Embed Size (px)
Citation preview

484 J. SCItORM~LLER und G. BRESSAU :
5Iach RIBI~REAU-GAYON soll Vitis berlandieri keine Diglueoside enthalten. Diese Art wurde yon uns nicht geprfift; sie dfirfte allerdings ffir die Weinproduktion keinerlei Bedeutung haben, da es davon keine Ertragshybriden gibt. Bei Vitis labrusca mani- festiert sich der Wildrebencharakter nur im Malvidindiglucosid ; die anderen Digl.uco- side fehlen.
Diese hier aufgeffihrten Verfahren sind ein sicherer Weg zum exakten Nachweis yon Wildrebenfarbstoffen in Beeren und Weinen der in Europa im Ertrag befindlichen roten Hybriden. Den mit der Prfifung betrauten staatlichen Stellen wird diese Methode zur Anwendung empfohlen.
Zusammen/assung
Es wird die Methode des Nachweises yon rotem Hybridenwein nach J. und P. R I B ~ A v - G A ¥ o N diskutiert. Dieses Verfahren hat sich bei unseren Untersuchungen bew~hrt, jedoch wurden einige methodische Erweiterungen angeffigt. Es zeigte sich bei den zweidimensional entwickelten Chromatogrammen~ dab neben den fiblichen Nachweisreagentien AICI a und 5IH a noch Benedicts Reagens notwendig ist, um alle Wildrebenanthocyane zu erfassen. Um vornehmlich bei ~lteren I%otweinen die stSrenden braunen Abbauprodukte auszuschalten, empfiehlt sich der n-Butanol- extrakt. Die Extraktionsmethode gew~hr]eistet auch eine schonende Anreicherung der Anthocyane yon farbsehwaehen Rotweinen und ist deshalb dem Eindampfen vor- zuziehen. Die Diglucoside der Wildrebenanthocyanidine sind in roten Hybriden- weinen wesentlich haltbarer als die Monoglucoside der Vitis vini/era-Sorten. Der Nachweis yon Malvin allein wird als nieht ausreichend ffir eine genaue Charakterisie- rung eines Hybridenweines oder eines Verschnittes angesehen, da die fibrig.~n charak- teristischen Anthocyane Aufschlu~ fiber den Grad des Wildrebenanteiles geben kSnnen. Aus diesen Grfinden wird auf den Wert der zweidimensionalen Papier- chromatographie nochmals hingewiesen.
Phosphate und organische Phosphorverbindungen in Lebensmitteln
V I I I . Mitteilung
Die pr~iparative Darstellung yon Inositphosphors~iureestern Von
J. SCHORM('TLLER und G. BaESSAU*
Mitteilung aus dem Institut ]i~r Lebensmittelchemie und Lebensmitteltechnologie der Technisehen Universitiit Berlin**
Mit 1 Text~bbfldung
(Eingeffangen am 12. August 1960)
In einer vorhergehenden Mitteilung 1 bat ten wir ein Trennverfahren zur Identi- fizierung der lnositphosphors~ureester besehrieben, die bei der Hydrolyse des Ino- sithexaphosphates auftreten. Die im folgenden gesehilderten Versuehe besehaftigen sieh mit der prgparativen Gewinnung der einzelnen Ester, insbesondere auch mit der
* Herrn Professor Dr. Dr. h. c. RIcgngw K~J~s zum 60. Geburtstag gewidmet. ** Die Untersuehungen wurden dureh den Verband der Chemisehen Industrie gef6rdert. Wir
danken hierfiir such an dieser Stelle. 1 SC~OgMOLLEg, J., u. G. BRESSAU: Diese Z. 113, 387 (1960).

Phosphate und organische Phosphorverbindungen in Lebensmitteln. VIII 485
Abtrennung yon Isomeren, wie sie yon uns frfiher (vgl. 7. Mitteilung) beobachtet worden waren. Da die enzymatisehe Hydrolyse des Hexaphosphates die Bildung des iso-Pentaphosphates in nennenswertem Ausmaft nicht erwarten lieft ~, das Verhalten dieses Esters gegeniiber Inositphosphors~Lureester spaltenden Fermenten aber yon besonderem Interesse sehien, wurde fiir die Gewinnung der einzelnen Inositphosphor- s/~ureester der ehemische Hydrolyseweg gew/ihlt.
a) Abhdngi!#ceit des Verlau]es der chemischen Phytinsiiurehydrolyse yore p~ des Realctionsmilieus
Bei systematischen Untersuchungen fiber die chemische Hydrolyse der Phytin- s/iure fanden FLOURY ~ sowie I)ESJOBE~T und FLEUI~EI~T 3, dab IHP* in alkalischer LSsung (pH 11,7) und bei Temperaturen bis zu ]00°C praktisch nicht angegriffen wird. Erhbht man die Wasserstoffionenkonzentration, so tr i t t Esterspaltung ein, die bei pH 3--4 ein Maximum erreicht. Mit zunehmender Aciditiit geht die Hydrolyse- geschwindigkeit bis zu einem Minimum bei pH 1 zurfick und steigt bei weiterer Acidit/~tserhShung wieder an. Ein gleichartiger Hydrolyseverlauf wurde auch bei einigen anderen Phosphors~ureestern beobachtet 4. Bei den niederen Estern, vor allem im alkalisehen Bereieh, verl/~uft die Hydrolyse jedoeh anders. Wie D~SJOBnl~T zeigte, erreieht die Spaltungsgesehwindigkeit ffir IMP bereits bei pl~ 7,5 den Null- wert. Darauf grfindet sieh tin Verfahren zur Gewinnung yon IMP aus Na-Phyta t 5, worauf sp/~ter (S. 487) noeh eingegangen wird. Der Diphosphorsaureester wird ober- halb yon pI~ etwa 7,7 nieht mehr dephoslohoryliert, so daft nach mehrt/igigem Kochen einer HexaphosphatlSsung bei p~ 9--8,3 a]s Hauptprodukt IDP entsteht (S. 488).
Versuche zur Darstellung von IHP-Abbauprodukten durch ehemisehe Hydrolyse haben wir zun/ichst naeh Angaben yon ARNOLD 6 durchgeifihrt. Das Veriahren, bei dem Phytins£urelSsungen unter Druck auf Temperaturen fiber 100 ° C erhitzt werden, hat den Nachteil, daft sich - - vor allem beim Einsatz grSfterer Substanzmengen -- der Hydrolyseverlauf schlecht beherrsehen l~Li~t. Neben Druck, Temperatur und Er- hitzungsdauer ist das pI~ der l%eaktionslSsung yon entseheidender Bedeutung f fir den IHP-Abbau.
Der Hydrolyseverlauf wurde mit Hilfe der Irtiher besehriebenen 7 Papierchroma- tographie verfolgt, die eine ausreiehende Orientierung erlaubt lind gegenfiber der zwar genaueren S/~ulenchromatographie Material und Zeit spart. Es zeigte sich, daft I H P bei prI 1 erheblich langsamer abgebaut wird als bei pit 3. Anderungen der Wasser- stoffionenkonzentration in dem genalmten Bereich bedingen vor allem zeitliche Ver- schiebungen im Hydrolyseverlauf.
Neuere Untersuehungen 6, s fiber die chemisehe IgP-Hydro lyse ergaben, daft bier /~hnlich wie bei dem in der vorhergehenden Arbeit erSrterten enzymatisehen Abbau
* Abkiirzungen der einzeinen Inositphosphors/~ureester, vgl. diese Z. 105, 400 (1957). Zit. S. 484, Anm. 1.
2 FLEImY, P.: C. I~. Acad. Sei. (Paris) 221, 416 (1945). 3 D~SJO~EI~T, A., u. P. FI~EVI~E~T: Bull. Soe. claim, biol. (Paris) 36, 475 (1954).
S~LIM, M., u. P. LEDUC: C. R. Acad. Sci. (Paris) 248, 1187 (1959). 5 DESZO~E~T, A. : Bull. Soc. china, biol. (Paris) 36, 1293 (1954).
All,COLD, P. W. : Bioehim. biophys. Aeta 19, 552 (1956). 7 Se~oI~fiiLLEI% J., u. G. WiYl~I)IG: Diese Z. 105, 397 (1957). - - G. W/3RDm: Untersuchungen
zur papierchromatographischen Trennung und Identifizierung der Inositphosphorsi£uren. Disser- tation, Techn. Univ. Berlin 1958 (D 83).
s D~SJO~RT, A., u. F. PET~.K: C. R. Aead. Sci. (Paris) 241, 1343 (1955); Bull. Soc. china. biol. (Paris) 38, 871 (1956).
32a

486 J. SC~On~iiLL~ und G. BlCESSAU:
intermedi£r eine Anreiehernng bes t immter Inosi tphosphors/ iureester eintr~tt. Welter- bin zeigte es sich, d~l~ die Bildung yon Pen t~phospha ten bis zur Abspal tung yon 2 0 - - 4 0 % des Esterphosphors zunimmt. Gleichzeitig werden bereits grSf~ere Mengen un I T e P gelunden, w£hrend niedere Inos i tphosphute in nennenswerten Mengen nicht ~nftreten. Bei Einhal tung des erw~hnten Hydrolysegrades miil~te ~lso die Darste]lung yon Pen t~phospha ten und I T e P in relutiv guter Ausbeute m6glich sein. Wie schon erw~hnt, ist die ~uBer yon ARNOLD auch yon anderen Antoren ~ benutz te Hydrolyse im Einschlul~rohr besonders bei f r o z e n Subs tanzmengen umst~ndl ich und schwer zu beherrschen. Dagegen ffihrte das Kochen unter l%fickflul~kfihlung zu gut repro- duzierb~ren Ergebnissen.
b) Darstellung und Isolierung von Inosithexa-, -isopenta-, -penta- und -tetraphosphat Zur Ermitt]ung der optimalen Hydi-olysedauer wurden einige Orientierungsversuehe dureh-
geffihrt. Papierchromatographisehe Prfifung des Hydrolyseganges zeigte, dal] ffir optimale Bildung der Tetra- und Pentaphosphate zweckm~l~ig eine 0,6 m-Phytins~urelSsung 100 min lang zu kochen ist.
Werden die Ester an der Chlorid/orm einer Dowex 2-S~ule (15 ml) getrennt, so wird nieht alles Phosphat fixiert. Dieser Nachteil ]iei~ sich durch vorhergehende Umwandlung etwa eines Drittels der Chloridform des Harzes in die Acetat/orm vermeiden.
Folgendes Verfahren erm6glichte schliel~lich die Isolierung der hochphosphorylierten Ester in grSl3erem Ausma~:
Eine S~ule der GrSl]e 18 × 2,5 cm wurde mit 80 ml Dowex 2 (Chloridform) ge~fillt und das I-Lrz mit m-Natriumacetutl6sung zu etwa l/3 der Gesamtk~pazit~t in die Acetatform fibergeffihrt. 5 ml Phytins~urehydrolysat (3,1 retool) wurden am Austauseher fixiert. Nach dem Waschen mit
je 200 bis 250 mlWasser und
s~hf $t~rk
st~rR
~ sehr ~/en/~
I,~P :P? f-lPf /#f 0,2 n-HC1 zur Entfernung
~ ~ / t t t v°n an°rganisehem Ph°s" phat und niederen Estern (einsehliel~lieh ITrP) wurde hinter diese S~ule eine 15 ml-Dowex 2-S~ule (Chlo- ridform) gesehaltet. Bei der ~~ / tt l_ Gradientelution enthielt das
r ] Mischgefgg anfangs 1] 0,25n- ¢0 ~0 :Z~ :~: HC1, das Vorratsgef~l~ n-
HC1. Die FlieBgesehwindig- fruktl~n keit betrug bis zu 1 ml/min;
das Eluat wurde in 8-ml- Fraktionen gesammelt. Der Phosphattest ffir die einzel-
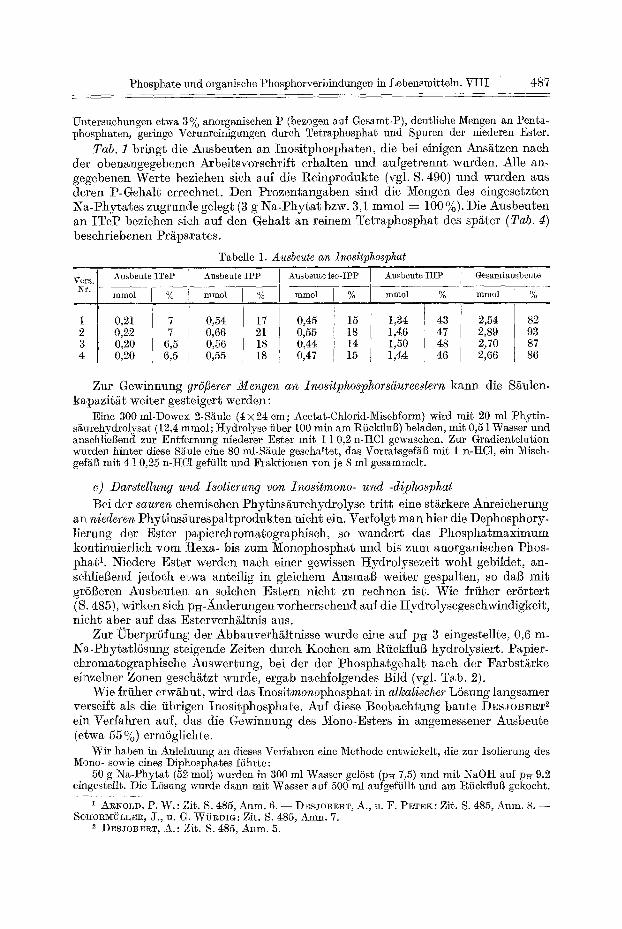
Abb. 1. Prdparative T~'ennung yon ITeP , I P P , iso-IPP und I H P dutch Gradiestelution mit Salzsgiure an Dowex 2
nen Proben zeigte, dal~ hier die Trennung yon ITeP, IPP, iso-IPP und IHP befriedigend gelang (Abb. 1). Bei Wiederholung des Versuches traten die Ester jeweils in den gleichen Fraktions- bereichen auf, wobei teilweise Versehiebungen um drei bis vier Fraktionen vorkamen.
Die P-freien oder nut Spuren an P enthaltenden Fraktionen wurden verworfen, die iibrigen zu einem Fraktionsbereich geh6renden vereinigt, mi~ NaOK neutralisiert, die Ester mit ges~tig- ter Ba-Aee~atlSsung gefgllt und abzentrifugiert. Die Ba-Inositphosphate wurden im ttomogeni- sator mit Wasser gewaschen und zentrifugiert, bis kein Chlorid mehr naehweisbar war. Die volumi- nSsen Niedersehli~ge wurden nach dem Zentrifugieren (14000 U/rain, 5 rain) im Vakuum fiber NaOH getrocknet.
Im Gange dieser Trennung wurde aueh das Hexaphosphat in reiner Form isoliert. Handels- pr~parate yon Phytin enthalten ngmlich neben anorganischem Phosphat meist betr~ichtliehe Mengen an Spaltprodukten, die wahrscheinlich im Laufe der teehnischen Aufarbeitung entstehen und die in einer sp~teren Arbeit beschriebenen Enzymversuche stSrend beeinflussen. Wie aus Angaben yon S~Iwg 3 hervorgeht, lassen sich die begleitenden Este r dutch t~einigung des Hexa- phosphates fiber das Eisensalz nieht entfernen. Na-Phytat des Handels enthielt nach nnseren
1 Zit. S. 4~5, Anm. 8. 2 S~nT~, D. It., u. F. E. CLARK: Soil Sei. Proe. 16, 170 (1952). - - S~ITK, D.H.: Iowt~ State
Coll. J. Sci. 26, 287 (1952).
Phosphate und organisehe Phosphorverbindunge~ in Lebensmitteln. VIII 487
Untersuehungen etwa 3% anorganisehen P (bezogen auf Gesamt-P), deuthehe Mengen an Penta- phosphaten, gerh~ge Verunreinigungen durch Tetraphosphat und Spuren der niederen Ester.
Tab. 1 bringt die Ansbeuten an Inositphosphaten, die bei einigen Ansgtzen naeh der obenangegebenen Arbeitsvorsehri{t erhMten und aufgetrennt win'den. Alle an- gegebenen Werte beziehen sieh auf die geinprodukte (vgl. S. 490) und wurden aus deren P-Gehalt erreehnet. Den Prozentangaben sind die ~engen des eingesetzten Na-Phytates zugrunde gelegt (3 g Na-Phyta t bzw. 3,1 mmol = 100 %). Dis Ausbeuten an ITeP beziehen sieh auf den Gehalt an reinem Tetraphosphat des spgter (Tab. ~) besehriebenen Pr/£parates.
Tabelle 1. Ausbeute an Inosit ~hosphat
I Ausbeute ITel ) Ausbeute IPP Vers.
mmo [ 1%
2 0,22 0,66 21 3 0,20 6,5 0,56 18 4 0,20 6,5 0,55 18
Ausbeute iso-IPP
mmol %
0,45 15 0,55 18 0,44 14 0,47 15
Ausbeute I t IP
mmol %
1,34 43 1,46 47 1,50 48 1,44 46
Gesamtausbeute
mmol %
2,54 82 2,89 93 2,70 87 2,66 86
Zur Gewinnung ~r6[3erer Mengen an Inositphosphorsi~ureestern kann die S~ulen- ka~pazit/~t weiter gesteigert werden:
Eine 300 ml-Dowex 2-S~ule (4 X 24 em; Aeetat-Chlorid-Misehform) wird mit 20 ml Phytin- s~urehydrolysat (12,4 retool; Hydrolyse fiber 100 min am Rfickttu{3) beladen, re_it 0,5 1 Wasser und ansehlief]end zur Entfernung niederer Ester mit 1 1 0,2 n-I-IC1 gewaschen. Zur Gradientelution wurden hinter diese S~ule eine 80 ml-S//ule geschMtet, das Vorratsgef~B mit 1 n-HC1, ein Misch- gef~g mit 41 0,25 n-IIC1 geffillt und Fraktionen yon je 8 ml gesammett.
c) Darstellung und Isolierung von Inositmono- und -diphosphat Bei der sauren chemisehen Phytins~urehydrolyse t r i t t eine st/£rkere Anreieherung
an niederen Phytins~,urespMtprodukten nieht ein. Verfolgt man hier die Dephosphory- lierung der Ester papierehroma~ographisch, so wandert das Phosphatmaximum kontinuierlieh veto :~Iexa- bis zum 3/fonophosphat und bis zum anorganisehen Phos- phatL Niedere Ester werden naeh einer gewissea I-Iydrolysezeit wohl gebfldct, an- sehliel3end jedoeh e'~wa anteilig in gleiehem Ausma6 welter gespMten, so dab mit grSBeren Ausbeuten an solehen Estern nieht zu reehnen ist. Wie friiher er6rtert (S. 485), wirken sieh prr-~nderungen vorherrsehend auf die Hydrolysegesehwindigkeit, nieht aber auf das Esterverh/~ltnis aus.
Zur f)berpr/ifung der Abbauverh/~ltnisse wurde eine auf io~ 3 eingestellte, 0,6 m- Na-Phytatl6sung steigende Zeiten dureh Koehen am l~/iekfiug hydrolysiert. Papier- chromatographisehe Auswertung, bei der der Phosphatgehalt naeh der Farbst~rke einzelner Zonen gesch£tzt wurde, ergab naehfolgendes Bild (vgl. Tab. 2).
Wie friiher erw/~hnt, wird das Inositmonophosphat in alkalischer LSsung langsamer verseift Ms die fibrigen Inositphosphate. Auf diese Beobaehtung baute DESJOB~T ~ ein Verfahren auf, das die Gewinnung des Mono-Esters in angemessener Ausbeute (etwa 55 %) erm6glichte.
Wir haben in Anlehnung an dieses Verfahren eine Methode entwiekelt, die zur Isolierung des Mono- sowie eines Diphosphates fiihrte-
50 g Na-Phytat (52, reel) wurden in 300 ml Wasser gel5st (p~ 7,5) und mit Na0H auf p~ 9,2 eh~gestellt. Die LSsung wurde dann mit Wasser auf 500 ml aufgeffillt und am Riiekflug gekocht.
An~oL~), P. W.." Zit. S. 485, Anm. 6. - - D~sJo~T, A., u. F. PETEK: Zit. S. 485, Anm. 8. - - Se~o~iiLL~, J., u. G. WiJ~Dm: Zit. S. 485, Anm. 7.
2 DES~O~Ea% A.: Zit. S. 485, Anm. 5.
4 8 8 J. SCKOR~CIULLER und G. BRESSAU :
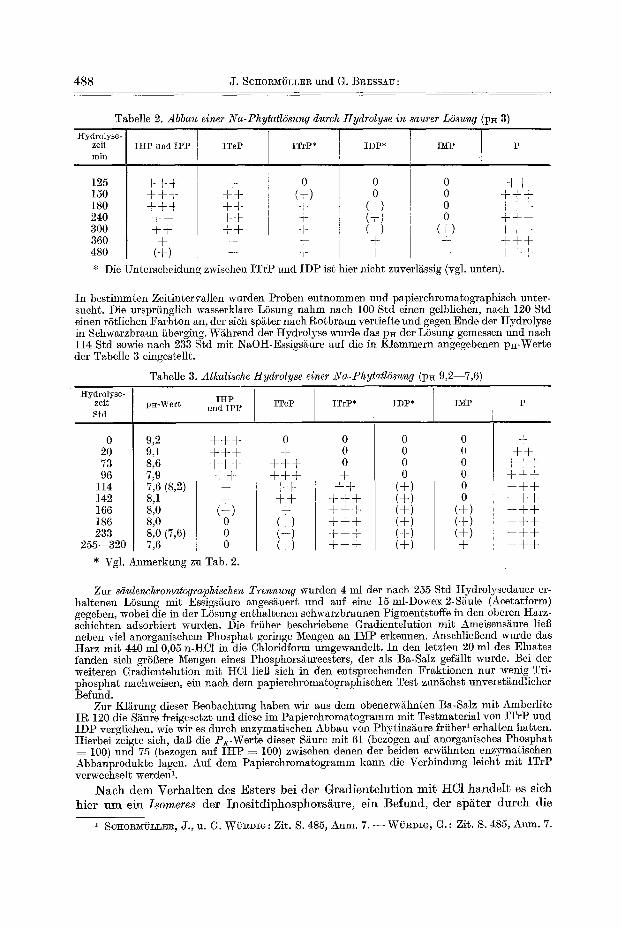
Tabelle 2. Abbau einer iVa-Phytatl6sung durch Hydrolyse in saurer LSsung (pE 3)
Hydrolyse- zeit rain
125 150 180 240 300 360 480
IKP und IPP
+++ ++÷ +++ ÷+ ++ + (+)
ITeP
÷ + + + + ÷ ÷ + + + +
ITrP*
0 (+) ÷ ÷ ÷ ÷ +
I I IDP*
0 0
(+) (+) (+) ÷ ÷
IMP
0 0 0 0
(+) + +
+ ÷ + ÷ ÷ + ÷ ÷ + ÷ ÷ + ÷ ÷ + + + + ÷ ÷
* Die Unterscheidung zwischen ITrP und IDP ist hier nicht zuverl£ssig (vgl. unten).
In bestimmten Zeitintervallen wurden Proben entnommen und papierchromatographiseh unter- sucht. Die ursprtinglich wasserklare LSsung nahm nach 100 Std einen gelblichen, nach 120 Std einen rStlichen Farbton an, der sieh sp£ter nach Rotbraun vertiefte und gegen Ende der Hydrolyse in Schwarzbraun fiberging. W~hrend der Hydro!yse wurde das pH der LSsung gemessen und nach 114 Std sowie nach 233 Std mit NaOg-Essigs~iure auf die in Klammern angegebenen p~-Werte der Tabelle 3 eingestellt.
Tabelle 3. Alkalische Hydrolyse einer Na-Phytatldsung (p~ 9,2--7,6)
Hydrolyse- zeit Std
0 20 73 96
114 142 166 186 233
255--320
IHP p~-Wert und IPF
9,2 ÷ + + 9,1 ÷ ÷ ÷ 8,6 ÷ ÷ + 7,9 ÷ + 7,6 (8,2) ÷ 8,5 + 8,o (+) 8,0 0 8,0 (7,6) 0 7,6 0
* Vgl. Anmerkung zu Tab. 2.
ITeP
o ÷
+ ÷ + + + + ÷ ÷ + + ÷
(+) (+) (+)
ITrP*
0 0 0 + ++ +++ +++ +++ +++ +++
IDP*
0 0 0 0
(+) (+) (+) (+) (+) (+)
IMP
0 0 0 0 0 0
(+) (+) (+) ÷
÷ + +
+ ÷ ÷ + + + + ÷ + + ÷ ÷ + + ÷ + ÷ ÷ ÷ ÷ ÷ + ÷ ÷
Zur 8iiulenchromatographischen Trennung wurden 4 ml der nach 255 Std Hydrolysedauer er- haltenen LSsung mit Essigs~iure anges~uert und auf eine 15 mLDowex 2-S£ule (Aeetatform) gegeben, wobei die in der LSsung enthaltenen sehwarzbraunen Pigmentstoffe in den oberen tIarz- schiehten adsorbiert wurden. Die friiher beschriebene Gradientelution mit Ameisens~iure ]iel~ neben viel anorganischem Phosphat geringe Mengen an IMP erkennen. AnschlieBend wurde das Harz mit 440 ml 0,05 n-HC1 in die Chlorid~orm umgewandelt. In den letzten 20 ml des Eluates fanden sieh grSl~ere Mengen eines Phosphorsitureesters, der als Ba-Salz gef~llt wurde. Bei der weiteren Gradientelution mit HC1 ]ieB sieh in den entsprechenden Fraktionen nut wenig Tri- phosphat naehweisen, ein nach dem papierehromatographisehen Test zun~ehst unverst~indlieher Befund.
Zur Kl~irung dieser Beobaehtung haben wir aus dem obenerw~hnten Ba-Sah mit Amberlite IR 120 die S~ure freigesetzt und diese im Papierehromatogramm mit Testmaterial yon ITrP und IDP verglichen, wie wir es durch enzymatischen Abbau yon Phytins£ure friiher I erhalten hatten. Hierbei zeigte sich, dal~ die Px-Werte dieser S~ure mit 61 (bezogen auf anorganisehes Phosphat = 100) und 75 (bezogen auf IHP ~ 100) zwischen denen der beiden erw~hnten enzymatischen Abbauprodukte lagen. Auf dem Papierehromatogramm kann die Verbindung leieht mit ITrP verweehselt werden 1.
N a c h d e m Verhalten des Esters bei der Gradientelut ion m i t HC1 handel t es sich hier u m ein Isomeres der Inositdiphosphors£ure, ein t~efund, der sp£ter durch die
1 SCHOBMI)LLER, J. , u. O. WI31%DIG : Zit. S. 485, Anm. 7. - -WOI~DIG, G.: Zit. S. 485, Anm. 7.

Phosphate und organische Phosphorverbindungen in Lebensmitteln. VIII 489
Elementaranalyse (vgl. S. 491) bestgtigt werden konnte. Chemisehe (alkalisehe) nnd enzymatisehe I-Iydrolyse unterseheiden sieh, ghnlieh wie im Falle der Pentaphos- phate, grunds~tzlieh voneinander; bei der alkalisehen Hydrolyse t r i t t ein i so- IDP auf, , ,normales" bei der enzymatisehen Spaltung gewonnenes Diphosphat entsteht bier in nur sehr geringen Mengen oder fiberhaupt nicht.
Ftir die Konst i tu t ionsermit t lung der Diphosphate ergeben sieh /thnliehe Sehwierig- keiten wie fiir die der Pentaphosphate . Coul~TOIS u. MitarbA nahmen an, dab die enzymatisehe Phyt insgurehydrolyse naeh Freisetzung einer Phosphatgruppe in meta- Stellung zu dieser ersten Spaltstelle fortsehreite. Falls dieses Prinzip ftir den gesamten enzymatisehen I tydrolysever lanf Gfiltigkeit bes~Be, m/il3te es sich bei dem, ,normalen" I D P nm ein Inosit-2,4- bzw. nln das enantiomorphe Inosit-2,6-diphosphat handeln. Die Phosphatgruppe in 2-Stellnng seheint n/~mlieh bis zuletzt am Inositmolektil zu verbleiben. F/Jr das i so-IDP kgme dann nnr die Konst i tu t ion eines Inosit- l ,2- bzw. -2,3- oder -2,5-diphosphates in l~rage. WeiCere ErSrternngen zu diesem Problem sollen einer folgenden Arbeit vorbehal ten bleiben, die sieh mit dem enzymatisehen Abbau definierter Inositphosphors/iureester beschgftigen wird.
Zur l~riiparativen Gewinnung von Inositmonophosphat und iso-Diphosphat wurden 180 ml (18,5 retool) eines dureh alkalische Spaltung iiber 277 Std gewonnenen IHP-tIydrolysates auI eine grol3e Sgule (300 ml Dowex 2 ; Acetatform) gegeben, das Harz anschliel3end mit Wasser gewaschen und mit 3 n-Ameisens~iure eluiert. Die im ttydrolysat enthaltenen sehwarzbraunen Pigmente wer- den auch hier quantitativ am Harz adsorbiert, beeintrgchtigen die Kapazitgt des Austausehers nicht und werden dureh Ameisen- oder SMzs~ure nicht vom Harz gelSst. Nachdem 480 ml phos- phorfreie Fliissigkeit die S~iule passiert hatten, wurden 750 ml monophosphathaltiges Eluat er- halten. Anschliegend entfernt man anorganisches Phospha~ mig 3,2 1 5 n-Ameisens~ure und gibt sehlieglieh 5 10,1 n-ttC1 durch die S/iule. Die letzten 700 ml dieses Eluates wurden im Vakuum bei Raumtemperatur auf 113,0 ml eingeengt, mit NaOH auf p~ 7 eingestellt nnd mit gesgttigter Ba- Acetatl6sung versetzt. Das ausgefgllte Ba-iso-IDP wurde, wie bei den hoehphos10horylier~en Estern besehrieben, gewasehen, zentrifugiert und getroeknet.
Ausbeute an reinem Ba-iso-IDP ~ 9,2 mmol (50%). Die Monolohosphat enthaltende LSsung wurde ebenfMls im Vakuum konzentriert, mit NaOI~I
neutralisiert, mit gesgttigter Pb-AeetatlSsung versegzt, der Niedersehlag abzentrifugiert, gewaschen und getroeknet.
Ausbeute an IMP-Reinprodu~t = 3,6 mmol (20%). Gesamtausbeute an IMP und iso-IDP also 70%.
Zur Darstellung yon I M P allein wurden in einem weiteren Versueh 500 ml einer 0,1 mol-Na- PhytatlSsung wie oben beschrieben hydrolysiert. Die Hydrolyse durehl~uft alle Ir/iher gesehil- derten Farbstufen und entspriehg weitgehend der in Tab. 3 gegebenen Darstellung. Zur vollst~n- digen Spaltung yon iso-lPP ist es jedoeh notwendig, das Reaktionsgemisch vor Beendigung der Hydrolyse noch etwa 80 Std bei p~ 7,5 zu kochen 2. Die ReaktionslSsung wurde iiber Nacht bei 0°C aufbewahrt, wobei ein Teil des anorganisehen Phosphates auskristallisierte. 200 ml der iiberstehenden Fliissigkeit (20 mmol Inositphosphat) wurden auf eine 300 ml-Dowex 2-Sgule ge- geben, IMP mig Ameisens/iure veMr/ingt und als Bleisalz isoliert. Ausbeute etwa 53 %.
Allem Anschein naeh handel~ es sieh bei dem isolier~en Produkt um das Inosi t- 2-monophosphat. A m Modell des m-Inosites (Sesselform) lggt sieh zeigen, dab sich nur die Hydroxylgruppe am C2-Atom in axialer Stellung befindet. Die tibrigen itinf Hydroxy lgruppen liegen in der Ebene des Kohlenstoffringes. B g o w ~ und HArm s nehmen an, dal~ die ffinf ~qnatorialen Phosphatgruppen im IHP,Molekii l raseher als die axiale Gruppe abgespalten werden und damit eine Anreicherung des 2-Isomeren eintritt. Diese Annahme wird dureh Ergebnisse gest/itzt, die bei enzymatisehen und
Cou~wom, J. : 2 ~ Journges Biochim. Franeo-Suisses, Gen~ve, 11.--13. Mai (1951). - - COUR- TOm, J., u. M. MASSO~r: Bull. Soe. ehim. biol. (Paris) ll~, 314; 326 (1950).- Cotr~wom, J., u. G. Jos~rm ]~ull. Soc. chim. biol. (Paris) 30, 610 (1948).
Vgl. auch D~sJOSEa~, A. : Zit. S. 485, Anm. 5. Bgow~r, D. M., u. G. E. HAnn: J. chem. Soe. 19~9, 357.

4 9 0 J . SCttOI~¢IiJLLER u n d G. BRESS~kU :
I I~-spekt rographisehen Unte rsuchungen versehiedener Monophospha tp rgpa ra t e er- ha l t en wurden und die in einer folgenden Arbe i t e rSr ter t werden sollen. Vgl. do r t die en tspreehenden Formelb i lder .
d) Versuche zur Darstellung yon Inosit-triphosphat Die Darstellung yon ITrF durch chemisehe Hydrolyse des Hexaphosphates bereitet groBe
Schwierigkeiten, da dieser Ester zwischen p~ 1 und 9 sehne]ler gespalten wird als das Tetra- phosphat 1. Eine 0,1 m-N~a-Phytatl6sung wurde wie schon gesehildert am RfiekfluB gekocht und das p~ wghrend der 54 Std dauernden Hydi'o]yse zwischen 8,3 und 9 reguliert. Das Endprodukt der Hydrolyse wurde zungehst papier- und s~ulenchromatographiseh fiberprfift. Neben IMP und geringen Mengen der h6heren Ester wurde iso-IDP als tIauptprocluk~ gefunden. Zur Trenaung wurden dann 200 ml Hydrolysat (21 mmol Inositphosphate) auf eine 300 ml-S~ule gegeben; mit 6 1 0,12 n-HC1 wurden anorganisches Phospbat und iso-IDP entfernt. Durch Elution mi~ 1 1 0,23 n-HC1 konnten aus den esterhaltigen Fraktionen lediglieh 900 mg ,,Inesit-Triphosphaf" in Form des Ba-Salzes isoliert werden. Wie die Elementaranalyse (Tab. 4) zeigt, bestand das Produkt etwa je zur Hglfte aus Ba-ITrP und Ba-ITeP. Die Ausbeute an Triphosphat betrug 0,42 mmol (2%), die an Tetraphosphat 0,39 mmol (2%).
In der Absicht, die Ausbeute an Triphosphat zu steigern, wurde ein enfsprechender Hydro]yse- versueh bei hSherer Hydroxylionenkonzentration durchgefiihrt.
Eine 0,1 mol-Na-Phytatl6sung wurde bei p~ 10,0--10,5 43 Sfd gekocht. Die sgulenehromato- graphisehe Analyse zeigte neben dem Vorkommen yon unvergndertem II-[P sowie der Bildung yon Penta-, Tetra- und Diphosi0haten, dal~ auch eine gewisse Anreicherung an ITrP eingetreten war. Eine 300 ml-S~ule wurde mit 200 ml dieses Hydrolysates (20 mmol Inositphosphate) beladen. Naeh Abtrennung yon anorganischem Phosphat und iso-IDP mit 41 0,13 n-]:IC1 wurde mit 0,25 n-HC1 duiert. Die ersten 500 ml des Eluates waren phosphorfrei; die folgenden, phosporhaltigen Frak- tionen(1200 ml) wurden mit NaOH neutralisiert. Der Ester wurde mit Ba-Aee~at gefgllt, die Fgllung abzen~ri~ugiert, gewasehen und getroeknet. Di~ Ausbeute an Reinprodukt (vgl. T~b. 4) betrug in zwei Versuchen 6 und 7 %, berechnet aus dem Phosphorgehalt. Die Elementaranalyse zeigte, dab aueh naeh diesem Verfahrensgang keine besseren Ergebnisse erzielt werden; die er- haltenen Produkte bestanden zu rund 42% aus ITrP und zu 58% ~us ITeP.
Wie frfiher erwghn~, ve rha l t en sieh einzelne Tri- und Te t r aphospha t e sgulen- chromafographiseh au~erordent l ich gleichart ig, so dal~ es n ieht mSglich ist, m i t Hilfe der yon uns bisher benn tz t en Trennungsmethode solche Es te r vone inander zu scheiden. Mit HiRe anderer E lu t ionsmi t t e l oder Aus tauseher lg[tt sieh jedoeh zweifel- los aneh die Trennung dieser I someren erreichen. Vor te i lhaf t dfir~te bier au~e rdem bei der E lu t ion die Verwendung entsprechender SalzlSsungen anstel le der Sguren sein; d a m i t wfirde du tch VerzSgerung der Es t e rhydro lyse wei tgehende Sehonung der Es t e r erreicht .
e) Reinigung und Analyse der Inositphosphate Die Inos i tphospha te , wie sie naeh der ehemisehen Phy t ins~urehydro lyse und n~eh
der sgulenehromatogr~phischen Trenmlng der t I y d r o l y s a t e als B~-S~lze ~usgefgllt worden w~ren, wurden nach ein und demselben Verfahren fiber ihre B~-Verbindungen gereinigt .
c~) Inosit-Monophosphat wurde in Form des Pb-Salzes (7 g) in Wasser unter Zusatz von Amberlite IIZ 120 (H+-Form, 16 ml) gel6st, das Gemisch auf eine mit 20 ml desselben Harzes geftillte Sgule (2,5 cm Z) gegeben mid mi~ Wasser naehgewasehen, bis das Einat nur noeh sehwaeh sauer (p~ 4) reagierte. Das bleifreie Eluat (Reaktion gegen Na2SO4-LSsung negativ) wurde mit gesgttigter Barytlauge neufralisiert (pH 7) und im Vakuum eingeeng~. Das Ba-IMIP wurde mit dem ffinlfachen Volumen an 96 %igem Xthanol gefgllt, abzentrifugiert und getroeknet.
Zur Isolierung der/reien Inosit-Monophosphors~iure wurden 5 g Ba-IMP unter Zusatz yon 15 ml Ambertife IR 120 wie oben gelSst und auf eine mit 15 ml Harz gefiillte Sgule gegeben. Diese wurde mit Wasser s~urefrei gewaschen. Die anfallende LSsung der freien S~ure (200 ml) wurde im Vakuum bei 3 5 ~ 0 ° C Badtemperatur auf 6 ml eingeengt, langsam unter Umsehfitteln mit 12 ml absolutem Athanol versetzt, die ausgefgll~e Sgure fiber eine Glasfri~te abgesaugf, mit
Cov~¢om, J. : Zit. S. 489, Anm. I.
Phosphate und organische Phosphorverbindungen in Lebensmitteln. VIII 491
Alkohol und ]4ther gewaschen und gebrocknetL Beim sehr langsamen Erhitzen (0,5* C/rain) sehmilzt die S~ture unter dem Sehmelzpunktsmikroskop (Mikroskopheiztiseh 350, E. Leitz, Wetzlar) unter Zersetzung bei 194--196 ° C. Angaben der Literatur liegen bei Stop = 195--197 ° C 2 und Stop = 196--I98 ° C a. Aus dem Fil~rat und der Wasehfliissigkeit .k.onn~en naeh Neutralisa~ion mit Bariumhydroxyd und Fallen mit dem doppelten Volumen an Athanol weitere Mengen an IMP in Form des Ba-Salzes gewommn werden.
fl) Die vorgereinigten Ba-Salze der i~brigen Ester wurden in etwas Wasser aufgesehlgmmt nnd mi~ soviel Amberlite I'_f~ 120 (H+-Form) verriihrt, dab sich das Salz 15ste. Die LSsung wurde fiber Blaubandfilter filtriert und zur Fiillung der Ester mit frisch bereiteter, gesgttigter Baryt- lauge versetzt (p~ 6--'7). Der Niederschlag wurde bei 15000 U/rain abzentrifugiert und im Vakuum getroeknet. Ba-iso-lDP ist in Wasser merklieh 15slieh; aus der Waschfliissigkeit dieses Esters kann nach dem Einengen im Vakuum eine weitere geringe Menge an Ba-Salz gewormen werden.
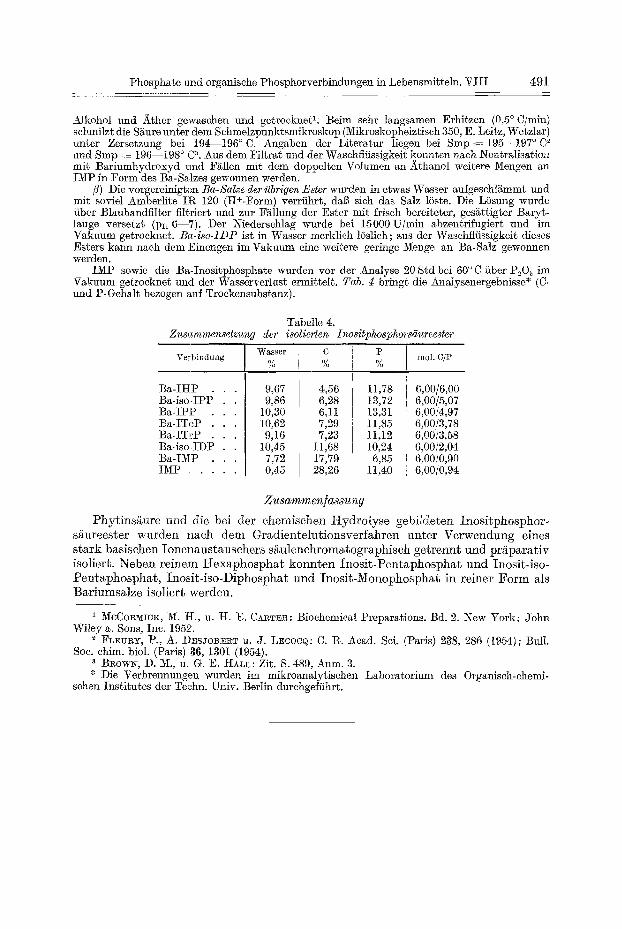
IMP sowie die Ba-Inositphospha~e wurden vor der Analyse 20 Std bei 60 ° C fiber PaO~ im Vakuum getrocknet und der Wasserverlust ermittelt. Tab. g bringt die Analysenergebni~sse* (C- und P-Gehalt bezogen auf Troekensubstanz).
Tabelle 4. Zusammensetzung der isolierten Inositphosphorsiiureester
Wasser C P Verbindung tool. C/P % % %
Ba-IHP Ba-isoJPP Ba-IPP Ba-ITeP Ba-ITrP Ba-iso JDP Ba-IMP IMP . . . . . .
9,67 9.86
10130 10,62 9,16
10,45 7,72 0,45
4,56 6,28 6,11 7,29 7,23
11,68 17,79 28,26
11,78 t3,72 13,31 11,85 11,12 10,24 6,85
11,40
6,00/6,00 6,00/5,07 6,00/4,97 6,00/3,78 6,00/3,58 6,00/2,04 6,00/0,90 6,oo/o,94
Zusammen]assung
Phyt insgure und die bei der chemischen Hydrolyse gebildeten Inosi tphosphor- s£ureester wurden nach dem Gradientelut ionsverfahren unte r Verwendung eines s tark basischen Ionenaustausehers s~ulenehromatographiseh ge t rennt u n d pr~parat iv isoliert. Neben reinem Hexaphosphat konn t en Inos i t -Pen taphospha t und Inosit-iso- Pentaphosphat , Inosi t - iso-Diphosphat und Inosi t -Monophosphat in reiner Fo rm als Bariumsalze isoliert werden.
1 McCoRMICK, M. H., U. H. E. CARTER: Biochemical Preparations. Bd. 2. New York: John Wiley a. Sons, Inc. 1952.
FLEV~r, P., A. DESaOB~T U. J. L~cOO~: C. ~. Acad. Sci. (Paris) 238, 286 (1954); Btfll. Soe. chim. biol. (Paris) 36, 1301 (1954).
B~ow~r, D. M., u. G. E. IL~LL: Zit. S. 489, Anm. 3. * Die Verbrennungen wurden im mikroanalytischen Laboratorinm des Organisch-chemi-
schen Institutes der Techn. Univ. Berlin durchgeffihrt.

![Phosphate Translocator of Isolated Guard-Cell Chloroplasts f ......["C]sorbitol as membrane-permeating and nonpermeating mark- ers and [32P]phosphate as tracer for phosphate. lhe affinities](https://img.pdfslide.org/doc/110x75/60796b9402c91a4d925da525/phosphate-translocator-of-isolated-guard-cell-chloroplasts-f-csorbitol.jpg)



























