliert wurde.
Neben der Arsenspeziesanalyse wird diese Kopplungstechnik bei der Chromspezies-
analyse eingesetzt. Da Chrom(III)-Verbindungen als essentiell gelten, hingegen
Chrom(VI)-Verbindungen sehr stark toxisch wirken können, sind Informationen über
den Gehalt bzw. der Konzentration und der Oxidationsstufe der Chromverbindungen
sehr wichtig [91-95]. Zoorob et al. setzten für ihre Arbeiten (Analyse von Chromspezies)
einen „Direkt-Injektions-Zerstäuber“ ein, sodass das Eluat direkt in der Plasmafackel
zerstäubt wurde. Auf diesem Wege erzielten sie absolute Nachweisgrenzen von bis zu 3
pg Chrom [91]. Riog-Navarro gelang es mithilfe der IEC / ICP-MS Kopplung simultan
Arsenspezies und Chrom(VI) zu analysieren [92].
Im Bereich der klinischen Forschung ist das Interesse an der Speziesanalyse essentieller
Elemente in humanen Seren groß. Hierzu setzen Sanz-Medel et al. ein doppelt-fokussie-
rendes ICP-MS ein. Die Quantifizierung der Spezies wurde mittels Isotopenverdün-
nungsanalyse durchgeführt [97]. Ein weiterer Bereich der klinischen Forschung be-
schäftigt sich mit der Klärung des Metabolismus von goldhaltigen Medikamenten6 in
menschlichem Blut. Hierzu wurden die Ergebnisse der Goldspeziesanalyse mittels IEC /
ICP-MS ausgewertet und bewertet [98].
Die Ionen-Austausch-Chromatographie auf Basis der Kationen spielt hinsichtlich ge-
koppelter Systeme eher eine untergeordnete Rolle und sei an dieser Stelle lediglich er-
wähnt.
2.2.2.4 Größenausschluss-Chromatographie gekoppelt mit der ICP-MS
Die online-Kopplung der SEC (angels.: size exclusion chromatography / Größenaus-
schluss-Chromatographie) ICP-MS ist eine leistungsfähige Trenntechnik, wenn sie mit
einer elementspezifischen Detektion verbunden ist. Bei dieser Trenntechnik erfolgt
keine chemische oder physikalische Wechselwirkung zwischen dem Analyten und der
stationären Phase [30]. Die Trennung erfolgt ausschließlich über das Verhältnis von
6 werden häufig bei Arthristiserkrankungen eingesetzt

Stand der Forschung 15
Analytgröße zu Porengröße der stationären Phase, insofern ist die Bezeichnung „Chro-
matographie“ streng genommen falsch. Es kann davon ausgegangen werden, dass die
native Form der Spezies während der Trennung erhalten bleibt. Aufgrund dieser scho-
nenden Trennbedingungen wird die SEC hauptsächlich in der Speziesanalytik von bio-
logischen Proben eingesetzt (z.B. bei der Analyse von Metallothioneinen (MT) in biolo-
gischer Matrices mit Tris-Puffer als mobile Phase) [101-110].
Dean et al. erreichten eine Separierung und Identifizierung der MTs mittels SEC / ICP-
MS in der Leber von Pferden und in den Nieren von Schweinen [101/106]. Wolf und
Richards analysierten ebenfalls MTs mithilfe der SEC / ICP-MS in biologischen Pro-
ben. Kernpunkt ihrer Arbeit war die Untersuchung der Verteilungsmuster der MT-Iso-
formen im Cytosol von humanen Hirnproben und Hirnproben von Alzheimerpatienten
[109-110]. Mason et al. beschränkten sich dagegen auf die Analyse von MT-Standardlö-
sungen und untersuchten die Elemente Cadmium, Zink und Kupfer. Hierbei stellten sie
fest, dass Zink während der chromatographischen Trennung durch Kupfer ersetzt wurde
(Reihenfolge des Einbaus als Ligand bei MTs: Cu > Cd > Zn) [102/104].
Shum und Houk vergrößerten das Einsatzgebiet der SEC / ICP-MS Analyse. Sie führten
unter Zuhilfenahme eines „Direkt-Injektions-Zerstäubers“ eine Proteintrennung in
humanen Serum ohne jegliche Probenvorbereitung durch. Das Hauptaugenmerk lag
hierbei auf Proteinen, die Blei, Cadmium, Kupfer, Eisen und Zink binden. Shum und
Houk erreichten mit der eingesetzten Kopplung eine absolute Nachweisgrenze von 0,5-
3 pg Metall [40].
2.2.3 Kapillarelektrophorese gekoppelt mit der ICP-MS
Durch die Kopplung der Kapillarelektrophorese (CE) mit der ICP-MS besteht die Mög-
lichkeit, sowohl die hohe Trennschärfe der CE (es können Anionen, Kationen und unge-
ladene Moleküle simultan getrennt werden) als auch die hohe Empfindlichkeit der ICP-
MS zur Bestimmung von Metallspezies zu verbinden. Ein weiterer Vorteil der CE ge-
genüber anderen Trennmethoden liegt in der Möglichkeit der Analyse kleinster Pro-
benmengen (im unteren nL-Bereich; einige Humanproben (Ausnahme: Blut, Urin,
extra-vasale Flüssigkeiten) stehen nur in sehr geringen Probenmengen zur Verfügung).
Bei der CE / ICP-MS Kopplung handelt es sich um eine relativ neue Kopplungstechnik,
die zum Beginn der 90iger Jahre eingeführt wurde. Erst gegen Ende der 90iger Jahre

Stand der Forschung 16
wurde in einer geringen Anzahl von Veröffentlichungen die Entwicklung und einige
Anwendungen der Methodik beschrieben.
Der Nachteil dieser Kopplung liegt darin, dass ein Interface für die online-Kopplung
von CE und ICP-MS benötigt wird. Es ist nicht möglich, wie bei den bisher beschrie-
benen Kopplungen, einen kommerziellen Zerstäuber mit Sprühkammer zu verwenden,
da die Flussrate der CE für die meisten kommerziellen Zerstäuber zu gering ist. An das
Interface sind folgende Anforderungen geknüpft:
1. der Stromkreis der CE muss über das Interface geschlossen werden können,
2. die Flussrate der CE muss an die Flussrate des Zerstäubers über das Interface ange-
passt werden können,
3. das Interface muss ein geringes Totvolumen besitzen, um keinen Sogeffekt auf die
Kapillare auszuüben,
4. es muss die Position der CE-Kapillare im Interface reproduzierbar sein.
In der Literatur sind verschiedenen Ansätze zur Entwicklung eines solchen Interface zu
finden. Tabelle 2.2-1 gibt einen Überblick über die Lösungsansätze und dem Einsatzge-
biet der CE / ICP-MS Kopplung.
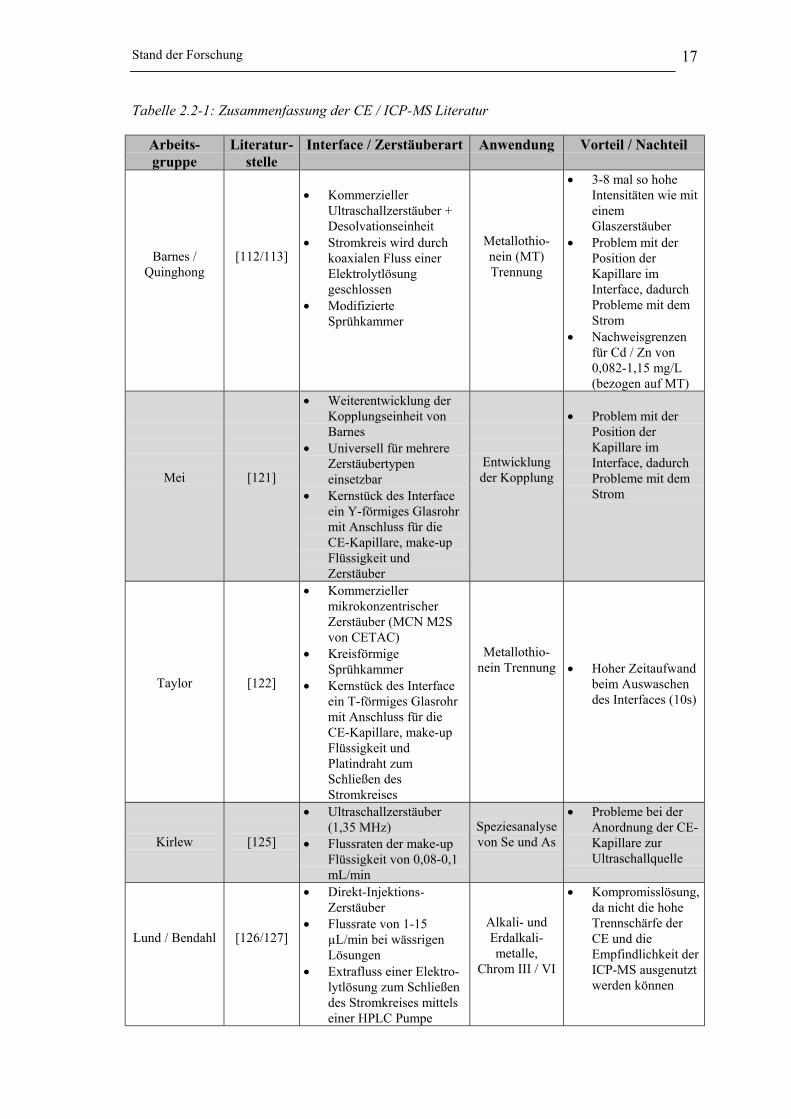
Stand der Forschung 17
Tabelle 2.2-1: Zusammenfassung der CE / ICP-MS Literatur
Arbeits-gruppe
Literatur-stelle
Interface / Zerstäuberart Anwendung Vorteil / Nachteil
Barnes /Quinghong
[112/113]
• KommerziellerUltraschallzerstäuber +Desolvationseinheit
• Stromkreis wird durchkoaxialen Fluss einerElektrolytlösunggeschlossen
• ModifizierteSprühkammer
Metallothio-nein (MT)Trennung
• 3-8 mal so hoheIntensitäten wie miteinemGlaszerstäuber
• Problem mit derPosition derKapillare imInterface, dadurchProbleme mit demStrom
• Nachweisgrenzenfür Cd / Zn von0,082-1,15 mg/L(bezogen auf MT)
Mei [121]
• Weiterentwicklung derKopplungseinheit vonBarnes
• Universell für mehrereZerstäubertypeneinsetzbar
• Kernstück des Interfaceein Y-förmiges Glasrohrmit Anschluss für dieCE-Kapillare, make-upFlüssigkeit undZerstäuber
Entwicklungder Kopplung
• Problem mit derPosition derKapillare imInterface, dadurchProbleme mit demStrom
Taylor [122]
• KommerziellermikrokonzentrischerZerstäuber (MCN M2Svon CETAC)
• KreisförmigeSprühkammer
• Kernstück des Interfaceein T-förmiges Glasrohrmit Anschluss für dieCE-Kapillare, make-upFlüssigkeit undPlatindraht zumSchließen desStromkreises
Metallothio-nein Trennung • Hoher Zeitaufwand
beim Auswaschendes Interfaces (10s)
Kirlew [125]
• Ultraschallzerstäuber(1,35 MHz)
• Flussraten der make-upFlüssigkeit von 0,08-0,1mL/min
Speziesanalysevon Se und As
• Probleme bei derAnordnung der CE-Kapillare zurUltraschallquelle
Lund / Bendahl [126/127]
• Direkt-Injektions-Zerstäuber
• Flussrate von 1-15µL/min bei wässrigenLösungen
• Extrafluss einer Elektro-lytlösung zum Schließendes Stromkreises mittelseiner HPLC Pumpe
Alkali- undErdalkali-metalle,
Chrom III / VI
• Kompromisslösung,da nicht die hoheTrennschärfe derCE und dieEmpfindlichkeit derICP-MS ausgenutztwerden können
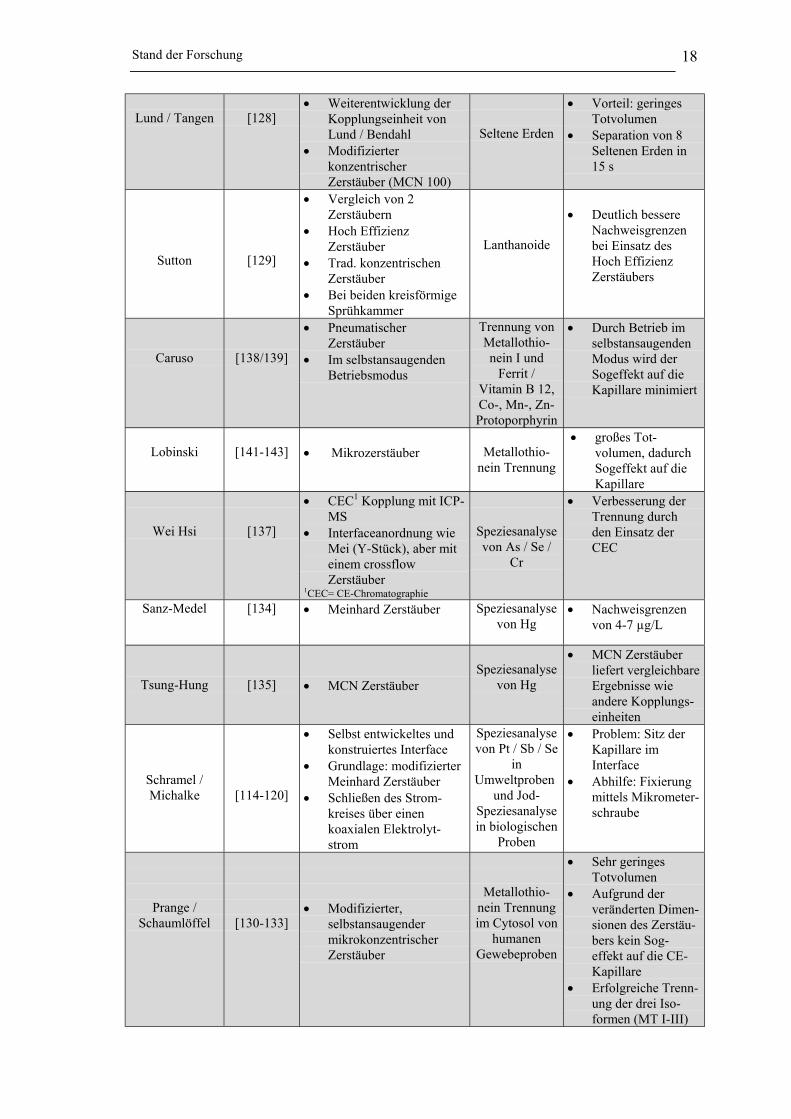
Stand der Forschung 18
Lund / Tangen [128]• Weiterentwicklung der
Kopplungseinheit vonLund / Bendahl
• ModifizierterkonzentrischerZerstäuber (MCN 100)
Seltene Erden
• Vorteil: geringesTotvolumen
• Separation von 8Seltenen Erden in15 s
Sutton [129]
• Vergleich von 2Zerstäubern
• Hoch EffizienzZerstäuber
• Trad. konzentrischenZerstäuber
• Bei beiden kreisförmigeSprühkammer
Lanthanoide
• Deutlich bessereNachweisgrenzenbei Einsatz desHoch EffizienzZerstäubers
Caruso [138/139]
• PneumatischerZerstäuber
• Im selbstansaugendenBetriebsmodus
Trennung vonMetallothio-nein I und
Ferrit /Vitamin B 12,Co-, Mn-, Zn-Protoporphyrin
• Durch Betrieb imselbstansaugendenModus wird derSogeffekt auf dieKapillare minimiert
Lobinski [141-143] • Mikrozerstäuber Metallothio-nein Trennung
• großes Tot-volumen, dadurchSogeffekt auf dieKapillare
Wei Hsi [137]
• CEC1 Kopplung mit ICP-MS
• Interfaceanordnung wieMei (Y-Stück), aber miteinem crossflowZerstäuber
1CEC= CE-Chromatographie
Speziesanalysevon As / Se /
Cr
• Verbesserung derTrennung durchden Einsatz derCEC
Sanz-Medel [134] • Meinhard Zerstäuber Speziesanalysevon Hg
• Nachweisgrenzenvon 4-7 µg/L
Tsung-Hung [135] • MCN ZerstäuberSpeziesanalyse
von Hg
• MCN Zerstäuberliefert vergleichbareErgebnisse wieandere Kopplungs-einheiten
Schramel /Michalke [114-120]
• Selbst entwickeltes undkonstruiertes Interface
• Grundlage: modifizierterMeinhard Zerstäuber
• Schließen des Strom-kreises über einenkoaxialen Elektrolyt-strom
Speziesanalysevon Pt / Sb / Se
inUmweltproben
und Jod-Speziesanalysein biologischen
Proben
• Problem: Sitz derKapillare imInterface
• Abhilfe: Fixierungmittels Mikrometer-schraube
Prange /Schaumlöffel [130-133]
• Modifizierter,selbstansaugendermikrokonzentrischerZerstäuber
Metallothio-nein Trennungim Cytosol von
humanenGewebeproben
• Sehr geringesTotvolumen
• Aufgrund derveränderten Dimen-sionen des Zerstäu-bers kein Sog-effekt auf die CE-Kapillare
• Erfolgreiche Trenn-ung der drei Iso-formen (MT I-III)

Stand der Forschung 19
Wie aus der Tabelle 2.2-1 zu entnehmen ist, wird die CE / ICP-MS Kopplung haupt-
sächlich in der bio-medizinischen bzw. klinischen Forschung eingesetzt.
Alle Arbeiten, die sich mit der CE / ICP-MS Kopplung beschäftigt haben, weisen eine
Gemeinsamkeit auf. In allen Fällen wird eine zusätzliche Elektrolytlösung in das Inter-
face geleitet, mit dem Ziel den CE-Stromkreis zu schließen. Die Wahl der Zerstäuber
variiert stark. Neben Ultraschallzerstäubern werden sehr häufig sowohl mikrokonzen-
trische wie auch Meinhard-Zerstäuber eingesetzt [112-120/122/125/187/130-135/141-143].
Bei allen Anwendungen stellte sich heraus, dass die Position der CE-Kapillare im
Interface bei der Kopplung eine bedeutende Rolle spielt. Einzig Michalke und
Schaumlöffel haben sich dieses Problems angenommen [114-120/130-133]. Michalke
positionierte die CE Kapillare mittels einer Mikrometerschraube am Interface,
Schaumlöffel hingegen löste das Problem durch den Einsatz eines T-Stücks, das
innerhalb der Zerstäuberkapillare konzentrisch zuläuft und so immer der optimale Sitz
der Kapillare garantiert ist.
Auf der Basis der Arbeiten von Prange und Schaumlöffel konnte die CE / ICP-MS
Kopplung optimiert werden [130/131]. Das von ihnen entwickelte Interface wurde stetig
verbessert und ist seit Anfang 2000 kommerziell bei der Firma CETAC als CEI 100
erhältlich (siehe Punkt 4.3 der Arbeit). Anwendung fand die von Schaumlöffel et al.
erarbeitete Methode in der Analyse von Metallothionein im Cytosol von humanen Hirn-
proben [132]. Weiterhin setzten Schaumlöffel et al. diese Kopplung zur Iostopen-Ver-
dünnungsanalyse ein, um die Metallzusammensetzung von MT-Standards, die aus
Kaninchenleber gewonnen wurden, zu bestimmen [133]. Es zeigte sich, dass die erar-
beitete Kopplungstechnik hervorragend zur Analyse biologischer Proben einsetzbar
war, sodass sie auch im Rahmen der vorliegenden Arbeit Anwendung fand.

Problemstellung 20
3 ProblemstellungNickel gehört neben Zinn, Vanadium und Arsen zu den Spurenelementen, deren Essen-
tialität für den Menschen nicht gesichert ist. Hingegen konnten die krebserregende und
cytotoxische Wirkung von Nickel und Nickelverbindungen nachgewiesen werden
[26/145-149]. Während die molekularen Mechanismen der Nickelkarzinogenese zum
großen Teil bekannt sind [149], sind die Bindungspartner von Nickel im Humangewebe
aufgrund ihrer sehr geringen Konzentrationen weitestgehend unbekannt. Erste Untersu-
chungen haben gezeigt, dass der Nickelgehalt von gesunden gegenüber malignen Ge-
webe des Intestinaltrakts Unterschiede aufweist [146]. Um sowohl die cytotoxische als
auch die Immunsystem anregende Wirkung von Nickel z.B. als Zusatz herkömmlicher
Chemotherapeutika, nutzen zu können, müssen die Nickelspezies in humanen Geweben
bekannt sein.
Im Rahmen dieser Arbeit sollte ein Analysenverfahren zur Nickelspeziesanalyse im
Cytosol humaner Gewebeproben entwickelt werden. Das Ziel sollte dabei sein, die
Nickelspezies im Cytosol von Normal- und Tumorgewebe des humanen Intestinaltrakts
zu trennen, zu charakterisieren und zu identifizieren. Weiter sollten mögliche Unter-
schiede der Nickelspezies im Cytosol normaler und maligner Zellen, die sowohl qualita-
tiver wie auch quantitativer Art sein können, aufgezeigt werden. Zu diesem Zweck
sollte eine CE / ICP-MS Kopplung (Kapillarelektrophorese gekoppelt an ein induktiv
gekoppeltes Plasma Massenspektrometer) eingesetzt und für die Trennung von cytosol-
ischen Nickelspezies optimiert werden. Die Ergebnisse sollten mittels SEC / ICP-MS
Kopplung verifiziert werden.
Dazu musste ein Konzept zur Reduzierung bzw. Eliminierung von Nickelkontami-
nationsquellen ausgearbeitet werden, da sonst eine Nickelspezies-Analyse unter den
gegebenen Bedingungen (geringe Nickelkonzentrationen) nicht möglich wäre. Nickel-
kontainationsquellen können die eingesetzten Chemikalien (Aufreinigung der Chemi-
kalien) sein; aber auch apparative Aspekte (Ausschalten von Kontaminationsquellen
durch eingesetzte Geräte bzw. Geräteparameter) sollten berücksichtigt werden. Neben
der Optimierung sämtlicher Trennverfahren (CE und SEC) kommt der
Probenpäparation und der Probenvorbehandlung eine bedeutende Rolle zu.

Analysenverfahren 21
4 Analysenverfahren
In diesem Kapitel werden die zur Nickelspezies-Analyse eingesetzten Analysenverfah-
ren beschrieben. Der Beschreibung der CE / ICP-MS Kopplung kommt eine besondere
Bedeutung zu [130-131].
4.1 Probenmaterial
Bei den zu analysierenden Gewebeproben handelte es sich jeweils um maligne (TG)
und normale Gewebeproben (NG) des humanen Intestinaltrakts. Im Anschluss an den
chirurgischen Eingriff wurde ein kleiner Teil des Resektats für die Speziesanalyse zur
Verfügung gestellt. Das verbleibende Resektat wurde pathologisch untersucht. Die
Gewebeproben wurden in Polyethylen-Folien eingeschweißt und bei –30°C gelagert.
Sowohl der pathologische Bericht wie auch die Patientendaten konnten unter
Einhaltung des Datenschutzes eingesehen werden. In Tabelle 4.1-1 sind die
individuellen Patientendaten zusammengefasst.
Tabelle 4.1-1: Kenndaten der analysierten Gewebeproben
Probe Entnahme-
ort
Gewebeart 1Tumorstadium (T)/
Differenzierungs-
grad (G)
Alter des
Patienten
/ Jahr
Geschlecht 2
1 Coecum3 TG / NG
T 4
G2-3 74 w
2 Rektum4 TG / NG
T 3
G3 84 w
3
Colon-
ascendens5 TG / NG
T 4
G2 60 m
4
Colon-
transversum6 entzünd.G /
NG
k.A. 60 m
5
Colon-
sigmoideum7 TG / NG
T 4
G3 53 m
Analysenverfahren 22
6-9 Bei diesen Proben waren nur Tumorgewebe vorhanden, sodass sie zur Opti-
mierung der Geräteparameter genutzt wurden
10 Rektum4 TG / NG Adenom8 67 m
11
Colon-
sigmoideum7 TG / NG
T 0
G 2 70
w
12
Colon-
sigmoideum7 TG / NG
T 3
G 3 58 m
13
Colon-
ascendens5 TG / NG
T 3
G 2 78
w
14
Colon-
transversum6 TG / NG9
T 2
G2 72 m
15
Colon-
sigmoideum7 TG / NG
T 2
G 2 72 m
1: TG = malignes Gewebe ; NG = normales Gewebe ; entzünd.G = entzündetes Gewebe2: w = weiblich ; m = männlich, k.A.= keine Angaben3: Blinddarm ; 4: Enddarm ; 5: aufsteigender Darm ; 6: Querdarm ; 7: Übergang zum Enddarm8: Adenom = Vorstufe des Karzinoms9: undefiniert, kann sich auch um Adenomgewebe handeln
Die folgende Abbildung 4.1-1 zeigt die Entnahmestellen der untersuchten Gewebepro-
ben.
Abbildung 4.1-1: Entnahmestellen der Gewebeproben [150]
Rektum(Probe 2 / 10)
Colon-transversum(Probe 4 / 14)
Colon-ascendens(Probe 3 / 13)
Coecum(Probe 1)
Appendix
Colon-sigmoideum(Probe 5 / 8 / 9 /11 / 12 / 15)
Colon-descendens

Analysenverfahren 23
4.2 Probenvorbereitung
Zur Herstellung des Cytosols aus humanen Gewebeproben können zwei unterschiedli-
che Verfahren (der mechanische Zellaufschluss oder der Zellaufschluss mittels Ultra-
schall) angewendet werden. Üblicherweise wird der mechanische Zellaufschluss mit
einem Potter-Eveljhem–Homogenisator und der Zellaufschluss mittels Ultraschall mit
einem kommerziell erhältlichen Ultraschalldesintegrator durchgeführt (siehe Abbildung
4.2-1). Vor dem Zellaufschluss wurden die gefrorenen Gewebeproben mit Quarzskal-
pellen in kleine Stücke geschnitten.
Abbildung 4.2-1: Potter-Eveljhem-Homogenisator (links), Ultraschalldesintegrator (rechts)
Bei der Herstellung der Cytosole mithilfe des Potter-Eveljhem Homogenisators wurden
die geschnitten Gewebeproben in einem Verhältnis Probe zu gereinigtem und entgastem
Tris-HNO3-Puffer 1:2 gemischt und mit einem Ultra Turrax unter Argonschutzgas-
atmosphäre und Eiskühlung vorhomogenisiert 7. Anschließend erfolgte der mechanische
Zellaufschluss im Eisbad unter Argon in einem Reinraum der Klasse 1000. Danach
wurde die homogenisierte Probe bei + 4°C und 25.000g 99 min. (Probe 10-15) bzw. 99
min. bei 100.000g (Probe 1-5) zentrifugiert. Das Cytosol wurde abpipettiert und unter
Argon bei –25°C gelagert.
Für den Zellaufschluss mittels Ultraschalldesintegrator wurde die geschnittene Probe
unter Argonschutzgasatmosphäre in ein „Eppendorfcap“ (Volumen 2 mL) überführt und
mit Puffer (gereinigt und entgast) versetzt (Verhältnis 1:2). Die Beschallungsdauer beim
Ultraschalldesintegrators betrug 15 min bei höchster Stufe mit einem Beschallungsin

Analysenverfahren 24
tervall von 50 %. Der Aufschluss wurde bei +4°C durchgeführt. Die anschließende
Zentrifugation über einen Zeitraum von 99 min. erfolgte bei 25.000g und +4°C. Das
hergestellte Cytosol wurde unter Argon bei –25°C gelagert.
Details beider Verfahren wurden mit Fotos dokumentiert und können im Anhang der
Arbeit eingesehen werden.
4.3 Eingesetzte Kopplungstechniken
Im Rahmen der angestrebten Nickelspeziesanalyse in Cytosolen humaner Gewebepro-
ben wurde sowohl mit einer CE / ICP-MS (nach Schaumlöffel) als auch mit einer SEC /
ICP-MS Kopplung gearbeitet.
Wie erste Messungen [164] mithilfe der TXRF8 gezeigt haben, lag die Gesamtnickel-
konzentration im Cytosol der humanen Darmproben im µg/L Bereich, sodass an das
Massenspektrometer hohe Ansprüche hinsichtlich der Nachweisstärke (vor allem für
Nickel) gestellt wurden. Um diesen Ansprüchen gerecht zu werden, wurde für beide
Kopplungen statt eines Quadrupolgerätes ein Sektorfeld-Massenspektrometer [Finnigan
MAT „Element 1“] verwendet. (Schema eines solchen Gerätes siehe Abbildung 4.3-1)
Um die größtmögliche Nachweisstärke zu erhalten, wurde während der gesamten Arbeit
mit der geringsten Auflösung von 300 gearbeitet. Diese Massenauflösung ist eine kon-
stante Größe und nicht wie bei einem Quadrupolgerät eine Funktion der Masse.
7 Zu Beginn der Arbeit (Probe 1-5) wurde noch Dithiotreitol (5mmol/L) als Antioxidants zugefügt, bei
den weiteren Proben wurde auf die Zugabe aus Gründen, die noch beschrieben werden (siehe Punkt
5.5.2), verzichtet.8 TXRF= Totalreflexionsröntgenfluoreszenz-Spektrometrie
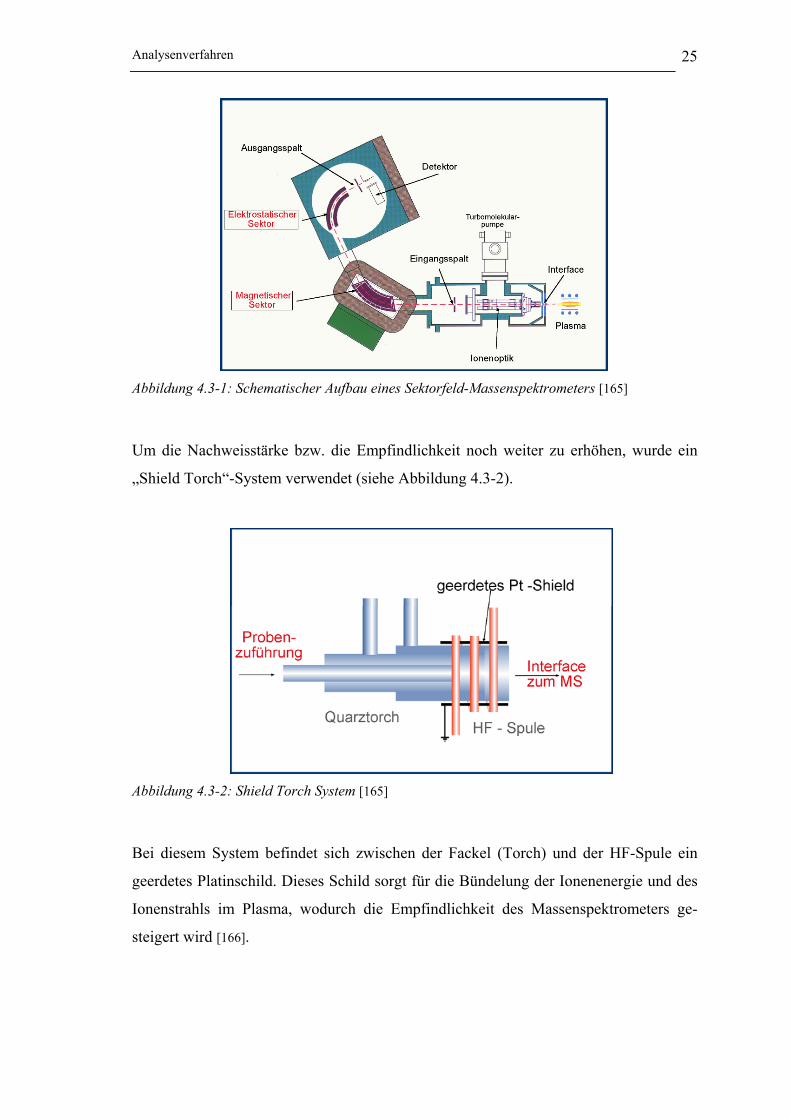
Analysenverfahren 25
Abbildung 4.3-1: Schematischer Aufbau eines Sektorfeld-Massenspektrometers [165]
Um die Nachweisstärke bzw. die Empfindlichkeit noch weiter zu erhöhen, wurde ein
„Shield Torch“-System verwendet (siehe Abbildung 4.3-2).
Abbildung 4.3-2: Shield Torch System [165]
Bei diesem System befindet sich zwischen der Fackel (Torch) und der HF-Spule ein
geerdetes Platinschild. Dieses Schild sorgt für die Bündelung der Ionenenergie und des
Ionenstrahls im Plasma, wodurch die Empfindlichkeit des Massenspektrometers ge-
steigert wird [166].

Analysenverfahren 26
4.3.1 CE / ICP-MS Kopplung
Das Kernstück der CE / ICP-MS Kopplung ist das Interface zwischen der CE und der
ICP-MS. Die Abbildung 4.3-3 zeigt den schematischen Aufbau der eingesetzten CE /
ICP-MS Kopplung.
Abbildung 4.3-3: Schema der CE / ICP-MS Kopplung
Nach der Konditionierung der Kapillare (siehe Tabelle 5.2-7) wird die Probe bzw. das
Cytosol hydrodynamisch in die CE Kapillare injiziert und anschließend Strom an die
CE Kapillare angelegt. Die Analyten werden aufgrund ihrer Mobilität getrennt und
gelangen über das Interface bzw. den Zerstäuber ins Plasma und werden mithilfe des
Massenspektrometers detektiert.
Um eine solche Kopplung betreiben zu können, wurde ein spezielles Interface einge-
setzt. Dieses Interface darf keine Sogeffekte (Bandenverbreiterung) besitzen, muss für
die Anpassung der CE-Flussrate (im unteren nL-Bereich) an den Zerstäuber (µL-
Bereich) sorgen und den Strom der CE schließen, so dass die hohe Trennschärfe der
Kapillarelektrophorese nicht negativ beeinflusst wird:
In Abbildung 4.3-4 ist ein solches Interface schematisch dargestellt. Dieses Interface
wurde von Schaumlöffel und Prange [130/131] entwickelt und wird kommerziell von der
Firma CETAC als CEI 100 vertrieben.
+ -+ -
Kapillare
Puffer
Hochspannung
Argon
Puffer
������������
�����������������
����������������
Interface
ICP - MS
Kapillarelektrophorese (CE)
Proben-vial
Kapillare
Puffer
Hochspannung
Argon
Puffer
������������
�����������������
����������������
Interface
ICP - MS
Proben-vial

Analysenverfahren 27
Abbildung 4.3-4: Schematischer Aufbau des Interface für die CE / ICP-MS Kopplung [130]
Wie aus der Abbildung hervorgeht, besteht das Interface aus zwei Teilstücken. Zum
einen aus dem Kreuzstück, das die Gegenelektrode (Platin) und den Anschluss für die
Make-up Flüssigkeit beinhaltet und zum anderen aus einem modifizierten, selbstansau-
genden mikrokonzentrischen Zerstäuber. Die CE-Kapillare befindet sich direkt über der
Zerstäuberkapillare, sodass nur ein sehr geringes Totvolumen im Zerstäuber vorhanden
ist [130].
Die CE-Kapillare wird von der Make-up Flüssigkeit (Ammoniumnitratpuffer pH 7,4
plus 5 µg Ge/L) umspült, um den Stromkreis der CE zu schließen. Gleichzeitig werden
die Analyten, die aus der Kapillare migrieren, mit dieser Make-up Flüssigkeit verdünnt
und sofort zerstäubt (Anpassung der Flussraten von CE und Zerstäuber). Aufgrund der
niedrigen Flussrate (7-12 µL/min) und der inneren Dimensionierung des Zerstäubers ist
praktisch kein Sogeffekt auf die Kapillare vorhanden. Anhand des konstanten German-
iumsignals der Make-up Flüssigkeit kann die Qualität der Zerstäubung während einer
Messung kontrolliert werden. Die Intensitäten der Signale in den Elektropherogrammen
wurden auf das Signal für Germanium in der Standardlösung normiert, um eine bessere
Vergleichbarkeit der Elektropherogramme zu gewährleisten. Dies wurde bei allen Elek-
troferrogrammen im weiteren Verlauf der Arbeit so gehandhabt.
Das trockene Aerosol gelangt durch eine Quarzsprühkammer über eine gegen
elektrostatische Aufladung geschützte Transferleitung direkt in die Plasmafackel des
ICP-MS. Die Abbildung 4.3-5 zeigt ein Foto des verwendeten Interface.

Analysenverfahren 28
Abbildung 4.3-5: Verwendetes Interface
Im unteren linken Bildrand ist die Transferleitung, die direkt in die Plasmafackel führt,
zu sehen. Im Vordergrund befinden sich zwei Röhrchen mit Reinstwasser für die Reini-
gung des Interface. Das mittlere Gefäß (PFA9-Gefäß) beinhaltet die Make-up Flüssig-
keit, die durch den Zerstäuber selbst angesogen wird.
4.3.2 SEC-Chromatographie mit UV-Detektion
Die SEC-Chromatographie mit UV-Detektion gehört zu den etablierten analytischen
Chromatographietechniken [6/30/167]. Sie wird im Verlauf der Arbeit zur Massenkali-
brierung der SEC-Säule benutzt. Dazu werden verschiedene UV-aktive Proteinstan-
dardlösungen mit bekannten Molmassen auf die Säule gegeben und bei 254 nm de-
tektiert. Über die Beziehung Retentionszeiten / Molmassen wird die Massenkalibrie-
rung der SEC-Säule durchgeführt. Mithilfe dieser Kalibriergeraden kann den aufge-
trennten Nickelspezies eine Molmasse zugeordnet werden. Der Aufbau der Kopplung
ist sehr einfach (siehe Abbildung 4.3-6).
9 PFA= Perfluor-Alkoxy-(Polymere)

Analysenverfahren 29
Abbildung 4.3-6: Schematischer Aufbau einer SEC mit einem UV-Detektor
Hierzu wird eine HPLC-Pumpe mit integrierter Probenschleife (500 µL) und eine SEC-
Säule benötigt. Das Vorfiltersystem dient zum Schutz der SEC-Säule vor möglichen
Bestandteilen des Cytosols, die das Säulenmaterial belegen und somit das Trennvermö-
gen der Säule verschlechtern. Ein solches Filtersystem ist im Anhang abgebildet. Das
Eluat gelangt aus der Säule in die Durchflußzelle des UV-Detektors, in dem die UV-
aktiven Probenbestandteile detektiert werden. In dieser Arbeit wurde eine präparative
SEC-Säule verwendet, da im weiteren Verlauf die Fraktionen der SEC analysiert wer-
den sollten. Die genauen Daten bezüglich Säule und Messparameter sind in der Tabelle
5.5-1 aufgelistet.
4.3.3 SEC / ICP-MS Kopplung
Zur Trennung der Elementspezies wurde eine Superdex Säule eingesetzt (Trennbe-
reich: 3000-70000 Da). Mithilfe dieser Säule können Proteine sehr schonend getrennt
werden. Die Abbildung 4.3-7 stellt den schematischen Aufbau der SEC / ICP-MS
Kopplung dar.
Injektions-
schleife
Vorfilter SEC-Säule Teflon
Kapillare

Analysenverfahren 30
Abbildung 4.3-7: Schematischer Aufbau einer SEC / ICP-MS Kopplung [168]
Im Falle der SEC / ICP-MS Kopplung kann die Trennsäule direkt mit dem Meinhard-
Zerstäuber (Leistung bis 2,7 mL/min) verbunden werden. Das gebildete Aerosol gelangt
durch eine gekühlte Sprühkammer in die Plasmafackel. Die Fotos in Abbildung 4.3-8
zeigen neben der Kopplung (A) eine solche gekühlte Sprühkammer (B).
A B
Abbildung 4.3-8: Kopplung SEC mit ICP-MS (A), Sprühkammer (B)
Um das Plasma nicht zu überlasten, wurde die Flussrate (2 mL/min) der SEC durch ei-
nen selbst konstruierten Split (Foto siehe Anhang) um den Faktor 5 (auf 400 µL/min)
reduziert. So konnten stabile Verhältnisse im Plasma erreicht werden. Der Split besteht
aus einem T-Stück (PTFE10), welches an der einen Seite mit einer peristaltischen
Pumpe und an der anderen Seite mit dem Meinhard-Zerstäuber verbunden ist. Über die
10 Polytetrafluorethylene
Injektions-schleife
Vorfilter SEC-SäuleTeflonKapillare
Zerstäuber
Sprüh-kammer
ICP-MS
Pumpe
Zerstäuber-gas
Split

Analysenverfahren 31
Pumpe kann das Splitverhältnis variiert und über gravimetrische Methoden bestimmt
werden.
Das Cytosol wurde über eine Probenschleife (Volumen 500 µL) und nach Passieren
eines Vorfilters auf die Trennsäule gegeben. Die optimierten Messparameter der SEC /
ICP-MS Kopplung sind in Tabelle 5.5-1 aufgelistet.
4.4 Totalreflexionsröntgenfluoreszensanalyse (TXRF)
Die TXRF wurde in der Arbeit zur Bestimmung der Gesamtnickelgehalte im Cytosol
eingesetzt. Sie wurde weiterhin für die Überprüfung der Reinheit der eingesetzten Che-
mikalien bzw. zur Identifizierung möglicher Kontaminationsquellen genutzt.
Hierzu wurden 10 µL des Cytosols jeder Probe auf eine vorher gereinigte und silikoni-
sierten TXRF-Träger (Quarzglas) gegebenen. Zu dem Cytosol wurden zusätzlich 10 µL
einer 10%igen HNO3 und 5 µL eines internen Yttrium-Standards (250 µg/L) auf den
Träger pipettiert und auf einer Heizplatte eingedampft. Die absolute Masse des Yttrium-
Standards auf der Quarzplatte betrug 5 ng. Die gesamte Probenvorbereitung fand in
einer Reinraumwerkbank der Klasse 1000 statt. Als Anregungsquelle diente eine
Molybdän-Röhre, wobei die Röhrenspannung 50 kV betrug. Es wurde mit einem Zr2O-
Filter bei einer Messzeit von 1000 s gearbeitet.
4.5 Gesamtproteinbestimmung nach Biuret-Lowry
Die Bestimmung der Gesamtproteingehalte der Cytosole erfolgte nach der Biuret-
Lowry-Methode [170]. Zu diesem Zweck wurde ein Analysen-Kit der Firma Sigma-
Aldrich verwendet. Die Detektion erfolgte photometrisch.
Das hergestellte Cytosol wurde mit dem Biuret-Reagenz (bestehend aus 0,75 mmol/ L
Kupfersulfat und 94 mmol/ L Natriumchlorid) versetzt und 10 Minuten inkubiert. Der
entstehende Komplex (in alkalischer Lösung) reduziert das zugesetzte Folin (0,1 mL)-
Ciocalteu-Phenol-Reagenz (2N laut Hersteller) [170]. Es entsteht eine intensive blaue
Färbung, die nach 30 Minuten bei 660 nm photometrisch detektiert wurde. Die Aus-
wertung der Messungen (nur für die Proben 1-5) wurde anhand einer Kalibriergerade
durchgeführt.

Optimierung der Analysenmethoden 32
5 Optimierung der Analysenmethoden
In den folgenden Abschnitten werden sowohl apparative wie auch messtechnische
Optimierungen der eingesetzten Kopplungstechniken CE / ICP-MS und SEC / ICP-MS
zur Nickelspeziesanalyse beschrieben.
5.1 Reduzierung von Nickelkontaminationen
Blindwerte spielen im spurenanalytischem Bereich eine bedeutende Rolle. Vor allem
ubiquitäre Elemente wie Zink, Kupfer und Nickel erschweren die Analytik im Ul-
traspurenbereich. Für diesen analytischen Bereich müssen sowohl alle benutzten Gefäße
und Verbrauchsmaterialien als auch die eingesetzten Chemikalien eine sehr hohe
Reinheit besitzen.
5.1.1 Reinigung der Gefäße
Um die unter 5.1 beschriebenen Kriterien erfüllen zu können, wurden nur Gefäße aus
PFA eingesetzt und sämtliche Verbrauchsmaterialien wie Pipettenspitzen mit ver-
dünnter unter dem Siedepunkt destillierter Salpetersäure gereinigt und anschließend mit
Reinstwasser (Wasseraufbereitungssystem Milli-Q-Element der Firma Millipore) ge-
spült. Die PFA Gefäße wurden vor jedem Gebrauch mithilfe einer Dampfreinigungsap-
paratur, die mit nachgereinigter Salpetersäure gefüllt wurde, gesäubert. In Abbildung
5.1-1 ist eine solche Dampfreinigungsapparatur abgebildet.

Optimierung der Analysenmethoden 33
Abbildung 5.1-1: Dampfreinigungsapparatur zur Reinigung der PFA-Gefäße
Nach der Reinigung wurden die Gefäße mit Reinstwasser unter einer Clean-Bench
mehrfach gespült und abschließend getrocknet.
5.1.2 Reinigung der Chemikalien
Bei der Reduzierung der Blindwerte kam den eingesetzten Chemikalien eine zentrale
Rolle zu (Liste der benutzen Chemikalien siehe Anhang). Es wurden, soweit sie kom-
merziell erhältlich waren, Chemikalien der Reinheit „suprapur“ eingesetzt. Die Salpe-
tersäure wurde vor ihrer Verwendung mithilfe einer „Subboiling“-Apparatur nachge-
reinigt. Trotz suprapurer Qualität musste auch der verwendete Tris-Puffer nachgereinigt
werden, wobei eine etwas aufwendigere Reinigungsprozedur entwickelt werden musste,
da der Tris-HNO3-Puffer starke Kontaminationen von Zink, Natrium und Kupfer
aufwies. Das enthaltene Natrium verursachte mit dem Plasmagas Argon, eine Inter-
ferenz auf der Masse von 63Cu, was zu erhöhten Blindwerten für Kupfer und zu einem
systematischen Fehler bei einer möglichen quantitativen Kupferbestimmung führte [6].
Mithilfe des Ionenaustauschermaterials Chelex 100 wurde der Tris-HNO3-Puffer in
mehreren Schritten gereinigt [6/172]. Im Anhang der Arbeit sind solche Reinigungssäu-
len abgebildet.
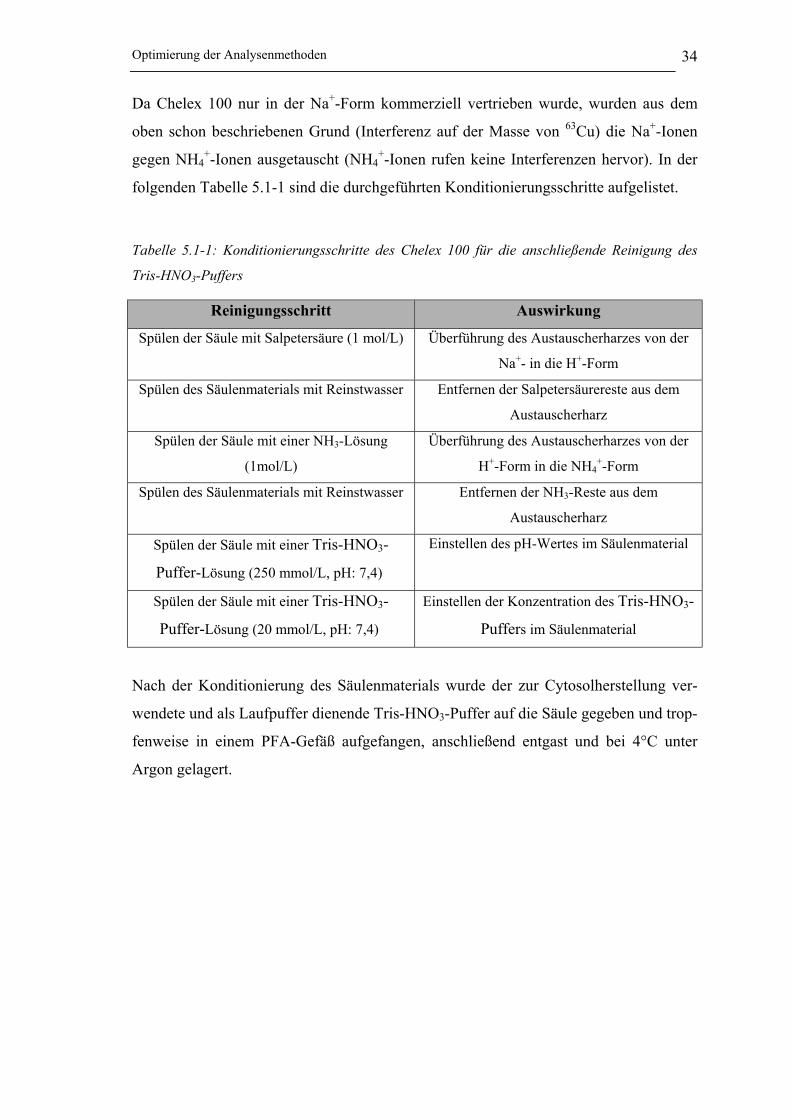
Optimierung der Analysenmethoden 34
Da Chelex 100 nur in der Na+-Form kommerziell vertrieben wurde, wurden aus dem
oben schon beschriebenen Grund (Interferenz auf der Masse von 63Cu) die Na+-Ionen
gegen NH4+-Ionen ausgetauscht (NH4
+-Ionen rufen keine Interferenzen hervor). In der
folgenden Tabelle 5.1-1 sind die durchgeführten Konditionierungsschritte aufgelistet.
Tabelle 5.1-1: Konditionierungsschritte des Chelex 100 für die anschließende Reinigung des
Tris-HNO3-Puffers
Reinigungsschritt Auswirkung
Spülen der Säule mit Salpetersäure (1 mol/L) Überführung des Austauscherharzes von der
Na+- in die H+-Form
Spülen des Säulenmaterials mit Reinstwasser Entfernen der Salpetersäurereste aus dem
Austauscherharz
Spülen der Säule mit einer NH3-Lösung
(1mol/L)
Überführung des Austauscherharzes von der
H+-Form in die NH4+-Form
Spülen des Säulenmaterials mit Reinstwasser Entfernen der NH3-Reste aus dem
Austauscherharz
Spülen der Säule mit einer Tris-HNO3-
Puffer-Lösung (250 mmol/L, pH: 7,4)
Einstellen des pH-Wertes im Säulenmaterial
Spülen der Säule mit einer Tris-HNO3-
Puffer-Lösung (20 mmol/L, pH: 7,4)
Einstellen der Konzentration des Tris-HNO3-
Puffers im Säulenmaterial
Nach der Konditionierung des Säulenmaterials wurde der zur Cytosolherstellung ver-
wendete und als Laufpuffer dienende Tris-HNO3-Puffer auf die Säule gegeben und trop-
fenweise in einem PFA-Gefäß aufgefangen, anschließend entgast und bei 4°C unter
Argon gelagert.

Optimierung der Analysenmethoden 35
5.1.3 Reduzierung der Nickelkontamination durch apparative Veränderungen
Da sowohl der „Sampler Cone“ wie auch der „Skimmer Cone“ der ICP-MS (siehe Abb.
5.1-4) üblicherweise aus Nickel gefertigt sind, waren beide Conen eine Hauptkontami-
nationsquelle für Nickel. Dies äusserte sich in einem hohen Hintergrundsignal für
Nickel.
Abbildung 5.1-2: Aufbau des Conensystems eines ICP-MS [167]
Sowohl der „Sample Cone“ als auch der „Skimmer Cone“ wurden durch Conen aus
Platin ersetzt, wodurch die Blindwerte bezüglich Nickel um den Faktor 10 reduziert
werden konnten.
Die Abbildung 5.1-3 zeigt eine Gegenüberstellung von zwei Elektroferrogrammen eines
Cytosols derselben Probe. Bei dieser Probe handelte es sich um eine normale Gewebe-
probe des humanen Darmtrakts. Die Detektion des Nickels erfolgte auf der Masse 60,
da das Hauptisotop 58 des Nickels durch starke Interferenzen (NaCl, Ar-O, Ar-O-H) ge-
stört wurde [171]. Anhand der beiden an den y-Achsen ablesbaren Blindwerte wird die
Reduzierung derselben bei Einsatz der Platinconen deutlich.
Bei Verwendung der Nickelconen war eine Nickelspeziesanalyse des Cytosols nicht
möglich, da der Untergrundwert für Nickel zu hoch war, sodass mögliche Nickelsignale
vom Untergrund überlagert wurden. Dagegen wurde durch den Einsatz der Platinconen
der Untergrundwert für Nickel so minimiert, dass gut aufgetrennte Peaks erkennbar
waren.
Optimierung der Analysenmethoden 36
Abbildung 5.1-3: Gegenüberstellung von zwei Elektroferrogrammen eines humanen Gewebe-
cytosols unter Einsatz verschiedener Conen; Detektion 60Ni
5.2 Optimierung der CE-Trennbedingungen mithilfe eines gemisch-
ten Metallothionein-Standards
Die Optimierung der kapillarelektrophoretischen Trennbedingungen wurde mithilfe
einer Standardmischung aus Cadmium beladenen Metallothionein (MT) der Isoform I
(MT I) und der Isoform II (MT II) durchgeführt. Die Wahl eines Standards für die Op-
timierung der Trennung fiel auf Metallothionein, da keine Nickel beladenen
Proteinstandards, sowohl auf dem amerikanischen wie auf dem europäischen Markt
kommerziell erhältlich waren. Alternativ wurde ein Standard gesucht, dessen
Eigenschaft wie Ladung, Größe und molare Masse ungefähr den im Cytosol der
Humanproben enthaltenen Nickelspezies entsprach. Die Abbildung 5.2-1 illustriert
einen Vergleich der Elektroferrogramme des ausgewählten Standards mit einem Cytosol
einer Humanprobe. Bei dieser Probe handelt es sich um das Cytosol einer malignen
Probe des Colon-transversums.
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,1
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
(Pt-C
onen
)
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
cps
Ni 6
0 / c
ps G
e 74
(Ni-C
onen
)
mit Pt-Conenmit Ni-Conen
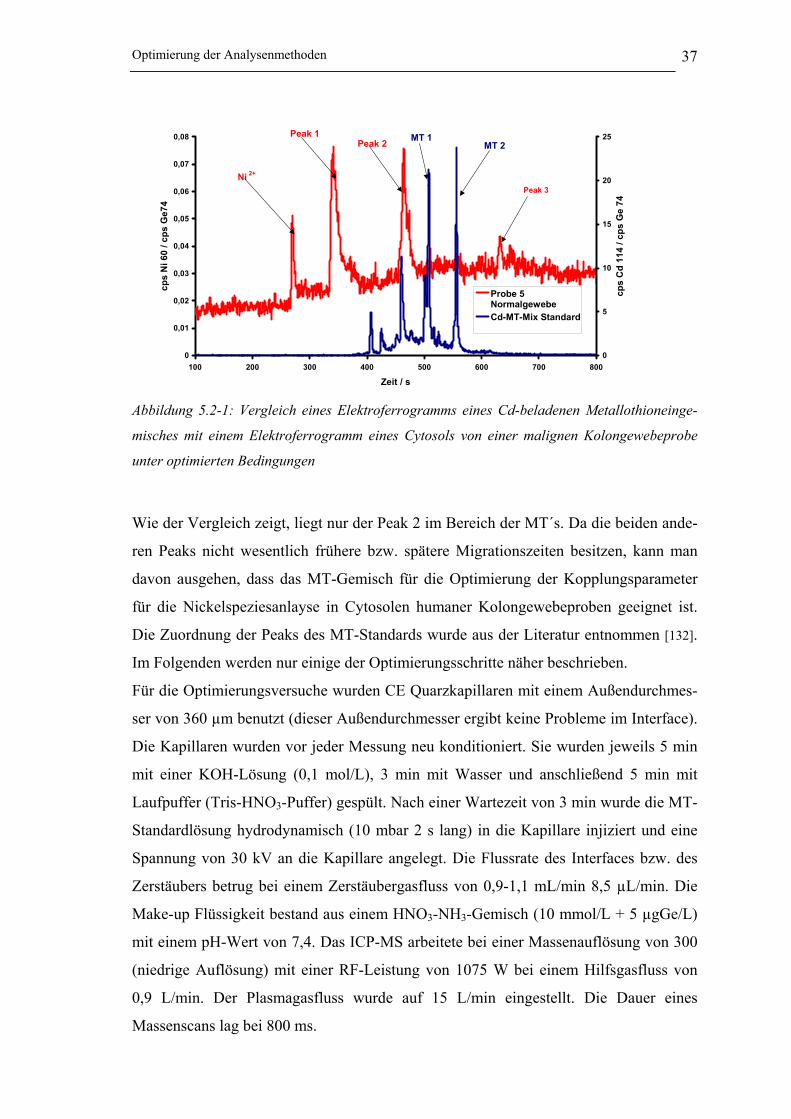
Optimierung der Analysenmethoden 37
Abbildung 5.2-1: Vergleich eines Elektroferrogramms eines Cd-beladenen Metallothioneinge-
misches mit einem Elektroferrogramm eines Cytosols von einer malignen Kolongewebeprobe
unter optimierten Bedingungen
Wie der Vergleich zeigt, liegt nur der Peak 2 im Bereich der MT´s. Da die beiden ande-
ren Peaks nicht wesentlich frühere bzw. spätere Migrationszeiten besitzen, kann man
davon ausgehen, dass das MT-Gemisch für die Optimierung der Kopplungsparameter
für die Nickelspeziesanlayse in Cytosolen humaner Kolongewebeproben geeignet ist.
Die Zuordnung der Peaks des MT-Standards wurde aus der Literatur entnommen [132].
Im Folgenden werden nur einige der Optimierungsschritte näher beschrieben.
Für die Optimierungsversuche wurden CE Quarzkapillaren mit einem Außendurchmes-
ser von 360 µm benutzt (dieser Außendurchmesser ergibt keine Probleme im Interface).
Die Kapillaren wurden vor jeder Messung neu konditioniert. Sie wurden jeweils 5 min
mit einer KOH-Lösung (0,1 mol/L), 3 min mit Wasser und anschließend 5 min mit
Laufpuffer (Tris-HNO3-Puffer) gespült. Nach einer Wartezeit von 3 min wurde die MT-
Standardlösung hydrodynamisch (10 mbar 2 s lang) in die Kapillare injiziert und eine
Spannung von 30 kV an die Kapillare angelegt. Die Flussrate des Interfaces bzw. des
Zerstäubers betrug bei einem Zerstäubergasfluss von 0,9-1,1 mL/min 8,5 µL/min. Die
Make-up Flüssigkeit bestand aus einem HNO3-NH3-Gemisch (10 mmol/L + 5 µgGe/L)
mit einem pH-Wert von 7,4. Das ICP-MS arbeitete bei einer Massenauflösung von 300
(niedrige Auflösung) mit einer RF-Leistung von 1075 W bei einem Hilfsgasfluss von
0,9 L/min. Der Plasmagasfluss wurde auf 15 L/min eingestellt. Die Dauer eines
Massenscans lag bei 800 ms.
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
100 200 300 400 500 600 700 800
Zeit / s
cps
Ni 6
0 / c
ps G
e74
0
5
10
15
20
25
cps
Cd
114
/ cps
Ge
74
Probe 5NormalgewebeCd-MT-Mix Standard
MT 1MT 2
Peak 1Peak 2
Peak 3Ni 2+
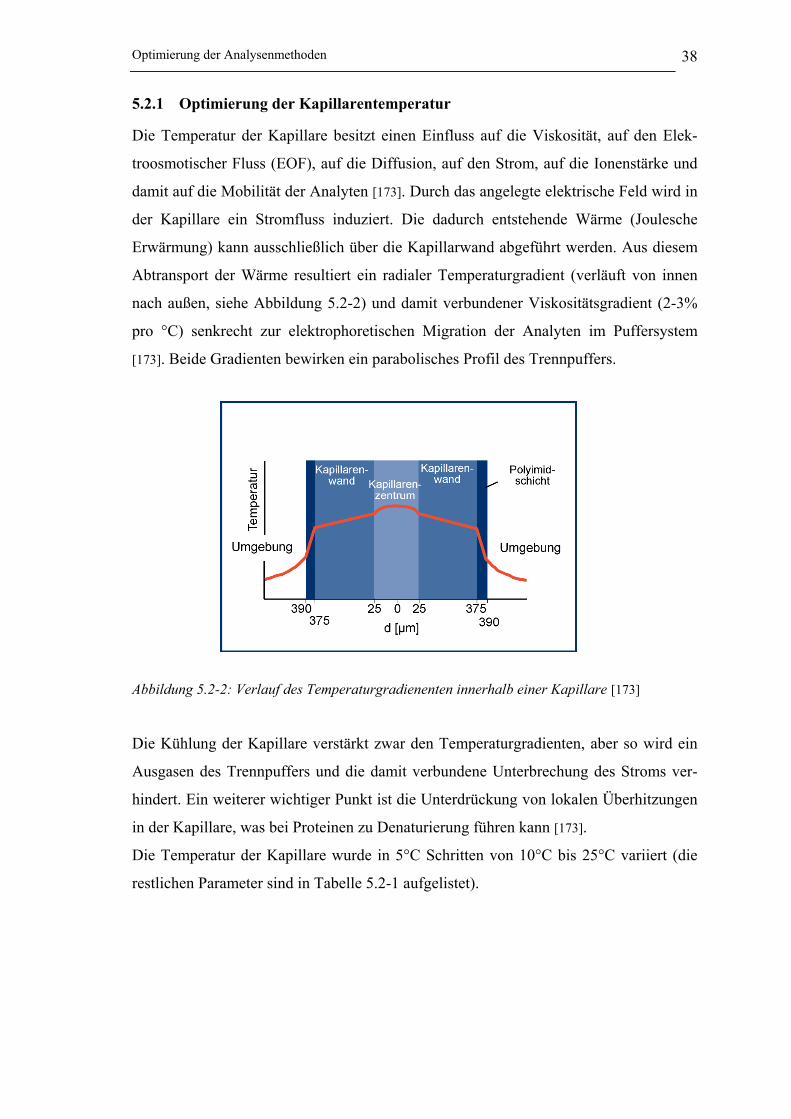
Optimierung der Analysenmethoden 38
5.2.1 Optimierung der Kapillarentemperatur
Die Temperatur der Kapillare besitzt einen Einfluss auf die Viskosität, auf den Elek-
troosmotischer Fluss (EOF), auf die Diffusion, auf den Strom, auf die Ionenstärke und
damit auf die Mobilität der Analyten [173]. Durch das angelegte elektrische Feld wird in
der Kapillare ein Stromfluss induziert. Die dadurch entstehende Wärme (Joulesche
Erwärmung) kann ausschließlich über die Kapillarwand abgeführt werden. Aus diesem
Abtransport der Wärme resultiert ein radialer Temperaturgradient (verläuft von innen
nach außen, siehe Abbildung 5.2-2) und damit verbundener Viskositätsgradient (2-3%
pro °C) senkrecht zur elektrophoretischen Migration der Analyten im Puffersystem
[173]. Beide Gradienten bewirken ein parabolisches Profil des Trennpuffers.
Abbildung 5.2-2: Verlauf des Temperaturgradienenten innerhalb einer Kapillare [173]
Die Kühlung der Kapillare verstärkt zwar den Temperaturgradienten, aber so wird ein
Ausgasen des Trennpuffers und die damit verbundene Unterbrechung des Stroms ver-
hindert. Ein weiterer wichtiger Punkt ist die Unterdrückung von lokalen Überhitzungen
in der Kapillare, was bei Proteinen zu Denaturierung führen kann [173].
Die Temperatur der Kapillare wurde in 5°C Schritten von 10°C bis 25°C variiert (die
restlichen Parameter sind in Tabelle 5.2-1 aufgelistet).

Optimierung der Analysenmethoden 39
Tabelle 5.2-1: Gewählte Messparameter für die Optimierung der Kapillarentemperatur
Parameter Einstellungen
Kapillare Quarz, Länge: 70 cm
Innendurchmesser: 75 µm
Puffer Tris-HNO3-Puffer; pH-Wert: 7,4;
Konzentration: 20 mmol/L
Angelegte Spannung 30 kV
Probenaufgabe 20 mbar×s
Bei einer Temperatur von 15°C wurde die beste Trennung des MT-Gemisches erzielt.
Bei dieser Temperatur war das aus Abbildung 5.2-1 schon bekannte
Aufspaltungsmuster des Standards erkennbar. Die entstehende Joulesche Wärme wurde
bei dieser Einstellung gut abgeführt und die Effizienzverluste der Trennung durch
Temperatureffekte innerhalb der Kapillare konnten als sehr gering eingestuft werden.
Bei niedrigeren Temperaturen nahm die Viskosität des Puffers zu und gleichzeitig sank
auch der EOF (elektroosmotischer Fluss). Eine direkte Folge war eine Verlängerung der
Retentionszeiten und eine Verringerung des Injektionsvolumens, was sich in der
geringeren Signalhöhe deutlich zeigte. Ab einer Temperatur von 20°C wurde der
Stromverlauf zunehmend instabiler und der Puffer begann auszugasen, was sich im
Zusammenbruch des Stromes äußerte.
Die optimale Temperatur der Kapillare unter den beschriebenen Bedingungen lag bei
15°C und wurde bei den weiteren Optimierungsschritten und den späteren Messun-
gen beibehalten.
5.2.2 Optimierung der Kapillarenlänge
Die Kapillarenlänge beeinflusst neben der Temperatur der Kapillare und dem
Injektionsvolumen auch den Strom. Der Strom nimmt mit zunehmender Länge der
Kapillare ab [173]. Zum Einsatz kamen Kapillaren mit verschiedenen Längen (55 cm, 70
cm und 100 cm). Kürzere Kapillaren als 55 cm konnten bei der eingesetzten CE / ICP-
MS Kopplung nicht verwendet werden, da der Abstand von der Kathode zum Interface
Mindestlänge verlangt [130].
Beim Einsatz der Kapillarlängen mit einer Länge von 55 bzw. 70 cm konnte eine ver-
gleichbar gute Auftrennung der MT-Isoformen erzielt werden, wobei die Retentions

Optimierung der Analysenmethoden 40
zeiten beim Einsatz der 55 cm Kapillare signifikant kürzer waren als bei den beiden
anderen Längen (70 bzw. 100 cm). Ein Problem bei der Verwendung kürzerer Kapilla-
ren lag in der Zunahme der Stromstärke. Als Folge des erhöhten Stromes konnte eine
Temperaturerhöhung innerhalb der Kapillare beobachtet werden, was sich wiederum
auf die Stabilität des Stromes negativ auswirkte (Ausgasen des Puffers) [173]. Ferner
konnte häufig ein Stromzusammenbruch bei der Verwendung der 55 cm Kapillare regi-
striert werden. Bei Einsatz der 100 cm Kapillare war die Peakverbreiterung nachteilig,
die sich negativ auf die Auflösung auswirkte.
Daher wurde im weiteren Verlauf eine 70 cm Kapillare für die elektrophoretische
Trennung der Cytosolspezies eingesetzt.
5.2.3 Optimierung des Innendurchmessers der Kapillare
Bei zunehmendem Innendurchmesser der Kapillare steigt neben Temperatur und EOF
auch die Stromstärke bei konstanter Spannung an. Die Folgen einer solchen Zunahme
wurden bereits unter Punkt 5.2.1 und 5.2.2 beschrieben.
Für diesen Optimierungsschritt wurden zwei verschiedene Innendurchmesser der Ka-
pillare (50 µm und 75 µm) gewählt. Die Verwendung der Kapillare mit einem Innen-
durchmesser von 50 µm wirkte sich positiv auf den Abtransport der Jouleschen Wärme
aus. Nachteilig wirkte sich die geringe Langzeitstabilität aus. Vor allem häufiges Kon-
ditionieren der Kapillare führte zu deren Bruch. Obwohl der Wärmeabtransport bei der
Verwendung der Kapillare mit einem Innendurchmesser von 75 µm geringer war,
konnte eine ausreichende Trennung des MT-MIX Standards (MT I und MT II) erreicht
werden. Diese war vergleichbar mit der Auftrennung, die mit einer Kapillare mit einem
Innendurchmesser von 50 µm erzielt wurde.
Aus diesen Gründen wurde im weiteren Verlauf der Arbeit eine Kapillare mit einem
Innendurchmesser von 75 µm eingesetzt.
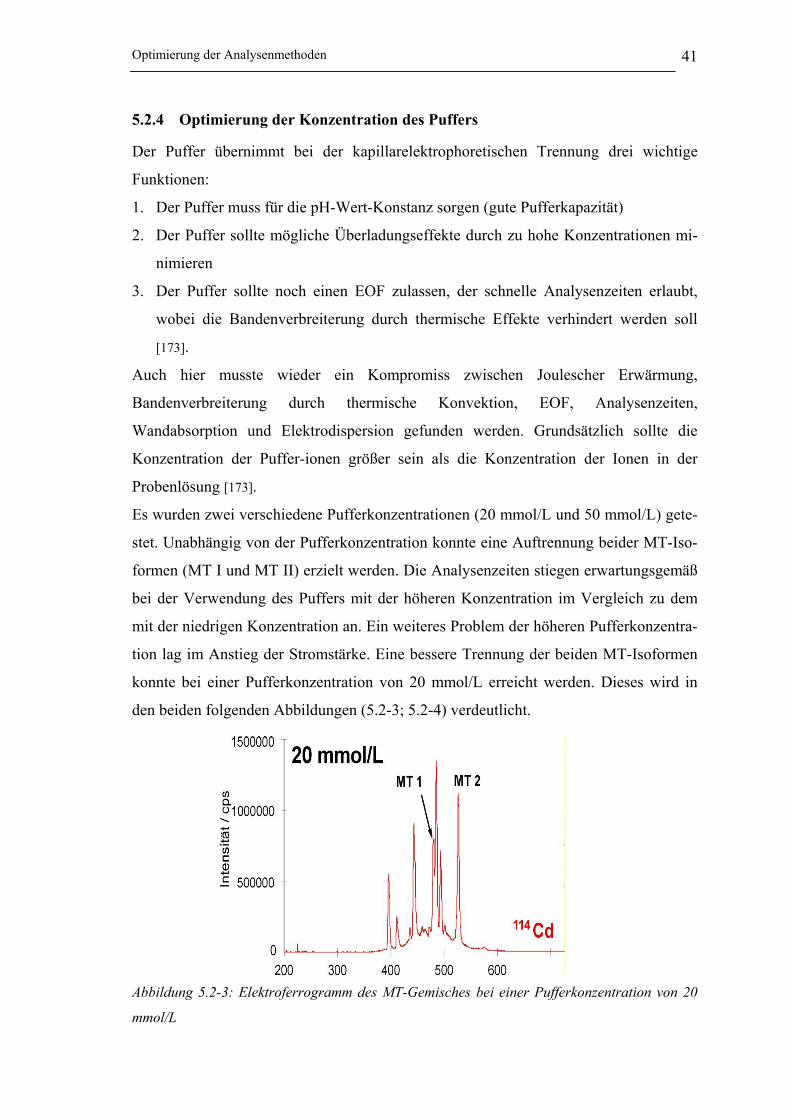
Optimierung der Analysenmethoden 41
5.2.4 Optimierung der Konzentration des Puffers
Der Puffer übernimmt bei der kapillarelektrophoretischen Trennung drei wichtige
Funktionen:
1. Der Puffer muss für die pH-Wert-Konstanz sorgen (gute Pufferkapazität)
2. Der Puffer sollte mögliche Überladungseffekte durch zu hohe Konzentrationen mi-
nimieren
3. Der Puffer sollte noch einen EOF zulassen, der schnelle Analysenzeiten erlaubt,
wobei die Bandenverbreiterung durch thermische Effekte verhindert werden soll
[173].
Auch hier musste wieder ein Kompromiss zwischen Joulescher Erwärmung,
Bandenverbreiterung durch thermische Konvektion, EOF, Analysenzeiten,
Wandabsorption und Elektrodispersion gefunden werden. Grundsätzlich sollte die
Konzentration der Puffer-ionen größer sein als die Konzentration der Ionen in der
Probenlösung [173].
Es wurden zwei verschiedene Pufferkonzentrationen (20 mmol/L und 50 mmol/L) gete-
stet. Unabhängig von der Pufferkonzentration konnte eine Auftrennung beider MT-Iso-
formen (MT I und MT II) erzielt werden. Die Analysenzeiten stiegen erwartungsgemäß
bei der Verwendung des Puffers mit der höheren Konzentration im Vergleich zu dem
mit der niedrigen Konzentration an. Ein weiteres Problem der höheren Pufferkonzentra-
tion lag im Anstieg der Stromstärke. Eine bessere Trennung der beiden MT-Isoformen
konnte bei einer Pufferkonzentration von 20 mmol/L erreicht werden. Dieses wird in
den beiden folgenden Abbildungen (5.2-3; 5.2-4) verdeutlicht.
Abbildung 5.2-3: Elektroferrogramm des MT-Gemisches bei einer Pufferkonzentration von 20
mmol/L
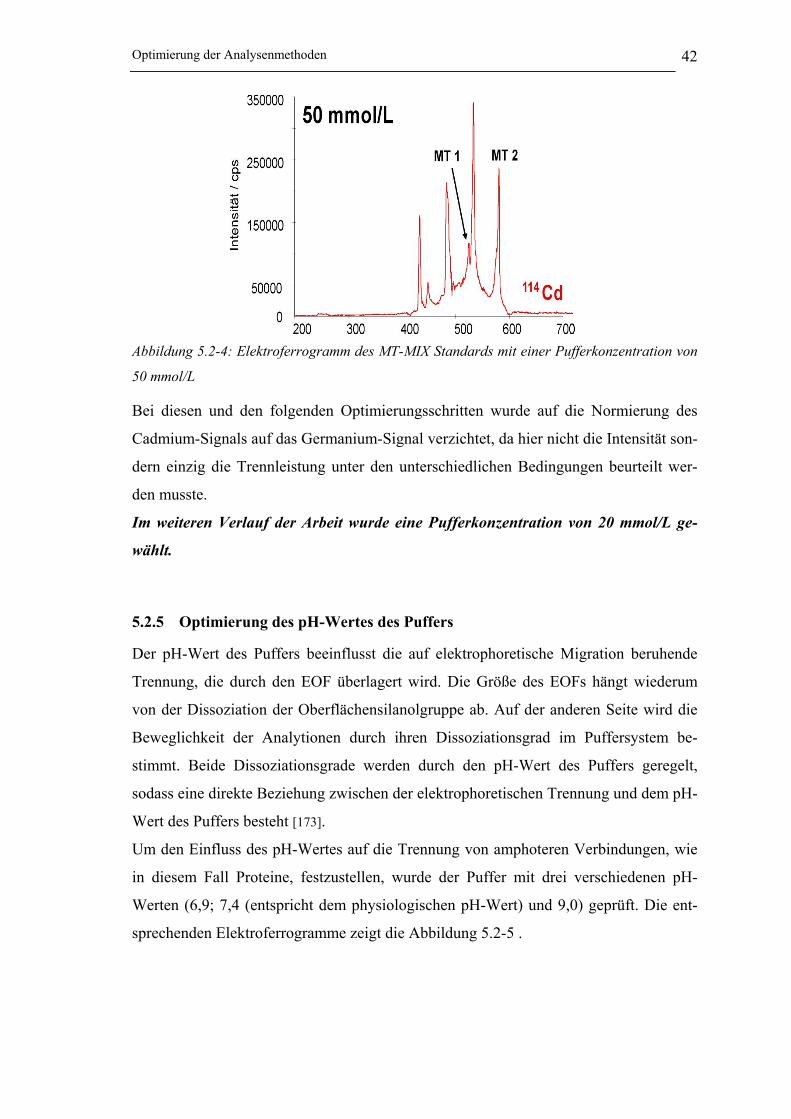
Optimierung der Analysenmethoden 42
Abbildung 5.2-4: Elektroferrogramm des MT-MIX Standards mit einer Pufferkonzentration von
50 mmol/L
Bei diesen und den folgenden Optimierungsschritten wurde auf die Normierung des
Cadmium-Signals auf das Germanium-Signal verzichtet, da hier nicht die Intensität son-
dern einzig die Trennleistung unter den unterschiedlichen Bedingungen beurteilt wer-
den musste.
Im weiteren Verlauf der Arbeit wurde eine Pufferkonzentration von 20 mmol/L ge-
wählt.
5.2.5 Optimierung des pH-Wertes des Puffers
Der pH-Wert des Puffers beeinflusst die auf elektrophoretische Migration beruhende
Trennung, die durch den EOF überlagert wird. Die Größe des EOFs hängt wiederum
von der Dissoziation der Oberflächensilanolgruppe ab. Auf der anderen Seite wird die
Beweglichkeit der Analytionen durch ihren Dissoziationsgrad im Puffersystem be-
stimmt. Beide Dissoziationsgrade werden durch den pH-Wert des Puffers geregelt,
sodass eine direkte Beziehung zwischen der elektrophoretischen Trennung und dem pH-
Wert des Puffers besteht [173].
Um den Einfluss des pH-Wertes auf die Trennung von amphoteren Verbindungen, wie
in diesem Fall Proteine, festzustellen, wurde der Puffer mit drei verschiedenen pH-
Werten (6,9; 7,4 (entspricht dem physiologischen pH-Wert) und 9,0) geprüft. Die ent-
sprechenden Elektroferrogramme zeigt die Abbildung 5.2-5 .
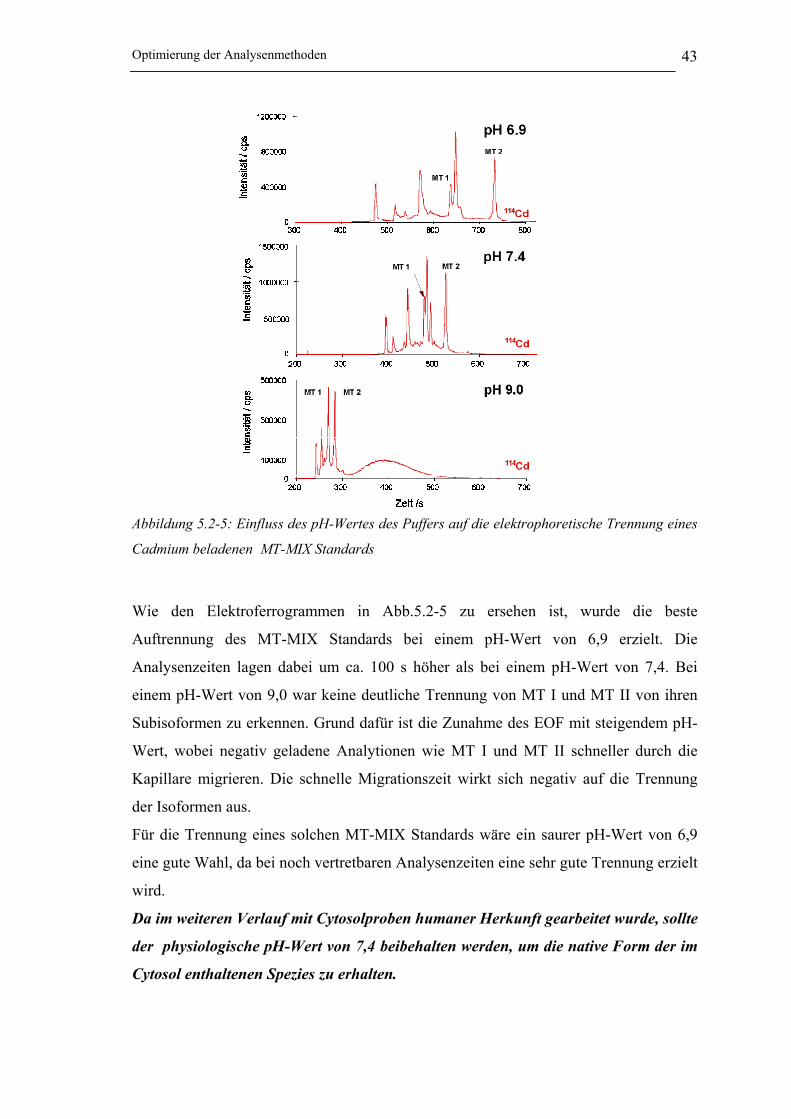
Optimierung der Analysenmethoden 43
Abbildung 5.2-5: Einfluss des pH-Wertes des Puffers auf die elektrophoretische Trennung eines
Cadmium beladenen MT-MIX Standards
Wie den Elektroferrogrammen in Abb.5.2-5 zu ersehen ist, wurde die beste
Auftrennung des MT-MIX Standards bei einem pH-Wert von 6,9 erzielt. Die
Analysenzeiten lagen dabei um ca. 100 s höher als bei einem pH-Wert von 7,4. Bei
einem pH-Wert von 9,0 war keine deutliche Trennung von MT I und MT II von ihren
Subisoformen zu erkennen. Grund dafür ist die Zunahme des EOF mit steigendem pH-
Wert, wobei negativ geladene Analytionen wie MT I und MT II schneller durch die
Kapillare migrieren. Die schnelle Migrationszeit wirkt sich negativ auf die Trennung
der Isoformen aus.
Für die Trennung eines solchen MT-MIX Standards wäre ein saurer pH-Wert von 6,9
eine gute Wahl, da bei noch vertretbaren Analysenzeiten eine sehr gute Trennung erzielt
wird.
Da im weiteren Verlauf mit Cytosolproben humaner Herkunft gearbeitet wurde, sollte
der physiologische pH-Wert von 7,4 beibehalten werden, um die native Form der im
Cytosol enthaltenen Spezies zu erhalten.
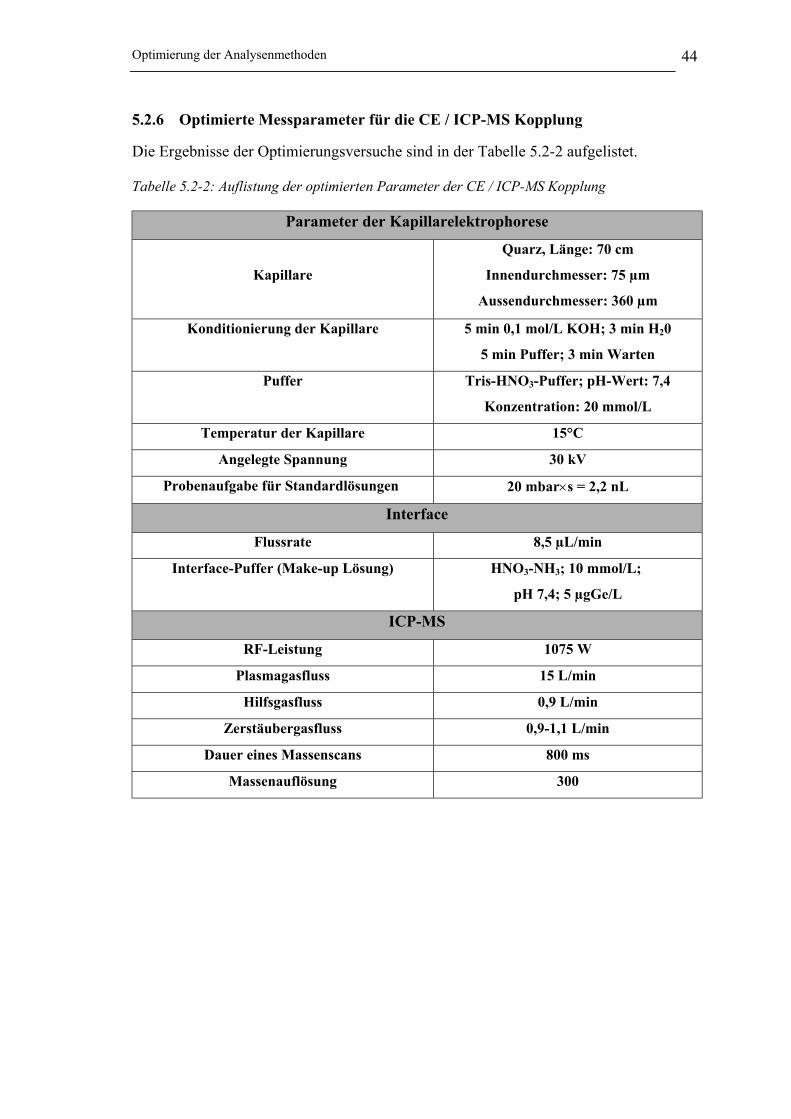
Optimierung der Analysenmethoden 44
5.2.6 Optimierte Messparameter für die CE / ICP-MS Kopplung
Die Ergebnisse der Optimierungsversuche sind in der Tabelle 5.2-2 aufgelistet.
Tabelle 5.2-2: Auflistung der optimierten Parameter der CE / ICP-MS Kopplung
Parameter der Kapillarelektrophorese
Kapillare
Quarz, Länge: 70 cm
Innendurchmesser: 75 µm
Aussendurchmesser: 360 µm
Konditionierung der Kapillare 5 min 0,1 mol/L KOH; 3 min H20
5 min Puffer; 3 min Warten
Puffer Tris-HNO3-Puffer; pH-Wert: 7,4
Konzentration: 20 mmol/L
Temperatur der Kapillare 15°C
Angelegte Spannung 30 kV
Probenaufgabe für Standardlösungen 20 mbar×s = 2,2 nL
Interface
Flussrate 8,5 µL/min
Interface-Puffer (Make-up Lösung) HNO3-NH3; 10 mmol/L;
pH 7,4; 5 µgGe/L
ICP-MS
RF-Leistung 1075 W
Plasmagasfluss 15 L/min
Hilfsgasfluss 0,9 L/min
Zerstäubergasfluss 0,9-1,1 L/min
Dauer eines Massenscans 800 ms
Massenauflösung 300
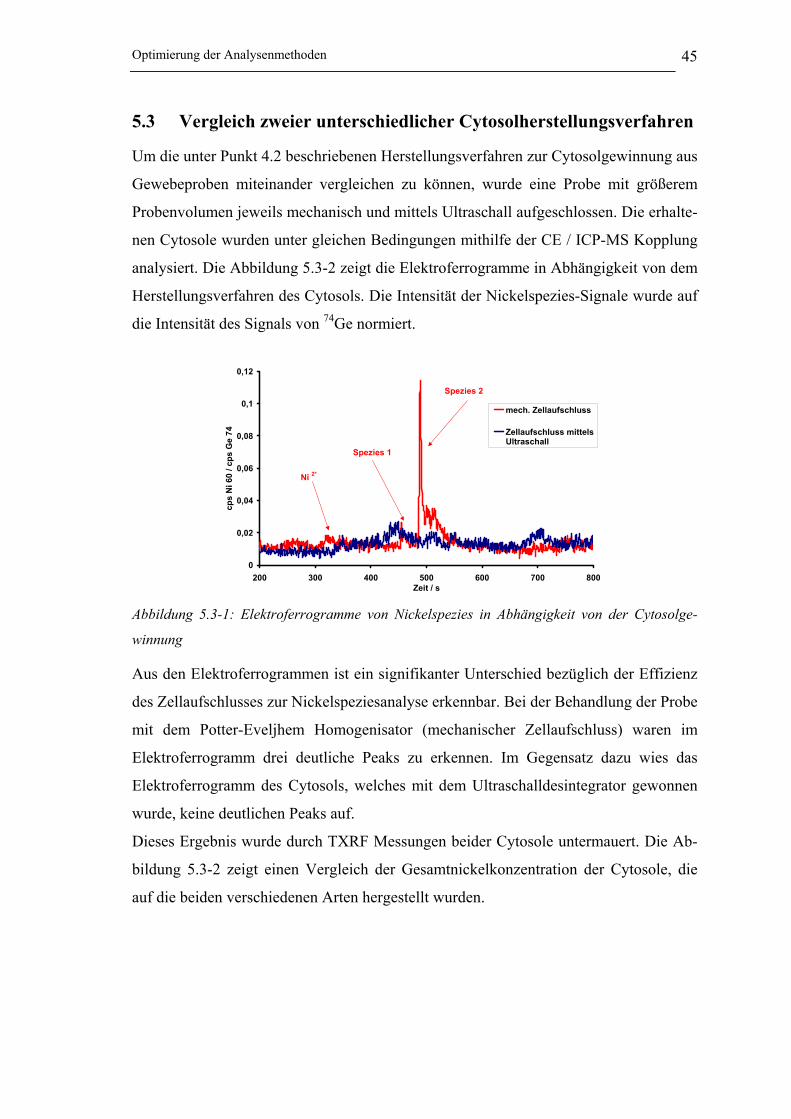
Optimierung der Analysenmethoden 45
5.3 Vergleich zweier unterschiedlicher Cytosolherstellungsverfahren
Um die unter Punkt 4.2 beschriebenen Herstellungsverfahren zur Cytosolgewinnung aus
Gewebeproben miteinander vergleichen zu können, wurde eine Probe mit größerem
Probenvolumen jeweils mechanisch und mittels Ultraschall aufgeschlossen. Die erhalte-
nen Cytosole wurden unter gleichen Bedingungen mithilfe der CE / ICP-MS Kopplung
analysiert. Die Abbildung 5.3-2 zeigt die Elektroferrogramme in Abhängigkeit von dem
Herstellungsverfahren des Cytosols. Die Intensität der Nickelspezies-Signale wurde auf
die Intensität des Signals von 74Ge normiert.
Abbildung 5.3-1: Elektroferrogramme von Nickelspezies in Abhängigkeit von der Cytosolge-
winnung
Aus den Elektroferrogrammen ist ein signifikanter Unterschied bezüglich der Effizienz
des Zellaufschlusses zur Nickelspeziesanalyse erkennbar. Bei der Behandlung der Probe
mit dem Potter-Eveljhem Homogenisator (mechanischer Zellaufschluss) waren im
Elektroferrogramm drei deutliche Peaks zu erkennen. Im Gegensatz dazu wies das
Elektroferrogramm des Cytosols, welches mit dem Ultraschalldesintegrator gewonnen
wurde, keine deutlichen Peaks auf.
Dieses Ergebnis wurde durch TXRF Messungen beider Cytosole untermauert. Die Ab-
bildung 5.3-2 zeigt einen Vergleich der Gesamtnickelkonzentration der Cytosole, die
auf die beiden verschiedenen Arten hergestellt wurden.
0
0,02
0,04
0,06
0,08
0,1
0,12
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
mech. Zellaufschluss
Zellaufschluss mittelsUltraschall
Ni 2*
Spezies 1
Spezies 2
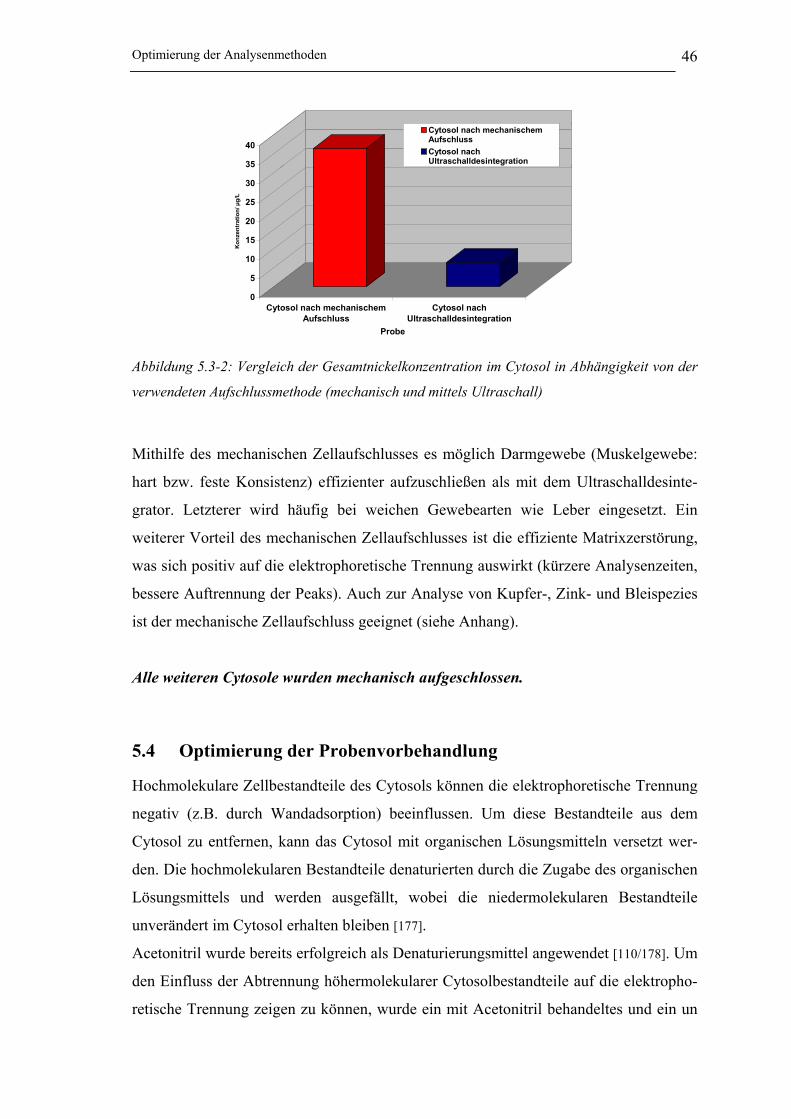
Optimierung der Analysenmethoden 46
Abbildung 5.3-2: Vergleich der Gesamtnickelkonzentration im Cytosol in Abhängigkeit von der
verwendeten Aufschlussmethode (mechanisch und mittels Ultraschall)
Mithilfe des mechanischen Zellaufschlusses es möglich Darmgewebe (Muskelgewebe:
hart bzw. feste Konsistenz) effizienter aufzuschließen als mit dem Ultraschalldesinte-
grator. Letzterer wird häufig bei weichen Gewebearten wie Leber eingesetzt. Ein
weiterer Vorteil des mechanischen Zellaufschlusses ist die effiziente Matrixzerstörung,
was sich positiv auf die elektrophoretische Trennung auswirkt (kürzere Analysenzeiten,
bessere Auftrennung der Peaks). Auch zur Analyse von Kupfer-, Zink- und Bleispezies
ist der mechanische Zellaufschluss geeignet (siehe Anhang).
Alle weiteren Cytosole wurden mechanisch aufgeschlossen.
5.4 Optimierung der Probenvorbehandlung
Hochmolekulare Zellbestandteile des Cytosols können die elektrophoretische Trennung
negativ (z.B. durch Wandadsorption) beeinflussen. Um diese Bestandteile aus dem
Cytosol zu entfernen, kann das Cytosol mit organischen Lösungsmitteln versetzt wer-
den. Die hochmolekularen Bestandteile denaturierten durch die Zugabe des organischen
Lösungsmittels und werden ausgefällt, wobei die niedermolekularen Bestandteile
unverändert im Cytosol erhalten bleiben [177].
Acetonitril wurde bereits erfolgreich als Denaturierungsmittel angewendet [110/178]. Um
den Einfluss der Abtrennung höhermolekularer Cytosolbestandteile auf die elektropho-
retische Trennung zeigen zu können, wurde ein mit Acetonitril behandeltes und ein un
0
5
10
15
20
25
30
35
40
Kon
zent
ratio
n/ µ
g/L
Cytosol nach mechanischemAufschluss
Cytosol nachUltraschalldesintegration
Probe
Cytosol nach mechanischemAufschlussCytosol nachUltraschalldesintegration
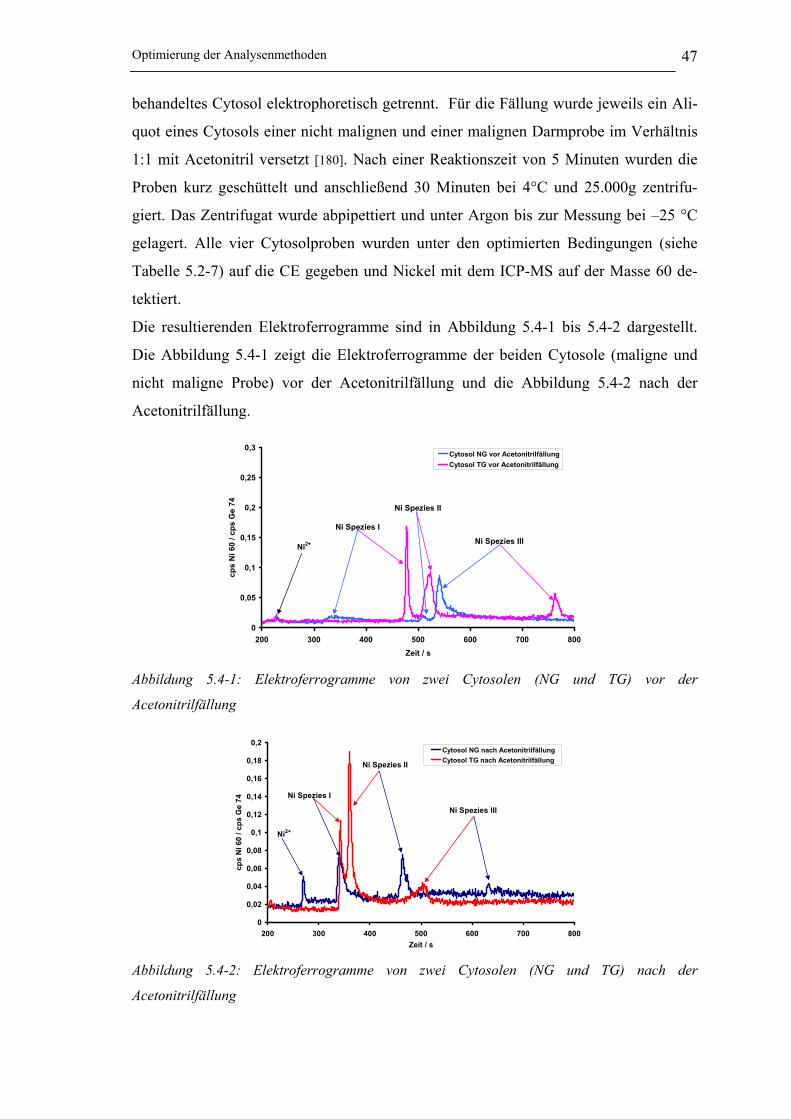
Optimierung der Analysenmethoden 47
behandeltes Cytosol elektrophoretisch getrennt. Für die Fällung wurde jeweils ein Ali-
quot eines Cytosols einer nicht malignen und einer malignen Darmprobe im Verhältnis
1:1 mit Acetonitril versetzt [180]. Nach einer Reaktionszeit von 5 Minuten wurden die
Proben kurz geschüttelt und anschließend 30 Minuten bei 4°C und 25.000g zentrifu-
giert. Das Zentrifugat wurde abpipettiert und unter Argon bis zur Messung bei –25 °C
gelagert. Alle vier Cytosolproben wurden unter den optimierten Bedingungen (siehe
Tabelle 5.2-7) auf die CE gegeben und Nickel mit dem ICP-MS auf der Masse 60 de-
tektiert.
Die resultierenden Elektroferrogramme sind in Abbildung 5.4-1 bis 5.4-2 dargestellt.
Die Abbildung 5.4-1 zeigt die Elektroferrogramme der beiden Cytosole (maligne und
nicht maligne Probe) vor der Acetonitrilfällung und die Abbildung 5.4-2 nach der
Acetonitrilfällung.
Abbildung 5.4-1: Elektroferrogramme von zwei Cytosolen (NG und TG) vor der
Acetonitrilfällung
Abbildung 5.4-2: Elektroferrogramme von zwei Cytosolen (NG und TG) nach der
Acetonitrilfällung
0
0,05
0,1
0,15
0,2
0,25
0,3
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
Cytosol NG vor AcetonitrilfällungCytosol TG vor Acetonitrilfällung
Ni2+
Ni Spezies I
Ni Spezies II
Ni Spezies III
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
Cytosol NG nach AcetonitrilfällungCytosol TG nach Acetonitrilfällung
Ni2+
Ni Spezies I
Ni Spezies II
Ni Spezies III

Optimierung der Analysenmethoden 48
In den Abbildungen 5.4-1 bis 5.4-2 ist erkennbar, dass durch die Acetonitrilfällung eine
signifikante Verkürzung der Analysenzeiten erreicht werden kann. Aufgrund der
Matrixabtrennung und der damit verbundenen geringen Wandadsorptionseffekte können
auch die Halbwertsbreiten der Peaks verringert werden [178/182]. Diese Effekte können
vor allem bei der Trennung der Nickelspezies im Cytosol der normalen Probe
beobachtet werden. Hingegen wird die Trennung der Nickelspezies im Cytosol der
malignen Probe eher negativ beeinflusst (schlechtere Auftrennung). Es kann trotzdem
eine klare Trennung der drei Spezies erreicht werden.
Aus den Abbildungen 5.4-1 und 5.4-2 kann aber nicht der Hauptvorteil der Acetonitril-
fällung abgeleitet werden. Dieser liegt in der Reduktion der Leitfähigkeit durch die
Matrixabtrennung, was sich positiv auf die Stromstärke und damit auf die Joulesche
Erwärmung auswirkt. Ein weiterer Vorteil der Fällung ist die Reduzierung der
Viskosität des Cytosols. Die Messungen der beiden Cytosole ohne Vorbehandlung
mussten aufgrund von Stromzusammenbrüchen oder Zerstäuberverstopfungen häufig
abgebrochen werden.
Um sicher zu gehen, dass nur die hochmolekularen Bestandteile des Cytosols denaturie-
ren [110/178], wurde die SEC / ICP-MS Kopplung (Optimierung siehe Punkt 5.5) zur
Verifizierung der Ergebnisse eingesetzt. Hierzu wurde ein Aliquot eines Cytosols un-
behandelt und eins nach der Acetonitrilfällung auf die SEC-Säule gegeben und Nickel
mithilfe des ICP-MS elementspezifisch detektiert. Um eine Vergleichbarkeit der Chro-
matogramme zu gewährleisten, wurde das unbehandelte Cytosol nochmals 1:1 mit
Puffer verdünnt, damit keine Verdünnungsunterschiede zwischen unbehandeltem Cyto-
sol und Cytosol nach der Acetonitrilfällung bestand. In der nachfolgenden Abbildung
5.4-3 sind die SEC-Chromatogramme gegenübergestellt.
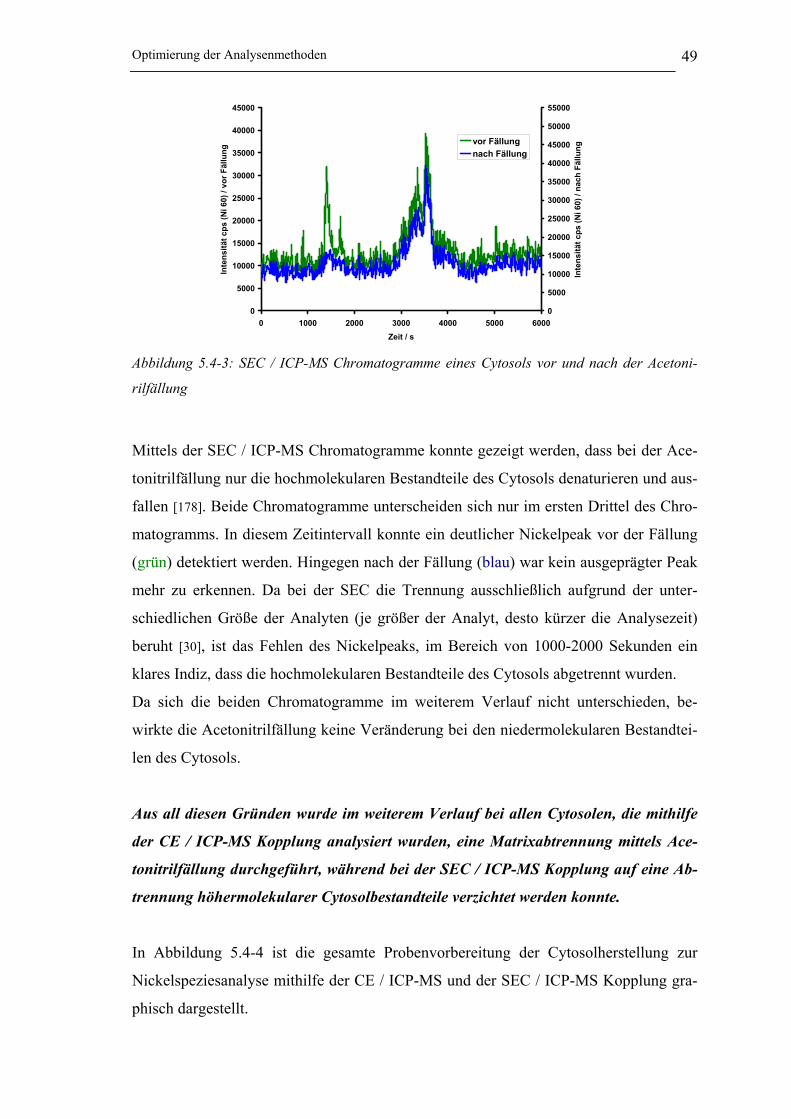
Optimierung der Analysenmethoden 49
Abbildung 5.4-3: SEC / ICP-MS Chromatogramme eines Cytosols vor und nach der Acetoni-
rilfällung
Mittels der SEC / ICP-MS Chromatogramme konnte gezeigt werden, dass bei der Ace-
tonitrilfällung nur die hochmolekularen Bestandteile des Cytosols denaturieren und aus-
fallen [178]. Beide Chromatogramme unterscheiden sich nur im ersten Drittel des Chro-
matogramms. In diesem Zeitintervall konnte ein deutlicher Nickelpeak vor der Fällung
(grün) detektiert werden. Hingegen nach der Fällung (blau) war kein ausgeprägter Peak
mehr zu erkennen. Da bei der SEC die Trennung ausschließlich aufgrund der unter-
schiedlichen Größe der Analyten (je größer der Analyt, desto kürzer die Analysezeit)
beruht [30], ist das Fehlen des Nickelpeaks, im Bereich von 1000-2000 Sekunden ein
klares Indiz, dass die hochmolekularen Bestandteile des Cytosols abgetrennt wurden.
Da sich die beiden Chromatogramme im weiterem Verlauf nicht unterschieden, be-
wirkte die Acetonitrilfällung keine Veränderung bei den niedermolekularen Bestandtei-
len des Cytosols.
Aus all diesen Gründen wurde im weiterem Verlauf bei allen Cytosolen, die mithilfe
der CE / ICP-MS Kopplung analysiert wurden, eine Matrixabtrennung mittels Ace-
tonitrilfällung durchgeführt, während bei der SEC / ICP-MS Kopplung auf eine Ab-
trennung höhermolekularer Cytosolbestandteile verzichtet werden konnte.
In Abbildung 5.4-4 ist die gesamte Probenvorbereitung der Cytosolherstellung zur
Nickelspeziesanalyse mithilfe der CE / ICP-MS und der SEC / ICP-MS Kopplung gra-
phisch dargestellt.
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t cps
(Ni 6
0) /
vor F
ällu
ng
0
5000
10000
15000
20000
25000
30000
35000
40000
45000
50000
55000
Inte
nsitä
t cps
(Ni 6
0) /
nach
Fäl
lung
vor Fällungnach Fällung
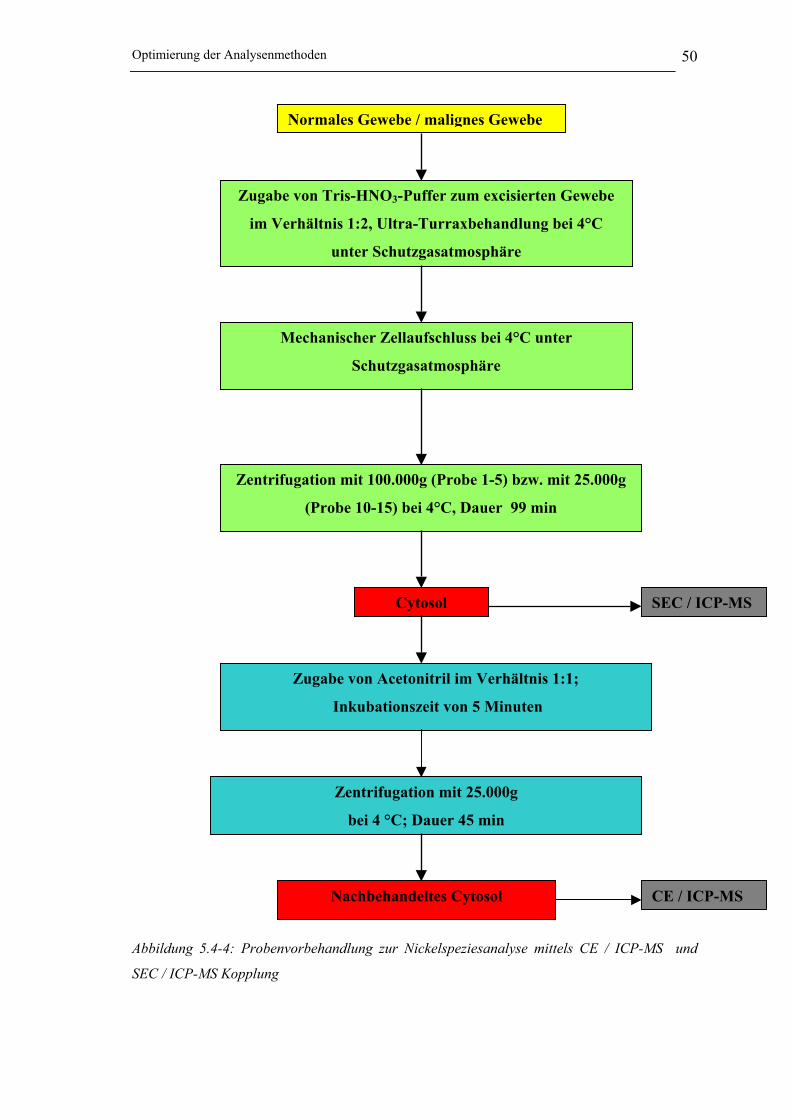
Optimierung der Analysenmethoden 50
Abbildung 5.4-4: Probenvorbehandlung zur Nickelspeziesanalyse mittels CE / ICP-MS und
SEC / ICP-MS Kopplung
Normales Gewebe / malignes Gewebe
Zugabe von Tris-HNO3-Puffer zum excisierten Gewebe
im Verhältnis 1:2, Ultra-Turraxbehandlung bei 4°C
unter Schutzgasatmosphäre
Mechanischer Zellaufschluss bei 4°C unter
Schutzgasatmosphäre
Zentrifugation mit 100.000g (Probe 1-5) bzw. mit 25.000g
(Probe 10-15) bei 4°C, Dauer 99 min
Cytosol
Zugabe von Acetonitril im Verhältnis 1:1;
Inkubationszeit von 5 Minuten
Zentrifugation mit 25.000g
bei 4 °C; Dauer 45 min
Nachbehandeltes Cytosol
SEC / ICP-MS
CE / ICP-MS
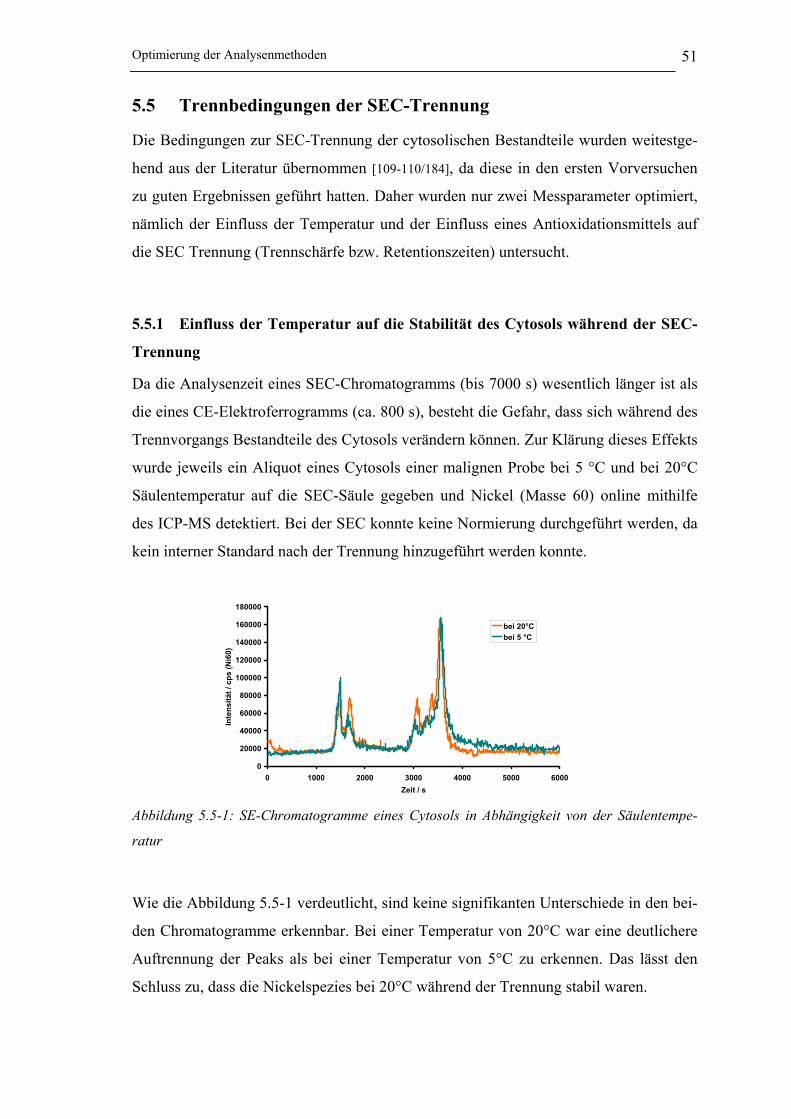
Optimierung der Analysenmethoden 51
5.5 Trennbedingungen der SEC-Trennung
Die Bedingungen zur SEC-Trennung der cytosolischen Bestandteile wurden weitestge-
hend aus der Literatur übernommen [109-110/184], da diese in den ersten Vorversuchen
zu guten Ergebnissen geführt hatten. Daher wurden nur zwei Messparameter optimiert,
nämlich der Einfluss der Temperatur und der Einfluss eines Antioxidationsmittels auf
die SEC Trennung (Trennschärfe bzw. Retentionszeiten) untersucht.
5.5.1 Einfluss der Temperatur auf die Stabilität des Cytosols während der SEC-
Trennung
Da die Analysenzeit eines SEC-Chromatogramms (bis 7000 s) wesentlich länger ist als
die eines CE-Elektroferrogramms (ca. 800 s), besteht die Gefahr, dass sich während des
Trennvorgangs Bestandteile des Cytosols verändern können. Zur Klärung dieses Effekts
wurde jeweils ein Aliquot eines Cytosols einer malignen Probe bei 5 °C und bei 20°C
Säulentemperatur auf die SEC-Säule gegeben und Nickel (Masse 60) online mithilfe
des ICP-MS detektiert. Bei der SEC konnte keine Normierung durchgeführt werden, da
kein interner Standard nach der Trennung hinzugeführt werden konnte.
Abbildung 5.5-1: SE-Chromatogramme eines Cytosols in Abhängigkeit von der Säulentempe-
ratur
Wie die Abbildung 5.5-1 verdeutlicht, sind keine signifikanten Unterschiede in den bei-
den Chromatogramme erkennbar. Bei einer Temperatur von 20°C war eine deutlichere
Auftrennung der Peaks als bei einer Temperatur von 5°C zu erkennen. Das lässt den
Schluss zu, dass die Nickelspezies bei 20°C während der Trennung stabil waren.
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t / c
ps (N
i60)
bei 20°Cbei 5 °C

Optimierung der Analysenmethoden 52
Da die Temperatur keinen Einfluss auf die Stabilität und auf die Trennung der
Nickelspezies hatte, wurden im weiteren Verlauf der Arbeit die SEC-Trennung bei
20°C durchgeführt.
Da die gesamten Arbeiten in einem klimatisierten (auf 20°C) Reinraum der Stufe 1000
durchgeführt wurden, konnte auf eine Kühlung der SEC-Säule verzichtet werden.
5.5.2 Einfluss eines Antioxidationsmittels auf die SEC-Trennung
Wie schon unter 5.5 erwähnt wurde, dauert eine SEC-Trennung bis zu 7000 Sekunden.
Aus diesem Grund stellte sich die Frage, ob alle Bestandteile eines Cytosols unter
diesen Bedingungen oxidationsstabil sind oder ob der Probe ein Antioxidationsmittel,
wie DTT (Dithiotreitol) hinzugefügt werden muss. Die Problematik ist, dass ein solches
Antioxidationsmittel auch als Komplexbildner fungieren kann und so lose gebundene
Metallionen von den Bindungspartnern entfernen kann.
Das Cytosol eines malignen Gewebes wurde folgendermaßen vorbereitet.
1. Cytosol und Tris-HNO3-Puffer ohne DTT Zusatz
2. Cytosol ohne DTT Zusatz und Laufpuffer mit DTT Zusatz (5 mmol/L)
3. Cytosol und Laufpuffer mit DTT Zusatz (5 mmol/L)
Abbildung 5.5-2 zeigt die drei erhaltenen Chromatogramme (es handelt sich hier zur
besseren Vergleichbarkeit um eine offset-Darstellung der Elektroferrogramme)
Abbildung 5.5-2: SE-Chromatogramme eines Cytosols in Abhängigkeit von der Zugabe eines
Antioxidationsmittels
0 1000 2000 3000 4000 5000 6000Zeit / s
Puffer und Probe ohne DTT
Puffer ohne / Probe mit DTT
Puffer und Probe mit DTT
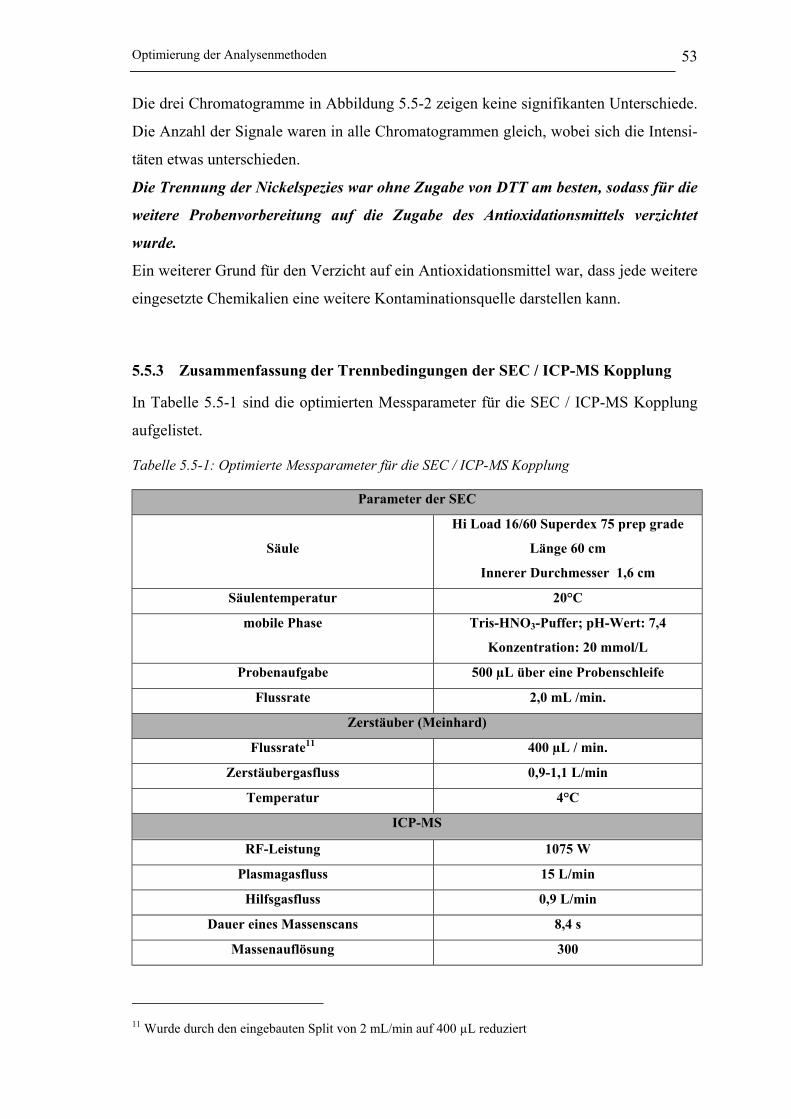
Optimierung der Analysenmethoden 53
Die drei Chromatogramme in Abbildung 5.5-2 zeigen keine signifikanten Unterschiede.
Die Anzahl der Signale waren in alle Chromatogrammen gleich, wobei sich die Intensi-
täten etwas unterschieden.
Die Trennung der Nickelspezies war ohne Zugabe von DTT am besten, sodass für die
weitere Probenvorbereitung auf die Zugabe des Antioxidationsmittels verzichtet
wurde.
Ein weiterer Grund für den Verzicht auf ein Antioxidationsmittel war, dass jede weitere
eingesetzte Chemikalien eine weitere Kontaminationsquelle darstellen kann.
5.5.3 Zusammenfassung der Trennbedingungen der SEC / ICP-MS Kopplung
In Tabelle 5.5-1 sind die optimierten Messparameter für die SEC / ICP-MS Kopplung
aufgelistet.
Tabelle 5.5-1: Optimierte Messparameter für die SEC / ICP-MS Kopplung
Parameter der SEC
Säule
Hi Load 16/60 Superdex 75 prep grade
Länge 60 cm
Innerer Durchmesser 1,6 cm
Säulentemperatur 20°C
mobile Phase Tris-HNO3-Puffer; pH-Wert: 7,4
Konzentration: 20 mmol/L
Probenaufgabe 500 µL über eine Probenschleife
Flussrate 2,0 mL /min.
Zerstäuber (Meinhard)
Flussrate11 400 µL / min.
Zerstäubergasfluss 0,9-1,1 L/min
Temperatur 4°C
ICP-MS
RF-Leistung 1075 W
Plasmagasfluss 15 L/min
Hilfsgasfluss 0,9 L/min
Dauer eines Massenscans 8,4 s
Massenauflösung 300
11 Wurde durch den eingebauten Split von 2 mL/min auf 400 µL reduziert

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 54
6 Nickelspeziesanalyse im Cytosol kolorektaler Humange-
webeproben
Im weiteren Verlauf der Arbeit werden alle durchgeführten Analysen und Speziesanaly-
sen beschrieben und die Ergebnisse der einzelnen Messungen vorgestellt und diskutiert.
Die Gesamtnickelgehalte der Cytosole wurde mithilfe der TXRF bestimmt. Neben der
Bestimmung der Gesamtproteingehalte werden die Ergebnisse der Nickelspeziesanalyse
mittels CE / ICP-MS und SEC / ICP-MS zusammengefasst und erörtert.
6.1 Bestimmung der Gesamtnickelgehalte im Cytosol mithilfe der
TXRF
Die Gesamtnickelkonzentration der Cytosole wurde sowohl vor als auch nach der Ace-
tonitrilfällung mithilfe der TXRF bestimmt (Durchführung siehe Punkt 4.3).
In Abbildung 6.1-1 und 6.1-2 sind die Ergebnisse der TXRF-Messungen für Nickel dar-
gestellt. Die Zahlen, die über den Balken zu sehen sind, geben den Wert der
gemessenen Nickelkonzentration in µg/L an (siehe Anhang: TXRF-Messungen). Die
Proben 6-9 wurden mittels TXRF untersucht, da bei diesen Proben nur Tumorproben
vorlagen (siehe Tabelle 4.1-1).
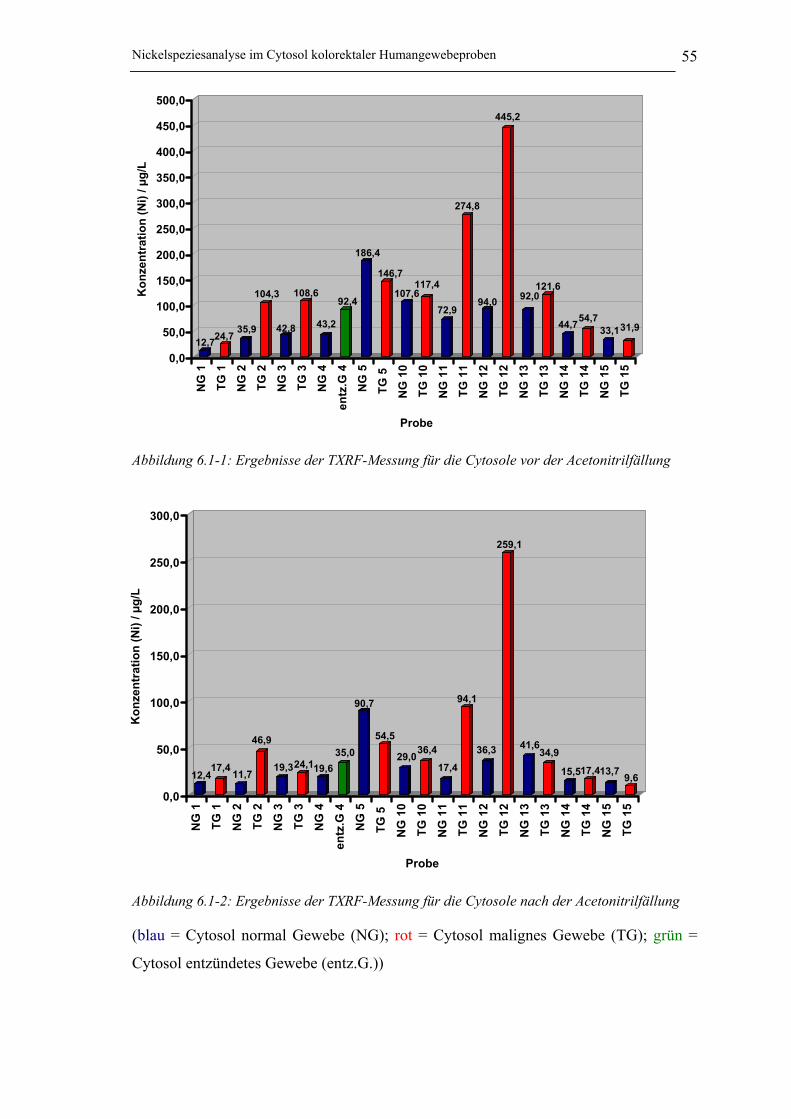
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 55
Abbildung 6.1-1: Ergebnisse der TXRF-Messung für die Cytosole vor der Acetonitrilfällung
Abbildung 6.1-2: Ergebnisse der TXRF-Messung für die Cytosole nach der Acetonitrilfällung
(blau = Cytosol normal Gewebe (NG); rot = Cytosol malignes Gewebe (TG); grün =
Cytosol entzündetes Gewebe (entz.G.))
12,724,7 35,9
104,3
42,8
108,6
43,2
92,4
186,4
146,7
107,6117,4
72,9
274,8
94,0
445,2
92,0121,6
44,754,733,131,9
0,0
50,0
100,0
150,0
200,0
250,0
300,0
350,0
400,0
450,0
500,0
Kon
zent
ratio
n (N
i) / µ
g/L
NG
1TG
1N
G 2
TG 2
NG
3TG
3N
G 4
entz
.G 4
NG
5TG
5
NG
10
TG 1
0N
G 1
1TG
11
NG
12
TG 1
2N
G 1
3TG
13
NG
14
TG 1
4N
G 1
5TG
15
Probe
12,417,4 11,7
46,9
19,324,119,635,0
90,7
54,5
29,036,417,4
94,1
36,3
259,1
41,634,9
15,517,413,7 9,6
0,0
50,0
100,0
150,0
200,0
250,0
300,0
Kon
zent
ratio
n (N
i) / µ
g/L
NG
1TG
1N
G 2
TG 2
NG
3TG
3N
G 4
entz
.G 4
NG
5TG
5
NG
10
TG 1
0N
G 1
1TG
11
NG
12
TG 1
2N
G 1
3TG
13
NG
14
TG 1
4N
G 1
5TG
15
Probe

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 56
6.1.1 Diskussion der Ergebnisse
Beim Vergleich der beiden Messreihen wird deutlich, dass die Nickelkonzentrationen
durch die Denaturierung der hochmolekularen Bestandteile aufgrund der Acetonitrilfäl-
lung zwar reduziert, aber das Verhältnis der Konzentrationen der Tumor- und Normal-
gewebeproben zueinander nicht stark verändert wurden. Dies stimmt mit den durchge-
führten elektrophoretischen und chromatographischen Messungen der Cytosole vor und
nach Acetonitrilfällung überein (siehe Kapitel 5.4 und Abb. 5.4-1 bis 2).
Als allgemeiner Trend der TXRF-Messungen lässt sich festhalten, dass die Nickelkon-
zentration in den Cytosolen der Tumorproben höher liegen als im Cytosol der Normal-
proben. Diese erhöhten Werte lassen sich durch die Störung der normalen Zellwachs-
tumskontrolle während der Karzinogenese erklären. Die Störungen führen zu einer un-
kontrollierten Steigerung der Zellvermehrung, die sowohl zu biochemischen wie auch
physikalischen Veränderungen in der Zelle führen, wodurch zelluläre und histologische
Abnormalitäten und eine Abnahme des Differenzierungsgrades der Tumorzelle bewirkt
werden. Gleichzeitig zeigen Tumorzellen einen gesteigerten Stoffwechsel, der zu einer
übersteigerten Produktion von Proteinen und Enzymen führt, sodass sich die Konzen-
tration möglicher Bindungspartner für Metalle erhöht.
Eine Ausnahme stellt die Probe 5 dar. Bei diesen Cytosolen ist die Nickelkonzentration
im Cytosol des Normalgewebes deutlich größer als in dem entsprechenden Cytosol des
malignen Gewebes. Ein möglicher Grund könnte hier das fortgeschrittene Stadium (T4)
des Tumors sein.
Bei den Proben 10, 14 und 15 sind keine großen Unterschiede in den Nickelkonzentra-
tionen zu erkennen. Ein möglicher Grund könnte auch hier das Stadium des Tumors
sein. Bei Probe 10 handelt es sich um ein Adenom, also einer Vorstufe des Karzinoms.
Das unkontrollierte Zellwachstum befindet sich im Anfangsstadium, sodass noch keine
großen Unterschiede in den Nickelkonzentrationen zu erwarten sind. Bei den beiden
anderen Proben befand sich der Tumor in einer recht frühen Phase (T2), sodass auch
hier das unkontrollierte Zellwachstum noch nicht soweit fortgeschritten war, dass große
Unterschiede in den Nickelkonzentrationen zwischen dem Cytosol des Normal- und des
Tumorgewebes zu erwarten waren. Bei Probe 14 dagegen liegt ein undefiniertes Nor-
malgewebe vor. Es könnte sich möglicherweise um adenomatöses Gewebe handeln,
wodurch der geringe Unterschied zwischen den beiden Gewebearten zu erklären wäre.
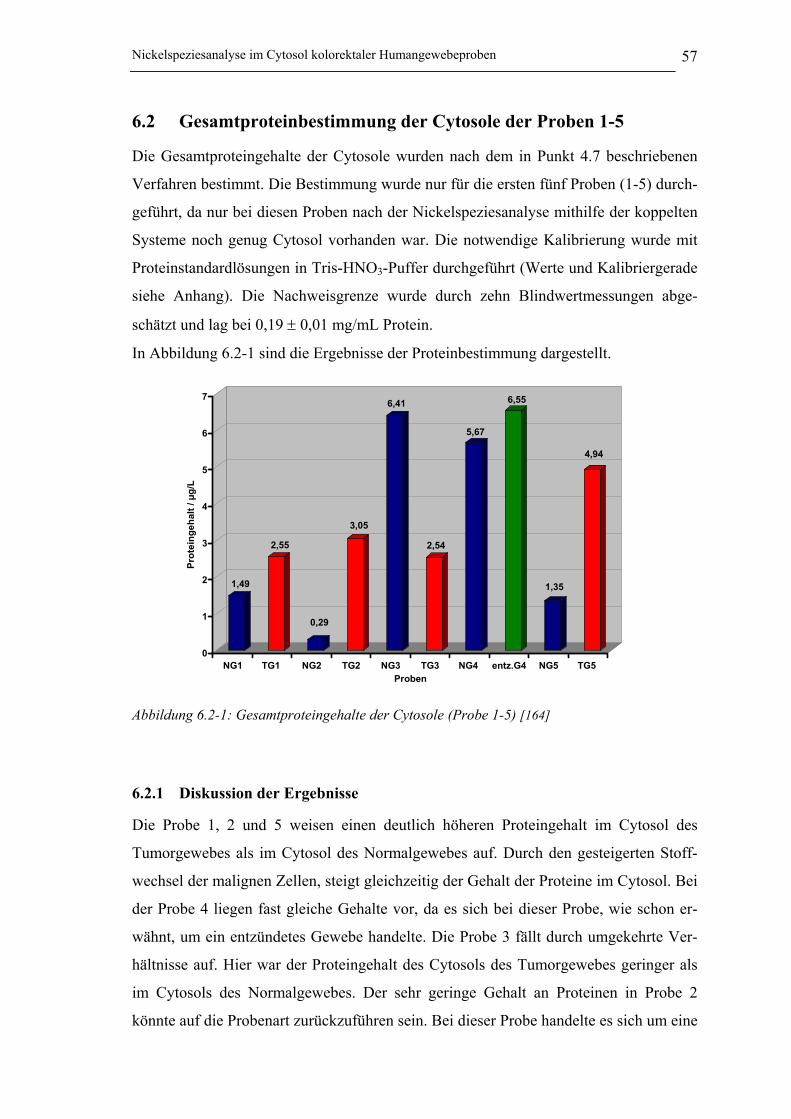
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 57
6.2 Gesamtproteinbestimmung der Cytosole der Proben 1-5
Die Gesamtproteingehalte der Cytosole wurden nach dem in Punkt 4.7 beschriebenen
Verfahren bestimmt. Die Bestimmung wurde nur für die ersten fünf Proben (1-5) durch-
geführt, da nur bei diesen Proben nach der Nickelspeziesanalyse mithilfe der koppelten
Systeme noch genug Cytosol vorhanden war. Die notwendige Kalibrierung wurde mit
Proteinstandardlösungen in Tris-HNO3-Puffer durchgeführt (Werte und Kalibriergerade
siehe Anhang). Die Nachweisgrenze wurde durch zehn Blindwertmessungen abge-
schätzt und lag bei 0,19 ± 0,01 mg/mL Protein.
In Abbildung 6.2-1 sind die Ergebnisse der Proteinbestimmung dargestellt.
Abbildung 6.2-1: Gesamtproteingehalte der Cytosole (Probe 1-5) [164]
6.2.1 Diskussion der Ergebnisse
Die Probe 1, 2 und 5 weisen einen deutlich höheren Proteingehalt im Cytosol des
Tumorgewebes als im Cytosol des Normalgewebes auf. Durch den gesteigerten Stoff-
wechsel der malignen Zellen, steigt gleichzeitig der Gehalt der Proteine im Cytosol. Bei
der Probe 4 liegen fast gleiche Gehalte vor, da es sich bei dieser Probe, wie schon er-
wähnt, um ein entzündetes Gewebe handelte. Die Probe 3 fällt durch umgekehrte Ver-
hältnisse auf. Hier war der Proteingehalt des Cytosols des Tumorgewebes geringer als
im Cytosols des Normalgewebes. Der sehr geringe Gehalt an Proteinen in Probe 2
könnte auf die Probenart zurückzuführen sein. Bei dieser Probe handelte es sich um eine
1,49
2,55
0,29
3,05
6,41
2,54
5,67
6,55
1,35
4,94
0
1
2
3
4
5
6
7
Prot
eing
ehal
t / µ
g/L
NG1 TG1 NG2 TG2 NG3 TG3 NG4 entz.G4 NG5 TG5 Proben

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 58
Rektumprobe, im Gegensatz zu den anderen Proben, bei denen es sich um Kolonproben
handelte.
6.3 Nickelspeziesanalyse mithilfe der CE / ICP-MS Kopplung
Die Nickelspeziesanalyse der Cytosole der Proben 1-5 und 10-15 wurden unter den zu-
vor optimierten Bedingungen (siehe Tabelle 5.2-7) mithilfe der CE / ICP-MS Kopplung
jeweils 5 mal durchgeführt. Statt einem Probeaufgabevolumen von 2,2 nL (20 mbar×s)
wurden bei den Cytosolproben 43,4 nL (400 mbar×s) in die Kapillare injiziert, da die
Konzentration der Nickelspezies im Cytosol so gering war, dass nur bei dieser Aufga-
bemenge eine Nickelspeziesanalyse überhaupt möglich war. Wie bereits erwähnt, wurde
der Make-up Flüssigkeit Germanium als interner Standard zugesetzt (siehe Tabelle 5.2-
7). Dies hatte zwei Gründe:
1. Während der Messung kann die Qualität der Zerstäubung kontrolliert werden
2. Es wurden alle Elektropherogramme auf das Germaniumsignal normiert, sodass
eine optimale Vergleichbarkeit der Elektropherogramme gegeben ist.
Jedes Cytosol wurde fünf Mal in die Kapillare injiziert, wobei permanent sowohl der
Strom wie die angelegte Spannung kontrolliert wurde. Es kamen keine Messung zur
Auswertung, bei denen Stromabbrüche oder Zerstäuberprobleme auftraten. Die Variati-
onskoeffizienten der Migrationszeiten der Nickelspezies eines Cytosols lagen zwischen
1-2 %. Die Variationskoeffizienten der Peakflächen der Nickelspezies betrugen 10-
12%, die der Peakflächenverhältnisse dagegen nur 1-2 % (Werte siehe Anhang). Die
Nachweisgrenze der gesamten Kopplung wurde mithilfe von verschieden konzentrierten
Ni2+-Standardlösungen abgeschätzt, da es keine kommerziell erhältlichen Ni-Pro-
teinstandards gab. Die Abschätzung erfolgte aus den Kalibrierdaten mit 3,7 µg/L. Die
Bestimmungsgrenze lag bei 14,7 µg/L. Die relative Verfahrensstandardabweichung
konnte mit 2,4 % ermittelt werden (Werte siehe Anhang).
Die nachfolgenden Abbildungen 6.3-1 bis 3 zeigen ausgesuchte Elektropherogramme
der Nickelspeziesanalyse (alle anderen Elektropherogramme siehe Anhang). Es sind
jeweils die Elektropherogramme von zwei Cytosolen (Normalgewebe (NG) /
Tumorgewebe (TG)) nach Acetonitrilfällung einer Probe in einer Abbildung
zusammengefasst.
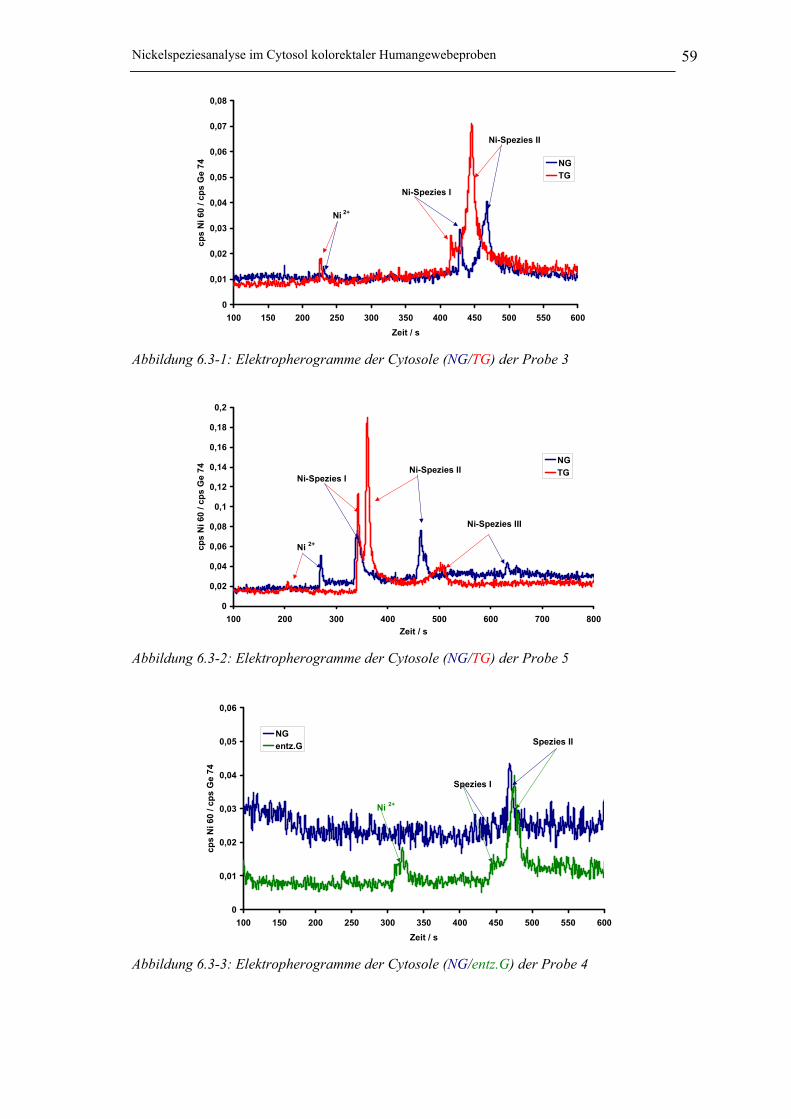
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 59
Abbildung 6.3-1: Elektropherogramme der Cytosole (NG/TG) der Probe 3
Abbildung 6.3-2: Elektropherogramme der Cytosole (NG/TG) der Probe 5
Abbildung 6.3-3: Elektropherogramme der Cytosole (NG/entz.G) der Probe 4
0
0,01
0,02
0,03
0,04
0,05
0,06
100 150 200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NGentz.G Spezies II
Ni 2+
Spezies I
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
100 150 200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74 NG
TG
Ni 2+
Ni-Spezies I
Ni-Spezies II
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
100 200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NGTG
Ni 2+
Ni-Spezies INi-Spezies II
Ni-Spezies III
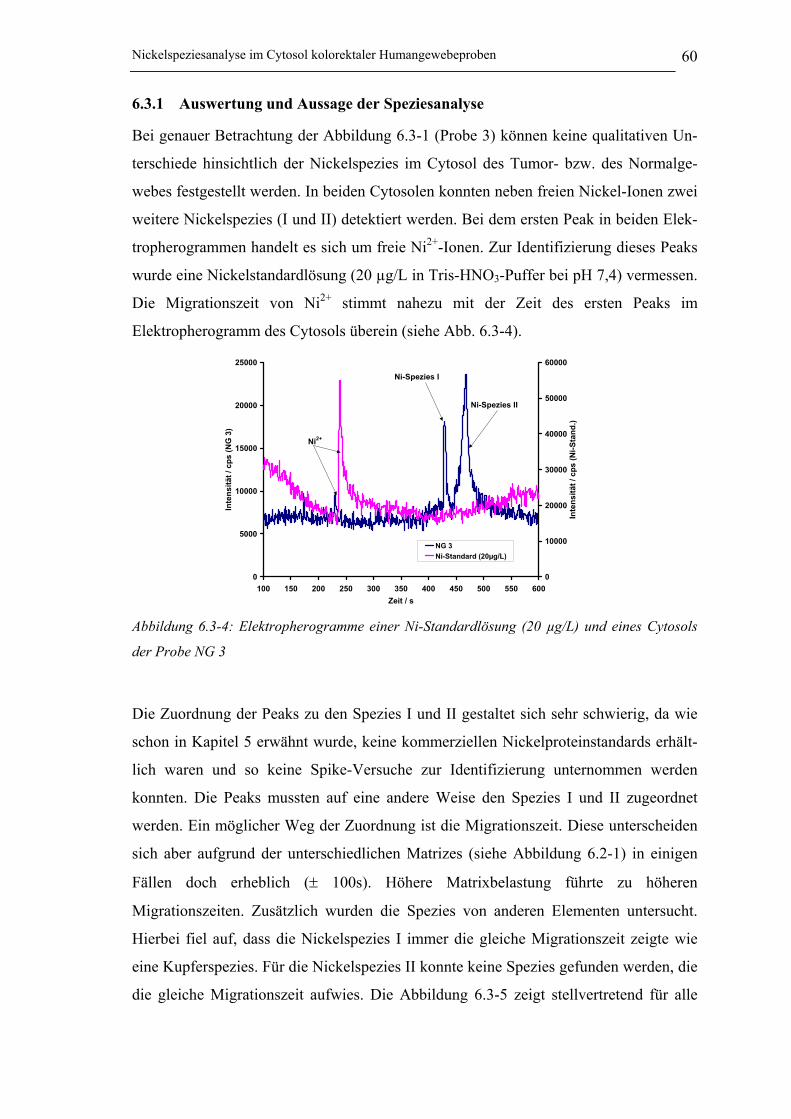
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 60
6.3.1 Auswertung und Aussage der Speziesanalyse
Bei genauer Betrachtung der Abbildung 6.3-1 (Probe 3) können keine qualitativen Un-
terschiede hinsichtlich der Nickelspezies im Cytosol des Tumor- bzw. des Normalge-
webes festgestellt werden. In beiden Cytosolen konnten neben freien Nickel-Ionen zwei
weitere Nickelspezies (I und II) detektiert werden. Bei dem ersten Peak in beiden Elek-
tropherogrammen handelt es sich um freie Ni2+-Ionen. Zur Identifizierung dieses Peaks
wurde eine Nickelstandardlösung (20 µg/L in Tris-HNO3-Puffer bei pH 7,4) vermessen.
Die Migrationszeit von Ni2+ stimmt nahezu mit der Zeit des ersten Peaks im
Elektropherogramm des Cytosols überein (siehe Abb. 6.3-4).
Abbildung 6.3-4: Elektropherogramme einer Ni-Standardlösung (20 µg/L) und eines Cytosols
der Probe NG 3
Die Zuordnung der Peaks zu den Spezies I und II gestaltet sich sehr schwierig, da wie
schon in Kapitel 5 erwähnt wurde, keine kommerziellen Nickelproteinstandards erhält-
lich waren und so keine Spike-Versuche zur Identifizierung unternommen werden
konnten. Die Peaks mussten auf eine andere Weise den Spezies I und II zugeordnet
werden. Ein möglicher Weg der Zuordnung ist die Migrationszeit. Diese unterscheiden
sich aber aufgrund der unterschiedlichen Matrizes (siehe Abbildung 6.2-1) in einigen
Fällen doch erheblich (± 100s). Höhere Matrixbelastung führte zu höheren
Migrationszeiten. Zusätzlich wurden die Spezies von anderen Elementen untersucht.
Hierbei fiel auf, dass die Nickelspezies I immer die gleiche Migrationszeit zeigte wie
eine Kupferspezies. Für die Nickelspezies II konnte keine Spezies gefunden werden, die
die gleiche Migrationszeit aufwies. Die Abbildung 6.3-5 zeigt stellvertretend für alle
0
5000
10000
15000
20000
25000
100 150 200 250 300 350 400 450 500 550 600Zeit / s
Inte
nsitä
t / c
ps (N
G 3
)
0
10000
20000
30000
40000
50000
60000
Inte
nsitä
t / c
ps (N
i-Sta
nd.)
NG 3Ni-Standard (20µg/L)
Ni2+
Ni-Spezies I
Ni-Spezies II
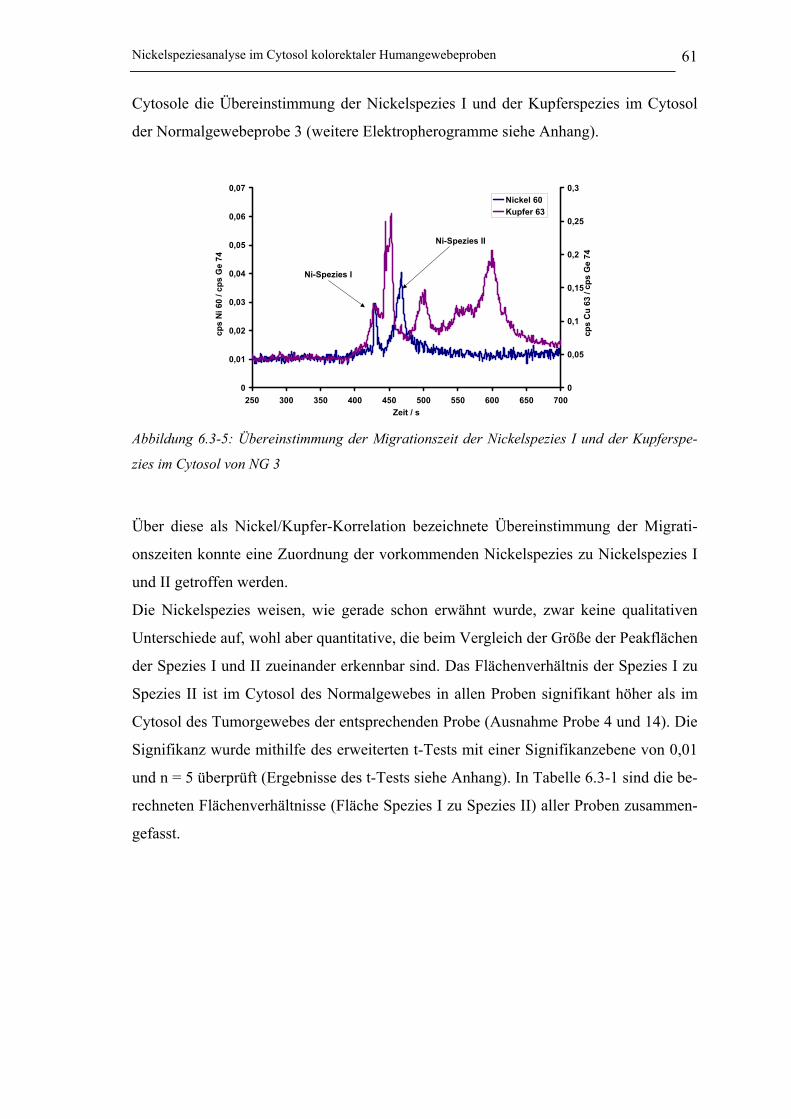
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 61
Cytosole die Übereinstimmung der Nickelspezies I und der Kupferspezies im Cytosol
der Normalgewebeprobe 3 (weitere Elektropherogramme siehe Anhang).
Abbildung 6.3-5: Übereinstimmung der Migrationszeit der Nickelspezies I und der Kupferspe-
zies im Cytosol von NG 3
Über diese als Nickel/Kupfer-Korrelation bezeichnete Übereinstimmung der Migrati-
onszeiten konnte eine Zuordnung der vorkommenden Nickelspezies zu Nickelspezies I
und II getroffen werden.
Die Nickelspezies weisen, wie gerade schon erwähnt wurde, zwar keine qualitativen
Unterschiede auf, wohl aber quantitative, die beim Vergleich der Größe der Peakflächen
der Spezies I und II zueinander erkennbar sind. Das Flächenverhältnis der Spezies I zu
Spezies II ist im Cytosol des Normalgewebes in allen Proben signifikant höher als im
Cytosol des Tumorgewebes der entsprechenden Probe (Ausnahme Probe 4 und 14). Die
Signifikanz wurde mithilfe des erweiterten t-Tests mit einer Signifikanzebene von 0,01
und n = 5 überprüft (Ergebnisse des t-Tests siehe Anhang). In Tabelle 6.3-1 sind die be-
rechneten Flächenverhältnisse (Fläche Spezies I zu Spezies II) aller Proben zusammen-
gefasst.
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
250 300 350 400 450 500 550 600 650 700Zeit / s
0
0,05
0,1
0,15
0,2
0,25
0,3
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
cps
Ni 6
0 / c
ps G
e 74
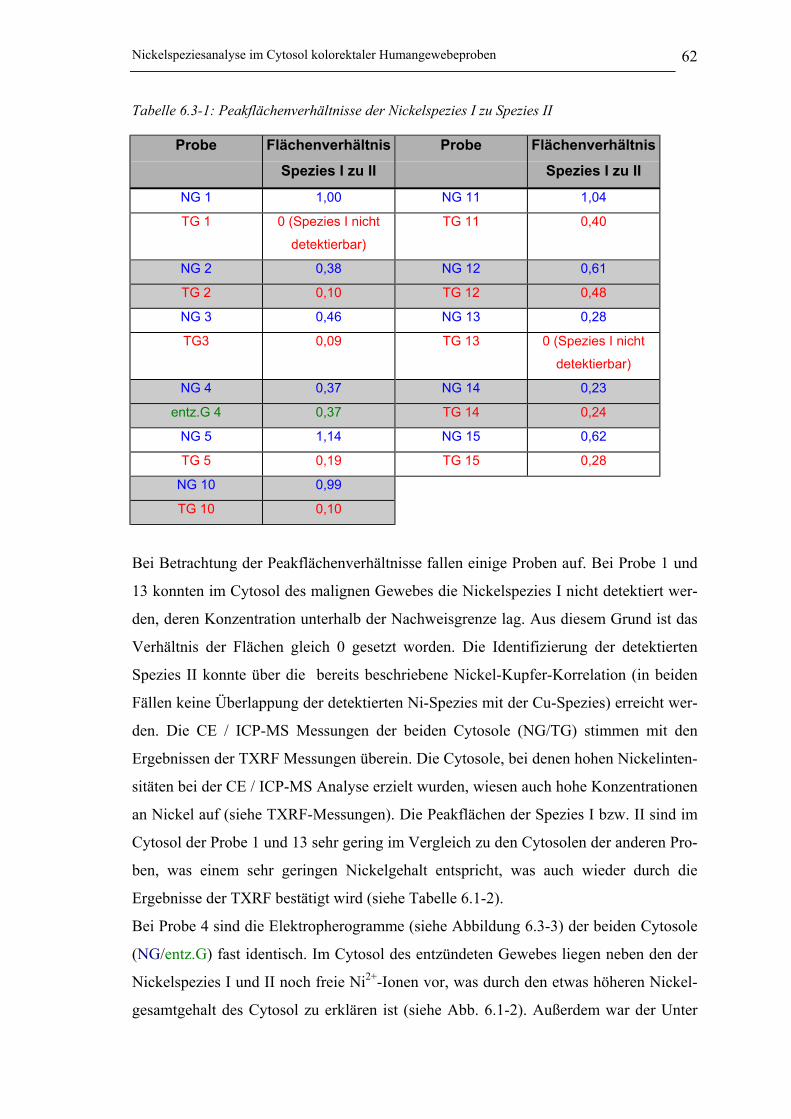
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 62
Tabelle 6.3-1: Peakflächenverhältnisse der Nickelspezies I zu Spezies II
Probe FlächenverhältnisSpezies I zu II
Probe FlächenverhältnisSpezies I zu II
NG 1 1,00 NG 11 1,04
TG 1 0 (Spezies I nicht
detektierbar)
TG 11 0,40
NG 2 0,38 NG 12 0,61
TG 2 0,10 TG 12 0,48
NG 3 0,46 NG 13 0,28
TG3 0,09 TG 13 0 (Spezies I nicht
detektierbar)
NG 4 0,37 NG 14 0,23
entz.G 4 0,37 TG 14 0,24
NG 5 1,14 NG 15 0,62
TG 5 0,19 TG 15 0,28
NG 10 0,99
TG 10 0,10
Bei Betrachtung der Peakflächenverhältnisse fallen einige Proben auf. Bei Probe 1 und
13 konnten im Cytosol des malignen Gewebes die Nickelspezies I nicht detektiert wer-
den, deren Konzentration unterhalb der Nachweisgrenze lag. Aus diesem Grund ist das
Verhältnis der Flächen gleich 0 gesetzt worden. Die Identifizierung der detektierten
Spezies II konnte über die bereits beschriebene Nickel-Kupfer-Korrelation (in beiden
Fällen keine Überlappung der detektierten Ni-Spezies mit der Cu-Spezies) erreicht wer-
den. Die CE / ICP-MS Messungen der beiden Cytosole (NG/TG) stimmen mit den
Ergebnissen der TXRF Messungen überein. Die Cytosole, bei denen hohen Nickelinten-
sitäten bei der CE / ICP-MS Analyse erzielt wurden, wiesen auch hohe Konzentrationen
an Nickel auf (siehe TXRF-Messungen). Die Peakflächen der Spezies I bzw. II sind im
Cytosol der Probe 1 und 13 sehr gering im Vergleich zu den Cytosolen der anderen Pro-
ben, was einem sehr geringen Nickelgehalt entspricht, was auch wieder durch die
Ergebnisse der TXRF bestätigt wird (siehe Tabelle 6.1-2).
Bei Probe 4 sind die Elektropherogramme (siehe Abbildung 6.3-3) der beiden Cytosole
(NG/entz.G) fast identisch. Im Cytosol des entzündeten Gewebes liegen neben den der
Nickelspezies I und II noch freie Ni2+-Ionen vor, was durch den etwas höheren Nickel-
gesamtgehalt des Cytosol zu erklären ist (siehe Abb. 6.1-2). Außerdem war der Unter

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 63
grund bei der Nickelspeziesanalyse des entzündeten Gewebes geringer als bei der des
Normalgewebes, so dass im letzteren Fall die freien Ni2+-Ionen nicht detektiert werden
konnten. Die Peakflächenverhältnisse der Cytosole beider Gewebearten sind ebenfalls
identisch. Da es sich bei entzündetem Gewebe um Normalgewebe mit erhöhtem Gehalt
an Enzymen, die am Entzündungsprozess beteiligt sind, jedoch nicht um malignes
Gewebe handelt, sind die übereinstimmden Ergebnisse der Nickelspeziesanalyse zu
erklären (siehe auch TXRF Messungen).
Eine ähnliche Auffälligkeit zeigt Probe 14, auch hier unterscheiden sich die Elektro-
pherogramme beider Gewebearten nur in den Migrationszeiten, aber nicht im
Peakmuster bzw. in den Peakflächenverhältnissen. Der pathologische Befund gibt an,
dass sich im Resektat weitere tumoröse Gewebeanteile befanden. Die CE / ICP-MS
Daten als auch die TXRF-Messungen (siehe Abb. 6.1-2) lassen den Schluss zu, dass es
sich bei den beiden Gewebeproben um eine ähnliche Gewebeart handelt,
möglicherweise auch um adenomatöses Gewebe. Dieser Eindruck wird durch das
Aussehen der beiden Gewebeproben bestätigt. Normalerweise kann aufgrund des
Aussehens ein Unterschied zwischen normalen und malignen Gewebe getroffen
werden. Das maligne Gewebe ist im Vergleich zum normalen Gewebe härter. Bei der
Probe 14 sahen beide Proben sehr ähnlich aus und unterschieden sich auch nicht in ihrer
Konsistenz, sodass schon aufgrund dieser Befunde davon ausgegangen werden konnte,
dass es sich um die gleiche Gewebearten handeln könnte.
Neben den bisher erwähnten zwei Nickelspezies wurden nur in beiden Cytosolen der
Probe 5 eine dritte Nickelspezies gefunden (siehe Abb. 6.3-2). Diese beiden Cytosole
wiesen eine sehr hohe Nickelgesamtkonzentration im Vergleich zu den ersten vier Pro-
ben auf (siehe Tab. 6.1-2). Da Dithiotreitol (DTT), wie schon in Kapitel 5 erwähnt
wurde, neben den antioxidativen Eigenschaften auch gute
Komplexierungseigenschaften besitzt, könnte es sich bei der dritten Nickelspezies um
eine Ni-DTT-Komplex handeln. Aufgrund des hohen Nickelgehaltes in den Cytosolen
konnte sich nur bei diesen beiden Cytosolen der Komplex in ausreichender,
detektierbarer Konzentration bilden. Um diese Vermutung bestätigen zu können, wurde
mithilfe der CE / ICP-MS Kopplung eine 20 µg/L Nickelstandardlösung mit 5 mmol
DTT-Lösung und einmal ohne DTT-Zusatz gemessen. Um Informationen über den
Ladungszustand der gefundenen Nickelspezies zu erhalten, wurde zusätzlich eine
Arsenobetain-Standardlösung in die CE-Kapillare injiziert, da Arsenobetain nach außen
neutral ist und durch den EOF aus der Säule eluiert wird. Damit konnte eine Aussage
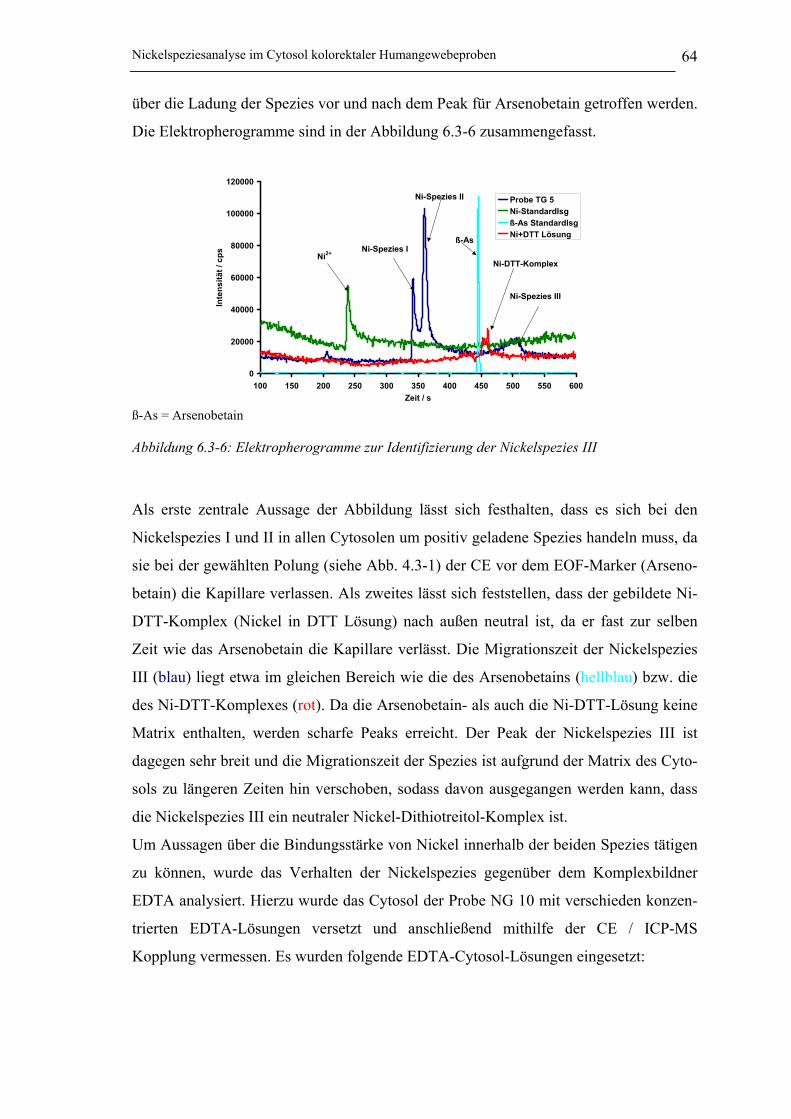
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 64
über die Ladung der Spezies vor und nach dem Peak für Arsenobetain getroffen werden.
Die Elektropherogramme sind in der Abbildung 6.3-6 zusammengefasst.
ß-As = Arsenobetain
Abbildung 6.3-6: Elektropherogramme zur Identifizierung der Nickelspezies III
Als erste zentrale Aussage der Abbildung lässt sich festhalten, dass es sich bei den
Nickelspezies I und II in allen Cytosolen um positiv geladene Spezies handeln muss, da
sie bei der gewählten Polung (siehe Abb. 4.3-1) der CE vor dem EOF-Marker (Arseno-
betain) die Kapillare verlassen. Als zweites lässt sich feststellen, dass der gebildete Ni-
DTT-Komplex (Nickel in DTT Lösung) nach außen neutral ist, da er fast zur selben
Zeit wie das Arsenobetain die Kapillare verlässt. Die Migrationszeit der Nickelspezies
III (blau) liegt etwa im gleichen Bereich wie die des Arsenobetains (hellblau) bzw. die
des Ni-DTT-Komplexes (rot). Da die Arsenobetain- als auch die Ni-DTT-Lösung keine
Matrix enthalten, werden scharfe Peaks erreicht. Der Peak der Nickelspezies III ist
dagegen sehr breit und die Migrationszeit der Spezies ist aufgrund der Matrix des Cyto-
sols zu längeren Zeiten hin verschoben, sodass davon ausgegangen werden kann, dass
die Nickelspezies III ein neutraler Nickel-Dithiotreitol-Komplex ist.
Um Aussagen über die Bindungsstärke von Nickel innerhalb der beiden Spezies tätigen
zu können, wurde das Verhalten der Nickelspezies gegenüber dem Komplexbildner
EDTA analysiert. Hierzu wurde das Cytosol der Probe NG 10 mit verschieden konzen-
trierten EDTA-Lösungen versetzt und anschließend mithilfe der CE / ICP-MS
Kopplung vermessen. Es wurden folgende EDTA-Cytosol-Lösungen eingesetzt:
0
20000
40000
60000
80000
100000
120000
100 150 200 250 300 350 400 450 500 550 600Zeit / s
Inte
nsitä
t / c
ps
Probe TG 5 Ni-Standardlsgß-As StandardlsgNi+DTT Lösung
Ni2+ Ni-Spezies I
Ni-Spezies II
Ni-Spezies III
Ni-DTT-Komplex
ß-As
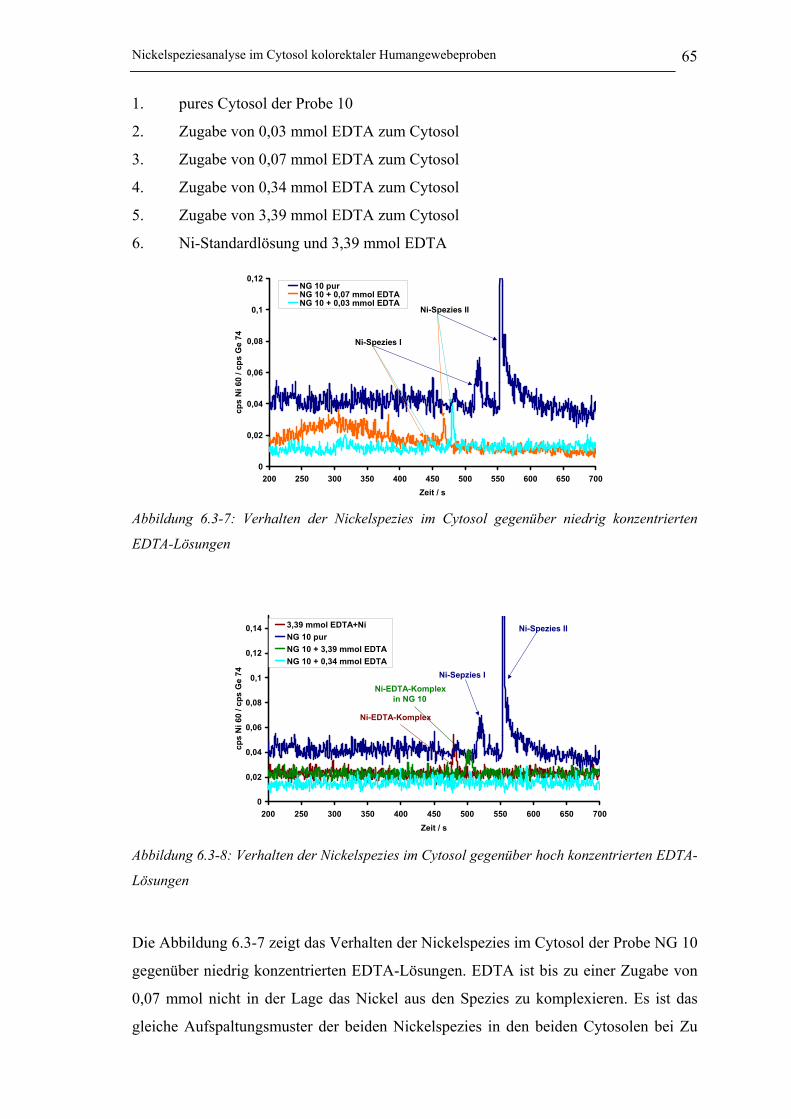
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 65
1. pures Cytosol der Probe 10
2. Zugabe von 0,03 mmol EDTA zum Cytosol
3. Zugabe von 0,07 mmol EDTA zum Cytosol
4. Zugabe von 0,34 mmol EDTA zum Cytosol
5. Zugabe von 3,39 mmol EDTA zum Cytosol
6. Ni-Standardlösung und 3,39 mmol EDTA
Abbildung 6.3-7: Verhalten der Nickelspezies im Cytosol gegenüber niedrig konzentrierten
EDTA-Lösungen
Abbildung 6.3-8: Verhalten der Nickelspezies im Cytosol gegenüber hoch konzentrierten EDTA-
Lösungen
Die Abbildung 6.3-7 zeigt das Verhalten der Nickelspezies im Cytosol der Probe NG 10
gegenüber niedrig konzentrierten EDTA-Lösungen. EDTA ist bis zu einer Zugabe von
0,07 mmol nicht in der Lage das Nickel aus den Spezies zu komplexieren. Es ist das
gleiche Aufspaltungsmuster der beiden Nickelspezies in den beiden Cytosolen bei Zu
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
200 250 300 350 400 450 500 550 600 650 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
3,39 mmol EDTA+NiNG 10 purNG 10 + 3,39 mmol EDTANG 10 + 0,34 mmol EDTA
Ni-Sepzies I
Ni-Spezies II
Ni-EDTA-Komplex
Ni-EDTA-Komplex in NG 10
0
0,02
0,04
0,06
0,08
0,1
0,12
200 250 300 350 400 450 500 550 600 650 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NG 10 purNG 10 + 0,07 mmol EDTANG 10 + 0,03 mmol EDTA
Ni-Spezies I
Ni-Spezies II

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 66
gabe von 0,03 und 0,07 mmol EDTA zu erkennen, sodass davon ausgegangen werden
kann, dass Nickel fest an die Spezies I und II gebunden ist. Die Migrationszeiten und
die Intensitäten der Spezies haben sich im Gegensatz zum Cytosol ohne Zugabe von
EDTA verändert, was an der Verdünnung des Cytosols, aber auch an der
Komplexierung von Cytosolbestandteilen durch die EDTA-Lösung zu erklären ist.
Abbildung 6.3-8 gewährt einen Einblick in das Verhalten der Spezies gegenüber höher
konzentrierten EDTA-Lösungen ( ≥ 0,34 mmol). Ab einer Zugabe von 0,34 mmol ist
EDTA in der Lage, das Nickel aus den beiden Spezies herauszulösen und zu komplexie-
ren. Die Konzentration des Ni-EDTA-Komplexes ist aber zu niedrig, sodass keine
Nickelspezies detektiert wurden. Erst ab einer Zugabe von 3,39 mmol EDTA wird das
gesamte Nickel der Probe komplexiert und der Ni-DTT-Komplex detektiert. Die höhere
Matrixbelastung im Cytosol im Gegensatz zur Ni-EDTA-Standardlösung sorgt für
längere Migrationszeiten.
Erstes Fazit:
Mit der vorgestellten CE / ICP-MS Kopplung ist eine Nickelspeziesanalyse in Cyto-
solen humaner Proben möglich. Es hat sich gezeigt, dass keine qualitativen Unter-
schiede im Cytosol von normalen und malignen Gewebeproben des humanen
Intestinaltrakts hinsichtlich der Nickelspezies festzustellen sind, aber quantitative
Unterschiede bestehen. Diese werden durch die Bildung der Peakflächenverhältnisse
der Nickelspezies I und II sichtbar. Dieses ist im Cytosol der normalen Gewebeproben
signifikant höher als im Cytosol der malignen Gewebeproben. Bei den ersten beiden
Nickelspezies (I und II) handelt es sich um positiv geladene Spezies, wobei Nickel fest
an die Spezies I und II gebunden ist.
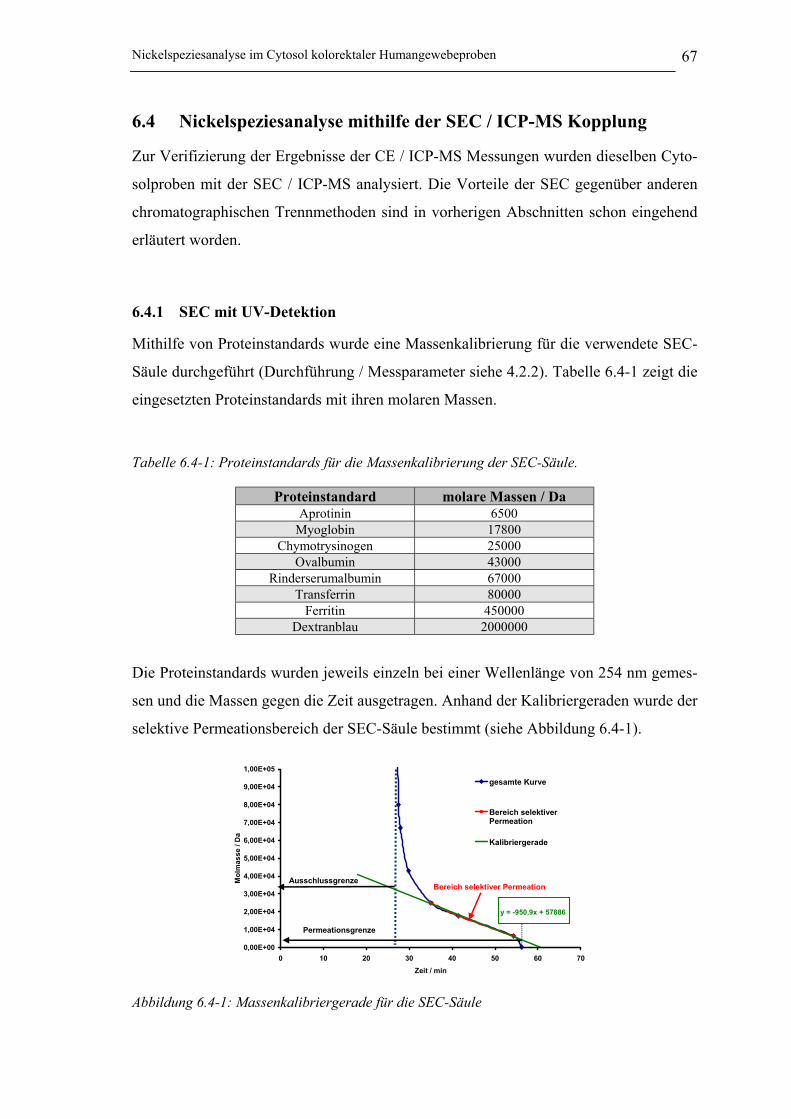
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 67
6.4 Nickelspeziesanalyse mithilfe der SEC / ICP-MS Kopplung
Zur Verifizierung der Ergebnisse der CE / ICP-MS Messungen wurden dieselben Cyto-
solproben mit der SEC / ICP-MS analysiert. Die Vorteile der SEC gegenüber anderen
chromatographischen Trennmethoden sind in vorherigen Abschnitten schon eingehend
erläutert worden.
6.4.1 SEC mit UV-Detektion
Mithilfe von Proteinstandards wurde eine Massenkalibrierung für die verwendete SEC-
Säule durchgeführt (Durchführung / Messparameter siehe 4.2.2). Tabelle 6.4-1 zeigt die
eingesetzten Proteinstandards mit ihren molaren Massen.
Tabelle 6.4-1: Proteinstandards für die Massenkalibrierung der SEC-Säule.
Proteinstandard molare Massen / DaAprotinin 6500
Myoglobin 17800Chymotrysinogen 25000
Ovalbumin 43000Rinderserumalbumin 67000
Transferrin 80000Ferritin 450000
Dextranblau 2000000
Die Proteinstandards wurden jeweils einzeln bei einer Wellenlänge von 254 nm gemes-
sen und die Massen gegen die Zeit ausgetragen. Anhand der Kalibriergeraden wurde der
selektive Permeationsbereich der SEC-Säule bestimmt (siehe Abbildung 6.4-1).
Abbildung 6.4-1: Massenkalibriergerade für die SEC-Säule
y = -950,9x + 57886
0,00E+00
1,00E+04
2,00E+04
3,00E+04
4,00E+04
5,00E+04
6,00E+04
7,00E+04
8,00E+04
9,00E+04
1,00E+05
0 10 20 30 40 50 60 70Zeit / min
Mol
mas
se /
Da
gesamte Kurve
Bereich selektiverPermeation
Kalibriergerade
Ausschlussgrenze
Permeationsgrenze
Bereich selektiver Permeation
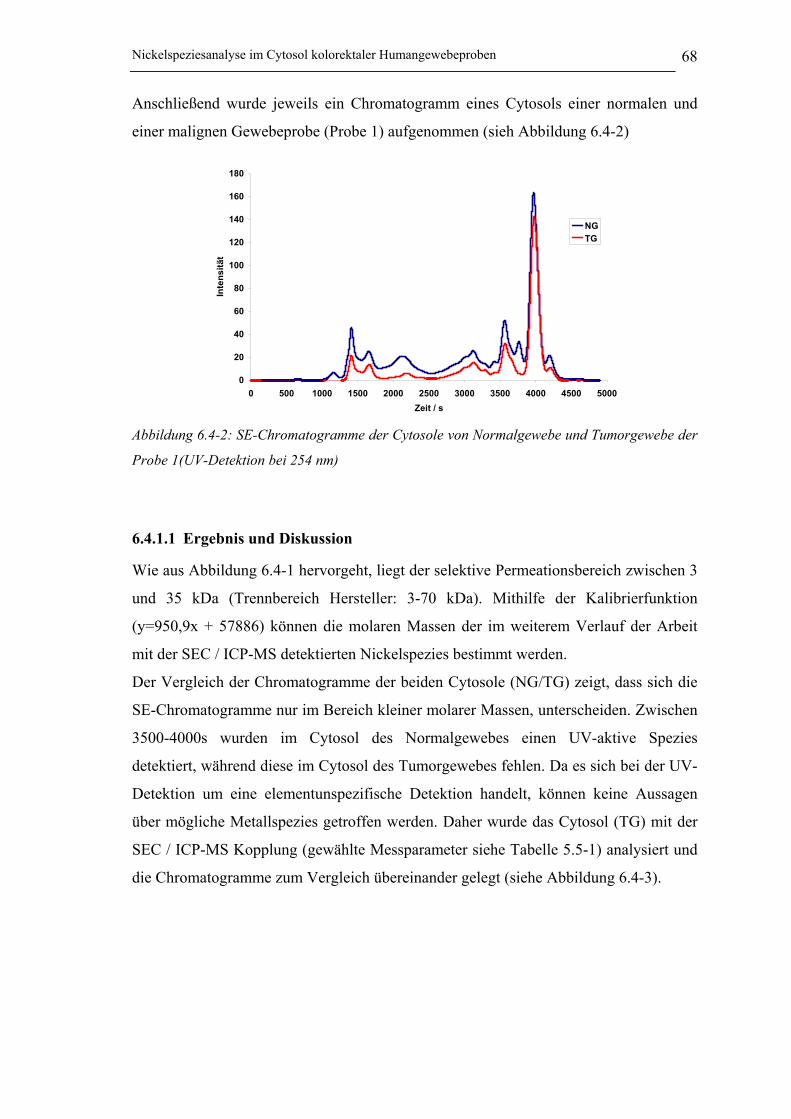
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 68
Anschließend wurde jeweils ein Chromatogramm eines Cytosols einer normalen und
einer malignen Gewebeprobe (Probe 1) aufgenommen (sieh Abbildung 6.4-2)
Abbildung 6.4-2: SE-Chromatogramme der Cytosole von Normalgewebe und Tumorgewebe der
Probe 1(UV-Detektion bei 254 nm)
6.4.1.1 Ergebnis und Diskussion
Wie aus Abbildung 6.4-1 hervorgeht, liegt der selektive Permeationsbereich zwischen 3
und 35 kDa (Trennbereich Hersteller: 3-70 kDa). Mithilfe der Kalibrierfunktion
(y=950,9x + 57886) können die molaren Massen der im weiterem Verlauf der Arbeit
mit der SEC / ICP-MS detektierten Nickelspezies bestimmt werden.
Der Vergleich der Chromatogramme der beiden Cytosole (NG/TG) zeigt, dass sich die
SE-Chromatogramme nur im Bereich kleiner molarer Massen, unterscheiden. Zwischen
3500-4000s wurden im Cytosol des Normalgewebes einen UV-aktive Spezies
detektiert, während diese im Cytosol des Tumorgewebes fehlen. Da es sich bei der UV-
Detektion um eine elementunspezifische Detektion handelt, können keine Aussagen
über mögliche Metallspezies getroffen werden. Daher wurde das Cytosol (TG) mit der
SEC / ICP-MS Kopplung (gewählte Messparameter siehe Tabelle 5.5-1) analysiert und
die Chromatogramme zum Vergleich übereinander gelegt (siehe Abbildung 6.4-3).
0
20
40
60
80
100
120
140
160
180
0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
tNGTG
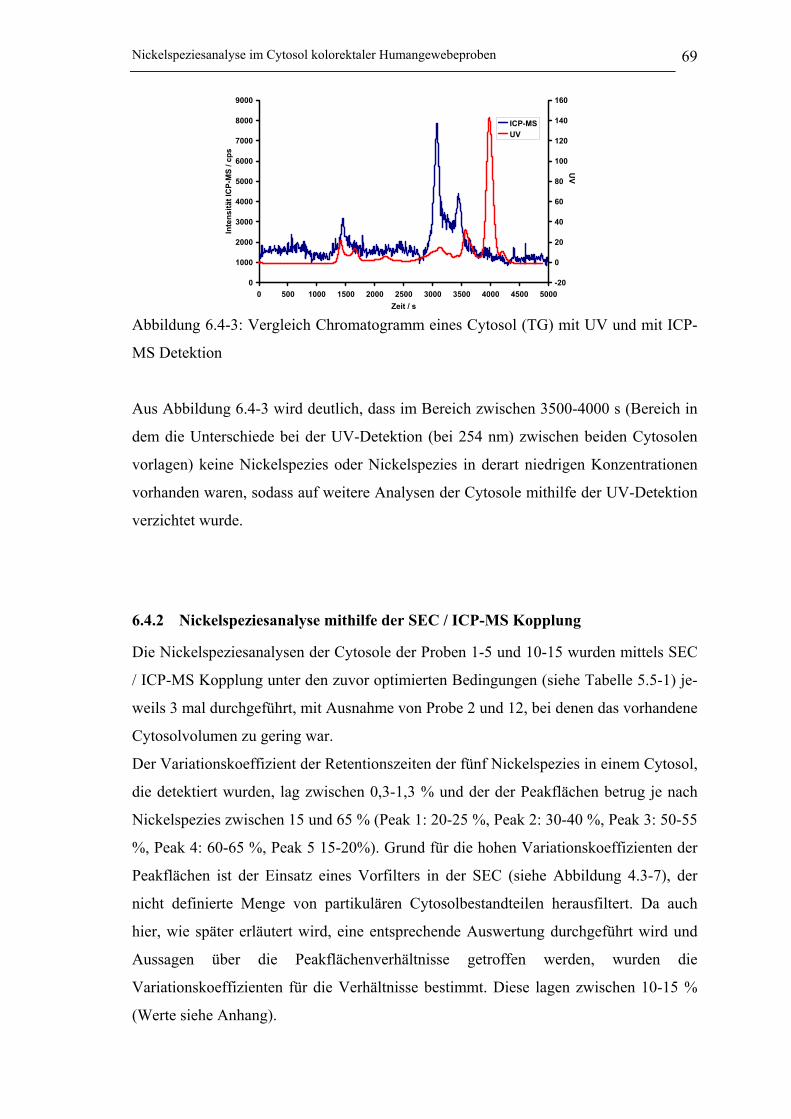
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 69
Abbildung 6.4-3: Vergleich Chromatogramm eines Cytosol (TG) mit UV und mit ICP-
MS Detektion
Aus Abbildung 6.4-3 wird deutlich, dass im Bereich zwischen 3500-4000 s (Bereich in
dem die Unterschiede bei der UV-Detektion (bei 254 nm) zwischen beiden Cytosolen
vorlagen) keine Nickelspezies oder Nickelspezies in derart niedrigen Konzentrationen
vorhanden waren, sodass auf weitere Analysen der Cytosole mithilfe der UV-Detektion
verzichtet wurde.
6.4.2 Nickelspeziesanalyse mithilfe der SEC / ICP-MS Kopplung
Die Nickelspeziesanalysen der Cytosole der Proben 1-5 und 10-15 wurden mittels SEC
/ ICP-MS Kopplung unter den zuvor optimierten Bedingungen (siehe Tabelle 5.5-1) je-
weils 3 mal durchgeführt, mit Ausnahme von Probe 2 und 12, bei denen das vorhandene
Cytosolvolumen zu gering war.
Der Variationskoeffizient der Retentionszeiten der fünf Nickelspezies in einem Cytosol,
die detektiert wurden, lag zwischen 0,3-1,3 % und der der Peakflächen betrug je nach
Nickelspezies zwischen 15 und 65 % (Peak 1: 20-25 %, Peak 2: 30-40 %, Peak 3: 50-55
%, Peak 4: 60-65 %, Peak 5 15-20%). Grund für die hohen Variationskoeffizienten der
Peakflächen ist der Einsatz eines Vorfilters in der SEC (siehe Abbildung 4.3-7), der
nicht definierte Menge von partikulären Cytosolbestandteilen herausfiltert. Da auch
hier, wie später erläutert wird, eine entsprechende Auswertung durchgeführt wird und
Aussagen über die Peakflächenverhältnisse getroffen werden, wurden die
Variationskoeffizienten für die Verhältnisse bestimmt. Diese lagen zwischen 10-15 %
(Werte siehe Anhang).
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t IC
P-M
S / c
ps
-20
0
20
40
60
80
100
120
140
160
ICP-MSUV
UV
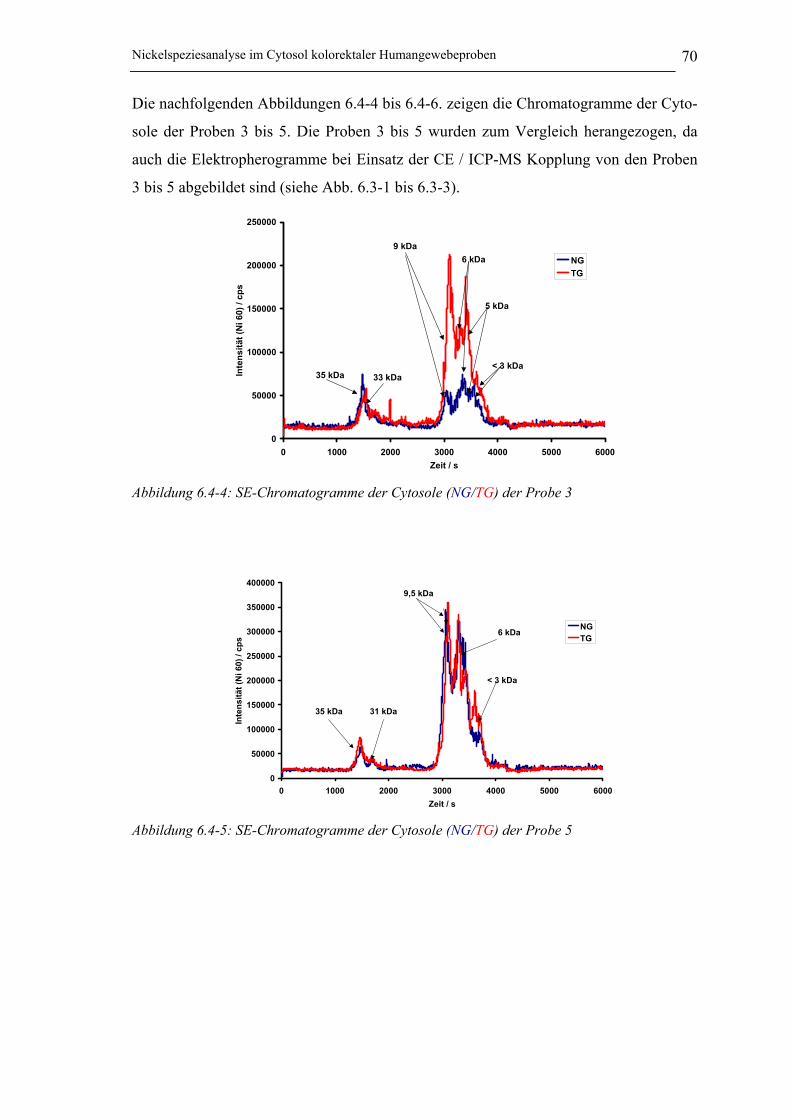
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 70
Die nachfolgenden Abbildungen 6.4-4 bis 6.4-6. zeigen die Chromatogramme der Cyto-
sole der Proben 3 bis 5. Die Proben 3 bis 5 wurden zum Vergleich herangezogen, da
auch die Elektropherogramme bei Einsatz der CE / ICP-MS Kopplung von den Proben
3 bis 5 abgebildet sind (siehe Abb. 6.3-1 bis 6.3-3).
Abbildung 6.4-4: SE-Chromatogramme der Cytosole (NG/TG) der Probe 3
Abbildung 6.4-5: SE-Chromatogramme der Cytosole (NG/TG) der Probe 5
0
50000
100000
150000
200000
250000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa 33 kDa
9 kDa6 kDa
5 kDa
< 3 kDa
0
50000
100000
150000
200000
250000
300000
350000
400000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa 31 kDa
9,5 kDa
6 kDa
< 3 kDa
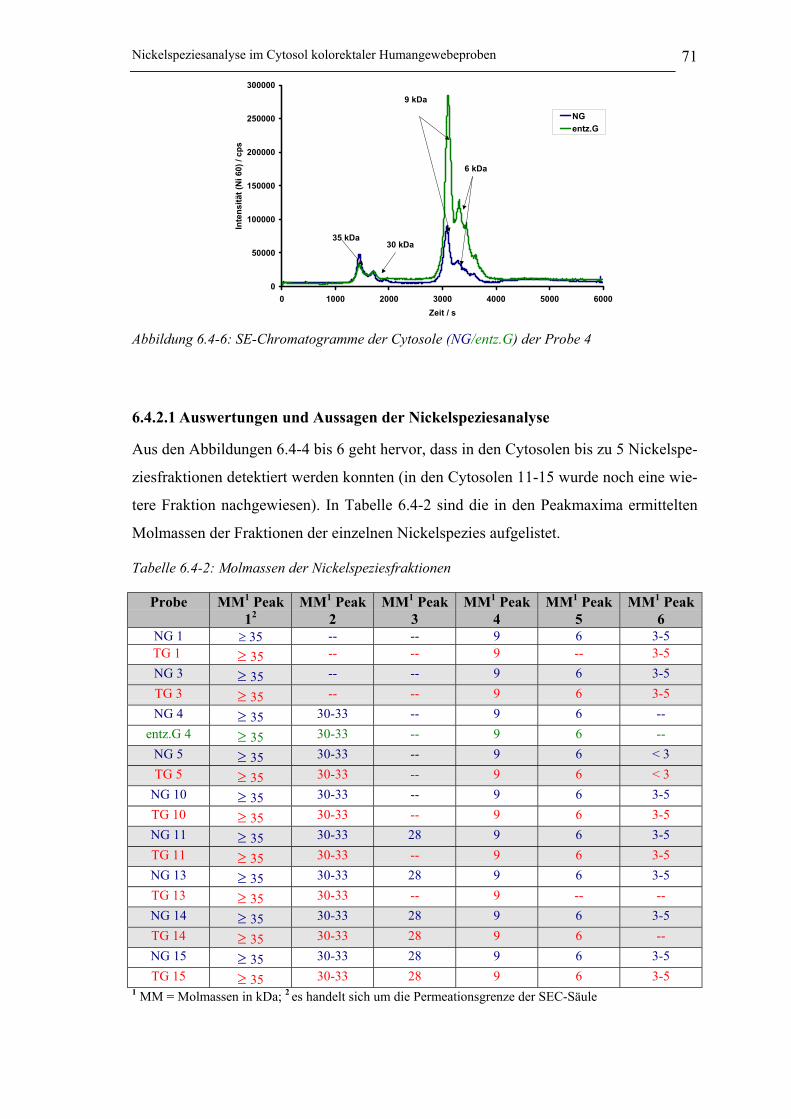
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 71
Abbildung 6.4-6: SE-Chromatogramme der Cytosole (NG/entz.G) der Probe 4
6.4.2.1 Auswertungen und Aussagen der Nickelspeziesanalyse
Aus den Abbildungen 6.4-4 bis 6 geht hervor, dass in den Cytosolen bis zu 5 Nickelspe-
ziesfraktionen detektiert werden konnten (in den Cytosolen 11-15 wurde noch eine wie-
tere Fraktion nachgewiesen). In Tabelle 6.4-2 sind die in den Peakmaxima ermittelten
Molmassen der Fraktionen der einzelnen Nickelspezies aufgelistet.
Tabelle 6.4-2: Molmassen der Nickelspeziesfraktionen
Probe MM1 Peak12
MM1 Peak2
MM1 Peak3
MM1 Peak4
MM1 Peak5
MM1 Peak6
NG 1 ≥ 35 -- -- 9 6 3-5TG 1 ≥ 35 -- -- 9 -- 3-5NG 3 ≥ 35 -- -- 9 6 3-5TG 3 ≥ 35 -- -- 9 6 3-5NG 4 ≥ 35 30-33 -- 9 6 --
entz.G 4 ≥ 35 30-33 -- 9 6 --NG 5 ≥ 35 30-33 -- 9 6 < 3TG 5 ≥ 35 30-33 -- 9 6 < 3
NG 10 ≥ 35 30-33 -- 9 6 3-5TG 10 ≥ 35 30-33 -- 9 6 3-5NG 11 ≥ 35 30-33 28 9 6 3-5TG 11 ≥ 35 30-33 -- 9 6 3-5NG 13 ≥ 35 30-33 28 9 6 3-5TG 13 ≥ 35 30-33 -- 9 -- --NG 14 ≥ 35 30-33 28 9 6 3-5TG 14 ≥ 35 30-33 28 9 6 --NG 15 ≥ 35 30-33 28 9 6 3-5TG 15 ≥ 35 30-33 28 9 6 3-5
1 MM = Molmassen in kDa; 2 es handelt sich um die Permeationsgrenze der SEC-Säule
0
50000
100000
150000
200000
250000
300000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGentz.G
35 kDa30 kDa
9 kDa
6 kDa

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 72
Da die Chromatogramme teilweise sehr breite Peaks bzw. breite Peakmaxima zeigen,
können die Molmassen nicht immer genau bestimmt sondern nur ein Molmassenbereich
angeben werden.
Die Cytosole weisen mit Ausnahme des Cytosols der Probe NG 4, entz.G 4, TG 13 und
TG 14 Nickelspezies mit den Molmassen von 4, 6 und 9 kDa auf. In den Cytosolen der
Proben 4 und 14 konnte die Spezies mit einer Molmasse von 4 kDa nicht detektiert
werden. Bei dem Cytosol der Probe TG 13 konnte wie schon bei der Nickelspeziesana-
lyse mithilfe der CE / ICP-MS Kopplung gezeigt werden, dass keine klare Auftrennung
der Speziesfraktion erreicht werden kann. Im höher molekularen Bereich konnte in allen
Cytosolen eine Nickelspezies mit einer molaren Masse von ≥ 35 kDa gefunden werden.
Hierbei muss beachtet werden, dass in diesem Molmassenbereich die Permea-
tionsgrenze der Säule lag und so auch alle höher molekularen Spezies (> 35 kDa) eluiert
werden. Daher kann über diese Fraktion keine konkrete Aussage gemacht werden. Bis
auf die Cytosole der Proben 1 und 3 enthalten alle Cytosolproben eine zusätzliche
Nickelspeziesfraktion im Bereich von 30-33 kDa. Bei den Proben 11-15 wird noch eine
weitere höher molekulare Nickelspeziesfraktion detektiert (MG= 28 kDa). Da bei
diesen Proben bei der Cytosolgewinnung nur mit 25.000g zentrifugiert wurde (Probe 1-
5 mit 100.000g), verblieben vermutlich mehr hochmolekulare Bestandteile im Cytosol.
Die Nickelspezies der beiden Cytosole der Probe 4 (siehe Abbildung 6.4-6) zeigen wie
schon bei der Nickelspeziesanalyse mittels CE / ICP-MS ein fast identisches Peakmu-
ster auf. Sowohl beim Cytosol der Probe TG 1 (Fehlen einer Spezies) wie auch bei den
Cytosolen der Probe 4 (identisches Peakmuster) stimmen die Ergebnisse der Nickel-
speziesanalyse mittels CE / ICP-MS mit denen, die mit der SEC / ICP-MS ermittelt
wurden, überein.
Die Zuordnung der Fraktionen ist bei der SEC-Trennung einfacher als bei der CE, da
bei der SEC die Fraktionen der Spezies über ihre Retentionszeit zugeordnet werden
können.
Die Chromatogramme weisen bei den Fraktionen der Nickelspezies wie bei den Elek-
tropherogrammen auch keine qualitativen Unterschiede zwischen dem Cytosol der ma-
lignen im Vergleich zu dem Cytosol der normalen Gewebeprobe auf. Die Anzahl der
Fraktionen von Nickelspezies sind bei der Betrachtung der korrespondierenden
Cytosole (NG/TG der Gewebeproben) gleich. Ausnahme ist die Probe 1, bei der beim
Cytosol des Tumorgewebes, wie schon durch die CE / ICP-MS Messungen gezeigt, eine
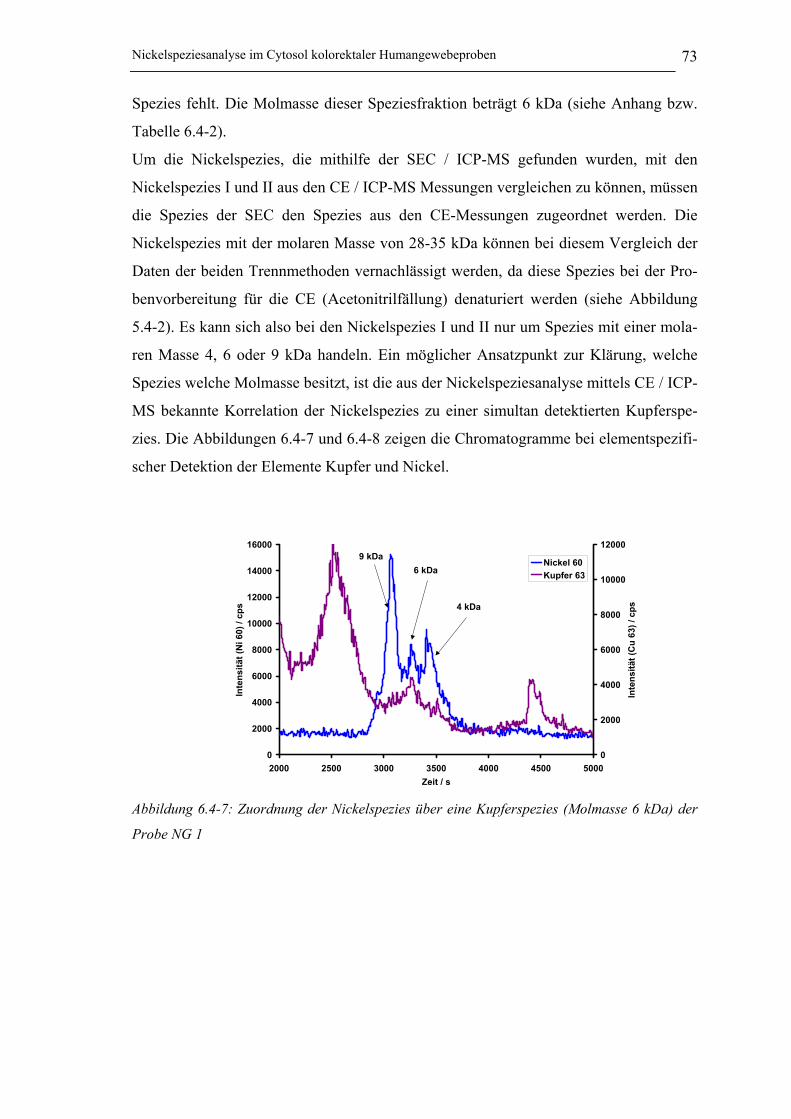
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 73
Spezies fehlt. Die Molmasse dieser Speziesfraktion beträgt 6 kDa (siehe Anhang bzw.
Tabelle 6.4-2).
Um die Nickelspezies, die mithilfe der SEC / ICP-MS gefunden wurden, mit den
Nickelspezies I und II aus den CE / ICP-MS Messungen vergleichen zu können, müssen
die Spezies der SEC den Spezies aus den CE-Messungen zugeordnet werden. Die
Nickelspezies mit der molaren Masse von 28-35 kDa können bei diesem Vergleich der
Daten der beiden Trennmethoden vernachlässigt werden, da diese Spezies bei der Pro-
benvorbereitung für die CE (Acetonitrilfällung) denaturiert werden (siehe Abbildung
5.4-2). Es kann sich also bei den Nickelspezies I und II nur um Spezies mit einer mola-
ren Masse 4, 6 oder 9 kDa handeln. Ein möglicher Ansatzpunkt zur Klärung, welche
Spezies welche Molmasse besitzt, ist die aus der Nickelspeziesanalyse mittels CE / ICP-
MS bekannte Korrelation der Nickelspezies zu einer simultan detektierten Kupferspe-
zies. Die Abbildungen 6.4-7 und 6.4-8 zeigen die Chromatogramme bei elementspezifi-
scher Detektion der Elemente Kupfer und Nickel.
Abbildung 6.4-7: Zuordnung der Nickelspezies über eine Kupferspezies (Molmasse 6 kDa) der
Probe NG 1
0
2000
4000
6000
8000
10000
12000
14000
16000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
2000
4000
6000
8000
10000
12000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa6 kDa
4 kDa
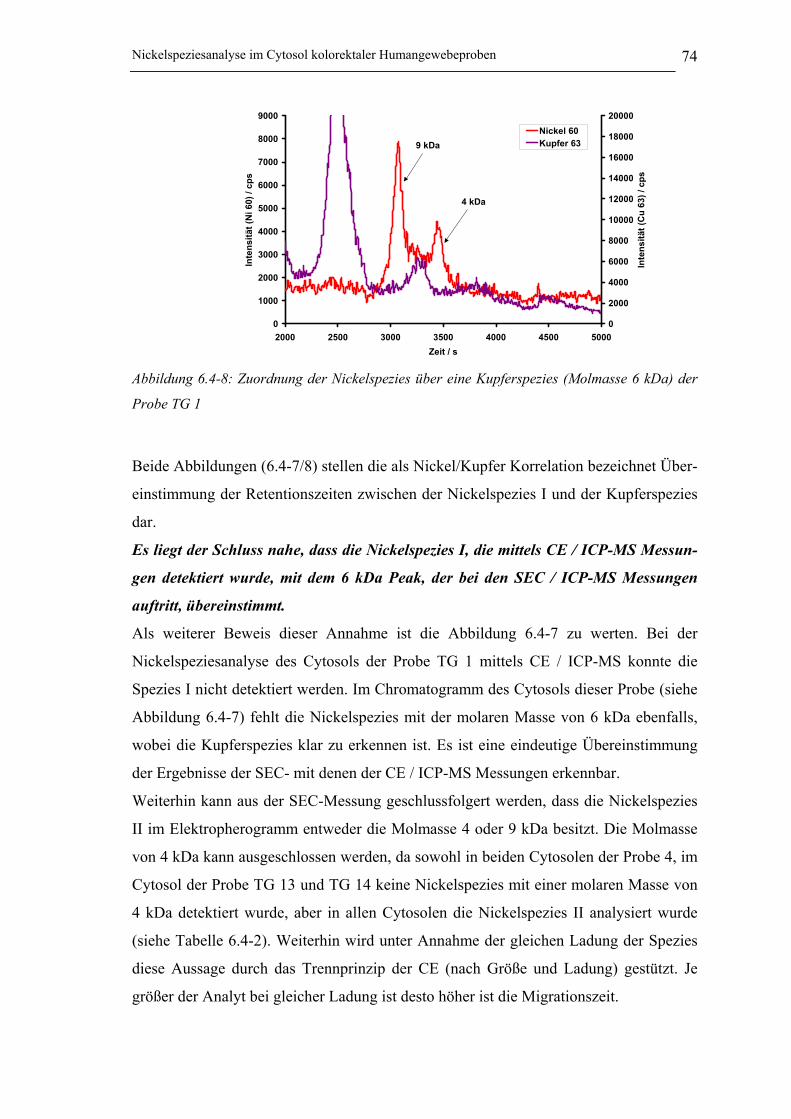
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 74
Abbildung 6.4-8: Zuordnung der Nickelspezies über eine Kupferspezies (Molmasse 6 kDa) der
Probe TG 1
Beide Abbildungen (6.4-7/8) stellen die als Nickel/Kupfer Korrelation bezeichnet Über-
einstimmung der Retentionszeiten zwischen der Nickelspezies I und der Kupferspezies
dar.
Es liegt der Schluss nahe, dass die Nickelspezies I, die mittels CE / ICP-MS Messun-
gen detektiert wurde, mit dem 6 kDa Peak, der bei den SEC / ICP-MS Messungen
auftritt, übereinstimmt.
Als weiterer Beweis dieser Annahme ist die Abbildung 6.4-7 zu werten. Bei der
Nickelspeziesanalyse des Cytosols der Probe TG 1 mittels CE / ICP-MS konnte die
Spezies I nicht detektiert werden. Im Chromatogramm des Cytosols dieser Probe (siehe
Abbildung 6.4-7) fehlt die Nickelspezies mit der molaren Masse von 6 kDa ebenfalls,
wobei die Kupferspezies klar zu erkennen ist. Es ist eine eindeutige Übereinstimmung
der Ergebnisse der SEC- mit denen der CE / ICP-MS Messungen erkennbar.
Weiterhin kann aus der SEC-Messung geschlussfolgert werden, dass die Nickelspezies
II im Elektropherogramm entweder die Molmasse 4 oder 9 kDa besitzt. Die Molmasse
von 4 kDa kann ausgeschlossen werden, da sowohl in beiden Cytosolen der Probe 4, im
Cytosol der Probe TG 13 und TG 14 keine Nickelspezies mit einer molaren Masse von
4 kDa detektiert wurde, aber in allen Cytosolen die Nickelspezies II analysiert wurde
(siehe Tabelle 6.4-2). Weiterhin wird unter Annahme der gleichen Ladung der Spezies
diese Aussage durch das Trennprinzip der CE (nach Größe und Ladung) gestützt. Je
größer der Analyt bei gleicher Ladung ist desto höher ist die Migrationszeit.
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 639 kDa
4 kDa
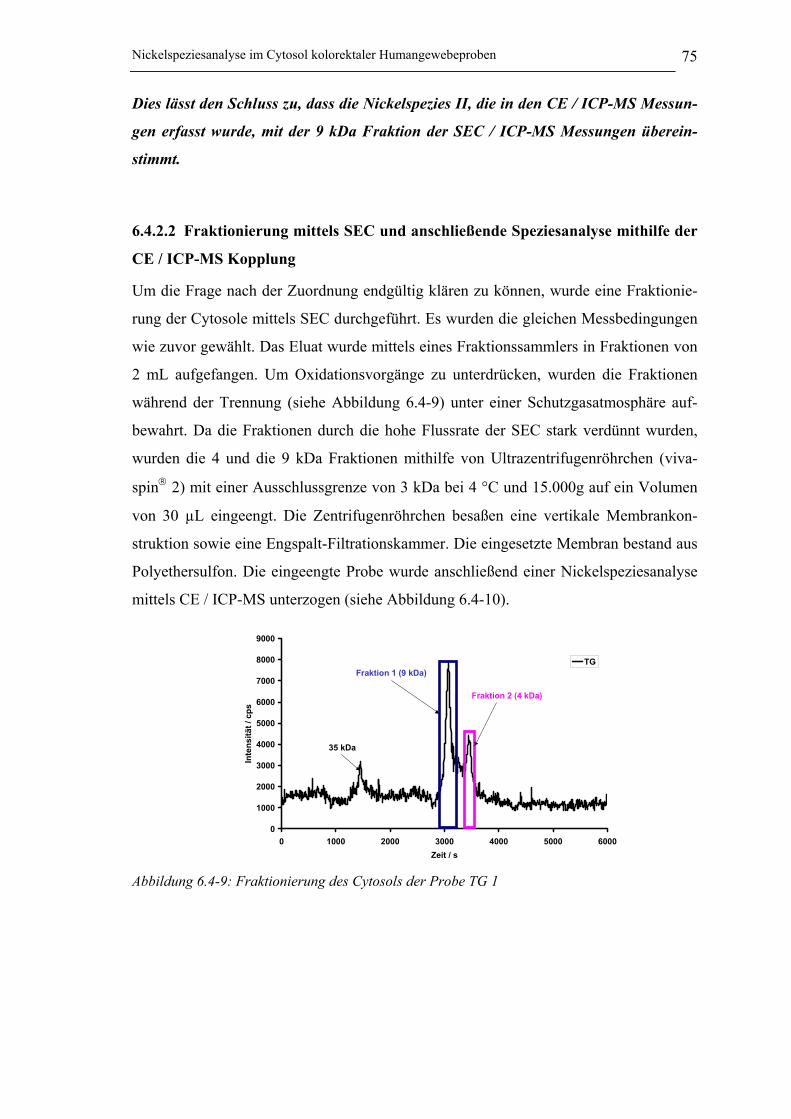
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 75
Dies lässt den Schluss zu, dass die Nickelspezies II, die in den CE / ICP-MS Messun-
gen erfasst wurde, mit der 9 kDa Fraktion der SEC / ICP-MS Messungen überein-
stimmt.
6.4.2.2 Fraktionierung mittels SEC und anschließende Speziesanalyse mithilfe der
CE / ICP-MS Kopplung
Um die Frage nach der Zuordnung endgültig klären zu können, wurde eine Fraktionie-
rung der Cytosole mittels SEC durchgeführt. Es wurden die gleichen Messbedingungen
wie zuvor gewählt. Das Eluat wurde mittels eines Fraktionssammlers in Fraktionen von
2 mL aufgefangen. Um Oxidationsvorgänge zu unterdrücken, wurden die Fraktionen
während der Trennung (siehe Abbildung 6.4-9) unter einer Schutzgasatmosphäre auf-
bewahrt. Da die Fraktionen durch die hohe Flussrate der SEC stark verdünnt wurden,
wurden die 4 und die 9 kDa Fraktionen mithilfe von Ultrazentrifugenröhrchen (viva-
spin 2) mit einer Ausschlussgrenze von 3 kDa bei 4 °C und 15.000g auf ein Volumen
von 30 µL eingeengt. Die Zentrifugenröhrchen besaßen eine vertikale Membrankon-
struktion sowie eine Engspalt-Filtrationskammer. Die eingesetzte Membran bestand aus
Polyethersulfon. Die eingeengte Probe wurde anschließend einer Nickelspeziesanalyse
mittels CE / ICP-MS unterzogen (siehe Abbildung 6.4-10).
Abbildung 6.4-9: Fraktionierung des Cytosols der Probe TG 1
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t / c
ps
TG
35 kDa
Fraktion 1 (9 kDa)
Fraktion 2 (4 kDa)
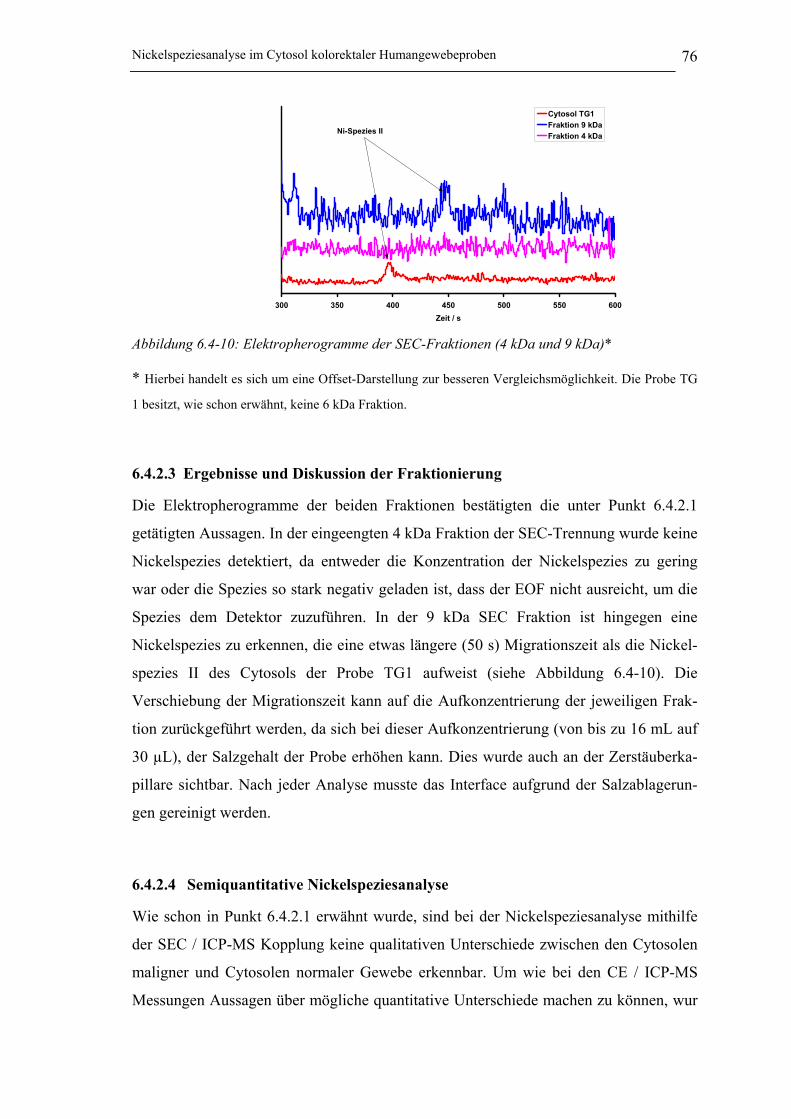
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 76
Abbildung 6.4-10: Elektropherogramme der SEC-Fraktionen (4 kDa und 9 kDa)*
* Hierbei handelt es sich um eine Offset-Darstellung zur besseren Vergleichsmöglichkeit. Die Probe TG
1 besitzt, wie schon erwähnt, keine 6 kDa Fraktion.
6.4.2.3 Ergebnisse und Diskussion der Fraktionierung
Die Elektropherogramme der beiden Fraktionen bestätigten die unter Punkt 6.4.2.1
getätigten Aussagen. In der eingeengten 4 kDa Fraktion der SEC-Trennung wurde keine
Nickelspezies detektiert, da entweder die Konzentration der Nickelspezies zu gering
war oder die Spezies so stark negativ geladen ist, dass der EOF nicht ausreicht, um die
Spezies dem Detektor zuzuführen. In der 9 kDa SEC Fraktion ist hingegen eine
Nickelspezies zu erkennen, die eine etwas längere (50 s) Migrationszeit als die Nickel-
spezies II des Cytosols der Probe TG1 aufweist (siehe Abbildung 6.4-10). Die
Verschiebung der Migrationszeit kann auf die Aufkonzentrierung der jeweiligen Frak-
tion zurückgeführt werden, da sich bei dieser Aufkonzentrierung (von bis zu 16 mL auf
30 µL), der Salzgehalt der Probe erhöhen kann. Dies wurde auch an der Zerstäuberka-
pillare sichtbar. Nach jeder Analyse musste das Interface aufgrund der Salzablagerun-
gen gereinigt werden.
6.4.2.4 Semiquantitative Nickelspeziesanalyse
Wie schon in Punkt 6.4.2.1 erwähnt wurde, sind bei der Nickelspeziesanalyse mithilfe
der SEC / ICP-MS Kopplung keine qualitativen Unterschiede zwischen den Cytosolen
maligner und Cytosolen normaler Gewebe erkennbar. Um wie bei den CE / ICP-MS
Messungen Aussagen über mögliche quantitative Unterschiede machen zu können, wur
300 350 400 450 500 550 600Zeit / s
Cytosol TG1 Fraktion 9 kDaFraktion 4 kDaNi-Spezies II
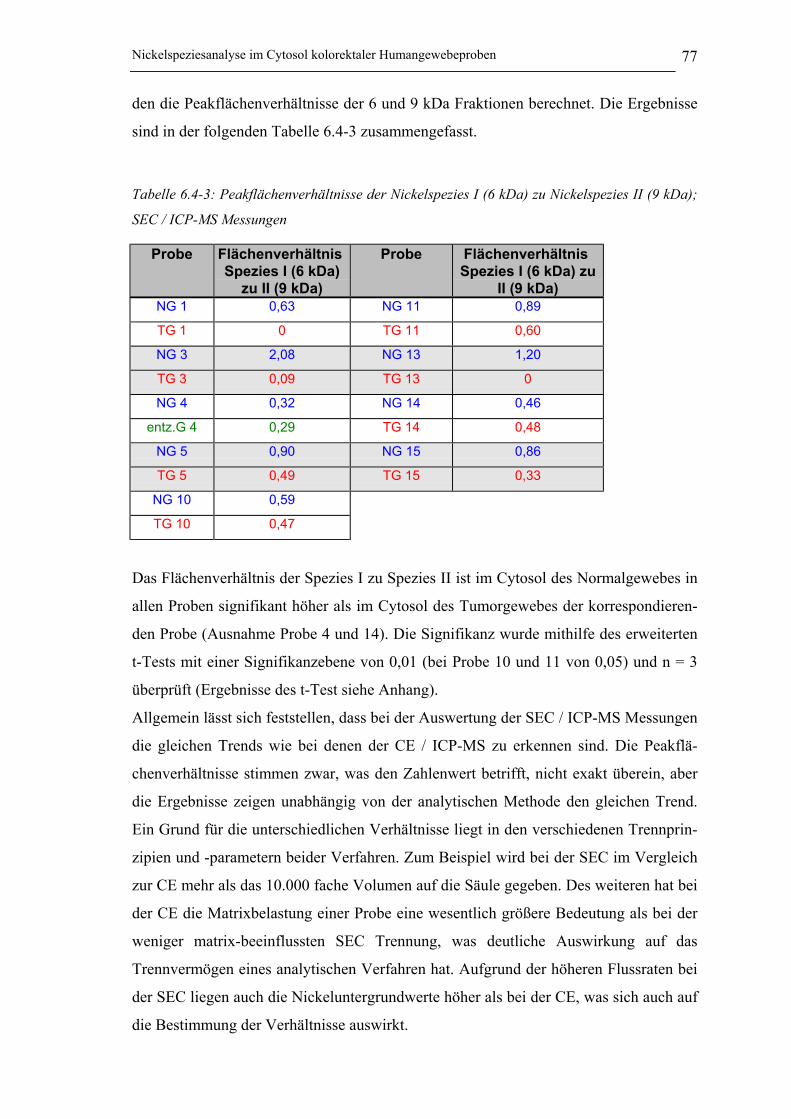
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 77
den die Peakflächenverhältnisse der 6 und 9 kDa Fraktionen berechnet. Die Ergebnisse
sind in der folgenden Tabelle 6.4-3 zusammengefasst.
Tabelle 6.4-3: Peakflächenverhältnisse der Nickelspezies I (6 kDa) zu Nickelspezies II (9 kDa);
SEC / ICP-MS Messungen
Probe Flächenverhältnis Spezies I (6 kDa)
zu II (9 kDa)
Probe Flächenverhältnis Spezies I (6 kDa) zu
II (9 kDa)NG 1 0,63 NG 11 0,89
TG 1 0 TG 11 0,60
NG 3 2,08 NG 13 1,20
TG 3 0,09 TG 13 0
NG 4 0,32 NG 14 0,46
entz.G 4 0,29 TG 14 0,48
NG 5 0,90 NG 15 0,86
TG 5 0,49 TG 15 0,33
NG 10 0,59
TG 10 0,47
Das Flächenverhältnis der Spezies I zu Spezies II ist im Cytosol des Normalgewebes in
allen Proben signifikant höher als im Cytosol des Tumorgewebes der korrespondieren-
den Probe (Ausnahme Probe 4 und 14). Die Signifikanz wurde mithilfe des erweiterten
t-Tests mit einer Signifikanzebene von 0,01 (bei Probe 10 und 11 von 0,05) und n = 3
überprüft (Ergebnisse des t-Test siehe Anhang).
Allgemein lässt sich feststellen, dass bei der Auswertung der SEC / ICP-MS Messungen
die gleichen Trends wie bei denen der CE / ICP-MS zu erkennen sind. Die Peakflä-
chenverhältnisse stimmen zwar, was den Zahlenwert betrifft, nicht exakt überein, aber
die Ergebnisse zeigen unabhängig von der analytischen Methode den gleichen Trend.
Ein Grund für die unterschiedlichen Verhältnisse liegt in den verschiedenen Trennprin-
zipien und -parametern beider Verfahren. Zum Beispiel wird bei der SEC im Vergleich
zur CE mehr als das 10.000 fache Volumen auf die Säule gegeben. Des weiteren hat bei
der CE die Matrixbelastung einer Probe eine wesentlich größere Bedeutung als bei der
weniger matrix-beeinflussten SEC Trennung, was deutliche Auswirkung auf das
Trennvermögen eines analytischen Verfahren hat. Aufgrund der höheren Flussraten bei
der SEC liegen auch die Nickeluntergrundwerte höher als bei der CE, was sich auch auf
die Bestimmung der Verhältnisse auswirkt.

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 78
Bei der Nickelspeziesanalyse mithilfe der SEC / ICP-MS zeigen die gleichen Cyto-
solproben Auffälligkeiten wie bei der Analyse mittels CE / ICP-MS. In beiden Fällen
konnte beim Cytosol der Probe TG 1 und TG 13 die Spezies I nicht detektiert werden.
Die Ergebnisse der SEC / ICP-MS Analyse der Cytosole der Proben 4 und 14 stimmen
ebenfalls mit den Ergebnissen der CE / ICP-MS Analyse überein. Dies unterstreicht die
in Punkt 6.3.1 getätigten Aussagen.
Es lässt sich festhalten, dass die Ergebnisse der Nickelspeziesanalyse mittels SEC /
ICP-MS Kopplung mit den Ergebnisse der Analyse mithilfe der CE / ICP-MS Kopp-
lung übereinstimmen.

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 79
6.5 Verdrängungsreaktionen der Ni2+- durch Zn2+-Ionen
Um weitere Informationen über die Nickelspezies im Cytosol von Gewebeproben des
humanen Intestinaltrakts zu bekommen, wurde ein Cytosol mit einer Zn2+ bzw. Ni2+
Lösung versetzt. Mit diesen Versuchen sollte geklärt werden, wie stark die Nickelbin-
dungen an den Bindungspartner sind. Weiterhin sollte das Verhalten der Nickelspezies
bei weiterer Zugabe von Ni2+-Ionen analysiert werden. In den Fällen, in denen Nickel-
Ionen nur locker an den organischen Bindungspartner gebunden sind, können sie durch
Zink-Ionen verdrängt werden. Für die Analyse wurden die beiden Cytosole (NG/TG)
der Probe 10 genutzt, um den Einfluss auf beide Gewebearten zu untersuchen.
6.5.1 Durchführung der Verdrängungsreaktion
Zum Ansetzen einer 1000 µg/L Zinklösung wurde die entsprechende Menge von
Zinknitrat eingewogen und in gereinigtem Tris-HNO3 Puffer (pH= 7,4) gelöst. Ebenso
wurde beim Ansetzten der Nickellösung verfahren (Nickelnitrat). Die erhaltende
Nickellösung wurde noch einmal 1:10 mit Tris-HNO3 Puffer verdünnt (wurde nur bei
dem Cytosol der malignen Probe verwendet). Von jeder Standardlösung wurden 50 µL
entnommen und auf 450 µL Cytosol gegeben. Die Probe ohne Zugabe von Stan-
dardlösung wurde in gleichem Verhältnis mit Tris-HNO3 Puffer verdünnt, damit die
Vergleichbarkeit der Proben gegeben war. Nach einer Inkubationszeit von 5 Minuten
wurden die sieben Cytosole (zwei Cytosole ohne Zugabe und fünf mit Zugabe von
Standardlösung) jeweils einer Speziesanalyse mittels SEC / ICP-MS unterzogen. Diese
Technik wurde gewählt, da bei der SEC Trennung alle Nickelspezies detektiert werden
und so der Einfluss der Standardlösung auf alle Spezies untersucht werden konnte und
nicht wie bei der CE / ICP-MS nur der Einfluss auf die Nickelspezies I und II. Es
wurden die schon bekannten Messparameter für die SEC / ICP-MS gewählt. Die folgen-
den Abbildungen 6.5-1 und 2 zeigen die Chromatogramme der Messreihe.
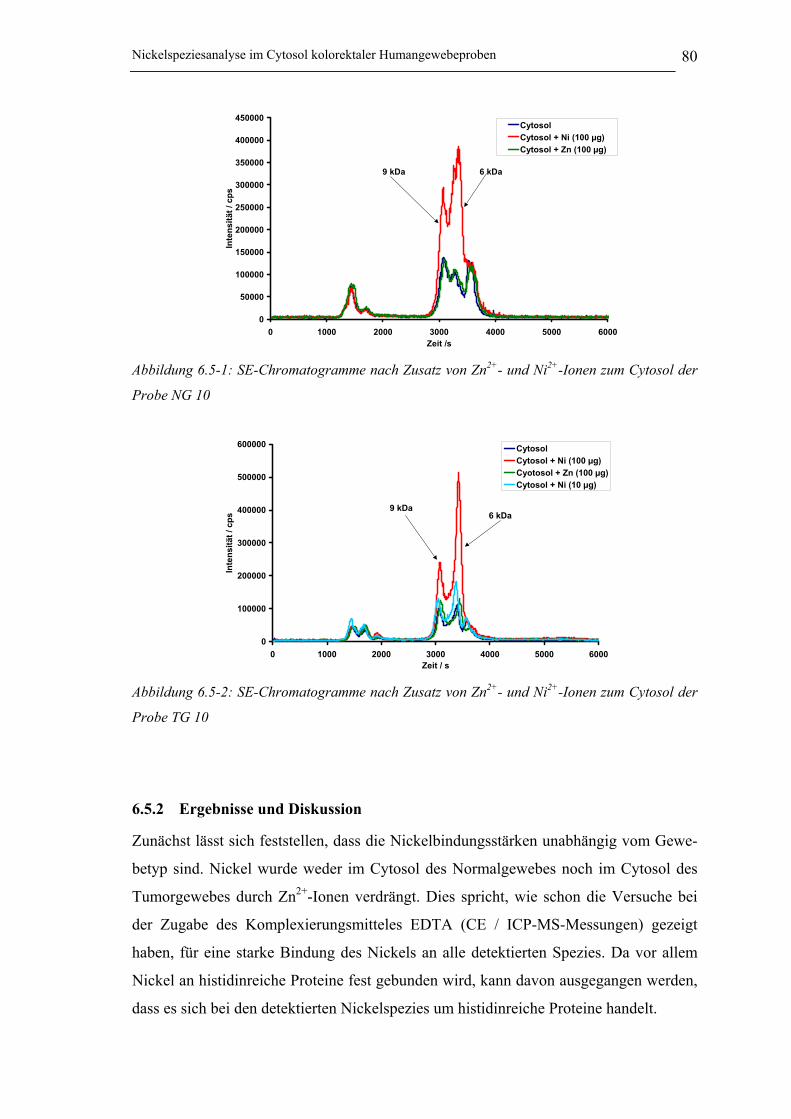
Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 80
Abbildung 6.5-1: SE-Chromatogramme nach Zusatz von Zn2+- und Ni2+-Ionen zum Cytosol der
Probe NG 10
Abbildung 6.5-2: SE-Chromatogramme nach Zusatz von Zn2+- und Ni2+-Ionen zum Cytosol der
Probe TG 10
6.5.2 Ergebnisse und Diskussion
Zunächst lässt sich feststellen, dass die Nickelbindungsstärken unabhängig vom Gewe-
betyp sind. Nickel wurde weder im Cytosol des Normalgewebes noch im Cytosol des
Tumorgewebes durch Zn2+-Ionen verdrängt. Dies spricht, wie schon die Versuche bei
der Zugabe des Komplexierungsmitteles EDTA (CE / ICP-MS-Messungen) gezeigt
haben, für eine starke Bindung des Nickels an alle detektierten Spezies. Da vor allem
Nickel an histidinreiche Proteine fest gebunden wird, kann davon ausgegangen werden,
dass es sich bei den detektierten Nickelspezies um histidinreiche Proteine handelt.
0
50000
100000
150000
200000
250000
300000
350000
400000
450000
0 1000 2000 3000 4000 5000 6000Zeit /s
Inte
nsitä
t / c
ps
CytosolCytosol + Ni (100 µg)Cytosol + Zn (100 µg)
9 kDa 6 kDa
0
100000
200000
300000
400000
500000
600000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t / c
ps
CytosolCytosol + Ni (100 µg)Cyotosol + Zn (100 µg)Cytosol + Ni (10 µg)
9 kDa6 kDa

Nickelspeziesanalyse im Cytosol kolorektaler Humangewebeproben 81
Möglicherweise ist die Konzentration der unspezifischen Bindungspartner im Cytosol
relativ hoch und erst durch weitere Zugabe von Ni2+-Ionen erfolgt eine quantitative
Bindungsbildung.
Diese Messungen (siehe Abb. 6.5-1 und 6.5.2) haben bestätigt, dass die
Bindungspartner für Nickel in Spezies I und II besonders starke Bindungen mit Ni2+-
Ionen eingehen. Um weitere Aussagen diesbezüglich treffen zu können, müsste das
molare Massenverhältnis genauer bestimmt werden.
Das relative Verhältnis der Spezies I zu Spezies II verändert sich nach Zugabe der Ni2+-
Ionen deutlich. Die Intensität der Spezies II (6 kDa) steigt stärker als der Spezies I (9
kDa) an. Diese Veränderung vollzieht sich sowohl im Cytosol des malignen Gewebes
als auch im Cytosol des normalen Gewebes (siehe Abb. 6.1-1 und 6.5-2), was als
weiteres Indiz gewertet werden kann, dass die Bindungspartner beider Spezies bevor-
zugte Nickelbindungspartner darstellen. Daher liegt es nahe, dass es sich sowohl bei
Spezies I als auch bei Spezies II um histidinreiche Proteine handelt.
Es lässt sich festhalten, dass die Bindungspartner der Spezies I und der Spezies II
bevorzugte Bindungspartner für Ni2+ Ionen sind.

Zusammenfassung 82
7 Zusammenfassung
In der vorliegenden Arbeit konnte gezeigt werden, dass mit den vorgestellten ICP-MS
Kopplungen (CE / ICP-MS und SEC / ICP-MS) eine Nickelspeziesanalyse im Cytosol
von humanen Gewebeproben des Intestinaltrakts möglich ist. Hierzu muss das verwen-
dete ICP-MS einigen Ansprüchen gerecht werden. Eine solche Speziesanalyse ist nur
mit dem Einsatz von Platinkonen und eines Shield-Torch-Systems durchführbar, da
sonst die Nachweisgrenze nicht ausreicht. Außerdem müssen alle Gefäße und Chemika-
lien speziell gereinigt und dadurch dekontaminiert werden.
Die Optimierung der Cytosolherstellung hat gezeigt, dass nur ein mechanischer Zellauf-
schluss eingesetzt werden kann, um das Gewebe des humanen Intestinaltrakts effizient
genug aufzuschließen. Zur Probenvorbereitung für die CE / ICP-MS-Kopplung hat sich
die Acetonitrilfällung als geeignet herausgestellt.
Der Vorteil der CE / ICP-MS Kopplung gegenüber der SEC / ICP-MS-Kopplung liegt
in dem geringen Probenaufgabenvolumen von wenigen Nanolitern, sodass auch sehr
kleine Mengen an Gewebeproben analysiert werden können. Nach der Optimierung
aller Messparameter konnten die Elektropherogramme von elf Cytosolen humaner
Gewebeproben aufgenommen werden. Sie wiesen keine qualitativen Unterschiede
zwischen dem Cytosol des malignen Gewebes und des normalen Gewebes auf. Es
wurden jeweils die gleiche Anzahl von Nickelspezies (Nickelspezies I und II) im
Cytosol beider Gewebearten gefunden. Die Zuordnung der Nickelspezies zu Spezies I
und II erfolgte über die Korrelation der Nickelspezies I zu einer Kupferspezies
(möglicherweise handelt es sich hierbei um kupferbeladenes Metallothionein). Nach
Auswertung der Peakflächen-verhältnisse von Nickelspezies I zu II zeigte sich bei neun
von elf Cytosolen ein signifi-kanter Unterschied zwischen dem Cytosol maligner
Gewebeproben und ihrer korrespon-dierenden Normalgewebeproben. Mittels
Nickelspeziesanalyse konnte das Cytosol einer entzündeten Gewebeprobe als das
Cytosol eines solchen Gewebetyps identifiziert werden. Denn sowohl die
Peakfächenverhältnisse als auch die Elektropherogramme beider Cytosole (normales
Gewebe und entzündetes Gewebe) waren nahezu gleich. Mithilfe der vorgestellten
Kopplung ist es also möglich, das Cytosol von malignem Gewebe von dem Cytosol von
normalem Gewebe zu unterscheiden.
Mithilfe eines EOF-Markers konnte die Ladung der beiden Nickelspezies bestimmt
werden. In beiden Fällen liegen die Spezies positiv geladen vor. Die Ergebnisse der

Zusammenfassung 83
Gesamtnickelbestimmung mittels TXRF stimmten gut mit den Ergebnissen der CE /
ICP-MS Analysen überein. Cytosole mit geringen Gesamtnickelkonzentrationen
wurden z.B. auch bei der CE / ICP-MS Messungen mit geringen Intensitäten für Nickel
analysiert.
Im weiteren Verlauf der Arbeit hat sich gezeigt, dass eine Nickelspeziesanalyse auch
mittels einer SEC / ICP-MS Kopplung möglich ist. Da die SEC sehr schonend und nur
nach Größe des Analyten trennt und eine Probenvorbereitung des Cytosols nicht nötig
war, konnten im Gegensatz zur CE-Trennung mehr Spezies (bis zu 6 Nickelspezies,
abhängig vom Permeationsbereich (4-35 kDa) der Säule) im Cytosol beider Gewebear-
ten detektiert werden.
Weiterhin stellte sich heraus, dass es sich bei den detektierten Nickelspezies um nicht
oxidationsempfindliche Substanzen handelte, da die Zugabe eines Antioxidants bei
einer Trennzeit von fast zwei Stunden keinen Einfluss auf das Aussehen der Chro-
matogramme hatte.
Bei der Auswertung der SE-Chromatogramme waren, wie schon bei den CE / ICP-MS
Messungen, keine qualitativen Unterschiede zwischen dem Cytosol der malignen und
der normalen Gewebeprobe erkennbar. Um die Ergebnisse der SEC / ICP-MS mit denen
der CE / ICP-MS vergleichen zu können, mussten die Nickelspezies I und II den ent-
sprechenden Peaks der SEC-Anaylse zugeordnet werden. Dabei stellte sich heraus, dass
die Nickelspezies I eine molare Masse von ca. 6 kDa und die Spezies II eine molare
Masse von ca. 9 kDa besitzt. Die Peakflächenverhältnisse von Spezies I zu Spezies II
waren im Cytosol des normalen Gewebes signifikant höher als im Cytosol des malignen
Gewebes, während sie im Cytosol des entzündeten Gewebes mit dem korrespondieren-
den Cytosol des Normalgewebes identisch waren. Mithilfe der SEC / ICP-MS Messun-
gen konnten die Ergebnisse der CE / ICP-MS Messungen verifiziert werden.
Um die Bindungsstärke von Nickel mit dem organischen Bindungspartner zu untersu-
chen, wurden Verdrängungsreaktionen durch Zn2+-Ionen initiiert. Auch bei der Zugabe
von hohen Zn2+-Konzentrationen konnte die Nickel nicht durch Zn2+-Ionen verdrängt
werden, was für eine starke Bindung des Nickels an den entsprechenden Bindungspart-
ner spricht. Ferner ließ sich Nickel erst nach der Zugabe konzentrierter EDTA-Lösung
aus dem Komplex entfernen, wodurch die Hypothese der hohen Bindungsstärke gestützt
wurde.

Zusammenfassung 84
Allgemein ist bekannt, dass histidinreiche Proteine privilegierte Bindungspartner für
Nickel sind. Zu diesen zählt ein großer Teil der Onkogene, die durch Genprodukte (so-
genannte Onkoproteine) an der Kontrolle von Wachstums- und Differenzierungsprozes-
sen von Zellen beteiligt sind, die in Tumorzellen ausser Kontrolle geraten sind (z.B.
undefinierter Zellwachstum). Es liegt der Schluss nahe, dass die Onkoproteine im
Cytosol von malignen Gewebeproben in anderen Verhältnissen zueinander vorliegen als
im Cytosol von normalen Gewebeproben, in denen ein normales Zellwachstum abläuft.
Dieses würde die unterschiedlichen Flächenverhältnisse der Nickelspezies I zu II erklä-
ren.
Mithilfe von Proteindatenbanken wurden nach in Frage kommenden Proteinen gesucht
[185]. Hierzu wurden folgende Kriterien berücksichtigt:
• Molare Masse zwischen 4 und 10 kDa
• Histidinreiche Proteine
• Onkogene
Eine große Anzahl von Transkriptions-Faktoren erfüllen diese Kriterien, hier vor allem
Ap-1 (C-Jun und C-Fos, Proto-Onkogen-Produkte). Hierbei handelt es sich um histidin-
reiche Proteine mit einer molaren Masse von 6549 Da (4832 Da C-Jun Homodimer)
bzw. 6694 Da. Die Proteine liegen im Massenbereich der Nickelspezies I. Das Onkogen
C-Myb, der humane Estrogen Rezeptor, und das Rezeptor Protein Rev-Erb(α) sind
ebenfalls histidinreiche Proteine mit molaren Massen von 6130, 9675 und 10863 Da.
Diese Proteine spielen eine zentrale Rolle bei der Entstehung von Darmkrebs.
Die Ergebnisse (molare Masse, Unterschiede der Flächenverhältnisse der Nickelspezies
I zu II) der Nickelspeziesanalyse mittels beider Kopplungen weisen stark darauf hin,
dass es sich bei den Nickelspezies I und II um die oben genannten Onkogene handelt.
Das gleiche Verfahren mit auf Kupfer abgestimmten Suchkriterien wurde zur Identifi-
zierung der Kupferspezies (6-7 kDa), die mit der Nickelspezies I in Korrelation steht,
verwendet. Die Suche ergab, dass es sich sehr wahrscheinlich um die Kupfer-Form des
Metallothionein I (MT 1) handeln muss, da sowohl das Vorkommen im Kolon als gesi-
chert gilt als auch die molare Masse von 6,2 kDa mit der im Experiment ermittelten
molaren Masse übereinstimmt.

Zusammenfassung 85
In dieser Arbeit konnte gezeigt werden, dass die eingesetzten Kopplungstechniken
nach Optimierung zur Nickelspeziesanalyse im Cytosol humaner Gewebeproben ge-
eignet sind. Unterschiede hinsichtlich Nickelspezies im Cytosol von malignen Gewebe
und Cytosol von normalen Gewebe konnten aufgezeigt werden und können als
Grundlage weiterer Untersuchungen zur Qualifizierung der entwickelten Methoden
als Diagnoseinstrument dienen .

Ausblick 86
8 AusblickMit dieser Arbeit wurde der Grundstein zur Nickelspeziesanalyse im Cytosol humaner
Gewebeproben gelegt. So könnten zum Beispiel spezielle Beschichtungen für die CE-
Kapillare zur Verbesserung der Trennung entwickelt werden, mit denen weitere
Nickelspezies detektiert werden könnten. Ein weiterer interessanter Punkt sind die
höher molekularen Bestandteile des Cytosols. Bei der eingesetzten SEC-Säule konnten
nur Spezies bis zu einer maximalen molaren Masse von 35 kDa detektiert werden.
Durch den Einsatz von SEC-Säulen mit einem höheren Permeationsbereich (z.B. 200
kDa) können die höher molekularen Spezies getrennt werden und es werden weitere
Informationen über die Zusammensetzung der Nickelspezies in den Gewebecytosolen
zugänglich. Der Einsatz von analytischen SEC-Säulen anstelle von präparativen Säulen
wird zudem zu einer verbesserten Auftrennung der Nickelspezies führen.
Eine weitere wichtige Frage stellt die Quantifizierung der Nickelspezies dar. Hierzu
müsste eine online Isotopenverdünnungsanalyse mittels CE / ICP-MS entwickelt wer-
den, um den Gehalt jeder einzeln Nickelspezies bestimmen zu können. Dafür muss ein
Komplexbildner für einen Nickelstandard gefunden werden, der Nickel-Ionen komple-
xiert und nach außen positiv geladen ist, da der Nickel-Komplex sonst bei der CE-
Trennung aufgrund der Polung der CE vom Detektor wegwandert.
Um die Identifizierung (mithilfe von Online-Proteindatenbanken) der Nickelspezies I
und II zu verifizieren, könnten die Spezies mithilfe einer SEC oder CE / ESI-MS/MS
Kopplung (ESI= Elektrosprayionisation) weitergehend charakterisiert und identifiziert
werden. Hierzu müssten allerdings beide Trennmethoden speziell auf die ESI-MS/MS
abgestimmt werden, d.h., es müssten gegebenenfalls neue Trennbedingungen für die
SEC bzw. CE entwickelt werden.
Um die Ergebnisse zu verifizieren, könnten kommerziell erhältliche Zelllinien genutzt
werden. Durch Inkubation mit Nickel-Ionen könnte geprüft werden, ob sich in den Zel-
len die gleichen Nickelspezies wie im Cytosol von realen Proben bilden. Anschließend
könnte eine CE oder SEC / ESI-MS/MS Analyse des Cytosols der Zelllinen durchge-
führt werden, um eine Identifizierung durchzuführen.
Ein langfristiges Ziel könnte in der medizinischen Diagnostik liegen. Wie die Arbeit
gezeigt hat, können Cytosole von malignen Gewebeproben vom Cytosol einer
normalen bzw. entzündeten Gewebeproben mithilfe der vorgestellten analytischen
Methoden unterschieden werden. Da es sich bei den Nickelspezies I und II mit hoher

Ausblick 87
Wahrscheinlichkeit um Onkoproteine handelt, kann davon ausgegangen werden, dass
sich schon bei der Entstehung von Krebs die Verhältnisse der Onkoproteine zueinander
ändern. Genau diese Veränderung kann mittels CE / ICP-MS bestimmt werden, sodass
eine Alternative zu molekularbiologischen Untersuchungen zur Verfügung stehen
würde. Weiterhin müsste geklärt werden, ob eine Abhängigkeit der
Peakflächenverhältnisse vom Tumorstadium vorliegt.
Ein weiteres langfristiges Anwendungsgebiet der durchgeführten Experimente könnte
der Einsatz der Nickelspezies als Additiv zu herkömmlichen Chemotherapeutika sein.
Nickel und Nickelverbindungen sind zwar karzinogen, ab einer bestimmten Konzentra-
tion aber auch cytotoxisch. Nach der gesicherten Identifizierung der Nickelspezies,
könnten diese in einer cytotoxischen Konzentration den herkömmlichen Chemothera-
peutika zugeführt werden und auf diesem Weg maligne Zellen zerstören.
Die Arbeit hat auch gezeigt, dass andere Elemente in diesem Zusammenhang wichtig
sein könnten. Hier kann als Beispiel Kupfer genannt werden. Es konnten drei Kupfer-
spezies detektiert werden. Eine davon ist die bereits erwähnte erste Isoform des
Metallothioneins (MT I). Bei den anderen beiden Spezies könnte es sich möglicher-
weise um die kupferbeladene Superoxid-Dismutase (SOD) und um das SOD assoziierte
Chaperon handeln. Die SODs sind im Organismus für die Entgiftung toxischer Sauer-
stoffspezies verantwortlich, die Chaperone gehören zu der Gruppe der intrazellulären
Proteine, die die Ausbildung einer definierten Raumstruktur (durch Faltung) neu-
synthetisierter Polypetidketten beschleunigt. Sowohl veränderte Raumstrukturen von
Proteinen, die zu Funktionsänderungen führen können wie auch toxische Sauer-
stoffspezies sind an der Entstehung von Krebs beteiligt. Mit der SEC / ICP-MS
Kopplung steht eine Methode zur Verfügung um mögliche Unterschiede bezüglich der
SOD und den Chaperonen im Cytosol beider Gewebearten zu bestimmen. Mit diesen
Informationen könnten vielleicht neue Behandlungsmethoden zur Krebsbekämpfung
entwickelt werden, da dann fundierte Informationen über Veränderung der SOD (Kup-
fer beladen) und der Chaperone im Cytosol der malignen Gewebeprobe zur Verfügung
stehen.
Diese Punkte zeigen, dass sich viele weitere Forschungsaspekte aus der vorgelegten
Arbeit ergeben können.

Literaturverzeichnis 88
9 Literaturverzeichnis
[1] Michalke, B., Schramel, P.
J Chromatogr A 750 (1996), 51
[2] Schwedt, G.
Chemie in unserer Zeit 31 (1997), 183
[3] Lobinski, R.
Appl Spectrom 51 (1997), 260
[4] Lobinski, R.
Spectrochim Acta 53 B (1998), 177
[5] Zoorob, G., McKiernan, W., Caruso, J.
Anal Chim Acta 128 (1998), 145
[6] Römpp :
Chemie Lexikon, 9. Auflage, Thieme Verlag,
Stuttgart, New York, (1995), 794
[7] Lin, S., Hopfer, S., Brennan, S., Sunderman Jr. F.
Res Comm Chem Path Pharm 65 (1989), 275
[8] Sunderman Jr. F., Costa, E., Fraser, C., Hui, G., Levine, J., Tse, T.
Annals Clin Lab Sci 11 (1981), 488
[9] Sunderman Jr. F., Varghese, A., Kroftova. O., Grbac-Ivankovic, Kotyza, J., Datta,
A., Davis, M., Bal, W., Kasprzak, K.
Mol Reprod Dev 44 (1996), 507
[10] Haspel, J., Sunderman Jr. F., Hopfer, S., Henjum, D., Brandt-Rauf, P., Weinstein,
B., Nishimura, S., Yamaizumi, Z., Pincus, M.
Res Comm Chem Path Pharm 79 (1993), 131
[11] Gilbert, J., Ramakrihna, J., Sunderman Jr. F., Wright, A., Plaut, A.
Infect Immun 63 (1995), 2682
[12] Sunderman Jr. F., Plowman, M., Kroftova, O., Grbac-Ivankovic, S., Foglia, L.,
Crivello, J.
Pharmacol Toxicol 76 (1995), 178
[13] Kotyza, J., Varghese, A., Korza, G., Sunderman Jr. F.
Biochim Biophys Acta 1382 (1998), 266

Literaturverzeichnis 89
[14] Templeton, D., Sunderman Jr. F., Herber, R.
Sci Total Environ 148 (1994), 243
[15] Grbac-Ivankovic, S., Antoijczuk, K., Varghese, A.
Mol Reprod Dev 38 (1994), 256
[16] Jacobsen, N., Brennhovd, I., Jonsen, J.
J Biol Buccale 5 (1977), 169
[17] Oskarsson, A., Tjalve, H.
Acta Pharmacol Toxicol 45 (1979), 306
[18] Sarkar, B.,
Nickel in blood and kidney, Nickel Toxicology
Jr. eds. London, Academic Press, (1980), 81
[19] Herlant-Peers, M., Hildebrand, F., Kerckaert, J.
Carcinogenesis 4 (1983), 387
[20] Templeton, D., Sarkar, B.
J Biochem 230 (1985), 35
[21] Cangul, H., Broday, L., Salnikow, K., Sutherland, J., Peng, W., Zhang, Q.
Poltaratsky, V., Yee, H., Zoroddu, M., Costa, M.
Toxicol Lett 127 (2002), 69
[22] Savolainen, H.
Rev Environ Health 11 (1996), 167
[23] Janknecht, R., Sander, C., Pongs, O.
FEBS Lett 295 (1991), 1
[24] Fu, C., Olson, J., Maier, R.
Proc Natl Acad Sci 92 (1995), 2333
[25] Berg, J.
Science 232 (1986), 485
[26] Denkhaus, E., Salnikow, K.
Crit Rev Oncol Hematol 42 (2002), 35
[27] Barceloux, D.
Clin Toxicol 37 (1999), 239
[28] Kondo, K., Ozaki, T., Nakamura, Y., Sakiyama, S.
Biochem Biophys Res Commun 216 (1995), 209

Literaturverzeichnis 90
[29] Ozaki, T., Nakamura, Y., Enomoto, H., Hirose, M., Sakiyama, S.
Cancer Res 55 (1993), 895
[30] Skoog, D.A., Leary, J.J.
Instrumentelle Analytik
Springer (1996) , Berlin, Heidelberg, New York, 213/254/712
[31] Byrdy, F., Caruso, J.
Environ Sci Technol 28 (1994), 528
[32] Byrdy, F., Caruso, J. in
Sievers (Ed.), Selective Detectors, Chemical Analysis Series, Vol. 131
Wiley, 1995, Weinheim, San Francisco
[33] Hill, S., Bloxham, M., Worsfold, P.
J Anal At Spectrom 8 (1993), 499
[34] Sutton, K., Sutton, M., Caruso, J.
J Chromatogr A 789 (1997), 85
[35] Jakubowski, N., Jepkens, B., Stüwer, D., Berndt, H.
J Anal At Spectrom 9 (1994), 196
[36] Koropchak, J., Winn, D.
Trends Anal Chem 6 (1987), 171
[37] Tomlinson, M., Caruso, J.
Anal Chim Acta 322 (1996), 1
[38] Shum, S., Neddersen, R., Houk, R.
Analyst 117 (1992), 577
[39] Shum, S., Pang, H, Houk, R.
Anal Chem 64 (1992), 2444
[40] Shum, S., Houk, R.
Anal Chem 65 (1995) 2972
[41] Hill,S.J., Pitts, L.J., Fisher, A.S.
Trends Anal Chem 19 (2000), 120
[42] Sutton, K.L., Caruso, J.
J Chromatogr A 856 (1999), 243
[43] Wind, M., Wesch, H., Lehmann, W.D.
Anal Chem 73 (2001), 117

Literaturverzeichnis 91
[44] Nicholson, J.K., Lindon, J.C., Scarfe, G.B., Wilson, I.D., Abou-Shakra, F.
Sage, A.B, Castro-Perez, J.
Anal Chem 73 (2001), 1491
[45] McSheehy, S., Pohl, P., Szpunar, J., Lobinski, R.
Anal Chim Acta 440 (2001), 3
[46] McSheehy, S., Pohl, P., Szpunar, J., Potin-Gautier, M., Lobinski, R.
J Anal At Spectrom 16 (2001), 68
[47] Polec, K., Garcia-Arribas, O., Perez-Calvo, M., Szpunar, J., Ribas-Ozonas, B.,
Lobinski, R.
J Anal At Spectrom 15 (2000), 1363
[48] Szpunar, J., Makarov, A., Pieper, T., Keppler, B.K., Lobinski, R.
Anal Chim Acta 387 (1999), 135
[49] Stadlober, M., Sager, M., Irgolic, K.J.
Food Chem 73 (2001), 357
[50] Ponce-de-Leon, C.A., Sutton, K.L., Caruso, J.A., Uden, P.C.
J Anal At Spectrom 15 (2000), 1103
[51] Quijano, M.A., Moreno, P., Gutierrez, A.M., Perez-Conde, M.C., Camara, C.
J Mass Spectrom 35 (2000), 878
[52] Madsen, A.D., Goessler, W., Pedersen, S.N., Francesconi, K.A.
J Anal At Spectrom 15 (2000), 657
[53] Inagaki, K., Umemura, T., Matsura, H., Haraguchi, H.
Anal Sci 16 (2000), 787
[54] Marchante-Gayon, J.M., Feldmann, I., Thomas, C., Jakubowski, N.
J Anal At Spectrom 16 (2001), 457
[55] Nagaoka, M.H., Maitani, T.
Analyst 125 (2000), 1962
[56] Dauchy, X., Cottier,R., Batel, A., Jeannot, R., Borsier, M., Astruc,A., Astruc, M.
J Chromatogr Sci 31 (1993), 416
[57] Al-Rashdam, A., Heitkemper, D., Caruso, J.
J Chromatogr Sci 29 (1991), 98
[58] Bushee, D.
Analyst, 113 (1988), 1167

Literaturverzeichnis 92
[59] Huang, C., Jinag, S.
J Anal At Spectrom 8 (1993), 681
[60] Takatera, K., Watanabe, T.
Anal Chem 65 (1993), 759
[61] Owen, L., Crews, H., Hutton, R., Walsh, A.
Analyst 117 (1992), 649
[62] Kumar, U., Doresy, J., Caruso, J., Evans, E.
J Chromatogr Sci 32 (1994), 282
[63] Carins, W., Ebdon, L., Hill, S.
Fresenius J Anal Chem 355 (1996), 202
[64] Wang, L., May, S., Browner, R., Pollock, J.
J Anal At Spectom 11 ( 1996), 1137
[65] Chassaigne, H., Lobinski, R.
Fresenius J Anal Chem 361 (1998), 267
[66] Beauchemin, D., Bednas, M., Berman, S., McLaren, J., Siu, K., Sturgeon, R.
Anal Chem 60 (1998), 2209
[67] Beauchemin, D., Siu, K., McLaren, J., Berman, S.
J Anal At Spectrom 4 (1989), 285
[68] Thompson, J., Houk, R.
Anal Chem 58 (1986), 2541
[69] Story, W., Caruso, J., Heitkemper, D., Perkins, L.
J Chromatogr Sci 30 (1992), 427
[70] Hwang, C., Jiang, S.
Anal Chim Acta 307 (1995), 109
[71] Thomas, P., Sniatecki, R.
J Anal At Spectrom 10 (1995), 615
[72] Yang, K., Jiang, S.
Anal Chim Acta 307 (1995), 109
[73] Cai, Y., Cabanas, M., Fernandez-Turiel, J., Abalos, M., Bayona, J.
Anal Chim Acta 314 (1995), 183
[74] Munoz, R., Donrad, O., Gilon, N., Potin-Gautier, J.
J Anal At Spectrom 11 (1996), 1171

Literaturverzeichnis 93
[75] Jiang, S., Houk, R.
Spectrochim Acta 43 B (1988), 405
[76] Al-Rashdan, A., Vela, N., Caruso, J., Heitkemper, J.
J Anal At Spectrom 7 (1992), 551
[77] Brown, A., Ebdon, L., Hill, S.
Anal Chim Acta 286 (1994), 391
[78] Suyani, H., Creed, J., Davidson, T, Caruso, J.
J Chromtogr Sci 27 (1989), 139
[79] Yang, H., Jiang, S, Yang, Y., Hwang, C.
Anal Chim Acta 312 (1995), 141
[80] Kumar, U., Dorsey, J., Caruso, J., Evans, E.
J Chromatogr A 654 (1993), 261
[81] Zhao, Z., Tepperman, K., Dorsey J., Elder, R.
J Chromatogr A 615 (1993), 83
[82] Heitkemper, D, Creed, J., Fircke, F.
J Anal At Spectrom 4 (1989), 279
[83] Sheppard, B., Shen, W., Caruso, J., Heitkemper, D., Fricke, F.
J Anal At Spectrom 5 (1990), 431
[84] Sheppard, B., Caruso, J., Heitkemper, D., Wollnik, K.
Analyst 117 ( 1992), 971
[85] Larsen, E., Pritzl, G., Honore Hansen, S.
J Anal At Spectrom 8 (1993), 1075
[86] Lintschinger, J., Schramel, P., Hatalak-Rauscher, A., Wendler, I., Michalke, B.
Fresenius J Anal Chem 362 (1998), 313
[87] Krachler, M., Emons, H.
J Anal At Spectrom 16 (2001), 20
[88] Lindemann, T., Prange, A., Neidhart, B., Dannecker, W.
Fresenius J Anal Chem 368 (2000), 214
[89] Chen, H., Yoshida, H., Wanibuchi, H., Fukushima, S., Inoue, Y., Endo, G.
Appl Organomet Chem 10 (1996), 741
[90] Goossens, J., Moens, L., Dams, R.
J Anal At Sectrom 9 (1993), 921

Literaturverzeichnis 94
[91] Zoorob, G., Tomlison, M., Wang, J, Caruso, J.
J Anal At Spetrom 10 (1995), 853
[92] Roig-Navarro, A.F., Martinez-Bravo, Y., Lopez, F.J., Hernandez, F.
J Chromatogr A 912 (2001), 319
[93] Bydry, F., Olson, L., Vela, N., Caruso, J.
J Chromatogr A. 712 (1995), 853
[94] Inoue, Y., Sakai, T., Kumagai, H.
J Chromatogr A 706 (1995), 127
[95] Ding, H., Olson, L., Caruso, J.
Spectrochim Acta 51 B (1996), 1801
[96] Creed, J., Magnuson, M., Pfaff, J., Brockhoff, C.
J Chromatogr A 753 (1996), 261
[97] Sariego-Muniz, C.,. Marchante-Gayon, J.M, Garcia-Alonso, J.I., Sanz-Medel, A.
J Anal At Spectrom 16 (2001), 587
[98] Matz, S., Elder, R., Tepperman, K.
J Anal At Spectrom 4 (1989), 767
[99] Owen, L, Crews, H, Hutton, R., Walsh, A.
Analyst 117 (1992), 649
[100] Takatera, K., Watanabe, T.
Anal. Sci 8 (1992), 469
[101] Dean, J., Munro, S., Ebdon, L., Crews, H., Massey, R.
J Anal At Spectrom 2 (1987), 607
[102] Mason, K., Stroms, S., Jenkins, K.
Anal Biochem 186 (1990), 187
[103] Lyon, T., Fell, G.
J Anal At Spectrom 5 (1990), 135
[104] Mason, A., Storms, S.
Mar Environ Res 35 (1993), 19
[105] Wang, J., Dreessen, D., Wiederin, D.R., Houk, R.S.
Anal Biochem 288 (2001), 89
[106] Crews, H., Dean, J., Ebdon, L., Massey, R.
Analyst, 114 (1989), 895

Literaturverzeichnis 95
[107] Takatera, K., Watanabe, T.
Anal Chem 65 (1993), 3644
[108] Szpunar, J., Makarov, A., Pieper, T., Keppler, B.K., Lobinski, R.
Anal Chim Acta 387 (1999), 135
[109] Wolf, C., Rösick, U., Brätter, P.
J Anal Chem 368 (2000), 839
[110] Wolf, C.
Dissertationsschrift im Fachbereich 5- Chemie der Technischen Universität
Berlin, (1999)
[111] Ferrarello, C.N., Fernandez-de-la-Campa, R., Muniz, C.S., Sanz-Medel, A.
Analyst 125 (2000), 2223
[112] Qinghong, L., Bird, S., Barnes, M.
Anal Chem 67 (1995), 2949
[113] Qinghong, L, Barne, R.
Microchem J 54 (1996), 129
[114] Michalke, B., Schramel, P.
Fresenius J Anal Chem 357 (1997), 554
[115] Michalke, B., Schramel, P.
Fresenius J Anal Chem 360 (1998), 18
[116] Michalke, B.
J Anal At Spectrom 14 (1999), 1297
[117] Michalke, B., Schramel, P.
J Chromatogr A 834 (1999), 341
[118] Michalke, B.
Spectroscopy 15 (2000), 30
[119] Michalke B., Schramel, P.
Electrophoresis 20 (1999),2547
[120] Michalke B., Schramel P., Kettrup, A.
Fresenius J Anal Chem 363 (1999), 456
[121] Mei, E., Ichihashi, H., Gu, W., Yamasaki, S.
Anal Chem 69 (1997), 2187
[122] Taylor, K., Sharp, B., Lewis, D., Crews, H.
J Anal At Spectrom 13 (1998), 1095

Literaturverzeichnis 96
[123] Majidi, V., Miller-Ihli, J.
Analyst 123 (1998), 809
[124] Barnes, R.
Fresenius J Anal Chem 361 (1998), 246
[125] Kirlew, P., Castillano, T., Caruso, J.
Spectrochim Acta B 53 (1998), 221
[126] Tangen, A., Lund, W., Josefsson, B., Borg, H.
J Chromatogr A 826 (1998), 87
[127] Bendahl,D., Gammelgaard, B., Joens, O., Farver, O., Hansen, S.H.
J Anal At Spectrom 16 (2001), 38
[128] Tangen, A., Lund, W.
J Chromatogr A 891 (2000), 129
[129] Sutton, K., B´Hymer, C., Caruso, J.
J Anal At Spectrom 13 (1998), 885
[130] Schaumlöffel, D., Prange, A.
J Anal Chem 364, (1999), 425
[131] Prange, A., Schaumlöffel, D.
J Anal At Spectrom 14 (1999), 1329
[132] Prange, A., Schaumlöffel, D., Brätter, P., Richarz, A., Wolf, C.
Fresenius J Anal Chem 371 (2001), 764
[133] Schaumlöffel D., Marx, G., Brätter, P., Heumann, K.
Anal Bioanal Chem 372 (2002), 155
[134] da Rocha, M., Soldado, A., Blanco-Gonzalez, E., Sanz-Medel, A.
J Anal At Spectrom 15 (2000), 513
[135] Tsung-Hung, L., Shiuh-Jen, J.
Anal Chim Acta, 413 (2000), 197
[136] Majid, V.
Mircochem J 66 (2000), 3
[137] Wei-Hsi, C., Shu-Yu, L., Cheun-Ying, L.
Anal Chim Acta 410 (2000), 25
[138] B´Hymer, C., Day, J., Caruso, J.
Appl Spectrosc 54 (2000), 1040

Literaturverzeichnis 97
[139] Ackely, K., Day, J., Caruso, J.
J Chromatogr A 888 (2000), 293
[140] Baker, S.A., Miller-Ihli, N.J
Spectrochim Acta 55B (2000), 1823
[141] Szpunar, J.
Analyst 125 (2000), 963
[142] Polec, K., Szpunar, J., Palacios, O., González-Duárte, P., Atrian, S., Lobinski, R.
J Anal At Spectrom 16 (2001), 567
[143] Mounicou, S., Polec, K., Chassaigne, H., Potin-Gautier, M., Lobinski, R.
J Anal At Spectrom 15 (2000), 653
[144] Lobinski R.
Appl Spectrosc, 51 No. 7 (1997), 260A
[145] International Agency on Cancer, IARC Monograhps on the evaluation of
carcinogenic risks to human, 49, (1990) Lyons
[146] Drake, E., Sky-Peck, H.
Cancer Res. 49 (1989), 4210
[147] Savolainen, H.
Rev Environ Health 11 (1996), 167
[148] Barceloux, D.
Clin Toxi 37 (1999), 239
[149] Cangul, H., Broday, L., Salnikow, K., Sutherland, J., Peng, W., Zhang, Q.,
Poltaratsky, V., Yee, H., Zoroddu, M., Costa, M.
Toxicol Letters 127 (2002), 39
[150] Pschyrembel
Klinisches Wörterbuch
neu bearbeitete Auflage (258.), Walter de Gryter, Berlin, New York (1998)
[151] Lobinski, R., Szunar, J.
Anal Chim Acta 400 (1999), 321
[152] Grüter, U., Kresimon, J., Hirner, A.
J Anal Chem 368 (2000), 67
[153] Suzuki, K.
Analusis Magazine 26 (1998),57

Literaturverzeichnis 98
[154] Lobinski, R.
Appl Spectrosc 51 (1997) 260
[155] Lobinski, R., Pereiro, I. R., Chassaigne, H., Wasik, A., Szpunar, J.
J Anal At Spectrom 13 (1998), 859
[156] Michalke, B., Schramel, P.
J Chromatogr A 750 (1996), 51
[157] Zoorob, G., MC Kiernan, J., Caruso, J.
Microchim Acta 128 (1998), 145
[158] Lu, Q., Bird, S., Barnes, R.
Anal Chem 67 (1995), 2949
[159] Barnes, R.
J Anal Chem 361 (1998), 246
[160] Liu, Y., Lopez-Avila, V.
Anal Chem 67 (1995), 2020
[161] Olesik, J., Kinzer, J., Olesik, V.
Anal Chem 67 (1995), 1
[162] Marchante-Gayon, J., Sariego Muniz, C., Grarcia Alonso, J, Sanz-Medel, A.
Anal Chim Acta 400 (1999), 307
[163] Lobinski, R.
Spectrochim Acta 53 B (1998), 177
[164] Volkmer, S.
Diplomarbeit, Gerhard-Mercator-Universität Duisburg Fachbereich 6 (Chemie-
Geographie), (2000)
[165] Informationsblatt der Firma Finnigan
Vorstellung des ICP-MS der Reihe Element, Bremen (1998)
[166] Option Instruction Manual für das Agilent 7500 ICP-MS
Agilent, Tokyo (2001)
[167] Schwedt, G.
Taschenaltas der Analytik
Georg Thieme Verlag, Stuttgart, (1992)
[168] Lindemann, T.
Dissertationsschrift im Fachbereich - Chemie der Hamburg, (1999)

Literaturverzeichnis 99
[169] Prange, A.
GIT 31 (1987), 513
[170] Holtzhauer
Biochemische Labormethoden
3. Aufl., Spinger, Berlin, Heidelberg, New York (1997)
[171] Interferenztabelle für die ICP-MS
Tabellenwerk der Firma Finnigan MAT, Bremen (1998)
[172] Informationsblatt von Bio Rad zu Chelex 100, München (2001)
[173] Engelhardt, H., Beck, W., Schmitt, T.
Kapillarelektrophorese
Vieweg & Sohn, Braunschweig/Wiesbaden (1994)
[174] Kägi, J.
Methods Enzym 205 (1991), 613
[175] Knudsen, C., Bjornsdottir, I., Jons, O., Hansen, S.
Anal Biochem 265 (1998), 167
[176] Lobinski, R., Chassaigne, H., Szunar, J.
Talanta, 46 (1998), 271
[177] Blannchard, J.
J Chromatgr A 226 (1981), 455
[178] Shihabi, Z.K.
J Chromatogr A 652 (1993), 471
[179] Sasongko, L., Ramzan, I., Williams, K.M., McLachlan, A.J.
J Chromatogr B 754 (2001), 467
[180] Camera, E., Rinaldi, M., Briganti, S., Picardo, M., Fanali, S.
J Chromatogr B 757 (2001), 69
[181] Meyer, Th., Böhler, J., Frahm, A.W.
J Pharm Biomed Anal 24 (2001), 495
[182] Connelly, J., Welch, J.
J Parenteral Sci Tech 47 (1993), 70
[183] Taylor, R., Toasaksiri, S., Reid, R.
Electrophoresis, 19 (1998), 2791

Literaturverzeichnis 100
[184] Richardz, A.
Diplomarbeit im Fachbereich 5- Chemie der Technischen Universität Berlin,
(1998)
[185] Proteindatenbank hptt//:www.rcsb.org/pdb
[186] Watt, R., Ludden, P.
Cell Mol Life Sci 56 (1999), 604

Anhang 101
10 Anhang
10.1 Geräteliste
• CE/ICP-MS Interface CEI 100 CETAC Omaha, USA
• CE-Vials 100-1000µL Agilent Waldbronn, Deutschland
• Cryoröhrchen 1-2 mL Roth Karlsruhe, Deutschland
• Dampfreinigung für PFA Gefäße Trabold Bern, Schweiz
• Diverse PFA Gefäße VitLab, Seeheim-Jugenheim,
Deutschland
• Diverse Pipetten Eppendorf Hamburg, Deutschland
• Potter-Eveljhem-Homogenisator Fa. B. Braun Biotech. International
Melsungen , Deutschland
• Fraktionssammler Foxy jr. ISCO Lincoln, USA
• Glove Beutel Roth Karlsruhe, Deutschland
• HPLC Anlage 626/606 S Waters Eschborn, Deutschland
• Induktiv gekoppeltes Plasma Sektorfeld Thermo-Finnigan Bremen
Massenspektrometer Element 1 Deutschland
• KPG-Rührer Fa. IKA Labortechnik Staufen,
Deutschland
• Kapillarelektrophoerese 3 D CE Agilent Waldbronn, Deutschland
• Konzentrator Vivaspin 2 PES Satorius Göttingen, Deutschland
• Konzentrator Vivaspin 6 PES Satorius Göttingen, Deutschland
• Kühler CETAC Omaha, USA
• Küvette, 1cm Schichtdicke mit Deckel Fa. Hellma Jena, Deutschland
• Meinhard Zerstäuber (2,7ml/min) Spetec Erdingen, Deutschland
• pH Meter inolab level 2 WTW Weilheim, Deutschland
• Pumpe Perimax 12 Spetec Erdingen, Deutschland
• Quarzskalpelle Kürner Rosenheim, Deutschland
• Reinstwasseranlage Milli-Q-Element Millpore Eschborn, Deutschland
• SEC Säule Hi Load 16/60 Superdex 75 Pharmacia Biotech, Uppsala,
prep grade Schweden
• Spektralphotometer CADAS 100 Dr. Lange Düsseldorf, Deutschland

Anhang 102
• Titrator TitroLine 96 Schott Mainz, Deutschland
• TXRF 8030 C Atomika Waldbronn, Deutschland
• Ultraschallbad Sonorex RK 102 H Bandelin Berlin, Deutschand
• Ultraschalldesintegrator Sonfier 450 Branson Carouge/Geneva, Schweiz
• Ultra Turax T 8 IKA Staufen, Deutschland
• UV Detektor 1100 Serie Agilent Waldbronn, Deutschland
• Vorreinigung zur Q-Element Elix 3 Millipore Eschborn, Deutschland
• Zentrifuge 5417 R Eppendorf Hamburg, Deutschland
• Zentrifuge 5804 R Eppendorf Hamburg, Deutschland
10.2 Verwendete Chemikalien
• α-Chymotrypsinogen A Sigma, Steinheim, Deutschland
• Acetonitril Uvasol Merck, Darmstadt, Deutschland
• Albumin Rinderserum Acros Organica, Geel, Belgien
• Albumin vom Ei Acros Organica, Geel, Belgien
• Ammoniaklösung 25% suprapur Merck, Darmstadt, Deutschland
• Aprotinin Sigma, Steinheim, Deutschland
• Arsenobetain Standard 1g/L Sigma, Steinheim, Deutschland
• Blue Dextran Sigma, Steinheim, Deutschland
• Chelex 100 Na+-Form Fluka, Buchs, Schweiz
• Cyanocobalamin 96% Acros Organica, Geel, Belgien
• DL-Dithiotreitol sigmaUltra Sigma, Steinheim, Deutschland
• Ethanol absolut p.a. Merck, Darmstadt, Deutschland
• Ethylendiamintetraacetic Acid
Trispotassium Salt Dihydrat Sigma, Steinheim, Deutschland
• Germanium Standard 1g/L Merck, Darmstadt, Deutschland
• Kaliumhydoxid-Monohydrat suprapur Merck, Darmstadt, Deutschland
• Kupfer 1g/L Standardlösung Merck, Darmstadt, Deutschland
• MT-Mix Standard Cd Form Sigma, Steinheim, Deutschland
• Myoglobin 90% Sigma, Steinheim, Deutschland
• Natriumchlorid, p.A. Merck, Darmstadt, Deutschland
• Natriumdibenzyldithiocarbamat Fluka, Buchs, Schweiz

Anhang 103
• Nickel Standard 1g/L Merck, Darmstadt, Deutschland
• Nickel Chlorid Hexahydrat Merck, Darmstadt, Deutschland
• Protein-Bestimmungs-Kit Sigma, Steinheim, Deutschland
• Salpetersäure konz. suprapur Merck, Darmstadt, Deutschland
• Transferrin 98 % Sigma, Steinheim, Deutschland
• Tris(hydroxymethyl)-aminomethan p.a. Merck, Darmstadt, Deutschland
• Tune-up Lösung für die ICP-MS Merck, Darmstadt, Deutschland
• Zink 1g/L Standardlösung Merck, Darmstadt, Deutschland
10.3 Verwendete Abkürzungen
• ICP Induktiv gekoppeltes Plasma
• ICP-MS Kopplung induktiv gekoppeltes
Plasma / Massenspektrometer
• CE Kapillarelektrophorese
• CE / ICP-MS online-Kopplung von CE u. ICP-MS
• EOF Elektroosmotischer Fluss
• HPLC Hochdruckflüssigkeitschromato-
graphie
• SEC Größenausschlusschromatographie
• SEC / ICP-MS online-Kopplung von SEC und ICP-
MS
• RPLC Reversed-Phase Liquid Chromato-
graphie
• IPC Ionenpaarchromatographie
• IEC Ionenaustauschchromatographie
• TXRF Total-Refections X-Ray Fluorescence
• Tris HNO3 Puffer eine Lösung aus Tris(hydroxymethyl)
Aminomethan und Salpetersäure
• DMA Dimethylarsensäure
• NWG Nachweisgrenze
• NG normales Gewebe

Anhang 104
• Entz.G entzündetes Gewebe
• TG malignes Gewebe (Tumorgewebe)
• L Liter
• mmol Milli-Mol
• nL Nano-Liter
• mL Milli-Liter
• µL Mikro-Liter
• nm Nano-Meter
• mA Milli-Ampere
• kV Kilo-Volt
• mbar Milli-Bar
• min Minute
• s Sekunde
• W Watt
• UV Detektion Detektion mit ultraviolettem Licht
• Da Dalton
• kDa Kilo Dalton
• PFA Perfluor-alkoxy
• MT-MIX Gemisch aus Metallothionein I und II
• MM Molmasse

Anhang 105
10.4 Experimetelles
Das Vorfiltersystem für die SEC-Chromatographie
Vorfilter an SEC-Säule Filtersystem auseinandergeschraubt
Das Spiltsystem
Der Splitter für die SEC / ICP-MS Kopplung
Aufbau der Pufferreinigung
Reinigungssäulen für den Tis-HNO3-Puffer
Anhang 106
10.5 Optimierung
10.5.1 Cytosolherstellung mit dem Potter Eveljhem Homogenisator
Ultra Turrax Probe Probe geschnitten Probe im Homogenisator
Probe + Puffer Ultra Turrax Probe nach Zentrifuge
10.5.2 Cytosolherstellung mit dem Ultraschalldesintegrator
Probe zerkleinern Probe + Puffer unter Argon Proben vor Ultraschalldes.

Anhang 107
Probe über dem Ultraschalldes. im Ultraschalldes. Proben in der Zentrifuge
Umfüllen des Cytosols unter Argon
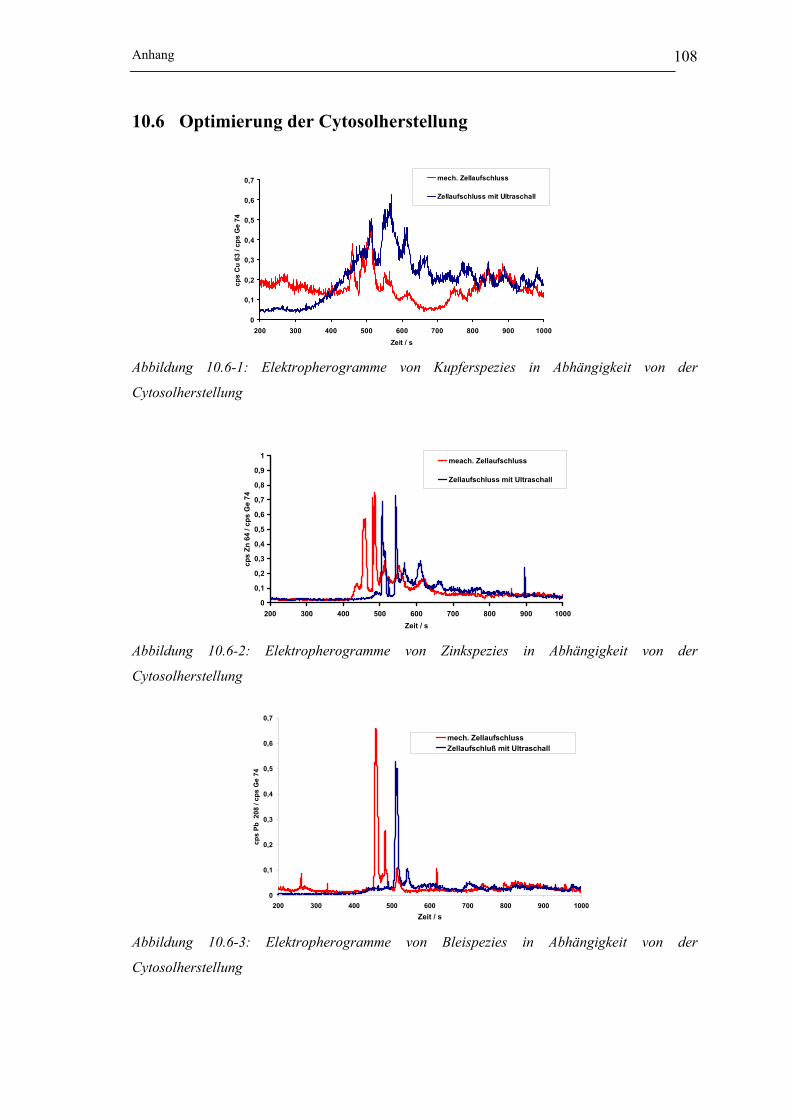
Anhang 108
10.6 Optimierung der Cytosolherstellung
Abbildung 10.6-1: Elektropherogramme von Kupferspezies in Abhängigkeit von der
Cytosolherstellung
Abbildung 10.6-2: Elektropherogramme von Zinkspezies in Abhängigkeit von der
Cytosolherstellung
Abbildung 10.6-3: Elektropherogramme von Bleispezies in Abhängigkeit von der
Cytosolherstellung
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
200 300 400 500 600 700 800 900 1000Zeit / s
cps
Cu
63 /
cps
Ge
74
mech. Zellaufschluss
Zellaufschluss mit Ultraschall
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
200 300 400 500 600 700 800 900 1000Zeit / s
cps
Zn 6
4 / c
ps G
e 74
meach. Zellaufschluss
Zellaufschluss mit Ultraschall
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
200 300 400 500 600 700 800 900 1000Zeit / s
cps
Pb 2
08 /
cps
Ge
74
mech. ZellaufschlussZellaufschluß mit Ultraschall
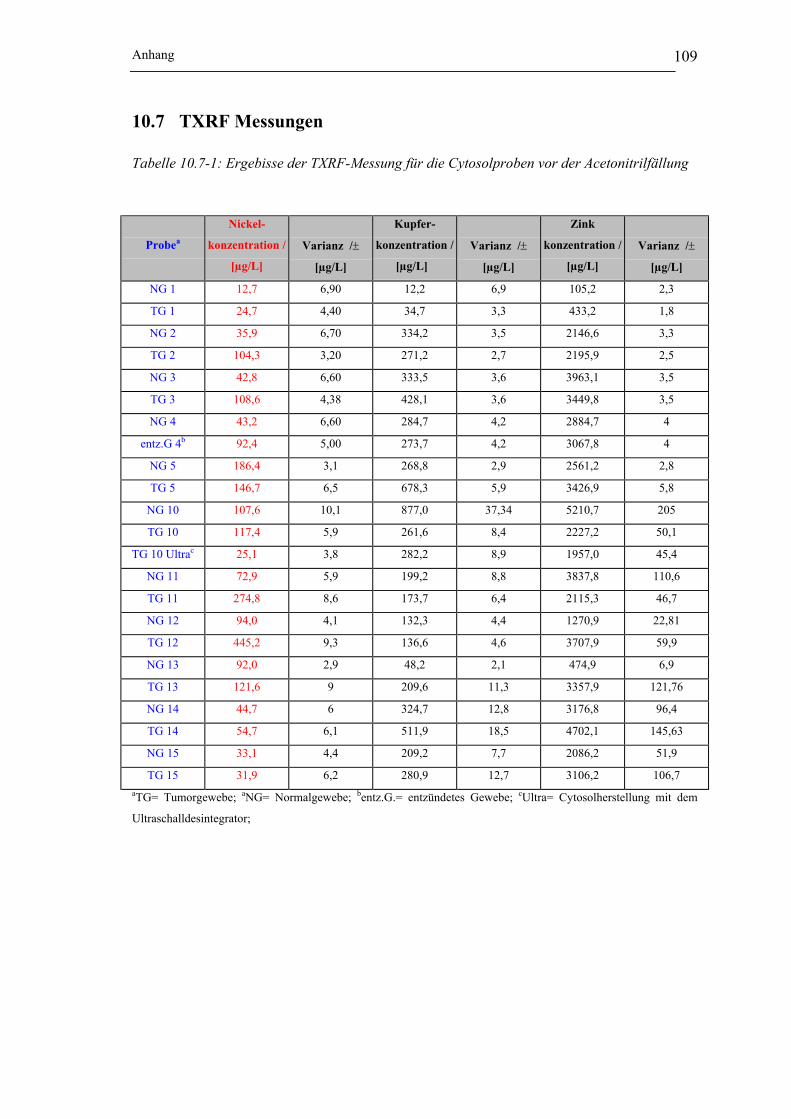
Anhang 109
10.7 TXRF Messungen
Tabelle 10.7-1: Ergebisse der TXRF-Messung für die Cytosolproben vor der Acetonitrilfällung
Probea
Nickel-
konzentration /
[µg/L]
Varianz /±
[µg/L]
Kupfer-
konzentration /
[µg/L]
Varianz /±
[µg/L]
Zink
konzentration /
[µg/L]
Varianz /±
[µg/L]
NG 1 12,7 6,90 12,2 6,9 105,2 2,3
TG 1 24,7 4,40 34,7 3,3 433,2 1,8
NG 2 35,9 6,70 334,2 3,5 2146,6 3,3
TG 2 104,3 3,20 271,2 2,7 2195,9 2,5
NG 3 42,8 6,60 333,5 3,6 3963,1 3,5
TG 3 108,6 4,38 428,1 3,6 3449,8 3,5
NG 4 43,2 6,60 284,7 4,2 2884,7 4
entz.G 4b 92,4 5,00 273,7 4,2 3067,8 4
NG 5 186,4 3,1 268,8 2,9 2561,2 2,8
TG 5 146,7 6,5 678,3 5,9 3426,9 5,8
NG 10 107,6 10,1 877,0 37,34 5210,7 205
TG 10 117,4 5,9 261,6 8,4 2227,2 50,1
TG 10 Ultrac 25,1 3,8 282,2 8,9 1957,0 45,4
NG 11 72,9 5,9 199,2 8,8 3837,8 110,6
TG 11 274,8 8,6 173,7 6,4 2115,3 46,7
NG 12 94,0 4,1 132,3 4,4 1270,9 22,81
TG 12 445,2 9,3 136,6 4,6 3707,9 59,9
NG 13 92,0 2,9 48,2 2,1 474,9 6,9
TG 13 121,6 9 209,6 11,3 3357,9 121,76
NG 14 44,7 6 324,7 12,8 3176,8 96,4
TG 14 54,7 6,1 511,9 18,5 4702,1 145,63
NG 15 33,1 4,4 209,2 7,7 2086,2 51,9
TG 15 31,9 6,2 280,9 12,7 3106,2 106,7aTG= Tumorgewebe; aNG= Normalgewebe; bentz.G.= entzündetes Gewebe; cUltra= Cytosolherstellung mit dem
Ultraschalldesintegrator;
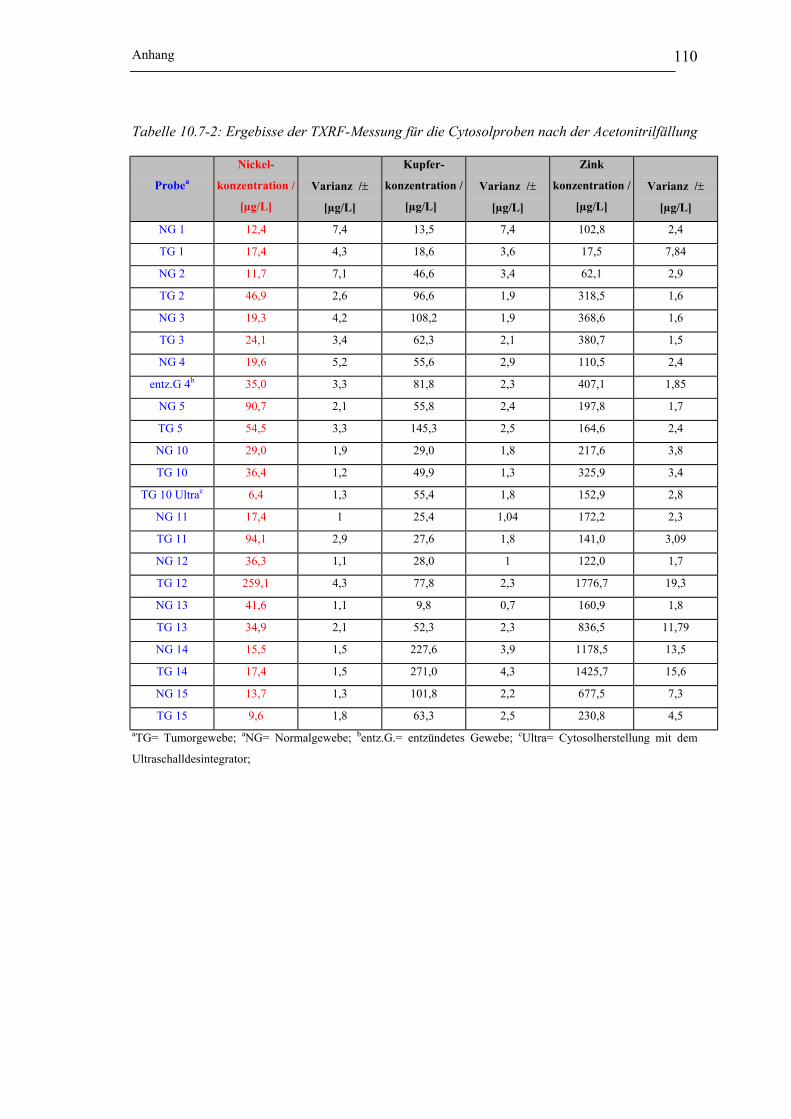
Anhang 110
Tabelle 10.7-2: Ergebisse der TXRF-Messung für die Cytosolproben nach der Acetonitrilfällung
Probea
Nickel-
konzentration /
[µg/L]
Varianz /±
[µg/L]
Kupfer-
konzentration /
[µg/L]
Varianz /±
[µg/L]
Zink
konzentration /
[µg/L]
Varianz /±
[µg/L]
NG 1 12,4 7,4 13,5 7,4 102,8 2,4
TG 1 17,4 4,3 18,6 3,6 17,5 7,84
NG 2 11,7 7,1 46,6 3,4 62,1 2,9
TG 2 46,9 2,6 96,6 1,9 318,5 1,6
NG 3 19,3 4,2 108,2 1,9 368,6 1,6
TG 3 24,1 3,4 62,3 2,1 380,7 1,5
NG 4 19,6 5,2 55,6 2,9 110,5 2,4
entz.G 4b 35,0 3,3 81,8 2,3 407,1 1,85
NG 5 90,7 2,1 55,8 2,4 197,8 1,7
TG 5 54,5 3,3 145,3 2,5 164,6 2,4
NG 10 29,0 1,9 29,0 1,8 217,6 3,8
TG 10 36,4 1,2 49,9 1,3 325,9 3,4
TG 10 Ultrac 6,4 1,3 55,4 1,8 152,9 2,8
NG 11 17,4 1 25,4 1,04 172,2 2,3
TG 11 94,1 2,9 27,6 1,8 141,0 3,09
NG 12 36,3 1,1 28,0 1 122,0 1,7
TG 12 259,1 4,3 77,8 2,3 1776,7 19,3
NG 13 41,6 1,1 9,8 0,7 160,9 1,8
TG 13 34,9 2,1 52,3 2,3 836,5 11,79
NG 14 15,5 1,5 227,6 3,9 1178,5 13,5
TG 14 17,4 1,5 271,0 4,3 1425,7 15,6
NG 15 13,7 1,3 101,8 2,2 677,5 7,3
TG 15 9,6 1,8 63,3 2,5 230,8 4,5aTG= Tumorgewebe; aNG= Normalgewebe; bentz.G.= entzündetes Gewebe; cUltra= Cytosolherstellung mit dem
Ultraschalldesintegrator;
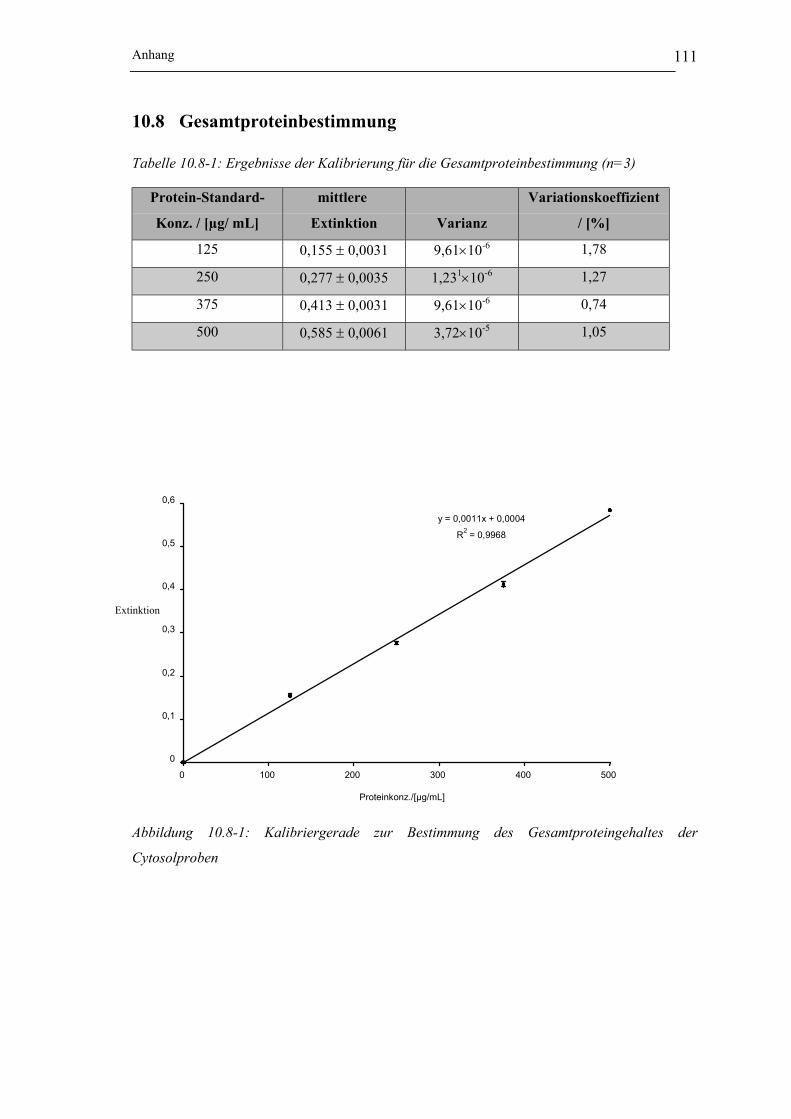
Anhang 111
10.8 Gesamtproteinbestimmung
Tabelle 10.8-1: Ergebnisse der Kalibrierung für die Gesamtproteinbestimmung (n=3)
Protein-Standard-
Konz. / [µg/ mL]
mittlere
Extinktion Varianz
Variationskoeffizient
/ [%]
125 0,155 ± 0,0031 9,61×10-6 1,78
250 0,277 ± 0,0035 1,231×10-6 1,27
375 0,413 ± 0,0031 9,61×10-6 0,74
500 0,585 ± 0,0061 3,72×10-5 1,05
y = 0,0011x + 0,0004
R2 = 0,9968
0
0,1
0,2
0,3
0,4
0,5
0,6
0 100 200 300 400 500
Proteinkonz./[µg/mL]
Abbildung 10.8-1: Kalibriergerade zur Bestimmung des Gesamtproteingehaltes der
Cytosolproben
Extinktion
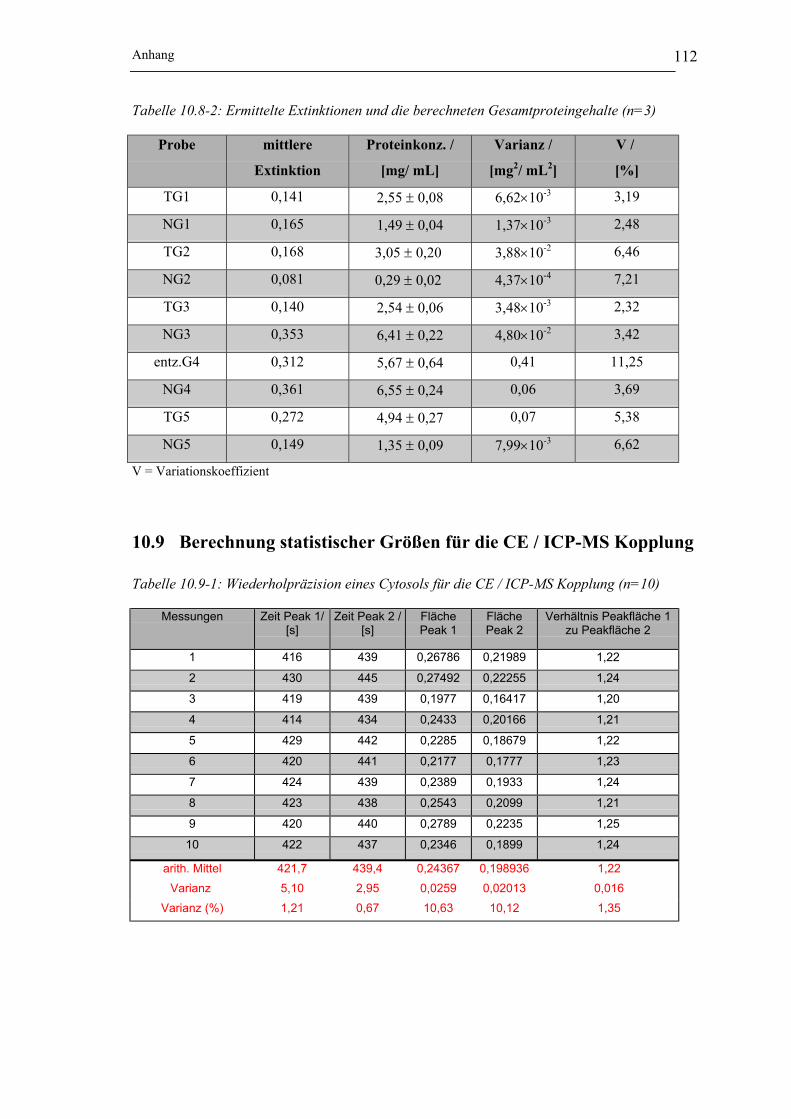
Anhang 112
Tabelle 10.8-2: Ermittelte Extinktionen und die berechneten Gesamtproteingehalte (n=3)
Probe mittlere
Extinktion
Proteinkonz. /
[mg/ mL]
Varianz /
[mg2/ mL2]
V /
[%]
TG1 0,141 2,55 ± 0,08 6,62×10-3 3,19
NG1 0,165 1,49 ± 0,04 1,37×10-3 2,48
TG2 0,168 3,05 ± 0,20 3,88×10-2 6,46
NG2 0,081 0,29 ± 0,02 4,37×10-4 7,21
TG3 0,140 2,54 ± 0,06 3,48×10-3 2,32
NG3 0,353 6,41 ± 0,22 4,80×10-2 3,42
entz.G4 0,312 5,67 ± 0,64 0,41 11,25
NG4 0,361 6,55 ± 0,24 0,06 3,69
TG5 0,272 4,94 ± 0,27 0,07 5,38
NG5 0,149 1,35 ± 0,09 7,99×10-3 6,62
V = Variationskoeffizient
10.9 Berechnung statistischer Größen für die CE / ICP-MS Kopplung
Tabelle 10.9-1: Wiederholpräzision eines Cytosols für die CE / ICP-MS Kopplung (n=10)
Messungen Zeit Peak 1/[s]
Zeit Peak 2 /[s]
FlächePeak 1
FlächePeak 2
Verhältnis Peakfläche 1zu Peakfläche 2
1 416 439 0,26786 0,21989 1,22
2 430 445 0,27492 0,22255 1,24
3 419 439 0,1977 0,16417 1,20
4 414 434 0,2433 0,20166 1,21
5 429 442 0,2285 0,18679 1,22
6 420 441 0,2177 0,1777 1,23
7 424 439 0,2389 0,1933 1,24
8 423 438 0,2543 0,2099 1,21
9 420 440 0,2789 0,2235 1,25
10 422 437 0,2346 0,1899 1,24
arith. Mittel 421,7 439,4 0,24367 0,198936 1,22Varianz 5,10 2,95 0,0259 0,02013 0,016
Varianz (%) 1,21 0,67 10,63 10,12 1,35
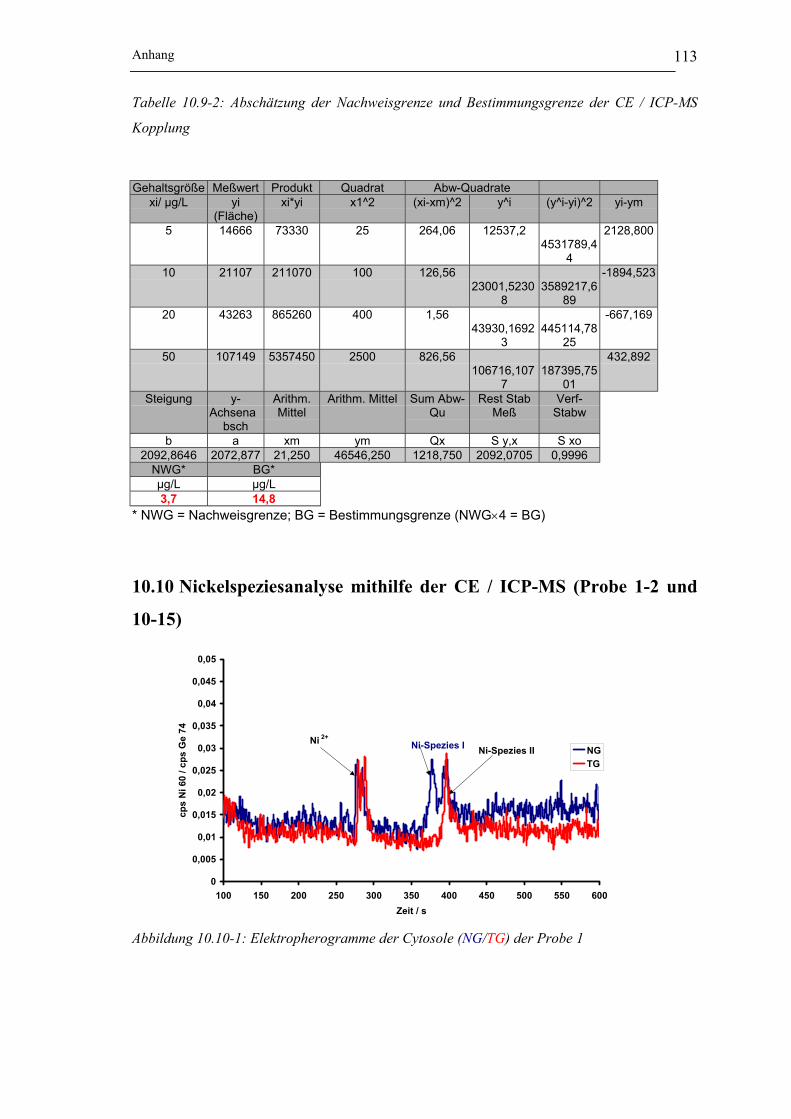
Anhang 113
Tabelle 10.9-2: Abschätzung der Nachweisgrenze und Bestimmungsgrenze der CE / ICP-MS
Kopplung
Gehaltsgröße Meßwert Produkt Quadrat Abw-Quadratexi/ µg/L yi
(Fläche)xi*yi x1^2 (xi-xm)^2 y^i (y^i-yi)^2 yi-ym
5 14666 73330 25 264,06 12537,24531789,4
4
2128,800
10 21107 211070 100 126,5623001,5230
83589217,6
89
-1894,523
20 43263 865260 400 1,5643930,1692
3445114,78
25
-667,169
50 107149 5357450 2500 826,56106716,107
7187395,75
01
432,892
Steigung y-Achsena
bsch
Arithm.Mittel
Arithm. Mittel Sum Abw-Qu
Rest StabMeß
Verf-Stabw
b a xm ym Qx S y,x S xo2092,8646 2072,877 21,250 46546,250 1218,750 2092,0705 0,9996
NWG* BG*µg/L µg/L3,7 14,8
* NWG = Nachweisgrenze; BG = Bestimmungsgrenze (NWG×4 = BG)
10.10 Nickelspeziesanalyse mithilfe der CE / ICP-MS (Probe 1-2 und
10-15)
Abbildung 10.10-1: Elektropherogramme der Cytosole (NG/TG) der Probe 1
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
0,045
0,05
100 150 200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NGTG
Ni 2+Ni-Spezies I Ni-Spezies II
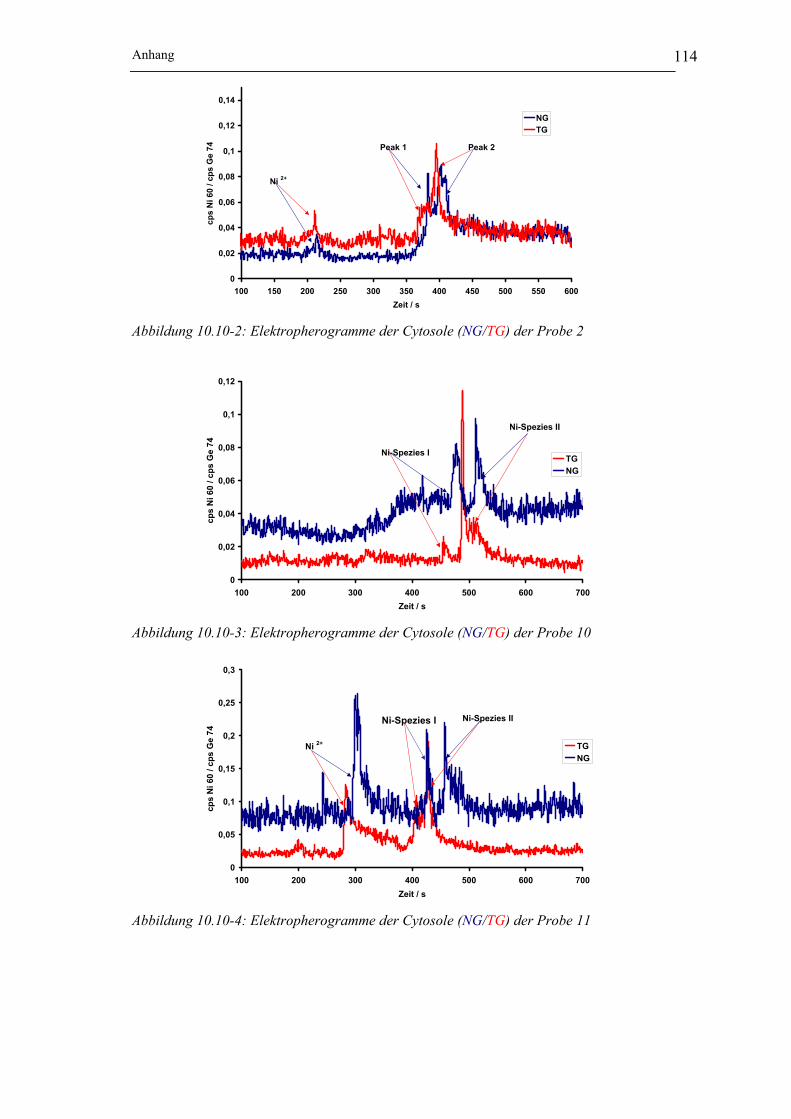
Anhang 114
Abbildung 10.10-2: Elektropherogramme der Cytosole (NG/TG) der Probe 2
Abbildung 10.10-3: Elektropherogramme der Cytosole (NG/TG) der Probe 10
Abbildung 10.10-4: Elektropherogramme der Cytosole (NG/TG) der Probe 11
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
100 150 200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NGTG
Ni 2+
Peak 1 Peak 2
0
0,02
0,04
0,06
0,08
0,1
0,12
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
TGNG
Ni-Spezies I
Ni-Spezies II
0
0,05
0,1
0,15
0,2
0,25
0,3
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
TGNG
Ni-Spezies I
Ni 2+
Ni-Spezies II
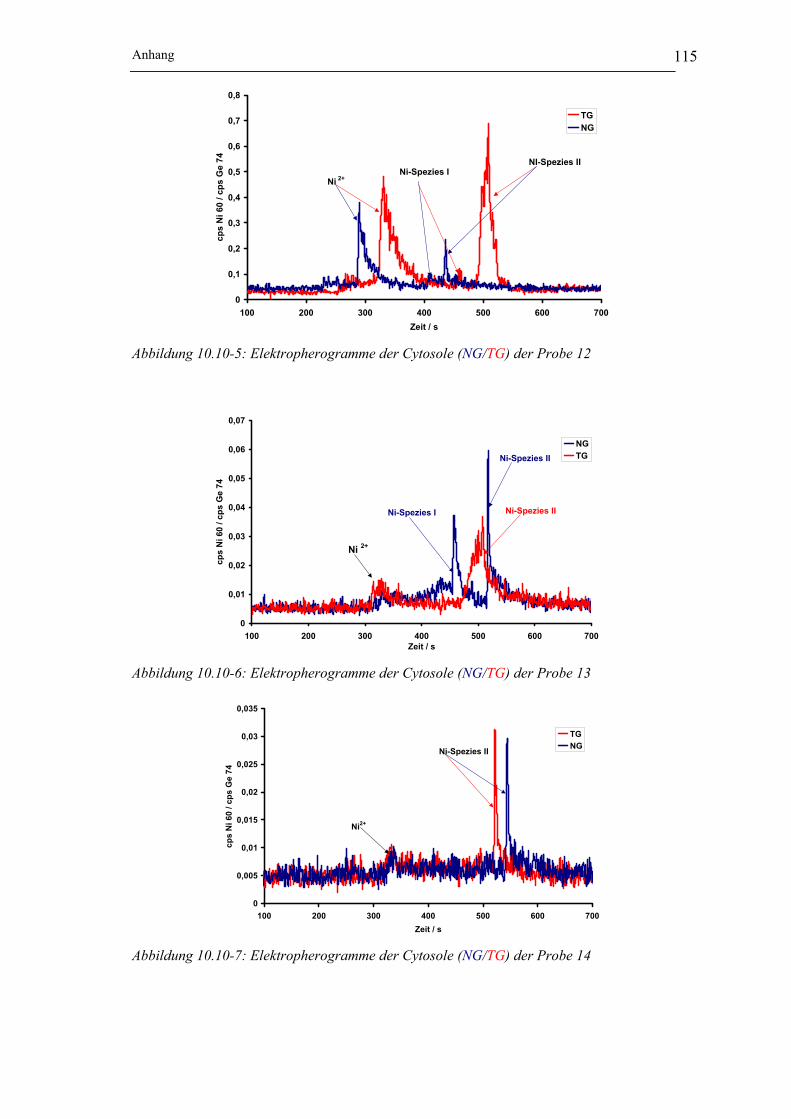
Anhang 115
Abbildung 10.10-5: Elektropherogramme der Cytosole (NG/TG) der Probe 12
Abbildung 10.10-6: Elektropherogramme der Cytosole (NG/TG) der Probe 13
Abbildung 10.10-7: Elektropherogramme der Cytosole (NG/TG) der Probe 14
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
NGTG
Ni-Spezies I
Ni-Spezies II
Ni-Spezies II
Ni 2+
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
TGNG
NI-Spezies II
Ni 2+Ni-Spezies I
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
TGNG
Ni2+
Ni-Spezies II
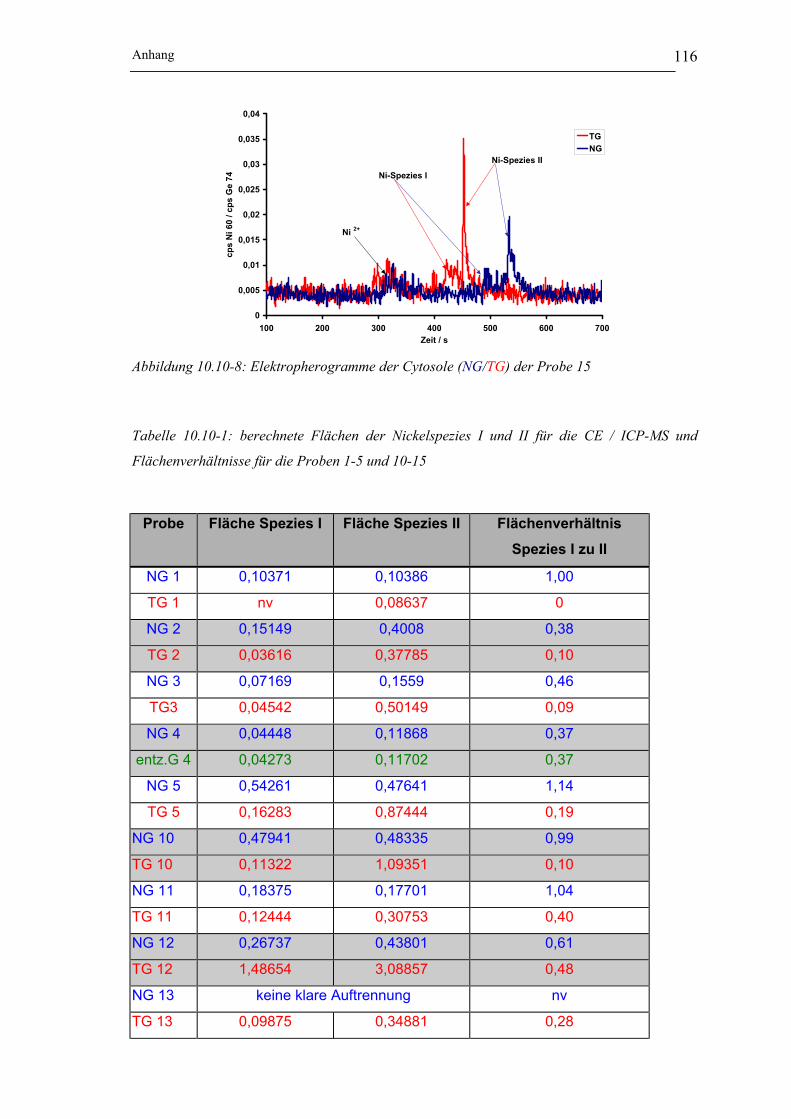
Anhang 116
Abbildung 10.10-8: Elektropherogramme der Cytosole (NG/TG) der Probe 15
Tabelle 10.10-1: berechnete Flächen der Nickelspezies I und II für die CE / ICP-MS und
Flächenverhältnisse für die Proben 1-5 und 10-15
Probe Fläche Spezies I Fläche Spezies II FlächenverhältnisSpezies I zu II
NG 1 0,10371 0,10386 1,00
TG 1 nv 0,08637 0
NG 2 0,15149 0,4008 0,38
TG 2 0,03616 0,37785 0,10
NG 3 0,07169 0,1559 0,46
TG3 0,04542 0,50149 0,09
NG 4 0,04448 0,11868 0,37
entz.G 4 0,04273 0,11702 0,37
NG 5 0,54261 0,47641 1,14
TG 5 0,16283 0,87444 0,19
NG 10 0,47941 0,48335 0,99
TG 10 0,11322 1,09351 0,10
NG 11 0,18375 0,17701 1,04
TG 11 0,12444 0,30753 0,40
NG 12 0,26737 0,43801 0,61
TG 12 1,48654 3,08857 0,48
NG 13 keine klare Auftrennung nv
TG 13 0,09875 0,34881 0,28
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
100 200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
TGNG
Ni-Spezies INi-Spezies II
Ni 2+
Anhang 117
NG 14 0,0256 0,10961 0,23
TG 14 0,03368 0,13954 0,24
NG 15 0,08506 0,13677 0,62
TG 15 0,06763 0,24257 0,28
Tabelle 10.10-2: Wertetabelle zur Durchführung des t-Test (auf der Ebene von 0,01) für die
Peakflächenverhältnisse (CE / ICP-MS)
Probe NG TG signifikantunterschiedlich
1
1,011,000,991,030,98
00000
ja
2
0,380,370,410,360,40
0,100,090,110,080,11
ja
3
0,460,420,480,450,49
0,090,080,100,070,11
ja
4
0,370,380,390,360,38
0,380,390,360,350,37
nein
5
1,151,141,161,141,13
0,180,170,200,210,19
ja
10
0,991,000,990,970,98
0,090,100,110,110,09
ja
11
1,031,051,041,061,04
0,390,420,400,410,39
ja
12
0,600,610,630,590,64
0,520,480,460,470,48
ja
130,300,290,270,280,27
00000
ja

Anhang 118
14
0,220,230,240,260,22
0,230,250,260,240,24
nein
15
0,600,640,630,620,61
0,270,280,290,300,26
ja
Tabelle 10.10-3: Ergbenisse des t-Test der Peakflächen
Probe 1
Daten Mittel Varianz N------------------------------------------------------------A 1,002 3,7E-4 5B 0 0 5------------------------------------------------------------t = -116,48013p = 3,29823E-14Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 2Daten Mittel Varianz N------------------------------------------------------------A 0,384 4,3E-4 5B 0,098 1,7E-4 5------------------------------------------------------------t = -26,10811p = 4,97474E-9Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 3Daten Mittel Varianz N------------------------------------------------------------A 0,46 7,5E-4 5B 0,09 2,5E-4 5------------------------------------------------------------t = -26,16295p = 4,89278E-9Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 4Daten Mittel Varianz N------------------------------------------------------------A 0,376 1,3E-4 5B 0,37 2,5E-4 5------------------------------------------------------------t = -0,68825p = 0,51076Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.

Anhang 119
Probe 5Daten Mittel Varianz N------------------------------------------------------------A 1,144 1,3E-4 5B 0,19 2,5E-4 5------------------------------------------------------------t = -109,43131p = 5,43302E-14Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 10Daten Mittel Varianz N------------------------------------------------------------A 0,986 1,3E-4 5B 0,1 1E-4 5------------------------------------------------------------t = -130,63357p = 1,3184E-14Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 11Daten Mittel Varianz N------------------------------------------------------------A 1,044 1,3E-4 5B 0,402 1,7E-4 5------------------------------------------------------------t = -82,88184p = 5,00868E-13Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 12Daten Mittel Varianz N------------------------------------------------------------A 0,614 4,3E-4 5B 0,482 5,2E-4 5------------------------------------------------------------t = -9,57629p = 1,17161E-5Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 13Daten Mittel Varianz N------------------------------------------------------------A 0,282 1,7E-4 5B 0 0 5------------------------------------------------------------t = -48,3626p = 3,6966E-11Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Anhang 120
Probe 14Daten Mittel Varianz N------------------------------------------------------------A 0,234 2,8E-4 5B 0,244 1,3E-4 5------------------------------------------------------------t = 1,10432p = 0,30156Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.
Probe 15Daten Mittel Varianz N------------------------------------------------------------A 0,62 2,5E-4 5B 0,28 2,5E-4 5------------------------------------------------------------t = -34p = 6,11792E-10Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
10.10.1 Nickel/Kupfer Korrelation bei der CE / ICP-MS
Abbildung 10.10-9: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 1
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
0,045
0,05
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies INi- Spezies II
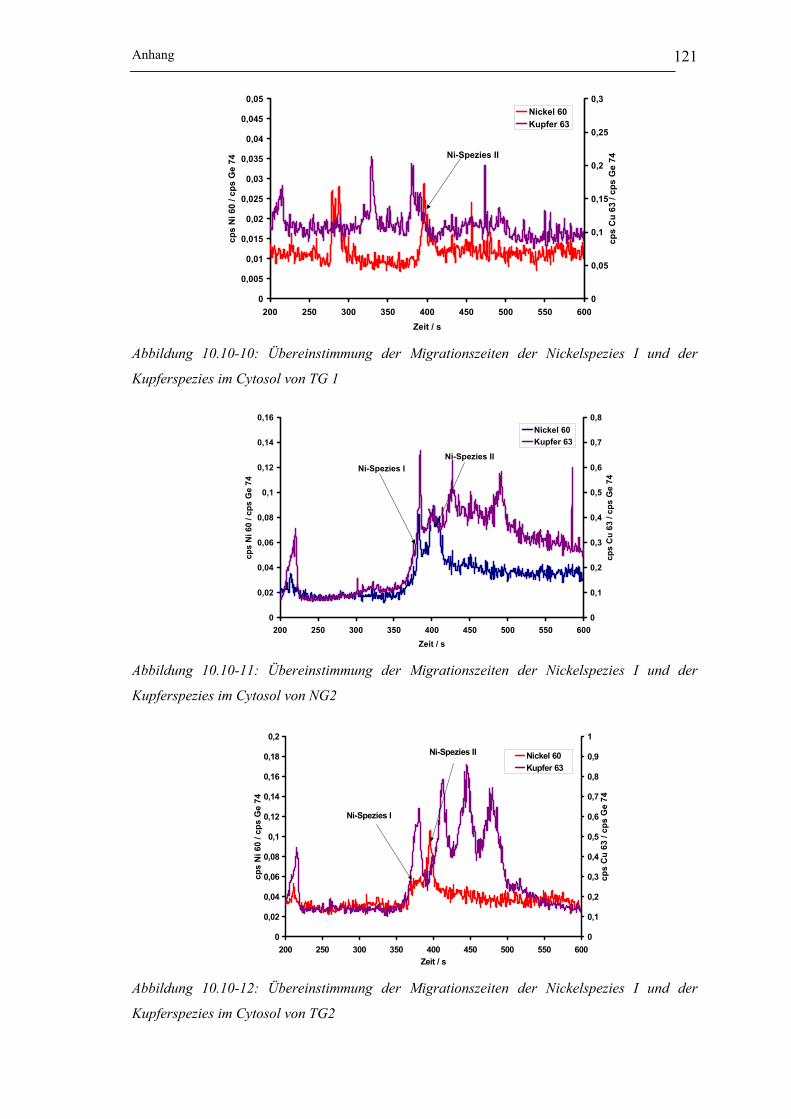
Anhang 121
Abbildung 10.10-10: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 1
Abbildung 10.10-11: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG2
Abbildung 10.10-12: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG2
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
0,045
0,05
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies II
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies INi-Spezies II
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
cps
Cu
63 /
cps
Ge
74
Nickel 60 Kupfer 63
Ni-Spezies I
Ni-Spezies II
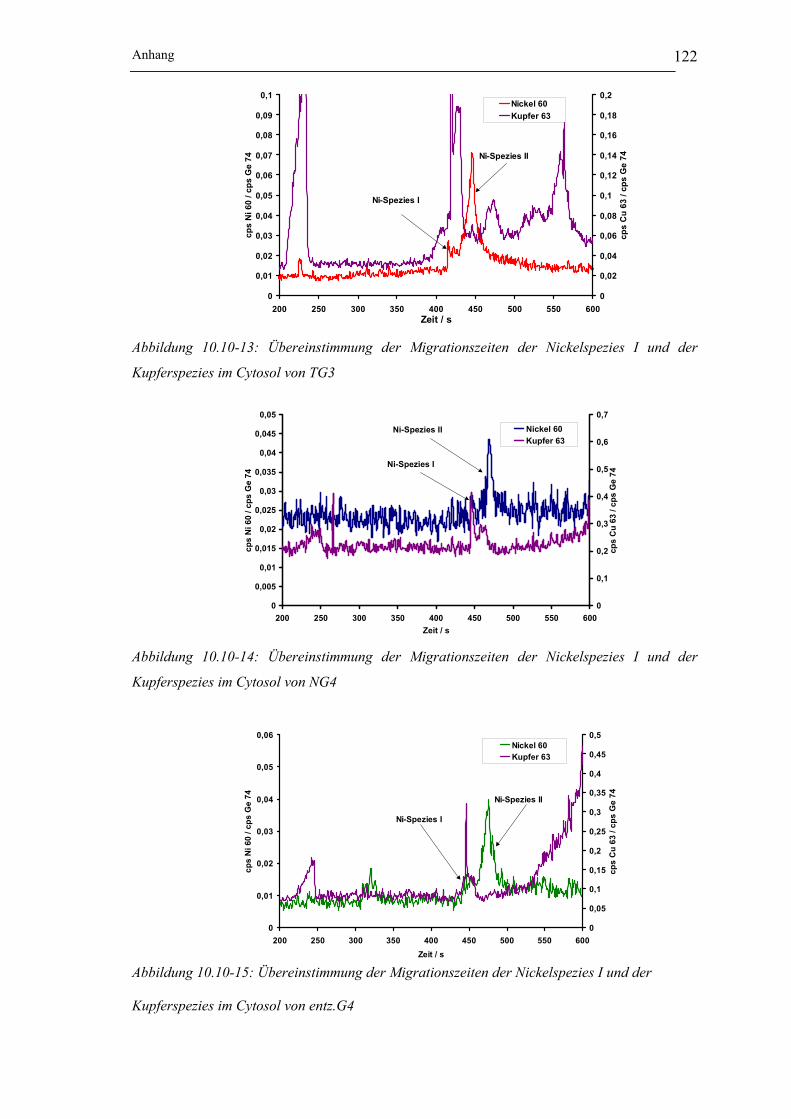
Anhang 122
Abbildung 10.10-13: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG3
Abbildung 10.10-14: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG4
Abbildung 10.10-15: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von entz.G4
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,1
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
0
0,01
0,02
0,03
0,04
0,05
0,06
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
cps
Cu
63 /
cps
Ge
74
Nickel 60 Kupfer 63
Ni-Spezies I
Ni-Spezies II
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
0,045
0,05
200 250 300 350 400 450 500 550 600Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
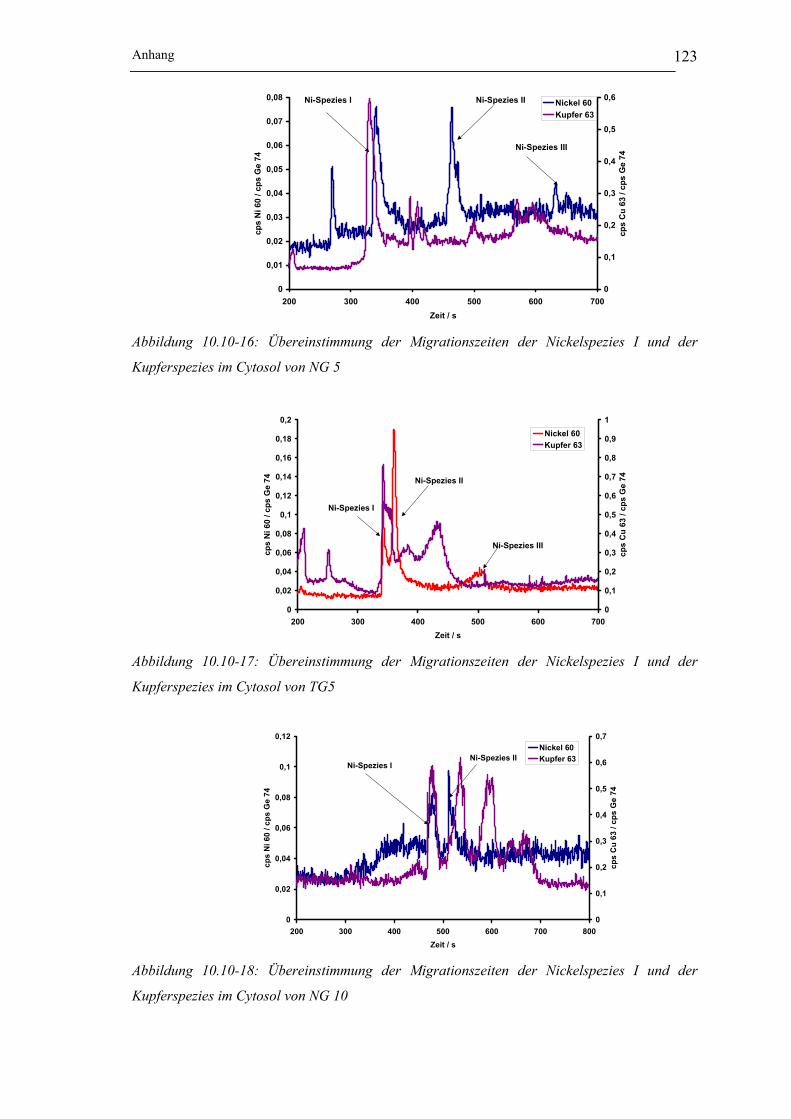
Anhang 123
Abbildung 10.10-16: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 5
Abbildung 10.10-17: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG5
Abbildung 10.10-18: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 10
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I Ni-Spezies II
Ni-Spezies III
0
0,02
0,04
0,06
0,08
0,1
0,12
0,14
0,16
0,18
0,2
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
cps
Cu
63 /
cps
Ge
74
Nickel 60 Kupfer 63
Ni-Spezies I
Ni-Spezies II
Ni-Spezies III
0
0,02
0,04
0,06
0,08
0,1
0,12
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies INi-Spezies II
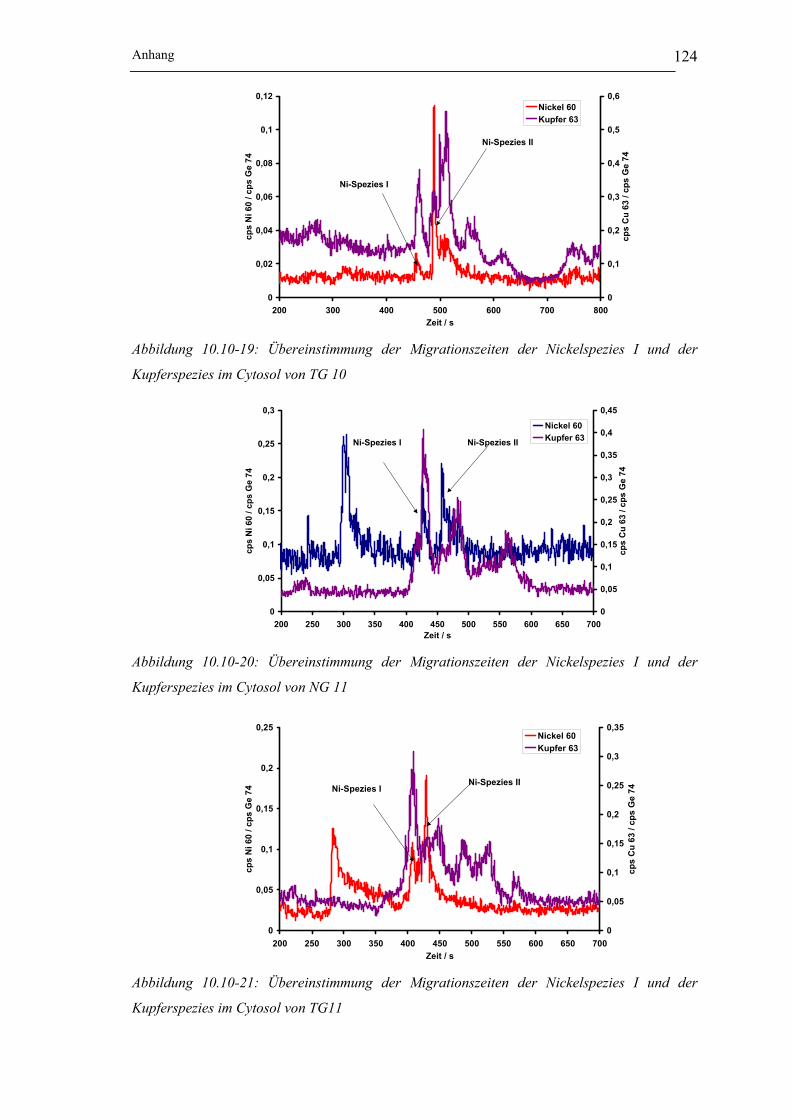
Anhang 124
Abbildung 10.10-19: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 10
Abbildung 10.10-20: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 11
Abbildung 10.10-21: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG11
0
0,02
0,04
0,06
0,08
0,1
0,12
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
0
0,05
0,1
0,15
0,2
0,25
0,3
200 250 300 350 400 450 500 550 600 650 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63Ni-Spezies I Ni-Spezies II
0
0,05
0,1
0,15
0,2
0,25
200 250 300 350 400 450 500 550 600 650 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies INi-Spezies II
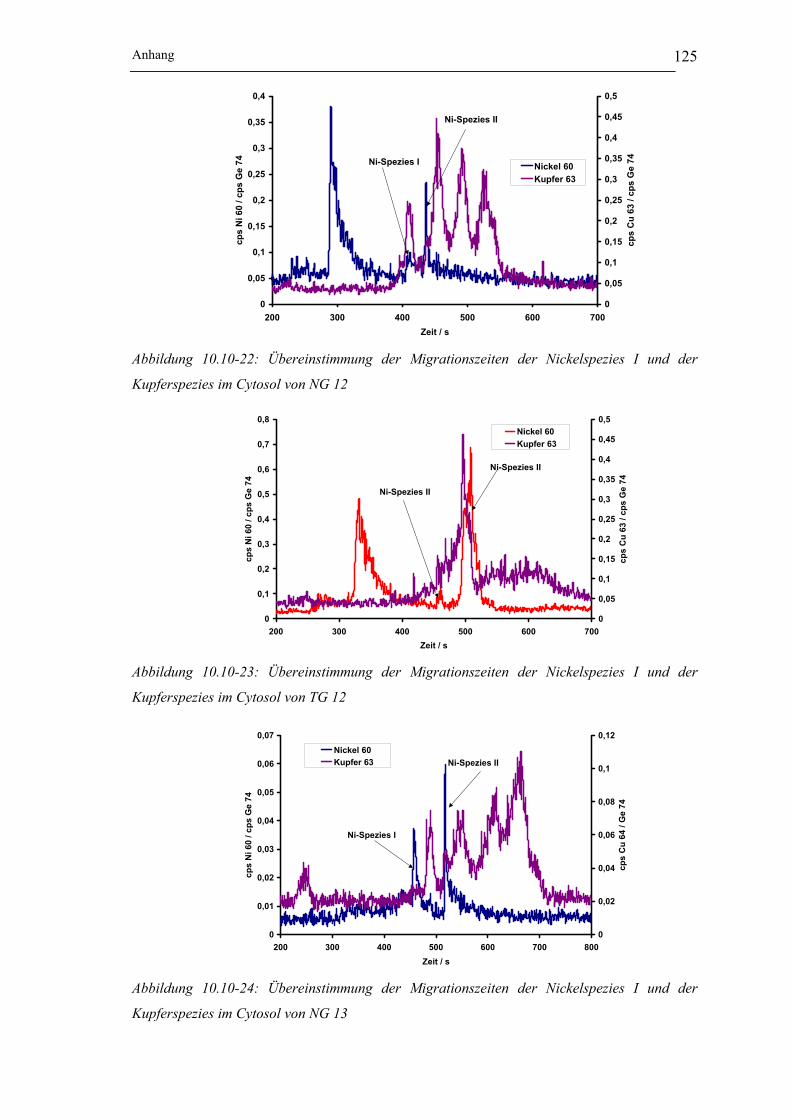
Anhang 125
Abbildung 10.10-22: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 12
Abbildung 10.10-23: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 12
Abbildung 10.10-24: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 13
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies II
Ni-Spezies II
0
0,01
0,02
0,03
0,04
0,05
0,06
0,07
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,02
0,04
0,06
0,08
0,1
0,12
cps
Cu
64 /
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
cps
Cu
63 /
cps
Ge
74Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
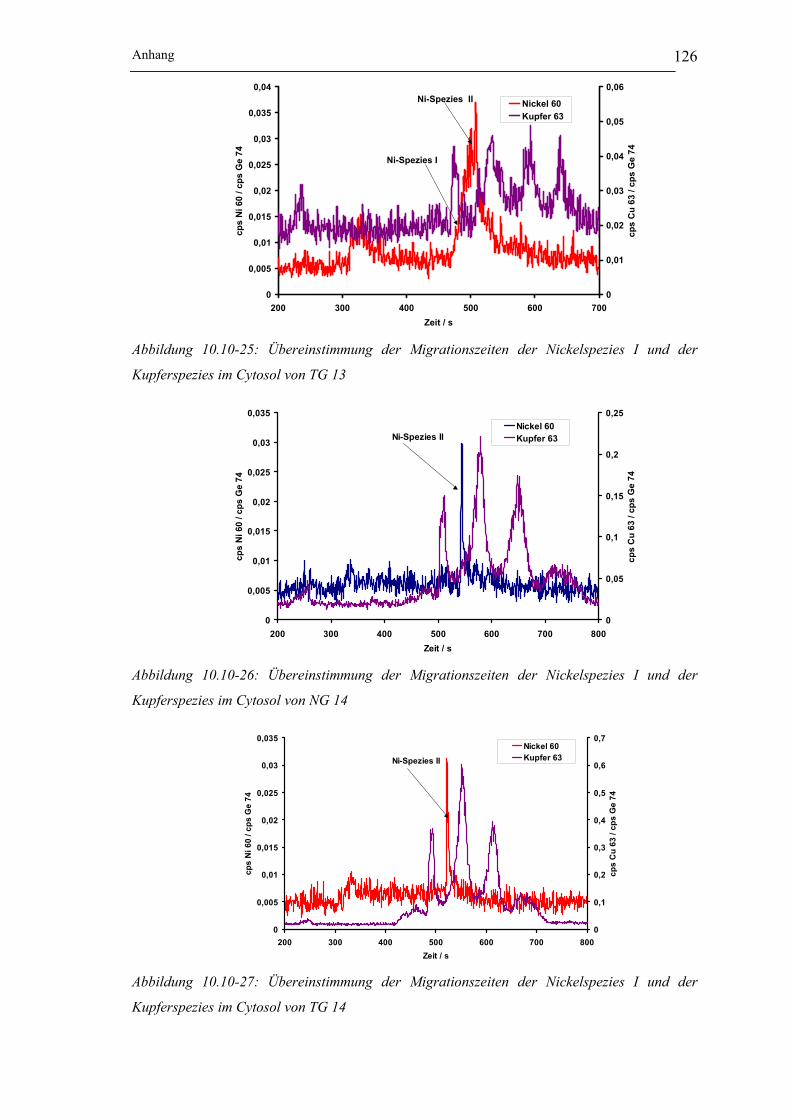
Anhang 126
Abbildung 10.10-25: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 13
Abbildung 10.10-26: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 14
Abbildung 10.10-27: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 14
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,01
0,02
0,03
0,04
0,05
0,06
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies II
Ni-Spezies I
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
cps
Cu
63 /
cps
Ge
74
Nickel 60 Kupfer 63Ni-Spezies II
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
200 300 400 500 600 700 800Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63Ni-Spezies II
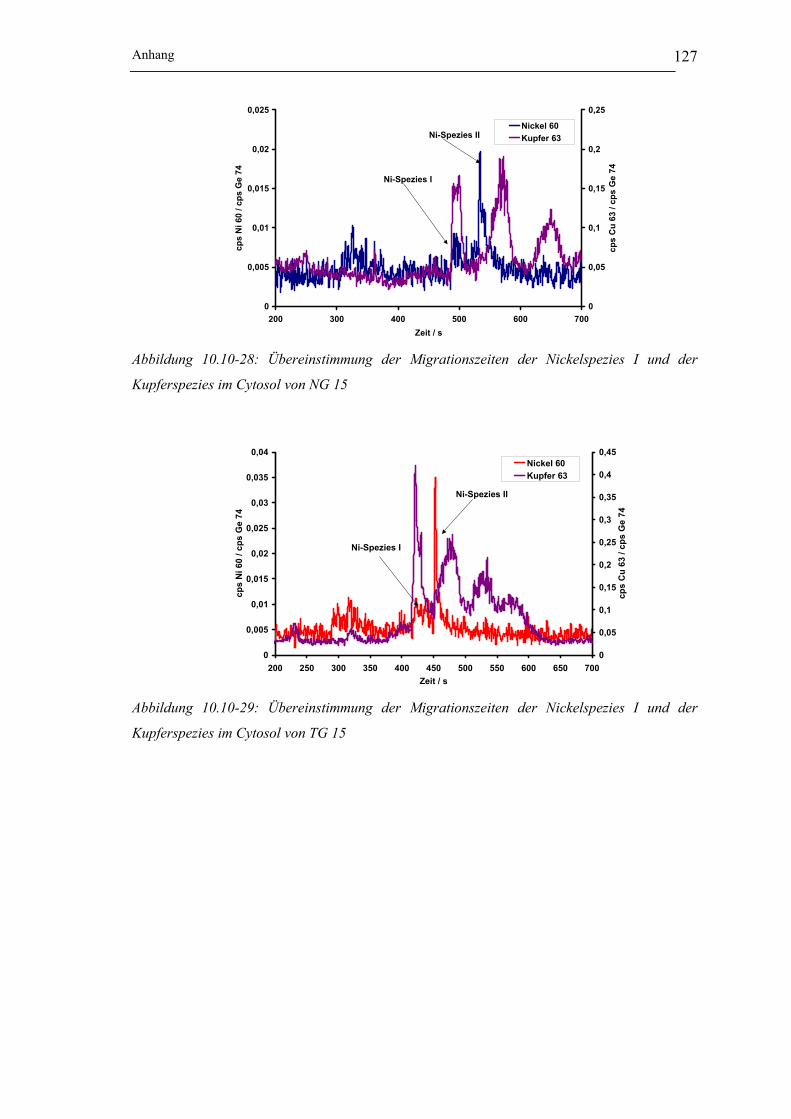
Anhang 127
Abbildung 10.10-28: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von NG 15
Abbildung 10.10-29: Übereinstimmung der Migrationszeiten der Nickelspezies I und der
Kupferspezies im Cytosol von TG 15
0
0,005
0,01
0,015
0,02
0,025
200 300 400 500 600 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
0
0,005
0,01
0,015
0,02
0,025
0,03
0,035
0,04
200 250 300 350 400 450 500 550 600 650 700Zeit / s
cps
Ni 6
0 / c
ps G
e 74
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
cps
Cu
63 /
cps
Ge
74
Nickel 60Kupfer 63
Ni-Spezies I
Ni-Spezies II
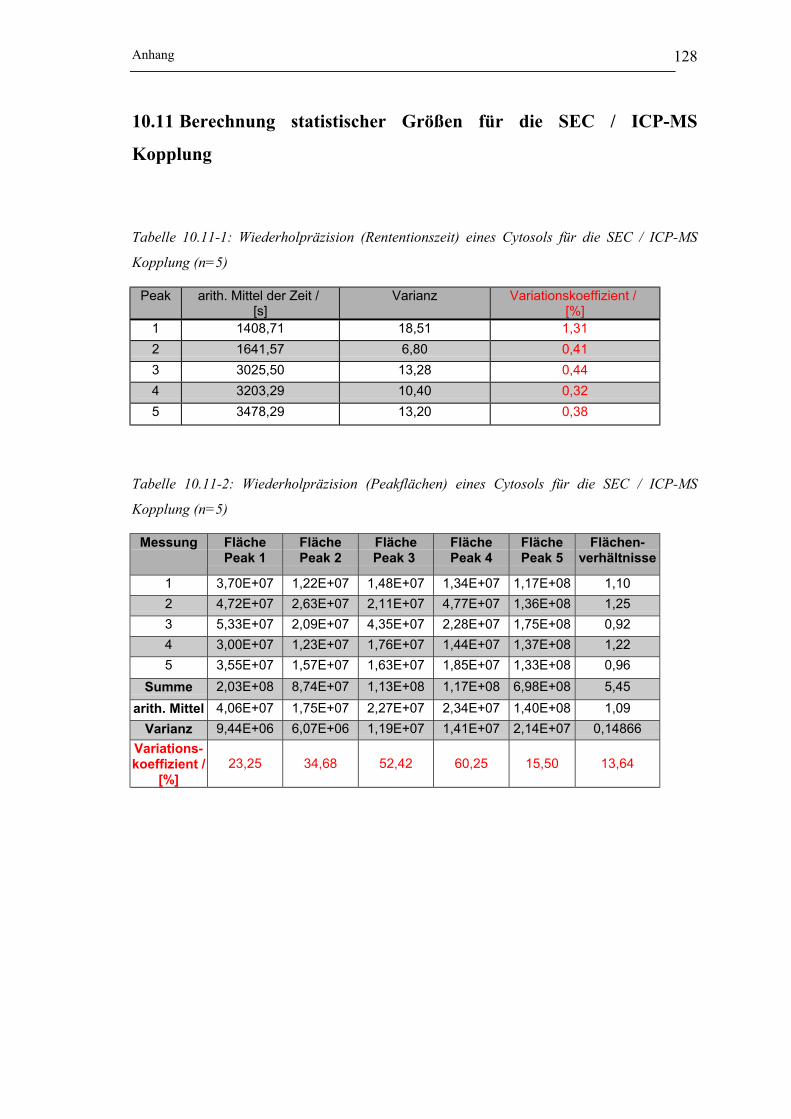
Anhang 128
10.11 Berechnung statistischer Größen für die SEC / ICP-MS
Kopplung
Tabelle 10.11-1: Wiederholpräzision (Rententionszeit) eines Cytosols für die SEC / ICP-MS
Kopplung (n=5)
Peak arith. Mittel der Zeit / [s]
Varianz Variationskoeffizient / [%]
1 1408,71 18,51 1,312 1641,57 6,80 0,413 3025,50 13,28 0,444 3203,29 10,40 0,325 3478,29 13,20 0,38
Tabelle 10.11-2: Wiederholpräzision (Peakflächen) eines Cytosols für die SEC / ICP-MS
Kopplung (n=5)
Messung FlächePeak 1
FlächePeak 2
FlächePeak 3
FlächePeak 4
FlächePeak 5
Flächen-verhältnisse
1 3,70E+07 1,22E+07 1,48E+07 1,34E+07 1,17E+08 1,102 4,72E+07 2,63E+07 2,11E+07 4,77E+07 1,36E+08 1,253 5,33E+07 2,09E+07 4,35E+07 2,28E+07 1,75E+08 0,924 3,00E+07 1,23E+07 1,76E+07 1,44E+07 1,37E+08 1,225 3,55E+07 1,57E+07 1,63E+07 1,85E+07 1,33E+08 0,96
Summe 2,03E+08 8,74E+07 1,13E+08 1,17E+08 6,98E+08 5,45arith. Mittel 4,06E+07 1,75E+07 2,27E+07 2,34E+07 1,40E+08 1,09
Varianz 9,44E+06 6,07E+06 1,19E+07 1,41E+07 2,14E+07 0,14866Variations-koeffizient /
[%]23,25 34,68 52,42 60,25 15,50 13,64
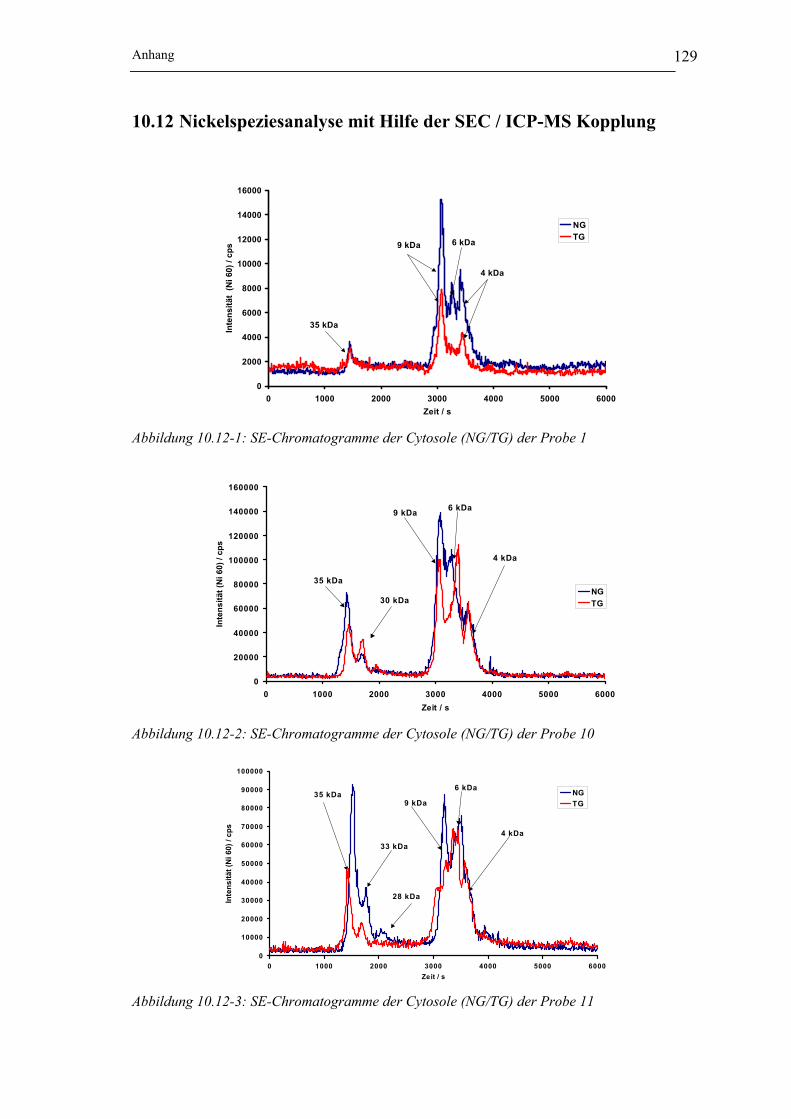
Anhang 129
10.12 Nickelspeziesanalyse mit Hilfe der SEC / ICP-MS Kopplung
Abbildung 10.12-1: SE-Chromatogramme der Cytosole (NG/TG) der Probe 1
Abbildung 10.12-2: SE-Chromatogramme der Cytosole (NG/TG) der Probe 10
Abbildung 10.12-3: SE-Chromatogramme der Cytosole (NG/TG) der Probe 11
0
2000
4000
6000
8000
10000
12000
14000
16000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
sNGTG
35 kDa
9 kDa 6 kDa
4 kDa
0
20000
40000
60000
80000
100000
120000
140000
160000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa
9 kDa 6 kDa
4 kDa
30 kDa
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa9 kDa
6 kDa
4 kDa33 kDa
28 kDa
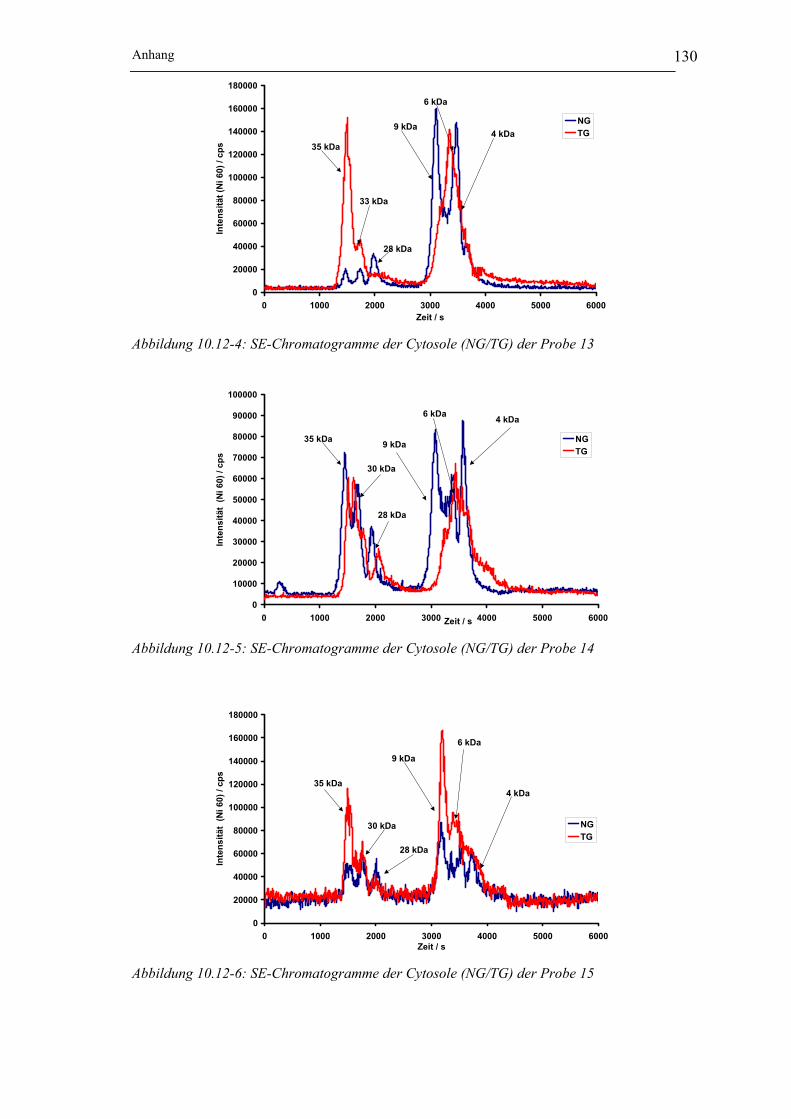
Anhang 130
Abbildung 10.12-4: SE-Chromatogramme der Cytosole (NG/TG) der Probe 13
Abbildung 10.12-5: SE-Chromatogramme der Cytosole (NG/TG) der Probe 14
Abbildung 10.12-6: SE-Chromatogramme der Cytosole (NG/TG) der Probe 15
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa
9 kDa
6 kDa
4 kDa
33 kDa
28 kDa
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa 9 kDa
6 kDa
30 kDa
28 kDa
4 kDa
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
0 1000 2000 3000 4000 5000 6000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
NGTG
35 kDa
9 kDa
6 kDa
4 kDa
30 kDa
28 kDa
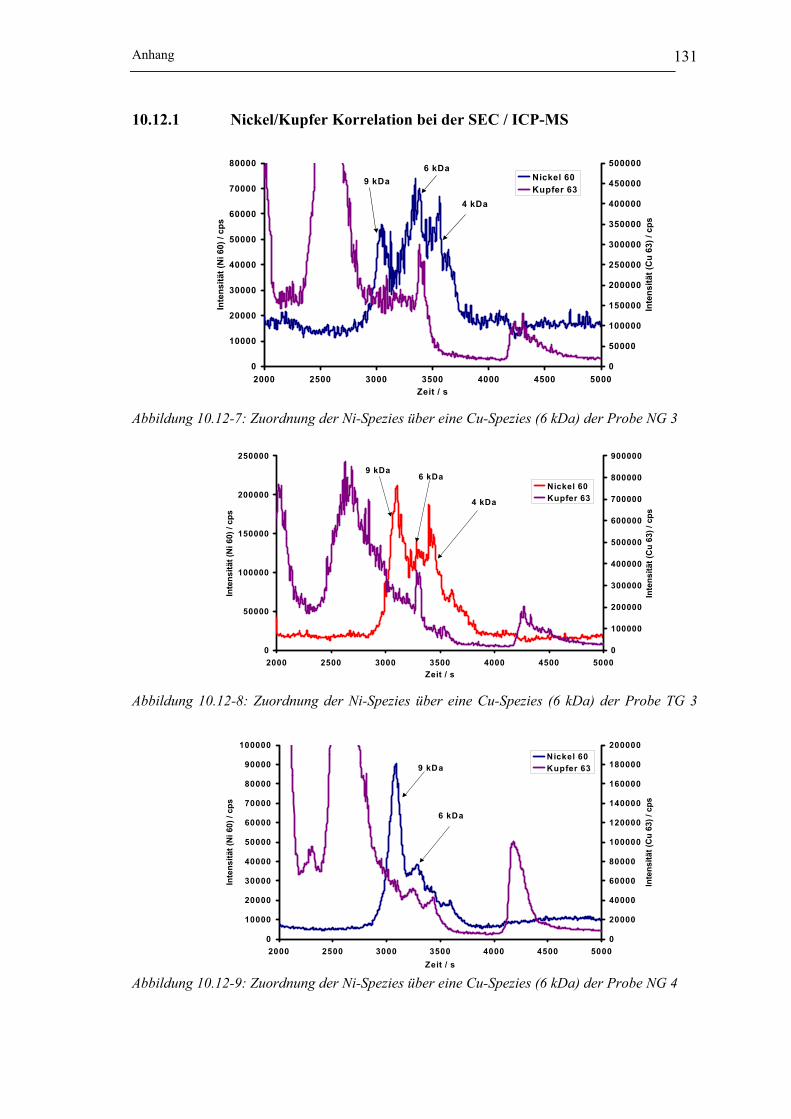
Anhang 131
10.12.1 Nickel/Kupfer Korrelation bei der SEC / ICP-MS
Abbildung 10.12-7: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 3
Abbildung 10.12-8: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 3
Abbildung 10.12-9: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 4
0
10000
20000
30000
40000
50000
60000
70000
80000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
50000
100000
150000
200000
250000
300000
350000
400000
450000
500000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60 Kupfer 63
9 kDa6 kDa
4 kDa
0
50000
100000
150000
200000
250000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
100000
200000
300000
400000
500000
600000
700000
800000
900000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa6 kDa
4 kDa
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
200000
Inte
nsitä
t (C
u 63
) / c
ps
N ickel 60Kupfer 639 kDa
6 kDa
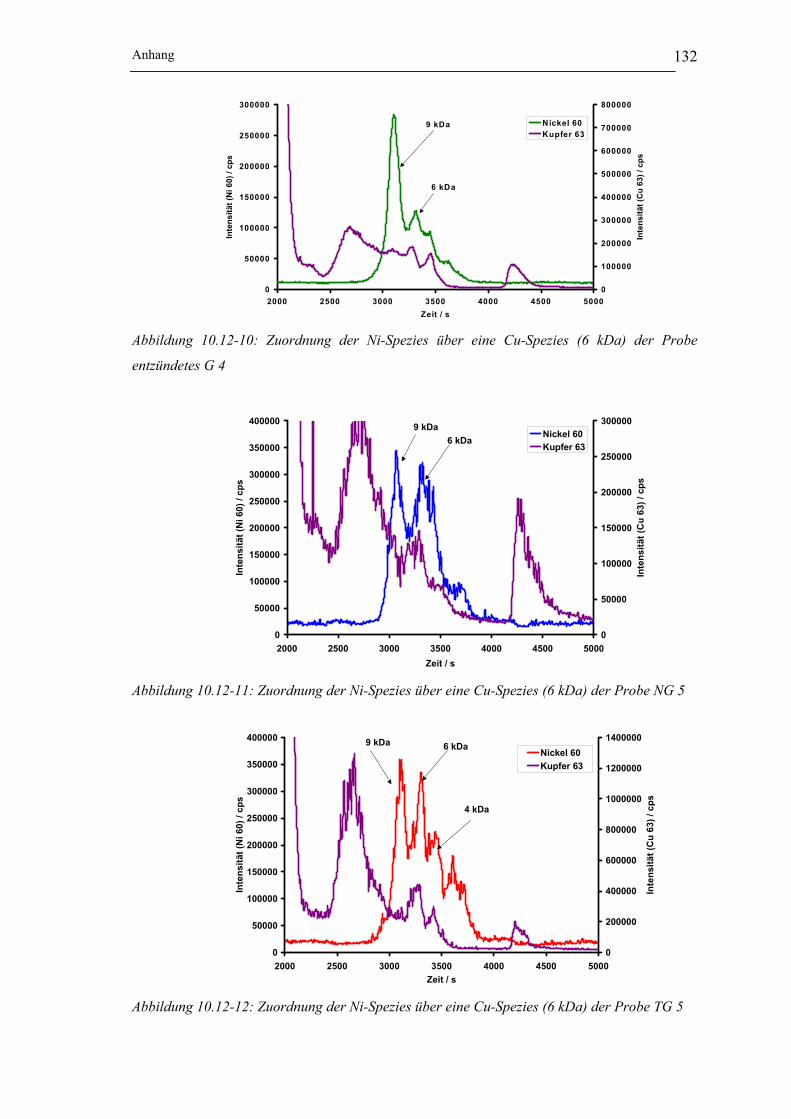
Anhang 132
Abbildung 10.12-10: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe
entzündetes G 4
Abbildung 10.12-11: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 5
Abbildung 10.12-12: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 5
0
50000
100000
150000
200000
250000
300000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
100000
200000
300000
400000
500000
600000
700000
800000
Inte
nsitä
t (C
u 63
) / c
ps
N ickel 60Kupfer 63
9 kDa
6 kDa
0
50000
100000
150000
200000
250000
300000
350000
400000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
50000
100000
150000
200000
250000
300000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa 6 kDa
0
50000
100000
150000
200000
250000
300000
350000
400000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
200000
400000
600000
800000
1000000
1200000
1400000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa 6 kDa
4 kDa
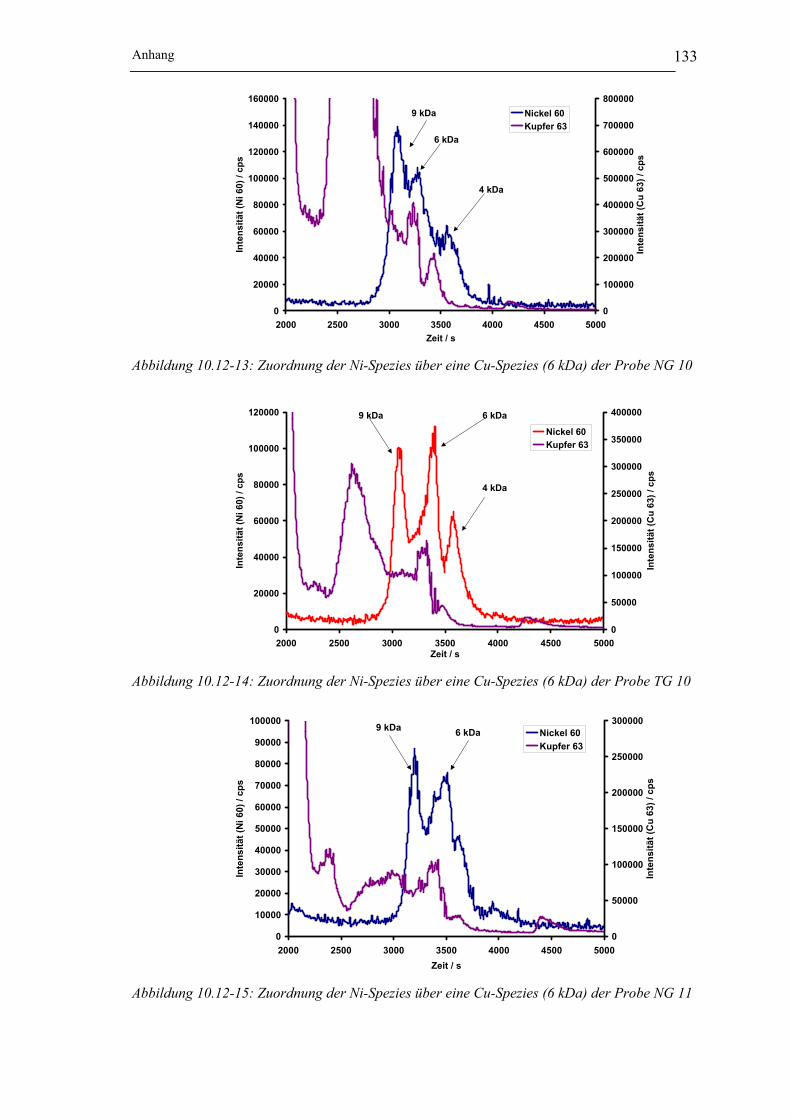
Anhang 133
Abbildung 10.12-13: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 10
Abbildung 10.12-14: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 10
Abbildung 10.12-15: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 11
0
20000
40000
60000
80000
100000
120000
140000
160000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
100000
200000
300000
400000
500000
600000
700000
800000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa
6 kDa
4 kDa
0
20000
40000
60000
80000
100000
120000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
50000
100000
150000
200000
250000
300000
350000
400000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa
4 kDa
6 kDa
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
50000
100000
150000
200000
250000
300000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa 6 kDa
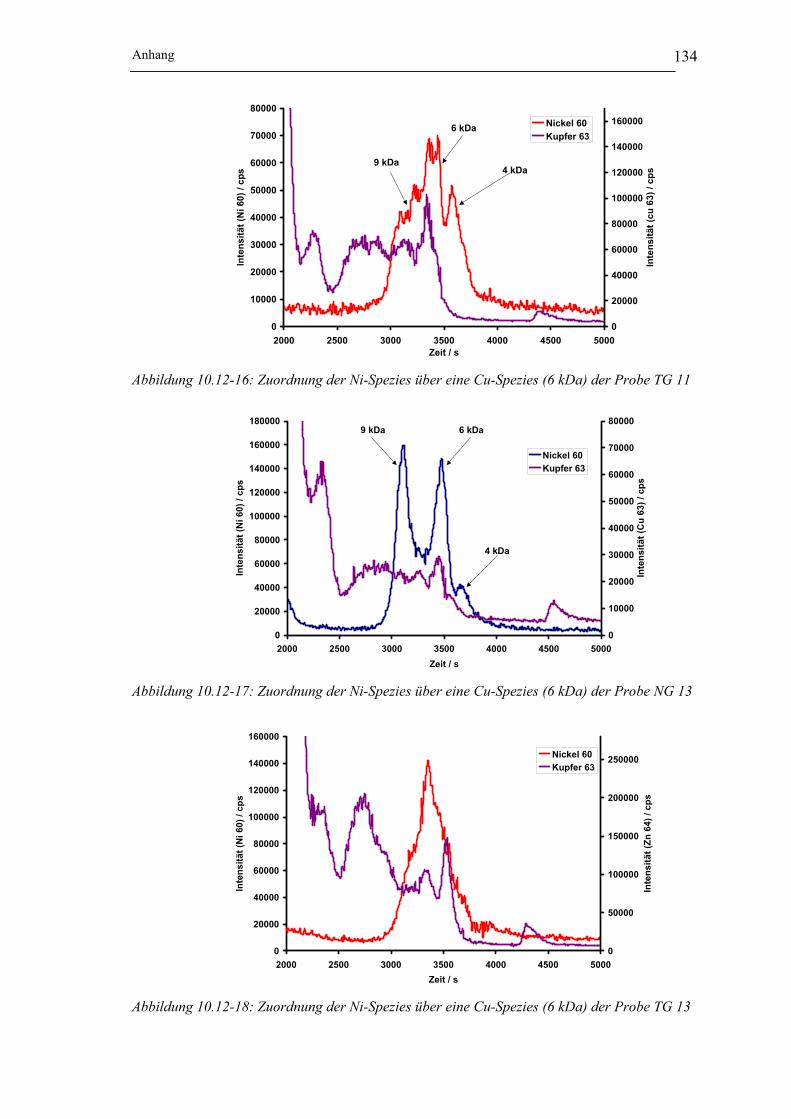
Anhang 134
Abbildung 10.12-16: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 11
Abbildung 10.12-17: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 13
Abbildung 10.12-18: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 13
0
10000
20000
30000
40000
50000
60000
70000
80000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
20000
40000
60000
80000
100000
120000
140000
160000
Inte
nsitä
t (cu
63)
/ cp
s
Nickel 60Kupfer 63
9 kDa
6 kDa
4 kDa
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
10000
20000
30000
40000
50000
60000
70000
80000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa 6 kDa
4 kDa
0
20000
40000
60000
80000
100000
120000
140000
160000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
50000
100000
150000
200000
250000
Inte
nsitä
t (Zn
64)
/ cp
s
Nickel 60Kupfer 63
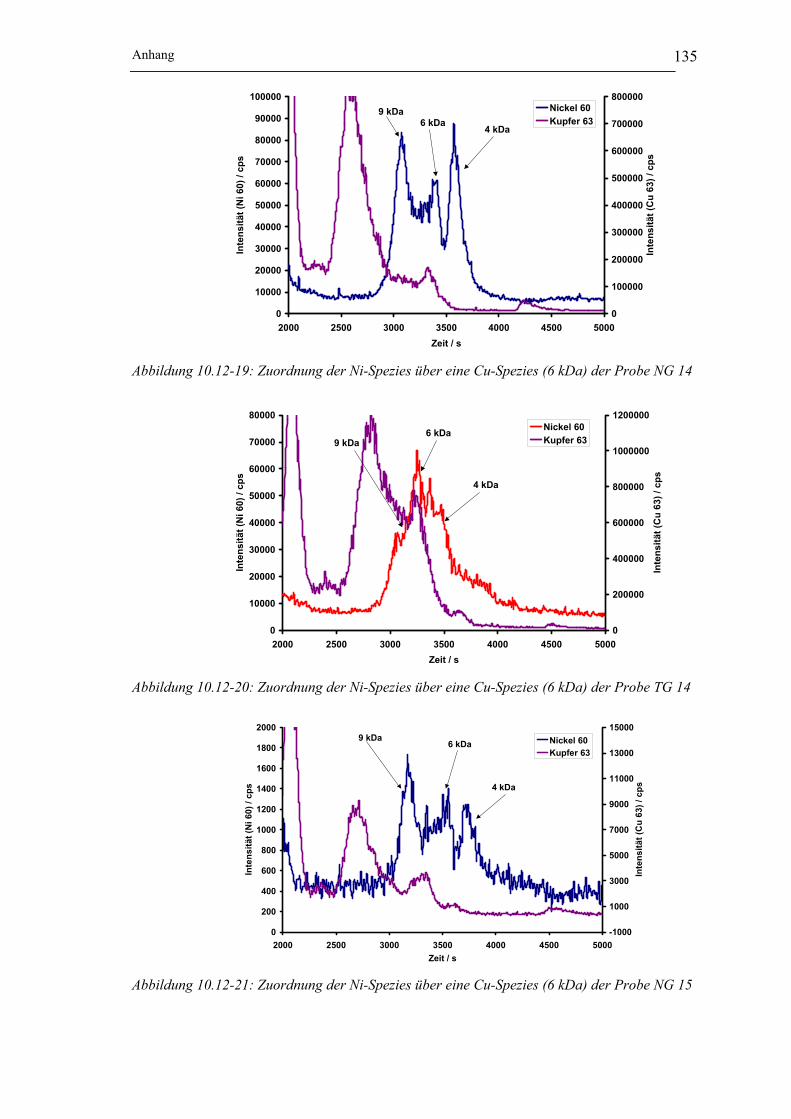
Anhang 135
Abbildung 10.12-19: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 14
Abbildung 10.12-20: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 14
Abbildung 10.12-21: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe NG 15
0
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
100000
200000
300000
400000
500000
600000
700000
800000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa6 kDa 4 kDa
0
10000
20000
30000
40000
50000
60000
70000
80000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
200000
400000
600000
800000
1000000
1200000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 639 kDa
6 kDa
4 kDa
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
-1000
1000
3000
5000
7000
9000
11000
13000
15000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa6 kDa
4 kDa
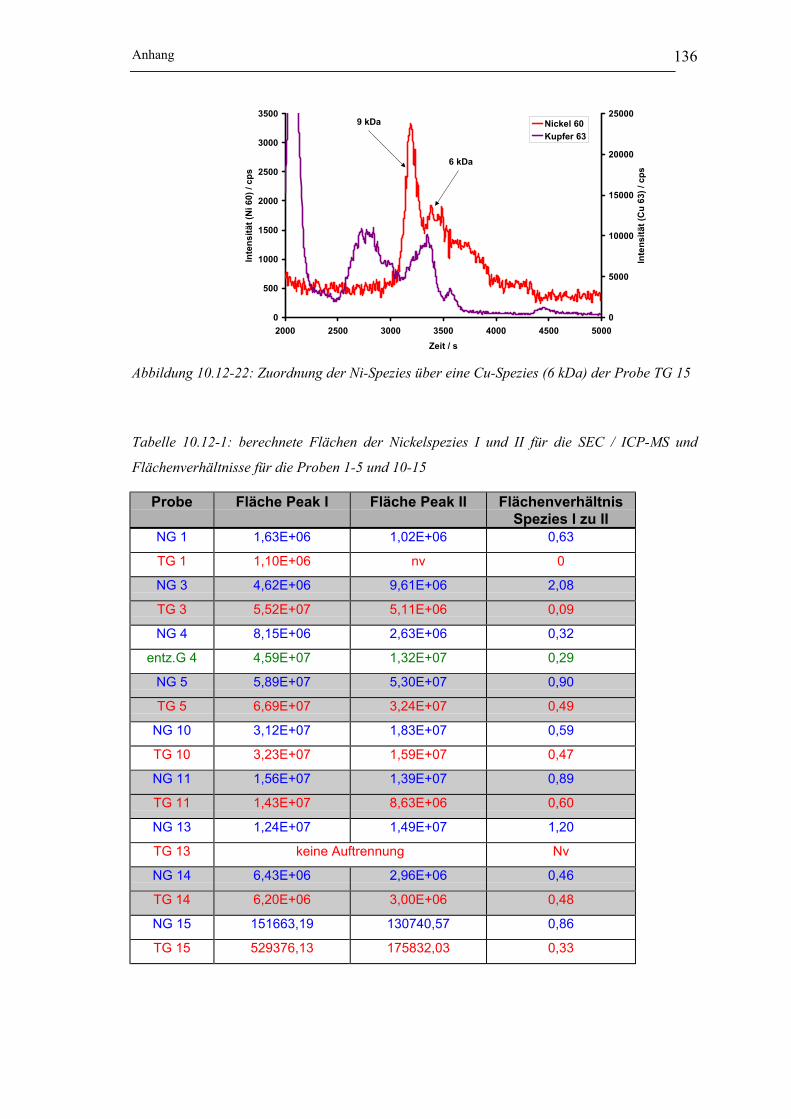
Anhang 136
Abbildung 10.12-22: Zuordnung der Ni-Spezies über eine Cu-Spezies (6 kDa) der Probe TG 15
Tabelle 10.12-1: berechnete Flächen der Nickelspezies I und II für die SEC / ICP-MS und
Flächenverhältnisse für die Proben 1-5 und 10-15
Probe Fläche Peak I Fläche Peak II FlächenverhältnisSpezies I zu II
NG 1 1,63E+06 1,02E+06 0,63
TG 1 1,10E+06 nv 0
NG 3 4,62E+06 9,61E+06 2,08
TG 3 5,52E+07 5,11E+06 0,09
NG 4 8,15E+06 2,63E+06 0,32
entz.G 4 4,59E+07 1,32E+07 0,29
NG 5 5,89E+07 5,30E+07 0,90
TG 5 6,69E+07 3,24E+07 0,49
NG 10 3,12E+07 1,83E+07 0,59
TG 10 3,23E+07 1,59E+07 0,47
NG 11 1,56E+07 1,39E+07 0,89
TG 11 1,43E+07 8,63E+06 0,60
NG 13 1,24E+07 1,49E+07 1,20
TG 13 keine Auftrennung Nv
NG 14 6,43E+06 2,96E+06 0,46
TG 14 6,20E+06 3,00E+06 0,48
NG 15 151663,19 130740,57 0,86
TG 15 529376,13 175832,03 0,33
0
500
1000
1500
2000
2500
3000
3500
2000 2500 3000 3500 4000 4500 5000Zeit / s
Inte
nsitä
t (N
i 60)
/ cp
s
0
5000
10000
15000
20000
25000
Inte
nsitä
t (C
u 63
) / c
ps
Nickel 60Kupfer 63
9 kDa
6 kDa
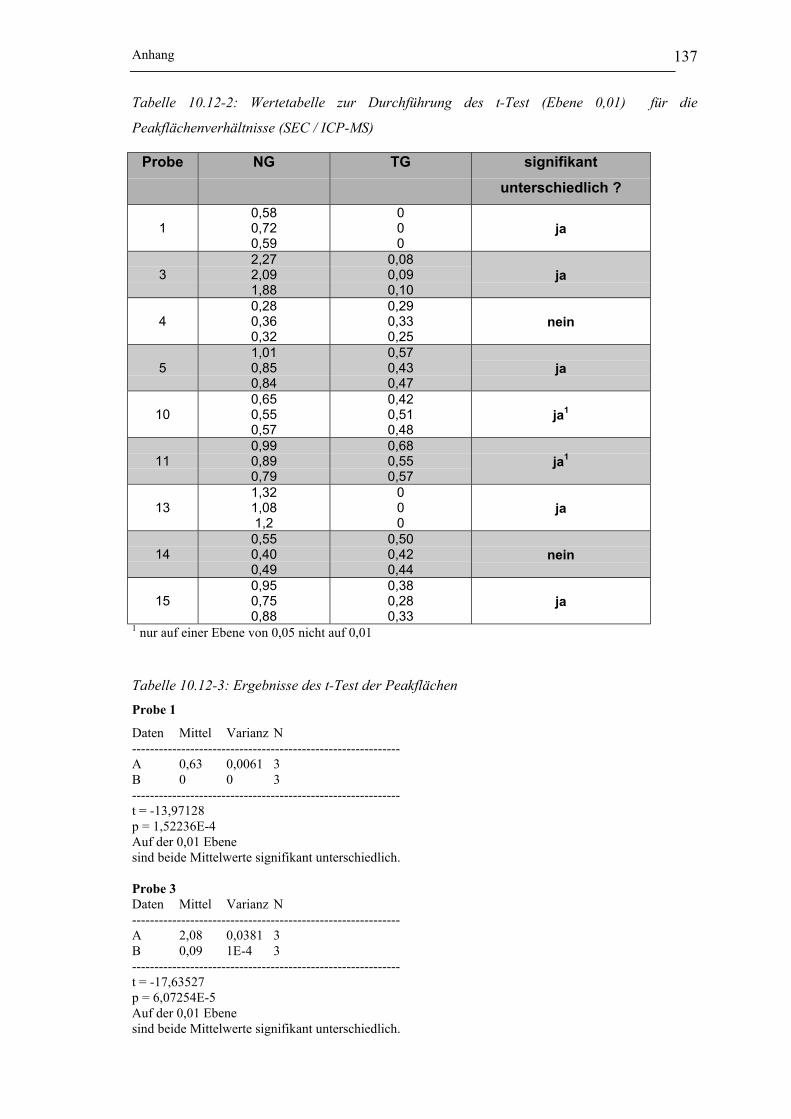
Anhang 137
Tabelle 10.12-2: Wertetabelle zur Durchführung des t-Test (Ebene 0,01) für die
Peakflächenverhältnisse (SEC / ICP-MS)
Probe NG TG signifikantunterschiedlich ?
10,580,720,59
000
ja
32,272,091,88
0,080,090,10
ja
40,280,360,32
0,290,330,25
nein
51,010,850,84
0,570,430,47
ja
100,650,550,57
0,420,510,48
ja1
110,990,890,79
0,680,550,57
ja1
131,321,081,2
000
ja
140,550,400,49
0,500,420,44
nein
150,950,750,88
0,380,280,33
ja
1 nur auf einer Ebene von 0,05 nicht auf 0,01
Tabelle 10.12-3: Ergebnisse des t-Test der PeakflächenProbe 1
Daten Mittel Varianz N------------------------------------------------------------A 0,63 0,0061 3B 0 0 3------------------------------------------------------------t = -13,97128p = 1,52236E-4Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 3Daten Mittel Varianz N------------------------------------------------------------A 2,08 0,0381 3B 0,09 1E-4 3------------------------------------------------------------t = -17,63527p = 6,07254E-5Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.

Anhang 138
Probe 4
Daten Mittel Varianz N------------------------------------------------------------A 0,32 0,0016 3B 0,29 0,0016 3------------------------------------------------------------t = -0,91856p = 0,4103Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.
Probe 5
Daten Mittel Varianz N------------------------------------------------------------A 0,9 0,0091 3B 0,49 0,0052 3------------------------------------------------------------t = -5,9385p = 0,00403Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 10
Daten Mittel Varianz N------------------------------------------------------------A 0,59 0,0028 3B 0,47 0,0021 3------------------------------------------------------------t = -2,96923p = 0,04118Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.
Daten Mittel Varianz N------------------------------------------------------------A 0,59 0,0028 3B 0,47 0,0021 3------------------------------------------------------------t = -2,96923p = 0,04118Auf der 0,05 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 11
Daten Mittel Varianz N------------------------------------------------------------A 0,89 0,01 3B 0,6 0,0049 3------------------------------------------------------------t = -4,11496p = 0,01467Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.

Anhang 139
Daten Mittel Varianz N------------------------------------------------------------A 0,89 0,01 3B 0,6 0,0049 3------------------------------------------------------------t = -4,11496p = 0,01467Auf der 0,05 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 13
Daten Mittel Varianz N------------------------------------------------------------A 1,2 0,0139 3B 0 0 3------------------------------------------------------------t = -17,62928p = 6,08071E-5Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.
Probe 14
Daten Mittel Varianz N------------------------------------------------------------A 0,48 0,0057 3B 0,47 0,0049 3------------------------------------------------------------t = -0,16823p = 0,87456Auf der 0,01 Ebenesind beide Mittelwerte NICHT signifikant unterschiedlich.
Probe 15Daten Mittel Varianz N------------------------------------------------------------A 0,86 0,0103 3B 0,33 0,0025 3------------------------------------------------------------t = -8,11393p = 0,00125Auf der 0,01 Ebenesind beide Mittelwerte signifikant unterschiedlich.





























